US20200055859A1 - Inhibitors of brutons tyrosine kinase - Google Patents
Inhibitors of brutons tyrosine kinase Download PDFInfo
- Publication number
- US20200055859A1 US20200055859A1 US16/361,494 US201916361494A US2020055859A1 US 20200055859 A1 US20200055859 A1 US 20200055859A1 US 201916361494 A US201916361494 A US 201916361494A US 2020055859 A1 US2020055859 A1 US 2020055859A1
- Authority
- US
- United States
- Prior art keywords
- substituted
- unsubstituted
- alkyl
- compound
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 [6*]/C([7*])=C(/[8*])C[Y]N1N=C(C2=CC=C(C[Ar])C=C2)C2=C(N)N=CN=C21 Chemical compound [6*]/C([7*])=C(/[8*])C[Y]N1N=C(C2=CC=C(C[Ar])C=C2)C2=C(N)N=CN=C21 0.000 description 49
- JFQJHJCTJYAIAM-UHFFFAOYSA-N CNC(C)(C)CC(C)C Chemical compound CNC(C)(C)CC(C)C JFQJHJCTJYAIAM-UHFFFAOYSA-N 0.000 description 5
- ZFHREWWRBHSDLZ-UHFFFAOYSA-N CN(CC[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1)C(C=C)=O Chemical compound CN(CC[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1)C(C=C)=O ZFHREWWRBHSDLZ-UHFFFAOYSA-N 0.000 description 3
- BGGXHOXATKKIEO-GPIIBWSTSA-N C#CC(=O)C(C)C.C/C=C/C(=O)C(C)C.C=CC(=O)C(C)C.C=CS(=O)(=O)C(C)C.CC(C)C(=O)/C=C/CN(C)C.COC/C=C/C(=O)C(C)C Chemical compound C#CC(=O)C(C)C.C/C=C/C(=O)C(C)C.C=CC(=O)C(C)C.C=CS(=O)(=O)C(C)C.CC(C)C(=O)/C=C/CN(C)C.COC/C=C/C(=O)C(C)C BGGXHOXATKKIEO-GPIIBWSTSA-N 0.000 description 2
- ZAZQUFKQXMNBTJ-UHFFFAOYSA-N C=CC(NCC[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1)=O Chemical compound C=CC(NCC[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1)=O ZAZQUFKQXMNBTJ-UHFFFAOYSA-N 0.000 description 2
- MWEQVPZDKGMOKD-UHFFFAOYSA-N CC(C)C1=NC(C(C)C)=C2C(N)=NC=NN12 Chemical compound CC(C)C1=NC(C(C)C)=C2C(N)=NC=NN12 MWEQVPZDKGMOKD-UHFFFAOYSA-N 0.000 description 2
- DBKRQICVRBRDJO-AVWXJUNHSA-N CC(C)C1CCCN(C(C)C)C1.CC(C)C1CCN(C(C)C)C1.CC(C)C1CCN(C(C)C)CC1.CC(C)CCN(C)C(C)C.CC(C)CCNC(C)C.CC(C)N[C@H]1CC[C@@H](C(C)C)CC1.CC(C)[C@@H]1CCCN(C(C)C)C1 Chemical compound CC(C)C1CCCN(C(C)C)C1.CC(C)C1CCN(C(C)C)C1.CC(C)C1CCN(C(C)C)CC1.CC(C)CCN(C)C(C)C.CC(C)CCNC(C)C.CC(C)N[C@H]1CC[C@@H](C(C)C)CC1.CC(C)[C@@H]1CCCN(C(C)C)C1 DBKRQICVRBRDJO-AVWXJUNHSA-N 0.000 description 2
- PCCAVXIMFOMEGI-UHFFFAOYSA-N NC1=NC=NC2=C1C(C1=CC=C(OC3=CC=CC=C3)C=C1)=CN2C1CCCNC1 Chemical compound NC1=NC=NC2=C1C(C1=CC=C(OC3=CC=CC=C3)C=C1)=CN2C1CCCNC1 PCCAVXIMFOMEGI-UHFFFAOYSA-N 0.000 description 2
- CNWZZPSLEUJBJW-UHFFFAOYSA-N *.B.C.CCC.C[Y]C Chemical compound *.B.C.CCC.C[Y]C CNWZZPSLEUJBJW-UHFFFAOYSA-N 0.000 description 1
- FPTUAFVEFQMPAS-SWSRPJROSA-N C#CC(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C#CC(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.CN(C)C/C=C/C(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 Chemical compound C#CC(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C#CC(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.CN(C)C/C=C/C(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 FPTUAFVEFQMPAS-SWSRPJROSA-N 0.000 description 1
- XAPPSYZUCBSHEU-GQCDPICSSA-N C#CC(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.CN(C)C/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.COC/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1 Chemical compound C#CC(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.CN(C)C/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.COC/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1 XAPPSYZUCBSHEU-GQCDPICSSA-N 0.000 description 1
- WMCJLVUCPLPRRE-HHPNXXKJSA-N C#CC(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.C/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.C=CS(=O)(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1 Chemical compound C#CC(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.C/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.C=CS(=O)(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1 WMCJLVUCPLPRRE-HHPNXXKJSA-N 0.000 description 1
- NRVDNNZXQMZCCS-JQCITGEHSA-N C.CN(C)C(OC(C)(C)C)N(C)C.N#CCNC1=CC=C(O[Ar])C=C1.N=CN.NC1=NC=NC2=C1N(C1=CC=C(O[Ar])C=C1)C=C2C1CCCNC1.[2H]CF.[C-]#[N+]/C(=C\O)C1CCCN(C(=O)OC(C)(C)C)C1.[C-]#[N+]C1=C(N)C(C2CCCN(C(=O)OC(C)(C)C)C2)=CN1C1=CC=C(O[Ar])C=C1.[C-]#[N+]CC1CCCN(C(=O)OC(C)(C)C)C1 Chemical compound C.CN(C)C(OC(C)(C)C)N(C)C.N#CCNC1=CC=C(O[Ar])C=C1.N=CN.NC1=NC=NC2=C1N(C1=CC=C(O[Ar])C=C1)C=C2C1CCCNC1.[2H]CF.[C-]#[N+]/C(=C\O)C1CCCN(C(=O)OC(C)(C)C)C1.[C-]#[N+]C1=C(N)C(C2CCCN(C(=O)OC(C)(C)C)C2)=CN1C1=CC=C(O[Ar])C=C1.[C-]#[N+]CC1CCCN(C(=O)OC(C)(C)C)C1 NRVDNNZXQMZCCS-JQCITGEHSA-N 0.000 description 1
- NBAGLHJWIPKOPZ-BABLKGGBSA-N C/C=C/C(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C=CS(=O)(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C=CS(=O)(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 Chemical compound C/C=C/C(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C=CS(=O)(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C=CS(=O)(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 NBAGLHJWIPKOPZ-BABLKGGBSA-N 0.000 description 1
- SYRKINONITVZHC-HAYHJZHISA-N C/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.CN(C)C/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.COC/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1 Chemical compound C/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.CN(C)C/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.COC/C=C/C(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1 SYRKINONITVZHC-HAYHJZHISA-N 0.000 description 1
- PKULTBBBDAIAOA-YINTVVIBSA-N C/C=C/C(=O)N1CCCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.CN(C)C/C=C/C(=O)N1CCCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.CN(C)C/C=C/C(=O)N[C@H]1CC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.COC/C=C/C(=O)N1CCCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1 Chemical compound C/C=C/C(=O)N1CCCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.CN(C)C/C=C/C(=O)N1CCCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1.CN(C)C/C=C/C(=O)N[C@H]1CC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.COC/C=C/C(=O)N1CCCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1 PKULTBBBDAIAOA-YINTVVIBSA-N 0.000 description 1
- BHQPOACREKDSLZ-QHHAFSJGSA-N C/C=C/C(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 Chemical compound C/C=C/C(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 BHQPOACREKDSLZ-QHHAFSJGSA-N 0.000 description 1
- JNNNPKPCKZZQAN-FGKWIGBISA-N C/C=C/C(=O)N[C@H]1CC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.C=CS(=O)(=O)N[C@H]1CC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.COC/C=C/C(=O)N[C@H]1CC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1 Chemical compound C/C=C/C(=O)N[C@H]1CC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.C=CS(=O)(=O)N[C@H]1CC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1.COC/C=C/C(=O)N[C@H]1CC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1 JNNNPKPCKZZQAN-FGKWIGBISA-N 0.000 description 1
- LJETUGTVRHSZOJ-QHHAFSJGSA-N C/C=C/C(N(CC1)CCC1[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1)=O Chemical compound C/C=C/C(N(CC1)CCC1[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1)=O LJETUGTVRHSZOJ-QHHAFSJGSA-N 0.000 description 1
- OPDJSSWXFCKILA-UHFFFAOYSA-N C1=CC2=C(C=C1)CCCC2.C1=CC2CCC1C2.C1=CC2CCCC2=C1.C1=CC2CCCCC2C1.C1=CCC=CC1.C1=CCCC1.C1=CCCC=C1.C1=CCCCC1.C1CC1.C1CC2CC(C1)C2.C1CC2CC12.C1CC2CC2C1.C1CC2CCC1C2.C1CC2CCC1CC2.C1CC2CCCC2C1.C1CCC1.C1CCC2CCCC2C1.C1CCC2CCCCC2C1.C1CCCC1.C1CCCCC1.C1CCCCCC1.C1CCCCCCC1 Chemical compound C1=CC2=C(C=C1)CCCC2.C1=CC2CCC1C2.C1=CC2CCCC2=C1.C1=CC2CCCCC2C1.C1=CCC=CC1.C1=CCCC1.C1=CCCC=C1.C1=CCCCC1.C1CC1.C1CC2CC(C1)C2.C1CC2CC12.C1CC2CC2C1.C1CC2CCC1C2.C1CC2CCC1CC2.C1CC2CCCC2C1.C1CCC1.C1CCC2CCCC2C1.C1CCC2CCCCC2C1.C1CCCC1.C1CCCCC1.C1CCCCCC1.C1CCCCCCC1 OPDJSSWXFCKILA-UHFFFAOYSA-N 0.000 description 1
- YJBQQODBBMQDFG-UHFFFAOYSA-N C1=CC2=C(C=C1)N=CC=N2.C1=CC2=C(C=CS2)S1.C1=CC=C2NC=CC2=C1.C1=CC=C2NC=NC2=C1.C1=CC=C2SC=CC2=C1.C1=CC=NC=C1.C1=CC=NC=C1.C1=CN=CC=N1.C1=CN=CC=N1.C1=CN=CN=C1.C1=CN=NC1.C1=CNC=C1.C1=CNC=N1.C1=CNN=C1.C1=COC=C1.C1=COC=N1.C1=CON=C1.C1=CSC=C1.C1=CSC=N1.C1=CSN=C1.C1=NC=NC=N1 Chemical compound C1=CC2=C(C=C1)N=CC=N2.C1=CC2=C(C=CS2)S1.C1=CC=C2NC=CC2=C1.C1=CC=C2NC=NC2=C1.C1=CC=C2SC=CC2=C1.C1=CC=NC=C1.C1=CC=NC=C1.C1=CN=CC=N1.C1=CN=CC=N1.C1=CN=CN=C1.C1=CN=NC1.C1=CNC=C1.C1=CNC=N1.C1=CNN=C1.C1=COC=C1.C1=COC=N1.C1=CON=C1.C1=CSC=C1.C1=CSC=N1.C1=CSN=C1.C1=NC=NC=N1 YJBQQODBBMQDFG-UHFFFAOYSA-N 0.000 description 1
- OMWAKOBMKASMFX-UHFFFAOYSA-N C1=CC2=C(C=C1)OCCO2.C1=CCNC1.C1=CNCCC1.C1=CNCCC1.C1=CSC=CN1.C1=NCCN1.C1=NCCO1.C1CC1.C1CC2CCC1O2.C1CCN2CCCCC2C1.C1CCNC1.C1CCNCC1.C1CCOC1.C1CN2CCC1NC2.C1CNNC1.C1COCCN1.C1CSCN1.C=S1(=O)CCCCCN1.O=C1CCCC1.O=C1CCCC1.O=C1CCCC1.O=C1CCCCN1.O=C1CCCN1.O=C1CCCO1.O=S1(=O)CCCC1 Chemical compound C1=CC2=C(C=C1)OCCO2.C1=CCNC1.C1=CNCCC1.C1=CNCCC1.C1=CSC=CN1.C1=NCCN1.C1=NCCO1.C1CC1.C1CC2CCC1O2.C1CCN2CCCCC2C1.C1CCNC1.C1CCNCC1.C1CCOC1.C1CN2CCC1NC2.C1CNNC1.C1COCCN1.C1CSCN1.C=S1(=O)CCCCCN1.O=C1CCCC1.O=C1CCCC1.O=C1CCCC1.O=C1CCCCN1.O=C1CCCN1.O=C1CCCO1.O=S1(=O)CCCC1 OMWAKOBMKASMFX-UHFFFAOYSA-N 0.000 description 1
- KLBZOFSTBACINB-UHFFFAOYSA-N C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(C)(C)OC(=O)N1CCCC(OS(C)(=O)=O)C1.CC(C)(C)OC(=O)NC1=NC=CC2=C1C(Br)=CN2.CC(C)(C)OC(=O)NC1=NC=CC2=C1C(C1=CC=C(OC3=CC=CC=C3)C=C1)=CN2.ClC1=CC=NC2=C1C=CN2.NC1=NC=CC2=C1C(C1=CC=C(OC3=CC=CC=C3)C=C1)=CN2C1CCCNC1.NC1=NC=CC2=C1C=CN2CC1=CC=CC=C1.NCC1=CC=CC=C1.OBOC1=CC=C(OC2=CC=CC=C2)C=C1.[Pd] Chemical compound C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(C)(C)OC(=O)N1CCCC(OS(C)(=O)=O)C1.CC(C)(C)OC(=O)NC1=NC=CC2=C1C(Br)=CN2.CC(C)(C)OC(=O)NC1=NC=CC2=C1C(C1=CC=C(OC3=CC=CC=C3)C=C1)=CN2.ClC1=CC=NC2=C1C=CN2.NC1=NC=CC2=C1C(C1=CC=C(OC3=CC=CC=C3)C=C1)=CN2C1CCCNC1.NC1=NC=CC2=C1C=CN2CC1=CC=CC=C1.NCC1=CC=CC=C1.OBOC1=CC=C(OC2=CC=CC=C2)C=C1.[Pd] KLBZOFSTBACINB-UHFFFAOYSA-N 0.000 description 1
- WLHSPVAXWDRPBV-UHFFFAOYSA-N C=CC(=O)N(C)CCC(C)C Chemical compound C=CC(=O)N(C)CCC(C)C WLHSPVAXWDRPBV-UHFFFAOYSA-N 0.000 description 1
- CXMKKEAJEPXAFD-UHFFFAOYSA-N C=CC(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C=CC(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C=CS(=O)(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1 Chemical compound C=CC(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C=CC(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.C=CS(=O)(=O)N1CCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)CC1 CXMKKEAJEPXAFD-UHFFFAOYSA-N 0.000 description 1
- CJEOCDTWPDPEBU-UHFFFAOYSA-N C=CC(=O)N1CCC(C(C)C)CC1 Chemical compound C=CC(=O)N1CCC(C(C)C)CC1 CJEOCDTWPDPEBU-UHFFFAOYSA-N 0.000 description 1
- MWNASUNUUFWMNK-UHFFFAOYSA-N C=CC(=O)N1CCCC1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C1N=CN=C2N Chemical compound C=CC(=O)N1CCCC1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C1N=CN=C2N MWNASUNUUFWMNK-UHFFFAOYSA-N 0.000 description 1
- KXEKOZMXMBGUQM-SNVBAGLBSA-N C=CC(=O)N1CCC[C@@H](C(C)C)C1 Chemical compound C=CC(=O)N1CCC[C@@H](C(C)C)C1 KXEKOZMXMBGUQM-SNVBAGLBSA-N 0.000 description 1
- XYFPWWZEPKGCCK-GOSISDBHSA-N C=CC(=O)N1CCC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1 Chemical compound C=CC(=O)N1CCC[C@@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C(N)N=CN=C32)C1 XYFPWWZEPKGCCK-GOSISDBHSA-N 0.000 description 1
- VHVZBJBHDIFWEG-SNVBAGLBSA-N C=CC(=O)N1CCC[C@@H]1CC(C)C Chemical compound C=CC(=O)N1CCC[C@@H]1CC(C)C VHVZBJBHDIFWEG-SNVBAGLBSA-N 0.000 description 1
- MWNASUNUUFWMNK-GOSISDBHSA-N C=CC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 Chemical compound C=CC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 MWNASUNUUFWMNK-GOSISDBHSA-N 0.000 description 1
- IKFFTYXJDCFYPF-WGKLFMOASA-N C=CC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.CC(C)(C)OC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.CC(C)(C)OC(=O)N1CCC[C@@H]1CO.NC1=C2C(=NC=N1)NN=C2C1=CC=C(OC2=CC=CC=C2)C=C1 Chemical compound C=CC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.CC(C)(C)OC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.CC(C)(C)OC(=O)N1CCC[C@@H]1CO.NC1=C2C(=NC=N1)NN=C2C1=CC=C(OC2=CC=CC=C2)C=C1 IKFFTYXJDCFYPF-WGKLFMOASA-N 0.000 description 1
- VHVZBJBHDIFWEG-JTQLQIEISA-N C=CC(=O)N1CCC[C@H]1CC(C)C Chemical compound C=CC(=O)N1CCC[C@H]1CC(C)C VHVZBJBHDIFWEG-JTQLQIEISA-N 0.000 description 1
- DCMZFZXOXBXCRU-UHFFFAOYSA-N C=CC(=O)NCCC(C)C Chemical compound C=CC(=O)NCCC(C)C DCMZFZXOXBXCRU-UHFFFAOYSA-N 0.000 description 1
- JXPYFPUCADHXEJ-PHIMTYICSA-N C=CC(=O)N[C@H]1CC[C@@H](C(C)C)CC1 Chemical compound C=CC(=O)N[C@H]1CC[C@@H](C(C)C)CC1 JXPYFPUCADHXEJ-PHIMTYICSA-N 0.000 description 1
- JXPYFPUCADHXEJ-XYPYZODXSA-N C=CC(=O)N[C@H]1CC[C@H](C(C)C)CC1 Chemical compound C=CC(=O)N[C@H]1CC[C@H](C(C)C)CC1 JXPYFPUCADHXEJ-XYPYZODXSA-N 0.000 description 1
- SSDFTYWIYVWCIE-WGSAOQKQSA-N C=CC(=O)N[C@H]1CC[C@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C2N=CN=C3N)CC1 Chemical compound C=CC(=O)N[C@H]1CC[C@H](N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C2N=CN=C3N)CC1 SSDFTYWIYVWCIE-WGSAOQKQSA-N 0.000 description 1
- ZGEMYXCQTVVYIT-QYQHSDTDSA-N C=CC(NCC[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1/C=C\S(N(CC1)CCC1[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1)(=O)=O)=O Chemical compound C=CC(NCC[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1/C=C\S(N(CC1)CCC1[n](c1ncnc(N)c11)nc1-c(cc1)ccc1Oc1ccccc1)(=O)=O)=O ZGEMYXCQTVVYIT-QYQHSDTDSA-N 0.000 description 1
- ZJPFXECQMIFGLO-UHFFFAOYSA-N C=CCC.C=CCOC(C)=O.CC.CC.CC.CC(=O)OCC(C)C.CC(=O)OCC1=CC=CC=C1.CC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2.CC(=O)OCCCCC(C)C.CC(C)=O.CCC.CCC(C)C.CCC1=CC=C(CO)C=C1.CCC1=CC=CC=C1.C[Si](C)(C)C Chemical compound C=CCC.C=CCOC(C)=O.CC.CC.CC.CC(=O)OCC(C)C.CC(=O)OCC1=CC=CC=C1.CC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2.CC(=O)OCCCCC(C)C.CC(C)=O.CCC.CCC(C)C.CCC1=CC=C(CO)C=C1.CCC1=CC=CC=C1.C[Si](C)(C)C ZJPFXECQMIFGLO-UHFFFAOYSA-N 0.000 description 1
- KEOXYNOFRLIAAM-UHFFFAOYSA-N CC#CC(=O)N1CCCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C2N=CN=C3N)C1 Chemical compound CC#CC(=O)N1CCCC(N2N=C(C3=CC=C(OC4=CC=CC=C4)C=C3)C3=C2N=CN=C3N)C1 KEOXYNOFRLIAAM-UHFFFAOYSA-N 0.000 description 1
- LWOBRLGIIVMUFF-LLVKDONJSA-N CC#CC(=O)N1CCC[C@@H](C(C)C)C1 Chemical compound CC#CC(=O)N1CCC[C@@H](C(C)C)C1 LWOBRLGIIVMUFF-LLVKDONJSA-N 0.000 description 1
- BLHJWGVNMPQXDY-LLVKDONJSA-N CC#CC(=O)N1CCC[C@@H]1CC(C)C Chemical compound CC#CC(=O)N1CCC[C@@H]1CC(C)C BLHJWGVNMPQXDY-LLVKDONJSA-N 0.000 description 1
- MCHPKOULFPNIOZ-LJQANCHMSA-N CC#CC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C1N=CN=C2N Chemical compound CC#CC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C1N=CN=C2N MCHPKOULFPNIOZ-LJQANCHMSA-N 0.000 description 1
- BLHJWGVNMPQXDY-NSHDSACASA-N CC#CC(=O)N1CCC[C@H]1CC(C)C Chemical compound CC#CC(=O)N1CCC[C@H]1CC(C)C BLHJWGVNMPQXDY-NSHDSACASA-N 0.000 description 1
- FFVGQPIBDNNCDG-TXEJJXNPSA-N CC#CC(=O)N[C@H]1CC[C@@H](C(C)C)CC1 Chemical compound CC#CC(=O)N[C@H]1CC[C@@H](C(C)C)CC1 FFVGQPIBDNNCDG-TXEJJXNPSA-N 0.000 description 1
- OHMIEQRWCWSFAY-JVOAEAJVSA-N CC(=O)C1=CC=C(OC2=CC=CC=C2)C=C1.CC(C)(C)OC(=O)N1CCCC(OS(C)(=O)=O)C1.N.N=CN.NC1=NC=NC2=C1C(C1=CC=C(OC3=CC=CC=C3)C=C1)=CN2.NC=O.[C-]#[N+]/C(C#N)=C(/CBr)C1=CC=C(OC2=CC=CC=C2)C=C1.[C-]#[N+]C1=C(N)NC=C1C1=CC=C(OC2=CC=CC=C2)C=C1.[C-]#[N+]CC#N Chemical compound CC(=O)C1=CC=C(OC2=CC=CC=C2)C=C1.CC(C)(C)OC(=O)N1CCCC(OS(C)(=O)=O)C1.N.N=CN.NC1=NC=NC2=C1C(C1=CC=C(OC3=CC=CC=C3)C=C1)=CN2.NC=O.[C-]#[N+]/C(C#N)=C(/CBr)C1=CC=C(OC2=CC=CC=C2)C=C1.[C-]#[N+]C1=C(N)NC=C1C1=CC=C(OC2=CC=CC=C2)C=C1.[C-]#[N+]CC#N OHMIEQRWCWSFAY-JVOAEAJVSA-N 0.000 description 1
- HZDSHYPUUXKELL-UHFFFAOYSA-N CC(C)(C)C1=CC=NC2=C1C(C(C)(C)C)=CN2C(C)(C)C.CC(C)(C)C1=CC=NN2C(C(C)(C)C)=CN(C(C)(C)C)C12.CC(C)(C)C1=CC=NN2C(C(C)(C)C)=NC(C(C)(C)C)=C12.CC(C)(C)C1=CN(C(C)(C)C)C2=C1C(C(C)(C)C)=NC=C2.CC(C)(C)C1=CN(C(C)(C)C)C2=C1C(C(C)(C)C)=NC=N2.CC(C)(C)C1=NC=CN2C(C(C)(C)C)=NC(C(C)(C)C)=C12.CC(C)(C)C1=NC=NC2=C1C(C(C)(C)C)=NN2C(C)(C)C.CC(C)(C)C1=NC=NN2C(C(C)(C)C)=NC(C(C)(C)C)=C12 Chemical compound CC(C)(C)C1=CC=NC2=C1C(C(C)(C)C)=CN2C(C)(C)C.CC(C)(C)C1=CC=NN2C(C(C)(C)C)=CN(C(C)(C)C)C12.CC(C)(C)C1=CC=NN2C(C(C)(C)C)=NC(C(C)(C)C)=C12.CC(C)(C)C1=CN(C(C)(C)C)C2=C1C(C(C)(C)C)=NC=C2.CC(C)(C)C1=CN(C(C)(C)C)C2=C1C(C(C)(C)C)=NC=N2.CC(C)(C)C1=NC=CN2C(C(C)(C)C)=NC(C(C)(C)C)=C12.CC(C)(C)C1=NC=NC2=C1C(C(C)(C)C)=NN2C(C)(C)C.CC(C)(C)C1=NC=NN2C(C(C)(C)C)=NC(C(C)(C)C)=C12 HZDSHYPUUXKELL-UHFFFAOYSA-N 0.000 description 1
- MAQSSRFNQPIBHQ-UHFFFAOYSA-N CC(C)(C)C1=CC=NC2=C1C(C(C)(C)C)=CN2C(C)(C)C.CC(C)(C)C1=CC=NN2C(C(C)(C)C)=CN(C(C)(C)C)C12.CC(C)(C)C1=CC=NN2C(C(C)(C)C)=NC(C(C)(C)C)=C12.CC(C)(C)C1=CN(C(C)(C)C)C2=C1C(C(C)(C)C)=NC=C2.CC(C)(C)C1=CN(C(C)(C)C)C2=C1C(C(C)(C)C)=NC=N2.CC(C)(C)C1=NC=CN2C(C(C)(C)C)=NC(C(C)(C)C)=C12.CC(C)(C)C1=NC=NC2C1C(C(C)(C)C)=NN2C(C)(C)C.CC(C)(C)C1=NC=NN2C(C(C)(C)C)=NC(C(C)(C)C)=C12 Chemical compound CC(C)(C)C1=CC=NC2=C1C(C(C)(C)C)=CN2C(C)(C)C.CC(C)(C)C1=CC=NN2C(C(C)(C)C)=CN(C(C)(C)C)C12.CC(C)(C)C1=CC=NN2C(C(C)(C)C)=NC(C(C)(C)C)=C12.CC(C)(C)C1=CN(C(C)(C)C)C2=C1C(C(C)(C)C)=NC=C2.CC(C)(C)C1=CN(C(C)(C)C)C2=C1C(C(C)(C)C)=NC=N2.CC(C)(C)C1=NC=CN2C(C(C)(C)C)=NC(C(C)(C)C)=C12.CC(C)(C)C1=NC=NC2C1C(C(C)(C)C)=NN2C(C)(C)C.CC(C)(C)C1=NC=NN2C(C(C)(C)C)=NC(C(C)(C)C)=C12 MAQSSRFNQPIBHQ-UHFFFAOYSA-N 0.000 description 1
- JWGLQBWGQWQHDY-UHFFFAOYSA-N CC(C)(C)OC(=O)N1CCCC(C(=O)NCC2=NN=CNC2=O)C1.CC(C)(C)OC(=O)N1CCCC(C(=O)O)C1.CC(C)(C)OC(=O)N1CCCC(C2=NC(I)=C3C(=O)NC=NN23)C1.CC(C)(C)OC(=O)N1CCCC(C2=NC=C3C(=O)NC=NN32)C1.CCOC(=O)C(=O)CN1C(=O)C2=C(C=CC=C2)C1=O.NCC1=NN=CNC1=O.NN.NNC(N)=S.O=C1NC(=S)NN=C1CN1C(=O)C2=C(C=CC=C2)C1=O.O=C1NC=NN=C1CN1C(=O)C2=C(C=CC=C2)C1=O.O=P(Cl)(Cl)Cl.SN=[IH] Chemical compound CC(C)(C)OC(=O)N1CCCC(C(=O)NCC2=NN=CNC2=O)C1.CC(C)(C)OC(=O)N1CCCC(C(=O)O)C1.CC(C)(C)OC(=O)N1CCCC(C2=NC(I)=C3C(=O)NC=NN23)C1.CC(C)(C)OC(=O)N1CCCC(C2=NC=C3C(=O)NC=NN32)C1.CCOC(=O)C(=O)CN1C(=O)C2=C(C=CC=C2)C1=O.NCC1=NN=CNC1=O.NN.NNC(N)=S.O=C1NC(=S)NN=C1CN1C(=O)C2=C(C=CC=C2)C1=O.O=C1NC=NN=C1CN1C(=O)C2=C(C=CC=C2)C1=O.O=P(Cl)(Cl)Cl.SN=[IH] JWGLQBWGQWQHDY-UHFFFAOYSA-N 0.000 description 1
- NTUKSDUNPUZVGR-UHFFFAOYSA-N CC(C)(C)OC(=O)N1CCCC(C(=O)O)C1.ClC1=CN=CC=N1.NC(C1=CC=C(OC2=CC=CC=C2)C=C1)C1=C(Cl)N=CC=N1.NC1=NC=CN2C(C3CCCNC3)=NC(C3=CC=C(OC4=CC=CC=C4)C=C3)=C12.O=CC1=CC=C(OC2=CC=CC=C2)C=C1.OC(C1=CC=C(OC2=CC=CC=C2)C=C1)C1=C(Cl)N=CC=N1.[Li]CCCC Chemical compound CC(C)(C)OC(=O)N1CCCC(C(=O)O)C1.ClC1=CN=CC=N1.NC(C1=CC=C(OC2=CC=CC=C2)C=C1)C1=C(Cl)N=CC=N1.NC1=NC=CN2C(C3CCCNC3)=NC(C3=CC=C(OC4=CC=CC=C4)C=C3)=C12.O=CC1=CC=C(OC2=CC=CC=C2)C=C1.OC(C1=CC=C(OC2=CC=CC=C2)C=C1)C1=C(Cl)N=CC=N1.[Li]CCCC NTUKSDUNPUZVGR-UHFFFAOYSA-N 0.000 description 1
- YBFZHUPLRUOMBF-UHFFFAOYSA-N CC(C)(C)OC(=O)N1CCCC(C2=NC(C3=CC=C(O[Ar])C=C3)=C3C(=O)NC=NN23)C1.NC1=NC=NN2C(C3CCCNC3)=NC(C3=CC=C(O[Ar])C=C3)=C12.OBOC1=CC=C(O[Ar])C=C1 Chemical compound CC(C)(C)OC(=O)N1CCCC(C2=NC(C3=CC=C(O[Ar])C=C3)=C3C(=O)NC=NN23)C1.NC1=NC=NN2C(C3CCCNC3)=NC(C3=CC=C(O[Ar])C=C3)=C12.OBOC1=CC=C(O[Ar])C=C1 YBFZHUPLRUOMBF-UHFFFAOYSA-N 0.000 description 1
- XYUOLGYAJMMXBB-PERWYRGUSA-N CC(C)(C)OC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=CC=C2)C2=C(N)N=CN=C21.CC(C)(C)OC(=O)N1CCC[C@@H]1CO.C[RaH].C[RaH].C[RaH].ClC(Cl)Cl.NC1=C2C(=NC=N1)NN=C2C1=CC=CC=C1.NC1=NC=NC2=C1C(I)=NN2.NC1=NC=NC2=C1C=NN2.OBOC1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@@H]1CN1N=C(C2=CC=CC=C2)C2=C(N)N=CN=C21.CC(C)(C)OC(=O)N1CCC[C@@H]1CO.C[RaH].C[RaH].C[RaH].ClC(Cl)Cl.NC1=C2C(=NC=N1)NN=C2C1=CC=CC=C1.NC1=NC=NC2=C1C(I)=NN2.NC1=NC=NC2=C1C=NN2.OBOC1=CC=CC=C1 XYUOLGYAJMMXBB-PERWYRGUSA-N 0.000 description 1
- KTBIREWDLOGBEX-UHFFFAOYSA-N CC(C)(C)[Y]N1N=C(C2=CC=C(C[Ar])C=C2)C2=C(N)N=CN=C21 Chemical compound CC(C)(C)[Y]N1N=C(C2=CC=C(C[Ar])C=C2)C2=C(N)N=CN=C21 KTBIREWDLOGBEX-UHFFFAOYSA-N 0.000 description 1
- VMBXHLRSUITXGL-UHFFFAOYSA-N CC(C)C(C(N)=C(N)N)=O Chemical compound CC(C)C(C(N)=C(N)N)=O VMBXHLRSUITXGL-UHFFFAOYSA-N 0.000 description 1
- HMVLRUJSAXEMJQ-UHFFFAOYSA-N CC(C)C1=CN(C(C)C)C2=C1C(N)=NC=C2 Chemical compound CC(C)C1=CN(C(C)C)C2=C1C(N)=NC=C2 HMVLRUJSAXEMJQ-UHFFFAOYSA-N 0.000 description 1
- PXIBTXSLYBMHNN-UHFFFAOYSA-N CC(C)C1=CN(C(C)C)C2=C1C(N)=NC=N2 Chemical compound CC(C)C1=CN(C(C)C)C2=C1C(N)=NC=N2 PXIBTXSLYBMHNN-UHFFFAOYSA-N 0.000 description 1
- PVFWHFKMZBRYHL-UHFFFAOYSA-N CC(C)C1=CN(C(C)C)C2=C1N=CN=C2N Chemical compound CC(C)C1=CN(C(C)C)C2=C1N=CN=C2N PVFWHFKMZBRYHL-UHFFFAOYSA-N 0.000 description 1
- SUJDRJPYHYZNRL-AATRIKPKSA-N CC(C)C1CCCN(C(=O)/C=C/C2=CNC=N2)C1 Chemical compound CC(C)C1CCCN(C(=O)/C=C/C2=CNC=N2)C1 SUJDRJPYHYZNRL-AATRIKPKSA-N 0.000 description 1
- GWURJMCJAVRNSN-FNORWQNLSA-N CC(C)C1CCCN(C(=O)/C=C/CN(C)CCO)C1 Chemical compound CC(C)C1CCCN(C(=O)/C=C/CN(C)CCO)C1 GWURJMCJAVRNSN-FNORWQNLSA-N 0.000 description 1
- IQROJIHHZBFSDV-GQCTYLIASA-N CC(C)C1CCCN(C(=O)/C=C/CN2CCOCC2)C1 Chemical compound CC(C)C1CCCN(C(=O)/C=C/CN2CCOCC2)C1 IQROJIHHZBFSDV-GQCTYLIASA-N 0.000 description 1
- IPGJGPMNBHWMSP-PPDFXLCASA-N CC(C)C1CCCN(C(C)C)C1.CC(C)C1CCN(C(C)C)C1.CC(C)C1CCN(C(C)C)CC1.CC(C)C1CCN(C(C)C)CC1.CC(C)CC1CCCN1C(C)C.CC(C)CCN(C)C(C)C.CC(C)CCNC(C)C.CC(C)N[C@H]1CC[C@@H](C(C)C)CC1.CC(C)[C@@H]1CCCN(C(C)C)C1 Chemical compound CC(C)C1CCCN(C(C)C)C1.CC(C)C1CCN(C(C)C)C1.CC(C)C1CCN(C(C)C)CC1.CC(C)C1CCN(C(C)C)CC1.CC(C)CC1CCCN1C(C)C.CC(C)CCN(C)C(C)C.CC(C)CCNC(C)C.CC(C)N[C@H]1CC[C@@H](C(C)C)CC1.CC(C)[C@@H]1CCCN(C(C)C)C1 IPGJGPMNBHWMSP-PPDFXLCASA-N 0.000 description 1
- MGFTWCRROMJFIY-AATRIKPKSA-N CC(C)C1CCN(C(=O)/C=C/CN(C)C)CC1 Chemical compound CC(C)C1CCN(C(=O)/C=C/CN(C)C)CC1 MGFTWCRROMJFIY-AATRIKPKSA-N 0.000 description 1
- CVEJIASBBWOZMY-ONEGZZNKSA-N CC(C)C1CCN(C(=O)/C=C/CN2CCOCC2)CC1 Chemical compound CC(C)C1CCN(C(=O)/C=C/CN2CCOCC2)CC1 CVEJIASBBWOZMY-ONEGZZNKSA-N 0.000 description 1
- CJPHAWNBCSHBJK-CFNZNRNTSA-N CC(C)C[C@@H]1CCCN1C(=O)/C=C/CN(C)C Chemical compound CC(C)C[C@@H]1CCCN1C(=O)/C=C/CN(C)C CJPHAWNBCSHBJK-CFNZNRNTSA-N 0.000 description 1
- COOFMVFBRKXNSC-DRDHIDPGSA-N CC(C)C[C@@H]1CCCN1C(=O)/C=C/CN1CCOCC1 Chemical compound CC(C)C[C@@H]1CCCN1C(=O)/C=C/CN1CCOCC1 COOFMVFBRKXNSC-DRDHIDPGSA-N 0.000 description 1
- CJPHAWNBCSHBJK-STMXVASLSA-N CC(C)C[C@H]1CCCN1C(=O)/C=C/CN(C)C Chemical compound CC(C)C[C@H]1CCCN1C(=O)/C=C/CN(C)C CJPHAWNBCSHBJK-STMXVASLSA-N 0.000 description 1
- GMBWBZHNAXFGKX-STMXVASLSA-N CC(C)[C@@H]1CCCN(C(=O)/C=C/CN(C)C)C1 Chemical compound CC(C)[C@@H]1CCCN(C(=O)/C=C/CN(C)C)C1 GMBWBZHNAXFGKX-STMXVASLSA-N 0.000 description 1
- IQROJIHHZBFSDV-CSPWOOARSA-N CC(C)[C@@H]1CCCN(C(=O)/C=C/CN2CCOCC2)C1 Chemical compound CC(C)[C@@H]1CCCN(C(=O)/C=C/CN2CCOCC2)C1 IQROJIHHZBFSDV-CSPWOOARSA-N 0.000 description 1
- NNPXLPFKADTTBY-DLPZHFSESA-N CC(C)[C@H]1CC[C@@H](NC(=O)/C=C/CN(C)C)CC1 Chemical compound CC(C)[C@H]1CC[C@@H](NC(=O)/C=C/CN(C)C)CC1 NNPXLPFKADTTBY-DLPZHFSESA-N 0.000 description 1
- YTMBSXSNGXSXEL-ZOSBBBERSA-N CC(C)[C@H]1CC[C@@H](NC(=O)/C=C/CN2CCOCC2)CC1 Chemical compound CC(C)[C@H]1CC[C@@H](NC(=O)/C=C/CN2CCOCC2)CC1 YTMBSXSNGXSXEL-ZOSBBBERSA-N 0.000 description 1
- NNPXLPFKADTTBY-GUUMZZRISA-N CC(C)[C@H]1CC[C@H](NC(=O)/C=C/CN(C)C)CC1 Chemical compound CC(C)[C@H]1CC[C@H](NC(=O)/C=C/CN(C)C)CC1 NNPXLPFKADTTBY-GUUMZZRISA-N 0.000 description 1
- YUGFMNZIROEBNV-WVLJPRKYSA-N CC1=CC(C)=N2C1=CC1=CC=C(CCC(=O)NCCN3CCN(C/C=C/C(=O)N4CCC[C@@H](N5N=C(C6=CC=C(OC7=CC=CC=C7)C=C6)C6=C(N)N=CN=C65)C4)CC3)N1B2(F)F Chemical compound CC1=CC(C)=N2C1=CC1=CC=C(CCC(=O)NCCN3CCN(C/C=C/C(=O)N4CCC[C@@H](N5N=C(C6=CC=C(OC7=CC=CC=C7)C=C6)C6=C(N)N=CN=C65)C4)CC3)N1B2(F)F YUGFMNZIROEBNV-WVLJPRKYSA-N 0.000 description 1
- MSRZCELFMRGUOI-ICHFDUKASA-N CC1=CC(C)=N2C1C/C1=C/C=C(/CCC(=O)NCCN3CCN(C/C=C/C(=O)N4CCC[C@@H](N5N=C(C6=CC=C(OC7=CC=CC=C7)C=C6)C6=C(N)N=CN=C65)C4)CC3)N1B2(F)F Chemical compound CC1=CC(C)=N2C1C/C1=C/C=C(/CCC(=O)NCCN3CCN(C/C=C/C(=O)N4CCC[C@@H](N5N=C(C6=CC=C(OC7=CC=CC=C7)C=C6)C6=C(N)N=CN=C65)C4)CC3)N1B2(F)F MSRZCELFMRGUOI-ICHFDUKASA-N 0.000 description 1
- FRLJPDWLFYJGPW-LXSSAFMLSA-N CC[C@H](CCC1)N1C(/C=C/CN(C)C)=O Chemical compound CC[C@H](CCC1)N1C(/C=C/CN(C)C)=O FRLJPDWLFYJGPW-LXSSAFMLSA-N 0.000 description 1
- NRRCZAZBMCWEAN-HYYQCAFJSA-N CN(C)C/C=C/C(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.COC/C=C/C(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.COC/C=C/C(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 Chemical compound CN(C)C/C=C/C(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.COC/C=C/C(=O)N(C)CCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21.COC/C=C/C(=O)NCCN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C(N)N=CN=C21 NRRCZAZBMCWEAN-HYYQCAFJSA-N 0.000 description 1
- DINVXRZOGZOFRM-FTUXVVSKSA-N CN(C)C/C=C/C(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C1N=CN=C2N Chemical compound CN(C)C/C=C/C(=O)N1CCC[C@@H]1CN1N=C(C2=CC=C(OC3=CC=CC=C3)C=C2)C2=C1N=CN=C2N DINVXRZOGZOFRM-FTUXVVSKSA-N 0.000 description 1
Images








Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
Definitions
- irreversible kinase inhibitor compounds Described herein are irreversible kinase inhibitor compounds, methods for synthesizing such irreversible inhibitors, and methods for using such irreversible inhibitors in the treatment of diseases. Further described herein are methods, assays and systems for determining an appropriate irreversible inhibitor of a protein, including a kinase.
- a kinase is a type of enzyme that transfers phosphate groups from high-energy donor molecules, such as ATP, to specific target molecules; the process is termed phosphorylation.
- Protein kinases which act on and modify the activity of specific proteins, are used to transmit signals and control complex processes in cells. Up to 518 different kinases have been identified in humans. Their enormous diversity and role in signaling makes them attractive targets for drug design.
- Btk Bruton's tyrosine kinase
- irreversible inhibitors of Btk Further described are irreversible inhibitors of Btk that form a covalent bond with a cysteine residue on Btk. Further described herein are irreversible inhibitors of other tyrosine kinases, wherein the other tyrosine kinases share homology with Btk by having a cysteine residue (including a Cys 481 residue) that forms a covalent bond with the irreversible inhibitor (such tyrosine kinases, are referred herein as “Btk tyrosine kinase cysteine homologs”).
- irreversible inhibitors of tyrosine kinases that have an accessible cysteine residue near an active site of the tyrosine kinase (referred herein as “Accessible Cysteine Kinases” or ACKs).
- irreversible inhibitors of any of the aforementioned tyrosine kinases in which the irreversible inhibitor includes a Michael acceptor moiety. Further described are such irreversible inhibitors in which the Michael acceptor moiety preferentially forms a covalent bond with the appropriate cysteine residue on the desired tyrosine kinase relative to forming a covalent bond with other biological molecules that contain an accessible SH moiety.
- Compounds described herein include those that have a structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), and pharmaceutically acceptable salts, solvates, esters, acids and prodrugs thereof.
- isomers and chemically protected forms of compounds having a structure represented by any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) are also provided.
- Formula (I) is as follows:
- salts of compounds of Formula (I) are provided pharmaceutically acceptable salts of compounds of Formula (I).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (I) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- substituents are optionally selected from among from a subset of the listed alternatives.
- L a is CH 2 , O, or NH.
- L a is O or NH.
- L a is O.
- Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- x is 2.
- Z is C( ⁇ O), OC( ⁇ O), NHC( ⁇ O), S( ⁇ O) x , OS( ⁇ O) x , or NHS( ⁇ O) x .
- Z is C( ⁇ O), NHC( ⁇ O), or NCH 3 C( ⁇ O).
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, and alkyleneheterocycloalkylene.
- Z is C( ⁇ O), NHC( ⁇ O), NR a C( ⁇ O), NR a S( ⁇ O) x , where x is 1 or 2, and R a is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl.
- R 7 and R 8 are H; and R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 8 alkylaminoalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted C 1 -C 8 alkylC 3 -C 6 cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C 2 -C 8 heterocycloalkyl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 8 alkylethers, C 1 -C 1 -C
- R 6 and R 8 are H; and R 7 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 8 alkylaminoalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted C 1 -C 8 alkylC 3 -C 6 cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C 2 -C 8 heterocycloalkyl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 8 alkylethers, C 1 -C 1 -C
- Y is an optionally substituted group selected from cycloalkylene or heterocycloalkylene.
- Z is C( ⁇ O), NHC( ⁇ O), NR a C( ⁇ O), NR a S( ⁇ O) x , where x is 1 or 2, and R a is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl.
- R 7 and R 8 are H; and R 6 is substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted C 1 -C 8 alkylC 3 -C 6 cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C 2 -C 8 heterocycloalkyl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 8 alkylethers, C 1 -C 8 alkylamides, or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 and R 8 are H; and R 7 is substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted C 1 -C 8 alkylC 3 -C 6 cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C 2 -C 8 heterocycloalkyl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 8 alkylethers, C 1 -C 8 alkylamides, or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- compositions which include a therapeutically effective amount of at least one of any of the compounds herein, or a pharmaceutically acceptable salt, pharmaceutically active metabolite, pharmaceutically acceptable prodrug, or pharmaceutically acceptable solvate.
- compositions provided herein further include a pharmaceutically acceptable diluent, excipient and/or binder.
- compositions formulated for administration by an appropriate route and means containing effective concentrations of one or more of the compounds provided herein, or pharmaceutically effective derivatives thereof, that deliver amounts effective for the treatment, prevention, or amelioration of one or more symptoms of diseases, disorders or conditions that are modulated or otherwise affected by tyrosine kinase activity, or in which tyrosine kinase activity is implicated, are provided.
- the effective amounts and concentrations are effective for ameliorating any of the symptoms of any of the diseases, disorders or conditions disclosed herein.
- a pharmaceutical composition containing: i) a physiologically acceptable carrier, diluent, and/or excipient; and ii) one or more compounds provided herein.
- provided herein are methods for treating a patient by administering a compound provided herein.
- a method of inhibiting the activity of tyrosine kinase(s), such as Btk, or of treating a disease, disorder, or condition, which benefit from inhibition of tyrosine kinase(s), such as Btk, in a patient which includes administering to the patient a therapeutically effective amount of at least one of any of the compounds herein, or pharmaceutically acceptable salt, pharmaceutically active metabolite, pharmaceutically acceptable prodrug, or pharmaceutically acceptable solvate.
- a compound disclosed herein for inhibiting Bruton's tyrosine kinase (Btk) activity or for the treatment of a disease, disorder, or condition, which benefit from inhibition of Bruton's tyrosine kinase (Btk) activity.
- compounds provided herein are administered to a human. In some embodiments, compounds provided herein are orally administered. In other embodiments, the pharmaceutical formulation that is formulated for a route of administration is selected from oral administration, parenteral administration, buccal administration, nasal administration, topical administration, or rectal administration.
- compounds provided herein are used for the formulation of a medicament for the inhibition of tyrosine kinase activity. In some other embodiments, compounds provided herein are used for the formulation of a medicament for the inhibition of Bruton's tyrosine kinase (Btk) activity.
- Btk Bruton's tyrosine kinase
- Articles of manufacture including packaging material, a compound or composition or pharmaceutically acceptable derivative thereof provided herein, which is effective for inhibiting the activity of tyrosine kinase(s), such as Btk, within the packaging material, and a label that indicates that the compound or composition, or pharmaceutically acceptable salt, pharmaceutically active metabolite, pharmaceutically acceptable prodrug, or pharmaceutically acceptable solvate thereof, is used for inhibiting the activity of tyrosine kinase(s), such as Btk, are provided.
- tyrosine kinases comprising a Bruton's tyrosine kinase, a Bruton's tyrosine kinase homolog, or a Btk tyrosine kinase cysteine homolog thereof covalently bound to an inhibitor having the structures:
- the inhibitor is covalently bound to a cysteine residue on the tyrosine kinase.
- a method for treating an autoimmune disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- the autoimmune disease is arthritis.
- the autoimmune disease is lupus.
- the autoimmune disease is inflammatory bowel disease (including Crohn's disease and ulcerative colitis), rheumatoid arthritis, psoriatic arthritis, osteoarthritis, Still's disease, juvenile arthritis, lupus, diabetes, myasthenia gravis, Hashimoto's thyroiditis, Ord's thyroiditis, Graves' disease Sjögren's syndrome, multiple sclerosis, Guillain-Barré syndrome, acute disseminated encephalomyelitis, Addison's disease, opsoclonus-myoclonus syndrome, ankylosing spondylitisis, antiphospholipid antibody syndrome, aplastic anemia, autoimmune hepatitis, coeliac disease, Goodpasture's syndrome, idiopathic thrombocytopenic purpura, optic neuritis, scleroderma, primary biliary cirrhosis, Reiter's syndrome, Takayasu's arteritis, temporal art
- a method for treating a heteroimmune condition or disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- the heteroimmune condition or disease is graft versus host disease, transplantation, transfusion, anaphylaxis, allergy, type I hypersensitivity, allergic conjunctivitis, allergic rhinitis, or atopic dermatitis.
- a method for treating an inflammatory disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- the inflammatory disease is asthma, inflammatory bowel disease (including Crohn's disease and ulcerative colitis), appendicitis, blepharitis, bronchiolitis, bronchitis, bursitis, cervicitis, cholangitis, cholecystitis, colitis, conjunctivitis, cystitis, dacryoadenitis, dermatitis, dermatomyositis, encephalitis, endocarditis, endometritis, enteritis, enterocolitis, epicondylitis, epididymitis, fasciitis, fibrositis, gastritis, gastroenteritis, hepatitis, hidradenitis suppurativa, laryngitis, mastitis, meningitis, myelitis myocarditis, myositis, nephritis, oophoritis, orchitis, osteitis, otitis
- a method for treating a cancer by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- the cancer is a B-cell proliferative disorder, e.g., diffuse large B cell lymphoma, follicular lymphoma, chronic lymphocytic lymphoma, chronic lymphocytic leukemia, B-cell prolymphocytic leukemia, lymphoplasmacytic lymphoma/Waldenström macroglobulinemia, splenic marginal zone lymphoma, plasma cell myeloma, plasmacytoma, extranodal marginal zone B cell lymphoma, nodal marginal zone B cell lymphoma, mantle cell lymphoma, mediastinal (thymic) large B cell lymphoma, intravascular large B cell lymphoma, primary effusion lymphoma, Burkitt's lymphoma/leukemia, or lymphomatoid granulomatosis.
- B-cell proliferative disorder e.g., diffuse large B cell lymphoma, follicular lymphoma, chronic lymphocytic lymph
- an anti-cancer agent is administered to the subject in addition to one of the above-mentioned compounds.
- the anti-cancer agent is an inhibitor of mitogen-activated protein kinase signaling, e.g., U0126, PD98059, PD184352, PD0325901, ARRY-142886, SB239063, SP600125, BAY 43-9006, wortmannin, or LY294002.
- a method for treating a thromboembolic disorder by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- the thromboembolic disorder is myocardial infarct, angina pectoris, reocclusion after angioplasty, restenosis after angioplasty, reocclusion after aortocoronary bypass, restenosis after aortocoronary bypass, stroke, transitory ischemia, a peripheral arterial occlusive disorder, pulmonary embolism, or deep venous thrombosis.
- a method for treating a mastocytosis by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- a method for treating a osteoporosis or bone resorption disorders by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- a method for treating lupus by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase.
- the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound.
- the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- a method for treating a heteroimmune condition or disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase.
- the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound.
- the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- a method for treating an inflammatory disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase.
- the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound.
- the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- a method for treating diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase.
- the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound.
- the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- a method for treating mastocytosis by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase.
- the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound.
- the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- a method for treating a osteoporosis or bone resorption disorders by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase.
- the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound.
- the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- an irreversible inhibitor of a kinase including a protein kinase, further including a tyrosine kinase.
- the inhibitor also has a moiety that binds an active site of the kinase.
- the kinases share homology with Btk by having a cysteine residue (including a Cys 481 residue) that forms a covalent bond with the irreversible inhibitor (such tyrosine kinases, are referred herein as “Btk kinase cysteine homologs”).
- Btk kinase cysteine homolog(s) are selected from the Tec family of kinases, the EGFR family of kinases, the Jak3 family of kinases and/or the Btk-Src family of kinases.
- the irreversible inhibitor is a selective irreversible inhibitor, including selectivity for a particular Btk kinase cysteine homolog over other Btk kinase cysteine homologs.
- the selective and irreversible inhibitor is an effective inhibitor for a kinase selected from Btk, a Btk homolog or a Btk kinase cysteine homolog, but is not an effective inhibitor for at least one other different kinase selected from kinase selected from Btk, a Btk homolog or a Btk kinase cysteine homolog.
- kinase inhibitors that selectively and irreversibly bind to a protein tyrosine kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog, in which the kinase inhibitor reversibly and non-selectively binds to a multiplicity of protein tyrosine kinases.
- the plasma half life of the kinase inhibitor is less than about 4 hours. In another embodiment the plasma half life of the kinase inhibitor is less than about 3 hours.
- kinase inhibitors that selectively and irreversibly bind to at least one of Btk, Jak3, Blk, Bmx, Tec, and Itk.
- kinase inhibitors that selectively and irreversibly bind to Btk.
- kinase inhibitors that selectively and irreversibly bind to Jak3.
- kinase inhibitors that selectively and irreversibly bind to Tec.
- kinase inhibitors that selectively and irreversibly bind to Btk and Tec.
- kinase inhibitors that selectively and irreversibly bind to Blk.
- kinase inhibitors that reversibly and non-selectively bind to a multiplicity of src-family protein kinase inhibitors.
- irreversible inhibitors that are identified using such methods, assays and systems.
- Such irreversible inhibitor comprise an active site binding moiety that binds to an active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog; a Michael acceptor moiety; and a moiety that links the active site binding moiety to the Michael acceptor moiety.
- the Michael acceptor moiety comprises and alkene and/or an alkyne moiety.
- the irreversible inhibitor is a selective irreversible inhibitor, including selectivity for a particular Btk kinase cysteine homolog over other Btk kinase cysteine homologs.
- the irreversible inhibitors have the structure of Formula (VII):
- kinase is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
- salts of compounds of Formula (VII) are provided pharmaceutically acceptable salts of compounds of Formula (VII).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (VII) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- Z is C( ⁇ O), NHC( ⁇ O), NCH 3 C( ⁇ O), or S( ⁇ O) 2 .
- x is 2.
- Z is C( ⁇ O), OC( ⁇ O), NHC( ⁇ O), S( ⁇ O) x , OS( ⁇ O) x , or NHS( ⁇ O) x .
- Z is C( ⁇ O), NHC( ⁇ O), or S( ⁇ O) 2 .
- R 7 and R 8 are independently selected from among H, unsubstituted C 1 -C 4 alkyl, substituted C 1 -C 4 alkyl, unsubstituted C 1 -C 4 heteroalkyl, and substituted C 1 -C 4 heteroalkyl; or R 7 and R 8 taken together form a bond. In yet other embodiments, each of R 7 and R 8 is H; or R and R 8 taken together form a bond.
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 8 alkylaminoalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 2 alkyl-N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —(C 1 -C 6 alkylamino), C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl containing 1 or 2 N atoms), or C 1 -C 4 alkyl(5- or 6-membered heterocycloalkyl containing 1 or 2 N atoms).
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, cycloalkylene, and heterocycloalkylene. In other embodiments, Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 4-, 5-, 6-, or 7-membered cycloalkylene, and 4-, 5-, 6-, or 7-membered heterocycloalkylene.
- Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 5- or 6-membered cycloalkylene, and 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- Y is a 5- or 6-membered cycloalkylene, or a 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- Y is a 4-, 5-, 6-, or 7-membered cycloalkylene ring; or Y is a 4-, 5-, 6-, or 7-membered heterocycloalkylene ring.
- test irreversible inhibitors in any of the aforementioned methods, assays and systems: such methods, assays and systems comprise a multiplicity of test irreversible inhibitors, in which the test irreversible inhibitors each have the same
- the multiplicity of test irreversible inhibitors is a panel of test irreversible inhibitors.
- the binding of the panel of test irreversible inhibitors to at least one kinase is determined (including a panel of kinases, further including a panel of kinases selected from Btk, Btk homologs, and Btk kinase cysteine homologs).
- the determined binding data is used to select and/or further design a selective irreversible inhibitor.
- Irreversible inhibitors described herein include those that have a structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), and pharmaceutically acceptable salts, solvates, esters, acids and prodrugs thereof.
- isomers and chemically protected forms of compounds having a structure represented by any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) are also provided.
- an irreversible inhibitor compound selected from among: 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one; 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)sulfonylethene; 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)sulfonylethene; 1-(3-(4
- compositions comprising the kinase inhibitors of any kinase inhibitor compound previously listed.
- the pharmaceutical formulation includes a pharmaceutical acceptable excipient.
- pharmaceutical formulations provided herein are administered to a human.
- the irreversible and/or selective kinase inhibitors provided herein are orally administered.
- the irreversible and/or selective kinase inhibitors provided herein are used for the formulation of a medicament for the inhibition of tyrosine kinase activity.
- the irreversible and/or selective kinase inhibitors provided herein are used for the formulation of a medicament for the inhibition of a kinase activity, including a tyrosine kinase activity, including a Btk activity, including a Btk homolog activity, including a Btk kinase cysteine homolog activity.
- any of the aforementioned aspects are further embodiments in which administration is enteral, parenteral, or both, and wherein (a) the effective amount of the compound is systemically administered to the mammal; (b) the effective amount of the compound is administered orally to the mammal; (c) the effective amount of the compound is intravenously administered to the mammal; (d) the effective amount of the compound administered by inhalation; (e) the effective amount of the compound is administered by nasal administration; or (f) the effective amount of the compound is administered by injection to the mammal; (g) the effective amount of the compound is administered topically (dermal) to the mammal; (h) the effective amount of the compound is administered by ophthalmic administration; or (i) the effective amount of the compound is administered rectally to the mammal.
- the pharmaceutical formulation is formulated for a route of administration selected from oral administration, parenteral administration, buccal administration, nasal administration, topical administration, or rectal administration.
- any of the aforementioned aspects are further embodiments comprising single administrations of the effective amount of the pharmaceutical formulation, including further embodiments in which (i) the pharmaceutical formulations is administered once; (ii) the pharmaceutical formulations is administered to the mammal once a day; (iii) the pharmaceutical formulations is administered to the mammal multiple times over the span of one day; (iv) continually; or (v) continuously.
- the method comprises a drug holiday, wherein the administration of the pharmaceutical formulations is temporarily suspended or the dose of the pharmaceutical formulations being administered is temporarily reduced; at the end of the drug holiday, dosing of the pharmaceutical formulations is resumed.
- the length of the drug holiday varies from 2 days to 1 year.
- test protein tyrosine kinase inhibitor that irreversibly and selectively binds to at least one protein kinase inhibitor selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- the test protein tyrosine kinase inhibitor is chemically modified to decrease the plasma half life to less than about 4 hours.
- the test protein tyrosine kinase inhibitor is chemically modified to decrease the plasma half life to about 3 hours.
- test protein tyrosine kinase inhibitor non-selectively and reversibly binds to a multiplicity of src-family protein tyrosine kinases.
- test protein kinase inhibitor has the structure of Formula (VII):
- kinase is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
- a method for treating a B-cell proliferative disorder or a mast cell proliferative disorder by administering to a subject in need thereof a test protein kinase inhibitor composition containing a therapeutically effective amount of a compound that forms a covalent bond (including an irreversible and/or selective covalent bond) with Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- the compound forms a covalent bound with the activated form of Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- the compound irreversibly inhibits Btk, a Btk homolog, or a Btk kinase cysteine homolog to which it is covalently bound.
- the compound forms a covalent bond (including an irreversible and/or selective covalent bond) with a cysteine residue on Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- a method for treating rheumatoid arthritis by administering to a subject in need thereof a test protein kinase inhibitor composition containing a therapeutically effective amount of a compound that forms a covalent bond (including an irreversible and/or selective covalent bond) with Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- the compound forms a covalent bound with the activated form of Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- the compound irreversibly inhibits Btk, a Btk homolog, or a Btk kinase cysteine homolog to which it is covalently bound.
- the compound forms a covalent bond (including an irreversible and/or selective covalent bond) with a cysteine residue on Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- a method for treating a disease characterized by hyperactive B-cells or hyperactive mast cells or both hyperactive B-cells and hyperactive mast cells by administering to a subject in need thereof a test protein kinase inhibitor composition containing a therapeutically effective amount of a compound that forms a covalent bond (including an irreversible and/or selective covalent bond) with Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- the compound forms a covalent bound with the activated form of Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- the compound irreversibly inhibits Btk, a Btk homolog, or a Btk kinase cysteine homolog to which it is covalently bound.
- the compound forms a covalent bond (including an irreversible and/or selective covalent bond) with a cysteine residue on Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- any of the aforementioned aspects are further embodiments comprising single administrations of the effective amount of the pharmaceutical formulation, including further embodiments in which (i) the pharmaceutical formulations is administered once; (ii) the pharmaceutical formulations is administered to the mammal once a day; (iii) the pharmaceutical formulations is administered to the mammal multiple times over the span of one day; (iv) continually; or (v) continuously.
- Also described herein is a method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog comprising:
- a method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog comprising:
- the at least one non-kinase molecule having at least one accessible SH group includes glutathione and/or hemoglobin.
- the desired irreversible inhibitor is selective for a particular kinase relative to other kinases, glutathione and hemoglobin.
- the methods, assays and systems for identifying an irreversible inhibitor of a kinase comprise contacting each kinase with an Activity Probe. In further embodiments, the methods, assays and systems for identifying an irreversible inhibitor of a kinase further comprise a panel of kinases comprising at least two kinases selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog.
- the panel of kinases comprises at least three such kinases, at least four such kinases, at least five such kinases, at least six such kinases, at least seven such kinases, at least eight such kinases, at least nine such kinases, or at least ten such kinases.
- steps (1) and (2) of the method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog is conducted in vivo.
- step (3) of the method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog is conducted in part using an Activity Probe.
- contacting a multiplicity of kinases selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog with a compound that comprises a Michael acceptor moiety is conducted in vivo.
- contacting at least one non-kinase molecule having at least one accessible SH group with the compound that comprises a Michael acceptor moiety is conducted in vivo.
- determining the covalent binding of the compound that comprises a Michael acceptor with the multiplicity of kinases and the at least one non-kinase molecule is conducted in part using an Activity Probe.
- the determining step uses mass spectrometry.
- the determining step uses fluorescence.
- a panel of kinases is contacted with at least one irreversible inhibitor.
- the panel of kinases is also contacted with an Activity Probe.
- the binding of an irreversible inhibitor to a kinase is determined from the binding of the Activity Probe to the kinase.
- the binding of the Activity Probe to a kinase is determined using fluorescence technique.
- the Activity Probe is compatible with flow cytometery.
- the binding of the irreversible inhibitor to one kinase is compared to the binding of the irreversible inhibitor to at least one other kinase.
- the panel of kinases is selected from Btk, Btk homologs, and Btk kinase cysteine homologs.
- the binding of an irreversible inhibitor to a kinase is determined by mass spectrometry.
- Activity Probes Bruton's tyrosine kinase (Btk), Btk homologs, and Btk kinase cysteine homologs. Further described are Activity Probes that include an irreversible inhibitor of Btk, a Btk homolog and/or a Btk kinase cysteine homolog; a linker moiety; and a reporter moiety. Further described are Activity Probes that include a Michael addition acceptor moiety in the structure of the Activity Probe.
- Activity Probes that form a covalent bond with a cysteine residue on Btk, a Btk homolog and/or a Btk kinase cysteine homolog. Also described herein are Activity Probes that form a non-covalent bond with a cysteine residue on Btk, a Btk homolog and/or a Btk kinase cysteine homolog.
- Activity Probes wherein the linker moiety is selected from a bond, an optionally substituted alkyl moiety, an optionally substituted heterocycle moiety, an optionally substituted amide moiety, a ketone moiety, an optionally substituted carbamate moiety, an ester moiety, or a combination thereof.
- the linker moiety comprises an optionally substituted heterocycle moiety.
- the optionally substituted heterocycle moiety comprises a piperazinyl-based moiety.
- the reporter moiety is selected from the group consisting of a label, a dye, a photocrosslinker, a cytotoxic compound, a drug, an affinity label, a photoaffinity label, a reactive compound, an antibody or antibody fragment, a biomaterial, a nanoparticle, a spin label, a fluorophore, a metal-containing moiety, a radioactive moiety, a novel functional group, a group that covalently or noncovalently interacts with other molecules, a photocaged moiety, an actinic radiation excitable moiety, a ligand, a photoisomerizable moiety, biotin, a biotin analogue, a moiety incorporating a heavy atom, a chemically cleavable group, a photocleavable group, a redox-active agent, an isotopically labeled moiety, a biophysical probe, a phosphorescent group, a chemiluminescent
- Activity Probes wherein the reporter moiety is a fluorophore.
- Activity Probes wherein the fluorophore is a Bodipy fluorophore.
- Activity Probes wherein the Bodipy fluorophore is a Bodipy FL fluorophore.
- Activity Probes wherein the inhibitor moiety is derived from an irreversible inhibitor of Btk, a Btk homolog and/or a Btk kinase cysteine homolog.
- the irreversible inhibitor is:
- Activity Probes wherein the probe selectively labels a phosphorylated conformation of Btk, a Btk homolog and/or a Btk kinase cysteine homolog.
- Activity Probes wherein the phosphorylated conformation of Btk, a Btk homolog and/or a Btk kinase cysteine homolog is either an active or inactive form of Btk, a Btk homolog and/or a Btk kinase cysteine homolog.
- Activity Probes wherein the phosphorylated conformation of Btk, a Btk homolog and/or a Btk kinase cysteine homolog is an active form of Btk, a Btk homolog and/or a Btk kinase cysteine homolog.
- a method for assessing the efficacy of a potential Btk, Btk homolog and/or Btk kinase cysteine homolog inhibitor in a mammal comprising administering a potential Btk, Btk homolog and/or Btk kinase cysteine homolog inhibitor to the mammal, administering the Activity Probe described herein to the mammal or to cells isolated from the mammal; measuring the activity of the reporter moiety of the Activity Probe, and comparing the activity of the reporter moiety to a standard.
- a method for assessing the pharmacodynamics of a Btk, Btk homolog and/or Btk kinase cysteine homolog inhibitor in a mammal comprising administering a Btk, Btk homolog and/or Btk kinase cysteine homolog inhibitor to the mammal, administering the Activity Probe presented herein to the mammal or to cells isolated from the mammal, and measuring the activity of the reporter moiety of the Activity Probe at different time points following the administration of the inhibitor.
- a method for in vitro labeling of Btk, a Btk homolog and/or a Btk kinase cysteine homolog comprising contacting an active Btk, Btk homolog and/or Btk kinase cysteine homolog with the Activity Probe described herein.
- a method for in vitro labeling of Btk, a Btk homolog and/or a Btk kinase cysteine homolog wherein the contacting step comprises incubating the active Btk, Btk homolog and/or Btk kinase cysteine homolog with the Activity Probe presented herein.
- a method for in vitro labeling of Btk, a Btk homolog and/or a Btk kinase cysteine homolog comprising contacting cells or tissues expressing the Btk, Btk homolog and/or Btk kinase cysteine homolog with an Activity Probe described herein.
- a method for detecting a labeled Btk, Btk homolog and/or Btk kinase cysteine homolog comprising separating proteins, the proteins comprising Btk, a Btk homolog and/or a Btk kinase cysteine homolog labeled by an Activity Probe described herein, by electrophoresis and detecting the Activity Probe by fluorescence.
- the irreversible inhibitor of a kinase further comprises an active site binding moiety. In yet further embodiments the irreversible inhibitor of a kinase further comprises a linker moiety that links the Michael acceptor moiety to the active binding moiety.
- the irreversible inhibitor of a kinase has the structure of Formula (VII):
- kinase is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
- the method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog comprising steps (1), (2), (3), and (4) further comprises analyzing the structure-function activity relationship between the structure of the linker moiety and/or the Michael acceptor moiety of each compound, and the binding and/or selectivity of each compound to at least one kinase.
- the method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog comprising steps (1), (2), (3), and (4) further comprises analyzing the structure-function activity relationship between the structure of Y—Z and/or
- each compound and the binding and/or selectivity of each compound to at least one kinase.
- the structure of the active site binding moiety of each compound is not varied. In another embodiment the structure of
- Also described herein is a method for improving the kinase selectivity of an inhibitor comprising use of any method previously listed.
- One aspect described herein is an assay comprising any of the methods previously listed. Another aspect described herein is system comprising any of the methods previously listed. In a further aspect described herein is an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog, wherein the inhibitor is identified using any methods described herein.
- the irreversible inhibitor is selective for one kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog over at least one other kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog.
- the irreversible inhibitor is selective for at least one kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog over at least one other non-kinase molecule having an accessible SH group.
- a pharmaceutical composition containing: i) a physiologically acceptable carrier, diluent, and/or excipient; and ii) one or more compounds provided herein.
- a method for treating an autoimmune disease or condition comprising administering to a patient in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- the autoimmune disease is selected from rheumatoid arthritis or lupus.
- a method for treating a B-cell proliferative disorder comprising administering to a patient in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- the B-cell proliferative disorder is diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia.
- a method for treating an inflammatory disease or condition comprising administering to a patient in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- a patient that has lymphoma is administered a pharmaceutical composition of any kinase inhibitor compound described herein which inhibits B cell receptor (BCR) signaling.
- BCR B cell receptor
- the inhibition of the BCR signaling by any kinase inhibitor compound described herein is correlated with the induction of apoptosis.
- a patient with lymphoma is selected for treatment with a pharmaceutical composition of any kinase inhibitor compound described herein based on a biomarker that indicates that the lymphoma in that patient has high levels of pErk or Erk transcriptional targets.
- the response to treatment with a pharmaceutical composition of any kinase inhibitor compound described herein is measured by a reduction in levels of pErk or Erk transcriptional targets.
- Standard techniques are optionally used for chemical syntheses, chemical analyses, pharmaceutical preparation, formulation, and delivery, and treatment of patients. Standard techniques are optionally used for recombinant DNA, oligonucleotide synthesis, and tissue culture and transformation (e.g., electroporation, lipofection). Reactions and purification techniques are performed using documented methodologies or as described herein.
- the terms used for complex moieties are to be read equivalently either from left to right or right to left.
- group alkylenecycloalkylene refers both to an alkylene group followed by a cycloalkylene group or as a cycloalkylene group followed by an alkylene group.
- a methylene is a diradical of a methyl group, that is, it is a —CH 2 — group; and an ethylene is a diradical of an ethyl group, i.e., —CH 2 CH 2 —.
- alkyl refers to an aliphatic hydrocarbon group.
- the alkyl moiety includes a “saturated alkyl” group, which means that it does not contain any alkene or alkyne moieties.
- the alkyl moiety also includes an “unsaturated alkyl” moiety, which means that it contains at least one alkene or alkyne moiety.
- An “alkene” moiety refers to a group that has at least one carbon-carbon double bond
- an “alkyne” moiety refers to a group that has at least one carbon-carbon triple bond.
- the alkyl moiety, whether saturated or unsaturated includes branched, straight chain, or cyclic moieties.
- an alkyl group includes a monoradical or a diradical (i.e., an alkylene group), and if a “lower alkyl” having 1 to 6 carbon atoms.
- C 1 -C x includes C 1 -C 2 , C 1 -C 3 . . . C 1 -C x .
- alkyl moiety optionally has 1 to 10 carbon atoms (whenever it appears herein, a numerical range such as “1 to 10” refers to each integer in the given range; e.g., “1 to 10 carbon atoms” means that the alkyl group is selected from a moiety having 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc., up to and including 10 carbon atoms, although the present definition also covers the occurrence of the term “alkyl” where no numerical range is designated).
- the alkyl group of the compounds described herein may be designated as “C 1 -C 4 alkyl” or similar designations.
- C 1 -C 4 alkyl indicates that there are one to four carbon atoms in the alkyl chain, i.e., the alkyl chain is selected from among methyl, ethyl, propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl, and t-butyl.
- C 1 -C 4 alkyl includes C 1 -C 2 alkyl and C 1 -C 3 alkyl.
- Alkyl groups are optionally substituted or unsubstituted.
- Typical alkyl groups include, but are in no way limited to, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tertiary butyl, pentyl, hexyl, ethenyl, propenyl, butenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like.
- alkenyl refers to a type of alkyl group in which the first two atoms of the alkyl group form a double bond that is not part of an aromatic group. That is, an alkenyl group begins with the atoms —C(R) ⁇ C(R)—R, wherein R refers to the remaining portions of the alkenyl group, which are either the same or different.
- the alkenyl moiety is optionally branched, straight chain, or cyclic (in which case, it is also known as a “cycloalkenyl” group).
- an alkenyl group includes a monoradical or a diradical (i.e., an alkenylene group). Alkenyl groups are optionally substituted.
- Non-limiting examples of an alkenyl group include —CH ⁇ CH 2 , —C(CH 3 ) ⁇ CH 2 , —CH ⁇ CHCH 3 , —C(CH 3 ) ⁇ CHCH 3 .
- Alkenylene groups include, but are not limited to, —CH ⁇ CH—, —C(CH 3 ) ⁇ CH—, —CH ⁇ CHCH 2 —, —CH ⁇ CHCH 2 CH 2 — and —C(CH 3 ) ⁇ CHCH 2 —.
- Alkenyl groups optionally have 2 to 10 carbons, and if a “lower alkenyl” having 2 to 6 carbon atoms.
- alkynyl refers to a type of alkyl group in which the first two atoms of the alkyl group form a triple bond. That is, an alkynyl group begins with the atoms —C ⁇ C—R, wherein R refers to the remaining portions of the alkynyl group, which is either the same or different.
- R refers to the remaining portions of the alkynyl group, which is either the same or different.
- the “R” portion of the alkynyl moiety may be branched, straight chain, or cyclic.
- an alkynyl group includes a monoradical or a diradical (i.e., an alkynylene group). Alkynyl groups are optionally substituted.
- Non-limiting examples of an alkynyl group include, but are not limited to, —C ⁇ CH, —C ⁇ CCH 3 , —C ⁇ CCH 2 CH 3 , —C ⁇ C—, and —C ⁇ CCH 2 —.
- Alkynyl groups optionally have 2 to 10 carbons, and if a “lower alkynyl” having 2 to 6 carbon atoms.
- alkoxy refers to a (alkyl)O— group, where alkyl is as defined herein.
- “Hydroxyalkyl” refers to an alkyl radical, as defined herein, substituted with at least one hydroxy group.
- Non-limiting examples of a hydroxyalkyl include, but are not limited to, hydroxymethyl, 2-hydroxyethyl, 2-hydroxypropyl, 3-hydroxypropyl, 1-(hydroxymethyl)-2-methylpropyl, 2-hydroxybutyl, 3-hydroxybutyl, 4-hydroxybutyl, 2,3-dihydroxypropyl, 1-(hydroxymethyl)-2-hydroxyethyl, 2,3-dihydroxybutyl, 3,4-dihydroxybutyl and 2-(hydroxymethyl)-3-hydroxypropyl.
- Alkoxyalkyl refers to an alkyl radical, as defined herein, substituted with an alkoxy group, as defined herein.
- Alkylaminoalkyl refers to an alkyl radical, as defined herein, substituted with an alkylamine, as defined herein.
- Hydroalkylaminoalkyl refers to an alkyl radical, as defined herein, substituted with an alkylamine, and alkylhydroxy, as defined herein.
- Alkoxyalkylaminoalkyl refers to an alkyl radical, as defined herein, substituted with an alkylamine and substituted with an alkylalkoxy, as defined herein.
- an “amide” is a chemical moiety with the formula —C(O)NHR or —NHC(O)R, where R is selected from among alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring carbon) and heteroalicyclic (bonded through a ring carbon).
- R is selected from among alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring carbon) and heteroalicyclic (bonded through a ring carbon).
- an amide moiety forms a linkage between an amino acid or a peptide molecule and a compound described herein, thereby forming a prodrug. Any amine, or carboxyl side chain on the compounds described herein can be amidified.
- esters refers to a chemical moiety with formula —COOR, where R is selected from among alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring carbon) and heteroalicyclic (bonded through a ring carbon). Any hydroxy, or carboxyl side chain on the compounds described herein can be esterified. The procedures and specific groups to make such esters are found in sources such as Greene and Wuts, Protective Groups in Organic Synthesis, 3 rd Ed., John Wiley & Sons, New York, N.Y., 1999, which is incorporated herein by reference for this disclosure.
- Rings refers to any covalently closed structure. Rings include, for example, carbocycles (e.g., aryls and cycloalkyls), heterocycles (e.g., heteroaryls and non-aromatic heterocycles), aromatics (e.g. aryls and heteroaryls), and non-aromatics (e.g., cycloalkyls and non-aromatic heterocycles). Rings can be optionally substituted. Rings can be monocyclic or polycyclic.
- ring system refers to one, or more than one ring.
- membered ring can embrace any cyclic structure.
- membered is meant to denote the number of skeletal atoms that constitute the ring.
- cyclohexyl, pyridine, pyran and thiopyran are 6-membered rings and cyclopentyl, pyrrole, furan, and thiophene are 5-membered rings.
- fused refers to structures in which two or more rings share one or more bonds.
- Carbocyclic or “carbocycle” refers to a ring wherein each of the atoms forming the ring is a carbon atom.
- Carbocycle includes aryl and cycloalkyl. The term thus distinguishes carbocycle from heterocycle (“heterocyclic”) in which the ring backbone contains at least one atom which is different from carbon (i.e a heteroatom).
- Heterocycle includes heteroaryl and heterocycloalkyl. Carbocycles and heterocycles can be optionally substituted.
- aromatic refers to a planar ring having a delocalized it-electron system containing 4n+2 ⁇ electrons, where n is an integer. Aromatic rings can be formed from five, six, seven, eight, nine, or more than nine atoms. Aromatics can be optionally substituted.
- aromatic includes both carbocyclic aryl (e.g., phenyl) and heterocyclic aryl (or “heteroaryl” or “heteroaromatic”) groups (e.g., pyridine).
- the term includes monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups.
- aryl refers to an aromatic ring wherein each of the atoms forming the ring is a carbon atom.
- Aryl rings can be formed by five, six, seven, eight, nine, or more than nine carbon atoms.
- Aryl groups can be optionally substituted. Examples of aryl groups include, but are not limited to phenyl, naphthalenyl, phenanthrenyl, anthracenyl, fluorenyl, and indenyl.
- an aryl group can be a monoradical or a diradical (i.e., an arylene group).
- aryloxy refers to an (aryl)O— group, where aryl is as defined herein.
- carbonyl refers to a group containing a moiety selected from the group consisting of —C(O)—, —S(O)—, —S(O)2-, and —C(S)—, including, but not limited to, groups containing a least one ketone group, and/or at least one aldehyde group, and/or at least one ester group, and/or at least one carboxylic acid group, and/or at least one thioester group.
- Such carbonyl groups include ketones, aldehydes, carboxylic acids, esters, and thioesters. In some embodiments, such groups are a part of linear, branched, or cyclic molecules.
- cycloalkyl refers to a monocyclic or polycyclic radical that contains only carbon and hydrogen, and is optionally saturated, partially unsaturated, or fully unsaturated. Cycloalkyl groups include groups having from 3 to 10 ring atoms. Illustrative examples of cycloalkyl groups include the following moieties:
- a cycloalkyl group is either a monoradical or a diradical (e.g., an cycloalkylene group), and if a “lower cycloalkyl” having 3 to 8 carbon atoms.
- Cycloalkylalkyl means an alkyl radical, as defined herein, substituted with a cycloalkyl group.
- Non-limiting cycloalkylalkyl groups include cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, and the like.
- heterocycle refers to heteroaromatic and heteroalicyclic groups containing one to four heteroatoms each selected from O, S and N, wherein each heterocyclic group has from 4 to 10 atoms in its ring system, and with the proviso that the ring of said group does not contain two adjacent O or S atoms.
- the number of carbon atoms in a heterocycle is indicated (e.g., C 1 -C 6 heterocycle), at least one other atom (the heteroatom) must be present in the ring.
- Designations such as “C 1 -C 6 heterocycle” refer only to the number of carbon atoms in the ring and do not refer to the total number of atoms in the ring.
- heterocylic ring can have additional heteroatoms in the ring.
- Designations such as “4-6 membered heterocycle” refer to the total number of atoms that are contained in the ring (i.e., a four, five, or six membered ring, in which at least one atom is a carbon atom, at least one atom is a heteroatom and the remaining two to four atoms are either carbon atoms or heteroatoms).
- those two or more heteroatoms can be the same or different from one another.
- Heterocycles can be optionally substituted. Binding to a heterocycle can be at a heteroatom or via a carbon atom.
- Non-aromatic heterocyclic groups include groups having only 4 atoms in their ring system, but aromatic heterocyclic groups must have at least 5 atoms in their ring system.
- the heterocyclic groups include benzo-fused ring systems.
- An example of a 4-membered heterocyclic group is azetidinyl (derived from azetidine).
- An example of a 5-membered heterocyclic group is thiazolyl.
- An example of a 6-membered heterocyclic group is pyridyl, and an example of a 10-membered heterocyclic group is quinolinyl.
- non-aromatic heterocyclic groups are pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 1,2,3,6-tetrahydropyridinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1,3-dioxolanyl, pyrazolinyl, dithio
- aromatic heterocyclic groups are pyridinyl, imidazolyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinox
- a group derived from pyrrole includes pyrrol-1-yl (N-attached) or pyrrol-3-yl (C-attached).
- a group derived from imidazole includes imidazol-1-yl or imidazol-3-yl (both N-attached) or imidazol-2-yl, imidazol-4-yl or imidazol-5-yl (all C-attached).
- heterocyclic groups include benzo-fused ring systems and ring systems substituted with one or two oxo ( ⁇ O) moieties such as pyrrolidin-2-one.
- a heterocycle group can be a monoradical or a diradical (i.e., a heterocyclene group).
- heteroaryl or, alternatively, “heteroaromatic” refers to an aromatic group that includes one or more ring heteroatoms selected from nitrogen, oxygen and sulfur.
- An N-containing “heteroaromatic” or “heteroaryl” moiety refers to an aromatic group in which at least one of the skeletal atoms of the ring is a nitrogen atom.
- heteroaryl groups include the following moieties:
- a heteroaryl group can be a monoradical or a diradical (i.e., a heteroarylene group).
- non-aromatic heterocycle refers to a non-aromatic ring wherein one or more atoms forming the ring is a heteroatom.
- a “non-aromatic heterocycle” or “heterocycloalkyl” group refers to a cycloalkyl group that includes at least one heteroatom selected from nitrogen, oxygen and sulfur.
- the radicals are fused with an aryl or heteroaryl.
- Heterocycloalkyl rings can be formed by three, four, five, six, seven, eight, nine, or more than nine atoms. Heterocycloalkyl rings can be optionally substituted.
- non-aromatic heterocycles contain one or more carbonyl or thiocarbonyl groups such as, for example, oxo- and thio-containing groups.
- heterocycloalkyls include, but are not limited to, lactams, lactones, cyclic imides, cyclic thioimides, cyclic carbamates, tetrahydrothiopyran, 4H-pyran, tetrahydropyran, piperidine, 1,3-dioxin, 1,3-dioxane, 1,4-dioxin, 1,4-dioxane, piperazine, 1,3-oxathiane, 1,4-oxathiin, 1,4-oxathiane, tetrahydro-1,4-thiazine, 2H-1,2-oxazine, maleimide, succinimide, barbituric acid, thiobarbituric acid, dioxopiperazine, hydanto
- heteroalicyclic also includes all ring forms of the carbohydrates, including but not limited to the monosaccharides, the disaccharides and the oligosaccharides.
- a heterocycloalkyl group can be a monoradical or a diradical (i.e., a heterocycloalkylene group).
- halo or, alternatively, “halogen” or “halide” means fluoro, chloro, bromo and iodo.
- haloalkyl refers to alkyl structures in which at least one hydrogen is replaced with a halogen atom. In certain embodiments in which two or more hydrogen atoms are replaced with halogen atoms, the halogen atoms are all the same as one another. In other embodiments in which two or more hydrogen atoms are replaced with halogen atoms, the halogen atoms are not all the same as one another.
- fluoroalkyl refers to alkyl group in which at least one hydrogen is replaced with a fluorine atom.
- fluoroalkyl groups include, but are not limited to, —CF 3 , —CH 2 CF 3 , —CF 2 CF 3 , —CH 2 CH 2 CF 3 and the like.
- heteroalkyl refers to optionally substituted alkyl radicals in which one or more skeletal chain atoms is a heteroatom, e.g., oxygen, nitrogen, sulfur, silicon, phosphorus or combinations thereof.
- the heteroatom(s) are placed at any interior position of the heteroalkyl group or at the position at which the heteroalkyl group is attached to the remainder of the molecule.
- Examples include, but are not limited to, —CH 2 —O—CH 3 , —CH 2 —CH 2 —O—CH 3 , —CH 2 —NH—CH 3 , —CH 2 —CH 2 —NH—CH 3 , —CH 2 —N(CH 3 )—CH 3 , —CH 2 —CH 2 —NH—CH 3 , —CH 2 —CH 2 —N(CH 3 )—CH 3 , —CH 2 —S—CH 2 —CH 3 , —CH 2 —CH 2 ,—S(O)—CH 3 , —CH 2 —CH 2 —S(O) 2 —CH 3 , —CH ⁇ CH—O—CH 3 , —Si(CH 3 ) 3 , —CH 2 —CH ⁇ N—OCH 3 , and —CH ⁇ CH—N(CH 3 )—CH 3 .
- up to two heteroatoms are consecutive, such as, by way
- heteroatom refers to an atom other than carbon or hydrogen. Heteroatoms are typically independently selected from among oxygen, sulfur, nitrogen, silicon and phosphorus, but are not limited to these atoms. In embodiments in which two or more heteroatoms are present, the two or more heteroatoms can all be the same as one another, or some or all of the two or more heteroatoms can each be different from the others.
- bond refers to a chemical bond between two atoms, or two moieties when the atoms joined by the bond are considered to be part of larger substructure.
- moiety refers to a specific segment or functional group of a molecule. Chemical moieties are often recognized chemical entities embedded in or appended to a molecule.
- a “thioalkoxy” or “alkylthio” group refers to a —S-alkyl group.
- a “SH” group is also referred to either as a thiol group or a sulfhydryl group.
- optionally substituted or “substituted” means that the referenced group may be substituted with one or more additional group(s) individually and independently selected from alkyl, cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, alkylthio, arylthio, alkylsulfoxide, arylsulfoxide, alkylsulfone, arylsulfone, cyano, halo, acyl, nitro, haloalkyl, fluoroalkyl, amino, including mono- and di-substituted amino groups, and the protected derivatives thereof.
- an optional substituents may be L s R s , wherein each L s is independently selected from a bond, —O—, —C( ⁇ O)—, —S—, —S( ⁇ O)—, —S( ⁇ O) 2 —, —NH—, —NHC(O)—, —C(O)NH—, S( ⁇ O) 2 NH—, —NHS( ⁇ O) 2 , —OC(O)NH—, —NHC(O)O—, -(substituted or unsubstituted C 1 -C 6 alkyl), or -(substituted or unsubstituted C 2 -C 6 alkenyl); and each R s is independently selected from H, (substituted or unsubstituted C 1 -C 4 alkyl), (substituted or unsubstituted C 3 -C 6 cycloalkyl), heteroaryl, or heteroalky
- Michael acceptor moiety refers to a functional group that can participate in a Michael reaction, wherein a new covalent bond is formed between a portion of the Michael acceptor moiety and the donor moiety.
- the Michael acceptor moiety is an electrophile and the “donor moiety” is a nucleophile.
- the “G” groups presented in any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) are non-limiting examples of Michael acceptor moieties.
- nucleophile refers to an electron rich compound, or moiety thereof.
- An example of a nucleophile includes, but in no way is limited to, a cysteine residue of a molecule, such as, for example Cys 481 of Btk.
- electrophile refers to an electron poor or electron deficient molecule, or moiety thereof.
- electrophiles include, but in no way are limited to, Micheal acceptor moieties.
- acceptable or “pharmaceutically acceptable”, with respect to a formulation, composition or ingredient, as used herein, means having no persistent detrimental effect on the general health of the subject being treated or does not abrogate the biological activity or properties of the compound, and is relatively nontoxic.
- agonist refers to a compound, the presence of which results in a biological activity of a protein that is the same as the biological activity resulting from the presence of a naturally occurring ligand for the protein, such as, for example, Btk.
- partial agonist refers to a compound the presence of which results in a biological activity of a protein that is of the same type as that resulting from the presence of a naturally occurring ligand for the protein, but of a lower magnitude.
- an antagonist refers to a compound, the presence of which results in a decrease in the magnitude of a biological activity of a protein.
- the presence of an antagonist results in complete inhibition of a biological activity of a protein, such as, for example, Btk.
- an antagonist is an inhibitor.
- amelioration of the symptoms of a particular disease, disorder or condition by administration of a particular compound or pharmaceutical composition refers to any lessening of severity, delay in onset, slowing of progression, or shortening of duration, whether permanent or temporary, lasting or transient that can be attributed to or associated with administration of the compound or composition.
- Bioavailability refers to the percentage of the weight of compounds disclosed herein, such as, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), dosed that is delivered into the general circulation of the animal or human being studied.
- the total exposure (AUC (0- ⁇ ) ) of a drug when administered intravenously is usually defined as 100% bioavailable (F %).
- Oral bioavailability refers to the extent to which compounds disclosed herein, such as, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are absorbed into the general circulation when the pharmaceutical composition is taken orally as compared to intravenous injection.
- biophysical probe refers to probes which detect or monitor structural changes in molecules (including biomolecules) in biological systems or in the presence of other biomolecules (e.g., ex vivo, in vivo or in vitro).
- molecules include, but are not limited to, proteins and the “biophysical probe” is used to detect or monitor interaction of proteins with other macromolecules.
- examples of biophysical probes include, but are not limited to, spin-labels, fluorophores, and photoactivatable groups.
- “Blood plasma concentration” refers to the concentration of compounds disclosed herein, such as, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), in the plasma component of blood of a subject. It is understood that the plasma concentration of compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), may vary significantly between subjects, due to variability with respect to metabolism and/or possible interactions with other therapeutic agents.
- the blood plasma concentration of the compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), does vary from subject to subject.
- values such as maximum plasma concentration (C max ) or time to reach maximum plasma concentration (T max ), or total area under the plasma concentration time curve (AUC (0- ⁇ ) ) may vary from subject to subject.
- the amount necessary to constitute “a therapeutically effective amount” of a compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), is expected to vary from subject to subject.
- Bruton's tyrosine kinase refers to Bruton's tyrosine kinase from Homo sapiens , as disclosed in, e.g., U.S. Pat. No. 6,326,469 (GenBank Accession No. NP_000052).
- Bruton's tyrosine kinase homolog refers to orthologs of Bruton's tyrosine kinase, e.g., the orthologs from mouse (GenBank Accession No. AAB47246), dog (GenBank Accession No. XP_549139.), rat (GenBank Accession No. NP_001007799), chicken (GenBank Accession No. NP_989564), or zebra fish (GenBank Accession No. XP_698117), and fusion proteins of any of the foregoing that exhibit kinase activity towards one or more substrates of Bruton's tyrosine kinase (e.g. a peptide substrate having the amino acid sequence “AVLESEEELYSSARQ”).
- chemiluminescent group refers to a group which emits light as a result of a chemical reaction without the addition of heat.
- luminol 5-amino-2,3-dihydro-1,4-phthalazinedione
- oxidants like hydrogen peroxide (H 2 O 2 ) in the presence of a base and a metal catalyst to produce an excited state product (3-aminophthalate, 3-APA).
- chromophore refers to a molecule which absorbs light of visible wavelengths, UV wavelengths or IR wavelengths.
- co-administration are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are administered by the same or different route of administration or at the same or different time.
- the term “detectable label,” as used herein, refers to a label which is observable using analytical techniques including, but not limited to, fluorescence, chemiluminescence, electron-spin resonance, ultraviolet/visible absorbance spectroscopy, mass spectrometry, nuclear magnetic resonance, magnetic resonance, and electrochemical methods.
- die refers to a soluble, coloring substance which contains a chromophore.
- an “effective amount” or “therapeutically effective amount,” as used herein, refer to a sufficient amount of an agent or a compound being administered which will relieve to some extent one or more of the symptoms of the disease or condition being treated. The result can be reduction and/or alleviation of the signs, symptoms, or causes of a disease, or any other desired alteration of a biological system.
- an “effective amount” for therapeutic uses is the amount of the composition including a compound as disclosed herein required to provide a clinically significant decrease in disease symptoms without undue adverse side effects.
- An appropriate “effective amount” in any individual case is optionally determined using techniques, such as a dose escalation study.
- the term “therapeutically effective amount” includes, for example, a prophylactically effective amount.
- an “effective amount” of a compound disclosed herein is an amount effective to achieve a desired pharmacologic effect or therapeutic improvement without undue adverse side effects. It is understood that “an effect amount” or “a therapeutically effective amount” can vary from subject to subject, due to variation in metabolism of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), age, weight, general condition of the subject, the condition being treated, the severity of the condition being treated, and the judgment of the prescribing physician.
- electrostatic group refers to a group which scatters electrons when irradiated with an electron beam.
- groups include, but are not limited to, ammonium molybdate, bismuth subnitrate cadmium iodide, 99%, carbohydrazide, ferric chloride hexahydrate, hexamethylene tetramine, 98.5%, indium trichloride anhydrous, lanthanum nitrate, lead acetate trihydrate, lead citrate trihydrate, lead nitrate, periodic acid, phosphomolybdic acid, phosphotungstic acid, potassium ferricyanide, potassium ferrocyanide, ruthenium red, silver nitrate, silver proteinate (Ag Assay: 8.0-8.5%) “Strong”, silver tetraphenylporphin (S-TPPS), sodium chloroaurate, sodium tungstate, thallium nitrate, thiosemicarbazide (TSC),
- the term “energy transfer agent,” as used herein, refers to a molecule which either donates or accepts energy from another molecule.
- fluorescence resonance energy transfer FRET is a dipole-dipole coupling process by which the excited-state energy of a fluorescence donor molecule is non-radiatively transferred to an unexcited acceptor molecule which then fluorescently emits the donated energy at a longer wavelength.
- “enhance” or “enhancing” means to increase or prolong either in potency or duration a desired effect.
- “enhancing” the effect of therapeutic agents refers to the ability to increase or prolong, either in potency or duration, the effect of therapeutic agents on during treatment of a disease, disorder or condition.
- An “enhancing-effective amount,” as used herein, refers to an amount adequate to enhance the effect of a therapeutic agent in the treatment of a disease, disorder or condition. When used in a patient, amounts effective for this use will depend on the severity and course of the disease, disorder or condition, previous therapy, the patient's health status and response to the drugs, and the judgment of the treating physician.
- fluorophore refers to a molecule which upon excitation emits photons and is thereby fluorescent.
- cysteine 482 is the homologous cysteine of the rat ortholog of Bruton's tyrosine kinase
- cysteine 479 is the homologous cysteine of the chicken ortholog
- cysteine 481 is the homologous cysteine in the zebra fish ortholog.
- the homologous cysteine of TXK is Cys 350.
- Other examples of kinases having homologous cysteines are shown in FIG. 7 . See also the sequence alignments of tyrosine kinases (TK) published on the world wide web at kinase.com/human/kinome/phylogeny.html.
- sequences or subsequences refers to two or more sequences or subsequences which are the same.
- substantially identical refers to two or more sequences which have a percentage of sequential units which are the same when compared and aligned for maximum correspondence over a comparison window, or designated region as measured using comparison algorithms or by manual alignment and visual inspection.
- two or more sequences are “substantially identical” if the sequential units are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region. Such percentages to describe the “percent identity” of two or more sequences.
- the identity of a sequence can exist over a region that is at least about 75-100 sequential units in length, over a region that is about 50 sequential units in length, or, where not specified, across the entire sequence.
- This definition also refers to the complement of a test sequence.
- two or more polypeptide sequences are identical when the amino acid residues are the same, while two or more polypeptide sequences are “substantially identical” if the amino acid residues are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region.
- the identity can exist over a region that is at least about 75-100 amino acids in length, over a region that is about 50 amino acids in length, or, where not specified, across the entire sequence of a polypeptide sequence.
- two or more polynucleotide sequences are identical when the nucleic acid residues are the same, while two or more polynucleotide sequences are “substantially identical” if the nucleic acid residues are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region.
- the identity can exist over a region that is at least about 75-100 nucleic acids in length, over a region that is about 50 nucleic acids in length, or, where not specified, across the entire sequence of a polynucleotide sequence.
- inhibitors refer to inhibition of enzymatic phosphotransferase activity.
- irreversible inhibitor refers to a compound that, upon contact with a target protein (e.g., a kinase) causes the formation of a new covalent bond with or within the protein, whereby one or more of the target protein's biological activities (e.g., phosphotransferase activity) is diminished or abolished notwithstanding the subsequent presence or absence of the irreversible inhibitor.
- a target protein e.g., a kinase
- biological activities e.g., phosphotransferase activity
- irreversible Btk inhibitor refers to an inhibitor of Btk that can form a covalent bond with an amino acid residue of Btk.
- the irreversible inhibitor of Btk can form a covalent bond with a Cys residue of Btk; in particular embodiments, the irreversible inhibitor can form a covalent bond with a Cys 481 residue (or a homolog thereof) of Btk or a cysteine residue in the homologous corresponding position of another tyrosine kinase, as shown in FIG. 7 .
- isolated refers to separating and removing a component of interest from at least some portion of components not of interest. Isolated substances can be in either a dry or semi-dry state, or in solution, including but not limited to an aqueous solution.
- the isolated component can be in a homogeneous state or the isolated component can be a part of a pharmaceutical composition that comprises additional pharmaceutically acceptable carriers and/or excipients.
- nucleic acids or proteins are “isolated” when such nucleic acids or proteins are free of at least some of the cellular components with which it is associated in the natural state, or that the nucleic acid or protein has been concentrated to a level greater than the concentration of its in vivo or in vitro production.
- a gene is isolated when separated from open reading frames which flank the gene and encode a protein other than the gene of interest.
- label refers to a substance which is incorporated into a compound and is readily detected, whereby its physical distribution is detected and/or monitored.
- linkages refer to bonds or a chemical moiety formed from a chemical reaction between the functional group of a linker and another molecule.
- bonds include, but are not limited to, covalent linkages and non-covalent bonds
- chemical moieties include, but are not limited to, esters, carbonates, imines, phosphate esters, hydrazones, acetals, orthoesters, peptide linkages, and oligonucleotide linkages.
- Hydrolytically stable linkages means that the linkages are substantially stable in water and do not react with water at useful pH values, including but not limited to, under physiological conditions for an extended period of time, perhaps even indefinitely.
- Hydrolytically unstable or degradable linkages means that the linkages are degradable in water or in aqueous solutions, including for example, blood.
- enzymatically unstable or degradable linkages means that the linkage is degraded by one or more enzymes.
- PEG and related polymers include degradable linkages in the polymer backbone or in the linker group between the polymer backbone and one or more of the terminal functional groups of the polymer molecule.
- Such degradable linkages include, but are not limited to, ester linkages formed by the reaction of PEG carboxylic acids or activated PEG carboxylic acids with alcohol groups on a biologically active agent, wherein such ester groups generally hydrolyze under physiological conditions to release the biologically active agent.
- hydrolytically degradable linkages include but are not limited to carbonate linkages; imine linkages resulted from reaction of an amine and an aldehyde; phosphate ester linkages formed by reacting an alcohol with a phosphate group; hydrazone linkages which are reaction product of a hydrazide and an aldehyde; acetal linkages that are the reaction product of an aldehyde and an alcohol; orthoester linkages that are the reaction product of a formate and an alcohol; peptide linkages formed by an amine group, including but not limited to, at an end of a polymer such as PEG, and a carboxyl group of a peptide; and oligonucleotide linkages formed by a phosphoramidite group, including but not limited to, at the end of a polymer, and a 5′ hydroxyl group of an oligonucleotide.
- the phrase “measuring the activity of the reporter moiety” refers to methods for quantifying (in absolute, approximate or relative terms) the reporter moiety in a system under study.
- such methods include any methods that quantify a reporter moiety that is a dye; a photocrosslinker; a cytotoxic compound; a drug; an affinity label; a photoaffinity label; a reactive compound; an antibody or antibody fragment; a biomaterial; a nanoparticle; a spin label; a fluorophore, a metal-containing moiety; a radioactive moiety; a novel functional group; a group that covalently or noncovalently interacts with other molecules; a photocaged moiety; an actinic radiation excitable moiety; a ligand; a photoisomerizable moiety; biotin; a biotin analogue; a moiety incorporating a heavy atom; a chemically cleavable group; a photocleav
- a “metabolite” of a compound disclosed herein is a derivative of that compound that is formed when the compound is metabolized.
- active metabolite refers to a biologically active derivative of a compound that is formed when the compound is metabolized.
- metabolized refers to the sum of the processes (including, but not limited to, hydrolysis reactions and reactions catalyzed by enzymes, such as, oxidation reactions) by which a particular substance is changed by an organism. Thus, enzymes produce specific structural alterations to a compound.
- cytochrome P450 catalyzes a variety of oxidative and reductive reactions while uridine diphosphate glucuronyl transferases catalyze the transfer of an activated glucuronic-acid molecule to aromatic alcohols, aliphatic alcohols, carboxylic acids, amines and free sulfhydryl groups. Further information on metabolism is obtained from The Pharmacological Basis of Therapeutics, 9th Edition, McGraw-Hill (1996). Metabolites of the compounds disclosed herein are optionally identified either by administration of compounds to a host and analysis of tissue samples from the host, or by incubation of compounds with hepatic cells in vitro and analysis of the resulting compounds. In some embodiments, metabolites of a compound are formed by oxidative processes and correspond to the corresponding hydroxy-containing compound. In some embodiments, a compound is metabolized to pharmacologically active metabolites.
- module means to interact with a target either directly or indirectly so as to alter the activity of the target, including, by way of example only, to enhance the activity of the target, to inhibit the activity of the target, to limit the activity of the target, or to extend the activity of the target.
- a modulator refers to a compound that alters an activity of a molecule.
- a modulator can cause an increase or decrease in the magnitude of a certain activity of a molecule compared to the magnitude of the activity in the absence of the modulator.
- a modulator is an inhibitor, which decreases the magnitude of one or more activities of a molecule.
- an inhibitor completely prevents one or more activities of a molecule.
- a modulator is an activator, which increases the magnitude of at least one activity of a molecule.
- the presence of a modulator results in an activity that does not occur in the absence of the modulator.
- molecular incorporating a heavy atom refers to a group which incorporates an ion of atom which is usually heavier than carbon.
- ions or atoms include, but are not limited to, silicon, tungsten, gold, lead, and uranium.
- nanoparticle refers to a particle which has a particle size between about 500 nm to about 1 nm.
- pERK refers to phosphorylated ERK1 and ERK2 at Thr202/Tyr 204 as detected by commercially available phospho-specific antibodies (e.g. Cell Signaling Technologies #4377).
- photoaffinity label refers to a label with a group, which, upon exposure to light, forms a linkage with a molecule for which the label has an affinity.
- a linkage is covalent or non-covalent.
- photocaged moiety refers to a group which, upon illumination at certain wavelengths, covalently or non-covalently binds other ions or molecules.
- photoisomerizable moiety refers to a group wherein upon illumination with light changes from one isomeric form to another.
- plasma half life refers to half-life in rat, dog or human as determined by measure drug concentration over time in plasma following a single dose and fitting data to standard pharmacokinetic models using software such as WinNonLin to determine the time at which drug has been 50% eliminated from plasma.
- prophylactically effective amount refers that amount of a composition applied to a patient which will relieve to some extent one or more of the symptoms of a disease, condition or disorder being treated. In such prophylactic applications, such amounts may depend on the patient's state of health, weight, and the like.
- radioactive moiety refers to a group whose nuclei spontaneously give off nuclear radiation, such as alpha, beta, or gamma particles; wherein, alpha particles are helium nuclei, beta particles are electrons, and gamma particles are high energy photons.
- selective binding compound refers to a compound that selectively binds to any portion of one or more target proteins.
- selective binds refers to the ability of a selective binding compound to bind to a target protein, such as, for example, Btk, with greater affinity than it binds to a non-target protein.
- specific binding refers to binding to a target with an affinity that is at least 10, 50, 100, 250, 500, 1000 or more times greater than the affinity for a non-target.
- selective modulator refers to a compound that selectively modulates a target activity relative to a non-target activity.
- specific modulator refers to modulating a target activity at least 10, 50, 100, 250, 500, 1000 times more than a non-target activity.
- spin label refers to molecules which contain an atom or a group of atoms exhibiting an unpaired electron spin (i.e. a stable paramagnetic group) that in some embodiments are detected by electron spin resonance spectroscopy and in other embodiments are attached to another molecule.
- spin-label molecules include, but are not limited to, nitryl radicals and nitroxides, and in some embodiments are single spin-labels or double spin-labels.
- substantially purified refers to a component of interest that may be substantially or essentially free of other components which normally accompany or interact with the component of interest prior to purification.
- a component of interest may be “substantially purified” when the preparation of the component of interest contains less than about 30%, less than about 25%, less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 4%, less than about 3%, less than about 2%, or less than about 1% (by dry weight) of contaminating components.
- a “substantially purified” component of interest may have a purity level of about 70%, about 75%, about 80%, about 85%, about 90%, about 95%, about 96%, about 97%, about 98%, about 99% or greater.
- subject refers to an animal which is the object of treatment, observation or experiment.
- a subject may be, but is not limited to, a mammal including, but not limited to, a human.
- target activity refers to a biological activity capable of being modulated by a selective modulator.
- Certain exemplary target activities include, but are not limited to, binding affinity, signal transduction, enzymatic activity, tumor growth, inflammation or inflammation-related processes, and amelioration of one or more symptoms associated with a disease or condition.
- target protein refers to a molecule or a portion of a protein capable of being bound by a selective binding compound.
- a target protein is Btk.
- treat include alleviating, abating or ameliorating a disease or condition symptoms, preventing additional symptoms, ameliorating or preventing the underlying metabolic causes of symptoms, inhibiting the disease or condition, e.g., arresting the development of the disease or condition, relieving the disease or condition, causing regression of the disease or condition, relieving a condition caused by the disease or condition, or stopping the symptoms of the disease or condition.
- the terms “treat,” “treating” or “treatment”, include, but are not limited to, prophylactic and/or therapeutic treatments.
- the IC 50 refers to an amount, concentration or dosage of a particular test compound that achieves a 50% inhibition of a maximal response, such as inhibition of Btk, in an assay that measures such response.
- EC 50 refers to a dosage, concentration or amount of a particular test compound that elicits a dose-dependent response at 50% of maximal expression of a particular response that is induced, provoked or potentiated by the particular test compound.
- FIG. 1A presents an illustrative table of GI 50 concentrations of Compound 1 that results in 50% decrease in cell proliferation.
- FIG. 1B presents an illustrative line graph showing inhibition of tumor growth in DLCL2 xenograft models.
- FIG. 1C presents an illustrative line graph showing inhibition of tumor growth in DOHH2 xenograft models.
- 5E6 DOHH2 or DLCL2 cells in 50% matrigel were implanted subcutaneously in SCID mice and dosed orally with Compound 1 beginning when tumor size reached 100 mm2.
- FIG. 2 presents an illustrative line graph showing inhibition of collagen-induced arthritis in male DBA/1O1aHsd mice.
- Compound 1 or vehicle was dosed orally once per day starting at day 1.
- Dexamethasone was included as a positive control.
- Paw inflammation was scored from 0-5 and averaged across all paws from all animals for each group in the study.
- Compound 1 at 12.5 mg/kg and 50 mg/kg regressed inflammation through the end of the study (day 11) while 3.125 mg/kg significantly reduced the increase in paw inflammation.
- FIG. 3 presents an illustrative line graph showing inhibition of disease progression in a mouse MRL/1 pr model of lupus.
- MRL/1 pr mice Jax strain 000485
- Compound 1 at 3.125 mg/kg, 12.5 mg/kg, and 50 mg/kg significantly reduced proteinuria, indicating amelioration of the progressive autoimmune renal failure seen in this mouse strain.
- FIG. 4 presents an illustrative bar graph showing inhibition of mast cell degranulation in a mouse passive cutaneous anaphylaxis model.
- Increasing doses of Compound 1 significantly decreased the amount of Evans Blue release, indicating decreased mast cell activation and vascular permeabilization.
- FIG. 5 presents an illustrative line graph showing in vivo plasma concentrations post-dosing of male jugular vein cannulated rats with Compounds 1, 7, 8, and 12. Blood samples were collected at 0.0833 (5 minutes), 0.333 (20 minutes), 1, 3, 6, 9, and 24 hours post-dosing from orally dosed rats.
- Compound 1 and Compound 12 have a short half-life in vivo.
- Compound 7 and Compound 8 have a significantly longer in vivo half-life.
- Compounds like 1 and 12 are predicted to have enhanced kinase selectivity in vivo because inhibition will be sustained only for those kinases that are irreversibly inhibited.
- FIG. 6 presents an illustrative bar graph showing brief exposure to Compound 1 in vitro is sufficient to inhibit B cell activation in normal human B cells.
- B cells were purified from blood from healthy donors by negative selecting using the RosetteSep Human B cell enrichment cocktail. Cells were plated in growth media and indicated concentrations of Compound 1 were added. After incubation for 1 hour at 37° C., cells were washed three times using an 8-fold dilution in growth media for each wash. Cells were then stimulated with IgM F(ab′) 2 for 18 hours at 37° C., stained with anti-CD69-PE antibody and analyzed by flow cytometry. This protocol mimics the predicted exposure of cells to Compound 1 in vivo and demonstrates that inhibition of B cells is sustained despite washing out of Compound 1.
- FIG. 7 presents illustrative ACKs, including Btk and Btk cysteine homologs.
- the inhibitor compounds described herein are selective for kinases having an accessible cysteine residue (such kinases are also known as Accessible Cysteine Kinases, or ACKs) that can form a covalent bond with a Michael acceptor moiety on the inhibitor compound.
- the cysteine residue is accessible or becomes accessible when the binding site moiety of the irreversible inhibitor binds to the kinase.
- the binding site moiety of the irreversible inhibitor binds to an active site of the ACK and the Michael acceptor moiety of irreversible inhibitor gains access (in one embodiment the step of binding leads to a conformational change in the ACK, thus exposing the cysteine) or is otherwise exposed to the cysteine residue of the ACK; as a result a covalent bond is formed between the “S” of the cysteine residue and the Michael acceptor of the irreversible inhibitor. Consequently, the binding site moiety of the irreversible inhibitor remains bound or otherwise blocks the active site of the ACK.
- the ACK is Btk, a homolog of Btk or a tyrosine kinase having a cysteine residue in an amino acid sequence position that is homologous to the amino acid sequence position of cysteine 481 in Btk. See, e.g., kinases in FIG. 7 .
- Inhibitor compounds described herein include a Michael acceptor moiety, a binding site moiety and a linker that links the binding site moiety and the Michael acceptor moiety (and in some embodiments, the structure of the linker provides a conformation, or otherwise directs the Michael acceptor moiety, so as to improve the selectivity of the irreversible inhibitor for a particular ACK).
- an irreversible inhibitor compound used in the methods described herein is identified or characterized in an in vitro assay, e.g., an a cellular biochemical assay or a cellular functional assay. Such assays are useful to determine an in vitro IC 50 for an irreversible inhibitor compound.
- an acellular kinase assay can be used to determine kinase activity after incubation of the kinase in the absence or presence of a range of concentrations of a candidate irreversible inhibitor compound. If the candidate compound is in fact an irreversible inhibitor, kinase activity will not be recovered by repeat washing with inhibitor-free medium. See, e.g., J. B. Smaill, et al. (1999), J. Med. Chem. 42(10): 1803-1815.
- covalent complex formation between a Kinase and a candidate irreversible inhibitor is a useful indicator of irreversible inhibition of the Kinase that can be readily determined by a number of methods (e.g., mass spectrometry). For example, some irreversible Kinase-inhibitor compounds can form a covalent bond with the aforenoted cysteine residue (e.g., via a Michael reaction).
- High throughput assays for many acellular biochemical assays e.g., kinase assays
- cellular functional assays e.g., calcium flux
- high throughput screening systems are commercially available (see, e.g., Zymark Corp., Hopkinton, Mass.; Air Technical Industries, Mentor, OH; Beckman Instruments, Inc. Fullerton, Calif.; Precision Systems, Inc., Natick, Mass., etc.). These systems typically automate entire procedures including all sample and reagent pipetting, liquid dispensing, timed incubations, and final readings of the microplate in detector(s) appropriate for the assay. Automated systems thereby allow the identification and characterization of a large number of irreversible compounds.
- Irreversible inhibitor compounds can used for the manufacture of a medicament for treating any of the foregoing conditions (e.g., autoimmune diseases, inflammatory diseases, allergy disorders, B-cell proliferative disorders, or thromboembolic disorders).
- autoimmune diseases e.g., inflammatory diseases, allergy disorders, B-cell proliferative disorders, or thromboembolic disorders.
- the irreversible inhibitor compound used for the methods described herein inhibits a Kinase activity with an in vitro IC 50 of less than 10 ⁇ M.
- an in vitro IC 50 of less than 10 ⁇ M.
- the irreversible inhibitor compound selectively and irreversibly inhibits an activated form of its target tyrosine kinase (e.g., a phosphorylated form of the tyrosine kinase).
- activated Btk is transphosphorylated at tyrosine 551.
- the irreversible Btk inhibitor inhibits the target kinase in cells only once the target kinase is activated by the signaling events.
- Described herein are compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Also described herein are pharmaceutically acceptable salts, pharmaceutically acceptable solvates, pharmaceutically active metabolites, and pharmaceutically acceptable prodrugs of such compounds. Pharmaceutical compositions that include at least one such compound or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, pharmaceutically active metabolite or pharmaceutically acceptable prodrug of such compound, are provided. In some embodiments, when compounds disclosed herein contain an oxidizable nitrogen atom, the nitrogen atom is optionally converted to an N-oxide.
- isomers and chemically protected forms of compounds having a structure represented by any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are also provided.
- salts of compounds of Formula (I) are provided pharmaceutically acceptable salts of compounds of Formula (I).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (I) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- substituents can be selected from among from a subset of the listed alternatives.
- L a is CH 2 , O, or NH.
- L a is O or NH.
- L a is O.
- Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- x is 2.
- Z is C( ⁇ O), OC( ⁇ O), NHC( ⁇ O), S( ⁇ O) x , OS( ⁇ O) x , or NHS( ⁇ O) x .
- Z is C( ⁇ O), NHC( ⁇ O), or NCH 3 C( ⁇ O).
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, and alkyleneheterocycloalkylene.
- Z is C( ⁇ O), NHC( ⁇ O), NR a C( ⁇ O), NR a S( ⁇ O) x , where x is 1 or 2, and R a is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl.
- R 7 and R 8 are H; and R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 8 alkylaminoalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted C 1 -C 8 alkylC 3 -C 6 cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C 2 -C 8 heterocycloalkyl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 8 alkylethers, C 1 -C 1 -C
- R 6 and R 8 are H; and R 7 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 8 alkylaminoalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted C 1 -C 8 alkylC 3 -C 6 cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C 2 -C 8 heterocycloalkyl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 8 alkylethers, C 1 -C 1 -C
- Y is an optionally substituted group selected from cycloalkylene or heterocycloalkylene.
- Z is C( ⁇ O), NHC( ⁇ O), NR a C( ⁇ O), NR a S( ⁇ O) x , where x is 1 or 2, and R a is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl.
- R 7 and R 8 are H; and R 6 is substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted C 1 -C 8 alkylC 3 -C 6 cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C 2 -C 8 heterocycloalkyl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 8 alkylethers, C 1 -C 8 alkylamides, or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 and R 8 are H; and R 7 is substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted C 1 -C 8 alkylC 3 -C 6 cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C 2 -C 8 heterocycloalkyl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 8 alkylethers, C 1 -C 8 alkylamides, or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- kinase is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
- salts of compounds of Formula (VII) are provided pharmaceutically acceptable salts of compounds of Formula (VII).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (VII) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- x is 2.
- Z is C( ⁇ O), OC( ⁇ O), NHC( ⁇ O), S( ⁇ O) x , OS( ⁇ O) x , or NHS( ⁇ O) x .
- Z is C( ⁇ O), NHC( ⁇ O), or S( ⁇ O) 2 .
- R 7 and R 8 are independently selected from among H, unsubstituted C 1 -C 4 alkyl, substituted C 1 -C 4 alkyl, unsubstituted C 1 -C 4 heteroalkyl, and substituted C 1 -C 4 heteroalkyl; or R 7 and R 8 taken together form a bond. In yet other embodiments, each of R 7 and R 8 is H; or R 7 and R 8 taken together form a bond.
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 8 alkylaminoalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 2 alkyl-N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —(C 1 -C 6 alkylamino), C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl containing 1 or 2 N atoms), or C 1 -C 4 alkyl(5- or 6-membered heterocycloalkyl containing 1 or 2 N atoms).
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene.
- Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 4-, 5-, 6-, or 7-membered cycloalkylene, and 4-, 5-, 6-, or 7-membered heterocycloalkylene.
- Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 5- or 6-membered cycloalkylene, and 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- Y is a 5- or 6-membered cycloalkylene, or a 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- Y is a 4-, 5-, 6-, or 7-membered cycloalkylene ring; or Y is a 4-, 5-, 6-, or 7-membered heterocycloalkylene ring.
- salts of compounds of Formula (A1) are provided pharmaceutically acceptable salts of compounds of Formula (A1).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (A1) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- the compound of Formula (A1) has the following structure of Formula (B1):
- R a is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl;
- G is selected from among
- R is H, alkyl, alkylhydroxy, heterocycloalkyl, heteroaryl, alkylalkoxy, alkylalkoxyalkyl.
- the compound of Formula (B1) has the following structure of Formula (C1):
- R a is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl;
- the “G” group of any of Formula (A1), Formula (B1), or Formula (C1) is any group that is used to tailor the physical and biological properties of the molecule. Such tailoring/modifications are achieved using groups which modulate Michael acceptor chemical reactivity, acidity, basicity, lipophilicity, solubility and other physical properties of the molecule.
- the physical and biological properties modulated by such modifications to G include, by way of example only, enhancing chemical reactivity of Michael acceptor group, solubility, in vivo absorption, and in vivo metabolism.
- in vivo metabolism includes, by way of example only, controlling in vivo PK properties, off-target activities, potential toxicities associated with cypP450 interactions, drug-drug interactions, and the like. Further, modifications to G allow for the tailoring of the in vivo efficacy of the compound through the modulation of, by way of example, specific and non-specific protein binding to plasma proteins and lipids and tissue distribution in vivo.
- salts of compounds of Formula (D1) are provided pharmaceutically acceptable salts of compounds of Formula (D1).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (D1) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- L a is O.
- Ar is phenyl
- Z is C( ⁇ O), NHC( ⁇ O), or NCH 3 C( ⁇ O).
- each of R 1 , R 2 , and R 3 is H.
- salts of compounds of Formula (D1) are provided pharmaceutically acceptable salts of compounds of Formula (D1).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (D1) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- substituents can be selected from among from a subset of the listed alternatives.
- L a is CH 2 , O, or NH.
- L a is O or NH.
- L a is O.
- Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- x is 2.
- Z is C( ⁇ O), OC( ⁇ O), NHC( ⁇ O), S( ⁇ O) x , OS( ⁇ O) x , or NHS( ⁇ O) x .
- Z is C( ⁇ O), NHC( ⁇ O), or S( ⁇ O) 2 .
- R 7 and R 8 are independently selected from among H, unsubstituted C 1 -C 4 alkyl, substituted C 1 -C 4 alkyl, unsubstituted C 1 -C 4 heteroalkyl, and substituted C 1 -C 4 heteroalkyl; or R 7 and R 8 taken together form a bond. In yet other embodiments, each of R 7 and R 8 is H; or R 7 and R 8 taken together form a bond.
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 2 alkyl-N(C 1 -C 3 alkyl) 2 , substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 2 alkyl-N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl containing 1 or 2 N atoms), or C 1 -C 4 alkyl(5- or 6-membered heterocycloalkyl containing 1 or 2 N atoms).
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, cycloalkylene, and heterocycloalkylene. In other embodiments, Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 4-, 5-, 6- or 7-membered cycloalkylene, and 4-, 5-, 6- or 7-membered heterocycloalkylene.
- Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 5-, or 6-membered cycloalkylene, and 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some other embodiments, Y is a 5-, or 6-membered cycloalkylene, or a 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- salts of compounds of Formula (A2-A6) are provided pharmaceutically acceptable salts of compounds of Formula (A2-A6).
- salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (A2-A6) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- the compound of Formula (A2-A6) has the following structure of Formula (B2-B6):
- G is selected from among
- the compound of Formula (B2-B6) has the following structure of Formula (C2-C6):
- the “G” group of any of Formula (A2-A6), Formula (B2-B6), or Formula (C2-C6) is any group that is used to tailor the physical and biological properties of the molecule. Such tailoring/modifications are achieved using groups which modulate Michael acceptor chemical reactivity, acidity, basicity, lipophilicity, solubility and other physical properties of the molecule.
- the physical and biological properties modulated by such modifications to G include, by way of example only, enhancing chemical reactivity of Michael acceptor group, solubility, in vivo absorption, and in vivo metabolism.
- in vivo metabolism includes, by way of example only, controlling in vivo PK properties, off-target activities, potential toxicities associated with cypP450 interactions, drug-drug interactions, and the like. Further, modifications to G allow for the tailoring of the in vivo efficacy of the compound through the modulation of, by way of example, specific and non-specific protein binding to plasma proteins and lipids and tissue distribution in vivo.
- salts of compounds of Formula (D2-D6) are provided pharmaceutically acceptable salts of compounds of Formula (D2-D6).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (D2-D6) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- L a is O.
- Ar is phenyl
- Z is C(O).
- each of R 1 , R 2 , and R 3 is H.
- salts of compounds of Formula (D2-D6) are provided pharmaceutically acceptable salts of compounds of Formula (D2-D6).
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid.
- Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palm
- esters of compounds of Formula (D2-D6) including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- substituents can be selected from among from a subset of the listed alternatives.
- L a is CH 2 , O, or NH.
- L a is O or NH.
- L a is O.
- Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- x is 2.
- Z is C( ⁇ O), OC( ⁇ O), NHC( ⁇ O), S( ⁇ O) x , OS( ⁇ O) x , or NHS( ⁇ O) x .
- Z is C( ⁇ O), NHC( ⁇ O), or S( ⁇ O) 2 .
- R 7 and R 8 are independently selected from among H, unsubstituted C 1 -C 4 alkyl, substituted C 1 -C 4 alkyl, unsubstituted C 1 -C 4 heteroalkyl, and substituted C 1 -C 4 heteroalkyl; or R 7 and R 8 taken together form a bond. In yet other embodiments, each of R 7 and R 8 is H; or R 7 and R 8 taken together form a bond.
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 2 alkyl-N(C 1 -C 3 alkyl) 2 , substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 2 alkyl-N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl).
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, —CH 2 —O—(C 1 -C 3 alkyl), —CH 2 —N(C 1 -C 3 alkyl) 2 , C 1 -C 4 alkyl(phenyl), or C 1 -C 4 alkyl(5- or 6-membered heteroaryl containing 1 or 2 N atoms), or C 1 -C 4 alkyl(5- or 6-membered heterocycloalkyl containing 1 or 2 N atoms).
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, cycloalkylene, and heterocycloalkylene. In other embodiments, Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 4-, 5-, 6- or 7-membered cycloalkylene, and 4-, 5-, 6- or 7-membered heterocycloalkylene.
- Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 5-, or 6-membered cycloalkylene, and 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some other embodiments, Y is a 5-, or 6-membered cycloalkylene, or a 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- compounds including irreversible inhibitors of ACKs, including Btk and its cysteine homologs having the structure of compounds of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), including, but are not limited to, compounds selected from the group consisting of:
- compounds including irreversible inhibitors of ACKs, including Btk and its cysteine homologs selected from among: (E)-4-(N-(2-hydroxyethyl)-N-methylamino)-1-(3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one (Compound 3); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3-(1H-imidazol-4-yl)prop-2-en-1-one (Compound 4); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 3); (E)
- the compounds of any of Formula (I), Formula (VII), Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), or Formula (D1-D6) irreversibly inhibit Btk and are optionally used to treat patients suffering from Bruton's tyrosine kinase-dependent or Bruton's tyrosine kinase mediated conditions or diseases, including, but not limited to, cancer, autoimmune and other inflammatory diseases.
- the reactions are optionally employed in a linear sequence to provide the compounds described herein or used to synthesize fragments which are subsequently joined by the methods described herein and/or documented elsewhere.
- the compounds described herein can be modified using various electrophiles or nucleophiles to form new functional groups or substituents.
- Table 1 entitled “Examples of Covalent Linkages and Precursors Thereof” lists selected examples of covalent linkages and precursor functional groups which yield and can be used as guidance toward the variety of electrophiles and nucleophiles combinations available.
- Precursor functional groups are shown as electrophilic groups and nucleophilic groups.
- each protective group be removable by a different means.
- Protective groups that are cleaved under totally disparate reaction conditions fulfill the requirement of differential removal.
- Protective groups can be removed by acid, base, and hydrogenolysis.
- Groups such as trityl, dimethoxytrityl, acetal and t-butyldimethylsilyl are acid labile and may be used to protect carboxy and hydroxy reactive moieties in the presence of amino groups protected with Cbz groups, which are removable by hydrogenolysis, and Fmoc groups, which are base labile.
- Carboxylic acid and hydroxy reactive moieties may be blocked with base labile groups such as, but not limited to, methyl, ethyl, and acetyl in the presence of amines blocked with acid labile groups such as t-butyl carbamate or with carbamates that are both acid and base stable but hydrolytically removable.
- Carboxylic acid and hydroxy reactive moieties may also be blocked with hydrolytically removable protective groups such as the benzyl group, while amine groups capable of hydrogen bonding with acids may be blocked with base labile groups such as Fmoc.
- Carboxylic acid reactive moieties may be protected by conversion to simple ester compounds as exemplified herein, or they may be blocked with oxidatively-removable protective groups such as 2,4-dimethoxybenzyl, while co-existing amino groups may be blocked with fluoride labile silyl carbamates.
- Allyl blocking groups are useful in then presence of acid- and base-protecting groups since the former are stable and can be subsequently removed by metal or pi-acid catalysts.
- an allyl-blocked carboxylic acid can be deprotected with a Pd 0 -catalyzed reaction in the presence of acid labile t-butyl carbamate or base-labile acetate amine protecting groups.
- Yet another form of protecting group is a resin to which a compound or intermediate may be attached. As long as the residue is attached to the resin, that functional group is blocked and cannot react. Once released from the resin, the functional group is available to react.
- blocking/protecting groups may be selected from:
- provided herein are methods of making and methods of using tyrosine kinase inhibitor compounds described herein.
- compounds described herein can be synthesized using the following synthetic schemes. Compounds may be synthesized using methodologies analogous to those described below by the use of appropriate alternative starting materials.
- Described herein are compounds that inhibit the activity of tyrosine kinase(s), such as Btk, and processes for their preparation. Also described herein are pharmaceutically acceptable salts, pharmaceutically acceptable solvates, pharmaceutically active metabolites and pharmaceutically acceptable prodrugs of such compounds. Pharmaceutical compositions that include at least one such compound or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, pharmaceutically active metabolite or pharmaceutically acceptable prodrug of such compound, are provided.
- the starting material used for the synthesis of the compounds described herein is either synthesized or obtained from commercial sources, such as, but not limited to, Aldrich Chemical Co. (Milwaukee, Wis.), Bachem (Torrance, Calif.), or Sigma Chemical Co. (St. Louis, Mo.).
- the compounds described herein, and other related compounds having different substituents are optionally synthesized using techniques and materials, such as described, for example, in March, A DVANCED O RGANIC C HEMISTRY 4 th Ed., (Wiley 1992); Carey and Sundberg, A DVANCED O RGANIC C HEMISTRY 4 th Ed., Vols.
- the products of the reactions are optionally isolated and purified, if desired, using conventional techniques, including, but not limited to, filtration, distillation, crystallization, chromatography and the like. Such materials are optionally characterized using conventional means, including physical constants and spectral data.
- Halogenation of commercially available 1H-pyrazolo[3,4-d]pyrimidin-4-amine provides an entry into the synthesis of compounds of Formula (A1-A6), (B1-B6), (C1-C6) and/or (D1-D6).
- 1H-pyrazolo[3,4-d]pyrimidin-4-amine is treated with N-iodosuccinamide to give 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine.
- Metal catalyzed cross coupling reactions are then carried out on 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine.
- palladium mediated cross-coupling of a suitably substituted phenyl boronic acid under basic conditions constructs intermediate 2.
- Intermediate 2 is coupled with N-Boc-3-hydroxypiperidine (as non-limiting example) via Mitsunobu reaction to give the Boc (tert-butyloxycarbonyl) protected intermediate 3.
- an acid chloride such as, but not limited to, acryloyl chloride
- tyrosine kinase inhibitors as disclosed herein are obtained in good yields and purity.
- the compounds prepared by the methods disclosed herein are purified by conventional means, such as, for example, filtration, recrystallization, chromatography, distillation, and combinations thereof.
- the compounds described herein may possess one or more stereocenters and each center may exist in the R or S configuration.
- the compounds presented herein include all diastereomeric, enantiomeric, and epimeric forms as well as the appropriate mixtures thereof.
- Stereoisomers may be obtained, if desired, by methods such as, for example, the separation of stereoisomers by chiral chromatographic columns.
- Diasteromeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods known, for example, by chromatography and/or fractional crystallization.
- enantiomers can be separated by chiral chromatographic columns.
- enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., alcohol), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereomers to the corresponding pure enantiomers. All such isomers, including diastereomers, enantiomers, and mixtures thereof are considered as part of the compositions described herein.
- the methods and formulations described herein include the use of N-oxides, crystalline forms (also known as polymorphs), or pharmaceutically acceptable salts of compounds described herein, as well as active metabolites of these compounds having the same type of activity.
- compounds exist as tautomers. All tautomers are included within the scope of the compounds presented herein.
- the compounds described herein can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. The solvated forms of the compounds presented herein are also considered to be disclosed herein.
- Compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) in unoxidized form can be prepared from N-oxides of compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) by treating with a reducing agent, such as, but not limited to, sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like in a suitable inert organic solvent, such as, but not limited to, acetonitrile, ethanol, aqueous dioxane, or the like at 0 to 80° C.
- a reducing agent such as, but not limited to, sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride
- compounds described herein are prepared as prodrugs.
- a “prodrug” refers to an agent that is converted into the parent drug in vivo. Prodrugs are often useful because, in some situations, they may be easier to administer than the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug.
- An example, without limitation, of a prodrug is a compound described herein, which is administered as an ester (the “prodrug”) to facilitate transmittal across a cell membrane where water solubility is detrimental to mobility but which then is metabolically hydrolyzed to the carboxylic acid, the active entity, once inside the cell where water-solubility is beneficial.
- a further example of a prodrug is a short peptide (polyaminoacid) bonded to an acid group where the peptide is metabolized to reveal the active moiety.
- a prodrug upon in vivo administration, is chemically converted to the biologically, pharmaceutically or therapeutically active form of the compound.
- a prodrug is enzymatically metabolized by one or more steps or processes to the biologically, pharmaceutically or therapeutically active form of the compound.
- a pharmaceutically active compound is modified such that the active compound will be regenerated upon in vivo administration.
- the prodrug can be designed to alter the metabolic stability or the transport characteristics of a drug, to mask side effects or toxicity, to improve the flavor of a drug or to alter other characteristics or properties of a drug.
- prodrugs of compounds can be designed (if desired) (for examples of this procedure applied to other compounds, see, e.g., Nogrady (1985) Medicinal Chemistry A Biochemical Approach , Oxford University Press, New York, pages 388-392; Silverman (1992), The Organic Chemistry of Drug Design and Drug Action, Academic Press, Inc., San Diego, pages 352-401, Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters , Vol. 4, p. 1985).
- Prodrug forms of the herein described compounds, wherein the prodrug is metabolized in vivo to produce a derivative as set forth herein are included within the scope of the claims. In some cases, some of the compounds herein-described are prodrugs for another derivative or active compound.
- Prodrugs are often useful because, in some situations, they are easier to administer than the parent drug. They are, for instance, bioavailable by oral administration whereas the parent is not.
- the prodrug optionally has improved solubility in pharmaceutical compositions over the parent drug.
- Prodrugs may be designed as reversible drug derivatives, for use as modifiers to enhance drug transport to site-specific tissues. In some embodiments, the design of a prodrug increases the effective water solubility. See, e.g., Fedorak et al., Am. J. Physiol., 269:G210-218 (1995); McLoed et al., Gastroenterol, 106:405-413 (1994); Hochhaus et al., Biomed.
- Sites on the aromatic ring portion of compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) can be susceptible to various metabolic reactions, therefore incorporation of appropriate substituents on the aromatic ring structures, such as, by way of example only, halogens can reduce, minimize or eliminate this metabolic pathway.
- Compounds described herein include isotopically-labeled compounds, which are identical to those recited in the various formulas and structures presented herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into the present compounds include isotopes of hydrogen, carbon, nitrogen, oxygen, fluorine and chlorine, such as 2 H, 3 H, 13 C, 14 C, 15 N, 18 O, 17 O, 35 S, 18 F, 36 Cl, respectively.
- isotopically-labeled compounds described herein for example those into which radioactive isotopes such as 3 H and 14 C are incorporated, are useful in drug and/or substrate tissue distribution assays. Further, substitution with isotopes such as deuterium, i.e., 2 H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements.
- the compounds described herein are metabolized upon administration to an organism in need to produce a metabolite that is then used to produce a desired effect, including a desired therapeutic effect.
- the type of pharmaceutical acceptable salts include, but are not limited to: (1) acid addition salts, formed) by reacting the free base form of the compound with a pharmaceutically acceptable: inorganic acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, metaphosphoric acid, and the like; or with an organic acid such as acetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, trifluoroacetic acid, tartaric acid, citric acid, benzoic acid, 3-(4-hydroxybenzoyl)benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2-ethanedisulfonic acid, 2-hydroxyethanesulfonic acid, benzenesulfonic acid
- organic bases include ethanolamine, diethanolamine, triethanolamine, tromethamine, N-methylglucamine, and the like.
- Acceptable inorganic bases include aluminum hydroxide, calcium hydroxide, potassium hydroxide, sodium carbonate, sodium hydroxide, and the like.
- the corresponding counterions of the pharmaceutically acceptable salts are optionally analyzed and identified using various methods including, but not limited to, ion exchange chromatography, ion chromatography, capillary electrophoresis, inductively coupled plasma, atomic absorption spectroscopy, mass spectrometry, or any combination thereof.
- the salts are recovered by using at least one of the following techniques: filtration, precipitation with a non-solvent followed by filtration, evaporation of the solvent, or, in the case of aqueous solutions, lyophilization.
- a reference to a pharmaceutically acceptable salt includes the solvent addition forms or crystal forms thereof, particularly solvates or polymorphs.
- Solvates contain either stoichiometric or non-stoichiometric amounts of a solvent, and are optionally formed during the process of crystallization with pharmaceutically acceptable solvents such as water, ethanol, and the like. Hydrates are formed when the solvent is water, or alcoholates are formed when the solvent is alcohol. Solvates of compounds described herein can be conveniently prepared or formed during the processes described herein.
- the compounds provided herein can exist in unsolvated as well as solvated forms. In general, the solvated forms are considered equivalent to the unsolvated forms for the purposes of the compounds and methods provided herein.
- a reference to a salt includes the solvent addition forms or crystal forms thereof, particularly solvates or polymorphs.
- Solvates contain either stoichiometric or non-stoichiometric amounts of a solvent, and are often formed during the process of crystallization with pharmaceutically acceptable solvents such as water, ethanol, and the like. Hydrates are formed when the solvent is water, or alcoholates are formed when the solvent is alcohol.
- Polymorphs include the different crystal packing arrangements of the same elemental composition of a compound. Polymorphs usually have different X-ray diffraction patterns, infrared spectra, melting points, density, hardness, crystal shape, optical and electrical properties, stability, and solubility. Various factors such as the recrystallization solvent, rate of crystallization, and storage temperature may cause a single crystal form to dominate.
- compounds described herein are optionally in various forms, including but not limited to, amorphous forms, milled forms and nano-particulate forms.
- compounds described herein include crystalline forms, also known as polymorphs.
- Polymorphs include the different crystal packing arrangements of the same elemental composition of a compound. Polymorphs usually have different X-ray diffraction patterns, infrared spectra, melting points, density, hardness, crystal shape, optical and electrical properties, stability, and solubility. Various factors such as the recrystallization solvent, rate of crystallization, and storage temperature may cause a single crystal form to dominate.
- the screening and characterization of the pharmaceutically acceptable salts, polymorphs and/or solvates may be accomplished using a variety of techniques including, but not limited to, thermal analysis, x-ray diffraction, spectroscopy, vapor sorption, and microscopy.
- Thermal analysis methods address thermo chemical degradation or thermo physical processes including, but not limited to, polymorphic transitions, and such methods are used to analyze the relationships between polymorphic forms, determine weight loss, to find the glass transition temperature, or for excipient compatibility studies.
- Such methods include, but are not limited to, Differential scanning calorimetry (DSC), Modulated Differential Scanning Calorimetry (MDCS), Thermogravimetric analysis (TGA), and Thermogravi-metric and Infrared analysis (TG/IR).
- DSC Differential scanning calorimetry
- MDCS Modulated Differential Scanning Calorimetry
- TGA Thermogravimetric analysis
- TG/IR Thermogravi-metric and Infrared analysis
- X-ray diffraction methods include, but are not limited to, single crystal and powder diffractometers and synchrotron sources.
- the various spectroscopic techniques used include, but are not limited to, Raman, FTIR, UVIS, and NMR (liquid and solid state).
- the various microscopy techniques include, but are not limited to, polarized light microscopy, Scanning Electron Microscopy (SEM) with Energy Dispersive X-Ray Analysis (EDX), Environmental Scanning Electron Microscopy with EDX (in gas or water vapor atmosphere), IR microscopy, and Raman microscopy.
- Protein kinases which act on and modify the activity of specific proteins, are used to transmit signals and control complex processes in cells. Up to 518 different kinases have been identified in humans. Many kinase inhibitor compounds non-selectively bind and/or inhibit these kinases because the active sites of some of these kinases are similar in structure. Such cross-reactivity is not a desired feature of a kinase inhibitor compound because of the potential for undesired side effects when such a compound is being administered to treat a disease or condition.
- kinase inhibitor compounds have profound effects in the selectivity of similarly-structured kinases (e.g., ACKs, including, Btk and the Btk kinase cysteine homologs).
- ACKs including, Btk and the Btk kinase cysteine homologs.
- the non-selective inhibitor compound is provided with a Michael acceptor moiety and a linker moiety that links the Michael acceptor moiety to the remainder of the non-selective inhibitor compound.
- a series of linker and Michael acceptor moieties provides a small library/panel of test inhibitor compounds.
- the inhibitor library/panel is contacted with a panel of structurally related kinases (e.g., Btk and the Btk kinase cysteine homologs).
- Binding is determined by a variety of means, included fluorescence detection (or via any other detectable label), mass spectrometry, or a combination of approaches.
- An Activity Probe is optionally used to detect binding of members of the inhibitor library/panel to the kinase library/panel.
- the binding data is then optionally collected and analyzed to provide a structure-activity relationship (SAR) between the structure of the members of the inhibitor panel/library (e.g., Michael acceptor and/or linker moieties) and the activity of binding to and/or inhibiting members of the kinase panel.
- SAR structure-activity relationship
- a similar approach can be use for converting a selective inhibitor compound for a group of similarly-structured ACKs (including, Btk and the Btk kinase cysteine homologs) into a more highly-selective inhibitor compound (e.g., more selective for a particular ACK over structurally-similar ACKs), or for converting a selective inhibitor compound for a particular ACK (e.g., Btk) into an even more selective inhibitor of that particular ACK.
- the selective inhibitor compound which, for example, contains an active-site binding moiety, a linker moiety and a Michael acceptor moiety is modified.
- a series of linker and Michael acceptor moieties provides a small library/panel of test inhibitor compounds.
- the inhibitor library/panel is contacted with a panel of structurally related kinases (e.g., Btk and the Btk kinase cysteine homologs). Binding is determined by a variety of means, included fluorescence detection (or via any other detectable label), mass spectrometry, or a combination of approaches.
- An Activity Probe is optionally used to detect binding of members of the inhibitor library/panel to the kinase library/panel.
- the binding data is then optionally collected and analyzed to provide a structure-activity relationship (SAR) between the structure of the members of the inhibitor panel/library (e.g., Michael acceptor and/or linker moieties) and the activity of binding to and/or inhibiting members of the kinase panel.
- SAR structure-activity relationship
- an electrophilic center capable of irreversibly inactivating the target enzyme, BTK. That is, to an active site binding moiety of a reversible inhibitor was added a linker moiety and a Michael acceptor moiety that achieved a high degree of potency and selectivity by (1) fitting the core scaffold into the active site ATP binding pocket of kinase enzymes, and (2) forming a covalent bond with Cysteine-481 located in BTK.
- the chemistry required for covalent bond formation involves an electrophilic moiety that acts as a Michael acceptor, which bonds with a nucleophile (such as Cys-481) present in a precise location within the active site.
- the linker and Michael acceptor moiety of Compound 1 was modified to provide Compound 9 which has a different selectivity pattern.
- Table 1 is a table showing the degree of inhibition of a panel of kinases for two example compounds. IC 50 s were determined using the in vitro HotSpot kinase assay (purified enzymes, 33P-ATP, an appropriate substrate and 1 uM ATP.)
- Compound 9 has similar potency toward Btk, but significantly less potency toward JAK-3, ITK, and EGFR and significantly more potency toward the src-family kinases lck, c-src, FGR, Fyn, Hck, and Lyn and Yes.
- subtle modifications in the linker moiety and the Michael acceptor moiety are important for the design of selective ACK inhibitors.
- Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the examples section provides further modifications of the linker moiety and/or the Michael acceptor moiety and the impact of such changes of inhibitor selectivity.
- kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog (or indeed, any ACK) comprising:
- the following steps are added: (4) comparing the covalent binding of the compound that comprises a Michael acceptor with the multiplicity of kinases and the at least one non-kinase molecule; and repeating steps (1), (2), (3) and (4) for at least one other compound that comprises a Michael acceptor moiety.
- the irreversible inhibitor compounds are also contacted with at least one non-ACK kinase in order to determine the selectivity of the irreversible inhibitor compound for the ACK relative to the non-ACK.
- non-kinase molecules with at least one accessible SH group are glutathione and/or hemoglobin. Because of the high abundance of these molecules in typical biological systems (e.g., in a patient), the desired irreversible inhibitor compounds have low selectivity/reactivity with these non-kinase molecules.
- an Activity Probe (described in more detail herein) is used as a rapid diagnostic method for determining whether a test inhibitor compound has irreversibly inhibited an ACK.
- the Activity Probe is itself an irreversible inhibitor of an ACK, and further, has a reporter moiety (e.g., a fluorescent moiety) as part of its structure.
- a reporter moiety e.g., a fluorescent moiety
- the absence of a ‘reporter’ signal on an ACK is one indication that the test irreversible inhibitor has prevented the Activity Probe from binding to the ACK (and that the test irreversible inhibitor has a higher binding affinity for the ACK than the Activity Probe).
- the Kinase Inhibitor Discovery Platform, steps (1) and (2) are conducted in vivo and step (3) is conducted in part using an Activity Probe. Further, in certain embodiments, the determining step uses mass spectrometry, fluorescence, or a combination thereof.
- the inhibitor tested with the Kinase Inhibitor Discovery Platform comprise an active site binding moiety, a Michael acceptor moiety, and a linker moiety that links the Michael acceptor moiety to the active site binding moiety.
- the following information is collected and analyzed: the structure-function activity relationship between the structure of the linker moiety and/or the Michael acceptor moiety of each compound, and the binding and/or selectivity of each compound to at least one kinase.
- structure of the active site binding moiety of each compound is not varied, whereas the structure of the linker moiety and/or the Michael acceptor moiety is varied.
- the inhibitors have the structure of Formula (VII):
- kinase is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
- each compound and the binding and/or selectivity of each compound to at least one kinase. Further, the structure of
- each compound is not varied, whereas the structure of the linker moiety (Y—Z) and/or the Michael acceptor moiety
- the resulting inhibitor is selective for one kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog over at least one other kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog.
- this selectivity is at least 5 ⁇ , at least 10 ⁇ , at least 20 ⁇ , at least 50 ⁇ , or at least 100 ⁇ .
- the resulting inhibitor is selective for at least one kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog over at least one other non-kinase molecule having an accessible SH group.
- this selectivity is at least 5 ⁇ , at least 10 ⁇ , at least 20 ⁇ , at least 50 ⁇ , or at least 100 ⁇ .
- the resulting inhibitor is used in the therapeutic methods described herein, or in the pharmaceutical compositions described herein.
- the Activity Probe compounds described herein are composed of a moiety comprising an inhibitor of Btk, a Btk homolog and/or a Btk kinase cysteine homolog (hereinafter, a “Kinase Inhibitor”), a linker moiety, and a reporter moiety.
- the Kinase Inhibitor is an irreversible inhibitor.
- the irreversible Kinase Inhibitor binds to a non-catalytic residue in the ATP binding pocket of Btk, a Btk homolog and/or a Btk kinase cysteine homolog (hereinafter a “Kinase”); in further embodiments, the non-catalytic residue is a cysteine residue.
- the Activity Probe forms a covalent bond with at least one non-catalytic residue of a Kinase.
- the Activity Probe forms a non-covalent bond with at least one non-catalytic residue of a Kinase.
- the Activity Probe forms hydrogen bonding within the ATP binding pocket of a Kinase.
- the Activity Probe has Van der Waals attractions with the Kinase.
- the Activity Probes described herein are activity dependent such that the probe binds only an active Kinase. In further embodiments, the Activity Probe binds a Kinase that has been switched on by phosphorylation by upstream kinases. In yet a further embodiment, the Activity Probes described herein are activity independent such that the probe binds Kinases that have not been switched on by phosphorylation by upstream kinases. In some embodiments, the Activity Probe labels a phosphorylated conformation of a Kinase. In other embodiments, the Activity Probe labels a Kinase in a non-phosphorylated conformation.
- the Activity Probe is permeable to cells.
- the linker moiety is selected from a bond, a substituted alkyl moiety, a substituted heterocycle moiety, a substituted amide moiety, a ketone moiety, a substituted carbamate moiety, an ester moiety, or any combination thereof.
- the reporter moiety is a moiety that is detected using standard or modified laboratory equipment.
- Activity Probe of Formula (I) comprising:
- the moiety comprising an irreversible Kinase Inhibitor is derived from an irreversible inhibitor of a Kinase.
- such irreversible Kinase Inhibitors should possess at least one of the following characteristics: potency, selectively and cell permeability.
- such irreversible Kinase Inhibitors possess at least two of the aforementioned characteristics, and in further embodiments, at least all of the aforementioned characteristics.
- the Kinase Inhibitor moiety is derived from a Btk inhibitor having the structure of Formula (II):
- L a is CH 2 , O, or NH. In other embodiments, L a is O or NH. In yet other embodiments, L a is O.
- Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene.
- Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene, 4-, 5-, 6-, or 7-membered cycloalkylene, and 4-, 5-, 6-, or 7-membered heterocycloalkylene.
- Y is an optionally substituted group selected from among C 1 -C 6 alkylene, C 1 -C 6 heteroalkylene 5- or 6-membered cycloalkylene, and 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- Y is a 5- or 6-membered cycloalkylene, or a 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- Y is a 4-, 5-, 6-, or 7-membered cycloalkylene ring; or Y is a 4-, 5-, 6-, or 7-membered heterocycloalkylene ring.
- the Kinase Inhibitor moiety is derived from a compound selected from among: 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one; 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)sulfonylethene; 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)sulf
- the linker moiety is selected from a bond, a polymer, a water soluble polymer, optionally substituted alkyl, optionally substituted heteroalkyl, optionally substituted heterocycloalkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkylalkyl, optionally substituted heterocycloalkylalkenyl, optionally substituted aryl, optionally substituted heteroaryl, and optionally substituted heterocycloalkylalkenylalkyl.
- the linker moiety is an optionally substituted heterocycle.
- the heterocycle is selected from aziridine, oxirane, episulfide, azetidine, oxetane, pyrroline, tetrahydrofuran, tetrahydrothiophene, pyrrolidine, pyrazole, pyrrole, imidazole, triazole, tetrazole, oxazole, isoxazole, oxirene, thiazole, isothiazole, dithiolane, furan, thiophene, piperidine, tetrahydropyran, thiane, pyridine, pyran, thiapyrane, pyridazine, pyrimidine, pyrazine, piperazine, oxazine, thiazine, dithiane, and dioxane.
- the heterocycle is piperazine.
- the linker moiety is optionally substituted with halogen, CN, OH, NO 2 , alkyl, S(O), and S(O) 2 .
- the water soluble polymer is a PEG group.
- the linker moiety provides sufficient spatial separation between the reporter moiety and the Kinase Inhibitor moiety. In further embodiments, the linker moiety is stable. In yet a further embodiment, the linker moiety does not substantially affect the response of the reporter moiety. In other embodiments the linker moiety provides chemical stability to the Activity Probe. In further embodiments, the linker moiety provides sufficient solubility to the Activity Probe.
- linkages such as water soluble polymers are coupled at one end to a Kinase Inhibitor moiety and to a reporter moiety at the other end.
- the water soluble polymers are coupled via a functional group or substituent of the Kinase Inhibitor moiety.
- the water soluble polymers are coupled via a functional group or substituent of the reporter moiety.
- covalent attachment of hydrophilic polymers to a Kinase Inhibitor moiety and a reporter moiety represents one approach to increasing water solubility (such as in a physiological environment), bioavailability, increasing serum half-life, increasing pharmacodynamic parameters, or extending the circulation time of the Activity Probe, including proteins, peptides, and particularly hydrophobic molecules.
- additional important features of such hydrophilic polymers include biocompatibility and lack of toxicity.
- the polymer is pharmaceutically acceptable for therapeutic use of the end-product preparation.
- examples of hydrophilic polymers include, but are not limited to: polyalkyl ethers and alkoxy-capped analogs thereof (e.g., polyoxyethylene glycol, polyoxyethylene/propylene glycol, and methoxy or ethoxy-capped analogs thereof, polyoxyethylene glycol, the latter is also known as polyethylene glycol or PEG); polyvinylpyrrolidones; polyvinylalkyl ethers; polyoxazolines, polyalkyl oxazolines and polyhydroxyalkyl oxazolines; polyacrylamides, polyalkyl acrylamides, and polyhydroxyalkyl acrylamides (e.g., polyhydroxypropylmethacrylamide and derivatives thereof); polyhydroxyalkyl acrylates; polysialic acids and analogs thereof; hydrophilic peptide sequences; polysaccharides and their derivatives, including dextran and dextran derivatives, e.g., carboxymethyldextran, dextran sul
- the water soluble polymer is any structural form including but not limited to linear, forked or branched.
- polymer backbones that are water-soluble, with from 2 to about 300 termini, are particularly useful.
- multifunctional polymer derivatives include, but are not limited to, linear polymers having two termini, each terminus being bonded to a functional group which is the same or different.
- the water polymer comprises a poly(ethylene glycol) moiety.
- the molecular weight of the polymer is of a wide range, including but not limited to, between about 100 Da and about 100,000 Da or more.
- the molecular weight of the polymer is between about 100 Da and about 100,000 Da, including but not limited to, about 100,000 Da, about 95,000 Da, about 90,000 Da, about 85,000 Da, about 80,000 Da, about 75,000 Da, about 70,000 Da, about 65,000 Da, about 60,000 Da, about 55,000 Da, about 50,000 Da, about 45,000 Da, about 40,000 Da, about 35,000 Da, 30,000 Da, about 25,000 Da, about 20,000 Da, about 15,000 Da, about 10,000 Da, about 9,000 Da, about 8,000 Da, about 7,000 Da, about 6,000 Da, about 5,000 Da, about 4,000 Da, about 3,000 Da, about 2,000 Da, about 1,000 Da, about 900 Da, about 800 Da, about 700 Da, about 600 Da, about 500 Da, about 400 Da, about 300 Da, about 200 Da, and about 100 Da.
- the molecular weight of the polymer is between about 100 Da and 50,000 Da. In some embodiments, the molecular weight of the polymer is between about 100 Da and 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 1,000 Da and 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 5,000 Da and 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 10,000 Da and 40,000 Da. In some embodiments, the poly(ethylene glycol) molecule is a branched polymer.
- the molecular weight of the branched chain PEG is between about 1,000 Da and about 100,000 Da, including but not limited to, about 100,000 Da, about 95,000 Da, about 90,000 Da, about 85,000 Da, about 80,000 Da, about 75,000 Da, about 70,000 Da, about 65,000 Da, about 60,000 Da, about 55,000 Da, about 50,000 Da, about 45,000 Da, about 40,000 Da, about 35,000 Da, about 30,000 Da, about 25,000 Da, about 20,000 Da, about 15,000 Da, about 10,000 Da, about 9,000 Da, about 8,000 Da, about 7,000 Da, about 6,000 Da, about 5,000 Da, about 4,000 Da, about 3,000 Da, about 2,000 Da, and about 1,000 Da.
- the molecular weight of the branched chain PEG is between about 1,000 Da and about 50,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 1,000 Da and about 40,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 5,000 Da and about 40,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 5,000 Da and about 20,000 Da.
- the foregoing list for substantially water soluble backbones is by no means exhaustive and is merely illustrative, and in some embodiments, the polymeric materials having the qualities described above suitable for use in methods and compositions described herein.
- the number of water soluble polymers linked to a Kinase Inhibitor moiety and a reporter moiety described herein is adjusted to provide an altered (including but not limited to, increased or decreased) pharmacologic, pharmacokinetic or pharmacodynamic characteristic such as in vivo half-life.
- the half-life of the Activity Probe is increased at least about 10, about 20, about 30, about 40, about 50, about 60, about 70, about 80, about 90 percent, about two fold, about five-fold, about 10-fold, about 50-fold, or at least about 100-fold over a Activity Probe without a water soluble linker.
- X is selected from the group consisting of: a bond, —O(C ⁇ O)—, —NR a (C ⁇ O)—, —NR a —,
- X is NR a (C ⁇ O). In another embodiment, X is a bond. In another embodiment, X is —O(C ⁇ O)—. In a further embodiment, Y is selected from the group consisting of: a bond, —O(C ⁇ O)—, —NR a (C ⁇ O)—, —NR a —,
- Y is a bond. In one embodiment, Y is —NR a (C ⁇ O)—. In yet another embodiment, R a is hydrogen. In yet a further embodiment, R a is alkyl.
- the reporter moiety is selected from the group consisting of a label, a dye, a photocrosslinker, a cytotoxic compound, a drug, an affinity label, a photoaffinity label, a reactive compound, an antibody or antibody fragment, a biomaterial, a nanoparticle, a spin label, a fluorophore, a metal-containing moiety, a radioactive moiety, a novel functional group, a group that covalently or noncovalently interacts with other molecules, a photocaged moiety, an actinic radiation excitable moiety, a ligand, a photoisomerizable moiety, biotin, a biotin analog, a moiety incorporating a heavy atom, a chemically cleavable group, a photocleavable group, a redox-active agent, an isotopically labeled moiety, a biophysical probe, a phosphorescent group, a chemiluminescent group, an electron dense group
- the reporter moiety is a fluorophore.
- the fluorophore is selected from the group consisting of: BODIPY 493/503, BODIPY FL, BODIPY R6G, BODIPY 530/550, BODIPY TMR, BODIPY 558/568, BODIPY 564/570, BODIPY 576/589, BODIPY 581/591, BODIPY TR, Fluorescein, 5(6)-Carboxyfluorescein, 2,7-Dichlorofluorescein, N,N-Bis(2,4,6-trimethylphenyl)-3,4:9,10-perylenebis(dicarboximide, HPTS, Ethyl Eosin, DY-490XL MegaStokes, DY-485XL MegaStokes, Adirondack Green 520, ATTO 465, ATTO 488, ATTO 495, YOYO-1, 5-FAM, BCECF, B
- the fluorophore is selected from the group consisting of: BODIPY 493/503, BODIPY FL, BODIPY R6G, BODIPY 530/550, BODIPY TMR, BODIPY 558/568, BODIPY 564/570, BODIPY 576/589, BODIPY 581/591, and BODIPY TR.
- the fluorophore is BODIPY FL.
- the fluorophore is not BODIPY 530.
- the fluorophore has an excitation maxima of between about 500 and about 600 nm.
- the fluorophore has an excitation maxima of between about 500 and about 550 nm. In another embodiments, the fluorophore has an excitation maxima of between about 550 and about 600 nm. In yet a further embodiment, the fluorophore has an excitation maxima of between about 525 and about 575 nm. In other embodiments, the fluorophore has an emission maxima of between about 510 and about 670 nm. In another embodiment, the fluorophore has an emission maxima of between about 510 and about 600 nm. In a further embodiment, the fluorophore has an emission maxima of between about 600 and about 670 nm. In another embodiment, the fluorophore has an emission maxima of between about 575 and about 625 nm.
- the observed potency, selectivity, and cell permeability of compounds such as Compound 2 are appropriate to incorporate these molecules into a Kinase-targeted, activity based probe that allows direct visualization of Kinase activity in intact cells.
- Compound 2 In vitro profiling against a panel of greater than 100 kinases showed Compound 2 to be a highly potent and selective inhibitor of Tec family kinases, including, Btk, as well as Src family kinases.
- the structural basis for the selectivity is covalent modification of a non-catalytic cysteine residue (Cys 481 in Btk) that is conserved in the ATP binding pocket of the Tec family and several other kinases.
- any irreversible Kinase Inhibitor that binds to the non-catalytic cysteine residue in the ATP binding pocket of a Kinase is used in the compounds and methods described herein.
- an illustrative probe was synthesized by attaching a bodipy FL fluorophore to an irreversible inhibitor via a piperazine linker.
- the piperazine linker served to maintain probe solubility and provided spatial separation between the fluorophore and the pyrazolopyrimidine core.
- the linkage formed is a stable linkage.
- the linker moiety forms a linkage, in some embodiments, a stable linkage, between the Kinase Inhibitor moiety and the reporter moiety.
- the linker moiety is stable and provides the means to control and determine the distance between the Kinase Inhibitor moiety and the report moiety.
- the linker moiety is selected such that the probe's solubility is maintained.
- the linker moiety is a piperazinyl moiety.
- a piperazinyl-based linkage is formed by using a piperazine containing compound.
- the number and order of units that comprise the linker moiety is selected such that the length between the first component and the second component, as well as the hydrophobic and hydrophilic characteristics of the linker is controlled.
- spatial separation means a thermochemically and photochemically non-active distance-making group and in some embodiments is used to join two or more different moieties of the types defined above.
- spacers are selected on the basis of a variety of characteristics including their hydrophobicity, hydrophilicity, molecular flexibility and length.
- the spacer thus, in some embodiments, comprises a chain of carbon atoms optionally interrupted or terminated with one or more heteroatoms, such as oxygen atoms, nitrogen atoms, and/or sulphur atoms.
- the spacer comprises one or more amide, ester, amino, ether, and/or thioether functionalities, and optionally aromatic or mono/polyunsaturated hydrocarbons, polyoxyethylene such as polyethylene glycol, oligo/polyamides such as poly- ⁇ -alanine, polyglycine, polylysine, and peptides in general, oligosaccharides, oligo/polyphosphates.
- the spacer consists of combined units thereof.
- the length of the spacer varies, taking into consideration the desired or necessary positioning and spatial orientation of the active/functional part of the Activity Probe.
- the reporter moiety is Bodipy.
- reporter moiety means a group which is detectable either by itself or as a part of a detection series.
- the labeled Activity Probes described herein are purified by one or more procedures including, but are not limited to, affinity chromatography; anion- or cation-exchange chromatography (using, including but not limited to, DEAE SEPHAROSE); chromatography on silica; reverse phase HPLC; gel filtration (using, including but not limited to, SEPHADEX G-75); hydrophobic interaction chromatography; size-exclusion chromatography, metal-chelate chromatography; ultrafiltration/diafiltration; ethanol precipitation; ammonium sulfate precipitation; chromatofocusing; displacement chromatography; electrophoretic procedures (including but not limited to preparative isoelectric focusing), differential solubility (including but not limited to ammonium sulfate precipitation), or extraction.
- affinity chromatography anion- or cation-exchange chromatography (using, including but not limited to, DEAE SEPHAROSE); chromatography on silica; reverse phase HPLC; gel filtration (using, including but not limited to, SE
- apparent molecular weight is estimated by GPC by comparison to globular protein standards (P ROTEIN P URIFICATION M ETHODS, A P RACTICAL A PPROACH (Harris & Angal, Eds.) IRL Press 1989, 293-306).
- the in vitro inhibitory potency of a probe against a panel of selected Kinases as a rapid means of confirming accessibility of the reactive moiety to the Kinase active site is tested.
- the illustrative probe of Compound 3 retains potency against Btk (IC 50 ⁇ 90 nM).
- Btk IC 50 ⁇ 90 nM
- the Activity Probes described herein label kinases at the non-catalytic Cys 481 (or a homologous cysteine) and that in some embodiments, probe labeling does not require the catalytic machinery per se. As such it differs from canonical activity-based probes that target the enzyme catalytic machinery directly.
- the Kinase undergoes a phosphorylation dependent conformational change that is tightly coupled to ATP binding and kinase activation.
- effective labeling by a probe requires the Kinase to be in its active conformation in order to directly detect Kinase activity in cells.
- effective labeling by a Activity Probe does not require the Kinase to be in its active conformation in order to directly detect Kinase activity in cells.
- Described herein are methods, compositions, uses and medicaments for the treatment of disorders comprising administering to a patient in need an irreversible inhibitor of an ACK.
- the ACK is Btk or a Btk homolog.
- the ACK is Blk or a Blk homolog.
- the ACK is tyrosine kinases that share homology with Btk by having a cysteine residue (including a Cys 481 residue) that can form a covalent bond with the irreversible inhibitor. See, e.g., protein kinases in FIG. 7 .
- the methods described herein (which includes uses of a pharmaceutical composition to treat a disease or disorder, or uses of a compound to form a medicament for treating a disease or disorder) include administering to a subject in need a composition containing a therapeutically effective amount of one or more irreversible Btk inhibitor compounds described herein.
- Btk signaling in various hematopoietic cell functions e.g., B-cell receptor activation
- small molecule Btk inhibitors are useful for reducing the risk of or treating a variety of diseases affected by or affecting many cell types of the hematopoietic lineage including, e.g., autoimmune diseases, heteroimmune conditions or diseases, inflammatory diseases, cancer (e.g., B-cell proliferative disorders), and thromboembolic disorders.
- autoimmune disease or condition comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Such an autoimmune disease or condition includes, but is not limited to, rheumatoid arthritis, psoriatic arthritis, osteoarthritis, Still's disease, juvenile arthritis, lupus, diabetes, myasthenia gravis, Hashimoto's thyroiditis, Ord's thyroiditis, Graves' disease Sjögren's syndrome, multiple sclerosis, Guillain-Barre syndrome, acute disseminated encephalomyelitis, Addison's disease, opsoclonus-myoclonus syndrome, ankylosing spondylitisis, antiphospholipid antibody syndrome, aplastic anemia, autoimmune hepatitis, coeliac disease, Goodpasture's syndrome, idiopathic thrombocytopenic purpura, optic neuritis, scleroderma, primary biliary cirrhosis, Reiter's syndrome, Takayasu's arteritis, temporal arteritis, warm autoimmune hemolytic anemia,
- any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- the autoimmune disease is selected from rheumatoid arthritis or lupus.
- are methods for treating a heteroimmune disease or condition comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Such a heteroimmune condition or disease includes, but is not limited to graft versus host disease, transplantation, transfusion, anaphylaxis, allergies (e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or cockroach calyx), type I hypersensitivity, allergic conjunctivitis, allergic rhinitis, and atopic dermatitis.
- allergies e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or cockroach calyx
- type I hypersensitivity e.g., allergic conjunctivitis, allergic rhinitis, and atopic dermatitis.
- any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- are methods for treating a cancer comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Such a cancer includes but is not limited to diffuse large B cell lymphoma, follicular lymphoma, chronic lymphocytic lymphoma, chronic lymphocytic leukemia, B-cell prolymphocytic leukemia, lymphoplasmacytic lymphoma/Waldenström macroglobulinemia, splenic marginal zone lymphoma, plasma cell myeloma, plasmacytoma, extranodal marginal zone B cell lymphoma, nodal marginal zone B cell lymphoma, mantle cell lymphoma, mediastinal (thymic) large B cell lymphoma, intravascular large B cell lymphoma, primary effusion lymphoma, burkitt lymphoma/leukemia, and lymphomatoid granulomatosis.
- diffuse large B cell lymphoma follicular lymphoma
- chronic lymphocytic lymphoma chronic lymphocytic leukemia
- B-cell prolymphocytic leukemia
- any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- the cancer is a B-cell proliferative disorder.
- the B-cell proliferative disorder is diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia.
- mastocytosis in some embodiments, are methods for treating mastocytosis comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Mastocytosis includes but is not limited to diseases characterized by hyperactive mast cells.
- any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- bone resorption disorders comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Bone resorption disorders include but are not limited to Paget's disease of bone, osteoporosis, and the bone changes secondary to cancer, such as occur in myeloma and metastases from breast cancer.
- any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- are methods for treating inflammatory diseases comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Inflammatory diseases include but are not limited to asthma, inflammatory bowel disease, appendicitis, blepharitis, bronchiolitis, bronchitis, bursitis, cervicitis, cholangitis, cholecystitis, colitis, conjunctivitis, cystitis, dacryoadenitis, dermatitis, dermatomyositis, encephalitis, endocarditis, endometritis, enteritis, enterocolitis, epicondylitis, epididymitis, fasciitis, fibrositis, gastritis, gastroenteritis, hepatitis, hidradenitis suppurativa, laryngitis, mastitis, meningitis, myelitis myocarditis, myositis, nephritis, oophoritis, orchitis, osteitis, otitis, pancreatitis, parotitis
- compositions for treating lupus comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- compositions for treating a heteroimmune disease or condition comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- Such a heteroimmune condition or disease includes, but is not limited to graft versus host disease, transplantation, transfusion, anaphylaxis, allergies (e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or cockroach calyx), type I hypersensitivity, allergic conjunctivitis, allergic rhinitis, and atopic dermatitis.
- allergies e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or cockroach calyx
- type I hypersensitivity e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or cockroach calyx
- type I hypersensitivity e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or
- compositions for treating diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- compositions for treating mastocytosis comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- compositions for treating osteoporosis or bone resorption disorders comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- compositions for treating an inflammatory disease or condition comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- Suitable compounds include compounds of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Such an autoimmune disease or condition includes, but is not limited to, rheumatoid arthritis, psoriatic arthritis, osteoarthritis, Still's disease, juvenile arthritis, lupus, diabetes, myasthenia gravis, Hashimoto's thyroiditis, Ord's thyroiditis, Graves' disease Sjögren's syndrome, multiple sclerosis, Guillain-Barré syndrome, acute disseminated encephalomyelitis, Addison's disease, opsoclonus-myoclonus syndrome, ankylosing spondylitisis, antiphospholipid antibody syndrome, aplastic anemia, autoimmune hepatitis, coeliac disease, Goodpasture's syndrome, idiopathic thrombocytopenic purpura, optic neuritis, scleroderma, primary biliary cirrhosis, Reiter's syndrome, Takayasu's arteritis, temporal arteritis, warm autoimmune hemolytic anemia,
- any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- the autoimmune disease is selected from rheumatoid arthritis or lupus.
- a B-cell proliferative disorder comprising administering to a patient in need a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Blk or a Blk homolog.
- Suitable compounds include compounds of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Such a B-cell proliferative disorder includes diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia.
- any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- Suitable compounds include compounds of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- Inflammatory diseases include but are not limited to asthma, inflammatory bowel disease, appendicitis, blepharitis, bronchiolitis, bronchitis, bursitis, cervicitis, cholangitis, cholecystitis, colitis, conjunctivitis, cystitis, dacryoadenitis, dermatitis, dermatomyositis, encephalitis, endocarditis, endometritis, enteritis, enterocolitis, epicondylitis, epididymitis, fasciitis, fibrositis, gastritis, gastroenteritis, hepatitis, hidradenitis suppurativa, laryngitis, mastitis, meningitis, myelitis myocarditis, myositis, nephritis, oophoritis, orchitis, osteitis, otitis, pancreatitis, parotitis
- the irreversible Btk inhibitor compounds described herein can be used to inhibit a small subset of other tyrosine kinases that share homology with Btk by having a cysteine residue (including a Cys 481 residue) that can form a covalent bond with the irreversible inhibitor. See, e.g., protein kinases in FIG. 7 .
- a subset of tyrosine kinases other than Btk are also expected to be useful as therapeutic targets in a number of health conditions, including:
- a number of animal models are useful for establishing a range of therapeutically effective doses of irreversible inhibitors, including irreversible Btk inhibitor compounds for treating any of the foregoing diseases.
- irreversible Btk inhibitor compounds for treating any of the foregoing diseases.
- dosing of irreversible inhibitor compounds for treating an autoimmune disease can be assessed in a mouse model of rheumatoid arthitis. In this model, arthritis is induced in Balb/c mice by administering anti-collagen antibodies and lipopolysaccharide. See Nandakumar et al. (2003), Am. J. Pathol 163:1827-1837.
- dosing of irreversible inhibitors for the treatment of B-cell proliferative disorders can be examined in, e.g., a human-to-mouse xenograft model in which human B-cell lymphoma cells (e.g. Ramos cells) are implanted into immunodefficient mice (e.g., “nude” mice) as described in, e.g., Pagel et al. (2005), Clin Cancer Res 11(13):4857-4866. Animal models for treatment of thromboembolic disorders are also known.
- the therapeutic efficacy of the compound for one of the foregoing diseases is optimized during a course of treatment.
- a subject being treated optionally undergoes a diagnostic evaluation to correlate the relief of disease symptoms or pathologies to inhibition of in vivo Btk activity achieved by administering a given dose of an irreversible Btk inhibitor.
- Cellular assays are used to determine in vivo activity of Btk in the presence or absence of an irreversible Btk inhibitor.
- activated Btk is phosphorylated at tyrosine 223 (Y223) and tyrosine 551 (Y551)
- phospho-specific immunocytochemical staining of P-Y223 or P-Y551-positive cells are used to detect or quantify activation of Bkt in a population of cells (e.g., by FACS analysis of stained vs unstained cells). See, e.g., Nisitani et al. (1999), Proc. Natl. Acad. Sci, USA 96:2221-2226.
- the amount of the Btk inhibitor inhibitor compound that is administered to a subject is optionally increased or decreased as needed so as to maintain a level of Btk inhibition optimal for treating the subject's disease state.
- an irreversible ACK inhibitor including, e.g., a compound of Formula (I)
- administering comprising administering to a test subject a composition containing an amount of the irreversible ACK inhibitor (including, e.g., a compound of Formula (I)) sufficient to inhibit B cell receptor signaling and correlating B cell receptor signaling with apoptosis.
- a patient for treatment for lymphoma with an irreversible ACK inhibitor comprising measuring pErk or Erk transcriptional target levels in a patient sample, and correlating a high level of transcriptional targets with a positive response to the treatment.
- in another or further embodiments are methods for measuring a patient's response to treatment comprising administering to the patient an irreversible ACK inhibitor (including, e.g., a compound of Formula (I)), measuring pErk or Erk transcriptional target levels in a patient sample, and correlating a reduced level of transcriptional targets with a positive response to the administration of the irreversible ACK inhibitor (including, e.g., a compound of Formula (I)).
- an irreversible ACK inhibitor including, e.g., a compound of Formula (I)
- the irreversible Btk inhibitor compositions described herein can also be used in combination with other well known therapeutic reagents that are selected for their therapeutic value for the condition to be treated.
- the compositions described herein and, in embodiments where combinational therapy is employed other agents do not have to be administered in the same pharmaceutical composition, and are optionally, because of different physical and chemical characteristics, have to be administered by different routes.
- the initial administration is made, for example, according to established protocols, and then, based upon the observed effects, the dosage, modes of administration and times of administration are modified.
- At least one irreversible Btk inhibitor compound described herein in combination with another therapeutic agent.
- another therapeutic agent if one of the side effects experienced by a patient upon receiving one of the irreversible Btk inhibitor compounds described herein is nausea, then it is appropriate to administer an anti-nausea agent in combination with the initial therapeutic agent.
- the therapeutic effectiveness of one of the compounds described herein is enhanced by administration of an adjuvant (i.e., by itself the adjuvant has minimal therapeutic benefit, but in combination with another therapeutic agent, the overall therapeutic benefit to the patient is enhanced).
- the benefit experienced by a patient is increased by administering one of the compounds described herein with another therapeutic agent (which also includes a therapeutic regimen) that also has therapeutic benefit.
- another therapeutic agent which also includes a therapeutic regimen
- the overall benefit experienced by the patient is in some embodiments simply additive of the two therapeutic agents or in other embodiments, the patient experiences a synergistic benefit.
- the particular choice of compounds used will depend upon the diagnosis of the attending physicians and their judgment of the condition of the patient and the appropriate treatment protocol.
- the compounds are optionally administered concurrently (e.g., simultaneously, essentially simultaneously or within the same treatment protocol) or sequentially, depending upon the nature of the disease, disorder, or condition, the condition of the patient, and the actual choice of compounds used.
- the determination of the order of administration, and the number of repetitions of administration of each therapeutic agent during a treatment protocol, is based on an evaluation of the disease being treated and the condition of the patient.
- Therapeutically-effective dosages can vary when the drugs are used in treatment combinations. Methods for experimentally determining therapeutically-effective dosages of drugs and other agents for use in combination treatment regimens are described in the literature. For example, the use of metronomic dosing, i.e., providing more frequent, lower doses in order to minimize toxic side effects, has been described extensively in the literature Combination treatment further includes periodic treatments that start and stop at various times to assist with the clinical management of the patient.
- dosages of the co-administered compounds will of course vary depending on the type of co-drug employed, on the specific drug employed, on the disease or condition being treated and so forth.
- the compound provided herein may be administered either simultaneously with the biologically active agent(s), or sequentially. If administered sequentially, the attending physician will decide on the appropriate sequence of administering protein in combination with the biologically active agent(s).
- the multiple therapeutic agents are optionally administered in any order or even simultaneously. If simultaneously, the multiple therapeutic agents are optionally provided in a single, unified form, or in multiple forms (by way of example only, either as a single pill or as two separate pills). One of the therapeutic agents may be given in multiple doses, or both may be given as multiple doses. If not simultaneous, the timing between the multiple doses may vary from more than zero weeks to less than four weeks.
- the combination methods, compositions and formulations are not to be limited to the use of only two agents; the use of multiple therapeutic combinations are also envisioned.
- the dosage regimen to treat, prevent, or ameliorate the condition(s) for which relief is sought can be modified in accordance with a variety of factors. These factors include the disorder from which the subject suffers, as well as the age, weight, sex, diet, and medical condition of the subject. Thus, the dosage regimen actually employed can vary widely and therefore can deviate from the dosage regimens set forth herein.
- the pharmaceutical agents which make up the combination therapy disclosed herein may be a combined dosage form or in separate dosage forms intended for substantially simultaneous administration.
- the pharmaceutical agents that make up the combination therapy may also be administered sequentially, with either therapeutic compound being administered by a regimen calling for two-step administration.
- the two-step administration regimen may call for sequential administration of the active agents or spaced-apart administration of the separate active agents.
- the time period between the multiple administration steps may range from, a few minutes to several hours, depending upon the properties of each pharmaceutical agent, such as potency, solubility, bioavailability, plasma half-life and kinetic profile of the pharmaceutical agent. Circadian variation of the target molecule concentration may also determine the optimal dose interval.
- the compounds described herein also are optionally used in combination with procedures that provide additional or synergistic benefit to the patient.
- patients are expected to find therapeutic and/or prophylactic benefit in the methods described herein, wherein pharmaceutical composition of a compound disclosed herein and/or combinations with other therapeutics are combined with genetic testing to determine whether that individual is a carrier of a mutant gene that is known to be correlated with certain diseases or conditions.
- the compounds described herein and combination therapies can be administered before, during or after the occurrence of a disease or condition, and the timing of administering the composition containing a compound can vary.
- the compounds can be used as a prophylactic and can be administered continuously to subjects with a propensity to develop conditions or diseases in order to prevent the occurrence of the disease or condition.
- the compounds and compositions can be administered to a subject during or as soon as possible after the onset of the symptoms.
- the administration of the compounds can be initiated within the first 48 hours of the onset of the symptoms, within the first 6 hours of the onset of the symptoms, or within 3 hours of the onset of the symptoms.
- the initial administration can be via any route practical, such as, for example, an intravenous injection, a bolus injection, infusion over 5 minutes to about 5 hours, a pill, a capsule, transdermal patch, buccal delivery, and the like, or combination thereof.
- a compound should be administered as soon as is practicable after the onset of a disease or condition is detected or suspected, and for a length of time necessary for the treatment of the disease, such as, for example, from about 1 month to about 3 months.
- the length of treatment can vary for each subject, and the length can be determined using the known criteria.
- the compound or a formulation containing the compound can be administered for at least 2 weeks, between about 1 month to about 5 years, or from about 1 month to about 3 years.
- an irreversible Btk inhibitor compound can be used in with one or more of the following therapeutic agents in any combination: immunosuppressants (e.g., tacrolimus, cyclosporin, rapamicin, methotrexate, cyclophosphamide, azathioprine, mercaptopurine, mycophenolate, or FTY720), glucocorticoids (e.g., prednisone, cortisone acetate, prednisolone, methylprednisolone, dexamethasone, betamethasone, triamcinolone, beclometasone, fludrocortisone acetate, deoxycorticosterone acetate, aldosterone), non-steroidal anti-inflammatory drugs (e.g., salicylates, arylalkanoic acids, 2-arylpropionic acids, N-arylan
- the subjected can be treated with an irreversible Btk inhibitor compound in any combination with one or more other anti-cancer agents.
- one or more of the anti-cancer agents are proapoptotic agents.
- anti-cancer agents include, but are not limited to, any of the following: gossyphol, genasense, polyphenol E, Chlorofusin, all trans-retinoic acid (ATRA), bryostatin, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), 5-aza-2′-deoxycytidine, all trans retinoic acid, doxorubicin, vincristine, etoposide, gemcitabine, imatinib (Gleevec®), geldanamycin, 17-N-Allylamino-17-Demethoxygeldanamycin (17-AAG), flavopiridol, LY294002, bortezomib, trastuzumab, BAY 11-7082, PKC412, or PD184352, TaxolTM, also referred to as “paclitaxel”, which is a well-known anti-cancer drug which acts by enhancing and stabilizing microtubule formation
- mitogen-activated protein kinase signaling e.g., U0126, PD98059, PD184352, PD0325901, ARRY-142886, SB239063, SP600125, BAY 43-9006, wortmannin, or LY294002
- Syk inhibitors e.g., mTOR inhibitors
- mTOR inhibitors e.g., rituxan
- anti-cancer agents that can be employed in combination with an irreversible Btk inhibitor compound include Adriamycin, Dactinomycin, Bleomycin, Vinblastine, Cisplatin, acivicin; aclarubicin; acodazole hydrochloride; acronine; adozelesin; aldesleukin; altretamine; ambomycin; ametantrone acetate; aminoglutethimide; amsacrine; anastrozole; anthramycin; asparaginase; asperlin; azacitidine; azetepa; azotomycin; batimastat; benzodepa; bicalutamide; bisantrene hydrochloride; bisnafide dimesylate; bizelesin; bleomycin sulfate; brequinar sodium; bropirimine; busulfan; cactinomycin; calusterone; caracemide; carbetimer; carboplatin
- anti-cancer agents that can be employed in combination with an irreversible Btk inhibitor compound include: 20-epi-1, 25 dihydroxyvitamin D3; 5-ethynyluracil; abiraterone; aclarubicin; acylfulvene; adecypenol; adozelesin; aldesleukin; ALL-TK antagonists; altretamine; ambamustine; amidox; amifostine; aminolevulinic acid; amrubicin; amsacrine; anagrelide; anastrozole; andrographolide; angiogenesis inhibitors; antagonist D; antagonist G; antarelix; anti-dorsalizing morphogenetic protein-1; antiandrogen, prostatic carcinoma; antiestrogen; antineoplaston; antisense oligonucleotides; aphidicolin glycinate; apoptosis gene modulators; apoptosis regulators; apurinic acid;
- nitrogen mustards e.g., mechloroethamine, cyclophosphamide, chlorambucil, etc.
- alkyl sulfonates e.g., busulfan
- nitrosoureas e.g., carmustine, lomusitne, ete.
- triazenes decarbazine, etc.
- antimetabolites include but are not limited to folic acid analog (e.g., methotrexate), or pyrimidine analogs (e.g., Cytarabine), purine analogs (e.g., mercaptopurine, thioguanine, pentostatin).
- folic acid analog e.g., methotrexate
- pyrimidine analogs e.g., Cytarabine
- purine analogs e.g., mercaptopurine, thioguanine, pentostatin.
- Examples of natural products useful in combination with an irreversible Btk inhibitor compound include but are not limited to vinca alkaloids (e.g., vinblastin, vincristine), epipodophyllotoxins (e.g., etoposide), antibiotics (e.g., daunorubicin, doxorubicin, bleomycin), enzymes (e.g., L-asparaginase), or biological response modifiers (e.g., interferon alpha).
- vinca alkaloids e.g., vinblastin, vincristine
- epipodophyllotoxins e.g., etoposide
- antibiotics e.g., daunorubicin, doxorubicin, bleomycin
- enzymes e.g., L-asparaginase
- biological response modifiers e.g., interferon alpha
- alkylating agents that can be employed in combination an irreversible Btk inhibitor compound include, but are not limited to, nitrogen mustards (e.g., mechloroethamine, cyclophosphamide, chlorambucil, meiphalan, etc.), ethylenimine and methylmelamines (e.g., hexamethlymelamine, thiotepa), alkyl sulfonates (e.g., busulfan), nitrosoureas (e.g., carmustine, lomusitne, semustine, streptozocin, etc.), or triazenes (decarbazine, ete.).
- nitrogen mustards e.g., mechloroethamine, cyclophosphamide, chlorambucil, meiphalan, etc.
- ethylenimine and methylmelamines e.g., hexamethlymelamine, thiote
- antimetabolites include, but are not limited to folic acid analog (e.g., methotrexate), or pyrimidine analogs (e.g., fluorouracil, floxouridine, Cytarabine), purine analogs (e.g., mercaptopurine, thioguanine, pentostatin.
- folic acid analog e.g., methotrexate
- pyrimidine analogs e.g., fluorouracil, floxouridine, Cytarabine
- purine analogs e.g., mercaptopurine, thioguanine, pentostatin.
- hormones and antagonists useful in combination with an irreversible Btk inhibitor compound include, but are not limited to, adrenocorticosteroids (e.g., prednisone), progestins (e.g., hydroxyprogesterone caproate, megestrol acetate, medroxyprogesterone acetate), estrogens (e.g., diethlystilbestrol, ethinyl estradiol), antiestrogen (e.g., tamoxifen), androgens (e.g., testosterone propionate, fluoxymesterone), antiandrogen (e.g., flutamide), gonadotropin releasing hormone analog (e.g., leuprolide).
- adrenocorticosteroids e.g., prednisone
- progestins e.g., hydroxyprogesterone caproate, megestrol acetate, medroxyprogesterone acetate
- platinum coordination complexes e.g., cisplatin, carboblatin
- anthracenedione e.g., mitoxantrone
- substituted urea e.g., hydroxyurea
- methyl hydrazine derivative e.g., procarbazine
- adrenocortical suppressant e.g., mitotane, aminoglutethimide
- anti-cancer agents which act by arresting cells in the G2-M phases due to stabilized microtubules and which can be used in combination with an irreversible Btk inhibitor compound include without limitation marketed drugs and drugs in development.
- anti-thromboembolic agents include, but are not limited any of the following: thrombolytic agents (e.g., alteplase anistreplase, streptokinase, urokinase, or tissue plasminogen activator), heparin, tinzaparin, warfarin, dabigatran (e.g., dabigatran etexilate), factor Xa inhibitors (e.g., fondaparinux, draparinux, rivaroxaban, DX-9065a, otamixaban, LY517717, or YM150), factor VIIa inhibitors, ticlopidine, clopidogrel, CS-747 (prasugrel, LY640
- compositions are formulated in a conventional manner using one or more physiologically acceptable carriers including excipients and auxiliaries which facilitate processing of the active compounds into preparations which can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen.
- a summary of pharmaceutical compositions described herein is found, for example, in Remington: The Science and Practice of Pharmacy , Nineteenth Ed (Easton, Pa.: Mack Publishing Company, 1995); Hoover, John E., Remington's Pharmaceutical Sciences , Mack Publishing Co., Easton, Pa. 1975; Liberman, H. A. and Lachman, L., Eds., Pharmaceutical Dosage Forms , Marcel Decker, New York, N.Y., 1980; and Pharmaceutical Dosage Forms and Drug Delivery Systems , Seventh Ed. (Lippincott Williams & Wilkins 1999).
- a pharmaceutical composition refers to a mixture of a compound described herein, such as, for example, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), with other chemical components, such as carriers, stabilizers, diluents, dispersing agents, suspending agents, thickening agents, and/or excipients.
- the pharmaceutical composition facilitates administration of the compound to an organism.
- therapeutically effective amounts of compounds described herein are administered in a pharmaceutical composition to a mammal having a disease, disorder, or condition to be treated.
- the mammal is a human.
- the compounds can be used singly or in combination with one or more therapeutic agents as components of mixtures.
- the pharmaceutical formulations described herein can be administered to a subject by multiple administration routes, including but not limited to, oral, parenteral (e.g., intravenous, subcutaneous, intramuscular), intranasal, buccal, topical, rectal, or transdermal administration routes.
- the pharmaceutical formulations described herein include, but are not limited to, aqueous liquid dispersions, self-emulsifying dispersions, solid solutions, liposomal dispersions, aerosols, solid dosage forms, powders, immediate release formulations, controlled release formulations, fast melt formulations, tablets, capsules, pills, delayed release formulations, extended release formulations, pulsatile release formulations, multiparticulate formulations, and mixed immediate and controlled release formulations.
- compositions including a compound described herein are optionally manufactured in a conventional manner, such as, by way of example only, by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or compression processes.
- the pharmaceutical compositions will include at least one compound described herein, such as, for example, a compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), as an active ingredient in free-acid or free-base form, or in a pharmaceutically acceptable salt form.
- the methods and pharmaceutical compositions described herein include the use of N-oxides, crystalline forms (also known as polymorphs), as well as active metabolites of these compounds having the same type of activity.
- compounds may exist as tautomers. All tautomers are included within the scope of the compounds presented herein.
- the compounds described herein can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. The solvated forms of the compounds presented herein are also considered to be disclosed herein.
- a “carrier” or “carrier materials” includes excipients in pharmaceutics and is selected on the basis of compatibility with compounds disclosed herein, such as, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C 1 -C 6 ), Formula (D1-D6), Formula (I), or Formula (VII), and the release profile properties of the desired dosage form.
- exemplary carrier materials include, e.g., binders, suspending agents, disintegration agents, filling agents, surfactants, solubilizers, stabilizers, lubricants, wetting agents, diluents, and the like.
- a “measurable serum concentration” or “measurable plasma concentration” describes the blood serum or blood plasma concentration, typically measured in mg, ⁇ g, or ng of therapeutic agent per ml, dl, or 1 of blood serum, absorbed into the bloodstream after administration. As used herein, measurable plasma concentrations are typically measured in ng/ml or ⁇ g/ml.
- “Pharmacodynamics” refers to the factors which determine the biologic response observed relative to the concentration of drug at a site of action. “Pharmacokinetics” refers to the factors which determine the attainment and maintenance of the appropriate concentration of drug at a site of action.
- Step state is when the amount of drug administered is equal to the amount of drug eliminated within one dosing interval resulting in a plateau or constant plasma drug exposure.
- compositions described herein which include a compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) can be formulated into any suitable dosage form, including but not limited to, aqueous oral dispersions, liquids, gels, syrups, elixirs, slurries, suspensions and the like, for oral ingestion by a patient to be treated, solid oral dosage forms, aerosols, controlled release formulations, fast melt formulations, effervescent formulations, lyophilized formulations, tablets, powders, pills, dragees, capsules, delayed release formulations, extended release formulations, pulsatile release formulations, multiparticulate formulations, and mixed immediate release and controlled release formulations.
- aqueous oral dispersions liquids, gels, syrups, elixirs, slurries, suspensions and the like
- solid oral dosage forms including but not limited to,
- the pharmaceutical solid dosage forms described herein optionally include a compound described herein and one or more pharmaceutically acceptable additives such as a compatible carrier, binder, filling agent, suspending agent, flavoring agent, sweetening agent, disintegrating agent, dispersing agent, surfactant, lubricant, colorant, diluent, solubilizer, moistening agent, plasticizer, stabilizer, penetration enhancer, wetting agent, anti-foaming agent, antioxidant, preservative, or one or more combination thereof.
- a compatible carrier such as a compatible carrier, binder, filling agent, suspending agent, flavoring agent, sweetening agent, disintegrating agent, dispersing agent, surfactant, lubricant, colorant, diluent, solubilizer, moistening agent, plasticizer, stabilizer, penetration enhancer, wetting agent, anti-foaming agent, antioxidant, preservative, or one or more combination thereof.
- a film coating is provided around the formulation of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- some or all of the particles of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) are coated.
- some or all of the particles of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are microencapsulated.
- the particles of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) are not microencapsulated and are uncoated.
- the compounds described herein can be used in the preparation of medicaments for the inhibition of Btk or a homolog thereof, or for the treatment of diseases or conditions that benefit, at least in part, from inhibition of Btk or a homolog thereof.
- a method for treating any of the diseases or conditions described herein in a subject in need of such treatment involves administration of pharmaceutical compositions containing at least one compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), described herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable N-oxide, pharmaceutically active metabolite, pharmaceutically acceptable prodrug, or pharmaceutically acceptable solvate thereof, in therapeutically effective amounts to said subject.
- compositions containing the compound(s) described herein can be administered for prophylactic and/or therapeutic treatments.
- the compositions are administered to a patient already suffering from a disease or condition, in an amount sufficient to cure or at least partially arrest the symptoms of the disease or condition. Amounts effective for this use will depend on the severity and course of the disease or condition, previous therapy, the patient's health status, weight, and response to the drugs, and the judgment of the treating physician.
- compositions containing the compounds described herein are administered to a patient susceptible to or otherwise at risk of a particular disease, disorder or condition. Such an amount is defined to be a “prophylactically effective amount or dose.”
- a patient susceptible to or otherwise at risk of a particular disease, disorder or condition is defined to be a “prophylactically effective amount or dose.”
- dose a pharmaceutically effective amount or dose.
- the precise amounts also depend on the patient's state of health, weight, and the like.
- effective amounts for this use will depend on the severity and course of the disease, disorder or condition, previous therapy, the patient's health status and response to the drugs, and the judgment of the treating physician.
- the irreversible kinase inhibitor is administered to the patient on a regular basis, e.g., three times a day, two times a day, once a day, every other day or every 3 days.
- the irreversible kinase inhibitor is administered to the patient on an intermittent basis, e.g., twice a day followed by once a day followed by three times a day; or the first two days of every week; or the first, second and third day of a week.
- intermittent dosing is as effective as regular dosing.
- the irreversible kinase inhibitor is administered only when the patient exhibits a particular symptom, e.g., the onset of pain, or the onset of a fever, or the onset of an inflammation, or the onset of a skin disorder.
- a particular symptom e.g., the onset of pain, or the onset of a fever, or the onset of an inflammation, or the onset of a skin disorder.
- the administration of the compounds may be administered chronically, that is, for an extended period of time, including throughout the duration of the patient's life in order to ameliorate or otherwise control or limit the symptoms of the patient's disease or condition.
- the administration of the compounds may be given continuously; alternatively, the dose of drug being administered may be temporarily reduced or temporarily suspended for a certain length of time (i.e., a “drug holiday”).
- the length of the drug holiday can vary between 2 days and 1 year, including by way of example only, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 10 days, 12 days, 15 days, 20 days, 28 days, 35 days, 50 days, 70 days, 100 days, 120 days, 150 days, 180 days, 200 days, 250 days, 280 days, 300 days, 320 days, 350 days, or 365 days.
- the dose reduction during a drug holiday may be from 10%-100%, including, by way of example only, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 100%.
- a maintenance dose is administered if necessary. Subsequently, the dosage or the frequency of administration, or both, can be reduced, as a function of the symptoms, to a level at which the improved disease, disorder or condition is retained. Patients can, however, require intermittent treatment on a long-term basis upon any recurrence of symptoms.
- the amount of a given agent that will correspond to such an amount will vary depending upon factors such as the particular compound, disease or condition and its severity, the identity (e.g., weight) of the subject or host in need of treatment, and is determined according to the particular circumstances surrounding the case, including, e.g., the specific agent being administered, the route of administration, the condition being treated, and the subject or host being treated.
- doses employed for adult human treatment will typically be in the range of 0.02-5000 mg per day, or from about 1-1500 mg per day.
- the desired dose may conveniently be presented in a single dose or as divided doses administered simultaneously (or over a short period of time) or at appropriate intervals, for example as two, three, four or more sub-doses per day.
- the pharmaceutical composition described herein may be in unit dosage forms suitable for single administration of precise dosages.
- the formulation is divided into unit doses containing appropriate quantities of one or more compound.
- the unit dosage may be in the form of a package containing discrete quantities of the formulation.
- Non-limiting examples are packaged tablets or capsules, and powders in vials or ampoules.
- Aqueous suspension compositions can be packaged in single-dose non-reclosable containers.
- multiple-dose reclosable containers can be used, in which case it is typical to include a preservative in the composition.
- formulations for parenteral injection may be presented in unit dosage form, which include, but are not limited to ampoules, or in multi-dose containers, with an added preservative.
- Toxicity and therapeutic efficacy of such therapeutic regimens can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, including, but not limited to, the determination of the LD 50 (the dose lethal to 50% of the population) and the ED 50 (the dose therapeutically effective in 50% of the population).
- the dose ratio between the toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio between LD 50 and ED 50 .
- Compounds exhibiting high therapeutic indices are preferred.
- the data obtained from cell culture assays and animal studies can be used in formulating a range of dosage for use in human.
- the dosage of such compounds lies preferably within a range of circulating concentrations that include the ED 50 with minimal toxicity.
- the dosage may vary within this range depending upon the dosage form employed and the route of administration utilized.
- Described herein are irreversible kinase inhibitors that are selective for one or more ACKs, including a Btk, a Btk homolog, and a Btk kinase cysteine homolog.
- the irreversible inhibitors described herein also bind reversibly to other kinases (some of which, in some embodiments, are also ACKs).
- such inhibitors are formulated (formulation includes chemical modifications of the inhibitor, use of excipients in a pharmaceutical composition, and combinations thereof) such that the pharmacokinetic profile favors enhanced selectivity of the inhibitors for an ACK over a non-ACK.
- an ACK is formulated to have a short plasma half-life. In other embodiments, an ACK is formulated to have an extended plasma half-life.
- Compound 1 and Compound 12 have a short half-life in vivo.
- Compound 7 and Compound 8 have a significantly longer in vivo half-life ( FIG. 5 ).
- Compounds like 1 and 12 are predicted to have enhanced kinase selectivity in vivo because inhibition will be sustained only for those kinases that are irreversibly inhibited.
- the irreversible kinase inhibitors described herein have both reversible (in general to non-ACKs) and irreversible (generally, to ACKs) activities, in vivo properties of absorption, distribution, metabolism and excretion (ADME) are selected in order to optimize the therapeutic index.
- ADME absorption, distribution, metabolism and excretion
- rapidly cleared compounds cause only brief inhibition of reversibly inhibited targets while maintaining sustained inhibition of irreversibly inhibited targets.
- sustained inhibition of particular targets results in therapeutic effects or toxicities.
- kinase inhibitors that selectively and irreversibly binds to a protein tyrosine kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog, in which the kinase inhibitor reversibly and non-selectively binds to a multiplicity of protein tyrosine kinases, and further in which the plasma half life of the kinase inhibitor is less than about 4 hours.
- the kinase inhibitor selectively and irreversibly binds to at least one of Btk, Jak3, Blk, Bmx, Tec, and Itk.
- the kinase inhibitor selectively and irreversibly binds to Btk. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Jak3. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Tec. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Btk and Tec. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Blk. In a further embodiment, the kinase inhibitor reversibly and non-selectively binds to a multiplicity of src-family protein kinase inhibitors. In a further embodiment, the plasma half life of the kinase inhibitor is less than about 3 hours. In a further embodiment, the plasma half life of the kinase inhibitor is less than about 2 hours.
- kinase inhibitors that selectively and irreversibly binds to a protein tyrosine kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog, in which the kinase inhibitor reversibly and non-selectively binds to a multiplicity of protein tyrosine kinases, and further in which the plasma half life of the kinase inhibitor is greater than about 12 hours.
- the kinase inhibitor selectively and irreversibly binds to at least one of Btk, Jak3, Blk, Bmx, Tec, and Itk.
- the kinase inhibitor selectively and irreversibly binds to Btk. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Jak3. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Tec. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Btk and Tec. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Blk.
- the kinase inhibitor reversibly and non-selectively binds to a multiplicity of src-family protein kinase inhibitors In a further embodiment, the kinase inhibitor the plasma half life of the kinase inhibitor is greater than about 16 hours.
- such kinase inhibitors have the structure of Formula (VII):
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
- Z is C( ⁇ O), OC( ⁇ O), NHC( ⁇ O), NCH 3 C( ⁇ O), C( ⁇ S), S( ⁇ O) x , OS( ⁇ O) x , NHS( ⁇ O) x , where x is 1 or 2;
- R 7 and R 8 are independently selected from among H, unsubstituted C 1 -C 4 alkyl, substituted C 1 -C 4 alkyl, unsubstituted C 1 -C 4 heteroal
- kinase inhibitor on the kinase inhibitor is a substituted fused biaryl moiety selected from
- Z is C( ⁇ O), NHC( ⁇ O), NCH 3 C( ⁇ O), or S( ⁇ O) 2 .
- the kinase inhibitor of Claim 49 wherein: each of R 7 and R 8 is H; or R 7 and R 8 taken together form a bond.
- R 6 is H, substituted or unsubstituted C 1 -C 4 alkyl, substituted or unsubstituted C 1 -C 4 heteroalkyl, C 1 -C 6 alkoxyalkyl, C 1 -C 8 alkylaminoalkyl, C 1 -C 8 hydroxyalkylaminoalkyl, C 1 -C 8 alkoxyalkylaminoalkyl, C 1 -C 4 alkyl(aryl), C 1 -C 4 alkyl(heteroaryl), C 1 -C 4 alkyl(C 3 -C 8 cycloalkyl), or C 1 -C 4 alkyl(C 2 -C 8 heterocycloalkyl).
- Y is a 4-, 5-, 6-, or 7-membered cycloalkylene ring; or Y is a 4-, 5-, 6-, or 7-membered heterocycloalkylene ring; or Y is a C 1 -C 4 alkylene, or 4-, 5-, 6-, or 7-membered heterocycloalkylene ring.
- such dosing methods are pharmaceutical formulations comprising any of the aforementioned ACK inhibitors and a pharmaceutically acceptable excipient.
- such pharmaceutical formulations are formulated for a route of administration selected from oral administration, parenteral administration, buccal administration, nasal administration, topical administration, or rectal administration.
- the pharmaceutical formulations are formulated for oral administration.
- dosing methods for treating rheumatoid arthritis comprising administering to a subject any of the aforementioned ACK inhibitors that selectively and irreversibly binds to Btk and Tec.
- test protein tyrosine kinase inhibitor that irreversibly and selectively binds to at least one protein kinase inhibitor selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog, in which the test protein tyrosine kinase inhibitor is chemically modified to decrease the plasma half life to less than about 4 hours. In some embodiments, the test protein tyrosine kinase inhibitor is chemically modified to decrease the plasma half life to less than about 3 hours.
- test protein tyrosine kinase inhibitor has the structure of Formula (VII):
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
- Z is C( ⁇ O), OC( ⁇ O), NHC( ⁇ O), NCH 3 C( ⁇ O), C( ⁇ S), S( ⁇ O) x , OS( ⁇ O) x , NHS( ⁇ O) x , where x is 1 or 2;
- R 7 and R 8 are independently selected from among H, unsubstituted C 1 -C 4 alkyl, substituted C 1 -C 4 alkyl, unsubstituted C 1 -C 4 heteroal
- test protein tyrosine kinase inhibitor non-selectively and reversibly binds to a multiplicity of src-family protein tyrosine kinases.
- dosing strategies are methods for treating a B-cell proliferative disorder or a mast cell proliferative disorder comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors.
- a pharmaceutical composition of any of the aforementioned ACK inhibitors for example, as presented in the Examples, brief exposure to Compound 1 in vitro is sufficient to inhibit B cell activation in normal human B cells. This protocol mimics the predicted exposure of cells to Compound 1 in vivo and demonstrates that inhibition of B cells is sustained despite washing out of Compound 1.
- dosing strategies are methods for treating a rheumatoid arthritis or condition comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors.
- methods for treating a disease characterized by hyperactive B cells comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors.
- methods for treating a disease characterized by hyperactive mast cells comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors.
- dosing strategies are methods for treating a disease characterized by both hyperactive B cells and hyperactive mast cells comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors.
- the pharmaceutical composition is administered once a day or less frequently than once a day.
- kits and articles of manufacture are also described herein.
- Such kits can include a carrier, package, or container that is compartmentalized to receive one or more containers such as vials, tubes, and the like, each of the container(s) including one of the separate elements to be used in a method described herein.
- Suitable containers include, for example, bottles, vials, syringes, and test tubes.
- the containers can be formed from a variety of materials such as glass or plastic.
- Packaging materials for use in packaging pharmaceutical products include, e.g., U.S. Pat. Nos. 5,323,907, 5,052,558 and 5,033,252.
- Examples of pharmaceutical packaging materials include, but are not limited to, blister packs, bottles, tubes, inhalers, pumps, bags, vials, containers, syringes, bottles, and any packaging material suitable for a selected formulation and intended mode of administration and treatment.
- a wide array of formulations of the compounds and compositions provided herein are contemplated as are a variety of treatments for any disease, disorder, or condition that benefit by inhibition of Btk, or in which Btk is a mediator or contributor to the symptoms or cause.
- the container(s) can include one or more compounds described herein, optionally in a composition or in combination with another agent as disclosed herein.
- the container(s) optionally have a sterile access port (for example the container can be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
- kits optionally comprising a compound with an identifying description or label or instructions relating to its use in the methods described herein.
- a kit will typically include one or more additional containers, each with one or more of various materials (such as reagents, optionally in concentrated form, and/or devices) desirable from a commercial and user standpoint for use of a compound described herein.
- materials include, but not limited to, buffers, diluents, filters, needles, syringes; carrier, package, container, vial and/or tube labels listing contents and/or instructions for use, and package inserts with instructions for use.
- a set of instructions will also typically be included.
- a label can be on or associated with the container.
- a label can be on a container when letters, numbers or other characters forming the label are attached, molded or etched into the container itself; a label can be associated with a container when it is present within a receptacle or carrier that also holds the container, e.g., as a package insert.
- a label can be used to indicate that the contents are to be used for a specific therapeutic application. The label can also indicate directions for use of the contents, such as in the methods described herein.
- the pharmaceutical compositions can be presented in a pack or dispenser device which can contain one or more unit dosage forms containing a compound provided herein.
- the pack can for example contain metal or plastic foil, such as a blister pack.
- the pack or dispenser device can be accompanied by instructions for administration.
- the pack or dispenser can also be accompanied with a notice associated with the container in form prescribed by a governmental agency regulating the manufacture, use, or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the drug for human or veterinary administration. Such notice, for example, can be the labeling approved by the U.S. Food and Drug Administration for prescription drugs, or the approved product insert.
- Compositions containing a compound provided herein formulated in a compatible pharmaceutical carrier can also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition.
- 4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL).
- 1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated.
- 1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
- 3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine.
- the solid was suspended in ethyl acetate (100 mL) and again washed with dilute aq. NaHCO 3 (100 mL). The ethyl acetate was dried (MgSO 4 ), filtered and concentrated to provide 0.43 g of a light yellow solid.
- the solid (0.14 g, 0.36 mmol) was stirred in THF (3 mL) and TEA (015 mL, 1.1 mmol) was added, followed by cooling the reaction with an ice bath for 30 min, then acryl chloride (30 ⁇ L, 0.36 mmol) added and the reaction was stirred for 2 hr.
- Compound 1 inhibits lymphoma tumor cell growth.
- a variety of lymphoma cell lines were incubated with a range of concentrations of Compound 1 to determine the GI50, the concentration that results in 50% decrease in cell proliferation ( FIG. 1A ).
- Compound 1 inhibits tumor growth in DOHH2 and DLCL2 xenograft models ( FIGS. 1B and 1C ).
- 5E6 DOHH2 or DLCL2 cells in 50% matrigel were implanted subcutaneously in SCID mice and dosed orally with Compound 1 beginning when tumor size reached 100 mm2.
- Compound 1 inhibits collagen-induced arthritis in the mouse.
- Male DBA/1O1 aHsd mice were injected intradermally with 150 microliters of 2 mg/mL Type II collagen in Freund's complete adjuvant with supplemental M. tuberculosis, 4 mg/mL and boosted with the same injection 21 days later.
- Compound 1 at 12.5 mg/kg and 50 mg/kg regressed inflammation through the end of the study (day 11) while 3.125 mg/kg significantly reduced the increase in paw inflammation ( FIG. 2 ).
- Dexamethasone was included as a positive control.
- Compound 1 was dosed at 12.5 mg/kg to such mice over: (a) each day of an 11-day period; (b) days 1, 2, and 3 of an 11-day period; or (c) days 9, 10, and 11 of an 11-day period. Intermittent dosing reduced the increase in paw inflammation.
- Compound 9 was dosed to such mice at a level of 12.5 mg/kg or 50 mg/kg each day of an 11-day period. Compound 9 reduced the increase in paw inflammation.
- Compound 1 inhibits disease progression in the mouse MRL/1 pr model of lupus.
- MRL/1pr mice (Jax strain 000485) were dosed orally once per day from 12 weeks of age until 20 weeks of age and urine protein levels were measured weekly using Clinitech Multistick dipstick.
- Compound 1 inhibits mast cell degranulation in a mouse passive cutaneous anaphylaxis model. Increasing doses of Compound 1 significantly decrease the amount of Evans Blue release, indicating decreased mast cell activation and vascular permeabilization. ( FIG. 4 )
- mice were sensitized with an intradermal injection of monoclonal anti-DNP-IgE in the back. 23 hours later they received a single oral dose of Compound 1 or vehicle. After one hour, animals were challenged with an intravenous injection of DNP-BSA and Evans Blue dye. Mast cell degranulation leads to vascular permeability and the distribution of the dye into the skin of the back. The area of extravasation after 1 hour is measured.
- compositions described below are presented with a compound of Formula (A1-A6) for illustrative purposes; any of the compounds of any of Formulas (A1-A6), (B1-B6), (C1-C6), or (D1-D6) are optionally used in such pharmaceutical compositions.
- Example 5a Parenteral Composition
- a parenteral pharmaceutical composition suitable for administration by injection 100 mg of a water-soluble salt of a compound of Formula (A1-A6) is dissolved in DMSO and then mixed with 10 mL of 0.9% sterile saline. The mixture is incorporated into a dosage unit form suitable for administration by injection.
- a pharmaceutical composition for oral delivery 100 mg of a compound of Formula (A1-A6) is mixed with 750 mg of starch.
- the mixture is incorporated into an oral dosage unit for, such as a hard gelatin capsule, which is suitable for oral administration.
- Example 5c Sublingual (Hard Lozenge) Composition
- a pharmaceutical composition for buccal delivery such as a hard lozenge
- a pharmaceutical composition for buccal delivery such as a hard lozenge
- the mixture is gently blended and poured into a mold to form a lozenge suitable for buccal administration.
- a pharmaceutical composition for inhalation delivery 20 mg of a compound of Formula (A1-A6) is mixed with 50 mg of anhydrous citric acid and 100 mL of 0.9% sodium chloride solution.
- the mixture is incorporated into an inhalation delivery unit, such as a nebulizer, which is suitable for inhalation administration.
- a pharmaceutical composition for rectal delivery 100 mg of a compound of Formula (A1-A6) is mixed with 2.5 g of methylcellulose (1500 mPa), 100 mg of methylparaben, 5 g of glycerin and 100 mL of purified water. The resulting gel mixture is then incorporated into rectal delivery units, such as syringes, which are suitable for rectal administration.
- a pharmaceutical topical gel composition 100 mg of a compound of Formula (A1-A6) is mixed with 1.75 g of hydroxypropyl cellulose, 10 mL of propylene glycol, 10 mL of isopropyl myristate and 100 mL of purified alcohol USP. The resulting gel mixture is then incorporated into containers, such as tubes, which are suitable for topical administration.
- a pharmaceutical ophthalmic solution composition 100 mg of a compound of Formula (A1-A6) is mixed with 0.9 g of NaCl in 100 mL of purified water and filtered using a 0.2 micron filter. The resulting isotonic solution is then incorporated into ophthalmic delivery units, such as eye drop containers, which are suitable for ophthalmic administration.
- ophthalmic delivery units such as eye drop containers
- phosphorylation events in the BCR signal transduction pathway were investigated.
- a panel of phospho-specific antibodies that recognize activating phosphorylation sites on Syk, Btk, BLNK, PLC-g1, PLC-g2, ERK, and AKT were used and tested the effects of Compound 4 on both basal phosphorylation and phosphorylation following BCR stimulation driven by anti-IgM or anti-IgG cross-linking.
- DOHH2 Compound 1 sensitive cell line
- Ramos Compound 1 resistant cell line
- Compound 1 inhibits most BCR-stimulus induced phosphorylation events with similar potency in both cell lines. However, when we examined basal phosphorylation levels, we found higher basal phosphorylation in DOHH2 compared to Ramos, with phospho-ERK in particular indicating higher levels of basal or tonic signaling in DOHH2. Furthermore, Compound 4 significantly decreased pERK levels in unstimulated DOHH2 cells (IC50 ⁇ 10 nM), but not in Ramos cells.
- a panel of nine Btk expressing B cell lymphoma cell lines was screened for basal pERK levels. Seven lines expressed significantly higher levels of basal pERK, and of these, 5 were sensitive to Compound 1 (GI50 ⁇ 1.3 uM), while the two cell lines with low pERK levels were resistant to Compound 1. This data shows that tonic BCR signaling contributes to the survival of a subset of lymphoma cell lines, and that inhibition of this signaling by Compound 4 is correlated with induction of apoptosis.
- Egr-1 pERK or ERK transcriptional targets such as Egr-1 serve as useful markers for lymphomas in which tonic BCR signaling is contributing to cell survival and that these lymphomas are particularly sensitive to BCR pathway inhibitors such as Compound 1.
- Example 1 Design of an Inhibitor
- BTK highly selective BTK inhibitor
- the approach employed structure based design to achieve a high degree of potency and selectivity by (1) fitting the core scaffold into the active site ATP binding pocket of kinase enzymes, and (2) forming a covalent bond with Cysteine-481 located in BTK.
- the unique chemistry required for covalent bond formation involves an electrophilic moiety that acts as a Michael acceptor, which bonds with a nucleophile (such as Cys-481) present in a precise location within the active site.
- a panel of 50-100 Cys-targeting kinase inhibitors is generated.
- the molecular orientation and positioning of the electrophilic group in these inhibitors in relation to the Cysteine residue will affect the potency and selectivity of a given inhibitor.
- Each inhibitor will then be profiled for kinetics of kinase inhibition (K i ) for each of the ten Cys-containing kinases, effect on tumor cell proliferation (GI 50 ), effect on relevant off-targets (hERG, CYPs), drug-like characteristics (solubility, clogP) and ability to block labeling by the active site probe.
- This panel of diverse inhibitors are then be used in cell assays (for example, inhibition of tumor growth) to screen for a phenotype of interest.
- the identification of additional inhibited kinases is determined using the active site probe and mass spectrometry.
- the linker and Michael acceptor moiety of Compound 1 was modified to provide Compound 9 which has a different selectivity pattern.
- Table 1 is a table showing the degree of inhibition of a panel of kinases for two example compounds. IC 50 s were determined using the in vitro HotSpot kinase assay (purified enzymes, 33 P-ATP, an appropriate substrate and 1 uM ATP.)
- Compound 9 has similar potency toward Btk, but significantly less potency toward JAK-3, ITK, and EGFR and significantly more potency toward the src-family kinases lck, c-src, FGR, Fyn, Hck, and Lyn and Yes.
- subtle modifications in the linker moiety and the Michael acceptor moiety are important for the design of selective ACK inhibitors.
- compounds are selected based on in vitro characteristics to optimize for potency of inhibition of particular kinases and degree of covalent binding to off-target cysteines such as glutathione.
- Table 2 Compound 9 and Compound 12 both inhibit Btk with a similar potency as Compound 1, but they are both significantly less potent inhibitors of EGFR, ITK, and JAK-3.
- Compound 11 is similar to Compound 1 for inhibition of Btk but does not bind glutathione as readily.
- a calculated value (e.g (1/Btk IC 50 )/Glutathione conjugation rate) as shown in the Table 2) is used to compare compounds for their ratio between potency at inhibiting their target and their non-specific binding to other SH groups, such as those in glutathione. As shown in Table 2, this calculated value is 4.7 for Compound 1 and for 239.6 for Compound 11. Calculated ratios such as these are used to quantitatively compare different compounds and select compounds for further study.
- Analogs are generated that are Btk inhibitors and that are cytotoxic to the lymphoma cell line DOHH2. See Table 2.
- DOHH2 cell proliferation assay cells were seeded in 96-well plates in standard growth media (RPMI+10% fetal calf serum) and compounds were added in a 9-point dilution series ranging from 10 uM to 0.04 uM with DMSO at 0.1% final concentration in all wells. After 72 hours, cell number was measured using Alamar Blue using manufacturer's protocol. A dilution series of untreated cells was run in parallel to verify that the Alamar Blue assay reliably reflected cell number and that growth conditions were not limiting. The GI 50 , the concentration that results in a 50% decrease in cell number, was calculated using Calcusyn to fit the dose-response curve.
- Compound 1 and Compound 12 have a short half-life in vivo. In contrast, Compound 7 and Compound 8 have a significantly longer in vivo half-life ( FIG. 5 ). Compounds like 1 and 12 are predicted to have enhanced kinase selectivity in vivo because inhibition will be sustained only for those kinases that are irreversibly inhibited.
- Plasma samples were thawed and 75 uL aliquots were transferred to centrifuge tubes to which 10 L aliquots of internal standard solution (1 ⁇ g/mL) were added. The samples were not diluted with blank plasma prior to further processing. Soluble proteins were precipitated by the addition of 200 ⁇ L of acetonitrile, followed by centrifugation (20 min at 16,000 ⁇ g). The samples were evaporated to dryness and reconstituted in 200 ⁇ L of water containing 0.2% formic acid and 10% methanol. All samples were loaded onto an autosampler maintained at 6° C. and evaluated for concentrations of test compounds using LC-MS/MS.
- B cells were purified from blood from healthy donors by negative selecting using the RosetteSep Human B cell enrichment cocktail.
- Cells were plated in growth media (10% RPMI+10% fetal calf serum) and indicated concentrations of Compound 1 were added. After incubation for 1 hour at 37° C., cells were washed three times using an 8-fold dilution in growth media for each wash. Cells were then stimulated with 10 ug/ml of IgM F(ab′) 2 for 18 hours at 37° C. Cells were then stained with anti-CD69-PE antibody and analyzed by flow cytometry using standard conditions.
- ADME absorption, distribution, metabolism and excretion
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physical Education & Sports Medicine (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Described herein are irreversible kinase inhibitor compounds, methods for synthesizing such irreversible inhibitors, and methods for using such irreversible inhibitors in the treatment of diseases. Further described herein are methods, assays and systems for determining an appropriate irreversible inhibitor of a protein, including a kinase.
Description
- This application is a continuation of U.S. application Ser. No. 13/543,065, filed Jul. 6, 2012; which is a continuation of U.S. application Ser. No. 12/594,805, filed Jun. 8, 2010, now U.S. Pat. No. 8,883,435, issued Nov. 11, 2014; which is a U.S. National Stage Entry of PCT/US2008/058528, filed Mar. 27, 2008; which claims the benefit of priority from U.S. patent application Ser. No. 11/692,870, filed Mar. 28, 2007, now U.S. Pat. No. 7,732,454, issued Jun. 8, 2010; U.S. patent application Ser. No. 11/964,285, filed Dec. 26, 2007, now U.S. Pat. No. 7,825,118, issued Nov. 2, 2010; and U.S. Provisional Patent Application No. 61/017,125, filed Dec. 27, 2007; all of which are herein incorporated by reference in their entirety.
- Described herein are irreversible kinase inhibitor compounds, methods for synthesizing such irreversible inhibitors, and methods for using such irreversible inhibitors in the treatment of diseases. Further described herein are methods, assays and systems for determining an appropriate irreversible inhibitor of a protein, including a kinase.
- A kinase, alternatively known as a phosphotransferase, is a type of enzyme that transfers phosphate groups from high-energy donor molecules, such as ATP, to specific target molecules; the process is termed phosphorylation. Protein kinases, which act on and modify the activity of specific proteins, are used to transmit signals and control complex processes in cells. Up to 518 different kinases have been identified in humans. Their enormous diversity and role in signaling makes them attractive targets for drug design.
- Described herein are inhibitors of Bruton's tyrosine kinase (Btk). Also described herein are irreversible inhibitors of Btk. Further described are irreversible inhibitors of Btk that form a covalent bond with a cysteine residue on Btk. Further described herein are irreversible inhibitors of other tyrosine kinases, wherein the other tyrosine kinases share homology with Btk by having a cysteine residue (including a
Cys 481 residue) that forms a covalent bond with the irreversible inhibitor (such tyrosine kinases, are referred herein as “Btk tyrosine kinase cysteine homologs”). Also described herein are irreversible inhibitors of tyrosine kinases that have an accessible cysteine residue near an active site of the tyrosine kinase (referred herein as “Accessible Cysteine Kinases” or ACKs). Also described herein are irreversible inhibitors of any of the aforementioned tyrosine kinases, in which the irreversible inhibitor includes a Michael acceptor moiety. Further described are such irreversible inhibitors in which the Michael acceptor moiety preferentially forms a covalent bond with the appropriate cysteine residue on the desired tyrosine kinase relative to forming a covalent bond with other biological molecules that contain an accessible SH moiety. Also described herein are methods for synthesizing such irreversible inhibitors, methods for using such irreversible inhibitors in the treatment of diseases (including diseases wherein irreversible inhibition of Btk provides therapeutic benefit to a patient having the disease). Further described are pharmaceutical formulations that include an irreversible inhibitor of Btk. - Compounds described herein include those that have a structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), and pharmaceutically acceptable salts, solvates, esters, acids and prodrugs thereof. In certain embodiments, isomers and chemically protected forms of compounds having a structure represented by any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are also provided.
- In one aspect, provided herein are compounds of Formula (I). Formula (I) is as follows:
-

- wherein
-
- La is CH2, O, NH or S;
- Ar is a substituted or unsubstituted aryl, or a substituted or unsubstituted heteroaryl; and either
- (a) Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene and alkyleneheterocycloalkylene;
- Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
- (i) R7 and R8 are H;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- (ii) R6 and R8 are H;
- R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- (iii) R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- (b) Y is an optionally substituted group selected from cycloalkylene or heterocycloalkylene;
- Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
- (i) R7 and R8 are H;
- R6 is substituted or unsubstituted C1-C4heteroalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- (ii) R6 and R8 are H;
- R7 is substituted or unsubstituted C1-C4heteroalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- (iii) R7 and R8 taken together form a bond;
- R6 is substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (I). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (I), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (I). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (I). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- For any and all of the embodiments, substituents are optionally selected from among from a subset of the listed alternatives. For example, in some embodiments, La is CH2, O, or NH. In other embodiments, La is O or NH. In yet other embodiments, La is O.
- In some embodiments, Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- In some embodiments, x is 2. In yet other embodiments, Z is C(═O), OC(═O), NHC(═O), S(═O)x, OS(═O)x, or NHS(═O)x. In some other embodiments, Z is C(═O), NHC(═O), or NCH3C(═O).
- In some embodiments Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, and alkyleneheterocycloalkylene.
- In some embodiments, Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl.
- In some embodiments, R7 and R8 are H; and R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl). In other embodiments, R6 and R8 are H; and R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl). In yet further embodiments, R7 and R8 taken together form a bond; and R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl).
- In some embodiments, Y is an optionally substituted group selected from cycloalkylene or heterocycloalkylene.
- In some embodiments, Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl.
- In some embodiments, R7 and R8 are H; and R6 is substituted or unsubstituted C1-C4heteroalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl). In other embodiments, R6 and R8 are H; and R7 is substituted or unsubstituted C1-C4heteroalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl). In further embodiments, R7 and R8 taken together form a bond; and R6 is substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl).
- Any combination of the groups described above for the various variables is contemplated herein.
- In one aspect, provided herein is a compound selected from among: (E)-4-(N-(2-hydroxyethyl)-N-methylamino)-1-(3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one (Compound 3); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3-(1H-imidazol-4-yl)prop-2-en-1-one (Compound 4); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 5); (E)-1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 7); (E)-N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)-4-(dimethylamino)but-2-enamide (Compound 8); N-((1r,4r)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)acrylamide (Compound 10); (E)-1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrolidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 11); (E)-1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrolidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 12); 1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 13); 1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 14); 1((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)but-2-yn-1-one (Compound 15); 1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)but-2-yn-1-one (Compound 16); 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-yn-1-one (Compound 17); (E)-N-((1,r,4r)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl-4-(dimethylamino)but-2-enamide (Compound 18); N-(2-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-N-methylacrylamide (Compound 19); (E)-1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 20); (E)-1-((S-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 21); N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)but-2-ynamide (Compound 22); N-(2-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)acrylamide (Compound 23); (E)-1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 24); (E)-N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)-4-morpholinobut-2-enamide (Compound 25).
- In a further aspect are provided pharmaceutical compositions, which include a therapeutically effective amount of at least one of any of the compounds herein, or a pharmaceutically acceptable salt, pharmaceutically active metabolite, pharmaceutically acceptable prodrug, or pharmaceutically acceptable solvate. In certain embodiments, compositions provided herein further include a pharmaceutically acceptable diluent, excipient and/or binder.
- Pharmaceutical compositions formulated for administration by an appropriate route and means containing effective concentrations of one or more of the compounds provided herein, or pharmaceutically effective derivatives thereof, that deliver amounts effective for the treatment, prevention, or amelioration of one or more symptoms of diseases, disorders or conditions that are modulated or otherwise affected by tyrosine kinase activity, or in which tyrosine kinase activity is implicated, are provided. The effective amounts and concentrations are effective for ameliorating any of the symptoms of any of the diseases, disorders or conditions disclosed herein.
- In certain embodiments, provided herein is a pharmaceutical composition containing: i) a physiologically acceptable carrier, diluent, and/or excipient; and ii) one or more compounds provided herein.
- In one aspect, provided herein are methods for treating a patient by administering a compound provided herein. In some embodiments, provided herein is a method of inhibiting the activity of tyrosine kinase(s), such as Btk, or of treating a disease, disorder, or condition, which benefit from inhibition of tyrosine kinase(s), such as Btk, in a patient, which includes administering to the patient a therapeutically effective amount of at least one of any of the compounds herein, or pharmaceutically acceptable salt, pharmaceutically active metabolite, pharmaceutically acceptable prodrug, or pharmaceutically acceptable solvate.
- In another aspect, provided herein is the use of a compound disclosed herein for inhibiting Bruton's tyrosine kinase (Btk) activity or for the treatment of a disease, disorder, or condition, which benefit from inhibition of Bruton's tyrosine kinase (Btk) activity.
- In some embodiments, compounds provided herein are administered to a human. In some embodiments, compounds provided herein are orally administered. In other embodiments, the pharmaceutical formulation that is formulated for a route of administration is selected from oral administration, parenteral administration, buccal administration, nasal administration, topical administration, or rectal administration.
- In other embodiments, compounds provided herein are used for the formulation of a medicament for the inhibition of tyrosine kinase activity. In some other embodiments, compounds provided herein are used for the formulation of a medicament for the inhibition of Bruton's tyrosine kinase (Btk) activity.
- Articles of manufacture including packaging material, a compound or composition or pharmaceutically acceptable derivative thereof provided herein, which is effective for inhibiting the activity of tyrosine kinase(s), such as Btk, within the packaging material, and a label that indicates that the compound or composition, or pharmaceutically acceptable salt, pharmaceutically active metabolite, pharmaceutically acceptable prodrug, or pharmaceutically acceptable solvate thereof, is used for inhibiting the activity of tyrosine kinase(s), such as Btk, are provided.
- In another aspect are inhibited tyrosine kinases comprising a Bruton's tyrosine kinase, a Bruton's tyrosine kinase homolog, or a Btk tyrosine kinase cysteine homolog thereof covalently bound to an inhibitor having the structures:
-


- whereinindicates the point of attachment between the inhibitor and the tyrosine kinase. In a further embodiment, the inhibitor is covalently bound to a cysteine residue on the tyrosine kinase.

- In a further aspect, provided herein is a method for treating an autoimmune disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). In one embodiment, the autoimmune disease is arthritis. In another embodiment, the autoimmune disease is lupus. In some embodiments, the autoimmune disease is inflammatory bowel disease (including Crohn's disease and ulcerative colitis), rheumatoid arthritis, psoriatic arthritis, osteoarthritis, Still's disease, juvenile arthritis, lupus, diabetes, myasthenia gravis, Hashimoto's thyroiditis, Ord's thyroiditis, Graves' disease Sjögren's syndrome, multiple sclerosis, Guillain-Barré syndrome, acute disseminated encephalomyelitis, Addison's disease, opsoclonus-myoclonus syndrome, ankylosing spondylitisis, antiphospholipid antibody syndrome, aplastic anemia, autoimmune hepatitis, coeliac disease, Goodpasture's syndrome, idiopathic thrombocytopenic purpura, optic neuritis, scleroderma, primary biliary cirrhosis, Reiter's syndrome, Takayasu's arteritis, temporal arteritis, warm autoimmune hemolytic anemia, Wegener's granulomatosis, psoriasis, alopecia universalis, Behcet's disease, chronic fatigue, dysautonomia, endometriosis, interstitial cystitis, neuromyotonia, scleroderma, or vulvodynia.
- In a further aspect, provided herein is a method for treating a heteroimmune condition or disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). In some embodiments, the heteroimmune condition or disease is graft versus host disease, transplantation, transfusion, anaphylaxis, allergy, type I hypersensitivity, allergic conjunctivitis, allergic rhinitis, or atopic dermatitis.
- In a further aspect, provided herein is a method for treating an inflammatory disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). In some embodiments, the inflammatory disease is asthma, inflammatory bowel disease (including Crohn's disease and ulcerative colitis), appendicitis, blepharitis, bronchiolitis, bronchitis, bursitis, cervicitis, cholangitis, cholecystitis, colitis, conjunctivitis, cystitis, dacryoadenitis, dermatitis, dermatomyositis, encephalitis, endocarditis, endometritis, enteritis, enterocolitis, epicondylitis, epididymitis, fasciitis, fibrositis, gastritis, gastroenteritis, hepatitis, hidradenitis suppurativa, laryngitis, mastitis, meningitis, myelitis myocarditis, myositis, nephritis, oophoritis, orchitis, osteitis, otitis, pancreatitis, parotitis, pericarditis, peritonitis, pharyngitis, pleuritis, phlebitis, pneumonitis, pneumonia, proctitis, prostatitis, pyelonephritis, rhinitis, salpingitis, sinusitis, stomatitis, synovitis, tendonitis, tonsillitis, uveitis, vaginitis, vasculitis, or vulvitis.
- In yet another aspect, provided herein is a method for treating a cancer by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). In one embodiment, the cancer is a B-cell proliferative disorder, e.g., diffuse large B cell lymphoma, follicular lymphoma, chronic lymphocytic lymphoma, chronic lymphocytic leukemia, B-cell prolymphocytic leukemia, lymphoplasmacytic lymphoma/Waldenström macroglobulinemia, splenic marginal zone lymphoma, plasma cell myeloma, plasmacytoma, extranodal marginal zone B cell lymphoma, nodal marginal zone B cell lymphoma, mantle cell lymphoma, mediastinal (thymic) large B cell lymphoma, intravascular large B cell lymphoma, primary effusion lymphoma, Burkitt's lymphoma/leukemia, or lymphomatoid granulomatosis. In some embodiments, where the subject is suffering from a cancer, an anti-cancer agent is administered to the subject in addition to one of the above-mentioned compounds. In one embodiment, the anti-cancer agent is an inhibitor of mitogen-activated protein kinase signaling, e.g., U0126, PD98059, PD184352, PD0325901, ARRY-142886, SB239063, SP600125, BAY 43-9006, wortmannin, or LY294002.
- In another aspect, provided herein is a method for treating a thromboembolic disorder by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). In some embodiments, the thromboembolic disorder is myocardial infarct, angina pectoris, reocclusion after angioplasty, restenosis after angioplasty, reocclusion after aortocoronary bypass, restenosis after aortocoronary bypass, stroke, transitory ischemia, a peripheral arterial occlusive disorder, pulmonary embolism, or deep venous thrombosis.
- In another aspect, provided herein is a method for treating a mastocytosis by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- In yet another aspect, provided herein is a method for treating a osteoporosis or bone resorption disorders by administering to a subject in need thereof a composition containing a therapeutically effective amount of at least one compound having the structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII).
- In a further aspect, provided herein is a method for treating lupus by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase. In further or alternative embodiments, the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- In a further aspect, provided herein is a method for treating a heteroimmune condition or disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase. In further or alternative embodiments, the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- In a further aspect, provided herein is a method for treating an inflammatory disease by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase. In further or alternative embodiments, the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- In a further aspect, provided herein is a method for treating diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase. In further or alternative embodiments, the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- In yet another aspect, provided herein is a method for treating mastocytosis by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase. In further or alternative embodiments, the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- In another aspect, provided herein is a method for treating a osteoporosis or bone resorption disorders by administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Bruton's tyrosine kinase. In further or alternative embodiments, the compound irreversibly inhibits the Bruton's tyrosine kinase to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond with a cysteine residue on Bruton's tyrosine kinase.
- Further described herein are methods, assays and systems for identifying an irreversible inhibitor of a kinase, including a protein kinase, further including a tyrosine kinase. Further described are methods, assays and systems for determining an appropriate irreversible inhibitor of a kinase, including a tyrosine kinase, in which the inhibitor forms a covalent bond with a cysteine residue on the kinase, further wherein the cysteine residue is near an active site of the kinase. In further embodiments, the inhibitor also has a moiety that binds an active site of the kinase. In some embodiments, the kinases share homology with Btk by having a cysteine residue (including a
Cys 481 residue) that forms a covalent bond with the irreversible inhibitor (such tyrosine kinases, are referred herein as “Btk kinase cysteine homologs”). In some embodiments the Btk kinase cysteine homolog(s) are selected from the Tec family of kinases, the EGFR family of kinases, the Jak3 family of kinases and/or the Btk-Src family of kinases. - In some embodiments, the irreversible inhibitor is a selective irreversible inhibitor, including selectivity for a particular Btk kinase cysteine homolog over other Btk kinase cysteine homologs. In some embodiments the selective and irreversible inhibitor is an effective inhibitor for a kinase selected from Btk, a Btk homolog or a Btk kinase cysteine homolog, but is not an effective inhibitor for at least one other different kinase selected from kinase selected from Btk, a Btk homolog or a Btk kinase cysteine homolog.
- Also described herein are kinase inhibitors that selectively and irreversibly bind to a protein tyrosine kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog, in which the kinase inhibitor reversibly and non-selectively binds to a multiplicity of protein tyrosine kinases. In one embodiment the plasma half life of the kinase inhibitor is less than about 4 hours. In another embodiment the plasma half life of the kinase inhibitor is less than about 3 hours.
- In a further embodiment are kinase inhibitors that selectively and irreversibly bind to at least one of Btk, Jak3, Blk, Bmx, Tec, and Itk. In another embodiment are kinase inhibitors that selectively and irreversibly bind to Btk. In another embodiment are kinase inhibitors that selectively and irreversibly bind to Jak3. In another embodiment are kinase inhibitors that selectively and irreversibly bind to Tec. In another embodiment are kinase inhibitors that selectively and irreversibly bind to Itk. In another embodiment are kinase inhibitors that selectively and irreversibly bind to Btk and Tec. In another embodiment are kinase inhibitors that selectively and irreversibly bind to Blk. In yet a further embodiment are kinase inhibitors that reversibly and non-selectively bind to a multiplicity of src-family protein kinase inhibitors.
- Also described herein are irreversible inhibitors that are identified using such methods, assays and systems. Such irreversible inhibitor comprise an active site binding moiety that binds to an active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog; a Michael acceptor moiety; and a moiety that links the active site binding moiety to the Michael acceptor moiety. In some embodiments, the Michael acceptor moiety comprises and alkene and/or an alkyne moiety. In some embodiments, the irreversible inhibitor is a selective irreversible inhibitor, including selectivity for a particular Btk kinase cysteine homolog over other Btk kinase cysteine homologs.
- In any of the aforementioned embodiments, the irreversible inhibitors have the structure of Formula (VII):
-

- wherein:
-
- wherein
-

- is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
-
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
- Z is C(═O), OC(═O), NHC(═O), NCH3C(═O), C(═S), S(═O)x, OS(═O)x, NHS(═O)x, where x is 1 or 2;
- R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl); and
- pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (VII). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (VII), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (VII). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (VII). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- In some embodiments,
-

- is a substituted fused biaryl moiety selected from
-

- In some embodiments Z is C(═O), NHC(═O), NCH3C(═O), or S(═O)2. In other embodiments, x is 2. In yet other embodiments, Z is C(═O), OC(═O), NHC(═O), S(═O)x, OS(═O)x, or NHS(═O)x. In some other embodiments, Z is C(═O), NHC(═O), or S(═O)2.
- In some embodiments, R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, and substituted C1-C4heteroalkyl; or R7 and R8 taken together form a bond. In yet other embodiments, each of R7 and R8 is H; or R and R8 taken together form a bond.
- In some embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). In some other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C2alkyl-N(C1-C3alkyl)2, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). In yet other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—N(C1-C3alkyl)2, C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl). In yet other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—(C1-C6alkylamino), C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl). In some embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—N(C1-C3alkyl)2, C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl containing 1 or 2 N atoms), or C1-C4alkyl(5- or 6-membered heterocycloalkyl containing 1 or 2 N atoms).
- In some embodiments, Y is an optionally substituted group selected from among alkylene, heteroalkylene, cycloalkylene, and heterocycloalkylene. In other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 4-, 5-, 6-, or 7-membered cycloalkylene, and 4-, 5-, 6-, or 7-membered heterocycloalkylene. In yet other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 5- or 6-membered cycloalkylene, and 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some other embodiments, Y is a 5- or 6-membered cycloalkylene, or a 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some embodiments, Y is a 4-, 5-, 6-, or 7-membered cycloalkylene ring; or Y is a 4-, 5-, 6-, or 7-membered heterocycloalkylene ring.
- Any combination of the groups described above for the various variables is contemplated herein.
- In any of the aforementioned methods, assays and systems: such methods, assays and systems comprise a multiplicity of test irreversible inhibitors, in which the test irreversible inhibitors each have the same
-

- moiety, but differ in at least one of Y, Z, R6, R7, or R8. In further embodiments, the multiplicity of test irreversible inhibitors is a panel of test irreversible inhibitors. In further embodiments, the binding of the panel of test irreversible inhibitors to at least one kinase is determined (including a panel of kinases, further including a panel of kinases selected from Btk, Btk homologs, and Btk kinase cysteine homologs). In further embodiments, the determined binding data is used to select and/or further design a selective irreversible inhibitor.
- Irreversible inhibitors described herein include those that have a structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), and pharmaceutically acceptable salts, solvates, esters, acids and prodrugs thereof. In certain embodiments, isomers and chemically protected forms of compounds having a structure represented by any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are also provided.
- In one aspect, provided herein is an irreversible inhibitor compound selected from among: 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one; 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)sulfonylethene; 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-yn-1-one; 1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)acrylamide; 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)pyrrolidin-1-yl)prop-2-en-1-one; 1-((S)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)pyrrolidin-1-yl)prop-2-en-1-one; 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; 1-((S)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; and (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-(dimethylamino)but-2-en-1-one; (E)-4-(N-(2-hydroxyethyl)-N-methylamino)-1-(3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one (Compound 3); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3-(1H-imidazol-4-yl)prop-2-en-1-one (Compound 4); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 5); (E)-1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 7); (E)-N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)-4-(dimethylamino)but-2-enamide (Compound 8); N-((1r,4r)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)acrylamide (Compound 10); (E)-1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrolidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 11); (E)-1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrolidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 12); 1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 13); 1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 14); 1((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)but-2-yn-1-one (Compound 15); 1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)but-2-yn-1-one (Compound 16); 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-yn-1-one (Compound 17); (E)-N-((1,r,4r)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl-4-(dimethylamino)but-2-enamide (Compound 18); N-(2-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-N-methylacrylamide (Compound 19); (E)-1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 20); (E)-1-((S-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 21); N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)but-2-ynamide (Compound 22); N-(2-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)acrylamide (Compound 23); (E)-1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 24); (E)-N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)-4-morpholinobut-2-enamide (Compound 25).
- Further described herein are pharmaceutical formulations comprising the kinase inhibitors of any kinase inhibitor compound previously listed. In one embodiment the pharmaceutical formulation includes a pharmaceutical acceptable excipient. In some embodiments, pharmaceutical formulations provided herein are administered to a human. In some embodiments, the irreversible and/or selective kinase inhibitors provided herein are orally administered. In other embodiments, the irreversible and/or selective kinase inhibitors provided herein are used for the formulation of a medicament for the inhibition of tyrosine kinase activity. In some other embodiments, the irreversible and/or selective kinase inhibitors provided herein are used for the formulation of a medicament for the inhibition of a kinase activity, including a tyrosine kinase activity, including a Btk activity, including a Btk homolog activity, including a Btk kinase cysteine homolog activity.
- In any of the aforementioned aspects are further embodiments in which administration is enteral, parenteral, or both, and wherein (a) the effective amount of the compound is systemically administered to the mammal; (b) the effective amount of the compound is administered orally to the mammal; (c) the effective amount of the compound is intravenously administered to the mammal; (d) the effective amount of the compound administered by inhalation; (e) the effective amount of the compound is administered by nasal administration; or (f) the effective amount of the compound is administered by injection to the mammal; (g) the effective amount of the compound is administered topically (dermal) to the mammal; (h) the effective amount of the compound is administered by ophthalmic administration; or (i) the effective amount of the compound is administered rectally to the mammal. In further embodiments the pharmaceutical formulation is formulated for a route of administration selected from oral administration, parenteral administration, buccal administration, nasal administration, topical administration, or rectal administration.
- In any of the aforementioned aspects are further embodiments comprising single administrations of the effective amount of the pharmaceutical formulation, including further embodiments in which (i) the pharmaceutical formulations is administered once; (ii) the pharmaceutical formulations is administered to the mammal once a day; (iii) the pharmaceutical formulations is administered to the mammal multiple times over the span of one day; (iv) continually; or (v) continuously.
- In any of the aforementioned aspects are further embodiments comprising multiple administrations of the effective amount of the pharmaceutical formulations, including further embodiments in which (i) the pharmaceutical formulations is administered in a single dose; (ii) the time between multiple administrations is every 6 hours; (iii) the pharmaceutical formulations is administered to the mammal every 8 hours. In further or alternative embodiments, the method comprises a drug holiday, wherein the administration of the pharmaceutical formulations is temporarily suspended or the dose of the pharmaceutical formulations being administered is temporarily reduced; at the end of the drug holiday, dosing of the pharmaceutical formulations is resumed. The length of the drug holiday varies from 2 days to 1 year.
- Further described herein is a method for increasing the selectivity of a test protein kinase inhibitor that irreversibly and selectively binds to at least one protein kinase inhibitor selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog. In one embodiment the test protein tyrosine kinase inhibitor is chemically modified to decrease the plasma half life to less than about 4 hours. In another embodiment the test protein tyrosine kinase inhibitor is chemically modified to decrease the plasma half life to about 3 hours. In yet another embodiment the test protein tyrosine kinase inhibitor non-selectively and reversibly binds to a multiplicity of src-family protein tyrosine kinases.
- In one embodiment the test protein kinase inhibitor has the structure of Formula (VII):
-

- wherein:
-
- wherein
-

- is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
-
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
- Z is C(═O), OC(═O), NHC(═O), NCH3C(═O), C(═S), S(═O)x, OS(═O)x, NHS(═O)x, where x is 1 or 2;
- R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl); and
- pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In a further aspect, provided herein is a method for treating a B-cell proliferative disorder or a mast cell proliferative disorder by administering to a subject in need thereof a test protein kinase inhibitor composition containing a therapeutically effective amount of a compound that forms a covalent bond (including an irreversible and/or selective covalent bond) with Btk, a Btk homolog, or a Btk kinase cysteine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Btk, a Btk homolog, or a Btk kinase cysteine homolog. In further or alternative embodiments, the compound irreversibly inhibits Btk, a Btk homolog, or a Btk kinase cysteine homolog to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond (including an irreversible and/or selective covalent bond) with a cysteine residue on Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- In a further aspect, provided herein is a method for treating rheumatoid arthritis by administering to a subject in need thereof a test protein kinase inhibitor composition containing a therapeutically effective amount of a compound that forms a covalent bond (including an irreversible and/or selective covalent bond) with Btk, a Btk homolog, or a Btk kinase cysteine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Btk, a Btk homolog, or a Btk kinase cysteine homolog. In further or alternative embodiments, the compound irreversibly inhibits Btk, a Btk homolog, or a Btk kinase cysteine homolog to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond (including an irreversible and/or selective covalent bond) with a cysteine residue on Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- In a further aspect, provided herein is a method for treating a disease characterized by hyperactive B-cells or hyperactive mast cells or both hyperactive B-cells and hyperactive mast cells by administering to a subject in need thereof a test protein kinase inhibitor composition containing a therapeutically effective amount of a compound that forms a covalent bond (including an irreversible and/or selective covalent bond) with Btk, a Btk homolog, or a Btk kinase cysteine homolog. In one embodiment, the compound forms a covalent bound with the activated form of Btk, a Btk homolog, or a Btk kinase cysteine homolog. In further or alternative embodiments, the compound irreversibly inhibits Btk, a Btk homolog, or a Btk kinase cysteine homolog to which it is covalently bound. In a further or alternative embodiment, the compound forms a covalent bond (including an irreversible and/or selective covalent bond) with a cysteine residue on Btk, a Btk homolog, or a Btk kinase cysteine homolog.
- In any of the aforementioned aspects are further embodiments comprising single administrations of the effective amount of the pharmaceutical formulation, including further embodiments in which (i) the pharmaceutical formulations is administered once; (ii) the pharmaceutical formulations is administered to the mammal once a day; (iii) the pharmaceutical formulations is administered to the mammal multiple times over the span of one day; (iv) continually; or (v) continuously.
- Also described herein is a method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog comprising:
-
- (1) contacting a multiplicity of kinases selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog with a compound that comprises a Michael acceptor moiety;
- (2) contacting at least one non-kinase molecule having at least one accessible SH group with the compound that comprises a Michael acceptor moiety; and
- (3) determining the covalent binding of the compound that comprises a Michael acceptor with the multiplicity of kinases and the at least one non-kinase molecule; and repeating steps (1), (2), and (3) for at least one other compound that comprises a Michael acceptor moiety.
- Further described herein is a method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog comprising:
-
- (1) contacting a multiplicity of kinases selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog with a compound that comprises a Michael acceptor moiety;
- (2) contacting at least one non-kinase molecule having at least one accessible SH group with the compound that comprises a Michael acceptor moiety; and
- (3) determining the covalent binding of the compound that comprises a Michael acceptor with the multiplicity of kinases and the at least one non-kinase molecule; and repeating steps (1), (2), and (3) for at least one other compound that comprises a Michael acceptor moiety; and
- (4) comparing the covalent binding of the compound that comprises a Michael acceptor with the multiplicity of kinases and the at least one non-kinase molecule; and
- repeating steps (1), (2), (3) and (4) for at least one other compound that comprises a Michael acceptor moiety.
- In one embodiment the at least one non-kinase molecule having at least one accessible SH group includes glutathione and/or hemoglobin. In another embodiment the desired irreversible inhibitor is selective for a particular kinase relative to other kinases, glutathione and hemoglobin.
- In some embodiments, the methods, assays and systems for identifying an irreversible inhibitor of a kinase comprise contacting each kinase with an Activity Probe. In further embodiments, the methods, assays and systems for identifying an irreversible inhibitor of a kinase further comprise a panel of kinases comprising at least two kinases selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog. In further embodiments, the panel of kinases comprises at least three such kinases, at least four such kinases, at least five such kinases, at least six such kinases, at least seven such kinases, at least eight such kinases, at least nine such kinases, or at least ten such kinases.
- In one embodiment steps (1) and (2) of the method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog is conducted in vivo. In another embodiment step (3) of the method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog is conducted in part using an Activity Probe.
- In one embodiment, contacting a multiplicity of kinases selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog with a compound that comprises a Michael acceptor moiety is conducted in vivo. In another embodiment contacting at least one non-kinase molecule having at least one accessible SH group with the compound that comprises a Michael acceptor moiety is conducted in vivo. In a further embodiment determining the covalent binding of the compound that comprises a Michael acceptor with the multiplicity of kinases and the at least one non-kinase molecule is conducted in part using an Activity Probe. In a further embodiment the determining step uses mass spectrometry. In yet further embodiments the determining step uses fluorescence.
- In further embodiments of methods and assays for identifying an irreversible inhibitor of a kinase, including a protein kinase, including a tyrosine kinase, a panel of kinases is contacted with at least one irreversible inhibitor. In a further embodiment, the panel of kinases is also contacted with an Activity Probe. In a further embodiment, the binding of an irreversible inhibitor to a kinase is determined from the binding of the Activity Probe to the kinase. In a further embodiment, the binding of the Activity Probe to a kinase is determined using fluorescence technique. In further or alternative methods and assays, the Activity Probe is compatible with flow cytometery. In further embodiments, the binding of the irreversible inhibitor to one kinase is compared to the binding of the irreversible inhibitor to at least one other kinase. In any of the aforementioned embodiments, the panel of kinases is selected from Btk, Btk homologs, and Btk kinase cysteine homologs. In a further or alternative embodiment, the binding of an irreversible inhibitor to a kinase is determined by mass spectrometry.
- Also described herein are activity probes of Bruton's tyrosine kinase (Btk), Btk homologs, and Btk kinase cysteine homologs (collectively “Activity Probes”). Further described are Activity Probes that include an irreversible inhibitor of Btk, a Btk homolog and/or a Btk kinase cysteine homolog; a linker moiety; and a reporter moiety. Further described are Activity Probes that include a Michael addition acceptor moiety in the structure of the Activity Probe. Further described are Activity Probes that form a covalent bond with a cysteine residue on Btk, a Btk homolog and/or a Btk kinase cysteine homolog. Also described herein are Activity Probes that form a non-covalent bond with a cysteine residue on Btk, a Btk homolog and/or a Btk kinase cysteine homolog. Also described herein are methods for synthesizing such Activity Probes, methods for using such Activity Probes in the study of the activity of Btk, a Btk homolog and/or a Btk kinase cysteine homolog, methods for using such Activity Probes in the study of inhibitors (including the development of new inhibitors) of Btk, a Btk homolog and/or a Btk kinase cysteine homolog, and methods for using such Activity Probes in the study of the pharmacodynamics of inhibitors of Btk, a Btk homolog and/or a Btk kinase cysteine homolog.
- In one embodiment are Activity Probes wherein the linker moiety is selected from a bond, an optionally substituted alkyl moiety, an optionally substituted heterocycle moiety, an optionally substituted amide moiety, a ketone moiety, an optionally substituted carbamate moiety, an ester moiety, or a combination thereof. In another embodiment are Activity Probes wherein the linker moiety comprises an optionally substituted heterocycle moiety. In a further embodiment are Activity Probes wherein the optionally substituted heterocycle moiety comprises a piperazinyl-based moiety.
- Also described herein are Activity Probes wherein the reporter moiety is selected from the group consisting of a label, a dye, a photocrosslinker, a cytotoxic compound, a drug, an affinity label, a photoaffinity label, a reactive compound, an antibody or antibody fragment, a biomaterial, a nanoparticle, a spin label, a fluorophore, a metal-containing moiety, a radioactive moiety, a novel functional group, a group that covalently or noncovalently interacts with other molecules, a photocaged moiety, an actinic radiation excitable moiety, a ligand, a photoisomerizable moiety, biotin, a biotin analogue, a moiety incorporating a heavy atom, a chemically cleavable group, a photocleavable group, a redox-active agent, an isotopically labeled moiety, a biophysical probe, a phosphorescent group, a chemiluminescent group, an electron dense group, a magnetic group, an intercalating group, a chromophore, an energy transfer agent, a biologically active agent, a detectable label, or a combination thereof. In another embodiment are Activity Probes wherein the reporter moiety is a fluorophore. In yet another embodiment are Activity Probes wherein the fluorophore is a Bodipy fluorophore. In yet a further embodiment are Activity Probes wherein the Bodipy fluorophore is a Bodipy FL fluorophore.
- Presented herein are Activity Probes wherein the inhibitor moiety is derived from an irreversible inhibitor of Btk, a Btk homolog and/or a Btk kinase cysteine homolog. In one embodiment, are Activity Probes wherein the irreversible inhibitor is:
-

- In another embodiment are Activity Probes having the structure:
-

- In a further embodiment are Activity Probes wherein the probe selectively labels a phosphorylated conformation of Btk, a Btk homolog and/or a Btk kinase cysteine homolog. In another embodiment are Activity Probes wherein the phosphorylated conformation of Btk, a Btk homolog and/or a Btk kinase cysteine homolog is either an active or inactive form of Btk, a Btk homolog and/or a Btk kinase cysteine homolog. In a further embodiment are Activity Probes wherein the phosphorylated conformation of Btk, a Btk homolog and/or a Btk kinase cysteine homolog is an active form of Btk, a Btk homolog and/or a Btk kinase cysteine homolog. In one embodiment are Activity Probes of wherein the probe is cell permeable.
- In one aspect is a method for assessing the efficacy of a potential Btk, Btk homolog and/or Btk kinase cysteine homolog inhibitor in a mammal, comprising administering a potential Btk, Btk homolog and/or Btk kinase cysteine homolog inhibitor to the mammal, administering the Activity Probe described herein to the mammal or to cells isolated from the mammal; measuring the activity of the reporter moiety of the Activity Probe, and comparing the activity of the reporter moiety to a standard.
- In another aspect is a method for assessing the pharmacodynamics of a Btk, Btk homolog and/or Btk kinase cysteine homolog inhibitor in a mammal, comprising administering a Btk, Btk homolog and/or Btk kinase cysteine homolog inhibitor to the mammal, administering the Activity Probe presented herein to the mammal or to cells isolated from the mammal, and measuring the activity of the reporter moiety of the Activity Probe at different time points following the administration of the inhibitor.
- In a further aspect is a method for in vitro labeling of Btk, a Btk homolog and/or a Btk kinase cysteine homolog comprising contacting an active Btk, Btk homolog and/or Btk kinase cysteine homolog with the Activity Probe described herein. In one embodiment is a method for in vitro labeling of Btk, a Btk homolog and/or a Btk kinase cysteine homolog wherein the contacting step comprises incubating the active Btk, Btk homolog and/or Btk kinase cysteine homolog with the Activity Probe presented herein.
- In another aspect is a method for in vitro labeling of Btk, a Btk homolog and/or a Btk kinase cysteine homolog comprising contacting cells or tissues expressing the Btk, Btk homolog and/or Btk kinase cysteine homolog with an Activity Probe described herein.
- In one aspect is a method for detecting a labeled Btk, Btk homolog and/or Btk kinase cysteine homolog comprising separating proteins, the proteins comprising Btk, a Btk homolog and/or a Btk kinase cysteine homolog labeled by an Activity Probe described herein, by electrophoresis and detecting the Activity Probe by fluorescence.
- In further embodiments the irreversible inhibitor of a kinase further comprises an active site binding moiety. In yet further embodiments the irreversible inhibitor of a kinase further comprises a linker moiety that links the Michael acceptor moiety to the active binding moiety.
- In one embodiment the irreversible inhibitor of a kinase has the structure of Formula (VII):
-

- wherein:
-
- wherein
-

- is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
-
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
- Z is C(═O), OC(═O), NHC(═O), NCH3C(═O), C(═S), S(═O)x, OS(═O)x, NHS(═O)x, where x is 1 or 2;
- R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl); and pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In one embodiment the method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog comprising steps (1), (2), (3), and (4) further comprises analyzing the structure-function activity relationship between the structure of the linker moiety and/or the Michael acceptor moiety of each compound, and the binding and/or selectivity of each compound to at least one kinase. In another embodiment the method of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog comprising steps (1), (2), (3), and (4) further comprises analyzing the structure-function activity relationship between the structure of Y—Z and/or
-

- of each compound, and the binding and/or selectivity of each compound to at least one kinase.
- In one embodiment the structure of the active site binding moiety of each compound is not varied. In another embodiment the structure of
-

- of each compound is not varied.
- Also described herein is a method for improving the kinase selectivity of an inhibitor comprising use of any method previously listed.
- One aspect described herein is an assay comprising any of the methods previously listed. Another aspect described herein is system comprising any of the methods previously listed. In a further aspect described herein is an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog, wherein the inhibitor is identified using any methods described herein.
- In some aspects described herein the irreversible inhibitor is selective for one kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog over at least one other kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog. In other aspects described herein the irreversible inhibitor is selective for at least one kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog over at least one other non-kinase molecule having an accessible SH group.
- In certain embodiments, provided herein is a pharmaceutical composition containing: i) a physiologically acceptable carrier, diluent, and/or excipient; and ii) one or more compounds provided herein.
- In a further aspect, provided herein is a method for treating an autoimmune disease or condition comprising administering to a patient in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog. In one embodiment the autoimmune disease is selected from rheumatoid arthritis or lupus.
- In a further aspect, provided herein is a method for treating a B-cell proliferative disorder comprising administering to a patient in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog. In one embodiment the B-cell proliferative disorder is diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia.
- In a further aspect, provided herein is a method for treating an inflammatory disease or condition comprising administering to a patient in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or Bruton's tyrosine homolog.
- Also described herein are methods to identify biomarkers for patient selection or patient monitoring prior to or during treatment with any kinase inhibitor compound described herein. In one embodiment, a patient that has lymphoma is administered a pharmaceutical composition of any kinase inhibitor compound described herein which inhibits B cell receptor (BCR) signaling. In another embodiment, the inhibition of the BCR signaling by any kinase inhibitor compound described herein is correlated with the induction of apoptosis. In another embodiment, a patient with lymphoma is selected for treatment with a pharmaceutical composition of any kinase inhibitor compound described herein based on a biomarker that indicates that the lymphoma in that patient has high levels of pErk or Erk transcriptional targets. In another embodiment, the response to treatment with a pharmaceutical composition of any kinase inhibitor compound described herein is measured by a reduction in levels of pErk or Erk transcriptional targets.
- Other objects, features and advantages of the methods and compositions described herein will become apparent from the following detailed description. It should be understood, however, that the detailed description and the specific examples, while indicating specific embodiments, are given by way of illustration only. The section headings used herein are for organizational purposes only and are not to be construed as limiting the subject matter described.
- It is to be understood that the foregoing general description and the following detailed description are exemplary and explanatory only and are not restrictive of any subject matter claimed. In this application, the use of the singular includes the plural unless specifically stated otherwise. It must be noted that, as used in the specification and the appended claims, the singular forms “a,” “an” and “the” include plural referents unless the context clearly dictates otherwise. In this application, the use of “or” means “and/or” unless stated otherwise. Furthermore, use of the term “including” as well as other forms, such as “include”, “includes,” and “included,” is not limiting.
- Definition of standard chemistry terms are found in reference works, including Carey and Sundberg “A
DVANCED ORGANIC CHEMISTRY 4TH ED .” Vols. A (2000) and B (2001), Plenum Press, New York. Unless otherwise indicated, conventional methods of mass spectroscopy, NMR, HPLC, protein chemistry, biochemistry, recombinant DNA techniques and pharmacology, within the skill of the art are employed. Unless specific definitions are provided, the nomenclature employed in connection with, and the laboratory procedures and techniques of, analytical chemistry, synthetic organic chemistry, and medicinal and pharmaceutical chemistry described herein are those known in the art. Standard techniques are optionally used for chemical syntheses, chemical analyses, pharmaceutical preparation, formulation, and delivery, and treatment of patients. Standard techniques are optionally used for recombinant DNA, oligonucleotide synthesis, and tissue culture and transformation (e.g., electroporation, lipofection). Reactions and purification techniques are performed using documented methodologies or as described herein. - It is to be understood that the methods and compositions described herein are not limited to the particular methodology, protocols, cell lines, constructs, and reagents described herein and as such optionally vary. It is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to limit the scope of the methods and compositions described herein, which will be limited only by the appended claims.
- Unless stated otherwise, the terms used for complex moieties (i.e., multiple chains of moieties) are to be read equivalently either from left to right or right to left. For example, the group alkylenecycloalkylene refers both to an alkylene group followed by a cycloalkylene group or as a cycloalkylene group followed by an alkylene group.
- The suffix “ene” appended to a group indicates that such a group is a diradical. By way of example only, a methylene is a diradical of a methyl group, that is, it is a —CH2— group; and an ethylene is a diradical of an ethyl group, i.e., —CH2CH2—.
- An “alkyl” group refers to an aliphatic hydrocarbon group. The alkyl moiety includes a “saturated alkyl” group, which means that it does not contain any alkene or alkyne moieties. The alkyl moiety also includes an “unsaturated alkyl” moiety, which means that it contains at least one alkene or alkyne moiety. An “alkene” moiety refers to a group that has at least one carbon-carbon double bond, and an “alkyne” moiety refers to a group that has at least one carbon-carbon triple bond. The alkyl moiety, whether saturated or unsaturated, includes branched, straight chain, or cyclic moieties. Depending on the structure, an alkyl group includes a monoradical or a diradical (i.e., an alkylene group), and if a “lower alkyl” having 1 to 6 carbon atoms.
- As used herein, C1-Cx includes C1-C2, C1-C3 . . . C1-Cx.
- The “alkyl” moiety optionally has 1 to 10 carbon atoms (whenever it appears herein, a numerical range such as “1 to 10” refers to each integer in the given range; e.g., “1 to 10 carbon atoms” means that the alkyl group is selected from a moiety having 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc., up to and including 10 carbon atoms, although the present definition also covers the occurrence of the term “alkyl” where no numerical range is designated). The alkyl group of the compounds described herein may be designated as “C1-C4 alkyl” or similar designations. By way of example only, “C1-C4 alkyl” indicates that there are one to four carbon atoms in the alkyl chain, i.e., the alkyl chain is selected from among methyl, ethyl, propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl, and t-butyl. Thus C1-C4 alkyl includes C1-C2 alkyl and C1-C3 alkyl. Alkyl groups are optionally substituted or unsubstituted. Typical alkyl groups include, but are in no way limited to, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tertiary butyl, pentyl, hexyl, ethenyl, propenyl, butenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like.
- The term “alkenyl” refers to a type of alkyl group in which the first two atoms of the alkyl group form a double bond that is not part of an aromatic group. That is, an alkenyl group begins with the atoms —C(R)═C(R)—R, wherein R refers to the remaining portions of the alkenyl group, which are either the same or different. The alkenyl moiety is optionally branched, straight chain, or cyclic (in which case, it is also known as a “cycloalkenyl” group). Depending on the structure, an alkenyl group includes a monoradical or a diradical (i.e., an alkenylene group). Alkenyl groups are optionally substituted. Non-limiting examples of an alkenyl group include —CH═CH2, —C(CH3)═CH2, —CH═CHCH3, —C(CH3)═CHCH3. Alkenylene groups include, but are not limited to, —CH═CH—, —C(CH3)═CH—, —CH═CHCH2—, —CH═CHCH2CH2— and —C(CH3)═CHCH2—. Alkenyl groups optionally have 2 to 10 carbons, and if a “lower alkenyl” having 2 to 6 carbon atoms.
- The term “alkynyl” refers to a type of alkyl group in which the first two atoms of the alkyl group form a triple bond. That is, an alkynyl group begins with the atoms —C≡C—R, wherein R refers to the remaining portions of the alkynyl group, which is either the same or different. The “R” portion of the alkynyl moiety may be branched, straight chain, or cyclic. Depending on the structure, an alkynyl group includes a monoradical or a diradical (i.e., an alkynylene group). Alkynyl groups are optionally substituted. Non-limiting examples of an alkynyl group include, but are not limited to, —C≡CH, —C≡CCH3, —C≡CCH2CH3, —C≡C—, and —C≡CCH2—. Alkynyl groups optionally have 2 to 10 carbons, and if a “lower alkynyl” having 2 to 6 carbon atoms.
- An “alkoxy” group refers to a (alkyl)O— group, where alkyl is as defined herein.
- “Hydroxyalkyl” refers to an alkyl radical, as defined herein, substituted with at least one hydroxy group. Non-limiting examples of a hydroxyalkyl include, but are not limited to, hydroxymethyl, 2-hydroxyethyl, 2-hydroxypropyl, 3-hydroxypropyl, 1-(hydroxymethyl)-2-methylpropyl, 2-hydroxybutyl, 3-hydroxybutyl, 4-hydroxybutyl, 2,3-dihydroxypropyl, 1-(hydroxymethyl)-2-hydroxyethyl, 2,3-dihydroxybutyl, 3,4-dihydroxybutyl and 2-(hydroxymethyl)-3-hydroxypropyl.
- “Alkoxyalkyl” refers to an alkyl radical, as defined herein, substituted with an alkoxy group, as defined herein.
- The term “alkylamine” refers to the —N(alkyl)xHy group, where x and y are selected from among x=1, y=1 and x=2, y=0. When x=2, the alkyl groups, taken together with the N atom to which they are attached, can optionally form a cyclic ring system.
- “Alkylaminoalkyl” refers to an alkyl radical, as defined herein, substituted with an alkylamine, as defined herein.
- “Hydroxyalkylaminoalkyl” refers to an alkyl radical, as defined herein, substituted with an alkylamine, and alkylhydroxy, as defined herein.
- “Alkoxyalkylaminoalkyl” refers to an alkyl radical, as defined herein, substituted with an alkylamine and substituted with an alkylalkoxy, as defined herein.
- An “amide” is a chemical moiety with the formula —C(O)NHR or —NHC(O)R, where R is selected from among alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring carbon) and heteroalicyclic (bonded through a ring carbon). In some embodiments, an amide moiety forms a linkage between an amino acid or a peptide molecule and a compound described herein, thereby forming a prodrug. Any amine, or carboxyl side chain on the compounds described herein can be amidified. The procedures and specific groups to make such amides are found in sources such as Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, New York, N.Y., 1999, which is incorporated herein by reference for this disclosure.
- The term “ester” refers to a chemical moiety with formula —COOR, where R is selected from among alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring carbon) and heteroalicyclic (bonded through a ring carbon). Any hydroxy, or carboxyl side chain on the compounds described herein can be esterified. The procedures and specific groups to make such esters are found in sources such as Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, New York, N.Y., 1999, which is incorporated herein by reference for this disclosure.
- As used herein, the term “ring” refers to any covalently closed structure. Rings include, for example, carbocycles (e.g., aryls and cycloalkyls), heterocycles (e.g., heteroaryls and non-aromatic heterocycles), aromatics (e.g. aryls and heteroaryls), and non-aromatics (e.g., cycloalkyls and non-aromatic heterocycles). Rings can be optionally substituted. Rings can be monocyclic or polycyclic.
- As used herein, the term “ring system” refers to one, or more than one ring.
- The term “membered ring” can embrace any cyclic structure. The term “membered” is meant to denote the number of skeletal atoms that constitute the ring. Thus, for example, cyclohexyl, pyridine, pyran and thiopyran are 6-membered rings and cyclopentyl, pyrrole, furan, and thiophene are 5-membered rings.
- The term “fused” refers to structures in which two or more rings share one or more bonds.
- The term “carbocyclic” or “carbocycle” refers to a ring wherein each of the atoms forming the ring is a carbon atom. Carbocycle includes aryl and cycloalkyl. The term thus distinguishes carbocycle from heterocycle (“heterocyclic”) in which the ring backbone contains at least one atom which is different from carbon (i.e a heteroatom). Heterocycle includes heteroaryl and heterocycloalkyl. Carbocycles and heterocycles can be optionally substituted.
- The term “aromatic” refers to a planar ring having a delocalized it-electron system containing 4n+2π electrons, where n is an integer. Aromatic rings can be formed from five, six, seven, eight, nine, or more than nine atoms. Aromatics can be optionally substituted. The term “aromatic” includes both carbocyclic aryl (e.g., phenyl) and heterocyclic aryl (or “heteroaryl” or “heteroaromatic”) groups (e.g., pyridine). The term includes monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups.
- As used herein, the term “aryl” refers to an aromatic ring wherein each of the atoms forming the ring is a carbon atom. Aryl rings can be formed by five, six, seven, eight, nine, or more than nine carbon atoms. Aryl groups can be optionally substituted. Examples of aryl groups include, but are not limited to phenyl, naphthalenyl, phenanthrenyl, anthracenyl, fluorenyl, and indenyl. Depending on the structure, an aryl group can be a monoradical or a diradical (i.e., an arylene group).
- An “aryloxy” group refers to an (aryl)O— group, where aryl is as defined herein.
- The term “carbonyl” as used herein refers to a group containing a moiety selected from the group consisting of —C(O)—, —S(O)—, —S(O)2-, and —C(S)—, including, but not limited to, groups containing a least one ketone group, and/or at least one aldehyde group, and/or at least one ester group, and/or at least one carboxylic acid group, and/or at least one thioester group. Such carbonyl groups include ketones, aldehydes, carboxylic acids, esters, and thioesters. In some embodiments, such groups are a part of linear, branched, or cyclic molecules.
- The term “cycloalkyl” refers to a monocyclic or polycyclic radical that contains only carbon and hydrogen, and is optionally saturated, partially unsaturated, or fully unsaturated. Cycloalkyl groups include groups having from 3 to 10 ring atoms. Illustrative examples of cycloalkyl groups include the following moieties:
-

- and the like. Depending on the structure, a cycloalkyl group is either a monoradical or a diradical (e.g., an cycloalkylene group), and if a “lower cycloalkyl” having 3 to 8 carbon atoms.
- “Cycloalkylalkyl” means an alkyl radical, as defined herein, substituted with a cycloalkyl group. Non-limiting cycloalkylalkyl groups include cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, and the like.
- The term “heterocycle” refers to heteroaromatic and heteroalicyclic groups containing one to four heteroatoms each selected from O, S and N, wherein each heterocyclic group has from 4 to 10 atoms in its ring system, and with the proviso that the ring of said group does not contain two adjacent O or S atoms. Herein, whenever the number of carbon atoms in a heterocycle is indicated (e.g., C1-C6 heterocycle), at least one other atom (the heteroatom) must be present in the ring. Designations such as “C1-C6 heterocycle” refer only to the number of carbon atoms in the ring and do not refer to the total number of atoms in the ring. It is understood that the heterocylic ring can have additional heteroatoms in the ring. Designations such as “4-6 membered heterocycle” refer to the total number of atoms that are contained in the ring (i.e., a four, five, or six membered ring, in which at least one atom is a carbon atom, at least one atom is a heteroatom and the remaining two to four atoms are either carbon atoms or heteroatoms). In heterocycles that have two or more heteroatoms, those two or more heteroatoms can be the same or different from one another. Heterocycles can be optionally substituted. Binding to a heterocycle can be at a heteroatom or via a carbon atom. Non-aromatic heterocyclic groups include groups having only 4 atoms in their ring system, but aromatic heterocyclic groups must have at least 5 atoms in their ring system. The heterocyclic groups include benzo-fused ring systems. An example of a 4-membered heterocyclic group is azetidinyl (derived from azetidine). An example of a 5-membered heterocyclic group is thiazolyl. An example of a 6-membered heterocyclic group is pyridyl, and an example of a 10-membered heterocyclic group is quinolinyl. Examples of non-aromatic heterocyclic groups are pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 1,2,3,6-tetrahydropyridinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1,3-dioxolanyl, pyrazolinyl, dithianyl, dithiolanyl, dihydropyranyl, dihydrothienyl, dihydrofuranyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, 3-azabicyclo[3.1.0]hexanyl, 3-azabicyclo[4.1.0]heptanyl, 3H-indolyl and quinolizinyl. Examples of aromatic heterocyclic groups are pyridinyl, imidazolyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinoxalinyl, naphthyridinyl, and furopyridinyl. The foregoing groups, as derived from the groups listed above, are optionally C-attached or N-attached where such is possible. For instance, a group derived from pyrrole includes pyrrol-1-yl (N-attached) or pyrrol-3-yl (C-attached). Further, a group derived from imidazole includes imidazol-1-yl or imidazol-3-yl (both N-attached) or imidazol-2-yl, imidazol-4-yl or imidazol-5-yl (all C-attached). The heterocyclic groups include benzo-fused ring systems and ring systems substituted with one or two oxo (═O) moieties such as pyrrolidin-2-one. Depending on the structure, a heterocycle group can be a monoradical or a diradical (i.e., a heterocyclene group).
- The terms “heteroaryl” or, alternatively, “heteroaromatic” refers to an aromatic group that includes one or more ring heteroatoms selected from nitrogen, oxygen and sulfur. An N-containing “heteroaromatic” or “heteroaryl” moiety refers to an aromatic group in which at least one of the skeletal atoms of the ring is a nitrogen atom. Illustrative examples of heteroaryl groups include the following moieties:
-

- and the like. Depending on the structure, a heteroaryl group can be a monoradical or a diradical (i.e., a heteroarylene group).
- As used herein, the term “non-aromatic heterocycle”, “heterocycloalkyl” or “heteroalicyclic” refers to a non-aromatic ring wherein one or more atoms forming the ring is a heteroatom. A “non-aromatic heterocycle” or “heterocycloalkyl” group refers to a cycloalkyl group that includes at least one heteroatom selected from nitrogen, oxygen and sulfur. In some embodiments, the radicals are fused with an aryl or heteroaryl. Heterocycloalkyl rings can be formed by three, four, five, six, seven, eight, nine, or more than nine atoms. Heterocycloalkyl rings can be optionally substituted. In certain embodiments, non-aromatic heterocycles contain one or more carbonyl or thiocarbonyl groups such as, for example, oxo- and thio-containing groups. Examples of heterocycloalkyls include, but are not limited to, lactams, lactones, cyclic imides, cyclic thioimides, cyclic carbamates, tetrahydrothiopyran, 4H-pyran, tetrahydropyran, piperidine, 1,3-dioxin, 1,3-dioxane, 1,4-dioxin, 1,4-dioxane, piperazine, 1,3-oxathiane, 1,4-oxathiin, 1,4-oxathiane, tetrahydro-1,4-thiazine, 2H-1,2-oxazine, maleimide, succinimide, barbituric acid, thiobarbituric acid, dioxopiperazine, hydantoin, dihydrouracil, morpholine, trioxane, hexahydro-1,3,5-triazine, tetrahydrothiophene, tetrahydrofuran, pyrroline, pyrrolidine, pyrrolidone, pyrrolidione, pyrazoline, pyrazolidine, imidazoline, imidazolidine, 1,3-dioxole, 1,3-dioxolane, 1,3-dithiole, 1,3-dithiolane, isoxazoline, isoxazolidine, oxazoline, oxazolidine, oxazolidinone, thiazoline, thiazolidine, and 1,3-oxathiolane. Illustrative examples of heterocycloalkyl groups, also referred to as non-aromatic heterocycles, include:
-

- and the like. The term heteroalicyclic also includes all ring forms of the carbohydrates, including but not limited to the monosaccharides, the disaccharides and the oligosaccharides. Depending on the structure, a heterocycloalkyl group can be a monoradical or a diradical (i.e., a heterocycloalkylene group).
- The term “halo” or, alternatively, “halogen” or “halide” means fluoro, chloro, bromo and iodo.
- The term “haloalkyl,” refers to alkyl structures in which at least one hydrogen is replaced with a halogen atom. In certain embodiments in which two or more hydrogen atoms are replaced with halogen atoms, the halogen atoms are all the same as one another. In other embodiments in which two or more hydrogen atoms are replaced with halogen atoms, the halogen atoms are not all the same as one another.
- The term “fluoroalkyl,” as used herein, refers to alkyl group in which at least one hydrogen is replaced with a fluorine atom. Examples of fluoroalkyl groups include, but are not limited to, —CF3, —CH2CF3, —CF2CF3, —CH2CH2CF3 and the like.
- As used herein, the term “heteroalkyl” refers to optionally substituted alkyl radicals in which one or more skeletal chain atoms is a heteroatom, e.g., oxygen, nitrogen, sulfur, silicon, phosphorus or combinations thereof. The heteroatom(s) are placed at any interior position of the heteroalkyl group or at the position at which the heteroalkyl group is attached to the remainder of the molecule. Examples include, but are not limited to, —CH2—O—CH3, —CH2—CH2—O—CH3, —CH2—NH—CH3, —CH2—CH2—NH—CH3, —CH2—N(CH3)—CH3, —CH2—CH2—NH—CH3, —CH2—CH2—N(CH3)—CH3, —CH2—S—CH2—CH3, —CH2—CH2,—S(O)—CH3, —CH2—CH2—S(O)2—CH3, —CH═CH—O—CH3, —Si(CH3)3, —CH2—CH═N—OCH3, and —CH═CH—N(CH3)—CH3. In addition, in some embodiments, up to two heteroatoms are consecutive, such as, by way of example, —CH2—NH—OCH3 and —CH2—O—Si(CH3)3.
- The term “heteroatom” refers to an atom other than carbon or hydrogen. Heteroatoms are typically independently selected from among oxygen, sulfur, nitrogen, silicon and phosphorus, but are not limited to these atoms. In embodiments in which two or more heteroatoms are present, the two or more heteroatoms can all be the same as one another, or some or all of the two or more heteroatoms can each be different from the others.
- The term “bond” or “single bond” refers to a chemical bond between two atoms, or two moieties when the atoms joined by the bond are considered to be part of larger substructure.
- The term “moiety” refers to a specific segment or functional group of a molecule. Chemical moieties are often recognized chemical entities embedded in or appended to a molecule.
- A “thioalkoxy” or “alkylthio” group refers to a —S-alkyl group.
- A “SH” group is also referred to either as a thiol group or a sulfhydryl group.
- The term “optionally substituted” or “substituted” means that the referenced group may be substituted with one or more additional group(s) individually and independently selected from alkyl, cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, alkylthio, arylthio, alkylsulfoxide, arylsulfoxide, alkylsulfone, arylsulfone, cyano, halo, acyl, nitro, haloalkyl, fluoroalkyl, amino, including mono- and di-substituted amino groups, and the protected derivatives thereof. By way of example an optional substituents may be LsRs, wherein each Ls is independently selected from a bond, —O—, —C(═O)—, —S—, —S(═O)—, —S(═O)2—, —NH—, —NHC(O)—, —C(O)NH—, S(═O)2NH—, —NHS(═O)2, —OC(O)NH—, —NHC(O)O—, -(substituted or unsubstituted C1-C6 alkyl), or -(substituted or unsubstituted C2-C6 alkenyl); and each Rs is independently selected from H, (substituted or unsubstituted C1-C4alkyl), (substituted or unsubstituted C3-C6cycloalkyl), heteroaryl, or heteroalkyl. The protecting groups that forms the protective derivatives of the above substituents include those found in sources such as Greene and Wuts, above.
- The term “Michael acceptor moiety” refers to a functional group that can participate in a Michael reaction, wherein a new covalent bond is formed between a portion of the Michael acceptor moiety and the donor moiety. The Michael acceptor moiety is an electrophile and the “donor moiety” is a nucleophile. The “G” groups presented in any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) are non-limiting examples of Michael acceptor moieties.
- The term “nucleophile” or “nucleophilic” refers to an electron rich compound, or moiety thereof. An example of a nucleophile includes, but in no way is limited to, a cysteine residue of a molecule, such as, for
example Cys 481 of Btk. - The term “electrophile”, or “electrophilic” refers to an electron poor or electron deficient molecule, or moiety thereof. Examples of electrophiles include, but in no way are limited to, Micheal acceptor moieties.
- The term “acceptable” or “pharmaceutically acceptable”, with respect to a formulation, composition or ingredient, as used herein, means having no persistent detrimental effect on the general health of the subject being treated or does not abrogate the biological activity or properties of the compound, and is relatively nontoxic.
- As used herein, the term “agonist” refers to a compound, the presence of which results in a biological activity of a protein that is the same as the biological activity resulting from the presence of a naturally occurring ligand for the protein, such as, for example, Btk.
- As used herein, the term “partial agonist” refers to a compound the presence of which results in a biological activity of a protein that is of the same type as that resulting from the presence of a naturally occurring ligand for the protein, but of a lower magnitude.
- As used herein, the term “antagonist” refers to a compound, the presence of which results in a decrease in the magnitude of a biological activity of a protein. In certain embodiments, the presence of an antagonist results in complete inhibition of a biological activity of a protein, such as, for example, Btk. In certain embodiments, an antagonist is an inhibitor.
- As used herein, “amelioration” of the symptoms of a particular disease, disorder or condition by administration of a particular compound or pharmaceutical composition refers to any lessening of severity, delay in onset, slowing of progression, or shortening of duration, whether permanent or temporary, lasting or transient that can be attributed to or associated with administration of the compound or composition.
- “Bioavailability” refers to the percentage of the weight of compounds disclosed herein, such as, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), dosed that is delivered into the general circulation of the animal or human being studied. The total exposure (AUC(0-∞)) of a drug when administered intravenously is usually defined as 100% bioavailable (F %). “Oral bioavailability” refers to the extent to which compounds disclosed herein, such as, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are absorbed into the general circulation when the pharmaceutical composition is taken orally as compared to intravenous injection.
- The term “biophysical probe,” as used herein, refers to probes which detect or monitor structural changes in molecules (including biomolecules) in biological systems or in the presence of other biomolecules (e.g., ex vivo, in vivo or in vitro). In some embodiments, such molecules include, but are not limited to, proteins and the “biophysical probe” is used to detect or monitor interaction of proteins with other macromolecules. In other embodiments, examples of biophysical probes include, but are not limited to, spin-labels, fluorophores, and photoactivatable groups.
- “Blood plasma concentration” refers to the concentration of compounds disclosed herein, such as, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), in the plasma component of blood of a subject. It is understood that the plasma concentration of compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), may vary significantly between subjects, due to variability with respect to metabolism and/or possible interactions with other therapeutic agents. In accordance with one embodiment disclosed herein, the blood plasma concentration of the compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), does vary from subject to subject. Likewise, values such as maximum plasma concentration (Cmax) or time to reach maximum plasma concentration (Tmax), or total area under the plasma concentration time curve (AUC(0-∞)) may vary from subject to subject. Due to this variability, the amount necessary to constitute “a therapeutically effective amount” of a compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), is expected to vary from subject to subject.
- The term “Bruton's tyrosine kinase,” as used herein, refers to Bruton's tyrosine kinase from Homo sapiens, as disclosed in, e.g., U.S. Pat. No. 6,326,469 (GenBank Accession No. NP_000052).
- The term “Bruton's tyrosine kinase homolog,” as used herein, refers to orthologs of Bruton's tyrosine kinase, e.g., the orthologs from mouse (GenBank Accession No. AAB47246), dog (GenBank Accession No. XP_549139.), rat (GenBank Accession No. NP_001007799), chicken (GenBank Accession No. NP_989564), or zebra fish (GenBank Accession No. XP_698117), and fusion proteins of any of the foregoing that exhibit kinase activity towards one or more substrates of Bruton's tyrosine kinase (e.g. a peptide substrate having the amino acid sequence “AVLESEEELYSSARQ”).
- The term “chemiluminescent group,” as used herein, refers to a group which emits light as a result of a chemical reaction without the addition of heat. By way of example only, luminol (5-amino-2,3-dihydro-1,4-phthalazinedione) reacts with oxidants like hydrogen peroxide (H2O2) in the presence of a base and a metal catalyst to produce an excited state product (3-aminophthalate, 3-APA).
- The term “chromophore,” as used herein, refers to a molecule which absorbs light of visible wavelengths, UV wavelengths or IR wavelengths.
- The terms “co-administration” or the like, as used herein, are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are administered by the same or different route of administration or at the same or different time.
- In other embodiments, the term “detectable label,” as used herein, refers to a label which is observable using analytical techniques including, but not limited to, fluorescence, chemiluminescence, electron-spin resonance, ultraviolet/visible absorbance spectroscopy, mass spectrometry, nuclear magnetic resonance, magnetic resonance, and electrochemical methods.
- The term “dye,” as used herein, refers to a soluble, coloring substance which contains a chromophore.
- The terms “effective amount” or “therapeutically effective amount,” as used herein, refer to a sufficient amount of an agent or a compound being administered which will relieve to some extent one or more of the symptoms of the disease or condition being treated. The result can be reduction and/or alleviation of the signs, symptoms, or causes of a disease, or any other desired alteration of a biological system. For example, an “effective amount” for therapeutic uses is the amount of the composition including a compound as disclosed herein required to provide a clinically significant decrease in disease symptoms without undue adverse side effects. An appropriate “effective amount” in any individual case is optionally determined using techniques, such as a dose escalation study. The term “therapeutically effective amount” includes, for example, a prophylactically effective amount. An “effective amount” of a compound disclosed herein is an amount effective to achieve a desired pharmacologic effect or therapeutic improvement without undue adverse side effects. It is understood that “an effect amount” or “a therapeutically effective amount” can vary from subject to subject, due to variation in metabolism of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), age, weight, general condition of the subject, the condition being treated, the severity of the condition being treated, and the judgment of the prescribing physician.
- The term “electron dense group,” as used herein, refers to a group which scatters electrons when irradiated with an electron beam. Such groups include, but are not limited to, ammonium molybdate, bismuth subnitrate cadmium iodide, 99%, carbohydrazide, ferric chloride hexahydrate, hexamethylene tetramine, 98.5%, indium trichloride anhydrous, lanthanum nitrate, lead acetate trihydrate, lead citrate trihydrate, lead nitrate, periodic acid, phosphomolybdic acid, phosphotungstic acid, potassium ferricyanide, potassium ferrocyanide, ruthenium red, silver nitrate, silver proteinate (Ag Assay: 8.0-8.5%) “Strong”, silver tetraphenylporphin (S-TPPS), sodium chloroaurate, sodium tungstate, thallium nitrate, thiosemicarbazide (TSC), uranyl acetate, uranyl nitrate, and vanadyl sulfate.
- In other embodiments, the term “energy transfer agent,” as used herein, refers to a molecule which either donates or accepts energy from another molecule. By way of example only, fluorescence resonance energy transfer (FRET) is a dipole-dipole coupling process by which the excited-state energy of a fluorescence donor molecule is non-radiatively transferred to an unexcited acceptor molecule which then fluorescently emits the donated energy at a longer wavelength.
- The terms “enhance” or “enhancing” means to increase or prolong either in potency or duration a desired effect. By way of example, “enhancing” the effect of therapeutic agents refers to the ability to increase or prolong, either in potency or duration, the effect of therapeutic agents on during treatment of a disease, disorder or condition. An “enhancing-effective amount,” as used herein, refers to an amount adequate to enhance the effect of a therapeutic agent in the treatment of a disease, disorder or condition. When used in a patient, amounts effective for this use will depend on the severity and course of the disease, disorder or condition, previous therapy, the patient's health status and response to the drugs, and the judgment of the treating physician.
- The term “fluorophore,” as used herein, refers to a molecule which upon excitation emits photons and is thereby fluorescent.
- The term “homologous cysteine,” as used herein refers to a cysteine residue found with in a sequence position that is homologous to that of
cysteine 481 of Bruton's tyrosine kinase, as defined herein. For example,cysteine 482 is the homologous cysteine of the rat ortholog of Bruton's tyrosine kinase;cysteine 479 is the homologous cysteine of the chicken ortholog; andcysteine 481 is the homologous cysteine in the zebra fish ortholog. In another example, the homologous cysteine of TXK, a Tec kinase family member related to Bruton's tyrosine, is Cys 350. Other examples of kinases having homologous cysteines are shown inFIG. 7 . See also the sequence alignments of tyrosine kinases (TK) published on the world wide web at kinase.com/human/kinome/phylogeny.html. - The term “identical,” as used herein, refers to two or more sequences or subsequences which are the same. In addition, the term “substantially identical,” as used herein, refers to two or more sequences which have a percentage of sequential units which are the same when compared and aligned for maximum correspondence over a comparison window, or designated region as measured using comparison algorithms or by manual alignment and visual inspection. By way of example only, two or more sequences are “substantially identical” if the sequential units are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region. Such percentages to describe the “percent identity” of two or more sequences. The identity of a sequence can exist over a region that is at least about 75-100 sequential units in length, over a region that is about 50 sequential units in length, or, where not specified, across the entire sequence. This definition also refers to the complement of a test sequence. By way of example only, two or more polypeptide sequences are identical when the amino acid residues are the same, while two or more polypeptide sequences are “substantially identical” if the amino acid residues are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region. The identity can exist over a region that is at least about 75-100 amino acids in length, over a region that is about 50 amino acids in length, or, where not specified, across the entire sequence of a polypeptide sequence. In addition, by way of example only, two or more polynucleotide sequences are identical when the nucleic acid residues are the same, while two or more polynucleotide sequences are “substantially identical” if the nucleic acid residues are about 60% identical, about 65% identical, about 70% identical, about 75% identical, about 80% identical, about 85% identical, about 90% identical, or about 95% identical over a specified region. The identity can exist over a region that is at least about 75-100 nucleic acids in length, over a region that is about 50 nucleic acids in length, or, where not specified, across the entire sequence of a polynucleotide sequence.
- The terms “inhibits”, “inhibiting”, or “inhibitor” of a kinase, as used herein, refer to inhibition of enzymatic phosphotransferase activity.
- The term “irreversible inhibitor,” as used herein, refers to a compound that, upon contact with a target protein (e.g., a kinase) causes the formation of a new covalent bond with or within the protein, whereby one or more of the target protein's biological activities (e.g., phosphotransferase activity) is diminished or abolished notwithstanding the subsequent presence or absence of the irreversible inhibitor.
- The term “irreversible Btk inhibitor,” as used herein, refers to an inhibitor of Btk that can form a covalent bond with an amino acid residue of Btk. In one embodiment, the irreversible inhibitor of Btk can form a covalent bond with a Cys residue of Btk; in particular embodiments, the irreversible inhibitor can form a covalent bond with a
Cys 481 residue (or a homolog thereof) of Btk or a cysteine residue in the homologous corresponding position of another tyrosine kinase, as shown inFIG. 7 . - The term “isolated,” as used herein, refers to separating and removing a component of interest from at least some portion of components not of interest. Isolated substances can be in either a dry or semi-dry state, or in solution, including but not limited to an aqueous solution. The isolated component can be in a homogeneous state or the isolated component can be a part of a pharmaceutical composition that comprises additional pharmaceutically acceptable carriers and/or excipients. By way of example only, nucleic acids or proteins are “isolated” when such nucleic acids or proteins are free of at least some of the cellular components with which it is associated in the natural state, or that the nucleic acid or protein has been concentrated to a level greater than the concentration of its in vivo or in vitro production. Also, by way of example, a gene is isolated when separated from open reading frames which flank the gene and encode a protein other than the gene of interest.
- In some embodiments, the term “label,” as used herein, refers to a substance which is incorporated into a compound and is readily detected, whereby its physical distribution is detected and/or monitored.
- The term “linkage,” as used herein to refer to bonds or a chemical moiety formed from a chemical reaction between the functional group of a linker and another molecule. In some embodiments, such bonds include, but are not limited to, covalent linkages and non-covalent bonds, while such chemical moieties include, but are not limited to, esters, carbonates, imines, phosphate esters, hydrazones, acetals, orthoesters, peptide linkages, and oligonucleotide linkages. Hydrolytically stable linkages means that the linkages are substantially stable in water and do not react with water at useful pH values, including but not limited to, under physiological conditions for an extended period of time, perhaps even indefinitely. Hydrolytically unstable or degradable linkages means that the linkages are degradable in water or in aqueous solutions, including for example, blood. In other embodiments, enzymatically unstable or degradable linkages means that the linkage is degraded by one or more enzymes. By way of example only, PEG and related polymers include degradable linkages in the polymer backbone or in the linker group between the polymer backbone and one or more of the terminal functional groups of the polymer molecule. Such degradable linkages include, but are not limited to, ester linkages formed by the reaction of PEG carboxylic acids or activated PEG carboxylic acids with alcohol groups on a biologically active agent, wherein such ester groups generally hydrolyze under physiological conditions to release the biologically active agent. Other hydrolytically degradable linkages include but are not limited to carbonate linkages; imine linkages resulted from reaction of an amine and an aldehyde; phosphate ester linkages formed by reacting an alcohol with a phosphate group; hydrazone linkages which are reaction product of a hydrazide and an aldehyde; acetal linkages that are the reaction product of an aldehyde and an alcohol; orthoester linkages that are the reaction product of a formate and an alcohol; peptide linkages formed by an amine group, including but not limited to, at an end of a polymer such as PEG, and a carboxyl group of a peptide; and oligonucleotide linkages formed by a phosphoramidite group, including but not limited to, at the end of a polymer, and a 5′ hydroxyl group of an oligonucleotide.
- The phrase “measuring the activity of the reporter moiety” (or a similarly worded phrase) refers to methods for quantifying (in absolute, approximate or relative terms) the reporter moiety in a system under study. In some embodiments, such methods include any methods that quantify a reporter moiety that is a dye; a photocrosslinker; a cytotoxic compound; a drug; an affinity label; a photoaffinity label; a reactive compound; an antibody or antibody fragment; a biomaterial; a nanoparticle; a spin label; a fluorophore, a metal-containing moiety; a radioactive moiety; a novel functional group; a group that covalently or noncovalently interacts with other molecules; a photocaged moiety; an actinic radiation excitable moiety; a ligand; a photoisomerizable moiety; biotin; a biotin analogue; a moiety incorporating a heavy atom; a chemically cleavable group; a photocleavable group; a redox-active agent; an isotopically labeled moiety; a biophysical probe; a phosphorescent group; a chemiluminescent group; an electron dense group; a magnetic group; an intercalating group; a chromophore; an energy transfer agent; a biologically active agent; a detectable label; and any combination of the above.
- A “metabolite” of a compound disclosed herein is a derivative of that compound that is formed when the compound is metabolized. The term “active metabolite” refers to a biologically active derivative of a compound that is formed when the compound is metabolized. The term “metabolized,” as used herein, refers to the sum of the processes (including, but not limited to, hydrolysis reactions and reactions catalyzed by enzymes, such as, oxidation reactions) by which a particular substance is changed by an organism. Thus, enzymes produce specific structural alterations to a compound. For example, cytochrome P450 catalyzes a variety of oxidative and reductive reactions while uridine diphosphate glucuronyl transferases catalyze the transfer of an activated glucuronic-acid molecule to aromatic alcohols, aliphatic alcohols, carboxylic acids, amines and free sulfhydryl groups. Further information on metabolism is obtained from The Pharmacological Basis of Therapeutics, 9th Edition, McGraw-Hill (1996). Metabolites of the compounds disclosed herein are optionally identified either by administration of compounds to a host and analysis of tissue samples from the host, or by incubation of compounds with hepatic cells in vitro and analysis of the resulting compounds. In some embodiments, metabolites of a compound are formed by oxidative processes and correspond to the corresponding hydroxy-containing compound. In some embodiments, a compound is metabolized to pharmacologically active metabolites.
- The term “modulate,” as used herein, means to interact with a target either directly or indirectly so as to alter the activity of the target, including, by way of example only, to enhance the activity of the target, to inhibit the activity of the target, to limit the activity of the target, or to extend the activity of the target.
- As used herein, the term “modulator” refers to a compound that alters an activity of a molecule. For example, a modulator can cause an increase or decrease in the magnitude of a certain activity of a molecule compared to the magnitude of the activity in the absence of the modulator. In certain embodiments, a modulator is an inhibitor, which decreases the magnitude of one or more activities of a molecule. In certain embodiments, an inhibitor completely prevents one or more activities of a molecule. In certain embodiments, a modulator is an activator, which increases the magnitude of at least one activity of a molecule. In certain embodiments the presence of a modulator results in an activity that does not occur in the absence of the modulator.
- The term “moiety incorporating a heavy atom,” as used herein, refers to a group which incorporates an ion of atom which is usually heavier than carbon. In some embodiments, such ions or atoms include, but are not limited to, silicon, tungsten, gold, lead, and uranium.
- The term “nanoparticle,” as used herein, refers to a particle which has a particle size between about 500 nm to about 1 nm.
- As used herein, the term “pERK” refers to phosphorylated ERK1 and ERK2 at Thr202/Tyr 204 as detected by commercially available phospho-specific antibodies (e.g. Cell Signaling Technologies #4377).
- The term “photoaffinity label,” as used herein, refers to a label with a group, which, upon exposure to light, forms a linkage with a molecule for which the label has an affinity. By way of example only, in some embodiments, such a linkage is covalent or non-covalent.
- The term “photocaged moiety,” as used herein, refers to a group which, upon illumination at certain wavelengths, covalently or non-covalently binds other ions or molecules.
- The term “photoisomerizable moiety,” as used herein, refers to a group wherein upon illumination with light changes from one isomeric form to another.
- The term “plasma half life,” as used herein refers to half-life in rat, dog or human as determined by measure drug concentration over time in plasma following a single dose and fitting data to standard pharmacokinetic models using software such as WinNonLin to determine the time at which drug has been 50% eliminated from plasma.
- The term “prophylactically effective amount,” as used herein, refers that amount of a composition applied to a patient which will relieve to some extent one or more of the symptoms of a disease, condition or disorder being treated. In such prophylactic applications, such amounts may depend on the patient's state of health, weight, and the like.
- The term “radioactive moiety,” as used herein, refers to a group whose nuclei spontaneously give off nuclear radiation, such as alpha, beta, or gamma particles; wherein, alpha particles are helium nuclei, beta particles are electrons, and gamma particles are high energy photons.
- As used herein, the term “selective binding compound” refers to a compound that selectively binds to any portion of one or more target proteins.
- As used herein, the term “selectively binds” refers to the ability of a selective binding compound to bind to a target protein, such as, for example, Btk, with greater affinity than it binds to a non-target protein. In certain embodiments, specific binding refers to binding to a target with an affinity that is at least 10, 50, 100, 250, 500, 1000 or more times greater than the affinity for a non-target.
- As used herein, the term “selective modulator” refers to a compound that selectively modulates a target activity relative to a non-target activity. In certain embodiments, specific modulator refers to modulating a target activity at least 10, 50, 100, 250, 500, 1000 times more than a non-target activity.
- The term “spin label,” as used herein, refers to molecules which contain an atom or a group of atoms exhibiting an unpaired electron spin (i.e. a stable paramagnetic group) that in some embodiments are detected by electron spin resonance spectroscopy and in other embodiments are attached to another molecule. Such spin-label molecules include, but are not limited to, nitryl radicals and nitroxides, and in some embodiments are single spin-labels or double spin-labels.
- The term “substantially purified,” as used herein, refers to a component of interest that may be substantially or essentially free of other components which normally accompany or interact with the component of interest prior to purification. By way of example only, a component of interest may be “substantially purified” when the preparation of the component of interest contains less than about 30%, less than about 25%, less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 4%, less than about 3%, less than about 2%, or less than about 1% (by dry weight) of contaminating components. Thus, a “substantially purified” component of interest may have a purity level of about 70%, about 75%, about 80%, about 85%, about 90%, about 95%, about 96%, about 97%, about 98%, about 99% or greater.
- The term “subject” as used herein, refers to an animal which is the object of treatment, observation or experiment. By way of example only, a subject may be, but is not limited to, a mammal including, but not limited to, a human.
- As used herein, the term “target activity” refers to a biological activity capable of being modulated by a selective modulator. Certain exemplary target activities include, but are not limited to, binding affinity, signal transduction, enzymatic activity, tumor growth, inflammation or inflammation-related processes, and amelioration of one or more symptoms associated with a disease or condition.
- As used herein, the term “target protein” refers to a molecule or a portion of a protein capable of being bound by a selective binding compound. In certain embodiments, a target protein is Btk.
- The terms “treat,” “treating” or “treatment”, as used herein, include alleviating, abating or ameliorating a disease or condition symptoms, preventing additional symptoms, ameliorating or preventing the underlying metabolic causes of symptoms, inhibiting the disease or condition, e.g., arresting the development of the disease or condition, relieving the disease or condition, causing regression of the disease or condition, relieving a condition caused by the disease or condition, or stopping the symptoms of the disease or condition. The terms “treat,” “treating” or “treatment”, include, but are not limited to, prophylactic and/or therapeutic treatments.
- As used herein, the IC50 refers to an amount, concentration or dosage of a particular test compound that achieves a 50% inhibition of a maximal response, such as inhibition of Btk, in an assay that measures such response.
- As used herein, EC50 refers to a dosage, concentration or amount of a particular test compound that elicits a dose-dependent response at 50% of maximal expression of a particular response that is induced, provoked or potentiated by the particular test compound.
-
FIG. 1A presents an illustrative table of GI50 concentrations ofCompound 1 that results in 50% decrease in cell proliferation. A variety of lymphoma cell lines incubated with a range of concentrations ofCompound 1.FIG. 1B presents an illustrative line graph showing inhibition of tumor growth in DLCL2 xenograft models.FIG. 1C presents an illustrative line graph showing inhibition of tumor growth in DOHH2 xenograft models. For in vivo lymphoma xenograft studies, 5E6 DOHH2 or DLCL2 cells in 50% matrigel were implanted subcutaneously in SCID mice and dosed orally withCompound 1 beginning when tumor size reached 100 mm2. -
FIG. 2 presents an illustrative line graph showing inhibition of collagen-induced arthritis in male DBA/1O1aHsd mice.Compound 1 or vehicle was dosed orally once per day starting atday 1. Dexamethasone was included as a positive control. Paw inflammation was scored from 0-5 and averaged across all paws from all animals for each group in the study.Compound 1 at 12.5 mg/kg and 50 mg/kg regressed inflammation through the end of the study (day 11) while 3.125 mg/kg significantly reduced the increase in paw inflammation. -
FIG. 3 presents an illustrative line graph showing inhibition of disease progression in a mouse MRL/1 pr model of lupus. MRL/1 pr mice (Jax strain 000485) were dosed orally once per day from 8 weeks of age until 20 weeks of age and urine protein levels were measured weekly.Compound 1 at 3.125 mg/kg, 12.5 mg/kg, and 50 mg/kg significantly reduced proteinuria, indicating amelioration of the progressive autoimmune renal failure seen in this mouse strain. -
FIG. 4 presents an illustrative bar graph showing inhibition of mast cell degranulation in a mouse passive cutaneous anaphylaxis model. 23 hours after mice were sensitized with an intradermal injection of monoclonal anti-DNP-IgE in the back, they received a single oral dose ofCompound 1 or vehicle. After one hour, animals were challenged with an intravenous injection of DNP-BSA and Evans Blue dye and the area of extravasation was measured. Increasing doses ofCompound 1 significantly decreased the amount of Evans Blue release, indicating decreased mast cell activation and vascular permeabilization. -
FIG. 5 presents an illustrative line graph showing in vivo plasma concentrations post-dosing of male jugular vein cannulated rats withCompounds Compound 1 and Compound 12 have a short half-life in vivo. In contrast,Compound 7 and Compound 8 have a significantly longer in vivo half-life. Compounds like 1 and 12 are predicted to have enhanced kinase selectivity in vivo because inhibition will be sustained only for those kinases that are irreversibly inhibited. -
FIG. 6 presents an illustrative bar graph showing brief exposure toCompound 1 in vitro is sufficient to inhibit B cell activation in normal human B cells. B cells were purified from blood from healthy donors by negative selecting using the RosetteSep Human B cell enrichment cocktail. Cells were plated in growth media and indicated concentrations ofCompound 1 were added. After incubation for 1 hour at 37° C., cells were washed three times using an 8-fold dilution in growth media for each wash. Cells were then stimulated with IgM F(ab′)2 for 18 hours at 37° C., stained with anti-CD69-PE antibody and analyzed by flow cytometry. This protocol mimics the predicted exposure of cells toCompound 1 in vivo and demonstrates that inhibition of B cells is sustained despite washing out ofCompound 1. -
FIG. 7 presents illustrative ACKs, including Btk and Btk cysteine homologs. - In the following description of irreversible kinase inhibitor compounds suitable for use in the methods described herein, definitions of referred-to standard chemistry terms may be found in reference works (if not otherwise defined herein), including Carey and Sundberg “Advanced Organic Chemistry 4th Ed.” Vols. A (2000) and B (2001), Plenum Press, New York. In addition, nucleic acid and amino acid sequences for Btk (e.g., human Btk) are disclosed in, e.g., U.S. Pat. No. 6,326,469. Unless specific definitions are provided, the nomenclature employed in connection with, and the laboratory procedures and techniques of, analytical chemistry, synthetic organic chemistry, and medicinal and pharmaceutical chemistry described herein are those known in the art. Standard techniques can be used for chemical syntheses, chemical analyses, pharmaceutical preparation, formulation, and delivery, and treatment of patients
- The inhibitor compounds described herein are selective for kinases having an accessible cysteine residue (such kinases are also known as Accessible Cysteine Kinases, or ACKs) that can form a covalent bond with a Michael acceptor moiety on the inhibitor compound. In some embodiments, the cysteine residue is accessible or becomes accessible when the binding site moiety of the irreversible inhibitor binds to the kinase. That is, the binding site moiety of the irreversible inhibitor binds to an active site of the ACK and the Michael acceptor moiety of irreversible inhibitor gains access (in one embodiment the step of binding leads to a conformational change in the ACK, thus exposing the cysteine) or is otherwise exposed to the cysteine residue of the ACK; as a result a covalent bond is formed between the “S” of the cysteine residue and the Michael acceptor of the irreversible inhibitor. Consequently, the binding site moiety of the irreversible inhibitor remains bound or otherwise blocks the active site of the ACK.
- In one embodiment, the ACK is Btk, a homolog of Btk or a tyrosine kinase having a cysteine residue in an amino acid sequence position that is homologous to the amino acid sequence position of
cysteine 481 in Btk. See, e.g., kinases inFIG. 7 . Inhibitor compounds described herein include a Michael acceptor moiety, a binding site moiety and a linker that links the binding site moiety and the Michael acceptor moiety (and in some embodiments, the structure of the linker provides a conformation, or otherwise directs the Michael acceptor moiety, so as to improve the selectivity of the irreversible inhibitor for a particular ACK). - Generally, an irreversible inhibitor compound used in the methods described herein is identified or characterized in an in vitro assay, e.g., an a cellular biochemical assay or a cellular functional assay. Such assays are useful to determine an in vitro IC50 for an irreversible inhibitor compound.
- For example, an acellular kinase assay can be used to determine kinase activity after incubation of the kinase in the absence or presence of a range of concentrations of a candidate irreversible inhibitor compound. If the candidate compound is in fact an irreversible inhibitor, kinase activity will not be recovered by repeat washing with inhibitor-free medium. See, e.g., J. B. Smaill, et al. (1999), J. Med. Chem. 42(10): 1803-1815. Further, covalent complex formation between a Kinase and a candidate irreversible inhibitor is a useful indicator of irreversible inhibition of the Kinase that can be readily determined by a number of methods (e.g., mass spectrometry). For example, some irreversible Kinase-inhibitor compounds can form a covalent bond with the aforenoted cysteine residue (e.g., via a Michael reaction).
- High throughput assays for many acellular biochemical assays (e.g., kinase assays) and cellular functional assays (e.g., calcium flux) are documented methodologies. In addition, high throughput screening systems are commercially available (see, e.g., Zymark Corp., Hopkinton, Mass.; Air Technical Industries, Mentor, OH; Beckman Instruments, Inc. Fullerton, Calif.; Precision Systems, Inc., Natick, Mass., etc.). These systems typically automate entire procedures including all sample and reagent pipetting, liquid dispensing, timed incubations, and final readings of the microplate in detector(s) appropriate for the assay. Automated systems thereby allow the identification and characterization of a large number of irreversible compounds.
- Irreversible inhibitor compounds can used for the manufacture of a medicament for treating any of the foregoing conditions (e.g., autoimmune diseases, inflammatory diseases, allergy disorders, B-cell proliferative disorders, or thromboembolic disorders).
- In some embodiments, the irreversible inhibitor compound used for the methods described herein inhibits a Kinase activity with an in vitro IC50 of less than 10 μM. (e.g., less than 1 μM, less than 0.5 μM, less than 0.4 μM, less than 0.3 μM, less than 0.1, less than 0.08 μM, less than 0.06 μM, less than 0.05 μM, less than 0.04 μM, less than 0.03 μM, less than less than 0.02 μM, less than 0.01, less than 0.008 μM, less than 0.006 μM, less than 0.005 μM, less than 0.004 μM, less than 0.003 μM, less than less than 0.002 μM, less than 0.001, less than 0.00099 μM, less than 0.00098 μM, less than 0.00097 μM, less than 0.00096 μM, less than 0.00095 μM, less than 0.00094 μM, less than 0.00093 μM, less than 0.00092, or less than 0.00090 μM).
- In one embodiment, the irreversible inhibitor compound selectively and irreversibly inhibits an activated form of its target tyrosine kinase (e.g., a phosphorylated form of the tyrosine kinase). For example, activated Btk is transphosphorylated at tyrosine 551. Thus, in these embodiments the irreversible Btk inhibitor inhibits the target kinase in cells only once the target kinase is activated by the signaling events.
- Described herein are compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Also described herein are pharmaceutically acceptable salts, pharmaceutically acceptable solvates, pharmaceutically active metabolites, and pharmaceutically acceptable prodrugs of such compounds. Pharmaceutical compositions that include at least one such compound or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, pharmaceutically active metabolite or pharmaceutically acceptable prodrug of such compound, are provided. In some embodiments, when compounds disclosed herein contain an oxidizable nitrogen atom, the nitrogen atom is optionally converted to an N-oxide. In certain embodiments, isomers and chemically protected forms of compounds having a structure represented by any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are also provided.
- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of Formula (I):
-

- wherein
-
- La is CH2, O, NH or S;
- Ar is a substituted or unsubstituted aryl, or a substituted or unsubstituted heteroaryl; and either
- (a) Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene and alkyleneheterocycloalkylene;
- Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
- (i) R7 and R8 are H;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- (ii) R6 and R8 are H;
- R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- (iii) R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- (b) Y is an optionally substituted group selected from cycloalkylene or heterocycloalkylene;
- Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
- (i) R7 and R8 are H;
- R6 is substituted or unsubstituted C1-C4heteroalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- (ii) R6 and R8 are H;
- R7 is substituted or unsubstituted C1-C4heteroalkyl, C1-C8 hydroxyalkylaminoalkyl, C1-C8 alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- (iii) R7 and R8 taken together form a bond;
- R6 is substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (I). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (I), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (I). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (I). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- For any and all of the embodiments, substituents can be selected from among from a subset of the listed alternatives. For example, in some embodiments, La is CH2, O, or NH. In other embodiments, La is O or NH. In yet other embodiments, La is O.
- In some embodiments, Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- In some embodiments, x is 2. In yet other embodiments, Z is C(═O), OC(═O), NHC(═O), S(═O)x, OS(═O)x, or NHS(═O)x. In some other embodiments, Z is C(═O), NHC(═O), or NCH3C(═O).
- In some embodiments Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, and alkyleneheterocycloalkylene.
- In some embodiments, Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl.
- In some embodiments, R7 and R8 are H; and R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl). In other embodiments, R6 and R8 are H; and R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl). In yet further embodiments, R7 and R8 taken together form a bond; and R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl).
- In some embodiments, Y is an optionally substituted group selected from cycloalkylene or heterocycloalkylene.
- In some embodiments, Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl.
- In some embodiments, R7 and R8 are H; and R6 is substituted or unsubstituted C1-C4heteroalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl). In other embodiments, R6 and R8 are H; and R7 is substituted or unsubstituted C1-C4heteroalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl). In further embodiments, R7 and R8 taken together form a bond; and R6 is substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl).
- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of Formula (VII):
-

-
- wherein
-

- is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
-
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
- Z is C(═O), OC(═O), NHC(═O), NCH3C(═O), C(═S), S(═O)x, OS(═O)x, NHS(═O)x, where x is 1 or 2;
- R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
- R7 and R8 taken together form a bond; and
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl); and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (VII). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (VII), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (VII). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (VII). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- In some embodiments, x is 2. In yet other embodiments, Z is C(═O), OC(═O), NHC(═O), S(═O)x, OS(═O)x, or NHS(═O)x. In some other embodiments, Z is C(═O), NHC(═O), or S(═O)2.
- In some embodiments, R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, and substituted C1-C4heteroalkyl; or R7 and R8 taken together form a bond. In yet other embodiments, each of R7 and R8 is H; or R7 and R8 taken together form a bond.
- In some embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). In some other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C2alkyl-N(C1-C3alkyl)2, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). In yet other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—N(C1-C3alkyl)2, C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl). In yet other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—(C1-C6alkylamino), C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl). In some embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—N(C1-C3alkyl)2, C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl containing 1 or 2 N atoms), or C1-C4alkyl(5- or 6-membered heterocycloalkyl containing 1 or 2 N atoms).
- In some embodiments, Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene. In other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 4-, 5-, 6-, or 7-membered cycloalkylene, and 4-, 5-, 6-, or 7-membered heterocycloalkylene. In yet other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 5- or 6-membered cycloalkylene, and 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some other embodiments, Y is a 5- or 6-membered cycloalkylene, or a 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some embodiments, Y is a 4-, 5-, 6-, or 7-membered cycloalkylene ring; or Y is a 4-, 5-, 6-, or 7-membered heterocycloalkylene ring.
- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of Formula (A1):
-

- wherein
-
- A is independently selected from N or CR5;
- R1 is H, L2-(substituted or unsubstituted alkyl), L2-(substituted or unsubstituted cycloalkyl), L2-(substituted or unsubstituted alkenyl), L2-(substituted or unsubstituted cycloalkenyl), L2-(substituted or unsubstituted heterocycle), L2-(substituted or unsubstituted heteroaryl), or L2-(substituted or unsubstituted aryl), where L2 is a bond, O, S, —S(═O), —S(═O)2, C(═O), -(substituted or unsubstituted C1-C6 alkyl), or -(substituted or unsubstituted C2-C6 alkenyl);
- R2 and R3 are independently selected from H, lower alkyl and substituted lower alkyl;
- R4 is L3-X-L4-G, wherein,
- L3 is optional, and when present is a bond, or an optionally substituted group selected from alkyl, heteroalkyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, or alkylheterocycloalkyl;
- X is optional, and when present is a bond, O, —C(═O), S, —S(═O), —S(═O)2, —NH, —NR9, —NHC(O), —C(O)NH, —NR9C(O), —C(O)NR9, —S(═O)2NH, —NHS(═O)2, —S(═O)2NR9—, —NR9S(═O)2, —OC(O)NH—, —NHC(O)O—, —OC(O)NR9—, —NR9C(O)O—, —CH═NO—, —ON═CH—, —NR10C(O)NR10—, heteroaryl, aryl, —NR10C(═NR11)NR10—, —NR10C(═NR11)—, —C(═NR11)NR10—, —OC(═NR11)—, or —C(═NR11)O—;
- L4 is optional, and when present is a bond, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocycle;
- or L3, X and L4 taken together form a nitrogen containing heterocyclic ring, or an optionally substituted group selected from alkyl, heteroalkyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, or alkylheterocycloalkyl;
- G is
-

-
-
-
- where Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
- R7 and R8 are H;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- R6 and R8 are H;
- R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
-
- R5 is H, halogen, -L6-(substituted or unsubstituted C1-C3 alkyl), -L6-(substituted or unsubstituted C2-C4 alkenyl), -L6-(substituted or unsubstituted heteroaryl), or -L6-(substituted or unsubstituted aryl), wherein L6 is a bond, O, S, —S(═O), S(═O)2, NH, C(O), —NHC(O)O, —OC(O)NH, —NHC(O), or —C(O)NH;
- each R9 is independently selected from among H, substituted or unsubstituted lower alkyl, and substituted or unsubstituted lower cycloalkyl;
- each R10 is independently H, substituted or unsubstituted lower alkyl, or substituted or unsubstituted lower cycloalkyl; or
- two R10 groups can together form a 5-, 6-, 7-, or 8-membered heterocyclic ring; or
- R9 and R10 can together form a 5-, 6-, 7-, or 8-membered heterocyclic ring; or
- each R11 is independently selected from H, —S(═O)2R8, —S(═O)2NH2, —C(O)R8, —CN, —NO2, heteroaryl, or heteroalkyl; and pharmaceutically active metabolites, pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
-
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (A1). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (A1), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (A1). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (A1). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- In a further or alternative embodiment, the compound of Formula (A1) has the following structure of Formula (B1):
-

- wherein:
-
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, and alkyleneheterocycloalkylene;
- each Ra is independently H, halogen, —CF3, —CN, —NO2, OH, NH2, -La-(substituted or unsubstituted alkyl), -La-(substituted or unsubstituted alkenyl), -La-(substituted or unsubstituted heteroaryl), or -La-(substituted or unsubstituted aryl), wherein La is a bond, O, S, —S(═O), —S(═O)2, NH, C(O), CH2, —NHC(O)O, —NHC(O), or —C(O)NH;
- G is
-

- where Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
-
- R7 and R8 are H;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- R6 and R8 are H;
- R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- R12 is H or lower alkyl; or
- Y and R12 taken together form a 4-, 5-, or 6-membered heterocyclic ring; and
- pharmaceutically acceptable active metabolites, pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- R7 and R8 are H;
- In further or alternative embodiments, G is selected from among
-

- where R is H, alkyl, alkylhydroxy, heterocycloalkyl, heteroaryl, alkylalkoxy, alkylalkoxyalkyl.
- In further or alternative embodiments,
-

- is selected from among
-

- In further or alternative embodiment, the compound of Formula (B1) has the following structure of Formula (C1):
-

-
- Y is an optionally substituted group selected from among alkyl, heteroalkyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, and alkylheterocycloalkyl;
- R12 is H or lower alkyl; or
- Y and R12 taken together form a 4-, 5-, or 6-membered heterocyclic ring;
- G is
-

- where Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
-
- R7 and R8 are H;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- R6 and R8 are H;
- R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); and
- pharmaceutically acceptable active metabolites, pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- R7 and R8 are H;
- In a further or alternative embodiment, the “G” group of any of Formula (A1), Formula (B1), or Formula (C1) is any group that is used to tailor the physical and biological properties of the molecule. Such tailoring/modifications are achieved using groups which modulate Michael acceptor chemical reactivity, acidity, basicity, lipophilicity, solubility and other physical properties of the molecule. The physical and biological properties modulated by such modifications to G include, by way of example only, enhancing chemical reactivity of Michael acceptor group, solubility, in vivo absorption, and in vivo metabolism. In addition, in vivo metabolism includes, by way of example only, controlling in vivo PK properties, off-target activities, potential toxicities associated with cypP450 interactions, drug-drug interactions, and the like. Further, modifications to G allow for the tailoring of the in vivo efficacy of the compound through the modulation of, by way of example, specific and non-specific protein binding to plasma proteins and lipids and tissue distribution in vivo.
- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of Formula (D1):
-

- wherein
-
- La is CH2, O, NH or S;
- Ar is an optionally substituted aromatic carbocycle or an aromatic heterocycle;
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, and alkyleneheterocycloalkylene, or combination thereof;
- Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O)x, where x is 1 or 2, and Ra is H, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
- R7 and R8 are H;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- R6 and R8 are H;
- R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- or combinations thereof; and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (D1). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (D1), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (D1). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (D1). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- In a further or alternative embodiment, La is O.
- In a further or alternative embodiment, Ar is phenyl.
- In a further or alternative embodiment, Z is C(═O), NHC(═O), or NCH3C(═O).
- In a further or alternative embodiment, each of R1, R2, and R3 is H.
- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of Formula (D1):
-

- wherein:
-
- La is CH2, O, NH or S;
- Ar is a substituted or unsubstituted aryl, or a substituted or unsubstituted heteroaryl;
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene and alkyleneheterocycloalkylene;
- Z is C(═O), NHC(═O), NRaC(═O), NRaS(═O), where x is 1 or 2, and Ra is substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl; and either
- R7 and R8 are H;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl);
- R6 and R8 are H;
- R7 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted C1-C8alkylC3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C8alkylethers, C1-C8alkylamides, or C1-C4alkyl(C2-C8heterocycloalkyl); and
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (D1). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (D1), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (D1). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (D1). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- For any and all of the embodiments, substituents can be selected from among from a subset of the listed alternatives. For example, in some embodiments, La is CH2, O, or NH. In other embodiments, La is O or NH. In yet other embodiments, La is O.
- In some embodiments, Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- In some embodiments, x is 2. In yet other embodiments, Z is C(═O), OC(═O), NHC(═O), S(═O)x, OS(═O)x, or NHS(═O)x. In some other embodiments, Z is C(═O), NHC(═O), or S(═O)2.
- In some embodiments, R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, and substituted C1-C4heteroalkyl; or R7 and R8 taken together form a bond. In yet other embodiments, each of R7 and R8 is H; or R7 and R8 taken together form a bond.
- In some embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C2alkyl-N(C1-C3alkyl)2, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). In some other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C2alkyl-N(C1-C3alkyl)2, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). In yet other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—N(C1-C3alkyl)2, C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl). In some embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—N(C1-C3alkyl)2, C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl containing 1 or 2 N atoms), or C1-C4alkyl(5- or 6-membered heterocycloalkyl containing 1 or 2 N atoms).
- In some embodiments, Y is an optionally substituted group selected from among alkylene, heteroalkylene, cycloalkylene, and heterocycloalkylene. In other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 4-, 5-, 6- or 7-membered cycloalkylene, and 4-, 5-, 6- or 7-membered heterocycloalkylene. In yet other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 5-, or 6-membered cycloalkylene, and 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some other embodiments, Y is a 5-, or 6-membered cycloalkylene, or a 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of Formula (A2-A6):
-

- wherein
-
- A is independently selected from N or CR5;
- R1 is H, L2-(substituted or unsubstituted alkyl), L2-(substituted or unsubstituted cycloalkyl), L2-(substituted or unsubstituted alkenyl), L2-(substituted or unsubstituted cycloalkenyl), L2-(substituted or unsubstituted heterocycle), L2-(substituted or unsubstituted heteroaryl), or L2-(substituted or unsubstituted aryl), where L2 is a bond, O, S, —S(═O), —S(═O)2, C(═O), -(substituted or unsubstituted C1-C6 alkyl), or -(substituted or unsubstituted C2-C6 alkenyl);
- R2 and R3 are independently selected from H, lower alkyl and substituted lower alkyl;
- R4 is L3-X-L4-G, wherein,
- L3 is optional, and when present is a bond, optionally substituted or unsubstituted alkyl, optionally substituted or unsubstituted cycloalkyl, optionally substituted or unsubstituted alkenyl, optionally substituted or unsubstituted alkynyl;
- X is optional, and when present is a bond, O, —C(═O), S, —S(═O), —S(═O)2, —NH, —NR9, —NHC(O), —C(O)NH, —NR9C(O), —C(O)NR9, —S(═O)2NH, —NHS(═O)2, —S(═O)2NR9—, —NR9S(═O)2, —OC(O)NH—, —NHC(O)O—, —OC(O)NR9—, —NR9C(O)O—, —CH═NO—, —ON═CH—, —NR10C(O)NR10—, heteroaryl, aryl, —NR10C(═NR11)NR10—, —NR10C(═NR11)—, —C(═NR11)NR10—, —OC(═NR11)—, or —C(═NR11)O—;
- L4 is optional, and when present is a bond, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocycle;
- or L3, X and L4 taken together form a nitrogen containing heterocyclic ring;
- G is
-

-
-
- wherein,
- R6, R7 and R8 are independently selected from among H, lower alkyl or substituted lower alkyl, lower heteroalkyl or substituted lower heteroalkyl, substituted or unsubstituted lower cycloalkyl, and substituted or unsubstituted lower heterocycloalkyl;
- wherein,
- R5 is H, halogen, -L6-(substituted or unsubstituted C1-C3 alkyl), -L6-(substituted or unsubstituted C2-C4 alkenyl), -L6-(substituted or unsubstituted heteroaryl), or -L6-(substituted or unsubstituted aryl), wherein L6 is a bond, O, S, —S(═O), S(═O)2, NH, C(O), —NHC(O)O, —OC(O)NH, —NHC(O), or —C(O)NH;
- each R9 is independently selected from among H, substituted or unsubstituted lower alkyl, and substituted or unsubstituted lower cycloalkyl;
- each R10 is independently H, substituted or unsubstituted lower alkyl, or substituted or unsubstituted lower cycloalkyl; or
- two R10 groups can together form a 5-, 6-, 7-, or 8-membered heterocyclic ring; or
- R9 and R10 can together form a 5-, 6-, 7-, or 8-membered heterocyclic ring; or
- each R11 is independently selected from H, —S(═O)2R8, —S(═O)2NH2, —C(O)R8, —CN, —NO2, heteroaryl, or heteroalkyl; and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
-
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (A2-A6). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (A2-A6), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (A2-A6). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (A2-A6). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- In a further or alternative embodiment, the compound of Formula (A2-A6) has the following structure of Formula (B2-B6):
-


- wherein:
-
- Y is alkylene or substituted alkylene, or a 4-, 5-, or 6-membered cycloalkylene ring;
- each Ra is independently H, halogen, —CF3, —CN, —NO2, OH, NH2, -La-(substituted or unsubstituted alkyl), -La-(substituted or unsubstituted alkenyl), -La-(substituted or unsubstituted heteroaryl), or -La-(substituted or unsubstituted aryl), wherein La is a bond, O, S, —S(═O), —S(═O)2, NH, C(O), CH2, —NHC(O)O, —NHC(O), or —C(O)NH;
- G is
-

- wherein,
-
- R6, R7 and R8 are independently selected from among H, lower alkyl or substituted lower alkyl, lower heteroalkyl or substituted lower heteroalkyl, substituted or unsubstituted lower cycloalkyl, and substituted or unsubstituted lower heterocycloalkyl;
- R12 is H or lower alkyl; or
- Y and R12 taken together form a 4-, 5-, or 6-membered heterocyclic ring; and
- pharmaceutically acceptable active metabolites, pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In further or alternative embodiments, G is selected from among
-

- In further or alternative embodiments,
-

- is selected from among
-

- In further or alternative embodiment, the compound of Formula (B2-B6) has the following structure of Formula (C2-C6):
-


-
- Y is alkylene or substituted alkylene, or a 4-, 5-, or 6-membered cycloalkylene ring;
- R12 is H or lower alkyl; or
- Y and R12 taken together form a 4-, 5-, or 6-membered heterocyclic ring;
- G is
-

- wherein,
-
- R6, R7 and R8 are independently selected from among H, lower alkyl or substituted lower alkyl, lower heteroalkyl or substituted lower heteroalkyl, substituted or unsubstituted lower cycloalkyl, and substituted or unsubstituted lower heterocycloalkyl; and
- pharmaceutically acceptable active metabolites, pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In a further or alternative embodiment, the “G” group of any of Formula (A2-A6), Formula (B2-B6), or Formula (C2-C6) is any group that is used to tailor the physical and biological properties of the molecule. Such tailoring/modifications are achieved using groups which modulate Michael acceptor chemical reactivity, acidity, basicity, lipophilicity, solubility and other physical properties of the molecule. The physical and biological properties modulated by such modifications to G include, by way of example only, enhancing chemical reactivity of Michael acceptor group, solubility, in vivo absorption, and in vivo metabolism. In addition, in vivo metabolism includes, by way of example only, controlling in vivo PK properties, off-target activities, potential toxicities associated with cypP450 interactions, drug-drug interactions, and the like. Further, modifications to G allow for the tailoring of the in vivo efficacy of the compound through the modulation of, by way of example, specific and non-specific protein binding to plasma proteins and lipids and tissue distribution in vivo.
- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of Formula (D2-D6):
-


- wherein
-
- La is CH2, O, NH or S;
- Ar is an optionally substituted aromatic carbocycle or an aromatic heterocycle;
- Y is an optionally substituted alkylene, heteroalkylene, carbocycloalkylene, heterocycloalkylene,
- or combination thereof;
- Z is C(O), OC(O), NHC(O), C(S), S(O)x, OS(O)x, NHS(O)x, where x is 1 or 2; and
- R6, R7, and R8 are independently selected from H, alkyl, heteroalkyl, carbocycle, heterocycle, or combinations thereof; and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (D2-D6). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (D2-D6), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (D2-D6). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (D2-D6). Examples of N-acyl groups include N-acetyl and N-ethoxycarbonyl groups.
- In a further or alternative embodiment, La is O.
- In a further or alternative embodiment, Ar is phenyl.
- In a further or alternative embodiment, Z is C(O).
- In a further or alternative embodiment, each of R1, R2, and R3 is H.
- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of Formula (D2-D6):
-

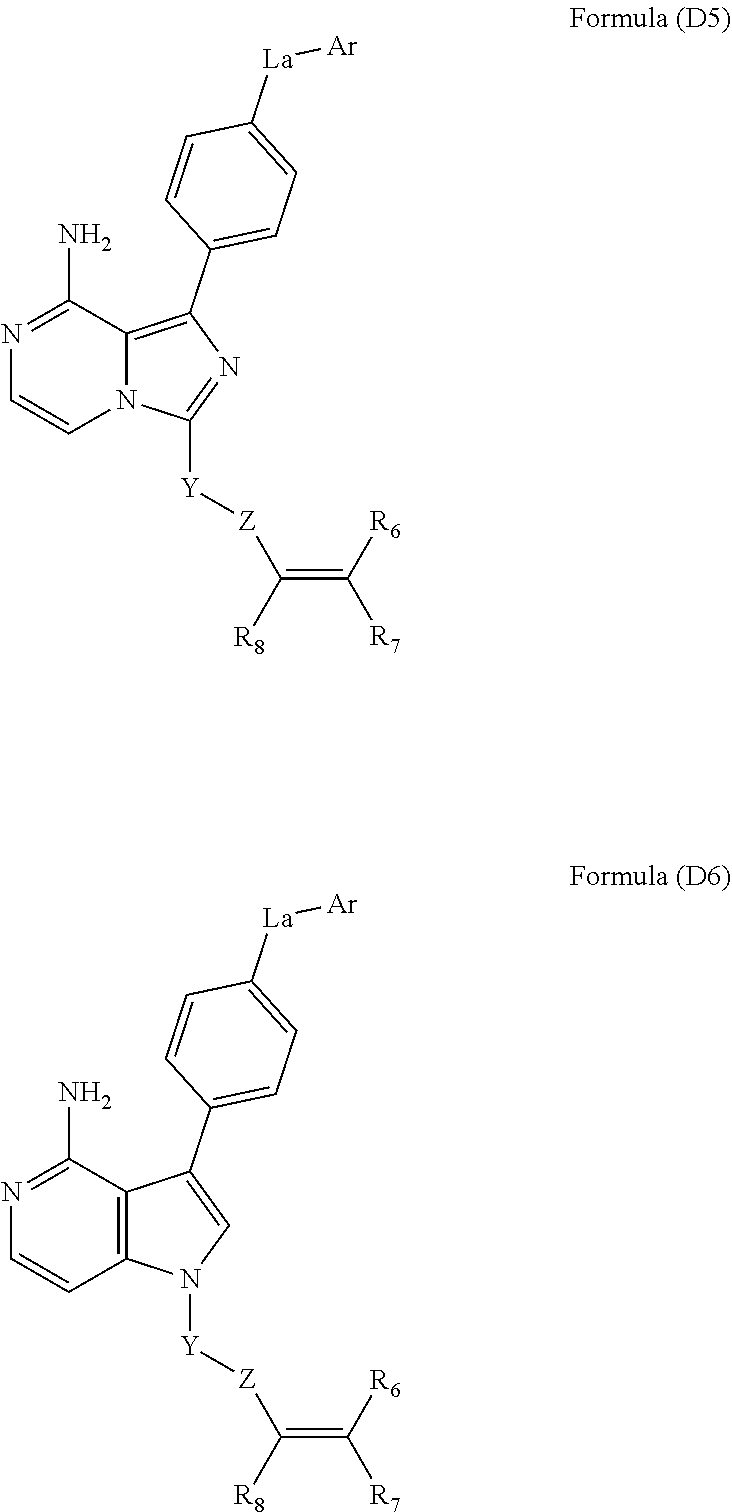
- wherein:
-
- La is CH2, O, NH or S;
- Ar is a substituted or unsubstituted aryl, or a substituted or unsubstituted heteroaryl;
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, cycloalkylene, heterocycloalkylene, arylene, and heteroarylene;
- Z is C(═O), OC(═O), NHC(═O), C(═S), S(═O)x, OS(═O)x, NHS(═O)x, where x is 1 or 2;
- R7 and R8 are independently selected from among H, unsubstituted C1-C4alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
- R7 and R8 taken together form a bond;
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl); and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof.
- In another embodiment are provided pharmaceutically acceptable salts of compounds of Formula (D2-D6). By way of example only, are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid. Further salts include those in which the counterion is an anion, such as adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, and valerate. Further salts include those in which the counterion is an cation, such as sodium, lithium, potassium, calcium, magnesium, ammonium, and quaternary ammonium (substituted with at least one organic moiety) cations.
- In another embodiment are pharmaceutically acceptable esters of compounds of Formula (D2-D6), including those in which the ester group is selected from a formate, acetate, propionate, butyrate, acrylate and ethylsuccinate.
- In another embodiment are pharmaceutically acceptable carbamates of compounds of Formula (D2-D6). In another embodiment are pharmaceutically acceptable N-acyl derivatives of compounds of Formula (D2-D6).
- For any and all of the embodiments, substituents can be selected from among from a subset of the listed alternatives. For example, in some embodiments, La is CH2, O, or NH. In other embodiments, La is O or NH. In yet other embodiments, La is O.
- In some embodiments, Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- In some embodiments, x is 2. In yet other embodiments, Z is C(═O), OC(═O), NHC(═O), S(═O)x, OS(═O)x, or NHS(═O)x. In some other embodiments, Z is C(═O), NHC(═O), or S(═O)2.
- In some embodiments, R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, and substituted C1-C4heteroalkyl; or R7 and R8 taken together form a bond. In yet other embodiments, each of R7 and R8 is H; or R7 and R8 taken together form a bond.
- In some embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C2alkyl-N(C1-C3alkyl)2, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). In some other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C2alkyl-N(C1-C3alkyl)2, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). In yet other embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—N(C1-C3alkyl)2, C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl). In some embodiments, R6 is H, substituted or unsubstituted C1-C4alkyl, —CH2—O—(C1-C3alkyl), —CH2—N(C1-C3alkyl)2, C1-C4alkyl(phenyl), or C1-C4alkyl(5- or 6-membered heteroaryl containing 1 or 2 N atoms), or C1-C4alkyl(5- or 6-membered heterocycloalkyl containing 1 or 2 N atoms).
- In some embodiments, Y is an optionally substituted group selected from among alkylene, heteroalkylene, cycloalkylene, and heterocycloalkylene. In other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 4-, 5-, 6- or 7-membered cycloalkylene, and 4-, 5-, 6- or 7-membered heterocycloalkylene. In yet other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 5-, or 6-membered cycloalkylene, and 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some other embodiments, Y is a 5-, or 6-membered cycloalkylene, or a 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
- Any combination of the groups described above for the various variables is contemplated herein.
- In further aspects are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) having the structure of compounds of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), including, but are not limited to, compounds selected from the group consisting of:
-










- In one aspect are compounds (including irreversible inhibitors of ACKs, including Btk and its cysteine homologs) selected from among: (E)-4-(N-(2-hydroxyethyl)-N-methylamino)-1-(3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one (Compound 3); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3-(1H-imidazol-4-yl)prop-2-en-1-one (Compound 4); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 5); (E)-1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 7); (E)-N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)-4-(dimethylamino)but-2-enamide (Compound 8); N-((1r,4r)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)acrylamide (Compound 10); (E)-1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrolidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 11); (E)-1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrolidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 12); 1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 13); 1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 14); 1((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)but-2-yn-1-one (Compound 15); 1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)but-2-yn-1-one (Compound 16); 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-yn-1-one (Compound 17); (E)-N-((1,r,4r)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl-4-(dimethylamino)but-2-enamide (Compound 18); N-(2-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-N-methylacrylamide (Compound 19); (E)-1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 20); (E)-1-((S-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 21); N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)but-2-ynamide (Compound 22); N-(2-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)acrylamide (Compound 23); (E)-1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 24); (E)-N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)-4-morpholinobut-2-enamide (Compound 25).
- The compounds of any of Formula (I), Formula (VII), Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), or Formula (D1-D6) irreversibly inhibit Btk and are optionally used to treat patients suffering from Bruton's tyrosine kinase-dependent or Bruton's tyrosine kinase mediated conditions or diseases, including, but not limited to, cancer, autoimmune and other inflammatory diseases.
- Compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) are optionally synthesized using standard synthetic techniques or using such methods known in combination with methods described herein. In additions, solvents, temperatures and other reaction conditions are presented herein for illustration only, and not to limit the scope of the methods and compositions described herein. As a further guide the following synthetic methods may also be utilized.
- The reactions are optionally employed in a linear sequence to provide the compounds described herein or used to synthesize fragments which are subsequently joined by the methods described herein and/or documented elsewhere.
- Formation of Covalent Linkages by Reaction of an Electrophile with a Nucleophile
- The compounds described herein can be modified using various electrophiles or nucleophiles to form new functional groups or substituents. Table 1 entitled “Examples of Covalent Linkages and Precursors Thereof” lists selected examples of covalent linkages and precursor functional groups which yield and can be used as guidance toward the variety of electrophiles and nucleophiles combinations available. Precursor functional groups are shown as electrophilic groups and nucleophilic groups.
-
TABLE 1 Examples of Covalent Linkages and Precursors Thereof Covalent Linkage Product Electrophile Nucleophile Carboxamides Activated esters amines/anilines Carboxamides acyl azides amines/anilines Carboxamides acyl halides amines/anilines Esters acyl halides alcohols/phenols Esters acyl nitriles alcohols/phenols Carboxamides acyl nitriles amines/anilines Imines Aldehydes amines/anilines Hydrazones aldehydes or ketones Hydrazines Oximes aldehydes or ketones Hydroxylamines Alkyl amines alkyl halides amines/anilines Esters alkyl halides carboxylic acids Thioethers alkyl halides Thiols Ethers alkyl halides alcohols/phenols Thioethers alkyl sulfonates Thiols Esters alkyl sulfonates carboxylic acids Ethers alkyl sulfonates alcohols/phenols Esters Anhydrides alcohols/phenols Carboxamides Anhydrides amines/anilines Thiophenols aryl halides Thiols Aryl amines aryl halides Amines Thioethers Azindines Thiols Boronate esters Boronates Glycols Carboxamides carboxylic acids amines/anilines Esters carboxylic acids Alcohols hydrazines Hydrazides carboxylic acids N-acylureas or Anhydrides carbodiimides carboxylic acids Esters diazoalkanes carboxylic acids Thioethers Epoxides Thiols Thioethers haloacetamides Thiols Ammotriazines halotriazines amines/anilines Triazinyl ethers halotriazines alcohols/phenols Amidines imido esters amines/anilines Ureas Isocyanates amines/anilines Urethanes Isocyanates alcohols/phenols Thioureas isothiocyanates amines/anilines Thioethers Maleimides Thiols Phosphite esters phosphoramidites Alcohols Silyl ethers silyl halides Alcohols Alkyl amines sulfonate esters amines/anilines Thioethers sulfonate esters Thiols Esters sulfonate esters carboxylic acids Ethers sulfonate esters Alcohols Sulfonamides sulfonyl halides amines/anilines Sulfonate esters sulfonyl halides phenols/alcohols Alkyl thiol α,β-unsaturated ester thiols Alkyl ethers α,β-unsaturated ester alcohols Alkyl amines α,β-unsaturated ester amines Alkyl thiol Vinyl sulfone thiols Alkyl ethers Vinyl sulfone alcohols Alkyl amines Vinyl sulfone amines Vinyl sulfide Propargyl amide thiol - In the reactions described, it may be necessary to protect reactive functional groups, for example hydroxy, amino, imino, thio or carboxy groups, where these are desired in the final product, to avoid their unwanted participation in the reactions. Protecting groups are used to block some or all reactive moieties and prevent such groups from participating in chemical reactions until the protective group is removed. In one embodiment, each protective group be removable by a different means. Protective groups that are cleaved under totally disparate reaction conditions fulfill the requirement of differential removal. Protective groups can be removed by acid, base, and hydrogenolysis. Groups such as trityl, dimethoxytrityl, acetal and t-butyldimethylsilyl are acid labile and may be used to protect carboxy and hydroxy reactive moieties in the presence of amino groups protected with Cbz groups, which are removable by hydrogenolysis, and Fmoc groups, which are base labile. Carboxylic acid and hydroxy reactive moieties may be blocked with base labile groups such as, but not limited to, methyl, ethyl, and acetyl in the presence of amines blocked with acid labile groups such as t-butyl carbamate or with carbamates that are both acid and base stable but hydrolytically removable.
- Carboxylic acid and hydroxy reactive moieties may also be blocked with hydrolytically removable protective groups such as the benzyl group, while amine groups capable of hydrogen bonding with acids may be blocked with base labile groups such as Fmoc. Carboxylic acid reactive moieties may be protected by conversion to simple ester compounds as exemplified herein, or they may be blocked with oxidatively-removable protective groups such as 2,4-dimethoxybenzyl, while co-existing amino groups may be blocked with fluoride labile silyl carbamates.
- Allyl blocking groups are useful in then presence of acid- and base-protecting groups since the former are stable and can be subsequently removed by metal or pi-acid catalysts. For example, an allyl-blocked carboxylic acid can be deprotected with a Pd0-catalyzed reaction in the presence of acid labile t-butyl carbamate or base-labile acetate amine protecting groups. Yet another form of protecting group is a resin to which a compound or intermediate may be attached. As long as the residue is attached to the resin, that functional group is blocked and cannot react. Once released from the resin, the functional group is available to react.
- Typically blocking/protecting groups may be selected from:
-

- Other protecting groups, plus a detailed description of techniques applicable to the creation of protecting groups and their removal are described in Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, New York, N.Y., 1999, and Kocienski, Protective Groups, Thieme Verlag, New York, N.Y., 1994, which are incorporated herein by reference for such disclosure.
- In certain embodiments, provided herein are methods of making and methods of using tyrosine kinase inhibitor compounds described herein. In certain embodiments, compounds described herein can be synthesized using the following synthetic schemes. Compounds may be synthesized using methodologies analogous to those described below by the use of appropriate alternative starting materials.
- Described herein are compounds that inhibit the activity of tyrosine kinase(s), such as Btk, and processes for their preparation. Also described herein are pharmaceutically acceptable salts, pharmaceutically acceptable solvates, pharmaceutically active metabolites and pharmaceutically acceptable prodrugs of such compounds. Pharmaceutical compositions that include at least one such compound or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, pharmaceutically active metabolite or pharmaceutically acceptable prodrug of such compound, are provided.
- The starting material used for the synthesis of the compounds described herein is either synthesized or obtained from commercial sources, such as, but not limited to, Aldrich Chemical Co. (Milwaukee, Wis.), Bachem (Torrance, Calif.), or Sigma Chemical Co. (St. Louis, Mo.). The compounds described herein, and other related compounds having different substituents are optionally synthesized using techniques and materials, such as described, for example, in March, A
DVANCED ORGANIC C HEMISTRY DVANCED ORGANIC C HEMISTRY ROTECTIVE GROUPS IN ORGANIC S YNTHESIS - The products of the reactions are optionally isolated and purified, if desired, using conventional techniques, including, but not limited to, filtration, distillation, crystallization, chromatography and the like. Such materials are optionally characterized using conventional means, including physical constants and spectral data.
- Compounds described herein are optionally prepared using the synthetic methods described herein as a single isomer or a mixture of isomers.
- A non-limiting example of a synthetic approach towards the preparation of compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) is shown in Scheme I.
-


- Halogenation of commercially available 1H-pyrazolo[3,4-d]pyrimidin-4-amine provides an entry into the synthesis of compounds of Formula (A1-A6), (B1-B6), (C1-C6) and/or (D1-D6). In one embodiment, 1H-pyrazolo[3,4-d]pyrimidin-4-amine is treated with N-iodosuccinamide to give 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine. Metal catalyzed cross coupling reactions are then carried out on 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine. In one embodiment, palladium mediated cross-coupling of a suitably substituted phenyl boronic acid under basic conditions constructs intermediate 2. Intermediate 2 is coupled with N-Boc-3-hydroxypiperidine (as non-limiting example) via Mitsunobu reaction to give the Boc (tert-butyloxycarbonyl) protected
intermediate 3. After deprotection with acid, coupling with, but not limited to, an acid chloride, such as, but not limited to, acryloyl chloride, completes the synthesis to giveCompound 13. - A non-limiting example of a synthetic approach towards the preparation of compounds containing the imidazotriazine moiety,
-

- is shown in Scheme II.
-


- A non-limiting example of a synthetic approach towards the preparation of compounds containing any imidazopyrazine moiety,
-

- is shown in Scheme III.
-

- A non-limiting example of a synthetic approach towards the preparation of compounds containing the pyrrolopyrimidine moiety,
-

- is shown in Scheme IV.
-


- A non-limiting example of a synthetic approach towards the preparation of compounds containing the Azaindole moiety,
-

- is shown in Scheme V.
-

- A non-limiting example of a synthetic approach towards the preparation of compounds containing the pyrrolopyrimidine moiety,
-

- is shown in Scheme VI.
-

- Using the synthetic methods described herein, tyrosine kinase inhibitors as disclosed herein are obtained in good yields and purity. The compounds prepared by the methods disclosed herein are purified by conventional means, such as, for example, filtration, recrystallization, chromatography, distillation, and combinations thereof.
- Any combination of the groups described above for the various variables is contemplated herein.
- Compounds disclosed herein have a structure of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). It is understood that when reference is made to compounds described herein, it is meant to include compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), as well as to all of the specific compounds that fall within the scope of these generic formulae, unless otherwise indicated.
- The compounds described herein may possess one or more stereocenters and each center may exist in the R or S configuration. The compounds presented herein include all diastereomeric, enantiomeric, and epimeric forms as well as the appropriate mixtures thereof. Stereoisomers may be obtained, if desired, by methods such as, for example, the separation of stereoisomers by chiral chromatographic columns.
- Diasteromeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods known, for example, by chromatography and/or fractional crystallization. In one embodiment, enantiomers can be separated by chiral chromatographic columns. In other embodiments, enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., alcohol), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereomers to the corresponding pure enantiomers. All such isomers, including diastereomers, enantiomers, and mixtures thereof are considered as part of the compositions described herein.
- The methods and formulations described herein include the use of N-oxides, crystalline forms (also known as polymorphs), or pharmaceutically acceptable salts of compounds described herein, as well as active metabolites of these compounds having the same type of activity. In some situations, compounds exist as tautomers. All tautomers are included within the scope of the compounds presented herein. In addition, the compounds described herein can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. The solvated forms of the compounds presented herein are also considered to be disclosed herein.
- Compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) in unoxidized form can be prepared from N-oxides of compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) by treating with a reducing agent, such as, but not limited to, sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like in a suitable inert organic solvent, such as, but not limited to, acetonitrile, ethanol, aqueous dioxane, or the like at 0 to 80° C.
- In some embodiments, compounds described herein are prepared as prodrugs. A “prodrug” refers to an agent that is converted into the parent drug in vivo. Prodrugs are often useful because, in some situations, they may be easier to administer than the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug. An example, without limitation, of a prodrug is a compound described herein, which is administered as an ester (the “prodrug”) to facilitate transmittal across a cell membrane where water solubility is detrimental to mobility but which then is metabolically hydrolyzed to the carboxylic acid, the active entity, once inside the cell where water-solubility is beneficial. A further example of a prodrug is a short peptide (polyaminoacid) bonded to an acid group where the peptide is metabolized to reveal the active moiety. In certain embodiments, upon in vivo administration, a prodrug is chemically converted to the biologically, pharmaceutically or therapeutically active form of the compound. In certain embodiments, a prodrug is enzymatically metabolized by one or more steps or processes to the biologically, pharmaceutically or therapeutically active form of the compound. To produce a prodrug, a pharmaceutically active compound is modified such that the active compound will be regenerated upon in vivo administration. The prodrug can be designed to alter the metabolic stability or the transport characteristics of a drug, to mask side effects or toxicity, to improve the flavor of a drug or to alter other characteristics or properties of a drug. By virtue of knowledge of pharmacodynamic processes and drug metabolism in vivo, once a pharmaceutically active compound is known, prodrugs of compounds can be designed (if desired) (for examples of this procedure applied to other compounds, see, e.g., Nogrady (1985) Medicinal Chemistry A Biochemical Approach, Oxford University Press, New York, pages 388-392; Silverman (1992), The Organic Chemistry of Drug Design and Drug Action, Academic Press, Inc., San Diego, pages 352-401, Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985).
- Prodrug forms of the herein described compounds, wherein the prodrug is metabolized in vivo to produce a derivative as set forth herein are included within the scope of the claims. In some cases, some of the compounds herein-described are prodrugs for another derivative or active compound.
- Prodrugs are often useful because, in some situations, they are easier to administer than the parent drug. They are, for instance, bioavailable by oral administration whereas the parent is not. The prodrug optionally has improved solubility in pharmaceutical compositions over the parent drug. Prodrugs may be designed as reversible drug derivatives, for use as modifiers to enhance drug transport to site-specific tissues. In some embodiments, the design of a prodrug increases the effective water solubility. See, e.g., Fedorak et al., Am. J. Physiol., 269:G210-218 (1995); McLoed et al., Gastroenterol, 106:405-413 (1994); Hochhaus et al., Biomed. Chrom., 6:283-286 (1992); J. Larsen and H. Bundgaard, Int. J Pharmaceutics, 37, 87 (1987); J. Larsen et al., Int. J. Pharmaceutics, 47, 103 (1988); Sinkula et al., J. Pharm. Sci., 64:181-210 (1975); T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series; and Edward B. Roche, Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, all incorporated by reference for such disclosure.
- Sites on the aromatic ring portion of compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) can be susceptible to various metabolic reactions, therefore incorporation of appropriate substituents on the aromatic ring structures, such as, by way of example only, halogens can reduce, minimize or eliminate this metabolic pathway.
- Compounds described herein include isotopically-labeled compounds, which are identical to those recited in the various formulas and structures presented herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be incorporated into the present compounds include isotopes of hydrogen, carbon, nitrogen, oxygen, fluorine and chlorine, such as 2H, 3H, 13C, 14C, 15N, 18O, 17O, 35S, 18F, 36Cl, respectively. Certain isotopically-labeled compounds described herein, for example those into which radioactive isotopes such as 3H and 14C are incorporated, are useful in drug and/or substrate tissue distribution assays. Further, substitution with isotopes such as deuterium, i.e., 2H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements.
- In additional or further embodiments, the compounds described herein are metabolized upon administration to an organism in need to produce a metabolite that is then used to produce a desired effect, including a desired therapeutic effect.
- Compounds described herein (for example, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII)) are optionally in the form of, and/or used as, pharmaceutically acceptable salts. The type of pharmaceutical acceptable salts, include, but are not limited to: (1) acid addition salts, formed) by reacting the free base form of the compound with a pharmaceutically acceptable: inorganic acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, metaphosphoric acid, and the like; or with an organic acid such as acetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, trifluoroacetic acid, tartaric acid, citric acid, benzoic acid, 3-(4-hydroxybenzoyl)benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2-ethanedisulfonic acid, 2-hydroxyethanesulfonic acid, benzenesulfonic acid, toluenesulfonic acid, 2-naphthalenesulfonic acid, 4-methylbicyclo-[2.2.2]oct-2-ene-1-carboxylic acid, glucoheptonic acid, 4,4′-methylenebis-(3-hydroxy-2-ene-1-carboxylic acid), 3-phenylpropionic acid, trimethylacetic acid, tertiary butylacetic acid, lauryl sulfuric acid, gluconic acid, glutamic acid, hydroxynaphthoic acid, salicylic acid, stearic acid, muconic acid, and the like; (2) salts formed when an acidic proton present in the parent compound either is replaced by a metal ion, e.g., an alkali metal ion (e.g. lithium, sodium, potassium), an alkaline earth ion (e.g. magnesium, or calcium), or an aluminum ion; or coordinates with an organic base. Acceptable organic bases include ethanolamine, diethanolamine, triethanolamine, tromethamine, N-methylglucamine, and the like. Acceptable inorganic bases include aluminum hydroxide, calcium hydroxide, potassium hydroxide, sodium carbonate, sodium hydroxide, and the like.
- The corresponding counterions of the pharmaceutically acceptable salts are optionally analyzed and identified using various methods including, but not limited to, ion exchange chromatography, ion chromatography, capillary electrophoresis, inductively coupled plasma, atomic absorption spectroscopy, mass spectrometry, or any combination thereof.
- The salts are recovered by using at least one of the following techniques: filtration, precipitation with a non-solvent followed by filtration, evaporation of the solvent, or, in the case of aqueous solutions, lyophilization.
- It should be understood that a reference to a pharmaceutically acceptable salt includes the solvent addition forms or crystal forms thereof, particularly solvates or polymorphs. Solvates contain either stoichiometric or non-stoichiometric amounts of a solvent, and are optionally formed during the process of crystallization with pharmaceutically acceptable solvents such as water, ethanol, and the like. Hydrates are formed when the solvent is water, or alcoholates are formed when the solvent is alcohol. Solvates of compounds described herein can be conveniently prepared or formed during the processes described herein. In addition, the compounds provided herein can exist in unsolvated as well as solvated forms. In general, the solvated forms are considered equivalent to the unsolvated forms for the purposes of the compounds and methods provided herein.
- It should be understood that a reference to a salt includes the solvent addition forms or crystal forms thereof, particularly solvates or polymorphs. Solvates contain either stoichiometric or non-stoichiometric amounts of a solvent, and are often formed during the process of crystallization with pharmaceutically acceptable solvents such as water, ethanol, and the like. Hydrates are formed when the solvent is water, or alcoholates are formed when the solvent is alcohol. Polymorphs include the different crystal packing arrangements of the same elemental composition of a compound. Polymorphs usually have different X-ray diffraction patterns, infrared spectra, melting points, density, hardness, crystal shape, optical and electrical properties, stability, and solubility. Various factors such as the recrystallization solvent, rate of crystallization, and storage temperature may cause a single crystal form to dominate.
- Compounds described herein are optionally in various forms, including but not limited to, amorphous forms, milled forms and nano-particulate forms. In addition, compounds described herein include crystalline forms, also known as polymorphs. Polymorphs include the different crystal packing arrangements of the same elemental composition of a compound. Polymorphs usually have different X-ray diffraction patterns, infrared spectra, melting points, density, hardness, crystal shape, optical and electrical properties, stability, and solubility. Various factors such as the recrystallization solvent, rate of crystallization, and storage temperature may cause a single crystal form to dominate.
- The screening and characterization of the pharmaceutically acceptable salts, polymorphs and/or solvates may be accomplished using a variety of techniques including, but not limited to, thermal analysis, x-ray diffraction, spectroscopy, vapor sorption, and microscopy. Thermal analysis methods address thermo chemical degradation or thermo physical processes including, but not limited to, polymorphic transitions, and such methods are used to analyze the relationships between polymorphic forms, determine weight loss, to find the glass transition temperature, or for excipient compatibility studies. Such methods include, but are not limited to, Differential scanning calorimetry (DSC), Modulated Differential Scanning Calorimetry (MDCS), Thermogravimetric analysis (TGA), and Thermogravi-metric and Infrared analysis (TG/IR). X-ray diffraction methods include, but are not limited to, single crystal and powder diffractometers and synchrotron sources. The various spectroscopic techniques used include, but are not limited to, Raman, FTIR, UVIS, and NMR (liquid and solid state). The various microscopy techniques include, but are not limited to, polarized light microscopy, Scanning Electron Microscopy (SEM) with Energy Dispersive X-Ray Analysis (EDX), Environmental Scanning Electron Microscopy with EDX (in gas or water vapor atmosphere), IR microscopy, and Raman microscopy.
- Protein kinases, which act on and modify the activity of specific proteins, are used to transmit signals and control complex processes in cells. Up to 518 different kinases have been identified in humans. Many kinase inhibitor compounds non-selectively bind and/or inhibit these kinases because the active sites of some of these kinases are similar in structure. Such cross-reactivity is not a desired feature of a kinase inhibitor compound because of the potential for undesired side effects when such a compound is being administered to treat a disease or condition.
- We have observed that small differences in the structure of kinase inhibitor compounds have profound effects in the selectivity of similarly-structured kinases (e.g., ACKs, including, Btk and the Btk kinase cysteine homologs).
- As a result, we have developed assays, methods, and systems for converting a non-selective inhibitor compound into a highly-selective inhibitor compound. In brief, the non-selective inhibitor compound is provided with a Michael acceptor moiety and a linker moiety that links the Michael acceptor moiety to the remainder of the non-selective inhibitor compound. A series of linker and Michael acceptor moieties provides a small library/panel of test inhibitor compounds. The inhibitor library/panel is contacted with a panel of structurally related kinases (e.g., Btk and the Btk kinase cysteine homologs). Binding is determined by a variety of means, included fluorescence detection (or via any other detectable label), mass spectrometry, or a combination of approaches. An Activity Probe is optionally used to detect binding of members of the inhibitor library/panel to the kinase library/panel. The binding data is then optionally collected and analyzed to provide a structure-activity relationship (SAR) between the structure of the members of the inhibitor panel/library (e.g., Michael acceptor and/or linker moieties) and the activity of binding to and/or inhibiting members of the kinase panel. Based on this information, further modifications are suggested if necessary. We have successfully used this approach to improve the binding and selectivity of Btk inhibitor compounds (see Examples herein, including “Kinase Inhibitor Discovery Platform” example section).
- A similar approach can be use for converting a selective inhibitor compound for a group of similarly-structured ACKs (including, Btk and the Btk kinase cysteine homologs) into a more highly-selective inhibitor compound (e.g., more selective for a particular ACK over structurally-similar ACKs), or for converting a selective inhibitor compound for a particular ACK (e.g., Btk) into an even more selective inhibitor of that particular ACK. For example, in brief, the selective inhibitor compound (which, for example, contains an active-site binding moiety, a linker moiety and a Michael acceptor moiety) is modified. In one embodiment, a series of linker and Michael acceptor moieties provides a small library/panel of test inhibitor compounds. The inhibitor library/panel is contacted with a panel of structurally related kinases (e.g., Btk and the Btk kinase cysteine homologs). Binding is determined by a variety of means, included fluorescence detection (or via any other detectable label), mass spectrometry, or a combination of approaches. An Activity Probe is optionally used to detect binding of members of the inhibitor library/panel to the kinase library/panel. The binding data is then optionally collected and analyzed to provide a structure-activity relationship (SAR) between the structure of the members of the inhibitor panel/library (e.g., Michael acceptor and/or linker moieties) and the activity of binding to and/or inhibiting members of the kinase panel. Based on this information, further modifications are suggested if necessary. We have also successfully used this approach to improve the binding and selectivity of Btk inhibitor compounds (see Examples herein, including “Kinase Inhibitor Discovery Platform” example section).
- Thus, for our highly selective
BTK inhibitor Compound 1, we engineered an electrophilic center capable of irreversibly inactivating the target enzyme, BTK. That is, to an active site binding moiety of a reversible inhibitor was added a linker moiety and a Michael acceptor moiety that achieved a high degree of potency and selectivity by (1) fitting the core scaffold into the active site ATP binding pocket of kinase enzymes, and (2) forming a covalent bond with Cysteine-481 located in BTK. The chemistry required for covalent bond formation involves an electrophilic moiety that acts as a Michael acceptor, which bonds with a nucleophile (such as Cys-481) present in a precise location within the active site. - In another example, the linker and Michael acceptor moiety of
Compound 1 was modified to provideCompound 9 which has a different selectivity pattern. Table 1 is a table showing the degree of inhibition of a panel of kinases for two example compounds. IC50s were determined using the in vitro HotSpot kinase assay (purified enzymes, 33P-ATP, an appropriate substrate and 1 uM ATP.) Compared toCompound 1,Compound 9 has similar potency toward Btk, but significantly less potency toward JAK-3, ITK, and EGFR and significantly more potency toward the src-family kinases lck, c-src, FGR, Fyn, Hck, and Lyn and Yes. Thus, subtle modifications in the linker moiety and the Michael acceptor moiety are important for the design of selective ACK inhibitors. -
TABLE 1 Compound 1Compound 9Kinase IC50 (nM) IC50 (nM) BTK 0.5 1.0 ITK 11.7 909.9 Bmx/ETK 0.8 1.1 TEC 77.8 108.0 EFGR 0.5 20.6 HER2 9.4 1536.0 HER4 0.1 3.2 LCK 2.0 1.0 BLK 0.5 0.2 C-src 262.6 14.3 FGR 2.3 0.4 Fyn 95.6 7.1 HCK 3.7 1.0 Lyn 16.2 1.2 YES 6.5 0.8 ABL 86.1 32.3 Brk 3.3 3.3 CSK 2.2 2.4 FER 8,070.0 3,346.0 JAK3 10.4 8,278.0 SYK >10,000 >10,000 - Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the examples section provides further modifications of the linker moiety and/or the Michael acceptor moiety and the impact of such changes of inhibitor selectivity.
- Thus, in one aspect described herein are methods of identifying an irreversible inhibitor of a kinase selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog (or indeed, any ACK) comprising:
-
- (1) contacting a multiplicity of kinases selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog (or indeed any ACK) with a compound that comprises a Michael acceptor moiety;
- (2) contacting at least one non-kinase molecule having at least one accessible SH group with the compound that comprises a Michael acceptor moiety (this step allows for the selection of inhibitors that have low selectivity for higher abundance biological molecules that have moieties that irreversibly react with the inhibitor; thus preventing the inhibitor from binding to the desire ACK when administered as a drug to a patient); and
- (3) determining the covalent binding of the compound that comprises a Michael acceptor with the multiplicity of kinases and the at least one non-kinase molecule; and repeating steps (1), (2), and (3) for at least one other compound that comprises a Michael acceptor moiety.
- In a further aspect, the following steps are added: (4) comparing the covalent binding of the compound that comprises a Michael acceptor with the multiplicity of kinases and the at least one non-kinase molecule; and repeating steps (1), (2), (3) and (4) for at least one other compound that comprises a Michael acceptor moiety.
- In a further aspect the irreversible inhibitor compounds are also contacted with at least one non-ACK kinase in order to determine the selectivity of the irreversible inhibitor compound for the ACK relative to the non-ACK.
- By way of certain relevant examples of non-kinase molecules with at least one accessible SH group are glutathione and/or hemoglobin. Because of the high abundance of these molecules in typical biological systems (e.g., in a patient), the desired irreversible inhibitor compounds have low selectivity/reactivity with these non-kinase molecules.
- In certain embodiments of the Kinase Inhibitor Discovery Platform, an Activity Probe (described in more detail herein) is used as a rapid diagnostic method for determining whether a test inhibitor compound has irreversibly inhibited an ACK. In one embodiment, the Activity Probe is itself an irreversible inhibitor of an ACK, and further, has a reporter moiety (e.g., a fluorescent moiety) as part of its structure. When used in competition with a test irreversible inhibitor, the absence of a ‘reporter’ signal on an ACK is one indication that the test irreversible inhibitor has prevented the Activity Probe from binding to the ACK (and that the test irreversible inhibitor has a higher binding affinity for the ACK than the Activity Probe).
- In certain embodiments, the Kinase Inhibitor Discovery Platform, steps (1) and (2) are conducted in vivo and step (3) is conducted in part using an Activity Probe. Further, in certain embodiments, the determining step uses mass spectrometry, fluorescence, or a combination thereof.
- As described herein, in one embodiment, the inhibitor tested with the Kinase Inhibitor Discovery Platform comprise an active site binding moiety, a Michael acceptor moiety, and a linker moiety that links the Michael acceptor moiety to the active site binding moiety. For example, in such a scheme, the following information is collected and analyzed: the structure-function activity relationship between the structure of the linker moiety and/or the Michael acceptor moiety of each compound, and the binding and/or selectivity of each compound to at least one kinase. Further, in certain embodiments, structure of the active site binding moiety of each compound is not varied, whereas the structure of the linker moiety and/or the Michael acceptor moiety is varied.
- In one example, the inhibitors have the structure of Formula (VII):
-

- wherein:
-
- wherein
-

- is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
-
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
- Z is C(═O), OC(═O), NHC(═O), NCH3C(═O), C(═S), S(═O)x, OS(═O)x, NHS(═O)x, where x is 1 or 2;
- R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
- R7 and R8 taken together form a bond; and
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl).
- In such a scheme, the following information is collected and analyzed: the structure-function activity relationship between the structure of Y—Z and/or
-

- of each compound, and the binding and/or selectivity of each compound to at least one kinase. Further, the structure of
-

- of each compound is not varied, whereas the structure of the linker moiety (Y—Z) and/or the Michael acceptor moiety
-

- is varied.
- In certain embodiments of the Kinase Inhibitor Discovery Platform, the resulting inhibitor is selective for one kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog over at least one other kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog. In some embodiments, this selectivity is at least 5×, at least 10×, at least 20×, at least 50×, or at least 100×. In further embodiments, the resulting inhibitor is selective for at least one kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog over at least one other non-kinase molecule having an accessible SH group. In some embodiments, this selectivity is at least 5×, at least 10×, at least 20×, at least 50×, or at least 100×.
- In further embodiments, the resulting inhibitor is used in the therapeutic methods described herein, or in the pharmaceutical compositions described herein.
- Because of the Kinase Inhibitor Discovery Platform described herein optionally utilizes an Activity Probe, the following section describes the design, structure and use of non-limiting examples of Activity Probes.
- The Activity Probe compounds described herein are composed of a moiety comprising an inhibitor of Btk, a Btk homolog and/or a Btk kinase cysteine homolog (hereinafter, a “Kinase Inhibitor”), a linker moiety, and a reporter moiety. In one embodiment, the Kinase Inhibitor is an irreversible inhibitor. In another embodiment, the irreversible Kinase Inhibitor binds to a non-catalytic residue in the ATP binding pocket of Btk, a Btk homolog and/or a Btk kinase cysteine homolog (hereinafter a “Kinase”); in further embodiments, the non-catalytic residue is a cysteine residue. In some embodiments, the Activity Probe forms a covalent bond with at least one non-catalytic residue of a Kinase. In other embodiments, the Activity Probe forms a non-covalent bond with at least one non-catalytic residue of a Kinase. In a further embodiment, the Activity Probe forms hydrogen bonding within the ATP binding pocket of a Kinase. In yet a further embodiment, the Activity Probe has Van der Waals attractions with the Kinase.
- In some other embodiments, the Activity Probes described herein are activity dependent such that the probe binds only an active Kinase. In further embodiments, the Activity Probe binds a Kinase that has been switched on by phosphorylation by upstream kinases. In yet a further embodiment, the Activity Probes described herein are activity independent such that the probe binds Kinases that have not been switched on by phosphorylation by upstream kinases. In some embodiments, the Activity Probe labels a phosphorylated conformation of a Kinase. In other embodiments, the Activity Probe labels a Kinase in a non-phosphorylated conformation.
- In some embodiments, the Activity Probe is permeable to cells.
- In further embodiments, the linker moiety is selected from a bond, a substituted alkyl moiety, a substituted heterocycle moiety, a substituted amide moiety, a ketone moiety, a substituted carbamate moiety, an ester moiety, or any combination thereof. In further embodiments, the reporter moiety is a moiety that is detected using standard or modified laboratory equipment.
- In one aspect is a Activity Probe of Formula (I) comprising:
-

- wherein:
-
- A is a Kinase Inhibitor moiety;
- X and Y are independently selected from the group consisting of: a bond, —O(C═O)—, —NRa(C═O)—, —NRa—,
-

- —O—, —S—, —S—S—, —O—NRa—, —O(C═O)O—, —O(C═O)NRa, —NRa(C═O)NRa—, —N═CRa—, —S(C═O)—, —S(O)—, and —S(O)2—;
-
- wherein
-

- forms a N-containing heterocycle;
-
- B is a linker moiety;
- C is a reporter moiety; and
- Ra is hydrogen or alkyl.
- In one embodiment, the moiety comprising an irreversible Kinase Inhibitor is derived from an irreversible inhibitor of a Kinase. In some embodiments, such irreversible Kinase Inhibitors should possess at least one of the following characteristics: potency, selectively and cell permeability. In further embodiments, such irreversible Kinase Inhibitors possess at least two of the aforementioned characteristics, and in further embodiments, at least all of the aforementioned characteristics.
- In another embodiment, the Kinase Inhibitor moiety is derived from a Btk inhibitor having the structure of Formula (II):
-

- wherein:
-
- La is CH2, O, NH or S;
- Ar is a substituted or unsubstituted aryl, or a substituted or unsubstituted heteroaryl; and
- Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene.
- In some embodiments, La is CH2, O, or NH. In other embodiments, La is O or NH. In yet other embodiments, La is O.
- In other embodiments, Ar is a substituted or unsubstituted aryl. In yet other embodiments, Ar is a 6-membered aryl. In some other embodiments, Ar is phenyl.
- In some embodiments, Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene. In other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 4-, 5-, 6-, or 7-membered cycloalkylene, and 4-, 5-, 6-, or 7-membered heterocycloalkylene. In yet other embodiments, Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene 5- or 6-membered cycloalkylene, and 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some other embodiments, Y is a 5- or 6-membered cycloalkylene, or a 5- or 6-membered heterocycloalkylene containing 1 or 2 N atoms. In some embodiments, Y is a 4-, 5-, 6-, or 7-membered cycloalkylene ring; or Y is a 4-, 5-, 6-, or 7-membered heterocycloalkylene ring.
- In some embodiments, the Kinase Inhibitor moiety is derived from a compound selected from among: 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one; 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)sulfonylethene; 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-yn-1-one; 1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; N-((1 s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)acrylamide; 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)pyrrolidin-1-yl)prop-2-en-1-one; 1-((S)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)pyrrolidin-1-yl)prop-2-en-1-one; 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; 1-((S)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one; and (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-(dimethylamino)but-2-en-1-one; (E)-4-(N-(2-hydroxyethyl)-N-methylamino)-1-(3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-en-1-one (Compound 3); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3-(1H-imidazol-4-yl)prop-2-en-1-one (Compound 4); (E)-1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 5); (E)-1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 7); (E)-N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)-4-(dimethylamino)but-2-enamide (Compound 8); N-((1r,4r)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)acrylamide (Compound 10); (E)-1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrolidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 11); (E)-1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrolidin-1-yl)-4-(dimethylamino)but-2-en-1-one (Compound 12); 1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 13); 1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 14); 1((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)but-2-yn-1-one (Compound 15); 1-((S)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)but-2-yn-1-one (Compound 16); 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)but-2-yn-1-one (Compound 17); (E)-N-((1,r,4r)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl-4-(dimethylamino)but-2-enamide (Compound 18); N-(2-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-N-methylacrylamide (Compound 19); (E)-1-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 20); (E)-1-((S-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 21); N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)but-2-ynamide (Compound 22); N-(2-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)acrylamide (Compound 23); (E)-1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-morpholinobut-2-en-1-one (Compound 24); (E)-N-((1s,4s)-4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexyl)-4-morpholinobut-2-enamide (Compound 25).
- In another embodiment, the linker moiety is selected from a bond, a polymer, a water soluble polymer, optionally substituted alkyl, optionally substituted heteroalkyl, optionally substituted heterocycloalkyl, optionally substituted cycloalkyl, optionally substituted heterocycloalkylalkyl, optionally substituted heterocycloalkylalkenyl, optionally substituted aryl, optionally substituted heteroaryl, and optionally substituted heterocycloalkylalkenylalkyl. In some embodiments, the linker moiety is an optionally substituted heterocycle. In other embodiments, the heterocycle is selected from aziridine, oxirane, episulfide, azetidine, oxetane, pyrroline, tetrahydrofuran, tetrahydrothiophene, pyrrolidine, pyrazole, pyrrole, imidazole, triazole, tetrazole, oxazole, isoxazole, oxirene, thiazole, isothiazole, dithiolane, furan, thiophene, piperidine, tetrahydropyran, thiane, pyridine, pyran, thiapyrane, pyridazine, pyrimidine, pyrazine, piperazine, oxazine, thiazine, dithiane, and dioxane. In some embodiments, the heterocycle is piperazine. In further embodiments, the linker moiety is optionally substituted with halogen, CN, OH, NO2, alkyl, S(O), and S(O)2. In other embodiments, the water soluble polymer is a PEG group.
- In other embodiments, the linker moiety provides sufficient spatial separation between the reporter moiety and the Kinase Inhibitor moiety. In further embodiments, the linker moiety is stable. In yet a further embodiment, the linker moiety does not substantially affect the response of the reporter moiety. In other embodiments the linker moiety provides chemical stability to the Activity Probe. In further embodiments, the linker moiety provides sufficient solubility to the Activity Probe.
- In some embodiments, linkages such as water soluble polymers are coupled at one end to a Kinase Inhibitor moiety and to a reporter moiety at the other end. In other embodiments, the water soluble polymers are coupled via a functional group or substituent of the Kinase Inhibitor moiety. In further embodiments, the water soluble polymers are coupled via a functional group or substituent of the reporter moiety. In other embodiments, covalent attachment of hydrophilic polymers to a Kinase Inhibitor moiety and a reporter moiety represents one approach to increasing water solubility (such as in a physiological environment), bioavailability, increasing serum half-life, increasing pharmacodynamic parameters, or extending the circulation time of the Activity Probe, including proteins, peptides, and particularly hydrophobic molecules. In further embodiments, additional important features of such hydrophilic polymers include biocompatibility and lack of toxicity. In other embodiments, for therapeutic use of the end-product preparation, the polymer is pharmaceutically acceptable.
- In some embodiments, examples of hydrophilic polymers include, but are not limited to: polyalkyl ethers and alkoxy-capped analogs thereof (e.g., polyoxyethylene glycol, polyoxyethylene/propylene glycol, and methoxy or ethoxy-capped analogs thereof, polyoxyethylene glycol, the latter is also known as polyethylene glycol or PEG); polyvinylpyrrolidones; polyvinylalkyl ethers; polyoxazolines, polyalkyl oxazolines and polyhydroxyalkyl oxazolines; polyacrylamides, polyalkyl acrylamides, and polyhydroxyalkyl acrylamides (e.g., polyhydroxypropylmethacrylamide and derivatives thereof); polyhydroxyalkyl acrylates; polysialic acids and analogs thereof; hydrophilic peptide sequences; polysaccharides and their derivatives, including dextran and dextran derivatives, e.g., carboxymethyldextran, dextran sulfates, aminodextran; cellulose and its derivatives, e.g., carboxymethyl cellulose, hydroxyalkyl celluloses; chitin and its derivatives, e.g., chitosan, succinyl chitosan, carboxymethylchitin, carboxymethylchitosan; hyaluronic acid and its derivatives; starches; alginates; chondroitin sulfate; albumin; pullulan and carboxymethyl pullulan; polyaminoacids and derivatives thereof, e.g., polyglutamic acids, polylysines, polyaspartic acids, polyaspartamides; maleic anhydride copolymers such as: styrene maleic anhydride copolymer, divinylethyl ether maleic anhydride copolymer; polyvinyl alcohols; copolymers thereof; terpolymers thereof; mixtures thereof; and derivatives of the foregoing. In other embodiments, the water soluble polymer is any structural form including but not limited to linear, forked or branched. In some embodiments, polymer backbones that are water-soluble, with from 2 to about 300 termini, are particularly useful. In further embodiments, multifunctional polymer derivatives include, but are not limited to, linear polymers having two termini, each terminus being bonded to a functional group which is the same or different. In some embodiments, the water polymer comprises a poly(ethylene glycol) moiety. In further embodiments, the molecular weight of the polymer is of a wide range, including but not limited to, between about 100 Da and about 100,000 Da or more. In yet further embodiments, the molecular weight of the polymer is between about 100 Da and about 100,000 Da, including but not limited to, about 100,000 Da, about 95,000 Da, about 90,000 Da, about 85,000 Da, about 80,000 Da, about 75,000 Da, about 70,000 Da, about 65,000 Da, about 60,000 Da, about 55,000 Da, about 50,000 Da, about 45,000 Da, about 40,000 Da, about 35,000 Da, 30,000 Da, about 25,000 Da, about 20,000 Da, about 15,000 Da, about 10,000 Da, about 9,000 Da, about 8,000 Da, about 7,000 Da, about 6,000 Da, about 5,000 Da, about 4,000 Da, about 3,000 Da, about 2,000 Da, about 1,000 Da, about 900 Da, about 800 Da, about 700 Da, about 600 Da, about 500 Da, about 400 Da, about 300 Da, about 200 Da, and about 100 Da. In some embodiments, the molecular weight of the polymer is between about 100 Da and 50,000 Da. In some embodiments, the molecular weight of the polymer is between about 100 Da and 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 1,000 Da and 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 5,000 Da and 40,000 Da. In some embodiments, the molecular weight of the polymer is between about 10,000 Da and 40,000 Da. In some embodiments, the poly(ethylene glycol) molecule is a branched polymer. In further embodiments, the molecular weight of the branched chain PEG is between about 1,000 Da and about 100,000 Da, including but not limited to, about 100,000 Da, about 95,000 Da, about 90,000 Da, about 85,000 Da, about 80,000 Da, about 75,000 Da, about 70,000 Da, about 65,000 Da, about 60,000 Da, about 55,000 Da, about 50,000 Da, about 45,000 Da, about 40,000 Da, about 35,000 Da, about 30,000 Da, about 25,000 Da, about 20,000 Da, about 15,000 Da, about 10,000 Da, about 9,000 Da, about 8,000 Da, about 7,000 Da, about 6,000 Da, about 5,000 Da, about 4,000 Da, about 3,000 Da, about 2,000 Da, and about 1,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 1,000 Da and about 50,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 1,000 Da and about 40,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 5,000 Da and about 40,000 Da. In some embodiments, the molecular weight of the branched chain PEG is between about 5,000 Da and about 20,000 Da. The foregoing list for substantially water soluble backbones is by no means exhaustive and is merely illustrative, and in some embodiments, the polymeric materials having the qualities described above suitable for use in methods and compositions described herein.
- In further embodiments, the number of water soluble polymers linked to a Kinase Inhibitor moiety and a reporter moiety described herein is adjusted to provide an altered (including but not limited to, increased or decreased) pharmacologic, pharmacokinetic or pharmacodynamic characteristic such as in vivo half-life. In some embodiments, the half-life of the Activity Probe is increased at least about 10, about 20, about 30, about 40, about 50, about 60, about 70, about 80, about 90 percent, about two fold, about five-fold, about 10-fold, about 50-fold, or at least about 100-fold over a Activity Probe without a water soluble linker.
- In another embodiment, X is selected from the group consisting of: a bond, —O(C═O)—, —NRa(C═O)—, —NRa—,
-

- —O—, —S—, —S—S—, —O—NRa—, —O(C═O)O—, —O(C═O)NRa, —NRa(C═O)NRa—, —N═CRa—, —S(C═O)—, —S(O)—, and —S(O)2—; wherein
-

- forms a N-containing heterocycle. In one embodiment, X is NRa(C═O). In another embodiment, X is a bond. In another embodiment, X is —O(C═O)—. In a further embodiment, Y is selected from the group consisting of: a bond, —O(C═O)—, —NRa(C═O)—, —NRa—,
-

- —O—, —S—, —S—S—, —O—NRa—, —O(C═O)O—, —O(C═O)NRa, —NRa(C═O)NRa—, —N═CRa—, —S(C═O)—, —S(O)—, and —S(O)2—; wherein
-

- forms a N-containing heterocycle. In yet a further embodiment, Y is a bond. In one embodiment, Y is —NRa(C═O)—. In yet another embodiment, Ra is hydrogen. In yet a further embodiment, Ra is alkyl.
- In a further embodiment, the reporter moiety is selected from the group consisting of a label, a dye, a photocrosslinker, a cytotoxic compound, a drug, an affinity label, a photoaffinity label, a reactive compound, an antibody or antibody fragment, a biomaterial, a nanoparticle, a spin label, a fluorophore, a metal-containing moiety, a radioactive moiety, a novel functional group, a group that covalently or noncovalently interacts with other molecules, a photocaged moiety, an actinic radiation excitable moiety, a ligand, a photoisomerizable moiety, biotin, a biotin analog, a moiety incorporating a heavy atom, a chemically cleavable group, a photocleavable group, a redox-active agent, an isotopically labeled moiety, a biophysical probe, a phosphorescent group, a chemiluminescent group, an electron dense group, a magnetic group, an intercalating group, a chromophore, an energy transfer agent, a biologically active agent, a detectable label, or a combination thereof.
- In another embodiment, the reporter moiety is a fluorophore. In a further embodiment, the fluorophore is selected from the group consisting of: BODIPY 493/503, BODIPY FL, BODIPY R6G, BODIPY 530/550, BODIPY TMR, BODIPY 558/568, BODIPY 564/570, BODIPY 576/589, BODIPY 581/591, BODIPY TR, Fluorescein, 5(6)-Carboxyfluorescein, 2,7-Dichlorofluorescein, N,N-Bis(2,4,6-trimethylphenyl)-3,4:9,10-perylenebis(dicarboximide, HPTS, Ethyl Eosin, DY-490XL MegaStokes, DY-485XL MegaStokes, Adirondack Green 520, ATTO 465, ATTO 488, ATTO 495, YOYO-1, 5-FAM, BCECF, BCECF, dichlorofluorescein, rhodamine 110, rhodamine 123, Rhodamine Green, YO-PRO-1, SYTOX Green, Sodium Green, SYBR Green I, Alexa Fluor 500, FITC, Fluo-3, Fluo-4, fluoro-emerald, YoYo-1 ssDNA, YoYo-1 dsDNA, YoYo-1, SYTO RNASelect, Diversa Green-FP, Dragon Green, EvaGreen, Surf Green EX, Spectrum Green, Oregon Green 488, NeuroTrace 500525, NBD-X, MitoTracker Green FM, LysoTracker Green DND-26, CBQCA, PA-GFP (post-activation), WEGFP (post-activation), FlASH-CCXXCC, Azami Green monomeric, Azami Green, EGFP (Campbell Tsien 2003), EGFP (Patterson 2001), Fluorescein, Kaede Green, 7-Benzylamino-4-Nitrobenz-2—Oxa-1,3-Diazole, Bexl, Doxorubicin, Lumio Green, and SuperGlo GFP.
- In a further embodiment, the fluorophore is selected from the group consisting of: BODIPY 493/503, BODIPY FL, BODIPY R6G, BODIPY 530/550, BODIPY TMR, BODIPY 558/568, BODIPY 564/570, BODIPY 576/589, BODIPY 581/591, and BODIPY TR. In yet a further embodiment, the fluorophore is BODIPY FL. In certain embodiments, the fluorophore is not BODIPY 530. In some embodiments, the fluorophore has an excitation maxima of between about 500 and about 600 nm. In some other embodiments, the fluorophore has an excitation maxima of between about 500 and about 550 nm. In another embodiments, the fluorophore has an excitation maxima of between about 550 and about 600 nm. In yet a further embodiment, the fluorophore has an excitation maxima of between about 525 and about 575 nm. In other embodiments, the fluorophore has an emission maxima of between about 510 and about 670 nm. In another embodiment, the fluorophore has an emission maxima of between about 510 and about 600 nm. In a further embodiment, the fluorophore has an emission maxima of between about 600 and about 670 nm. In another embodiment, the fluorophore has an emission maxima of between about 575 and about 625 nm.
- By way of example only and in some embodiments, the observed potency, selectivity, and cell permeability of compounds such as
Compound 2 are appropriate to incorporate these molecules into a Kinase-targeted, activity based probe that allows direct visualization of Kinase activity in intact cells. In vitro profiling against a panel of greater than 100 kinases showedCompound 2 to be a highly potent and selective inhibitor of Tec family kinases, including, Btk, as well as Src family kinases. Without limiting the scope of the compositions and methods described herein, it is postulated that the structural basis for the selectivity is covalent modification of a non-catalytic cysteine residue (Cys 481 in Btk) that is conserved in the ATP binding pocket of the Tec family and several other kinases. - However, in other embodiments, any irreversible Kinase Inhibitor that binds to the non-catalytic cysteine residue in the ATP binding pocket of a Kinase is used in the compounds and methods described herein.
- Without limiting the scope of the compositions described herein, an illustrative probe was synthesized by attaching a bodipy FL fluorophore to an irreversible inhibitor via a piperazine linker. The piperazine linker served to maintain probe solubility and provided spatial separation between the fluorophore and the pyrazolopyrimidine core.
-

- In some embodiments, the linkage formed is a stable linkage. In other embodiments, in the case where the conjugate comprises two components, the linker moiety forms a linkage, in some embodiments, a stable linkage, between the Kinase Inhibitor moiety and the reporter moiety. In some embodiments, the linker moiety is stable and provides the means to control and determine the distance between the Kinase Inhibitor moiety and the report moiety. Further, in some embodiments, the linker moiety is selected such that the probe's solubility is maintained. In some embodiments, the linker moiety is a piperazinyl moiety. In further embodiments, a piperazinyl-based linkage is formed by using a piperazine containing compound. In other embodiments, the number and order of units that comprise the linker moiety is selected such that the length between the first component and the second component, as well as the hydrophobic and hydrophilic characteristics of the linker is controlled.
- In the present context, spatial separation means a thermochemically and photochemically non-active distance-making group and in some embodiments is used to join two or more different moieties of the types defined above. In other embodiments, spacers are selected on the basis of a variety of characteristics including their hydrophobicity, hydrophilicity, molecular flexibility and length. The spacer, thus, in some embodiments, comprises a chain of carbon atoms optionally interrupted or terminated with one or more heteroatoms, such as oxygen atoms, nitrogen atoms, and/or sulphur atoms. Thus, in some embodiments, the spacer comprises one or more amide, ester, amino, ether, and/or thioether functionalities, and optionally aromatic or mono/polyunsaturated hydrocarbons, polyoxyethylene such as polyethylene glycol, oligo/polyamides such as poly-α-alanine, polyglycine, polylysine, and peptides in general, oligosaccharides, oligo/polyphosphates. Moreover, in other embodiments, the spacer consists of combined units thereof. In further embodiments, the length of the spacer varies, taking into consideration the desired or necessary positioning and spatial orientation of the active/functional part of the Activity Probe.
- Without limiting the scope of the compositions described herein, in some embodiments the reporter moiety is Bodipy. In the present context, the term reporter moiety means a group which is detectable either by itself or as a part of a detection series.
- In some embodiments, the labeled Activity Probes described herein are purified by one or more procedures including, but are not limited to, affinity chromatography; anion- or cation-exchange chromatography (using, including but not limited to, DEAE SEPHAROSE); chromatography on silica; reverse phase HPLC; gel filtration (using, including but not limited to, SEPHADEX G-75); hydrophobic interaction chromatography; size-exclusion chromatography, metal-chelate chromatography; ultrafiltration/diafiltration; ethanol precipitation; ammonium sulfate precipitation; chromatofocusing; displacement chromatography; electrophoretic procedures (including but not limited to preparative isoelectric focusing), differential solubility (including but not limited to ammonium sulfate precipitation), or extraction. In other embodiments, apparent molecular weight is estimated by GPC by comparison to globular protein standards (P
ROTEIN PURIFICATION METHODS, A PRACTICAL APPROACH (Harris & Angal, Eds.) IRL Press 1989, 293-306). - In one aspect, the in vitro inhibitory potency of a probe against a panel of selected Kinases as a rapid means of confirming accessibility of the reactive moiety to the Kinase active site is tested. By way of example only, although less potent than the
parent Compound 2, the illustrative probe ofCompound 3 retains potency against Btk (IC50˜90 nM). Thus, the piperazine linker and bodipy fluorophore do not seriously compromise accessibility of the illustrative probe to the enzyme active site. - The Activity Probes described herein label kinases at the non-catalytic Cys 481 (or a homologous cysteine) and that in some embodiments, probe labeling does not require the catalytic machinery per se. As such it differs from canonical activity-based probes that target the enzyme catalytic machinery directly. In some embodiments, the Kinase undergoes a phosphorylation dependent conformational change that is tightly coupled to ATP binding and kinase activation. In some embodiments, effective labeling by a probe requires the Kinase to be in its active conformation in order to directly detect Kinase activity in cells. In other embodiments, effective labeling by a Activity Probe does not require the Kinase to be in its active conformation in order to directly detect Kinase activity in cells.
- Described herein are methods, compositions, uses and medicaments for the treatment of disorders comprising administering to a patient in need an irreversible inhibitor of an ACK. In some embodiments, the ACK is Btk or a Btk homolog. In further embodiments, the ACK is Blk or a Blk homolog. In yet further embodiments, the ACK is tyrosine kinases that share homology with Btk by having a cysteine residue (including a
Cys 481 residue) that can form a covalent bond with the irreversible inhibitor. See, e.g., protein kinases inFIG. 7 . - The methods described herein (which includes uses of a pharmaceutical composition to treat a disease or disorder, or uses of a compound to form a medicament for treating a disease or disorder) include administering to a subject in need a composition containing a therapeutically effective amount of one or more irreversible Btk inhibitor compounds described herein. Without being bound by theory, the diverse roles played by Btk signaling in various hematopoietic cell functions, e.g., B-cell receptor activation, show that small molecule Btk inhibitors are useful for reducing the risk of or treating a variety of diseases affected by or affecting many cell types of the hematopoietic lineage including, e.g., autoimmune diseases, heteroimmune conditions or diseases, inflammatory diseases, cancer (e.g., B-cell proliferative disorders), and thromboembolic disorders.
- In some embodiments, are methods for treating an autoimmune disease or condition comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Such an autoimmune disease or condition includes, but is not limited to, rheumatoid arthritis, psoriatic arthritis, osteoarthritis, Still's disease, juvenile arthritis, lupus, diabetes, myasthenia gravis, Hashimoto's thyroiditis, Ord's thyroiditis, Graves' disease Sjögren's syndrome, multiple sclerosis, Guillain-Barre syndrome, acute disseminated encephalomyelitis, Addison's disease, opsoclonus-myoclonus syndrome, ankylosing spondylitisis, antiphospholipid antibody syndrome, aplastic anemia, autoimmune hepatitis, coeliac disease, Goodpasture's syndrome, idiopathic thrombocytopenic purpura, optic neuritis, scleroderma, primary biliary cirrhosis, Reiter's syndrome, Takayasu's arteritis, temporal arteritis, warm autoimmune hemolytic anemia, Wegener's granulomatosis, psoriasis, alopecia universalis, Behcet's disease, chronic fatigue, dysautonomia, endometriosis, interstitial cystitis, neuromyotonia, scleroderma, and vulvodynia. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor. In some embodiments, the autoimmune disease is selected from rheumatoid arthritis or lupus.
- In some embodiments, are methods for treating a heteroimmune disease or condition comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Such a heteroimmune condition or disease includes, but is not limited to graft versus host disease, transplantation, transfusion, anaphylaxis, allergies (e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or cockroach calyx), type I hypersensitivity, allergic conjunctivitis, allergic rhinitis, and atopic dermatitis. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- In some embodiments, are methods for treating a cancer comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Such a cancer, e.g., B-cell proliferative disorders, includes but is not limited to diffuse large B cell lymphoma, follicular lymphoma, chronic lymphocytic lymphoma, chronic lymphocytic leukemia, B-cell prolymphocytic leukemia, lymphoplasmacytic lymphoma/Waldenström macroglobulinemia, splenic marginal zone lymphoma, plasma cell myeloma, plasmacytoma, extranodal marginal zone B cell lymphoma, nodal marginal zone B cell lymphoma, mantle cell lymphoma, mediastinal (thymic) large B cell lymphoma, intravascular large B cell lymphoma, primary effusion lymphoma, burkitt lymphoma/leukemia, and lymphomatoid granulomatosis. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor. In some embodiments, the cancer is a B-cell proliferative disorder. In further embodiments, the B-cell proliferative disorder is diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia.
- In some embodiments, are methods for treating mastocytosis comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Mastocytosis includes but is not limited to diseases characterized by hyperactive mast cells. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- In some embodiments, are methods for treating osteoporosis or bone resorption disorders comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Bone resorption disorders include but are not limited to Paget's disease of bone, osteoporosis, and the bone changes secondary to cancer, such as occur in myeloma and metastases from breast cancer. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- In some embodiments, are methods for treating inflammatory diseases comprising administering to a patient in need a pharmaceutical formulation of any irreversible inhibitor of Btk (or a Btk homolog) of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Inflammatory diseases include but are not limited to asthma, inflammatory bowel disease, appendicitis, blepharitis, bronchiolitis, bronchitis, bursitis, cervicitis, cholangitis, cholecystitis, colitis, conjunctivitis, cystitis, dacryoadenitis, dermatitis, dermatomyositis, encephalitis, endocarditis, endometritis, enteritis, enterocolitis, epicondylitis, epididymitis, fasciitis, fibrositis, gastritis, gastroenteritis, hepatitis, hidradenitis suppurativa, laryngitis, mastitis, meningitis, myelitis myocarditis, myositis, nephritis, oophoritis, orchitis, osteitis, otitis, pancreatitis, parotitis, pericarditis, peritonitis, pharyngitis, pleuritis, phlebitis, pneumonitis, pneumonia, proctitis, prostatitis, pyelonephritis, rhinitis, salpingitis, sinusitis, stomatitis, synovitis, tendonitis, tonsillitis, uveitis, vaginitis, vasculitis, and vulvitis. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- In further embodiments are methods for treating lupus comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- In still further embodiments are methods for treating a heteroimmune disease or condition comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog. Such a heteroimmune condition or disease includes, but is not limited to graft versus host disease, transplantation, transfusion, anaphylaxis, allergies (e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or cockroach calyx), type I hypersensitivity, allergic conjunctivitis, allergic rhinitis, and atopic dermatitis.
- In still further embodiments are methods for treating diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- In still further embodiments are methods for treating mastocytosis, comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- In still further embodiments are methods for treating osteoporosis or bone resorption disorders comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- In still further embodiments are methods for treating an inflammatory disease or condition comprising administering to a subject in need thereof a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Bruton's tyrosine kinase or a Bruton's tyrosine kinase homolog.
- In further embodiments are methods for treating an autoimmune disease or condition comprising administering to a patient in need a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Blk or a Blk homolog. Suitable compounds include compounds of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Such an autoimmune disease or condition includes, but is not limited to, rheumatoid arthritis, psoriatic arthritis, osteoarthritis, Still's disease, juvenile arthritis, lupus, diabetes, myasthenia gravis, Hashimoto's thyroiditis, Ord's thyroiditis, Graves' disease Sjögren's syndrome, multiple sclerosis, Guillain-Barré syndrome, acute disseminated encephalomyelitis, Addison's disease, opsoclonus-myoclonus syndrome, ankylosing spondylitisis, antiphospholipid antibody syndrome, aplastic anemia, autoimmune hepatitis, coeliac disease, Goodpasture's syndrome, idiopathic thrombocytopenic purpura, optic neuritis, scleroderma, primary biliary cirrhosis, Reiter's syndrome, Takayasu's arteritis, temporal arteritis, warm autoimmune hemolytic anemia, Wegener's granulomatosis, psoriasis, alopecia universalis, Behcet's disease, chronic fatigue, dysautonomia, endometriosis, interstitial cystitis, neuromyotonia, scleroderma, and vulvodynia. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor. In some embodiments, the autoimmune disease is selected from rheumatoid arthritis or lupus.
- In further embodiments are methods for treating a B-cell proliferative disorder comprising administering to a patient in need a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Blk or a Blk homolog. Suitable compounds include compounds of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Such a B-cell proliferative disorder includes diffuse large B cell lymphoma, follicular lymphoma or chronic lymphocytic leukemia. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- In further embodiments are methods for treating an inflammatory disease or condition comprising administering to a patient in need a composition containing a therapeutically effective amount of a compound that forms a covalent bond with a cysteine sidechain of a Blk or a Blk homolog. Suitable compounds include compounds of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). Inflammatory diseases include but are not limited to asthma, inflammatory bowel disease, appendicitis, blepharitis, bronchiolitis, bronchitis, bursitis, cervicitis, cholangitis, cholecystitis, colitis, conjunctivitis, cystitis, dacryoadenitis, dermatitis, dermatomyositis, encephalitis, endocarditis, endometritis, enteritis, enterocolitis, epicondylitis, epididymitis, fasciitis, fibrositis, gastritis, gastroenteritis, hepatitis, hidradenitis suppurativa, laryngitis, mastitis, meningitis, myelitis myocarditis, myositis, nephritis, oophoritis, orchitis, osteitis, otitis, pancreatitis, parotitis, pericarditis, peritonitis, pharyngitis, pleuritis, phlebitis, pneumonitis, pneumonia, proctitis, prostatitis, pyelonephritis, rhinitis, salpingitis, sinusitis, stomatitis, synovitis, tendonitis, tonsillitis, uveitis, vaginitis, vasculitis, and vulvitis. In some embodiments, any of the compounds presented in Table 2 of Example 1c in the “Kinase Discovery Platform and Pulse Dosing” section of the Examples is the aforementioned irreversible inhibitor.
- Further, the irreversible Btk inhibitor compounds described herein can be used to inhibit a small subset of other tyrosine kinases that share homology with Btk by having a cysteine residue (including a
Cys 481 residue) that can form a covalent bond with the irreversible inhibitor. See, e.g., protein kinases inFIG. 7 . Thus, a subset of tyrosine kinases other than Btk are also expected to be useful as therapeutic targets in a number of health conditions, including: -
- autoimmune diseases, which include, but are not limited to, rheumatoid arthritis, psoriatic arthritis, osteoarthritis, Still's disease, juvenile arthritis, lupus, diabetes, myasthenia gravis, Hashimoto's thyroiditis, Ord's thyroiditis, Graves' disease Sjögren's syndrome, multiple sclerosis, Guillain-Barré syndrome, acute disseminated encephalomyelitis, Addison's disease, opsoclonus-myoclonus syndrome, ankylosing spondylitisis, antiphospholipid antibody syndrome, aplastic anemia, autoimmune hepatitis, coeliac disease, Goodpasture's syndrome, idiopathic thrombocytopenic purpura, optic neuritis, scleroderma, primary biliary cirrhosis, Reiter's syndrome, Takayasu's arteritis, temporal arteritis, warm autoimmune hemolytic anemia, Wegener's granulomatosis, psoriasis, alopecia universalis, Behcet's disease, chronic fatigue, dysautonomia, endometriosis, interstitial cystitis, neuromyotonia, scleroderma, and vulvodynia.
- heteroimmune conditions or diseases, which include, but are not limited to graft versus host disease, transplantation, transfusion, anaphylaxis, allergies (e.g., allergies to plant pollens, latex, drugs, foods, insect poisons, animal hair, animal dander, dust mites, or cockroach calyx), type I hypersensitivity, allergic conjunctivitis, allergic rhinitis, and atopic dermatitis.
- inflammatory diseases, which include, but are not limited to asthma, inflammatory bowel disease, appendicitis, blepharitis, bronchiolitis, bronchitis, bursitis, cervicitis, cholangitis, cholecystitis, colitis, conjunctivitis, cystitis, dacryoadenitis, dermatitis, dermatomyositis, encephalitis, endocarditis, endometritis, enteritis, enterocolitis, epicondylitis, epididymitis, fasciitis, fibrositis, gastritis, gastroenteritis, hepatitis, hidradenitis suppurativa, laryngitis, mastitis, meningitis, myelitis myocarditis, myositis, nephritis, oophoritis, orchitis, osteitis, otitis, pancreatitis, parotitis, pericarditis, peritonitis, pharyngitis, pleuritis, phlebitis, pneumonitis, pneumonia, proctitis, prostatitis, pyelonephritis, rhinitis, salpingitis, sinusitis, stomatitis, synovitis, tendonitis, tonsillitis, uveitis, vaginitis, vasculitis, and vulvitis.
- a cancer, e.g., B-cell proliferative disorders, which include, but are not limited to diffuse large B cell lymphoma, follicular lymphoma, chronic lymphocytic lymphoma, chronic lymphocytic leukemia, B-cell prolymphocytic leukemia, lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia, splenic marginal zone lymphoma, plasma cell myeloma, plasmacytoma, extranodal marginal zone B cell lymphoma, nodal marginal zone B cell lymphoma, mantle cell lymphoma, mediastinal (thymic) large B cell lymphoma, intravascular large B cell lymphoma, primary effusion lymphoma, burkitt lymphoma/leukemia, and lymphomatoid granulomatosis.
- thromboembolic disorders, which include, but are not limited to myocardial infarct, angina pectoris (including unstable angina), reocclusions or restenoses after angioplasty or aortocoronary bypass, stroke, transitory ischemia, peripheral arterial occlusive disorders, pulmonary embolisms, and deep venous thromboses.
- mastocytosis, which include but are not limited to diseases characterized by hyperactive mast cells.
- bone resorption disorders, which include but are not limited to Paget's disease of bone, osteoporosis, and the bone changes secondary to cancer, such as occur in myeloma and metastases from breast cancer.
- Symptoms, diagnostic tests, and prognostic tests for each of the above-mentioned conditions included, e.g., Harrison's Principles of Internal Medicine©,” 16th ed., 2004, The McGraw-Hill Companies, Inc. Dey et al. (2006), Cytojournal 3(24), and the “Revised European American Lymphoma” (REAL) classification system (see, e.g., the website maintained by the National Cancer Institute).
- A number of animal models are useful for establishing a range of therapeutically effective doses of irreversible inhibitors, including irreversible Btk inhibitor compounds for treating any of the foregoing diseases. For example, refer to Examples 1-4 of the “Therapeutic Uses” section of the Examples included herein. Also, for example, dosing of irreversible inhibitor compounds for treating an autoimmune disease can be assessed in a mouse model of rheumatoid arthitis. In this model, arthritis is induced in Balb/c mice by administering anti-collagen antibodies and lipopolysaccharide. See Nandakumar et al. (2003), Am. J. Pathol 163:1827-1837. In another example, dosing of irreversible inhibitors for the treatment of B-cell proliferative disorders can be examined in, e.g., a human-to-mouse xenograft model in which human B-cell lymphoma cells (e.g. Ramos cells) are implanted into immunodefficient mice (e.g., “nude” mice) as described in, e.g., Pagel et al. (2005), Clin Cancer Res 11(13):4857-4866. Animal models for treatment of thromboembolic disorders are also known.
- In one embodiment, the therapeutic efficacy of the compound for one of the foregoing diseases is optimized during a course of treatment. For example, a subject being treated optionally undergoes a diagnostic evaluation to correlate the relief of disease symptoms or pathologies to inhibition of in vivo Btk activity achieved by administering a given dose of an irreversible Btk inhibitor. Cellular assays are used to determine in vivo activity of Btk in the presence or absence of an irreversible Btk inhibitor. For example, since activated Btk is phosphorylated at tyrosine 223 (Y223) and tyrosine 551 (Y551), phospho-specific immunocytochemical staining of P-Y223 or P-Y551-positive cells are used to detect or quantify activation of Bkt in a population of cells (e.g., by FACS analysis of stained vs unstained cells). See, e.g., Nisitani et al. (1999), Proc. Natl. Acad. Sci, USA 96:2221-2226. Thus, the amount of the Btk inhibitor inhibitor compound that is administered to a subject is optionally increased or decreased as needed so as to maintain a level of Btk inhibition optimal for treating the subject's disease state.
- In one embodiment are methods for identifying biomarkers suitable for determining patient response to an irreversible ACK inhibitor (including, e.g., a compound of Formula (I)) comprising administering to a test subject a composition containing an amount of the irreversible ACK inhibitor (including, e.g., a compound of Formula (I)) sufficient to inhibit B cell receptor signaling and correlating B cell receptor signaling with apoptosis. In another or further embodiment are methods for selecting a patient for treatment for lymphoma with an irreversible ACK inhibitor (including, e.g., a compound of Formula (I)) comprising measuring pErk or Erk transcriptional target levels in a patient sample, and correlating a high level of transcriptional targets with a positive response to the treatment. In another or further embodiments are methods for measuring a patient's response to treatment comprising administering to the patient an irreversible ACK inhibitor (including, e.g., a compound of Formula (I)), measuring pErk or Erk transcriptional target levels in a patient sample, and correlating a reduced level of transcriptional targets with a positive response to the administration of the irreversible ACK inhibitor (including, e.g., a compound of Formula (I)).
- The irreversible Btk inhibitor compositions described herein can also be used in combination with other well known therapeutic reagents that are selected for their therapeutic value for the condition to be treated. In general, the compositions described herein and, in embodiments where combinational therapy is employed, other agents do not have to be administered in the same pharmaceutical composition, and are optionally, because of different physical and chemical characteristics, have to be administered by different routes. The initial administration is made, for example, according to established protocols, and then, based upon the observed effects, the dosage, modes of administration and times of administration are modified.
- In certain instances, it is appropriate to administer at least one irreversible Btk inhibitor compound described herein in combination with another therapeutic agent. By way of example only, if one of the side effects experienced by a patient upon receiving one of the irreversible Btk inhibitor compounds described herein is nausea, then it is appropriate to administer an anti-nausea agent in combination with the initial therapeutic agent. Or, by way of example only, the therapeutic effectiveness of one of the compounds described herein is enhanced by administration of an adjuvant (i.e., by itself the adjuvant has minimal therapeutic benefit, but in combination with another therapeutic agent, the overall therapeutic benefit to the patient is enhanced). Or, by way of example only, the benefit experienced by a patient is increased by administering one of the compounds described herein with another therapeutic agent (which also includes a therapeutic regimen) that also has therapeutic benefit. In any case, regardless of the disease, disorder or condition being treated, the overall benefit experienced by the patient is in some embodiments simply additive of the two therapeutic agents or in other embodiments, the patient experiences a synergistic benefit.
- The particular choice of compounds used will depend upon the diagnosis of the attending physicians and their judgment of the condition of the patient and the appropriate treatment protocol. The compounds are optionally administered concurrently (e.g., simultaneously, essentially simultaneously or within the same treatment protocol) or sequentially, depending upon the nature of the disease, disorder, or condition, the condition of the patient, and the actual choice of compounds used. The determination of the order of administration, and the number of repetitions of administration of each therapeutic agent during a treatment protocol, is based on an evaluation of the disease being treated and the condition of the patient.
- Therapeutically-effective dosages can vary when the drugs are used in treatment combinations. Methods for experimentally determining therapeutically-effective dosages of drugs and other agents for use in combination treatment regimens are described in the literature. For example, the use of metronomic dosing, i.e., providing more frequent, lower doses in order to minimize toxic side effects, has been described extensively in the literature Combination treatment further includes periodic treatments that start and stop at various times to assist with the clinical management of the patient.
- For combination therapies described herein, dosages of the co-administered compounds will of course vary depending on the type of co-drug employed, on the specific drug employed, on the disease or condition being treated and so forth. In addition, when co-administered with one or more biologically active agents, the compound provided herein may be administered either simultaneously with the biologically active agent(s), or sequentially. If administered sequentially, the attending physician will decide on the appropriate sequence of administering protein in combination with the biologically active agent(s).
- In any case, the multiple therapeutic agents (one of which is a compound of Formula (A1-A6), (B1-B6), (C1-C6), or (D1-D6) described herein) are optionally administered in any order or even simultaneously. If simultaneously, the multiple therapeutic agents are optionally provided in a single, unified form, or in multiple forms (by way of example only, either as a single pill or as two separate pills). One of the therapeutic agents may be given in multiple doses, or both may be given as multiple doses. If not simultaneous, the timing between the multiple doses may vary from more than zero weeks to less than four weeks. In addition, the combination methods, compositions and formulations are not to be limited to the use of only two agents; the use of multiple therapeutic combinations are also envisioned.
- It is understood that the dosage regimen to treat, prevent, or ameliorate the condition(s) for which relief is sought, can be modified in accordance with a variety of factors. These factors include the disorder from which the subject suffers, as well as the age, weight, sex, diet, and medical condition of the subject. Thus, the dosage regimen actually employed can vary widely and therefore can deviate from the dosage regimens set forth herein.
- The pharmaceutical agents which make up the combination therapy disclosed herein may be a combined dosage form or in separate dosage forms intended for substantially simultaneous administration. The pharmaceutical agents that make up the combination therapy may also be administered sequentially, with either therapeutic compound being administered by a regimen calling for two-step administration. The two-step administration regimen may call for sequential administration of the active agents or spaced-apart administration of the separate active agents. The time period between the multiple administration steps may range from, a few minutes to several hours, depending upon the properties of each pharmaceutical agent, such as potency, solubility, bioavailability, plasma half-life and kinetic profile of the pharmaceutical agent. Circadian variation of the target molecule concentration may also determine the optimal dose interval.
- In addition, the compounds described herein also are optionally used in combination with procedures that provide additional or synergistic benefit to the patient. By way of example only, patients are expected to find therapeutic and/or prophylactic benefit in the methods described herein, wherein pharmaceutical composition of a compound disclosed herein and/or combinations with other therapeutics are combined with genetic testing to determine whether that individual is a carrier of a mutant gene that is known to be correlated with certain diseases or conditions.
- The compounds described herein and combination therapies can be administered before, during or after the occurrence of a disease or condition, and the timing of administering the composition containing a compound can vary. Thus, for example, the compounds can be used as a prophylactic and can be administered continuously to subjects with a propensity to develop conditions or diseases in order to prevent the occurrence of the disease or condition. The compounds and compositions can be administered to a subject during or as soon as possible after the onset of the symptoms. The administration of the compounds can be initiated within the first 48 hours of the onset of the symptoms, within the first 6 hours of the onset of the symptoms, or within 3 hours of the onset of the symptoms. The initial administration can be via any route practical, such as, for example, an intravenous injection, a bolus injection, infusion over 5 minutes to about 5 hours, a pill, a capsule, transdermal patch, buccal delivery, and the like, or combination thereof. A compound should be administered as soon as is practicable after the onset of a disease or condition is detected or suspected, and for a length of time necessary for the treatment of the disease, such as, for example, from about 1 month to about 3 months. The length of treatment can vary for each subject, and the length can be determined using the known criteria. For example, the compound or a formulation containing the compound can be administered for at least 2 weeks, between about 1 month to about 5 years, or from about 1 month to about 3 years.
- Exemplary Therapeutic Agents for Use in Combination with an Irreversible Inhibitor Compound
- Where the subject is suffering from or at risk of suffering from an autoimmune disease, an inflammatory disease, or an allergy disease, an irreversible Btk inhibitor compound can be used in with one or more of the following therapeutic agents in any combination: immunosuppressants (e.g., tacrolimus, cyclosporin, rapamicin, methotrexate, cyclophosphamide, azathioprine, mercaptopurine, mycophenolate, or FTY720), glucocorticoids (e.g., prednisone, cortisone acetate, prednisolone, methylprednisolone, dexamethasone, betamethasone, triamcinolone, beclometasone, fludrocortisone acetate, deoxycorticosterone acetate, aldosterone), non-steroidal anti-inflammatory drugs (e.g., salicylates, arylalkanoic acids, 2-arylpropionic acids, N-arylanthranilic acids, oxicams, coxibs, or sulphonanilides), Cox-2-specific inhibitors (e.g., valdecoxib, celecoxib, or rofecoxib), leflunomide, gold thioglucose, gold thiomalate, aurofin, sulfasalazine, hydroxychloroquinine, minocycline, TNF-α binding proteins (e.g., infliximab, etanercept, or adalimumab), abatacept, anakinra, interferon-β, interferon-γ, interleukin-2, allergy vaccines, antihistamines, antileukotrienes, beta-agonists, theophylline, anticholinergics or other selective kinase inhibitors (e.g p38 inhibitors, Syk inhibitors, PKC inhibitors).
- Where the subject is suffering from or at risk of suffering from a B-cell proliferative disorder (e.g., plasma cell myeloma), the subjected can be treated with an irreversible Btk inhibitor compound in any combination with one or more other anti-cancer agents. In some embodiments, one or more of the anti-cancer agents are proapoptotic agents. Examples of anti-cancer agents include, but are not limited to, any of the following: gossyphol, genasense, polyphenol E, Chlorofusin, all trans-retinoic acid (ATRA), bryostatin, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), 5-aza-2′-deoxycytidine, all trans retinoic acid, doxorubicin, vincristine, etoposide, gemcitabine, imatinib (Gleevec®), geldanamycin, 17-N-Allylamino-17-Demethoxygeldanamycin (17-AAG), flavopiridol, LY294002, bortezomib, trastuzumab, BAY 11-7082, PKC412, or PD184352, Taxol™, also referred to as “paclitaxel”, which is a well-known anti-cancer drug which acts by enhancing and stabilizing microtubule formation, and analogs of Taxol™, such as Taxotere™. Compounds that have the basic taxane skeleton as a common structure feature, have also been shown to have the ability to arrest cells in the G2-M phases due to stabilized microtubules and may be useful for treating cancer in combination with the compounds described herein.
- Further examples of anti-cancer agents for use in combination with an irreversible Btk inhibitor compound include inhibitors of mitogen-activated protein kinase signaling, e.g., U0126, PD98059, PD184352, PD0325901, ARRY-142886, SB239063, SP600125, BAY 43-9006, wortmannin, or LY294002; Syk inhibitors; mTOR inhibitors; and antibodies (e.g., rituxan).
- Other anti-cancer agents that can be employed in combination with an irreversible Btk inhibitor compound include Adriamycin, Dactinomycin, Bleomycin, Vinblastine, Cisplatin, acivicin; aclarubicin; acodazole hydrochloride; acronine; adozelesin; aldesleukin; altretamine; ambomycin; ametantrone acetate; aminoglutethimide; amsacrine; anastrozole; anthramycin; asparaginase; asperlin; azacitidine; azetepa; azotomycin; batimastat; benzodepa; bicalutamide; bisantrene hydrochloride; bisnafide dimesylate; bizelesin; bleomycin sulfate; brequinar sodium; bropirimine; busulfan; cactinomycin; calusterone; caracemide; carbetimer; carboplatin; carmustine; carubicin hydrochloride; carzelesin; cedefingol; chlorambucil; cirolemycin; cladribine; crisnatol mesylate; cyclophosphamide; cytarabine; dacarbazine; daunorubicin hydrochloride; decitabine; dexormaplatin; dezaguanine; dezaguanine mesylate; diaziquone; doxorubicin; doxorubicin hydrochloride; droloxifene; droloxifene citrate; dromostanolone propionate; duazomycin; edatrexate; eflornithine hydrochloride; elsamitrucin; enloplatin; enpromate; epipropidine; epirubicin hydrochloride; erbulozole; esorubicin hydrochloride; estramustine; estramustine phosphate sodium; etanidazole; etoposide; etoposide phosphate; etoprine; fadrozole hydrochloride; fazarabine; fenretinide; floxuridine; fludarabine phosphate; fluorouracil; flurocitabine; fosquidone; fostriecin sodium; gemcitabine; gemcitabine hydrochloride; hydroxyurea; idarubicin hydrochloride; ifosfamide; iimofosine; interleukin Il (including recombinant interleukin II, or rlL2), interferon alfa-2a; interferon alfa-2b; interferon alfa-n1; interferon alfa-n3; interferon beta-1 a; interferon gamma-1 b; iproplatin; irinotecan hydrochloride; lanreotide acetate; letrozole; leuprolide acetate; liarozole hydrochloride; lometrexol sodium; lomustine; losoxantrone hydrochloride; masoprocol; maytansine; mechlorethamine hydrochloride; megestrol acetate; melengestrol acetate; melphalan; menogaril; mercaptopurine; methotrexate; methotrexate sodium; metoprine; meturedepa; mitindomide; mitocarcin; mitocromin; mitogillin; mitomalcin; mitomycin; mitosper; mitotane; mitoxantrone hydrochloride; mycophenolic acid; nocodazoie; nogalamycin; ormaplatin; oxisuran; pegaspargase; peliomycin; pentamustine; peplomycin sulfate; perfosfamide; pipobroman; piposulfan; piroxantrone hydrochloride; plicamycin; plomestane; porfimer sodium; porfiromycin; prednimustine; procarbazine hydrochloride; puromycin; puromycin hydrochloride; pyrazofurin; riboprine; rogletimide; safingol; safingol hydrochloride; semustine; simtrazene; sparfosate sodium; sparsomycin; spirogermanium hydrochloride; spiromustine; spiroplatin; streptonigrin; streptozocin; sulofenur; talisomycin; tecogalan sodium; tegafur; teloxantrone hydrochloride; temoporfin; teniposide; teroxirone; testolactone; thiamiprine; thioguanine; thiotepa; tiazofurin; tirapazamine; toremifene citrate; trestolone acetate; triciribine phosphate; trimetrexate; trimetrexate glucuronate; triptorelin; tubulozole hydrochloride; uracil mustard; uredepa; vapreotide; verteporfin; vinblastine sulfate; vincristine sulfate; vindesine; vindesine sulfate; vinepidine sulfate; vinglycinate sulfate; vinleurosine sulfate; vinorelbine tartrate; vinrosidine sulfate; vinzolidine sulfate; vorozole; zeniplatin; zinostatin; zorubicin hydrochloride.
- Other anti-cancer agents that can be employed in combination with an irreversible Btk inhibitor compound include: 20-epi-1, 25 dihydroxyvitamin D3; 5-ethynyluracil; abiraterone; aclarubicin; acylfulvene; adecypenol; adozelesin; aldesleukin; ALL-TK antagonists; altretamine; ambamustine; amidox; amifostine; aminolevulinic acid; amrubicin; amsacrine; anagrelide; anastrozole; andrographolide; angiogenesis inhibitors; antagonist D; antagonist G; antarelix; anti-dorsalizing morphogenetic protein-1; antiandrogen, prostatic carcinoma; antiestrogen; antineoplaston; antisense oligonucleotides; aphidicolin glycinate; apoptosis gene modulators; apoptosis regulators; apurinic acid; ara-CDP-DL-PTBA; arginine deaminase; asulacrine; atamestane; atrimustine; axinastatin 1; axinastatin 2; axinastatin 3; azasetron; azatoxin; azatyrosine; baccatin III derivatives; balanol; batimastat; BCR/ABL antagonists; benzochlorins; benzoylstaurosporine; beta lactam derivatives; beta-alethine; betaclamycin B; betulinic acid; bFGF inhibitor; bicalutamide; bisantrene; bisaziridinylspermine; bisnafide; bistratene A; bizelesin; breflate; bropirimine; budotitane; buthionine sulfoximine; calcipotriol; calphostin C; camptothecin derivatives; canarypox IL-2; capecitabine; carboxamide-amino-triazole; carboxyamidotriazole; CaRest M3; CARN 700; cartilage derived inhibitor; carzelesin; casein kinase inhibitors (ICOS); castanospermine; cecropin B; cetrorelix; chlorlns; chloroquinoxaline sulfonamide; cicaprost; cis-porphyrin; cladribine; clomifene analogues; clotrimazole; collismycin A; collismycin B; combretastatin A4; combretastatin analogue; conagenin; crambescidin 816; crisnatol; cryptophycin 8; cryptophycin A derivatives; curacin A; cyclopentanthraquinones; cycloplatam; cypemycin; cytarabine ocfosfate; cytolytic factor; cytostatin; dacliximab; decitabine; dehydrodidemnin B; deslorelin; dexamethasone; dexifosfamide; dexrazoxane; dexverapamil; diaziquone; didemnin B; didox; diethylnorspermine; dihydro-5-azacytidine; 9-dioxamycin; diphenyl spiromustine; docosanol; dolasetron; doxifluridine; droloxifene; dronabinol; duocarmycin SA; ebselen; ecomustine; edelfosine; edrecolomab; eflornithine; elemene; emitefur; epirubicin; epristeride; estramustine analogue; estrogen agonists; estrogen antagonists; etanidazole; etoposide phosphate; exemestane; fadrozole; fazarabine; fenretinide; filgrastim; finasteride; flavopiridol; flezelastine; fluasterone; fludarabine; fluorodaunorunicin hydrochloride; forfenimex; formestane; fostriecin; fotemustine; gadolinium texaphyrin; gallium nitrate; galocitabine; ganirelix; gelatinase inhibitors; gemcitabine; glutathione inhibitors; hepsulfam; heregulin; hexamethylene bisacetamide; hypericin; ibandronic acid; idarubicin; idoxifene; idramantone; ilmofosine; ilomastat; imidazoacridones; imiquimod; immunostimulant peptides; insulin-like growth factor-1 receptor inhibitor; interferon agonists; interferons; interleukins; iobenguane; iododoxorubicin; ipomeanol, 4-; iroplact; irsogladine; isobengazole; isohomohalicondrin B; itasetron; jasplakinolide; kahalalide F; lamellarin-N triacetate; lanreotide; leinamycin; lenograstim; lentinan sulfate; leptolstatin; letrozole; leukemia inhibiting factor; leukocyte alpha interferon; leuprolide+estrogen+progesterone; leuprorelin; levamisole; liarozole; linear polyamine analogue; lipophilic disaccharide peptide; lipophilic platinum compounds; lissoclinamide 7; lobaplatin; lombricine; lometrexol; lonidamine; losoxantrone; lovastatin; loxoribine; lurtotecan; lutetium texaphyrin; lysofylline; lytic peptides; maitansine; mannostatin A; marimastat; masoprocol; maspin; matrilysin inhibitors; matrix metalloproteinase inhibitors; menogaril; merbarone; meterelin; methioninase; metoclopramide; MIF inhibitor; mifepristone; miltefosine; mirimostim; mismatched double stranded RNA; mitoguazone; mitolactol; mitomycin analogues; mitonafide; mitotoxin fibroblast growth factor-saporin; mitoxantrone; mofarotene; molgramostim; monoclonal antibody, human chorionic gonadotrophin; monophosphoryl lipid A+myobacterium cell wall sk; mopidamol; multiple drug resistance gene inhibitor; multiple tumor suppressor 1-based therapy; mustard anticancer agent; mycaperoxide B; mycobacterial cell wall extract; myriaporone; N-acetyldinaline; N-substituted benzamides; nafarelin; nagrestip; naloxone+pentazocine; napavin; naphterpin; nartograstim; nedaplatin; nemorubicin; neridronic acid; neutral endopeptidase; nilutamide; nisamycin; nitric oxide modulators; nitroxide antioxidant; nitrullyn; O6-benzylguanine; octreotide; okicenone; oligonucleotides; onapristone; ondansetron; ondansetron; oracin; oral cytokine inducer; ormaplatin; osaterone; oxaliplatin; oxaunomycin; palauamine; palmitoylrhizoxin; pamidronic acid; panaxytriol; panomifene; parabactin; pazelliptine; pegaspargase; peldesine; pentosan polysulfate sodium; pentostatin; pentrozole; perflubron; perfosfamide; perillyl alcohol; phenazinomycin; phenylacetate; phosphatase inhibitors; picibanil; pilocarpine hydrochloride; pirarubicin; piritrexim; placetin A; placetin B; plasminogen activator inhibitor; platinum complex; platinum compounds; platinum-triamine complex; porfimer sodium; porfiromycin; prednisone; propyl bis-acridone; prostaglandin J2; proteasome inhibitors; protein A-based immune modulator; protein kinase C inhibitor; protein kinase C inhibitors, microalgal; protein tyrosine phosphatase inhibitors; purine nucleoside phosphorylase inhibitors; purpurins; pyrazoloacridine; pyridoxylated hemoglobin polyoxyethylerie conjugate; raf antagonists; raltitrexed; ramosetron; ras farnesyl protein transferase inhibitors; ras inhibitors; ras-GAP inhibitor; retelliptine demethylated; rhenium Re 186 etidronate; rhizoxin; ribozymes; RII retinamide; rogletimide; rohitukine; romurtide; roquinimex; rubiginone B1; ruboxyl; safingol; saintopin; SarCNU; sarcophytol A; sargramostim; Sdi 1 mimetics; semustine; senescence derived inhibitor 1; sense oligonucleotides; signal transduction inhibitors; signal transduction modulators; single chain antigen-binding protein; sizofiran; sobuzoxane; sodium borocaptate; sodium phenylacetate; solverol; somatomedin binding protein; sonermin; sparfosic acid; spicamycin D; spiromustine; splenopentin; spongistatin 1; squalamine; stem cell inhibitor; stem-cell division inhibitors; stipiamide; stromelysin inhibitors; sulfinosine; superactive vasoactive intestinal peptide antagonist; suradista; suramin; swainsonine; synthetic glycosaminoglycans; tallimustine; tamoxifen methiodide; tauromustine; tazarotene; tecogalan sodium; tegafur; tellurapyrylium; telomerase inhibitors; temoporfin; temozolomide; teniposide; tetrachlorodecaoxide; tetrazomine; thaliblastine; thiocoraline; thrombopoietin; thrombopoietin mimetic; thymalfasin; thymopoietin receptor agonist; thymotrinan; thyroid stimulating hormone; tin ethyl etiopurpurin; tirapazamine; titanocene bichloride; topsentin; toremifene; totipotent stem cell factor; translation inhibitors; tretinoin; triacetyluridine; triciribine; trimetrexate; triptorelin; tropisetron; turosteride; tyrosine kinase inhibitors; tyrphostins; UBC inhibitors; ubenimex; urogenital sinus-derived growth inhibitory factor; urokinase receptor antagonists; vapreotide; variolin B; vector system, erythrocyte gene therapy; velaresol; veramine; verdins; verteporfin; vinorelbine; vinxaltine; vitaxin; vorozole; zanoterone; zeniplatin; zilascorb; and zinostatin stimalamer.
- Yet other anticancer agents that can be employed in combination with an irreversible Btk inhibitor compound include alkylating agents, antimetabolites, natural products, or hormones, e.g., nitrogen mustards (e.g., mechloroethamine, cyclophosphamide, chlorambucil, etc.), alkyl sulfonates (e.g., busulfan), nitrosoureas (e.g., carmustine, lomusitne, ete.), or triazenes (decarbazine, etc.). Examples of antimetabolites include but are not limited to folic acid analog (e.g., methotrexate), or pyrimidine analogs (e.g., Cytarabine), purine analogs (e.g., mercaptopurine, thioguanine, pentostatin).
- Examples of natural products useful in combination with an irreversible Btk inhibitor compound include but are not limited to vinca alkaloids (e.g., vinblastin, vincristine), epipodophyllotoxins (e.g., etoposide), antibiotics (e.g., daunorubicin, doxorubicin, bleomycin), enzymes (e.g., L-asparaginase), or biological response modifiers (e.g., interferon alpha).
- Examples of alkylating agents that can be employed in combination an irreversible Btk inhibitor compound include, but are not limited to, nitrogen mustards (e.g., mechloroethamine, cyclophosphamide, chlorambucil, meiphalan, etc.), ethylenimine and methylmelamines (e.g., hexamethlymelamine, thiotepa), alkyl sulfonates (e.g., busulfan), nitrosoureas (e.g., carmustine, lomusitne, semustine, streptozocin, etc.), or triazenes (decarbazine, ete.). Examples of antimetabolites include, but are not limited to folic acid analog (e.g., methotrexate), or pyrimidine analogs (e.g., fluorouracil, floxouridine, Cytarabine), purine analogs (e.g., mercaptopurine, thioguanine, pentostatin.
- Examples of hormones and antagonists useful in combination with an irreversible Btk inhibitor compound include, but are not limited to, adrenocorticosteroids (e.g., prednisone), progestins (e.g., hydroxyprogesterone caproate, megestrol acetate, medroxyprogesterone acetate), estrogens (e.g., diethlystilbestrol, ethinyl estradiol), antiestrogen (e.g., tamoxifen), androgens (e.g., testosterone propionate, fluoxymesterone), antiandrogen (e.g., flutamide), gonadotropin releasing hormone analog (e.g., leuprolide). Other agents that can be used in the methods and compositions described herein for the treatment or prevention of cancer include platinum coordination complexes (e.g., cisplatin, carboblatin), anthracenedione (e.g., mitoxantrone), substituted urea (e.g., hydroxyurea), methyl hydrazine derivative (e.g., procarbazine), adrenocortical suppressant (e.g., mitotane, aminoglutethimide).
- Examples of anti-cancer agents which act by arresting cells in the G2-M phases due to stabilized microtubules and which can be used in combination with an irreversible Btk inhibitor compound include without limitation marketed drugs and drugs in development.
- Where the subject is suffering from or at risk of suffering from a thromboembolic disorder (e.g., stroke), the subject can be treated with an irreversible Btk inhibitor compound in any combination with one or more other anti-thromboembolic agents. Examples of anti-thromboembolic agents include, but are not limited any of the following: thrombolytic agents (e.g., alteplase anistreplase, streptokinase, urokinase, or tissue plasminogen activator), heparin, tinzaparin, warfarin, dabigatran (e.g., dabigatran etexilate), factor Xa inhibitors (e.g., fondaparinux, draparinux, rivaroxaban, DX-9065a, otamixaban, LY517717, or YM150), factor VIIa inhibitors, ticlopidine, clopidogrel, CS-747 (prasugrel, LY640315), ximelagatran, or BIBR 1048.
- Pharmaceutical compositions are formulated in a conventional manner using one or more physiologically acceptable carriers including excipients and auxiliaries which facilitate processing of the active compounds into preparations which can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen. A summary of pharmaceutical compositions described herein is found, for example, in Remington: The Science and Practice of Pharmacy, Nineteenth Ed (Easton, Pa.: Mack Publishing Company, 1995); Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa. 1975; Liberman, H. A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; and Pharmaceutical Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams & Wilkins 1999).
- A pharmaceutical composition, as used herein, refers to a mixture of a compound described herein, such as, for example, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), with other chemical components, such as carriers, stabilizers, diluents, dispersing agents, suspending agents, thickening agents, and/or excipients. The pharmaceutical composition facilitates administration of the compound to an organism. In practicing the methods of treatment or use provided herein, therapeutically effective amounts of compounds described herein are administered in a pharmaceutical composition to a mammal having a disease, disorder, or condition to be treated. Preferably, the mammal is a human. The compounds can be used singly or in combination with one or more therapeutic agents as components of mixtures.
- The pharmaceutical formulations described herein can be administered to a subject by multiple administration routes, including but not limited to, oral, parenteral (e.g., intravenous, subcutaneous, intramuscular), intranasal, buccal, topical, rectal, or transdermal administration routes. The pharmaceutical formulations described herein include, but are not limited to, aqueous liquid dispersions, self-emulsifying dispersions, solid solutions, liposomal dispersions, aerosols, solid dosage forms, powders, immediate release formulations, controlled release formulations, fast melt formulations, tablets, capsules, pills, delayed release formulations, extended release formulations, pulsatile release formulations, multiparticulate formulations, and mixed immediate and controlled release formulations.
- Pharmaceutical compositions including a compound described herein are optionally manufactured in a conventional manner, such as, by way of example only, by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or compression processes.
- The pharmaceutical compositions will include at least one compound described herein, such as, for example, a compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), as an active ingredient in free-acid or free-base form, or in a pharmaceutically acceptable salt form. In addition, the methods and pharmaceutical compositions described herein include the use of N-oxides, crystalline forms (also known as polymorphs), as well as active metabolites of these compounds having the same type of activity. In some situations, compounds may exist as tautomers. All tautomers are included within the scope of the compounds presented herein. Additionally, the compounds described herein can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. The solvated forms of the compounds presented herein are also considered to be disclosed herein.
- A “carrier” or “carrier materials” includes excipients in pharmaceutics and is selected on the basis of compatibility with compounds disclosed herein, such as, compounds of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), and the release profile properties of the desired dosage form. Exemplary carrier materials include, e.g., binders, suspending agents, disintegration agents, filling agents, surfactants, solubilizers, stabilizers, lubricants, wetting agents, diluents, and the like. See, e.g., Remington: The Science and Practice of Pharmacy, Nineteenth Ed (Easton, Pa.: Mack Publishing Company, 1995); Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa. 1975; Liberman, H. A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; and Pharmaceutical Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams & Wilkins 1999).
- A “measurable serum concentration” or “measurable plasma concentration” describes the blood serum or blood plasma concentration, typically measured in mg, μg, or ng of therapeutic agent per ml, dl, or 1 of blood serum, absorbed into the bloodstream after administration. As used herein, measurable plasma concentrations are typically measured in ng/ml or μg/ml.
- “Pharmacodynamics” refers to the factors which determine the biologic response observed relative to the concentration of drug at a site of action. “Pharmacokinetics” refers to the factors which determine the attainment and maintenance of the appropriate concentration of drug at a site of action.
- “Steady state,” as used herein, is when the amount of drug administered is equal to the amount of drug eliminated within one dosing interval resulting in a plateau or constant plasma drug exposure.
- Moreover, the pharmaceutical compositions described herein, which include a compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII) can be formulated into any suitable dosage form, including but not limited to, aqueous oral dispersions, liquids, gels, syrups, elixirs, slurries, suspensions and the like, for oral ingestion by a patient to be treated, solid oral dosage forms, aerosols, controlled release formulations, fast melt formulations, effervescent formulations, lyophilized formulations, tablets, powders, pills, dragees, capsules, delayed release formulations, extended release formulations, pulsatile release formulations, multiparticulate formulations, and mixed immediate release and controlled release formulations.
- The pharmaceutical solid dosage forms described herein optionally include a compound described herein and one or more pharmaceutically acceptable additives such as a compatible carrier, binder, filling agent, suspending agent, flavoring agent, sweetening agent, disintegrating agent, dispersing agent, surfactant, lubricant, colorant, diluent, solubilizer, moistening agent, plasticizer, stabilizer, penetration enhancer, wetting agent, anti-foaming agent, antioxidant, preservative, or one or more combination thereof. In still other aspects, using standard coating procedures, such as those described in Remington's Pharmaceutical Sciences, 20th Edition (2000), a film coating is provided around the formulation of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII). In one embodiment, some or all of the particles of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are coated. In another embodiment, some or all of the particles of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are microencapsulated. In still another embodiment, the particles of the compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), are not microencapsulated and are uncoated.
- The compounds described herein can be used in the preparation of medicaments for the inhibition of Btk or a homolog thereof, or for the treatment of diseases or conditions that benefit, at least in part, from inhibition of Btk or a homolog thereof. In addition, a method for treating any of the diseases or conditions described herein in a subject in need of such treatment, involves administration of pharmaceutical compositions containing at least one compound of any of Formula (A1-A6), Formula (B1-B6), Formula (C1-C6), Formula (D1-D6), Formula (I), or Formula (VII), described herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable N-oxide, pharmaceutically active metabolite, pharmaceutically acceptable prodrug, or pharmaceutically acceptable solvate thereof, in therapeutically effective amounts to said subject.
- The compositions containing the compound(s) described herein can be administered for prophylactic and/or therapeutic treatments. In therapeutic applications, the compositions are administered to a patient already suffering from a disease or condition, in an amount sufficient to cure or at least partially arrest the symptoms of the disease or condition. Amounts effective for this use will depend on the severity and course of the disease or condition, previous therapy, the patient's health status, weight, and response to the drugs, and the judgment of the treating physician.
- In prophylactic applications, compositions containing the compounds described herein are administered to a patient susceptible to or otherwise at risk of a particular disease, disorder or condition. Such an amount is defined to be a “prophylactically effective amount or dose.” In this use, the precise amounts also depend on the patient's state of health, weight, and the like. When used in a patient, effective amounts for this use will depend on the severity and course of the disease, disorder or condition, previous therapy, the patient's health status and response to the drugs, and the judgment of the treating physician.
- In some embodiments, the irreversible kinase inhibitor is administered to the patient on a regular basis, e.g., three times a day, two times a day, once a day, every other day or every 3 days. In other embodiments, the irreversible kinase inhibitor is administered to the patient on an intermittent basis, e.g., twice a day followed by once a day followed by three times a day; or the first two days of every week; or the first, second and third day of a week. In some embodiments, intermittent dosing is as effective as regular dosing. In further or alternative embodiments, the irreversible kinase inhibitor is administered only when the patient exhibits a particular symptom, e.g., the onset of pain, or the onset of a fever, or the onset of an inflammation, or the onset of a skin disorder.
- In the case wherein the patient's condition does not improve, upon the doctor's discretion the administration of the compounds may be administered chronically, that is, for an extended period of time, including throughout the duration of the patient's life in order to ameliorate or otherwise control or limit the symptoms of the patient's disease or condition.
- In the case wherein the patient's status does improve, upon the doctor's discretion the administration of the compounds may be given continuously; alternatively, the dose of drug being administered may be temporarily reduced or temporarily suspended for a certain length of time (i.e., a “drug holiday”). The length of the drug holiday can vary between 2 days and 1 year, including by way of example only, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 10 days, 12 days, 15 days, 20 days, 28 days, 35 days, 50 days, 70 days, 100 days, 120 days, 150 days, 180 days, 200 days, 250 days, 280 days, 300 days, 320 days, 350 days, or 365 days. The dose reduction during a drug holiday may be from 10%-100%, including, by way of example only, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 100%.
- Once improvement of the patient's conditions has occurred, a maintenance dose is administered if necessary. Subsequently, the dosage or the frequency of administration, or both, can be reduced, as a function of the symptoms, to a level at which the improved disease, disorder or condition is retained. Patients can, however, require intermittent treatment on a long-term basis upon any recurrence of symptoms.
- The amount of a given agent that will correspond to such an amount will vary depending upon factors such as the particular compound, disease or condition and its severity, the identity (e.g., weight) of the subject or host in need of treatment, and is determined according to the particular circumstances surrounding the case, including, e.g., the specific agent being administered, the route of administration, the condition being treated, and the subject or host being treated. In general, however, doses employed for adult human treatment will typically be in the range of 0.02-5000 mg per day, or from about 1-1500 mg per day. The desired dose may conveniently be presented in a single dose or as divided doses administered simultaneously (or over a short period of time) or at appropriate intervals, for example as two, three, four or more sub-doses per day.
- The pharmaceutical composition described herein may be in unit dosage forms suitable for single administration of precise dosages. In unit dosage form, the formulation is divided into unit doses containing appropriate quantities of one or more compound. The unit dosage may be in the form of a package containing discrete quantities of the formulation. Non-limiting examples are packaged tablets or capsules, and powders in vials or ampoules. Aqueous suspension compositions can be packaged in single-dose non-reclosable containers. Alternatively, multiple-dose reclosable containers can be used, in which case it is typical to include a preservative in the composition. By way of example only, formulations for parenteral injection may be presented in unit dosage form, which include, but are not limited to ampoules, or in multi-dose containers, with an added preservative.
- The foregoing ranges are merely suggestive, as the number of variables in regard to an individual treatment regime is large, and considerable excursions from these recommended values are not uncommon. Such dosages may be altered depending on a number of variables, not limited to the activity of the compound used, the disease or condition to be treated, the mode of administration, the requirements of the individual subject, the severity of the disease or condition being treated, and the judgment of the practitioner.
- Toxicity and therapeutic efficacy of such therapeutic regimens can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, including, but not limited to, the determination of the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population). The dose ratio between the toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio between LD50 and ED50. Compounds exhibiting high therapeutic indices are preferred. The data obtained from cell culture assays and animal studies can be used in formulating a range of dosage for use in human. The dosage of such compounds lies preferably within a range of circulating concentrations that include the ED50 with minimal toxicity. The dosage may vary within this range depending upon the dosage form employed and the route of administration utilized.
- Described herein are irreversible kinase inhibitors that are selective for one or more ACKs, including a Btk, a Btk homolog, and a Btk kinase cysteine homolog. In some embodiments, the irreversible inhibitors described herein also bind reversibly to other kinases (some of which, in some embodiments, are also ACKs). As a means of enhancing the selectivity profile, such inhibitors are formulated (formulation includes chemical modifications of the inhibitor, use of excipients in a pharmaceutical composition, and combinations thereof) such that the pharmacokinetic profile favors enhanced selectivity of the inhibitors for an ACK over a non-ACK. By way of example only, an ACK is formulated to have a short plasma half-life. In other embodiments, an ACK is formulated to have an extended plasma half-life.
- For example, as shown in the Examples,
Compound 1 and Compound 12 have a short half-life in vivo. In contrast,Compound 7 and Compound 8 have a significantly longer in vivo half-life (FIG. 5 ). Compounds like 1 and 12 are predicted to have enhanced kinase selectivity in vivo because inhibition will be sustained only for those kinases that are irreversibly inhibited. Further, given that the irreversible kinase inhibitors described herein have both reversible (in general to non-ACKs) and irreversible (generally, to ACKs) activities, in vivo properties of absorption, distribution, metabolism and excretion (ADME) are selected in order to optimize the therapeutic index. Specifically, in some embodiments, rapidly cleared compounds cause only brief inhibition of reversibly inhibited targets while maintaining sustained inhibition of irreversibly inhibited targets. Depending on the degree to which sustained inhibition of particular targets results in therapeutic effects or toxicities, we identify compounds with an optimal combination of in vitro selectivity profiles and in vivo ADME properties. - In one embodiment are kinase inhibitors that selectively and irreversibly binds to a protein tyrosine kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog, in which the kinase inhibitor reversibly and non-selectively binds to a multiplicity of protein tyrosine kinases, and further in which the plasma half life of the kinase inhibitor is less than about 4 hours. In such an embodiment, the kinase inhibitor selectively and irreversibly binds to at least one of Btk, Jak3, Blk, Bmx, Tec, and Itk. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Btk. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Jak3. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Tec. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Btk and Tec. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Blk. In a further embodiment, the kinase inhibitor reversibly and non-selectively binds to a multiplicity of src-family protein kinase inhibitors. In a further embodiment, the plasma half life of the kinase inhibitor is less than about 3 hours. In a further embodiment, the plasma half life of the kinase inhibitor is less than about 2 hours.
- In one embodiment are kinase inhibitors that selectively and irreversibly binds to a protein tyrosine kinase selected from Btk, a Btk homolog, and a Btk kinase cysteine homolog, in which the kinase inhibitor reversibly and non-selectively binds to a multiplicity of protein tyrosine kinases, and further in which the plasma half life of the kinase inhibitor is greater than about 12 hours. In such an embodiment, the kinase inhibitor selectively and irreversibly binds to at least one of Btk, Jak3, Blk, Bmx, Tec, and Itk. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Btk. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Jak3. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Tec. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Btk and Tec. In a further embodiment, the kinase inhibitor selectively and irreversibly binds to Blk. In a further embodiment, the kinase inhibitor reversibly and non-selectively binds to a multiplicity of src-family protein kinase inhibitors In a further embodiment, the kinase inhibitor the plasma half life of the kinase inhibitor is greater than about 16 hours.
- In one particular embodiment of any of the aforementioned kinase inhibitors, such kinase inhibitors have the structure of Formula (VII):
-
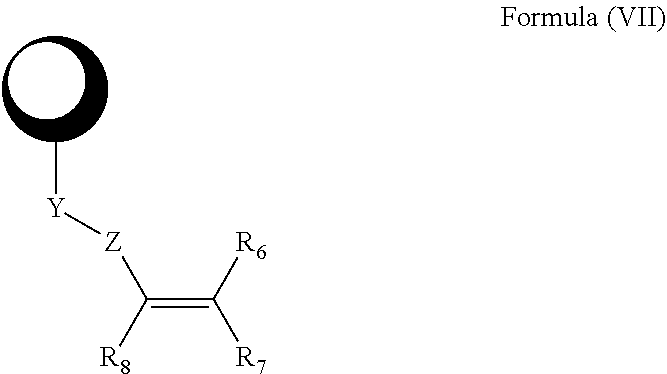
- wherein:
wherein -

- is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
Z is C(═O), OC(═O), NHC(═O), NCH3C(═O), C(═S), S(═O)x, OS(═O)x, NHS(═O)x, where x is 1 or 2;
R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
R7 and R8 taken together form a bond;
R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl); and
pharmaceutically active metabolites, or pharmaceutically acceptable solvates, pharmaceutically acceptable salts, or pharmaceutically acceptable prodrugs thereof. - In a further embodiment,
-

- on the kinase inhibitor is a substituted fused biaryl moiety selected from
-

- In a further embodiment of such kinases:
- Z is C(═O), NHC(═O), NCH3C(═O), or S(═O)2.
The kinase inhibitor of Claim 49, wherein:
each of R7 and R8 is H; or
R7 and R8 taken together form a bond. - In a further embodiment of such kinases:
- R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl).
- In a further embodiment of such kinases:
- Y is a 4-, 5-, 6-, or 7-membered cycloalkylene ring; or
Y is a 4-, 5-, 6-, or 7-membered heterocycloalkylene ring; or
Y is a C1-C4 alkylene, or 4-, 5-, 6-, or 7-membered heterocycloalkylene ring. - In another aspect of such dosing methods are pharmaceutical formulations comprising any of the aforementioned ACK inhibitors and a pharmaceutically acceptable excipient. In some embodiments, such pharmaceutical formulations are formulated for a route of administration selected from oral administration, parenteral administration, buccal administration, nasal administration, topical administration, or rectal administration. In certain embodiments, the pharmaceutical formulations are formulated for oral administration.
- In another aspect of such dosing methods are methods for treating rheumatoid arthritis comprising administering to a subject any of the aforementioned ACK inhibitors that selectively and irreversibly binds to Btk and Tec.
- In yet another aspect of such dosing strategies are methods for increasing the selectivity of a test protein tyrosine kinase inhibitor that irreversibly and selectively binds to at least one protein kinase inhibitor selected from Btk, a Btk homolog, or a Btk kinase cysteine homolog, in which the test protein tyrosine kinase inhibitor is chemically modified to decrease the plasma half life to less than about 4 hours. In some embodiments, the test protein tyrosine kinase inhibitor is chemically modified to decrease the plasma half life to less than about 3 hours.
- In further embodiments, the test protein tyrosine kinase inhibitor has the structure of Formula (VII):
- wherein:
-

- wherein
-

- is a moiety that binds to the active site of a kinase, including a tyrosine kinase, further including a Btk kinase cysteine homolog;
Y is an optionally substituted group selected from among alkylene, heteroalkylene, arylene, heteroarylene, heterocycloalkylene, cycloalkylene, alkylenearylene, alkyleneheteroarylene, alkylenecycloalkylene, and alkyleneheterocycloalkylene;
Z is C(═O), OC(═O), NHC(═O), NCH3C(═O), C(═S), S(═O)x, OS(═O)x, NHS(═O)x, where x is 1 or 2;
R7 and R8 are independently selected from among H, unsubstituted C1-C4 alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
R7 and R8 taken together form a bond; and
R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, C1-C8hydroxyalkylaminoalkyl, C1-C8alkoxyalkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl). - In a further embodiment, the test protein tyrosine kinase inhibitor non-selectively and reversibly binds to a multiplicity of src-family protein tyrosine kinases.
- In a further aspect of such dosing strategies are methods for treating a B-cell proliferative disorder or a mast cell proliferative disorder comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors. For example, as presented in the Examples, brief exposure to
Compound 1 in vitro is sufficient to inhibit B cell activation in normal human B cells. This protocol mimics the predicted exposure of cells toCompound 1 in vivo and demonstrates that inhibition of B cells is sustained despite washing out ofCompound 1. - In a further aspect of such dosing strategies are methods for treating a rheumatoid arthritis or condition comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors. In a further aspect of such dosing strategies are methods for treating a disease characterized by hyperactive B cells comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors. In a further aspect of such dosing strategies are methods for treating a disease characterized by hyperactive mast cells comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors. In a further aspect of such dosing strategies are methods for treating a disease characterized by both hyperactive B cells and hyperactive mast cells comprising administering to a patient in need a pharmaceutical composition of any of the aforementioned ACK inhibitors. In any of the aforementioned treatment methods using such dosing strategies, the pharmaceutical composition is administered once a day or less frequently than once a day.
- For use in the therapeutic applications described herein, kits and articles of manufacture are also described herein. Such kits can include a carrier, package, or container that is compartmentalized to receive one or more containers such as vials, tubes, and the like, each of the container(s) including one of the separate elements to be used in a method described herein. Suitable containers include, for example, bottles, vials, syringes, and test tubes. The containers can be formed from a variety of materials such as glass or plastic.
- The articles of manufacture provided herein contain packaging materials. Packaging materials for use in packaging pharmaceutical products include, e.g., U.S. Pat. Nos. 5,323,907, 5,052,558 and 5,033,252. Examples of pharmaceutical packaging materials include, but are not limited to, blister packs, bottles, tubes, inhalers, pumps, bags, vials, containers, syringes, bottles, and any packaging material suitable for a selected formulation and intended mode of administration and treatment. A wide array of formulations of the compounds and compositions provided herein are contemplated as are a variety of treatments for any disease, disorder, or condition that benefit by inhibition of Btk, or in which Btk is a mediator or contributor to the symptoms or cause.
- For example, the container(s) can include one or more compounds described herein, optionally in a composition or in combination with another agent as disclosed herein. The container(s) optionally have a sterile access port (for example the container can be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle). Such kits optionally comprising a compound with an identifying description or label or instructions relating to its use in the methods described herein.
- A kit will typically include one or more additional containers, each with one or more of various materials (such as reagents, optionally in concentrated form, and/or devices) desirable from a commercial and user standpoint for use of a compound described herein. Non-limiting examples of such materials include, but not limited to, buffers, diluents, filters, needles, syringes; carrier, package, container, vial and/or tube labels listing contents and/or instructions for use, and package inserts with instructions for use. A set of instructions will also typically be included.
- A label can be on or associated with the container. A label can be on a container when letters, numbers or other characters forming the label are attached, molded or etched into the container itself; a label can be associated with a container when it is present within a receptacle or carrier that also holds the container, e.g., as a package insert. A label can be used to indicate that the contents are to be used for a specific therapeutic application. The label can also indicate directions for use of the contents, such as in the methods described herein.
- In certain embodiments, the pharmaceutical compositions can be presented in a pack or dispenser device which can contain one or more unit dosage forms containing a compound provided herein. The pack can for example contain metal or plastic foil, such as a blister pack. The pack or dispenser device can be accompanied by instructions for administration. The pack or dispenser can also be accompanied with a notice associated with the container in form prescribed by a governmental agency regulating the manufacture, use, or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the drug for human or veterinary administration. Such notice, for example, can be the labeling approved by the U.S. Food and Drug Administration for prescription drugs, or the approved product insert. Compositions containing a compound provided herein formulated in a compatible pharmaceutical carrier can also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition.
- The following specific and non-limiting examples are to be construed as merely illustrative, and do not limit the present disclosure in any way whatsoever.
- 4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) is added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture is stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue is taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gives a semisolid residue which is treated with a little ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gives 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which is sufficiently pure for further use.
- 1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
- 1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
- 3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine.
-

- Synthesis of
Compound 13; a) triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) TFA/CH2Cl2; then acryloyl chloride, diisopropylethylamine (DIPEA), tetrahydrofuran (THF). - Compounds described herein were synthesized by following the steps outlined in
Scheme 1. A detailed illustrative example of the reaction conditions shown inScheme 1 is described for the synthesis of 1-((R)-2-((4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyrrolidin-1-yl)prop-2-en-1-one (Compound 13). - 0.5 g of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine and 0.65 g of triphenylphosphine (TPP) were mixed together with 15 mL of tetrahydrofuran (THF). (R)-tert-butyl 2-(hydroxymethyl)pyrrolidine-1-carboxylate (0.5 g; 1.5 equivalents) was added to the mixture followed by the addition of diisopropyl diazodicarboxylate (0.5 mL). The reaction mixture was stirred at room temperature for 4 hr. The reaction mixture was concentrated and purified by flash chromatography (acetone/CH2Cl2=1/1) to give intermediate 3 (1.49 g).
- Intermediate 3 (1.49 g) was treated with 4 mL of TFA and 5 mL of CH2Cl2 and stirred overnight at room temperature and then concentrated to dryness. The residue was dissolved in ethyl acetate (100 mL) and then washed with dilute aq. NaHCO3(100 mL). The ethyl acetate layer was dried (MgSO4), filtered and concentrated to ˜20 mL and then 4.0 M HCl\dioxane (1 mL) was added and a yellow precipitate formed. The solid was collected by filtration and washed with ethyl acetate (20 mL). The solid was suspended in ethyl acetate (100 mL) and again washed with dilute aq. NaHCO3(100 mL). The ethyl acetate was dried (MgSO4), filtered and concentrated to provide 0.43 g of a light yellow solid. The solid (0.14 g, 0.36 mmol) was stirred in THF (3 mL) and TEA (015 mL, 1.1 mmol) was added, followed by cooling the reaction with an ice bath for 30 min, then acryl chloride (30 μL, 0.36 mmol) added and the reaction was stirred for 2 hr. The reaction mixture was diluted with ethyl acetate (75 mL) and washed with dilute aq. NaHCO3(100 mL). The organic layer was dried (MgSO4), filtered and concentrated. Flash chromatography (with CH2Cl2/MeOH=20/1) gave 90 mg of
compound 4 as a white solid. EM (calc)=440.2; MS (M+1): 441.2. -

- The synthesis of
Compound 14 was accomplished using a procedure analogous to that described in Example 2. EM (calc.): 440.2; MS (M+1H): 441.2. -

- The synthesis of this compound was accomplished using a procedure analogous to that described for Example 2 EM (calc.): 454.21; MS (M+1): 455.2.
-

- The synthesis of this compound was accomplished using a procedure analogous to that described for Example 2. EM (calc.): 414.18; MS (M+1H): 415.2.
-

- The synthesis of this compound was accomplished using a procedure analogous to that described for Example 2. EM (calc.): 400.16; MS (M+1H): 401.2.
-

- The synthesis of this compound was accomplished using a procedure analogous to that described for Example 2. EM (calc.): 452.2; MS (M+1H): 453.2.
-
- The synthesis of this compound was accomplished using a procedure analogous to that described for Example 2. EM (calc.): 452.2; MS (M+1H): 453.2.
-

- The synthesis of this compound was accomplished using a procedure analogous to that described for Example 2. EM (calc.): 497.25; MS (M+1H): 498.2.
-
Compound 1 inhibits lymphoma tumor cell growth. A variety of lymphoma cell lines were incubated with a range of concentrations ofCompound 1 to determine the GI50, the concentration that results in 50% decrease in cell proliferation (FIG. 1A ).Compound 1 inhibits tumor growth in DOHH2 and DLCL2 xenograft models (FIGS. 1B and 1C ). - For in vitro cell proliferation assays, cells were seeded in 96-well plates in standard growth media (in most cases RPMI+10% fetal calf serum) and
Compound 1 was added in a 9-point dilution series ranging from 10 uM to 0.04 uM with DMSO at 0.1% final concentration in all wells. After 72 hours, cell number was measured using Alamar Blue using manufacturer's protocol. A dilution series of untreated cells was run in parallel to verify that the Alamar Blue assay reliably reflected cell number and that growth conditions were not limiting. The GI50, the concentration that results in a 50% decrease in cell number, was calculated using Calcusyn to fit the dose-response curve. GI50 values were confirmed in two or more separate experiments for each cell line. - For in vivo lymphoma xenograft studies, 5E6 DOHH2 or DLCL2 cells in 50% matrigel were implanted subcutaneously in SCID mice and dosed orally with
Compound 1 beginning when tumor size reached 100 mm2. -
Compound 1 inhibits collagen-induced arthritis in the mouse. Male DBA/1O1 aHsd mice were injected intradermally with 150 microliters of 2 mg/mL Type II collagen in Freund's complete adjuvant with supplemental M. tuberculosis, 4 mg/mL and boosted with the same injection 21 days later. After paw inflammation was established, animals were randomized andCompound 1 or vehicle was dosed orally once per day starting atday 1. Paw inflammation was scored from 0-5 and averaged across all paws from all animals for each group in the study.Compound 1 at 12.5 mg/kg and 50 mg/kg regressed inflammation through the end of the study (day 11) while 3.125 mg/kg significantly reduced the increase in paw inflammation (FIG. 2 ). Dexamethasone was included as a positive control. - In another study,
Compound 1 was dosed at 12.5 mg/kg to such mice over: (a) each day of an 11-day period; (b)days days Compound 9 was dosed to such mice at a level of 12.5 mg/kg or 50 mg/kg each day of an 11-day period.Compound 9 reduced the increase in paw inflammation. -
Compound 1 inhibits disease progression in the mouse MRL/1 pr model of lupus.Compound 1 at 3.125 mg/kg, 12.5 mg/kg, and 50 mg/kg significantly reduced proteinuria, indicating amelioration of the progressive autoimmune renal failure seen in this mouse strain (FIG. 3 ). MRL/1pr mice (Jax strain 000485) were dosed orally once per day from 12 weeks of age until 20 weeks of age and urine protein levels were measured weekly using Clinitech Multistick dipstick. -
Compound 1 inhibits mast cell degranulation in a mouse passive cutaneous anaphylaxis model. Increasing doses ofCompound 1 significantly decrease the amount of Evans Blue release, indicating decreased mast cell activation and vascular permeabilization. (FIG. 4 ) - Mice were sensitized with an intradermal injection of monoclonal anti-DNP-IgE in the back. 23 hours later they received a single oral dose of
Compound 1 or vehicle. After one hour, animals were challenged with an intravenous injection of DNP-BSA and Evans Blue dye. Mast cell degranulation leads to vascular permeability and the distribution of the dye into the skin of the back. The area of extravasation after 1 hour is measured. - The compositions described below are presented with a compound of Formula (A1-A6) for illustrative purposes; any of the compounds of any of Formulas (A1-A6), (B1-B6), (C1-C6), or (D1-D6) are optionally used in such pharmaceutical compositions.
- To prepare a parenteral pharmaceutical composition suitable for administration by injection, 100 mg of a water-soluble salt of a compound of Formula (A1-A6) is dissolved in DMSO and then mixed with 10 mL of 0.9% sterile saline. The mixture is incorporated into a dosage unit form suitable for administration by injection.
- To prepare a pharmaceutical composition for oral delivery, 100 mg of a compound of Formula (A1-A6) is mixed with 750 mg of starch. The mixture is incorporated into an oral dosage unit for, such as a hard gelatin capsule, which is suitable for oral administration.
- To prepare a pharmaceutical composition for buccal delivery, such as a hard lozenge, mix 100 mg of a compound of Formula (A1-A6), with 420 mg of powdered sugar mixed, with 1.6 mL of light corn syrup, 2.4 mL distilled water, and 0.42 mL mint extract. The mixture is gently blended and poured into a mold to form a lozenge suitable for buccal administration.
- To prepare a pharmaceutical composition for inhalation delivery, 20 mg of a compound of Formula (A1-A6) is mixed with 50 mg of anhydrous citric acid and 100 mL of 0.9% sodium chloride solution. The mixture is incorporated into an inhalation delivery unit, such as a nebulizer, which is suitable for inhalation administration.
- To prepare a pharmaceutical composition for rectal delivery, 100 mg of a compound of Formula (A1-A6) is mixed with 2.5 g of methylcellulose (1500 mPa), 100 mg of methylparaben, 5 g of glycerin and 100 mL of purified water. The resulting gel mixture is then incorporated into rectal delivery units, such as syringes, which are suitable for rectal administration.
- To prepare a pharmaceutical topical gel composition, 100 mg of a compound of Formula (A1-A6) is mixed with 1.75 g of hydroxypropyl cellulose, 10 mL of propylene glycol, 10 mL of isopropyl myristate and 100 mL of purified alcohol USP. The resulting gel mixture is then incorporated into containers, such as tubes, which are suitable for topical administration.
- To prepare a pharmaceutical ophthalmic solution composition, 100 mg of a compound of Formula (A1-A6) is mixed with 0.9 g of NaCl in 100 mL of purified water and filtered using a 0.2 micron filter. The resulting isotonic solution is then incorporated into ophthalmic delivery units, such as eye drop containers, which are suitable for ophthalmic administration.
- To identify biomarkers that correlate with response to
Compound 1, phosphorylation events in the BCR signal transduction pathway were investigated. A panel of phospho-specific antibodies that recognize activating phosphorylation sites on Syk, Btk, BLNK, PLC-g1, PLC-g2, ERK, and AKT were used and tested the effects ofCompound 4 on both basal phosphorylation and phosphorylation following BCR stimulation driven by anti-IgM or anti-IgG cross-linking. We examined phosphorylation patterns in both aCompound 1 sensitive cell line (DOHH2) and aCompound 1 resistant cell line (Ramos). -
Compound 1 inhibits most BCR-stimulus induced phosphorylation events with similar potency in both cell lines. However, when we examined basal phosphorylation levels, we found higher basal phosphorylation in DOHH2 compared to Ramos, with phospho-ERK in particular indicating higher levels of basal or tonic signaling in DOHH2. Furthermore,Compound 4 significantly decreased pERK levels in unstimulated DOHH2 cells (IC50<10 nM), but not in Ramos cells. - A panel of nine Btk expressing B cell lymphoma cell lines was screened for basal pERK levels. Seven lines expressed significantly higher levels of basal pERK, and of these, 5 were sensitive to Compound 1 (GI50<1.3 uM), while the two cell lines with low pERK levels were resistant to
Compound 1. This data shows that tonic BCR signaling contributes to the survival of a subset of lymphoma cell lines, and that inhibition of this signaling byCompound 4 is correlated with induction of apoptosis. - Two additional experiments demonstrate that sensitivity to
Compound 1 is correlated with high levels of pERK. First 1 uM ofCompound 4 reduces expression of the known ERK transcriptional target Egr-1 within 1 hr, with maximal downregulation (10-fold) achieved by 4 hr. Second, in the lymphoma cell line WSU-DLCL2, BCR cross-linking by anti-IgG (30 ug/ml) overcomes inhibition of pERK byCompound 4, showing that strong BCR stimulus activates parallel pathways to pERK that do not require Btk. BCR stimulus also rescues WSU-DLCL2 fromCompound 1 induced cytotoxicity, further confirming that inhibition of pERK is correlated with apoptosis induction byCompound 1. Taken together these data show high levels of pERK or ERK transcriptional targets such as Egr-1 serve as useful markers for lymphomas in which tonic BCR signaling is contributing to cell survival and that these lymphomas are particularly sensitive to BCR pathway inhibitors such asCompound 1. - Because the ATP binding sites of the >500 kinases in the human genome are highly conserved, it has proven difficult to engineer selectivity for individual kinases using conventional reversible binding inhibitors. For our highly selective
BTK inhibitor Compound 1, we engineered an electrophilic center capable of irreversibly inactivating the target enzyme, BTK. The approach employed structure based design to achieve a high degree of potency and selectivity by (1) fitting the core scaffold into the active site ATP binding pocket of kinase enzymes, and (2) forming a covalent bond with Cysteine-481 located in BTK. The unique chemistry required for covalent bond formation involves an electrophilic moiety that acts as a Michael acceptor, which bonds with a nucleophile (such as Cys-481) present in a precise location within the active site. - By way of example only, a panel of 50-100 Cys-targeting kinase inhibitors is generated. The molecular orientation and positioning of the electrophilic group in these inhibitors in relation to the Cysteine residue will affect the potency and selectivity of a given inhibitor. Each inhibitor will then be profiled for kinetics of kinase inhibition (Ki) for each of the ten Cys-containing kinases, effect on tumor cell proliferation (GI50), effect on relevant off-targets (hERG, CYPs), drug-like characteristics (solubility, clogP) and ability to block labeling by the active site probe. This panel of diverse inhibitors are then be used in cell assays (for example, inhibition of tumor growth) to screen for a phenotype of interest. With the phenotype, the identification of additional inhibited kinases is determined using the active site probe and mass spectrometry.
- In another example, the linker and Michael acceptor moiety of Compound 1 was modified to provide
Compound 9 which has a different selectivity pattern. Table 1 is a table showing the degree of inhibition of a panel of kinases for two example compounds. IC50s were determined using the in vitro HotSpot kinase assay (purified enzymes, 33P-ATP, an appropriate substrate and 1 uM ATP.) Compared to Compound 1, Compound 9 has similar potency toward Btk, but significantly less potency toward JAK-3, ITK, and EGFR and significantly more potency toward the src-family kinases lck, c-src, FGR, Fyn, Hck, and Lyn and Yes. Thus, subtle modifications in the linker moiety and the Michael acceptor moiety are important for the design of selective ACK inhibitors. -
TABLE 1 Compound 1Compound 9 Kinase IC50 (nM) IC50 (nM) BTK 0.5 1.0 ITK 11.7 909.9 Bmx/ETK 0.8 1.1 TEC 77.8 108.0 EFGR 0.5 20.6 HER2 9.4 1536.0 HER4 0.1 3.2 LCK 2.0 1.0 BLK 0.5 0.2 C-src 262.6 14.3 FGR 2.3 0.4 Fyn 95.6 7.1 HCK 3.7 1.0 Lyn 16.2 1.2 YES 6.5 0.8 ABL 86.1 32.3 Brk 3.3 3.3 CSK 2.2 2.4 FER 8,070.0 3,346.0 JAK3 10.4 8,278.0 SYK >10,000 >10,000 - In this example, compounds are selected based on in vitro characteristics to optimize for potency of inhibition of particular kinases and degree of covalent binding to off-target cysteines such as glutathione. For example, in Table 2,
Compound 9 and Compound 12 both inhibit Btk with a similar potency asCompound 1, but they are both significantly less potent inhibitors of EGFR, ITK, and JAK-3. As another example,Compound 11 is similar toCompound 1 for inhibition of Btk but does not bind glutathione as readily. - A calculated value (e.g (1/Btk IC50)/Glutathione conjugation rate) as shown in the Table 2) is used to compare compounds for their ratio between potency at inhibiting their target and their non-specific binding to other SH groups, such as those in glutathione. As shown in Table 2, this calculated value is 4.7 for
Compound 1 and for 239.6 forCompound 11. Calculated ratios such as these are used to quantitatively compare different compounds and select compounds for further study. - For enzyme inhibition assays, compounds were tested in range of ten concentrations from 10 uM to 0.0005 uM using purified enzymes and the Hotspot kinase assay. Reaction conditions were 1 uM ATP, one hour incubation with inhibitor, and kinase activity detected using 33-ATP phosphorylation of an appropriately selected peptide substrate. Dose-response curves were fit using Prism, and the IC50, the concentration at which enzyme inhibition is 50% of maximal inhibition, was determined. See Table 2.
- For the glutathione binding assays, 5 mM glutathione, 10 μM Btk inhibitor in DMSO (10 μL) and 6 equivalents of N′N′ Diisopropyl ethyl amine were combined in 1 mL potassium phosphate buffer. The mixture was incubated for 0, 15, 60 minutes at room temperature and the reaction was stopped with 10 equivalents of formic acid. 50 L of each reaction mixture was injected on HPLC (Mobil Phase A: 0.2% formic acid in water, Mobile Phase B: 0.2% formic acid in acetonitrile, HPLC Column: Metasil Basic 3μ, 150×4.6 mm, 10% B, Gradient: 10% to 90% B, Detection: UV/Vis 260 nM). Rate of reaction was reported as nmole GSH conjugate conversion per minute from the normalized ratio for area under the curve from HPLC chromatograms for both GSH conjugate and the parent.
- Analogs are generated that are Btk inhibitors and that are cytotoxic to the lymphoma cell line DOHH2. See Table 2. For the DOHH2 cell proliferation assay, cells were seeded in 96-well plates in standard growth media (RPMI+10% fetal calf serum) and compounds were added in a 9-point dilution series ranging from 10 uM to 0.04 uM with DMSO at 0.1% final concentration in all wells. After 72 hours, cell number was measured using Alamar Blue using manufacturer's protocol. A dilution series of untreated cells was run in parallel to verify that the Alamar Blue assay reliably reflected cell number and that growth conditions were not limiting. The GI50, the concentration that results in a 50% decrease in cell number, was calculated using Calcusyn to fit the dose-response curve.
-
TABLE 2 (1/BTK BTK ITK EGFR LCK JAK3 Glutathione IC50)/ DOHH2 Compound IC50 IC50 IC50 IC50 IC50 Conj Rate Glutathione GI50 # Structure (nM) (nM) (nM) (nM) (nM) (nmol/min) Rate (μM) 1 
0.5 11.7 0.5 2.0 10.4 0.398 4.7 0.1 2 
1.1 48.8 0.32 3 
21 74.5 4 
22.2 487.6 5 
5.6 326.0 0.004 44.5 6 
3.1 60.9 0.39 0.8 7 
6.3 6,123 268.7 2.6 >10,000 0.01 15.9 0.317 8 
1.4 83.4 9 
1.0 909.9 20.6 1.0 8278.0 0.011 10 
1.31 1954 44.5 0.88 >10,000 <0.03 11 
0.92 6891 18.85 2.43 >10,000 0.004525 239.6 >10 12 
1.33 14290 698.3 5.97 >10,000 0.004361 172.2 >10 13 
0.67 3013 18.75 1.56 12980 0.24 14 
0.39 592.3 2.298 9.24 1456 0.37 15 
4.16 21100 289.4 5.90 >10,000 0.59 16 
3.14 >10,000 2807 3.82 >10,000 0.21 17 
2.00 2333 435.3 2.07 >10,000 0.0243 20.6 0.21 18 
1.38 2536 22.53 0.76 >10,000 <0.03 19 
1.58 534.6 28.22 6.62 5997 0.69 20 
4.07 7993 303.60 98.59 >10,000 0.39 21 
4.15 >10,000 6238.00 1346 >10,000 1.53 22 
1.57 3691 156.30 22.12 >10,000 0.014 45.4 <0.04 23 
0.32 830 70.49 208.00 3306.00 0.11 24 
0.89 476 383.70 235.40 9077.00 0.44 25 
3.48 >10,000 272.90 25.81 >10,000 0.05 -
Compound 1 and Compound 12 have a short half-life in vivo. In contrast,Compound 7 and Compound 8 have a significantly longer in vivo half-life (FIG. 5 ). Compounds like 1 and 12 are predicted to have enhanced kinase selectivity in vivo because inhibition will be sustained only for those kinases that are irreversibly inhibited. - Male jugular vein cannulated rats were administered a single dose of all test compounds at 8 mg/kg each, in combination by oral gavage. Dose volumes were adjusted based on body weight data collected immediately prior to dosing. Blood samples were collected at 0.0833 (5 minutes), 0.333 (20 minutes), 1, 3, 6, 9, and 24 hours post-dosing from orally dosed rats. The samples were collected into plasma separator Microtainer tubes with anticoagulant (lithium heparin). Plasma samples were prepared by centrifugation (5 min at 5000×g), and at least 100 L were transferred to storage tubes and stored frozen at −80° C. Plasma samples were thawed and 75 uL aliquots were transferred to centrifuge tubes to which 10 L aliquots of internal standard solution (1 μg/mL) were added. The samples were not diluted with blank plasma prior to further processing. Soluble proteins were precipitated by the addition of 200 μL of acetonitrile, followed by centrifugation (20 min at 16,000×g). The samples were evaporated to dryness and reconstituted in 200 μL of water containing 0.2% formic acid and 10% methanol. All samples were loaded onto an autosampler maintained at 6° C. and evaluated for concentrations of test compounds using LC-MS/MS.
- Brief exposure to
Compound 1 in vitro is sufficient to inhibit B cell activation in normal human B cells (FIG. 6 ). This protocol mimics the predicted exposure of cells toCompound 1 in vivo and demonstrates that inhibition of B cells is sustained despite washing out ofCompound 1. - B cells were purified from blood from healthy donors by negative selecting using the RosetteSep Human B cell enrichment cocktail. Cells were plated in growth media (10% RPMI+10% fetal calf serum) and indicated concentrations of
Compound 1 were added. After incubation for 1 hour at 37° C., cells were washed three times using an 8-fold dilution in growth media for each wash. Cells were then stimulated with 10 ug/ml of IgM F(ab′)2 for 18 hours at 37° C. Cells were then stained with anti-CD69-PE antibody and analyzed by flow cytometry using standard conditions. - Given that kinase inhibitors described above will have both reversible and irreversible activities, we select their in vivo properties of absorption, distribution, metabolism and excretion (ADME) in order to optimize the therapeutic index. Specifically, rapidly cleared compounds are expected to cause only brief inhibition of reversibly inhibited targets while maintaining sustained inhibition of irreversibly inhibited targets. Depending on the degree to which sustained inhibition of particular targets results in therapeutic effects or toxicities, we identify compounds with an optimal combination of in vitro selectivity profiles and in vivo ADME properties.
Claims (20)
1-100. (canceled)
101. A compound of Formula (B2) having the structure:

wherein:
Y is alkylene or substituted alkylene, or a 4-, 5-, or 6-membered cycloalkylene ring;
each Ra is independently H, halogen, —CF3, —CN, —NO2, OH, NH2, -La-(substituted or unsubstituted alkyl), -La-(substituted or unsubstituted alkenyl), -La-(substituted or unsubstituted heteroaryl), or -La-(substituted or unsubstituted aryl), wherein La is a bond, O, S, —S(═O), —S(═O)2, NH, C(O), CH2, —NHC(O)O, —NHC(O), or —C(O)NH;
G is

wherein,
R6, R7 and R8 are independently selected from among H, lower alkyl or substituted lower alkyl, lower heteroalkyl or substituted lower heteroalkyl, substituted or unsubstituted lower cycloalkyl, and substituted or unsubstituted lower heterocycloalkyl;
R12 is H or lower alkyl; or
Y and R12 taken together form a 4-, 5-, or 6-membered heterocyclic ring; or
or a pharmaceutically acceptable salt thereof.
102. The compound according to claim 101 , wherein G is selected from among

103. The compound according to claim 101 , wherein

is selected from among

104. The compound according to claim 101 , wherein the compound of Formula (B2) has the following structure of Formula (C2):

wherein:
Y is alkylene or substituted alkylene, or a 4-, 5-, or 6-membered cycloalkylene ring;
R12 is H or lower alkyl; or
Y and R12 taken together form a 4-, 5-, or 6-membered heterocyclic ring;
G is

wherein,
R6, R7 and R8 are independently selected from among H, lower alkyl or substituted lower alkyl, lower heteroalkyl or substituted lower heteroalkyl, substituted or unsubstituted lower cycloalkyl, and substituted or unsubstituted lower heterocycloalkyl; or
a pharmaceutically acceptable salt thereof.
105. The compound of claim 101 , wherein the compound is of formula (D2):

wherein:
La is CH2, O, NH or S;
Ar is a substituted or unsubstituted aryl, or a substituted or unsubstituted heteroaryl;
Y is an optionally substituted group selected from among alkylene, heteroalkylene, cycloalkylene, heterocycloalkylene, arylene, and heteroarylene;
Z is C(═O), OC(═O), NHC(═O), C(═S), S(═O)x, OS(═O)x, or NHS(═O)x, where x is 1 or 2;
R7 and R8 are independently selected from among H, unsubstituted C1-C4alkyl, substituted C1-C4alkyl, unsubstituted C1-C4heteroalkyl, substituted C1-C4heteroalkyl, unsubstituted C3-C6cycloalkyl, substituted C3-C6cycloalkyl, unsubstituted C2-C6heterocycloalkyl, and substituted C2-C6heterocycloalkyl; or
R7 and R8 taken together form a bond;
R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C8alkylaminoalkyl, substituted or unsubstituted C3-C6cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted C2-C8heterocycloalkyl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl); or
a pharmaceutically acceptable salt thereof.
106. The compound of claim 105 having the following structure of Formula (D2):

wherein
La is CH2, O, NH or S;
Ar is an optionally substituted aryl or heteroaryl;
Y is an optionally substituted alkylene, heteroalkylene, carbocycloalkylene, or heterocycloalkylene, or a combination thereof;
Z is C(═O), OC(═O), NHC(═O), C(═S), S(═O)x, OS(═O)x, or NHS(═O)x, where x is 1 or 2; and
R6, R7, and R8 are independently selected from H, alkyl, heteroalkyl, carbocycle, and heterocycle, or combinations thereof; or
a pharmaceutically acceptable salt thereof.
107. The compound according to claim 105 , La is O.
108. The compound according to claim 105 , wherein Ar is phenyl.
109. The compound according to claim 105 , wherein Z is C(O).
110. The compound according to claim 105 , wherein Y is an optionally substituted group selected from among C1-C6alkylene, C1-C6heteroalkylene, 4-, 5-, 6- or 7-membered cycloalkylene, and 4-, 5-, 6- or 7-membered heterocycloalkylene.
111. The compound according to claim 105 , wherein Y is a 5-, or 6-membered cycloalkylene, or a 5-, or 6-membered heterocycloalkylene containing 1 or 2 N atoms.
112. The compound according to claim 105 , wherein each of R7 and R8 is H; or R7 and R8 taken together form a bond.
113. The compound according to claim 105 , wherein R6 is H, substituted or unsubstituted C1-C4alkyl, substituted or unsubstituted C1-C4heteroalkyl, C1-C6alkoxyalkyl, C1-C2alkyl-N(C1-C3alkyl)2, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, C1-C4alkyl(aryl), C1-C4alkyl(heteroaryl), C1-C4alkyl(C3-C8cycloalkyl), or C1-C4alkyl(C2-C8heterocycloalkyl).
114. A pharmaceutical composition comprising a therapeutically effective amount of a compound of claim 101 , and a pharmaceutically acceptable carrier, diluent and/or excipient.
115. A method of treating an autoimmune disorder, a heteroimmune disorder, inflammation, or a combination thereof in a subject, comprising administering a therapeutically effective amount a compound of claim 101 to the subject.
116. The method of claim 115 , wherein the autoimmune disorder is rheumatoid arthritis, lupus, or a combination thereof.
117. A method of treating an allergy, type I hypersensitivity, allergic conjunctivitis, allergic rhinitis, atopic dermatitis, or a combination thereof in a subject, comprising a therapeutically effective amount of claim 101 to the subject.
118. A method of B-cell proliferative disorder in a subject, comprising a therapeutically effective amount of a compound of claim 101 to the subject.
119. The method of claim 118 , wherein the B-cell proliferative disorder is diffuse large B cell lymphoma, follicular lymphoma, chronic lymphocytic leukemia, or a combination thereof.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/361,494 US20200055859A1 (en) | 2007-03-28 | 2019-03-22 | Inhibitors of brutons tyrosine kinase |
| US17/403,589 US20220213106A1 (en) | 2006-09-22 | 2021-08-16 | Inhibitors of Bruton's Tyrosine Kinase |
| US18/375,645 US20240317756A1 (en) | 2006-09-22 | 2023-10-02 | Inhibitors of bruton's tyrosine kinase |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/692,870 US7732454B2 (en) | 2006-09-22 | 2007-03-28 | Inhibitors of Bruton's tyrosine kinase |
| US11/964,285 US7825118B2 (en) | 2006-09-22 | 2007-12-26 | Inhibitors of bruton's tyrosine kinase |
| US1712507P | 2007-12-27 | 2007-12-27 | |
| PCT/US2008/058528 WO2008121742A2 (en) | 2007-03-28 | 2008-03-27 | Inhibitors of bruton's tyrosine kinase |
| US59480510A | 2010-06-08 | 2010-06-08 | |
| US13/543,065 US9079908B2 (en) | 2007-03-28 | 2012-07-06 | Inhibitors of Bruton'S tyrosine kinase |
| US14/794,685 US9556182B2 (en) | 2007-03-28 | 2015-07-08 | Inhibitors of Bruton's tyrosine kinase |
| US201715412738A | 2017-01-23 | 2017-01-23 | |
| US15/692,261 US20170362246A1 (en) | 2007-03-28 | 2017-08-31 | Inhibitors of bruton's tyrosine kinase |
| US16/361,494 US20200055859A1 (en) | 2007-03-28 | 2019-03-22 | Inhibitors of brutons tyrosine kinase |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/692,261 Continuation US20170362246A1 (en) | 2006-09-22 | 2017-08-31 | Inhibitors of bruton's tyrosine kinase |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US202017122814A Continuation | 2006-09-22 | 2020-12-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20200055859A1 true US20200055859A1 (en) | 2020-02-20 |
Family
ID=45973505
Family Applications (21)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/340,276 Active US8809273B2 (en) | 2007-03-28 | 2011-12-29 | Inhibitors of Bruton's tyrosine kinase |
| US13/340,621 Abandoned US20120101114A1 (en) | 2007-03-28 | 2011-12-29 | Inhibitors of bruton's tyrosine kinase |
| US13/340,559 Abandoned US20120101113A1 (en) | 2007-03-28 | 2011-12-29 | Inhibitors of bruton's tyrosine kinase |
| US13/404,422 Abandoned US20120165328A1 (en) | 2007-03-28 | 2012-02-24 | Inhibitors of b lymphocyte kinase |
| US13/410,110 Abandoned US20120184013A1 (en) | 2007-03-28 | 2012-03-01 | Inhibitors of BMX non-receptor tyrosine kinase |
| US13/430,173 Abandoned US20120184567A1 (en) | 2007-03-28 | 2012-03-26 | Inhibitors of bruton's tyrosine kinase |
| US13/439,775 Abandoned US20120202264A1 (en) | 2007-03-28 | 2012-04-04 | Inhibitors of il2-inducible t-cell kinase |
| US13/543,394 Active 2027-08-22 US9139591B2 (en) | 2007-03-28 | 2012-07-06 | Inhibitors of bruton's tyrosine kinase |
| US13/543,399 Abandoned US20120296089A1 (en) | 2007-03-28 | 2012-07-06 | Inhibitors of bruton's tyrosine kinase |
| US13/543,065 Active 2027-06-10 US9079908B2 (en) | 2006-09-22 | 2012-07-06 | Inhibitors of Bruton'S tyrosine kinase |
| US13/606,949 Active US8940750B2 (en) | 2007-03-28 | 2012-09-07 | Inhibitors of bruton's tyrosine kinase |
| US13/607,036 Abandoned US20120329130A1 (en) | 2007-03-28 | 2012-09-07 | Inhibitors of bruton's tyrosine kinase |
| US13/612,143 Abandoned US20130018060A1 (en) | 2007-03-28 | 2012-09-12 | Inhibitors of bruton's tyrosine kinase |
| US14/069,222 Abandoned US20140057907A1 (en) | 2007-12-27 | 2013-10-31 | Inhibitors of bruton's tyrosine kinase |
| US14/179,457 Abandoned US20140163027A1 (en) | 2007-03-28 | 2014-02-12 | Inhibitors of bruton's tyrosine kinase |
| US14/605,854 Active US9181263B2 (en) | 2007-03-28 | 2015-01-26 | Inhibitors of bruton's tyrosine kinase |
| US14/605,857 Abandoned US20150265618A1 (en) | 2007-03-28 | 2015-01-26 | Inhibitors of bruton's tyrosine kinase |
| US14/703,750 Abandoned US20150307500A1 (en) | 2007-12-27 | 2015-05-04 | Inhibitors of bruton's tyrosine kinase |
| US14/794,685 Active US9556182B2 (en) | 2006-09-22 | 2015-07-08 | Inhibitors of Bruton's tyrosine kinase |
| US15/692,261 Abandoned US20170362246A1 (en) | 2006-09-22 | 2017-08-31 | Inhibitors of bruton's tyrosine kinase |
| US16/361,494 Abandoned US20200055859A1 (en) | 2006-09-22 | 2019-03-22 | Inhibitors of brutons tyrosine kinase |
Family Applications Before (20)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/340,276 Active US8809273B2 (en) | 2007-03-28 | 2011-12-29 | Inhibitors of Bruton's tyrosine kinase |
| US13/340,621 Abandoned US20120101114A1 (en) | 2007-03-28 | 2011-12-29 | Inhibitors of bruton's tyrosine kinase |
| US13/340,559 Abandoned US20120101113A1 (en) | 2007-03-28 | 2011-12-29 | Inhibitors of bruton's tyrosine kinase |
| US13/404,422 Abandoned US20120165328A1 (en) | 2007-03-28 | 2012-02-24 | Inhibitors of b lymphocyte kinase |
| US13/410,110 Abandoned US20120184013A1 (en) | 2007-03-28 | 2012-03-01 | Inhibitors of BMX non-receptor tyrosine kinase |
| US13/430,173 Abandoned US20120184567A1 (en) | 2007-03-28 | 2012-03-26 | Inhibitors of bruton's tyrosine kinase |
| US13/439,775 Abandoned US20120202264A1 (en) | 2007-03-28 | 2012-04-04 | Inhibitors of il2-inducible t-cell kinase |
| US13/543,394 Active 2027-08-22 US9139591B2 (en) | 2007-03-28 | 2012-07-06 | Inhibitors of bruton's tyrosine kinase |
| US13/543,399 Abandoned US20120296089A1 (en) | 2007-03-28 | 2012-07-06 | Inhibitors of bruton's tyrosine kinase |
| US13/543,065 Active 2027-06-10 US9079908B2 (en) | 2006-09-22 | 2012-07-06 | Inhibitors of Bruton'S tyrosine kinase |
| US13/606,949 Active US8940750B2 (en) | 2007-03-28 | 2012-09-07 | Inhibitors of bruton's tyrosine kinase |
| US13/607,036 Abandoned US20120329130A1 (en) | 2007-03-28 | 2012-09-07 | Inhibitors of bruton's tyrosine kinase |
| US13/612,143 Abandoned US20130018060A1 (en) | 2007-03-28 | 2012-09-12 | Inhibitors of bruton's tyrosine kinase |
| US14/069,222 Abandoned US20140057907A1 (en) | 2007-12-27 | 2013-10-31 | Inhibitors of bruton's tyrosine kinase |
| US14/179,457 Abandoned US20140163027A1 (en) | 2007-03-28 | 2014-02-12 | Inhibitors of bruton's tyrosine kinase |
| US14/605,854 Active US9181263B2 (en) | 2007-03-28 | 2015-01-26 | Inhibitors of bruton's tyrosine kinase |
| US14/605,857 Abandoned US20150265618A1 (en) | 2007-03-28 | 2015-01-26 | Inhibitors of bruton's tyrosine kinase |
| US14/703,750 Abandoned US20150307500A1 (en) | 2007-12-27 | 2015-05-04 | Inhibitors of bruton's tyrosine kinase |
| US14/794,685 Active US9556182B2 (en) | 2006-09-22 | 2015-07-08 | Inhibitors of Bruton's tyrosine kinase |
| US15/692,261 Abandoned US20170362246A1 (en) | 2006-09-22 | 2017-08-31 | Inhibitors of bruton's tyrosine kinase |
Country Status (1)
| Country | Link |
|---|---|
| US (21) | US8809273B2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10751342B2 (en) | 2010-06-03 | 2020-08-25 | Pharmacyclics Llc | Use of inhibitors of Bruton's tyrosine kinase (Btk) |
| US10954567B2 (en) | 2012-07-24 | 2021-03-23 | Pharmacyclics Llc | Mutations associated with resistance to inhibitors of Bruton's Tyrosine Kinase (BTK) |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2529621B1 (en) * | 2006-09-22 | 2016-10-05 | Pharmacyclics LLC | Inhibitors of bruton's tyrosine kinase |
| US8809273B2 (en) | 2007-03-28 | 2014-08-19 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US20090301928A1 (en) * | 2008-06-05 | 2009-12-10 | United Comb & Novelty Corporation | Packaging For Lipped Containers |
| US11351168B1 (en) | 2008-06-27 | 2022-06-07 | Celgene Car Llc | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| AU2009262068C1 (en) | 2008-06-27 | 2015-07-02 | Celgene Car Llc | Heteroaryl compounds and uses thereof |
| EP2307025B1 (en) | 2008-07-16 | 2017-09-20 | Pharmacyclics LLC | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors |
| CN102482277B (en) | 2009-05-05 | 2017-09-19 | 达纳-法伯癌症研究所有限公司 | Epidermal growth factor receptor inhibitor and the method for treating obstacle |
| WO2012021444A1 (en) | 2010-08-10 | 2012-02-16 | Avila Therapeutics, Inc. | Besylate salt of a btk inhibitor |
| JP5956999B2 (en) | 2010-11-01 | 2016-07-27 | セルジーン アヴィロミクス リサーチ, インコーポレイテッド | Heteroaryl compounds and uses thereof |
| US8975249B2 (en) | 2010-11-01 | 2015-03-10 | Celgene Avilomics Research, Inc. | Heterocyclic compounds and uses thereof |
| US8796255B2 (en) | 2010-11-10 | 2014-08-05 | Celgene Avilomics Research, Inc | Mutant-selective EGFR inhibitors and uses thereof |
| FR2974576B1 (en) * | 2011-04-29 | 2013-07-19 | Sanofi Aventis | N - [(1H-PYRAZOL-1-YL) ARYL] -1H-INDOLE OR 1H-INDAZOLE-3-CARBOXAMIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATIONS |
| CA2841080A1 (en) | 2011-07-13 | 2013-01-17 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| CA2841886C (en) | 2011-07-19 | 2016-08-16 | Merck Sharp & Dohme B.V. | 4-imidazopyridazin-1-yl-benzamides and 4-imidazotriazin-1-yl-benzamides as btk-inhibitors |
| EP2548877A1 (en) | 2011-07-19 | 2013-01-23 | MSD Oss B.V. | 4-(5-Membered fused pyridinyl)benzamides as BTK-inhibitors |
| EP3409278B8 (en) | 2011-07-21 | 2020-11-04 | Sumitomo Dainippon Pharma Oncology, Inc. | Heterocyclic protein kinase inhibitors |
| CA2853498A1 (en) | 2011-10-28 | 2013-05-02 | Celgene Avilomics Research, Inc. | Methods of treating a bruton's tyrosine kinase disease or disorder |
| US8377946B1 (en) | 2011-12-30 | 2013-02-19 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| MX356179B (en) | 2012-03-15 | 2018-05-17 | Celgene Avilomics Res Inc | Salts of an epidermal growth factor receptor kinase inhibitor. |
| ES2880109T3 (en) | 2012-03-15 | 2021-11-23 | Celgene Car Llc | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| CA3218491A1 (en) | 2012-06-04 | 2013-12-12 | Pharmacyclics Llc | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| MX2015006168A (en) | 2012-11-15 | 2015-08-10 | Pharmacyclics Inc | Pyrrolopyrimidine compounds as kinase inhibitors. |
| CN103848810A (en) | 2012-11-30 | 2014-06-11 | 北京赛林泰医药技术有限公司 | Bruton's tyrosine kinases inhibitor |
| US9126950B2 (en) | 2012-12-21 | 2015-09-08 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| PE20151274A1 (en) | 2013-02-08 | 2015-09-12 | Celgene Avilomics Res Inc | ERK INHIBITORS AND THEIR USES |
| WO2014130856A2 (en) * | 2013-02-21 | 2014-08-28 | Wayne Rothbaum | Treatment of skeletal-related disorders |
| CN108997225A (en) | 2013-03-14 | 2018-12-14 | 特雷罗药物股份有限公司 | JAK2 and ALK2 inhibitor and its application method |
| BR112015021995A2 (en) * | 2013-03-14 | 2017-07-18 | Pharmacyclics Llc | combinations of bruton tyrosine kinase inhibitors and cyp3a4 inhibitors |
| US20160138077A1 (en) * | 2013-06-20 | 2016-05-19 | The General Hospital Corporation | Monitoring and assessing deacetylase enzyme activity |
| JP6458018B2 (en) | 2013-07-02 | 2019-01-23 | ファーマサイクリックス エルエルシー | Prinone compounds as kinase inhibitors |
| EP3027192A4 (en) | 2013-08-02 | 2017-03-22 | Pharmacyclics, LLC | Methods for the treatment of solid tumors |
| US9415050B2 (en) | 2013-08-12 | 2016-08-16 | Pharmacyclics Llc | Methods for the treatment of HER2 amplified cancer |
| US9492471B2 (en) | 2013-08-27 | 2016-11-15 | Celgene Avilomics Research, Inc. | Methods of treating a disease or disorder associated with Bruton'S Tyrosine Kinase |
| KR20160062103A (en) | 2013-09-30 | 2016-06-01 | 파마싸이클릭스 엘엘씨 | Inhibitors of bruton's tyrosine kinase |
| WO2015061751A1 (en) | 2013-10-25 | 2015-04-30 | Pharmacyclics, Inc. | Methods of treating and preventing graft versus host disease |
| CN105979948A (en) | 2013-12-05 | 2016-09-28 | 安塞塔制药公司 | Therapeutic combination of a pi3k inhibitor and a btk inhibitor |
| AU2014362231B2 (en) * | 2013-12-11 | 2019-04-04 | Biogen Ma Inc. | Biaryl compounds useful for the treatment of human diseases in oncology, neurology and immunology |
| US9415049B2 (en) | 2013-12-20 | 2016-08-16 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US10272083B2 (en) | 2014-01-21 | 2019-04-30 | Acerta Pharma B.V. | Methods of treating chronic lymphocytic leukemia and small lymphocytic leukemia using a BTK inhibitor |
| WO2015143400A1 (en) | 2014-03-20 | 2015-09-24 | Pharmacyclics, Inc. | Phospholipase c gamma 2 and resistance associated mutations |
| US9937171B2 (en) | 2014-04-11 | 2018-04-10 | Acerta Pharma B.V. | Methods of blocking the CXCR-4/SDF-1 signaling pathway with inhibitors of bruton's tyrosine kinase |
| WO2015193740A2 (en) | 2014-06-17 | 2015-12-23 | Acerta Pharma B.V. | Therapeutic combinations of a btk inhibitor, a pi3k inhibitor and/or a jak-2 inhibitor |
| AU2015296215A1 (en) | 2014-08-01 | 2017-03-23 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| CN106573002A (en) | 2014-08-07 | 2017-04-19 | 药品循环有限责任公司 | Novel formulations of a bruton's tyrosine kinase inhibitor |
| RS62713B1 (en) | 2014-08-11 | 2022-01-31 | Acerta Pharma Bv | Therapeutic combinations of a btk inhibitor and a bcl-2 inhibitor |
| ES2741785T3 (en) | 2014-08-13 | 2020-02-12 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| TW201702218A (en) | 2014-12-12 | 2017-01-16 | 美國杰克森實驗室 | Compositions and methods relating to the treatment of cancer, autoimmune disease, and neurodegenerative disease |
| IL315294A (en) | 2015-03-03 | 2024-10-01 | Pharmacyclics Llc | Pharmaceutical formulations of bruton's tyrosine kinase inhibitor |
| WO2016161571A1 (en) | 2015-04-08 | 2016-10-13 | Merck Sharp & Dohme Corp. | Indazole and azaindazole btk inhibitors |
| PT3954690T (en) | 2015-07-02 | 2023-06-06 | Acerta Pharma Bv | Solid forms and formulations of (s)-4-(8-amino-3-(1-(but-2-ynoyl)pyrrolidin-2-yl)imidazo[1,5-a]pyrazin-1-yl)-n-(pyridin-2-yl)benzamide |
| CN106995446B (en) * | 2016-01-22 | 2021-07-02 | 山东新时代药业有限公司 | Preparation method of Bruton's tyrosine kinase inhibitor |
| JP6972002B2 (en) * | 2016-03-11 | 2021-11-24 | エンジェル・ファーマシューティカル・カンパニー・リミテッド | Compounds and methods for regulating Bruton's tyrosine kinase |
| CN106432249B (en) * | 2016-09-30 | 2018-12-04 | 陕西科技大学 | The synthesis of a kind of pyrrolo- [2,1-f] [1,2,4] triazine parent nucleus compound and its medical usage |
| EP3773591A4 (en) | 2018-04-05 | 2021-12-22 | Sumitomo Dainippon Pharma Oncology, Inc. | Axl kinase inhibitors and use of the same |
| AU2019310590A1 (en) | 2018-07-26 | 2021-01-14 | Sumitomo Pharma Oncology, Inc. | Methods for treating diseases associated with abnormal acvr1 expression and acvr1 inhibitors for use in the same |
| EP3924351A4 (en) | 2019-02-12 | 2022-12-21 | Sumitomo Pharma Oncology, Inc. | Formulations comprising heterocyclic protein kinase inhibitors |
| WO2024097653A1 (en) | 2022-10-31 | 2024-05-10 | Sumitomo Pharma America, Inc. | Pim1 inhibitor for treating myeloproliferative neoplasms |
Family Cites Families (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5052558A (en) | 1987-12-23 | 1991-10-01 | Entravision, Inc. | Packaged pharmaceutical product |
| US5033252A (en) | 1987-12-23 | 1991-07-23 | Entravision, Inc. | Method of packaging and sterilizing a pharmaceutical product |
| JPH01167840A (en) * | 1987-12-24 | 1989-07-03 | Konica Corp | Novel photographic cyan coupler |
| US5323907A (en) | 1992-06-23 | 1994-06-28 | Multi-Comp, Inc. | Child resistant package assembly for dispensing pharmaceutical medications |
| GB9226855D0 (en) | 1992-12-23 | 1993-02-17 | Erba Carlo Spa | Vinylene-azaindole derivatives and process for their preparation |
| US6326469B1 (en) | 1994-04-22 | 2001-12-04 | Sugen, Inc. | Megakaryocytic protein tyrosine kinases |
| US5593997A (en) | 1995-05-23 | 1997-01-14 | Pfizer Inc. | 4-aminopyrazolo(3-,4-D)pyrimidine and 4-aminopyrazolo-(3,4-D)pyridine tyrosine kinase inhibitors |
| CH690773A5 (en) | 1996-02-01 | 2001-01-15 | Novartis Ag | Pyrrolo (2,3-d) pyrimides and their use. |
| AU3176297A (en) | 1996-06-25 | 1998-01-14 | Novartis Ag | Substituted 7-amino-pyrrolo{3,2-d}pyrimidines and the use thereof |
| WO1998041525A1 (en) | 1997-03-19 | 1998-09-24 | Basf Aktiengesellschaft | Pyrrolo[2,3d]pyrimidines and their use as tyrosine kinase inhibitors |
| US6303652B1 (en) | 1998-08-21 | 2001-10-16 | Hughes Institute | BTK inhibitors and methods for their identification and use |
| MXPA00010150A (en) | 1998-04-17 | 2002-05-14 | Parker Hughes Inst | Btk inhibitors and methods for their identification and use. |
| US20050287596A9 (en) | 1998-06-26 | 2005-12-29 | Braisted Andrew C | Novel ligands and libraries of ligands |
| US6998233B2 (en) | 1998-06-26 | 2006-02-14 | Sunesis Pharmaceuticals, Inc. | Methods for ligand discovery |
| US6335155B1 (en) | 1998-06-26 | 2002-01-01 | Sunesis Pharmaceuticals, Inc. | Methods for rapidly identifying small organic molecule ligands for binding to biological target molecules |
| US6306897B1 (en) | 1999-03-19 | 2001-10-23 | Parker Hughes Institute | Calanolides for inhibiting BTK |
| US6921763B2 (en) | 1999-09-17 | 2005-07-26 | Abbott Laboratories | Pyrazolopyrimidines as therapeutic agents |
| CN1390219A (en) | 1999-09-17 | 2003-01-08 | 艾博特股份有限两合公司 | Pyrazolopyrimidines as therapeutic agents |
| US6506769B2 (en) | 1999-10-06 | 2003-01-14 | Boehringer Ingelheim Pharmaceuticals, Inc. | Heterocyclic compounds useful as inhibitors of tyrosine kinases |
| JP5036112B2 (en) | 1999-10-06 | 2012-09-26 | ベーリンガー インゲルハイム ファーマシューティカルズ インコーポレイテッド | Heterocyclic compounds useful as inhibitors of tyrosine kinases |
| AU4508601A (en) | 1999-11-30 | 2001-06-18 | Parker Hughes Institute | Inhibitors of collagen-induced platelet aggregation |
| AU2439701A (en) | 1999-12-17 | 2001-06-25 | Ariad Pharmaceuticals, Inc. | Novel heterocycles |
| GB0005345D0 (en) | 2000-03-06 | 2000-04-26 | Mathilda & Terence Kennedy Ins | Methods of treating sepsis septic shock and inflammation |
| AU2002236692A1 (en) | 2000-10-23 | 2002-05-21 | Bristol-Myers Squibb Company | Modulators of bruton's tyrosine kinase and bruton's tyrosine kinase, their identification and use |
| JP2004514732A (en) | 2000-12-06 | 2004-05-20 | ファルマシア・コーポレーション | Rapidly dispersing pharmaceutical composition |
| MXPA03008560A (en) | 2001-03-22 | 2004-06-30 | Abbot Gmbh & Co Kg | Single-stage pfc + ballast control circuit/general purpose power converter. |
| US8306897B2 (en) | 2001-05-04 | 2012-11-06 | Stockshield, Inc. | Method and system for insuring against investment loss |
| WO2003000187A2 (en) | 2001-06-21 | 2003-01-03 | Ariad Pharmaceuticals, Inc. | Novel pyrazolo-and pyrrolo-pyrimidines and uses thereof |
| EP2258371A1 (en) | 2001-08-10 | 2010-12-08 | Novartis AG | Use of c-Src inhibitors alone or in combination with STI571 for the treatment of leukaemia |
| WO2003016338A1 (en) | 2001-08-15 | 2003-02-27 | Parker Hughes Institute | Crystal structure of the btk kinase domain |
| WO2003046200A2 (en) | 2001-11-21 | 2003-06-05 | Sunesis Pharmaceuticals, Inc. | Methods for ligand discovery |
| US20050084905A1 (en) | 2002-03-21 | 2005-04-21 | Prescott John C. | Identification of kinase inhibitors |
| GB2388594A (en) | 2002-05-16 | 2003-11-19 | Bayer Ag | Imidazo-triazine PDE 4 inhibitors |
| WO2004014905A1 (en) | 2002-08-08 | 2004-02-19 | Boehringer Ingelheim Pharmaceuticals, Inc. | Substituted benzimidazole compounds |
| GB0303910D0 (en) | 2003-02-20 | 2003-03-26 | Merck Sharp & Dohme | Therapeutic agents |
| US7687506B2 (en) | 2003-04-11 | 2010-03-30 | The Regents Of The University Of California | Selective serine/threonine kinase inhibitors |
| WO2004100868A2 (en) | 2003-04-23 | 2004-11-25 | Abbott Laboratories | Method of treating transplant rejection |
| EP1473039A1 (en) | 2003-05-02 | 2004-11-03 | Centre National De La Recherche Scientifique (Cnrs) | Use of inhibitors and antisense oligonucleotides of BTK for the treatment of proliferative mastocytosis |
| US7405295B2 (en) | 2003-06-04 | 2008-07-29 | Cgi Pharmaceuticals, Inc. | Certain imidazo[1,2-a]pyrazin-8-ylamines and method of inhibition of Bruton's tyrosine kinase by such compounds |
| WO2005005429A1 (en) | 2003-06-30 | 2005-01-20 | Cellular Genomics, Inc. | Certain heterocyclic substituted imidazo[1,2-a]pyrazin-8-ylamines and methods of inhibition of bruton’s tyrosine kinase by such compounds |
| US8131475B2 (en) | 2003-09-03 | 2012-03-06 | The United States Of America As Represented By The Secretary, Department Of Health And Human Services | Methods for identifying, diagnosing, and predicting survival of lymphomas |
| CN1897950A (en) | 2003-10-14 | 2007-01-17 | 惠氏公司 | Fused-aryl and heteroaryl derivatives and methods of their use |
| UA83509C2 (en) | 2003-10-15 | 2008-07-25 | Оси Фармасьютикалз, Инк. | Imidazopyrazines as tyrosine kinase inhibitors |
| WO2005060956A1 (en) | 2003-12-12 | 2005-07-07 | University Of Maryland, Baltimore | IMMUNOMODULATORY COMPOUNDS THAT TARGET AND INHIBIT THE pY+3 BINDING SITE OF TYROSENE KINASE p56 LCK SH2 DOMAIN |
| BRPI0418031A (en) | 2003-12-22 | 2007-04-17 | Gilead Sciences Inc | phosphonate-substituted kinase inhibitors |
| JP2007519742A (en) | 2004-01-26 | 2007-07-19 | バーテックス ファーマシューティカルズ インコーポレイテッド | Compositions useful as inhibitors of protein kinases |
| EP1730148A4 (en) | 2004-02-03 | 2009-08-19 | Abbott Lab | Aminobenzoxazoles as therapeutic agents |
| AR053090A1 (en) | 2004-07-20 | 2007-04-25 | Osi Pharm Inc | IMIDAZOTRIAZINS AS PROTEIN KINASE INHIBITORS AND THEIR USE FOR THE PREPARATION OF MEDICINES |
| CA2581375A1 (en) | 2004-09-27 | 2006-04-06 | Kosan Biosciences Incorporated | Specific kinase inhibitors |
| KR20070063562A (en) | 2004-09-28 | 2007-06-19 | 얀센 파마슈티카 엔.브이. | Substituted dipiperidine ccr2 antagonists |
| WO2006053121A2 (en) | 2004-11-10 | 2006-05-18 | Cgi Pharmaceuticals, Inc. | Imidazo[1 , 2-a] pyrazin-8-ylamines useful as modulators of kinase activity |
| GB0425035D0 (en) | 2004-11-12 | 2004-12-15 | Novartis Ag | Organic compounds |
| KR100735639B1 (en) * | 2004-12-29 | 2007-07-04 | 한미약품 주식회사 | Quinazoline derivatives inhibiting the growth of cancer cell and preparation thereof |
| TW200716551A (en) | 2005-03-10 | 2007-05-01 | Cgi Pharmaceuticals Inc | Certain substituted amides, method of making, and method of use thereof |
| PL1891066T3 (en) | 2005-05-13 | 2011-05-31 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| TWI473808B (en) | 2005-06-22 | 2015-02-21 | Plexxikon Inc | Compounds and methods for kinase modulation, and indications therefor |
| US20070065449A1 (en) | 2005-09-16 | 2007-03-22 | Claire Verschraegen | Method of treating cancer, especially soft tissue sarcoma utilizing gemcitabine in combination with docetaxel and anti-VEGF therapy (bevacizumab) |
| EP1957491A2 (en) | 2005-11-12 | 2008-08-20 | Boehringer Ingelheim International GmbH | Pyrrolo (2,3-b) pyridine derivatives useful as tec kinase inhibitors |
| WO2007087068A2 (en) | 2006-01-13 | 2007-08-02 | Pharmacyclics, Inc. | Inhibitors of tyrosine kinases and uses thereof |
| ES2420834T3 (en) | 2006-01-30 | 2013-08-27 | The Scripps Research Institute | Methods of detection of circulating tumor cells and methods of diagnosis of cancer in a mammalian subject |
| PT2004654E (en) | 2006-04-04 | 2013-08-27 | Univ California | Pyrazolopyrimidine derivatives for use as kinase antagonists |
| MX2008014450A (en) | 2006-05-18 | 2009-03-09 | Mannkind Corp | Intracellular kinase inhibitors. |
| EP2529621B1 (en) | 2006-09-22 | 2016-10-05 | Pharmacyclics LLC | Inhibitors of bruton's tyrosine kinase |
| CA2668286C (en) | 2006-11-03 | 2014-09-16 | Pharmacyclics, Inc. | Bruton's tyrosine kinase activity probe and method of using |
| NL2000640C2 (en) | 2007-03-05 | 2008-09-08 | Stichting Wetsus Ct Of Excelle | Method and system for purifying a liquid. |
| US8809273B2 (en) | 2007-03-28 | 2014-08-19 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US20120065201A1 (en) | 2007-03-28 | 2012-03-15 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| CA3001152A1 (en) | 2007-03-28 | 2008-10-09 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US20090010911A1 (en) | 2007-04-06 | 2009-01-08 | Iowa State University Research Foundation, Inc. | Methods and compositions for affecting cyclophilin a regulation of kinases in modulating cellular activities |
| CN105367503A (en) | 2007-10-19 | 2016-03-02 | 阿维拉制药公司 | Heteroaryl compounds and uses thereof |
| US7989465B2 (en) | 2007-10-19 | 2011-08-02 | Avila Therapeutics, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| JP5587193B2 (en) | 2007-10-23 | 2014-09-10 | エフ.ホフマン−ラ ロシュ アーゲー | Novel kinase inhibitors |
| US8426441B2 (en) | 2007-12-14 | 2013-04-23 | Roche Palo Alto Llc | Inhibitors of bruton's tyrosine kinase |
| US20150152115A1 (en) | 2007-12-27 | 2015-06-04 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| AU2009262068C1 (en) | 2008-06-27 | 2015-07-02 | Celgene Car Llc | Heteroaryl compounds and uses thereof |
| EP2307025B1 (en) | 2008-07-16 | 2017-09-20 | Pharmacyclics LLC | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors |
| CN105574346A (en) | 2008-09-05 | 2016-05-11 | 新基阿维罗米克斯研究公司 | Design method and detection method for polypeptide conjugate and irreversible inhibitor |
| US8426428B2 (en) | 2008-12-05 | 2013-04-23 | Principia Biopharma, Inc. | EGFR kinase knockdown via electrophilically enhanced inhibitors |
| WO2010126960A1 (en) | 2009-04-29 | 2010-11-04 | Locus Pharmaceuticals, Inc. | Pyrrolotriazine compounds |
| CN105063001A (en) | 2009-09-16 | 2015-11-18 | 新基阿维罗米克斯研究公司 | Protein kinase complex, pharmaceutical composition containing the same and use thereof |
| US7718662B1 (en) | 2009-10-12 | 2010-05-18 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of bruton's tyrosine kinase |
| MY189483A (en) | 2010-05-31 | 2022-02-16 | Ono Pharmaceutical Co | Purinone derivative |
| KR101580714B1 (en) | 2010-06-03 | 2016-01-04 | 파마싸이클릭스 엘엘씨 | The use of inhibitors of bruton's tyrosine kinase (btk) |
| US20120071497A1 (en) | 2010-06-03 | 2012-03-22 | Pharmacyclics, Inc. | Methods of treating abc-dlbcl using inhibitors of bruton's tyrosine kinase |
| CN102947316B (en) | 2010-06-23 | 2016-08-10 | 韩美科学株式会社 | For suppressing the novel fused pyrimidine derivatives of tyrosine kinase activity |
| WO2012021444A1 (en) | 2010-08-10 | 2012-02-16 | Avila Therapeutics, Inc. | Besylate salt of a btk inhibitor |
| CN103534258B (en) | 2011-05-17 | 2016-09-14 | 普林斯匹亚生物制药公司 | Tyrosine kinase inhibitor |
| CA2841886C (en) * | 2011-07-19 | 2016-08-16 | Merck Sharp & Dohme B.V. | 4-imidazopyridazin-1-yl-benzamides and 4-imidazotriazin-1-yl-benzamides as btk-inhibitors |
| US20130179327A1 (en) | 2011-12-09 | 2013-07-11 | Mortgage Market Analytics Llc | Adaptable assumable mortgage |
| US8377946B1 (en) | 2011-12-30 | 2013-02-19 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| US8501724B1 (en) | 2012-01-31 | 2013-08-06 | Pharmacyclics, Inc. | Purinone compounds as kinase inhibitors |
| CA3218491A1 (en) | 2012-06-04 | 2013-12-12 | Pharmacyclics Llc | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| BR112015001690A2 (en) | 2012-07-24 | 2017-11-07 | Pharmacyclics Inc | mutations associated with resistance to bruton tyrosine kinase inhibitors (btk) |
| MX2015006168A (en) | 2012-11-15 | 2015-08-10 | Pharmacyclics Inc | Pyrrolopyrimidine compounds as kinase inhibitors. |
| CA2914284A1 (en) | 2013-05-30 | 2014-12-04 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using pi3 kinase isoform modulators |
| EP3027192A4 (en) | 2013-08-02 | 2017-03-22 | Pharmacyclics, LLC | Methods for the treatment of solid tumors |
| TWI617309B (en) | 2013-10-25 | 2018-03-11 | 製藥公司 | Treatment using bruton's tyrosine kinase inhibitors and immunotherapy |
| CN105949197A (en) | 2014-01-29 | 2016-09-21 | 苏州晶云药物科技有限公司 | Novel crystal forms of Ibrutinib and preparation method of novel crystal forms |
| WO2015127261A1 (en) | 2014-02-21 | 2015-08-27 | Pharmacyclics, Inc. | Biomarkers for predicting response of dlbcl to treatment with ibrutinib |
-
2011
- 2011-12-29 US US13/340,276 patent/US8809273B2/en active Active
- 2011-12-29 US US13/340,621 patent/US20120101114A1/en not_active Abandoned
- 2011-12-29 US US13/340,559 patent/US20120101113A1/en not_active Abandoned
-
2012
- 2012-02-24 US US13/404,422 patent/US20120165328A1/en not_active Abandoned
- 2012-03-01 US US13/410,110 patent/US20120184013A1/en not_active Abandoned
- 2012-03-26 US US13/430,173 patent/US20120184567A1/en not_active Abandoned
- 2012-04-04 US US13/439,775 patent/US20120202264A1/en not_active Abandoned
- 2012-07-06 US US13/543,394 patent/US9139591B2/en active Active
- 2012-07-06 US US13/543,399 patent/US20120296089A1/en not_active Abandoned
- 2012-07-06 US US13/543,065 patent/US9079908B2/en active Active
- 2012-09-07 US US13/606,949 patent/US8940750B2/en active Active
- 2012-09-07 US US13/607,036 patent/US20120329130A1/en not_active Abandoned
- 2012-09-12 US US13/612,143 patent/US20130018060A1/en not_active Abandoned
-
2013
- 2013-10-31 US US14/069,222 patent/US20140057907A1/en not_active Abandoned
-
2014
- 2014-02-12 US US14/179,457 patent/US20140163027A1/en not_active Abandoned
-
2015
- 2015-01-26 US US14/605,854 patent/US9181263B2/en active Active
- 2015-01-26 US US14/605,857 patent/US20150265618A1/en not_active Abandoned
- 2015-05-04 US US14/703,750 patent/US20150307500A1/en not_active Abandoned
- 2015-07-08 US US14/794,685 patent/US9556182B2/en active Active
-
2017
- 2017-08-31 US US15/692,261 patent/US20170362246A1/en not_active Abandoned
-
2019
- 2019-03-22 US US16/361,494 patent/US20200055859A1/en not_active Abandoned
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10751342B2 (en) | 2010-06-03 | 2020-08-25 | Pharmacyclics Llc | Use of inhibitors of Bruton's tyrosine kinase (Btk) |
| US11672803B2 (en) | 2010-06-03 | 2023-06-13 | Pharmacyclics Llc | Use of inhibitors of Brutons tyrosine kinase (Btk) |
| US10954567B2 (en) | 2012-07-24 | 2021-03-23 | Pharmacyclics Llc | Mutations associated with resistance to inhibitors of Bruton's Tyrosine Kinase (BTK) |
Also Published As
| Publication number | Publication date |
|---|---|
| US20120184567A1 (en) | 2012-07-19 |
| US20120202264A1 (en) | 2012-08-09 |
| US9556182B2 (en) | 2017-01-31 |
| US20120165328A1 (en) | 2012-06-28 |
| US20120178753A1 (en) | 2012-07-12 |
| US8940750B2 (en) | 2015-01-27 |
| US20150265618A1 (en) | 2015-09-24 |
| US20120277255A1 (en) | 2012-11-01 |
| US20150133661A1 (en) | 2015-05-14 |
| US20170362246A1 (en) | 2017-12-21 |
| US20140163027A1 (en) | 2014-06-12 |
| US8809273B2 (en) | 2014-08-19 |
| US20120296089A1 (en) | 2012-11-22 |
| US9079908B2 (en) | 2015-07-14 |
| US20120277225A1 (en) | 2012-11-01 |
| US20120101114A1 (en) | 2012-04-26 |
| US20150306106A1 (en) | 2015-10-29 |
| US20140057907A1 (en) | 2014-02-27 |
| US20130035334A1 (en) | 2013-02-07 |
| US20130018060A1 (en) | 2013-01-17 |
| US20120184013A1 (en) | 2012-07-19 |
| US20120329130A1 (en) | 2012-12-27 |
| US9139591B2 (en) | 2015-09-22 |
| US20150307500A1 (en) | 2015-10-29 |
| US20120101113A1 (en) | 2012-04-26 |
| US9181263B2 (en) | 2015-11-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20200055859A1 (en) | Inhibitors of brutons tyrosine kinase | |
| US8883435B2 (en) | Inhibitors of Bruton's tyrosine kinase | |
| EP2139487B1 (en) | Inhibitors of bruton's tyrosine kinase | |
| US20120065201A1 (en) | Inhibitors of bruton's tyrosine kinase | |
| US20150152115A1 (en) | Inhibitors of bruton's tyrosine kinase | |
| US20110224235A1 (en) | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors | |
| US20240317756A1 (en) | Inhibitors of bruton's tyrosine kinase | |
| AU2013276959A1 (en) | Inhibitors of Bruton's tyrosine kinase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: NON FINAL ACTION MAILED |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: NON FINAL ACTION MAILED |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: RESPONSE TO NON-FINAL OFFICE ACTION ENTERED AND FORWARDED TO EXAMINER |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |