US20100035891A1 - Pharmaceutically Active Compounds - Google Patents
Pharmaceutically Active Compounds Download PDFInfo
- Publication number
- US20100035891A1 US20100035891A1 US12/469,311 US46931109A US2010035891A1 US 20100035891 A1 US20100035891 A1 US 20100035891A1 US 46931109 A US46931109 A US 46931109A US 2010035891 A1 US2010035891 A1 US 2010035891A1
- Authority
- US
- United States
- Prior art keywords
- ethyl
- ylsulphonyl
- pyrazolo
- dihydro
- pyrimidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 [1*]C.[2*]c1nnc2c1N=C(C1=CC([4*])=CN=C1[13*])NC2=O Chemical compound [1*]C.[2*]c1nnc2c1N=C(C1=CC([4*])=CN=C1[13*])NC2=O 0.000 description 42
- QWTDNUCVQCZILF-UHFFFAOYSA-N CCC(C)C Chemical compound CCC(C)C QWTDNUCVQCZILF-UHFFFAOYSA-N 0.000 description 9
- VNKYTQGIUYNRMY-UHFFFAOYSA-N CCCOC Chemical compound CCCOC VNKYTQGIUYNRMY-UHFFFAOYSA-N 0.000 description 5
- FOTXAJDDGPYIFU-UHFFFAOYSA-N CCC1CC1 Chemical compound CCC1CC1 FOTXAJDDGPYIFU-UHFFFAOYSA-N 0.000 description 4
- GHKYDKKHTDOYEI-UHFFFAOYSA-N CCC1=C(N)C(C(N)=O)=NN1CCOC Chemical compound CCC1=C(N)C(C(N)=O)=NN1CCOC GHKYDKKHTDOYEI-UHFFFAOYSA-N 0.000 description 3
- NEZRFXZYPAIZAD-UHFFFAOYSA-N CCC1CCC1 Chemical compound CCC1CCC1 NEZRFXZYPAIZAD-UHFFFAOYSA-N 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N CCCCC Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- FCVTUHVLKGWQKP-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCOC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCOC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 FCVTUHVLKGWQKP-UHFFFAOYSA-N 0.000 description 3
- YPFZMBHKIVDSNR-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 YPFZMBHKIVDSNR-UHFFFAOYSA-N 0.000 description 3
- DUVULSZACJKOQF-UHFFFAOYSA-N CC1(C)C2CCC1(CS(=O)(=O)O)C(=O)C2.CCOC1=NC=C(S(=O)(=O)N2CCN(CC)CC2)C=C1C1=NC2=C(CC)N(CCOC)N=C2C(=O)N1 Chemical compound CC1(C)C2CCC1(CS(=O)(=O)O)C(=O)C2.CCOC1=NC=C(S(=O)(=O)N2CCN(CC)CC2)C=C1C1=NC2=C(CC)N(CCOC)N=C2C(=O)N1 DUVULSZACJKOQF-UHFFFAOYSA-N 0.000 description 2
- XKIIVXRFPFFJLJ-UHFFFAOYSA-N CC1=NC=C([N+](=O)[O-])C=C1C(=O)O.CC1=NC=C([N+](=O)[O-])C=C1C(=O)O.CC1=NC=C([N+](=O)[O-])C=C1C(=O)O Chemical compound CC1=NC=C([N+](=O)[O-])C=C1C(=O)O.CC1=NC=C([N+](=O)[O-])C=C1C(=O)O.CC1=NC=C([N+](=O)[O-])C=C1C(=O)O XKIIVXRFPFFJLJ-UHFFFAOYSA-N 0.000 description 2
- GDOPTJXRTPNYNR-UHFFFAOYSA-N CC1CCCC1 Chemical compound CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 2
- UAEPNZWRGJTJPN-UHFFFAOYSA-N CC1CCCCC1 Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 2
- TVSMLBGFGKLKOO-UHFFFAOYSA-N CC1CCN(C)CC1 Chemical compound CC1CCN(C)CC1 TVSMLBGFGKLKOO-UHFFFAOYSA-N 0.000 description 2
- OVRKATYHWPCGPZ-UHFFFAOYSA-N CC1CCOCC1 Chemical compound CC1CCOCC1 OVRKATYHWPCGPZ-UHFFFAOYSA-N 0.000 description 2
- ILBZSNOGPTUKHC-UHFFFAOYSA-N CC1CN(C)C1 Chemical compound CC1CN(C)C1 ILBZSNOGPTUKHC-UHFFFAOYSA-N 0.000 description 2
- GBNIXNFORFLIJY-INIZCTEOSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1[C@@H](C)CC Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1[C@@H](C)CC GBNIXNFORFLIJY-INIZCTEOSA-N 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N CCC1=CC=CC=C1 Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- IFTRQJLVEBNKJK-UHFFFAOYSA-N CCC1CCCC1 Chemical compound CCC1CCCC1 IFTRQJLVEBNKJK-UHFFFAOYSA-N 0.000 description 2
- DJEQZVQFEPKLOY-UHFFFAOYSA-N CCCCN(C)C Chemical compound CCCCN(C)C DJEQZVQFEPKLOY-UHFFFAOYSA-N 0.000 description 2
- GKOWDLWWAZINFZ-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)N(CCN(C)S(C)(=O)=O)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)N(CCN(C)S(C)(=O)=O)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 GKOWDLWWAZINFZ-UHFFFAOYSA-N 0.000 description 2
- ZUHZZVMEUAUWHY-UHFFFAOYSA-N CCCN(C)C Chemical compound CCCN(C)C ZUHZZVMEUAUWHY-UHFFFAOYSA-N 0.000 description 2
- DUZUGIAXDBVLBS-UHFFFAOYSA-N CCN1CC(C)C1 Chemical compound CCN1CC(C)C1 DUZUGIAXDBVLBS-UHFFFAOYSA-N 0.000 description 2
- GMRKCXFZJBCTEM-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 GMRKCXFZJBCTEM-UHFFFAOYSA-N 0.000 description 2
- CZNILTGORLWQMB-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CN(C)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CN(C)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 CZNILTGORLWQMB-UHFFFAOYSA-N 0.000 description 2
- UNXPOOHHOFQHOO-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CC4CCCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CC4CCCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 UNXPOOHHOFQHOO-UHFFFAOYSA-N 0.000 description 2
- UPSZJGWEONPGGZ-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(N)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(N)C=N1 UPSZJGWEONPGGZ-UHFFFAOYSA-N 0.000 description 2
- KCIISMUOZNFOOJ-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(C(C)C)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(C(C)C)CC2)C=N1 KCIISMUOZNFOOJ-UHFFFAOYSA-N 0.000 description 2
- ZUWITCSMJULFEU-UHFFFAOYSA-N OC(c(cc(cn1)S(O)(=O)=O)c1O)=O Chemical compound OC(c(cc(cn1)S(O)(=O)=O)c1O)=O ZUWITCSMJULFEU-UHFFFAOYSA-N 0.000 description 2
- UEYQJQVBUVAELZ-UHFFFAOYSA-N OC(c(cccn1)c1O)=O Chemical compound OC(c(cccn1)c1O)=O UEYQJQVBUVAELZ-UHFFFAOYSA-N 0.000 description 2
- ARCPUQZCMKBNGO-UHFFFAOYSA-N C.CCCC(C)OC1=C(C2=NC3=C(CC)N(C4CNC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound C.CCCC(C)OC1=C(C2=NC3=C(CC)N(C4CNC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 ARCPUQZCMKBNGO-UHFFFAOYSA-N 0.000 description 1
- RFDNHUQVWQFWKT-UHFFFAOYSA-N C.CCCOC1=C(C2=NC3=C(CC)N(CCNC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound C.CCCOC1=C(C2=NC3=C(CC)N(CCNC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 RFDNHUQVWQFWKT-UHFFFAOYSA-N 0.000 description 1
- NAZVAXYLJSVIFQ-UHFFFAOYSA-N C.CCOC1=C(C(=O)NC2=C(CC)N(C3CCNCC3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound C.CCOC1=C(C(=O)NC2=C(CC)N(C3CCNCC3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 NAZVAXYLJSVIFQ-UHFFFAOYSA-N 0.000 description 1
- SVJSSHDFKKDAFW-UHFFFAOYSA-N C.CCOC1=C(C2=NC3=C(CC)N(C4=NC=CC=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound C.CCOC1=C(C2=NC3=C(CC)N(C4=NC=CC=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 SVJSSHDFKKDAFW-UHFFFAOYSA-N 0.000 description 1
- OTYTXMXJQFYCRW-UHFFFAOYSA-N C.CCOC1=C(C2=NC3=C(CC)N(C4CCNCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound C.CCOC1=C(C2=NC3=C(CC)N(C4CCNCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 OTYTXMXJQFYCRW-UHFFFAOYSA-N 0.000 description 1
- SPBHFHMMWKMJFT-UHFFFAOYSA-N C.CCOC1=C(C2=NC3=C(CC)N(C4CNC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound C.CCOC1=C(C2=NC3=C(CC)N(C4CNC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 SPBHFHMMWKMJFT-UHFFFAOYSA-N 0.000 description 1
- XPDIKRMPZNLBAC-UHFFFAOYSA-N CC(C)(C)OC(=O)N1CC(I)C1 Chemical compound CC(C)(C)OC(=O)N1CC(I)C1 XPDIKRMPZNLBAC-UHFFFAOYSA-N 0.000 description 1
- VROBVTOKXNHTBI-UHFFFAOYSA-N CC1=CC=C(S(=O)(=O)OCC2CCCC2)C=C1 Chemical compound CC1=CC=C(S(=O)(=O)OCC2CCCC2)C=C1 VROBVTOKXNHTBI-UHFFFAOYSA-N 0.000 description 1
- ZFZWXJXSISUYHJ-UHFFFAOYSA-N CC1=NC=C(N)C=C1C(=O)O.CC1=NC=C(N)C=C1C(=O)O.CC1=NC=C(N)C=C1C(=O)O Chemical compound CC1=NC=C(N)C=C1C(=O)O.CC1=NC=C(N)C=C1C(=O)O.CC1=NC=C(N)C=C1C(=O)O ZFZWXJXSISUYHJ-UHFFFAOYSA-N 0.000 description 1
- FPJAOGIVTMGBBH-UHFFFAOYSA-K CC1=NC=C(S(=O)O[Y])C=C1C(=O)O.CC1=NC=C(S(=O)O[Y])C=C1C(=O)O.CC1=NC=C(S(=O)O[Y])C=C1C(=O)O Chemical compound CC1=NC=C(S(=O)O[Y])C=C1C(=O)O.CC1=NC=C(S(=O)O[Y])C=C1C(=O)O.CC1=NC=C(S(=O)O[Y])C=C1C(=O)O FPJAOGIVTMGBBH-UHFFFAOYSA-K 0.000 description 1
- RSAHOMRSRWAPAF-UHFFFAOYSA-N CC1CN(C(=O)OC(C)(C)C)C1 Chemical compound CC1CN(C(=O)OC(C)(C)C)C1 RSAHOMRSRWAPAF-UHFFFAOYSA-N 0.000 description 1
- KHRLPNHFWHYDSC-UHFFFAOYSA-N CCC1=C(N)C(C(N)=O)=NN1CCN(C)C(=O)OCC1=CC=CC=C1 Chemical compound CCC1=C(N)C(C(N)=O)=NN1CCN(C)C(=O)OCC1=CC=CC=C1 KHRLPNHFWHYDSC-UHFFFAOYSA-N 0.000 description 1
- PCZZXKZPFCDOBR-UHFFFAOYSA-N CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1C1CN(C(=O)OC(C)(C)C)C1 Chemical compound CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1C1CN(C(=O)OC(C)(C)C)C1 PCZZXKZPFCDOBR-UHFFFAOYSA-N 0.000 description 1
- UKBJFKLSSOFLFY-UHFFFAOYSA-N CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCN(C)C(=O)OCC1=CC=CC=C1 Chemical compound CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCN(C)C(=O)OCC1=CC=CC=C1 UKBJFKLSSOFLFY-UHFFFAOYSA-N 0.000 description 1
- GYWKKQHZMMSMKE-UHFFFAOYSA-N CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCOC Chemical compound CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCOC GYWKKQHZMMSMKE-UHFFFAOYSA-N 0.000 description 1
- FGHIYSSINMKTOB-UHFFFAOYSA-N CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCOC.CCC1=NN(CCOC)C(C(N)=O)=C1[N+](=O)[O-] Chemical compound CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCOC.CCC1=NN(CCOC)C(C(N)=O)=C1[N+](=O)[O-] FGHIYSSINMKTOB-UHFFFAOYSA-N 0.000 description 1
- LKMWWOKLUDPKDP-UHFFFAOYSA-N CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCO[Si](C)(C)C(C)(C)C Chemical compound CCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCO[Si](C)(C)C(C)(C)C LKMWWOKLUDPKDP-UHFFFAOYSA-N 0.000 description 1
- MRVKISAJENLFTB-UHFFFAOYSA-N CCC1=C2N=C(C3=C(N4CCCC4)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CC1CCC1 Chemical compound CCC1=C2N=C(C3=C(N4CCCC4)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CC1CCC1 MRVKISAJENLFTB-UHFFFAOYSA-N 0.000 description 1
- PRFXQKMNCGVJTO-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCO.CCOC1=C(C2=NC3=C(CC)N(CCO)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCO.CCOC1=C(C2=NC3=C(CC)N(CCO)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 PRFXQKMNCGVJTO-UHFFFAOYSA-N 0.000 description 1
- WYEMIYXBIBIAQO-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC.CCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC Chemical compound CCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC.CCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC WYEMIYXBIBIAQO-UHFFFAOYSA-N 0.000 description 1
- UZTBWIADHKSPTG-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCC(C)C)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CCOC Chemical compound CCC1=C2N=C(C3=C(OCC(C)C)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CCOC UZTBWIADHKSPTG-UHFFFAOYSA-N 0.000 description 1
- SISGKOAGBAJWPA-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCC(C)C)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1CCN(C)CC1 Chemical compound CCC1=C2N=C(C3=C(OCC(C)C)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1CCN(C)CC1 SISGKOAGBAJWPA-UHFFFAOYSA-N 0.000 description 1
- KPSBFMNMIOHYPY-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CC(C)C Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CC(C)C KPSBFMNMIOHYPY-UHFFFAOYSA-N 0.000 description 1
- ROTQJGWQFIKDMZ-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CC1CC1 Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CC1CC1 ROTQJGWQFIKDMZ-UHFFFAOYSA-N 0.000 description 1
- JQXAVRJTJOJMSE-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CC1CCC1 Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CC1CCC1 JQXAVRJTJOJMSE-UHFFFAOYSA-N 0.000 description 1
- GBNIXNFORFLIJY-MRXNPFEDSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1[C@H](C)CC Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1[C@H](C)CC GBNIXNFORFLIJY-MRXNPFEDSA-N 0.000 description 1
- BZOGTWGGSZIOKW-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1 Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1 BZOGTWGGSZIOKW-UHFFFAOYSA-N 0.000 description 1
- VMKMXEFVKPDPDC-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C(C)CC Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C(C)CC VMKMXEFVKPDPDC-UHFFFAOYSA-N 0.000 description 1
- QQXLMOBJFYPLBM-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C(C)COC Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C(C)COC QQXLMOBJFYPLBM-UHFFFAOYSA-N 0.000 description 1
- FOOKSXFTEWJFEU-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1=CC=CC=C1 Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1=CC=CC=C1 FOOKSXFTEWJFEU-UHFFFAOYSA-N 0.000 description 1
- MVINGZXKOVPFKX-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1=NC2=C(C=CC=C2)O1 Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1=NC2=C(C=CC=C2)O1 MVINGZXKOVPFKX-UHFFFAOYSA-N 0.000 description 1
- RPEWDRLCUSXLLE-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1=NC=CC=N1 Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1=NC=CC=N1 RPEWDRLCUSXLLE-UHFFFAOYSA-N 0.000 description 1
- XKRXMWPVDJKAJI-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1CCC1 Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1C1CCC1 XKRXMWPVDJKAJI-UHFFFAOYSA-N 0.000 description 1
- NLNFVNNWOWUWAC-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CC(C)C Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CC(C)C NLNFVNNWOWUWAC-UHFFFAOYSA-N 0.000 description 1
- ZOSSPVVBOIYOBG-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CC1CCC1 Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CC1CCC1 ZOSSPVVBOIYOBG-UHFFFAOYSA-N 0.000 description 1
- VYFHWUGIGFPUHL-UHFFFAOYSA-N CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC Chemical compound CCC1=C2N=C(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC VYFHWUGIGFPUHL-UHFFFAOYSA-N 0.000 description 1
- NRGGMCIBEHEAIL-UHFFFAOYSA-N CCC1=NC=CC=C1 Chemical compound CCC1=NC=CC=C1 NRGGMCIBEHEAIL-UHFFFAOYSA-N 0.000 description 1
- QYDDQYQBBQASQB-UHFFFAOYSA-N CCC1=NN(CCOC)C(C(N)=O)=C1N Chemical compound CCC1=NN(CCOC)C(C(N)=O)=C1N QYDDQYQBBQASQB-UHFFFAOYSA-N 0.000 description 1
- WCQYJWRMYFJLRQ-UHFFFAOYSA-N CCC1=NNC(C(=O)O)=C1 Chemical compound CCC1=NNC(C(=O)O)=C1 WCQYJWRMYFJLRQ-UHFFFAOYSA-N 0.000 description 1
- UDYWTLMYXXTHKR-UHFFFAOYSA-N CCC1=NNC(C(=O)O)=C1[N+](=O)[O-] Chemical compound CCC1=NNC(C(=O)O)=C1[N+](=O)[O-] UDYWTLMYXXTHKR-UHFFFAOYSA-N 0.000 description 1
- XTPUQGFKKAURLH-UHFFFAOYSA-N CCC1=NNC(C(N)=O)=C1[N+](=O)[O-] Chemical compound CCC1=NNC(C(N)=O)=C1[N+](=O)[O-] XTPUQGFKKAURLH-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N CCCC Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- XHLCOJUEKSXUTP-UHFFFAOYSA-N CCCC(C)OC1=C(C2=NC3=C(CC)N(C4CN(C(=O)OC(C)(C)C)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCC(C)OC1=C(C2=NC3=C(CC)N(C4CN(C(=O)OC(C)(C)C)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 XHLCOJUEKSXUTP-UHFFFAOYSA-N 0.000 description 1
- IRDSMINGIWFYDX-UHFFFAOYSA-N CCCC1=C(NC(=O)C2=C(OCC)N=CC(S(=O)(=O)N3CCN(C)CC3)=C2)C(C(N)=O)=NN1CCOC Chemical compound CCCC1=C(NC(=O)C2=C(OCC)N=CC(S(=O)(=O)N3CCN(C)CC3)=C2)C(C(N)=O)=NN1CCOC IRDSMINGIWFYDX-UHFFFAOYSA-N 0.000 description 1
- XWAPCZPLSIPCBO-UHFFFAOYSA-N CCCC1=C(NC(=O)C2=C(OCC)N=CC(S(=O)(=O)N3CCN(CC)CC3)=C2)C(C(N)=O)=NN1CCOC Chemical compound CCCC1=C(NC(=O)C2=C(OCC)N=CC(S(=O)(=O)N3CCN(CC)CC3)=C2)C(C(N)=O)=NN1CCOC XWAPCZPLSIPCBO-UHFFFAOYSA-N 0.000 description 1
- JJCYXUXTWLSDOJ-UHFFFAOYSA-N CCCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCOC.CCCC1=NN(CCOC)C(C(N)=O)=C1[N+](=O)[O-] Chemical compound CCCC1=C([N+](=O)[O-])C(C(N)=O)=NN1CCOC.CCCC1=NN(CCOC)C(C(N)=O)=C1[N+](=O)[O-] JJCYXUXTWLSDOJ-UHFFFAOYSA-N 0.000 description 1
- CCBQKQVKDGJXEU-UHFFFAOYSA-N CCCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC.CCCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC Chemical compound CCCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC.CCCC1=C2N=C(C3=C(OC(C)COC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC CCBQKQVKDGJXEU-UHFFFAOYSA-N 0.000 description 1
- WHZLAQCYTKWPJL-UHFFFAOYSA-N CCCC1=C2N=C(C3=C(OCC(C)C)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC Chemical compound CCCC1=C2N=C(C3=C(OCC(C)C)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC WHZLAQCYTKWPJL-UHFFFAOYSA-N 0.000 description 1
- IPKJZIIPYPKKDR-UHFFFAOYSA-N CCCC1=C2N=C(C3=C(OCC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CCOC Chemical compound CCCC1=C2N=C(C3=C(OCC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)NC(=O)C2=NN1CCOC IPKJZIIPYPKKDR-UHFFFAOYSA-N 0.000 description 1
- USCAQVLUGMUUDT-UHFFFAOYSA-N CCCC1=C2N=C(C3=C(OCC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC Chemical compound CCCC1=C2N=C(C3=C(OCC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)NC(=O)C2=NN1CCOC USCAQVLUGMUUDT-UHFFFAOYSA-N 0.000 description 1
- ODLMAHJVESYWTB-UHFFFAOYSA-N CCCC1=CC=CC=C1 Chemical compound CCCC1=CC=CC=C1 ODLMAHJVESYWTB-UHFFFAOYSA-N 0.000 description 1
- YZLURMSOHHDSEI-UHFFFAOYSA-N CCCC1=NN(CCOC)C(C(N)=O)=C1N Chemical compound CCCC1=NN(CCOC)C(C(N)=O)=C1N YZLURMSOHHDSEI-UHFFFAOYSA-N 0.000 description 1
- VSQRXHMQCZYIRM-UHFFFAOYSA-N CCCC1=NN(CCOC)C(C(N)=O)=C1NC(=O)C1=C(OCC)N=CC(S(=O)(=O)N2CCN(C)CC2)=C1 Chemical compound CCCC1=NN(CCOC)C(C(N)=O)=C1NC(=O)C1=C(OCC)N=CC(S(=O)(=O)N2CCN(C)CC2)=C1 VSQRXHMQCZYIRM-UHFFFAOYSA-N 0.000 description 1
- UKUOUKZDQRZMPM-UHFFFAOYSA-N CCCC1=NN(CCOC)C2=C1N=C(C1=C(OCC)N=CC(S(=O)(=O)N3CCN(C)CC3)=C1)NC2=O Chemical compound CCCC1=NN(CCOC)C2=C1N=C(C1=C(OCC)N=CC(S(=O)(=O)N3CCN(C)CC3)=C1)NC2=O UKUOUKZDQRZMPM-UHFFFAOYSA-N 0.000 description 1
- IJUGCQRVRBBAFH-UHFFFAOYSA-N CCCCN1N=C2C(=O)NC(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)=NC2=C1CC Chemical compound CCCCN1N=C2C(=O)NC(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(C)CC4)=C3)=NC2=C1CC IJUGCQRVRBBAFH-UHFFFAOYSA-N 0.000 description 1
- CXBDYQVECUFKRK-UHFFFAOYSA-N CCCCOC Chemical compound CCCCOC CXBDYQVECUFKRK-UHFFFAOYSA-N 0.000 description 1
- PMZAAIHNCKIVAB-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(C(=O)N2)N(CC(=O)N(C)C)N=C3CC)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1.CCCCOC1=C(C2=NC3=C(CC)N(CC(=O)N(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(C(=O)N2)N(CC(=O)N(C)C)N=C3CC)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1.CCCCOC1=C(C2=NC3=C(CC)N(CC(=O)N(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 PMZAAIHNCKIVAB-UHFFFAOYSA-N 0.000 description 1
- HETKPZZEMFLZLD-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)N(C4CCN(C(=O)OC(C)(C)C)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)N(C4CCN(C(=O)OC(C)(C)C)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 HETKPZZEMFLZLD-UHFFFAOYSA-N 0.000 description 1
- XHEWFWLSKNTLNI-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)N(C4CCN(S(C)(=O)=O)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)N(C4CCN(S(C)(=O)=O)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 XHEWFWLSKNTLNI-UHFFFAOYSA-N 0.000 description 1
- DFPRTXSFCFCKAF-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)N(C4CN(C(=O)OC(C)(C)C)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)N(C4CN(C(=O)OC(C)(C)C)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 DFPRTXSFCFCKAF-UHFFFAOYSA-N 0.000 description 1
- DYDBSYBLOUPXRA-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)N(C4CN(C(C)=O)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)N(C4CN(C(C)=O)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 DYDBSYBLOUPXRA-UHFFFAOYSA-N 0.000 description 1
- NBKPLXZORUBWGA-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)N(CCN(C)C(=O)OC(C)(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)N(CCN(C)C(=O)OC(C)(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 NBKPLXZORUBWGA-UHFFFAOYSA-N 0.000 description 1
- KYLOFMQLLNEPBT-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)N(CCN(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)N(CCN(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 KYLOFMQLLNEPBT-UHFFFAOYSA-N 0.000 description 1
- IEKBOZAFRXXLHO-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 IEKBOZAFRXXLHO-UHFFFAOYSA-N 0.000 description 1
- ADSGEEZYZAYDJB-UHFFFAOYSA-N CCCCOC1=C(C2=NC3=C(CC)NN=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCCOC1=C(C2=NC3=C(CC)NN=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 ADSGEEZYZAYDJB-UHFFFAOYSA-N 0.000 description 1
- RKBLAXHVQYRXFV-UHFFFAOYSA-N CCCN(C)C(=O)OC(C)(C)C Chemical compound CCCN(C)C(=O)OC(C)(C)C RKBLAXHVQYRXFV-UHFFFAOYSA-N 0.000 description 1
- ADKDIYPITQHYAA-UHFFFAOYSA-N CCCN1CCN(S(=O)(=O)C2=CC(C3=NC4=C(CC)N(CCOC)N=C4C(=O)N3)=C(OCC)N=C2)CC1 Chemical compound CCCN1CCN(S(=O)(=O)C2=CC(C3=NC4=C(CC)N(CCOC)N=C4C(=O)N3)=C(OCC)N=C2)CC1 ADKDIYPITQHYAA-UHFFFAOYSA-N 0.000 description 1
- BTODKUJZXRUYSX-UHFFFAOYSA-N CCCOC1=C(C2=NC3=C(CC)N(CCN(C)C(=O)OC(C)(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCOC1=C(C2=NC3=C(CC)N(CCN(C)C(=O)OC(C)(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 BTODKUJZXRUYSX-UHFFFAOYSA-N 0.000 description 1
- JISQHCORFFRLLW-UHFFFAOYSA-N CCCOC1=C(C2=NC3=C(CC)N(CCN(C)C(C)=O)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCCOC1=C(C2=NC3=C(CC)N(CCN(C)C(C)=O)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 JISQHCORFFRLLW-UHFFFAOYSA-N 0.000 description 1
- FPTCHHKCUSVAOO-UHFFFAOYSA-N CCCO[Si](C)(C)C(C)(C)C Chemical compound CCCO[Si](C)(C)C(C)(C)C FPTCHHKCUSVAOO-UHFFFAOYSA-N 0.000 description 1
- XWPYRBXBZULFKE-UHFFFAOYSA-N CCN1CCN(S(=O)(=O)C2=CC(Br)=C(Cl)N=C2)CC1 Chemical compound CCN1CCN(S(=O)(=O)C2=CC(Br)=C(Cl)N=C2)CC1 XWPYRBXBZULFKE-UHFFFAOYSA-N 0.000 description 1
- ZFMHNOYNFAKUDY-UHFFFAOYSA-N CCOC(=O)C1=C(OCC)N=CC(S(=O)(=O)N2CCN(C)CC2)=C1 Chemical compound CCOC(=O)C1=C(OCC)N=CC(S(=O)(=O)N2CCN(C)CC2)=C1 ZFMHNOYNFAKUDY-UHFFFAOYSA-N 0.000 description 1
- YOXICXXCICYQKS-UHFFFAOYSA-N CCOC(=O)C1=C(OCC)N=CC(S(=O)(=O)N2CCN(CC)CC2)=C1 Chemical compound CCOC(=O)C1=C(OCC)N=CC(S(=O)(=O)N2CCN(CC)CC2)=C1 YOXICXXCICYQKS-UHFFFAOYSA-N 0.000 description 1
- YFONSBZEAHNVCW-UHFFFAOYSA-N CCOC(=O)C1=CC(CC)=NN1 Chemical compound CCOC(=O)C1=CC(CC)=NN1 YFONSBZEAHNVCW-UHFFFAOYSA-N 0.000 description 1
- RDXXMEQXCWOIFC-UHFFFAOYSA-N CCOC1=C(Br)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 Chemical compound CCOC1=C(Br)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 RDXXMEQXCWOIFC-UHFFFAOYSA-N 0.000 description 1
- ZMCLXTAKNVTOJO-UHFFFAOYSA-N CCOC1=C(Br)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(Br)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 ZMCLXTAKNVTOJO-UHFFFAOYSA-N 0.000 description 1
- KVMKNIIHJGVQMJ-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(C(N)=O)NN=C2CC)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(C(N)=O)NN=C2CC)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 KVMKNIIHJGVQMJ-UHFFFAOYSA-N 0.000 description 1
- KPTDZKRDBROYPO-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(C(N)=O)NN=C2CC)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(C(N)=O)NN=C2CC)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 KPTDZKRDBROYPO-UHFFFAOYSA-N 0.000 description 1
- TWHYQBGXUSCAFH-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(C(C)COC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(C(C)COC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 TWHYQBGXUSCAFH-UHFFFAOYSA-N 0.000 description 1
- FEXJCGDDOPQBHD-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(C3=CC=C([N+](=O)[O-])C=C3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(C3=CC=C([N+](=O)[O-])C=C3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 FEXJCGDDOPQBHD-UHFFFAOYSA-N 0.000 description 1
- DXSREGBEMSUZNY-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(C3CCC3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(C3CCC3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 DXSREGBEMSUZNY-UHFFFAOYSA-N 0.000 description 1
- KZNMXDFZWNHYHT-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(C3CCN(C(=O)OC(C)(C)C)CC3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(C3CCN(C(=O)OC(C)(C)C)CC3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 KZNMXDFZWNHYHT-UHFFFAOYSA-N 0.000 description 1
- ZIQKEGFLCSLVPN-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(C3CCN(C)CC3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(C3CCN(C)CC3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 ZIQKEGFLCSLVPN-UHFFFAOYSA-N 0.000 description 1
- JXSPDAHWDJOGIL-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(C3CN(C(=O)OC(C)(C)C)C3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(C3CN(C(=O)OC(C)(C)C)C3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 JXSPDAHWDJOGIL-UHFFFAOYSA-N 0.000 description 1
- LUZJBGZNFKGQOP-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(C3CN(C)C3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(C3CN(C)C3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 LUZJBGZNFKGQOP-UHFFFAOYSA-N 0.000 description 1
- AKWUTPRXCPBDNB-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CC(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CC(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 AKWUTPRXCPBDNB-UHFFFAOYSA-N 0.000 description 1
- XRGNFVWDSQGSMZ-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CC3CCCO3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CC3CCCO3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 XRGNFVWDSQGSMZ-UHFFFAOYSA-N 0.000 description 1
- TZKBUYPFCDOSQG-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCCN(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCCN(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 TZKBUYPFCDOSQG-UHFFFAOYSA-N 0.000 description 1
- OZSRJKKMXJGGNQ-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCN(C)C(=O)OC(C)(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCN(C)C(=O)OC(C)(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 OZSRJKKMXJGGNQ-UHFFFAOYSA-N 0.000 description 1
- YSJZFNFOMOEMIZ-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCN(C)C(=O)OCC3=CC=CC=C3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCN(C)C(=O)OCC3=CC=CC=C3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 YSJZFNFOMOEMIZ-UHFFFAOYSA-N 0.000 description 1
- XZAIMZAICXXTGH-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCN(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCN(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 XZAIMZAICXXTGH-UHFFFAOYSA-N 0.000 description 1
- OQLRNKXTBJYVLP-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCN3C=CC=N3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCN3C=CC=N3)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 OQLRNKXTBJYVLP-UHFFFAOYSA-N 0.000 description 1
- NUQFHGCKUGOKNE-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCNC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCNC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 NUQFHGCKUGOKNE-UHFFFAOYSA-N 0.000 description 1
- DGVGAVRTXDOPSX-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCOC)N=C2C(N)=O)C=C(N)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCOC)N=C2C(N)=O)C=C(N)C=N1 DGVGAVRTXDOPSX-UHFFFAOYSA-N 0.000 description 1
- ZVTUZTWTEYSRLV-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCOC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCOC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 ZVTUZTWTEYSRLV-UHFFFAOYSA-N 0.000 description 1
- XSHQBXPJATYRDZ-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCOC)N=C2C(N)=O)C=C([N+](=O)[O-])C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCOC)N=C2C(N)=O)C=C([N+](=O)[O-])C=N1 XSHQBXPJATYRDZ-UHFFFAOYSA-N 0.000 description 1
- SEGAVHBORNGYGW-UHFFFAOYSA-N CCOC1=C(C(=O)NC2=C(CC)N(CCO[Si](C)(C)C(C)(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N(CCO[Si](C)(C)C(C)(C)C)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 SEGAVHBORNGYGW-UHFFFAOYSA-N 0.000 description 1
- TWHYQBGXUSCAFH-INIZCTEOSA-N CCOC1=C(C(=O)NC2=C(CC)N([C@@H](C)COC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N([C@@H](C)COC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 TWHYQBGXUSCAFH-INIZCTEOSA-N 0.000 description 1
- TWHYQBGXUSCAFH-MRXNPFEDSA-N CCOC1=C(C(=O)NC2=C(CC)N([C@H](C)COC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)NC2=C(CC)N([C@H](C)COC)N=C2C(N)=O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 TWHYQBGXUSCAFH-MRXNPFEDSA-N 0.000 description 1
- VSYZKMGXNCJWBT-UHFFFAOYSA-N CCOC1=C(C(=O)O)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1.Cl Chemical compound CCOC1=C(C(=O)O)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1.Cl VSYZKMGXNCJWBT-UHFFFAOYSA-N 0.000 description 1
- PCRIWLRXFJTJPJ-UHFFFAOYSA-N CCOC1=C(C(=O)O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C(=O)O)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 PCRIWLRXFJTJPJ-UHFFFAOYSA-N 0.000 description 1
- GLSREOLUIPGEMD-UHFFFAOYSA-N CCOC1=C(C(=O)O)C=C([N+](=O)[O-])C=N1 Chemical compound CCOC1=C(C(=O)O)C=C([N+](=O)[O-])C=N1 GLSREOLUIPGEMD-UHFFFAOYSA-N 0.000 description 1
- UZINTNOLWNTVTM-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(C(=O)N2)N(CCOC)N=C3CC)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(C(=O)N2)N(CCOC)N=C3CC)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 UZINTNOLWNTVTM-UHFFFAOYSA-N 0.000 description 1
- YTHWHVYWWVOQGG-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C(C)CC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C(C)CC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 YTHWHVYWWVOQGG-UHFFFAOYSA-N 0.000 description 1
- RMTOBEKVSJFANN-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=CC=C(C#N)C=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=CC=C(C#N)C=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 RMTOBEKVSJFANN-UHFFFAOYSA-N 0.000 description 1
- JFTKLBDUXIPJPG-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=CC=C(N)C=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=CC=C(N)C=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 JFTKLBDUXIPJPG-UHFFFAOYSA-N 0.000 description 1
- PJDAICIQEZBJEV-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=CC=C(NS(C)(=O)=O)C=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=CC=C(NS(C)(=O)=O)C=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 PJDAICIQEZBJEV-UHFFFAOYSA-N 0.000 description 1
- WMEHLBHAZNTVML-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=CC=C([N+](=O)[O-])C=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=CC=C([N+](=O)[O-])C=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 WMEHLBHAZNTVML-UHFFFAOYSA-N 0.000 description 1
- NMEFXGPLRHHGTK-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=CC=CC=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=CC=CC=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 NMEFXGPLRHHGTK-UHFFFAOYSA-N 0.000 description 1
- SMONQQFCQSWFTN-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=CN=CC=N4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=CN=CC=N4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 SMONQQFCQSWFTN-UHFFFAOYSA-N 0.000 description 1
- QJHYJZXHHUUKFL-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=NC=CC=N4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=NC=CC=N4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 QJHYJZXHHUUKFL-UHFFFAOYSA-N 0.000 description 1
- TUTZELSNMPOKOF-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=NC=CS4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=NC=CS4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 TUTZELSNMPOKOF-UHFFFAOYSA-N 0.000 description 1
- XKUCRVCDTWWQTL-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4=NC=NC(Cl)=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4=NC=NC(Cl)=C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 XKUCRVCDTWWQTL-UHFFFAOYSA-N 0.000 description 1
- AWTKWYNUEPYTKP-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CCCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CCCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 AWTKWYNUEPYTKP-UHFFFAOYSA-N 0.000 description 1
- XTUONRBNDFFKMH-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CCCCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CCCCC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 XTUONRBNDFFKMH-UHFFFAOYSA-N 0.000 description 1
- KYXVMIBCUDMYOA-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CCN(C(=O)OC(C)(C)C)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CCN(C(=O)OC(C)(C)C)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 KYXVMIBCUDMYOA-UHFFFAOYSA-N 0.000 description 1
- HOSHWJFJTDRLSY-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CCN(C(C)=O)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CCN(C(C)=O)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 HOSHWJFJTDRLSY-UHFFFAOYSA-N 0.000 description 1
- GTEPPOTUMRRYNS-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CCN(S(C)(=O)=O)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CCN(S(C)(=O)=O)CC4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 GTEPPOTUMRRYNS-UHFFFAOYSA-N 0.000 description 1
- NRNAOKTYLMOYEF-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CN(C(=O)OC(C)(C)C)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CN(C(=O)OC(C)(C)C)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 NRNAOKTYLMOYEF-UHFFFAOYSA-N 0.000 description 1
- OWUDRRZUBFXXDN-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CN(C(C)=O)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CN(C(C)=O)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 OWUDRRZUBFXXDN-UHFFFAOYSA-N 0.000 description 1
- KXXCQTLFOCTZGD-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(C4CN(CC)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(C4CN(CC)C4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 KXXCQTLFOCTZGD-UHFFFAOYSA-N 0.000 description 1
- RCTAONFWLXDUQT-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CC4CCCO4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CC4CCCO4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 RCTAONFWLXDUQT-UHFFFAOYSA-N 0.000 description 1
- UXDCRIGPYLLISI-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CCCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CCCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 UXDCRIGPYLLISI-UHFFFAOYSA-N 0.000 description 1
- OUOAQISPLRGBEK-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CCN4C=CC=N4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CCN4C=CC=N4)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 OUOAQISPLRGBEK-UHFFFAOYSA-N 0.000 description 1
- CVJJVQHDUKHESY-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CCNC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CCNC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 CVJJVQHDUKHESY-UHFFFAOYSA-N 0.000 description 1
- AJOWMAMISRGPEP-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CCOC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(C)CC2)C=N1 AJOWMAMISRGPEP-UHFFFAOYSA-N 0.000 description 1
- YBWFBUINTRROIH-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)N(CCO[Si](C)(C)C(C)(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N(CCO[Si](C)(C)C(C)(C)C)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 YBWFBUINTRROIH-UHFFFAOYSA-N 0.000 description 1
- JMIOIYJBQYLMRL-INIZCTEOSA-N CCOC1=C(C2=NC3=C(CC)N([C@@H](C)COC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N([C@@H](C)COC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 JMIOIYJBQYLMRL-INIZCTEOSA-N 0.000 description 1
- JMIOIYJBQYLMRL-MRXNPFEDSA-N CCOC1=C(C2=NC3=C(CC)N([C@H](C)COC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)N([C@H](C)COC)N=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 JMIOIYJBQYLMRL-MRXNPFEDSA-N 0.000 description 1
- JKLIOVZYYDNLKM-UHFFFAOYSA-N CCOC1=C(C2=NC3=C(CC)NN=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 Chemical compound CCOC1=C(C2=NC3=C(CC)NN=C3C(=O)N2)C=C(S(=O)(=O)N2CCN(CC)CC2)C=N1 JKLIOVZYYDNLKM-UHFFFAOYSA-N 0.000 description 1
- KBJCCONHJLAGMM-UHFFFAOYSA-N CCOC1=NC=C(S(=O)(=O)N2CCN(CC)CC2)C=C1C1=NC2=C(CC)N(CC)N=C2C(=O)N1.CCS(=O)(=O)O Chemical compound CCOC1=NC=C(S(=O)(=O)N2CCN(CC)CC2)C=C1C1=NC2=C(CC)N(CC)N=C2C(=O)N1.CCS(=O)(=O)O KBJCCONHJLAGMM-UHFFFAOYSA-N 0.000 description 1
- STFRDYSZKVPPQF-UHFFFAOYSA-N CCOC1=NC=C(S(=O)(=O)N2CCN(CC)CC2)C=C1C1=NC2=C(CC)N(CCOC)N=C2C(=O)N1.O=S(=O)(O)C1=CC=CC=C1 Chemical compound CCOC1=NC=C(S(=O)(=O)N2CCN(CC)CC2)C=C1C1=NC2=C(CC)N(CCOC)N=C2C(=O)N1.O=S(=O)(O)C1=CC=CC=C1 STFRDYSZKVPPQF-UHFFFAOYSA-N 0.000 description 1
- OSFKPGUKCKGFJN-UHFFFAOYSA-N CCOCCN1N=C(C(N)=O)C(NC(=O)C2=C(OCC)N=CC(S(=O)(=O)N3CCN(CC)CC3)=C2)=C1CC Chemical compound CCOCCN1N=C(C(N)=O)C(NC(=O)C2=C(OCC)N=CC(S(=O)(=O)N3CCN(CC)CC3)=C2)=C1CC OSFKPGUKCKGFJN-UHFFFAOYSA-N 0.000 description 1
- IQVOLDCMCJDUFR-UHFFFAOYSA-N CCOCCN1N=C2C(=O)NC(C3=C(OCC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)=NC2=C1CC Chemical compound CCOCCN1N=C2C(=O)NC(C3=C(OCC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)=NC2=C1CC IQVOLDCMCJDUFR-UHFFFAOYSA-N 0.000 description 1
- FDNDOIVWZKHWRI-UHFFFAOYSA-N CCOCCN1N=C2C(=O)NC(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)=NC2=C1CC Chemical compound CCOCCN1N=C2C(=O)NC(C3=C(OCCOC)N=CC(S(=O)(=O)N4CCN(CC)CC4)=C3)=NC2=C1CC FDNDOIVWZKHWRI-UHFFFAOYSA-N 0.000 description 1
- BQWQGEBNCNNFCI-RXMQYKEDSA-N CC[C@@H](C)OS(C)(=O)=O Chemical compound CC[C@@H](C)OS(C)(=O)=O BQWQGEBNCNNFCI-RXMQYKEDSA-N 0.000 description 1
- BQWQGEBNCNNFCI-YFKPBYRVSA-N CC[C@H](C)OS(C)(=O)=O Chemical compound CC[C@H](C)OS(C)(=O)=O BQWQGEBNCNNFCI-YFKPBYRVSA-N 0.000 description 1
- TVJGGJVHQXLHAV-UHFFFAOYSA-N CCc([n](CC)nc12)c1N=C(c1cc(S(N3CCN(CC)CC3)(=O)=O)cnc1OCC)NC2=O Chemical compound CCc([n](CC)nc12)c1N=C(c1cc(S(N3CCN(CC)CC3)(=O)=O)cnc1OCC)NC2=O TVJGGJVHQXLHAV-UHFFFAOYSA-N 0.000 description 1
- VDYDFKWVLSGUTO-UHFFFAOYSA-O CCc([n](CCO[SH+](C)(C)C(C)(C)C)nc1C(N)=O)c1NC(c1cc(S(N2CCN(CC)CC2)(=O)=O)cnc1OCC)=O Chemical compound CCc([n](CCO[SH+](C)(C)C(C)(C)C)nc1C(N)=O)c1NC(c1cc(S(N2CCN(CC)CC2)(=O)=O)cnc1OCC)=O VDYDFKWVLSGUTO-UHFFFAOYSA-O 0.000 description 1
- DMXGFDSGVSISBY-UHFFFAOYSA-N CN1CCN(S(=O)(=O)C2=CC(Br)=C(Cl)N=C2)CC1 Chemical compound CN1CCN(S(=O)(=O)C2=CC(Br)=C(Cl)N=C2)CC1 DMXGFDSGVSISBY-UHFFFAOYSA-N 0.000 description 1
- ZYVYEJXMYBUCMN-UHFFFAOYSA-N COCC(C)C Chemical compound COCC(C)C ZYVYEJXMYBUCMN-UHFFFAOYSA-N 0.000 description 1
- OFLUAKABUQZNOY-UHFFFAOYSA-N COCC(C)OS(C)(=O)=O Chemical compound COCC(C)OS(C)(=O)=O OFLUAKABUQZNOY-UHFFFAOYSA-N 0.000 description 1
- GZZJNGDFHRBPDN-UHFFFAOYSA-N COCCN(C)C(=O)OC(C)(C)C.O=S=O Chemical compound COCCN(C)C(=O)OC(C)(C)C.O=S=O GZZJNGDFHRBPDN-UHFFFAOYSA-N 0.000 description 1
- ARXJGSRGQADJSQ-SCSAIBSYSA-N COC[C@@H](C)O Chemical compound COC[C@@H](C)O ARXJGSRGQADJSQ-SCSAIBSYSA-N 0.000 description 1
- OFLUAKABUQZNOY-RXMQYKEDSA-N COC[C@@H](C)OS(C)(=O)=O Chemical compound COC[C@@H](C)OS(C)(=O)=O OFLUAKABUQZNOY-RXMQYKEDSA-N 0.000 description 1
- OFLUAKABUQZNOY-YFKPBYRVSA-N COC[C@H](C)OS(C)(=O)=O Chemical compound COC[C@H](C)OS(C)(=O)=O OFLUAKABUQZNOY-YFKPBYRVSA-N 0.000 description 1
- KOGOBKOHQTZGIS-UHFFFAOYSA-N CS(=O)(=O)OC1CCCCC1 Chemical compound CS(=O)(=O)OC1CCCCC1 KOGOBKOHQTZGIS-UHFFFAOYSA-N 0.000 description 1
- GSEZHCLWHDZJAB-UHFFFAOYSA-N CS(=O)(=O)OC1CCOCC1 Chemical compound CS(=O)(=O)OC1CCOCC1 GSEZHCLWHDZJAB-UHFFFAOYSA-N 0.000 description 1
- AQPIHLQSXPSMHZ-UHFFFAOYSA-N NC1=C(Br)C=C(S(=O)(=O)O)C=N1 Chemical compound NC1=C(Br)C=C(S(=O)(=O)O)C=N1 AQPIHLQSXPSMHZ-UHFFFAOYSA-N 0.000 description 1
- YXFLGKSXYWHALA-UHFFFAOYSA-N NC1=CC=C(S(=O)(=O)O)C=N1 Chemical compound NC1=CC=C(S(=O)(=O)O)C=N1 YXFLGKSXYWHALA-UHFFFAOYSA-N 0.000 description 1
- TURGMVYIESHZBE-UHFFFAOYSA-N O=S(=O)(Cl)C1=CC(Br)=C(Cl)N=C1 Chemical compound O=S(=O)(Cl)C1=CC(Br)=C(Cl)N=C1 TURGMVYIESHZBE-UHFFFAOYSA-N 0.000 description 1
- DSCWWJIQVMMRBI-MNYXATJNSA-N [3H]OC(=O)[V] Chemical compound [3H]OC(=O)[V] DSCWWJIQVMMRBI-MNYXATJNSA-N 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/80—Acids; Esters in position 3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/06—Antiabortive agents; Labour repressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/12—Drugs for genital or sexual disorders; Contraceptives for climacteric disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D231/38—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- This invention relates to a series of pyrazolo[4,3-d]pyrimidin-7-ones, which inhibit cyclic guanosine 3′,5′-monophosphate phosphodiesterases (cGMP PDEs). More notably, the compounds of the invention are potent and selective inhibitors of type 5 cyclic guanosine 3′,5′-monophosphate phosphodiesterase (cGMP PDE5) and have utility therefore in a variety of therapeutic areas.
- the compounds of the invention are of value for the curative or prophylactic treatment of mammalian sexual disorders.
- the compounds are of value in the treatment of mammalian sexual dysfunctions such as male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder or female sexual orgasmic dysfunction (FSOD) as well as sexual dysfunction due to spinal cord injury or selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction but, clearly, will be useful also for treating other medical conditions for which a potent and selective cGMP PDE5 inhibitor is indicated.
- MED male erectile dysfunction
- FSD female sexual dysfunction
- clitoral dysfunction female hypoactive sexual desire disorder
- female sexual arousal disorder female sexual pain disorder or female sexual orgasmic dysfunction
- SSRI selective serotonin re-uptake inhibitor
- Such conditions include premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, e.g.
- post-PTCA post-percutaneous transluminal coronary angioplasty
- peripheral vascular disease stroke, nitrate induced tolerance, bronchitis, allergic asthma, chronic asthma, allergic rhinitis, diseases and conditions of the eye such as glaucoma, optic neuropathy, macular degeneration, elevated intra-occular pressure, retinal or arterial occulsion and diseases characterised by disorders of gut motility, e.g. irritable bowel syndrome (IBS).
- IBS irritable bowel syndrome
- Particularly preferred conditions include MED and FSD.
- PCT/IB99/00519 relates to a series of pyrazolo[4,3-d]pyrimidin-7-ones, which inhibit cyclic guanosine 3′,5′-monophosphate phosphodiesterases (cGMP PDEs).
- R 1 , R 2 , R 4 and R 13 are as defined hereinbefore.
- alkyl, alkoxy and alkenyl groups having three or more carbon atoms, and alkanoyl groups having four or more carbon atoms may be straight chain or branched chain.
- a C 4 alkyl substituent can be in the form of normal-butyl (n-butyl), iso-butyl (i-butyl), secondary-butyl (sec-butyl) or tertiary-butyl (t-butyl).
- halo atom includes Cl, Br, F, and I.
- Haloalkyl and haloalkoxy are preferably —CF 3 and —OCF 3 respectively.
- aromatic as defined herein means a fully unsaturated system.
- a compound of the formula (I) contains one or more asymmetric carbon atoms and therefore exists in two or more stereoisomeric forms. Where a compound of the formula (I) contains an alkenyl or alkenylene group, cis (E) and trans (Z) isomerism may also occur.
- the present invention includes the individual stereoisomers of the compounds of the formula (I) and, where appropriate, the individual tautomeric forms thereof, together with mixtures thereof. Separation of diastereoisomers or cis and trans isomers may be achieved by conventional techniques, e.g. by fractional crystallisation, chromatography or H.P.L.C. of a stereoisomeric mixture of a compound of the formula (I) or a suitable salt or derivative thereof.
- An individual enantiomer of a compound of the formula (I) may also be prepared from a corresponding optically pure intermediate or by resolution, such as by H.P.L.C. of the corresponding racemate using a suitable chiral support or by fractional crystallisation of the diastereoisomeric salts formed by reaction of the corresponding racemate with a suitable optically active acid or base, as appropriate.
- the compounds of formulae (IA) and (IB) may also exist in tautomeric forms and the invention includes both mixtures thereof and the individual tautomers.
- radiolabelled derivatives of compounds of formulae (I), (IA) and (IB) which are suitable for biological studies.
- the pharmaceutically or veterinarily acceptable salts of the compounds of the invention which contain a basic centre are, for example, non-toxic acid addition salts formed with inorganic acids such as hydrochloric, hydrobromic, hydroiodic, sulphuric and phosphoric acid, with carboxylic acids or with organo-sulphonic acids.
- Examples include the HCl, HBr, HI, sulphate or bisulphate, nitrate, phosphate or hydrogen phosphate, acetate, benzoate, succinate, saccarate, fumarate, maleate, lactate, citrate, tartrate, gluconate, camsylate, methanesulphonate, ethanesulphonate, benzenesulphonate, p-toluenesulphonate and pamoate salts.
- Compounds of the invention can also provide pharmaceutically or veterinarily acceptable metal salts, in particular non-toxic alkali and alkaline earth metal salts, with bases. Examples include the sodium, potassium, aluminium, calcium, magnesium, zinc and diethanolamine salts.
- the pharmaceutically acceptable solvates of the compounds of the invention include the hydrates thereof. Also included within the scope of the compound and various salts of the invention are polymorphs thereof.
- a preferred group of compounds of formulae (I), (IA) and (IB) is that wherein, R 1 is C 1 to C 6 alkyl or C 3 to C 6 alkenyl wherein said alkyl or alkenyl groups may be branched chain or straight chain or R 1 is C 3 to C 6 cycloalkyl or C 4 to C 6 cycloalkenyl and wherein when R 1 is C 1 to C 3 alkyl said alkyl group is substituted by; and wherein when R 1 is C 4 to C 6 alkyl, C 3 to C 6 alkenyl, C 3 to C 6 cycloalkyl or C 4 to C 6 cycloalkenyl said alkyl, alkenyl, cycloalkyl or cycloalkenyl group is optionally substituted by;
- substituents selected from: hydroxy; C 1 to C 4 alkoxy; C 3 to C 4 cycloalkyl; phenyl substituted with one or more substitutents selected from C 1 to C 3 alkyl, C 1 to C 4 alkoxy, C 1 to C 4 haloalkyl or C 1 to C 4 haloalkoxy, halo, CN, NO 2 , NHR 11 , NHCOR 12 , NHSO 2 R 12 , SO 2 R 12 , SO 2 NHR 11 , COR 11 , CO 2 R 11 wherein said haloalkyl and haloalkoxy groups contain one or more halo atoms;
- a Het 1 group which is an N-linked 4-membered N-containing heterocyclic group
- a Het 2 group which is a C-linked 5-membered heterocyclic group containing an O, S or N heteroatom optionally containing one or more heteroatoms selected from N, O or S
- a Het 3 group which is a C-linked 6-membered heterocyclic group containing an O or S heteroatom optionally containing one or more heteroatoms selected from O, S or N or a Het 3 group which is a C-linked 6-membered heterocyclic group containing three N heteroatoms
- R 7 , R 8 , R 11 and R 12 are as previously defined herein or R 1 is a Het 4 group which is a C-linked 4- or 5-membered heterocyclic group containing one heteroatom selected from S, O or N
- a Het 4 group which is a C-linked 6-membered heterocyclic group containing one, two or three heteroatoms selected from S or O
- R 13 is OR 3 ;
- R 3 is C 1 to C 6 alkyl optionally substituted with one or two substituents selected from C 3 to C 5 cycloalkyl, hydroxy, C 1 to C 4 alkoxy, benzyloxy, NR 5 R 6 , phenyl, furanyl, tetrahydrofuranyl or pyridinyl wherein said C 1 to C 6 alkyl and C 1 to C 4 alkoxy groups may optionally be terminated by a haloalkyl group such as CF 3 ; or R 3 is C 3 to C 6 cycloalkyl, 1-(C 1 to C 4 alkyl)piperidinyl, tetrahydrofuranyl or tetrahydropyranyl; R 4 is a piperazin-1-ylsulphonyl group having a substituent R 10 at the 4-position of the piperazinyl group wherein said piperazinyl group is optionally substituted with one or two C 1 to C 4 alkyl groups and is optionally in the form
- a further preferred group of compounds of formulae (I), (IA) and (IIB) is that wherein, R 1 is C 1 to C 6 alkyl wherein said alkyl may be branched or straight chain or R 1 is C 3 to C 6 cycloalkyl
- R 1 is C 1 to C 3 alkyl said alkyl group is substituted by; and wherein when R 1 is C 4 to C 6 alkyl or C 3 to C 6 cycloalkyl said alkyl or cycloalkyl group is optionally substituted by; one or more substituents selected from: hydroxy; C 1 to C 2 alkoxy; C 3 to C 5 cycloalkyl; NR 7 R 8 , NR 7 COR 11 or COR 11 wherein R 7 and R 8 are each independently selected from H, C 1 to C 4 alkyl or CO 2 R 9 wherein R 9 and R 11 are as previously defined herein; a Het 1 group which is an N-linked 4-membered N-containing heterocyclic group; a Het 3 group which is a C-linked 6-membered heterocyclic group containing an O or S heteroatom optionally containing one or more heteroatoms selected from O, S or N or a Het 3 group which is a C-linked 6-membered heterocyclic group
- R 1 is a Het 4 group which is a C-linked 4-membered heterocyclic group containing one heteroatom selected from S, O or N or R 1 is a Het 4 group which is a C-linked 6-membered heterocyclic group containing one, two or three heteroatoms selected from S or O
- any of said heterocyclic groups Het 1 , Het 2 , Het 3 or Het 4 is saturated, partially unsaturated or aromatic and is optionally substituted with one or more substituents selected from C 1 to C 4 alkyl, C 1 to C 4 alkoxy, —CO 2 R 11 , —SO 2 R 12 , —COR 11 or NHR 11 wherein R 11 and R 12 are as defined hereinbefore and/or wherein any of said heterocyclic groups is benzo-fused;
- R 1 is phenyl substituted by one or more substituents selected from CF 3 , —OCF 3 , —SO 2 R 12 , —COR 11 , —CO 2 R 11 wherein R 11 and R 12 are as defined hereinbefore
- R 2 is C 1 to C 6 alkyl
- R 13 is OR 3 ;
- R 3 is methyl, ethyl, n-propyl, i-propyl, n-butyl, sec-butyl, i-butyl or t-butyl alkyl optionally substituted with one or two substituents selected from cyclopropyl, cyclobutyl, hydroxy, methoxy, ethoxy, benzyloxy, phenyl, benzyl, furan-3-yl, tetrahydrofuran-2-ylmethyl, tetrahydrofuran-3-ylmethyl, pyridin-2-yl, pyridin-3-yl or NR 5 R 6 wherein R 5 and R 6 are each independently selected from H and C 1 to C 2 alkyl; R 4 is a piperazin-1-ylsulphonyl group having a substituent, R 10 at the 4-position of the piperazinyl group wherein said piperazinyl group is optionally substituted with one or two C 1 to C 4 alky
- a particularly preferred group of compounds of formulae (I), (IA) or (IB) is that wherein
- R 1 is —(CH 2 ) n (C 3 -C 4 )cycloalkyl wherein n is 1 or 2; or R 1 is —(CH 2 ) n (C 3 -C 6 )cycloalkyl wherein n is 0; or R 1 is -cyclopentyl methyl; or R 1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or R 1 is i-, n-, sec- or t-butyl; R 2 is C 2 to C 4 alkyl; R 13 is OR 3 wherein the R 3 alkyl group is methyl, ethyl, n-propyl,
- R 1 is —(CH 2 ) n (C 3 -C 4 )cycloalkyl wherein n is 1 or 2; or R 1 is —(CH 2 ) n (C 3 -C 5 )cycloalkyl wherein n is 0; or R 1 is -cyclopentylmethyl; or R 1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or R 1 is i-, n-, sec- or t-butyl; R 2 is C 2 to C 4 alkyl; R 13 is OR 3 wherein the R 3
- Highly preferred compounds according to the present invention include: 1- ⁇ 6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl ⁇ -4-ethylpiperazine and salts and polymorphs thereof.
- Preferred salts of 1- ⁇ 6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl ⁇ -4-ethylpiperazine are sulphonic acid salts, more preferably the p-toluenesulfonate, benzenesulfonate, camphorsulfonate and ethanesulfonate salts respectively, and especially the benzenesulfonate.
- the present invention provides processes for the preparation of compounds of formulae (I), (IA) and (IB), their pharmaceutically and veterinarily acceptable salts, and pharmaceutically and veterinarily acceptable solvates of either entity, as illustrated below.
- the order of the synthetic steps employed may be varied and will depend inter alia on factors such as the nature of other functional groups present in a particular substrate, the availability of key intermediates and the protecting group strategy (if any) to be adopted. Clearly, such factors will also influence the choice of reagent for use in the said synthetic steps.
- Illustrative of a protecting group strategy is the route to the azetidine analogues (Examples 18, 19 and 20), the precursor to which (preparations 63, 66 and 61 respectively) contain t-butoxycarbonyl (Boc) as the nitrogen protecting group.
- FIG. 1 is a reaction scheme depicting a general route for the synthesis of compounds (I) via compounds (IXB).
- FIG. 2 is a reaction scheme depicting a route for the preparation of compounds of general formula (XB).
- FIG. 3 is a reaction scheme depicting a preferred embodiment of FIG. 2 in which X is an —OR 3 alkoxy group.
- formula (I) may equally be represented by general formulae (IA) and (IB) and wherein R 1 , R 2 , R 4 and R 13 are as previously defined herein may be prepared from a compound of general formula (IX):
- R P is R 13 (i.e. OR 3 or NR 5 R 6 ) or X wherein R 13 , R 3 , R 5 and R 6 are as defined hereinbefore and X is a leaving group and wherein general formula (IX) can be represented by formulae (IXA), (IXB) or (IXC) respectively:
- R 1 , R 2 , R 3 , R 4 , R 5 and R 6 are as previously defined herein and wherein X is a leaving group and may be any group which is displaceable by an amino group of the formula —NR 5 R 6 or by an alkoxy group and wherein the intermediate compounds of general formulae (IXA) and (IXB) can be represented by their regioisomeric general formulae as previously illustrated for compounds having the general formulae (I).
- Suitable leaving groups, X, for use herein include halogen, alkoxy, amino, tosylate groups and further groups are detailed hereinafter.
- the cyclisation is base-mediated, using an alkali metal salt of a sterically hindered alcohol or amine.
- the required cyclisation may be effected using about a 1- to 5-, preferably a 1.2- to 3.5-fold excess of potassium t-butoxide, potassium bis(trimethylsilyl)amide or cesium carbonate, optionally in the presence of molecular sieves, in a suitable solvent, such as for example an inert solvent e.g.
- R 1 , R 2 and R 4 are as previously defined herein for compounds of the formula (I), (IA) and (IB) and wherein X is a leaving group as defined hereinbefore, by reaction in the presence of —OR 3 and a hydroxide trapping agent.
- the conversion (IXB) to (I) can be undertaken in either a stepwise process or a one-pot process. A number of stepwise permutations are feasible, some of which are subsets of others. These include
- OR 3a is an alkoxy group which is different from and displaceable by the desired OR 3 group on the final compounds of general formula (I) and wherein R 3a is selected from C 1 to C 6 alkyl optionally substituted with one or two substituents selected from C 3 to C 5 cycloalkyl, hydroxy, C 1 to C 4 alkoxy, benzyloxy, NR 5 R 6 , phenyl, Het 1 , Het 2 , Het 3 or Het 4 wherein the C 1 to C 6 alkyl and C 1 to C 4 alkoxy groups may optionally be terminated by a haloalkyl group such as CF 3 and wherein the C 3 -C 5 cycloalkyl group may optionally be substituted by C 1 -C 4 alkyl, hydroxy or halo; C 3 to C 6 cycloalkyl; Het 1 , Het 2 , Het 3 or Het
- R 3 is C 1 to C 6 alkyl.
- the displacement with ⁇ OR 3 (in (iii) or (iv)) is carried out in the range of from about 80° C. to about 90° C. to provide a compound of the general formula (IXC).
- Subsequent cyclisation to a compound of general formula (I) is generally carried out at a temperature greater than about 115° C.
- the reaction is conducted at a temperature greater than about 110° C. with —OR 3a in R 3a OH.
- the temperature of the reaction of compounds of the general formula (IXB) to compounds of the general formula (I) is preferably at least about 80° C., more preferably about 80 to about 130° C., more preferably still about 100 to about 130° C. and most preferably about 115 to about 125° C. These temperatures are also applicable for the conversion of compounds (XXX) to (I), although the temperature in this case could also probably be lower (e.g. about 60° C.) since there is no cyclisation taking place.
- R 1 is —(CH 2 ) n (C 3 -C 4 )cycloalkyl wherein n is 1 or 2; or R 1 is —(CH 2 ) n (C 3 -C 6 )cycloalkyl wherein n is 0; or R 1 is -cyclopentylmethyl; or R 1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or R 1 is i-, n-, sec- or t-butyl; R 2 is C 2 to C 4 alkyl; R 13 is OR 3 wherein the R 3 alkyl
- R 1 is —(CH 2 ) n (C 3 -C 4 )cycloalkyl wherein n is 1 or 2; or R 1 is —(CH 2 ) n (C 3 -C 6 )cycloalkyl wherein n is 0; or R 1 is -cyclopentylmethyl; or R 1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or R 1 is i-, n-, sec- or t-butyl; R 2 is C 2 to C 4 alkyl; R 13 is OR 3 wherein the R 3 alkyl group is
- hydroxide trapping agent is an ester. More preferably said hydroxide trapping agent is an ester of the formula:
- OT is OR 3 or the residue of a bulky alcohol or a non-nucleophilic alcohol or TOH is an alcohol which can be azeotropically removed during the reaction; and C(O)V is the residue of a carboxylic acid.
- OR 3 is OEt in compound (IXC)
- the hydroxide trapping agent (TOC(O)V) could be e.g. ethyl acetate or ethyl pivalate.
- V is a C 1 to C 4 alkyl group.
- X is selected from the group consisting of —OR 3 , halo, optionally substituted arylsulphonyloxy, preferably phenylsulphonyloxy, more preferably a para-substituted aryl (phenyl) such as by a C 1 -C 4 alkyl group e.g. p-toluenesulphonyloxy; C 1 -C 4 alkylsulphonyloxy e.g. methanesulphonyloxy; nitro or halo substituted benzenesulphonyloxy preferably para-substituted e.g.
- C 1 -C 4 perfluoroalkylsulphonyloxy e.g. trifluoromethylsulphonyloxy; optionally substituted aroyloxy such as benzoyloxy; C 1 -C 4 perfluoroalkanoyloxy such as trifluoroacetyloxy; C 1 -C 4 alkanoyloxy such as acetyloxy; diazonium; quatenaryammonium C 1 -C 4 alkylsulphonyloxy; halosulphonyloxy e.g.
- ⁇ OR 3 can act both as a nucleophile (to displace the leaving group by nucleophilic substitution) and as a base (to bring about the cyclisation).
- ⁇ OR 3 can be generated in solution from, for example, a salt ZOR 3 (wherein Z is a cation) such as a metal salt.
- ⁇ OR 3 is formed in situ from R 3 OH plus an auxiliary base (i.e. a base other than ⁇ OR 3 ).
- auxiliary base i.e. a base other than ⁇ OR 3
- ZOR 3 could be used in the reaction system with an auxiliary base.
- the solvent in which the reaction takes place can be R 3 OH or an inert solvent (or a mixture of both).
- inert solvent we mean a solvent which will not form a nucleophile under the reaction conditions or if a nucleophile is formed it is sufficiently hindered or unreactive such that it does not substantially compete in the displacement reaction.
- R 3 OH When R 3 OH is used as a source of ⁇ OR 3 , then a separate solvent is not essentially required but an (auxiliary) inert solvent (i.e. a solvent other than R 3 OH) may be used as a co-solvent in the reaction.
- an (auxiliary) inert solvent i.e. a solvent other than R 3 OH
- Suitable solvents are as follows: R 3 OH, a secondary or tertiary C 4 -C 12 alkanol, a C 3 -C 12 cycloalkanol, a tertiary C 4 -C 12 cycloalkanol, a secondary or tertiary (C 3 -C 7 cycloalkyl)C 2 -C 6 alkanol, a C 3 -C 8 alkanone, 1,2-dimethoxyethane, 1,2-diethoxyethane, diglyme, tetrahydrofuran, 1,4-dioxan, toluene, xylene, chlorobenzene, 1,2-dichlorobenzene, acetonitrile, dimethyl sulphoxide, sulpholane, dimethylformamide, N-methylpyrrolidin-2-one, pyridine, and mixtures thereof.
- the solvent is R 3 OH, a tertiary C 4 -C 12 alkanol, a tertiary C 4 -C 12 cycloalkanol, a tertiary (C 3 -C 7 cycloalkyl)C 2 -C 6 alkanol, a C 3 -C 8 alkanone, 1,2-dimethoxyethane, 1,2-diethoxyethane, diglyme, tetrahydrofuran, 1,4-dioxan, toluene, xylene, chlorobenzene, 1,2-dichlorobenzene, acetonitrile, dimethyl sulphoxide, sulpholane, dimethylformamide, N-methylpyrrolidin-2-one, pyridine, and mixtures thereof.
- the solvent is R 3 OH, which means that ⁇ OR 3 is formed in situ, such as in the presence of an auxiliary base.
- auxiliary bases can be used in the process of the invention. Typically the bases would not substantially compete with ⁇ OR 3 in the nucleophilic substitution of X (i.e. they would be non nucleophilic) such as by suitably being sterically hindered.
- the auxiliary base is selected from the group consisting of a sterically hindered base, a metal hydride, metal oxide, metal carbonate and metal bicarbonate.
- the sterically hindered base is advantageously a metal salt of a sterically hindered alcohol or amine.
- auxiliary bases in accordance with the invention are selected from the group consisting of metal salts of a sterically hindered alcohol or amine such as a secondary or tertiary C 4 -C 12 alkanol, a C 3 -C 12 cycloalkanol and a secondary or tertiary (C 3 -C 8 cycloalkyl)C 1 -C 6 alkanol, a N-(secondary or tertiary C 3 -C 6 alkyl)-N-(primary, secondary or tertiary C 3 -C 6 alkyl)amine, a N—(C 3 -C 8 cycloalkyl)-N-(primary, secondary or tertiary C 3 -C 6 alkyl)amine, a di(C 3 -C 8 cycloalkyl)amine or hexamethyldisilazane; 1,5-diazabicyclo[4,3,0]non-5-ene and 1,8
- the auxiliary bases in accordance with the invention are selected from the group consisting of metal salts of a sterically hindered alcohol or amine such as a tertiary C 4 -C 12 alkanol, a C 3 -C 12 cycloalkanol and a tertiary (C 3 -C 8 cycloalkyl)C 1 -C 6 alkanol, a N-(secondary or tertiary C 3 -C 6 alkyl)-N-(primary, secondary or tertiary C 3 -C 6 alkyl)amine, a N—(C 3 -C 8 cycloalkyl)-N-(primary, secondary or tertiary C 3 -C 6 alkyl)amine, a di(C 3 -C 8 cycloalkyl)amine or hexamethyldisilazane; 1,5-diazabicyclo[4,3,0]non-5-ene and 1,8-di
- the auxiliary base is selected from the sterically hindered bases of the previous paragraph (i.e. all of them except the metal hydride, oxide, carbonate and bicarbonate).
- the auxiliary base is the metal salt of a tertiary C 4 -C 6 alcohol such as the alkali or alkaline earth metal salts (e.g. Na/K) of t-butanol or t-amyl alcohol, or the base is KHMDS.
- the auxiliary base is the alkali metal salt of t-butanol (e.g. potassium t-butoxide).
- the metal of the salt of ZOR 3 and the auxiliary base can be independently selected from alkali metals (lithium, sodium, potassium, rubidium, cesium) or alkaline earth metals (beryllium, magnesium, calcium, strontium, barium).
- the metal is sodium, potassium, lithium or magnesium. More preferably the metal is sodium or potassium.
- X is any group hereinbefore defined except —OR 3 , then at least about 1 molecular equivalent of auxiliary base and ⁇ OR 3 are used. If ⁇ OR 3 also functions as a base (i.e. there is no auxiliary base present) then preferably at least about 2 equivalents of ⁇ OR 3 are present.
- At least about 1 equivalent of trapping agent (preferably at least about 2 equivalents) is present.
- at least 1 equivalent of base is required, wherein said base may be —OR 3 or auxiliary base.
- the temperature of the reaction of compounds of the general formula (IXC) to compounds of the general formula (I) is preferably at least about 80° C., more preferably about 80 to about 130° C., more preferably still about 100 to about 130° C. and most preferably about 115 to about 125° C.
- the reaction temperature attainable to effect the conversion of compounds of the general formulae (IXB), (IXC) or (XXX) to compounds of the general formula (I) depends on the solvent, the nature of ⁇ OR 3 and X.
- X is OR 3a (wherein OR 3a and OR 3 are not the same), i.e. a compound of the formula (IXC a ) and R 3 OH is the solvent, preferably XH (such as C 1 -C 6 alkohol) is removed azeotropically (of course the reaction vessel must be configured to distill over the azeotrope mixture) with R 3 OH by running the reaction at the azeotrope temperature of XH and R 3 OH.
- XH such as C 1 -C 6 alkohol
- R 1 is —(CH 2 ) n (C 3 -C 4 )cycloalkyl wherein n is 1 or 2; or R 1 is —(CH 2 ) n (C 3 -C 6 )cycloalkyl wherein n is 0; or R 1 is —(CH 2 ) n (C 5 )cycloalkyl wherein n is 1; or R 1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups; or R 1 is i-,
- R 1 , R 2 , R 3 and R 4 are as previously defined herein for compounds of the formula (I), (IA) and (IB).
- the appropriate alcohol of formula R 3 OH should be employed as the solvent in order to obviate potential problems associated with alkoxide exchange at the 2-position of the pyridine ring or an inert solvent or a mixture of the two.
- the appropriate alcohol as defined herein means that the solvent alcohol should be of the same alkyl chain length as the alkoxy (—OR 3 ) substituent, for example, where —OR 3 is ethoxy, ethanol is the appropriate alcohol.
- said cyclisation is base-mediated, using an alkali metal salt of a sterically hindered alcohol or amine.
- the required cyclisation may be effected using about a 1- to 8, preferably about a 1- to 5-, more preferably a 1.2- to 3.5-fold excess of potassium t-butoxide or potassium bis(trimethylsilyl)amide, optionally under suitable drying conditions i.e. in the presence of molecular sieves or under azeotroping conditions, in a suitable solvent as described above at the reflux temperature of the reaction mixture optionally in the presence of about 1 to 2 molar equivalents of a hydroxide trapping agent such as ethyl acetate or ethyl pivalate, or, the reaction can optionally be carried out in a sealed vessel at about 100-130° C. optionally in the presence of about 1 to 2 molar equivalents of a hydroxide trapping agent such as ethyl acetate or ethyl pivalate.
- suitable drying conditions i.e. in the presence of molecular sieves or under azeotroping conditions
- a suitable solvent as described above
- Alternative reaction conditions for the cyclisation reactions of compounds of (IXC) wherein X is OR 3 are to conduct the reaction with about 1.2 to 4.5 molecular equivalents of sterically hindered base such as potassium t-butoxide or KHMDS, optionally in a sealed vessel at from about 100° C. to about 150° C. with, rather than an alcohol of formula R 3 OH as solvent, a sterically hindered alcohol, e.g. 3-methylpentan-3-ol, as solvent optionally in the presence of about 1 or 2 molar equivalents of ethyl acetate or ethyl pivalate.
- sterically hindered base such as potassium t-butoxide or KHMDS
- a compound of formula (IXA) or a compound of general (IXB) wherein X is OR 3 i.e. a compound of general formula (IXC)
- X is OR 3
- a compound of general formula (IXC) may be prepared by a coupling reaction between a compound of formula (VII):
- R 1 and R 2 are as previously defined for formulae (IXA), (IXB) or (IXC) with a compound of formula (XA), (XB) or (XC) respectively:
- R 3 , R 4 , R 5 , R 6 and X are also as previously defined for formulae (IXA), (IXB) or (IXC).
- R 5 and/or R 6 in the —NR 5 R 6 group of formula (XA) are H, then a suitable N-protecting group strategy may be advantageously employed. Any known suitable protecting group strategy may be used.
- the coupling reaction may be carried out using conventional amide bond-forming techniques, e.g.
- acyl chloride derivative of (XA) or (XB) in the presence of up to about a five-fold excess of a tertiary amine such as triethylamine or pyridine to act as scavenger for the acid by-product (HY), optionally in the presence of a catalyst such as 4-dimethylaminopyridine, in a suitable solvent such as dichloromethane, at from about 0° C. to about room temperature.
- a catalyst such as 4-dimethylaminopyridine
- a suitable solvent such as dichloromethane
- pyridine may also be used as the solvent.
- any one of a host of amino acid coupling variations may be used.
- the acid of formula (XA), (XB) or (XC) or a suitable salt e.g.
- sodium salt) thereof may be activated using a carbodiimide such as 1,3-dicyclohexylcarbodiimide or 1-ethyl-3-(3-dimethylaminoprop-1-yl)carbodiimide optionally in the presence of 1-hydroxybenzotriazole hydrate and/or a catalyst such as 4-dimethylaminopyridine, or by using a halotrisaminophosphonium salt such as bromotris(pyrrolidino)phosphonium hexafluorophosphate or by using a suitable pyridinium salt such as 2-chloro-1-methylpyridinium iodide.
- a carbodiimide such as 1,3-dicyclohexylcarbodiimide or 1-ethyl-3-(3-dimethylaminoprop-1-yl)carbodiimide optionally in the presence of 1-hydroxybenzotriazole hydrate and/or a catalyst such as 4-dimethylaminopyridine,
- Either type of coupling is conducted in a suitable solvent such as dichloromethane, tetrahydrofuran or N,N-dimethylformamide, optionally in the presence of a tertiary amine such as triethylamine or N-ethyldiisopropylamine (for example when either the compound of formula (VII), or the activating reagent, is presented in the form of an acid addition salt), at from about 0° C. to about room temperature.
- a suitable solvent such as dichloromethane, tetrahydrofuran or N,N-dimethylformamide
- a tertiary amine such as triethylamine or N-ethyldiisopropylamine (for example when either the compound of formula (VII), or the activating reagent, is presented in the form of an acid addition salt)
- a tertiary amine such as triethylamine or N-ethyldiisopropyl
- the carboxylic acid function of (XA), (XB) or (XC) may first of all be activated using up to about a 5% excess of a reagent such as N,N-carbonyldiimidazole in a suitable solvent, e.g. ethyl acetate or butan-2-one, at from about room temperature to about 80° C., followed by reaction of the intermediate imidazolide with (VII) at from about 20° C. to about 90° C.
- a reagent such as N,N-carbonyldiimidazole
- a suitable solvent e.g. ethyl acetate or butan-2-one
- R q is selected from OH, C 1 -C 6 alkoxy or NR 5 R 6 wherein R 5 and R 6 are as hereinbefore defined, according to the procedures detailed in the preparations section herein and as particularly described in Preparations 96(a) to (h).
- Compounds having the general formulae (XA) or (XC) may be prepared from the carboxylic acid compounds of the general formulae (VIIIA), (VIIIB) or (VIIIC) respectively:
- R 3 , R 5 and R 6 are as defined for compounds of the general formulae (I), (IA) and (IB) by reaction with a 4-R 10 -piperizinyl compound, such as for example 4-methylpiperizine.
- a 4-R 10 -piperizinyl compound such as for example 4-methylpiperizine.
- Such reaction can be conducted at from about 0° C. to about room temperature, preferably in the presence of an appropriate solvent such as a C 1 to C 3 alkanol or dichloromethane optionally in the presence of a suitable base such as triethylamine to scavenge the acid by-product (HY).
- HY acid by-product
- R 5 or R 6 is H a suitable amino protecting group strategy may be employed as detailed hereinbefore.
- Compounds of the general formulae (VIIIA), (VIIIB) or (VIIIC) may be prepared from compounds of the general formulae (XIA), (XIB) or (XIC) respectively:
- R 3 , R 5 , R 6 and X are as defined for compounds of the general formulae (I), (IA) and (IB) by the application of known methods for converting amino to an SO 2 Y group, wherein Y is halo, preferably chloro.
- Y is chloro
- Y by the action of about a two-fold excess of sodium nitrite in a mixture of concentrated hydrochloric acid and glacial acetic acid at from about ⁇ 25° C. to about 0° C., followed by treatment with excess liquid sulphur dioxide and a solution of about a three-fold excess of cupric chloride in aqueous acetic acid at from about ⁇ 15° C. to about room temperature.
- R 13 contains a primary or secondary amino group
- protection of the said amino group with an acid stable group such as acetyl or benzyl will generally be advantageous.
- Compounds of the general formula (XIA), (XIB) and (XIC) may be prepared by reduction of compounds of the general formulae (XIIA), (XIIB) and (XIIC) respectively:
- compound (XB) can be formed from compounds (XIV) (wherein Q ⁇ OH and W ⁇ OH) and an appropriate derivatising agent, more particularly an appropriate sulphonylating agent such as arylsulfonylhalide, C 1 -C 4 alkylsulfonylhalide, C 1 -C 4 perfluoroalkylsulfonylhalide, arylhalide, C 1 -C 4 perfluoroalkanoylhalide, C 1
- Compounds of formula (XIV) can be formed from compounds (XV) (wherein P is hydrolisable group) via use of a hydrolising agent, preferably a hydroxide base (ideally 2 molar equivalents), more preferably a metal hydroxide such as sodium hydroxide, in an appropriate solvent, such as water.
- a hydrolising agent preferably a hydroxide base (ideally 2 molar equivalents), more preferably a metal hydroxide such as sodium hydroxide, in an appropriate solvent, such as water.
- the metal of the hydroxide base can be as defined hereinbefore for Z (in ZOR). This will also apply for other reactions of scheme 2 and 3 hereafter where hydroxide base/hydrolising agent is used.
- P is group which is not hydrolisable by hydroxide then a suitable de-protection strategy should be employed according to standard literature practise.
- the deprotecting agent as used herein in accordance with the invention is a hydrolysing agent, more preferably a hydroxide nucleophile, advantageously a hydroxide base (ideally 1 molar equivalent), such as sodium hydroxide preferably in an appropriate solvent, such as water.
- a hydrolysing agent more preferably a hydroxide nucleophile, advantageously a hydroxide base (ideally 1 molar equivalent), such as sodium hydroxide preferably in an appropriate solvent, such as water.
- an appropriate derivatising agent preferably an appropriate sulphonylating agent such as arylsulphonylhalide, preferably arysulfonylchloride (ideally at least 2 molar equivalents) and preferably in the presence of a base (ideally 2 molar equivalents thereof), such as triethylamine in an appropriate solvent.
- the compounds of formula (XV) can be formed from compounds of formula (XVI) by reaction with a mono-N-substituted piperazine group wherein the mono-substituent R 10 as defined herein before, optionally in the presence of a supplementary base (which does not react irreversibly with the sulphonyl chloride moiety) such as triethylamine preferably in an appropriate solvent, such as toluene.
- a supplementary base which does not react irreversibly with the sulphonyl chloride moiety
- triethylamine preferably in an appropriate solvent, such as toluene.
- “D” in compounds (XV) and (XVI) is Cl or Br.
- the monosubstituted piperazine group may also be the base where more than one equivalent of monosubstituted piperazine is present. Preferably about 2 equivalents are used.
- a supplementary base it either does not react with the sulphonyl chloride moiety (such as a metal oxide, carbonate or bicarbonate) or it reacts with the sulphonyl chloride moiety in such a way as to keep it activated to nucleophilic attack (e.g. a tertiary amine such as triethylamine).
- the amine NH(R3)(R4) may also act as a base, in which case preferably more than one equivalent is present, more preferably about 2 equivalents (or more).
- the compounds of formula (XVI) can be formed from compounds of formula (XX) in the presence of a chlorinating or brominating agent such as thionyl chloride or thionyl bromide more preferably in the presence of a halogenation catalyst, more preferably still thionyl chloride or thionyl bromide in the presence of dimethylformamide.
- a chlorinating or brominating agent such as thionyl chloride or thionyl bromide more preferably in the presence of a halogenation catalyst, more preferably still thionyl chloride or thionyl bromide in the presence of dimethylformamide.
- the thionyl chloro/bromo can also act as the solvent, but more preferably the reaction takes place or in an appropriate other solvent such as toluene. In such case only stoicheometric amounts of thionyl chloride/bromide would be required, preferably at least 2 molar equivalents, more preferably
- steps (XX) to (XB) can be telescoped together using a single solvent such as a water immiscible inert organic solvent.
- a hydrocarbon solvent such as toluene, xylene, anisole, chlorobenzene, hexane, heptane, octane, nonane, decane, cyclohexane, methylcyclohexane
- ethers such as dibutyl ether, diphenyl ether
- ketones such as methylisobutylketone, methylethylketone
- esters such as ethyl acetate, butyl acetate
- a hydrocarbon solvent such as toluene, xylene, anisole, chlorobenzene, octane, nonane, decane, methylcyclohexane
- ethers such as dibutyl ether, diphenyl ether
- esters such as ethyl acetate, butyl acetate
- the telescoping solvent is toluene.
- the intermediate of formula (XX) is formed from a compound of formula (XVII) in the presence of an agent which will form a protecting group (P) for the carboxylic acid (i.e. to form the —COP group).
- said agent is an esterification agent, to form a carboxylic acid ester (wherein, e.g. P will be alkoxy and the protecting forming agent will be an alcohol) such as a C 1 -C 6 carboxylic acid ester which will be carried through the reaction scheme and hydrolised under basic conditions to the carboxylic acid function of compound (XB).
- a carboxylic acid ester wherein, e.g. P will be alkoxy and the protecting forming agent will be an alcohol
- the esterification agent is ethanol.
- An additional solvent such as toluene may be appropriate.
- the intermediate of formula (XVII) is formed from 2-hydroxynicotinic acid or a salt thereof in the presence of a sulphonylating agent, more preferably an agent comprising SO 3 (ideally at least 1 molar equivalent of SO 3 ), for example using SO 3 in an organic solvent (e.g. THF, dioxan and heptane) or an aprotic solvent (e.g. nitrobenzene, nitromethane, 1,4-dioxane, dichloromethane) or a mineral acid as solvent (e.g. sulphuric acid) or in a liquid carboxylic acid as solvent (e.g. acetic acid) or THF or heptane.
- a sulphonylating agent more preferably an agent comprising SO 3 (ideally at least 1 molar equivalent of SO 3 ), for example using SO 3 in an organic solvent (e.g. THF, dioxan and heptane) or an aprotic solvent (e.
- the sulphonylating agent is oleum (SO 3 in sulphuric acid) such as about 20% to 30% oleum.
- Compounds of the general formula (IXB) are formed by the reaction of intermediates of general formula (XB) with compounds of the general formula (VII), as detailed hereinbefore in the presence of a coupling agent, such as N,N′-carbonyldiimidazole and a suitable solvent, such as ethyl acetate. Methods for the preparation of compounds of the general formula (VII) are described hereinafter.
- X is an —OR 3 alkoxy group and so Q in compound (XIV) represents OR 3 .
- OR 3 is a C 1 to C 6 alkoxy group, more preferably a C 1 to C 4 primary or secondary alkoxy group and especially ethoxy.
- This preferred embodiment of Scheme 2 is illustrated in Scheme 3.
- the intermediate of formula (XB) is formed from a compound of formula (XIV) by removal of protecting group P by a deprotecting agent, advantageously by saponification in the presence of a hydroxide base such as sodium hydroxide, preferably in an appropriate solvent such as water and toluene.
- the intermediate of formula (XIV) is formed from a compound of formula (XV) in the presence of an appropriate C 1 -C 6 alkoxide nucleophile (—OR 3 ), (such as a primary or secondary alkoxide), preferably a metal alkoxide of the formula ZOR 3 , wherein the metal (Z) is as defined hereinbefore for ZOR, such as sodium ethoxide, preferably in an appropriate solvent such as toluene or R 3 OH, wherein R 3 OH is as defined hereinbefore and is preferably ethoxy.
- D in compounds of formulae (XV) and (XVI) is Cl or Br, more preferably D is Cl.
- the intermediate of formula (XV) is formed from a compound of formula (XVI) by reaction with N—R 10 piperazine, preferably in the presence of a base, such as triethylamine or excess N—R 10 piperazine, preferably in an appropriate solvent such as toluene.
- the intermediate of formula (XVI) is formed from a compound of formula (XX) in the presence of a chlorinating or brominating agent as defined for the same step in Scheme 2 such as thionyl chloride or bromide, preferably thionyl chloride or bromide/dimethylformamide.
- a chlorinating or brominating agent as defined for the same step in Scheme 2
- thionyl chloride or bromide preferably thionyl chloride or bromide/dimethylformamide.
- the former can also act as the solvent, but more preferably the reaction takes place in an appropriate other solvent, such as toluene. In such a case only stoicheiometric amounts of thionyl chloride/bromide would be required, preferably as at least 2 molar equivalents more preferably at least 5 molar equivalents.
- the intermediate of formula (XX) is formed from a compound of formula (XVII) in the presence of an agent which will form a protecting group (P) for the carboxylic acid (i.e. to form the —COP group) as defined herein before.
- said agent is an esterification agent, to form a carboxylic acid ester such as a C 1 -C 6 carboxylic acid ester which will be carried through the reaction scheme and hydrolysed under basic conditions to the carboxylic acid function of compound (XB).
- the esterification agent is ethanol.
- An additional solvent such as toluene may be utilised as appropriate.
- the intermediate of formula (XVII) is formed from 2-hydroxynicotinic acid with a sulphonylating agent such as 30% oleum.
- a sulphonylating agent such as 30% oleum.
- the HBr/HCl (i.e. HD) salts which are formed could be washed out (in aqueous) or filtered from the reaction vessel and the remainder of the aqueous solvent (where applicable) azeotroped off with some of the telescoping solvent.
- solvent such as ethanol
- this solvent could again be azeotroped off with some of the telescoping solvent. If solid alkoxide is used then this latter azeotroping step is not required.
- the telescoping solvent for any telescoped steps of scheme 3 is toluene.
- salts of the compounds of Schemes 1 to 3 can be formed in accordance with the invention by converting the relevant compound to a salt thereof (either in situ or as a separate step). Also an acid addition salt of the compound of formula (I) can be formed in accordance with the invention. 1.4.
- compounds of the general formula (I), (IA) or (IB) may be prepared from “alternative” compounds of the general formula (I), (IA) or (IB) wherein said process may comprise either interconversion of differing —OR 3 groups, interconversion of X and —OR 3 groups or interconversion of —OR 3 and —NR 5 R 6 groups wherein X, R 3 and NR 5 R 6 are as defined hereinbefore.
- certain compounds of formulae (I), (IA) and (IB) can be interconverted by inducing alkoxide exchange or displacement at the 2-position of the 5-(pyridin-3-yl) substituent.
- an alkali metal salt of a sterically hindered alcohol or amine in order to generate the required alkoxide anion which then reacts with the substrate.
- a mixture of from about 1 to about 8, more preferably from about 5 to about 8, and especially from about 4 to about 8 molecular equivalents of potassium bis(trimethylsilyl)amide and the required alcohol (of formula R 3a OH) as solvent is heated at from about 80° C. to about 100° C.
- the substrate may be treated directly, in the required alcohol as solvent, with from about 1.2 to about 6, preferably from about 4 to about 6 molecular equivalents of, for example, potassium bis(trimethylsilyl)amide, potassium t-butoxide or cesium carbonate at from about 80° C. to about 130° C.
- a hydroxide trapping agent may be optionally included in such alkoxide exchange reactions.
- certain compounds of the general formula (I), (IA) or (IB) wherein R 13 is —OR 3 may be obtained from compounds of the general formula (XXX):
- the substrate may be treated with an excess of R 5 R 6 NH, or a suitable acid addition salt thereof, in the presence of an excess of a non-nucleophilic base such as a sterically hindered amine or a suitable inorganic base in a suitable solvent.
- a non-nucleophilic base such as a sterically hindered amine or a suitable inorganic base in a suitable solvent.
- R 5 R 6 NH is used as the free base with about a 3-fold excess (over the substrate) of potassium bis(trimethylsilyl)amide (KHMDS) in dimethylformamide (DMF) as solvent at about 100° C.
- KHMDS potassium bis(trimethylsilyl)amide
- DMF dimethylformamide
- an excess of R 5 R 6 NH may be used as the solvent and the reaction conducted in the presence of about a 50% excess of copper(II) sulphate at up to the reflux temperature of the reaction medium.
- the exchange reaction may be carried out by refluxing with the appropriate amine, and copper(II) sulphate penta- or hepta-hydrate or anhydrous copper (II) sulphate or KHDMS in DMF.
- the OR 3 group for alternative amines of the formula NHR 5 R 6 , such as compounds wherein R 5 or R 6 are selected from aliphatic or cyclic amines, optionally including oxygen (e.g.
- a compound of the general formula (I) may be prepared from a compound of general formulae (IIA) or (IIC) respectively:
- Y is halo, preferably chloro
- R 1 , R 2 , R 3 , R 5 and R 6 are as previously defined for formulae (IXA) and (IXC), by a reaction with a 4-R 10 -piperazinyl compound as described for the preparation of compounds of formula (XA) and (XB) from compounds of formula (VIIA) and (VIIIB) respectively.
- a compound of the general formula (I), (IA) or (IB) may be prepared from a compound of the general formula (IIB):
- R 1 , R 2 , R 4 and X are as previously defined herein via reaction with a 4-R 10 piperazinyl compound followed by an optional displacement reaction in the presence of a hydroxide trapping agent and —OR 3 ⁇ as detailed hereinbefore for the preparation of compound (I) from compound (IXB) or (XXX).
- a compound of general formulae (IIA) or (IIB) or (IIC) may be prepared from a compound of general formula (IVA) or (IVB) or (IVC) respectively:
- R 1 , R 2 , R 3 , R 5 , R 6 and X are as previously herein, by the application of known methods for converting amino to a SO 2 Y group wherein Y is also as previously defined for formulae (IIA), (IIB) and (IIC).
- Y is also as previously defined for formulae (IIA), (IIB) and (IIC).
- Such reactions are previously described for the preparation of compounds of the general formulae (VIIIA) and (VIIIB) from compounds of the general formulae (XIA) and (XIB) respectively.
- a compound of the general formula (IVA) or (IVB) or (IVC) may be prepared by cyclisation of a compound of the general formula (VA) or (VB) or (VC) respectively:
- R 1 , R 2 , R 3 , R 5 , R 6 and X are as previously defined herein and wherein the conditions for cyclisation are analogous to those previously described for cyclisation of the compounds of general formulae (IXA), (IXB) or (IXC).
- a compound of formula (VA) or (VB) or (VC) may be prepared by reduction of a compound of formula (VIA) or (VIB) or (IVC) respectively:
- R 1 , R 2 , R 3 , R 5 , R 6 and X are as previously defined for compounds of the general formulae (VA), (VB) and (VC), by conventional catalytic or catalytic transfer hydrogenation procedures as previously detailed for preparation of compounds of the general formulae (XIA) or (XIB) from compounds of the general formulae (XIIA) or (XIIB) respectively.
- a compound of formula (VIA), (VIB) or (VIC) may be prepared by reaction of a compound of formula (VII) as defined previously herein with a compound of formula (XIIA) or (XIIB) or (XIIC) respectively:
- R 3 , R 5 , R 6 and X are as previously defined for compounds of the general formulae (VIA) or (VIB) or (VIC).
- a conventional amine protecting group strategy is preferred for (XIIA) when NR 5 R 6 is a primary or secondary amino group.
- the coupling reaction is analogous to the reactions of (VII) with the compounds of general formulae (XA) or (XB) or (XC) already described herein.
- a compound of general formulae (IIA) or (IIB) or (IIC) may be prepared from a compound of formula (IVA) or (IVB) or (IVC) respectively as described hereinbefore wherein said compound of the general formulae (IVA) or (IVB) or (IVC) may be prepared by direct cyclisation of a compound of the general formula (VIA) or (VIB) or (VIC) respectively:
- R 1 , R 2 , R 3 , R 5 , R 6 and X are as previously herein and wherein the conditions for said direct cyclisation are analogous to the previously described cyclisation for compounds of the general formulae (IXA) or (IXB) or (IXC) and wherein said cyclisation is followed by reduction of the resultant intermediate compounds according to the methods previously detailed herein to provide compounds of the general formulae (IVA) or (IVB) or (IVC) from compounds of the general formulae (VA) or (VB) or (VC).
- Compounds of the general formula (XIIC) wherein X is Cl may be prepared from 2-hydroxy nicotinic acid via nitration followed by esterification then chlorination of the suitably protected nicotinic acid and subsequent ester hydrolysis.
- Compounds of the general formula (XIIIC) i.e. compounds of general formula (XIIIB wherein X is —OR 3
- a further, generally applicable, synthetic route to compounds of the general formula (I), (IA) or (IB) involves incorporation of the R 1 substituent in the final step of the synthesis.
- a compound of the general formula (I), (IA) or (IB) may be prepared by alkylation of a compound of formula (I a ), (IA a ) or (IB a ) wherein R 1 is hydrogen and R 2 , R 13 and R 4 are as previously defined for formulae (I), (IA) and (IB), using one or more of a plethora of well-known methods, such as:
- the present invention also includes all suitable isotopic variations of a compound of the formula (I) or a pharmaceutically acceptable salt thereof.
- An isotopic variation of a compound of the formula (I) or a pharmaceutically acceptable salt thereof is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature.
- isotopes that can be incorporated into compounds of the formula (I) and pharmaceutically acceptable salts thereof include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine and chlorine such as 2 H, 3 H, 13 C, 14 C, 15 N, 17 O, 18 O, 31 P, 32 P, 35 S, 18 F and 36 Cl, respectively.
- isotopic variations of the compounds of the formula (I) and pharmaceutically acceptable salts thereof are useful in drug and/or substrate tissue distribution studies.
- Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability.
- substitution with isotopes such as deuterium, i.e., 2 H may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements and hence may be preferred in some circumstances.
- Isotopic variations of the compounds of formula (I) and pharmaceutically acceptable salts thereof of this invention can generally be prepared by conventional procedures such as by the illustrative methods or by the preparations described in the Examples and Preparations hereafter using appropriate isotopic variations of suitable reagents.
- the biological activities of the compounds of the present invention were determined by the following test methods.
- the compounds of the present invention are potent and selective cGMP PDE5 inhibitors.
- In vitro PDE inhibitory activities against cyclic guanosine 3′,5′-monophosphate (cGMP) and cyclic adenosine 3′,5′-monophosphate (cAMP) phosphodiesterases were determined by measurement of their IC 50 values (the concentration of compound required for 50% inhibition of enzyme activity).
- the required PDE enzymes were isolated from a variety of sources, including human corpus cavernosum, human and rabbit platelets, human cardiac ventricle, human skeletal muscle and bovine retina, essentially by the method of W. J. Thompson and M. M. Appleman (Biochem., 1971, 10, 311).
- the cGMP-specific PDE (PDE5) and the cGMP-inhibited cAMP PDE (PDE3) were obtained from human corpus cavernosum tissue, human platelets or rabbit platelets; the cGMP-stimulated PDE (PDE2) was obtained from human corpus cavernosum; the calcium/calmodulin (Ca/CAM)-dependent PDE (PDE1) from human cardiac ventricle; the cAMP-specific PDE (PDE4) from human skeletal muscle; and the photoreceptor PDE (PDE6) from bovine retina.
- Phosphodiesterases 7-11 were generated from full length human recombinant clones transfected into SF9 cells.
- the final assay volume was made up to 100 ⁇ l with assay buffer [20 mM Tris-HCl pH 7.4, 5 mM MgCl 2 , 1 mg/ml bovine serum albumin]. Reactions were initiated with enzyme, incubated for 30-60 min at 30° C. to give ⁇ 30% substrate turnover and terminated with 50 ⁇ l yttrium silicate SPA beads (containing 3 mM of the respective unlabelled cyclic nucleotide for PDEs 9 and 11).
- Preferred compounds of the present invention such as those of Examples 3-12, 14-17, 19, 21-30, 32, 33, 35-46, 48-59, 61, 62, 65-75, 77, 79-102 have IC 50 values of less than about 10 nM for the PDE5 enzyme. More preferred compounds, such as those of Examples 3-12, 14, 15, 17, 23-30, 32, 33, 35-46, 48, 50-59, 61, 62, 65, 69-74, 79-102 have IC 50 values of less than about 5 nM for the PDE5 enzyme.
- Especially preferred compounds such as those of Examples 4-10, 15, 17, 23-28, 30, 32, 33, 35-42, 44, 45, 46, 50, 52-56, 58, 59, 61, 62, 65, 69-74, 79-93, 96, 98-102 have IC 50 values of less than about 2 nM for the PDE5 enzyme.
- Especially preferred herein are compounds which have an IC 50 value of less than about 10, more preferably less than about 5, and most preferably less than about 2 nM for the PDE5 enzyme in combination with selectivity of greater than 10-fold, more preferably greater than 50-fold, more preferably greater than 100-fold and especially greater than 200-fold selectivity for the PDE5 enzyme versus the PDE6 enzyme
- the compounds of formulae (I), (IA) or (1B), their pharmaceutically acceptable salts, and pharmaceutically acceptable solvates of either entity can be administered alone but, in human therapy will generally be administered in admixture with a suitable pharmaceutical excipient diluent or carrier selected with regard to the intended route of administration and standard pharmaceutical practice.
- the compounds of formulae (I), (IA) or (1B) or salts or solvates thereof can be administered orally, buccally or sublingually in the form of tablets, capsules (including soft gel capsules), ovules, elixirs, solutions or suspensions, which may contain flavouring or colouring agents, for immediate-, delayed-, modified-, or controlled-release such as sustained-, dual-, or pulsatile delivery applications.
- the compounds of the invention may also be administered via intracavernosal injection.
- the compounds of the invention may also be administered via fast dispersing or fast dissolving dosages forms or in the form of a high energy dispersion or as coated particles.
- Suitable pharmaceutical formulations of the compounds of the invention may be in coated or un-coated form as desired.
- Such tablets may contain excipients such as microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate, glycine and starch (preferably corn, potato or tapioca starch), disintegrents such as sodium starch glycollate, croscarmellose sodium and certain complex silicates, and granulation binders such as polyvinylpyrrolidone, hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), sucrose, gelatin and acacia. Additionally, lubricating agents such as magnesium stearate, stearic acid, glyceryl behenate and talc may be included.
- excipients such as microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate, glycine and starch (preferably corn, potato or tapioca starch), disintegrents such as sodium starch glycollate, croscarmellose sodium and certain complex silicates, and
- Solid compositions of a similar type may also be employed as fillers in gelatin capsules.
- Preferred excipients in this regard include lactose, starch, a cellulose, milk sugar or high molecular weight polyethylene glycols.
- the compounds of the formula (I), (IA) or (IB) may be combined with various sweetening or flavouring agents, colouring matter or dyes, with emulsifying and/or suspending agents and with diluents such as water, ethanol, propylene glycol and glycerin, and combinations thereof.
- Modified release and pulsatile release dosage forms may contain excipients such as those detailed for immediate release dosage forms together with additional excipients that act as release rate modifiers, these being coated on and/or included in the body of the device.
- Release rate modifiers include, but are not exclusively limited to, hydroxypropylmethyl cellulose, methyl cellulose, sodium carboxymethylcellulose, ethyl cellulose, cellulose acetate, polyethylene oxide, Xanthan gum, Carbomer, ammonio methacrylate copolymer, hydrogenated castor oil, carnauba wax, paraffin wax, cellulose acetate phthalate, hydroxypropylmethyl cellulose phthalate, methacrylic acid copolymer and mixtures thereof.
- Modified release and pulsatile release dosage forms may contain one or a combination of release rate modifying excipients.
- Release rate modifying excipients maybe present both within the dosage form i.e. within the matrix, and/or on the dosage form i.e. upon the surface or coating.
- Fast dispersing or dissolving dosage formulations may contain the following ingredients: aspartame, acesulfame potassium, citric acid, croscarmellose sodium, crospovidone, diascorbic acid, ethyl acrylate, ethyl cellulose, gelatin, hydroxypropylmethyl cellulose, magnesium stearate, mannitol, methyl methacrylate, mint flavouring, polyethylene glycol, fumed silica, silicon dioxide, sodium starch glycolate, sodium stearyl fumarate, sorbitol, xylitol.
- dispersing or dissolving as used herein to describe FDDFs are dependent upon the solubility of the drug substance used i.e. where the drug substance is insoluble a fast dispersing dosage form can be prepared and where the drug substance is soluble a fast dissolving dosage form can be prepared.
- the compounds of the invention can also be administered parenterally, for example, intracavernosally, intravenously, intra-arterially, intraperitoneally, intrathecally, intraventricularly, intraurethrally intrasternally, intracranially, intramuscularly or subcutaneously, or they may be administered by infusion or needless injection techniques.
- parenteral administration they are best used in the form of a sterile aqueous solution which may contain other substances, for example, enough salts or glucose to make the solution isotonic with blood.
- the aqueous solutions should be suitably buffered (preferably to a pH of from 3 to 9), if necessary.
- the preparation of suitable parenteral formulations under sterile conditions is readily accomplished by standard pharmaceutical techniques well-known to those skilled in the art.
- the daily dosage level of the compounds of formula (I), (IA) or (1B) or salts or solvates thereof will usually be from 10 to 500 mg (in single or divided doses).
- tablets or capsules of the compounds of formulae (I), (IA) or (IB) or salts or solvates thereof may contain from 5 mg to 250 mg of active compound for administration singly or two or more at a time, as appropriate.
- the physician in any event will determine the actual dosage which will be most suitable for any individual patient and it will vary with the age, weight and response of the particular patient.
- the above dosages are exemplary of the average case. There can, of course, be individual instances where higher or lower dosage ranges are merited and such are within the scope of this invention.
- compounds of the invention may be taken as a single dose on an “as required” basis (i.e. as needed or desired).
- Such tablets can be manufactured by standard processes, for example, direct compression or a wet or dry granulation process.
- the tablet cores may be coated with appropriate overcoats.
- the compounds of the invention can also be administered intranasally or by inhalation and are conveniently delivered in the form of a dry powder inhaler or an aerosol spray presentation from a pressurised container, pump, spray or nebuliser with the use of a suitable propellant, e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, a hydrofluoroalkane such as 1,1,1,2-tetrafluoroethane (HFA 134A [trade mark] or 1,1,1,2,3,3,3-heptafluoropropane (HFA 227EA [trade mark]), carbon dioxide or other suitable gas.
- a suitable propellant e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, a hydrofluoroalkane such as 1,1,1,2-tetrafluoroethane (HFA 134
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- the pressurised container, pump, spray or nebuliser may contain a solution or suspension of the active compound, e.g. using a mixture of ethanol and the propellant as the solvent, which may additionally contain a lubricant, e.g. sorbitan trioleate.
- Capsules and cartridges (made, for example, from gelatin) for use in an inhaler or insufflator may be formulated to contain a powder mix of a compound of the formula (I), (IA) or (IB) and a suitable powder base such as lactose or starch.
- Aerosol or dry powder formulations are preferably arranged so that each metered dose or “puff” contains from 1 to 50 mg of a compound of the formula (I), (IA) or (IB) for delivery to the patient.
- the overall daily dose with an aerosol will be in the range of from 1 to 50 mg which may be administered in a single dose or, more usually, in divided doses throughout the day.
- the compounds of the invention may also be formulated for delivery via an atomiser.
- Formulations for atomiser devices may contain the following ingredients as solubilisers, emulsifiers or suspending agents: water, ethanol, glycerol, propylene glycol, low molecular weight polyethylene glycols, sodium chloride, fluorocarbons, polyethylene glycol ethers, sorbitan trioleate, oleic acid.
- the compounds of the formulae (I), (IA) or (IB) or salts or solvates thereof can be administered in the form of a suppository or pessary, or they may be applied topically in the form of a gel, hydrogel, lotion, solution, cream, ointment or dusting powder.
- the compounds of the formulae (IA) and (IB) or salts or solvates thereof may also be dermally administered.
- the compounds of the formulae (I), (IA) or (IB) or salts or solvates thereof may also be transdermally administered, for example, by the use of a skin patch. They may also be administered by the ocular, pulmonary or rectal routes.
- the compounds can be formulated as micronised suspensions in isotonic, pH adjusted, sterile saline, or, preferably, as solutions in isotonic, pH adjusted, sterile saline, optionally in combination with a preservative such as a benzylalkonium chloride.
- a preservative such as a benzylalkonium chloride.
- they may be formulated in an ointment such as petrolatum.
- the compounds of the formulae (I), (IA) or (IB) or salts or solvates thereof can be formulated as a suitable ointment containing the active compound suspended or dissolved in, for example, a mixture with one or more of the following: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene polyoxypropylene compound, emulsifying wax and water.
- ком ⁇ онентs can be formulated as a suitable lotion or cream, suspended or dissolved in, for example, a mixture of one or more of the following: mineral oil, sorbitan monostearate, a polyethylene glycol, liquid paraffin, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the compounds of the formulae (I), (IA) or (IB) may also be used in combination with a cyclodextrin.
- Cyclodextrins are known to form inclusion and non-inclusion complexes with drug molecules. Formation of a drug-cyclodextrin complex may modify the solubility, dissolution rate, bioavailability and/or stability property of a drug molecule. Drug-cyclodextrin complexes are generally useful for most dosage forms and administration routes.
- the cyclodextrin may be used as an auxiliary additive, e.g. as a carrier, diluent or solubiliser.
- Alpha-, beta- and gamma-cyclodextrins are most commonly used and suitable examples are described in WO-A-91/11172, WO-A-94/02518 and WO-A-98/55148.
- oral administration of the compounds of the invention is the preferred route, being the most convenient and, for example in MED, avoiding the well-known disadvantages associated with intracavernosal (i.c.) administration.
- a preferred oral dosing regimen in MED for a typical man is from 5 to 250 mg of compound when required.
- the drug may be administered parenterally, sublingually or buccally.
- a compound of formula (I), (IA) or (IB), or a veterinarily acceptable salt thereof, or a veterinarily acceptable solvate or pro-drug thereof is administered as a suitably acceptable formulation in accordance with normal veterinary practice and the veterinary surgeon will determine the dosing regimen and route of administration which will be most appropriate for a particular animal.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), (IA) or (IB), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate or pro-drug thereof, together with a pharmaceutically acceptable diluent or carrier.
- a veterinary formulation comprising a compound of formula (I), (IA) or (IB), or a veterinarily acceptable salt thereof, or a veterinarily acceptable solvate or pro-drug thereof, together with a veterinarily acceptable diluent or carrier.
- the invention also provides a compound of formula (I), (IA) or (IB), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate or pro-drug thereof, or a pharmaceutical composition containing any of the foregoing, for use as a human medicament.
- the invention provides the use of a compound of formula (I), (IA) or (IB), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate or pro-drug thereof, for the manufacture of a human medicament for the curative, palliative or prophylactic treatment of a medical condition for which a cGMP PDE5 inhibitor is indicated.
- a compound of formula (I), (IA) or (IB) or a suitable salt, solvate or pro-drug thereof in the manufacture of a medicament for the treatment of a medical condition in which inhibition of a cGMP PDE5 is desirable.
- the invention provides the use of a compound of formula (I), (IA) or (IB), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate or pro-drug thereof, for the manufacture of a human medicament for the curative, palliative or prophylactic treatment of male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder, female sexual orgasmic dysfunction (FSOD), sexual dysfunction due to spinal cord injury, selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction, premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, peripheral vascular disease, stroke,
- gastroparesis Alzheimer's disease, acute respiratory failure, psoriasis, skin necrosis, cancer, metastasis, baldness, nutcracker oesophagus, anal fissure, haemorrhoids, hypoxic vasoconstriction or blood pressure stabilisation during haemodialysis.
- Particularly preferred conditions include MED and FSD.
- a compound of formula (I), (IA) or (IB), or a veterinarily acceptable salt thereof, or a veterinarily acceptable solvate or pro-drug thereof for the manufacture of an animal medicament for the curative, palliative or prophylactic treatment of male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder, female sexual orgasmic dysfunction (FSOD), sexual dysfunction due to spinal cord injury, selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction, premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, peripheral vascular disease, stroke,
- MED male
- the invention provides a method of treating or preventing a medical condition for which a cGMP PDE5 inhibitor is indicated, in a mammal (including a human being), which comprises administering to said mammal a therapeutically effective amount of a compound of formula (I), (IA) or (IB), or a pharmaceutically or veterinarily acceptable salt thereof, or a pharmaceutically or veterinarily acceptable solvate or pro-drug thereof, or a pharmaceutical veterinary formulation containing any of the foregoing.
- the invention provides a method of treating or preventing male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder, female sexual orgasmic dysfunction (FSOD), sexual dysfunction due to spinal cord injury, selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction, premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, peripheral vascular disease, stroke, nitrate induced tolerance, bronchitis, allergic asthma, chronic asthma, allergic rhinitis, diseases and conditions of the eye, diseases characterised by disorders of gut motility, pre-eclampsia, Kawasaki's syndrome,
- a mammal including a human being
- a mammal which comprises administering to said mammal a therapeutically effective amount of a compound of formula (I), (IA) or (IB), or a pharmaceutically or veterinarily acceptable salt thereof, or a pharmaceutically or veterinarily acceptable solvate or pro-drug thereof, or a pharmaceutical composition or veterinary formulation containing any of the foregoing.
- a yet further aspect of the present invention provides a combination of compounds of the general formula (I), (IA) or (IB) with further compounds useful in the inhibition of PDE 5 wherein said combination is useful for the treatment or prevention of male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder, female sexual orgasmic dysfunction (FSOD), sexual dysfunction due to spinal cord injury, selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction, premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, peripheral vascular disease, stroke, nitrate induced tolerance, bronchitis, allergic asthma, chronic asthma
- gastroparesis peripheral diabetic neuropathy, Alzheimer's disease, acute respiratory failure, psoriasis, skin necrosis, cancer, metastasis, baldness, nutcracker oesophagus, anal fissure, haemorrhoids, hypoxic vasoconstriction or blood pressure stabilisation of during haemodialysis in a mammal (including a human being).
- the invention also includes any novel intermediates described herein, for example those of formulae (IXA), (IXB), (VIIA), (VIIB), (VIII), (VIIIA) and (X).
- the present invention additionally comprises the combined administration of a cGMP PDE 5 inhibitor of the general formula (I), wherein said combined administration can be in the form of simultaneous, sequential or joint administration with: (a) one or more naturally occurring or synthetic prostaglandins or esters thereof.
- Suitable prostaglandins for use herein include compounds such as alprostadil, prostaglandin E 1 , prostaglandin E 0 , 13, 14-dihydroprosta glandin E 1 , prostaglandin E 2 , eprostinol, natural synthetic and semi-synthetic prostaglandins and derivatives thereof including those described in U.S. Pat. No. 6,037,346 issued on 14 Mar.
- PGE 0 PGE 1 , PGA 1 , PGB 1 , PGF 1 ⁇ , 19-hydroxy PGA 1 , 19-hydroxy —PGB 1 , PGE 2 , PGB 2 , 19-hydroxy-PGA 2 , 19-hydroxy-PGB 2 , PGE 3 ⁇ , carboprost tromethamine dinoprost, tromethamine, dinoprostone, lipo prost, gemeprost, metenoprost, sulprostune, tiaprost and moxisylate; and/or (b) one or more ⁇ -adrenergic receptor antagonist compounds also known as ⁇ -adrenoceptors or ⁇ -receptors or ⁇ -blockers.
- ⁇ -adrenergic receptor antagonist compounds also known as ⁇ -adrenoceptors or ⁇ -receptors or ⁇ -blockers.
- Suitable compounds for use herein include: the ⁇ -adrenergic receptors as described in PCT application WO99/30697 published on 14 Jun. 1998, the disclosures of which relating to ⁇ -adrenergic receptors are incorporated herein by reference and include, selective ⁇ 1 -adrenoceptors or ⁇ 2 -adrenoceptors and non-selective adrenoceptors, suitable ⁇ 1 -adrenoceptors include: phentolamine, phentolamine mesylate, trazodone, alfuzosin, indoramin, naftopidil, tamsulosin, dapiprazole, phenoxybenzamine, idazoxan, efaraxan, yohimbine, rauwolfa alkaloids, Recordati 15/2739, SNAP 1069, SNAP 5089, RS17053, SL 89.0591, doxazos
- ⁇ -adrenergic receptors as described in U.S. Pat. Nos. 4,188,390; 4,026,894; 3,511,836; 4,315,007; 3,527,761; 3,997,666; 2,503,059; 4,703,063; 3,381,009; 4,252,721 and 2,599,000 each of which is incorporated herein by reference;
- ⁇ 2 -Adrenoceptors include: clonidine, papaverine, papaverine hydrochloride, optionally in the presence of a cariotonic agent such as pirxamine; and/or (c) one or more NO-donor (NO-agonist) compounds.
- Suitable NO-donor compounds for use herein include organic nitrates, such as mono- di or tri-nitrates or organic nitrate esters including glyceryl brinitrate (also known as nitroglycerin), isosorbide 5-mononitrate, isosorbide dinitrate, pentaerythritol tetranitrate, erythrityl tetranitrate, sodium nitroprusside (SNP), 3-morpholinosydnonimine molsidomine, S-nitroso-N-acetyl penicilliamine (SNAP) S-nitroso-N-glutathione (SNO-GLU), N-hydroxy-L-arginine, amylnitrate, linsidomine, linsidomine chlorohydrate, (SIN-1) S-nitroso—N-cysteine, diazenium diolates, (NONOates), 1,5-pentanedinitrate, L-
- Suitable potassium channel openers for use herein include nicorandil, cromokalim, levcromakalim, lemakalim, pinacidil, cliazoxide, minoxidil, charybdotoxin, glyburide, 4-amini pyridine, BaCl 2 ; and/or (e) one or more dopaminergic agents.
- Suitable dopaminergic compounds for use herein include D 2 -agonists such as, pramipexol; apomorphine; and/or (f) one or more vasodilator agents.
- Suitable vasodilator agents for use herein include nimodepine, pinacidil, cyclandelate, isoxsuprine, chloroprumazine, halo peridol, Rec 15/2739, trazodone, pentoxifylline; and/or (g) one or more thromboxane A2 agonists; and/or (h) one or more CNS active agents; and/or (i) one or more ergot alkoloids; Suitable ergot alkaloids are described in U.S. Pat. No. 6,037,346 issued on 14 Mar.
- insulin sensitising agents such as rezulin and hypoglycaemic agents such as glipizide
- L-DOPA or carbidopa and/or (u) one or more acetylcholinesterase inhibitors such as donezipil; and/or (v) one or more steroidal or non-steroidal anti-inflammatory agents.
- Mass spectra (m/z) were recorded using a Fisons Instruments Trio mass spectrometer in the thermospray ionisation mode.
- Room temperature means 20 to 25° C.
- LRMS m/z 516 (M + 1) + 7 1 48 ⁇ (CDCl 3 ): 1.01 (3 H, t), 1.20 (3 H, t), 1.40 (3 H, t), 1.56 (4 H, m), 1.88 (4 H, m), 2.07 (2 H, m), 2.40 (2 H, q), 2.56 (4 H, m), 3.00 (2 H, m), 3.15 (4 H, m), 4.34 (2 H, d), 4.76 (2 H, q), 8.61 (1 H, s), 9.02 (1 H, s), 10.60 (1 H, s).
- LRMS m/z 530 (M + 1) + 8 2 27 Found: C, 53.18; H, 6.48; N, 18.14.
- the crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 90:10), and triturated with ethyl acetate to afford the title compound as a white solid, 80 mg.
- the residual gum was purified by column chromatography on silica gel twice, using dichloromethane:methanol:0.88 ammonia (89:10:1) as eluant and repeated using ethyl acetate:methanol:diethylamine (78:20:2) as eluant to afford the title compound, 32 mg, as a beige foam.
- Trifluoroacetic acid (3 ml) was added to a solution of the title compound from preparation 63 (350 mg, 0.57 mmol) in dichloromethane (3 ml), and the reaction stirred at room temperature for 21 ⁇ 2 hours. The reaction was concentrated under reduced pressure and the residual gum was triturated several times with ether. The resulting suspension was sonicated for a minute, then the solid filtered, washed with ether, and dried to give the title compound as a white powder, 280 mg.
- the title compound was obtained as a beige-coloured powder, (51%), from the title compound of preparation 66, following a similar procedure to that described in example 18.
- the title compound was obtained as a white solid, (79%) from the title compound from preparation 61 and trifluoroacetic acid, following the procedure described in example 18.
- Acetyl chloride (6 mg, 0.076 mmol) was added to a mixture of the title compound from example 18 (43 mg, 0.056 mmol) and triethylamine (8.5 mg, 0.086 mmol) in dichloromethane (2 ml), and the reaction stirred for 36 hours at room temperature. The mixture was treated with aqueous saturated sodium bicarbonate solution and extracted with ethyl acetate (2 ⁇ ). The combined organic extracts were dried (MgSO 4 ), and evaporated under reduced pressure. The residual gum was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (97:3 to 95:5) to give the title compound, 19 mg.
- the title compound was obtained (33%) from the title compound from preparation 68 and methanesulphonic anhydride, following a similar procedure to that described in example 22.
- Trifluoroacetic acid (0.5 ml) was added to a solution of the title compound from preparation 65 (28 mg, 0.043 mmol) in dichloromethane (0.5 ml), and the solution stirred for 21 ⁇ 2 hours at room temperature. The mixture was evaporated under reduced pressure and the residue triturated with ether several times. The resulting precipitate was filtered off, washed with ether and dried, to give a beige-coloured solid.
- LRMS m/z 534 (M + 1) + 36 ⁇ (CDCl 3 ): 1.04 (6 H, 2xt), 1.40 (3 H, t), 1.55 (2 H, m), 1.95 (2 H, m), 2.42 (2 H, q), 2.55 (4 H, m), 3.07 (2 H, q), 3.15 (4 H, m), 3.30 (3 H, s), 3.92 (2 H, t), 4.46 (2 H, t), 4.66 (2 H, t), 8.62 (1 H, s), 9.02 (1 H, s), 10.60 (1 H, s).
- LRMS m/z 548 (M + 1) + 37 Found: C, 54.77; H, 6.82; N, 17.75.
- the title compound was obtained as a white solid (64%) from the title compound from example 7 and 2-methoxyethanol, following the procedure described in example 41.
- the title compound was obtained as a yellow foam (57%) from the title compound from example 9 and 2-methoxyethanol, following the procedure described in example 41.
- Potassium bis(trimethylsilyl)amide (359 mg, 1.8 mmol) was added to a solution of the title compound from preparation 70 (250 mg, 0.45 mmol) in 2-methoxyethanol (5 ml), and the reaction heated under reflux for 6 hours. Tlc analysis showed starting material remaining, so additional potassium bis(trimethylsilyl)amide (90 mg, 0.45 mmol) was added to the cooled mixture, and the reaction stirred for a further 4 hours under reflux. The cooled mixture was concentrated under reduced pressure and the residue purified by column chromatography on silica gel using dichloromethane:methanol (95:5) as eluant. The product was triturated with ether and pentane to afford the title compound as a crystalline solid, 75 mg.
- Potassium bis(trimethylsilyl)amide (732 mg, 3.68 mmol) was added to a solution of the title compound from preparation 40 (958 mg, 1.84 mmol) in 2-methoxyethanol (20 ml) and the reaction stirred for 16 hours at 120° C. The cooled mixture was concentrated under reduced pressure, the residue dissolved in water (25 ml) and the pH adjusted to 2 using hydrochloric acid (2N). The solution was washed with ethyl acetate, neutralised and the resulting precipitate filtered off.
- the title compound was obtained as a beige solid (31%) from the title compound from preparation 41 and 2-methoxyethanol, using a similar procedure to that described 52.
- Trifluoroacetic acid (1 ml) was added to a solution of the title compound from preparation 62 (76 mg, 0.117 mmol) in dichloromethane (1 ml), and the solution stirred for 21 ⁇ 2 hours at room temperature. The mixture was evaporated under reduced pressure, the residue triturated well with ether, and the resulting precipitate, filtered and dried to give a white powder.
- Methanesulphonyl chloride (20 ⁇ l, 0.26 mmol) was added to a solution of this intermediate in dichloromethane (2 ml) and triethylamine (65 ⁇ l, 0.47 mmol), and the reaction stirred at room temperature for 11 ⁇ 2 hours.
- the mixture was treated with saturated aqueous sodium bicarbonate solution (10 ml), and extracted with ethyl acetate (2 ⁇ 10 ml).
- the combined organic extracts were dried (MgSO 4 ) and evaporated under reduced pressure to give a gum.
- the crude product was purified by column chromatography on silica gel using dichloromethane:methanol (96:4) as eluant to afford the title compound as a beige foam, 30 mg.
- the title compound was obtained as a white solid, (34%), from the title compound from preparation 67 and methanesulphonyl chloride, following the procedure described in example 61.
- Methanesulphonyl chloride (15 ⁇ l, 0.19 mmol) was added to an ice-cooled solution of the title compound from example 64 (93 mg, 0.17 mmol) in pyridine (2 ml), and the reaction allowed to warm to room temperature, and stirred for 90 minutes. Tlc analysis showed starting material remaining, so additional methanesulphonyl chloride (15 ⁇ l, 0.19 mmol) was added, and the reaction stirred for a further hour. The reaction was quenched by the addition of aqueous ammonium chloride solution, and extracted with ethyl acetate. The combined organic extracts were dried (MgSO 4 ) and concentrated under reduced pressure.
- the residual solid was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (90:10:1) as eluant, then repeated using dichloromethane:methanol:0.88 ammonia (95:5:1) to afford the title compound, 36 mg.
- Tris(dibenzylideneacetone)dipalladium (0) (8 mg, 0.009 mmol), R-BINAP (8 mg, 0.013 mmol), sodium tert-butoxide (41 mg, 0.43 mmol) and 2-bromopyridine (50 ⁇ l, 0.52 mmol) were added to a solution of the title compound from preparation 58 (200 mg, 0.43 mmol) in toluene (3 ml), and the reaction heated at 70° C. for 16 hours. The cooled mixture was evaporated under reduced pressure and the residue filtered through silica gel, using dichloromethane:methanol (80:20) as eluant.
- the product was purified by reverse phase HPLC on silica gel, using an elution gradient of acetonitrile:0.1% aqueous trifluoroacetic acid (5:95 to 85:15) to afford the title compound, as a solid, 13 mg.
- Potassium bis(trimethylsilyl)amide (450 mg, 2.25 mmol) was added to a suspension of the compound from preparation 84 (243 mg, 0.45 mmol) in ethanol (5 ml), and the mixture heated at 100° C. in a Reactivial® for 24 hours. Tlc analysis showed starting material remaining, so additional potassium bis(trimethylsilyl)amide (250 mg, 1.25 mmol) and ethyl acetate (3 drops) were added, and the reaction heated at 111° C. for 18 hours. The cooled mixture was partitioned between ethyl acetate and sodium bicarbonate solution, and the phases separated.
- the title compound was obtained as a white powder in 41% yield from the compound from preparation 85, following the procedure described in example 80.
- Potassium bis(trimethylsilyl)amide (2.10 g, 10.5 mmol) was added to a solution of the compound from preparation 90 (1.20 g, 2.17 mmol) and ethyl acetate (200 ⁇ l, 2.02 mmol) in ethanol (40 ml), and the reaction heated in a sealed vessel at 130° C. for 6 hours. The cooled mixture was concentrated under reduced pressure and the residue partitioned between ethyl acetate and water, and neutralised by the addition of solid carbon dioxide. The layers were separated, the aqueous phase extracted with ethyl acetate, and the combined organic solutions dried (Na 2 SO 4 ) and evaporated under reduced pressure.
- the crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (99:1 to 96:4), and the product crystallised from ether/pentane to afford the title compound, 250 mg.
- Potassium bis(trimethylsilyl)amide (123 mg, 0.62 mmol) was added to a solution of the compound from preparation 27 (162 mg, 0.31 mmol) in n-butanol, and the reaction mixture heated at 120° C. for 18 hours. The cooled mixture was concentrated under reduced pressure and the residual yellow oil purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol:0.88 ammonia (98:2:0.2 to 95:5:0.5). The product was triturated with ether to give the title compound as a white solid, 78 mg.
- the title compound was obtained as a white powder in 23% yield from the compound from preparation 94 and 2-methoxyethanol, following the procedure described in example 94.
- Example 103 water and pharmaceutically acceptable alcohols such as methanol, ethanol, propanol, butanol and mixtures thereof can be used to prepare the compound of Examples 8 and 102.
- the powder X-ray diffraction (PXRD) pattern for this salt having Mpt 242-244° C., was determined using a Siemens D5000 powder X-ray diffractometer fitted with a theta-theta goniometer, automatic beam divergence slits, a secondary monochromator and a scintillation counter.
- the same besylate salt, as defined by the XRD pattern described in Table 1, when made via alternative routes can have a melting point in the range of from 235-246° C. (measured using a Perkin Elmer DSC7 at a heating rate of 20° C./minute).
- IC 50 measurements for cGMP PDE 5 were based upon data generated on human corpus cavernosum tissue and the IC 50 measurements for rod cGMP PDE 6 were based upon data generated on bovine retina tissue and wherein the selectivity ratio for cGMP PDE 5 to cGMP PDE 6 quoted is based upon IC 50 PDE 5 /IC 50 PDE 6 .
- the crude product was purified by column chromatography on silica gel, using ethyl acetate as eluant, and repeated using an elution gradient of pentane:ethyl acetate (50:50 to 0:100), to give some of the desired compound.
- N-Methylpiperazine (7.65 ml, 69.0 mmol) was added dropwise to a solution of the title compound of preparation 18 (10.0 g, 34.5 mmol) in ethanol (200 ml), and the reaction stirred at room temperature for 3 hours. The mixture was concentrated under reduced pressure and the residue partitioned between dichloromethane (200 ml) and water (100 ml) and the layers separated. The organic phase was dried (Na 2 SO 4 ), and evaporated under reduced pressure to afford the title compound, 10.53 g, as a yellow solid.
- Oxalyl chloride (500 ml, 5.73 mmol) was added dropwise to an ice-cooled solution of the title compound of preparation 26 (522 mg, 1.43 mmol) and N,N-dimethylformamide (1 drop) in dichloromethane (20 ml), and the reaction stirred for 2 hours. The mixture was concentrated under reduced pressure and azeotroped several times with dichloromethane to give the intermediate acid chloride. A solution of this product in dichloromethane (20 ml) was added to a solution of the title compound of preparation 9 (250 mg, 1.18 mmol) and triethylamine (500 ml, 3.18 mmol) in dichloromethane (20 ml), and the reaction stirred at room temperature for 18 hours.
- the title compound was obtained as a white solid (79%), from the title compounds from preparation 10 and 26, following the procedure described in preparation 27.
- the reaction mixture was diluted with dichloromethane (100 ml), washed with water (100 ml), saturated aqueous sodium bicarbonate solution (100 ml), and brine (100 ml), dried (MgSO 4 ), and evaporated under reduced pressure.
- the crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to give the title compound as a foam, 10.05 g.
- N-Diisopropylethylamine (0.92 ml, 5.3 mmol) was added to a solution of the title compounds from preparation 25 (1.0 g, 2.65 mmol), and preparation 10 (600 mg, 2.65 mmol), 1-hydroxybenzotriazole hydrate (465 mg, 3.45 mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (660 mg, 3.45 mmol) in dichloromethane (20 ml), and the reaction stirred for 18 hours. The reaction mixture was washed with brine, dried (MgSO 4 ), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 740 mg.
- Triethylamine (1.0 ml, 7.2 mmol) was added to a solution of the title compounds from preparation 25 (1.5 g, 4.5 mmol), and preparation 13 (1.7 g, 4.95 mmol), 1-hydroxybenzotriazole hydrate (833 mg, 5.44 mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.28 g, 6.68 mmol) in dichloromethane (50 ml), and the reaction stirred for 3 days at room temperature. The reaction mixture was concentrated under reduced pressure and the residue partitioned between saturated aqueous sodium bicarbonate solution and ethyl acetate, and the layers separated.
- the aqueous phase was extracted with ethyl acetate (2 ⁇ 50 ml), and the combined organic solutions, dried (MgSO 4 ), and evaporated under reduced pressure.
- the crude product was purified by column chromatography on silica gel using dichloromethane:methanol (95:5) as eluant to afford the title compound, 3.0 g.
- Oxalyl chloride (9.5 ml, 108 mmol) was added dropwise to an ice-cold solution of the title compound from preparation 26 (10.0 g, 27.0 mmol) and N,N-dimethylformamide (160 ⁇ l) in dichloromethane (150 ml), and once addition was complete, the reaction was stirred at room temperature for 51 ⁇ 2 hours. The mixture was evaporated under reduced pressure and the residue azeotroped with toluene, to give a yellow solid.
- Triethylamine (11.2 ml, 81.0 mmol) was added to a solution of the intermediate acid chloride (10.5 g, 27.3 mmol) and 4-amino-3-ethyl-1H-pyrazole-5-carboxamide (WO, 9849166), (4.2 g, 27.3 mmol) in dichloromethane (150 ml), and the reaction stirred at room temperature for 18 hours. The mixture was diluted with water, and the layers separated. The aqueous phase was extracted with dichloromethane (2 ⁇ ), and the combined organic solutions dried (Na 2 SO 4 ) and evaporated under reduced pressure. The crude product was triturated with ether, and the resulting solid filtered to give the title compound, 10.1 g.
- Methanesulphonyl chloride (2.86 ml, 36.9 mmol) was added dropwise to an ice-cooled solution of 1-methoxy-2-propanol (3 ml, 30.7 mmol) and triethylamine (10.27 ml, 73.7 mmol) in dichloromethane (150 ml), and the reaction stirred at room temperature for 18 hours. The mixture was washed with water, then 2M hydrochloric acid, dried (MgSO 4 ) and evaporated under reduced pressure to give the title compound as a yellow oil, 5.24 g.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Pulmonology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Dermatology (AREA)
- Diabetes (AREA)
- Gynecology & Obstetrics (AREA)
- Ophthalmology & Optometry (AREA)
- Epidemiology (AREA)
- Pregnancy & Childbirth (AREA)
- Hospice & Palliative Care (AREA)
- Vascular Medicine (AREA)
- Obesity (AREA)
- Psychiatry (AREA)
- Oncology (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Otolaryngology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compounds of the formula (I):

wherein R1, R2, R4 and R13 are as defined or a pharmaceutically or veterinarily acceptable salt or polymorph thereof, or a pharmaceutically or veterinarily acceptable solvate or pro-drug thereof: are potent and selective inhibitors of type 5 cyclic guanosine 3′,5′-monophosphate phosphodiesterase (cGMP PDE5) and have utility in the treatment of, inter alia, male erectile dysfunction (MED) and female sexual dysfunction (FSD).
Description
- This invention relates to a series of pyrazolo[4,3-d]pyrimidin-7-ones, which inhibit
cyclic guanosine 3′,5′-monophosphate phosphodiesterases (cGMP PDEs). More notably, the compounds of the invention are potent and selective inhibitors of type 5cyclic guanosine 3′,5′-monophosphate phosphodiesterase (cGMP PDE5) and have utility therefore in a variety of therapeutic areas. - The compounds of the invention are of value for the curative or prophylactic treatment of mammalian sexual disorders. In particular, the compounds are of value in the treatment of mammalian sexual dysfunctions such as male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder or female sexual orgasmic dysfunction (FSOD) as well as sexual dysfunction due to spinal cord injury or selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction but, clearly, will be useful also for treating other medical conditions for which a potent and selective cGMP PDE5 inhibitor is indicated. Such conditions include premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, e.g. post-percutaneous transluminal coronary angioplasty (post-PTCA), peripheral vascular disease, stroke, nitrate induced tolerance, bronchitis, allergic asthma, chronic asthma, allergic rhinitis, diseases and conditions of the eye such as glaucoma, optic neuropathy, macular degeneration, elevated intra-occular pressure, retinal or arterial occulsion and diseases characterised by disorders of gut motility, e.g. irritable bowel syndrome (IBS).
- Further medical conditions for which a potent and selective cGMP PDE5 inhibitor is indicated, and for which treatment with compounds of the present invention may be useful include pre-eclampsia, Kawasaki's syndrome, nitrate tolerance, multiple sclerosis, diabetic nephropathy, neuropathy including autonomic and peripheral neuropathy and in particular diabetic neuropathy and symptoms thereof e.g. gastroparesis, peripheral diabetic neuropathy, Alzheimer's disease, acute respiratory failure, psoriasis, skin necrosis, cancer, metastasis, baldness, nutcracker oesophagus, anal fissure, haemorrhoids, hypoxic vasoconstriction as well as the stabilisation of blood pressure during haemodialysis.
- Particularly preferred conditions include MED and FSD.
- PCT application PCT/IB99/00519 relates to a series of pyrazolo[4,3-d]pyrimidin-7-ones, which inhibit
cyclic guanosine 3′,5′-monophosphate phosphodiesterases (cGMP PDEs). - Thus the present invention provides compounds of the formula (I):
-

- or a pharmaceutically or veterinarily acceptable salt thereof, or a pharmaceutically or veterinarily acceptable solvate of either entity, wherein
-
- R1 is C1 to C6 alkyl or C3 to C6 alkenyl, C3 to C6 cycloalkyl or C4 to C6 cycloalkenyl wherein said alkyl group may be branched or straight chain and wherein
- when R1 is C1 to C3 alkyl said alkyl group is substituted by;
- and wherein when R1 is C4 to C6 alkyl, C3 to C6 alkenyl or C3 to C6 cycloalkyl said alkyl, alkenyl or cycloalkyl group is optionally substituted by;
- one or more substituents selected from:
- hydroxy;
- C1 to C4 alkoxy;
- C3 to C6 cycloalkyl;
- phenyl substituted with one or more substitutents selected from C1 to C3 alkyl, C1 to C4 alkoxy, C1 to C4 haloalkyl, C1 to C4 haloalkoxy, halo, CN, NO2, NHR11, NHCOR12, NHSO2R12, SO2R12, SO2NHR11, COR11 or CO2R11 wherein said haloalkyl and haloalkoxy groups contain one or more halo atoms;
- NR7R8, CONR7R8 or NR7COR11 wherein R7 and R8 are each independently selected from H, C1 to C4 alkyl, C3 to C4 alkenyl, CO2R9 or SO2R9 and wherein said alkyl or alkenyl groups are optionally substituted by C1 to C4 haloalkyl or C1 to C4 haloalkoxy;
- Het1;
- Het2 or Het3;
- or R1 is Het4 or phenyl wherein said phenyl group is optionally substituted by one or more substituents selected from C1 to C4 alkyl, C3 to C4 alkenyl, C1 to C4 alkoxy, halo, CN, CF3, OCF3, NO2, NHR11, NHCOR12, NHSO2R12, SO2R12, SO2NHR11, COR11, CO2R11;
- R2 is C1 to C6 alkyl, C3 to C6 alkenyl or (CH2)n(C3 to C6 cycloalkyl) wherein n is 0, 1 or 2;
- R13 is OR3 or NR5R6;
- R3 is C1 to C6 alkyl optionally substituted with one or two substituents selected from C3 to C5 cycloalkyl, hydroxy, C1 to C4 alkoxy, benzyloxy, NR5R6, phenyl, Het1, Het2, Het3 or Het4 wherein the C1 to C6 alkyl and C1 to C4 alkoxy groups may optionally be terminated by a haloalkyl group such as CF3 and wherein the C3-C5 cycloalkyl group may optionally be substituted by C1-C4 alkyl, hydroxy or halo;
- C3 to C6 cycloalkyl; Het1, Het2, Het3 or Het4;
- R4 is a piperazin-1-ylsulphonyl group having a substituent R10 at the 4-position of the piperazinyl group wherein said piperazinyl group is optionally substituted with one or two C1 to C4 alkyl groups and is optionally in the form of its 4-N-oxide;
- R5 and R6 are each independently selected from H and C1 to C4 alkyl optionally substituted with C3 to C5 cycloalkyl or C1 to C4 alkoxy, or, together with the nitrogen atom to which they are attached, form an azetidinyl, pyrrolidinyl, piperidinyl or morpholinyl group;
- R7 and R8 are each independently selected from H, C1 to C4 alkyl, C3 to C4 alkenyl, CO2R9 or SO2R9;
- R9 is C1 to C4 alkyl optionally substituted with C1 to C4 haloalkyl, C1 to C4 haloalkoxy or phenyl wherein said phenyl group is optionally substituted by one or more substituents selected from C1 to C4 alkyl optionally substituted by C1 to C4 haloalkyl or C1 to C4 haloalkoxy, C1 to C4 alkoxy, halo, CN, NO2, NHR11, NHCOR12, NHSO2R12, SO2R12, SO2NHR11, COR11 or CO2R11;
- R10 is H; C1 to C4 alkyl optionally substituted with one or two substituents selected from hydroxy, NR5R6, CONR5R6, phenyl optionally substituted with C1 to C4 alkyl or C1 to C4 alkoxy; C3 to C6 alkenyl or Het4;
- R11 is H, C1 to C4 alkyl, C3 to C4 alkenyl, CO(C1 to C4 alkyl) or C1 to C4 haloalkyl;
- R12 is C1 to C4 alkyl, C3 to C4 alkenyl, C1 to C4 haloalkyl or C1 to C4 haloalkoxy;
- Het1 is an N-linked 4-, 5- or 6-membered nitrogen-containing heterocyclic group optionally containing one or more further heteroatoms selected from S, N or O;
- Het2 is a C-linked 5-membered heterocyclic group containing an O, S or N heteroatom optionally containing one or more heteroatoms selected from N, O or S;
- Het3 is a C-linked 6-membered heterocyclic group containing an O or S heteroatom optionally containing one or more heteroatoms selected from O, S or N or Het3 is a C-linked 6-membered heterocyclic group containing three N heteroatoms;
- Het4 is a C-linked 4-, 5- or 6-membered heterocyclic group containing one, two or three heteroatoms selected from S, O or N; and
- wherein any of said heterocyclic groups Het1, Het2, Het3 or Het4 may be saturated, partially unsaturated or aromatic and wherein any of said heterocyclic groups may be optionally substituted with one or more substituents selected from C1 to C4 alkyl, C3 to C4 alkenyl, C1 to C4 alkoxy, halo, CF3, CO2R11, COR11, SO2R12, NHR11 or NHCOR12 and/or wherein any of said heterocyclic groups is benzo-fused;
with the provisos that (a) when R1 is C1 to C3 alkyl then Het1 is not morpholinyl or piperidinyl and (b) when R1 is C1 to C3 alkyl substituted by phenyl then said phenyl group is not substituted by C1 to C4 alkoxy, halo, CN, CF3, OCF3 or C1 to C4 alkyl.
As will be recognised by the skilled chemist, the general formula (I) can be represented by the regio-isomeric general formulae (IA) and (IB). Thus the present invention provides compounds of formulae (IA) and (IB):
-
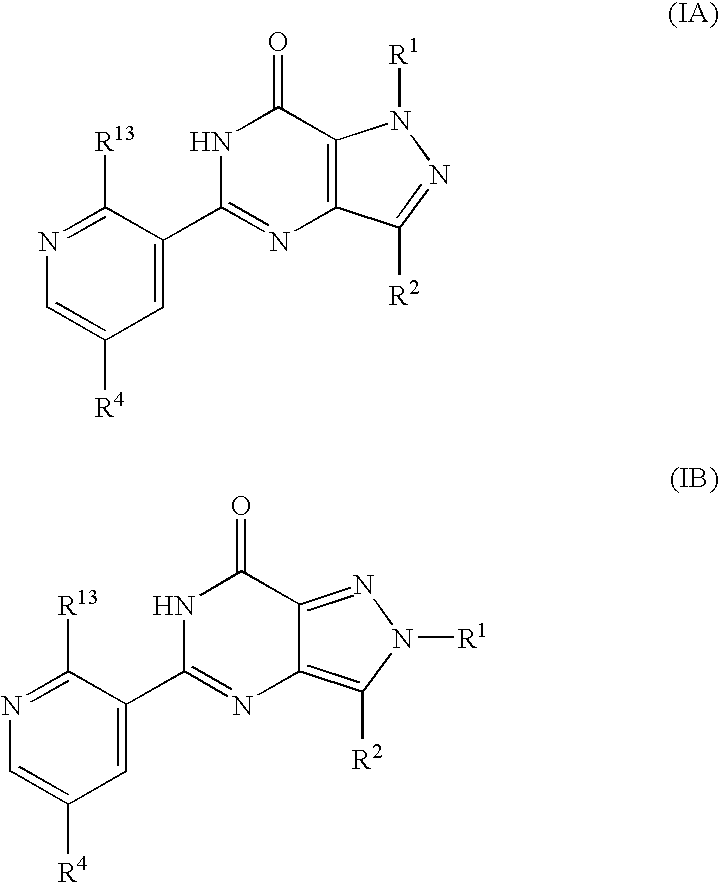
- wherein R1, R2, R4 and R13 are as defined hereinbefore.
- In the above definitions, unless otherwise indicated, alkyl, alkoxy and alkenyl groups having three or more carbon atoms, and alkanoyl groups having four or more carbon atoms, may be straight chain or branched chain. For example, a C4 alkyl substituent can be in the form of normal-butyl (n-butyl), iso-butyl (i-butyl), secondary-butyl (sec-butyl) or tertiary-butyl (t-butyl). The term halo atom includes Cl, Br, F, and I. Haloalkyl and haloalkoxy are preferably —CF3 and —OCF3 respectively. The term aromatic as defined herein means a fully unsaturated system.
- A compound of the formula (I) contains one or more asymmetric carbon atoms and therefore exists in two or more stereoisomeric forms. Where a compound of the formula (I) contains an alkenyl or alkenylene group, cis (E) and trans (Z) isomerism may also occur. The present invention includes the individual stereoisomers of the compounds of the formula (I) and, where appropriate, the individual tautomeric forms thereof, together with mixtures thereof. Separation of diastereoisomers or cis and trans isomers may be achieved by conventional techniques, e.g. by fractional crystallisation, chromatography or H.P.L.C. of a stereoisomeric mixture of a compound of the formula (I) or a suitable salt or derivative thereof. An individual enantiomer of a compound of the formula (I) may also be prepared from a corresponding optically pure intermediate or by resolution, such as by H.P.L.C. of the corresponding racemate using a suitable chiral support or by fractional crystallisation of the diastereoisomeric salts formed by reaction of the corresponding racemate with a suitable optically active acid or base, as appropriate.
- All stereoisomers are included within the scope of the invention.
- The compounds of formulae (IA) and (IB) may also exist in tautomeric forms and the invention includes both mixtures thereof and the individual tautomers.
- Also included in the invention are radiolabelled derivatives of compounds of formulae (I), (IA) and (IB) which are suitable for biological studies.
- The pharmaceutically or veterinarily acceptable salts of the compounds of the invention which contain a basic centre are, for example, non-toxic acid addition salts formed with inorganic acids such as hydrochloric, hydrobromic, hydroiodic, sulphuric and phosphoric acid, with carboxylic acids or with organo-sulphonic acids. Examples include the HCl, HBr, HI, sulphate or bisulphate, nitrate, phosphate or hydrogen phosphate, acetate, benzoate, succinate, saccarate, fumarate, maleate, lactate, citrate, tartrate, gluconate, camsylate, methanesulphonate, ethanesulphonate, benzenesulphonate, p-toluenesulphonate and pamoate salts. Compounds of the invention can also provide pharmaceutically or veterinarily acceptable metal salts, in particular non-toxic alkali and alkaline earth metal salts, with bases. Examples include the sodium, potassium, aluminium, calcium, magnesium, zinc and diethanolamine salts. For a review on suitable pharmaceutical salts see Berge et al, J. Pharm, Sci., 66, 1-19, 1977.
- The pharmaceutically acceptable solvates of the compounds of the invention include the hydrates thereof.
Also included within the scope of the compound and various salts of the invention are polymorphs thereof. - A preferred group of compounds of formulae (I), (IA) and (IB) is that wherein, R1 is C1 to C6 alkyl or C3 to C6 alkenyl wherein said alkyl or alkenyl groups may be branched chain or straight chain or R1 is C3 to C6 cycloalkyl or C4 to C6 cycloalkenyl and wherein when R1 is C1 to C3 alkyl said alkyl group is substituted by; and wherein when R1 is C4 to C6 alkyl, C3 to C6 alkenyl, C3 to C6 cycloalkyl or C4 to C6 cycloalkenyl said alkyl, alkenyl, cycloalkyl or cycloalkenyl group is optionally substituted by;
- one or more substituents selected from:
hydroxy;
C1 to C4 alkoxy;
C3 to C4 cycloalkyl;
phenyl substituted with one or more substitutents selected from C1 to C3 alkyl, C1 to C4 alkoxy, C1 to C4 haloalkyl or C1 to C4 haloalkoxy, halo, CN, NO2, NHR11, NHCOR12, NHSO2R12, SO2R12, SO2NHR11, COR11, CO2R11 wherein said haloalkyl and haloalkoxy groups contain one or more halo atoms; - a Het1 group which is an N-linked 4-membered N-containing heterocyclic group;
a Het2 group which is a C-linked 5-membered heterocyclic group containing an O, S or N heteroatom optionally containing one or more heteroatoms selected from N, O or S;
a Het3 group which is a C-linked 6-membered heterocyclic group containing an O or S heteroatom optionally containing one or more heteroatoms selected from O, S or N or a Het3 group which is a C-linked 6-membered heterocyclic group containing three N heteroatoms;
wherein R7, R8, R11 and R12 are as previously defined herein
or R1 is a Het4 group which is a C-linked 4- or 5-membered heterocyclic group containing one heteroatom selected from S, O or N; a Het4 group which is a C-linked 6-membered heterocyclic group containing one, two or three heteroatoms selected from S or O; a Het4 group which is a C-linked 6-membered heterocyclic group containing three nitrogen heteroatoms; a Het4 group which is a C-linked 6-membered heterocyclic group containing one or two nitrogen heteroatoms which is substituted by one or more substitutents selected from C1 to C4 alkyl, C1 to C4 alkoxy, CO2R11, SO2R12, COR11, NHR11 or NHCOR12 and optionally including a further heteroatom selected from S, O or N
wherein any of said heterocyclic groups Het1, Het2, Het3 or Het4 is saturated, partially unsaturated or aromatic as appropriate and wherein any of said heterocyclic groups is optionally substituted with one or more substituents selected from C1 to C4 alkyl, C3 to C4 alkenyl, C1 to C4 alkoxy, halo, CO2R11, SO2R12, COR11 or NHR11 wherein R11 is as defined hereinbefore and/or wherein any of said heterocyclic groups is benzo-fused;
or R1 is phenyl substituted by one or more substituents selected from CF3, OCF3, SO2R12 or CO2R12 wherein R12 is C1 to C4 alkyl which is optionally substituted by phenyl, C1 to C4 haloalkyl or C1 to C4 haloalkoxy wherein said haloalkyl and haloalkoxy groups contain one or more halo atoms;
R2 is C1 to C6 alkyl; - R3 is C1 to C6 alkyl optionally substituted with one or two substituents selected from C3 to C5 cycloalkyl, hydroxy, C1 to C4 alkoxy, benzyloxy, NR5R6, phenyl, furanyl, tetrahydrofuranyl or pyridinyl wherein said C1 to C6 alkyl and C1 to C4 alkoxy groups may optionally be terminated by a haloalkyl group such as CF3; or R3 is C3 to C6 cycloalkyl, 1-(C1 to C4 alkyl)piperidinyl, tetrahydrofuranyl or tetrahydropyranyl;
R4 is a piperazin-1-ylsulphonyl group having a substituent R10 at the 4-position of the piperazinyl group wherein said piperazinyl group is optionally substituted with one or two C1 to C4 alkyl groups and is optionally in the form of its 4-N-oxide;
R5 and R6 are each independently selected from H and C1 to C4 alkyl optionally substituted with C3 to C5 cycloalkyl or C1 to C4 alkoxy, or, together with the nitrogen atom to which they are attached, form an azetidinyl, pyrrolidinyl, piperidinyl or morpholinyl group; and
R10 is H; C1 to C4 alkyl optionally substituted with one or two substituents selected from hydroxy, NR5R6, CONR5R6, phenyl optionally substituted with C1 to C4 alkyl or C1 to C4 alkoxy; C3 to C6 alkenyl; Het4;
with the proviso that when R1 is C1 to C3 alkyl substituted by phenyl then said phenyl group is not substituted by C1 to C4 alkoxy; CN; halo; CF3; OCF3; or C1 to C4 alkyl. - A further preferred group of compounds of formulae (I), (IA) and (IIB) is that wherein, R1 is C1 to C6 alkyl wherein said alkyl may be branched or straight chain or R1 is C3 to C6 cycloalkyl
- and wherein when R1 is C1 to C3 alkyl said alkyl group is substituted by; and wherein when R1 is C4 to C6 alkyl or C3 to C6 cycloalkyl said alkyl or cycloalkyl group is optionally substituted by;
one or more substituents selected from:
hydroxy;
C1 to C2 alkoxy;
C3 to C5 cycloalkyl;
NR7R8, NR7COR11 or COR11 wherein R7 and R8 are each independently selected from H, C1 to C4 alkyl or CO2R9 wherein R9 and R11 are as previously defined herein;
a Het1 group which is an N-linked 4-membered N-containing heterocyclic group;
a Het3 group which is a C-linked 6-membered heterocyclic group containing an O or S heteroatom optionally containing one or more heteroatoms selected from O, S or N or a Het3 group which is a C-linked 6-membered heterocyclic group containing three N heteroatoms; - or R1 is a Het4 group which is a C-linked 4-membered heterocyclic group containing one heteroatom selected from S, O or N or R1 is a Het4 group which is a C-linked 6-membered heterocyclic group containing one, two or three heteroatoms selected from S or O
- wherein any of said heterocyclic groups Het1, Het2, Het3 or Het4 is saturated, partially unsaturated or aromatic and is optionally substituted with one or more substituents selected from C1 to C4 alkyl, C1 to C4 alkoxy, —CO2R11, —SO2R12, —COR11 or NHR11 wherein R11 and R12 are as defined hereinbefore and/or wherein any of said heterocyclic groups is benzo-fused;
- or R1 is phenyl substituted by one or more substituents selected from CF3, —OCF3, —SO2R12, —COR11, —CO2R11 wherein R11 and R12 are as defined hereinbefore
- R2 is C1 to C6 alkyl;
- R3 is methyl, ethyl, n-propyl, i-propyl, n-butyl, sec-butyl, i-butyl or t-butyl alkyl optionally substituted with one or two substituents selected from cyclopropyl, cyclobutyl, hydroxy, methoxy, ethoxy, benzyloxy, phenyl, benzyl, furan-3-yl, tetrahydrofuran-2-ylmethyl, tetrahydrofuran-3-ylmethyl, pyridin-2-yl, pyridin-3-yl or NR5R6 wherein R5 and R6 are each independently selected from H and C1 to C2 alkyl;
R4 is a piperazin-1-ylsulphonyl group having a substituent, R10 at the 4-position of the piperazinyl group wherein said piperazinyl group is optionally substituted with one or two C1 to C4 alkyl groups and is optionally in the form of its 4-N-oxide; and
R10 is H, C1 to C3 alkyl optionally substituted with one or two substituents selected from hydroxy, NR5R5, CONR5R6 wherein R5 and R6 are each independently selected from H, C1 to C4 alkyl and C3 alkenyl.
Preferred compounds of the present invention include: - 5-[2-Ethoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-[2-methoxyethyl]-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-[2-methoxyethyl]-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(sec-Butyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(iso-Butyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(Cyclopropylmethyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(Cyclobutylmethyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxy-1-methylethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-(methylamino)ethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(2-Dimethylaminoethyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-methylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-dimethylaminoethyl-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-ethylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-{2-[Acetyl(methyl)amino]ethyl}-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-n-propoxypyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(1-Acetylazetidin-3-yl)-5-[2-n-butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-iso-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-(2-methoxyethyl)-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-methylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-ethylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Benzyloxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-ethylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-iso-Butoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-n-propoxypyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-iso-propoxypyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[(S)-2-sec-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[(R)-2-sec-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-{(pyridin-2-yl)methyl}pyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-sec-Butyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-Cyclobutylmethyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(S)-(2-methoxy-1-methylethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(R)-(2-methoxy-1-methylethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[5-(4-Ethylpiperazin-1-ylsulphonyl)-2-(S)-(2-methoxy-1-methylethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[5-(4-Ethylpiperazin-1-ylsulphonyl)-2-(R)-(2-methoxy-1-methylethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-hydroxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(2-Dimethylaminoethyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-iso-Butyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-iso-Butyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-Cyclobutylmethyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-[2-(dimethylamino)-2-oxoethyl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-{2-[methyl(methylsulphonyl)amino]ethyl}-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-Cyclobutylpropylmethyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-n-Butyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(2-Ethoxyethyl)-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(3-methoxypropyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(S)-(2-methoxypropyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(R)-(2-methoxypropyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(S)-sec-Butyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-1-(2-methoxyethyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-(R)-sec-Butyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-Cyclobutyl-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-Cyclopentyl-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-Cyclopentylmethyl-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-Cyclohexyl-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-(2-ethoxyethyl)-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[(1S)-1-methyl-2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[(1R)-1-methyl-2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(3-methoxy-n-propyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 2-Cyclobutyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
- 5-[2-n-Butoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone,
- 3-Ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-[(1S)-1-methylpropyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone,
- 3-Ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-[(1R)-1-methylpropyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone,
- 2-n-Butyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone,
- 2-Cyclopropylmethyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone,
- 2-Cyclobutylmethyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(tetrahydro-2-furanylmethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone,
- 3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2-(2-methoxyethoxy)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone,
- 5-[2-Ethoxy-5-(4-iso-propylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone and
- 5-[2-Ethoxy-5-(4-n-propylpiperazin-1-ylsulphonyl)pyridin-3-yl)-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidinone.
A yet further preferred group of compounds of formulae (I), (IA) or (IB) is that wherein
R1 is —(CH2)n(C3-C6)cycloalkyl wherein n is 0, 1, 2 or 3; or
R1 is methyl, ethyl, iso-propyl or n-propyl substituted by one or more C1 to C4 alkoxy substituents wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or
R1 is a C4 alkyl group selected from i-, n-, sec- or t-butyl optionally substituted by one or more substituents selected from C1 to C4 alkoxy or C3 to C4 cycloalkyl;
R2 is C1 to C4 alkyl;
R13 is OR3 wherein R3 is C1 to C4 alkyl optionally substituted with one or two C1 to C4 alkoxy substituents wherein said C1 to C4 alkyl and C1 to C4 alkoxy groups may optionally be terminated by a haloalkyl group such as CF3;
R4 is a piperazin-1-ylsulphonyl group having a single substituent, R10 at the 4-position of the piperazinyl group and is optionally in the form of its 4-N-oxide; and R10 is methyl, ethyl, n-propyl or i-propyl. - A particularly preferred group of compounds of formulae (I), (IA) or (IB) is that wherein
- R1 is —(CH2)n(C3-C4)cycloalkyl wherein n is 1 or 2; or
R1 is —(CH2)n(C3-C6)cycloalkyl wherein n is 0; or
R1 is -cyclopentyl methyl; or
R1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or
R1 is i-, n-, sec- or t-butyl;
R2 is C2 to C4 alkyl;
R13 is OR3 wherein the R3 alkyl group is methyl, ethyl, n-propyl, i-propyl, i-butyl, n-butyl, sec-butyl or t-butyl optionally substituted with one or two methoxy, ethoxy, n-propoxy or i-propoxy substituents; and R4 is a 4-methyl, 4-ethyl, 4-n-propyl or 4-i-propylpiperazin-1-ylsulphonyl group.
In highly preferred embodiment of the present invention there is provided a compound of the formula (IB) wherein
R1 is —(CH2)n(C3-C4)cycloalkyl wherein n is 1 or 2; or
R1 is —(CH2)n(C3-C5)cycloalkyl wherein n is 0; or
R1 is -cyclopentylmethyl; or
R1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or
R1 is i-, n-, sec- or t-butyl;
R2 is C2 to C4 alkyl; R13 is OR3 wherein the R3 alkyl group is methyl, ethyl, n-propyl, i-propyl, i-butyl, n-butyl, sec-butyl or t-butyl; and R4 is a 4-methyl or 4-ethylpiperazin-1-ylsulphonyl group.
Highly preferred compounds according to the present invention include: 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine and salts and polymorphs thereof. Preferred salts of 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine are sulphonic acid salts, more preferably the p-toluenesulfonate, benzenesulfonate, camphorsulfonate and ethanesulfonate salts respectively, and especially the benzenesulfonate. - According to a further aspect of the present invention there are provided compounds of the general formula (I):
-
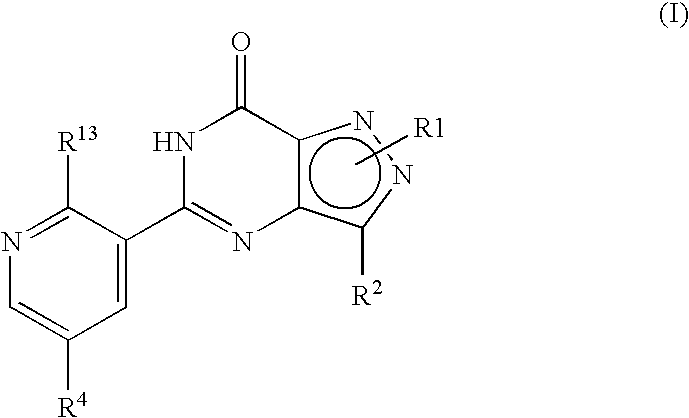
- or a pharmaceutically or veterinarily acceptable salt thereof, or a pharmaceutically or veterinarily acceptable solvate of either entity, wherein
-
- R1 is C1 to C6 alkyl or C3 to C6 alkenyl, C3 to C6 cycloalkyl or C3 to C6 cycloalkenyl wherein said alkyl group may be branched or straight chain and wherein
- when R1 is C1 to C3 alkyl said alkyl group is substituted by; and wherein when R1 is C4 to C6 alkyl, C3 to C6 alkenyl or C3 to C6 cycloalkyl said alkyl, alkenyl or cycloalkyl group is optionally substituted by; one or more substituents selected from: hydroxy; C1 to C4 alkoxy; C3 to C6 cycloalkyl; phenyl substituted with one or more substitutents selected from C1 to C3 alkyl, C1 to C4 alkoxy, C1 to C4 haloalkyl or C1 to C4 haloalkoxy wherein said haloalkyl and haloalkoxy groups contain one or more halo atoms, halo, CN, NO2, NHR11, NHSO2R12, SO2R12, SO2NHR11, COR11, CO2R11 wherein R11 is H, C1 to C4 alkyl, C2 to C4 alkenyl, C1 to C4 alkanoyl, C1, to C4 haloalkyl or C1 to C4 haloalkoxy and wherein R12 is C1 to C4 alkyl, C2 to C4 alkenyl, C1 to C4 alkanoyl, C1 to C4 haloalkyl or C1 to C4 haloalkoxy; NR7R8, CONR7R8 or NR7COR11 wherein R7 and R8 are each independently selected from H, C1 to C4 alkyl, C2 to C4 alkenyl, C1 to C4 alkoxy, CO2R9, SO2R9 wherein said alkyl, alkenyl or alkoxy groups are optionally substituted by C1 to C4 haloalkyl or C1 to C4 haloalkoxy and wherein R9 is C1 to C4 alkyl which is optionally substituted with phenyl wherein said phenyl group is optionally substituted by one or more substituents selected from C1 to C4 alkyl optionally substituted by C1 to C4 haloalkyl or C1 to C4 haloalkoxy, C1 to C4 alkoxy, halo, CN, NO2, NHR11, NHSO2R12, SO2R12, SO2NHR11, COR11 or CO2R11; Het1; Het2 or Het3; or R1 is Het4 or phenyl wherein said phenyl group is optionally substituted by one or more substituents selected from C1 to C4 alkyl, C2 to C4 alkenyl, C1 to C4 alkoxy, halo, CN, CF3, OCF3, NO2, NHR11, NHSO2R12, SO2R12, SO2NHR11, COR11, CO2R11;
- R2 is C1 to C6 alkyl, C3 to C6 alkenyl or (CH2)n(C3 to C6 cycloalkyl) wherein n is 0, 1 or 2;
- R13 is OR3 or NR5R6;
- R3 is C1 to C6 alkyl optionally substituted with one or two substituents selected from C3 to C5 cycloalkyl, hydroxy, C1 to C4 alkoxy, benzyloxy, NR5R6, phenyl, Het1, Het2, Het3 or Het4 wherein the C1 to C6 alkyl and C1 to C4 alkoxy groups may optionally be terminated by a haloalkyl group such as CF3; C3 to C6 cycloalkyl; Het1, Het2, Het3 or Het4;
- R4 is a piperazin-1-ylsulphonyl group having a substituent, R10 at the 4-position of the piperazinyl group wherein said piperazinyl group is optionally substituted with one or two C1 to C4 alkyl groups and is optionally in the form of its 4-N-oxide;
- R5 and R6 are each independently selected from H and C1 to C4 alkyl optionally substituted with C3 to C5 cycloalkyl or C1 to C4 alkoxy, or, together with the nitrogen atom to which they are attached, form an azetidinyl, pyrrolidinyl, piperidinyl or morpholinyl group;
- R10 is H; C1 to C4 alkyl optionally substituted with one or two substituents selected from hydroxy, NR5R6, CONR5R6, phenyl optionally substituted with C1 to C4 alkyl or C1 to C4 alkoxy; C2 to C6 alkenyl or Het4;
- Het1 is an N-linked 4-, 5- or 6-membered nitrogen-containing heterocyclic group optionally containing one or more further heteroatoms selected from S, N or O;
- Het2 is a C-linked 5-membered heterocyclic group containing an O, S or N heteroatom optionally containing one or more heteroatoms selected from O or S;
- Het3 is a C-linked 6-membered heterocyclic group containing an O or S heteroatom optionally containing one or more heteroatoms selected from O, S or N or Het3 is a C-linked 6-membered heterocyclic group containing three N heteroatoms;
- Het4 is a C-linked 4-, 5- or 6-membered heterocyclic group containing one, two or three heteroatoms selected from S, O or N; and
- wherein any of said heterocyclic groups Het1, Het2, Het3 or Het4 may be saturated, partially unsaturated or aromatic and wherein any of said heterocyclic groups may be optionally substituted with one or more substituents selected from C1 to C4 alkyl, C2 to C4 alkenyl, C1 to C4 alkoxy, halo, CO2R11, COR11, SO2R12 or NHR11 and/or wherein any of said heterocyclic groups is benzo-fused;
with the provisos that (a) when R1 is C1 to C3 then Het1 is not morpholinyl or piperidinyl and (b) when R1 is C1 to C3 substituted by phenyl then said phenyl group is not substituted by C1 to C4 alkoxy, halo, CN, CF3, OCF3 or C1 to C4 alkyl.
- In a further aspect, the present invention provides processes for the preparation of compounds of formulae (I), (IA) and (IB), their pharmaceutically and veterinarily acceptable salts, and pharmaceutically and veterinarily acceptable solvates of either entity, as illustrated below. It will be appreciated by persons skilled in the art that, within certain of the processes described, the order of the synthetic steps employed may be varied and will depend inter alia on factors such as the nature of other functional groups present in a particular substrate, the availability of key intermediates and the protecting group strategy (if any) to be adopted. Clearly, such factors will also influence the choice of reagent for use in the said synthetic steps. Illustrative of a protecting group strategy is the route to the azetidine analogues (Examples 18, 19 and 20), the precursor to which (preparations 63, 66 and 61 respectively) contain t-butoxycarbonyl (Boc) as the nitrogen protecting group.
- It will also be appreciated that various standard substituent or functional group interconversions and transformations within certain compounds of formulae (I), (IA) or (IB) will provide other compounds of formulae (I), (IA) or (IB). Examples include alkoxide exchange at the 2-position of the 5-(pyridin-3-yl) substituent (see conversions of Example 3 to Examples 27, Example 8 to Example 28 and 29, Example 21 to Example 32 and 33, Example 4 to Examples 41, Example 9 to Example 43, and Example 66 to Example 75), amine exchange at the 2-position of the 5-(pyridin-3-yl) substituent (see conversions of Example 7 to Examples 78), reactions at a nitrogen containing substituent, such as reductive alkylation (Example 18 to Example 21), acetamide formation (Examples 18, and 20 to Examples 22 and 24 respectively) or sulphonamide formation (Preparations 68, 67 to Examples 25 to 62 respectively), and reduction of a nitro functionality to provide an amino group (Example 63 to Example 64). The deprotection and transformations described herein and as illustrated in the Examples and Preparations sections may be effected in a “one-pot” procedure (see for example the conversion of the compound of preparation 65 into the compound of example 26).
-
FIG. 1 is a reaction scheme depicting a general route for the synthesis of compounds (I) via compounds (IXB). -
FIG. 2 is a reaction scheme depicting a route for the preparation of compounds of general formula (XB). -
FIG. 3 is a reaction scheme depicting a preferred embodiment ofFIG. 2 in which X is an —OR3 alkoxy group. - The following processes are illustrative of the general synthetic procedures which may be adopted in order to obtain the compounds of the invention.
- 1. A compound of formula (I):
-
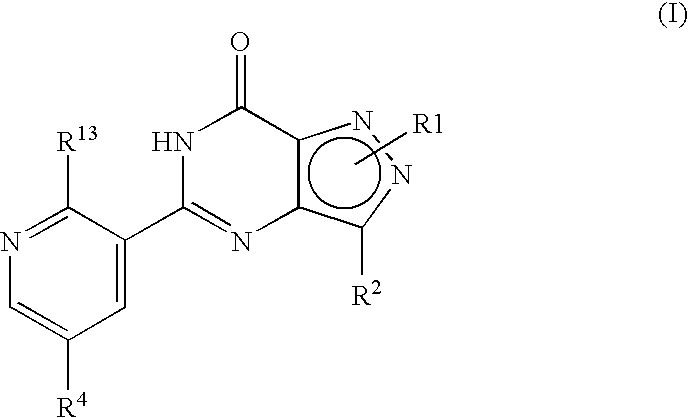
- wherein formula (I) may equally be represented by general formulae (IA) and (IB) and wherein R1, R2, R4 and R13 are as previously defined herein may be prepared from a compound of general formula (IX):
-

- wherein RP is R13 (i.e. OR3 or NR5R6) or X wherein R13, R3, R5 and R6 are as defined hereinbefore and X is a leaving group and wherein general formula (IX) can be represented by formulae (IXA), (IXB) or (IXC) respectively:
-

- wherein R1, R2, R3, R4, R5 and R6 are as previously defined herein and wherein X is a leaving group and may be any group which is displaceable by an amino group of the formula —NR5R6 or by an alkoxy group and wherein the intermediate compounds of general formulae (IXA) and (IXB) can be represented by their regioisomeric general formulae as previously illustrated for compounds having the general formulae (I). Suitable leaving groups, X, for use herein include halogen, alkoxy, amino, tosylate groups and further groups are detailed hereinafter.
1.1 A compound of formula (I) wherein R13═NR5R6 may be prepared by cyclisation of a compound of general formula (IXA): -

- wherein R1, R2, R4, R5 and R6 are as previously defined herein for compounds of the formula (I), (IA) or (IB). Preferably, the cyclisation is base-mediated, using an alkali metal salt of a sterically hindered alcohol or amine. For example, the required cyclisation may be effected using about a 1- to 5-, preferably a 1.2- to 3.5-fold excess of potassium t-butoxide, potassium bis(trimethylsilyl)amide or cesium carbonate, optionally in the presence of molecular sieves, in a suitable solvent, such as for example an inert solvent e.g. DMF or NHR5R6 or mixtures thereof, at the reflux temperature of the reaction mixture optionally in the presence of about a 1 molar equivalent of ethyl acetate or ethyl pivalate, or, the reaction can optionally be carried out in a sealed vessel at about 100-130° C. optionally in the presence of about a 1 molar equivalent of ethyl acetate or ethyl pivalate.
1.2 A general route for the synthesis of compounds (I) via compounds (IXB) is illustrated inScheme 1 wherein said intermediate compounds (IXB) have the general formula: -
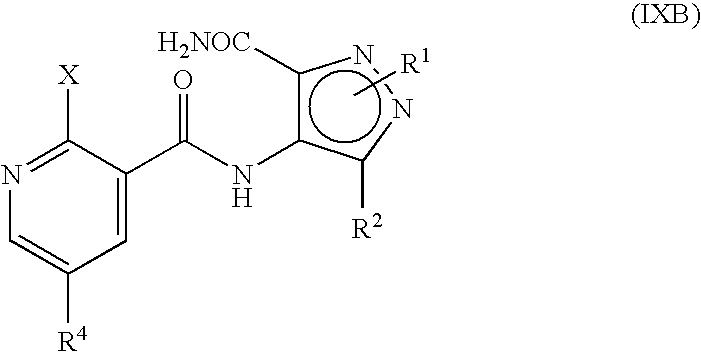
- wherein R1, R2 and R4 are as previously defined herein for compounds of the formula (I), (IA) and (IB) and wherein X is a leaving group as defined hereinbefore, by reaction in the presence of —OR3 and a hydroxide trapping agent. The conversion (IXB) to (I) can be undertaken in either a stepwise process or a one-pot process. A number of stepwise permutations are feasible, some of which are subsets of others. These include
- i) cyclisation (IXB to XXX) followed by displacement (XXX to I);
- ii) cyclisation (IXCa to XXX) followed by displacement (XXX to I);
- iii) displacement (IXB to IXC) followed by cyclisation (IXC to I); and
- iv) displacement (IXCa to IXC) followed by cyclisation (IXC to I) wherein compounds (XXX) and (IXCa) have the general formulae:
-
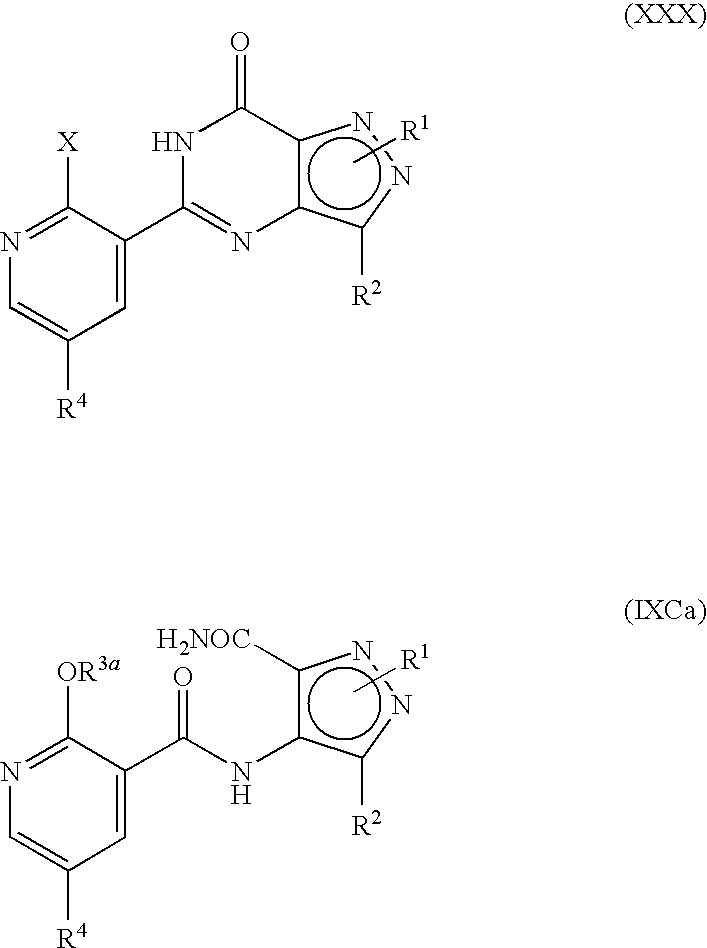
- wherein R1, R2, R4 and X are as defined herein before and OR3a is an alkoxy group which is different from and displaceable by the desired OR3 group on the final compounds of general formula (I) and wherein R3a is selected from C1 to C6 alkyl optionally substituted with one or two substituents selected from C3 to C5 cycloalkyl, hydroxy, C1 to C4 alkoxy, benzyloxy, NR5R6, phenyl, Het1, Het2, Het3 or Het4 wherein the C1 to C6 alkyl and C1 to C4 alkoxy groups may optionally be terminated by a haloalkyl group such as CF3 and wherein the C3-C5 cycloalkyl group may optionally be substituted by C1-C4 alkyl, hydroxy or halo; C3 to C6 cycloalkyl; Het1, Het2, Het3 or Het4. Preferably R3, is C1 to C6 alkyl.
To effect initial displacement without significant simultaneous cyclisation it is preferred that the displacement with −OR3 (in (iii) or (iv)) is carried out in the range of from about 80° C. to about 90° C. to provide a compound of the general formula (IXC). Subsequent cyclisation to a compound of general formula (I) is generally carried out at a temperature greater than about 115° C.
To effect initial cyclisation without significant simultaneous displacement it is preferred that, for (IXCa) to (XXX) (in (ii)), the reaction is conducted at a temperature greater than about 110° C. with —OR3a in R3aOH. Subsequent displacement to a compound of general formula (I) is generally carried out with −OR3 in R3OH in the range of from about 80° C. to about 90° C.
For conversion of (IXB) to (I) (ie. (i) above), it may be preferred to obtain compounds of general formula (I) directly from compounds of general formula (IXB) since both the cyclisation and displacement components of this reaction can be carried out in a “one-pot” reaction. Such a “one-pot” process can be run at lower pressures (ie. nearer ambient pressure) than say a stepwise cyclisation and displacement process (ie. (ii) above) if the boiling point of R3OH is higher than that of R3aOH and where the ambient boiling point of R3aOH is less than about 115° C. (ie. too low to effect cyclisation at ambient pressure). It should be noted that is may still be necessary to operate such processes at higher temperatures than the boiling point of HOR3, i.e. at higher pressure.
In the case of compounds of general formula (IXC) as detailed hereinafter wherein X is OR3, compounds of general formula (I) can be obtained by direct cyclisation by reacting in the presence of an auxiliary base, a hydroxide trapping agent and an appropriate solvent R3OH or an inert solvent or a combination thereof.
The temperature of the reaction of compounds of the general formula (IXB) to compounds of the general formula (I) (such as the corresponding formation of compounds (IA) and (IB)) is preferably at least about 80° C., more preferably about 80 to about 130° C., more preferably still about 100 to about 130° C. and most preferably about 115 to about 125° C. These temperatures are also applicable for the conversion of compounds (XXX) to (I), although the temperature in this case could also probably be lower (e.g. about 60° C.) since there is no cyclisation taking place.
Preferably compounds of formula (I), or (IA), or (IB) wherein
R1 is —(CH2)n(C3-C4)cycloalkyl wherein n is 1 or 2; or
R1 is —(CH2)n(C3-C6)cycloalkyl wherein n is 0; or
R1 is -cyclopentylmethyl; or
R1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or
R1 is i-, n-, sec- or t-butyl;
R2 is C2 to C4 alkyl; R13 is OR3 wherein the R3 alkyl group is methyl, ethyl, n-propyl, i-propyl, i-butyl, n-butyl, sec-butyl or t-butyl optionally substituted with one or two methoxy, ethoxy, n-propoxy or i-propoxy substituents; and R4 is a 4-methyl, 4-ethyl, 4-n-propyl or 4-i-propylpiperazin-1-ylsulphonyl group are prepared from compounds of general formula (IXB) wherein X is OR3 (i.e. compounds of general formula (IXC) as detailed hereinbefore and after).
Thus, according to a further aspect of the present invention there is provided a further process for the preparation of a compound of general formula (I): -
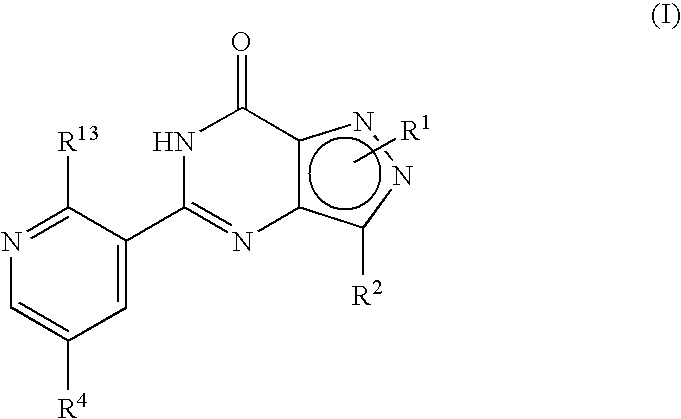
- or a compound of general formula (IA), or (IB) wherein
R1 is —(CH2)n(C3-C4)cycloalkyl wherein n is 1 or 2; or
R1 is —(CH2)n(C3-C6)cycloalkyl wherein n is 0; or
R1 is -cyclopentylmethyl; or
R1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups other than the C-atom directly linked to the pyrazole ring; or
R1 is i-, n-, sec- or t-butyl;
R2 is C2 to C4 alkyl; R13 is OR3 wherein the R3 alkyl group is methyl, ethyl, n-propyl, i-propyl, i-butyl, n-butyl, sec-butyl or t-butyl optionally substituted with one or two methoxy, ethoxy, n-propoxy or i-propoxy substituents; and R4 is a 4-methyl, 4-ethyl, 4-n-propyl or 4-i-propylpiperazin-1-ylsulphonyl group comprising reacting a compounds of general formula (IXC): -

- wherein R1, R2, R3 and R4 are as defined previously herein, wherein said reaction is carried out in the presence of −OR3 and a hydroxide trapping agent, or alternatively reacting in the presence of hydroxide trapping agent and an auxiliary base.
Intermediates of the general formula (IXC) and more specifically (IXCA) and (IXCB) form further aspects of the invention. -

- A particular advantage of the use of the hydroxide trapping agent is that a higher yield of final product (compounds of general formula (I), (IA) or (IB)) can be obtained than for the same reaction where the trapping agent is not present.
Preferably the hydroxide trapping agent is an ester. More preferably said hydroxide trapping agent is an ester of the formula: -
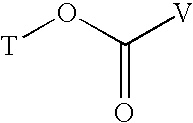
- wherein OT is OR3 or the residue of a bulky alcohol or a non-nucleophilic alcohol or TOH is an alcohol which can be azeotropically removed during the reaction; and C(O)V is the residue of a carboxylic acid. For example, where OR3 is OEt in compound (IXC) the hydroxide trapping agent (TOC(O)V) could be e.g. ethyl acetate or ethyl pivalate. Preferably V is a C1 to C4 alkyl group.
Preferably X is selected from the group consisting of —OR3, halo, optionally substituted arylsulphonyloxy, preferably phenylsulphonyloxy, more preferably a para-substituted aryl (phenyl) such as by a C1-C4 alkyl group e.g. p-toluenesulphonyloxy; C1-C4 alkylsulphonyloxy e.g. methanesulphonyloxy; nitro or halo substituted benzenesulphonyloxy preferably para-substituted e.g. p-bromobenzenesulfonyloxy or p-nitrobenzenesulphonyloxy; C1-C4 perfluoroalkylsulphonyloxy e.g. trifluoromethylsulphonyloxy; optionally substituted aroyloxy such as benzoyloxy; C1-C4 perfluoroalkanoyloxy such as trifluoroacetyloxy; C1-C4 alkanoyloxy such as acetyloxy; diazonium; quatenaryammonium C1-C4 alkylsulphonyloxy; halosulphonyloxy e.g. fluorosulphonyloxy and other fluorinated leaving groups; and diarylsulphonylamino e.g. ditosyl (NTs2).
More preferably, X is a C1-C6 primary or secondary alkoxy and is especially a C1-C4 alkoxy group such as ethoxy or methoxy.
−OR3 can act both as a nucleophile (to displace the leaving group by nucleophilic substitution) and as a base (to bring about the cyclisation).
−OR3 can be generated in solution from, for example, a salt ZOR3 (wherein Z is a cation) such as a metal salt. More particularly an alkali (such as sodium or potassium) or alkaline earth metal salt of −OR3 in a suitable solvent would give rise to −OR3 in solution. In another embodiment, −OR3 is formed in situ from R3OH plus an auxiliary base (i.e. a base other than −OR3). However, in another system, ZOR3 could be used in the reaction system with an auxiliary base.
As will be appreciated the solvent in which the reaction takes place can be R3OH or an inert solvent (or a mixture of both). By inert solvent we mean a solvent which will not form a nucleophile under the reaction conditions or if a nucleophile is formed it is sufficiently hindered or unreactive such that it does not substantially compete in the displacement reaction. When R3OH is used as a source of −OR3, then a separate solvent is not essentially required but an (auxiliary) inert solvent (i.e. a solvent other than R3OH) may be used as a co-solvent in the reaction.
Suitable solvents are as follows: R3OH, a secondary or tertiary C4-C12 alkanol, a C3-C12 cycloalkanol, a tertiary C4-C12 cycloalkanol, a secondary or tertiary (C3-C7 cycloalkyl)C2-C6 alkanol, a C3-C8 alkanone, 1,2-dimethoxyethane, 1,2-diethoxyethane, diglyme, tetrahydrofuran, 1,4-dioxan, toluene, xylene, chlorobenzene, 1,2-dichlorobenzene, acetonitrile, dimethyl sulphoxide, sulpholane, dimethylformamide, N-methylpyrrolidin-2-one, pyridine, and mixtures thereof.
More preferably, the solvent is R3OH, a tertiary C4-C12 alkanol, a tertiary C4-C12 cycloalkanol, a tertiary (C3-C7 cycloalkyl)C2-C6 alkanol, a C3-C8 alkanone, 1,2-dimethoxyethane, 1,2-diethoxyethane, diglyme, tetrahydrofuran, 1,4-dioxan, toluene, xylene, chlorobenzene, 1,2-dichlorobenzene, acetonitrile, dimethyl sulphoxide, sulpholane, dimethylformamide, N-methylpyrrolidin-2-one, pyridine, and mixtures thereof.
Most preferably the solvent is R3OH, which means that −OR3 is formed in situ, such as in the presence of an auxiliary base.
A wide range of auxiliary bases can be used in the process of the invention. Typically the bases would not substantially compete with −OR3 in the nucleophilic substitution of X (i.e. they would be non nucleophilic) such as by suitably being sterically hindered.
Preferably the auxiliary base is selected from the group consisting of a sterically hindered base, a metal hydride, metal oxide, metal carbonate and metal bicarbonate.
The sterically hindered base is advantageously a metal salt of a sterically hindered alcohol or amine.
More preferably the auxiliary bases in accordance with the invention are selected from the group consisting of metal salts of a sterically hindered alcohol or amine such as a secondary or tertiary C4-C12 alkanol, a C3-C12 cycloalkanol and a secondary or tertiary (C3-C8 cycloalkyl)C1-C6 alkanol, a N-(secondary or tertiary C3-C6 alkyl)-N-(primary, secondary or tertiary C3-C6 alkyl)amine, a N—(C3-C8 cycloalkyl)-N-(primary, secondary or tertiary C3-C6 alkyl)amine, a di(C3-C8 cycloalkyl)amine or hexamethyldisilazane; 1,5-diazabicyclo[4,3,0]non-5-ene and 1,8-diazabicyclo[5,4,0]undec-7-ene; a metal hydride, oxide, carbonate, and bicarbonate.
Yet more preferably the auxiliary bases in accordance with the invention are selected from the group consisting of metal salts of a sterically hindered alcohol or amine such as a tertiary C4-C12 alkanol, a C3-C12 cycloalkanol and a tertiary (C3-C8 cycloalkyl)C1-C6 alkanol, a N-(secondary or tertiary C3-C6 alkyl)-N-(primary, secondary or tertiary C3-C6 alkyl)amine, a N—(C3-C8 cycloalkyl)-N-(primary, secondary or tertiary C3-C6 alkyl)amine, a di(C3-C8 cycloalkyl)amine or hexamethyldisilazane; 1,5-diazabicyclo[4,3,0]non-5-ene and 1,8-diazabicyclo[5,4,0]undec-7-ene; a metal hydride, oxide, carbonate, and bicarbonate.
More preferably still, the auxiliary base is selected from the sterically hindered bases of the previous paragraph (i.e. all of them except the metal hydride, oxide, carbonate and bicarbonate).
Most preferably still, the auxiliary base is the metal salt of a tertiary C4-C6 alcohol such as the alkali or alkaline earth metal salts (e.g. Na/K) of t-butanol or t-amyl alcohol, or the base is KHMDS.
Most preferably, the auxiliary base is the alkali metal salt of t-butanol (e.g. potassium t-butoxide).
The metal of the salt of ZOR3 and the auxiliary base can be independently selected from alkali metals (lithium, sodium, potassium, rubidium, cesium) or alkaline earth metals (beryllium, magnesium, calcium, strontium, barium). Preferably the metal is sodium, potassium, lithium or magnesium. More preferably the metal is sodium or potassium.
To maximise yields, it is further preferred that when X is any group hereinbefore defined except —OR3, then at least about 1 molecular equivalent of auxiliary base and −OR3 are used. If −OR3 also functions as a base (i.e. there is no auxiliary base present) then preferably at least about 2 equivalents of −OR3 are present. Suitably, at least about 1 equivalent of trapping agent (preferably at least about 2 equivalents) is present. In the case where X═OR3 (i.e. starting from (IXC) rather than (IXB) then, in theory, at least 1 equivalent of base is required, wherein said base may be —OR3 or auxiliary base.
The temperature of the reaction of compounds of the general formula (IXC) to compounds of the general formula (I) (such as the corresponding formation of compounds (IA) and (IB)) is preferably at least about 80° C., more preferably about 80 to about 130° C., more preferably still about 100 to about 130° C. and most preferably about 115 to about 125° C.
The reaction temperature attainable to effect the conversion of compounds of the general formulae (IXB), (IXC) or (XXX) to compounds of the general formula (I) depends on the solvent, the nature of −OR3 and X. When X is OR3a (wherein OR3a and OR3 are not the same), i.e. a compound of the formula (IXCa) and R3OH is the solvent, preferably XH (such as C1-C6 alkohol) is removed azeotropically (of course the reaction vessel must be configured to distill over the azeotrope mixture) with R3OH by running the reaction at the azeotrope temperature of XH and R3OH. In this way the yield and quality of the final product can be further improved. For example, (where X is an alkoxy, preferably ethanol) the conversion of compound (XXX), (IXB) or (IXC) to (I) is preferably carried out at the azeotrope temperature of the alcohol (i.e. XH (preferably ethanol)) with R3OH. When X═OR3 and the solvent is R3OH there is no requirement to azeotrope out R3OH.
Thus in a preferred embodiment of the present invention there is provided a process for the synthesis of compounds of general formula (I), (IA) or (IB) and in particular compounds of general formula (I), (IA) or (IB) wherein R1 is —(CH2)n(C3-C4)cycloalkyl wherein n is 1 or 2; or R1 is —(CH2)n(C3-C6)cycloalkyl wherein n is 0; or R1 is —(CH2)n(C5)cycloalkyl wherein n is 1; or
R1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups; or R1 is i-, n-, sec- or t-butyl; R2 is C2 to C4 alkyl; R13 is OR3 wherein the R3 alkyl group is methyl, ethyl, n-propyl, i-propyl, i-butyl, n-butyl, sec-butyl or t-butyl optionally substituted with one or two methoxy, ethoxy, n-propoxy or i-propoxy substituents; and R4 is a 4-methyl, 4-ethyl, 4-n-propyl or 4-i-propylpiperazin-1-ylsulphonyl group wherein said process comprises reacting a compound of general formula (XIB), (XIC) or (XID) respectively:
a) with R3OH and auxiliary base, optionally in an inert solvent and in the presence of said trapping agent; or
b) with ZOR3 and an auxiliary base in R3OH or an inert solvent or both, in the presence of said trapping agent; or
c) with ZOR3 and R3OH or an inert solvent or both, in the presence of said trapping agent; or
d) with auxiliary base, inert solvent or R3OH or a combination thereof and a hydroxide trapping agent for compounds of the general formula (IXC).
1.3 For compounds of the general formula (IXB) wherein X is OR3 and an alcohol is selected as solvent a compound of formula (I) may be prepared by cyclisation of a compound of general formula (IXC): -

- wherein R1, R2, R3 and R4 are as previously defined herein for compounds of the formula (I), (IA) and (IB). In said reaction the appropriate alcohol of formula R3OH should be employed as the solvent in order to obviate potential problems associated with alkoxide exchange at the 2-position of the pyridine ring or an inert solvent or a mixture of the two. The appropriate alcohol as defined herein means that the solvent alcohol should be of the same alkyl chain length as the alkoxy (—OR3) substituent, for example, where —OR3 is ethoxy, ethanol is the appropriate alcohol. Preferably, said cyclisation is base-mediated, using an alkali metal salt of a sterically hindered alcohol or amine. For example, the required cyclisation may be effected using about a 1- to 8, preferably about a 1- to 5-, more preferably a 1.2- to 3.5-fold excess of potassium t-butoxide or potassium bis(trimethylsilyl)amide, optionally under suitable drying conditions i.e. in the presence of molecular sieves or under azeotroping conditions, in a suitable solvent as described above at the reflux temperature of the reaction mixture optionally in the presence of about 1 to 2 molar equivalents of a hydroxide trapping agent such as ethyl acetate or ethyl pivalate, or, the reaction can optionally be carried out in a sealed vessel at about 100-130° C. optionally in the presence of about 1 to 2 molar equivalents of a hydroxide trapping agent such as ethyl acetate or ethyl pivalate.
- Alternative reaction conditions for the cyclisation reactions of compounds of (IXC) wherein X is OR3 are to conduct the reaction with about 1.2 to 4.5 molecular equivalents of sterically hindered base such as potassium t-butoxide or KHMDS, optionally in a sealed vessel at from about 100° C. to about 150° C. with, rather than an alcohol of formula R3OH as solvent, a sterically hindered alcohol, e.g. 3-methylpentan-3-ol, as solvent optionally in the presence of about 1 or 2 molar equivalents of ethyl acetate or ethyl pivalate.
- A compound of formula (IXA) or a compound of general (IXB) wherein X is OR3 (i.e. a compound of general formula (IXC)) may be prepared by a coupling reaction between a compound of formula (VII):
-

- wherein R1 and R2 are as previously defined for formulae (IXA), (IXB) or (IXC) with a compound of formula (XA), (XB) or (XC) respectively:
-

- wherein R3, R4, R5, R6 and X are also as previously defined for formulae (IXA), (IXB) or (IXC). Where either R5 and/or R6 in the —NR5R6 group of formula (XA) are H, then a suitable N-protecting group strategy may be advantageously employed. Any known suitable protecting group strategy may be used.
The coupling reaction may be carried out using conventional amide bond-forming techniques, e.g. via the acyl chloride derivative of (XA) or (XB) in the presence of up to about a five-fold excess of a tertiary amine such as triethylamine or pyridine to act as scavenger for the acid by-product (HY), optionally in the presence of a catalyst such as 4-dimethylaminopyridine, in a suitable solvent such as dichloromethane, at from about 0° C. to about room temperature. For convenience pyridine may also be used as the solvent.
In particular, any one of a host of amino acid coupling variations may be used. For example, the acid of formula (XA), (XB) or (XC) or a suitable salt (e.g. sodium salt) thereof may be activated using a carbodiimide such as 1,3-dicyclohexylcarbodiimide or 1-ethyl-3-(3-dimethylaminoprop-1-yl)carbodiimide optionally in the presence of 1-hydroxybenzotriazole hydrate and/or a catalyst such as 4-dimethylaminopyridine, or by using a halotrisaminophosphonium salt such as bromotris(pyrrolidino)phosphonium hexafluorophosphate or by using a suitable pyridinium salt such as 2-chloro-1-methylpyridinium iodide. Either type of coupling is conducted in a suitable solvent such as dichloromethane, tetrahydrofuran or N,N-dimethylformamide, optionally in the presence of a tertiary amine such as triethylamine or N-ethyldiisopropylamine (for example when either the compound of formula (VII), or the activating reagent, is presented in the form of an acid addition salt), at from about 0° C. to about room temperature. Preferably, from 1 to 2 molecular equivalents of the activating reagent and from 1 to 3 molecular equivalents of any tertiary amine present are employed. - In a further variation, the carboxylic acid function of (XA), (XB) or (XC) may first of all be activated using up to about a 5% excess of a reagent such as N,N-carbonyldiimidazole in a suitable solvent, e.g. ethyl acetate or butan-2-one, at from about room temperature to about 80° C., followed by reaction of the intermediate imidazolide with (VII) at from about 20° C. to about 90° C.
- It will be appreciated that the general formula (VII) can also be represented by the regioisomeric formulae (VIIA) and (VIIB):
-
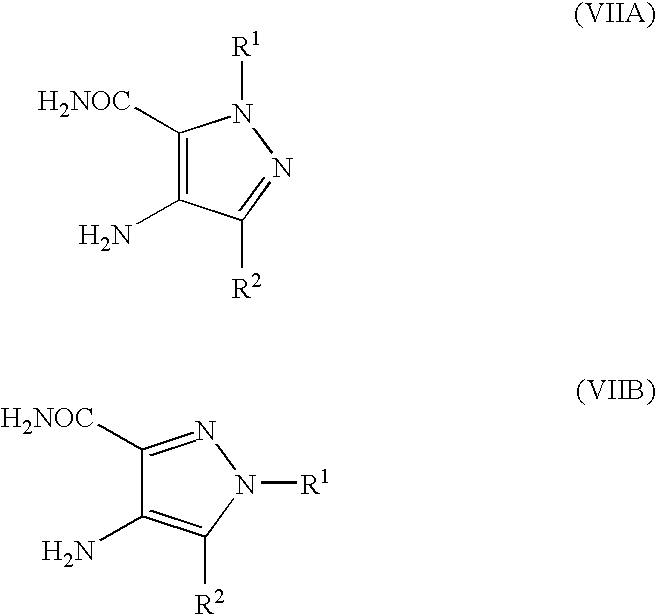
- wherein R1 and R2 are as previously defined herein.
The 4-aminopyrazole-5-carboxamide compounds having the general formulae (VII), (VIIA) or (VIIB) may be prepared from pyrazole compounds of the general formula (XIII): -

- wherein Rq is selected from OH, C1-C6 alkoxy or NR5R6 wherein R5 and R6 are as hereinbefore defined, according to the procedures detailed in the preparations section herein and as particularly described in Preparations 96(a) to (h).
Compounds having the general formulae (XA) or (XC) may be prepared from the carboxylic acid compounds of the general formulae (VIIIA), (VIIIB) or (VIIIC) respectively: -

- wherein R3, R5 and R6 are as defined for compounds of the general formulae (I), (IA) and (IB) by reaction with a 4-R10-piperizinyl compound, such as for example 4-methylpiperizine. Such reaction can be conducted at from about 0° C. to about room temperature, preferably in the presence of an appropriate solvent such as a C1 to C3 alkanol or dichloromethane optionally in the presence of a suitable base such as triethylamine to scavenge the acid by-product (HY). Where either R5 or R6 is H a suitable amino protecting group strategy may be employed as detailed hereinbefore.
Compounds of the general formulae (VIIIA), (VIIIB) or (VIIIC) may be prepared from compounds of the general formulae (XIA), (XIB) or (XIC) respectively: -

- wherein R3, R5, R6 and X are as defined for compounds of the general formulae (I), (IA) and (IB) by the application of known methods for converting amino to an SO2Y group, wherein Y is halo, preferably chloro. For example, when Y is chloro, by the action of about a two-fold excess of sodium nitrite in a mixture of concentrated hydrochloric acid and glacial acetic acid at from about −25° C. to about 0° C., followed by treatment with excess liquid sulphur dioxide and a solution of about a three-fold excess of cupric chloride in aqueous acetic acid at from about −15° C. to about room temperature. When R13 contains a primary or secondary amino group, protection of the said amino group with an acid stable group such as acetyl or benzyl will generally be advantageous.
Compounds of the general formula (XIA), (XIB) and (XIC) may be prepared by reduction of compounds of the general formulae (XIIA), (XIIB) and (XIIC) respectively: -

- wherein R3, R5, R6 and X are as previously defined. Such conversion of compounds of the general formulae (XIIA), (XIIB) and (XIIC) to compounds of the general formulae (XIA), (XIB) and (XIC) can be achieved by conventional catalytic or catalytic transfer hydrogenation procedures. Typically, the hydrogenation is achieved using a Raney® nickel catalyst or a palladium catalyst such as 10% Pd on charcoal, in a suitable solvent such as ethanol at a hydrogen pressure of from about 345 kPa (50 psi) to about 414 kPa (60 psi) at from about room temperature to about 60° C., preferably from about 40° C. to about 50° C.
- Intermediates of the general formula (IXC) as described in 1.2 and 1.3 hereinbefore can be prepared via a coupling reaction between a compound of the general formula (XB) and a compound of the general formula (VII) wherein said coupling may be achieved by any of the methods described hereinbefore. Compounds of general formula (XB) may be prepared according to the route outlined in
Scheme 2. - With reference to
Scheme 2, the intermediate of formula (XB) is formed from a compound of formula (XIV), the exact process being dependent on leaving group X. - For compounds of formula (XB) wherein X=arylsulfonyloxy, C1-C4 alkylsulfonyloxy, C1-C4 perfluoroalkylsulfonyloxy, aryloxy, C1-C4 perfluoroalkanoyloxy, C1-C4 alkanoyloxy, quarternaryammonium C1-C4 alkylsulfonyloxy or halosulfonyloxy, compound (XB) can be formed from compounds (XIV) (wherein Q═OH and W═OH) and an appropriate derivatising agent, more particularly an appropriate sulphonylating agent such as arylsulfonylhalide, C1-C4 alkylsulfonylhalide, C1-C4 perfluoroalkylsulfonylhalide, arylhalide, C1-C4 perfluoroalkanoylhalide, C1-C4 alkanoylhalide, quarternary ammonium C1-C4 alkylsulfonylhalide or halosulfonylhalide, or an appropriate arylating agent such as arylhalide, or an appropriate acylating agent such as C1-C4 perfluoroalkanoylhalide, or C1-C4 alkanoylhalide), respectively (preferably the halide substituent of the above is chloride), in an appropriate solvent. Compounds of formula (XIV) (wherein Q=OH and W=OH) can be formed from compounds (XV) (wherein P is hydrolisable group) via use of a hydrolising agent, preferably a hydroxide base (ideally 2 molar equivalents), more preferably a metal hydroxide such as sodium hydroxide, in an appropriate solvent, such as water. The metal of the hydroxide base can be as defined hereinbefore for Z (in ZOR). This will also apply for other reactions of
scheme - Compounds of formula (XB) where X=chloro, can be formed from (XIV) wherein Q=Cl and W=P (such as OEt) (i.e. formula XV) and a hydroxide base (ideally 1 molar equivalent), such as sodium hydroxide preferably in an appropriate solvent, such as water and a deprotecting agent.
- Preferably the deprotecting agent as used herein in accordance with the invention is a hydrolysing agent, more preferably a hydroxide nucleophile, advantageously a hydroxide base (ideally 1 molar equivalent), such as sodium hydroxide preferably in an appropriate solvent, such as water.
Compounds of formula (XB) wherein X=diazonium, can be formed from (XIV) (wherein Q=NH2, W=OH) and nitrous acid. Compounds of formula (XIV) (wherein Q=NH2, W=OH) can be formed from compounds of formula (XIV) (wherein Q=NH2, W=P, e.g. OEt) and a deprotecting agent such as a hydroxide base e.g. sodium hydroxide, in an appropriate solvent, such as water. Intermediate (XIV) (Q=NH2, W=P, e.g. OEt) is formed from (XV) and an ammoniating agent, such as ammonia, in an appropriate solvent, such as water.
Compounds of formula (XB) wherein X=diarylsulfonylamino, can be formed from (XIV) (wherein Q=NH2, W=OH) and an appropriate derivatising agent, preferably an appropriate sulphonylating agent such as arylsulphonylhalide, preferably arysulfonylchloride (ideally at least 2 molar equivalents) and preferably in the presence of a base (ideally 2 molar equivalents thereof), such as triethylamine in an appropriate solvent.
Compounds of formula (XB) wherein X═C1-C6 (preferably C1-C4) preferably primary or secondary alkoxy, can be formed from (XIV) (wherein Q=C1-C6 (preferably C1-C4) primary or secondary alkoxy and W=P, such as OEt) and a deprotecting agent (for P=OEt), preferably a hydroxide base, such as sodium hydroxide, in an appropriate solvent, such as water. Compounds of formula (XIV) (wherein Q=C1-C6 (preferably C1-C4) primary or secondary alkoxy, W=P e.g. OEt) can be formed from (XV) and an appropriate alkoxide, OR− wherein R is C1-C6 alkyl more preferably C1-C4 primary or secondary alkyl, such as sodium ethoxide in an appropriate solvent such as toluene. Most preferably P=X (wherein X is an alkoxy) since this avoids trans-esterification issues.
The compounds of formula (XV) can be formed from compounds of formula (XVI) by reaction with a mono-N-substituted piperazine group wherein the mono-substituent R10 as defined herein before, optionally in the presence of a supplementary base (which does not react irreversibly with the sulphonyl chloride moiety) such as triethylamine preferably in an appropriate solvent, such as toluene. “D” in compounds (XV) and (XVI) is Cl or Br. The monosubstituted piperazine group may also be the base where more than one equivalent of monosubstituted piperazine is present. Preferably about 2 equivalents are used.
Where a supplementary base is used it either does not react with the sulphonyl chloride moiety (such as a metal oxide, carbonate or bicarbonate) or it reacts with the sulphonyl chloride moiety in such a way as to keep it activated to nucleophilic attack (e.g. a tertiary amine such as triethylamine). The amine NH(R3)(R4) may also act as a base, in which case preferably more than one equivalent is present, more preferably about 2 equivalents (or more). - The compounds of formula (XVI) can be formed from compounds of formula (XX) in the presence of a chlorinating or brominating agent such as thionyl chloride or thionyl bromide more preferably in the presence of a halogenation catalyst, more preferably still thionyl chloride or thionyl bromide in the presence of dimethylformamide. The thionyl chloro/bromo can also act as the solvent, but more preferably the reaction takes place or in an appropriate other solvent such as toluene. In such case only stoicheometric amounts of thionyl chloride/bromide would be required, preferably at least 2 molar equivalents, more preferably at least 5 molar equivalents.
- It is possible to undertake the four step conversion of (XX) to (XB) in a single telescoped step, without intermediate product isolation, using the same solvent throughout (hereinafter the “telescoping solvent”). Thus where X is an alkoxy group (—OR3 group), steps (XX) to (XB) can be telescoped together using a single solvent such as a water immiscible inert organic solvent. More preferably a hydrocarbon solvent (such as toluene, xylene, anisole, chlorobenzene, hexane, heptane, octane, nonane, decane, cyclohexane, methylcyclohexane) or ethers (such as dibutyl ether, diphenyl ether) or ketones (such as methylisobutylketone, methylethylketone) or esters (such as ethyl acetate, butyl acetate) or dimethylformamide. More preferably still a hydrocarbon solvent (such as toluene, xylene, anisole, chlorobenzene, octane, nonane, decane, methylcyclohexane) or ethers (such as dibutyl ether, diphenyl ether) or esters (such as ethyl acetate, butyl acetate). More preferably still the telescoping solvent is toluene.
The intermediate of formula (XX) is formed from a compound of formula (XVII) in the presence of an agent which will form a protecting group (P) for the carboxylic acid (i.e. to form the —COP group). Preferably said agent is an esterification agent, to form a carboxylic acid ester (wherein, e.g. P will be alkoxy and the protecting forming agent will be an alcohol) such as a C1-C6 carboxylic acid ester which will be carried through the reaction scheme and hydrolised under basic conditions to the carboxylic acid function of compound (XB). Most preferably the esterification agent is ethanol. An additional solvent such as toluene may be appropriate.
The intermediate of formula (XVII) is formed from 2-hydroxynicotinic acid or a salt thereof in the presence of a sulphonylating agent, more preferably an agent comprising SO3 (ideally at least 1 molar equivalent of SO3), for example using SO3 in an organic solvent (e.g. THF, dioxan and heptane) or an aprotic solvent (e.g. nitrobenzene, nitromethane, 1,4-dioxane, dichloromethane) or a mineral acid as solvent (e.g. sulphuric acid) or in a liquid carboxylic acid as solvent (e.g. acetic acid) or THF or heptane. More preferably still, the sulphonylating agent is oleum (SO3 in sulphuric acid) such as about 20% to 30% oleum.
Compounds of the general formula (IXB) are formed by the reaction of intermediates of general formula (XB) with compounds of the general formula (VII), as detailed hereinbefore in the presence of a coupling agent, such as N,N′-carbonyldiimidazole and a suitable solvent, such as ethyl acetate.
Methods for the preparation of compounds of the general formula (VII) are described hereinafter.
In a preferred embodiment ofScheme 2, X is an —OR3 alkoxy group and so Q in compound (XIV) represents OR3. Preferably OR3 is a C1 to C6 alkoxy group, more preferably a C1 to C4 primary or secondary alkoxy group and especially ethoxy. However for other leaving groups the general method forScheme 2 would apply.
This preferred embodiment ofScheme 2 is illustrated inScheme 3. InScheme 3 the intermediate of formula (XB) is formed from a compound of formula (XIV) by removal of protecting group P by a deprotecting agent, advantageously by saponification in the presence of a hydroxide base such as sodium hydroxide, preferably in an appropriate solvent such as water and toluene.
The intermediate of formula (XIV) is formed from a compound of formula (XV) in the presence of an appropriate C1-C6 alkoxide nucleophile (—OR3), (such as a primary or secondary alkoxide), preferably a metal alkoxide of the formula ZOR3, wherein the metal (Z) is as defined hereinbefore for ZOR, such as sodium ethoxide, preferably in an appropriate solvent such as toluene or R3OH, wherein R3OH is as defined hereinbefore and is preferably ethoxy. D in compounds of formulae (XV) and (XVI) is Cl or Br, more preferably D is Cl. - The intermediate of formula (XV) is formed from a compound of formula (XVI) by reaction with N—R10piperazine, preferably in the presence of a base, such as triethylamine or excess N—R10piperazine, preferably in an appropriate solvent such as toluene.
- The intermediate of formula (XVI) is formed from a compound of formula (XX) in the presence of a chlorinating or brominating agent as defined for the same step in
Scheme 2 such as thionyl chloride or bromide, preferably thionyl chloride or bromide/dimethylformamide. The former can also act as the solvent, but more preferably the reaction takes place in an appropriate other solvent, such as toluene. In such a case only stoicheiometric amounts of thionyl chloride/bromide would be required, preferably as at least 2 molar equivalents more preferably at least 5 molar equivalents.
The intermediate of formula (XX) is formed from a compound of formula (XVII) in the presence of an agent which will form a protecting group (P) for the carboxylic acid (i.e. to form the —COP group) as defined herein before. Preferably said agent is an esterification agent, to form a carboxylic acid ester such as a C1-C6 carboxylic acid ester which will be carried through the reaction scheme and hydrolysed under basic conditions to the carboxylic acid function of compound (XB). Most preferably the esterification agent is ethanol. An additional solvent such as toluene may be utilised as appropriate.
The intermediate of formula (XVII) is formed from 2-hydroxynicotinic acid with a sulphonylating agent such as 30% oleum.
Again it is possible to undertake the four step conversion of (XX) to (XB) in a single telescoped step (as set out hereinbefore) in the same pot, without intermediate product isolation, using the same solvent (herein the “telescoping” solvent) throughout. The list of solvents described with respect toScheme 2 are directly applicable here. Most preferably the solvent is toluene.
For example after formation of compound (XVI), the excess chlorinating/brominating agent could be azeotroped off at the azeotrope temperature of the said agent and the telescope solvent. After formation of compound (XV), the HBr/HCl (i.e. HD) salts which are formed could be washed out (in aqueous) or filtered from the reaction vessel and the remainder of the aqueous solvent (where applicable) azeotroped off with some of the telescoping solvent. In the formation of compound (XIV), if the alkoxide used to introduce OR3 is dissolved in solvent (such as ethanol), then this solvent could again be azeotroped off with some of the telescoping solvent. If solid alkoxide is used then this latter azeotroping step is not required. Most preferably the telescoping solvent for any telescoped steps ofscheme 3 is toluene.
It will be appreciated that salts of the compounds ofSchemes 1 to 3 can be formed in accordance with the invention by converting the relevant compound to a salt thereof (either in situ or as a separate step). Also an acid addition salt of the compound of formula (I) can be formed in accordance with the invention.
1.4. Clearly, for certain compounds of formulae (I), (IA) or (IB) wherein R13 is OR3, by exploiting the cyclisation and alkoxide exchange methodology described in sections 1.2 and 2.1 herein, it may be particularly advantageous to generate a compound of formula (I), (IA) or (IB) from a compound of the general formula (IXCa), wherein the 2-alkoxy group of the 5-(pyridin-3-yl) substituent in the former is different from that in the latter, directly in a “one-pot reaction”. To achieve this an alternative alcohol (R3OH) should be used wherein the alkyl chain of the —R3 group of the alcohol is different from that of the —R3a group on the starting compound of general formula (IXCa). When the alcohol which is to provide the alternative 2-alkoxy group (—OR3) is too scarce or expensive to be employed as the reaction solvent, then it will be expedient to use a suitable alternative such as 1,4-dioxan as reaction solvent with the required alcohol (R3aOH) present in an amount sufficient to effect the desired conversion, typically from about 1 to about 2 molecular equivalents. (IXCa) and R3a are as defined hereinbefore.
2. In a further generally applicable process, compounds of the general formula (I), (IA) or (IB) may be prepared from “alternative” compounds of the general formula (I), (IA) or (IB) wherein said process may comprise either interconversion of differing —OR3 groups, interconversion of X and —OR3 groups or interconversion of —OR3 and —NR5R6 groups wherein X, R3 and NR5R6 are as defined hereinbefore.
2.1 As mentioned earlier, certain compounds of formulae (I), (IA) and (IB) can be interconverted by inducing alkoxide exchange or displacement at the 2-position of the 5-(pyridin-3-yl) substituent. This may be achieved, by treating the appropriate alcohol (of formula R3aOH wherein the R3a alkyl group is as defined hereinbefore and is different from the R3 group on the starting material (I), (IA) or (IB) with an alkali metal salt of a sterically hindered alcohol or amine in order to generate the required alkoxide anion which then reacts with the substrate. Typically, in a two-step procedure, a mixture of from about 1 to about 8, more preferably from about 5 to about 8, and especially from about 4 to about 8 molecular equivalents of potassium bis(trimethylsilyl)amide and the required alcohol (of formula R3aOH) as solvent is heated at from about 80° C. to about 100° C. for about 25 minutes to about 1 hour, followed by addition of the compound of formula (IA) or (IB) and heating of the reaction mixture at from about 100° C. to about 130° C. for from about 6 to about 24 hours. Alternatively, in a one-step procedure, the substrate may be treated directly, in the required alcohol as solvent, with from about 1.2 to about 6, preferably from about 4 to about 6 molecular equivalents of, for example, potassium bis(trimethylsilyl)amide, potassium t-butoxide or cesium carbonate at from about 80° C. to about 130° C. A hydroxide trapping agent may be optionally included in such alkoxide exchange reactions.
2.2 Alternatively, certain compounds of the general formula (I), (IA) or (IB) wherein R13 is —OR3 may be obtained from compounds of the general formula (XXX): -

- wherein R1, R2, R4 are as defined previously herein and wherein X is anything other than —OR3 by reaction in the presence of —OR3− optionally in the presence of a hydroxide trapping agent as defined hereinbefore.
2.3 In a yet further alternative synthesis compounds of the general formula (I), (IA) or (IB) wherein R13 is NR5R6 may be generated directly from a compound of general formula (I) wherein R13═R3. When R13 is OR3, the substrate may be treated with an excess of R5R6NH, or a suitable acid addition salt thereof, in the presence of an excess of a non-nucleophilic base such as a sterically hindered amine or a suitable inorganic base in a suitable solvent. Typically, R5R6NH is used as the free base with about a 3-fold excess (over the substrate) of potassium bis(trimethylsilyl)amide (KHMDS) in dimethylformamide (DMF) as solvent at about 100° C. Alternatively, an excess of R5R6NH may be used as the solvent and the reaction conducted in the presence of about a 50% excess of copper(II) sulphate at up to the reflux temperature of the reaction medium. Where the desired amino substituent on the compound of the formula (I), (IA) or (IB) is —NR5R6 and one of either R5 or R6 is H, then the exchange reaction may be carried out by refluxing with the appropriate amine, and copper(II) sulphate penta- or hepta-hydrate or anhydrous copper (II) sulphate or KHDMS in DMF. Typically, to exchange the OR3 group for alternative amines of the formula NHR5R6, such as compounds wherein R5 or R6 are selected from aliphatic or cyclic amines, optionally including oxygen (e.g. morpholine), then the reaction is preferably carried out by treating with the appropriate amine and about 3 equivalents of potassium bis(trimethylsilyl)amide in DMF for about 18 hours at 100° C.
3. In a yet further alternative process, a compound of the general formula (I) may be prepared from a compound of general formulae (IIA) or (IIC) respectively: -
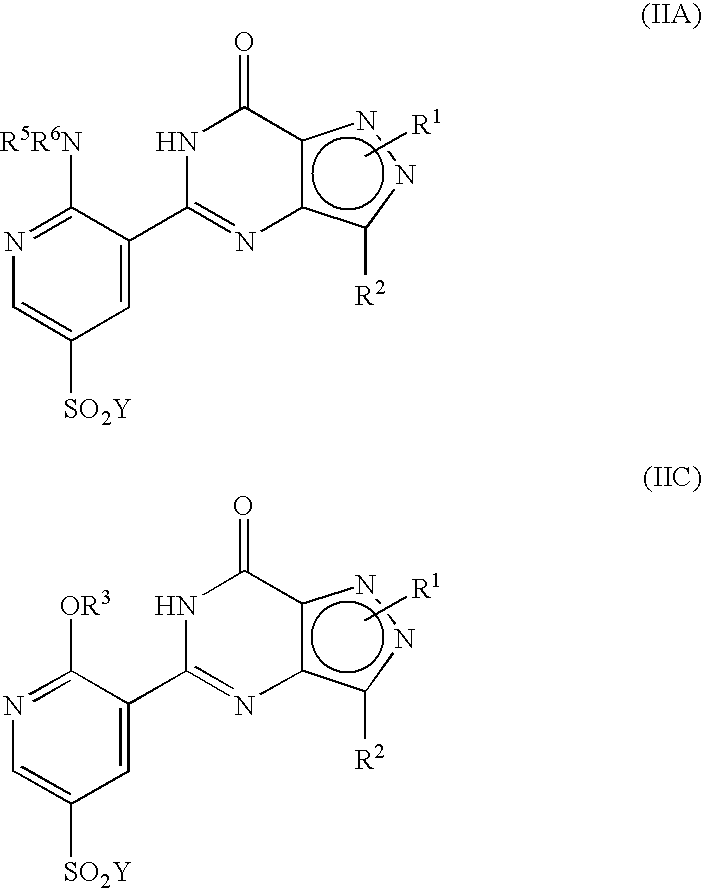
- wherein Y is halo, preferably chloro, and R1, R2, R3, R5 and R6 are as previously defined for formulae (IXA) and (IXC), by a reaction with a 4-R10-piperazinyl compound as described for the preparation of compounds of formula (XA) and (XB) from compounds of formula (VIIA) and (VIIIB) respectively.
Alternatively, a compound of the general formula (I), (IA) or (IB) may be prepared from a compound of the general formula (IIB): -

- wherein R1, R2, R4 and X are as previously defined herein via reaction with a 4-R10 piperazinyl compound followed by an optional displacement reaction in the presence of a hydroxide trapping agent and —OR3− as detailed hereinbefore for the preparation of compound (I) from compound (IXB) or (XXX).
3.1 A compound of general formulae (IIA) or (IIB) or (IIC) may be prepared from a compound of general formula (IVA) or (IVB) or (IVC) respectively: -

- wherein R1, R2, R3, R5, R6 and X are as previously herein, by the application of known methods for converting amino to a SO2Y group wherein Y is also as previously defined for formulae (IIA), (IIB) and (IIC). Such reactions are previously described for the preparation of compounds of the general formulae (VIIIA) and (VIIIB) from compounds of the general formulae (XIA) and (XIB) respectively.
- A compound of the general formula (IVA) or (IVB) or (IVC) may be prepared by cyclisation of a compound of the general formula (VA) or (VB) or (VC) respectively:
-

- wherein R1, R2, R3, R5, R6 and X are as previously defined herein and wherein the conditions for cyclisation are analogous to those previously described for cyclisation of the compounds of general formulae (IXA), (IXB) or (IXC).
- A compound of formula (VA) or (VB) or (VC) may be prepared by reduction of a compound of formula (VIA) or (VIB) or (IVC) respectively:
-
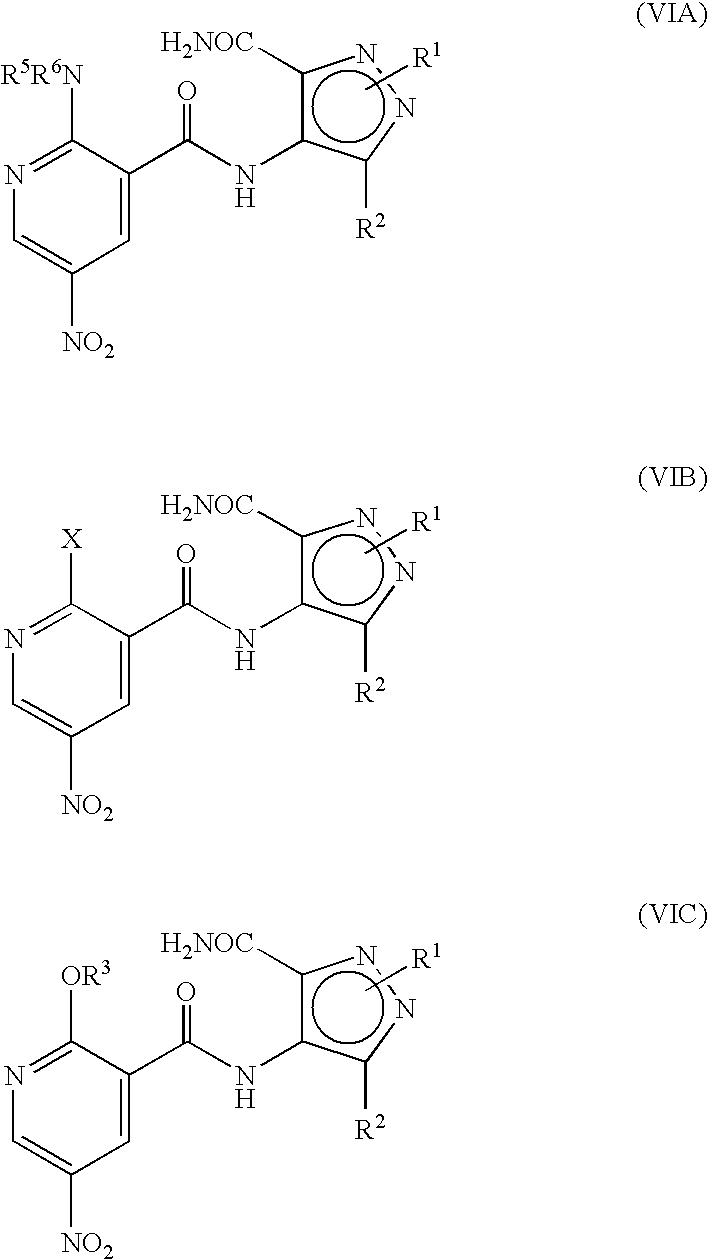
- wherein R1, R2, R3, R5, R6 and X are as previously defined for compounds of the general formulae (VA), (VB) and (VC), by conventional catalytic or catalytic transfer hydrogenation procedures as previously detailed for preparation of compounds of the general formulae (XIA) or (XIB) from compounds of the general formulae (XIIA) or (XIIB) respectively.
- A compound of formula (VIA), (VIB) or (VIC) may be prepared by reaction of a compound of formula (VII) as defined previously herein with a compound of formula (XIIA) or (XIIB) or (XIIC) respectively:
-
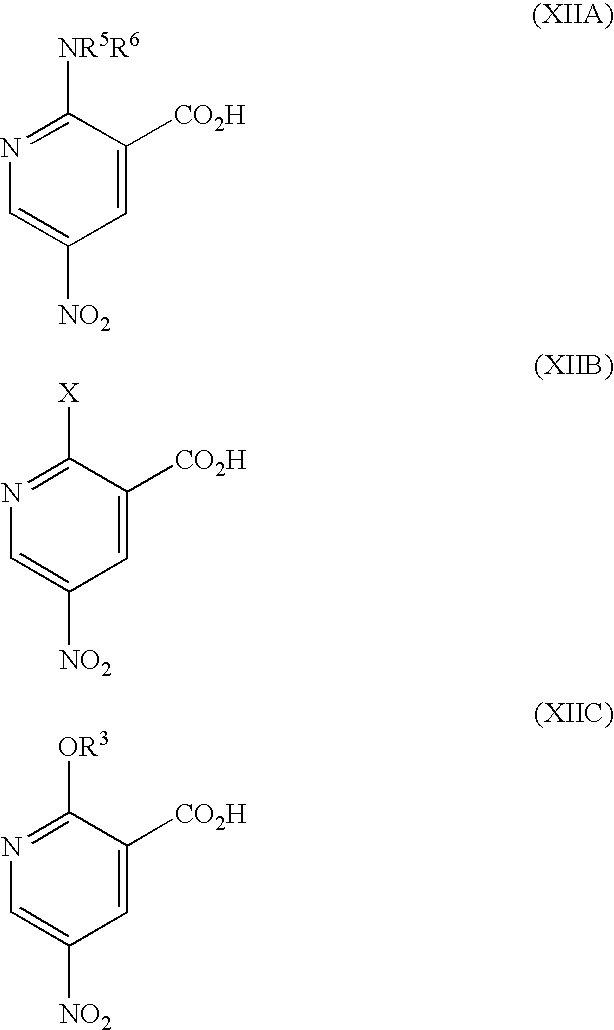
- wherein R3, R5, R6 and X are as previously defined for compounds of the general formulae (VIA) or (VIB) or (VIC). Again, as previously detailed a conventional amine protecting group strategy is preferred for (XIIA) when NR5R6 is a primary or secondary amino group. The coupling reaction is analogous to the reactions of (VII) with the compounds of general formulae (XA) or (XB) or (XC) already described herein.
3.2 A compound of general formulae (IIA) or (IIB) or (IIC) may be prepared from a compound of formula (IVA) or (IVB) or (IVC) respectively as described hereinbefore wherein said compound of the general formulae (IVA) or (IVB) or (IVC) may be prepared by direct cyclisation of a compound of the general formula (VIA) or (VIB) or (VIC) respectively: -

- wherein R1, R2, R3, R5, R6 and X are as previously herein and wherein the conditions for said direct cyclisation are analogous to the previously described cyclisation for compounds of the general formulae (IXA) or (IXB) or (IXC) and wherein said cyclisation is followed by reduction of the resultant intermediate compounds according to the methods previously detailed herein to provide compounds of the general formulae (IVA) or (IVB) or (IVC) from compounds of the general formulae (VA) or (VB) or (VC).
Compounds of the general formula (XIIC) wherein X is Cl may be prepared from 2-hydroxy nicotinic acid via nitration followed by esterification then chlorination of the suitably protected nicotinic acid and subsequent ester hydrolysis.
Compounds of the general formula (XIIIC) (i.e. compounds of general formula (XIIIB wherein X is —OR3) can be prepared by analogy with the methods detailed previously herein.
4. A further, generally applicable, synthetic route to compounds of the general formula (I), (IA) or (IB) involves incorporation of the R1 substituent in the final step of the synthesis. Thus a compound of the general formula (I), (IA) or (IB) may be prepared by alkylation of a compound of formula (Ia), (IAa) or (IBa) wherein R1 is hydrogen and R2, R13 and R4 are as previously defined for formulae (I), (IA) and (IB), using one or more of a plethora of well-known methods, such as: - (i) reaction with a compound of formula R1J, wherein R1 is as previously defined for compounds of general formulae (I), (IA) and (IB), and J is a suitable leaving group, e.g. halo (preferably chloro, bromo or iodo), C1-C4 alkanesulphonyloxy, trifluoromethanesulphonyloxy or arylsulphonyloxy (such as benzenesulphonyloxy or p-toluenesulphonyloxy), in the presence of an appropriate base, optionally in the presence of sodium iodide or potassium iodide, at from about −70° C. to about 100° C. Preferably the alkylation is conducted at from about room temperature to about 120° C. Suitable base-solvent combinations may be selected from:
- (a) sodium, potassium or cesium carbonate, sodium or potassium bicarbonate, or a tertiary amine such as triethylamine or pyridine, together with a C1 to C4 alkanol, 1,2-dimethoxyethane, tetrahydrofuran, 1,4-dioxan, acetonitrile, pyridine, N,N-dimethylformamide or N,N-dimethylacetamide;
- (b) sodium or potassium hydroxide, or a sodium or potassium C1 to C4 alkoxide, together with a C1 to C4 alkanol, water or mixtures thereof;
- (c) lithium, sodium or potassium hydride, lithium, sodium or potassium bis(trimethylsilyl)amide, lithium diisopropylamide or butyllithium, together with toluene, ether, 1,2-dimethoxyethane, tetrahydrofuran or 1,4-dioxan; or
- (d) under phase transfer catalysis conditions, a tetraalkylammonium halide or hydroxide, together with a mixture of an aqueous solution of sodium or potassium hydroxide and dichloromethane, 1,2-dichloroethane or chloroform;
- Typically, either about a 10% excess of sodium hydride is added to a solution of the substrate in a suitable solvent, e.g. anhydrous tetrahydrofuran or cesium carbonate in dimethylformamide (DMF) is employed, and the resulting anion treated with about a 10% excess of the required R1J.
- (ii) reaction with a compound of formula R10H, wherein R1 is as previously defined for compounds of the general formulae (I), (IA) and (IB), using classical Mitsunobu methodology. Typical reaction conditions involve treating the substrate with the alkanol in the presence of a triarylphosphine and a di(C1 to C4)alkyl azodicarboxylate, in a suitable solvent such as tetrahydrofuran or 1,4-dioxan, at from about −5° C. to about room temperature.
- (iii) reaction with a compound of formula R1M, wherein R1 represents optionally substituted phenyl, Het2, Het3 or Het4 and wherein said Het groups are either aromatic or partially unsaturated at the C atom that is attached to M, and wherein M represents an optionally substituted metal or boron group wherein said metal or boron group is suitable for cross-coupling reactions (of metal or boron compounds), for example a dihydroxyborane, in the presence of an appropriate catalyst system (e.g. copper (II) acetate) or under so-called “Goldberg” conditions. Such cross-coupling is preferably carried out in the presence of a suitable base (e.g. pyridine), and a drying agent, typically 4 Å molecular sieves, in a suitable solvent such as dichloromethane or N-methylpyrrolidine, and optionally under microwave irradiation.
- (iv) reaction with a compound of formula R1E, where E is halo, preferably bromo, under conditions suitable for cross-coupling of halogenated compounds, where R1 is as defined in (iii). Such reaction is typically carried out in the presence of an appropriate catalyst system (e.g. Palladium catalyst), in the presence of a suitable base, such as for example sodium t-butoxide, in a suitable solvent, such as toluene, with heating, typically at about 70° C.
4.1 Thus, a compound of general formula (Ia), (IAa) or (IBa), wherein R1 is hydrogen and R2, R13 and R4 are as previously defined for compounds of general formulae (I), (IA) or (IB), may be obtained from a compound of formula (IXAa) or (IXBa) or (IXCa) respectively wherein R1 is hydrogen, and R2, R3, R5, R6 and R4 and X are as previously defined for formulae (IXA), (IXB) or (IXC), under the same conditions as those used for the conversion of a compound of the general formula (IXA), (IXB) or (IXC) to a compound of the general formula (I), (IA) or (IB) respectively when R1 is other than hydrogen, followed by acidification of the reaction mixture to a pH of about 6.
4.2 In a further alternative, generally applicable synthetic route the compounds of the present invention may be prepared by cyclisation of compounds of the general formulae (IXA), (IXB) or (IXC) wherein said compounds of the general formulae (IXA), (IXB) or (IXC) are obtained from compounds of the general formulae (IXAa), (IXBa) or (IXCa) wherein R1 is hydrogen and R2, R3, R5, R6 and R4 are as previously defined herein, using one or more of a plethora of well-known methods such as are detailed hereinbefore for conversion of compounds of the general formulae (Ia), (IAa) and (IBa) to compounds of the general formulae (I), (IA) and (IB). Any of the previously detailed methods for such general conversion may be used. Preferred conditions for such conversion use either from about 1.0 to 1.3 equivalents of sodium hydride in tetrahydrofuran solvent at from about −78° to about room temperature and from about 1.1 to about 2.3 equivalents of alkylating agent at from about 60° C. to about 70° C., or from about 2.2 equivalents of cesium carbonate as base in dimethylformamide as solvent and about 1.1. equivalent of alkylating agent at about 60° C.
5. In a yet further alternative synthesis, compounds of the general formula (I), (IA) or (IB) can be obtained from compounds of the general formula (I) wherein R10 is H, via a suitable alkylation reaction such as for example with an alkyl halide and a suitable base e.g. cesium carbonate and methyl chloride.
In a preferred process for the preparation of the compounds according to the present invention compounds of general formula (VIIB) are prepared from compounds of the general formula (XIIIB) according to the process detailed in Preparations 96(a) to (h). These compounds of general formula (VIIB) are coupled with compounds of general formula (XC) according to the process detailed in Preparations 29 and 96(i) to provide a compound of general formula (IXC), wherein said compound of general formula (IXC) is prepared according to the process detailed in Preparation 95. The compound of general formula (IXC) is then preferably cyclised under basic conditions according to the process detailed in Examples 8 and 102 to form compounds of general formula (IB) wherein R13 is OR3.
The 4-aminopyrazole-5-carboxamides of general formulae (VII), (VIIA) and (VIIB), the pyrazoles of general formula (XIII), the carboxylic acids of formulae (XA), (XB), (XIIA), (XIIB), (XIIC), (VIIA), (VIIB), (VIIC) and (X), or the compounds of the general formula R1J and R1E when neither commercially available nor subsequently described, can be obtained either by analogy with the processes described in the Preparations section or by conventional synthetic procedures, in accordance with standard textbooks on organic chemistry or literature precedent, from readily accessible starting materials using appropriate reagents and reaction conditions.
Moreover, persons skilled in the art will be aware of variations of, and alternatives to, those processes described hereinafter in the Examples and Preparations sections which allow the compounds defined by formulae (I), (IA) or (IB) to be obtained.
The pharmaceutically acceptable acid addition salts of the compounds of formulae (I), (IA) or (IB) which contain a basic centre may also be prepared in a conventional manner. By way of illustration, acid addition salts of compounds of formula (I) (more particularly IA and IB) can be formed by reacting a compound of formula (I) with an equimolar or excess amount of the appropriate acid, either neat or in a suitable solvent. The salt may then be precipitated out of solution and isolated by filtration or the reaction solvent can be stripped off by conventional means such as by evaporation under vacuum. Typical salts which can be used in the schemes of 1 to 3 are given in PCT/IB99/00519. Example of salts of compounds IA and IB are the p-toluenesulfonate, benzenesulfonate, camphorsulfonate and ethanesulfonate respectively.
Pharmaceutically acceptable base addition salts can be obtained in an analogous manner by treating a solution of a compound of formula (I), (IA) or (IB) with the appropriate base. Both types of salt may be formed or interconverted using ion-exchange resin techniques. - The present invention also includes all suitable isotopic variations of a compound of the formula (I) or a pharmaceutically acceptable salt thereof. An isotopic variation of a compound of the formula (I) or a pharmaceutically acceptable salt thereof is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature. Examples of isotopes that can be incorporated into compounds of the formula (I) and pharmaceutically acceptable salts thereof include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine and chlorine such as 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F and 36Cl, respectively. Certain isotopic variations of the compounds of the formula (I) and pharmaceutically acceptable salts thereof, for example, those in which a radioactive isotope such as 3H or 14C is incorporated, are useful in drug and/or substrate tissue distribution studies. Tritiated, i.e., 3H, and carbon-14, i.e., 14C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with isotopes such as deuterium, i.e., 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements and hence may be preferred in some circumstances. Isotopic variations of the compounds of formula (I) and pharmaceutically acceptable salts thereof of this invention can generally be prepared by conventional procedures such as by the illustrative methods or by the preparations described in the Examples and Preparations hereafter using appropriate isotopic variations of suitable reagents.
- It will be appreciated by those skilled in the art that certain protected derivatives of compounds of the formulae (I), (IA) or (IB), which may be made prior to a final de-protection stage, may not possess pharmacological activity as such, but may, in certain instances, be administered orally or parenterally and thereafter metabolised in the body to form compounds of the invention which are pharmacologically active. Such derivatives may therefore be described as “pro-drugs”. Further, certain compounds of the formulae (I), (IA) or (IB) may act as pro-drugs of other compounds of the formulae (I), (IA) or (IB).
All protected derivatives, and pro-drugs, of compounds of general formulae (I), (IA) or (IB) are included within the scope of the invention. Suitable protecting groups for use in accordance with the invention can be found in “Protecting Groups” edited by P. J. Kocienski, Thieme, New York, 1994-see particularly chapter 4, page 118-154 for carboxy protecting groups; and “Protective Groups in Organic Synthesis” 2nd edition, T. W. Greene & P. G. M. Wutz, Wiley-Interscience (1991)—see particularly chapter 5 for carboxy protecting groups. Examples of suitable pro-drugs for the compounds of the present invention are described in Drugs of Today, Volume 19, Number 9, 1983, pp 499-538 and in Topics in Chemistry, Chapter 31, pp 306-316. - The biological activities of the compounds of the present invention were determined by the following test methods.
- The compounds of the present invention are potent and selective cGMP PDE5 inhibitors. In vitro PDE inhibitory activities against
cyclic guanosine 3′,5′-monophosphate (cGMP) andcyclic adenosine 3′,5′-monophosphate (cAMP) phosphodiesterases were determined by measurement of their IC50 values (the concentration of compound required for 50% inhibition of enzyme activity). - The required PDE enzymes were isolated from a variety of sources, including human corpus cavernosum, human and rabbit platelets, human cardiac ventricle, human skeletal muscle and bovine retina, essentially by the method of W. J. Thompson and M. M. Appleman (Biochem., 1971, 10, 311). In particular, the cGMP-specific PDE (PDE5) and the cGMP-inhibited cAMP PDE (PDE3) were obtained from human corpus cavernosum tissue, human platelets or rabbit platelets; the cGMP-stimulated PDE (PDE2) was obtained from human corpus cavernosum; the calcium/calmodulin (Ca/CAM)-dependent PDE (PDE1) from human cardiac ventricle; the cAMP-specific PDE (PDE4) from human skeletal muscle; and the photoreceptor PDE (PDE6) from bovine retina. Phosphodiesterases 7-11 were generated from full length human recombinant clones transfected into SF9 cells.
- Assays were performed either using a modification of the “batch” method of W. J. Thompson et al. (Biochem., 1979, 18, 5228) or using a scintillation proximity assay for the direct detection of AMP/GMP using a modification of the protocol described by Amersham plc under product code TRKQ7090/7100. In summary, the effect of PDE inhibitors was investigated by assaying a fixed amount of enzyme in the presence of varying inhibitor concentrations and low substrate, (CGMP or cAMP in a 3:1 ratio unlabelled to [3H]-labeled at a conc ˜1/3 Km) such that IC50≈Ki. The final assay volume was made up to 100 μl with assay buffer [20 mM Tris-HCl pH 7.4, 5 mM MgCl2, 1 mg/ml bovine serum albumin]. Reactions were initiated with enzyme, incubated for 30-60 min at 30° C. to give <30% substrate turnover and terminated with 50 μl yttrium silicate SPA beads (containing 3 mM of the respective unlabelled cyclic nucleotide for PDEs 9 and 11). Plates were re-sealed and shaken for 20 min, after which the beads were allowed to settle for 30 min in the dark and then counted on a TopCount plate reader (Packard, Meriden, Conn.) Radioactivity units were converted to % activity of an uninhibited control (100%), plotted against inhibitor concentration and inhibitor IC50 values obtained using the ‘Fit Curve’ Microsoft Excel extension. Results from these tests show that the compounds of the present invention are potent and selective inhibitors of cGMP-specific PDE5.
- Preferred compounds of the present invention, such as those of Examples 3-12, 14-17, 19, 21-30, 32, 33, 35-46, 48-59, 61, 62, 65-75, 77, 79-102 have IC50 values of less than about 10 nM for the PDE5 enzyme. More preferred compounds, such as those of Examples 3-12, 14, 15, 17, 23-30, 32, 33, 35-46, 48, 50-59, 61, 62, 65, 69-74, 79-102 have IC50 values of less than about 5 nM for the PDE5 enzyme. Especially preferred compounds, such as those of Examples 4-10, 15, 17, 23-28, 30, 32, 33, 35-42, 44, 45, 46, 50, 52-56, 58, 59, 61, 62, 65, 69-74, 79-93, 96, 98-102 have IC50 values of less than about 2 nM for the PDE5 enzyme.
Especially preferred herein are compounds which have an IC50 value of less than about 10, more preferably less than about 5, and most preferably less than about 2 nM for the PDE5 enzyme in combination with selectivity of greater than 10-fold, more preferably greater than 50-fold, more preferably greater than 100-fold and especially greater than 200-fold selectivity for the PDE5 enzyme versus the PDE6 enzyme - This was assessed in vitro by determining the capacity of a compound of the invention to enhance sodium nitroprusside-induced relaxation of pre-contracted rabbit corpus cavernosum tissue strips, as described by S. A. Ballard et al. (Brit. J. Pharmacol., 1996, 118 (suppl.), abstract 153P).
- In vivo Activity
- Compounds were screened in anaesthetised dogs to determine their capacity, after i.v. administration, to enhance the pressure rises in the corpora cavernosa of the penis induced by intracavernosal injection of sodium nitroprusside, using a method based on that described by Trigo-Rocha et al. (Neurourol. and Urodyn., 1994, 13, 71).
- The compounds of formulae (I), (IA) or (1B), their pharmaceutically acceptable salts, and pharmaceutically acceptable solvates of either entity can be administered alone but, in human therapy will generally be administered in admixture with a suitable pharmaceutical excipient diluent or carrier selected with regard to the intended route of administration and standard pharmaceutical practice. For example, the compounds of formulae (I), (IA) or (1B) or salts or solvates thereof can be administered orally, buccally or sublingually in the form of tablets, capsules (including soft gel capsules), ovules, elixirs, solutions or suspensions, which may contain flavouring or colouring agents, for immediate-, delayed-, modified-, or controlled-release such as sustained-, dual-, or pulsatile delivery applications. The compounds of the invention may also be administered via intracavernosal injection. The compounds of the invention may also be administered via fast dispersing or fast dissolving dosages forms or in the form of a high energy dispersion or as coated particles. Suitable pharmaceutical formulations of the compounds of the invention may be in coated or un-coated form as desired.
- Such tablets may contain excipients such as microcrystalline cellulose, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate, glycine and starch (preferably corn, potato or tapioca starch), disintegrents such as sodium starch glycollate, croscarmellose sodium and certain complex silicates, and granulation binders such as polyvinylpyrrolidone, hydroxypropylmethyl cellulose (HPMC), hydroxypropylcellulose (HPC), sucrose, gelatin and acacia. Additionally, lubricating agents such as magnesium stearate, stearic acid, glyceryl behenate and talc may be included.
- Solid compositions of a similar type may also be employed as fillers in gelatin capsules. Preferred excipients in this regard include lactose, starch, a cellulose, milk sugar or high molecular weight polyethylene glycols. For aqueous suspensions and/or elixirs, the compounds of the formula (I), (IA) or (IB) may be combined with various sweetening or flavouring agents, colouring matter or dyes, with emulsifying and/or suspending agents and with diluents such as water, ethanol, propylene glycol and glycerin, and combinations thereof.
- Modified release and pulsatile release dosage forms may contain excipients such as those detailed for immediate release dosage forms together with additional excipients that act as release rate modifiers, these being coated on and/or included in the body of the device. Release rate modifiers include, but are not exclusively limited to, hydroxypropylmethyl cellulose, methyl cellulose, sodium carboxymethylcellulose, ethyl cellulose, cellulose acetate, polyethylene oxide, Xanthan gum, Carbomer, ammonio methacrylate copolymer, hydrogenated castor oil, carnauba wax, paraffin wax, cellulose acetate phthalate, hydroxypropylmethyl cellulose phthalate, methacrylic acid copolymer and mixtures thereof. Modified release and pulsatile release dosage forms may contain one or a combination of release rate modifying excipients. Release rate modifying excipients maybe present both within the dosage form i.e. within the matrix, and/or on the dosage form i.e. upon the surface or coating.
- Fast dispersing or dissolving dosage formulations (FDDFs) may contain the following ingredients: aspartame, acesulfame potassium, citric acid, croscarmellose sodium, crospovidone, diascorbic acid, ethyl acrylate, ethyl cellulose, gelatin, hydroxypropylmethyl cellulose, magnesium stearate, mannitol, methyl methacrylate, mint flavouring, polyethylene glycol, fumed silica, silicon dioxide, sodium starch glycolate, sodium stearyl fumarate, sorbitol, xylitol. The terms dispersing or dissolving as used herein to describe FDDFs are dependent upon the solubility of the drug substance used i.e. where the drug substance is insoluble a fast dispersing dosage form can be prepared and where the drug substance is soluble a fast dissolving dosage form can be prepared.
- The compounds of the invention can also be administered parenterally, for example, intracavernosally, intravenously, intra-arterially, intraperitoneally, intrathecally, intraventricularly, intraurethrally intrasternally, intracranially, intramuscularly or subcutaneously, or they may be administered by infusion or needless injection techniques. For such parenteral administration they are best used in the form of a sterile aqueous solution which may contain other substances, for example, enough salts or glucose to make the solution isotonic with blood. The aqueous solutions should be suitably buffered (preferably to a pH of from 3 to 9), if necessary. The preparation of suitable parenteral formulations under sterile conditions is readily accomplished by standard pharmaceutical techniques well-known to those skilled in the art.
- For oral and parenteral administration to human patients, the daily dosage level of the compounds of formula (I), (IA) or (1B) or salts or solvates thereof will usually be from 10 to 500 mg (in single or divided doses).
- Thus, for example, tablets or capsules of the compounds of formulae (I), (IA) or (IB) or salts or solvates thereof may contain from 5 mg to 250 mg of active compound for administration singly or two or more at a time, as appropriate. The physician in any event will determine the actual dosage which will be most suitable for any individual patient and it will vary with the age, weight and response of the particular patient. The above dosages are exemplary of the average case. There can, of course, be individual instances where higher or lower dosage ranges are merited and such are within the scope of this invention. The skilled person will also appreciate that, in the treatment of certain conditions (including MED and FSD), compounds of the invention may be taken as a single dose on an “as required” basis (i.e. as needed or desired).
-
-
Ingredient % w/w Besylate salt of Example 103 13.038* Lactose 62.222 Starch 20.740 Croscarmellose Sodium 3.000 Magnesium Stearate 1.000 *Quantity adjusted in accordance with drug activity.
Such tablets can be manufactured by standard processes, for example, direct compression or a wet or dry granulation process. The tablet cores may be coated with appropriate overcoats. - The compounds of the invention can also be administered intranasally or by inhalation and are conveniently delivered in the form of a dry powder inhaler or an aerosol spray presentation from a pressurised container, pump, spray or nebuliser with the use of a suitable propellant, e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, a hydrofluoroalkane such as 1,1,1,2-tetrafluoroethane (HFA 134A [trade mark] or 1,1,1,2,3,3,3-heptafluoropropane (HFA 227EA [trade mark]), carbon dioxide or other suitable gas. In the case of a pressurised aerosol, the dosage unit may be determined by providing a valve to deliver a metered amount. The pressurised container, pump, spray or nebuliser may contain a solution or suspension of the active compound, e.g. using a mixture of ethanol and the propellant as the solvent, which may additionally contain a lubricant, e.g. sorbitan trioleate. Capsules and cartridges (made, for example, from gelatin) for use in an inhaler or insufflator may be formulated to contain a powder mix of a compound of the formula (I), (IA) or (IB) and a suitable powder base such as lactose or starch.
- Aerosol or dry powder formulations are preferably arranged so that each metered dose or “puff” contains from 1 to 50 mg of a compound of the formula (I), (IA) or (IB) for delivery to the patient. The overall daily dose with an aerosol will be in the range of from 1 to 50 mg which may be administered in a single dose or, more usually, in divided doses throughout the day.
- The compounds of the invention may also be formulated for delivery via an atomiser. Formulations for atomiser devices may contain the following ingredients as solubilisers, emulsifiers or suspending agents: water, ethanol, glycerol, propylene glycol, low molecular weight polyethylene glycols, sodium chloride, fluorocarbons, polyethylene glycol ethers, sorbitan trioleate, oleic acid.
- Alternatively, the compounds of the formulae (I), (IA) or (IB) or salts or solvates thereof can be administered in the form of a suppository or pessary, or they may be applied topically in the form of a gel, hydrogel, lotion, solution, cream, ointment or dusting powder. The compounds of the formulae (IA) and (IB) or salts or solvates thereof may also be dermally administered. The compounds of the formulae (I), (IA) or (IB) or salts or solvates thereof may also be transdermally administered, for example, by the use of a skin patch. They may also be administered by the ocular, pulmonary or rectal routes.
- For ophthalmic use, the compounds can be formulated as micronised suspensions in isotonic, pH adjusted, sterile saline, or, preferably, as solutions in isotonic, pH adjusted, sterile saline, optionally in combination with a preservative such as a benzylalkonium chloride. Alternatively, they may be formulated in an ointment such as petrolatum.
- For application topically to the skin, the compounds of the formulae (I), (IA) or (IB) or salts or solvates thereof can be formulated as a suitable ointment containing the active compound suspended or dissolved in, for example, a mixture with one or more of the following: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene polyoxypropylene compound, emulsifying wax and water. Alternatively, they can be formulated as a suitable lotion or cream, suspended or dissolved in, for example, a mixture of one or more of the following: mineral oil, sorbitan monostearate, a polyethylene glycol, liquid paraffin, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- The compounds of the formulae (I), (IA) or (IB) may also be used in combination with a cyclodextrin. Cyclodextrins are known to form inclusion and non-inclusion complexes with drug molecules. Formation of a drug-cyclodextrin complex may modify the solubility, dissolution rate, bioavailability and/or stability property of a drug molecule. Drug-cyclodextrin complexes are generally useful for most dosage forms and administration routes. As an alternative to direct complexation with the drug the cyclodextrin may be used as an auxiliary additive, e.g. as a carrier, diluent or solubiliser. Alpha-, beta- and gamma-cyclodextrins are most commonly used and suitable examples are described in WO-A-91/11172, WO-A-94/02518 and WO-A-98/55148.
- Generally, in humans, oral administration of the compounds of the invention is the preferred route, being the most convenient and, for example in MED, avoiding the well-known disadvantages associated with intracavernosal (i.c.) administration. A preferred oral dosing regimen in MED for a typical man is from 5 to 250 mg of compound when required. In circumstances where the recipient suffers from a swallowing disorder or from impairment of drug absorption after oral administration, the drug may be administered parenterally, sublingually or buccally.
- For veterinary use, a compound of formula (I), (IA) or (IB), or a veterinarily acceptable salt thereof, or a veterinarily acceptable solvate or pro-drug thereof, is administered as a suitably acceptable formulation in accordance with normal veterinary practice and the veterinary surgeon will determine the dosing regimen and route of administration which will be most appropriate for a particular animal.
- It is to be appreciated that all references herein to treatment include curative, palliative and prophylactic treatment.
- Thus the invention provides a pharmaceutical composition comprising a compound of formula (I), (IA) or (IB), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate or pro-drug thereof, together with a pharmaceutically acceptable diluent or carrier.
- It further provides a veterinary formulation comprising a compound of formula (I), (IA) or (IB), or a veterinarily acceptable salt thereof, or a veterinarily acceptable solvate or pro-drug thereof, together with a veterinarily acceptable diluent or carrier.
- The invention also provides a compound of formula (I), (IA) or (IB), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate or pro-drug thereof, or a pharmaceutical composition containing any of the foregoing, for use as a human medicament.
- In addition, it provides a compound of formula (I), (IA) or (IB), or a veterinarily acceptable salt thereof, or a veterinarily acceptable solvate or pro-drug thereof, or a veterinary formulation containing any of the foregoing, for use as an animal medicament.
- In yet another aspect, the invention provides the use of a compound of formula (I), (IA) or (IB), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate or pro-drug thereof, for the manufacture of a human medicament for the curative, palliative or prophylactic treatment of a medical condition for which a cGMP PDE5 inhibitor is indicated. There is further provided the use of a compound of formula (I), (IA) or (IB) or a suitable salt, solvate or pro-drug thereof, in the manufacture of a medicament for the treatment of a medical condition in which inhibition of a cGMP PDE5 is desirable.
- It also provides the use of a compound of formula (I), (IA) or (IB), or a veterinarily acceptable salt thereof, or a veterinarily acceptable solvate or pro-drug thereof, for the manufacture of an animal medicament for the curative, palliative or prophylactic treatment of a medical condition for which a cGMP PDE5 inhibitor is indicated.
- Moreover, the invention provides the use of a compound of formula (I), (IA) or (IB), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate or pro-drug thereof, for the manufacture of a human medicament for the curative, palliative or prophylactic treatment of male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder, female sexual orgasmic dysfunction (FSOD), sexual dysfunction due to spinal cord injury, selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction, premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, peripheral vascular disease, stroke, nitrate induced tolerance, bronchitis, allergic asthma, chronic asthma, allergic rhinitis, diseases and conditions of the eye, diseases characterised by disorders of gut motility, pre-eclampsia, Kawasaki's syndrome, nitrate tolerance, multiple sclerosis, diabetic nephropathy, neuropathy including autonomic and peripheral neuropathy and in particular diabetic neuropathy and symptoms thereof e.g. gastroparesis, Alzheimer's disease, acute respiratory failure, psoriasis, skin necrosis, cancer, metastasis, baldness, nutcracker oesophagus, anal fissure, haemorrhoids, hypoxic vasoconstriction or blood pressure stabilisation during haemodialysis. Particularly preferred conditions include MED and FSD.
- It also provides the use of a compound of formula (I), (IA) or (IB), or a veterinarily acceptable salt thereof, or a veterinarily acceptable solvate or pro-drug thereof, for the manufacture of an animal medicament for the curative, palliative or prophylactic treatment of male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder, female sexual orgasmic dysfunction (FSOD), sexual dysfunction due to spinal cord injury, selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction, premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, peripheral vascular disease, stroke, nitrate induced tolerance, bronchitis, allergic asthma, chronic asthma, allergic rhinitis, diseases and conditions of the eye, diseases characterised by disorders of gut motility, pre-eclampsia, Kawasaki's syndrome, nitrate tolerance, multiple sclerosis, diabetic nephropathy, neuropathy including autonomic and peripheral neuropathy and in particular diabetic neuropathy and symptoms thereof, Alzheimer's disease, acute respiratory failure, psoriasis, skin necrosis, cancer, metastasis, baldness, nutcracker oesophagus, anal fissure, haemorrhoids, hypoxic vasoconstriction or blood pressure stabilisation during haemodialysis. Particularly preferred conditions include MED and FSD.
- Additionally, the invention provides a method of treating or preventing a medical condition for which a cGMP PDE5 inhibitor is indicated, in a mammal (including a human being), which comprises administering to said mammal a therapeutically effective amount of a compound of formula (I), (IA) or (IB), or a pharmaceutically or veterinarily acceptable salt thereof, or a pharmaceutically or veterinarily acceptable solvate or pro-drug thereof, or a pharmaceutical veterinary formulation containing any of the foregoing.
- Still further, the invention provides a method of treating or preventing male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder, female sexual orgasmic dysfunction (FSOD), sexual dysfunction due to spinal cord injury, selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction, premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, peripheral vascular disease, stroke, nitrate induced tolerance, bronchitis, allergic asthma, chronic asthma, allergic rhinitis, diseases and conditions of the eye, diseases characterised by disorders of gut motility, pre-eclampsia, Kawasaki's syndrome, nitrate tolerance, multiple sclerosis, diabetic nephropathy, neuropathy including autonomic and peripheral neuropathy and in particular diabetic neuropathy and symptoms thereof e.g. gastroparesis, peripheral diabetic neuropathy, Alzheimer's disease, acute respiratory failure, psoriasis, skin necrosis, cancer, metastasis, baldness, nutcracker oesophagus, anal fissure, haemorrhoids, hypoxic vasoconstriction or blood pressure stabilisation of during haemodialysis in a mammal (including a human being), which comprises administering to said mammal a therapeutically effective amount of a compound of formula (I), (IA) or (IB), or a pharmaceutically or veterinarily acceptable salt thereof, or a pharmaceutically or veterinarily acceptable solvate or pro-drug thereof, or a pharmaceutical composition or veterinary formulation containing any of the foregoing.
- In a yet further aspect of the present invention provides a combination of compounds of the general formula (I), (IA) or (IB) with further compounds useful in the inhibition of PDE5 wherein said combination is useful for the treatment or prevention of male erectile dysfunction (MED), impotence, female sexual dysfunction (FSD), clitoral dysfunction, female hypoactive sexual desire disorder, female sexual arousal disorder, female sexual pain disorder, female sexual orgasmic dysfunction (FSOD), sexual dysfunction due to spinal cord injury, selective serotonin re-uptake inhibitor (SSRI) induced sexual dysfunction, premature labour, dysmenorrhoea, benign prostatic hyperplasia (BPH), bladder outlet obstruction, incontinence, stable, unstable and variant (Prinzmetal) angina, hypertension, pulmonary hypertension, chronic obstructive pulmonary disease, coronary artery disease, congestive heart failure, atherosclerosis, conditions of reduced blood vessel patency, peripheral vascular disease, stroke, nitrate induced tolerance, bronchitis, allergic asthma, chronic asthma, allergic rhinitis, diseases and conditions of the eye, diseases characterised by disorders of gut motility, pre-eclampsia, Kawasaki's syndrome, nitrate tolerance, multiple sclerosis, diabetic nephropathy, neuropathy including autonomic and peripheral neuropathy and in particular diabetic neuropathy and symptoms thereof e.g. gastroparesis, peripheral diabetic neuropathy, Alzheimer's disease, acute respiratory failure, psoriasis, skin necrosis, cancer, metastasis, baldness, nutcracker oesophagus, anal fissure, haemorrhoids, hypoxic vasoconstriction or blood pressure stabilisation of during haemodialysis in a mammal (including a human being).
- The invention also includes any novel intermediates described herein, for example those of formulae (IXA), (IXB), (VIIA), (VIIB), (VIII), (VIIIA) and (X).
- The present invention additionally comprises the combined administration of a cGMP PDE5 inhibitor of the general formula (I), wherein said combined administration can be in the form of simultaneous, sequential or joint administration with:
(a) one or more naturally occurring or synthetic prostaglandins or esters thereof. Suitable prostaglandins for use herein include compounds such as alprostadil, prostaglandin E1, prostaglandin E0, 13, 14-dihydroprosta glandin E1, prostaglandin E2, eprostinol, natural synthetic and semi-synthetic prostaglandins and derivatives thereof including those described in U.S. Pat. No. 6,037,346 issued on 14 Mar. 2000 and incorporated herein by reference, PGE0, PGE1, PGA1, PGB1, PGF1 α, 19-hydroxy PGA1, 19-hydroxy —PGB1, PGE2, PGB2, 19-hydroxy-PGA2, 19-hydroxy-PGB2, PGE3α, carboprost tromethamine dinoprost, tromethamine, dinoprostone, lipo prost, gemeprost, metenoprost, sulprostune, tiaprost and moxisylate; and/or
(b) one or more α-adrenergic receptor antagonist compounds also known as α-adrenoceptors or α-receptors or α-blockers. Suitable compounds for use herein include: the α-adrenergic receptors as described in PCT application WO99/30697 published on 14 Jun. 1998, the disclosures of which relating to α-adrenergic receptors are incorporated herein by reference and include, selective α1-adrenoceptors or α2-adrenoceptors and non-selective adrenoceptors, suitable α1-adrenoceptors include: phentolamine, phentolamine mesylate, trazodone, alfuzosin, indoramin, naftopidil, tamsulosin, dapiprazole, phenoxybenzamine, idazoxan, efaraxan, yohimbine, rauwolfa alkaloids, Recordati 15/2739, SNAP 1069, SNAP 5089, RS17053, SL 89.0591, doxazosin, terazosin, abanoquil and prazosin; α2-blockers from U.S. Pat. No. 6,037,346 [14 Mar. 2000] dibenamine, tolazoline, trimazosin and dibenamine; α-adrenergic receptors as described in U.S. Pat. Nos. 4,188,390; 4,026,894; 3,511,836; 4,315,007; 3,527,761; 3,997,666; 2,503,059; 4,703,063; 3,381,009; 4,252,721 and 2,599,000 each of which is incorporated herein by reference; α2-Adrenoceptors include: clonidine, papaverine, papaverine hydrochloride, optionally in the presence of a cariotonic agent such as pirxamine; and/or
(c) one or more NO-donor (NO-agonist) compounds. Suitable NO-donor compounds for use herein include organic nitrates, such as mono- di or tri-nitrates or organic nitrate esters including glyceryl brinitrate (also known as nitroglycerin), isosorbide 5-mononitrate, isosorbide dinitrate, pentaerythritol tetranitrate, erythrityl tetranitrate, sodium nitroprusside (SNP), 3-morpholinosydnonimine molsidomine, S-nitroso-N-acetyl penicilliamine (SNAP) S-nitroso-N-glutathione (SNO-GLU), N-hydroxy-L-arginine, amylnitrate, linsidomine, linsidomine chlorohydrate, (SIN-1) S-nitroso—N-cysteine, diazenium diolates, (NONOates), 1,5-pentanedinitrate, L-arginene, ginseng, zizphi fructus, molsidomine, Re-2047, nitrosylated maxisylyte derivatives such as NMI-678-11 and NMI-937 as described in published PCT application WO 0012075; and/or
(d) one or more potassium channel openers. Suitable potassium channel openers for use herein include nicorandil, cromokalim, levcromakalim, lemakalim, pinacidil, cliazoxide, minoxidil, charybdotoxin, glyburide, 4-amini pyridine, BaCl2; and/or
(e) one or more dopaminergic agents. Suitable dopaminergic compounds for use herein include D2-agonists such as, pramipexol; apomorphine; and/or
(f) one or more vasodilator agents. Suitable vasodilator agents for use herein include nimodepine, pinacidil, cyclandelate, isoxsuprine, chloroprumazine, halo peridol, Rec 15/2739, trazodone, pentoxifylline; and/or
(g) one or more thromboxane A2 agonists; and/or
(h) one or more CNS active agents; and/or
(i) one or more ergot alkoloids; Suitable ergot alkaloids are described in U.S. Pat. No. 6,037,346 issued on 14 Mar. 2000 and include acetergamine, brazergoline, bromerguride, cianergoline, delorgotrile, disulergine, ergonovine maleate, ergotamine tartrate, etisulergine, lergotrile, lysergide, mesulergine, metergoline, metergotamine, nicergoline, pergolide, propisergide, proterguride, terguride; and/or
(k) one or more compounds which modulate the action of atrial natruretic factor (also known as atrial naturetic peptide), such as inhibitors or neutral endopeptidase; and/or
(l) one or more compounds which inhibit angiotensin-converting enzyme such as enapril, and combined inhibitors of angiotensin-converting enzyme and neutral endopeptidase such as omapatrilat; and/or
(m) one or more angiotensin receptor antagonists such as losartan; and/or
(n) one or more substrates for NO-synthase, such as L-arginine; and/or
(o) one or more calcium channel blockers such as amlodipine; and/or
(p) one or more antagonists of endothelin receptors and inhibitors or endothelin-converting enzyme; and/or
(q) one or more cholesterol lowering agents such as statins and fibrates; and/or
(r) one or more antiplatelet and antithrombotic agents, e.g. tPA, uPA, warfarin, hirudin and other thrombin inhibitors, heparin, thromboplastin activating factor inhibitors; and/or
(s) one or more insulin sensitising agents such as rezulin and hypoglycaemic agents such as glipizide; and/or
(t) L-DOPA or carbidopa; and/or
(u) one or more acetylcholinesterase inhibitors such as donezipil; and/or
(v) one or more steroidal or non-steroidal anti-inflammatory agents. - The syntheses of the compounds of the invention and of the intermediates for use therein are illustrated by the following Examples and Preparations. A number of the compounds included in the Preparations section are compounds of the formula (I), (IA) or (IB) and are thereby examples of compounds according to the present invention.
- 1H Nuclear magnetic resonance (NMR) spectra were recorded using either a Varian Unity 300 or a Varian Inova 400 spectrometer and were in all cases consistent with the proposed structures. Characteristic chemical shifts (δ) are given in parts-per-million downfield from tetramethylsilane using conventional abbreviations for designation of major peaks: e.g. s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad.
- Mass spectra (m/z) were recorded using a Fisons Instruments Trio mass spectrometer in the thermospray ionisation mode.
- Room temperature means 20 to 25° C.
-
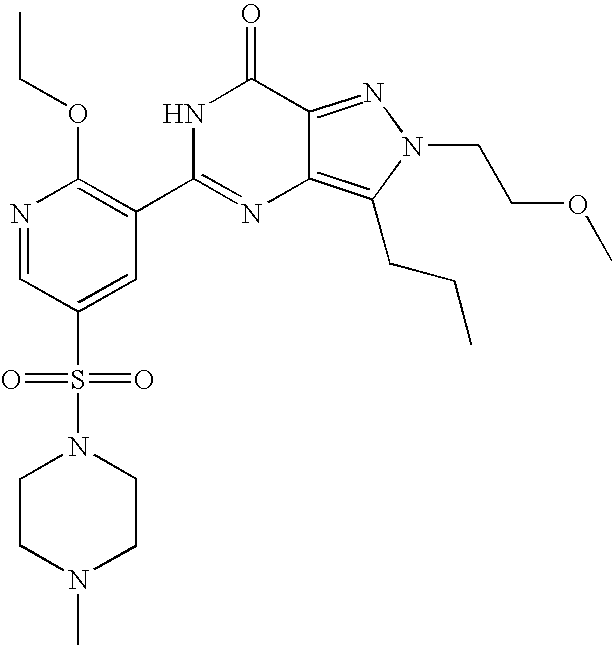
- A mixture of the title compound from preparation 28 (560 mg, 1.04 mmol) and potassium tert-butoxide (292 mg, 2.4 mmol) in ethanol (20 ml) was heated at 100° C. in a sealed vessel for 18 hours. The cooled mixture was concentrated under reduced pressure and the residue partitioned between ethyl acetate and water. The organic phase was separated, dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 220 mg.
- Found: C, 52.65; H, 6.43; N, 18.39. C23H33N7O5S; 0.3H2O requires C, 53.16; H, 6.40; N, 18.87%.
- LRMS: m/z 520 (M+1)+
-

- A mixture of the title compound from preparation 27 (420 mg, 0.80 mmol) and potassium bis(trimethylsilyl)amide (240 mg, 1.20 mmol) in ethanol (40 ml) was heated at 100° C. for 18 hours in a sealed vessel. The cooled mixture was concentrated under reduced pressure and the residue purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 90:10) to give the title compound, 130 mg.
- δ (CDCl3): 1.40 (3H, t), 1.58 (3H, t), 2.27 (3H, s), 2.50 (4H, m), 3.10 (6H, m), 3.30 (3H, s), 3.92 (2H, t), 4.45 (2H, t), 4.75 (2H, q), 8.62 (1H, d), 9.02 (1H, d), 10.65 (1H, s).
- LRMS: m/z 506 (M+1)+
-

- A mixture of the title compound from preparation 30 (740 mg, 1.34 mmol) and potassium bis(trimethylsilyl)amide (321.5 mg, 1.61 mmol) in ethanol (40 ml) was heated at 100° C. for 18 hours in a sealed vessel. Tlc analysis showed starting material remaining, so additional potassium bis(trimethylsilyl)amide (321.5 mg, 1.61 mmol) was added, and the reaction continued for a further 18 hours. The cooled mixture was concentrated under reduced pressure and the residue partitioned between water and ethyl acetate, and the layers separated. The organic phase was evaporated under reduced pressure and the crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 90:10) to give the title compound, 150 mg.
- δ (CDCl3): 1.02 (6H, m), 1.58 (3H, t), 1.83 (2H, m), 2.41 (2H, q), 2.56 (4H, m), (2H, t), 3.14 (4H, m), 3.29 (3H, s), 3.90 (2H, t), 4.44 (2H, t), 4.75 (2H, q), (1H, s), 9.02 (1H, s), 10.61 (1H, s).
- LRMS: m/z 534 (M+1)+
-

- A mixture of the title compound from preparation 39 (400 mg, 0.75 mmol), potassium bis(trimethylsilyl)amide (298 mg, 1.50 mmol) and ethyl acetate (73 μl, 0.75 mmol) in ethanol (10 ml) was heated at 120° C. in a sealed vessel for 12 hours. The cooled mixture was partitioned between ethyl acetate and aqueous sodium bicarbonate solution, and the layers separated. The organic phase was dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (98:2) as eluant to afford the title compound, 164 mg.
- δ (CDCl3): 0.79 (3H, t), 1.02 (3H, t), 1.38 (3H, t), 1.56 (6H, m), 1.90 (1H, m), 2.21 (1H, m), 2.41 (2H, q), 2.57 (4H, m), 2.98-3.18 (6H, m), 4.41 (1H, m), 4.75 (2H, q), 8.61 (1H, s), 9.02 (1H, s), 10.58 (1H, s).
- The compounds of the following tabulated examples, of general structure:
-
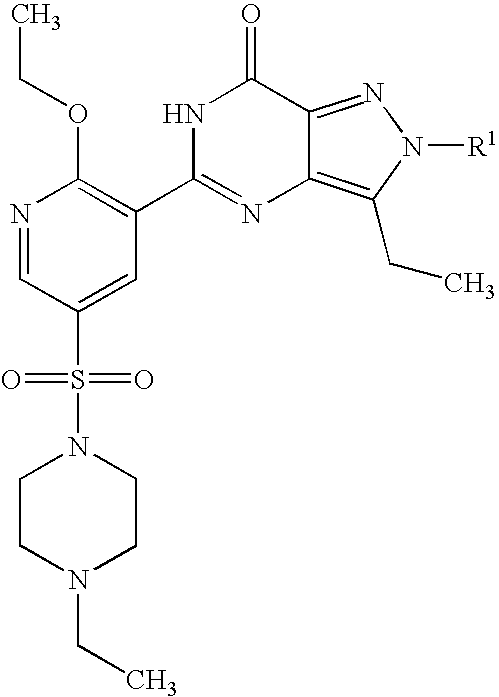
- were prepared from the corresponding carboxamides, following a similar procedure to that described in example 4.
-
Exam- Yield ple R1 (%) Data 5 
23 δ (CDCl3): 0.97 (6 H, d), 1.02 (3 H, t), 1.40 (3 H, t), 1.58 (3 H, t), 2.41 (3 H, m), 2.56 (4 H, m), 3.01 (2 H, q), 3.14 (4 H, m), 4.10 (2 H, d), 4.75 (2 H, q), 8.61 (1 H, s), 9.02 (1 H, s), 10.61 (1 H, s). 61 
28 δ (CDCl3): 0.47 (2 H, m), 0.63 (2 H, m), 1.01 (3 H, t), 1.40 (3 H, t), 1.48-1.72 (4 H, m), 2.45 (2 H, q), 2.56 (4 H, m), 3.04 (2 H, q), 3.15 (4 H, m), 3.47 (2 H, q), 4.20 (2 H, d), 4.76 (2 H, q), 8.61 (1 H, s), 9.02 (1 H, s), 10.60 (1 H, s). LRMS: m/z 516 (M + 1)+ 71 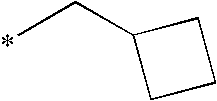
48 δ (CDCl3): 1.01 (3 H, t), 1.20 (3 H, t), 1.40 (3 H, t), 1.56 (4 H, m), 1.88 (4 H, m), 2.07 (2 H, m), 2.40 (2 H, q), 2.56 (4 H, m), 3.00 (2 H, m), 3.15 (4 H, m), 4.34 (2 H, d), 4.76 (2 H, q), 8.61 (1 H, s), 9.02 (1 H, s), 10.60 (1 H, s). LRMS: m/z 530 (M + 1)+ 82 
27 Found: C, 53.18; H, 6.48; N, 18.14. C23H33N7O5S; 0.20C2H5CO2CH3 requires C, 53.21; H, 6.49; N, 18.25%. δ (CDCl3): 1.04 (3 H, t), 1.40 (3 H, t), 1.58 (3 H, t), 2.41 (2 H, q), 2.57 (4 H, m), 3.08 (2 H, q), 3.14 (4 H, m), 3.30 (3 H, s), 3.92 (2 H, t), 4.46 (2 H, t), 4.75 (2 H, q), 8.62 (1 H, d), 9.04 (1 H, d), 10.61 (1 H, s). LRMS: m/z 520 (M + 1)+ mp 161-162° C. 9 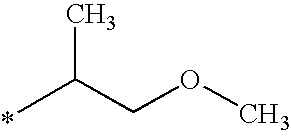
47 δ (CDCl3): 1.02 (3 H, t), 1.38 (3 H, t), 1.58 (6 H, m), 2.41 (2 H, q), 2.57 (4 H, m), 3.05 (2 H, m), 3.14 (4 H, m), 3.22 (3 H, s), 3.72 (1 H, m), 3.96 (1 H, dd), 4.73 (3 H, m), 8.61 (1 H, s), 9.02 (1 H, s), 10.56 (1 H, s). LRMS: m/z 534 (M + 1)+ 1= Purified by ether trituration 2= additionally, recrystallised from ethyl acetate -
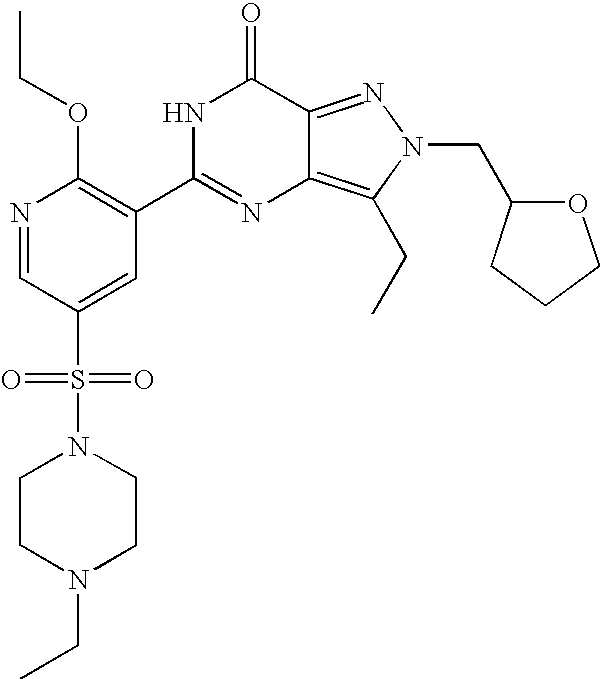
- A mixture of the title compound from preparation 42 (250 mg, 0.44 mmol), potassium bis(trimethylsilyl)amide (132 mg, 0.66 mmol) and ethyl acetate (40 μl, 0.41 mmol) in 3-methyl-3-pentanol (4 ml) was heated at 120° C. in a sealed vessel for 18 hours. Tlc analysis showed starting material remaining, so additional potassium bis(trimethylsilyl)amide (132 mg, 0.66 mmol) was added and the reaction heated under reflux for a further 24 hours. The cooled mixture was evaporated under reduced pressure, and the residue purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to give the title compound, 60 mg.
- δ (CDCl3): 1.03 (3H, t), 1.40 (3H, t), 1.58 (3H, t), 1.84 (3H, m), 2.08 (1H, m), 2.41 (2H, q), 2.56 (4H, m), 3.14 (6H, m), 3.70-3.90 (2H, m), 4.30-4.50 (3H, m), 4.75 (2H, q), 8.62 (1H, s), 9.02 (1H, s), 10.62 (1H, s).
- LRMS: m/z 546 (M+1)+
-

- A mixture of the title compound from preparation 48 (300 mg, 0.52 mmol), potassium bis(trimethylsilyl)amide (320 mg, 1.57 mmol) and ethyl acetate (50 μl, 0.52 mmol) in ethanol (40 ml) was heated at 130° C. in a sealed vessel for 18 hours. The cooled mixture was concentrated under reduced pressure and the residue partitioned between water and dichloromethane and the layers separated. The aqueous phase was extracted with dichloromethane, and the combined organic solutions were dried (Na2SO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 90:10), and triturated with ethyl acetate to afford the title compound as a white solid, 80 mg.
- δ (CDCl3): 1.01 (3H, t), 1.18 (3H, t), 1.57 (3H, t), 2.41 (2H, q), 2.58 (6H, m), 3.14 (4H, m), 4.77 (6H, m), 6.08 (1H, m), 6.96 (1H, d), 7.57 (1H, d), 8.62 (1H, d), 9.00 (1H, d), 10.67 (1H, s).
- LRMS: m/z 556 (M+1)+
-
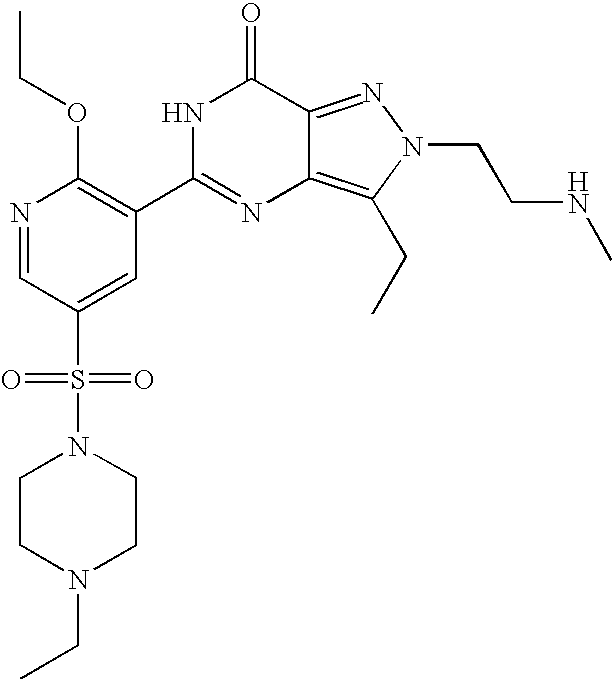
- A mixture of the title compound of preparation 54 (130 mg, 0.24 mmol) and potassium bis(trimethylsilyl)amide (58 mg, 0.29 mmol) in ethanol (6 ml) was heated at 130° C. for 16 hours in a sealed vessel. The cooled mixture was concentrated under reduced pressure, the residue suspended in sodium bicarbonate solution (15 ml) and extracted with ethyl acetate (3×15 ml). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The residual gum was purified by column chromatography on silica gel twice, using dichloromethane:methanol:0.88 ammonia (89:10:1) as eluant and repeated using ethyl acetate:methanol:diethylamine (78:20:2) as eluant to afford the title compound, 32 mg, as a beige foam.
- δ CDCl3): 1.02 (3H, t), 1.41 (3H, t), 1.58 (3H, t), 2.41 (2H, q), 2.56 (7H, m), 3.10 (6H, m), 3.27 (2H, t), 4.47 (2H, t), 4.77 (2H, q), 8.61 (1H, s), 9.00 (1H, s), 10.50-10.80 (1H, br s).
- LRMS: m/z 519 (M+1)+
- The following tabulated examples of the general structure:
-

- were prepared from the corresponding carboxamides, following a similar procedure to that described in example 12.
-
Exam- Yield ple R1 (%) 1Hnmr 131 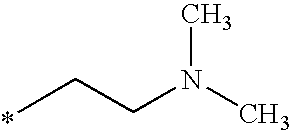
81 δ (CDCl3): 1.02 (3 H, t), 1.42 (3 H, t), 1.58 3 H, t), 2.30 (6 H, s), 2.41 (2 H, q), 2.56 (4 H, m), 2.90 (2 H, t), 3.05 (2 H, q), 3.14 (4 H, m), 4.40 (2 H, t), 4.75 (2 H, q), 8.61 (1 H, s), 9.02 (1 H, s), 10.62 (1 H, s). 141 
21 δ (CDCl3): 1.03 (3 H, t), 1.40 (3 H, t), 1.44 (9 H, s), 1.58 (3 H, t), 2.41 (2 H, q), 2.54-2.68 (7 H, m), 3.01 (2 H, q), 3.16 (4 H, m), 3.78 (2 H, t), 4.47 (2 H, m), 4.78 (2 H, q), 8.63 (1 H, s), 9.04 (1 H, s), 10.66 (1 H, br s). 151 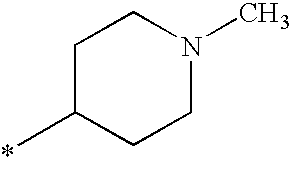
58 δ (CDCl3): 1.02 (3 H, t), 1.40 (3 H, t), 1.58 (3 H, t), 1.93 (2 H, m), 2.16 (2 H, m), 2.36 (3 H, s), 2.41 (2 H, q), 2.56 (6 H, m), 3.04 (4 H, m), 3.14 (4 H, m), 4.22 (1 H, m), 4.77 (2 H, q), 8.62 (1 H, d), 9.01 (1 H, d), 10.54 (1 H, s). 1= column eluant of dichloromethane:methanol -

- A mixture of the title compound from preparation 53 (470 mg, 0.86 mmol) and potassium bis(trimethylsilyl)amide (600 mg, 3.0 mmol) in ethanol (45 ml) was heated at 130° C. for 16 hours. The cooled mixture was concentrated under reduced pressure, the solution diluted with aqueous sodium bicarbonate solution to give pH 8, and extracted with ethyl acetate (3×). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (91.75:7.5:0.75) as eluant to give the title compound, 170 mg.
- δ (CDCl3): 1.02 (3H, t), 1.38 (3H, t), 1.58 (3H, m), 2.40 (2H, q), 2.50 (3H, s), 2.57 (4H, m), 3.01 (2H, q), 3.16 (4H, m), 3.79 (2H, t), 3.90 (2H, t), 4.78 (2H, q), 5.12 (1H, m), 8.62 (1H, d), 9.01 (1H, d), 10.62 (1H, s).
-

- A mixture of the title compound from preparation 55 (150 mg, 0.27 mmol) and potassium bis(trimethylsilyl)amide (109 mg, 0.55 mmol) in n-butanol (5 ml) was heated at 120° C. for 16 hours in a sealed vessel. The cooled reaction mixture was poured into saturated aqueous sodium bicarbonate solution, and extracted with ethyl acetate. The combined organic extracts were dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (90:10) as eluant to afford the title compound as a white foam, 27 mg.
- δ (CDCl3): 1.02 (6H, m), 1.42 (3H, t), 1.57 (2H, m), 1.95 (2H, m), 2.30 (6H, s), 2.41 (2H, q), 2.57 (4H, m), 2.90 (2H, t), 3.05 (2H, q), 3.16 (4H, m), 4.40 (2H, t), 4.66 (2H, t), 8.61 (1H, d), 9.01 (1H, t), 10.60 (1H, s).
-
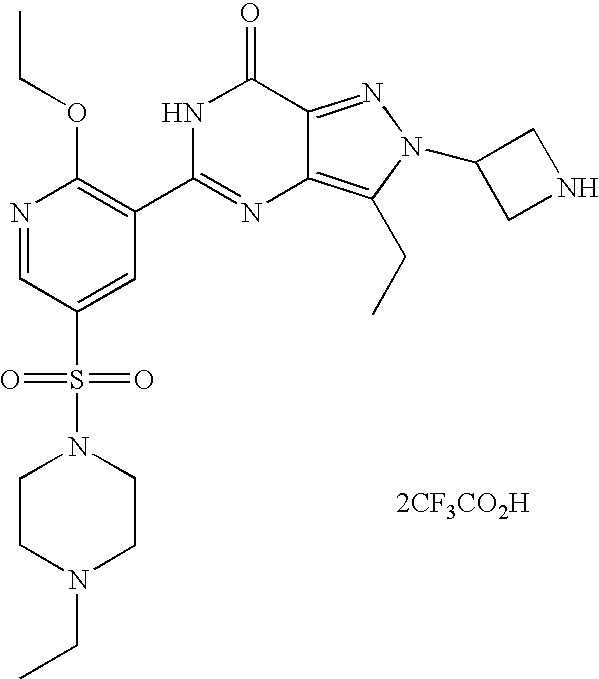
- Trifluoroacetic acid (3 ml) was added to a solution of the title compound from preparation 63 (350 mg, 0.57 mmol) in dichloromethane (3 ml), and the reaction stirred at room temperature for 2½ hours. The reaction was concentrated under reduced pressure and the residual gum was triturated several times with ether. The resulting suspension was sonicated for a minute, then the solid filtered, washed with ether, and dried to give the title compound as a white powder, 280 mg.
- Found: C, 42.82; H, 4.80; N, 14.92. C23H32N8O4S; 2CF3CO2H; H2O requires C, 42.52; H, 4.76; N, 14.69%.
-

- The title compound was obtained as a beige-coloured powder, (51%), from the title compound of preparation 66, following a similar procedure to that described in example 18.
- δ (DMSOd6): 0.86 (3H, t), 1.07-1.46 (12H, m), 2.41-3.50 (12H, m), 4.49 (4H, m), 5.38 (1H, m), 5.68 (1H, m), 8.26 (1H, s), 8.74 (1H, s), 9.00 (1H, m), 9.26 (1H, m), 11.96 (1H, s).
-

- The title compound was obtained as a white solid, (79%) from the title compound from preparation 61 and trifluoroacetic acid, following the procedure described in example 18.
- δ (DMSOd6): 0.94 (3H, t), 1.12 (3H, m), 1.26 (3H, t), 1.73 (2H, m), 2.41 (6H, m), 2.60 (3H, s), 2.68-3.60 (7H, m), 4.39 (2H, t), 4.60 (2H, t), 8.23 (1H, s), 8.57 (2H, m), 8.74 (1H, s), 11.94 (1H, s).
-

- Sodium triacetoxyborohydride (81 mg, 0.38 mmol) was added to a solution of the title compound from example 18 (215 mg, 0.28 mmol), acetaldehyde (17.3 μl, 0.31 mmol), acetic acid (16 μl, 0.28 mmol) and triethylamine (7.9%, 0.28 mmol) in tetrahydrofuran (6 ml), and the reaction stirred at room temperature for 16 hours. The reaction mixture was diluted with saturated aqueous sodium bicarbonate solution (30 ml), and this mixture extracted with ethyl acetate (2×30 ml). The combined organic extracts were dried (MgSO4), and evaporated under reduced pressure. The residual gum was purified by column chromatography on silica gel using dichloromethane:methanol (90:10) as eluant, to give the title compound, 120 mg.
- δ (CDCl3): 1.04 (6H, m), 1.38 (3H, t), 1.58 (3H, t), 2.41 (2H, q), 2.57 (4H, m), 2.68 (2H, q), 3.01 (2H, q), 3.15 (4H, m), 3.76 (2H, m), 3.95 (2H, m), 4.76 (2H, q), 5.16 (1H, m), 8.63 (1H, d), 9.02 (1H, d), 10.68 (1H, s).
-
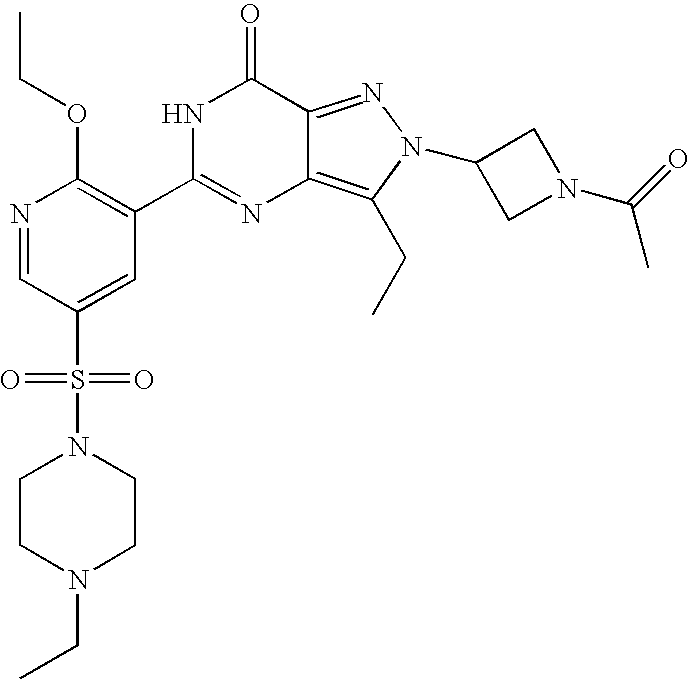
- Acetyl chloride (6 mg, 0.076 mmol) was added to a mixture of the title compound from example 18 (43 mg, 0.056 mmol) and triethylamine (8.5 mg, 0.086 mmol) in dichloromethane (2 ml), and the reaction stirred for 36 hours at room temperature. The mixture was treated with aqueous saturated sodium bicarbonate solution and extracted with ethyl acetate (2×). The combined organic extracts were dried (MgSO4), and evaporated under reduced pressure. The residual gum was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (97:3 to 95:5) to give the title compound, 19 mg.
- δ(CDCl3): 1.02 (3H, t), 1.38 (3H, t), 1.60 (3H, t), 1.98 (3H, s), 2.42 (2H, q), 2.58 (4H, m), 3.02 (2H, q), 3.16 (4H, m), 4.50 (2H, m), 4.59 (1H, m), 4.78 (2H, q), 5.05 (1H, m), 5.31 (1H, m), 8.62 (1H, d), 9.01 (1H, d), 10.70 (1H, s).
-

- The title compound was obtained (30%) from the compound of preparation 68, and acetyl chloride, following the procedure described in example 22.
- δ(CDCl3): 1.02 (3H, t), 1.40 (3H, t), 1.56 (3H, t), 2.00 (2H, m), 2.17 (3H, s), 2.23-2.44 (4H, m), 2.55 (4H, m), 2.78 (1H, m), 3.09 (6H, m), 3.27 (1H, m), 4.06 (1H, m), 4.50 (1H, m), 4.70-4.90 (3H, m), 8.62 (1H, d), 9.02 (1H, d), 10.60 (1H, s).
-

- The title compound was obtained (74%) from the compound of example 20, and acetyl chloride, following the procedure described in example 22.
- δ (CDCl3): 1.02 (3H, t), 1.14 (3H, t), 1.40 (3H, t), 1.99 (2H, m), 2.06 (3H, s), 2.42 (2H, q), 2.57 (4H, m), 2.80 (3H, s), 3.01 (2H, q), 3.16 (4H, m), 3.93 (2H, t), 4.50 (2H, t), 4.62 (2H, t), 8.62 (1H, d), 9.04 (1H, d), 10.66 (1H, s).
-

- The title compound was obtained (33%) from the title compound from preparation 68 and methanesulphonic anhydride, following a similar procedure to that described in example 22.
- δ (CDCl3): 1.02 (3H, t), 1.40 (3H, t), 1.58 (3H, t), 2.10 (2H, m), 2.40 (2H, q), 2.56 (6H, m), 2.90 (3H, s), 3.00-3.20 (8H, m), 4.01 (2H, m), 4.21 (1H, m), 4.78 (2H, q), 8.62 (1H, d), 9.01 (1H, s), 10.61 (1H, s).
-

- Trifluoroacetic acid (0.5 ml) was added to a solution of the title compound from preparation 65 (28 mg, 0.043 mmol) in dichloromethane (0.5 ml), and the solution stirred for 2½ hours at room temperature. The mixture was evaporated under reduced pressure and the residue triturated with ether several times. The resulting precipitate was filtered off, washed with ether and dried, to give a beige-coloured solid.
- Acetyl chloride (16 μl, 0.22 mmol) was added to a solution of this intermediate in dichloromethane (3 ml) and triethylamine (61 μl, 0.44 mmol), and the reaction stirred at room temperature for 16 hours. Aqueous saturated sodium bicarbonate solution (10 ml) was added, and the mixture extracted with ethyl acetate. The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure to give a gum. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (98:2 to 95:5) to give the title compound, 7 mg.
- δ (CDCl3): 1.02 (6H, m), 1.38 (3H, t), 1.57 (2H, m), 1.94 (5H, m), 2.40 (2H, q), 2.47 (4H, m), 3.02 (2H, q), 3.14 (4H, m), 4.50 (2H, m), 4.59 (1H, m), 4.67 (2H, m), 5.06 (1H, m), 5.31 (1H, m), 8.62 (1H, d), 9.01 (1H, d), 10.68 (1H, s).
-

- Potassium bis(trimethylsilyl)amide (149.7 mg, 0.75 mmol) was added to a solution of the title compound of example 3 (80 mg, 0.15 mmol) in 2-methyl-n-propanol (5 ml) and the reaction stirred at 120° C. for 18 hours. The cooled mixture was concentrated under reduced pressure and the residue suspended in water (10 ml), and extracted with ethyl acetate (3×10 ml). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel, using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 67 mg, as a solid.
- Found: C, 54.92; H, 7.08; N, 16.92. C26H39N7O5S; 0.7H2O requires C, 54.38; H, 7.09; N, 17.07%.
- δ (CDCl3): 1.03 (6H, m), 1.14 (6H, d), 1.83 (2H, m), 2.30 (1H, m), 2.41 (2H, q), 2.55 (4H, m), 3.01 (2H, t), 3.13 (4H, m), 3.30 (3H, s), 3.90 (2H, t), 4.46 (4H, m), 8.61 (1H, s), 9.01 (1H, s), 10.60 (1H, s).
- LRMS: m/z 562 (M+1)+
- The compounds of the following tabulated examples of general formula:
-
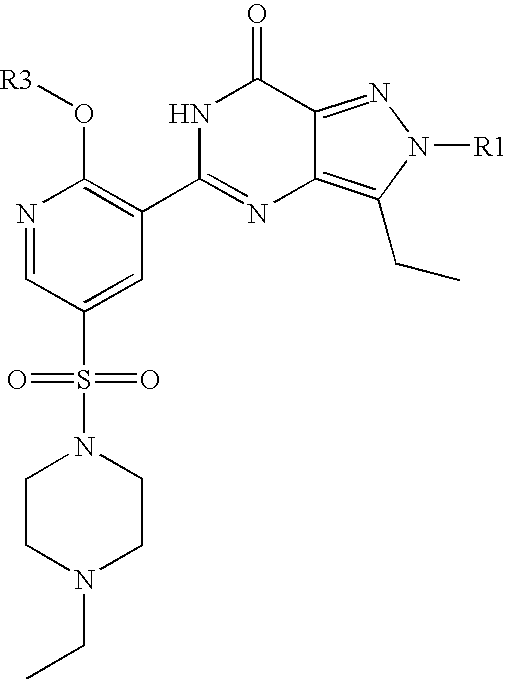
- were prepared from the appropriate 2-ethoxypyridin-3-ylpyrazolo[4,3-d]pyrimidinone and alcohol, following similar procedures to that described in example 27.
-
Ex R1 R3 Data 28 

Found: C, 54.88; H, 7.08; N, 17.13, C26H39N7O5S; 0.6H2O requires C, 54.55; H, 7.08; N, 17.13% δ (CDCl3): 1.02 (6 H, 2xt), 1.40 (3 H, t), 1.56 (2 H, m), 1.83 (2 H, m), 1.94 (2 H, m), 2.41 (2 H, q), 2.55 (4 H, m), 3.00 (2 H, t), 3.16 (4 H, m), 3.30 (3 H, s), 3.92 (2 H, t), 4.45 (2 H, t), 4.67 (2 H, t), 8.61 (1 H, s), 9.01 (1 H, s), 10.60 (1 H, s). LRMS: m/z 562 (M + 1)+ 29 

Found: C, 52.90; H, 6.79; N, 16.86, C25H37N7O6S requires C, 53.27; H, 6.62; N, 17.36% δ (CDCl3): 1.02 (6 H, m), 1.84 (2 H, m), 2.42 (2 H, q), 2.56 (4 H, m), 3.01 (2 H, t), 3.15 (4 H, m), 3.29 (3 H, s), 3.57 (3 H, s), 3.88 (4 H, m), 4.44 (2 H, t), 4.78 (2 H, t), 8.61 (1 H, s), 8.98 (1 H, s), 10.76 (1 H, s). LRMS: m/z 564 (M + 1)+ 30 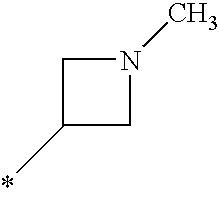
n-Bu δ (CDCl3): 1.02 (6 H, t), 1.38 (3 H, t), 1.57 (2 H, m), 1.96 (2 H, m), 2.41 (2 H, q), 2.50 (3 H, s), 2.56 (4 H, m), 3.00 (2 H, q), 3.15 (4 H, m), 3.79 (2 H, t), 3.94 (2 H, t), 4.68 (2 H, t), 5.12 (1 H, m), 8.62 (1 H, d), 9.01 (1 H, d), 10.61 (1 H, s). 31 

δ (CDCl3): 1.01 (3 H, t), 1.37 (3 H, t), 2.40 (2 H, q), 2.55 (7 H, m), 3.00 (2 H, q), 3.13 (4 H, m), 3.25 (2 H, t), 3.80 (2 H, t), 3.95 (2 H, t), 4.88 (2 H, t), 5.12 (1 H, m), 7.22 (2 H, m), 7.38 (3 H, m), 8.62 (1 H, d), 9.00 (1 H, d), 10.49 (1 H, s). 32 
n-Bu δ (CDCl3): 1.02 (9 H, t), 1.38 (3 H, t), 1.57 (2 H, m), 1.96 (2 H, m), 2.41 (2 H, q), 2.56 (4 H, m), 2.67 (2 H, q), 3.01 (2 H, q), 3.15 (4 H, m), 3.74 (2 H, t), 3.90 (2 H, t), 4.68 (2 H, t), 5.17 (1 H, m), 8.62 (1 H, d), 9.01 (1 H, d), 10.60 (1 H, s). 33 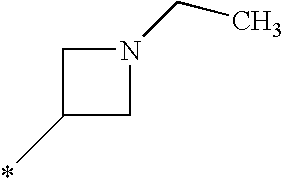

δ (CDCl3): 1.02 (6 H, m), 1.37 (3 H, t), 2.41 (2 H, q), 2.57 (4 H, m), 2.69 (2 H, q), 3.01 (2 H, q), 3.15 (4 H, m), 3.76 (2 H, t), 3.95 (2 H, t), 5.18 (1 H, m), 5.77 (2 H, s), 7.38 (3 H, m), 7.50 (2 H, m), 8.63 (1 H, d), 9.00 (1 H, d), 10.59 (1 H, br s).
wherein for examples 28 and 29 R2 is n-propyl and for examples 30 to 33 R2 is ethyl. -

- Potassium bis(trimethylsilyl)amide (306 mg, 1.54 mmol) was added to a solution of the title compound of example 2 (155 mg, 0.31 mmol) in 2-methyl-n-propanol (10 ml) and the reaction stirred under reflux for 24 hours. The cooled mixture was evaporated under reduced pressure and the residue purified by column chromatography on silica gel, using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 88 mg, as a solid.
- Found: C, 52.45; H, 6.43; N, 17.33. C24H35N7O5S; 1.1H2O requires C, 52.08; H, 6.77; N, 17.71%.
- δ (CDCl3): 1.14 (6H, d), 1.41 (3H, t), 2.30 (4H, m), 2.52 (4H, m), 3.07 (2H, q), 3.15 (4H, m), 3.30 (3H, s), 3.92 (2H, t), 4.46 (4H, m), 8.62 (1H, s), 9.03 (1H, S).
- LRMS: m/z 534 (M+1)+
- The compounds of the following tabulated examples of the general formula:
-
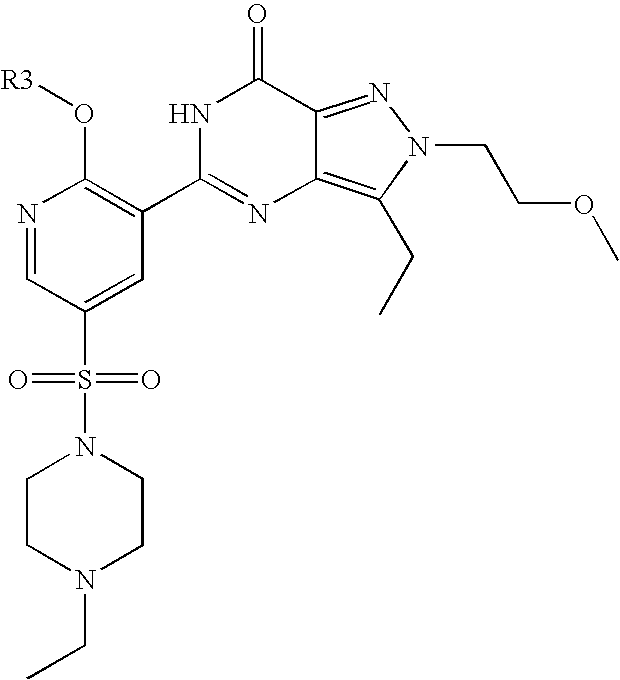
- were prepared from the title compound of example 8 and the appropriate alcohol, following similar procedures to that described in example 34.
-
Ex R3 Data 35 
δ (CDCl3): 1.02 (3 H, t), 1.40 (3 H, t), 1.52 (3 H, t), 1.98 (2 H, m), 2.40 (2 H, q), 2.57 (4 H, m), 3.14 (6 H, m), 3.32 (3 H, s), 3.94 (2 H, t), 4.46 (2 H, t), 4.62 (2 H, t), 8.61 (1 H, s), 9.02 (1 H, s), 10.62 (1 H, s). LRMS: m/z 534 (M + 1)+ 36 
δ (CDCl3): 1.04 (6 H, 2xt), 1.40 (3 H, t), 1.55 (2 H, m), 1.95 (2 H, m), 2.42 (2 H, q), 2.55 (4 H, m), 3.07 (2 H, q), 3.15 (4 H, m), 3.30 (3 H, s), 3.92 (2 H, t), 4.46 (2 H, t), 4.66 (2 H, t), 8.62 (1 H, s), 9.02 (1 H, s), 10.60 (1 H, s). LRMS: m/z 548 (M + 1)+ 37 
Found: C, 54.77; H, 6.82; N, 17.75. C25H37N7O5S requires C, 54.83; H, 6.81; N, 17.90% δ (CDCl3): 1.02 (3 H, t), 1.12 (6 H, d), 1.40 (3 H, t), 2.30 (1 H, m), 2.42 (2 H, q), 2.57 (4 H, m), 3.08 (2 H, q), 3.13 (4 H, m), 3.30 (3 H, s), 3.90 (2 H, t), 4.46 (4 H, m), 8.62 (1 H, s), 9.02 (1 H, s), 10.60 (1 H, s). LRMS: m/z 548 (M + 1)+ 381 
Found: C, 54.76; H, 6.79; N, 17.72. C25H37N7O5S requires C, 54.83; H, 6.81; N, 17.90% δ (CDCl3): 1.03 (6 H, m), 1.40 (3 H, t), 1.50 (3 H, d), 1.85 (1 H, m), 1.98 (1 H, m), 2.41 (2 H, q), 2.58 (4 H, m), 3.07 (2 H, q), 3.15 (4 H, m), 3.30 (3 H, s), 3.92 (2 H, t), 4.47 (2 H, t), 5.57 (1 H, m), 8.61 (1 H, s), 9.03 (1 H, s), 10.65 (1 H, s). LRMS: m/z 548 (M + 1)+ 391 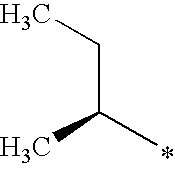
Found: C, 55.03; H, 6.97; N, 16.84. C25H37N7O5S requires C, 54.83; H, 6.81; N, 17.90% δ (CDCl3): 1.04 (6 H, t), 1.40 (3 H, t), 1.50 (3 H, d), 1.83 (1 H, m), 1.98 (1 H, m), 2.42 (2 H, q), 2.58 (4 H, m), 3.07 (2 H, q), 3.15 (4 H, m), 3.30 (3 H, s), 3.92 (2 H, t), 4.46 (2 H, t), 5.55 (1 H, m), 8.61 (1 H, s), 9.04 (1 H, s), 10.64 (1 H, s). LRMS: m/z 548 (M + 1)+ 40 
Found: C, 54.91; H, 5.91; N, 18.85. C27H34N8O5S; 0.5H2O requires C, 54.81; H, 5.96; N, 18.94% δ (CDCl3): 1.02 (3 H, t), 1.42 (3 H, t), 2.42 (2 H, q), 2.57 (4 H, m), 3.12 (6 H, m), 3.30 (3 H, s), 3.94 (2 H, t), 4.46 (2 H, t), 5.90 (2 H, s), 7.35 (2 H, m), 7.78 (1 H, m), 8.59 (1 H, s), 8.84 (2 H, m), 12.70 (1 H, s). LRMS: m/z 583 (M + 1)+ 1= purified using an elution gradient of ethyl acetate:methanol (95:5 to 90:10), followed by ether trituration -

- A solution of the title compound from example 4 (129 mg, 0.25 mmol) in 2-methoxyethanol (10 ml) was heated at 110° C. for 15 minutes, then cooled. Potassium bis(trimethylsilyl)amide (249 mg, 1.50 mmol) was added and the reaction stirred at 130° C. for 22 hours. The cooled mixture was partitioned between ethyl acetate and aqueous sodium bicarbonate solution, and the layers separated. The organic phase was dried (MgSO4) and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using dichloromethane:methanol (98:2) as eluant to afford the title compound as a yellow foam, 59 mg.
- δ (CDCl3): 0.79 (3H, t), 1.03 (3H, t), 1.39 (3H, t), 1.60 (3H, d), 1.90 (1H, m), 2.22 (1H, m), 2.41 (2H, q), 2.57 (4H, m), 2.97-3.18 (6H, m), 3.57 (3H, s), 3.85 (2H, m), 4.40 (1H, m), 4.78 (2H, m), 8.62 (1H, s), 8.98 (1H, s), 10.76 (1H, s).
- LRMS: m/z 548 (M+1)+
-

- The title compound was obtained as a white solid (64%) from the title compound from example 7 and 2-methoxyethanol, following the procedure described in example 41.
- δ (CDCl3): 1.01 (3H, t), 1.40 (3H, t), 1.80-1.98 (5H, m), 2.05 (2H, m), 2.40 (2H, q), 2.54 (4H, m), 3.00 (2H, m), 3.15 (4H, m), 3.55 (3H, s), 3.83 (2H, t), 4.30 (2H, d), 4.76 (2H, t), 8.60 (1H, s), 8.96 (1H, d), 10.74 (1H, br s).
-

- The title compound was obtained as a yellow foam (57%) from the title compound from example 9 and 2-methoxyethanol, following the procedure described in example 41.
- δ (CDCl3): 1.02 (3H, t), 1.38 (3H, t), 1.59 (3H, d), 2.41 (2H, q), 2.56 (4H, m), 3.05 (6H, m), 3.22 (3H, s), 3.56 (3H, s), 3.72 (1H, m), 3.84 (2H, m), 3.96 (1H, dd), 4.71 (1H, m), 4.78 (2H, m), 8.61 (1H, s), 8.97 (1H, s), 10.78 (1H, br s).
-

- A mixture of the title compound from example 8 (250 mg, 0.48 mmol) and potassium bis(trimethylsilyl)amide (480 mg, 2.41 mmol) in 1-methoxy-2-propanol (20 ml) was heated at 120° C. for 18 hours. The cooled mixture was evaporated under reduced pressure and the residue purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to give a white solid. This material was purified by HPLC, using a Chiralpak AD 250 column, with hexane:1% diethylamine in iso-propanol (85:15) as eluant, to give the title compound of example 44, 49 mg,
- δ (CDCl3): 1.02 (3H, t), 1.40 (3H, t), 1.50 (3H, m), 2.42 (2H, q), 2.57 (4H, m), 3.06 (2H, m), 3.15 (4H, m), 3.30 (3H, s), 3.55 (3H, s), 3.64 (1H, m), 3.76 (1H, m), 3.92 (2H, t), 4.45 (2H, t), 5.60 (1H, m), 8.60 (1H, s), 8.90 (1H, s), 10.80 (1H, s).
- LRMS: m/z 564 (M+1)+
- and the title compound of example 45, 39 mg.
- δ (CDCl3): 1.04 (3H, t), 1.40 (3H, t), 1.50 (3H, d), 2.42 (2H, q), 2.57 (4H, m), 3.07 (2H, q), 3.16 (4H, m), 3.29 (3H, s), 3.56 (3H, s), 3.64 (1H, m), 3.75 (1H, m), 3.90 (2H, t), 4.45 (2H, t), 5.60 (1H, m), 8.60 (1H, s), 8.90 (1H, s), 10.80 (1H, s).
- LRMS: m/z 564 (M+1)+
-

- A mixture of the title compound from example 3 (345 mg, 0.65 mmol) and potassium bis(trimethylsilyl)amide (645 mg, 3.24 mmol) in 1-methoxy-2-propanol (2.5 ml) was heated at 110° C. for 16 hours. The cooled mixture was diluted with ethyl acetate, then washed with aqueous ammonium chloride solution, then water, dried (MgSO4), and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using dichloromethane:methanol (97:3) as eluant to give a yellow gum.
- This material was purified by HPLC, using a Chiralpak AD 250 column, with hexane:1% diethylamine in iso-propanol (85:15) as eluant, to give the title compound of example 46, 17 mg,
- δ (CDCl3): 1.02 (6H, m), 1.50 (3H, d), 1.81 (2H, m), 2.41 (2H, q), 2.56 (4H, m), 3.00 (2H, m), 3.14 (4H, m), 3.28 (3H, s), 3.55 (3H, s), 3.62-3.78 (2H, m), 3.90 (2H, t), 4.44 (2H, t), 5.60 (1H, m), 8.60 (1H, s), 8.89 (1H, s), 10.80 (1H, s).
- LRMS: m/z 578 (M+1)+
- and the title compound of example 47, 64 mg.
- δ (CDCl3): 1.01 (6H, m), 1.48 (3H, d), 1.81 (2H, m), 2.40 (2H, q), 2.54 (4H, m), 2.99 (2H, t), 3.10 (4H, m), 3.27 (3H, s), 3.51 (3H, s), 3.60-3.76 (2H, m), 4.87 (2H, t), 4.44 (2H, t), 5.59 (1H, m), 8.60 (1H, s), 8.86 (1H, s).
- LRMS: m/z 578 (M+1)+
-
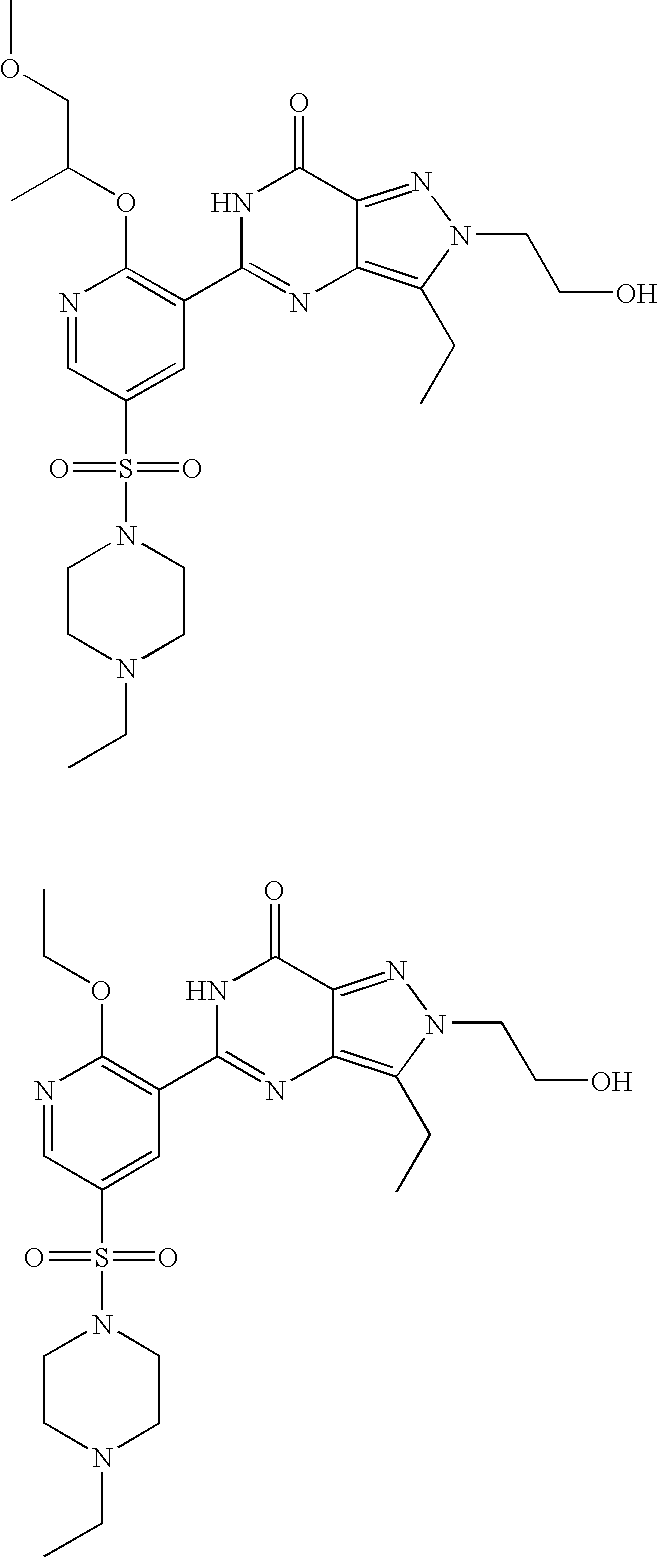
- Potassium bis(trimethylsilyl)amide (200 mg, 1.0 mmol) was added to a solution of the title compound from preparation 60 (120 mg, 0.2 mmol) in 1-methoxy-2-propanol (10 ml), and the reaction heated under reflux for 18 hours. The cooled mixture was evaporated under reduced pressure and the residue purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 90:10) to give the title compound of example 48, 8 mg.
- δ (CDCl3): 1.02 (3H, t), 1.40 (3H, t), 1.50 (3H, d), 2.41 (2H, q), 2.58 (4H, m), 3.10 (7H, m), 3.58 (3H, s), 3.70 (2H, m), 4.20 (2H, m), 4.40 (2H, m), 5.59 (1H, m), 8.61 (1H, d), 8.88 (1H, d), 10.90 (1H, s).
- LRMS: m/z 550 (M+1)+
- and the title compound of example 49 as a white solid.
- δ (CDCl3): 1.02 (3H, t), 1.40 (3H, t), 1.58 (3H, t), 2.41 (2H, q), 2.56 (4H, m), 2.87 (1H, br s), 3.02-3.19 (6H, m), 4.22 (2H, m), 4.42 (2H, t), 4.77 (2H, q), 8.62 (1H, s), 9.02 (1H, s), 10.66 (1H, s).
- LRMS: m/z 506 (M+1)+
-

- Potassium bis(trimethylsilyl)amide (359 mg, 1.8 mmol) was added to a solution of the title compound from preparation 70 (250 mg, 0.45 mmol) in 2-methoxyethanol (5 ml), and the reaction heated under reflux for 6 hours. Tlc analysis showed starting material remaining, so additional potassium bis(trimethylsilyl)amide (90 mg, 0.45 mmol) was added to the cooled mixture, and the reaction stirred for a further 4 hours under reflux. The cooled mixture was concentrated under reduced pressure and the residue purified by column chromatography on silica gel using dichloromethane:methanol (95:5) as eluant. The product was triturated with ether and pentane to afford the title compound as a crystalline solid, 75 mg.
- Found: C, 52.88; H, 6.59; N, 17.39. C25H37N7O6S requires C, 53.27; H, 6.62; N, 17.39%.
- d (CDCl3): 1.02 (3H, t), 1.12 (3H, t), 1.40 (3H, t), 2.41 (2H, q), 2.57 (5H, m), 3.06 (2H, q), 3.15 (4H, m), 3.42 (2H, q), 3.57 (3H, s), 3.85 (2H, t), 3.94 (2H, t), 4.44 (2H, t), 4.78 (2H, t), 8.61 (1H, s), 8.98 (1H, s), 10.78 (1H, s).
-

- Potassium bis(trimethylsilyl)amide (732 mg, 3.68 mmol) was added to a solution of the title compound from preparation 40 (958 mg, 1.84 mmol) in 2-methoxyethanol (20 ml) and the reaction stirred for 16 hours at 120° C. The cooled mixture was concentrated under reduced pressure, the residue dissolved in water (25 ml) and the pH adjusted to 2 using hydrochloric acid (2N). The solution was washed with ethyl acetate, neutralised and the resulting precipitate filtered off. The solid was dissolved in ethyl acetate, evaporated under reduced pressure and the crude product was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (95:5:0.5) as eluant to give the title compound, 53 mg.
- δ (CDCl3): 0.97 (6H, d), 1.40 (3H, t), 2.28 (4H, m), 2.52 (4H, m), 3.02 (2H, q), 3.16 (4H, m), 3.57 (3H, s), 3.86 (2H, t), 4.10 (2H, d), 4.78 (2H, t), 8.61 (1H, d), 8.98 (1H, d), 10.79 (1H, s).
- LRMS: m/z 534 (M+1)+
-

- Potassium bis(trimethylsilyl)amide (1.85 g, 9.35 mmol) was added to a solution of the title compound from preparation 36 (1.0 g, 1.89 mmol) in 2-methoxyethanol (8 ml) and the reaction stirred for 18 hours at 120° C. The cooled mixture was concentrated under reduced pressure, and the residue partitioned between water (200 ml) and dichloromethane (200 ml). The resulting precipitate was filtered off, and the layers separated. The aqueous phase was extracted with dichloromethane (2×200 ml), and the combined organic solutions evaporated under reduced pressure, to give a cream solid. The isolated solids were combined and purified by column chromatography on silica gel using dichloromethane:methanol (90:10) as eluant to give the title compound as a pale yellow solid, 220 mg.
- δ (CDCl3): 0.95 (6H, d), 1.05 (3H, t), 1.40 (3H, d), 2.40 (3H, m), 2.55 (4H, m), 3.00 (2H, q), 3.10 (4H, m), 3.55 (3H, s), 3.85 (2H, t), 5.05 (2H, d), 4.80 (2H, t), 8.60 (1H, s), 8.95 (1H, s), 10.80 (1H, s).
- LRMS: m/z 549 (M+1)+
-
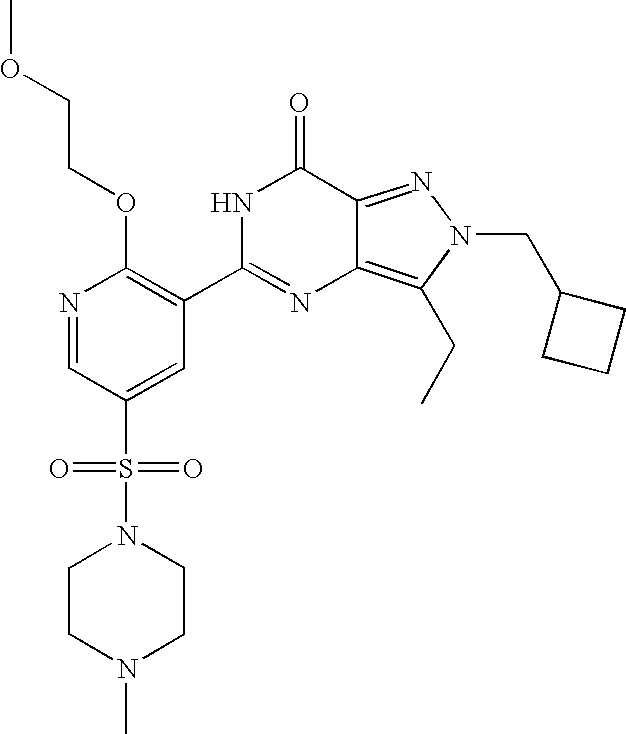
- The title compound was obtained as a beige solid (31%) from the title compound from preparation 41 and 2-methoxyethanol, using a similar procedure to that described 52.
- δ (CDCl3): 1.41 (3H, t), 1.88 (4H, m), 2.07 (2H, m), 2.26 (3H, s), 2.52 (4H, m), 3.00 (3H, m), 3.15 (4H, m), 3.57 (3H, s), 3.86 (2H, m), 4.33 (2H, d), 4.79 (2H, t), 8.62 (1H, s), 8.98 (1H, s), 10.75 (1H, s).
-

- A mixture of the title compound from preparation 52 (90 mg, 0.156 mmol), potassium bis(trimethylsilyl)amide (156 mg, 0.78 mmol) and ethyl acetate (14 mg, 0.156 mmol) in iso-propanol (12 ml) was stirred at 130° C. for 6 hours in a sealed vessel. The cooled reaction mixture was poured into saturated aqueous sodium bicarbonate solution (60 ml), and extracted with ethyl acetate (60 ml). The combined organic extracts were dried (MgSO4), and evaporated under reduced pressure to give a gum. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (92.6:6.6:0.6) to afford the title compound as a beige foam, 36 mg.
- δ (CDCl3): 1.01 (3H, t), 1.12 (6H, d), 1.39 (3H, t), 1.94 (2H, m), 2.15 (2H, m), 2.22-2.44 (6H, m), 2.55 (6H, m), 3.02 (4H, m), 3.14 (4H, m), 4.22 (1H, m), 4.43 (2H, d), 8.60 (1H, d), 9.00 (1H, d), 10.54 (1H, s).
- The compounds of the following tabulated examples of general formula:
-

- were prepared from the appropriate carboxamide and alcohol, following similar procedures to that described in example 54.
-
Ex. R1 R3 Data 55 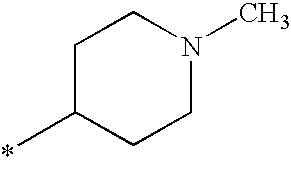
n-Bu δ (CDCl3): 1.02 (6 H, m), 1.40 (3 H, t), 1.57 (2 H, m), 1.94 (4 H, m), 2.16 (2 H, m), 2.37 (3 H, s), 2.41 (2 H, q), 2.56 (6 H, m), 3.03 (4 H, m), 3.15 (4 H, m), 4.22 (1 H, m), 4.66 (2 H, t), 8.62 (1 H, d), 9.01 (1 H, d), 10.55 (1 H, s). 56 

δ (CDCl3): 1.02 (3 H, t), 1.12 (6 H, d), 1.42 (3 H, t), 2.31 (7 H, m), 2.42 (2 H, q), 2.57 (4 H, m), 2.90 (2 H, t), 3.06 (2 H, q), 3.16 (4 H, m), 4.38-4.47 (4 H, m), 8.61 (1 H, d), 9.01 (1 H, d), 10.60 (1 H, s). 57 
n-Bu δ (CDCl3): 1.01 (6 H, t), 1.40 (3 H, t), 1.56 (2 H, m), 1.95 (2 H, m), 2.17 (2 H, m), 2.21 (6 H, s), 2.24 (2 H, t), 2.40 (2 H, q), 2.57 (4 H, m), 3.06 (2 H, q), 3.17 (4 H, m), 4.37 (2 H, t), 4.65 (2 H, t), 8.61 (1 H, d), 9.02 (1 H, d), 10.59 (1 H, s). 58 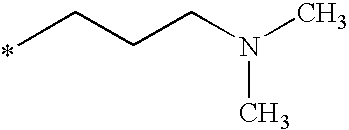
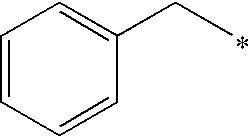
δ (CDCl3): 1.02 (3 H, t), 1.40 (3 H, t), 2.17 (2 H, m), 2.21 (6 H, s), 2.27 (2 H, t), 2.40 (2 H, q), 2.57 (4 H, m), 3.05 (2 H, q), 3.17 (4 H, m), 4.37 (2 H, t), 5.77 (2 H, s), 7.39 (3 H, m), 7.52 (2 H, m), 8.63 (1 H, d), 9.01 (1 H, d), 10.54 (1 H, s). -

- Sodium hydride (13 mg, 60% dispersion in mineral oil, 0.33 mmol) was added to a solution of the title compound from preparation 59 (145 mg, 0.30 mmol) in tetrahydrofuran (2 ml) and the solution stirred for 30 minutes. 2-Chloro-N,N-dimethylacetamide (40 mg, 0.034 mmol) was added and the reaction stirred at room temperature for 16 hours, followed by a further 16 hours at 60° C. The cooled mixture was treated with aqueous sodium bicarbonate solution (15 ml) and extracted with ethyl acetate (2×15 ml). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (96.5:3.5) as eluant, and repeated using ethyl acetate:diethylamine (90:10) as eluant, to afford the title compound of example 59, 20 mg,
- δ (CDCl3): 1.03 (6H, t), 1.41 (3H, t), 1.59 (2H, m), 1.95 (2H, m), 2.41 (2H, q), 2.57 (4H, m), 3.00 (5H, m), 3.15 (7H, m), 4.66 (2H, t), 5.44 (2H, s), 8.63 (1H, d), 9.10 (1H, d), 10.85 (1H, s).
- and the title compound of example 60, 45 mg.
- δ (CDCl3): 1.01 (6H, t), 1.42 (3H, t), 1.55 (2H, m), 1.94 (2H, m), 2.40 (2H, q), 2.55 (4H, m), 3.00 (5H, m), 3.14 (7H, m), 4.64 (2H, t), 5.19 (2H, s), 8.61 (1H, d), 9.01 (1H, d), 10.58 (1H, s).
-
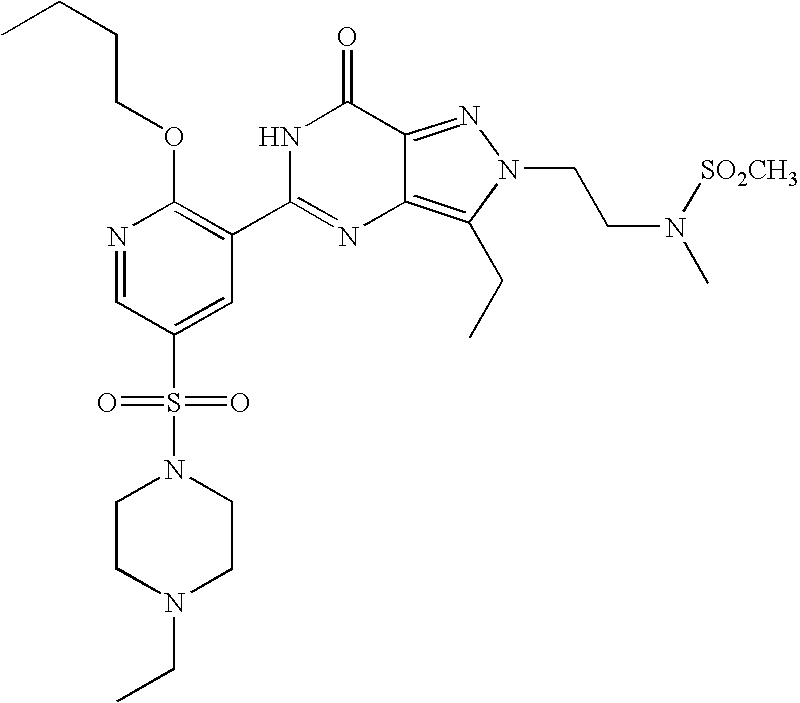
- Trifluoroacetic acid (1 ml) was added to a solution of the title compound from preparation 62 (76 mg, 0.117 mmol) in dichloromethane (1 ml), and the solution stirred for 2½ hours at room temperature. The mixture was evaporated under reduced pressure, the residue triturated well with ether, and the resulting precipitate, filtered and dried to give a white powder.
- Methanesulphonyl chloride (20 μl, 0.26 mmol) was added to a solution of this intermediate in dichloromethane (2 ml) and triethylamine (65 μl, 0.47 mmol), and the reaction stirred at room temperature for 1½ hours. The mixture was treated with saturated aqueous sodium bicarbonate solution (10 ml), and extracted with ethyl acetate (2×10 ml). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure to give a gum.
The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (96:4) as eluant to afford the title compound as a beige foam, 30 mg. - δ (CDCl3): 1.02 (6H, t), 1.42 (3H, t), 1.54 (2H, m), 1.94 (2H, m), 2.41 (2H, q), 2.57 (4H, m), 2.65 (3H, s), 2.80 (3H, s), 3.13 (6H, m), 3.76 (2H, t), 4.52 (2H, t), 4.67 (2H, t), 8.62 (1H, d), 9.04 (1H, d), 10.68 (1H, s).
-

- The title compound was obtained as a white solid, (34%), from the title compound from preparation 67 and methanesulphonyl chloride, following the procedure described in example 61.
- δ (CDCl3): 1.01 (6H, t), 1.40 (3H, t), 1.55 (2H, m), 1.95 (2H, m), 2.08 (2H, m), 2.42 (2H, q), 2.57 (6H, m), 2.90 (3H, s), 3.01-3.18 (8H, m), 4.01 (2H, m), 4.42 (1H, m), 4.66 (2H, t), 8.62 (1H, d), 9.01 (1H, d), 10.60 (1H, s).
-

- Potassium bis(trimethylsilyl)amide (134 mg, 0.67 mmol) was added to a suspension of the title compound from preparation 49 (200 mg, 0.33 mmol) and ethyl acetate (50 μl, 0.51 mmol) in ethanol (5 ml) and the reaction mixture heated at 120° C. in a sealed vessel for 12 hours. The cooled reaction was concentrated under reduced pressure and the residue partitioned between ethyl acetate and water, and the layers separated. The aqueous phase was extracted with ethyl acetate, the combined organic solutions dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (98:2) as eluant to give the title compound as a yellow oil, 10 mg.
- δ (CDCl3): 1.02 (3H, t), 1.36 (3H, t), 1.60 (3H, t), 2.41 (2H, q), 2.57 (4H, m), 3.17 (6H, m), 4.78 (2H, q), 7.82 (2H, d), 8.42 (2H, d), 8.66 (1H, d), 9.07 (1H, d), 10.78 (1H, br s).
- LRMS: m/z 583 (M+1)+
-

- A solution of the title compound from example 63 (100 mg, 0.17 mmol) in methanol (2 ml) was added to a suspension of iron powder (29 mg, 0.52 mmol) in ammonium chloride (45 mg, 0.85 mmol) in water (2 ml), and the reaction heated at 60° C. for 1 hour. The cooled mixture was filtered, and the filtrate evaporated under reduced pressure to give the title compound as a pale brown solid, 93 mg.
- δ (CDCl3): 1.02 (3H, t), 1.26 (3H, t), 1.59 (3H, t), 2.41 (2H, q), 2.57 (4H, m), 3.03 (2H, q), 3.16 (4H, m), 3.94 (2H, s), 4.77 (2H, q), 6.78 (2H, d), 7.27 (2H, d), 8.63 (1H, d), 9.07 (1H, d), 10.66 (1H, s).
- LRMS: m/z 553 (M+1)+
-

- Methanesulphonyl chloride (15 μl, 0.19 mmol) was added to an ice-cooled solution of the title compound from example 64 (93 mg, 0.17 mmol) in pyridine (2 ml), and the reaction allowed to warm to room temperature, and stirred for 90 minutes. Tlc analysis showed starting material remaining, so additional methanesulphonyl chloride (15 μl, 0.19 mmol) was added, and the reaction stirred for a further hour. The reaction was quenched by the addition of aqueous ammonium chloride solution, and extracted with ethyl acetate. The combined organic extracts were dried (MgSO4) and concentrated under reduced pressure. The residual solid was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (90:10:1) as eluant, then repeated using dichloromethane:methanol:0.88 ammonia (95:5:1) to afford the title compound, 36 mg.
- δ (CDCl3): 1.03 (3H, t), 1.34 (3H, t), 1.59 (3H, t), 2.42 (2H, q), 2.58 (4H, m), 3.14 (9H, m), 4.78 (2H, q), 6.92 (1H, s), 7.44 (2H, d), 7.58 (2H, d), 8.65 (1H, d), 9.07 (1H, d), 10.75 (1H, s).
- LRMS: m/z 631 (M+1)+
-

- Pyridine (0.1 ml, 1.08 mmol) was added to a mixture of the title compound from preparation 58 (250 mg, 0.54 mmol), copper (II) acetate monohydrate (145 mg, 0.72 mmol), benzeneboronic acid (132 mg, 1.08 mmol) and 4 Å molecular sieves (392 mg) in dichloromethane (5 ml), and the reaction stirred at room temperature for 4 days. The reaction mixture was filtered and the filtrate evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (97:3:0.5) as eluant, and triturated with ether:hexane. The resulting solid was filtered and recrystallised from iso-propanol:dichloromethane to give the title compound as a solid, 200 mg.
- δ (CDCl3): 1.02 (3H, t), 1.47 (3H, t), 1.60 (3H, t), 2.42 (2H, q), 2.58 (4H, m), 3.10 (2H, q), 3.17 (4H, m), 4.76 (2H, q), 7.40 (1H, m), 7.51 (2H, m), 7.80 (2H, d), 8.67 (1H, d), 9.16 (1H, s), 10.90 (1H, s).
- LRMS: m/z 538 (M+1)+
-

- A mixture of the title compound from preparation 58 (100 mg, 0.22 mmol), copper (II) acetate monohydrate (58 mg, 0.29 mmol), 4-cyanobenzeneboronic acid (63 mg, 0.44 mmol) and 4 Å molecular sieves (156 mg) in pyridine (1 ml) and N-methylpyrrolidine (1 ml) was irradiated by microwave at full power for 3×10 seconds, followed by 2×20 seconds. The mixture was evaporated under reduced pressure and the residue purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (95:5:0.5) as eluant. The product was recrystallised from dichloromethane:iso-propanol to give the title compound, 45 mg.
- δ (CDCl3): 1.03 (3H, t), 1.49 (3H, t), 1.62 (3H, t), 2.42 (2H, q), 2.58 (4H, m), 3.08 (2H, q), 3.17 (4H, m), 4.58 (2H, q), 7.79 (2H, d), 8.14 (2H, d), 8.70 (1H, d), 9.16 (1H, d), 11.09 (1H, s).
- LRMS: m/z 563 (M+1)+
-

- Tris(dibenzylideneacetone)dipalladium (0) (8 mg, 0.009 mmol), R-BINAP (8 mg, 0.013 mmol), sodium tert-butoxide (41 mg, 0.43 mmol) and 2-bromopyridine (50 μl, 0.52 mmol) were added to a solution of the title compound from preparation 58 (200 mg, 0.43 mmol) in toluene (3 ml), and the reaction heated at 70° C. for 16 hours. The cooled mixture was evaporated under reduced pressure and the residue filtered through silica gel, using dichloromethane:methanol (80:20) as eluant. The product was purified by reverse phase HPLC on silica gel, using an elution gradient of acetonitrile:0.1% aqueous trifluoroacetic acid (5:95 to 85:15) to afford the title compound, as a solid, 13 mg.
- δ (CDCl3): 1.36 (3H, t), 1.48 (3H, t), 1.57 (3H, t), 3.00 (2H, m), 3.14 (6H, m), 3.70 (2H, m), 3.96 (2H, m), 4.77 (2H, q), 7.52 (1H, m), 8.15-8.26 (2H, m), 8.69 (2H, m), 8.92 (1H, d), 10.80-11.00 (1H, s).
- LRMS: m/z 539 (M+1)+
-
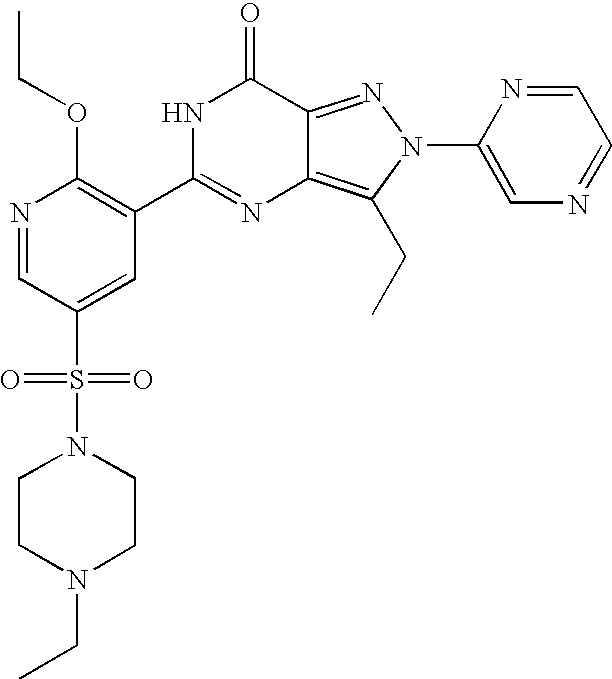
- Cesium carbonate (353 mg, 1.09 mmol) followed by 2-chloropyrazine (100 μl, 1.12 mmol) were added to a solution of the title compound from preparation 58 (500 mg, 1.08 mmol) in N,N-dimethylformamide (10 ml), and the reaction heated at 120° C. for 18 hours. The cooled mixture was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (98:2:1) as eluant. The product was further purified by reverse phase HPLC on silica gel, using an elution gradient of acetonitrile: 0.1% aqueous trifluoroacetic acid (5:95 to 50:50) to afford the title compound, 86 mg.
- δ (CDCl3): 1.38 (6H, 2xt), 1.58 (3H, t), 2.98-3.22 (6H, m), 3.54 (2H, q), 3.76 (2H, m), 4.00 (2H, m), 4.78 (2H, q), 8.57-8.74 (3H, m), 8.98 (1H, d), 9.57 (1H, s).
- LRMS: m/z 540 (M+1)+
-
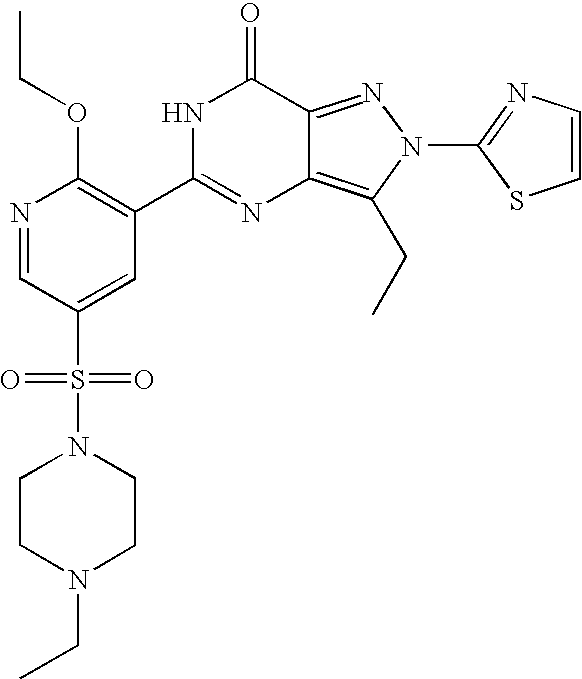
- The title compound was obtained, (7%) from the title compound from preparation 58 and 2-bromothiazole, following a similar procedure to that described in example 69.
- δ (CD3OD): 1.28-1.41 (6H, m), 1.48 (3H, t), 3.20-3.34 (6H, m), 3.34-3.60 (6H, m), 4.65 (2H, q), 7.59 (1H, d), 7.78 (1H, d), 8.58 (1H, d), 8.78 (1H, d).
- LRMS: m/z 545 (M+1)+
-
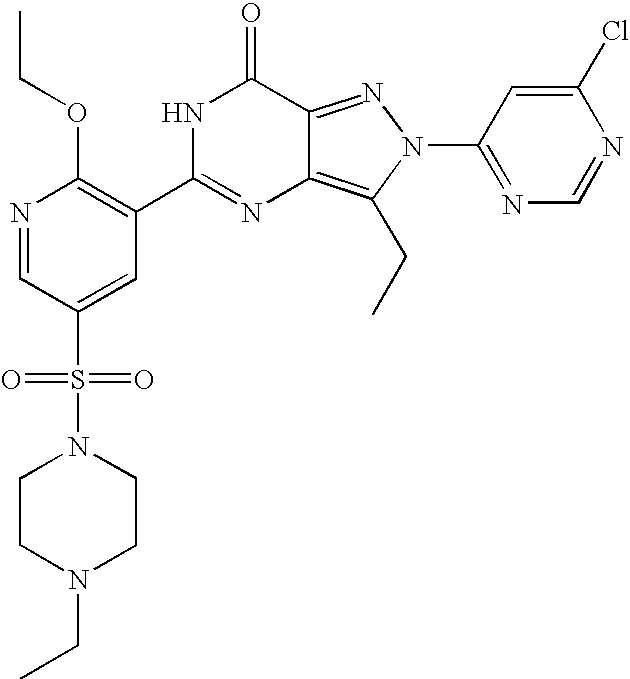
- Sodium hydride (22 mg, 60% dispersion in mineral oil, 0.55 mmol) was added to an ice-cooled solution of the title compound from preparation 58 (250 mg, 0.54 mmol) in tetrahydrofuran (5 ml), and the solution then allowed to warm to room temperature. 4,6-Dichloropyrimidine (80 mg, 0.54 mmol) was added, and the reaction stirred at 65° C. for 18 hours. The cooled mixture was evaporated under reduced pressure and the residue purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (95:5:0.5) as eluant. The product was triturated with dichloromethane to afford the title compound as a pale yellow solid, 5 mg.
- δ (CDCl3): 1.02 (3H, t), 1.40 (3H, t), 1.60 (3H, t), 2.42 (2H, q), 2.58 (4H, m), 3.16 (4H, m), 3.62 (2H, q), 4.78 (2H, q), 8.40 (1H, s), 8.67 (1H, d), 8.97 (1H, s), 9.10 (1H, d), 10.79 (1H, s).
- LRMS: m/z 574, 576 (M+1)+
-

- The title compound was obtained (8%), from the compound from preparation 58 and 2-chloropyrimidine, following a similar procedure to that described in example 71.
- Found: C, 53.33; H, 5.36; N, 23.12. C24H29N9O4S requires C, 53.42; H, 5.42; N, 23.36%.
- δ (CDCl3): 1.03 (3H, t), 1.37 (3H, t), 1.59 (3H, t), 2.41 (2H, q), 2.58 (4H, m), 3.17 (4H, m), 3.55 (2H, q), 4.78 (2H, q), 7.42 (1H, m), 8.64 (1H, d), 8.95 (2H, d), 9.11 (1H, d), 10.73 (1H, s).
- LRMS: m/z 540 (M+1)+
-
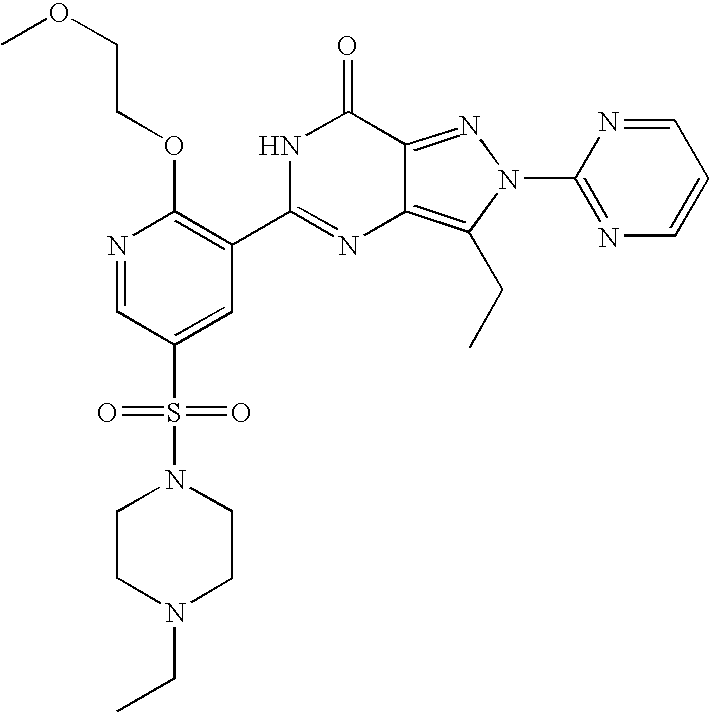
- Sodium hydride (19 mg, 60% dispersion in mineral oil, 0.48 mmol) was added to an ice-cold solution of the title compound from preparation 69 (200 mg, 0.41 mmol) in tetrahydrofuran (4 ml), and the solution stirred for 30 minutes. 2-Chloropyrimidine (56 mg, 0.48 mmol) was added, and the reaction heated under reflux for 18 hours. The mixture was evaporated under reduced pressure, the residue diluted with water, and extracted with dichloromethane. The combined organic extracts were dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to give the title compound, 31 mg.
- δ (CDCl3): 1.02 (3H, t), 1.36 (3H, t), 2.42 (2H, q), 2.58 (4H, m), 3.18 (4H, m), 3.50 (2H, q), 3.58 (3H, s), 3.88 (2H, t), 4.80 (2H, t), 7.42 (1H, m), 8.64 (1H, d), 8.95 (2H, d), 9.02 (1H, d), 10.82 (1H, s).
- LRMS: m/z 570 (M+1)+
-

- The title compound was obtained (35%), from the title compound from preparation 69 and 2-chlorobenzoxazole, following the procedure described in example 73.
- δ (CDCl3): 1.02 (3H, t), 1.50 (3H, t), 2.42 (2H, q), 2.58 (4H, m), 3.18 (4H, m), 3.59 (3H, s), 3.62 (2H, q), 3.87 (2H, t), 4.80 (2H, t), 7.43 (2H, m), 7.64 (1H, m), 7.80 (1H, m), 8.65 (1H, d), 9.02 (1H, d), 10.98 (1H, s).
- LRMS: m/z 609 (M+1)+
-
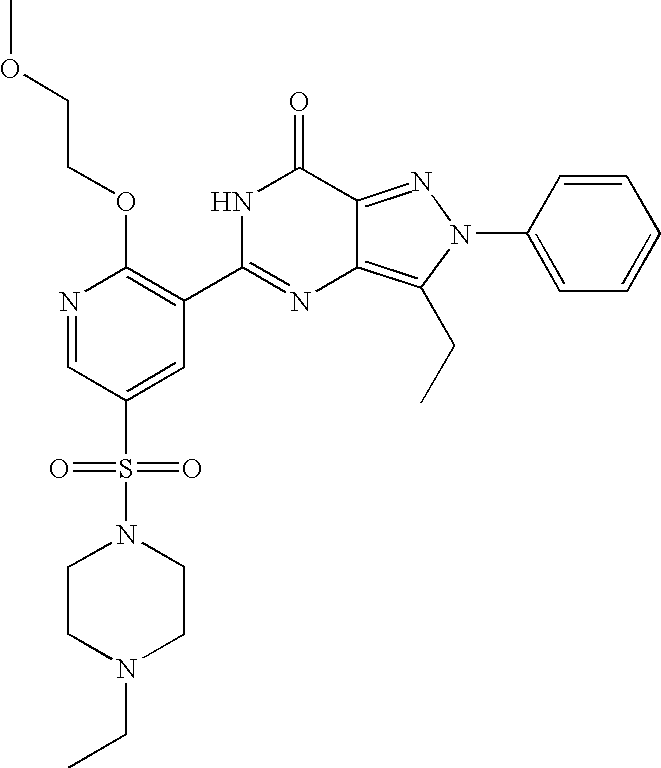
- Potassium bis(trimethylsilyl)amide (294 mg, 1.47 mmol) was added to a solution of the compound from example 66 (200 mg, 0.37 mmol) in 2-methoxyethanol (10 ml), and the reaction heated under reflux for 18 hours. The mixture was evaporated under reduced pressure and the residue purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (97:3:0.5) as eluant. The product was recrystallised from dichloromethane:iso-propanol to give the desired compound as a white solid, 82 mg.
- Found: C, 57.06; H, 5.83; N, 17.27. C27H35N7O5S requires C, 57.13; H, 5.86; N, 17.27%.
- δ (CDCl3): 1.02 (3H, t), 1.46 (3H, t), 2.42 (2H, q), 2.57 (4H, m), 3.16 (2H, q), 3.17 (4H, m), 3.56 (3H, s), 3.84 (2H, t), 4.58 (2H, t), 7.38 (1H, m), 7.48 (2H, m), 7.80 (2H, m), 8.64 (1H, m), 9.04 (1H, m), 11.10 (1H, br s).
- LRMS: m/z 568 (M+1)+
-

- A mixture of the title compound of preparation 57 (440 mg, 0.82 mmol), and potassium bis(trimethylsilyl)amide (196 mg, 0.98 mmol) in ethanol (15 ml) was heated at 100° C. for 18 hours, in a sealed vessel. The cooled mixture was concentrated under reduced pressure, the residue partitioned between ethyl acetate (20 ml) and brine (10 ml), and the layers separated. The organic phase was separated, dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel, using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 275 mg, as a pale yellow solid.
- δ (CDCl3): 1.02 (3H, t), 1.60 (3H, t), 1.86 (2H, m), 2.29 (3H, s), 2.52 (4H, m), 2.95 (2H, t), 3.16 (4H, m), 3.35 (3H, s), 3.87 (2H, t), 4.78 (4H, m), 8.64 (1H, s), 9.09 (1H, s), 10.81 (1H, s).
- LRMS: m/z 520 (M+1)+
-
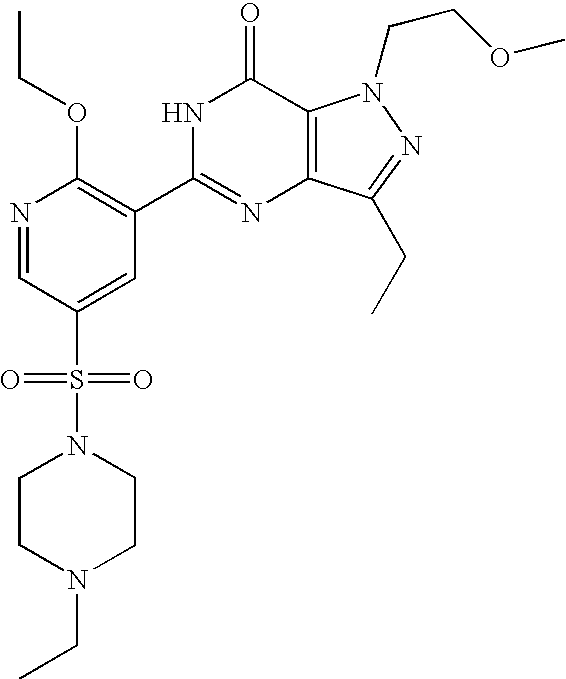
- A mixture of the title compound of preparation 56 (1.02 g, 1.9 mmol) and potassium tert-butoxide (533 mg, 4.75 mmol) in ethanol (40 ml) was heated at 100° C. in a sealed vessel for 18 hours. The cooled mixture was concentrated under reduced pressure and the residue partitioned between ethyl acetate (50 ml) and brine (25 ml), and the layers separated. The organic phase was washed with brine (25 ml), dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel, using an elution gradient of ethyl acetate:methanol (100:0 to 90:10) to afford the title compound, 698 mg, as a pale yellow solid.
- Found: C, 53.00; H, 6.39; N, 18.87 C23H33N7O5S requires C, 53.16; H, 6.40; N, 18.87%.
- δ (CDCl3): 1.03 (3H, t), 1.40 (3H, t), 1.59 (3H, t), 2.41 (2H, q), 2.57 (4H, m), 3.00 (2H, q), 3.15 (4H, m), 3.35 (3H, s), 3.88 (2H, t), 4.78 (4H, m), 8.63 (1H, s), 9.09 (1H, s), 10.83 (1H, s).
- LRMS: m/z 520 (M+1)+
-
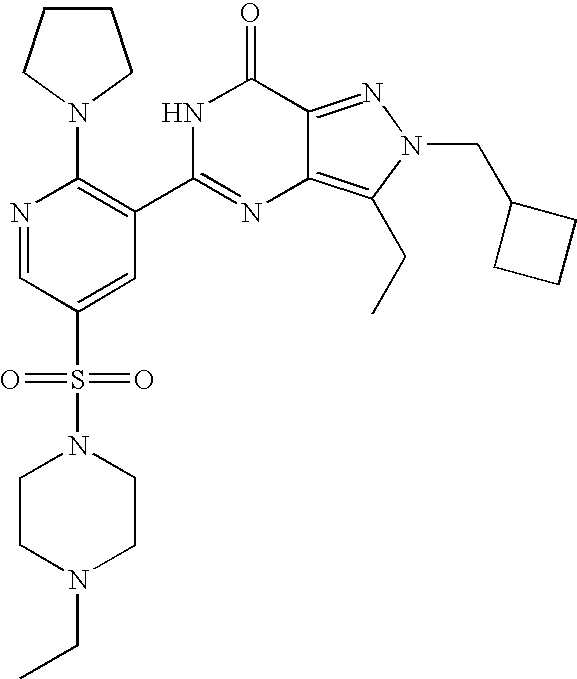
- A mixture of the title compound from example 7 (200 mg, 0.38 mmol) and copper sulphate pentahydrate (74 mg, 0.30 mmol) in pyrrolidine (4 ml) was heated under reflux for 18 hours. The cooled mixture was concentrated under reduced pressure and the residue purified by column chromatography on silica gel twice using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to give the title compound as a pale brown solid, 109 mg.
- δ (CDCl3): 1.04 (3H, m), 1.38 (3H, t), 1.90 (8H, m), 2.10 (2H, m), 2.37-2.68 (5H, m), 3.00 (2H, q), 3.14 (4H, m), 3.42 (4H, m), 4.32 (2H, d), 8.00 (1H, s), 8.58 (1H, s).
- LRMS: m/z 555 (M+1)+
-

- A mixture of the compound from preparation 83 (440 mg, 0.83 mmol), potassium bis(trimethylsilyl)amide (500 mg, 2.51 mmol) and ethyl acetate (100 μl, 1.0 mmol) in ethanol (10 ml) was heated at 120° C. in a sealed vessel for 18 hours. The cooled mixture was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 263 mg.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.01 (3H, t), 1.35 (3H, t), 1.58 (3H, t), 1.96 (2H, m), 2.38-2.60 (8H, m), 2.98 (4H, m), 3.14 (4H, m), 4.76 (2H, q), 4.96 (1H, m), 8.61 (1H, d), 9.02 (1H, d), 10.59 (1H, s).
- LRMS: m/z 516 (MH+)
-

- Potassium bis(trimethylsilyl)amide (450 mg, 2.25 mmol) was added to a suspension of the compound from preparation 84 (243 mg, 0.45 mmol) in ethanol (5 ml), and the mixture heated at 100° C. in a Reactivial® for 24 hours. Tlc analysis showed starting material remaining, so additional potassium bis(trimethylsilyl)amide (250 mg, 1.25 mmol) and ethyl acetate (3 drops) were added, and the reaction heated at 111° C. for 18 hours. The cooled mixture was partitioned between ethyl acetate and sodium bicarbonate solution, and the phases separated. The aqueous layer was extracted with ethyl acetate (2×), the combined organic solutions washed with brine, dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using methanol:dichloromethane (2:98) as eluant, and triturated with ether to afford the title compound as a white powder, 55 mg.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.02 (3H, t), 1.39 (3H, t), 1.55 (3H, t), 1.72 (2H, m), 2.05 (2H, m), 2.17 (2H, m), 2.30 (2H, m), 2.40 (2H, q), 2.56 (4H, m), 3.04 (2H, q), 3.16 (4H, m), 4.76 (2H, q), 4.82 (1H, m), 8.61 (1H, s), 9.02 (1H, s), 10.55 (1H, s).
- LRMS: m/z 530.8 (MH+)
- Anal. Found: C, 57.17; H, 6.65; N, 18.14. C25H35N7O4S requires C, 56.69; H, 6.66; N, 18.51%.
-

- The title compound was obtained as a white powder in 41% yield from the compound from preparation 85, following the procedure described in example 80.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.01 (3H, t), 1.30 (4H, m), 1.40 (3H, t), 1.54 (5H, m), 1.70 (2H, m), 2.40 (2H, q), 2.56 (4H, m), 2.63 (1H, m), 3.02 (2H, q), 3.12 (4H, m), 4.20 (2H, d), 4.74 (2H, q), 8.61 (1H, d), 9.03 (1H, d), 10.60 (1H, s).
- LRMS: m/z 547.7 (MH+)
-
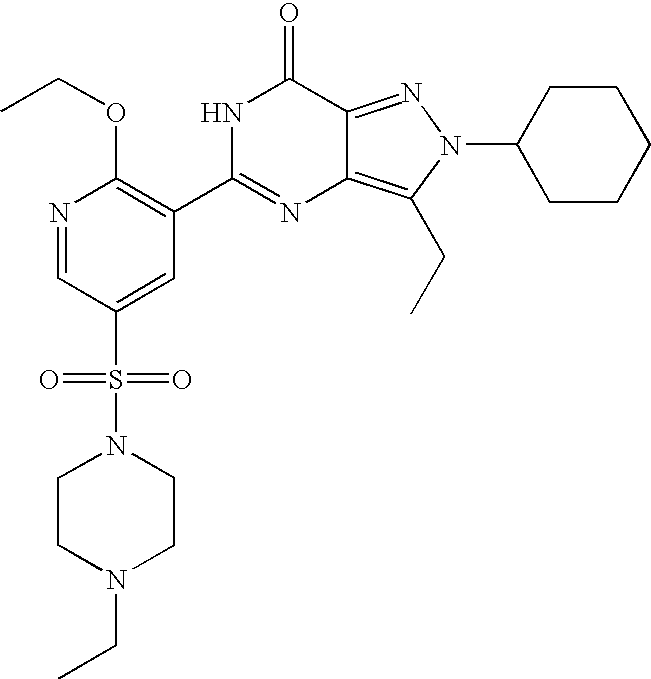
- The title compound was obtained as a white solid in 35% yield, from the compound of preparation 86, following a similar procedure to that described in example 80.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.02 (3H, t), 1.30-1.50 (6H, m), 1.58 (3H, t), 1.78 (1H, m), 1.98 (4H, m), 2.22 (2H, m), 2.41 (2H, q), 2.55 (4H, m), 3.05 (2H, q), 3.16 (4H, m), 4.23 (1H, m), 4.75 (2H, q), 8.61 (1H, s), 9.01 (1H, s), 10.54 (1H, s).
- LRMS: m/z 548.8 (MH+)
- Anal. Found: C, 57.23; H, 6.96; N, 17.54. C26H37N7O4S requires C, 57.44; H, 6.86; N, 18.03%.
-
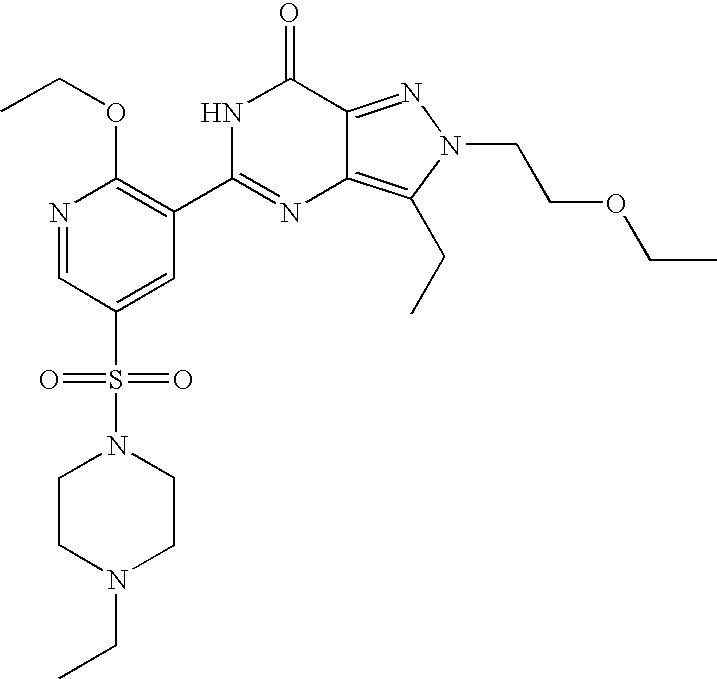
- Potassium bis(trimethylsilyl)amide (256 mg, 1.28 mmol) was added to a solution of the compound from preparation 70 (170 mg, 0.30 mmol) and ethyl acetate (30 mg, 0.33 mmol) in ethanol (5 ml), and the reaction heated at 130° C. for 6 hours. The cooled mixture was evaporated under reduced pressure and the residual yellow solid was purified by column chromatography on silica gel using dichloromethane:methanol (97:3) as eluant. The product was triturated with isopropyl ether then re-purified by column chromatography using an elution gradient of dichloromethane:methanol (100:0 to 90:10) to afford the title compound, 20 mg.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.00 (3H, t), 1.10 (3H, t), 1.40 (3H, t), 1.54 (3H, t), 2.40 (2H, q), 2.50 (4H, m), 3.05 (2H, q), 3.10 (4H, m), 3.40 (2H, q), 3.90 (2H, t), 4.42 (2H, t), 4.70 (2H, q), 8.60 (1H, s), 9.00 (1H, s), 10.60 (1H, s).
- LRMS: m/z 535 (MH+)
- Anal. Found: C, 53.97; H, 6.64; N, 18.14. C24H35N7O5S requires C, 54.02; H, 6.61; N, 18.37%.
-

- Potassium bis(trimethylsilyl)amide (2.10 g, 10.5 mmol) was added to a solution of the compound from preparation 90 (1.20 g, 2.17 mmol) and ethyl acetate (200 μl, 2.02 mmol) in ethanol (40 ml), and the reaction heated in a sealed vessel at 130° C. for 6 hours. The cooled mixture was concentrated under reduced pressure and the residue partitioned between ethyl acetate and water, and neutralised by the addition of solid carbon dioxide. The layers were separated, the aqueous phase extracted with ethyl acetate, and the combined organic solutions dried (Na2SO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (99:1 to 96:4), and the product crystallised from ether/pentane to afford the title compound, 250 mg.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.02 (3H, t), 1.39 (3H, t), 1.58 (6H, m), 2.41 (2H, q), 2.56 (4H, m), 3.08 (6H, m), 3.22 (3H, s), 3.74 (1H, m), 3.98 (1H, m), 4.74 (3H, m), 8.62 (1H, d), 9.02 (1H, d), 10.58 (1H, s).
- Anal. Found: C, 53.79; H, 6.61; N, 18.26. C24H35N7O5S requires C, 54.02; H, 6.61; N, 18.38%.
-

- The title compound was obtained as a crystalline solid in 17% yield from the compound from preparation 89, following a similar procedure to that described in example 84.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.02 (3H, t), 1.39 (3H, t), 1.58 (6H, m), 2.40 (2H, q), 2.55 (4H, m), 3.08 (6H, m), 3.22 (3H, s), 3.70 (1H, m), 3.98 (1H, m), 4.72 (3H, m), 8.61 (1H, d), 9.02 (1H, d), 10.58 (1H, s).
- LRMS: m/z 534.4 (MH+)
- Anal. Found: C, 53.67; H, 6.62; N, 18.27. C24H35N7O5S requires C, 54.02; H, 6.61; N, 18.38%.
-
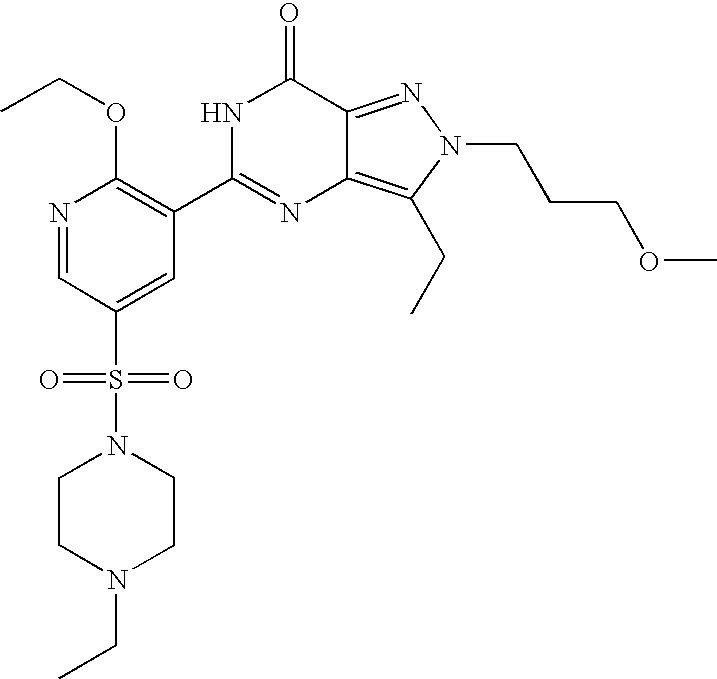
- Potassium bis(trimethylsilyl)amide (145 mg, 0.72 mmol) was added to a solution of the compound from preparation 88 (200 mg, 0.36 mmol) in 3-methyl-3-pentanol (4 ml), and the reaction heated at 130° C. for 10 hours, then cooled. The mixture was evaporated under reduced pressure and the residue purified twice by column chromatography on silica gel using dichloromethane:methanol (97:3) as eluant, to afford the title compound, 40 mg.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.00 (3H, t), 1.40 (3H, t), 1.57 (3H, t), 2.20 (2H, m), 2.42 (2H, m), 2.60 (4H, m), 3.03 (2H, q), 3.15 (4H, m), 3.30 (3H, s), 3.35 (2H, t), 4.40 (2H, t), 4.72 (2H, q), 8.60 (1H, s), 9.00 (1H, s), 10.60 (1H, br s).
- LRMS: m/z 535 (MH+)
-
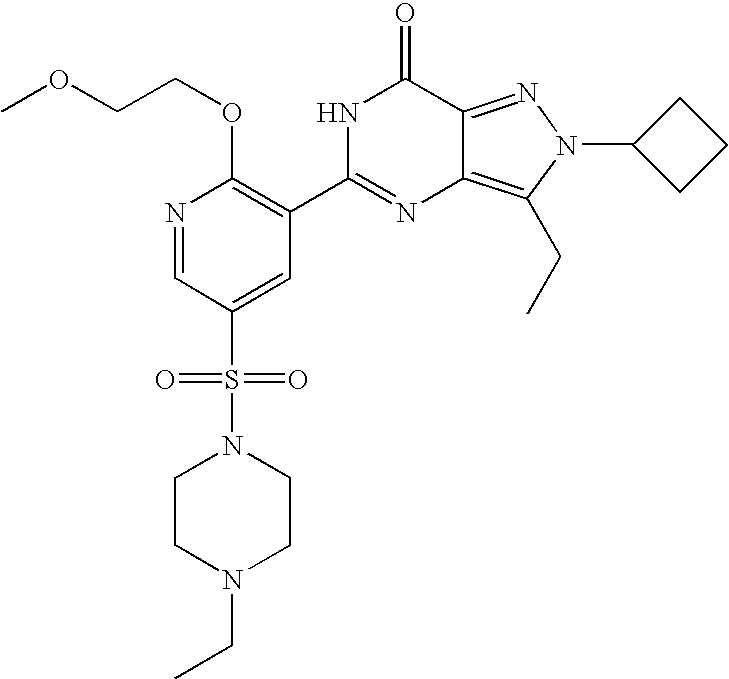
- A mixture of the compound from preparation 83 (238 mg, 0.45 mmol) and potassium bis(trimethylsilyl)amide (450 mg, 2.25 mmol) in 2-methoxyethanol (5 ml) was stirred under reflux for 6 hours. The cooled mixture was partitioned between ethyl acetate and sodium bicarbonate solution, and the layers separated. The organic phase was washed with brine, dried (MgSO4) and evaporated under reduced pressure. The residual orange oil was purified by column chromatography on silica gel using dichloromethane:methanol (98:2) as eluant to afford the title compound as an off-white foam, 150 mg.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.00 (3H, t), 1.38 (3H, t), 1.85-2.05 (2H, m), 2.40 (2H, q), 2.45 (2H, m), 2.54 (4H, m), 2.90-3.05 (4H, m), 3.15 (4H, m), 3.55 (3H, s), 3.80 (2H, m), 4.74 (2H, m), 4.95 (1H, m), 8.60 (1H, s), 8.98 (1H, s), 10.75 (1H, s).
- LRMS: m/z 546.4 (MH+)
- Anal. Found: C, 54.53; H, 6.59; N, 17.77. C25H35N7O5S requires C, 55.03; H, 6.47; N, 17.97%.
- The compounds of the following general structure:
-
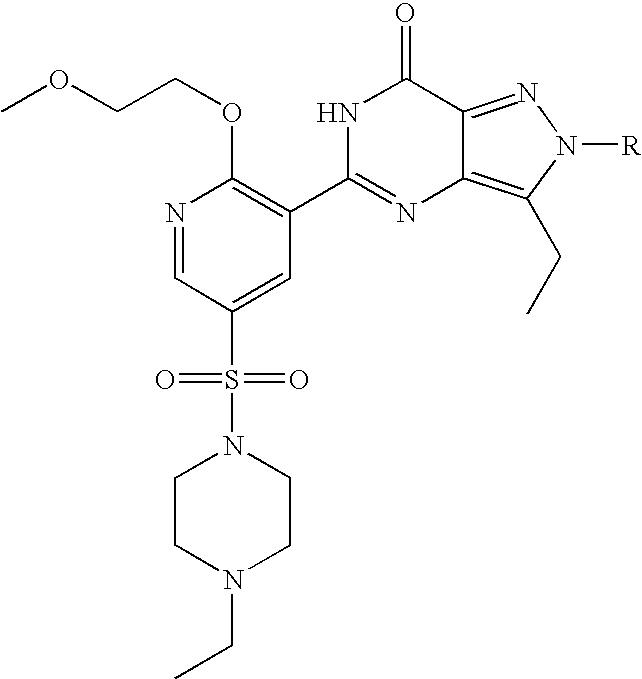
- were prepared from the corresponding pyrazole carboxamide and 2-methoxyethanol, following a similar method to that described in example 87.
-
Ex Yield no. R (%) Data 881 
15 1Hnmr (CDCl3, 400 MHz) δ: 0.43 (2 H, m), 0.60 (2 H, m), 0.80 (1 H, m), 1.00 (3 H, t), 1.40 (3 H, t), 2.40 (2 H, q), 2.54 (4 H, m), 3.00 (2 H, q), 3.07 (4 H, m), 3.50 (3 H, s), 3.80 (2 H, m), 4.20 (2 H, d), 4.78 (2 H, m), 8.60 (1 H, s), 8.97 (1 H, s), 10.57 (1 H, br s). Anal. Found: C, 52.68; H, 6.27; N, 17.19. C25H35N7O5S•H2O requires C, 53.27; H, 6.26; N, 17.39%. 892 
39 1Hnmr (CDCl3, 400 MHz) δ: 1.02 (3 H, t), 1.38 (3 H, t), 1.72 (2 H, m), 2.00-2.19 (4 H, m), 2.28 (2 H, m), 2.40 (2 H, q), 2.56 (4 H, m), 3.04 (2 H, q), 3.16 (4 H, m), 3.57 (3 H, s), 3.86 (2 H, t), 4.78 (2 H, t), 4.82 (1 H, m), 8.61 (1 H, d), 8.98 (1 H, d), 10.73 (1 H, s). LRMS: m/z 560.4 (MH+) Anal. Found: C, 55.30; H, 6.79; N, 17.49. C26H37N7O5S requires C, 55.80; H, 6.66; N, 17.52%. 902 
40 1Hnmr (CDCl3, 400 MHz) δ: 1.02 (3 H, t), 1.32 (2 H, m), 1.40 (3 H, t), 1.58 (2 H, m), 1.70 (4 H, m), 2.40 (2 H, q), 2.56 (4 H, m), 2.62 (1 H, m), 3.01 (2 H, q), 3.16 (4 H, m), 3.57 (3 H, s), 3.86 (2 H, t), 4.21 (2 H, d), 4.79 (2 H, t), 8.61 (1 H, s), 8.98 (1 H, s), 10.74 (1 H, s). LRMS: m/z 574.8 (MH+) Anal. Found: C, 54.88; H, 6.89; N, 16.63. C27H39N7O5S; H2O requires C, 54.80; H, 6.98; N, 16.57%. 91 
46 1Hnmr (CDCl3, 400 MHz) δ: 1.02 (3 H, t), 1.38 (6 H, m), 1.77 (1 H, m), 1.98 (4 H, m), 2.22 (2 H, m), 2.41 (2 H, q), 2.57 (4 H, m), 3.02 (2 H, q), 3.14 (4 H, m), 3.56 (3 H, s), 3.84 (2 H, t), 4.22 (1 H, m), 4.78 (2 H, t), 8.61 (1 H, d), 8.98 (1 H, d), 10.71 (1 H, s). 92 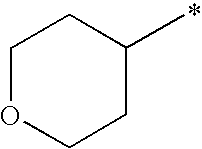
21 1Hnmr (CDCl3, 300 MHz) δ: 1.00 (3 H, t), 1.40 (3 H, t), 1.83 (2 H, m), 2.40 (2 H, q), 2.55 (6 H, m), 3.06 (2 H, q), 3.10 (4 H, m), 3.55 (3 H, s), 3.60 (2 H, t), 3.80 (2 H, t), 4.20 (2 H, m), 4.48 (1 H, m), 4.80 (2 H, t), 8.60 (1 H, s), 9.00 (1 H, s), 10.80 (1 H, s). LRMS: m/z 576.6 (MH+) 1= additionally purified by ether trituration 2= purified by ether trituration -
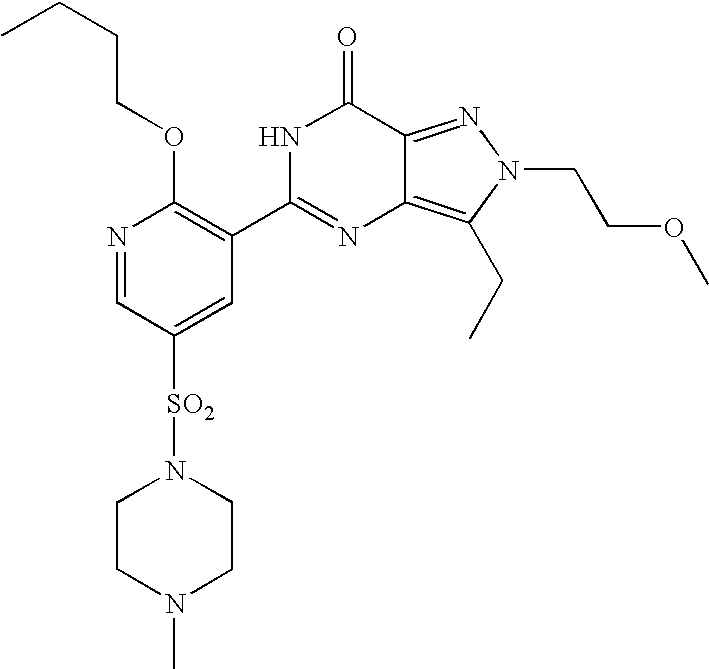
- Potassium bis(trimethylsilyl)amide (123 mg, 0.62 mmol) was added to a solution of the compound from preparation 27 (162 mg, 0.31 mmol) in n-butanol, and the reaction mixture heated at 120° C. for 18 hours. The cooled mixture was concentrated under reduced pressure and the residual yellow oil purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol:0.88 ammonia (98:2:0.2 to 95:5:0.5). The product was triturated with ether to give the title compound as a white solid, 78 mg.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.03 (3H, t), 1.41 (3H, t), 1.54 (2H, m), 1.94 (2H, m), 2.28 (3H, s), 2.51 (4H, m), 3.07 (2H, m), 3.14 (4H, m), 3.30 (3H, s), 3.95 (2H, t), 4.46 (2H, t), 4.67 (2H, t), 8.63 (1H, m), 9.04 (1H, m), 10.60 (1H, m).
- Anal. Found: C, 53.64; H, 6.64; N, 18.15. C24H35N7O5S requires C, 54.02; H, 6.61; N, 18.37%.
-

- Potassium bis(trimethylsilyl)amide (960 mg, 4.8 mmol) was added to a solution of the compound from preparation 93 (500 mg, 0.96 mmol) in 2-methoxyethanol (15 ml), and the reaction heated at 130° C. for 5 hours. The cooled reaction mixture was partitioned between ethyl acetate and water, and the mixture neutralised using solid carbon dioxide. The layers were separated, the organic phase washed with water, dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (99:1 to 96:4) to give an oil. This was triturated with ether, to afford the title compound as a white powder, 170 mg.
- 1Hnmr (CDCl3, 400 MHz) δ: 0.80 (3H, t), 1.40 (3H, t), 1.60 (3H, d), 1.90 (1H, m), 2.20 (1H, m), 2.22 (3H, s), 2.50 (4H, m), 3.00 (2H, m), 3.10 (4H, m), 3.58 (3H, s), 3.80 (2H, m), 4.40 (1H, m), 4.80 (2H, m), 8.60 (1H, s), 9.00 (1H, s), 10.70 (1H, s).
- LRMS: m/z 534.6 (MH+)
- Anal. Found: C, 54.20; H, 6.68; N, 18.39. C24H35N7O5S requires C, 54.08; H, 6.71; N, 18.40%.
- [α]D+26.0° (c=0.1, methanol).
-

- The title compound was obtained as a white powder in 23% yield from the compound from preparation 94 and 2-methoxyethanol, following the procedure described in example 94.
- 1Hnmr (CDCl3, 400 MHz) δ: 0.80 (3H, t), 1.40 (3H, t), 1.60 (3H, d), 1.90 (1H, m), 2.20 (1H, m), 2.22 (3H, s), 2.50 (4H, m), 3.00 (2H, m), 3.10 (4H, m), 3.58 (3H, s), 3.80 (2H, m), 4.40 (1H, m), 4.80 (2H, m), 8.60 (1H, s), 9.00 (1H, s), 10.70 (1H, s).
- LRMS: m/z 534.6 (MH+)
-

- The title compound was obtained as a solid in 54% yield from the compound from preparation 91 and 2-methoxyethanol, following a similar procedure to that described in example 95.
- 1Hnmr (CDCl3, 400 MHz) δ: 0.95 (3H, t), 1.40 (5H, m), 1.97 (2H, m), 2.35 (3H, s), 2.58 (4H, m), 3.01 (2H, q), 3.18 (4H, m), 3.56 (3H, s), 3.85 (2H, t), 4.28 (2H, t), 4.78 (2H, t), 8.62 (1H, d), 8.98 (1H, d), 10.75 (1H, s).
- LRMS: m/z 535 (MH+)
-
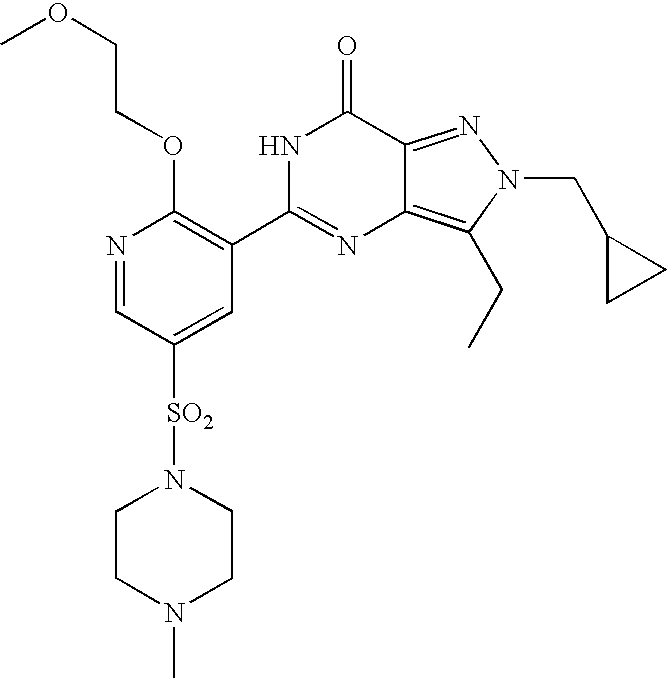
- The title compound was obtained as a solid in 41% yield from the compound from preparation 92 and 2-methoxyethanol, following a similar procedure to that described in example 95.
- 1Hnmr (CDCl3, 400 MHz) δ: 0.46 (2H, m), 0.62 (2H, m), 1.40 (4H, m), 2.27 (3H, s), 2.50 (4H, m), 3.05 (2H, q), 3.16 (4H, m), 3.57 (3H, s), 3.84 (2H, t), 4.20 (2H, d), 4.58 (2H, t), 8.61 (1H, d), 8.98 (1H, d), 10.77 (1H, s).
- LRMS: m/z 532.2 (MH+)
-

- A mixture of the compound from example 7 (200 mg, 0.38 mmol) and potassium bis(trimethylsilyl)amide (371 mg, 1.86 mmol) in tetrahydrofurfuryl alcohol (2.5 ml) was heated under reflux for 18 hours. The cooled mixture was concentrated under reduced pressure, and the residue purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 90:10). The product was recrystallised from ether to afford the title compound, 20 mg.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.01 (3H, t), 1.40 (3H, t), 1.75-2.18 (10H, m), 2.40 (2H, q), 2.55 (4H, m), 3.00 (3H, m), 3.15 (4H, m), 3.88 (1H, m), 4.16 (1H, m), 4.30 (2H, d), 4.38 (1H, m), 4.59 (1H, m), 4.75 (1H, m), 8.60 (1H, d), 8.98 (1H, d), 10.73 (1H, s).
- LRMS: m/z 587 (MH+)
-
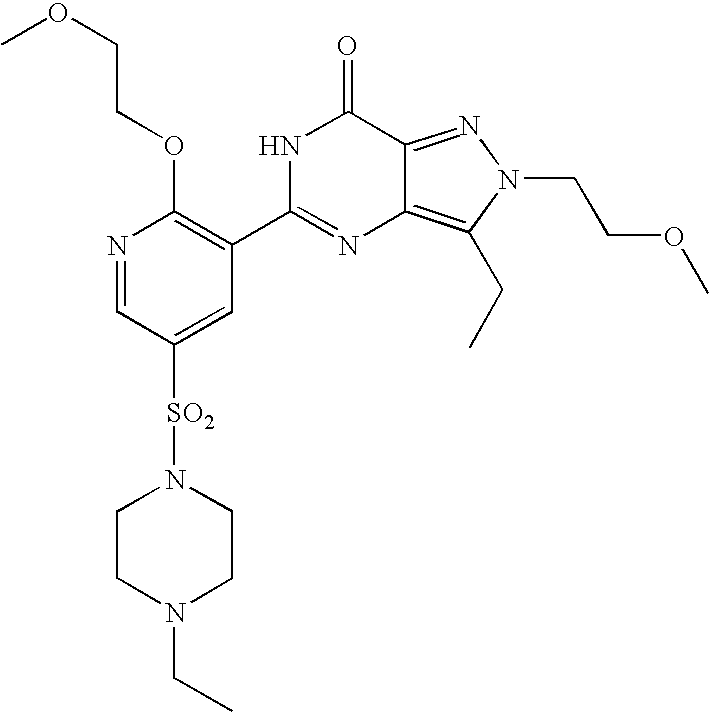
- The title compound was obtained as a solid, from the compound from example 8 and 2-methoxyethanol, following a similar procedure to that described in example 98.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.02 (6H, m), 1.84 (2H, m), 2.42 (2H, q), 2.56 (4H, m), 3.01 (2H, t), 3.15 (4H, m), 3.29 (3H, s), 3.57 (3H, s), 3.88 (2H, m), 4.44 (2H, t), 4.78 (2H, t), 8.61 (1H, s), 8.98 (1H, s), 10.76 (1H, s).
- LRMS: m/z 564 (MH+)
-

- Sodium nitrite (116 mg, 1.68 mmol) was added to a cooled (−20° C.) solution of the amine from preparation 82 (400 mg, 1.12 mmol) in acetic acid (5 ml) and concentrated hydrochloric acid (5 ml), and the solution allowed to warm to room temperature over 4 hours. The solution was then re-cooled to −15° C., liquid sulphur dioxide (3 ml) added followed by a solution of copper (II) chloride (450 mg, 3.34 mmol) in water (2 ml), and the solution stirred for 2 hours, then allowed to warm to room temperature. The reaction was diluted with water and extracted with dichloromethane (100 ml). The combined organic extracts were dried (Na2SO4), concentrated under reduced pressure and the residue azeotroped with toluene. The product was dissolved in ethanol (5 ml), N-isopropylpiperazine (500 μl, 3.56 mmol) added and the reaction stirred at room temperature for 18 hours. The reaction mixture was evaporated under reduced pressure and the crude product purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (96:4:0.5) as eluant. The resulting pale yellow solid was recrystallised from isopropyl ether:dichloromethane to give the title compound, 211 mg.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.00 (6H, 2×s), 1.40 (3H, t), 1.56 (3H, t), 2.60 (4H, m), 2.66 (1H, m), 3.08 (6H, m), 3.27 (3H, s), 3.92 (2H, t), 4.45 (2H, t), 4.75 (2H, q), 8.61 (1H, d), 9.02 (1H, d), 10.61 (1H, s).
- LRMS: m/z 534.5 (MH+)
- Anal. Found: C, 54.00; H, 6.69; N, 18.24. C24H35N7O5S requires C, 54.02; H, 6.61; N, 18.37%.
-

- The title compound was obtained in 21% yield from the amine of preparation 82 and n-propyl piperazine (prepared from the hydrobromide salt, in the presence of excess triethylamine), following the procedure described in Example 100.
- 1Hnmr (CDCl3, 300 MHz) δ: 0.84 (3H, t), 1.40 (3H, t), 1.55 (5H, m), 2.30 (2H, m), 2.55 (4H, m), 3.08 (6H, m), 3.28 (3H, s), 3.94 (2H, t), 4.44 (2H, t), 4.75 (2H, q), 8.62 (1H, d), 9.03 (1H, d), 10.61 (1H, s).
- LRMS: m/z 534.4 (MH+)
-
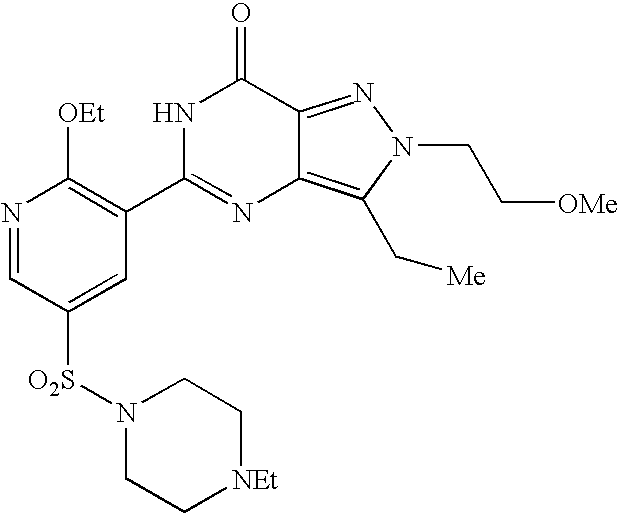
- To prepare the compound of Example 8 a mixture of N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl)nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120° C. for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55° C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4 L. The precipitated solids were granulated at 5° C. for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p.=157° C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N7O5S. 0.2 C2H5CO2CH3 requires C, 53.21; H, 6.49; N, 18.25%.
- δ (CDCl3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1H, d), 9.06 (1H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.
- LRMS: m/z=520 (M+1)+
-
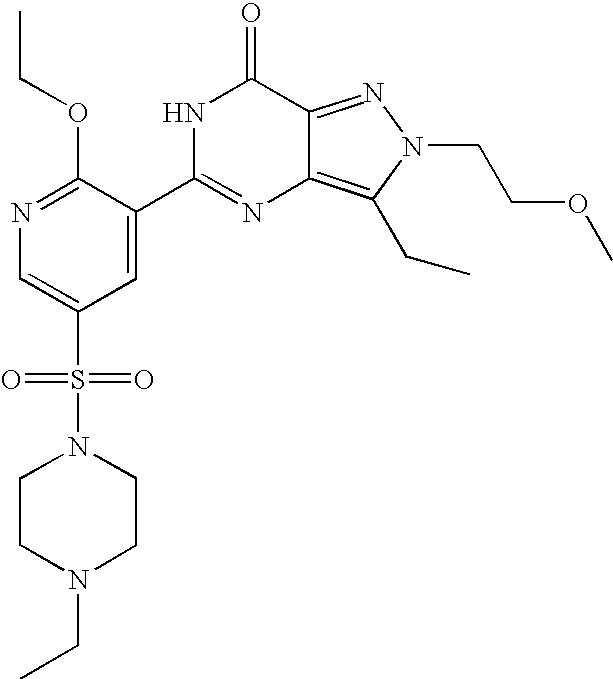
- 10 g (0.019 mol) of the compound of Example 8 and Example 102, 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine ethyl acetate solvate, was charged followed by 12 ml/g (120 mls) of 16% water in ethyl alcohol. The slurry was heated to reflux to yield a solution and 6 ml/g (60 mls) distilled off at atmospheric pressure. The solution was then cooled to room temperature with crystallisation occurring at 40° C. The slurry was then cooled to 5-10° C. and granulated for 30 minutes following which it was filtered and washed with 2 ml/g ethyl alcohol (20 mls). The damp solid was dried in vacuo overnight at 55-60° C. to yield a white crystalline solid. (Yield 7.6 g, 76%). Melting Point 162-165° C.
- δ (CDCl3): 1.05 (3H, t), 1.42 (3H, t), 1.58 (3H, t), 2.43 (2H, q), 2.57 (4H, t), 3.09 (2H, t), 3.15 (4H, t), 3.30 (3H, s), 3.93 (2H, t), 4.48 (2H, t), 4.90 (2H, q), 8.65 (1H, d), 9.05 (1H, d), 10.65 (1H, s).
- In the process of Example 103, water and pharmaceutically acceptable alcohols such as methanol, ethanol, propanol, butanol and mixtures thereof can be used to prepare the compound of Examples 8 and 102.
-

- 170 g (0.33 mol) of the compound of Example 103, 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine, was charged followed by a water/2-butanone (4% v/v) at 10 ml/g (1.7 litres) and warmed to reflux. 53 g (0.33 mol) of benzene sulfonic acid dissolved in water (23 mls, resulting in 70% w/w solution) was added to the refluxing solution over 30 minutes. 5.3 ml/g (0.9 litres) of 2-butanone were striped and replaced and the slurry cooled. The slurry was cooled to 5-10° C. and granulated for 2 hours after which it was filtered and washed with 2 ml/g (0.3 litres) of 2-butanone. The salt was dried overnight in vacuo at 55-60° C. to yield a white crystalline solid. Yield 215 g, 96.4%. Mpt 242-244° C.
- δ (DMSO): 1.17 (3H, t), 1.28 (3H, t), 1.35 (3H, t), 2.73 (2H, q), 2.97 (2H, q), 3.2 (3H, s), 3.58 (2H, t), 3.78 (3H, t), 3.81 (2H, t), 4.49 (2H, t) 4.51 (2H, q), 7.29-7.33 (3H, m), 7.57-7.60 (2H, m), 8.28 (1H, d), 8.73 (1H, d), 9.13 (1H, s), 11.90 (1H, s).
- The powder X-ray diffraction (PXRD) pattern for this salt, having Mpt 242-244° C., was determined using a Siemens D5000 powder X-ray diffractometer fitted with a theta-theta goniometer, automatic beam divergence slits, a secondary monochromator and a scintillation counter. The specimen was rotated whilst being irradiated with copper K-alpha1 X-rays (Wavelength=1.5046 Angstroms) filtered with a graphite monochromator (λ=0.15405 nm) with the X-ray tube operated at 40 kV/mA.
- The main peaks (in degrees θ) of the PXRD pattern are illustrated in Table I.
-
TABLE I Angle Intensity % 2-Theta ° % 4.208 8.6 7.292 52.5 8.153 12.6 8.422 4.1 9.426 10.2 10.957 100.0 12.645 11.4 14.150 18.6 14.639 3.1 14.928 2.7 15.080 4.9 15.363 1.8 16.070 4.5 16.245 5.4 16.351 11.4 16.892 33.9 17.554 35.1 18.178 11.8 18.562 3.2 18.903 3.0 19.174 3.1 19.591 31.6 20.392 43.3 20.598 6.8 20.965 12.8 21.136 7.8 21.485 32.9 22.000 24.0 22.294 91.9 22.708 13.4 23.414 12.6 23.682 4.7 24.132 4.6 24.361 13.3 24.554 12.9 24.844 6.9 24.902 7.6 25.444 15.2 25.854 43.0 26.054 16.4 26.369 12.5 27.016 9.5 27.706 4.8 28.302 7.2 28.504 10.9 28.998 4.0 29.615 16.1 30.197 5.2 31.039 12.5 31.445 7.7 32.094 6.5 32.611 6.4 32.734 9.3 33.014 6.5 33.110 7.2 33.740 3.5 34.255 3.4 34.952 5.5 35.497 5.6 35.830 5.4 36.507 4.5 36.816 8.4 37.047 16.0 37.641 5.5 38.362 8.7 38.582 17.7 39.203 8.8 40.549 7.8 41.277 6.7 41.487 11.9 42.376 8.4 42.759 7.1 43.450 8.0 44.400 4.5 45.043 8.3 45.888 6.2 46.393 6.2 46.897 7.3 48.197 7.8 48.373 7.9 49.163 5.3 50.501 6.0 50.619 5.9 52.248 14.6 52.746 5.7 54.668 5.1 - The same besylate salt, as defined by the XRD pattern described in Table 1, when made via alternative routes can have a melting point in the range of from 235-246° C. (measured using a Perkin Elmer DSC7 at a heating rate of 20° C./minute).
-

- 5 g (0.0096 mol) of the compound of Example 103, 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine, was charged followed by 10 ml/g (50 mls) of ethyl alcohol and warmed to reflux. 1.86 g (0.0097 mol) of p-toluene sulfonic acid dissolved in 10 mls ethyl alcohol was added to the refluxing solution over 15 seconds. The solution was allowed cool and allowed granulate for 1 hour at <R.T. The slurry was filtered and washed with 3 ml/g (15 mls) of ethyl alcohol. The salt was dried overnight in vacuo at 55-60° C. to yield a white crystalline solid. Yield 6.12 g, 92.3%. Mpt 208° C.
- δ (DMSO): 1.18 (3H, t), 1.28 (3H, t), 1.36 (3H, t), 2.28 (3H, s), 2.78 (2H, q), 2.99 (2H, q), 3.23 (4H, t) 3.25 (3H, s), 3.55 (2H, t), 3.80 (2H, t), 3.82 (2H, t), 4.51 (2H, t), 4.53 (2H, q), 7.11 (2H, d), 7.47 (2H, d), 8.30 (1H, d), 8.73 (1H, d), 9.2 (1H, s), 11.90 (1H, s).
-

- 3 g (0.006 mol) of the compound of Example 103, 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine, was charged followed by a 2-butanone/water (4% v/v) at 10 ml/g (30 mls) and warmed to reflux. 1.48 g (0.006 mol) of (+)-camphor sulphonic acid dissolved in 5 mls 2-butanone and 1 ml water was added to the refluxing solution in <1 minute. 3.3 ml/g (10 mls) were azeotroped out and the solution cooled with crystallisation occurring at 45° C. approximately. The slurry was cooled to 5-10° C. and granulated for 0.5 hours after which it was filtered and washed with 5 ml/g (15 mls) of 2-butanone. The salt was dried overnight in vacuo at 55-60° C. to yield a white crystalline solid. (Yield 3.4 g, 77%). Mpt 222-225° C.
- δ (DMSO): 0.75 (3H, s), 1.03 (3H, s), 1.18 (3H, t), 1.28 (3H, t), 1.36 (3H, t), 1.20-1.40 (2H, m), 1.79-198 (3H, m), 2.2-2.3 (1H, m), 2.5-2.62 (2H, m), 2.78 (2H, q), 2.99 (2H, q), 3.02 (1H, d), 3.23 (4H, t) 3.25 (3H, s), 3.55 (2H, t), 3.79 (2H, t), 3.82 (2H, t), 4.51 (2H, t), 4.50 (2H, q), 8.29 (1H, d), 8.73 (1H, d), 9.33 (1H, s), 11.85 (1H, s).
-
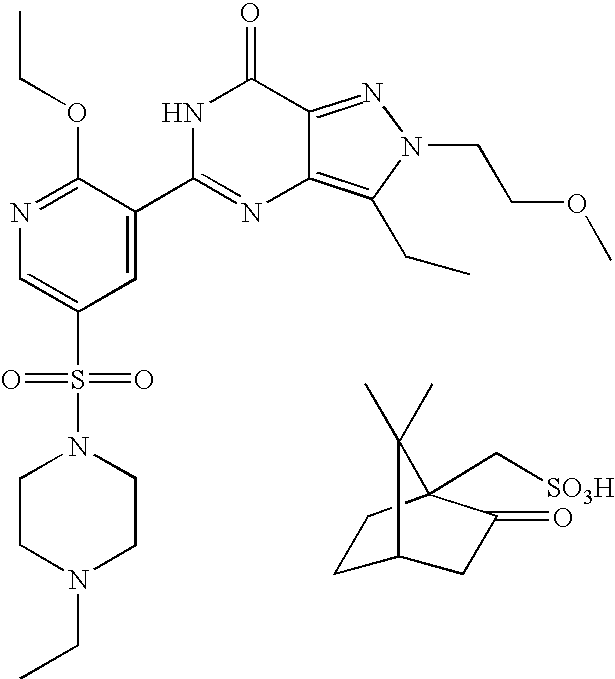
- 17 g (0.033 mol) of the compound of Example 103, 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine, was charged followed by ethyl alcohol at 10 ml/g (170 mls) and warmed to reflux. 7.75 g (0.035 mol) of racemic camphor sulphonic acid dissolved in 30 mls ethyl alcohol was added to the refluxing solution instantaneously. The solution was allowed cool and crystallisation occurred at 65-66° C. The slurry was cooled to 5-10° C. and granulated for 1 hours after which it was filtered and washed with 3 ml/g (51 mls) of ethyl alcohol. The salt was dried overnight in vacuo at 55-60° C. to yield a white crystalline solid. (Yield 22.1 g, 89.8%).
- δ (DMSO): 0.75 (3H, s), 1.03 (3H, s), 1.18 (3H, t), 1.28 (3H, t), 1.36 (3H, t), (2H, m), 1.79-198 (3H, m), 2.2-2.3 (1H, m), 2.5-2.62 (2H, m), 2.78 (2H, q), 2.99 (2H, q), 3.02 (1H, d), 3.23 (4H, t), 3.25 (3H, s), 3.55 (2H, t), 3.79 (2H, t), 3.82 (2H, t) 4.51 (2H, t), 4.50 (2H, q), 8.29 (1H, d), 8.73 (1H, d), 9.33 (1H, s), 11.85 (1H, s).
-
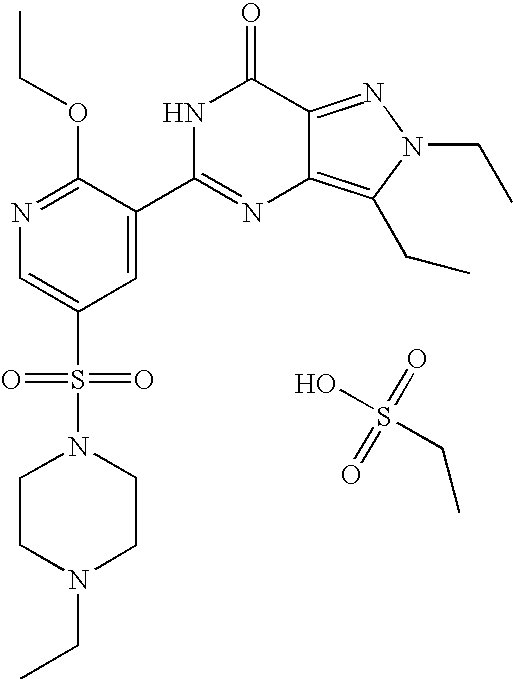
- 5 g (9.6 mmol) of the title compound of Example 102, 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine, was charged followed by a 10 ml/g (0.05 litres) Ethanol and warmed to reflux. 1.1 g (10.5 mmol) of ethane sulfonic acid diluted in 2 ml ethanol was added to the refluxing solution. The slurry was cooled with crystallisation occurring at 26-30° C. The slurry was granulated filtered and washed with 2 ml/g (0.01 litres) of ethanol. The salt was dried overnight in vacuo at 55-60° C. to yield a white crystalline solid. Yield 5.2 g, 86.1%. Mpt 205-210° C. δ (CDCl3): 1.16 (3H, t), 1.39 (3H, t), 1.41 (3H, t), 1.52 (3H, t), 2.73 (2H, q), 3.03 (2H, t), 3.09 (2H, q, 3.16 (2H, t), 3.30 (3H, s), 3.35 (2H, t), 3.65 (2H, t), 3.89 (2H, t), 3.90 (2H, q), 4.46 (2H, t), 4.71 (2H, q), 8.63 (1H, d), 8.71 (1H, d), 10.76 (1H, s), 11.29 (1H, s).
- The following Table illustrates both the in vitro activities for a range of the compounds of the invention as inhibitors of cGMP PDE5 as well as their selectivity for cGMP PDE5 versus cGMP PDE6.
- The IC50 measurements for cGMP PDE5 were based upon data generated on human corpus cavernosum tissue and the IC50 measurements for rod cGMP PDE6 were based upon data generated on bovine retina tissue and wherein the selectivity ratio for cGMP PDE5 to cGMP PDE6 quoted is based upon IC50 PDE5/IC50 PDE6.
-
TABLE EXAMPLE IC50 (nM) Selectivity (PDE 5/6) 5 1.0 — 8 1.68 223.8 17 0.90 254.1 22 6.4 325.3 24 1.52 134.9 27 0.85 161 53 1.09 — 60 0.45 343.7 -

- A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (WO, 9849166), (1.7 g, 8.8 mmol), 2-bromoethyl methyl ether (0.85 ml, 8.85 mmol) and cesium carbonate (2.9 g, 9.0 mmol) in N,N-dimethylformamide (20 ml) was stirred at room temperature for 20 hours. The reaction mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (125 ml) and brine (100 ml). The phases were separated, and the organic layer was dried (Na2SO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel, using ethyl acetate:methanol (97:3) as eluant to afford the title compound of
preparation 1, 831 mg, - δ (DMSOd6): 1.19 (3H, t), 2.82 (2H, q), 3.20 (3H, s), 3.68 (2H, t), 4.22 (2H, t), 8.18 (1H, s), 8.38 (1H, s).
- LRMS: m/z 260 (M+18)+
- and the title compound of
preparation 2, 793 mg. - δ (CDCl3): 1.18 (3H, t), 2.98 (2H, q), 3.22 (3H, s), 3.70 (2H, t), 4.28 (2H, t), 7.65 (1H, s), 7.94 (1H, s).
- LRMS: m/z 243 (M+1)+
-
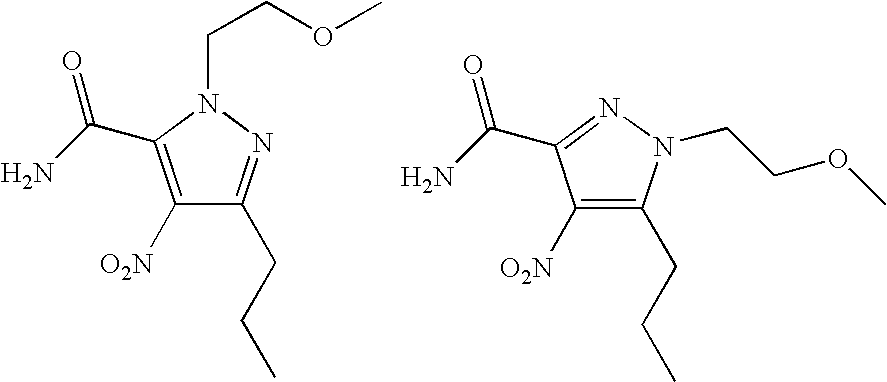
- A mixture of 4-nitro-3-n-propyl-1H-pyrazole-5-carboxamide (WO, 9849166), (7.3 g, 37.0 mmol), 2-bromoethyl methyl ether (3.85 ml, 41.0 mmol) and cesium carbonate (24.0 g, 74.0 mmol) in N,N-dimethylformamide (300 ml) was heated at 70° C. for 4 hours. The cooled mixture was concentrated under reduced pressure and the residue partitioned between ethyl acetate (100 ml) and brine (100 ml) and the phases separated. The aqueous layer was extracted with ethyl acetate (2×100 ml), the combined organic solutions dried (Na2SO4) and evaporated under reduced pressure. The residue was triturated with ether and the resulting precipitate filtered and dried, to give some of the N2 isomer. The filtrate was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel, using an elution gradient of ethyl acetate:methanol (100:0 to 99:1). The product of
preparation 3 was suspended in ether, the mixture filtered and the filtrate evaporated under reduced pressure to afford the title compound ofpreparation 3, 1.07 g, δ (CDCl3): 1.00 (3H, t), 1.74 (2H, m), 2.88 (2H, t), 3.35 (3H, s), 3.78 (2H, t), 4.47 (2H, t), 6.06 (1H, s), 7.24 (1H, s). - LRMS: m/z 257 (M+1)+
- More of the N2 isomer (preparation 4) was also obtained to give a total of 3.85 g.
- δ (DMSOd6): 1.04 (3H, t), 1.68 (2H, m), 2.98 (2H, t), 3.30 (3H, s), 3.79 (2H, t), 4.29 (2H, t), 5.85 (1H, s), 7.35 (1H, s).
- LRMS: m/z 257 (M+1)+
-
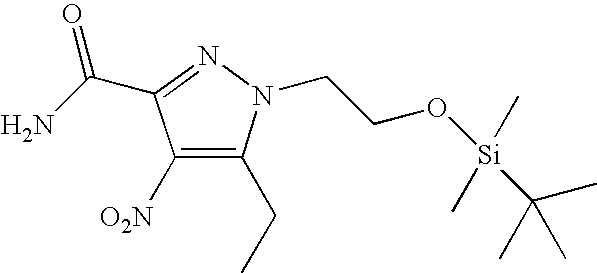
- A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (WO, 9849166), (4.9 g, 26.6 mmol), cesium carbonate (21.0 g, 64.5 mmol) and (2-bromoethoxy)-tert-butyldimethylsilane (7.0 g, 29.0 mmol) in acetonitrile (400 ml) was stirred at 80° C. for 20 hours. The cooled mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (200 ml) and water (100 ml). The layers were separated, the organic phase washed with water (3×50 ml), dried (Na2SO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel, using ethyl acetate as eluant, and repeated using an elution gradient of pentane:ethyl acetate (50:50 to 0:100), to give some of the desired compound.
- The crude product containing both the N1 and N2 isomers was triturated with pentane, the resulting precipitate filtered and dried to afford the title compound as a solid (1.7 g, in total)
- δ (CDCl3): −0.05 (6H, s), 0.81 (9H, s), 1.28 (3H, t), 3.08 (2H, q), 4.03 (2H, t), 4.24 (2H, t), 5.80 (1H, s), 7.34 (1H, s).
- LRMS: m/z 343 (M+1)+
-

- A mixture of tert-butyl 3-[(methylsulphonyl)oxy]-1-azetidinecarboxylate (Synlett; 1998; 379), (5.0 g, 19.9 mmol), and potassium iodide (16.5 g, 99.4 mmol) in N,N-dimethylformamide (25 ml), was heated at 100° C. for 42 hours. The cooled mixture was partitioned between water and ethyl acetate, and the layers separated. The organic phase was dried (MgSO4), concentrated under reduced pressure and the residue azeotroped with xylene. The crude product was purified by column chromatography on silica gel using dichloromethane as eluant, to give the title compound, 3.26 g. δ (CDCl3): 1.43 (9H, s), 4.28 (2H, m), 4.46 (1H, m), 4.62 (2H, m),
- LRMS: m/z 284 (M+1)+
-

- A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (WO, 9849166), (6.59 g, 35.8 mmol), cesium carbonate (12.25 g, 37.6 mmol), and the title compound of preparation 6 (10.3 g, 37.6 mmol) in N,N-dimethylformamide (60 ml) was heated at 60° C. for 3 days. The cooled reaction was poured into 2% aqueous sodium bicarbonate solution (250 ml), and extracted with ethyl acetate (1×230 ml, 1×100 ml). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The residual oil was purified by column chromatography on silica gel, using an elution gradient of ethyl acetate:pentane (50:50 to 100:0) to give the N1-isomer (5.0 g) and the title compound of preparation 7, 4.1 g. δ (CDCl3): 1.25 (3H, t), 1.46 (9H, s), 2.96 (2H, q), 4.37 (2H, m), 4.44 (2H, m), 5.06 (1H, m), 5.82 (1H, s), 6.63 (1H, s).
-

- Obtained (25%) from 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (WO, 9849166), and 2-[(benzyloxy)carbonyl](methyl)amino]ethyl methanesulphonate (J. Med. Chem. 37; 23; 1994; 3977) following a similar procedure to that described in preparation 7.
- δ (CDCl3): (rotamers in 0.42:0.58 ratio) 1.03 and 1.20 (3H, t), 2.69 and 2.87 (2H, q), 2.80 and 2.92 (3H, s), 3.72 (2H, m), 4.20 and 4.33 (2H, t), 5.02 and 5.14 (2H, s), 5.86 (1H, m), 7.35 (6H, m).
-

- A mixture of the title compound from preparation 2 (500 mg, 2.07 mmol) and 10% palladium on charcoal (50 mg) in ethanol (20 ml) was hydrogenated at 50 psi and room temperature for 18 hours. The reaction mixture was filtered through Arbocel®, and the filtrate evaporated under reduced pressure to afford the title compound as a white solid.
- δ (DMSOd6): 1.03 (3H, t), 2.57 (2H, q), 3.20 (3H, s), 3.63 (2H, t), 4.09 (2H, t), 4.39 (2H, s), 6.90 (1H, s), 7.01 (1H, s).
- LRMS: m/z 213 (M+1)+
- The compounds of the general structure:
-
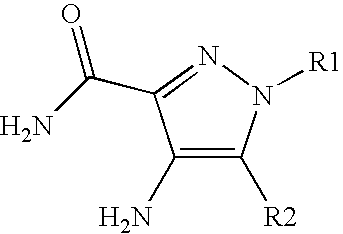
- were prepared from the corresponding nitropyrazole, following a similar procedure to that described in preparation 9.
-
Prep Yield No. R1 R2 (%) m/z 1Hnmr 10 
(CH2)2CH3 95 227 (M + 1)+ δ (CDCl3): 0.98 (3 H, t), 1.60 (2 H, m), 2.47 (2 H, t), 3.30 (3 H, s), 3.74 (2 H, t), 3.94 (2 H, s), 4.15 (2 H, t), 5.20 (1 H, s), 6.58 (1 H, s). 11 
CH2CH3 335 (M + 23)+ δ (CDCl3): −0.03 (6 H, s), 0.85 (9 H, s), 1.18 (3 H, t), 2.63 (2 H, q), 3.94 (4 H, m), 4.08 (2 H, t), 5.15 (1 H, s), 6.57 (1 H, s). 12 
CH2CH3 73 δ (CDCl3): 1.14 (3 H, t), 1.46 (9 H, s), 2.55 (2 H, q), 3.98 (2 H, s), 4.29 (2 H, m), 4.40 (2 H, m), 4.94 (1 H, m), 5.23 (1 H, s), 6.64 (1 H, s). -

- A mixture of the title compound of preparation 8 (1.92 g, 5.28 mmol), iron powder (3.04 g) and water (2.5 ml) in acetic acid (50 ml) was stirred at room temperature for 25 minutes. The reaction mixture was filtered through Arbocel®, and the filtrate poured slowly into saturated sodium bicarbonate solution (400 ml). The pH of the solution was adjusted to 8 using solid sodium carbonate, and this solution was then extracted with ethyl acetate (2×350 ml). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure to afford the title compound, 1.5 g.
- δ (CDCl3): (rotamers in a 0.46:0.54 ratio), 1.00 and 1.14 (3H, t), 2.38 and 2.50 (2H, q), 2.68 and 2.80 (3H, s), 3.63 (2H, m), 3.95 (2H, s), 4.04 and 4.17 (2H, t), 5.10 and 5.14 (2H, s), 5.14 (1H, s), 6.53 (1H, s), 7.36 (5H, m).
-
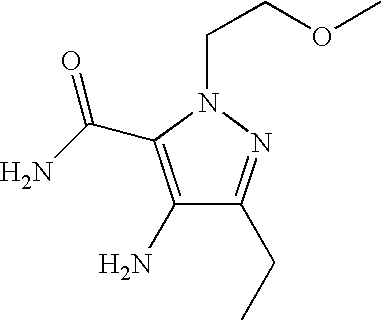
- Obtained from the title compound of preparation 1 (95%), using a similar procedure to that described in preparation 9, and after purification by column chromatography using dichloromethane:methanol (90:10) as eluant.
- δ (CDCl3): 1.26 (3H, t), 2.58 (2H, q), 3.37 (3H, s), 3.60 (2H, s), 3.82 (2H, t), 4.50 (2H, t).
- LRMS: m/z 213 (M+1)+
-

- Obtained as a solid (99%) from the title compound of
preparation 3, using the procedure described in preparation 9. - δ (CDCl3): 0.95 (3H, t), 1.63 (2H, m), 2.48 (2H, t), 3.30 (3H, s), 3.78 (2H, t), 4.46 (2H, t).
- LRMS: m/z 227 (M+1)+
-

- 2-Aminopyridine (80 g, 0.85 mol) was added portionwise over 30 minutes to oleum (320 g) and the resulting solution heated at 140° C. for 4 hours. On cooling, the reaction was poured onto ice (200 g) and the mixture stirred in an ice/salt bath for a further 2 hours. The resulting suspension was filtered, the solid washed with ice water (200 ml) and cold IMS (200 ml) and dried under suction to afford the title compound as a solid, 111.3 g.
- LRMS: m/z 175 (M+1)+
-

- Bromine (99 g, 0.62 mol) was added dropwise over an hour, to a hot solution of the title compound of preparation 16 (108 g, 0.62 mol) in water (600 ml) so as to maintain a steady reflux. Once the addition was complete the reaction was cooled and the resulting mixture filtered. The solid was washed with water and dried under suction to afford the title compound, 53.4 g.
- δ (DMSOd6, 300 MHz): 8.08 (1H, s), 8.14 (1H, s).
- LRMS: m/z 253 (M)+
-
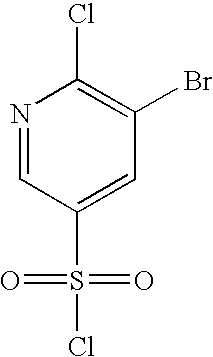
- A solution of sodium nitrite (7.6 g, 110.0 mmol) in water (30 ml) was added dropwise to an ice-cooled solution of the title compound of preparation 17 (25.3 g, 100.0 mmol) in aqueous hydrochloric acid (115 ml, 20%), so as to maintain the temperature below 6° C. The reaction was stirred for 30 minutes at 0° C. and for a further hour at room temperature. The reaction mixture was evaporated under reduced pressure and the residue dried under vacuum at 70° C. for 72 hours. A mixture of this solid, phosphorus pentachloride (30.0 g, 144.0 mmol) and phosphorus oxychloride (1 ml, 10.8 mmol) was heated at 125° C. for 3 hours, and then cooled. The reaction mixture was poured onto ice (100 g) and the resulting solid filtered, and washed with water. The product was dissolved in dichloromethane, dried (MgSO4), and evaporated under reduced pressure to afford the title compound as a yellow solid, 26.58 g.
- δ (CDCl3, 300 MHz): 8.46 (1H, s), 8.92 (1H, s).
-

- A solution of 1-ethylpiperazine (11.3 ml, 89.0 mmol) and triethylamine (12.5 ml, 89.0 mmol) in dichloromethane (150 ml) was added dropwise to an ice-cooled solution of the title compound of preparation 18 (23.0 g, 79.0 mmol) in dichloromethane (150 ml) and the reaction stirred at 0° C. for an hour. The reaction mixture was concentrated under reduced pressure and the residual brown oil was purified by column chromatography on silica gel, using an elution gradient of dichloromethane:methanol (99:1 to 97:3) to afford the title compound as an orange solid, 14.5 g.
- δ (CDCl3, 300 MHz): 1.05 (3H, t), 2.42 (2H, q), 2.55 (4H, m), 3.12 (4H, m), 8.24 (1H, s), 8.67 (1H, s).
-

- N-Methylpiperazine (7.65 ml, 69.0 mmol) was added dropwise to a solution of the title compound of preparation 18 (10.0 g, 34.5 mmol) in ethanol (200 ml), and the reaction stirred at room temperature for 3 hours. The mixture was concentrated under reduced pressure and the residue partitioned between dichloromethane (200 ml) and water (100 ml) and the layers separated. The organic phase was dried (Na2SO4), and evaporated under reduced pressure to afford the title compound, 10.53 g, as a yellow solid.
- δ (CDCl3): 2.28 (3H, s), 2.51 (4H, m), 3.14 (4H, m), 8.24 (1H, s), 8.67 (1H, s).
-

- A mixture of the title compound of preparation 19 (6.60 g, 17.9 mmol) and sodium ethoxide (6.09 g, 89.55 mmol) in ethanol (100 ml) was heated under reflux for 18 hours, then cooled. The reaction mixture was concentrated under reduced pressure, the residue partitioned between water (100 ml) and ethyl acetate (100 ml), and the layers separated. The aqueous phase was extracted with ethyl acetate (2×100 ml), the combined organic solutions dried (MgSO4) and evaporated under reduced pressure to afford the title compound as a brown solid, 6.41 g.
- Found: C, 41.27; H, 5.33; N, 11.11. C13H20BrN3O3S requires C, 41.35; H, 5.28; N, 10.99%.
- δ (CDCl3, 300 MHz): 1.06 (3H, t), 1.48 (3H, t), 2.42 (2H, q), 2.56 (4H, m), 3.09 (4H, m), 4.54 (2H, q), 8.10 (1H, s), 8.46 (1H, s).
- LRMS: m/z 378, 380 (M+1)+
-

- A mixture of the title compound of preparation 20 (10.0 g, 39.1 mmol), potassium bis(trimethylsilyl)amide (5.92 g, 29.7 mmol) and ethanol (3.5 ml) in tetrahydrofuran (150 ml) was stirred at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure and the residue partitioned between ethyl acetate (150 ml) and brine (50 ml). The layers were separated, and the organic phase dried (Na2SO4), filtered and evaporated under reduced pressure, to afford the title compound, 9.1 g.
- δ (CDCl3): 1.44 (3H, t), 2.29 (3H, s), 2.51 (4H, m), 3.08 (4H, m), 4.54 (2H, q), 8.10 (1H, s), 8.44 (1H, s).
- LRMS: m/z 365 (M+1)+
-

- A mixture of the title compound of preparation 21 (6.40 g, 16.92 mmol), triethylamine (12 ml, 86.1 mmol), and palladium (0) tris(triphenylphosphine) in ethanol (60 ml) was heated at 100° C. and 200 psi, under a carbon monoxide atmosphere, for 18 hours, then cooled. The reaction mixture was evaporated under reduced pressure and the residue purified by column chromatography on silica gel, using an elution gradient of dichloromethane:methanol (100:0 to 97:3) to afford the title compound as an orange oil, 6.2 g.
- δ (CDCl3, 300 MHz): 1.02 (3H, t), 1.39 (3H, t), 1.45 (3H, t), 2.40 (2H, q), 2.54 (4H, m), 3.08 (4H, m), 4.38 (2H, q), 4.55 (2H, q), 8.37 (1H, s), 8.62 (1H, s).
- LRMS: m/z 372 (M+1)+
-
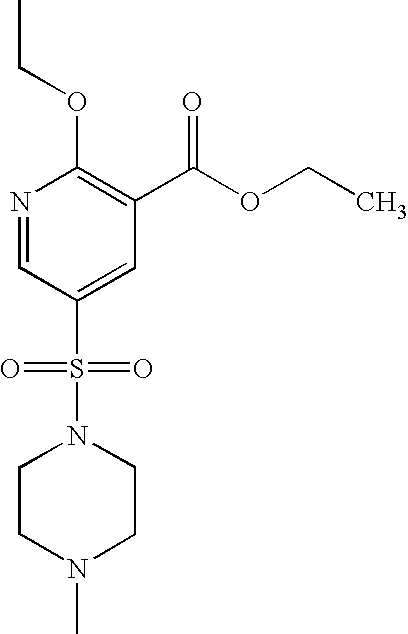
- Obtained (85%) as an orange solid, from the title compound of preparation 22 using a similar procedure to that described in preparation 23.
- δ (CDCl3): 1.40 (3H, t), 1.46 (3H, t), 2.28 (3H, s), 2.50 (4H, m), 3.09 (4H, m), 4.40 (2H, q), 4.57 (2H, q), 8.40 (1H, s), 8.63 (1H, s).
- LRMS: m/z 358 (M+1)+
-

- A mixture of the title compound of preparation 23 (4.96 g, 13.35 mmol) and aqueous sodium hydroxide solution (25 ml, 2N, 50.0 mmol) in ethanol (25 ml) was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure to half it's volume, washed with ether and acidified to pH 5 using 4N hydrochloric acid. The aqueous solution was extracted with dichloromethane (3×30 ml), the combined organic extracts dried (MgSO4) and evaporated under reduced pressure to afford the title compound as a tan coloured solid, 4.02 g.
- δ (DMSOd6, 300 MHz): 1.18 (3H, t), 1.37 (3H, t), 3.08 (2H, q), 3.17-3.35 (8H, m), 4.52 (2H, q), 8.30 (1H, s), 8.70 (1H, s).
-
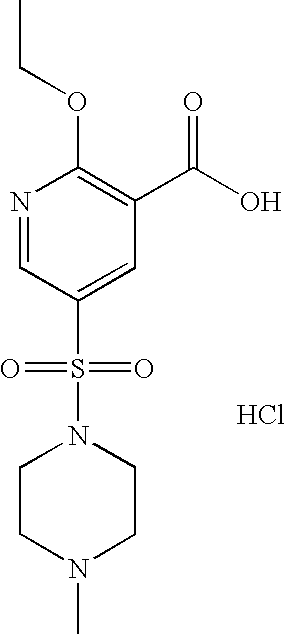
- Sodium hydroxide solution (21 ml, 2M, 42.0 mmol) was added to a solution of the title compound of preparation 24 (7.57 g, 21.0 mmol) in dioxan (150 ml) and the reaction stirred at room temperature for 18 hours. The mixture was neutralised using hydrochloric acid, the dioxan removed under reduced pressure and the remaining aqueous solution acidified to
pH 2, using hydrochloric acid. The solution was evaporated under reduced pressure, the residue re-suspended in hot ethanol, filtered, and the filtrate re-evaporated to afford the title compound, 5.46 g. - δ (DMSOd6): 1.37 (3H, t), 2.50 (4H, m), 2.72 (3H, s), 3.13-3.39 (4H, m), 4.53 (2H, q), 8.30 (1H, s), 8.75 (1H, s).
- LRMS: m/z 330 (M+1)+
-
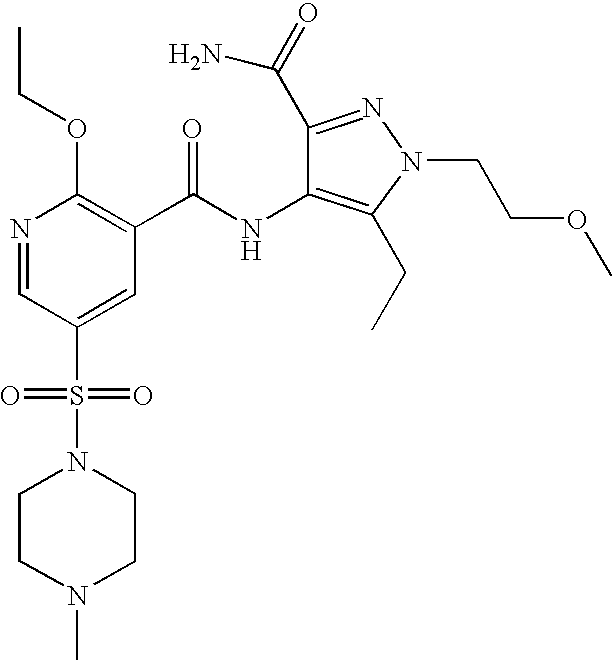
- Oxalyl chloride (500 ml, 5.73 mmol) was added dropwise to an ice-cooled solution of the title compound of preparation 26 (522 mg, 1.43 mmol) and N,N-dimethylformamide (1 drop) in dichloromethane (20 ml), and the reaction stirred for 2 hours. The mixture was concentrated under reduced pressure and azeotroped several times with dichloromethane to give the intermediate acid chloride. A solution of this product in dichloromethane (20 ml) was added to a solution of the title compound of preparation 9 (250 mg, 1.18 mmol) and triethylamine (500 ml, 3.18 mmol) in dichloromethane (20 ml), and the reaction stirred at room temperature for 18 hours. The mixture was washed with water, dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel, using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 428 mg.
- δ (CDCl3): 1.20 (3H, t), 1.59 (3H, t), 2.28 (3H, s), 2.50 (4H, m), 2.95 (2H, m), 3.10 (4H, m), 3.36 (3H, s), 3.80 (2H, t), 4.25 (2H, t), 4.78 (2H, q), 5.26 (1H, s), 6.65 (1H, s), 8.65 (1H, s), 8.85 (1H, s), 10.51 (1H, s).
- LRMS: m/z 524 (M+1)+
-
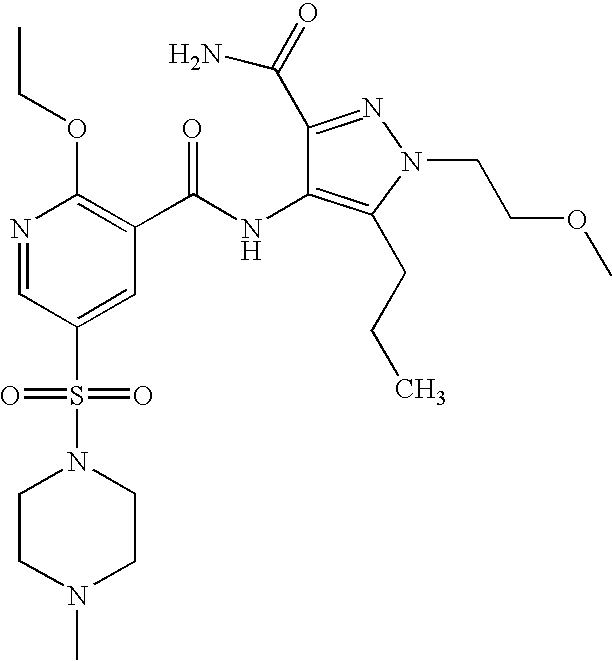
- The title compound was obtained as a white solid (79%), from the title compounds from preparation 10 and 26, following the procedure described in preparation 27.
- δ (CDCl3): 0.92 (3H, t), 1.58 (5H, m), 2.24 (3H, s), 2.47 (4H, m), 2.90 (2H, t), 3.10 (4H, m), 3.35 (3H, s), 3.78 (2H, t), 4.23 (2H, t), 4.78 (2H, q), 5.42 (1H, br s), 6.68 (1H, br s), 8.62 (1H, d), 8.82 (1H, d), 10.48 (1H, s).
- LRMS: m/z 538 (M+1)+
-

- 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (5.26 g, 27.4 mmol) was added to a solution of the title compounds from preparation 25 (7.25 g, 21.1 mmol), and preparation 9 (4.45 g, 20.9 mmol), 1-hydroxybenzotriazole hydrate (3.71 g, 27.4 mmol), and N-diisopropylethylamine (10.96 ml, 63.3 mmol) in dichloromethane (70 ml), and the reaction stirred for 18 hours. The reaction mixture was diluted with dichloromethane (100 ml), washed with water (100 ml), saturated aqueous sodium bicarbonate solution (100 ml), and brine (100 ml), dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to give the title compound as a foam, 10.05 g.
- δ (CDCl3): 1.03 (3H, t), 1.20 (3H, t), 1.58 (3H, t), 2.40 (2H, q), 2.54 (4H, m), 2.95 (2H, q), 3.10 (4H, m), 3.37 (3H, s), 3.80 (2H, t), 4.26 (2H, t), 4.78 (2H, q), 5.27 (1H, s), 6.66 (1H, s), 8.65 (1H, s), 8.85 (1H, s), 10.51 (1H, s).
- LRMS: m/z 538 (M+1)+
-

- N-Diisopropylethylamine (0.92 ml, 5.3 mmol) was added to a solution of the title compounds from preparation 25 (1.0 g, 2.65 mmol), and preparation 10 (600 mg, 2.65 mmol), 1-hydroxybenzotriazole hydrate (465 mg, 3.45 mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (660 mg, 3.45 mmol) in dichloromethane (20 ml), and the reaction stirred for 18 hours. The reaction mixture was washed with brine, dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 740 mg.
- δ (CDCl3): 0.94 (3H, t), 1.03 (3H, t), 1.59 (5H, m), 2.40 (2H, q), 2.54 (4H, m), 2.92 (2H, t), 3.11 (4H, m), 3.37 (3H, s), 3.80 (2H, t), 4.25 (2H, t), 4.78 (2H, q), 5.26 (1H, s), 6.66 (1H, s), 8.65 (1H, s), 8.83 (1H, s), 10.48 (1H, s).
- LRMS: m/z 552 (M+1)+
-

- Obtained as a white solid (67%) from the title compounds of preparations 25 and 11 following a similar procedure to that described in preparation 27.
- δ (CDCl3): 0.00 (6H, s), 0.85 (9H, s), 1.04 (3H, t), 1.22 (3H, t), 1.57 (3H, t), 2.40 (2H, q), 2.53 (4H, m), 2.94 (2H, q), 3.10 (4H, m), 4.02 (2H, t), 4.19 (2H, t), 4.78 (2H, q), 5.39 (1H, s), 6.66 (1H, s), 8.64 (1H, s), 8.83 (1H, s), 10.49 (1H, s).
- LRMS: m/z 638 (M+1)+
-

- Triethylamine (1.0 ml, 7.2 mmol) was added to a solution of the title compounds from preparation 25 (1.5 g, 4.5 mmol), and preparation 13 (1.7 g, 4.95 mmol), 1-hydroxybenzotriazole hydrate (833 mg, 5.44 mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.28 g, 6.68 mmol) in dichloromethane (50 ml), and the reaction stirred for 3 days at room temperature. The reaction mixture was concentrated under reduced pressure and the residue partitioned between saturated aqueous sodium bicarbonate solution and ethyl acetate, and the layers separated. The aqueous phase was extracted with ethyl acetate (2×50 ml), and the combined organic solutions, dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (95:5) as eluant to afford the title compound, 3.0 g.
- δ (CDCl3): 1.00-1.20 (6H, m), 1.58 (3H, t), 2.40 (2H, q), 2.54 (4H, m), 2.70-2.91 (5H, m), 3.10 (4H, m), 3.70 (2H, m), 4.16-4.32 (2H, m), 4.79 (2H, q), 5.12 (2H, m), 5.24 (1H, s), 6.62 (1H, s), 7.37 (5H, m), 8.64 (1H, s), 8.82 (1H, s), 10.50 (1H, s).
-

- The title compound was obtained (72%) from the title compounds from preparation 25 and preparation 12, following a similar procedure to that described in preparation 32.
- δ (CDCl3): 1.01 (3H, t), 1.19 (3H, t), 1.47 (9H, s), 1.58 (3H, t), 2.40 (2H, q), 2.54 (4H, m), 2.86 (2H, q), 3.10 (4H, m), 4.38 (2H, m), 4.41 (2H, m), 4.79 (2H, q), 5.10 (1H, m), 5.30 (1H, br s), 6.77 (1H, br s), 8.63 (1H, d), 8.82 (1H, d), 10.57 (1H, s).
-

- A solution of 3-ethyl-1H-pyrazole-5-carboxamide (WO, 9849166) (9.2 g, 59.8 mmol) in N,N-dimethylformamide (60 ml) was added to a solution of the title compound from preparation 25 (21.7 g, 62.9 mmol), 1-hydroxybenzotriazole hydrate (10.1 g, 66.0 mmol) and triethylamine (13.15 ml, 94.3 mmol) in dichloromethane (240 ml). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (13.26 g, 69.2 mmol) was added and the reaction stirred at room temperature for 6 hours. The dichloromethane was removed under reduced pressure, the remaining solution poured into ethyl acetate (400 ml), and this mixture washed with aqueous sodium bicarbonate solution (400 ml). The resulting crystalline precipitate was filtered, washed with ethyl acetate and dried under vacuum, to afford the title compound, as a white powder, 22 g.
- δ (CDCl3+1 drop DMSOd6) 0.96 (3H, t), 1.18 (3H, t), 1.50 (3H, t), 2.25-2.56 (6H, m), 2.84 (2H, q), 3.00 (4H, m), 4.70 (2H, q), 5.60 (1H, br s), 6.78 (1H, br s), 8.56 (1H, d), 8.76 (1H, d), 10.59 (1H, s), 12.10-12.30 (1H, s).
- LRMS: m/z 480 (M+1)+
-

- Oxalyl chloride (9.5 ml, 108 mmol) was added dropwise to an ice-cold solution of the title compound from preparation 26 (10.0 g, 27.0 mmol) and N,N-dimethylformamide (160 μl) in dichloromethane (150 ml), and once addition was complete, the reaction was stirred at room temperature for 5½ hours. The mixture was evaporated under reduced pressure and the residue azeotroped with toluene, to give a yellow solid.
- Triethylamine (11.2 ml, 81.0 mmol) was added to a solution of the intermediate acid chloride (10.5 g, 27.3 mmol) and 4-amino-3-ethyl-1H-pyrazole-5-carboxamide (WO, 9849166), (4.2 g, 27.3 mmol) in dichloromethane (150 ml), and the reaction stirred at room temperature for 18 hours. The mixture was diluted with water, and the layers separated. The aqueous phase was extracted with dichloromethane (2×), and the combined organic solutions dried (Na2SO4) and evaporated under reduced pressure. The crude product was triturated with ether, and the resulting solid filtered to give the title compound, 10.1 g.
- δ (CDCl3): 1.21 (3H, t), 1.59 (3H, t), 2.26 (3H, s), 2.50 (4H, m), 2.94 (2H, q), 3.10 (4H, m), 4.79 (2H, q), 5.50 (1H, br s), 6.80 (1H, br s), 8.64 (1H, d), 8.84 (1H, d), 10.65 (1H, s).
-
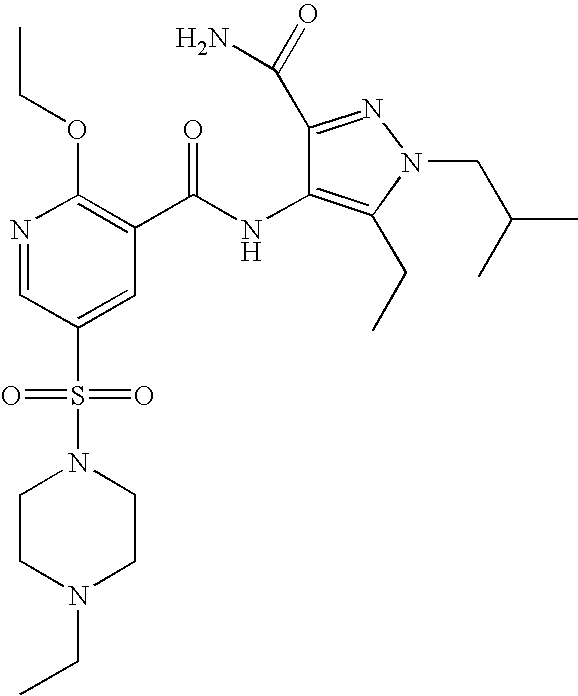
- 1-Bromo-2-methylpropane (187 μl, 1.72 mmol) was added to a solution of the title compound from preparation 34 (750 mg, 1.56 mmol) and cesium carbonate (1.12 g, 3.44 mmol) in N,N-dimethylformamide (15 ml) and the reaction stirred at 60° C. for 18 hours. The cooled mixture was partitioned between water and ethyl acetate, and the layers separated. The organic layer was dried (MgSO4), concentrated under reduced pressure and azeotroped with toluene to give a solid. This product was recrystallised from ether, to afford the title compound as a white solid, 152 mg.
- δ (CDCl3): 0.96 (6H, d), 1.02 (3H, t), 1.19 (3H, t), 1.58 (3H, t), 2.26 (1H, m), 2.40 (2H, q), 2.52 (4H, m), 2.94 (2H, q), 3.10 (4H, m), 3.88 (2H, d), 4.78 (2H, q), 5.25 (1H, s), 6.65 (1H, s), 8.64 (1H, d), 8.83 (1H, d), 10.54 (1H, s).
- LRMS: m/z 536 (M+1)+
- The following tabulated compounds of the general formula:
-

- were prepared from the title compound from preparation 34 and the appropriate bromide, following a similar procedure to that described in preparation 36.
-
Prep Yield m/z no R1 R10 (%) (M + 1)+ 1Hnmr 37 
Et 48 534 δ (CDCl3): 0.42 (2 H, m), 0.63 (2 H, m), 1.02 (3 H, t), 1.20 (3 H, t), 1.58 (3 H, t), 2.40 (2 H, q), 2.54 (4 H, m), 2.95 (2 H, q), 3.10 (4 H, m), 3.47 (1 H, m), 3.98 (2 H, d), 4.78 (2 H, q), 5.22 (1 H, br s), 6.65 (1 H, br s), 8.63 (1 H, s), 8.83 (1 H, s), 10.57 (1 H, s). 38 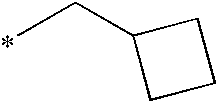
Et 51 548 δ (CDCl3): 1.01 (3 H, t), 1.18 (3 H, m), 1.58 (3 H, t), 1.80-1.97 (4 H, m), 2.08 (2 H, m), 2.40 (2 H, q), 2.54 (4 H, m), 2.80-2.97 (3 H, m), 3.10 (4 H, m), 4.10 (2 H, d), 4.78 (2 H, q), 5.11 (1 H, br s), 6.63 (1 H, br s), 8.63 (1 H, s), 8.83 (1 H, s), 10.53 (1 H, s). 39 
Et 51 536 δ (CDCl3): 0.83 (3 H, t), 1.03 (3 H, t), 1.21 (3 H, t), 1.48 (3 H, d), 1.60 (3 H, t), 1.80 (1 H, m), 2.00 (1 H, m), 2.40 (2 H, q), 2.55 (4 H, m), 2.90 (2 H, m), 3.12 (4 H, m), 4.24 (1 H, m), 4.78 (2 H, q), 5.22 (1 H, br s), 6.70 (1 H, br s), 8.64 (1 H, s), 8.83 (1 H, s), 10.50 (1 H, s). 40 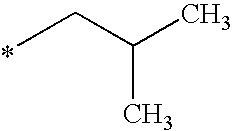
Me 44 522 δ (CDCl3): 0.96 (6 H, d), 1.17 (3 H, t), 1.59 (3 H, t), 2.27 (4 H, m), 2.48 (4 H, m), 2.91 (2 H, q), 3.09 (4 H, m), 3.88 (2 H, d), 4.78 (2 H, q), 5.24 (1 H, br s), 6.67 (1 H, br s), 8.65 (1 H, m), 8.84 (1 H, m), 10.54 (1 H, s). 41 
Me 33 546 δ (CDCl3): 1.19 (3 H, t), 1.58 (3 H, m), 1.87 (4 H, m), 2.10 (2 H, m), 2.26 (3 H, s), 2.48 (4 H, m), 2.92 (3 H, m), 3.10 (4 H, m), 4.10 (2 H, d), 4.79 (2 H, q), 5.24 (1 H, br s), 6.65 (1 H, br s), 8.64 (1 H, d), 8.84 (1 H, d), 10.55 (1 H, s). -
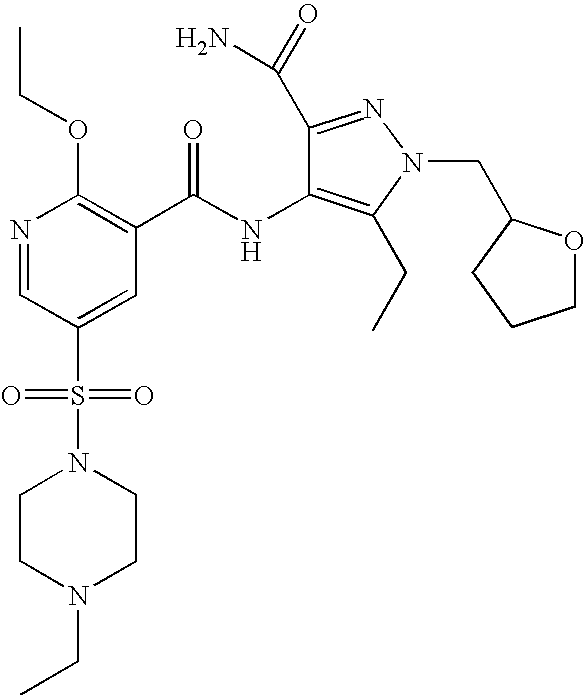
- Cesium carbonate (1.63 g, 5.0 mmol) was added to an ice-cold solution of the title compound from preparation 34 (2.0 g, 4.18 mmol) in N,N-dimethylformamide (40 ml), and the solution stirred for 30 minutes. Tetrahydrofuryl bromide (0.6 ml, 5.28 mmol) was added, and the reaction heated at 60° C. for 72 hours. The cooled mixture was evaporated under reduced pressure, and the residue partitioned between water and dichloromethane. The phases were separated, and the organic layer was dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 1.20 g.
- δ (CDCl3): 1.01 (3H, t), 1.18 (3H, t), 1.58 (3H, t), 1.70-2.12 (4H, m), 2.40 (2H, q), 2.54 (4H, m), 2.97 (2H, m), 3.10 (4H, m), 3.74-3.94 (2H, m), 4.16 (2H, m), 4.32 (1H, m), 4.78 (2H, q), 5.32 (1H, br s), 6.64 (1H, br s), 8.63 (1H, s), 8.82 (1H, s), 10.50 (1H, s).
- LRMS: m/z 564 (M+1)+
-

- Methanesulphonyl chloride (2.86 ml, 36.9 mmol) was added dropwise to an ice-cooled solution of 1-methoxy-2-propanol (3 ml, 30.7 mmol) and triethylamine (10.27 ml, 73.7 mmol) in dichloromethane (150 ml), and the reaction stirred at room temperature for 18 hours. The mixture was washed with water, then 2M hydrochloric acid, dried (MgSO4) and evaporated under reduced pressure to give the title compound as a yellow oil, 5.24 g.
- δ (CDCl3): 1.39 (3H, d), 3.03 (3H, s), 3.39 (3H, s), 3.46 (2H, m), 4.88 (1H, m).
- LRMS: m/z 186 (M+18)+
-
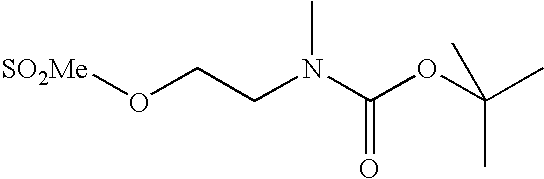
- Methanesulphonyl chloride (2.98 ml, 38.6 mmol) was added to an ice-cold solution of tert-butyl 2-hydroxyethyl(methyl)carbamate (Synth. Commun. 1993; 23(17); 2443) (4.5 g, 25.7 mmol) in pyridine (40 ml), and the reaction stirred for 2 hours. The solution was poured into water (150 ml), and extracted with ethyl acetate (2×50 ml). The combined organic extracts were washed with 10% aqueous citric acid solution, dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of ethyl acetate:pentane (34:66 to 40:60) to give the title compound, 1.0 g.
- δ (CDCl3): 1.46 (9H, s), 2.96 (3H, s), 3.02 (3H, s), 3.56 (2H, m), 4.34 (2H, m).
-
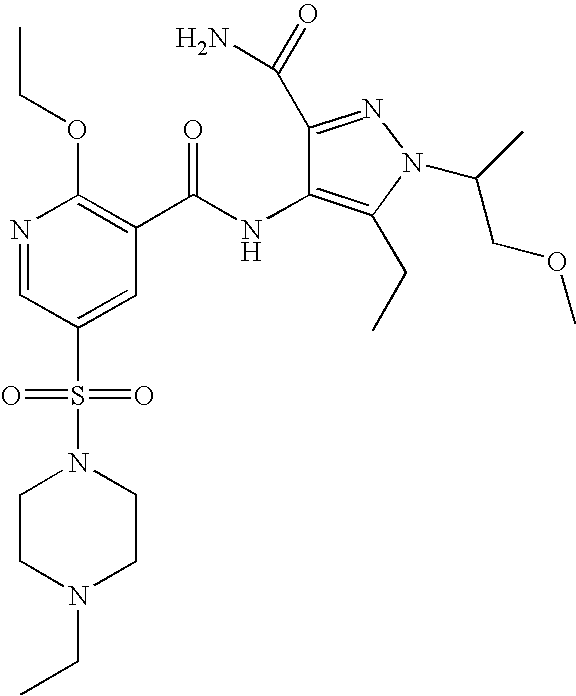
- Sodium hydride (64 mg, 60% dispersion in mineral oil, 1.6 mmol) was added to a solution of the title compound from preparation 34 (700 mg, 1.46 mmol) in tetrahydrofuran (10 ml), and the solution stirred for 10 minutes. The title compound from preparation 43 (270 mg, 1.60 mmol) was added and the reaction stirred at 60° C. for 3 days. The cooled mixture was partitioned between ethyl acetate and aqueous sodium bicarbonate solution, and the phases separated. The aqueous layer was extracted with ethyl acetate, the combined organic solutions dried (MgSO4) and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using dichloromethane:methanol (98:2) as eluant to afford the title compound as a white foam, 310 mg.
- δ (CDCl3): 1.02 (3H, t), 1.22 (3H, m), 1.50 (3H, d), 1.59 (3H, t), 2.40 (2H, q), 2.55 (4H, m), 2.92 (2H, m), 3.10 (4H, m), 3.30 (3H, s), 3.60 (1H, m), 3.78 (1H, m), 4.57 (1H, m), 4.78 (2H, q), 5.25 (1H, br s), 6.68 (1H, br s), 8.64 (1H, s), 8.83 (1H, s), 10.48 (1H, s).
- LRMS: m/z 552 (M+1)+
-

- The title compound was obtained (43%), from the title compound from preparation 34 and tert-butyl 4-[(methylsulphonyl)oxy]-1-piperidinecarboxylate (WO, 9319059), following the procedure described in preparation 45.
- δ (CDCl3): 1.02 (3H, t), 1.23 (3H, t), 1.49 (9H, s), 1.57 (3H, m), 1.93 (2H, m), 2.16 (2H, m), 2.40 (2H, q), 2.54 (4H, m), 2.82-2.97 (4H, m), 3.10 (4H, m), 4.30 (3H, m), 4.79 (2H, q), 5.23 (1H, s), 6.65 (1H, s), 8.63 (1H, d), 8.82 (1H, d), 10.57 (1H, s).
-

- The title compound was prepared from the title compounds from preparation 34 and 44 following a similar procedure to that described in preparation 45. The crude product was purified by column chromatography on silica gel using ethyl acetate:diethylamine (95:5) as eluant to give the title compound, 30%.
- δ (CDCl3): 1.02 (3H, t), 1.20 (3H, t), 1.46 (9H, s), 1.57 (3H, t), 2.40 (2H, q), 2.53 (4H, m), 2.88 (3H, s), 3.10 (4H, m), 3.58 (1H, m), 3.64 (2H, m), 4.22 (2H, m), 4.30 (1H, m), 4.79 (2H, q), 5.24 (1H, s), 6.65 (1H, s), 8.62 (1H, d), 8.82 (1H, d), 10.53 (1H, s).
-
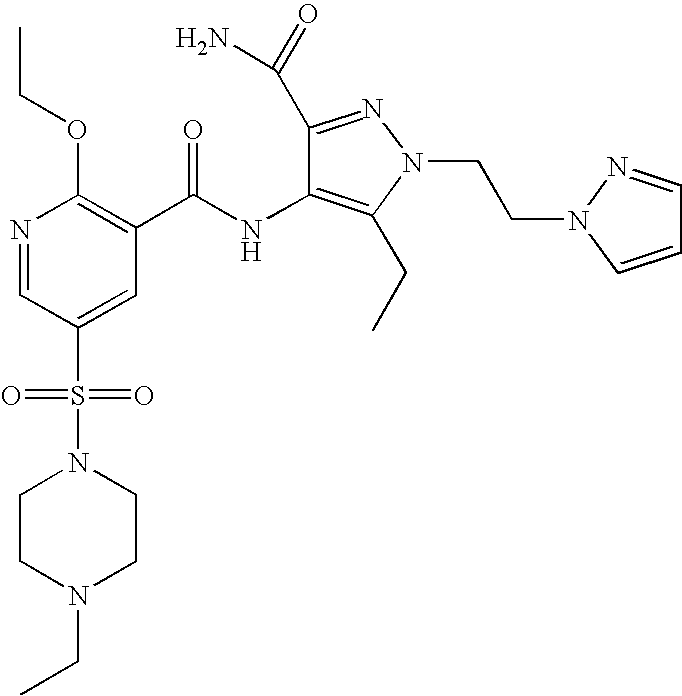
- Sodium hydride (88 mg, 60% dispersion in mineral oil, 2.19 mmol) was added to an ice-cold solution of the title compound from preparation 34 (1.0 g, 2.09 mmol) in tetrahydrofuran (25 ml), and the solution stirred for an hour. 1-(2-Chloroethyl)pyrazole (WO 9849166) (410 mg, 3.14 mmol) was added and the reaction heated under reflux for 18 hours. The cooled mixture was concentrated under reduced pressure and the residue partitioned between water and ethyl acetate and the layers separated. The aqueous phase was extracted with ethyl acetate, the combined organic solutions dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 90:10) to give the title compound, 300 mg.
- δ (CDCl3): 1.02 (6H, m), 1.58 (3H, t), 2.40 (2H, q), 2.56 (6H, m), 3.10 (4H, m), 4.50 (2H, t), 4.63 (2H, t), 4.78 (2H, q), 6.20 (1H, m), 7.06 (1H, m), 7.58 (1H, m), 8.63 (1H, d), 8.80 (1H, d), 10.46 (1H, s).
-
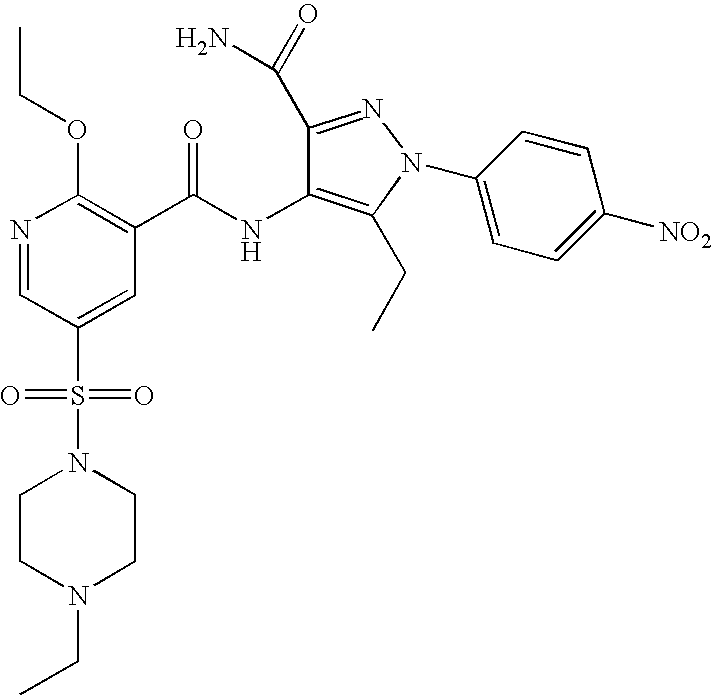
- Sodium hydride (80 mg, 80% dispersion in mineral oil, 2.67 mmol) was added to a cooled (−78° C.) solution of the title compound from preparation 34 (1.0 g, 2.08 mmol) in tetrahydrofuran (10 ml), and the mixture allowed to warm slowly to room temperature. 4-Fluoronitrobenzene (0.5 ml, 4.7 mmol) was added, and the reaction heated at 65° C. for 72 hours. The cooled mixture was partitioned between aqueous ammonium chloride solution and ethyl acetate, and the layers separated. The aqueous phase was extracted with ethyl acetate, the combined organic solutions washed with water, then brine, dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 95:5) to afford the title compound, 630 mg.
- δ (CDCl3): 0.93 (6H, m), 1.52 (3H, t), 2.32 (2H, m), 2.44 (4H, m), 2.98 (6H, m), 4.72 (2H, q), 5.96 (1H, s), 6.76 (1H, s), 7.62 (2H, d), 8.32 (2H, d), 8.58 (1H, d), 8.75 (1H, d), 10.63 (1H, s).
- LRMS: m/z 601 (M+1)+
-

- Methanesulphonyl chloride (4.95 ml, 64.0 mmol) was added to an ice-cold solution of 3-dimethylamino-1-propanol (6 g, 58.2 mmol) and triethylamine (9.7 ml, 69.8 mmol) in dichloromethane (200 ml), and the reaction stirred at room temperature for 16 hours. The mixture was partitioned between ethyl acetate and aqueous sodium bicarbonate solution, and the phases separated. The aqueous layer was extracted with ethyl acetate, and the combined organic solutions dried (Na2SO4) and evaporated under reduced pressure. The residue was immediately purified by column chromatography on silica gel using dichloromethane:methanol (90:10) as eluant to give an oily solid, 1.5 g. This was immediately re-dissolved in dichloromethane (3 ml), filtered, and the filtrate diluted with tetrahydrofuran (10 ml).
- Sodium hydride (70 mg, 60% dispersion in mineral oil, 1.75 mmol) was added portionwise to an ice-cooled solution of the title compound from preparation 34 (760 mg, 1.59 mmol) in tetrahydrofuran (15 ml), and once addition was complete, the solution was stirred at room temperature for an hour.
- The previously prepared solution of mesylate was then added, and the reaction stirred at 70° C. for 16 hours. The cooled mixture was poured into saturated sodium bicarbonate solution (120 ml), and extracted with ethyl acetate (2×100 ml). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (89:10:1) as eluant to afford the title compound, 140 mg.
- δ (CDCl3): 1.02 (3H, t), 1.21 (3H, t), 1.58 (3H, t), 2.32 (6H, s), 2.40 (2H, q), 2.54 (4H, m), 2.78 (2H, t), 2.92 (2H, q), 3.08 (4H, m), 4.18 (2H, t), 4.78 (2H, q), 5.25 (1H, s), 6.66 (1H, s), 8.64 (1H, s), 8.83 (1H, s), 10.54 (1H, s).
-
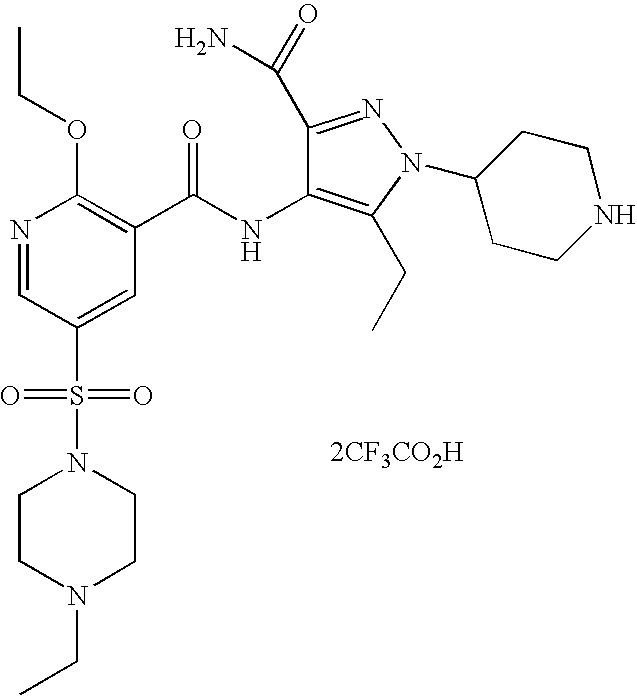
- Trifluoroacetic acid (3 ml) was added to a solution of the title compound from preparation 46 (309 mg, 0.47 mmol) in dichloromethane (4 ml), and the solution stirred for 2½ hours. The reaction was evaporated under reduced pressure and the residue triturated well with ether. The resulting solid was sonicated in ether for 1 minute, the resulting precipitate filtered and dried to afford the title compound as a white solid, 278 mg.
- δ (DMSOd6): 1.15 (6H, m), 1.46 (3H, t), 2.04 (2H, m), 2.20 (2H, m), 2.40-2.84 (6H, m), 3.00-3.22 (6H, m), 3.25-3.60 (4H, m), 3.76 (1H, m), 4.62 (4H, m), 7.27 (1H, s), 7.40 (1H, s), 8.41 (2H, m), 8.70 (2H, m), 10.24 (1H, s).
-

- Trifluoroacetic acid (1.5 ml) was added to a solution of the title compound from preparation 46 (320 mg, 0.48 mmol) in dichloromethane (2 ml) and the solution stirred at room temperature for 2½ hours. The reaction mixture was evaporated under reduced pressure and the residue triturated well with ether and dried under vacuum, to provide a white solid.
- Formaldehyde (217 μl, 37% aqueous, 2.90 mmol) was added to a solution of the intermediate amine in dichloromethane (8 ml), and the solution stirred vigorously for 30 minutes. Acetic acid (88 μl, 1.69 mmol) was added, the solution stirred for a further 30 minutes, then sodium triacetoxyborohydride (169 mg, 0.80 mmol) was added and the reaction stirred at room temperature for 16 hours. The reaction mixture was poured into aqueous sodium bicarbonate solution, and extracted with ethyl acetate. The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (91.75:7.5:0.75) as eluant to afford the title compound, 70 mg.
- δ (CDCl3): 1.02 (3H, t), 1.22 (3H, t), 1.58 (3H, t), 1.92 (2H, m), 2.14 (2H, m), 2.25-2.45 (7H, m), 2.54 (4H, m), 2.91 (2H, q), 2.99-3.16 (6H, m), 4.08 (1H, m), 4.78 (2H, q), 5.11 (1H, br s), 6.65 (1H, br s), 8.63 (1H, d), 8.83 (1H, d), 10.53 (1H, s).
-

- Trifluoroacetic acid (2.5 ml) was added to a solution of the title compound from preparation 33 (700 mg, 1.1 mmol) in dichloromethane (3.5 ml) and the solution stirred at room temperature for 2½ hours. The reaction mixture was evaporated under reduced pressure and the residue triturated well with ether and dried under vacuum. The solid was suspended in saturated aqueous sodium bicarbonate solution, extracted with ethyl acetate, and the combined organic extracts evaporated under reduced pressure.
- Formaldehyde (280 μl, 37% aqueous, 4.4 mmol) was added to a solution of the intermediate amine in dichloromethane (8 ml), and the solution stirred vigorously for 30 minutes. Acetic acid (53 μl, 1.1 mmol) was added, the solution stirred for a further 30 minutes, then sodium triacetoxyborohydride (238 mg, 1.12 mmol) was added and the reaction stirred at room temperature for 16 hours. The reaction mixture was poured into aqueous sodium bicarbonate solution (30 ml), and extracted with ethyl acetate (2×30 ml). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (91.75:7.5:0.75) to afford the title compound, 470 mg.
- δ (CDCl3): 1.01 (3H, t), 1.18 (3H, t), 1.58 (3H, t), 2.40 (2H, q), 2.48 (3H, s), 2.54 (4H, m), 2.85 (2H, q), 3.10 (4H, m), 3.59 (2H, t), 3.82 (2H, t), 4.79 (2H, q), 4.96 (1H, m), 5.32 (1H, br s), 6.79 (1H, br s), 8.64 (1H, d), 8.82 (1H, d), 10.52 (1H, s).
-
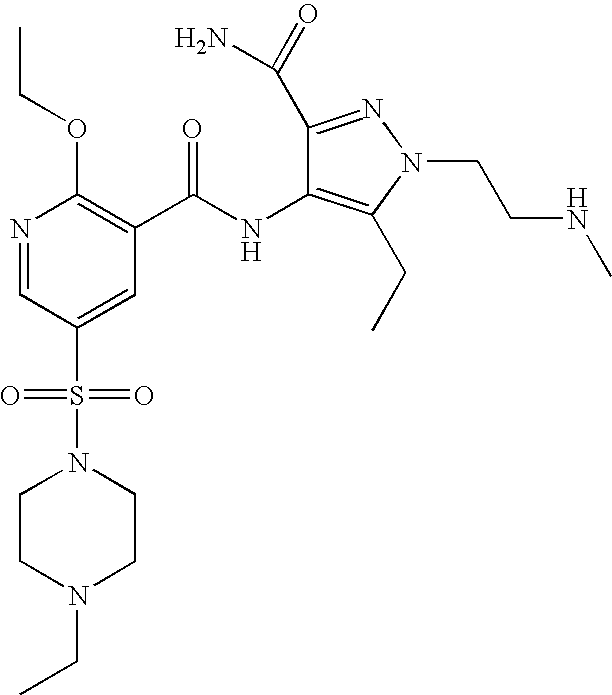
- A mixture of the title compound of preparation 32 (250 mg, 0.37 mmol) and 10% palladium on charcoal (35 mg) in methanol (3 ml) was hydrogenated at 60 psi and room temperature for 16 hours. The reaction mixture was filtered through Arbocel®, the filter pad washed with methanol and the combined filtrates evaporated under reduced pressure. The residue was purified by column chromatography on silica gel, using an elution gradient of dichloromethane:methanol:0.88 ammonia (90:10:0 to 89:10:1) to afford the title compound (135 mg, 68%) as a white foam.
- δ (CDCl3): 1.02 (3H, t), 1.20 (3H, t), 1.60 (3H, t), 2.40 (2H, q), 2.48 (3H, s), 2.52 (4H, m), 2.94 (2H, q), 3.10 (6H, m), 4.22 (2H, t), 4.79 (2H, q), 5.28 (1H, s), 6.67 (1H, s), 8.64 (1H, s), 8.83 (1H, s), 10.54 (1H, s).
-

- Sodium hydride (88 mg, 60% dispersion in mineral oil, 2.2 mmol) was added portionwise to an ice-cold solution of the title compound from preparation 34 (1.0 g, 2.1 mmol) in tetrahydrofuran (25 ml), and the solution stirred for 30 minutes.
- 2-Dimethylaminoethylchloride hydrochloride (451 mg, 3.15 mmol) was treated with saturated aqueous sodium bicarbonate solution, and this mixture extracted with dichloromethane (2×15 ml). The combined extracts were concentrated under reduced pressure at room temperature to a volume of about 2 ml, and this solution diluted with tetrahydrofuran (10 ml). This was then added to the previously prepared solution, and the reaction heated under reflux for 20 hours. The cooled mixture was poured into aqueous saturated sodium bicarbonate solution, and extracted with ethyl acetate (100 ml). The organic extract was evaporated under reduced pressure, and the residual foam was purified by column chromatography on silica gel using ethyl acetate:diethylamine (95:5) as eluant to afford the title compound, 300 mg.
- δ (CDCl3): 1.02 (3H, t), 1.22 (3H, t), 1.59 (9H, m), 2.40 (2H, q), 2.54 (4H, m), 2.78 (2H, t), 2.94 (2H, q), 3.09 (4H, m), 4.19 (2H, t), 4.78 (2H, q), 5.25 (1H, s), 6.65 (1H, s), 8.62 (1H, s), 8.83 (1H, s), 10.54 (1H, s).
-

- The title compound was obtained (70%) from the title compounds of preparations 25 and 14, following a similar procedure to that described in preparation 27.
- δ (CDCl3): 1.04 (3H, t), 1.27 (3H, t), 1.59 (3H, t), 2.42 (2H, q), 2.57 (4H, m), 2.72 (2H, q), 3.12 (4H, m), 3.38 (3H, s), 3.85 (2H, t), 4.55 (2H, t), 4.77 (2H, q), 5.57 (1H, s), 7.92 (1H, s), 8.68 (1H, s), 8.86 (1H, s), 9.82 (1H, s).
- LRMS: m/z 538 (M+1)+
-

- A mixture of the title compounds of preparations 26 (585 mg, 1.77 mmol) and 15 (300 mg, 1.32 mmol), 1-hydroxybenzotriazole hydrate (189 mg, 1.40 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (267 mg, 1.40 mmol) and N-ethyldiisopropylamine (0.39 ml, 2.25 mmol) in dichloromethane (20 ml) was stirred at room temperature for 18 hours. The mixture was washed with brine (10 ml), then water (10 ml) and then extracted with hydrochloric acid (1M, 3×20 ml). The combined acidic extracts were neutralised using sodium bicarbonate solution, and this aqueous solution extracted with dichloromethane (3×30 ml). The combined organic extracts were dried (Na2SO4), and evaporated under reduced pressure to afford the title compound as a white solid, 446 mg.
- δ (CDCl3): 0.97 (3H, t), 1.67 (5H, m), 2.28 (3H, s), 2.50 (4H, m), 2.65 (2H, t), 3.10 (4H, m), 3.37 (3H, s), 3.82 (2H, t), 4.52 (2H, t), 4.76 (2H, q), 5.57 (1H, s), 7.87 (1H, s), 8.67 (1H, s), 8.85 (1H, s), 9.77 (1H, s).
- LRMS: m/z 538 (M+1)+
-

- Potassium bis(trimethylsilyl)amide (8.28 g, 41.6 mmol) was added to a solution of the title compound from preparation 34 (10.0 g, 20.8 mmol) and ethyl acetate (2 ml, 20 mmol) in ethanol (160 ml), and the reaction mixture heated at 120° C. for 12 hours in a sealed vessel. The cooled mixture was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (95:5:0.5) as eluant, to give the title compound, 3.75 g.
- δ (CDCl3): 1.03 (3H, t), 1.42 (3H, t), 1.60 (3H, t), 2.42 (2H, q), 2.58 (4H, m), 3.02 (2H, q), 3.16 (4H, m), 4.78 (2H, q), 8.66 (1H, d), 9.08 (1H, d), 11.00 (1H, s) 11.05-11.20 (1H, br s).
- LRMS: m/z 462 (M+1)+
-

- A mixture of the title compound from preparation 34 (500 mg, 1.04 mmol), and potassium bis(trimethylsilyl)amide (436 mg, 2.19 mmol) in n-butanol (12 ml) was heated at 130° C. for 16 hours in a sealed vessel. The cooled mixture was poured into saturated aqueous sodium bicarbonate solution, and extracted with ethyl acetate, and the combined organic extracts dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (96:4) as eluant to afford the title compound, 128 mg.
- δ (CDCl3): 1.04 (6H, m), 1.42 (3H, t), 1.59 (2H, m), 1.96 (2H, m), 2.46 (2H, m), 2.60 (4H, m), 3.01 (2H, q), 3.19 (4H, m), 4.70 (2H, t), 8.64 (1H, d), 9.03 (1H, d), 11.09 (1H, s).
- LRMS: m/z 490 (M+1)+
-

- A mixture of the title compound of preparation 31 (2.02 g, 3.17 mmol), and potassium bis(trimethylsilyl)amide (950 mg, 4.76 mmol) in 3-methyl-3-pentanol (50 ml) was stirred under reflux for 8 hours. The cooled mixture was concentrated under reduced pressure, the residue suspended in ethyl acetate (100 ml), washed with water (50 ml) and brine (50 ml), dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel, using an elution gradient of dichloromethane:methanol (100:0 to 90:10) to afford the title compound, 124 mg.
- δ (CDCl3): −0.08 (6H, s), 0.81 (9H, s), 1.02 (3H, t), 1.40 (3H, t), 1.57 (3H, t) 2.41 (2H, q), 2.56 (4H, m), 3.14 (6H, m), 4.15 (2H, t), 4.40 (2H, t), 4.74 (2H, q), 8.62 (1H, s), 9.03 (1H, s), 10.68 (1H, s).
- LRMS: m/z 620 (M+1)+
-
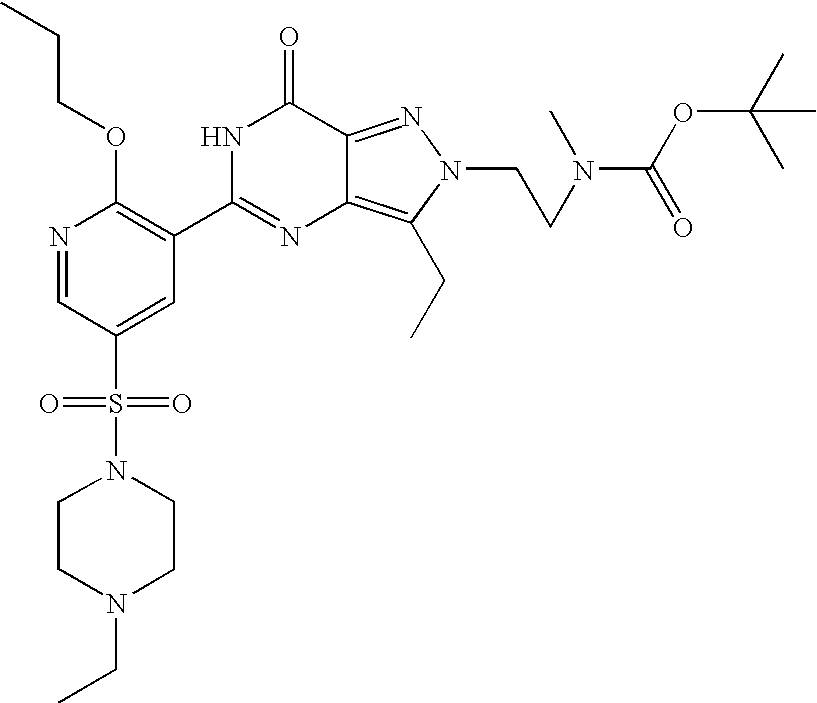
- A mixture of the title compound from example 14 (100 mg, 0.16 mmol) and potassium bis(trimethylsilyl)amide (161 mg, 0.81 mmol) in n-propanol (3 ml) was heated at 100° C. for 16 hours. The cooled reaction mixture was poured into saturated sodium bicarbonate solution (20 ml), extracted with ethyl acetate (2×30 ml), and the combined organic extracts evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (97:3) as eluant to afford the title compound, 71 mg.
- δ (CDCl3): 1.03 (3H, t), 1.14 (3H, t), 1.41 (3H, t), 1.45 (9H, s), 2.00 (2H, m), 2.42 (2H, q), 2.58 (7H, m), 3.01 (2H, q), 3.16 (4H, m), 3.78 (2H, t), 4.46 (2H, m), 4.63 (2H, t), 8.63 (1H, d), 9.04 (1H, d), 10.66 (1H, br s).
-
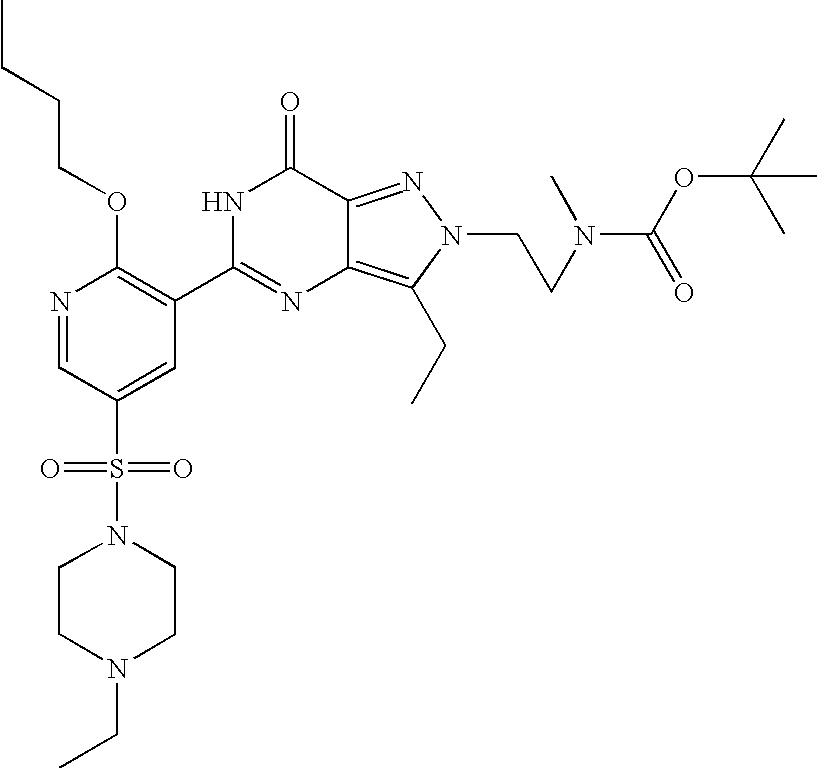
- A mixture of the title compound from example 14 (123 mg, 0.20 mmol), potassium bis(trimethylsilyl)amide (198 mg, 1.0 mmol) and ethyl acetate (18 mg, 0.20 mmol) in n-butanol (12 ml) was heated at 110° C. for 8 hours in a sealed vessel. The cooled mixture was poured into aqueous saturated sodium bicarbonate solution (60 ml), and extracted with ethyl acetate (2×60 ml). The combined organic extracts were dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (97:3) as eluant to give the title compound as a beige foam, 36 mg.
- δ (CDCl3): 1.02 (6H, t), 1.40 (3H, t), 1.45 (9H, s), 1.55 (2H, m), 1.95 (2H, m), 2.41 (2H, q), 2.58 (7H, m), 3.01 (2H, q), 3.16 (4H, m), 3.78 (2H, t), 4.45 (2H, m), 4.67 (2H, t), 8.63 (1H, d), 9.03 (1H, d), 10.64 (1H, s).
-
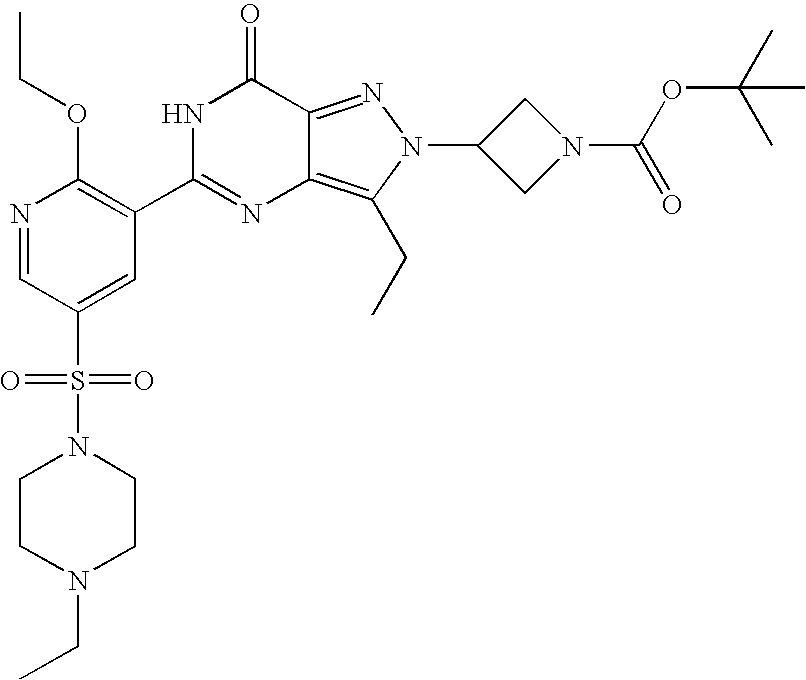
- A mixture of the title compound of preparation 33 (1.3 g, 2.05 mmol) and potassium bis(trimethylsilyl)amide (490 mg, 2.46 mmol) in ethanol (35 ml) was heated at 130° C. in a sealed vessel for 16 hours. The cooled reaction mixture was concentrated under reduced pressure, the residue dissolved in water (15 ml), the solution neutralised using hydrochloric acid (2N), and then saturated sodium bicarbonate added. This aqueous solution was extracted with dichloromethane (5×30 ml), the combined organic extracts dried (MgSO4) and evaporated under reduced pressure. The residual gum was purified by column chromatography on silica gel, using ethyl acetate:diethylamine (96:4) as eluant to afford the title compound, 350 mg.
- δ (CDCl3): 1.02 (3H, t), 1.38 (3H, t), 1.48 (9H, s), 1.58 (3H, t), 2.40 (2H, q), 2.57 (4H, m), 3.02 (2H, q), 3.14 (4H, m), 4.37 (2H, t), 4.42 (2H, m), 4.77 (2H, q), 5.25 (1H, m), 8.64 (1H, s), 8.81 (1H, s), 10.57 (1H, s).
-

- The title compound was prepared from the title compound from preparation 46, following a similar procedure to that described in preparation 63. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol (95:5) as eluant to give the title compound (62%).
- δ (CDCl3): 1.03 (3H, t), 1.38-1.60 (15H, m), 1.94 (2H, m), 2.41 (4H, m), 2.57 (4H, m), 2.90 (2H, m), 3.10 (6H, m), 4.26-4.48 (3H, m), 4.77 (2H, q), 8.62 (1H, d), 9.02 (1H, d), 10.60 (1H, s).
-
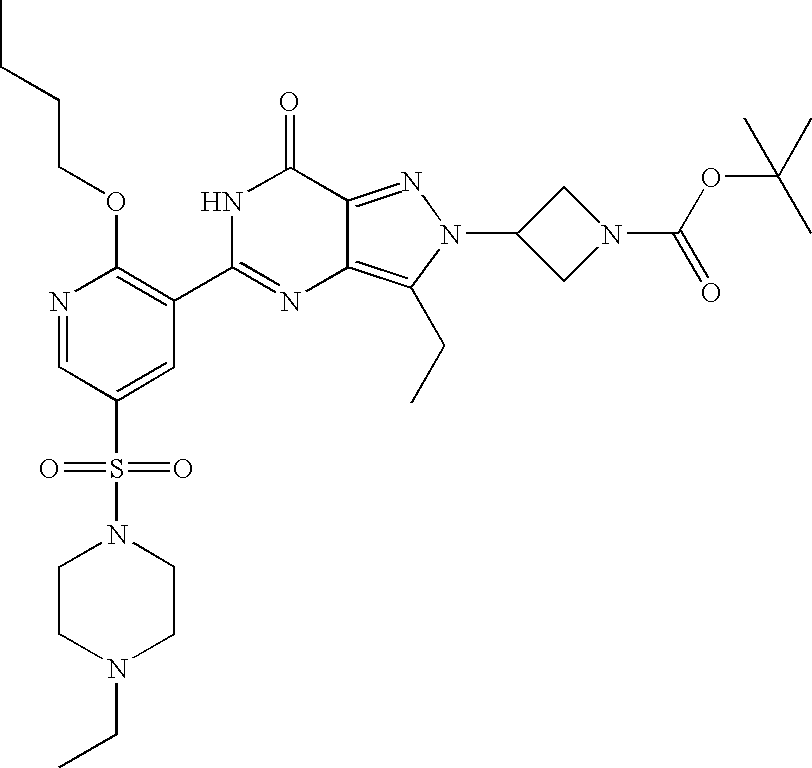
- The title compound was obtained (67%) from the title compound from preparation 63 and n-butanol, following a similar procedure to that described in preparation 61.
- δ (CDCl3): 1.02 (6H, t), 1.38 (3H, t), 1.48 (9H, s), 1.57 (2H, m), 1.96 (2H, m), 2.41 (2H, q), 2.57 (4H, m), 3.02 (2H, q), 3.15 (4H, m), 4.39 (2H, m), 4.68 (4H, m), 5.26 (1H, m), 8.62 (1H, m), 9.02 (1H, m), 10.67 (1H, s).
-

- A mixture of the title compound from preparation 63 (100 mg, 0.16 mmol) and potassium bis(trimethylsilyl)amide (157 mg, 0.79 mmol) in (R)-pentan-2-ol (1 ml), and the mixture heated at 120° C. for 4 days. The cooled mixture was suspended in aqueous saturated sodium bicarbonate solution (35 ml) and extracted with ethyl acetate (2×35 ml). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane:methanol:0.88 ammonia (95:4.7:0.3) as eluant to give the title compound, 14 mg.
- δ (CDCl3): 1.02 (6H, m), 1.38 (3H, t), 1.48 (12H, m), 1.80 (1H, m), 1.98 (1H, m), 2.42 (2H, q), 2.58 (4H, m), 3.02 (2H, q), 3.16 (4H, m), 4.40 (2H, t), 4.67 (2H, m), 5.25 (1H, m), 5.62 (1H, m), 8.62 (1H, s), 9.02 (1H, s), 10.70 (1H, s).
-
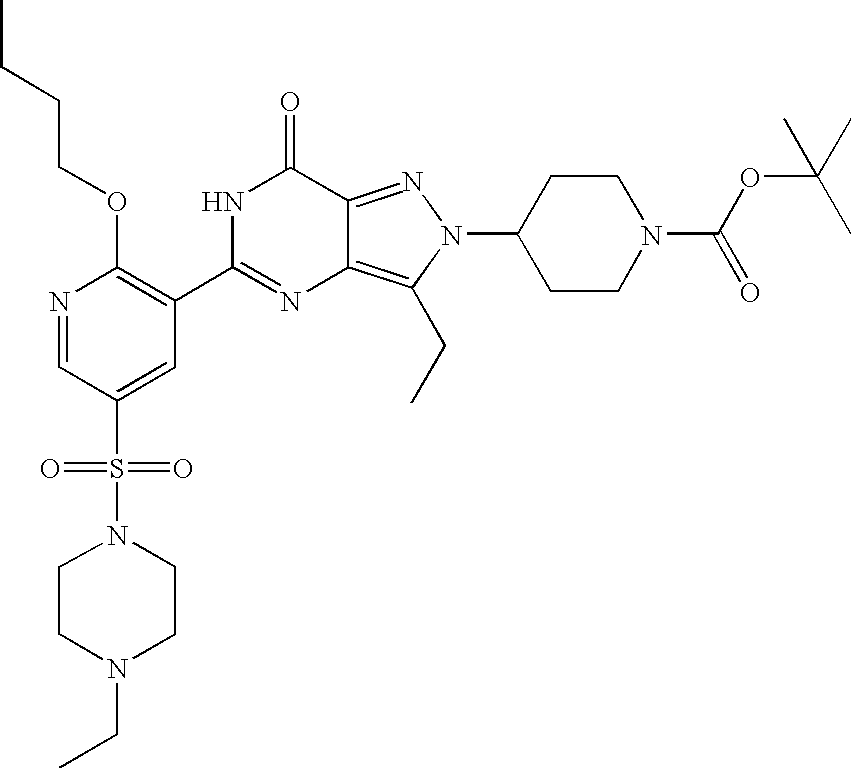
- The title compound was obtained (69%) from the title compound from preparation 46 and n-butanol, following a similar procedure to that described in preparation 62.
- δ (CDCl3): 1.01 (6H, t), 1.34-1.60 (14H, m), 1.93 (4H, m), 2.41 (4H, m), 2.57 (4H, m), 2.90 (2H, m), 3.00-3.20 (6H, m), 4.38 (3H, m), 4.66 (2H, t), 8.61 (1H, d), 9.00 (1H, s), 10.58 (1H, s).
-

- A solution of the title compound from preparation 64 (48 mg, 0.075 mmol) in trifluoroacetic acid (0.5 ml) and dichloromethane (0.5 ml) was stirred at room temperature for 2½ hours. The mixture was concentrated under reduced pressure and the residue triturated well with ether. The solid was then sonicated in ether for a minute, and the resulting precipitate filtered and dried to give the title compound, 54 mg.
- δ (DMSOd6): 1.16 (3H, t), 1.22-1.38 (6H, m), 2.10 (2H, m), 2.38 (2H, m), 3.00 (2H, q), 3.07-3.54 (14H, m), 4.50 (2H, q), 5.85 (1H, m), 8.24 (1H, s), 8.44 (1H, br s), 8.74 (2H, m), 11.90 (1H, s).
-

- A mixture of the title compound from preparation 58 (1.0 g, 2.2 mmol), and potassium bis(trimethylsilyl)amide (2.16 g, 10.8 mmol) in 2-methoxyethanol (20 ml) was heated under reflux for 18 hours. The cooled mixture was evaporated under reduced pressure and the residue purified by column chromatography on silica gel using an elution gradient of dichloromethane:methanol (100:0 to 90:10) to give the title compound, 860 mg.
- δ (CDCl3): 1.03 (3H, t), 1.42 (3H, t), 2.43 (2H, q), 2.59 (4H, m), 3.02 (2H, q), 3.18 (4H, m), 3.59 (3H, s), 4.80 (2H, t), 8.63 (1H, d), 9.00 (1H, d), 11.25 (1H, br s).
- LRMS: m/z 492 (M+1)+
-
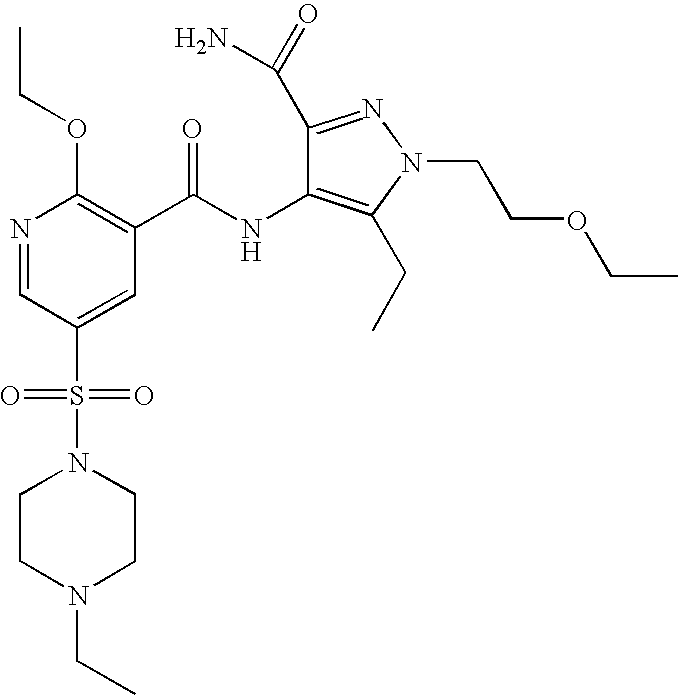
- 2-Bromoethyl ethyl ether (0.28 ml, 2.50 mmol) was added to a mixture of the title compound from preparation 34 (1.0 g, 2.09 mmol) and cesium carbonate (816 mg, 2.50 mmol) in N,N-dimethylformamide (20 ml), and the reaction stirred at 60° C. for 12 hours. The mixture was diluted with water (100 ml), and extracted with ethyl acetate (2×100 ml). The combined organic extracts were dried (MgSO4), evaporated under reduced pressure and the residue azeotroped with toluene. The crude product was triturated with ether, the resulting solid filtered and dried to afford the title compound as a crystalline solid, 550 mg.
- d (DMSOd6): 0.92 (3H, t), 1.10 (6H, m), 1.44 (3H, t), 2.30 (2H, q), 2.42 (4H, m), 2.80 (2H, q), 2.96 (4H, m), 3.40 (2H, q), 3.78 (2H, t), 4.24 (2H, t), 4.63 (2H, q), 7.29 (1H, s), 7.40 (1H, s), 8.40 (1H, d), 8.66 (1H, d), 10.40 (1H, s).
- LRMS: m/z 552 (M+1)+
-
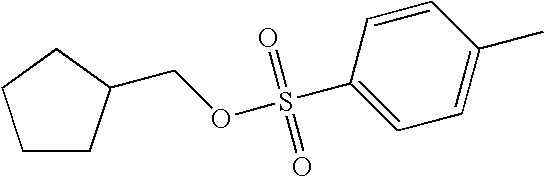
- p-Toluenesulphonyl chloride (2.12 g, 11.1 mmol) was added to a solution of cyclopentanemethanol (1 ml, 9.25 mmol) in ether (25 ml), and the solution cooled in an ice/salt bath. Freshly powdered potassium hydroxide (4.7 g, 83.3 mmol) was added and the reaction mixture allowed to warm to room temperature, over 2 hours. The reaction was diluted with water, the phases separated, and the aqueous layer extracted with ether. The combined organic solutions were dried (MgSO4), and evaporated under reduced pressure, to give the title compound as a clear oil, 2.18 g.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.20 (2H, m), 1.55 (4H, m), 1.74 (2H, m), 2.20 (1H, m), 2.43 (3H, s), 3.92 (2H, d), 7.36 (2H, d), 7.80 (2H, d).
- LRMS: m/z 277 (MNa+)
-

- Methanesulphonyl chloride (1.82 ml, 23.5 mmol) was added dropwise over 10 minutes to an ice-cold solution of tetrahydro-2H-pyran-4-ol (2.0 g, 19.6 mmol) and triethylamine (3.56 ml, 25.5 mmol) in dichloromethane (20 ml), and the reaction then stirred at room temperature for 72 hours. The reaction was washed with saturated aqueous sodium bicarbonate solution (10 ml), dried (MgSO4) and evaporated under reduced pressure to give an orange oil, that solidified on standing, 3.1 g.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.88 (2H, m), 2.03 (2H, m), 3.01 (3H, s), 3.55 (2H, m), 3.95 (2H, m), 4.90 (1H, m).
- LRMS: m/z 198 (MNH4)+
- Anal. Found: C, 39.90; H, 6.74. C6H12O4S requires C, 39.99; H, 6.71%.
-

- The title compound was prepared according to the method described in Tetrahedron 41; 17; 1985; 3447.
-

- A solution of methanesulphonic anhydride (8.33 g, 47.8 mmol) in dichloromethane (30 ml) was added dropwise over 30 minutes to an ice-cooled solution of (R)-2-butanol (4.0 ml, 43.5 mmol) and triethylamine (6.65 ml, 47.8 mmol) in dichloromethane (70 ml). The reaction was then allowed to warm to room temperature and stirred for 18 hours. The mixture was then washed with water, 2N hydrochloric acid, then dried (Na2SO4) and evaporated under reduced pressure to give the title compound as a pale yellow oil, 7.0 g.
- 1Hnmr (CDCl3, 300 MHz) δ: 0.98 (3H, t), 1.40 (3H, d), 1.62-1.80 (2H, m), 3.00 (3H, s), 4.76 (1H, m).
-

- The title compound was obtained as an oil, in 54% yield from (S)-2-butanol and methanesulphonic anhydride, following the procedure described in preparation 74.
- 1Hnmr (CDCl3, 300 MHz) δ: 0.96 (3H, t), 1.38 (3H, d), 1.60-1.76 (2H, m), 2.96 (3H, s), 4.70 (1H, m).
-

- Sodium methoxide (54 g, 1.0 mol) was added portionwise to ice-cooled methanol (1000 ml), and the resulting solution stirred for 20 minutes in an ice-bath. (R)-Propylene oxide (58 g, 1 mol) was added dropwise over 30 minutes, and once addition was complete, the reaction was stirred at room temperature for 18 hours. The mixture was concentrated under reduced pressure, and acidified, with ice-cooling, using (1M) ethereal hydrochloric acid, and the resulting mixture stirred for an hour, then filtered. The filtrate was dried (K2CO3), filtered and evaporated under reduced pressure. The residue was heated to 70° C. over dried calcium oxide for 30 minutes, then distilled at atmospheric pressure to afford the title compound as an oil, 25.4 g.
- b.p. 118-120° C.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.16 (3H, d), 2.28 (1H, d), 3.20 (1H, m), 3.36 (1H, m), 3.40 (3H, s), 3.97 (1H, m).
- [α]D−20.83° (c=1.02, dichloromethane)
-

- Triethylamine (8.5 ml, 61 mmol) was added to a solution of the alcohol from preparation 76 (5.0 g, 55 mmol) in dichloromethane (100 ml), and the solution cooled in an ice/acetone bath. A solution of methanesulphonic anhydride (10.64 g, 61 mmol) in dichloromethane (50 ml) was added dropwise over 30 minutes, then the reaction stirred at room temperature for 18 hours. The reaction mixture was washed with water, and 2M hydrochloric acid, then dried (Na2SO4), and evaporated under reduced pressure to give the title compound, 2.77 g.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.39 (3H, d), 3.03 (3H, s), 3.38 (3H, s), 3.44 (2H, m), 4.87 (1H, m).
-

- S(−)-propylene oxide (17.58 g, 0.30 mol) was added dropwise over 45 minutes, to a freshly prepared solution of sodium (7.0 g, 0.30 mol) in methanol (100 ml), and the mixture stirred at room temperature for 18 hours. The reaction was diluted with pentane (150 ml), then acetic acid (17 ml, 0.30 mol) added slowly. The resulting mixture was filtered through Celite®, and the filtrate concentrated under reduced pressure. The residual oil was distilled at 30 Torr, and fractions boiling at 30° C. were collected, to give 3.3 g of an oil, containing about 30% methanol.
- Triethylamine (5.56 ml, 0.04 mol) was added to a solution of this oil in dichloromethane (60 ml), then the solution cooled in ice. A solution of methanesulphonic anhydride (7.03 g, 0.04 mol) in dichloromethane (30 ml) was added dropwise over 30 minutes, then the reaction stirred at room temperature for 18 hours. The mixture was washed with water, and 2M hydrochloric acid, then dried (MgSO4), and evaporated under reduced pressure to give the title compound, 3.3 g, which was used without further purification.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.39 (3H, d), 3.03 (3H, s), 3.38 (3H, s), 3.44 (2H, m), 4.87 (1H, m).
-

- A suspension of 2-ethoxy-3-pyridinecarboxylic acid (16.4 g, 98 mmol), and cesium carbonate (32 g, 98 mmol) in N,N-dimethylformamide (240 ml) was stirred at room temperature for 2 hours. Ethyl iodide (7.85 ml, 98 mmol) was added and the reaction stirred for a further 24 hours. The reaction mixture was concentrated under reduced pressure and the residue partitioned between aqueous sodium carbonate solution (100 ml) and ethyl acetate (100 ml). The phases were separated and the aqueous layer extracted with ethyl acetate (2×100 ml). The combined organic solutions were washed with brine, dried (Na2SO4) and evaporated under reduced pressure to afford the ethyl ester, 18.0 g, as a pale yellow oil.
- Ammonium nitrate (5.36 g, 66 mmol) was added portionwise to an ice-cooled solution of the oil (4.66 g, 22.3 mmol) in trifluoroacetic anhydride (50 ml) and the reaction stirred for 18 hours at room temperature. The reaction mixture was carefully poured into ice water (200 ml) and the resulting suspension stirred for an hour. The precipitate was filtered off, washed with water and dried under suction to afford the nitro ester as a solid, 3.29 g.
Aqueous sodium hydroxide solution (4 ml, 5N, 20 mmol) was added dropwise to a solution of the solid (5.1 g, 20 mmol) in ethanol (100 ml) and the reaction stirred at room temperature for 18 hours. The reaction mixture was concentrated under reduced pressure, the residue suspended in water (50 ml) and acidified topH 3 with hydrochloric acid. This aqueous solution was extracted with ethyl acetate (3×100 ml), the combined organic layers washed with brine (100 ml), dried (Na2SO4) and evaporated under reduced pressure to give a beige solid. The crude product was recrystallised from ethyl acetate/hexane to afford the title compound, 3.32 g, as beige crystals. - 1Hnmr (CDCl3, 300 MHz) δ: 1.55 (3H, t), 4.78 (2H, q), 9.17 (1H, s), 9.23 (1H, s).
-

- A mixture of the acid from preparation 79 (4.46 g, 21.0 mmol), the pyrazole from preparation 9 (4.15 g, 19.6 mmol), 1-hydroxybenzotriazole hydrate (3.51 g, 26.0 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (4.98 g, 26.0 mmol) and N-ethyldiisopropylamine (10.38 ml, 60.0 mmol) in dichloromethane (110 ml) was stirred at room temperature for 18 hours. The reaction was diluted with dichloromethane (100 ml), then washed consecutively with water (70 ml), 10% aqueous sodium bicarbonate solution (70 ml), and brine (70 ml), then dried (Na2SO4) and concentrated under reduced pressure. The residual yellow solid was purified by column chromatography on silica gel using dichloromethane:methanol (95:5) as eluant. The product was recrystallised from ethyl acetate to afford the title compound as a pale yellow crystalline solid, 3.96 g.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.21 (3H, t), 1.59 (3H, t), 2.94 (2H, q), 3.35 (3H, s), 3.80 (2H, t), 4.27 (2H, t), 4.83 (2H, q), 5.29 (1H, br s), 6.62 (1H, br s), 9.15 (1H, d), 9.32 (1H, d), 10.51 (1H, br s).
- LRMS: m/z 407.5 (MH+)
- Anal. Found: C, 50.21; H, 5.39; N, 20.66. C17H22N6O6 requires C, 50.24; H, 5.46; N, 20.68%.
-
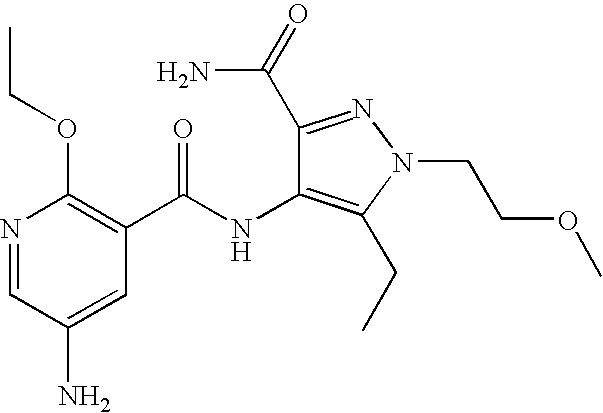
- A mixture of the nitro compound from preparation 80 (3.86 g, 9.50 mmol), and 10% palladium on charcoal (200 mg) in dichloromethane (75 ml) and ethanol (25 ml) was hydrogenated at 50 psi and room temperature for 2 hours. The mixture was diluted with dichloromethane, then filtered through Solkafloc®, and the filtrate evaporated under reduced pressure to give the title compound, 3.63 g.
- 1Hnmr (DMSOd6, 400 MHz) δ: 1.06 (3H, t), 1.37 (3H, t), 2.75 (2H, q), 3.23 (3H, s), 3.72 (2H, t), 4.24 (2H, t), 4.39 (2H, q), 5.02 (2H, br s), 7.25 (1H, br s), 7.37 (1H, br s), 7.70 (2H, m), 10.33 (1H, s).
- LRMS: m/z 377.2 (MH+)
-
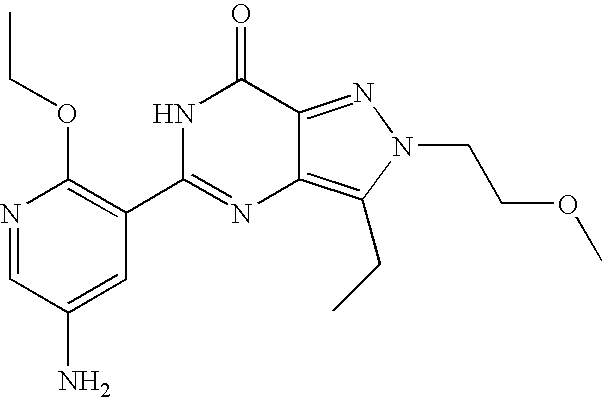
- A mixture of the amine from preparation 81 (2.53 g, 6.72 mmol), and potassium bis(trimethylsilyl)amide (5.56 g, 27.9 mmol) in ethanol (50 ml) was heated at 120° C. in a sealed vessel for 8 hours. The cooled reaction was evaporated under reduced pressure and the residue purified by column chromatography on silica gel using an elution gradient of ethyl acetate:ethanol (100:0 to 96:4) to afford the title compound, 1.96 g.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.40 (3H, t), 1.51 (3H, t), 3.06 (2H, q), 3.30 (3H, s), 3.57 (2H, br s), 3.90 (2H, t), 4.45 (2H, t), 4.55 (2H, q), 7.77 (1H, d), 8.18 (1H, d), 11.03 (1H, br s).
- LRMS: m/z 359.1 (MH+)
-

- Cesium carbonate (2.7 g, 8.31 mmol) was added to a solution of the compound from preparation 34 (1.8 g, 3.76 mmol) in N,N-dimethylformamide (40 ml), followed by cyclobutyl bromide (388 μl, 4.13 mmol), and the reaction mixture stirred at 60° C. for 3 days. The cooled solution was partitioned between ethyl acetate and sodium bicarbonate solution, and the layers separated. The aqueous phase was extracted with ethyl acetate (3×), the combined organic solutions dried (MgSO4), and evaporated under reduced pressure. The residual yellow solid was triturated with ether to afford the title compound as a pale yellow powder, 762 mg.
- 1Hnmr (CDCl3, 400 MHz) δ: 1.00 (3H, t), 1.20 (3H, t), 1.57 (3H, t), 1.88 (2H, m), 2.40 (4H, m), 2.52 (4H, m), 2.70 (2H, m), 2.82 (2H, q), 3.08 (4H, m), 4.78 (3H, m), 5.24 (1H, br s), 6.75 (1H, br s), 8.62 (1H, s), 8.81 (1H, s), 10.50 (1H, s).
- The compounds of the following general structure:
-

- were prepared from the compound from preparation 34 and the appropriate alkylating agent, following a similar method to that described in preparation 83.
-
Prep. Alkylating Yield No. R agent (%) Data 84 
bromide 54 1Hnmr (CDCl3, 400 MHz) δ: 1.02 (3 H, t), 1.22 (3 H, t), 1.58 (3 H, t), 1.71 (2 H, m), 1.97 (2 H, m), 2.08 (4 H, m), 2.40 (2 H, q), 2.52 (4 H, m), 2.92 (2 H, q), 3.10 (4 H, m), 4.65 (1 H, m), 4.78 (2 H, q), 5.21 (1 H, br s, 6.66 (1 H, br s), 8.64 (1 H, d), 8.82 (1 H, d), 10.50 (1 H, s). LRMS: m/z 548 (MH+) 85 
tosylate 52 1Hnmr (CDCl3, 400 MHz) δ: 1.02 (3 H, t), 1.15-1.38 (6 H, m), 1.58-1.72 (6 H, m), 2.37-2.57 (7 H, m), 2.94 (2 H, m), 3.16 (4 H, m), 4.00 (2 H, d), 4.78 (2 H, q), 5.20 (1 H, br s), 6.64 (1 H, br s), 8.64 (1 H, s), 8.83 (1 H, s), 10.54 (1 H, s). LRMS: m/z 561.8 (M+) 861 
mesylate 25 1Hnmr (CDCl3, 300 MHz) δ: 1.01 (3 H, t), 1.21 (3 H, t), 1.42 (2 H, m), 1.58 (3 H, t), 1.94 (6 H, m), 2.40 (4 H, m), 2.52 (4 H, m), 2.88 (2 H, q), 3.08 (4 H, m), 4.76 (3 H, m), 5.20 (1 H, br s), 6.66 (1 H, br s), 8.62 (1 H, d), 8.82 (1 H, d), 10.60 (1 H, s). LRMS: m/z 562.3 (MH+) 87 
mesylate 21 1Hnmr (CDCl3, 300 MHz) δ: 1.00 (3 H, t), 1.30 (3 H, t), 1.55 (3 H, t), 1.80 (2 H, m), 2.35 (2 H, m), 2.40 (2 H, q), 2.55 (4 H, m), 2.90 (2 H, q), 3.10 (4 H, m), 3.50 (2 H, t), 4.14 (2 H, m), 4.30 (1 H, m), 4.80 (2 H, q), 5.22 (1 H, br s), 6.66 (1 H, br s), 8.60 (1 H, s), 8.80 (1 H, s), 10.50 (1 H, s). LRMS: m/z 565 (MH+) 88 
bromide 52 1Hnmr (CDCl3, 300 MHz) δ: 1.00 (3 H, t), 1.20 (3 H, t), 1.60 (3 H, t), 2.18 (2 H, m), 2.40 (2 H, q), 2.50 (4 H, m), 2.90 (2 H, q), 3.08 (4 H, m), 3.32 (3 H, s), 3.40 (2 H, t), 4.20 (2 H, t), 4.80 (2 H, q), 5.22 (1 H, br s), 6.64 (1 H, br s), 8.60 (1 H, s), 8.80 (1 H, s), 10.50 (1 H, br s). LRMS: m/z 553 (MH+) 1= product purified by column chromatography eluting with dichloromethane:methanol (97:3). -
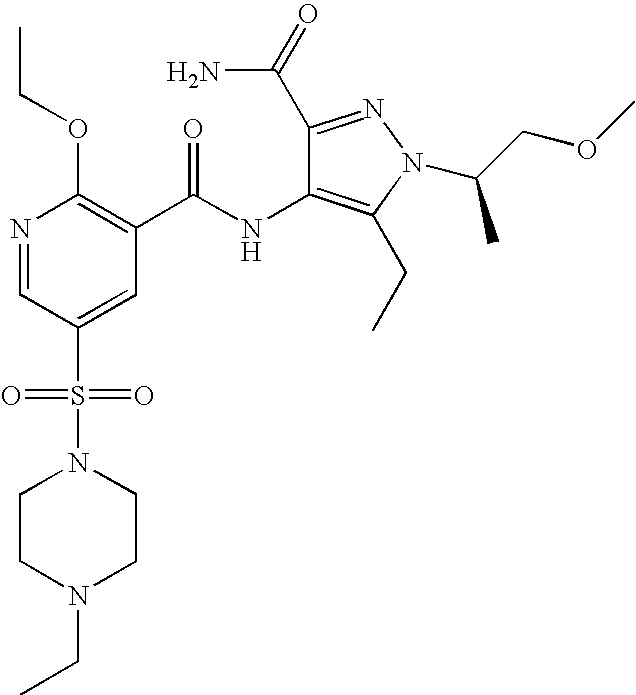
- Cesium carbonate (3.00 g, 9.20 mmol) was added to a solution of the compound from preparation 34 (2.0 g, 4.17 mmol) in N,N-dimethylformamide (30 ml), and the mixture stirred for 30 minutes. The mesylate from preparation 78 (0.77 g, 4.58 mmol) was added and the reaction stirred at 60° C. for 8 hours. The cooled mixture was partitioned between ethyl acetate and water, and the pH adjusted to 8, using solid carbon dioxide. The layers were separated, and the aqueous phase extracted with ethyl acetate (2×). The combined organic extracts were dried (Na2SO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using an elution gradient of methanol:dichloromethane (1:99 to 8:92) to afford the title compound, 300 mg.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.02 (3H, t), 1.23 (3H, t), 1.48 (3H, d), 1.58 (3H, t), 2.40 (2H, q), 2.52 (4H, m), 2.90 (2H, m), 3.08 (4H, m), 3.30 (3H, s), 3.60 (1H, m), 3.78 (1H, m), 4.56 (1H, m), 4.78 (2H, q), 5.30 (1H, br s), 6.66 (1H, br s), 8.63 (1H, d), 8.82 (1H, d), 10.48 (1H, s).
- LRMS: m/z 552.3 (MH+)
-

- The title compound was obtained as an oil in 52% yield, from the compound from preparation 34 and the mesylate from preparation 77, following the procedure described in preparation 89.
- 1Hnmr (CDCl3, 300 MHz) δ: 1.01 (3H, t), 1.22 (3H, t), 1.48 (3H, d), 1.58 (3H, t), 2.40 (2H, q), 2.54 (4H, m), 2.90 (2H, m), 3.08 (4H, m), 3.30 (3H, s), 3.61 (1H, m), 3.78 (1H, m), 4.56 (1H, m), 4.78 (2H, q), 5.25 (1H, br s), 6.66 (1H, br s), 8.63 (1H, d), 8.82 (1H, d), 10.48 (1H, s).
- LRMS: m/z 552.4 (MH+)
- The compounds of the following general structure:
-
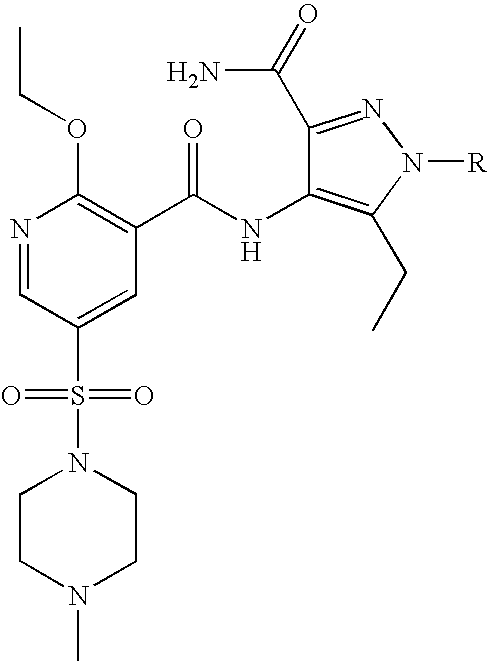
- were prepared from the compound from preparation 35 and the appropriate alkylating agent, following a similar method to that described in preparation 83.
-
Prep. Alkylating Yield No. R agent (%) Data 911 
bromide 34 1Hnmr (CDCl3, 400 MHz) δ: 0.95 (3 H, t), 1.20 (3 H, t), 1.40 (2 H, m), 1.60 (3 H, t), 1.86 (2 H, m), 2.25 (3 H, s), 2.46 (4 H, m), 2.88 (2 H, q), 3.09 (4 H, m), 4.05 (2 H, t), 4.75 (2 H, t), 5.25 (1 H, br s), 6.65 (1 H, br s), 8.65 (1 H, s), 8.85 (1 H, s), 10.55 (1 H, s). 92 
bromide 47 1Hnmr (CDCl3, 400 MHz) δ: 0.41 (2 H, m), 0.62 (2 H, m), 1.22 (4 H, m), 1.59 (3 H, t), 2.26 (3 H, s), 2.48 (4 H, m), 2.98 (2 H, q), 3.10 (4 H, m), 3.98 (2 H, d), 4.78 (2 H, q), 5.27 (1 H, br s), 6.68 (1 H, br s), 8.65 (1 H, d), 8.85 (1 H, d), 10.57 (1 H, s). 93 
mesylate 45 1Hnmr (CDCl3, 400 MHz) δ: 0.82 (3 H, t), 1.22 (3 H, t), 1.60 (6 H, m), 1.80 (1 H, m), 2.00 (1 H, m), 2.23 (3 H, s), 2.50 (4 H, m), 2.85 (2 H, m), 3.10 (4 H, m), 4.22 (1 H, m), 4.80 (2 H, q), 5.20 (1 H, br s), 6.70 (1 H, br s), 8.60 (1 H, s), 8.82 (1 H, s), 10.50 (1 H, s). LRMS: m/z 522.0 (MH+) 94 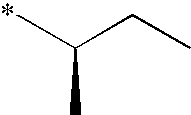
mesylate 42 1Hnmr (CDCl3, 400 MHz) δ: 0.82 (3 H, t), 1.22 (3 H, t), 1.60 (6 H, m), 1.80 (1 H, m), 2.00 (1 H, m), 2.23 (3 H, s), 2.50 (4 H, m), 2.85 (2 H, m), 3.10 (4 H, m), 4.22 (1 H, m), 4.80 (2 H, q), 5.20 (1 H, br s), 6.70 (1 H, br s), 8.60 (1 H, s), 8.82 (1 H, s), 10.50 (1 H, s). LRMS: m/z 522.0 (MH+) 1= purified by column chromatography on silica gel, using dichloromethane:methanol (100:0 to 98:2). - 2-Hydroxynicotinic acid (27 Kg, 194.2 mol) was added portionwise to 30% oleum (58.1 Kg) at 50° C. over 1 hr. This caused an exotherm to 82° C. The reaction mixture was heated further to 140° C. After maintaining this temperature for 12 hrs the reactor contents were cooled to 15 C and filtered. The filter cake was then re-slurried with acetone (33 Kg) at room temperature, filtered and dried to afford the title compound (35.3 Kg, 83%) as a white solid. Decomposition pt 273° C. δ (DMSOd6): 7.93 (1H, d), 8.42 (1H, d). m/z
- (Found:220 [M+H]+, 100%. C6H6NO6S requires 220.17).
- 2-Hydroxy-5-sulfonicotinic acid (500 g, 2.28 mol) was dissolved in ethanol (2.5 L) with stirring and heated to 80° C. After 30 mins 0.5 L of solvent was distilled off, then replaced with fresh ethanol (0.5 L) and taken back to 80° C. After a further 60 mins 1.0 L of solvent was distilled off, then replaced with fresh ethanol (1.0 L) and taken back to 80° C. After a further 60 mins 1.0 L of solvent was distilled off, the reaction cooled to 22° C. and stirred for 16 hr. The precipitated product was filtered, washed with ethanol (0.5 L) and dried at 50° C. under vacuum to afford the title compound (416 g, 74%) as a white solid. Decomposition pt 237° C. δ (DMSOd6): 1.25 (3H, t), 4.19 (2H, q), 7.66 (1H, d), 8.13 (1H, d). m/z (Found:248 [M+H]+, 100%. C8H6NO6S requires 248.22).
- Ethyl 2-hydroxy-5-sulfonicotioate (24.7 g, 0.1 mol) was slurried in thionyl chloride (238 g, 2.0 mol) and dimethylformamide (1.0 mL) with stirring. The reaction mixture was then heated to reflux for 2.5 hr. The bulk of the thionyl chloride was removed under vacuum with residual thionyl chloride removed with a toluene azeotrope to afford the crude title compound (30.7 g, 108%) as a yellow oil. d (CDCl3): 1.46 (3H, t), 4.50 (2H, q), 8.72 (1H, d), 9.09 (1H, d). This was taken directly onto the next step.
- Crude ethyl 2-chloro-5-chlorosulfonicotinoate (30.7 g, 0.1 mol assumed) was dissolved in ethyl acetate (150 mL) with stirring then ice cooled. To this was added a solution of N-ethylpiperazine (11.4 g, 0.1 mol) and triethylamine (22.5 g, 0.22 mol) in ethyl acetate (50 mL), carefully over 30 mins, keeping the internal temperature below 10° C. Once the addition was complete the reaction was allowed to warm to 22° C. and stir for 1 hr. The solid was filtered off and the remaining filtrate was concentrated under vacuum to afford the crude title compound (37.1 g, 103%) as a crude yellow gum. δ (CDCl3): 1.10 (3H, t), 1.42 (3H, m), 2.50 (2H, m), 2.60 (4H, m), 3.19 (4H, m), 4.43 (2H, q), 8.40 (1H, d), 8.80 (1H, d). m/z (Found:362 [M+H]+, 100%. C14H21ClN3O4S requires 362.85).
- A solution of Ethyl 2-chloro-5-(4-ethyl-1-piperazinylsulfonyl)nicotinoate (36.1 g, 0.1 mol) in ethanol (180 mL) was cooled to 10° C. with stirring. Sodium ethoxide (10.2 g, 0.15 mol) was added portionwise keeping the temperature below 20° C. The reaction mixture was then stirred at ambient temperature for 18 hours. The precipitate was filtered off and water (180 mL) added to the filtrate. The filtrate was then heated to 40° C. for 1 hour. Ethanol (180 mL) was then distilled off at ambient pressure and the remaining aqueous solution allowed to cool to ambient temperature. The precipitated product was then filtered off, washed with water and dried under vacuo at 50° C. to afford the title compound (12.6 g, 34%) as a light brown solid. M.p. 66-68° C. δ (CDCl3): 1.04 (3H, t), 1.39 (3H, t), 1.45 (3H, t), 2.41 (2H, q), 2.52 (4H, m), 3.08 (4H, m), 4.38 (2H, q), 2.57 (2H, q), 8.38 (1H, d), 8.61 (1H, d). m/z (Found:372 [M+H]+, 100%. C16H26N3O5S requires 372.46).
- Ethyl 2-ethoxy-5-(4-ethyl-1-piperazinylsulfonyl)nicotinoate (10.2 g, 0.0275 mol) was dissolved in toluene (50 mL) and a solution of sodium hydroxide (1.1 g, 0.0275 mol) in water (20 mL) added to it. This two phase mixture was then stirred vigorously at ambient temperature overnight. The aqueous phase was separated off and adjusted to pH=5.6 by addition of c. hydrochloric acid. The precipitated product was slurried with ice cooling for 15 minutes, filtered, water washed and dried under vacuo at 50° C. to afford the title compound as an off-white solid. Mpt 206-207° C. δ (CDCl3): 1.25 (3H, t), 1.39 (3H, t), 2.82 (2H, q), 3.03 (4H, m), 3.25 (4H, m), 4.50 (2H, q), 8.25 (1H, d), 8.56 (1H, d). m/z (Found:344 [M+H]+, 100%. C14H22N3O5S requires 344.38).
- This step 95(f) is already set out in preparation 23 of PCT/IB99/00519 (herein incorporated by reference) and the yield obtained is 88%.
-
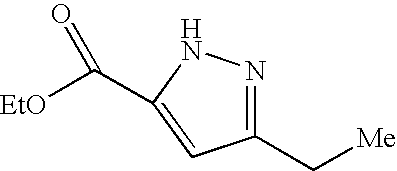
- To a stirred solution of 2,2-dimethoxybutane (10 g, 84.7 mMol) in CH2Cl2 (50 mL) under a nitrogen atmosphere at 0° C. was added pyridine (13.7 mL, 169.5 mMol). The reaction mixture was maintained at 0° C. and a solution of trichloroacetyl chloride (18.9 mL, 169.5 mMol) in CH2CL2 (35 mL) was added over 1 hour with constant stirring. The yellow-orange solution begins to precipitate a white solid as the reaction progresses. The reaction mixture is allowed to warm to room temperature over 20 h. The reaction mixture was diluted with ethanol (150 mL) and re-cooled to 0° C. before treatment with hydrazine hydrate (8.2 mL, 169.5 mMol) as a solution in ethanol (35 mL) over 30 min. The reaction was heated to 50° C. and solvent was distilled at atmospheric pressure. The temperature was increased until the head temperature reached 78° C. Reflux was maintained for a further 2 h, before cooling to room temperature. The reaction mixture was diluted with water (250 mL) and ethanol was removed by evaporation at reduced pressure. The resultant mixture was extracted with CH2Cl2 (3×200 mL). The combined organics were dried (MgSO4), filtered and evaporated at reduced pressure to afford the title compound as a brown oil, 12.05 g, 85%.
- 1H NMR (300 MHz, CDCl3): δ=1.20 (3H, t), 1.28 (3H, t), 2.67 (2H, q), 4.29 (2H, q), 6.55 (1H, s), 12.56 (1H, s).
- LRMS m/z=167.1 [M−H]+, C8H12N2O2 requires 168.2.
-
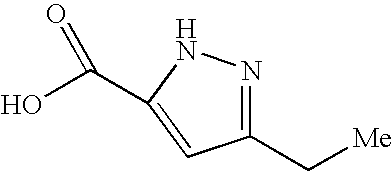
- Aqueous sodium hydroxide solution (10M; 100 ml, 1.0 mol) was added dropwise to a stirred suspension of the title compound of Preparation 96(a) (66.0 g, 0.39 mol) in methanol and the resulting solution heated under reflux for 4 hours. The cool reaction mixture was concentrated under reduced pressure to ca. 200 ml, diluted with water (200 ml) and this mixture washed with toluene (3×100 ml). The resulting aqueous phase was acidified with concentrated hydrochloric acid to pH 4 and the white precipitate collected and dried by suction to provide the title compound (34.1 g). δ (DMSOd6): 1.13 (3H, t), 2.56 (2H, q), 6.42 (1H, s).
- Fuming sulphuric acid (17.8 ml) was added dropwise to stirred, ice-cooled fuming nitric acid (16.0 ml), the resulting solution heated to 50° C., then 3-n-propyl-1H-pyrazole-5-carboxylic acid (Chem. Pharm. Bull., 1984, 32, 1568; 16.4 g, 0.106 mol) added portionwise over 30 minutes whilst maintaining the reaction temperature below 60° C. The resulting solution was heated for 18 hours at 60° C., allowed to cool, then poured onto ice. The white precipitate was collected, washed with water and dried by suction to yield the title compound (15.4 g), m.p. 170-172° C. Found: C, 42.35; H, 4.56; N, 21.07. C7H9N3O4 requires C, 42.21; H, 4.55; N, 21.10%. δ (DMSOd6): 0.90 (3H, t), 1.64 (2H, m), 2.83 (2H, m), 14.00 (1H, s).
-

- Obtained from the title compound of Preparation 96(b), by analogy with the process of Preparation 96(c), as a brown solid (64%). δ (DMSOd6): 1.18 (3H, t), 2.84 (2H, m), 13.72 (1H, s).
- A solution of the title compound of Preparation 96(c) (15.4 g, 0.077 mol) in thionyl chloride (75 ml) was heated under reflux for 3 hours and then the cool reaction mixture evaporated under reduced pressure. The residue was azeotroped with tetrahydrofuran (2×50 ml) and subsequently suspended in tetrahydrofuran (50 ml), then the stirred suspension ice-cooled and treated with gaseous ammonia for 1 hour. Water (50 ml) was added and the resulting mixture evaporated under reduced pressure to give a solid which, after trituration with water and drying by suction, furnished the title compound (14.3 g), m.p. 197-199° C. Found: C, 42.35; H, 5.07; N, 28.38. C7H10N4O3 requires C, 42.42; H, 5.09; N, 28.27%. δ (DMSOd6): 0.90 (3H, t), 1.68 (2H, m), 2.86 (2H, t), 7.68 (1H, s), 8.00 (1H, s).
-

- Obtained from the title compound of Preparation 96(d), by analogy with Preparation 96(e), as a white solid (90%). δ (DMSOd6): 1.17 (3H, t), 2.87 (2H, m), 7.40 (1H, s), 7.60 (1H, s), 7.90 (1H, s). LRMS: m/z 185 (M+1)+.
-

- A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (2.5 kg, 13.6 Mol), sodium carbonate (1.8 Kg, 17.0 Mol) and 2-bromoethyl methyl ether (1.98 kg, 14.2 Mol) in THF (22.5 L) and water (2.5 L) was heated under reflux and stirred for 20 hours. The mixture was cooled to ambient temperature and CH2Cl2 (67.5 L) and water (22.5 L) were added. The resultant organic and aqueous layers were separated. The aqueous phase was extracted with CH2Cl2 (22.5 L) and the combined organic solution was distilled under atmospheric pressure and replaced with ethyl acetate (33 L) to a final volume of 17 L. The cooled mixture was granulated at ambient temperature for 2 hours, filtered and washed with ethyl acetate (2.5 L). This afforded 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a white crystalline solid, 2.10 kg, 57%. m.p.=140° C. Found: C, 44.46; H, 5.79; N, 23.01. C9H14N4O4 requires C, 44.63; H, 5.79; N, 23.14%.
- δ (CDCl3): 1.18 (3H, t), 2.98 (2H, q), 3.22 (3H, s), 3.77 (2H, t), 4.28 (2H, q), 6.03 (1H, s), 7.36 (1H, s).
- LRMS: m/z=243 (M+1)+
- A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (25 g, 0.136 Mol), sodium carbonate (18 g, 0.17 Mol) and sodium iodide (20.4 g, 0.136 Mol) were suspended in ethyl methyl ketone (125 mL) at room temperature. 2-bromoethyl methyl ether (12.8 mL, 0.142 Mol) was added and the mixture was heated to reflux and stirred for 70 hours. The mixture was cooled to ambient temperature and water (250 mL) was added. The resultant slurry was warmed to reflux and held at that temperature for 30 min before cooling to room temperature. The resultant precipitate was granulated at room temperature for 3 h, filtered and vacuum dried to afford 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a yellow crystalline solid 24.3 g, 74%. Data as reported for Example (b)(i).
-

- A mixture of 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide (20 g, 82.6 mMol) and 5% Pd/C (1 g) in methanol (200 mL) was pressurised at 50 psi/25° C. in a sealed vessel and stirred for 15 hours. At the end of the reaction the mixture was filtered through arbocel and the filter cake was washed with methanol. The methanolic solution was distilled at atmospheric pressure and replaced with ethyl acetate to a final volume of 100 mL. The cooled mixture was granulated at ambient temperature for 2 h filtered and washed with ethyl acetate (20 mL) to afford 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide as a white crystalline solid, 15 g, 88%. m.p.=131° C. Found: C, 50.75; H, 7.62; N, 26.38. C9H16N4O2 requires C, 50.94; H, 7.55; N, 26.42%.
- δ (CDCl3): 1.20 (3H, t), 2.63 (2H, q), 3.32 (3H, s), 3.74 (2H, t), 3.95 (2H, s), 4.15 (2H, t), 5.27 (1H, s), 6.59 (1H, s).
- LRMS: m/z=213 (M+1)+
-
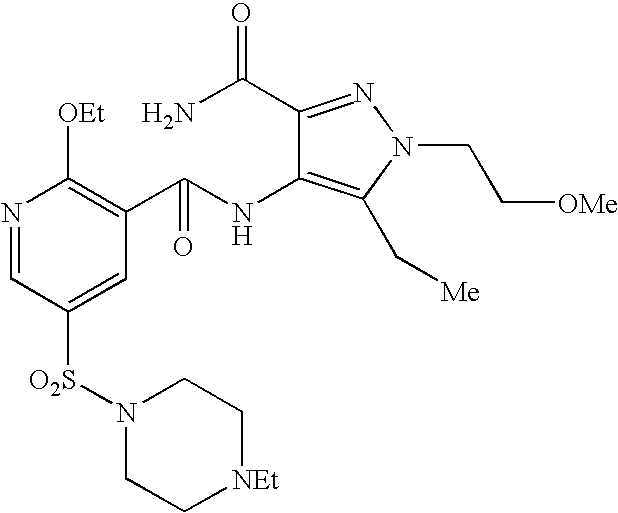
- 2-ethoxy-5-(4-ethyl-1-piperazinylsulfonyl)nicotinic acid (2.31 kg, 6.73 Mol) was suspended in ethyl acetate (16.2 L) and 1,1-carbonyldimidazole (1.09 kg, 6.73 Mol) was added at room temperature. The reaction mixture was heated at 45° C. for 40 minutes and then the reaction was stirred for a further 40 minutes at reflux. After cooling to ambient temperature 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (1.5 kg, 7.06 Mol) was added to the cooled mixture, and the reaction stirred for a further 15 hours under reflux. The mixture was cooled filtered and the filter cake was washed with 90% water/10% ethyl acetate, (2 mL/g) to afford N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl)nicotinamide as an off white crystalline solid, 3.16 kg, 88%. m.p.=156° C.
- Found: C, 51.33; H, 6.56; N, 18.36. C23H35N7O6S requires C, 51.40; H, 6.53; N, 18.25%.
- δ (CDCl3): 1.04 (3H, t), 1.22 (3H, t), 1.60 (3H, t), 2.44 (2H, q), 2.54 (4H, m), 2.96 (2H, q), 3.12 (4H, m), 3.36 (3H, s), 3.81 (2H, t), 4.27 (2H, t), 4.80 (2H, q), 5.35 (1H, s), 6.68 (1H, s), 8.66 (1H, d), 8.86 (1H, d), 10.51 (1H, s).
- LRMS: m/z=539 (M+1)+
- Additionally, in accordance with the invention, the intermediate compounds (XIV) and (XB) (as illustrated in
Schemes 2 and 3) can be prepared from commercially available starting materials (2-hydroxy nicotinic acid) in better yield than the corresponding reaction sequence in PCT/IB99/00519. For example, compound (XIV) (wherein Q and W are OEt) is formed in a yield of 14.5% in preparation 18 of PCT/IB99/00519 (i.e. from a reaction sequence ofpreparation - It will be appreciated that formation of compounds of formula (XB) and (XIV) from (XV) respectively is an independent invention and is preferably prepared from 2-hydroxynicotinic acid as outlined herein. Likewise each and every step (and telescoped step) in
Schemes Schemes - Thus in a further aspect of the invention a compound of formula (XVII) is formed by reacting 2-hydroxynicotinic acid or a salt thereof in the presence of SO3 in a solvent.
Claims (6)
1. A method of treating or preventing benign prostatic hyperplasia, bladder outlet obstruction or incontinence comprising administering to a patient in need thereof a therapeutically effective amount of a compound of the formula (I):
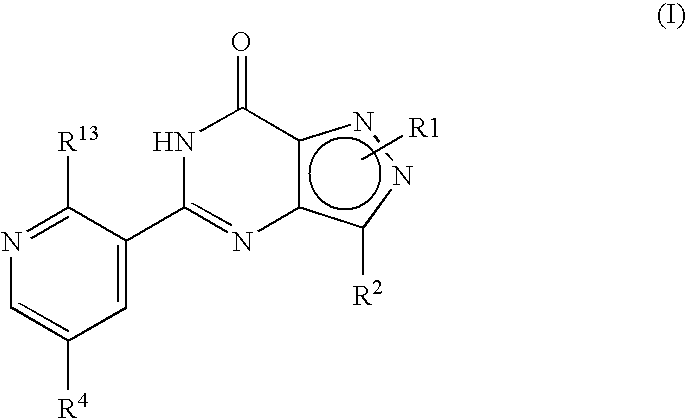
or a pharmaceutically or veterinarily acceptable salt, or a pharmaceutically or veterinarily acceptable solvate or pro-drug thereof, wherein
R1 is C1 to C6 alkyl or C3 to C6 alkenyl, C3 to C6 cycloalkyl or C4 to C6 cycloalkenyl wherein said alkyl group may be branched or straight chain and wherein
when R1 is C1 to C3 alkyl said alkyl group is substituted by;
and wherein when R1 is C4 to C6 alkyl, C3 to C6 alkenyl or C3 to C6 cycloalkyl said alkyl, alkenyl or cycloalkyl group is optionally substituted by;
one or more substituents selected from: hydroxy; C1 to C4 alkoxy; C3 to C6 cycloalkyl; phenyl substituted with one or more substitutents selected from C1 to C3 alkyl, C1 to C4 alkoxy, C1 to C4 haloalkyl, C1 to C4 haloalkoxy, halo, CN, NO2, NHR11, NHCOR12, NHSO2R12, SO2R12, SO2NHR11, COR11 or CO2R11 wherein said haloalkyl and haloalkoxy groups contain one or more halo atoms; NR7R8, CONR7R8 or NR7COR11 wherein R7 and R8 are each independently selected from H, C1 to C4 alkyl, C3 to C4 alkenyl, CO2R9 or SO2R9 and wherein said alkyl or alkenyl groups are optionally substituted by C1 to C4 haloalkyl or C1 to C4 haloalkoxy; Het1; Het2 or Het3;
or R1 is Het4 or phenyl wherein said phenyl group is optionally substituted by one or more substituents selected from C1 to C4 alkyl, C3 to C4 alkenyl, C1 to C4 alkoxy, halo, CN, CF3, OCF3, NO2, NHR11, NHCOR12, NHSO2R12, SO2R12, SO2NHR11, COR11, CO2R11;
R2 is C1 to C6 alkyl, C3 to C6 alkenyl or (CH2)n(C3 to C6 cycloalkyl) wherein n is 0, 1 or 2;
R13 is OR3 or NR5R6;
R3 is C1 to C6 alkyl optionally substituted with one or two substituents selected from C3 to C5 cycloalkyl, hydroxy, C1 to C4 alkoxy, benzyloxy, NR5R6, phenyl, Het1, Het2, Het3 or Het4 wherein the C1 to C6 alkyl and C1 to C4 alkoxy groups may optionally be terminated by a haloalkyl group such as CF3 and wherein the C3-C5 cycloalkyl group may optionally be substituted by C1-C4 alkyl, hydroxy or halo;
C3 to C6 cycloalkyl; Het1, Het2, Het3 or Het4;
R4 is a piperazin-1-ylsulphonyl group having a substituent R10 at the 4-position of the piperazinyl group wherein said piperazinyl group is optionally substituted with one or two C1 to C4 alkyl groups and is optionally in the form of its 4-N-oxide;
R5 and R6 are each independently selected from H and C1 to C4 alkyl optionally substituted with C3 to C5 cycloalkyl or C1 to C4 alkoxy, or, together with the nitrogen atom to which they are attached, form an azetidinyl, pyrrolidinyl, piperidinyl or morpholinyl group;
R7 and R8 are each independently selected from H, C1 to C4 alkyl, C3 to C4 alkenyl, CO2R9 or SO2R9;
R9 is C1 to C4 alkyl optionally substituted with C1 to C4 haloalkyl, C1 to C4 haloalkoxy or phenyl wherein said phenyl group is optionally substituted by one or more substituents selected from C1 to C4 alkyl optionally substituted by C1 to C4 haloalkyl or C1 to C4 haloalkoxy, C1 to C4 alkoxy, halo, CN, NO2, NHR11, NHCOR12, NHSO2R12, SO2R12, SO2NHR11, COR11 or CO2R11,
R10 is H; C1 to C4 alkyl optionally substituted with one or two substituents selected from hydroxy, NR5R6, CONR5R6, phenyl optionally substituted with C1 to C4 alkyl or C1 to C4 alkoxy; C3 to C6 alkenyl or Het4;
R11 is H, C1 to C4 alkyl, C3 to C4 alkenyl, CO(C1 to C4 alkyl) or C1 to C4 haloalkyl;
R12 is C1 to C4 alkyl, C3 to C4 alkenyl, C1 to C4 haloalkyl or C1 to C4 haloalkoxy;
Het1 is an N-linked 4-, 5- or 6-membered nitrogen-containing heterocyclic group optionally containing one or more further heteroatoms selected from S, N or O;
Het2 is a C-linked 5-membered heterocyclic group containing an O, S or N heteroatom optionally containing one or more heteroatoms selected from N, O or S;
Het3 is a C-linked 6-membered heterocyclic group containing an O or S heteroatom optionally containing one or more heteroatoms selected from O, S or N or Het3 is a C-linked 6-membered heterocyclic group containing three N heteroatoms;
Het4 is a C-linked 4-, 5- or 6-membered heterocyclic group containing one, two or three heteroatoms selected from S, O or N; and
wherein any of said heterocyclic groups Het1, Het2, Het3 or Het4 may be saturated, partially unsaturated or aromatic and wherein any of said heterocyclic groups may be optionally substituted with one or more substituents selected from C1 to C4 alkyl, C3 to C4 alkenyl, C1 to C4 alkoxy, halo, CF3, CO2R11, COR11, SO2R12, NHR11 or NHCOR12 and/or wherein any of said heterocyclic groups is benzo-fused;
with the provisos that (a) when R1 is C1 to C3 alkyl then Het1 is not morpholinyl or piperidinyl and (b) when R1 is C1 to C3 alkyl substituted by phenyl then said phenyl group is not substituted by C1 to C4 alkoxy, CN, halo, CF3, OCF3 or C1 to C4 alkyl.
2-4. (canceled)
5. The method of claim 1 , wherein the compound has the general formula (IA) or (IB):
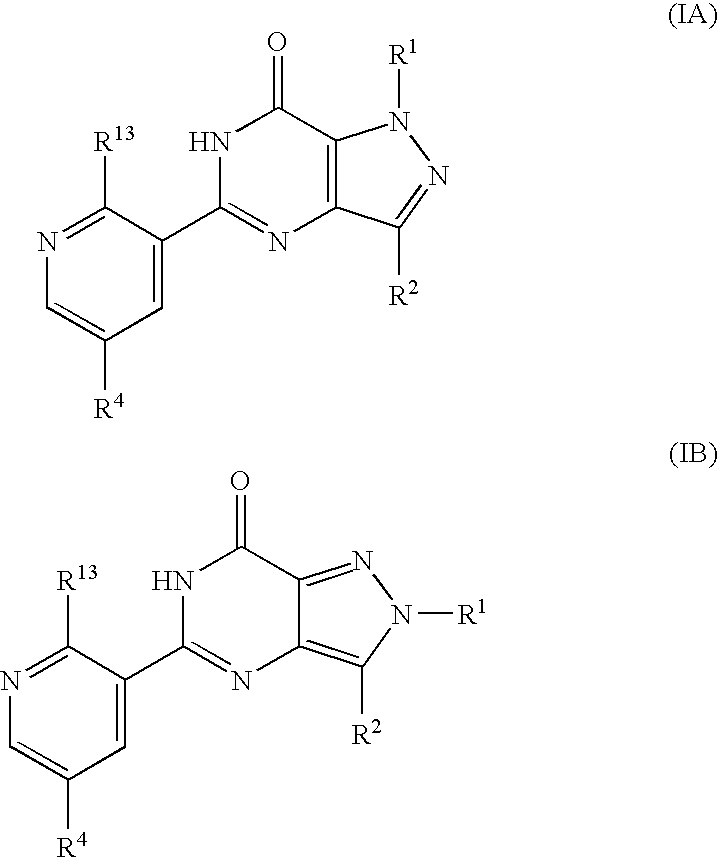
wherein R1 is —(CH2)n(C3-C4)cycloalkyl wherein n is 1 or 2; or R1 is —(CH2)n(C3-C5)cycloalkyl wherein n is 0; or R1 is —(CH2)n(C5)cycloalkyl wherein
n is 1; or R1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups; or R1 is i-, n-, sec- or t-butyl;
R2 is C2 to C4 alkyl;
R13 is OR3 wherein the R3 alkyl group is methyl, ethyl, n-propyl, i-propyl, i-butyl, n-butyl, sec-butyl or t-butyl optionally substituted with one or two methoxy, ethoxy, n-propoxy or i-propoxy substituents; and
R4 is a 4-methyl or 4-ethylpiperazin-1-ylsulphonyl group.
6. A method according to claim 5 , wherein the compound has the general formula (IB):

wherein R1 is —(CH2)n(C3-C4)cycloalkyl wherein n is 1 or 2; or R1 is —(CH2)n(C3-C5)cycloalkyl wherein n is 0; or R1 is —(CH2)n(C5)cycloalkyl wherein n is 1; or R1 is methyl, ethyl, i-propyl or n-propyl substituted by methoxy, ethoxy, n-propoxy or i-propoxy wherein said alkoxy substituent may be directly attached to any C-atom within the ethyl, iso-propyl or n-propyl groups; or R1 is i-n-, sec- or t-butyl;
R2 is C2 to C4 alkyl;
R13 is OR3 wherein the R3 alkyl group is methyl, ethyl, n-propyl, i-propyl, i-butyl, n-butyl, sec-butyl or t-butyl optionally substituted with one or two methoxy, ethoxy, n-propoxy or i-propoxy substituents; and
R4 is a 4-methyl or 4-ethylpiperazin-1-ylsulphonyl group.
7. The method according to claims 1 , 5 , or 6, wherein the compound is selected from:
5-[2-Ethoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-[2-methoxyethyl]-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-[2-methoxyethyl]-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(sec-Butyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(iso-Butyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(Cyclopropylmethyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(Cyclobutylmethyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxy-1-methylethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-(methylamino)ethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(2-Dimethylaminoethyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-methylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-dimethylaminoethyl-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-ethylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-{2-[Acetyl(methyl)amino]ethyl}-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-n-propoxypyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(1-Acetylazetidin-3-yl)-5-[2-n-butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-iso-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-(2-methoxyethyl)-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-methylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-ethylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Benzyloxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(1-ethylazetidin-3-yl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-iso-Butoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethanol)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-n-propoxypyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethanol)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-iso-propoxypyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[(S)-2-sec-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethanol)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[(R)-2-sec-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethanol)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-{(pyridin-2-yl)methyl}pyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-sec-Butyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-Cyclobutylmethyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(S)-(2-methoxy-1-methylethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
3-Ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(R)-(2-methoxy-1-methylethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[5-(4-Ethylpiperazin-1-ylsulphonyl)-2-(S)-(2-methoxy-1-methylethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[5-(4-Ethylpiperazin-1-ylsulphonyl)-2-(R)-(2-methoxy-1-methylethoxy)pyridin-3-yl]-2-(2-methoxyethyl)-3-n-propyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-hydroxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(2-Dimethylaminoethyl)-5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-iso-Butyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-iso-Butyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-Cyclobutylmethyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2-[2-(dimethylamino)-2-oxoethyl]-3-ethyl-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-n-Butoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-{2-[methyl(methylsulphonyl)amino]ethyl}-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-Cyclobutylpropylmethyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-n-Butyl-3-ethyl-5-[2-(2-methoxyethoxy)-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-n-Butoxy-5-(4-methylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(2-Ethoxyethyl)-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(3-methoxypropyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(S)-(2-methoxypropyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-(R)-(2-methoxypropyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
2-(S)-sec-Butyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-1-(2-methoxyethyl)-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one or
2-(R)-sec-Butyl-3-ethyl-5-[5-(4-ethylpiperazin-1-ylsulphonyl)-2-(2-methoxyethoxy)pyridin-3-yl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one and pharmaceutically acceptable salts or polymorphs thereof.
8-38. (canceled)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/469,311 US20100035891A1 (en) | 1999-10-11 | 2009-05-20 | Pharmaceutically Active Compounds |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9924063.2 | 1999-10-11 | ||
| GBGB9924063.2A GB9924063D0 (en) | 1999-10-11 | 1999-10-11 | Pharmaceutically active compounds |
| US16805999P | 1999-11-30 | 1999-11-30 | |
| GB0018656A GB0018656D0 (en) | 2000-07-28 | 2000-07-28 | Pharmaceutically active compounds |
| GB0018656.9 | 2000-07-28 | ||
| US23112200P | 2000-09-08 | 2000-09-08 | |
| US09/685,080 US6677335B1 (en) | 1999-10-11 | 2000-10-06 | Pharmaceutically active compounds |
| US10/753,199 US7176311B2 (en) | 1999-10-11 | 2004-01-06 | Process for preparing pharmaceutically active compounds |
| US11/463,931 US20060293347A1 (en) | 1999-10-11 | 2006-08-11 | Pharmaceutically active compounds |
| US12/469,311 US20100035891A1 (en) | 1999-10-11 | 2009-05-20 | Pharmaceutically Active Compounds |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/463,931 Continuation US20060293347A1 (en) | 1999-10-11 | 2006-08-11 | Pharmaceutically active compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20100035891A1 true US20100035891A1 (en) | 2010-02-11 |
Family
ID=26244757
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/685,080 Expired - Fee Related US6677335B1 (en) | 1999-10-11 | 2000-10-06 | Pharmaceutically active compounds |
| US10/753,199 Expired - Fee Related US7176311B2 (en) | 1999-10-11 | 2004-01-06 | Process for preparing pharmaceutically active compounds |
| US11/463,931 Abandoned US20060293347A1 (en) | 1999-10-11 | 2006-08-11 | Pharmaceutically active compounds |
| US12/469,311 Abandoned US20100035891A1 (en) | 1999-10-11 | 2009-05-20 | Pharmaceutically Active Compounds |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/685,080 Expired - Fee Related US6677335B1 (en) | 1999-10-11 | 2000-10-06 | Pharmaceutically active compounds |
| US10/753,199 Expired - Fee Related US7176311B2 (en) | 1999-10-11 | 2004-01-06 | Process for preparing pharmaceutically active compounds |
| US11/463,931 Abandoned US20060293347A1 (en) | 1999-10-11 | 2006-08-11 | Pharmaceutically active compounds |
Country Status (49)
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10435405B2 (en) | 2016-09-09 | 2019-10-08 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US10722495B2 (en) | 2017-09-08 | 2020-07-28 | Incyte Corporation | Cyanoindazole compounds and uses thereof |
| US10745388B2 (en) | 2018-02-20 | 2020-08-18 | Incyte Corporation | Indazole compounds and uses thereof |
| US10752635B2 (en) | 2018-02-20 | 2020-08-25 | Incyte Corporation | Indazole compounds and uses thereof |
| US10800761B2 (en) | 2018-02-20 | 2020-10-13 | Incyte Corporation | Carboxamide compounds and uses thereof |
| US10899755B2 (en) | 2018-08-08 | 2021-01-26 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| US11014929B2 (en) | 2016-09-09 | 2021-05-25 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11066394B2 (en) | 2019-08-06 | 2021-07-20 | Incyte Corporation | Solid forms of an HPK1 inhibitor |
| US11111247B2 (en) | 2018-09-25 | 2021-09-07 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11242343B2 (en) | 2016-09-09 | 2022-02-08 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11299473B2 (en) | 2018-04-13 | 2022-04-12 | Incyte Corporation | Benzimidazole and indole compounds and uses thereof |
| US11406624B2 (en) | 2017-02-15 | 2022-08-09 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
Families Citing this family (86)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7235625B2 (en) | 1999-06-29 | 2007-06-26 | Palatin Technologies, Inc. | Multiple agent therapy for sexual dysfunction |
| TWI265925B (en) | 1999-10-11 | 2006-11-11 | Pfizer | Pyrazolo[4,3-d]pyrimidin-7-ones useful in inhibiting type 5 cyclic guanosine 3',5'-monophosphate phosphodiesterases(cGMP PDE5), process and intermediates for their preparation, their uses and composition comprising them |
| GB0000561D0 (en) * | 2000-01-11 | 2000-03-01 | Pfizer Ltd | Treatment of diabetic ulcers |
| WO2001078781A2 (en) * | 2000-04-19 | 2001-10-25 | Johns Hopkins University | Methods for prevention and treatment of gastrointestinal disorders |
| US6271228B1 (en) * | 2000-04-28 | 2001-08-07 | Pfizer Inc. | Blood pressure stabilization during hemodialysis |
| US6420557B1 (en) | 2000-07-28 | 2002-07-16 | Pfizer Inc. | Crystalline therapeutic agent |
| EP1176142A1 (en) * | 2000-07-28 | 2002-01-30 | Pfizer Inc. | Process for the preparation of pyrazoles |
| US6407259B1 (en) | 2000-07-28 | 2002-06-18 | Pfizer Inc. | Process for the preparation of pyrazoles |
| ES2231521T3 (en) * | 2000-07-28 | 2005-05-16 | Pfizer Inc. | CRYSTAL THERAPEUTIC AGENT. |
| US6548508B2 (en) | 2000-10-20 | 2003-04-15 | Pfizer, Inc. | Use of PDE V inhibitors for improved fecundity in mammals |
| GB0025782D0 (en) * | 2000-10-20 | 2000-12-06 | Pfizer Ltd | Use of inhibitors |
| FR2820319B3 (en) * | 2001-02-08 | 2003-12-05 | Ellipse Pharmaceuticals | PROCESS FOR PRODUCING A FLOATING TABLET INCLUDING ALFUZOSINE AND TABLET OBTAINED |
| DE10135815A1 (en) * | 2001-07-23 | 2003-02-06 | Bayer Ag | Use of imidazo-triazinone derivative phosphodiesterase 5 inhibitors e.g. for treatment of cardiac insufficiency, psoriasis, diabetes, cancer, glaucoma, bladder disease, Parkinson's disease or pain |
| AU2003223071A1 (en) * | 2002-05-23 | 2003-12-12 | Pfizer Inc. | Pharmaceutical combination of PDE5 inhibitors with ACE inhibitors |
| GB0214784D0 (en) * | 2002-06-26 | 2002-08-07 | Pfizer Ltd | Novel combination |
| DE10229778A1 (en) * | 2002-07-03 | 2004-01-29 | Bayer Ag | New use of imidazotriazinones |
| GB0219961D0 (en) | 2002-08-28 | 2002-10-02 | Pfizer Ltd | Oxytocin inhibitors |
| US7323462B2 (en) | 2002-12-10 | 2008-01-29 | Pfizer Inc. | Morpholine dopamine agonists |
| EP1572173B1 (en) | 2002-12-13 | 2010-04-28 | Warner-Lambert Company LLC | Alpha-2-delta ligand to treat lower urinary tract symptoms |
| WO2004076450A1 (en) | 2003-02-27 | 2004-09-10 | J. Uriach Y Compañia S.A. | Pyrazolopyridine derivates |
| MXPA05011643A (en) | 2003-04-29 | 2005-12-15 | Pfizer Ltd | 5,7-diaminopyrazolo`4,3-d!pyrimidines useful in the treatment of hypertension. |
| BRPI0411347A (en) * | 2003-06-10 | 2006-07-11 | Pfizer | therapeutic combinations comprising pde inhibitors and vasopressin receptor antagonists for the treatment of dysmenorrhea |
| JP2006219373A (en) * | 2003-06-13 | 2006-08-24 | Daiichi Asubio Pharma Co Ltd | Pyridinylpyrazolopyrimidinone derivative having pde 7 inhibition |
| US7291640B2 (en) | 2003-09-22 | 2007-11-06 | Pfizer Inc. | Substituted triazole derivatives as oxytocin antagonists |
| US7572799B2 (en) | 2003-11-24 | 2009-08-11 | Pfizer Inc | Pyrazolo[4,3-d]pyrimidines as Phosphodiesterase Inhibitors |
| DE10360835A1 (en) * | 2003-12-23 | 2005-07-21 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | New bicyclic imidazole derivatives are dipeptidylpeptidase-IV inhibitors useful to treat e.g. arthritis, obesity, allograft transplantation and calcitonin-induced osteoporosis |
| US7649002B2 (en) | 2004-02-04 | 2010-01-19 | Pfizer Inc | (3,5-dimethylpiperidin-1yl)(4-phenylpyrrolidin-3-yl)methanone derivatives as MCR4 agonists |
| US7569572B2 (en) | 2004-04-07 | 2009-08-04 | Pfizer Inc | Pyrazolo[4,3-D]pyrimidines |
| EP1863795B1 (en) | 2005-03-21 | 2008-10-29 | Pfizer Limited | Substituted triazole derivatives as oxytocin antagonists |
| NZ564187A (en) | 2005-05-12 | 2010-03-26 | Pfizer | Anhydrous crystalline forms of N-[1-(2-ethoxyethyl)-5-(N-ethyl-N-methylamino)-7-(4-methylpyridin-2-yl-amino)-1H-pyrazolo[4,3-d]pyrimidine-3-carbonyl]methanesulfonamide |
| US8273750B2 (en) | 2005-06-06 | 2012-09-25 | Takeda Pharmaceutical Company Limited | Organic compounds |
| EP1928437A2 (en) | 2005-08-26 | 2008-06-11 | Braincells, Inc. | Neurogenesis by muscarinic receptor modulation |
| EP2258357A3 (en) | 2005-08-26 | 2011-04-06 | Braincells, Inc. | Neurogenesis with acetylcholinesterase inhibitor |
| AU2006304787A1 (en) | 2005-10-21 | 2007-04-26 | Braincells, Inc. | Modulation of neurogenesis by PDE inhibition |
| CA2625210A1 (en) | 2005-10-31 | 2007-05-10 | Braincells, Inc. | Gaba receptor mediated modulation of neurogenesis |
| EP1965863A2 (en) * | 2005-12-20 | 2008-09-10 | Pfizer Products Inc. | Pharmaceutical combination for the treatment of luts comprising a pde5 inhibitor and a muscarinic antagonist |
| US20080261995A1 (en) * | 2005-12-21 | 2008-10-23 | Pfizer Inc. | Pharmaceutical Combination of a Pde-5 Inhibitor and a 5-Alpha Reductase Inhibitor |
| UY30111A1 (en) * | 2006-02-03 | 2007-04-30 | Recordati Ireland Ltd | ACID ADDITION SALTS OF N- (3- (4- (2-METOXIFENIL) -1-PIPERAZINIL) -PROPIL) -7-OXO-5-TRIFLUORMETIL-7H-TIENO (3,2-B) PIRAN-3- CARBOXAMIDE |
| CA2643199A1 (en) * | 2006-03-08 | 2007-09-13 | Braincells, Inc. | Modulation of neurogenesis by nootropic agents |
| US20100216734A1 (en) | 2006-03-08 | 2010-08-26 | Braincells, Inc. | Modulation of neurogenesis by nootropic agents |
| EP2377531A2 (en) | 2006-05-09 | 2011-10-19 | Braincells, Inc. | Neurogenesis by modulating angiotensin |
| US20100009983A1 (en) * | 2006-05-09 | 2010-01-14 | Braincells, Inc. | 5 ht receptor mediated neurogenesis |
| US7678808B2 (en) | 2006-05-09 | 2010-03-16 | Braincells, Inc. | 5 HT receptor mediated neurogenesis |
| US9255099B2 (en) | 2006-06-06 | 2016-02-09 | Intra-Cellular Therapies, Inc. | Pyrazolo[3,4-D]pyrimidine-4,6(5H,7H)-diones as phosphodiesterase 1 inhibitors |
| US20080051413A1 (en) * | 2006-08-25 | 2008-02-28 | Kaohsiung Medical University | Nitrophenylpiperazine derivative of xanthine which relaxes tracheal airway and increases respiratory performance |
| MX2009002496A (en) * | 2006-09-08 | 2009-07-10 | Braincells Inc | Combinations containing a 4-acylaminopyridine derivative. |
| US20100184806A1 (en) | 2006-09-19 | 2010-07-22 | Braincells, Inc. | Modulation of neurogenesis by ppar agents |
| WO2008036678A2 (en) * | 2006-09-19 | 2008-03-27 | Braincells, Inc. | Combination comprising a peroxisome proliferator activated receptor agent and a second neurogenic agent for treating a nervous system disorder, increasing neurodifferentiation and increasing neurogenesis |
| US20080081816A1 (en) * | 2006-10-03 | 2008-04-03 | Kaohsiung Medical University | Anti-inflammation activity of newly synthesized xanthine derivatives kmup-1 and kmup-3 |
| WO2008070095A1 (en) | 2006-12-05 | 2008-06-12 | Intra-Cellular Therapies, Inc. | Novel uses |
| KR20100094551A (en) | 2007-12-06 | 2010-08-26 | 인트라-셀룰라 써래피스, 인코퍼레이티드. | Organic compounds |
| ES2551325T3 (en) | 2008-12-06 | 2015-11-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| SG171777A1 (en) | 2008-12-06 | 2011-07-28 | Intra Cellular Therapies Inc | Organic compounds |
| WO2010065147A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| JP5784501B2 (en) | 2008-12-06 | 2015-09-24 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | Organic compounds |
| GEP20146030B (en) | 2008-12-06 | 2014-02-10 | Intracellular Therapies Inc | Organic compounds |
| CA2740391A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| JP2012513464A (en) | 2008-12-23 | 2012-06-14 | ザ トラスティーズ オブ コロンビア ユニヴァーシティ イン ザ シティ オブ ニューヨーク | Phosphodiesterase inhibitors and uses thereof |
| WO2010099217A1 (en) | 2009-02-25 | 2010-09-02 | Braincells, Inc. | Modulation of neurogenesis using d-cycloserine combinations |
| GB0903493D0 (en) | 2009-02-27 | 2009-04-08 | Vantia Ltd | New compounds |
| US9468637B2 (en) | 2009-05-13 | 2016-10-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| EP2590657A4 (en) | 2010-05-31 | 2014-02-12 | Intra Cellular Therapies Inc | Organic compounds |
| TW201206937A (en) | 2010-05-31 | 2012-02-16 | Intra Cellular Therapies Inc | Organic compounds |
| JP5879336B2 (en) | 2010-05-31 | 2016-03-08 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | Organic compounds |
| EP2576551A4 (en) | 2010-05-31 | 2014-04-16 | Intra Cellular Therapies Inc | Organic compounds |
| JP6051210B2 (en) | 2011-06-10 | 2016-12-27 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | Organic compounds |
| US9402877B2 (en) | 2011-11-04 | 2016-08-02 | Xion Pharmaceuticals Corporation | Methods and compositions for oral administration of melanocortin receptor agonist compounds |
| US20150119399A1 (en) | 2012-01-10 | 2015-04-30 | President And Fellows Of Harvard College | Beta-cell replication promoting compounds and methods of their use |
| WO2013109738A1 (en) | 2012-01-17 | 2013-07-25 | The Trustees Of Columbia University In The City Of New York | Novel phosphodiesterase inhibitors and uses thereof |
| AR091507A1 (en) | 2012-06-21 | 2015-02-11 | Intra Cellular Therapies Inc | SALTS OF (6aR, 9aS) -5.6a, 7,8,9,9a-HEXAHYDRO-5-METHYL-3- (PHENYLAMINE) -2 - ((4- (6-FLUOROPIRIDIN-2-IL) PHENYL) METAL ) -CICLOPENT [4,5] IMIDAZO [1,2-a] PIRAZOLO [4,3-e] PYRIMIDIN-4 (2H) -ONA |
| WO2014127331A1 (en) | 2013-02-17 | 2014-08-21 | Intra-Cellular Therapies, Inc. | Novel uses |
| WO2014145617A2 (en) | 2013-03-15 | 2014-09-18 | Intra-Cellular Therapies, Inc. | Novel uses |
| WO2014151409A1 (en) | 2013-03-15 | 2014-09-25 | Intra-Cellular Therapies, Inc. | Organic compounds |
| EP3702358A1 (en) * | 2013-06-21 | 2020-09-02 | Intra-Cellular Therapies, Inc. | Process for preparing salts of a known pde1 inhibitor |
| US10899756B2 (en) | 2013-07-17 | 2021-01-26 | The Trustees Of Columbia University In The City Of New York | Phosphodiesterase inhibitors and uses thereof |
| AR102315A1 (en) * | 2014-04-02 | 2017-02-22 | Bayer Cropscience Ag | DERIVATIVES OF PIRAZOLIL-NICOTIN (UNCLE) AMIDA SUBSTITUTED AND ITS USE AS FUNGICIDES |
| EP3157926B1 (en) | 2014-06-20 | 2019-05-15 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9546175B2 (en) | 2014-08-07 | 2017-01-17 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2016022893A1 (en) | 2014-08-07 | 2016-02-11 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10285992B2 (en) | 2014-08-07 | 2019-05-14 | Intra-Cellular Therapies, Inc. | Combinations of PDE1 inhibitors and NEP inhibitors and associated methods |
| MX2017003646A (en) | 2014-09-17 | 2017-07-13 | Intra Cellular Therapies Inc | Compounds and methods. |
| WO2017172795A1 (en) | 2016-03-28 | 2017-10-05 | Intra-Cellular Therapies, Inc. | Novel compositions and methods |
| WO2018002673A1 (en) | 2016-07-01 | 2018-01-04 | N4 Pharma Uk Limited | Novel formulations of angiotensin ii receptor antagonists |
| ES2906107T3 (en) | 2016-09-12 | 2022-04-13 | Intra Cellular Therapies Inc | novel uses |
| WO2019152697A1 (en) | 2018-01-31 | 2019-08-08 | Intra-Cellular Therapies, Inc. | Novel uses |
| CN113493459B (en) * | 2020-04-07 | 2022-12-13 | 广州白云山医药集团股份有限公司白云山制药总厂 | PDE5 inhibitor compound, preparation method and application thereof |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2089971A (en) * | 1937-08-17 | Condensation products of | ||
| US4653326A (en) * | 1984-01-12 | 1987-03-31 | Commissariat A L'energie Atomique | Directional accelerometer and its microlithographic fabrication process |
| US4666908A (en) * | 1985-04-05 | 1987-05-19 | Warner-Lambert Company | 5-Substituted pyrazolo[4,3-d]pyrimidine-7-ones and methods of use |
| US4871843A (en) * | 1983-10-18 | 1989-10-03 | Dropic-Societe Civile De Gestion De Droits De Propriete Industrielle | Cyclic benzenesulfonamides, process for their preparation and their use as active substance of pharmaceutical compositions |
| US5250534A (en) * | 1990-06-20 | 1993-10-05 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| US5272147A (en) * | 1991-07-09 | 1993-12-21 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| US5294612A (en) * | 1992-03-30 | 1994-03-15 | Sterling Winthrop Inc. | 6-heterocyclyl pyrazolo [3,4-d]pyrimidin-4-ones and compositions and method of use thereof |
| US5734053A (en) * | 1992-06-26 | 1998-03-31 | Pfizer Inc | Purinone antianginal agents |
| US5736548A (en) * | 1995-03-10 | 1998-04-07 | Sanofi | 6-aryl pyrazolo 3,4-D! pyrimidin-4-ones and compositions and method of use thereof |
| US5955611A (en) * | 1996-06-14 | 1999-09-21 | Pfizer Inc. | Process for preparing sildenafil |
| US6251904B1 (en) * | 1998-04-20 | 2001-06-26 | Pfizer Inc. | Pyrazolopyrimidinone cGMP PDE5 inhibitors for the treatment of sexual dysfunction |
| US6420557B1 (en) * | 2000-07-28 | 2002-07-16 | Pfizer Inc. | Crystalline therapeutic agent |
| US6723719B1 (en) * | 1997-04-25 | 2004-04-20 | Pfizer Inc | Pyrazolopyrimidinones which inhibit type 5 cyclic guanosine 3′,5′—monophosphate phosphodiesterase (cGMP PDE5) for the treatment of sexual dysfunction |
Family Cites Families (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE425921C (en) * | 1923-08-11 | 1926-02-27 | Ernst Otto Baum | Compressed air tractor for mines |
| US4663326A (en) | 1985-04-04 | 1987-05-05 | Warner-Lambert Company | Pyrazolo[4,3-d]pyrimidine-5,7-(4H,6H)dione or -5-thione-7-one analogs |
| US5075310A (en) | 1988-07-01 | 1991-12-24 | Smith Kline & French Laboratories, Ltd. | Pyrimidone derivatives as bronchodilators |
| GB8817651D0 (en) | 1988-07-25 | 1988-09-01 | Smith Kline French Lab | Chemical compounds |
| IL95975A (en) | 1989-10-24 | 1997-06-10 | Takeda Chemical Industries Ltd | N-benzyl- 2-alkylbenzimidazole derivatives, their production and pharmaceutical compositions containing them |
| GB9013750D0 (en) * | 1990-06-20 | 1990-08-08 | Pfizer Ltd | Therapeutic agents |
| GB9119704D0 (en) | 1991-09-14 | 1991-10-30 | Pfizer Ltd | Therapeutic agents |
| GB9121028D0 (en) | 1991-10-03 | 1991-11-13 | Pfizer Ltd | Therapeutic agents |
| GB9126260D0 (en) | 1991-12-11 | 1992-02-12 | Pfizer Ltd | Therapeutic agents |
| GB9202238D0 (en) | 1992-02-03 | 1992-03-18 | Wellcome Found | Compounds |
| GB9213623D0 (en) | 1992-06-26 | 1992-08-12 | Pfizer Ltd | Therapeutic agents |
| GB9218322D0 (en) | 1992-08-28 | 1992-10-14 | Pfizer Ltd | Therapeutic agents |
| GB9301192D0 (en) | 1993-06-09 | 1993-06-09 | Trott Francis W | Flower shaped mechanised table |
| GB9315017D0 (en) | 1993-07-20 | 1993-09-01 | Glaxo Lab Sa | Chemical compounds |
| US5728705A (en) | 1993-10-04 | 1998-03-17 | The Trustees Of Columbia University In The City Of New York | Method of inducing vasorelaxation to treat pulmonary hypertension |
| GB9423910D0 (en) | 1994-11-26 | 1995-01-11 | Pfizer Ltd | Therapeutic agents |
| GB9423911D0 (en) | 1994-11-26 | 1995-01-11 | Pfizer Ltd | Therapeutic agents |
| US5656629A (en) | 1995-03-10 | 1997-08-12 | Sanofi Winthrop, Inc. | 6-substituted pyrazolo (3,4-d)pyrimidin-4-ones and compositions and methods of use thereof |
| US5952006A (en) | 1995-09-29 | 1999-09-14 | L.A.M. Pharmaceuticals, Llc | Drug preparations for treating impotency |
| DE19540642A1 (en) | 1995-11-01 | 1997-05-07 | Stief Christian Georg Priv Doz | Use of phosphodiesterase I, IV and V inhibitors |
| PT996447E (en) | 1997-05-19 | 2006-12-29 | Zonagen Inc | Combination therapy for modulating the human sexual response |
| CA2291256A1 (en) | 1997-05-29 | 1998-12-03 | Mochida Pharmaceutical Co., Ltd. | Therapeutic agent for erection dysfunction |
| YU33700A (en) | 1997-12-16 | 2004-05-12 | Pfizer Products Inc. | Combination effective for the treatment of impotence |
| WO1999059584A1 (en) | 1998-05-20 | 1999-11-25 | Schering Corporation | Combination of phentolamine and cyclic gmp phosphodiesterase inhibitors for the treatment of sexual dysfunction |
| US6087368A (en) | 1998-06-08 | 2000-07-11 | Bristol-Myers Squibb Company | Quinazolinone inhibitors of cGMP phosphodiesterase |
| EP1120120A4 (en) | 1998-10-05 | 2009-04-29 | Eisai R&D Man Co Ltd | Tablets immediately disintegrating in the oral cavity |
| JP2000119198A (en) | 1998-10-16 | 2000-04-25 | Eisai Co Ltd | Phosphodiesterase inhibitor-containing peroral preparation hiding bitterness or the like |
| CA2251255A1 (en) | 1998-10-20 | 2000-04-20 | Mcgill University | The use of dopaminergic agents in the management of sexual dysfunction |
| IL132406A0 (en) | 1998-10-21 | 2001-03-19 | Pfizer Prod Inc | Treatment of bph with cgmp elevators |
| GB9823101D0 (en) | 1998-10-23 | 1998-12-16 | Pfizer Ltd | Pharmaceutically active compounds |
| WO2000024745A1 (en) | 1998-10-23 | 2000-05-04 | Pfizer Limited | PYRAZOLOPYRIMIDINONE cGMP PDE5 INHIBITORS FOR THE TREATMENT OF SEXUAL DYSFUNCTION |
| GB9828340D0 (en) | 1998-12-22 | 1999-02-17 | Novartis Ag | Organic compounds |
| JP3199115B2 (en) * | 1998-12-24 | 2001-08-13 | モトローラ株式会社 | Digital-to-analog conversion circuit |
| DE19903087A1 (en) | 1999-01-27 | 2000-08-10 | Forssmann Wolf Georg | Treatment of erectile dysfunction with C-type natriuretic polypeptide (CNP) as monotherapy or in combination with phosphodiesterase inhibitors |
| WO2000053148A2 (en) | 1999-03-08 | 2000-09-14 | Merck & Co., Inc. | Methods and compositions for treating erectile dysfunction |
| IL137429A0 (en) | 1999-07-28 | 2001-07-24 | Pfizer Prod Inc | Methods and compsitions for treating diseases and conditions of the eye |
| WO2001008659A1 (en) | 1999-07-30 | 2001-02-08 | Lakefield Minerals Ltd. | Pharmaceutical compositions for topical application to treat erectile dysfunction |
| EP1246605A2 (en) | 1999-08-10 | 2002-10-09 | The Board Of Regents, The University Of Texas System | Facilitating the preservation of sight by increasing optic nerve, choroidal and retinal blood flow |
| US6436944B1 (en) | 1999-09-30 | 2002-08-20 | Pfizer Inc. | Combination effective for the treatment of impotence |
| TWI265925B (en) | 1999-10-11 | 2006-11-11 | Pfizer | Pyrazolo[4,3-d]pyrimidin-7-ones useful in inhibiting type 5 cyclic guanosine 3',5'-monophosphate phosphodiesterases(cGMP PDE5), process and intermediates for their preparation, their uses and composition comprising them |
| YU59100A (en) | 1999-10-11 | 2003-10-31 | Pfizer Inc. | Process for the preparation of the pyrazolo (4,3-d) pyrimidin-7-ones-3-pyridylsulphonyl compounds and intermediates thereof |
| US6407259B1 (en) * | 2000-07-28 | 2002-06-18 | Pfizer Inc. | Process for the preparation of pyrazoles |
-
2000
- 2000-09-14 TW TW089118861A patent/TWI265925B/en not_active IP Right Cessation
- 2000-10-06 US US09/685,080 patent/US6677335B1/en not_active Expired - Fee Related
- 2000-10-09 PH PH12000002783A patent/PH12000002783B1/en unknown
- 2000-10-09 MY MYPI20004712A patent/MY130319A/en unknown
- 2000-10-09 EG EG20001281A patent/EG24411A/en active
- 2000-10-10 GT GT200000171A patent/GT200000171A/en unknown
- 2000-10-10 TN TNTNSN00199A patent/TNSN00199A1/en unknown
- 2000-10-10 PA PA20008504501A patent/PA8504501A1/en unknown
- 2000-10-11 CO CO00077467A patent/CO5251477A1/en not_active Application Discontinuation
- 2000-10-11 CN CNB2004100384099A patent/CN100383141C/en not_active Expired - Fee Related
- 2000-10-11 CA CA002387357A patent/CA2387357C/en not_active Expired - Fee Related
- 2000-10-11 ES ES00966347T patent/ES2232502T3/en not_active Expired - Lifetime
- 2000-10-11 CN CNB008151954A patent/CN1204138C/en not_active Expired - Fee Related
- 2000-10-11 BR BR0014698-6A patent/BR0014698A/en not_active Application Discontinuation
- 2000-10-11 EE EEP200200195A patent/EE05181B1/en not_active IP Right Cessation
- 2000-10-11 DK DK00966347T patent/DK1220856T3/en active
- 2000-10-11 IL IL14902600A patent/IL149026A0/en unknown
- 2000-10-11 SK SK460-2002A patent/SK4602002A3/en unknown
- 2000-10-11 RS YUP-314/02A patent/RS50476B/en unknown
- 2000-10-11 HU HU0203450A patent/HUP0203450A3/en unknown
- 2000-10-11 GE GEAP20006408A patent/GEP20033142B/en unknown
- 2000-10-11 SV SV2000000195A patent/SV2002000195A/en unknown
- 2000-10-11 EP EP00966347A patent/EP1220856B1/en not_active Expired - Lifetime
- 2000-10-11 KR KR1020027004643A patent/KR100549152B1/en not_active IP Right Cessation
- 2000-10-11 TR TR2002/00989T patent/TR200200989T2/en unknown
- 2000-10-11 GE GEAP20005578A patent/GEP20033068B/en unknown
- 2000-10-11 AU AU76784/00A patent/AU779065B2/en not_active Ceased
- 2000-10-11 PT PT00966347T patent/PT1220856E/en unknown
- 2000-10-11 OA OA1200200104A patent/OA12061A/en unknown
- 2000-10-11 AP APAP/P/2002/002456A patent/AP1238A/en active
- 2000-10-11 WO PCT/IB2000/001457 patent/WO2001027113A2/en active Application Filing
- 2000-10-11 EA EA200200337A patent/EA004982B1/en not_active IP Right Cessation
- 2000-10-11 NZ NZ518078A patent/NZ518078A/en unknown
- 2000-10-11 DZ DZ003220A patent/DZ3220A1/en active
- 2000-10-11 SI SI200030593T patent/SI1220856T1/en unknown
- 2000-10-11 PE PE2000001090A patent/PE20010737A1/en not_active Application Discontinuation
- 2000-10-11 CZ CZ20021158A patent/CZ20021158A3/en unknown
- 2000-10-11 JP JP2001530331A patent/JP3834236B2/en not_active Expired - Fee Related
- 2000-10-11 AT AT00966347T patent/ATE284404T1/en not_active IP Right Cessation
- 2000-10-11 CN CNB2005100625827A patent/CN100378100C/en not_active Expired - Fee Related
- 2000-10-11 DE DE60016615T patent/DE60016615T2/en not_active Expired - Lifetime
- 2000-10-11 PL PL00357536A patent/PL357536A1/en not_active Application Discontinuation
-
2002
- 2002-03-21 IS IS6320A patent/IS2181B/en unknown
- 2002-03-21 CU CU20020060A patent/CU23198A3/en not_active IP Right Cessation
- 2002-03-28 IN IN385MU2002 patent/IN2002MU00385A/en unknown
- 2002-04-05 MA MA26586A patent/MA26825A1/en unknown
- 2002-04-08 IL IL149026A patent/IL149026A/en not_active IP Right Cessation
- 2002-04-08 CR CR6617A patent/CR6617A/en not_active Application Discontinuation
- 2002-04-10 NO NO20021694A patent/NO322856B1/en not_active IP Right Cessation
- 2002-04-10 MX MXPA02003627 patent/MX228340B/en active IP Right Grant
- 2002-04-11 HR HR20020317A patent/HRP20020317B1/en not_active IP Right Cessation
- 2002-05-09 BG BG106678A patent/BG106678A/en unknown
-
2003
- 2003-04-10 HK HK03102607A patent/HK1050367A1/en not_active IP Right Cessation
-
2004
- 2004-01-06 US US10/753,199 patent/US7176311B2/en not_active Expired - Fee Related
-
2005
- 2005-01-28 IN IN80MU2005 patent/IN2005MU00080A/en unknown
- 2005-03-11 HK HK05102106A patent/HK1069577A1/en not_active IP Right Cessation
- 2005-11-18 HK HK05110418A patent/HK1078574A1/en not_active IP Right Cessation
-
2006
- 2006-03-08 IN IN264MU2006 patent/IN2006MU00264A/en unknown
- 2006-08-11 US US11/463,931 patent/US20060293347A1/en not_active Abandoned
-
2007
- 2007-04-19 IN IN567MU2007 patent/IN2007MU00567A/en unknown
-
2008
- 2008-08-08 CR CR10191A patent/CR10191A/en not_active Application Discontinuation
-
2009
- 2009-05-20 US US12/469,311 patent/US20100035891A1/en not_active Abandoned
Patent Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2089971A (en) * | 1937-08-17 | Condensation products of | ||
| US4871843A (en) * | 1983-10-18 | 1989-10-03 | Dropic-Societe Civile De Gestion De Droits De Propriete Industrielle | Cyclic benzenesulfonamides, process for their preparation and their use as active substance of pharmaceutical compositions |
| US4653326A (en) * | 1984-01-12 | 1987-03-31 | Commissariat A L'energie Atomique | Directional accelerometer and its microlithographic fabrication process |
| US4666908A (en) * | 1985-04-05 | 1987-05-19 | Warner-Lambert Company | 5-Substituted pyrazolo[4,3-d]pyrimidine-7-ones and methods of use |
| US5346901A (en) * | 1990-06-20 | 1994-09-13 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| US5250534A (en) * | 1990-06-20 | 1993-10-05 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| US5719283A (en) * | 1990-06-20 | 1998-02-17 | Pfizer Inc. | Intermediates useful in the synthesis of pyrazolopyrimidinone antianginal agents |
| US5426107A (en) * | 1991-07-09 | 1995-06-20 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| US5272147A (en) * | 1991-07-09 | 1993-12-21 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| US5294612A (en) * | 1992-03-30 | 1994-03-15 | Sterling Winthrop Inc. | 6-heterocyclyl pyrazolo [3,4-d]pyrimidin-4-ones and compositions and method of use thereof |
| US5734053A (en) * | 1992-06-26 | 1998-03-31 | Pfizer Inc | Purinone antianginal agents |
| US5736548A (en) * | 1995-03-10 | 1998-04-07 | Sanofi | 6-aryl pyrazolo 3,4-D! pyrimidin-4-ones and compositions and method of use thereof |
| US5955611A (en) * | 1996-06-14 | 1999-09-21 | Pfizer Inc. | Process for preparing sildenafil |
| US6723719B1 (en) * | 1997-04-25 | 2004-04-20 | Pfizer Inc | Pyrazolopyrimidinones which inhibit type 5 cyclic guanosine 3′,5′—monophosphate phosphodiesterase (cGMP PDE5) for the treatment of sexual dysfunction |
| US6251904B1 (en) * | 1998-04-20 | 2001-06-26 | Pfizer Inc. | Pyrazolopyrimidinone cGMP PDE5 inhibitors for the treatment of sexual dysfunction |
| US6420557B1 (en) * | 2000-07-28 | 2002-07-16 | Pfizer Inc. | Crystalline therapeutic agent |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11242343B2 (en) | 2016-09-09 | 2022-02-08 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11891388B2 (en) | 2016-09-09 | 2024-02-06 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11795166B2 (en) | 2016-09-09 | 2023-10-24 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US10435405B2 (en) | 2016-09-09 | 2019-10-08 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11542265B2 (en) | 2016-09-09 | 2023-01-03 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US10934288B2 (en) | 2016-09-09 | 2021-03-02 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11014929B2 (en) | 2016-09-09 | 2021-05-25 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11406624B2 (en) | 2017-02-15 | 2022-08-09 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US10722495B2 (en) | 2017-09-08 | 2020-07-28 | Incyte Corporation | Cyanoindazole compounds and uses thereof |
| US11731958B2 (en) | 2018-02-20 | 2023-08-22 | Incyte Corporation | Carboxamide compounds and uses thereof |
| US11492354B2 (en) | 2018-02-20 | 2022-11-08 | Incyte Corporation | Indazole compounds and uses thereof |
| US10800761B2 (en) | 2018-02-20 | 2020-10-13 | Incyte Corporation | Carboxamide compounds and uses thereof |
| US10752635B2 (en) | 2018-02-20 | 2020-08-25 | Incyte Corporation | Indazole compounds and uses thereof |
| US10745388B2 (en) | 2018-02-20 | 2020-08-18 | Incyte Corporation | Indazole compounds and uses thereof |
| US11299473B2 (en) | 2018-04-13 | 2022-04-12 | Incyte Corporation | Benzimidazole and indole compounds and uses thereof |
| US10899755B2 (en) | 2018-08-08 | 2021-01-26 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| US11866426B2 (en) | 2018-08-08 | 2024-01-09 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| US11111247B2 (en) | 2018-09-25 | 2021-09-07 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11066394B2 (en) | 2019-08-06 | 2021-07-20 | Incyte Corporation | Solid forms of an HPK1 inhibitor |
| US11787784B2 (en) | 2019-08-06 | 2023-10-17 | Incyte Corporation | Solid forms of an HPK1 inhibitor |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7176311B2 (en) | Process for preparing pharmaceutically active compounds | |
| EP1372656B1 (en) | Pyrazolo[4,3-d]pyrimidinone compounds as cgmp pde inhibitors | |
| JP3727525B2 (en) | Pharmaceutically active compounds | |
| US6784185B2 (en) | Pharmaceutically active compounds | |
| US20030064990A1 (en) | Pharmaceutically active compounds | |
| JP2004523585A (en) | Cyclic guanosine 3 ', 5'-monophosphate phosphodiesterase inhibitor | |
| US6831074B2 (en) | Pharmaceutically active compounds | |
| EP1395589A1 (en) | Imidazo-triazine derivatives as pde 5 inhibitors | |
| US6794387B2 (en) | Pharmaceutically active compounds | |
| EP1241170A2 (en) | Pyrazolopyrimidine derivatives | |
| EP1380293A1 (en) | Bone metabolism improving agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |