US20220340613A1 - Cyclic di-nucleotide compounds and methods of use - Google Patents
Cyclic di-nucleotide compounds and methods of use Download PDFInfo
- Publication number
- US20220340613A1 US20220340613A1 US17/697,247 US202217697247A US2022340613A1 US 20220340613 A1 US20220340613 A1 US 20220340613A1 US 202217697247 A US202217697247 A US 202217697247A US 2022340613 A1 US2022340613 A1 US 2022340613A1
- Authority
- US
- United States
- Prior art keywords
- mmol
- compound
- alkyl
- amino
- added
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 59
- 125000004122 cyclic group Chemical group 0.000 title claims description 54
- 150000001875 compounds Chemical class 0.000 claims abstract description 240
- 150000003839 salts Chemical class 0.000 claims description 134
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 121
- 150000003573 thiols Chemical class 0.000 claims description 102
- 229910052736 halogen Inorganic materials 0.000 claims description 101
- 150000002367 halogens Chemical class 0.000 claims description 101
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 94
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 92
- 206010028980 Neoplasm Diseases 0.000 claims description 88
- -1 poly(ethylene glycol) Polymers 0.000 claims description 85
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 84
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 83
- 125000005113 hydroxyalkoxy group Chemical group 0.000 claims description 83
- 125000006619 (C1-C6) dialkylamino group Chemical group 0.000 claims description 81
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 77
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 claims description 73
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 72
- 201000011510 cancer Diseases 0.000 claims description 61
- 229910052739 hydrogen Inorganic materials 0.000 claims description 33
- 239000001257 hydrogen Substances 0.000 claims description 28
- 229920001223 polyethylene glycol Polymers 0.000 claims description 27
- 230000004913 activation Effects 0.000 claims description 9
- 230000037361 pathway Effects 0.000 claims description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 5
- 101710196623 Stimulator of interferon genes protein Proteins 0.000 claims 2
- 239000000203 mixture Substances 0.000 abstract description 261
- 239000008194 pharmaceutical composition Substances 0.000 abstract description 84
- 239000002773 nucleotide Substances 0.000 abstract description 6
- 238000013160 medical therapy Methods 0.000 abstract description 3
- 230000002194 synthesizing effect Effects 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 229
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 177
- 238000003756 stirring Methods 0.000 description 151
- 239000000243 solution Substances 0.000 description 140
- 235000002639 sodium chloride Nutrition 0.000 description 119
- 239000007787 solid Substances 0.000 description 114
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 109
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 100
- 238000010898 silica gel chromatography Methods 0.000 description 84
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 78
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 74
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 69
- 239000007864 aqueous solution Substances 0.000 description 64
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 57
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 47
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 46
- 238000011282 treatment Methods 0.000 description 46
- 239000004698 Polyethylene Substances 0.000 description 43
- 239000003814 drug Substances 0.000 description 43
- 239000003112 inhibitor Substances 0.000 description 42
- 239000012044 organic layer Substances 0.000 description 42
- 239000003795 chemical substances by application Substances 0.000 description 39
- 238000002360 preparation method Methods 0.000 description 37
- 0 [1*][C@@H]1C[C@H](C)[C@H]([3*])C1.[2*]C1CC(C)[C@@H]([5*])C[C@H]1[4*] Chemical compound [1*][C@@H]1C[C@H](C)[C@H]([3*])C1.[2*]C1CC(C)[C@@H]([5*])C[C@H]1[4*] 0.000 description 35
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 33
- 239000002246 antineoplastic agent Substances 0.000 description 33
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 32
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 30
- 229940124597 therapeutic agent Drugs 0.000 description 28
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 27
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 27
- RFCBNSCSPXMEBK-INFSMZHSSA-N c-GMP-AMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]3[C@@H](O)[C@H](N4C5=NC=NC(N)=C5N=C4)O[C@@H]3COP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 RFCBNSCSPXMEBK-INFSMZHSSA-N 0.000 description 26
- 201000010099 disease Diseases 0.000 description 26
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 25
- 108020004414 DNA Proteins 0.000 description 24
- 229940034982 antineoplastic agent Drugs 0.000 description 24
- 239000012267 brine Substances 0.000 description 24
- 239000012071 phase Substances 0.000 description 24
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 24
- 239000000427 antigen Substances 0.000 description 23
- 102000036639 antigens Human genes 0.000 description 23
- 108091007433 antigens Proteins 0.000 description 23
- 150000002431 hydrogen Chemical class 0.000 description 23
- 239000000546 pharmaceutical excipient Substances 0.000 description 23
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 22
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 22
- 101000643024 Homo sapiens Stimulator of interferon genes protein Proteins 0.000 description 22
- 102100035533 Stimulator of interferon genes protein Human genes 0.000 description 22
- 239000000843 powder Substances 0.000 description 22
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 19
- 230000000694 effects Effects 0.000 description 17
- 230000019491 signal transduction Effects 0.000 description 17
- 229960005215 dichloroacetic acid Drugs 0.000 description 16
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 15
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 15
- 239000010410 layer Substances 0.000 description 15
- 239000003921 oil Substances 0.000 description 15
- 235000017557 sodium bicarbonate Nutrition 0.000 description 15
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 15
- 239000000725 suspension Substances 0.000 description 15
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 14
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 14
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 14
- 239000011734 sodium Substances 0.000 description 14
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 13
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 13
- 239000002671 adjuvant Substances 0.000 description 13
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 13
- 235000019198 oils Nutrition 0.000 description 13
- 210000004881 tumor cell Anatomy 0.000 description 13
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 12
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 12
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 12
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 12
- 238000003786 synthesis reaction Methods 0.000 description 12
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 12
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 11
- 108091000080 Phosphotransferase Proteins 0.000 description 11
- 239000012530 fluid Substances 0.000 description 11
- 239000007788 liquid Substances 0.000 description 11
- 238000004519 manufacturing process Methods 0.000 description 11
- 102000020233 phosphotransferase Human genes 0.000 description 11
- 239000003826 tablet Substances 0.000 description 11
- 239000003039 volatile agent Substances 0.000 description 11
- BHVPSGYYQHPKFP-ROHFMWTOSA-N *.*.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound *.*.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 BHVPSGYYQHPKFP-ROHFMWTOSA-N 0.000 description 10
- VHPJEIGORCRPDZ-FHGLABGWSA-N *.*.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound *.*.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 VHPJEIGORCRPDZ-FHGLABGWSA-N 0.000 description 10
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 10
- 102100031256 Cyclic GMP-AMP synthase Human genes 0.000 description 10
- 101710118064 Cyclic GMP-AMP synthase Proteins 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 10
- 229910004373 HOAc Inorganic materials 0.000 description 10
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 230000015572 biosynthetic process Effects 0.000 description 10
- 229910052799 carbon Inorganic materials 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 230000022131 cell cycle Effects 0.000 description 10
- 230000005855 radiation Effects 0.000 description 10
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 10
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 9
- 239000000908 ammonium hydroxide Substances 0.000 description 9
- 229910000085 borane Inorganic materials 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 239000003085 diluting agent Substances 0.000 description 9
- 235000019439 ethyl acetate Nutrition 0.000 description 9
- 229940088597 hormone Drugs 0.000 description 9
- 239000005556 hormone Substances 0.000 description 9
- 210000001331 nose Anatomy 0.000 description 9
- 229910052711 selenium Inorganic materials 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 229910052717 sulfur Inorganic materials 0.000 description 9
- 238000002560 therapeutic procedure Methods 0.000 description 9
- 229960005486 vaccine Drugs 0.000 description 9
- PQPIVQWIPINCPT-FHGLABGWSA-N *.*.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound *.*.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 PQPIVQWIPINCPT-FHGLABGWSA-N 0.000 description 8
- 208000035473 Communicable disease Diseases 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 8
- NVOTUSOAWRDABP-LOTMFFQUSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 NVOTUSOAWRDABP-LOTMFFQUSA-N 0.000 description 8
- 230000009286 beneficial effect Effects 0.000 description 8
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 8
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 230000002163 immunogen Effects 0.000 description 8
- 239000002105 nanoparticle Substances 0.000 description 8
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 8
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 8
- JBWYRBLDOOOJEU-UHFFFAOYSA-N 1-[chloro-(4-methoxyphenyl)-phenylmethyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC(OC)=CC=1)C1=CC=CC=C1 JBWYRBLDOOOJEU-UHFFFAOYSA-N 0.000 description 7
- 102000009465 Growth Factor Receptors Human genes 0.000 description 7
- 108010009202 Growth Factor Receptors Proteins 0.000 description 7
- 241000282414 Homo sapiens Species 0.000 description 7
- WEVYAHXRMPXWCK-FIBGUPNXSA-N acetonitrile-d3 Chemical compound [2H]C([2H])([2H])C#N WEVYAHXRMPXWCK-FIBGUPNXSA-N 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- 239000006260 foam Substances 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 210000003928 nasal cavity Anatomy 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 239000003755 preservative agent Substances 0.000 description 7
- 102000005962 receptors Human genes 0.000 description 7
- 108020003175 receptors Proteins 0.000 description 7
- 230000011664 signaling Effects 0.000 description 7
- 229940124931 vaccine adjuvant Drugs 0.000 description 7
- 239000012646 vaccine adjuvant Substances 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- BQDFHACIOUBBDF-UHFFFAOYSA-N 3-(8-chloro-2,3-dimethoxy-10-phenothiazinyl)-N,N-dimethyl-1-propanamine Chemical compound S1C2=CC=C(Cl)C=C2N(CCCN(C)C)C2=C1C=C(OC)C(OC)=C2 BQDFHACIOUBBDF-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- XRILCFTWUCUKJR-GJDXYYIKSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 XRILCFTWUCUKJR-GJDXYYIKSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 6
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 6
- 229940122803 Vinca alkaloid Drugs 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 230000030833 cell death Effects 0.000 description 6
- 230000008859 change Effects 0.000 description 6
- 150000004141 diterpene derivatives Chemical class 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 230000012010 growth Effects 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 230000028993 immune response Effects 0.000 description 6
- 230000001939 inductive effect Effects 0.000 description 6
- 210000004072 lung Anatomy 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 230000002265 prevention Effects 0.000 description 6
- NRTYMEPCRDJMPZ-UHFFFAOYSA-N pyridine;2,2,2-trifluoroacetic acid Chemical compound C1=CC=NC=C1.OC(=O)C(F)(F)F NRTYMEPCRDJMPZ-UHFFFAOYSA-N 0.000 description 6
- 238000001959 radiotherapy Methods 0.000 description 6
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 6
- 239000012312 sodium hydride Substances 0.000 description 6
- 229910000104 sodium hydride Inorganic materials 0.000 description 6
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 6
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- VOEOIPMHJGHRNC-MDKWFLGWSA-N *.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O Chemical compound *.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O VOEOIPMHJGHRNC-MDKWFLGWSA-N 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 5
- 206010006187 Breast cancer Diseases 0.000 description 5
- 208000026310 Breast neoplasm Diseases 0.000 description 5
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 5
- HVMZSPLJAJJQQD-RCRHYTMJSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 HVMZSPLJAJJQQD-RCRHYTMJSA-N 0.000 description 5
- 229910019142 PO4 Inorganic materials 0.000 description 5
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 5
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 5
- 239000000443 aerosol Substances 0.000 description 5
- 229940100198 alkylating agent Drugs 0.000 description 5
- 239000002168 alkylating agent Substances 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 239000011230 binding agent Substances 0.000 description 5
- 229940127089 cytotoxic agent Drugs 0.000 description 5
- 230000001419 dependent effect Effects 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 239000003937 drug carrier Substances 0.000 description 5
- 239000012091 fetal bovine serum Substances 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000000796 flavoring agent Substances 0.000 description 5
- 230000003054 hormonal effect Effects 0.000 description 5
- 208000026278 immune system disease Diseases 0.000 description 5
- 239000004615 ingredient Substances 0.000 description 5
- 102000037979 non-receptor tyrosine kinases Human genes 0.000 description 5
- 108091008046 non-receptor tyrosine kinases Proteins 0.000 description 5
- 244000052769 pathogen Species 0.000 description 5
- 229910052697 platinum Inorganic materials 0.000 description 5
- 238000002953 preparative HPLC Methods 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 230000001737 promoting effect Effects 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 5
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 5
- 239000011669 selenium Substances 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 239000008107 starch Substances 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 5
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 4
- XGLVDUUYFKXKPL-UHFFFAOYSA-N 2-(2-methoxyethoxy)-n,n-bis[2-(2-methoxyethoxy)ethyl]ethanamine Chemical compound COCCOCCN(CCOCCOC)CCOCCOC XGLVDUUYFKXKPL-UHFFFAOYSA-N 0.000 description 4
- QWTBDIBOOIAZEF-UHFFFAOYSA-N 3-[chloro-[di(propan-2-yl)amino]phosphanyl]oxypropanenitrile Chemical compound CC(C)N(C(C)C)P(Cl)OCCC#N QWTBDIBOOIAZEF-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- 108010074708 B7-H1 Antigen Proteins 0.000 description 4
- 102000008096 B7-H1 Antigen Human genes 0.000 description 4
- OBGWDQZBROYYQL-FQKFWMNRSA-N C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 OBGWDQZBROYYQL-FQKFWMNRSA-N 0.000 description 4
- HGGYXKHFLGFOJP-WSDAMQLNSA-N CCO[C@@H]1C2OP(=O)(S)OC[C@H]3O[C@@H](n4ccc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(S)OC[C@H]3O[C@@H](n4ccc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 HGGYXKHFLGFOJP-WSDAMQLNSA-N 0.000 description 4
- MCMOOSBZOLVSMO-KFLZFNJTSA-N CP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@@H]2CC(O1)[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 Chemical compound CP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@@H]2CC(O1)[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 MCMOOSBZOLVSMO-KFLZFNJTSA-N 0.000 description 4
- XZCVEQXNLDAJPE-QPTCJMCJSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 XZCVEQXNLDAJPE-QPTCJMCJSA-N 0.000 description 4
- 102000001301 EGF receptor Human genes 0.000 description 4
- 108060006698 EGF receptor Proteins 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- 101001011382 Homo sapiens Interferon regulatory factor 3 Proteins 0.000 description 4
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 4
- 102000002227 Interferon Type I Human genes 0.000 description 4
- 108010014726 Interferon Type I Proteins 0.000 description 4
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Substances IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 4
- NIZKBRRCHBUIKM-MQVIZUGASA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 NIZKBRRCHBUIKM-MQVIZUGASA-N 0.000 description 4
- BMPJFJCUDFHNST-RCRHYTMJSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@@H](O)[C@@H](COP(=O)(O)OC3[C@@H]2O)O[C@H]4n2ccc3c(N)ncnc32)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@@H](O)[C@@H](COP(=O)(O)OC3[C@@H]2O)O[C@H]4n2ccc3c(N)ncnc32)c(=O)[nH]1 BMPJFJCUDFHNST-RCRHYTMJSA-N 0.000 description 4
- LTGWLWDFLBDKHQ-JLYFVSOXSA-N Nc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 LTGWLWDFLBDKHQ-JLYFVSOXSA-N 0.000 description 4
- LTQXSWOHMBWBJW-GXKBJAMNSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 LTQXSWOHMBWBJW-GXKBJAMNSA-N 0.000 description 4
- UCFPSVAEYVIGOC-KENIHOTESA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 UCFPSVAEYVIGOC-KENIHOTESA-N 0.000 description 4
- MIOBZNRITVVGDB-MDKWFLGWSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 MIOBZNRITVVGDB-MDKWFLGWSA-N 0.000 description 4
- WYLZVEUIJAQMPZ-PQLDZLGPSA-N Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(O)OC3[C@H](O)[C@H](n4ccc5c(N)ncnc54)O[C@@H]3COP(=O)(S)OC1[C@H]2O Chemical compound Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(O)OC3[C@H](O)[C@H](n4ccc5c(N)ncnc54)O[C@@H]3COP(=O)(S)OC1[C@H]2O WYLZVEUIJAQMPZ-PQLDZLGPSA-N 0.000 description 4
- 102000014400 SH2 domains Human genes 0.000 description 4
- 108050003452 SH2 domains Proteins 0.000 description 4
- 102000000395 SH3 domains Human genes 0.000 description 4
- 108050008861 SH3 domains Proteins 0.000 description 4
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical group [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- PYMYPHUHKUWMLA-LMVFSUKVSA-N aldehydo-D-ribose Chemical compound OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 4
- 239000004037 angiogenesis inhibitor Substances 0.000 description 4
- 230000000340 anti-metabolite Effects 0.000 description 4
- 229940100197 antimetabolite Drugs 0.000 description 4
- 239000002256 antimetabolite Substances 0.000 description 4
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 4
- 239000006172 buffering agent Substances 0.000 description 4
- 230000004700 cellular uptake Effects 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- 230000001086 cytosolic effect Effects 0.000 description 4
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 4
- 150000002009 diols Chemical class 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 235000013355 food flavoring agent Nutrition 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011187 glycerol Nutrition 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 150000004677 hydrates Chemical class 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- 230000001717 pathogenic effect Effects 0.000 description 4
- 239000010452 phosphate Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 108700042226 ras Genes Proteins 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 238000004007 reversed phase HPLC Methods 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- 239000004094 surface-active agent Substances 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 4
- XRILCFTWUCUKJR-INFSMZHSSA-N 2'-3'-cGAMP Chemical compound C([C@H]([C@H]1O)O2)OP(O)(=O)O[C@H]3[C@@H](O)[C@H](N4C5=NC=NC(N)=C5N=C4)O[C@@H]3COP(O)(=O)O[C@H]1[C@@H]2N1C=NC2=C1NC(N)=NC2=O XRILCFTWUCUKJR-INFSMZHSSA-N 0.000 description 3
- GVZJRBAUSGYWJI-UHFFFAOYSA-N 2,5-bis(3-dodecylthiophen-2-yl)thiophene Chemical compound C1=CSC(C=2SC(=CC=2)C2=C(C=CS2)CCCCCCCCCCCC)=C1CCCCCCCCCCCC GVZJRBAUSGYWJI-UHFFFAOYSA-N 0.000 description 3
- 208000011594 Autoinflammatory disease Diseases 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- FCAHNMGTSNUTLR-UHFFFAOYSA-N C.CC(C)Cc1cn(CC(C)C)nn1.CC(C)Cn1cc(COC(C)C)nn1 Chemical compound C.CC(C)Cc1cn(CC(C)C)nn1.CC(C)Cn1cc(COC(C)C)nn1 FCAHNMGTSNUTLR-UHFFFAOYSA-N 0.000 description 3
- NXXZIIPBSMWIQX-ZPUBDCECSA-N CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 NXXZIIPBSMWIQX-ZPUBDCECSA-N 0.000 description 3
- OPZCDJODFCYBKD-SNYGBICDSA-N CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1O Chemical compound CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1O OPZCDJODFCYBKD-SNYGBICDSA-N 0.000 description 3
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 3
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 3
- 230000006820 DNA synthesis Effects 0.000 description 3
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 3
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 3
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 3
- 102000014150 Interferons Human genes 0.000 description 3
- 108010050904 Interferons Proteins 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 102000029749 Microtubule Human genes 0.000 description 3
- 108091022875 Microtubule Proteins 0.000 description 3
- XCOBLONWWXQEBS-KPKJPENVSA-N N,O-bis(trimethylsilyl)trifluoroacetamide Chemical compound C[Si](C)(C)O\C(C(F)(F)F)=N\[Si](C)(C)C XCOBLONWWXQEBS-KPKJPENVSA-N 0.000 description 3
- AKLHSFPTGFZFCL-DDYBOZTLSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5ccc6c(=O)[nH]c(N)nc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5ccc6c(=O)[nH]c(N)nc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 AKLHSFPTGFZFCL-DDYBOZTLSA-N 0.000 description 3
- GTWMOORQETVGGV-RXWLUYGTSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O)c(=O)[nH]1 GTWMOORQETVGGV-RXWLUYGTSA-N 0.000 description 3
- 206010060862 Prostate cancer Diseases 0.000 description 3
- 108091005682 Receptor kinases Proteins 0.000 description 3
- 230000018199 S phase Effects 0.000 description 3
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 3
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 3
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 3
- 102000004243 Tubulin Human genes 0.000 description 3
- 108090000704 Tubulin Proteins 0.000 description 3
- 102100029823 Tyrosine-protein kinase BTK Human genes 0.000 description 3
- 238000002441 X-ray diffraction Methods 0.000 description 3
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 3
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 3
- 229940044684 anti-microtubule agent Drugs 0.000 description 3
- 229940041181 antineoplastic drug Drugs 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 230000001363 autoimmune Effects 0.000 description 3
- 208000037979 autoimmune inflammatory disease Diseases 0.000 description 3
- 235000012216 bentonite Nutrition 0.000 description 3
- 239000000440 bentonite Substances 0.000 description 3
- 229910000278 bentonite Inorganic materials 0.000 description 3
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 3
- 230000003115 biocidal effect Effects 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 238000010549 co-Evaporation Methods 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 235000008504 concentrate Nutrition 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 239000006184 cosolvent Substances 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- RAFNCPHFRHZCPS-UHFFFAOYSA-N di(imidazol-1-yl)methanethione Chemical compound C1=CN=CN1C(=S)N1C=CN=C1 RAFNCPHFRHZCPS-UHFFFAOYSA-N 0.000 description 3
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000006196 drop Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 229940029575 guanosine Drugs 0.000 description 3
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 3
- 229940047124 interferons Drugs 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000003211 malignant effect Effects 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 210000004688 microtubule Anatomy 0.000 description 3
- 230000011278 mitosis Effects 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 210000002345 respiratory system Anatomy 0.000 description 3
- 229910001961 silver nitrate Inorganic materials 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 229960002920 sorbitol Drugs 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- 239000001993 wax Substances 0.000 description 3
- KWUUETQWUHYXKZ-KFQTYFLOSA-N *.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1.S Chemical compound *.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1.S KWUUETQWUHYXKZ-KFQTYFLOSA-N 0.000 description 2
- GZTYTTPPCAXUHB-UHFFFAOYSA-N 1,2-benzodithiol-3-one Chemical compound C1=CC=C2C(=O)SSC2=C1 GZTYTTPPCAXUHB-UHFFFAOYSA-N 0.000 description 2
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- CFIBTBBTJWHPQV-UHFFFAOYSA-N 2-methyl-n-(6-oxo-3,7-dihydropurin-2-yl)propanamide Chemical compound N1C(NC(=O)C(C)C)=NC(=O)C2=C1N=CN2 CFIBTBBTJWHPQV-UHFFFAOYSA-N 0.000 description 2
- DGMOBVGABMBZSB-UHFFFAOYSA-N 2-methylpropanoyl chloride Chemical compound CC(C)C(Cl)=O DGMOBVGABMBZSB-UHFFFAOYSA-N 0.000 description 2
- WRMNZCZEMHIOCP-UHFFFAOYSA-N 2-phenylethanol Chemical compound OCCC1=CC=CC=C1 WRMNZCZEMHIOCP-UHFFFAOYSA-N 0.000 description 2
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 2
- CFKMVGJGLGKFKI-UHFFFAOYSA-N 4-chloro-m-cresol Chemical compound CC1=CC(O)=CC=C1Cl CFKMVGJGLGKFKI-UHFFFAOYSA-N 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 2
- 206010000830 Acute leukaemia Diseases 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 102100022014 Angiopoietin-1 receptor Human genes 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- KHNGOJZZLHWTMC-LETJFLAOSA-N BB(B)B(B)B.Cc1nc2c(ccn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1 Chemical compound BB(B)B(B)B.Cc1nc2c(ccn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1 KHNGOJZZLHWTMC-LETJFLAOSA-N 0.000 description 2
- KFMVLWLFEZUPHW-LXLBQDSMSA-N BB.Cc1nc2c(cnn2[C@@H]2O[C@H](CO)C(O)[C@@H]2O)c(=O)[nH]1 Chemical compound BB.Cc1nc2c(cnn2[C@@H]2O[C@H](CO)C(O)[C@@H]2O)c(=O)[nH]1 KFMVLWLFEZUPHW-LXLBQDSMSA-N 0.000 description 2
- TZLGDGGRQRZDOU-BUFGSZFNSA-N BB.O=C(Nc1ncnc2c1nnn2[C@@H]1O[C@H](CO)[C@H](O)C1O)c1ccccc1 Chemical compound BB.O=C(Nc1ncnc2c1nnn2[C@@H]1O[C@H](CO)[C@H](O)C1O)c1ccccc1 TZLGDGGRQRZDOU-BUFGSZFNSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- QFOHBWFCKVYLES-UHFFFAOYSA-N Butylparaben Chemical compound CCCCOC(=O)C1=CC=C(O)C=C1 QFOHBWFCKVYLES-UHFFFAOYSA-N 0.000 description 2
- NQXASBNBPUCFHR-RFMLDLTKSA-N C#CB(C#C)C#C.OC[C@H]1O[C@@H](c2cccc3ccccc23)C[C@H]1O Chemical compound C#CB(C#C)C#C.OC[C@H]1O[C@@H](c2cccc3ccccc23)C[C@H]1O NQXASBNBPUCFHR-RFMLDLTKSA-N 0.000 description 2
- JCBIYUSFPHXOOE-JFEVQYLNSA-N C#CCO[C@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)C(OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)[C@H]3O Chemical compound C#CCO[C@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)C(OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)[C@H]3O JCBIYUSFPHXOOE-JFEVQYLNSA-N 0.000 description 2
- LMLPEUMAMSPCKI-MBYUAGNBSA-N C#CCOc1nc(N)nc2c1ncn2[C@@H]1O[C@@H]2COP(=O)(O)OC3[C@H](O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)OC1[C@H]2O Chemical compound C#CCOc1nc(N)nc2c1ncn2[C@@H]1O[C@@H]2COP(=O)(O)OC3[C@H](O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)OC1[C@H]2O LMLPEUMAMSPCKI-MBYUAGNBSA-N 0.000 description 2
- LDVNLVPCODHISR-VKUYVQLPSA-N C#CCOc1nc(N)nc2c1ncn2[C@@H]1O[C@@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@H]1C2O.CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(C)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3)c(=O)[nH]1.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP([BH3-])(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O Chemical compound C#CCOc1nc(N)nc2c1ncn2[C@@H]1O[C@@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@H]1C2O.CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(C)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3)c(=O)[nH]1.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP([BH3-])(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O LDVNLVPCODHISR-VKUYVQLPSA-N 0.000 description 2
- JHEDEBBSRIRIFG-NZKFWXDFSA-N C.C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CC)[C@H](C)C1OP(C)N(C(C)C)C(C)C Chemical compound C.C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CC)[C@H](C)C1OP(C)N(C(C)C)C(C)C JHEDEBBSRIRIFG-NZKFWXDFSA-N 0.000 description 2
- OQHCXPAVJDYPOJ-FQLIIPEFSA-N C.CC[C@@H]1CC(OP(C)N(C(C)C)C(C)C)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1 Chemical compound C.CC[C@@H]1CC(OP(C)N(C(C)C)C(C)C)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1 OQHCXPAVJDYPOJ-FQLIIPEFSA-N 0.000 description 2
- CFTDJXZLTQNNJZ-BBVUDJRFSA-N C.CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C Chemical compound C.CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C CFTDJXZLTQNNJZ-BBVUDJRFSA-N 0.000 description 2
- PHZJADRTTVDLIH-IWVXOWFHSA-N C.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C Chemical compound C.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C PHZJADRTTVDLIH-IWVXOWFHSA-N 0.000 description 2
- NQJYNHWZJBJYTD-KAMJXADWSA-N C.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C Chemical compound C.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C NQJYNHWZJBJYTD-KAMJXADWSA-N 0.000 description 2
- IBMKWRHXVGNSHH-APHJIHOJSA-N C.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C Chemical compound C.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C IBMKWRHXVGNSHH-APHJIHOJSA-N 0.000 description 2
- OGDQPDCLAPYLIF-QJZNNZKGSA-N C.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C Chemical compound C.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C OGDQPDCLAPYLIF-QJZNNZKGSA-N 0.000 description 2
- MPWOPYYYOXYMPU-VVRVNSBJSA-N C.[H]P(=O)(O)OC1C[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.[H]P(=O)(O)OC1C[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 MPWOPYYYOXYMPU-VVRVNSBJSA-N 0.000 description 2
- XBCJRXUMFINSQA-DJNVOPTESA-N C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CO Chemical compound C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CO XBCJRXUMFINSQA-DJNVOPTESA-N 0.000 description 2
- QRFQPHZBOIOVTL-DEGJWMOCSA-N C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CO Chemical compound C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CO QRFQPHZBOIOVTL-DEGJWMOCSA-N 0.000 description 2
- HGRBRTTVGKPDPV-UHFFFAOYSA-N CC(C)Cc1cn(CC(C)C)nn1.CC(C)Cn1cc(COC(C)C)nn1 Chemical compound CC(C)Cc1cn(CC(C)C)nn1.CC(C)Cn1cc(COC(C)C)nn1 HGRBRTTVGKPDPV-UHFFFAOYSA-N 0.000 description 2
- QWOJMRHUQHTCJG-UHFFFAOYSA-N CC([CH2-])=O Chemical compound CC([CH2-])=O QWOJMRHUQHTCJG-UHFFFAOYSA-N 0.000 description 2
- FTGZQZBPFSNNNG-PRLVFJKXSA-N CCCC1[C@H](OC(C)=O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CC Chemical compound CCCC1[C@H](OC(C)=O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CC FTGZQZBPFSNNNG-PRLVFJKXSA-N 0.000 description 2
- KFVCMRVFKGOAQO-JNCVGDKRSA-N CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](c4cccc5ccccc45)CC3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](c4cccc5ccccc45)CC3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 KFVCMRVFKGOAQO-JNCVGDKRSA-N 0.000 description 2
- PURATGKXEOHGJI-WDGKGQDKSA-N CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4ccc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4ccc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 PURATGKXEOHGJI-WDGKGQDKSA-N 0.000 description 2
- BAFAILBNKNMEPP-MCFSLQMXSA-N CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4ccc5ccccc54)CC3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4ccc5ccccc54)CC3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 BAFAILBNKNMEPP-MCFSLQMXSA-N 0.000 description 2
- RONPGFNPLFLTBO-ABMWOVQLSA-N CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21 RONPGFNPLFLTBO-ABMWOVQLSA-N 0.000 description 2
- RRRZRFWNLXVXNU-GVEVCTRBSA-N CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](C)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](C)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 RRRZRFWNLXVXNU-GVEVCTRBSA-N 0.000 description 2
- RJFBFZZWNDRNIP-NLEPYMBVSA-N CCO[C@@H]1C2OP(=O)(S)OC[C@H]3O[C@@H](n4ccc5c(N)ncnc54)[C@@H](O)C3OP(=O)(S)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(S)OC[C@H]3O[C@@H](n4ccc5c(N)ncnc54)[C@@H](O)C3OP(=O)(S)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 RJFBFZZWNDRNIP-NLEPYMBVSA-N 0.000 description 2
- NWYUBDYFZAWYJW-BCLRGNAFSA-N CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N)[C@H]1C Chemical compound CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N)[C@H]1C NWYUBDYFZAWYJW-BCLRGNAFSA-N 0.000 description 2
- MBDRXLJZENEYOB-PKDUTCDESA-N CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1OC Chemical compound CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1OC MBDRXLJZENEYOB-PKDUTCDESA-N 0.000 description 2
- HIMFPZVJOLMQGQ-FWKUWYBHSA-N CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2CC1C Chemical compound CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2CC1C HIMFPZVJOLMQGQ-FWKUWYBHSA-N 0.000 description 2
- CJCDFXWLBXPIFC-MSIMALESSA-N C[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound C[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 CJCDFXWLBXPIFC-MSIMALESSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- CLRVKDJSLJULQJ-IJXPTTLFSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@@H](O)[C@@H](COP(=O)(S)O[C@H]2C3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@@H](O)[C@@H](COP(=O)(S)O[C@H]2C3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@@H](O)[C@@H](COP(=O)(O)O[C@H]2C3OCC(F)(F)F)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@@H](O)[C@@H](COP(=O)(S)O[C@H]2C3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@@H](O)[C@@H](COP(=O)(S)O[C@H]2C3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@@H](O)[C@@H](COP(=O)(O)O[C@H]2C3OCC(F)(F)F)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1 CLRVKDJSLJULQJ-IJXPTTLFSA-N 0.000 description 2
- PLGNXLRRNBEAPU-YCJRFTQYSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(=O)[nH]c(N)nc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@H]4C(O)[C@@H](COP(=O)(O)O[C@@H]3C2O)O[C@H]4n2ccc3c(N)ccnc32)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(=O)[nH]c(N)nc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@H]4C(O)[C@@H](COP(=O)(O)O[C@@H]3C2O)O[C@H]4n2ccc3c(N)ccnc32)c(=O)[nH]1 PLGNXLRRNBEAPU-YCJRFTQYSA-N 0.000 description 2
- GEJPZXPDKKWPEY-AAGHADDOSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 GEJPZXPDKKWPEY-AAGHADDOSA-N 0.000 description 2
- IRZDJWPMCQNZNE-PNDMDGRVSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@H]4C(O)[C@@H](COP(=O)(O)O[C@@H]3C2O)O[C@H]4n2ccc3c(N)ccnc32)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@H]4C(O)[C@@H](COP(=O)(O)O[C@@H]3C2O)O[C@H]4n2ccc3c(N)ccnc32)c(=O)[nH]1 IRZDJWPMCQNZNE-PNDMDGRVSA-N 0.000 description 2
- GPSTXXMQCXDPML-SPANPNNGSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 GPSTXXMQCXDPML-SPANPNNGSA-N 0.000 description 2
- XVYMPIOJOOLIKP-SNMPAJOKSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(NCCO)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(NCCO)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 XVYMPIOJOOLIKP-SNMPAJOKSA-N 0.000 description 2
- YDIQMYDEYQOMRP-UTWRNRKDSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 YDIQMYDEYQOMRP-UTWRNRKDSA-N 0.000 description 2
- YESFRWJCJFACBA-MOLHOFDHSA-N Cc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 YESFRWJCJFACBA-MOLHOFDHSA-N 0.000 description 2
- PKWPUYJDGVXBET-ZMVNUZMWSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)O[C@H]2C3O)c(=O)[nH]1.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)O[C@H]2C3O)c(=O)[nH]1.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O PKWPUYJDGVXBET-ZMVNUZMWSA-N 0.000 description 2
- GJMFWSFIHBJNEH-NYQAICHPSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(C)(=O)O[C@H]2C3)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(C)(=O)O[C@H]2C3)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(C)(=O)O[C@H]2C3)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(C)(=O)O[C@H]2C3)c(=O)[nH]1 GJMFWSFIHBJNEH-NYQAICHPSA-N 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 208000004449 DNA Virus Infections Diseases 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000007317 Farnesyltranstransferase Human genes 0.000 description 2
- 108010007508 Farnesyltranstransferase Proteins 0.000 description 2
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 2
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 2
- 102000016621 Focal Adhesion Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108010067715 Focal Adhesion Protein-Tyrosine Kinases Proteins 0.000 description 2
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 description 2
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 description 2
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 2
- 101000834898 Homo sapiens Alpha-synuclein Proteins 0.000 description 2
- 101000753291 Homo sapiens Angiopoietin-1 receptor Proteins 0.000 description 2
- 101000611936 Homo sapiens Programmed cell death protein 1 Proteins 0.000 description 2
- 101000652359 Homo sapiens Spermatogenesis-associated protein 2 Proteins 0.000 description 2
- 101000864342 Homo sapiens Tyrosine-protein kinase BTK Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 108030003815 Inositol 3-kinases Proteins 0.000 description 2
- 102100029843 Interferon regulatory factor 3 Human genes 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 239000005089 Luciferase Substances 0.000 description 2
- 108010058398 Macrophage Colony-Stimulating Factor Receptor Proteins 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 101150111783 NTRK1 gene Proteins 0.000 description 2
- 101150117329 NTRK3 gene Proteins 0.000 description 2
- ZWKUWBHNJVBBRL-XFPWQAPDSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 ZWKUWBHNJVBBRL-XFPWQAPDSA-N 0.000 description 2
- NJAORQKAJZSNOX-NXKMACRTSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(NCCO)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(NCCO)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 NJAORQKAJZSNOX-NXKMACRTSA-N 0.000 description 2
- PPRDYPVGZIHATQ-UKIGVWQRSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@H]4C(O)[C@@H](COP(=O)(S)OC2[C@H]3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@H]4C(O)[C@@H](COP(=O)(S)OC2[C@H]3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1 PPRDYPVGZIHATQ-UKIGVWQRSA-N 0.000 description 2
- RFDHWTXDKIGDJD-YAQCIAJZSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4C[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4C[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 RFDHWTXDKIGDJD-YAQCIAJZSA-N 0.000 description 2
- UCTYAYGKEVAKRJ-LVJKKOKJSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)NC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)NC2[C@H]3O)c(=O)[nH]1 UCTYAYGKEVAKRJ-LVJKKOKJSA-N 0.000 description 2
- UCQKSXCNYMPTRH-FLLSPZAJSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2C3)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2C3)c(=O)[nH]1 UCQKSXCNYMPTRH-FLLSPZAJSA-N 0.000 description 2
- YGPYEXZAPJWGGH-OPHCBYCYSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3OCO)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3OCO)c(=O)[nH]1 YGPYEXZAPJWGGH-OPHCBYCYSA-N 0.000 description 2
- PYOLGYGHVZCQAJ-FXCUJRLGSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cc4cn(nn4)[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cc4cn(nn4)[C@H]2C3O)c(=O)[nH]1 PYOLGYGHVZCQAJ-FXCUJRLGSA-N 0.000 description 2
- XJRAFYDRKPMVDU-SNGCHKTOSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@H]4C(O)[C@@H](COP(=O)(O)OC2[C@H]3OCC(F)(F)F)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@H]4C(O)[C@@H](COP(=O)(O)OC2[C@H]3OCC(F)(F)F)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1 XJRAFYDRKPMVDU-SNGCHKTOSA-N 0.000 description 2
- AZOJZAVVRXFKRZ-PXCTXCFFSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@H]4C(O)[C@@H](COP(=O)(S)OC2[C@H]3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@H]4C(O)[C@@H](COP(=O)(S)OC2[C@H]3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1 AZOJZAVVRXFKRZ-PXCTXCFFSA-N 0.000 description 2
- JJWOITPMMIENLI-MDKWFLGWSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 JJWOITPMMIENLI-MDKWFLGWSA-N 0.000 description 2
- ZGZUCWLVVNSRDJ-JLYFVSOXSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 ZGZUCWLVVNSRDJ-JLYFVSOXSA-N 0.000 description 2
- QTBCBMYLPDHQOM-ZKAQCFDUSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5nnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5nnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 QTBCBMYLPDHQOM-ZKAQCFDUSA-N 0.000 description 2
- 101150056950 Ntrk2 gene Proteins 0.000 description 2
- 108091008606 PDGF receptors Proteins 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- 108091008611 Protein Kinase B Proteins 0.000 description 2
- 108090000315 Protein Kinase C Proteins 0.000 description 2
- 102000003923 Protein Kinase C Human genes 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 101100313260 Schizosaccharomyces pombe (strain 972 / ATCC 24843) tea3 gene Proteins 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 102000004357 Transferases Human genes 0.000 description 2
- 108090000992 Transferases Proteins 0.000 description 2
- 108091008605 VEGF receptors Proteins 0.000 description 2
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 2
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 2
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 2
- 240000008042 Zea mays Species 0.000 description 2
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 2
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 2
- PAKRGPXAQBMIBM-JGURTLBNSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3O1)C2O.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3O1)C2O.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O PAKRGPXAQBMIBM-JGURTLBNSA-N 0.000 description 2
- QCZMVVOZESZHJC-MDKWFLGWSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3O1)[C@H]2O Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3O1)[C@H]2O QCZMVVOZESZHJC-MDKWFLGWSA-N 0.000 description 2
- YMIIBMNOLHFVQM-GQNCLNMLSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(OP([BH3-])(=O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3O1)[C@H]2O Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(OP([BH3-])(=O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3O1)[C@H]2O YMIIBMNOLHFVQM-GQNCLNMLSA-N 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 229930183665 actinomycin Natural products 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 102000035181 adaptor proteins Human genes 0.000 description 2
- 108091005764 adaptor proteins Proteins 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 2
- 150000008052 alkyl sulfonates Chemical class 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 229940046836 anti-estrogen Drugs 0.000 description 2
- 230000001833 anti-estrogenic effect Effects 0.000 description 2
- 230000000118 anti-neoplastic effect Effects 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 210000000612 antigen-presenting cell Anatomy 0.000 description 2
- 239000004599 antimicrobial Substances 0.000 description 2
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000000074 antisense oligonucleotide Substances 0.000 description 2
- 238000012230 antisense oligonucleotides Methods 0.000 description 2
- 230000005975 antitumor immune response Effects 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical class N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 201000008275 breast carcinoma Diseases 0.000 description 2
- PKFDLKSEZWEFGL-MHARETSRSA-N c-di-GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]3[C@@H](O)[C@H](N4C5=C(C(NC(N)=N5)=O)N=C4)O[C@@H]3COP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 PKFDLKSEZWEFGL-MHARETSRSA-N 0.000 description 2
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 2
- 235000013539 calcium stearate Nutrition 0.000 description 2
- 239000008116 calcium stearate Substances 0.000 description 2
- 229940127093 camptothecin Drugs 0.000 description 2
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 229920003123 carboxymethyl cellulose sodium Polymers 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 229940105329 carboxymethylcellulose Drugs 0.000 description 2
- 229940063834 carboxymethylcellulose sodium Drugs 0.000 description 2
- XMPZTFVPEKAKFH-UHFFFAOYSA-P ceric ammonium nitrate Chemical compound [NH4+].[NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O XMPZTFVPEKAKFH-UHFFFAOYSA-P 0.000 description 2
- OGEBRHQLRGFBNV-RZDIXWSQSA-N chembl2036808 Chemical compound C12=NC(NCCCC)=NC=C2C(C=2C=CC(F)=CC=2)=NN1C[C@H]1CC[C@H](N)CC1 OGEBRHQLRGFBNV-RZDIXWSQSA-N 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- KQIADDMXRMTWHZ-UHFFFAOYSA-N chloro-tri(propan-2-yl)silane Chemical compound CC(C)[Si](Cl)(C(C)C)C(C)C KQIADDMXRMTWHZ-UHFFFAOYSA-N 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 235000005822 corn Nutrition 0.000 description 2
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- KUMNEOGIHFCNQW-UHFFFAOYSA-N diphenyl phosphite Chemical compound C=1C=CC=CC=1OP([O-])OC1=CC=CC=C1 KUMNEOGIHFCNQW-UHFFFAOYSA-N 0.000 description 2
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 2
- 229910000397 disodium phosphate Inorganic materials 0.000 description 2
- 210000004696 endometrium Anatomy 0.000 description 2
- 239000000328 estrogen antagonist Substances 0.000 description 2
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 2
- 229940126864 fibroblast growth factor Drugs 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 229940028334 follicle stimulating hormone Drugs 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 125000002686 geranylgeranyl group Chemical group [H]C([*])([H])/C([H])=C(C([H])([H])[H])/C([H])([H])C([H])([H])/C([H])=C(C([H])([H])[H])/C([H])([H])C([H])([H])/C([H])=C(C([H])([H])[H])/C([H])([H])C([H])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 2
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- AFQIYTIJXGTIEY-UHFFFAOYSA-N hydrogen carbonate;triethylazanium Chemical compound OC(O)=O.CCN(CC)CC AFQIYTIJXGTIEY-UHFFFAOYSA-N 0.000 description 2
- 230000007124 immune defense Effects 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 239000002955 immunomodulating agent Substances 0.000 description 2
- 238000009169 immunotherapy Methods 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- 239000000411 inducer Substances 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229960000367 inositol Drugs 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 108020001756 ligand binding domains Proteins 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007523 nucleic acids Chemical group 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 150000004713 phosphodiesters Chemical class 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- 230000000861 pro-apoptotic effect Effects 0.000 description 2
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- BBEAQIROQSPTKN-UHFFFAOYSA-N pyrene Chemical compound C1=CC=C2C=CC3=CC=CC4=CC=C1C2=C43 BBEAQIROQSPTKN-UHFFFAOYSA-N 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000021014 regulation of cell growth Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 235000019204 saccharin Nutrition 0.000 description 2
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 2
- 229940081974 saccharin Drugs 0.000 description 2
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 2
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 2
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 2
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 2
- 238000009491 slugging Methods 0.000 description 2
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 2
- 235000010234 sodium benzoate Nutrition 0.000 description 2
- 239000004299 sodium benzoate Substances 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 239000001488 sodium phosphate Substances 0.000 description 2
- 229910000162 sodium phosphate Inorganic materials 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 235000010199 sorbic acid Nutrition 0.000 description 2
- 239000004334 sorbic acid Substances 0.000 description 2
- 229940075582 sorbic acid Drugs 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 239000006068 taste-masking agent Substances 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 150000003536 tetrazoles Chemical class 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 238000001890 transfection Methods 0.000 description 2
- 150000004654 triazenes Chemical class 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 229940124676 vascular endothelial growth factor receptor Drugs 0.000 description 2
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 2
- 229960002066 vinorelbine Drugs 0.000 description 2
- 239000000230 xanthan gum Substances 0.000 description 2
- 229920001285 xanthan gum Polymers 0.000 description 2
- 235000010493 xanthan gum Nutrition 0.000 description 2
- 229940082509 xanthan gum Drugs 0.000 description 2
- 235000010447 xylitol Nutrition 0.000 description 2
- 239000000811 xylitol Substances 0.000 description 2
- 229960002675 xylitol Drugs 0.000 description 2
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 2
- NOOLISFMXDJSKH-UTLUCORTSA-N (+)-Neomenthol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O NOOLISFMXDJSKH-UTLUCORTSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- DSSYKIVIOFKYAU-XCBNKYQSSA-N (R)-camphor Chemical compound C1C[C@@]2(C)C(=O)C[C@@H]1C2(C)C DSSYKIVIOFKYAU-XCBNKYQSSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- HCZDHZUYQHPVOL-FCZQXEIOSA-N *.*.*.*.*.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@@H](O)[C@@H](COP(=O)(S)O[C@H]2C3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.S Chemical compound *.*.*.*.*.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@@H](O)[C@@H](COP(=O)(S)O[C@H]2C3O)O[C@H]4n2cnc3c(N)ncnc32)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.S HCZDHZUYQHPVOL-FCZQXEIOSA-N 0.000 description 1
- KHDANRMCDVAPPL-UWGXXXKKSA-N *.*.*.*.*.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.S.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O Chemical compound *.*.*.*.*.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.S.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O KHDANRMCDVAPPL-UWGXXXKKSA-N 0.000 description 1
- UNZBUSNUAMBLPH-HLUZHKLHSA-N *.*.*.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.S.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O Chemical compound *.*.*.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.S.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O UNZBUSNUAMBLPH-HLUZHKLHSA-N 0.000 description 1
- ADXTXDBUFOGNSY-BYYCBPJZSA-N *.*.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound *.*.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 ADXTXDBUFOGNSY-BYYCBPJZSA-N 0.000 description 1
- VDCFKLMPKBPVPD-IPZLXZRKSA-N *.*.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(=O)[nH]cnc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)ncnc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CC(F)(F)P(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)C[C@H]2C3O)c(=O)[nH]1 Chemical compound *.*.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(=O)[nH]cnc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)ncnc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CC(F)(F)P(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)C[C@H]2C3O)c(=O)[nH]1 VDCFKLMPKBPVPD-IPZLXZRKSA-N 0.000 description 1
- BITDZJGTOUYDEV-MPIPNPPBSA-N *.*.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.S.S Chemical compound *.*.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.S.S BITDZJGTOUYDEV-MPIPNPPBSA-N 0.000 description 1
- BHVPSGYYQHPKFP-LTQOJAQDSA-N *.*.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound *.*.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 BHVPSGYYQHPKFP-LTQOJAQDSA-N 0.000 description 1
- QTPPPXLNYGCCQJ-HAMFXCLTSA-N *.CNP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)cccc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N(C)C)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(N)(=O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound *.CNP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)cccc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N(C)C)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(N)(=O)O[C@H]2C3O)c(=O)[nH]1 QTPPPXLNYGCCQJ-HAMFXCLTSA-N 0.000 description 1
- NIZKBRRCHBUIKM-WDLCLOIXSA-N *.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound *.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 NIZKBRRCHBUIKM-WDLCLOIXSA-N 0.000 description 1
- VPASTJUSNHXJKW-XYXLKDEHSA-N *.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(=O)[nH]cnc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound *.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(=O)[nH]cnc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 VPASTJUSNHXJKW-XYXLKDEHSA-N 0.000 description 1
- JHSMMEAUZBQBCU-GRHSDFQJSA-N *.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)cccc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound *.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)cccc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 JHSMMEAUZBQBCU-GRHSDFQJSA-N 0.000 description 1
- IDRYETHYNAVVLA-XYXLKDEHSA-N *.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)ncnc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound *.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)ncnc56)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 IDRYETHYNAVVLA-XYXLKDEHSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- TWQVZFCNOMQQQV-UHFFFAOYSA-N 1,2,4-dithiazole-3-thione Chemical group S=C1N=CSS1 TWQVZFCNOMQQQV-UHFFFAOYSA-N 0.000 description 1
- JCIIKRHCWVHVFF-UHFFFAOYSA-N 1,2,4-thiadiazol-5-amine;hydrochloride Chemical compound Cl.NC1=NC=NS1 JCIIKRHCWVHVFF-UHFFFAOYSA-N 0.000 description 1
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 1
- HBQSFSHZXMBAKL-UHFFFAOYSA-N 1-chlorosulfonyloxy-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(OS(Cl)(=O)=O)C=C1 HBQSFSHZXMBAKL-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- RTMMSCJWQYWMNK-UHFFFAOYSA-N 2,2,2-trifluoroethyl trifluoromethanesulfonate Chemical compound FC(F)(F)COS(=O)(=O)C(F)(F)F RTMMSCJWQYWMNK-UHFFFAOYSA-N 0.000 description 1
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- IKMXRUOZUUKSON-UHFFFAOYSA-N 2-(4-nitrophenyl)ethanol Chemical compound OCCC1=CC=C([N+]([O-])=O)C=C1 IKMXRUOZUUKSON-UHFFFAOYSA-N 0.000 description 1
- OEICGMPRFOJHKO-UHFFFAOYSA-N 2-(ethoxymethylidene)propanedinitrile Chemical compound CCOC=C(C#N)C#N OEICGMPRFOJHKO-UHFFFAOYSA-N 0.000 description 1
- YJYAGNPMQVHYAH-UHFFFAOYSA-N 2-[tert-butyl(dimethyl)silyl]oxyethanol Chemical compound CC(C)(C)[Si](C)(C)OCCO YJYAGNPMQVHYAH-UHFFFAOYSA-N 0.000 description 1
- VOXBZHOHGGBLCQ-UHFFFAOYSA-N 2-amino-3,7-dihydropurine-6-thione;hydrate Chemical compound O.N1C(N)=NC(=S)C2=C1N=CN2.N1C(N)=NC(=S)C2=C1N=CN2 VOXBZHOHGGBLCQ-UHFFFAOYSA-N 0.000 description 1
- VZMUCIBBVMLEKC-UHFFFAOYSA-N 2-chloro-5,5-dimethyl-1,3,2$l^{5}-dioxaphosphinane 2-oxide Chemical compound CC1(C)COP(Cl)(=O)OC1 VZMUCIBBVMLEKC-UHFFFAOYSA-N 0.000 description 1
- QSKPIOLLBIHNAC-UHFFFAOYSA-N 2-chloro-acetaldehyde Chemical compound ClCC=O QSKPIOLLBIHNAC-UHFFFAOYSA-N 0.000 description 1
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 description 1
- XNPUQBBDBSSZQX-UHFFFAOYSA-M 2-methyl-1-tetradecylpyridin-1-ium;chloride Chemical compound [Cl-].CCCCCCCCCCCCCC[N+]1=CC=CC=C1C XNPUQBBDBSSZQX-UHFFFAOYSA-M 0.000 description 1
- ZZVDXRCAGGQFAK-UHFFFAOYSA-N 2h-oxazaphosphinine Chemical class N1OC=CC=P1 ZZVDXRCAGGQFAK-UHFFFAOYSA-N 0.000 description 1
- WUACDRFRFTWMHE-UHFFFAOYSA-N 3,4-diaminocyclobut-3-ene-1,2-dione Chemical compound NC1=C(N)C(=O)C1=O WUACDRFRFTWMHE-UHFFFAOYSA-N 0.000 description 1
- SZBNZTGCAMLMJY-UHFFFAOYSA-N 3,4-dimethoxycyclobut-3-ene-1,2-dione Chemical compound COC1=C(OC)C(=O)C1=O SZBNZTGCAMLMJY-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- HRDCVMSNCBAMAM-UHFFFAOYSA-N 3-prop-2-ynoxyprop-1-yne Chemical compound C#CCOCC#C HRDCVMSNCBAMAM-UHFFFAOYSA-N 0.000 description 1
- 238000004679 31P NMR spectroscopy Methods 0.000 description 1
- NIGDWBHWHVHOAD-UHFFFAOYSA-N 4,6-dichloropyrimidin-5-amine Chemical compound NC1=C(Cl)N=CN=C1Cl NIGDWBHWHVHOAD-UHFFFAOYSA-N 0.000 description 1
- BPTCCCTWWAUJRK-UHFFFAOYSA-N 4-chloro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=NC=NC2=C1C=CN2 BPTCCCTWWAUJRK-UHFFFAOYSA-N 0.000 description 1
- SGOOQMRIPALTEL-UHFFFAOYSA-N 4-hydroxy-N,1-dimethyl-2-oxo-N-phenyl-3-quinolinecarboxamide Chemical compound OC=1C2=CC=CC=C2N(C)C(=O)C=1C(=O)N(C)C1=CC=CC=C1 SGOOQMRIPALTEL-UHFFFAOYSA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 1
- XZLIYCQRASOFQM-UHFFFAOYSA-N 5h-imidazo[4,5-d]triazine Chemical compound N1=NC=C2NC=NC2=N1 XZLIYCQRASOFQM-UHFFFAOYSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- WBZFUFAFFUEMEI-UHFFFAOYSA-M Acesulfame k Chemical compound [K+].CC1=CC(=O)[N-]S(=O)(=O)O1 WBZFUFAFFUEMEI-UHFFFAOYSA-M 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 108010029445 Agammaglobulinaemia Tyrosine Kinase Proteins 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 102400000068 Angiostatin Human genes 0.000 description 1
- 108010079709 Angiostatins Proteins 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- YCGIKTWOEXFANB-FPBMXWMCSA-N B.C.CC.CC.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO[P+]([BH3-])(C)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound B.C.CC.CC.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO[P+]([BH3-])(C)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 YCGIKTWOEXFANB-FPBMXWMCSA-N 0.000 description 1
- YAGVOXPLOFJVRE-VEBCUZQZSA-N B.CC(C)(C)[Si](C)(C)Cl.CC1(C)OC2C(O)O[C@H](CO)[C@@H]2O1.CC1(C)OC2[C@@H](O1)[C@@H](CO)O[C@H]2n1ccc2c(N)ncnc21.CCN(CC)CC.CC[C@H]1OC(O)C2OC(C)(C)O[C@H]21.CC[C@H]1O[C@@H](n2ccc3c(Cl)ncnc32)C2OC(C)(C)O[C@H]21.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C2OC(C)(C)O[C@H]21.Clc1ncnc2[nH]ccc12.O=C(Nc1ncnc2c1ccn2[C@@H]1O[C@H](CO)[C@H](O)C1O)c1ccccc1.O=S(=O)(O)O.OC[C@H]1OC(O)[C@@H](O)C1O Chemical compound B.CC(C)(C)[Si](C)(C)Cl.CC1(C)OC2C(O)O[C@H](CO)[C@@H]2O1.CC1(C)OC2[C@@H](O1)[C@@H](CO)O[C@H]2n1ccc2c(N)ncnc21.CCN(CC)CC.CC[C@H]1OC(O)C2OC(C)(C)O[C@H]21.CC[C@H]1O[C@@H](n2ccc3c(Cl)ncnc32)C2OC(C)(C)O[C@H]21.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C2OC(C)(C)O[C@H]21.Clc1ncnc2[nH]ccc12.O=C(Nc1ncnc2c1ccn2[C@@H]1O[C@H](CO)[C@H](O)C1O)c1ccccc1.O=S(=O)(O)O.OC[C@H]1OC(O)[C@@H](O)C1O YAGVOXPLOFJVRE-VEBCUZQZSA-N 0.000 description 1
- VIDSPBGUWLWOSH-MQHJEUPDSA-N B.O=C(Nc1ncnc2c1ccn2[C@@H]1O[C@H](CO)[C@H](O)C1O)c1ccccc1 Chemical compound B.O=C(Nc1ncnc2c1ccn2[C@@H]1O[C@H](CO)[C@H](O)C1O)c1ccccc1 VIDSPBGUWLWOSH-MQHJEUPDSA-N 0.000 description 1
- KHNGOJZZLHWTMC-VNZAFQORSA-N BB(B)B(B)B.Cc1nc2c(ccn2[C@@H]2O[C@H](CO)C(O)[C@@H]2O)c(=O)[nH]1 Chemical compound BB(B)B(B)B.Cc1nc2c(ccn2[C@@H]2O[C@H](CO)C(O)[C@@H]2O)c(=O)[nH]1 KHNGOJZZLHWTMC-VNZAFQORSA-N 0.000 description 1
- OQSFSYVBUULSGB-AHBUDECNSA-N BB(B)B.CO[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO Chemical compound BB(B)B.CO[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO OQSFSYVBUULSGB-AHBUDECNSA-N 0.000 description 1
- WQJZFYFAFCEUHR-LCUCIILCSA-N BB(B)C(c1ccccc1)(c1ccc(OC)cc1)c1ccc(OC)cc1.CC[C@@H]1CC(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1 Chemical compound BB(B)C(c1ccccc1)(c1ccc(OC)cc1)c1ccc(OC)cc1.CC[C@@H]1CC(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1 WQJZFYFAFCEUHR-LCUCIILCSA-N 0.000 description 1
- BDIZMVXHTXDWFF-IRBLJYCHSA-N BB.C.C.CC(C)(C)[Si](C)(C)Cl.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)C(O)[C@H]1C.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)C(O)[C@H]1O.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C.COc1ccc(C(Cl)(c2ccccc2)c2ccc(OC)cc2)cc1.Cc1nc2c(cnn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1.O=[N+]([O-])O[Ag].[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ncc2c(=O)[nH]c(C)nc21 Chemical compound BB.C.C.CC(C)(C)[Si](C)(C)Cl.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)C(O)[C@H]1C.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)C(O)[C@H]1O.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C.COc1ccc(C(Cl)(c2ccccc2)c2ccc(OC)cc2)cc1.Cc1nc2c(cnn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1.O=[N+]([O-])O[Ag].[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ncc2c(=O)[nH]c(C)nc21 BDIZMVXHTXDWFF-IRBLJYCHSA-N 0.000 description 1
- KFMVLWLFEZUPHW-STXPVALESA-N BB.Cc1nc2c(cnn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1 Chemical compound BB.Cc1nc2c(cnn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1 KFMVLWLFEZUPHW-STXPVALESA-N 0.000 description 1
- VGTMUDYWOREQNZ-JRWLJZFTSA-M BBB(B)B.C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CC)[C@H](OC(C)=O)C1OC(C)=O.C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CO)[C@H](O)C1O.C=CCBr.CC(=O)OC(C)=O.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OC(C)=O)[C@H]1OC(C)=O.Cc1nc2c(ncn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1.O[Na] Chemical compound BBB(B)B.C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CC)[C@H](OC(C)=O)C1OC(C)=O.C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CO)[C@H](O)C1O.C=CCBr.CC(=O)OC(C)=O.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OC(C)=O)[C@H]1OC(C)=O.Cc1nc2c(ncn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1.O[Na] VGTMUDYWOREQNZ-JRWLJZFTSA-M 0.000 description 1
- KDEMVSCMHQLNHD-WEYDQNNMSA-N BBB(B)B.C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CO)[C@H](O)C1O Chemical compound BBB(B)B.C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CO)[C@H](O)C1O KDEMVSCMHQLNHD-WEYDQNNMSA-N 0.000 description 1
- LJBBMHKMBGORHN-NXRNPVCNSA-N BC.C#CCOC1[C@@H](O)[C@@H](CO)O[C@H]1n1cnc2c(NC(=O)c3ccccc3)ncnc21 Chemical compound BC.C#CCOC1[C@@H](O)[C@@H](CO)O[C@H]1n1cnc2c(NC(=O)c3ccccc3)ncnc21 LJBBMHKMBGORHN-NXRNPVCNSA-N 0.000 description 1
- JUFJAXANSQXRSR-NVXOOKNLSA-N BC.C.C.C#CCBr.C#CCO[C@H]1C(O)[C@@H](CO)O[C@H]1n1cnc2c(N)ncnc21.C#CCO[C@H]1C(O)[C@@H](CO)O[C@H]1n1cnc2c(NC(=O)c3ccccc3)ncnc21.N.Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O.O Chemical compound BC.C.C.C#CCBr.C#CCO[C@H]1C(O)[C@@H](CO)O[C@H]1n1cnc2c(N)ncnc21.C#CCO[C@H]1C(O)[C@@H](CO)O[C@H]1n1cnc2c(NC(=O)c3ccccc3)ncnc21.N.Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O.O JUFJAXANSQXRSR-NVXOOKNLSA-N 0.000 description 1
- XUMHYWNUXRDXJO-LUTDUAEUSA-N BC.C.C.C.C.C.C=O.CC1(C)OC2[C@H](O1)[C@H](n1cnc3c(NC(=O)c4ccccc4)ncnc31)O[C@@H]2CN=[N+]=[N-].CC1(C)OC2[C@H](O1)[C@H](n1cnc3c(NC(=O)c4ccccc4)ncnc31)O[C@@H]2CO.COC.COS(=O)(=O)OC.NC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O)C1O.O=C(Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 Chemical compound BC.C.C.C.C.C.C=O.CC1(C)OC2[C@H](O1)[C@H](n1cnc3c(NC(=O)c4ccccc4)ncnc31)O[C@@H]2CN=[N+]=[N-].CC1(C)OC2[C@H](O1)[C@H](n1cnc3c(NC(=O)c4ccccc4)ncnc31)O[C@@H]2CO.COC.COS(=O)(=O)OC.NC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O)C1O.O=C(Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 XUMHYWNUXRDXJO-LUTDUAEUSA-N 0.000 description 1
- TUMBBNIYDFKIKD-XMGVGMEQSA-N BC.C.C.C.C.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1.O=c1ssc2ccccc12.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21 Chemical compound BC.C.C.C.C.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1.Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1.O=c1ssc2ccccc12.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21 TUMBBNIYDFKIKD-XMGVGMEQSA-N 0.000 description 1
- XXEGWHGZWVZENV-AGLGHCLDSA-M BC.C.C.CC[C@H]1OC(OC(C)=O)[C@@H](OC(=O)c2ccccc2)C1OC(=O)c1ccccc1.CC[C@H]1O[C@@H](n2ncc3c(N)ncnc32)[C@@H](OC(=O)c2ccccc2)C1OC(=O)c1ccccc1.CC[C@H]1O[C@@H](n2ncc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](OC(=O)c2ccccc2)C1OC(=O)c1ccccc1.COc1c(Cl)ncnc1Cl.Clc1ncnc2[nH]ncc12.N.NN.Nc1ncnc2[nH]ncc12.O.O=C(Cl)c1ccccc1.O=C(Nc1ncnc2c1cnn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1.[Li]O Chemical compound BC.C.C.CC[C@H]1OC(OC(C)=O)[C@@H](OC(=O)c2ccccc2)C1OC(=O)c1ccccc1.CC[C@H]1O[C@@H](n2ncc3c(N)ncnc32)[C@@H](OC(=O)c2ccccc2)C1OC(=O)c1ccccc1.CC[C@H]1O[C@@H](n2ncc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](OC(=O)c2ccccc2)C1OC(=O)c1ccccc1.COc1c(Cl)ncnc1Cl.Clc1ncnc2[nH]ncc12.N.NN.Nc1ncnc2[nH]ncc12.O.O=C(Cl)c1ccccc1.O=C(Nc1ncnc2c1cnn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1.[Li]O XXEGWHGZWVZENV-AGLGHCLDSA-M 0.000 description 1
- RROQUCUJEOXLOF-FQKFWMNRSA-N BC.Cc1nc2c(ncn2[C@@H]2O[C@H](CO)[C@H](C)C2O)c(=O)[nH]1 Chemical compound BC.Cc1nc2c(ncn2[C@@H]2O[C@H](CO)[C@H](C)C2O)c(=O)[nH]1 RROQUCUJEOXLOF-FQKFWMNRSA-N 0.000 description 1
- IZQQOQFQSZODBG-VSCDLRQESA-N BC.NC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O)[C@H]1O Chemical compound BC.NC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O)[C@H]1O IZQQOQFQSZODBG-VSCDLRQESA-N 0.000 description 1
- IZQQOQFQSZODBG-KGEQFWMKSA-N BC.NC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O)C1O Chemical compound BC.NC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O)C1O IZQQOQFQSZODBG-KGEQFWMKSA-N 0.000 description 1
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 206010006143 Brain stem glioma Diseases 0.000 description 1
- CASOAIGICNSQFR-MUHHGIEPSA-N Brc1ccc2ccc3cccc4ccc1c2c34.C.C.C.CB(C)C.CC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)CC1C.CO.CO[Na].OC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)CC1O Chemical compound Brc1ccc2ccc3cccc4ccc1c2c34.C.C.C.CB(C)C.CC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)CC1C.CO.CO[Na].OC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)CC1O CASOAIGICNSQFR-MUHHGIEPSA-N 0.000 description 1
- ZSLRBJFLRZBZOE-ZBPDOEELSA-N C#C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N)[C@H]1C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(Nc2c(OC)c(=O)c2=O)[C@H]1C.COc1c(C)c(=O)c1=O.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCc1c(NC2[C@@H](C)[C@@H](CO)O[C@H]2n2cnc3c(C)nc(C)nc32)c(=O)c1=O Chemical compound C#C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N)[C@H]1C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(Nc2c(OC)c(=O)c2=O)[C@H]1C.COc1c(C)c(=O)c1=O.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCc1c(NC2[C@@H](C)[C@@H](CO)O[C@H]2n2cnc3c(C)nc(C)nc32)c(=O)c1=O ZSLRBJFLRZBZOE-ZBPDOEELSA-N 0.000 description 1
- KRXWYBDPUJWFMT-UIHWPTKESA-N C#C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCc1c(NC2[C@@H](C)[C@@H](CO)O[C@H]2n2cnc3c(C)nc(C)nc32)c(=O)c1=O Chemical compound C#C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCc1c(NC2[C@@H](C)[C@@H](CO)O[C@H]2n2cnc3c(C)nc(C)nc32)c(=O)c1=O KRXWYBDPUJWFMT-UIHWPTKESA-N 0.000 description 1
- XHKALRCYGKBHST-RDXZFXAWSA-N C#C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1COP(C)(=S)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C#C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1COP(C)(=S)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 XHKALRCYGKBHST-RDXZFXAWSA-N 0.000 description 1
- QPMRRAKBBGWIDG-RDLXJQSLSA-N C#CB(C#C)C#CC.CCC[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO Chemical compound C#CB(C#C)C#CC.CCC[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO QPMRRAKBBGWIDG-RDLXJQSLSA-N 0.000 description 1
- XSIDZHTXMJKSFV-JNBPBJDWSA-N C#CB(C#C)C#CC.CCC[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(NCC(C)C)nc32)O[C@@H]1CO Chemical compound C#CB(C#C)C#CC.CCC[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(NCC(C)C)nc32)O[C@@H]1CO XSIDZHTXMJKSFV-JNBPBJDWSA-N 0.000 description 1
- YLMDWFKCKFTRLI-ZZQBSQCGSA-N C#CB(C)C#C.Cc1nc(C)c2ncn([C@@H]3O[C@H](CO)[C@H](O)C3N=[N+]=[N-])c2n1 Chemical compound C#CB(C)C#C.Cc1nc(C)c2ncn([C@@H]3O[C@H](CO)[C@H](O)C3N=[N+]=[N-])c2n1 YLMDWFKCKFTRLI-ZZQBSQCGSA-N 0.000 description 1
- HKERMEVTJBHRGC-RDLXJQSLSA-N C#CB(C)C.CCC[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO Chemical compound C#CB(C)C.CCC[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO HKERMEVTJBHRGC-RDLXJQSLSA-N 0.000 description 1
- QNLQWFJKAHCUPJ-BIPOCJNZSA-N C#CB(C)C.CCC[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(O)[C@H]1CCO[Si](C(C)C)(C(C)C)C(C)C.COc1ccc(C(Cl)(c2ccccc2)c2ccc(OC)cc2)cc1.[H]P(=O)(O)OC1[C@@H](CCO[Si](C(C)C)(C(C)C)C(C)C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C#CB(C)C.CCC[C@@H]1C(O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(O)[C@H]1CCO[Si](C(C)C)(C(C)C)C(C)C.COc1ccc(C(Cl)(c2ccccc2)c2ccc(OC)cc2)cc1.[H]P(=O)(O)OC1[C@@H](CCO[Si](C(C)C)(C(C)C)C(C)C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 QNLQWFJKAHCUPJ-BIPOCJNZSA-N 0.000 description 1
- OMTZMAXQCLLUNS-VXMWBYSOSA-N C#CC.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N)[C@H]1C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(NS(=O)(=O)Oc2ccc([N+](=O)[O-])cc2)[C@H]1C.O=[N+]([O-])c1ccc(OS(=O)(=O)Cl)cc1.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCS(=O)(=O)NC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(C)nc(C)nc21 Chemical compound C#CC.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N)[C@H]1C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(NS(=O)(=O)Oc2ccc([N+](=O)[O-])cc2)[C@H]1C.O=[N+]([O-])c1ccc(OS(=O)(=O)Cl)cc1.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCS(=O)(=O)NC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(C)nc(C)nc21 OMTZMAXQCLLUNS-VXMWBYSOSA-N 0.000 description 1
- KVIHTXDWGRNOLZ-NMUBBIOJSA-N C#CC.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C#CC.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 KVIHTXDWGRNOLZ-NMUBBIOJSA-N 0.000 description 1
- JEOFHRIIWZIGMY-LCVVHCRQSA-N C#CC.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCS(=O)(=O)NC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(C)nc(C)nc21 Chemical compound C#CC.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCS(=O)(=O)NC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(C)nc(C)nc21 JEOFHRIIWZIGMY-LCVVHCRQSA-N 0.000 description 1
- ZVAROBSXUGSBAK-HOAMCTTLSA-N C#CCOC1NC(N)=Nc2c1ncn2[C@@H]1O[C@@H]2COP(=O)(S)OC3[C@H](O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(S)OC1[C@H]2O Chemical compound C#CCOC1NC(N)=Nc2c1ncn2[C@@H]1O[C@@H]2COP(=O)(S)OC3[C@H](O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(S)OC1[C@H]2O ZVAROBSXUGSBAK-HOAMCTTLSA-N 0.000 description 1
- ASKTUHLOVSYQKF-CLQUMNPUSA-N C#CCO[C@H]1CC(O)[C@@H](CO)O[C@H]1n1cnc2c(N)ncnc21 Chemical compound C#CCO[C@H]1CC(O)[C@@H](CO)O[C@H]1n1cnc2c(N)ncnc21 ASKTUHLOVSYQKF-CLQUMNPUSA-N 0.000 description 1
- BEIBDYFRZBJQAC-YGNZUQDKSA-N C#CCO[C@H]1CC(O)[C@@H](CO)O[C@H]1n1cnc2c(NC(=O)c3ccccc3)ncnc21 Chemical compound C#CCO[C@H]1CC(O)[C@@H](CO)O[C@H]1n1cnc2c(NC(=O)c3ccccc3)ncnc21 BEIBDYFRZBJQAC-YGNZUQDKSA-N 0.000 description 1
- FHNHCRMQGCEOTO-OMOUFDEYSA-N C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CC)C(OC(C)=O)[C@@H]1OC(C)=O Chemical compound C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CC)C(OC(C)=O)[C@@H]1OC(C)=O FHNHCRMQGCEOTO-OMOUFDEYSA-N 0.000 description 1
- SQAOFRBQOGDEGB-PODXTCKDSA-N C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O Chemical compound C#CCOc1nc(C)nc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O SQAOFRBQOGDEGB-PODXTCKDSA-N 0.000 description 1
- OLEJDBHBCGQBIY-LLTZYFPTSA-N C.C#C.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CO.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1COP(C)(=S)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.C#C.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CO.[H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1COP(C)(=S)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 OLEJDBHBCGQBIY-LLTZYFPTSA-N 0.000 description 1
- FZCZASQYXJUMHD-FFYCHGNNSA-N C.C#CC.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.C#CC.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 FZCZASQYXJUMHD-FFYCHGNNSA-N 0.000 description 1
- NMEQJONRQRONJW-BVJJQNNASA-N C.C.C#CB(C)C#C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N)[C@H]1C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N=[N+]=[N-])[C@H]1O.COc1ccc(C(Cl)(c2ccccc2)c2ccc(OC)cc2)cc1.Cc1nc(C)c2ncn([C@@H]3O[C@H](CO)[C@H](O)C3N=[N+]=[N-])c2n1 Chemical compound C.C.C#CB(C)C#C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N)[C@H]1C.CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N=[N+]=[N-])[C@H]1O.COc1ccc(C(Cl)(c2ccccc2)c2ccc(OC)cc2)cc1.Cc1nc(C)c2ncn([C@@H]3O[C@H](CO)[C@H](O)C3N=[N+]=[N-])c2n1 NMEQJONRQRONJW-BVJJQNNASA-N 0.000 description 1
- YGQYNSXNWSSMKO-CIAANCMPSA-N C.C.C#CC.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5cnc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O)c(=O)[nH]1.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.C.C#CC.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5cnc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O)c(=O)[nH]1.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 YGQYNSXNWSSMKO-CIAANCMPSA-N 0.000 description 1
- LOSHHWORHJKZSZ-RJONDGNXSA-N C.C.C#CC.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)C(C)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)C(O1)[C@H]2O[Si](C)(C)C(C)(C)C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.C.C#CC.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)C(C)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)C(O1)[C@H]2O[Si](C)(C)C(C)(C)C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 LOSHHWORHJKZSZ-RJONDGNXSA-N 0.000 description 1
- PBLBFJRQVHMETG-UYHHTHHGSA-N C.C.C.C.C#CC#CC.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(C)(=S)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1.O=c1ssc2ccccc12.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=S)O[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.C.C.C.C#CC#CC.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(C)(=S)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1.O=c1ssc2ccccc12.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=S)O[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 PBLBFJRQVHMETG-UYHHTHHGSA-N 0.000 description 1
- SFUXLTXJWFXJGI-TWRQOUPVSA-N C.C.C.C.C.C.C.CBI.CC1(C)O[C@H]2C(O)[C@@H](CO)O[C@@H]2O1.CCC1[C@@H]2OC(C)(C)O[C@H]2O[C@@H]1CC.CCCC1[C@@H](CO)O[C@@H]2OC(C)(C)O[C@@H]12.CCCC1[C@@H]2OC(C)(C)O[C@H]2O[C@@H]1CC.CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1=O.CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1CCO.CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1O.COC(=O)CP(C)(C)=O.O=C(Cl)c1ccccc1.O=C=O.[AlH3].[LiH] Chemical compound C.C.C.C.C.C.C.CBI.CC1(C)O[C@H]2C(O)[C@@H](CO)O[C@@H]2O1.CCC1[C@@H]2OC(C)(C)O[C@H]2O[C@@H]1CC.CCCC1[C@@H](CO)O[C@@H]2OC(C)(C)O[C@@H]12.CCCC1[C@@H]2OC(C)(C)O[C@H]2O[C@@H]1CC.CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1=O.CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1CCO.CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1O.COC(=O)CP(C)(C)=O.O=C(Cl)c1ccccc1.O=C=O.[AlH3].[LiH] SFUXLTXJWFXJGI-TWRQOUPVSA-N 0.000 description 1
- OBURNQGTVPWLES-RLXUOSSRSA-N C.C.C.C.C.C.C.CC(=O)O.CC(C)C(=O)Cl.CC1(C)OC2[C@H](O1)C(O)O[C@@H]2CO.CC1(C)OC2[C@H](O1)[C@H](n1ncc(C#N)c1N)O[C@@H]2CO.CC1(C)OC2[C@H](O1)[C@H](n1ncc(C(N)=O)c1NC(=S)NC(=O)c1ccccc1)O[C@@H]2CO.CC=C(C#N)C#N.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)[C@H]2OC(C)(C)OC21.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(N)nc32)[C@H]2OC(C)(C)OC21 Chemical compound C.C.C.C.C.C.C.CC(=O)O.CC(C)C(=O)Cl.CC1(C)OC2[C@H](O1)C(O)O[C@@H]2CO.CC1(C)OC2[C@H](O1)[C@H](n1ncc(C#N)c1N)O[C@@H]2CO.CC1(C)OC2[C@H](O1)[C@H](n1ncc(C(N)=O)c1NC(=S)NC(=O)c1ccccc1)O[C@@H]2CO.CC=C(C#N)C#N.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)[C@H]2OC(C)(C)OC21.CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(N)nc32)[C@H]2OC(C)(C)OC21 OBURNQGTVPWLES-RLXUOSSRSA-N 0.000 description 1
- PIQRWOGOLBQJKL-RMQCECSZSA-N C.C.C.C.C.C.C.CC(C)(C)[Si](C)(C)Cl.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](O)C1C.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](O)C1O.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](OP(C)N(C(C)C)C(C)C)C1C.[H]P(=O)(O)O[C@H]1C(C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.C.C.C.C.C.C.CC(C)(C)[Si](C)(C)Cl.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](O)C1C.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](O)C1O.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](OP(C)N(C(C)C)C(C)C)C1C.[H]P(=O)(O)O[C@H]1C(C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 PIQRWOGOLBQJKL-RMQCECSZSA-N 0.000 description 1
- JOKVBYXDWMDNAV-XGZGQIPJSA-N C.C.C.C.CC(C)(C)[Si](C)(C)Cl.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O)C1O.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1O.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1OP(C)C.COc1ccc(C(Cl)(c2ccccc2)c2ccc(OC)cc2)cc1.C[Si](C)(C)Cl.Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O.O=C(Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 Chemical compound C.C.C.C.CC(C)(C)[Si](C)(C)Cl.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O)C1O.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1O.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1OP(C)C.COc1ccc(C(Cl)(c2ccccc2)c2ccc(OC)cc2)cc1.C[Si](C)(C)Cl.Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O.O=C(Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 JOKVBYXDWMDNAV-XGZGQIPJSA-N 0.000 description 1
- MXTADOIXRUSBRJ-PPAHTOFYSA-N C.C.C.CC#CC.CC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)C(OP(=O)(O)OC[C@H]2O[C@H]1n1ccc2c(N)ncnc21)[C@H]3O[Si](C)(C)C(C)(C)C.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(C)(=O)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1.N.O.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.C.C.CC#CC.CC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)C(OP(=O)(O)OC[C@H]2O[C@H]1n1ccc2c(N)ncnc21)[C@H]3O[Si](C)(C)C(C)(C)C.CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(C)(=O)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1.N.O.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)O[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 MXTADOIXRUSBRJ-PPAHTOFYSA-N 0.000 description 1
- OJWZOONBZHHHKV-LVDKDRQHSA-N C.C.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=S)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.C.CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=S)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 OJWZOONBZHHHKV-LVDKDRQHSA-N 0.000 description 1
- GQYPSHGALWLJMX-UACGDTGUSA-N C.C/C=C\c1c(C)cc2c(C(C)C)cccc2c1C.CC(C)C1C=Cc2c(N)ncnc21.Cc1cc(C)c2c(c1)C(C(C)C)C=C2.Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32.Cc1nc2c(c(=O)[nH]1)C=CC2C(C)C.Cc1nc2c(c3nccn13)C=CC2C(C)C Chemical compound C.C/C=C\c1c(C)cc2c(C(C)C)cccc2c1C.CC(C)C1C=Cc2c(N)ncnc21.Cc1cc(C)c2c(c1)C(C(C)C)C=C2.Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32.Cc1nc2c(c(=O)[nH]1)C=CC2C(C)C.Cc1nc2c(c3nccn13)C=CC2C(C)C GQYPSHGALWLJMX-UACGDTGUSA-N 0.000 description 1
- CKRGMADKBYBJQB-IRAIOCNNSA-N C.CBC.CC[C@H]1O[C@@H](n2ccc3ccccc32)CC1C.CC[C@H]1O[C@H](Cl)CC1C.CO.CO[Na].OC[C@H]1O[C@@H](n2ccc3ccccc32)CC1O.[NaH] Chemical compound C.CBC.CC[C@H]1O[C@@H](n2ccc3ccccc32)CC1C.CC[C@H]1O[C@H](Cl)CC1C.CO.CO[Na].OC[C@H]1O[C@@H](n2ccc3ccccc32)CC1O.[NaH] CKRGMADKBYBJQB-IRAIOCNNSA-N 0.000 description 1
- BWRRKTAYBSYFIG-UHFFFAOYSA-N C.CC(C)C1C=Cc2c(N)ncnc21.Cc1cc(C)c2c(c1)C(C(C)C)C=C2.Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1nc2c(c(=O)[nH]1)C=CC2C(C)C.Cc1nc2c(c3nccn13)C=CC2C(C)C Chemical compound C.CC(C)C1C=Cc2c(N)ncnc21.Cc1cc(C)c2c(c1)C(C(C)C)C=C2.Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1nc2c(c(=O)[nH]1)C=CC2C(C)C.Cc1nc2c(c3nccn13)C=CC2C(C)C BWRRKTAYBSYFIG-UHFFFAOYSA-N 0.000 description 1
- SLHTVWMODWRRIF-MOIMYFGXSA-N C.CC1(C)OC2[C@H](O1)[C@H](n1cnc3c(NC(=O)c4ccccc4)ncnc31)O[C@@H]2CN=[N+]=[N-] Chemical compound C.CC1(C)OC2[C@H](O1)[C@H](n1cnc3c(NC(=O)c4ccccc4)ncnc31)O[C@@H]2CN=[N+]=[N-] SLHTVWMODWRRIF-MOIMYFGXSA-N 0.000 description 1
- HLSFYXCYPWUCBE-MOIMYFGXSA-N C.CC1(C)OC2[C@H](O1)[C@H](n1cnc3c(NC(=O)c4ccccc4)ncnc31)O[C@@H]2CO Chemical compound C.CC1(C)OC2[C@H](O1)[C@H](n1cnc3c(NC(=O)c4ccccc4)ncnc31)O[C@@H]2CO HLSFYXCYPWUCBE-MOIMYFGXSA-N 0.000 description 1
- KBDBAQPWCDZOFH-JZWSEXQVSA-N C.CC1(C)OC2[C@H](O[C@H](CO)[C@H]2O)O1 Chemical compound C.CC1(C)OC2[C@H](O[C@H](CO)[C@H]2O)O1 KBDBAQPWCDZOFH-JZWSEXQVSA-N 0.000 description 1
- PQDXXCJFXLJMJK-OEWADGBISA-N C.CCO[C@@H]1C(OP(C)N(C(C)C)C(C)C)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CC Chemical compound C.CCO[C@@H]1C(OP(C)N(C(C)C)C(C)C)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CC PQDXXCJFXLJMJK-OEWADGBISA-N 0.000 description 1
- OYXDDKVRFUUYCE-AOOLDZOASA-N C.CC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)CC1C Chemical compound C.CC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)CC1C OYXDDKVRFUUYCE-AOOLDZOASA-N 0.000 description 1
- CRQSQIISAJPORQ-PTFPZXFRSA-N C.CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C Chemical compound C.CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C CRQSQIISAJPORQ-PTFPZXFRSA-N 0.000 description 1
- PFUDBASRFNVVFT-OXFGIQQGSA-N C.CC[C@H]1O[C@@H](n2ccc3ccccc32)CC1C Chemical compound C.CC[C@H]1O[C@@H](n2ccc3ccccc32)CC1C PFUDBASRFNVVFT-OXFGIQQGSA-N 0.000 description 1
- WXUQDQAHIFYSLL-RIDBKFJTSA-N C.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OP(C)NC(C)C)[C@H]1C Chemical compound C.CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OP(C)NC(C)C)[C@H]1C WXUQDQAHIFYSLL-RIDBKFJTSA-N 0.000 description 1
- DCNMWDUXKQPIIO-LTDKPLRPSA-N C.CC[C@H]1O[C@@H](n2nnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1CP(C)C Chemical compound C.CC[C@H]1O[C@@H](n2nnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1CP(C)C DCNMWDUXKQPIIO-LTDKPLRPSA-N 0.000 description 1
- UICYHLKXSLRGCO-YAFYIJHYSA-N C.CC[C@H]1O[C@@H](n2nnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C Chemical compound C.CC[C@H]1O[C@@H](n2nnc3c(NC(=O)c4ccccc4)ncnc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(C)C UICYHLKXSLRGCO-YAFYIJHYSA-N 0.000 description 1
- WEGRFHZKXLPGIT-SCEIGXRVSA-N C.NC1NC=Nc2c1ncn2[C@@H]1O[C@H](CO)[C@H](O)C1O Chemical compound C.NC1NC=Nc2c1ncn2[C@@H]1O[C@H](CO)[C@H](O)C1O WEGRFHZKXLPGIT-SCEIGXRVSA-N 0.000 description 1
- FJWZKLWIDIPXQD-RPIBNWFZSA-N C.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound C.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 FJWZKLWIDIPXQD-RPIBNWFZSA-N 0.000 description 1
- HPNHQIIEXZWLQB-AXJWPUBPSA-N C.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1.O=CCCl Chemical compound C.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1.Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1.O=CCCl HPNHQIIEXZWLQB-AXJWPUBPSA-N 0.000 description 1
- GPFNVQKESXHOJT-SVHUWJRNSA-N C.Nc1nc2c(ncn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1 Chemical compound C.Nc1nc2c(ncn2[C@@H]2O[C@H](CO)[C@H](O)C2O)c(=O)[nH]1 GPFNVQKESXHOJT-SVHUWJRNSA-N 0.000 description 1
- IEILELYDTRZODO-JJLCOGIFSA-N C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21 Chemical compound C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21 IEILELYDTRZODO-JJLCOGIFSA-N 0.000 description 1
- LMKQHSWWVIOVAQ-XRPJDSSCSA-N C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ncc2c(=O)[nH]c(C)nc21 Chemical compound C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ncc2c(=O)[nH]c(C)nc21 LMKQHSWWVIOVAQ-XRPJDSSCSA-N 0.000 description 1
- XRPYVUGZTFOJNM-ONGRONPVSA-N C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=S)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.[H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=S)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 XRPYVUGZTFOJNM-ONGRONPVSA-N 0.000 description 1
- QCSOBLRGHXNXIF-RIEHPMEPSA-N C.[H]P(=O)(O)OC1[C@@H](CCC)[C@@H](CC)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.[H]P(=O)(O)OC1[C@@H](CCC)[C@@H](CC)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 QCSOBLRGHXNXIF-RIEHPMEPSA-N 0.000 description 1
- JXNXYJVGLBXAIM-RDLXJQSLSA-N C.[H]P(=O)(O)OC1[C@@H](CCC)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.[H]P(=O)(O)OC1[C@@H](CCC)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 JXNXYJVGLBXAIM-RDLXJQSLSA-N 0.000 description 1
- BAILJXZRAIFWLF-CMDLMSATSA-N C.[H]P(=O)(O)OC1[C@@H](OCC)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound C.[H]P(=O)(O)OC1[C@@H](OCC)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 BAILJXZRAIFWLF-CMDLMSATSA-N 0.000 description 1
- QRFQPHZBOIOVTL-MMXBPUGVSA-N C.[H]P(=O)(O)OC1[C@H](O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CO Chemical compound C.[H]P(=O)(O)OC1[C@H](O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CO QRFQPHZBOIOVTL-MMXBPUGVSA-N 0.000 description 1
- XQMVUZXMZXAHOE-MOBZPFQKSA-N C/C=C\c1c(C)cc2c(C(C)C)cccc2c1C.Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32.Cc1nc2c(c3nccn13)C=CC2C(C)C Chemical compound C/C=C\c1c(C)cc2c(C(C)C)cccc2c1C.Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32.Cc1nc2c(c3nccn13)C=CC2C(C)C XQMVUZXMZXAHOE-MOBZPFQKSA-N 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- HIEZIDKCCFPTOC-VTXZZLSLSA-N CB(C)C.OC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)CC1O Chemical compound CB(C)C.OC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)CC1O HIEZIDKCCFPTOC-VTXZZLSLSA-N 0.000 description 1
- HIEZIDKCCFPTOC-OYXQGUJPSA-N CB(C)C.OC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)C[C@H]1O Chemical compound CB(C)C.OC[C@H]1O[C@@H](c2ccc3ccc4cccc5ccc2c3c45)C[C@H]1O HIEZIDKCCFPTOC-OYXQGUJPSA-N 0.000 description 1
- HJZNWMQERUDTAN-OELQWNMESA-N CBC.OC[C@H]1O[C@@H](n2ccc3ccccc32)CC1O Chemical compound CBC.OC[C@H]1O[C@@H](n2ccc3ccccc32)CC1O HJZNWMQERUDTAN-OELQWNMESA-N 0.000 description 1
- HJZNWMQERUDTAN-NLPVPVDASA-N CBC.OC[C@H]1O[C@@H](n2ccc3ccccc32)C[C@H]1O Chemical compound CBC.OC[C@H]1O[C@@H](n2ccc3ccccc32)C[C@H]1O HJZNWMQERUDTAN-NLPVPVDASA-N 0.000 description 1
- FAGCCLYZTRLDMY-GPSUDNJJSA-N CC(=O)OCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.COP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(N=[N+]=[N-])[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(OCCO)O[C@H]2C3O)c(=O)[nH]1 Chemical compound CC(=O)OCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.COP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(N=[N+]=[N-])[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(OCCO)O[C@H]2C3O)c(=O)[nH]1 FAGCCLYZTRLDMY-GPSUDNJJSA-N 0.000 description 1
- IEWQSCWTZXQJPH-PRUHYNRCSA-N CC(=O)OCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound CC(=O)OCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O IEWQSCWTZXQJPH-PRUHYNRCSA-N 0.000 description 1
- HENJIXPUSSGWCC-VKGMVAHQSA-M CC(C)(C)[Si](C)(C)Cl.CC(C)C(=O)Cl.CC1(C)OC2[C@H](O1)[C@H](n1ccc3c(=O)[nH]c(N)nc31)O[C@@H]2CO.CC[C@H]1OC(O)[C@H]2OC(C)(C)OC21.CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)[C@H]2OC(C)(C)OC21.CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(N)nc32)[C@H]2OC(C)(C)OC21.CC[C@H]1O[C@@H](n2ccc3c(Cl)nc(N)nc32)[C@H]2OC(C)(C)OC21.Nc1nc(Cl)c2cc[nH]c2n1.O[Na] Chemical compound CC(C)(C)[Si](C)(C)Cl.CC(C)C(=O)Cl.CC1(C)OC2[C@H](O1)[C@H](n1ccc3c(=O)[nH]c(N)nc31)O[C@@H]2CO.CC[C@H]1OC(O)[C@H]2OC(C)(C)OC21.CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)[C@H]2OC(C)(C)OC21.CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(N)nc32)[C@H]2OC(C)(C)OC21.CC[C@H]1O[C@@H](n2ccc3c(Cl)nc(N)nc32)[C@H]2OC(C)(C)OC21.Nc1nc(Cl)c2cc[nH]c2n1.O[Na] HENJIXPUSSGWCC-VKGMVAHQSA-M 0.000 description 1
- NEXZZOKYLVPDQS-UHFFFAOYSA-N CC(C)C1C=CC2=C1N=CCC2N.CC1=CC2=C(C=CC2C(C)C)C(C)C1.Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1nc2c(c(=O)[nH]1)C=CC2C(C)C.Cc1nc2c(c3nccn13)C=CC2C(C)C Chemical compound CC(C)C1C=CC2=C1N=CCC2N.CC1=CC2=C(C=CC2C(C)C)C(C)C1.Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1nc2c(c(=O)[nH]1)C=CC2C(C)C.Cc1nc2c(c3nccn13)C=CC2C(C)C NEXZZOKYLVPDQS-UHFFFAOYSA-N 0.000 description 1
- FJVBHHFARQSITF-UHFFFAOYSA-N CC(C)C1CCc2c(N)ccnc21.CC1=CC(C)Cc2c1cccc2C(C)C.CC1Cc2c(cccc2C(C)C)C2=C1C=CCC2.Cc1cc(C)c2c(c1)C(C(C)C)CC2.Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32.Cc1nc2c(c(=O)[nH]1)CCC2C(C)C.Cc1nc2c(c3nccn13)CCC2C(C)C Chemical compound CC(C)C1CCc2c(N)ccnc21.CC1=CC(C)Cc2c1cccc2C(C)C.CC1Cc2c(cccc2C(C)C)C2=C1C=CCC2.Cc1cc(C)c2c(c1)C(C(C)C)CC2.Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32.Cc1nc2c(c(=O)[nH]1)CCC2C(C)C.Cc1nc2c(c3nccn13)CCC2C(C)C FJVBHHFARQSITF-UHFFFAOYSA-N 0.000 description 1
- MUOPWBRCECSFFW-TUULINNZSA-N CC(C)OC(=O)OCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound CC(C)OC(=O)OCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O MUOPWBRCECSFFW-TUULINNZSA-N 0.000 description 1
- MYJVMOGNFHXJEX-ILLDJXIASA-N CC(C)OC(=O)OCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O.CCCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](OP(=O)(OCCOC)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3O1)C2O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(NCCO)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(NCCO)O[C@H]2C3O)c(=O)[nH]1.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP([BH3-])(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O Chemical compound CC(C)OC(=O)OCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O.CCCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](OP(=O)(OCCOC)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3O1)C2O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(NCCO)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(NCCO)O[C@H]2C3O)c(=O)[nH]1.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP([BH3-])(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O MYJVMOGNFHXJEX-ILLDJXIASA-N 0.000 description 1
- TZTQIOAVSCHJDG-UONRGADFSA-N CC.CC[C@@H]1CC[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1.CO Chemical compound CC.CC[C@@H]1CC[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1.CO TZTQIOAVSCHJDG-UONRGADFSA-N 0.000 description 1
- LXRYBHNLEXRODJ-UONRGADFSA-N CC.CC[C@@H]1CC[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1.COP(C)N(C(C)C)C(C)C Chemical compound CC.CC[C@@H]1CC[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1.COP(C)N(C(C)C)C(C)C LXRYBHNLEXRODJ-UONRGADFSA-N 0.000 description 1
- BYOXLBHYJMMOQF-DCXXCVGISA-N CC.CC[C@@H]1CC[C@H](n2ncc3c(=O)[nH]c(C)nc32)O1.CO Chemical compound CC.CC[C@@H]1CC[C@H](n2ncc3c(=O)[nH]c(C)nc32)O1.CO BYOXLBHYJMMOQF-DCXXCVGISA-N 0.000 description 1
- GBFHOAJWUAFEGM-DCXXCVGISA-N CC.CC[C@@H]1CC[C@H](n2ncc3c(=O)[nH]c(C)nc32)O1.COP(C)N(C(C)C)C(C)C Chemical compound CC.CC[C@@H]1CC[C@H](n2ncc3c(=O)[nH]c(C)nc32)O1.COP(C)N(C(C)C)C(C)C GBFHOAJWUAFEGM-DCXXCVGISA-N 0.000 description 1
- VCTBPZIRJNSWPX-CJGWJYNLSA-N CC.Cc1nc2c(cnn2[C@H]2CC[C@@H](CO)O2)c(=O)[nH]1.[H]P(=O)(O)OC Chemical compound CC.Cc1nc2c(cnn2[C@H]2CC[C@@H](CO)O2)c(=O)[nH]1.[H]P(=O)(O)OC VCTBPZIRJNSWPX-CJGWJYNLSA-N 0.000 description 1
- HRWSMTKVASCVJH-OXOJUWDDSA-N CC.Cc1nc2c(ncn2[C@H]2CC[C@@H](CO)O2)c(=O)[nH]1.[H]P(=O)(O)OC Chemical compound CC.Cc1nc2c(ncn2[C@H]2CC[C@@H](CO)O2)c(=O)[nH]1.[H]P(=O)(O)OC HRWSMTKVASCVJH-OXOJUWDDSA-N 0.000 description 1
- OYYTWUSIDMJZCP-OZRTXJSJSA-N CC1(C)OC2C(O)O[C@H](CO)[C@@H]2O1 Chemical compound CC1(C)OC2C(O)O[C@H](CO)[C@@H]2O1 OYYTWUSIDMJZCP-OZRTXJSJSA-N 0.000 description 1
- GDFBYNTYPOKYAC-CATGTTOSSA-N CC1(C)OC2C[C@H](O1)[C@H](n1ncc(C#N)c1N)O[C@@H]2CO Chemical compound CC1(C)OC2C[C@H](O1)[C@H](n1ncc(C#N)c1N)O[C@@H]2CO GDFBYNTYPOKYAC-CATGTTOSSA-N 0.000 description 1
- UFRIIQBAKRDMBD-KIMSHKSISA-N CC1(C)OC2C[C@H](O1)[C@H](n1ncc(C(N)=O)c1NC(=S)NC(=O)c1ccccc1)O[C@@H]2CO Chemical compound CC1(C)OC2C[C@H](O1)[C@H](n1ncc(C(N)=O)c1NC(=S)NC(=O)c1ccccc1)O[C@@H]2CO UFRIIQBAKRDMBD-KIMSHKSISA-N 0.000 description 1
- SUMIWWKGIVTNKL-UXSTWLAOSA-N CC1(C)OC2[C@H](O1)[C@H](n1ccc3c(=O)[nH]c(N)nc31)O[C@@H]2CO Chemical compound CC1(C)OC2[C@H](O1)[C@H](n1ccc3c(=O)[nH]c(N)nc31)O[C@@H]2CO SUMIWWKGIVTNKL-UXSTWLAOSA-N 0.000 description 1
- AIFHSVXRAUJJGF-STJJODOGSA-N CC1(C)OC2[C@H](O1)[C@H](n1ccc3c(N)ncnc31)O[C@@H]2CO Chemical compound CC1(C)OC2[C@H](O1)[C@H](n1ccc3c(N)ncnc31)O[C@@H]2CO AIFHSVXRAUJJGF-STJJODOGSA-N 0.000 description 1
- CJCDFXWLBXPIFC-OCMUNMMHSA-N CC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 Chemical compound CC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 CJCDFXWLBXPIFC-OCMUNMMHSA-N 0.000 description 1
- GIOQBVWVEWRUEM-YVUOAEARSA-N CC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)C(OP(=O)(O)OC[C@H]2O[C@H]1n1ccc2c(N)ncnc21)[C@H]3O[Si](C)(C)C(C)(C)C Chemical compound CC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)C(OP(=O)(O)OC[C@H]2O[C@H]1n1ccc2c(N)ncnc21)[C@H]3O[Si](C)(C)C(C)(C)C GIOQBVWVEWRUEM-YVUOAEARSA-N 0.000 description 1
- ZKHMQVPUAWTSQT-DUZXFSJDSA-N CC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)[C@@H](OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)C3O Chemical compound CC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)[C@@H](OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)C3O ZKHMQVPUAWTSQT-DUZXFSJDSA-N 0.000 description 1
- UFNLTANGOMVBHK-HDVBOMOJSA-N CCC1[C@@H]2OC(C)(C)O[C@H]2O[C@@H]1CC.O=C=O Chemical compound CCC1[C@@H]2OC(C)(C)O[C@H]2O[C@@H]1CC.O=C=O UFNLTANGOMVBHK-HDVBOMOJSA-N 0.000 description 1
- YIGGKUVHRJIXOX-NLOFOUMPSA-N CCC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 Chemical compound CCC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 YIGGKUVHRJIXOX-NLOFOUMPSA-N 0.000 description 1
- KUMQGBLJINGFHJ-WKCDVJHYSA-N CCCC1[C@@H](CO)O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@H]1O Chemical compound CCCC1[C@@H](CO)O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@H]1O KUMQGBLJINGFHJ-WKCDVJHYSA-N 0.000 description 1
- VHSSGNCPNKPTOY-FWKUWYBHSA-N CCCC1[C@@H](CO)O[C@@H]2OC(C)(C)O[C@@H]12 Chemical compound CCCC1[C@@H](CO)O[C@@H]2OC(C)(C)O[C@@H]12 VHSSGNCPNKPTOY-FWKUWYBHSA-N 0.000 description 1
- JWWMROMXAVQVAY-HUAZRZQGSA-N CCCC1[C@@H]2OC(C)(C)O[C@H]2O[C@@H]1CC Chemical compound CCCC1[C@@H]2OC(C)(C)O[C@H]2O[C@@H]1CC JWWMROMXAVQVAY-HUAZRZQGSA-N 0.000 description 1
- ROYBJABSZPLLCY-QBHFIFKDSA-N CCCC1[C@H](OC(C)=O)C(OC(C)=O)O[C@@H]1CC Chemical compound CCCC1[C@H](OC(C)=O)C(OC(C)=O)O[C@@H]1CC ROYBJABSZPLLCY-QBHFIFKDSA-N 0.000 description 1
- KKDCIAILNPGNJH-YRQGFPBVSA-N CCCC1[C@H](OC(C)=O)[C@H](n2cnc3c(OC(=O)N(c4ccccc4)c4ccccc4)nc(C)nc32)O[C@@H]1CC Chemical compound CCCC1[C@H](OC(C)=O)[C@H](n2cnc3c(OC(=O)N(c4ccccc4)c4ccccc4)nc(C)nc32)O[C@@H]1CC KKDCIAILNPGNJH-YRQGFPBVSA-N 0.000 description 1
- ILYNTRNYYHGLFN-UJPJIPQWSA-N CCCC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(C)nc31)O2.CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c4ncn4ccnc54)C(O)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound CCCC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(C)nc31)O2.CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c4ncn4ccnc54)C(O)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 ILYNTRNYYHGLFN-UJPJIPQWSA-N 0.000 description 1
- LEMPCUXPNPHXAD-YNPJMKPMSA-N CCCC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 Chemical compound CCCC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 LEMPCUXPNPHXAD-YNPJMKPMSA-N 0.000 description 1
- NYPAGNAJCUGKRP-QJGCKTFHSA-N CCCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](OP(=O)(OCCOC)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3O1)C2O Chemical compound CCCOP1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](OP(=O)(OCCOC)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3O1)C2O NYPAGNAJCUGKRP-QJGCKTFHSA-N 0.000 description 1
- QSVZIYJUONPAOM-UONRGADFSA-N CCCO[Si](C(C)C)(C(C)C)C(C)C.CC[C@@H]1CC[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1.CO Chemical compound CCCO[Si](C(C)C)(C(C)C)C(C)C.CC[C@@H]1CC[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1.CO QSVZIYJUONPAOM-UONRGADFSA-N 0.000 description 1
- HMWMEOKLOZTWIQ-WRCMBJAESA-N CCCO[Si](C(C)C)(C(C)C)C(C)C.Cc1nc2c(ncn2[C@H]2CC[C@@H](CO)O2)c(=O)[nH]1.[H]P(=O)(O)OC Chemical compound CCCO[Si](C(C)C)(C(C)C)C(C)C.Cc1nc2c(ncn2[C@H]2CC[C@@H](CO)O2)c(=O)[nH]1.[H]P(=O)(O)OC HMWMEOKLOZTWIQ-WRCMBJAESA-N 0.000 description 1
- XPKBSTDKMZAWQQ-QJNRBXNLSA-N CCC[C@@H]1C2NP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCC[C@@H]1C2NP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 XPKBSTDKMZAWQQ-QJNRBXNLSA-N 0.000 description 1
- LEMPCUXPNPHXAD-TXPXMQKESA-N CCC[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCC[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 LEMPCUXPNPHXAD-TXPXMQKESA-N 0.000 description 1
- ZXVGZYBIVDJZES-XKUKUCHXSA-N CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](c4cccc5ccccc45)C[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](c4cccc5ccccc45)C[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 ZXVGZYBIVDJZES-XKUKUCHXSA-N 0.000 description 1
- XYUZDOICBLPPJE-ACSLQPSXSA-N CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](c4cccc5ccccc45)C[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](c4cccc5ccccc45)C[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 XYUZDOICBLPPJE-ACSLQPSXSA-N 0.000 description 1
- PZSQNUQJKOWVTO-UAYUOIFVSA-N CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4ccc5ccccc54)C[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4ccc5ccccc54)C[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 PZSQNUQJKOWVTO-UAYUOIFVSA-N 0.000 description 1
- GVDJEMYOKYVXJE-CVKJPFSYSA-N CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4ccc5ccccc54)C[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5nnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4ccc5ccccc54)C[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](n5ccc6ccccc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5nnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 GVDJEMYOKYVXJE-CVKJPFSYSA-N 0.000 description 1
- QIICZBQAHQJEOI-PDJIUWQDSA-N CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c4ncn4ccnc54)C(O)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCc4c(c(=O)c4=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound CCOC1[C@@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c4ncn4ccnc54)C(O)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCc4c(c(=O)c4=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1 QIICZBQAHQJEOI-PDJIUWQDSA-N 0.000 description 1
- JJBJIJBZHLGNSN-AUHPQJTISA-N CCO[C@@H]1C2NP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c4ncn4ccnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2NP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c4ncn4ccnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 JJBJIJBZHLGNSN-AUHPQJTISA-N 0.000 description 1
- JRDJZPVRCIFVES-ROSBPPHPSA-N CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c4ncn4ccnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 Chemical compound CCO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c4ncn4ccnc54)[C@@H](O)C3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(N)nc21 JRDJZPVRCIFVES-ROSBPPHPSA-N 0.000 description 1
- JHFUPRLTCTZHMC-QMDUSEKHSA-N CC[C@@H]1CC[C@H](n2ccc3ccccc32)O1.CO Chemical compound CC[C@@H]1CC[C@H](n2ccc3ccccc32)O1.CO JHFUPRLTCTZHMC-QMDUSEKHSA-N 0.000 description 1
- RRBHMESBBSJRJY-QMDUSEKHSA-N CC[C@@H]1CC[C@H](n2ccc3ccccc32)O1.COP(C)C Chemical compound CC[C@@H]1CC[C@H](n2ccc3ccccc32)O1.COP(C)C RRBHMESBBSJRJY-QMDUSEKHSA-N 0.000 description 1
- GWLLNTIELNIPBT-KFWOVWKUSA-N CC[C@@H]1CC[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O1.CO.CO Chemical compound CC[C@@H]1CC[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O1.CO.CO GWLLNTIELNIPBT-KFWOVWKUSA-N 0.000 description 1
- IGXJULRWKKHEFE-KFWOVWKUSA-N CC[C@@H]1CC[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O1.CO.CO[Si](C)(C)C(C)(C)C Chemical compound CC[C@@H]1CC[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O1.CO.CO[Si](C)(C)C(C)(C)C IGXJULRWKKHEFE-KFWOVWKUSA-N 0.000 description 1
- WMRHLHYLEVSGQF-DCXXCVGISA-N CC[C@@H]1CC[C@H](n2ncc3c(=O)[nH]c(C)nc32)O1.CO.CO Chemical compound CC[C@@H]1CC[C@H](n2ncc3c(=O)[nH]c(C)nc32)O1.CO.CO WMRHLHYLEVSGQF-DCXXCVGISA-N 0.000 description 1
- RYXLUWZPKUEUMI-RGNHYFCHSA-N CC[C@@H]1C[C@H](O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1 Chemical compound CC[C@@H]1C[C@H](O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1 RYXLUWZPKUEUMI-RGNHYFCHSA-N 0.000 description 1
- FEORLTVMJKHXAF-IIYDPXPESA-N CC[C@@H]1C[C@H](O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1 Chemical compound CC[C@@H]1C[C@H](O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O1 FEORLTVMJKHXAF-IIYDPXPESA-N 0.000 description 1
- CYQOLMJDVCSUOR-NYOFMCRDSA-N CC[C@H]1OC(O)C2OC(C)(C)O[C@H]21 Chemical compound CC[C@H]1OC(O)C2OC(C)(C)O[C@H]21 CYQOLMJDVCSUOR-NYOFMCRDSA-N 0.000 description 1
- PCYFYJYYXINZMM-VKAADOFISA-N CC[C@H]1OC(OC(C)=O)[C@@H](OC(C)=O)C1OC Chemical compound CC[C@H]1OC(OC(C)=O)[C@@H](OC(C)=O)C1OC PCYFYJYYXINZMM-VKAADOFISA-N 0.000 description 1
- FSQYOBHLPVCCSX-BXVAXIAFSA-N CC[C@H]1OC(OC(C)=O)[C@@H](OC(C)=O)CC1C Chemical compound CC[C@H]1OC(OC(C)=O)[C@@H](OC(C)=O)CC1C FSQYOBHLPVCCSX-BXVAXIAFSA-N 0.000 description 1
- ZXEYJHLNQVENFB-QFCBBHACSA-N CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C Chemical compound CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)C(OP(C)N(C(C)C)C(C)C)[C@H]1C ZXEYJHLNQVENFB-QFCBBHACSA-N 0.000 description 1
- CQSMGSKIYBJDDG-CFMBFPBTSA-N CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(OCCC#N)N(C(C)C)C(C)C Chemical compound CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)C(O[Si](C)(C)C(C)(C)C)[C@H]1OP(OCCC#N)N(C(C)C)C(C)C CQSMGSKIYBJDDG-CFMBFPBTSA-N 0.000 description 1
- NMNMTJYTFDVBOJ-GSNLGQFWSA-N CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)[C@H]2OC(C)(C)OC21 Chemical compound CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(C)nc32)[C@H]2OC(C)(C)OC21 NMNMTJYTFDVBOJ-GSNLGQFWSA-N 0.000 description 1
- SMGCODLEJJUGMP-STJJODOGSA-N CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(N)nc32)[C@H]2OC(C)(C)OC21 Chemical compound CC[C@H]1O[C@@H](n2ccc3c(=O)[nH]c(N)nc32)[C@H]2OC(C)(C)OC21 SMGCODLEJJUGMP-STJJODOGSA-N 0.000 description 1
- MMJFNLLVTXNQMM-STJJODOGSA-N CC[C@H]1O[C@@H](n2ccc3c(Cl)nc(N)nc32)[C@H]2OC(C)(C)OC21 Chemical compound CC[C@H]1O[C@@H](n2ccc3c(Cl)nc(N)nc32)[C@H]2OC(C)(C)OC21 MMJFNLLVTXNQMM-STJJODOGSA-N 0.000 description 1
- VKSBTKQSARSMGA-VTKXWYFXSA-N CC[C@H]1O[C@@H](n2ccc3c(Cl)ncnc32)C2OC(C)(C)O[C@H]21 Chemical compound CC[C@H]1O[C@@H](n2ccc3c(Cl)ncnc32)C2OC(C)(C)O[C@H]21 VKSBTKQSARSMGA-VTKXWYFXSA-N 0.000 description 1
- SYZYZRBNCNFPNQ-OJTYORENSA-N CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)[C@H]2OC(C)(C)OC21 Chemical compound CC[C@H]1O[C@@H](n2ccc3c(NC(=O)c4ccccc4)ncnc32)[C@H]2OC(C)(C)OC21 SYZYZRBNCNFPNQ-OJTYORENSA-N 0.000 description 1
- WJFQDQGQWYWDEP-MTOMTMTDSA-N CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OP(C)NC(C)C)[C@H]1C Chemical compound CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)C(OP(C)NC(C)C)[C@H]1C WJFQDQGQWYWDEP-MTOMTMTDSA-N 0.000 description 1
- WOSMEXZCNCTMII-BWIWHEPQSA-N CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](OC(C)=O)C1OC Chemical compound CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](OC(C)=O)C1OC WOSMEXZCNCTMII-BWIWHEPQSA-N 0.000 description 1
- PMNYGGNUASQCDL-ZIWBQIBKSA-N CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](OC(C)=O)C1OC(C)=O Chemical compound CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](OC(C)=O)C1OC(C)=O PMNYGGNUASQCDL-ZIWBQIBKSA-N 0.000 description 1
- ACTMWEMCRLSJRH-KAWVDDRJSA-N CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](OC(C)=O)CC1C Chemical compound CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](OC(C)=O)CC1C ACTMWEMCRLSJRH-KAWVDDRJSA-N 0.000 description 1
- ZGBCJRHCGQWNNL-OVHGWZCWSA-N CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1O Chemical compound CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1O ZGBCJRHCGQWNNL-OVHGWZCWSA-N 0.000 description 1
- CZYCZAUCSQNZJF-OMOUFDEYSA-N CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1OC(C)=S Chemical compound CC[C@H]1O[C@@H](n2cnc3c(=O)[nH]c(C)nc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1OC(C)=S CZYCZAUCSQNZJF-OMOUFDEYSA-N 0.000 description 1
- PWXHAKIOBGXPQZ-IZSYJUOESA-N CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N=[N+]=[N-])[C@H]1O Chemical compound CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(N=[N+]=[N-])[C@H]1O PWXHAKIOBGXPQZ-IZSYJUOESA-N 0.000 description 1
- ODMPTWJENBYHOZ-YJFSCPGTSA-N CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(NS(=O)(=O)Oc2ccc([N+](=O)[O-])cc2)[C@H]1C Chemical compound CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(NS(=O)(=O)Oc2ccc([N+](=O)[O-])cc2)[C@H]1C ODMPTWJENBYHOZ-YJFSCPGTSA-N 0.000 description 1
- FSSFKVPOJHXBPC-MVMLMGAKSA-N CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(Nc2c(OC)c(=O)c2=O)[C@H]1C Chemical compound CC[C@H]1O[C@@H](n2cnc3c(C)nc(C)nc32)C(Nc2c(OC)c(=O)c2=O)[C@H]1C FSSFKVPOJHXBPC-MVMLMGAKSA-N 0.000 description 1
- JKNSMWOUKDYBFL-XKYDNLKZSA-N CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(OC)[C@H]1OP(OCCC#N)N(C(C)C)C(C)C Chemical compound CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C(OC)[C@H]1OP(OCCC#N)N(C(C)C)C(C)C JKNSMWOUKDYBFL-XKYDNLKZSA-N 0.000 description 1
- AFFDSZUZTWNOQD-JASKIIDBSA-N CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C[C@H]1OP(OCCC#N)N(C(C)C)C(C)C Chemical compound CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)C[C@H]1OP(OCCC#N)N(C(C)C)C(C)C AFFDSZUZTWNOQD-JASKIIDBSA-N 0.000 description 1
- PFAADTNJTXCQCE-UBHSXOEASA-N CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O)C1O Chemical compound CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O)C1O PFAADTNJTXCQCE-UBHSXOEASA-N 0.000 description 1
- QGFXFMKVVZOBSJ-LRYMFLOJSA-N CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1O Chemical compound CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1O QGFXFMKVVZOBSJ-LRYMFLOJSA-N 0.000 description 1
- NCUFAIURNDNOCR-IFWJREMNSA-N CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1OP(C)C Chemical compound CC[C@H]1O[C@@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](O[Si](C)(C)C(C)(C)C)C1OP(C)C NCUFAIURNDNOCR-IFWJREMNSA-N 0.000 description 1
- YKQHQBGKLCZHLU-OAYJICASSA-N CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)[C@H]2OC(C)(C)OC21 Chemical compound CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(C)nc32)[C@H]2OC(C)(C)OC21 YKQHQBGKLCZHLU-OAYJICASSA-N 0.000 description 1
- IKGSCAZAYPKAES-UXSTWLAOSA-N CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(N)nc32)[C@H]2OC(C)(C)OC21 Chemical compound CC[C@H]1O[C@@H](n2ncc3c(=O)[nH]c(N)nc32)[C@H]2OC(C)(C)OC21 IKGSCAZAYPKAES-UXSTWLAOSA-N 0.000 description 1
- DWWCXIAACKTFLA-LYUBYTMKSA-N CC[C@H]1O[C@@H](n2ncc3c(N)ncnc32)[C@@H](OC(=O)c2ccccc2)CC1OC(=O)c1ccccc1 Chemical compound CC[C@H]1O[C@@H](n2ncc3c(N)ncnc32)[C@@H](OC(=O)c2ccccc2)CC1OC(=O)c1ccccc1 DWWCXIAACKTFLA-LYUBYTMKSA-N 0.000 description 1
- UYFDIKMLKXPCEW-HHZRXBBFSA-N CC[C@H]1O[C@@H](n2ncc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](OC(=O)c2ccccc2)CC1OC(=O)c1ccccc1 Chemical compound CC[C@H]1O[C@@H](n2ncc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](OC(=O)c2ccccc2)CC1OC(=O)c1ccccc1 UYFDIKMLKXPCEW-HHZRXBBFSA-N 0.000 description 1
- YPBSCAYCVVMPGF-OAIPGLHFSA-N CC[C@H]1O[C@@H](n2nnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](OC(=O)c2ccccc2)C1OC(=O)c1ccccc1 Chemical compound CC[C@H]1O[C@@H](n2nnc3c(NC(=O)c4ccccc4)ncnc32)[C@@H](OC(=O)c2ccccc2)C1OC(=O)c1ccccc1 YPBSCAYCVVMPGF-OAIPGLHFSA-N 0.000 description 1
- SNBJJITUERNNHG-MHSYXAOVSA-N CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1=O Chemical compound CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1=O SNBJJITUERNNHG-MHSYXAOVSA-N 0.000 description 1
- BYUGFDOEJRJMPA-FWKUWYBHSA-N CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1CCO Chemical compound CC[C@H]1O[C@@H]2OC(C)(C)O[C@H]2C1CCO BYUGFDOEJRJMPA-FWKUWYBHSA-N 0.000 description 1
- NDECYCAOLLFPDR-QYNIQEEDSA-N CC[C@H]1O[C@H](Cl)C[C@H]1C Chemical compound CC[C@H]1O[C@H](Cl)C[C@H]1C NDECYCAOLLFPDR-QYNIQEEDSA-N 0.000 description 1
- UQABWPSYPMMXBY-PFSYLSIKSA-N CN(C)P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound CN(C)P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O UQABWPSYPMMXBY-PFSYLSIKSA-N 0.000 description 1
- XLTIIBBMQATQEE-FKFVKSHNSA-N CN(C)P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(C)(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound CN(C)P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(C)(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O XLTIIBBMQATQEE-FKFVKSHNSA-N 0.000 description 1
- QWMFETMWVBGITH-IKXGZXCVSA-N CNP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound CNP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O QWMFETMWVBGITH-IKXGZXCVSA-N 0.000 description 1
- FZJRCPOHCUCAGA-ONYAPDETSA-N CNP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(C)(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.COC(=O)C(C)NP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(NC(C)C(=O)OC)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(C)(=O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N(C)C)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(N)(=O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(N)(=O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound CNP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(C)(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.COC(=O)C(C)NP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(NC(C)C(=O)OC)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)[C@@H](O1)C2O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(C)(=O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N(C)C)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(N)(=O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(N)(=O)O[C@H]2C3O)c(=O)[nH]1 FZJRCPOHCUCAGA-ONYAPDETSA-N 0.000 description 1
- QPIFHEHSRBSQOD-RHFVVLBQSA-N CNP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(C)(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound CNP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(C)(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O QPIFHEHSRBSQOD-RHFVVLBQSA-N 0.000 description 1
- RIBCYFJCVCMPGT-HVXHQEPFSA-N COC(=O)C(C)NP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(NC(C)C(=O)OC)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound COC(=O)C(C)NP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(NC(C)C(=O)OC)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O RIBCYFJCVCMPGT-HVXHQEPFSA-N 0.000 description 1
- XYVJLDJKBWHEQJ-HRLNAYTHSA-N COC1[C@H](O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO Chemical compound COC1[C@H](O)[C@H](n2cnc3c(=O)[nH]c(C)nc32)O[C@@H]1CO XYVJLDJKBWHEQJ-HRLNAYTHSA-N 0.000 description 1
- FHTILWPDDTYZPJ-SUFOIOTQSA-N COC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(C)nc54)[C@@H](OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)C3O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3N=[N+]=[N-])c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(OCCO)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound COC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(C)nc54)[C@@H](OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)C3O.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3N=[N+]=[N-])c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(OCCO)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 FHTILWPDDTYZPJ-SUFOIOTQSA-N 0.000 description 1
- YVUQSOFRLPMPJE-SNGCHKTOSA-N COC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)[C@@H](OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)C3O Chemical compound COC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)[C@@H](OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)C3O YVUQSOFRLPMPJE-SNGCHKTOSA-N 0.000 description 1
- CVOLKFILGNNKMD-XFZKIOFTSA-N COP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound COP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O CVOLKFILGNNKMD-XFZKIOFTSA-N 0.000 description 1
- QKZPEWZIBNKYQH-LLFNNVKUSA-N CO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21 Chemical compound CO[C@@H]1C2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3OP(=O)(O)OC[C@H]1O[C@H]2n1cnc2c(=O)[nH]c(C)nc21 QKZPEWZIBNKYQH-LLFNNVKUSA-N 0.000 description 1
- FOIRXEYCIQVWIU-CQSOCPNPSA-N CO[Si](C)(C)C(C)(C)C.NC[C@@H]1CC[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O1.[H]P(=O)(O)OC Chemical compound CO[Si](C)(C)C(C)(C)C.NC[C@@H]1CC[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O1.[H]P(=O)(O)OC FOIRXEYCIQVWIU-CQSOCPNPSA-N 0.000 description 1
- FKMCPHNVRPTQLT-CQSOCPNPSA-N CO[Si](C)(C)C(C)(C)C.O=C(Nc1ncnc2c1ncn2[C@H]1CC[C@@H](CO)O1)c1ccccc1.[H]P(=O)(O)OC Chemical compound CO[Si](C)(C)C(C)(C)C.O=C(Nc1ncnc2c1ncn2[C@H]1CC[C@@H](CO)O1)c1ccccc1.[H]P(=O)(O)OC FKMCPHNVRPTQLT-CQSOCPNPSA-N 0.000 description 1
- SHOSNIYMWFNTIT-UHFFFAOYSA-N COc1ccc(Cn2nnc3c(Cl)ncnc32)cc1 Chemical compound COc1ccc(Cn2nnc3c(Cl)ncnc32)cc1 SHOSNIYMWFNTIT-UHFFFAOYSA-N 0.000 description 1
- DXTMNSACRHJXOU-UHFFFAOYSA-N COc1ccc(Cn2nnc3c(N)ncnc32)cc1 Chemical compound COc1ccc(Cn2nnc3c(N)ncnc32)cc1 DXTMNSACRHJXOU-UHFFFAOYSA-N 0.000 description 1
- YNYYLORKRJLUJA-UHFFFAOYSA-N COc1ccc(Cn2nnc3c(NC(=O)c4ccccc4)ncnc32)cc1 Chemical compound COc1ccc(Cn2nnc3c(NC(=O)c4ccccc4)ncnc32)cc1 YNYYLORKRJLUJA-UHFFFAOYSA-N 0.000 description 1
- HEFDTWILGQKLEL-GSGSRNIKSA-N CP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O Chemical compound CP1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O HEFDTWILGQKLEL-GSGSRNIKSA-N 0.000 description 1
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 1
- 102000008203 CTLA-4 Antigen Human genes 0.000 description 1
- 229940045513 CTLA4 antagonist Drugs 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- UWKGKOBEBIAVMY-UHFFFAOYSA-N Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32.Cc1cc2c(C(C)C)cccc2c2c1C=CCC2.Cc1nc2c(c3nccn13)C=CC2C(C)C Chemical compound Cc1cc(C)c2cccc(C(C)C)c2c1.Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32.Cc1cc2c(C(C)C)cccc2c2c1C=CCC2.Cc1nc2c(c3nccn13)C=CC2C(C)C UWKGKOBEBIAVMY-UHFFFAOYSA-N 0.000 description 1
- NDOPPSIZXYSTAI-UHFFFAOYSA-N Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32 Chemical compound Cc1cc2c(C(C)C)ccc3ccc4cccc1c4c32 NDOPPSIZXYSTAI-UHFFFAOYSA-N 0.000 description 1
- POZSUVRXUXGTGQ-VSIZLFLHSA-N Cc1nc(C)c2ncn([C@@H]3O[C@@H]4CO[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)OC4[C@@H]3N=[N+]=[N-])c2n1 Chemical compound Cc1nc(C)c2ncn([C@@H]3O[C@@H]4CO[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)OC4[C@@H]3N=[N+]=[N-])c2n1 POZSUVRXUXGTGQ-VSIZLFLHSA-N 0.000 description 1
- XXHSJVQUPRMSMR-VSIZLFLHSA-N Cc1nc(C)c2ncn([C@@H]3O[C@@H]4CO[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)OC4[C@@H]3O)c2n1 Chemical compound Cc1nc(C)c2ncn([C@@H]3O[C@@H]4CO[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)OC4[C@@H]3O)c2n1 XXHSJVQUPRMSMR-VSIZLFLHSA-N 0.000 description 1
- XXHSJVQUPRMSMR-LHTGEOHTSA-N Cc1nc(C)c2ncn([C@@H]3O[C@@H]4CO[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)OC4[C@H]3O)c2n1 Chemical compound Cc1nc(C)c2ncn([C@@H]3O[C@@H]4CO[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)OC4[C@H]3O)c2n1 XXHSJVQUPRMSMR-LHTGEOHTSA-N 0.000 description 1
- BCMLAQGQTNRTQW-RQRPQDJZSA-N Cc1nc(C)c2ncn([C@@H]3O[C@H](CO)C(O)[C@@H]3N=[N+]=[N-])c2n1 Chemical compound Cc1nc(C)c2ncn([C@@H]3O[C@H](CO)C(O)[C@@H]3N=[N+]=[N-])c2n1 BCMLAQGQTNRTQW-RQRPQDJZSA-N 0.000 description 1
- RDSNTCXRKJWFNI-XJHPQARYSA-N Cc1nc2c([C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]5C(O)[C@H](n6cnc7c(N)ncnc76)O[C@@H]5COP(=O)(O)O[C@H]3C4O)cncc2c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)ncnc56)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c([C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]5C(O)[C@H](n6cnc7c(N)ncnc76)O[C@@H]5COP(=O)(O)O[C@H]3C4O)cncc2c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)ncnc56)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 RDSNTCXRKJWFNI-XJHPQARYSA-N 0.000 description 1
- FGFWOKZIDDTMEI-CEWRSHTGSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(=O)[nH]c(N)nc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(S)O[C@H]3C(O)[C@@H](COP(=O)(O)O[C@@H]2C1O)O[C@H]3n1ccc2c(N)ccnc21 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(=O)[nH]c(N)nc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(S)O[C@H]3C(O)[C@@H](COP(=O)(O)O[C@@H]2C1O)O[C@H]3n1ccc2c(N)ccnc21 FGFWOKZIDDTMEI-CEWRSHTGSA-N 0.000 description 1
- WCPSDSAATGGIJF-OADKIORYSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(=O)[nH]c(N)nc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(S)O[C@H]3C(O)[C@@H](COP(=O)(O)O[C@@H]2C1O)O[C@H]3n1ccc2c(N)ccnc21.Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(S)O[C@H]3C(O)[C@@H](COP(=O)(O)O[C@@H]2C1O)O[C@H]3n1ccc2c(N)ccnc21 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(=O)[nH]c(N)nc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(S)O[C@H]3C(O)[C@@H](COP(=O)(O)O[C@@H]2C1O)O[C@H]3n1ccc2c(N)ccnc21.Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(S)O[C@H]3C(O)[C@@H](COP(=O)(O)O[C@@H]2C1O)O[C@H]3n1ccc2c(N)ccnc21 WCPSDSAATGGIJF-OADKIORYSA-N 0.000 description 1
- SWGYBZABTTWGTG-AAZCZVOYSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1.S.S Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1.S.S SWGYBZABTTWGTG-AAZCZVOYSA-N 0.000 description 1
- BOTLJSVDVHMLAM-QPTCJMCJSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 BOTLJSVDVHMLAM-QPTCJMCJSA-N 0.000 description 1
- KQMAYXHZEITBAF-CNJYWWHQSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 KQMAYXHZEITBAF-CNJYWWHQSA-N 0.000 description 1
- PJNCHVZRJRBZDZ-YHXGCQAVSA-N Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(S)O[C@H]3C(O)[C@@H](COP(=O)(O)O[C@@H]2C1O)O[C@H]3n1ccc2c(N)ccnc21 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Nc1ncnc2c1ccn2[C@@H]1O[C@@H]2COP(=O)(S)O[C@H]3C(O)[C@@H](COP(=O)(O)O[C@@H]2C1O)O[C@H]3n1ccc2c(N)ccnc21 PJNCHVZRJRBZDZ-YHXGCQAVSA-N 0.000 description 1
- IIOMTZAHFPMMRR-XKNZDDLCSA-N Cc1nc2c(ccn2[C@@H]2O[C@H](CO)C(O)C[C@@H]2O)c(=O)[nH]1 Chemical compound Cc1nc2c(ccn2[C@@H]2O[C@H](CO)C(O)C[C@@H]2O)c(=O)[nH]1 IIOMTZAHFPMMRR-XKNZDDLCSA-N 0.000 description 1
- UAQNZBSZVYAFPG-CMGGGZSGSA-N Cc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(N4CCOCC4)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(nnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(N4CCOCC4)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(nnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 UAQNZBSZVYAFPG-CMGGGZSGSA-N 0.000 description 1
- DMOWVYPXGRXMLW-ZUYVBGECSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=N)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=N)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=S)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=S)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=N)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=N)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=S)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=S)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1 DMOWVYPXGRXMLW-ZUYVBGECSA-N 0.000 description 1
- YGVLIQNVPIKWLA-XHOBUEJNSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=O)C[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)C[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)C[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CNc4c(c(=O)c4=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNc4c(c(=O)c4=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=O)C[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)C[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)C[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CNc4c(c(=O)c4=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNc4c(c(=O)c4=O)N[C@H]2C3O)c(=O)[nH]1 YGVLIQNVPIKWLA-XHOBUEJNSA-N 0.000 description 1
- GBQVPDZMKZQECW-SOYFKUMESA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCC(=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)C(F)(F)[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3Cc4cn(nn4)[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cc4cn(nn4)[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCC(=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)C(F)(F)[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3Cc4cn(nn4)[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cc4cn(nn4)[C@H]2C3O)c(=O)[nH]1 GBQVPDZMKZQECW-SOYFKUMESA-N 0.000 description 1
- ORHUMQCBPYKBLW-RJVCOGEISA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1 ORHUMQCBPYKBLW-RJVCOGEISA-N 0.000 description 1
- JPUWEYHYSPKCDC-DKPAQZBISA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1 JPUWEYHYSPKCDC-DKPAQZBISA-N 0.000 description 1
- UWLOCCSTJKKITH-CUDHWODHSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5cnc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(C)[C@H](n5cnc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1 UWLOCCSTJKKITH-CUDHWODHSA-N 0.000 description 1
- NUAKMGIOLHXLQD-TXPMOIIDSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCc4c(c(=O)c4=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCc4c(c(=O)c4=O)N[C@H]2C3O)c(=O)[nH]1 NUAKMGIOLHXLQD-TXPMOIIDSA-N 0.000 description 1
- PERHJRSFVLHMCJ-LLFNNVKUSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3OCO)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3OCO)c(=O)[nH]1 PERHJRSFVLHMCJ-LLFNNVKUSA-N 0.000 description 1
- ZLOWBAIRMHCNCF-QSDLLSNPSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3C)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3CCO)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3OCCO)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3OCCOCCO)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3C)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3CCO)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3OCCO)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3OCCOCCO)c(=O)[nH]1 ZLOWBAIRMHCNCF-QSDLLSNPSA-N 0.000 description 1
- YNDBYRRJVSUGEM-MLZUDDORSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cc4cn(nn4)[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cn4cc(nn4)CO[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cc4cn(nn4)[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cn4cc(nn4)CO[C@H]2C3O)c(=O)[nH]1 YNDBYRRJVSUGEM-MLZUDDORSA-N 0.000 description 1
- VCBMPGFEGZOERM-LTOHBJIQSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C[C@H](c5ccc6ccc7cccc8ccc5c6c78)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 VCBMPGFEGZOERM-LTOHBJIQSA-N 0.000 description 1
- ZYKNFIHHJKHBLB-MOLHOFDHSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 ZYKNFIHHJKHBLB-MOLHOFDHSA-N 0.000 description 1
- CNOISFDSEXSYBR-JTKGTCKXSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5nnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ncc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5nnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 CNOISFDSEXSYBR-JTKGTCKXSA-N 0.000 description 1
- PPGQWXOYKXZHAK-KPFSPWNZSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(C)(=S)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3COP(C)(=S)O[C@@H]4C(C)[C@H](n5ccc6c(NC(=O)c7ccccc7)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O[Si](C)(C)C(C)(C)C)c(=O)[nH]1 PPGQWXOYKXZHAK-KPFSPWNZSA-N 0.000 description 1
- ZFOHRIHAINAIML-BVLVASOUSA-N Cc1nc2c(ncn2[C@@H]2O[C@@H]3CO[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)OC3[C@@H]2O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@@H]3CO[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)OC3[C@@H]2O)c(=O)[nH]1 ZFOHRIHAINAIML-BVLVASOUSA-N 0.000 description 1
- YEKFUIDNJQMJEH-TVEGWDGASA-N Cc1nc2c(ncn2[C@@H]2O[C@H](CO)C(C)C[C@@H]2O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2O[C@H](CO)C(C)C[C@@H]2O)c(=O)[nH]1 YEKFUIDNJQMJEH-TVEGWDGASA-N 0.000 description 1
- DTIWCZCOIGIZAM-PZHPKLTDSA-N Cc1nc2c(ncn2[C@@H]2S[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)S[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2S[C@@H]3COP(O)(=S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)S[C@@H]4COP(O)(=S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2[Se][C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)[Se][C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2[Se][C@@H]3COP(O)(=S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)[Se][C@@H]4COP(O)(=S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Cc1nc2c(ncn2[C@@H]2S[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)S[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2S[C@@H]3COP(O)(=S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)S[C@@H]4COP(O)(=S)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2[Se][C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)[Se][C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1.Cc1nc2c(ncn2[C@@H]2[Se][C@@H]3COP(O)(=S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)[Se][C@@H]4COP(O)(=S)O[C@H]2C3O)c(=O)[nH]1 DTIWCZCOIGIZAM-PZHPKLTDSA-N 0.000 description 1
- VPTJKMDVCFDSIL-UHFFFAOYSA-N Cc1ncnc(Cl)c1N Chemical compound Cc1ncnc(Cl)c1N VPTJKMDVCFDSIL-UHFFFAOYSA-N 0.000 description 1
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 1
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 1
- GHXZTYHSJHQHIJ-UHFFFAOYSA-N Chlorhexidine Chemical compound C=1C=C(Cl)C=CC=1NC(N)=NC(N)=NCCCCCCN=C(N)N=C(N)NC1=CC=C(Cl)C=C1 GHXZTYHSJHQHIJ-UHFFFAOYSA-N 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- 241000723346 Cinnamomum camphora Species 0.000 description 1
- YMXQUFUYCADCFL-UHFFFAOYSA-N Clc1ncnc2[nH]ncc12 Chemical compound Clc1ncnc2[nH]ncc12 YMXQUFUYCADCFL-UHFFFAOYSA-N 0.000 description 1
- 102100031162 Collagen alpha-1(XVIII) chain Human genes 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 108010024986 Cyclin-Dependent Kinase 2 Proteins 0.000 description 1
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 1
- 108010025468 Cyclin-Dependent Kinase 6 Proteins 0.000 description 1
- 102100036239 Cyclin-dependent kinase 2 Human genes 0.000 description 1
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 description 1
- 102100026804 Cyclin-dependent kinase 6 Human genes 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- NOOLISFMXDJSKH-UHFFFAOYSA-N DL-menthol Natural products CC(C)C1CCC(C)CC1O NOOLISFMXDJSKH-UHFFFAOYSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 102000003915 DNA Topoisomerases Human genes 0.000 description 1
- 108090000323 DNA Topoisomerases Proteins 0.000 description 1
- 230000005778 DNA damage Effects 0.000 description 1
- 231100000277 DNA damage Toxicity 0.000 description 1
- 231100001074 DNA strand break Toxicity 0.000 description 1
- 241000450599 DNA viruses Species 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- ZQZFYGIXNQKOAV-OCEACIFDSA-N Droloxifene Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=C(O)C=CC=1)\C1=CC=C(OCCN(C)C)C=C1 ZQZFYGIXNQKOAV-OCEACIFDSA-N 0.000 description 1
- 101100015729 Drosophila melanogaster drk gene Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- OVBJJZOQPCKUOR-UHFFFAOYSA-L EDTA disodium salt dihydrate Chemical compound O.O.[Na+].[Na+].[O-]C(=O)C[NH+](CC([O-])=O)CC[NH+](CC([O-])=O)CC([O-])=O OVBJJZOQPCKUOR-UHFFFAOYSA-L 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 1
- 102100031706 Fibroblast growth factor 1 Human genes 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 230000010337 G2 phase Effects 0.000 description 1
- 230000004668 G2/M phase Effects 0.000 description 1
- 241000237858 Gastropoda Species 0.000 description 1
- 239000001828 Gelatine Substances 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 108010069236 Goserelin Proteins 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101000904173 Homo sapiens Progonadoliberin-1 Proteins 0.000 description 1
- 101000579425 Homo sapiens Proto-oncogene tyrosine-protein kinase receptor Ret Proteins 0.000 description 1
- 101000665442 Homo sapiens Serine/threonine-protein kinase TBK1 Proteins 0.000 description 1
- 101001074035 Homo sapiens Zinc finger protein GLI2 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 108010047852 Integrin alphaVbeta3 Proteins 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 206010024612 Lipoma Diseases 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 229920000715 Mucilage Polymers 0.000 description 1
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- GVLHXHKLGNIMCC-RFCBMDAISA-N N=C1CC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](NC(=N)NC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3N1)C2O Chemical compound N=C1CC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](NC(=N)NC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)C(O)[C@H]3N1)C2O GVLHXHKLGNIMCC-RFCBMDAISA-N 0.000 description 1
- LAJTUKCIFPIRBL-IDBXARSASA-N NC1=Nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@@H](O)[C@@H](COP(=O)(O)OC3[C@@H]2O)O[C@H]4n2ccc3c(=O)[nH]c(N)nc32)C(N)N1 Chemical compound NC1=Nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@@H](O)[C@@H](COP(=O)(O)OC3[C@@H]2O)O[C@H]4n2ccc3c(=O)[nH]c(N)nc32)C(N)N1 LAJTUKCIFPIRBL-IDBXARSASA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- CSHRFWOHUJULMU-BNLYFJINSA-N Nc1nc2c([C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]5C(O)[C@H](n6cnc7c(N)ncnc76)O[C@@H]5COP(=O)(O)O[C@H]3C4O)cncc2c(=O)[nH]1 Chemical compound Nc1nc2c([C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]5C(O)[C@H](n6cnc7c(N)ncnc76)O[C@@H]5COP(=O)(O)O[C@H]3C4O)cncc2c(=O)[nH]1 CSHRFWOHUJULMU-BNLYFJINSA-N 0.000 description 1
- KWAKDVHBJNEYMA-JNNYHLCPSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 KWAKDVHBJNEYMA-JNNYHLCPSA-N 0.000 description 1
- VUIJRGZOVHFLTM-JMKQKTBBSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1.S Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1.S VUIJRGZOVHFLTM-JMKQKTBBSA-N 0.000 description 1
- QODOEPRTKNAVCW-YRIIINANSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(NCCO)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)OC4[C@H](O)[C@H](n5ccc6c(NCCO)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 QODOEPRTKNAVCW-YRIIINANSA-N 0.000 description 1
- PQPIVQWIPINCPT-WZUCXXBOSA-N Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ccn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 PQPIVQWIPINCPT-WZUCXXBOSA-N 0.000 description 1
- LTGWLWDFLBDKHQ-GZYNHYCXSA-N Nc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(cnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 LTGWLWDFLBDKHQ-GZYNHYCXSA-N 0.000 description 1
- BGCZLZNORZUYND-WZTRFBLOSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=O)C[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=O)C[C@H]2C3O)c(=O)[nH]1 BGCZLZNORZUYND-WZTRFBLOSA-N 0.000 description 1
- VMAHWOQNJRKPAJ-VOVGDBHBSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCC(=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCC(=O)N[C@H]2C3O)c(=O)[nH]1 VMAHWOQNJRKPAJ-VOVGDBHBSA-N 0.000 description 1
- NLRUKFPXHHTZFV-RFCBMDAISA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=O)N[C@H]2C3O)c(=O)[nH]1 NLRUKFPXHHTZFV-RFCBMDAISA-N 0.000 description 1
- YLXXUHLAEJIOQM-RFCBMDAISA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=S)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=S)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCC(=S)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNC(=S)N[C@H]2C3O)c(=O)[nH]1 YLXXUHLAEJIOQM-RFCBMDAISA-N 0.000 description 1
- XGAVTTCRBSDEOE-KYMVQTTMSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)C[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)C[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)C[C@H]2C3O)c(=O)[nH]1 XGAVTTCRBSDEOE-KYMVQTTMSA-N 0.000 description 1
- KYUJDTOAXZEFBV-PLTJGGTGSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1 KYUJDTOAXZEFBV-PLTJGGTGSA-N 0.000 description 1
- YSUKNDHLQOBRKV-FQEDZZQKSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CCS(=O)(=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNS(=O)(=O)N[C@H]2C3O)c(=O)[nH]1 YSUKNDHLQOBRKV-FQEDZZQKSA-N 0.000 description 1
- DNIZIBMTDSCLLQ-PLTJGGTGSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CN=C=N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CN=C=N[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CN=C=N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CN=C=N[C@H]2C3O)c(=O)[nH]1 DNIZIBMTDSCLLQ-PLTJGGTGSA-N 0.000 description 1
- SGDIDCSDFZYXFQ-TWFBIKPLSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3CNc4c(c(=O)c4=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNc4c(c(=O)c4=O)N[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3CNc4c(c(=O)c4=O)N[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNc4c(c(=O)c4=O)N[C@H]2C3O)c(=O)[nH]1 SGDIDCSDFZYXFQ-TWFBIKPLSA-N 0.000 description 1
- FKGKHXUORMUKBN-XXOAXBTDSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(N4CCOCC4)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(N4CCOCC4)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)O[C@H]2C3O)c(=O)[nH]1 FKGKHXUORMUKBN-XXOAXBTDSA-N 0.000 description 1
- KIAWNOMSRZQLEW-OBFBJLTLSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(NCCO)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(NCCO)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(NCCO)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(NCCO)O[C@H]2C3O)c(=O)[nH]1 KIAWNOMSRZQLEW-OBFBJLTLSA-N 0.000 description 1
- UTYHBKZTJXGVRM-XFPWQAPDSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 UTYHBKZTJXGVRM-XFPWQAPDSA-N 0.000 description 1
- AGKCBKGLLKTVGM-LNUBRJLUSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCc4c(c(=O)c4=O)NC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CCc4c(c(=O)c4=O)NC2[C@H]3O)c(=O)[nH]1 AGKCBKGLLKTVGM-LNUBRJLUSA-N 0.000 description 1
- KXXQEYIDFCANIV-GDCMQRIJSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNc4c(c(=O)c4=O)NC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CNc4c(c(=O)c4=O)NC2[C@H]3O)c(=O)[nH]1 KXXQEYIDFCANIV-GDCMQRIJSA-N 0.000 description 1
- HTJDGTMSADJTQQ-TXPXMQKESA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3CCCS)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3CCCS)c(=O)[nH]1 HTJDGTMSADJTQQ-TXPXMQKESA-N 0.000 description 1
- DZIBBHIDDLPARP-DYAHWJSYSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)NC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)NC2[C@H]3O)c(=O)[nH]1 DZIBBHIDDLPARP-DYAHWJSYSA-N 0.000 description 1
- GZWGNHWTLOUWIE-KTDVQVRBSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)OC4[C@H](O)[C@H](n5cnc6c5ncn5ccnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 GZWGNHWTLOUWIE-KTDVQVRBSA-N 0.000 description 1
- GNTUHQUEAZWYJF-BNLYFJINSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)ncnc56)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](c5cncc6c(N)ncnc56)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 GNTUHQUEAZWYJF-BNLYFJINSA-N 0.000 description 1
- UTYHBKZTJXGVRM-QWTHJVQCSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(O)OC2[C@H]3O)c(=O)[nH]1 UTYHBKZTJXGVRM-QWTHJVQCSA-N 0.000 description 1
- HRKIQOZBILGGFA-BILJAGFLSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CC(F)(F)P(=O)(O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4CC(F)(F)P(=O)(O)O[C@H]2C3O)c(=O)[nH]1 HRKIQOZBILGGFA-BILJAGFLSA-N 0.000 description 1
- GTWMOORQETVGGV-YKOZUWMESA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(N4CCOCC4)OC2[C@H]3O)c(=O)[nH]1 GTWMOORQETVGGV-YKOZUWMESA-N 0.000 description 1
- GOHMMLWJWIRSPM-VHRHROOBSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)C(F)(F)[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)C(F)(F)[C@H]2C3O)c(=O)[nH]1 GOHMMLWJWIRSPM-VHRHROOBSA-N 0.000 description 1
- HZMILZKTLSFJNU-RGVKKHIASA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)C[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)C[C@H]2C3O)c(=O)[nH]1 HZMILZKTLSFJNU-RGVKKHIASA-N 0.000 description 1
- POXFSBZEVXHWPH-YNPJMKPMSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3CCCO)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3CCCO)c(=O)[nH]1 POXFSBZEVXHWPH-YNPJMKPMSA-N 0.000 description 1
- JKFQOODHZIJUTQ-NLOFOUMPSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3CCO)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3CCO)c(=O)[nH]1 JKFQOODHZIJUTQ-NLOFOUMPSA-N 0.000 description 1
- WKFFSEOIKYTXCF-NLOFOUMPSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3CCS)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3CCS)c(=O)[nH]1 WKFFSEOIKYTXCF-NLOFOUMPSA-N 0.000 description 1
- CKVIFAMKXHGFAN-UDUPATSFSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3OCCOCCO)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3OCCOCCO)c(=O)[nH]1 CKVIFAMKXHGFAN-UDUPATSFSA-N 0.000 description 1
- KFDWGKBFQHNESJ-UTFWSIBGSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(OCCO)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(OCCO)O[C@H]2C3O)c(=O)[nH]1 KFDWGKBFQHNESJ-UTFWSIBGSA-N 0.000 description 1
- ZPXACAYHVPLDRG-VVGDMVEUSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(N)(=O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(N)(=O)O[C@H]2C3O)c(=O)[nH]1 ZPXACAYHVPLDRG-VVGDMVEUSA-N 0.000 description 1
- HFMFYPFXASFSSE-RSINILKMSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cn4cc(nn4)CO[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cn4cc(nn4)CO[C@H]2C3O)c(=O)[nH]1 HFMFYPFXASFSSE-RSINILKMSA-N 0.000 description 1
- OEZJKFUYSYXIOU-XJZNRSMASA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(OCCO)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(O)O[C@@H]4C(OCCO)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 OEZJKFUYSYXIOU-XJZNRSMASA-N 0.000 description 1
- VHPJEIGORCRPDZ-DIOWXFCESA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5ccc6c(N)ncnc65)O[C@@H]4COP(=O)(S)OC2[C@H]3O)c(=O)[nH]1 VHPJEIGORCRPDZ-DIOWXFCESA-N 0.000 description 1
- NVOTUSOAWRDABP-AJVGHTOPSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 NVOTUSOAWRDABP-AJVGHTOPSA-N 0.000 description 1
- XTERZONMCYRPCE-QBQYHPQLSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(N)(=O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(N)(=O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3COP(N)(=O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(N)(=O)O[C@H]2C3O)c(=O)[nH]1 XTERZONMCYRPCE-QBQYHPQLSA-N 0.000 description 1
- SQXDEULJNXYVMQ-YDBBEIBMSA-N Nc1nc2c(ncn2[C@@H]2O[C@@H]3Cc4cn(nn4)[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cc4cn(nn4)[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2O[C@@H]3Cc4cn(nn4)[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4Cc4cn(nn4)[C@H]2C3O)c(=O)[nH]1 SQXDEULJNXYVMQ-YDBBEIBMSA-N 0.000 description 1
- SGDNUUFVAPJCHI-BILJAGFLSA-N Nc1nc2c(ncn2[C@@H]2S[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)S[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2S[C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)S[C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 SGDNUUFVAPJCHI-BILJAGFLSA-N 0.000 description 1
- ZUDKJQMOEPRARL-FHUDRKDOSA-N Nc1nc2c(ncn2[C@@H]2S[C@@H]3COP(O)(=S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)S[C@@H]4COP(O)(=S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2S[C@@H]3COP(O)(=S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)S[C@@H]4COP(O)(=S)O[C@H]2C3O)c(=O)[nH]1 ZUDKJQMOEPRARL-FHUDRKDOSA-N 0.000 description 1
- LWLQPWHMYYFKGS-BILJAGFLSA-N Nc1nc2c(ncn2[C@@H]2[Se][C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)[Se][C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2[Se][C@@H]3COP(=O)(O)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)[Se][C@@H]4COP(=O)(O)O[C@H]2C3O)c(=O)[nH]1 LWLQPWHMYYFKGS-BILJAGFLSA-N 0.000 description 1
- UBWJEGTUCUMHBO-FHUDRKDOSA-N Nc1nc2c(ncn2[C@@H]2[Se][C@@H]3COP(O)(=S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)[Se][C@@H]4COP(O)(=S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(ncn2[C@@H]2[Se][C@@H]3COP(O)(=S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)[Se][C@@H]4COP(O)(=S)O[C@H]2C3O)c(=O)[nH]1 UBWJEGTUCUMHBO-FHUDRKDOSA-N 0.000 description 1
- YEWOJDRGKAVNDR-BBEZKFMKSA-N Nc1nc2c(nnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 Chemical compound Nc1nc2c(nnn2[C@@H]2O[C@@H]3COP(=O)(S)O[C@@H]4C(O)[C@H](n5cnc6c(N)ncnc65)O[C@@H]4COP(=O)(S)O[C@H]2C3O)c(=O)[nH]1 YEWOJDRGKAVNDR-BBEZKFMKSA-N 0.000 description 1
- LHCPRYRLDOSKHK-UHFFFAOYSA-N Nc1ncnc2[nH]ncc12 Chemical compound Nc1ncnc2[nH]ncc12 LHCPRYRLDOSKHK-UHFFFAOYSA-N 0.000 description 1
- 239000004384 Neotame Substances 0.000 description 1
- 241000183666 Nepsera aquatica Species 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 102100039306 Nucleotide pyrophosphatase Human genes 0.000 description 1
- 102000003832 Nucleotidyltransferases Human genes 0.000 description 1
- 108090000119 Nucleotidyltransferases Proteins 0.000 description 1
- BPERWUJJTXWDQL-UHFFFAOYSA-N O=C(Nc1ncnc2[nH]nnc12)c1ccccc1 Chemical compound O=C(Nc1ncnc2[nH]nnc12)c1ccccc1 BPERWUJJTXWDQL-UHFFFAOYSA-N 0.000 description 1
- OWRNOLMZLOHLGL-FEWYLQRLSA-N O=C(Nc1ncnc2c1ccn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 Chemical compound O=C(Nc1ncnc2c1ccn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 OWRNOLMZLOHLGL-FEWYLQRLSA-N 0.000 description 1
- LGUKXAQOLMZVJI-FOSLCNLBSA-N O=C(Nc1ncnc2c1cnn2[C@@H]1O[C@H](CO)C(O)C[C@@H]1O)c1ccccc1 Chemical compound O=C(Nc1ncnc2c1cnn2[C@@H]1O[C@H](CO)C(O)C[C@@H]1O)c1ccccc1 LGUKXAQOLMZVJI-FOSLCNLBSA-N 0.000 description 1
- NZDWTKFDAUOODA-LXAXPIHBSA-N O=C(Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 Chemical compound O=C(Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 NZDWTKFDAUOODA-LXAXPIHBSA-N 0.000 description 1
- PWSSOQNCOQRAIF-RDMACPANSA-N O=C(Nc1ncnc2c1nnn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 Chemical compound O=C(Nc1ncnc2c1nnn2[C@@H]1O[C@H](CO)C(O)[C@@H]1O)c1ccccc1 PWSSOQNCOQRAIF-RDMACPANSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102000038030 PI3Ks Human genes 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 1
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 1
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 1
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 1
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 1
- 201000005746 Pituitary adenoma Diseases 0.000 description 1
- 206010061538 Pituitary tumour benign Diseases 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 244000236480 Podophyllum peltatum Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920002556 Polyethylene Glycol 300 Polymers 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 108010040201 Polymyxins Proteins 0.000 description 1
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 1
- 102100024028 Progonadoliberin-1 Human genes 0.000 description 1
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102100028286 Proto-oncogene tyrosine-protein kinase receptor Ret Human genes 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 102000009609 Pyrophosphatases Human genes 0.000 description 1
- 108010009413 Pyrophosphatases Proteins 0.000 description 1
- 102000003901 Ras GTPase-activating proteins Human genes 0.000 description 1
- 108090000231 Ras GTPase-activating proteins Proteins 0.000 description 1
- 208000015634 Rectal Neoplasms Diseases 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 208000005678 Rhabdomyoma Diseases 0.000 description 1
- KKPPROWYBVJUQS-ZWWYFVOISA-N S.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O Chemical compound S.[BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O KKPPROWYBVJUQS-ZWWYFVOISA-N 0.000 description 1
- 108060006706 SRC Proteins 0.000 description 1
- 102000001332 SRC Human genes 0.000 description 1
- 229940044665 STING agonist Drugs 0.000 description 1
- 102100038192 Serine/threonine-protein kinase TBK1 Human genes 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 239000004376 Sucralose Substances 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101000996723 Sus scrofa Gonadotropin-releasing hormone receptor Proteins 0.000 description 1
- 108091005735 TGF-beta receptors Proteins 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- GNVMUORYQLCPJZ-UHFFFAOYSA-M Thiocarbamate Chemical compound NC([S-])=O GNVMUORYQLCPJZ-UHFFFAOYSA-M 0.000 description 1
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 description 1
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 description 1
- 102000014384 Type C Phospholipases Human genes 0.000 description 1
- 108010079194 Type C Phospholipases Proteins 0.000 description 1
- 108010046308 Type II DNA Topoisomerases Proteins 0.000 description 1
- 102000007537 Type II DNA Topoisomerases Human genes 0.000 description 1
- 101150071882 US17 gene Proteins 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 241000863480 Vinca Species 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 102100035558 Zinc finger protein GLI2 Human genes 0.000 description 1
- WOTQVEKSRLZRSX-HYSGBLIFSA-N [(2r,3r,4s,5r,6s)-4,5,6-triacetyloxy-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxyoxan-2-yl]methyl acetate Chemical compound CC(=O)OC[C@H]1O[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](OC(C)=O)[C@@H]1O[C@H]1[C@H](OC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1 WOTQVEKSRLZRSX-HYSGBLIFSA-N 0.000 description 1
- XVOMVJURLZFCAD-NGLCSSBQSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(OP(=O)(S)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3O1)[C@H]2O Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(OP(=O)(S)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3O1)[C@H]2O XVOMVJURLZFCAD-NGLCSSBQSA-N 0.000 description 1
- PYLIKACMRGCLDB-UNCNNLGBSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O[PH]([BH3-])(S)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3O1)[C@H]2O Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O[PH]([BH3-])(S)OC[C@H]3O[C@@H](n4cnc5c(N)ncnc54)[C@@H](O)C3O1)[C@H]2O PYLIKACMRGCLDB-UNCNNLGBSA-N 0.000 description 1
- UDUNYNOAPYLNIL-RGEINWCBSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2CCC Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2CCC UDUNYNOAPYLNIL-RGEINWCBSA-N 0.000 description 1
- SDSHZUYSVVEPDJ-KUHMAFEQSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2OCC Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2OCC SDSHZUYSVVEPDJ-KUHMAFEQSA-N 0.000 description 1
- YMIIBMNOLHFVQM-QBQYHPQLSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP([BH3-])(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)C(O)[C@H]2OP([BH3-])(=O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)[C@@H](O1)C2O YMIIBMNOLHFVQM-QBQYHPQLSA-N 0.000 description 1
- WAVDNJWFEXUGJW-VENPIBPCSA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O[Si](C)(C)C(C)(C)C Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(N)ncnc43)[C@@H](O)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(N)nc43)C(O1)[C@H]2O[Si](C)(C)C(C)(C)C WAVDNJWFEXUGJW-VENPIBPCSA-N 0.000 description 1
- GYEHPONFGZVDKT-NBTDUMQESA-N [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)[C@@H](C)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)C(O1)[C@H]2O[Si](C)(C)C(C)(C)C Chemical compound [BH3-]P1(=O)OC[C@H]2O[C@@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)[C@@H](C)C2OP(=O)(O)OC[C@H]2O[C@@H](n3cnc4c(=O)[nH]c(C)nc43)C(O1)[C@H]2O[Si](C)(C)C(C)(C)C GYEHPONFGZVDKT-NBTDUMQESA-N 0.000 description 1
- WBWWWOHNERYEHD-ZMRISJLTSA-N [H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21 Chemical compound [H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21 WBWWWOHNERYEHD-ZMRISJLTSA-N 0.000 description 1
- LZBPLPCAYJIBMI-SNVPNBKQSA-N [H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(NC(=O)c3ccccc3)ncnc21 Chemical compound [H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(NC(=O)c3ccccc3)ncnc21 LZBPLPCAYJIBMI-SNVPNBKQSA-N 0.000 description 1
- VGOFBRVDNHNQCD-POIVVALJSA-N [H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ncc2c(=O)[nH]c(C)cc21 Chemical compound [H]P(=O)(O)OC1[C@@H](C)[C@@H](CO)O[C@H]1n1ncc2c(=O)[nH]c(C)cc21 VGOFBRVDNHNQCD-POIVVALJSA-N 0.000 description 1
- FKSKVBPUEVVRBB-CKMGIZSISA-N [H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)C[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound [H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)C[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 FKSKVBPUEVVRBB-CKMGIZSISA-N 0.000 description 1
- NERIAVSNHCCLGJ-NBARGWHUSA-N [H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21 Chemical compound [H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=O)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1ccc2c(=O)[nH]c(C)nc21 NERIAVSNHCCLGJ-NBARGWHUSA-N 0.000 description 1
- XPVMHYRQJUOSTN-TVGYTVOTSA-N [H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=S)O[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound [H]P(=O)(O)OC1[C@@H](C)[C@@H](COP(C)(=S)O[C@@H]2C(C)[C@H](n3ccc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 XPVMHYRQJUOSTN-TVGYTVOTSA-N 0.000 description 1
- BLKFZVQQHGPMOI-GUBVXQJLSA-N [H]P(=O)(O)OC1[C@@H](C)[C@@H](CO[P+]([BH3-])(C)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound [H]P(=O)(O)OC1[C@@H](C)[C@@H](CO[P+]([BH3-])(C)OC2[C@H](C)[C@H](n3cnc4c(NC(=O)c5ccccc5)ncnc43)O[C@@H]2CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 BLKFZVQQHGPMOI-GUBVXQJLSA-N 0.000 description 1
- SAOINCOPNZSDQD-KAMMAAHDSA-N [H]P(=O)(O)OC1[C@@H](CCC)[C@@H](CC)O[C@H]1n1cnc2c(=O)[nH]c(NCC(C)C)cc21 Chemical compound [H]P(=O)(O)OC1[C@@H](CCC)[C@@H](CC)O[C@H]1n1cnc2c(=O)[nH]c(NCC(C)C)cc21 SAOINCOPNZSDQD-KAMMAAHDSA-N 0.000 description 1
- HBPHWXDWTSPCIG-WEWZKHDXSA-N [H]P(=O)(O)OC1[C@@H](CCO[Si](C(C)C)(C(C)C)C(C)C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound [H]P(=O)(O)OC1[C@@H](CCO[Si](C(C)C)(C(C)C)C(C)C)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 HBPHWXDWTSPCIG-WEWZKHDXSA-N 0.000 description 1
- WCQRWTROZCROLE-ZIUXOXTBSA-N [H]P(=O)(O)OC1[C@@H](OCC)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 Chemical compound [H]P(=O)(O)OC1[C@@H](OCC)[C@@H](CO)O[C@H]1n1cnc2c(=O)[nH]c(C)nc21 WCQRWTROZCROLE-ZIUXOXTBSA-N 0.000 description 1
- DMIQNHSBQYFKBR-XMVDTUKRSA-N [H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCS(=O)(=O)NC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(C)nc(C)nc21 Chemical compound [H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCS(=O)(=O)NC1[C@@H](C)[C@@H](CO)O[C@H]1n1cnc2c(C)nc(C)nc21 DMIQNHSBQYFKBR-XMVDTUKRSA-N 0.000 description 1
- VXDAGDWWQWMMKC-KNTCECPOSA-N [H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCc1c(NC2[C@@H](C)[C@@H](CO)O[C@H]2n2cnc3c(C)nc(C)nc32)c(=O)c1=O Chemical compound [H]P(=O)(O)O[C@@H]1C(O[Si](C)(C)C(C)(C)C)[C@H](n2cnc3c(NC(=O)c4ccccc4)ncnc32)O[C@@H]1CCc1c(NC2[C@@H](C)[C@@H](CO)O[C@H]2n2cnc3c(C)nc(C)nc32)c(=O)c1=O VXDAGDWWQWMMKC-KNTCECPOSA-N 0.000 description 1
- XUEIBXCAJDYYBE-NKFZLVBVSA-N [N-]=[N+]=NC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 Chemical compound [N-]=[N+]=NC1[C@H]2COP(=O)(O)O[C@@H]3C(O)[C@H](n4cnc5c(N)ncnc54)O[C@@H]3COP(=O)(O)O[C@@H]1[C@H](n1cnc3c(=O)[nH]c(N)nc31)O2 XUEIBXCAJDYYBE-NKFZLVBVSA-N 0.000 description 1
- IEPMBCXBGRFAAK-NKFZLVBVSA-N [N-]=[N+]=NC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)[C@@H](OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)C3O Chemical compound [N-]=[N+]=NC1[C@H]2OP(=O)(O)OC[C@H]3O[C@@H](n4cnc5c(=O)[nH]c(N)nc54)[C@@H](OP(=O)(O)OC[C@H]2O[C@H]1n1cnc2c(N)ncnc21)C3O IEPMBCXBGRFAAK-NKFZLVBVSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010358 acesulfame potassium Nutrition 0.000 description 1
- 239000000619 acesulfame-K Substances 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000012082 adaptor molecule Substances 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 150000001398 aluminium Chemical class 0.000 description 1
- 239000005030 aluminium foil Substances 0.000 description 1
- 159000000013 aluminium salts Chemical class 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000003432 anti-folate effect Effects 0.000 description 1
- 230000001946 anti-microtubular Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 230000002137 anti-vascular effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940127074 antifolate Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 229940045686 antimetabolites antineoplastic purine analogs Drugs 0.000 description 1
- 239000003080 antimitotic agent Substances 0.000 description 1
- 229940045688 antineoplastic antimetabolites pyrimidine analogues Drugs 0.000 description 1
- 229940027983 antiseptic and disinfectant quaternary ammonium compound Drugs 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 239000008122 artificial sweetener Substances 0.000 description 1
- 235000021311 artificial sweeteners Nutrition 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 229940092782 bentonite Drugs 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- CPEKAXYCDKETEN-UHFFFAOYSA-N benzoyl isothiocyanate Chemical compound S=C=NC(=O)C1=CC=CC=C1 CPEKAXYCDKETEN-UHFFFAOYSA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960004217 benzyl alcohol Drugs 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- IBNQLYMPUGQNLN-UHFFFAOYSA-M benzyl-[2-(4-dodecanoylphenoxy)ethyl]-dimethylazanium;chloride Chemical compound [Cl-].C1=CC(C(=O)CCCCCCCCCCC)=CC=C1OCC[N+](C)(C)CC1=CC=CC=C1 IBNQLYMPUGQNLN-UHFFFAOYSA-M 0.000 description 1
- 208000036815 beta tubulin Diseases 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- WOTQVEKSRLZRSX-UHFFFAOYSA-N beta-D-cellobioside octaacetate Natural products CC(=O)OCC1OC(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(COC(C)=O)O1 WOTQVEKSRLZRSX-UHFFFAOYSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 238000011243 body radiation therapy Methods 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- BYKCUMSOQIPHSR-UHFFFAOYSA-N boron;n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound [B].CCN(C(C)C)C(C)C BYKCUMSOQIPHSR-UHFFFAOYSA-N 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 201000002143 bronchus adenoma Diseases 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940067596 butylparaben Drugs 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229960000846 camphor Drugs 0.000 description 1
- 229930008380 camphor Natural products 0.000 description 1
- 238000002619 cancer immunotherapy Methods 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 229960002798 cetrimide Drugs 0.000 description 1
- 229960001927 cetylpyridinium chloride Drugs 0.000 description 1
- NFCRBQADEGXVDL-UHFFFAOYSA-M cetylpyridinium chloride monohydrate Chemical compound O.[Cl-].CCCCCCCCCCCCCCCC[N+]1=CC=CC=C1 NFCRBQADEGXVDL-UHFFFAOYSA-M 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960003260 chlorhexidine Drugs 0.000 description 1
- DDYAZDRFUVZBMM-UHFFFAOYSA-N chloro-[chloro-di(propan-2-yl)silyl]oxy-di(propan-2-yl)silane Chemical compound CC(C)[Si](Cl)(C(C)C)O[Si](Cl)(C(C)C)C(C)C DDYAZDRFUVZBMM-UHFFFAOYSA-N 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 229960002242 chlorocresol Drugs 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000024207 chronic leukemia Diseases 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- PDXMFTWFFKBFIN-XPWFQUROSA-N cyclic di-AMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]3[C@@H](O)[C@H](N4C5=NC=NC(N)=C5N=C4)O[C@@H]3COP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 PDXMFTWFFKBFIN-XPWFQUROSA-N 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229960000978 cyproterone acetate Drugs 0.000 description 1
- UWFYSQMTEOIJJG-FDTZYFLXSA-N cyproterone acetate Chemical compound C1=C(Cl)C2=CC(=O)[C@@H]3C[C@@H]3[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 UWFYSQMTEOIJJG-FDTZYFLXSA-N 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 230000016396 cytokine production Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 229950004203 droloxifene Drugs 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 229960004199 dutasteride Drugs 0.000 description 1
- JWJOTENAMICLJG-QWBYCMEYSA-N dutasteride Chemical compound O=C([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)N[C@@H]4CC3)C)CC[C@@]21C)NC1=CC(C(F)(F)F)=CC=C1C(F)(F)F JWJOTENAMICLJG-QWBYCMEYSA-N 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 150000002085 enols Chemical class 0.000 description 1
- 108060002566 ephrin Proteins 0.000 description 1
- 102000012803 ephrin Human genes 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 102000015694 estrogen receptors Human genes 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 229960001617 ethyl hydroxybenzoate Drugs 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 235000010228 ethyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004403 ethyl p-hydroxybenzoate Substances 0.000 description 1
- NUVBSKCKDOMJSU-UHFFFAOYSA-N ethylparaben Chemical compound CCOC(=O)C1=CC=C(O)C=C1 NUVBSKCKDOMJSU-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 239000010642 eucalyptus oil Substances 0.000 description 1
- 229940044949 eucalyptus oil Drugs 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 206010016629 fibroma Diseases 0.000 description 1
- 229960004039 finasteride Drugs 0.000 description 1
- DBEPLOCGEIEOCV-WSBQPABSSA-N finasteride Chemical compound N([C@@H]1CC2)C(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)NC(C)(C)C)[C@@]2(C)CC1 DBEPLOCGEIEOCV-WSBQPABSSA-N 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- GVEPBJHOBDJJJI-UHFFFAOYSA-N fluoranthrene Natural products C1=CC(C2=CC=CC=C22)=C3C2=CC=CC3=C1 GVEPBJHOBDJJJI-UHFFFAOYSA-N 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- KGPPDNUWZNWPSI-UHFFFAOYSA-N flurotyl Chemical compound FC(F)(F)COCC(F)(F)F KGPPDNUWZNWPSI-UHFFFAOYSA-N 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 229960002258 fulvestrant Drugs 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- LPLVUJXQOOQHMX-QWBHMCJMSA-N glycyrrhizinic acid Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@H](O[C@@H]1O[C@@H]1C([C@H]2[C@]([C@@H]3[C@@]([C@@]4(CC[C@@]5(C)CC[C@@](C)(C[C@H]5C4=CC3=O)C(O)=O)C)(C)CC2)(C)CC1)(C)C)C(O)=O)[C@@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O LPLVUJXQOOQHMX-QWBHMCJMSA-N 0.000 description 1
- XLXSAKCOAKORKW-UHFFFAOYSA-N gonadorelin Chemical compound C1CCC(C(=O)NCC(N)=O)N1C(=O)C(CCCN=C(N)N)NC(=O)C(CC(C)C)NC(=O)CNC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 XLXSAKCOAKORKW-UHFFFAOYSA-N 0.000 description 1
- 229960003690 goserelin acetate Drugs 0.000 description 1
- 101150098203 grb2 gene Proteins 0.000 description 1
- 229920000591 gum Polymers 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 150000004688 heptahydrates Chemical class 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 210000000244 kidney pelvis Anatomy 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 1
- 229950007325 lauralkonium chloride Drugs 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- GZQKNULLWNGMCW-PWQABINMSA-N lipid A (E. coli) Chemical compound O1[C@H](CO)[C@@H](OP(O)(O)=O)[C@H](OC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCCCC)[C@@H](NC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H]1OC[C@@H]1[C@@H](O)[C@H](OC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](OP(O)(O)=O)O1 GZQKNULLWNGMCW-PWQABINMSA-N 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 229960003511 macrogol Drugs 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 208000006178 malignant mesothelioma Diseases 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 229960004296 megestrol acetate Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- 229940041616 menthol Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000031864 metaphase Effects 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 229940071648 metered dose inhaler Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- SIGOIUCRXKUEIG-UHFFFAOYSA-N methyl 2-dimethoxyphosphorylacetate Chemical compound COC(=O)CP(=O)(OC)OC SIGOIUCRXKUEIG-UHFFFAOYSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 229960002900 methylcellulose Drugs 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 230000035773 mitosis phase Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000002625 monoclonal antibody therapy Methods 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 208000009091 myxoma Diseases 0.000 description 1
- BLCLNMBMMGCOAS-UHFFFAOYSA-N n-[1-[[1-[[1-[[1-[[1-[[1-[[1-[2-[(carbamoylamino)carbamoyl]pyrrolidin-1-yl]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-[(2-methylpropan-2-yl)oxy]-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amin Chemical compound C1CCC(C(=O)NNC(N)=O)N1C(=O)C(CCCN=C(N)N)NC(=O)C(CC(C)C)NC(=O)C(COC(C)(C)C)NC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 BLCLNMBMMGCOAS-UHFFFAOYSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- XVDBWWRIXBMVJV-UHFFFAOYSA-N n-[bis(dimethylamino)phosphanyl]-n-methylmethanamine Chemical group CN(C)P(N(C)C)N(C)C XVDBWWRIXBMVJV-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-M naphthalene-1-sulfonate Chemical compound C1=CC=C2C(S(=O)(=O)[O-])=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-M 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical class C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 238000002663 nebulization Methods 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 235000019412 neotame Nutrition 0.000 description 1
- HLIAVLHNDJUHFG-HOTGVXAUSA-N neotame Chemical compound CC(C)(C)CCN[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 HLIAVLHNDJUHFG-HOTGVXAUSA-N 0.000 description 1
- 108010070257 neotame Proteins 0.000 description 1
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 1
- 229960002653 nilutamide Drugs 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 125000005440 p-toluyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C(*)=O)C([H])([H])[H] 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 210000002990 parathyroid gland Anatomy 0.000 description 1
- 239000011236 particulate material Substances 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 210000003899 penis Anatomy 0.000 description 1
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 239000008180 pharmaceutical surfactant Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229940067107 phenylethyl alcohol Drugs 0.000 description 1
- 229940096826 phenylmercuric acetate Drugs 0.000 description 1
- PDTFCHSETJBPTR-UHFFFAOYSA-N phenylmercuric nitrate Chemical compound [O-][N+](=O)O[Hg]C1=CC=CC=C1 PDTFCHSETJBPTR-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 208000021310 pituitary gland adenoma Diseases 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 239000011253 protective coating Substances 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- IGFXRKMLLMBKSA-UHFFFAOYSA-N purine Chemical compound N1=C[N]C2=NC=NC2=C1 IGFXRKMLLMBKSA-UHFFFAOYSA-N 0.000 description 1
- DOTPSQVYOBAWPQ-UHFFFAOYSA-N pyrazolo[4,3-d]pyrimidin-3-one Chemical compound N1=CN=C2C(=O)N=NC2=C1 DOTPSQVYOBAWPQ-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 238000010833 quantitative mass spectrometry Methods 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 108010077182 raf Kinases Proteins 0.000 description 1
- 102000009929 raf Kinases Human genes 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 229960003522 roquinimex Drugs 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 229960003885 sodium benzoate Drugs 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 239000004296 sodium metabisulphite Substances 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- JXKPEJDQGNYQSM-UHFFFAOYSA-M sodium propionate Chemical compound [Na+].CCC([O-])=O JXKPEJDQGNYQSM-UHFFFAOYSA-M 0.000 description 1
- 235000010334 sodium propionate Nutrition 0.000 description 1
- 239000004324 sodium propionate Substances 0.000 description 1
- 229960003212 sodium propionate Drugs 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 102000009076 src-Family Kinases Human genes 0.000 description 1
- 108010087686 src-Family Kinases Proteins 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 235000019408 sucralose Nutrition 0.000 description 1
- BAQAVOSOZGMPRM-QBMZZYIRSA-N sucralose Chemical compound O[C@@H]1[C@@H](O)[C@@H](Cl)[C@@H](CO)O[C@@H]1O[C@@]1(CCl)[C@@H](O)[C@H](O)[C@@H](CCl)O1 BAQAVOSOZGMPRM-QBMZZYIRSA-N 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-M sulfamate Chemical compound NS([O-])(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-M 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- MHYGQXWCZAYSLJ-UHFFFAOYSA-N tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1 MHYGQXWCZAYSLJ-UHFFFAOYSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 239000000892 thaumatin Substances 0.000 description 1
- 235000010436 thaumatin Nutrition 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 230000008467 tissue growth Effects 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 229940100611 topical cream Drugs 0.000 description 1
- 229940100615 topical ointment Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- SIOVKLKJSOKLIF-HJWRWDBZSA-N trimethylsilyl (1z)-n-trimethylsilylethanimidate Chemical compound C[Si](C)(C)OC(/C)=N\[Si](C)(C)C SIOVKLKJSOKLIF-HJWRWDBZSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 210000000626 ureter Anatomy 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 210000003905 vulva Anatomy 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
- C07H19/213—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids containing cyclic phosphate
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7084—Compounds having two nucleosides or nucleotides, e.g. nicotinamide-adenine dinucleotide, flavine-adenine dinucleotide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
Definitions
- the present invention provides novel cyclic di-nucleotide cGAMP analogs, pharmaceutical compositions thereof, their synthetic methods and their use in medical therapy.
- the compounds of the invention enhance the body's immune responses by activating STING (Stimulator of Interferon Genes) and are useful for the immunotherapy of cancer, infectious diseases and immune disorders.
- STING Stimulator of Interferon Genes
- the compounds are also useful as adjuvants for developing vaccines against cancer and infectious diseases.
- Cytosolic DNA induces type-I interferons and other cytokines that are important for immune defense against microbial infections and malignant cells but can also result in autoimmunity.
- This DNA signaling pathway requires the adaptor protein STING (Stimulator of Interferon Genes) and the transcription factor IRF3, but the mechanism of DNA sensing was unclear until recently.
- WO 2014099824 to The University of Texas disclosed that mammalian cytosolic extracts synthesized cyclic-GMP-AMP (cGAMP) in vitro from ATP and GTP in the presence of DNA but not RNA. DNA transfection or DNA virus infection of mammalian cells also triggered cGAMP production.
- cGAMP bound to STING, lead to the activation of IRF3 and induction of type-I interferons including interferon-0 (IFN-(3).
- IFN-(3) interferon-0
- cGAMP represents the first cyclic di-nucleotide in metazoa and it functions as an endogenous second messenger that triggers interferon production in response to cytosolic DNA.
- cGAS cGAMP synthase
- the inventors on WO 2014099824 also determined that the second messenger cGAMP they isolated and synthesized contains two phosphodiester linkages, one between the 2′-OH of GMP and 5′-phosphate of AMP, and the other between the 3′-OH of AMP and 5′-phosphate of GMP; this molecule is referred to as 2′3′-cGAMP.
- CDN cyclic-di-nucleotide
- WO 2015077354 A1 to The University of Chicago discloses Methods and compositions for treating cancer by intratumorally administering a stimulator of interferon genes (STING) agonist.
- STING interferon genes
- compositions and methods concerning methods for treating cancer in a subject comprising administering to the subject an effective amount of a stimulator of interferon genes (STING) agonist, wherein the STING agonist is administered intratumorally.
- WO 2015161762 to Fudan University discloses the use of cyclic dinucleotide cGAMP for preparing antitumor drugs, wherein the tumor is gastric cancer, lung cancer, colon cancer, liver cancer, prostate cancer or pancreatic cancer. cGAMP was shown to inhibit the growth of human tumor cell lines in immune compromised mice.
- WO 2015185565 to GlaxoSmithKline discloses a class of cyclic dinucleotide compounds, or a pharmaceutically acceptable salt and tautomers thereof, compositions, combinations and medicaments containing said compounds and processes for their preparation.
- the invention also relates to the use of said compounds, combinations, compositions and medicaments, in the treatment of diseases and conditions in which modulation of STING is beneficial, for example inflammation, allergic and autoimmune diseases, infectious diseases, cancer and as vaccine adjuvants.
- WO 2014179335 to Memorial Sloan Kettering Cancer Center discloses compositions, methods, kits, and assays related to the use and/or exploitation of isomers of cGAMP as well as the structure of the enzyme cGAS.
- cGAMP analogues with better potency, stability and specificity than endogenous cGAMP are still needed.
- Formula I encompasses Formula Ia-Ii.
- the present invention provides a compound of Formula Ia
- X 1 and X 2 are independently O, S or Se in a five-membered ring;
- L 1 starting from the carbon alpha to X 1
- L 2 starting from the carbon alpha to X 2
- L 1 is independently —CH 2 O—P(O)R 6 —O—, —CH 2 O—P(S)R 6 —O—, —C(Y 1 )(Y 2 )O—P(O)R 6 —C(Y 3 )(Y 4 )—, —CH 2 NHSO 2 NH—, —CH 2 NHC(O)NH—, —CH 2 NHC(S)NH—, —CH 2 NHC(NH)NH—, —CH 2 NHC(O)CH 2 —, —CH 2 NHSO 2 CH 2 —, —CH 2 CH 2 C(O)NH—, —CH 2 CH 2 SO 2 NH—, —CH 2 NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
- R 1 and R 2 are independently aromatic rings or heteroaromatic rings with the following general structure including its tautomeric forms:
- R 3 , R 4 , and R 5 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1 -6alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R
- the present invention provides a compound of Formula Ib
- X 1 and X 2 are independently O, S or Se;
- Z 12 , Z 13 , Z 14 , Z 15 , Z 16 and Z 17 are independently CH or N;
- L 1 starting from the carbon alpha to X 1
- L 2 starting from the carbon alpha to X 2
- L 1 is independently —CH 2 O—P(O)R 6 —O—, —CH 2 O—P(S)R 6 —O—, —C(Y 1 )(Y 2 )O—P(O)R 6 —C(Y 3 )(Y 4 )—, —CH 2 NHSO 2 NH—, —CH 2 NHC(O)NH—, —CH 2 NHC(S)NH—, —CH 2 NHC(NH)NH—, —CH 2 NHC(O)CH 2 —, —CH 2 NHSO 2 CH 2 —, —CH 2 CH 2 C(O)NH—, —CH 2 CH 2 SO 2 NH—, —CH 2 NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
- R 3 and R 4 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R 8 ;
- the present invention provides a compound of Formula Ic
- Z 12 , Z 13 , Z 14 , Z 15 , Z 16 and Z 17 are independently CH or N;
- R 3 and R 4 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R 8 ;
- R 9 and R 19 are independently hydroxyl, thiol, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1 -6alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH 3 ), or —NR 7 R 8 ;
- oxygen atom in one or both of the tetrahydrofuranyl rings of Formula Ic is replaced by a sulfur or a selenium atom.
- the present invention provides a compound of Formula Id
- X 1 and X 2 are independently O, S or Se;
- L 1 starting from the carbon alpha to X 1
- L 2 starting from the carbon alpha to X 2
- L 1 is independently —CH 2 O—P(O)R 6 —O—, —CH 2 O—P(S)R 6 —O—, —C(Y 1 )(Y 2 )O—P(O)R 6 —C(Y 3 )(Y 4 )—, —CH 2 NHSO 2 NH—, —CH 2 NHC(O)NH—, —CH 2 NHC(S)NH—, —CH 2 NHC(NH)NH—, —CH 2 NHC(O)CH 2 —, —CH 2 NHSO 2 CH 2 —, —CH 2 CH 2 C(O)NH—, —CH 2 CH 2 SO 2 NH—, —CH 2 NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
- R 3 and R 4 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R 8 ;
- R 11 and R 12 are independently selected from the group consisting of:
- the present invention provides a compound of Formula Ie
- X 3 , X 4 , X 5 , and X 6 are independently O, NH, CH 2 , CHF, or CF2;
- R 13 and R H are independently selected from the group consisting of:
- R 3 and R 4 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R 8 ; and
- R 15 and R 16 are independently hydroxyl, thiol, methoxy, ethoxy, amino, N-methylamino, N,N-dimethylamino, N-ethylamino, N,N-diethylamino, N-morpholino, or borano (—BH 3 );
- oxygen atom in one or both of the tetrahydrofuranyl rings of Formula Ie is replaced by a sulfur or a selenium atom.
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the compound is:
- the present invention provides a compound of Formula If
- X 1 and X 2 are independently O, S or Se;
- Z 12 , Z 13 , Z 14 , Z 15 , Z 16 and Z 17 are independently CH or N;
- L 1 starting from the carbon alpha to X 1
- L 2 starting from the carbon alpha to X 2
- L 1 is independently —CH 2 O—P(O)R 6 —O—, —CH 2 O—P(S)R 6 —O—, —C(Y 1 )(Y 2 )O—P(O)R 6 —C(Y 3 )(Y 4 )—, —CH 2 NHSO 2 NH—, —CH 2 NHC(O)NH—, —CH 2 NHC(S)NH—, —CH 2 NHC(NH)NH—, —CH 2 NHC(O)CH 2 —, —CH 2 NHSO 2 CH 2 —, —CH 2 CH 2 C(O)NH—, —CH 2 CH 2 SO 2 NH—, —CH 2 NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
- R 3 and R 5 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R 8 ;
- the present invention provides a compound of Formula Ig
- Z 12 , Z 13 , Z 14 , Z 15 , Z 16 and Z 17 are independently CH or N;
- R 3 and R 5 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R 8 ;
- R 9 and R 19 are independently hydroxyl, thiol, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1 -6alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH 3 ), or —NR 7 R 8 ;
- the oxygen atom in one or both of the tetrahydrofuranyl rings of Formula Ig is replaced by a sulfur or a selenium atom.
- the present invention provides a compound of Formula Ih
- X 1 and X 2 are independently O, S or Se;
- L 1 starting from the carbon alpha to X 1
- L 2 starting from the carbon alpha to X 2
- L 1 is independently —CH 2 O—P(O)R 6 —O—, —CH 2 O—P(S)R 6 —O—, —C(Y 1 )(Y 2 )O—P(O)R 6 —C(Y 3 )(Y 4 )—, —CH 2 NHSO 2 NH—, —CH 2 NHC(O)NH—, —CH 2 NHC(S)NH—, —CH 2 NHC(NH)NH—, —CH 2 NHC(O)CH 2 —, —CH 2 NHSO 2 CH 2 —, —CH 2 CH 2 C(O)NH—, —CH 2 CH 2 SO 2 NH—, —CH 2 NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
- R 3 and R 5 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R 8 ;
- R 11 and R 12 are independently selected from the group consisting of:
- W 3 , W 4 , W 5 , W 6 , W 7 , W 8 , and W 9 are independently hydrogen, halogen, hydroxyl, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1 -6alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(
- the present invention provides a compound of Formula Ii
- X 3 , X 5 , X 6 , and X 7 are independently O, NH, CH 2 , CHF, or CF2;
- R 13 and R H are independently selected from the group consisting of:
- R 3 and R 5 are independently hydrogen, halogen, hydroxyl, amino, C 1-6 alkyl, C 1-6 alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 alkoxy, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 1-6 alkoxy, C 1-6 alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C 1-6 hydroxyalkoxy, amino, C 1-6 alkylamino, di(C 1-6 alkyl)amino, or azido groups, C 3-5 alkenyl-O—, C 3-5 alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR 7 R 8 ; and
- oxygen atom in one or both of the tetrahydrofuranyl rings of Formula Ii is replaced by a sulfur or a selenium atom.
- the compound is:
- the compound is:
- the present invention provides a method of treating a disease or condition in which modulation of STING is beneficial comprising: administering to a patient in need thereof a compound of Formula I or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in the treatment of a disease or condition in which modulation of STING is beneficial.
- the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in therapy.
- the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, or a pharmaceutically composition thereof, such as a nanoparticle or a delivery vehicles that enhances the cellular uptake, stability and efficacy of a compound of Formula I for use in the treatment of cancer.
- the present invention provides a method of treating cancer comprising: administering a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof, in the manufacture of a medication for the treatment of cancer.
- the present invention provides pharmaceutical composition
- a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, at least one further therapeutic agent, and one or more of pharmaceutically acceptable excipients.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in therapy.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of a disease or condition for which modulation of STING is beneficial.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of cancer.
- the present invention provides a method for treating a disease or condition for which modulation of STING is beneficial comprising: administering to a patient in need thereof a therapeutically effective amount of a combination comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- the therapeutic agent includes but is not limited to immune checkpoint inhibitors, such as humanized antibodies against PD1, PD-L1, CTLA4 and other molecules that block effective anti-tumor immune responses.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of cancer.
- the therapeutic agent includes radiation, such as high-dose radiation, which directly kills tumor cells, enhances presentation of tumor antigens and activates the STING pathway.
- the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- the therapeutic agent includes radiation, such as high-dose radiation, which directly kills tumor cells, enhances presentation of tumor antigens and activates the STING pathway.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of cancer.
- the therapeutic agent includes another chemotherapeutic agent that selectively kills tumor cells and enhances presentation of tumor antigens.
- the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- the therapeutic agent includes another chemotherapeutic agent that selectively kills tumor cells and enhances presentation of tumor antigens.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, a pharmaceutical formulation including a nanoparticle, and at least one further therapeutic agent for use in the treatment of cancer.
- the therapeutic agent includes radiation and/or another chemotherapeutic agent.
- the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, a pharmaceutical formulation including a nanoparticle, and at least one further therapeutic agent for use in the treatment of cancer.
- the therapeutic agent includes radiation and/or another chemotherapeutic agent.
- the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, a pharmaceutical formulation including a nanoparticle, and at least one further therapeutic agent for use in the treatment of cancer.
- the compound of Formula I may be injected directly to tumors, or systemically, including injection into muscles (intramuscular), skins (subcutaneous and intra-dermal), peritoneal (intraperitoneal), lymph nodes (intralymphatic) or veins (intravenous).
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use as a vaccine adjuvant.
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, such as a nanoparticle or a delivery vehicles that enhances the cellular uptake, stability and efficacy of a compound of Formula I, for use as a vaccine adjuvant.
- the pharmaceutical composition is a vaccine.
- the present invention provides a method of inducing or promoting an immune response comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, as an adjuvant and a tumor antigen.
- the present invention provides a method of inducing or promoting an immune response comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutical composition thereof, as an adjuvant, a tumor antigen, or a pharmaceutical composition thereof, such as a nanoparticle or a delivery vehicles that enhances the cellular uptake of the adjuvant and tumor antigen.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, as an adjuvant and an immunogen for a target pathogen.
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use as a vaccine adjuvant.
- the present invention provides a method of inducing or promoting an immune response comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, as an adjuvant and an immunogen for a target pathogen.
- the present invention provides a vaccine adjuvant comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- the present invention provides an immunogenic composition
- an immunogenic composition comprising: an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- the present invention provides an immunogenic composition
- an immunogenic composition comprising: an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use in the treatment or prevention of a disease, including cancer and infectious diseases.
- the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof, for the manufacture of an immunogenic composition comprising an antigen or antigen composition, for the treatment or prevention of a disease, including cancer and infectious diseases.
- the present invention provides a method of treating or preventing a disease comprising: administering to a patient suffering from or susceptible to the disease, an immunogenic composition comprising an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- the present invention provides a vaccine composition
- a vaccine composition comprising: an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use in the treatment or prevention of a disease, including cancer and infectious diseases.
- the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof, for the manufacture of a vaccine composition comprising an antigen or antigen composition for the treatment or prevention of a disease, including cancer and infectious diseases.
- the present invention provides a method of treating or preventing disease comprising the administration to a patient suffering from or susceptible to the disease, a vaccine composition comprising an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use in the treatment of immune disorders, including autoimmune and autoinflammatory diseases.
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, or a pharmaceutically composition thereof, such as a nanoparticle or a delivery vehicles that enhances the cellular uptake, stability and efficacy of a compound of Formula I, for use in the treatment of immune disorders, including autoimmune and autoinflammatory diseases.
- the present invention provides a method of treating immune disorders comprising: administering a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof, in the manufacture of a medication for the treatment of immune disorders, including autoimmune and autoinflammatory diseases.
- the present invention provides novel cGAMP analogs, pharmaceutical compositions thereof, and uses thereof in therapy.
- 2′3′-cGAMP is an endogenous second messenger produced by mammalian cells, it is a high affinity ligand for STING, inducing conformational changes therein, and a potent inducer of type-1 interferons.
- cGAS and the cGAS-cGAMP pathway is important for triggering inflammatory responses to self and foreign DNA. As such, cGAS is important for immune defense against microbial pathogens that contain DNA and require DNA in their life cycles. These pathogens include DNA viruses, retroviruses including HIV, bacteria including Mycobacterium tuberculosis , fungi and parasites.
- cGAS can also detect tumor DNA and is important for the body's intrinsic immunity against malignant cells. Activation of the cGAS-cGAMP-STING pathway is important for cancer immunotherapy.
- cGAMP As a potent inducer of type-I interferons, cGAMP (and hence the cGAMP analogs of the present invention) provides a rational immune adjuvant.
- a compound of Formula I or a pharmaceutically acceptable salt thereof may be used as a vaccine adjuvant, particularly with mucosal vaccines, and may be formulated with immunogens and delivered as have been cyclic-di-GMP and c-di-AMP as vaccine adjuvants (see, e.g. Pedersen, et al.
- the invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in the treatment of cancer.
- the pharmaceutical composition is a compound of Formula I.
- the pharmaceutical composition is a compound of Formula I in a pharmaceutical formulation including a nanoparticle or another delivery vehicle.
- the pharmaceutical composition is a compound of Formula I in combination with at least one further therapeutic agent, which includes but is not limited to immune checkpoint inhibitors such as antibodies against PD-1, PD-L1 or CTLA-4.
- the therapeutic agent used in combination with a compound of Formula I also includes radiation of tumors or a chemotherapeutic agent that targets tumor cells.
- the invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, as an adjuvant and an immunogen for a target pathogen.
- the pharmaceutical composition is a vaccine.
- the present invention provides a method of inducing or promoting an immune response comprising: administering to a patient in need thereof an effective amount a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, as an adjuvant and an immunogen for a target pathogen.
- halo and “halogen”, alone or in combination with other groups, refers to fluoro-, chloro-, bromo- and iodo-.
- C 1-6 alkyl refers to monovalent, linear chain or branched chain alkyl groups containing from 1 to 6 carbon atoms.
- Exemplary C 1-6 alkyl groups include but not limited to metheyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl and tert-butyl groups. More preferred are C 1-4 alkyls.
- C 1-6 alkoxy refers to, alone or in combination with other groups, R′—O—, where R′ is C 1-6 alkyl.
- haloC 1-6 alkyl refers to a C 1-6 alkyl group subsituted with one or more halo suctsitutents, for example CF 3 and CH 2 CF 3 .
- a compound of the invention or “a compound of Formula I” includes all solvates, complexes, polymorphs, radiolabeled derivatives, tautomers, stereoisomers, and optical isomers of the compounds of Formula I and salts thereof, unless otherwise specified.
- the term “effective amount” means that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system, animal or human that is being sought, for instance, by a researcher or clinician.
- terapéuticaally effective amount means any amount which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder.
- the term also includes within its scope amounts effective to enhance normal physiological function.
- prophylaxis includes prevention and refers to a measure or procedure which is to prevent rather than cure or treat a disease. Preventing refers to a reduction in risk of acquiring or developing a disease causing at least one clinical symptom of the disease not to developing a subject that may be exposed to a disease-causing agent or a subject predisposed to the disease in advance of disease outset.
- pharmaceutically acceptable refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable excipients includes all diluents, carriers, binders, glidants, and other components of pharmaceutical formulations with which the compound of the invention is administered.
- the compounds of the invention may exist in solid or liquid form.
- compound of the invention may exist in a continuum of solid states ranging from fully amorphous to fully crystalline.
- amorphous refers to a state in which the material lacks long range order at the molecular level and, depending upon the temperature, may exhibit the physical properties of a solid or a liquid. Typically, such materials do not give distinctive X-ray diffraction patterns and, while exhibiting the properties of a solid, are more formally described as a liquid. Upon heating, a change from solid to liquid properties occurs which is characterized by a change of state, typically second order (‘glass transition’).
- crystalline refers to a solid phase in which the material has a regular ordered internal structure at the molecular level and gives a distinctive X-ray diffraction pattern with defined peaks. Such materials when heated sufficiently will also exhibit the properties of a liquid, but the change from solid to liquid is characterized by a phase change, typically first order (‘melting point’).
- the compounds of the invention may have the ability to crystallize in more than one form, a characteristic, which is known as polymorphism, and it is understood that such polymorphic forms (“polymorphs”) are within the scope of the invention.
- Polymorphism generally can occur as a response to changes in temperature or pressure or both and can also result from variations in the crystallization process.
- Polymorphs can be distinguished by various physical characteristics known in the art such as x-ray diffraction patterns, solubility and melting point.
- the compound of Formula I may exist in solvated and unsolvated forms.
- solvate refers to a complex of variable stoichiometry formed by a solute (in this invention, a compound of Formula I or a salt) and a solvent. Such solvents for the purpose of the invention may not interfere with the biological activity of the solute.
- pharmaceutically acceptable solvates may be formed for crystalline compounds wherein solvent molecules are incorporated into the crystalline lattice during crystallization.
- the incorporated solvent molecules may be water molecules or non-aqueous such as ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and ethyl acetate molecules. Crystalline lattice incorporated with water molecules are typically referred to as “hydrates”. Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The present invention includes all such solvates.
- Tautomers refer to compounds that are interchangeable forms of a particular compound structure, and that vary in the displacement of hydrogen atoms and electrons. Thus, two structures may be in equilibrium through the movement of re electrons and an atom (usually H). For example, enols and ketones are tautomers because they are rapidly interconverted by treatment with either acid or base. It is understood that all tautomers and mixtures of tautomers of the compounds of the present invention are included within the scope of the compounds of the present invention.
- the compounds of Formula I may be in the form of a salt.
- the salts of the present invention are pharmaceutically acceptable salts.
- Salts encompassed within the term “pharmaceutically acceptable salts” refer to non-toxic salts of the compounds of this invention.
- suitable salts see e.g., Berge et al, J. Pharm. Sci. 1977, 66, 1-19.
- suitable pharmaceutically acceptable salts can include acid addition salts.
- a pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of Formula I with a suitable inorganic or organic acid (such as hydrobromic, hydrochloric, sulfuric, nitric, phosphoric, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic), optionally in a suitable solvent such as an organic solvent, to give the salt which is usually isolated for example by crystallization and filtration.
- a suitable inorganic or organic acid such as hydrobromic, hydrochloric, sulfuric, nitric, phosphoric, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic
- a suitable solvent such as an organic solvent
- a pharmaceutically acceptable acid addition salt of a compound of Formula I can be, for example, a hydrobromide, hydrochloride, sulfate, nitrate, phosphate, p-toluenesulfonate, benzenesulfonate, methanesulfonate, ethanesulfonate, or naphthalenesulfonate (e.g. 2-naphthalenesulfonate) salt.
- Other non-pharmaceutically acceptable salts e.g. trifluoroacetates, may be used, for example in the isolation of compounds of the invention, and are included within the scope of this invention.
- the invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the compounds of Formula I.
- compositions can be prepared in a manner well known in the pharmaceutical art and comprise at least one active compound. Accordingly, the invention further provides pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically acceptable excipients.
- the excipient(s) must be acceptable in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof.
- a process for the preparation of a pharmaceutical composition including the agent, or pharmaceutically acceptable salts thereof, with one or more pharmaceutically acceptable excipients.
- the pharmaceutical composition can be for use in the treatment and/or prophylaxis of any of the conditions described herein.
- the compound of the invention is administered in a pharmaceutically effective amount.
- the amount of the compound actually administered will typically be determined by a physician, in the light of the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered, the age, weight, and response of the individual patient, the severity of the patient's symptoms, and the like.
- Pharmaceutical compositions may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose.
- unit dosage forms refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient, vehicle or carrier.
- Typical unit dosage forms include prefilled, premeasured ampules or syringes of the liquid compositions or pills, tablets, capsules or the like in the case of solid compositions.
- Preferred unit dosage compositions are those containing a daily dose or sub-dose, or an appropriate fraction thereof, of an active ingredient. Such unit doses may therefore be administered once or more than once a day.
- Such pharmaceutical compositions may be prepared by any of the methods well known in the pharmacy art.
- compositions may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual), rectal, inhaled, intranasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal) route.
- Such compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier(s) or excipient(s).
- compositions adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids, edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert excipient such as ethanol, glycerol, water and the like.
- an oral, non-toxic pharmaceutically acceptable inert excipient such as ethanol, glycerol, water and the like.
- Powders are prepared by reducing the compound to a suitable fine size and mixing with a similarly prepared pharmaceutical excipient such as an edible carbohydrate, as, for example, starch or mannitol. Flavoring, preservative, dispersing and coloring agent can also be present.
- Capsules are made by preparing a powder mixture, as described above, and filling formed gelatin sheaths. Excipients including glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation. A disintegrating or solubilizing agent such as agar-agar, calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
- Excipients including glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation.
- a disintegrating or solubilizing agent such as agar-agar, calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
- excipients including suitable binders, glidants, lubricants, sweetening agents, flavors, disintegrating agents and coloring agents can also be incorporated into the mixture.
- suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like.
- Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
- Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like. Tablets are formulated, for example, by preparing a powder mixture, granulating or slugging, adding a lubricant and disintegrant and pressing into tablets.
- a powder mixture is prepared by mixing the compound, suitably comminuted, with a diluent or base as described above, and optionally, with a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone, a solution retardant such as paraffin, a resorption accelerator such as a quaternary salt and/or an absorption agent such as bentonite, kaolin or dicalcium phosphate.
- the powder mixture can be granulated by wetting with a binder such as syrup, starch paste, acadia mucilage or solutions of cellulosic or polymeric materials and forcing through a screen.
- the powder mixture can be run through the tablet machine and the result is imperfectly formed slugs broken into granules.
- the granules can be lubricated to prevent sticking to the tablet forming dies by means of the addition of stearic acid, a stearate salt, talc or mineral oil.
- the lubricated mixture is then compressed into tablets.
- the compounds of the present invention can also be combined with a free flowing inert carrier and compressed into tablets directly without going through the granulating or slugging steps.
- a clear or opaque protective coating consisting of a sealing coat of shellac, a coating of sugar or polymeric material and a polish coating of wax can be provided. Dyestuffs can be added to these coatings to distinguish different unit dosages.
- Oral fluids such as solution, suspensions, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of the compound.
- Syrups can be prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound in a non-toxic vehicle.
- Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
- dosage unit compositions for oral administration can be microencapsulated.
- the composition can also be prepared to prolong or sustain the release as for example by coating or embedding particulate material in polymers, wax or the like.
- the compounds of the invention may also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
- Pharmaceutical compositions adapted for transdermal administration may be presented as discrete patches intended to remain in intimate contact with the epidermis of the recipient for a prolonged period of time.
- compositions adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- compositions are preferably applied as a topical ointment or cream.
- the active ingredient may be employed with either a paraffinic or a water-miscible ointment base.
- the active ingredient may be formulated in a cream with an oil-in-water cream base or a water-in-oil base.
- compositions adapted for topical administrations to the eye include eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent.
- compositions adapted for topical administration in the mouth include lozenges, pastilles and mouth washes.
- compositions adapted for rectal administration may be presented as suppositories or as enemas.
- Dosage forms for nasal or inhaled administration may conveniently be formulated as aerosols, solutions, suspension drops, gels or dry powders.
- compositions for intranasal administration include aqueous compositions administered to the nose by drops or by pressurised pump. Suitable compositions contain water as the diluent or carrier for this purpose.
- Compositions for administration to the lung or nose may contain one or more excipients, for example one or more suspending agents, one or more preservatives, one or more surfactants, one or more tonicity adjusting agents, one or more co-solvents, and may include components to control the pH of the composition, for example a buffer system. Further, the compositions may contain other excipients such as antioxidants, for example sodium metabisulphite, and taste-masking agents. Compositions may also be administered to the nose or other regions of the respiratory tract by nebulization.
- Intranasal compositions may permit the compound(s) of Formula I or (a) pharmaceutically acceptable salt(s) thereof to be delivered to all areas of the nasal cavities (the target tissue) and further, may permit the compound(s) of Formula I or (a) pharmaceutically acceptable salt(s) thereof to remain in contact with the target tissue for longer periods of time.
- a suitable dosing regime for intranasal compositions would be for the patient to inhale slowly through the nose subsequent to the nasal cavity being cleared. During inhalation, the composition would be administered to one nostril while the other is manually compressed. This procedure would then be repeated for the other nostril. Typically, one or two sprays per nostril would be administered by the above procedure one, two, or three times each day, ideally once daily.
- intranasal compositions suitable for once-daily administration.
- the suspending agent(s), if included, will typically be present in an amount of from 0.1 to 5% (w/w), such as from 1.5% to 2.4% (w/w), based on the total weight of the composition.
- pharmaceutically acceptable suspending agents include, but are not limited to, Avicef (microcrystalline cellulose and carboxymethylcellulose sodium), carboxymethylcellulose sodium, veegum, tragacanth, bentonite, methylcellulose, xanthan gum, carbopol and polyethylene glycols.
- compositions for administration to the lung or nose may contain one or more excipients may be protected from microbial or fungal contamination and growth by inclusion of one or more preservatives.
- pharmaceutically acceptable anti-microbial agents or preservatives include, but are not limited to, quaternary ammonium compounds (for example benzalkonium chloride, benzethonium chloride, cetrimide, cetylpyridinium chloride, lauralkonium chloride and myristyl picolinium chloride), mercurial agents (for example phenylmercuric nitrate, phenylmercuric acetate and thimerosal), alcoholic agents (for example chlorobutanol, phenylethyl alcohol and benzyl alcohol), antibacterial esters (for example esters of para-hydroxybenzoic acid), chelating agents such as disodium edetate (EDTA) and other anti-microbial agents such as chlorhexidine, chlorocresol, sorbic acid and its salts (such
- compositions may include one or more surfactants which functions to facilitate dissolution of the medicament particles in the aqueous phase of the composition.
- the amount of surfactant used is an amount which will not cause foaming during mixing.
- Examples of pharmaceutically acceptable surfactants include fatty alcohols, esters and ethers, such as polyoxyethylene (20) sorbitan monooleate (Polysorbate 80), macrogol ethers, and poloxamers.
- the surfactant may be present in an amount of between about 0.01 to 10% (w/w), such as from 0.01 to 0.75% (w/w), for example about 0.5% (w/w), based on the total weight of the composition.
- One or more tonicity-adjusting agent(s) may be included to achieve tonicity with body fluids e.g. fluids of the nasal cavity, resulting in reduced levels of irritancy.
- pharmaceutically acceptable tonicity-adjusting agents include, but are not limited to, sodium chloride, dextrose, xylitol, calcium chloride, glucose, glycerine and sorbitol.
- a tonicity-adjusting agent, if present, may be included in an amount of from 0.1 to 10% (w/w), such as from 4.5 to 5.5% (w/w), for example about 5.0% (w/w), based on the total weight of the composition.
- compositions of the invention may be buffered by the addition of suitable buffering agents such as sodium citrate, citric acid, trometamol, phosphates such as disodium phosphate (for example the dodecahydrate, heptahydrate, dihydrate and anhydrous forms), or sodium phosphate and mixtures thereof.
- suitable buffering agents such as sodium citrate, citric acid, trometamol, phosphates such as disodium phosphate (for example the dodecahydrate, heptahydrate, dihydrate and anhydrous forms), or sodium phosphate and mixtures thereof.
- a buffering agent if present, may be included in an amount of from 0.1 to 5% (w/w), for example 1 to 3% (w/w) based on the total weight of the composition.
- taste-masking agents include sucralose, sucrose, saccharin or a salt thereof, fructose, dextrose, glycerol, corn syrup, aspartame, acesulfame-K, xylitol, sorbitol, erythritol, ammonium glycyrrhizinate, thaumatin, neotame, mannitol, menthol, eucalyptus oil, camphor, a natural flavouring agent, an artificial flavouring agent, and combinations thereof.
- co-solvent(s) may be included to aid solubility of the medicament compound(s) and/or other excipients.
- pharmaceutically acceptable co-solvents include, but are not limited to, propylene glycol, dipropylene glycol, ethylene glycol, glycerol, ethanol, polyethylene glycols (for example PEG300 or PEG400), and methanol.
- the co-solvent is propylene glycol.
- Co-solvent(s), if present, may be included in an amount of from 0.05 to 30% (w/w), such as from 1 to 25% (w/w), for example from 1 to 10% (w/w) based on the total weight of the composition.
- compositions for inhaled administration include aqueous, organic or aqueous/organic mixtures, dry powder or crystalline compositions administered to the respiratory tract by pressurised pump or inhaler, for example, reservoir dry powder inhalers, unit-dose dry powder inhalers, pre-metered multi-dose dry powder inhalers, nasal inhalers or pressurised aerosol inhalers, nebulisers or insufflators.
- Suitable compositions contain water as the diluent or carrier for this purpose and may be provided with conventional excipients such as buffering agents, tonicity modifying agents and the like.
- Aqueous compositions may also be administered to the nose and other regions of the respiratory tract by nebulisation.
- Such compositions may be aqueous solutions or suspensions or aerosols delivered from pressurised packs, such as a metered dose inhaler, with the use of a suitable liquefied propellant.
- compositions for administration topically to the nose (for example, for the treatment of rhinitis) or to the lung include pressurised aerosol compositions and aqueous compositions delivered to the nasal cavities by pressurised pump.
- Compositions which are non-pressurised and are suitable for administration topically to the nasal cavity are of particular interest. Suitable compositions contain water as the diluent or carrier for this purpose.
- Aqueous compositions for administration to the lung or nose may be provided with conventional excipients such as buffering agents, tonicity-modifying agents and the like. Aqueous compositions may also be administered to the nose by nebulisation.
- a fluid dispenser may typically be used to deliver a fluid composition to the nasal cavities.
- the fluid composition may be aqueous or non-aqueous, but typically aqueous.
- Such a fluid dispenser may have a dispensing nozzle or dispensing orifice through which a metered dose of the fluid composition is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser.
- Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid composition, the doses being dispensable upon sequential pump actuations.
- the dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid composition into the nasal cavity.
- Dry powder compositions for topical delivery to the lung by inhalation may, for example, be presented in capsules and cartridges of for example gelatine, or blisters of for example laminated aluminium foil, for use in an inhaler or insufflator.
- Powder blend compositions generally contain a powder mix for inhalation of the compound of Formula 1 or a pharmaceutically acceptable salt thereof and a suitable powder base (carrier/diluent/excipient substance) such as mono-, di-, or polysaccharides (for example lactose or starch).
- Dry powder compositions may also include, in addition to the drug and carrier, a further excipient (for example a ternary agent such as a sugar ester for example cellobiose octaacetate, calcium stearate, or magnesium stearate.
- a further excipient for example a ternary agent such as a sugar ester for example cellobiose octaacetate, calcium stearate, or magnesium stearate.
- compositions adapted for parental administration include aqueous and nonaqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the composition isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the compositions may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- compositions may include other agents conventional in the art having regard to the type of formulation in question, for example, those suitable for oral administration may include flavoring agents.
- compositions may contain antibody(ies) or antibody fragment(s) or an antigenic component including but not limited to protein, DNA, live or dead bacteria and/or viruses or virus-like particles, together with one or more components with adjuvant activity including but not limited to aluminium salts, oil and water emulsions, heat shock proteins, lipid A preparations and derivatives, glycolipids, other TLR agonists such as CpG DNA or similar agents, cytokines such as GM-CSF or IL-12 or similar agents.
- antibody(ies) or antibody fragment(s) or an antigenic component including but not limited to protein, DNA, live or dead bacteria and/or viruses or virus-like particles, together with one or more components with adjuvant activity including but not limited to aluminium salts, oil and water emulsions, heat shock proteins, lipid A preparations and derivatives, glycolipids, other TLR agonists such as CpG DNA or similar agents, cytokines such as GM-CSF or IL-12 or similar agents.
- a therapeutically effective amount of the agent will depend upon a number of factors including, for example, the age and weight of the subject, the precise condition requiring treatment and its severity, the nature of the formulation, and the route of administration, and will ultimately be at the discretion of the attendant physician or veterinarian.
- the subject to be treated is a mammal, particularly a human.
- the agent may be administered in a daily dose. This amount may be given in a single dose per day or more usually in a number (such as two, three, four, five or six) of sub-doses per day such that the total daily dose is the same.
- the amount of the compound of the invention administered according to the present invention will be an amount selected from about 0.01 mg to about 1 g per day (calculated as the free or unsalted compound).
- the compounds of Formula I and pharmaceutically acceptable salts thereof may be employed alone or in combination with other therapeutic agents.
- the compounds of Formula I and pharmaceutically acceptable salts thereof and the other pharmaceutically active agent(s) may be administered together or separately and, when administered separately, administration may occur simultaneously or sequentially, in any order, by any convenient route in separate or combined pharmaceutical compositions.
- the amounts of the compound(s) of Formula I or pharmaceutically acceptable salt(s) thereof and the other pharmaceutically active agent(s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect.
- the compound(s) of Formula I or pharmaceutically acceptable salt(s) thereof and further therapeutic agent(s) may be employed in combination by administration simultaneously in a unitary pharmaceutical composition including both compounds.
- the combination may be administered separately in separate pharmaceutical compositions, each including one of the compounds in a sequential manner wherein, for example, the compound of the invention is administered first and the other second and vice versa.
- Such sequential administration may be close in time (e.g. simultaneously) or remote in time.
- the compounds are administered in the same dosage form, e.g. one compound may be administered topically and the other compound may be administered orally.
- both compounds are administered orally.
- combination kit or kit of parts as used herein is meant the pharmaceutical composition or compositions that are used to administer the combination according to the invention.
- the combination kit can contain both compounds in a single pharmaceutical composition, such as a tablet, or in separate pharmaceutical compositions.
- the combination kit will contain each compound in separate pharmaceutical compositions either in a single package or in separate pharmaceutical compositions in separate packages.
- the combination kit can also be provided by instruction, such as dosage and administration instructions.
- dosage and administration instructions can be of the kind that are provided to a doctor, for example by a drug product label, or they can be of the kind that are provided by a doctor, such as instructions to a patient.
- such sequential administration may be close in time or remote in time.
- administration of the other agent several minutes to several dozen minutes after the administration of the first agent, and administration of the other agent several hours to several days after the administration of the first agent are included, wherein the lapse of time is not limited.
- one agent may be administered once a day, and the other agent may be administered 2 or 3 times a day, or one agent may be administered once a week, and the other agent may be administered once a day and the like.
- the other therapeutic ingredients(s) may be used in the form of salts, for example as alkali metal or amine salts or as acid addition salts, or prodrugs, or as esters, for example lower alkyl esters, or as solvates, for example hydrates, to optimize the activity and/or stability and/or physical characteristics, such as solubility, of the therapeutic ingredient. It will be clear also that, where appropriate, the therapeutic ingredients may be used in optically pure form.
- the two compounds When combined in the same composition it will be appreciated that the two compounds must be stable and compatible with each other and the other components of the composition and may be formulated for administration. When formulated separately they may be provided in any convenient composition, conveniently, in such a manner as known for such compounds in the art.
- the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art.
- the patient in the methods and uses of the present invention is a mammal. In another embodiment, the patient is a human.
- the compounds of the invention are useful in the treatment of diseases and conditions in which modulation of STING is beneficial, including cancer. As modulators of the immune response, the compounds of Formula I and pharmaceutically acceptable salts thereof may also be useful, as stand-alone, in combination or as adjuvants, in the treatment of diseases and conditions in which modulation of STING is beneficial.
- the disease or condition to be treated is cancer.
- cancer diseases and conditions in which a compound of Formula I or pharmaceutically acceptable salt thereof, may have potentially beneficial anti-tumor effects include cancers of the lung, bone, pancreas, skin, head, neck, uterus, ovaries, stomach, colon, breast, esophagus, small intestine, bowel, endocrine system, thyroid gland, parathyroid gland, adrenal gland, urethra, prostate, penis, testes, ureter, bladder, kidney or liver; rectal cancer; cancer of the anal region; carcinomas of the fallopian tubes, endometrium, cervix, vagina, vulva, renal pelvis, renal cell; sarcoma of soft tissue; myxoma; rhabdomyoma; fibroma; lipoma; teratoma; cholangiocarcinoma; hepatoblastoma; angiosarcoma; hemagioma; hepatom
- the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in the treatment of cancer.
- the present invention provides a method of treating cancer comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula I or a pharmaceutically acceptable salt thereof.
- the present invention provides the use of a compound of Formula I or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of cancer.
- a compound of the invention may be employed with other therapeutic methods of cancer treatment, e.g., in anti-neoplastic therapy, combination therapy with immune checkpoint inhibitors, other chemotherapeutic, hormonal, antibody agents as well as surgical and/or radiation treatments.
- Immune checkpoint inhibitors such as humanized antibodies against PD-1, PD-L1 and CTLA4, have recently been shown to be highly successful in treating several types of metastatic cancer, including melanoma, non-small cell lung cancers, renal cell carcinoma and bladder cancer (Sharma and Allison, 2015, Science 348, 56).
- checkpoint inhibitor therapies in part because insufficient number of anti-tumor immune cells, such as CD8 T cells, are generated and/or infiltrated into the tumors.
- Activation of the cGAS-STING pathway activates anti-tumor immunity, including the production and infiltration of tumor-specific CD8 T cells. Therefore, cGAMP analogues are expected to function synergistically with immune checkpoint inhibitors and the combination therapies are likely to bring therapeutic benefits to a larger percentage of cancer patients.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one immune checkpoint inhibitor.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one immune checkpoint inhibitor for use in therapy.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or pharmaceutically acceptable salt thereof, and at least one immune checkpoint inhibitor for use in treating cancer.
- the present invention provides the use of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one immune checkpoint inhibitor in the manufacture of a medicament for the treatment of cancer.
- the present invention provides a method of treating cancer, comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least immune checkpoint inhibitor.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, at least one immune checkpoint inhibitor, and one or more of pharmaceutically acceptable carriers, diluents and excipients.
- cGAMP analogues are expected to function synergistically with radiation therapies to benefit a larger percentage of cancer patients.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in combination with radiation therapy such as SBRT.
- the present invention provides a pharmaceutical composition comprising a compound of Formula I or pharmaceutically acceptable salt thereof, in combination with radiation therapy such as SBRT for use in treating cancer.
- the present invention provides the use of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in combination with radiation therapy such as SBRT in the manufacture of a medicament for the treatment of cancer.
- the present invention provides a method of treating cancer, comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in combination with radiation therapy such as SBRT.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and one or more of pharmaceutically acceptable carriers, diluents and excipients, in combination with radiation therapy such as SBRT for the treatment of cancer.
- Anti-neoplastic agents include chemical compounds and antibodies that kill tumor cells by inhibiting cell cycle, signal transduction, DNA metabolism and angiogenesis and/or by promoting DNA damage, apoptosis and necrosis. These agents comprise that largest class of molecules currently used for cancer therapies. Anti-neoplastic agents selectively kill tumor cells, although many of them also kill normal cells, thereby generating severe side effects. Processing of dead tumor cell associated antigens by antigen presenting cells leads to the generation of tumor-specific cytotoxic T cells. This process can be enhanced by cGAMP analogues. Therefore, combination of cGAMP analogues with anti-neoplastic agents are likely to generate synergistic effects that benefit a larger percentage of patients.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent for use in therapy.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent for use in treating cancer.
- the present invention provides the use of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent in the manufacture of a medicament for the treatment of cancer.
- the present invention provides a method of treating cancer, comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, at least one anti-neoplastic agent, and one or more of pharmaceutically acceptable carriers, diluents and excipients.
- anti-neoplastic agent that has activity versus a susceptible tumor being treated may be utilized in the combination.
- Typical anti-neoplastic agents useful include anti-microtubule agents such as diterpenoids and vinca alkaloids; platinum coordination complexes; alkylating agents such as nitrogen mustards, oxazaphosphorines, alkylsulfonates, nitrosoureas, and triazenes; antibiotic agents such as anthracycline, actinomycins and bleomycins; topoisomerase II inhibitors such as epipodophyllotoxins; antimetabolites such as purine and pyrimidine analogues and anti-folate compounds; topoisomerase I inhibitors such as camptothecins; hormones and hormonal analogues; signal transduction pathway inhibitors; non-receptor tyrosine angiogenesis inhibitors; immunotherapeutic agents; proapoptotic agents; and cell cycle signaling inhibitors.
- Anti-microtubule or anti-mitotic agents are phase specific agents active against the microtubules of tumor cells during M or the mitosis phase of the cell cycle.
- Examples of anti-microtubule agents include diterpenoids and vinca alkaloids.
- Diterpenoids which are derived from natural sources, are phase specific anti-cancer agents that operate at the G 2 /M phases of the cell cycle. It is believed that the diterpenoids stabilize the ⁇ -tubulin subunit of the microtubules, by binding with this protein. Disassembly of the protein appears then to be inhibited with mitosis being arrested and cell death following. Examples of diterpenoids include paclitaxel and its analog docetaxel.
- Vinca alkaloids are phase specific anti-neoplastic agents derived from the periwinkle plant. Vinca alkaloids act at the M phase (mitosis) of the cell cycle by binding specifically to tubulin. Consequently, the bound tubulin molecule is unable to polymerize into microtubules. Mitosis is believed to be arrested in metaphase with cell death following. Examples of vinca alkaloids include vinblastine, vincristine, and vinorelbine.
- Platinum coordination complexes are non-phase specific anti-cancer agents, which are interactive with DNA.
- the platinum complexes enter tumor cells, undergo, aquation and form intra- and interstrand crosslinks with DNA causing adverse biological effects to the tumor.
- Examples of platinum coordination complexes include oxaliplatin, cisplatin and carboplatin.
- Alkylating agents are non-phase anti-cancer specific agents and strong electrophiles. Typically, alkylating agents form covalent linkages, by alkylation, to DNA through nucleophilic moieties of the DNA molecule such as phosphate, amino, sulfhydryl, hydroxyl, carboxyl, and imidazole groups. Such alkylation disrupts nucleic acid function leading to cell death.
- alkylating agents include nitrogen mustards such as cyclophosphamide, melphalan, and chlorambucil; alkyl sulfonates such as busulfan; nitrosoureas such as carmustine; and triazenes such as dacarbazine.
- Antibiotic anti-neoplastics are non-phase specific agents, which bind or intercalate with DNA. Typically, such action results in stable DNA complexes or strand breakage, which disrupts ordinary function of the nucleic acids leading to cell death.
- antibiotic anti-neoplastic agents include actinomycins such as dactinomycin, anthrocyclins such as daunorubicin and doxorubicin; and bleomycins.
- Topoisomerase II inhibitors include epipodophyllotoxins.
- Epipodophyllotoxins are phase specific anti-neoplastic agents derived from the mandrake plant. Epipodophyllotoxins typically affect cells in the S and G 2 phases of the cell cycle by forming a ternary complex with topoisomerase II and DNA causing DNA strand breaks. The strand breaks accumulate and cell death follows. Examples of epipodophyllotoxins include etoposide and teniposide.
- Antimetabolite neoplastic agents are phase specific anti-neoplastic agents that act at S phase (DNA synthesis) of the cell cycle by inhibiting DNA synthesis or by inhibiting purine or pyrimidine base synthesis and thereby limiting DNA synthesis. Consequently, S phase does not proceed and cell death follows.
- Examples of antimetabolite anti-neoplastic agents include fluorouracil, methotrexate, cytarabine, mecaptopurine, thioguanine, and gemcitabine.
- Camptothecins including, camptothecin and camptothecin derivatives are available or under development as Topoisomerase I inhibitors. Camptothecins cytotoxic activity is believed to be related to its Topoisomerase I inhibitory activity. Examples of camptothecins include, but are not limited to irinotecan, topotecan, and the various optical forms of 7-(4-methylpiperazino-methylene)-10,11-ethylenedioxy-20-camptothecin described below.
- Hormones and hormonal analogues are useful compounds for treating cancers in which there is a relationship between the hormone(s) and growth and/or lack of growth of the cancer.
- hormones and hormonal analogues useful in cancer treatment include adrenocorticosteroids such as prednisone and prednisolone which are useful in the treatment of malignant lymphoma and acute leukemia in children; aminoglutethimide and other aromatase inhibitors such as anastrozole, letrazole, vorazole, and exemestane useful in the treatment of adrenocortical carcinoma and hormone dependent breast carcinoma containing estrogen receptors; progestrins such as megestrol acetate useful in the treatment of hormone dependent breast cancer and endometria 1 carcinoma; estrogens, and anti-estrogens such as fulvestrant, flutamide, nilutamide, bicalutamide, cyproterone acetate and 5a-reductases such as finasteride and
- Signal transduction pathway inhibitors are those inhibitors, which block or inhibit a chemical process which evokes an intracellular change. As used herein this change is cell proliferation or differentiation.
- Signal transduction inhibitors useful in the present invention include inhibitors of receptor tyrosine kinases, non-receptor tyrosine kinases, SH2/SH3 domain blockers, serine/threonine kinases, phosphotidyl inositol-3 kinases, myoinositol signaling, and Ras oncogenes.
- protein tyrosine kinases catalyze the phosphorylation of specific tyrosyl residues in various proteins involved in the regulation of cell growth.
- protein tyrosine kinases can be broadly classified as receptor or non-receptor kinases.
- Receptor tyrosine kinases are transmembrane proteins having an extracellular ligand binding domain, a transmembrane domain, and a tyrosine kinase domain. Receptor tyrosine kinases are involved in the regulation of cell growth and are generally termed growth factor receptors. Inappropriate or uncontrolled activation of many of these kinases, i.e. aberrant kinase growth factor receptor activity, for example by over-expression or mutation, has been shown to result in uncontrolled cell growth. Accordingly, the aberrant activity of such kinases has been linked to malignant tissue growth. Consequently, inhibitors of such kinases could provide cancer treatment methods.
- Growth factor receptors include, for example, epidermal growth factor receptor (EGFr), platelet derived growth factor receptor (PDGFr), erbB2, erbB4, ret, vascular endothelial growth factor receptor (VEGFr), tyrosine kinase with immunoglobulin-like and epidermal growth factor homology domains (TIE-2), insulin growth factor-I (IGFI) receptor, macrophage colony stimulating factor (cfms), BTK, ckit, cmet, fibroblast growth factor (FGF) receptors, Trk receptors (TrkA, TrkB, and TrkC), ephrin (eph) receptors, and the RET protooncogene.
- EGFr epidermal growth factor receptor
- PDGFr platelet derived growth factor receptor
- erbB2 erbB2
- VEGFr vascular endothelial growth factor receptor
- TIE-2 vascular endothelial growth factor receptor
- inhibitors of growth receptors include ligand antagonists, antibodies, tyrosine kinase inhibitors and anti-sense oligonucleotides.
- Growth factor receptors and agents that inhibit growth factor receptor function are described, e.g., in Kath, John C, Exp. Opin. Ther. Patents (2000) 10(6):803-818; Shawver et al. DDT Vol 2, No. 2 Feb. 1997; and Lofts, F. J. et al in “Growth factor receptors as targets”, New Molecular Targets for Cancer Chemotherapy, ed. Workman, Paul and Kerr, David, CRC press 1994, London.
- Non-receptor tyrosine kinases which are not growth factor receptor kinases are termed nonreceptor tyrosine kinases.
- Non-receptor tyrosine kinases useful in the present invention include cSrc, Lck, Fyn, Yes, Jak, cAbl, FAK (Focal adhesion kinase), Brutons tyrosine kinase, and Bcr-Abl.
- Such non-receptor kinases and agents which inhibit non-receptor tyrosine kinase function are described, e.g., in Sinh, S. and Corey, S. J., (1999) Journal of Hematotherapy and Stem Cell Research 8 (5): 465-80; and Bolen, J. B., Brugge, J. S., (1997) Annual review of Immunology. 15: 371-404.
- SH2/SH3 domain blockers are agents that disrupt SH2 or SH3 domain binding in a variety of enzymes or adaptor proteins including PI3-K p85 subunit, Src family kinases, adaptor molecules (She, Crk, Nek, Grb2) and Ras-GAP.
- SH2/SH3 domains as targets for anti-cancer drugs are discussed in Smithgall, T. E. (1995), Journal of Pharmacological and Toxicological Methods. 34(3) 125-32.
- Inhibitors of Serine/Threonine Kinases include MAP kinase cascade blockers which include blockers of Raf kinases (rafk), Mitogen or Extracellular Regulated Kinase (M EKs), and Extracellular Regulated Kinases (ERKs); and Protein kinase C family member blockers including blockers of PKCs (alpha, beta, gamma, epsilon, mu, lambda, iota, zeta).
- I kB kinase family I KKa, I KKb
- PKB family kinases akt kinase family members
- TGF beta receptor kinases TGF beta receptor kinases.
- Serine/Threonine kinases and inhibitors thereof are described, e.g., in Yamamoto, T. et al., (1999), Journal of Biochemistry. 126 (5) 799-803; Brodt, P et al. (2000), Biochemical Pharmacology, 60. 1101-1107; Massague, J., and Weis-Garcia, F. (1996) Cancer Surveys. 27:41-64; Philip, P. A.; and Harris, A. L. (1995), Cancer Treatment and Research. 78: 3-27; Lackey, K. et al Bioorganic and Medicinal Chemistry Letters, (10), 2000, 223-226; U.S. Pat. No. 6,268,391; and Martinez-lacaci, L, et al., Int. J. Cancer (2000), 88(1), 44-52.
- Inhibitors of Phosphotidyl inositol-3 Kinase family members including blockers of PI3-kinase, ATM, DNA-PK, and Ku are also useful in the present invention.
- Such kinases are discussed, e.g., in Abraham, R. T. (1996), Current Opinion in Immunology. 8 (3) 412-8; Canman, C. E., Lim, D. S. (1998), Oncogene 17 (25) 3301-3308; Jackson, S. P. (1997), International Journal of Biochemistry and Cell Biology. 29 (7):935-8; and Zhong, H. et al. Cancer Res., (2000) 60(6), 1541-1545.
- Myo-inositol signaling inhibitors such as phospholipase C blockers and Myoinositol analogues.
- signal inhibitors are described, e.g., in Powis, G., and Kozikowski A., (1994) New Molecular Targets for Cancer Chemotherapy ed., Paul Workman and David Kerr, CRC press 1994, London.
- Ras Oncogene Another group of signal transduction pathway inhibitors are inhibitors of Ras Oncogene.
- Such inhibitors include inhibitors of farnesyltransferase, geranyl-geranyl transferase, and CAAX proteases as well as anti-sense oligonucleotides, ribozymes and immunotherapy.
- Such inhibitors have been shown to block ras activation in cells containing wild type mutant ras, thereby acting as antiproliferation agents.
- Ras oncogene inhibition is discussed in Scharovsky, O. G., et al. (2000), Journal of Biomedical Science. 7(4) 292-8; Ashby, M. N. (1998), Current Opinion in Lipidology. 9 (2) 99-102; and BioChim. Biophys. Acta, (19899) 1423(3):19-30.
- Antibody antagonists to receptor kinase ligand binding may also serve as signal transduction inhibitors.
- This group of signal transduction pathway inhibitors includes the use of humanized antibodies to the extracellular ligand binding domain of receptor tyrosine kinases. Examples include Imclone C 225 EGFR specific antibody (see Green, M. C. et al, Monoclonal Antibody Therapy for Solid Tumors, Cancer Treat. Rev., (2000), 26(4), 269-286); Herceptin® erbB2 antibody (see Tyrosine Kinase Signalling in Breast cancer:erbB Family Receptor Tyrosine Kinases, Breast cancer Res., 2000, 2(3), 176-183); and 2CB VEGFR2 specific antibody (see Brekken, R. A. et al, Selective Inhibition of VEGFR2 Activity by a monoclonal Anti-VEGF antibody blocks tumor growth in mice, Cancer Res. (2000) 60, 5117-5124).
- Anti-angiogenic agents such as non-receptor MEK angiogenesis inhibitors may also be useful, as well as those which inhibit the effects of vascular endothelial growth factor (e.g., the anti-vascular endothelial cell growth factor antibody bevacizumab [AvastinTM]), and compounds that work by other mechanisms (e.g., linomide, inhibitors of integrin ⁇ v ⁇ 3 function, endostatin and angiostatin).
- vascular endothelial growth factor e.g., the anti-vascular endothelial cell growth factor antibody bevacizumab [AvastinTM]
- linomide inhibitors of integrin ⁇ v ⁇ 3 function, endostatin and angiostatin
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent which is an anti-microtubule agent, platinum coordination complex, alkylating agent, antibiotic agent, topoisomerase II inhibitor, antimetabolite, topoisomerase I inhibitor, hormones and hormonal analogue, signal transduction pathway inhibitor, non-receptor tyrosine MEK angiogenesis inhibitor, immunotherapeutic agent, proapoptotic agent, or cell cycle signaling inhibitor.
- an anti-microtubule agent platinum coordination complex, alkylating agent, antibiotic agent, topoisomerase II inhibitor, antimetabolite, topoisomerase I inhibitor, hormones and hormonal analogue, signal transduction pathway inhibitor, non-receptor tyrosine MEK angiogenesis inhibitor, immunotherapeutic agent, proapoptotic agent, or cell cycle signaling inhibitor.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent selected from diterpenoids and vinca alkaloids.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent which is a platinum coordination complex.
- at least one anti-neoplastic agent is paclitaxel, carboplatin, or vinorelbine.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent which is a signal transduction pathway inhibitor.
- the signal transduction pathway inhibitor is an inhibitor of a growth factor receptor kinase VEGFR2, TIE2, PDGFR, BTK, erbB2, EGFr, IGFR-1, TrkA, TrkB, TrkC, or c-fms.
- the signal transduction pathway inhibitor is an inhibitor of a serine/threonine kinase rafk, akt, or PKC-zeta.
- the signal transduction pathway inhibitor is an inhibitor of a non-receptor tyrosine kinase selected from the src family of kinases. In another embodiment, the signal transduction pathway inhibitor is an inhibitor of c-src. In another embodiment, the signal transduction pathway inhibitor is an inhibitor of Ras oncogene selected from inhibitors of farnesyl transferase and geranylgeranyl transferase. In another embodiment, the signal transduction pathway inhibitor is an inhibitor of a serine/threonine kinase selected from the group consisting of PI3K.
- the signal transduction pathway inhibitor is a dual EGFr/erbB2 inhibitor, for example N- ⁇ 3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl ⁇ -6-[5-( ⁇ [2-(methanesulphonyl) ethyl]amino ⁇ methyl)-2-furyl]-4-quinazolinamine.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent which is a cell cycle signaling inhibitor.
- the cell cycle signaling inhibitor is an inhibitor of CDK2, CDK4 or CDK6.
- Compounds of Formula I may be prepared by methods known in the art of organic synthesis as set forth in the schemes below and/or the specific Examples described below. In all of the methods, it is well understood that protecting groups for sensitive or reactive groups may be employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Green and P. G. M. Wuts (1999) Protective Groups in Organic Synthesis, 3′ edition, John Wiley & Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection of processes as well as the reaction conditions and order of their execution shall be consistent with the preparation of compounds of Formula I.
- Step 3 EB1 and EB2
- 2′3′-cGAMP can be degraded by the enzyme ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP1) which is present in fetal bovine serum (FBS) (Li et al., 2015, Nat Chem Biol, 11, 235).
- ENPP1 ecto-nucleotide pyrophosphatase/phosphodiesterase
- FBS fetal bovine serum
- Example compounds in Table 9 are also compounds contemplated by the present disclosure.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Biomedical Technology (AREA)
- Physics & Mathematics (AREA)
- Nanotechnology (AREA)
- Optics & Photonics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicinal Preparation (AREA)
- Cephalosporin Compounds (AREA)
Abstract
Description
- This application is a continuation of U.S. application Ser. No. 15/953,492, filed Apr. 15, 2018, which is a continuation of International Application No, PCT/US17/023093, filed on Mar. 17, 2017, which claims the benefit of U.S. Provisional Application No. 62/310,364, filed Mar. 18, 2016, U.S. Provisional Application No. 62/355,382, filed Jun. 28, 2016, and U.S. Provisional Application No. 62/396,140, filed Sep. 17, 2016, the entire contents of each of which is hereby incorporated by reference herein.
- The present invention provides novel cyclic di-nucleotide cGAMP analogs, pharmaceutical compositions thereof, their synthetic methods and their use in medical therapy. In particular, the compounds of the invention enhance the body's immune responses by activating STING (Stimulator of Interferon Genes) and are useful for the immunotherapy of cancer, infectious diseases and immune disorders. The compounds are also useful as adjuvants for developing vaccines against cancer and infectious diseases.
- Cytosolic DNA induces type-I interferons and other cytokines that are important for immune defense against microbial infections and malignant cells but can also result in autoimmunity. This DNA signaling pathway requires the adaptor protein STING (Stimulator of Interferon Genes) and the transcription factor IRF3, but the mechanism of DNA sensing was unclear until recently. WO 2014099824 to The University of Texas disclosed that mammalian cytosolic extracts synthesized cyclic-GMP-AMP (cGAMP) in vitro from ATP and GTP in the presence of DNA but not RNA. DNA transfection or DNA virus infection of mammalian cells also triggered cGAMP production. cGAMP bound to STING, lead to the activation of IRF3 and induction of type-I interferons including interferon-0 (IFN-(3). Thus, cGAMP represents the first cyclic di-nucleotide in metazoa and it functions as an endogenous second messenger that triggers interferon production in response to cytosolic DNA.
- Through biochemical fractionation and quantitative mass spectrometry, the inventors on WO 2014099824 also identified a cGAMP synthase (cGAS), which belongs to the nucleotidyltransferase family. Overexpression of cGAS activated the transcription factor IRF3 and induced IFN in a STING-dependent manner. Knockdown of cGAS inhibited IRF3 activation and IFN induction by DNA transfection or DNA virus infection. cGAS bound to DNA in the cytoplasm and catalyzed cGAMP synthesis. These results indicate that cGAS is a cytosolic DNA sensor that induces interferons by producing the second messenger cGAMP. The inventors on WO 2014099824 also determined that the second messenger cGAMP they isolated and synthesized contains two phosphodiester linkages, one between the 2′-OH of GMP and 5′-phosphate of AMP, and the other between the 3′-OH of AMP and 5′-phosphate of GMP; this molecule is referred to as 2′3′-cGAMP.
- Several additional patents applications in this field have henceforth published:
- US20140205653 and US 20140341976 to Aduro Biotech disclose cyclic-di-nucleotide (CDN) compounds that activate and inhibit STING, respectively. In particular, the CDNs of the invention are provided in the form of a composition comprising one or more cyclic purine dinucleotides which activate or inhibit STING-dependent TBK1 activation and the resulting production of type I interferon.
- WO 2015077354 A1 to The University of Chicago discloses Methods and compositions for treating cancer by intratumorally administering a stimulator of interferon genes (STING) agonist. In some embodiments, there are provided compositions and methods concerning methods for treating cancer in a subject comprising administering to the subject an effective amount of a stimulator of interferon genes (STING) agonist, wherein the STING agonist is administered intratumorally.
- WO 2015161762 to Fudan University discloses the use of cyclic dinucleotide cGAMP for preparing antitumor drugs, wherein the tumor is gastric cancer, lung cancer, colon cancer, liver cancer, prostate cancer or pancreatic cancer. cGAMP was shown to inhibit the growth of human tumor cell lines in immune compromised mice.
- WO 2015185565 to GlaxoSmithKline discloses a class of cyclic dinucleotide compounds, or a pharmaceutically acceptable salt and tautomers thereof, compositions, combinations and medicaments containing said compounds and processes for their preparation. The invention also relates to the use of said compounds, combinations, compositions and medicaments, in the treatment of diseases and conditions in which modulation of STING is beneficial, for example inflammation, allergic and autoimmune diseases, infectious diseases, cancer and as vaccine adjuvants.
- WO 2014179335 to Memorial Sloan Kettering Cancer Center discloses compositions, methods, kits, and assays related to the use and/or exploitation of isomers of cGAMP as well as the structure of the enzyme cGAS.
- There is still a need for the discovery and development of new cyclic di-nucleotide cGAMP analogs for use in medical therapy. Specifically, cGAMP analogues with better potency, stability and specificity than endogenous cGAMP are still needed. cGAMP analogues with superior safety and efficacy in animal models of human diseases, including cancer and infectious diseases, have yet to be developed.
- Formula I encompasses Formula Ia-Ii.
- In one aspect, the present invention provides a compound of Formula Ia
-

- wherein:
- a and b are independently 0 or 1 and a+b=1, when a is 1, b is 0 and R5 is not present; and when a is 0, b is 1 and R4 is not present;
- X1 and X2 are independently O, S or Se in a five-membered ring;
- L1, starting from the carbon alpha to X1, and L2, starting from the carbon alpha to X2, are independently —CH2O—P(O)R6—O—, —CH2O—P(S)R6—O—, —C(Y1)(Y2)O—P(O)R6—C(Y3)(Y4)—, —CH2NHSO2NH—, —CH2NHC(O)NH—, —CH2NHC(S)NH—, —CH2NHC(NH)NH—, —CH2NHC(O)CH2—, —CH2NHSO2CH2—, —CH2CH2C(O)NH—, —CH2CH2SO2NH—, —CH2NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
-

-
- cis 0, 1, or 2;
- d, e, and f are independently 0 or 1;
- Y1, Y2, Y3 and Y4 are independently H or F;
- R6 is hydroxyl, thiol, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6 alkylamino, di(C1-6 alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH3 −), or —NR7R8;
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6 alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups;
- R1 and R2 are independently aromatic rings or heteroaromatic rings with the following general structure including its tautomeric forms:
-

-
- g and h are independently 0 or 1;
- W1 and W2 are independently hydrogen, halogen, hydroxyl, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- Z1, Z2, Z3, Z4, Z5, and Z6 are independently CH or N;
- if present, Z7, Z8, Z9, Z10 and Z11 are independently CH or N, and then W1 is CH or N; and
- R3, R4, and R5 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- or a pharmaceutically acceptable salt thereof.
- In one embodiment, the present invention provides a compound of Formula Ib
-
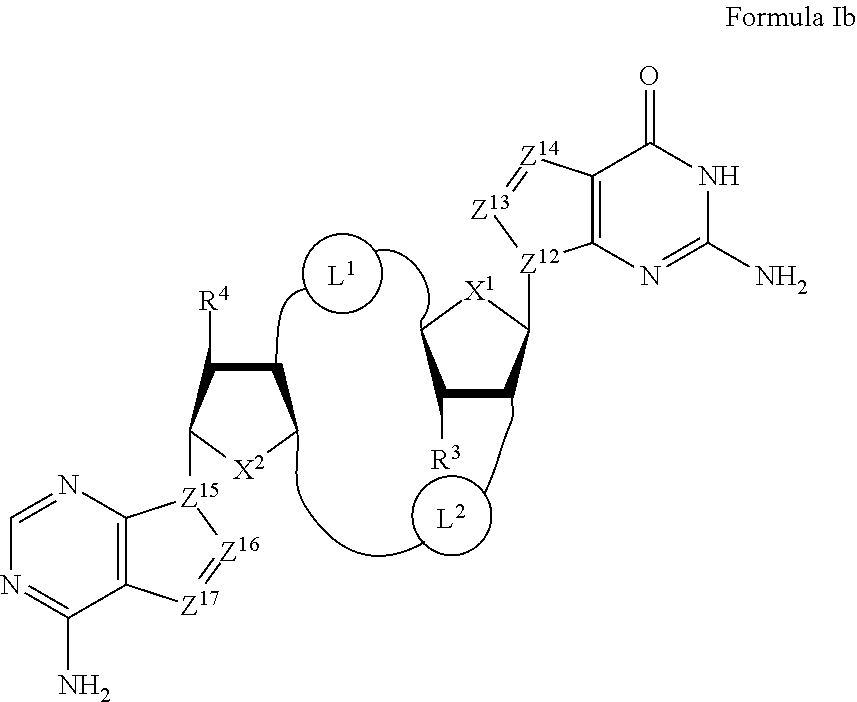
- wherein:
- X1 and X2 are independently O, S or Se;
- Z12, Z13, Z14, Z15, Z16 and Z17 are independently CH or N;
- L1, starting from the carbon alpha to X1, and L2, starting from the carbon alpha to X2, are independently —CH2O—P(O)R6—O—, —CH2O—P(S)R6—O—, —C(Y1)(Y2)O—P(O)R6—C(Y3)(Y4)—, —CH2NHSO2NH—, —CH2NHC(O)NH—, —CH2NHC(S)NH—, —CH2NHC(NH)NH—, —CH2NHC(O)CH2—, —CH2NHSO2CH2—, —CH2CH2C(O)NH—, —CH2CH2SO2NH—, —CH2NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
-

-
- c is 0, 1, or 2;
- d, e, and f are independently 0 or 1;
- Y1, Y2, Y3 and Y4 are independently H or F;
- R6 is hydroxyl, thiol, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH3), or —NR7R8;
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups; and
- R3 and R4 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- or a pharmaceutically acceptable salt thereof.
- In another embodiment, the present invention provides a compound of Formula Ic
-

- wherein:
- Z12, Z13, Z14, Z15, Z16 and Z17 are independently CH or N;
- R3 and R4 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
-
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6 alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6 alkoxy, C1-6hydroxyalkoxy, amino, C1-6 alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups; and
- R9 and R19 are independently hydroxyl, thiol, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6 alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH3), or —NR7R8;
- or a pharmaceutically acceptable salt thereof.
- In another embodiment, the oxygen atom in one or both of the tetrahydrofuranyl rings of Formula Ic is replaced by a sulfur or a selenium atom.
- In another embodiment, the present invention provides a compound of Formula Id
-

- wherein:
- X1 and X2 are independently O, S or Se;
- L1, starting from the carbon alpha to X1, and L2, starting from the carbon alpha to X2, are independently —CH2O—P(O)R6—O—, —CH2O—P(S)R6—O—, —C(Y1)(Y2)O—P(O)R6—C(Y3)(Y4)—, —CH2NHSO2NH—, —CH2NHC(O)NH—, —CH2NHC(S)NH—, —CH2NHC(NH)NH—, —CH2NHC(O)CH2—, —CH2NHSO2CH2—, —CH2CH2C(O)NH—, —CH2CH2SO2NH—, —CH2NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
-

-
- cis 0, 1, or 2;
- d, e, and f are independently 0 or 1;
- Y1, Y2, Y3 and Y4 are independently H or F;
- R6 is hydroxyl, thiol, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH3), or —NR7R8;
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups;
- R3 and R4 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- R11 and R12 are independently selected from the group consisting of:
-

- with at least one of R11 and R12 being
-

-
- Z12, Z13, Z14, Z15, Z16, Z17, Z18, Z19, Z20, Z21, Z22, Z23, Z24, Z25, Z26, Z27, Z28, Z29, Z30, Z31, Z32, Z33, Z34, Z35, Z36, and Z37 are each independently CH or N; and
- W3, W4, W5, W6, W7, W8, and W9 are independently hydrogen, halogen, hydroxyl, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- or a pharmaceutically acceptable salt thereof.
- In another embodiment, the present invention provides a compound of Formula Ie
-

- wherein:
- X3, X4, X5, and X6 are independently O, NH, CH2, CHF, or CF2;
- R13 and RH are independently selected from the group consisting of:
-

-
- Z12, Z13, Z14, Z15, Z16, Z17, Z18, Z19, Z20, Z21, Z22, Z23, Z24, Z25, Z26, Z27, and Z28, are each independently CH or N; and
- W3, W4, W5, W6, and W7 are independently hydrogen, halogen, hydroxyl, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6 alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups;
- R3 and R4 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6 alkoxy, C1-6hydroxyalkoxy, amino, C1-6 alkylamino, di(C1-6 alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8; and
- R15 and R16 are independently hydroxyl, thiol, methoxy, ethoxy, amino, N-methylamino, N,N-dimethylamino, N-ethylamino, N,N-diethylamino, N-morpholino, or borano (—BH3);
- or a pharmaceutically acceptable salt thereof.
- In another embodiment, the oxygen atom in one or both of the tetrahydrofuranyl rings of Formula Ie is replaced by a sulfur or a selenium atom.
- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- [In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In one embodiment, the present invention provides a compound of Formula If
-

- wherein:
- X1 and X2 are independently O, S or Se;
- Z12, Z13, Z14, Z15, Z16 and Z17 are independently CH or N;
- L1, starting from the carbon alpha to X1, and L2, starting from the carbon alpha to X2, are independently —CH2O—P(O)R6—O—, —CH2O—P(S)R6—O—, —C(Y1)(Y2)O—P(O)R6—C(Y3)(Y4)—, —CH2NHSO2NH—, —CH2NHC(O)NH—, —CH2NHC(S)NH—, —CH2NHC(NH)NH—, —CH2NHC(O)CH2—, —CH2NHSO2CH2—, —CH2CH2C(O)NH—, —CH2CH2SO2NH—, —CH2NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
-

-
- cis 0, 1, or 2;
- d, e, and f are independently 0 or 1;
- Y1, Y2, Y3 and Y4 are independently H or F;
- R6 is hydroxyl, thiol, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH3), or —NR7R8;
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups; and
- R3 and R5 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- or a pharmaceutically acceptable salt thereof.
- In one embodiment, the present invention provides a compound of Formula Ig
-

- wherein:
- Z12, Z13, Z14, Z15, Z16 and Z17 are independently CH or N;
- R3 and R5 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
-
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups; and
- R9 and R19 are independently hydroxyl, thiol, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH3), or —NR7R8;
- or a pharmaceutically acceptable salt thereof.
- In another embodiment, the oxygen atom in one or both of the tetrahydrofuranyl rings of Formula Ig is replaced by a sulfur or a selenium atom.
- In another embodiment, the present invention provides a compound of Formula Ih
-

- wherein:
- X1 and X2 are independently O, S or Se;
- L1, starting from the carbon alpha to X1, and L2, starting from the carbon alpha to X2, are independently —CH2O—P(O)R6—O—, —CH2O—P(S)R6—O—, —C(Y1)(Y2)O—P(O)R6—C(Y3)(Y4)—, —CH2NHSO2NH—, —CH2NHC(O)NH—, —CH2NHC(S)NH—, —CH2NHC(NH)NH—, —CH2NHC(O)CH2—, —CH2NHSO2CH2—, —CH2CH2C(O)NH—, —CH2CH2SO2NH—, —CH2NH(3,4-dioxocyclobuten-1,2-diyl)NH—,
-

-
- c is 0, 1, or 2;
- d, e, and f are independently 0 or 1;
- Y1, Y2, Y3 and Y4 are independently H or F;
- R6 is hydroxyl, thiol, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6 alkylamino, di(C1-6 alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), borano (—BH3), or —NR7R8;
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6 alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups;
- R3 and R5 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6 alkoxy, C1-6hydroxyalkoxy, amino, C1-6 alkylamino, di(C1-6 alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- R11 and R12 are independently selected from the group consisting of:
-

- with at least one of R11 and R12 being
-
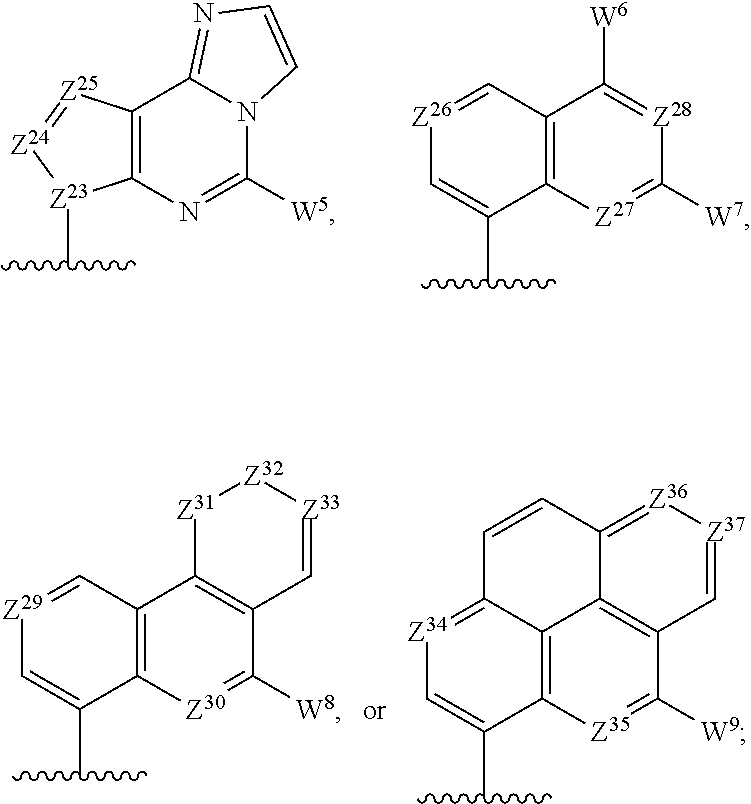
-
- Z12, Z13, Z14, Z15, Z16, Z17, Z18, Z19, Z20, Z21, Z22, Z23, Z24, Z25, Z26, Z27, Z28, Z29, Z30, Z31, Z32, Z33, Z34, Z35, Z36, and Z37 are each independently CH or N; and
- W3, W4, W5, W6, W7, W8, and W9 are independently hydrogen, halogen, hydroxyl, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- or a pharmaceutically acceptable salt thereof.
- In another embodiment, the present invention provides a compound of Formula Ii
-

- wherein:
- X3, X5, X6, and X7 are independently O, NH, CH2, CHF, or CF2;
- R13 and RH are independently selected from the group consisting of:
-

-
- Z12, Z13, Z14, Z15, Z16, Z17, Z18, Z19, Z20, Z21, Z22, Z23, Z24, Z25, Z26, Z27, and Z28, are each independently CH or N; and
- W4, W5, W6, and W7 are independently hydrogen, halogen, hydroxyl, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8;
- R7 and R8 are independently hydrogen, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, cyclic —(C1-6alkyl)-, cyclic —(C1-6alkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6 alkoxy, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups, cyclic —(C1-6oxaalkyl)-, or cyclic —(C1-6oxaalkyl)-selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, or di(C1-6alkyl)amino groups;
- R3 and R5 are independently hydrogen, halogen, hydroxyl, amino, C1-6alkyl, C1-6alkyl selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6 alkoxy, C1-6hydroxyalkoxy, amino, C1-6 alkylamino, di(C1-6 alkyl)amino, or azido groups, C1-6alkoxy, C1-6alkoxy selectively functionalized with one or more halogen, thiol, hydroxyl, carbonyl, carboxyl, carbonyloxyl, C1-6hydroxyalkoxy, amino, C1-6alkylamino, di(C1-6alkyl)amino, or azido groups, C3-5alkenyl-O—, C3-5alkynyl-O—, oligo(ethylene glycol), poly(ethylene glycol), azido, or —NR7R8; and
- R15 and R16 are independently hydroxyl, thiol, methoxy, ethoxy, amino, N-methylamino, N,N-dimethylamino, N-ethylamino, N,N-diethylamino, N-morpholino, or borano (—BH3);
- or a pharmaceutically acceptable salt thereof.
- In another embodiment, the oxygen atom in one or both of the tetrahydrofuranyl rings of Formula Ii is replaced by a sulfur or a selenium atom.
- In another embodiment, the compound is:
-

- In another embodiment, the compound is:
-

- In another aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable excipients.
- In another aspect, the present invention provides a method of treating a disease or condition in which modulation of STING is beneficial comprising: administering to a patient in need thereof a compound of Formula I or a pharmaceutically acceptable salt thereof.
- In another aspect, the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in the treatment of a disease or condition in which modulation of STING is beneficial.
- In another aspect, the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in therapy.
- In another aspect, the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in the treatment of cancer.
- In another aspect, the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, or a pharmaceutically composition thereof, such as a nanoparticle or a delivery vehicles that enhances the cellular uptake, stability and efficacy of a compound of Formula I for use in the treatment of cancer.
- In another aspect, the present invention provides a method of treating cancer comprising: administering a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- In another aspect, the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof, in the manufacture of a medication for the treatment of cancer.
- In another aspect, the present invention provides pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, at least one further therapeutic agent, and one or more of pharmaceutically acceptable excipients.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in therapy.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of a disease or condition for which modulation of STING is beneficial.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of cancer.
- In another aspect, the present invention provides a method for treating a disease or condition for which modulation of STING is beneficial comprising: administering to a patient in need thereof a therapeutically effective amount of a combination comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- In another aspect, the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of cancer. The therapeutic agent includes but is not limited to immune checkpoint inhibitors, such as humanized antibodies against PD1, PD-L1, CTLA4 and other molecules that block effective anti-tumor immune responses.
- In another aspect, the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent. The therapeutic agent includes but is not limited to immune checkpoint inhibitors, such as humanized antibodies against PD1, PD-L1, CTLA4 and other molecules that block effective anti-tumor immune responses.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of cancer. The therapeutic agent includes radiation, such as high-dose radiation, which directly kills tumor cells, enhances presentation of tumor antigens and activates the STING pathway.
- In another aspect, the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent. The therapeutic agent includes radiation, such as high-dose radiation, which directly kills tumor cells, enhances presentation of tumor antigens and activates the STING pathway.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent for use in the treatment of cancer. The therapeutic agent includes another chemotherapeutic agent that selectively kills tumor cells and enhances presentation of tumor antigens.
- In another aspect, the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one further therapeutic agent. The therapeutic agent includes another chemotherapeutic agent that selectively kills tumor cells and enhances presentation of tumor antigens.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, a pharmaceutical formulation including a nanoparticle, and at least one further therapeutic agent for use in the treatment of cancer. The therapeutic agent includes radiation and/or another chemotherapeutic agent.
- In another aspect, the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, a pharmaceutical formulation including a nanoparticle, and at least one further therapeutic agent for use in the treatment of cancer. The therapeutic agent includes radiation and/or another chemotherapeutic agent.
- In another aspect, the present invention provides a method of treating cancer comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, a pharmaceutical formulation including a nanoparticle, and at least one further therapeutic agent for use in the treatment of cancer. The compound of Formula I, may be injected directly to tumors, or systemically, including injection into muscles (intramuscular), skins (subcutaneous and intra-dermal), peritoneal (intraperitoneal), lymph nodes (intralymphatic) or veins (intravenous).
- In another aspect, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use as a vaccine adjuvant.
- In another aspect, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, such as a nanoparticle or a delivery vehicles that enhances the cellular uptake, stability and efficacy of a compound of Formula I, for use as a vaccine adjuvant.
- In one embodiment, the pharmaceutical composition is a vaccine.
- In another embodiment, the present invention provides a method of inducing or promoting an immune response comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, as an adjuvant and a tumor antigen.
- In another embodiment, the present invention provides a method of inducing or promoting an immune response comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutical composition thereof, as an adjuvant, a tumor antigen, or a pharmaceutical composition thereof, such as a nanoparticle or a delivery vehicles that enhances the cellular uptake of the adjuvant and tumor antigen.
- In another aspect, the present invention provides a pharmaceutical composition comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof, as an adjuvant and an immunogen for a target pathogen.
- In another aspect, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use as a vaccine adjuvant.
- In another embodiment, the present invention provides a method of inducing or promoting an immune response comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, as an adjuvant and an immunogen for a target pathogen.
- In another aspect, the present invention provides a vaccine adjuvant comprising: a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- In another aspect, the present invention provides an immunogenic composition comprising: an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- In another aspect, the present invention provides an immunogenic composition comprising: an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use in the treatment or prevention of a disease, including cancer and infectious diseases.
- In another aspect, the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof, for the manufacture of an immunogenic composition comprising an antigen or antigen composition, for the treatment or prevention of a disease, including cancer and infectious diseases.
- In another aspect, the present invention provides a method of treating or preventing a disease comprising: administering to a patient suffering from or susceptible to the disease, an immunogenic composition comprising an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- In another aspect, the present invention provides a vaccine composition comprising: an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use in the treatment or prevention of a disease, including cancer and infectious diseases.
- In another aspect, the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof, for the manufacture of a vaccine composition comprising an antigen or antigen composition for the treatment or prevention of a disease, including cancer and infectious diseases.
- In another aspect, the present invention provides a method of treating or preventing disease comprising the administration to a patient suffering from or susceptible to the disease, a vaccine composition comprising an antigen or antigen composition and a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- In another aspect, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, for use in the treatment of immune disorders, including autoimmune and autoinflammatory diseases.
- In another aspect, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt thereof, or a pharmaceutically composition thereof, such as a nanoparticle or a delivery vehicles that enhances the cellular uptake, stability and efficacy of a compound of Formula I, for use in the treatment of immune disorders, including autoimmune and autoinflammatory diseases.
- In another aspect, the present invention provides a method of treating immune disorders comprising: administering a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- In another aspect, the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt thereof, in the manufacture of a medication for the treatment of immune disorders, including autoimmune and autoinflammatory diseases.
- It will be appreciated that all combinations of the above aspects/embodiments, and other aspects/embodiments disclosed elsewhere herein, are contemplated and are further embodiments of the invention.
- The present invention provides novel cGAMP analogs, pharmaceutical compositions thereof, and uses thereof in therapy. 2′3′-cGAMP is an endogenous second messenger produced by mammalian cells, it is a high affinity ligand for STING, inducing conformational changes therein, and a potent inducer of type-1 interferons. cGAS and the cGAS-cGAMP pathway is important for triggering inflammatory responses to self and foreign DNA. As such, cGAS is important for immune defense against microbial pathogens that contain DNA and require DNA in their life cycles. These pathogens include DNA viruses, retroviruses including HIV, bacteria including Mycobacterium tuberculosis, fungi and parasites. cGAS can also detect tumor DNA and is important for the body's intrinsic immunity against malignant cells. Activation of the cGAS-cGAMP-STING pathway is important for cancer immunotherapy.
- As a potent inducer of type-I interferons, cGAMP (and hence the cGAMP analogs of the present invention) provides a rational immune adjuvant. As such, a compound of Formula I or a pharmaceutically acceptable salt thereof, may be used as a vaccine adjuvant, particularly with mucosal vaccines, and may be formulated with immunogens and delivered as have been cyclic-di-GMP and c-di-AMP as vaccine adjuvants (see, e.g. Pedersen, et al. PLoS ONE; November 2011, 6, 11, e26973; Ebensen et al., Vaccine 29, 2011, 5210-5220; Chen et al., Vaccine 28, 2010, 3080-3085). In fact, such adjuvants are often more effective because cGAMP (and the cGAMP analogs of the present invention) is more potent than c-di-GMP in inducing interferons.
- In one aspect, the invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in the treatment of cancer. In one embodiment, the pharmaceutical composition is a compound of Formula I. In another embodiment, the pharmaceutical composition is a compound of Formula I in a pharmaceutical formulation including a nanoparticle or another delivery vehicle. In another embodiment, the pharmaceutical composition is a compound of Formula I in combination with at least one further therapeutic agent, which includes but is not limited to immune checkpoint inhibitors such as antibodies against PD-1, PD-L1 or CTLA-4. The therapeutic agent used in combination with a compound of Formula I also includes radiation of tumors or a chemotherapeutic agent that targets tumor cells.
- In another aspect, the invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, as an adjuvant and an immunogen for a target pathogen. In one embodiment, the pharmaceutical composition is a vaccine. In another embodiment, the present invention provides a method of inducing or promoting an immune response comprising: administering to a patient in need thereof an effective amount a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, as an adjuvant and an immunogen for a target pathogen.
- As used herein:
- The terms “halo” and “halogen”, alone or in combination with other groups, refers to fluoro-, chloro-, bromo- and iodo-.
- The term “C1-6 alkyl”, alone or in combination with other groups, refers to monovalent, linear chain or branched chain alkyl groups containing from 1 to 6 carbon atoms. Exemplary C1-6 alkyl groups include but not limited to metheyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl and tert-butyl groups. More preferred are C1-4 alkyls.
- The term “C1-6 alkoxy” refers to, alone or in combination with other groups, R′—O—, where R′ is C1-6 alkyl.
- The term “haloC1-6 alkyl”, alone or in combination with other groups, refers to a C1-6 alkyl group subsituted with one or more halo suctsitutents, for example CF3 and CH2CF3.
- The term “a compound of the invention” or “a compound of Formula I” includes all solvates, complexes, polymorphs, radiolabeled derivatives, tautomers, stereoisomers, and optical isomers of the compounds of Formula I and salts thereof, unless otherwise specified.
- The term “effective amount” means that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system, animal or human that is being sought, for instance, by a researcher or clinician.
- The term “therapeutically effective amount” means any amount which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder. The term also includes within its scope amounts effective to enhance normal physiological function.
- The term “prophylaxis” includes prevention and refers to a measure or procedure which is to prevent rather than cure or treat a disease. Preventing refers to a reduction in risk of acquiring or developing a disease causing at least one clinical symptom of the disease not to developing a subject that may be exposed to a disease-causing agent or a subject predisposed to the disease in advance of disease outset.
- The term “pharmaceutically acceptable” refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- The term “pharmaceutically acceptable excipients” includes all diluents, carriers, binders, glidants, and other components of pharmaceutical formulations with which the compound of the invention is administered.
- The compounds of the invention may exist in solid or liquid form. In solid form, compound of the invention may exist in a continuum of solid states ranging from fully amorphous to fully crystalline.
- The term ‘amorphous’ refers to a state in which the material lacks long range order at the molecular level and, depending upon the temperature, may exhibit the physical properties of a solid or a liquid. Typically, such materials do not give distinctive X-ray diffraction patterns and, while exhibiting the properties of a solid, are more formally described as a liquid. Upon heating, a change from solid to liquid properties occurs which is characterized by a change of state, typically second order (‘glass transition’).
- The term ‘crystalline’ refers to a solid phase in which the material has a regular ordered internal structure at the molecular level and gives a distinctive X-ray diffraction pattern with defined peaks. Such materials when heated sufficiently will also exhibit the properties of a liquid, but the change from solid to liquid is characterized by a phase change, typically first order (‘melting point’).
- The compounds of the invention may have the ability to crystallize in more than one form, a characteristic, which is known as polymorphism, and it is understood that such polymorphic forms (“polymorphs”) are within the scope of the invention. Polymorphism generally can occur as a response to changes in temperature or pressure or both and can also result from variations in the crystallization process. Polymorphs can be distinguished by various physical characteristics known in the art such as x-ray diffraction patterns, solubility and melting point.
- The compound of Formula I may exist in solvated and unsolvated forms. As used herein, the term “solvate” refers to a complex of variable stoichiometry formed by a solute (in this invention, a compound of Formula I or a salt) and a solvent. Such solvents for the purpose of the invention may not interfere with the biological activity of the solute. The skilled artisan will appreciate that pharmaceutically acceptable solvates may be formed for crystalline compounds wherein solvent molecules are incorporated into the crystalline lattice during crystallization. The incorporated solvent molecules may be water molecules or non-aqueous such as ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and ethyl acetate molecules. Crystalline lattice incorporated with water molecules are typically referred to as “hydrates”. Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The present invention includes all such solvates.
- it is also noted that some compounds may form tautomers. ‘Tautomers’ refer to compounds that are interchangeable forms of a particular compound structure, and that vary in the displacement of hydrogen atoms and electrons. Thus, two structures may be in equilibrium through the movement of re electrons and an atom (usually H). For example, enols and ketones are tautomers because they are rapidly interconverted by treatment with either acid or base. It is understood that all tautomers and mixtures of tautomers of the compounds of the present invention are included within the scope of the compounds of the present invention.
- The compounds of Formula I may be in the form of a salt. Typically, the salts of the present invention are pharmaceutically acceptable salts. Salts encompassed within the term “pharmaceutically acceptable salts” refer to non-toxic salts of the compounds of this invention. For a review on suitable salts, see e.g., Berge et al, J. Pharm. Sci. 1977, 66, 1-19. Suitable pharmaceutically acceptable salts can include acid addition salts. A pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of Formula I with a suitable inorganic or organic acid (such as hydrobromic, hydrochloric, sulfuric, nitric, phosphoric, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic), optionally in a suitable solvent such as an organic solvent, to give the salt which is usually isolated for example by crystallization and filtration. A pharmaceutically acceptable acid addition salt of a compound of Formula I can be, for example, a hydrobromide, hydrochloride, sulfate, nitrate, phosphate, p-toluenesulfonate, benzenesulfonate, methanesulfonate, ethanesulfonate, or naphthalenesulfonate (e.g. 2-naphthalenesulfonate) salt. Other non-pharmaceutically acceptable salts, e.g. trifluoroacetates, may be used, for example in the isolation of compounds of the invention, and are included within the scope of this invention.
- The invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the compounds of Formula I.
- While it is possible that, for use in therapy, the compound of the invention may be administered as the raw chemical, it is possible to present the compound of the invention as the active ingredient in a pharmaceutical composition. Such compositions can be prepared in a manner well known in the pharmaceutical art and comprise at least one active compound. Accordingly, the invention further provides pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically acceptable excipients. The excipient(s) must be acceptable in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof. In accordance with another aspect of the invention there is also provided a process for the preparation of a pharmaceutical composition including the agent, or pharmaceutically acceptable salts thereof, with one or more pharmaceutically acceptable excipients. The pharmaceutical composition can be for use in the treatment and/or prophylaxis of any of the conditions described herein.
- Generally, the compound of the invention is administered in a pharmaceutically effective amount. The amount of the compound actually administered will typically be determined by a physician, in the light of the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered, the age, weight, and response of the individual patient, the severity of the patient's symptoms, and the like. Pharmaceutical compositions may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose. The term “unit dosage forms” refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient, vehicle or carrier. Typical unit dosage forms include prefilled, premeasured ampules or syringes of the liquid compositions or pills, tablets, capsules or the like in the case of solid compositions.
- Preferred unit dosage compositions are those containing a daily dose or sub-dose, or an appropriate fraction thereof, of an active ingredient. Such unit doses may therefore be administered once or more than once a day. Such pharmaceutical compositions may be prepared by any of the methods well known in the pharmacy art.
- Pharmaceutical compositions may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual), rectal, inhaled, intranasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal) route. Such compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier(s) or excipient(s).
- Pharmaceutical compositions adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids, edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- For instance, for oral administration in the form of a tablet or capsule, the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert excipient such as ethanol, glycerol, water and the like. Powders are prepared by reducing the compound to a suitable fine size and mixing with a similarly prepared pharmaceutical excipient such as an edible carbohydrate, as, for example, starch or mannitol. Flavoring, preservative, dispersing and coloring agent can also be present.
- Capsules are made by preparing a powder mixture, as described above, and filling formed gelatin sheaths. Excipients including glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation. A disintegrating or solubilizing agent such as agar-agar, calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
- Moreover, when desired or necessary, excipients including suitable binders, glidants, lubricants, sweetening agents, flavors, disintegrating agents and coloring agents can also be incorporated into the mixture. Suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like. Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like. Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like. Tablets are formulated, for example, by preparing a powder mixture, granulating or slugging, adding a lubricant and disintegrant and pressing into tablets. A powder mixture is prepared by mixing the compound, suitably comminuted, with a diluent or base as described above, and optionally, with a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone, a solution retardant such as paraffin, a resorption accelerator such as a quaternary salt and/or an absorption agent such as bentonite, kaolin or dicalcium phosphate. The powder mixture can be granulated by wetting with a binder such as syrup, starch paste, acadia mucilage or solutions of cellulosic or polymeric materials and forcing through a screen. As an alternative to granulating, the powder mixture can be run through the tablet machine and the result is imperfectly formed slugs broken into granules. The granules can be lubricated to prevent sticking to the tablet forming dies by means of the addition of stearic acid, a stearate salt, talc or mineral oil. The lubricated mixture is then compressed into tablets. The compounds of the present invention can also be combined with a free flowing inert carrier and compressed into tablets directly without going through the granulating or slugging steps. A clear or opaque protective coating consisting of a sealing coat of shellac, a coating of sugar or polymeric material and a polish coating of wax can be provided. Dyestuffs can be added to these coatings to distinguish different unit dosages.
- Oral fluids such as solution, suspensions, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of the compound. Syrups can be prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle. Suspensions can be formulated by dispersing the compound in a non-toxic vehicle. Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
- Where appropriate, dosage unit compositions for oral administration can be microencapsulated. The composition can also be prepared to prolong or sustain the release as for example by coating or embedding particulate material in polymers, wax or the like.
- The compounds of the invention may also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles. Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines. Pharmaceutical compositions adapted for transdermal administration may be presented as discrete patches intended to remain in intimate contact with the epidermis of the recipient for a prolonged period of time.
- Pharmaceutical compositions adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- For treatments of the eye or other external tissues, for example mouth and skin, the compositions are preferably applied as a topical ointment or cream. When formulated in an ointment, the active ingredient may be employed with either a paraffinic or a water-miscible ointment base. Alternatively, the active ingredient may be formulated in a cream with an oil-in-water cream base or a water-in-oil base.
- Pharmaceutical compositions adapted for topical administrations to the eye include eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent.
- Pharmaceutical compositions adapted for topical administration in the mouth include lozenges, pastilles and mouth washes.
- Pharmaceutical compositions adapted for rectal administration may be presented as suppositories or as enemas.
- Dosage forms for nasal or inhaled administration may conveniently be formulated as aerosols, solutions, suspension drops, gels or dry powders.
- Compositions for intranasal administration include aqueous compositions administered to the nose by drops or by pressurised pump. Suitable compositions contain water as the diluent or carrier for this purpose. Compositions for administration to the lung or nose may contain one or more excipients, for example one or more suspending agents, one or more preservatives, one or more surfactants, one or more tonicity adjusting agents, one or more co-solvents, and may include components to control the pH of the composition, for example a buffer system. Further, the compositions may contain other excipients such as antioxidants, for example sodium metabisulphite, and taste-masking agents. Compositions may also be administered to the nose or other regions of the respiratory tract by nebulization. Intranasal compositions may permit the compound(s) of Formula I or (a) pharmaceutically acceptable salt(s) thereof to be delivered to all areas of the nasal cavities (the target tissue) and further, may permit the compound(s) of Formula I or (a) pharmaceutically acceptable salt(s) thereof to remain in contact with the target tissue for longer periods of time. A suitable dosing regime for intranasal compositions would be for the patient to inhale slowly through the nose subsequent to the nasal cavity being cleared. During inhalation, the composition would be administered to one nostril while the other is manually compressed. This procedure would then be repeated for the other nostril. Typically, one or two sprays per nostril would be administered by the above procedure one, two, or three times each day, ideally once daily. Of particular interest are intranasal compositions suitable for once-daily administration.
- The suspending agent(s), if included, will typically be present in an amount of from 0.1 to 5% (w/w), such as from 1.5% to 2.4% (w/w), based on the total weight of the composition. Examples of pharmaceutically acceptable suspending agents include, but are not limited to, Avicef (microcrystalline cellulose and carboxymethylcellulose sodium), carboxymethylcellulose sodium, veegum, tragacanth, bentonite, methylcellulose, xanthan gum, carbopol and polyethylene glycols.
- Compositions for administration to the lung or nose may contain one or more excipients may be protected from microbial or fungal contamination and growth by inclusion of one or more preservatives. Examples of pharmaceutically acceptable anti-microbial agents or preservatives include, but are not limited to, quaternary ammonium compounds (for example benzalkonium chloride, benzethonium chloride, cetrimide, cetylpyridinium chloride, lauralkonium chloride and myristyl picolinium chloride), mercurial agents (for example phenylmercuric nitrate, phenylmercuric acetate and thimerosal), alcoholic agents (for example chlorobutanol, phenylethyl alcohol and benzyl alcohol), antibacterial esters (for example esters of para-hydroxybenzoic acid), chelating agents such as disodium edetate (EDTA) and other anti-microbial agents such as chlorhexidine, chlorocresol, sorbic acid and its salts (such as potassium sorbate) and polymyxin. Examples of pharmaceutically acceptable anti-fungal agents or preservatives include, but are not limited to, sodium benzoate, sorbic acid, sodium propionate, methylparaben, ethylparaben, propylparaben and butylparaben. The preservative(s), if included, may be present in an amount of from 0.001 to 1% (w/w), such as from 0.015% to 0.5% (w/w) based on the total weight of the composition. Compositions (for example wherein at least one compound is in suspension) may include one or more surfactants which functions to facilitate dissolution of the medicament particles in the aqueous phase of the composition. For example, the amount of surfactant used is an amount which will not cause foaming during mixing. Examples of pharmaceutically acceptable surfactants include fatty alcohols, esters and ethers, such as polyoxyethylene (20) sorbitan monooleate (Polysorbate 80), macrogol ethers, and poloxamers. The surfactant may be present in an amount of between about 0.01 to 10% (w/w), such as from 0.01 to 0.75% (w/w), for example about 0.5% (w/w), based on the total weight of the composition.
- One or more tonicity-adjusting agent(s) may be included to achieve tonicity with body fluids e.g. fluids of the nasal cavity, resulting in reduced levels of irritancy. Examples of pharmaceutically acceptable tonicity-adjusting agents include, but are not limited to, sodium chloride, dextrose, xylitol, calcium chloride, glucose, glycerine and sorbitol. A tonicity-adjusting agent, if present, may be included in an amount of from 0.1 to 10% (w/w), such as from 4.5 to 5.5% (w/w), for example about 5.0% (w/w), based on the total weight of the composition.
- The compositions of the invention may be buffered by the addition of suitable buffering agents such as sodium citrate, citric acid, trometamol, phosphates such as disodium phosphate (for example the dodecahydrate, heptahydrate, dihydrate and anhydrous forms), or sodium phosphate and mixtures thereof.
- A buffering agent, if present, may be included in an amount of from 0.1 to 5% (w/w), for example 1 to 3% (w/w) based on the total weight of the composition.
- Examples of taste-masking agents include sucralose, sucrose, saccharin or a salt thereof, fructose, dextrose, glycerol, corn syrup, aspartame, acesulfame-K, xylitol, sorbitol, erythritol, ammonium glycyrrhizinate, thaumatin, neotame, mannitol, menthol, eucalyptus oil, camphor, a natural flavouring agent, an artificial flavouring agent, and combinations thereof.
- One or more co-solvent(s) may be included to aid solubility of the medicament compound(s) and/or other excipients. Examples of pharmaceutically acceptable co-solvents include, but are not limited to, propylene glycol, dipropylene glycol, ethylene glycol, glycerol, ethanol, polyethylene glycols (for example PEG300 or PEG400), and methanol. In one embodiment, the co-solvent is propylene glycol.
- Co-solvent(s), if present, may be included in an amount of from 0.05 to 30% (w/w), such as from 1 to 25% (w/w), for example from 1 to 10% (w/w) based on the total weight of the composition.
- Compositions for inhaled administration include aqueous, organic or aqueous/organic mixtures, dry powder or crystalline compositions administered to the respiratory tract by pressurised pump or inhaler, for example, reservoir dry powder inhalers, unit-dose dry powder inhalers, pre-metered multi-dose dry powder inhalers, nasal inhalers or pressurised aerosol inhalers, nebulisers or insufflators. Suitable compositions contain water as the diluent or carrier for this purpose and may be provided with conventional excipients such as buffering agents, tonicity modifying agents and the like. Aqueous compositions may also be administered to the nose and other regions of the respiratory tract by nebulisation. Such compositions may be aqueous solutions or suspensions or aerosols delivered from pressurised packs, such as a metered dose inhaler, with the use of a suitable liquefied propellant.
- Compositions for administration topically to the nose (for example, for the treatment of rhinitis) or to the lung, include pressurised aerosol compositions and aqueous compositions delivered to the nasal cavities by pressurised pump. Compositions which are non-pressurised and are suitable for administration topically to the nasal cavity are of particular interest. Suitable compositions contain water as the diluent or carrier for this purpose. Aqueous compositions for administration to the lung or nose may be provided with conventional excipients such as buffering agents, tonicity-modifying agents and the like. Aqueous compositions may also be administered to the nose by nebulisation.
- A fluid dispenser may typically be used to deliver a fluid composition to the nasal cavities. The fluid composition may be aqueous or non-aqueous, but typically aqueous. Such a fluid dispenser may have a dispensing nozzle or dispensing orifice through which a metered dose of the fluid composition is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser. Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid composition, the doses being dispensable upon sequential pump actuations. The dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid composition into the nasal cavity.
- Dry powder compositions for topical delivery to the lung by inhalation may, for example, be presented in capsules and cartridges of for example gelatine, or blisters of for example laminated aluminium foil, for use in an inhaler or insufflator. Powder blend compositions generally contain a powder mix for inhalation of the compound of Formula 1 or a pharmaceutically acceptable salt thereof and a suitable powder base (carrier/diluent/excipient substance) such as mono-, di-, or polysaccharides (for example lactose or starch). Dry powder compositions may also include, in addition to the drug and carrier, a further excipient (for example a ternary agent such as a sugar ester for example cellobiose octaacetate, calcium stearate, or magnesium stearate.
- Pharmaceutical compositions adapted for parental administration include aqueous and nonaqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the composition isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. The compositions may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- It should be understood that in addition to the ingredients particularly mentioned above, the compositions may include other agents conventional in the art having regard to the type of formulation in question, for example, those suitable for oral administration may include flavoring agents.
- The compounds of Formula I and pharmaceutically acceptable salts thereof may also be formulated with other adjuvants to modulate their activity. Such compositions may contain antibody(ies) or antibody fragment(s) or an antigenic component including but not limited to protein, DNA, live or dead bacteria and/or viruses or virus-like particles, together with one or more components with adjuvant activity including but not limited to aluminium salts, oil and water emulsions, heat shock proteins, lipid A preparations and derivatives, glycolipids, other TLR agonists such as CpG DNA or similar agents, cytokines such as GM-CSF or IL-12 or similar agents.
- A therapeutically effective amount of the agent will depend upon a number of factors including, for example, the age and weight of the subject, the precise condition requiring treatment and its severity, the nature of the formulation, and the route of administration, and will ultimately be at the discretion of the attendant physician or veterinarian. In particular, the subject to be treated is a mammal, particularly a human.
- The agent may be administered in a daily dose. This amount may be given in a single dose per day or more usually in a number (such as two, three, four, five or six) of sub-doses per day such that the total daily dose is the same.
- Suitably, the amount of the compound of the invention administered according to the present invention will be an amount selected from about 0.01 mg to about 1 g per day (calculated as the free or unsalted compound).
- The compounds of Formula I and pharmaceutically acceptable salts thereof may be employed alone or in combination with other therapeutic agents. The compounds of Formula I and pharmaceutically acceptable salts thereof and the other pharmaceutically active agent(s) may be administered together or separately and, when administered separately, administration may occur simultaneously or sequentially, in any order, by any convenient route in separate or combined pharmaceutical compositions. The amounts of the compound(s) of Formula I or pharmaceutically acceptable salt(s) thereof and the other pharmaceutically active agent(s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect. The compound(s) of Formula I or pharmaceutically acceptable salt(s) thereof and further therapeutic agent(s) may be employed in combination by administration simultaneously in a unitary pharmaceutical composition including both compounds. Alternatively, the combination may be administered separately in separate pharmaceutical compositions, each including one of the compounds in a sequential manner wherein, for example, the compound of the invention is administered first and the other second and vice versa. Such sequential administration may be close in time (e.g. simultaneously) or remote in time. Furthermore, it does not matter if the compounds are administered in the same dosage form, e.g. one compound may be administered topically and the other compound may be administered orally. Suitably, both compounds are administered orally.
- The combinations may be presented as a combination kit. By the term “combination kit” “or kit of parts” as used herein is meant the pharmaceutical composition or compositions that are used to administer the combination according to the invention. When both compounds are administered simultaneously, the combination kit can contain both compounds in a single pharmaceutical composition, such as a tablet, or in separate pharmaceutical compositions. When the compounds are not administered simultaneously, the combination kit will contain each compound in separate pharmaceutical compositions either in a single package or in separate pharmaceutical compositions in separate packages. The combination kit can also be provided by instruction, such as dosage and administration instructions. Such dosage and administration instructions can be of the kind that are provided to a doctor, for example by a drug product label, or they can be of the kind that are provided by a doctor, such as instructions to a patient.
- When the combination is administered separately in a sequential manner wherein one is administered first and the other second or vice versa, such sequential administration may be close in time or remote in time. For example, administration of the other agent several minutes to several dozen minutes after the administration of the first agent, and administration of the other agent several hours to several days after the administration of the first agent are included, wherein the lapse of time is not limited. For example, one agent may be administered once a day, and the other agent may be administered 2 or 3 times a day, or one agent may be administered once a week, and the other agent may be administered once a day and the like. It will be clear to a person skilled in the art that, where appropriate, the other therapeutic ingredients(s) may be used in the form of salts, for example as alkali metal or amine salts or as acid addition salts, or prodrugs, or as esters, for example lower alkyl esters, or as solvates, for example hydrates, to optimize the activity and/or stability and/or physical characteristics, such as solubility, of the therapeutic ingredient. It will be clear also that, where appropriate, the therapeutic ingredients may be used in optically pure form.
- When combined in the same composition it will be appreciated that the two compounds must be stable and compatible with each other and the other components of the composition and may be formulated for administration. When formulated separately they may be provided in any convenient composition, conveniently, in such a manner as known for such compounds in the art.
- When the compound of Formula I is used in combination with a second therapeutic agent active against the same disease, condition or disorder, the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art.
- In one embodiment, the patient in the methods and uses of the present invention is a mammal. In another embodiment, the patient is a human. The compounds of the invention are useful in the treatment of diseases and conditions in which modulation of STING is beneficial, including cancer. As modulators of the immune response, the compounds of Formula I and pharmaceutically acceptable salts thereof may also be useful, as stand-alone, in combination or as adjuvants, in the treatment of diseases and conditions in which modulation of STING is beneficial.
- In one aspect, the disease or condition to be treated is cancer. Examples of cancer diseases and conditions in which a compound of Formula I or pharmaceutically acceptable salt thereof, may have potentially beneficial anti-tumor effects include cancers of the lung, bone, pancreas, skin, head, neck, uterus, ovaries, stomach, colon, breast, esophagus, small intestine, bowel, endocrine system, thyroid gland, parathyroid gland, adrenal gland, urethra, prostate, penis, testes, ureter, bladder, kidney or liver; rectal cancer; cancer of the anal region; carcinomas of the fallopian tubes, endometrium, cervix, vagina, vulva, renal pelvis, renal cell; sarcoma of soft tissue; myxoma; rhabdomyoma; fibroma; lipoma; teratoma; cholangiocarcinoma; hepatoblastoma; angiosarcoma; hemagioma; hepatoma; fibrosarcoma; chondrosarcoma; myeloma; chronic or acute leukemia; lymphocytic lymphomas; primary CNS lymphoma; neoplasms of the CNS; spinal axis tumours; squamous cell carcinomas; synovial sarcoma; malignant pleural mesotheliomas; brain stem glioma; pituitary adenoma; bronchial adenoma; chondromatous hanlartoma; inesothelioma; Hodgkin's Disease; or a combination of one or more of the foregoing cancers.
- In a further aspect, the present invention provides a compound of Formula I or a pharmaceutically acceptable salt thereof, for use in the treatment of cancer.
- In a further aspect, the present invention provides a method of treating cancer comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula I or a pharmaceutically acceptable salt thereof.
- In a further aspect, the present invention provides the use of a compound of Formula I or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of cancer.
- A compound of the invention may be employed with other therapeutic methods of cancer treatment, e.g., in anti-neoplastic therapy, combination therapy with immune checkpoint inhibitors, other chemotherapeutic, hormonal, antibody agents as well as surgical and/or radiation treatments.
- Immune checkpoint inhibitors, such as humanized antibodies against PD-1, PD-L1 and CTLA4, have recently been shown to be highly successful in treating several types of metastatic cancer, including melanoma, non-small cell lung cancers, renal cell carcinoma and bladder cancer (Sharma and Allison, 2015, Science 348, 56). However, still only a small percentage of cancer patients benefit from the checkpoint inhibitor therapies, in part because insufficient number of anti-tumor immune cells, such as CD8 T cells, are generated and/or infiltrated into the tumors. Activation of the cGAS-STING pathway activates anti-tumor immunity, including the production and infiltration of tumor-specific CD8 T cells. Therefore, cGAMP analogues are expected to function synergistically with immune checkpoint inhibitors and the combination therapies are likely to bring therapeutic benefits to a larger percentage of cancer patients.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one immune checkpoint inhibitor.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one immune checkpoint inhibitor for use in therapy.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or pharmaceutically acceptable salt thereof, and at least one immune checkpoint inhibitor for use in treating cancer.
- In a further aspect, the present invention provides the use of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one immune checkpoint inhibitor in the manufacture of a medicament for the treatment of cancer.
- In a further aspect, the present invention provides a method of treating cancer, comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least immune checkpoint inhibitor.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, at least one immune checkpoint inhibitor, and one or more of pharmaceutically acceptable carriers, diluents and excipients.
- Radiation of tumors, especially high-dose radiation such as stereotatic body radiation therapy (SBRT), kills tumor cells with a high degree of precision. Dead tumor cells not only provide tumor antigens to generate tumor-specific cytotoxic T cells, but also release tumor DNA into antigen presenting cells to activate the cGAS-STING pathway (Deng et al., 2014, Immunity 41, 843). Therefore, cGAMP analogues are expected to function synergistically with radiation therapies to benefit a larger percentage of cancer patients.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in combination with radiation therapy such as SBRT.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or pharmaceutically acceptable salt thereof, in combination with radiation therapy such as SBRT for use in treating cancer.
- In a further aspect, the present invention provides the use of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in combination with radiation therapy such as SBRT in the manufacture of a medicament for the treatment of cancer.
- In a further aspect, the present invention provides a method of treating cancer, comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in combination with radiation therapy such as SBRT.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and one or more of pharmaceutically acceptable carriers, diluents and excipients, in combination with radiation therapy such as SBRT for the treatment of cancer.
- Anti-neoplastic agents include chemical compounds and antibodies that kill tumor cells by inhibiting cell cycle, signal transduction, DNA metabolism and angiogenesis and/or by promoting DNA damage, apoptosis and necrosis. These agents comprise that largest class of molecules currently used for cancer therapies. Anti-neoplastic agents selectively kill tumor cells, although many of them also kill normal cells, thereby generating severe side effects. Processing of dead tumor cell associated antigens by antigen presenting cells leads to the generation of tumor-specific cytotoxic T cells. This process can be enhanced by cGAMP analogues. Therefore, combination of cGAMP analogues with anti-neoplastic agents are likely to generate synergistic effects that benefit a larger percentage of patients.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent for use in therapy.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent for use in treating cancer.
- In a further aspect, the present invention provides the use of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent in the manufacture of a medicament for the treatment of cancer.
- In a further aspect, the present invention provides a method of treating cancer, comprising: administering to a patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent.
- In a further aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, at least one anti-neoplastic agent, and one or more of pharmaceutically acceptable carriers, diluents and excipients.
- Any anti-neoplastic agent that has activity versus a susceptible tumor being treated may be utilized in the combination. Typical anti-neoplastic agents useful include anti-microtubule agents such as diterpenoids and vinca alkaloids; platinum coordination complexes; alkylating agents such as nitrogen mustards, oxazaphosphorines, alkylsulfonates, nitrosoureas, and triazenes; antibiotic agents such as anthracycline, actinomycins and bleomycins; topoisomerase II inhibitors such as epipodophyllotoxins; antimetabolites such as purine and pyrimidine analogues and anti-folate compounds; topoisomerase I inhibitors such as camptothecins; hormones and hormonal analogues; signal transduction pathway inhibitors; non-receptor tyrosine angiogenesis inhibitors; immunotherapeutic agents; proapoptotic agents; and cell cycle signaling inhibitors.
- Anti-microtubule or anti-mitotic agents are phase specific agents active against the microtubules of tumor cells during M or the mitosis phase of the cell cycle. Examples of anti-microtubule agents include diterpenoids and vinca alkaloids.
- Diterpenoids, which are derived from natural sources, are phase specific anti-cancer agents that operate at the G2/M phases of the cell cycle. It is believed that the diterpenoids stabilize the β-tubulin subunit of the microtubules, by binding with this protein. Disassembly of the protein appears then to be inhibited with mitosis being arrested and cell death following. Examples of diterpenoids include paclitaxel and its analog docetaxel.
- Vinca alkaloids are phase specific anti-neoplastic agents derived from the periwinkle plant. Vinca alkaloids act at the M phase (mitosis) of the cell cycle by binding specifically to tubulin. Consequently, the bound tubulin molecule is unable to polymerize into microtubules. Mitosis is believed to be arrested in metaphase with cell death following. Examples of vinca alkaloids include vinblastine, vincristine, and vinorelbine.
- Platinum coordination complexes are non-phase specific anti-cancer agents, which are interactive with DNA. The platinum complexes enter tumor cells, undergo, aquation and form intra- and interstrand crosslinks with DNA causing adverse biological effects to the tumor. Examples of platinum coordination complexes include oxaliplatin, cisplatin and carboplatin.
- Alkylating agents are non-phase anti-cancer specific agents and strong electrophiles. Typically, alkylating agents form covalent linkages, by alkylation, to DNA through nucleophilic moieties of the DNA molecule such as phosphate, amino, sulfhydryl, hydroxyl, carboxyl, and imidazole groups. Such alkylation disrupts nucleic acid function leading to cell death. Examples of alkylating agents include nitrogen mustards such as cyclophosphamide, melphalan, and chlorambucil; alkyl sulfonates such as busulfan; nitrosoureas such as carmustine; and triazenes such as dacarbazine.
- Antibiotic anti-neoplastics are non-phase specific agents, which bind or intercalate with DNA. Typically, such action results in stable DNA complexes or strand breakage, which disrupts ordinary function of the nucleic acids leading to cell death. Examples of antibiotic anti-neoplastic agents include actinomycins such as dactinomycin, anthrocyclins such as daunorubicin and doxorubicin; and bleomycins.
- Topoisomerase II inhibitors include epipodophyllotoxins. Epipodophyllotoxins are phase specific anti-neoplastic agents derived from the mandrake plant. Epipodophyllotoxins typically affect cells in the S and G2 phases of the cell cycle by forming a ternary complex with topoisomerase II and DNA causing DNA strand breaks. The strand breaks accumulate and cell death follows. Examples of epipodophyllotoxins include etoposide and teniposide.
- Antimetabolite neoplastic agents are phase specific anti-neoplastic agents that act at S phase (DNA synthesis) of the cell cycle by inhibiting DNA synthesis or by inhibiting purine or pyrimidine base synthesis and thereby limiting DNA synthesis. Consequently, S phase does not proceed and cell death follows. Examples of antimetabolite anti-neoplastic agents include fluorouracil, methotrexate, cytarabine, mecaptopurine, thioguanine, and gemcitabine.
- Camptothecins, including, camptothecin and camptothecin derivatives are available or under development as Topoisomerase I inhibitors. Camptothecins cytotoxic activity is believed to be related to its Topoisomerase I inhibitory activity. Examples of camptothecins include, but are not limited to irinotecan, topotecan, and the various optical forms of 7-(4-methylpiperazino-methylene)-10,11-ethylenedioxy-20-camptothecin described below.
- Hormones and hormonal analogues are useful compounds for treating cancers in which there is a relationship between the hormone(s) and growth and/or lack of growth of the cancer. Examples of hormones and hormonal analogues useful in cancer treatment include adrenocorticosteroids such as prednisone and prednisolone which are useful in the treatment of malignant lymphoma and acute leukemia in children; aminoglutethimide and other aromatase inhibitors such as anastrozole, letrazole, vorazole, and exemestane useful in the treatment of adrenocortical carcinoma and hormone dependent breast carcinoma containing estrogen receptors; progestrins such as megestrol acetate useful in the treatment of hormone dependent breast cancer and endometria 1 carcinoma; estrogens, and anti-estrogens such as fulvestrant, flutamide, nilutamide, bicalutamide, cyproterone acetate and 5a-reductases such as finasteride and dutasteride, useful in the treatment of prostatic carcinoma and benign prostatic hypertrophy; anti-estrogens such as tamoxifen, toremifene, raloxifene, droloxifene, iodoxyfene, as well as selective estrogen receptor modulators (SERMS) useful in the treatment of hormone dependent breast carcinoma and other susceptible cancers; and gonadotropin-releasing hormone (GnRH) and analogues thereof which stimulate the release of leutinizing hormone (LH) and/or follicle stimulating hormone (FSH) for the treatment prostatic carcinoma, for instance, LHRH agonists and antagagonists such as goserelin acetate and luprolide.
- Signal transduction pathway inhibitors are those inhibitors, which block or inhibit a chemical process which evokes an intracellular change. As used herein this change is cell proliferation or differentiation. Signal transduction inhibitors useful in the present invention include inhibitors of receptor tyrosine kinases, non-receptor tyrosine kinases, SH2/SH3 domain blockers, serine/threonine kinases, phosphotidyl inositol-3 kinases, myoinositol signaling, and Ras oncogenes.
- Several protein tyrosine kinases catalyze the phosphorylation of specific tyrosyl residues in various proteins involved in the regulation of cell growth. Such protein tyrosine kinases can be broadly classified as receptor or non-receptor kinases.
- Receptor tyrosine kinases are transmembrane proteins having an extracellular ligand binding domain, a transmembrane domain, and a tyrosine kinase domain. Receptor tyrosine kinases are involved in the regulation of cell growth and are generally termed growth factor receptors. Inappropriate or uncontrolled activation of many of these kinases, i.e. aberrant kinase growth factor receptor activity, for example by over-expression or mutation, has been shown to result in uncontrolled cell growth. Accordingly, the aberrant activity of such kinases has been linked to malignant tissue growth. Consequently, inhibitors of such kinases could provide cancer treatment methods. Growth factor receptors include, for example, epidermal growth factor receptor (EGFr), platelet derived growth factor receptor (PDGFr), erbB2, erbB4, ret, vascular endothelial growth factor receptor (VEGFr), tyrosine kinase with immunoglobulin-like and epidermal growth factor homology domains (TIE-2), insulin growth factor-I (IGFI) receptor, macrophage colony stimulating factor (cfms), BTK, ckit, cmet, fibroblast growth factor (FGF) receptors, Trk receptors (TrkA, TrkB, and TrkC), ephrin (eph) receptors, and the RET protooncogene. Several inhibitors of growth receptors are under development and include ligand antagonists, antibodies, tyrosine kinase inhibitors and anti-sense oligonucleotides. Growth factor receptors and agents that inhibit growth factor receptor function are described, e.g., in Kath, John C, Exp. Opin. Ther. Patents (2000) 10(6):803-818; Shawver et al. DDT Vol 2, No. 2 Feb. 1997; and Lofts, F. J. et al in “Growth factor receptors as targets”, New Molecular Targets for Cancer Chemotherapy, ed. Workman, Paul and Kerr, David, CRC press 1994, London.
- Tyrosine kinases which are not growth factor receptor kinases are termed nonreceptor tyrosine kinases. Non-receptor tyrosine kinases useful in the present invention, which are targets or potential targets of anti-cancer drugs, include cSrc, Lck, Fyn, Yes, Jak, cAbl, FAK (Focal adhesion kinase), Brutons tyrosine kinase, and Bcr-Abl. Such non-receptor kinases and agents which inhibit non-receptor tyrosine kinase function are described, e.g., in Sinh, S. and Corey, S. J., (1999) Journal of Hematotherapy and Stem Cell Research 8 (5): 465-80; and Bolen, J. B., Brugge, J. S., (1997) Annual review of Immunology. 15: 371-404.
- SH2/SH3 domain blockers are agents that disrupt SH2 or SH3 domain binding in a variety of enzymes or adaptor proteins including PI3-K p85 subunit, Src family kinases, adaptor molecules (She, Crk, Nek, Grb2) and Ras-GAP. SH2/SH3 domains as targets for anti-cancer drugs are discussed in Smithgall, T. E. (1995), Journal of Pharmacological and Toxicological Methods. 34(3) 125-32.
- Inhibitors of Serine/Threonine Kinases include MAP kinase cascade blockers which include blockers of Raf kinases (rafk), Mitogen or Extracellular Regulated Kinase (M EKs), and Extracellular Regulated Kinases (ERKs); and Protein kinase C family member blockers including blockers of PKCs (alpha, beta, gamma, epsilon, mu, lambda, iota, zeta). I kB kinase family (I KKa, I KKb), PKB family kinases, akt kinase family members, and TGF beta receptor kinases. Such Serine/Threonine kinases and inhibitors thereof are described, e.g., in Yamamoto, T. et al., (1999), Journal of Biochemistry. 126 (5) 799-803; Brodt, P et al. (2000), Biochemical Pharmacology, 60. 1101-1107; Massague, J., and Weis-Garcia, F. (1996) Cancer Surveys. 27:41-64; Philip, P. A.; and Harris, A. L. (1995), Cancer Treatment and Research. 78: 3-27; Lackey, K. et al Bioorganic and Medicinal Chemistry Letters, (10), 2000, 223-226; U.S. Pat. No. 6,268,391; and Martinez-lacaci, L, et al., Int. J. Cancer (2000), 88(1), 44-52.
- Inhibitors of Phosphotidyl inositol-3 Kinase family members including blockers of PI3-kinase, ATM, DNA-PK, and Ku are also useful in the present invention. Such kinases are discussed, e.g., in Abraham, R. T. (1996), Current Opinion in Immunology. 8 (3) 412-8; Canman, C. E., Lim, D. S. (1998), Oncogene 17 (25) 3301-3308; Jackson, S. P. (1997), International Journal of Biochemistry and Cell Biology. 29 (7):935-8; and Zhong, H. et al. Cancer Res., (2000) 60(6), 1541-1545.
- Also useful in the present invention are Myo-inositol signaling inhibitors such as phospholipase C blockers and Myoinositol analogues. Such signal inhibitors are described, e.g., in Powis, G., and Kozikowski A., (1994) New Molecular Targets for Cancer Chemotherapy ed., Paul Workman and David Kerr, CRC press 1994, London.
- Another group of signal transduction pathway inhibitors are inhibitors of Ras Oncogene. Such inhibitors include inhibitors of farnesyltransferase, geranyl-geranyl transferase, and CAAX proteases as well as anti-sense oligonucleotides, ribozymes and immunotherapy. Such inhibitors have been shown to block ras activation in cells containing wild type mutant ras, thereby acting as antiproliferation agents. Ras oncogene inhibition is discussed in Scharovsky, O. G., et al. (2000), Journal of Biomedical Science. 7(4) 292-8; Ashby, M. N. (1998), Current Opinion in Lipidology. 9 (2) 99-102; and BioChim. Biophys. Acta, (19899) 1423(3):19-30.
- Antibody antagonists to receptor kinase ligand binding may also serve as signal transduction inhibitors. This group of signal transduction pathway inhibitors includes the use of humanized antibodies to the extracellular ligand binding domain of receptor tyrosine kinases. Examples include Imclone C225 EGFR specific antibody (see Green, M. C. et al, Monoclonal Antibody Therapy for Solid Tumors, Cancer Treat. Rev., (2000), 26(4), 269-286); Herceptin® erbB2 antibody (see Tyrosine Kinase Signalling in Breast cancer:erbB Family Receptor Tyrosine Kinases, Breast cancer Res., 2000, 2(3), 176-183); and 2CB VEGFR2 specific antibody (see Brekken, R. A. et al, Selective Inhibition of VEGFR2 Activity by a monoclonal Anti-VEGF antibody blocks tumor growth in mice, Cancer Res. (2000) 60, 5117-5124).
- Anti-angiogenic agents such as non-receptor MEK angiogenesis inhibitors may also be useful, as well as those which inhibit the effects of vascular endothelial growth factor (e.g., the anti-vascular endothelial cell growth factor antibody bevacizumab [Avastin™]), and compounds that work by other mechanisms (e.g., linomide, inhibitors of integrin αvβ3 function, endostatin and angiostatin).
- In one aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent which is an anti-microtubule agent, platinum coordination complex, alkylating agent, antibiotic agent, topoisomerase II inhibitor, antimetabolite, topoisomerase I inhibitor, hormones and hormonal analogue, signal transduction pathway inhibitor, non-receptor tyrosine MEK angiogenesis inhibitor, immunotherapeutic agent, proapoptotic agent, or cell cycle signaling inhibitor.
- In another aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent selected from diterpenoids and vinca alkaloids.
- In another aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent which is a platinum coordination complex. In one embodiment, at least one anti-neoplastic agent is paclitaxel, carboplatin, or vinorelbine.
- In another aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent which is a signal transduction pathway inhibitor. In one embodiment, the signal transduction pathway inhibitor is an inhibitor of a growth factor receptor kinase VEGFR2, TIE2, PDGFR, BTK, erbB2, EGFr, IGFR-1, TrkA, TrkB, TrkC, or c-fms. In another embodiment, the signal transduction pathway inhibitor is an inhibitor of a serine/threonine kinase rafk, akt, or PKC-zeta. In another embodiment, the signal transduction pathway inhibitor is an inhibitor of a non-receptor tyrosine kinase selected from the src family of kinases. In another embodiment, the signal transduction pathway inhibitor is an inhibitor of c-src. In another embodiment, the signal transduction pathway inhibitor is an inhibitor of Ras oncogene selected from inhibitors of farnesyl transferase and geranylgeranyl transferase. In another embodiment, the signal transduction pathway inhibitor is an inhibitor of a serine/threonine kinase selected from the group consisting of PI3K. In another embodiment, the signal transduction pathway inhibitor is a dual EGFr/erbB2 inhibitor, for example N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl) ethyl]amino}methyl)-2-furyl]-4-quinazolinamine.
- In another aspect, the present invention provides a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, and at least one anti-neoplastic agent which is a cell cycle signaling inhibitor. In one embodiment, the cell cycle signaling inhibitor is an inhibitor of CDK2, CDK4 or CDK6.
- Compounds of Formula I may be prepared by methods known in the art of organic synthesis as set forth in the schemes below and/or the specific Examples described below. In all of the methods, it is well understood that protecting groups for sensitive or reactive groups may be employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Green and P. G. M. Wuts (1999) Protective Groups in Organic Synthesis, 3′ edition, John Wiley & Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection of processes as well as the reaction conditions and order of their execution shall be consistent with the preparation of compounds of Formula I.
- It is understood that the examples and embodiments described herein are for illustrative purposes only and that various modifications or changes in light thereof will be suggested to persons skilled in the art and are to be included within the spirit and purview of this application and scope of the appended claims.
- The following list provides definitions of certain abbreviations as used herein. It will be appreciated that the list is not exhaustive, but the meaning of those abbreviations not herein below defined will be readily apparent to those skilled in the art: Ac is acetyl; AcOH is acetic acid: Ac2O is acetic anhydride: AIBN is 2,2′-azobisisobutyronitrile; Bn is benzyl; BSA is N,O-bis(trimethylsilyl)acetamide: BSTFA is N,O-bis(trimethylsilyl)trifluoroacetamide; Bu is butyl: Bz is benzoyl: CAN is ceric ammonium nitrate: CE is 2-cyanoethyl; DCA is dichloroacetic acid; DCM is dichloromethane: DDTT is 1,2,4-dithiazole-5-thione: DEAD is diethyl azodicarboxylate; DIAD is diisopropyl azodicarboxylate; DIPEA is N,N-diisopropylethylamine: DMAP is 4-(dimethylamino)pyridine; DMF is N,N-dimethvlformamide; DMOCP is 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide; DMSO is dimethylsulphoxide; DMTr is 4,4′-dimethoxytrityl; EtOAc is ethyl acetate; EtOH is ethanol; HMPT is hexamethylphosphorous triamide; HPLC is high performance liquid chromatography; ibu is isobutyryl; IBX is 2-iodoxvbenzoic acid: Imid is imidazole; iPr is isopropyl: KOH is potassium hydroxide: Me is methyl; MeCN is acetonitrile; MeOH is methanol; MTBE is methyl tert-butyl ether: Ms is methanesulfonyl; Pd/C is palladium on activated charcoal; NIS is N-iodosuccinimide; NPE is 2-(4-nitrophenyl)ethyl; PE is petroleum ether: Ph is phenyl; PMB is p-methoxybenzyl; PPh3 is triphenylphosphine; Py is pyridine; TBAF is tetra-n-butylammonium fluoride: TBAI is tetrabutylammonium iodide; TBDPS is tert-butyldiphenylsilyl; TBHP is tert-Butyl hydroperoxide; TBS is tert-butyldimethylsilyl; TCDI is 1,1′-thiocarbonyldiimidazole: TDA-1 is tris[2-(2-methoxyethoxy)ethyl]amine; TEA is triethylamine: Tf is trifluoromethanesulfonyl; TFA is trifluoroacetic acid: TFE is 2,2,2-trifluoroethyl; THF is tetrahydrofuran: TIPS is triisopropylsilyl: TLC is thin-layer chromatography; TMS is trimethylsilyl; TMSOTf is trimethylsilyl trifluoromethanesulfonate; Tol is p-toluoyl; Tr is trityl.
-

-

- To a suspension of D-Ribose (1) (160 g, 1.07 mol) in acetone (2.0 L) is added concentrated sulfuric acid (10.7 g, 107 mmol, 5.8 mL) at 27° C. dropwise. After stirring for 12 hours, solid Sodium bicarbonate (100 g) is added. The mixture is then filtered and the filtrate is concentrated to give crude 2 (215.0 g).
-

- To a solution of crude 2 (215 g, 1.13 mol) in DCM (1.5 L) is added TBSCl (170 g, 1.13 mol) and TEA (172 g, 1.69 mol) at 0° C. After stirring at 27° C. for 12 hours, the mixture is filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/100 to 1/50) to give 3 as a colorless oil (285 g, 83% yield).
-

- To a solution of 3 (60.0 g, 197.08 mmol) and carbon tetrachloride (67.3 g, 438 mmol, 42 mL) in THF (1.2 L) is added HMPT (63.0 g, 386 mmol, 70 mL) dropwise at −78° C. and stirred at 27° C. for 2 hours. To another solution of 6-chloro-7-deazapurine (24.2 g, 158 mmol) and KOH (16.6 g, 296 mmol) in MeCN (1.2 L) is added TDA-1 (6.37 g, 19.7 mmol) at 27° C. followed by the THF solution obtained above. After stirring at 27° C. for 12 hours, the reaction mixture is filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/50 to 1/10) to give 4 as a yellow oil (15.3 g, 18% yield).
-

- A solution of 4 (28.6 g, 64.9 mmol) in dioxane (150 mL) and ammonium hydroxide aqueous solution (500 mL) is stirred at 120° C. for 30 hours in a sealed autoclave. The volatiles are then removed and the aqueous solution is extracted with EA (300 mL×3). The combined organic layers are washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and partially purified by silica gel column chromatography (EA/DCM=1/1) to give a yellow foam (9.65 g). This residue is then dissolved in THF (50 mL) and treated with TBAF trihydate (10.9 g, 34.4 mmol) at 27° C. After stirring for 2 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/DCM=1/1 to 7/1) to give 5 (5.56 g, 79% yield) as yellow solid.
-

- To a solution of 5 (7.26 g, 23.7 mmol) in DCM (60 mL) is added Imid (4.84 g, 71.1 mmol) and TBSCl (5.36 g, 35.6 mmol) at 27° C. After stirring at 27° C. for 1.5 hours, water (100 mL) is added and the mixture is extracted with DCM (200 mL). The organic layer is washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered, concentrated to give crude TBS-5. To a solution of the crude TBS-5 obtained above in DCM (100 mL) is added benzoyl chloride (5.14 g, 36.6 mmol) at 27° C. After stirring for 12 hours, water (200 mL) is added the mixture is extracted with DCM (500 mL). The organic layer is dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/50 to 1/10) to give 6 as a yellow foam (8.12 g, 64% yield).
-

- A solution of 6 (15.2 g, 28.9 mmol) in TFA (90 mL) and DCM (20 mL) is stirred at 27° C. for 12 hours. The volatiles are then removed and the residue is purified by silica gel column chromatography (MeOH/DCM=1/100 to 1/10) to B1 as a yellow solid (10.16 g, 95% yield).
-

-

- To a solution of 5-amino-4,6-dichloropyrimidine (63.0 g, 384 mmol) in n-BuOH (300.0 mL) is added p-methoxybenzylamine (58.0 g, 423 mmol, 55 mL) and DIPEA (99.3 g, 768 mmol, 134 mL). After stirring at 100-110° C. for 15 hours, the volatiles are removed before MTBE (100 mL) is added. The solid is collected by filtration and washed with EA to give 8 as an off-white solid (55.0 g, 54% yield). (MS: [M+H]+ 265.0)
-

- To a solution of 8 (10.0 g, 37.8 mmol) in a mixture of DCM (200 mL), AcOH (100 mL), and water (100 mL) is added sodium nitrite (2.87 g, 41.6 mmol, 2.3 mL) at 0° C. After stirring at 0-25° C. for 1 hour, DCM (30 mL) and saturated sodium bicarbonate aqueous solution (30 mL) are added. The layers are then separated and the aqueous phase is extracted with DCM (150 mL×3). The combined organic phases are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/3) to give 9 as a light yellow solid (6.0 g, 88% yield). (MS: [M+H]+ 276.0)
-

- To a solution of 9 (6.0 g, 21.8 mmol) in 1,4-dioxane (30 mL) is added ammonium hydroxide aqueous solution (30 mL). After stirring at 30-40° C. for 5 hours, the solid is collected by filtration to give 10 as a white solid (4.0 g, 70% yield). (MS: [M+H]+257.1)
-

- To a solution of 10 (17.0 g, 66.3 mmol) in Py (100 mL) is added DMAP (8.92 g, 73.0 mmol), Imid (13.6 g, 199 mmol) and benzoyl chloride (14.0 g, 99.5 mmol, 11.6 mL). After stirring at 110-120° C. for 18 hours, the volatiles are removed and DCM (300 mL) and water (300 mL) are added. The layers are separated and the organic phase is dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1:1) to give 11 as an off-white solid (17.0 g, 68% yield). (MS: [M+H]+ 361.2)
-

- To a suspension of 11 (6.40 g, 17.8 mmol) in MeCN (60 mL) is added a solution of CAN (29.2 g, 53.3 mmol) and sodium bicarbonate (1.49 g, 17.76 mmol) in water (60 mL) at 0° C. After stirring at 0-25° C. for 12 hours, the mixture is neutralized with sodium bicarbonate to ˜pH 7. The solid is collected by filtration to give 12 (2.6 g, 57% yield). (MS: [M+H]+ 241.1)
-

- To a solution of 12 (9.30 g, 38.7 mmol) and 13 (20.5 g, 40.7 mmol) in MeCN (350 mL) is added tin(IV) chloride (30.3 g, 116 mmol, 13.6 mL) at 0° C. After stirring at 0-25° C. for 24 hours, the reaction mixture is poured into saturated sodium bicarbonate aqueous solution (300 mL). The solid is filtered off and washed with water (100 mL). The filtrate is extracted with DCM (150 mL×4) and the combined organic layers are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/DCM=1/10) to give 14 as an off-white gum (6.10 g, 21% yield). (MS: [M+H]+ 684.9)
-

- To a solution of 14 (6.1 g, 8.9 mmol) in a mixture of THF (35 mL) and MeOH (28 mL) is added lithium hydroxide aqueous solution (1M, 16.0 mL) at 0° C. After stirring at 0-25° C. for 3 hours, the mixture is neutralized with citric acid aqueous solution (1M) to ˜pH 7 and then concentrated and purified by silica gel column chromatography (MeOH/DCM=1/20) to give B2 as an off-white solid (2.9 g, 87% yield). (MS: [M+H]+ 373.1)
-

-

- To a solution of 15 (880 mg, 2.5 mmol) in Py (10 mL) is added a solution of DMTrCl (940 mg, 2.6 mmol) in Py (5 mL). After stirring for 3 hours, the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/20 to 1/10) to provide 16 as a white foam (1.23 g, 75% yield). (MS: [M+H]+ 656.2)
-

- To a solution of 16 (900 mg, 1.4 mmol) and Imid (280 mg, 4.15 mmol) in Py (15 mL) is added TBSCl (310 mg, 2.05 mmol). After stirring for 4 hours, the volatiles are removed and the residue is dissolved in DCM (50 mL), washed with saturated sodium bicarbonate aqueous solution and brine, dried over anhydrous sodium sulfate, concentrated, and purified by silica gel column chromatography (EA/toluene=1/3 to 2/3) to provide 17 as a white solid (480 mg, 45% yield). (MS: [M+H]+ 770.2)
-

- To a solution of 17 (500 mg, 0.65 mmol) in DMF (6 mL) is added TCDI (350 mg, 1.94 mmol). After stirring for 2 days, EA (40 mL) and water (25 mL) are added and the layers are separated. The aqueous layer is extracted with ethyl acetate (25 mL×3). The combined organic layers are washed with water (20 mL), brine (20 mL×2), dried over anhydrous sodium sulfate, and concentrated to give crude 18. (MS: [M+H]+ 880.2)
-

- To a degassed solution of crude 18 in toluene (10 mL) at 110° C. is added a degassed solution of AIBN (57 mg, 0.34 mmol), tributyltin hydride (0.51 mL, 1.94 mmol) in toluene (3 mL) over 30 minutes. After stirring at 110° C. for 6 hours, the mixture is cooled to room temperature, concentrated, and purified by silica gel column chromatography (EA/hexanes=1/5 to 2/1) to give 19 as a yellow oil (195 mg, 40% yield over two steps). (MS: [M+H]+ 754.2)
-

- To a solution of 19 (190 mg, 0.252 mmol) in THF (5 mL) is added TBAF (1 M in THF, 0.50 mL). After stirring at room temperature for 2 hours, water (5 mL) is added and the mixture is extracted with EA (8 mL×3), dried over anhydrous sodium sulfate, concentrated, and purified by silica gel column chromatography (MeOH/DCM=1/20) to give DMTr-B3 as a white solid (132 mg, 82% yield). (MS: [M+H]+ 640.2)
-

-

- To a solution of 20 (12.8 g, 67.0 mmol) in Py (300 mL) is added TBDPSCl (21.0 mL, 80.4 mmol). After stirring for 3 h, MeOH (25 mL) is added and the mixture is concentrated. The residue is dissolved in diethyl ether (200 mL), washes with sodium bicarbonate aqueous solution (10%, 100 mL) and water (100 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (diethyl ether/PE=1/2) to give 21 as a white solid (27.2 g, 95% yield). (MS: [M+Na]+ 451.2)
-

- A solution of 21 (27.2 g, 63.7 mmol) in DMSO (200 mL) and Ac2O (50 mL) is stirred for 16 hour before pouring into ice water (200 mL). The mixture is extracted with diethyl ether (100 mL×3) and the combined organic layers are washed with sodium bicarbonate aqueous solution (10%, 100 mL) and water (100 mL), and concentrated. The residue is then dissolved in MeOH (250 mL) and DCM (250 mL) at 0° C. followed by addition of sodium borohydride (12.0 g) in 10 portions. After stirring for 5 minutes, water (100 mL) is added and the layers are separated. The organic layer is then concentrated and purified by silica gel column chromatography (diethyl ether/PE=1/2) to give 22 as a white solid (20.4 g, 75% yield over two steps). (MS: [M+Na]+ 451.2)
-

- To a solution of 22 (4.0 g, 9.33 mmol) in DMF (45 mL) is added sodium hydride (484 mg, 12.1 mmol) at 0° C. and stirred for 30 minutes before methyl iodide (0.64 mL, 10.3 mmol) is added slowly. After stirring for 3 hours, water (3 mL) is added and the volatiles are removed and purified by silica gel column chromatography (EA/PE=1/10) to give 23 as a white solid (3.8 g, 92% yield). (MS: [M+Na]+ 465.2)
-

- To a solution of 23 (3.1 g, 7.0 mmol) in THF (50 mL) is added TBAF (8.4 mL, 8.4 mmol) at 0° C. After stirring for 4 h at room temperature, water (5 mL) and EA are added. The layers are separated and the organic layer is washed with water and brine, concentrated, and the resulting residue is dissolved in DCM followed by addition of TEA (4.9 mL, 35 mmol) and benzoyl chloride (0.98 mL, 8.4 mmol). After stirring for 1 hour, water (3 mL) is added and the volatiles are removed. The residue is purified by silica gel column chromatography (EA/PE=1/5) to give 24 as a white solid (1.9 g, 88% yield). (MS: [M+Na]+331.0)
-

- A solution of 24 (0.71 g, 2.3 mmol) in HOAc (14 mL) and water (6 mL) is heated under reflux for 30 minutes. After cooling to room temperature, the mixture is co-evaporated with toluene (10 mL×4) and the resulting residue is dissolved in Py/Ac2O (10/1 v/v, 10 mL) followed by addition of DMAP (50 mg, 0.46 mmol). After stirring for 4 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/3) to give 25 as a white solid (0.75 g, 92% yield). (MS: [M+Na]+ 375.0)
-

- To a suspension of 25 (500 mg, 1.42 mmol) and N2-isobutyrylguanine (500 mg, 2.13 mmol) in DCM (20 mL) at 80° C. is added BSA (1.8 mL, 7.4 mmol) and stirred for 1 hour before addition of TMSOTf (0.77 mL, 4.26 mmol). After stirring at 80° C. for 3 hours, the mixture is cooled to room temperature before sodium bicarbonate aqueous solution (50 mL) is added. The mixture is then extracted with DCM (50 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, concentrated, and purified by silica gel column chromatography (MeOH/DCM=1/20 to 1/10) to give 26 as a white powder (624 mg, 85% yield). (MS: [M+H]+ 514.2)
-

- To a solution of 27 (0.49 g, 0.96 mmol) in MeOH/THF/water (4/5/1 v/v/v, 20 mL) is added sodium hydroxide aqueous solution (10 M, 0.25 mL, 2.5 mmol) at 0° C. After stirring for 30 minutes, HOAc is added and the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/10 to 1/5) to give B4 as an oil (322 mg, 92% yield). (MS: [M+H]+ 368.2)
-

-

- To a solution of 15 (7.0 g, 20 mmol) in MeCN (100 mL) is added DMAP (1.2 g, 10 mmol) and Ac2O (7.5 mL, 80 mmol) at 0° C. After stirring at room temperature overnight, the mixture is concentrated and purified by silica gel flash chromatography (MeOH/DCM=1/20 to 1/10) to give 27 as a white solid (8.77 g, 92% yield). (MS: [M+H]+480.0)
-

- To a solution of 27 (480 mg, 1.0 mmol) in 1,4-dioxane (1 mL) is added PPh3 (656 mg, 2.5 mmol), propargyl bromide (0.15 mL, 2 mmol) and a solution of DEAD (0.49 mL, 2.5 mmol) in dioxane (1 mL) at 0° C. After stirring for 2 hours, the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/50 to 1/20) to give 28 as a white solid (440 mg, 47% yield). (MS: [M+H]+ 518.2)
-

- To a solution of 28 (150 mg, 0.17 mmol) in THF (4.5 mL) and MeOH (0.5 mL) is added sodium hydroxide aqueous solution (1 M, 0.5 mL) at 0° C. After stirring for 1 hour, HOAc (0.1 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/20 to 1/10) to give B5 as a white solid (40 mg, 64% yield). (MS: [M+H]+ 392.0)
-


-

- To a solution of 3 (40.0 g, 131 mmol) and carbon tetrachloride (33.6 g, 218 mmol, 21 mL) in THF (500 mL) at −78° C. is added HMPT (22.5 g, 138 mmol, 25 mL) over 15 min. After stirring for 2 hours with brief periods of slight warming to prevent gel formation, the mixture is concentrated to about 70 mL. To a suspension of KOH (25.8 g, 460 mmol) in MeCN (600 mL) is added TDA-1 (4.25 g, 13.14 mmol, 4.2 mL). After stirring at 25° C. for 10 minutes, 2-amino-6-chloro-7-deazapurine (22.2 g, 131 mmol) is added. The mixture is stirred for another 10 minutes before the THF solution obtained above is added. After stirring for 2 hours, the mixture is filtered, concentrated, and purified by silica gel column chromatography (EA/PE=3/17) to give 29 (9.20 g, 15% yield). (MS: [M+H]+ 455.3)
-

- To a mixture of 29 (13.7 g, 30.1 mmol) in dioxane (10 mL) is added a solution of sodium hydroxide (11.7 g, 291 mmol) in water (100 mL) at 25° C. After stirring at 80° C. for 64 hours, the mixture is cooled to 0° C., neutralized with AcOH to ˜pH 7, and extracted with EtOAc (100 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered, and concentrated to give crude 30. (MS: [M+1]+ 323.1)
-

- To a solution of crude 30 (9.7 g, 30.1 mmol) and Imid (4.1 g, 60.3 mmol) in DCM (10 mL) is added TBSCl (9.08 g, 60.3 mmol) at 25° C. After stirring for 16 hours, the mixture is diluted with DCM (100 mL), washed with brine (80 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/4 tot/1 then MeOH/DCM=1/50) to give 31 (9.0 g, 68% yield) as a solid. (MS: [M+H]+ 437.2)
-

- To a solution of 31 (9.0 g, 20.6 mmol) and TEA (4.2 g, 41.2 mmol) in DCM (80 mL) is added isobutyryl chloride (3.29 g, 30.9 mmol) at 0° C. After stirring at 25° C. for 16 hours, the mixture is diluted with DCM (100 mL), washed with saturated sodium bicarbonate aqueous solution (50 mL×2) and brine (50 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/5 to 1/2) to give 32 as a white solid (4.2 g, 40% yield). (MS: [M+H]+ 507.2)
-

- A solution of 32 (4.2 g, 8.29 mmol) in DCM (6 mL) and TFA (24 mL) is stirred at 25° C. for 1 hour before concentrated. The residue is then treated with hydrogen chloride (4M in MeOH, 10 mL) at 0° C. After stirring at 25° C. for 10 minutes, the mixture is concentrated to give crude B6 as a white solid (2.92 g, 99% yield). (MS: [M+H]+ 353.0)
-

-

- To a solution of adenosine (33) (5.0 g, 18.7 mmol) in DMF (200 mL) at 0° C. is added sodium hydride (60% dispersion in mineral oil, 1.0 g, 25 mmol) followed by the TBAI (1.5 g, 4.06 mmol) and propargyl bromide (2.12 mL, 20.9 mmol). After stirring at 55° C. for 2 days, the mixture is purified by silica gel column chromatography (MeOH/DCM=7/93) followed by re-crystallization from ethanol to give 34 as a pale yellow solid (2.56 g, 45%).
-

- To a solution of 34 (1.4 g, 4.59 mmol, co-evaporated twice with Py) in Py (20 mL) is added TMSCl (2.4 mL, 18.9 mmol). After stirring for 30 minutes, benzoyl chloride (0.7 mL, 6.0 mmol) is added and the mixture is stirred for 3 hours before addition of water (10 mL) and ammonium hydroxide aqueous solution (15 mL) at 0° C. After stirring for 20 minutes at room temperature, the mixture is extracted with DCM (25 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, concentrated, and purified by silica gel column chromatography (MeOH/DCM=5:95) to give BA1 as a white foam (1.73 g, 92%). (MS: [M+H]+ 410.2)
-

-

- To a solution of 35 (10.0 g, 56.5 mmol) in THF (80 mL) is added DIPEA (7.3 g, 56.5 mmol, 9.9 mL). After stirring at 0° C. for 10 minutes, a solution of hydrazine (1.81 g, 56.5 mmol, 2.0 mL) in THF (20 mL) is added. The mixture is then stirred at 20° C. for 2 hours before concentrated. After addition of DCM (100 mL) and H2O (100 mL) to the residue, the layers are separated and the aqueous layer is extracted with DCM (100 mL×3). The combined organic layers are washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/15 to 1/8) to give 36 as a yellow solid (3.10 g, 35% yield). (MS: [M+H]+ 155.1)
-

- To a solution of 36 (200 mg, 1.29 mmol) in THF (2.0 mL) is added ammonium hydroxide (2.0 mL). After stirring at 20-30° C. for 2 hours, the mixture is concentrated, triturated with MeCN (0.5 mL), and collected by filtration to give 37 as a red solid (100 mg, 57% yield).
-

- To a suspension of 37 (20.0 g, 148 mmol) and 13 (101 g, 200 mmol) in MeCN (1.2 L) is added boron trifluoride diethyl etherate (30.5 g, 215 mmol, 26.5 mL). After stirring at 75-85° C. for 2 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/5 to 2/1) to give 38 as a yellow solid (35.0 g, 40% yield). (MS: [M+H]+ 580.3)
-

- To a solution of 38 (10.0 g, 17.3 mmol) in DCM (100 mL) is added DMAP (421 mg, 3.45 mmol) and TEA (5.24 g, 51.8 mmol, 7.2 mL) followed by benzoyl chloride (2.91 g, 20.7 mmol, 2.4 mL) dropwise. After stirring at 20-25° C. for 8 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/5 to 11/) to give 39 as a white solid (9.0 g, 76% yield). (MS: [M+H]+ 684.1)
-

- To a solution of 39 (1.0 g, 1.46 mmol) in THF (1.5 mL), MeOH (1.2 mL) and H2O (0.3 mL) is added lithium hydroxide aqueous solution (5 M, 0.53 mL). After stirring at 0-25° C. for 2 hours, the mixture is neutralized with citric acid (1 M) to ˜pH 7 before removal of the volatiles. The solid in the aqueous solution is then collected by filtration to give BA2 as an off-white solid (300 mg, 54% yield). (MS: [M+H]+ 372.2)
-

-

- To a solution of 22 (0.4 g, 0.93 mmol) in DMF (8 mL) is added sodium hydride (48 mg, 1.12 mmol) at 0° C. and the mixture is stirred for 30 minutes before 2,2,2-trifluoroethyl trifluoromethanesulfonate (0.165 mL, 1.12 mmol) is added slowly. After stirring at 0° C. for 3 hours, water (3 mL) is added the mixture is concentrated and purified by silica gel column chromatography (EA/hexanes=1/10) to give 40 as a white solid (218 mg, 46% yield).
-

- To a solution of 40 (1.4 g, 2.74 mmol) in THF (25 mL) is added TBAF (3.3 mL, 3.3 mmol) at 0° C. After stirring at room temperature for 4 hours, water (2 mL) is added and the mixture is extracted with EA. The organic layer is washed with water and brine, and concentrated. The residue is then dissolved in DCM followed by addition of and TEA (1.92 mL, 13.8 mmol) benzoyl chloride (0.42 mL, 3.6 mmol). After stirring for 1 hour, water (1 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/5) to give 41 as a white solid (0.795 g, 77% yield).
-

- A solution of 41 (0.79 g, 2.1 mmol) in HOAc (17.5 mL) and water (7.5 mL) is stirred at reflux for 30 minutes before cooled to room temperature, and co-evaporated with toluene (10 mL×4). The residue is then dissolved in Py (12 mL) followed by addition of Ac2O (0.8 mL, 8.4 mmol). After stirring for 6 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/4) to give 42 as a white solid (0.82 g, 91% yield).
-

- To a suspension of 42 (800 mg, 1.9 mmol) and N2-isobutyrylguanine (633 mg, 2.86 mmol) in dichloroethane (25 mL) at 80° C. is added BSA (2.74 mL, 10.1 mmol) and stirred for 1 hour before addition of TMSOTf (1.03 mL, 5.7 mmol). After stirring for 3 hours at 100° C., the mixture is poured into sodium bicarbonate aqueous solution (60 mL) and extracted with DCM (60 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, concentrated, and purified by silica gel column chromatography (MeOH/DCM=1/20 to 1/10) to give 43 as a white solid (938 mg, 85% yield).
-

- To a solution of 43 (0.5 g, 0.86 mmol) in MeOH (8 mL), THF (10 mL) and water (2 mL) is added sodium hydroxide aqueous solution (10 M, 0.34 mL) at 0° C. After stirring for 30 minutes, HOAc is added and the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/10 to 1/5) to afford BA3 as an oil (348 mg, 93% yield).
-


-

- To a solution of 2 (75 g, 395 mmol) in MeOH (600 mL) is added hydrazine hydrate (120 mL) at 25° C. After stirring for 2 hours, the mixture is concentrated and the residue is dissolved in EtOH (600 mL) before addition of (ethoxymethylene)malononitrile (110 g, 901 mmol). After stirring at 78° C. for 30 minutes, the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/20 to 1/15) to give 44 as a pale yellow solid (42 g, 38% yield).
-

- To a solution of 44 (16.0 g, 57 mmol) in MeOH (75 mL) and water (25 mL) is added ammonium hydroxide (280 mL) and hydrogen peroxide (150 mL). After stirring at 25° C. for 16 hours, the mixture is poured into sodium sulfite aqueous solution (2 L) and then extracted with EA (700 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered, and concentrated. The residue is then dissolved in acetone (90 mL) before benzoyl isothiocyanate (6.96 g, 42.7 mmol, 5.75 mL) is added at 25° C. After stirring at 60° C. for 4 hours, the mixture is concentrated to give crude 45 as a yellow solid.
-

- To a solution of the crude 45 obtained above in MeOH (150 mL) is added sodium hydroxide aqueous solution (0.7 M, 80 mL) followed by methyl iodide (6.8 g, 47.9 mmol, 3.0 mL). After stirring at 20° C. for 2 hours, the mixture is neutralized with HOAc to ˜pH 6 followed by addition of water (80 mL), and extracted with EA (100 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered, and concentrated. The residue is then dissolved in MeOH (30 mL) and sodium hydroxide aqueous solution (1.4 M, 250 mL) is added. After stirring at 100° C. for 2 hours, the mixture is concentrated and the residue is co-evaporated with toluene (200 mL×3) and dissolved in DCM (500 mL). Imid (18.5 g, 271 mmol), DMAP (1.66 g, 13.6 mmol), and TBSCl (40.9 g, 271 mmol) are then added. After stirring at 25° C. for 18 hours, saturated sodium bicarbonate aqueous solution (1 L) is added and the mixture is extracted with EA (500 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (MeOH/DCM=1/60 to 1/30) to give 46 as a white solid (8.50 g, 34% yield over five steps).
-

- To a solution of 46 (23.4 g, 53.5 mmol) in Py (120 mL) is added isobutyryl chloride (11.4 g, 107 mmol, 11.2 mL) at 25° C. After stirring at 25° C. for 16 hours, ammonium hydroxide (0.5 mL) is added and the mixture is stirred for 30 minutes before concentrated. The residue is then dissolved in EtOAc (1.5 L), washed with saturated ammonium hydroxide aqueous solution (500 mL×3) and brine (500 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/10 to 1/3) to give 47 as a light yellow solid (24.0 g, 88% yield). (MS: [M+Na]+ 530.1)
-

- A solution of 47 (10.0 g, 19.7 mmol) in HOAc (6 mL) and water (3 mL) is stirred at 65° C. for 5 hours. The reaction mixture is then concentrated and triturated with DCM (15 mL). The solid is collected by filtration to give curde BB1 as a white solid (4.0 g). (MS: [M+H]+ 354.0)
-

-

- To a solution of 48 (50 g, 135 mmol) in acetone (500 mL) is added 2,2-dimethoxypropane (85 g, 816 mmol, 100 mL) and concentrated sulfuric acid (1.32 g, 13.5 mmol, 0.72 mL). After stirring at 25° C. for 30 minute, saturated sodium bicarbonate aqueous solution (30 mL) is added. The solution is filtered, concentrated, and purified by silica gel column chromatography (MeOH/DCM=1/200 to 1/50) to give 49 as a white solid (35 g, 63% yield). (MS: [M+H]+ 412.1)
-

- To a solution of 49 (5.0 g, 12.2 mmol) in Py (50 mL) is added methanesulfonyl chloride (2.1 g, 18 mmol, 1.4 mL) at 0° C. After stirring at 25° C. for 1 hour, DCM (200 mL) is added, and the solution is washed with saturated sodium bicarbonate aqueous solution followed by brine, dried over anhydrous sodium sulfate, and concentrated. The residue is then dissolved in DMF (50 mL) followed by addition of sodium azide (3.4 g, 52.3 mmol). After stirring at 50° C. for 16 hours, DCM (400 mL) is added. The mixture is washed with water (300 mL), brine, dried over sodium sulfate, concentrate, and purified by silica gel column chromatography (EA/PE=1/1) to give 50 as a light yellow solid (4.0 g). (MS: [M+H]+ 437.1)
-

- A solution of 50 (50 g, 115 mmol) in TFA (125 mL) and water (125 mL) is stirred at 25° C. for 5 hours before concentrated, co-evaporated twice with toluene, and dissolved in MeOH (50 mL). The mixture is then neutralized by sodium bicarbonate aqueous solution (1%) and triturated with MTBE. The solid is collected, washed with MTBE, dried, and dissolved in DMF (400 mL). Pd/C (10% w/w, 10 g) is then added and the mixture is stirred under an atmosphere of hydrogen (15 psi) at 25° C. for 6 hours before filtered and concentrated to give crude BC1 as a yellow oil (39 g). (MS: [M+H]+ 371.1)
-

-

- To a solution of indole (305 mg, 2.6 mmol) in MeCN (10 mL) is added sodium hydride (160 mg, 4.0 mmol) at 0° C. and stirred for 30 minutes before 51 (1.0 g, 2.6 mmol) is added. After stirring for 1 hour, saturated sodium bicarbonate aqueous solution (5 mL) is added and the mixture is extracted with EA (20 mL×3). The combined organic layers are washed with saturated sodium bicarbonate aqueous solution and brine, concentrated, and purified by silica gel column chromatography (EA/hexanes=1/4) to give 52 as a yellow oil (886 mg, 71% yield). (MS: [M+H]+ 470.2)
-

- To a solution of 52 (610 mg, 1.3 mmol) in MeOH (9 mL) is added sodium methoxide (5.4 M in methanol, 0.54 mL). After stirring for 1 hour, hydrochloric acid (5 M, 0.5 mL) is added at 0° C. and the solution is stirred for 10 minutes before concentrated and purified by silica gel column chromatography (MeOH/DCM=1/9) to give the desired product as a white solid (197 mg, 92% yield). (MS: [M+H]+ 234.1)
-

-

- To a solution of 53 (560 mg, 2 mmol) in THF is added magnesium (54 mg, 2.3 mmol) followed by a small amount of iodine. After stirring at 55° C. for 3 hours, copper(I) iodide (213 mg, 1.1 mmol) is added at 0° C. The mixture is stirred at room temperature for 45 minutes before 51 (367 mg, 0.98 mmol) is added at 40° C. After stirring for 2 hours, saturated ammonium chloride aqueous solution (2 mL) and DCM (20 mL) are added. The layers are separated and the organic layer is washed by saturated sodium bicarbonate aqueous solution and brine, dried anhydride sodium sulfate, concentrated, and purified by silica gel column chromatography (EA/hexanes=1/10 to 1/5) to give 54 as a white solid (52 mg, 10%).
-

- To a solution of 54 (230 mg, 0.4 mmol) in MeOH (5 mL) is added sodium methoxide (30% in MeOH, 0.23 mL, 1.2 mmol) at room temperature. After stirring for 1 hour, saturated ammonium chloride (5 mL) is added and the mixture is extracted by EA (10 mL×3). The organic layers are washed with brine, dried over anhydrous sodium sulfate, and concentrated to give crude BC3 as a white solid (150 mg).
-


-

- To a solution of 55 (91.4 g, 481 mmol) in Py (600 mL) is added trityl chloride (160.7 g, 577 mmol). After stirring at 60° C. for 16 hours, the mixture is concentrated and co-evaporated with toluene for three times. The residue is partitioned between DCM (400 mL) and saturated sodium bicarbonate aqueous solution (750 mL). The layers are separated and the aqueous phase is extracted with DCM (400 mL×2). The combined organic layers are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/10 to 1/5) to afford 56 as a white solid (180.5 g, 87% yield). (MS: [M+Na]+ 455.0)
-

- To a solution of 56 (176 g, 407 mmol) in MeCN (1.0 L) is added IBX (228 g, 814 mmol). After stirring at 90° C. for 6 hours, the mixture is filtered and concentrated to give crude 57 as a light yellow oil (175 g). (MS: [M+Na]+ 453.0)
-

- To a solution of sodium hydride (20.1 g, 502 mmol) in THF (1.0 L) is added methyl 2-dimethoxyphosphorylacetate (96.3 g, 529 mmol, 76.5 mL) at 0° C. dropwise over 15 minutes. After stirring for 60 minutes, crude 57 (175 g) obtained above in THF (500 mL) is added dropwise at 0° C. After stirring at 25° C. for 16 hours, water (50 mL) is added at 0° C. and the volatiles are removed and brine (500 mL) is added. The mixture is then extracted with DCM (500 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered, and concentrated. The residue (198 g of 216 g obtained above) is then dissolved in EA (500 mL) and Pd/C (10% w/w, 10 g) is added. After stirring under a hydrogen atmosphere (20 psi) at 25° C. for 16 hours, the mixture is filtered and the filtrate is concentrated and purified by silica gel column chromatography (EA/PE=1/15 to 1/10) to give 58 as a white solid (120 g, 66% yield) (MS: [M+Na]+ 511.1)
-

- To a solution of lithium aluminum hydride (6.21 g, 164 mmol) in THF (200 mL) is added 58 (20.0 g, 40.9 mmol) in THF (50 mL) slowly at 0° C. After stirring at 25° C. for 16 hours, the reaction is quenched by sequential addition of water (6.2 mL), sodium hydroxide aqueous solution (15%, 6.2 mL), and water (18.6 mL). The mixture is then dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/10 to 1/4) to give 59 as a white solid. (MS: [M+Na]+ 483.2)
-

- To a solution of sodium hydride (60% w/w, 6.95 g, 174 mmol) in THF (200 mL) is added 59 (20.0 g, 43.4 mmol) in THF (80 mL) at −20° C. dropwise over 5 minutes. After stirring at 25° C. for 2 hours, benzyl bromide (22.3 g, 130 mmol, 15.5 mL) is added dropwise and the mixture is stirred at 80° C. for 16 hours before water (2 mL) is added at 0° C. The mixture is diluted with water (200 mL) and extracted with DCM (200 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered and concentrated. The residue is then dissolved in DCM (200 mL) and DCA (5.48 g, 42.5 mmol, 12.0 mL) is added. After stirring at 25° C. for 3 hours, saturated sodium bicarbonate aqueous solution is added at 0° C. The mixture is then extracted with DCM (150 mL×3). The combined organic solvent are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/5 to 1/2) to give 60 as a yellow oil (12.1 g, 90% yield).
-

- To a solution of 60 (24.0 g, 78 mmol) in DCM (500 mL) is added benzoyl chloride (16.4 g, 116.7 mmol, 13.6 mL) and TEA (23.6 g, 233.5 mmol, 32.4 mL). After stirring at 25° C. for 16 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/15 to 1/10) to give 61 as a light yellow oil (30.0 g, 93% yield). (MS: [M+Na]+ 435.1)
-

- A mixture of 61 (29.0 g, 70.3 mmol) and water (3.0 mL) in HOAc (220 mL) is stirred at 70° C. for 16 hours before saturated sodium bicarbonate aqueous solution is added. The mixture is then extracted with DCM (400 mL×3). The combined organic layers are concentrated and the residue is dissolved in Py (30 mL) followed by addition of Ac2O (28.5 g, 280 mmol, 26 mL). After stirring at 20° C. for 16 hours, saturated sodium bicarbonate aqueous solution is added and the mixture is then extracted with DCM (500 mL×3). The combined organic layers are concentrated and purified by silica gel column chromatography (EA/PE=1/10 to 1/5) to give 62 as a white solid (31.1 g, 97% yield). (MS: [M+Na]+ 479.1)
-

- To a suspension of O6-diphenylcarbamoyl-N2-isobutyrylguanine (5.47 g, 13.1 mmol) in MeCN (150 mL) is added BSA (11.6 g, 57.0 mmol, 14.1 mL) at 20° C. After stirring at 63° C. for 30 minutes, the volatiles are removed and the residue is dissolved in MeCN (200 mL) before 62 (5.00 g, 11.0 mmol) in MeCN (50 mL) and TMSOTf (3.65 g, 16.4 mmol, 3.0 mL) are added at −15° C. After stirring at 63° C. for 50 minutes, the mixture is cooled to 0° C., poured into saturated sodium bicarbonate aqueous solution and extracted with EA (150 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, and concentrated, and purified by silica gel flash chromatography (EA/PE=1/3 to 1/1) to give 63 as a white solid. (MS: [M+H]+ 813.1)
-

- A solution of 63 (16.2 g, 19.9 mmol) in 90% TFA aqueous solution (60 mL) is stirred at 20° C. for 30 minutes before poured into saturated sodium bicarbonate aqueous solution at 0° C. and extracted with EA (100 mL×4). The combined organic layers are dried over anhydrous sodium sulfate, concentrated, and purified by silica gel flash chromatography (EA/PE=1/1 to 1/0) to give 64 as a white solid (11.4 g, 93% yield). (MS: [M+H]+ 618.1)
-

- To a solution of 64 (15.0 g, 24.3 mmol) in EtOH (500 mL) is added Pd/C (10% w/w, 2.0 g) and concentrated hydrochloric acid (10 drops). After stirring at 50° C. under an atmosphere of hydrogen (45 psi) for 15 hours, the mixture is filtered and solid is washed with EtOH (100 mL×3). The filtrate is concentrated and one-third of the residue is dissolved in DMF (60 mL) followed by addition of Imid (1.57 g, 23.0 mmol), DMAP (46.9 mg, 0.38 mmol) and triisopropylsilyl chloride (2.22 g, 11.5 mmol, 2.5 mL). After stirring at 20° C. for 16 hours, saturated sodium bicarbonate aqueous solution (20 mL) and water (100 mL) are added. The mixture is then extracted with EA (100 mL×2). The combined organic layers are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/5 to 2/1) to give 65 as a white solid (4.52 g, 86% yield). (MS: [M+H]+ 684.4)
-

- To a solution of 65 (3.0 g, 4.4 mmol) in EtOH (30 mL) is added sodium hydroxide aqueous solution (2 M, 31 mL) at 0° C. After stirring at 0° C. for 30 minute, the mixture is neutralized by addition of hydrochloric acid solution (1 N) and HOAc at 0° C. Toluene (30 mL) is then added and mixture is concentrated to give crude BC4 as a white solid (3.0 g). (MS: [M+H]+ 538.2)
-
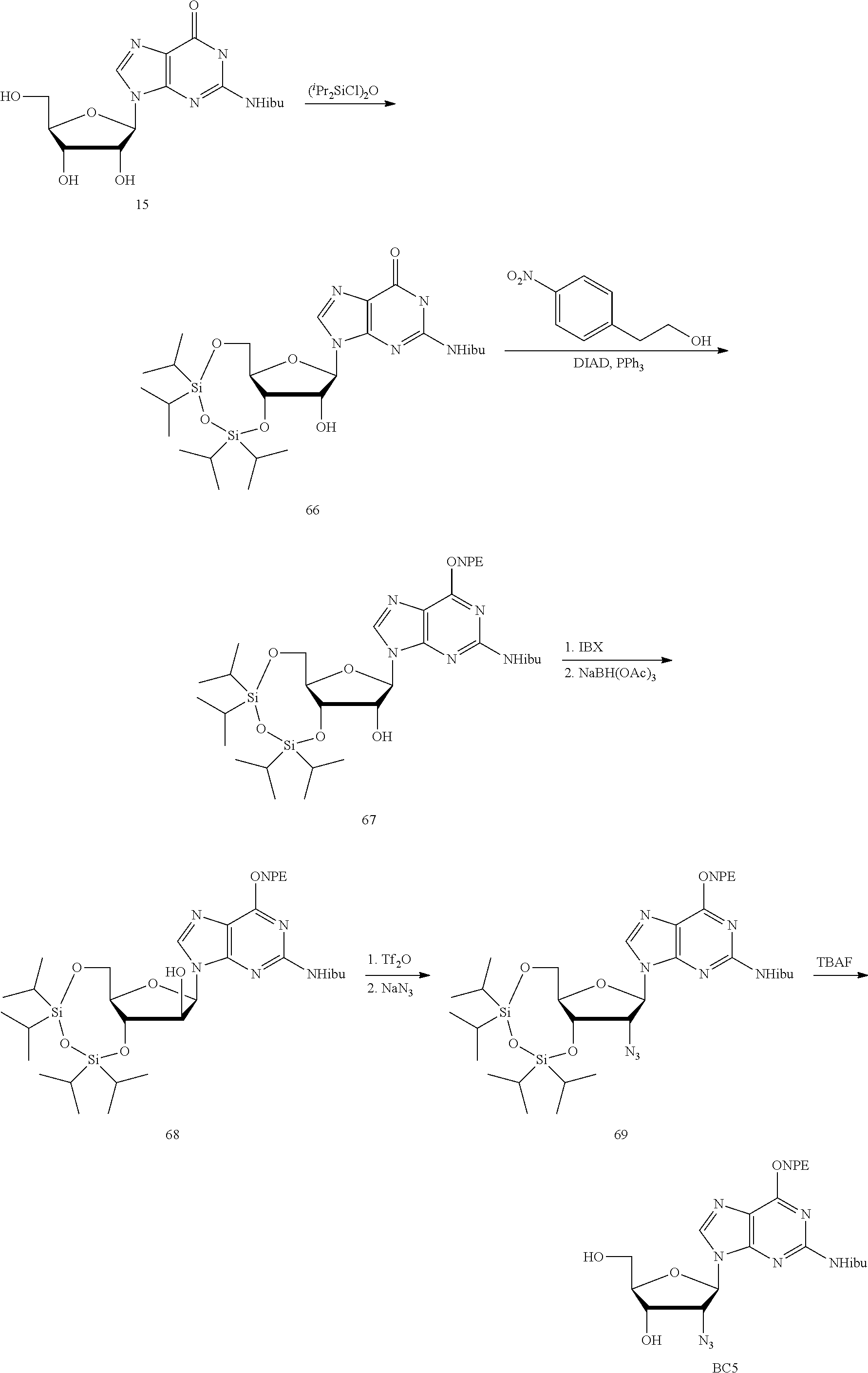
-

- To a solution of 15 (2.0 g, 5.66 mmol) in Py (56 mL) at 0° C. is added 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (1.79 g, 5.66 mmol, 1.8 mL) slowly. After stirring at 0° C. for 30 minutes and 25° C. for 12 hours, the solution is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/20) to give 66 (1.6 g, 47% yield). (MS: [M+H]+ 596.3)
-

- To a solution of 66 (8.0 g, 13.5 mmol) and 2-(4-nitrophenyl)ethanol (3.37 g, 20.2 mmol) in THF (100 mL) is added DIAD (6.81 g, 33.7 mmol, 6.6 mL) and PPh3 (8.83 g, 33.7 mmol) at 25° C. slowly. After stirring at 25° C. for 12 hours, water (5 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/20) to give 67 as a pale yellow solid (4.5 g, 44% yield). (MS: [M+H]+ 745.3)
-

- To a solution of 67 (4.20 g, 5.64 mmol) in MeCN (40 mL) at 25° C. is added IBX (3.16 g, 11.3 mmol). After stirring at 80° C. for 12 hours, the mixture is filtered and concentrated, and dissolved in THF (50 mL). Sodium triacetoxyborohydride (5.7 g, 27.0 mmol) is then added at 0° C. slowly. After stirring at 25° C. for 6 hours, water (5 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/20) to give 68 as a pale yellow oil (1.0 g, 33% yield). (MS: [M+H]+ 745.3)
-

- To a solution of 68 (2.2 g, 2.95 mmol) and DMAP (1.44 g, 11.8 mmol) in DCM (140 mL) and Py (10 mL) is added trifluoromethanesulfonic anhydride (1.33 g, 4.72 mmol, 0.78 mL) at 0° C. slowly. After stirring at 0° C. for 1.5 hours, the mixture is concentrated. The residue is then dissolved in DMF (10 mL) and sodium azide (0.49 g, 7.53 mmol) is added. After stirring at 60° C. for 6 hours, the solution is concentrated and purified by preparative HPLC (MeOH/water with 0.1% HCOOH: 40-100%) to give 69 as a pale yellow solid (1.50 g, 79% yield). (MS: [M+H]+ 770.4)
-

- To a solution of 69 (2.50 g, 3.25 mmol) in THF (12 mL) is added TBAF (13.1 g, 50.1 mmol) and HOAc (1.50 g, 25.0 mmol, 1.43 mL) at 15° C. slowly. After stirring at 15° C. for 12 hours, the mixture is concentrated. The residue is then dissolved in DCM (20 mL), washed with water (5 mL×2), dried over anhydrous sodium sulfate, filtered, and purified by silica gel column chromatography (MeOH/DCM=1/20) to give BC5 as a pale yellow solid (900 mg, 53% yield). (MS: [M+H]+ 528.2)
- The following compounds are prepared essentially by the method for Intermediates BC3 and BC4 above.
-
TABLE 1 Intermediates BC6 and BC7 Reference of Starting Material Product Preparation 

BC3 

BC4 -


-

- To a solution of 33 (120 g, 449 mmol) in Py (1.0 L) is added TMSCl (390 g, 3.59 mol, 454 mL). After stirring at 0° C. for 2 hours, benzoyl chloride (316 g, 2.25 mol, 261 mL) is added dropwise and the mixture is stirred at 25° C. for 14 hours before cooled to 0° C. Water (240 mL) is then added and the mixture is stirred at 25° C. for 30 minutes before ammonium hydroxide (460 mL) is added at 0° C. After stirring for 2 hours, the mixture is concentrated to give 70 as a white solid (150 g, 90% yield).
-

- To a solution of 70 (150 g, 404 mmol) in Py (500 mL) is added DMTrCl (274 g, 808 mmol), TEA (81.8 g, 808 mmol, 112 mL) and DMAP (4.93 g, 40.4 mmol) at 0° C. After stirring at 25° C. for 16 hours, saturated sodium bicarbonate aqueous solution (1 L) is added and the mixture is extracted with EtOAc (600 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE 1/4 to ½ then MeOH/DCM 1/100 to 1/20) to give 71 as a white solid (65.0 g, 24% yield).
-

- To a solution of 71 (65.0 g, 96.5 mmol) in Py (500 mL) is added silver nitrate (32.8 g, 193 mmol) and TBSCl (29.1 g, 193 mmol) at 25° C. After stirring at 25° C. for 1 hour, saturated sodium bicarbonate aqueous solution (1 L) is added and the mixture is extracted with EtOAc (600 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/4 to 1/1) to give 72 as a white solid (20.0 g, 26% yield).
-

- To a solution of 72 (12.0 g, 15.2 mmol) in DIPEA (15 mL) and DCM (30 mL) is added DMAP (744 mg, 6.09 mmol) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (5.41 g, 22.9 mmol) at 25° C. After stirring for 2 hours, the mixture is purified directly by basic silica gel column chromatography (EA/PE=1/4 to 1/1) to give A2 as a white solid (13.0 g, 86% yield).
-

- To a solution of A2 (3.8 g, 3.9 mmol) in MeCN (20 mL) is added water (0.1 mL) and pyridinium trifluoroacetate (1.1 g, 5.8 mmol) at 25° C. and stirred for 5 minutes before tert-butylamine (14.0 g, 0.19 mmol) is added. After stirring for 15 minutes, the volatiles are removed and the residue is dissolved in DCM (20 mL). A solution of DCA (1.9 g, 14.6 mmol) in DCM (20 mL) is then added. After stirring for 30 minutes, TEA (3 mL) is added and the mixture is concentrated and purified by reverse-phase silica gel column chromatography (MeCN with 0.1% TEA/water=0% to 100%) to give AlTEA salt as a white solid (1.5 g, 71% yield). (MS: [M+H]+ 549.2)
-

- To a solution of A2 (494 mg, 0.5 mmol) and triphenylphosphine (197 mg, 0.75 mmol), and 2-(tert-butyldimethylsilyloxy)ethanol (132 mg, 0.75 mmol) in THF (5 mL) is added DIAD (0.15 mL, 0.75 mmol). After stirring at room temperature for 5 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/hexanes=1/9 to 1/4) to give AB1 as a white solid (230 mg, 40% yield).
-

-

- To a solution of crude BC1 (39 g) in Py (40 mL) is added DMTrCl (35.9 g, 106 mmol) at 0° C. After stirring at 25° C. for 16 hours, MeOH (50 mL) is added and the mixture is concentrated. The residue is then dissolved in DCM (600 mL), washed with saturated sodium bicarbonate aqueous solution and brine, concentrated, and purified by silica gel column chromatography (MeOH/DCM=1/100 to 1/50) to give 73 as a pale yellow solid (34.0 g, 48% yield over two steps). (MS: [M+H]+ 673.2)
-

- To a solution of 73 (1.0 g, 1.49 mmol) in Py (10 mL) is added silver nitrate (380 mg, 2.24 mmol, 0.38 mL) at 0° C. After stirring for 15 minutes, TBSCl (270 mg, 1.79 mmol) is added and the mixture is stirred at 25° C. for 2 hours before saturated sodium bicarbonate aqueous solution is added. The mixture is then extracted with EA (10 mL) and the organic layer is washed with brine, concentrated, and purified by silica gel column chromatography (EA/PE=1/5) to give 74 as a pale yellow solid (400 mg, 34% yield). (MS: [M+4]+ 787.3)
-

- To a solution of 74 (1.0 g, 1.27 mmol) in Py (10 mL) is added diphenyl phosphite (80%, 744 mg, 2.54 mmol, 0.61 mL). After stirring at 20° C. for 1 hour, EA (2 mL) and saturated sodium bicarbonate aqueous solution (2 mL) are added and the mixture is stirred for 1 hour. The layers are separated and the organic layer is concentrated. The residue is then dissolved in DCM (1.0 mL) and DCA (164 mg, 1.27 mmol, 0.1 mL) is added. After stirring at 25° C. for 30 minutes, TEA (1 mL) is added and the solution is concentrated and purified by reverse-phase silica gel column chromatography (MeCN with 0.1% TEA/water=0% to 100%) to give AC1 as a white solid (500 mg, 72% yield). (MS: [M+H]+ 549.2)
-

-

- To a solution of BC2 (348 mg, 1.49 mmol) in Py (15 mL) is added DMAP (18 mg, 0.15 mmol) and DMTrCl (0.66 g, 1.94 mmol). After stirring overnight, MeOH (3 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (EA/hexanes=1/4) to give 75 (662 mg, 83% yield). (MS: [M+H]+ 536.2)
-

- To a solution of 75 (0.2 g, 0.37 mmol) in DCM (4 mL) is added DIEPA (0.15 g, 1.2 mmol, 0.2 mL) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (0.14 g, 0.56 mmol, 0.13 mL). After stirring for 4 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/hexanes with 1% TEA=1/4) to give AC2 (232 mg, 85% yield). (MS: [M-NiPr2+H2O]+ 653.2)
-

-

- To a solution of 16 (11.0 g, 16.8 mmol) in DCM (80 mL) is added TBSCl (7.59 g, 50.3 mmol) and Imid (3.43 g, 50.3 mmol). After stirring at 25° C. for 16 hours, sodium bicarbonate aqueous solution (5%, 30 mL) is added and the mixture is extracted with DCM (60 mL×3). The combined organic layers are washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by basic silica gel column chromatography (EA/PE=1/5 to 1/1) to 76 as a white solid (2.1 g, 16% yield).
-

- To a solution of 76 (900 mg, 1.17 mmol) in THF (4.0 mL) and DIEPA (4.0 mL) is added DMAP (14.3 mg, 0.12 mmol) followed by 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (415 mg, 1.76 mmol) dropwise at 0° C. After stirring at 20-25° C. for 2 hours, sodium bicarbonate aqueous solution (5%, 15 mL) is added at 0° C. The mixture is then diluted with water (15 mL) and extracted with EA (15 mL×3). The combined organic layers are washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (acetone/PE=1/10 to 1/3) to give G2 as a white solid (600 mg, 53% yield).
-

- To a solution of G1 (7.0 g, 7.22 mmol) in MeCN (30 mL) is added water (0.11 mL) and pyridinium trifluoroacetate (4.18 g, 21.7 mmol) at 25° C. After stirring at 25° C. for 15 minutes, tert-butylamine (37 mL) is added and the mixture is stirred at 25° C. for 45 minutes before concentrated. The residue is then dissolved in DCM (30 mL) and a solution of DCA in DCM (6% v/v, 30 mL) is added dropwise. After stirring at 20-25° C. for 30 minutes, DCM (30 mL) and TEA (4 mL) are added. The mixture is then concentrated, dissolved in a mixture of MeCN (5 mL) and water (5 mL), and purified by C18 reverse-phase medium pressure liquid chromatography (MeCN with 0.1% TEA/water=0% to 60%) to give G1.TEA salt as a yellow solid (2.30 g, 56% yield). (MS: [M+H]+ 532.3)
-

-

- To a solution of two batches of the crude BB1 (8.0 g) obtained above in Py (50 mL) is added DMTrCl (9.2 g, 27.2 mmol). After stirring at 20-30° C. for 1 hour, MeOH (10 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/5 to MeOH/DCM=1/20) to give 77 as a yellow solid (11.0 g, 30% yield over two steps). (MS: [M+Na]+ 678.2)
-

- To a solution of 77 (9.0 g, 14.0 mmol) in Py (50 mL) is added TBSCl (2.48 g, 16.5 mmol) and silver nitrate (5.83 g, 34.3 mmol). After stirring at 25-30° C. for 30 minutes, saturated sodium bicarbonate aqueous solution is added. The mixture is then extracted with DCM (200 mL×2), and the combined organic layers are washed with water (50 mL), brine (50 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE=1/10 to 1/5 to 1/3) to give 78 as a white foam (1.50 g, 14% yield). (MS: [M+Na]+ 792.2)
-

- To a solution of 78 (2.50 g, 3.3 mmol) in DIEPA (5 mL) and DCM (5 mL) is added DMAP (200 mg, 1.62 mmol) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (1.0 g, 4.22 mmol). After stirring at 20-25° C. for 2 hours, the mixture is concentrated and purified by silica gel column chromatography (EA/PE=1/10 to 1/4) to give GB1 as a white foam (2.30 g, 73% yield).
-

- To a solution of GB1 (2.30 g, 2.4 mmol) in MeCN (2.0 mL) is added water (0.11 mL, 6.1 mmol) and pyridinium trifluoroacetate (687 mg, 3.56 mmol) at 25° C. After stirring at 25-30° C. for 30 minutes, the mixture is concentrated and the residue is dissolved in MeCN (20 mL) before tert-butylamine (10.5 g, 144 mmol, 15.0 mL) is added. After stirring at 25-30° C. for 30 minutes, the mixture is concentrated and DCM (20 mL) followed by addition of a solution of DCA in DCM (6% v/v, 18.2 mL). The mixture is stirred at 25-30° C. for 30 minutes before neutralized by TEA to ˜pH 7, concentrated, and purified by C18 reverse-phase silica gel column chromatography (MeCN with 0.1% TEA/water=0% to 40%) to give GB2 as a white solid (800 mg, 63% yield). (MS: [M+H]+ 532.0)
-

-

- To a solution of BC4 (1.97 g, 2.4 mmol) in Py (20 mL) is added DMTrCl (984 mg, 2.90 mmol) at 25° C. After stirring for 3 hours, MeOH (30 mL) is added and the mixture is concentrated, and purified by basic silica gel flash chromatography (EA/PE=1/5 to 4/1) to give 79 as a light yellow powder (1.65 g, 82% yield). (MS: [M+H]+ 840.2)
-

- To a solution of 79 (2.0 g, 2.38 mmol) in Py (15 mL) is added diphenyl phosphite (1.7 g, 7.1 mmol, 1.4 mL). After stirring at 20° C. for 30 minutes, saturated sodium bicarbonate aqueous solution (30 mL) is added and the mixture is stirred for 1 hour. The mixture is then extracted with EA (30 mL×3). The combined organic layers are washed with saturated sodium bicarbonate aqueous solution (20 mL) and brine (20 mL), dried over anhydrous sodium sulfate, and concentrated. The residue is then dissolved in DCM (40 mL) followed by addition of water (0.4 mL) and DCA (6% v/v in DCM, 40 mL). After stirring at 20° C. for 15 minutes, the mixture is neutralized to ˜pH 7 by TEA, concentrated, and purified by reverse-phase silica gel column chromatography (MeCN/water=25% to 90%) to give GC1 as a white solid (1.3 g, 90% yield). (MS: [M+H]+ 602.1)
-

-

- To a solution of BC5 (900 mg, 1.71 mmol) in Py (10 mL) is added DMTrCl (809 mg, 2.39 mmol) at 15° C. After stirring at 15° C. for 12 hours, MeOH (0.5 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/50) to give 80 as a yellow oil (1.0 g, 71% yield). (MS: [M+H]+ 528.2)
-
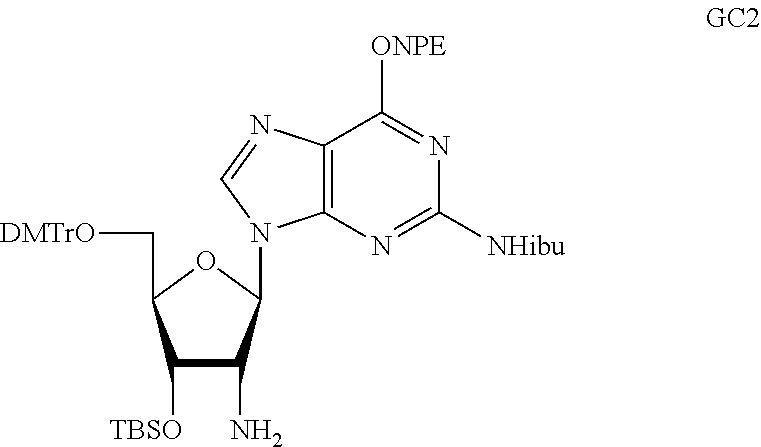
- To a solution of 80 (2.05 g, 2.47 mmol) in DMF (5.0 mL) is added Imid (673 mg, 9.88 mmol) and TBSCl (745 mg, 4.94 mmol, 0.61 mL) at 15° C. After stirring at 15° C. for 12 hours, the mixture is concentrated and the residue is triturated with water (10 mL). The solid is then collected and washed with water (10 mL×2), PE (10 mL×2), dried, and dissolved in THF (18 mL) before PPh3 (1.11 g, 4.24 mmol) is added at 15° C. After stirring at 15° C. for 2.5 hours, water (0.16 mL) is added and the mixture is stirred at 50° C. for 12 hours. The solution is then concentrated and purified by reverse-phase preparative-HPLC (MeOH with 0.1% TEA/water=20% to 80%), to give GC2 as a white solid (900 mg, 58% yield). (MS: [M+H]+ 918.1)
- The following compounds are prepared essentially by the method for Intermediates A1, A2, AC1, AC2, G1, G2, GC1, and GC2 above.
-
TABLE 2 Intermediates A1 to A5, AA1, AA2, AB1, AC1 to AC6, G1 to G7, GA1, GB1 to GB3, and GC1 to GC5 Reference of Starting Material Product Preparation 

A1 33 
A2 

A1 B1 
A2 

A2 

A2 

A2 A2 
AB1 

AC1 

AC2 B1 
G1 33 
G1 

AC2 

AC2 

G1 37 
G2 

G1 

G1 

G2 DMTr-B3 
G2 

G2 

G2 

GB1 

GB2 

GB2 

GC1 

GC2 

G1 

A2 

GC1 -

- To a solution of crude G1 (obtained from 187 mg of G2.TEA salt, 0.2 mmol, containing PyDCA salt) in MeCN (0.5 mL) is added a solution of A2 (0.26 g, 0.26 mmol) in MeCN (0.2 mL). After stirring for 30 minutes, DDTT (46 mg, 0.22 mmol) is added and the mixture is stirred for 1 hour before concentrated. The residue is dissolved in DCM (4.8 mL) and water (0.036 mL) and DCA (6% in DCM, 4.8 mL) are added. After stirring for 10 minutes, Py (1 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/10 to 1/5) to give C1Py salt as a white solid (86 mg, 35% yield). (MS: [M+H]+ 1148.0)
-

- To a mixture of A1 (1.0 g, 1.82 mmol, co-evaporated MeCN 20 mL×3) and G2 (2.3 g, 2.37 mmol, co-evaporated with MeCN 20 mL×3) is added tetrazole (0.45 M in MeCN, 10 mL) at 25° C. and stirred for 1 hour before elemental sulfur (1.75 g, 6.84 mmol) is added. After stirring for 1 hour, MeCN (20 mL) is added and the mixture is filtered and concentrated. The residue is dissolved in DCM (100 mL) and DCA (1.96 g, 15.2 mmol, 1.25 mL) is added. After stirring at 25° C. for 2 hours, saturated sodium bicarbonate aqueous solution (100 mL) is added. The layers are separated and the aqueous layer is extracted with EA (100 mL×3). The combined organic layers are dried over anhydrous sodium sulfate, filtered, concentrated, and purified by reverse-phase silica gel column chromatography (MeCN with 0.1% TEA/water=0% to 100%) to give C2.TEA salt as a white solid (100 mg, 5% yield). (MS: {[M+2H]2+}/2 574.6)
-

- To a solution of A2 (510 mg, 0.52 mmol, co-evaporated with MeCN 5 mL×3) in MeCN (1 mL) treated with 3 Å MS (100 mg) for 30 minutes is added a mixture of G1 (250 mg, 0.47 mmol, co-evaporated with MeCN 5 mL×3) and pyridinium trifluoroacetate (109 mg, 0.56 mmol, co-evaporated with MeCN 5 mL×3) in MeCN (1.5 mL) treated with 3 Å MS (50 mg) for 30 minutes. After stirring for 4 hours, TBHP (5.5 M in decane, 0.26 mL) is added and the mixture is stirred for 30 minutes before sodium bisulfite aqueous solution (33%, 0.24 mL) is added at 0° C. The mixture is then stirred at room temperature for 10 minutes before filtered and concentrated. The residue is dissolved in DCM (6.2 mL) followed by addition of water (0.09 mL) and DCA (0.37 mL) in DCM (6.2 mL). After stirring at room temperature for 10 minutes, Py (0.73 mL, 9.05 mmol) and DCM (35 mL) are added. The mixture is washed with water (10 mL×2) and the combined aqueous layers are extracted by dichloromethane (10 mL×2). The combined organic layers are dried over anhydrous sodium sulfate, concentrated, and purified by silica gel column chromatography (MeOH/CH2C12/Py=10:89.5:0.5 to 25:74.5:0.5) to give C3 as a white solid (250 mg, 47%). (MS: [M+H]+ 1132.2)
-

- To a solution of G1 (500 mg, 0.94 mmol, co-evaporated with MeCN 10 mL×3) and pyridinium trifluoroacetate (218 mg, 1.13 mmol, co-evaporation with MeCN 10 mL×3) in MeCN (3 mL) treated with 3 Å MS (100 mg) for 30 minutes is added a solution of A2 (976 mg, 0.99 mmol, co-evaporated with MeCN 10 mL×3) in MeCN (2 mL) treated with 3 Å MS (200 mg) for 30 minutes. After stirring at room temperature for 2.5 hours, the mixture is concentrated and co-evaporated with MeCN (10 mL×2). The residue is then dissolved in DCM (20 mL) followed by addition of borane dimethyl sulfide complex (2 M in THF, 0.94 mL, 1.88 mmol) dropwise. After stirring at room temperature for 1 hour, MeOH (0.17 mL) is added at 0° C. and stirred for 20 minutes before concentrated to give crude CA1.
-

-

- To a solution of GC2 (802 mg, 0.87 mmol) in DMF (5 mL) is added 3,4-dimethoxy-3-cyclobutene-1,2-dione (186 mg, 1.31 mmol) at 15° C. slowly. After stirring at 15° C. for 2 hours, the mixture is concentrated and purified by reverse-phase preparative HPLC (MeCN with 0.1% TEA/water=0% to 100%) to give a mixture of the desired product and an unidentified byproduct (0.7 g, ca. 84% purity). (MS: [M+4]+ 1028.4)
-

- To a solution of 81 (ca. 84% pure, 0.6 g) obtained above and AC1 (0.64 g, 1.17 mmol) in DMF (5.0 mL) is added TEA (177 mg, 1.75 mmol, 0.24 mL) at 15° C. After stirring at 15° C. for 12 hours, the mixture is concentrated and the residue is dissolved in DCM (5.0 mL) before DCA (470 mg, 3.65 mmol, 0.3 mL) is added. The mixture is then stirred at 15° C. for 15 minutes before concentrated. The residue is purified by reverse-phase preparative HPLC (MeCN with 0.1% TEA/water=0% to 100%) to give CC1 as a while solid (0.54 g, 40% yield over two steps). (MS: [M+H]+ 1242.3)
-

-

- To a solution of 4-nitrophenyl chlorosulfate (0.31 g, 1.31 mmol) in DCM (1.0 mL) is added a solution of GC2 (0.40 g, 0.44 mmol), 4-nitrophenol (0.61 g, 4.4 mmol) and TEA (0.73 mL, 5.23 mmol) in DCM (5 mL) at −78° C. After stirring for 30 minutes, the mixture is warmed to room temperature, diluted with DCM (20 mL), and washed with water (20 mL×3). The combined aqueous layers are extracted with DCM (20 mL×2) and the combined organic layers are dried over anhydrous magnesium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/PE with 1% TEA=1/5 to 1/2) to give 82 (0.30 g, 59% yield) as a white solid. (MS: [M+H]+ 1118.9)
-

- To a solution of 82 (0.2 g, 0.18 mmol) in THF (1.0 mL), 4 Å MS (0.05 g) and TEA (0.12 mL, 0.89 mmol) is added AC1 (0.12 g, 0.21 mmol). After stirring for 12 hours, the mixture is diluted with THF (2 mL), filtered, and concentrated. The residue is then dissolved in DCM (5.0 mL) before water (0.1 mL) and a solution of DCA (0.46 mL) in DCM (5.0 mL) are added. After stirring for 15 minutes, the mixture is neutralized with TEA to ˜pH 7 before concentrated and purified by reversed-phase C18 silica gel column chromatography (MeCN with 0.5% TEA/water=0% to 40%) to give CC2 (0.11 g, 44% yield) as a white solid. (MS: [M+H]+ 1226.0)
-


-

- To a solution of A4 (1.0 g, 1.01 mmol) and G1 (1.07 g, 2.02 mmol) in MeCN (10 mL) is added Py.DCA (420 mg, 2.02 mmol). After stirring at 20° C. for 2 hours, TBHP (70% in water, 0.65 mL, 5.05 mmol) is added and the mixture is stirred for 1 hour before sodium bicarbonate aqueous solution (50 mL) is added. The mixture is extracted with ethyl acetate (100 mL) and the organic layer is washed with brine (30 mL), dried with anhydrous sodium sulfate, filtered, and concentrated. The residue is then dissolved in a mixture of DCM (20 mL), TFA (1.0 mL) and triethylsilane (5.0 mL). After stirring for 2 hours, the mixture is neutralized with solid sodium bicarbonate to ˜pH 7. The mixture is then filtered and the solid is washed with EA (50 mL×3). The filtrate is concentrate and purified by preparative HPLC (MeCN with 0.1% TEA/water=0% to 30%) to give C4.TEA as a white solid (620 mg, 49% yield). (MS: [M+4]+ 1131.1)
-

- To a solution of C4 (583 mg, 0.515 mmol) in Py (10 mL) is added DMOCP (583 mg, 3.16 mmol) at 20° C. After stirring for 2 hours, iodine (654 mg, 2.58 mmol) is added and the mixture is stirred for 1 hour before saturated sodium sulfate aqueous solution (30 mL) and saturated sodium bicarbonate aqueous solution (30 mL) is adde. The mixture is then extracted with EA (100 mL), washed with brine (60 mL), dried over anhydrous sodium sulfate, filtered, concentrated, and purified by reversed-phase silica gel column chromatography (MeCN with 0.1% TEA/water=20% to 40%) to give 83.TEA as a white solid (172.0 mg, 31% yield). (MS: [M+H]+ 1076.1)
-

- A solution of 83 (100 mg, 0.093 mmol) in MeOH (1.0 mL) and ammonium hydroxide (1.0 mL) is stirred at 50° C. for 12 hours. The mixture is then concentrated and purified by reverse-phase silica gel column chromatography (MeCN with 0.1% TEA/water=20% to 40%) to give 84 as a yellow solid (27.0 mg, 32% yield). (MS: [M+H]+ 902.5)
-

- A solution of 84 (27 mg, 0.030 mmol) in TEA3.HF (10 mL) is stirred at 50° C. for 3 hours. The mixture is then neutralized with cold triethylammonium bicarbonate to ˜pH 7, concentrated, and purified by a C18 reverse-phase silica gel column chromatography (MeCN with 0.1% TEA/water=0% to 20%) to give E5.TEA as a white solid (5.2 mg, 26% yield). (MS: [M+H]+ 673.7)
-

-

- To a mixture of A4 (500 mg, 0.50 mmol, co-evaporated with MeCN 5 mL×1 and toluene 10 mL×2) and G1 (322 mg, 0.61 mmol, co-evaporated with MeCN 5 mL×1 and toluene 10 mL×2) is added tetrazole (0.45 M in MeCN, 4.0 mL). After stirring at 25° C. for 2 hours, DDTT (240 mg, 1.2 mmol) is added and the mixture is stirred for 16 hour before filtered and concentrated. The residue is then dissolved in DCM (10 mL) followed by addition of water (0.1 mL) and DCA (0.21 mL). After stirring for 10 minutes, TEA (1 mL) is added and the mixture is concentrate and purified by reverse-phase silica gel column chromatography (MeCN with 0.1% TEA/water=0% to 100%) to give C5.TEA as a white solid (250 mg, 35% yield). (MS: {[M+2H]2+)/2 574.6)
-
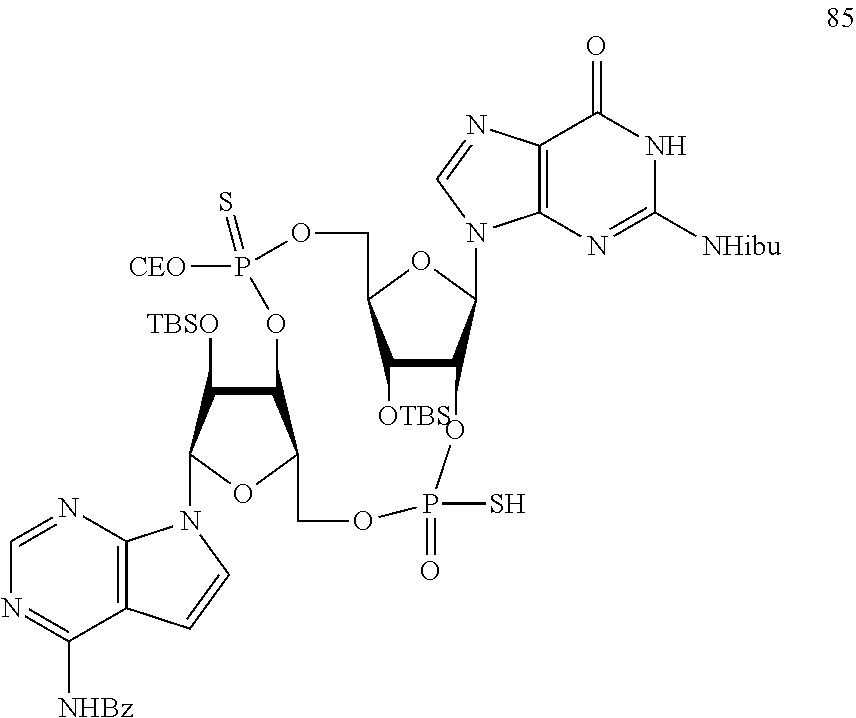
- To a solution of C5 (600 mg, 0.423 mmol, co-evaporated with Py 3 mL×2) in Py (5.0 mL) is added DMOCP (313 mg, 1.69 mmol) at 25° C. After stirring for 2 hours, 3H-1,2-benzodithiol-3-one (142 mg, 0.85 mmol) is added and the mixture is stirred for 2 hours before sodium bicarbonate aqueous solution (5%, 10 mL) is added. The mixture is then extracted with EA (10 mL×3). The combined organic layers are dried with anhydrous sodium sulfate, filtered, concentrated, and purified by reverse-phase HPLC (MeCN with 0.1% TEA/water=0 to 100%) to give four diastereomers of 85.TEA as white solids. Isomer 1 (28 mg) (MS: [M+H]+ 1160.9); Isomer 2 (25 mg) (MS: [M+H]+ 1160.9); Isomer 3 (50 mg) (MS: [M+H]+ 1160.9); Isomer 4 (52 mg) (MS: [M+1]+ 1160.9)
-

- Each of the isomers of 85.TEA (25 mg, 0.022 mmol) in ammonium hydroxide (5.6 mL) and MeOH (4.0 mL) is stirred at 50° C. for 16 hours. The mixture is then purged with nitrogen at room temperature for 5 minutes before concentrated. The residue is dissolved in TEA (0.5 mL) and Py (0.2 mL), and TEA.3HF (0.7 mL) is added. After stirring at 50° C. for 24 hours, triethylammonium bicarbonate aqueous solution (1M, 5 mL) is added and the mixture is purified by a reverse-phase silica gel column chromatorgraphy (MeCN with 0.1% TEA/water=0% to 30%) to give E15-E18 as white solids.
-

-

- To a solution of C3 (16 mg, 0.014 mmol, co-evaporation with Py 1 mL×3) in Py (0.5 mL) is added DMOCP (10.4 mg, 0.056 mmol). After stirring for 15 minutes, NIS (4.1 mg, 0.0183 mmol) and morpholine (0.012 mL, 0.141 mmol) are added and the mixture is stirred for 1 hour before sodium bisulfite aqueous solution (0.14%, 1 mL) and sodium bicarbonate (80 mg) is added. The mixture is then extracted with DCM (5 mL×3), dried over anhydrous sodium sulfate, and concentrated. The residue is then stirred in MeCN (0.5 mL) and t-butylamine (0.5 mL) at room temperature for 15 minutes before concentrated. The resulting residue is then co-evaporated with MeCN (1 mL×3) and purified by HPLC to give 86 as a white solid (2.4 mg, 15%). (MS: [M+H]+ 1146.2)
-

- To 86 (2.4 mg, 0.0021 mmol) is added methylamine (33% in EtOH, 0.3 mL). After stirring at room temperature for 16 hours, the mixture is concentration and the residue is stirred in a mixture of TEA and TEA.3HF in THF (0.036 mL/0.018 mL/0.3 mL) at 35° C. for 18 hours. MeCN (1.0 mL) is then added and the solid is collected by centrifugation, washed with MeCN (1 mL×2) to give E24 as a white solid (0.6 mg, 38% yield). (MS: [M+H]+ 744.0)
-

-

- To a solution of C3 (100 mg, 0.088 mmol, co-evaporated with Py 4 mL×3) in Py (3 mL) is added DMOCP (57 mg, 0.337 mmol). After stirring for 15 minutes, BSTFA (0.10 mL, 0.371 mmol) is added dropwise and the mixture is stirred for 20 minutes before borane N,N-diisopropylethylamine complex (0.092 mL, 0.530 mmol) is added. The mixture is then stirred for 3 hours before concentrated and purified by silica gel column chromatography (MeOH/DCM=1/19 to 1/9) to give semi-pure CE-protected 87 as a yellow solid. The semi-pure CE-protected 87 obtained above is stirred in a mixture of MeCN (1 mL) and t-butylamine (0.5 mL) for 10 minutes before concentrated. The residue is then co-evaporated with MeCN (4 mL×3) and purified by reverse-phase HPLC (MeCN with 0.1% TEA/water=40% to 90%) to give 87 as a white solid (11 mg, 12% over two steps). (MS: [M]− 1073.2)
-

- To 87 (5.7 mg, 0.0053 mmol) is added methylamine (33% in EtOH, 1 mL). After stirring at room temperature for 18 hours, the mixture is concentration and the residue is stirred in a mixture of TEA (0.08 mL) and TEA.3HF (0.04 mL) in THF (0.5 mL) at 35° C. for 18 hours. MeCN (1.2 mL) is then added and the solid is collected by centrifugation, purified by reverse-phase HPLC (MeCN with 0.1% TFA/water=0% to 20%) to give E25 as a white solid (2.5 mg, 61% yield) (MS: [M]− 671.2)
-

-

- To a solution of GB3 (160 mg, 0.25 mmol, co-evaporated with MeCN 1 mL×3) and pyridinium trifluoroacetate (35 mg, 0.39 mmol, co-evaporation with MeCN 1 mL×3) in MeCN (1 mL) treated with 3 Å MS (500 mg) for 30 minutes is added a solution of A4 (355 mg, 0.36 mmol, co-evaporated with MeCN 1 mL×3) in MeCN (1 mL) treated with 3 Å MS (700 mg) for 30 minutes. After stirring at room temperature for 2 hours, TBHP (5.5 M in decane, 0.164 mL, 0.9 mmol) is added and the mixture is stirred for 30 minutes before sodium bisulfite aqueous solution (33%, 0.15 mL) is added at 0° C. The mixture is then concentrated and the residue is dissolved in DCM (4.8 mL) followed by addition of water (0.054 mL) and dichloroacetic acid (6% in methylene chloride, 4.8 mL). After stirring for 10 min, the Py (1.5 mL) is added and the mixture is concentrated and purified by silica gel column chromatography (MeOH/DCM=1/10 to 1/4 with 1% Py) to give CB1.Py as a white solid (213 mg, 66% yield).
-

- To a solution of CB1 (60 mg, 53 μmol) in Py (1 mL) is added DMOCP (30 mg, 162.5 μmol). After stirring for 10 minutes, water (0.027 mL) and 3H-1,2-benzodithiol-3-one (13 mg, 0.077 mmol) are added. The mixture is stirred for 5 minutes before pouring into a solution of sodium bicarbonate (210 mg) in water (7.5 mL). After stirring for 5 minutes, the mixture is extracted by EA/diethyl ether (1:1, 10 mL×3). The combined organic layers are concentrated to give a yellow solid (100 mg). To a solution of the yellow solid obtained above in MeCN (0.5 mL) is added tert-butylamine (0.5 mL). After stirring for 10 minutes, the mixture is concentrated and purified by HPLC (MeCN/water with 0.1% TFA: 50% to 100%) to give two diastereomers of 88. Isomer 1 (7 mg) ([M+H]+ 977.0); Isomer 2 (16 mg) (MS: [M+H]+ 977.0)
-

- To the Isomer 1 of 88 (7 mg) is added methylamine (33% in ethanol, 1 mL). After stirring at room temperature for 12 hours, the mixture is concentrated and the residue is dissolved in TFA aqueous solution (3% v/v, 1 mL). After stirring for 2 hours, the mixture is concentrated and purified by HPLC (MeCN/water with 0.1% TFA, 0% to 45%) to give EB1 as a white solid (2.5 mg, 57% yield). (MS: [M+H]+ 689.0)
- To the Isomer 2 of 63 (16 mg) is added methylamine (33% in ethanol, 2 mL) at 0° C. After stirring at room temperature for 12 hours, the mixture is concentrated and the residue is co-evaporated with a mixture of Py/TEA (5 mL/2 mL×3) before dissolved in Py (0.04 mL). TEA (0.25 mL) and TEA3.HF (0.15 mL are then added. After stirring at 55° C. for 3 hours, acetone (2 mL) is added. The solid is collected (10 mg) by filtration and purified by HPLC (MeCN/water with 0.1% TFA, 0% to 30%) to give EB2 as a white solid (5 mg, 45% yield). (MS: [M+H]+ 689.0, [M−H]− 687.0)
-

- To a solution of E1.TEA salt (10 mg, 0.0114 mmol) in water (0.3 mL) is added 2-chloroacetaldehyde (0.015 mL, 0.118 mmol) and sodium hydroxide aqueous solution (1 M, 0.012 mL, 0.012 mmol). After stirring at 37° C. for 18 hours, the mixture is concentrated and purified by reverse-phase HPLC (MeCN/water with 0.1% TFA=0% to 30%) to give EC26 as a white solid. (MS: [M]− 697.1)
- The following compounds are prepared essentially by the methods above.
-
TABLE 3 Examples E1 to E25, EA1 to EA11, EB1 to EB7, and EC1 to EC24 Reference of Example Intermediate 1 Intermediate 2 Structure Preparation E1 


Example A E2 
A2 
Example A E3 
A2 
Example A E4 


Example A E5 G1 

Example A E6 

Example A E7 C1 
Example A E8 

Example A E9 C2 
Example A E10 
A2 
Example A E11 GC6 A2 
Example A E12 
A1 
Example B E13 G2 A1 
Example B E14 G2 A1 
Example B E15 G1 A4 
Example B E16 G1 A4 
Example B E17 G1 A4 
Example B E18 G1 A4 
Example B E19 G1 

Example B E20 


Example B E21 G7 A3 
Example B E22 G7 A3 
Example B E23 G7 A3 
Example B E24 

Example C E25 C3 
Example D EA1 G7 A3 
Example A EA2 
A1 
Example A EA3 G1 

Example A EA4 G1 

Example B EA5 G1 AA2 
Example B EA6 G7 A1 
Example B EA7 G7 A1 
Example B EA8 G7 A1 
Example B EA9 C3 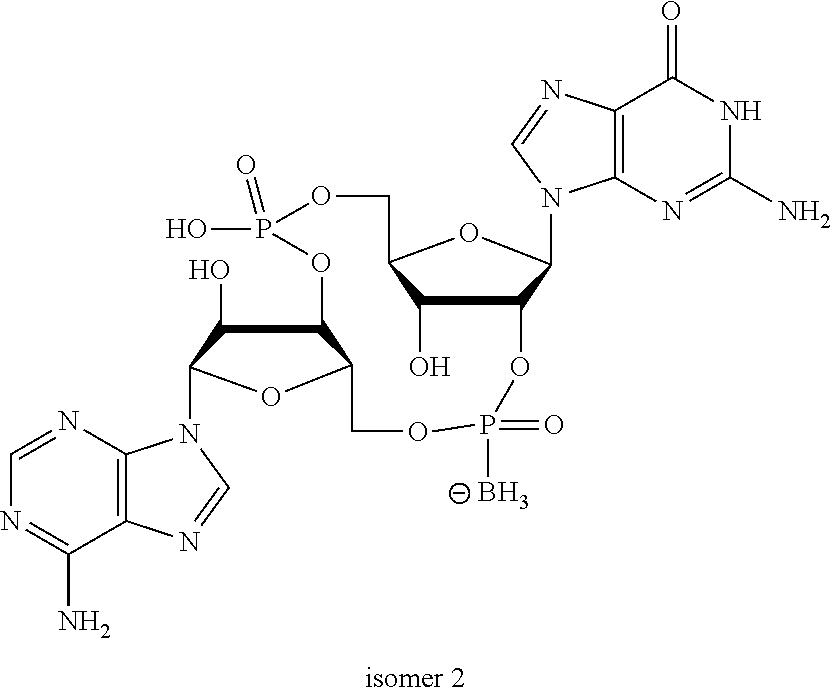
Example D EA10 

Example A EA11 CA1 
Example D EB1 
A4 
Example E EB GB3 A4 
Example E EB3 GB3 A2 
Example E EB4 GB3 A2 
Example E EB5 GB3 

Example E EB6 
A2 
Example E EB7 GB2 A2 
Example E EC1 
A4 
Example E EC2 GC3 A4 
Example E EC3 GC3 A4 
Example A EC4 GB3 

Example E EC5 GB3 GC4 
Example E EC6 
A4 
Example E EC7 AC3 A4 
Example E EC8 AC3 GC4 
Example E EC9 AC3 GC4 
Example E EC10 GC3 

Example A EC11 G1 
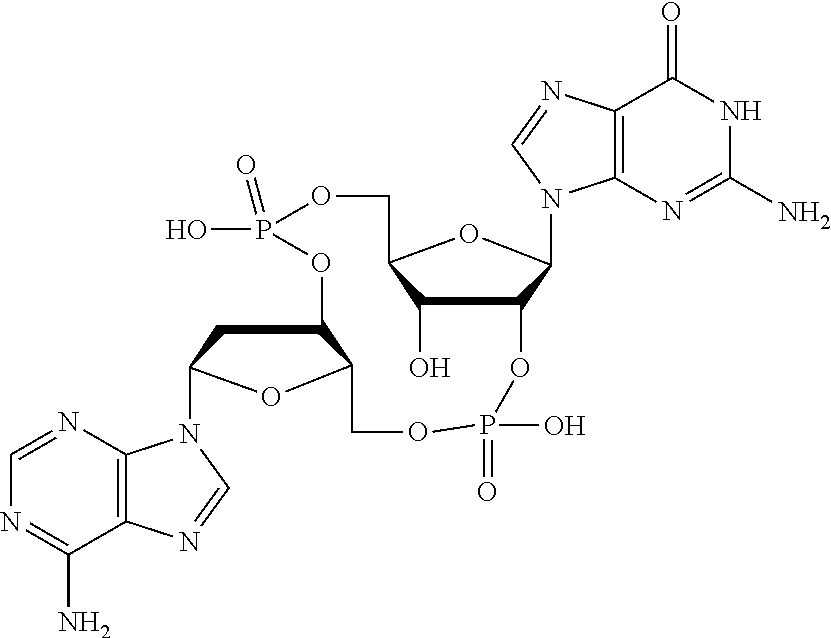
Example A EC12 
A2 
Example A EC13 GA1 

Example A EC14 G7 AC4 
Example E EC15 G7 AC4 
Example E EC16 GA1 

Example A EC17 G1 AC2 
Example E EC18 G1 AC2 
Example E EC19 G1 

Example A EC20 G1 

Example E EC21 GC3 AC6 
Example E EC22 

Example A EC23 

Example A EC24 
A2 
Example A EC25 

Example F EC26 

Example F - Selected physical data of the example compounds are summarized below.
-
TABLE 4 Physical data of cyclic dinucleotide and analogs characteristic 31P NMR 1H NMR data data δ MS data Example Structure δ (ppm)* (ppm)* m/z E1 
8.58 (s, 1H) 8.56 (s, 1H) 8.16 (s, 1H) 6.45 (s, 1H) 6.22 (d, J = 8.5 Hz, 1H) 5.89 (m, 1H) 5.31 (m, 1H) 50° C. 0.1 −0.9 50° C. [M + H]+ 675.1 E2 
[M + H]+ 659.0 E3 
8.31 (s, 1H) 8.29 (s, 1H) 7.88 (s, 1H) 6.19 (s, 1H) 5.92 (d, J = 8.7 Hz, 1H) 5.71 (ddd, J = 8.4, 8.3, 4.3 Hz, 1H) 5.09 (ddd, J = 9.8, 6.9, 4.2 Hz, 1H) Na+ salt in D2O −1.2 −2.5 Na+ salt in D2O [M − H]− 687.2 E4 
8.24 (s, 1H) 8.18 (s, 1H) 7.97 (s, 1H) 6.10 (d, J = 1.4 Hz, 1H) 5.97 (d, J = 8.4 Hz, 1H) 5.63 (ddd, J = 8.1, 7.9, 4.2 Hz, 1H) 5.09 (m, 1H) −1.5 −2.4 [M + H]+ 713.2 E5 
8.20 (s, 1H) 7.95 (s, 1H) 7.47 (d, J = 3.8 Hz, 1H) 6.77 (d, J = 3.8 Hz, 1H) 6.08 (d, J = 4.7 Hz, 1H) 5.80 (d, J = 4.9 Hz, 1H) in D2O—CH3CN [M + H]+ 673.7 E6 
8.25 (s, 1H) 8.23 (s, 1H) 8.02 (s, 1H) 6.18 (s, 1H) 5.96 (d, J = 8.6 Hz, 1H) 5.43 (td, J = 8.1, 3.9 Hz, 1H) 52.4 −2.4 [M + H]+ 691.0 E7 
8.29 (s, 1H) 8.26 (s, 1H) 7.85 (s, 1H) 6.17 (s, 1H) 5.92 (d, J = 8.5 Hz, 1H) 5.61 (ddd, J = 7.9, 7.9, 4.0 Hz, 1H) 5.20 (ddd, J = 8.8, 8.8, 4.1 Hz, 1H) 55.1 −2.5 [M + H]+ 691.0 E8 
8.59 (s, 1H) 8.18 (s, 1H) 8.01 (s, 1H) 6.18 (s, 1H) 5.98 (s, 1H) 55.1 −1.5 [M + H]+ 691.0 E9 
8.35 (s, 1H) 8.13 (s, 1H) 7.95 (s, 1H) 6.12 (s, 1H) 5.92 (s, 1H) 54.4 −1.6 [M + H]+ 691.0 E10 
8.19 (s, 1H) 8.10 (s, 1H) 7.83 (s, 1H) 6.00 (d, J = 3.7 Hz, 1H) 5.84 (d, J = 5.0 Hz, 1H) 33.8 −0.4 [M + H]+ 657.2 E11 
8.08 (s, 1H) 8.01 (s, 1H) 7.85 (s, 1H) 6.00 (1H) 5.76 (1H) 32.1 −0.3 [M + H]+ 657.2 E12 
[M + H]+ 707.0 E13 
[M + H]+ 707.0 E14 
[M + H]+ 707.0 E15 
[M + H]2+ 2 354.0 E16 
8.06 (s, 1H) 8.03 (s, 1H) 7.23 (d, J = 3.7 Hz, 1H) 6.59 (d, J = 3.7 Hz, 1H) 6.10 (d, J = 6.4 Hz, 1H) 5.87 (d, J = 8.5 Hz, 1H) 5.26 (ddd, J = 8.5, 6.8, 4.2 Hz, 1H) 5.06 (ddd, J = 8.0, 4.7, 2.8 Hz, 1H) in DMSO-d6 57.9 51.7 in DMSO- d6 [M + H]+ 706.1 E17 
[M + H]+ 706.1 E18 
8.23 (s, 1H) 8.19 (s, 1H) 7.43 (d, J = 3.8 Hz, 1H) 6.52 (d, J = 3.8 Hz, 1H) 6.19 (s, 1H) 5.89 (d, J = 8.5 Hz, 1H) 5.23 (m, 1H) 4.95 (m, 1H) in CD3CN 51.3 49.1 in CD3CN [M + H]+ 706.1 E19 
8.84 (s, 1H) 8.58 (s, 1H) 6.58 (d, J = 4.6 Hz, 1H) 6.24 (d, J = 8.5 Hz, 1H) 5.79 (m, 1H) 5.60 (m, 1H) in D2O 59.9 56.7 in D2O [M + H]+ 708.2 E20 
8.57 (s, 1H) 7.83 (d, J = 3.8 Hz, 1H) 7.62 (d, J = 3.8 Hz, 1H) 7.06 (d, J = 3.8 Hz, 1H) 6.84 (d, J = 3.8 Hz, 1H) 6.61 (d, J = 6.2 Hz, 1H) 6.48 (d, J = 8.4 Hz, 1H) 5.69 (ddd, J = 8.0, 4.6, 3.1 Hz, 1H) 5.63 (ddd, J = 12.1, 8.4, 4.3 Hz, 1H) in D2O) 59.3 57.8 in D2O [M + H]+ 705.3 E21 
8.34 (s, 1H) 7.62 (d, J = 3.8 Hz, 1H) 7.33 (d, J = 3.8 Hz, 1H) 6.94 (d, J = 3.8 Hz, 1H) 6.28 (d, J = 3.8 Hz, 1H) 6.13 (d, J = 5.4 Hz, 1H) 6.10 (d, J = 8.4 Hz, 1H) in DMSO-d6 54.9 50.1 in DMSO- d6 [M + H]+ 705.1 E22 
8.35 (s, 1H) 7.65 (d, J = 3.8 Hz, 1H) 7.09 (d, J = 3.8 Hz, 1H) 6.97 (d, J = 3.7 Hz, 1H) 6.28 (d, J = 3.7 Hz, 1H) 6.11 (d, J = 7.9 Hz, 1H) 6.07 (d, J = 8.5 Hz, 1H) 5.28 (dd, J = 8.6, 4.4 Hz, 1H) 5.01 (m, 1H) in DMSO- d6 59.7 58.9 in DMSO- d6 [M + H]+ 705.1 E23 
8.35 (s, 1H) 7.69 (d, J = 3.8 Hz, 1H) 7.07 (d, J = 3.8 Hz, 1H) 6.95 (d, J = 3.7 Hz, 1H) 6.31 (d, J = 3.7 Hz, 1H) 6.13 (d, J = 7.2 Hz, 1H) 6.06 (d, J = 8.5 Hz, 1H) 5.05 (m, 1H) 4.99 (m, 1H) in DMSO-d6 58.5 50.9 in DMSO- d6 [M + H]+ 705.1 E24 
8.32 (s, 1H) 8.02 (s, 1H) 7.98 (s, 1H) 6.18 (s, 1H) 6.07 (d, J = 8.7 Hz, 1H) 5.63 (ddd, J = 9.1, 5.0, 5.0, 1H) 5.6 −1.4 [M + H]+ 744.0 E25 
8.55 (s, 1H) 8.45 (s, 1H) 8.29 (s, 1H) 6.27 (s, 1H) 5.96 (d, J = 8.3 Hz, 1H) 5.56 (m, 1H) 5.03 (ddd, J = 9.4, 6.5, 4.2 Hz, 1H) −1.3 [M]− 671.2 EA1 
8.07 (s, 1H) 7.37 (d, J = 3.8 Hz, 1H) 7.09 (d, J = 3.8 Hz, 1H) 6.65 (d, J = 3.7 Hz, 1H) 6.35 (d, J = 3.7 Hz, 1H) 6.08 (d, J = 7.4 Hz, 1H) 6.04 (d, J = 8.4 Hz, 1H) 4.85 (m, 1H) 4.81 (m, 1H) in DMSO-d6 0.98 0.96 in DMSO- d6 [M + H]+ 673.1 EA2 
8.29 (s, 1H) 8.25 (s, 1H) 8.25 (s, 1H) 7.89 (s, 1H) 6.17 (s, 1H) 6.01 (d, J = 8.6 Hz, 1H) 5.69 (ddd, J = 8.7, 8.7, 4.0 Hz, 1H) 5.09 (m, 1H) Na+ salt in D2O −1.1 −2.3 Na+ salt in D2O [M + H]+ 757.0 [M − H− 755.0 EA3 
8.26 (s, 1H) 8.23 (s, 1H) 7.85 (s, 1H) 6.27 (s, 1H) 5.92 (d, J = 8.6 Hz, 1H) 5.63 (m, 1H) 5.10 (m, 1H) −1.6 −2.3 [M + H]+ 713.0 [M − H]− 711.0 EA4 
8.19 (s, 1H) 8.18 (s, 1H) 8.15 (brs, 1H) 6.25 (d, J = 3.3 Hz, 1H) 5.92 (d, J = 8.5 Hz, 1H) 5.24 (m, 2H) in CD3CN 55.4 53.5 in CD3CN [M + H]+ 707.1 EA5 
59.6 58.1 in CD3CN [M + H]+ 707.1 EA6 
8.62 (s, 1H) 8.55 (s, 1H) 7.69 (d, J = 3.7 Hz, 1H) 6.59 (d, J = 3.7 Hz, 1H) 6.41 (d, J = 8.4 Hz, 1H) 6.37 (d, J = 4.6 Hz, 1H) 5.59 (m, 1H) 5.51 (m, 1H) in D2O 58.1 56.0 in D2O [M + H]+ 706.0 EA7 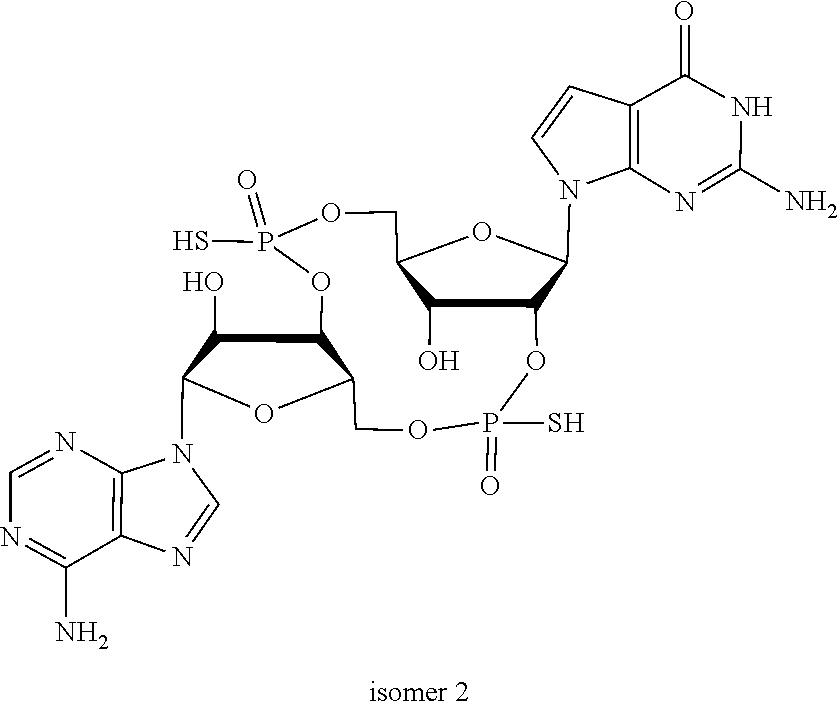
56.7 55.6 in CD3CN [M + H]+ 706.0 EA8 
53.4 48.7 in CD3CN [M + H]+ 705.9 EA9 
8.41 (s, 1H) 8.26 (s, 1H) 7.85 (s, 1H) 6.17 (s, 1H) 5.90 (d, J = 8.6 Hz, 1H) 5.76 (ddd, J = 8.8, 8.6, 4.1 Hz, 1H) 5.08 (m, 1H) [M]− 671.2 EA10 
8.32 (s, 1H) 8.26 (s, 1H) 7.86 (s, 1H) 6.16 (s, 1H) 5.92 (d, J = 8.5 Hz, 1H) 5.67 (m, 1H) 5.21 (m, 1H) [M]− 671.2 EA11 
8.43 (s, 1H) 8.41 (s, 1H) 6.19 (s, 1H) 6.07 (d, J = 8.2 Hz, 1H) [M + H]− 669.2 EB1 
8.16 (s, 1H) 7.48 (d, J = 3.3 Hz, 1H) 7.18 (d, J = 3.3 Hz, 1H) 6.50 (d, J = 3.5 Hz, 1H) 6.49 (d, J = 3.5 Hz, 1H) 6.29 (d, J = 5.1 Hz, 1H) 6.09 (d, J = 8.7 Hz, 1H) 5.49 (m, 1H) 5.17 (m, 1H) 57.7 −0.2 [M + H]+ 689.0 EB2 
8.16 (s, 1H) 7.33 (d, J = 3.8 Hz, 1H) 7.07 (d, J = 3.8 Hz, 1H) 6.44 (d, J = 3.8 Hz, 1H) 6.33 (d, J = 3.8 Hz, 1H) 6.27 (d, J = 2.5 Hz, 1H) 6.01 (d, J = 8.5 Hz, 1H) 5.48 (dd, J = 8.6, 8.5, 4.1 Hz, 1H) 5.00 (ddd, J = 7.1, 7.1, 4.7 Hz, 1H) 52.0 −1.1 [M + H]+ 689.0 EB3 
8.26 (s, 1H) 8.25 (s, 1H) 6.95 (d, J = 3.8 Hz, 6.29 (d, J = 3.8 Hz, 1H) 6.15 (s, 1H) 5.93 (d, J = 8.7 Hz, 1H) 5.55 (m, 1H) 4.99 (m, 1H) 51.5 −1.3 [M + H]+ 690.0 EB4 
8.46 (s, 1H) 8.26 (s, 1H) 7.03 (d, J = 3.7 Hz, 1H) 6.38 (d, J = 3.7 Hz, 1H) 6.17 (d, J = 2.7 Hz, 1H) 5.99 (d, J = 8.4 Hz, 1H) 5.63 (m, 1H) 5.11 (ddd, J = 6.7, 6.6, 4.5 Hz, 1H) 56.1 −0.7 [M + H]+ 690.0 EB5 
8.26 (s, 1H) 8.23 (s, 1H) 6.94 (d, J = 3.7 Hz, 1H) 6.26 (d, J = 3.7 Hz, 1H) 6.14 (s, 1H) 5.92 (d, J = 8.5 Hz, 5.55 (m, 1H) 1H) 5.00 (m, 1H) 51.5 −1.3 [M + H]+ 734.0 [M − H]− 732.0 EB6 
8.55 (s, 1H) 8.26 (s, 1H) 7.55 (s, 1H) 6.22 (s, 1H) 6.08 (d, J = 8.6 Hz, 1H) 5.81 (ddd, J = 10.4, 8.6, 4.0 Hz, 1H) 5.32 (ddd, J = 8.9, 8.8, 4.3 Hz, 1H) 54.1 52.5 [M − H]− 705.0 EB7 
8.59 (s, 1H) 8.26 (s, 1H) 7.79 (s, 1H) 6.21 (s, 1H) 6.05 (d, J = 8.6 Hz, 1H) 5.88 (ddd, J = 10.5, 8.6, 4.1 Hz, 1H) 5.31 (ddd, J = 8.4, 8.4, 4.1 Hz, 1H) 55.4 54.2 [M − H]− 705.0 EC1 
8.15 (s, 1H) 7.89 (s, 1H) 7.35 (d, J = 3.8 Hz, 1H) 6.27 (s, 1H) 6.26 (d, J = 3.8 Hz, 1H) 5.98 (d, J = 8.6 Hz, 1H) 5.75 (ddd, J = 9.2, 9.1, 4.1 Hz, 1H) 5.07 (m, 1H) 52.0 −1.3 [M + H]+ 772.1 EC2 
8.15 (s, 1H) 7.97 (s, 1H) 7.56 (d, J = 3.8 Hz, 1H) 6.37 (d, J = 3.8 Hz, 1H) 6.30 (d, J = 3.1 Hz, 1H) 6.02 (d, J = 8.5 Hz, 1H) 5.78 (ddd, J = 12.6, 8.5, 4.1 Hz, 1H) 5.16 (m, 1H) 56.3 −0.6 [M + H]+ 772.1 EC3 
8.15 (s, 1H) 7.87 (s, 1H) 7.33 (d, J = 3.8 Hz, 1H) 6.27 (s, 1H) 6.24 (d, J = 3.8 Hz, 1H) 5.98 (d, J = 8.6 Hz, 1H) 5.68 (ddd, J = 8.5, 8.45, 4.0 Hz, 1H) 5.08 (ddd, J = 7.7, 7.5, 4.5 Hz, 1H) −1.4 −2.4 [M + H]+ 756.2 EC4 
7.03 (d, J = 3.7 Hz, 1H) 6.88 (m, 1H) 6.34 (d, J = 3.7 Hz, 6.31 (d, J = 3.7 Hz, 1H) 6.08 (m, 1H) 5.93 (rn, 1H) 5.05 (m, 1H) 4.98 (m, 1H) in DMSO-d6/D2O 61.1 −0.4 [M + H]+ 705.0 EC5 
7.02 (d, J = 3.7 Hz, 1H) 6.92 (d, J = 3.7 Hz, 1H) 6.33 (m, 2H) 6.09 (d, J = 8.7 Hz, 1H) 5.92 (d, J = 7.9 Hz, 1H) 4.94 (m, 1H) 4.81 (m, 1H) 4.60 (m, 1H) 4.43 (m, 1H) in DMSO-d6 /D2O 53.1 −0.8 [M + H]+ 704.9 EC6 
8.11 (s, 1H) 8.11 (s, 1H) 7.59 (d, J = 3.8 Hz, 1H) 7.20 (d, J = 3.9 Hz, 1H) 6.40 (m, 2H) 6.22 (m, 2H) 5.23 (ddd, J = 9.0, 8.8, 4.2 Hz, 1H) 4.94 (m, 1H) 52.9 −1.0 [M + H]+ 673.0 EC7 
8.103 (s, 1H) 8.102 (s, 1H) 7.67 (d, J = 3.9 Hz, 1H) 7.34 (d, J = 3.9 Hz, 1H) 6.58 (d, J = 3.8 Hz, 1H) 6.51 (d, J = 3.8 Hz, 1H) 6.42 (d, J = 8.4 Hz, 1H) 6.23 (d, J = 6.5 Hz, 1H) 59.1 0.1 [M + H]+ 673.0 EC8 
8.27 (s, 2H) 8.05 (s, 1H) 7.47 (d, J = 3.8 Hz, 1H) 6.92 (d, J = 3.7 Hz, 1H) 6.65 (d, J = 3.7 Hz, 1H) 6.30 (m, 2H) 5.99 (d, J = 8.2 Hz, 1H) 5.01 (m, 1H) 4.85 (m, 1H) in DMSO-d6 49.5 0.9 in DMSO- d6 [M + H]+ 689.2 EC9 
8.29 (s, 2H) 8.04 (s, 1H) 7.50 (d, J = 3.8 Hz, 1H) 6.82 (d, J = 3.7 Hz, 1H) 6.59 (d, J = 3.8 Hz, 1H) 6.35 (d, 1H) J = 3.7 Hz, 1H) 6.29 (d, J = 8.5 Hz, 1H) 5.98 (d, J = 8.5 Hz, 1H) 5.11 (m, 1H) 4.40 (m, 1H) in DMSO- d6 57.1 1.4 in DMSO- d6 [M + H]+ 689.1 EC10 
8.28 (s, 1H) 8.26 (s, 1H) 7.83 (s, 1H) 6.26 (s, 1H) 5.96 (d, J = 8.6 Hz, 1H) 5.76 (ddd, J = 8.6, 8.6, 4.1 Hz, 1H) 5.09 (m,1H) −1.6 −2.7 [M + H]+ 771.2 EC11 
8.32 (s, 1H) 8.27 (s, 1H) 7.93 (s, 1H) 6.47 (m, 1H) 5.97 (d, J = 8.3 Hz, 1H) 5.58 (m, 1H) 5.19 (m, 1H) [M + H]+ 659.0 [M − H]− 656.8 EC12 
8.30 (s, 1H) 8.25 (s, 1H) 7.84 (s, 1H) 6.16 (s, 1H) 5.88 (d, J = 8.4 Hz, 1H) 5.73 (ddd, J = 8.0, 8.0, 8.0 Hz, 1H) 5.08 (m, 1H) −1.0 −2.4 [M + H]+ 703.0 EC13 
8.36 (s, 1H) 8.18 (s, 1H) 7.84 (s, 1H) 6.29 (d, J = 8.1 Hz, 1H) 5.99 (d, J = 8.4 Hz, 1H) 5.38 (ddd, J = 8.1, 7.9, 4.4 Hz, 1H) 5.09 (ddd, J = 8.3, 4.2, 4.2 Hz, 1H) −1.3 −1.8 [M + H]+ 757.0 EC14 
8.69 (s, 1H) 8.13 (s, 1H) 6.88 (d, J = 3.8 Hz, 1H) 6.34 (d, J = 3.8 Hz, 1H) 6.28 (d, J = 8.1 Hz, 1H) 6.10 (d, J = 8.1 Hz, 1H) 5.17 (m, 2H) 56.7 −1.5 [M + H]+ 689.9 EC15 
8.35 (s, 1H) 7.93 (s, 1H) 6.77 (d, J = 3.8 Hz, 1H) 6.25 (d, J = 8.1 Hz, 1H) 6.17 (d, J = 3.8 Hz, 1H) 6.10 (d, J = 8.1 Hz, 1H) 5.19 (ddd, J = 8.6, 4.6, 4.6 Hz, 1H) 5.14 (ddd, J = 8.5, 4.4, 4.4 Hz, 1H) 52.1 −1.6 [M + H]+ 690.0 EC16 
8.03 (s, 1H) 7.68 (m, 1H) 7.58 (d, J = 8.0 Hz, 1H) 7.44 (d, J = 3.4 Hz, 1H) 7.20 (m, 2H) 6.59 (t, J = 6.5 Hz, 1H) 6.55 (d, J = 3.4 Hz, 1H) 6.05 (d, J = 8.5 Hz, 1H) 5.55 (m, 1H) 5.20 (m, 1H) −1.0 −1.1 [M + H]+ 723.1 EC17 
8.03 (s, 1H) 7.67 (m, 1H) 7.58 (m, 1H) 7.44 (d, J = 3.4 Hz, 1H) 7.18 (m, 2H) 6.57 (t, J = 6.4 Hz, 1H) 6.52 (m, 1H) 6.02 (d, J = 8.5 Hz, 1H) 5.57 (m, 1H) 5.20 (m, 1H) 53.0 −1.0 [M + H]+ 657.0 [M − H]− 655.0 EC18 
8.16 (s, 1H) 7.68 (d, J = 7.8 Hz, 1H) 7.62 (d, J = 8.1 Hz, 1H) 7.52 (d, J = 3.5 Hz, 1H) 7.26 (m, 1H) 7.19 (m, 1H) 6.61 (m, 2H) 6.05 (d, J = 8.3 Hz, 1H) 5.50 (m, 1H) 5.31 (m, 1H) 59.5 −1.0 [M + H]+ 657.0 [M − H]− 655.0 EC19 
8.06 (m, 1H) 7.99 (m, 1H) 7.97 (s, 1H) 7.84 (d, J = 8.3 Hz, 1H) 7.69 (d, J = 7.2 Hz, 1H) 7.59 (m, 2H) 7.32 (t, J = 7.7 Hz, 1H) 6.02 (m, 2H) 5.52 (ddd, J = 8.7, 8.7, 4.2 Hz, 1H) 5.08 (m, 1H) −1.0 −1.2 [M + H]+ 734.0 EC20 
9.12 (s, 1H) 8.29 (d, J = 9.3 Hz, 1H) 8.12 (m, 5H) 8.00 (s, 2H) 7.93 (t, J = 7.6 Hz, 1H) 6.18 (m, 2H) 5.36 (m, 2H) 62.8 −0.7 [M + H]+ 742.1 EC21 
9.09 (s, 1H) 8.28 (d, J = 9.3 Hz, 1H) 8.11 (m, 5H) 7.98 (s, 2H) 7.93 (t, J = 7.6 Hz, 1H) 6.17 (dd, J = 10.5, 5.3 Hz, 1H) 6.13 (d, J = 8.0 Hz, 1H) 5.46 (m, 1H) 5.13 (m, 1H) 56.5 −0.8 [M + H]+ 742.0 EC22 
8.21 (s, 1H) 8.14 (s, 1H) 7.82 (s, 1H) 6.19 (d, J = 7.7 Hz, 1H) 6.00 (d, J = 3.1 Hz, 1H) −0.1 [M + H]+ 689.0 EC23 
8.15 (s, 1H) 7.91 (s, 1H) 7.44 (s, 1H) 6.17 (s, 1H) 5.88 (d, J = 9.0 Hz, 1H) 5.13 (m, 2H) −1.24 [M + H]+ 672.9 EC24 
8.31 (s, 1H) 8.26 (s, 1H) 7.84 (s, 1H) 6.17 (s, 1H) 5.86 (d, J = 8.4 Hz, 1H) 5.68 (m, 1H) 5.08 (m, 1H) −1.0 −2.2 [M + H]+ 701.2 [M − H]− 699.0 EC25 
9.08 (s, 1H) 8.38 (s, 1H) 7.96 (s, 1H) 7.90 (s, 1H) 7.57 (s, 1H) 6.28 (s, 1H) 5.95 (d, J = 8.5 Hz, 1H) 5.64 (ddd, J = 8.3, 8.3, 4.2 Hz, 1H) 5.08 (ddd, J = 8.8, 6.5, 4.3 Hz, 1H) −1.3 −2.0 [M − H]− 697.1 EC26 
9.15 (s, 1H) 8.40 (s, 1H) 8.02 (s, 1H) 7.90 (s, 1H) 7.63 (s, 1H) 6.32 (s, 1H) 5.98 (d, J = 8.6 Hz, 1H) 5.72 (ddd, J = 8.7, 8.7, 4.1 Hz, 1H) 5.11 (ddd, J = 8.6, 6.8, 4.3 Hz, 1H) −1.3 −2.3 [M − H]− 779.0 *In NaH2PO4/Na2HPO4/D2O unless otherwise mentioned. - Stereochemical information of represented compounds is given below.
-
TABLE 5 P-Configuration of Examples E23 and EB3 E23 [Rp,Rp] 
EB3 [Rp] 
- Serial dilutions of cGAMP analog compounds in phosphate buffer saline (PBS) are mixed with THP1 luciferase reporter cells in a 96-well plate at 0.2×106/well, in the presence or absence of 1 nM of Perfringolysin 0 (PFO), which can facilitate compound uptake by forming open channels on the plasma membrane. 16 hours later, 15 μL of the media from each well is transferred to a new plate, and luminescence is measured. Fold increase in luminescence compared to PBS stimulated cells is plotted against logarithm of concentrations of each compound, and EC50 is calculated using Graphpad.
-
TABLE 6 Activity of cyclic dinucleotides and analogs THP1-ISG-Luc THP-ISG-Luc with PFO, without PFO, Example EC50, nM EC50, μM E1 A A E2 A A E3 A A E4 A A E5 C C E6 A A E7 A A E8 B C E9 A A E10 C C E11 B C E12 A A E13 B C E14 B C E15 C C E16 A A E17 C C E18 A A E19 C C E20 B A E21 B A E22 B A E23 A A E24 B A E25 A A EA1 B C EA2 A A EA3 C C EA4 B A EA5 C C EA6 C C EA7 B A EA8 B A EA9 A A EA10 A A EA11 A A EB1 C B EB2 A A EB3 A A EB4 C B EB5 B C EB6 C C EB7 C C EC1 A A EC2 B B EC3 B A EC4 C C EC5 B B EC6 C C EC7 C C EC8 C C EC9 C C EC10 C C EC11 A C EC12 A A EC13 B A EC14 C C EC15 B A EC16 C C EC17 C C EC18 C C EC19 C C EC20 C C EC21 C C EC22 C C EC23 C C EC24 A A EC26 B C Activity A ≤100 nM A ≤30 μM Code B 100-1000 nM B 30-100 μM C >1000 nM C >100 μM - 2′3′-cGAMP can be degraded by the enzyme ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP1) which is present in fetal bovine serum (FBS) (Li et al., 2015, Nat Chem Biol, 11, 235). To test if cGAMP analogues have improved stability, 5 μL of synthetic cGAMP analogues (100 μM stock) were incubated with 45 μL of FBS in a final volume of 50 μL at 37° C. for 1, 2 and 4 hours. At the indicated time, 10 μL of the reaction mixture was taken out and mixed with 10 μL phosphate buffered saline (PBS), then heated at 95° C. to denature proteins, which were removed by centrifugation at 13000 g for 5 minutes. The supernatants were delivered to THP1-ISG-luciferase cell line in the presence of PFO to measure the activity of remaining cGAMP analogues, as described above. Category A indicates less than 10% decrease of activity after 4-hour incubation, B indicates 10-75% decrease of activity after 4-hour incubation, and C indicates more than 75% loss of activity after 4-hour incubation.
-
TABLE 7 Stability of cyclic dinucleotides and analogues in fetal bovine serum Reduction of activity after FBS Example incubation E1 C E2 C E3 A E6 C E7 C E8 A E9 A E16 B E18 B E20 A E21 B E22 A E23 B E25 A EA2 A EA4 A EA6 A EA7 A EA8 A EA10 C EA11 A EB1 A EB2 B EB3 A EB4 A EB5 A EC1 A EC2 A EC3 B EC5 A EC13 A EC15 A EC24 A EC26 A Activity A <10% Code B >10% C >75% - The following series of prophetic examples are also compounds of the present invention:
-
TABLE 8 Structures of P1 to P15 
P1 
P2 
P3 
P4 
P5 
P6 
P7 
P8 
P9 
P10 
P11 
P12 
P13 
P14 
P15 - The following Example compounds in Table 9 are also compounds contemplated by the present disclosure.
-
TABLE 9 Additional Compounds Compound Structure PA1 
PA2 
PA3 
PA4 
PA5 
PA6 
PA7 
PA8 
PA9 
PA10 
PA11 
PA12 
PB1 
PB2 
PB3 
PB4 
PB5 
PB6 
PB7 
PB8 
PB9 
PB10 
PB11 
PB12 
PB13 
PB14 
PC1 
PC2 
PC3 
PC4 
PC5 
PC6 
PC7 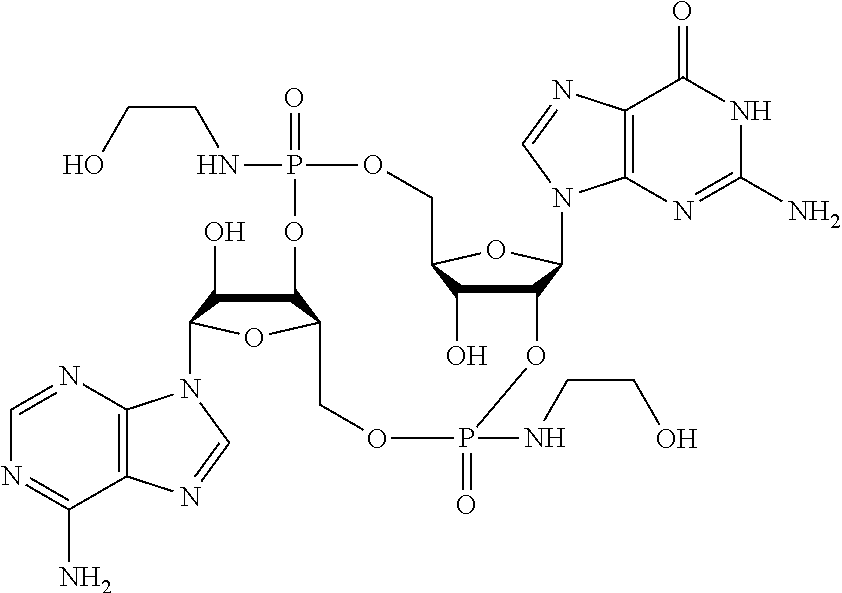
PC8 
PC9 
PC10 
PC11 
PC12 
PD1 
PD2 
PD3 
PD4 
PD5 
PD6 
Claims (29)











































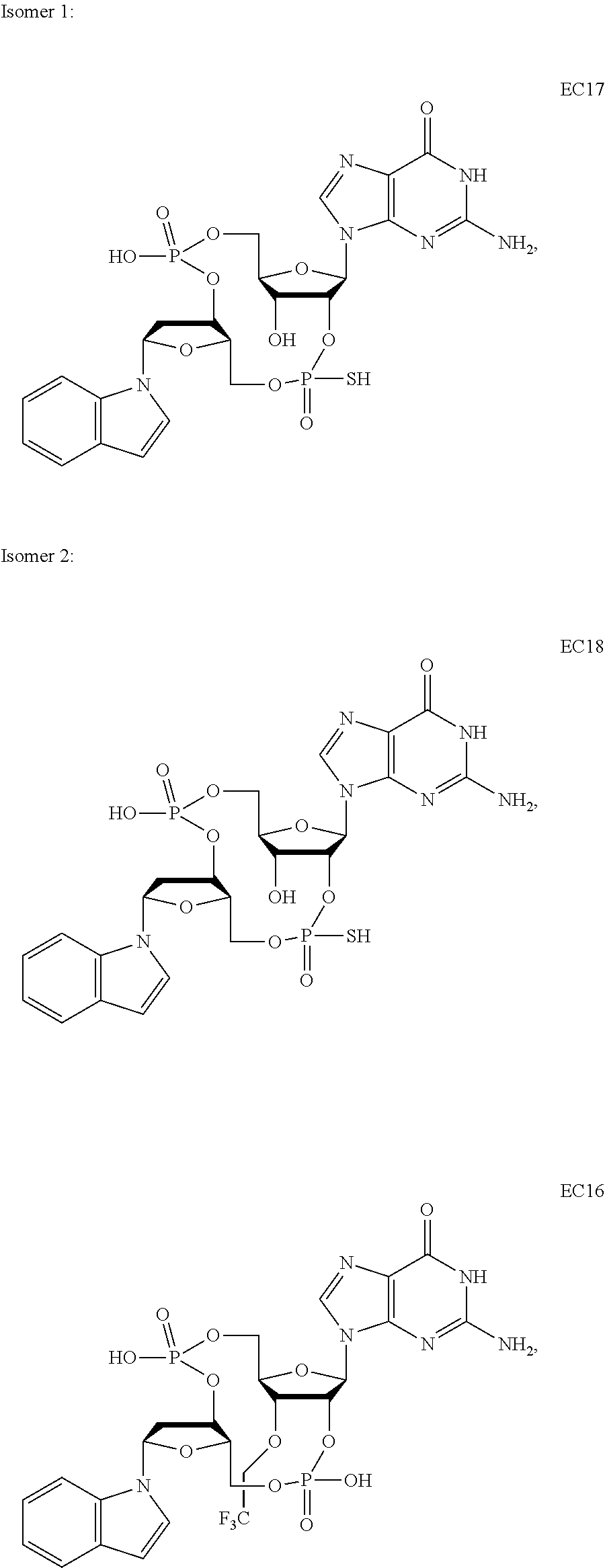




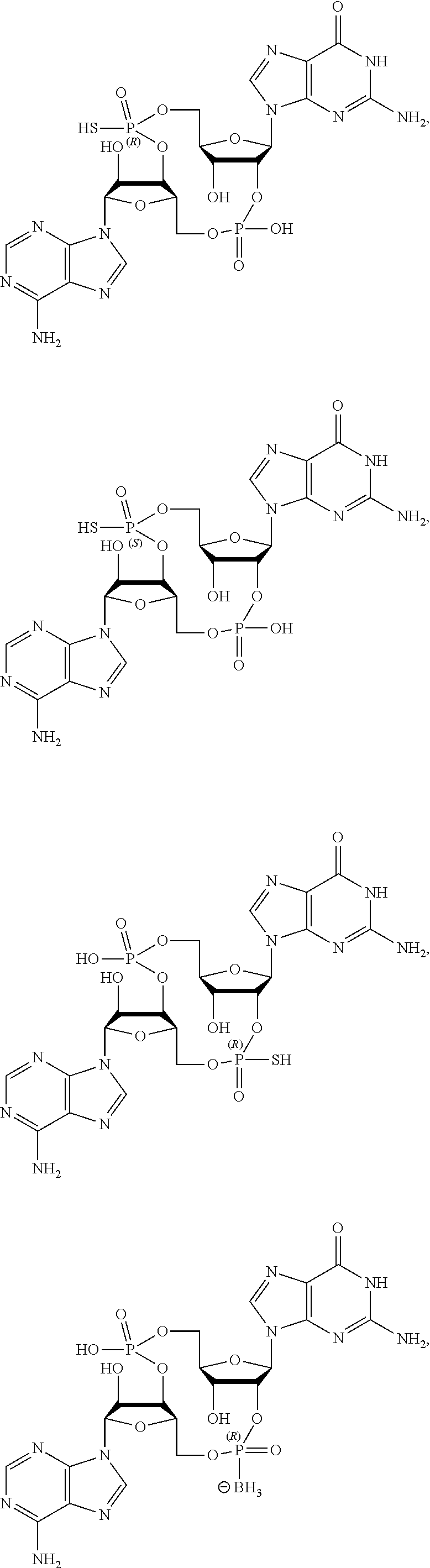























Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/697,247 US20220340613A1 (en) | 2016-03-18 | 2022-03-17 | Cyclic di-nucleotide compounds and methods of use |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662310364P | 2016-03-18 | 2016-03-18 | |
| US201662355382P | 2016-06-28 | 2016-06-28 | |
| US201662396140P | 2016-09-17 | 2016-09-17 | |
| PCT/US2017/023093 WO2017161349A1 (en) | 2016-03-18 | 2017-03-17 | Cyclic di-nucleotide compounds and methods of use |
| US15/953,492 US10519188B2 (en) | 2016-03-18 | 2018-04-15 | Cyclic di-nucleotide compounds and methods of use |
| US16/438,153 US11299512B2 (en) | 2016-03-18 | 2019-06-11 | Cyclic di-nucleotide compounds and methods of use |
| US17/697,247 US20220340613A1 (en) | 2016-03-18 | 2022-03-17 | Cyclic di-nucleotide compounds and methods of use |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/438,153 Continuation US11299512B2 (en) | 2016-03-18 | 2019-06-11 | Cyclic di-nucleotide compounds and methods of use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20220340613A1 true US20220340613A1 (en) | 2022-10-27 |
Family
ID=59851227
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/953,492 Active US10519188B2 (en) | 2016-03-18 | 2018-04-15 | Cyclic di-nucleotide compounds and methods of use |
| US16/438,153 Active 2038-06-29 US11299512B2 (en) | 2016-03-18 | 2019-06-11 | Cyclic di-nucleotide compounds and methods of use |
| US17/697,247 Pending US20220340613A1 (en) | 2016-03-18 | 2022-03-17 | Cyclic di-nucleotide compounds and methods of use |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/953,492 Active US10519188B2 (en) | 2016-03-18 | 2018-04-15 | Cyclic di-nucleotide compounds and methods of use |
| US16/438,153 Active 2038-06-29 US11299512B2 (en) | 2016-03-18 | 2019-06-11 | Cyclic di-nucleotide compounds and methods of use |
Country Status (22)
| Country | Link |
|---|---|
| US (3) | US10519188B2 (en) |
| EP (2) | EP3429596B1 (en) |
| JP (3) | JP6980198B2 (en) |
| KR (1) | KR102530488B1 (en) |
| CN (3) | CN114230625A (en) |
| AU (1) | AU2017233068C1 (en) |
| BR (1) | BR112018068748B1 (en) |
| CA (1) | CA3017524A1 (en) |
| DK (1) | DK3429596T3 (en) |
| ES (1) | ES2929628T3 (en) |
| HR (1) | HRP20221263T1 (en) |
| HU (1) | HUE060396T2 (en) |
| IL (2) | IL280430B2 (en) |
| LT (1) | LT3429596T (en) |
| MA (1) | MA43827A (en) |
| MX (1) | MX2022001755A (en) |
| NZ (1) | NZ746112A (en) |
| PL (1) | PL3429596T3 (en) |
| PT (1) | PT3429596T (en) |
| SG (2) | SG10201912074PA (en) |
| WO (1) | WO2017161349A1 (en) |
| ZA (1) | ZA201806074B (en) |
Families Citing this family (111)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11453697B1 (en) | 2015-08-13 | 2022-09-27 | Merck Sharp & Dohme Llc | Cyclic di-nucleotide compounds as sting agonists |
| SG10202010609QA (en) | 2015-08-13 | 2020-11-27 | Merck Sharp & Dohme | Cyclic di-nucleotide compounds as sting agonists |
| TWI704154B (en) | 2015-12-03 | 2020-09-11 | 英商葛蘭素史克智慧財產發展有限公司 | Novel compounds |
| JP6867395B2 (en) | 2016-01-11 | 2021-04-28 | イネイト・テューマー・イミュニティ・インコーポレイテッドInnate Tumor Immunity, Inc. | Circular dinucleotides for treating symptoms associated with STING activity, such as cancer |
| NZ746112A (en) * | 2016-03-18 | 2023-01-27 | Immune Sensor Llc | Cyclic di-nucleotide compounds and methods of use |
| TN2020000158A1 (en) | 2016-10-04 | 2022-04-04 | Merck Sharp & Dohme | BENZO[b]THIOPHENE COMPOUNDS AS STING AGONISTS |
| BR112019007450A2 (en) | 2016-10-14 | 2020-07-07 | Precision Biosciences, Inc. | modified meganucleases specific for recognition sequences in the hepatitis b virus genome |
| JOP20170188A1 (en) * | 2016-11-25 | 2019-01-30 | Janssen Biotech Inc | Cyclic dinucleotides as sting agonists |
| JOP20170192A1 (en) | 2016-12-01 | 2019-01-30 | Takeda Pharmaceuticals Co | Cyclic dinucleotide |
| JP2018090562A (en) * | 2016-12-01 | 2018-06-14 | 武田薬品工業株式会社 | Cyclic dinucleotide |
| US20200113924A1 (en) * | 2016-12-20 | 2020-04-16 | Merck Sharp & Dohme Corp. | Cyclic dinucleotide sting agonists for cancer treatment |
| AU2018212787B2 (en) | 2017-01-27 | 2023-10-26 | Janssen Biotech, Inc. | Cyclic dinucleotides as sting agonists |
| JP7275031B2 (en) * | 2017-01-27 | 2023-05-17 | ヤンセン バイオテツク,インコーポレーテツド | Cyclic dinucleotides as STING agonists |
| US20200055883A1 (en) | 2017-02-17 | 2020-02-20 | Eisai R&D Management Co., Ltd. | Cyclic di-nucleotides derivative for the treatment of cancer |
| CA3053568A1 (en) | 2017-02-21 | 2018-08-30 | Board Of Regents, The University Of Texas System | Cyclic dinucleotides as agonists of stimulator of interferon gene dependent signalling |
| WO2018198084A1 (en) * | 2017-04-27 | 2018-11-01 | Lupin Limited | Cyclic di-nucleotide compounds with tricyclic nucleobases |
| US11466047B2 (en) | 2017-05-12 | 2022-10-11 | Merck Sharp & Dohme Llc | Cyclic di-nucleotide compounds as sting agonists |
| EP3661498A4 (en) | 2017-08-04 | 2021-04-21 | Merck Sharp & Dohme Corp. | BENZO[b]THIOPHENE STING AGONISTS FOR CANCER TREATMENT |
| MA49773A (en) | 2017-08-04 | 2021-04-21 | Merck Sharp & Dohme | COMBINATIONS OF PD-1 ANTAGONISTS AND STING BENZO AGONISTS [B |
| JP7311514B2 (en) | 2017-08-30 | 2023-07-19 | ベイジン シュエンイー ファーマサイエンシズ カンパニー, リミテッド | Cyclic Dinucleotides as Interferon Gene Stimulator Modulators |
| EP3675859A4 (en) | 2017-08-31 | 2021-06-30 | Sperovie Biosciences, Inc. | Compounds, compositions, and methods for the treatment of disease |
| CN111344297B (en) | 2017-10-10 | 2023-10-20 | 百时美施贵宝公司 | Cyclic dinucleotides as anticancer agents |
| WO2019079261A1 (en) * | 2017-10-16 | 2019-04-25 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019084060A1 (en) | 2017-10-24 | 2019-05-02 | Silverback Therapeutics, Inc. | Conjugates and methods of use thereof for selective delivery of immune-modulatory agents |
| CA3082351A1 (en) * | 2017-11-10 | 2019-05-16 | Takeda Pharmaceutical Company Limited | Sting modulator compounds, and methods of making and using |
| EA038805B1 (en) * | 2017-11-21 | 2021-10-21 | Такеда Фармасьютикал Компани Лимитед | Cyclic dinucleotides as sting (stimulator of interferon genes) agonists |
| JP7317014B2 (en) * | 2017-12-15 | 2023-07-28 | ヤンセン バイオテツク,インコーポレーテツド | Cyclic dinucleotides as STING agonists |
| US11685761B2 (en) | 2017-12-20 | 2023-06-27 | Merck Sharp & Dohme Llc | Cyclic di-nucleotide compounds as sting agonists |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US20190185509A1 (en) * | 2017-12-20 | 2019-06-20 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| AU2019223182B2 (en) | 2018-02-26 | 2021-08-19 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds as HBV replication inhibitors |
| JP7250808B2 (en) * | 2018-03-08 | 2023-04-03 | ブリストル-マイヤーズ スクイブ カンパニー | Cyclic dinucleotides as anticancer agents |
| EP3768685A1 (en) * | 2018-03-23 | 2021-01-27 | Takeda Pharmaceutical Company Limited | Sting modulator compounds with sulfamate linkages, and methods of making and using |
| US11702430B2 (en) | 2018-04-03 | 2023-07-18 | Merck Sharp & Dohme Llc | Aza-benzothiophene compounds as STING agonists |
| WO2019195124A1 (en) | 2018-04-03 | 2019-10-10 | Merck Sharp & Dohme Corp. | Benzothiophenes and related compounds as sting agonists |
| US10870691B2 (en) | 2018-04-05 | 2020-12-22 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis B virus protein X |
| WO2019195658A1 (en) | 2018-04-05 | 2019-10-10 | Dana-Farber Cancer Institute, Inc. | Sting levels as a biomarker for cancer immunotherapy |
| TWI833744B (en) * | 2018-04-06 | 2024-03-01 | 捷克科學院有機化學與生物化學研究所 | 3'3'-cyclic dinucleotides |
| TWI818007B (en) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-cyclic dinucleotides |
| TW202005654A (en) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2'2'-cyclic dinucleotides |
| TW201945388A (en) | 2018-04-12 | 2019-12-01 | 美商精密生物科學公司 | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis B virus genome |
| CN110407879A (en) * | 2018-04-28 | 2019-11-05 | 杭州星鳌生物科技有限公司 | The chemical composition of TXS-WX class compound, preparation method and its application in antitumor |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| CA3101368A1 (en) | 2018-05-25 | 2019-11-28 | Incyte Corporation | Tricyclic heterocyclic compounds as sting activators |
| EP3820477A4 (en) * | 2018-07-10 | 2022-07-13 | Sperovie Biosciences, Inc. | Compounds, compositions, and methods for the treatment of disease |
| TW202030199A (en) * | 2018-07-17 | 2020-08-16 | 美商健生生物科技公司 | Cyclic dinucleotides as sting agonists |
| US11008344B2 (en) | 2018-07-31 | 2021-05-18 | Incyte Corporation | Tricyclic heteroaryl compounds as STING activators |
| WO2020028566A1 (en) | 2018-07-31 | 2020-02-06 | Incyte Corporation | Heteroaryl amide compounds as sting activators |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| WO2020036199A1 (en) | 2018-08-16 | 2020-02-20 | Eisai R&D Management Co., Ltd. | Salts of compounds and crystals thereof |
| US20220008549A1 (en) | 2018-09-06 | 2022-01-13 | Daiichi Sankyo Company, Limited | Novel cyclic dinucleotide derivative and antibody-drug conjugate thereof |
| KR20210081332A (en) | 2018-09-12 | 2021-07-01 | 실버백 테라퓨틱스, 인크. | Compositions for Treatment of Diseases Using Immunostimulatory Conjugates |
| EP3854799B1 (en) * | 2018-09-21 | 2024-07-17 | Shanghai de Novo Pharmatech Co., Ltd. | Cyclic dinucleotide analogue, pharmaceutical composition thereof, and application |
| WO2020074004A1 (en) * | 2018-10-12 | 2020-04-16 | 上海济煜医药科技有限公司 | Cyclic dinucleotide compound and uses thereof |
| US11161864B2 (en) | 2018-10-29 | 2021-11-02 | Venenum Biodesign, LLC | Sting agonists |
| US11110106B2 (en) | 2018-10-29 | 2021-09-07 | Venenum Biodesign, LLC | Sting agonists for treating bladder cancer and solid tumors |
| JP7273172B2 (en) | 2018-10-31 | 2023-05-12 | ギリアード サイエンシーズ, インコーポレイテッド | Substituted 6-azabenzimidazole compounds with HPK1 inhibitory activity |
| LT3873903T (en) | 2018-10-31 | 2024-05-10 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| TWI827720B (en) * | 2018-11-02 | 2024-01-01 | 大陸商上海濟煜醫藥科技有限公司 | Cyclic dinucleotide compounds and uses thereof |
| US11596692B1 (en) | 2018-11-21 | 2023-03-07 | Incyte Corporation | PD-L1/STING conjugates and methods of use |
| CN111349132B (en) * | 2018-12-21 | 2021-06-04 | 上海济煜医药科技有限公司 | Tumor immunity compound and application thereof |
| US12129267B2 (en) | 2019-01-07 | 2024-10-29 | Incyte Corporation | Heteroaryl amide compounds as sting activators |
| JP7350871B2 (en) * | 2019-03-07 | 2023-09-26 | インスティチュート オブ オーガニック ケミストリー アンド バイオケミストリー エーエスシーアール,ヴイ.ヴイ.アイ. | 2'3'-cyclic dinucleotide and its prodrug |
| KR20210137518A (en) | 2019-03-07 | 2021-11-17 | 인스티튜트 오브 오가닉 케미스트리 앤드 바이오케미스트리 에이에스 씨알 브이.브이.아이. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| EP3935065A1 (en) | 2019-03-07 | 2022-01-12 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| US20220168415A1 (en) | 2019-03-21 | 2022-06-02 | Codiak Biosciences, Inc. | Extracellular vesicles for vaccine delivery |
| US20220387906A1 (en) | 2019-03-21 | 2022-12-08 | Codiak Biosciences, Inc. | Process for preparing extracellular vesicles |
| WO2020210938A1 (en) * | 2019-04-15 | 2020-10-22 | Bioardis Llc | Quinazoline derivatives as cd73 inhibitors |
| TWI751517B (en) | 2019-04-17 | 2022-01-01 | 美商基利科學股份有限公司 | Solid forms of a toll-like receptor modulator |
| TWI751516B (en) | 2019-04-17 | 2022-01-01 | 美商基利科學股份有限公司 | Solid forms of a toll-like receptor modulator |
| CN109929894B (en) * | 2019-04-17 | 2021-06-01 | 中国农业科学院兰州兽医研究所 | Preparation and activity identification method of porcine second messenger molecule 2 '3' -cGAMP |
| CA3137119A1 (en) * | 2019-05-09 | 2020-11-12 | Aligos Therapeutics, Inc. | Modified cyclic dinucleoside compounds as sting modulators |
| EP3972695A1 (en) | 2019-05-23 | 2022-03-30 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| CR20210687A (en) | 2019-06-25 | 2022-03-03 | Gilead Sciences Inc | Flt3l-fc fusion proteins and methods of use |
| WO2021003445A1 (en) | 2019-07-03 | 2021-01-07 | Codiak Biosciences, Inc. | Extracellular vesicles targeting t cells and uses thereof |
| US20230181758A1 (en) | 2019-07-03 | 2023-06-15 | Codiak Biosciences, Inc. | Extracellular vesicles targeting dendritic cells and uses thereof |
| MX2022000815A (en) | 2019-07-19 | 2022-05-03 | Immunesensor Therapeutics Inc | Antibody-sting agonist conjugates and their use in immunotherapy. |
| US20220296619A1 (en) | 2019-08-19 | 2022-09-22 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| WO2021041532A1 (en) | 2019-08-26 | 2021-03-04 | Dana-Farber Cancer Institute, Inc. | Use of heparin to promote type 1 interferon signaling |
| MX2022003570A (en) | 2019-09-25 | 2022-07-11 | Codiak Biosciences Inc | Extracellular vesicle compositions. |
| KR20220074917A (en) | 2019-09-30 | 2022-06-03 | 길리애드 사이언시즈, 인코포레이티드 | HBV vaccines and methods of treating HBV |
| CA3151322A1 (en) | 2019-10-01 | 2021-04-08 | Silverback Therapeutics, Inc. | Combination therapy with immune stimulatory conjugates |
| US20230270773A1 (en) * | 2019-10-14 | 2023-08-31 | Immunesensor Therapeutics, Inc. | Methods of treating cancer with a sting agonist |
| EP4069729A1 (en) | 2019-12-06 | 2022-10-12 | Precision BioSciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| EP4106819A1 (en) | 2020-02-21 | 2022-12-28 | Silverback Therapeutics, Inc. | Nectin-4 antibody conjugates and uses thereof |
| IL296124A (en) | 2020-03-06 | 2022-11-01 | Daiichi Sankyo Co Ltd | Antibody-drug conjugate including novel cyclic dinucleotide derivative |
| US20230114434A1 (en) | 2020-03-13 | 2023-04-13 | Codiak Biosciences, Inc. | Extracellular vesicles for treating neurological disorders |
| JP2023518414A (en) | 2020-03-20 | 2023-05-01 | コディアック バイオサイエンシーズ, インコーポレイテッド | Extracellular vesicles for therapy |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| TW202200136A (en) | 2020-04-10 | 2022-01-01 | 日商小野藥品工業股份有限公司 | Cancer treatment method |
| MX2022014110A (en) * | 2020-05-15 | 2023-01-30 | Immunesensor Therapeutics Inc | Sting agonist combination treatments with immune checkpoint inhibitors. |
| AU2021300362A1 (en) | 2020-07-01 | 2023-02-23 | ARS Pharmaceuticals, Inc. | Anti-ASGR1 antibody conjugates and uses thereof |
| IL299980A (en) | 2020-08-07 | 2023-03-01 | Gilead Sciences Inc | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| JPWO2022050300A1 (en) | 2020-09-02 | 2022-03-10 | ||
| WO2022066928A2 (en) | 2020-09-23 | 2022-03-31 | Codiak Biosciences, Inc. | Process for preparing extracellular vesicles |
| US20240082389A1 (en) | 2020-09-23 | 2024-03-14 | Lonza Sales Ag | Methods of producing extracellular vesicles |
| WO2022066883A1 (en) | 2020-09-23 | 2022-03-31 | Codiak Biosciences, Inc. | Extracellular vesicles comprising kras antigens and uses thereof |
| WO2022066934A2 (en) | 2020-09-23 | 2022-03-31 | Codiak Biosciences, Inc. | Process for preparing extracellular vesicles |
| US20220168330A1 (en) | 2020-11-09 | 2022-06-02 | Takeda Pharmaceutical Company Limited | Antibody drug conjugates |
| CA3217107A1 (en) | 2021-05-13 | 2022-11-17 | Daniel J. CLOUTIER | Combination of a tlr8 modulating compound and anti-hbv sirna therapeutics |
| US20220389394A1 (en) | 2021-05-18 | 2022-12-08 | Gilead Sciences, Inc. | METHODS OF USING FLT3L-Fc FUSION PROTEINS |
| JP2024522594A (en) | 2021-06-23 | 2024-06-21 | ギリアード サイエンシーズ, インコーポレイテッド | Diacylglycerol kinase modulating compounds |
| US11976072B2 (en) | 2021-06-23 | 2024-05-07 | Gilead Sciences, Inc. | Diacylglycerol kinase modulating compounds |
| IL309378A (en) | 2021-06-23 | 2024-02-01 | Gilead Sciences Inc | Diacylglyercol kinase modulating compounds |
| US11999733B2 (en) | 2021-06-23 | 2024-06-04 | Gilead Sciences, Inc. | Diacylglycerol kinase modulating compounds |
| JP2024526874A (en) | 2021-07-23 | 2024-07-19 | イミューンセンサー セラピューティクス、インコーポレイテッド | STING agonist combination therapy with cytokines |
| EP4137499A1 (en) | 2021-08-17 | 2023-02-22 | Ustav organicke chemie a biochemie AV CR, v.v.i. | 7-substituted 7-deazaadenine-containing 2,3 -cyclic dinucleotides |
| AU2023229142A1 (en) | 2022-03-02 | 2024-10-03 | Daiichi Sankyo Company, Limited | METHOD FOR PRODUCING Fc-CONTAINING MOLECULE |
| CN115417906B (en) * | 2022-09-14 | 2024-02-27 | 杭州星鳌生物科技有限公司 | Cyclic dinucleotide metal compound and preparation method and application thereof |
| GB202304385D0 (en) | 2023-03-24 | 2023-05-10 | Prostate Cancer Res | Combinatorial IL-15 therapy |
Family Cites Families (96)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9716557D0 (en) | 1997-08-06 | 1997-10-08 | Glaxo Group Ltd | Benzylidene-1,3-dihydro-indol-2-one derivatives having anti-cancer activity |
| AU2004275696B2 (en) | 2003-07-28 | 2010-02-18 | Karagen Pharmaceuticals, Inc. | Method for attenuating virulence of microbial pathogens and for inhibiting microbial biofilm formation |
| US7569555B2 (en) | 2004-03-15 | 2009-08-04 | Karaolis David K R | Method for stimulating the immune, inflammatory or neuroprotective response |
| EP1782826A1 (en) | 2005-11-08 | 2007-05-09 | GBF Gesellschaft für Biotechnologische Forschung mbH | PQS and c-diGMP and its conjugates as adjuvants and their uses in pharmaceutical compositions |
| WO2010017248A2 (en) | 2008-08-04 | 2010-02-11 | University Of Miami | Sting (stimulator of interferon genes), a regulator of innate immune responses |
| WO2011003025A1 (en) | 2009-07-01 | 2011-01-06 | Rutgers, The State University Of New Jersey | Synthesis of cyclic diguanosine monophosphate and thiophosphate analogs thereof |
| US9061048B2 (en) | 2010-12-15 | 2015-06-23 | The Regents Of The University Of California | Cyclic di-AMP induction of type I interferon |
| JP5650780B2 (en) | 2012-04-04 | 2015-01-07 | 日東電工株式会社 | Vaccine composition |
| KR20150004416A (en) | 2012-04-30 | 2015-01-12 | 글렌 엔. 바버 | Modulating immune responses |
| CN104507538B (en) | 2012-06-08 | 2018-04-06 | 艾杜罗生物科技公司 | The composition and method of immunotherapy for cancer |
| CN105008381B (en) | 2012-12-13 | 2018-08-07 | 艾杜罗生物科技公司 | Including composition and its preparation and application with the stereochemical ring purine dinucleotides of determination |
| WO2014099824A1 (en) | 2012-12-19 | 2014-06-26 | Board Of Regents, The University Of Texas System | Pharmaceutical targeting of a mammalian cyclic di-nucleotide signaling pathway |
| EA201590397A8 (en) * | 2012-12-27 | 2016-08-31 | Адуро Биотек, Инк. | CONTRIBUTING TO ANTIGENIC SEQUENCE IN LISTERIA PARTNERS FOR HYBRIDIZATION, PRESENTING SIGNAL PEPTIDES, AND METHODS FOR THEIR RECEIVING AND APPLICATION |
| JP6153116B2 (en) | 2013-01-09 | 2017-06-28 | 国立大学法人東北大学 | Triazole-linked cyclic dinucleotide analogs |
| WO2014144666A2 (en) * | 2013-03-15 | 2014-09-18 | The University Of Chicago | Methods and compositions related to t-cell activity |
| WO2014179335A1 (en) | 2013-04-29 | 2014-11-06 | Memorial Sloan Kettering Cancer Center | Compositions and methods for altering second messenger signaling |
| ES2822584T3 (en) * | 2013-05-03 | 2021-05-04 | Univ California | Induction of cyclic dinucleotides of type I interferon |
| CN105188373B (en) * | 2013-05-18 | 2017-09-22 | 艾杜罗生物科技公司 | Suppress the composition and method of " interferon gene stimulates the protein " dependent signals conduction |
| US9549944B2 (en) | 2013-05-18 | 2017-01-24 | Aduro Biotech, Inc. | Compositions and methods for inhibiting “stimulator of interferon gene”—dependent signalling |
| SI2996473T1 (en) * | 2013-05-18 | 2019-12-31 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene"-dependent signalling |
| US10176292B2 (en) | 2013-07-31 | 2019-01-08 | Memorial Sloan-Kettering Cancer Center | STING crystals and modulators |
| WO2015061294A2 (en) | 2013-10-21 | 2015-04-30 | Philadelphia Health & Education Corporation D/B/A/ | Use of sting agonists to treat chronic hepatitis b virus infection |
| JP2016538344A (en) | 2013-11-19 | 2016-12-08 | ザ・ユニバーシティ・オブ・シカゴThe University Of Chicago | Use of STING agonists as cancer treatments |
| US10421971B2 (en) * | 2014-01-15 | 2019-09-24 | The University Of Chicago | Anti-tumor therapy |
| CN103908468B (en) | 2014-04-21 | 2017-02-08 | 上海捌加壹医药科技有限公司 | Application of cyclic dinucleotide cGAMP in preparing anti-tumor medicaments |
| WO2015185565A1 (en) | 2014-06-04 | 2015-12-10 | Glaxosmithkline Intellectual Property Development Limited | Cyclic di-nucleotides as modulators of sting |
| WO2016079899A1 (en) | 2014-11-20 | 2016-05-26 | 国立研究開発法人医薬基盤・健康・栄養研究所 | NOVEL Th1-INDUCING ADJUVANT COMPRISING COMBINATION OF DIFFERENT NUCLEIC ACID ADJUVANTS, AND USE OF SAME |
| EP3233191A1 (en) | 2014-12-16 | 2017-10-25 | Invivogen | Combined use of a chemotherapeutic agent and a cyclic dinucleotide for cancer treatment |
| EP3546473A1 (en) | 2014-12-16 | 2019-10-02 | Kayla Therapeutics | Cyclic [(2',5')p(3',5')p]-dinucleotides for cytokine induction |
| EP3233089A4 (en) | 2014-12-17 | 2018-11-14 | Lipogen LLC | Method of treating cancer with cgamp or cgasmp |
| GB201501462D0 (en) | 2015-01-29 | 2015-03-18 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| EP3268035A4 (en) | 2015-03-10 | 2018-10-31 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
| WO2016176222A1 (en) | 2015-04-30 | 2016-11-03 | University Of Washington | Cgas in systemic lupus erythematosus (sle) |
| WO2016201450A2 (en) | 2015-06-11 | 2016-12-15 | University Of Miami | Cancer treatment and diagnosis |
| CN104962561A (en) * | 2015-06-25 | 2015-10-07 | 河南农业大学 | RNA aptamer and detection method for enzymatic cGAMP generation amount detection |
| CN106318997A (en) | 2015-07-03 | 2017-01-11 | 聊城市奥润生物医药科技有限公司 | Efficient preparation and purification method of thio- (seleno-) phosphoric acid cyclic di-nucleotide cGAMP |
| WO2017011444A1 (en) | 2015-07-13 | 2017-01-19 | The Wistar Institute Of Anatomy And Biology | Methods and compositions for treating b cell cancers |
| TW201717968A (en) | 2015-07-14 | 2017-06-01 | 春季銀行製藥公司 | Compounds and compositions that induce RIG-I and other pattern recognition receptors |
| SG10202010609QA (en) | 2015-08-13 | 2020-11-27 | Merck Sharp & Dohme | Cyclic di-nucleotide compounds as sting agonists |
| AU2016343993A1 (en) | 2015-10-28 | 2018-05-10 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene"-dependent signalling |
| US20170146519A1 (en) | 2015-11-20 | 2017-05-25 | Oregon Health & Science University | Sting agonists and methods of selecting sting agonists |
| TWI704154B (en) | 2015-12-03 | 2020-09-11 | 英商葛蘭素史克智慧財產發展有限公司 | Novel compounds |
| WO2017100305A2 (en) | 2015-12-07 | 2017-06-15 | Opi Vi - Ip Holdco Llc | Composition of antibody construct-agonist conjugates and methods of use thereof |
| US20170158772A1 (en) | 2015-12-07 | 2017-06-08 | Opi Vi - Ip Holdco Llc | Compositions of antibody construct - agonist conjugates and methods of use thereof |
| CN106540260A (en) | 2015-12-09 | 2017-03-29 | 聊城市奥润生物医药科技有限公司 | Interferon gene stimulatory protein(SP)(STING)Application of the agonist in anti-alzheimer's disease |
| CN106554416B (en) | 2015-12-09 | 2019-03-15 | 聊城市奥润生物医药科技有限公司 | A kind of application of anti-PD-L1 Humanized monoclonal antibodies joint interferon gene stimulates the protein (STING) agonist in antitumor |
| US20180369268A1 (en) | 2015-12-16 | 2018-12-27 | Aduro Biotech, Inc. | Methods for identifying inhibitors of "stimulator of interferon gene"- dependent interferon production |
| WO2017123657A1 (en) | 2016-01-11 | 2017-07-20 | Gary Glick | Cyclic dinucleotides for treating conditions associated with sting activity such as cancer |
| JP6867395B2 (en) | 2016-01-11 | 2021-04-28 | イネイト・テューマー・イミュニティ・インコーポレイテッドInnate Tumor Immunity, Inc. | Circular dinucleotides for treating symptoms associated with STING activity, such as cancer |
| US20170239283A1 (en) | 2016-02-23 | 2017-08-24 | Providence Health & Services - Oregon | Use of sting agonists to treat virally-induced and pre-malignant growths |
| AU2017225769B2 (en) | 2016-03-02 | 2023-01-05 | The Board Of Regents Of The University Of Texas System | Sting activating nanovaccine for immunotherapy |
| US20190070212A1 (en) | 2016-03-11 | 2019-03-07 | Spring Bank Pharmaceuticals, Inc. | Compounds and compositions for the treatment of infections |
| JP6724156B2 (en) | 2016-03-16 | 2020-07-15 | アンスティテュ・クリー | Method for preparing viral particles comprising cyclic dinucleotides and use of said particles for treating cancer |
| NZ746112A (en) * | 2016-03-18 | 2023-01-27 | Immune Sensor Llc | Cyclic di-nucleotide compounds and methods of use |
| CN106539757A (en) | 2016-03-20 | 2017-03-29 | 聊城市奥润生物医药科技有限公司 | Application of the ring dinucleotide cGAMP- liposomees in antitumor |
| PE20181920A1 (en) | 2016-04-07 | 2018-12-11 | Glaxosmithkline Ip Dev Ltd | USEFUL HETEROCYCLIC AMIDES AS PROTEIN MODULATORS |
| AU2017247806B2 (en) | 2016-04-07 | 2019-11-14 | Glaxosmithkline Intellectual Property Development Limited | Heterocyclic amides useful as protein modulators |
| EP3448393A1 (en) | 2016-04-25 | 2019-03-06 | Invivogen | Novel complexes of immunostimulatory compounds, and uses thereof |
| CN107412260B (en) | 2016-05-23 | 2022-07-19 | 北京大学 | cGAS-STING pathway activators and uses thereof |
| WO2017218358A1 (en) | 2016-06-13 | 2017-12-21 | The Regents Of The University Of California | FLUORESCENT BIOSENSOR FOR 2', 3'-cGAMP |
| CN106552265A (en) | 2016-06-21 | 2017-04-05 | 聊城市奥润生物医药科技有限公司 | STING agonist and application of the IDO1 inhibitor drug combinations in antitumor |
| WO2017223422A1 (en) | 2016-06-24 | 2017-12-28 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| WO2018009466A1 (en) | 2016-07-05 | 2018-01-11 | Aduro Biotech, Inc. | Locked nucleic acid cyclic dinucleotide compounds and uses thereof |
| TW201803886A (en) | 2016-07-06 | 2018-02-01 | 史貝羅威生物科學有限公司 | Compounds, compositions, and methods for the treatment of disease |
| IL264049B2 (en) | 2016-07-06 | 2023-11-01 | Sperovie Biosciences Inc | Compounds, compositions, and methods for the treatment of disease |
| WO2018013887A1 (en) | 2016-07-15 | 2018-01-18 | Sperovie Biosciences, Inc. | Compounds, compositions, and methods for the treatment of disease |
| WO2018029256A1 (en) | 2016-08-09 | 2018-02-15 | Aarhus Universitet | Modulation of ifi16 and sting activity |
| EP3922279A1 (en) | 2016-08-30 | 2021-12-15 | Dana Farber Cancer Institute, Inc. | Drug delivery compositions and uses thereof |
| US20190345191A1 (en) | 2016-08-31 | 2019-11-14 | Innate Tumor Immunity, Inc. | Cyclic dinucleotide analogs for treating conditions associated with sting (stimulator of interferon genes) activity |
| US11052149B2 (en) | 2016-09-19 | 2021-07-06 | The University Of North Carolina At Chapel Hill | Methods and compositions for inducing an immune response |
| US10537590B2 (en) | 2016-09-30 | 2020-01-21 | Boehringer Ingelheim International Gmbh | Cyclic dinucleotide compounds |
| TN2020000158A1 (en) | 2016-10-04 | 2022-04-04 | Merck Sharp & Dohme | BENZO[b]THIOPHENE COMPOUNDS AS STING AGONISTS |
| WO2018065360A1 (en) | 2016-10-07 | 2018-04-12 | Biolog Life Science Institute Forschungslabor Und Biochemica-Vertrieb Gmbh | Cyclic dinucleotides containing benzimidazole, method for the production of same, and use of same to activate stimulator of interferon genes (sting)-dependent signaling pathways |
| AU2017341735B2 (en) | 2016-10-11 | 2022-01-13 | Helmholtz Center for Infection Research | Hepatitis C virus immunogenic compositions comprising as an adjuvant a cyclic dinucleotide or an archaeosome and methods of use thereof |
| JOP20170188A1 (en) | 2016-11-25 | 2019-01-30 | Janssen Biotech Inc | Cyclic dinucleotides as sting agonists |
| JOP20170192A1 (en) | 2016-12-01 | 2019-01-30 | Takeda Pharmaceuticals Co | Cyclic dinucleotide |
| US20200113924A1 (en) | 2016-12-20 | 2020-04-16 | Merck Sharp & Dohme Corp. | Cyclic dinucleotide sting agonists for cancer treatment |
| JP2020511420A (en) | 2016-12-20 | 2020-04-16 | メルク・シャープ・アンド・ドーム・コーポレーションMerck Sharp & Dohme Corp. | Combination of PD-1 antagonists with cyclic dinucleotide STING agonists for the treatment of cancer |
| WO2018119117A1 (en) | 2016-12-22 | 2018-06-28 | The Regents Of The University Of California | Methods of producing cyclic dinucleotides |
| US20200085782A1 (en) | 2016-12-22 | 2020-03-19 | Mavupharma, Inc. | Compositions and methods of enhancing or augmenting type i ifn production |
| CA3047589A1 (en) | 2016-12-22 | 2018-06-28 | Mavupharma, Inc. | Phosphodiesterase inhibitors and methods of microbial treatment |
| WO2018140831A2 (en) | 2017-01-27 | 2018-08-02 | Silverback Therapeutics, Inc. | Tumor targeting conjugates and methods of use thereof |
| JP7275031B2 (en) | 2017-01-27 | 2023-05-17 | ヤンセン バイオテツク,インコーポレーテツド | Cyclic dinucleotides as STING agonists |
| AU2018212787B2 (en) | 2017-01-27 | 2023-10-26 | Janssen Biotech, Inc. | Cyclic dinucleotides as sting agonists |
| NZ755780A (en) | 2017-02-01 | 2023-10-27 | Modernatx Inc | Rna cancer vaccines |
| JP2018131427A (en) | 2017-02-17 | 2018-08-23 | 国立研究開発法人理化学研究所 | Technology for controlling immune cells |
| CA3053568A1 (en) | 2017-02-21 | 2018-08-30 | Board Of Regents, The University Of Texas System | Cyclic dinucleotides as agonists of stimulator of interferon gene dependent signalling |
| JOP20190218A1 (en) | 2017-03-22 | 2019-09-22 | Boehringer Ingelheim Int | Modified cyclic dinucleotide compounds |
| WO2018198084A1 (en) | 2017-04-27 | 2018-11-01 | Lupin Limited | Cyclic di-nucleotide compounds with tricyclic nucleobases |
| US11466047B2 (en) | 2017-05-12 | 2022-10-11 | Merck Sharp & Dohme Llc | Cyclic di-nucleotide compounds as sting agonists |
| EP3431484A1 (en) | 2017-07-21 | 2019-01-23 | Ludwig-Maximilians-Universität München | A fluorescent cyclic dinucleotide and its use in methods of identifying substances having an ability to modulate the cgas/sting pathway |
| CN107335049B (en) | 2017-08-18 | 2019-10-18 | 中国药科大学 | Application of the composite family type cyclic peptide compounds as cGAS-STING signal pathway inhibitor |
| JP7311514B2 (en) | 2017-08-30 | 2023-07-19 | ベイジン シュエンイー ファーマサイエンシズ カンパニー, リミテッド | Cyclic Dinucleotides as Interferon Gene Stimulator Modulators |
| EP3675859A4 (en) | 2017-08-31 | 2021-06-30 | Sperovie Biosciences, Inc. | Compounds, compositions, and methods for the treatment of disease |
| EP3768685A1 (en) | 2018-03-23 | 2021-01-27 | Takeda Pharmaceutical Company Limited | Sting modulator compounds with sulfamate linkages, and methods of making and using |
| CN108498529A (en) | 2018-06-20 | 2018-09-07 | 福建师范大学 | Dnmt rna inhibitor and cGAMP pharmaceutical compositions for tumor prevention treatment |
-
2017
- 2017-03-17 NZ NZ746112A patent/NZ746112A/en unknown
- 2017-03-17 DK DK17767683.0T patent/DK3429596T3/en active
- 2017-03-17 EP EP17767683.0A patent/EP3429596B1/en active Active
- 2017-03-17 SG SG10201912074PA patent/SG10201912074PA/en unknown
- 2017-03-17 MA MA043827A patent/MA43827A/en unknown
- 2017-03-17 PT PT177676830T patent/PT3429596T/en unknown
- 2017-03-17 CA CA3017524A patent/CA3017524A1/en active Pending
- 2017-03-17 LT LTEPPCT/US2017/023093T patent/LT3429596T/en unknown
- 2017-03-17 BR BR112018068748-0A patent/BR112018068748B1/en active IP Right Grant
- 2017-03-17 WO PCT/US2017/023093 patent/WO2017161349A1/en active Application Filing
- 2017-03-17 AU AU2017233068A patent/AU2017233068C1/en active Active
- 2017-03-17 HR HRP20221263TT patent/HRP20221263T1/en unknown
- 2017-03-17 HU HUE17767683A patent/HUE060396T2/en unknown
- 2017-03-17 EP EP19218894.4A patent/EP3692996A1/en active Pending
- 2017-03-17 CN CN202111504995.1A patent/CN114230625A/en active Pending
- 2017-03-17 IL IL280430A patent/IL280430B2/en unknown
- 2017-03-17 KR KR1020187029283A patent/KR102530488B1/en active IP Right Grant
- 2017-03-17 PL PL17767683.0T patent/PL3429596T3/en unknown
- 2017-03-17 SG SG11201807660QA patent/SG11201807660QA/en unknown
- 2017-03-17 CN CN201780030376.9A patent/CN109475570B/en active Active
- 2017-03-17 ES ES17767683T patent/ES2929628T3/en active Active
- 2017-03-17 JP JP2018568181A patent/JP6980198B2/en active Active
- 2017-03-17 CN CN202210558244.6A patent/CN114751950A/en active Pending
-
2018
- 2018-04-15 US US15/953,492 patent/US10519188B2/en active Active
- 2018-09-11 ZA ZA2018/06074A patent/ZA201806074B/en unknown
- 2018-09-14 MX MX2022001755A patent/MX2022001755A/en unknown
- 2018-09-17 IL IL261827A patent/IL261827B/en active IP Right Grant
-
2019
- 2019-06-11 US US16/438,153 patent/US11299512B2/en active Active
-
2020
- 2020-02-27 JP JP2020031617A patent/JP6980200B2/en active Active
-
2021
- 2021-11-05 JP JP2021180838A patent/JP2022017500A/en active Pending
-
2022
- 2022-03-17 US US17/697,247 patent/US20220340613A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11299512B2 (en) | Cyclic di-nucleotide compounds and methods of use | |
| US12065416B2 (en) | cGAS antagonist compounds | |
| JP6462006B2 (en) | Cyclic dinucleotides as modulators of STING | |
| US9988376B2 (en) | Benzothiophene derivatives as estrogen receptor inhibitors | |
| US20190292215A1 (en) | Compounds, compositions, and methods for the treatment of disease | |
| US12091387B2 (en) | Quinoline cGAS antagonist compounds | |
| EA037513B1 (en) | Cyclic dinucleotide compounds and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: IMMUNE SENSOR, LLC, TEXAS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:ZHONG, BOYU;REEL/FRAME:059951/0363 Effective date: 20190406 Owner name: IMMUNESENSOR THERAPEUTICS, INC., TEXAS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:IMMUNE SENSOR, LLC;REEL/FRAME:059951/0408 Effective date: 20190601 Owner name: THE BOARD OF REGENTS OF THE UNIVERSITY OF TEXAS SYSTEM, TEXAS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:SUN, LIJUN;SHI, HEPING;WEI, QI;AND OTHERS;SIGNING DATES FROM 20190610 TO 20190612;REEL/FRAME:059951/0395 |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: DOCKETED NEW CASE - READY FOR EXAMINATION |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: NON FINAL ACTION MAILED |