WO2007123225A1 - オキサジアゾリジンジオン化合物 - Google Patents
オキサジアゾリジンジオン化合物 Download PDFInfo
- Publication number
- WO2007123225A1 WO2007123225A1 PCT/JP2007/058694 JP2007058694W WO2007123225A1 WO 2007123225 A1 WO2007123225 A1 WO 2007123225A1 JP 2007058694 W JP2007058694 W JP 2007058694W WO 2007123225 A1 WO2007123225 A1 WO 2007123225A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- esi
- methoxy
- compound
- fab
- oxadiazolidine
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
- C07D271/07—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles with oxygen, sulfur or nitrogen atoms, directly attached to ring carbon atoms, the nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to a novel oxadiazolidinedione compound or a pharmaceutically acceptable salt thereof useful as a pharmaceutical, particularly an insulin secretagogue, a prophylactic or therapeutic agent for diabetes.
- Diabetes mellitus is a disease whose main feature is chronic hyperglycemia and develops due to an absolute or relative lack of insulin action.
- insulin-dependent glycouria IDDM
- non-insulin-dependent diabetes mellitus NIDDM
- 8 cells is one of the main causes of the disease, especially postprandial hyperglycemia due to early insulin secretion disorder.
- sulfonylurea is the mainstream insulin secretagogue. It is known to cause hypoglycemia and to cause secondary ineffectiveness due to exhaustion of the spleen in long-term administration. . Moreover, it is difficult to suppress hyperglycemia after a power meal, which is effective for controlling blood sugar between meals.
- GPR40 is a G protein-coupled receptor that is highly expressed in ⁇ -cells of the spleen identified as a fatty acid receptor and reported to be involved in the insulin secretion of fatty acids.
- GPR40 receptor agonists are expected to correct postprandial hyperglycemia based on the insulin secretion-promoting action, so insulin-dependent diabetes mellitus (IDDM), insulin-independent Diabetes mellitus (NIDDM) and its boundary type (glucose tolerance / fasting blood glucose level abnormality) It is useful as an agent for the prevention / treatment of mild diabetes.
- IDDM insulin-dependent diabetes mellitus
- NIDDM insulin-independent Diabetes mellitus
- boundary type glucose tolerance / fasting blood glucose level abnormality
- Patent Document 1 it is reported that the compounds represented by the formula (A) including a wide range of compounds have GPR40 receptor modulatory activity and are useful as insulin secretagogues and preventive / therapeutic agents for diabetes. ing. However, there is no specific disclosure of a compound having an oxadiazolidinedione structure.
- ring P represents an optionally substituted aromatic ring
- ring Q represents
- Patent Document 2 reports that the compound represented by the formula (B) has a GPR40 receptor-modulating action and is useful as an insulin secretion promoter or a prophylactic or therapeutic agent for diabetes. However, there is no specific disclosure of a compound having an oxadiazolidinedione structure! /.
- Patent Document 3 it is reported that the compound represented by the formula (C) has a GPR40 receptor-modulating action and is useful as an insulin secretion promoter or a prophylactic or therapeutic agent for diabetes.
- the compound represented by the formula (C) has a GPR40 receptor-modulating action and is useful as an insulin secretion promoter or a prophylactic or therapeutic agent for diabetes.
- an oxadiazolidinedione compound represented by the formula (D) has a plasminogen activation inhibitor (PAI) -l inhibitory action, and is a thrombus and atrial fibrillation. It has been reported to be useful for the treatment of myocardial ischemia, diabetes and the like. However, the structure of part X is different from the compound of the present invention. Moreover, there is no description about the action on the GPR40 receptor.
- PAI plasminogen activation inhibitor
- Patent Document 5 reports that a compound having two oxadiazolidinedione structures represented by the formula (E) has an insulin sensitivity enhancing action and is useful for the treatment of diabetes. . However, there is no description about the action on the GPR40 receptor.
- Patent Document 6 it is reported that the oxazolidinedione compound represented by the formula (F) has a blood glucose lowering action and a blood lipid lowering action and is useful for the treatment of diabetes. ing.
- the ring corresponding to oxadiazolidinedione of the compound of the present invention is oxazolidindione.
- Patent Document 7 it is reported that the oxadiazolidinedione compound represented by the formula (G) has a hypoglycemic action and is useful for the treatment of diabetes.
- the ring corresponding to the A ring of the compound of the present invention is an oxadiazole ring. Also, the effect on GPR40 receptor is not described.
- Patent Document 8 it is reported that the compound represented by the formula (H) has a blood glucose lowering effect and is useful for the treatment of diabetes. However, there is no description of effects on the GPR40 receptor.
- Patent Document 9 reports that an oxadiazolidinedione compound represented by the formula CO has a hypoglycemic action and is useful for the treatment of diabetes.
- the ring corresponding to the heel ring of the compound of the present invention is oxazole or thiazole.
- GPR40 The description of the
- Non-Patent Document 2 an oxadiazolidinedione compound represented by the formula (L) is used. It has been reported that the product has a hypoglycemic effect and is useful in the treatment of diabetes. However, the ring corresponding to the A ring of the compound of the present invention is a (di) azole ring. Moreover, there is no description about the action to GPR40 receptor.
- Non-Patent Document 1 “Nature” (UK), 2003, 422 ⁇ , p.173-176
- Non-Patent Document 2 "European Journal of Medicinal Chemistry", (France), 2001, 36 ⁇ , p.31-42
- Patent Document 1 International Publication No. 2004/041266 Pamphlet
- Patent Document 2 International Publication No. 2005/063729 Pamphlet
- Patent Document 3 International Publication No. 2005/063725 Pamphlet
- Patent Document 4 International Publication No. 2005/030203 Pamphlet
- Patent Document 5 Pamphlet of International Publication No. 94/25448
- Patent Document 6 Japanese Patent Laid-Open No. 2000-212174
- Patent Document 7 WO95 / 30664 pamphlet
- Patent Document 8 International Publication No. 97/41097 Pamphlet
- Patent Document 9 US Pat. No. 5,480,896
- Patent Document 10 Japanese Patent Laid-Open No. 7-2848
- An object of the present invention is to provide a novel compound having a GPR40 receptor agonist action useful as an insulin secretagogue and a preventive or therapeutic agent for diabetes. Means for solving the problem
- the present inventors have intensively studied a compound having a GPR40 receptor agonist action, and found that a novel oxadiazolidinedione compound or a salt thereof is excellent in GPR40 receptor agonist- It was found to have a strike action. Furthermore, the present inventors have found that these oxadiazolidinedione compounds have an excellent insulin secretion promoting action and strongly suppress the increase in blood glucose after glucose loading, and completed the present invention.
- the present invention relates to an oxadiazolidinedione compound represented by the following formula (I) or a pharmaceutically acceptable salt thereof.
- R 1 —H, halogen, —R °, halogeno lower alkyl, —OR—S—R. Or -0-halogeno lower amino quinole.
- R ° lower alkyl.
- R z the same or different from each other, —H or lower alkyl.
- Ring A benzene, pyridine, thiophene, piperidine, dihydropyridine, pyrimidine or tetrahydroquinoline.
- Ring B benzene or pyridine.
- R 2 the same or different from each other, -norogen, -R °, halogeno lower alkyl, -OR -SR °, -O-halogeno lower alkyl, -0-lower alkylene-aryl or oxo; n: 0, 1 or 2.
- R 3 -halogen, -R °, -halogeno lower alkyl, -0-R °, -S-R °, -0-halogeno lower alkyl, -X- (substituted, may be phenyl ) Or -X- (which may be substituted or heteroaryl).
- R 4 -H or lower alkyl.
- R 1 and R 4 may form a lower alkylene to form a lower alkylene!
- the present application also relates to a pharmaceutical, particularly GPR40 agonist, comprising an oxadiazolidinedione compound represented by the general formula (I) or a salt thereof as an active ingredient.
- the present application relates to a compound represented by formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of GPR40 agonist, insulin secretagogue, or diabetes prevention and Z or therapeutic agent. And a method for preventing and / or treating diabetes, comprising administering to a patient an effective amount of a compound represented by formula (I) or a pharmaceutically acceptable salt thereof. To do.
- a pharmaceutical composition comprising a compound according to formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
- composition according to (1) which is a diabetes preventive and Z or therapeutic agent.
- a method for preventing and / or treating diabetes comprising administering to a patient an effective amount of the compound according to formula (I) or a salt thereof.
- Test method 1 GPR40 agonist activity measurement
- the full-length sequence of GPR40 was obtained by the PCR method using human genomic DNA (Clontech) as a template.
- the oligonucleotide consisting of the base sequence represented by SEQ ID NO: 1 was used as a forward primer
- the oligonucleotide consisting of the base sequence represented by SEQ ID NO: 2 was used as a reverse primer.
- a base sequence containing an Xbal recognition site is added to the 5 ′ end of each of the forward primer and the reverse primer.
- PCR was performed using Taq DNA polymerase (Ex Taq DNA polymerase; Takara Bio Inc.) in the presence of 5% dimethyl sulfoxide (DMSO) at 94 ° C (15 seconds) / 55 ° C (30 seconds) / 12 .
- C (1 minute) force cycle was repeated 30 times. As a result, a DNA fragment of about 0.9 kbp was amplified.
- This DNA fragment was digested with Xbal and then inserted into the Xbal site of plasmid pEF-BOS-dhfr (Nucleic Acids Research, 18, 5322, 1990) to obtain plasmid pEF-BOS-dhfr-GPR40.
- the nucleotide sequence of the GPR40 gene in the plasmid pEF-BOS-dhfr-GPR40 is the DNA seek It was determined by the dideoxy terminator method using an ensemble (ABI377 DNA Sequencer; Applied Biosystems).
- the base sequence of GPR40 gene was as shown in the base sequence represented by SEQ ID NO: 3.
- the base sequence represented by SEQ ID NO: 3 has an open reading frame (ORF) of 903 bases, and the amino acid sequence (300 amino acid) predicted from this ORF is represented by SEQ ID NO: 4. It was as the amino acid sequence.
- CHO dhfr cells (CHO cells lacking the dihydrofolate reductase (dhfr) gene) were used as cells that express GPR40 protein. Further, the plasmid pEF-BOS-dhfr-GPR40 obtained in the above 0 was used as an expression plasmid for expressing the GPR40 protein. Seed CHO dhfr—cells in ⁇ -MEM medium containing 10% fetal calf serum (FCS) to 6-well plates (Asahi Techno Glass) to 80-90% confluence.
- FCS fetal calf serum
- plasmid pEF-BOS-dhfr-GPR40 per well was introduced using a transfection reagent (Lipof ectamine 2000; Invitrogen). After 24 hours of culturing after gene transfer, the cells were diluted and reseeded. At that time, the sputum medium containing 10% FCS was changed to the ⁇ -MEM medium containing no nucleic acid containing 10% FCS. After culturing for 20 days, the formed cell colonies were individually collected and cultured to obtain CHO cells stably expressing GPR40. From these cells, cells having high reactivity to the endogenous ligands oleic acid and linoleic acid were selected.
- a CHO cell line expressing human GPR40 was seeded in a 384-well black plate (Betaton Decktonson) at 6 ⁇ 10 3 per well and cultured overnight in a CO incubator.
- the luminescent dye was dissolved in 10 ml of HBSS-HEPES buffer (PH7.4, 1 X HBSS, 20 mM HEPES, Invitrogen) per bottle using Calcium-3 Atsey Kit (Molecular Device).
- Probenecid (Sigma) 35.68 mg was dissolved in 1M NaOH 250 1 and adjusted by adding HBSS-HEPES buffer 250 1.
- the fluorescent dye solution was prepared by mixing 16 ml of HB SS-HEPES buffer and fluorescent dyes 640 1 and 32 1 probenecid per plate. The plate medium was removed, and the fluorochrome solution was dispensed at 40 1 min per well and then incubated for 2 hours at room temperature.
- test compound was dissolved in DMSO, diluted with HBSS-HEPES buffer, and 101 1 was dispensed onto the plate to start the reaction.
- the change in intracellular calcium concentration was measured with FLIPR.
- the EC value of the test compound was determined from the dose response curve of the change in fluorescence intensity after 1 min.
- Test Method 2 Insulin secretion promoting action using MIN6 cells
- MIN6 cells a mouse spleen j8 cell line
- the test method is shown below.
- MIN6 cells were seeded in a 96-well plate at 5xl0 4 / hole (200 1).
- the medium used was DMEM (25 mM glucose) containing 10% FBS, 55 2-mercaptoethanol, lOOU / ml penicillin 100 ⁇ g / ml streptomycin. After 2 days, remove the medium with an aspirator, KRB-HEPES containing 2.8 mM glucose warmed to 37 ° C (116 mM NaCl, 4.7 mM KC1, 1.2 mM KH PO, 1.2 mM MgSO, 0.25 mM CaCl, 25 mM NaHCO, 0.005% FFA Free
- Test Method 3 Normal Mouse Single Oral Glucose Tolerance Test
- mice Male ICR mice (6 weeks old) preliminarily raised for 1 week were fasted overnight and used as test animals.
- the test compound was a 0.5% methylcellulose suspension and was orally administered at 10 mg / kg 30 minutes before glucose (2 g / kg) loading. In the control group, 0.5% methylcellulose was administered. The blood glucose lowering rate (%) relative to the control group at 30 minutes of glucose load was calculated.
- the compound of the present invention has an excellent GPR40 agonist action, so that it is an insulin secretagogue, diabetes (insulin-dependent diabetes (IDDM), non-insulin-dependent diabetes (NIDDM) It is clear that it is useful as a prophylactic / therapeutic agent for diseases in which GPR40 is involved such as glucose tolerance, fasting blood glucose level abnormality, and mild diabetes.
- IDDM insulin-dependent diabetes
- NIDDM non-insulin-dependent diabetes
- alkyl and “alkylene” mean a linear or branched hydrocarbon chain.
- “Lower alkyl” is preferably an alkyl group having 1 to 6 carbon atoms (hereinafter abbreviated as C).
- it is C alkyl, and still more preferably methyl and ethyl.
- the "lower alkynyl” is preferably linear or branched, C alkynyl,
- they are ethynyl, probyl, butynyl, pentyl, 1-methyl-2-probyl, 1,3-butadiyl, 1,3-pentadiyl group and the like. More preferably C alkyl.
- ethynyl and propynyl are particularly preferred.
- “Lower alkylene” means a divalent group (C alkylene) formed by removing any one hydrogen atom of the above “lower alkyl”, preferably C alkylene, more preferably me.
- Tylene ethylene, trimethylene, propylene and dimethylmethylene, more preferably methylene and ethylene.
- Halogen refers to F, Cl, Br and I.
- Halogeno lower alkyl is preferably a C-alkyl substituted with one or more halogens.
- 1-6 means alkyl, more preferably halogeno C alkyl, still more preferably fluoro
- Cycloalkyl is a C 3 saturated hydrocarbon ring group, which may have a bridge.
- C cycloalkyl Preferably C cycloalkyl
- cyclopropyl More preferred are cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- Cycloalkenyl is C cycloalkenyl, a double chain that may have a bridge.
- a ring group fused to a benzene ring at the binding site includes cyclopentane, cyclopentaenyl, cyclohexenyl, cyclohexadenyl, 1-tetrahydronaphthyl, 1-indul, 9-fluorenyl group and the like.
- cyclopental More preferred are cyclopental, cyclohexyl, 1-indul, and 1-tetrahydronaphthyl.
- Aryl is an aromatic hydrocarbon group of C, preferably phenyl, naphthyl and
- tetrahydronaphthyl more preferably a phenol.
- Heteroaryl is a monocyclic 5- to 6-membered aromatic heterocycle having 1 to 4 heteroatoms selected from 0, S and N, and ii) the heterocycle shown in 0 above A condensed bicyclic heterocycle, provided that the condensed rings may be the same or different from each other; and iii) the heterocyclic ring shown in 0 above and a benzene ring or a 5- to 7-membered cycloalkane is condensed 2 It means a group that also has a ring force selected from a cyclic heterocycle.
- Examples of the ring constituting the group include: 0 pyridine, virazine, pyrimidine, pyridazine, imidazole, pyrrole, thiophene, furan, triazine, triazole, thiazole, thiadiazole, oxadiazole, pyrazole, isothiazole, oxazole, isoxazole, ii) naphthyridine , Imidazopyridine, pyrrolopyrimidine, naphthyridine, chenoviridine, thienopyrroline, m) quinoline, benzimidazole Nore, benzofuran, benzothiphene, benzothiadiazonole, benzothiazonole, benzoisothiazole, benzoxazole, benzisoxazole, quinoline, isoquinoline, 5,6,7,8-tetrahydro Examples include quinoline, 5,6,7,8-tetrahydroiso
- Heterocycle and “heterocyclic group” are 0 to 0, S and N forces monocyclic 4 to 8 members, preferably 5 to 7 members having 1 to 4 selected heteroatoms, A saturated, unsaturated or partially unsaturated heterocycle, i) a bicyclic heterocycle condensed with the heterocycle shown in 0 above, provided that the condensed rings may be the same or different from each other; and iii) A group consisting of a ring selected from the heterocycle shown in 0 above and a benzene ring or a bicyclic heterocycle condensed with a 5- to 7-membered cycloalkane.
- Examples of the ring constituting the group include 0-azetidine, piperidine, pyrrolidine, piperazine, azepane, diazepan, morpholine, thiomorpholine, dioxane, dixolan, virazolidine, piperidine, piperazine, Xetane, tetrahydrofuran, tetrahydropyran, dihydropyridine, pyridine, pyrazine, pyrimidine, pyridazine, imidazole, pyrrole, thiophene, furan, triazine, triazole, thiazole, thiadiazol, oxadiazole, pyrazole, isothiazole, oxazole, isoxazole, ii ) Quinutaridin, naphthyridine, imidazopyridine, pyropyrimidine, naphthyridine, chenoviridine, thienopyrroline, iii
- Optionally substituted means “unsubstituted” or “1 to 5 substituents which are the same or different. It has ". “Substituted” means “having 1 to 5 identical or different substituents”.
- Substituents allowed in “substituted or phenyl” and “substituted or heteroaryl” in R 3 are preferably groups shown in the following group G. .
- Group G Nono androgenic, - CN, - R °, halogeno-lower alkyl, - OR z, -0- halogeno-lower alkyl, - N (R z) CO- R z, -CO R - CON (R z), - CO-heterocyclic group, -C0N (R z ) -lower alkyl
- lower alkylene halogen or - 0R Z Yogu cycloalkyl optionally substituted with, Shikuroaruke - Le, heterocyclic group Ariru and to is substituted with a group selected the following Group G 1 power! Hey! /.
- G 1 group neurogen, sheared -R °, halogeno lower alkyl, -0R -0-halogeno lower alkyl, -N (R Z ), -S-R °, -SO -R °, -SO N ( R Z), - CO- R z , - C0N (R z), - C0N (R z) - lower ⁇
- Alkylene - 0R Z, - N (R z) C0- R z, Okiso, lower alkylene - CN, lower alkylene - 0R Z, - Ariru, - (- 0R Z may be substituted with lower alkylene) - Ariru, Lower alkylene-0-aryl, heterocyclic group, and lower alkylene-heterocyclic group.
- the aryl and heterocyclic groups in the G 1 group may be substituted with a group selected from the following G 2 group.
- G 2 group Nono androgenic, ShiaIri R °, halogeno-lower alkyl, -0R -0- halogeno-lower alkyl, and, Okiso.
- G 3 groups: neurogen, -R °, halogeno lower alkyl, -0R -C0N (R Z ), -C0N (R z ) -heterocycle
- the lower alkylene in the G 3 group may be substituted with halogen or —0R Z
- the cycloalkyl and the heterocyclic group may be substituted with a group selected from the group G 1 force. Yes.
- R 3 As an acceptable substituent in “substituted or phenyl” and “substituted or heteroaryl” in R 3 , still more preferably, —0-lower alkylene-OR 0-lower alkylene-CON (R Z ) or -0-lower alkylene- (substituted with -OR Z
- R 1 is preferably —H, —halogen or —R. And more preferably -H.
- R 2 is preferably -norogen, -0-R. Or -R. More preferably, it is halogen or R °.
- n is preferably 0 or 1.
- R 3 is preferably -X- (substituted or optional) or -X- (optionally substituted or heteroaryl), more preferably each substituted a le or pyridyl, more preferably, may Hue substituted - - which may also be Hue is Le, preferably Ri by further be substituted with a group selected from the group G 3 Good fibers, especially preferred are: -0-lower alkylene -OR Z , -0-lower alkylene -CON (R Z ) and -0-low
- R 4 is preferably —H.
- the A ring is preferably a benzene ring, a pyridine ring or a thiophene ring, and more preferably a benzene ring.
- the B ring is preferably a benzene ring.
- L is preferably * -lower alkylene-0- or * -lower alkylene-NH- More preferably, it is * —CH 2 —O— or * —CH 2 —NH— (* represents a bond to the A ring.)
- the position of substitution in the B ring of L is preferably the 4-position with respect to —CH (R 4 ) — (3,5-dioxo-1,2,4-oxadiazolidine-2-yl).
- R 2 is halogen or R Q (6) compounds according.
- R 3 is -0-lower alkylene-OR z , -0-lower alkylene-CON (R z ) and -0-lower alkylene
- Lucylene- (substituted with —OR z !, may! /, Cycloalkyl) substituted with a group selected from the group consisting of 1 to 2 lower alkyls, halogens or —OR °
- the compound of (7) which is a fail may be a fail.
- the compound of the present invention represented by the formula (I) may form a salt and is included in the compound of the present invention as long as the salt to be obtained is a pharmaceutically acceptable salt.
- inorganic salts such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid Acid, lactic acid, malic acid, tartaric acid, citrate, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, aspartic acid, glutamic acid and other acid addition salts with sodium, potassium, calcium
- inorganic bases containing metals such as gnesium, addition salts with organic bases such as methylamine, ethylamine, ethanolamine, lysine, and orthotin, and ammoni
- the compound of the present invention may contain an asymmetric carbon atom depending on the type of the substituent, and optical isomers based on this may exist.
- the present invention encompasses all of these optical isomer mixtures and isolated ones.
- the compounds of the present invention exist in tautomers.
- the present invention includes separated or mixed isomers.
- a label that is, a compound obtained by substituting one or more atoms of the compound of the present invention with a radioactive isotope or a non-radioactive isotope is also included in the present invention.
- the present invention includes various hydrates and solvates of the compound of the present invention and substances having crystal polymorphs. It should be understood that the compounds of the present invention include all compounds represented by formula (I) and pharmaceutically acceptable salts thereof, which are not limited to the compounds described in the Examples below. It is.
- the compounds of the present invention include all compounds that are metabolized in vivo and converted into the compounds of the present invention, so-called prodrugs.
- the group that forms a prodrug of the compound of the present invention is described in “Progress in Medicine”, Life Sciences, Medica, 1985, p. 2157-2161. And the groups described in Yodogawa Shoten 1999 “Pharmaceutical Development”, 7th Molecular Design, pages 163-198.
- the compound of the present invention and a pharmaceutically acceptable salt thereof can be produced by applying various known synthetic methods using characteristics based on the basic skeleton or the type of substituent.
- a typical production method is illustrated below.
- it is effective in terms of production technology to replace the functional group with a suitable protecting group at the raw material or intermediate stage, that is, a group that can be easily converted to the functional group. There are cases. Thereafter, the protecting group can be removed as necessary to obtain the desired compound.
- functional groups include a hydroxyl group, a carboxyl group, and an amino group.
- protective groups for these functional groups include “Protective”, “Groups”, “In” and “Organic” by Greene and Wuts.
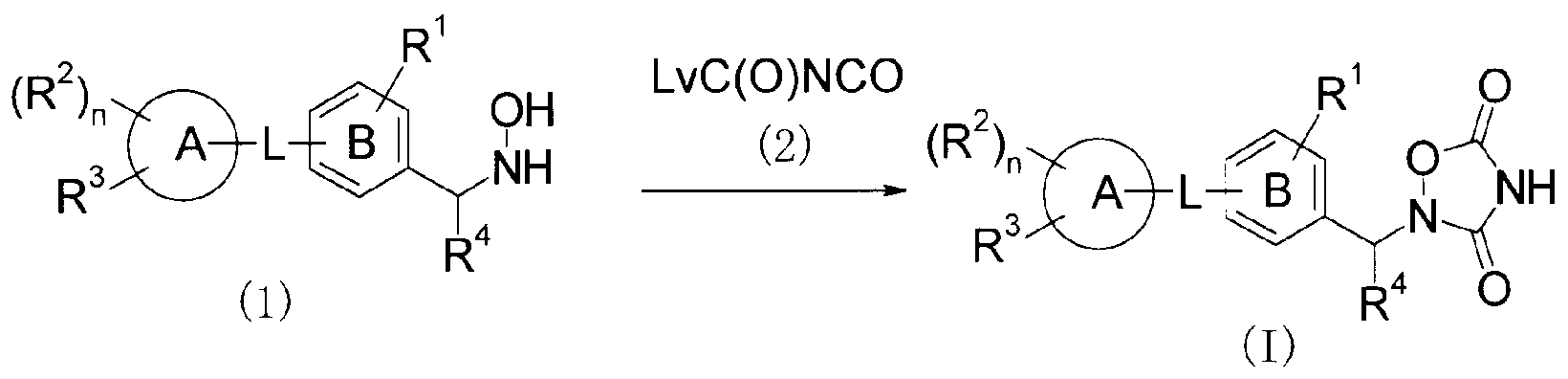
- This production method is a method for producing the compound (I) of the present invention by a cyclization reaction between the compound (1) and the compound (2).
- the leaving group for Lv halogens such as black mouth and bromo, and alkoxy groups such as methoxy and ethoxy are preferred.
- the reaction is carried out by using an equivalent amount of compound (1) and compound (2) or an excess of one of them, ethers such as jetyl ether, tetrahydrofuran (THF), dioxane or dimethoxyethane (DME), dichloromethane, 1,2-
- ethers such as jetyl ether, tetrahydrofuran (THF), dioxane or dimethoxyethane (DME), dichloromethane, 1,2-
- ethers such as jetyl ether, tetrahydrofuran (THF), dioxane or dimethoxyethane (DME), dichloromethane, 1,2-
- THF tetrahydrofuran
- DME dimethoxyethane
- dichloromethane 1,2-
- the reaction can be carried out in a solvent such as halogenated hydrocarbons such as dichloroethane or chloroform, and aromatic hydrocarbons such as benzene, toluen
- the hydroxyl group may be rubbed.
- the removal of the strong rubermoyl group can be carried out by a person skilled in the art by a method usually used for weak ruby moistle.
- the reaction can be performed under cooling, at room temperature or under heating using a base such as sodium methoxide, sodium ethoxide, sodium hydroxide in an alcohol such as methanol or ethanol or a solvent such as water.
- Lv 1 and Lv are either halogen or trifluoromethylsulfo-oxy group, and the other is -B (OH), -B (OR °°) or -SnR Q , Ar Are each replaced
- phenyl or heteroaryl may be optionally substituted, and R is lower alkyl, or two R ° degrees are lower alkylene. The same applies below.
- This production method is a method for producing the compound (I-a) of the present invention by a coupling reaction of the compound (3) and the compound (4).
- the reaction is carried out using a palladium complex such as tetrakistriphenylphosphine palladium or palladium acetate as a catalyst in a solvent such as ethers, alcohols, halogenated hydrocarbons, aromatic hydrocarbons or water, or a mixture thereof.
- a solvent such as ethers, alcohols, halogenated hydrocarbons, aromatic hydrocarbons or water, or a mixture thereof.
- the reaction can be carried out in an equivalent amount of compound (3) and compound (4) or in excess of one of them in a solvent, under cooling, at room temperature and under heating.
- a base such as sodium carbonate, cesium carbonate, or sodium tert-butoxide, or a lithium salt such as lithium chloride or lithium bromide may be advantageous for the smooth progress of the reaction. is there.
- R 1-5 1-5 means alkyl. However, R 1Q and R 11 have a total of 0 to 5 carbon atoms. The same applies below. )
- This production method is a method for producing the compound (i-b) of the present invention by reductive amination of the compound (5) and the compound (6).
- the compound (5) and the compound (6) are used in the same amount or in an excess amount, and in the presence of a reducing agent, in a solvent inert to the reaction, from ⁇ 45 ° C. to heating under reflux, preferably 0 ° C to room temperature Usually, stirring is performed for 0.1 hour to 5 days.
- a reducing agent in a solvent inert to the reaction, from ⁇ 45 ° C. to heating under reflux, preferably 0 ° C to room temperature
- stirring is performed for 0.1 hour to 5 days.
- the solvent include alcohols, ethers, and mixtures thereof.
- the reducing agent include sodium cyanide sodium borohydride, sodium triacetoxyborohydride, sodium borohydride and the like.
- a dehydrating agent such as molecular sieves, or an acid such as acetic acid, hydrochloric acid, or titanium (IV) isopropoxide complex.
- a reduction reaction may be separately performed after obtaining the imine.
- This production method is a method for producing the compound (I c) of the present invention by amidating the compound (7) and the compound (6).
- a reactive derivative thereof can also be used.
- the reaction is carried out by using an equivalent amount of the carboxylic acid compound (7) or its reactive derivative and the amino compound (6), or an excess of one, and using aromatic hydrocarbons, halogenated hydrocarbons, ethers, ⁇ , ⁇ -Cooling in a solvent such as dimethylformamide (DMF), ⁇ , ⁇ -dimethylacetamide (DMA), 1-methylpyrrolidin-2-one (NMP), dimethylsulfoxide (DMSO), ethyl acetate, pyridine, or acetonitrile
- DMF dimethylformamide
- DMA 1-methylpyrrolidin-2-one
- DMSO dimethylsulfoxide
- ethyl acetate pyridine
- acetonitrile acetonitrile
- Examples of reactive derivatives of the carboxylic acid compound (7) include acid halides (acid chloride, acid bromide, etc.), acid anhydrides (ethyl carbonate, carbonic acid benzyl, carbonic acid carbonate, P- Mixed acid anhydride obtained by reaction with toluenesulfonic acid, isovaleric acid, etc., or symmetrical acid anhydride), active ester (phenol, HOBt, which may be substituted with an electron-withdrawing group such as nitro group or fluorine atom) Esters prepared using HONSu, etc.), lower alkyl esters, acid azides and the like. These reactive derivatives can be produced by conventional methods.
- reaction force in the presence of a base such as triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4- (N, N-dimethylamino) pyridine (DMAP) It may be advantageous for progress.
- a base such as triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4- (N, N-dimethylamino) pyridine (DMAP) It may be advantageous for progress.
- Manufacturing method 5 Other manufacturing methods
- Amido can be carried out in the same manner as in production method 4.
- a sulfoxide compound or a sulfone compound can be produced by oxidizing the S atom of the sulfide compound with various oxidizing agents.
- the reaction can be carried out, for example, by using m-chloroperbenzoic acid, peracetic acid, hydrogen peroxide solution, Dess-Martin reagent (1,1,1-triaceal). Toxic-1,1-dihydro-1,2-benzodoxol-3 (1H) -one) etc. as an oxidant and using an excess amount in excess of solvents such as halogenated hydrocarbons, acetic acid, water, etc. Medium, under cooling, under room temperature or under heating.
- a compound having a carboxyl group By hydrolyzing the compound having an ester group, a compound having a carboxyl group can be produced.
- an inert solvent such as aromatic hydrocarbons, ethers, halogenated hydrocarbons, alcohols, DMF, DMA, NMP, DMSO, pyridine, water, sulfuric acid, hydrochloric acid, hydrobromic acid, etc.
- an organic acid such as mineral acid, formic acid or acetic acid
- a base such as lithium hydroxide, sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, cesium carbonate or ammonia
- the raw materials used for the production of the compound of the present invention can be produced, for example, by applying the following methods, the methods described in the production examples described later, known methods or methods obvious to those skilled in the art, or modifications thereof. Can do.
- Lv 3 represents —OH or a leaving group such as halogen, methanesulfo-loxy, p-toluenesulfo-loxy, and so on.
- compound (8) is 0-alkylated with compound (9) to obtain compound (10).
- the compound (8) having Lv 3 of —OH can be carried out by using a conventional Mitsunobu reaction method commonly used by those skilled in the art.
- a conventional Mitsunobu reaction method commonly used by those skilled in the art.
- an activator or cyano compound prepared from a phosphorus compound such as tributylphosphine or triphenylphosphine and an azodicarbonyl compound such as jetyl azodicarboxylate or 1,1 ′-(azodicarbol) dipiperidine.
- a reagent such as methylenetributylphosphorane
- the reaction can be carried out in a solvent such as halogenated hydrocarbons, ethers, aromatic hydrocarbons, under cooling, at room temperature or under heating.
- This step is a step of obtaining compound (12) by oximation of compound (11).
- an oximation method commonly used by those skilled in the art can be applied.
- compound (11) and hydroxylamine or a salt thereof in an equivalent amount or one of them in excess and in a solvent such as alcohols, acetic acid, pyridine, water, etc., under cooling, at room temperature to under heating. It can be carried out.
- addition of sodium acetate, P-toluenesulfonic acid, etc. may be advantageous for the progress of the reaction.
- Second step reduction
- This step is a step of obtaining compound (1) by reducing compound (12).
- the oxime reduction method commonly used by those skilled in the art can be applied.
- a reducing agent such as compound (12) and borane-pyridine complex or sodium cyanoborohydride or an excess amount of one, and a solvent such as ethers, alcohols, aromatic hydrocarbons, acetic acid, etc.
- a solvent such as ethers, alcohols, aromatic hydrocarbons, acetic acid, etc.
- the compound of the present invention produced as described above is isolated or purified as a salt by leaving it as it is or subjecting it to a conventional salt formation treatment. Isolation 'Purification is performed by applying ordinary chemical operations such as extraction, concentration, distillation, crystallization, filtration, recrystallization, and various chromatography.
- Various isomers can be isolated by conventional methods utilizing the difference in physicochemical properties between isomers. For example, a racemic mixture can be converted to an optically pure heteroisomer by a general racemic resolution method, such as a method of optical resolution by diastereomeric salt with a general optically active acid such as tartaric acid. The diastereomeric mixture can be separated by, for example, fractional crystallization or various types of chromatography.
- An optically active compound can also be produced by using an appropriate optically active raw material.
- a pharmaceutical composition containing the compound of the present invention or one or more of pharmaceutically acceptable salts thereof as an active ingredient contains a carrier carrier, excipient, and other additives for commonly used formulations. It is prepared into tablets, powders, fine granules, granules, capsules, pills, solutions, injections, suppositories, ointments, patches, etc. and administered orally or parenterally.
- the clinical dose of the compound of the present invention for humans is appropriately determined in consideration of the symptoms, body weight, age, sex, etc. of the patient to which it is applied.
- the daily dose is usually about 0.0001 per body weight. ⁇ 50 mg / kg, preferably about 0.001 to 10 mg / kg, more preferably 0.01 to 1 mg / kg, which is administered once or divided into 2 to 4 times.
- the daily dose is about 0.0001 to 1 mg / kg per body weight, preferably about 0.0001 to 0.1 mg / kg, and is administered once to several times a day. Dosage is seed Since it varies depending on various conditions, it may be less than the above-mentioned dose range!
- Tablets, powders, granules and the like are used as the solid composition for oral administration according to the present invention.
- one or more active substances are present in at least one inert diluent such as lactose, mannitol, glucose, hydroxypropyl cellulose, microcrystalline cellulose, starch, poly Mixed with bull pyrrolidone, magnesium metasilicate aluminate, etc.
- the composition contains additives other than inert diluents, for example, lubricants such as magnesium stearate, disintegrating agents such as calcium calcium glycolate, stabilizers, solubilizing agents and the like according to conventional methods. It may be. If necessary, tablets or pills may be coated with sugar coating such as sucrose, gelatin, hydroxypropylcellulose, hydroxypropylmethylcellulose phthalate, or a gastric or enteric film.
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, elixirs and the like, commonly used inert diluents, For example, purified water and ethanol (EtOH) are included.
- the composition may contain adjuvants such as wetting agents and suspending agents, sweeteners, flavors, fragrances and preservatives.
- Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions.
- aqueous solutions and suspensions include distilled water for injection and physiological saline.
- non-aqueous solutions and suspensions include propylene glycol, polyethylene glycol, vegetable oils such as olive oil, alcohols such as EtOH, and polysorbate 80.
- Such a composition may further contain auxiliary agents such as preservatives, wetting agents, emulsifiers, dispersants, stabilizers, and solubilizing agents. These are sterilized, for example, by filtration through a bacteria-retaining filter, blending with a bactericide, or irradiation. These can also be used by producing a sterile solid composition and dissolving it in sterile water or a sterile solvent for injection before use.
- External preparations include ointments, plasters, creams, jellies, poultices, sprays, lotions, eye drops, eye ointments and the like.
- ointment bases include commonly used ointment bases, lotion bases, aqueous or non-aqueous solutions, suspensions, emulsions, and the like.
- ointment or lotion bases include polyethylene glycol, propylene glycol, white petrolatum, And wax, polyoxyethylene hydrogenated castor oil, glyceryl monostearate, stearyl alcohol, cetyl alcohol, lauromacrogol, sorbitan sesquioleate and the like.
- Transmucosal agents such as inhalants and nasal agents are used in solid, liquid or semi-solid form and can be produced according to conventionally known methods.
- known excipients, and further pH adjusters, preservatives, surfactants, lubricants, stabilizers, thickeners and the like may be appropriately added.
- an appropriate device for inhalation or insufflation can be used.
- a known device such as a metered dose inhalation device or a nebulizer
- the compound is administered alone or as a powder in a formulated mixture or as a solution or suspension in combination with a pharmaceutically acceptable carrier. be able to.
- the dry powder inhaler or the like can use a dry powder or a powder-containing capsule which can be used for single or multiple administrations.
- a dry powder or a powder-containing capsule which can be used for single or multiple administrations.
- it may be in the form of an appropriate propellant such as a pressurized aerosol spray using a suitable gas such as black mouth fluoroalkane, hydrofluoroalkane or carbon dioxide.
- the raw material mixture used in the examples includes a new substance, and a method for producing such a raw material compound will be described as a production example.
- the following abbreviations are used.
- REx Production example number
- Ex Example number
- No Compound number
- Str Structural formula (when HC1 is in the structural formula, it means that the compound is hydrochloride)
- Syn Production method (In the case of only numbers, the example numbers produced in the same manner are shown, and in the case where R precedes the numbers, the number of the production examples produced in the same manner is shown.)
- trifluoromethanesulfonic acid 4-cyano-2,6-dimethylphenol, (3-formylphenol) boronic acid, palladium acetate, dicyclohexyl (2 ', 6 dimethoxybiphenyl-
- 2-yl) phosphine tripotassium phosphate
- toluene was stirred at room temperature for 6 hours to obtain 3′-formyl-2,6-dimethylbiphenyl-4-carbotolyl.
- trifluoromethanesulfonic anhydride was added dropwise to a mixture of 4-[(4'-hydroxy-2,2'-dimethylbiphenyl-3-yl) methoxy] benzaldehyde, pyridine and dichloromethane. , 0.
- trifluoromethanesulfonic acid 3 was obtained to give [(4-formylphenoxy) methyl] -2,2′-dimethylbiphenyl-4-yl.
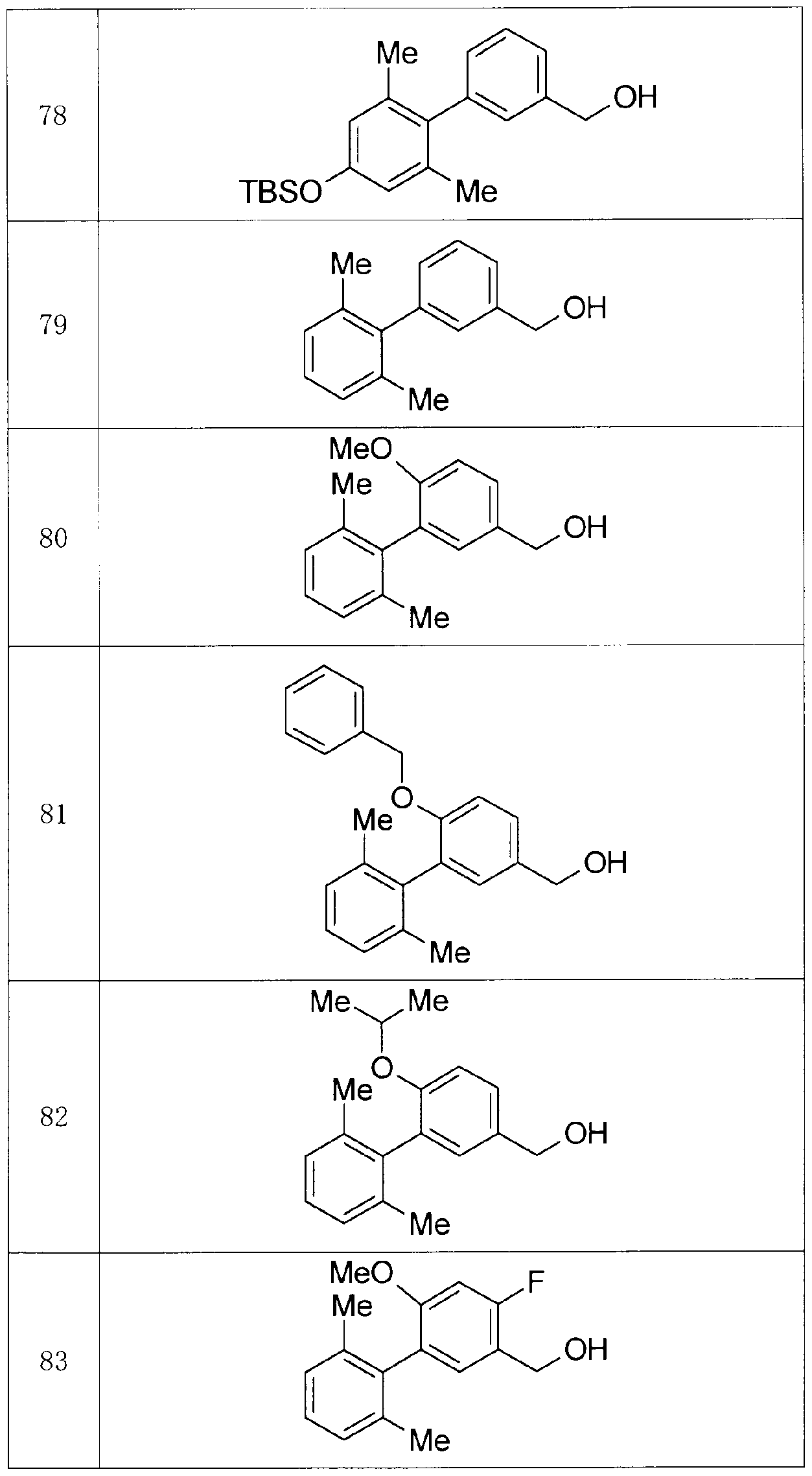
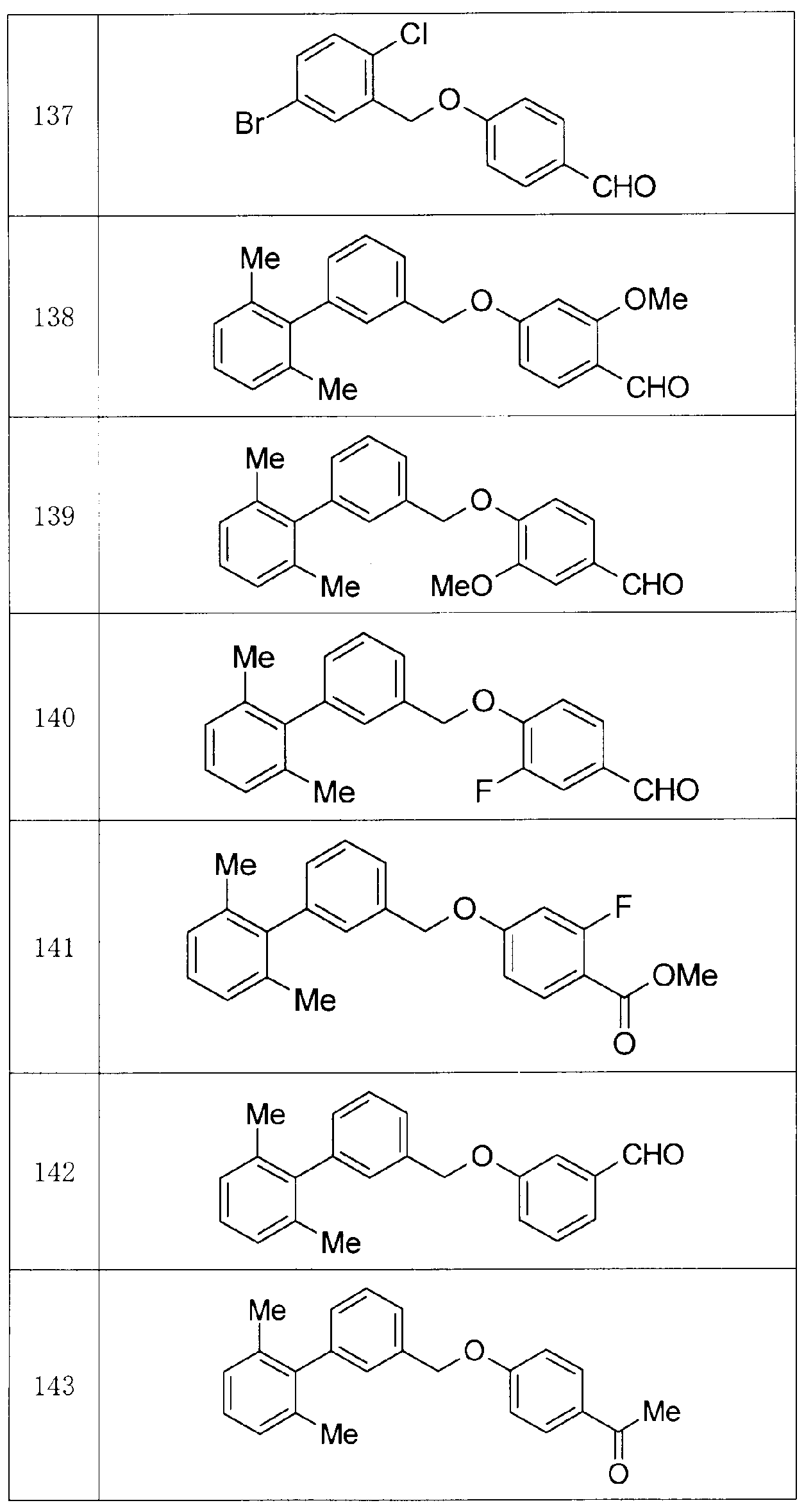
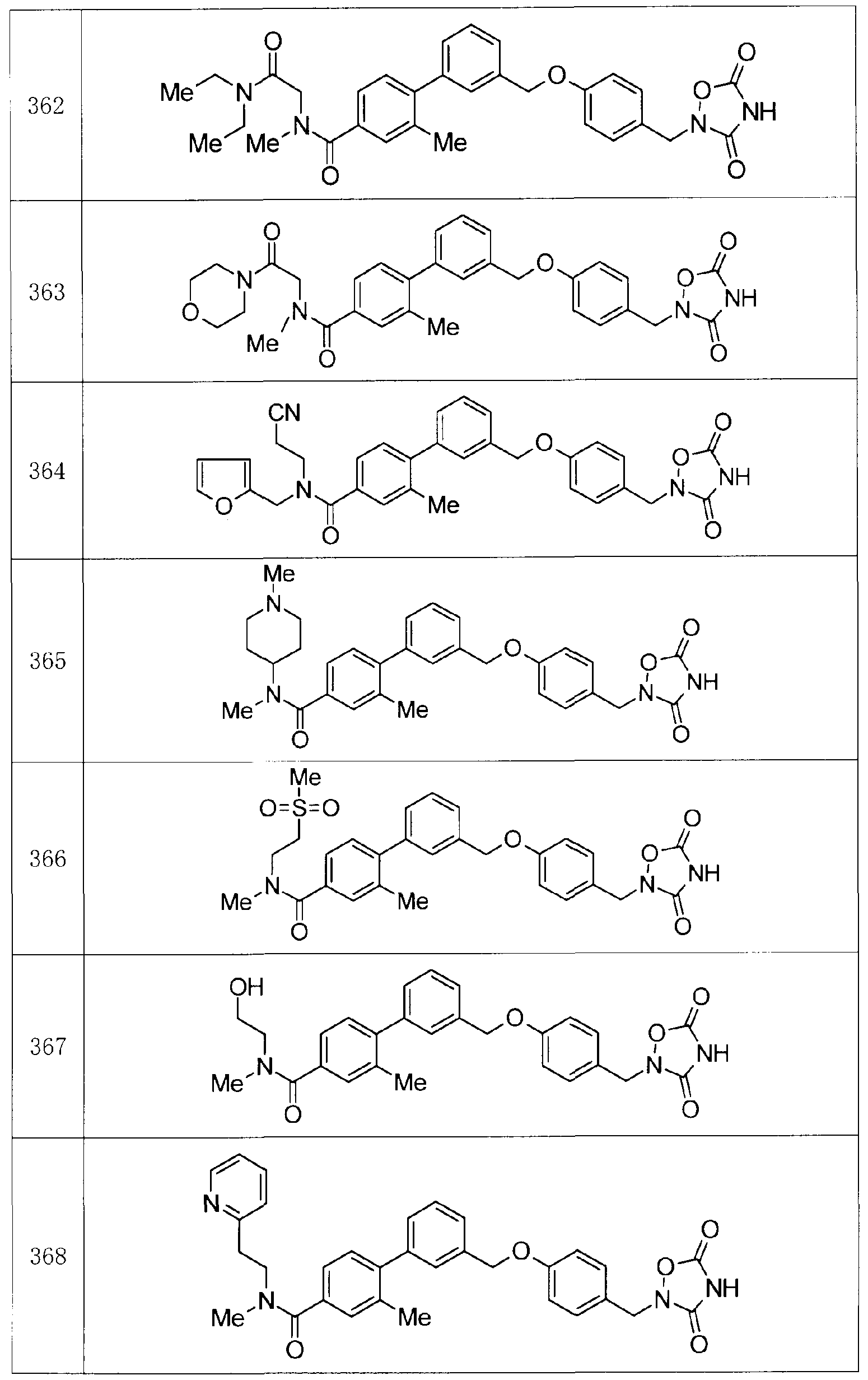
- Production Example compounds 39 to 299 were produced using the corresponding starting materials.
- Tables 4 to 44 show the structures of the production example compounds, and Tables 45 to 52 show the production methods and physicochemical data.
- the obtained foam was dissolved in ethanol (5 ml), 1M aqueous sodium hydroxide solution (1.06 ml) was added, and the mixture was concentrated under reduced pressure.
- the obtained residue was recrystallized from water-isopropanol to give sodium 2- ⁇ 4-[( 4′-Chloro-2′-methylbiphenyl-3-yl) methoxy] benzyl ⁇ -3,5-dioxo-1,2,4-oxodiazolidine-4-ide (347 mg) was obtained as colorless crystals.
- the organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (black mouth form-methanol) to obtain a colorless foam (777 mg).
- Sodium methoxide (50 mg) was added to a methanol (10 ml) solution of the obtained foam (116 mg), and the mixture was stirred at room temperature for 30 minutes. Thereafter, sodium methoxide (200 mg) was added to the reaction mixture, and the mixture was stirred at room temperature for 1 hour.
- the reaction mixture was heated to 60 ° C, stirred for 2 hours, and then allowed to cool to room temperature.
- the organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (black mouth form-methanol) to obtain a colorless foam (777 mg). Black mouth of the obtained foam (600mg) Under cooling in an ice-methanol bath, m-cloperbenzoic acid (630 mg) was added to the form (20 ml) solution and stirred for 30 minutes. Water (20 ml) was added to the reaction mixture, and the mixture was extracted with black mouth form. The organic layer was dried over anhydrous magnesium sulfate and the solvent was distilled off under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (black mouth form-methanol) to obtain a colorless foam (510 mg).
- the obtained foam 51 Omg was washed with diisopropyl ether-ethyl acetate and dried under reduced pressure to obtain a slightly yellow solid (432mg).
- Sodium methoxide 800 mg was added to a solution of the obtained pale yellow solid (387 mg) in methanol (30 ml), stirred at 60 ° C. for 2 hours, and then allowed to cool to room temperature.
- To the reaction mixture was added 1M hydrochloric acid (30 ml) and water (50 ml), and the mixture was extracted with black mouth form.
- the obtained support was purified by silica gel column chromatography (black mouth form-methanol) to obtain a pale yellow foam (614 mg).
- the resulting foam (614 mg)
- the product was dissolved in THF (5 ml) -ethanol (5 ml), 1M aqueous sodium hydroxide solution (1.32 ml) was added, and the mixture was concentrated under reduced pressure.
- the residue obtained was recrystallized from isopropanol-water to give sodium 2- ⁇ 4-[(2 ', 6difluor-4'-methoxybiphenyl-3-yl) methoxy] benzyl ⁇ - 3,5-Dioxo-1,2,4-oxadiazolidine-4-id (366 mg) was obtained as a colorless solid.
- the reaction mixture was diluted with chloroform / methanol (4/1), washed with saturated aqueous sodium chloride and saturated aqueous sodium chloride, and the solvent was evaporated under reduced pressure.
- the residue was purified by silica gel column chromatography (chloroform-methanol), and the resulting solid was recrystallized from ethyl acetate-hexane-ethyl ether to give 2- ( 4 - ⁇ [4,-( 2 -Hydroxyethoxy) -3 -biphenylyl] methoxy ⁇ benzyl) -1,2,4-oxadiazolidine-3,5-dione (171 mg) was obtained as white crystals.
- the organic layer was washed with a saturated sodium chloride aqueous solution, the solvent was distilled off under reduced pressure, toluene was added to the residue, and the solvent was distilled off again under reduced pressure.
- the residue was purified by silica gel column chromatography (hexane-ethyl acetate) to a pale yellow foam (620 mg), THF (5 ml), methanol (5 ml) and 1M aqueous sodium hydroxide (1.47 ml) The mixture was stirred and stirred at room temperature for 5 minutes.
- the organic layer was washed with a saturated sodium chloride aqueous solution and then dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure, toluene was added to the residue, the solvent was distilled off again under reduced pressure, and the residue was purified by silica gel column chromatography (hexane-ethyl acetate) to obtain a pale yellow foam. (406mg) to methanol (1 0 ml) and sodium methoxide (52 mg) were added, and the mixture was heated with stirring at 60 ° C for 2 hours.
- the solvent was distilled off under reduced pressure, and black residue was added to the residue, washed with water and saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate.
- the pale yellow foam (373 mg) obtained by distilling off the solvent under reduced pressure was charged with THF (5 ml), methanol (5 ml) and 1M aqueous sodium hydroxide solution (0.81 ml), and stirred at room temperature for 5 minutes. did.
- the solvent was distilled off under reduced pressure, and the residue was purified by ODS column chromatography (water-acetonitrile) to obtain a solid by adding jetyl ether to the pale yellow foam obtained by filtration.
- a mixture of 4-oxadiazolidine-3,5-dione (340 mg) and acetic acid (6 ml) was stirred at room temperature for 20 hours.
- To the reaction solution was added sodium triacetoxyborohydride (573 mg), and the mixture was stirred at room temperature for 2 hours. After distilling off the solvent under reduced pressure, water was added to the residue and extracted with black mouth form. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (hexane-ethyl acetate), and the resulting pale yellow foam (719 mg), THF (5 ml), methanol (5 ml) and 1M sodium hydroxide A mixture of aqueous solution (4 ml) was stirred at 50 ° C. for 4 hours. 1M hydrochloric acid was added to the reaction mixture to adjust the pH to 4 to 5, and then extracted with black mouth form. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (hexane-ethyl acetate), and a mixture of the obtained pale yellow oil (448 mg), THF (3 ml) and methanol (3 ml) was added with 1M aqueous sodium hydroxide solution ( 0.8 9 ml) was added and stirred for 10 minutes. After distilling off the solvent under reduced pressure, the resulting residue was washed with jetyl ether and sodium 2- [4-( ⁇ [4,-(3-hydroxy-3-methylbutoxy) -2,2, -dimethyl]. Rubiphenyl-3-yl] methyl ⁇ amino) benzyl] -3,5-dioxo-1,2,4-oxadiazolidin-4-ide (398 mg) was obtained as a white solid.
- Acetic acid 3-( ⁇ 3,-[(4-formylphenoxy) methyl] -2,6-dimethylbiphenyl-4-yl ⁇ oxy ) A mixture of propyl (675 mg), hydroxylamine hydrochloride (217 mg), sodium acetate (307 mg), ethanol (15 ml) and water (4 ml) was stirred at room temperature for 18 hours. The solvent was distilled off under reduced pressure, water was added to the residue, and the mixture was extracted with black mouth form. The organic layer was washed with a saturated sodium chloride aqueous solution and dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure, and acetic acid (5 ml) and sodium cyanobium borohydride (196 mg) were added to the residue, followed by stirring at room temperature for 7 hours.
- a saturated aqueous sodium carbonate solution was added to the reaction system to make it alkaline, and the mixture was extracted with black mouth form.
- the organic layer was washed with a saturated sodium chloride aqueous solution, and then the solvent was distilled off under reduced pressure.
- the residue was purified by silica gel chromatography (black form-methanol), colorless oil (256 mg) obtained, THF (lOml) was added and ice-cooled, and black-colored carboxylic isocyanate ( 0.05 ml) was added dropwise and stirred at room temperature for 15.5 hours.
- the solvent was distilled off under reduced pressure, and chloroform was added to the residue, followed by washing with 1M hydrochloric acid and saturated sodium chloride aqueous solution.
- the solvent was distilled off under reduced pressure, and the residue was purified by silica gel chromatography (hexane-ethyl acetate) to a colorless oil (242 mg), methanol (10 ml) and sodium methoxide (92 mg).
- the mixture was heated and stirred at 60 ° C for 2 hours.
- the solvent was distilled off under reduced pressure, water was added to the residue, extracted with chloroform, and washed with a saturated aqueous sodium chloride solution.
- the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (hexane-ethyl acetate) to a colorless oil (152 mg), THF (5 ml), methanol (5 ml) and 1M hydroxylation.
- Aqueous sodium solution (0.33 ml) was added and stirred at room temperature for 5 minutes.
- a saturated aqueous sodium hydrogen carbonate solution (20 ml) was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane-ethyl acetate) to obtain a colorless oil (0.50 g). To the mixture of the obtained oil and THF (5 ml), black-mouthed carbonic acid (90 ml) was added, and the mixture was stirred at room temperature for 15 minutes and allowed to stand overnight. Water (10 ml) was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the obtained residue was purified by silica gel column chromatography (hexane-ethyl acetate) to obtain a colorless oil (33 lmg).
- THF 10 ml
- methanol l ml
- 1M aqueous sodium hydroxide solution 0.7 ml
- a solid obtained by filtering the jetyl ether in the residue was collected by filtration, dried under reduced pressure at 60 ° C, and sodium 2- (4- ⁇ [4 '-(2-hydroxyethoxy) -2'-methylbiphenol. -L-3-yl] methoxy ⁇ benzyl) -3,5-dioxo-1,2,4-oxadiazolidine-4-id (243 mg) was obtained as a colorless solid.
- the organic layer was washed with a saturated sodium chloride aqueous solution and then dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (hexane-ethyl acetate and chloroform-form-methanol) .
- the pale yellow oil (178 mg) obtained was purified by adding methanol (5 ml), THF (5 ml) and 1M aqueous sodium hydroxide solution (0.3 6 ml) was added and stirred at room temperature for 5 minutes.
- Acetic acid 2,2,2-trifluoro-1-[( ⁇ 3,-[(4-formylphenoxy) methyl] -2,6-dimethylbiphenyl-4-yl ⁇ oxy) methyl] ethyl A mixture of (590 mg), hydroxylamine hydrochloride (253 mg), sodium acetate (378 mg), ethanol (15 ml) and water (4 ml) was stirred at room temperature for 21 hours. The solvent was distilled off under reduced pressure, water was added to the residue, and the mixture was extracted with black mouth form. The organic layer was washed with a saturated aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (hexane-ethyl acetate) to a colorless oil (310 mg), acetic acid (8 ml) and sodium cyanoborohydride (127 mg). And stirred at room temperature for 4 hours.
- the solvent was distilled off under reduced pressure, and the residue was made alkaline with 1M aqueous sodium hydroxide solution and extracted with black mouth form.
- the organic layer was washed with a saturated aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (chloroform formaldehyde-methanol).
- the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (hexane-ethyl acetate) to obtain a colorless foam (16 4 mg), methanol (4 ml), THF (4 ml), 1M An aqueous potassium hydroxide solution (0.31 ml) was added and stirred at room temperature for 10 minutes.
- the solvent was distilled off under reduced pressure, ethyl acetate was added to the residue, the solvent was distilled off again under reduced pressure, and the residue was charged with jetyl ether and stirred at room temperature. The precipitated solid was collected by filtration and then dried by heating under reduced pressure.
- the residue was purified by silica gel column chromatography (chloroform form-methanol and hexane-ethyl acetate), and colorless foam (196 mg) was added to methanol (3 ml), THF (3 ml) and 1M hydroxide. Aqueous sodium solution (0.41 ml) was stirred at room temperature for 10 minutes. The solvent was distilled off under reduced pressure, the residue was purified by ODS column chromatography (water-acetonitrile), and the solid foam was obtained by adding jetyl ether to the colorless foam, and the solid was collected by filtration.
- the solvent was distilled off under reduced pressure, and acetic acid (10 ml) and sodium cyanoborohydride (167 mg) were added to the residue, followed by stirring at room temperature for 4 hours.
- the reaction mixture was made alkaline by adding 1M sodium hydroxide aqueous solution and extracted with chloroform. The organic layer was washed with a saturated sodium chloride aqueous solution and then dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure, and THF (8 ml) was added to the resulting colorless foam (528 mg), ice-cooled, and black-mouthed carbonate isocyanate (0.094 ml) was added dropwise, and the mixture was stirred at room temperature for 14.5 hours. Stir.
- the obtained residue was purified by silica gel column chromatography (black mouth form-methanol), and the resulting colorless foam (1.17 g) in THF (15 ml) was added to a THF (15 ml) solution under ice cooling with a black mouth carboisocyanate ( 0.234 ml) was added and stirred at room temperature for 24 hours.
- 1M Hydrochloric acid was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
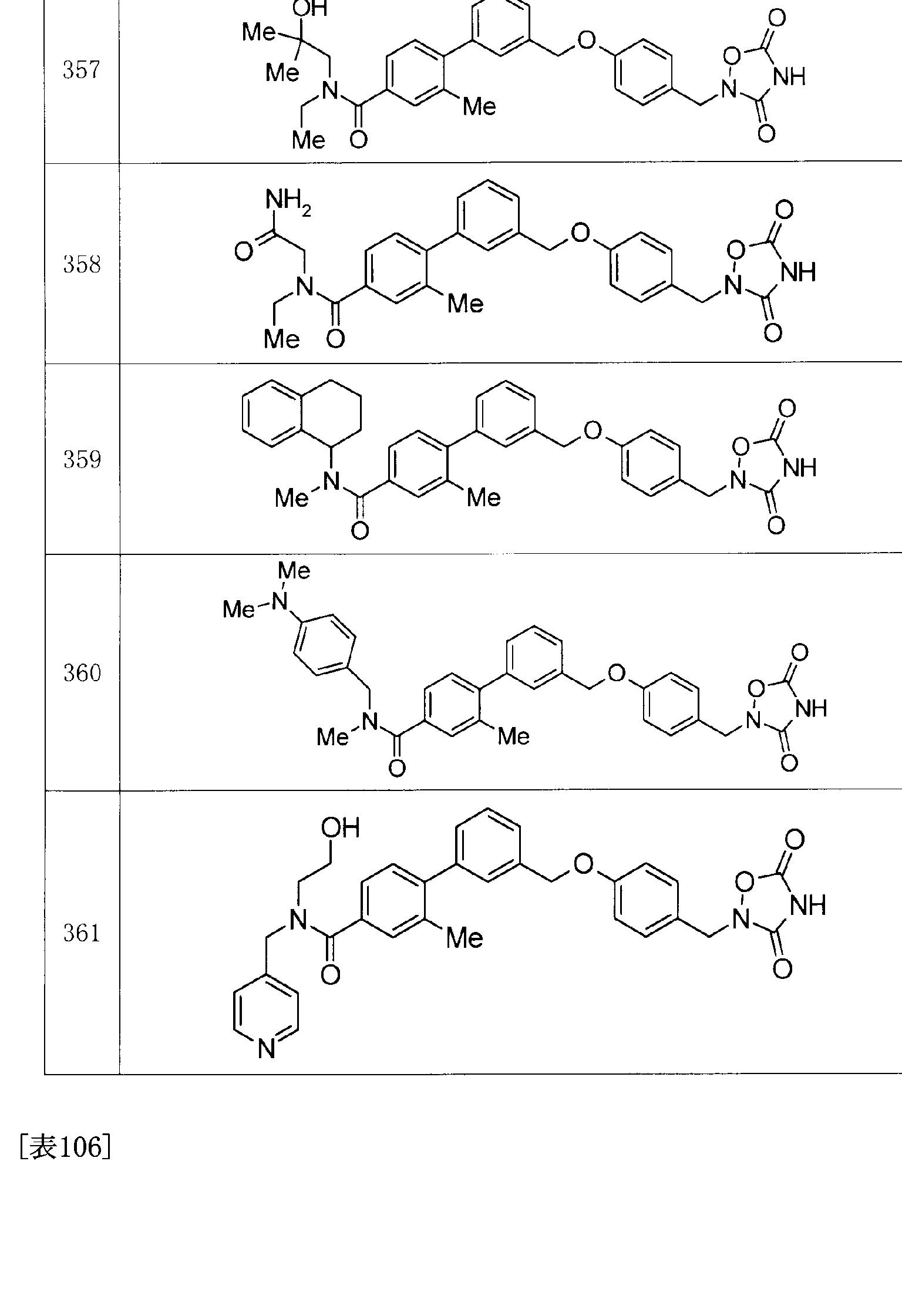
- Tables 136 to 138 show structures of other compounds of the present invention. These can be easily synthesized by using the above-described production methods, the methods described in the examples, methods obvious to those skilled in the art, or variations thereof. [0129] [Table 4]
- Thigh 1 1.92 (6H, s), 3.69-3.75 (2H, m), 3.97-4.00 (2H, m), 4.32 (2 H, s), 4.84-4.87 (IH, m), 5.13 (2H, s ), 6.66 (2H, s), 6.94-6.96 (2
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Description




























































































































Claims

Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2007241759A AU2007241759B2 (en) | 2006-04-24 | 2007-04-23 | Oxadiazolidinedione compound |
| JP2008512175A JP5309991B2 (ja) | 2006-04-24 | 2007-04-23 | オキサジアゾリジンジオン化合物 |
| NZ572146A NZ572146A (en) | 2006-04-24 | 2007-04-23 | Oxadiazolidinedione compound |
| BRPI0710531-2A BRPI0710531A2 (pt) | 2006-04-24 | 2007-04-23 | composto oxadiazolidinodiona. |
| US12/298,522 US7968552B2 (en) | 2006-04-24 | 2007-04-23 | Oxadiazolidinedione compound |
| CA002650124A CA2650124A1 (en) | 2006-04-24 | 2007-04-23 | Oxadiazolidinedione compound |
| EP07742129A EP2011788A4 (en) | 2006-04-24 | 2007-04-23 | OXADIAZOLIDINDIONVERBINDUNG |
| CN2007800146618A CN101426775B (zh) | 2006-04-24 | 2007-04-23 | *二唑烷二酮化合物 |
| MX2008013660A MX2008013660A (es) | 2006-04-24 | 2007-04-23 | Compuesto de oxiadiazolidinodiona. |
| ZA2008/08639A ZA200808639B (en) | 2006-04-24 | 2008-10-14 | Oxadiazolidinedione compound |
| NO20084919A NO20084919L (no) | 2006-04-24 | 2008-11-21 | Oksadiazolidindionforbindelse |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006118630 | 2006-04-24 | ||
| JP2006-118630 | 2006-04-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007123225A1 true WO2007123225A1 (ja) | 2007-11-01 |
Family
ID=38625122
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/058694 WO2007123225A1 (ja) | 2006-04-24 | 2007-04-23 | オキサジアゾリジンジオン化合物 |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US7968552B2 (ja) |
| EP (1) | EP2011788A4 (ja) |
| JP (1) | JP5309991B2 (ja) |
| KR (1) | KR20090005208A (ja) |
| CN (1) | CN101426775B (ja) |
| AR (1) | AR060628A1 (ja) |
| AU (1) | AU2007241759B2 (ja) |
| BR (1) | BRPI0710531A2 (ja) |
| CA (1) | CA2650124A1 (ja) |
| MX (1) | MX2008013660A (ja) |
| NO (1) | NO20084919L (ja) |
| NZ (1) | NZ572146A (ja) |
| RU (1) | RU2440994C2 (ja) |
| TW (1) | TW200815377A (ja) |
| UA (1) | UA94455C2 (ja) |
| WO (1) | WO2007123225A1 (ja) |
| ZA (1) | ZA200808639B (ja) |
Cited By (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7465804B2 (en) | 2005-05-20 | 2008-12-16 | Amgen Inc. | Compounds, pharmaceutical compositions and methods for their use in treating metabolic disorders |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2009054423A1 (ja) | 2007-10-24 | 2009-04-30 | Astellas Pharma Inc. | オキサジアゾリジンジオン化合物 |
| US7572934B2 (en) | 2007-04-16 | 2009-08-11 | Amgen Inc. | Substituted biphenyl GPR40 modulators |
| US7582803B2 (en) | 2005-09-14 | 2009-09-01 | Amgen Inc. | Conformationally constrained 3-(4-hydroxy-phenyl)-substituted-propanoic acids useful for treating metabolic disorders |
| US7687526B2 (en) | 2006-09-07 | 2010-03-30 | Amgen Inc. | Benzo-fused compounds for use in treating metabolic disorders |
| US7714008B2 (en) | 2006-09-07 | 2010-05-11 | Amgen Inc. | Heterocyclic GPR40 modulators |
| US7834037B2 (en) | 2005-11-04 | 2010-11-16 | Amira Pharmaceuticals, Inc. | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| WO2011052756A1 (ja) | 2009-10-30 | 2011-05-05 | 持田製薬株式会社 | 新規3-ヒドロキシ-5-アリールイソキサゾール誘導体 |
| WO2011078371A1 (ja) | 2009-12-25 | 2011-06-30 | 持田製薬株式会社 | 新規3-ヒドロキシ-5-アリールイソチアゾール誘導体 |
| US7977359B2 (en) | 2005-11-04 | 2011-07-12 | Amira Pharmaceuticals, Inc. | 5-lipdxygenase-activating protein (FLAP) inhibitors |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| US8030354B2 (en) | 2007-10-10 | 2011-10-04 | Amgen Inc. | Substituted biphenyl GPR40 modulators |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012046869A1 (ja) | 2010-10-08 | 2012-04-12 | 持田製薬株式会社 | 環状アミド誘導体 |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012147518A1 (ja) | 2011-04-27 | 2012-11-01 | 持田製薬株式会社 | 新規3-ヒドロキシイソチアゾール 1-オキシド誘導体 |
| WO2012147516A1 (ja) | 2011-04-28 | 2012-11-01 | 持田製薬株式会社 | 環状アミド誘導体 |
| WO2012150221A2 (de) | 2011-05-04 | 2012-11-08 | Bayer Cropscience Ag | Neue halogenierte benzylalkoholester der cyclopropancarbonsäure als schädlingsbekämpfungsmittel |
| WO2012150208A1 (de) | 2011-05-04 | 2012-11-08 | Bayer Cropscience Ag | Verwendung von substituierten benzylalkoholestern der cyclopropancarbonsäure zur bekämpfung von insektizid-resistenten insekten |
| US8399666B2 (en) | 2005-11-04 | 2013-03-19 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8450522B2 (en) | 2008-03-06 | 2013-05-28 | Amgen Inc. | Conformationally constrained carboxylic acid derivatives useful for treating metabolic disorders |
| US8546431B2 (en) | 2008-10-01 | 2013-10-01 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| WO2014011926A1 (en) | 2012-07-11 | 2014-01-16 | Elcelyx Therapeutics, Inc. | Compositions comprising statins, biguanides and further agents for reducing cardiometabolic risk |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| EP2674417A3 (en) * | 2007-11-21 | 2014-04-09 | Decode Genetics EHF | Biaryl PDE4 inhibitors for treating inflammation |
| US8697730B2 (en) | 2007-10-26 | 2014-04-15 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase activating protein (FLAP) inhibitor |
| JP2014509600A (ja) * | 2011-03-14 | 2014-04-21 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Gpr119モジュレーターとしてのn−シクロプロピル−n−ピペリジニルベンズアミド |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| US8748462B2 (en) | 2008-10-15 | 2014-06-10 | Amgen Inc. | Spirocyclic GPR40 modulators |
| US8772495B2 (en) | 2008-05-23 | 2014-07-08 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein inhibitor |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2015020184A1 (ja) * | 2013-08-09 | 2015-02-12 | 武田薬品工業株式会社 | 芳香環化合物 |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2015095256A1 (en) | 2013-12-19 | 2015-06-25 | Merck Sharp & Dohme Corp. | Antidiabetic substituted heteroaryl compounds |
| WO2015097713A1 (en) | 2013-11-14 | 2015-07-02 | Cadila Healthcare Limited | Novel heterocyclic compounds |
| JP2015524808A (ja) * | 2012-07-25 | 2015-08-27 | ユニバーシティ・オブ・シンシナティ | アポリポタンパク質aivを用いたi型糖尿病の治療法 |
| WO2015176640A1 (en) | 2014-05-22 | 2015-11-26 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2016022446A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | [5,6]-fused bicyclic antidiabetic compounds |
| WO2016022448A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2016019863A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | [7,6]-fused bicyclic antidiabetic compounds |
| WO2016022742A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| KR20160048946A (ko) * | 2013-09-04 | 2016-05-04 | 브리스톨-마이어스 스큅 컴퍼니 | 면역조절제로서 유용한 화합물 |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| US9957219B2 (en) | 2013-12-04 | 2018-05-01 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| US10059667B2 (en) | 2014-02-06 | 2018-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic compounds |
| US10519115B2 (en) | 2013-11-15 | 2019-12-31 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US10676458B2 (en) | 2016-03-29 | 2020-06-09 | Merch Sharp & Dohne Corp. Rahway | Antidiabetic bicyclic compounds |
| US10710986B2 (en) | 2018-02-13 | 2020-07-14 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10774071B2 (en) | 2018-07-13 | 2020-09-15 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10899735B2 (en) | 2018-04-19 | 2021-01-26 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11225471B2 (en) | 2017-11-16 | 2022-01-18 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US11236085B2 (en) | 2018-10-24 | 2022-02-01 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
| US11512065B2 (en) | 2019-10-07 | 2022-11-29 | Kallyope, Inc. | GPR119 agonists |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103864754B (zh) * | 2012-12-10 | 2016-12-21 | 中国科学院上海药物研究所 | 五元唑类杂环化合物及其制备方法、药物组合物和用途 |
| JP6387023B2 (ja) | 2013-02-28 | 2018-09-05 | ティウムバイオ カンパニー、リミテッドTiumBio Co., Ltd. | 三環式化合物およびその使用 |
| US9850225B2 (en) | 2014-04-14 | 2017-12-26 | Bristol-Myers Squibb Company | Compounds useful as immunomodulators |
| US10745382B2 (en) * | 2015-10-15 | 2020-08-18 | Bristol-Myers Squibb Company | Compounds useful as immunomodulators |
| CA3029991A1 (en) * | 2016-07-08 | 2018-01-11 | Bristol-Myers Squibb Company | 1,3-dihydroxy-phenyl derivatives useful as immunomodulators |
| US10144706B2 (en) | 2016-09-01 | 2018-12-04 | Bristol-Myers Squibb Company | Compounds useful as immunomodulators |
| KR102599339B1 (ko) | 2016-12-20 | 2023-11-08 | 브리스톨-마이어스 스큅 컴퍼니 | 면역조정제로서 유용한 화합물 |
| ES2961550T3 (es) | 2017-03-27 | 2024-03-12 | Bristol Myers Squibb Co | Derivados de isoquinolina sustituidos como inmunomoduladores |
| JP7214752B2 (ja) | 2018-01-23 | 2023-01-30 | ブリストル-マイヤーズ スクイブ カンパニー | 免疫調節剤として有用な2,8-ジアシル-2,8-ジアザスピロ[5.5]ウンデカン化合物 |
| ES2933601T3 (es) | 2018-03-01 | 2023-02-10 | Bristol Myers Squibb Co | Compuestos útiles como inmunomoduladores |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0625448A (ja) | 1991-08-15 | 1994-02-01 | Oji Kako Kk | 紫外線遮蔽フィルムの製造方法 |
| WO1994025448A1 (en) * | 1993-04-30 | 1994-11-10 | Yamanouchi Pharmaceutical Co., Ltd. | Novel bisoxadiazolidine derivative |
| JPH072848A (ja) | 1993-04-23 | 1995-01-06 | Sankyo Co Ltd | モルホリンおよびチオモルホリン誘導体 |
| JPH0730664A (ja) | 1993-07-08 | 1995-01-31 | Nippon Telegr & Teleph Corp <Ntt> | 音声ダイヤル方式 |
| US5480896A (en) | 1994-01-27 | 1996-01-02 | American Home Products Corporation | Aralkyl-1,2,4-oxadiazolidine-3,5-diones as antihyperglycemic agents |
| JPH0859638A (ja) * | 1994-08-26 | 1996-03-05 | Yamanouchi Pharmaceut Co Ltd | 新規なヘテロ環誘導体又はその塩 |
| JPH0941097A (ja) | 1995-07-28 | 1997-02-10 | Nisshin Steel Co Ltd | 吸収式冷暖房機機用オーステナイト系ステンレス鋼 |
| JPH10502907A (ja) * | 1994-05-10 | 1998-03-17 | アメリカン・ホーム・プロダクツ・コーポレイション | 抗高血糖症薬としての新規なジ−オキサジアゾール誘導体 |
| JP2000212174A (ja) | 1993-08-09 | 2000-08-02 | Takeda Chem Ind Ltd | 2,4−オキサゾリジンジオン誘導体、その製造法およびそれを含んでなる医薬組成物 |
| JP2002503255A (ja) * | 1997-06-17 | 2002-01-29 | アストラ・アクチエボラーグ | 新しいチアゾリジンジオン、オキサゾリジンジオンおよびオキサジアゾリジンジオン誘導体 |
| JP2002515874A (ja) * | 1996-12-31 | 2002-05-28 | ドクター・レディーズ・リサーチ・ファウンデーション | 新規なヘテロ環化合物、これらの製造方法及びこれらを含有する薬学的組成物、並びに糖尿病及び関連疾患の治療におけるこれらの使用 |
| JP2005015461A (ja) * | 2002-11-08 | 2005-01-20 | Takeda Chem Ind Ltd | 受容体機能調節剤 |
| JP2005030203A (ja) | 2003-07-10 | 2005-02-03 | Doris Engineering | 例えばメタンタンカーのような荷積み降ろし船のためのフローティングターミナル |
| JP2005063725A (ja) | 2003-08-08 | 2005-03-10 | Fujitsu Hitachi Plasma Display Ltd | フラットパネルディスプレイの製造方法 |
| JP2005063729A (ja) | 2003-08-08 | 2005-03-10 | Takao Makabe | 静電気被災防止マット |
| WO2005030203A1 (en) * | 2003-09-25 | 2005-04-07 | Wyeth | Substituted oxadiazolidinediones als pai-1 inhibitors |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5885997A (en) * | 1996-07-01 | 1999-03-23 | Dr. Reddy's Research Foundation | Heterocyclic compounds, process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases |
| CN1735408A (zh) * | 2002-11-08 | 2006-02-15 | 武田药品工业株式会社 | 受体机能调节剂 |
| US7960369B2 (en) * | 2002-11-08 | 2011-06-14 | Takeda Pharmaceutical Company Limited | Receptor function regulator |
| TW200523247A (en) | 2003-12-25 | 2005-07-16 | Takeda Pharmaceutical | 3-(4-benzyloxyphenyl) propanoic acid derivatives |
| WO2005063725A1 (ja) | 2003-12-26 | 2005-07-14 | Takeda Pharmaceutical Company Limited | フェニルプロパン酸誘導体 |
| US7816367B2 (en) | 2004-02-27 | 2010-10-19 | Amgen Inc. | Compounds, pharmaceutical compositions and methods for use in treating metabolic disorders |
| US7786165B2 (en) | 2004-03-15 | 2010-08-31 | Takeda Pharmaceutical Company Limited | Aminophenylpropanoic acid derivative |
-
2007
- 2007-04-20 TW TW096114040A patent/TW200815377A/zh unknown
- 2007-04-23 AU AU2007241759A patent/AU2007241759B2/en not_active Expired - Fee Related
- 2007-04-23 WO PCT/JP2007/058694 patent/WO2007123225A1/ja active Application Filing
- 2007-04-23 CN CN2007800146618A patent/CN101426775B/zh not_active Expired - Fee Related
- 2007-04-23 MX MX2008013660A patent/MX2008013660A/es active IP Right Grant
- 2007-04-23 EP EP07742129A patent/EP2011788A4/en not_active Withdrawn
- 2007-04-23 KR KR1020087028510A patent/KR20090005208A/ko not_active Application Discontinuation
- 2007-04-23 US US12/298,522 patent/US7968552B2/en not_active Expired - Fee Related
- 2007-04-23 UA UAA200813478A patent/UA94455C2/ru unknown
- 2007-04-23 NZ NZ572146A patent/NZ572146A/en not_active IP Right Cessation
- 2007-04-23 JP JP2008512175A patent/JP5309991B2/ja not_active Expired - Fee Related
- 2007-04-23 BR BRPI0710531-2A patent/BRPI0710531A2/pt not_active IP Right Cessation
- 2007-04-23 RU RU2008146088/04A patent/RU2440994C2/ru not_active IP Right Cessation
- 2007-04-23 CA CA002650124A patent/CA2650124A1/en not_active Abandoned
- 2007-04-24 AR ARP070101751A patent/AR060628A1/es not_active Application Discontinuation
-
2008
- 2008-10-14 ZA ZA2008/08639A patent/ZA200808639B/en unknown
- 2008-11-21 NO NO20084919A patent/NO20084919L/no not_active Application Discontinuation
Patent Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0625448A (ja) | 1991-08-15 | 1994-02-01 | Oji Kako Kk | 紫外線遮蔽フィルムの製造方法 |
| JPH072848A (ja) | 1993-04-23 | 1995-01-06 | Sankyo Co Ltd | モルホリンおよびチオモルホリン誘導体 |
| WO1994025448A1 (en) * | 1993-04-30 | 1994-11-10 | Yamanouchi Pharmaceutical Co., Ltd. | Novel bisoxadiazolidine derivative |
| JPH0730664A (ja) | 1993-07-08 | 1995-01-31 | Nippon Telegr & Teleph Corp <Ntt> | 音声ダイヤル方式 |
| JP2000212174A (ja) | 1993-08-09 | 2000-08-02 | Takeda Chem Ind Ltd | 2,4−オキサゾリジンジオン誘導体、その製造法およびそれを含んでなる医薬組成物 |
| US5480896A (en) | 1994-01-27 | 1996-01-02 | American Home Products Corporation | Aralkyl-1,2,4-oxadiazolidine-3,5-diones as antihyperglycemic agents |
| JPH10502907A (ja) * | 1994-05-10 | 1998-03-17 | アメリカン・ホーム・プロダクツ・コーポレイション | 抗高血糖症薬としての新規なジ−オキサジアゾール誘導体 |
| JPH0859638A (ja) * | 1994-08-26 | 1996-03-05 | Yamanouchi Pharmaceut Co Ltd | 新規なヘテロ環誘導体又はその塩 |
| JPH0941097A (ja) | 1995-07-28 | 1997-02-10 | Nisshin Steel Co Ltd | 吸収式冷暖房機機用オーステナイト系ステンレス鋼 |
| JP2002515874A (ja) * | 1996-12-31 | 2002-05-28 | ドクター・レディーズ・リサーチ・ファウンデーション | 新規なヘテロ環化合物、これらの製造方法及びこれらを含有する薬学的組成物、並びに糖尿病及び関連疾患の治療におけるこれらの使用 |
| JP2002503255A (ja) * | 1997-06-17 | 2002-01-29 | アストラ・アクチエボラーグ | 新しいチアゾリジンジオン、オキサゾリジンジオンおよびオキサジアゾリジンジオン誘導体 |
| JP2005015461A (ja) * | 2002-11-08 | 2005-01-20 | Takeda Chem Ind Ltd | 受容体機能調節剤 |
| JP2005030203A (ja) | 2003-07-10 | 2005-02-03 | Doris Engineering | 例えばメタンタンカーのような荷積み降ろし船のためのフローティングターミナル |
| JP2005063725A (ja) | 2003-08-08 | 2005-03-10 | Fujitsu Hitachi Plasma Display Ltd | フラットパネルディスプレイの製造方法 |
| JP2005063729A (ja) | 2003-08-08 | 2005-03-10 | Takao Makabe | 静電気被災防止マット |
| WO2005030203A1 (en) * | 2003-09-25 | 2005-04-07 | Wyeth | Substituted oxadiazolidinediones als pai-1 inhibitors |
Non-Patent Citations (10)
| Title |
|---|
| "Bunshi Sekkei", vol. 7, 1990, HIROKAWA SHOTEN, article "Iyakuhin no Kaihatsu", pages: 163 - 198 |
| "Progress in Medicine", LIFESCIENCE MEDICA, vol. 5, 1985, pages 2157 - 2161 |
| "Protective Groups in Organic Synthesis" (USA", 1999, JOHN WILEY & SONS |
| BRIGHT S.W. ET AL.: "Monoclonal Antibodies as Surragate Receptors in A High Throughput Screen for Compounds That Enhance Insulin Sensitivity", LIFE SCIENCES, vol. 61, no. 23, 1997, pages 2305 - 2315, XP002192657 * |
| EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, (FRANCE, vol. 36, 2001, pages 31 - 42 |
| MADHAVAN G.R. ET AL.: "Synthesis and Biological Activity of Novel Pyrimidione Containing Thiazolidinedione Derivatives", BIOORGANIC & MEDICINAL CHEMISTRY, vol. 10, no. 8, 2002, pages 2671 - 2680, XP003012941 * |
| MALAMAS M.S. ET AL.: "Antihyperglycemic activity of new 1,2,4-oxadiazolidine-3,5-diones", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 36, no. 1, January 2001 (2001-01-01), pages 31 - 42, XP002211978 * |
| NATURE, (ENGLAND, vol. 422, 2003, pages 173 - 176 |
| NUCLEIC ACIDS RESEARCH, vol. 18, 1990, pages 5322 |
| See also references of EP2011788A4 * |
Cited By (105)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7465804B2 (en) | 2005-05-20 | 2008-12-16 | Amgen Inc. | Compounds, pharmaceutical compositions and methods for their use in treating metabolic disorders |
| US7582803B2 (en) | 2005-09-14 | 2009-09-01 | Amgen Inc. | Conformationally constrained 3-(4-hydroxy-phenyl)-substituted-propanoic acids useful for treating metabolic disorders |
| US8841295B2 (en) | 2005-11-04 | 2014-09-23 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US7834037B2 (en) | 2005-11-04 | 2010-11-16 | Amira Pharmaceuticals, Inc. | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US8710081B2 (en) | 2005-11-04 | 2014-04-29 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US8399666B2 (en) | 2005-11-04 | 2013-03-19 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US7977359B2 (en) | 2005-11-04 | 2011-07-12 | Amira Pharmaceuticals, Inc. | 5-lipdxygenase-activating protein (FLAP) inhibitors |
| US8003648B2 (en) | 2006-09-07 | 2011-08-23 | Amgen Inc. | Heterocyclic GPR40 modulators |
| US7714008B2 (en) | 2006-09-07 | 2010-05-11 | Amgen Inc. | Heterocyclic GPR40 modulators |
| US7687526B2 (en) | 2006-09-07 | 2010-03-30 | Amgen Inc. | Benzo-fused compounds for use in treating metabolic disorders |
| US7572934B2 (en) | 2007-04-16 | 2009-08-11 | Amgen Inc. | Substituted biphenyl GPR40 modulators |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US8030354B2 (en) | 2007-10-10 | 2011-10-04 | Amgen Inc. | Substituted biphenyl GPR40 modulators |
| US20100267775A1 (en) * | 2007-10-24 | 2010-10-21 | Kenji Negoro | Oxadiazolidinedione compound |
| EP2216330A4 (en) * | 2007-10-24 | 2011-09-14 | Astellas Pharma Inc | OXADIAZOLIDINEDIONE COMPOUND |
| WO2009054423A1 (ja) | 2007-10-24 | 2009-04-30 | Astellas Pharma Inc. | オキサジアゾリジンジオン化合物 |
| EP2216330A1 (en) * | 2007-10-24 | 2010-08-11 | Astellas Pharma Inc. | Oxadiazolidinedione compound |
| US8697730B2 (en) | 2007-10-26 | 2014-04-15 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase activating protein (FLAP) inhibitor |
| AU2008326309C1 (en) * | 2007-11-21 | 2015-03-12 | Decode Genetics Ehf | Biaryl PDE4 inhibitors for treating pulmonary and cardiovascular disorders |
| EP2674417A3 (en) * | 2007-11-21 | 2014-04-09 | Decode Genetics EHF | Biaryl PDE4 inhibitors for treating inflammation |
| US8791267B2 (en) | 2007-11-21 | 2014-07-29 | Decode Genetics Ehf | Biaryl PDE4 inhibitors for treating inflammatory, cardiovascular and CNS disorders |
| AU2008326309B2 (en) * | 2007-11-21 | 2014-10-02 | Decode Genetics Ehf | Biaryl PDE4 inhibitors for treating pulmonary and cardiovascular disorders |
| US8450522B2 (en) | 2008-03-06 | 2013-05-28 | Amgen Inc. | Conformationally constrained carboxylic acid derivatives useful for treating metabolic disorders |
| US8772495B2 (en) | 2008-05-23 | 2014-07-08 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein inhibitor |
| US8546431B2 (en) | 2008-10-01 | 2013-10-01 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US8748462B2 (en) | 2008-10-15 | 2014-06-10 | Amgen Inc. | Spirocyclic GPR40 modulators |
| WO2011052756A1 (ja) | 2009-10-30 | 2011-05-05 | 持田製薬株式会社 | 新規3-ヒドロキシ-5-アリールイソキサゾール誘導体 |
| US8455500B2 (en) | 2009-10-30 | 2013-06-04 | Mochida Pharmaceutical Co., Ltd. | 3-hydroxy-5-arylisoxazole derivative |
| US8476287B2 (en) | 2009-12-25 | 2013-07-02 | Mochida Pharmaceutical Co., Ltd. | 3-hydroxy-5-arylisothiazole derivative |
| WO2011078371A1 (ja) | 2009-12-25 | 2011-06-30 | 持田製薬株式会社 | 新規3-ヒドロキシ-5-アリールイソチアゾール誘導体 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US9040525B2 (en) | 2010-10-08 | 2015-05-26 | Mochida Pharmaceutical Co., Ltd. | Cyclic amide derivative |
| WO2012046869A1 (ja) | 2010-10-08 | 2012-04-12 | 持田製薬株式会社 | 環状アミド誘導体 |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| JP2014509600A (ja) * | 2011-03-14 | 2014-04-21 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Gpr119モジュレーターとしてのn−シクロプロピル−n−ピペリジニルベンズアミド |
| US8765752B2 (en) | 2011-04-27 | 2014-07-01 | Mochida Pharmaceutical Co., Ltd. | 3-hydroxyisothiazole 1-oxide derivatives |
| WO2012147518A1 (ja) | 2011-04-27 | 2012-11-01 | 持田製薬株式会社 | 新規3-ヒドロキシイソチアゾール 1-オキシド誘導体 |
| US8629102B2 (en) | 2011-04-27 | 2014-01-14 | Mochida Pharmaceutical Co., Ltd. | 3-hydroxyisothiazole 1-oxide derivatives |
| US8557766B2 (en) | 2011-04-27 | 2013-10-15 | Mochida Pharmaceutical Co., Ltd. | 3-hydroxyisothiazole 1-oxide derivatives |
| US9072758B2 (en) | 2011-04-28 | 2015-07-07 | Mochida Pharmaceutical Co., Ltd. | Cyclic amide derivative |
| WO2012147516A1 (ja) | 2011-04-28 | 2012-11-01 | 持田製薬株式会社 | 環状アミド誘導体 |
| WO2012150221A2 (de) | 2011-05-04 | 2012-11-08 | Bayer Cropscience Ag | Neue halogenierte benzylalkoholester der cyclopropancarbonsäure als schädlingsbekämpfungsmittel |
| WO2012150208A1 (de) | 2011-05-04 | 2012-11-08 | Bayer Cropscience Ag | Verwendung von substituierten benzylalkoholestern der cyclopropancarbonsäure zur bekämpfung von insektizid-resistenten insekten |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014011926A1 (en) | 2012-07-11 | 2014-01-16 | Elcelyx Therapeutics, Inc. | Compositions comprising statins, biguanides and further agents for reducing cardiometabolic risk |
| JP2015524808A (ja) * | 2012-07-25 | 2015-08-27 | ユニバーシティ・オブ・シンシナティ | アポリポタンパク質aivを用いたi型糖尿病の治療法 |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US9527875B2 (en) | 2012-08-02 | 2016-12-27 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US9840512B2 (en) | 2013-02-22 | 2017-12-12 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| JPWO2015020184A1 (ja) * | 2013-08-09 | 2017-03-02 | 武田薬品工業株式会社 | 芳香環化合物 |
| WO2015020184A1 (ja) * | 2013-08-09 | 2015-02-12 | 武田薬品工業株式会社 | 芳香環化合物 |
| US9776962B2 (en) * | 2013-08-09 | 2017-10-03 | Takeda Pharmaceutical Company Limited | Aromatic compounds with GPR40 agonistic activity |
| US20160115128A1 (en) * | 2013-08-09 | 2016-04-28 | Takeda Pharmaceutical Company Limited | Aromatic compound |
| KR102276644B1 (ko) | 2013-09-04 | 2021-07-13 | 브리스톨-마이어스 스큅 컴퍼니 | 면역조절제로서 유용한 화합물 |
| US9872852B2 (en) | 2013-09-04 | 2018-01-23 | Bristol-Myers Squibb Company | Compounds useful as immunomodulators |
| KR20160048946A (ko) * | 2013-09-04 | 2016-05-04 | 브리스톨-마이어스 스큅 컴퍼니 | 면역조절제로서 유용한 화합물 |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US9932311B2 (en) | 2013-10-08 | 2018-04-03 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US10011609B2 (en) | 2013-11-14 | 2018-07-03 | Cadila Healthcare Limited | Heterocyclic compounds |
| WO2015097713A1 (en) | 2013-11-14 | 2015-07-02 | Cadila Healthcare Limited | Novel heterocyclic compounds |
| US10246470B2 (en) | 2013-11-14 | 2019-04-02 | Cadila Healthcare Limited | Heterocyclic compounds |
| US10519115B2 (en) | 2013-11-15 | 2019-12-31 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US9957219B2 (en) | 2013-12-04 | 2018-05-01 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| EP3974413A1 (en) | 2013-12-19 | 2022-03-30 | Merck Sharp & Dohme Corp. | Antidiabetic substituted heteroaryl compounds |
| WO2015095256A1 (en) | 2013-12-19 | 2015-06-25 | Merck Sharp & Dohme Corp. | Antidiabetic substituted heteroaryl compounds |
| US9834563B2 (en) | 2013-12-19 | 2017-12-05 | Merck Sharp & Dohme Corp. | Antidiabetic substituted heteroaryl compounds |
| US10059667B2 (en) | 2014-02-06 | 2018-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic compounds |
| WO2015176640A1 (en) | 2014-05-22 | 2015-11-26 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US10000454B2 (en) | 2014-05-22 | 2018-06-19 | Merck Sharp & Dohme | Antidiabetic tricyclic compounds |
| US10100042B2 (en) | 2014-08-08 | 2018-10-16 | Merck Sharp & Dohme Corp. | [5,6]—fused bicyclic antidiabetic compounds |
| US10131651B2 (en) | 2014-08-08 | 2018-11-20 | Merck Sharp & Dohme Corp. | [7,6]-fused bicyclic antidiabetic compounds |
| WO2016022742A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US10968193B2 (en) | 2014-08-08 | 2021-04-06 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US10662171B2 (en) | 2014-08-08 | 2020-05-26 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2016022448A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2016019863A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | [7,6]-fused bicyclic antidiabetic compounds |
| WO2016022446A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | [5,6]-fused bicyclic antidiabetic compounds |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| US10676458B2 (en) | 2016-03-29 | 2020-06-09 | Merch Sharp & Dohne Corp. Rahway | Antidiabetic bicyclic compounds |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| US11072602B2 (en) | 2016-12-06 | 2021-07-27 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| US10968232B2 (en) | 2016-12-20 | 2021-04-06 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| US11225471B2 (en) | 2017-11-16 | 2022-01-18 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US11555029B2 (en) | 2018-02-13 | 2023-01-17 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10710986B2 (en) | 2018-02-13 | 2020-07-14 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10899735B2 (en) | 2018-04-19 | 2021-01-26 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10774071B2 (en) | 2018-07-13 | 2020-09-15 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11236085B2 (en) | 2018-10-24 | 2022-02-01 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11512065B2 (en) | 2019-10-07 | 2022-11-29 | Kallyope, Inc. | GPR119 agonists |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11851429B2 (en) | 2020-05-19 | 2023-12-26 | Kallyope, Inc. | AMPK activators |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2007123225A1 (ja) | 2009-09-10 |
| CA2650124A1 (en) | 2007-11-01 |
| RU2008146088A (ru) | 2010-05-27 |
| CN101426775B (zh) | 2012-10-31 |
| MX2008013660A (es) | 2008-11-10 |
| RU2440994C2 (ru) | 2012-01-27 |
| TW200815377A (en) | 2008-04-01 |
| NZ572146A (en) | 2010-09-30 |
| EP2011788A1 (en) | 2009-01-07 |
| ZA200808639B (en) | 2009-12-30 |
| KR20090005208A (ko) | 2009-01-12 |
| CN101426775A (zh) | 2009-05-06 |
| BRPI0710531A2 (pt) | 2012-06-19 |
| AU2007241759B2 (en) | 2012-05-17 |
| US20090186909A1 (en) | 2009-07-23 |
| NO20084919L (no) | 2009-01-20 |
| UA94455C2 (en) | 2011-05-10 |
| JP5309991B2 (ja) | 2013-10-09 |
| AR060628A1 (es) | 2008-07-02 |
| US7968552B2 (en) | 2011-06-28 |
| AU2007241759A1 (en) | 2007-11-01 |
| EP2011788A4 (en) | 2010-10-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007123225A1 (ja) | オキサジアゾリジンジオン化合物 | |
| JPWO2009054423A1 (ja) | オキサジアゾリジンジオン化合物 | |
| RU2293721C2 (ru) | Сложноэфирные соединения и их применение в медицине | |
| JP5564947B2 (ja) | キノロン誘導体 | |
| JP5820056B2 (ja) | Cetp阻害剤としてのシクロアルケニルアリール誘導体 | |
| US8153658B2 (en) | Piperidine derivative or salt thereof | |
| WO2008066097A1 (en) | Carboxylic acid derivative | |
| JP2013532185A (ja) | 化合物 | |
| WO2010123017A1 (ja) | テトラゾール化合物 | |
| TWI465439B (zh) | 三氮唑衍生物或其鹽 | |
| CA2646430A1 (en) | Bicyclic carboxylic acid derivatives useful for treating metabolic disorders | |
| WO2007085136A1 (fr) | Dérivés de 1,3-benzodioxolecyclopentène, procédé de préparation et applications médicales | |
| EP2154130A1 (en) | Pyridone compound | |
| WO2012102254A1 (ja) | インドール誘導体、またはその薬理学的に許容される塩 | |
| JP2011016722A (ja) | チアゾリジンジオン化合物 | |
| CN104125960A (zh) | 氮杂金刚烷的衍生物及其用途 | |
| JP4832897B2 (ja) | エステル誘導体及びその医薬用途 | |
| TW202308992A (zh) | Tead抑制劑 | |
| TW201920107A (zh) | 作為ttx-s阻斷劑之雙芳氧基衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07742129 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2008512175 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008502251 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 194675 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007241759 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 572146 Country of ref document: NZ Ref document number: 2007742129 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2650124 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780014661.8 Country of ref document: CN Ref document number: MX/a/2008/013660 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12298522 Country of ref document: US Ref document number: 5756/CHENP/2008 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007241759 Country of ref document: AU Date of ref document: 20070423 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087028510 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008146088 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0710531 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081020 |