SYNTHESIS OF DISCODERMOLIDE AND ANALOGS
BACKGROUND OF THE INVENTION
1. Field of the Invention
The present invention relates generally to the synthesis of discodermo- lide and analogs thereof. More particularly, the present invention involves the synthesis of three specific precursors of discodermolide which are combined together using the chelation controlled alkylation reaction to form the final discodermolide product.
2. Description of Related Art
The publications and other reference materials referred to herein to describe the background of the invention and to provide additional details regarding its practice are hereby incorporated by reference. For convenience, the reference materials are numerically referenced and identified in the appended bibliography.
The polyhydroxylated lactone discodermolide is a potent microtubule stabilizing agent showing activity similar to that of taxol. Discodermolide was first isolated from the marine sponge (Discodermia dissoluta) in the early 1990's by scientists at the Harbor Branch Oceanographic Institute. Discoder- molide was initially found to be a promising candidate for immunosuppressive therapy because of its ability to inhibit the proliferation of cultured lymphocytes.
Discodermolide is similar to Taxol (Ref. 1) in that it arrests the cell cycle at the G2/M boundary (Ref. 2). When breast carcinoma cells were treated with discodermolide at a concentration of lOnM, extensive microtubule bundling was observed. To create the same effect, a factor of 100 higher concentration (1 mM) of taxol was required. Under a variety of other assay conditions, discodermolide has shown higher potency than taxol. Furthermore, the toxicity of taxol has been found to be quite high (Ref. 3). The medicinal potential of discodermolide as an immunosuppressive and antimitotic agent have made it the object of substantial synthetic interest. The microtubule stabilizing ability of discodermolide has further intensified efforts to produce this drug synthetically. Several partial syntheses of discodermolide have been developed (Refs. 5, 6 and 7). Total syntheses of discodermolide have also been described by Schreiber and coworkers (Ref. 8) and Smith and coworkers (Ref.
9).
In view of the importance of discodermolide as a therapeutic agent, there is a continuing need to develop procedures for synthesizing this new drug in large quantities which are suitable for widespread pharmaceutical use.
SUMMARY OF THE INVENTION In accordance with the present invention, a method is provided for synthesizing discodermolide. The invention is based on a highly convergent strategy for synthesizing discodermolide which involves disconnecting the carbon backbone of discodermolide at the C-7 to C-8 allylic bond and the C-15 to C-16 allylic bond to provide three subunits which can be synthesized and combined together to form discodermolide. The present invention is particularly well suited for synthesizing relatively large quantities of discodermolide for use as a pharmaceutical or investigative agent. The method may be used to make both the plus and minus enantiomers of discodermolide as well as numerous analogs thereof.
As a feature of the present invention, a first precursor having the formula:
is provided for use in synthesizing discodermolide. In addition, a second precursor having the formula:
is provided in accordance with the present invention for use in preparing discodermolide.
A third precursor having the formula:
is further provided which is combined with the first and second precursors to form discodermolide.
As an additional feature of the present invention, the first and second precursors are initially combined to form an intermediate compound having the formula:
This intermediate compound is then combined with the third precursor to form discodermolide. The highly convergent synthesis of discodermolide in accordance with the present invention takes advantage of a chelation-controlled alkylation reaction to achieve high selection in the key bond coupling reaction. The method and precursors (including analogs) provide an efficient procedure for making discodermolide and structural analogs thereof. The above described features and other advantages of the present invention will become better understood by reference to the following detailed description when taken in conjunction with the accompanying drawings.
BRIEF DESCRIPTION OF THE DRAWINGS FIG. 1 is a diagrammatic representation showing discodermolide and the location at which the carbon backbone is disconnected (C-7 to C-8 allylic bond, C- 15 to C-16 allylic bond and C-21 to C-22 alkyl bond) to thus divide the compound into three key subunit structures which form the basis for synthesis of the complete discodermolide compound. The three key subunits are referenced in FIG. 1 as compounds 2, 3 and 4 or, alternatively, subparts
A, B and C, respectively.
FIG. 2 is a schematic representation of the synthesis of a first precursor (compound 9) which corresponds to subunit B of the discodermolide compound.
FIG. 3 is a diagrammatic representation of a synthesis protocol for making an intermediate compound (compound 14) which corresponds to a
combination of subparts B and C of discodermolide as represented in FIG. 1. Also, FIG. 3 shows the synthesis of a second precursor (compound 12) which corresponds to subunit C of the discodermolide compound as shown in FIG. 1.
FIG. 4 is a diagram of the synthesis of a third precursor for use in making discodermolide which corresponds to subunit A of the total discodermolide compound.
FIG. 5 is a diagrammatic representation of the overall synthesis of discodermolide utilizing intermediate compound 14 (combination of precursors 1 and 3) with the third precursor (compound 17).
FIG. 6 depicts exemplary analogs of (-) or (+)-Discodermolide which may be made in accordance with the present invention.
FIG. 7 depicts additional exemplary analogs of (-) or (+)-Discodermolide which may be made in accordance with the present invention.
FIG. 8 depicts exemplary analogs of (-) or (+)-Discodermolide which may be made in accordance with the present invention.
DETAILED DESCRIPTION OF THE INVENTION In accordance with the present invention, a method is provided for synthesizing the immunosuppressive agent discodermolide. The natural form of discodermolide which is isolated from the sponge discodermia dissoluta is
(+)-discodermolide. The term "discodermolide," when used in this specification is intended to cover both (+) -discodermolide and (-)-discodermolide. The method of the present invention is applicable to synthesizing discodermolide (i.e., both the positive and negative enantiomers) . In addition, the present invention may be utilized to synthesize analogs of discodermolide. FIGS. 6-8 set forth exemplary analogs of discodermolide which may be made in accordance with the present invention. The following detailed description will be limited to demonstrating the synthesis of (-)-discodermolide, with it being understood by those skilled in the art that the method and precursor compounds disclosed herein may be modified and adapted using known procedures to prepare (+)-discodermolide and analogs thereof as exemplified in FIGS. 6-8.
Referring to FIG. 1, (-) -discodermolide is shown as compound 1. The carbon backbone of discodermolide is disconnected to form three target synthetic subunits (2, 3 and 4) which are alternatively referred to as subunits "A," "B" and "C," respectively. The locations where the discodermolide backbone is disconnected are shown by ' w-" Discodermolide has a carbon backbone which includes 24 carbon atoms ranging from C-1 to C-24 as best shown in FIG. 1. In accordance with the present invention, the synthesis strategy involves making three precursors which correspond to the three subunits which result from disconnecting the carbon backbone at the C-7 to C-8 allylic bond and the C-15 to C-16 allylic bond. The C-22 and C-23 backbone positions for discodermolide are added during the synthesis procedure. Accordingly, the three subunits A, B and C correspond to the C-1 to C-7, C-8 to C-15 and C-16 to C-21 locations on the backbone, respectively. The basic synthetic process can be viewed as either a combination of the three precursors (9, 12 and 17) to form the final product or a combination of the first two precursors (9, 12) to form an intermediate product (14) which is then combined with the third precursor (17) to form discodermolides and analogs thereof.
The key coupling reaction (C-15 to C-16) occurs by way of a distereo- selective alkylation reaction between the anion of ethyl ketone 4 and iodine 3.
A metal-promoted coupling of a C-8 Z-vinyl iodide with a C-7 aldehyde completes the carbon backbone to form the discodermolide product. The steps in the method of the present invention are set forth in FIGS. 2-5. The synthetic procedure is also disclosed in reference 19. An exemplary synthesis of the first precursor in accordance with the present invention is shown in
FIG. 2. The first precursor (compound 9) is prepared utilizing dihydro-4- pyrone 7 which is obtained via the diene-aldehyde cyclocondensation reaction of diene 6 and aldehyde 5 (Ref. 10). Dihydropyrone 7, obtained in greater than 95% ee (Ref. 11) from homochiral 5, serves as the template for the establishment of the required Z-trisubstituted alkene of discodermolide.
Reduction of the carbonyl of pyrone 7 is achieved using sodium borohydride in the presence of cerium trichloride to afford the corresponding alcohol as a mixture of diastereomers. Sequential treatment of this material with aqueous p-toluenesulfonic acid followed by lithium borohydride leads to reductive opening of the resultant hemiacetal to afford the desired Z- allylic alcohol 8 in
62% yield from pyrone 7. Protecting group manipulation and conversion of the allylic alcohol to its corresponding iodide (PhO)3P/MeI, DMF) (Ref. 1 1) produces compound 9 (first precursor) in good yield.
Referring to FIG. 2, BnO is benzyloxy, TMS is trimethylsilyl, PvCl is pivaloly chloride and TIPS is triisopropylsilyl.
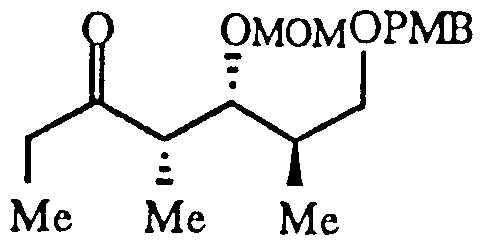
The second precursor is an ethyl ketone having the formula:
The second precursor corresponds to subpart "C" and was prepared directly by aldol condensation of the lithium anion of 3-pentanone and i?-3-benzyloxy- 2-methylpropionaldehyde, followed by protection of the resulting alcohol as the methoxymethyl ether (see FIG. 3) (Ref. 13). The methoxymethyl (MOM) protecting group for the alcohol β to the ketone promotes the desired chelation of the metal counter ion of the enolate. In addition, this protecting group facilitates introduction of the C-19 carbamate function at a late stage in the synthesis. The second precursor can also be made by diastereoselective aldol condensation of aldehyde 11 and oxazolidionone 15 followed by protection of the resulting alcohol as its methoxymethyl ether and homologation to produce compound 12.
Treatment of the second precursor 12 with lithium diisopropylamide and tetramethylethylenediamine in THF at -78°C followed by addition of the first precursor 9 leads to smooth alkylation. The diastereo selectivity of the chelation controlled alkylation shows a remarkable solvent dependence. For the coupling of the first precursor 9 and second precursor 12, the diastereo- selectivity is preferably optimized to a level of 6: 1 in the mixed solvent system 45:55 hexanes:THF, favoring the desired diastereomer. Referring to FIG. 3, chelation controlled reduction of ketone 13 with lithium aluminum hydride (LAH) and lithium iodide in ether at -100°C establishes the C-17 alcohol stereochemistry with excellent diastereo selectivity, and facilitates separation of all minor diastereomers from the major product. Silylation of the secondary alcohol produces an intermediate product having the formula 14. This intermediate product is then combined with the third precursor to form discodermolide as described below. As shown in FIG. 4, the synthesis of the third precursor corresponding to the C- 1 to C-7 fragment of discodermolide is achieved starting from aldehyde 11 using the chemistry of allylic boranes developed by Brown and coworkers (Ref. 13). Thus, treatment of 11 with the E-crotyl borane reagent,
followed by oxidative work up and silylation produces the crotyl adduct with excellent diastereoselectivity. It is necessary to install C-5 stereochemistry via a second addition of an allylic borane to a C-5 aldehyde. A systematic study of a variety of C- 1 functionality revealed that the diastereoselectivity of allyl boronation at C-5 was highly dependent on the nature of this functionality.
The highest diastereoselectivity (>8: 1) for the allylboration was observed for the C-1 methyl ester. However, following allylboration, the adduct spontaneously lactonized. Upon ozonolysis of the alkenelactone, epimerization of C-5 was observed. It was found that the C-1 pivalate afforded the desired adduct in 5: 1 selectivity. In contrast, the C-1 benzyl afforded only cα. 2: 1 selectively.
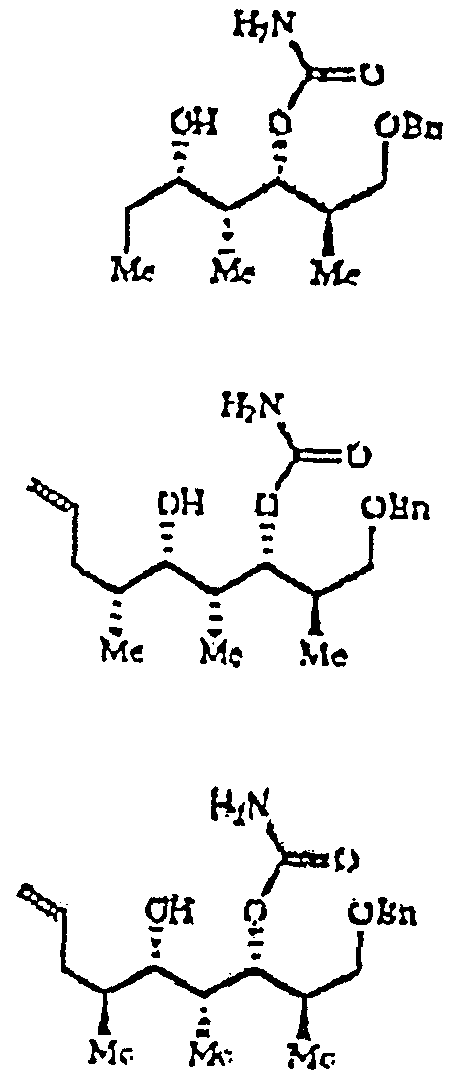
Thus it is necessary, at a minimum, to exchange the benzyl protecting group at C- 1 for a pivaloyl ester. Reductive cleavage of the benzyl ether followed by pivaloylation and ozonolysis gives 16. Treatment of aldehyde 16 with allyl-bis- isopinocamphylborane, followed by oxidative work up, provides the homo- allylic alcohol containing the required C-5 stereochemistry. Protection and ozonolysis gives the third precursor 17. Third precursor 17 is an aldehyde which corresponds to subpart A and sets the stage for the coupling of the C-7 to C-8 bond to assemble the complete chiral array of discodermolide.
After extensive investigation into alternative strategies, the Nozaki-Kishi coupling (Ref. 14) of a C-8 Z- vinyl iodide to the third precursor 17 emerged as the preferred synthetic route (see FIG. 5). Selective reductive deben-jylation at C-9 was achieved using Raney nickel and hydrogen in ethanol. Reductive cleavage of the C-21 PMB ether was minimized under these conditions. No reduction of the trisubstituted double bond was observed. Oxidation of the C-9 alcohol was achieved using tetrapropylamonium perruthenate (TPAP) in acetonitrile (Ref. 15) to afford the expected aldehyde. The vinyl iodide moiety was prepared from this material by treatment with iodomethlenetriphenyl- phosphorane (Ref. 16). Oxidative cleavage of the C-21 PMB ether using dichlorodicyanoquinone (DDQ) proceeds smoothly to afford alcohol 18. After TPAP oxidation, the Z-diene is prepared following the conditions of Roush (Ref.
17) to afford the desired alkene in >20:1 Z:E selectivity.
Model experiments have shown that catecholboron chloride is the reagent of choice for the removal of the methoxymethyl ether. This reagent proved equally effective in the fully elaborated system, affording the desired alcohol in ca. 60%. A two-step protocol furnishes carbamate 19 in excellent yield (Ref. 18). The transition metal promoted addition of the vinyl iodide 19 to aldehyde 17 proceeds to afford the C-1 to C-24 fragment of discodermolide in ca. 50% yield. The balance of the material recovered was starting materials.
The diastereoselectivity of this coupling process is ca. 2.5: 1. As with diastereoselective alkylation, it is not necessary to prove the stereochemistry of the major isomer at this point. This sequence completes the installation of all stereocenters and carbon atoms required for the synthesis of discoder- molide. Hydrolysis of the silyl ethers and simultaneous lactonization is all that is required to produce discodermolide. Indeed, when the C-1 to C-24 chiral array (as a 2.5:1 mixture at C-7) is subjected to HF under a variety of conditions, the major product co-eluted on TLC (10% MeOH in dichloro- methane) is discodermolide. Optimized conditions (HF/pyridine in aceto- nitrile) afforded (-) -discodermolide in ca. 60% yield after 36 hours. The material obtained from this sequence was identical in all respects (XH NMR, 1 C NMR, HRMS, TLC) to its enantiomer, (+)-discodermolide (Ref. 19).
The above described total synthesis method is highly convergent and can be easily modified using known synthetic procedures to synthesize analogs of discodermolide of the type shown in FIGS. 6-8.
The following specific procedures were followed in conducting the above-described synthetic procedure. As will be recognized by those skilled in the art, the specific procedures may be modified, if desired, provided that the overall synthesis procedure set forth above is followed.
General Experimental: Air- and moisture-sensitive reactions were carried out under argon atmosphere. When necessary, solvents and reagents were purified and dried before use by standard methods. Routine monitoring of reactions was performed using precoated silica gel TLC plates (Baker Si250F-254). Spots were visualized by UV and/ or dipping the TLC plate into a vanillin solution and heating with a hot plate. The normal processing of organic extracts consisted of washing the extract with saturated NaCl solution, drying over MgS04 or Na2S0 , filtration, and concentration with a rotary evaporator. Flash chromatography was carried out on silica gel 60 (230-400 mesh Scienti- fie Adsorbents) according to the method of Still. Preparative TLC was performed using precoated TLC plates (silica gel F-254, 0.5 mm, Baker). Thin layer chromatography with Chromatron was carried out on silica gel plates of various thicknesses (silica gel 60, PF254, containing gypsum).
Synthesis of Intermediate Compound 14: To a flame dried 250 mL round bottom flask equipped with a magnetic stir bar and rubber septum was placed lithium hexamethyldisilazide (solid, 97% Aldrich, 820 mg, 4.68 mmol) inside a N2 atmosphere dry box. The round bottom flask was sealed and removed from
dry box before adding THF (5 mL) and hexane (5 mL). The reaction flask was placed on -78°C bath before adding dry (2 x 3 mL benzene azeotrope) ketone 12 (second precursor) (1267 mg, 3.75 mmol) in THF (4 mL, followed by 2 x 1 mL THF rinses) dropwise over 15 minutes. The reaction mixture was stirred at -78°C for 45 minutes before adding tetramethylethyllenediamine (0.850 mL,
5.63 mmol) in THF (3 mL) dropwise over 5 minutes. The resulting reaction mixture was allowed to stir for 30 minutes at -78°C before adding dry (3 x 3 mL benzene azeotrope) allyl iodide 9 (first precursor) (1020 mg, 1.875 mmol) in hexane (5 mL, followed by 2 x 1 mL hexane rinses and l x l mL THF rinse) dropwise. The reaction mixture was stirred in the dark at -78°C for 49 hours and then quenched by addition of aqueous NaHC03 (1M, 50 mL). The resulting mixture was extracted with diethyl ether (4 x 50 mL). The combined organic layers were washed with sat. NaCl and dried with MgS0 . The dried organic was filtered through a short plug of silica gel and concentrated in vacuo providing a colorless, viscous oil. The crude material was purified by column chromatography (20 cm silica gel on a 3 cm diameter column, 7% EA in hexane) providing clean intermediate compound 14 (980 mg, 1.30 mmol) in 70% yield as ca. 6: 1 ratio of epimers at C-16. Rf = 0.5 20% EA in hexane.
Synthesis of Compound 18: To a stirred solution of C-16 isomers 14 (300 mg,
0.398 mmol) in dry diethyl ether (5 ml) at 0°C, was added Lil (532 mg, 3.98 mmol) in diethyl ether (5 mL). The solution was stirred until the Lil was completely dissolved. The mixture was then cooled to -78°C for 10 minutes. A fresh solution of LAH in THF (1.0M, 4 mL, centrifuged) was added dropwise. The stirring was continued at -78°C for 30 minutes, and then sodium potassium tartrate solution (IN, 30 mL) was added. The mixture was stirred at room temperature for 30 minutes, and the phases were separated. The aqueous layer was extracted with EtOAc (4 x 30 mL). The combined organic extracts were then washed with sat. NaCl (100 mL), dried (Na2S0 ), filtered, and concentrated. The material was purified by column chromatography (20 cm silica gel on 3 cm diameter column, 8% EtOAc in hexanes). Concentration in vacuo of the appropriate fractions provided a chelation controlled alcohol (240 mg) in 80% yield as pale yellow oil. Additionally, the non-chelation isomer at C- 17 was isolated (30 mg) in 10% yield. To a stirred solution of the yellow oil (2050 mg, 2.71 mmol) in CH2C12
(17 mL), was added excess triethyl amine (0.84 mL, 6.0 mmol), Tips-OTf (1.0 mL, 4.0 mmol), and catalytic 4-dimethylamino pyridine (ca. 20 mg). The stirring was continued at room temperature for 48 hours, at which time
NaHC03 (1 M, 20 mL) was added. The phases were separated and the aqueous layer was extracted with CH2C1 (5 x 20 mL). The combined organic extracts were then washed with sat. NaCl (200 mL), dried with Na2S0 filtered through plug silica gel and concentrated in vacuo to provide crude residue (2.95 g) as a viscous oil. The resulting residue was used in the following procedure.
To the solution containing the residue from the preceding procedure in absolute EtOH (20 mL) was added 2-day old W - 2 Raney Ni (400 mg). The mixture was stirred at room temperature under 1 atmosphere of H2 for 24 hours. The Raney Ni was then removed by filtration. The filtrate was concentrated in vacuo. Flash chromatography (10% EA in hexanes) of the residue gave a clear oil (1.59 g, 1.93 mmol) in 75% yield from the yellow oil.
Dry benzene (3 mL) was added to the clear oil (540 mg, 0.657 mmol) in a 50 ml round bottom flask equipped with a magnetic stir bar. To remove water azetropically, the solution was concentrated in vacuo. To the dried alcohol were added CH2CI2 (4.5 mL), acetonitrile (0.7 mL) and 4A molecular sieves (powdered, 330 mg). After allowing the solution to stir for 5 minutes, anhydrous 4-methyl morpholine n-oxide (130 mg, 1.11 mmol) was added in one portion, followed by tetrapropylammonium perruthenate (10 mg, 28 μmmol), causing an instant color change from colorless to green. The oxidation reaction was stirred for 2 hours, during which time the color changed from green to black. At that time, the reaction was judged to be complete by TLC (10% EtOAC in hexanes). The crude mixture was then concentrated in vacuo. The crude residue was purified by column chromatography (4 cm silica gel on a 3 cm diameter column, elute CH2CI2). The eluate was concentrated in vacuo providing clean aldehyde (475 mg) which was dried azeotropically with benzene (1 x 3 mL) in vacuo and then dissolved in THF (10 mL). This solution was immediately used in the following procedure.
To a flame dried, single neck 500 ml round bottom flask equipped with a magnetic stir bar and rubber septum were added solid, white idodmethyl- enetriphenylphosphonium iodide (930 mg, 1.75 mmol) and THF (20 mL) to give a white suspension. The suspension was placed in a -20°C bath for 10 minutes before adding dropwise a sodium hexamethyldisilazide (solid, 334 mg, 1.85 mmol) solution in THF (10 mL) transforming the white suspension to a bright yellow solution. Upon completion of the addition of sodium hexamethyldisilazide, the reaction mixture was allowed to stir at -20°C for an additional 5 minutes. The reaction mixture was then placed in a -78°C bath for 3 minutes before adding the above freshly prepared THF (10 mL) solution
of aldehyde via cannula. Care was taken to cool the aldehyde in THF solution by passing it down the side of the reaction flask during the addition. The reaction mixture was stirred for 10 minutes and then quenched by pouring into aqueous NH C1 (1M, 50 mL). The resulting mixture was extracted with EA (3 x 50 mL) and CH2CI2 (1 x 20 mL). The combined organic layers were washed with sat. NaCl (1 x 200 mL) and dried with anhydrous Na2S0 . The dried organic layers were filtered through a short plug of silica gel and concentrated in vacuo to provide the crude product (1.75 g). The crude residue was purified by column chromatography (20 cm silica gel on a 3 cm diameter column, 5% EtOAc in hexane) providing clean vinyl iodide as a colorless foamy oil (500 mg, 0.529 mmol) in 80% from the clean oil.
Final synthesis of compound 18 (see FIG. 5) was accomplished by adding CH2C12 (10 mL) and H2O (0.5 mL) to a 50 ml round bottom flask containing the colorless foamy oil (540 mg, 0.571 mmol) and a stir bar was added. The solution was allowed to stir for 5 minutes before adding DDQ (190 mg, 0.837 mmol) in one portion, causing an instant color change from colorless to green. The reaction mixture was allowed to stir at room temperature for 45 minutes before being quenched by pouring into aqueous NaHC03 (1M, 100 mL). Upon quenching the reaction, the aqueous layer became burgundy in color. The layers were partitioned, and the aqueous layer was extracted with CH2CI2 (5 x 30 mL). The combined organic layers were washed with sat. NaCl (1 x 200 mL) and dried with anhydrous Na2S04. The organic layer was filtered and concentrated in vacuo to provide crude product 18 (495 mg) as a colorless oil. This material was purified by column chromatography (20 cm silica gel on a 3 cm diameter column, 5% EA in hexanes, gradient to 10% EA in hexanes) providing clean alcohol 18 (412 mg, 0.500 mmol) in 87% yield.
Synthesis of Compound 19: To alcohol 18 (190 mg, 0.230 mmol) in a 50 mL round bottom flask equipped with a magnetic stir bar was added CH2CI2 (4 mL), acetonitrile (0.4 mL) and 4A molecular sieves (powdered, 120 mg). After allowing the solution to stir for 5 minutes, 4-methylmorpholine n-oxide (40 mg, 0.345 mmol) was added in one portion, followed by tetrapropylammonium perruthernate (5 mg, 1.42 μmol) which causes an instant color change from colorless to green. The oxidation reaction was allowed to stir for 40 minutes and then judged to be complete (TLC, 10% EtOAc in hexanes) over the 40 minutes of reaction time the color turned from green to black. After 1 hour, the reaction mixture was concentrated in vacuo. The crude residue was purified by column chromatography (5 cm silica gel on a 2 cm diameter column,
elute 5% EA in CH2CI2, 100 mL). The eluate was concentrated in vacuo providing clean aldehyde (170 mg), which was immediately used in the following procedure.
To the above freshly prepared aldehyde in a 100 ml round bottom flask was added toluene (3 mL), a stir bar and 4A molecular sieves (powdered, 100 mg). The reaction mixture was placed in a 0°C bath and allowed to stir for 5 minutes before adding (E)-γ-(trimethylsilyl) allylboronate (2 mL of a crude ca. 1M solution in toluene) dropwise over 2 minutes. The reaction mixture was allowed to stir for 1 hour and then judged to be complete by TLC (5% EA in hexane). At that time, the reaction mixture was purified by column chromatography (20 cm silica gel on a 3 cm diameter column, 5% EA in hexane) by loading reaction mixture directly to the column. The eluate was concentrated in vacuo in a 100 mL round bottom flask providing the expected allylboration adduct, which was immediately used in the following procedure. To the alloboration adduct in a 100 mL round bottom flask equipped with a magnetic stir bar was added THF (10 mL). The reaction mixture was placed in a 0°C bath and stirred for 10 minutes before adding KH (ca. 100 mg, from 20-25 wt. % dispersion in mineral oil, rinsed and decanted 3 x 50 mL hexanes). After 10 minutes the reaction was quenched by the addition of aqueous NaHC03 (1M, 20 mL). The resulting mixture was extracted with diethyl ether (4 x 20 mL). The combined organic layers were washed with sat. NaCl (1 x 50 mL) and dried with anhydrous Na2S0 . The dried organic layers were filtered and concentrated in vacuo, furnishing the crude product (172 mg) as a colorless viscous oil. The crude product was further purified by column chromatography (20 cm silica gel on a 3 cm diameter column, 3%
EtOAc in hexanes). The eluate was concentrated in vacuo providing clean terminal diene (141 mg, 0.161 mmol) in 70% yield from alcohol 18.
In a flame dried 50 mL round bottom flask equipped with a magnetic stir bar and rubber septum were placed the clean terminal diene (58 mg, 0.066 mmol) and CH2CI2 (1.0 mL). The resulting solution was placed in a 0°C bath. In a separate flame dried 100 mL round bottom flask were placed catecholboron chloride/0.5 eq. H2O that was clear and colorless. This catecholboron chloride solution (0.5 mL) was added dropwise to the clean terminal diene over 3 minutes at 0°C. After 30 minutes of reaction time, TLC analysis showed no reaction occurred and more catecholboron chloride 0.5 eq.
H2O in CH2CI2 solution was added (0.8 mL) and the reaction mixture was allowed to warm to room temperature. The reaction was monitored by TLC and periodically (every 12 hours) more reagent (1 mL) was added. The
reaction was allowed to stir for a total of 48 hours at room temperature and then quenched by pouring into aqueous NaHC03 (1M, 20 mL). The layers were separated and the aqueous layer was extracted with CH2CI2 (5 x 10 mL). The combined organic layers were washed with sat. NaCl and dried with Na2S04. The dried organic layers were filtered and concentrated in vacuo providing crude residue (61 mg). The crude residue was purified by column chromatography (18 cm silica gel on a 2 cm diameter column, 3% EtOAc in hexanes) . The eluate was concentrated in vacuo providing a clean alcohol (28 mg, 0.0336 mmol) in a 51% yield. Rf = 0.2 5% EtOAc in hexanes. The final synthesis of the carbonate 19 was accomplished as follows.
In a flame dried 50 ml round bottom flask equipped with a magnetic stir bar and rubber septum was placed the clean alcohol isolated in the previous step (78 mg, 0.094 mmol) which was then azeotropically dried with benzene (2 x 1 mL). CH2CI2 (4 mL) was added to the dried alcohol, followed by dropwise addition of trichloroacetyl isocyanate (100 μL, 0.937 mmol) over 1 minute.
The reaction was allowed to stir at room temperature for 30 minutes and then directly placed on a short pad of AI2O3 (neutral, activity II) that was pre-wetted with a 1 : 1 benzene:CH2Cl2 solution. The reaction mixture was allowed to soak on the column of AI2O3 for 30 minutes before eluting with 1 :1 benzene:CH2Cl2 solution (60 mL). Concentration of the eluate provided crude residue (88 mg) which was purified by column chromatography (18 cm silica gel on 2 cm diameter column, 8% EtOAc in hexanes) . Concentration of the eluate in vacuo provided carbamate 19 (62 mg) as a white foamy solid in 78% yield.
Discodermolide was synthesized as follows: Aldehyde 17 (19 mg, 0.038 mmol) and the vinyl iodide 19 (36 mg, 0.043 mmol) were added to a flame dried 100 mL pear bottom flask equipped with a magnetic stir bar and a teflon stopcock vacuum adapter was added. The mixture was azeotropically dried with benzene (4 x 0.7 mL) and then placed under high vacuum (2 mm Hg) for 30 minutes. At this time the flask was flushed with argon and evacuated for 2 minutes. This process was repeated several times before ultimately leaving the reaction flask under vacuum while transporting it into an N22 atmosphere dry box. To the reaction flask was then added dry DMSO (ca. 0.5 mL) followed by 1% NiCb/CrCh (ca. 35 mg). After a total of 100 hours the reaction was quenched by removal from dry box followed by addition of aqueous NH C1 (1M, 15 mL) and EtOAc (15 mL). The resulting mixture was stirred for 1 hour before separating layers. The aqueous layer was extracted with EtOAc (5 x 15 mL) . The combined organic layers were washed with sat. NaCl and dried with Na2S0 . The dried organic layer was filtered and concentrated in vacuo
providing crude residue (60 mg) which was purified by column chromatography (18 cm of silica gel on 2 cm diameter column, gradient 6% EtOAc up to 10% EtOAc in hexanes). Concentration of the appropriate fractions in vacuo resulted in isolation of recovered starting material 19 (11.1 mg, 0.013 mmol) in 31% yield and clean product ester (19 mg, 0.015 mmol) in a 40% yield as an approximately 2.5 to 1 ratio of epimers at C-7 Rf = 0.25 (10% EtOAc in hexanes) as a white, foamy solid. This material was routinely used in its impure form.
To a clean, dry high density polyethylene vial equipped with a stir bar was added the ester product of the previous step (15 mg, 12 μmol) as a solution in CH2CI2. The CH2CI2 was evaporated using a stream of air followed by pumping vial under high vacuum (2 mm Hg) for 5 minutes. To the resulting white, foamy solid was added 10% aqueous HF in acetonitrile (1.5 mL). The reaction was allowed to stir at room temperature for 18 hours and then quenched by pouring into aqueous NaHC03 (sat. 15 mL). An additional amount of solid NaHC03 (ca. 50 to 100 mg) was added to separatory funnel. The aqueous layer was extracted with EtOAc (7 x 10 mL). The combined organic layers were washed with sat. NaCl (1 x 30 mL) an dried with Na2S0 . Filtration and concentration of the dried organic layer in vacuo provided a crude yellowish solid residue (10 mg) that was used directly in the next procedure.
The crude yellowish solid residue from above was dissolved in 10% MeOH in CH2CI2 and transferred to a high density polyethylene vial equipped with a magnetic stir bar. The CH2CI2 was evaporated using a stream of air and then the vial was placed under high vacuum (2 mm Hg) for 5 minutes.
The reaction vial and solid was azeotropically dried with benzene (1 x 0.1 mL) and then THF (0.3 mL) was added before placing vial in 0°C bath. To the cooled reaction vial was added 0.2 mL of a pre-mixed solution of 1 : 1 THF to HF-Pyr (70% anhydrous HF to 30% anhydrous pyridine). The reaction mixture was allowed to stir at 0°C for 30 minutes before an additional portion
(0.2 mL) of 1: 1 70% HF-Pyr/THF solution was added. The reaction mixture was allowed to stir at room temperature for 14 hours before another portion (0.1 mL) of 1 : 1 70% HF-Pyr/THF was added. The reaction mixture was stirred a total of 18 hours and then quenched by pouring into sat. NaHC03 (10 mL). Additional solid NaHC03 was added to the aqueous layer until gas evolution stopped. The resulting mixture was extracted with EtOAc (6 x 10 mL). The combined organic layers were washed with sat. NaCl. The sat. aqueous NaCl was back-extracted with EtOAc (2 x 15 mL). The combined organic layers
were dried with Na2S0 . Filtration and concentration in vacuo provided crude brown, oily residue (14 mg). The crude residue was purified by chromatography (Pasteur pipet, 5% MeOH in CH2CI2, 25 drop fractions) resulting in the isolation of (-) -discodermolide 1 (2.8 mg, 4.7 μmol) in 40% yield from the ester product of the previous step as a white amorphous solid.
Rf = 0.25 10% MeOH in CH2C12. In addition C-7-epi-discodermolide (1.5 mg 2.5 μmol) was isolated in 20% yield.
Having thus described exemplary embodiments of the present invention, it should be noted by those skilled in the art that the within disclosures are exemplary only and that various other alternatives, adaptations, and modifications may be made within the scope of the present invention. Accordingly, the present invention is not limited to the specific embodiments as illustrated herein, but is only limited by the following claims.
BIBLIOGRAPHY
1. Schiff, P.B., Frant, J., Horwitz, S.B., "Promotion of microtubule assembly in vitro, by taxol," Nature, 1979, 665.
2. ter Harr, E., Kowalski, R.J., Hamel, E., Lin, CM., Longley, R.E., Gunasekera, S.P., Rosenkranz, H.S., and Day, B.W., "Discodermolide, a cytotoxic marine agent that stabilizes microtubules more potently than Taxol," Biochemistry, 1996, 35(l):243-250 and references therein.
3. Rowinsky, E.K., Eisenhauer, E.A., Chaudhry, V., Arbuck, S.G., Donehower, R.C., "Clinical toxicities encountered with paclitaxel (TAXOL)," Semin. Oncol, 1993, 20, 1.
4. Yang, G., Myles, D.C., "An Alkylative Strategy to the C-13 to C-21 Sector of Discodermolide," Tetrahedron Lett., 1994, 35, 1313.
5. Yang, G., Myles, D.C., "The Synthesis of the C-9 to C-21 Sector of Discodermolide: An Efficient Route to the C-13 C-14 Z-trisubstituted Alkene," Tetrahedron Lett, 1994, 35, 2503.
6. a) Paterson, I., Wren, S.P., "Studies Towards the Total Synthesis of the Marine-Derived Immunosuppressant Discodermolide: Asymmetric Synthesis of a C1-C8 Delta- Lactone Unit," J. Chem. Soc. Chem. Commun., 1993, 1790. b) Clark, D.L., Heathcock, C.H., "Studies on the Alkylation of Chiral Enolates-Application Toward the Total Synthesis of Discodermolide," J. Org. Chem., 1993, 58, 5878. c) Golec, J.M.C., Jones, S.D., "The Synthesis of a C-l-C-8 Lactone Fragment of Discodermolide," Tetrahedron Lett, 1993, 34, 8159. d) Evans, P.L., Golec, J.M.C., Gillespie, R.J., "The Synthesis of a C-9-C-17 Fragment of Discodermolide," Tetrahedron Lett, 1993, 34, 8163. e) Golec, J.M.C., Gillespie, R.J., "An Approach to the Synthesis of a C-9-C-15 Fragment of Discodermolide, " Tetrahedron Lett, 1993, 34, 8167. f) Paterson, I., Schlapbach, A., "Studies Towards the Total Synthesis of the Marine- Derived Immunosuppressant Discodermolide," Synlet, 1995, 5, 498.
7. Nerenberg, J.B., Hung, D.T., Somers, P.K., Schreiber, S.L., "The Total Synthesis of the Immunosuppressive Agent Discodermolide," J. Am. Chem. Soc, 1993, 115, 12621.
8. Smith, A.B., Qui, Y., Jones, D.R., Kobayashi, K., "The Total Synthesis of Discodermolide," J. Am. Chem. Soc, 1995, 117, 12011.
9. Danishefsky, S.J., Larson, E., Askin, D., Kato, N., "On the Scope, Mechanism, and Stereochemistry of the Lewis-Acid Catalyzed Cyclocondensation of Activated Dene with Aldehydes: An Application to the Erythronolide Problem," J. Am. Chem. Soc, 1985, 107, 1246.
10. The enantiomeric excess of 11 and 12 were determined by derivitizing alcolic analogs of these materials using the method of Mosher. See Dale, J.D., Mosher, H.S.J., J. Am. Chem. Soc, 1973, 95, 512.
1 1. Gemal, A.L., Luche, J.L., "Lanthanoids in Organic Synthesis. 6. The Reduction of alpha-Eneones by Sodium Borohydride in the Presence of Lanthanoid Chlorides: Synthetic and Mechanistic Aspects," J. Am. Chem. Soc, 1981, 103, 5454.
12. Masamune, S., Ellingboe, J.W., Choy, W., "Aldol Strategy: Coordination of the Lithium Cation with an Alkoxy Substituent," J. Am. Chem. Soc, 1982, 104, 5526.
13. Brown, H.C., Bhat, K.S., Randad, R.S., "Chiral Synthesis via Organo- boranes. 21. Ally- and Crotylboration of alpha-Chiral Aldehydes with Diisopinocamphenylboron as the Chiral Auxiliary," J. Org. Chem., 1989, 54, 1570 and references therein.
14. Jin, H., Uenishi, J.-I, Christ, W.J., Kishi, Y., "Effects of Nickel(II) Chloride and Palladium(II) Acetate on Chromium(II)-Mediated Coupling Reaction of IodoOlefins with Aldehydes," J. Am. Chem. Soc, 1986, 5644. b) Takai, K., Tagashira, M., Kuroda, T., Oshima, K., Utimoto, Nozaki, H., "Reactions of Alkenylchromium Reagents Prepared from Alkenyl Trifluoromethanesulfonates with Chromium(II) Chloride under Cickel Catalysis," J. Am. Chem. Soc, 1986, 108, 6048.
15. Ley, S.V., Norman, J., Griffith, W.P., Marsden, S.P., "Tetrapropyl- amonium Perruthenate, TPAP: A Catalytic Oxidant for Organic Synthesis," Tetrahedron, 1994, 639.
16. Stork, G., Zhao, K., "A Stereoselective Synthesis of Z-l-iodo-1 Alkenes," Tetrahedron Lett., 1989, 30, 2173.
17. Roush, W.R., Grover, P.T., "Diiopropyl Tartrate Modified (E)-[(Cyclo- hexyloxy)dimethysilyl]- and (E)-(Dimethylphenylsilyl)allylboronates: Chiral Reagents for the Enantio- and Diastereoselective Synthesis of Anti 1,2-Diols and Butene-l,4-diols via the Formεd alpha- and gamma- hydroxyallylation of Aldehydes," Tetrahedron, 1992, 48, 1981.
18. Kocovsky, P., Tetrahedron Lett, 1986, 27, 5521.
19. Harried, S.S., Yang, G., Strawn, M.A., Myles, D.C., "Total Synthesis of (-)-Discodermolide: An Application of a Chelation- Controlled Alkylation Reaction," J. Org. Chem., 1997, 62, 6098-6099.