USE OF CALPAIN INHIBITORS IN THE INHIBITION
AND TREATMENT OF MEDICAL CONDITIONS ASSOCIATED WITH
INCREASED CALPAIN ACTIVITY
Background of the Invention The present invention relates generally to medical treatments involving the inhibition of calcium-activated proteases, such as Calpain. More specifically, the present invention relates to the treatment of neurodegenerative conditions, coronary disease, circulatory pathology, cataract formation, and other medical conditions associated with calcium-activated protease activity using inhibitors of these proteases.
Neural tissues, including brain, are known to possess a large variety of proteases, including at least two calcium-stimulated proteases, termed calpain I and calpain II, which are activated by micromolar and millimolar Ca2+ concentrations,
respectively. Calpains are a family of calcium activated thiol proteases that are present in many tissues and use a cysteine residue in their catalytic mechanism. Calpain II is the predominant form, but calpain I is found at synapses and is thought to be the form involved in long term potentiation, synaptic plasticity and cell death. Thiol proteases are distinguished from serine proteases, metalloproteases and other proteases by their mechanism of action and by the amino acid residue (cysteine) that participates in substrate attack. Although several thiol proteases are produced by plants, these proteases are not common in mammals, with cathepsin B (a lysosomal enzyme), other cathepsins and the calpains being among the few representatives of this family that have been described in mammals. Calpain I and calpain II are the best described of these, but several other members of the calpain family have been reported.
Other Ca2+ -activated thiol proteases may exist, such as those reported by Yoshihara et al, in /. Biol Chem., 265:5809-5815 (1990). The term "Calpain" is used hereinafter to refer to any Ca2+ -activated thiol proteases including the Yoshihara enzyme and calpains I and II.
Although Calpains degrade a wide variety of protein substrates, cytoskeletal proteins seem to be particularly susceptible to attack. In at least some cases, the products of the proteolytic digestion of these proteins by Calpain are distinctive and persistent over time. Since cytoskeletal proteins are major components of certain types of cells, this provides a simple method of detecting Calpain activity in cells and tissues.
Specifically, the accumulation of the breakdown products ("BDP's") of spectrin, a cytoskeletal protein, has been associated with the activation of Calpain. Thus, calpain activation can be measured indirectly by assaying the proteolysis of the cytoskeletal protein spectrin, which produces a large, distinctive and biologically persistent breakdown product when attacked by calpain (Siman, Baudry, and Lynch, Proc. Natl.
Acad. Set USA 81:3572-3576 (1984); incorporated herein by reference). In neural tissues, activation of Calpains, as evidenced by accumulation of these BDP's, has been observed in many neurodegenerative conditions. For example, these phenomena have been observed after denervation resulting from focal electrolytic lesions, in genetic abnormalities, after excitotoxicity, following ischemia in gerbils and rats, following administration of the toxins kainate and colchicine in rats, an in human Alzheimer's disease. Calpains have also been shown to degrade the lens proteins alpha-crystallin,
vimentin, and actin in vitro, and have been implicated in the degradation of cardiac muscle proteins and other tissues.
Commercially available in vitro inhibitors of Calpain include peptide aldehydes such as leupeptin (Ac-Leu-Leu-Arg-H) and Ac-Leu-Leu-Nle-H, as well as epoxysuccinates such as E-64. These compounds are not useful in inhibiting Calpain in
Central Nervous System ("CNS") tissue in vivo because they are poorly membrane permeant and, accordingly, do not cross the blood brain barrier very well. Some of these compounds have also been found to have other adverse side effects. For example, leupeptin has been found to be harmful to heart cells and to adversely affect blood clotting (Toyo-Oka, et al., Jpn. Heart J., 23(5):829 (1982)). Also, many of these inhibitors are poorly specific and will inhibit a wide variety of proteases in addition to Calpain. Thus, no effective therapy has yet been developed for most neurodegenerative diseases and conditions. Millions of individuals suffer from neurodegenerative diseases and thus, there is a need for therapies effective in treating and preventing these diseases and conditions.
Cathepsin B is involved in muscular dystrophy, myocardial tissue damage, tumor metastasis, and bone resorption. In addition, a number of viral processing enzymes, which are essential for viral infection* are cysteine proteases. Inhibitors of cysteine proteases would thus have multiple therapeutic uses. These commercially available compounds are based upon peptide structures that are believed to interact with the substrate binding site of Calpain. Active groups associated with the Calpain inhibitors then either block or attack the catalytic moiety of Calpain in order to inhibit the enzyme.
In addition, other types of compounds that are not commercially available which inhibit cysteine proteases and are thought to possess in vitro Calpain inhibitory activity have been reported. Examples of such compounds include the peptide diazomethanes and peptide diazomethyl ketones. See Rich, D.H., Inhibitors of cysteine proteinases, in Protease Inhibitors, pp 153-178 (AJ. Barrett and G. Salversen, Eds., Elsevier, New York, 1986), the disclosure of which is hereby incorporated by reference. Peptide diazomethyl ketones are potentially carcinogenic and along with peptide diazomethanes are thought to be poorly membrane permeant and to have low specificity.
There is some evidence that certain particular inhibitors of Calpain have certain therapeutic utilities. For example, leupeptin can facilitate nerve repair in primates. Loxastatin (also known as EST, Ep-460 or E-64d), a derivative of E-64, is believed to have utility in the treatment of muscular dystrophy. E-64d, while not having significant protease inhibitory activity itself, is believed to be converted to more potent forms, such as to E-64c, inside a mammalian body.
Evidence from electrophysiological studies suggests that one of the earliest factors in the chain of reactions leading to cell death is an increase in intracellular-free calcium as a consequence of Ca2+ channel opening and/or energy depletion. Intracellular calcium is likely to produce a large number of consequences, including the activation of a large number of enzymes, including proteases, such as Calpain, upases and kinases. An increase in intracellular calcium is also thought to induce changes in gene expression.
Ischemia, head trauma and stroke have all been associated with the release of glutamate in amounts large enough to lead to excitotoxicity, the toxicity resulting from the actions of certain amino acids on neurons of the CNS. The excess glutamate and other factors, such as free radical damage of membranes or energy depletion, cause an increase in intracellular Ca2+. It is known that an excess of intracellular Ca2+ leads to several effects believed to be associated with neuronal cell damage, including destruction of cell structures through activation of phospholipase and Calpain, as well as free radical production resulting from activation of phospholipase and xanthine oxidase. Many other factors have been associated with neurotoxicity. For example, reductions in action potentials and changes in a wide variety of chemical markers are known to be associated with neurons exposed to ischemic conditions. The excitotoxic death of nerve cells following ischemia is the result of a cascade of events which begins with energy depletion, followed by release of glutamate, stimulation of glutamate receptors, and an elevation of intracellular calcium. See, e.g., Meldrum, "Excitotoxicity in Ischemia: An Overview," in Cerebrovascular Diseases, Ginsberg et al. (eds.), Raven Press, New York, pp. 47-60 (1989). Since many researchers believe that excitotoxicity plays a large role in the pathology of stroke and ischemia, much recent research has focused upon developing drugs which reduce excitotoxicity by acting at specific stages of the excitotoxic cascade.
Elevations in intracellular calcium have been proposed to play a central role in the induction of excitotoxic cell death. See, e.g. Meldrum et al, Trends Pharmacol Sci., 11:379-387 (1990). Many attempts to prevent excitotoxicity have focused upon blocking the NMDA subtype of glutamate receptor, which functions as a calcium channel. Although glutamate toxicity is calcium dependent, it is clear that calcium influx through the NMDA receptor is not the sole culprit in excitotoxicity. The correlation between NMDA antagonist mediated reduction in glutamate-induced intracellular Ca2+ and cell rescue is poor. Further, agents acting at non-NMDA type calcium channels are effective inhibitors of glutamate toxicity and excitotoxicity appears to involve not only calcium influx through both NMDA and non-NMDA calcium channels but also the release of Ca from intracellular stores. Thus, the mechanism by which Ca2+ becomes elevated is still unknown.
It is clear that elevated Ca2+ is a prime intracellular mediator of excitotoxicity. Elevations of intracellular calcium modulate many effects, including the activation of the calcium-dependent thiol proteases calpain I and II. Calpain has been shown to be activated during excitotoxicity, and calpain activation can be detected early following ischemia.
Calpain action results in the irreversible cleavage of cellular proteins and alterations in their function, and this degradative function fits in well with a possible role in cell death. Further, leupeptin, a calpain inhibitor, has been shown to reduce ischemic damage in gerbils and to reduce hypoxic damage in rat hippocampal slices.
Much of what is known about excitotoxicity derives from studies of neurons in vitro. Primary cultures of cerebral cortex, hippocampal and cerebellar neurons are killed by exposure to glutamate or glutamate analogs. Recently, glutamate has been reported to kill pheochromocytoma PC12 cells in a calcium-dependent manner.
Increases in intracellular calcium and subsequent calpain activity have also been linked to other pathological conditions. It has been found, for example, that in experimental cataracts induced in mice, increased calcium levels have been recorded just before the onset of cataract formation. The size of infarcted heart tissue in ischemic myocardium can also be reduced by the administration of calpain inhibitors
(Toda, et al, Jpn. Heart , 30:375-86 (1989); Toyo-Oka, Drug Res., 36(l):671-75 (1986)). Notwithstanding the foregoing understanding of certain aspects of neurotoxicity,
no effective therapy has yet been developed for most neurodegenerative diseases and conditions of the CNS. Millions of individuals suffer from these diseases and conditions. Thus, there is a need for therapies effective in treating and preventing these diseases and conditions. In addition to being involved in cytotoxicity, proteases such as calpain have also been linked to the regulation of cellular growth. However, the mechanisms of such regulation have not been well studied. Some protease inhibitors inhibit cellular proliferation, for example, while others enhance it. Because calpains are ubiquitously distributed in mammalian cells but apparently do not contribute to normal protein catabolism or general protein turnover, they appear to serve a regulatory role in such cells. However, the mechanisms of such regulation have not been well studied. While some calpain inhibitors have been shown to inhibit cellular proliferation and thus cell cycling, the specific point in the reproductive cycle at which such inhibition occurs is not yet known. An understanding of the regulation of cell cycling is relevant to the development of treatments for cancer, because cancer cells grow without regulation of such cell cycling. Chemotherapy treatments for cancer sometime take the form of administering chemicals which will kill cells that are passing through the cell cycle and actively dividing while sparing those cells which are not dividing. In one such form of chemotherapy, drugs which interfere with the replication of the DNA of cells during the "S" (synthesis) phase of the cell cycle are administered to a patient. This treatment, however, will only be effective in killing cells in the S phase. Thus, a drug must be present in a patient's body for long enough so that all of the cancer cells in the patient progress through the S phase. Since chemotherapeutic agents kill non-cancerous cells which are dividing as well as cancerous cells, the timing and duration of chemotherapeutic drug administration is critical to successful therapy.
There exists, therefore, a need for compounds which are capable of manipulating the cell cycle, resulting in a shortened duration of chemotherapy and greater efficacy of the chemotherapeutic agent. The processes of angiogenesis and vascular repair both depend upon smooth muscle cell proliferation, since smooth muscle cells play an essential part in the functioning of blood vessels as well as other organs. Smooth muscle cells are
stimulated to proliferate following vascular injury by a number of different factors, including PDGF (platelet derived growth factor). This is normally a desirable process which is necessary for healing. However, following therapeutic angioplasty for the opening of obstructed arteries, the proliferation of the smooth muscle cells can result in restenosis, the blockage of the previously opened artery. Austin G.E., et al., /. Am.
Coli Cardiology, 6:369-377 (1985). This is a significant problem in the clinical use of angioplasty, and a need therefore exists for a drug which can inhibit the proliferation of the smooth muscle cells.
Additionally, proteases such as calpain have also been linked to the regulation of smooth muscle contraction. However, the mechanism by which contractility and the maintenance of the tonically contracted state is regulated in smooth muscle is not well understood. Many agents which act to decrease contractility of smooth muscle have little or no efficacy at inhibiting the establishment of the tonic state or reversing the tonic contractile state once established. The tonic contraction of smooth muscle is a normal process. In some cases, however, such tonic contraction can lead to serious pathological conditions. For example, contraction of the bronchial smooth muscle leads to shortness of breath and other symptoms of asthma. Contraction of the coronary arteries can lead to angina, partial coronary hypoxia and subsequent loss of coronary function. Contraction of the smooth muscle in cerebral arteries can lead to cerebral vasospasm and hypoxia of the brain tissue, a serious condition that can leave patients mentally disabled and permanently brain damaged.
Summary of the Invention One aspect of the present invention is a method of synchronizing the reproductive cycle of actively dividing cells. In this method, an amount of a Calpain
Inhibitor which is pharmacologically effective to block the progression of cells from G1 phase into S phase is administered to the cells. The Calpain Inhibitor can be one of the Peptide Keto-Compounds, the Halo-Ketone Peptides, or the Substituted Heterocyclic Compounds. In one embodiment, the cells to be treated in this method are located in vivo in a mammal, so that the administering step of the method comprises administering a Calpain Inhibitor to cells in a mammal. Alternatively, the administering step can comprise administering a Calpain Inhibitor to cells in vitro. In
one preferred embodiment, the administering step of this method comprises administering a Peptide Keto-Compound. Calpain Inhibitors can be administered in this method either intravenously, intramuscularly, intraperitoneally, topically, orally, or by direct application to cells. In another aspect, the present invention comprises a method of blocking the progression of the cell cycle from Gj phase to S phase in actively dividing cells in a mammal. In this method, a mammal is administered an amount of a Calpain Inhibitor which is pharmacologically effective to block the progression of the cell cycles of actively dividing cells in the mammal from Gj phase into S phase. The Calpain Inhibitor can be one of the Peptide Keto-Compounds, the Halo-Ketone Peptides, or the Substituted Heterocyclic Compounds. In one preferred embodiment, the Calpain Inhibitor is a Peptide Keto-Compound. Calpain Inhibitors can be administered according to this method either intravenously, intramuscularly, intraperitoneally, topically, orally, or by direct application to living cells. In one embodiment, the Calpai Inhibitor is administered by direct application, where such direct application can comprise either applying a gel to an area of living cells, driving microspheres loaded with the Calpain Inhibitor into tissue comprising the living cells, or injecting a solution containing the Calpain Inhibitor directly into tissue comprising such living cells.
In yet another aspect, the present invention comprises a method of enhancing the efficacy of chemotherapy in the treatment of cancer in a human patient. This method comprises administering to the cancerous cells of the patient an amount of a Calpain Inhibitor which is pharmacologically effective to block the progression of the cell cycles of such cancerous cells from Gj phase to S phase, and thereafter administering to the cells a chemotherapeutic agent. The Calpain Inhibitor in this method is selected from the group consisting of Peptide Keto-Compounds, Halo-
Ketone Peptides, and Substituted Heterocyclic Compounds. In one preferred embodiment, the Calpain Inhibitor is a Peptide Keto-Compound. The Calpain Inhibitor in this method can be administered intravenously, intramuscularly, intraperitoneally, topically, orally, or by direct application to the cancerous cells. The chemotherapeutic agent can be administered beginning 24-48 hours after the administration of the Calpain Inhibitor, at which time the cell cycles of the patient's cancerous cells which were treatable with the Calpain Inhibitor will be synchronized.
A further aspect of the present invention includes a method of determining the effectiveness of a chemotherapeutic agent, comprising growing cancerous cells in vitro, administering to such cancerous cells an amount of a Calpain Inhibitor which is effective to block the progression of the cells from G1 phase into S phase, administering to the cells the chemotherapeutic agent in an amount sufficient to kill the cells, and thereafter determining the amount of cell death that occurs. The amount of cell death that occurs in this method is indicative of the effectiveness the chemotherapeutic agent tested.
Another aspect of the present invention is a method of increasing the efficiency of cell transformation and thus increasing the efficiency of integration of foreign DNA into living cells. This method comprises administering to a population of cells comprising actively dividing cells an amount of a Calpain Inhibitor which is pharmacologically effective to block the progression of the cell cycles of the cells from Gj phase into S phase, discontinuing the administration of the Calpain Inhibitor, and thereafter introducing foreign DNA into the population of cells. The Calpain Inhibitor in this method is selected from the group consisting of Peptide Keto-Compounds, Halo- Ketone Peptides, and Substituted Heterocyclic Compounds. In one embodiment, the Calpain Inhibitor is a Peptide Keto-Compound. The administration of the Calpain Inhibitor in this method can continue for the length of one cell cycle in the population of living cells. The target of the Calpain Inhibitor can be a population of cells located in a mammal, which can be administered a Calpain Inhibitor intravenously, intramuscularly, intraperitoneally, topically, orally, or by direct application to the population of cells in the mammal. In another embodiment, the Calpain Inhibitor is administered instead to a population of cells in vitro. The present invention provides methods of treating a variety of medical conditions associated with calcium-activated protease activity in a mammal by administering the Calpain inhibitors of the present invention to that mammal. These Calpain inhibitors are Peptide Keto-Compounds, Halo-Ketone Peptides, and Substituted Heterocyclic Compounds. Particularly preferred compounds for this use include the Peptide Ketoamides, such as Z-Leu-Abu-CONH-Et, Z-Leu-Phe-CONH-Et and Z-Leu-Phe-CONH(CH2)2C6H5. Administration of the inhibitors can be through any of a variety of routes. These routes include all of the following types of
administration: intravenous, intraperitoneal, intramuscular, oral, topical treatment such as through ointments (including ophthalmic ointments), eye drops, contact lenses, catheter, directly onto tissues such as blood vessels or cardiac tissue during surgery, or injection into the pericardial space. Specific medical conditions which can be treated with these Calpain Inhibitors include cardiac muscle tissue damage. After a mammal with cardiac muscle tissue damage has been identified, that mammal can be treated with a Calpain Inhibitor. Mammals at risk for developing cardiac muscle tissue damage can also be treated with the present Calpain Inhibitors. Administering these Inhibitors to such mammals protects them from the cardiac tissue damage experienced by mammals which are not so protected.
In another embodiment of the present invention, cataracts are treated by the administration of a Calpain Inhibitor. If a mammal has already developed cataracts, the development of the cataracts can be slowed or arrested through the administration of a Calpain Inhibitor. On the other hand, if a mammal has been identified as being a risk for developing cataracts in the future, the development of cataracts in such a mammal can be prevented or slowed through the administration of a Calpain Inhibitor.
A variety of other tissues and conditions can also be treated with the novel Calpain Inhibitors of the present invention. Skeletal and smooth muscle damage, for example, can be treated by identifying a mammal with such tissue damage and administering a Calpain Inhibitor to that mammal. Vasospasm, a condition of a particular kind of smooth muscle, the vascular tissue, can also be reversed in a mamma identified as having this condition by the administration of Calpain Inhibitors. Erythrocytes damaged by the proteolytic activity of Calpain in hypertensive mammals can also be treated with the Calpain Inhibitors of the present invention.
In one aspect, the present invention provides methods of halting or inhibiting the proliferation of smooth muscle cells both in vivo and in vitro by administering a Calpain Inhibitor. These Calpain Inhibitors are Peptide Keto-Compounds, Halo- Ketone Peptides, and Substituted Heterocyclic Compounds. Particularly preferred compounds for this use include the Peptide Ketoamides, such as Z-Leu-Phe-CONH-Et
Administration of the inhibitors can be through any of a variety of routes. These routes include all of the following types of administration: intravenous, intramuscular,
intraperitoneal, topical, oral, or by direct application. Preferred Peptide Keto- Compounds useful in the present invention include (Ph)2CHCO-Leu-Phe-CONH-CH2- 2-Py; Z-Leu-Nva-CONH-CH2-2-Py; Z-Leu-Phe-CONH-CH2CH(OH)Ph; (Ph)2CHCO- Leu-Abu-CONH-CH2CH(OH)Ph; Z-Leu-Phe-CONH2; Z-Leu-Abu-CONH- CH2CH(OH)Ph; and Z-Leu-Phe-CONHEt.
Direct application of the Calpain Inhibitors can be through various means. Such means include using a gel or ointment containing the inhibitor to coat the surface of the balloon of a balloon catheter or onto another surgical instrument that is inserted into the blood vessel during angioplasty. Alternatively, the gel may be applied directly to an area of vascular tissue which has been treated by angioplasty during the surgical procedure. Another route of administration comprises driving microspheres which have been loaded with a Calpain Inhibitor directly into the mammal's blood vessel. This can be accomplished by applying the microspheres to the surface of the balloon or other surgical instrument used during the angioplasty procedure. The microspheres are driven into the arterial wall, where they lodge and release the Calpain Inhibitor over time.
Specific medical conditions which can be treated with these Calpain Inhibitors include the treatment of a mammal to prevent restenosis of a blood vessel following angioplasty. After a mammal which has undergone angioplasty has been identified, that mammal can be treated with a Calpain Inhibitor. Mammals at risk for developing restenosis can also be treated with the present Calpain Inhibitors. Administering these Inhibitors to such mammals protects them from the smooth muscle cell proliferation experienced by mammals which are not so protected.
In another aspect, the present invention provides a method of inhibiting tonic smooth muscle contraction in a mammal susceptible to inappropriate contraction in a smooth muscle thereof. The method includes administering to the smooth muscle an amount of a Calpain Inhibitor that is pharmacologically effective to suppress the contraction thereof. The Calpain Inhibitor is one of the Peptide Keto-Compounds, Halo-Ketone Peptides or Substituted Heterocylic Compounds. Preferably, the inhibitor is administered intravenously, intramuscularly, intraperitoneally, topically, orally, by injection into cerebrospinal fluid, by inhalation, or by direct application to the smooth muscle, such as by applying directly to an area of smooth muscle. Direct application
can also be by driving microspheres loaded with the Calpain Inhibitor into the smooth muscle. Relaxation of the smooth muscle is preferably induced.
In an additional aspect, the present invention provides a method of treating coronary vasospasm in a mammal. In this aspect, the method includes administering t the mammal an amount of a Calpain Inhibitor which is pharmacologically effective to stop vasospasm of coronary tissue in the mammal. The Calpain Inhibitor is one of the Peptide Keto-Compounds, Halo-Ketone Peptides or Substituted Heterocylic Compounds. In a preferred embodiment, the coronary tissue is surgically exposed and a solution of Calpain Inhibitor is applied directly to the tissue. Preferably, the coronar tissue comprises a coronary artery. In a preferred embodiment of this aspect, the mammal is suffering from angina and the method comprises a treatment for the angin
In still another aspect of the invention, there is provided a method of treating bronchial vasospasm in a mammal. This method includes administering to the mamm an amount of a Calpain Inhibitor which is pharmacologically effective to stop vasospasm of bronchial tissue in the mammal. The Calpain Inhibitor is one of the
Peptide Keto-Compounds, Halo-Ketone Peptides or Substituted Heterocylic Compounds. The bronchial tissue can be surgically exposed and a solution of Calpain Inhibitor applied directly to the tissue. In a preferred embodiment of the method, the mammal is suffering from asthma and the method comprises a treatment for the asthma.
Yet another aspect of the invention relates to a method of treating cerebral vasospasm in a mammal. This method includes administering to the mammal an amount of a Calpain Inhibitor which is pharmacologically effective to stop vasospasm cerebral tissue in the mammal. The Calpain Inhibitor is one of the Peptide Keto- Compounds, Halo-Ketone Peptides and Substituted Heterocylic Compounds. The cerebral tissue can be surgically exposed and a solution of Calpain Inhibitor applied directly to the tissue. In one embodiment of this aspect of the invention, the Calpain Inhibitor can be injected into the mammal's cerebrospinal fluid.
One aspect of the present invention provides a method of medical treatment f a medical condition in a mammal. In this method, a pharmaceutical composition containing a morpholine Peptide Keto-Compound is administered to the mammal. Th composition is administered in an amount that is pharmacologically effective to treat
the condition. The condition is one which is associated with increased proteolytic activity of Calpain. The morpholine Peptide Keto-Compound can be either a C-terminal or N-terminal morpholine Peptide Keto-Compound, such as cardiac muscle tissue damage, cataracts, skeletal muscle damage, vasospasm or restenosis following cardiac angioplasty.
Another aspect of the present invention also provides a method of medical treatment for a medical condition in a mammal. In this method, a pharmaceutical composition containing a Peptide Ketoamide, Subclass C is administered to the mammal. This composition is administered in an amount that is pharmacologically effective to treat the condition. The condition that can be treated with this method is also one associated with increased proteolytic activity of Calpain, such as cardiac muscle tissue damage, cataracts, skeletal muscle damage, vasospasm or restenosis following cardiac angioplasty.
One of skill in the art will recognize that the present Calpain Inhibitors can be used to counteract the harmful effects associated with calpain activity which arise in a number of medical conditions and diseases. Therefore, the treatment of such conditions with the present Calpain Inhibitors is within the scope of the present invention.
These and other features and advantages of the present invention will become apparent from the detailed description which follows, considered together with the attached drawings and claims.
Brief Description of the Figures Figure 1 shows the percentage of inhibition of glutamate-induced cell death through the addition of glutamate and various Calpain Inhibitors relative to control where no glutamate was added.
Figure 2 shows that Calpain inhibitor reduces cell death following glutamate exposure. PC12 cells were exposed to 7.5mM glutamate with the indicated concentration of inhibitor, as described in the text, for 24 hours. Cell viability was assayed using the Mi 1 assay. Values are expressed as % of naive control ± sem. Figure 3 shows the dependence of the ability of Calpain inhibitors to reduce cel death on glutamate concentration. PC 12 cells were incubated with the indicated concentration of glutamate and no inhibitor (circles), 20uM Z-Leu-Nva-CONH(CH2)3
morpholine (triangles), or 30uM Z-Leu-Phe-CONHCH2CH (squares) for 24 hours an cell viability was assayed by MTT. Values expressed as % of naive control ± sem.
Figure 4. Delayed addition of calpain inhibitor. Glutamate (7.5mM) was adde at 0 time and Z-Leu-Phe-CONHCH2CH3 (squares) or Z-Leu-Nva-CONH(CH2)3 morpholine (triangles) added at the indicated times to final concentrations of lOOuM each. Cell viability was measured 24 hours after the addition of glutamate by the MT assay. Values expressed as % of naive control ± sem.
Figure 5 graphically depicts the effects of Z-Leu-Phe-CONH-Et and Z-Leu- Abu-CONH-Et on the size of infarction produced upon MCA occlusion in male rats. Figure 6 shows the effects of Z-Leu-Abu-C02Et, a Peptide Keto-Compound, and CIl (Ac-Leu-Leu-Nle-H) relative to control slices on survival of hippocampal slice exposed to 10 minutes exposure of anoxic atmosphere where both of these compounds were added at their optimal inhibitory concentration at both 1 hour and 2 hour incubation times. Figure 7 shows the evoked potential amplitude for control, CIl treated and Z-
Leu-Abu-C02Et treated hippocampal slices over a time course during which the slices are exposed to anoxic atmosphere.
Figure 8 shows the percent recovery of EPSP from severe hypoxia over the course of one hour incubation for Z-Leu-Phe-CONH-Et and Z-Leu-Phe-C02Et. Figure 9 shows a comparison of the effect of the presence of CIl or Z-Leu-Ph
C02Et on survival of hippocampal slices expressed as the duration of anoxia (in minutes) before fiber volley disappearance.
Figure 10 shows the effects of CIl compared with control on the behavioral an convulsive effects of kainic acid. Figure 11 shows the amount of spectrin BDP's in rat brains exposed to kainate for control and CIl treated rats.
Figure 12 graphically depicts the effect of several different Calpain Inhibitors contraction of isolated arteries induced by endothelin (ET-1). Drug A is Z-Leu-Abu- CONHEt, Drug B is Z-Leu-Phe-CONHEt, Drug C is 1,10-Phenanthroline and Drug is TLCK (Tosyl-Lysine-chloromethylketone).
Figure 13 graphically depicts the effect of several other Calpain Inhibitors on contraction of isolated arteries induced by endothelin (ET-1). Drug E is Z-Leu-Phe,
Drug F is Z-Leu-Phe-CONHEt (the same as drug B), Drug G is Z-Leu-Phe- CONH(CH2)2Ph, Drug H is Ac-Leu-Leu-Nle-H (Calpain Inhibitor I), Drug I is Gly- Gly-Gly and Drug J is (Ph)2CHCO-Leu-Abu-CONH-CH2CH(OH)Ph.
Figure 14 shows the effect of Calpain Inhibitors on contraction of isolated arteries induced by phorbol dibutyrate (PDB). Drugs E through J are as in Figure 15.
Figure 15 graphically depicts the effect of Calpain Inhibitors on smooth muscle resting tension. Drugs E through J are as in Figure 13.
Figure 16 shows the dose-dependent inhibition of oxyhemoglobin-induced constriction by a Calpain Inhibitor, Z-Leu-Phe-CONH(CH2)3, of the present invention. Figure 17 shows an example of the time course of artery constriction in an artery constricted by subarachnoid hemorrhage (SAH) and treated with a Calpain Inhibitor, Z-Leu-Phe-CONH(CH2)3, of the present invention.
Figure 18 shows the summary of data from three animals in which a Calpain Inhibitor, Z-Leu-Phe-CONH(CH2)3, of the present invention reversed constrictions caused by SAH.
Figure 19 graphically depicts the effects of Z-Leu-Phe-CONHEt and Ph2CHCO-Leu-Abu-CONH-CH2CH(OH)Ph on the proliferation of cultured bovine smooth muscle cells.
Figure 20 shows the continued viability of smooth muscle cells after treatment with a Calpain Inhibitor, despite a complete inhibition of cell proliferation.
Figure 21 graphically depicts the blocking of the progression into S phase of bovine aortic smooth muscle cells (BASMC) after treatment with the Calpain Inhibitor Ph2-CHCO-Leu-Abu-CONH-CH2CH(OH)Ph. In this graph, "Drug C" is Ph2-CHCO- Leu-Abu-CONH-CH2CH(OH)Ph ("Drug C" elsewhere may be a different compound). Figure 22 graphically depicts the synchronous progression into S phase of HeLa and AT-2 cells after the Calpain Inhibitor Ph2-CHCO-Leu-Abu-CONH-CH2CH(OH)Ph was washed out of the medium in which such cells were maintained. In this graph, "Drug C" is also Ph2-CHCO-Leu-Abu-CONH-CH2CH(OH)Ph (though "Drug C" elsewhere may be a different compound).
Detailed Description of the Invention A. INTRODUCTION
We have discovered that Calpain activation is an event central to many cases o brain atrophy and degeneration and that inhibition of Calpain alone is sufficient to inhibit or prevent cell deterioration and loss. Thus, we have further discovered that inhibition of Calpain provides protection from neurotoxicity associated with many neurodegenerative conditions and diseases.
In accordance with the foregoing discoveries, we believe that the elevation of intracellular calcium associated with neuropathological conditions in neuronal cells activates Calpain and sets in motion the digestion of neuronal cells from within. We believe there may be other mechanisms of activation of Calpains associated with these conditions. Accordingly, one aspect of the present invention is directed to inhibition and treatment of the neurodegeneration and other diseases associated with this digestion through the inhibition of Calpain activity. Thus, part of this aspect of the present invention is to prevent the neurodegeneration and other pathology caused by this digestion through the in vivo administration of Calpain inhibitors. By way of example, and not of limitation, diseases and conditions which can be treated using this aspect of the present invention include neurodegeneration following excitotoxicity, HI induced neuropathy, ischemia, denervation following ischemia or injury, subarachnoid hemorrhage, stroke, multiple infarction dementia, Alzheimer's Disease (AD),
Parkinson's Disease, Huntington's Disease, surgery-related brain damage and other neuropathological conditions.
As stated above, spectrin BDP's have been found to be associated with Calpain activation in vivo. We have observed that in each instance of neurodegeneration in which BDP's characteristic of Calpain activation are detected, Calpain activation is localized to the brain areas most vulnerable to the particular pathogenic manipulation. In addition, as judged by histological methods, Calpain activation precedes overt evidence of neurodegeneration. Accordingly, Calpain activation is spatially and temporally linked to impending or ongoing cell death in the brain. Thus, we believe that Calpain activation is an important mechanism of cell damage and death in many pathological conditions, including neuropathological conditions. Moreover, there is evidence that the activation of Calpains is an early event in the death of cells includin
neural cells. This is in contrast to other known proteases which are activated at later stages of cell death. Thus, we believe that, advantageously, inhibition of Calpain activity provides intervention at an early stage of cell death, prior to significant deterioration of cellular machinery. Another aspect of the involvement of Calpains in neurodegeneration is the involvement of these proteins in regenerating systems. It is known that developing or regenerating axons are somehow inhibited from further development in a stabilization process called the "stop pathway." This stabilization can occur when axons have reached their targets; however, in some systems stabilization can also occur at inappropriate places. One researcher has developed evidence that this stop pathway operates at least in part by the activation of intracellular Calpain and that inhibition of Calpain can interfere with stabilization (Luizzi, 1990). We believe that Calpain inhibitors, when used in accordance with the present invention, can advantageously aid regeneration and recovery of neural tissue after injury, in addition to inhibiting neurodegeneration.
Another aspect of the present invention is our discovery that at least three classes of compounds, the substituted isocoumarins, the peptide keto-compounds and the Halo-Ketone Peptides have Calpain inhibitory activity. We have further discovered, as will be described hereinbelow, that these three classes of compounds exhibit additional properties that render them especially useful as therapeutically effective compounds in the treatment of neurodegenerative conditions and diseases.
Calpain has also been implicated in the pathogenesis of a number of other medical conditions. The inhibition of Calpain is capable of slowing the progress of these diseases and of preventing certain conditions altogether. The formation of cataracts, for exarasle, has been linked to Calpain activity in mammalian lenses. In in vivo models of cataract formation, increased Calpain activity has been documented just before the onset of detectable cataract formation. Calpain activity has also been observed to decrease after a cataract has formed in a lens, leading to the inference that calpain activity is involved in the formation of cataracts. Moreover, we have shown that there are increased levels of spectrin breakdown products found in in vitro models of cataract formation. The presence of such spectrin breakdown products is known to be reflective of increased Calpain activity. Thus, we
believe that by administering the Calpain Inhibitors of the present invention, the formation of cataracts can be prevented or slowed.
Calpain activity has also been implicated in producing myocardial infarctions. Calpain activity is regulated by intracellular calcium concentrations, and increased intracellular calcium in myocardial tissues has been observed when the myocardium is cut off from its supply of oxygen due to ischemia. Cell damage and ultimately cell death results from such ischemia. The increased proteolytic activity of Calpain due to increased levels of intracellular calcium during ischemia is therefore a contributor to o direct cause of cell death during cardiac ischemia. Cardiac tissue damage can thus be prevented or minimized with the present Calpain Inhibitors.
Calpain is also believed to be an important regulator of cell growth. Several Calpain Inhibitors have been found, for example, to inhibit smooth muscle cell proliferation. Such proliferation is in fact necessary to repair injured smooth muscle tissue. Following therapeutic angioplasty, however, smooth muscle cell proliferation may result in restenosis of the opened blood vessel. Calpain Inhibitors may thus be used to prevent the smooth muscle cell proliferation which results in the restenosis of blood vessels.
Other disease conditions can be treated with Calpain Inhibitors as well. Calpa has been shown to degrade the constituents of skeletal and smooth muscle cells, and has been implicated in causing vasospasm. Increased Calpain activity has also been shown in the blood cells of hypertensive patients, and has been shown to be five times as active in degrading proteins in such cells as in the cells of non-hypertensive patients Calpain Inhibitors therefore can reduce or eliminate the harmful effects of Calpain activity in these tissues. We have also found that Calpain Inhibitors inhibit tonic smooth muscle contraction. These compounds are useful in the treatment of animals or humans for the purpose of preventing or reducing the smooth muscle contraction associated with vasospasm and bronchospasm.
The present invention includes the use of a variety of Calpain Inhibitors and methods for using these inhibitors to treat disease conditions. Specifically, Substituted
Heterocyclic Compounds, Peptide Keto-Compounds, and Halo-Ketone Peptides have been found to be effective in treating the foregoing conditions as well as other disease
Unless otherwise stated, the Calpain Inhibitors of the present invention refers to the novel Substituted Heterocyclic Compounds, Peptide Keto-Compounds, and Halo- Ketone Peptides described herein.
Several Calpain Inhibitors have also been found to play a role in the regulation of the reproductive cycle of the cell. These compounds can be used in the treatment of cancer in animals or humans along with other chemotherapeutic agents in order to enhance the effectiveness of such agents. By synchronizing the growth of rapidly dividing cells, these compounds can increase the effectiveness of chemotherapeutics that act at a specific stage in the cell cycle, such as at DNA replication. By synchronizing the cell cycles of cells, Calpain Inhibitors are also useful in increasing the efficiency of cell transformation. Transformation results from the incorporation of foreign DNA into a cell. Such incorporation is increased when cells are synthesizing DNA. Thus, by synchronizing cells to the DNA synthetic portion of the cell cycle, the cells will be more efficiently transformed by foreign DNA introduced into the cells.
B. SUBSTITUTED HETEROCYCLIC COMPOUNDS
One particular class of compounds exhibiting Calpain inhibitory activity, when used in accordance with the present invention, are the substituted heterocyclic compounds. These compounds include the substituted isocoumarins. The substituted heterocyclic compounds are known to be excellent inhibitors of serine proteases. As discussed hereinbelow, we have now discovered that these compounds are also inhibitors of calpain I and calpain II, and also of other Calpains. Additionally, as also discussed below, we have found that, unlike most known inhibitors of Calpains, these substituted heterocyclic compounds are not effective as inhibitors of papain or cathepsin B. Thus, we believe that the substituted heterocyclic compounds provide a relatively specific means of inhibiting Calpains while not affecting other thiol proteases.
One particular class of substituted heterocyclic compounds with Calpain inhibitory activity are the isocoumarins having cationic substituents. These substituted heterocyclic compounds are referred to herein as the "Class I Substituted
Isocoumarins." The Class I Substituted Isocoumarins are known to be excellent inhibitors of several serine proteases, including bovine thrombin, human thrombin,
human factor Xa, human factor XIa, human factor Xlla, bovine trypsin, human plasm plasmin, human tissue plasminogen activator, human lung tryptase, rat skin tryptase, human leukocyte elastase, porcine pancreatic elastase, bovine chymotrypsin and huma leukocyte cathepsin G. The Class I Substituted Isocoumarins inhibit the serine proteases by reaction with the active site serine to form an acyl enzyme, which in som cases may further react with another active site nucleophile to form an additional covalent bond. We have discovered that the Class I Substituted Isocoumarins also react with Calpain. We believe that the mechanism of action of Calpain inhibition is similar to that of the inhibition of serine proteases since the reaction mechanism of Calpains is similar to that of the serine proteases.
The Class I Substituted Isocoumarins having Calpain inhibitory activity have th following structural formula:
or a pharmaceutically acceptable salt, wherein
Z is selected from the group consisting of C1 6 alkoxy with an amino group attached to the alkoxy group, C 6 alkoxy with an isothiureido group attached to the alkoxy group, Cw alkoxy with a guanidino group attached to the alkoxy group, C1 6 alkoxy with an amidino group attached to the alkoxy group, C^g alkyl with an amino group attached to the alkyl group, C^g alkyl with an isothiureido group attached to th alkyl group, C]_6 alkyl with an guanidino group attached to the alkyl group, C _6 alkyl with an amidino group attached to the alkyl group, R is selected from the group consisting of 0 = C=N-, S = C=N-, AA-NH-, AA-
AA-NH-, AA-O, AA-AA-O-, M-NH-, M-AA-NH, M-AA-AA-NH-, M-O-, M-AA-O-,
M-AA-AA-O-,
wherein AA represents alanine, valine, leucine, isoleucine, proline, methionine, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, beta-alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine or sarcosine, wherein M represents NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X-NH-CS,
X-NH-S02, X-CO-, X-CS-, X-S02-, X-O-CO-, or X-O-CS-, wherein X represents C^_6 alkyl, Cj_g fluoroalkyl, Cχ_6 alkyl substituted with K, Cw fluoroalkyl substituted with K, phenyl, phenyl substituted with J, phenyl disubstituted with J, phenyl trisubstituted with J, naphthyl, naphthyl substituted with J, naphthyl disubstituted with J, naphthyl trisubstituted with J, C^g alkyl with an attached phenyl group, C^g alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with J, or C1_6 alkyl with two attached phenyl groups substituted with J, wherein J represents halogen, COOH, OH, CN, N02, C^g alkyl, C^g alkoxy, Cj_ alkylamine, Cl 6 dialkylamine, or C^g alkyl-O-CO-, wherein K represents halogen, COOH, OH, CN, N02, NH2, Cj.g alkylamine, Cj.g dialkylamine, or C^g alkyl-O-CO-,
Y is selected from the group consisting of H, halogen, trifluoromethyl, methyl, OH and methoxy. The compounds of Formula (I) can also contain one or more substituents at position B as shown in the following structure:
wherein electronegative substituents such as N02, CN, Cl, COOR, and COOH will increase the reactivity of the isocoumarin, and electropositive substituents such as
NH2, OH, alkoxy, thioalkyl, alkyl, alkylamino, and dialkylamino will increase its stability. Neutral substituents could also increase the stability of acyl enzyme and improve the effectiveness of the inhibitors.
The following compounds are representative of the Class I Substituted Isocoumarins of the present invention:
4-chloro-3-(3-isothiureidopropoxy)isocoumarin (CiTPrOIC)
7-(benzylcarbamoylamino)-4-chloro-3-(3- isothiureidopropoxy)isocoumarin (PhCH2NHCONH-CiTPrOIC)
7-(phenylcarbamoylamino)-4-chloro-3-(3- isothiureidopropoxy)isocoumarin (PhNHCONH-CiTPrOIC) 7-(acetylamino)-4-chloro-3-(3- isothiureidopropoxy)isocoumarin (CH3CONH-CiTPrOIC)
7-(3-phenylpropionylamino)-4-chloro-3-(3- isothiureidopropoxy)isocoumarin (PhCH2CH2CONH-CiTPrOIC)
7-(phenylacetylamino)-4-chloro-3-(3- isothiureidopropoxy)isocoumarin (PhCH2CONH-CiTPrOIC)
7-(L-phenylalanylamino)-4-chloro-3-(3- isothiureidopropoxy)isocoumarin (L-Phe-NH-CiTPrOIC)
7-(N-t-butyloxycarbonyl-L-phenylalanylamino)-4-chloro-3-(3- isothiureidopropoxy)isocoumarin (Boc-L-Phe-NH-CiTPrOIC) 7-(D-phenylalanylamino)-4-chloro-3-(3- isothiureidopropoxy)isocoumarin (D-Phe-NH-CiTPrOIC)
7-(N-t-butyloxycarbonyl-D-phenylalanylamino)-4-chloro-3- (3-isothiureidopropoxy)isocoumarin (Boc-D-Phe-NH-CiTPrOIC)
7-(benzylcarbamoylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (PhCH2NHCONH-CiTEtOIC)
7-(phenylcarbamoylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (PhNHCONH-CiTEtOIC)
7-(isopropylcarbamoylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin ((CH3)2CHNHCONH-CiTEtOIC) 7-(phenylacetylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (PhCH2CONH-CiTEtOIC)
7-(L-phenylalanylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (L-Phe-NH-CiTEtOIC)
7-(N-t-butyloxycarbonyl-L-phenylalanylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (Boc-L-Phe-NH-CiTEtOIC)
7-(D-phenylalanylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (D-Phe-NH-CiTEtOIC) 7-(N-t-butyloxycarbonyl-D-phenylalanylamino)-4-chloro-3-(2~ isothiureidoethoxy)isocoumarin (Boc-D-Phe-NH-CiTEtOIC)
7-(N-t-butyloxycarbonyl-L-alanyl-L-alanylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (Boc-Ala-Ala-NH-CiTEtOIC)
7-(L-alanyl-L-alanylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (Ala-Ala-NH-CiTEtOIC)
7-(l-naphthylcarbamoylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (NaphthylNH-CiTEtOIC)
7-((S)-α-methylbenzylcarbamoylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (S-C6H5(CH3)CHNHCONH-CiTEtOIC) 7-((R)-α-methylbenzylcarbamoylamino)-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (R-C6H5(CH3)CHNHCONH-CiTEtOIC)
7-dansylamino-4-chloro-3-(2-isothiureidoethoxy)isocoumarin (DansylNH-CiTEtOIC) 7-phenylthiocarbamoylamino-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (PhNHCSNH-CiTEtOIC)
7-(m-carboxyphenylthiocarbamoyl)amino-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (m-COOH-PhNHCSNH-CiTEtOIC)
7-(p-carboxyphenylthiocarbamoyl)amino-4-chloro-3-(2- isothiureidoethoxy)isocoumarin (p-COOH-PhNHCSNH-CiTEtOIC)
7-amino-4-chloro-3-(3-isothiureidopropoxy)isocoumarin (ACΓΠC)
Isocoumarins with basic substituents are also known to be effective inhibitors of serine proteases. See Powers et al, U.S. Patent No. 4,845,242, the disclosure of which is hereby incorporated by reference. This class of compounds, referred to herein as the "Class II Substituted Isocoumarins," along with the other substituted heterocyclic compounds, is believed to be effective in the use of the present invention.
The Class II Substituted Isocoumarins have the following structural formula:
or a pharmaceutically acceptable salt, wherein:
R is selected from the group consisting of -N-H-C(=NH)-NH2, -C(=NH)NH2, C1_6 alkyl with an attached amino, and C^ alkyl with an attached isothiureido of the formula -S-C(+NH2+)NH2,
Z is selected from the group consisting of H, halogen, C _6 alkyl, C-^g alkyl wit an attached phenyl, C^ fluorinated alkyl, Cj_6 alkyl with an attached hydroxyl, Cj. alkyl with an attached C1__(t alkoxy, Cχ_6 alkoxy, C^g fluorinated alkoxy, Cj.g alkoxy wit an attached phenyl, benzyloxy, 4-fluorobenzyloxy, -OCH2C6H 4R' (2-substituent), - OCH2C6H4R' (3-substituent), -OCH2C6H4R' (4-substituent), -OCH2C6H3R2' (2,3- substituents), -OCH2C6H3R2' (2,4-substituents), -OCH2C6H3R2' (2,5-substituents), - OCH2C6H3R2' (2,6-substituents), -OCH2C6H3R2' (3,4-substituents), and OCH2C6H3R
(3,5-substituents) .
R' is selected from the group consisting of H, halogen, trifluoromethyl, N02, cyano, methyl, methoxy, acetyl, carboxyl, OH, and amino.
Y is selected from the group consisting of H, halogen, trifluoromethyl, methyl, OH, and methoxy.
Alternately, the Class II Substituted Isocoumarins are represented by structure (II) where,
Z is selected from the group consisting of C^g alkoxy with an attached isothiureido, Cw alkoxy with an attached guanidino, C^ alkoxy with an attached amidino, C^ alkyl with an attached amino, C _6 alkyl with an attached isothiureido,
Cj. alkyl with an attached guanidino, C^g alkyl with an attached amidino,
R is selected from the group consisting of H, OH, NH2, N02 halogen, Cj.g alkoxy, Cl 6 fluorinated alkoxy, C^g alkyl, C-_6 alkyl with an attached amino, M-AA- NH-, M-AA-O-, wherein AA represents alanine, valine, leucine, isoleucine, proline, methionine, phenylalanine, tryptophan, glycine-- §ferine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, beta-alanine, norleucine, norvaline, alpha-aminobutyric and epsilon-aminocaponic acid, citrulline, hydroxyproline, ornithine and sarcosine, wherein M represents H, lower alkanoyl having 1 to 6 carbons, carboxyalkanoyl, hydroxyalkanoyl, amin-alkanoyl, benzene sulfonyl, tosyl, benzoyl, and lower alkyl sulfonyl having 1 to 6 carbons,
Y is selected from the group consisting of H, halogen, trifluoromethyl, methyl, OH and methoxy.
As a further alternative, the Class II Substituted Isocoumarins are represented by structure (II) where
R is selected from the group consisting of -N-H-C(=NH)-NH2, -C(=NH)NH2, Cj. alkyl with an attached amino, Cj_6 alkyl with an attached isothiureido,
Z is selected from the group consisting of C^6 alkoxy with an attached amino, Cj_ alkoxy with an attached isothiureido, C-^g alkoxy with an attached guanidino, C^g alkoxy with an attached amidino, Cj_6 alkyl with an attached amino, Cj_6 alkyl with an attached guanidino, Cj.g alkyl with an attached amidino,
Y is selected from the group consisting of H, halogen, trifluoromethyl, methyl, OH and methoxy.
The following compounds are representative of the Class II Substituted Isocoumarins:
3-(3-aminopropoxy)isocoumarin,
3-(3-aminopropoxy)-4-chloroisocoumarin,
3-(2-isothiureidoethoxy)-4-chloroisocoumarin,
3-(3-isothiureidopropoxy)-4-chloroisocoumarin, 7-amino-3-(3-isothiureidopropoxy)-4-chloroisocoumarin,
7-guanidino-3-methoxyisocoumarin,*
7-guanidino-3-methoxy-4-chloroisocoumarin,
7-guanidino-3-ethoxy isocoumarin,
7-guanidino-3-ethoxy-4-chloroisocoumarin,
7-guanidino-3-(2-phenylethoxy)isocoumarin,
7-guanidino-3-(2-phenylethoxy)-4-chloroisocoumarin.
Still another class of susbstituted heterocyclic compounds useful in the present invention is referred to herein as the "Class III Heterocyclic Compounds" and have the following structural formula:
wherein
Z is selected from the group consisting of CO, SO, S02, CC1 and CF,
Y is selected from the group consisting of O, S and NH, X is selected from the group consisting of N and CH, and
R is selected from the group consisting of C^g alkyl (such as methyl, ethyl and propyl), Cμ alkyl containing a phenyl (such as benzyl), and C^6 fluoroalkyl (such as trifluoromethyl, pentafluoroethyl, and heptafluoropropyl).
The Z group must be electrophilic since it interacts with the active site serine OH group of the serine protease. The R group must be uncharged and hydrophobic.
One or more of the carbons in the R group could be replaced by O, S, NH and other such atomic groups as long as the R group maintains its hydrophobic character.
The following compounds are representative of the Class III Heterocyclic Compounds: 2-trifluoromethyl-4H-3, l-benzoxazine-4-one,
2-pentafluoroethyl-4H-3,l-benzoxazine-4-one,
2-heptafluoropropyl-4H-3,l-benzoxazine-4-one,
2-methyl-4H-3,l-benzoaxazine-4-one,
2-propyl-4H-3, l-benzoaxazine-4-one,
2-benzyl-4H-3,l-benxoaxazine-4-one,
2-heptafluoropropyl-4-quinazolinone, 2-propyl-4-quinazolinone,
2-benzyl-4-quinazolinone,
2-(C6H5CCl2)-4-chloroquinazoline, and
2-propyl-4-chloroquinazoline. The Class III Heterocyclic Compounds are disclosed in Powers et al., U.S. Patent No. 4,847,202, the disclosure of which is hereby incorporated by reference.
Other substituted heterocyclic compounds have been prepared earlier for other purposes, such as 3-chloroisocoumarin, Davies and Poole, /. Chem. Soc, pp. 1616-1629 (1928); 3-chloro and 3,4-dichloroisocoumarin, Milevskaya, et al., Zhur. Org. KJiim., 9:2145-2149 (1973); 3-methyl and 4-carboxy-3-methylisocoumarin, Tirodkar and Usgaonkar, IncL J. Chem., 7:1114-1116 (1969); 7-nitro and 7-aminoisocoumarin,
Choksey and Usgaonkar, Ind J. Chem., 14B:596-598 (1976). The disclosures of all of the preceding articles are hereby incorporated by reference. These other substituted isocoumarins are also believed to exhibit Calpain inhibitory activity when used in accordance with the present invention. Still other substituted isocoumarins which have been prepared recently for inhibition of serine proteases are 3-chloroisocoumarin, Harper, et al., J. A. Chem. Soc, 105:6518-6520 (1983); 3,4-dichloroisocoumarin, Harper, et al., Biochemistry, 24:1831- 1841 (1985); 3-alkoxy-7-amino-4-chloroisocoumarin, Harper and Powers, /. Am. Chem. Soc, 106:7618-7619 (1984), Harper and Powers, Biochemistry, 24:7200-7213 (1983); additional substituted isocoumarins with basic groups (aminoalkoxy, guanidino or isothiureidoalkoxy), Kam, et al, Biochemistry, 27:2547-2557 (1988); 7-substituted 3- alkoxy-4-chloroisocoumarins, Powers, et al., J. Cell Biochem., 39:33-46 (1989) and Powers, et al. Biochemistry, 29:3108-3118 (1990). The disclosures of all of the preceding articles are hereby incorporated by reference. We believe that the foregoing compounds, which exhibit serine protease inhibitory activity, also exhibit Calpain inhibitory activity when used in accordance with the present invention. All of the foregoing isocoumarin compounds, including the Class I and II Substituted
Isocoumarins, the Class III Substituted Heterocyclic Compounds and the other substituted heterocyclic compounds useful in the practice of the present invention shal be referred to collectively hereinafter as the "Substituted Heterocyclic Compounds." The term "Substituted Heterocyclic Compound" shall be used to refer to any particula species of these compounds.
The preparation of the various Substituted Heterocyclic Compounds is illustrated by Examples SHC1-SHC9.
EXAMPLE SHC1 Preparation of 7-(phenylcarbamoylamino)-4-chloroisocoumarin was synthesize as previously described in Powers, et al., Biochemistry, 29:3108-3118 (1990). This compound (0.32 g, 1 mmole) was mixed with phenyl isocyanate (0.12g, 1 mmole) in 5 ml of THF and the reaction mixture was stirred at r.t. overnight. The product 7-(phenylcarbamoylamino)-4-chloro-3-(2-bromoethoxy)isocoumarin precipitated out, yield 40%, m.p. 215-217°C, mass spectrum m/e = 437.9 (M+)> Anal. Calc. for C18H14N204ClBr: C, 49.40; H, 3.22; N, 6.40; Cl, 8.10. Found: Q49.48; H, 3.25; N,6.3
Cl, 8.12. The phenylcarbamoylamino compound (0.1 g, 0.23 mmole) was heated with 0.02 g of thiourea (0.26 mmole) in 10 ml of THF at 70° C overnight. The final produ precipitated out, yield 0.04 g, 36%, m.p. 161-163° C (dec), mass spectrum (FAB + ) m = 433 (M-Br). Anal. Calc. for C19H18N4O4ClBrS:0.25 THF: C, 45.12; H, 3.86; N, 10.53; Cl, 6.67. Found: C, 44.83; H, 3.92; N, 10.12; Cl, 6.41.
7-(Ethylcarbamoylamino)-4-chloro-3-(2-isothiureidoethoxy)isocoumarin, 7-(t-butylcarbamoylamino)-4-chloro-3-(2-isothiureidoethoxy)isocoumarin, 7-(benzylthiocarbamoylamino)-4-chloro-3-(2-isothiureidoethoxy)isocoumarin, 7- (ethylthiocarbamoylamino)-4-chloro-3-(2-isothiureidoethoxy)isocoumarin, 7-(4- fluorobenzyl) thiocarbamoylamino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, and
(2,5-dimethylbenzyl) thiocarbamoylamino-4-chloro-3-(2-isothiureidoethoxy) isocoumar can be prepared by the same procedure.
EXAMPLE SHC2
Preparation of 7-(acetylamino)-4-chloro-3-(3-isothiureidopropoxy) isocoumari 7-Amino-3(3-bromopropoxy)-4-chloroisocoumarin was synthesized as previous described (Kam, et al., supra). This compound (0.33 g, 1 mmole) was heated with 0.1 g of acetic anhydride (1.5 mmole) in 20 ml of dry THF. After a few minutes, a yello
solid precipitated out. After 3 hrs, the solution was concentrated to 5 ml, and the solid was filtered to give 0.37 g of 7-(acetylamino)-4-chloro-3-(3-bromopropoxy) isocoumarin, m.p. 170-172°C; mass spectrum: m/e = 375 (M+). The acetylated isocoumarin (0.15 g, 0.4 mmole) was treated with thiourea (0.036 g, 0.47 mmole) to give 0.9 g of the final product, (yield 50%), m.p. 180-181°C, mass spectrum m/e = 370 (M+-Br). Anal. Calc. for C15H17N304ClBrS: C, 39.97; H, 3.80; N, 9.32; Cl 7.87. Found: C, 39.86; H 3.83; N, 9.29; Cl, 7.85.
7-trifluoroacetylamino-4-chloro-3-(3-isothiureidopropoxy) isocoumarin, 7- heptafluorobutyroylamino-4-chloro-3-(3-isothiureidopropoxy) isocoumarin, 7- succinylamino-4-chloro-3-(3-isothiureidopropoxy) isocoumarin, and 7-(o-phthalyl)amino-
4-chloro-3-(3-isothiureidopropoxy) isocoumarin can be prepared by the same procedure.
EXAMPLE SHC3 Preparation of 7-(benzylcarbamoylamino)-4-chloro-3-(3-isothiureidopropoxy) isocoumarin: 7-(benzylcarbamoylamino)-4-chloro-3(3-bromopropoxy) isocoumarin was prepared from the reaction of benzyl isocyanate with 7-amino-4-chloro-3-(3- bromopropoxy) isocoumarin as described above, m.p. 188-189° C, mass spectrum: m/e = 359 (M+ -benzyl). The final product was obtained from the reaction of 7- (benzylcarbamoylamino)-4-chloro-3-(3-bromopropoxy) isocoumarin with thiourea as described above (yield 74%), m.p. 165-166°C; mass spectrum (FAB + ) m/e = 461
(M+-Br). Anal. Calc. for C21H22N4O4ClBrS:0.75 THF: C, 48.36; H, 4.70; N, 9.40; Cl, 6.56. Found: C, 48.13; H, 4.87; N, 9.65; Cl, 6.15.
EXAMPLE SHC4 Preparation of 7-(phenylacetylamino)-4-chloro-3-(2-isothiureidoethoxy) isocoumarin:
7-Amino-4-chloro-3-(2-bromoethoxy) isocoumarin (0.15 g, 0.47 mmole) was first mixed with phenylacetyl chloride (0.09 g, 0.55 mmole) in 10 ml of THF, triethylamine (0.05 g, 0.47 mmole) was then added and the reaction mixture was stirred at r.t. overnight. After Et3NHCl salt was removed by filtration, the product 7- (phenylacetylamino)-4-chloro-3-(2-bromoethoxy) isocoumarin was crystallized from
THF and Pet. ether (yield, 73%), m.p. 165-169° C; mass spectrum; m/e = 436.7 (M+). The phenylacetyamino derivative (0.1 g) was heated with thiourea (0.02 g) to give the
product 0.05 g (yield, 40%), m.p. 115-120° C; mass spectrum (FAB + ) m/e = 432 (M+ -Br). Anal. Calc. for C20H19N3O4ClBrSO.5 H20: C 45.99; H, 3.83; N, 8.05; Cl, 6.80. Found: C, 46.09; H, 4.17; N, 8.02; Cl, 6.79.
EXAMPLE SHC5 Preparation of 7-(R-α-methylbenzylcarbamoylamino)-4-chloro-3-(2- isothiureidoethoxy) isocoumarin:
7-(R-α-methylbenzylcarbamoylamino)-4-chloro-3-(2-bromoethoxy) isocoumarin was synthesized in the same manner as described above, m.p. 183-185° C; mass spectrum m/e = 464 (M+). This compound (0.1 g) reacted with thiourea (0.02 g) under the same condition described above to form the final product 7-(R-α- methylbenzylcarbamoylamino)-4-chloro-3-(2-isothiureidoethoxy) isocoumarin (0.078 g), m.p. 143- 150° C; mass spectrum (FAB + ) m/e = 461 (M+ -Br). Anal. Calc. for C21H22N4O4ClBrS 0.5H2O: C, 45.75; H, 4.35; N, 10.17; Cl, 6.44. Found: C, 44.95; H, 4.31; N, 10.02; Cl, 6.36. EXAMPLE SHC6
Preparation of 7-(D-phenylalanylamino)-4-chloro-3 (2-isothiureidoethoxy) isocoumarin:
Boc-D-Phe (0.33 g, 1.2 mmole) reacted with 1,3-dicyclohexylcarbodiimide (0.13 g, 0.6 mmole) in 10 ml THF at 0°C for 1 hour to form the symmetric anhydride, and then 7-amino-4-chloro-3(2-bromoethoxy) isocoumarin (0.2g, 0.6 mmole) was added.
The reaction was stirred at r.t. overnight and the precipitate 7-(Boc-D-Phe-amino)-4- chloro-3-(2-bromoethoxy) isocoumarin was formed (0.29 g, 71%). TLC one spot, m.p. 180-182° C; mass spectrum m/3 = 566(M+). Anal. Calc. for C25H26N2OgClBr: C, 53.07; H, 4.63; N, 4.95; Cl 6.27. Found: C, 53.25: H, 4.66; N, 4.87; Cl, 6.24. Boc-D-Ph compound (0.2 g, 0.35 mmole) was reacted with thiourea (0.027 g, 0.35 mmole) in the same manner to give 7-(Boc-D-phenylalanylamino)-4-chloro-3-(2-isothiureidoethoxy) isocoumarin (0.14 g), yield 62%, mass spectrum (FAB + ) m/e = 561 (M+ -Br). This compound (0.1 g) was dissolved in 3 ml of THF at 0°C and then the solvent was evaporated to dryness. The final product precipitated out after addition of ether, one spot on TLC (CH3CN:H20:Ac0H = 8:1:1); mass spectrum (FAB + ) m/e = 462 (M+ -
Br -CF3C00).
7-Boc-alanylamino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, 7- benzoylamino-Ala-4-chloro-3(2-isothiureidoethoxy) isocoumarin, 7-benzoylamino-Phe-4- chloro-3-(2-isothiureidoethoxy) isocoumarin and 7-Boc-valylamino-4-chloro-3-(2- isothiureidoethoxy) isocoumarin can be prepared by the same procedure. EXAMPLE SHC7
Preparation of 7-(Boc-alanylalanylamino)-4-chloro-3-(2-isothiureidoethoxy) isocoumarin:
7-(Boc-alanylalanylamino)-4-chloro-3-(2-bromoethoxy) isocoumarin was synthesized in the same manner, m.p. 147-151°C; mass spectrum m/e = 561 (M+). Anal. Calc: C, 47.12: H, 4.85. Found: C, 47.18; H, 4.87. This compound (0.2 g) was reacted with thiourea (0.03 g) by the same procedure to form 7-(Boc- alanylalanylamino)-4-chloro-3-(2-isothiureidoethoxy) isocoumarin (0.04 g), mass spectrum m/e = 556 (M+ -Br).
7-(Alanylalanylamino)-4-chloro-3 (2-isothiureidoethoxy ) isocoumarin was prepared by deblocking of Boc-Ala-Ala-NH-CiTEtOIC with trifluoroacetic acid, mass spectrum (FAB+) m/e = 456 (M+ -Br -CF3COO).
EXAMPLE SHC8 Preparation of 7-(phenylthiocarbamoylamino)-4-chloro-3-(2-isothiureidoethoxy) isocoumarin: 7-(Phenylthiocarbamoylamino)-4-chloro-3-(2-bromoethoxy) isocoumarin was prepared from the reaction of phenyl isothiocyanate with 7-amino-4-chloro-3-(2- bromoethoxy) isocoumarin, yield 59%, m.p. 157-158° C; mass spectrum m/e = 361 (M+ -PhNH+l). Anal. Calc: C, 48.36; H, 3.39. Found: C, 48.26; H, 3.40. The bromoethoxy compound was then reacted with thiourea by the same procedure to give the final product, yield 32%; mass spectrum (FAB+) m/e 449 (M+ -Br).
EXAMPLE SHC9 Preparation of 7-(m-carboxyphenylthiocarbamoylamino)-4-chloro-3-(2- bromoethoxy) isocoumarin was prepared from the reaction of m-carboxyphenyl isothiocyanate with 7-amino-4-chloro-3-(2-bromoethoxy) isocoumarin, yield 64%, m.p. 157-158°C; mass spectrum m/e 361 (M+ -(COOH)PhNH+-Br).
7-(3-Fluorobenzoyl)amino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, 7-(3- nitrobenzoyl) amino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, 7-
diphenylacetylamino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, 7- diphenylpropionylamino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, 7-(p- toluenesulfonyl) amino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, and 7-(α- toluenesulfonyl) amino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin can be prepared from the reaction of corresponding 7-substituted-4-chloro-3-(2-bromoethoxy) isocoumarin with thiourea as described above. 7-substituted-4-chloro-3-(2- bromoethoxy) isocoumarin can be synthesized by reacting 7-amino-4-chloro-3-(2- bromoethoxy) isocoumarin with appropriate acid chloride or sulfonyl chloride in the presence of Et3N. 7-Ethoxycarbonylamino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, 7- benzyloxycarbonylamino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin, and 7- phenoxycarbonylamino-4-chloro-3-(2-isothiureidoethoxy) isocoumarin can be prepared from the reaction of 7-substituted-4-chloro-3-(2-bromoethoxy) isocoumarin with thiourea. 7-Ethoxycarbonylamino-4-chloro-3-(2-bromoethoxy) isocoumarin, 7- benzyloxycarbonylamino-4-chloro-3-(2-bromoethoxy) isocoumarin and 7- phenoxycarbonylamino-4-chloro-3-(2-bromoethoxy) isocoumarin can be synthesized by reacting 7-amino-4-chloro-3-(2-bromoethoxy) isocoumarin with the corresponding chloroformate.
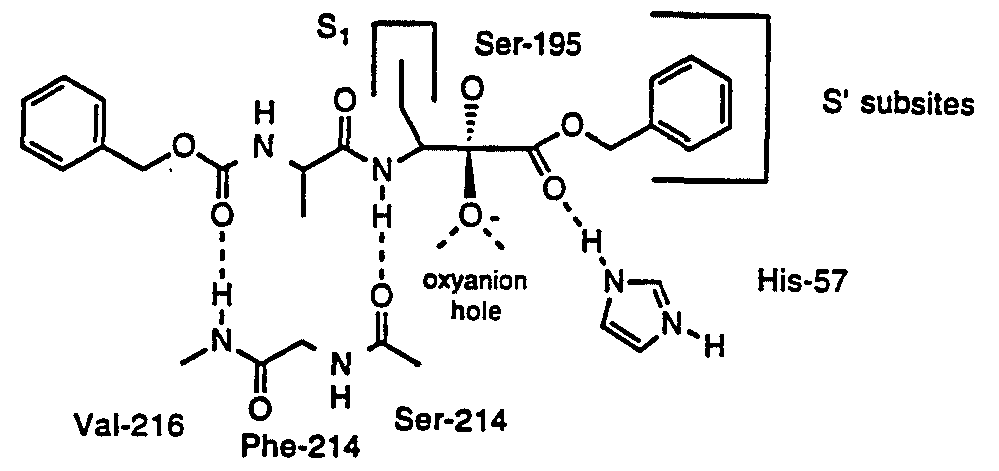
C. PEPTIDE KETO-COMPOUNDS Peptide α-ketoesters, peptide α-ketoacids, and peptide α-ketoamides are transition state analog inhibitors for serine proteases and cysteine proteases. While these subclasses of compounds are chemically distinguishable, for simplicity, all of these compounds will be referred to collectively herein as the "Peptide Keto-Compounds". The interactions of peptides with serine and cysteine proteases are designated herein using the nomenclature of Schechter and Berger, Biochem. Biophys. Res.
Commun., 27:157-162 (1967), incoφorated herein by reference. The individual amino acid residues of a substrate or inhibitor are designated PI, P2, etc. and the corresponding subsites of the enzyme are designated SI, S2, etc. The scissile bond of the substrate is Pl-Pl'. The primary recognition site of serine proteases is SI. The most important recognition subsites of cysteine proteases are SI and S2. There are additional recognition sites at the prime subsites such as SI' and S2'.
Amino acid residues and blocking groups are designated using standard abbreviations using nomenclature rules presented in /. Biol. Chem., 260:14-42 (1985), incoφorated herein by reference. An amino acid residue (AA) in a peptide or inhibitor structure refers to the part structure -NH-CHRj-CO-, where Rj is the side chain of the amino acid AA. A peptide α-ketoester residue would be designated
-AA-CO-OR which represents the part structure -NH-CHRrCO-CO-OR. Thus, the ethyl ketoester derived from benzoyl alanine would be designated Bz-Ala-CO-OEt which represents C6H5CO-NH-CHMe-CO-CO-OEt. Likewise, peptide ketoacid residues would be designated -AA-CO-OH. Further, peptide ketoamide residues are designated -AA-CO-NH-R. Thus, the ethyl keto amide derived from Z-Leu-Phe-OH would be designated Z-Leu-Phe-CO-NH-Et which represents C6H5CH2OCO-NH- CH(CH2CHMe2)-CO-NH-CH(CH2Ph)-CO-CO-NH-Et.
Peptide α-ketoesters containing amino acid residues with hydrophobic side chain at the PI site have also been found to be excellent inhibitors of several cysteine proteases including papain, cathepsin B and calpain. Calpains can be inhibited by peptide inhibitors having several different active groups. Structure-activity relationships with the commercially available in vitro inhibitors of Calpain, such as peptide aldehydes, have revealed that Calpains strongly prefer Leu or Val in the P2 position. These enzymes are inhibited by inhibitors having a wide variety of amino acids in the PI position, but are generally more effectively inhibited by inhibitors having amino acids with nonpolar or hydrophobic side chains in the PI position. Thus, we have discovered that another particular class of compounds exhibiting Calpain inhibitory activity, when used in accordance with the present invention, are the Peptide Keto-Compounds. These are compounds of the general structure:
O
M-(aa)n-C-Q-R
or a pharmaceutically acceptable salt, wherein:
M represents NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X-NH-CS-, X-NH-SOr, X-CO-, X-CS-, X-S02-, X-O-CO-, or X-O-CS-, H, acetyl, carbobenzoxy, succinyl, methyloxysuccinyl, butyloxycarbonyl;
X is selected from the group consisting of Cw alkyl, C^ fluoroalkyl, Cw alkyl substituted with J, C^ fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^ alkyl with an attached phenyl group, Cj.g alkyl with two attached phenyl groups, Cj.g alkyl with an attached phenyl group substituted with K, and C^ alkyl with two attached phenyl groups substituted with K;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, C^ alkoxy, Cj.g alkylamine, Cλ_6 dialkylamine, Cj_g alkyl-O-CO-,
Cw alkyl-O-CO-NH, and C 6 alkyl-S-;
K is selected from the group consisting of halogen, Cj.6 alkyl, Cl-6 perfluoroalkyl, C^g alkoxy, N02, CN, OH, C02H, amino, Cj. alkylamino, C2.1 dialkylamino, C^ acyl, and C^ alkoxy-CO-, and C^ alkyl-S-; aa represents a blocked or unblocked amino acid of the L or D configuration, preferably selected from the group consisting of: alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine (nle), norvaline (nva), alpha-aminobutyric acid (abu), epsilon-aminocaproic acid, citrulline, hydroxyproline, homoarginine, ornithine o sarcosine; n is a number from 1 to 20; Q is O or NH, R represents H, C^ alkyl, C1_6 fluoroalkyl, C^g chloroalkyl, benzyl, C-μ alkyl substituted with phenyl, C1_6 alkyl with an attached phenyl group substituted with K.
Thus, the Peptide Keto-Compounds can be divided into the Peptide Ketoesters Peptide Ketoacids and Peptide Ketoamides. Each of the compounds can also be classified based on the number of amino acids contained within the compound, such a an amino acid peptide, dipeptide, tripeptide, tetrapeptide, pentapeptide and so on.
We have found certain subclasses of Peptide α-Ketoester compounds to be particularly useful as Calpain Inhibitors when used in accordance with the present invention. These subclasses are referred to herein as the Dipeptide α-Ketoesters (Subclass A), the Dipeptide α-Ketoesters (Subclass B), the Tripeptide α-Ketoesters (Subclass A), the Tripeptide α-Ketoesters (Subclass B), the Tetrapeptide α-Ketoesters and the Amino Acid Peptide α-Ketoesters. All of these subclasses are considered to be to be within the class of Peptide Keto-Compounds.
The Dipeptide α-Ketoesters (Subclass A) are compounds of the formula: M1-AA2-AA1-CO-0-R1 or a pharmaceutically acceptable salt, wherein
M- represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-SO , X2N-S02-, X-CO-, X-CS-, X-SOr, X-O-CO-, or X- O-CS-;
X is selected from the group consisting of C^jg alkyl, 1 0 fluoroalkyl, C^. alkyl substituted with J, Cj.-g fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, 1 0 alkyl with an attached phenyl group, C^ alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, and C1 0 alkyl with two attached phenyl groups substituted with K, ^g alkyl with an attached phenoxy group, and C-^g alkyl with an attached phenoxy group substituted with K on the phenoxy group;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, Cj^Q alkoxy, C^ alkylamine, C2.12 dialkylamine, C^g alkyl-O-CO-, C^g alkyl-O-CO- NH-, and Cw0 alkyl-S-;
K is selected from the group consisting of halogen, C^g alkyl, C^ perfluoroalkyl, C-^g alkoxy, N02, CN, OH, C02H, amino, C^g alkylamino, C2_12 dialkylamino, C1-C10 acyl, and C^g alkoxy-CO-, and C^ alkyl-S-;
AAj is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, serine, threonine, cysteine, tyrosine, asparagine, glutamine,
aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta-alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2- azetidinecarboxylic acid, pipecolinic acid (2-piρeridine carboxylic acid), O-methylserin O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-
CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COO NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine; AA2 is a side chain blocked or unblocked amino acid with the L configuration,
D configuration, or no chirality at the α-carbon selected from the group consisting of leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspart acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta-alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2- azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O-methylserin O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COO NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine;
Rx is selected from the group consisting of H, C^ alkyl, C^g alkyl with a phenyl group attached to the C^g alkyl, and C-^g alkyl with an attached phenyl grou substituted with K.
The Dipeptide α-Ketoesters (Subclass B) are compounds of the structure: MrAA-NH-CHR2-CO-CO-0-R or a pharmaceutically acceptable salt, wherein
Mχ represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-SQ2-, X2N-SOr, X-CO-, X-CS-, X-S02-, X-O-CO-, or X
O-CS-;
X is selected from the group consisting of C-^g alkyl, C^ fluoroalkyl, Cj_10 alkyl substituted with J, l l0 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C-^g alkyl with an attached phenyl group, C^g alkyl with two attached phenyl groups, Cj.j alkyl with an attached phenyl group substituted with K, and Cj_10 alkyl with two attached phenyl groups substituted with K, C^g alkyl with an attached phenoxy group, and C^ alkyl with an attached phenoxy group substituted with K on the phenoxy group; J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2,
Cj.j alkoxy, Cωo alkylamine, C2.12 dialkylamine, Cj.jg alkyl-O-CO-, C^ alkyl-O-CO- NH-, and Cω0 alkyl-S-;
K is selected from the group consisting of halogen, C^ alkyl, C^ perfluoroalkyl, C1 0 alkoxy, N02, CN, OH, C02H, amino, C^g alkylamino, C2.12 dialkylamino, Cj-Cj acyl, and C^ alkoxy-CO-, and Cj.10 alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-naρthyl)-COOH,
NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine;
R2 represents C^g branched and unbranched alkyl, C 8 branched and unbranched cyclized alkyl, or C^g branched and unbranched fluoroalkyl;
R is selected from the group consisting of H, C^g alkyl, C^ alkyl with a phenyl group attached to the Cj_20 alkyl, and C^g alkyl with an attached phenyl grou substituted with K.
The Tripeptide α-Ketoesters (Subclass A) are compounds of the structure: M3-AA-AA-AA-CO-0-R or a pharmaceutically acceptable salt, wherein
M3 represents H, NH2-CO-, NH2-CS-, NH2-SOr, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-SOr, X2N-S02-, X-CO-, X-CS-, X-SO , T-O-CO-, or X- O-CS-; X is selected from the group consisting of Cj_10 alkyl, C^g fluoroalkyl, C1Λ0 alkyl substituted with J, C^g fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, C^j alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, and Cj.jg alkyl with two attached phenyl groups substituted with K, C 0 alkyl with an attached phenoxy group, and C]_10 alkyl with an attached phenoxy group substituted with K on the phenoxy group;
T is selected from the group consisting of C1Λ0 alkyl, C 0 fluoroalkyl, Cj.10 alkyl substituted with J, C^g fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C2.10 alkyl with an attached phenyl group, C^g alkyl with two attached phenyl groups, Cj_10 alkyl with an attached phenyl group substituted with K, and CM0 alkyl with two attached phenyl groups substituted with K;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH Cj^g alkoxy, C^g alkylamine, C^^ dialkylamine, C^JQ alkyl-O-CO-, C^g alkyl-O-C NH-, and Cwo alkyl-S-; ,
K is selected from the group consisting of halogen, C^g alkyl, C1_10 perfluoroalkyl, C^Q alkoxy, N02, CN, OH, C02H, amino, C-^g alkylamino, C2.12 dialkylamino, Cj^-C^ acyl, and C^ alkoxy-CO-, and C^g alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α -carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH,
NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine;
R is selected from the group consisting of H, C2.20 alkyl, Cwo alkyl with a phenyl group attached to the Cj_20 alkyl, and C^g alkyl with an attached phenyl group substituted with K.
The Tripeptide α-Ketoesters (Subclass B) are compounds of the structure: M3-AA-AA-NH-CHR2-CO-CO-0-R or a pharmaceutically acceptable salt, wherein M3 represents H, NH2-CO-, NH2-CS-, NH2-SOr, X-NH-CO-, X2N-CO-,
X-NH-CS-, X2N-CS-, X-NH-SOr, X2N-S02-, X-CO-, X-CS-, X-S02-, T-O-CO-, or X O-CS-;
X is selected from the group consisting of C^g alkyl, Cj.10 fluoroalkyl, C-^g alkyl substituted with J, C^g fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, Cj.jg alkyl with an attached phenyl group, Cj.jg alkyl with two attached phenyl groups, C^ alkyl with an attached phenyl group substituted with K, and Cj.jg alkyl with two attached phenyl groups substituted with K, C^ alkyl with an attached phenoxy group, and Cw0 alkyl with an attached phenoxy group substituted with K on the phenoxy group;
T is selected from the group consisting of C1Λ0 alkyl, x_w fluoroalkyl, C]_10 alkyl substituted with J, Cj.j fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C2.10 alkyl with an attached phenyl group, C1.10 alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, and Cj_10 alkyl with two attached phenyl groups substituted with K;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, C^g alkoxy, C^ alkylamine, C-^g dialkylamine, C^g alkyl-O-CO-, C^g alkyl-O-CO- NH-, and Cwo alkyl-S-;
K is selected from the group consisting of halogen, Cj.jg alkyl, C^jg perfluoroalkyl, Cl l0 alkoxy, N02, CN, OH, C02H, amino, Cu0 alkylamino, C2.12 dialkylamino, Cj-Cjg acyl, and C^g alkoxy-CO-, and C1.10 alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, configuration, or no chirality at the α -carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid
2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-
COOH, trifluoroleucine, and hexafluoroleucine;
R2 represents C^g branched and unbranched alkyl, Cj. branched and unbranched cyclized alkyl, or C^g branched and unbranched fluoroalkyl;
R is selected from the group consisting of H, C]_20 alkyl, C^g alkyl with a phenyl group attached to the C^g alkyl, and C^ alkyl with an attached phenyl group substituted with K.
The Tetrapeptide α-Ketoesters are compounds of the structure: M3-AA4-AA-AA-AA-CO-0-R or a pharmaceutically acceptable salt, wherein
M3 represents H, NH2-CO-, NH2-CS-, NH2-SOr, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-S02-, X2N-SO , X-CO-, X-CS-, X-SOr, T-O-CO-, or X-
O-CS-;
X is selected from the group consisting of 1 0 alkyl, Cj.jg fluoroalkyl, C^g alkyl substituted with J, C^g fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^jg alkyl with an attached phenyl group, i l0 alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, and Cj.jg alkyl with two attached phenyl groups substituted with K, 1 0 alkyl with an attached phenoxy group, and C^g alkyl with an attached phenoxy group substituted with K on the phenoxy group;
T is selected from the group consisting of C^JQ alkyl, Cj.j fluoroalkyl, C^ alkyl substituted with J, Cj_10 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C2.10 alkyl with an attached phenyl group, Cj.10 alkyl with two attached phenyl groups, Cl l0 alkyl with an attached phenyl group substituted with K, and CJ.JQ alkyl with two attached phenyl groups substituted with K;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, C1 0 alkoxy, C^g alkylamine, C2.12 dialkylamine, C1Λ0 alkyl-O-CO-, C^g alkyl-O-CO- NH-, and Cu0 alkyl-S-;
K is selected from the group consisting of halogen, ^g alkyl, C^ perfluoroalkyl, C-^g alkoxy, N02, CN, OH, C02H, amino, C^g alkylamino, C2 12 dialkylamino, C^Cjg acyl, and Cj.10 alkoxy-CO-, and C^j alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α -carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine,
glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-
CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine;; AA is a side chain blocked or unblocked amino acid with the L configuration,
D configuration, or no chirality at the α-carbon selected from the group consisting of leucine, isoleucine, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta-alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2- azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine;
R is selected from the group consisting of H, C^o alkyl, C^g alkyl with a phenyl group attached to the C1_20 alkyl, and C^g alkyl with an attached phenyl group substituted with K.
The Amino Acid Peptide α-Ketoesters are compounds of the structure:
MΓAA-CO-O-R or a pharmaceutically acceptable salt, wherein
M represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-S02-, X2N-SOr, Y-CO-, X-CS-, X-S02-, X-O-CO-, or X-
O-CS-;
X is selected from the group consisting of C-_1Q alkyl, C^ fluoroalkyl, C1.10 alkyl substituted with J, C^g fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, C^ alkyl with two attached phenyl groups, Cj_10 alkyl with an attached phenyl group substituted with K, and CJ.JQ alkyl with two attached phenyl groups substituted with K, C^g alkyl with an attached phenoxy group, and C^g alkyl with an attached phenoxy group substituted with K on the phenoxy group; Y is selected from the group consisting of Cg_10 alkyl, C^g fluoroalkyl, l W alkyl substituted with J, Cj_10 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, ^_10 alkyl with an attached phenyl group, C^ alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K and C^Q alkyl with two attached phenyl groups substituted with K;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, Cj.jg alkoxy, C^ alkylamine, C2.12 dialkylamine, lΛ0 alkyl-O-CO-, C _10 alkyl-O-CO- NH-, and C^g alkyl-S-; K is selected from the group consisting of halogen, 1Λ0 alkyl, C^g perfluoroalkyl, Cj.jg alkoxy, N02, CN, OH, C02H, amino, Cj.10 alkylamino, C2_12 dialkylamino, Cj-Cjg acyl, and C^j alkoxy-CO-, and C^jg alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α -carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH,
NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine;
R is selected from the group consisting of H, C^g alkyl, C^g alkyl with a phenyl group attached to the C^g alkyl, and C1 2o al yl with an attached phenyl group substituted with K.
A few amino acid and peptide ketoesters and ketoacids have been previously reported. Cornforth and Cornforth in /. Chem. Soc, 93-96 (1953), incoφorated herein by reference, report the synthesis of the ketoacids PhCH2CO-Gly-CO-OH and Ac-Gly-CO-OH upon hydrolysis of heterocyclic molecules. Charles et al. in /. Chem.
Soc. Perkin 1:1139-1146 (1980), incoφorated herein by reference, use ketoesters for th synthesis of bicyclic heterocycles. They report the synthesis of n-Bu-CO-Ala-CO-OEt, Pr-CO-Ala-CO-OEt, cyclopentyl-CO-Ala-CO-OEt, Pr-CO-Phg-CO-OEt, and Bz-Ala-CO-OEt. Hori et al. in Peptides: Structure and Function-Proceedings of the Nint American Peptide Symposium (Deber, Hruby, and Kopple, Eds., Pierce Chemical Co.), pp 819-822 (1985), incoφorated herein by reference, report Bz-Ala-CO-OEt, Bz-Ala-CO-OH, Z-Ala-Ala-Abu-CO-OEt, Z-Ala-Ala-Abu-CO-OBzl, and Z-Ala-Ala-Ala-Ala-CO-OEt (Abu = 2-aminobutanoic acid or a-aminobutyric acid) and report that these compounds inhibit elastase. Trainer in Trends Pharm. Sci. 8:303-307 (1987), incoφorated herein by reference, comments on one of these compounds.
Burkhart, J. et al in Tetrahedron Lett. 29:3433-3436 (1988), incoφorated herein by reference, report the synthesis of Z-Val-Phe-CO-OMe and Bz-Phe-CO-OMe.
Angelastro et al in /. Med Chem. 33:13-16 (1990), incorporated herein by reference, report some a-ketoesters which are inhibitors of calpain and chymotrypsin. Hu and Abeles in Arch. Biochem. Biophys. 281:271-274 (1990), incoφorated herein by reference, report some peptidyl a-ketoamides and a-ketoacids which are inhibitors of cathepsin B and papain. Peet et al in /. Med Chem. 33:394-407 (1990), incoφorated herein by reference, report some peptidyl a-ketoesters which are inhibitors of porcine pancreatic elastase, human neutrophil elastase, and rat & human neutrophil cathepsin G.
The following Peptide Ketoester compounds are representative of the Peptide Keto-Compounds found to be useful as Calpain inhibitors within the context of the present invention:
Bz-DL-Ala-COOEt Bz-DL-Ala-COOBzl
Bz-DL-Ala-COOnBu
Bz-DL-Phe-COOEt
Bz-DL-Ala-COOCH2-C6H4-CF3 (para)
Bz-DL-Arg-COOEt Bz-DL-Lys-COOEt
Z-Ala-DL-Ala-COOEt
Z-Ala-DL-Ala-COOBzl
Z-Ala-DL-Ala-COOnBu
MeO-Suc-Ala-DL-Ala-COOMe Z-Leu-Nva-COOEt
Z-Leu-Nle-COOEt
Z-Leu-Phe-COOEt
Z-Leu-Abu-COOEt
Z-Leu-Met-COOEt Z-Phe-DL-Phe-COOEt
H-Gly-DL-Lys-COOEt
H-Ala-DL-Lys-COOEt
H-Pro-DL-Lys-COOEt
H-Phe-DL-Lys-COOEt Z-Ala-Ala-DL-Ala-COOEt
Z-Ala-Pro-DL-Ala-COOEt
Z-Ala-Ala-DL-Abu-COOEt
Z-Ala-Ala-DL-Abu-COOBzl
Z-Ala-Ala-DL-Abu-COOCH2-C6H4-CF3 (para) MeO-Suc-Val-Pro-DL-Phe-COOMe
H-Leu-Ala-DL-Lys-COOEt
Z-Ala-Ala-Ala-DL-Ala-COOEt
MeO-Suc-Ala-Ala-Pro-DL-Abu-COOMe. Z-Leu-Phe-COOEt PhCO-Abu-COOEt (CH3)2CH(CH2)2CO-Abu-COOEt CH3CH2CH)2CHCO-Abu-COOEt
Ph(CH2)6CO-Abu-COOEt Z-Leu-4-Cl-Phe-COOEt Z-Leu-Leu-Abu-COOEt Z-Leu-Leu-Phe-COOEt 2-NapSOrLeu-Abu-COOEt
2-NapS02-Leu-Leu-Abu-COOEt Z-Leu-NLeu-C02Et Z-Leu-Phe-C02Bu Z-Leu-Abu-C02Bu Z-Leu-Phe-C02Bzl
Z-Leu-Abu-C02Bzl. We have found certain subclasses of Peptide Ketoacid Compounds to be particularly useful when used in accordance with the present invention. These are subclasses are the Dipeptide α-Ketoacids (Subclass A), the Dipeptide α-Ketoacids (Subclass B), the Tripeptide α-Ketoacids, the Tetrapeptide α-Ketoacids and the Amin
Acid peptide α-Ketoacids. All of these are considered to be within the class of Peptid Keto-Compounds.
The Dipeptide α-Ketoacids (Subclass A) are compounds of the structure: MrAA-NH-CHR2-CO-CO-OH or a pharmaceutically acceptable salt, wherein
Mt represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-SOr, X2N-SOr, X-CO-, X-CS-, X-SO , X-O-CO-, or X- O-CS-;
X is selected from the group consisting of Cj.jg alkyl, Cwo fluoroalkyl, C^ alkyl substituted with J, C^ fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl
trisubstituted with K, C^g alkyl with an attached phenyl group, Cj.10 alkyl with two attached phenyl groups, Cj_10 alkyl with an attached phenyl group substituted with K, C]_10 alkyl with two attached phenyl groups substituted with K, Cλ_10 alkyl with an attached phenoxy group, and C^g alkyl with an attached phenoxy group substituted with K on the phenoxy group;
J is selected from the group consisting of halogen, COOH, OH, CN Ν02, NH2> Cj.jg alkoxy, Cj.j alkylamine, C2.1 dialkylamine, C 0 alkyl-O-CO-, ^_10 alkyl-O-CO- NH-, and Cw0 alkyl-S-;
K is selected from the group consisting of halogen, C^ alkyl, Cj.10 perfluoroalkyl, C^ alkoxy, N02, CN, OH, C02H, amino, C 0 alkylamino, C2.12 dialkylamino, ^-C^ acyl, and C^g alkoxy-CO-, and Cj.10 alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-
CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine; R2 represents C1.g branched and unbranched alkyl, C^g branched and unbranched cyclized alkyl, or C^g branched and unbranched fluoroalkyl.
The Dipeptide α-Ketoacids (Subclass B) are compounds of the structure: M1-AA2-AAι-CO-OH or a pharmaceutically acceptable salt, wherein Mx represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-,
X-NH-CS-, X2N-CS-, X-NH-SO , X2N-S02-, X-CO-, X-CS-, X-SO , X-O-CO-, or X- O-CS-;
X is selected from the group consisting of C^j alkyl, Cj.jg fluoroalkyl, C^g alkyl substituted with J, C-^ fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, Cj.jg alkyl with an attached phenyl group, C1.10 alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, and Cj.jβ alkyl with two attached phenyl groups substituted with K, C1Λ0 alkyl with an attached phenoxy group, and Cl l0 alkyl with an attached phenoxy group substituted with K on the phenoxy group; J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2,
Cj.jg alkoxy, C^ alkylamine, C2.12 dialkylamine, C^g alkyl-O-CO-, Cj.jg alkyl-O-CO- NH-, and C1A0 alkyl-S-;
K is selected from the group consisting of halogen, Cj.jg alkyl, C^g perfluoroalkyl, Cl lQ alkoxy, N02, CN, OH, C02H, amino, C^g alkylamino, C2.12 dialkylamino, ^C^ acyl, and C^g alkoxy-CO-, and C _10 alkyl-S-;
AAj is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta-alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2- azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH,
NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine;
AA2 is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine,
glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-
CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine. The Tripeptide α-Ketoacids are compounds of the structure:
MrAA-AA-AA-CO-OH or a pharmaceutically acceptable salt, wherein
Mj represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-S02-, X2N-SOr, X-CO-, X-CS-, X-S02-, X-O-CO-, or X- O-CS-;
X is selected from the group consisting of Cj.10 alkyl, C^.g fluoroalkyl, C^g alkyl substituted with J, C1.10 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, C 0 alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, and C^g alkyl with two attached phenyl groups substituted with K, C^g alkyl with an attached phenoxy group, and C^JQ alkyl with an attached phenoxy group substituted with K on the phenoxy group; J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2,
Cj.jg alkoxy, C^g alkylamine, C^.^ dialkylamine, C]_10 alkyl-O-CO-, C^g alkyl-O-CO- NH-, and C 0 alkyl-S-;
K is selected from the group consisting of halogen, C^g alkyl, C-^g perfluoroalkyl, C^g alkoxy, N02, CN, OH, C02H, amino, C1.10 alkylamino, C2.12 dialkylamino, CrC10 acyl, and Cj_10 alkoxy-CO-, and C 10 alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of
alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid,
2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-
COOH, trifluoroleucine, and hexafluoroleucine.
The Tetrapeptide α-Ketoacids are compounds of the structure: MrAA-AA-AA-AA-CO-OH or a pharmaceutically acceptable salt, wherein Mχ represents H, NH2-CO-, NH2-CS-, NH2-SOr, X-NH-CO-, X2N-CO-,
X-NH-CS-, X2N-CS-, X-NH-S02-, X2N-SOr, YrCO-, X-CS-, X-S02-, X-O-CO-, or X- O-CS-;
X is selected from the group consisting of C 0 alkyl, Cj_10 fluoroalkyl, Cl i0 alkyl substituted with J, C1.10 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, ^g alkyl with an attached phenyl group, Cj.10 alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, and C^g alkyl with two attached phenyl groups substituted with K, C^g alkyl with an attached phenoxy group, and C^g alkyl with an attached phenoxy group substituted with K on the phenoxy group;
Y1 is selected from the group consisting of C2.10 alkyl, C^jg fluoroalkyl, C^jg alkyl substituted with J, C1.10 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, C^jg alkyl with two
attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, and Cj.jg alkyl with two attached phenyl groups substituted with K;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, Cj.jg alkoxy, C^g alkylamine, C2-_2 dialkylamine, C1.10 alkyl-O-CO-, C^g alkyl-O-CO- NH-, and Cw0 alkyl-S-;
K is selected from the group consisting of halogen, 1Λ0 alkyl, C^Q perfluoroalkyl, C^g alkoxy, N02, CN, OH, C02H, amino, C^g alkylamino, C2.12 dialkylamino, Cj-C^ acyl, and 1 0 alkoxy-CO-, and C^jg alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid,
2-azetidinecarboxylic acid, pipecolinic acid (2-pipεridine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2" CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-
COOH, trifluoroleucine, and hexafluoroleucine.
The Amino Acid Peptide α-Ketoacids are compounds of the structure: MrAA-CO-OH or a pharmaceutically acceptable salt, wherein Mχ represents H, NH2-CO-, NH2-CS-, NH2-SOr, X-NH-CO-, X2N-CO-,
X-NH-CS-, X2N-CS-, X-NH-S02-, X2N-S02-, Y2-CO-, X-CS-, X-S02-, X-O-CO-, or X- O-CS-;
X is selected from the group consisting of ^_10 alkyl, C^JQ fluoroalkyl, Cj.j alkyl substituted with J, C1.10 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, i 0 alkyl with an attached phenyl group, C^g alkyl with two
attached phenyl groups, C-^ alkyl with an attached phenyl group substituted with K, and Cj.j alkyl with two attached phenyl groups substituted with K, C^jg alkyl with an attached phenoxy group, and C^g alkyl with an attached phenoxy group substituted with K on the phenoxy group; Y2 is selected from the group consisting of Cj.j alkyl, Cj.jg fluoroalkyl, C^Q alkyl substituted with J, Cj.jg fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, Cj_10 alkyl with two attached phenyl groups, C^Q alkyl with an attached phenyl group substituted with K, and Cj.jg alkyl with two attached phenyl groups substituted with K;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, Cj.jg alkoxy, Cj_10 alkylamine, C2.12 dialkylamine, l0 alkyl-O-CO-, C1 0 alkyl-O-CO- NH-, and Cl ,10 alkyl-S-; K is selected from the group consisting of halogen, C 0 alkyl, Cj.10 perfluoroalkyl, Cj.jg alkoxy, N02, CN, OH, C02H, amino, C1 0 alkylamino, C2.12 dialkylamino, j-C1Q acyl, and C^g alkoxy-CO-, and C^g alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, alpha-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, -NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine.
The following Peptide Ketoacid compounds are representative of the Peptide Keto-Compounds found to be useful as Calpain inhibitors within the context of the present invention:
Bz-DL-Lys-COOH Bz-DL-Ala-COOH
Z-Leu-Phe-COOH Z-Leu-Abu-COOH. The peptide α-ketoamides are transition state analogue inhibitors for cysteine proteases, such as Calpain. We have found that Peptide α-ketoamides containing amino acid residues with hydrophobic side chains at the Pj site are excellent inhibitors of several cysteine proteases including calpain I and calpain II.
We have found six subclasses of the peptide ketoamides to be particularly effective in inhibiting Calpain. These subclasses are referred to herein as Dipeptide α-Ketoamides (Subclass A), Dipeptide α-Ketoamides (Subclass B), Dipeptide α-Ketoamides (Subclass C, Types 1 through 6), Tripeptide α-Ketoamides, Tetrapeptide α-Ketoamides and Amino Acid α-Ketoamides. All of these subclasses are considered herein to be within the class of Peptide Keto-Compounds.
The Dipeptide α-Ketoamides (Subclass A) have the following structural formula: MrAA-NH-CHR2-CO-CO-NR3R4 or a pharmaceutically acceptable salt, wherein
Mj represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-SO , X2N-S02-, X-CO-, X-CS-, X-S02-, X-O-CO-, or X- O-CS-; X is selected from the group consisting of C1.10 alkyl, C^-g fluoroalkyl, C^ alkyl substituted with J, Cj_10 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, C1.10 alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K,
C-^Q alkyl with two attached phenyl groups substituted with K, Cj_10 alkyl with an
attached phenoxy group, and C^g alkyl with an attached phenoxy group substituted with K on the phenoxy group;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, CJ.JO alkoxy, C1 0 alkylamine, C2.12 dialkylamine, C^g alkyl-O-CO-, Cj_10 alkyl-O-CO- NH-, and Cuo alkyl-S-;
K is selected from the group consisting of halogen, C^ alkyl, 1 0 perfluoroalkyl, C1 0 alkoxy, N02, CN, OH, C02H, amino, C^g alkylamino, C2.12 dialkylamino, Cj-Cj acyl, C1 0 alkoxy-CO-, and C^g alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, α-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid
2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, α-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-
COOH, trifluoroleucine, and hexafluoroleucine;
R2 is selected from the group consisting of C^g branched and unbranched alkyl Cj. branched and unbranched cyclized alkyl, and C-^g branched and unbranched fluoroalkyl; R3 and R4 are selected independently from the group consisting of H, C^ alkyl, C1 2Q cyclized alkyl, C^g alkyl with a phenyl group attached to the C1.20 alkyl, C-l-20 cyclized alkyl with an attached phenyl group, Cj.20 alkyl with an attached phenyl group substituted with K, C1.20 alkyl with an attached phenyl group disubstituted with K, Cj.20 alkyl with an attached phenyl group trisubstituted with K, Cj_20 cyclized alkyl with an attached phenyl group substituted with K, C^g alkyl with a moφholine [-
N(CH2CH2)0] ring attached through nitrogen to the alkyl, C^ alkyl with a piperidin ring attached through nitrogen to the alkyl, ^_-0 alkyl with a pyrrolidine ring attached
through nitrogen to the alkyl, C^ alkyl with an OH group attached to the alkyl, - CH2CH2OCH2CH2OH, C^g with an attached 4-pyridyl group, C^g with an attached 3-pyridyl group, CJ.JQ with an attached 2-pyridyl group, C^Q with an attached cyclohexyl group, -NH-CH2CH2-(4-hydroxyphenyl), and -NH-CH2CH2-(3-indolyl). The Dipeptide α-Ketoamides (Subclass B) have the following structural formula:
M1-AA2-AA1-CO-NR3R4 or a pharmaceutically acceptable salt, wherein
Mj represents H, NH2-CO-, NH2-CS-, NR2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-S02-, X2N-SOr, X-CO-, X-CS-, X-S02-, X-O-CO-, or X- O-CS-;
X is selected from the group consisting of C^jg alkyl, Cw0 fluoroalkyl, 1 0 alkyl substituted with J, C^g fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, Cj.j alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, Cj^g alkyl with two attached phenyl groups substituted with K, Cj.jg alkyl with an attached phenoxy group, and C^g alkyl with an attached phenoxy group substituted with K on the phenoxy group; J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2,
C^g alkoxy, Cj.jg alkylamine, C2.12 dialkylamine, Cj.j alkyl-O-CO-, C 0 alkyl-O-CO- NH-, and CM0 alkyl-S-;
K is selected from the group consisting of halogen, Cj.jg alkyl, C^_10 perfluoroalkyl, C1.10 alkoxy, N02, CN, OH, C02H, amino, CM0 alkylamino, C2_12 dialkylamino, Cj-Cjg acyl, and ClA0 alkoxy-CO-, and Cj.jg alkyl-S-;
AAj is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta-alanine, norleucine, norvaline, α-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-
azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, α-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-
COOH, trifluoroleucine, and hexafluoroleucine;
AA2 is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, α-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2"
CH(CH2CHEt2)-COOH, α-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine; R3 and R are selected independently from the group consisting of H, C^g alkyl, C^ cyclized alkyl, C^g alkyl with a phenyl group attached to the C^g alkyl, Cj.20 cyclized alkyl with an attached phenyl group, C1 20 alkyl with an attached phenyl group substituted with K, C^ alkyl with an attached phenyl group disubstituted with K, C^g alkyl with an attached phenyl group trisubstituted with K, C^g cyclized alkyl with an attached phenyl group substituted with K, C^Q alkyl with a moφholine
[-N(CH2CH2)0] ring attached through nitrogen to the alkyl, Cj_10 alkyl with a piperidine ring attached through nitrogen to the alkyl, C1A0 alkyl with a pyrrolidine rin attached through nitrogen to the alkyl, C^Q alkyl with an OH group attached to the alkyl, -CH2CH2OCH2CH2OH, Cj_10 with an attached 4-pyridyl group, C^ with an attached 3-pyridyl group, C^g with an attached 2-pyridyl group, Cj.10 with an attached cyclohexyl group, -NH-CH2CH2-(4-hydroxyphenyl), and -NH-CH2CH2-(3-indolyl).
The Dipeptide α-Ketoamides (Subclass C, Type 1) have the following structural formula:
M1CO-AA2-AA1-CO-NH-CH2CH(OH)-R1 or a pharmaceutically acceptable salt, wherein Mj is selected from the group consisting of Cl4 alkyl monosubstituted with phenyl, C1-4 alkyl disubstituted with phenyl, C^ alkyl monosubstituted with 1-naphthyl, C1-4 alkyl monosubstituted with 2-naphthyl, C1- alkoxy monosubstituted with phenyl, CM alkoxy disubstituted with phenyl, ArCH20-, rO-, ArCH2NH-, and ArNH-; wherein Ar is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, 1-naphthyl, 1-naphthyl monosubstituted with J, 2-naphthyl, and 2-naphthyl monosubstituted with J;
J is selected from the group consisting of halogen, OH, CN, N02, NH2, COOH, C02Me, C02Et, CF3, C1-4 alkoxy, CM alkylamine, C2.8 dialkylamine, Cw perfluoroalkyl, and -N(CH2CH2)20; AA2 is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid,
NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
AAj is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, arginine, lysine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH,
NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
Rj is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, phenyl trisubstituted with J, pentafluorophenyl,
1-naphthyl, 1-naphthyl monosubstituted with J, 1-naphthyl disubstituted with J,
2-naphthyl, 2-naphthyl monosubstituted with J, 2-naphthyl disubstituted with J, 2-pyridyl, 2-quinolinyl, and 1-isoquinolinyl;
R2 represents C^ alkyl substituted with phenyl, phenyl and phenyl substituted with J. Dipeptide α-Ketoamides (Subclass C, Type 2) have the following structural formula:
M1CO-AA2-AA1-CO-NH-(CH2)n-R3 or a pharmaceutically acceptable salt, wherein
Mχ is selected from the group consisting of Cw alkyl monosubstituted with phenyl, C^ alkyl disubstituted with phenyl, Cw alkyl monosubstituted with 1-naphthyl,
Cw alkyl monosubstituted with 2-naphthyl, Cw alkoxy monosubstituted with phenyl, CM alkoxy disubstituted with phenyl, ArCH20-, ArO-, ArCH2NH-, and ArNH-; wherein Ar is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, 1-naphthyl, 1-naphthyl monosubstituted with J, 2-naphthyl, and 2-naphthyl monosubstituted with J;
J is selected from the group consisting of halogen, OH, CN, N02, NH2, COOH, C02Me, C02Et, CF3, C1^} alkoxy, C1-4 alkylamine, C2.8 dialkylamine, C1_4 perfluoroalkyl, and -N(CH2CH2)20;
AA2 is an amino acid with the L configuration, D c" duration, or DL configuration at the a-carbon selected from the group con g of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid,
O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
AAj is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, arginine, lysine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH,
5,5,5-trifluoroleucine, and hexafluoroleucine; n = 1-3;
R3 is selected from the group consisting of 2-furyl, 2-furyl monosubstituted with J, 2-pyridyl, 2-pyridyl monosubstituted with J, 3-pyridyl, 3-pyridyl monosubstituted with J, 4-pyridyl, 4-pyridyl monosubstituted with J, 2-quinolinyl, 2-quinolinyl monosubstituted with J, 1-isoquinolinyl, 1-isoquinolinyl monosubstituted with J,
-.ς -(CH2)4CONH(CH2)2N
Dipeptide α-Ketoamides (Subclass C, Type 3) have the following structural formula:
M3-(CH2)q-CO-AA2-AA1-CO-NH-CH2CH(OH)-R1 or a pharmaceutically acceptable salt, wherein M3 is selected from the group consisting of 2-furyl, 2-tetrahydrofuryl, 2-pyridyl,
3-pyridyl, 4-pyridyl, 2-pyrazinyl, 2-quinolinyl, 1-tetrahydroquinolinyl, 1-isoquinolinyl, 2-tetrahydroisoquinolinyl, and -N(CH2CH2)20; q = 0-2;
AA2 is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH,
NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
AAχ is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, arginine, lysine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH,
NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
Rj is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, phenyl trisubstituted with J, pentafluorophenyl,
l-naphthyl, 1-naphthyl monosubstituted with J, 1-naphthyl disubstituted with J, 2-naphthyl 2-naphthyl monosubstituted with J, 2-naphthyl disubstituted with J, 2-pyridyl, 2-quinolinyl, and 1-isoquinolinyl;
R2 represents Cl4 alkyl substituted with phenyl, phenyl and phenyl substituted with J.
J is selected from the group consisting of halogen, OH, CN, N02, NH2, COOH, C02Me, C02Et, CF3, Cw alkoxy, C1- alkylamine, C2.8 dialkylamine, C1_4 perfluoroalkyl, and N(CH2CH2)20;
Dipeptide α-Ketoamides (Subclass C, Type 4) have the following structural formula:
M3-(CH2)q-CO-AA2-AArCO-NH-(CH2)n-R3 or a pharmaceutically acceptable salt, wherein M3 is selected from the group consisting of 2-furyl, 2-tetrahydrofuryl, 2-pyridyl,
3-pyridyl, 4-pyridyl, 2-pyrazinyl, 2-quinolinyl, 1-tetrahydroquinolinyl, 1-isoquinolinyl, 2-tetrahydroisoquinolinyl, and -N(CH2CH2)20; q = 0-2;
AA2 is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-cH(CH2 HEt2)" OOH' alpha-aminoheptanoic acid, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH,
NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
AAj is an amino acid with the L configuration, D configuration, or DL configuration at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, arginine, lysine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH,
5,5,5-trifluoroleucine, and hexafluoroleucine; n = 1-3;
R3 is selected from the group consisting of 2-furyl, 2-furyl monosubstituted with J, 2-pyridyl, 2-pyridyl monosubstituted with J, 3-pyridyl, 3-pyridyl monosubstituted with J, 4-pyridyl, 4-pyridyl monosubstituted with J, 2-quinolinyl, 2-quinolinyl monosubstituted with J, 1-isoquinolinyl, 1-isoquinolinyl monosubstituted with J,
H
2)
4CONH(CH
2)
2 J is selected from the group consisting of halogen, OH, CN, N0
2, NH
2, COOH, C0
2Me, C0
2Et, CF
3, C^ alkoxy, C
w alkylamine, C
2.
8 dialkylamine, C
M perfluoroalkyl, and N(CH
2CH
2)
20;
Dipeptide α-Ketoamides (Subclass C, Type 5) have the following structural formula:
M4-(CH2)q-0-CO-AA2-AA1-CO-NH-CH2CH(OH)-R1 or a pharmaceutically acceptable salt, wherein M4 is selected from the group consisting of 2-furyl, 2-tetrahydrofuryl, 2-pyridyl,
2-pyrazinyl, 2-quinolinyl, 2-tetrahydroquinolinyl, 1-isoquinolinyl, and 1-tetrahydroisoquinolinyl; q = 0-2;
AA2 is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH,
NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
AAj^ is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, arginine, lysine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH,
NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
Rj is selected from the group consisting of phenyl, phenyl monosubstituted wit J, phenyl disubstituted with J, phenyl trisubstituted with J, pentafluorophenyl,
1-naphthyl, 1-naphthyl monosubstituted with J, 1-naphthyl disubstituted with J, 2-naphthyl 2-naphthyl monosubstituted with J, 2-naphthyl disubstituted with J, 2-pyridyl, 2-quinolinyl, and 1-isoquinolinyl;
R2 represents C1_4 alkyl substituted with phenyl, phenyl and phenyl substituted with J.
J is selected from the group consisting of halogen, OH, CN, N02, NH2, COOH C02Me, C02Et, CF3, ClJ} alkoxy, C1_4 alkylamine, C2.8 dialkylamine, C1-4 perfluoroalkyl, and N(CH2CH2)20;
Dipeptide α-Ketoamides (Subclass C, Type 6) have the following structural formula:
M4-(CH2)q-0-CO-AA2-AArCO-NH-(CH2)n-R3 or a pharmaceutically acceptable salt, wherein M4 is selected from the group consisting of 2-furyl, 2-tetrahydrofuryl, 2-pyridyl,
2-pyrazinyl, 2-quinolinyl, 2-tetrahydroquinolinyl, 1-isoquinolinyl, and 1-tetrahydroisoquinolinyl; q = 0-2;
AA2 is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH,
NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
AAj is an amino acid with the L configuration, D configuration, or DL configuration at the a-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, histidine, methionine, methionine sulfoxide, phenylalanine, arginine, lysine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, phenylglycine, norleucine, norvaline, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH,
5,5,5-trifluoroleucine, and hexafluoroleucine; n = 1-3;
R3 is selected from the group consisting of 2-furyl, 2-furyl monosubstituted with J, 2-pyridyl, 2-pyridyl monosubstituted with J, 3-pyridyl, 3-pyridyl monosubstituted with J, 4-pyridyl, 4-pyridyl monosubstituted with J, 2-quinolinyl, 2-quinolinyl monosubstituted with J, 1-isoquinolinyl, 1-isoquinolinyl monosubstituted with J,
J is selected from, the group consisting of halogen, OH, CN, N02, NH2, COOH, C02Me, C02Et, CF3, C1_4 alkoxy, C^ alkylamine, C2.8 dialkylamine, C-^ perfluoroalkyl, and N(CH2CH2)20.
The Tripeptide α-Ketoamides have the following structural formula: MrAA-AA-AA-CO-NR3R4 or a pharmaceutically acceptable salt, wherein
M1 represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-S02-, X2N-SO , X-CO-, X-CS-, X-S02-, X-O-CO-, or X-
O-CS-;
X is selected from the group consisting of C^g alkyl, C1.10 fluoroalkyl, C^g alkyl substituted with J, Cl l0 fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, C^g alkyl with two attached phenyl groups, C^g alkyl with an attached phenyl group substituted with K, Cj.jg alkyl with two attached phenyl groups substituted with K, C^g alkyl with an attached phenoxy group, and C^ alkyl with an attached phenoxy group substituted with K on the phenoxy group;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, Cωo alkoxy, 10 alkylamine, C2.12 dialkylamine, C-^g alkyl-O-CO-, C^g alkyl-O-CO- NH-, and C^g alkyl-S-;
K is selected from the group consisting of halogen, Cj.jg alkyl, C^jg perfluoroalkyl, C1 0 alkoxy, N02, CN, OH, C02H, amino, C1 o alkylamino, C2.12 dialkylamino, Cj-Cj acyl, and Cj.j alkoxy-CO-, and C^g alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, α-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid, 2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-
CH(CH2CHEt2)-COOH, α-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-
cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)- COOH, trifluoroleucine, and hexafluoroleucine;
R3 and R4 are selected independently from the group consisting of H, C^ alkyl, Cj.^ cyclized alkyl, C^g alkyl with a phenyl group attached to the C^ alkyl, C-i-20 cyclized alkyl with an attached phenyl group, Cj_20 alkyl with an attached phenyl group substituted with K, C^ alkyl with an attached phenyl group disubstituted with K, Cj.20 alkyl with an attached phenyl group trisubstituted with K, C-^g cyclized alkyl with an attached phenyl group substituted with K, C-^g alkyl with a moφholine [-N(CH2CH2)0] ring attached through nitrogen to the alkyl, Cj_10 alkyl with a piperidine ring attached through nitrogen to the alkyl, Cj^g alkyl with a pyrrolidine ring attached through nitrogen to the alkyl, Cι_20 alkyl with an OH group attached to the alkyl, -CH2CH2OCH2CH2OH, C^g with an attached 4-pyridyl group, C^g with an attached 3-pyridyl group, CJ.JQ with an attached 2-pyridyl group, C 0 with an attached cyclohexyl group, -NH-CH2CH2-(4-hydroxyphenyl), and -NH-CH2CH2-(3-indolyl). The Tetrapeptide α-Ketoamides have the following structural formula:
M1-AA-AA-AA-AA-CO-NR3R4 or a pharmaceutically acceptable salt, wherein
M1 represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-SOr, X2N-SOr, X-CO-, X-CS-, X-SOr, X-O-CO-, or X- O-CS-;
X is selected from the group consisting of C .jg alkyl, CM0 fluoroalkyl, C^Q alkyl substituted with J, C^g fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, C^g alkyl with an attached phenyl group, C1.10 alkyl with two attached phenyl groups, Cj_10 alkyl with an attached phenyl group substituted with K, C^g alkyl with two attached phenyl groups substituted with K, C g alkyl with an attached phenoxy group, and ^j alkyl with an attached phenoxy group substituted with K on the phenoxy group; J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2,
Cj.10 alkoxy, C^g alkylamine, C2.12 dialkylamine, C^g alkyl-O-CO-, Cχ_w alkyl-O-CO- NH-, and Cw0 alkyl-S-;
K is selected from the group consisting of halogen, C^g alkyl, Cj.10 perfluoroalkyl, Cj.j alkoxy, N02, CN, OH, C02H, amino, C^g alkylamino, C2.12 dialkylamino, Cj-Cjg acyl, and C^ alkoxy-CO-, and C^ alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, α-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid,
2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, α-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-
COOH, trifluoroleucine, and hexafluoroleucine;
R3 and R4 are selected independently from the group consisting of H, C^g alkyl, CJ.-Q cyclized alkyl, C^g alkyl with a phenyl group attached to the C1 2β alkyl, ^1-20 cyclized alkyl with an attached phenyl group, C^o alkyl with an attached phenyl group substituted with K, C^g alkyl with an attached phenyl group disubstituted with
K, Cj.jg alkyl with an attached phenyl group trisubstituted with K, Cj.20 cyclized alkyl with an attached phenyl group substituted with K, C^g alkyl with a moφholine [-N(CH2CH2)0] ring attached through nitrogen to the alkyl, C1 Q alkyl with a piperidine ring attached through nitrogen to the alkyl, C^ alkyl with a pyrrolidine ring attached through nitrogen to the alkyl, C^ alkyl with an OH group attached to the alkyl, -CH2CH2OCH2CH2OH, C1.10 with an attached 4-pyridyl group, C^g with an attached 3-pyridyl group, C^g with an attached 2-pyridyl group, C^g with an attached cyclohexyl group, -NH-CH2CH2-(4-hydroxyphenyl), and -NH-CH2CH2-(3-indolyl). The Amino Acid α-Ketoamides have the following structural formula: MrAA-CO-NR3R4 or a pharmaceutically acceptable salt, wherein
M. represents H, NH2-CO-, NH2-CS-, NH2-S02-, X-NH-CO-, X2N-CO-, X-NH-CS-, X2N-CS-, X-NH-S02-, X2N-SOr, X-CO-, X-CS-, X-SOr, X-O-CO-, or X- O-CS-;
X is selected from the group consisting of Cj.10 alkyl, C^g fluoroalkyl, C^g alkyl substituted with J, C^ fluoroalkyl substituted with J, 1-admantyl, 9-fluorenyl, phenyl, phenyl substituted with K, phenyl disubstituted with K, phenyl trisubstituted with K, naphthyl, naphthyl substituted with K, naphthyl disubstituted with K, naphthyl trisubstituted with K, Cj_10 alkyl with an attached phenyl group, Cj.j alkyl with two attached phenyl groups, C^ alkyl with an attached phenyl group substituted with K, C1-io alkyl with two attached phenyl groups substituted with K, C^Q alkyl with an attached phenoxy group, and CJ^Q alkyl with an attached phenoxy group substituted with K on the phenoxy group;
J is selected from the group consisting of halogen, COOH, OH, CN, N02, NH2, C1 0 alkoxy, C-^g alkylamine, C2.12 dialkylamine, C 0 alkyl-O-CO-, Cl 0 alkyl-O-CO- NH-, and C1AQ alkyl-S-;
K is selected from the group consisting of halogen, C1_10 alkyl, C w perfluoroalkyl, CJ.JQ alkoxy, N02, CN, OH, C02H, amino, C1A0 alkylamino, C2.12 dialkylamino, Cj-Cjg acyl, and C^jg alkoxy-CO-, and Cj.10 alkyl-S-;
AA is a side chain blocked or unblocked amino acid with the L configuration, D configuration, or no chirality at the α-carbon selected from the group consisting of alanine, valine, leucine, isoleucine, proline, methionine, methionine sulfoxide, phenylalanine, tryptophan, glycine, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, phenylglycine, beta- alanine, norleucine, norvaline, α-aminobutyric acid, epsilon-aminocaproic acid, citrulline, hydroxyproline, ornithine, homoarginine, sarcosine, indoline 2-carboxylic acid,
2-azetidinecarboxylic acid, pipecolinic acid (2-piperidine carboxylic acid), O- methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2- CH(CH2CHEt2)-COOH, α-aminoheptanoic acid, NH2-CH(CH2-l-napthyl)-COOH, NH2-CH(CH2-2-napthyl)-COOH, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2- cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-
COOH, trifluoroleucine, and hexafluoroleucine;
R3 and R are selected independently from the group consisting of H, C1.20 alkyl, C1 2Q cyclized alkyl, Cl 20 alkyl with a phenyl group attached to the C^g alkyl, C--1-20 cyclized alkyl with an attached phenyl group, C^g alkyl with an attached phenyl group substituted with K, C^Q alkyl with an attached phenyl group disubstituted with K, Cj^ alkyl with an attached phenyl group trisubstituted with K, Cj.2 cyclized alkyl with an attached phenyl group substituted with K, Cj.j alkyl with a moφholine [-N(CH2CH2)0] ring attached through nitrogen to the alkyl, Cl l0 alkyl with a piperidine ring attached through nitrogen to the alkyl, l W alkyl with a pyrrolidine ring attached through nitrogen to the alkyl, C^g alkyl with an OH group attached to the alkyl, -CH2CH2OCH2CH2OH, C^g with an attached 4-pyridyl group, C 0 with an attached 3-pyridyl group, C^g with an attached 2-pyridyl group, Cj.jg with an attached cyclohexyl group, -NH-CH2CH2-(4-hydroxyphenyl), and -NH-CH2CH2-(3-indolyl).
, The Applicants are aware of only a single peptide ketoamide reported in the literature. This compound is Z-Phe-NHCH2CO-CO-NH-Et (Z-Phe-Gly-CO-NH-Et). The compound is reported by Hu and Abeles (supra) to be an inhibitor of papain (Kj =
1.5 mM) and cathepsin B (K_j = 4 mM).
The following Peptide Ketoamide compounds are representative of the Peptide Keto-Compounds found to be useful as Calpain inhibitors within the context of the present invention: Z-Leu-Phe-CONH-Et
Z-Leu-Phe-CONH-nPr Z-Leu-Phe-CONH-nBu Z-Leu-Phe-CONH-iBu Z-Leu-Phe-CONH-Bzl Z-Leu-Phe-CONH-(CH2)2Ph
Z-Leu-Abu-CONH-Et Z-Leu-Abu-CONH-nPr Z-Leu-Abu-CONH-nBu Z-Leu-Abu-CONH-iBu Z-Leu-Abu-CONH-Bzl
Z-Leu-Abu-CONH-(CH2)2Ph Z-Leu-Abu-CONH-(CH2)3-N(CH2CH2)20
Z-Leu-Abu-CONH-(CH2)7CH3 Z-Leu-Abu-CONH-(CH2)2OH Z-Leu-Abu-CONH-(CH2)20(CH2)2OH Z-Leu-Abu-CONH-(CH2)17CH3 5 Z-Leu-Abu-CONH-CH2-C6H3[3,5-(OCH3)2]
Z-Leu-Abu-CONH-CH2-C4H4N Z-Leu-Abu-CONH-(CH2)5OH Z-Leu-Abu-CONH-CH2CH(OCH3)2 Z-Leu-Abu-CONH-CH2CH(OC2H5)2
10 Z-Leu-Abu-CONH-CH2-CgHg[l,3,3-(CH3)3-5-OH]
Z-Leu-Abu-CONH-(CH2)2C6H4(4-OH) Z-Leu-Abu-CONH-(CH2)2CgH4(2-OCH3) Z-Leu-Abu-CONH-(CH2)2C6H4(3-OCH3) Z-Leu-Abu-CONH-(CH2)2C6H4(4-OCH3)
15 Z-Leu-Abu-CONH-CH2CH(OH)Ph
Z-Leu-Abu-CONH-CH2CH(OH)C6H4(4-OCH3) Z-Leu-Abu-CONH-CH2CH(OH)C6H2[2,4,6-(OCH3)3] Z-Leu-Abu-CONH-CH2CH(OH)C6H4[4-N(CH3)2] Z-Leu-Abu-CONH-CH2CH(OH)C6F5
20 Z-Leu-Abu-CONH-CH2CH(OH)C6H4(3-CF3)
Z-Leu-Abu-CONH-CH2CH(OH)C6H4(3-OPh) Z-Leu-Abu-CONH-CH2CH(OH)C6H4(4-OPh) Z-Leu-Abu-CONH-CH2CH(OH)C6H4(4-OCH2Ph) Z-Leu-Abu-CONH-CH2CH(OH)C6H4-3-OC6H4(3-CF3)
25 Z-Leu-Abu-CONH-CH2CH(OH)C6H4-3-OCgH3(3,4-Cl2)
Z-Leu-Abu-CO-NH-CH2CH(OH)C6H3[3,4-(OCH2Ph)2] Z-Leu-Abu-.CONH-CH2CH(OH)-1-C10H7 Z-Leu-Abu-CONH-CH2CH(OH)-2-C10H7 Z-Leu-Phe-CONH-CH2CH(OH)Ph
30 Z-Leu-Phe-CONH-CH2CH(OH)C6H4[4-N(CH3)2]
Z-Leu-Phe-CONH-CH2CH(OH)C6F5 Z-Leu-Phe-CONH-CH2CH(OH)C6H4(3-CF3)
Z-Leu-Phe-CONH-CH2CH(OH)C6H4(3-OPh) Z-Leu-Phe-CONH-CH2CH(OH)C6H4(4-OPh) Z-Leu-Phe-CONH-CH2CH(OH)C6H4(4-OCH2Ph) Z-Leu-Phe-CONH-CH2CH(OH)CgH4-3-OC6H4(3-CF3) 5 Z-Leu-Phe-CONH-CH2CH(OH)C6H4-3-OCgH3(3,4-Cl2)
Z-Leu-Phe-CONH-CH2CH(OH)C6H3(3,4-(OCH2Ph)2) Z-Leu-Abu-CONH-CH2-2-furyl Z-Leu-Abu-CONH-CH2-2-tetrahydrofuryl Z-Leu-Abu-CONH-CH2-2-pyridyl
10 Z-Leu-Abu-CONH-CH2-3-pyridyl
Z-Leu-Abu-CONH-CH2-4-pyridyl Z-Leu-Abu-CONH-(CH2)2-2-pyridyl Z-Leu-Abu-CONH-CH2-2-pyridyl(3-COOCH3) Z-Leu-Abu-CONH-CH2-2-pyridyl(5-COOCH3)
15 Z-Leu-Abu-CONH-(CH2)2-2-(N-methylpyrrolyl)
Z-Leu-Abu-CONH-(CH2)3-l-imidazolyl Z-Leu-Abu-CONH-(CH2)2-4-morpholinyl Z-Leu-Abu-CONH-(CH2)3-4-moφholinyl Z-Leu-Abu-CONH-(CH2)3-l-pyrrolidinyl-2-one
20 Z-Leu-Abu-CONH-CH2)2-3-indolyl
Z-Leu-Abu-CONH-CH2-2-quinolinyl Z-Leu-Abu-CONH-CH2-l-isoquinoline Z-Leu-Abu-CONH-(CH2)3-l-tetrahydroquinolinyl Z-Leu-Abu-CONH-(CH2)3-2-tetrahydroisoquinolinyl
25 Z-Leu-Abu-CONH-CH2-8-caffeinyl
Z-Leu-Abu-CONH-CH2-2-(4-methyl-2-thiazolyl) Z-Leu-Abu-CONH-CONH-(CH2)2NH-biotinyl Z-Leu-Abu-CONH-CH2-3-pyridyl-N-oxide Z-Leu-Abu-CONH-CH2-6-uracil
30 Z-Leu-Phe-CONH-CH2-2-pyridyl
Z-Leu-Phe-CONH-(CH2)3-4-moφholinyl Z-Leu-Phe-CONH-CH2-2-quinolinyl
Z-Leu-Phe-CONH-CH2-l-isoquinolinyl
Z-Leu-Phe-CONH-(CH2)3-l-tetrahydroquinolinyl
Z-Leu-Phe-CONH-(CH2)3-2-tetrahydroisoquinolinyl
Z-Leu-Phe-CONH-(CH2)2-NH-biotinyl Z-Leu-Nva-CONH-CH2CH(OH)Ph
Z-Leu-Nva-CONH-CH2-2-pyridyl
Z-Leu-Nva-CONH-(CH2)3-4-moφholinyl
CH3OCO(CH2)2CO-Leu-Abu-CONHEt
2-furyl-CO-Leu-Abu-CONHEt 2-tetrahydrofuryl-CO-Leu-Abu-CONHEt
3-pyridyl-CO-Leu-Abu-CONHEt
2-pyrazyl-CO-Leu-Abu-CONHEt
2-quinolinyl-CO-Leu-Abu-CONHEt
1-isoquinolinyl-CO-Leu-Abu-CONHEt 4-moφholinyl-CO-Leu-Abu-CONHEt
Ph(CH2)2CO-Leu-Abu-CONHEt l-C10H7CH2CO-Leu-Abu-CONHEt
Ph2CHCO-Leu-Abu-CONHEt
Ph2CHCO-Leu-Abu-CONH-CH2CH(OH)Ph Ph2CHCO-Leu-Abu-CONH-CH2-2-pyridyl
Ph2CHCO-Leu-Abu-CONH-(CH2)3-4-morpholinyl
Ph2CHCO-Leu-Phe-CONH-CH2CH(OH)Ph
Ph2CHCO-Leu-Phe-CONH-CH2-2-pyridyl
Ph2CHCO-Leu-Phe-CONH-(CH2)3-4-moφholinyl We studied the inhibition mechanism of the Peptide Keto-Compounds in both serine and thiol proteases. A crystal structure of one α -ketoester bound into the active site of the serine protease, porcine pancreatic elastase, has been completed. The active site Ser-195 oxygen of the enzyme adds to the carbonyl group of the ketoester to form a tetrahedral intermediate which is stabilized by interactions with the oxyanion hole. This structure resembles the tetrahedral intermediate involved in peptide bond hydrolysis and proves that α -ketoesters are transition-state analogs. His-57 is hydrogen bonded to the carbonyl group of the ester functional group, the peptide backbone on a
section of the PPE polypeptide backbone hydrogen bonds to the inhibitor to form a β-sheet, and the benzyl ester is directed toward the S' subsites. The side chain of the PI amino acid residue is located in the SI pocket of the enzyme. Interactions with ketoamides would be similar except that there is the possibility of forming an additional hydrogen bond with the NH group of the ketoamide functional group. If R is a longer substituent, then it would make favorable interactions with the S' subsites of the enzyme.
In the case of ketoacids, there would be no R group to interact with the S' subsites. Therefore, these inhibitors would be expected to be slightly less potent than the ketoesters and ketoamides. However, unexpectedly, certain ketoacid compounds have been found to have suφrisingly high activity when used in the context of the present invention. In particular, Z-Leu-Phe-COOH and Z-Leu-Abu-COOH have been found to be extremely potent inhibitors of Calpains. The active site of cysteine proteases shares several features in common with serine proteases including an active site histidine residue. In place of the Ser-195, cysteine proteases have an active site cysteine residue which would add to the ketonic carbonyl group of the peptide ketoacids, ketoesters, or ketoamides to form an adduct very similar to the structure described above except with a cysteine residue replacing the serine-195 residue. Additional interactions would occur between the extended substrate binding site of the cysteine protease and the inhibitor that would increase the binding affinity and specificity of the inhibitors.
The Peptide Keto-Compounds bind to the proteases inhibited thereby using many of the interactions that are found in complexes of a particular individual enzyme with its substrates. In order to design an inhibitor for a particular cysteine protease, it is necessary to: 1) find the amino acid sequences of good peptide substrates for that enzyme, and 2) place those or similar amino acid sequences into a Peptide Keto-
Compound. This design strategy will also work when other classes of peptide inhibitors are used in place of the peptide substrate to gain information on the appropriate sequence to place in the Peptide Keto-Compound inhibitor. Thus, we are able to predict the structure of new inhibitors for other proteases based on knowledge of their substrate specificities. Once a good inhibitor structure for a particular enzyme is found, it is then possible to change other characteristics such as solubility or hydrophobicity by adding substituents to the M or R groups.
Additional interactions with the enzyme can be obtained by tailoring the R group of the inhibitor to imitate the amino acid residues which are preferred by an individual protease at the SI' and S2' subsites. For example, ketoamides with R = alkyl substituted with phenyl would interact effectively with serine and cysteine proteases which prefer Phe, Tyr, Tφ residues at PI' and/or P2\ Likewise, the Ml group can be tailored to interact with the S subsites of the enzyme. This design strategy will also work when other classes of peptide inhibitors are used in place of the peptide substrate to gain information on the appropriate sequence to place in the ketoamide inhibitor. Thus, we are able to predict the structure of new inhibitors for other serine and cysteine proteases based on knowledge of their substrate specificities. Once a good inhibitor structure for a particular enzyme is found, it is then possible to change other characteristics such as solubility or hydrophobicity by adding substituents to the Ml or R groups.
In the case of Calpain, a cysteine protease, a known inhibitor sequence is the peptide aldehyde, Ac-Leu-Leu-Nle-H (also known as Calpain Inhibitor 1 and hereinafter designated as "CH"). This inhibitor, in addition to a related peptide aldehyde inhibitor Ac-Leu-Leu-Nme-H (also known as Calpain Inhibitor II) are commercially available from Calbiochem of La Jolla, California. We have discovered that peptide α -ketoesters with aromatic amino acid residues in PI are good inhibitors of the thiol proteases, cathepsin B, papain and Calpain. Additionally, we have discovered that peptide α-ketoester and peptide α-ketoamides with either aromatic amino acid residues or small hydrophobic alkyl amino acid residues at PI are good inhibitors of Calpain.
Our discovery of Peptide Keto-Compounds effective as Calpain Inhibitors was made through assay of the Peptide Keto-Compounds as reversible inhibitors. Various
concentrations of inhibitors in dimethylsulfoxide (DMSO) were added to the assay mixture, which contained buffer and substrate. The reaction was started by the addition of the enzyme and the hydrolysis rates were followed spectrophotometrically or fluorimetrically. 88 mM KH2P04, 12 mM Na2HP04, 1.33 mM EDTA, 2.7 mM cysteine, pH 6.0 was used as a buffer for cathepsin B; and 20 mM Hepes, 10 mM
CaCl2, 10 mM β-mercaptoethanol, pH 7.2 buffer was utilized for calpain I and calpain π.
All peptide thioester hydrolysis rates were measured with assay mixtures containing 4,4'-dithiodipyridine (e324 = 19800 M^cm"1; Grasetti & Murray, Arch. Biochem. Biophys., 119:41-49 (1967)). Papain was assayed with Bz-Arg-AMC or
Bz-Arg-NA (Kanaoka et al., Chem. Phann. Bull, 25:3126-3128 (1977)), and the AMC (7-amino-4-methylcoumarin) release was followed fluorimetrically (excitation at 380 nm, and emission at 460 nm). Cathepsin B was assayed with Z-Arg-Arg-AFC (Barrett and Kirschke, Methods Enτymol, 80:535-561 (1981)), and the AFC (7-amino-4- trifluoromethylcoumarin) release was followed fluorimetrically (excitation at 400 nm, and emission at 505 nm). Calpain I from human erythrocytes and calpain II from rabbit were assayed using Suc-Leu-Tyr-AMC (Sasaki et al., /. Biol. Chem. 259:12489-12494 (1984), hereby incoφorated by reference), and the AMC (7-amino-4- methylcoumarin) release was followed fluorimetrically (excitation at 380 nm, and emission at 460 nm). Enzymatic hydrolysis rates were measured at various substrate and inhibitor concentrations, and Kj values were determined by Dixon plot.
Table PKC1 shows the inhibition constants (K ) for papain, cathepsin B, calpain I, and calpain II.
The inhibition constants for papain shown in Table PKC1 were measured in 0.05 M Tris-HCl, pH 7.5 buffer, containing 2mM EDTA, 5mM cysteine (freshly prepared), 1% DMSO, at 25° C, using Ne-Benzoyl- Arg-AMC as a substrate, except that those values of inhibition constants for papain marked with an "e" in Table PKC1 were measured in 50 mM Tris-HCl, pH 7.5 buffer, containing 20 mM EDTA, 5 mM cysteine, 9% DMSO, at 25° C, using Nα-Benzoyl-Arg-NA as a substrate.
TABLE PKC1
Inhibition of Cvsteine Proteases by Peptide Ketoesters and Ketoacids
j(μM)
Compounds
CBD CIC CIId
Z-Leu-Abu-COOEt
Z-Leu-Phe-COOEt
Z-Leu-Nle-COOEt
Z-Leu-Nva-COOEt
Bz-DL-Phe-COOEt
Z-Phe-DL-Phe-COOEt
Z-Phe-DL-Ala-COOEt
Z-Ala-Ala-DL-Ala-COOEt
Z-Ala-Ala-DL-Abu-COOEt
Z-Ala-Ala-DL-Abu-COOBzl
Z-Ala-Ala-DL-Nva-COOEt
Z-Ala-Pro-DL-Ala-COOEt
MeO-Suc-Val-Pro-DL-Phe-COOMe
Z-Ala-Ala-Ala-DL-Ala-COOEt MeO-Suc-Ala-Ala-Pro-Abϋ-COOMe
aP = Papain CCI = Calpain I bCB = Cathepsin B dCII = Calpain II
It can be seen from the data in Table PKC1 that the dipeptide ketoesters with Abu, Phe, or Nle in the PI site and Leu in the P2 site are potent inhibitors of calpain I and calpain II. Tripeptides with Abu or Ala in the PI site and Ala in the P2 site are also seen to be inhibitors of Calpain, albeit somewhat weaker inhibitors than the dipeptides. Thus, in accordance with the foregoing description of the design of Peptide
Keto-Compound inhibitors, we believe that Peptide Keto-Compounds based on these and similar structures will exhibit Calpain inhibitory activity.
Assay of Inhibitory Potency of Peptide a-ketoamides. HEPES, heparin, and A23187 were obtained from Calbiochem. Suc-Leu-Tyr-AMC and chromogenic substrates were obtained from Sigma. Calpain I was purified from human erythrocytes according to the method of Kitahara (Kitahara, et al, J. Biochem. 95:1759-1766 (1984)) omitting the Blue-Sepharose step. Calpain II from rabbit muscle and cathepsin B were purchased from Sigma. Papain was purchased from Calbiochem.
Peptide α-ketoamides were assayed as reversible enzyme inhibitors. Various concentrations of inhibitors in Me2SO were added to the assay mixture which contained buffer and substrate. The reaction was started by the addition of the enzyme and the hydrolysis rates were followed spectrophotometrically or fluorimetrically.
Calpain I from human erythrocytes and calpain II from rabbit were assayed using Suc-Leu-Tyr-AMC (Sasaki et al, J. Biol Chem. 259:12489-12494 (1984); incoφorated herein by reference), and the AMC (7-amino-4-methylcoumarin) release was followed fluorimetrically (excitation at 380 nm, and emmision at 460 nm). Calpains were assayed in 25 mM Tris pH = 8.0, 10 mM CaC12. Fluorescence was followed using a Gilson FL-1A fluorometer or a Perkin-Elmer 203 Fluorescence spectrometer. Cathepsin B was assayed in 20 mM sodium acetate pH = 5.2, 0.5 mM dithiothreitol using Bz-Phe-Val-Arg-/7-nitroanilide as substrate. Alternately, cathepsin B was assayed with Z-Arg-Arg-AFC (Barrett and Kirschke, Methods Enzymol 80:535-561 (1981); incoφorated herein by reference), and the AFC (7-amino-4-trifluoromethylcoumarin) release was followed fluorimetrically (excitation at 400 nm and emmision at 505 nm). Papain was assayed in 100 mM KP0 , 1 mM EDTA, 2.5 mM cysteine pH = 6.0 using Bz-Arg-AMC or Bz-Arg-NA (Kanaoka et al, Chem. Pharm. Bull 25:3126-3128 (1977); incoφorated herein by reference) as a substrate. The AMC (7-amino-4-methylcoumarin) release was followed fluorimetrically (excitation at 380 nm,
and emmision at 460 nm). Enzymatic hydrolysis rates were measured at various substrate and inhibitor concentrations, and Kj values were determined by either Lineweaver-Burk plots or Dixon plots.
A 0.1 M Hepes, 0.5 M NaCl, pH 7.5 buffer was utilized for human leukocyte elastase (HLE), porcine pancreatic elastase (PPE), chymotrypsin and cathepsin G. A
0.1 Hepes, 0.01 M CaCl2, pH 7.5 buffer was utilized for trypsin, plasmin, and coagulation enzymes. A 50 mM Tris.HCl, 2 mM EDTA, 5 mM cysteine, pH 7.5 was used as a buffer for papain. A 88 mM KH2P04, 12 mM Na2HP04, 1.33 mM EDTA, 2.7 mM cysteine, pH 6.0 solution was used as a buffer for cathepsin B. A 20 mM Hepes, 10 mM CaCl2, 10 mM mercatoethanol, pH 7.2 buffer was utilized for calpain I and calpain II.
HLE and PPE were assayed with MeO-Suc-Ala-Ala-Pro-Val-NA and Suc-Ala-Ala-Ala-NA, respectively (Nakajima et al, J. Biol Chem. 254:4027-4032 (1979); incoφorated herein by reference). Human leukocyte cathepsin G and chymotrypsin Aa were assayed with Suc-Val-Pro-Phe-NA (Tanaka et al, Biochemistry 24:2040-2047
(1985); incoφorated herein by reference). The hydrolysis of peptide 4-nitroanilides was measured at 410 nm (e410 = 8800 M^cm"1; Erlanger et al., Arch. Biochem. Biophys. 95:271-278 (1961); incorporated herein by reference). Trypsin, thrombin, human plasma kallikrein, porcine pancreatic kallikrein, human factor XIa, and human plasmin were assayed with Z-Arg-SBzl or Z-Gly-Arg-SBu-i (McRae et al, Biochemistry
20:7196-7206 (1981); incoφorated herein by reference). All peptide thioester hydrolysis rates were measured with assay mixtures containing 4,4'-dithiodipyridine (e324 = 19800 M^cm"1; Grasetti & Murray, Arch. Biochem. Biophys. 119:41-49 (1967); incoφorated herein by reference). Structure-Activity Relationships. Table PKC2 shows the inhibition constants (Kj) for calpain I, calpain II and cathepsin B. Changing the R group on the amide significantly improves the inhibitory potency toward calpains. Dipeptide α-ketoamides with Abu, Phe, and Nva in the PI site and Leu in the P2 site are potent inhibitors of these cysteine proteases. The presence of a hydrogen bond donor in the SI' subsite of the cysteine proteases which may be interacting with the N-H on the ketoamide functional group is indicated since disubstituted amides were much less effective inhibitors. Derivatives of Z-Leu-AA-CONHR where the R group contained a hydroxy
or alkoxy group, such as (CH2)5OH and CH2CH(OC2H5)2, are very good inhibitors of the calpains. The prescence of an aromatic group in PI' position of the peptide ketoamide inhibitor resulted in improved inhibitory potency for calpains which indicates the prescence of hydrophobic residues in the S' subsites of both calpains. The derivatives Z-Leu-AA-CO-NOH^CH^R where R was phenyl, phenyl substituted with hydroxy or alkoxy groups and naphthyl, are also very good inhibitors of calpains and cathepsin B. Derivatives of Z-Leu-Abu-CONH(CH2)nR where the R group contained a heterocylic group which has both a hydrophobic moiety with an electronnegative atom, are among the best inhibitors for calpains and cathepsin B. For example Z-Leu-Nva-CONHCH2-2-pyridyl is the best inhibitor of calpain I.
Z-Leu-Abu-CONHCH2-2-pyridyl is the best inhibitor of calpain II respectively in this series, but its isomers, Z-Leu-Abu-CONH-CH2-3-pyridyl and Z-Leu-Abu-CONH-CH2-4-pyridyl, are substantially poorer inhibitors.
TABLE PKC2. Inhibition of Cysteine Proteases by Peptide a-Ketoamides with the
Structures Z-Leu-AA-CONHR.
R i (μM)
Cal I Cal II Cat B
AA = a-aminobutyric acid (CH2)2OH 0.8 0.078
4.5
(CH2)5OH 0.5 0.051
0.28
(CH2)20(CH2)2OH 0.65 0.16 2.0
CH2CH(OCH3)2
CH2CH(OC2H5)2
CH2-C6Hg(l,3,3-(CH3)3-5-OH)
0.63
(CH2)2C6H4(3-OCH3) 0.11 0.086
0.31
(CH2)2C6H4(4-OCH3) 0.12 0.046
0.44 CH2C6H3(3,5-(OCH3)2) 2.3 0.022
1.8
CH2-2-furyl 0.80 0.033
6.0
CH2-2-tetrahydrofuryl 0.33 0.066 4.5
CH2-2-pyridyl 0.64 0.017
3.0
CH2-3-pyridyl 0.12
1.2 CH2-4-pyridyl 1.1 0.11
6.4
(CH2)2-2-pyridyl 0.41 0.47
0.20
CH2-2-pyridyl(3-COOCH3) ca.110
CH2-2-pyridyl(5-COOCH3) ca.28
(CH2)2-2-(N-methylpyrrole) 0.16 0.076
1.2
(CH2)3- 1-imidazolyl 0.29 0.068
9.9
(CH2)2-4-moφholinyl 1.0 0.16
2.5
(CH2)3-4-moφholinyl 0.14 0.041
6.9
(CH2)3-l-pyrrolidine-2-one 1.2 0.27 2.0
(CH2)2-3-indolyl 0.3 0.05
CH2-2-quinolinyl 0.13
CH2-l-isoquinolinyl 0.25
0.3
(CH2)3-l-tetrahydroquinolinyl 0.37
(CH2)3-2-tetrahydroisoquinolinyl 0.31 8
CH2-8-caffeine 32.0
CH2-2-(4-methylthiazole) 34.0
(CH2)2NH-biotinyl 0.65
CH2-3-pyridyl-N-oxide 9.5 CH2-6-uracil 9.0
AA = phenylalanine
CH2-2-pyridyl 0.65
0.27
(CH2)3-4-moφholinyl 0.22 CH2-2-quinolinyl 0.11 0.023
0.34
CH2-l-isoquinolinyl 2.4
9.6
(CH2)3- 1-tetrahydroquinolinyl (CH2)3-2-tetrahydroisoquinolinyl
(CH2)2NH-biotinyl
AA = Norvaline
CH2-2-pyridyl
Table PKC3 shows the inhibition constants (Kj) of Z-Leu-AA- CONH-CH2CH(OH)R. The hydrophobic moiety substituted with CH2CH-X (X = electronegative atoms such as O, N) resulted in good inhibitor structures. Z-Leu-Abu-CONH-CH2CH(OH)C6F5 is the best inhibitor for calpain I, and
Z-Leu-Abu- CONH-CH2CH(OH)Ph is the best inhibitor for calpain II respectively in this series.
TABLE PKC3. Inhibition of Cysteine Proteases by Peptide a-Ketoamides with the Structures Z-Leu-AA-CONH-CH2CH(OH)-R.
R i
Cal I Cal IlCat B AA = a-aminobutyric acid
Ph 1.1 0.0150.37
C6H4(4-OCH3) 0.24
C6H2(2,4,6-(OCH3)3) 0.38
C6H4(4-N(CH3)2) 0.33 C6F5 0.05
C6H4(3-CF3) 0.35
C6H4(3-OPh) 0.90
C6H4(4-OPh) 0.10
C6H4(4-OCH2Ph) 0.08 CgH4-3-OC6H4(3-CF3) 0.07
C6H4-3-OC6H3(3,4-Cl2) 0.27
C6H3(3,4-(OCH2Ph)2) 0.23
1-C10H7 0.12
2-C10H7 0.35 AA = phenylalanine
Ph 1.3 0.052.1
C6H4(4-N(CH3)2) 0.62
C6F5 0.70
C6H4(3-CF3) 0.46 C6H4(3-OPh) 0.60
C6H4(4-OPh) 0.20
C6H4(4-OCH2Ph) 0.20
CgH4-3-OC6H4(3-CF3) _ 0.18
CgH4-3-OC6H3(3,4-Cl2) 0.59 C6H3(3,4-(OCH2Ph)2)
AA = Norvaline
Ph 7.8 11
In general, replacement of the Z group (PhCH2OCO-) by related aromatic groups also resulted in good inhibitor structures (Table PKC4).
Table PKC4. Inhibition of Cysteine Proteases by Peptide α-ketoamides with the Structures MjCO-Leu-AA-CONH-R.
M, R KI (μM)
Cal l Cal ll
Ph2CH (CH2)3-4-moφholinyl 0.76 0.0743.8
Preparation of peptide a-ketoesters. The peptide α -ketoesters are prepared by a two step Dakin-West procedure. This procedure can be utilized with either amino acid derivatives, dipeptide derivatives, tripeptide derivatives, or tetrapeptide derivatives as shown in the following scheme: O
M-(AA)n-OH --> Enol Ester --> M-(AA)n-CO-R. The precursor peptide ((AA)n) can be prepared using standard peptide chemistry procedures, including those that are well described in publications such as The Peptides. Analysis. Synthesis. Biology. 1-9 (1979-1987), published by Academic
Press ("The Peptides") and Synthese von Peptiden in Houben-Weyl Methoden der Organischen Che ie. 15, Parts 1 and 2, (1974) published by Georg Thieme Verlag ("Houben-Weyl"): both references hereby incorporated herein by reference.
The M group can be introduced using a number of different reaction schemes. For example, it could be introduced directly on an amino acid as shown in the following scheme:
H-(AA)n-OH -> M-(AA)n-OH. Alternatively, the M group can be introduced by reaction with an amino acid ester, followed by removal of the ester group to give the same product, as shown in the following scheme:
H-(AA)n-OR' -> M-(AA)n-OR' -- > M-(AA)n-OH. These and other techniques for introduction of the M group are well documented in the The Peptides. Houben-Weyl. and many other textbooks on organic synthesis. For example reaction with cyanate or p-nitrophenyl cyanate would introduce a carbamyl group (M = NH2CO-). Reaction with p-nitrophenyl thiocarbamate would introduce a thio carbamyl group (M = NH2CS-). Reaction with NH2S402C1 would introduce the NH2S02- group. Reaction with a substituted alkyl or aryl isocyanate would introduce the X-NH-CO- group where X is a substituted alkyl or aryl group. Reaction with a substituted alkyl or aryl isothiocyanate would introduce the X-NH-CS- group where X is a substituted alkyl or aryl group. Reaction with X-S02-C1 would introduce the X-S02- group. Reaction with a substituted alkyl or aryl acid chloride would introduce an acyl group (M = Y-CO-). For example, reaction with MeO-CO-CH2CH2-CO-Cl would give the Y-CO- group when Y is a C2 alkyl
substituted with a Cl alkyl-OCO- group. Reaction with a substituted alkyl or aryl thioacid chloride would introduce a thioacyl group (M = Y-CS-). Reaction with an a substituted alkyl or aryl sulfonyl chloride would introduce an X-S02- group. For example reaction with dansyl chloride would give the X-S02- derivative where X was a napthyl group monosubstituted with a dimethylamino group. Reaction with a substituted alkyl or aryl chloroformate would introduce a X-O-CO- group. Reaction with a substituted alkyl or aryl chlorothioformate would introduce a X-O-CS-. There are many alternate reaction schemes which could be used to introduce all of the above M groups to give either M-AA-OH or M-AA-OR'. The M-AA-OH derivatives could then be used directly in the Dakin-West reaction or could be converted into the dipeptides, tripeptides, and tetrapeptides M-AA-AA-OH, M-AA-AA-AA-OH, or M-AA-AA-AA-AA-OH which could be be used in the Dakin-West reaction. The substituted peptides M-AA-AA-OH, M-AA-AA-AA-OH, or M-AA-AA-AA-AA-OH could also be prepared directly from H-AA-AA-OH, H-AA-AA-AA-OH, or H-AA-AA-AA-AA-OH using the reactions described above for introduction of the M group. Alternately, the M group could be introduced by reaction with carboxyl blocked peptides M-AA-AA-OR', M-AA-AA-AA-OR', or M-AA-AA-AA-AA-OR', followed by the removal of the blocking group R'. The R group in the ketoester structures is introduced during the Dakin-West reaction by reaction with an oxalyl chloride Cl-CO-CO-O-R. For example, reaction of M-AA-AA-OH with ethyl oxaiyl chloride Cl-CO-CO-O-Et gives the keto ester M-AA-AA-CO-O-Et. Reaction of M-AA-AA-AA-AA-OH with Cl-CO-CO-O-Bzl would give the ketoester M-AA-AA-AA-AA-CO-O-Bzl. Clearly a wide variety of R groups can be introduced into the ketoester structure by reaction with various alkyl or arylalkyl oxalyl chlorides (Cl-CO-CO-O-R).
The oxalyl chlorides are easily prepared by reaction of an alkyl or arylalkyl alcohol with oxalyl chloride C1-CO-CO-C1. For example, Bzl-O-CO-CO-Cl and n-Bu-O-CO-CO-Cl are prepared by reaction of benzyl alcohol and butanol, respectively, with oxalyl chloride in yields of 50% and 80% (Warren and Malee, /. Chromat,
64:219-222 (1972); incoφorated herein by reference).
Ketoacids M-AA-CO-OH, M-AA-AA-CO-OH, M-AA-AA-AA-CO-OH, M-AA-AA-AA-AA-CO-OH, are generally prepared from the corresponding ketoesters M-AA-CO-OR, M-AA-AA-CO-OR, M-AA-AA-AA-CO-OR,
M-AA-AA-AA-AA-CO-OR by alkaline hydrolysis. In some cases, it may be necessary to use other methods such as hydrogenolysis of a benzyl group (R = Bzl) or acid cleavage (R = t-butyl) to obtain the ketoacid. The alternate methods would be used when the M group was labile to alkaline hydrolysis.
The various peptide ketoamide subclasses, including M-AA-NH-CHR2-CO-CO- NR3R4 (Dipeptide Ketoamides, Subclass A), M-AA-AA-CO-NR3R4 (Dipeptide Ketoamides, Subclass B), M1CO-AA2-AA1-CO-NH-CH2CH(OH)-R1 and five others presented above (Dipeptide α-Ketoamides, Subclass C, Types 1 through 6), M-AA-AA- AA-CO-NR3R4 (Tripeptide Ketoamides), M-AA-AA-AA-AA-CO-NR3R4 (Tetrapeptide Ketoamides) and M1-AA-CO-NR3R4 (Amino Acid Ketoamides), were prepared indirectly from the corresponding ketoesters. The ketone carbonyl group was first protected as shown in the following scheme and then the ketoamide was prepared by reaction with an amine H-NR3R4. The illustrated procedure should also work with other protecting groups.
1-NR3R4
In addition to the scheme outlined above, a ketoacid could be used as a precursor to produce a corresponding ketoamide. Blocking the ketone carbonyl group of the ketoacid and then coupling with an amine H-NR3R4 using standard peptide
coupling reagents would yield an intermediate which could then be deblocked to form the ketoamide.
Ketoamides MjCO-AA-AA-CONHR were prepared indirectly from the ketoesters. The ketone carbonyl group is first protected as shown in the following scheme and then the ketoamide is prepared by reaction with an amine RNH2. The product is easily isolated from the reaction mixture when using this procedure. This procedure will also work with other ketone protecting groups. In addition, the corresponding ketoacid can be used as a precursor to the a-ketoamide via coupling with an amine RNH2 using standard peptide coupling reagents would result in formation of the peptide a-
General Synthetic Methods for Peptide Keto-Compounds
The techniques for synthesis of a wide variety of amines are described in many publications. For example, Evans et al. in J.Org.Chem. 39:914 (1974) reported the syntheses of phenylethanol derivatives with alkylamino, alkoxyamino and phenyloxyamino groups. Katrizky et al. in /. Chem. Soc :2404-2408 (1956), Fife et al. in Heterocycles
22(l):93-96 (1984), and Heterocycles 22(5):1121-1124 (1984), and Isoda et al. in Chemical and Pharmaceutical Bulletin 28:1408-1414 (1980) reported the syntheses of pyridine derivatives with alkylamino and COOR groups. Nagata et al. in Yakugaku Zasshi 83:679-682 (1963) reported the syntheses of quinoline derivatives with alkylamino groups. Zimmer et al. in Tetrahedron Letters 24:2805-2807 (1968) reported the syntheses of isoquinoline derivatives with alkylamino groups. Aroyan et al. in Izv. Akad Nauk Arm. SSR, Kliim. Nauki 18(l):76-82 (1965) reported the syntheses of tetrahydroquinoline derivatives with alkylamino groups. Yonan, (U.S. 3,245,997 (Cl. 260-288), April 12, 1966. 2 pp) reported the syntheses of tetrahydroisoquinoline derivatives with alkylamino groups. Rybar et al. in Chem. Commun. 35:1415-1433 (1970), Golovchinskaya et al. in /. General
Chem. 22:599-603 (1952), and Nantka-Na irski et al in Acta. Polon. Pharm. 1:5-12 (1974) reported the syntheses of caffeine derivatives with alkylamino groups. Goldberg et al. in
J.Chem.Soc :l372 (1947) reported the syntheses of methylthiazole derivatives with alkylamino groups. Mizuno et al. mJ.Org.Chem. 39:1250 (1974) reported the syntheses of pyridine-N-oxide derivatives with alkylamino groups. Wade in /. Heterocyclic Chem. 23:981 (1986) reported the syntheses of uracil derivatives with alkylamino groups. All of the above citations are incoφorated herein by reference.
Unless otherwise noted, materials were obtained from commercial suppliers and used without further purification. Melting points were taken with a Buchi capillary apparatus and are uncorrected. 1H NMR spectra were determined on a Varian Gemini 300. Chemical shifts are expressed in ppm (δ) relative to internal tetramethylsilane. Flash column chromatography was performed with Universal Scientific Inc. silica gel 0-63.
Electron-impact mass spectra (MS) of novel compounds were determined with a Varian MAT 112S spectrometer. The purity of all compounds was checked by thin- layer chromatography on Baker Si250F silica gel plates using the following solvent system: A, CHCl3:MeOH = 20:1 v/v; B, CHCl3:MeOH = 100: 1 v/v; C, AcOEt; D, CHCl3:MeOH = 10:1 v/v; E, n-BuOH:AcOH;py:H20 = 4:1:1:2 v/v; F, CHCl3:MeOH = 5:1 v/v; G,
AcOEt:MeOH = 10:1 v/v; H, (i-Pr)20; I, CHCl3:MeOH:AcOH = 80:10:5 v/v; J, CHCl3:MeOH:AcOH = 95:5:3 v/v; K, AcOEt:AcOH = 200:1 v/v; L, CHC13; M, CHCl3:MeOH = 50:1 v/v.
Amino acid methyl ester hydrochlorides were prepared according to M. Brenner et al., Helv. Chem. Acta 33:568 (1950); 36:1109 (1953) in a scale over 10 mmol or according to Rachele, /. Org. Chem. 28:2898 (1963) in a scale of 0.1-1.0 mmol.
Yield (%) mp (°C) m.p. (literature)
DL-Nva-OCH3 HCl, 100 113-116 116-117
L-Ile-OCH3 HCl, 98 90-91 98-100 L-Phe-OCH3 HCl, 98 159-161 158-160
DL-Abu-OCH3 HCl, 100 148-150 150-151
L-Leu-OCH3 HCl 100 145.5-146.5 147
DL-Nle-OCH3ΗCl 93 120-121 122-123
4-Cl-Phe-OCH
3 HCl 98 184-185 (decomp.) 185-186 N-Acylamino acids was synthesized via Schotten-Baumann reaction as in Bergmann and Zervas, Chem. Ber., 65:1192 (1932) in the case when the acyl group was phenylsulphonyl, 2- naphthylsulphonyl or benzoyl.
N-Acylamino acids with 4-methylpentanoic, 2-(l- propyl)pentanoic and 7-phenylheptanoic group was synthesized in a two step synthesis. The N-acylamino acid methyl ester was obtained first and then was hydrolysed to the free N-acylamino acid. N-Acylamino Acid Methyl Esters (General Procedure). To a chilled (10 °C) slurry of the appropriate amino acid methyl ester hydrochloride (20 mmol) in 100 ml benzene was added slowly (temp. 10-15 °C) 40 mmol triethylamine or N- methylmoφholine and then the reaction mixture was stirred for 30 minutes at this temperature. Then 18 mmol of appropriate acid chloride (temp. 10-15 °C) was added slowly to the reaction mixture and the reaction mixture was stirred overnight at room temperature. The precipiatated hydrochloride was filtered, washed on a funnel with 2 x 20 ml benzene, and the collected filtrate was washed successively with 2 x 50 ml 1 M HCl, 2 x 50 ml 5% NaHC03, 1 x 100 ml H20, 2 x 50 ml satd. NaCl and dried over MgS04. After evaporation of the solvent in vacuo (rotavaporator), the residue was checked for purity (TLC) and used for the next step (hydrolysis).
Yield (%) mp (°C) (CH3)2CH(CH2)2CO-DL-Abu-OCH3 80 oil
(CH3CH2CH2)2CHCO-DL-Abu-OCH3 96 117-118
Ph(CH2)6CO-DL-Abu-OCH3 72 oil Hydrolysis (General Procedure). To a solution of 10 mmole of the appropriate
N-acylamino acid methyl ester in 100 ml of methanol was added in one portion 11.25 ml of 1 M NaOH (11.25 mmol) and the reaction mixture was stirred three hours at room temperature. Then the reaction mixture was cooled to 0 °C (ice- salt bath) and acidified to pH = 2 with 1 M HCl aq. To this reaction mixture was added 100 ml ethyl acetate, transferred to a separatory funnel and organic layer separated. The water layer was saturated with solid NaCl or (NH4)2S04 and reextracted with 2 x 50 ml AcOEt. The collected organic layer was washed with 2 x 50 ml H20, decolorized with carbon, and dried
over MgS04. After evaporation of the solvent in vacuo (rotavaporator), the residue was checked for purity (TLC) and in the case of contamination was crystallized from an appropriate solvent.
Yield (%) mp (°C)
(CH3)2CH(CH2)2CO-DL-Abu-OH 92 110.5-112
(CH3CH2CH2)2CHCO-DL-Abu-OH 99 126-127 (n-octane)
Ph(CH2)6CO-DL-Abu-OH 89 110-112 (n-octane)
N-Acyldipeptide methyl esters were synthesized via the HOBt-DCC method in a DMF solution as in Kδnig and Geiger, Chem. Ber., 103:788 (1970).
Z-Leu-DL-NVa-OCH3 Z-Leu-L-Phe-OCH,
Z-Leu-L-Ile-OCH,
Z-Leu-DL-Abu-OCH,
Z-Leu-L-Leu-OCH3
Z-Leu-DL-NLeu-OCH3
(liquid crystal?) 0.68 K
2-NapSOrLeu-DL-Abu-OCH3
99 oil 0.59 A
2-NapS02-Leu-L-Leu-OCH3
90 97-98.5 0.63 A
N-Acyldipeptides were obtained by hydrolysis of the appropriate methyl esters via a general hydrolysis procedure. In the case of N-sulphonyldipeptide methyl esters, 1 equivalent of the methyl ester was hydrolyzed with 2.25 equivalent of 1 molar NaOH because of form a sulfonamide sodium salt.
N-Acytripeptide methyl esters were synthesized via HOBt- DCC method in DMF solution as in Kδnig and Geiger, supra.
Yield (%) mp (°C) TLC (Rf, eluent)
Z-Leu-Leu-Abu-OCH3 87 140-141.5 0.50 A Z-Leu-Leu-Phe-OCH3 76 158-159 0.83 J 2-NapSOrLeu-Leu-Abu-OCH3
97 >200 0.52 A
N-Acyltripeptide were obtained through hydrolysis of the appropriate methyl esters via general hydrolysis procedure. In the case of N-sulphonyltripeptide methyl ester, 1 equivalent of methyl ester was hydrolyzed with 2.25 equivalent of 1 molar NaOH to form the sulfonamide sodium salt.
We have also discovered a process for the synthesis of a-ketoamides with the structures
M-CO-AA2-AA1-CO-NH-R and M-CO-AA3-AA2-AArCO-NH-R, wherein
M is selected from the group consisting of C1-4 alkyl monosubstituted with phenyl,
Cw alkyl disubstituted with phenyl, C1-4 alkyl monosubstituted with 1-naphthyl, C1-4 alkyl monosubstituted with 2-naphthyl, Cw alkoxy monosubstituted with phenyl, Cl4 alkoxy disubstituted with phenyl, Ar1CH20-, A^O-, ArjCH^H-, ArjNH- and
Heterocycle1(CH2)q-;
AT- is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, 1-naphthyl, 1-naphthyl monosubstituted with J, 2-naphthyl, and 2-naphthyl monosubstituted with J; J is selected from the group consisting of halogen, OH, CN, N02, NH2, COOH,
C02Me, C02Et, CF3, C1-4 alkoxy, C1-4 alkylamine, C28 dialkylamine, Cw perfluoroalkyl, and -N(CH2CH2)20;
Heterocyclβj is selected from the group consisting of 2-furyl, 2-tetrahydrofuryl, 2-pyrazinyl, 3-pyridyl, 4-pyridyl, 2-quinolinyl, 1-tetrahydroquinolinyl, 1-isoquinolinyl, 2-tetrahydroisoquinolinyl, and -N(CH2CH2)20; q = 0-2;
AAj, AA2 and AA3 are side chain blocked or unblocked a-amino acids with the L configuration, D configuration, or DL configuration at the a-carbon selected independently from the group consisting of alanine, valine, leucine, isoleucine, histidine, proline, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, arginine, lysine, tryptophan, glycine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH,
NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH,
NH2-CH(CH2- l-naphthyl)-COOH, NH2-CH( CH2-2-n aphthyl) -COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
R is selected from the group consisting of H, C^g alkyl, C^o cyclized alkyl, C^ alkyl with a phenyl group attached to the Cj_2g alkyl, Cj.2 cyclized alkyl with an attached phenyl group, C^ alkyl with an attached phenyl group substituted with K, C1.2g alkyl with an attached phenyl group disubstituted with K, C^g alkyl with an attached phenyl group trisubstituted with K, C^g cyclized alkyl with an attached phenyl group substituted with K, Cj.jg alkyl with a moφholine [-N(CH2CH2)0] ring attached through nitrogen to the alkyl, Cl l0 alkyl with a piperidine ring attached through nitrogen to the alkyl, C^g alkyl with a pyrrolidine ring attached through nitrogen to the alkyl, C-^Q alkyl with an OH group attached to the alkyl, -CH2CH2OCH2CH2OH, C^Q with an attached 4-pyridyl group, C-^g with an attached 3-pyridyl group, Cj.jg with an attached 2-pyridyl group, Cj.jg with an attached cyclohexyl group, -NH-CH2CH2-(4-hydroxyphenyl), -NH-CH2CH2-(3-indolyl), CH2CH(OH)-Ar2 and (CH2)n-Heterocycle2; K is selected from the group consisting of halogen, C^g alkyl, C^ perfluoroalkyl,
Cj.jg alkoxy, N02, CN, OH, C02H, amino, Cj.jg alkylamino, C2.12 dialkylamino, C^jg acyl, and Cj.jg alkoxy-CO-, and C^g alkyl-S-;
Ar2 is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, phenyl trisubstituted with J, pentafluorophenyl, C6H4(3-OR2), C6H4(4-OR2), C6H3(3,4-(OR2)2, C6H2(2,4,6-(OR2)3, 1-naphthyl, 1-naphthyl monosubstituted with J, 1-naphthyl disubstituted with J, 2-naphthyl, 2-naphthyl monosubstituted with J, 2-naphthyl disubstituted with J, 2-pyridyl, 2-quinolinyl, and 1-isoquinolinyl;
R2 represents C1-4 alkyl substituted with phenyl, phenyl and phenyl substituted with J.
Heterocycle2 is selected from the group consisting of 2-furyl, 2-furyl monosubstituted with J, 2-tetrahydrofuryl, 2-pyridyl, 2-pyridyl monosubstituted with J, 3-pyridyl, 3-pyridyl monosubstituted with J, 4-pyridyl, 4-pyridyl monosubstituted with J, 2-pyrazinyl, 2-quinolinyl, 2-quinr,1-nyl monosubstituted with J, 1-isoquinolinyl, 1-isoquinolinyl monosubstituted wit'r „, 1-tetrahydroquinolinyl, 2-tetrahydroisoquinolinyl,
3-indolyl, 2-pyridyl-N-oxide, 3-pyridyl-N-oxide, 4-pyridyl-N-oxide, 2-(N-methyl-2-pyrrolyl), 1-imidazolyl, l-pyrrolidinyl-2-one, 2-(5-methyl-3-thiazolyl), (CH2)2-NH-biotin;
(a) Protecting the α-ketone carbonyl of a peptidyl α-ketoester with the structures
M-CO-AA2-AArCOOR6 and M-CO-AA3-AA2-AA1-COOR6, wherein
R6 is selected from the group consisting of C 6 alkyls and C^g alkyls monosubstituted with phenyl, by treatment with a blocking reagent in the presence of a Lewis acid in an organic solvent at 0-100 °C for 1-48 hours, wherein the preferred blocking reagent is 1,2-ethanedithiol; the preferred Lewis acids are selected from the group consisting of BF3.Et20, 4-toluene sulfonic acid, A1C13 and ZnCl2; the preferred organic solvents are selected from the group consisting of CH2C12, CHC13, Et20 and THF;
(b) Treating the product with a primary amine RNH2 in an organic solvent a 0-100 °C for 1-72 hours, wherein the preferred organic solvents are selected from the group consisting of EtOH,
THF, CH2C12 and DMF;
(c) Removing the blocking group from the α -carbonyl to give the desire peptidyl α -ketoamide.
We have also discovered another process for the synthesis of peptidyl α-ketoamide with the structures
M-CO-AA2-AArCO-NH-R and M-CO-AA3-AA2-AArCO-NH-R,
wherein
M is selected from the group consisting of C1-4 alkyl monosubstituted with phenyl,
Cj- alkyl disubstituted with phenyl, C^ alkyl monosubstituted with 1-naphthyl, Cj.4 alkyl monosubstituted with 2-naphthyl, Cj_ alkoxy monosubstituted with phenyl, Cw alkoxy disubstituted with phenyl, Ar1CH20-, Ar10-, Ar.CH^H-, Ar1NH- and
Heterocycle^CH^ -;
Arj is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, 1-naphthyl, 1-naphthyl monosubstituted with J, 2-naphthyl, and 2-naphthyl monosubstituted with J; J is selected from the group consisting of halogen, OH, CN, N02, NH2, COOH,
C02Me, C02Et, CF3, Cw alkoxy, CM alkylamine, C2.8 dialkylamine, C1-4 perfluoroalkyl, and -N(CH2CH2)20;
Heterocycle1 is selected from the group consisting of 2-furyl, 2-tetrahydrofuryl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrazinyl, 2-quinolinyl, 1-tetrahydroquinolinyl, 1-isoquinolinyl, 2-tetrahydroisoquinolinyl, and -N(CH2CH2)20; q = 0-2;
AAj, AA2 and AA3 are side chain blocked or unblocked a-amino acids with the L configuration, D configuration, or DL configuration at the a-carbon selected independently from the group consisting of alanine, valine, leucine, isoleucine, histidine, proline, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, arginine, lysine, tryptophan, glycine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid, NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH,
NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, NH2-CH(CH2-l-naphthyl)-COOH, NH2-CH(CH2-2-naphthyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine;
R is selected from the group consisting of H, C._20 alkyl, Cj^ cyclized alkyl, C^ alkyl with a phenyl group attached to the C^g alkyl, Cj_20 cyclized alkyl with an attached phenyl group, C^Q alkyl with an attached phenyl group substituted with K, C^ alkyl with an attached phenyl group disubstituted with K, C 20 alkyl with an attached phenyl group
trisubstituted with K, C^ cyclized alkyl with an attached phenyl group substituted with K, Cj.j alkyl with a moφholine [-N(CH2CH2)0] ring attached through nitrogen to the alkyl, C-^ alkyl with a piperidine ring attached through nitrogen to the alkyl, C^jg alkyl with a pyrrolidine ring attached through nitrogen to the alkyl, Cj.20 alkyl with an OH group attached to the alkyl, -CH2CH2OCH2CH2OH, Cw0 with an attached 4-pyridyl group, C^g with an attached 3-pyridyl group, C^g with an attached 2-pyridyl group, C^g with an attached cyclohexyl group, -NH-CH2CH2-(4-hydroxyphenyl), -NH-CH2CH2-(3-indolyl), CH2CH(OH)-Ar2 and (CH2)n-Heterocycle2;
K is selected from the group consisting of halogen, Cj.10 alkyl, C^g perfluoroalkyl, C1-io alkoxy, N02, CN, OH, C02H, amino, Cj.jg alkylamino, C2.]2 dialkylamino, C^jg acyl, and Cj.jg alkoxy-CO-, and Cj.jg alkyl-S-;
Ar2 is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, phenyl trisubstituted with J, pentafluorophenyl, C6H4(3-OR2), C6H4(4-OR2), C6H3(3,4-(OR2)2, C6H2(2,4,6-(OR2)3, 1-naphthyl, 1-naphthyl monosubstituted with J, 1-naphthyl disubstituted with J, 2-naphthyl, 2-naphthyl monosubstituted with J, 2-naphthyl disubstituted with J, 2-pyridyl, 2-quinolinyl, and 1-isoquinolinyl;
R2 represents Cw alkyl substituted with phenyl, phenyl and phenyl substituted with J. Heterocycle2 is selected from the group consisting of 2-furyl, 2-furyl monosubstituted with J, 2-tetrahydrofuryl, 2-pyridyl, 2-pyridyl monosubstituted with J, 3-pyridyl, 3-pyridyl monosubstituted with J, 4-pyridyl, 4-pyridyl monosubstituted with J, 2-pyrazinyl, 2-quinolinyl, 2-quinolinyl monosubstituted with J, 1-isoquinolinyl, 1-isoquinolinyl monosubstituted with J, 1-tetrahydroquinolinyl, 2-tetrahydroisoquinolinyl, 3-indolyl, 2-pyridyl-N-oxide, 3-pyridyl-N-oxide, 4-pyridyl-N-oxide, 2-(N-methyl-2-pyrrolyl),
1-imidazolyl, l-pyrrolidinyl-2-one, 2-(5-methyl-3-thiazolyl), (CH2)2-NH-biotin;
(a) Hydrolyzing a peptidyl α-ketoester with the structures
M-CO-AA2-AArCOOR6 and
M-CO-AA3-AA2-AArCOOR6, wherein
R6 is selected from the group consisting of C^ alkyls and Cj_6 alkyls monosubstituted with phenyl; by treating the peptidyl α -ketoester with a hydrolysis reagent in an appropriate solvent at
0-100 °C for 1-24 hours to give the corresponding peptidyl α-ketoacid, wherein the preferred hydrolysis reagents are selected from the group consisting of NaOH, KOH, EtONa and EtOK; the preferred solvent are selected from the group consisting of water, MeOH, EtOH, THF and DMF;
(b) Coupling the product peptidyl α-ketoacid with a primary amine RNH2 in an organic solvent at 0-100 °C for 1-72 hours to give the desired peptidyl α-ketoamide, wherein the preferred coupling conditions are selected from the group consisting of treatment with 1,1-carbonyldiimidazole, treatment with dicyclohexylcarbodiimide, and treatment with dicyclohexylcarbodiimide- 1-hydroxybenzotriazole; the preferred organic solvents are selected from the group consisting of CH2C12, CHC13, DMF and THF.
We have also discovered a process for the synthesis of peptidyl α-ketoamides with the structures
M-CO-AA2-AArCO-NH-R and
M-CO AA3-AA2-AArCO-NH-R, wherein
M is selected from the group consisting of C
4 alkyl monosubstituted with phenyl, ^ alkyl disubstituted with phenyl, C
j_
4 alkyl monosubstituted with 1-naphthyl, C^ alkyl monosubstituted with 2-naphthyl, C^
4 alkoxy monosubstituted with phenyl, C
w alkoxy disubstituted with phenyl,
and
Heterocycle^CHj) -;
Aτ- is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, 1-naphthyl, 1-naphthyl monosubstituted with J, 2-naphthyl, and 2-naphthyl monosubstituted with J;
J is selected from the group consisting of halogen, OH, CN, N02, NH2, COOH, C02Me, C02Et, CF3, C1^} alkoxy, CM alkylamine, C2_8 dialkylamine, CM perfluoroalkyl, and -N(CH2CH2)20;
Heterocyclej^ is selected from the group consisting of 2-furyl, 2-tetrahydrofuryl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrazinyl, 2-quinolinyl, 2-tetrahydroquinolinyl,
1-isoquinolinyl, 1-tetrahydroisoquinolinyl, and -N(CH2CH2)20; q = 0-2;
AA^ AA2 and AA3 are side chain blocked or unblocked a-amino acids with the L configuration, D configuration, or DL configuration at the a-carbon selected independently from the group consisting of alanine, valine, leucine, isoleucine, histidine, proline, methionine, methionine sulfoxide, phenylalanine, serine, threonine, phenylglycine, norleucine, norvaline, arginine, lysine, tryptophan, glycine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, alpha-aminobutyric acid, O-methylserine, O-ethylserine, S-methylcysteine, S-ethylcysteine, S-benzylcysteine, NH2-CH(CH2CHEt2)-COOH, alpha-aminoheptanoic acid,
NH2-CH(CH2-cyclohexyl)-COOH, NH2-CH(CH2-cyclopentyl)-COOH, NH2-CH(CH2-cyclobutyl)-COOH, NH2-CH(CH2-cyclopropyl)-COOH, NH2-CH(CH2-l-naphthyl)-COOH, NH2-CH(CH2-2-naphthyl)-COOH, 5,5,5-trifluoroleucine, and hexafluoroleucine; R is selected from the group consisting of H, Cj.20 alkyl, Cj_2g cyclized alkyl, C^ alkyl with a phenyl group attached to the C1.20 alkyl, C^g cyclized alkyl with an attached phenyl group, Cj.^ alkyl with an attached phenyl group substituted with K, Cj_20 alkyl with an attached phenyl group disubstituted with K, C^g alkyl with an attached phenyl group trisubstituted with K, Cj_20 cyclized alkyl with an attached phenyl group substituted with K, C^g alkyl with a morpholine [-N(CH2CH2)0] ring attached through nitrogen to the alkyl, C^ alkyl with a piperidine ring attached through nitrogen to the alkyl, CJ.JQ alkyl with a pyrrolidine ring attached through nitrogen to the alkyl, Cj^g alkyl with an OH group attached to the alkyl, -CH2CH2OCH2CH2OH, Cl-10 with an attached 4-pyridyl group, 1 Q with an attached 3-pyridyl group, CΪ 0 with an attached 2-pyridyl group, C-^ with an attached cyclohexyl group, -NH-CH2CH2-(4-hydroxyphenyl),
-NH-CH2CH2-(3-indolyl), CH2CH(OH)-Ar2 and (CH2)n-Heterocycle2;
K is selected from the group consisting of halogen, Cj.jg alkyl, C1Λ0 perfluoroalkyl, Cj.jg alkoxy, N02, CN, OH, C02H, amino, CiΛ0 alkylamino, C2.12 dialkylamino, Cw0 acyl, and C1 Q alkoxy-CO-, and Cj.jg alkyl-S-;
Ar2 is selected from the group consisting of phenyl, phenyl monosubstituted with J, phenyl disubstituted with J, phenyl trisubstituted with J, pentafluorophenyl,
C6H4(3-OR2), C6H4(4-OR2), C6H3(3,4-(OR2)2, C6H2(2,4,6-(OR2)3, 1-naphthyl, 1-naphthyl monosubstituted with J, 1-naphthyl disubstituted with J, 2-naphthyl, 2-naphthyl monosubstituted with J, 2-naphthyl disubstituted with J, 2-pyridyl, 2-quinolinyl, and 1-isoquinolinyl; R2 represents C1-4 alkyl substituted with phenyl, phenyl and phenyl substituted with
J.
Heterocycle2 is selected from the group consisting of 2-furyl, 2-furyl monosubstituted with J, 2-tetrahydrofuryl, 2-pyridyl, 2-pyridyl monosubstituted with J,
3-pyridyl, 3-pyridyl monosubstituted with J, 4-pyridyl, 4-pyridyl monosubstituted with J, 2-ρyrazinyl, 2-quinolinyl, 2-quinolinyl monosubstituted with J, 1-isoquinolinyl,
1-isoquinolinyl monosubstituted with J, 1-tetrahydroquinoiinyl, 2-tetrahydroisoquinolinyl,
3-indolyl, 2-pyridyl-N-oxide, 3-pyridyl-N-oxide, 4-pyridyl-N-oxide, 2-(N-methyl-2-pyrrolyl),
1
consisting of treating a peptidyl a-enolester derived from a peptidyl a-ketoester with the structures
M-CO-AA2-AArCOOR6 and M-CO-AA3-AA2-AArCOORg, wherein
Rg is selected from the group consisting of C^g alkyls and C^ alkyl monosubstituted with phenyl; with a primary amine RNH2 in an organic solvent at 0-100 °C for 1-72 hours to give th desired peptidyl α -ketoamide, wherein the preferred organic solvents are selected from the group consisting of CH2C12 EtOH, DMF and THF.
The following examples, Examples PKC1-PKC65, are given to illustrate the synthesis of Peptide Keto-Compounds:
EXAMPLE PKC1 Z-AIa-DL-AIa-COOEt. This compound was synthesized by a modified Dakin-West procedure as in Charles et al, /. Chem. Soc. Perkin 1:1139-1146 (1980). To a stirred solution of Z-Ala-Ala-OH (880 mg, 3 mmole), 4-dimethylaminopyridine (15 mg, 0.31 mmole), and pyridine (0.8 mL, 10 mmole) in tetrahydrofuran (3 mL) was added ethyl oxalyl chloride (0.7 mL, 6 mmole) at a rate sufficient to initiate refluxing. The mixture was gently refluxed for 3.5 h. The mixture was treated with water (3 mL) and stirred vigorously at room temperature for 30 min. The mixture was extracted with ethyl acetate.
The organic extracts were dried and evaporated to obtain the residue (1.45 g). The residue was chromatographed on silica gel and eluted with CH2C12 to give the enol ester product, oil (500 mg, 37%); single spot on tic, Rf 2 = 0.67 (CHCl3:MeOH = 9:1); MS, m/e = 451 (M + + l). To a stirred suspension of the enol ester (210 mg, 0.47 mmol) in anhydrous ethanol (1 mL) at room temperature was added dropwise a solution of sodium ethoxide in ethanol until a clear yellow solution resulted. The ethanol was then removed and the residue was treated with ether. The ether solution was washed with water, dried, and evaporated to give a residue. This residue was chromatographed on a silica gel and the product was eluted with methylene chloride. The solvent was removed, and the peptide ketoester Z-Ala-DL-Ala-C02Et was obtained as an semi-solid (150 mg, 92 %); single spot on tic, R 0.58 (CHCl3:MeOH = 5:1); MS, m/e = 351 (M+ + 1). Anal. Calcd. for C17H22θgN2 l/3 H20: C, 57.29; H, 6.22; N, 7.86. Found: C, 57.23; H, 6.36; N, 8.17.
EXAMPLE PKC2 Z-Ala-AIa-DL-Ala-C02Et. This compound was prepared from Z-Ala-Ala-Ala- OH using the same procedure as described in Example PKC1. The product was crystallized from ethyl ether in 23% yield; single spot on tic, Rf 2 = 0.31 (CHCl3:MeOH = 9:1); mp 143-144 °C; MS, m/e = 421 (M+). Anal. Calcd. for C20H27O7N3: C, 56.99; H, 6.46; N, 9.97. Found: C, 56.96; H, 6.49; N, 9.92.
EXAMPLE PKC3 Z-Ala-Ala-D Abu-C02Et. This compound was prepared from Z-Ala-Ala-DL-
Abu-OH in 11% yield by the procedure described in Example PKC1; single spot on tic, Rf 2 = 0.60 (CHCl3:MeOH = 9:1); mp 111-113 °C; MS, m/e = 436 (M+ + l). Anal.
Calcd. for C21H2907N3 l/3 H20: C, 57.13; H, 6.75; N, 9.51. Found: C, 57.38; H, 6.82; N, 9.62.
EXAMPLE PKC4 Z-Ala-Ala-DL-Nva-C02Et. This compound was prepared from Z-Ala-Nva-OH in 20% yield by the procedure described in Example PKC1; single spot on tic, Rf 1 =
0.64 (CHCl3:MeOH = 5:1); MS, m/e = 450 (M+ + l). Anal. Calcd. for C22H3107N3Η20: C, 56.51; H, 7.11; N, 8.99. Found: C, 56.42; H, 7.08; N, 9.06.
EXAMPLE PKC5 Z-Ala-Pro-DL-Ala-C02Et. This compound was prepared from Z-Ala-Pro-Ala- OH dicyclohexylamine in 19% yield by the procedure described in Example PKC1; single spot on tic, Rf 2 = 0.55 (CHCl3:MeOH = 9:1); MS, m/e = 447 (M + ). Anal. Calcd. for C^H^OyN l/tt H20: C, 57.88; H, 6.62; N, 9.21. Found: C, 57.65; H, 6.68; N, 9.17.
EXAMPLE PKC6 Z-Ala-Ala-Ala-DL-AIa-C02Et. The compound was prepared from Z-Ala-Ala-
Ala-Ala-OH in 7% yield by the procedure described in Example PKC1; single spot on tic, Rf 2 =0.40 (CHCl3:MeOH = 9:1); mp. 163-165 °C; MS, m/e = 493 (M+ + l). Anal. Calcd. for C^H^O^ l^ H20: C, 55.08; H, 6.63; N, 11.17. Found: C, 54.85; H, 6.53; N, 11.14. EXAMPLE PKC7
Bz-DL-Phe-C02Et. This compound was prepared from Bz-Phe-OH in 36% yield by the procedure described in Example PKC1, oil, single spot on tic, Rf 2 = 0.61 (CHCl3:MeOH = 9:1); MS, m/e = 325 (M+). Anal. Calcd. for C19H1904N-l/3 H20: C, 68.86; H, 5.98; N, 4.22. Found: C, 69.10; H, 6.09; N, 4.38. EXAMPLE PKC8
MeO-Suc-Ala-D AIa-C02Me. This compound was prepared from MeO-Suc- Ala-Ala-OH in 22% yield by the same procedure as described in Example PKC1, except that sodium methoxide in methanol was used for enol ester hydrolysis, single spot on tic, Rf 2 = 0.43 (CHCl3:MeOH = 9:1); MS, m/e = 317 (M+ + l). Anal. Calcd. for C13H20O7N4 l/3 H20: C, 48.44; H, 6.46; N, 8.69. Found: C, 48.56; H, 6.39; N, 8.69.
EXAMPLE PKC9 MeO-Suc-Ala-Ala-Pro-DL-Abu-C02Me. This compound was prepared from MeO-Suc-Ala-Ala-Pro-DL-Abu-OH in 22% yield by the procedure described in Example PKC8; foam, single spot on tic, R = 0.66 (CHCl3:MeOH = 5:1). Anal. Calcd. for C22H3409N4Η20: C, 51.53; H, 7.02; N, 10.85. Found: C. 51.11; H, 7.03; N,
10.88.
EXAMPLE PKC10 MeO-Suc-Val-Pro-DL-Phe-C02Me. This compound was prepared from MeO- Suc-Val-Pro-Phe-OH in 42% yield by the same procedure as described in Example PKC8; foam, single spot on tic, Rf 2 0.57 (CHCl3:MeOH = 9:1); MS, m/e = 517 (M+).
Anal. Calcd. for C26H3508N32/3 H20: C, 58.96; H, 6.90; N, 7.93. Found: C, 58.92; H, 6.96; N, 7.89.
EXAMPLE PKC11 Bz-DL-Ala-C02-n-Bu. This compound was prepared from Bz-Ala-OH in 45% yield by the procedure described in Example PKC1, except that /i-butyl oxalylchloride was used for the Dakin-West reaction and sodium .z-butoxide in .z-butanol was used for enol ester hydrolysis; colorless oil, single spot on tic, Rf 2 = 0.72 (CHCl3:MeOH = 9:1); MS, m/e = 277 (M+).
EXAMPLE PKC12 Bz-DL-Ala-C02BzI. This compound was prepared from Bz-Ala-OH in 26% yield by the procedure described in Example PKC1, except that benzyl oxalyl chloride was used in place of ethyl oxayl chloride and sodium benzyloxide in benzyl alcohol was used for enol ester hydrolysis; single spot on tic, Rf 2 =0.69 (CHCl3:MeOH = 9:1); mp 95-97 °C; MS, m/e = 312 (M+ + l). Anal. Calcd. for C18H1704N.l/2 H20: C, 67.48; H, 5.66; N, 4.37. Found: C, 67.78; H, 5.55; N, 4.66.
EXAMPLE PKC13 Z-Ala-DL-Ala-C02-ϋ-Bu. This compound was prepared from Z-Ala-Ala-OH in 14% yield by the procedure described in Example PKC1, except that /:-butyl oxalyl chloride was used in the Dakin-West reaction and sodium π-butoxide was used for enol ester hydrolysis; oil, single spot on tic, Rf 2 = 0.45 (CHCl3:MeOH = 9:1); MS, m/e =
378 (M+). Anal. Calcd. for C19H26OgN2T/3 H20: C, 59.35; H, 7.00; N, 7.29. Found: C, 59.41; H, 7.03; N, 7.10.
EXAMPLE PKC14 Z-Ala-DL-Ala-C02BzI. This compound was prepared from Z-Ala-Ala-OH in 36% yield by the procedure described in Example PKC1, except that benzyl oxalyl chloride was used in the Dakin-West reaction and sodium benzyloxide in benzyl alcohol was used for enol ester hydrolysis; single spot on tic, Rf 2 = 0.55 (CHCl3:MeOH = 9:1);
MS, m/e = 413 (M+ + l). Anal. Calcd. for C22H24OgN2: C, 64.06; H, 5.87; N, 6.79. Found: C, 63.79; H, 5.95; N, 6.72.
EXAMPLE PKC15 Z-Ala-Ala-DL-Abu-C02BzI. This compound was prepared from Z-Ala-Ala-Abu- OH in 31% yield by the procedure described in Example PKC1, except that benzyl oxalyl chloride was used in the Dakin-West reaction and sodium benzyloxide in benzyl alcohol was used for enol ester hydrolysis; single spot on tic, Rf 2 = 0.40 (CHCl3:MeOH = 9:1); mp 124-125 °C; MS, m/e = 498 (M + + l). Anal. Calcd. for C26H3107N32/3 H20: C, 61.28; H, 6.39; N, 8.24. Found: C, 61.14; H, 6.65; N, 7.94. EXAMPLE PKC16
Bz-DL-Ala-COOH. The hydrolysis procedure of Tsushima et al., /. Org. Chem., 49:1163-1169 (1984) was used. Bz-DL-Ala-C02Et (540 mg, 2.2 mmol) was added to a solution of 650 mg of sodium bicarbonate in an aqueous 50% 2-propanol solution (7.5 mL of H20 and 2-propanol) and stirred at 40 °C under nitrogen. After adding ethyl acetate and a saline solution to the reaction mixture, the aqueous layer was separated and acidified with 2N HCl and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude hydrolysis product was chromatographed on silica gel and eluted with methylene chloride and methanol to obtain an oil (150 mg, 31%); single spot on tic, Rf 4 = 0.68 («-butanol:acetic acid:pyridine:H20 = 4:1:1:2). Anal. Calcd. for CnHπ04N.3/4
H20: C, 56.28; H, 5.37; N, 5.97. Found: C, 56.21; H, 5.46; 5.66.
EXAMPLE PKC17 Z-Leu-DL-Nva-COOEt. This compound was prepared from Z-Leu-Nva-OH in 60 % yield by the procedure described in Example PKC1; oil, one spot on tic, Rf = 0.49 (CHCl3:MeOH = 20: 1). NMR (CDC13) δ: 0.91 (t, 9H), CH3; 1.25 (t, 3H), CH3;
1.38 (q, 2H), OCH2CH3; 1.64 (m, 6H), CH2; 1.85 (m, IH), CH(CH3)2; 4.34 (m, IH)
CH2CH(NHCOOCH2Ph)CONH; 5.12 (d, 3H) NHCH(CO)CH2 and OCH2Ph; 5.32 (d,
IH) NH; 6.71 (d, IH) NH; 7.36 (s, 5H) Ph.
Z-Leu-DL-Nva-enol ester, the precursor of Z-Leu-DL-Nva-COOEt was synthesized by the same procedure as described in Example PKC1 and purified by column chromatography, oil, one spot on tic. NMR (CDC13) δ: 0.96 (t, 9H); 1.25 (t,
3H); 1.41 (t, 2H); 1.54 (m, 4H); 1.72 (m, 3H); 2.80 (t, 2H); 4.20 (q, 2H); 4.43 (q, 2H);
5.16 (q, 2H); 5.23 (s, IH); 7.37 (m, 5H); 11.33 (s, IH).
EXAMPLE PKC18 Z-Leu-DL-Phe-COOEt. This compound was prepared from Z-Leu-Phe-OH in 30 % yield by the procedure described in Example PKC1; oil, one spot on tic, Rf =
0.47 (CHCl3:MeOH = 50:1). NMR (CDC13) δ: 0.88 (d, 9H), OCH2CH3 and
(CH3)2CH; 1.35 (q, 2H), OCH2CH3; 1.56 (q, 2H), (CH3)2CHCH2CH; 3.03 (m, IH),
(CH3)2CH; 4.32 (m, 2H), NHCH(CO)CH2; 5.08 (s, 4H) CH2Ph; 5.40 (m, IH) NH; 6.61
(d, IH) NH; 7.31 (s, 5H) Ph; 7.35.(s, 5H) Ph. Z-Leu-DL-Phe-enol ester, the precursor of Z-Leu-DL-Phe-COOEt was synthesized by the same procedure as described in Example PKC1 and purified by column chromatography, oil, one spot on tic. NMR (CDC13) δ: 0.86 (t, 3H); 0.99 (t,
3H); 1.24 (t, 3H); 1.40 (t, 3H); 1.52 (m, 2H); 1.83 (m, 2H); 4.23 (m, 4H); 4.39 (q, 2H);
5.10 (t, 2H); 5.18 (s, IH); 7.26 (m, 5H); 7.34 (m, 5H); 8.89 (s, IH). EXAMPLE PKC19
Z-Leu-DL-Abu-COOEt. This compound was prepared from Z-Leu-Abu-OH in
33 % yield by the procedure described in Example PKC1; oil, one spot on tic, Rf =
0.66 (CHCl3:MeOH = 20:1). NMR (CDC13) δ: 0.96 (t, 9H), OCH2CH3 and
(CH3)2CH; 1.26 (t, 3H), CH2CH2CH3; 1.37 (q, 2H), OCH2CH3; 1.66 (q, 2H), (CH3)2CHCH2CH ; 2.00 (m, IH), CH(CH3)2; 4.12 (q, 2H) CHCH2CH3; 4.34 (m, IH)
NHCH(CONH)CH2CH(CH3)2; 5.12 (q, 3H) CH2Ph and CONH(Et)CHCOCOO; 5.29
(t, IH) NH; 6.79 (d, IH) NH; 7.35 (s, 5H) Ph.
Z-Leu-DL-Abu-enol ester, the precursor of Z-Leu-DL-Abu-COOEt was synthesized by the same procedure as described in Example PKC1 and purified by column chromatography, oil, one spot on tic. NMR (CDC13) δ: 0.98 (t, 6H); 1.12 (t,
3H); 1.24 (t, 3H); 1.41 (t, 3H); 1.73 (m, 4H); 2.86 (q, 2H); 4.20 (q, 2H); 4.31 (m, IH);
4.42 (q, 2H); 5.15 (q, 2H); 5.21 (s, IH); 7.34 (m, 5H); 11.29 (s, IH).
EXAMPLE PKC20 Ala-DL-Lys-COOEfHCl. To a solution of N-carbobenzyloxyalanyl-Ne- carbobenzyloxylysine (1.88 g, 3.9 mmol), 4-dimethylaminopyridine (21 mg, 0.17 mmol), and pyridine (1.0 mL, 12.4 mmol) in THF (7 mL) was added ethyl oxalyl chloride (0.9 mL, 8.0 mmol) at a rate sufficient to start refluxing. The mixture was refluxed gently for 3 hr, treated with water (4 mL), and stirred vigorously at room temperature for 30 min. The mixture was extracted with ethyl acetate, the organic extracts were washed with water, dried over MgS04 and evaporated to give an oily residue (1.56 g). To a solution of the enol ester (1.56 g, 2.7 mmol) in anhydrous ethanol was added dropwise a solution of sodium ethoxide in ethanol at room temperature until the solution turned clear yellow. Ethanol was removed and the residue was dissolved in ethyl acetate. The organic solution was washed with water, dried over MgS04, and evaporated to give a residue. This residue was then purified by column chromatography and the product was eluted with chloroform-methanol. The solvent was removed and Z-Ala-DL-Lys(Z)- C02Et was obtained as a hygroscopic powder (328 mg, 16 %), single spot on tic, Rf 2 =
0.53 (CHCl3:MeOH = 9:1); MS, m/e = 542 (M + + l).
N-Carbobenzoxyalanyl-DL-Necarbobenzoxylysine keto ethyl ester, Z-Ala-DL- Lys(Z)-C02Et (328 mg, 0.61 mmol) was deprotected with liquid HF containing anisole at 0 °C for 30 min. The HF was removed under reduced pressure. The residual oil was dissolved in absolute ethanol. HCl/ethanol was added to the solution, and ethanol was removed in vacuo. The residue was washed by decantation with ether to give a semi solid (216 mg, 100 %); single spot on tic (>ι-butanol:acetic acid:pyridine:H20 = 4:1:1:2).
EXAMPLE PKC21 Bz-DL-Lys-COOEtHCl. This compound was prepared from Bz-DL-Lys(Z)-
COOEt in 62% yield by the procedure described in Example PKC20; one spot on tic, Rf 4 = 0.57 («-butanol:acetic acid:pyridine:H20 = 4:1:1:2). The precursor, Bz-DL- Lys(Z)-COOEt was prepared from Bz-Lys(Z)-OH in 100% yield by the procedure described in Example PKC1; powder, one spot on tic, Rf 2 = 0.75 (CHCl3:MeOH = 9:1); MS, m/e = 440 (M+). Anal, Calcd. for C^H^OgN-,^ H20: C, 63.70; H, 6.53;
N, 6.19. Found: C, 63.49; H, 6.51; N, 5.92.
EXAMPLE PKC22 Bz-DL-Arg-COOEfHCI. This compound was prepared from Bz-DL-Arg(Z)- COOEt in 99% yield by the procedure described in Example PKC20; one spot on tic, Rf 4 = 0.71 (n-butanol:acetic acid:pyridine:H20 = 4:1:1:2), Sakaguchi reagent positive. Bz-DL-Arg(Z)-COOEt was prepared from Bz-DL-Arg(Z)-OH in 19% yield by the procedure described in Example PKC20, Rf 2 = 0.38 (CHC /.MeOH = 9:1); mp 140- 142 °C; MS, m/e = 468 (M+). Anal. Calcd. for C^H^OgN.,: C, 61.53; H, 6.02; N, 11.96. Found: C, 61.96; H, 6.48; N, 12.34.
EXAMPLE PKC23 H-Gly-DL-Lys-COOEt2HCI. This compound was prepared from Z-Gly-DL-
Lys(Z)-COOEt in 92% yield by the procedure described in Example PKC20; Rf 4 = 0.21 («-butanol:acetic acid:pyridine:H20 = 4:1:1:2). Z-Gly-DL-Lys(Z)-COOEt was prepared from Z-Gly-Lys(Z)-OH in 9% yield by the procedure described in Example PKC20, one spot on tic, R = 0.68 (CHCl3:MeOH = 5: 1); MS, m/e = 528 (M + + l). EXAMPLE PKC24
H-Pro-DL-Lys-COOEf2HCI. This compound was prepared from Z-Pro-DL- Lys(Z)-COOEt in 100% yield by the procedure described in Example PKC20; one spot on tic (n-butanol:acetic acid:pyridine:H20 = 4:1:1:2). Z-Pro-DL-Lys(Z)-COOEt was prepared from Z-Pro-Lys(Z)-OH in 15% yield by the procedure described in Example PKC20; Rf2 = 0.73 (CHCl3:MeOH = 9:1); MS, m/e 568 (M+ + 1).
EXAMPLE PKC25 H-Phe-DL-Lys-COOEt2HCl. This compound was prepared from Z-Phe-DL- Lys(Z)-COOEt in 39% yield by the procedure described in Example PKC20; one spot on tic («-butanol:acetic acid:pyridine:H20 = 4:1: 1:2). Z-Phe-DL-Lys(Z)-COOEt was prepared from Z-Phe-Lys(Z)-OH as previously described in 9% yield, Rf 2 = 0.68
(CHCl3:MeOH = 9:1); MS, m/e = 482 (M+).
EXAMPLE PKC26 H-Leu-AIa-D Lys-COOEt'2HCl. This compound was prepared from Z-Leu- Ala-DL-Lys(Z)-COOEt in 52% yield by the procedure described in Example PKC20; one spot on tic («-butanol:acetic acid:pyridine:H20 = 4:1:1:2).
Z-Leu-Ala-DL-Lys(Z)-COOEt was prepared from Z-Leu-Ala-DL-Lys(Z)-OH in 5% yield by the previously described Dakin West reaction, Rf 3 = 0.34 (CHCl3:MeOH = 19:1); MS, m/e = 609 (M+-OCH2CH3).
EXAMPLE PKC27 Simple Amino Acid, Di- and Tripeptide Enol Esters (General Procedure). A modified Dakin-West procedure was used (Charles et al., supra) and is illustrated with the synthesis of Z-Leu-DL-Phe-EE. To a stirred solution of Z-Leu-Phe-OH (6.19 g, 15.0 mmol), 4- dimethylaminopyndine (0.183 g; 1,5 mmol) and pyridine (4.75 g, 4,85 ml, 60 mmol) in tetrahydrofuran (45 ml) warmed 50 ° C was added ethyl oxalyl chloride (4.30 g, 3.52 ml, 31.5 mmol) at a rate sufficient to initiate refluxing. The mixture was then heated at a gentle reflux for 4 h. After cooling to room temperature the mixture was treated with water (25 ml) and stirred vigorously at room temperature for 30 min. The mixture was extracted with ethyl acetate (150 ml) and after separation of the organic layer, the water layer was saturated with solid (NH4)2S04 and re-extracted 2-times with 25 ml ethyl acetate. The combined organic phases were washed 2-times with 75 ml water, 2-times with 50 ml of satd. NaCl, decolorized with carbon and dried over MgS04. After evaporation of the solvent, the crude enol ester (8,36 g, 98%) was flash-chromatographed on silica gel and the product was eluted with a AcOEt. The solvent was evaporated in vacuo (rotavaporator) and the pure enol ester was obtained as a oil (7.22 g, 85%); single spot on TLC, Rf = 0.84, A; 0.68, C.
Z-Leu-Nva-EE. This compound was prepared from Z-Leu-Nva- OH using the general procedure and purified by flash chromatography on silica gel using CHCl3:MeOH = 50:1 v/v as eluent. Yield 95%, single spot on TLC, Rf = 0.92, C; 0.28, L. Z-Leu-Abu-EE. This compound was prepared from Z-Leu-Abu- OH in 78% yield the general procedure described above. Purification by flash obramatography on silica gel. Eluent, CHCl3:MeOH = 50:1 v/v, single spot on TLC, Rf = 0.86, A.
PhCO-Abu-EE. This compound was prepared from PhCO-Abu-OH in 26% yield by the general procedure as described above. Purification by flash chromatography on silica gel. Eluent CHC13, single spot on TLC, Rf = 0.60, M.
(CH3) CH(CH2)2CO-Abu-EE. This compound was prepared from (CH3)2CH(CH2)2CO-Abu-OH in 82% yield by the general procedure as described
above. Purification by flash chromatography on silica gel. Eluent AcOEt, single spot on TLC, Rf = 0.72, C.
(CH3CH2CH2)2 CH CO-Abu-EE. This compound was prepared from (CΗ3CΗ2CΗ2)2CΗ CO-Abu-OH in 100% yield by the general procedure described above. Purification by flash chromatography on silica gel. Eluent AcOEt, single spot on TLC, Rf = 0.78, C; 0.81, K.
Ph(CH^)6CO-Abu-EE. This compound was prepared from Ph(CH2)6CO-Abu-OH in 86% yield by the general procedure described above. Purification by flash chromatography on silica gel. Eluent AcOEt. Single spot on TLC, Rf =0.74, C.
Z-Leu-4-Cl-Phe-EE. This compound was prepared from Z-Leu-4-Cl-Phe-OH in 69% yield by the general procedure described above. Purification by flash chromatography on silica gel. Eluent AcOEt, single spot on TLC, Rf = 0.77, C; 0.78, K. Z-Leu-Leu-Abu-EE. This compound was prepared from Z2-Leu-Leu-Abu-OH in 62% yield by the general procedure described above. Purification by flash chromatography on silica gel. Element CHCl3:MeOH = 50:1 v/v. Single spot on TLC, Rf = 0.89, A; 0.75, M.
Z-Leu-Leu-Phe-EE. This compound was prepared from Z-Leu-Leu-Phe-OH in 60% yield by the general procedure described above. Purification by flash chromatography on silica gel. Eluent CHCl3:MeOH = 50:1 v/v. Single spot on TLC, Rf = 0.80, K; 0.70, M.
2-NapS02-Leu-Leu-Abu-EE. This compound was prepared from 2-NapS02-Leu-Abu-OH in 73% yield by the general procedure described above. Purification by flash chromatography on silica gel. Eluent AcOEt, single spot on TLC,
Rf = 0.71, K; 0.54, C.
2-NapS02-Leu-Leu-Abu-EE. This compound was prepared from 2-NapS02-Leu-Leu-Abu-OH in 74% yield by the general procedure described above. Purification by flash chromatography on silica gel. Eluent AcOEt: AcOH = 200:1 v/v. Single spot on TLC, Rf = 0.69. K. .
EXAMPLE PKC28 Z-Leu-Phe-COOEt. Single Aminoacid, Di-and Tripeptide- ketoesters (General Procedure). To a stirred solution of 8.53 g (15.0 mmol) of Z-Leu-Phe-EE in 40 ml anhydrous ethanol at room temperature was added dropwise a solution of sodium ethoxide (0.204 g; 3.0 mmol) in 20.0 ml anhydrous ethanol. The color of the reaction mixture change from colorless or pall yellow to deep yellow or orange dependent on enol-ester. Then the reaction mixture was stirred at room temperature for 4-5 hours, the ethanol was then evaporated in vacuo (rotavaporator) and the residue treated with 200 ml ethyl ether (or 200 ml ethyl acetate in the case of the tripeptide). The ether (ethyl acetate) solution was washed with 2 x 75 ml H20, 2 x 75 ml satd. NaCl, decolorized with carbon and dried over MgS04. After evaporation of solvent, the crude product 6.09 g (89.7%) was flash chromatographed on silica gel using CHC13: MeOH = 50:1 v/v. Evaporation of solvent give pure Z-Leu-Phe-COOEt (4.08 g; 58.0%) as a thick oil. Single spot on TLC, Rf = 0.60, A; 0.47, M. Mass spectrum, FB-MS [(M+ 1)/Z] = 469.
EXAMPLE PKC29 Z-Leu-Nva-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent CHC13: MeOH = 100:1 v/v, yield 86.6%, thick, colorless oil, single spot on TLC, Rf = 0.49, A; 0.37, M. Mass spectrum FB-MS [(M+ l)/Z] = 421.
EXAMPLE PKC30 Z-Len-Abu-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent CHC13, yield 82%, thick, pale yellow oil, single spot on TLC, Rf = 0.66, A. Mass spectrum, CI-MS [(M+ 1)/Z] = 407.
EXAMPLE PKC31 PhCO-Abu-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent CHCl3:MeOH = 50:1 v/v, yield 83%, oil, single spot on TLC, Rf = 0.44, M. Mass spectrum, M/Z 263 (M+); CI-MS, 264 ((M+ l)/Z).
EXAMPLE PKC32 (CH3)2CH(CH2)2CO-Abu-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent AcOEt, yield 43%, oil, single spot on TLC, Rf = 0.56, C. Mass spectrum EI-MS M/Z 257 (M+); FB-MS, [(M+ 1)/Z] = 258.
EXAMPLE PKC33 CH3CH2CH)2CHCO-Abu-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent CHCl3:MeOH = 50: 1 v/v, thick, yellowish oil, yield 66%, single spot on TLC, Rf = 0.80, C; 0.66, M. Mass spectrum EI-MS M/Z = 285 (M+); CI-MS, [(M+ 1)/Z] = 286.
EXAMPLE PKC34 Ph(CH2)6CO-Abu-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent CHCl3:MeOH = 50:1 v/v, yield 64%, pale yellow oil, single spot on TLC, Rf = 0.29, M. Mass spectrum EI-MS M/Z = 347 (M+), FB-MS, [(M+ 1)/Z] = 348.
EXAMPLE PKC35 Z-Leu-4-Cl-Phe-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent AcOEt, yield 100%, colorless oil, single spot on TLC, Rf = 0.71, C. Mass spectrum FB-MS M/Z = 503(M+).
EXAMPLE PKC36 Z-Leu-Leu-Abu-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent CHCl3:MeOH = 50:1 v/v, yield 79.2%, very thick, colorless oil, single spot on TLC, Rf = 0.28. M. Mass spectrum FB-MS, [(M+ 1)/Z] = 520.
EXAMPLE PKC37 Z-Leu-Leu-Phe-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent CHCl3:MeOH = 50:1 v/v, yield 33%, oil, single spot on TLC, Rf = 0.56, M. Mass spectrum, FB-MS, [(M+ l)/Z] = 582.
EXAMPLE PKC38 2-NapS02-Leu-Abu-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent CHCl3:MeOH = 50:1 v/v, yield 38%, thick oil, single spot on TLC, Rf = 0.71, K; 0.54, A. Mass spectrum FB-MS, [(M+ l)/Z] = 463.
EXAMPLE PKC39 2-NapS02-Leu-Leu-Abu-COOEt. This was prepared by the preceding general procedure. Purification by flash chromatography on silica gel, eluent AcOEt:AcOH = 200:1 v/v, yield 61%, semi-solid, single spot on TLC, Rf = 0.67, K. Mass spectrum FB-MS, [(M+ l)/Z] = 576.
EXAMPLE PKC40 Z-Leu-Met-C02Et. This compound was prepared by the above procedure. Yellow oil, single spot on TLC, Rf = 0.52 (CHC13:CH3OH = 50: 1), yield 46% (from dipeptide), MS (FAB) 454 (m+ 1). EXAMPLE PKC41
Z-Leu-NLeu-C02Et. This compound was prepared by the above procedure. Pale yellow oil, single spot on TLC, Rf = 0.57 (CHCl3:CH3OH = 50:1), yield 53% (from dipeptide), MS (FAB) 434 (m+ 1).
EXAMPLE PKC42 Synthesis of n-Butyl Oxalyl Chloride. This was prepared by a literature procedure
(Warren and Malee, supra). N-Butanol (0.1 mol. 7.41 g) was added dropwise to oxalyl chloride (0.5 mol. 63.5 g) at -10 °C. After the addition was completed, the reaction mixture was stirred for 20 min. at r.t. and distilled, giving 15.0 g (91.18 mol. 91%) of the product n-butyl oxalyl chloride, bp 58-60 °C (0.6 mm Hg). Z-Leu-Phe-C02Bu. This compound was prepared from Z-Leu- Phe-OH and butyl oxalyl chloride in 43% yield by the procedure described for the synthesis of Z-Leu-Phe-C02Et, except that butyl oxalyl chloride was used in place of ethyl oxalyl chloride and sodium butyloxide in butanol was used for enol ester hydrolysis. Single spot on TLC, Rf = 0.54 (CHCl3:CH3OH = 50:1) MS(FAB) m/e = 497 (m+ 1), lR NMR (CDC13) ok.
EXAMPLE PKC43 Z-Leu-Abu-C02Bu. This compound was prepared by the above procedure. Single spot on TLC, Rf = 0.53 (CHCl3:CH3OH = 50:1), yield = 36%, pale yellow oil, MS (FAB) m/e = 435 (M+ l), 1H NMR (CDC13) ok. EXAMPLE PKC44
Synthesis of Benzyl Oxalyl Chloride. Benzyl alcohol (0.15 mol. 16 g) was added dropwise to oxalyl chloride (0.75 mol. 95 g) at 5-10 °C. After the addition was complete, the reaction was stirred for 20 min. at r.t. The excess oxalyl chloride was distilled and recycled. Then the mixture was distilled under vacuo, giving 26 g (0.12 mol. 86%) of benzyl oxalyl chloride, bp. 110-112 °C (0.6 mm-Hg). H!NMR (CDC13)
7.39 (s, 5H), 5,33 (s.2H).
Z-Leu-Phe-C02Bzl. This compound was prepared from Z-Leu- Phe-OH and benzyl oxalyl chloride in 17% yield by the procedure described in the synthesis of Z-Leu-Phe-C02Et, except that benzyl oxalyl chloride was used in place of ethyl oxalyl chloride and sodium benzyloxide in benzyl alcohol was used for enol ester hydrolysis.
Single spot on TLC, Rf = 0.63 (CHCl3:CH3OH = 50:1). Pale yellow solid, mp 117-119 °C. MS(FAB) m/e = 532 (m+ l). HJNMR ok.
EXAMPLE PKC45 Z-Leu-Abu-C02Bzl. This compound was prepared by the above procedure. Single spot on TLC. Rf = 0.51 (CHCl3:CH3OH = 50:1), pale yellow oil, MS(FAB) m/e
= 469 (m+ l), yield = 26%.
EXAMPLE PKC46 Z-Leu-Phe-COOH. Dipeptide Ketoacids (General Procedure). To a stirred solution of 0.53g (1,13 mmol) Z-Leu-Phe-COOEt in 6.0 ml methanol was added 1.27 ml (1.27 mmol) 1M NaOH. The color of the reaction mixture turned dark yellow and a small amount of solid was deposited. The reaction was run at room temperature and progress of the hydrolysis was checked on TLC. After 24 h. no more substrate was detected. The reaction mixture was chilled in one ice bath at 5 °C, acidified with 1M HCl to pH = 3 and extracted with AcOEt (2 x 50 mL). The organic extract were washed with 2 x 50 ml H20 and if necessary, decolorized with carbon and dried over
MgS04. After evaporation of the solvent (rotavaporator), the residue (thick oil) were titurated with 2 x 25 ml n-hexane and dried in vacuo. Yield 0.39 g (78%) of colorless,
very thick oil. TLC, main spot at Rf = 0.24, trace of impurity at Rf = 0.78, 1. Mass spectrum, FB-MS [(M+ l)/Z] = 441.
EXAMPLE PKC47 Z-Leu-Abu-COOH. This compound was prepared from Z-L-Leu- Abu-COOEt in 83% yield by the general procedure as described above; TLC, main spot at Rf =
0.14, trace of impurity at Rf = 0.73, 1. Mass spectrum, FB-MS [(M+ l)/Z] = 379.
EXAMPLE PKC48 Z-Leu-Phe-CONH-Et. To a stirred solution of Z-Leu-Phe-OH (20 g, 48.5 mmole), 4-dimethylaminopyridine (0.587 g, 4.8 mmole),and pyridine (15.7 ml, 194 mmole) in anhydrous THF (100 ml) was added ethyl oxalyl chloride (11.4 ml, 101.8 mmole) at a rate sufficient to initiate refluxing. The mixture was gently refluxed for 4 hours, cooled to room temperature, and water (80 ml) was added. The reaction mixture was stirred vigorously for 30 min, and extracted with ethyl acetate (3 x 100 ml). The combined organic layers were washed with water (2 x 100 ml), saturated sodium chloride (2 x 100 ml), decolorized with decolorizing carbon, dried over magnesium sulfate, and concentrated, leaving a dark orange oil. Chromatography on a silica gel column with CHCl3/CH3OH (50: 1 v/v) afforded 14.63 g (y = 53 %) of Z-Leu-Phe- enolester. The product was a yellow oil. Single spot on TLC, Rf = 0.77 (CHCL3/CH3OH 50:1). NMR (CDC13) ok. To a stirred pale yellow solution of the Z-Leu-Phe-enolester (14.63 g, 25.73 mmole) in anhydrous ethanol (50 ml) was added a solution of sodium ethoxide (0.177 g, 2.6 mmole) in ethanol (5 ml). The orange solution was stirred for 3 hours at room temperature, then the ethanol was evaporated and the residue was treated with ethyl ether (300 ml). The ether layer was washed with water (2 x 100 ml), saturated sodium chloride (2 x 100 ml), dried over magnesium sulfate, and concentrated, leaving a orange oil. Chromatography on a silica gel column with CHCl3/CH3OH (50:1 v/v) afforded 7.76 g (y = 64 %) of the α -ketoester Z-Leu-Phe-COOEt. The product was a yellow oil. Single spot on TLC, Rf = 0.44 (CHCl3/CH3OH 50:1). NMR (CDC13) ok. MS (FAB, calcd. for C26H32N206: 468.6), m/e = 469 (M+ l). The α -carbonyl group of Z-Leu-Phe-COOEt was protected by following procedure. A solution of Z-Leu-Phe-COOEt (1 g, 2.13 mmole) in 5 ml of CH2C12 was added 1,2-ethanedithiol (0.214 ml, 2.55 mmole), followed by 0.5 ml of boron trifluoride
etherate. The solution was stirred overnight at room temperature. Water (20 ml) and ethyl ether (20 ml) were added. The organic layer was separated, washed with water (2 x 10 ml), saturated sodium chloride (2 x 10 ml), dried over magnesium sulfate, and evaporated to afford 0.98 g (y = 84 %) yellow semisolid. The protected α-ketoester (0.98 g, 1.8 mmole) was dissolved in ethanol (5 ml), cooled to 0-5 °C in a ice bath, and ethylamine was bubbled through the solution until 2.43 g (54 mmole) had been added. The reaction mixture was allowed to warm to room temperature slowly, and stirred overnight. The mixture was filtered, a white precipitate was removed, leaving a yellow semisolid. Chromatography on a silica gel column with CHCl3/CH3OH (30:1 v/v) afford 0.63 g (y = 75 %) of Z-Leu-Phe-CONH¬
Et. The product was a pale yellow solid. Single spot on TLC, Rf = 0.60 (CHCl3/CH3OH 20:1); mp 145-147 °C. Anal, calcd. for C26H33N305. 467.56; C, 66.79; H, 7.11; N,8.99; found: C, 66.59; H, 7.09; N, 8.95. NMR (CDC13) ok. MS (FAB) m/e = 468 (M+ l). EXAMPLE PKC49
Z-Leu-Phe-CONH-nPr. This compound was synthesized from the protected α- ketoester and propylamine in 92 % yield by the procedure described in Example PKC48. Single spot on TLC, Rf = 0.50 (CHCl3/CH3OH 50:1); mp 152-153 °C. Anal, calcd. for C27H35N305: 481.57; C, 67.33; H, 7.33; N, 8.72. Found: C, 67.21; H, 7.38; N, 8.64. NMR (CDC13) ok. MS (FAB) m/e = 482 (M+ 1).
EXAMPLE PKC50 Z-Leu-Phe-CONH-nBu. This compound was synthesized from the protected α- ketoester and butylamine in 67 % yield by the procedure described in Example PKC48. Single spot on TLC, Rf = 0.50.(CHC13/CH3OH 50: 1); mp 152-153 °C. Anal, calcd. for C2gH37N305: 495.59; C, 67.85; H, 7.52; N, 8.48. Found: C, 67.70; H, 7.57; N, 8.43.
NMR (CDC13) ok. MS (FAB) m/e = 496 (M+ 1).
EXAMPLE PKC51 Z-Leu-Phe-CONH-iBu. This compound was synthesized from the protected α- ketoester and isobutylamine in 53 % yield by the procedure described in Example PKC48. Single spot on TLC, Rf = 0.54 (CHCl3/CH3OH 50:1); mp 152 °C. Anal. calcd. for C^H^N^: 495.59; C, 67.85; H, 7.52; N, 8.48. Found: C, 67.77; H, 7.56; N, 8.40. NMR (CDC13) ok. MS (FAB) m/e = 496 (M+ l).
EXAMPLE PKC52 Z-Leu-Phe-CONH-BzI. This compound was synthesized from the protected α- ketoester and benzylamine in 40 % yield by the procedure described in Example PKC48. After reacting overnight, ethyl acetate (60 ml) was added. The mixture was filtered to remove a white precipitate. The solution was washed with cooled 1 N HCl
(3 x 25 ml), water (1 x 20 ml), saturated sodium chloride (2 x 20 ml), and dried over magnesium sulfate. The solution was evaporated leaving a yellow solid. Chromatography on a silica gel column with CHCl3/CH3OH 30:1 v/v) afforded a yellow solid. Single spot on TLC, Rf = 0.45 (CHCl3/CH3OH 30:1); mp 160-162 °C. Anal, calcd. for C31H35N305: 529.61; C, 70.30; H, 6.66; N, 7.93. Found: C, 70.18; H,
6.67; N, 7.99. NMR (CDC13) ok. MS (FAB) m/e = 530 (M+ l).
EXAMPLE PKC53 Z-Leu-Phe-CONH-(CH2)2Ph. This compound was synthesized from the protected α-ketoester and phenethylamine in 50 % yield by the procedure described in Example PKC52. Single spot on TLC, Rf = 0.50 (CHCl3/CH3OH 30:1); mp 151-153
°C. Anal, calcd. for C32H37N305: 543.66; C, 70.70; H, 6.86; N, 7.73. Found: C, 70.54; H, 6.88; N, 7.74. NMR (CDC13) ok. MS (FAB) m/e = 544 (M+ l).
EXAMPLE PKC54 Z-Leu-Abu-CONH-Et. This compound was synthesized from protected α- ketoester derived from Z-Leu-Abu-C02Et and ethylamine in 64 % yield by the procedure described in Example PKC48. Single spot on TLC, Rf = 0.36 (CHCl3/CH3OH 50;1); mp 130-132 °C. Anal, calcd. for C21H31N305: 405.45; C, 62.20; H, 7.71; N, 10.36. Found: C, 61.92; H, 7.62; N, 10.31. NMR (CDC13) ok. MS (FAB) m/e = 406 (M+ l). EXAMPLE PKC55
Z-Leu-Abu-CONH-nPr. This compound was synthesized from the corresponding protected α-ketoester and propylamine in 47 % yield by the procedure described in Example PKC48. Single spot on TLC, Rf = 0.28 (CHCl3/CH3OH 50:1); mp 134-135 °C. Anal, calcd. for C22H33N305: 419.50; C, 62.98; H, 7.93; N, 10.02. Found: C, 62.84; H, 7.97; N, 9.94. NMR (CDC13) ok. MS (FAB) m/e = 420 (M+ l).
EXAMPLE PKC56 Z-Leu-Abu-CONH-nBu. This compound was synthesized from the corresponding protected α-ketoester and butylamine in 42 % yield by the procedure described in Example PKC48. Single spot on TLC, Rf = 0.54 (CHCl3/CH3OH 50:1); mp 135-136 °C. Anal, calcd. for C^I^^Oj: 433.53; C, 63.71; H, 8.13; N, 9.69.
Found: C, 63.48; H, 8.07; N, 9.67. NMR (CDC13) ok. MS (FAB) m/e = 434 (M+ l).
EXAMPLE PKC57 Z-Leu-Abu-CONH-iBu. This compound was synthesized from the corresponding protected α-ketoester and isobutylamine in 65 % yield by the procedure described in Example PKC48. Single spot on TLC, Rf = 0.25 (CHCl3/CH3OH 50:1); mp 133-135 °C. Anal, calcd. for C^H^Oj: 433.52; C, 63.72; H, 8.14; N, 9.69. Found: C, 63.46; H, 8.10; N, 9.60. NMR (CDC13) ok. MS (FAB) m/e = 434 (M+ l).
EXAMPLE PKC58 Z-Leu-Abu-CONH-Bzl. This compound was synthesized from the corresponding protected α-ketoester and benzylamine in 29 % yield by the procedure described in
Example PKC52. Single spot on TLC, Rf = 0.56 (CHCl3/CH3OH 30:1); mp 140-141 °C. Anal, calcd. for C2gH33N305: 467.54; C, 66.79; H, 7.11; N, 8.99. Found: C, 66.65; H, 7.07; N, 8.93. NMR (CDC13) ok. MS (FAB) m/e = 468 (M+ l).
EXAMPLE PKC59 Z-Leu-Abu-C0NH-(CH2)2Ph. This compound was synthesized from the corresponding protected α-ketoester and phenethylamine in 51 % yield by the procedure described in Example PKC52. Single spot on TLC, Rf = 0.44 (CHCl3/CH3OH 30:1); mp 156-157 °C. Anal, calcd. for C27H35N305: 481.59; C, 67.34; H, 7.33; N, 8.72. Found: C, 67.38; H, 7.33; N, 8.78. NMR (CDC13) ok. MS (FAB) m/e = 482 (M+ l).
EXAMPLE PKC60 Z-Leu-Abu-C0NH-(CH2)3-N(CH2CH2)20. This compound was synthesized from protected α-ketoester and 4(3-aminopropyl)moφholine in 33 % yield by the procedure described in Example PKC48. After reacting overnight, ethyl acetate (80 ml) was added. The mixture was filtered to remove a white precipitate. The solution was washed with water (3 x 20 ml), saturated sodium chloride (2 x 20 ml), and dried over magnesium sulfate. The solution was evaporated leaving a yellow oil. Chromatography
on a silica gel column with CHCl3/CH3OH (10:1 v/v) afforded a yellow semisolid, which was recrystallized from ethyl acetate/hexane to obtain a pale yellow solid. Single spot on TLC, Rf = 0.42 (CHCl3/CH3OH 10:1); mp 125-126 °C. Anal, calcd. for C^H^Og: 504.63; C, 61.88; H, 7.99; N, 11.10. Found: C, 61,69; H, 7.95; N, 11.07. NMR (CDC13) ok. MS (FAB) m/e = 505 (M+ 1).
EXAMPLE PKC61 Z-Leu-Abu-CONH-(CH2)7CH3 This compound was synthesized from the corresponding protected α-ketoester and octylamine in 67 % yield by the procedure described in Example PKC52. It was white solid. Single spot on TLC, Rf = 0.55 (CHCl3/CH3OH 30:1); p 134-135 °C. Anal, calcd. for C27H43N305: 489.66; C, 66.23;
H, 8.85; N, 8.58. Found: C, 66.19; H, 8.81; N, 8.61. NMR (CDC13) ok. MS (FAB) m/e = 490 (M+ l).
EXAMPLE PKC62 Z-Leu-Abu-CONH-(CH2)2OH. This compound was synthesized from the corresponding protected α-ketoester and ethanolamine in 29 % yield by the procedure described in Example PKC60. The product was a white sticky solid. Single spot on TLC, Rf = 0.42 (CHCl3/CH3OH 10:1); mp 151-153 °C. Anal: calcd. for C21H31N306: 421.49; C, 59.84; H, 7.41; N, 9.97. Found: C, 59.11; H, 7.44; N, 9.81. NMR (CDC13) ok. MS (FAB) m/e = 422 (M+ l). EXAMPLE PKC63
Z-Leu-Abu-C0NH-(CH2)20(CH2)20H. This compound was synthesized from the corresponding protected α-ketoester and 2-(2-aminoethoxy)ethanol in 34 % yield by the procedure described in Example PKC60. The product was white sticky solid. Single spot on TLC, Rf = 0.42 (CHCl3/CH3OH 10:1); mp 103-105 °C. Anal.: calcd. for C^H^N-O-,: 465.55; C, 59.34; H, 7.58; N, 9.03. Found: C, 59.23; H, 7.58; N, 9.01.
NMR (CDC13) ok. MS (FAB) m/e = 466 (M+ l).
EXAMPLE PKC64 Z-Leu-Abu-CONH-(CH2)17CH3. This compound was synthesized from the corresponding protected α-ketoester and octadecylamine in 12 % yield by the procedure described in Example PKC52. The product was a pale yellow solid. Single spot on TLC, Rf = 0.54 (CHCl3/CH3OH 30:1); mp 134-136 °C. Anal: calcd. for
C37H63N305: 629.92; C, 70.55; H, 10.08; N, 6.67. Found: C, 70.71; H, 10.14; N, 6.75. NMR (CDC13) ok. MS (FAB) m/e = 630.2 (M+ l).
EXAMPLE PKC65 Z-Leu-Abu-CONH-CH2-C6H3(OCH3)2. This compound was synthesized from the corresponding protected α-ketoester and 3,5-dimethoxybenzylamine in 45 % yield by the procedure described in Example PKC52. The product was yellow sticky solid. Single spot on TLC, Rf = 0.44 (CHCl3/CH3OH 30:1); mp 153-155 °C. Anal.: calcd. for C^H^N-O.-: 527.62; C, 63.74; H, 7.07; N, 7.96. Found: C, 63.66; H, 7.09; N, 7.92. NMR (CDC13) ok. MS (FAB) m/e = 528.8 (M+ l). EXAMPLE PKC66
Z-Leu-Abu-CONH-CH2-C4H4N. This compound was synthesized from the corresponding protected α-ketoester and 4-(aminomethyl)pyridine in 45 % yield by the procedure described in Example PKC60. The product was greenish yellow solid. Single spot on TLC, Rf = 0.55 (CHCl3/CH3OH 10: 1); mp 124-126 °C. Anal: calcd. for C25H32N405: 468.55; C, 64.08; H, 6.88; N, 11.96. Found: C, 63.88; H, 6.87; N, 11.96.
NMR (CDC13) ok. MS (FAB) m/e =469 (M+ 1).
EXAMPLE PKC67 Z-Leu-Abu-CONH-(CH2)5OH. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 5-amino-l-pentanol. To a solution of protected a-ketoester (1 mmol) in ethanol (3 mL) was added 5-amino-l-pentanol (3 mmol) and stirred overnight at r.t. To the mixture was added AcOEt (25 mL) and white precipitate was filtered. The filtrate was washed with cold IN HCl (2 x 10 mL), water (1 x 10 mL), saturated NaCl (2 x 10 mL) and dried over MgS04. After evaporation of the solvent, chromatography on a silica gel column using solvent CHCl3/CH3OH 10:1 followed by precipitation from AcOEt/hexane afforded a white solid (42% yield). Single spot on TLC, Rf = 0.54 (CHCl3/CH3OH 10:1), mp 122-123
C. *H NMR (CDC13) ok, MS (FAB) m/e = 464 (M+ 1). Anal: calcd. for C^H^NjOg, 463; C, 62.18; H, 8.04; N, 9.06. Found: C, 61.52; H, 7.96; N, 8.98.
EXAMPLE PKC68 Z-Leu-Abu-CONH-(CH2)2OH. This is an alternative synthesis for the compound designated in Example PKC 62. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and ethanolamine by the procedure
described in Example PKC67, and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (40% yield). White solid, single spot on TLC, Rf = 0.42 (CHCl3/CH3OH 10:1), mp 151-154 C. 1H NMR (CDC13) ok, MS (FAB) m/e = 422 (M+ l). Anal: calcd. for C21H31N3Og, 421; C, 59.84; H, 7.41; N, 9.97. Found: C, 59.11; H, 7.44; N, 9.81.
EXAMPLE PKC69 Z-Leu-Abu-CONH-(CH2)20(CH2)2OH. This is an alternative synthesis for the compound designated in Example PKC 63. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 2-(2-aminoethoxy)ethanol by the procedure described in Example PKC67, and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (34% yield). White solid, single spot on TLC, Rf = 0.42 (CHCl3/CH3OH 10:1), mp 103-105 C. 1H NMR (CDC13) ok, MS (FAB) m/e = 466 (M+ l). Anal: calcd. for C^H^N^, 465; C, 59.30; H, 7.58; N, 9.02. Found: C, 59.23; H, 7.58; N, 9.01. EXAMPLE PKC70
Z-Leu-Abu-CONH-CH2CH(OCH3)2. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and aminoacetaldehyde dimethylacetal by the procedure described in Example PKC67, and purified by column chromatography using solvent CHCl3/CH3OH 20: 1 (25% yield). White solid, single spot on TLC, Rf = 0.47 (CHCl3/CH3OH 20:1), mp 99-102 C. 1H NMR (CDC13) ok,
MS (FAB) m/e = 466 (M+ l). Anal: calcd. for C^H^NjO-?, 465; C, 59.30; H, 7.58; N, 9.02. Found: C, 58.95; H, 7.71; N, 9.00.
EXAMPLE PKC71 Z-Leu-Abu-CONH-CH2CH(OC2Hs)2. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and aminoacetaldehyde diethylacetal, and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (36% yield). White solid, single spot on TLC, Rf = 0.37 (CHCl3/CH3OH 20:1), mp 100-103 C. 1H NMR (CDC13) ok, MS (FAB) m/e =494 (12%, M+ 1), 448 (100%, M+ 1-45). Anal: calcd. for C25H39N307, 493; C, 60.83; H, 7.96; N, 8.51. Found: C, 60.73; H, 7.98; N, 8.42.
EXAMPLE PKC72 Z-Leu-Abu-CONH-CH2-C6H8(l 3^3-(CH3)3-5-OH). This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 3-aminomethyl-3,5,5-trimethyl-cyclohexanol, and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (51% yield). White solid, single spot on TLC, Rf =
0.55 (CHCl3/CH3OH 30:1), mp 59-61 C. ~R NMR (CDC13) ok, MS (FAB) m/e = 532 (M+ l). Anal; calcd. for C29H45N306^31; C, 65.51; H, 8.53; N, 7.90. Found, C, 65.21; H, 8.55, N, 7.81.
EXAMPLE PKC73 Z-Leu-Abu-CONH-(CH2)2C6H4(4-OH). This compound was synthesized from
1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 4-(2-aminoethyl)phenol, and purified by column chromatography using solvent CHCl /CH3OH 30:1 (60% yield). White solid, single spot on TLC, Rf = 0.56 (CHCl3/CH3OH 30:1), mp 151-153 C. -R NMR (CDC13) ok, MS (FAB) m/e = 498 (M+ 1). Anal: calcd. for C27H35N3Og, 497; C, 65.17; H, 7.09; N, 8.45. Found, C, 65.16; H, 7.13, N, 8.52.
EXAMPLE PKC74 Z-Leu-Abu-CONH-(CH2)2C6H4(2-OCH3). This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 2-methoxyphenethylamine, and purified by column chromatography using solvent CHCl3/CH3OH 50:1 (71% yield). Yellow solid, single spot on TLC, Rf = 0.47 (CHCl3/CH3OH 50:1), mp 101-103 C.
-K NMR (CDC13) ok, MS (FAB) m/e = 512 (M+ 1). Anal; calcd. for jgH^Og, 511; C, 65.73; H, 7.29; N, 8.21. Found, C, 65.50; H, 7.31; N, 8.19.
EXAMPLE PKC75 Z-Leu-Abu-CONH-(CH2)2C6H4(3-OCH3). This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 3-methoxyphenethylamine, and purified by column chromatography using solvent CHCl3/CH3OH 50:1 (56% yield). YeUow solid, single spot on TLC, Rf = 0.46 (CHCl3/CH3OH 50:1), mp 99-100 C. *H NMR (CDC13) ok, MS (FAB) m/e =512 (M+ l). Anal: calcd. for C^H^Og, 511; C, 65.73; H, 7.29; N, 8.21. Found, C, 65.62; H, 7.34; N, 8.16. EXAMPLE PKC76
Z-Leu-Abu-CONH-(CH2)2CgH4(4-OCH3). This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 4-methoxyphenethylamine, and
purified by column chromatography using solvent CHCl3/CH3OH 50:1 (50% yield). White solid, single spot on TLC, Rf = 0.46 (CHCl3/CH3OH 50:1), mp 152-155 C. *H NMR (CDC13) ok, MS (FAB) m/e = 512 (M+ 1). Anal: calcd. for C^H^NjOg, 511; C, 65.73; H, 7.29; N, 8.21. Found, C, 65.64; H, 7.30; N, 8.19. EXAMPLE PKC77
Z-Leu-Abu-CONH-CH2C6H3(3,5-(OCH3)2. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 3,5-dimethoxyphenethylamine, and purified by column chromatography using solvent CHCl3/CH3OH 50:1 (50% yield). White solid, single spot on TLC, Rf = 0.46 (CHCl3/CH3OH 50:1), mp 153-155 _\_C. -R NMR (CDC13) ok, MS (FAB) m/e = 528 (M+ l). Anal: calcd. for
C^H^O-,, 527; C, 63.74; H, 7.07; N, 7.96. Found, C, 63.66; H, 7.09; N, 7.92.
EXAMPLE PKC78 Z-Leu-Abu-CONH-CH2CH(OH)Ph. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 2-amino- 1-phenylethanol by the procedure described in Example PKC67, and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (50% yield). White solid, single spot on TLC, Rf = 0.48 (CHCl3/CH3OH 10:1), mp 152-154 C. -R NMR (CDC13) ok, MS (FAB) m/e = 498 (M+ l). Anal: calcd. for C27H35N306, 497; C, 65.17; H, 7.09; N, 8.44. Found, C, 65.06; H, 7.05; N, 8.50. EXAMPLE PKC79
Z-Leu-Abu-CONH-CH2CH(OH)C6H4(4-OCH3). This compound was synthesized using 2-amino-l(4-methoxy)phenylethanol and purified by column chromatography using solvent AcOEt/hexane 3:2 (26% yield). Yellow solid, single spot on TLC, Rf = 0.56 (AcOEt/hexane 1:1), mp 128-129 C. -R NMR (CDC13) ok, MS (FAB) m/e = 528 (M+ 1). Anal: calcd. for C-^H^O-,, 527; C, 63.74; H, 7.07; N,
7.96. Found, C, 63.44; H, 7.08; N, 7.82.
EXAMPLE PKC80 Z-Leu-Abu-CONH-CH2CH(OH)C6H2(2,4,6-(OCH3)3). This compound was synthesized using 2-amino-l(2,4,6-trimethoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 foUowed by CHCl3/CH3OH 10:1
(29% yield). YeUow solid, single spot on TLC, Rf = 0.54 (CHCl3/CH3OH 10:1), mp 170-172 XC. ~H NMR (CDC13) ok, MS (FAB) m/e = 588 (90%, M+ 1), 570 (100%,
M+ l-18). Anal: calcd. for C30H41N3O9, 587; C, 61.31; H, 7.03; N, 7.15. Found, C, 60.86; H, 7.29; N, 6.95.
EXAMPLE PKC81 Z-Leu-Abu-CONH-CH2CH(OH)C6H4(4-N(CH3)2). This compound was synthesized using 2-amino-l(4-dimethylamino)phenylethanol and purified by column chromatography using solvent AcOEt/hexane 6:1 (23% yield). YeUow solid, single spot on TLC, Rf = 0.41 (AcOEt/hexane 6:1), mp 104 C (dec). XH NMR (CDC13) ok, MS (FAB) m/e = 523 (M+ 1-18). Anal: calca. for C29H40N4O6, 540; C, 64.42; H, 7.45; N, 10.36. Found, C, 64.27, H, 7.42; N, 10.34. EXAMPLE PKC82
Z-Leu-Abu-CONH-CH2CH(OH)C6F5. This compound was synthesized using 2-amino-l-pentafluorophenylethanol and purified by column chromatography using solvent CHCL3/CH3OH 10:1 (66% yield). White solid, single spot on TLC, Rf = 0.28 (CHCl3/CH3OH 10:1), mp 167-171 C. -R NMR (DMSO-d6) ok, MS (FAB) m/e = 570 (M+ l-18). Anal: calcd. for C27H30N3O6F5, 587; C, 55.19; H, 5.14; N, 7.15. Found,
C, 56.13; H, 5.58; N, 7.20.
EXAMPLE PKC83 Z-Leu-Abu-CONH-CH2CH(OH)C6H4(3-CF3). This compound was synthesized using 2-amino-l(3-trifluoromethyl)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (72% yield). Dark yeUow semisolid, single spot on TLC, Rf = 0.48 (CHCl3/CH3OH 10:1). JH NMR (CDC13) ok, MS (FAB) m/e = 566 (M+ l). Anal: calcd. for CjgH^NjOgF^ 565; C, 59.46; H, 6.06; N, 7.42. Found, C, 59.12; H, 6.18; N, 7.14.
EXAMPLE PKC84 Z-Leu-Abu-CONH-CH2CH(OH)C6H4(3-OPh). This compound was synthesized using 2-amino-l(3-phenoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (67% yield). YeUow oU, single spot on TLC, Rf = 0.54 (CHCl3/CH3OH 10:1). *H NMR (CDC13) ok, MS (FAB) m/e = 590 (53%, M+ l), 572 (100%, M+ l-18). Anal: Calcd. for C33H39N307, 589; C, 67.21; H, 6.66; N, 7.12. Found, C, 66.76; H, 6.25; N, 7.06.
EXAMPLE PKC85 Z-Leu-Abu-CONH-CH2CH(OH)C6H4(4-OPh). This compound was synthesized using 2-amino-l(4-phenoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (48% yield). YeUow semisolid, single spot on TLC, Rf = 0.22 (CHCl3/CH3OH 20:1), mp 55-60 . C. *H NMR (CDC13) ok, MS (FAB) m/e
= 590 (47%, M+ l),572 (100%, M+ l-18). Anal: calcd. for C33H39N307, 589; C, 67.21; H, 6.66; N, 7.12. Found, C, 67.30; H, 6.67; N, 7.10.
EXAMPLE PKC86 Z-leu-Abu-CONH-CH2CH(OH)C6H4(4-OCH2Ph). This compound was synthesized using 2-amino-l(4-benzyloxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (39% yield). YeUow solid, single spot on TLC, Rf = 0.40 (CHCl3/CH3OH 20:1), mp 59-62 C. -R NMR (CDC13) ok, MS (FAB) m/e = 604 (M+ l). Anal: calcd. for C34H41N307, 603; C, 67.64; H, 6.84; N, 6.96. Found, C, 67.50; H, 6.87; N, 6.90. EXAMPLE PKC87
Z-Leu-Abu-CONH-CH2CH(OH)C6H4-3-OC6H4(3-CF3). This compound was synthesized using 2-amino-l(3-(3'-trifluoromethyl)phenoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (57% yield). YeUow solid, single spot on TLC, Rf = 0.40 (CHCl3/CH3OH 30:1), mp 97-101 C. -R NMR (CDC13) ok, MS (FAB) m/e = 658 (M+ 1). Anal: calcd. for C34H38N307F3, 657; C,
62.09; H, 5.82; N, 6.39. Found, C, 62.05; H, 5.84; N, 6.42.
EXAMPLE PKC88 Z-Leu-Abu-CONH-CH2CH(OH)CgH4-3-OC6H3(3,4-Cl2). This compound was synthesized using 2-amino-l(3-(3',4'-dichloro)phenoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (55% yield). YeUow solid, single spot on TLC, Rf = 0.28 (CHCl3/CH3OH 20:1), mp 63-67 C. -R NMR (CDC13) ok, MS (FAB) m/e = 659 (M+ l). Anal: calcd. for C33H37N307C12, 658; C, 60.18; H, 5.66; N, 6.38. Found, C, 59.37; H, 5.12; N, 6.16.
EXAMPLE PKC89 Z-Leu-Abu-CONH-CH2CH(OH)C6H3(3,4-(OCH2Ph)2). This compound was synthesized using 2-amino-l(3,4-dibenzyloxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (60% yield). White solid, single
spot on TLC, Rf = 0.48 (CHCl3/CH3OH 10:1), mp 101-104 C. -R NMR (CDC13) ok, MS (FAB) m/e = 710 (M+ l). Anal: calcd. for C41H47N308, 709; C, 69.37; H, 6.67; N, 5.92. Found, C, 68.23; H, 6.70; N, 6.08.
EXAMPLE PKC90 Z-Leu-Abu-CONH-CH2CH(OH)-1-C10H7. This compound was synthesized using
2-amino-l(l-naphthyl)phenylethanol and purified by column chromatography using solvent AcOEt/hexane 1:1 (15% yield). Pale orange solid, single spot on TLC, Rf = 0.48 (AcOEt/hexane 1:1), mp 63-71 C. *H NMR (CDC13) ok, MS (FAB) m/e = 548 (M+ l). Anal: calcd. for C31H37N306, 547; C, 67.99; H, 6.81; N, 7.67. Found, C, 67.73; H, 7.03; N, 7.40.
EXAMPLE PKC91 Z-Leu-Abu-CONH-CH2CH(OH)-2-C10H7. This compound was synthesized using 2-amino-l(2-naphthyl)phenylethanol and purified by column chromatography using solvent AcOEt/hexane 3:2 (17% yield). Orange solid, single spot on TLC, Rf = 0.39 (AcOEt/hexane 3:1), mp 137-140 C. -R NMR (CDC13) ok, MS (FAB) m/e = 548
(M+ l). Anal: calcd. for C31H37N306, 547; C, 67.99; H, 6.81; N, 7.67. Found, C, 68.15; H, 6.83; N, 7.43.
EXAMPLE PKC92 Z-Leu-Phe-CONH-CH2CH(OH)Ph. This compound was synthesized using 2-amino-l-phenylethanol and purified by column chromatography using solvent
CHCl3/CH3OH 10:1 (46% yield). White solid, single spot on TLC, Rf = 0.72 (CHC13/CH30H 10:1), mp 164-166 C. 1H NMR (CDC13) ok, MS (FAB) m/e = 560 (M+ l). Anal: calcd. for C32H37N3Og, 559; C,68.67; H, 6.66; N, 7.51. Found, C, 68.46, H, 6.68, N, 7.50. EXAMPLE PKC93
Z-Leu-Phe-CONH-CH2CH(OH)C6H4(4-N(CH3)2). This compound was prepared using 2-amino-l(4-dimethylamino)phenylethanol and purified by column chromatography with solvent CHCl3/CH3OH 10:1 (22% yield). YeUow solid, single spot on TLC, Rf = 0.68 (CHCl3/CH3OH 10:1), mp 130 C (dec). -R NMR (CDC13) ok, MS (FAB) m/e = 603 (35%, M+ 1), 585 (100%, M+ 1-18). Anal: calcd. for
C34H42N406, 602; C, 67.75, H, 7.02, N, 9.29. Found, C, 66.43; H, 7.06; N, 9.22.
EXAMPLE PKC94 Z-Leu-Phe-CONH-CH2CH(OH)C6F5. This compound was prepared using 2-amino-l-pentafluorophenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (47% yield). White solid, single spot on TLC, Rf = 0.45 (CHCl3/CH3OH 20:1), mp 191-192 C. -R NMR (DMSO-d6) ok, MS (FAB) m/e =
632 (100%, M+ l-18). Anal: calcd. for C32H32N3OgF5, 649; C, 59.16; H, 4.96; N, 6.46. Found, C, 61.18; H, 5.37; N, 6.68.
EXAMPLE PKC95 Z-Leu-Phe-CONH-CH2CH(OH)C6H4(3-CF3). This compound was prepared using 2-amino-l(3-trifluoromethyl)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (42% yield). Dark yeUow semisolid, single spot on TLC, Rf = 0.48 (CHCl3/CH3OH 10:1). 1H NMR (CDC13) ok, MS (FAB) m/e = 628 (M+ l). Anal: calcd. for C33H36N306F3, 627; C, 63.15; H, 5.78; N, 6.69. Found, C, 63.24; H, 5.82; N, 6.65. EXAMPLE PKC96
Z-Leu-Phe-CONH-CH2CH(OH)C6H4(3-OPh). This compound was prepared using 2-amino-l(3-phenoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (50% yield). YeUow semisolid, single spot on TLC, Rf = 0.25 (CHCl3/CH3OH 20:1). lR NMR (CDC13) ok, MS (FAB) m/e = 652 (M+ l). Anal: Calcd. for C38H41N307, 651; C, 70.02; H, 6.34; N, 6.44. Found, 69.67;
H, 6.60; N, 6.23.
EXAMPLE PKC97 Z-Leu-Phe-CONH-CH2CH(OH)C6H4(4-OPh). This compound was prepared using 2-amino-l(4-phenoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (30% yield). YeUow semisolid, single spot on TLC,
Rf = 0.20 (CHCl3/CH3OH 30:1), mp 146-149 C. -R NMR (CDC13) ok, MS (FAB) m/e = 652 (25%, M+ l), 634 (100 %, M+ l-18). Anal: calcd. for C38H41N307, 651; C, 70.02; H, 6.34; N, 6.44. Found, 70.14; H, 6.36; N, 6.38.
EXAMPLE PKC98 Z-Leu-Phe-CONH-CH2CH(OH)C6H4(4-OCH2Ph). This compound was prepared using 2-amino-l(4-benzyloxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (49% yield). YeUow solid, single
spot on TLC, Rf = 0.45 (CHCl3/CH3OH 20:1), mp 133-134 C. 1H NMR (CDC13) ok, MS (FAB) m/e = 666 (M+ l). Anal: calcd. for C39H43N307, 665; C, 70.35; H, 6.51; N, 6.31. Found, 69.55; H, 6.46; N, 6.25.
EXAMPLE PKC99 Z-Leu-Phe-CONH-CH2CH(OH)C6H4-3-OC6H4(3-CF3). This compound was prepared using 2-amino-l(3-trifluoromethyl)phenoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (52% yield). YeUow solid, single spot on TLC, Rf = 0.23 (CHCl3/CH3OH 20:1), mp 142-143 C. αH NMR (CDC13) ok, MS (FAB) m/e = 720 (M+ 1). Anal: calcd. for C39H40N3O7F3, 719; C, 65.08; H, 5.60; N, 5.72. Found, C, 64.66; H, 5.58; N, 5.72.
EXAMPLE PKC100 Z-Leu-Phe-CONH-CH2CH(OH)C6H4-3-OC6H3(3,4-Cl2). This compound was prepared using 2-amino-l(3-(3',4'-dichloro)phenoxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (41% yield). YeUow solid, single spot on TLC, Rf = 0.40 (CHCl3/CH3OH 20:1), mp 136-137 C. *H NMR
(CDC13) ok, MS (FAB) m/e = 721 (M+ l). Anal: calcd. for C38H39N307C12, 720; C, 63.33; H, 5.45; N, 5.83. Found, C, 62.78; H, 5.09; N, 5.42.
EXAMPLE PKC101 Z-Leu-Phe-CONH-CH2CH(OH)C6H3(3,4-(OCH2Ph)2). This compound was prepared using 2-amino-l(3,4-dibenzyloxy)phenylethanol and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (45% yield). YeUow solid, single spot on TLC, Rf = 0.42 (CHCl3/CH3OH 20:1), mp 149-152 C. -R NMR (CDC13) ok, MS (FAB) m/e = 772 (M+ l). Anal: calcd. for C46H49N3Os, 771; C, 71.57; H, 6.39; N, 5.44. Found, C, 71.33; H, 6.45; N, 5.41. EXAMPLE PKC102
Z-Leu-Abu-CONH-CH2-2-FuryI. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 2-furfurylamine by the procedure described in Example PKC67, and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (43% yield). White solid, single spot on TLC, Rf = 0.68 (CHCl3/CH3OH 10:1), mp 138-139 C. 1H NMR (CDC13) ok, MS (FAB) m/e = 458
(M+ l). Anal: calcd. for C24H31N306, 457; C, 63.00; H, 6.83; N, 9.18. Found, C, 62.22; H, 6.72; N, 9.00.
EXAMPLE PKC103 Z-Leu-Abu-CONH-CH2-2-Tetrahydrofuryl. This compound was synthesized using 2-tetrahydrofurfurylamine and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (35% yield). YeUow solid, single spot on TLC, Rf = 0.59 (CHCl3/CH3OH 20:1), mp 126-128 C. -R NMR (CDC13) ok, MS (FAB) m/e = 462
(M+ 1). Anal: calcd. for C-^H^Og, 461; C, 62.45; H, 7.64; N, 9.10. Found, C, 62.37; H, 7.63; N, 9.19.
EXAMPLE PKC104 Z-Leu-Abu-CONH-CH2-2-Pyridyl. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 2-aminomethylpyridine. After reacting overnight at r.t., to the mixture was added AcOEt (25 mL) and white precipitate was filtered. The filtrate was washed with water (3 x 10 mL), saturated NaCl (2 x 10 mL) and dried over MgS0 . After evaporation of the solvent, chromatography on a silica gel column using solvent CHCl3/CH3OH 10:1 foUowed by precipitation from AcOEt/hexane afforded a yeUow solid (50% yield).
Single spot on TLC, Rf = 0.50 (CHCl3/CH3OH 10:1), mp 117-119 C. -R NMR (CDC13) ok, MS (FAB) m/e = 469 (M+ l). Anal: calcd. for C25H32N405, 468; C, 64.08; H, 6.88; N, 11.96. Found, C, 63.93; H, 6.86; N, 11.85.
EXAMPLE PKC105 Z-Leu-Abu-CONH-CH2-3-Pyridyl. This compound was synthesized from
1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 3-aminomethylpyridine by the procedure described in Example PKC104, and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (35% yield). YeUow solid, single spot on TLC, Rf = 0.54 (CHCl3/CH3OH 10:1), mp 122-123 C. -R NMR (CDC13) ok, MS (FAB) m/e = 469 (M+ 1). Anal: calcd. for C^H^N^, 468; C, 64.08; H, 6.88; N, 11.96. Found, C,
63.98; H, 6.91; N, 11.97.
EXAMPLE PKC106 Z-Leu-Abu-CONH-CH2-4-Pyridyl. This compound was synthesized using 4-aminomethyl-pyridine and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (45% yield). YeUow solid, single spot on TLC, Rf = 0.55
(CHCl3/CH3OH 10:1), mp 124-126 C. -R NMR (CDC13) ok, MS (FAB) m/e = 469
(M+ l). Anal: calcd. for C^H^N^, 468; C, 64.08; H, 6.88; N, 11.96. Found, C, 63.88; H, 6.87; N, 11.96.
EXAMPLE PKC107 Z-Leu-Abu-CONH-(CH2)2-2-Pyridyl. This compound was synthesized using 2-(2-aminoethyl)pyridine and purified by column chromatography using solvent
CHCl3/CH3OH 10:1 (53% yield). YeUow solid, single spot on TLC, Rf = 0.60 (CHCl3/CH3OH 10:1), mp 128-130 C. ~R NMR (CDC13) ok, MS (FAB) m/e = 483 (M+ l). Anal: calcd. for GjgH^N^, 482; C, 64.71; H, 7.10; N, 11.61. Found, C, 64.04; H, 7.05; N, 11.49. EXAMPLE PKC108
Z-Leu-Abu-CONH-(CH2)2-2-(N-Methylpyrrole). This compound was synthesized from protected Z-Leu-Abu-COOEt and 2(2-aminoethyl)-l-methylpyrrole by the procedure described in Example PKC104, and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (16% yield). Orange semisolid, single spot on TLC, Rf = 0.34 (CHCl3/CH3OH 30:1), mp 120-123 C. 1H NMR (CDC13) ok, MS (FAB) m/e = 485 (M+ l). Anal: calcd. for C26H36N405, 484; C. 64.44; H, 7.48; N, 11.56. Found, C, 64.02; H, 7.26; N, 11.21.
EXAMPLE PKC109 Z-Leu-Abu-CONH-(CH2)3-1-Imidazolyl. his compound was synthesized using l(3-aminopropyl)imidazole by the procedure described in Example PKC104, and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (27% yield). YeUow semisolid, single spot on TLC, Rf = 0.33 (CHCl3/CH3OH 10:1), mp 52-55 C. 1H NMR (CDC13) ok, MS (FAB) m/e = 486 (M+ 1). Anal: calcd. for C25H35N5Os, 485; C, 61.83; H, 7.26; N, 14.42. Found, C, 60.90; H, 7.21; N, 13.87. EXAMPLE PKC110
Z-Leu-Abu-CONH-(CH2)2-4-Morpholinyl. This compound was synthesized using 4-(2-aminoethyl)moφholine and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (55% yield). YeUow semisolid, single spot on TLC, Rf = 0.49 (CHCl3/CH3OH 10:1), mp 124-126 C. lR NMR (CDC13) ok, MS (FAB) m/e = 491 (M+ l). Anal: calcd. for C25H38N4Og, 490; C, 61.15; H, 7.81; N, 11.42. Found, C,
61,08; H, 7.86; N, 11.34.
EXAMPLE PKC111 Z-Leu-Abu-CONH-(CH2)3-4-Morpholinyl. This compound was synthesized using 4-(3-aminopropyl)moφholine and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (42% yield). YeUow semisolid, single spot on TLC, Rf = 0.50 (CHCl3/CH3OH 10:1), mp 125-126 C. -R NMR (CDC13) ok, MS (FAB) m/e =
505 (M+ l). Anal: calcd. for C26H40N4Og, 504; C, 61.88; H, 7.99; N, 11.10. Found, C, 61,69; H, 7.95; N, 11.07.
EXAMPLE PKC112 Z-Leu-Abu-CONH-(CH2)3-l-PyrroIidinyl-2-one. This compound was prepared from Z-Leu-Abu-COOH and l-(3-aminopropyl)2-pyrrolidinone, and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (33% yield). White semisolid, single spot on TLC, Rf = 0.51 (CHCl3/CH3OH 10:1). 1H NMR (CDC13) ok, MS (FAB), m/e = 503 (M+ l). Anal: calcd. for C26H38N406, 502; C, 62.13; H, 7.62; N, 11.14. Found, C, 62.02; H, 7.71; N, 10.52. EXAMPLE PKC113
Z-Leu-Abu-CONH-(CH2)2-3-IndolyI. This compound was prepared from Z-Leu-Abu-COOH and 3-(2-aminoethyl)indole and purified by column chromatography using solvent CHCl3/CH3OH 30: 1 (18% yield). White semisolid, single spot on TLC, Rf = 0.47 (CHCl3/CH3OH 30:1). -R NMR (CDC13) ok, MS (exact FAB), m/e = 521 2745.
EXAMPLE PKC114 Z-Leu-Abu-CONH-CH2-2-QuinolinyI. This compound was prepared from 1,3-dithiolane derivative of Z-Leu-Abu-COOEt and 2-aminomethylquinoline by the procedure described in Example PKC104, and purified by column chromatography using solvent AcOEt/hexane 2:1 (16% yield). YeUow solid, single spot on TLC, Rf =
0.27 (AcOEt/hexane 2:1), mp 135-138 C. *H NMR (CDCI3) ok, MS (FAB) m/e = 519 (M+ l). Anal: calcd. for C29H34N405, 518; C, 67.16; H, 6.60; N, 10.80. Found, C, 66.89; H, 6.68; N, 10.61.
EXAMPLE PKC115 Z-Leu-Abu-CONH-CH2-1-Isoquinolinyl. This compound was prepared using
1-aminomethylisoquinoline and purified by column chromatography using solvent AcOEt/hexane 2:1 (12% yield). YeUow solid, single spot on TLC, Rf = 0.34
(AcOEt/hexanel:l), mp 121-125 C. -R NMR (CDC13) ok, MS (FAB) m/e = 519 (M+ l). Anal: calcd. for C29H34N405, 518; C, 67.16; H, 6.60; N, 10.80. Found, C, 67.11; H, 6.61; N, 10.83.
EXAMPLE PKC116 Z-Leu-Abu-CONH-(CH2)3-1-Tetrahydroquinolinyl. This compound was synthesized using N-aminopropyltetraquinoline and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (40% yield). OU, single spot on TLC, Rf = 0.26 (CHCl3/CH3OH 20:1). 1H NMR (CDC13) ok, MS (FAB) m/e = 551 (M+ l). Anal: calcd. for C31H42N405, 550; C, 67.61; H, 7.69; N, 10.17. Found, C, 67.15; H, 7.42; N, 10.02.
EXAMPLE PKC117 Z-Leu-Abu-CONH-(CH2)3-2-TetrahydroisoquinolinyI. This compound was synthesized using N-aminopropylisotetraquinoline and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (20% yield). YeUow semisolid, single spot on TLC, Rf = 0.51 (CHCl3/CH3OH 20:1). 1H NMR (CDC13) ok, MS
(FAB) m/e = 551 (M+ l). Anal: calcd. for C31H42N405, 550; C, 67.61; H, 7.69; N, 10.17. Found, C, 67.23; H, 7.32; N, 9.98.
EXAMPLE PKC118 Z-Leu-Abu-CONH-CH2-8-Ca-Teine. This compound was synthesized using 8-aminomethylcaffeine and purified by column chromatography using solvent
CHCl
3/CH
3OH 20:1 (30% yield). YeUow solid, single spot on TLC, R
f = 0.35 (CHCl
3/CH
3OH 10:1), mp 171-177 C (dec). -R NMR (CDC1
3) ok, MS (FAB) m/e = 556 (16%, M+ l-28), 471 (100%, M+ 1-113). Anal: calcd. for
583; C, 57.62; H, 6.39; N, 16.79. Found, C, 57.70; H, 6.48; N, 16.69. EXAMPLE PKC119
Z-Leu-Abu-CONH-CH2-2-(4-MethyI-2-thiazoIyl). This compound was prepared using synthesized 2-aminomethyl-4-methylthiazole and purified by column chromatography using solvent AcOEt/hexane 6:1 (26% yield). Orange semisolid, single spot on TLC, Rf = 0.40 (AcOEt/hexane 6:1). -R NMR (CDCI3) ok, MS (FAB, calcd. for C24H32N405S, 488) m/e = 489 (3%, M+ l), 376 (100%, M+ 1-113).
EXAMPLE PKC120 Z-Leu-Abu-CONH-(CH2)2NH-Biotinyl. This compound was prepared from Z-Leu-Abu-COOH and biotmylethylenediamine hydrochloride. Biotin (1 g, 4.1 mmol was dissolved in 20 mL of DMF at 70 _C and cooled to 40 _C, CDI (0.97 g, 6 mmole in 3 mL of DMF was then added and white precipitate were appeared. After stirring at r.t. for two hours, ethylenediamine (1.34 mL, 20 mmole) in 10 mL of DMF was added and stirred for another 3 hours. After evaporating DMF, the semisolid residu was dissolved in 50 mL of refluxed methanol and the unreacted biotin was removed b filtration. The solution was evaporated to dryness, the residue was washed with CHC to remove the imidazole, dissolved in 6 mL of water, acidified to pH 3.0 with IN HCl and evaporated to dryness. The crude product was crystaUized from methanol to give 1.04 g of biotinylethylenediamine hydrochloride (79% yield). Long spot on TLC, Rf 0.21 (butanol:AcOH:H20 = 4:1:1), mp 241-242 _C. 1H NMR is consistent with the structure. To a stirred solution of Z-Leu-Abu-COOH (0.6 g, 1.58 mmol) in DMF (15 m was added HOBt (0.22 g, 1.58 mmol), DCC (0.49 g, 2.38 mmol), and stirring continue for 2 hours at r.t.(mixture A). To a stirred solution of biotinylethylenediamine hydrochloride (0.6 g, 1.85 mmol) in DMF (10 mL) was added TEA (0.28 mL, 2.03 mmol) at 0-5 C and stirred for 2 hours at r.t.(mixture B). To the stirred mixture A was added mixture B and stirred 3 days. After fUtering, the filtrate was evaporated to get a semisolid which was washed with H20 (30 mL), 1M HCl (30 mL), H20 (30 mL and dried under vacuum. Chromatography on a sUica gel column using solvent CHCl3/CH3OH 5:1 afforded a yeUow solid (42 % yield). Long spot on TLC, Rf = 0. (CHCl3/CH3OH 5:1), mp 188-192 C (dec). JH NMR (DMSO-d6) ok, MS (FAB) m/e = 647 (M+ 1). Anal: calcd. for C31H46N607S, 646; C, 57.56; H, 7.17; N, 12.99.
Found, C, 57.04; H, 7.21; N, 13.29.
EXAMPLE PKC121 Z-Leu-Abu-CONH-CH2-3-Pyridyl-N-oxide. This compound was prepared fro Z-Leu-Abu-COOH and 3-aminomethylpyridine-N-oxide, and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (15% yield). YeUow oil, long spo on TLC, Rf = 0.40 (CHCl3/CH3OH 5:1). 1H NMR (CDC13) ok, MS (FAB, calcd. fo C25H32N4°6> 484) m/e = 485 ( 2%~ M + 1)> 372 (100%' + 1-113).
EXAMPLE PKC122 Z-Leu-Abu-CONH-CH2-6-Uracil. This compound was prepared from Z-Leu-Abu-COOH and 6-aminomethyluracU and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (1.5% yield). Brown oU, long spot on TLC, Rf = 0.28 (CHCl3/CH3OH 10:1). -R NMR (CDC13) ok, MS (FAB, calcd. for C24H31N507,
501) m/e = 389 (100%, M+ 1-113).
EXAMPLE PKC123 Z-Leu-Phe-CONH-CH2-2-Pyridyl. This compound was prepared from 1,3-dithiolane derivative of Z-Leu-Phe-COOEt and 2-aminomethylpyridine by the procedure described in Example PKC104, and purified by column chromatography using solvent CHCl3/CH3OH 20:1(41% yield) . YeUow solid, long spot on TLC, Rf = 0.40 (CHCl3/CH3OH 20:1), mp 144-146 C. *H NMR (CDC13) ok, MS (FAB) m/e = 531 (M+ l). Anal: calcd. for C30H34N4O^, 530; C, 67.91; H, 6.46; N, 10.56. Found, C, 67.64; H, 6.50; N, 10.64. EXAMPLE PKC124
Z-Leu-Phe-CONH-(CH2)3-4-MorphoIinyl. This compound was prepared from 1,3-dithiolane derivative of Z-Leu-Phe-COOEt and 4-(3-aminopropyl)moφholine, and purified by column chromatography using solvent CHCl3/CH3OH 10:1(40% yield) . YeUow solid, long spot on TLC, Rf = 0.55 (CHCl3/CH3OH 10: 1), mp 155-156 C. *H NMR (CDC13) ok, MS (FAB) m/e = 581 (M+ 1). Anal: calcd. for C3]H42N406, 566;
C, 65.70; H, 7.47; N, 9.89. Found, C, 65.64; H, 7.49; N, 9.84.
EXAMPLE PKC125 Z-Leu-Phe-CONH-CH2-2-Quinolinyl. This compound was prepared using 2-aminomethylquinoline and purified by column chromatography using solvent AcOEt/hexane 1:1 (33% yield) . YeUow solid, long spot on TLC, Rf = 0.30
(AcOEt/hexane 1:1), mp 131-135 C. -R NMR (CDC13) ok, MS (FAB) m/e = 581 (M+ l). Anal: calcd. for C34H36N405, 580; C, 70.32; H, 6.25; N, 9.65. Found, C, 70.31; H, 6.27; N, 9.63. .
EXAMPLE PKC126 Z-Leu-Phe-CONH-CH2-1-Isoquinolinyl. This compound was prepared using
1-aminomethylisoquinoline and purified by column chromatography using solvent AcOEt/hexane 1:1 (7% yield). YeUow solid, single spot on TLC, Rf = 0.45
(AcOEt/hexane 1:1), mp 169-173 C. -R NMR (CDC13) ok, MS (FAB) m/e = 581 (M+ 1). Anal: calcd. for C34H36N405, 580; C, 70.32; H, 6.25; N, 9.65. Found, C, 70.05; H, 6.29; N, 9.47.
EXAMPLE PKC127 Z-Leu-Phe-CONH-(CH2)3-1-Tetrahydroquinolinyl. This compound was prepared using N-aminopropyltetraquinoline and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (40% yield). YeUow solid, single spot on TLC, Rf = 0.58 (CHCl3/CH3OH 20:1), mp 115-120 C. -R NMR (CDC13) ok, MS (FAB) m/e = 613 (M+ l). Anal: calcd. for jgH^N^, 612; C, 70.56; H, 7.24; N, 9.14. Found, C, 70.46; H, 7.26; N, 9.19.
EXAMPLE PKC128 Z-Leu-Phe-CONH-(CH2)3-2-Tetrahydroisoquinolinyl. This compound was prepared using N-aminopropylisotetraquinoline and purified by column chromatography using solvent CHCl3/CH3OH 20:1 (51% yield). YeUow solid, single spot on TLC, Rf = 0.62 (CHCl3/CH3OH 10: 1), mp 107-111 C. -R NMR (CDC13) ok,
MS (FAB) m/e = 613 (M+ l). Anal: calcd. for C36H44N405, 612; C, 70.56; H, 7.24; N, 9.14. Found, C, 69.61; H, 7.25; N, 9.05.
EXAMPLE PKC129 Z-Leu-Phe-CONH-(CH2)2NH-biotinyl. This compound was prepared from Z-Leu-Phe-COOH and synthesized biotinylethylenediamine hydrochloride by the procedure described for Example PKC120, and purified by column chromatography using solvent CHCl3/CH3OH 5:1 (35% yield). White solid, long spot on TLC, Rf = 0.42 (CHCl3/CH3OH 5:1), mp 204-206 C (dec). *H NMR (DMSO-d6) ok, MS (FAB) m/e = 709 (M+ l). Anal: calcd. for C36H48N607S, 708; C, 60.99; H, 6.82; N, 11.85. Found, C, 61.03; H, 6.83; N, 11.77.
EXAMPLE PKC130 Z-Leu-Nva-CONH-CH2CH(OH)Ph. This compound was synthesized from 1,3-dithiolane derivative of Z-Leu-Nva-COOEt and 2-amino-l-phenylethanol by the procedure described in Example PKC67, and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (54% yield). White solid, single spot on TLC, Rf = 0.56
(CHCl3/CH3OH 10:1), mp 75-77 C. -R NMR (CDC13) ok, MS (FAB, calcd. for C28H37N306, 511) m/e = 512 (M+ l).
EXAMPLE PKC131 Z-Leu-Nva-CONH-CH2-2-Pyridyl. This compound was prepared from 1,3-dithiolane derivative of Z-Leu-Nva-COOEt and 2-aminomethylpyridine by the procedure described in Example PKC104, and purified by column chromatography using solvent CHCl3/CH3OH 10:1(50% yield) . YeUow solid, long spot on TLC, Rf =
0.55 (CHCl3/CH3OH 10:1), mp 65-70 C. -R NMR (CDC13) ok, MS (FAB, calcd. for C2gH34N405,482) m/e = 483 (M+ l).
EXAMPLE PKC132 Z-Leu-Nva-CONH-(CH2)3-4-MorpholinyI. This compound was prepared from 1,3-dithiolane derivative of Z-Leu-Nva-COOEt and 4-(3-aminopropyl)moφholine, and purified by column chromatography using solvent CHCl3/CH3OH 10:1(37% yield) . YeUow solid, long spot on TLC, Rf = 0.23 (CHCl3/CH3OH 10:1), mp 108-110 C. -R NMR (CDC13) ok, MS (FAB, calcd. for C27H42N406, 518) m/e = 519 (M + l).
EXAMPLE PKC133 CH3OCO(CH2)2CO-Leu-Abu-CONHEt. To a solid Z-Leu-Abu-CONHEt (1 g,
2.47 mmol) was added a solution of hydrogen bromide in acetic acid (30 wt%, 1.52 mL, 7.40 mmol) at r.t. The mixture was vigorously stirred for 1 hour during this time aU of the ketoamide dissolved in acetic acid. The reaction was quenched with Et20 (30 mL) then separated. The semisolid was triturated and washed successively with Et20 (5 x 30 mL). After removing solvent, the residue was dried under vacuum, leaving a very hydroscopic solid. 1H NMR (CDC13) showed loss of Z group. The yield was 70-80%. To a stirred solution of mono-methylsuccinate (0.28 g, 2.13 mmol) in DMF (10 mL) was added DCC (0.44 g, 2.13 mmol) and HOBt (0.29 g, 2.13 mmol). The mixture was stirred for 2 hours at r.t.(mixture A). To a stirred solution of Leu-Abu-CONHEt.HBr (0.5 g, 1.42 mmol) in DMF (5 mL) was added TEA (0.2 mL,
1.42 mmol) at 0-5 C and stirred for 30 min (mixture B). To the stirred mixture B was added mixture A at 0-5 C and the reaction was stirred overnight at r.t. After evaporation of the solvent, AcOEt (40 mL) was added, the precipitate was fUtered, the fUtrate was washed with 0.25 N HCl (10 mL), H20 (20 mL), 10% Na2C03 (3 x 20 mL), H20 (20 mL), satd. NaCl (2 x 20 mL), dried over MgS04, and concentrated.
Chromatography on a sUica gel column with solvent CHCl3/CH3OH 10:1 afforded a
yeUow semisolid (42% yield). Single spot on TLC, Rf = 0.43 (CHCl3/CH3OH 10:1). 1H NMR (CDC13) ok, MS (FAB, calcd. for C18H31N3Oδ, 385) m/e = 386 (M+ l).
EXAMPLE PKC134 2-Furyl-CO-Leu-Abu-CONHEt. This compound was synthesized using 2-furoic acid by the procedure described for compound 67 and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (39% yield). YeUow solid, single spot on TLC, Rf = 0.51 (CHCl3/CH3OH 10:1), mp 58-59 C. JH NMR (CDC13) ok, MS (FAB) m/e = 366 (M+ l). Anal: calcd. for C18H27N305, 365; C, 59.16; H, 7.44; N, 11.50. Found, C, 58.12; H, 7.53; N, 11.64. EXAMPLE PKC135
2-Tetrahydrofuryl-CO-Leu-Abu-CONHEt. This compound was synthesized using 2-tetrahydrofuroic acid and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (41% yield). YeUow oU, single spot on TLC, Rf = 0.54 (CHCl3/CH3OH 10:1). 1H NMR (CDC13) ok, MS (FAB, calcd. for C18H31N305, 369) m/e = 370 (M+ 1).
EXAMPLE PKC136 3-Pyridyl-CO-Leu-Abu-CONHEt. This compound was synthesized using nicotinic acid and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (49% yield). YeUow solid, single spot on TLC, Rf = 0.56 (CHCl3/CH3OH 10:1), mp 57-61 C. -R NMR (CDC13) ok, MS (FAB) m/e = 377 (M+ 1). Anal: calcd. for
C 19 H28 N 4°4' 31β~' C' 60-58; H> 7A9~' N- 14-92- Found, C, 60.05; H, 7.51; N, 14.58.
EXAMPLE PKC137 2-Pyrazinyl-CO-Leu-Abu-CONHEt. This compound was synthesized using 2-pyrazinecarboxylic acid and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (18% yield). YeUow solid, single spot on TLC, Rf = 0.33
(CHCl3/CH3OH 10:1), mp 51-56 C. -R NMR (CDC13) ok, MS (FAB) m/e = 378 (M+ l). Anal: calcd. for C18H27N504, 377; C, 57.29; H, 7.16; N, 18.56. Found, C, 56.74; H, 7.28; N, 18.32.
EXAMPLE PKC138 2-QuinolinyI-CO-Leu-Abu-CONHEt. This compound was synthesized using quinaldic acid and purified by column chromatography using solvent AcOEt/hexane 1:1 (45% yield). Orange solid, single spot on TLC, Rf = 0.48 (AcOEt/hexane 1:1), mp
56-59 C. -R NMR (CDC13) ok, MS (FAB) m/e = 427 (M+ l). Anal: calcd. for C^H^O 426; C, 64.79; H, 7.09; N, 13.13. Found, C, 64.98; H, 7.45; N, 12.48.
EXAMPLE PKC139 1-IsoquinolinyI-CO-Leu-Abu-CONHEt. This compound was synthesized using 1-isoquinoline carboxylic acid and purified by column chromatography with solvent
AcOEt/hexane 1:1 (46% yield). Red solid, single spot on TLC, Rf = 0.47 (AcOEt/hexane 1:1), mp 104-106 C. -R NMR (CDC13) ok, MS (FAB) m/e = 427 (M+ l). Anal: calcd. for C23H30N4O4, 426; C, 64.79; H, 7.09; N, 13.13. Found, C, 65.00; H, 7.31; N, 12.96. EXAMPLE PKC140
4-Morpholinyl-CO-Leu-Abu-CONHEt. This compound was synthesized from 4-moφholinecarbonyl chloride (1 mmol), Leu-AbuCONH-EtHBr (1 mmol) and TEA (2.5 mmol), and purified by column chromatography using solvent CHCl3/CH3OH 10:1 (33% yield). YeUow oU, single spot on TLC, Rf = 0.45 (CHCl3/CH3OH 10:1). JH NMR (CDC13) ok, MS (FAB, calcd. for C18H32N405, 384) m/e = 385 (M+ l).
EXAMPLE PKC141 Ph(CH2)2CO-Leu-Abu-CONHEt. This compound was synthesized from 1,3-dithiolane derivative of Ph(CH2)2CO-Leu-Abu-COOEt and EtNH2, and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (72% yield). YeUow solid, single spot on TLC, Rf = 0.23 (CHCl3/CH3OH 30:1), mp 134-136 C. 1H NMR
(CDC13) ok, MS (FAB) m/e = 404 (M+ l). Anal: calcd. for C22H33N304, 403; C, 65.48; H, 8.24; N, 9.60. Found, C, 65.52; H, 8.30; N, 9.42.
EXAMPLE PKC142 l-C10H7CH2CO-Leu-Abu-CONHEt. This compound was synthesized from 1,3-dithiolane derivative of l-C10H7CO-Leu-Abu-COOEt and EtNH2, and purified by column chromatography using solvent CHCl3/CH3OH 30:1 (67% yield). YeUow solid, single spot on TLC, Rf = 0.47 (CHCl3/CH3OH 30:1), mp 201-203 C. ~R NMR . (CDC13) ok, MS (FAB) m/e = 440 (M+ l). Anal: calcd. for C^H^N^, 439; C, 68.31; H, 7.57; N, 9.56. Found, C, 68.19; H, 7.52; N, 9.49. EXAMPLE PKC143
Ph2CHCO-Leu-Abu-CONHEt. This compound was synthesized from 1,3-dithiolane derivative of Ph2CHCO-Leu-Abu-COOEt and EtNH2, and purified by
column chromatography using solvent CHCl3/CH3OH 10:1 (24% yield). YeUow solid, single spot on TLC, Rf = 0.40 (CHCl3/CH3OH 10:1), mp 78-83 C. *H NMR (CDC13) ok, MS (FAB) m/e = 467 (M+ l). Anal: calcd. for C27H35N304, 466; C, 69.65; H, 7.58; N, 9.02. Found, C, 70.04; H, 7.72; N, 8.72. EXAMPLE PKC144
Ph2CHCO-Leu-Abu-CONH-CH2CH(OH)Ph. This compound was synthesized from 1,3-dithiolane derivative of Ph2CHCO-Leu-Abu-COOEt and 2-amino-l-phenylethanol, and purified by column chromatography using CHC13 foUowed by solvent CHCl3/CH3OH 30:1 (30% yield). YeUow solid, single spot on TLC, Rf =0.40 (AcOEt/hexane 1:1), mp 178-180 C. -R NMR (CDC13) ok, MS
(FAB) m/e = 558 (M+ l). Anal: calcd. for C33H39N305, 557; C, 71.07; H, 7.05; N, 7.53. Found, C, 70.93; H, 7.10; N, 7.46.
EXAMPLE PKC145 Ph2CHCO-Leu-Abu-CONH-2-CH2-Pyridyl. This compound was prepared from 1,3-dithiolane derivative of Ph2CHCO-Leu-Abu-COOEt and 2-aminomethylpyridine, and purified by column chromatography using CHC13 foUowing by solvent CHCl3/AcOEt 7:3 (9% yield), mp 161-163. YeUow solid, single spot on TLC, Rf = 0.30 (CHCl3/CH3OH 10:1). 1H NMR (CDC13) ok, MS (FAB) m/e = 529 (M+ l). Anal: calcd. for C31H36N404, 528; C, 70.43; H, 6.86; N, 10.60. Found, C, 70.42; H, 6.91 N, 10.47.
EXAMPLE PKC146 Ph2CHCO-Leu-Abu-CONH-N-(CH2)3-Morpholinyl. This compound was prepared from 1,3-dithiolane derivative of Ph2CHCO-Leu-Abu-COOEt and N-aminopropylmoφholine, and purified by column chromatography using CHC13 foUowed by solvent CHCl3/AcOEt 7:3 (25 % yield), mp 170-174. YeUow solid, single spot on TLC, Rf = 0.25 (CHCl3/CH3OH 10:1). -R NMR (CDC13) ok, MS (FAB) m/ = 565 (M+ l). Anal: calcd. for C32H44N405, 564; C, 68.06; H, 7.85; N, 9.92. Found, C 67.22; H, 7.77; N, 9.75.
EXAMPLE PKC147 Ph2CHCO-Leu-Phe-CONH-CH2CH(OH)Ph. This compound was prepared from 1,3-dithiolane derivative of Ph2CHCO-Leu-Phe-COOEt and 2-amino-l-phenylethanol, and purified by crystaUization from CHCl3/ether (16% yield)
YeUow solid, single spot on TLC, Rf = 0.41 (AcOET/CH3OH 9:1), mp 192-196 C. -R NMR (CDC13) ok, MS (FAB) m/e = 620 (M+ 1). Anal: calcd. for C38H41N305, 619; C, 73.64; H, 6.67; N, 6.78. Found, C, 72.00; H, 6.62; N, 6.41.
EXAMPLE PKC148 Ph2CHCO-Leu-Phe-CONH-CH2-2-Pyridyl. This compound was synthesized from 1,3-dithiolane derivative of Ph2CHCO-Leu-Phe-COOEt and 2-aminomethylpyridine, and purified by column chromatography using CHC13 foUowing by solvent CHCl3/AcOEt 9:1 (9% yield). YeUow solid, single spot on TLC, Rf = 0.33 (AcOET/CH3OH 9:1), mp 160-162 C. 1H NMR (CDC13) ok, MS (FAB) m/e = 591 (M+ l). Anal: calcd. for C36H38N404, 590; C, 73.20; H, 6.48; N, 9.48. Found, C, 69.91;
H, 6.29; N, 8.98.
EXAMPLE PKC149 Ph2CHCO-Leu-Phe-CONH-(CH2)3-4-MorpholinyI. This compound was synthesized from 1,3-dithiolane derivative of Ph2CHCO-Leu-Phe-COOEt and N-aminopropylmoφholine, and purified by column chromatography using AcOEt foUowing by crystallization from AcOEt/ether (20 % yield). YeUow solid, single spot on TLC, Rf = 0.45 (AcOET/CH3OH 9:1), mp 158-160 C. *H NMR (CDC13) ok, MS (FAB) m/e = 627 (M+ l). Anal: calcd. for C37H46N405, 626; C, 70.90; H, 7.40; N, 8.94. Found, C, 70.05; H, 7.43; N, 8.68. A variety of techniques for certain synthetic steps in the synthesis of the
Peptide Keto-Compounds can be used. Additional synthetic procedures are provided in the foUowing two Examples.
EXAMPLE PKC150 DimethyIurea-(L)-Leu-(L)-Abu-CONH-Et. The structure of Dimethylurea-(L)-Leu-(L)- Abu-CONH-Et is shown below:
This compound was produced through synthesis of the reactant Dimethylurea-(L)-Leu- (L)-Abu hydroxy ethyl amide.
(L)-Leucine (1.31 g (10 mmoles)) was placed in a 3 neck round-bottomed flask, equipped with two pressure equalizing dropping funnels. 12.5 mL of 1.0 N NaOH (12.5 mmoles) was added to the flask and then the mixture was cooled on ice, 12.5 mL of 1.0
N NaOH was placed in one dropping funnel and 1.15 mL (12.5 mmoles) of dimethylcarbamoyl chloride was placed in the other. The contents of the addition funnels were added to the flask simultaneously over ten minutes. The mixture was aUowed to react for an additional fifteen minutes. The reaction was then washed twice with 15 mL of ethyl acetate. The aqueous layer was cooled on ice and acidified to a pH of 2 with 1.0 N HCl. The aqueous layer was extracted three times with 15 mL of ethyl acetate. The combined organics were dried over magnesium sulfate, filtered and concentrated in vacuo. There remained 0.10 g of a white solid (5%) which possessed an Rf value of 0.31 using 91:8:1 chloroform:methanol:acetic acid as the eluent. Boc-Abu hydroxy ethyl amide (0.233 g, 0.894 mmoles) was dissolved in 5 mL of dioxane foUowed by the addition of 20 mL of 4N HCl/dioxane. The reaction mixture was aUowed to react for two hours. After this time, the reaction mixture was concentrated in vacuo and used immediately in the next step. The HCl-Abu hydroxy ethyl amide was dissolved in 30 mL of anhydrous DMF and cooled on an ice bath for ten minutes. To this solution was added 0.217 g (1.07 mmoles) of moφholineleucine urea, 0.46 mL (2.68 mmoles) of diisopropylethylamine and 0.133 g (0.984 mmoles) of 1- hydroxybenzotriazole (HOBt) and aUowed to equUibrate for thirty minutes. After this time, 0.188 g (0.984 mmoles) of EDC suspended in 10 mL of anhydrous DMF was added and the reaction mixture was aUowed to react overnight. The reaction mixture was concentrated in vacuo and the resulting residue was purified by sUica gel column chromatography employing 90:10 chloroform:methanol as the eluent. There remained 0.2044 g (66.56% yield) of a white solid with Rf value of 0.38 in the solvent system detaUed above.
0.100 g (0.291 mmoles) of Dimethylurea-Leu-Abu hydroxy ethyl amide was dissolved in 10 mL of methylene chloride and cooled in an ice bath. To this mixture was added 0.487 mg (.003 mmoles) of 2,2,6,6-tetramethyl-l-piperidinyloxy, free radical (TEMPO) and .014 mL (0.291 mmoles) of an aqueous KBr solution (5.95 g of KBr
dissolved in 25 mL of water). The reaction mix was stirred vigorously whUe four 80 microliter portions of a 1M aqueous sodium hypochlorite (pH 9.5) were added at 15 minute intervals. After this time the reaction mixture was analyzed by TLC employing 90:10 chloroform:methanol to check for completeness of the reaction. If the reaction was not complete another portion of TEMPO and another regimen of the sodium hypochlorite solution should be added. This reaction required three additional regimens of TEMPO and sodium hypochlorite.
When the reaction was deemed complete by TLC, the layers were separated. The aqueous layer was extracted with methylene chloride (3 x 10 mL). The combined organic layer was washed with 10% HCl (1 x 10 mL), 30 mL of a 100 mL stock solutio of 10% HCl containing 1.6 g of KI, 10% sodium thiosulfate (2 x 30 mL) and brine (1 x 30 mL). The organic layer was then dried over magnesium sulfate, fUtered, and concentrated in vacuo. The crude material was triturated with petroleum ether to give an off-white solid which was recrystaUized from ethyl acetate:hexane. There remained 0.048 g (48.5% yield) of a white solid with an Rf value of 0.43 in the solvent system detaUed above.
TLC analysis of the product on sUica gel gave an Rf value of 0.43 in the solven system detailed above. HPLC analysis was performed on a Vydac C4 column (4.6 x 250 mm) at 60° C using a gradient of 15-25% B/30 minutes (A = 0.1% TFA in water, B = 0.1% TFA in acetonitrile). The product had a retention time of 14.49 minutes and a purity of 97%.
Analyses of the final product provided the foUowing results: Mass spectrum analysis found (M+H)+ at m/z 343. Elemental analysis for C16H30N4O4 found 55.80 C, 8.70 H and 15.97N whUe calculated values were 56.12 C, 8.83 H and 16.36N. For 1HNMR (600 MHz, d6-DMSO) analysis, the shifts observed were 8.65(t,lH),
8.10(d,lH), 6.07(d,lH), 4.85(m,lH), 4.20(m,lH), 3.12(m,2H), 2.77(s,6H), 1.77(m,lH), 1.63(m,lH), 1.48(m,2H), 1.40(m, IH), 1.02(t,lH), 0.85(m,9H).
EXAMPLE PKC151 Boc-(L)-Leu-(L)-Abu-CONH-Et. The structure of Boc-(L)-Leu-(L)-Abu-CONH-Et is shown below:
This compound was produced by synthesis of the reactant Boc-(L)-Leu-(L)-Abu hydroxy ethyl amide.
Boc-Abu hydroxy ethyl amide (0.233 g, 894 mmoles) was dissolved in 5 mL of dioxane foUowed by the addition of 20 mL of 4N HCl/dioxane. The reaction mixture was aUowed to react for two hours. After this time, the reaction mixture was concentrated in vacuo and used immediately in the next step. The HCl -Abu hydroxy ethyl amide prepared above, was dissolved in 25 mL of anhydrous DMF and cooled on an ice bath for ten minutes. To this solution was added 0.267 g (1.07 mmoles) of moφholineleucine urea, 0.46 mL (2.68 mmoles) of diisopropylethylamine and 0.133 g (0.984 mmoles) of 1-hydroxybenzotriazole (HOBt) and aUowed to equUibrate for thirty minutes. After this time 0.188 g (0.984 mmoles) of EDC suspended in 10 mL of anhydrous DMF was added and the reaction mixture was aUowed to react overnight. The reaction mixture was concentrated in vacuo and the resulting residue redissolved in 100 mL of chloroform. The solution was washed twice with 50 mL of both saturated sodium bicarbonate and brine. The organic layer was dried over magnesium sulfate, filtered and concentrated in vacuo. The crude material was purified by sUica gel column chromatography employing 90:10 chlorofornrmethanol as the eluent. There remained 0.1841 g (55.12% yield) of a white solid with and Rf value of 0.42 in the solvent system detaUed above.
Boc-Leu-Abu hydroxy ethyl amide (.0823 g, 0.22 mmoles) was dissolved in 10 mL of methylene chloride and cooled in an ice bath. To this mixture was added 0.325 mg (.002 mmoles) of 2,2,6,6-tetramethyl-l-piperidinyloxy, free radical (TEMPO) and .011 mL of an aqueous KBr solution (5.95 g of KBr dissolved in 25 mL of water). The
reaction mix was stirred vigorously whUe four 60 microliter portions of an IM aqueous sodium hypochlorite (pH 9.5) were added at 15 minute intervals. After this time the reaction mixture was analyzed by TLC employing 90:10 chloroform:methanol to check for completeness of the reaction. If the reaction was not complete another portion of TEMPO and another regimen of the sodium hypochlorite solution should be added.
This reaction required one additional regiment of TEMPO and sodium hypochlorite. When the reaction was deemed complete by TLC, the layers were separated. The aqueous layer was extracted with methylene chloride (3 x 10 mL). The combined organic layer was washed with 10% HCl (1 x 10 mL), 30 mL of a 100 mL stock solutio of 10% HCl containing 1.6 g of KI, 10% sodium thiosulfate (2 x 30 mL) and brine (1 x
30 mL). The organic layer was then dried over magnesium sulfate, fUtered and Concentrated in vacuo. The crude material was triturated with petroleum ether to give an off-white solid which was recrystaUized from ethyl acetate:hexane. There remained 0.067 g (82.3% yield) of a white solid with an Rf value of 0.52 in the solvent system detailed above.
TLC analysis of the product on sUica gel gave an Rf value of 0.52 in the solvent system detaUed above. HPLC analysis was performed on a Vydac C4 column (4.6 x 250 mm) at 60° C using a gradient of 25-35% B/30 minutes (A = 0.1% TFA in water, B = 0.1% TFA in acetonitrUe). The product had a retention time of 21.05 minutes and a purity of 99.14%.
Analyses of the final product provided the foUowing results: Mass spectrum analysis found (M+H)+ at m/z 372. Elemental analysis for C18H33N305 found 57.84 C, 8.84 H and 11.05 N whUe calculated values were 58.20 C, 8.95 H and 11.05 N. For 1HNMR (600 MHz, d6-DMSO) analysis, the shifts observed were 8.66(t,lH), 8.06(d,lH), 6.85(d,lH), 4.88(m,lH), 3.99(m,lH), 3.12(m,2H), 1.77(m,lH), 1.77(m,lH),
1.60(m,lH), 1.51(m,lH), 1.35(br s,HH), 1.02(t,3H), 0.86(m,9H). Moφholine Peptide Keto-Compounds
As is clear from the foregoing description of the Peptide Keto-Compounds, the term Peptide Keto-Compound as used herein also includes the moφholine Peptide Keto-Compounds. These morpholine compounds can be classified in any of the variou classes or subclasses and types of Peptide Keto-Compounds referred to hereinabove. Thus, for example these compounds include the moφholine Peptide Ketoacids, the
moφholine Peptide Ketoamides and the moφholine Peptide Ketoesters. The moφholine Peptide Keto-Compounds can include either N-terminal or C-terminal moφholine groups. In the N-terminal moφholine Peptide Keto-Compounds, the M group (or M-., M2, M3, M4 group) includes a moφholine ring that can, in some circumstances, include the nitrogen of the N-terminal amino acid. The C-terminal moφholine Peptide Keto-Compounds include a moφholine ring that is part of the C-terminal R (or Rj etc.) group of the compound. In certain examples of these compounds, the R-group includes an alkyl moφholine, as in the compound described above in Example PKC140. The moφholine Peptide Keto-Compounds can be produced using synthesis techniques generaUy simUar to those used for synthesis of other Peptide Keto- Compounds.
The C-terminal moφholine Peptide Keto-Compounds can be produced using the general method of production of Peptide Ketoamides, which are derived from the corresponding Peptide Ketoesters. In the case of C-terminal Peptide Keto-Compounds, the Peptide Ketoesters can be reacted with N-amino alkyl moφholine to produce the C-terminal N-alkyl moφholine derivative of the Peptide Ketoester. One such procedure is shown hereinabove as Example PKC140.
The N-terminal moφholine Peptide Keto-Compounds can be produced using the general scheme outlined above, wherein a moφholine compound is substitued for other N-terminal blocking groups. However, other methods of synthesis can also be used. The foUowing Example shows one exemplary procedure for production of N-terminal moφholine compounds.
Example PKC152 Morpholineurea-(L)-Leu-(L)-Abu-CONH-Et. The structure of Moφholineurea-(L)- Leu-(L)-Abu-CONH-Et is shown below:
Ten grams of N-t-butyloxycarboxy-(L)-α-aminobutric acid (N-Boc-(L)-Abu) was dissolved in 100 ml of anhydrous tetrahydrofuran (THF). To this solution was added 9.4 mL of dϋsopropylethylamine and 25.61 g (49.2 mmoles) of PyBOP. The solution was aUowed to equUibrate for 10 minutes. FoUowing equUibration, a solution of 5.28 g
(54.1 mmoles) of N.O-dimethylhydroxylamine hydrochloride dissolved in 5 mL of acetonitrUe and containing 25.6 mL of N,N-diisopropylethylamine (54.1 mmoles) was added. The reaction was stirred overnight at room temperature.
The reaction mixture was then concentrated in vacuo and redissolved in 200 mL of ethyl acetate. The ethyl acetate layer was washed three times with 1.0 N HCl (100 mL), three times with saturated sodium bicarbonate (100 mL) and three times with brine (100 mL). The reaction mixture was dried over magnesium sulfate, f tered and concentrated in vacuo giving a yeUow oU. The crude product was purified by sUica gel chromatography using 2:1 ethyl acetate:hexane as the eluent. The product was isolated as a white solid (77% yield) with an Rf of 0.77 on sUica employing the same solvent system used above.
Anhydrous ethyl ether (75 mL) and 0.9 g (23.7 mmoles) of lithium aluminum hydride were placed in a 500 mL round-bottomed flask. The suspension was cooled in an ice bath for ten minutes. A pressure equalizing dropping funnel, containing 4.5 g (18.4 mmoles) of Boc-Abu hydroxamate dissolved in 75 mL of anhydrous ethyl ether, was attached to the round bottom flask and the contents were added dropwise over one
hour, with continued cooling. The reaction mixture was aUowed to react for an additional two hours at room temperature.
The reaction mixture was then cooled in an ice bath and a cold solution of potassium hydrogen sulfate (5.4 g in 230 mL of water) was slowly added to the reaction flask and aUowed to react for an additional 10 minutes. The aqueous and organic layers were separated and the aqueous layer was extracted with anhydrous ethyl ether (3 x 100 mLs). The combined organic layer was washed 3 x 100 mLs each with 1.0 N HCl, saturated sodium bicarbonate and brine and then dried over magnesium sulfate, filtered and concentrated in vacuo. The product was isolate as a white solid (63% yield), with an
Rf of 0.90 on silica, using 2:1 ethyl acetate:hexane as the eluent.
N-Boc abuinal (4.00 g (21.39 mmoles)) was dissolved in 26 mL of methanol and cooled on ice. To this was added a cold solution of 2.67 g of sodium bisulfite dissolved in 54 mL of water. This reaction was stirred overnight at 4° C. 265 mL of ethyl acetate was then added to the above reaction mix foUowed by a solution of 1.08 g (22 mmoles) of sodium cyanide dissolved in 80 mL of water, and then stirred overnight at 4°C. The aqueous and organic layers were separated and the aqueous layer was extracted twice with 50 mL of ethyl acetate. The combined organics were dried over magnesium sulfate, fUtered and evaporated in vacuo leaving a clear colorless oU (70% yield). TLC analysis on sUica using 1:1 ethyl acetate:hexane as the eluent showed the product to have an Rf of 0.69. The Boc-Abu cyanohydrin was used without further purification.
The Boc-Abu cyanohydrin isolated was dissolved in 120 mL of 4N HCl/dioxane. 60 mL of water was then added to the reaction mixture and it was refluxed overnight. The reaction mixture was rotavapped to dryness leaving a brown solid. The solid was dissolved in water and extracted three times with 100 mL of ethyl acetate. The aqueous layer was then concentrated in vacuo and rotavapped from ethyl ether three times. This material was used without further purification.
HCl -Abu hydroxy acid (2.9 g (17.16 mmoles)) was dissolved in 51 mL of 2:1 dioxane:water and placed in an ice bath. To this was added 42.5 mL (42.5 mmoles) of IN sodium hydroxide. The reaction was aUowed to cool and 6.12 g (28.04 mmoles) of di-tert-butyl dicarbonate was then added. The pH of the reaction was maintained between 9.5 and 10 by the addition of base. FoUowing an overnight reaction time it
was worked up as foUows. The dioxane was rotavapped off and an additional 15 mL of water was added to the reaction mixture. The water was covered with a layer of ethyl acetate and cooled on ice. The pH of the aqueous layer was adjusted to 2.5 with 3N HCl. The organic and aqueous layers were separated and the aqueous layer was extracted twice with 50 mL of ethyl acetate. The combined organic layers were dried over magnesium sulfate, fUtered, and evaporated in vacuo leaving a brown viscous oU. The crude material was purified by sUica gel chromatography using 91:8:1 chloroform:methanol:acetic acid as the eluent. There remained 1.140 g Boc-Abu hydroxy acid (26.3% yield from Boc-Abuinal). TLC analysis on sUica using the same system detaUed above showed the product to be one spot with an Rf value of
0.22.
Boc-Abu hydroxy acid (0.96 g (4.13 mmoles)) was dissolved in 35 mL of dimethylformamide (DMF) and cooled in an ice bath. 0.78 mL (12.4 mmoles) of 70% triethylamine and 0.84 g (6.2 mmoles) of 1-hydroxybenzotriazole (HOBT) were added and aUowed to equUibrate for thirty minutes. After this time 1.0 g (5.22 mmoles) of 1-
(3-dimethylaminopropy)-3-ethylcarbodiimide hydrochloride (EDC) suspended in 10 mL of DMF was added. The reaction was aUowed react at room temperature overnight.
The reaction was then rotavapped to dryness and redissolved in 100 mL of chloroform and washed three times with 35 ml of saturated sodium bicarbonate and then brine. The mixture was dried over magnesium sulfate, fUtered and concentrated in vacuo. The crude material was purified by sUica gel column chromatography employing 9:1 ethyl acetate:hexane. 0.938 g (85% yield) of product was isolated which possessed and Rf value of 0.55 in the above solvent system.
Boc-Abu hydroxy ethylamide (0.233 g, 0.894 mmoles) was dissolved in 5mL of dioxane foUowed by the addition of 20 mL of 4N HCl/dioxane. The reaction mixture was aUowed to react for two hours. After this time, the reaction mixture was concentrated in vacuo and used immediately in the next step.
(L)-Leucine (1.31 g (10 mmoles)) was placed in a 3-neck round-bottom flask, equipped with two pressure equalizing dropping funnels. 12.5 mL of l.ON NaOH (12.5 mmoles) was added to the flask and then the mixture was cooled on ice. 12.5 mL of
1.0 N NaOH was placed in one dropping funnel and 1.46 mL (12.5 mmoles) of moφholinecarbonyl chloride was placed in the other. The contents of the addition
funnels were added to the flask simultaneously over ten minutes. The mixture was aUowed to react for an additional twenty minutes. The reaction mixture was then washed twice with 15 mL of ethyl acetate. The aqueous layer was cooled on ice and acidified to a pH of 2 with 1.0 N HCl. The aqueous layer was extracted three times with 15 mL of ethyl acetate. The combined organics were dried over magnesium sulfate, fUtered and concentrated in vacuo. There remained 0.48 g of a white solid (20% yield) which possessed an Rf value of 0.45 using 91:8:1 chloroform:methanol:acetic acid as the eluent.
Boc-Abu hydroxy ethyl amide (0.266g) was dissolved in 5 mL of dioxane foUowed by the addition of 20 mL of 4N HCl/dioxane. The reaction mixture was aUowed to react for two hours. After this time, the reaction mixture was concentrated in vacuo and used immediately in the next step. The HCl-Abu hydroxy ethyl amide was dissolved in 30 mL of anhydrous DMF and cooled on an ice bath for ten minutes. To this solution was added 0.30 g (1.23 mmoles) of moφholineleucine urea, 0.55 mL (3.07 mmoles) of diisopropylethylamine and 0.152 g (1.13 mmoles) of 1- hydroxybenzotriazole and aUowed to equUibrate for thirty minutes. After this time, 0.218 g (21.13 mmoles) of EDC suspended in 10 mL anhydrous DMF was added and the reaction mixture was aUowed to react overnight. The reaction mixture was concentrated in vacuo and the resulting residue was purified by sUica gel column chromatography employing 90:10 chloroform:methanol as the eluent. There remained
0.2414 g (61.04% yield) of a white solid with an Rf value of 0.36 in the solvent system detaUed above.
Boc-(L)-Leu-(L)-Abu hydroxy ethyl amide (0.1225 g (0.317 mmoles)) was dissolved in 10 mL of methylene chloride and cooled in an ice bath. To this mixture was added 0.5 mg (.00317 mmoles) of 2,2,6,6-tetramethyl-l-piperidinyloxy, free radical
(TEMPO) and .0159 mL (.0317 mmoles) of an aqueous KBr solution (5.95 g of KBr dissolved in 25 mL of water), The reaction mix was stirred vigorously whUe four 87 microliter portions of a IM aqueous sodium hypochlorite (pH 9.5) were added at 15 minute intervals. After this time the reaction mixture was analyzed by TLC employing 90:10 chloroform:methanol to check for completeness of the reaction. If the reaction was not complete another portion of TEMPO and another regimen of the sodium
hypochlorite solution should be added. This reaction required three additional regimens of TEMPO and sodium hypochlorite.
When the reaction was deemed complete by TLC, the layers were separated. The aqueous layer was extracted with methylene chloride (3 x 10 mL). The combined organic layer was washed with 10% HCl (1 x 10 mL), 30 mL of a 100 mL stock solution of 10% HCl containing 1.6 g of KI, 10% sodium thiosulfate (2 x 30 mL) and brine (1 x 30 mL). The organic layer was then dried over magnesium sulfate, fUtered and concentrated in vacuo. The crude material was triturated with petroleum ether to give an off-white solid which was recrystaUized from ethyl acetate hexane. There remained 0.048 g (39.6% yield) of a white solid with an Rf value of 0.32 in the solvent system detaUed above.
TLC analysis of the product on silica gel gave an Rf value of 0.32 in the solvent system detaUed above. HPLC analysis was performed on a Vydac C4 column (4.6 x 250 mm) at 60° C using a gradient of 15-25% B/30 minutes (A =0.1% TFA in water, B = 0.1% TFA in acetonitrUe). The product had a retention time of 14 minutes and a purity of 97.8%. Analyses of the final product provided the foUowing results: Mass spectrum analysis (FABMS) found (M+H)+ at m/z 385. Elemental analysis for C18H32N405 found 56.14 C, 8.24 H and 14.36 N whUe calculated values were 56.23 C, 8.39 H and 14.57 N. For *HNMR (600 MHz, d6-DMSO) analysis, the shifts observed were 8.65(t,lH), 8.10(d,lH), 6.41(d,lH), 4.85(m,lH), 4.20(m,lH), 3.51(m,4H),
3.26(m,4H), 3.12(m,2H), 1.75(m,lH), 1.62(m,lH), 1.48(m,2H), 1.40(m,lH), 1.02(t,3H), 0.85(m,9H).
D. HALO-KETONE PEPTIDES
Halomethyl ketone peptides are irreversible inhibitors for serine proteases and cysteine proteases. This class of compounds includes peptides having a variety of halomethyl groups at their C-terminus. These halomethyl groups include -CH2X, - CHX2 and CX3, where X represents any halogen. A number of analogous compounds have been synthesized, including the amino-halo ketones and the diazo-ketone peptides.
Although these analogous compounds are chemicaUy distinguishable, aU of these
haloketone compounds are believed to have a simUar mechanism of action. Accordingly, for simplicity, aU of the foregoing compounds wiU be referred to coUectively herein as the "Halo-Ketone Peptides."
The reactivity of haloketones has generaUy been found to be in the order I > Br > Cl > F. However, increasing the reactivity of the haloketone in this way can lead to acceleration of competing side effects. Thus, it is preferable to increase the reactivity of the halomethyl ketone peptides by altering the peptide structure.
In selecting a proper inhibitor for Calpain, the same basic peptide structure selection techniques as used for the Peptide Keto-Compounds can be used. Once a peptide structure has been identified, the most effective C-terminus grouping can be empiricaUy determined through kinetic inhibition studies of each of the compounds with Calpain.
Many of the Halo-Ketone Peptides are avaUable commerciaUy. For example, Leu-CH2C1, Phe-CH2C1, Z-lys-CH2Cl, Tosyl-LysCH2Cl (TLCK), Tosyl-PheCH2Cl (TPCK), Z-Gly-Leu-Phe-CH2C1, Z-Phe-Ala-CH2C1, z-Phe-Phe-CH2Cl, D-Phe-Pro-Arg-
CH2C1, MeoSuc-Phe-Gly-Gly-Ala-CH2Cl, MeoSuc-Ala-Ala-Pro-Ala-CH2Cl, MeoSuc- Ala-Ala-Pro-Val-CH2Cl, Ala-Ala-Pro- Val-CH2C1, Ala-Ala-Phe-CH2C1, Suc-Ala-Ala-Pro- Phe-CH2C1 and D-Val-Leu-Lys-CH2C1 are aU avaUable from suppliers such as Enzyme Systems Products of Livermore, California. From the same suppliers, the foUowing diazomethyl ketone peptides are avaUable: Leu-CHN2, Z-Phe-Phe-CHN2, Z-Phe-Ala-
CHN2, Z-Phe-Pro-CHN2, Z-Lys-CHN2 and Gly-Phe-CHN2. In addition, the production of α-amino fluoro ketone peptides has been described in United States Patent No. 4,518,528 to David W. Rasnick, the disclosure of which is hereby incoφorated by this reference. The preparation of various Halo-Ketone Peptides is reviewed in Methods in
Enzymology, 46:197-208 (1977), the disclosure of which is hereby incoφorated by reference. Briefly, halomethyl ketone derivatives of blocked amino acids are readUy prepared by the reaction of mineral acids (hydrohalic) with the corresponding diazomethyl ketone. Iodomethyl ketones are prepared by reaction of a halo-ketone with Nal, since reaction with HI with a diazomethyl ketone yields the methyl ketone.
A number of different blocking groups can be used, including benzyloxycarbonyl (Z) and t-butyloxycarbonyl (Boc). The diazomethyl ketone is prepared by reaction of
diazomethane with the appropriate acid activated by means of dicyclohexylcarbodiimide (DCCI), by the mixed anhydride method.
Unblocked amino acid chloromethyl ketones can be prepared by reaction of benzyloxycarbonyl blocked derivatives with HBr or HOAc, trifluoroacetic acid, or by hydrogenation.
Synthesis of peptide chloromethyl ketones can be accomplished simply by coupUng an appropriate peptide or amino acid with an unblocked amino acid chloromethyl ketone. A few dipeptides can be converted directly to the chloromethyl ketone using the mixed anhydride and CH2N2 foUowed by HCl. Various synthetic problems are encountered in the preparation of chloromethyl ketone derivatives of basic amino acids. The side chain usuaUy must be blocked during synthesis, and difficulties are often encountered during removal of the blocking group. Use of trifluoroacetic acid or HF was eventuaUy found to give a good conversion to product. A number of examples of the preparation of Halo-Ketone Peptides have been reported in the literature, including a comprehensive review of over 100 amino acid derivatives and approximately 60 peptide derivatives listed in J.C. Powers, in "Chemistry and Biochemistry of Amino Acids, Peptides and Proteins," Vol. 4, Dekker, New York (1977), the disclosure of which is hereby incorporated by reference. Those of skill in the art will recognize how to locate a multitude of examples of the production of the Halo-Ketone Peptides. Accordingly, no additional examples are provided herein.
E. IN VITRO USES
In addition to the foregoing classes of compounds now discovered to possess Calpain inhibitory activity, we believe that a large number of other such compounds exist. In view of the large number of inhibitors of Calpain of different classes we disclose herein, aU of the known, newly discovered and yet undiscovered inhibitors of Calpain shaU be referred to hereinafter coUectively, using the term "Calpain Inhibitor." The Calpain Inhibitors may be used in vitro for a variety of puφoses to inhibit unwanted Calpain activity. For example, the Calpain Inhibitors may be used in vitro to prevent proteolysis that occurs in the process of production, isolation, purification, storage or transport of peptides and proteins.
The Calpain Inhibitors described herein can also be used in vitro to prevent further degradation of tissue samples from occurring after preparation of the samples. This in vitro prevention of degradation can be especiaUy useful in the preparation of assays for neurodegeneration wherein the assay comprises a test for the products of Calpain activity in the tissues, such as assays for breakdown products (BDP's) of cytoskeletal components such as spectrin, MAP2, actin binding protein and tau. P. Seubert et al, Neuroscience, 31:195 (1989), the disclosure of which is hereby incoφorated by reference, disclose an exemplary method of quantitating the amount of spectrin BDP's as an indication of Calpain activity. The Calpain Inhibitors of this invention are also useful in a variety of other experimental procedures where proteolysis due to Calpains is a significant problem. For example, inclusion of the Calpain Inhibitors in radioimmunoassay experiments can result in higher sensitivity. The use of the Calpain Inhibitors in plasma fractionation procedures can result in higher yields of valuable plasma proteins and can make purification of the proteins easier. The Calpain Inhibitors disclosed here can be used in cloning experiments utUizing recombinant or transfected bacterial or eukaryotic ceU cultures in order to increase yield of purified recombinant product.
To use the Calpain Inhibitors in vitro, the Calpain Inhibitors are dissolved in an organic acid, such as dimethylsulfoxide (DMSO) or ethanol, and are added to an aqueous solution containing the protease which is to be inhibited, such that the final concentration of organic solvent is 25% or less. The Calpain Inhibitors may also be added as solids or in suspension. F. TREATMENT OF NEURODEGENERATION
We have discovered that the Calpain Inhibitors are useful in vivo to treat pathologies in which excess proteolysis by Calpains is involved. Such pathologies are believed to include neuropathologies such as neurodegeneration resulting from excitotoxicity, HIV-induced neuropathy, ischemia, denervation, injury, subarachnoid hemorrhage, stroke, multiple infarction dementia, Alzheimer's Disease (AD), Huntington's Disease, surgery-related brain damage, Parkinson's Disease, and other pathological conditions.
In additional in vivo uses, peptide α-ketoamide can be used to control protein turnover, muscular dystrophy, myocardial tissue damage, and bone resorption as shown
in Tables PKC2, PKC3, and PKC4 by effective inhibition of lysosomal cathepsin B. Peptide α-ketoamides can also be used as neuroprotectants or for the treatment of ischemia, stroke, restenosis or Alzheimer's disease as shown in Tables PKC2, PKC3, and PKC4 by effective inhibiton of calpain I and calpain II. 1. Identification of Inhibitors
In order to identify Calpain Inhibitors that are useful in the practice of the present invention for treatment or inhibition of neurodegenerative conditions and diseases, it is important to identify those inhibitors posessing significant Calpain inhibitory activity. It is also important to identify those Calpain Inhibitors having a high degree of specificity for inhibition of Calpain, in order to avoid interference with other biological processes when the Calpain Inhibitor is introduced into a mammal requiring treatment for neurodegeneration. Because aU thiol proteases are believed to exert their effect through a simUar mechanism of action, our primary concern was to identify those Calpain Inhibitors having substantial inhibitory activity against Calpain, but relatively weak or no activity against other thiol proteases. Accordingly, in order to identify such Calpain Inhibitors, we tested a variety of Calpain Inhibitors for their ability to inhibit calpains I and II, and compared this data with the abUity of the same Calpain Inhibitors to inhibit Cathepsin B, another thiol protease. Those Calpain Inhibitors with high in vitro inhibitory activity against Calpain and a relatively lower activity against Cathepsin B are believed to be most useful for in vivo therapy.
Examples IA through IC show the results of these studies for a variety of Calpain Inhibitors.
EXAMPLE IA Inhibition by Substituted Heterocyclic Compounds The isocoumarins are irreversible inhibitors of Calpain. We obtained IC50 values for a variety of these Calpain Inhibitors as a kinetic analysis of these compounds. Purified Calpains can be assayed using the fluorogenic substrate succinyl- leucine-tyrosine-methylaminocoumarin (avaUable commerciaUy) or by measuring the release of acid-soluble peptides from casein because we have found that the isocoumarins inhibit casein proteolysis by Calpain.
Calpains I and II were purified by the method of (Yoshimura, et al. 1983). (Kitahara, supra) provides an alternative purification scheme. Calpain II may
alternatively be purchased from Sigma Chemical Co. as "Calcium Activated Neutral Protease." In this assay, purified Calpain was incubated with 1 C-methylated casein in the presence of various Heterocyclic Compounds and the amount of acid-soluble radioactivity released by the action of Calpain was measured. The IC50 values were determined as the concentration of Heterocyclic Compound compound at which 50% of the Calpain activity was inhibited. Table IA shows IC50 values for various Isocoumarin Compounds.
TABLE IA INHIBITION OF CALPAINS BY SUBSTITUTED ISOCOUMARINS
CiTPrOIC NH2-CiTPrOIC (ACITIC) PhCH2NHCONH-CiTPrOIC
CH3CONH-CiTPrOIC L-Phe-NH-CiTPrOIC BOC-L-Phe-NH-CiTPrOIC PhCH2NHCONH-CiTEtOIC PhCH2CONH-CiTEtOIC
Thus, it can be seen from Table IA that a variety of the Isocoumarin Compounds have significant Calpain inhibitory activity at low concentrations. EXAMPLE lB(i)
Protease Inhibition by Peptide Keto-Compounds The Peptide Keto-Compounds are reversible inhibitors of Calpains and other thiol proteases. The 1^ values for the inhibition of calpain I, calpain II and Cathepsin B were determined for several Peptide Keto-Compounds. Inhibition of calpain I from human erythrocytes and calpain II from rabbit muscle were assayed using
Suc-Leu-Tyr-amidomcthylcoumarin as substrate in an assay buffer of 20mM HEPES
pH=7.2, lOmM CaCl2, lOmM β-mercaptoethanol. Cathepsin B from bovine spleen was assayed using Z-Lys-4-nitrophenylphosphate as substrate.
Table lB(i) shows the results of the studies of Example lB(i). The Ki value for the inhibition of Calpains and cathepsin B by several Peptide Keto-Compounds are shown in μM (micromolar). The values for leupeptin, which is commerciaUy avaUable from Calbiochem of La JoUa, California, are shown for comparison.
Table lB(i) Kj VALUES FOR PEPTIDE KETO-COMPOUNDS
Inhibitor Calpain I Calpain II Cathepsin B
10
It can be seen from the results in Table lB(i) that the Peptide Keto-Compounds inhibit Calpain with Ki values similar or superior to leupeptin. In particular, Z-Leu- 15 Phe-C02Et, Z-Leu-Nle-C02Et and Z-Leu-Abu-C02Et were found to possess greater
Calpain inhibitory activity than leupeptin. In addition, these particular compounds were highly specific to Calpain, with lower inhibitory activity toward Cathepsin B than leupeptin.
EXAMPLE lB(ii) 20 Protease Inhibition of Peptide Keto-Compounds
We tested the abUity of an additional group of Peptide Keto-Compounds to inhibit several proteases in order to evaluate their specificity for Calpain. The results of these studies are shown in Table lB(ii). Table lB(ii). Inhibition of Calpain I, Calpain II, Cathepsin B (CathB), Chymotrypsin (Chym), PP Elastase and Papain
Table lB(ii) shows the inhibition constants (Kj) for cathepsin B, calpain I, and calpain II with peptide ketoamides. Dipeptide Ketoamides with Abu and Phe in the Pχ site and Leu in the P2 site are potent inhibitors of calpain I and calpain II. Z-Leu- Abu-CONH-Et is a better inhibitor of calpain I than Z-Leu-Phe-CONH-Et by 14 fold. Replacement of the Z group (PhCH2OCO-) by simUar groups such as PhCH2CH2CO-, PhCH2CH2S02-, PhCH2NHCO-, and PhCH2NHCS- would also result in good inhibitor
structures. One good inhibitor of calpain II is Z-Leu-Abu-CONH-(CH2)2-Ph. Changing the R3 and R4 groups significantly improves the inhibitory potency toward calpain II. The best Dipeptide Ketoamide inhibitors are those which have long alkyl side chains (e.g. Z-Leu-Abu-CONH-(CH2)7CH3), alkyl side chains with phenyl substituted on the alkyl group (e.g. Z-Leu-Abu-CONH-(CH2)2-Ph), or alkyl groups with a moφholine ring substituted on the alkyl group (e.g. Z-Leu-Abu-CONH-(CH2)3-Mpl, Mpl = -N(CH2CH2)20). Dipeptide α-ketoamides with a s aU aliphatic amino acid residue or a Phe in the Pj site are also good inhibitors for cathepsin B. The best inhibitor is Z-Leu-Abu-CONHEt and replacement of the Z (PhCH2OCO-) by PhCH2CH2CO-, PhCH2CH2S02-, PhCH2NHCO-, and PhCH2NHCS- would also result in good inhibitor structures.
EXAMPLE lB(iii) Stability of Peptide Keto-Compounds We determined the half-life in minutes of several Peptide Keto-Compounds in both plasma and liver homogenates. The results of the determinations of stabUity of the compounds in plasma and liver homogenates are shown in Table lB(iii).
Table lB(iii). Stability in Plasma and in Liver of Peptide Keto-Compounds.
It can be seen from the data in Table lB(iii) that the Peptide Keto-Compounds are generaUy quite stable in plasma and liver homogenates. However, it is also shown that the Peptide α-ketoamides were substantiaUy more stable in both plasma and liver than the corresponding peptide α -ketoesters
EXAMPLE IC
Protease Inhibition by Halo-Ketone Peptides
The Halo-Ketone Peptides, like the substituted isocoumarins, are irreversible inhibitors of Calpain. We determined the Kapp/[I] values for various members of this class of compounds against Calpains I and II. For comparison, we also determined these values against the additional thiol proteases Papain and Cathepsin B for at least one Halo-Ketone Peptide. These Kgpp values are not directly comparable to the 1^ or
IC50 values determined above for other classes of inhibitors.
We assayed Calpain I and II using Suc-leu-tyr-amidomethylcoumarin. Papain was assayed using benzoyl-arg-4-nitroanUide, and Cathepsin B (bovine) was assayed using CBZ-lys-4-nitrophenyl ester. We foUowed the progress curve method of Tian and
Tsou, Biochemistry, 21:1028-1032 (1982), the disclosure of which is hereby incoφorated by reference, to derive kinetic data. Briefly, this method makes use of the equation:
[P J = V[S]/K (1 + [S]/K)A[Y] where [PJ represents the concentration of product formed at a time approaching infinity, A is the Kapp in the presence of substrate (S), K is the Michaelis constant and
[Y] is the concentration of the inhibitor. Since [S] and [Y] are known and V and K can be determined, Kapp can be readUy determined. The K /[I] for various Halo-Ketone Peptides are shown in Table IC.
TABLE IC KINETIC PARAMETERS OF HALO-KETONE PEPTIDES
Inhibitor Cl CIl3 P CB
Z-Gly-Leu-Phe-CH2C1 2840001 946000
Boc-Gly-Leu-Phe-CH2Cl 902000 540000 290000
Z-Leu-Phe-CH2C1 2250002 585000
Z-Gly-Leu-Ala-CH2C1 210000
Ac-Leu-Phe-CH2C1 25900 1 33400
Z-Val-Phe-CH2C1 27200
Z-Ala-Phe-CH2C1 2400
Ac-Ala-Ala-Ala-Ala-CH2C1 1300
Cl = Calpain I ! - Rat CIl = Calpain II 2 - Human
P = Papain 3 - Rabbit CB = Cathepsin B
It can be seen from the results in Table IC that the Halo-Ketone Peptides inhibit Calpain with relatively high K /[I] values. In particular, Z-gly-leu-phe-CH2Cl, Boc-gly-leu-phe-CH2Cl, Z-leu-phe-CH2Cl and Z-gly-leu-ala-CH2Cl were found to possess significant Calpain inhibitory activity. In addition, Boc-gly-leu-phe-CH2Cl was shown to be somewhat specific to Calpain, with lower inhibitory activity toward Cathepsin B or Papain than toward Calpain. The results shown in the table reveal that Z-gly-leu-phe-CH2Cl and Boc-gly-leu-phe-CH2Cl produce simUar inhibitory effects. Thus, the blocking group is not shown to have a great effect on Calpain inhibitory activity.
The kinetic constants of other irreversible Calpain Inhibitors include the foUowing with Kapp/[I] in parentheses: E-64 (7500), E64-d (23000) and Z-leu-leu-tyr- CHN2 (230000). E-64 is commerciaUy avaUable from Sigma Chemical Co., and is shown here to be a poor inhibitor of Calpain. Z-leu-leu-tyr-CHN2 is a diazomethyl peptide compound, here shown to possess significant Calpain inhibitory activity.
2. Inhibition of Calpain in Neural Tissues
In order to evaluate the inhibition of Calpain by the various Calpain Inhibitors in neural tissues, we assayed the Calpain Inhibitors using the known abUity of Calpain to cleave spectrin, a protein component of neuronal and other tissue, into BDP's. In this assay, more effective Calpain Inhibitors wiU prevent the conversion of spectrin into
BDP's. Example 2 is one example of such an assay.
EXAMPLE 2
Inhibition of Calpain in Crude Brain Extracts bv Calpain Inhibitors The activity of Calpain in crude brain extracts was measured by examining the
Ca2+ -stimulated proteolysis of the endogenous substrate spectrin. Brain tissue was homogenized in lOmM Tris pH = 7.4, 0.32M sucrose, ImM EGTA, ImM dithiothreitol a nd the nuclei and debris removed by low speed centrifugation. Various Calpain Inhibitors were added to the supernatant in a DMSO vehicle and a calcium salt (final effective concentration about 1.2mM) added to start the reaction. Proteolysis of spectrin was evaluated by western blot as described by Seubert, et al, Brain Res., 459:226-232 (1988), the disclosure of which is hereby incoφorated by reference. Briefly, a known quantity of a spectrin-containing sample treated with Calpain is separated by SDS-PAGE and immunoblotted with anti-spectrin antibody. The amount of spectrin immunoreactivity found corresponding to the characteristic BDP's is indicative of the amount of spectrin activity present in the sample. An examplary method for quantitating BDP's is to assay Spectrin BDP's by homogenizing brain parts in 20mM Tris pH=7.2, .32M sucrose, 50μM Ac-Leu-Leu-nLeu-H on ice. Homogenates are then mixed 1:1 with 10% SDS, 5% β-mercaptoethanol, 10% glycerol, lOmM Tris pH = 8.0, 0.5% bromophenolblue, heated to 95° C, and subjected to electrophoresis in
4-1/2 % polyacrylamide gels. The proteins in the gels are transferred to nitroceUulose and the spectrin and BDP's detected using a rabbit polyclonal anti-spectrin antibody and established immunodetection methods. The amount of spectrin and BDP's in each sample can be quantitated by densitrometric scanning of the developed nitroceUulose. Due to Calpain's requirement for Ca , in the absence of Ca little or no spectrin proteolysis occurred, regardless of the presence of inhibitor, whUe in the
presence of Ca2+ the spectrin was >95% cleaved to BDP's within 40 min. if no Calpain Inhibitor is added.
Both leupeptin and CIl showed inhibition in the system of Example 2. In addition, the foUowing compounds of the Substituted Heterocyclic Compounds were found to produce significant inhibition at 100 μM:
3-chloroisocoumarin 3,4-dichloroisocoumarin 3-benzyloxy-4-chloroisocoumarin 7-(acetylamino)-4-chloro-3-(propoxy)-isocoumarin 4-chloro-3-(3-isothiureidopropoxy)isocoumarin
7-amino-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(benzylcarbamoylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(phenylcarbamoylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(acetylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(3-phenylpropionylamino)-4-chloro-3- (3-isothiureidopropoxy)isocoumarin
7-(phenylacetylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(L-phenylalanylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(benzylcarbamoylamino)-4-chloro-3-
(3-isothiureidoethoxy)isocoumarin 7-(phenylcarbamoylamino)-4-chloro-3-
(3-isothiureidoethoxy)isocoumarin 7-(D-phenylalanylamino)-4-chloro-3- (3-isothiureidoethoxy)isocoumarin.
The foUowing compounds of the Halo-Ketone Peptides were also found to produce significant inhibition at 100 μM: Z-Leu-Phe-CH2C1 Ac-Leu-Phe-CH2C1 Z-Gly-Leu-Phe-CH2C1
Boc-Gly-Leu-Phe-CH2Cl Ac-Val-Phe-CH2C1 Z-Gly-Leu-Ala-CH2C1.
In addition, the foUowing compounds of the Peptide Keto-Compounds were found to produce significant inhibition at 100 μM:
Bz-DL-Phe-COOEt Z-Leu-Nva-COOEt Z-Leu-Nle-COOEt Z-Leu-Phe-COOEt Z-Leu-Abu-COOEt
Z-Leu-Met-COOEt Z-Ala-Ala-DL-Abu-COOEt MeO-Suc-Val-Pro-DL-Phe-COOMe Z-Ala-Ala-Ala-DL-Ala-COOEt MeO-Suc-Ala-Ala-Pro-DL-Abu-COOMe.
Z-Leu-Phe-COOEt
Thus, the Substituted Heterocyclic Compounds, Peptide Keto-Compounds and Halo-Ketone Peptides, in addition to leupeptin and CIl, provide inhibition in brain homogenates. 3. In vivo Inhibition of Neurodegeneration through Infusion Techniques
In order to demonstrate that the inhibition of Calpain activity alone is sufficient to inhibit neurodegeneration in vivo, we tested the abUity of the Calpain Inhibitor, leupeptin, to inhibit neurodegeneration in gerbUs subjected to transient ischemia.
As stated above, leupeptin is poorly membrane permeant. Therefore, leupeptin is not expected to cross the blood-brain barrier ("BBB") very weU. Accordingly, in order to provide the brain with sufficient leupeptin to adequately inhibit Calpain activation, we used brain infusion techniques. Through the use of these techniques we
were able to subject brain tissues to intimate contact with leupeptin for sustained periods of time. Example 3A is provided to show the in vivo protection from neurodegeneration found during one such study.
EXAMPLE 3A In Vivo Protection Against Neurodegeneration
A smaU cannula was implanted in the right lateral ventricle of adult gerbUs, and secured to the skuU with dental cement. An Alzet micro-osmotic pump was attached to the cannula for intracerebroventricular perfusion. The pump was fiUed with either saline alone (control) or leupeptin (20 mg/ml in saline). After three days perfusion with either the control solution or with the leupeptin solution, transient ischemia was induced by bUateraUy clamping the carotid arteries for a period of ten minutes. Core temperatures were taken during and foUowing ischemia, with no differences noted between control and leupeptin treated animals. Fourteen days later, the animals were sacrificed by Nembutal overdose and transcardial perfusion of a 10% solution of paraformaldehyde in PBS. Coronal sections of the brain were stained with cresyl violet and were examined for the extent of neuronal loss. The control gerbUs exhibited the typical damage found in the CA1 field foUowing ischemia, with a 72% loss of neurons. However, the leupeptin treated gerbUs showed far less neurodegeneration, with only a 15 % loss of neurons. The results of Example 3A cannot be explained by changes in thermoregulation, since core temperatures did not differ between the groups. Accordingly, we believe that the Calpain inhibitory activity of leupeptin is responsible for the observed differences in neuronal ceU loss. In order to further quantitate the differences, and verify that leupeptin produced a Calpain inhibitory effect within the observed regions of the brain, we performed a related series of experiments. In this series of experiments, spectrin BDP's were measured in the leupeptin treated and control animals. As discussed above, these BDP's are indicative of the amount of Calpain activity occurring within the tissue. Example 3B is provided to demonstrate the results of these experiments.
EXAMPLE 3B
In Vivo Inhibition of Calpain Activity Implantation surgeries and clamping of the carotid arteries were performed as above with a control-ischemia group (n=4) and a leupeptin-ischemia group (n = 5). A third group of animals (n=4) received implantation with pumping of saline, but was not subjected to ischemia. Animals were sacrificed by decapitation 30 minutes after clamping of the arteries. The brains were rapidly removed and placed in cold homogenization buffer (0.32 M sucrose, 10 mM Tris-HCl, 2 mM EDTA, 1 mM EGTA, 100 μM leupeptin and 1 μg/ml of the Halo-Ketone Compound, tos-phe-CH2Cl (TPCK)). The CA1 region of the hippocampus was then dissected. The samples from both control and leupeptin treated animals were then prepared for SDS-PAGE and immunoblotting with labeled anti-spectrin antibody, as described above in connection with in vitro uses of the Calpain Inhibitors. The control animals exhibited a marked increase in the levels of BDP's relative to the gerbUs not subjected to ischemia. These BDP's co-migrated with BDP's observed after in vitro proteolysis of spectrin with
Calpain. The brain tissue from the leupeptin treated gerbUs exhibited approximately 25% of the BDP's observed in the control ischemia treated gerbUs.
Another group of gerbUs (n = 3) were sacrificed immediately after ischemia without leupeptin treatment in order to observe the effects of ischemia without reoxygenation. These gerbUs exhibited a simUar amount of increase of BDP's as the control-ischemic gerbUs observed after a 30 minute reperfusion period.
Thus, the results of Example 3B indicate that leupeptin exerts its neuroprotective effect through the inhibition of Calpain activation. The results also indicate that the observed proteolysis of spectrin was an effect of ischemia, and not secondary to the reoxygenation. Accordingly, the results indicate that inhibition of
Calpain activity in vivo produces a neuroprotective effect.
Although the foregoing studies demonstrate that leupeptin can inhibit neurodegeneration in vivo, leupeptin is not the therapeutic drug of choice because of the need to infuse the drug directly into the brain for an extended period of time to exert its neuroprotective effect. This is due to the relatively poor abUity of this compound to cross the BBB. Accordingly, it is believed that a more therapeuticaUy
practical way to inhibit neurodegeneration would be to use more membrane permeant Inhibitor of Calpain.
4. Platelet Permeability
In accordance with our discoveries demonstrated in Examples 3 and 3A, we believe that having a compound cross the BBB and enter CNS tissue is a key characteristic of a therapeuticaUy useful approach to treat or inhibit neurodegeneration within the CNS. Use of Calpain inhibitors that have enhanced membrane permeabUity is one such approach. Thus, we measured the abUity of various Calpain inhibitors to penetrate the platelet membrane and inhibit Calpain that is normaUy contained in platelets. As shown below in the foUowing examples, our results indicate that particular compounds of the Heterocyclic Compounds, Peptide Keto-Compounds and Halo- Ketone Peptides, in addition to the Peptide Aldehyde, CIl, exhibit good membrane permeabUity.
As an indication of the membrane permeabUity of the various Calpain Inhibitors, we measured the abUity of various Calpain Inhibitors to penetrate platelet membranes and inhibit the Calpain normaUy found within platelets. The membrane of platelets is believed to have many simUarities to the BBB and accordingly, such experiments are believed to provide a good indication of the abUity of the various Calpain Inhibitors to cross the BBB. Example 4 shows the results of some of these platelet experiments using the Calpain Inhibitors of the present invention.
EXAMPLE 4A Membrane Permeation of Calpain Inhibitors Platelets were isolated by a modification of the method of FerreU and Martin, /. Biol Chem., 264:20723-20729 (1989), the disclosure of which is hereby incoφorated by reference. Blood (15-20 ml) was drawn from male Sprague-Dawley rats into lOOmM
EDTA-citrate containing 10 units heparin, and centrifuged 30 minutes at 1600 φm at room temperature. The plasma was resuspended in 15ml buffer 1 (136mM NaCl, 2.7mM KCl, 0.42mM NaR2P04, 12mM NaHC03, 2mM MgCl2, 2 mg/ml BSA (Sigma), 5.6mM glucose, 22mM Na3Citrate pH 6.5) and platelets were isolated at 2200 φm at room temperature of 25 minutes.. Platelets were resuspended to 107 ceUs/ml in buffer
2 (136mM NaCl, 2.7mM KCl, 0.42 NaH2PO 12mM NaHCOs, 2mM MgCl, 1 mg/ml
BSA, 5.6mM glucose, 20mM HEPES pH 7.4) and aUowed to "rest" for a minimum of 10 minutes at room temperature before use.
Platelets were incubated for 5 minutes in the presence of inhibitor. In order to provide sufficient intraceUular calcium to activate Calpain, the calcium ionophore A23187 was added to a final concentration of lμM. After a further 5 minute incubation, the platelets were harvested by centrifugation (1 min 10,000 x g) and resuspended in 10% sodium dodecyl sulfate, lOmM Tris pH = 8.0, 5% β-mercaptoethanol, 0.02% bromophenol blue, and heated to 95° C for 5 min. Samples were subjected to SDS-PAGE on 6% mini gels and transferred to nitroceUulose (Schleicher and SchueU BA83) for 2 hours at lOOmA/gel in an LKB Novablot. FUters were blocked for 10 minutes in 0.25% gelatin, 1% BSA, 0.25% Triton X100, 0.9% NaCl, lOmM Tris-Cl pH 7.5, incubated overnight in the same solution containing antibody to rat spectrin, washed 3 X 10 minutes with lOmM Tris-Cl pH 7.5, 0.5% Triton X100, incubated 4 hours in wash buffer plus alkaline phosphatase conjugated goat anti-rabbit antibody (Biorad), and washed as above. FUters were developed using the Biorad AP conjugate substrate kit. Spectrin immunoreactivity on the fUters was quantitated by densitometry.
The inhibition of Calpain within platelets as measured by the proteolysis of the endogenous Calpain substrate spectrin in the presence of inhibitors was assayed for a variety of Calpain Inhibitors. The poorly permeant inhibitors leupeptin and E-64 had little effect on intraceUular Calpain. In contrast, the highly membrane permeant Heterocyclic Compounds, Peptide Keto-Compounds, and Halo-Ketone Peptides effectively inhibited platelet Calpain.
The foUowing Heterocyclic Compounds were found to produce significant inhibition at 100 μM in the system of Example 4:
3-chloroisocoumarin
4-chloro-3-(3-isothiureidopropoxy)isocoumarin 7-amino-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(benzylcarbamoylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin
7-(phenylcarbamoylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(acetylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(3-phenylpropionylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(phenylacetylamino)-4-chloro-3-
(3-isothiureidopropoxy)isocoumarin 7-(L-phenylalanylamino)-4-chloro-3- (3-isothiureidopropoxy)isocoumarin
7-(benzylcarbamoylamino)-4-chloro-3-
(3-isothiureidoethoxy)isocoumarin 7-(phenylcarbamoylamino)-4-chloro-3-
(3-isothiureidoethoxy)isocoumarin 7-(D-phenylalanylamino)-4-chloro-3-
(3-isothiureidoethoxy)isocoumarin. The foUowing Halo-Ketone Peptides were found to produce significant inhibition at 100 μM in the system of Example 4: Z-Leu-Phe-CH2C1 Ac-Leu-Phe-CH2C1
Z-Gly-Leu-Phe-CH2C1 Boc-Gly-Leu-Phe-CH2Cl.
The foUowing Peptide Keto-Compounds were found to produce significant inhibition at 100 μM in the system of Example 4: Z-Ala-Ala-D,L-Abu-COOEt
Z-Ala-Ala-Ala-D,L-Ala-COOEt MeO-Suc-Ala-Ala-Pro-D,L-Abu-COOMe Z-Leu-Phe-COOEt Z-Leu-Nle-COOEt Z-Leu-Nva-COOEt
Z-Leu-Abu-COOEt Z-Leu-4-Cl-Phe-COOEt
Z-Leu-Leu-Abu-COOEt Z-Leu-Leu-Phe-COOEt 2-NapS02-Leu-Abu-COOEt 2-NapS02-Leu-Leu-Abu-COOEt Z-Leu-Met-C02Et
Z-Leu-NLeu-C02Et Z-Leu-Phe-C02Bu Z-Leu-Abu-C02Bu Z-Leu-Phe-C02Bzl Z-Leu-Abu-C02Bzl
Z-Ala-Ala-D,L-Abu-COOBzl Z-Leu-Phe-COOH Z-Leu-Abu-COOH. Among those compounds found to exhibit Calpain inhibitory activity in the homogenate system of Example 2, we found at least three compounds which faUed to exhibit Calpain inhibitory activity in the platelet system of Example 4. These compounds are leupeptin, MeO-Suc-Val-Pro-D,L-Phe-COOMe and Bz-D,L-Phe- COOEt. Leupeptin is known to be poorly membrane permeant, thus confirming that the platelet assay wiU exclude known poorly membrane permeant compounds. Accordingly, the two Peptide Ketocompounds found not to provide Calpain inhibitory activity within platelets are also believed to be poorly membrane permeant, and would not be expected to cross the BBB.
EXAMPLE 4B Quantitative Studies of Platelet Membrane Permeability We performed additional quantitative or semi-quantitative studies on several
Peptide Keto-Compounds using the assay of Example 4A, except that IC50 values were determined as the concentration at which 50% of the Calpain activation present in controls occurred. Results are shown in Table 4B. For the semi-quantitative assays, indicated with +'s in Table 4B, "+" indicates detectable inhibition at 100 μM, "+ +" indicates significantly more inhibition than "+", and "+ + +" indicates no detectable activation of Calpain detected.
TABLE 4B Platelet Assay of Peptide Ketoamides, Ketoesters and Ketoacids
Table 4B shows that peptide α-ketoamides and ketoacids were much more effective than corresponding peptide ketoesters in this platelet assay. Extending the R
3 group to an alkyl group or an alkyl group substituted with a phenyl group increased the membrane permeabUity of the inhibitors as indicated by increased potency in the platelet assay. In view of these results, Applicants believe that extending the R group to include longer alkyl groups or alkyl groups substituted with phenyl groups would increase the membrane permeabUity of a given inhibitor.
In view of the foregoing, the results of Examples 4A and 4B support our belief that CIl and the Substituted Heterocyclic Compounds, Peptide Keto-Compounds and Halo-Ketone Peptides are believed to be membrane permeant and therefore, are expected to be effective in crossing the BBB subsequent to in vivo administration of the compounds.
5. Reduction of Glutamate Toxicity
To further identify those Calpain Inhibitors likely to possess pharmacologicaUy active neuroprotective abUity, we tested the abUity of the Calpain Inhibitors to protect against glutamate excitotoxicity. Excess extraceUular glutamate is thought to play a key
role in the induction of neuropathology in ischemia, which is accompanied by Calpain activation. In support of this role for excess glutamate, cultured N18-RE-105 (a neuroblastoma-retinal hybrid) ceUs can be kiUed by the addition of glutamate into the culture medium. This glutamate-mediated cytotoxicity is calcium dependent and can be reduced through a number of mechanisms, including free radical scavengers, blockers of the N-type voltage-sensitive calcium channel, and quisqualate-subtype glutamate antagonists. Thus, glutamate-mediated killing of N18-RE-105 ceUs is an in vitro model for neuropathology.
Accordingly, we tested the abUity of the Calpain Inhibitors to inhibit glutamate- induced ceU death in these ceUs in order to establish that the Calpain Inhibitors can decrease or prevent glutamate-induced death Of N18-RE-105 ceUs. Some of these tests are shown in Example 5A.
EXAMPLE 5A Inhibition of Glutamate-induced Cell Death Stock cultures of N18-RE-105 ceUs were maintained in Dulbecco's modified
Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and supplemented with hypoxanthine, aminopterin and thymidine (HAT). Subconfluent cultures were split and plated into 96-weU plates. Twenty-four hours after plating the ceUs were exposed to fresh media containing glutamate and various concentrations of Calpain inhibitors. Control ceUs were not treated with glutamate. The treated ceUs received 5mM glutamate and leupeptin (5μg/ml) or the other Calpain Inhibitors listed in Figure 1 at 3μg/ml. Conversion of MTT was measured 19 hours later as described. Nineteen hours after the onset of exposure, ceU viabUity was quantitated by measuring the extent to which the ceUs convert 3(4,5-dimethylthiazol-2-yl)-2-5-diphenyltetrazolium bromide (Mi l) to a blue formazan product, which occurs in the mitochondria of living but not dead ceUs (Pauwels et al.; 1988). A higher absorbance is indicative of greater ceU viabUity.
Figure 1 shows the percent of blue formazan product remaining after treatment with glutamate, relative to control where no glutamate was added. Thus, it can be seen that with vehicle plus glutamate but no inhibitor, less than 70% of the mitochondrial activity remains. However, Figure 1 shows that several Calpain inhibitors, including leupeptin, CIl and representatives of the Heterocyclic Compounds, Peptide Keto-
Compounds and Halo-Ketone Peptides protect N18-RE-105 ceUs against glutamate toxicity. The Peptide Keto-Compound Calpain inhibitor, Z-Ala-Ala-Abu-C02Et, the Substituted Heterocyclic Compounds, CITPrOIC and ACITIC, and the Halo-Ketone Peptide, TPCK completely blocked the toxic effects of glutamate, resulting in 100% or greater of the formazan product as seen with ceUs not treated with glutamate. Thus,
Example 5 shows that these Calpain Inhibitors effectively block ceU death in an in vitro model for neuropathology. Accordingly, this data further supports our discovery that Calpain Inhibitors are neuroprotective in vivo.
We have discovered that glutamate-induced ceU death in pheochromocytoma PC12 ceUs can be prevented by the membrane permeant calpain inhibitors
Z-Leu-Phe-CONHCH2CH3 and Z-Leu-Nva-CONH(CH2)3 moφholine. These inhibitors rescue a greater proportion of the PC12 ceUs than calpain inhibitor 1 (Ac- Leu-Leu-Norleucinal) although higher concentrations can be required in certain instances. Z-Leu-Nva-CONH(CH2)3 moφholine also induces short processes in both the presence and absence of glutamate. We observed reduction in glutamate-induced ceU death is observed even when calpain inhibitors are added several hours after glutamate. These observations are the first demonstration that Calpain inhibitors can rescue ceUs in vitro from glutamate toxicity and support the critical role of calpain activation in excitotoxicity. These experiments provide still further support that inhibition of calpain is useful in the treatment of neurodegeneration, such as stroke and ischemia.
The rat pheochromocytoma PC 12 is described by Greene et al, Proc. Natl. Acad Sci USA, 73:2424-2427 (1976), the disclosure of which is hereby incoφorated by reference. This tissue expresses the NMDA subtype of glutamate receptors. Treatment of PC12 ceUs with glutamate for 24 hours produces death of 80% of the ceUs as measured by conversion of Glutamic acid, 3-[4,5-dimethylthazol-2-yl]-2,5- diphenyltetrazolium bromide (MTT) into its blue formazan product. When PC12 ceUs are exposed simultaneously to glutamate and calpain inhibitor, ceU death is reduced. Thus, these ceUs can be used as an effective model to determine the effectiveness of the various calpain inhibitors to aUeviate ceU death. The experimental procedures we used to evaluate glutamate toxicity are described below in Example 5B.
Example 5B
Glutamate Toxicity Assay in Pheochromocytoma Cells
CeUs of the RC72 subclone of the rat pheochromocytoma PC12 were grown in high glucose Dulbecco's modified Eagle's medium (DME) without glutamine with 10% fetal bovine serum and 5% horse serum and gentamycin. These ceUs are avaUable from Dr.
David Schubert of the Salk Institute. Media, horse serum, dialyzed fetal calf serum, and gentamycin were from Irvine Scientific. Fetal calf serum was from BioCeU.
Prior to plating, ceUs were cultured for 2 passages (4 days) in the same media with glutamine. CeUs were plated at 10,000 ceUs/weU into 96 weU plates coated with coUagen and grown for 24 hours prior to experiments.
Exposure to glutamate was performed in DME without glutamine with 10% dialyzed fetal calf serum and 50 ug/ml gentamycin. Calpain inhibitors were added to cultures from DMSO stocks. Final DMSO concentrations did not exceed 0.1%.
After 24 hours exposure to glutamate and inhibitors, 20ul of 7.5 mg/ml Glutamic acid, 3-[4,5-dimethylthazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) in PBS was added to each weU. MTT is avaUable from Sigma. The cultures were incubated for 60 minutes and the media carefuUy removed. Detergent (10% Triton X-100, 0.4% concentrated HCl in isopropanol) was added and incubated for 10 minutes on a shaking table before the plates were read using a microplate reader. The difference between the absorbance at 655 and 595 nm was used as a measure of viabUity. AU experiments were normalized to untreated ceUs in the same plate.
We tested a number of Calpain inhibitors for their abUity to rescue PC12 ceUs from glutamate toxicity using the experimental protocol described in the foregoing example. The results are shown in Table 5B. TABLE SB
Inhibitor Concentration μM % of control
Ac-Leu-Leu-Arg-H (leupeptin) 5000 6.9
Ac-Leu-Leu-Nle-H (calpain inhibitor 1)
E64 TPCK
Cystatin C
Z-Leu-Phe-CONHCH2CH
3 Z-Leu-Abu-CONHCH
2CH
3 Z-Leu-Abu-CONH(CH
2)
3 moφholine Z-Leu-Abu-CONH(CH
2)
2 phenyl Z-Leu-Abu-CONH
2 Z-Leu-Nva-CONHCH
2CH
3 Z-Leu-Phe-CONH(CH
2)
3 moφholine Z-Leu-Nva-CONH(CH
2)
3 moφholine
Reduction of glutamate-mediated ceU death is produced by several different calpain inhibitors. Z-Leu-Phe-CONHCH2CH3 and Z-Leu-Nva-CONH(CH2)3 moφhoHne appeared to provide the best results in rescuing ceUs from glutamate toxicity. Inhibitors related to Z-Leu-Phe-CONHCH2CH3 also rescue PC 12 ceUs from glutamate toxicity, although with varying efficacy. Substitution of Abu for Phe or Nva in the PI position decreases the efficacy of the compounds. Several calpain inhibitors, including leupeptin and E64, did not rescue the cells. Leupeptin and E64 are known to be poorly ceU- penetrating, providing further support for membrane-permeance as an important factor in the pharmacological effectiveness of the calpain inhibitors as used in the present invention. The poor calpain inhibitors cystatin C and TPCK also did not rescue the ceUs. This result is consistent with our conclusion that ceU death is specificaUy the result of calpain activation and does not involve another protease with related specificity.
We further studied the more effective compounds, Z-Leu-Phe-CONHCH2CH3 and Z-Leu-Nva-CONH(CH2)3 moφholine. Figure 2 shows the results obtained using the procedures of the foregoing Example using different concentration of these two compounds along with calpain inhibitor 1 (CIl), a peptide aldehyde. The concentrations we used are as indicated. CIl prevents glutamate-induced ceU death at concentrations as low as 3uM but its efficacy does not increase but rather decreases with increasing concentration. We have observed toxicity by CIl at higher concentrations; however, CIl is not toxic by itself at concentrations below lOuM. Thus, we believe that this toxicity explains the observed increase in ceU death at higher concentrations of CIl. The calpain inhibitors Z-Leu-Phe-CONHCH2CH3 and Z-Leu-Nva-CONH(CH2)3 moφholine exhibit
typical sigmoidal dose-response curves for ceU rescue and produce nearly complete rescue at high concentrations.
We observed no toxicity of Z-Leu-Phe-CONHCH2CH3 or Z-Leu-Nva-CONH(CH2)3 moφholine at any concentration tested. This reduction of ceU death by these compounds is dose-dependent with IC50 values of 20-50uM. The IC50 values of 20-50uM for Z-Leu-Phe-CONHCH2CH3 and Z-Leu-Nva-CONH(CH2)3 moφholine are significantly above the Ki's for calpain I or calpain II for these compounds. The i values for Z-Leu-Phe-CONHCH2CH3 and Z-Leu-Nva-CONH(CH2)3 moφholine are 200 nM and 250nM, respectively, using human erythrocyte calpain I, and 22nM and lOOnM, respectively, using rabbit muscle calpain II. There are two possible explanations for the difference between the IC50 values we measured and the I^'s for these compounds using purified Calpain: less than complete penetration of the ceU or the metabolism of the inhibitors by the ceUs. As discussed above, the poorly permeant inhibitor leupeptin is ineffective at preventing ceU death. Thus, we believe that the difference between the Ki and IC50 values is due to membrane permeance effects.
We also evaluated the effect of glutamate concentration on the abUity o Z-Leu-Phe-CONHCH2CH3 or Z-Leu-Nva-CONH(CH2)3 moφholine to aUeviate ceU death. Figure 3 shows the results obtained when PC12 ceUs were incubated with the indicated concentration of glutamate and no inhibitor (circles), 20u Z-Leu-Nva-CONH(CH2)3 morpholine (triangles), or 30uM Z-Leu-Phe-CONHCH2CH3
(squares) for 24 hours and ceU viabUity was assayed by MTT, as described in the Example. Values are expressed as % of naive control ± sem. At submaximal concentrations of these compounds, the rescuing effect can be overcome by high concentrations of glutamate. Thus, it is clear that the rescue of PC12 ceUs from glutamate toxicity is related to both th concentration of glutamate and to the concentration of inhibitor. We believe that th dependence on glutamate concentration is the result of the activation of multiple pathway of ceU damage by the high concentration of glutamate. There is ample evidence fo calpain-independent mechanisms of excitotoxic ceU death. Observations by others tha antioxidants such as vitamin E also rescue these ceUs from glutamate toxicity provid support for the idea that such calpain-independent mechanisms are operative. However we have shown that inhibition of calpain alone is sufficient to aUeviate ceU death. Thus
the use of Calpain inhibitors in accordance with the present invention is a signficant unexpected finding.
We also studied the abUity of Calpain inhibitors to aUeviate ceU death after the ceUs have been exposed to glutamate. The results of this analysis are shown in Figure 4. Glutamate at 7.5mM was added at 0 time and Z-Leu-Phe-CONHCH2CH3 (squares) or
Z-Leu-Nva-CONH(CH2)3 moφholine (triangles) added at the indicated times to final concentrations of lOOμM each. CeU viabUity was measured 24 hours after the addition of glutamate by the MTT assay. Values are expressed in the Figure as % of naive control ± sem. It can be seen that addition of calpain inhibitors after the ceUs have been exposed to glutamate is only partiaUy effective. Advantageously, some rescue of ceU death is still observed if inhibitor is added as long as 2 hours after glutamate. However, by 8 hours after the glutamate addition, inhibitors no longer have an effect. Accordingly, it is advantageous to administer the Calpain inhibitors of the present invention within two hours of glutamate activation. Interestingly, at 8 hours after the addition of glutamate, the ceUs are stUl largely normal in appearance and are 100% viable by MTT assay regardless of the presence of inhibitor. This suggests that Calpain cleaves one or more ceUular proteins which are essential for ceUular functioning, and this perturbation results in ceU death some hours later. Thus, it is still desirable to administer Calpain inhibitors within two hours of glutamate release, even where ceU or tissue morphology remains normal. We also evaluated moφhological changes produced by the Calpain inhibitors. We exposed ceUs to 100 μM Z-Leu-Nva-CONH(CH2)3 moφholine alone or to 100 μM Z-Leu-Nva-CONH(CH2) moφholine and 7.5 μM glutamate. CeUs exposed to both treatments show an altered moφhology with short processes extending from the ceU body which are rarely seen in the absence of inhibitor but are occasionaUy seen with glutamate alone in surviving ceUs. Inhibitor at high concentrations does not prevent some of the glutamate-induced decrease in the number of adherent ceUs, but these non-adherent ceUs remain viable as measured by MTT conversion. Incubation of PC12 ceUs with Z-Leu-Nva-CONH(CH2)3 for longer times (up to 72 hours) does not cause the expansion of these short processes into longer neurites, nor does it cause cytotoxicity. This moφhological effect is not seen consistently with different calpain inhibitors, and is not caused by CIl.
Excitotoxicity in vivo, as weU as other forms of neurodegeration, are accompanie by the breakdown of the cytoskeletal protein spectrin, which we believe is mediated b calpain. The breakdown of spectrin in vivo, as weU as the digestion of spectrin by calpai in vivo, produces not only the reduction in the amount of intact spectrin but also characteristic doublet of spectrin breakdown products (BDP's) of molecular weight 150 an
155kDa. These BDP's appear to be unusuaUy persistent in vivo. The detection of spectri BDP's can be used as an assay for ceUular degeneration, especiaUy neurodegeneration See U.S. Patent No. 5,118,606 to Lynch et al., the disclosure of which is hereb incoφorated by reference. We exposed PC12 ceUs to glutamate for 24 hours. CeUs were extracted wit
CHAPS and analyzed for spectrin breakdown by western assay, as described in U.S. Paten No. 5,118,606. Analysis of the PC12 ceUs after glutamate toxicity reveals a decrease in th amount of intact spectrin but no striking increase in the 150 and 155kDa BDP's. Th decrease in the amount of spectrin immunoreactivity cannot be accounted for by loss o protein from the samples as equal amounts of protein were loaded in each lane. Thus, i this assay the BDP's that are usuaUy seen upon proteolysis appear to be degraded int smaU fragments not recognized in the western assay either directly or through the SBDP' more rapidly than is observed in vivo. We also added either Z-Leu-Phe-CONHCH2CH or Z-Leu-Nva-CONH(CH2)3 moφholine to the samples exposed to glutamate, an included in the western assay. The loss of spectrin immunoreactivity was prevented by th addition of either calpain inhibitor.
Our results show that calpain inhibitors can rescue PC12 ceUs from glutamat toxicity. Thus, inhibition of calpain represents an exciting new approach to th amelioration of ischemic and excitotoxic damage in stroke and other neurodegenerativ processes.
6. Reduction of Infarction upon MCA Occlusion
Stroke is a significant health problem in the human population. Strokes ar occlusions of cerebral arteries producing a decreased blood flow to brain regions, whic cause ceU death through oxygen and nutrient deprivation. This type of lesion can b modeled in rats by surgical occlusion of the middle cerebral artery (MCA). Several model for MCA occlusion have been developed, and aU give substantiaUy simUar results.
MCA occlusion produces a large volume of infarcted brain tissue 24 hours after occlusion. Previous studies have shown that the size of the infarct as judged by TTC staining does not increase after the first 24 hours post-occlusion. Thus, we used an MCA occlusion model in order to test the abUity of Calpain inhibitors to prevent neurodegeneration. This model is described in Example 6.
EXAMPLE 6 MCA Occlusion Model for Neurodegeneration Male Sprague-Dawley albino rats weighing approximately 250-300 grams were anesthetized with pentobarbital (70 mg/kg, i.p.). The neck region was shaven and a 2 cm incision was made. The superficial fascia was teased away with tissue forceps and blunt tip tissue scissors using a spread method. The right common carotid artery was isolated away from the vagus nerve and tied off with a single suture (3.0 sUk). The external carotid was permanently occluded by suturing. The bifurcation of the internal carotid and pterygopalatine arteries was exposed and a single microaneurysm clip was placed on the pterygopalatine. Another microaneurysm clip was placed on the common carotid just proximal to the external/internal bifurcation. A suture was placed loosely around the common carotid and a lumen was made in the vessel with the tip of a 25g needle. A 40 mm nylon suture was prepared by melting the tip to smooth the pointed end and marked with a dot exactly 17.5 mm from the melted end. The suture was inserted into the lumen of the artery as far as the vessel clip, the clip is removed and the suture advanced untU the marking was at the bifurcation of the internal and external carotid arteries. This places the end of the suture in the circle of WiUis just beyond the source of the middle cerebral artery and occludes this artery. The loose suture around the carotid is tied lightly to keep the nylon suture in place. The microaneurysm clip on the pterygopalatine artery was removed, the incision is closed and the animals are aUowed to recover in heated recovery cages.
Twenty-four hours after occlusion, the brains of these animals were removed and sUced into 2mm sections. The sections were stained using 2,3,5-triphenyltetrazolium chloride as in Lundy, et al., /. Pharmacol. Meth., 16:201-214 (1986). Absence of red color development indicated tissue damage or death. The sizes of the infarcted tissue zone
(area with red stain) and impaired zone (area with partial development of red color) were evaluated using quantitative moφhometry.
Drugs or vehicle were administered by infusion into the femoral vein. AU animals received the same volume of drug or vehicle (20% dimethyl sulfoxide/80% propylene glycol) via a catheter attached to an Alzet osmotic minipump (24 hr pump, 8 μl/hr, 90 ul total volume). The model of Example 6 was used to determine the size of infarcted area for control (vehicle, i.v.) and with administration of each of two Calpain inhibitors: Z-Leu- Phe-CONH-Et and Z-Leu-Abu-CONH-Et. These results are depicted graphicaUy in Figure 5. It can be seen that administration of either of the Calpain inhibitors Z-Leu-Phe- CONH-Et or Z-Leu-Abu-CONH-Et produces a reduction in the size of the infarcted area. 7. Inhibition of Anoxic and Hypoxic Damage
The CA1 region of hippocampus is a brain area particularly vulnerable to ischemic damage and other insults involving excitatory amino acids. The hippocampus is also a major focus of ceU degeneration in Alzheimer's disease. Neural ceUs in shces in vitro degenerate foUowing hypoxia through the same chain of events (including reperfusion effects) observed in vivo during and after ischemia. We believe that studies of degeneration of neural slices in the presence of the various Calpain Inhibitors is an effective indicator of the membrane permeance of the Calpain Inhibitors. Accordingly, we believe that these studies provide a model for the treatment and inhibition of neurodegeneration in vivo. SimUar studies for determining the efficacy of compounds useful in the treatment of neurodegeneration in accordance with the present invention can be performed using other models, such as protection against degeneration in platelets or ceUs in culture.
It is believed that hypoxia is a major cause of neurotoxicity in a variety of neurodegenerative diseases and conditions, such as stroke and head injury. Thus, we conducted further studies using hippocampal slices to show that the various Calpain inhibitors, advantageously, can increase survival of hippocampal nerve ceUs during exposure to hypoxic or anoxic conditions. An initial screening procedure was first used to qualitatively determine whether the various Calpain Inhibitors can provide neuroprotection from anoxia in hippocampal slices. An example of these initial screening procedures is shown by Example 7A.
EXAMPLE 7A
Initial Screen for Inhibition of Anoxic Damage
Hippocampal slices (400 um) were prepared from Sprague Dawley rats (6 to 7 weeks) and maintained in an interface chamber at 35° C using conventional techniques, i.e., the lower surface of the slice received a constant perfusion (0.5 ml/min) of ACSF, whUe the upper surface was exposed to a moist atmosphere of 02:C02 (95%:5%) exchanged at a rate of 2 L/min. The ACSF medium contains (in mM): NaCl (124), KCl (3), KHP04
(2.5), CaCl2 (3.4), NaHCOs (26) and D-Glucose (10). Field excitatory post-synaptic responses were recorded from stratum radiatum of CAlb in response to stimulation of Schaffer-commissural fibers in CAla or CAlc. The depth of the recording electrode was optimized and evoked responses were coUected at a rate of one evoked response every 30 seconds.
For the initial screening procedure, 14 to 16 slices are harvested from the hippocampus of a single rat and placed in a common ACSF bath. Each slice is tested in sequence to determine the magnitude of its pre-anoxic evoked response. Five stimulation pulses (each 0.1 ms (mUlisecond) in duration) were presented over a 15 second interval.
The largest evoked response was noted and recorded for each slice.
FoUowing this, the slices were incubated for one hour, with either drug or vehicle alone added to the ACSF. After the one hour drug incubation period, the oxygen-enriched atmosphere of the chamber was made anoxic by substituting nitrogen for oxygen (N2 =
95%; C02 = 5%). The slices were retained in this anoxic environment for 10 minutes, foUowing which the oxygen-enriched atmosphere (02 = 95%; C02 = 5%) was reestabUshed.
The slices were given the opportunity to recover for 30 minutes foUowing reoxygenation whereupon each was stimulated and the maximum evoked potential determined, as described above during the pre-anoxia period. Those slices which, after anoxia, produced a maximum evoked potential of greater than 50% of that observed prior to anoxia were defined as surviving slices.
Results of the studies of Example 7A are shown in Figure 6. Figure 6 shows the effects of Z-Leu-Abu-C02Et, a Peptide Keto-Compound, and Cl 1 relative to control slices on survival of hippocampal slices exposed to 10 minutes exposure of anoxic atmosphere.
As seen in this figure, when the control slices are deprived of oxygen for 10 minutes in th
absence of drug, virtuaUy aU faU to survive, as measured by their abUity to elicit 50% of their pre-anoxia evoked response. In accordance with this finding, few if any recover upon reoxygenation. Figure 6 also shows that when CIl or Z-Leu-Abu-C02Et are added to the ACSF, the slices are protected from the effects of anoxia, evidenced by a substantial proportion of slices eliciting evoked potentials.
FinaUy, it can be seen that Z-Leu-Abu-C02Et is significantly more effective in protecting against anoxia and preventing degradation of slices at the minimal 1 hour incubation time, and at lower concentrations than CIl. This effect is believed to be due to the superior membrane permeance of the Peptide Keto-Compounds.
Table 7A shows further data from the studies of Example 7A.
TABLE 7A PERCENT OF SLICES SURVIVING TEN MINUTES ANOXIA
It can be seen from the data in Table 7A that aU of the Calpain Inhibitors tested provide increased survival. SHC, a Substituted Heterocyclic Compound is ACITIC; HKP, a Halo-Ketone Peptide, is Boc-Gly-Leu-Phe-CH2Cl; and PKC, a Peptide Keto-Compound, is Z-Leu-Abu-C02Et. AU are shown to be highly effective in influencing survival times. Leupeptin is seen to be the least effective neuroprotectant. Thus, we believe that ACITIC, Boc-Gly-Leu-Phe-CH2Cl and Z-Leu-Abu-C02Et are more effective in influencing survival because of their membrane permeabUity. Accordingly, the results shown in Table 7A support our beUef that Calpain Inhibitors with membrane permeabUity are effective neuroprotectants.
To further elucidate the abUity of Calpain Inhibitors to provide neuroprotection to hippocampal slices, and to provide a more quantitative indication of the membrane permeability of these Calpain Inhibitors, we measured the effect of various Calpain
Inhibitors on the evoked response on a single neuronal slice before, during and after anoxia. These studies are shown in Example 7B.
EXAMPLE 7B Inhibition of Anoxic Damage As in Example 7A, hippocampal sUces (400 μm) were prepared from Sprague
Dawley rats (6-7 weeks) and maintained in an interface chamber at 35° C using conventional techniques, i.e. the lower surface of the slice received a constant perfusion (0.5 ml/min) of an artificial cerebrospinal fluid (ACSF), whUe the upper surface was exposed to a moist atmosphere of 02:C02 (95%:5%) exchanged at a rate of 2 L/min. The ACSF medium contains (in mM): NaCl (124), KCl (3), KHP04 (1.25), MgS04 (2.5), CaCl2
(3.4), NaHC03 (26) and D-Glucose (10). Field excitatory post-synaptic responses were recorded from stratum radiatum of CAlb in response to stimulation of Schaffer- commissural fibers in CAla or CAlc. The depth of the recording electrode was optimized and evoked responses were coUected at a rate of one evoked response every 30 seconds.
After establishing a stable baseline of evoked responses (approximately 10 minutes), ACSF containing Calpain Inhibitor was washed into the chamber and slices were incubated for a period of one hour. After incubation, evoked responses were again recorded and the change in the amplitude of the responses from baseline levels was noted. No effect of the inhibitors tested on baseline evoked responses was observed.
For anoxia experiments, incubation in the drug-containing medium was foUowed by replacement of the 02:C02 (95%:5%) atmosphere with N2:C02 (95%:5%). Slices were exposed to this anoxic environment untU disappearance of the pre-synaptic fiber voUey and for two minutes (severe anoxia) longer (total time in anoxic environment approximately 7-8 minutes in control case). Effects of Calpain Inhibitors on the functional recovery of the sUces after the anoxic episode were then measured. Recovery of the evoked potential (EPSP) slope and amplitude by the drug treated slices can be compared to control slices to determine the relative efficacy of various Calpain Inhibitors.
Figure 7 shows the EPSP amplitude in millivolts for control, CIl treated and Z- Leu-Abu-C02Et (a Peptide Keto-Compound) treated hippocampal slices in the studies of
Example 7B. The periods of anoxia are represented by the black bars under the graph. It can be seen in Figure 7 that the control slices deprived of oxygen in the absence of drug
display a gradual reduction of EPSP and abruptly lose fiber voUey activity about 5-6 minutes after the beginning of anoxia. Reoxygenation at or before this point leads to complete functional recovery after about 20 minutes of reoxygenation, but reoxygenation after this point does not. In the latter case the recovered EPSP slope and amplitude become progressively reduced as the duration of anoxia post-fiber voUey disappearance
(post-FVD) increases. After severe anoxia (2 minutes post-FVD), slices recover only 15% of the EPSP slope.
In contrast to the control shces, recovery begins to occur shortly after the end of anoxia for the treated shces. Figure 7 shows a comparison of the effects on EPSP amplitude produced in the presence of no inhibitor; the Peptide Keto-Compound, Z-Leu-
Abu-C02Et and CIl. Z-Leu-Abu-C02Et produces a recovery from severe anoxia superior to that seen with CIl.
Figure 8 shows the percent recovery of EPSP from severe hypoxia using the peptide ketoester Z-Leu-Phe-C02Et and its corresponding peptide ketoamide Z-Leu-Phe- CONH-Et. These studies were performed in a manner simUar to that of Example 7B, except using a hypoxic environment in place of the anoxia of Example 7B. It can be seen that use of the peptide ketoamide results in essentiaUy complete (near 100%) recovery from hypoxia whUe the peptide ketoester produces a partial recovery. The control slices experienced little or no recovery. An interesting characteristic that we have discovered for certain Calpain Inhibitors is their abUity to lengthen the period of exposure to anoxia required to produce fiber voUey disappearance (FVD). TypicaUy, under control anoxia conditions, fiber voUey disappearance occurs in less than six minutes (Figure 9). The Peptide Keto-Compound, Z-Leu-Phe-C02Et, substantiaUy lengthens the period of exposure to anoxia required to produce FVD. This is an important advantage of the use of this Peptide Keto-Compound for neuroprotection because slices can be expected to recover completely if reoxygenated before fiber voUey disappearance. Thus, treatment with this Peptide Keto-Compound is expected to produce a greater percentage of recovery of ceUs from incipient neurodegenerative conditions. It is believed that other representatives of the Peptide Keto-Compounds as weU as of other classes of Calpain Inhibitors also provide this effect.
Table 7B shows the perecentage of recovery of pre-anoxia synaptic transmission (evoked potential amplitude) of slices treated with various Calpain Inhibitors or of control
shces. AU of these slices were exposed to ten minutes of anoxia according to the protocol of Example 7B.
TABLE 7B PERCENT RECOVERY OF SYNAPTIC TRANSMISSION AFTER ANOXIA
The results shown in Table 7B provide further evidence that the peptide aldehyde, CIl, as weU as the Substituted Heterocyclic Compounds (SHC) represented by ACITIC, Halo-Ketone Peptides (HKP) represented by Boc-Gly-Leu-Phe-CH2Cl, and Peptide Keto- Compounds (PKC) represented by Z-Leu-Phe-C02Et (PKC-1) and Z-Leu-Abu-C02Et
(PKC-2) are sufficiently membrane permeant to provide neuroprotection through Calpain inhibition.
CIl, which is at least partiaUy membrane permeant, produces some effect, however, does not significantly lengthen the period of anoxia required to suppress electrical activity. For example, see Figure 9. Thus, compared to control, or even compared to leupeptin and
CIl, the Substituted Heterocyclic Compounds, Peptide Keto-Compounds and Halo-Ketone Peptides can increase the degree of recovery after anoxic episodes whUe producing the additional advantage of extending the amount of time slices can tolerate anoxia and thereby recover completely. An important effect of the Peptide Keto-Compounds and other membrane permeant Calpain Inhibitors is that they are significantly more effective in lower doses than less permeable Calpain Inhibitors such as CIl. Although CIl is shown to be at least somewhat membrane permeant due to its abUity to affect slice survival, the more membrane-permeant inhibitors provide significantly increased protection. Thus, the more highly membrane-permeant Calpain Inhibitors are believed to be especiaUy effective in treating and inhibiting neurodegeneration.
The results of the studies of Examples 7A and 7B show that the Substituted Heterocy ic Compounds, Peptide Keto-Compounds and Halo-Ketone Peptides are membrane-permeant Calpain Inhibitors which are believed to be especiaUy effective in treating and inhibiting neurodegeneration. The results also show that Peptide Keto- Compounds, and perhaps representatives of other classes, can extend the duration of anoxia required to suppress electrical activity in hippocampal slices. As discussed above, these effects are important advantages of these compounds. 8. in vivo Neuroprotection by Calpain Inhibitors
As discussed above, therapeutics useful for influencing the function of ceUs within the CNS must cross the BBB to reach their targets within the CNS. Non-BBB permeant compounds might, in addition to the brain infusion techniques described above, be administered via intraventricular administration, but this also severely Umits their usefulness in practice. In order to test the in vivo effectiveness of the Calpain Inhibitors to cross the BBB and become therapeuticaUy useful, we tested the abUity of intraperitoneal injection of the Calpain Inhibitors to protect against excitotoxic damage in vivo. Protection was measured by evaluating changes in behavior of rats after injection with kainate. These studies are shown in Example 8A.
EXAMPLE 8A Protection Against Behavioral Changes from Excitotoxic Damage by Peripherally Administered Calpain Inhibitors
Rats (male Sprague-Dawley, 200±5 gms) were injected intraperitoneaUy with 12mg/kg kainic acid in saline vehicle and either 200μl DMSO (dimethylsulfoxide) or 4.6mg calpain inhibitor dissolved in the same volume of DMSO. The rats were observed for six hours foUowing the injections and the kainate-induced behavioral symptoms and convulsions scored on a scale of 0-6 (0 = no symptoms; l =wet dog shakes; 2=saUvation and chewing; 3 = at least one convulsive episode; 4 = repeated or sustained convulsions; 5 = convulsions, including rearing and faUing; 6 = convulsions foUowed by death within the 6 hrs post injection).
Figure 10 shows the effects of CIl on the behavioral and convulsive effects of kainic acid. In the control group, over half the animals showed symptoms greater than mUd behavioral symptoms, and many exhibited overt convulsions, presumably reflecting seizure activity within the brain. Unexpectedly, in the inhibitor treated group, the
-co¬ incidence and severity of convulsions was reduced. Thus, this data suggests that Calpain Inhibitors have an anti-convulsive effect. This effect is a distinct advantage in the use of Calpain Inhibitors in epUepsy-related neurodegenerative conditions and in stroke, which is often accompanied by seizures. In order to more clearly demonstrate that the behavioral and anti-convulsive effects seen with the Calpain Inhibitors result from inhibition of Calpain we tested the brain tissues of the rats from Example 8A for accumulation of spectrin BDP's. As discussed above, these BDP's are associated with Calpain activity and with the neurodegeneration associated therewith. EXAMPLE 8B
Protection Against Spectrin Breakdown from Excitotoxic Damage by Peripherally Administered Calpain Inhibitors Four days foUowing the injection of kainate in the rats from Example 8A, the brains of the rats were removed and assayed for spectrin BDP's. Spectrin BDP's were assayed by homogenizing brain parts in 20mM Tris pH = 7.2, .32M sucrose, 50μM Ac-
Leu-Leu-nLeu-H on ice. Homogenates were mixed 1:1 with 10% SDS, 5% β-mercaptoethanol, 10% glycerol, lOmM Tris pH = 8.0, 0.5% bromophenolblue, heated to 95° C, and subjected to electrophoresis in 4-1/2% polyacrylamide gels. The proteins in the gels were transferred to nitroceUulose and the spectrin and BDP's detected using a rabbit polyclonal anti-spectrin antibody and established immunodetection methods.
The amount of spectrin and BDP's in each sample was quantitated by densitometric scanning of the developed nitroceUulose.
Figure 11 shows the results of Example 8B. It can be seen that kainate stimulated the breakdown of spectrin in both Calpain Inhibitor treated and control rats. However, treated rats exhibited significantly less BDP's. These results verify that
Calpain activity in the brains of the treated rats was reduced. An unexpected observation was that even those treated animals that exhibited severe seizures had significantly less spectrin breakdown than untreated animals subjected to kainate. Thus Calpain Inhibitor treatment reduced both the behavioral/convulsive effects of kainate and the activation of calpain in the most severely affected animals.
9. Conclusion
AU of the foregoing studies support our discovery that Calpain Inhibitors provide in vivo protection against neurodegeneration associated with anoxia, excitotoxicity and other causes. Thus, these Calpain inhibitors possess neuroprotective activity against a variety of in vivo neurodegenerative diseases and conditions, including excitotoxicity, HIV-induced neuropathy, ischemia foUowing denervation or injury, subarachnoid hemorrhage, stroke, multiple infarction dementia, Alzheimer's Disease (AD), Huntington's Disease, Parkinson's Disease, surgery-related brain damage and other pathological conditions. Those Calpain Inhibitors which possess significant Calpain Inhibitory activity in vitro and also meet at least one of the foregoing or different tests for membrane permeabUity are exceUent candidates for treatment of neurodegeneration. G. TREATMENT OF NON-NEUROLOGICAL CONDITIONS
A number of medical conditions associated with increased Calpain activity can be treated with the Calpain Inhibitors of the present invention. These Inhibitors are administered to a mammal having a medical condition which is caused at least in part by the proteolytic activity of Calpain. Specific medical conditions are described below which benefit from the administration of Calpain Inhibitors. 1. Treatment of Cardiac Muscle Tissue Damage Damage to the cardiac muscle tissue of a mammal can be slowed or prevented by the administration of Calpain Inhibitors. To treat a mammal, such as a human patient, who has cardiac muscle tissue damage with Calpain Inhibitors, that patient is first identified by screening patients for those with symptoms of having cardiac tissue damage. Examples of such patients include those who have experienced heart attacks. Other groups likely to have experienced cardiac tissue damage include victims of violent assault whose thoracic cavities have received a physical insult, as weU as those who have suffered from viruses or other pathogenic agents known to attack the heart muscle. After identifying such people, routine tests, such as ultrasound and magnetic resonance imaging, are then used to determine whether or not they actuaUy have suffered cardiac muscle tissue damage.
Calpain Inhibitors can be administered to people with damaged myocardial tissue in a number of ways. The most direct method of administration is the injection
of up to a liter of a Calpain Inhibitor solution directly into the damaged tissue, where the Calpain Inhibitor is at a concentration in the range of between 0.001 mg/ml and 10 mg/ml, and preferably 0.01 mg/ml, of a Calpain Inhibitor. Any pharmaceuticaUy acceptable carrier vehicle may be used to carry the Calpain Inhibitor. This method of administration is normaUy used only when the myocardium is exposed and undergoing treatment, as during surgery. The direct infusion of a Calpain Inhibitor into the heart in the foregoing concentrations with a catheter, or the injection of such a Calpain Inhibitor into the pericardial space, in order to achieve the local administration of the Inhibitor is another way of treating the heart muscle when tissue damage is occurring. The intravenous or intramuscular administration of a Calpain Inhibitor is preferred, however, when a patient is not undergoing surgery or other invasive treatment. A Calpain Inhibitor in a pharmaceuticaUy acceptable solution is injected once daUy in a dosage of between approximately 0.001 mg/kg of bodyweight and 100 mg/kg, and preferably between 0.01 mg/kg and 10 mg/kg, to treat cardiac muscle tissue damage. More preferably, a Calpain Inhibitor is administered several times per day at appropriately reduced dosages. More preferably stiU, a Calpain Inhibitor is infused slowly into a patient by drip infusion, to ensure that the Calpain Inhibitor is present in the bloodstream in a relatively constant concentration. Of course, oral formulations of a Calpain Inhibitor can also be administered, and other methods of administration known to the art can be used as weU.
Calpain Inhibitors can also be used to treat a mammal at risk for suffering damage to that mammal's cardiac tissue. Thus, myocardial infarctions can be prevented or decreased in size through the administration of a Calpain Inhibitor to such a mammal. A mammal, such as a person, at risk for suffering damage to its myocardial tissue is identified by screening a population for people with symptoms that indicate a higher-than-average risk for suffering a heart attack, including shortness of breath, obesity, high blood pressure, and high levels of cholesterol in the blood. Preferably, people who have been diagnosed as having cardiac ischemia, who have experienced a mUd heart attack, or who have other symptoms which indicate that they are at risk for suffering a serious heart attack in the near future are identified. Those at risk for suffering cardiac tissue damage can be at least partiaUy protected from such damage by taking Calpain Inhibitors prophylacticaUy. The screening of a population
for people who would benefit from Calpain Inhibitor therapy is normaUy done by a physician in the course of examining that physician's patients, but might also be done through a health screening program sponsored by an employer or a school.
Once an individual who would benefit from the administration of a Calpain Inhibitor has been identified, an Inhibitor can be administered in a number of ways.
One method of administration is to inject an Inhibitor only once at a relatively large dosage in order to treat a person suffering from acute myocardial ischemia (a heart attack). In this case, between approximately 0.001 mg/kg and 100 mg/kg, and preferably between 0.01 mg/kg and 1 mg/kg, of an Inhibitor suspended or dissolved in an appropriate pharmaceutical carrier can be injected intravenously into such an individual. In this way, the acute damage to the myocardium suffered by heart attack victims can be avoided or reduced.
Inhibitors can also be injected several times over the course of a period of time, or they can be infused intravenously at a steady rate for a period of time in order to protect an individual at risk for suffering cardiac tissue damage. Individuals are administered between approximately 0.001 mg/kg and 100 mg/kg, and preferably between 0.01 mg/kg and 10 mg/kg, of a Calpain Inhibitor daUy. This route of administration is preferred for individuals who are at high risk for suffering a myocardial infarction in the near future, and such administration may be continued as long as such a risk of having a heart attack remains.
In addition, other methods of administering a Calpain Inhibitor are possible to protect an individual from myocardial tissue damage. The oral administration of a Calpain Inhibitor in tablet or liquid form is preferable for long term Calpain Inhibitor therapy, because such routes of administration are easier for an individual to administer to him or herself. Between approximately 0.001 mg/kg and 100 mg/kg, and preferably between 0.01 mg/kg and 10 mg/kg, of a Calpain Inhibitor is administered daUy in this form of Calpain Inhibitor therapy.
EXAMPLE 9 The effect of a Calpain Inhibitor is tested on rabbits by inducing a region of ischemic myocardium and administering a Calpain Inhibitor. Cardiac ischemia is induced in rabbit myocardium by ligating the coronary artery foUowed by ligation of the branches of the left circumflex artery adjacent to the ischemic (cyanotic) area when the
epicardial cyanotic area reaches 0.75 to 0.80 of the length of the long axis of the left ventricle (measured between the atrioventricular groove and the cardiac apex). This produces a coronary infarct of regular size. A Calpain Inhibitor is administered intravenously at 10 mg/kg/hour for two hours beginning either 5 minutes before or just after the ligations. In both cases, groups treated with a Calpain Inhibitor exhibit smaUer infarcts than those treated with a pharmaceutical carrier alone without a Calpain Inhibitor.
EXAMPLE 10 A human diagnosed as having suffered a mUd heart attack is prescribed an oral formulation of a Dipeptide α-Ketoamide (Subclass A) Calpain Inhibitor providing 2 mg/kg of the inhibitor. The formulation is taken once per day to protect the individual in the event he or she suffers a further heart attack.
EXAMPLE 11 A human who has been picked up by an emergency team within several minutes of suffering a heart attack is administered an injectable composition providing 10 mg/kg of a Dipeptide α-Ketoamide (Subclass B) Calpain Inhibitor. In this way permanent tissue damage to the myocardial tissue is avoided. 2. Treatment of Skeletal Muscle Tissue Damage
Damage to muscle tissues can also be prevented or reduced through the administration of Calpain Inhibitors. Research has shown that Calpain is involved in the degeneration of muscle tissues. During the autolysis of muscle fibers, for example, Calpain degrades the Z-disc of skeletal muscle myofibrUs. Calpain also degrades myosin, a muscle protein, over a wide range of protease concentrations. Since Calpain is activated by elevated intraceUular levels of calcium, any rise in such levels can lead to damage to a muscle ceU. Any condition in which intraceUular calcium levels become elevated, therefore, can be treated with a Calpain Inhibitor in order to prevent or limit the damage to skeletal muscle tissue due to the activity of Calpain.
Calpain Inhibitors can be used to treat skeletal muscle tissue damage caused by a variety of factors. A physical insult to skeletal muscle tissue that damages the muscle ceU membrane and results in increased intraceUular levels of calcium, for example, can be treated with a Calpain Inhibitor, which blocks the proteolysis of muscle ceU
constituents. Muscular dystrophy, a condition characterized by elevated intraceUular calcium levels, can also be treated with Calpain Inhibitors.
Identifying an individual with skeletal muscle tissue damage is normaUy done by a physician. A physician, for example, can identify an individual with the clinical symptoms of muscular dystrophy. Other damage to skeletal muscle tissue can likewise be diagnosed.
As with the treatment of myocardial tissue, the route of administration of Calpain Inhibitors to skeletal muscle tissue wiU vary, depending on the site of the damaged tissue. An injury to the bicep is most directly treated through an intramuscular injection of a Calpain Inhibitor into the bicep muscle of the arm, the site of injury. A more systemic condition, such as muscular dystrophy, however, is better treated through a systemic injection of a Calpain Inhibitor. Such an injection can be intramuscular, intraperitoneal, or intravenous. Alternatively, the oral administration of Calpain Inhibitors can effect systemic administration of such Inhibitors. EXAMPLE 12
An individual is diagnosed with Duchenne's muscular dystrophy by a physician. To treat that condition, between approximately 0.1 mg/kg and 10 mg/kg of a Calpain Inhibitor in a phosphate-buffered saline solution is injected intravenously into the individual once per day for the course of the treatment. 3. Treatment of Smooth Muscle Injury
Calpain is involved as weU in the breakdown of smooth muscle ceUs. It has been reported that certain smooth muscle ceU proteins, such as calponin, are degraded by Calpain. As indicated by studies of the proteolytic proclivity of Calpain in other tissues, Calpain poses a threat to smooth muscle ceUs whenever those ceUs experience elevated intraceUular calcium levels. In particular, mammals which have experienced physical damage to their smooth muscle tissue or whose blood circulation to their smooth muscle tissue has been cut off would benefit from the administration of Calpain Inhibitors. Prophylactic administration of such Inhibitors reduces the damage done by such conditions. Administration of Calpain Inhibitors after smooth muscle tissue has been damaged, however, is also beneficial since such Inhibitors can slow or stop the progress of the proteolytic activity of Calpain in such tissues.
The identification of an individual with smooth muscle tissue damage, such as an intestinal ulcer, is normaUy done by a physician. After diagnosing an individual as having damage to her smooth muscle tissue, the physician can give a Calpain Inhibitor to that individual to slow further damage to such smooth muscle tissue. The administration of a Calpain Inhibitor to smooth muscle tissue can be accomplished by any method known to the art. WhUe intramuscular injection is a possible route of delivery, in most cases intravenous, intraperitoneal, or oral delivery wUl be preferred, depending on the location of the damaged tissue. For example, someone with an ulcer is more effectively treated with an oral delivery of a Calpain Inhibitor, since the Inhibitor can then coat the affected area directly in the course of passing through the digestive system. Intravenous delivery can also be used, however, and would be preferable if the Inhibitor is itself upsetting to the digestive system of the individual or if the low pH of the stomach interferes with the effectiveness of the Inhibitor. Between approximately 0.001 mg/kg and 100 mg/kg, and preferably between 0.01 mg/kg and 10 mg/kg is used to treat smooth muscle tissue damage.
EXAMPLE 13 An individual is diagnosed as having an ulcer by her physician, and is prescribed a Calpain Inhibitor along with other ulcer medications. Approximately 2 mg/kg of the Calpain Inhibitor is administered oraUy in tablet form to the individual with every meal as long as the individual is experiencing the ulcer.
4. Treatment of Smooth Muscle Contraction
The tonic contraction of smooth muscle in appropriate circumstances is a normal process. However, in inappropriate circumstances, tonic contraction can lead to serious pathological conditions. For example, contraction of the bronchial smooth muscle leads to the shortness of breath and other symptoms of asthma. Contraction of the coronary arteries can lead to angina, partial coronary hypoxia and subsequent loss of coronary function. Contraction of the smooth muscle in cerebral arteries can lead to cerebral vasospasm and hypoxia of the brain tissue, a serious condition that can leave patients mentaUy disabled and permanently brain damaged. Angina generaUy results from both an occlusive and a spastic component. In some patients, angina is largely a result of either the occlusive or spastic component.
Advantageously, the Calpain Inhibitors of the present invention can be used to treat both components of angina.
Vasospasm is a condition which affects smooth muscles, particularly blood vessels. Vasospasm is a sustained spastic or tonic contraction of the vascular tissue. Such contractions are associated with a rise in intraceUular calcium levels. Calpain activity has been linked to these contractions, and contracted blood vessels can be dUated with Calpain Inhibitors.
An individual experiencing vasospasm is normaUy identified by a physician. A physician might, for example, detect vasospasm during surgery, or might detect vasospasm indirectly through the observation of external symptoms. Vasospasm is also often detected by angiogram.
Vasospasms frequently occur as a result of subarachnoid hemorrhage, which causes blood clots. Such blood clots are believed to provide factors that promote vasospasm. Such hemorrhaging is therefore an indication that an individual is at risk for vasospasm.
It is one of the suφrising discoveries of the present invention that Calpain Inhibitors can be used to block the establishment of the tonic state, or to relax tonicaUy contracted smooth muscle. We have found Calpain Inhibitors of several classes to be particularly useful in this regard, including the Substituted Heterocyclic Compounds, Peptide Keto-Compounds, Peptide Aldehydes, Halo-Ketone Peptides. Various subclasses of compounds within these broad classes that we have found to be useful in the inhibition of tonic smooth muscle contraction include the peptide ketoamides, chloromethyl ketone peptides, epoxysuccinates, diazomethane peptides and peptide aldehydes. The Calpain Inhibitors of the present invention provide a number of advantages in the treatment of smooth muscle contraction. These inhibitors provide unexpectedly high efficacy in the treatment of smooth muscle contraction, such as in the treatment of vasospasm disorders of many types. Furthermore, we have shown that Calpain Inhibitors of the present invention are additionaUy beneficial because they have relatively little effect on resting tension in smooth muscle. Thus, we consider these
Calpain Inhibitors to be particularly useful in the treatment of human subjects without adverse side effects.
The administration of Calpain Inhibitors to prevent smooth muscle tonic contraction or to relax tonicaUy contracted smooth muscle can be by any of a number of methods known to those of skiU in the art. Such methods include the systemic deUvery of Calpain Inhibitors through intravenous, intraperitoneal, or intramuscular injection, or through oral deUvery, as described hereinbelow. Administration of the
Calpain Inhibitor for this puφose can also be through use of a catheter system such as wiU be readUy known to those having skiU in the art. A solution of Calpain Inhibitor can be injected directly into the cerebrospinal fluid. Aerosolization of solutions containing a Calpain Inhibitor is a preferred mode of administration. Finely dispersed dry powders can also be used successfuUy. Other known methods of delivery are also acceptable. Thus, formulations including Calpain Inhibitors can be of many forms, including tablets, troches, solutions, powders and the like, as described herein.
In order to induce dUation of a spastic or tonicaUy contracted blood vessel and thereby reverse vasospasm, a Calpain Inhibitor can be administered by direct topical application to the blood vessel. This method, of course, necessitates the exposure of the blood vessel so that it can be physicaUy manipulated, and thus requires surgery. A Calpain Inhibitor at a concentration of approximately 1 - 500 μM can be topicaUy applied to achieve vasodUation.
However, when it is desired to treat vasospasm without surgery, a Calpain Inhibitor can be administered intravenously. In this event, between approximately
0.001 mg/kg and 100 mg/kg, and preferably between 0.01 mg/kg and 10 mg/kg, of a Calpain Inhibitor suspended or dissolved in a pharmaceuticaUy acceptable carrier is administered once to an individual. The oral administration of a Calpain Inhibitor in like amounts is also possible, although this route of administration would not be as fast- acting as intravenous administration.
For inhibition of other tonic smooth muscle contractions, between approximately 0.001 mg/kg body weight and 100 mg/kg body weight of a Calpain Inhibitor can be administered daUy, divided into one to eight doses, or via continuous infusion intravenously. More preferably, the daUy dosage to an individual to prevent or relax tonic smooth muscle contraction would provide between 0.01 mg/kg to 10 mg/kg body weight. Optimum dosages can be determined for each particular Calpain Inhibitor using techniques known to those having ordinary skill in the art.
Higher concentrations of Calpain Inhibitors can be administered through the direct application of such Inhibitors to the smooth muscle tissue. For example, the Calpain Inhibitor can be loaded into a microsphere and the microsphere driven into the smooth muscle to effect direct application. For the relaxation of spastic arteries, the artery can be surgicaUy exposed and a solution of Calpain Inhibitor appUed directly to the artery. Calpain Inhibitors can also be delivered through the use of a slow- release compound, such as a gel or ointment, applied directly to the smooth muscle tissue during surgery. Local administration strategies include inhalation formulations for use in treating bronchospasm. In the foUowing examples, certain specific Calpain Inhibitor compounds were tested in order to verify the results of the present invention. These drugs are specificaUy identified hereinabove in the Brief Description of the Figures, upon reference to the figures referred to in the Examples. Drugs A, B, F (the same drug as B), G, H, J and CX are aU Peptide Keto-Compounds that are inhibitors of Calpain. Drug H is another inhibitor of Calpain. Drugs C, E and I are not inhibitors. Drug D is a relatively poor inhibitor that is a compound of the Halo-Ketone Peptide class.
EXAMPLE 14 Isolated arteries in vitro were treated with 10"8M endothelin (ET) to induce contraction of the smooth muscle. The arteries were then treated with Calpain Inhibitor at a concentration of between 10"7M and lO^M. The results of this procedure are shown in Figures 12 and 13 for Calpain Inhibitors A-J, as described hereinabove in the Brief Description of the Figures. These Figures show that administration of a Calpain Inhibitor can effectively reduce endothelin-induced contraction of isolated arteries in vitro. EXAMPLE 15
Isolated arteries in vitro were treated with 10"7M phorbol dibutyrate (PDB) to induce contraction of the smooth muscle. The arteries were then treated with compounds E-J (as described hereinabove in the Brief Description of the Figures) at a concentration of between 10'6M and lO^M. The results of this procedure are shown in Figure 14. This Example demonstrates that administration of a Calpain Inhibitor in accordance with the present invention can effectively reduce PDB-induced contraction of isolated arteries in vitro.
EXAMPLE 16
Isolated arteries in vitro were treated with Calpain Inhibitors E-J (as described hereinabove in the Brief Description of the Figures) at concentrations of 3x10 M, 10"5M, and 3xlO"5M. The tension in the arteries, in mg, was then measured. The results are shown in Figure 15. This example verifies that Calpain Inhibitors of the present invention have relatively little effect on resting tension in smooth muscle.
EXAMPLE 17 The protective effect of Z-Leu-Phe-CONH(CH2)3 on the acute phase of vasospasm was evaluated in live rabbits. Arterial contraction measured for approximately 10 minutes in order to establish a baseline. Z-Leu-Phe-CONH(CH2)3 at concentrations ranging from 100 μM to 300 μM, or vehicle was then administered to the animals. At a time approximately 60 minutes after establishment of a baseline, oxyhemoglobin was administered in order to induce constriction. Figure 16 shows that higher concentrations of Calpain Inhibitor resulted in less constriction, with virtuaUy no constriction occurring in the animal receiving the Calpain Inhibitor at 300 μM.
Constriction was reversed by administration with aCSF.
EXAMPLE 18 Figure 17 provides an example of the effect of a Calpain Inhibitor of the present invention on an artery in a live animal that was constricted by subarachnoid hemorrhage (SAH). The "normal" resting size of the vessel, as estimated from an age- matched control animal, was approximately 750 μm. The vessel was constricted to approximately 400 μm foUowing hemorrhage. Perfusion of aCSF alone for 90 minutes had no effect. Z-Leu-Phe-CONH(CH2)3 at 100 μM reversed the SAH-induced constriction by approximmately 60% to about 600 μm. EXAMPLE 19
Three rabbits were subjected to SAH resulting in constriction of cerebral arteries. Z-Leu-Phe-CONH(CH2)3 was then administered to produce a concentration of lOOμM and the amount of relaxation measured. Figure 18 shows the summary of data from aU three animals. In aU three, Z-Leu-Phe-CONH(CH2)3 reversed constrictions induced by SAH. The constricted diameter is taken as the "100% value" in this graph (unlike in Figure 16) and the relaxation is expressed as a percentage of
the 100 % value. The data were analyzed in this fashion because the absolute amount of consstriction foUowing SAH varies from animal to animal.
EXAMPLE 20 A human diagnosed as suffering from tonic smooth muscle contraction is intramuscularly administered approximately 10 mg of a Calpain Inhibitor, such as Z-
Leu-Phe-CONH(CH2)2Ph, in a phosphate buffered saline solution by intravenous injection at least once per day for approximately one week or untU it is determined that the risk of tonic contraction has subsided.
EXAMPLE 21 A human diagnosed as suffering from angina associated with coronary artery vasospasm is oraUy administered approximately 100 mg of Z-Leu-Phe-CONHEt. The Calpain Inhibitor is delivered by surgicaUy exposing the artery and applying a solution of Calpain Inhibitor in a phosphate buffered saline solution directly to the artery.
EXAMPLE 22 A human diagnosed as suffering from asthma associated with bronchospasm is administered between approximately 100 mg of (Ph)2CHCO-Leu-Abu-CONH- CH2CH(OH)Ph by inhalation. The Calpain Inhibitor is delivered by inhalation of a formulation containing the Calpain Inhibitor directly into the patient's lungs.
EXAMPLE 23 A human diagnosed a suffering from cerebral vasospasm is administered approximately 100 mg of Z-Leu-Phe-CONHEt into the CSF. The Calpain Inhibitor is delivered by injecting said Calpain Inhibitor in a phosphate buffered saline solution directly into the patient's cerebrospinal fluid.
EXAMPLE 24 During surgery, a blood vessel is discovered to be experiencing tonic smooth muscle contraction. A solution containing a lOOμM solution of a Calpain Inhibitor is topicaUy applied to the contracted blood vessel. If one application faUs to produce fuU dUation of the blood vessel, further topical applications are performed.
EXAMPLE 25 Cerebral vasospasm in an individual is detected, and a solution containing approximately 2 mg/kg of a Calpain Inhibitor is dissolved in phosphate buffered saline and then injected intravenously into the individual.
EXAMPLE 26
We tested the ability of several Calpain Inhibitors to dUate ex vivo blood vessels treated to induce vasospasm. We found that Calpain Inhibitor- 1 provided low activity in inducing vasodUation, whUe at least three Peptide Ketoamide Calpain Inhibitors within the scope of the present invention provided appreciably greater activity. We found that highly HpophUic compounds were particularly effective. Thus, we found that each of Z-Leu-Phe-CONH(CH2)2C6H5, Z-Leu-Phe-CONHEt, and Z-Leu-Abu- CONHEt were more effective than Calpain Inhibitor- 1; however these compounds are Hsted in decreasing order of effectiveness. 5. Treatment of Hypertension-Related Injury
The activity of calpastatin, a natural inhibitor of Calpain, is significantly reduced in the erythrocytes of individuals who have hypertension. The activity of Calpain in such ceUs concomitantly increased, and both functional and structural lesions have been observed in the erythrocytes of hypertensive mammals. Calpain Inhibitors can therefore be administered to a hypertensive mammal in order to counteract the harmful effects of hypertension on such a mammal's erythrocytes caused by the increased proteolytic activity of Calpain in such ceUs.
An individual with hypertension is diagnosed by a physician, and in conjunction with other therapies to lower that individual's blood pressure a Calpain Inhibitor is administered to that individual. Approximately 1 - 10 mg/kg of a Calpain Inhibitor is administered to the individual daUy, preferably in an oral formulation for ease of administration.
EXAMPLE 27 An individual is diagnosed as having hypertension. Approximately 2 mg/kg of a Calpain Inhibitor is administered daUy in tablet form to that individual untU the individual's blood pressure returns to a normal range. 6. Treatment of Cataracts
Calpain has been implicated in the causation of cataracts. In a murine model of cataractogenesis, Calpain was found to have a very high activity in the lenses of mice just before visible cataracts appeared. The activity of Calpain then decreased as cataracts formed on the lenses of such mice. Other indicators of Calpain activity include the fact that the concentration of calcium increases markedly in the lenses of
such mice as cataracts begin to develop. Proteins simUar to the in vitro reaction products of the degradation of the lens protein crystaUin by Calpain are also found in the lenses of such mice.
The formation of cataracts is linked to a number of external and internal factors, including age, diabetes, hereditary diseases, UV radiation, drugs such as steroids, and toxic chemicals. Calpain Inhibitors can protect people exposed to agents known to cause cataracts, or who show signs of developing cataracts, through the administration of a Calpain Inhibitor to such people before the development of a cataract. Calpain Inhibitors can also be administered to people who have not developed fuUy mature cataracts, in order to halt or slow the progress of cataract development. Such treatments can potentiaUy save mUlions of doUars in medical care costs. In one study, it was estimated that if cataract development in individuals could be slowed for 10 years or more, over $600 miUion (in 1973 doUars) could be saved in annual medical care costs in the U.S. alone.
A person having a cataract who would benefit from treatment with a Calpain Inhibitor is easUy identified by a physician trained in diagnosing cataracts. Such a person is then treated through the administration of Calpain Inhibitors to slow or halt the progress of cataract formation. Individuals who would benefit from protection from cataract formation using
Calpain Inhibitors can also be identified by a physician. For example, patients whose families have a history of developing cataracts are candidates for treatment with Calpain Inhibitors. Preventative therapy with Calpain Inhibitors is also indicated for people with diabetes or who are regularly exposed to agents, such as steroids, which can cause cataracts.
The administration of Calpain Inhibitors for the treatment or prevention of cataracts can be by any of a number of methods. Cataracts can be treated by the systemic delivery of Calpain Inhibitors through intravenous, intraperitoneal, or intramuscular injection or through oral delivery. Between approximately 0.001 mg/kg and 100 mg/kg of a Calpain Inhibitor is administered daUy to an individual to prevent or slow cataract development.
Higher concentrations of Calpain Inhibitors can be delivered, however, through the direct application of such Inhibitors to the eyes. Direct application can be done either through the injection of Calpain Inhibitors into the eye, or through the topical application of Calpain Inhibitors which have been suspended in or mixed with eye drops, ointments, or solutions which are then placed on the eye. Calpain Inhibitor solutions can also be soaked into contact lenses which then deliver a Calpain Inhibitor to the eye slowly over time. Between approximately 0.00001 mg/kg and 1 mg/kg, and preferably between 0.0001 mg/kg and 0.1 mg/kg, are administered daUy in such direct applications. EXAMPLE 28
An elderly individual with partiaUy developed cataracts is treated with eye drops containing Calpain Inhibitors. One drop in each eye is administered twice daUy. The eye drop solution is formulated so that each drop contains between 0.001 mg/ml and 1 mg/ ml of a Calpain Inhibitor. EXAMPLE 29
An individual whose famUy has a history of cataract development is administered a Calpain Inhibitor in tablet form to be taken once da y. Each tablet contains approximately 2 mg of a Calpain Inhibitor approximately per kUogram of bodyweight. 7. Treatment of Restenosis
When blood vessels become blocked by arterial plaques and fatty deposits, therapeutic angioplasty can be used to open such stenotic regions. One of the most commonly used angioplasty procedures, coronary baUoon angioplasty, makes use of a catheter which has an inflatable baUoon at its distal end. In this procedure, the catheter is inserted into the arterial lumen, and the distal end of the catheter is guided to the stenotic region. Once positioned within the stenotic area, the baUoon is expanded in order to flatten the arterial plaque against the waU of the vessel. Other types of mechanical procedures for opening stenoses within the vasculature have also been used. These include the use of lasers or atherectomy devices to remove occlusions. SimUar procedures for mechanicaUy opening stenoses are also performed in heart valves (valvuloplasty) and peripheral vessels (peripheral angioplasty).
Smooth muscle ceU prohferation of the vascular waU is a normal response to various physiological stimuU, including those associated with procedures for mechanicaUy opening stenoses. Such stimuli can trigger the proliferation of smooth muscle ceUs and the migration of these ceUs to the vascular subintima, where they continue to proliferate. Smooth muscle ceU proliferation is normaUy a desirable process, one in fact that is often necessary for healing, but which is not necessary for recovery from angioplasty.
However, foUowing therapeutic angioplasty for the opening of obstructed arteries, the proliferation of smooth muscle ceUs can result in restenosis and blockage of the opened artery. As many as 50% of the patients who undergo successful coronary angioplasty can develop recurrent coronary artery obstructions foUowing the procedure due to such restenosis. It would therefore be of great medical value to be able to inhibit smooth muscle ceU proliferation in order to prevent restenosis foUowing angioplasty. It is one of the suφrising discoveries of the present invention that Calpain
Inhibitors can be used to inhibit smooth muscle ceU proliferation and thereby prevent restenosis foUowing angioplasty. A class of Calpain Inhibitors which are particularly useful in this application are the Peptide Keto-Compounds. These inhibitors are potent inhibitors of calpain. For example, Z-Leu-Phe-CONHEt inhibits the proliferation of cultured bovine smooth muscle ceUs with an IC50 of around 150 μM. Ph2CHCO-Leu-
Abu-CONH-CH2CH(OH)Ph has been found to inhibit the proliferation of cultured bovine smooth muscle ceUs with an IC50 of around 60 μM. Other Calpain Inhibitors which are effective in preventing restenosis can be determined by routine experimentation, such as through thymidine incorporation into cultured bovine aortic smooth muscle ceUs. Additional Calpain Inhibitors believed to be particularly effective in this regard are the Halo-Ketone Peptides, and the Substituted heterocyclic Compounds.
The administration of Calpain Inhibitors to inhibit smooth muscle ceU proliferation and to prevent restenosis can be by any of a number of methods known to those of skiU in the art. Such methods include the systemic delivery of Calpain
Inhibitors through intravenous, intraperitoneal, or intramuscular injection, or through oral delivery. Formulations used for injection may also contain elements such as
ethanol, ethoxylated castor oU, and N,N'-dimethyl acetamide, which serve to solubUize the hydrophobic Calpain Inhibitors. Other art-known methods of delivery are also, of course, possible. Between approximately 0.001 mg/kg and 100 mg/kg of a Calpain Inhibitor can be administered daUy to an individual to inhibit smooth muscle ceU proliferation and to prevent restenosis.
Higher concentrations of Calpain Inhibitors can also be administered through the direct application of such Inhibitors to the vascular tissue. For example, during a coronary baUoon angioplasty procedure, a baUoon catheter can be inserted into the desired blood vessel and Calpain Inhibitors can be delivered through the catheter. One such method of appUcation involves passing a baUoon through the lumen of a vessel to the site of a vascular lesion or stenosis, after which the baUoon is inflated in order to flatten plaque against the waU of the vessel. In this method, a Calpain Inhibitor is directly applied to the blood vessel through the angioplasty baUoon. In another method, the angioplasty baUoon or another tool used during the angioplasty procedure is coated with the Calpain Inhibitor so that the Calpain Inhibitor is applied directly to the site of stenosis. Alternatively, the Calpain Inhibitor can be loaded into a microsphere and the microsphere can be driven into the injured tissue to effect direct appUcation. A Calpain Inhibitor can also be delivered through the use of a slow- release compound, such as a gel or ointment, applied directly to the injured tissue during surgery.
EXAMPLE 30 Two cultures of bovine aortic smooth muscle ceUs at low density were serum starved. One ceU culture was treated with about 10-100 μM Z-Leu-Phe-CONHEt, and the other culture was treated with about 10-100 μM Ph2CHCO-Leu-Abu-CONH- CH2CH(OH)Ph. Treatment with Calpain Inhibitor began 6 hours prior to stimulation with serum. The ceUs were then stimulated to divide by changing the old, serum free media to the same media containing 10% fetal bovine serum. The results of this procedure for two Calpain Inhibitor compounds are shown in Figure 19. This Figure shows that the peptide keto compounds Z-Leu-Phe-CONHEt and Ph2CHCO-Leu-Abu- CONH-CH2CH(OH)Ph effectively inhibit the proliferation of cultured bovine smooth muscle ceUs with an IC50 of around 150 μM and 60 μM, respectively. Results of this same testing using several different Calpain Inhibitors are shown in Table 30.
TABLE 30
Inhibition of BASMC Proliferation Structure-Activity Relationship structure IC50. μM
(Ph)2CHCO-Leu-Phe-CONH-CH2-2-Py 30 Z-Leu-Nva-CONH-CH2-2-Py 30
Z-Leu-Phe-CONH-CH2CH(OH)Ph 40
(Ph)2CHCO-Leu-Abu-CONH-CH2CH(OH)Ph 60
Z-Leu-Phe-CONH2 75
Z-Leu-Abu-CONH-CH2CH(OH)Ph 130 Z-Leu-Phe-CONHEt 150
Z-Leu-Abu-CONHEt >200
(Ph)2CHCO-Leu-Abu-COOEt no inhib
EXAMPLE 31
To ensure that the inhibition of ceU proliferation in Example 30 was not due to a toxic effect on the ceUs, the bovine aortic smooth muscle ceUs treated according to the procedure of Example 30 were further treated for one week with about 10-100 μM Ph2CHCO-Leu-Abu-CONH-CH2CH(OH)Ph. Figure 20 shows that treatment with Calpain Inhibitor did not cause significant ceU death despite a complete inhibition of proliferation. In addition, no increase in trypan blue permeabUity was seen after treatment for 1 week with the aforementioned Calpain Inhibitor, thus indicating the continued viabUity of the ceUs. Furthermore, the antiproliferative effect of the Calpain Inhibitor (Ph)2CHCO-Leu-Abu-CONH-CH2CH(OH)Ph was found to be rapidly reversed upon washout of the drug, another indication of the continued viabUity of the smooth muscle ceUs. EXAMPLE 32
A human diagnosed as undergoing restenosis is administered between approximately between 0.01 mg/kg and 100 mg/kg of a Calpain Inhibitor in a phosphate buffered saline solution by intravenous injection at least once per day for approximately one week or untU it is determined that the risk of restenosis has subsided.
EXAMPLE 33
A human who is determined to be at risk for developing restenosis, such as someone who has undergone angioplasty, is administered between about 0.01 mg/kg and 100 mg/kg of a Calpain Inhibitor in phosphate buffered saline by intravenous injection once per day for approximately one week or untU it is determined that the risk of restenosis has passed.
EXAMPLE 34 A human undergoing coronary baUoon angioplasty is administered a Calpain Inhibitor to prevent restenosis. The Calpain Inhibitor is delivered by means of an ointment containing between about 0.2 - 10% (2 g/kg to 100 g/kg) of a Calpain
Inhibitor. The ointment is coated on the surface of the baUoon used in the angioplasty procedure. The Calpain Inhibitor is thus delivered directly to the injured tissue when the baUoon is inflated during the procedure.
8. Synchronization of the CeU Cycle The ubiquitous distribution of calpain in mammalian ceUs, taken together with the fact that the cleavage of substrate proteins by calpain does not appear to be part of protein catabolism or general protein turnover, points to a regulatory role for calpain in ceUs. WhUe not wishing to be bound by any particular theory, we believe that a ceU's progression through the ceU cycle is dependent upon the calpain mediated cleavage of regulatory proteins. This theory has been deduced from our discovery that the inhibition of calpain in a ceU with the Calpain Inhibitors of the present invention prevents the progression of the ceU's reproductive cycle from the Gj phase to the S phase. Thus, we have found that Calpain Inhibitors can be used to synchronize the reproductive cycles of ceUs. An experiment which was conducted to show that Calpain Inhibitors block the passage of ceUs from G- to S phase is shown in Figure 21. By serum starving the ceUs used in this experiment, these ceUs were aU blocked at the G1 phase due to lack of sufficient nutrients to aUow passage into the S phase. In this experiment, serum was added to the ceUs in order to aUow them to pass into S phase, and the DNA content of the ceUs was analyzed with a flow cytometer. The ceUs were divided into several groups, and the Calpain Inhibitor Ph2-CHCO-Leu-Abu-CONH-CH2CH(OH)Ph was added to each of these groups at the various points in time foUowing serum addition
shown above the top row of squares (at 0, 12, 18, 21, 24„ 30, or 48 hours after the addition of serum to the ceUs). The peaks shown in each of the individual squares depict the amount of DNA in the ceUs of the analyzed group at the designated point in time, where the leftmost peak in each of these squares represents the amount of DNA in ceUs which are in the Gj phase. The peak to the right of this Gj peak represents the amount of DNA which has accumulated in ceUs which have passed through the S phase.
The numbers shown along the diagonal axis of the graph represent the time after the addition of serum that the Calpain Inhibitor Ph2-CHCO-Leu-Abu-CONH- CH2CH(OH)Ph was added to each of the samples of the ceUs. Thus, the leftmost square in each row represents the amount of DNA in ceUs which have not been exposed to a Calpain Inhibitor. As can be seen from the first row of squares, when Calpain Inhibitor was added at the time of serum addition, the ceUs thus treated remained in G- phase and did not progress through to the S phase (as shown by the absence of a second, right-shifted peak). However, if Calpain Inhibitor is added after some ceUs have already begun passing into S phase, such as at 18 or 21 hours after serum addition, then these ceUs wUl continue on in the ceU cycle. When such ceUs again reach Gχ phase, however, they will be prevented from progressing, as shown in the squares representing ceUs sampled at 48 hours, in which only a very smaU right- shifted peak is visible in aU of the ceU cultures.
The discovery that Calpain Inhibitors can block the ceU cycle has been utUized to devise a treatment for cancer. This treatment involves the synchronization of the ceU cycles of cancer ceUs, foUowed by a course of chemotherapy. According to this embodiment of the present invention, a patient is first treated with a Calpain Inhibitor, which blocks the patient's actively dividing ceUs, including cancer ceUs, from passing from the Gj phase (the "gap" between mitosis and the beginning of DNA synthesis) to the S phase. After the patient has been treated with the Calpain Inhibitor for the length of one ceU cycle, aU of the patient's cancerous ceUs wiU be in the G1 phase. Treatment with the Calpain Inhibitor is then stopped, thereby aUowing the actively dividing ceUs to enter the S phase. All of the cancer ceUs which have been exposed to the Calpain Inhibitor wiU then progress synchronously into the S phase. At this point, a chemotherapeutic agent which interferes with proper DNA replication is
administered to the patient. Since aU of the cancerous ceUs exposed to the calpain inhibitor wiU be progressing synchronously into the S phase when the chemotherapeutic agent is administered, aU of these ceUs will be affected by the agent. This course of treatment can be repeated in order to treat ceUs which did not get previously exposed to the calpain inhibitor.
Figure 22 shows that cancer ceUs in particular are susceptible to having their ceU cycles blocked by Calpain Inhibitors, and that such blockage can be reversed by removing the Calpain Inhibitor. In the experiment illustrated by Figure 22, the ceU cycles of ceUs from AT-2 and HeLa ceU lines were synchronized through the use of a the Calpain Inhibitor Ph2-CHCO-Leu-Abu-CONH-CH2CH(OH)Ph. However, we have also found that P388 leukemia ceUs, L1210 leukemia ceUs, and human myeloma ceUs can also be simUarly synchronized through the use of a Calpain Inhibitor. Thus, it is beUeved that aU cancer ceUs can be treated according to the methods of the present invention As in Figure 21, the squares in Figure 22 depict the results of subjecting ceUs to flow cytometry analysis, in which the amount of DNA in ceUs is quantitated. The ceUs used in this experiment were first exposed to the Calpain Inhibitor Ph2-CHCO-Leu- Abu-CONH-CH2CH(OH)Ph for 48 hours, after which the ceU medium (which contained this Calpain Inhibitor) in which the ceUs were suspended was changed. At time 0 in this figure, when the ceU medium was changed, most of the AT-2 (top row) and HeLa (bottom row) ceUs were in Gj phase, as shown by the presence of a large left-shifted peak and only a very smaU right-shifted peak. After washing out the medium containing the Calpain Inhibitor, the ceUs began to progress through S phase, untU after 30 hours quite a number of ceUs had progressed through the ceU cycle (as shown by the significantly larger right-shifted peak). Thus, Calpain Inhibitors can be used to synchronize the ceU cycles of cancer ceUs and aUow them to synchronously pass into S phase so that they can be effectively treated with a chemotherapeutic agent.
One of the benefits of using Calpain Inhibitors in conjunction with a chemotherapeutic agent is that the use of the chemotherapeutic agent can be discontinued after the length of the S phase of a patient's cancer ceUs, rather than requiring the agent to be administered for a fuU ceU cycle in order to affect aU treatable cancer ceUs. This results in a shorter duration of treatment and therefore a
lessening of the discomfort and side effects of the chemotherapy. The efficacy of the chemotherapeutic agent is also increased.
In order to determine the length of a cancerous ceU's ceU cycle or the length of such a ceU's S phase, the type of cancer ceU should first be determined. This can be done, for example, by taking a biopsy of the tissue which is or is believed to be cancerous. Once the type of cancer ceU is determined, the length of that ceU's ceU cycle and of the S phase of that ceU's cycle can be approximately determined by reference to information known to those of skUl in the art. Alternatively, cancerous tissue extracted during the biopsy can be observed in order to determine the length of the ceU cycle of the ceUs in such cancerous tissue and the length of the S phase of the ceU cycle of such ceUs. Since the S phase of a ceU's reproductive cycle typicaUy makes up a relatively short period in the ceU's reproductive cycle, it is anticipated that standard chemotherapy regimens which target the DNA synthesis of actively dividing cancer ceUs can be considerably shortened through the use of Calpain Inhibitors according to this aspect of the present invention.
The administration of Calpain Inhibitors to synchronize the ceU cycle can be by any of a number of methods known to those of skiU in the art. Such methods include the systemic deUvery of a Calpain Inhibitor through intravenous, intraperitoneal, or intramuscular injection, or through oral delivery. Administration of a Calpain Inhibitor can also be accomplished through the use of a catheter. Other methods of delivery known to those of skiU in the art are as weU possible.
A pharmacologicaUy effective does of a calpain inhibitor for blocking the ceU cycle of ceUs from progressing from G phase to S phase of between approximately 0.001 mg/kg and 100 mg/kg Calpain Inhibitor can be administered daUy to an individual to cause synchronization of the ceU cycle. Preferably, between 1 and 50 mg/kg of a Calpain Inhibitor are administered to such an individual. In one possible course of treatment, a patient can be administered 1 mg/kg of a Calpain Inhibitor. Approximately 24-48 hours after the patient has received this dose of the Calpain Inhibitor, the patient is administered between about 60-75 mg/m2 of adriamycin, a chemotherapeutic drug sold by Adria Laboratories. Dublin, Ohio. This course of treatment can then be repeated approximately every 21 days or otherwise as needed untU the cancer is eradicated or in remission.
Higher concentrations of Calpain Inhibitors than those discussed above can also be administered through the direct application of such Inhibitors to living ceUs. For example, the Calpain Inhibitor can be loaded into a microsphere, and the microsphere can then be driven into tissue to effect direct application to cancer ceUs. A Calpain Inhibitor can also be deUvered through the use of a slow-release compound, such as a gel or ointment, which is applied directly to ceUs. Other strategies for the local administration of a Calpain Inhibitor include the injection of a solution containing a Calpain Inhibitor directly into a malignant tumor to effect synchronization of the ceU cycles of the cancerous ceUs of the tumor. The Calpain Inhibitors of the present invention can also be used in assessing the effectiveness of a chemotherapeutic agent. By synchronizing the ceU cycles of ceUs grown in vitro, chemotherapeutic agents that interfere with DNA synthesis can be assayed most effectively. Thus, to growing cancerous ceUs, an amount of a Calpain Inhibitor is administered to synchronize the ceUs at the end of G- phase. The Calpain Inhibitor can then be rinsed or washed out in a manner weU known to those having ordinary skiU in the art. Thereafter, the ceUs are aUowed to enter S phase, and a potential chemotherapeutic agent is administered to the ceUs in an amount believed to kill cancerous ceUs. The number of kUled ceUs can be determined using any of a number of techniques known in the art, such as by measuring the ceUs' abUity to convert MTT into its blue product. The more effective chemotherapeutic agents wiU be more effective at kUling ceUs at low dosages.
EXAMPLE 35 Determining the Lengths of the Phases of a CeU Cycle A biopsy is performed on a patient who has been determined to have cancer, and cancerous ceUs are thereby removed from the patient. These ceUs are given a brief pulse of H-thymidine and are then washed. Samples of these ceUs are taken at various times over the course of approximately 24 hours. Autoradiographs are then prepared from these samples. InitiaUy, the ceUs that are in the S phase are radiolabeled, whUe ceUs in the G2, M and G- phases are not labeled. After a length of time equal to the length of the G2 phase, the labeled ceUs wiU enter the M phase. By monitoring when labeled ceUs pass into the M phase, and then eventuaUy re-enter the M phase, one can
determine the average durations of the G2, M, S and Gj phases of the ceU cycle, as weU as the length of the entire ceU cycle.
EXAMPLE 36 Inhibition of the Passage of CeUs into S Phase with a Calpain Inhibitor Bovine aortic smooth muscle ceUs (BASMC) were grown in tissue culture medium for at least 48 hours. These ceUs were then serum starved for an additional 48 hours, resulting in a population of ceUs arrested in Gj. The ceUs were then exposed to media containing about 10% fetal bovine serum to stimulate ceU cycle progression and division. At various times foUowing the addition of the serum, the Calpain Inhibitor Ph2-CHCO- Leu-Abu-CONH-CH2CH(OH)Ph was added to the culture medium to a final concentration of 70mM. CeUs from these cultures were removed and stained with DNA dye and analyzed for DNA content using fluorescence activated ceU counting.
The results of this experiment are shown in Figure 21. This experiment demonstrates that ceUs exposed to a Calpain Inhibitor at the time of serum addition do not increase their DNA content over the course of the experiment, which means that they do not progress into the S phase of the ceU cycle. The addition of serum to serum-starved ceUs normaUy aUows ceUs to progress synchronously into the S phase. The addition of a Calpain Inhibitor 18 or more hours after the addition of serum, however, does not inhibit the increase in DNA content and the subsequent division of the ceUs. Thus, Calpain Inhibitors act to block the progression of the ceU cycle into the S phase.
EXAMPLE 37
Removal of a Calpain Inhibitor from a Cell Culture Arrested in Gl Phase WiU Allowthe Culture to Progress to S Phase Two ceU cultures, one of HeLa ceUs and one of AT-2 ceUs, were each grown in the presence of serum and the Calpain Inhibitor Ph2-CHCO-Leu-Abu-CONH- CH2CH(OH)Ph at a final concentration of 70mM for 48 hours. The culture media was then replaced with media lacking this Calpain Inhibitor. This aUowed the ceUs to progress through the ceU cycle. At various times after the removal of the Calpain Inhibitor the ceUs were stained with DNA dye and analyzed using fluorescence activated ceU counting.
The results of this experiment are lustrated in Figure 21. Both ceU types were predominantly in the G- phase after 48 hours of treatment with Calpain Inhibitor, as
shown by their normal DNA content. After washout of the Calpain Inhibitor, both ceU types progressed into the S phase, as shown by the increase in their DNA content. Thus, it was shown that ceUs can be made to synchronously progress into the S phase of the ceU cycle after being treated with a Calpain Inhibitor after removal of the Calpain Inhibitor.
EXAMPLE 38 Use of Substituted Isocoumarins In Chemotherapy A human diagnosed as having a cancerous tumor is administered a Substituted Isocoumarin. Such administration is performed by injecting directly into the tumor a solution containing approximately 1 mg/kg of a Substituted Isocoumarin in phosphate buffered saUne. Beginning 24-48 hours after administration of the Substituted Isocoumarin, 70 mg/m2 of adriamycin (Adria Laboratories, Dublin, OH) is administered to the patient. This treatment is repeated at 21. day intervals untU the tumor is eradicated or in remission.
EXAMPLE 39 Use of Peptide Ketoamides in Chemotherapy A human diagnosed as having a cancerous tumor is administered a Peptide Ketoamide. Such administration is performed by injecting directly into the tumor a solution containing approximately 1 mg/kg of a Peptide Ketoamide in phosphate buffered saline. Beginning 24-48 hours after administration of the Peptide Ketoamide, 70 mg/m2 of adriamycin (Adria Laboratories, Dublin, OH) is administered to the patient. This treatment is repeated at 21 day intervals untU the tumor is eradicated or in remission.
EXAMPLE 40
Use of Peptide Ketoacids in Chemotherapy A human diagnosed as having a cancerous tumor is administered a Peptide Ketoacid. Such administration is performed by injecting directly into the tumor a solution containing approximately 1 mg/kg of a Peptide Ketoacid in phosphate buffered saline. Beginning 24-48 hours after administration of the Peptide Ketoacid. 70 mg/m2 of adriamycin (Adria Laboratories, Dublin, OH) is administered to the patient. This treatment is repeated at 21 day intervals untU the tumor is eradicated or in remission.
EXAMPLE 41
Use of Peptide Ketoesters in Chemotherapy A human diagnosed as having a cancerous tumor is administered a Peptide Ketoester. Such administration is performed by injecting directly into the tumor a solution containing approximately 1 mg/kg of a Peptide Ketoester in phosphate buffered saline.
Beginning 24-48 hours after administration of the Peptide Ketoester, 70 mg/m2 of adriamycin (Adria Laboratories, Dublin, OH) is administered to the patient. This treatment is repeated at 21 day intervals untU the tumor is eradicated or in remission.
*. Increasing the Efficiency of Cell Transformation
With the discovery that Calpain Inhibitors can prevent a ceU from entering the S phase of the ceU cycle, we have found that such Inhibitors can be used to increase the efficiency with which ceUs are transformed with DNA. When foreign DNA is introduced (transformed) into a ceU, such DNA can be incoφorated into the genome of that ceU. Whether such incoφoration takes place depends upon the presence of DNA splicing and replication enzymes which are most active during the S phase of the ceU cycle. Thus, the efficiency of the incoφoration of foreign DNA can be increased by introducing the foreign DNA into a population of ceUs which have been synchronized in the S phase using a Calpain Inhibitor. In this aspect of the present invention, cells in vitro, such as mammalian ceUs in culture, can first be synchronized as described above by administering a Calpain Inhibitor to such ceUs. A dose of a Calpain Inhibitor which is pharmacologicaUy effective to block the ceU cycle of ceUs form progressing from Gj phase to S phase is between approximately 10 μM and 500 mM. Such a dose can be administered to the ceUs by adding the Calpain Inhibitor in solution to the media in which the ceUs are suspended. FoUowing the addition of the Calpain Inhibitor, the ceUs wiU pass into the Gj phase and remain in that state untU the Calpain Inhibitor is washed out of the ceU media or untU it is used up by the ceUs.
Once the ceUs are thus synchronized in the Gj phase, the Calpain Inhibitor can be removed from the ceU media by removing the ceU media and adding fresh medium. After aUowing sufficient time to aUow the ceUs to pass into the S phase, the ceUs can be transformed by methods known to those of skill in the art. For example, the methods disclosed in Molecular Cloning (Sambrook, Jr., Fritsch, E.F., and Maniatis, T., Molecular
Cloning: A Laboratory Manual, Cold Spring Harbor Press, Cold Spring Harbor, NY (1989)) or in Short Protocols in Molecular Biology, (Ausbel, et al. eds., Short Protocols in Molecular Biology, John WUey & Sons (1989)), which are hereby incoφorated by reference, can be used to transform the ceUs. In a preferred embodiment, exogenous oligonucleotides are included in vector sequences for transformation of the ceUs." In this embodiment, the exogenous oligonucleotides preferably code for protein are operatively linked to a promoter sequence for transcription and later translation. FoUowing transformation, the ceUs can be used in a variety of ways known to those having skill in the art.
In one embodiment, this method can be used to treat a mammal which has a disease caused by a genetic mutation that results in a protein deficiency in a particular tissue. In this embodiment, ceUs of the affected tissue can be removed from a patient synchronized in the G, phase as described above. After allowing the ceUs to pass into the S phase, these ceUs are transformed by methods known to the art, for example by introduction of a viral vector carrying exogenous nucleotide sequences. The ceUs transformed with oligonucleotides coding for a normal gene can be retransplanted into the patient from whom the ceUs were taken, where they wUl then be able to function normaUy due to the incoφoration of the normal gene.
EXAMPLE 42 Use of Calpain Inhibitors to Increase Efficiency of Transformation in Gene Therapy
A human is diagnosed with sickle ceU anemia, which is caused by a genetic mutation that results in a deficiency of normal hemoglobin in red blood ceUs.
Hematopoietic bone marrow ceUs are removed from the patient and put into culture in vitro in the presence of 100 mM of a Calpain Inhibitor, which causes the ceUs to synchronize at the G1 phase. After synchronization, the Calpain Inhibitor is washed out, aUowing the ceUs to proceed to the S phase. The ceUs are then transformed with foreign DNA which includes the normal gene coding for hemoglobin. After transformation with such foreign DNA, the ceUs are reintroduced into the patient, where they wUl repopulate the bone marrow and produce normal hemoglobin protein in red blood ceUs.
10. Other Non-Neurological Uses
It is known that a large number of medical conditions and diseases are associated with an increase in the activity of Calpain and other calcium-activated proteases. We therefore believe that the compositions of the present invention are beneficial in treating a large number of these other conditions, and the treatment of these other conditions can properly be considered within the scope of the present invention. H. DRUG DELIVERY
The ability of the various Calpain Inhibitors to penetrate plasma membranes is a significant advantage of these compounds from a pharmaceutical perspective. We believe that this abUity, advantageously, aUows the Calpain Inhibitors to provide exceUent permeation of the blood-brain barrier. This is in contrast to many pharmaceuticals, especiaUy peptides, which often exhibit poor permeation of the blood-brain barrier. Thus, we believe that the Calpain Inhibitors wiU exhibit exceUent results as pharmaceuticaUy neuroprotective agents. For treatment of neurodegeneration and other medical conditions, the Calpain
Inhibitors can be administered oraUy, topicaUy, intraperitoneaUy or parenteraUy. The term "parenteral" as used herein includes aU non-oral delivery techniques including transdermal administration, subcutaneous injection, intravenous, intramuscular or intrasternal injection, intrathecal injection (directly into the CNS) or infusion techniques. The dosage depends primarUy on the specific formulation and on the object of the therapy or prophylaxis. The amount of the individual doses as weU as the administration is best determined by individuaUy assessing the particular case. However, in preferred compositions, the dosages of Calpain Inhibitors per day are preferably in the range of 1 μg/kg total body mass to 100 mg/kg total body mass, more preferably in the range of 10 μg/kg total body mass to 10 mg/kg total body mass.
The pharmaceutical compositions containing the active ingredient may be in a form suitable for oral use, for example as tablets, troches, lozenges, aqueous or oUy suspensions, dispersible powders or granules, emulsions, hard or soft capsules or syrups or elixirs. Dosage levels of the order to 0.2 mg to 140 mg per kUogram of body weight per day are useful in the treatment of above-indicated conditions (10 mg to 7 gms per patient per day).
The amount of active ingredient that may be combined with carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode
of administration. However, typicaUy, a single dose wiU contain sufficient Calpain Inhibitor to provide a complete day's dosage in a single oraUy acceptable form.
For injection, the therapeutic amount of the Calpain Inhibitors or their pharmaceuticaUy acceptable salts wiU normaUy be made by subcutaneous injection, intravenous, intramuscular, intraperitoneal or intrasternal injection, or by intrathecal injection (directly into the brain). For injection, the therapeutic amount of the peptide a-ketoamides or their pharmaceuticaUy acceptable salts wiU normaUy be in the dosage range from 0.2 to 140 mg/kg of body weight. In order to provide a single day's dose with a single injection, the pharmaceutical compositions for parenteral administration wiU contain, in a single dosage form, from about 70 μg to about 7 g of Calpain Inhibitor per dose of from about 0.5 ml to about 1 liter of carrier solution. In addition to the active ingredient, these pharmaceutical compositions wiU usuaUy contain a buffer, e.g. a phosphate buffer that keeps the pH in the range from 3.5 to 7 and also sodium chloride, and can also contain mannitol or sorbitol for adjusting the isotonic pressure. In a preferred form of these compositions, DMSO or other organic solvent is added in order to assist the introduction of the Calpain Inhibitor across membranes.
AdditionaUy, lipids can be introduced into the pharmaceutical compositions in order to facUitate entry of the Calpain inhibitor compounds into tissue of the CNS. These compositions are prepared in accordance with methods known to those of skiU in the art. Briefly, a Hpid such as, phosphatidyl choline, cholesterol, other weU-known lipid carrier or mixtures thereof, is mixed with the active compound along with a solvent, the solvent is dried off and the material reconstituted in saline. The compositions can also include other ingredients known to those of ordinary skUl in the art, such as detergents, surfactants or emulsifying agents. A composition for topical application or infusion can be formulated as an aqueous solution, lotion, jeUy or an oUy solution or suspension. A composition in the form of an aqueous solution is obtained by dissolving the Calpain Inhibitor in aqueous buffer solution of pH 4 to 6.5 and, if desired, adding a polymeric binder. An oUy formulation for topical appUcation is obtained by suspending the Calpain Inhibitor in an oU, optionaUy with the addition of a sweUing agent such as aluminium stearate and/or a surfactant. The addition of DMSO to these topical compositions is believed to aUow at least partial penetration of
the active Calpain Inhibitor into the blood stream after application of the composition to the skin of a patient to aUow for transdermal administration.
For treatment of neurodegeneration resulting from excitotoxicity, HIV-induced neuropathy, ischemia foUowing denervation or injury, subarachnoid hemorrhage, stroke, multiple infarction dementia, Alzheimer's Disease (AD), Huntington's Disease, surgery- related brain damage, Parkinson's Disease, and other pathological conditions, the Calpain Inhibitors or pharmaceuticaUy acceptable salts thereof may be administered oraUy or parenteraUy. The dosage depends primarUy on the specific formulation and on the object of the therapy or prophylaxis. The amount of the individual doses as weU as the administration is best determined by individuaUy assessing the particular case.
In many acute neurodegenerative conditions and events, such as stroke and head injury, it is important to deliver the Calpain Inhibitor as soon after injury as is practicable. Thus, it is preferable to identify those individuals who have suffered stroke, head injury or other injury in which neurodegeneration is associated or is likely to occur, and to begin administration of a Calapin Inhibitor within 1 minute to 2 hours after the event, in order to prevent as much neurodegeneration as possible.
A particular application of the Calpain Inhibitors within the scope of the present invention is the application of these compounds during surgery to prevent neurodegeneration associated therewith. For example, for surgeries performed under general anesthesia, hypoxic conditions can occur through inadequate perfusion of the CNS whUe under anesthesia. AdditionaUy, many major surgeries of the cardiovascular system require that a patient's heart be stopped and that perfusion be maintained through artificial means. In such surgeries, there is an increased danger of hypoxia occurring within the CNS, which can also result in neurodegeneration. Moreover, during neurosurgeries, there is an inherent risk of neurodegeneration resulting from inflammation, bleeding, hemorrhaging and the like. Such neurodegeneration can be inhibited by infusion with a solution containing Calpain Inhibitor. However, neurodegeneration resulting from neurosurgery can also be reduced prophylacticaUy by administration of a Calpain Inhibitor through any of the foregoing administration techniques. Such administration is also beUeved to inhibit or prevent neurodegeneration associated with the use of anesthesia or with the use of artifical means of perfusion during
major surgeries. A surgical patient can also have Calpain Inhibitor administered throughout surgery through intravenous drip.
The foUowing examples are intended to Ulustrate certain neuroprotective uses of the Calpain Inhibitors within the scope of the present invention. As such, they are not meant to limit the invention in any way.
EXAMPLE 35 A Neuroprotective Composition for Intravenous Injection 500 μg CH3CONH-CiTPrOIC from Example SHC2 4 ml Propylene Glycol 1 ml DMSO
EXAMPLE 36 A Neuroprotective Composition for Intravenous Drip 250 mg Z-Leu-Phe-CONH-Et from Example PKC 48
1000 ml Phosphate Buffered Saline (pH 6.0) 10 ml DMSO
EXAMPLE 37 A Neuroprotective Composition for Transdermal Application 25 mg Z-Leu-DL-Abu-COOEt from Example PKC19
3 ml Phosphate Buffered Saline (pH 6.0) 2 ml DMSO
EXAMPLE 38 Neuroprotection after Head Injury
A first group of patients who are victims of head trauma is given 2 ml of the injectable composition of Example 30 intravenously within ten minutes of the time of injury. A second group of simUarly matched patients does not receive the composition. The first group of patients exhibits markedly fewer and less severe outward symptoms of neurodegeneration, such as dementia, memory loss and paralysis.
EXAMPLE 39
Neuroprotection During Surgery A patient about to undergo a triple bypass heart surgery is administered 500 ml of the composition of Example 31 per hour using an intravenous drip system. During surgery, the patient's heart is stopped and perfusion continued through artificial means.
Although complications develop whUe restarting the heart and disconnecting the patient from the artificial means of perfusion, the patient becomes conscious within several hours of surgery. Within a few days, the patient's mental status is normal with no indications of neurodegeneration. For the inhibition of smooth muscle ceU proliferation in the treatment or prevention of restenosis, Calpain Inhibitor can also be administered directly to the site of injured smooth muscle tissue. Such administration can be accomplished, for example, by means of an ointment or gel applied to the surface of a baUoon or other surgical tool used in an angioplasty procedure. In this way, if damage is done during angioplasty that would otherwise result in restenosis, restenosis can be prevented.
The direct administration of a Calpain Inhibitor to the site of injured tissue can also be accomplished by loading the Calpain Inhibitor into microspheres and imbedding the microspheres into the injured tissue. This can be accomplished by applying the microspheres to the surface of the baUoon used in the angioplasty procedure. When the baUoon is inflated inside the artery, the force of the expansion drives the microspheres into the arterial waU, where they become lodged. The microspheres then release the Calpain Inhibitor slowly over time and provide local application to the injured tissue.
For the treatment of numerous medical conditions, Calpain Inhibitors can be injected in solutions either intravenously, intraocularly, intramuscularly; intraperitoneaUy, or intrasternaUy. These solutions wiU contain a Calpain Inhibitor in the range of from about 70 μg to about 7 g per dose in about 0.5 ml to 1 liter of a pharmaceuticaUy acceptable carrier solution. The solution preferably contains a buffer, such as a phosphate buffer, that keeps the pH in the range of about 3.5 to 7. The solution also preferably contains approximately 9000 mg/1 of sodium chloride (0.9% saline), as weU as mannitol or sorbitol for adjusting the isotonio pressure. DMSO at 0.01 to 10 ml/liter can also be used in injectable solutions of the present invention in order to potentiate the Calpain
Inhibitor, or help it to penetrate membranes. Other additives such as ethanol or ethoxylated oUs can also be used.
When using Calpain Inhibitors to protect cardiac, skeletal, or smooth muscle tissue from damage, intravenous drip infusion is preferred to the periodic injection of such Inhibitors. A solution suitable for intravenous infusion can be prepared by suspending approximately 250 mg of a Calpain Inhibitor in 1000 ml of an aqueous solution of phosphate buffered saline containing 10 ml DMSO. However, in the treatment of muscular dystrophy, or when treating a condition for a long period of time with Calpain Inhibitors, an oral formulation of a Calpain Inhibitor is preferred. Such an oral formulation can be in the form of a tablet, in which a powdered form of a Calpain
Inhibitor is mixed with a pharmaceuticaUy acceptable fUler material capable of being formed into a tablet.
In the treatment of cataracts, the injectable solutions referred to above can be administered by soaking them into a contact lens, which is then worn for a period of time long enough to aUow the solution to diffuse into the eye from the lens. Other methods of delivering Calpain Inhibitors to an eye with the injectable solutions described above include the administration of eye drops comprising the solution.
Calpain Inhibitors can also be formulated in an ointment for administration to the eye. This can be accomplished by dissolving a Calpain Inhibitor in an aqueous solution and then adding a pharmaceutically acceptable polymeric binder. A Calpain Inhibitor can also be directly dissolved or suspended in such a polymeric binder.
For the treatment or prevention of tonic smooth muscle contraction, Calpain Inhibitor can also be administered directly to the smooth muscle, including application to coronary tissue. Such administration can be accomplished by means of an ointment, gel or solution appUed directly to the smooth muscle during surgery. Direct administration can also be accompUshed by loading the Calpain Inhibitor into a microsphere and imbedding the microsphere into the smooth muscle tissue. The microsphere then releases the Calpain Inhibitor slowly over time and provides local appliction to the tissue.
For the treatment of cerebral vasospasm, a solution of a Calpain Inhibitor can be injected directly into the cerebrospinal fluid of the patient. For the treatment of bronchospasm, such as that which occurs in asthmatic patients, a solution containing a Calpain Inhibitor can be inhaled directly into the patient's lungs.
The foUowing additional examples are provided to further Ulustrate certain embodiments of pharmaceutical compositions within the scope of the present invention.
EXAMPLE 40 An Injectable Composition for Non-Neurological Uses 1 mg Z-Leu-Abu-CONH-Bzl from Example PKC58
4 ml Propylene Glycol 1 ml DMSO
EXAMPLE 41 An Ophthalmic Solution for Treating Cataracts 500 μg Z-Leu-Abu-CONH-iBu from Example PKC57
5 ml sterUe phosphate buffered saline
EXAMPLE 42 A Solution for Topical Application to a Tonic. Contracted Blood Vessel 2 mg Z-Leu-Abu-CONH-(CH2)3-N(CHCH2)20 from Example PKC60
1 ml DMSO
10 ml sterUe phosphate buffered saline
It wiU be appreciated that certain variations may suggest themselves to those skUled in the art. The foregoing detaUed description is to be clearly understood as given by way of illustration, the spirit and scope of this invention being interpreted through reference to the appended claims.