WO1996010572A1 - Neue heterocyclische substituierte imidazolo-chinoxalinone, ihre herstellung und verwendung - Google Patents
Neue heterocyclische substituierte imidazolo-chinoxalinone, ihre herstellung und verwendung Download PDFInfo
- Publication number
- WO1996010572A1 WO1996010572A1 PCT/EP1995/003686 EP9503686W WO9610572A1 WO 1996010572 A1 WO1996010572 A1 WO 1996010572A1 EP 9503686 W EP9503686 W EP 9503686W WO 9610572 A1 WO9610572 A1 WO 9610572A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- trifluoromethyl
- phenyl
- nitro
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/26—Psychostimulants, e.g. nicotine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- the present invention relates to new heterocyclically substituted imidazoloquinoxalinones, processes for their preparation and their use for combating diseases.
- excitatory amino acids especially glutamic acid
- This excitatory amino acid acts as a transmitter substance for glutamate receptors, of which various subtypes are known.
- a subtype is e.g. named after the specific agonist N-methyl-D-aspartate NMDA receptor.
- This NMDA receptor has different binding sites for agonists or antagonists.
- the amino acid glycine also binds to the NMDA receptor and modulates the action of the natural agonist glutamic acid. Antagonists at this glycine binding site can then show antagonistic effects on the NMDA receptor and inhibit "overexcitation" of this receptor.
- AMPA 2-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- kainic acid Analogous to the NMDA receptor already mentioned, antagonists of these receptors could also inhibit "overexcitation”.
- a number of neurodegenerative diseases or mental disorders have elevated glutamate levels, which can lead to states of overexcitation or toxic effects in the CNS.
- Glutamate antagonists including in particular NMDA antagonists or their modulators (such as glycine antagonists) and the AMPA antagonists, are therefore suitable for therapeutic use as agents against neurodegenerative diseases (Huntington's disease and Parkinson's disease), neurotoxic disorders Hypoxia, anoxia or ischemia, as they occur after "Stroke”, or as antiepileptics, antidepressants and anxiolytics (cf. Medicinal Research 1990, 40, 511-514; TIPS, 1990, 11, 334-338 and Drugs of the Future 1989, 14 (11), 1059-1071.
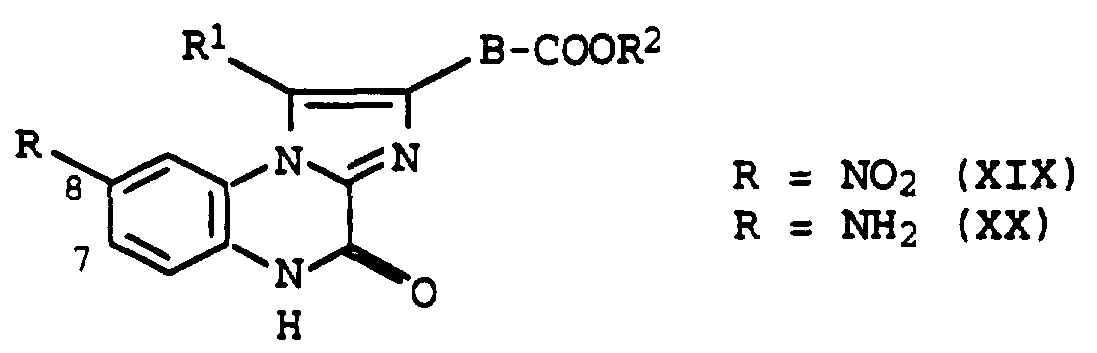
- a number of imidazoloquinoxalinones of the formula II are already known:
- the compounds published as glutamate antagonists in the fused imidazole ring are only characterized by alkyl, trifluoromethyl or phenyl substituents.
- the invention relates to new imidazolo-quinoxalinones of the formula I. wherein
- R 1 is hydrogen, branched or rectilinear C 1-5 alkyl or a phenyl, pyridyl or thienyl group which is optionally substituted by one or two chlorine atoms, a trifluoromethyl, a nitro or methylenedioxy group,
- R 2 is hydrogen, C 1-5 alkyl or C 3-8 dialkylaminoalkyl
- R 3 is a chlorine or bromine atom, a trifluoromethyl, cyano or nitro group
- A is a five-membered heterocycle, optionally substituted by R 4 and R 5, with 1-4 nitrogen atoms or with 1-2 nitrogen atoms and one oxygen or sulfur atom, each of the radicals R 4 and R 5 , which may be the same or different, being hydrogen, C 1-5 alkyl, C 1-5 hydroxyethyl, phenyl, phenyl substituted by a chlorine atom or a trifluoromethyl or nitro group, -COOH, -COO-C 1-5 alkyl,
- B represents a bond or a C 1-5 alkylene chain
- Preferred compounds of formula I are those in which R 1 represents a methyl, ethyl or phenyl group.
- R 2 preferably represents a methyl or ethyl group or a hydrogen atom. If R 2 is hydrogen, these are
- Acids capable of salt formation with alkali metal and alkaline earth metal hydroxides or organic nitrogen bases.
- salt formation with, for example, sodium hydroxide or tris (hydroxymethyl) methylamine, the acids can, if desired, be converted into a water-soluble form.
- Preferred Substituents for R 3 are electron-withdrawing groups such as the nitro or trifluoromethyl group in position 7.
- Pyrrol and its derivatives are preferably mentioned as 5-ring heterocycles for A.
- Preferred pyrrole derivatives are 3-formylpyrrole, acyl derivatives of 3-aminomethylpyrrole, such as the benzoyl or pyridoyl derivatives or those which have an arylurea group, substitution of the benzoyl group with a nitro or CF 3 group being particularly emphasized.
- 5-ring heterocycles with 3 and 4 N atoms are, for example, 1,2,3-triazole, 1,2, 4-triazole and its derivatives, as well as the tetrazole system.
- B preferably represents a bond.
- the present heterocyclically substituted imidazole quinoxalinones surprisingly show advantages over known imidazole quinoxalinones, in particular higher effectiveness.
- R 1 , R 3 and B have the meaning given above and R 2 represents an alkyl group, with 1,4-dicarbonyl compounds or amber dialdehyde derivatives, or cyclic or acyclic acetals derived therefrom, for example formula IV
- Pyrrolyl compounds V according to the invention can be prepared by using appropriately substituted diketones or acetals of the formula IV.
- substitution R 4 or R 5 can be changed in a suitable manner.
- an aldehyde group can be converted into a hydroxyalkyl group or into an aminoalkyl group by reductive amination.
- the reductive amination is in general at temperatures of 5 to 80 ° C, preferably 10 to 30 ° C, in the presence of reducing agents such as sodium cyanoborohydride or hydrogen in the presence of hydrogenation catalysts such as Pd / carbon, Pt / carbon or Raney-Nikkel, advantageously in polar organic solvents such as alcohols or dimethylformamide.
- An aldehyde can by conventional methods, for. B. in RC Larock, "Comprehensive Organic Transformations", 1989, VCH Publisher, p. 838 f.
- To the carboxylic acid according to the invention preferably the oxidation with potassium permanganate in solvents such as acetone is carried out at temperatures of 25 ° C.
- the starting compounds of the formula VI, in which R 3 has the meaning given above but does not include the nitro group can be prepared analogously to a process according to EP 400 583 with subsequent nitration and reduction of the nitro group according to Scheme 1:
- a double-activated carbonic acid derivative such as phosgene, diphenyl carbonate or, preferably, N, N'-carbonyldiimidazole in an inert aprotic solvent at an elevated temperature of 150-200 ° C.
- the ring is closed to form imidazolo-quinoxalinone X.
- Suitable solvents are decalin, tetralin, 1,2 -Dichlorobenzene or 1,3-dimethylethylene or propylene urea.
- a process for the preparation of the nitro compounds XI is characterized in that compounds X (R 3 as above, but without affecting the nitro group) are nitrated with nitric acid, sulfuric acid, nitric acid or sulfuric acid potassium nitrate at temperatures between -10 ° and 20 °.
- a further process for the preparation of the substances according to the invention is that, as described above, a nitrobenzene derivative XII, which has two interchangeable halogen atoms, is initially admixed with an imidazole derivative VII
- R 1 -R 5 , A and B have the meaning given above.
- Suitable heterocycles of the formula XIII are in particular
- Suitable heterocycles can also contain another hetero atom, such as an oxygen or sulfur atom.
- the process can optionally also be designed such that a corresponding nitrobenzene with two interchangeable halogen atoms and a protected amino group XV which is in the correct position for the final cyclization is first reacted with the desired heterocycle XIII, then with the desired imidazole derivative VII to XVI and after removal of the amino protecting group to XVII, the ring closure is carried out as described above:
- the hydrolysis is preferably carried out under alkaline conditions, for example in the presence of an alkali metal hydroxide or of sodium hydrogen carbonate in a solvent such as water, a lower alcohol, tetrahydrofuran or mixtures thereof.
- a solvent such as water, a lower alcohol, tetrahydrofuran or mixtures thereof.
- the organic acids obtained in this way are optionally converted into a physiologically compatible amine or metal salt.
- Tris (hydroxymethyl) aminomethane are antagonists of the excitatory amino acid glutamate, in particular antagonists of the glycine binding side of the NMDA receptor, the AMPA receptor and the kainate receptor. They are suitable as active pharmaceutical ingredients in human medicine and can be used to produce medicaments for the treatment of neurodegenerative diseases and neutoxic disorders of the central nervous system and for the production of antispasmodics, antiepileptics, anxiolytics and antidepressants.
- the pharmacological activity of the compounds I was investigated on isolated membrane material from rat cerebrum. For this, the membranes were in the presence of the invention
- 3H-2-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid ( 3 H-AMPA), [ 3 H] -glycine and [ 3 H] -cainate, respectively, specifically on AMPA-, NMDA and kainate, respectively Bind receptors.
- 3 H-AMPA 3H-2-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- [ 3 H] -glycine and [ 3 H] -cainate respectively, specifically on AMPA-, NMDA and kainate, respectively Bind receptors.
- the extent of the binding of the radioactive receptor ligands mentioned to the membrane receptors was determined on the basis of the radioactivity which was measured by scintillation counting.
- the concentration-dependent displacement of this bond by the
- the compounds according to the invention could be used to calculate the affinity of the compounds according to the invention for the corresponding receptors.
- the dissociation constant K l (as a measure of the affinity) was determined using an iterative nonlinear regression analysis with the Statistical Analysis System (SAS), similar to the program "Ligand” by PJ Munson and D. Rodbard (Analytical Biochem. 107, 220 (1980) , Ligand: Versatile Computerized Approach for Characterization of Ligand Binding Systems).
- a buffer solution A consisting of 30 mM ⁇ , ⁇ , ⁇ -tris (hydroxymethyl) methylamine hydrochloride (TRIS-HCl) and 0.5 mM ethylenediaminetetraacetic acid (EDTA) - pH 7.4 - homogenized using an Ultra-Turrax ® stirrer.
- the suspension was centrifuged at 48,000 g for 20 min. After separation of the supernatant liquid, the proteinaceous membrane material contained in the sediment was washed three times by suspending it in buffer solution A and then centrifuging at 48,000 g for 20 minutes each. The membrane material was then suspended in a 15-fold volume of buffer solution A and incubated at 37 ° C. for 30 min. The protein material was then washed twice by centrifugation and suspension and frozen at -70 ° C until used.
- the protein material thawed at 37 ° C was centrifuged twice at 48,000 g (20 min) and then suspended in a buffer solution B of 50 mM TRIS-HCl, 0.1 M potassium thiocyanate and 2.5 mM calcium chloride - pH 7 , 1 - washed. Then 0.25 mg of membrane material, 0.1 ⁇ Ci 3 H-AMPA (60 Ci / mmol) and compound I dissolved in 1 ml of buffer solution B and incubated on ice for 60 min. The incubated solution was filtered through a CF / B filter (Whatman), which had previously been treated with a 0.5% aqueous solution of polyethyleneimine for at least 2 hours.
- a buffer solution B 50 mM TRIS-HCl, 0.1 M potassium thiocyanate and 2.5 mM calcium chloride - pH 7 , 1 - washed. Then 0.25 mg of membrane material, 0.1 ⁇ Ci 3 H-AMPA (60 Ci / mmol)
- the membrane residue was then washed with 5 ml of cold buffer solution B in order to separate bound and free 3H-AMPA from one another. After measuring the radioactivity of the bound 3 H-AMPA in the membrane material by scintillation counting, the K 1 value was determined by evaluating the displacement curves by means of regression analysis.
- rat hippocampi were homogenized in a 10-fold volume of preparation buffer (50 mM Tris-HCl, 10 mM EDTA) using a Potter homogenizer. The homogenate was centrifuged at 48,000 xg for 20 min. The supernatant was discarded and the membranes obtained in the pellet were washed twice by resuspending and centrifuging at 48000 xg (20 min each). The resuspended membranes were frozen in liquid nitrogen and thawed again at 37 ° C. After a further washing step, the membrane suspension was incubated for 15 min at 37 ° C. in a shaking water bath.
- preparation buffer 50 mM Tris-HCl, 10 mM EDTA
- the membranes were frozen at -70 ° C. until further use.
- the frozen membranes were thawed at 37 ° C. and washed twice by centrifugation at 48,000 ⁇ g (20 min) and then resuspending in binding buffer (50 mM Tris-HCl pH 7.4; 10 mM MgCl 2 ).
- An incubation mixture contained 0.25 mg protein (membranes), 25 nM 3 H-glycine (16 Ci / mmol) and the substances to be tested in a total of 0.5 ml binding buffer. Non-specific binding was determined by adding 1 mM glycine.
- the frozen membranes were thawed at 37 ° C, suspended in binding buffer (50 mM Tris-HCl pH 7.4) and centrifuged at 48,000 x g for 20 min. The membranes in the pellet were resuspended in binding buffer. An incubation batch contained 0.25 mg protein (membranes),
- the pharmaceutical preparations according to the invention contain a therapeutically effective amount of the compounds I.
- the active ingredients are present in an amount of 0.0001 to 1% by weight, preferably 0.001 to 0.1% by weight.
- the preparations are administered in single doses. 0.1 to 100 mg per kg body weight are given in a single dose.
- the preparations can be in a daily or several doses depending on the type and severity of the diseases.
- the pharmaceutical preparations according to the invention contain, in addition to the active ingredient, the customary carriers and diluents.
- pharmaceutical technical auxiliaries such as ethanol, isopropanol, ethoxylated castor oil, ethoxylated hydrogenated castor oil, polyacrylic acid, polyethylene glycol, polyethylene glycol stearate, ethoxylated fatty alcohols, paraffin oil, petroleum jelly and wool fat can be used.
- z As milk sugar, propylene glycol, ethanol, starch, talc and polyvinyl pyrrolidone.
- Antioxidants such as tocopherol and butylated hydroxyanisole and butylated hydroxytoluene, taste-improving additives, stabilizers, emulsifiers and lubricants can also be present.
- the substances contained in the preparation in addition to the active substance and the substances used in the manufacture of the pharmaceutical preparation are toxicologically safe and compatible with the respective active substance.
- the preparation of the pharmaceutical preparations is carried out in a conventional manner, e.g. B. by mixing the active ingredient with the other conventional carriers and diluents.
- the pharmaceutical preparations can be administered in various modes of administration, such as orally, parenterally, subcutaneously, intraperitoneally and topically.
- forms of preparation such as tablets, emulsions, infusion and injection solutions, pastes, ointments, gels, creams, lotions, powders and sprays are possible.
- Example 1 4,5-Dihydro-1-methyl-8 (pyrrol-1-yl) -7-trifluoromethyl-4-oxo-imidazolo [1,2-a] quinoxaline-2-carboxylic acid ethyl ester a. 1- (2-nitro-4-trifluoromethylphenyl) -4-carbethoxy-5-methylimidazole
- the cooled reaction mixture was mixed with 1000 ml of water, extracted with 250 ml of methylene chloride and the methylene chloride phase was dried with magnesium sulfate. The dried solution was evaporated and the residue was crystallized by trituration with ether.
- Ethyl 4,5-dihydro-1-methyl-4-oxo-imidazolo- [1,2-a] quinoxaline-2-carboxylate was obtained by reacting 2-fluoronitrobenzene with 4 (5) -carbethoxy-5 (4) methylimidazole, followed by Hydrogenation and by subsequent ring closure with N, N'-carbonyldiimidazole.
- the above compound was obtained by hydrolysis of the compound according to Example 27 with lithium hydroxide according to Example 10.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description

















Claims

Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU36506/95A AU3650695A (en) | 1994-09-30 | 1995-09-19 | New heterocyclic substituted imidazoloquinoxalinones, their preparation and their use |
| SI9520108A SI9520108A (en) | 1994-09-30 | 1995-09-19 | New heterocyclic substituted imidazoloquinoxalinones, their preparation and their use |
| EP95934074A EP0783506A1 (de) | 1994-09-30 | 1995-09-19 | Neue heterocyclische substituierte imidazolo-chinoxalinone, ihre herstellung und verwendung |
| KR1019970702087A KR970706289A (ko) | 1994-09-30 | 1995-09-19 | 헤테로고리 치환 신규 이미다졸로퀴녹살리논, 그의 제조 방법 및 용도(New Heterocyclic Substituted Imidazoloquinoxalinones, Their Preparation and Their Use) |
| JP8511334A JPH10513435A (ja) | 1994-09-30 | 1995-09-19 | 複素環で置換された新規イミダゾロキノキサリノン、その製造および使用 |
| US08/809,170 US6121265A (en) | 1994-09-30 | 1995-09-19 | Heterocyclic substituted imidazoloquinoxalinones, their preparation and their use |
| CZ97942A CZ94297A3 (en) | 1994-09-30 | 1995-09-19 | Novel heterocyclic substituted imidazoloquinoxalinones, process of their preparation and use |
| BR9509055A BR9509055A (pt) | 1994-09-30 | 1995-09-19 | Imidazoloquinoxalinona uso da mesma e composiçao farmacêutica |
| BG101328A BG101328A (en) | 1994-09-30 | 1997-03-14 | New derivatives of imidazoloquinaxalon with heterocyclic substituents, their preparation and use |
| NO971424A NO971424L (no) | 1994-09-30 | 1997-03-25 | Nye heterocyklisk substituerte imidazolo-kinoksalinoner, deres fremstilling og anvendelse |
| FI971317A FI971317A0 (fi) | 1994-09-30 | 1997-03-27 | Uusia heterosyklisiä, substituoituja imidatsolokinoksalinoneja, niiden valmistus ja käyttö |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE4434941 | 1994-09-30 | ||
| DEP4434941.6 | 1994-09-30 | ||
| DE19503825.8 | 1995-02-06 | ||
| DE19503825A DE19503825A1 (de) | 1994-09-30 | 1995-02-06 | Neue heterocyclische substituierte Imidazolo-chinoxalinone, ihre Herstellung und Verwendung |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1996010572A1 true WO1996010572A1 (de) | 1996-04-11 |
Family
ID=25940620
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1995/003686 WO1996010572A1 (de) | 1994-09-30 | 1995-09-19 | Neue heterocyclische substituierte imidazolo-chinoxalinone, ihre herstellung und verwendung |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US6121265A (de) |
| EP (1) | EP0783506A1 (de) |
| JP (1) | JPH10513435A (de) |
| CN (1) | CN1046518C (de) |
| AU (1) | AU3650695A (de) |
| BG (1) | BG101328A (de) |
| BR (1) | BR9509055A (de) |
| CA (1) | CA2200358A1 (de) |
| CZ (1) | CZ94297A3 (de) |
| FI (1) | FI971317A0 (de) |
| HU (1) | HUT73969A (de) |
| IL (1) | IL115444A0 (de) |
| NO (1) | NO971424L (de) |
| PL (1) | PL319406A1 (de) |
| SI (1) | SI9520108A (de) |
| WO (1) | WO1996010572A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997034896A1 (de) * | 1996-03-22 | 1997-09-25 | Basf Aktiengesellschaft | Neue heterocyclische substituierte imidazolo-chinoxalinone, ihre herstellung und verwendung |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000051590A2 (en) * | 1999-03-04 | 2000-09-08 | The Board Of Trustees Of The University Of Illinois | Neuropharmacological treatment of sleep-related breathing disorders |
| US7542773B2 (en) * | 2002-03-29 | 2009-06-02 | At&T Intellectual Property I, L.P. | Customized alerts for incoming data messages |
| CN100386326C (zh) * | 2003-10-22 | 2008-05-07 | 中国人民解放军军事医学科学院毒物药物研究所 | 1-羟甲基咪唑并[1,2-a]喹喔啉化合物及其应用 |
| JP2009523820A (ja) * | 2006-01-23 | 2009-06-25 | アミラ ファーマシューティカルス,インコーポレーテッド | 5−リポキシゲナーゼの三環系抑制剤 |
| EP2666775A1 (de) * | 2012-05-21 | 2013-11-27 | Domain Therapeutics | Substituierte Pyrazolochinazolinone und Pyrrolochinazolinone als allosterische Modulatoren von metabotropen Glutamatrezeptoren der Gruppe II |
| FR3031105B1 (fr) * | 2014-12-31 | 2018-04-06 | Universite De Montpellier | Nouveaux imidazo[1,2-a]quinoxalines et derives pour le traitement des cancers |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3004750A1 (de) * | 1979-02-09 | 1980-08-21 | Roussel Uclaf | Neue heterotricyclische derivate, verfahren zu deren herstellung, ihre verwendung als arzneimittel und sie enthaltenden pharmazeutischen zusammensetzungen |
| EP0518530A2 (de) * | 1991-06-05 | 1992-12-16 | Eli Lilly And Company | Verbesserungen von oder in Bezug auf exzitatorische Aminosäurenrezeptorantagonisten |
| US5196421A (en) * | 1991-06-05 | 1993-03-23 | Eli Lilly And Company | Excitatory amino acid receptor antagonists in methods for the use thereof |
| WO1993020077A1 (en) * | 1992-04-03 | 1993-10-14 | Yamanouchi Pharmaceutical Co., Ltd. | Fused quinoxalinone derivative and pharmaceutical composition containing the same |
| WO1994022447A1 (de) * | 1993-03-31 | 1994-10-13 | Basf Aktiengesellschaft | Imidazolochinoxalinone zur behandlung zentralnervöser erkrankungen |
| WO1995021842A1 (en) * | 1994-02-11 | 1995-08-17 | Novo Nordisk A/S | Heterocyclic compounds and their preparation and use |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2448541A1 (fr) * | 1979-02-09 | 1980-09-05 | Roussel Uclaf | Nouveaux derives de l'oxoimidazoquinoxaline et leurs sels, leur procede de preparation, leur application comme medicaments et les compositions pharmaceutiques les renfermant |
| SE462338B (sv) * | 1982-04-28 | 1990-06-11 | Roussel Uclaf | Nya imidazo /1,2-a/ kinoxaliner och deras salter, foerfarande foer framstaellning daerav samt farmaceutiska kompositioner innehaallande densamma |
| IE66149B1 (en) * | 1986-09-16 | 1995-12-13 | Novo Nordisk As | Quinoxaline compounds and their preparation and use |
| DK716188D0 (da) * | 1988-12-22 | 1988-12-22 | Ferrosan As | Quinoxalinforbindelser, deres fremstilling og anvendelse |
| US5055465A (en) * | 1989-05-31 | 1991-10-08 | Berlex Laboratories, Inc. | Imidazoquinoxalinones, their aza analogs and process for their preparation |
| US5182386A (en) * | 1991-08-27 | 1993-01-26 | Neurogen Corporation | Certain imidazoquinoxalinones; a new class of gaba brain receptor ligands |
| US5306819A (en) * | 1992-08-27 | 1994-04-26 | Neurogen Corporation | Certain aryl a cycloalkyl fused imidazopyrazinols; and new class of GABA brain receptor ligands |
| US5536718A (en) * | 1995-01-17 | 1996-07-16 | American Cyanamid Company | Tricyclic benzazepine vasopressin antagonists |
-
1995
- 1995-09-19 WO PCT/EP1995/003686 patent/WO1996010572A1/de not_active Application Discontinuation
- 1995-09-19 AU AU36506/95A patent/AU3650695A/en not_active Abandoned
- 1995-09-19 JP JP8511334A patent/JPH10513435A/ja active Pending
- 1995-09-19 CN CN95195396A patent/CN1046518C/zh not_active Expired - Fee Related
- 1995-09-19 CZ CZ97942A patent/CZ94297A3/cs unknown
- 1995-09-19 PL PL95319406A patent/PL319406A1/xx unknown
- 1995-09-19 CA CA002200358A patent/CA2200358A1/en not_active Abandoned
- 1995-09-19 EP EP95934074A patent/EP0783506A1/de not_active Withdrawn
- 1995-09-19 BR BR9509055A patent/BR9509055A/pt not_active Application Discontinuation
- 1995-09-19 SI SI9520108A patent/SI9520108A/sl unknown
- 1995-09-19 US US08/809,170 patent/US6121265A/en not_active Expired - Fee Related
- 1995-09-28 IL IL11544495A patent/IL115444A0/xx unknown
- 1995-09-29 HU HU9502847A patent/HUT73969A/hu unknown
-
1997
- 1997-03-14 BG BG101328A patent/BG101328A/xx unknown
- 1997-03-25 NO NO971424A patent/NO971424L/no unknown
- 1997-03-27 FI FI971317A patent/FI971317A0/fi unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3004750A1 (de) * | 1979-02-09 | 1980-08-21 | Roussel Uclaf | Neue heterotricyclische derivate, verfahren zu deren herstellung, ihre verwendung als arzneimittel und sie enthaltenden pharmazeutischen zusammensetzungen |
| EP0518530A2 (de) * | 1991-06-05 | 1992-12-16 | Eli Lilly And Company | Verbesserungen von oder in Bezug auf exzitatorische Aminosäurenrezeptorantagonisten |
| US5196421A (en) * | 1991-06-05 | 1993-03-23 | Eli Lilly And Company | Excitatory amino acid receptor antagonists in methods for the use thereof |
| WO1993020077A1 (en) * | 1992-04-03 | 1993-10-14 | Yamanouchi Pharmaceutical Co., Ltd. | Fused quinoxalinone derivative and pharmaceutical composition containing the same |
| WO1994022447A1 (de) * | 1993-03-31 | 1994-10-13 | Basf Aktiengesellschaft | Imidazolochinoxalinone zur behandlung zentralnervöser erkrankungen |
| WO1995021842A1 (en) * | 1994-02-11 | 1995-08-17 | Novo Nordisk A/S | Heterocyclic compounds and their preparation and use |
Non-Patent Citations (1)
| Title |
|---|
| L. MCQUAID ET AL.: "Synthesis and Excitatory Amino Acid Pharmacology of a Series of Heterocyclic-Fused Quinoxalinones and Quinazolinones", J. MED. CHEM., vol. 35, no. 18, pages 3319 - 3324 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997034896A1 (de) * | 1996-03-22 | 1997-09-25 | Basf Aktiengesellschaft | Neue heterocyclische substituierte imidazolo-chinoxalinone, ihre herstellung und verwendung |
Also Published As
| Publication number | Publication date |
|---|---|
| IL115444A0 (en) | 1995-12-31 |
| EP0783506A1 (de) | 1997-07-16 |
| SI9520108A (en) | 1997-10-31 |
| HU9502847D0 (en) | 1995-11-28 |
| BG101328A (en) | 1997-08-29 |
| HUT73969A (en) | 1996-10-28 |
| NO971424L (no) | 1997-05-22 |
| CN1159807A (zh) | 1997-09-17 |
| JPH10513435A (ja) | 1998-12-22 |
| US6121265A (en) | 2000-09-19 |
| BR9509055A (pt) | 1998-06-23 |
| FI971317A (fi) | 1997-03-27 |
| CZ94297A3 (en) | 1997-08-13 |
| NO971424D0 (no) | 1997-03-25 |
| PL319406A1 (en) | 1997-08-04 |
| CA2200358A1 (en) | 1996-04-11 |
| CN1046518C (zh) | 1999-11-17 |
| FI971317A0 (fi) | 1997-03-27 |
| AU3650695A (en) | 1996-04-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69132340T2 (de) | Kondensiertes pyrazinderivat | |
| EP0077024B1 (de) | Neue Imidazol-Derivate, Verfahren zu ihrer Herstellung und diese enthaltende pharmazeutische Präparate | |
| EP0572852B1 (de) | Chinoxalin-2,3(1H,4H)-dione als Arzneimittel | |
| DE4242675A1 (de) | Neue Hydroxyiminoalkylindolcarbonsäure-Derivate, ihre Herstellung und Verwendung | |
| WO1996010572A1 (de) | Neue heterocyclische substituierte imidazolo-chinoxalinone, ihre herstellung und verwendung | |
| EP0691970B1 (de) | Imidazolochinoxalinonderivate als eaa antagonisten | |
| WO1997049701A1 (de) | Pyrrolylchinoxalindione, ihre herstellung und verwendung als ampa-rezeptorantagonisten | |
| DE3346575A1 (de) | Neue benzimidazole, ihre herstellung und diese verbindungen enthaltende arzneimittel | |
| EP0785932B1 (de) | Pyrrolyl-tetrahydrobenzochinoxalindione, ihre herstellung und verwendung als glutamatrezeptor-antagonisten | |
| DE4340045A1 (de) | Neue Chinoxaline und Arzneimittel daraus | |
| DE2550959B2 (de) | Tetrazolyl-imidazole und Tetrazolyl--benzimidazole, Verfahren zu deren Herstellung und diese enthaltende Arzneimittel | |
| DE4428152A1 (de) | Neue Amido-chinoxalindione, ihrer Herstellung und Verwendung | |
| DE19503825A1 (de) | Neue heterocyclische substituierte Imidazolo-chinoxalinone, ihre Herstellung und Verwendung | |
| EP0673377B1 (de) | Neue triazolochinazoline, ihre herstellung und verwendung | |
| DE2650231A1 (de) | Neue imidazolverbindungen, verfahren zu ihrer herstellung und diese enthaltende arzneimittel | |
| DE19611476A1 (de) | Neue heterocyclische substituierte Imidazolo-chinoxalinone, ihre Herstellung und Verwendung | |
| DE2651580A1 (de) | Neue imidazolverbindungen, verfahren zu deren herstellung und diese enthaltende arzneimittel | |
| DE4329970A1 (de) | Neue Imidazolo-chinoxalinone, ihre Hestellung und Verwendung | |
| EP0847395A1 (de) | Pyrrolyl-tetrahydrochinoxalindione, ihre herstellung und verwendung zur bekämpfung von krankheiten | |
| AT358033B (de) | Verfahren zur herstellung neuer imidazol- derivate und ihrer salze | |
| AT358032B (de) | Verfahren zur herstellung neuer imidazol- derivate und ihrer salze | |
| DE2340709A1 (de) | Kombinationspraeparat aus einem benzisothiozolinon und einem 2-nitrofuryl- oder 2-nitrothienylderivat | |
| EP0086450A2 (de) | Substituierte Phenylpyrazolderivate, Verfahren zu ihrer Herstellung, Arzneimittel auf Basis dieser Verbindungen, sowie deren Verwendung | |
| DE2846125A1 (de) | 7-halogen-4-alkyl-1,2-dihydro-5,6- aethano-6h- eckige klammer auf 1,4 eckige klammer zu -diazepino- eckige klammer auf 4,5-a eckige klammer zu -benzimidazole, deren saeureadditionssalze, verfahren zu deren herstellung und diese enthaltende arzneimittel |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 95195396.6 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BG BR BY CA CN CZ FI HU JP KR KZ MX NO NZ PL RU SG SI SK UA US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1995934074 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2200358 Country of ref document: CA Ref document number: 2200358 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 293934 Country of ref document: NZ Ref document number: 08809170 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1997-942 Country of ref document: CZ Ref document number: 971317 Country of ref document: FI |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019970702087 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 1995934074 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1997-942 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019970702087 Country of ref document: KR |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV1997-942 Country of ref document: CZ |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1019970702087 Country of ref document: KR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1995934074 Country of ref document: EP |