WO2002060867A2 - Carbazole derivatives and their uses as heparanase inhibitors - Google Patents
Carbazole derivatives and their uses as heparanase inhibitors Download PDFInfo
- Publication number
- WO2002060867A2 WO2002060867A2 PCT/IL2002/000079 IL0200079W WO02060867A2 WO 2002060867 A2 WO2002060867 A2 WO 2002060867A2 IL 0200079 W IL0200079 W IL 0200079W WO 02060867 A2 WO02060867 A2 WO 02060867A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- heteroaryl
- alkyl
- nrr
- Prior art date
Links
- 108010037536 heparanase Proteins 0.000 title claims abstract description 95
- 102100024025 Heparanase Human genes 0.000 title claims abstract description 90
- 125000000609 carbazolyl group Chemical class C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 title claims abstract description 15
- 239000003112 inhibitor Substances 0.000 title abstract description 17
- -1 3-(substituted)amino-2-hydroxypropyl group Chemical group 0.000 claims abstract description 41
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 38
- 230000003197 catalytic effect Effects 0.000 claims abstract description 17
- 208000023275 Autoimmune disease Diseases 0.000 claims abstract description 16
- 208000037765 diseases and disorders Diseases 0.000 claims abstract description 9
- 201000011510 cancer Diseases 0.000 claims abstract description 8
- 150000001875 compounds Chemical class 0.000 claims description 177
- 125000001072 heteroaryl group Chemical group 0.000 claims description 111
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 95
- 239000008194 pharmaceutical composition Substances 0.000 claims description 84
- 229910052736 halogen Inorganic materials 0.000 claims description 83
- 150000002367 halogens Chemical class 0.000 claims description 83
- 125000005915 C6-C14 aryl group Chemical group 0.000 claims description 76
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 73
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 72
- 150000003254 radicals Chemical class 0.000 claims description 70
- 210000004027 cell Anatomy 0.000 claims description 66
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 62
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 61
- 229910006069 SO3H Inorganic materials 0.000 claims description 54
- 238000000034 method Methods 0.000 claims description 53
- 229910052739 hydrogen Inorganic materials 0.000 claims description 36
- 239000001257 hydrogen Substances 0.000 claims description 36
- 125000005842 heteroatom Chemical group 0.000 claims description 35
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 claims description 34
- 229940126214 compound 3 Drugs 0.000 claims description 34
- 208000035475 disorder Diseases 0.000 claims description 31
- 150000003839 salts Chemical class 0.000 claims description 31
- 201000010099 disease Diseases 0.000 claims description 30
- 229940122588 Heparanase inhibitor Drugs 0.000 claims description 29
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 28
- 230000002401 inhibitory effect Effects 0.000 claims description 25
- 150000002431 hydrogen Chemical class 0.000 claims description 21
- 201000001441 melanoma Diseases 0.000 claims description 21
- 206010027476 Metastases Diseases 0.000 claims description 20
- 230000009401 metastasis Effects 0.000 claims description 19
- 206010061218 Inflammation Diseases 0.000 claims description 17
- 230000002757 inflammatory effect Effects 0.000 claims description 17
- 230000004054 inflammatory process Effects 0.000 claims description 17
- 230000005764 inhibitory process Effects 0.000 claims description 17
- 125000004432 carbon atom Chemical group C* 0.000 claims description 15
- 125000002950 monocyclic group Chemical group 0.000 claims description 13
- 125000003367 polycyclic group Polymers 0.000 claims description 13
- 229910052799 carbon Inorganic materials 0.000 claims description 12
- 208000024891 symptom Diseases 0.000 claims description 12
- 125000003545 alkoxy group Chemical group 0.000 claims description 11
- 239000000203 mixture Substances 0.000 claims description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 9
- 210000004072 lung Anatomy 0.000 claims description 9
- 230000002062 proliferating effect Effects 0.000 claims description 9
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 8
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 8
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 8
- 201000009030 Carcinoma Diseases 0.000 claims description 8
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 claims description 8
- 206010020751 Hypersensitivity Diseases 0.000 claims description 8
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 8
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 8
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 8
- 201000004681 Psoriasis Diseases 0.000 claims description 8
- 206010039710 Scleroderma Diseases 0.000 claims description 8
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 8
- 201000000448 autoimmune hemolytic anemia Diseases 0.000 claims description 8
- 229940125904 compound 1 Drugs 0.000 claims description 8
- 229940125782 compound 2 Drugs 0.000 claims description 8
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 claims description 8
- 208000002780 macular degeneration Diseases 0.000 claims description 8
- 201000006417 multiple sclerosis Diseases 0.000 claims description 8
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 8
- 208000035408 type 1 diabetes mellitus 1 Diseases 0.000 claims description 8
- 229940125898 compound 5 Drugs 0.000 claims description 7
- 229920006395 saturated elastomer Polymers 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 125000003118 aryl group Chemical group 0.000 claims description 6
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 6
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N o-biphenylenemethane Natural products C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 claims description 6
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 claims description 6
- 210000003491 skin Anatomy 0.000 claims description 6
- 206010061309 Neoplasm progression Diseases 0.000 claims description 5
- 210000000481 breast Anatomy 0.000 claims description 5
- 210000001072 colon Anatomy 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 230000014399 negative regulation of angiogenesis Effects 0.000 claims description 5
- 210000002307 prostate Anatomy 0.000 claims description 5
- 239000007787 solid Substances 0.000 claims description 5
- 230000005751 tumor progression Effects 0.000 claims description 5
- 210000003932 urinary bladder Anatomy 0.000 claims description 5
- 125000004070 6 membered heterocyclic group Chemical group 0.000 claims description 4
- 208000002874 Acne Vulgaris Diseases 0.000 claims description 4
- 201000003076 Angiosarcoma Diseases 0.000 claims description 4
- 206010002556 Ankylosing Spondylitis Diseases 0.000 claims description 4
- 208000022211 Arteriovenous Malformations Diseases 0.000 claims description 4
- 201000001320 Atherosclerosis Diseases 0.000 claims description 4
- 206010003805 Autism Diseases 0.000 claims description 4
- 208000020706 Autistic disease Diseases 0.000 claims description 4
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 4
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 4
- 208000027496 Behcet disease Diseases 0.000 claims description 4
- 208000009137 Behcet syndrome Diseases 0.000 claims description 4
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 claims description 4
- 208000033222 Biliary cirrhosis primary Diseases 0.000 claims description 4
- 201000006390 Brachial Plexus Neuritis Diseases 0.000 claims description 4
- 208000011691 Burkitt lymphomas Diseases 0.000 claims description 4
- 208000002691 Choroiditis Diseases 0.000 claims description 4
- 208000032544 Cicatrix Diseases 0.000 claims description 4
- 206010010741 Conjunctivitis Diseases 0.000 claims description 4
- 208000011231 Crohn disease Diseases 0.000 claims description 4
- 201000004624 Dermatitis Diseases 0.000 claims description 4
- 206010012468 Dermatitis herpetiformis Diseases 0.000 claims description 4
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 4
- 208000005917 Exostoses Diseases 0.000 claims description 4
- 208000024869 Goodpasture syndrome Diseases 0.000 claims description 4
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 claims description 4
- 208000003807 Graves Disease Diseases 0.000 claims description 4
- 208000015023 Graves' disease Diseases 0.000 claims description 4
- 208000035895 Guillain-Barré syndrome Diseases 0.000 claims description 4
- 208000001204 Hashimoto Disease Diseases 0.000 claims description 4
- 208000030836 Hashimoto thyroiditis Diseases 0.000 claims description 4
- 208000001258 Hemangiosarcoma Diseases 0.000 claims description 4
- 208000002250 Hematologic Neoplasms Diseases 0.000 claims description 4
- 201000004331 Henoch-Schoenlein purpura Diseases 0.000 claims description 4
- 206010019617 Henoch-Schonlein purpura Diseases 0.000 claims description 4
- 208000017604 Hodgkin disease Diseases 0.000 claims description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 4
- 208000031814 IgA Vasculitis Diseases 0.000 claims description 4
- 206010022941 Iridocyclitis Diseases 0.000 claims description 4
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 4
- 201000010743 Lambert-Eaton myasthenic syndrome Diseases 0.000 claims description 4
- 206010049567 Miller Fisher syndrome Diseases 0.000 claims description 4
- 208000003250 Mixed connective tissue disease Diseases 0.000 claims description 4
- 208000003452 Multiple Hereditary Exostoses Diseases 0.000 claims description 4
- 208000034578 Multiple myelomas Diseases 0.000 claims description 4
- 208000029027 Musculoskeletal and connective tissue disease Diseases 0.000 claims description 4
- 206010028424 Myasthenic syndrome Diseases 0.000 claims description 4
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 4
- 208000003435 Optic Neuritis Diseases 0.000 claims description 4
- 208000005141 Otitis Diseases 0.000 claims description 4
- 206010034277 Pemphigoid Diseases 0.000 claims description 4
- 241000721454 Pemphigus Species 0.000 claims description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 4
- 206010035603 Pleural mesothelioma Diseases 0.000 claims description 4
- 208000007048 Polymyalgia Rheumatica Diseases 0.000 claims description 4
- 208000003971 Posterior uveitis Diseases 0.000 claims description 4
- 208000012654 Primary biliary cholangitis Diseases 0.000 claims description 4
- 206010037778 Radiculitis brachial Diseases 0.000 claims description 4
- 208000033464 Reiter syndrome Diseases 0.000 claims description 4
- 201000000582 Retinoblastoma Diseases 0.000 claims description 4
- 206010038933 Retinopathy of prematurity Diseases 0.000 claims description 4
- 206010039491 Sarcoma Diseases 0.000 claims description 4
- 206010039705 Scleritis Diseases 0.000 claims description 4
- 208000034189 Sclerosis Diseases 0.000 claims description 4
- 208000021386 Sjogren Syndrome Diseases 0.000 claims description 4
- 208000021712 Soft tissue sarcoma Diseases 0.000 claims description 4
- 208000007107 Stomach Ulcer Diseases 0.000 claims description 4
- 206010047115 Vasculitis Diseases 0.000 claims description 4
- 206010000210 abortion Diseases 0.000 claims description 4
- 231100000176 abortion Toxicity 0.000 claims description 4
- 206010000496 acne Diseases 0.000 claims description 4
- 230000001154 acute effect Effects 0.000 claims description 4
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 4
- 208000026935 allergic disease Diseases 0.000 claims description 4
- 210000004141 ampulla of vater Anatomy 0.000 claims description 4
- 210000002255 anal canal Anatomy 0.000 claims description 4
- 201000004612 anterior uveitis Diseases 0.000 claims description 4
- 230000005744 arteriovenous malformation Effects 0.000 claims description 4
- 208000006673 asthma Diseases 0.000 claims description 4
- 230000009286 beneficial effect Effects 0.000 claims description 4
- 210000000988 bone and bone Anatomy 0.000 claims description 4
- 210000004556 brain Anatomy 0.000 claims description 4
- 208000000594 bullous pemphigoid Diseases 0.000 claims description 4
- 210000003679 cervix uteri Anatomy 0.000 claims description 4
- 210000000795 conjunctiva Anatomy 0.000 claims description 4
- 230000007812 deficiency Effects 0.000 claims description 4
- 201000001981 dermatomyositis Diseases 0.000 claims description 4
- 208000019258 ear infection Diseases 0.000 claims description 4
- 210000003238 esophagus Anatomy 0.000 claims description 4
- 210000003020 exocrine pancreas Anatomy 0.000 claims description 4
- 201000010934 exostosis Diseases 0.000 claims description 4
- 210000002603 extrahepatic bile duct Anatomy 0.000 claims description 4
- 201000002788 eyelid carcinoma Diseases 0.000 claims description 4
- 210000000232 gallbladder Anatomy 0.000 claims description 4
- 201000005917 gastric ulcer Diseases 0.000 claims description 4
- 201000007116 gestational trophoblastic neoplasm Diseases 0.000 claims description 4
- 201000009277 hairy cell leukemia Diseases 0.000 claims description 4
- 201000011066 hemangioma Diseases 0.000 claims description 4
- 230000009033 hematopoietic malignancy Effects 0.000 claims description 4
- 208000006454 hepatitis Diseases 0.000 claims description 4
- 231100000283 hepatitis Toxicity 0.000 claims description 4
- 230000009610 hypersensitivity Effects 0.000 claims description 4
- 230000001969 hypertrophic effect Effects 0.000 claims description 4
- 208000015446 immunoglobulin a vasculitis Diseases 0.000 claims description 4
- 230000001939 inductive effect Effects 0.000 claims description 4
- 210000003734 kidney Anatomy 0.000 claims description 4
- 210000000244 kidney pelvis Anatomy 0.000 claims description 4
- 201000009314 lacrimal gland carcinoma Diseases 0.000 claims description 4
- 210000000867 larynx Anatomy 0.000 claims description 4
- 208000032839 leukemia Diseases 0.000 claims description 4
- 210000000088 lip Anatomy 0.000 claims description 4
- 210000004185 liver Anatomy 0.000 claims description 4
- 201000002576 malignant conjunctival melanoma Diseases 0.000 claims description 4
- 230000003211 malignant effect Effects 0.000 claims description 4
- 208000000516 mast-cell leukemia Diseases 0.000 claims description 4
- 210000000214 mouth Anatomy 0.000 claims description 4
- 206010028417 myasthenia gravis Diseases 0.000 claims description 4
- 201000006098 orbit sarcoma Diseases 0.000 claims description 4
- 210000001672 ovary Anatomy 0.000 claims description 4
- 210000003101 oviduct Anatomy 0.000 claims description 4
- 210000003695 paranasal sinus Anatomy 0.000 claims description 4
- 210000003899 penis Anatomy 0.000 claims description 4
- 210000003800 pharynx Anatomy 0.000 claims description 4
- 201000006292 polyarteritis nodosa Diseases 0.000 claims description 4
- 208000005987 polymyositis Diseases 0.000 claims description 4
- 230000035935 pregnancy Effects 0.000 claims description 4
- 208000002574 reactive arthritis Diseases 0.000 claims description 4
- 230000010410 reperfusion Effects 0.000 claims description 4
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 4
- 210000003079 salivary gland Anatomy 0.000 claims description 4
- 231100000241 scar Toxicity 0.000 claims description 4
- 230000037387 scars Effects 0.000 claims description 4
- 208000017520 skin disease Diseases 0.000 claims description 4
- 210000000813 small intestine Anatomy 0.000 claims description 4
- 210000000278 spinal cord Anatomy 0.000 claims description 4
- 210000002784 stomach Anatomy 0.000 claims description 4
- 230000001629 suppression Effects 0.000 claims description 4
- 208000011580 syndromic disease Diseases 0.000 claims description 4
- 210000001550 testis Anatomy 0.000 claims description 4
- 206010043554 thrombocytopenia Diseases 0.000 claims description 4
- 210000001685 thyroid gland Anatomy 0.000 claims description 4
- 206010043778 thyroiditis Diseases 0.000 claims description 4
- 230000005740 tumor formation Effects 0.000 claims description 4
- 210000000626 ureter Anatomy 0.000 claims description 4
- 210000003708 urethra Anatomy 0.000 claims description 4
- 210000004291 uterus Anatomy 0.000 claims description 4
- 210000001745 uvea Anatomy 0.000 claims description 4
- 210000001215 vagina Anatomy 0.000 claims description 4
- 230000002792 vascular Effects 0.000 claims description 4
- 210000003905 vulva Anatomy 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 2
- 229910052717 sulfur Inorganic materials 0.000 claims description 2
- CIUQDSCDWFSTQR-UHFFFAOYSA-N [C]1=CC=CC=C1 Chemical group [C]1=CC=CC=C1 CIUQDSCDWFSTQR-UHFFFAOYSA-N 0.000 claims 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims 3
- 125000004429 atom Chemical group 0.000 claims 2
- 125000000623 heterocyclic group Chemical group 0.000 claims 2
- 208000027866 inflammatory disease Diseases 0.000 abstract description 8
- 125000005518 carboxamido group Chemical group 0.000 abstract 1
- 125000003983 fluorenyl group Chemical class C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 abstract 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 33
- 230000000694 effects Effects 0.000 description 31
- 238000003556 assay Methods 0.000 description 28
- 241000699670 Mus sp. Species 0.000 description 26
- 239000000758 substrate Substances 0.000 description 23
- 229920002971 Heparan sulfate Polymers 0.000 description 15
- 239000002253 acid Substances 0.000 description 15
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 14
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 14
- 230000033115 angiogenesis Effects 0.000 description 14
- 210000002744 extracellular matrix Anatomy 0.000 description 14
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- 102000003896 Myeloperoxidases Human genes 0.000 description 12
- 108090000235 Myeloperoxidases Proteins 0.000 description 12
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- 229960002897 heparin Drugs 0.000 description 11
- 229920000669 heparin Polymers 0.000 description 11
- 239000007924 injection Substances 0.000 description 10
- 238000002347 injection Methods 0.000 description 10
- 239000004372 Polyvinyl alcohol Substances 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- 206010061289 metastatic neoplasm Diseases 0.000 description 9
- 229920002451 polyvinyl alcohol Polymers 0.000 description 9
- 102000008055 Heparan Sulfate Proteoglycans Human genes 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 108090000054 Syndecan-2 Proteins 0.000 description 8
- 230000002491 angiogenic effect Effects 0.000 description 8
- 210000002469 basement membrane Anatomy 0.000 description 8
- 230000001394 metastastic effect Effects 0.000 description 8
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 7
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 7
- 150000001716 carbazoles Chemical class 0.000 description 7
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 7
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 239000002953 phosphate buffered saline Substances 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- MUUHXGOJWVMBDY-UHFFFAOYSA-L tetrazolium blue Chemical compound [Cl-].[Cl-].COC1=CC(C=2C=C(OC)C(=CC=2)[N+]=2N(N=C(N=2)C=2C=CC=CC=2)C=2C=CC=CC=2)=CC=C1[N+]1=NC(C=2C=CC=CC=2)=NN1C1=CC=CC=C1 MUUHXGOJWVMBDY-UHFFFAOYSA-L 0.000 description 7
- VAJIZAPXBKMPRO-UHFFFAOYSA-N 9-(oxiran-2-ylmethyl)carbazole Chemical compound C12=CC=CC=C2C2=CC=CC=C2N1CC1CO1 VAJIZAPXBKMPRO-UHFFFAOYSA-N 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 238000002835 absorbance Methods 0.000 description 6
- 238000010171 animal model Methods 0.000 description 6
- 229940088598 enzyme Drugs 0.000 description 6
- 239000000284 extract Substances 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 102000004169 proteins and genes Human genes 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 210000004881 tumor cell Anatomy 0.000 description 6
- 230000004614 tumor growth Effects 0.000 description 6
- 229920002684 Sepharose Polymers 0.000 description 5
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 5
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 238000007398 colorimetric assay Methods 0.000 description 5
- 150000002220 fluorenes Chemical class 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 238000007912 intraperitoneal administration Methods 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 4
- 229930182566 Gentamicin Natural products 0.000 description 4
- 210000000709 aorta Anatomy 0.000 description 4
- 230000004071 biological effect Effects 0.000 description 4
- 210000004204 blood vessel Anatomy 0.000 description 4
- 230000015556 catabolic process Effects 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 230000014509 gene expression Effects 0.000 description 4
- 229960002518 gentamicin Drugs 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 210000002865 immune cell Anatomy 0.000 description 4
- 238000000099 in vitro assay Methods 0.000 description 4
- 108010082117 matrigel Proteins 0.000 description 4
- 239000011159 matrix material Substances 0.000 description 4
- 210000000440 neutrophil Anatomy 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- 229920001184 polypeptide Polymers 0.000 description 4
- 102000004196 processed proteins & peptides Human genes 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 238000011725 BALB/c mouse Methods 0.000 description 3
- 102000004266 Collagen Type IV Human genes 0.000 description 3
- 108010042086 Collagen Type IV Proteins 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 239000003146 anticoagulant agent Substances 0.000 description 3
- 229940127219 anticoagulant drug Drugs 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 239000000287 crude extract Substances 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- IPZJQDSFZGZEOY-UHFFFAOYSA-N dimethylmethylene Chemical group C[C]C IPZJQDSFZGZEOY-UHFFFAOYSA-N 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000012894 fetal calf serum Substances 0.000 description 3
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 239000003055 low molecular weight heparin Substances 0.000 description 3
- 229940127215 low-molecular weight heparin Drugs 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 239000007981 phosphate-citrate buffer Substances 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 150000003335 secondary amines Chemical class 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- 0 *CC(C[n](c(ccc(Cl)c1)c1c1c2)c1ccc2Cl)O Chemical compound *CC(C[n](c(ccc(Cl)c1)c1c1c2)c1ccc2Cl)O 0.000 description 2
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 2
- DGMRGIDWASSLGY-UHFFFAOYSA-N 3,6-dibromo-9-(oxiran-2-ylmethyl)carbazole Chemical compound C12=CC=C(Br)C=C2C2=CC(Br)=CC=C2N1CC1CO1 DGMRGIDWASSLGY-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- ODLMAHJVESYWTB-UHFFFAOYSA-N CCCc1ccccc1 Chemical compound CCCc1ccccc1 ODLMAHJVESYWTB-UHFFFAOYSA-N 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 2
- 108010008951 Chemokine CXCL12 Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 108010085895 Laminin Proteins 0.000 description 2
- 102000007547 Laminin Human genes 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- XUMBMVFBXHLACL-UHFFFAOYSA-N Melanin Chemical compound O=C1C(=O)C(C2=CNC3=C(C(C(=O)C4=C32)=O)C)=C2C4=CNC2=C1C XUMBMVFBXHLACL-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 238000010240 RT-PCR analysis Methods 0.000 description 2
- 102100021669 Stromal cell-derived factor 1 Human genes 0.000 description 2
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 210000001772 blood platelet Anatomy 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- 230000004709 cell invasion Effects 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000002299 complementary DNA Substances 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 239000007857 degradation product Substances 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 150000004676 glycans Chemical class 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 210000003630 histaminocyte Anatomy 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 230000028709 inflammatory response Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 210000004088 microvessel Anatomy 0.000 description 2
- NOPBLWOROFDLCR-UHFFFAOYSA-N n-(3,4-dimethoxyphenyl)-2-piperazin-1-ylquinazolin-4-amine Chemical compound C1=C(OC)C(OC)=CC=C1NC1=NC(N2CCNCC2)=NC2=CC=CC=C12 NOPBLWOROFDLCR-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 125000000466 oxiranyl group Chemical group 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 238000011158 quantitative evaluation Methods 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 230000005747 tumor angiogenesis Effects 0.000 description 2
- 230000029663 wound healing Effects 0.000 description 2
- RDAFNSMYPSHCBK-QPJJXVBHSA-N (e)-3-phenylprop-2-en-1-amine Chemical compound NC\C=C\C1=CC=CC=C1 RDAFNSMYPSHCBK-QPJJXVBHSA-N 0.000 description 1
- 125000004529 1,2,3-triazinyl group Chemical group N1=NN=C(C=C1)* 0.000 description 1
- SDALHPNZAOPIDL-UHFFFAOYSA-N 1-carbazol-9-yl-3-[4-(3-carbazol-9-yl-2-hydroxypropyl)piperazin-1-yl]propan-2-ol Chemical compound C12=CC=CC=C2C2=CC=CC=C2N1CC(O)CN1CCN(CC(O)CN2C3=CC=CC=C3C3=CC=CC=C32)CC1 SDALHPNZAOPIDL-UHFFFAOYSA-N 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- WGTASENVNYJZBK-UHFFFAOYSA-N 3,4,5-trimethoxyamphetamine Chemical compound COC1=CC(CC(C)N)=CC(OC)=C1OC WGTASENVNYJZBK-UHFFFAOYSA-N 0.000 description 1
- 125000003762 3,4-dimethoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C(OC([H])([H])[H])C([H])=C1* 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- UXTIAFYTYOEQHV-UHFFFAOYSA-N 4-(4-amino-3-methoxyphenyl)-2-methoxyaniline;hydron;dichloride Chemical compound [Cl-].[Cl-].C1=C([NH3+])C(OC)=CC(C=2C=C(OC)C([NH3+])=CC=2)=C1 UXTIAFYTYOEQHV-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 241001019659 Acremonium <Plectosphaerellaceae> Species 0.000 description 1
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 208000010667 Carcinoma of liver and intrahepatic biliary tract Diseases 0.000 description 1
- 102000004225 Cathepsin B Human genes 0.000 description 1
- 108090000712 Cathepsin B Proteins 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 208000009386 Experimental Arthritis Diseases 0.000 description 1
- 206010015866 Extravasation Diseases 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 102000013382 Gelatinases Human genes 0.000 description 1
- 108010026132 Gelatinases Proteins 0.000 description 1
- 108010060309 Glucuronidase Proteins 0.000 description 1
- 102000053187 Glucuronidase Human genes 0.000 description 1
- 229920002683 Glycosaminoglycan Polymers 0.000 description 1
- 206010073069 Hepatic cancer Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 208000006552 Lewis Lung Carcinoma Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 206010027458 Metastases to lung Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 101000648740 Mus musculus Tumor necrosis factor Proteins 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- VXZNOIQBPJUPAE-UHFFFAOYSA-N NC(NN=C(c1c2)c3cc(S(NCCO)(=O)=O)ccc3-c1ccc2S(NCCO)(=O)=O)=O Chemical compound NC(NN=C(c1c2)c3cc(S(NCCO)(=O)=O)ccc3-c1ccc2S(NCCO)(=O)=O)=O VXZNOIQBPJUPAE-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010029113 Neovascularisation Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- OJOCUCQSVCQTNW-UHFFFAOYSA-N OC(CN(CCc1ccccc1)C(Nc(cc1)ccc1S(O)(=O)=O)=S)C[n](c(c(c1c2)c3)ccc3Br)c1ccc2Br Chemical compound OC(CN(CCc1ccccc1)C(Nc(cc1)ccc1S(O)(=O)=O)=S)C[n](c(c(c1c2)c3)ccc3Br)c1ccc2Br OJOCUCQSVCQTNW-UHFFFAOYSA-N 0.000 description 1
- UTVAPCQDVLRLDF-UHFFFAOYSA-N OC(CNCCc1ccccc1)C[n](c(c(c1c2)c3)ccc3Br)c1ccc2Br Chemical compound OC(CNCCc1ccccc1)C[n](c(c(c1c2)c3)ccc3Br)c1ccc2Br UTVAPCQDVLRLDF-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 108010067372 Pancreatic elastase Proteins 0.000 description 1
- 102000016387 Pancreatic elastase Human genes 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 108010001014 Plasminogen Activators Proteins 0.000 description 1
- 102000001938 Plasminogen Activators Human genes 0.000 description 1
- 208000037062 Polyps Diseases 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 239000004280 Sodium formate Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- JRMSLDWZFJZLAS-UHFFFAOYSA-M [7-(dimethylamino)-1,9-dimethylphenothiazin-3-ylidene]-dimethylazanium;chloride Chemical compound [Cl-].CC1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC(C)=C3N=C21 JRMSLDWZFJZLAS-UHFFFAOYSA-M 0.000 description 1
- ATNKHRJTLMRVJV-UHFFFAOYSA-N [N-]=C=S.O.[Na+] Chemical compound [N-]=C=S.O.[Na+] ATNKHRJTLMRVJV-UHFFFAOYSA-N 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- AEMOLEFTQBMNLQ-BKBMJHBISA-N alpha-D-galacturonic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-BKBMJHBISA-N 0.000 description 1
- OCIBBXPLUVYKCH-QXVNYKTNSA-N alpha-maltohexaose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)O[C@H](O[C@@H]2[C@H](O[C@H](O[C@@H]3[C@H](O[C@H](O[C@@H]4[C@H](O[C@H](O[C@@H]5[C@H](O[C@H](O)[C@H](O)[C@H]5O)CO)[C@H](O)[C@H]4O)CO)[C@H](O)[C@H]3O)CO)[C@H](O)[C@H]2O)CO)[C@H](O)[C@H]1O OCIBBXPLUVYKCH-QXVNYKTNSA-N 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 159000000032 aromatic acids Chemical class 0.000 description 1
- 230000006472 autoimmune response Effects 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 201000001531 bladder carcinoma Diseases 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 201000008274 breast adenocarcinoma Diseases 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 239000003710 calcium ionophore Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002975 chemoattractant Substances 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 125000005805 dimethoxy phenyl group Chemical group 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000002224 dissection Methods 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000012154 double-distilled water Substances 0.000 description 1
- 230000023117 embryonic morphogenesis Effects 0.000 description 1
- 201000002491 encephalomyelitis Diseases 0.000 description 1
- 230000008519 endogenous mechanism Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- CJAONIOAQZUHPN-KKLWWLSJSA-N ethyl 12-[[2-[(2r,3r)-3-[2-[(12-ethoxy-12-oxododecyl)-methylamino]-2-oxoethoxy]butan-2-yl]oxyacetyl]-methylamino]dodecanoate Chemical compound CCOC(=O)CCCCCCCCCCCN(C)C(=O)CO[C@H](C)[C@@H](C)OCC(=O)N(C)CCCCCCCCCCCC(=O)OCC CJAONIOAQZUHPN-KKLWWLSJSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000010228 ex vivo assay Methods 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 230000036251 extravasation Effects 0.000 description 1
- 150000002222 fluorine compounds Chemical class 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 239000002634 heparin fragment Substances 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 239000012433 hydrogen halide Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- QWPPOHNGKGFGJK-UHFFFAOYSA-N hypochlorous acid Chemical compound ClO QWPPOHNGKGFGJK-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000011532 immunohistochemical staining Methods 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 201000002250 liver carcinoma Diseases 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- DJMVHSOAUQHPSN-UHFFFAOYSA-N malto-hexaose Natural products OC1C(O)C(OC(C(O)CO)C(O)C(O)C=O)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(OC4C(C(O)C(O)C(CO)O4)O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 DJMVHSOAUQHPSN-UHFFFAOYSA-N 0.000 description 1
- LUEWUZLMQUOBSB-OUBHKODOSA-N maltotetraose Chemical class O[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@H](CO)O[C@@H](O[C@@H]2[C@@H](O[C@@H](O[C@@H]3[C@@H](O[C@@H](O)[C@H](O)[C@H]3O)CO)[C@H](O)[C@H]2O)CO)[C@H](O)[C@H]1O LUEWUZLMQUOBSB-OUBHKODOSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 238000003358 metastasis assay Methods 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 238000010232 migration assay Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- FVSUYFWWFUVGRG-UHFFFAOYSA-N naphthalen-1-ylurea Polymers C1=CC=C2C(NC(=O)N)=CC=CC2=C1 FVSUYFWWFUVGRG-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 230000014511 neuron projection development Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 229940043138 pentosan polysulfate Drugs 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 229940117803 phenethylamine Drugs 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 229940127126 plasminogen activator Drugs 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000008057 potassium phosphate buffer Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 201000001514 prostate carcinoma Diseases 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000004309 pyranyl group Chemical class O1C(C=CC=C1)* 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 125000004260 quinazolin-2-yl group Chemical group [H]C1=NC(*)=NC2=C1C([H])=C([H])C([H])=C2[H] 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 229940116351 sebacate Drugs 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- DQTKLICLJUKNCG-ZTYPAOSTSA-N siastatin b Chemical class CC(=O)N[C@H]1NC[C@H](C(O)=O)[C@H](O)[C@@H]1O DQTKLICLJUKNCG-ZTYPAOSTSA-N 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- HLBBKKJFGFRGMU-UHFFFAOYSA-M sodium formate Chemical compound [Na+].[O-]C=O HLBBKKJFGFRGMU-UHFFFAOYSA-M 0.000 description 1
- 235000019254 sodium formate Nutrition 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 235000014347 soups Nutrition 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- FIAFUQMPZJWCLV-UHFFFAOYSA-N suramin Chemical compound OS(=O)(=O)C1=CC(S(O)(=O)=O)=C2C(NC(=O)C3=CC=C(C(=C3)NC(=O)C=3C=C(NC(=O)NC=4C=C(C=CC=4)C(=O)NC=4C(=CC=C(C=4)C(=O)NC=4C5=C(C=C(C=C5C(=CC=4)S(O)(=O)=O)S(O)(=O)=O)S(O)(=O)=O)C)C=CC=3)C)=CC=C(S(O)(=O)=O)C2=C1 FIAFUQMPZJWCLV-UHFFFAOYSA-N 0.000 description 1
- 229960005314 suramin Drugs 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940066771 systemic antihistamines piperazine derivative Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 230000017423 tissue regeneration Effects 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 208000010570 urinary bladder carcinoma Diseases 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000037314 wound repair Effects 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/88—Carbazoles; Hydrogenated carbazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the ring system
Definitions
- the present invention relates to heparanase inhibitors, particularly to certain carbazole and fluorene derivatives, and to their use in the treatment of diseases and disorders caused by or associated with heparanase catalytic activity such as cancer, inflammatory disorders and autoimmune diseases.
- Heparan sulfate proteoglycans are ubiquitous macromolecules associated with the cell surface and with the extracellular matrix (ECM) of various tissues. They consist of a protein core to which several linear heparan sulfate (HS) chains are covalently attached.
- ECM extracellular matrix
- HSPGs are also prominent components of blood vessels. In capillaries they are found mainly in the subendothelial basement membrane, where they support proliferating and migrating endothelial cells and stabilize the structure of the capillary wall.
- heparanase an endo- ⁇ -D-glucuronidase that cleaves HS at specific intrachain sites
- Heparanase released from cells removes HS molecules from the basement membrane resulting in increase of basement membrane permeability.
- Heparanase also facilitates proteolytic degradation of the core structural components such as type IV collagen in collaboration with gelatinases.
- blood-borne cells accomplish penetration through the basement membrane.
- HS catabolism is observed in wound repair, inflammation, and in diabetes.
- heparanase was found to correlate with the metastatic potential of mouse lymphoma (Vlodavsky et al., 1983), fibrosarcoma and melanoma cells (Nakajima et al., 1988). Similar correlation was observed in human breast, colon, bladder, prostate, and liver carcinomas (Vlodavsky et al., 1999). Moreover, elevated levels of heparanase were detected in sera of metastatic tumor bearing animals (Nakajima et al., 1988) and of cancer patients, in urine of highly metastatic patients (Vlodavsky et al., 1997), and in tumor biopsies (Vlodavsky et al., 1988).
- heparanase substrates or inhibitors e.g., non-anticoagulant species of low molecular weight heparin and polysulfated saccharides
- VEGF vascular endothelial growth factor
- bFGF basic fibroblast growth factor
- bFGF binds to HSPG in the ECM and can be released in an active form by HS-degrading enzymes.
- Heparanase expressed by platelets, mast cells, neutrophils, and lymphoma cells was found to be involved in the release of active bFGF from ECM and basement membranes, suggesting that heparanase activity may not only function in cell migration and invasion, but may also elicit an indirect neovascular response (Elkin et al., 2001).
- Heparanase catalytic activity correlates with the ability of activated cells of the immune system to leave the circulation and elicit both inflammatory and autoimmune responses.
- Interaction of platelets, granulocytes, T and B lymphocytes, macrophages, and mast cells with the subendothelial ECM is associated with degradation of HS by heparanase (Vlodavsky et al., 1992).
- the enzyme is released from intracellular compartments (e.g., lysosomes, specific granules) in response to various activation signals (e.g., thrombin, calcium ionophore, immune complexes, antigens, mitogens), suggesting its regulated involvement in inflammatory sites and in autoimmune diseases.
- heparanase substrates e.g., non- anticoagulant species of low molecular weight heparin
- EAE experimental autoimmune encephalomyelitis
- graft rejection indicating that heparanase inhibitors may inhibit autoimmune and inflammatory diseases
- Heparanase inhibitors have been proposed for treatment of human metastasis, for example, derivatives of siastatin B (Nishimura et al., 1994; Kawase et al., 1995), a pyran derivative isolated from the fungal strain Acremonium sp.
- MT70646 PCT/KR00/01493
- suramin a polysulfonated naphthylurea
- sulfated oligosaccharides e.g., sulfated maltotetraose and maltohexaose (Parish et al., 1999)
- sulfated polysaccharides parish et al, 1987; Lapierre et al., 1996.
- U.S. Patent No. 5,968,822 discloses a polynucleotide encoding a polypeptide having heparanase catalytic activity and host cells, particularly insect cells, expressing said polypeptide.
- the recombinant polypeptide having heparanase activity is said to be useful for potential treatment of several diseases and disorders such as wound healing, angiogenesis, restenosis, inflammation and neurodegenerative diseases as well as for development of new drugs that inhibit tumor cell metastasis, inflammation and autoimmunity.
- International Patent Publication No. WO 99/57244 of the present applicants discloses bacterial, yeast and animal cells and methods for over expressing recombinant heparanase in cellular systems.
- EP 1094063 discloses certain piperazine derivatives of carbazoles, particularly 9-(piperazinylalkyl)carbazoles, for use in the treatment of disorders associated with the modulation of the Bax function and/or the Bax activation. None of the above-mentioned publications discloses or suggests the heparanase inhibitors of the present invention.
- the present invention provides, in one aspect, a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and at least one heparanase inhibitor selected from a carbazole or fluorene derivative of the general Formula I hereinafter or a pharmaceutically acceptable salt thereof.
- the pharmaceutical composition of the invention is particularly useful for the treatment of diseases and disorders caused by or associated with heparanase catalytic activity such as, but not being limited to, cancer, inflammatory disorders and autoimmune diseases.
- the present invention relates to the use of a carbazole or fluorene derivative of the general Formula I for the manufacture of a pharmaceutical composition.
- said compositions are for treatment of diseases and disorders caused by or associated with heparanase catalytic activity such as, but not being limited to, cancer, inflammatory disorders and autoimmune diseases.
- the present invention provides certain novel carbazole derivatives of the general Formula I.
- the present invention relates to a method for treatment of a patient suffering from a disease or disorder caused by or associated with heparanase catalytic activity such as cancer, an inflammatory disorder or an autoimmune disease, which comprises administering to said patient an effective amount of a carbazole or fluorene derivative of the general Formula I.
- Fig. 1 is a graph showing the total number of cells extracted from three polyvinyl alcohol (PVA) sponges implanted in BALB/c mice untreated (control) or treated with Compound 3 (at doses of 1 mg/ml/mouse and 0.2 mg/ml/mouse).
- Fig. 2 is a graph showing the myeloperoxidase (MPO) activity in a suspension of cells squeezed from the PVA sponges implanted in BALB/c mice untreated (control) or treated with Compound 3 (at doses of 1 mg/ml/mouse and 0.2 mg/ml/mouse).
- MPO myeloperoxidase
- Control 3 is a graph showing changes in TNF- ⁇ concentration in the supernatant extracted from the PVA sponges implanted in BALB/c mice untreated (Control 2) or treated with Compound 3 (at doses of 1 mg/ml/mouse and 0.2 mg/ml/mouse).
- Control 1
- compositions are provided, particularly for treatment of diseases and disorders caused by or associated with heparanase catalytic activity, said compositions comprising a pharmaceutically acceptable carrier and at least one heparanase inhibitor which is a carbazole or fluorene compound of the general Formula I:
- Y and Y' each independently represents hydrogen, halogen, C1-C6 alkyl, C2-C6 alkenyl, C6-C14 aryl, nitro, -OR, -SR, -CONRR', -NRCONRR', -NRR', -SO 3 H, or -SO 2 NRR';
- R2 and R3 each independently represents hydrogen, C1-C6 alkyl, C2-C6 alkenyl, C6-C14 aryl, or heteroaryl; or R2 is hydrogen, C1-C6 alkyl, C2-C6 alkenyl, C6-C14 aryl, or heteroaryl and R3 is -CONRR', -CSNRR', or -CONHNRR'; or R2 and R3 together with the N atom to which they are attached form a saturated 5-7 membered heterocyclic ring optionally containing at least one further heteroatom selected from N, O, and/or S, said at least one further N atom
- R4 is -CONRR', -CSNRR' or -CONHNRR'
- R5 is C1-C6 alkyl substituted by carbazolyl at the terminal carbon atom and by a further group selected from halogen, -OH, -SH, -NH 2 , C1-C6 alkoxy or C1-C6 alkylthio; or heteroaryl derived from a bicyclic heteroaromatic ring containing one to three heteroatoms selected from N, O and/or S;
- R and R' each independently represents (i) hydrogen; (ii) C1-C6 alkyl optionally substituted by halogen, -OH, -SH, -NH 2 , C1-C6 alkoxy, C1-C6 alkylthio, C6-C14 aryl, and/or heteroaryl; (iii) C2-C6 alkenyl optionally substituted by halogen, -OH, -SH, -NH 2 , C1-C6 alkoxy, C1-C6 alkylthio, C6-C14 aryl and/or heteroaryl; (iv) C6-C14 aryl optionally substituted by halogen, -OH, -SH, -NH 2 , -SO 3 H, C1-C6 alkyl, C2-C6 alkenyl, C1-C6 alkoxy, and/or C1-C6 alkylthio; or (v) heteroaryl optionally substituted by halogen, -
- heteroaryl in radicals R, R', R2, and R3 is a radical derived from a mono- or poly- cyclic heteroaromatic ring containing one to three heteroatoms selected from N, O and/or S; any "C1-C6 alkyl” or “C2-C6 alkenyl” in radicals R2 and R3 may be substituted by at least one radical selected from halogen, -OH, -SH, C1-C6 alkoxy, C1-C6 alkylthio, C6-C14 aryl, heteroaryl, -NRR', -COOR, -CONRR', -SO 3 H, or -SO 2 NRR'; any "C6-C14 aryl” and “heteroaryl” in radicals R2, R3 and R5 may be substituted by at least one radical selected from halogen, -OH, -SH, C1-C6 alkyl, C2-C6 alkenyl, Cl- C6 alk
- C1-C6 alkyl typically refers to a straight or branched alkyl radical having 1-6 carbon atoms and includes for example methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert-butyl, n-heptyl, 2,2-dimethylpropyl, n-hexyl and the like.
- C2-C6 alkenyl refers to straight or branched hydrocarbon radicals having 2-6 carbon atoms and one double bond, preferably a terminal double bond, and includes for example vinyl, prop-2-en-l-yl, but-3-en-l-yl, pent-4-en-l-yl, and hex-5-en- l-yl.
- C1-C6 alkoxy refers to the group C1-C6 alkyl-O-, wherein C1-C6 alkyl is as defined above. Examples of alkoxy are methoxy, ethoxy, hexoxy and the like.
- C1-C6 alkylthio refers to the group C1-C6 alkyl-S-, wherein C1-C6 alkyl is as defined above. Examples of alkylthio are methylthio, ethylthio, butylthio and the like.
- C6-C14 aryl refers to an aromatic carbocyclic group having 6 to 14 carbon atoms consisting of a single ring or multiple condensed rings such as phenyl, naphthyl, and phenanthryl optionally substituted by C1-C6 alkyl.
- heteroaryl in radicals R, R', R2, and R3 refers to a radical derived from a mono- or poly-cyclic heteroaromatic ring containing one to three heteroatoms selected from N, O and/or S.
- Particular examples are pyridyl, pyrrolyl, furyl, thienyl, imidazolyl, oxazolyl, quinolinyl, thiazolyl, pyrazolyl, 1,3,4-triazinyl, 1,2,3-triazinyl, benzofuryl, isobenzofuryl, indolyl, imidazo[l,2-a]pyridyl, benzimidazolyl, benzthiazolyl and benzoxazolyl.
- heteroaryl in radical R5 refers to a radical derived from a bicyclic heteroaromatic ring containing one to three heteroatoms selected from N, O and/or S, such as, but not limited to, quinazolinyl, quinolinyl, benzofuryl, isobenzofuryl, indolyl, imidazo[l,2-a]pyridyl, benzimidazolyl, benzthiazolyl and benzoxazolyl, that may be substituted as defined above.
- R is quinazolinyl substituted by -NRR', wherein R is H and R' is dimethoxyphenyl.
- halogen refers to fluoro, chloro, bromo or iodo.
- the group -NR2R3 may be -NH 2 , when R2 and R3 are both hydrogen, or R2 is hydrogen, C1-C6 alkyl, C2-C6 alkenyl, C6-C14 aryl or heteroaryl and R3 is -CONRR', - CSNRR', or -CONHNRR', as defined above, or R2 and R3 together with the nitrogen atom to which they are attached form a saturated 5-7 membered heterocyclic ring, preferably a 6-membered ring, optionally containing at least one further heteroatom selected from nitrogen, oxygen and/or sulfur.
- Such rings may be substituted, for example with one or two C1-C6 alkyl groups, preferably at the N atom.
- Examples of such rings include, without being limited to, pyrrolidino, piperidino, morpholino, thiomorpholino, piperazino, N-C1-C6 alkylpiperazino, e.g. N-methylpiperazino and the like.
- compositions of formula I are also contemplated by the present invention, both salts formed by any carboxy or sulfo groups present in the molecule and a base as well as acid addition and/or base salts.
- Pharmaceutically acceptable salts are formed with metals or amines, such as alkali and alkaline earth metals or organic amines.
- metals used as cations are sodium, potassium, magnesium, calcium, and the like.
- suitable amines are N,N'-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, N-methylglucamine, and procaine (see, for example, Berge S. M., et al., "Pharmaceutical Salts," (1977) J. of Pharmaceutical Science, 66:1-19).
- the salts can also be pharmaceutically acceptable quaternary salts such as a quaternary salt of the formula - NRR'R" + Z' wherein R, R' and R" each is independently hydrogen, alkyl or benzyl and Z is a counterion, including chloride, bromide, iodide, O-alkyl, toluenesulfonate, methylsulfonate, sulfonate, phosphate, or carboxylate.
- quaternary salts such as a quaternary salt of the formula - NRR'R" + Z' wherein R, R' and R" each is independently hydrogen, alkyl or benzyl and Z is a counterion, including chloride, bromide, iodide, O-alkyl, toluenesulfonate, methylsulfonate, sulfonate, phosphate, or carboxylate.
- Pharmaceutically acceptable acid addition salts of the compounds include salts derived from inorganic acids such as hydrochloric, nitric, phosphoric, sulfuric, hydrobromic, hydriodic, phosphorous, and the like, as well as salts derived from organic acids such as aliphatic mono- and dicarboxylic acids, phenyl-substituted alkanoic acids, hydroxy alkanoic acids, alkanedioic acids, aromatic acids, aliphatic and aromatic sulfonic acids, etc.
- inorganic acids such as hydrochloric, nitric, phosphoric, sulfuric, hydrobromic, hydriodic, phosphorous, and the like
- organic acids such as aliphatic mono- and dicarboxylic acids, phenyl-substituted alkanoic acids, hydroxy alkanoic acids, alkanedioic acids, aromatic acids, aliphatic and aromatic sulfonic acids, etc.
- Such salts thus include sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, nitrate, phosphate, monohydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, caprylate, isobutyrate, oxalate, malonate, succinate, suberate, sebacate, fumarate, maleate, mandelate, benzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, phthalate, benzenesulfonate, toluenesulfonate, phenylacetate, citrate, lactate, maleate, tartrate, methanesulfonate, and the like.
- salts of amino acids such as arginate and the like and gluconate or galacturonate (see, for example, Berge S. M., et al., "Pharmaceutical Salts,” (1977) J. of Pharmaceutical Science, 66:1-19).
- the acid addition salts of said basic compounds are prepared by contacting the free base form with a sufficient amount of the desired acid to produce the salt in the conventional manner.
- the free base form may be regenerated by contacting the salt form with a base and isolating the free base in the conventional manner.
- the free base forms differ from their respective salt forms somewhat in certain physical properties such as solubility in polar solvents, but otherwise the salts are equivalent to their respective free base for purposes of the present invention.
- the base addition salts of said acidic compounds are prepared by contacting the free acid form with a sufficient amount of the desired base to produce the salt in the conventional manner.
- the free acid form may be regenerated by contacting the salt form with an acid and isolating the free acid in the conventional manner.
- the free acid forms differ from their respective salt forms somewhat in certain physical properties such as solubility in polar solvents, but otherwise the salts are equivalent to their respective free acid for purposes of the present invention.
- the pharmaceutical composition comprises a compound of the general Formula I wherein X is N and Rl is a 3-amino-2-hydroxy-propyl radical, as exemplified by a compound of the formula la:
- Y, Y', R2, R3, m and n are as defined above.
- R2 is H and R3 is selected from C1-C6 alkyl, C2-C6 alkenyl, C6-C14 aryl, or heteroaryl
- said Cl- C6 alkyl and C2-C6 alkenyl may be substituted by at least one radical selected from halogen, -OH, -SH, C1-C6 alkoxy, C1-C6 alkylthio, C6-C14 aryl, heteroaryl, -NRR', - COOR, -CONRR', -SO 3 H, or -SO 2 NRR'
- said C6-C14 aryl and heteroaryl may be substituted by at least one radical selected from halogen, -OH, -SH, C1-C6 alkyl, C2-C6 alkenyl, C1-C6 alkoxy, C1-C6 alkylthio, -NRR', -COOR, -CONRR',
- R3 is preferably C1-C6 alkyl, most preferably ethyl, substituted by C6-C14 aryl, preferably phenyl, as identified in the formula lb below:
- R2 is C1-C6 alkyl optionally substituted by at least one radical selected from halogen, -OH, -SH, Cl- C6 alkoxy, C1-C6 alkylthio, C6-C14 aryl, heteroaryl, -NRR', -COOR, -CONRR', -SO 3 H, or -SO 2 NRR', and R3 is -CONRR', -CSNRR', or -CONHNRR'.
- R2 is ethyl substituted by C6-C14 aryl, preferably phenyl
- R3 is -CSNRR', as depicted in formula Ic:
- R, R', Y, Y', m and n are as described hereinabove.
- R is H and R' is phenyl substituted at the para position by -SO 3 H, Y and Y' are Br and m and n are 1, as exemplified by the novel compound herein identified as Compound 3 in the Appendix A.
- the pharmaceutical composition comprises a compound of the formula la, wherein R2 and R3 together with the N atom to which they are attached form a saturated 6-membered heterocyclic ring containing at least one further heteroatom selected from N, O, and/or S, preferably piperazino substituted at the further N atom by R5, as depicted in formula Id:
- the pharmaceutical composition comprises a compound of the formula Id, wherein R5 is heteroaryl derived from a bicyclic ring containing two N atoms, preferably quinazolinyl substituted by - NRR', wherein R is hydrogen and R' is C6-C14 aryl substituted by at least one radical selected from halogen, -OH, -SH, -NH 2 , -SO 3 H, C1-C6 alkyl, C2-C6 alkenyl, C1-C6 alkoxy, or C1-C6 alkylthio.
- the pharmaceutical composition comprises a compound of formula Id, wherein R5 is C1-C6 alkyl substituted by carbazolyl at the terminal carbon atom and by a further group selected from halogen, - OH, -SH, -NH 2 , C1-C6 alkoxy or C1-C6 alkylthio, preferably a 3-carbazolyl-2-hydroxy- propyl group as exemplified by the compound herein identified as Compound 5 in Appendix A.
- R5 is C1-C6 alkyl substituted by carbazolyl at the terminal carbon atom and by a further group selected from halogen, - OH, -SH, -NH 2 , C1-C6 alkoxy or C1-C6 alkylthio, preferably a 3-carbazolyl-2-hydroxy- propyl group as exemplified by the compound herein identified as Compound 5 in Appendix A.
- This compound is described in the literature [CAS No. 300392-
- the pharmaceutical composition comprises a fluorine compound of formula Ie:
- R4 is -CONRR', as depicted in formula If:
- the Compounds 1, 2, 4 and 5 may be prepared from unsubstituted or the appropriately substituted carbazole as depicted in Scheme 1.
- the carbazole is reacted with epichlorohydrin under basic conditions, and the resulting 9-oxiranylmethyl- 9H-carbazole is reacted with a suitable primary or secondary amine, the oxiranyl ring is therefore opened and the desired ⁇ -amino alcohol is formed.
- Compound 3 was prepared in one step from Compound 2 as shown in Scheme 2 by stirring a mixture of Compound 2 and/?-sulfophenylisothiocyanate.
- Compound 4 was prepared in one step by reacting 3,6-dibromo-9- oxiranylmethyl-9H-carbazole with 2-piperazino-4-(3 ,4-dimethoxyphenyl)amino- quinazoline, as shown in Scheme 3.
- the inhibitory effect of the compounds of the present invention on heparanase activity can be evaluated by several methods carried out in vitro, ex vivo, or in vivo. Some of the in vitro assays used according to the present invention were described in US 6,190,875. In these assays, heparanase is incubated with a heparanase substrate in the presence and in the absence of a compound of the present invention, and the inhibitory effect of the compound on the catalytic activity of the heparanase on its substrate is evaluated.
- the heparanase may be natural mammalian heparanase, such as human heparanase purified as described in U.S.
- Patent 5,362,641 or, preferably, recombinant mammalian, e.g. human or mouse recombinant heparanase as described in US 5,968,822, US 6,190,875, and WO 99/57244, in purified or non-purified form.
- a source of non- purified recombinant heparanase is, for example, an extract of cells in which mammalian heparanase cDNA is expressed.
- the heparanase substrate may be a natural heparan sulfate substrate, or an alternative substrate of the enzyme as described in U.S. 6,190,875, for example, heparin (e.g. heparin immobilized on a gel such as Sepharose), heparin fragments (e.g. several species of low molecular weight heparin), modified non-anticoagulant species of heparin, other sulfated polysaccharides (e.g. pentosan polysulfate), soluble HSPG or ECM.
- heparin e.g. heparin immobilized on a gel such as Sepharose
- heparin fragments e.g. several species of low molecular weight heparin
- modified non-anticoagulant species of heparin e.g. pentosan polysulfate
- soluble HSPG soluble HSPG or ECM.
- Evaluation of the inhibitory effect can be carried out, for example, as described in US 6,190,875, by a size separation assay adapted for detection of degradation products of the heparanase substrate.
- assays include gel electrophoresis and column chromatography.
- Colorimetric assays Any colorimetric assay based on any color producing reaction is envisaged by the invention, be it a simple color reaction, which is readily detectable, or a fluorimetric or a luminiscent (e.g., chemiluminiscent) reaction, which are readily detectable by fluorescence detecting techniques.
- suitable colorimetric assays include, but are not limited to, the dimethylmethylene blue (DMB), tetrazolium blue and carbazole assays.
- Qualitative colorimetric assays include the dimethylmethylene blue (DMB) assay, which yields color shift in the presence of polyanionic compounds such as sulfated glycosaminoglycans having different sizes that are released from the substrate (soluble or immobilized), and the carbazole assay, which detects uronic acid derivatives present in complete hydrolyzates of products released from an immobilized substrate, both assays being applicable for crude extracts of heparanase and for the purified enzyme as well.
- DMB dimethylmethylene blue
- a quantitative evaluation is desired and the preferred in vitro assays are those which are adapted for detection of reducing moieties associated with degradation products of the heparanase substrate, preferably a reducing sugar assay.
- An example of a quantitative colorimetric assay is the tetrazolium blue assay which allows colorimetric detection of reducing moieties released from the substrate, e.g. heparan sulfate, which may be present either in soluble or immobilized form.
- Another possibility although less preferred, consists in evaluating the catalytic activity of heparanase on the substrate by radioactive techniques, in which case the substrate used is radiolabeled, either in vitro or metabolically.
- the ex vivo assays for evaluating the inhibitory effect of the compounds on heparanase activity include angiogenic sprout formation and transmigration assays.
- the angiogenic sprout formation assay is carried out in the rat aorta model (Nicosia et al., 1997; Nicosia and Ottinetti, 1990), whereby rat aorta rings are embedded in a basement membrane-like matrix composed of ECM-derived proteins such as laminin and collagen type IV, and HSPG, thus constituting a relevant heparanase substrate.
- the rings then develop angiogenic sprouts and angiogenesis can be quantitated.
- the compounds to be tested are added to the embedded aortic rings and their effect on angiogenic sprout formation is then evaluated.
- immune cell migration is evaluated, optionally in the presence of a chemoattractant factor such as stromal cell-derived factor 1 (SDF-1), a process which mimics in vivo extravasation of immune cells from the vasculature to sites of inflammation.
- a chemoattractant factor such as stromal cell-derived factor 1 (SDF-1)
- SDF-1 stromal cell-derived factor 1
- immune cells such as lymphocytes are let to migrate from the upper to the lower chamber through a transwell filter coated with a basement membrane-like matrix composed of ECM-derived proteins.
- the migration rate of the cells through the filter is then evaluated by counting the number of cells migrated through the filter (e.g. using a FACSort) compared to the number of cells added on top of the upper chamber.
- Over expression of heparanase in the immune cells results in an increase in the transmigration rate of the cells while addition of a heparanase inhibitor reduces the transmigration rate of the cells.
- the inhibitory effect of the compounds on heparanase activity may be also assayed in vivo, for example, using the primary tumor growth or metastasis animal models or the sponge inflammation assay.
- primary tumor animal model animals are injected subcutaneously (s.c.) with tumor cells and treated with the heparanase inhibitors. Tumor growth is measured when animals in untreated control group start to die.
- primary tumors may be generated with B16-F1 melanoma cells or with a highly metastatic subclone thereof injected s.c. into the flanks of mice.
- the mice are treated with heparanase inhibitors injected intraperitoneally (i.p.) twice a day starting 4 days after cell injection and are sacrificed and the tumor measured about 3 weeks after cell injection.
- metastasis animal model animals are injected intravenously (i.v.) with tumor cells and treated with the heparanase inhibitors.
- the number of lung metastasis is counted when animals in untreated control group start to die or about 3 weeks after cell injection.
- metastasis may be generated with B16-F1 melanoma cells or with a highly metastatic subclone thereof injected i.v. to mice.
- the mice are treated with heparanase inhibitors injected i.p. at certain times following cell injection, and are then sacrificed and the number of lung metastasis is counted.
- PVA polyvinyl alcohol
- MPO myeloperoxidase
- heparanase inhibitors of the present invention can be used for the treatment of diseases and disorders caused by or associated with heparanase catalytic activity such as, but not limited to, cancer, inflammatory disorders and autoimmune diseases.
- the compounds can be used for inhibition of angiogenesis, and are thus useful for the treatment of diseases and disorders associated with angiogenesis or neovascularization such as, but not limited to, tumor angiogenesis, ophthalmologic disorders such as diabetic retinopathy and macular degeneration, particularly age-related macular degeneration, reperfusion of gastric ulcer, and also for contraception or for inducing abortion at early stages of pregnancy.
- diseases and disorders associated with angiogenesis or neovascularization such as, but not limited to, tumor angiogenesis, ophthalmologic disorders such as diabetic retinopathy and macular degeneration, particularly age-related macular degeneration, reperfusion of gastric ulcer, and also for contraception or for inducing abortion at early stages of pregnancy.
- the compounds of the general formula I are useful for treatment or inhibition of a malignant cell proliferative disease or disorder.
- non-solid cancers e.g hematopoietic malignancies such as all types of leukemia, e.g. acute lymphocytic leukemia (ALL), acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), myelodysplastic syndrome (MDS), mast cell leukemia, hairy cell leukemia, Hodgkin's disease, non-Hodgkin's lymphomas, Burkitt's lymphoma and multiple myeloma, as well as for the treatment or inhibition of solid tumors such as tumors in lip and oral cavity, pharynx, larynx, paranasal sinuses, major salivary glands, thyroid gland, esophagus, stomach, small intestine, colon, colorectum, anal canal, liver, gallbla
- ALL acute lymphocytic leukemia
- AML acute myelogenous leukemia
- the compounds of the general formula I are useful for treating or inhibiting tumors at all stages, namely tumor formation, primary tumors, tumor progression or tumor metastasis.
- the compounds of general formula I are also useful for inhibiting or treating other cell proliferative diseases or disorders such as psoriasis, hypertrophic scars, acne and sclerosis/scleroderma, and for inhibition or treatment of other diseases or disorders such as polyps, multiple exostosis, hereditary exostosis, retrolental fibroplasia, hemangioma, and arteriovenous malformation.
- the compounds of general formula I are useful for treatment of or amelioration of inflammatory symptoms in any disease, condition or disorder where immune and/or inflammation suppression is beneficial such as, but not limited to, treatment of or amelioration of inflammatory symptoms in the joints, musculoskeletal and connective tissue disorders, or of inflammatory symptoms associated with hypersensitivity, allergic reactions, asthma, atherosclerosis, otitis and other otorhinolaryngological diseases, dermatitis and other skin diseases, posterior and anterior uveitis, conjunctivitis, optic neuritis, scleritis and other immune and/or inflammatory ophthalmic diseases.
- any disease, condition or disorder where immune and/or inflammation suppression is beneficial such as, but not limited to, treatment of or amelioration of inflammatory symptoms in the joints, musculoskeletal and connective tissue disorders, or of inflammatory symptoms associated with hypersensitivity, allergic reactions, asthma, atherosclerosis, otitis and other otorhinolaryngological diseases, dermatitis and other skin diseases, posterior and anterior uveit
- the compounds of general formula I are useful for treatment of or amelioration of an autoimmune disease such as, but not limited to, Eaton-Lambert syndrome, Goodpasture's syndrome, Grave's disease, Guillain-Barre syndrome, autoimmune hemolytic anemia (AIHA), hepatitis, insulin-dependent diabetes mellitus (IDDM), systemic lupus erythematosus (SLE), multiple sclerosis (MS), myasthenia gravis, plexus disorders e.g. acute brachial neuritis, polyglandular deficiency syndrome, primary biliary cirrhosis, rheumatoid arthritis, scleroderma, thrombocytopenia, thyroiditis e.g.
- an autoimmune disease such as, but not limited to, Eaton-Lambert syndrome, Goodpasture's syndrome, Grave's disease, Guillain-Barre syndrome, autoimmune hemolytic anemia (AIHA), hepatitis, insulin-dependent diabetes mellit
- Hashimoto's disease Sjogren's syndrome, allergic purpura, psoriasis, mixed connective tissue disease, polymyositis, dermatomyositis, vasculitis, polyarteritis nodosa, polymyalgia rheumatica, Wegener's granulomatosis, Reiter's syndrome, Behcet's syndrome, ankylosing spondylitis, pemphigus, bullous pemphigoid, dermatitis herpetiformis, Crohn's disease and autism.
- compositions for use in accordance with the present invention may be formulated in conventional manner using one or more physiologically acceptable carriers or excipients.
- the carrier(s) must be acceptable in the sense that it is compatible with the other ingredients of the composition and it is not deleterious to the recipient thereof.
- carrier refers to a diluent, adjuvant, excipient, or any other suitable vehicle.
- Such pharmaceutical carriers can be sterile liquids such as water and oils.
- the pharmaceutical composition can be administered systemically, for example by parenteral, e.g. intravenous , intraperitoneal or intramuscular injection.
- parenteral e.g. intravenous , intraperitoneal or intramuscular injection.
- the pharmaceutical composition can be introduced to a site by any suitable route including intravenous, subcutaneous, transcutaneous, topical, intramuscular, intraarticular, subconjunctival, or mucosal, e.g. oral, intranasal, or intraocular.
- the pharmaceutical composition is administered to the area in need of treatment. This may be achieved by, for example, local infusion during surgery, topical application, direct injection into the inflammed joint, directly onto the eye, etc.
- the pharmaceutical preparation may be in liquid form, for example, solutions, syrups or suspensions, or in solid form as tablets, capsules and the like.
- the compositions are conveniently delivered in the form of drops or aerosol sprays.
- the formulations may be presented in unit dosage form, e.g. in ampoules or in multidose containers with an added preservative.
- compositions of the invention can also be delivered in a vesicle, in particular in liposomes.
- the compositions can be delivered in a controlled release system.
- the amount of the therapeutic or pharmaceutical composition of the invention which is effective in the treatment of a particular disease, condition or disorder will depend on the nature of the disease, condition or disorder and can be determined by standard clinical techniques. In general, the dosage ranges from about 0.01 mg/kg to about 50-100 mg/kg. In addition, in vitro assays as well in vivo experiments may optionally be employed to help identify optimal dosage ranges. The precise dose to be employed in the formulation will also depend on the route of administration, and the seriousness of the disease, condition or disorder, and should be decided according to the judgment of the practitioner and each patient's circumstances. Effective doses may be extrapolated from dose-response curves derived from in vitro or animal model test systems. For example, in order to obtain an effective mg/kg dose for humans based on data generated from mice or rat studies, the effective mg/kg dosage in mice or rats is divided by twelve or six, respectively.
- the invention will now be illustrated by the following non-limiting examples.
- the carbazole compounds may be readily synthesized from the appropriate carbazole derivative, as shown in Scheme 1.
- the carbazole is reacted with epichlorohydrin in the present of sodium hydride, thus obtaining 9-oxiranylmethyl-9H- carbazole.
- the oxiranyl ring opening is achieved in the presence of a primary or secondary amine, which produces a secondary or tertiary ⁇ -amino alcohol.
- Such reactive primary or secondary amine may be, for example, phenethylamine, 3-phenylallyl amine, and N-substituted piperazines.
- Compounds 1 and 2 are prepared by reacting 3 -phenethylamine with dihalo 9- oxiranylmethyl-9H-carbazole.
- Compounds 4 and 5 are prepared by reacting the 9- oxiranylmethyl-9H-carbazole with the appropriate N-substituted piperazine.
- Heparin Sepharose CL-6B was purchased from Pharmacia (Amersham Pharmacia Biotech) Uppsala, Sweden ; 1 ,9-Dimethylmethylene blue (DMB), tetrazolium blue and heparan sulfate were purchased from Sigma- Aldrich (Rehovot, Israel); MCDB 131 medium was purchased from Clonetics (San Diego, CA, USA); DMEM and fetal calf serum were purchased from Gibco BRL (InVitrogen Co ⁇ oration, CA, USA) ; glutamine and gentamicin were purchased from Biological Industries (Bet Haemek, Israel). Matrigel was kindly provided by Dr. H. Kleinmann, NIDR, NIH, Bethesda, MD, USA.
- Heparin Sepharose CL-6B beads were added up to the top of the wells of a multiscreen column loader (Millipore).
- a 96-well multiscreen plate containing 0.65 ⁇ m hydrophilic, low protein binding, Durapore membrane (Millipore) was placed, upside down, on top of the multiscreen column loader.
- the column loader and the multiscreen plate were held together, turned over, and the beads were uniformly transferred from the column loader to the multiscreen plate.
- Double-distilled water (DDW) was then added to the beads, which were allowed to swell for one minute, and then washed (three times) with DDW under vacuum. Heparin concentration was estimated to be 20 ⁇ M/well.
- Human recombinant heparanase of at least 50% purity was obtained by expression in the CHO cells SI -11 subclone (generated as described for CHO clones S1PPT-4 and S1PPT-8 in WO 99/57244).
- Active human recombinant heparanase purified from the CHO cell extracts by ion exchange chromatography (as described for the CHO 2TT1-8 subclone in WO 99/57244), was added (5 ng/well) to a reaction mixture containing 20 mM phosphate citrate buffer, pH 5.4, 1 mM CaCl 2 , 1 mM NaCl, and 1 mM dithiothreitol (DTT; total volume of 100 ⁇ l).
- DTT dithiothreitol
- heparanase reaction products were filtered under vacuum and collected into a 96-well polystyrene flat bottom plate (Greiner Cat. No. 655101).
- PBS phosphate-buffered saline
- BSA bovine serum albumin
- DMB 32 mg of DMB were dissolved in 5 ml ethanol, diluted to 1 liter with formate buffer containing 4 g sodium formate and 4 ml formic acid; 125 ⁇ l /well) were added.
- crude extracts of CHO cells SI -11 subclone expressing human recombinant or crude extracts of CHO cells mhG9 clone expressing mouse recombinant heparanase (generated with the mouse heparanase cDNA as described for CHO clones expressing human recombinant heparanase in WO 99/57244) were used.
- the cell extracts were centrifuged and resuspended in 20 mM phosphate citrate buffer, pH 5.4 containing 50 mM NaCl.
- the cells were lysed by three cycles of freezing and thawing.
- the cell lysates were centrifuged (lOOOOxg for 5 min), supernatants were collected and then assayed for heparanase activity using the DMB assay.
- each compound was dissolved in dimethylsulfoxide (DMSO) and added, at a concentration range of 1-30 ⁇ M, to the heparin Sepharose swollen beads in the 96-multiscreen plate.
- DMSO dimethylsulfoxide
- the partially purified human recombinant heparanase or the crude cell extracts expressing either human or mouse recombinant heparanase was added for a 3-hour incubation and the reaction continued as described above. Color was developed and the absorbance was measured as described above. The IC 50 value (the concentration at which the heparanase activity was inhibited by 50%) for each compound was evaluated.
- IC 50 value the concentration at which the heparanase activity was inhibited by 50%
- Human recombinant heparanase of at least 50% purity obtained by expression in the CHO cells Sl-11 subclone as described in (a) above was added (4 ng) to each well of a 96-well microplate and incubated in a reaction mixture containing 20 mM phosphate citrate buffer, pH 5.4, 1 mM CaCl 2 , 1 mM NaCl, and 4 ⁇ M heparan sulfate (final volume of 100 ⁇ l).
- thoracic aortas were excised from 2- to 3-month-old Fischer 344 male rats, rinsed in serum-free MCDB 131 growth medium containing 50 ⁇ g/ml gentamicin, cleaned of periadventitial fibroadipose tissue, and cross-sectioned at ⁇ 1 mm intervals.
- Freshly cut aortic rings were rinsed in serum-free MCDB 131 medium and each ring was embedded in Matrigel (a basement membrane-like matrix composed of ECM-derived proteins such as laminin and collagen type IV and others, and HSPG, thus constituting a relevant heparanase substrate).
- Matrigel a basement membrane-like matrix composed of ECM-derived proteins such as laminin and collagen type IV and others, and HSPG, thus constituting a relevant heparanase substrate.
- Matrigel cultures were transferred to 18-mm wells of 4- well plates (Nunc) and grown at 35.5°C in 0.5 ml of serum-free MCDB131 medium that was changed 3 times a week.
- Angiogenesis was quantitated by counting the number of neovessels according to published criteria (Nicosia and Ottinetti, 1990).
- a test compound was added to the Matrigel aortic ring cultures and its effect on reduction of the number of new microvessels was determined in comparison with untreated cultures.
- primary tumor was generated in C57BL mice by cells herein designated FOR cells, which were generated as follows: B16-F1 mouse melanoma cells (ATCC No. 6326) were grown in DMEM containing 10% fetal calf serum, 2 mM glutamine, and 50 ⁇ g/ml gentamicin. A subclone of the B16- Fl cell line, Fl-J, produced large amounts of melanin and exhibited a highly metastasis potential. These highly metastatic Fl-J cells were injected to syngeneic mice (100,000 cells, s.c). Cells from metastases that were formed were cultured in different conditions.
- a clone, Fl-LG, designated herein FOR was selected by its high heparanase expression and activity using the reverse transcriptase-polymerase chain reaction (RT-PCR) and the radiolabeled ECM degradation analyses, respectively, as previously described (Vlodavsky et al., 1999; U.S. 6,190,875).
- FOR cells were grown in DMEM containing 10% fetal calf serum, 2 mM glutamine, and 50 ⁇ g/ml gentamicin until they reached confluence (typically 4-5 days) and then splitted (1:5).
- This splitting yielded subconfluent and growing cells at day 7, the day of cell injection, at which the cells were trypsinized, washed with PBS and counted to yield a cell suspension of 10 6 cells/ml in PBS.
- Male C57BL mice (-20 gram each; at least 10 mice/group) were injected s.c. on the flank with a suspension of the FOR cells (100 ⁇ l/mouse).
- a test compound dissolved in DMSO was injected (100 ⁇ l) i.p to the mice, twice a day (morning and evening). Each compound was injected at concentration of 1 mg/mouse/day).
- Control mice were injected i.p. with DMSO only (100 ⁇ l). Mice were observed daily, and usually three weeks after cell injection, mice were sacrificed, the tumors were harvested and weighted.
- FOR cells were cultured as described in (d) above. After trypsinization, the cells were washed with PBS and counted to yield a cell suspension of 1.5xl0 6 cells/ml in PBS.
- Male C57BL mice ( ⁇ 20 gram each; at least 10 mice/group) were injected i.v. with a suspension of the FOR cells (100 ⁇ l/mouse).
- a test compound dissolved in DMSO was injected (100 ⁇ l) i.p to the mice 4 and 8 hours after cell injection. The compound was injected at concentration of 0.5 mg/mouse/day). Control mice were injected i.p. with DMSO only. Mice were observed daily, and three weeks after cell injecion, mice were sacrificed, the lungs were fixed in Bouen's solution and scored for the number of metastatic nodules as previously described (Vlodavsky et al., 1994).
- the sponge inflammation assay mimics the inflammatory reaction resulting from the presence of a foreign body in the organism. It was carried out by placing 3 polyvinyl alcohol (PVA) sponges (10 x 0.4 mm) under the skin of BALB/c male mice. The compound to be tested was then injected to the mouse in the following order: (i) immediately following sponge implantation, (ii) 4 hours after sponge implantation, and (iii) 8 hours after sponge implantation. The total volume of all three injections is 1 ml. The starting concentration of the tested compound was lmg/ml/mice. At the next morning, the sponges were taken out and all three were squeezed into an Eppendorf tube (the total volume collected was about 0.6 ml).
- PVA polyvinyl alcohol
- TNF- ⁇ concentrations were determined in the supernatant (in pg/ml) with mouse TNF- ⁇ ELISA kit according to the manufacturer's instructions (Bender Medsystems, Vienna, Austria).
- MPO is an oxidoreductase that catalyzes the reaction of hydrogen peroxide and halide ions to produce cytotoxic acids (such as hypochlorous acid) and other intermediates; these play a role in oxygen-dependent killing of microorganisms and tumor cells.
- MPO is a green hemoprotein found in the azurophil granules of neutrophils and its quantification serves as an index of neutrophils infiltration.
- the cells in the cell pellets obtained from the squeezed sponges were suspended in ice cold 0.5% hexadecyltrimethylammonium bromide (HTAB) in 50 mM potassium phosphate buffer, pH 6.0, frozen at -80°C and then warmed at 60°C for 24 minutes.
- MPO was measured spectrophotometrically: 10 ⁇ l of the cell homogenate were mixed with 380 ⁇ l of a 50 mM phosphate buffer, at pH 6.0 and containing 0.167 mg/ml o-dianisidine dihydrochloride and 0.0005% H 2 O 2 .
- the reaction was quenched with 10 ⁇ l H 2 SO 4 (final concentration 3.3M) and then the change in absorbance at 460 nm was measured by an ELISA reader (MRX, Dinatec).
- Example II In vitro inhibition of heparanase activity by compounds of the invention.
- the inhibition of heparanase activity by the compounds of the present invention was first detected in two colorimetric in vitro assays, i.e., the DMB assay and the tetrazolium blue assay as described in Methods (a) and (b) above.
- the human recombinant heparanase (designated h-hepa) expressed in CHO cells Sl-11 subclone was used herein either in its partially purified form (50% purity) or in crude cell extracts, and the mouse recombinant heparanase (designated m-hepa) expressed in the CHO cells mhG9 clone was used herein in crude cell extracts only.
- IC 0 values of the different compounds are shown in Table 1. All the tested compounds were found to inhibit heparanase activity at micromolar concentrations. However, Compound 3 was shown to be potent (IC 50 values in the range of 2.2 to 12 ⁇ M compared to IC 50 values in the range of 6 to 36 ⁇ M for the other compounds). Table 1. IC 50 values of the tested compounds for inhibition of heparanase as detected by the in vitro DMB and tetrazolium assays.
- the angiogenesis inhibitory effect of Compound 3 was assayed using the angiogenic sprout formation assay described in Method (c) above.
- Compound 3 showed inhibitory concentration of 120 ⁇ M.
- Example II Inhibition of mouse melanoma primary tumor growth and of metastasis by Compound 3
- Example II (4). Measurement of myeloperoxidase (MPO) activity.
- Sponge inflammation assay was carried out with the heparanase inhibitor
- Figs. 1-3 10 mice in each group.
- administration of 1 mg/ml/mouse of Compound 3 resulted in about 25% decrease in the total number of cells extracted from the sponges (in comparison to control, untreated mice).
- Fig. 2 shows that there was a decrease in the MPO activity in the animal treated with 1 mg/ml of Compound 3
- the results of Figs. 1 and 2 indicate that fewer neutrophils were recruited to the sponge, indicating that treatment with Compound 3 resulted in a weaker inflammatory reaction.
- the heparanase inhibitor Compound 3 at concentrations of 1 mg/ml/mouse and 0.2 mg/ml/mouse, dramatically reduced the amount of the pro-inflammatory TNF- ⁇ in the supernatant extracted from sponge samples in a manner that correlates well with the total number of cells extracted from the sponges (Fig. 1).
- the heparanase inhibitor Compound 3 was shown to reduce the inflammatory response.
- Nicosia, R.F., Lin, Y.J., Hazelton, D., and Qian, X. (1997) Endogenous regulation of angiogenesis in the rat aorta model. Amer. J. Pathol. 151 : 1379-1386.
- Vlodavsky I., Hua-Quan Miao., Benezra, M., Lider, O., Bar-Shavit, R., Schmidt,
- Vlodavsky I., Mohsen, M., Lider, O., Svahn, CM., Ekre, H.P., Vigoda, M., Ishai- Michaeli, R., and Peretz, T. (1994) Inhibition of tumor metastasis by heparanase inhibiting species of heparin. Invasion Metastasis 14:290-302.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Indole Compounds (AREA)
Abstract
Description






















Claims





























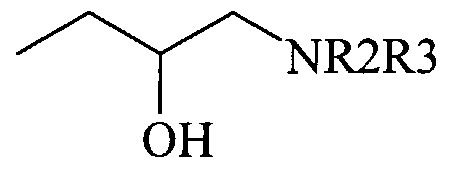









































Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2002228316A AU2002228316A1 (en) | 2001-01-29 | 2002-01-29 | Carbazole derivatives and their uses as heparanase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US26430401P | 2001-01-29 | 2001-01-29 | |
| US60/264,304 | 2001-01-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002060867A2 true WO2002060867A2 (en) | 2002-08-08 |
| WO2002060867A3 WO2002060867A3 (en) | 2004-03-18 |
Family
ID=23005445
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IL2002/000079 WO2002060867A2 (en) | 2001-01-29 | 2002-01-29 | Carbazole derivatives and their uses as heparanase inhibitors |
Country Status (2)
| Country | Link |
|---|---|
| AU (1) | AU2002228316A1 (en) |
| WO (1) | WO2002060867A2 (en) |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004020422A1 (en) * | 2002-08-30 | 2004-03-11 | De Novo Pharmaceuticals Limited | 1, 4- substituted piperidine derivatives and use thereof as base-inhibitors in treatment of alzheimer's disease |
| WO2004089940A1 (en) * | 2003-04-11 | 2004-10-21 | Glenmark Pharmaceuticals S.A. | Novel heterocyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| WO2005074375A2 (en) * | 2004-02-06 | 2005-08-18 | Insight Biopharmaceuticals Ltd | Heparanase inhibitors and uses thereof |
| WO2005074971A1 (en) * | 2004-01-29 | 2005-08-18 | Cellzome Ag | Treatment of neurodegenerative diseases by the use of cgi-13 interacting molecules |
| WO2005118528A2 (en) * | 2004-06-04 | 2005-12-15 | The University Court Of The University Of Aberdeen | Aryl alkyl sulfonamides as therapeutic agents for the treatment of bone conditions |
| US7238725B2 (en) | 2002-10-23 | 2007-07-03 | Glenmark Pharmaceuticals Ltd. | Tricyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| EP1896035A2 (en) * | 2004-06-16 | 2008-03-12 | Jack Arbiser | Carbazole formulations for the treatment of psoriasis and angiogenesis |
| US7563900B2 (en) | 2004-10-13 | 2009-07-21 | Glenmark Pharmaceuticals S.A. | Process for the preparation N-(3,5-dichloropyrid-4-yl)-4-difluoromethoxy-8-methane sulfonamido-dibenzo[b,d]furan-1-carboxamide |
| EP2086553A1 (en) * | 2006-10-20 | 2009-08-12 | The Australian National University | Inhibition of degradation of extracellular matrix |
| US7943634B2 (en) | 2004-12-17 | 2011-05-17 | Glenmark Pharmaceuticals S.A. | Substituted benzo[4,5]furo[3,2-c]pyridine derivatives as PDE 4 inhibitors |
| US8129401B2 (en) | 2004-12-17 | 2012-03-06 | Glenmark Pharmaceuticals S.A. | Heterocyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| US8207167B2 (en) | 2008-09-19 | 2012-06-26 | Pimco 2664 Limited | Aryl-phenyl-sulfonamide-phenylene compounds and their use |
| EP2484666A1 (en) * | 2008-08-15 | 2012-08-08 | Georgetown University | Fluorescent regulators of RASSF1A expression and human cancer cell proliferation |
| US8362277B2 (en) | 2009-01-09 | 2013-01-29 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US8435968B2 (en) | 2008-09-19 | 2013-05-07 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| EP2526942A3 (en) * | 2005-06-08 | 2013-07-17 | The University of North Carolina at Chapel Hill | Methods of facilitating neural cell survival using non-peptide and peptide BDNF neurotrophin mimetics |
| US20130190273A1 (en) * | 2009-01-09 | 2013-07-25 | Board Of Regents Of The University Of Texas System | Methods for Treating Amyotrophic Lateral Sclerosis Using Pro-Neurogenic Compounds |
| US8524778B2 (en) | 2007-03-21 | 2013-09-03 | Pimco 2664 Limited | Biphenyl-4-yl-sulfonic acid arylamides and their use as therapeutic agents |
| US8604074B2 (en) | 2009-01-09 | 2013-12-10 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| WO2014108168A1 (en) | 2013-01-10 | 2014-07-17 | Merck Patent Gmbh | Piperidinylcarbazole as antimalarial |
| US9095572B2 (en) | 2009-01-09 | 2015-08-04 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US9243281B2 (en) | 2013-11-11 | 2016-01-26 | Board Of Regents Of The University Of Texas System | Neuroprotective chemicals and methods for identifying and using same |
| EP2925129A4 (en) * | 2012-08-24 | 2016-06-08 | Univ Texas | Pro-neurogenic compounds |
| KR20160094802A (en) * | 2015-02-02 | 2016-08-10 | 충남대학교산학협력단 | Composition containing the carbazole urea derivative for preventing or treating vascular disease |
| WO2016203468A1 (en) * | 2015-06-15 | 2016-12-22 | Raziel Therapeutics Ltd. | Carbazole derivatives for the treatment of fibrotic diseases and related symptoms, and conditions thereof |
| US9616048B2 (en) | 2009-01-09 | 2017-04-11 | Board Of Regents Of The University Of Texas System | Anti-depression compounds |
| US9624167B2 (en) | 2013-06-26 | 2017-04-18 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| US9701676B2 (en) | 2012-08-24 | 2017-07-11 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| EP3052190A4 (en) * | 2013-10-01 | 2017-07-19 | New York University | Amino, amido, and heterocyclic compounds as modulators of rage activity and uses thereof |
| US9828330B2 (en) | 2013-03-15 | 2017-11-28 | Pharmatrophix, Inc. | Non-peptide BDNF neurotrophin mimetics |
| US9828332B2 (en) | 2013-03-15 | 2017-11-28 | PharmatorophiX, Inc. | Non-peptide BDNF neurotrophin mimetics |
| EP3188738A4 (en) * | 2014-09-02 | 2018-01-24 | The Children's Hospital of Philadelphia | Compositions and methods for the inhibition of chondrogenesis |
| US9902713B2 (en) | 2013-11-11 | 2018-02-27 | Board Of Regents Of The University Of Texas System | Neuroprotective compounds and use thereof |
| US9994577B2 (en) | 2014-07-04 | 2018-06-12 | Merck Patent Gmbh | Azepanyl-derivatives and pharmaceutical compositions comprising the same with antiparasitic activity |
| WO2018107200A1 (en) * | 2016-12-13 | 2018-06-21 | Beta Therapeutics Pty Ltd | Heparanase inhibitors and use thereof |
| US10005733B2 (en) | 2014-12-17 | 2018-06-26 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamide and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)-benzenesulfonamide compounds and their therapeutic use |
| EP3381906A1 (en) | 2017-03-27 | 2018-10-03 | Leadiant Biosciences SA | Compounds for use as heparanase inhibitors |
| CN111217741A (en) * | 2019-03-01 | 2020-06-02 | 广东省中医院(广州中医药大学第二附属医院、广州中医药大学第二临床医学院、广东省中医药科学院) | Fluorine-substituted monocarbazole derivative, preparation method and application thereof |
| US11053255B2 (en) | 2015-06-22 | 2021-07-06 | Georgetown University | Synthesis of mahanine and related compounds |
| US11787783B2 (en) | 2016-12-13 | 2023-10-17 | Beta Therapeutics Pty Ltd | Heparanase inhibitors and use thereof |
| CN117486782A (en) * | 2023-12-29 | 2024-02-02 | 中国医学科学院药用植物研究所 | N-substituted carbazole derivative and preparation method and application thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5407940A (en) * | 1992-10-02 | 1995-04-18 | Adir Et Compagnie | New ellipticine compounds |
| US5679694A (en) * | 1992-07-20 | 1997-10-21 | The Wellcome Foundation Ltd. | Tetracyclic compounds, intermediates for their preparation and their use as antitumor agents |
-
2002
- 2002-01-29 WO PCT/IL2002/000079 patent/WO2002060867A2/en not_active Application Discontinuation
- 2002-01-29 AU AU2002228316A patent/AU2002228316A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5679694A (en) * | 1992-07-20 | 1997-10-21 | The Wellcome Foundation Ltd. | Tetracyclic compounds, intermediates for their preparation and their use as antitumor agents |
| US5407940A (en) * | 1992-10-02 | 1995-04-18 | Adir Et Compagnie | New ellipticine compounds |
Cited By (87)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004020422A1 (en) * | 2002-08-30 | 2004-03-11 | De Novo Pharmaceuticals Limited | 1, 4- substituted piperidine derivatives and use thereof as base-inhibitors in treatment of alzheimer's disease |
| US7238725B2 (en) | 2002-10-23 | 2007-07-03 | Glenmark Pharmaceuticals Ltd. | Tricyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| JP2006522789A (en) * | 2003-04-11 | 2006-10-05 | グレンマーク・ファーマシューティカルズ・エス・エー | Novel heterocyclic compounds useful for the treatment of inflammatory and allergic disorders; methods for their preparation and pharmaceutical compositions |
| US7223789B2 (en) | 2003-04-11 | 2007-05-29 | Glenmark Pharmaceuticals S.A. | Heterocyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| WO2004089940A1 (en) * | 2003-04-11 | 2004-10-21 | Glenmark Pharmaceuticals S.A. | Novel heterocyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| US7384962B2 (en) | 2003-04-11 | 2008-06-10 | Glenmark Pharmaceuticals S.A. | Heterocyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| US7393846B2 (en) | 2003-04-11 | 2008-07-01 | Glenmark Pharmaceuticals, S.A. | Heterocyclic compounds useful for the treatment of inflammatory and allergic disorders |
| EA010634B1 (en) * | 2003-04-11 | 2008-10-30 | Гленмарк Фармасьютикалс С.А. | Novel heterocyclic compounds useful for thr treatment of inflammatory and allergic disorders:process for their preparation and pharmaceutical compositions containing them |
| AP2008A (en) * | 2003-04-11 | 2009-06-29 | Glenmark Pharmaceuticals Sa | Novel heterocyclic compounds useful for the treatment of inflammatory and allergic disorders; process for their preparation and pharmaceutical compositions containing them |
| WO2005074971A1 (en) * | 2004-01-29 | 2005-08-18 | Cellzome Ag | Treatment of neurodegenerative diseases by the use of cgi-13 interacting molecules |
| WO2005074375A2 (en) * | 2004-02-06 | 2005-08-18 | Insight Biopharmaceuticals Ltd | Heparanase inhibitors and uses thereof |
| WO2005074375A3 (en) * | 2004-02-06 | 2009-04-23 | Insight Biopharmaceuticals Ltd | Heparanase inhibitors and uses thereof |
| WO2005118528A2 (en) * | 2004-06-04 | 2005-12-15 | The University Court Of The University Of Aberdeen | Aryl alkyl sulfonamides as therapeutic agents for the treatment of bone conditions |
| WO2005118528A3 (en) * | 2004-06-04 | 2006-01-26 | Univ Aberdeen | Aryl alkyl sulfonamides as therapeutic agents for the treatment of bone conditions |
| US7964643B2 (en) | 2004-06-04 | 2011-06-21 | The University Court Of The University Of Aberdeen | Aryl alkyl sulfonamides as therapeutic agents for the treatment of bone conditions |
| EP1896035A4 (en) * | 2004-06-16 | 2010-09-15 | Jack L Arbiser | Carbazole formulations for the treatment of psoriasis and angiogenesis |
| EP1896035A2 (en) * | 2004-06-16 | 2008-03-12 | Jack Arbiser | Carbazole formulations for the treatment of psoriasis and angiogenesis |
| US9289414B2 (en) | 2004-06-16 | 2016-03-22 | Jack L. Arbiser | Carbazole formulations for the treatment of psoriasis and angiogenesis |
| US7563900B2 (en) | 2004-10-13 | 2009-07-21 | Glenmark Pharmaceuticals S.A. | Process for the preparation N-(3,5-dichloropyrid-4-yl)-4-difluoromethoxy-8-methane sulfonamido-dibenzo[b,d]furan-1-carboxamide |
| US8129401B2 (en) | 2004-12-17 | 2012-03-06 | Glenmark Pharmaceuticals S.A. | Heterocyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| US7943634B2 (en) | 2004-12-17 | 2011-05-17 | Glenmark Pharmaceuticals S.A. | Substituted benzo[4,5]furo[3,2-c]pyridine derivatives as PDE 4 inhibitors |
| US8686045B2 (en) | 2005-06-08 | 2014-04-01 | The University Of North Carolina At Chapel Hill | Methods of facilitating neural cell survival using non-peptide and peptide BDNF neurotrophin mimetics |
| US9604907B2 (en) | 2005-06-08 | 2017-03-28 | The University Of North Carolina At Chapel Hill | Methods of facilitating neural cell survival using non-peptide and peptide BDNF neurotrophin mimetics |
| EP2526942A3 (en) * | 2005-06-08 | 2013-07-17 | The University of North Carolina at Chapel Hill | Methods of facilitating neural cell survival using non-peptide and peptide BDNF neurotrophin mimetics |
| JP2010506858A (en) * | 2006-10-20 | 2010-03-04 | ジ オーストラリアン ナショナル ユニバーシティ | Inhibition of extracellular matrix degradation |
| EP2086553A1 (en) * | 2006-10-20 | 2009-08-12 | The Australian National University | Inhibition of degradation of extracellular matrix |
| EP2086553A4 (en) * | 2006-10-20 | 2010-12-29 | Univ Australian | Inhibition of degradation of extracellular matrix |
| US8524778B2 (en) | 2007-03-21 | 2013-09-03 | Pimco 2664 Limited | Biphenyl-4-yl-sulfonic acid arylamides and their use as therapeutic agents |
| US10457639B2 (en) | 2008-08-15 | 2019-10-29 | Georgetown University | Fluorescent regulators of RASSF1A expression and human cancer cell proliferation |
| EP2484666A1 (en) * | 2008-08-15 | 2012-08-08 | Georgetown University | Fluorescent regulators of RASSF1A expression and human cancer cell proliferation |
| US9616037B2 (en) | 2008-09-19 | 2017-04-11 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US8435968B2 (en) | 2008-09-19 | 2013-05-07 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US9302984B2 (en) | 2008-09-19 | 2016-04-05 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US8822507B2 (en) | 2008-09-19 | 2014-09-02 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US8207167B2 (en) | 2008-09-19 | 2012-06-26 | Pimco 2664 Limited | Aryl-phenyl-sulfonamide-phenylene compounds and their use |
| US9050329B1 (en) | 2008-09-19 | 2015-06-09 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US9278923B2 (en) | 2009-01-09 | 2016-03-08 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US10183011B2 (en) | 2009-01-09 | 2019-01-22 | Board Of Regents Of The University Of Texas System | Anti-depression compounds |
| US8791149B2 (en) | 2009-01-09 | 2014-07-29 | Board Of Regents Of The University Of Texas System | Methods of treating traumatic brain injury using pro-neurogenic compounds |
| US9095571B2 (en) | 2009-01-09 | 2015-08-04 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US9095572B2 (en) | 2009-01-09 | 2015-08-04 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US9156787B2 (en) | 2009-01-09 | 2015-10-13 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US8362277B2 (en) | 2009-01-09 | 2013-01-29 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US8877797B2 (en) | 2009-01-09 | 2014-11-04 | Board Of Regents Of The University Of Texas System | Methods for treating Parkinson's disease using pro-neurogenic compounds |
| US8748473B2 (en) * | 2009-01-09 | 2014-06-10 | Board Of The Regents Of The University Of Texas System | Methods of treating post-traumatic stress disorder using pro-neurogenic compounds |
| US8735440B2 (en) * | 2009-01-09 | 2014-05-27 | Board Of Regents Of The University Of Texas System | Methods for treating amyotrophic lateral sclerosis using pro-neurogenic compounds |
| US10172827B2 (en) | 2009-01-09 | 2019-01-08 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US9962368B2 (en) | 2009-01-09 | 2018-05-08 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US9884820B2 (en) | 2009-01-09 | 2018-02-06 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US9446042B2 (en) | 2009-01-09 | 2016-09-20 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US9446022B2 (en) | 2009-01-09 | 2016-09-20 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US20130190273A1 (en) * | 2009-01-09 | 2013-07-25 | Board Of Regents Of The University Of Texas System | Methods for Treating Amyotrophic Lateral Sclerosis Using Pro-Neurogenic Compounds |
| US8604074B2 (en) | 2009-01-09 | 2013-12-10 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| US9616048B2 (en) | 2009-01-09 | 2017-04-11 | Board Of Regents Of The University Of Texas System | Anti-depression compounds |
| US9701676B2 (en) | 2012-08-24 | 2017-07-11 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| EP2925129A4 (en) * | 2012-08-24 | 2016-06-08 | Univ Texas | Pro-neurogenic compounds |
| WO2014108168A1 (en) | 2013-01-10 | 2014-07-17 | Merck Patent Gmbh | Piperidinylcarbazole as antimalarial |
| US9828330B2 (en) | 2013-03-15 | 2017-11-28 | Pharmatrophix, Inc. | Non-peptide BDNF neurotrophin mimetics |
| US9828332B2 (en) | 2013-03-15 | 2017-11-28 | PharmatorophiX, Inc. | Non-peptide BDNF neurotrophin mimetics |
| US9796670B2 (en) | 2013-06-26 | 2017-10-24 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| US10233147B2 (en) | 2013-06-26 | 2019-03-19 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| US9624167B2 (en) | 2013-06-26 | 2017-04-18 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| US10029979B2 (en) | 2013-06-26 | 2018-07-24 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| EP3052190A4 (en) * | 2013-10-01 | 2017-07-19 | New York University | Amino, amido, and heterocyclic compounds as modulators of rage activity and uses thereof |
| US9645139B2 (en) | 2013-11-11 | 2017-05-09 | Board Of Regents Of The University Of Texas System | Neuroprotective chemicals and methods for identifying and using same |
| US9243281B2 (en) | 2013-11-11 | 2016-01-26 | Board Of Regents Of The University Of Texas System | Neuroprotective chemicals and methods for identifying and using same |
| US9902713B2 (en) | 2013-11-11 | 2018-02-27 | Board Of Regents Of The University Of Texas System | Neuroprotective compounds and use thereof |
| EP3459944A1 (en) | 2014-07-04 | 2019-03-27 | Merck Patent GmbH | Azepanyl-derivatives and pharmaceutical compositions comprising the same with antiparasitic activity |
| US9994577B2 (en) | 2014-07-04 | 2018-06-12 | Merck Patent Gmbh | Azepanyl-derivatives and pharmaceutical compositions comprising the same with antiparasitic activity |
| EP3188738A4 (en) * | 2014-09-02 | 2018-01-24 | The Children's Hospital of Philadelphia | Compositions and methods for the inhibition of chondrogenesis |
| US10005733B2 (en) | 2014-12-17 | 2018-06-26 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamide and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)-benzenesulfonamide compounds and their therapeutic use |
| KR20160094802A (en) * | 2015-02-02 | 2016-08-10 | 충남대학교산학협력단 | Composition containing the carbazole urea derivative for preventing or treating vascular disease |
| KR101652305B1 (en) | 2015-02-02 | 2016-08-30 | 충남대학교산학협력단 | Composition containing the carbazole urea derivative for preventing or treating vascular disease |
| US11191748B2 (en) | 2015-06-15 | 2021-12-07 | Raziel Therapeutics Ltd | Carbazole derivatives for the treatment of fibrotic diseases and related symptoms, and conditions thereof |
| US12097179B2 (en) | 2015-06-15 | 2024-09-24 | Raziel Therapeutics Ltd. | Carbazole derivatives for the treatment of fibrotic diseases and related symptoms, and conditions thereof |
| US10632102B2 (en) | 2015-06-15 | 2020-04-28 | Raziel Therapeutics Ltd | Carbazole derivatives for the treatment of fibrotic diseases and related symptoms, and conditions thereof |
| WO2016203468A1 (en) * | 2015-06-15 | 2016-12-22 | Raziel Therapeutics Ltd. | Carbazole derivatives for the treatment of fibrotic diseases and related symptoms, and conditions thereof |
| US11053255B2 (en) | 2015-06-22 | 2021-07-06 | Georgetown University | Synthesis of mahanine and related compounds |
| JP2020503377A (en) * | 2016-12-13 | 2020-01-30 | ベータ セラピューティクス プロプライアタリー リミティド | Heparanase inhibitors and uses thereof |
| CN110291073A (en) * | 2016-12-13 | 2019-09-27 | 贝塔医疗私人有限公司 | Heparanase inhibitors and application thereof |
| US11718609B2 (en) | 2016-12-13 | 2023-08-08 | Beta Therapeutics Pty Ltd | Heparanase inhibitors and use thereof |
| US11787783B2 (en) | 2016-12-13 | 2023-10-17 | Beta Therapeutics Pty Ltd | Heparanase inhibitors and use thereof |
| WO2018107200A1 (en) * | 2016-12-13 | 2018-06-21 | Beta Therapeutics Pty Ltd | Heparanase inhibitors and use thereof |
| WO2018177865A1 (en) | 2017-03-27 | 2018-10-04 | Leadiant Biosciences Sa In Liquidazione | Compounds for use as heparanase inhibitors |
| EP3381906A1 (en) | 2017-03-27 | 2018-10-03 | Leadiant Biosciences SA | Compounds for use as heparanase inhibitors |
| CN111217741A (en) * | 2019-03-01 | 2020-06-02 | 广东省中医院(广州中医药大学第二附属医院、广州中医药大学第二临床医学院、广东省中医药科学院) | Fluorine-substituted monocarbazole derivative, preparation method and application thereof |
| CN117486782A (en) * | 2023-12-29 | 2024-02-02 | 中国医学科学院药用植物研究所 | N-substituted carbazole derivative and preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2002228316A1 (en) | 2002-08-12 |
| WO2002060867A3 (en) | 2004-03-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2002060867A2 (en) | Carbazole derivatives and their uses as heparanase inhibitors | |
| WO2002060374A2 (en) | Benz-1,3-azole derivatives and their uses as heparanase inhibitors | |
| EP0891187B1 (en) | Use of derivatives of tetrahydro-beta-carbolines as antimetastatic agents | |
| US20070185176A1 (en) | Heparanase inhibitors and uses thereof | |
| US20080287469A1 (en) | Phosphoinositide 3-Kinase Inhibitors for Inhibiting Leukocyte Accumulation | |
| WO2002060375A2 (en) | Diphenyl ether derivatives and their uses as heparanase inhibitors | |
| US20050054614A1 (en) | Methods of inhibiting leukocyte accumulation | |
| CN104997774A (en) | Novel anti-aging agent and method to identify them | |
| EA009383B1 (en) | SOLUBLE HYALURONIDASE (sHASEGP), PROCESS FOR PREPARING THE SAME, USES AND PHARMACEUTICAL COMPOSITIONS COMPRISING THEREOF | |
| MXPA04010441A (en) | PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVE AND NAD(P)H OXIDASE INHIBITOR CONTAINING THE SAME. | |
| JP2007525494A6 (en) | Heparanase inhibitors and uses thereof | |
| US20220040177A1 (en) | Compounds, Compositions and Methods of Treating or Preventing Acute Lung Injury | |
| US20230321056A1 (en) | Methods for Treating Metastasis with Cathepsin C Inhibitors | |
| US9040673B2 (en) | Synthesis and identification of novel RSK-specific inhibitors | |
| WO2002060373A2 (en) | Indole derivatives and their uses as heparanase inhibitors | |
| WO2014100113A2 (en) | Syk kinase inhibitors as treatment for malaria | |
| CN101155591B (en) | Glucosamine and derivatives thereof useful as tg inhibitors | |
| KR100830018B1 (en) | Preventives and remedies for complications of diabetes | |
| CN113368249A (en) | OGT inhibitor and application thereof | |
| Cuzzocrea et al. | The tyrosine kinase inhibitor tyrphostin AG126 reduces the development of acute and chronic inflammation | |
| SK972013A3 (en) | Use of 5-carboxymethyl-3-mercapto-1,2,4-triazino-[5,6-b]indoles and pharmaceutical preparation containing them | |
| US20150094294A1 (en) | Compounds and methods for treating malaria | |
| CN115590861B (en) | Application of tripterygium wilfordii chlorolide | |
| US20050192209A1 (en) | Eosinophil Major Basic Protein as a natural heparanase-inhibiting protein, compositions, methods and uses thereof | |
| KR20020045821A (en) | Apicidin derivatives which inhibit histone deacetylase activity and cancer metastasis, pharmaceutical compositions containing the same and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| 122 | Ep: pct application non-entry in european phase | ||
| NENP | Non-entry into the national phase in: |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: JP |