US9701608B2 - Process for the synthesis of 7-methoxy-naphthalene-1-carbaldehyde and application in the synthesis of agomelatine - Google Patents
Process for the synthesis of 7-methoxy-naphthalene-1-carbaldehyde and application in the synthesis of agomelatine Download PDFInfo
- Publication number
- US9701608B2 US9701608B2 US15/101,120 US201415101120A US9701608B2 US 9701608 B2 US9701608 B2 US 9701608B2 US 201415101120 A US201415101120 A US 201415101120A US 9701608 B2 US9701608 B2 US 9701608B2
- Authority
- US
- United States
- Prior art keywords
- formula
- compound
- process according
- conversion
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 40
- 230000008569 process Effects 0.000 title claims abstract description 38
- 230000015572 biosynthetic process Effects 0.000 title claims abstract description 18
- 238000003786 synthesis reaction Methods 0.000 title claims abstract description 18
- YJYPHIXNFHFHND-UHFFFAOYSA-N agomelatine Chemical compound C1=CC=C(CCNC(C)=O)C2=CC(OC)=CC=C21 YJYPHIXNFHFHND-UHFFFAOYSA-N 0.000 title claims description 18
- 229960002629 agomelatine Drugs 0.000 title claims description 15
- WYDUFXRPFJPXGH-UHFFFAOYSA-N 7-methoxynaphthalene-1-carbaldehyde Chemical compound C1=CC=C(C=O)C2=CC(OC)=CC=C21 WYDUFXRPFJPXGH-UHFFFAOYSA-N 0.000 title description 9
- 150000001875 compounds Chemical class 0.000 claims abstract description 81
- 238000006243 chemical reaction Methods 0.000 claims description 35
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 18
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 10
- 229910052723 transition metal Inorganic materials 0.000 claims description 10
- 150000003624 transition metals Chemical class 0.000 claims description 9
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-bis(diphenylphosphino)propane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 claims description 8
- 239000003638 chemical reducing agent Substances 0.000 claims description 8
- 238000006392 deoxygenation reaction Methods 0.000 claims description 8
- UNFNRIIETORURP-UHFFFAOYSA-N 7-methoxynaphthalen-2-ol Chemical compound C1=CC(O)=CC2=CC(OC)=CC=C21 UNFNRIIETORURP-UHFFFAOYSA-N 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical group [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 7
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 7
- 125000003944 tolyl group Chemical group 0.000 claims description 7
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 6
- 230000009471 action Effects 0.000 claims description 5
- 229910052763 palladium Inorganic materials 0.000 claims description 5
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 4
- 239000007787 solid Substances 0.000 claims description 4
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 claims description 4
- FWZRNGJNGNRONB-UHFFFAOYSA-N (1-formyl-7-methoxynaphthalen-2-yl) 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)C=O FWZRNGJNGNRONB-UHFFFAOYSA-N 0.000 claims description 3
- UFMVAMMCYVBZAS-UHFFFAOYSA-N (1-formyl-7-methoxynaphthalen-2-yl) methanesulfonate Chemical compound CS(=O)(=O)OC1=C(C2=CC(=CC=C2C=C1)OC)C=O UFMVAMMCYVBZAS-UHFFFAOYSA-N 0.000 claims description 3
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 claims description 3
- 238000000354 decomposition reaction Methods 0.000 claims description 3
- 229910052759 nickel Inorganic materials 0.000 claims description 3
- 150000002940 palladium Chemical class 0.000 claims description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 2
- 150000008064 anhydrides Chemical class 0.000 claims description 2
- 229910052697 platinum Inorganic materials 0.000 claims description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 claims description 2
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims 2
- LVQCQPJRMITAJV-UHFFFAOYSA-N 1-methoxynaphthalen-2-ol Chemical compound C1=CC=C2C(OC)=C(O)C=CC2=C1 LVQCQPJRMITAJV-UHFFFAOYSA-N 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 238000004611 spectroscopical analysis Methods 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- 239000000243 solution Substances 0.000 description 7
- 0 *S(=O)(=O)OC1=C(C=O)C2=C(C=CC(C)=C2)C=C1 Chemical compound *S(=O)(=O)OC1=C(C=O)C2=C(C=CC(C)=C2)C=C1 0.000 description 6
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 6
- KPTJFNWVWKKZHP-UHFFFAOYSA-N CC1=CC2=C(C=CC=C2C=O)C=C1 Chemical compound CC1=CC2=C(C=CC=C2C=O)C=C1 KPTJFNWVWKKZHP-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- GABLTKRIYDNDIN-UHFFFAOYSA-N 7-methoxy-3,4-dihydro-2h-naphthalen-1-one Chemical compound C1CCC(=O)C2=CC(OC)=CC=C21 GABLTKRIYDNDIN-UHFFFAOYSA-N 0.000 description 3
- DNCGBNFOSLTQQB-UHFFFAOYSA-N CC1=CC2=C(C=C1)C=CC(O)=C2 Chemical compound CC1=CC2=C(C=C1)C=CC(O)=C2 DNCGBNFOSLTQQB-UHFFFAOYSA-N 0.000 description 3
- GDJNKVIORHVPCB-UHFFFAOYSA-N CC1=CC2=C(C=C1)C=CC(O)=C2C=O Chemical compound CC1=CC2=C(C=C1)C=CC(O)=C2C=O GDJNKVIORHVPCB-UHFFFAOYSA-N 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- KVHHMYZBFBSVDI-UHFFFAOYSA-N 8-aminonaphthalen-2-ol Chemical compound C1=C(O)C=C2C(N)=CC=CC2=C1 KVHHMYZBFBSVDI-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 150000001342 alkaline earth metals Chemical class 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 238000005899 aromatization reaction Methods 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 2
- 150000004678 hydrides Chemical class 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- WQGADZACBGKOHW-UHFFFAOYSA-N 2-hydroxy-7-methoxynaphthalene-1-carbaldehyde Chemical compound C1=CC(O)=C(C=O)C2=CC(OC)=CC=C21 WQGADZACBGKOHW-UHFFFAOYSA-N 0.000 description 1
- 208000027559 Appetite disease Diseases 0.000 description 1
- PCRSJNQCJJKQOJ-UHFFFAOYSA-N CC(=O)NCCC1=CC=CC2=C1C=C(C)C=C2 Chemical compound CC(=O)NCCC1=CC=CC2=C1C=C(C)C=C2 PCRSJNQCJJKQOJ-UHFFFAOYSA-N 0.000 description 1
- LRQYSMQNJLZKPS-UHFFFAOYSA-N CC1=CC2=C(C=C1)C=CC(C)=C2 Chemical compound CC1=CC2=C(C=C1)C=CC(C)=C2 LRQYSMQNJLZKPS-UHFFFAOYSA-N 0.000 description 1
- 208000019888 Circadian rhythm sleep disease Diseases 0.000 description 1
- 208000019454 Feeding and Eating disease Diseases 0.000 description 1
- 208000001456 Jet Lag Syndrome Diseases 0.000 description 1
- MPIMZAKRTOXRAO-UHFFFAOYSA-N N[C]N Chemical class N[C]N MPIMZAKRTOXRAO-UHFFFAOYSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001343 alkyl silanes Chemical class 0.000 description 1
- TVJORGWKNPGCDW-UHFFFAOYSA-N aminoboron Chemical compound N[B] TVJORGWKNPGCDW-UHFFFAOYSA-N 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- RJTANRZEWTUVMA-UHFFFAOYSA-N boron;n-methylmethanamine Chemical compound [B].CNC RJTANRZEWTUVMA-UHFFFAOYSA-N 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000007333 cyanation reaction Methods 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- XYYQWMDBQFSCPB-UHFFFAOYSA-N dimethoxymethylsilane Chemical compound COC([SiH3])OC XYYQWMDBQFSCPB-UHFFFAOYSA-N 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000006170 formylation reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 206010022437 insomnia Diseases 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 208000033915 jet lag type circadian rhythm sleep disease Diseases 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 208000024714 major depressive disease Diseases 0.000 description 1
- 230000001193 melatoninergic effect Effects 0.000 description 1
- 150000002815 nickel Chemical class 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 208000012672 seasonal affective disease Diseases 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 238000012306 spectroscopic technique Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- -1 transition metal salt Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/65—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by splitting-off hydrogen atoms or functional groups; by hydrogenolysis of functional groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/11—Aldehydes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/26—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids
- C07C303/28—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids by reaction of hydroxy compounds with sulfonic acids or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/64—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms
- C07C309/65—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms of a saturated carbon skeleton
- C07C309/66—Methanesulfonates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/72—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/73—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton to carbon atoms of non-condensed six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
- C07C45/455—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation with carboxylic acids or their derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/78—Separation; Purification; Stabilisation; Use of additives
- C07C45/81—Separation; Purification; Stabilisation; Use of additives by change in the physical state, e.g. crystallisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/78—Separation; Purification; Stabilisation; Use of additives
- C07C45/86—Use of additives, e.g. for stabilisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/52—Compounds having —CHO groups bound to carbon atoms of six—membered aromatic rings
- C07C47/575—Compounds having —CHO groups bound to carbon atoms of six—membered aromatic rings containing ether groups, groups, groups, or groups
Definitions
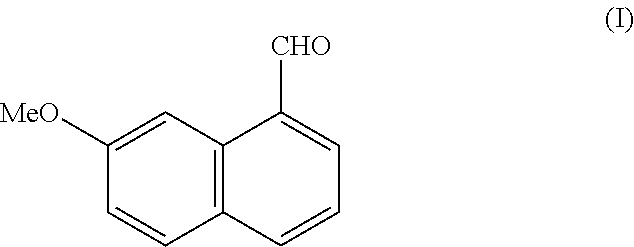
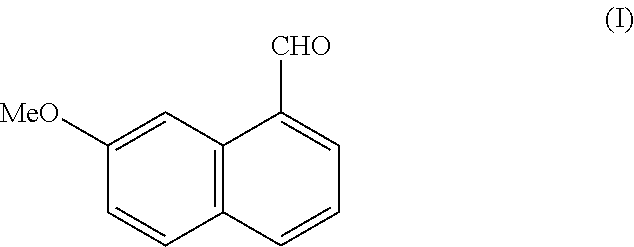
- the present invention relates to a new process for the industrial synthesis of (7-methoxy-1-naphthalene-1-carbaldehyde and to its application in the industrial production of agomelatine, or N-[2-(7-methoxy-1-naphthyl)ethyl]acetamide.
- the present invention relates to a process for the industrial synthesis of the compound of formula (I):
- the compound of formula (I) obtained according to the process of the invention is useful in the synthesis of agomelatine, or N-[2-(7-methoxy-1-naphthyl)ethyl]acetamide, of formula (II):
- Agomelatine or N-[2-(7-methoxy-1-naphthyl)ethyl]acetamide, has valuable pharmacological properties.
- Patent specification EP 0 447 285 describes obtaining agomelatine in eight steps starting from 7-methoxy-1-tetralone. When transferred to the industrial scale, however, difficulties in implementing that process rapidy came to light.
- the Applicant has continued his investigations and has developed a new industrial synthesis which, in reproducible manner and without the need for laborious purification, yields agomelatine with a purity which is compatible with its use as a pharmaceutical active ingredient, starting from a less costly and more readily obtainable starting material.
- the Applicant has now developed a new industrial synthesis process making it possible to obtain 7-methoxy-naphthalene-1-carbaldehyde in reproducible manner without the need for laborious purification, using 7-methoxy-naphthalen-2-ol as starting material.
- This new starting material has the advantage of being simple and readily obtainable in large amounts at less cost.
- 7-Methoxy-naphthalen-2-ol also has the advantage of having in its structure a naphthalene ring system, which avoids incorporating an aromatisation step in the synthesis—a step which is always problematic from an industrial point of view.
- the present invention relates to a process for the industrial synthesis of the compound of formula (I):
- R represents a —CH 3 , —(CH 2 ) 2 —CH 3 , —CF 3 or tolyl group
- R preferably represents a —CH 3 or tolyl group.
- conversion of the compound of formula (III) into the compound of formula (IV) consists of the action of ethyl orthoformate in the presence of aniline followed by hydrolysis of the intermediate imine obtained.
- conversion of the compound of formula (IV) into the compound of formula (V) consists of a sulphonylation step carried out by means of the action of a sulphonyl chloride, a sulphonic anhydride or a sulphonimide.
- this sulphonylation step is carried out by means of the action of a sulphonyl chloride and, especially, tosyl chloride or mesyl chloride.
- conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of a transition metal and a reducing agent.
- the transition metal is nickel, palladium or platinum.
- the transition metal can be either in the form of a salt or in the form of a simple substance.
- the transition metal salt is a nickel salt or a palladium salt, more preferably a palladium salt.
- the reducing agent is either a hydride such as sodium borohydride or lithium aluminium hydride; or an aminoborane such as dimethylamine borane; or an alkoxysilane such as dimethoxymethylsilane; or an alkylsilane such as triethylsilane; or an alkaline earth metal such as magnesium; or dihydrogen.
- the reducing agent is dihydrogen which is used directly in its gaseous form or is indirectly obtained by decomposition of an ammonium formate.
- the reducing agent is preferably dihydrogen obtained by decomposition of an ammonium formate.
- conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of nickel, especially a nickel salt, and a hydride, preferably sodium borohydride.
- conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of palladium and dihydrogen.
- conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of palladium and an alkaline earth metal, preferably magnesium.
- reaction converting the compound of formula (V) into the compound of formula (I) is carried out in dimethylformamide, dioxane, tetrahydrofuran and toluene, and more preferably dimethylformamide.
- reaction converting the compound of formula (V) into the compound of formula (I) is carried out between 25° C. and 110° C., more especially between 40° C. and 95° C.
- conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of a transition metal, a reducing agent and a ligand.
- the ligand can be either a phosphine ligand or a diaminocarbene ligand, more preferably a phosphine ligand and, more specifically, 1,3-bis(diphenylphosphino)propane or (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane).
- a variant which is advantageous for the industrial synthesis process consists of conversion of the compound of formula (IV) being carried out to form the compound of formula (I) directly, said sulphonylation reaction and said deoxygenation reaction in the presence of a transition metal being carried out as a “one-pot” procedure.
- Preferred compounds of formula (V) are the following:
- the compound of formula (I) hereby obtained is subsequently subjected to a series of customary chemical reactions (for example: reduction of the aldehyde into a primary alcohol, cyanation, reduction and acetylation of the primary amine obtained) to yield agomelatine of formula (II).
- Step B 1-formyl-7-methoxynaphthalen-2-yl 4-methylbenzenesulphonate
- Step B above The product of Step B above (356 mg; 1 mmol), palladium acetate (4.5 mg; 0.02 mmol), 1,3-bis(diphenylphosphino)propane (8.2 mg; 0.02 mmol), dimethylformamide (2 mL), triethylamine (556 ⁇ L; 4 mmol) and formic acid (150 ⁇ L; 4 mmol) are introduced into a flask placed in an oven and purged with argon. The flask is placed in a bath heated to 90° C. for 1.5 hours.
- the mixture is diluted with ethyl acetate and the organic phase is washed with 1M aqueous hydrochloric acid solution and with brine, dried over sodium sulphate and filtered. After evaporating off the solvent, the crude product is purified by filtration over neutral alumina to yield the title product (139 mg; 75%).
- Step A 1-formyl-7-methoxynaphthalen-2-yl methanesulphonate
- Step B 7-methoxynaphthalene-1-carbaldehyde
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Process for the industrial synthesis of the compound of formula (I):

Description
The present invention relates to a new process for the industrial synthesis of (7-methoxy-1-naphthalene-1-carbaldehyde and to its application in the industrial production of agomelatine, or N-[2-(7-methoxy-1-naphthyl)ethyl]acetamide.
More specifically, the present invention relates to a process for the industrial synthesis of the compound of formula (I):

The compound of formula (I) obtained according to the process of the invention is useful in the synthesis of agomelatine, or N-[2-(7-methoxy-1-naphthyl)ethyl]acetamide, of formula (II):

Agomelatine, or N-[2-(7-methoxy-1-naphthyl)ethyl]acetamide, has valuable pharmacological properties.
It does, in fact, have the double characteristic of being, on the one hand, an agonist of receptors of the melatoninergic system and, on the other hand, an antagonist of the 5-HT2C receptor. These properties provide it with activity in the central nervous system and, more especially, in the treatment of major depression, seasonal affective disorder, sleep disorders, cardiovascular pathologies, pathologies of the digestive system, insomnia and fatigue due to jet-lag, appetite disorders and obesity.
Agomelatine, its preparation and its use in therapeutics have been described in European Patent specifications EP 0 447 285 and EP 1 564 202.
In view of the pharmaceutical value of this compound, it is important to be able to obtain it by an effective synthesis process that is readily transferable to the industrial scale and that results in agomelatine in a good yield and with excellent purity, starting from economical and readily obtainable starting materials.
Patent specification EP 0 447 285 describes obtaining agomelatine in eight steps starting from 7-methoxy-1-tetralone. When transferred to the industrial scale, however, difficulties in implementing that process rapidy came to light.
The literature describes obtaining 7-methoxy-naphthalene-1-carbaldehyde in 5 steps starting from 8-amino-naphthalen-2-ol (Kandagatla et al., Tetrahedron Lett. 2012, 53, 7125-7127). The preparation of 7-methoxy-naphthalene-1-carbaldehyde in 4 steps starting from 7-methoxy-tetralone has also been described (Garipati et al., Bioorg. Med. Chem. Lett. 1997, 7, 1421-1426). 7-Methoxy-1-tetralone and 8-amino-naphthalen-2-ol are, however, costly starting materials and consequently the search for new synthesis routes, especially starting from less expensive reagents, is still ongoing.
The Applicant has continued his investigations and has developed a new industrial synthesis which, in reproducible manner and without the need for laborious purification, yields agomelatine with a purity which is compatible with its use as a pharmaceutical active ingredient, starting from a less costly and more readily obtainable starting material.
More especially, the Applicant has now developed a new industrial synthesis process making it possible to obtain 7-methoxy-naphthalene-1-carbaldehyde in reproducible manner without the need for laborious purification, using 7-methoxy-naphthalen-2-ol as starting material. This new starting material has the advantage of being simple and readily obtainable in large amounts at less cost. 7-Methoxy-naphthalen-2-ol also has the advantage of having in its structure a naphthalene ring system, which avoids incorporating an aromatisation step in the synthesis—a step which is always problematic from an industrial point of view.
More specifically, the present invention relates to a process for the industrial synthesis of the compound of formula (I):

characterised in that 7-methoxy-naphthalen-2-ol of formula (III):

is used for reaction, a formylation reaction being carried out at position 1 of the compound of formula (III) to yield the compound of formula (IV):

which compound of formula (IV) is subjected to a sulphonylation reaction to yield the compound of formula (V):
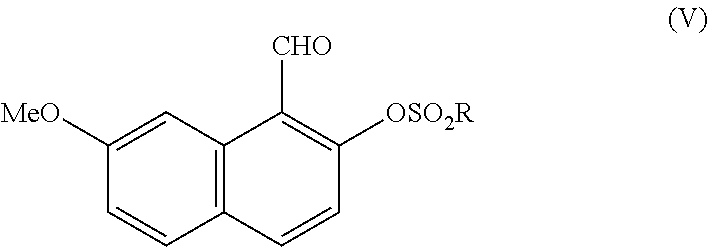
wherein R represents a —CH3, —(CH2)2—CH3, —CF3 or tolyl group;
which compound of formula (V) undergoes a deoxygenation reaction in the presence of a transition metal and a reducing agent to yield the compound of formula (I), which is isolated in the form of a solid.
The compound of formula (III) is commercially available or readily obtainable by the skilled person using chemical reactions that are customary or described in the literature.
R preferably represents a —CH3 or tolyl group.
In the process according to the invention, conversion of the compound of formula (III) into the compound of formula (IV) consists of the action of ethyl orthoformate in the presence of aniline followed by hydrolysis of the intermediate imine obtained.
In the process according to the invention, conversion of the compound of formula (IV) into the compound of formula (V) consists of a sulphonylation step carried out by means of the action of a sulphonyl chloride, a sulphonic anhydride or a sulphonimide. In a preferred embodiment, this sulphonylation step is carried out by means of the action of a sulphonyl chloride and, especially, tosyl chloride or mesyl chloride.
In the process according to the invention, conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of a transition metal and a reducing agent.
Preferably, the transition metal is nickel, palladium or platinum. The transition metal can be either in the form of a salt or in the form of a simple substance. Preferably, the transition metal salt is a nickel salt or a palladium salt, more preferably a palladium salt.
Advantageously, the reducing agent is either a hydride such as sodium borohydride or lithium aluminium hydride; or an aminoborane such as dimethylamine borane; or an alkoxysilane such as dimethoxymethylsilane; or an alkylsilane such as triethylsilane; or an alkaline earth metal such as magnesium; or dihydrogen. Preferably, the reducing agent is dihydrogen which is used directly in its gaseous form or is indirectly obtained by decomposition of an ammonium formate. The reducing agent is preferably dihydrogen obtained by decomposition of an ammonium formate.
In accordance with another preferred embodiment, conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of nickel, especially a nickel salt, and a hydride, preferably sodium borohydride.
In accordance with another preferred embodiment, conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of palladium and dihydrogen.
In accordance with another preferred embodiment, conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of palladium and an alkaline earth metal, preferably magnesium.
Advantageously, the reaction converting the compound of formula (V) into the compound of formula (I) is carried out in dimethylformamide, dioxane, tetrahydrofuran and toluene, and more preferably dimethylformamide.
Preferably, the reaction converting the compound of formula (V) into the compound of formula (I) is carried out between 25° C. and 110° C., more especially between 40° C. and 95° C.
In accordance with another preferred embodiment, conversion of the compound of formula (V) into the compound of formula (I) consists of a deoxygenation step in the presence of a transition metal, a reducing agent and a ligand.
The ligand can be either a phosphine ligand or a diaminocarbene ligand, more preferably a phosphine ligand and, more specifically, 1,3-bis(diphenylphosphino)propane or (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane).
A variant which is advantageous for the industrial synthesis process consists of conversion of the compound of formula (IV) being carried out to form the compound of formula (I) directly, said sulphonylation reaction and said deoxygenation reaction in the presence of a transition metal being carried out as a “one-pot” procedure.
This process is especially advantageous for the following reasons:
-
- it makes it possible to obtain the compound of formula (I) on the industrial scale in good yields starting from a simple and low-cost starting material;
- it makes it possible to avoid an aromatisation reaction because the naphthalene ring system is present in the starting substrate;
- it makes it possible to obtain agomelatine starting from 7-methoxy-naphthalen-2-ol in a reduced number of steps.
The compounds of formula (V) obtained in accordance with the process of the invention are new and useful as intermediates in the synthesis of agomelatine and the compound of formula (I).
Preferred compounds of formula (V) are the following:
-
- 1-formyl-7-methoxynaphthalen-2-yl 4-methylbenzenesulphonate;
- 1-formyl-7-methoxynaphthalen-2-yl methanesulphonate.
The compound of formula (I) hereby obtained is subsequently subjected to a series of customary chemical reactions (for example: reduction of the aldehyde into a primary alcohol, cyanation, reduction and acetylation of the primary amine obtained) to yield agomelatine of formula (II).
The Examples hereinbelow illustrate the invention but do not limit it in any way. In order to properly validate the reaction routes, the synthesis intermediates were systematically isolated and characterised. However, it is possible to considerably optimise the procedures by limiting the number of intermediates isolated.
The structures of the compounds described were confirmed by the usual spectroscopic techniques: proton NMR (s=singlet; d=doublet; dd=doublet of doublets); carbon NMR (s=singlet; d=doublet; q=quadruplet).
7-Methoxy-naphthalen-2-ol (3.5 g; 20.11 mmol), ethyl orthoformate (3.51 mL; 21.12 mmol) and aniline (1.83 mL; 20.11 mmol) are introduced into a flask equipped with a condenser. After stirring for 20 hours at reflux and cooling, the solid is ground in a 2M ethanolic solution of hydrochloric acid (20 mL). After stirring for 30 minutes at 60° C. and cooling, the solid is collected by filtration and then washed with water and dried by azeotropic distillation with ethanol and used directly without any other purification (2.95 g; 73%).
1H NMR spectroscopic analysis (CDCl3, δ in ppm): 13.17 (s, 1H); 10.74 (s, 1H); 7.88 (d, J=9.1 Hz, 1H); 7.69 (d, J=8.9 Hz, 1H); 7.65 (d, J=2.4 Hz, 1H); 7.07 (dd, J=8.9 and 2.4 Hz, 1H); 6.97 (d, J=9.1 Hz, 1H); 3.95 (s, 3H).
To a solution of the product of Step A above (1 g; 4.95 mmol) in dichloromethane (20 mL) there are added triethylamine (826 μL; 5.94 mmol) and tosyl chloride (0.99 g; 5.2 mmol). After stirring for 24 hours, the solvent is evaporated off and then the residue is taken up in a mixture of water/ethyl acetate. The organic phase is washed with a dilute solution of hydrochloric acid, water and brine, and then dried over sodium sulphate and filtered. Evaporating off the solvents results in a crude product, which is purified by recrystallised from hot ethyl acetate to yield the title product (1.132 g; 65%).
Melting point: 147-148° C.
1H NMR spectroscopic analysis (CDCl3, δ in ppm): 10.41 (s, 1H); 8.68 (d, J=2.6 Hz, 1H); 7.95 (d, J=8.9 Hz, 1H); 7.74 (d, J=8.2 Hz, 2H); 7.72 (d, J=8.9 Hz, 1H); 7.33 (d, J=8.2 Hz, 2H); 7.19 (dd, J=8.9 and 2.6 Hz, 1H); 7.15 (d, J=8.9 Hz, 1H); 3.93 (s, 3H); 2.45 (s, 3H).
13C NMR spectroscopic analysis (CDCl3, δ in ppm): 190.3 (d); 161.5 (s); 154.3 (s); 146.4 (s); 136.4 (d); 132.8 (s); 131.5 (s); 130.3 (2×d); 129.9 (d); 128.6 (2×d); 127.8 (s); 121.5 (s); 120.1 (d); 118.6 (d); 104.1 (d); 55.6 (q); 21.9 (q).
The product of Step B above (356 mg; 1 mmol), palladium acetate (4.5 mg; 0.02 mmol), 1,3-bis(diphenylphosphino)propane (8.2 mg; 0.02 mmol), dimethylformamide (2 mL), triethylamine (556 μL; 4 mmol) and formic acid (150 μL; 4 mmol) are introduced into a flask placed in an oven and purged with argon. The flask is placed in a bath heated to 90° C. for 1.5 hours. After cooling, the mixture is diluted with ethyl acetate and the organic phase is washed with 1M aqueous hydrochloric acid solution and with brine, dried over sodium sulphate and filtered. After evaporating off the solvent, the crude product is purified by filtration over neutral alumina to yield the title product (139 mg; 75%).
Melting point: 65-67° C.
1H NMR spectroscopic analysis (CDCl3, 300.13 MHz, δ in ppm): 10.29 (s, 1H); 8.75 (d, J=2.6 Hz, 1H); 7.99 (d, J=8.1 Hz, 1H); 7.9 (d, J=7.1 Hz, 1H); 7.77 (d, J=8.9 Hz, 1H); 7.45 (dd, J=8.1 and 7.1 Hz, 1H); 7.23 (dd, J=8.9 and 2.6 Hz, 1H); 3.98 (s, 3H).
13C NMR spectroscopic analysis (CDCl3, 75.5 MHz, δ in ppm): 194.1 (d); 160.7 (s); 138.3 (d); 135.1 (d); 132.2 (s); 130.2 (s); 129.9 (d); 129.3 (s); 122.5 (d); 119.8 (d); 103.6 (d); 55.6 (q).
To a solution of the compound obtained in Step A of Example 1 (300 mg; 1.485 mmol) in dichloromethane (5 mL) there are added triethylamine (250 μL; 1.782 mmol) and mesyl chloride (120 μL). After stirring for one hour, the solvent is evaporated off and the residue is taken up in a mixture of ethyl acetate/water. The organic fraction is washed twice with water and then with brine, dried over sodium sulphate and filtered. Evaporating off the solvent yields the clean title product (416 mg; 95%) without the need for purification.
1H NMR spectroscopic analysis (CDCl3, δ in ppm): 10.74 (s, 1H); 8.72 (d, J=2.4 Hz, 1H); 8.03 (d, J=8.9 Hz, 1H); 7.75 (d, J=8.9 Hz, 1H); 7.36 (d, J=8.9 Hz, 1H); 7.22 (dd, J=8.9 and 2.4 Hz, 1H); 3.97 (s, 3H); 3.32 (s, 3H).
13C NMR spectroscopic analysis (CDCl3, δ in ppm): 190.4 (d); 161.6 (s); 153.2 (s); 136.8 (d); 133.1 (s); 130.0 (d); 128.0 (s); 121.6 (s); 120.3 (d); 118.2 (d); 104.0 (d); 55.7 (q); 38.5 (q).
The title product (84%) is obtained in accordance with the process described in Step C of Example 1 starting from the product of Step A above and with a reaction time of 4 hours at 90° C. instead of 1.5 hours.
Melting point: 65-67° C.
1H NMR spectroscopic analysis (CDCl3, 300.13 MHz, δ in ppm): 10.29 (s, 1H); 8.75 (d, J=2.6 Hz, 1H); 7.99 (d, J=8.1 Hz, 1H); 7.9 (d, J=7.1 Hz, 1H); 7.77 (d, J=8.9 Hz, 1H); 7.45 (dd, J=8.1 and 7.1 Hz, 1H); 7.23 (dd, J=8.9 and 2.6 Hz, 1H); 3.98 (s, 3H).
13C NMR spectroscopic analysis (CDCl3, 75.5 MHz, δ in ppm): 194.1 (d); 160.7 (s); 138.3 (d); 135.1 (d); 132.2 (s); 130.2 (s); 129.9 (d); 129.3 (s); 122.5 (d); 119.8 (d); 103.6 (d); 55.6 (q).
Sodium hydride (60%; 17 mg; 0.415 mmol) is added, in several portions, to a solution of 7-methoxy-naphthalen-2-ol (70 mg; 0.35 mmol) in anhydrous dimethylformamide (1 mL) in a flask purged with argon. After stirring for 30 minutes at ambient temperature, tosyl chloride is then added in several portions (190.5 mg; 0.36 mmol). After stirring for 4 hours at ambient temperature, 1,3-bis(diphenylphosphino)propane (7.1 mg; 0.017 mmol), palladium acetate (3.9 mg; 0.073 mmol), triethylamine (192 μL; 1.38 mmol) and formic acid (150 μL; 4 mmol) are added and the reaction mixture is heated at 90° C. for 1.5 hours. After cooling, the mixture is diluted with ethyl acetate and the organic phase is washed with 1M aqueous hydrochloric acid solution and then with brine, dried over sodium sulphate and filtered. After evaporating off the solvent, the crude product is filtered over neutral alumina (eluant: ethyl acetate) to yield the title product (61.6 mg; 95%).
Melting point: 65-67° C.
Claims (22)
1. A process for the synthesis of a compound of formula (I):
wherein 7-methoxy-naphthalen-2-ol of formula (III):

is used for reaction, the formyl group being introduced at position 1 to yield a compound of formula (IV):

which compound of formula (IV) is subjected to a sulphonylation reaction to yield a compound of formula (V):

wherein R represents —CH3, —(CH2)2—CH3, —CF3 or tolyl;
which compound of formula (V) undergoes a deoxygenation reaction in the presence of a transition metal and a reducing agent to yield the compound of formula (I), which is isolated in the form of a solid.
2. The process according to claim 1 , wherein R represents —CH3 or tolyl.
3. The process according to claim 1 , wherein the conversion of the compound of formula (IV) into the compound of formula (V) is carried out by means of the action of a sulphonyl chloride, a sulphonic anhydride or a sulphonimide.
4. The process according to claim 3 , wherein the conversion of the compound of formula (IV) into the compound of formula (V) is carried out by means of the action of a sulphonyl chloride.
5. The process according to claim 1 , wherein, in the conversion of the compound of formula (V) into the compound of formula (I), the transition metal is nickel, palladium or platinum.
6. The process according to claim 1 , wherein, in the conversion of the compound of formula (V) into the compound of formula (I), the transition metal is a palladium salt.
7. The process according to claim 1 , wherein the conversion of the compound of formula (V) into the compound of formula (I) is carried out in dimethylformamide, dioxane, tetrahydrofuran or toluene.
8. The process according to claim 7 , wherein the conversion of the compound of formula (V) into the compound of formula (I) is carried out in dimethylformamide.
9. The process according to claim 1 , wherein the conversion of the compound of formula (V) into the compound of formula (I) is carried out between 25° C. and 110° C.
10. The process according to claim 9 , wherein the conversion of the compound of formula (V) into the compound of formula (I) is carried out between 40° C. and 95° C.
11. The process according to claim 1 , wherein, in the conversion of the compound of formula (V) into the compound of formula (I), the reducing agent is dihydrogen.
12. The process according to claim 11 , wherein the dihydrogen is obtained by decomposition of an ammonium formate.
13. The process according to claim 1 , wherein the conversion of the compound of formula (V) into the compound of formula (I) is carried out in the presence of palladium and dihydrogen.
14. The process according to claim 1 , wherein the conversion of the compound of formula (V) into the compound of formula (I) is carried out in the presence of (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) or 1,3-bis(diphenylphosphino)propane.
15. A compound of formula (V):

wherein R represents —(CH2)2—CH3, —CF3 or tolyl.
16. A process for the synthesis of agomelatine employing a compound of formula (V)

wherein R represents —CH3, —(CH2)2—CH3, —CF3 or tolyl.
17. The compound according to claim 15 , which is selected from the following compounds:
1-formyl-7-methoxynaphthalen-2-yl 4-methylbenzenesulphonate and
1-formyl-7-methoxynaphthalen-2-yl methanesulphonate.
18. A process for the synthesis of the compound of formula (I):

employing a compound of formula (V)

wherein R represents —CH3, —(CH2)2—CH3, —CF3 or tolyl.
19. The process according to claim 18 , wherein the process further comprises subjecting the compound of formula (I) to a series of reactions to provide agomelatine.
20. A process for the synthesis of a compound of formula (I)
employing a compound of formula (III):

21. The process according to claim 20 , wherein the process further comprises subjecting the compound of formula (I) to a series of reactions to provide agomelatine.
22. The process according to claim 16 , wherein methoxy-naphthalen-2-ol of formula (III):

is used for reaction, and a formyl group is introduced at position 1 to yield a compound of formula (IV):

which compound of formula (IV) is subjected to a sulphonylation reaction to yield the compound of formula (V), which compound of formula (V) is subjected to a series of reactions to provide agomelatine.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR1362200 | 2013-12-05 | ||
| FR1362200A FR3014434B1 (en) | 2013-12-05 | 2013-12-05 | NOVEL PROCESS FOR THE SYNTHESIS OF 7-METHOXY-NAPHTHALENE-1-CARBALDEHYDE AND APPLICATION TO THE SYNTHESIS OF AGOMELATIN |
| PCT/FR2014/053159 WO2015082849A2 (en) | 2013-12-05 | 2014-12-04 | Novel method for the synthesis of 7-methoxy-naphthalene-1-carbaldehyde and use thereof in the synthesis of agomelatine |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20160304429A1 US20160304429A1 (en) | 2016-10-20 |
| US9701608B2 true US9701608B2 (en) | 2017-07-11 |
Family
ID=50482960
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/101,120 Active US9701608B2 (en) | 2013-12-05 | 2014-12-04 | Process for the synthesis of 7-methoxy-naphthalene-1-carbaldehyde and application in the synthesis of agomelatine |
Country Status (29)
| Country | Link |
|---|---|
| US (1) | US9701608B2 (en) |
| EP (1) | EP3077355B1 (en) |
| JP (1) | JP2016539169A (en) |
| KR (1) | KR20170018799A (en) |
| CN (1) | CN105793226B (en) |
| AU (1) | AU2014358967B2 (en) |
| CA (1) | CA2932196C (en) |
| CY (1) | CY1120160T1 (en) |
| DK (1) | DK3077355T3 (en) |
| EA (1) | EA031684B1 (en) |
| ES (1) | ES2668527T3 (en) |
| FR (1) | FR3014434B1 (en) |
| GE (1) | GEP20186847B (en) |
| HR (1) | HRP20180693T1 (en) |
| HU (1) | HUE036874T2 (en) |
| LT (1) | LT3077355T (en) |
| MA (1) | MA39062B1 (en) |
| MD (1) | MD20160072A2 (en) |
| ME (1) | ME03048B (en) |
| MX (1) | MX2016007130A (en) |
| NO (1) | NO3077355T3 (en) |
| PL (1) | PL3077355T3 (en) |
| PT (1) | PT3077355T (en) |
| RS (1) | RS56933B1 (en) |
| RU (1) | RU2680243C1 (en) |
| SI (1) | SI3077355T1 (en) |
| TR (1) | TR201802197T4 (en) |
| UA (1) | UA117940C2 (en) |
| WO (1) | WO2015082849A2 (en) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2918369A1 (en) | 2007-07-02 | 2009-01-09 | Servier Lab | New substituted naphthalene derivatives are melatonin 1 receptor binders useful e.g. to treat sleep disorders, stress, anxiety, schizophrenia, panic attacks, appetite disorders, obesity, insomnia, epilepsy, diabetes and Parkinson's disease |
| US8217032B2 (en) * | 2004-01-22 | 2012-07-10 | Eli Lilly And Company | Selective estrogen receptor modulators for the treatment of vasomotor symptoms |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2658818B1 (en) * | 1990-02-27 | 1993-12-31 | Adir Cie | NOVEL DERIVATIVES WITH NAPHTHALENIC STRUCTURE, PROCESS FOR THEIR PREPARATION AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| FR2866335B1 (en) | 2004-02-13 | 2006-05-26 | Servier Lab | NEW PROCESS FOR THE SYNTHESIS OF AGOMELATIN |
| FR2866337B1 (en) * | 2004-02-13 | 2006-05-26 | Servier Lab | NOVEL PROCESS FOR THE SYNTHESIS OF (7-METHOXY-1-NAPHTHYL) ACETONITRILE AND APPLICATION TO THE SYNTHESIS OF AGLOMELATIN |
| CN101638376B (en) * | 2008-07-29 | 2011-04-27 | 江苏恩华药业股份有限公司 | Method for preparing agomelatine and intermediate of agomelatine |
| FR2934859B1 (en) * | 2008-08-05 | 2010-08-13 | Servier Lab | NEW PROCESS FOR THE SYNTHESIS OF AGOMELATIN |
-
2013
- 2013-12-05 FR FR1362200A patent/FR3014434B1/en not_active Expired - Fee Related
-
2014
- 2014-04-12 UA UAA201606901A patent/UA117940C2/en unknown
- 2014-12-04 GE GEAP201414198A patent/GEP20186847B/en unknown
- 2014-12-04 MD MDA20160072A patent/MD20160072A2/en not_active Application Discontinuation
- 2014-12-04 LT LTEP14821796.1T patent/LT3077355T/en unknown
- 2014-12-04 US US15/101,120 patent/US9701608B2/en active Active
- 2014-12-04 ME MEP-2018-43A patent/ME03048B/en unknown
- 2014-12-04 AU AU2014358967A patent/AU2014358967B2/en not_active Ceased
- 2014-12-04 MX MX2016007130A patent/MX2016007130A/en unknown
- 2014-12-04 CA CA2932196A patent/CA2932196C/en not_active Expired - Fee Related
- 2014-12-04 HU HUE14821796A patent/HUE036874T2/en unknown
- 2014-12-04 CN CN201480066210.9A patent/CN105793226B/en not_active Expired - Fee Related
- 2014-12-04 RS RS20180247A patent/RS56933B1/en unknown
- 2014-12-04 DK DK14821796.1T patent/DK3077355T3/en active
- 2014-12-04 NO NO14821796A patent/NO3077355T3/no unknown
- 2014-12-04 ES ES14821796.1T patent/ES2668527T3/en active Active
- 2014-12-04 MA MA39062A patent/MA39062B1/en unknown
- 2014-12-04 PL PL14821796T patent/PL3077355T3/en unknown
- 2014-12-04 TR TR2018/02197T patent/TR201802197T4/en unknown
- 2014-12-04 RU RU2016126660A patent/RU2680243C1/en active
- 2014-12-04 WO PCT/FR2014/053159 patent/WO2015082849A2/en active Application Filing
- 2014-12-04 SI SI201430642T patent/SI3077355T1/en unknown
- 2014-12-04 EA EA201600439A patent/EA031684B1/en not_active IP Right Cessation
- 2014-12-04 PT PT148217961T patent/PT3077355T/en unknown
- 2014-12-04 EP EP14821796.1A patent/EP3077355B1/en active Active
- 2014-12-04 JP JP2016536761A patent/JP2016539169A/en not_active Ceased
- 2014-12-04 KR KR1020167017809A patent/KR20170018799A/en not_active Application Discontinuation
-
2018
- 2018-05-02 CY CY20181100451T patent/CY1120160T1/en unknown
- 2018-05-03 HR HRP20180693TT patent/HRP20180693T1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8217032B2 (en) * | 2004-01-22 | 2012-07-10 | Eli Lilly And Company | Selective estrogen receptor modulators for the treatment of vasomotor symptoms |
| FR2918369A1 (en) | 2007-07-02 | 2009-01-09 | Servier Lab | New substituted naphthalene derivatives are melatonin 1 receptor binders useful e.g. to treat sleep disorders, stress, anxiety, schizophrenia, panic attacks, appetite disorders, obesity, insomnia, epilepsy, diabetes and Parkinson's disease |
Non-Patent Citations (4)
| Title |
|---|
| Garigipati, et al., Bioorganic and Medicinal Chemistry Letters, vol. 7, No. 11, p. 1421-1426, Jun. 3, 1997. |
| International Preliminary Report for PCT/FR2014/053159 on Sep. 6, 2016. |
| International Search Report with Written Opinion for PCT/FR2014/053159 of Oct. 6, 2015. |
| Kandagatla, et al., Tetrahedron Letters, vol. 53, p. 7125-7127, Oct. 26, 2012. |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8212077B2 (en) | Process for the synthesis of agomelatine | |
| US8329947B2 (en) | Process for the synthesis of agomelatine | |
| US8436206B2 (en) | Process for the synthesis of (7-methoxy-1-naphthyl) acetonitrile and application in the synthesis of agomelatine | |
| BG64986B1 (en) | Method for the preparation of 5-cyanophthalide | |
| US8779199B2 (en) | Agomelatine intermediates and preparation method thereof | |
| US8143449B2 (en) | Process for the synthesis of agomelatine | |
| US9604911B2 (en) | Process for the synthesis of agomelatine | |
| US9701608B2 (en) | Process for the synthesis of 7-methoxy-naphthalene-1-carbaldehyde and application in the synthesis of agomelatine | |
| TWI438180B (en) | New process for the synthesis of agomelatine | |
| WO2015082848A2 (en) | Novel method for the synthesis of 7-methoxy-naphthalene-1-carbaldehyde and use thereof in the synthesis of agomelatine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: LES LABORATOIRES SERVIER, FRANCE Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:BRIERE, JEAN-FRANCOIS;LEBEUF, RAPHAEL;LEVACHER, VINCENT;AND OTHERS;REEL/FRAME:039578/0932 Effective date: 20160530 |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| CC | Certificate of correction | ||
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 4TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1551); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 4 |