US20030144360A1 - Composition and method for modulating BAR/FXR receptor activity - Google Patents
Composition and method for modulating BAR/FXR receptor activity Download PDFInfo
- Publication number
- US20030144360A1 US20030144360A1 US10/104,385 US10438502A US2003144360A1 US 20030144360 A1 US20030144360 A1 US 20030144360A1 US 10438502 A US10438502 A US 10438502A US 2003144360 A1 US2003144360 A1 US 2003144360A1
- Authority
- US
- United States
- Prior art keywords
- leu
- bar
- glu
- agn
- ser
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 238000000034 method Methods 0.000 title claims abstract description 35
- 239000000203 mixture Substances 0.000 title claims description 13
- 102100038495 Bile acid receptor Human genes 0.000 title abstract description 233
- 230000000694 effects Effects 0.000 title abstract description 60
- 206010009944 Colon cancer Diseases 0.000 claims abstract description 27
- 208000029742 colonic neoplasm Diseases 0.000 claims abstract description 22
- 241000124008 Mammalia Species 0.000 claims abstract description 15
- 210000004027 cell Anatomy 0.000 claims description 76
- 239000003613 bile acid Substances 0.000 claims description 71
- 239000003446 ligand Substances 0.000 claims description 51
- 230000014509 gene expression Effects 0.000 claims description 37
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 claims description 24
- 230000001404 mediated effect Effects 0.000 claims description 13
- 230000002401 inhibitory effect Effects 0.000 claims description 7
- 230000001575 pathological effect Effects 0.000 claims 6
- 206010020961 Hypocholesterolaemia Diseases 0.000 claims 2
- 230000005907 cancer growth Effects 0.000 claims 1
- 230000006882 induction of apoptosis Effects 0.000 claims 1
- 210000004962 mammalian cell Anatomy 0.000 claims 1
- 150000001875 compounds Chemical class 0.000 abstract description 43
- 230000003042 antagnostic effect Effects 0.000 abstract description 2
- 230000000871 hypocholesterolemic effect Effects 0.000 abstract 1
- 108070000005 Bile acid receptors Proteins 0.000 description 132
- 108010015722 farnesoid X-activated receptor Proteins 0.000 description 97
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 81
- 239000000243 solution Substances 0.000 description 77
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 68
- RUDATBOHQWOJDD-UHFFFAOYSA-N (3beta,5beta,7alpha)-3,7-Dihydroxycholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)CC2 RUDATBOHQWOJDD-UHFFFAOYSA-N 0.000 description 51
- RUDATBOHQWOJDD-BSWAIDMHSA-N chenodeoxycholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)CC1 RUDATBOHQWOJDD-BSWAIDMHSA-N 0.000 description 49
- 229960001091 chenodeoxycholic acid Drugs 0.000 description 49
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 48
- 108010038912 Retinoid X Receptors Proteins 0.000 description 47
- 102000034527 Retinoid X Receptors Human genes 0.000 description 47
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 38
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 36
- 108090000623 proteins and genes Proteins 0.000 description 31
- 230000015572 biosynthetic process Effects 0.000 description 27
- 102000005962 receptors Human genes 0.000 description 27
- 108020003175 receptors Proteins 0.000 description 27
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 24
- 235000012000 cholesterol Nutrition 0.000 description 24
- 229940107161 cholesterol Drugs 0.000 description 24
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 23
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 23
- 239000002904 solvent Substances 0.000 description 22
- 238000013518 transcription Methods 0.000 description 22
- 230000035897 transcription Effects 0.000 description 22
- 102100030426 Gastrotropin Human genes 0.000 description 21
- 101001062849 Homo sapiens Gastrotropin Proteins 0.000 description 21
- 239000012190 activator Substances 0.000 description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 20
- 241000282414 Homo sapiens Species 0.000 description 20
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 19
- 239000012267 brine Substances 0.000 description 18
- 239000010410 layer Substances 0.000 description 18
- 108020001756 ligand binding domains Proteins 0.000 description 18
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 18
- 102100039556 Galectin-4 Human genes 0.000 description 17
- 230000002829 reductive effect Effects 0.000 description 17
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 16
- 238000003556 assay Methods 0.000 description 16
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 16
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 108091027981 Response element Proteins 0.000 description 14
- 239000000556 agonist Substances 0.000 description 14
- 238000003786 synthesis reaction Methods 0.000 description 14
- 238000005160 1H NMR spectroscopy Methods 0.000 description 13
- 101000608765 Homo sapiens Galectin-4 Proteins 0.000 description 13
- 108060001084 Luciferase Proteins 0.000 description 13
- 239000005089 Luciferase Substances 0.000 description 13
- 230000004913 activation Effects 0.000 description 13
- 238000009739 binding Methods 0.000 description 13
- 239000013612 plasmid Substances 0.000 description 13
- 230000001105 regulatory effect Effects 0.000 description 13
- 150000001413 amino acids Chemical group 0.000 description 12
- 230000027455 binding Effects 0.000 description 12
- 235000019341 magnesium sulphate Nutrition 0.000 description 12
- 108010003814 member 2 group B nuclear receptor subfamily 0 Proteins 0.000 description 12
- 108020004017 nuclear receptors Proteins 0.000 description 12
- 125000003729 nucleotide group Chemical group 0.000 description 12
- 230000007115 recruitment Effects 0.000 description 12
- 102100023172 Nuclear receptor subfamily 0 group B member 2 Human genes 0.000 description 11
- 102000027507 nuclear receptors type II Human genes 0.000 description 11
- 108091008686 nuclear receptors type II Proteins 0.000 description 11
- 102000003702 retinoic acid receptors Human genes 0.000 description 11
- 108090000064 retinoic acid receptors Proteins 0.000 description 11
- 108700008625 Reporter Genes Proteins 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- 239000002773 nucleotide Substances 0.000 description 10
- 101001093899 Homo sapiens Retinoic acid receptor RXR-alpha Proteins 0.000 description 9
- FOIVPCKZDPCJJY-JQIJEIRASA-N arotinoid acid Chemical compound C=1C=C(C(CCC2(C)C)(C)C)C2=CC=1C(/C)=C/C1=CC=C(C(O)=O)C=C1 FOIVPCKZDPCJJY-JQIJEIRASA-N 0.000 description 9
- 238000002474 experimental method Methods 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- 108010037462 Cyclooxygenase 2 Proteins 0.000 description 8
- 108020004414 DNA Proteins 0.000 description 8
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 8
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 8
- 102100035178 Retinoic acid receptor RXR-alpha Human genes 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 8
- 230000006907 apoptotic process Effects 0.000 description 8
- 229910052786 argon Inorganic materials 0.000 description 8
- 239000012091 fetal bovine serum Substances 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 238000010898 silica gel chromatography Methods 0.000 description 8
- -1 steroids Chemical class 0.000 description 8
- 108090000943 Cholesterol 7-alpha-monooxygenases Proteins 0.000 description 7
- 102000004410 Cholesterol 7-alpha-monooxygenases Human genes 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- 239000005557 antagonist Substances 0.000 description 7
- 230000015556 catabolic process Effects 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 235000019441 ethanol Nutrition 0.000 description 7
- 239000000284 extract Substances 0.000 description 7
- 102000039446 nucleic acids Human genes 0.000 description 7
- 108020004707 nucleic acids Proteins 0.000 description 7
- 150000007523 nucleic acids Chemical class 0.000 description 7
- 108010026333 seryl-proline Proteins 0.000 description 7
- 230000000638 stimulation Effects 0.000 description 7
- 102100029077 3-hydroxy-3-methylglutaryl-coenzyme A reductase Human genes 0.000 description 6
- 230000004568 DNA-binding Effects 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- YBAFDPFAUTYYRW-UHFFFAOYSA-N N-L-alpha-glutamyl-L-leucine Natural products CC(C)CC(C(O)=O)NC(=O)C(N)CCC(O)=O YBAFDPFAUTYYRW-UHFFFAOYSA-N 0.000 description 6
- 238000000636 Northern blotting Methods 0.000 description 6
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 6
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 230000007423 decrease Effects 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 108020001507 fusion proteins Proteins 0.000 description 6
- 102000037865 fusion proteins Human genes 0.000 description 6
- 239000000833 heterodimer Substances 0.000 description 6
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 6
- 230000023603 positive regulation of transcription initiation, DNA-dependent Effects 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- CRDAMVZIKSXKFV-YFVJMOTDSA-N (2-trans,6-trans)-farnesol Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\CO CRDAMVZIKSXKFV-YFVJMOTDSA-N 0.000 description 5
- 239000000260 (2E,6E)-3,7,11-trimethyldodeca-2,6,10-trien-1-ol Substances 0.000 description 5
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 5
- 241000701022 Cytomegalovirus Species 0.000 description 5
- LFSQWRSVPNKJGP-WDCWCFNPSA-N Leu-Thr-Glu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@H](C(O)=O)CCC(O)=O LFSQWRSVPNKJGP-WDCWCFNPSA-N 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- 108010005233 alanylglutamic acid Proteins 0.000 description 5
- 108010047495 alanylglycine Proteins 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 238000006731 degradation reaction Methods 0.000 description 5
- 229940121360 farnesoid X receptor (fxr) agonists Drugs 0.000 description 5
- 229930002886 farnesol Natural products 0.000 description 5
- 229940043259 farnesol Drugs 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 229940005657 pyrophosphoric acid Drugs 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- CRDAMVZIKSXKFV-UHFFFAOYSA-N trans-Farnesol Natural products CC(C)=CCCC(C)=CCCC(C)=CCO CRDAMVZIKSXKFV-UHFFFAOYSA-N 0.000 description 5
- KJTLQQUUPVSXIM-ZCFIWIBFSA-M (R)-mevalonate Chemical compound OCC[C@](O)(C)CC([O-])=O KJTLQQUUPVSXIM-ZCFIWIBFSA-M 0.000 description 4
- LCBSSOCDWUTQQV-SDDRHHMPSA-N Arg-Met-Pro Chemical compound CSCC[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CCCN=C(N)N)N LCBSSOCDWUTQQV-SDDRHHMPSA-N 0.000 description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 4
- KJTLQQUUPVSXIM-UHFFFAOYSA-N DL-mevalonic acid Natural products OCCC(O)(C)CC(O)=O KJTLQQUUPVSXIM-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 241000588724 Escherichia coli Species 0.000 description 4
- 108010001515 Galectin 4 Proteins 0.000 description 4
- 102100031181 Glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 4
- 101000602930 Homo sapiens Nuclear receptor coactivator 2 Proteins 0.000 description 4
- 108090000895 Hydroxymethylglutaryl CoA Reductases Proteins 0.000 description 4
- ZJZNLRVCZWUONM-JXUBOQSCSA-N Leu-Thr-Ala Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C)C(O)=O ZJZNLRVCZWUONM-JXUBOQSCSA-N 0.000 description 4
- 102100037226 Nuclear receptor coactivator 2 Human genes 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- 101100187476 Rattus norvegicus Nr1h4 gene Proteins 0.000 description 4
- 102000006601 Thymidine Kinase Human genes 0.000 description 4
- 108020004440 Thymidine kinase Proteins 0.000 description 4
- KOSRFJWDECSPRO-UHFFFAOYSA-N alpha-L-glutamyl-L-glutamic acid Natural products OC(=O)CCC(N)C(=O)NC(CCC(O)=O)C(O)=O KOSRFJWDECSPRO-UHFFFAOYSA-N 0.000 description 4
- 108010013835 arginine glutamate Proteins 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 230000003081 coactivator Effects 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- XPPKVPWEQAFLFU-UHFFFAOYSA-N diphosphoric acid Chemical compound OP(O)(=O)OP(O)(O)=O XPPKVPWEQAFLFU-UHFFFAOYSA-N 0.000 description 4
- 108010057988 ecdysone receptor Proteins 0.000 description 4
- 229960001484 edetic acid Drugs 0.000 description 4
- 239000013604 expression vector Substances 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 230000004927 fusion Effects 0.000 description 4
- 108010078144 glutaminyl-glycine Proteins 0.000 description 4
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 230000000968 intestinal effect Effects 0.000 description 4
- 108010034529 leucyl-lysine Proteins 0.000 description 4
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 108090000865 liver X receptors Proteins 0.000 description 4
- 102000004311 liver X receptors Human genes 0.000 description 4
- 108010054155 lysyllysine Proteins 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- QJPQVXSHYBGQGM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical group [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QJPQVXSHYBGQGM-UHFFFAOYSA-N 0.000 description 4
- WEEMDRWIKYCTQM-UHFFFAOYSA-N 2,6-dimethoxybenzenecarbothioamide Chemical compound COC1=CC=CC(OC)=C1C(N)=S WEEMDRWIKYCTQM-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- YJHKTAMKPGFJCT-NRPADANISA-N Ala-Val-Glu Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O YJHKTAMKPGFJCT-NRPADANISA-N 0.000 description 3
- HJVGMOYJDDXLMI-AVGNSLFASA-N Arg-Arg-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](N)CCCNC(N)=N HJVGMOYJDDXLMI-AVGNSLFASA-N 0.000 description 3
- URAUIUGLHBRPMF-NAKRPEOUSA-N Arg-Ser-Ile Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O URAUIUGLHBRPMF-NAKRPEOUSA-N 0.000 description 3
- 108091026890 Coding region Proteins 0.000 description 3
- 206010048832 Colon adenoma Diseases 0.000 description 3
- WVLZTXGTNGHPBO-SRVKXCTJSA-N Cys-Leu-Leu Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O WVLZTXGTNGHPBO-SRVKXCTJSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 101001051083 Homo sapiens Galectin-12 Proteins 0.000 description 3
- 101001034009 Homo sapiens Glutamate receptor-interacting protein 1 Proteins 0.000 description 3
- PXKACEXYLPBMAD-JBDRJPRFSA-N Ile-Ser-Ser Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)O)N PXKACEXYLPBMAD-JBDRJPRFSA-N 0.000 description 3
- SENJXOPIZNYLHU-UHFFFAOYSA-N L-leucyl-L-arginine Natural products CC(C)CC(N)C(=O)NC(C(O)=O)CCCN=C(N)N SENJXOPIZNYLHU-UHFFFAOYSA-N 0.000 description 3
- 241000880493 Leptailurus serval Species 0.000 description 3
- GRZSCTXVCDUIPO-SRVKXCTJSA-N Leu-Arg-Gln Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(O)=O GRZSCTXVCDUIPO-SRVKXCTJSA-N 0.000 description 3
- SBANPBVRHYIMRR-UHFFFAOYSA-N Leu-Ser-Pro Natural products CC(C)CC(N)C(=O)NC(CO)C(=O)N1CCCC1C(O)=O SBANPBVRHYIMRR-UHFFFAOYSA-N 0.000 description 3
- AIMGJYMCTAABEN-GVXVVHGQSA-N Leu-Val-Glu Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O AIMGJYMCTAABEN-GVXVVHGQSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 101100187475 Mus musculus Nr1h4 gene Proteins 0.000 description 3
- KZNQNBZMBZJQJO-UHFFFAOYSA-N N-glycyl-L-proline Natural products NCC(=O)N1CCCC1C(O)=O KZNQNBZMBZJQJO-UHFFFAOYSA-N 0.000 description 3
- FEPSEIDIPBMIOS-QXEWZRGKSA-N Pro-Gly-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)CNC(=O)[C@@H]1CCCN1 FEPSEIDIPBMIOS-QXEWZRGKSA-N 0.000 description 3
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical class C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 3
- LHEZGZQRLDBSRR-WDCWCFNPSA-N Thr-Glu-Leu Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(O)=O LHEZGZQRLDBSRR-WDCWCFNPSA-N 0.000 description 3
- MGJLBZFUXUGMML-VOAKCMCISA-N Thr-Lys-Lys Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)O)N)O MGJLBZFUXUGMML-VOAKCMCISA-N 0.000 description 3
- 108010086434 alanyl-seryl-glycine Proteins 0.000 description 3
- 230000008485 antagonism Effects 0.000 description 3
- 108010059459 arginyl-threonyl-phenylalanine Proteins 0.000 description 3
- 108010077245 asparaginyl-proline Proteins 0.000 description 3
- 102000030904 bile acid binding Human genes 0.000 description 3
- 108091022863 bile acid binding Proteins 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- MHPINZYYYIJFET-UHFFFAOYSA-N ethyl 2-hydroxy-3,5-di(propan-2-yl)benzoate Chemical compound CCOC(=O)C1=CC(C(C)C)=CC(C(C)C)=C1O MHPINZYYYIJFET-UHFFFAOYSA-N 0.000 description 3
- 239000012634 fragment Substances 0.000 description 3
- 108010063718 gamma-glutamylaspartic acid Proteins 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 108010055341 glutamyl-glutamic acid Proteins 0.000 description 3
- 108010050848 glycylleucine Proteins 0.000 description 3
- 108010040030 histidinoalanine Proteins 0.000 description 3
- 239000000710 homodimer Substances 0.000 description 3
- 108010051673 leucyl-glycyl-phenylalanine Proteins 0.000 description 3
- 108010000761 leucylarginine Proteins 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- OVEHNNQXLPJPPL-UHFFFAOYSA-N lithium;n-propan-2-ylpropan-2-amine Chemical compound [Li].CC(C)NC(C)C OVEHNNQXLPJPPL-UHFFFAOYSA-N 0.000 description 3
- 108010003700 lysyl aspartic acid Proteins 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 229940056360 penicillin g Drugs 0.000 description 3
- 108010051242 phenylalanylserine Proteins 0.000 description 3
- 229920001184 polypeptide Polymers 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 108700042769 prolyl-leucyl-glycine Proteins 0.000 description 3
- 108010090894 prolylleucine Proteins 0.000 description 3
- 238000011808 rodent model Methods 0.000 description 3
- 238000002390 rotary evaporation Methods 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 150000003431 steroids Chemical class 0.000 description 3
- 230000004936 stimulating effect Effects 0.000 description 3
- 229960002385 streptomycin sulfate Drugs 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 230000002103 transcriptional effect Effects 0.000 description 3
- 238000003146 transient transfection Methods 0.000 description 3
- 241001529453 unidentified herpesvirus Species 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- 108050000156 vitamin D receptors Proteins 0.000 description 3
- 102000009310 vitamin D receptors Human genes 0.000 description 3
- VTUYATVOWGSQFU-AQIGWABESA-N (2e,4e,6z)-7-[2-hexoxy-3,5-di(propan-2-yl)phenyl]-3-methylocta-2,4,6-trienoic acid Chemical compound CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1\C(C)=C/C=C/C(/C)=C/C(O)=O VTUYATVOWGSQFU-AQIGWABESA-N 0.000 description 2
- CABVTRNMFUVUDM-VRHQGPGLSA-N (3S)-3-hydroxy-3-methylglutaryl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C[C@@](O)(CC(O)=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 CABVTRNMFUVUDM-VRHQGPGLSA-N 0.000 description 2
- KSOFVRZDRXNHFP-WQRHYEAKSA-N (z)-3-[2-hexoxy-3,5-di(propan-2-yl)phenyl]but-2-enal Chemical compound CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1\C(C)=C/C=O KSOFVRZDRXNHFP-WQRHYEAKSA-N 0.000 description 2
- NWWAOAALQDNCMN-WQRHYEAKSA-N (z)-3-[2-hexoxy-3,5-di(propan-2-yl)phenyl]but-2-enenitrile Chemical compound CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1\C(C)=C/C#N NWWAOAALQDNCMN-WQRHYEAKSA-N 0.000 description 2
- PDNCYOUVFRTRDB-UHFFFAOYSA-N 1-ethynyl-2-hexoxy-3,5-di(propan-2-yl)benzene Chemical compound CCCCCCOC1=C(C#C)C=C(C(C)C)C=C1C(C)C PDNCYOUVFRTRDB-UHFFFAOYSA-N 0.000 description 2
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 2
- NEFBXJYCHRKCLA-UHFFFAOYSA-N 2-hexoxy-3,5-di(propan-2-yl)benzoic acid Chemical compound CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C(O)=O NEFBXJYCHRKCLA-UHFFFAOYSA-N 0.000 description 2
- VWFJDQUYCIWHTN-YFVJMOTDSA-N 2-trans,6-trans-farnesyl diphosphate Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\CO[P@](O)(=O)OP(O)(O)=O VWFJDQUYCIWHTN-YFVJMOTDSA-N 0.000 description 2
- 101710158485 3-hydroxy-3-methylglutaryl-coenzyme A reductase Proteins 0.000 description 2
- ZCDMIFHZKQEGHB-UHFFFAOYSA-N 6-iodo-1,1,4,4-tetramethyl-2,3-dihydronaphthalene Chemical compound IC1=CC=C2C(C)(C)CCC(C)(C)C2=C1 ZCDMIFHZKQEGHB-UHFFFAOYSA-N 0.000 description 2
- 108700001666 APC Genes Proteins 0.000 description 2
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 2
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 2
- GGNHBHYDMUDXQB-KBIXCLLPSA-N Ala-Glu-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)N GGNHBHYDMUDXQB-KBIXCLLPSA-N 0.000 description 2
- UHMQKOBNPRAZGB-CIUDSAMLSA-N Ala-Glu-Met Chemical compound C[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCSC)C(=O)O)N UHMQKOBNPRAZGB-CIUDSAMLSA-N 0.000 description 2
- XYTNPQNAZREREP-XQXXSGGOSA-N Ala-Glu-Thr Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O XYTNPQNAZREREP-XQXXSGGOSA-N 0.000 description 2
- WGDNWOMKBUXFHR-BQBZGAKWSA-N Ala-Gly-Arg Chemical compound C[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CCCN=C(N)N WGDNWOMKBUXFHR-BQBZGAKWSA-N 0.000 description 2
- MEFILNJXAVSUTO-JXUBOQSCSA-N Ala-Leu-Thr Chemical compound C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O MEFILNJXAVSUTO-JXUBOQSCSA-N 0.000 description 2
- RTZCUEHYUQZIDE-WHFBIAKZSA-N Ala-Ser-Gly Chemical compound C[C@H](N)C(=O)N[C@@H](CO)C(=O)NCC(O)=O RTZCUEHYUQZIDE-WHFBIAKZSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 102100032187 Androgen receptor Human genes 0.000 description 2
- LVMUGODRNHFGRA-AVGNSLFASA-N Arg-Leu-Arg Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O LVMUGODRNHFGRA-AVGNSLFASA-N 0.000 description 2
- GIMTZGADWZTZGV-DCAQKATOSA-N Arg-Lys-Cys Chemical compound C(CCN)C[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CCCN=C(N)N)N GIMTZGADWZTZGV-DCAQKATOSA-N 0.000 description 2
- HCAUEJAQCXVQQM-ACZMJKKPSA-N Asn-Glu-Asp Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(O)=O HCAUEJAQCXVQQM-ACZMJKKPSA-N 0.000 description 2
- SOYOSFXLXYZNRG-CIUDSAMLSA-N Asp-Arg-Gln Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(O)=O SOYOSFXLXYZNRG-CIUDSAMLSA-N 0.000 description 2
- WSOKZUVWBXVJHX-CIUDSAMLSA-N Asp-Arg-Glu Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(O)=O WSOKZUVWBXVJHX-CIUDSAMLSA-N 0.000 description 2
- KIJLEFNHWSXHRU-NUMRIWBASA-N Asp-Gln-Thr Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O KIJLEFNHWSXHRU-NUMRIWBASA-N 0.000 description 2
- GIKOVDMXBAFXDF-NHCYSSNCSA-N Asp-Val-Leu Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O GIKOVDMXBAFXDF-NHCYSSNCSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 102000008169 Co-Repressor Proteins Human genes 0.000 description 2
- 108010060434 Co-Repressor Proteins Proteins 0.000 description 2
- MBPKYKSYUAPLMY-DCAQKATOSA-N Cys-Arg-Leu Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(O)=O MBPKYKSYUAPLMY-DCAQKATOSA-N 0.000 description 2
- MBILEVLLOHJZMG-FXQIFTODSA-N Cys-Gln-Glu Chemical compound C(CC(=O)N)[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)O)NC(=O)[C@H](CS)N MBILEVLLOHJZMG-FXQIFTODSA-N 0.000 description 2
- UXUSHQYYQCZWET-WDSKDSINSA-N Cys-Glu-Gly Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(O)=O UXUSHQYYQCZWET-WDSKDSINSA-N 0.000 description 2
- GUKYYUFHWYRMEU-WHFBIAKZSA-N Cys-Gly-Asp Chemical compound [H]N[C@@H](CS)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(O)=O GUKYYUFHWYRMEU-WHFBIAKZSA-N 0.000 description 2
- LHMSYHSAAJOEBL-CIUDSAMLSA-N Cys-Lys-Asn Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(O)=O LHMSYHSAAJOEBL-CIUDSAMLSA-N 0.000 description 2
- GQNZIAGMRXOFJX-GUBZILKMSA-N Cys-Val-Met Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCSC)C(O)=O GQNZIAGMRXOFJX-GUBZILKMSA-N 0.000 description 2
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 2
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 108010007005 Estrogen Receptor alpha Proteins 0.000 description 2
- VWFJDQUYCIWHTN-UHFFFAOYSA-N Farnesyl pyrophosphate Natural products CC(C)=CCCC(C)=CCCC(C)=CCOP(O)(=O)OP(O)(O)=O VWFJDQUYCIWHTN-UHFFFAOYSA-N 0.000 description 2
- 108090000331 Firefly luciferases Proteins 0.000 description 2
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 2
- GLZPCOQZEFWAFX-UHFFFAOYSA-N Geraniol Chemical compound CC(C)=CCCC(C)=CCO GLZPCOQZEFWAFX-UHFFFAOYSA-N 0.000 description 2
- SNLOOPZHAQDMJG-CIUDSAMLSA-N Gln-Glu-Glu Chemical compound NC(=O)CC[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O SNLOOPZHAQDMJG-CIUDSAMLSA-N 0.000 description 2
- BVELAHPZLYLZDJ-HGNGGELXSA-N Gln-His-Ala Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](C)C(O)=O BVELAHPZLYLZDJ-HGNGGELXSA-N 0.000 description 2
- FKXCBKCOSVIGCT-AVGNSLFASA-N Gln-Lys-Leu Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O FKXCBKCOSVIGCT-AVGNSLFASA-N 0.000 description 2
- NHMRJKKAVMENKJ-WDCWCFNPSA-N Gln-Thr-Leu Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(O)=O NHMRJKKAVMENKJ-WDCWCFNPSA-N 0.000 description 2
- NCWOMXABNYEPLY-NRPADANISA-N Glu-Ala-Val Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(O)=O NCWOMXABNYEPLY-NRPADANISA-N 0.000 description 2
- ILGFBUGLBSAQQB-GUBZILKMSA-N Glu-Glu-Arg Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O ILGFBUGLBSAQQB-GUBZILKMSA-N 0.000 description 2
- VSRCAOIHMGCIJK-SRVKXCTJSA-N Glu-Leu-Arg Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O VSRCAOIHMGCIJK-SRVKXCTJSA-N 0.000 description 2
- LZMQSTPFYJLVJB-GUBZILKMSA-N Glu-Leu-Cys Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CCC(=O)O)N LZMQSTPFYJLVJB-GUBZILKMSA-N 0.000 description 2
- QDMVXRNLOPTPIE-WDCWCFNPSA-N Glu-Lys-Thr Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O QDMVXRNLOPTPIE-WDCWCFNPSA-N 0.000 description 2
- QMOSCLNJVKSHHU-YUMQZZPRSA-N Glu-Met-Gly Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCSC)C(=O)NCC(O)=O QMOSCLNJVKSHHU-YUMQZZPRSA-N 0.000 description 2
- HVKAAUOFFTUSAA-XDTLVQLUSA-N Glu-Tyr-Ala Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](C)C(O)=O HVKAAUOFFTUSAA-XDTLVQLUSA-N 0.000 description 2
- LGQZOQRDEUIZJY-YUMQZZPRSA-N Gly-Cys-Lys Chemical compound NCCCC[C@H](NC(=O)[C@H](CS)NC(=O)CN)C(O)=O LGQZOQRDEUIZJY-YUMQZZPRSA-N 0.000 description 2
- MBOAPAXLTUSMQI-JHEQGTHGSA-N Gly-Glu-Thr Chemical compound [H]NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O MBOAPAXLTUSMQI-JHEQGTHGSA-N 0.000 description 2
- KMSGYZQRXPUKGI-BYPYZUCNSA-N Gly-Gly-Asn Chemical compound NCC(=O)NCC(=O)N[C@H](C(O)=O)CC(N)=O KMSGYZQRXPUKGI-BYPYZUCNSA-N 0.000 description 2
- HFPVRZWORNJRRC-UWVGGRQHSA-N Gly-Pro-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)CN HFPVRZWORNJRRC-UWVGGRQHSA-N 0.000 description 2
- ABPRMMYHROQBLY-NKWVEPMBSA-N Gly-Ser-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CO)NC(=O)CN)C(=O)O ABPRMMYHROQBLY-NKWVEPMBSA-N 0.000 description 2
- JBCLFWXMTIKCCB-UHFFFAOYSA-N H-Gly-Phe-OH Natural products NCC(=O)NC(C(O)=O)CC1=CC=CC=C1 JBCLFWXMTIKCCB-UHFFFAOYSA-N 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- KZTLOHBDLMIFSH-XVYDVKMFSA-N His-Ala-Asp Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(O)=O)C(O)=O KZTLOHBDLMIFSH-XVYDVKMFSA-N 0.000 description 2
- STOOMQFEJUVAKR-KKUMJFAQSA-N His-His-His Chemical compound C([C@H](N)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC=1N=CNC=1)C(O)=O)C1=CNC=N1 STOOMQFEJUVAKR-KKUMJFAQSA-N 0.000 description 2
- SAPLASXFNUYUFE-CQDKDKBSSA-N His-Phe-Ala Chemical compound C[C@@H](C(=O)O)NC(=O)[C@H](CC1=CC=CC=C1)NC(=O)[C@H](CC2=CN=CN2)N SAPLASXFNUYUFE-CQDKDKBSSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- VAXBXNPRXPHGHG-BJDJZHNGSA-N Ile-Ala-Leu Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)O)N VAXBXNPRXPHGHG-BJDJZHNGSA-N 0.000 description 2
- OONBGFHNQVSUBF-KBIXCLLPSA-N Ile-Gln-Cys Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CS)C(O)=O OONBGFHNQVSUBF-KBIXCLLPSA-N 0.000 description 2
- PARSHQDZROHERM-NHCYSSNCSA-N Ile-Lys-Gly Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)O)N PARSHQDZROHERM-NHCYSSNCSA-N 0.000 description 2
- IALVDKNUFSTICJ-GMOBBJLQSA-N Ile-Met-Asp Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(=O)O)C(=O)O)N IALVDKNUFSTICJ-GMOBBJLQSA-N 0.000 description 2
- SAEWJTCJQVZQNZ-IUKAMOBKSA-N Ile-Thr-Asn Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(=O)N)C(=O)O)N SAEWJTCJQVZQNZ-IUKAMOBKSA-N 0.000 description 2
- DLEBSGAVWRPTIX-PEDHHIEDSA-N Ile-Val-Ile Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(O)=O)[C@@H](C)CC DLEBSGAVWRPTIX-PEDHHIEDSA-N 0.000 description 2
- 208000032177 Intestinal Polyps Diseases 0.000 description 2
- QVJMXSGZTCGLHZ-VWADHSNXSA-N Juvenile hormone III Natural products O=C(OC)/C=C(\CC/C=C(\CC[C@H]1C(C)(C)O1)/C)/C QVJMXSGZTCGLHZ-VWADHSNXSA-N 0.000 description 2
- HGCNKOLVKRAVHD-UHFFFAOYSA-N L-Met-L-Phe Natural products CSCCC(N)C(=O)NC(C(O)=O)CC1=CC=CC=C1 HGCNKOLVKRAVHD-UHFFFAOYSA-N 0.000 description 2
- 102000007330 LDL Lipoproteins Human genes 0.000 description 2
- 108010007622 LDL Lipoproteins Proteins 0.000 description 2
- YOZCKMXHBYKOMQ-IHRRRGAJSA-N Leu-Arg-Lys Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCCN)C(=O)O)N YOZCKMXHBYKOMQ-IHRRRGAJSA-N 0.000 description 2
- UCOCBWDBHCUPQP-DCAQKATOSA-N Leu-Arg-Ser Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(O)=O UCOCBWDBHCUPQP-DCAQKATOSA-N 0.000 description 2
- KUIDCYNIEJBZBU-AJNGGQMLSA-N Leu-Ile-Leu Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(O)=O KUIDCYNIEJBZBU-AJNGGQMLSA-N 0.000 description 2
- IFMPDNRWZZEZSL-SRVKXCTJSA-N Leu-Leu-Cys Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(O)=O IFMPDNRWZZEZSL-SRVKXCTJSA-N 0.000 description 2
- FAELBUXXFQLUAX-AJNGGQMLSA-N Leu-Leu-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC(C)C FAELBUXXFQLUAX-AJNGGQMLSA-N 0.000 description 2
- LVTJJOJKDCVZGP-QWRGUYRKSA-N Leu-Lys-Gly Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(O)=O LVTJJOJKDCVZGP-QWRGUYRKSA-N 0.000 description 2
- VVQJGYPTIYOFBR-IHRRRGAJSA-N Leu-Lys-Met Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCSC)C(=O)O)N VVQJGYPTIYOFBR-IHRRRGAJSA-N 0.000 description 2
- KWLWZYMNUZJKMZ-IHRRRGAJSA-N Leu-Pro-Leu Chemical compound CC(C)C[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CC(C)C)C(O)=O KWLWZYMNUZJKMZ-IHRRRGAJSA-N 0.000 description 2
- SBANPBVRHYIMRR-GARJFASQSA-N Leu-Ser-Pro Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CO)C(=O)N1CCC[C@@H]1C(=O)O)N SBANPBVRHYIMRR-GARJFASQSA-N 0.000 description 2
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 2
- ZXFRGTAIIZHNHG-AJNGGQMLSA-N Lys-Ile-Leu Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(C)C)C(=O)O)NC(=O)[C@H](CCCCN)N ZXFRGTAIIZHNHG-AJNGGQMLSA-N 0.000 description 2
- YPLVCBKEPJPBDQ-MELADBBJSA-N Lys-Leu-Pro Chemical compound CC(C)C[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CCCCN)N YPLVCBKEPJPBDQ-MELADBBJSA-N 0.000 description 2
- AFFKUNVPPLQUGA-DCAQKATOSA-N Met-Leu-Ala Chemical compound [H]N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(O)=O AFFKUNVPPLQUGA-DCAQKATOSA-N 0.000 description 2
- JQHYVIKEFYETEW-IHRRRGAJSA-N Met-Phe-Ser Chemical compound CSCC[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CO)C(O)=O)CC1=CC=CC=C1 JQHYVIKEFYETEW-IHRRRGAJSA-N 0.000 description 2
- WOGNGBROIHHFAO-JYJNAYRXSA-N Met-Tyr-Met Chemical compound CSCC[C@@H](C(=O)N[C@@H](CC1=CC=C(C=C1)O)C(=O)N[C@@H](CCSC)C(=O)O)N WOGNGBROIHHFAO-JYJNAYRXSA-N 0.000 description 2
- 101000741798 Mus musculus Peroxisome proliferator-activated receptor delta Proteins 0.000 description 2
- 101000741806 Mus musculus Peroxisome proliferator-activated receptor gamma Proteins 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- XMBSYZWANAQXEV-UHFFFAOYSA-N N-alpha-L-glutamyl-L-phenylalanine Natural products OC(=O)CCC(N)C(=O)NC(C(O)=O)CC1=CC=CC=C1 XMBSYZWANAQXEV-UHFFFAOYSA-N 0.000 description 2
- SEQKRHFRPICQDD-UHFFFAOYSA-N N-tris(hydroxymethyl)methylglycine Chemical compound OCC(CO)(CO)[NH2+]CC([O-])=O SEQKRHFRPICQDD-UHFFFAOYSA-N 0.000 description 2
- 108091008784 NR1I4 Proteins 0.000 description 2
- 101100342977 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) leu-1 gene Proteins 0.000 description 2
- KAHUBGWSIQNZQQ-KKUMJFAQSA-N Phe-Asn-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 KAHUBGWSIQNZQQ-KKUMJFAQSA-N 0.000 description 2
- OXKJSGGTHFMGDT-UFYCRDLUSA-N Phe-Phe-Arg Chemical compound C([C@H](N)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CC=CC=C1 OXKJSGGTHFMGDT-UFYCRDLUSA-N 0.000 description 2
- GLJZDMZJHFXJQG-BZSNNMDCSA-N Phe-Ser-Phe Chemical compound [H]N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O GLJZDMZJHFXJQG-BZSNNMDCSA-N 0.000 description 2
- PTDAGKJHZBGDKD-OEAJRASXSA-N Phe-Thr-Lys Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CC1=CC=CC=C1)N)O PTDAGKJHZBGDKD-OEAJRASXSA-N 0.000 description 2
- SHUFSZDAIPLZLF-BEAPCOKYSA-N Phe-Thr-Pro Chemical compound C[C@H]([C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CC2=CC=CC=C2)N)O SHUFSZDAIPLZLF-BEAPCOKYSA-N 0.000 description 2
- DBNGDEAQXGFGRA-ACRUOGEOSA-N Phe-Tyr-Lys Chemical compound C1=CC=C(C=C1)C[C@@H](C(=O)N[C@@H](CC2=CC=C(C=C2)O)C(=O)N[C@@H](CCCCN)C(=O)O)N DBNGDEAQXGFGRA-ACRUOGEOSA-N 0.000 description 2
- XZGWNSIRZIUHHP-SRVKXCTJSA-N Pro-Arg-Met Chemical compound CSCC[C@@H](C(=O)O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H]1CCCN1 XZGWNSIRZIUHHP-SRVKXCTJSA-N 0.000 description 2
- WFIVLLFYUZZWOD-RHYQMDGZSA-N Pro-Lys-Thr Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O WFIVLLFYUZZWOD-RHYQMDGZSA-N 0.000 description 2
- YDTUEBLEAVANFH-RCWTZXSCSA-N Pro-Val-Thr Chemical compound C[C@@H](O)[C@@H](C(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H]1CCCN1 YDTUEBLEAVANFH-RCWTZXSCSA-N 0.000 description 2
- 230000010799 Receptor Interactions Effects 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- YRBGKVIWMNEVCZ-WDSKDSINSA-N Ser-Glu-Gly Chemical compound OC[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(O)=O YRBGKVIWMNEVCZ-WDSKDSINSA-N 0.000 description 2
- NNFMANHDYSVNIO-DCAQKATOSA-N Ser-Lys-Arg Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O NNFMANHDYSVNIO-DCAQKATOSA-N 0.000 description 2
- GYDFRTRSSXOZCR-ACZMJKKPSA-N Ser-Ser-Glu Chemical compound OC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CCC(O)=O GYDFRTRSSXOZCR-ACZMJKKPSA-N 0.000 description 2
- OLKICIBQRVSQMA-SRVKXCTJSA-N Ser-Ser-Tyr Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O OLKICIBQRVSQMA-SRVKXCTJSA-N 0.000 description 2
- 229930182558 Sterol Natural products 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- BIENEHRYNODTLP-HJGDQZAQSA-N Thr-Glu-Met Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCSC)C(=O)O)N)O BIENEHRYNODTLP-HJGDQZAQSA-N 0.000 description 2
- BNGDYRRHRGOPHX-IFFSRLJSSA-N Thr-Glu-Val Chemical compound CC(C)[C@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)[C@@H](C)O)C(O)=O BNGDYRRHRGOPHX-IFFSRLJSSA-N 0.000 description 2
- CSNBWOJOEOPYIJ-UVOCVTCTSA-N Thr-Thr-Lys Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(O)=O CSNBWOJOEOPYIJ-UVOCVTCTSA-N 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- ZJPSMXCFEKMZFE-IHPCNDPISA-N Trp-Tyr-Ser Chemical compound [H]N[C@@H](CC1=CNC2=C1C=CC=C2)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(O)=O ZJPSMXCFEKMZFE-IHPCNDPISA-N 0.000 description 2
- OSXNCKRGMSHWSQ-ACRUOGEOSA-N Tyr-His-Tyr Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O OSXNCKRGMSHWSQ-ACRUOGEOSA-N 0.000 description 2
- WSFXJLFSJSXGMQ-MGHWNKPDSA-N Tyr-Ile-Lys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CC1=CC=C(C=C1)O)N WSFXJLFSJSXGMQ-MGHWNKPDSA-N 0.000 description 2
- SOAUMCDLIUGXJJ-SRVKXCTJSA-N Tyr-Ser-Asn Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(O)=O SOAUMCDLIUGXJJ-SRVKXCTJSA-N 0.000 description 2
- SYFHQHYTNCQCCN-MELADBBJSA-N Tyr-Ser-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CO)NC(=O)[C@H](CC2=CC=C(C=C2)O)N)C(=O)O SYFHQHYTNCQCCN-MELADBBJSA-N 0.000 description 2
- XGJLNBNZNMVJRS-NRPADANISA-N Val-Glu-Ala Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(O)=O XGJLNBNZNMVJRS-NRPADANISA-N 0.000 description 2
- FTKXYXACXYOHND-XUXIUFHCSA-N Val-Ile-Leu Chemical compound CC(C)[C@H](N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(O)=O FTKXYXACXYOHND-XUXIUFHCSA-N 0.000 description 2
- OFTXTCGQJXTNQS-XGEHTFHBSA-N Val-Thr-Ser Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CO)C(=O)O)NC(=O)[C@H](C(C)C)N)O OFTXTCGQJXTNQS-XGEHTFHBSA-N 0.000 description 2
- RLVTVHSDKHBFQP-ULQDDVLXSA-N Val-Tyr-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)CC1=CC=C(O)C=C1 RLVTVHSDKHBFQP-ULQDDVLXSA-N 0.000 description 2
- ZHWZDZFWBXWPDW-GUBZILKMSA-N Val-Val-Cys Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CS)C(O)=O ZHWZDZFWBXWPDW-GUBZILKMSA-N 0.000 description 2
- GLELYEBJESCJJV-XYGWBWBKSA-N [4-[(z)-2-(3,5,5,8,8-pentamethyl-6,7-dihydronaphthalen-2-yl)-2-trimethylsilylethenyl]phenyl]methanol Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1\C([Si](C)(C)C)=C\C1=CC=C(CO)C=C1 GLELYEBJESCJJV-XYGWBWBKSA-N 0.000 description 2
- 208000009956 adenocarcinoma Diseases 0.000 description 2
- 108010050025 alpha-glutamyltryptophan Proteins 0.000 description 2
- 108010080146 androgen receptors Proteins 0.000 description 2
- YCOXTKKNXUZSKD-UHFFFAOYSA-N as-o-xylenol Natural products CC1=CC=C(O)C=C1C YCOXTKKNXUZSKD-UHFFFAOYSA-N 0.000 description 2
- 108010038633 aspartylglutamate Proteins 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 108010005774 beta-Galactosidase Proteins 0.000 description 2
- 230000006696 biosynthetic metabolic pathway Effects 0.000 description 2
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 239000003610 charcoal Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 150000001841 cholesterols Chemical class 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 238000012761 co-transfection Methods 0.000 description 2
- 230000004736 colon carcinogenesis Effects 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- 108010016616 cysteinylglycine Proteins 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 238000006471 dimerization reaction Methods 0.000 description 2
- 238000006073 displacement reaction Methods 0.000 description 2
- 210000002919 epithelial cell Anatomy 0.000 description 2
- OQIXXLWNAGCVKR-OQRBYTRCSA-N ethyl (2e,4e,6z)-7-[2-hexoxy-3,5-di(propan-2-yl)phenyl]-3-methylocta-2,4,6-trienoate Chemical compound CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1\C(C)=C/C=C/C(/C)=C/C(=O)OCC OQIXXLWNAGCVKR-OQRBYTRCSA-N 0.000 description 2
- GLELCBUFSZVPJP-UHFFFAOYSA-N ethyl 4-hex-1-ynylbenzoate Chemical compound CCCCC#CC1=CC=C(C(=O)OCC)C=C1 GLELCBUFSZVPJP-UHFFFAOYSA-N 0.000 description 2
- 239000013613 expression plasmid Substances 0.000 description 2
- 125000004030 farnesyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])C([H])([H])C([H])=C(C([H])([H])[H])C([H])([H])C([H])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 108010049041 glutamylalanine Proteins 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- XBGGUPMXALFZOT-UHFFFAOYSA-N glycyl-L-tyrosine hemihydrate Natural products NCC(=O)NC(C(O)=O)CC1=CC=C(O)C=C1 XBGGUPMXALFZOT-UHFFFAOYSA-N 0.000 description 2
- 108010082286 glycyl-seryl-alanine Proteins 0.000 description 2
- 108010037850 glycylvaline Proteins 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- SNHMUERNLJLMHN-UHFFFAOYSA-N iodobenzene Chemical compound IC1=CC=CC=C1 SNHMUERNLJLMHN-UHFFFAOYSA-N 0.000 description 2
- 108010044374 isoleucyl-tyrosine Proteins 0.000 description 2
- 108010027338 isoleucylcysteine Proteins 0.000 description 2
- QVJMXSGZTCGLHZ-HONBPKQLSA-N juvenile hormone III Chemical compound COC(=O)\C=C(/C)CC\C=C(/C)CC[C@H]1OC1(C)C QVJMXSGZTCGLHZ-HONBPKQLSA-N 0.000 description 2
- 108010083708 leucyl-aspartyl-valine Proteins 0.000 description 2
- IHLVCKWPAMTVTG-UHFFFAOYSA-N lithium;carbanide Chemical compound [Li+].[CH3-] IHLVCKWPAMTVTG-UHFFFAOYSA-N 0.000 description 2
- 108010064235 lysylglycine Proteins 0.000 description 2
- 108010038320 lysylphenylalanine Proteins 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 108010005942 methionylglycine Proteins 0.000 description 2
- 108010068488 methionylphenylalanine Proteins 0.000 description 2
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- CWHFDTWZHFRTAB-UHFFFAOYSA-N phenyl cyanate Chemical compound N#COC1=CC=CC=C1 CWHFDTWZHFRTAB-UHFFFAOYSA-N 0.000 description 2
- 229920002401 polyacrylamide Polymers 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 108010070643 prolylglutamic acid Proteins 0.000 description 2
- 150000003180 prostaglandins Chemical class 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 150000004492 retinoid derivatives Chemical class 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 239000003270 steroid hormone Substances 0.000 description 2
- 150000003432 sterols Chemical class 0.000 description 2
- 235000003702 sterols Nutrition 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- TVOQECFXQDURLC-UHFFFAOYSA-N tert-butyl-diphenyl-[[4-(2-trimethylsilylethynyl)phenyl]methoxy]silane Chemical compound C=1C=CC=CC=1[Si](C=1C=CC=CC=1)(C(C)(C)C)OCC1=CC=C(C#C[Si](C)(C)C)C=C1 TVOQECFXQDURLC-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 108010061238 threonyl-glycine Proteins 0.000 description 2
- 108091008762 thyroid hormone receptors ß Proteins 0.000 description 2
- 238000001890 transfection Methods 0.000 description 2
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 108010003137 tyrosyltyrosine Proteins 0.000 description 2
- 239000013598 vector Substances 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- YHRUHBBTQZKMEX-YFVJMOTDSA-N (2-trans,6-trans)-farnesal Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\C=O YHRUHBBTQZKMEX-YFVJMOTDSA-N 0.000 description 1
- YHRUHBBTQZKMEX-UHFFFAOYSA-N (2E,6E)-3,7,11-trimethyl-2,6,10-dodecatrien-1-al Natural products CC(C)=CCCC(C)=CCCC(C)=CC=O YHRUHBBTQZKMEX-UHFFFAOYSA-N 0.000 description 1
- WJHFZYAELPOJIV-UHFFFAOYSA-N (2E,6E)-3,7,11-trimethyl-2,6,10-dodecatrienoic acid Natural products CC(C)=CCCC(C)=CCCC(C)=CC(O)=O WJHFZYAELPOJIV-UHFFFAOYSA-N 0.000 description 1
- WJHFZYAELPOJIV-IJFRVEDASA-N (2E,6E)-farnesoic acid Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\C(O)=O WJHFZYAELPOJIV-IJFRVEDASA-N 0.000 description 1
- NTUPOKHATNSWCY-PMPSAXMXSA-N (2s)-2-[[(2s)-1-[(2r)-2-amino-3-phenylpropanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound C([C@@H](N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O)C1=CC=CC=C1 NTUPOKHATNSWCY-PMPSAXMXSA-N 0.000 description 1
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 1
- TYWSGDVBXZNFND-UHFFFAOYSA-N (4-bromophenyl)methoxy-tert-butyl-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](C=1C=CC=CC=1)(C(C)(C)C)OCC1=CC=C(Br)C=C1 TYWSGDVBXZNFND-UHFFFAOYSA-N 0.000 description 1
- QURIGKMFLBTLHH-UHFFFAOYSA-N (4-bromophenyl)methoxy-tert-butyl-diphenylsilane;tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1.C=1C=CC=CC=1[Si](C=1C=CC=CC=1)(C(C)(C)C)OCC1=CC=C(Br)C=C1 QURIGKMFLBTLHH-UHFFFAOYSA-N 0.000 description 1
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 description 1
- OJISWRZIEWCUBN-QIRCYJPOSA-N (E,E,E)-geranylgeraniol Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\CC\C(C)=C\CO OJISWRZIEWCUBN-QIRCYJPOSA-N 0.000 description 1
- GMRQFYUYWCNGIN-ZVUFCXRFSA-N 1,25-dihydroxy vitamin D3 Chemical compound C1([C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=CC=C1C[C@@H](O)C[C@H](O)C1=C GMRQFYUYWCNGIN-ZVUFCXRFSA-N 0.000 description 1
- CGHIBGNXEGJPQZ-UHFFFAOYSA-N 1-hexyne Chemical compound CCCCC#C CGHIBGNXEGJPQZ-UHFFFAOYSA-N 0.000 description 1
- ANOOTOPTCJRUPK-UHFFFAOYSA-N 1-iodohexane Chemical compound CCCCCCI ANOOTOPTCJRUPK-UHFFFAOYSA-N 0.000 description 1
- KSXTUUUQYQYKCR-LQDDAWAPSA-M 2,3-bis[[(z)-octadec-9-enoyl]oxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC KSXTUUUQYQYKCR-LQDDAWAPSA-M 0.000 description 1
- HSTAGCWQAIXJQM-UHFFFAOYSA-N 2,5-dichloro-2,5-dimethylhexane Chemical compound CC(C)(Cl)CCC(C)(C)Cl HSTAGCWQAIXJQM-UHFFFAOYSA-N 0.000 description 1
- XUFUYOGWFZSHGE-UHFFFAOYSA-N 2-hydroxy-3,5-di(propan-2-yl)benzoic acid Chemical compound CC(C)C1=CC(C(C)C)=C(O)C(C(O)=O)=C1 XUFUYOGWFZSHGE-UHFFFAOYSA-N 0.000 description 1
- INBGSXNNRGWLJU-ZHHJOTBYSA-N 25-hydroxycholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@@H](CCCC(C)(C)O)C)[C@@]1(C)CC2 INBGSXNNRGWLJU-ZHHJOTBYSA-N 0.000 description 1
- INBGSXNNRGWLJU-UHFFFAOYSA-N 25epsilon-Hydroxycholesterin Natural products C1C=C2CC(O)CCC2(C)C2C1C1CCC(C(CCCC(C)(C)O)C)C1(C)CC2 INBGSXNNRGWLJU-UHFFFAOYSA-N 0.000 description 1
- ZGIGZINMAOQWLX-NCZFFCEISA-N 3,7,11-Trimethyl-2,6,10-dodecatrienyl acetate Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\COC(C)=O ZGIGZINMAOQWLX-NCZFFCEISA-N 0.000 description 1
- HFXUKPRUHVBGHX-UHFFFAOYSA-N 3-[2-hexoxy-3,5-di(propan-2-yl)phenyl]prop-2-ynenitrile Chemical compound CCCCCCOC1=C(C#CC#N)C=C(C(C)C)C=C1C(C)C HFXUKPRUHVBGHX-UHFFFAOYSA-N 0.000 description 1
- NYYSVYAQWWOZFG-UHFFFAOYSA-N 3-bromo-6-methyl-1h-pyridin-2-one Chemical compound CC1=CC=C(Br)C(=O)N1 NYYSVYAQWWOZFG-UHFFFAOYSA-N 0.000 description 1
- VVVPQLIUALZTGS-UHFFFAOYSA-N 4-[2-(5,5,8,8-tetramethyl-6,7-dihydronaphthalen-2-yl)hex-1-enyl]benzoic acid Chemical compound C=1C=C(C(CCC2(C)C)(C)C)C2=CC=1C(CCCC)=CC1=CC=C(C(O)=O)C=C1 VVVPQLIUALZTGS-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- ONNHBALCPUEXBT-UHFFFAOYSA-N 6-bromo-1,1,4,4,7-pentamethyl-2,3-dihydronaphthalene Chemical compound CC1(C)CCC(C)(C)C2=C1C=C(Br)C(C)=C2 ONNHBALCPUEXBT-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- 208000003200 Adenoma Diseases 0.000 description 1
- 241000349731 Afzelia bipindensis Species 0.000 description 1
- RLMISHABBKUNFO-WHFBIAKZSA-N Ala-Ala-Gly Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(=O)NCC(O)=O RLMISHABBKUNFO-WHFBIAKZSA-N 0.000 description 1
- CXRCVCURMBFFOL-FXQIFTODSA-N Ala-Ala-Pro Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O CXRCVCURMBFFOL-FXQIFTODSA-N 0.000 description 1
- YYSWCHMLFJLLBJ-ZLUOBGJFSA-N Ala-Ala-Ser Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(O)=O YYSWCHMLFJLLBJ-ZLUOBGJFSA-N 0.000 description 1
- DAEFQZCYZKRTLR-ZLUOBGJFSA-N Ala-Cys-Asp Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(O)=O)C(O)=O DAEFQZCYZKRTLR-ZLUOBGJFSA-N 0.000 description 1
- IXTPACPAXIOCRG-ACZMJKKPSA-N Ala-Glu-Cys Chemical compound C[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CS)C(=O)O)N IXTPACPAXIOCRG-ACZMJKKPSA-N 0.000 description 1
- WKOBSJOZRJJVRZ-FXQIFTODSA-N Ala-Glu-Glu Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O WKOBSJOZRJJVRZ-FXQIFTODSA-N 0.000 description 1
- SMCGQGDVTPFXKB-XPUUQOCRSA-N Ala-Gly-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)CNC(=O)[C@H](C)N SMCGQGDVTPFXKB-XPUUQOCRSA-N 0.000 description 1
- KMGOBAQSCKTBGD-DLOVCJGASA-N Ala-His-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@H](C)N)CC1=CN=CN1 KMGOBAQSCKTBGD-DLOVCJGASA-N 0.000 description 1
- CBCCCLMNOBLBSC-XVYDVKMFSA-N Ala-His-Ser Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CO)C(O)=O CBCCCLMNOBLBSC-XVYDVKMFSA-N 0.000 description 1
- LNNSWWRRYJLGNI-NAKRPEOUSA-N Ala-Ile-Val Chemical compound C[C@H](N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C(C)C)C(O)=O LNNSWWRRYJLGNI-NAKRPEOUSA-N 0.000 description 1
- YHKANGMVQWRMAP-DCAQKATOSA-N Ala-Leu-Arg Chemical compound C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(O)=O)CCCN=C(N)N YHKANGMVQWRMAP-DCAQKATOSA-N 0.000 description 1
- IAUSCRHURCZUJP-CIUDSAMLSA-N Ala-Lys-Cys Chemical compound NCCCC[C@H](NC(=O)[C@@H](N)C)C(=O)N[C@@H](CS)C(O)=O IAUSCRHURCZUJP-CIUDSAMLSA-N 0.000 description 1
- OMDNCNKNEGFOMM-BQBZGAKWSA-N Ala-Met-Gly Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CCSC)C(=O)NCC(O)=O OMDNCNKNEGFOMM-BQBZGAKWSA-N 0.000 description 1
- DGLQWAFPIXDKRL-UBHSHLNASA-N Ala-Met-Phe Chemical compound C[C@@H](C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)O)N DGLQWAFPIXDKRL-UBHSHLNASA-N 0.000 description 1
- BTRULDJUUVGRNE-DCAQKATOSA-N Ala-Pro-Lys Chemical compound C[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCCCN)C(O)=O BTRULDJUUVGRNE-DCAQKATOSA-N 0.000 description 1
- PEEYDECOOVQKRZ-DLOVCJGASA-N Ala-Ser-Phe Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O PEEYDECOOVQKRZ-DLOVCJGASA-N 0.000 description 1
- XQNRANMFRPCFFW-GCJQMDKQSA-N Ala-Thr-Asn Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](C)N)O XQNRANMFRPCFFW-GCJQMDKQSA-N 0.000 description 1
- WNHNMKOFKCHKKD-BFHQHQDPSA-N Ala-Thr-Gly Chemical compound [H]N[C@@H](C)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(O)=O WNHNMKOFKCHKKD-BFHQHQDPSA-N 0.000 description 1
- KTXKIYXZQFWJKB-VZFHVOOUSA-N Ala-Thr-Ser Chemical compound [H]N[C@@H](C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(O)=O KTXKIYXZQFWJKB-VZFHVOOUSA-N 0.000 description 1
- OMSKGWFGWCQFBD-KZVJFYERSA-N Ala-Val-Thr Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O OMSKGWFGWCQFBD-KZVJFYERSA-N 0.000 description 1
- UISQLSIBJKEJSS-GUBZILKMSA-N Arg-Arg-Ser Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CO)C(O)=O UISQLSIBJKEJSS-GUBZILKMSA-N 0.000 description 1
- RVDVDRUZWZIBJQ-CIUDSAMLSA-N Arg-Asn-Glu Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O RVDVDRUZWZIBJQ-CIUDSAMLSA-N 0.000 description 1
- OTCJMMRQBVDQRK-DCAQKATOSA-N Arg-Asp-Leu Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(O)=O OTCJMMRQBVDQRK-DCAQKATOSA-N 0.000 description 1
- QIWYWCYNUMJBTC-CIUDSAMLSA-N Arg-Cys-Gln Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(O)=O QIWYWCYNUMJBTC-CIUDSAMLSA-N 0.000 description 1
- GIVWETPOBCRTND-DCAQKATOSA-N Arg-Gln-Arg Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O GIVWETPOBCRTND-DCAQKATOSA-N 0.000 description 1
- MTANSHNQTWPZKP-KKUMJFAQSA-N Arg-Gln-Tyr Chemical compound C1=CC(=CC=C1C[C@@H](C(=O)O)NC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CCCN=C(N)N)N)O MTANSHNQTWPZKP-KKUMJFAQSA-N 0.000 description 1
- LMPKCSXZJSXBBL-NHCYSSNCSA-N Arg-Gln-Val Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(O)=O LMPKCSXZJSXBBL-NHCYSSNCSA-N 0.000 description 1
- OGUPCHKBOKJFMA-SRVKXCTJSA-N Arg-Glu-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CCCN=C(N)N OGUPCHKBOKJFMA-SRVKXCTJSA-N 0.000 description 1
- DJAIOAKQIOGULM-DCAQKATOSA-N Arg-Glu-Met Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCSC)C(O)=O DJAIOAKQIOGULM-DCAQKATOSA-N 0.000 description 1
- OOIMKQRCPJBGPD-XUXIUFHCSA-N Arg-Ile-Leu Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(O)=O OOIMKQRCPJBGPD-XUXIUFHCSA-N 0.000 description 1
- GNYUVVJYGJFKHN-RVMXOQNASA-N Arg-Ile-Pro Chemical compound CC[C@H](C)[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CCCN=C(N)N)N GNYUVVJYGJFKHN-RVMXOQNASA-N 0.000 description 1
- NMRHDSAOIURTNT-RWMBFGLXSA-N Arg-Leu-Pro Chemical compound CC(C)C[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CCCN=C(N)N)N NMRHDSAOIURTNT-RWMBFGLXSA-N 0.000 description 1
- JEOCWTUOMKEEMF-RHYQMDGZSA-N Arg-Leu-Thr Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O JEOCWTUOMKEEMF-RHYQMDGZSA-N 0.000 description 1
- MJINRRBEMOLJAK-DCAQKATOSA-N Arg-Lys-Asp Chemical compound OC(=O)C[C@@H](C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CCCN=C(N)N MJINRRBEMOLJAK-DCAQKATOSA-N 0.000 description 1
- NPAVRDPEFVKELR-DCAQKATOSA-N Arg-Lys-Ser Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(O)=O NPAVRDPEFVKELR-DCAQKATOSA-N 0.000 description 1
- JOADBFCFJGNIKF-GUBZILKMSA-N Arg-Met-Ala Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C)C(O)=O JOADBFCFJGNIKF-GUBZILKMSA-N 0.000 description 1
- KXOPYFNQLVUOAQ-FXQIFTODSA-N Arg-Ser-Ala Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(O)=O KXOPYFNQLVUOAQ-FXQIFTODSA-N 0.000 description 1
- FRBAHXABMQXSJQ-FXQIFTODSA-N Arg-Ser-Ser Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(O)=O FRBAHXABMQXSJQ-FXQIFTODSA-N 0.000 description 1
- MOGMYRUNTKYZFB-UNQGMJICSA-N Arg-Thr-Phe Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H]([C@H](O)C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MOGMYRUNTKYZFB-UNQGMJICSA-N 0.000 description 1
- XRNXPIGJPQHCPC-RCWTZXSCSA-N Arg-Thr-Val Chemical compound CC(C)[C@H](NC(=O)[C@@H](NC(=O)[C@@H](N)CCCNC(N)=N)[C@@H](C)O)C(O)=O XRNXPIGJPQHCPC-RCWTZXSCSA-N 0.000 description 1
- QHUOOCKNNURZSL-IHRRRGAJSA-N Arg-Tyr-Ser Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(O)=O QHUOOCKNNURZSL-IHRRRGAJSA-N 0.000 description 1
- PSUXEQYPYZLNER-QXEWZRGKSA-N Arg-Val-Asn Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O PSUXEQYPYZLNER-QXEWZRGKSA-N 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- XYOVHPDDWCEUDY-CIUDSAMLSA-N Asn-Ala-Leu Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(O)=O XYOVHPDDWCEUDY-CIUDSAMLSA-N 0.000 description 1
- QEYJFBMTSMLPKZ-ZKWXMUAHSA-N Asn-Ala-Val Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(O)=O QEYJFBMTSMLPKZ-ZKWXMUAHSA-N 0.000 description 1
- HUAOKVVEVHACHR-CIUDSAMLSA-N Asn-Asp-His Chemical compound C1=C(NC=N1)C[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CC(=O)N)N HUAOKVVEVHACHR-CIUDSAMLSA-N 0.000 description 1
- DMLSCRJBWUEALP-LAEOZQHASA-N Asn-Glu-Val Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(O)=O DMLSCRJBWUEALP-LAEOZQHASA-N 0.000 description 1
- FVKHEKVYFTZWDX-GHCJXIJMSA-N Asn-Ile-Cys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CC(=O)N)N FVKHEKVYFTZWDX-GHCJXIJMSA-N 0.000 description 1
- BXUHCIXDSWRSBS-CIUDSAMLSA-N Asn-Leu-Asp Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(O)=O BXUHCIXDSWRSBS-CIUDSAMLSA-N 0.000 description 1
- WIDVAWAQBRAKTI-YUMQZZPRSA-N Asn-Leu-Gly Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(O)=O WIDVAWAQBRAKTI-YUMQZZPRSA-N 0.000 description 1
- BZWRLDPIWKOVKB-ZPFDUUQYSA-N Asn-Leu-Ile Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O BZWRLDPIWKOVKB-ZPFDUUQYSA-N 0.000 description 1
- JEEFEQCRXKPQHC-KKUMJFAQSA-N Asn-Leu-Phe Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O JEEFEQCRXKPQHC-KKUMJFAQSA-N 0.000 description 1
- FTSAJSADJCMDHH-CIUDSAMLSA-N Asn-Lys-Asp Chemical compound C(CCN)C[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)O)NC(=O)[C@H](CC(=O)N)N FTSAJSADJCMDHH-CIUDSAMLSA-N 0.000 description 1
- LZLCLRQMUQWUHJ-GUBZILKMSA-N Asn-Lys-Gln Chemical compound C(CCN)C[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)O)NC(=O)[C@H](CC(=O)N)N LZLCLRQMUQWUHJ-GUBZILKMSA-N 0.000 description 1
- NYGILGUOUOXGMJ-YUMQZZPRSA-N Asn-Lys-Gly Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(O)=O NYGILGUOUOXGMJ-YUMQZZPRSA-N 0.000 description 1
- ZYPWIUFLYMQZBS-SRVKXCTJSA-N Asn-Lys-Lys Chemical compound C(CCN)C[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CC(=O)N)N ZYPWIUFLYMQZBS-SRVKXCTJSA-N 0.000 description 1
- MYVBTYXSWILFCG-BQBZGAKWSA-N Asn-Met-Gly Chemical compound CSCC[C@@H](C(=O)NCC(=O)O)NC(=O)[C@H](CC(=O)N)N MYVBTYXSWILFCG-BQBZGAKWSA-N 0.000 description 1
- HZZIFFOVHLWGCS-KKUMJFAQSA-N Asn-Phe-Leu Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC(C)C)C(O)=O HZZIFFOVHLWGCS-KKUMJFAQSA-N 0.000 description 1
- PLTGTJAZQRGMPP-FXQIFTODSA-N Asn-Pro-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@@H]1CCCN1C(=O)[C@@H](N)CC(N)=O PLTGTJAZQRGMPP-FXQIFTODSA-N 0.000 description 1
- XMHFCUKJRCQXGI-CIUDSAMLSA-N Asn-Pro-Gln Chemical compound C1C[C@H](N(C1)C(=O)[C@H](CC(=O)N)N)C(=O)N[C@@H](CCC(=O)N)C(=O)O XMHFCUKJRCQXGI-CIUDSAMLSA-N 0.000 description 1
- NJSNXIOKBHPFMB-GMOBBJLQSA-N Asn-Pro-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CC(=O)N)N NJSNXIOKBHPFMB-GMOBBJLQSA-N 0.000 description 1
- IIQIOFVDFOLCHP-UHFFFAOYSA-N Asn-Pro-Ser-Ser Chemical compound NC(=O)CC(N)C(=O)N1CCCC1C(=O)NC(CO)C(=O)NC(CO)C(O)=O IIQIOFVDFOLCHP-UHFFFAOYSA-N 0.000 description 1
- LTDGPJKGJDIBQD-LAEOZQHASA-N Asn-Val-Gln Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O LTDGPJKGJDIBQD-LAEOZQHASA-N 0.000 description 1
- LMIWYCWRJVMAIQ-NHCYSSNCSA-N Asn-Val-Lys Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CC(=O)N)N LMIWYCWRJVMAIQ-NHCYSSNCSA-N 0.000 description 1
- GHWWTICYPDKPTE-NGZCFLSTSA-N Asn-Val-Pro Chemical compound CC(C)[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CC(=O)N)N GHWWTICYPDKPTE-NGZCFLSTSA-N 0.000 description 1
- OERMIMJQPQUIPK-FXQIFTODSA-N Asp-Arg-Ala Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(O)=O OERMIMJQPQUIPK-FXQIFTODSA-N 0.000 description 1
- IXIWEFWRKIUMQX-DCAQKATOSA-N Asp-Arg-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](N)CC(O)=O IXIWEFWRKIUMQX-DCAQKATOSA-N 0.000 description 1
- JDHOJQJMWBKHDB-CIUDSAMLSA-N Asp-Asn-Lys Chemical compound C(CCN)C[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CC(=O)O)N JDHOJQJMWBKHDB-CIUDSAMLSA-N 0.000 description 1
- RDRMWJBLOSRRAW-BYULHYEWSA-N Asp-Asn-Val Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(O)=O RDRMWJBLOSRRAW-BYULHYEWSA-N 0.000 description 1
- FRSGNOZCTWDVFZ-ACZMJKKPSA-N Asp-Asp-Gln Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(O)=O FRSGNOZCTWDVFZ-ACZMJKKPSA-N 0.000 description 1
- QXHVOUSPVAWEMX-ZLUOBGJFSA-N Asp-Asp-Ser Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(O)=O QXHVOUSPVAWEMX-ZLUOBGJFSA-N 0.000 description 1
- LJRPYAZQQWHEEV-FXQIFTODSA-N Asp-Gln-Gln Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(O)=O LJRPYAZQQWHEEV-FXQIFTODSA-N 0.000 description 1
- PMEHKVHZQKJACS-PEFMBERDSA-N Asp-Gln-Ile Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O PMEHKVHZQKJACS-PEFMBERDSA-N 0.000 description 1
- PDECQIHABNQRHN-GUBZILKMSA-N Asp-Glu-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CC(O)=O PDECQIHABNQRHN-GUBZILKMSA-N 0.000 description 1
- OVPHVTCDVYYTHN-AVGNSLFASA-N Asp-Glu-Phe Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 OVPHVTCDVYYTHN-AVGNSLFASA-N 0.000 description 1
- RRKCPMGSRIDLNC-AVGNSLFASA-N Asp-Glu-Tyr Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O RRKCPMGSRIDLNC-AVGNSLFASA-N 0.000 description 1
- CRNKLABLTICXDV-GUBZILKMSA-N Asp-His-Glu Chemical compound C1=C(NC=N1)C[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)O)NC(=O)[C@H](CC(=O)O)N CRNKLABLTICXDV-GUBZILKMSA-N 0.000 description 1
- ODNWIBOCFGMRTP-SRVKXCTJSA-N Asp-His-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC(O)=O)CC1=CN=CN1 ODNWIBOCFGMRTP-SRVKXCTJSA-N 0.000 description 1
- YFSLJHLQOALGSY-ZPFDUUQYSA-N Asp-Ile-Lys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CC(=O)O)N YFSLJHLQOALGSY-ZPFDUUQYSA-N 0.000 description 1
- DWOGMPWRQQWPPF-GUBZILKMSA-N Asp-Leu-Glu Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O DWOGMPWRQQWPPF-GUBZILKMSA-N 0.000 description 1
- UJGRZQYSNYTCAX-SRVKXCTJSA-N Asp-Leu-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC(O)=O UJGRZQYSNYTCAX-SRVKXCTJSA-N 0.000 description 1
- UZFHNLYQWMGUHU-DCAQKATOSA-N Asp-Lys-Arg Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O UZFHNLYQWMGUHU-DCAQKATOSA-N 0.000 description 1
- QNIACYURSSCLRP-GUBZILKMSA-N Asp-Lys-Gln Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(N)=O)C(O)=O QNIACYURSSCLRP-GUBZILKMSA-N 0.000 description 1
- HSGOFISJLFDMBJ-CIUDSAMLSA-N Asp-Met-Gln Chemical compound CSCC[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)O)NC(=O)[C@H](CC(=O)O)N HSGOFISJLFDMBJ-CIUDSAMLSA-N 0.000 description 1
- VWWAFGHMPWBKEP-GMOBBJLQSA-N Asp-Met-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(=O)O)N VWWAFGHMPWBKEP-GMOBBJLQSA-N 0.000 description 1
- WQSXAPPYLGNMQL-IHRRRGAJSA-N Asp-Met-Tyr Chemical compound CSCC[C@@H](C(=O)N[C@@H](CC1=CC=C(C=C1)O)C(=O)O)NC(=O)[C@H](CC(=O)O)N WQSXAPPYLGNMQL-IHRRRGAJSA-N 0.000 description 1
- RPUYTJJZXQBWDT-SRVKXCTJSA-N Asp-Phe-Ser Chemical compound C1=CC=C(C=C1)C[C@@H](C(=O)N[C@@H](CO)C(=O)O)NC(=O)[C@H](CC(=O)O)N RPUYTJJZXQBWDT-SRVKXCTJSA-N 0.000 description 1
- QSFHZPQUAAQHAQ-CIUDSAMLSA-N Asp-Ser-Leu Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(O)=O QSFHZPQUAAQHAQ-CIUDSAMLSA-N 0.000 description 1
- OTKUAVXGMREHRX-CFMVVWHZSA-N Asp-Tyr-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC(O)=O)CC1=CC=C(O)C=C1 OTKUAVXGMREHRX-CFMVVWHZSA-N 0.000 description 1
- MFDPBZAFCRKYEY-LAEOZQHASA-N Asp-Val-Gln Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O MFDPBZAFCRKYEY-LAEOZQHASA-N 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- XKRUKZLQEAHJBD-COKSUYGGSA-M C#CCCCC.CC(C)(Cl)CCC(C)(C)Cl.CC1(C)CCC(C)(C)C2=C1C=CC(I)=C2.CC1=CC=C(I)C=C1.CCCC/C(=C\C1=CC=C(C(=O)O)C=C1)C1=CC2=C(C=C1)C(C)(C)CCC2(C)C.CCCC/C(=C\C1=CC=C(C)C=C1)C1=CC2=C(C=C1)C(C)(C)CCC2(C)C.CCCCC#CC1=CC=C(C)C=C1.CCCCC#CC1=CC=C(C)C=C1.IC1=CC=CC=C1.O[K] Chemical compound C#CCCCC.CC(C)(Cl)CCC(C)(C)Cl.CC1(C)CCC(C)(C)C2=C1C=CC(I)=C2.CC1=CC=C(I)C=C1.CCCC/C(=C\C1=CC=C(C(=O)O)C=C1)C1=CC2=C(C=C1)C(C)(C)CCC2(C)C.CCCC/C(=C\C1=CC=C(C)C=C1)C1=CC2=C(C=C1)C(C)(C)CCC2(C)C.CCCCC#CC1=CC=C(C)C=C1.CCCCC#CC1=CC=C(C)C=C1.IC1=CC=CC=C1.O[K] XKRUKZLQEAHJBD-COKSUYGGSA-M 0.000 description 1
- JMFVEXOOEZOBID-UHFFFAOYSA-N C.C#CC1=CC(C(C)C)=CC(C(C)C)=C1OCCCCCC.CC(C)C1=CC(C(C)C)=C(O)C(C(=O)O)=C1.CC1=CC(C(C)C)=CC(C(C)C)=C1O.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C#CC#N.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C(=O)O.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C(C)=O.N#COC1=CC=CC=C1 Chemical compound C.C#CC1=CC(C(C)C)=CC(C(C)C)=C1OCCCCCC.CC(C)C1=CC(C(C)C)=C(O)C(C(=O)O)=C1.CC1=CC(C(C)C)=CC(C(C)C)=C1O.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C#CC#N.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C(=O)O.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C(C)=O.N#COC1=CC=CC=C1 JMFVEXOOEZOBID-UHFFFAOYSA-N 0.000 description 1
- ZTUPZDHRRGTKFV-ILOMPSDQSA-N C/C=C(C)/C=C/C=C(/C)C1=CC(C(C)C)=CC(C(C)C)=C1OCCCCCC.C/C=C(\C)CC=O.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1/C(C)=C\C#N.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1/C(C)=C\C=C\C(C)=C\C(=O)O.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1/C(C)=C\C=O Chemical compound C/C=C(C)/C=C/C=C(/C)C1=CC(C(C)C)=CC(C(C)C)=C1OCCCCCC.C/C=C(\C)CC=O.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1/C(C)=C\C#N.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1/C(C)=C\C=C\C(C)=C\C(=O)O.CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1/C(C)=C\C=O ZTUPZDHRRGTKFV-ILOMPSDQSA-N 0.000 description 1
- KXJOGQVGIGUXDY-AATRIKPKSA-N CC1(C)CCC(C)(C)C2=C1C=CC(/C=C/C1=CC=C(C(=O)O)C=C1)=C2 Chemical compound CC1(C)CCC(C)(C)C2=C1C=CC(/C=C/C1=CC=C(C(=O)O)C=C1)=C2 KXJOGQVGIGUXDY-AATRIKPKSA-N 0.000 description 1
- HSNQBLMIEGEVOR-JLPGSUDCSA-N CC1=CC2=C(C=C1/C(=C/C1=CC=C(C(=O)O)C=C1)[Si](C)(C)C)C(C)(C)CCC2(C)C Chemical compound CC1=CC2=C(C=C1/C(=C/C1=CC=C(C(=O)O)C=C1)[Si](C)(C)C)C(C)(C)CCC2(C)C HSNQBLMIEGEVOR-JLPGSUDCSA-N 0.000 description 1
- VVVPQLIUALZTGS-HEHNFIMWSA-N CCCC/C(=C\C1=CC=C(C(=O)O)C=C1)C1=CC2=C(C=C1)C(C)(C)CCC2(C)C Chemical compound CCCC/C(=C\C1=CC=C(C(=O)O)C=C1)C1=CC2=C(C=C1)C(C)(C)CCC2(C)C VVVPQLIUALZTGS-HEHNFIMWSA-N 0.000 description 1
- 101150071146 COX2 gene Proteins 0.000 description 1
- 101100505161 Caenorhabditis elegans mel-32 gene Proteins 0.000 description 1
- XZFRIPGNUQRGPI-WBQKLGIQSA-N Carbaprostacyclin Chemical compound C1\C(=C/CCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@H]21 XZFRIPGNUQRGPI-WBQKLGIQSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- 108010022102 Cholestanetriol 26-monooxygenase Proteins 0.000 description 1
- 102000012272 Cholestanetriol 26-monooxygenase Human genes 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 239000004380 Cholic acid Substances 0.000 description 1
- 241001112696 Clostridia Species 0.000 description 1
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 1
- 108091033380 Coding strand Proteins 0.000 description 1
- ACTIUHUUMQJHFO-UHFFFAOYSA-N Coenzym Q10 Natural products COC1=C(OC)C(=O)C(CC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)C)=C(C)C1=O ACTIUHUUMQJHFO-UHFFFAOYSA-N 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- FMDCYTBSPZMPQE-JBDRJPRFSA-N Cys-Ala-Ile Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O FMDCYTBSPZMPQE-JBDRJPRFSA-N 0.000 description 1
- ABLJDBFJPUWQQB-DCAQKATOSA-N Cys-Leu-Arg Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CCCN=C(N)N)C(=O)O)NC(=O)[C@H](CS)N ABLJDBFJPUWQQB-DCAQKATOSA-N 0.000 description 1
- VPQZSNQICFCCSO-BJDJZHNGSA-N Cys-Leu-Ile Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O VPQZSNQICFCCSO-BJDJZHNGSA-N 0.000 description 1
- CIVXDCMSSFGWAL-YUMQZZPRSA-N Cys-Lys-Gly Chemical compound C(CCN)C[C@@H](C(=O)NCC(=O)O)NC(=O)[C@H](CS)N CIVXDCMSSFGWAL-YUMQZZPRSA-N 0.000 description 1
- VOBMMKMWSIVIOA-SRVKXCTJSA-N Cys-Lys-His Chemical compound C1=C(NC=N1)C[C@@H](C(=O)O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CS)N VOBMMKMWSIVIOA-SRVKXCTJSA-N 0.000 description 1
- YXPNKXFOBHRUBL-BJDJZHNGSA-N Cys-Lys-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CS)N YXPNKXFOBHRUBL-BJDJZHNGSA-N 0.000 description 1
- ALTQTAKGRFLRLR-GUBZILKMSA-N Cys-Val-Val Chemical compound CC(C)[C@@H](C(=O)N[C@@H](C(C)C)C(=O)O)NC(=O)[C@H](CS)N ALTQTAKGRFLRLR-GUBZILKMSA-N 0.000 description 1
- 102000004594 DNA Polymerase I Human genes 0.000 description 1
- 108010017826 DNA Polymerase I Proteins 0.000 description 1
- 101100202237 Danio rerio rxrab gene Proteins 0.000 description 1
- 101100309320 Danio rerio rxrga gene Proteins 0.000 description 1
- YHRUHBBTQZKMEX-FBXUGWQNSA-N E,E-Farnesal Natural products CC(C)=CCC\C(C)=C/CC\C(C)=C/C=O YHRUHBBTQZKMEX-FBXUGWQNSA-N 0.000 description 1
- 239000006145 Eagle's minimal essential medium Substances 0.000 description 1
- UPEZCKBFRMILAV-JNEQICEOSA-N Ecdysone Natural products O=C1[C@H]2[C@@](C)([C@@H]3C([C@@]4(O)[C@@](C)([C@H]([C@H]([C@@H](O)CCC(O)(C)C)C)CC4)CC3)=C1)C[C@H](O)[C@H](O)C2 UPEZCKBFRMILAV-JNEQICEOSA-N 0.000 description 1
- 241000701832 Enterobacteria phage T3 Species 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- ZGIGZINMAOQWLX-UHFFFAOYSA-N Farnesyl acetate Natural products CC(C)=CCCC(C)=CCCC(C)=CCOC(C)=O ZGIGZINMAOQWLX-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 239000005792 Geraniol Substances 0.000 description 1
- GLZPCOQZEFWAFX-YFHOEESVSA-N Geraniol Natural products CC(C)=CCC\C(C)=C/CO GLZPCOQZEFWAFX-YFHOEESVSA-N 0.000 description 1
- YJIUYQKQBBQYHZ-ACZMJKKPSA-N Gln-Ala-Ala Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(O)=O YJIUYQKQBBQYHZ-ACZMJKKPSA-N 0.000 description 1
- XXLBHPPXDUWYAG-XQXXSGGOSA-N Gln-Ala-Thr Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O XXLBHPPXDUWYAG-XQXXSGGOSA-N 0.000 description 1
- DLOHWQXXGMEZDW-CIUDSAMLSA-N Gln-Arg-Asn Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(O)=O DLOHWQXXGMEZDW-CIUDSAMLSA-N 0.000 description 1
- WOACHWLUOFZLGJ-GUBZILKMSA-N Gln-Arg-Gln Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(O)=O WOACHWLUOFZLGJ-GUBZILKMSA-N 0.000 description 1
- KJRXLVZYJJLUCV-DCAQKATOSA-N Gln-Arg-Met Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(O)=O KJRXLVZYJJLUCV-DCAQKATOSA-N 0.000 description 1
- AAOBFSKXAVIORT-GUBZILKMSA-N Gln-Asn-Leu Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(O)=O AAOBFSKXAVIORT-GUBZILKMSA-N 0.000 description 1
- KYFSMWLWHYZRNW-ACZMJKKPSA-N Gln-Asp-Cys Chemical compound C(CC(=O)N)[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CS)C(=O)O)N KYFSMWLWHYZRNW-ACZMJKKPSA-N 0.000 description 1
- CRRFJBGUGNNOCS-PEFMBERDSA-N Gln-Asp-Ile Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O CRRFJBGUGNNOCS-PEFMBERDSA-N 0.000 description 1
- VNCLJDOTEPPBBD-GUBZILKMSA-N Gln-Cys-Lys Chemical compound C(CCN)C[C@@H](C(=O)O)NC(=O)[C@H](CS)NC(=O)[C@H](CCC(=O)N)N VNCLJDOTEPPBBD-GUBZILKMSA-N 0.000 description 1
- IVCOYUURLWQDJQ-LPEHRKFASA-N Gln-Gln-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CCC(=O)N)N)C(=O)O IVCOYUURLWQDJQ-LPEHRKFASA-N 0.000 description 1
- NPTGGVQJYRSMCM-GLLZPBPUSA-N Gln-Gln-Thr Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O NPTGGVQJYRSMCM-GLLZPBPUSA-N 0.000 description 1
- KDXKFBSNIJYNNR-YVNDNENWSA-N Gln-Glu-Ile Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O KDXKFBSNIJYNNR-YVNDNENWSA-N 0.000 description 1
- VOLVNCMGXWDDQY-LPEHRKFASA-N Gln-Glu-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CCC(=O)N)N)C(=O)O VOLVNCMGXWDDQY-LPEHRKFASA-N 0.000 description 1
- FTIJVMLAGRAYMJ-MNXVOIDGSA-N Gln-Ile-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H](N)CCC(N)=O FTIJVMLAGRAYMJ-MNXVOIDGSA-N 0.000 description 1
- ITZWDGBYBPUZRG-KBIXCLLPSA-N Gln-Ile-Ser Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(O)=O ITZWDGBYBPUZRG-KBIXCLLPSA-N 0.000 description 1
- YPMDZWPZFOZYFG-GUBZILKMSA-N Gln-Leu-Ser Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(O)=O YPMDZWPZFOZYFG-GUBZILKMSA-N 0.000 description 1
- ATTWDCRXQNKRII-GUBZILKMSA-N Gln-Lys-Cys Chemical compound C(CCN)C[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CCC(=O)N)N ATTWDCRXQNKRII-GUBZILKMSA-N 0.000 description 1
- HHRAEXBUNGTOGZ-IHRRRGAJSA-N Gln-Phe-Gln Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CCC(N)=O)C(O)=O HHRAEXBUNGTOGZ-IHRRRGAJSA-N 0.000 description 1
- OZEQPCDLCDRCGY-SOUVJXGZSA-N Gln-Phe-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CC2=CC=CC=C2)NC(=O)[C@H](CCC(=O)N)N)C(=O)O OZEQPCDLCDRCGY-SOUVJXGZSA-N 0.000 description 1
- PBYFVIQRFLNQCO-GUBZILKMSA-N Gln-Pro-Gln Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(O)=O PBYFVIQRFLNQCO-GUBZILKMSA-N 0.000 description 1
- HMIXCETWRYDVMO-GUBZILKMSA-N Gln-Pro-Glu Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(O)=O)C(O)=O HMIXCETWRYDVMO-GUBZILKMSA-N 0.000 description 1
- HLRLXVPRJJITSK-IFFSRLJSSA-N Gln-Thr-Val Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(O)=O HLRLXVPRJJITSK-IFFSRLJSSA-N 0.000 description 1
- UBRQJXFDVZNYJP-AVGNSLFASA-N Gln-Tyr-Ser Chemical compound C1=CC(=CC=C1C[C@@H](C(=O)N[C@@H](CO)C(=O)O)NC(=O)[C@H](CCC(=O)N)N)O UBRQJXFDVZNYJP-AVGNSLFASA-N 0.000 description 1
- ZZLDMBMFKZFQMU-NRPADANISA-N Gln-Val-Ala Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C)C(O)=O ZZLDMBMFKZFQMU-NRPADANISA-N 0.000 description 1
- ZFBBMCKQSNJZSN-AUTRQRHGSA-N Gln-Val-Gln Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O ZFBBMCKQSNJZSN-AUTRQRHGSA-N 0.000 description 1
- LKDIBBOKUAASNP-FXQIFTODSA-N Glu-Ala-Glu Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(O)=O LKDIBBOKUAASNP-FXQIFTODSA-N 0.000 description 1
- HUWSBFYAGXCXKC-CIUDSAMLSA-N Glu-Ala-Met Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCSC)C(O)=O HUWSBFYAGXCXKC-CIUDSAMLSA-N 0.000 description 1
- KBKGRMNVKPSQIF-XDTLVQLUSA-N Glu-Ala-Tyr Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O KBKGRMNVKPSQIF-XDTLVQLUSA-N 0.000 description 1
- VTTSANCGJWLPNC-ZPFDUUQYSA-N Glu-Arg-Ile Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O VTTSANCGJWLPNC-ZPFDUUQYSA-N 0.000 description 1
- SBYVDRJAXWSXQL-AVGNSLFASA-N Glu-Asn-Phe Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O SBYVDRJAXWSXQL-AVGNSLFASA-N 0.000 description 1
- LXAUHIRMWXQRKI-XHNCKOQMSA-N Glu-Asn-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CC(=O)N)NC(=O)[C@H](CCC(=O)O)N)C(=O)O LXAUHIRMWXQRKI-XHNCKOQMSA-N 0.000 description 1
- VAIWPXWHWAPYDF-FXQIFTODSA-N Glu-Asp-Gln Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(O)=O VAIWPXWHWAPYDF-FXQIFTODSA-N 0.000 description 1
- JVSBYEDSSRZQGV-GUBZILKMSA-N Glu-Asp-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](N)CCC(O)=O JVSBYEDSSRZQGV-GUBZILKMSA-N 0.000 description 1
- GZWOBWMOMPFPCD-CIUDSAMLSA-N Glu-Asp-Met Chemical compound CSCC[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CCC(=O)O)N GZWOBWMOMPFPCD-CIUDSAMLSA-N 0.000 description 1
- PBFGQTGPSKWHJA-QEJZJMRPSA-N Glu-Asp-Trp Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(O)=O PBFGQTGPSKWHJA-QEJZJMRPSA-N 0.000 description 1
- RQNYYRHRKSVKAB-GUBZILKMSA-N Glu-Cys-Leu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(C)C)C(O)=O RQNYYRHRKSVKAB-GUBZILKMSA-N 0.000 description 1
- PVBBEKPHARMPHX-DCAQKATOSA-N Glu-Gln-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CCC(O)=O PVBBEKPHARMPHX-DCAQKATOSA-N 0.000 description 1
- WLIPTFCZLHCNFD-LPEHRKFASA-N Glu-Gln-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CCC(=O)O)N)C(=O)O WLIPTFCZLHCNFD-LPEHRKFASA-N 0.000 description 1
- QQLBPVKLJBAXBS-FXQIFTODSA-N Glu-Glu-Asn Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O QQLBPVKLJBAXBS-FXQIFTODSA-N 0.000 description 1
- YLJHCWNDBKKOEB-IHRRRGAJSA-N Glu-Glu-Phe Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O YLJHCWNDBKKOEB-IHRRRGAJSA-N 0.000 description 1
- PXXGVUVQWQGGIG-YUMQZZPRSA-N Glu-Gly-Arg Chemical compound OC(=O)CC[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CCCN=C(N)N PXXGVUVQWQGGIG-YUMQZZPRSA-N 0.000 description 1
- WVTIBGWZUMJBFY-GUBZILKMSA-N Glu-His-Ser Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CO)C(O)=O WVTIBGWZUMJBFY-GUBZILKMSA-N 0.000 description 1
- WTMZXOPHTIVFCP-QEWYBTABSA-N Glu-Ile-Phe Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 WTMZXOPHTIVFCP-QEWYBTABSA-N 0.000 description 1
- ZCFNZTVIDMLUQC-SXNHZJKMSA-N Glu-Ile-Trp Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC1=CNC2=CC=CC=C21)C(=O)O)NC(=O)[C@H](CCC(=O)O)N ZCFNZTVIDMLUQC-SXNHZJKMSA-N 0.000 description 1
- IVGJYOOGJLFKQE-AVGNSLFASA-N Glu-Leu-Lys Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CCC(=O)O)N IVGJYOOGJLFKQE-AVGNSLFASA-N 0.000 description 1
- NJCALAAIGREHDR-WDCWCFNPSA-N Glu-Leu-Thr Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O NJCALAAIGREHDR-WDCWCFNPSA-N 0.000 description 1
- ILWHFUZZCFYSKT-AVGNSLFASA-N Glu-Lys-Leu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O ILWHFUZZCFYSKT-AVGNSLFASA-N 0.000 description 1
- SUIAHERNFYRBDZ-GVXVVHGQSA-N Glu-Lys-Val Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(O)=O SUIAHERNFYRBDZ-GVXVVHGQSA-N 0.000 description 1
- ZQYZDDXTNQXUJH-CIUDSAMLSA-N Glu-Met-Ala Chemical compound C[C@@H](C(=O)O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(=O)O)N ZQYZDDXTNQXUJH-CIUDSAMLSA-N 0.000 description 1
- CBEUFCJRFNZMCU-SRVKXCTJSA-N Glu-Met-Leu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(O)=O CBEUFCJRFNZMCU-SRVKXCTJSA-N 0.000 description 1
- UMHRCVCZUPBBQW-GARJFASQSA-N Glu-Met-Pro Chemical compound CSCC[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CCC(=O)O)N UMHRCVCZUPBBQW-GARJFASQSA-N 0.000 description 1
- FGSGPLRPQCZBSQ-AVGNSLFASA-N Glu-Phe-Ser Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CO)C(O)=O FGSGPLRPQCZBSQ-AVGNSLFASA-N 0.000 description 1
- SYWCGQOIIARSIX-SRVKXCTJSA-N Glu-Pro-Leu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CC(C)C)C(O)=O SYWCGQOIIARSIX-SRVKXCTJSA-N 0.000 description 1
- WIKMTDVSCUJIPJ-CIUDSAMLSA-N Glu-Ser-Arg Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CCCN=C(N)N WIKMTDVSCUJIPJ-CIUDSAMLSA-N 0.000 description 1
- VNCNWQPIQYAMAK-ACZMJKKPSA-N Glu-Ser-Ser Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(O)=O VNCNWQPIQYAMAK-ACZMJKKPSA-N 0.000 description 1
- JWNZHMSRZXXGTM-XKBZYTNZSA-N Glu-Ser-Thr Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(O)=O JWNZHMSRZXXGTM-XKBZYTNZSA-N 0.000 description 1
- VHPVBPCCWVDGJL-IRIUXVKKSA-N Glu-Thr-Tyr Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O VHPVBPCCWVDGJL-IRIUXVKKSA-N 0.000 description 1
- HJTSRYLPAYGEEC-SIUGBPQLSA-N Glu-Tyr-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CC1=CC=C(C=C1)O)NC(=O)[C@H](CCC(=O)O)N HJTSRYLPAYGEEC-SIUGBPQLSA-N 0.000 description 1
- KIEICAOUSNYOLM-NRPADANISA-N Glu-Val-Ala Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C)C(O)=O KIEICAOUSNYOLM-NRPADANISA-N 0.000 description 1
- LZEUDRYSAZAJIO-AUTRQRHGSA-N Glu-Val-Glu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O LZEUDRYSAZAJIO-AUTRQRHGSA-N 0.000 description 1
- 108091009426 Glutamate receptor-interacting protein 1 Proteins 0.000 description 1
- 102100039770 Glutamate receptor-interacting protein 1 Human genes 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 102000005720 Glutathione transferase Human genes 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- YMUFWNJHVPQNQD-ZKWXMUAHSA-N Gly-Ala-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)CN YMUFWNJHVPQNQD-ZKWXMUAHSA-N 0.000 description 1
- LJPIRKICOISLKN-WHFBIAKZSA-N Gly-Ala-Ser Chemical compound NCC(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(O)=O LJPIRKICOISLKN-WHFBIAKZSA-N 0.000 description 1
- OCQUNKSFDYDXBG-QXEWZRGKSA-N Gly-Arg-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)CN)CCCN=C(N)N OCQUNKSFDYDXBG-QXEWZRGKSA-N 0.000 description 1
- OVSKVOOUFAKODB-UWVGGRQHSA-N Gly-Arg-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)CN)CCCN=C(N)N OVSKVOOUFAKODB-UWVGGRQHSA-N 0.000 description 1
- KRRMJKMGWWXWDW-STQMWFEESA-N Gly-Arg-Phe Chemical compound NC(=N)NCCC[C@H](NC(=O)CN)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 KRRMJKMGWWXWDW-STQMWFEESA-N 0.000 description 1
- DJTXYXZNNDDEOU-WHFBIAKZSA-N Gly-Asn-Cys Chemical compound C([C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)CN)C(=O)N DJTXYXZNNDDEOU-WHFBIAKZSA-N 0.000 description 1
- FMVLWTYYODVFRG-BQBZGAKWSA-N Gly-Asn-Met Chemical compound CSCC[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)CN FMVLWTYYODVFRG-BQBZGAKWSA-N 0.000 description 1
- IWAXHBCACVWNHT-BQBZGAKWSA-N Gly-Asp-Arg Chemical compound NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(O)=O)CCCN=C(N)N IWAXHBCACVWNHT-BQBZGAKWSA-N 0.000 description 1
- BULIVUZUDBHKKZ-WDSKDSINSA-N Gly-Gln-Asn Chemical compound NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O BULIVUZUDBHKKZ-WDSKDSINSA-N 0.000 description 1
- LXXANCRPFBSSKS-IUCAKERBSA-N Gly-Gln-Leu Chemical compound [H]NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(O)=O LXXANCRPFBSSKS-IUCAKERBSA-N 0.000 description 1
- YYPFZVIXAVDHIK-IUCAKERBSA-N Gly-Glu-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CN YYPFZVIXAVDHIK-IUCAKERBSA-N 0.000 description 1
- FQKKPCWTZZEDIC-XPUUQOCRSA-N Gly-His-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)CN)CC1=CN=CN1 FQKKPCWTZZEDIC-XPUUQOCRSA-N 0.000 description 1
- HAXARWKYFIIHKD-ZKWXMUAHSA-N Gly-Ile-Ser Chemical compound NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(O)=O HAXARWKYFIIHKD-ZKWXMUAHSA-N 0.000 description 1
- ULZCYBYDTUMHNF-IUCAKERBSA-N Gly-Leu-Glu Chemical compound NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O ULZCYBYDTUMHNF-IUCAKERBSA-N 0.000 description 1
- LLZXNUUIBOALNY-QWRGUYRKSA-N Gly-Leu-Lys Chemical compound NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(O)=O)CCCCN LLZXNUUIBOALNY-QWRGUYRKSA-N 0.000 description 1
- LOEANKRDMMVOGZ-YUMQZZPRSA-N Gly-Lys-Asp Chemical compound NCCCC[C@H](NC(=O)CN)C(=O)N[C@@H](CC(O)=O)C(O)=O LOEANKRDMMVOGZ-YUMQZZPRSA-N 0.000 description 1
- YHYDTTUSJXGTQK-UWVGGRQHSA-N Gly-Met-Leu Chemical compound CSCC[C@H](NC(=O)CN)C(=O)N[C@@H](CC(C)C)C(O)=O YHYDTTUSJXGTQK-UWVGGRQHSA-N 0.000 description 1
- OMOZPGCHVWOXHN-BQBZGAKWSA-N Gly-Met-Ser Chemical compound CSCC[C@@H](C(=O)N[C@@H](CO)C(=O)O)NC(=O)CN OMOZPGCHVWOXHN-BQBZGAKWSA-N 0.000 description 1
- YYXJFBMCOUSYSF-RYUDHWBXSA-N Gly-Phe-Gln Chemical compound [H]NCC(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CCC(N)=O)C(O)=O YYXJFBMCOUSYSF-RYUDHWBXSA-N 0.000 description 1
- FEUPVVCGQLNXNP-IRXDYDNUSA-N Gly-Phe-Phe Chemical compound C([C@H](NC(=O)CN)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CC=CC=C1 FEUPVVCGQLNXNP-IRXDYDNUSA-N 0.000 description 1
- QAMMIGULQSIRCD-IRXDYDNUSA-N Gly-Phe-Tyr Chemical compound C([C@H](NC(=O)C[NH3+])C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C([O-])=O)C1=CC=CC=C1 QAMMIGULQSIRCD-IRXDYDNUSA-N 0.000 description 1
- IRJWAYCXIYUHQE-WHFBIAKZSA-N Gly-Ser-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)CN IRJWAYCXIYUHQE-WHFBIAKZSA-N 0.000 description 1
- YABRDIBSPZONIY-BQBZGAKWSA-N Gly-Ser-Met Chemical compound [H]NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(O)=O YABRDIBSPZONIY-BQBZGAKWSA-N 0.000 description 1
- NWOSHVVPKDQKKT-RYUDHWBXSA-N Gly-Tyr-Gln Chemical compound [H]NCC(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CCC(N)=O)C(O)=O NWOSHVVPKDQKKT-RYUDHWBXSA-N 0.000 description 1
- BAYQNCWLXIDLHX-ONGXEEELSA-N Gly-Val-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](C(C)C)NC(=O)CN BAYQNCWLXIDLHX-ONGXEEELSA-N 0.000 description 1
- IZVICCORZOSGPT-JSGCOSHPSA-N Gly-Val-Tyr Chemical compound [H]NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O IZVICCORZOSGPT-JSGCOSHPSA-N 0.000 description 1
- BIAKMWKJMQLZOJ-ZKWXMUAHSA-N His-Ala-Ala Chemical compound C[C@H](NC(=O)[C@H](C)NC(=O)[C@@H](N)Cc1cnc[nH]1)C(O)=O BIAKMWKJMQLZOJ-ZKWXMUAHSA-N 0.000 description 1
- CIWILNZNBPIHEU-DCAQKATOSA-N His-Arg-Asn Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(O)=O CIWILNZNBPIHEU-DCAQKATOSA-N 0.000 description 1
- JBJNKUOMNZGQIM-PYJNHQTQSA-N His-Arg-Ile Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O JBJNKUOMNZGQIM-PYJNHQTQSA-N 0.000 description 1
- SYMSVYVUSPSAAO-IHRRRGAJSA-N His-Arg-Leu Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(O)=O SYMSVYVUSPSAAO-IHRRRGAJSA-N 0.000 description 1
- NELVFWFDOKRTOR-SDDRHHMPSA-N His-Gln-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CC2=CN=CN2)N)C(=O)O NELVFWFDOKRTOR-SDDRHHMPSA-N 0.000 description 1
- SDTPKSOWFXBACN-GUBZILKMSA-N His-Glu-Asp Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(O)=O SDTPKSOWFXBACN-GUBZILKMSA-N 0.000 description 1
- KWBISLAEQZUYIC-UWJYBYFXSA-N His-His-Ala Chemical compound C[C@@H](C(=O)O)NC(=O)[C@H](CC1=CN=CN1)NC(=O)[C@H](CC2=CN=CN2)N KWBISLAEQZUYIC-UWJYBYFXSA-N 0.000 description 1
- BZKDJRSZWLPJNI-SRVKXCTJSA-N His-His-Ser Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CO)C(O)=O BZKDJRSZWLPJNI-SRVKXCTJSA-N 0.000 description 1
- IGBBXBFSLKRHJB-BZSNNMDCSA-N His-Lys-Phe Chemical compound C([C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CN=CN1 IGBBXBFSLKRHJB-BZSNNMDCSA-N 0.000 description 1
- YXASFUBDSDAXQD-UWVGGRQHSA-N His-Met-Gly Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CCSC)C(=O)NCC(O)=O YXASFUBDSDAXQD-UWVGGRQHSA-N 0.000 description 1
- YEKYGQZUBCRNGH-DCAQKATOSA-N His-Pro-Ser Chemical compound C1C[C@H](N(C1)C(=O)[C@H](CC2=CN=CN2)N)C(=O)N[C@@H](CO)C(=O)O YEKYGQZUBCRNGH-DCAQKATOSA-N 0.000 description 1
- SWBUZLFWGJETAO-KKUMJFAQSA-N His-Tyr-Asn Chemical compound C1=CC(=CC=C1C[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](CC2=CN=CN2)N)O SWBUZLFWGJETAO-KKUMJFAQSA-N 0.000 description 1
- WSXNWASHQNSMRX-GVXVVHGQSA-N His-Val-Gln Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)O)NC(=O)[C@H](CC1=CN=CN1)N WSXNWASHQNSMRX-GVXVVHGQSA-N 0.000 description 1
- 102100038885 Histone acetyltransferase p300 Human genes 0.000 description 1
- 101000836540 Homo sapiens Aldo-keto reductase family 1 member B1 Proteins 0.000 description 1
- 101000809450 Homo sapiens Amphiregulin Proteins 0.000 description 1
- 101000882390 Homo sapiens Histone acetyltransferase p300 Proteins 0.000 description 1
- 101000928259 Homo sapiens NADPH:adrenodoxin oxidoreductase, mitochondrial Proteins 0.000 description 1
- 101100187477 Homo sapiens NR1I2 gene Proteins 0.000 description 1
- 101000640876 Homo sapiens Retinoic acid receptor RXR-beta Proteins 0.000 description 1
- 101000640882 Homo sapiens Retinoic acid receptor RXR-gamma Proteins 0.000 description 1
- 101100482127 Homo sapiens TRB gene Proteins 0.000 description 1
- 101000641550 Homo sapiens Vitamin D3 receptor Proteins 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- BOTVMTSMOUSDRW-GMOBBJLQSA-N Ile-Arg-Asn Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC(N)=O)C(O)=O BOTVMTSMOUSDRW-GMOBBJLQSA-N 0.000 description 1
- YOTNPRLPIPHQSB-XUXIUFHCSA-N Ile-Arg-Lys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCCN)C(=O)O)N YOTNPRLPIPHQSB-XUXIUFHCSA-N 0.000 description 1
- UAVQIQOOBXFKRC-BYULHYEWSA-N Ile-Asn-Gly Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(O)=O UAVQIQOOBXFKRC-BYULHYEWSA-N 0.000 description 1
- SJIGTGZVQGLMGG-NAKRPEOUSA-N Ile-Cys-Arg Chemical compound N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)O SJIGTGZVQGLMGG-NAKRPEOUSA-N 0.000 description 1
- LGMUPVWZEYYUMU-YVNDNENWSA-N Ile-Glu-Gln Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCC(=O)N)C(=O)O)N LGMUPVWZEYYUMU-YVNDNENWSA-N 0.000 description 1
- KFVUBLZRFSVDGO-BYULHYEWSA-N Ile-Gly-Asp Chemical compound CC[C@H](C)[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CC(O)=O KFVUBLZRFSVDGO-BYULHYEWSA-N 0.000 description 1
- GQKSJYINYYWPMR-NGZCFLSTSA-N Ile-Gly-Pro Chemical compound CC[C@H](C)[C@@H](C(=O)NCC(=O)N1CCC[C@@H]1C(=O)O)N GQKSJYINYYWPMR-NGZCFLSTSA-N 0.000 description 1
- HPCFRQWLTRDGHT-AJNGGQMLSA-N Ile-Leu-Leu Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O HPCFRQWLTRDGHT-AJNGGQMLSA-N 0.000 description 1
- TVYWVSJGSHQWMT-AJNGGQMLSA-N Ile-Leu-Lys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)O)N TVYWVSJGSHQWMT-AJNGGQMLSA-N 0.000 description 1
- PHRWFSFCNJPWRO-PPCPHDFISA-N Ile-Leu-Thr Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)N PHRWFSFCNJPWRO-PPCPHDFISA-N 0.000 description 1
- RMNMUUCYTMLWNA-ZPFDUUQYSA-N Ile-Lys-Asp Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)O)C(=O)O)N RMNMUUCYTMLWNA-ZPFDUUQYSA-N 0.000 description 1
- GLYJPWIRLBAIJH-UHFFFAOYSA-N Ile-Lys-Pro Natural products CCC(C)C(N)C(=O)NC(CCCCN)C(=O)N1CCCC1C(O)=O GLYJPWIRLBAIJH-UHFFFAOYSA-N 0.000 description 1
- JZNVOBUNTWNZPW-GHCJXIJMSA-N Ile-Ser-Asp Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(=O)O)C(=O)O)N JZNVOBUNTWNZPW-GHCJXIJMSA-N 0.000 description 1
- QGXQHJQPAPMACW-PPCPHDFISA-N Ile-Thr-Lys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)O)N QGXQHJQPAPMACW-PPCPHDFISA-N 0.000 description 1
- ANTFEOSJMAUGIB-KNZXXDILSA-N Ile-Thr-Pro Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@@H]1C(=O)O)N ANTFEOSJMAUGIB-KNZXXDILSA-N 0.000 description 1
- HZVRQFKRALAMQS-SLBDDTMCSA-N Ile-Trp-Asp Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC1=CNC2=CC=CC=C21)C(=O)N[C@@H](CC(=O)O)C(=O)O)N HZVRQFKRALAMQS-SLBDDTMCSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108010065920 Insulin Lispro Proteins 0.000 description 1
- 208000005016 Intestinal Neoplasms Diseases 0.000 description 1
- PWWVAXIEGOYWEE-UHFFFAOYSA-N Isophenergan Chemical compound C1=CC=C2N(CC(C)N(C)C)C3=CC=CC=C3SC2=C1 PWWVAXIEGOYWEE-UHFFFAOYSA-N 0.000 description 1
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical group CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 1
- PMGDADKJMCOXHX-UHFFFAOYSA-N L-Arginyl-L-glutamin-acetat Natural products NC(=N)NCCCC(N)C(=O)NC(CCC(N)=O)C(O)=O PMGDADKJMCOXHX-UHFFFAOYSA-N 0.000 description 1
- FADYJNXDPBKVCA-UHFFFAOYSA-N L-Phenylalanyl-L-lysin Natural products NCCCCC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FADYJNXDPBKVCA-UHFFFAOYSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- RCFDOSNHHZGBOY-UHFFFAOYSA-N L-isoleucyl-L-alanine Natural products CCC(C)C(N)C(=O)NC(C)C(O)=O RCFDOSNHHZGBOY-UHFFFAOYSA-N 0.000 description 1
- KFKWRHQBZQICHA-STQMWFEESA-N L-leucyl-L-phenylalanine Natural products CC(C)C[C@H](N)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 KFKWRHQBZQICHA-STQMWFEESA-N 0.000 description 1
- TYYLDKGBCJGJGW-UHFFFAOYSA-N L-tryptophan-L-tyrosine Natural products C=1NC2=CC=CC=C2C=1CC(N)C(=O)NC(C(O)=O)CC1=CC=C(O)C=C1 TYYLDKGBCJGJGW-UHFFFAOYSA-N 0.000 description 1
- CQQGCWPXDHTTNF-GUBZILKMSA-N Leu-Ala-Glu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@H](C(O)=O)CCC(O)=O CQQGCWPXDHTTNF-GUBZILKMSA-N 0.000 description 1
- JUWJEAPUNARGCF-DCAQKATOSA-N Leu-Arg-Ala Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(O)=O JUWJEAPUNARGCF-DCAQKATOSA-N 0.000 description 1
- QUAAUWNLWMLERT-IHRRRGAJSA-N Leu-Arg-Leu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC(C)C)C(O)=O QUAAUWNLWMLERT-IHRRRGAJSA-N 0.000 description 1
- STAVRDQLZOTNKJ-RHYQMDGZSA-N Leu-Arg-Thr Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(O)=O STAVRDQLZOTNKJ-RHYQMDGZSA-N 0.000 description 1
- OIARJGNVARWKFP-YUMQZZPRSA-N Leu-Asn-Gly Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(O)=O OIARJGNVARWKFP-YUMQZZPRSA-N 0.000 description 1
- ZDSNOSQHMJBRQN-SRVKXCTJSA-N Leu-Asp-His Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CC1=CN=CN1)C(=O)O)N ZDSNOSQHMJBRQN-SRVKXCTJSA-N 0.000 description 1
- QLQHWWCSCLZUMA-KKUMJFAQSA-N Leu-Asp-Tyr Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 QLQHWWCSCLZUMA-KKUMJFAQSA-N 0.000 description 1
- NFHJQETXTSDZSI-DCAQKATOSA-N Leu-Cys-Arg Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O NFHJQETXTSDZSI-DCAQKATOSA-N 0.000 description 1
- KWURTLAFFDOTEQ-GUBZILKMSA-N Leu-Cys-Glu Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(=O)O)C(=O)O)N KWURTLAFFDOTEQ-GUBZILKMSA-N 0.000 description 1
- ZTLGVASZOIKNIX-DCAQKATOSA-N Leu-Gln-Glu Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CCC(=O)O)C(=O)O)N ZTLGVASZOIKNIX-DCAQKATOSA-N 0.000 description 1
- DZQMXBALGUHGJT-GUBZILKMSA-N Leu-Glu-Ala Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(O)=O DZQMXBALGUHGJT-GUBZILKMSA-N 0.000 description 1
- RVVBWTWPNFDYBE-SRVKXCTJSA-N Leu-Glu-Arg Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O RVVBWTWPNFDYBE-SRVKXCTJSA-N 0.000 description 1
- IWTBYNQNAPECCS-AVGNSLFASA-N Leu-Glu-His Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H](C(O)=O)CC1=CN=CN1 IWTBYNQNAPECCS-AVGNSLFASA-N 0.000 description 1
- WQWSMEOYXJTFRU-GUBZILKMSA-N Leu-Glu-Ser Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(O)=O WQWSMEOYXJTFRU-GUBZILKMSA-N 0.000 description 1
- HVJVUYQWFYMGJS-GVXVVHGQSA-N Leu-Glu-Val Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(O)=O HVJVUYQWFYMGJS-GVXVVHGQSA-N 0.000 description 1
- FIYMBBHGYNQFOP-IUCAKERBSA-N Leu-Gly-Gln Chemical compound CC(C)C[C@@H](C(=O)NCC(=O)N[C@@H](CCC(=O)N)C(=O)O)N FIYMBBHGYNQFOP-IUCAKERBSA-N 0.000 description 1
- KEVYYIMVELOXCT-KBPBESRZSA-N Leu-Gly-Phe Chemical compound CC(C)C[C@H]([NH3+])C(=O)NCC(=O)N[C@H](C([O-])=O)CC1=CC=CC=C1 KEVYYIMVELOXCT-KBPBESRZSA-N 0.000 description 1
- XBCWOTOCBXXJDG-BZSNNMDCSA-N Leu-His-Phe Chemical compound C([C@H](NC(=O)[C@@H](N)CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CN=CN1 XBCWOTOCBXXJDG-BZSNNMDCSA-N 0.000 description 1
- WRLPVDVHNWSSCL-MELADBBJSA-N Leu-His-Pro Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CC1=CN=CN1)C(=O)N2CCC[C@@H]2C(=O)O)N WRLPVDVHNWSSCL-MELADBBJSA-N 0.000 description 1
- SGIIOQQGLUUMDQ-IHRRRGAJSA-N Leu-His-Val Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CC1=CN=CN1)C(=O)N[C@@H](C(C)C)C(=O)O)N SGIIOQQGLUUMDQ-IHRRRGAJSA-N 0.000 description 1
- PDQDCFBVYXEFSD-SRVKXCTJSA-N Leu-Leu-Asp Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(O)=O PDQDCFBVYXEFSD-SRVKXCTJSA-N 0.000 description 1
- QNBVTHNJGCOVFA-AVGNSLFASA-N Leu-Leu-Glu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(O)=O)CCC(O)=O QNBVTHNJGCOVFA-AVGNSLFASA-N 0.000 description 1
- YOKVEHGYYQEQOP-QWRGUYRKSA-N Leu-Leu-Gly Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)NCC(O)=O YOKVEHGYYQEQOP-QWRGUYRKSA-N 0.000 description 1
- LXKNSJLSGPNHSK-KKUMJFAQSA-N Leu-Leu-Lys Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)O)N LXKNSJLSGPNHSK-KKUMJFAQSA-N 0.000 description 1
- HDHQQEDVWQGBEE-DCAQKATOSA-N Leu-Met-Ser Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CO)C(O)=O HDHQQEDVWQGBEE-DCAQKATOSA-N 0.000 description 1
- BIZNDKMFQHDOIE-KKUMJFAQSA-N Leu-Phe-Asn Chemical compound CC(C)C[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(O)=O)CC1=CC=CC=C1 BIZNDKMFQHDOIE-KKUMJFAQSA-N 0.000 description 1
- INCJJHQRZGQLFC-KBPBESRZSA-N Leu-Phe-Gly Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)NCC(O)=O INCJJHQRZGQLFC-KBPBESRZSA-N 0.000 description 1
- MJWVXZABPOKJJF-ACRUOGEOSA-N Leu-Phe-Phe Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O MJWVXZABPOKJJF-ACRUOGEOSA-N 0.000 description 1
- YWKNKRAKOCLOLH-OEAJRASXSA-N Leu-Phe-Thr Chemical compound CC(C)C[C@H](N)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)O)C(O)=O)CC1=CC=CC=C1 YWKNKRAKOCLOLH-OEAJRASXSA-N 0.000 description 1
- WMIOEVKKYIMVKI-DCAQKATOSA-N Leu-Pro-Ala Chemical compound [H]N[C@@H](CC(C)C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C)C(O)=O WMIOEVKKYIMVKI-DCAQKATOSA-N 0.000 description 1
- UCBPDSYUVAAHCD-UWVGGRQHSA-N Leu-Pro-Gly Chemical compound CC(C)C[C@H](N)C(=O)N1CCC[C@H]1C(=O)NCC(O)=O UCBPDSYUVAAHCD-UWVGGRQHSA-N 0.000 description 1
- PWPBLZXWFXJFHE-RHYQMDGZSA-N Leu-Pro-Thr Chemical compound CC(C)C[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)O)C(O)=O PWPBLZXWFXJFHE-RHYQMDGZSA-N 0.000 description 1
- JIHDFWWRYHSAQB-GUBZILKMSA-N Leu-Ser-Glu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CCC(O)=O JIHDFWWRYHSAQB-GUBZILKMSA-N 0.000 description 1
- BRTVHXHCUSXYRI-CIUDSAMLSA-N Leu-Ser-Ser Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(O)=O BRTVHXHCUSXYRI-CIUDSAMLSA-N 0.000 description 1
- ZDJQVSIPFLMNOX-RHYQMDGZSA-N Leu-Thr-Arg Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@H](C(O)=O)CCCN=C(N)N ZDJQVSIPFLMNOX-RHYQMDGZSA-N 0.000 description 1
- VDIARPPNADFEAV-WEDXCCLWSA-N Leu-Thr-Gly Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(O)=O VDIARPPNADFEAV-WEDXCCLWSA-N 0.000 description 1
- HGLKOTPFWOMPOB-MEYUZBJRSA-N Leu-Thr-Tyr Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 HGLKOTPFWOMPOB-MEYUZBJRSA-N 0.000 description 1
- VHTIZYYHIUHMCA-JYJNAYRXSA-N Leu-Tyr-Gln Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CCC(N)=O)C(O)=O VHTIZYYHIUHMCA-JYJNAYRXSA-N 0.000 description 1
- VJGQRELPQWNURN-JYJNAYRXSA-N Leu-Tyr-Glu Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CCC(O)=O)C(O)=O VJGQRELPQWNURN-JYJNAYRXSA-N 0.000 description 1
- VKVDRTGWLVZJOM-DCAQKATOSA-N Leu-Val-Ser Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(O)=O VKVDRTGWLVZJOM-DCAQKATOSA-N 0.000 description 1
- KCXUCYYZNZFGLL-SRVKXCTJSA-N Lys-Ala-Leu Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(O)=O KCXUCYYZNZFGLL-SRVKXCTJSA-N 0.000 description 1
- NQCJGQHHYZNUDK-DCAQKATOSA-N Lys-Arg-Ser Chemical compound NCCCC[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CO)C(O)=O)CCCN=C(N)N NQCJGQHHYZNUDK-DCAQKATOSA-N 0.000 description 1
- DGAAQRAUOFHBFJ-CIUDSAMLSA-N Lys-Asn-Ala Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(O)=O DGAAQRAUOFHBFJ-CIUDSAMLSA-N 0.000 description 1
- ABHIXYDMILIUKV-CIUDSAMLSA-N Lys-Asn-Asn Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O ABHIXYDMILIUKV-CIUDSAMLSA-N 0.000 description 1
- HQVDJTYKCMIWJP-YUMQZZPRSA-N Lys-Asn-Gly Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(O)=O HQVDJTYKCMIWJP-YUMQZZPRSA-N 0.000 description 1
- LZWNAOIMTLNMDW-NHCYSSNCSA-N Lys-Asn-Val Chemical compound CC(C)[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CCCCN)N LZWNAOIMTLNMDW-NHCYSSNCSA-N 0.000 description 1
- AAORVPFVUIHEAB-YUMQZZPRSA-N Lys-Asp-Gly Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(O)=O AAORVPFVUIHEAB-YUMQZZPRSA-N 0.000 description 1
- SVJRVFPSHPGWFF-DCAQKATOSA-N Lys-Cys-Arg Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O SVJRVFPSHPGWFF-DCAQKATOSA-N 0.000 description 1
- BYEBKXRNDLTGFW-CIUDSAMLSA-N Lys-Cys-Ser Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CO)C(O)=O BYEBKXRNDLTGFW-CIUDSAMLSA-N 0.000 description 1
- WTZUSCUIVPVCRH-SRVKXCTJSA-N Lys-Gln-Arg Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(O)=O)CCCN=C(N)N WTZUSCUIVPVCRH-SRVKXCTJSA-N 0.000 description 1
- PBIPLDMFHAICIP-DCAQKATOSA-N Lys-Glu-Glu Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O PBIPLDMFHAICIP-DCAQKATOSA-N 0.000 description 1
- IMAKMJCBYCSMHM-AVGNSLFASA-N Lys-Glu-Lys Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H](C(O)=O)CCCCN IMAKMJCBYCSMHM-AVGNSLFASA-N 0.000 description 1
- GPJGFSFYBJGYRX-YUMQZZPRSA-N Lys-Gly-Asp Chemical compound NCCCC[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CC(O)=O GPJGFSFYBJGYRX-YUMQZZPRSA-N 0.000 description 1
- GQFDWEDHOQRNLC-QWRGUYRKSA-N Lys-Gly-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN GQFDWEDHOQRNLC-QWRGUYRKSA-N 0.000 description 1
- JZMGVXLDOQOKAH-UWVGGRQHSA-N Lys-Gly-Met Chemical compound [H]N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCSC)C(O)=O JZMGVXLDOQOKAH-UWVGGRQHSA-N 0.000 description 1
- PBLLTSKBTAHDNA-KBPBESRZSA-N Lys-Gly-Phe Chemical compound [H]N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O PBLLTSKBTAHDNA-KBPBESRZSA-N 0.000 description 1
- WOEDRPCHKPSFDT-MXAVVETBSA-N Lys-His-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CC1=CN=CN1)NC(=O)[C@H](CCCCN)N WOEDRPCHKPSFDT-MXAVVETBSA-N 0.000 description 1
- ZMMDPRTXLAEMOD-BZSNNMDCSA-N Lys-His-Phe Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O ZMMDPRTXLAEMOD-BZSNNMDCSA-N 0.000 description 1
- OIYWBDBHEGAVST-BZSNNMDCSA-N Lys-His-Tyr Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O OIYWBDBHEGAVST-BZSNNMDCSA-N 0.000 description 1
- XREQQOATSMMAJP-MGHWNKPDSA-N Lys-Ile-Tyr Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O XREQQOATSMMAJP-MGHWNKPDSA-N 0.000 description 1
- PINHPJWGVBKQII-SRVKXCTJSA-N Lys-Leu-Cys Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CCCCN)N PINHPJWGVBKQII-SRVKXCTJSA-N 0.000 description 1
- ONPDTSFZAIWMDI-AVGNSLFASA-N Lys-Leu-Gln Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O ONPDTSFZAIWMDI-AVGNSLFASA-N 0.000 description 1
- AIRZWUMAHCDDHR-KKUMJFAQSA-N Lys-Leu-Leu Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O AIRZWUMAHCDDHR-KKUMJFAQSA-N 0.000 description 1
- ZJWIXBZTAAJERF-IHRRRGAJSA-N Lys-Lys-Arg Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(O)=O)CCCN=C(N)N ZJWIXBZTAAJERF-IHRRRGAJSA-N 0.000 description 1
- HVAUKHLDSDDROB-KKUMJFAQSA-N Lys-Lys-Leu Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O HVAUKHLDSDDROB-KKUMJFAQSA-N 0.000 description 1
- YXPJCVNIDDKGOE-MELADBBJSA-N Lys-Lys-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)N)C(=O)O YXPJCVNIDDKGOE-MELADBBJSA-N 0.000 description 1
- URBJRJKWSUFCKS-AVGNSLFASA-N Lys-Met-Arg Chemical compound CSCC[C@@H](C(=O)N[C@@H](CCCN=C(N)N)C(=O)O)NC(=O)[C@H](CCCCN)N URBJRJKWSUFCKS-AVGNSLFASA-N 0.000 description 1
- IPSDPDAOSAEWCN-RHYQMDGZSA-N Lys-Met-Thr Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CCSC)C(=O)N[C@@H]([C@@H](C)O)C(O)=O IPSDPDAOSAEWCN-RHYQMDGZSA-N 0.000 description 1
- BOJYMMBYBNOOGG-DCAQKATOSA-N Lys-Pro-Ala Chemical compound [H]N[C@@H](CCCCN)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C)C(O)=O BOJYMMBYBNOOGG-DCAQKATOSA-N 0.000 description 1
- CTJUSALVKAWFFU-CIUDSAMLSA-N Lys-Ser-Cys Chemical compound C(CCN)C[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CS)C(=O)O)N CTJUSALVKAWFFU-CIUDSAMLSA-N 0.000 description 1
- ZUGVARDEGWMMLK-SRVKXCTJSA-N Lys-Ser-Lys Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CCCCN ZUGVARDEGWMMLK-SRVKXCTJSA-N 0.000 description 1
- IEIHKHYMBIYQTH-YESZJQIVSA-N Lys-Tyr-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CC2=CC=C(C=C2)O)NC(=O)[C@H](CCCCN)N)C(=O)O IEIHKHYMBIYQTH-YESZJQIVSA-N 0.000 description 1
- VKCPHIOZDWUFSW-ONGXEEELSA-N Lys-Val-Gly Chemical compound OC(=O)CNC(=O)[C@H](C(C)C)NC(=O)[C@@H](N)CCCCN VKCPHIOZDWUFSW-ONGXEEELSA-N 0.000 description 1
- 108010093662 Member 11 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 102000001479 Member 11 Subfamily B ATP Binding Cassette Transporter Human genes 0.000 description 1
- YNOVBMBQSQTLFM-DCAQKATOSA-N Met-Asn-Leu Chemical compound [H]N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(O)=O YNOVBMBQSQTLFM-DCAQKATOSA-N 0.000 description 1
- WGBMNLCRYKSWAR-DCAQKATOSA-N Met-Asp-Lys Chemical compound CSCC[C@H](N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(O)=O)CCCCN WGBMNLCRYKSWAR-DCAQKATOSA-N 0.000 description 1
- TZLYIHDABYBOCJ-FXQIFTODSA-N Met-Asp-Ser Chemical compound CSCC[C@H](N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(O)=O TZLYIHDABYBOCJ-FXQIFTODSA-N 0.000 description 1
- MCNGIXXCMJAURZ-VEVYYDQMSA-N Met-Asp-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CCSC)N)O MCNGIXXCMJAURZ-VEVYYDQMSA-N 0.000 description 1
- RAAVFTFEAUAVIY-DCAQKATOSA-N Met-Glu-Met Chemical compound CSCC[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCSC)C(=O)O)N RAAVFTFEAUAVIY-DCAQKATOSA-N 0.000 description 1
- MVBZBRKNZVJEKK-DTWKUNHWSA-N Met-Gly-Pro Chemical compound CSCC[C@@H](C(=O)NCC(=O)N1CCC[C@@H]1C(=O)O)N MVBZBRKNZVJEKK-DTWKUNHWSA-N 0.000 description 1
- BEZJTLKUMFMITF-AVGNSLFASA-N Met-Lys-Arg Chemical compound CSCC[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(O)=O)CCCNC(N)=N BEZJTLKUMFMITF-AVGNSLFASA-N 0.000 description 1
- HSJIGJRZYUADSS-IHRRRGAJSA-N Met-Lys-Leu Chemical compound [H]N[C@@H](CCSC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O HSJIGJRZYUADSS-IHRRRGAJSA-N 0.000 description 1
- OIFHHODAXVWKJN-ULQDDVLXSA-N Met-Phe-Leu Chemical compound CSCC[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C(O)=O)CC1=CC=CC=C1 OIFHHODAXVWKJN-ULQDDVLXSA-N 0.000 description 1
- QEDGNYFHLXXIDC-DCAQKATOSA-N Met-Pro-Gln Chemical compound CSCC[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(O)=O QEDGNYFHLXXIDC-DCAQKATOSA-N 0.000 description 1
- BJPQKNHZHUCQNQ-SRVKXCTJSA-N Met-Pro-Val Chemical compound CC(C)[C@@H](C(=O)O)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCSC)N BJPQKNHZHUCQNQ-SRVKXCTJSA-N 0.000 description 1
- MNGBICITWAPGAS-BPUTZDHNSA-N Met-Ser-Trp Chemical compound [H]N[C@@H](CCSC)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(O)=O MNGBICITWAPGAS-BPUTZDHNSA-N 0.000 description 1
- GGXZOTSDJJTDGB-GUBZILKMSA-N Met-Ser-Val Chemical compound [H]N[C@@H](CCSC)C(=O)N[C@@H](CO)C(=O)N[C@@H](C(C)C)C(O)=O GGXZOTSDJJTDGB-GUBZILKMSA-N 0.000 description 1
- SPSSJSICDYYTQN-HJGDQZAQSA-N Met-Thr-Gln Chemical compound CSCC[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@H](C(O)=O)CCC(N)=O SPSSJSICDYYTQN-HJGDQZAQSA-N 0.000 description 1
- NBEFNGUZUOUGFG-KKUMJFAQSA-N Met-Tyr-Gln Chemical compound CSCC[C@@H](C(=O)N[C@@H](CC1=CC=C(C=C1)O)C(=O)N[C@@H](CCC(=O)N)C(=O)O)N NBEFNGUZUOUGFG-KKUMJFAQSA-N 0.000 description 1
- CKAVKDJBSNTJDB-SRVKXCTJSA-N Met-Val-Met Chemical compound CSCC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(O)=O)CCSC CKAVKDJBSNTJDB-SRVKXCTJSA-N 0.000 description 1
- 241000713333 Mouse mammary tumor virus Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101000714513 Mus musculus Carbonic anhydrase-related protein Proteins 0.000 description 1
- 101000978776 Mus musculus Neurogenic locus notch homolog protein 1 Proteins 0.000 description 1
- 101100187479 Mus musculus Nr1i2 gene Proteins 0.000 description 1
- 101000741778 Mus musculus Peroxisome proliferator-activated receptor alpha Proteins 0.000 description 1
- 101100464833 Mus musculus Ppara gene Proteins 0.000 description 1
- 108010002311 N-glycylglutamic acid Proteins 0.000 description 1
- 108010077850 Nuclear Localization Signals Proteins 0.000 description 1
- 108090001146 Nuclear Receptor Coactivator 1 Proteins 0.000 description 1
- 108090001145 Nuclear Receptor Coactivator 3 Proteins 0.000 description 1
- 108010062309 Nuclear Receptor Interacting Protein 1 Proteins 0.000 description 1
- 102000010839 Nuclear Receptor Interacting Protein 1 Human genes 0.000 description 1
- 102100037223 Nuclear receptor coactivator 1 Human genes 0.000 description 1
- 102100022883 Nuclear receptor coactivator 3 Human genes 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- 102000016978 Orphan receptors Human genes 0.000 description 1
- 108070000031 Orphan receptors Proteins 0.000 description 1
- 102000023984 PPAR alpha Human genes 0.000 description 1
- 108010015181 PPAR delta Proteins 0.000 description 1
- 108010044210 PPAR-beta Proteins 0.000 description 1
- MDHZEOMXGNBSIL-DLOVCJGASA-N Phe-Ala-Cys Chemical compound C[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CC1=CC=CC=C1)N MDHZEOMXGNBSIL-DLOVCJGASA-N 0.000 description 1
- OXUMFAOVGFODPN-KKUMJFAQSA-N Phe-Asn-His Chemical compound C1=CC=C(C=C1)C[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CC2=CN=CN2)C(=O)O)N OXUMFAOVGFODPN-KKUMJFAQSA-N 0.000 description 1
- XMPUYNHKEPFERE-IHRRRGAJSA-N Phe-Asp-Arg Chemical compound NC(N)=NCCC[C@@H](C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 XMPUYNHKEPFERE-IHRRRGAJSA-N 0.000 description 1
- PDUVELWDJZOUEI-IHRRRGAJSA-N Phe-Cys-Arg Chemical compound [H]N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O PDUVELWDJZOUEI-IHRRRGAJSA-N 0.000 description 1
- NKLDZIPTGKBDBB-HTUGSXCWSA-N Phe-Gln-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CC1=CC=CC=C1)N)O NKLDZIPTGKBDBB-HTUGSXCWSA-N 0.000 description 1
- DOXQMJCSSYZSNM-BZSNNMDCSA-N Phe-Lys-Leu Chemical compound [H]N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O DOXQMJCSSYZSNM-BZSNNMDCSA-N 0.000 description 1
- CZQZSMJXFGGBHM-KKUMJFAQSA-N Phe-Pro-Gln Chemical compound C1C[C@H](N(C1)C(=O)[C@H](CC2=CC=CC=C2)N)C(=O)N[C@@H](CCC(=O)N)C(=O)O CZQZSMJXFGGBHM-KKUMJFAQSA-N 0.000 description 1
- AFNJAQVMTIQTCB-DLOVCJGASA-N Phe-Ser-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC1=CC=CC=C1 AFNJAQVMTIQTCB-DLOVCJGASA-N 0.000 description 1
- BONHGTUEEPIMPM-AVGNSLFASA-N Phe-Ser-Glu Chemical compound [H]N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(O)=O BONHGTUEEPIMPM-AVGNSLFASA-N 0.000 description 1
- IAOZOFPONWDXNT-IXOXFDKPSA-N Phe-Ser-Thr Chemical compound [H]N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(O)=O IAOZOFPONWDXNT-IXOXFDKPSA-N 0.000 description 1
- MVIJMIZJPHQGEN-IHRRRGAJSA-N Phe-Ser-Val Chemical compound CC(C)[C@@H](C([O-])=O)NC(=O)[C@H](CO)NC(=O)[C@@H]([NH3+])CC1=CC=CC=C1 MVIJMIZJPHQGEN-IHRRRGAJSA-N 0.000 description 1
- SJRQWEDYTKYHHL-SLFFLAALSA-N Phe-Tyr-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CC2=CC=C(C=C2)O)NC(=O)[C@H](CC3=CC=CC=C3)N)C(=O)O SJRQWEDYTKYHHL-SLFFLAALSA-N 0.000 description 1
- KUSYCSMTTHSZOA-DZKIICNBSA-N Phe-Val-Gln Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)O)NC(=O)[C@H](CC1=CC=CC=C1)N KUSYCSMTTHSZOA-DZKIICNBSA-N 0.000 description 1
- 241001505332 Polyomavirus sp. Species 0.000 description 1
- 208000037062 Polyps Diseases 0.000 description 1
- KIZQGKLMXKGDIV-BQBZGAKWSA-N Pro-Ala-Gly Chemical compound OC(=O)CNC(=O)[C@H](C)NC(=O)[C@@H]1CCCN1 KIZQGKLMXKGDIV-BQBZGAKWSA-N 0.000 description 1
- IFMDQWDAJUMMJC-DCAQKATOSA-N Pro-Ala-Leu Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(O)=O IFMDQWDAJUMMJC-DCAQKATOSA-N 0.000 description 1
- ZSKJPKFTPQCPIH-RCWTZXSCSA-N Pro-Arg-Thr Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(O)=O ZSKJPKFTPQCPIH-RCWTZXSCSA-N 0.000 description 1
- SMCHPSMKAFIERP-FXQIFTODSA-N Pro-Asn-Asp Chemical compound OC(=O)C[C@@H](C(O)=O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@@H]1CCCN1 SMCHPSMKAFIERP-FXQIFTODSA-N 0.000 description 1
- ZCXQTRXYZOSGJR-FXQIFTODSA-N Pro-Asp-Ser Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(O)=O ZCXQTRXYZOSGJR-FXQIFTODSA-N 0.000 description 1
- FISHYTLIMUYTQY-GUBZILKMSA-N Pro-Gln-Gln Chemical compound NC(=O)CC[C@@H](C(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H]1CCCN1 FISHYTLIMUYTQY-GUBZILKMSA-N 0.000 description 1
- UPJGUQPLYWTISV-GUBZILKMSA-N Pro-Gln-Glu Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O UPJGUQPLYWTISV-GUBZILKMSA-N 0.000 description 1
- CMOIIANLNNYUTP-SRVKXCTJSA-N Pro-Gln-His Chemical compound C1C[C@H](NC1)C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CC2=CN=CN2)C(=O)O CMOIIANLNNYUTP-SRVKXCTJSA-N 0.000 description 1
- WGAQWMRJUFQXMF-ZPFDUUQYSA-N Pro-Gln-Ile Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O WGAQWMRJUFQXMF-ZPFDUUQYSA-N 0.000 description 1
- UAYHMOIGIQZLFR-NHCYSSNCSA-N Pro-Gln-Val Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(O)=O UAYHMOIGIQZLFR-NHCYSSNCSA-N 0.000 description 1
- KIPIKSXPPLABPN-CIUDSAMLSA-N Pro-Glu-Asn Chemical compound NC(=O)C[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H]1CCCN1 KIPIKSXPPLABPN-CIUDSAMLSA-N 0.000 description 1
- MGDFPGCFVJFITQ-CIUDSAMLSA-N Pro-Glu-Asp Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(O)=O MGDFPGCFVJFITQ-CIUDSAMLSA-N 0.000 description 1
- FRKBNXCFJBPJOL-GUBZILKMSA-N Pro-Glu-Glu Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O FRKBNXCFJBPJOL-GUBZILKMSA-N 0.000 description 1
- SSWJYJHXQOYTSP-SRVKXCTJSA-N Pro-His-Gln Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CCC(N)=O)C(O)=O SSWJYJHXQOYTSP-SRVKXCTJSA-N 0.000 description 1
- AQSMZTIEJMZQEC-DCAQKATOSA-N Pro-His-Ser Chemical compound C1C[C@H](NC1)C(=O)N[C@@H](CC2=CN=CN2)C(=O)N[C@@H](CO)C(=O)O AQSMZTIEJMZQEC-DCAQKATOSA-N 0.000 description 1
- XYSXOCIWCPFOCG-IHRRRGAJSA-N Pro-Leu-Leu Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O XYSXOCIWCPFOCG-IHRRRGAJSA-N 0.000 description 1
- VTFXTWDFPTWNJY-RHYQMDGZSA-N Pro-Leu-Thr Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O VTFXTWDFPTWNJY-RHYQMDGZSA-N 0.000 description 1
- YAZNFQUKPUASKB-DCAQKATOSA-N Pro-Lys-Cys Chemical compound C1C[C@H](NC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)O YAZNFQUKPUASKB-DCAQKATOSA-N 0.000 description 1
- BLJMJZOMZRCESA-GUBZILKMSA-N Pro-Met-Asn Chemical compound CSCC[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@@H]1CCCN1 BLJMJZOMZRCESA-GUBZILKMSA-N 0.000 description 1
- WIPAMEKBSHNFQE-IUCAKERBSA-N Pro-Met-Gly Chemical compound CSCC[C@@H](C(=O)NCC(=O)O)NC(=O)[C@@H]1CCCN1 WIPAMEKBSHNFQE-IUCAKERBSA-N 0.000 description 1
- CGSOWZUPLOKYOR-AVGNSLFASA-N Pro-Pro-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@H]1NCCC1 CGSOWZUPLOKYOR-AVGNSLFASA-N 0.000 description 1
- QHSSUIHLAIWXEE-IHRRRGAJSA-N Pro-Tyr-Asn Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CC(N)=O)C(O)=O QHSSUIHLAIWXEE-IHRRRGAJSA-N 0.000 description 1
- VEUACYMXJKXALX-IHRRRGAJSA-N Pro-Tyr-Ser Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(O)=O VEUACYMXJKXALX-IHRRRGAJSA-N 0.000 description 1
- IMNVAOPEMFDAQD-NHCYSSNCSA-N Pro-Val-Glu Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O IMNVAOPEMFDAQD-NHCYSSNCSA-N 0.000 description 1
- FIODMZKLZFLYQP-GUBZILKMSA-N Pro-Val-Ser Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(O)=O FIODMZKLZFLYQP-GUBZILKMSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 108010003201 RGH 0205 Proteins 0.000 description 1
- 101150050070 RXRA gene Proteins 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 102100034253 Retinoic acid receptor RXR-beta Human genes 0.000 description 1
- 102100034262 Retinoic acid receptor RXR-gamma Human genes 0.000 description 1
- 102100023606 Retinoic acid receptor alpha Human genes 0.000 description 1
- 102100033909 Retinoic acid receptor beta Human genes 0.000 description 1
- 102100033912 Retinoic acid receptor gamma Human genes 0.000 description 1
- 229940121908 Retinoid X receptor agonist Drugs 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- MWMKFWJYRRGXOR-ZLUOBGJFSA-N Ser-Ala-Asn Chemical compound N[C@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)O)CC(N)=O)C)CO MWMKFWJYRRGXOR-ZLUOBGJFSA-N 0.000 description 1
- SRTCFKGBYBZRHA-ACZMJKKPSA-N Ser-Ala-Glu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(O)=O SRTCFKGBYBZRHA-ACZMJKKPSA-N 0.000 description 1
- HBZBPFLJNDXRAY-FXQIFTODSA-N Ser-Ala-Val Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(O)=O HBZBPFLJNDXRAY-FXQIFTODSA-N 0.000 description 1
- FTVRVZNYIYWJGB-ACZMJKKPSA-N Ser-Asp-Glu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O FTVRVZNYIYWJGB-ACZMJKKPSA-N 0.000 description 1
- BGOWRLSWJCVYAQ-CIUDSAMLSA-N Ser-Asp-Leu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(O)=O BGOWRLSWJCVYAQ-CIUDSAMLSA-N 0.000 description 1
- WTPKKLMBNBCCNL-ACZMJKKPSA-N Ser-Cys-Glu Chemical compound C(CC(=O)O)[C@@H](C(=O)O)NC(=O)[C@H](CS)NC(=O)[C@H](CO)N WTPKKLMBNBCCNL-ACZMJKKPSA-N 0.000 description 1
- SQBLRDDJTUJDMV-ACZMJKKPSA-N Ser-Glu-Asn Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O SQBLRDDJTUJDMV-ACZMJKKPSA-N 0.000 description 1
- GRSLLFZTTLBOQX-CIUDSAMLSA-N Ser-Glu-Met Chemical compound CSCC[C@@H](C(=O)O)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)N GRSLLFZTTLBOQX-CIUDSAMLSA-N 0.000 description 1
- IXCHOHLPHNGFTJ-YUMQZZPRSA-N Ser-Gly-His Chemical compound C1=C(NC=N1)C[C@@H](C(=O)O)NC(=O)CNC(=O)[C@H](CO)N IXCHOHLPHNGFTJ-YUMQZZPRSA-N 0.000 description 1
- IOVHBRCQOGWAQH-ZKWXMUAHSA-N Ser-Gly-Ile Chemical compound [H]N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(O)=O IOVHBRCQOGWAQH-ZKWXMUAHSA-N 0.000 description 1
- JFWDJFULOLKQFY-QWRGUYRKSA-N Ser-Gly-Phe Chemical compound [H]N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O JFWDJFULOLKQFY-QWRGUYRKSA-N 0.000 description 1
- OQPNSDWGAMFJNU-QWRGUYRKSA-N Ser-Gly-Tyr Chemical compound OC[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 OQPNSDWGAMFJNU-QWRGUYRKSA-N 0.000 description 1
- QBUWQRKEHJXTOP-DCAQKATOSA-N Ser-His-Arg Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O QBUWQRKEHJXTOP-DCAQKATOSA-N 0.000 description 1
- CICQXRWZNVXFCU-SRVKXCTJSA-N Ser-His-Leu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC(C)C)C(O)=O CICQXRWZNVXFCU-SRVKXCTJSA-N 0.000 description 1
- ZUDXUJSYCCNZQJ-DCAQKATOSA-N Ser-His-Val Chemical compound CC(C)[C@@H](C(=O)O)NC(=O)[C@H](CC1=CN=CN1)NC(=O)[C@H](CO)N ZUDXUJSYCCNZQJ-DCAQKATOSA-N 0.000 description 1
- SFTZTYBXIXLRGQ-JBDRJPRFSA-N Ser-Ile-Ala Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O SFTZTYBXIXLRGQ-JBDRJPRFSA-N 0.000 description 1
- IFPBAGJBHSNYPR-ZKWXMUAHSA-N Ser-Ile-Gly Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(O)=O IFPBAGJBHSNYPR-ZKWXMUAHSA-N 0.000 description 1
- XNCUYZKGQOCOQH-YUMQZZPRSA-N Ser-Leu-Gly Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)NCC(O)=O XNCUYZKGQOCOQH-YUMQZZPRSA-N 0.000 description 1
- OCWWJBZQXGYQCA-DCAQKATOSA-N Ser-Lys-Met Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCSC)C(O)=O OCWWJBZQXGYQCA-DCAQKATOSA-N 0.000 description 1
- FBLNYDYPCLFTSP-IXOXFDKPSA-N Ser-Phe-Thr Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H]([C@@H](C)O)C(O)=O FBLNYDYPCLFTSP-IXOXFDKPSA-N 0.000 description 1
- PJIQEIFXZPCWOJ-FXQIFTODSA-N Ser-Pro-Asp Chemical compound [H]N[C@@H](CO)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CC(O)=O)C(O)=O PJIQEIFXZPCWOJ-FXQIFTODSA-N 0.000 description 1
- GZGFSPWOMUKKCV-NAKRPEOUSA-N Ser-Pro-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@@H](N)CO GZGFSPWOMUKKCV-NAKRPEOUSA-N 0.000 description 1
- HHJFMHQYEAAOBM-ZLUOBGJFSA-N Ser-Ser-Ala Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(O)=O HHJFMHQYEAAOBM-ZLUOBGJFSA-N 0.000 description 1
- BMKNXTJLHFIAAH-CIUDSAMLSA-N Ser-Ser-Leu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(O)=O BMKNXTJLHFIAAH-CIUDSAMLSA-N 0.000 description 1
- CUXJENOFJXOSOZ-BIIVOSGPSA-N Ser-Ser-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CO)NC(=O)[C@H](CO)N)C(=O)O CUXJENOFJXOSOZ-BIIVOSGPSA-N 0.000 description 1
- JURQXQBJKUHGJS-UHFFFAOYSA-N Ser-Ser-Ser-Ser Chemical compound OCC(N)C(=O)NC(CO)C(=O)NC(CO)C(=O)NC(CO)C(O)=O JURQXQBJKUHGJS-UHFFFAOYSA-N 0.000 description 1
- NADLKBTYNKUJEP-KATARQTJSA-N Ser-Thr-Leu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(O)=O NADLKBTYNKUJEP-KATARQTJSA-N 0.000 description 1
- WMZVVNLPHFSUPA-BPUTZDHNSA-N Ser-Trp-Arg Chemical compound C1=CC=C2C(C[C@H](NC(=O)[C@H](CO)N)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O)=CNC2=C1 WMZVVNLPHFSUPA-BPUTZDHNSA-N 0.000 description 1
- PQEQXWRVHQAAKS-SRVKXCTJSA-N Ser-Tyr-Asn Chemical compound NC(=O)C[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CO)N)CC1=CC=C(O)C=C1 PQEQXWRVHQAAKS-SRVKXCTJSA-N 0.000 description 1
- VVKVHAOOUGNDPJ-SRVKXCTJSA-N Ser-Tyr-Ser Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(O)=O VVKVHAOOUGNDPJ-SRVKXCTJSA-N 0.000 description 1
- BEBVVQPDSHHWQL-NRPADANISA-N Ser-Val-Glu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O BEBVVQPDSHHWQL-NRPADANISA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UZMAPBJVXOGOFT-UHFFFAOYSA-N Syringetin Natural products COC1=C(O)C(OC)=CC(C2=C(C(=O)C3=C(O)C=C(O)C=C3O2)O)=C1 UZMAPBJVXOGOFT-UHFFFAOYSA-N 0.000 description 1
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 description 1
- JTEICXDKGWKRRV-HJGDQZAQSA-N Thr-Asn-Lys Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CCCCN)C(=O)O)N)O JTEICXDKGWKRRV-HJGDQZAQSA-N 0.000 description 1
- YBXMGKCLOPDEKA-NUMRIWBASA-N Thr-Asp-Glu Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O YBXMGKCLOPDEKA-NUMRIWBASA-N 0.000 description 1
- APIQKJYZDWVOCE-VEVYYDQMSA-N Thr-Asp-Met Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCSC)C(O)=O APIQKJYZDWVOCE-VEVYYDQMSA-N 0.000 description 1
- NRUPKQSXTJNQGD-XGEHTFHBSA-N Thr-Cys-Arg Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O NRUPKQSXTJNQGD-XGEHTFHBSA-N 0.000 description 1
- DHPPWTOLRWYIDS-XKBZYTNZSA-N Thr-Cys-Glu Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(O)=O)C(O)=O DHPPWTOLRWYIDS-XKBZYTNZSA-N 0.000 description 1
- VUVCRYXYUUPGSB-GLLZPBPUSA-N Thr-Gln-Glu Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CCC(=O)O)C(=O)O)N)O VUVCRYXYUUPGSB-GLLZPBPUSA-N 0.000 description 1
- DKDHTRVDOUZZTP-IFFSRLJSSA-N Thr-Gln-Val Chemical compound CC(C)[C@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)[C@@H](C)O)C(O)=O DKDHTRVDOUZZTP-IFFSRLJSSA-N 0.000 description 1
- WDFPMSHYMRBLKM-NKIYYHGXSA-N Thr-Glu-His Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC1=CN=CN1)C(=O)O)N)O WDFPMSHYMRBLKM-NKIYYHGXSA-N 0.000 description 1
- JMGJDTNUMAZNLX-RWRJDSDZSA-N Thr-Glu-Ile Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O JMGJDTNUMAZNLX-RWRJDSDZSA-N 0.000 description 1
- ONNSECRQFSTMCC-XKBZYTNZSA-N Thr-Glu-Ser Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(O)=O ONNSECRQFSTMCC-XKBZYTNZSA-N 0.000 description 1
- KCRQEJSKXAIULJ-FJXKBIBVSA-N Thr-Gly-Arg Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(O)=O KCRQEJSKXAIULJ-FJXKBIBVSA-N 0.000 description 1
- RRRRCRYTLZVCEN-HJGDQZAQSA-N Thr-Leu-Asp Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(O)=O RRRRCRYTLZVCEN-HJGDQZAQSA-N 0.000 description 1
- RFKVQLIXNVEOMB-WEDXCCLWSA-N Thr-Leu-Gly Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)O)N)O RFKVQLIXNVEOMB-WEDXCCLWSA-N 0.000 description 1
- MEJHFIOYJHTWMK-VOAKCMCISA-N Thr-Leu-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)[C@@H](C)O MEJHFIOYJHTWMK-VOAKCMCISA-N 0.000 description 1
- BDGBHYCAZJPLHX-HJGDQZAQSA-N Thr-Lys-Asn Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(O)=O BDGBHYCAZJPLHX-HJGDQZAQSA-N 0.000 description 1
- KZURUCDWKDEAFZ-XVSYOHENSA-N Thr-Phe-Asn Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC(=O)N)C(=O)O)N)O KZURUCDWKDEAFZ-XVSYOHENSA-N 0.000 description 1
- WNQJTLATMXYSEL-OEAJRASXSA-N Thr-Phe-Leu Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC(C)C)C(O)=O WNQJTLATMXYSEL-OEAJRASXSA-N 0.000 description 1
- NDXSOKGYKCGYKT-VEVYYDQMSA-N Thr-Pro-Asp Chemical compound C[C@@H](O)[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CC(O)=O)C(O)=O NDXSOKGYKCGYKT-VEVYYDQMSA-N 0.000 description 1
- VTMGKRABARCZAX-OSUNSFLBSA-N Thr-Pro-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@@H](N)[C@@H](C)O VTMGKRABARCZAX-OSUNSFLBSA-N 0.000 description 1
- DEGCBBCMYWNJNA-RHYQMDGZSA-N Thr-Pro-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@@H](N)[C@@H](C)O DEGCBBCMYWNJNA-RHYQMDGZSA-N 0.000 description 1
- BDENGIGFTNYZSJ-RCWTZXSCSA-N Thr-Pro-Met Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCSC)C(O)=O BDENGIGFTNYZSJ-RCWTZXSCSA-N 0.000 description 1
- BCYUHPXBHCUYBA-CUJWVEQBSA-N Thr-Ser-His Chemical compound C[C@@H](O)[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1cnc[nH]1)C(O)=O BCYUHPXBHCUYBA-CUJWVEQBSA-N 0.000 description 1
- VUXIQSUQQYNLJP-XAVMHZPKSA-N Thr-Ser-Pro Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CO)C(=O)N1CCC[C@@H]1C(=O)O)N)O VUXIQSUQQYNLJP-XAVMHZPKSA-N 0.000 description 1
- RVMNUBQWPVOUKH-HEIBUPTGSA-N Thr-Ser-Thr Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(O)=O RVMNUBQWPVOUKH-HEIBUPTGSA-N 0.000 description 1
- ZESGVALRVJIVLZ-VFCFLDTKSA-N Thr-Thr-Pro Chemical compound C[C@H]([C@@H](C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@@H]1C(=O)O)N)O ZESGVALRVJIVLZ-VFCFLDTKSA-N 0.000 description 1
- MNYNCKZAEIAONY-XGEHTFHBSA-N Thr-Val-Ser Chemical compound C[C@@H](O)[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(O)=O MNYNCKZAEIAONY-XGEHTFHBSA-N 0.000 description 1
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical compound IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 1
- 239000007997 Tricine buffer Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- HYVLNORXQGKONN-NUTKFTJISA-N Trp-Ala-Lys Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(O)=O)=CNC2=C1 HYVLNORXQGKONN-NUTKFTJISA-N 0.000 description 1
- ICNFHVUVCNWUAB-SZMVWBNQSA-N Trp-Arg-Val Chemical compound CC(C)[C@@H](C(=O)O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC1=CNC2=CC=CC=C21)N ICNFHVUVCNWUAB-SZMVWBNQSA-N 0.000 description 1
- IXEGQBJZDIRRIV-QEJZJMRPSA-N Trp-Asn-Glu Chemical compound [H]N[C@@H](CC1=CNC2=C1C=CC=C2)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O IXEGQBJZDIRRIV-QEJZJMRPSA-N 0.000 description 1
- RERIQEJUYCLJQI-QRTARXTBSA-N Trp-Asp-Val Chemical compound CC(C)[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CC1=CNC2=CC=CC=C21)N RERIQEJUYCLJQI-QRTARXTBSA-N 0.000 description 1
- JWGRSJCYCXEIKH-QEJZJMRPSA-N Trp-Glu-Cys Chemical compound C1=CC=C2C(=C1)C(=CN2)C[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CS)C(=O)O)N JWGRSJCYCXEIKH-QEJZJMRPSA-N 0.000 description 1
- TVOGEPLDNYTAHD-CQDKDKBSSA-N Tyr-Ala-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 TVOGEPLDNYTAHD-CQDKDKBSSA-N 0.000 description 1
- LGEYOIQBBIPHQN-UWJYBYFXSA-N Tyr-Ala-Ser Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 LGEYOIQBBIPHQN-UWJYBYFXSA-N 0.000 description 1
- PZXUIGWOEWWFQM-SRVKXCTJSA-N Tyr-Asn-Asn Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O PZXUIGWOEWWFQM-SRVKXCTJSA-N 0.000 description 1
- KLGFILUOTCBNLJ-IHRRRGAJSA-N Tyr-Cys-Arg Chemical compound C1=CC(=CC=C1C[C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCN=C(N)N)C(=O)O)N)O KLGFILUOTCBNLJ-IHRRRGAJSA-N 0.000 description 1
- NZFCWALTLNFHHC-JYJNAYRXSA-N Tyr-Glu-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 NZFCWALTLNFHHC-JYJNAYRXSA-N 0.000 description 1
- BXPOOVDVGWEXDU-WZLNRYEVSA-N Tyr-Ile-Thr Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(O)=O BXPOOVDVGWEXDU-WZLNRYEVSA-N 0.000 description 1
- GZUIDWDVMWZSMI-KKUMJFAQSA-N Tyr-Lys-Cys Chemical compound NCCCC[C@@H](C(=O)N[C@@H](CS)C(O)=O)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 GZUIDWDVMWZSMI-KKUMJFAQSA-N 0.000 description 1
- PGEFRHBWGOJPJT-KKUMJFAQSA-N Tyr-Lys-Ser Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(O)=O PGEFRHBWGOJPJT-KKUMJFAQSA-N 0.000 description 1
- ARMNWLJYHCOSHE-KKUMJFAQSA-N Tyr-Pro-Gln Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(O)=O ARMNWLJYHCOSHE-KKUMJFAQSA-N 0.000 description 1
- NHOVZGFNTGMYMI-KKUMJFAQSA-N Tyr-Ser-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 NHOVZGFNTGMYMI-KKUMJFAQSA-N 0.000 description 1
- AGDDLOQMXUQPDY-BZSNNMDCSA-N Tyr-Tyr-Ser Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(O)=O AGDDLOQMXUQPDY-BZSNNMDCSA-N 0.000 description 1
- AUMNPAUHKUNHHN-BYULHYEWSA-N Val-Asn-Asp Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CC(=O)O)C(=O)O)N AUMNPAUHKUNHHN-BYULHYEWSA-N 0.000 description 1
- ZMDCGGKHRKNWKD-LAEOZQHASA-N Val-Asn-Glu Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CCC(=O)O)C(=O)O)N ZMDCGGKHRKNWKD-LAEOZQHASA-N 0.000 description 1
- VUTHNLMCXKLLFI-LAEOZQHASA-N Val-Asp-Gln Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CCC(=O)N)C(=O)O)N VUTHNLMCXKLLFI-LAEOZQHASA-N 0.000 description 1
- CPTQYHDSVGVGDZ-UKJIMTQDSA-N Val-Gln-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](C(C)C)N CPTQYHDSVGVGDZ-UKJIMTQDSA-N 0.000 description 1
- AGKDVLSDNSTLFA-UMNHJUIQSA-N Val-Gln-Pro Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)N1CCC[C@@H]1C(=O)O)N AGKDVLSDNSTLFA-UMNHJUIQSA-N 0.000 description 1
- ZXAGTABZUOMUDO-GVXVVHGQSA-N Val-Glu-Lys Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCCCN)C(=O)O)N ZXAGTABZUOMUDO-GVXVVHGQSA-N 0.000 description 1
- FOADDSDHGRFUOC-DZKIICNBSA-N Val-Glu-Phe Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)O)N FOADDSDHGRFUOC-DZKIICNBSA-N 0.000 description 1
- WFENBJPLZMPVAX-XVKPBYJWSA-N Val-Gly-Glu Chemical compound CC(C)[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CCC(O)=O WFENBJPLZMPVAX-XVKPBYJWSA-N 0.000 description 1
- HGJRMXOWUWVUOA-GVXVVHGQSA-N Val-Leu-Gln Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)O)NC(=O)[C@H](C(C)C)N HGJRMXOWUWVUOA-GVXVVHGQSA-N 0.000 description 1
- AEMPCGRFEZTWIF-IHRRRGAJSA-N Val-Leu-Lys Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(O)=O AEMPCGRFEZTWIF-IHRRRGAJSA-N 0.000 description 1
- SYSWVVCYSXBVJG-RHYQMDGZSA-N Val-Leu-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)N)O SYSWVVCYSXBVJG-RHYQMDGZSA-N 0.000 description 1
- WBAJDGWKRIHOAC-GVXVVHGQSA-N Val-Lys-Gln Chemical compound [H]N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(N)=O)C(O)=O WBAJDGWKRIHOAC-GVXVVHGQSA-N 0.000 description 1
- OJOMXGVLFKYDKP-QXEWZRGKSA-N Val-Met-Asp Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(=O)O)C(=O)O)N OJOMXGVLFKYDKP-QXEWZRGKSA-N 0.000 description 1
- XBJKAZATRJBDCU-GUBZILKMSA-N Val-Pro-Ala Chemical compound CC(C)[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C)C(O)=O XBJKAZATRJBDCU-GUBZILKMSA-N 0.000 description 1
- ZXYPHBKIZLAQTL-QXEWZRGKSA-N Val-Pro-Asp Chemical compound CC(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)N[C@@H](CC(=O)O)C(=O)O)N ZXYPHBKIZLAQTL-QXEWZRGKSA-N 0.000 description 1
- VIKZGAUAKQZDOF-NRPADANISA-N Val-Ser-Glu Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CCC(O)=O VIKZGAUAKQZDOF-NRPADANISA-N 0.000 description 1
- DOBHJKVVACOQTN-DZKIICNBSA-N Val-Tyr-Gln Chemical compound NC(=O)CC[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)CC1=CC=C(O)C=C1 DOBHJKVVACOQTN-DZKIICNBSA-N 0.000 description 1
- STTYIMSDIYISRG-UHFFFAOYSA-N Valyl-Serine Chemical compound CC(C)C(N)C(=O)NC(CO)C(O)=O STTYIMSDIYISRG-UHFFFAOYSA-N 0.000 description 1
- 229930003316 Vitamin D Natural products 0.000 description 1
- QYSXJUFSXHHAJI-XFEUOLMDSA-N Vitamin D3 Natural products C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)CCCC(C)C)=C/C=C1\C[C@@H](O)CCC1=C QYSXJUFSXHHAJI-XFEUOLMDSA-N 0.000 description 1
- LGEZQZUVPMAASL-UHFFFAOYSA-N [2-hexoxy-3,5-di(propan-2-yl)phenyl]-phenylmethanone Chemical compound CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C(=O)C1=CC=CC=C1 LGEZQZUVPMAASL-UHFFFAOYSA-N 0.000 description 1
- MYTNAKVQLVOPOA-FCRXEQRPSA-N [O-2].[O-2].[Mn+4].CC=1C(=CC=2C(CCC(C2C1)(C)C)(C)C)/C(=C/C1=CC=C(C(=O)OCC)C=C1)/[Si](C)(C)C Chemical compound [O-2].[O-2].[Mn+4].CC=1C(=CC=2C(CCC(C2C1)(C)C)(C)C)/C(=C/C1=CC=C(C(=O)OCC)C=C1)/[Si](C)(C)C MYTNAKVQLVOPOA-FCRXEQRPSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 231100000569 acute exposure Toxicity 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 230000008484 agonism Effects 0.000 description 1
- 108010070944 alanylhistidine Proteins 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- UPEZCKBFRMILAV-UHFFFAOYSA-N alpha-Ecdysone Natural products C1C(O)C(O)CC2(C)C(CCC3(C(C(C(O)CCC(C)(C)O)C)CCC33O)C)C3=CC(=O)C21 UPEZCKBFRMILAV-UHFFFAOYSA-N 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 108010008355 arginyl-glutamine Proteins 0.000 description 1
- 108010069926 arginyl-glycyl-serine Proteins 0.000 description 1
- 108010062796 arginyllysine Proteins 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 108010010430 asparagine-proline-alanine Proteins 0.000 description 1
- 108010093581 aspartyl-proline Proteins 0.000 description 1
- 108010047857 aspartylglycine Proteins 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-FPRJBGLDSA-N beta-D-galactose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-FPRJBGLDSA-N 0.000 description 1
- 210000000941 bile Anatomy 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 231100000357 carcinogen Toxicity 0.000 description 1
- 239000003183 carcinogenic agent Substances 0.000 description 1
- 235000021466 carotenoid Nutrition 0.000 description 1
- 150000001747 carotenoids Chemical class 0.000 description 1
- 230000006652 catabolic pathway Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 235000020974 cholesterol intake Nutrition 0.000 description 1
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 1
- 235000019416 cholic acid Nutrition 0.000 description 1
- 229960002471 cholic acid Drugs 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 239000013599 cloning vector Substances 0.000 description 1
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 1
- 239000005516 coenzyme A Substances 0.000 description 1
- ACTIUHUUMQJHFO-UPTCCGCDSA-N coenzyme Q10 Chemical compound COC1=C(OC)C(=O)C(C\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CCC=C(C)C)=C(C)C1=O ACTIUHUUMQJHFO-UPTCCGCDSA-N 0.000 description 1
- 235000017471 coenzyme Q10 Nutrition 0.000 description 1
- 229940093530 coenzyme a Drugs 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 230000001447 compensatory effect Effects 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- LNDJVIYUJOJFSO-UHFFFAOYSA-N cyanoacetylene Chemical compound C#CC#N LNDJVIYUJOJFSO-UHFFFAOYSA-N 0.000 description 1
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 1
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000019007 dietary guidelines Nutrition 0.000 description 1
- LGTLXDJOAJDFLR-UHFFFAOYSA-N diethyl chlorophosphate Chemical compound CCOP(Cl)(=O)OCC LGTLXDJOAJDFLR-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- KCFYHBSOLOXZIF-UHFFFAOYSA-N dihydrochrysin Natural products COC1=C(O)C(OC)=CC(C2OC3=CC(O)=CC(O)=C3C(=O)C2)=C1 KCFYHBSOLOXZIF-UHFFFAOYSA-N 0.000 description 1
- FSXRLASFHBWESK-UHFFFAOYSA-N dipeptide phenylalanyl-tyrosine Natural products C=1C=C(O)C=CC=1CC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FSXRLASFHBWESK-UHFFFAOYSA-N 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 1
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 description 1
- 150000002031 dolichols Chemical class 0.000 description 1
- UPEZCKBFRMILAV-JMZLNJERSA-N ecdysone Chemical compound C1[C@@H](O)[C@@H](O)C[C@]2(C)[C@@H](CC[C@@]3([C@@H]([C@@H]([C@H](O)CCC(C)(C)O)C)CC[C@]33O)C)C3=CC(=O)[C@@H]21 UPEZCKBFRMILAV-JMZLNJERSA-N 0.000 description 1
- 150000002066 eicosanoids Chemical class 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 108010030074 endodeoxyribonuclease MluI Proteins 0.000 description 1
- 210000001842 enterocyte Anatomy 0.000 description 1
- 230000008556 epithelial cell proliferation Effects 0.000 description 1
- 230000008508 epithelial proliferation Effects 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 239000012259 ether extract Substances 0.000 description 1
- BHJOMCDSJVVGAV-UHFFFAOYSA-N ethyl 2-hexoxy-3,5-di(propan-2-yl)benzoate Chemical compound CCCCCCOC1=C(C(C)C)C=C(C(C)C)C=C1C(=O)OCC BHJOMCDSJVVGAV-UHFFFAOYSA-N 0.000 description 1
- KHOLZIBBOJZRHQ-ITYLOYPMSA-N ethyl 4-[(z)-2-(3,5,5,8,8-pentamethyl-6,7-dihydronaphthalen-2-yl)-2-trimethylsilylethenyl]benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1\C=C([Si](C)(C)C)\C1=CC(C(CCC2(C)C)(C)C)=C2C=C1C KHOLZIBBOJZRHQ-ITYLOYPMSA-N 0.000 description 1
- YCBJOQUNPLTBGG-UHFFFAOYSA-N ethyl 4-iodobenzoate Chemical compound CCOC(=O)C1=CC=C(I)C=C1 YCBJOQUNPLTBGG-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 150000003620 farnesol derivatives Chemical class 0.000 description 1
- 229940007703 farnesyl acetate Drugs 0.000 description 1
- 108091005640 farnesylated proteins Proteins 0.000 description 1
- 230000002550 fecal effect Effects 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 208000001130 gallstones Diseases 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000003736 gastrointestinal content Anatomy 0.000 description 1
- 238000010363 gene targeting Methods 0.000 description 1
- 229940113087 geraniol Drugs 0.000 description 1
- GVVPGTZRZFNKDS-JXMROGBWSA-N geranyl diphosphate Chemical compound CC(C)=CCC\C(C)=C\CO[P@](O)(=O)OP(O)(O)=O GVVPGTZRZFNKDS-JXMROGBWSA-N 0.000 description 1
- XWRJRXQNOHXIOX-UHFFFAOYSA-N geranylgeraniol Natural products CC(C)=CCCC(C)=CCOCC=C(C)CCC=C(C)C XWRJRXQNOHXIOX-UHFFFAOYSA-N 0.000 description 1
- OJISWRZIEWCUBN-UHFFFAOYSA-N geranylnerol Natural products CC(C)=CCCC(C)=CCCC(C)=CCCC(C)=CCO OJISWRZIEWCUBN-UHFFFAOYSA-N 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 108010057083 glutamyl-aspartyl-leucine Proteins 0.000 description 1
- 108010079547 glutamylmethionine Proteins 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 108010090037 glycyl-alanyl-isoleucine Proteins 0.000 description 1
- 108010028188 glycyl-histidyl-serine Proteins 0.000 description 1
- 108010081551 glycylphenylalanine Proteins 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 108010028295 histidylhistidine Proteins 0.000 description 1
- 108010025306 histidylleucine Proteins 0.000 description 1
- 108010092114 histidylphenylalanine Proteins 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 102000046818 human AR Human genes 0.000 description 1
- 102000046668 human RXRA Human genes 0.000 description 1
- 102000051544 human VDR Human genes 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 201000009019 intestinal benign neoplasm Diseases 0.000 description 1
- 210000002490 intestinal epithelial cell Anatomy 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 229940125425 inverse agonist Drugs 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 239000012500 ion exchange media Substances 0.000 description 1
- 229930000772 juvenile hormones III Natural products 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 108010044056 leucyl-phenylalanine Proteins 0.000 description 1
- 108010057821 leucylproline Proteins 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000001638 lipofection Methods 0.000 description 1
- WRUGWIBCXHJTDG-UHFFFAOYSA-L magnesium sulfate heptahydrate Chemical compound O.O.O.O.O.O.O.[Mg+2].[O-]S([O-])(=O)=O WRUGWIBCXHJTDG-UHFFFAOYSA-L 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000003159 mammalian two-hybrid assay Methods 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000002395 mineralocorticoid Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 230000000869 mutational effect Effects 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- 230000006654 negative regulation of apoptotic process Effects 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 231100001083 no cytotoxicity Toxicity 0.000 description 1
- 230000036963 noncompetitive effect Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 108010040421 oxysterol binding protein Proteins 0.000 description 1
- 102000044160 oxysterol binding protein Human genes 0.000 description 1
- VEDDBHYQWFOITD-UHFFFAOYSA-N para-bromobenzyl alcohol Chemical compound OCC1=CC=C(Br)C=C1 VEDDBHYQWFOITD-UHFFFAOYSA-N 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000003614 peroxisome proliferator Substances 0.000 description 1
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 108010065135 phenylalanyl-phenylalanyl-phenylalanine Proteins 0.000 description 1
- 108010089198 phenylalanyl-prolyl-arginine Proteins 0.000 description 1
- 108010073025 phenylalanylphenylalanine Proteins 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 150000004032 porphyrins Chemical class 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000026341 positive regulation of angiogenesis Effects 0.000 description 1
- 230000029279 positive regulation of transcription, DNA-dependent Effects 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 239000000186 progesterone Substances 0.000 description 1
- 229960003387 progesterone Drugs 0.000 description 1
- 239000000583 progesterone congener Substances 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 108010077112 prolyl-proline Proteins 0.000 description 1
- 108010004914 prolylarginine Proteins 0.000 description 1
- 108010029020 prolylglycine Proteins 0.000 description 1
- 108010053725 prolylvaline Proteins 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 229940048084 pyrophosphate Drugs 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- NPCOQXAVBJJZBQ-UHFFFAOYSA-N reduced coenzyme Q9 Natural products COC1=C(O)C(C)=C(CC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)C)C(O)=C1OC NPCOQXAVBJJZBQ-UHFFFAOYSA-N 0.000 description 1
- 230000024774 regulation of lipid catabolic process Effects 0.000 description 1
- 230000028503 regulation of lipid metabolic process Effects 0.000 description 1
- 102000037983 regulatory factors Human genes 0.000 description 1
- 108091008025 regulatory factors Proteins 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 108091008726 retinoic acid receptors α Proteins 0.000 description 1
- 108091008761 retinoic acid receptors β Proteins 0.000 description 1
- 108091008760 retinoic acid receptors γ Proteins 0.000 description 1
- 102000027483 retinoid hormone receptors Human genes 0.000 description 1
- 108091008679 retinoid hormone receptors Proteins 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 108010048818 seryl-histidine Proteins 0.000 description 1
- 239000013605 shuttle vector Substances 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000001476 sodium potassium tartrate Substances 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- JAJWGJBVLPIOOH-IZYKLYLVSA-M sodium taurocholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 JAJWGJBVLPIOOH-IZYKLYLVSA-M 0.000 description 1
- 229940031439 squalene Drugs 0.000 description 1
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 239000005495 thyroid hormone Substances 0.000 description 1
- 229940036555 thyroid hormone Drugs 0.000 description 1
- 102000004217 thyroid hormone receptors Human genes 0.000 description 1
- 108090000721 thyroid hormone receptors Proteins 0.000 description 1
- 108091008023 transcriptional regulators Proteins 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- 229940035722 triiodothyronine Drugs 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- 108010044292 tryptophyltyrosine Proteins 0.000 description 1
- 108010020532 tyrosyl-proline Proteins 0.000 description 1
- 229940035936 ubiquinone Drugs 0.000 description 1
- 108010073969 valyllysine Proteins 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000011710 vitamin D Substances 0.000 description 1
- 235000019166 vitamin D Nutrition 0.000 description 1
- 150000003710 vitamin D derivatives Chemical class 0.000 description 1
- 229940046008 vitamin d Drugs 0.000 description 1
Images




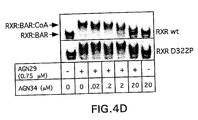

Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/192—Carboxylic acids, e.g. valproic acid having aromatic groups, e.g. sulindac, 2-aryl-propionic acids, ethacrynic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/20—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids
- A61K31/202—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids having three or more double bonds, e.g. linolenic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/20—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids
- A61K31/203—Retinoic acids ; Salts thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/695—Silicon compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
Definitions
- the present invention is relevant to the fields of human and veterinary medicine, physiology and biochemistry, particularly in the regulation of lipid metabolism and catabolism and cholesterol synthesis and breakdown.
- nuclear hormone receptors are able to bind to cis-acting regulatory elements present in the promoters of the target genes.
- the glucocorticoid, estrogen, androgen, progestin, and mineralcorticoid receptors have been found to bind as homodimers to specific response elements organized as inverted repeats.
- RAR retinoid receptor
- RXR retinoid X receptor
- Another class of nuclear hormone receptors which includes the retinoid receptor RAR (retinoic acid receptor), the thyroid receptor, the vitamin D receptor, the peroxisome proliferator receptor, and the insect ecdysone receptor bind their response element as a heterodimer in conjunction with the retinoid X receptor (RXR), which in turn is positively activated by 9-cis retinoic acid.
- RXR retinoid X receptor
- RAR and RXR like many nuclear receptors, exist as a number of subtypes (RAR ⁇ , RAR ⁇ , RAR ⁇ , and RXR ⁇ , RXR ⁇ , and RXR ⁇ ). Additionally, each subtype may exist in different isoforms.
- nuclear hormone receptors referenced above have all been shown to have specific ligand partners
- nucleic acid and amino acid sequencing experiments and sequence alignment and comparison have revealed a class of protein molecules retaining significant sequence homology and structural similarity to the nuclear hormone receptor superfamily, but for which no corresponding ligand has yet been discovered.
- some of these “receptors” have been discovered to require no ligand binding to exhibit transcriptional activity.
- these unassigned receptors have been collectively termed “orphan” receptors.
- Farnesol is an isoprenoid involved in the mevalonate biosynthetic pathway, which leads to the synthesis of cholesterol, bile acids, porphyrin, dolichol, ubiquinone, carotenoids, retinoids, vitamin D, steroid hormones, and farnesylated proteins.
- Farnesyl pyrophosphate a derivative of farnesol, is the last common intermediate in the mevalonate biosynthetic pathway.
- FXR farnesoid X-activated receptor
- BAR Bile Acid Receptor
- BAR/FXR The amino acid sequence of BAR/FXR reveals a conserved DNA-binding domain (DBD) and ligand-binding domain (LBD).
- the LBD comprises subdomains responsible for ligand binding, receptor dimerization, and transactivation.
- cells expressing chimeric proteins that contain the LBD of BAR/FXR fused to the DBD of the yeast GAL4 transcription activator did not transcribe a reporter gene containing a GAL4 response element unless the BAR/FXR construct was coexpressed with another protein comprising the dimerization and ligand binding subdomains of RXR.
- BAR/FXR and RXR interact to form a transcriptionally active dimer. No interaction was seen between BAR/FXR and any other nuclear hormone receptors that were tested. Id.
- the BAR-RXR ⁇ complex was found to be activated by juvenile hormone III (JH III) incubation of cells transfected with RXR and BAR.
- the cells were also transfected with a reporter plasmid containing 5 copies of the hsp27 response element within a portion of the mouse mammary tumor virus (MTV) promoter; the promoter was positioned upstream of the firefly luciferase gene. Activation of this gene results in the expression of luciferase, which is easily quantifiable as a measure of transactivation activity.
- Other potential ligands, including selected steroids, and eicosanoids were found to have no effect in this system. JH III failed to activate other nuclear hormone receptors, and does not activate either BAR/FXR or RXR alone. Forman et al., Cell 81:687-693 (1995).
- JH III is a derivative of farnesyl pyrophosphate.
- Other farnesyl derivatives have been tested for the ability to activate the BAR-RXR complex. Farnesol was demonstrated to strongly activate the heterodimer.
- Other derivatives such as farnesal, farnesyl acetate, farnesoic acid and geranylgeraniol activated the BAR-RXR complex somewhat less strongly; the farnesyl metabolites geraniol, squalene and cholesterol did not activate BAR-RXR. Id.
- Cholesterol synthesis is closely regulated by modulation of the levels of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA), which regulates the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A to mevalonate.
- HMG-CoA 3-hydroxy-3-methylglutaryl-coenzyme A reductase
- mevalonate is converted into 3-isopentenyl pyrophosphoric acid, which isomerizes to 3,3-dimethylallyl pyrophosphoric acid.
- HMG-CoA reductase The levels of HMG-CoA reductase are governed in part by controlling the gene transcription, translation, and by degradation of the enzyme. Farnesol has been shown to be involved in the regulation of HMG-CoA reductase degradation. Evidence exists for the synergistic promotion of HMG-CoA reductase degradation by farnesol and a sterol component, such as 25-hydroxycholesterol. See e.g., Meigs et al., J. Biol. Chem. 271:7916-7922 (1996), hereby incorporated by reference herein.
- Cholesterol is the precursor of various compounds such as sterols, bile acids such as cholic acid, and the steroid hormones such as testosterone and progesterone. All these compounds retain the basic cholesterol nucleus. The more polar bile acids are formed in the liver and secreted into the small intestine, where they aid in the absorption of lipids. The formation of bile acids from cholesterol is therefore an important degradation pathway for cholesterol, and is a key determinant of the steady-state concentration of cholesterol in the body.
- LDL low density lipoprotein
- COX-2 cyclooxygenase-2
- bile acids such as CDCA and DCA activate cyclooxygenase-2 (COX-2) transcription.
- COX-2 is overexpressed in many cancer cell lines and results in the production of prostaglandins capable of inhibiting apoptosis (an important element in the body's defense against cancers) and which have been implicated in the stimulation of angiogenesis and invasiveness.
- Inhibitors of COX-2 expression are known to decrease the size and occurrence of intestinal polyps. Thus, the maintenance of bile acid concentrations within the body may be very important.
- ion exchange media such as colestipol and cholestyramine. These drugs function by sequestering bile acids in the gut; the bile acids are then excreted in the feces. Because the intestine does not reabsorb the sequestered bile acids, the bile acids are no longer available to inhibit the formation of bile acids by cholesterol degradation. As a result, bile acid synthesis is “derepressed” with the result that the steady-state concentration of cholesterol is lowered.
- BAR/FXR is integrally involved in bile acid biology.
- Bile acid-activated BAR/FXR induces expression of SHP (small heterodimer partner), a molecule lacking a DNA binding site which can bind many nuclear receptors.
- BAR/FXR regulates the expression of ileal bile acid binding protein (IBABP), a protein which helps to prevent bile acids from exerting cytotoxic activity when they are shuttled within cells.
- IBABP ileal bile acid binding protein
- Specific bile acid transporters are required in order for bile acids to enter cells; the expression of at least one such transporter, BSEP, is increased by bile acid-activated BAR.
- BAR/FXR is also involved in the bile acid mediated control of triglyceride levels; CDCA reduces triglyceride levels in humans, and BAR/FXR mediates the synthesis of CDCA.
- bile acids have been shown to promote the growth of colon cancer in various rodent models.
- oral or intrarectal administration of bile acids induced significantly greater numbers of colon adenomas and adenocarcinomas in a carcinogen-induced model of colon cancer.
- mutational inactivation of the APC gene initiates most colon carcinomas.
- Another useful rodent model of colon carcinogenesis is the APC/Min (multiple intestinal neoplasia) mouse, which develops intestinal polyps due to a mutation the APC gene.
- Administration of bile acids to these mice result in increased numbers of ampullary tumors.
- experimental results from these rodent models support the epidemiological studies linking elevated bile acid levels to an increased risk of developing colon cancer.
- apoptosis is also critical to the development of colon cancer as the progression from colon adenomas to adenocarcinomas is associated with an inhibition of apoptosis.
- Several bile acids have been shown to induce apoptosis in colon cancer cell lines.
- bile acids decreased apoptosis in a cell line derived from a benign colon adenoma. While the role of bile acids in regulating apoptosis in vivo remains to be studied, these studies suggest that bile acids may have different effects on benign adenomas as compared to the less differentiated adenocarcinomas. The precise mechanisms governing how bile acids transduce signals that ultimately regulate cell proliferation and apoptosis remain to be elucidated.
- Bile acids have also been shown to regulate transcriptional events critical to colon carcinogenesis.
- chenodeoxycholic acid (CDCA) and DCA activate cyclooxygenase-2 (COX-2) transcription in gastrointestinal cell lines.
- COX-2 which is over expressed in many colon cancers, produces prostaglandins that inhibit apoptosis and stimulate angiogenesis and invasiveness.
- selective COX-2 inhibitors decrease the number and size of polyps in APC/Min mice and are currently being evaluated in clinical trials.
- the ability of bile acids derived from dietary cholesterol to regulate transcription may have important implications for the development of colon cancer.
- FIG. 1A Shows the activation of CV-1 cells transfected with CMX-BAR, CMX-RXR and the EcRE ⁇ 6 TK luc reporter. Reporter activation was measured in transfectant cells alone and upon treatment with 100 gM CDCA, 5 ⁇ M AGN 29, 5 ⁇ M AGN 31, 5 ⁇ M TTNPB and 100 nM LG268.
- FIG. 1B shows the activation of CV-1 cells transfected with CMX-BAR, CMX-RXR and the EcRE ⁇ 6 TK luc reporter.
- Cells were also transfected with the RAR fusion vector Gal-L-RAR. Reporter activation was measured in transfectant cells alone and upon treatment with 100 ⁇ M CDCA, 5 ⁇ M AGN 29, 5 ⁇ M AGN 31, 5 ⁇ M TTNPB and 100 nM LG268.
- FIG. 1C shows the activation of CV-1 cells transfected with CMX-BAR, CMX-RXR and the EcRE ⁇ 6 TK luc reporter.
- Cells were also transfected with the RXRA fusion vector Gal-L-RXR. Reporter activation was measured in transfectant cells alone and upon treatment with 100 ⁇ M CDCA, 5 ⁇ M AGN 29, 5 ⁇ M AGN 31, 5 ⁇ M TTNPB and 100 nM LG268.
- FIG. 2A shows the activation of CV-1 cells transfected with full length BAR/FXR upon treatment with increasing doses of CDCA, AGN 29 and AGN 31.
- FIG. 2B shows the change in polyacrylamide gel electrophoresis migration of BAR:RXRM heterodimers upon incubation with the receptor interaction domain of the co-activator GRIP 1 and differing amounts of either AGN 29 or AGN 31.
- FIG. 3A shows a Northern blot analysis of IBABP expression in Caco-2 cells upon incubation alone or in the presence of CCDA, AGN 29, or AGN 31.
- FIG. 3B shows a Northern blot analysis of CYP7A, SHP and GAPDH expression in HepG2 cells upon incubation alone or in the presence of CCDA, AGN 29, or AGN 31.
- FIG. 4A shows the results of a co-transfection assay conducted in which the combined effects of AGN 34 and CDCA on Bar/FXR activity were determined.
- FIG. 4B shows a dose-response curve of transactivation activity when cells are incubated in the presence of a constant amount of CDCA and increasing amounts of AGN 34.
- FIG. 4C shows the effect upon the transactivational activity of a variety of nuclear receptors of incubation with AGN 34.
- FIG. 4D shows a “gel shift” co-activator (GRIP) recruitment assay of AGN 34 and AGN 29, alone and together, in which the concentration of AGN 34 is increased.
- GRIP gel shift co-activator
- FIG. 5A is a Western blot of IBABP, SHP and GAPDH RNA expression upon treatment of Caco-2 cells with CDCA, AGN34 and CDCA and AGN 34.
- FIG. 5B is a Western blot of IBABP, SHP and GAPDH RNA expression upon treatment of HepG2 cells with CDCA, AGN34 and CDCA and AGN 34.
- the present invention is directed to methods for modulating the transcriptional activity of BAR/FXR through the use of synthetic ligands of the BAR:RXR heterodimer. Such ligands are able to cause BAR, preferably in combination with another nuclear hormone receptor such as RXR, to suppress, inhibit, or stimulate the transcription of a given target gene.
- the invention is directed to methods for stimulating BAR/FXR activity comprising administering an effective dose of a synthetic agonist of BAR/FXR activity.
- Preferred synthetic agonists are identified as AGN 29 and AGN31.
- the invention is directed to methods for inhibiting the BAR-mediated stimulation of Intestinal Bile Acid Binding Protein (IBABP) gene expression, comprising administering an effective dose of AGN 34.
- IBABP Intestinal Bile Acid Binding Protein
- Contemplated by the present invention are methods for regulating the concentration of bile acids in a mammal.
- a heightened concentration of bile acids in mammals has been associated with an increased occurrence of colon cancer; thus, the use of BAR/FXR ligands which do not significantly increase, or which decrease Cyp7a expression may effectively lower abnormally high bile acid concentrations therefore providing a therapeutic and/or prophylactic effect for this indication.
- proteins other than Cyp7a are regulated by bile acids; these include Intestinal Bile Acid Binding Protein and Cyclooxygenase 2 (both up-regulated by CDCA), and sterol-27-hydroxylase, Intestinal Bile Acid Transporter, and Liver Bile Acid Transporter (these proteins are down regulated by CDCA).
- CDCA Intestinal Bile Acid Binding Protein
- Cyclooxygenase 2 both up-regulated by CDCA
- sterol-27-hydroxylase Intestinal Bile Acid Transporter
- Liver Bile Acid Transporter these proteins are down regulated by CDCA.
- the methods of the present invention are therefore useful in modulating the expression of these proteins as well.
- the present invention is directed to a method of treating colorectal cancer through the administration to a patient in need thereof of a pharmaceutically effective dose of a BARIFXR ligand which causes a decrease or inhibition of the formation of a IBABP:bile acid complex.
- a BARIFXR ligand which causes a decrease or inhibition of the formation of a IBABP:bile acid complex.
- the BAR/FXR ligand decreases the expression of IBABP without antagonizing at least one other activity characteristic of agonism of the BAR/FXR receptor.
- Particularly preferred as a BAR/FXR ligand is AGN 34.
- BAR/FXR ligand binds either to the BAR/FXR receptor or to a complex intermolecular complex or multimer which comprises the BAR/FXR receptor and which modulates an activity associated with the BAR/FXR receptor.
- the BAR/FXR ligands of the present invention may be BAR/FXR antagonists, BAR/FXR inverse agonists, or have attributes of more than one of these.
- agonist is meant that the ligand stimulates a ligand-dependent BAR/FXR activity above any baseline levels present in the absence of ligand.
- antagonist is meant that the ligand binds to BAR, and functions as a competitive or non-competitive inhibitor of BAR/FXR agonist activity.
- inverse agonist is meant that the ligand will bind to BAR/FXR and cause the suppression of an BAR/FXR activity to a level lower than seen in the absence of any BAR/FXR ligand.
- modulating an activity associated with the BAR/FXR receptor is meant that the ligand affects an activity associated primarily with the BAR/FXR receptor alone or in combination with another factor, with the BAR:RXR heterodimer, but not with the RXR homodimer.
- a ligand may exert its activity by binding the BARIFXR subunit, by binding the RXR subunit, or by binding both the BAR/FXR and RXR subunit. The mechanism of modulation is irrelevant to this invention.
- the present invention pertains to methods of stimulating or inhibiting an activity, or stimulating one activity and inhibiting another activity associated with a BARIFXR receptor of a mammal by treating such a mammal with a pharmaceutically acceptable composition comprising a compound selected from the group consisting of: AGN 29, AGN 31 and AGN 34.
- AGN 29 has the following structure:
- AGN 31 has the following structure:
- AGN 34 has the following structure:
- the present invention is directed to methods for modulating the activity of a mammalian BAR/FXR receptor, preferably the human BAR/FXR protein.
- Such methods involve the use of a BAR/FXR ligand which will bind the BAR/FXR receptor or a complex containing the BAR/FXR receptor, thereby affecting the ability of BAR/FXR to exert its biological effects, either directly or by blocking the ability of a naturally occurring ligand to exert its affects.
- the BAR/FXR ligands of the present invention may be BAR/FXR antagonists, BAR/FXR agonists, or BAR/FXR inverse agonists.
- the BAR/FXR ligands have substantially no activity at the retinoid nuclear receptors, RAR and RXR.
- the BAR/FXR ligand may be a bi-specific compound able to bind and modulate both RXR and BAR.
- aspects of the invention directed to methods for increasing the plasma concentration of cholesterol in a mammal pathologically deficient in cholesterol through the use of an BAR/FXR agonist.
- the BAR/FXR receptor when bound by an BAR/FXR agonist, may inhibit the transcription of the oxysterol receptor LXR ⁇ , which in turn activates transcription of Cyp7a. Repression of transcription of this key enzyme in the biosynthesis of bile acids therefore results in a lower concentration of bile acids within the body; high bile acid concentrations have been associated with a heightened risk of colon cancer.
- All mammalian expression vectors were derivatives of the bacterial/mammalian shuttle vector pCMX, an expression vector containing the cytomegalovirus (CMV) promoter/enhancer, followed by a bacteriophage T7 promoter for transcription of the cloned gene in vitro.
- Plasmid pCMX also contains the SV40 small t intron/poly adenylation signal sequence, polyoma virus enhancer/origin and the SV40 enhancer/origin of plasmid CDM8 (see Seed, Nature 329:840-842 (1987), hereby incorporated by reference herein) cloned into the large Pvu II fragment of pUC 19.
- PUC 19 is a commonly used cloning vector available from New England Biolabs, Inc. This Pvu II fragment contains a Col El origin of replication and an ampicillin resistance gene for plasmid selection, but lacks the pUCl 9 polylinker cloning site.
- a synthetic polylinker comprising the following restriction sites: 5′-KpnI, EcoRV, BamHI, MscI, NheI-3′ followed by a translational termination sequence inserted in all three reading frames. See Umesono et al., Cell 65:1255-1266 (1991), hereby incorporated by reference herein.
- nucleic acid regions encoding the following full-length proteins were cloned into a CMV expression vector.
- the sequences of these genes and/or their corresponding polypeptides have the indicated GenBank accession numbers: rat BARIFXR (accession #U18374), human RXRA (accession #X52773), human TR ⁇ (accession #X04707), human LXR ⁇ (accession #U22662), mouse CARP (accession #AF009327), mouse PPARa (accession #X57638), mouse PPAR ⁇ (accession #U10375), mouse PPAR ⁇ (accession #U10374), VDR (accession #NM — 000376).
- Gal4 fusions containing the indicated protein fragments were fused to the C-terminal end of the yeast Gal4 DNA binding domain (amino acids 1-147, accession #X85976): Gal-RAR (human RAR ligand binding domain, Glu 156-Ser 463, accession #X06614), Gal-RXR (human RXRa igand binding domain, Glu 203-Thr 462, accession #X52773).
- the ⁇ gal contains the E. coli P-galactosidase coding sequences derived from pCH110 (accession #U02445).
- RXRm contains a single point mutation (Asp-322-Pro) in the LBD of human RXR ⁇ .
- Luciferase reporter constructs contain the Herpes virus thymidine kinase promoter ( ⁇ 105/+51) linked to the indicated number of copies of the following response elements: hsp27 EcRE ⁇ 6 (see Yao et al., 71 Cell 63-72 (1992)); IBABP IR-1 ⁇ 3 (CCTTA AGGTGA A TAACCT TGGGGCTCC) (SEQ ID NO: 13); UAS G ⁇ 4 (MH100 ⁇ 4); PPRE ⁇ 3 (see Forman et al., 81 Cell 687 (1995)); ⁇ RE2 ⁇ 3 (see Forman et al., 395 Nature 612 (1998); LXREx3 (see Willy et al., 9 Genes Dev.
- GAL4 fusion proteins were constructed using standard molecular biological methods (see e.g., Sambrook et al., Molecular Cloning: A Laboratory Manual (2d ed. Cold Spring Harbor Laboratory Press 1989), incorporated by reference herein in its entirety) by inserting a nucleotide sequence encoding the indicated polypeptide immediately downstream of the yeast GAL4 DNA-binding domain in plasmid pSG424, described in Sadowski et al., Nucleic Acids Research 17:7539, hereby incorporated by reference herein.
- the amino acid sequence of the yeast GAL4 DBD is as follows: NH2-MKLLSSIEQA CDICRLKKLK CSKEKPKCAK CLKNNWECRY SPKTKRSPLT RAHLTEVESR LERLEQLFLL IFPREDLDM ILKMDSLQD IKALLTGLF VQDNVNKDAV TDRLASVETD MPLTLRQHRI SATSSSEESS NKGQRQLTVS-COOH
- Fusion proteins were made, as indicated above, using common molecular biological techniques by creation of open nucleic acid reading frames encoding the indicated polypeptides, and cloning into the polylinker portion of pCMX.
- the plasmid nucleic acids encoded amino acids Glu 203 to Thr 462 of human RXR ⁇ (SEQ ID NO: 4) fused to the GAL4 sequences.
- the junction between the carboxyl terminal section of GAL4 and the amino terminal portion of the RXR LBD had the following structure: EcoRI Asp718 Sal/Xho GTA-TCG-CCG- GAA-TTC-GGT-ACC-GTC-GAG -GCC-GTG-CAG- GAG-.. Val-Ser Glu-Ala-Val-Gln- GLu-..
- 5′GTATCGCCGGAATTCGGTACCGTCGAGGCCGTGCAGGAG3′ is hereby designated SEQ ID NO: 6.
- the plasmid nucleic acids encoded amino acids Leu 181 to Gln 469 of rat BAR/FXR (SEQ ID NO: 1) fused to the GAL4 sequences.
- the junction between the carboxyl terminal section of GAL4 and the amino terminal portion of the BAR/FXR LBD had the following structure: EcoRI former
- RXR ligand binding domain (LBD) expression construct L-RXR contains nucleotide residues encoding the SV40 Tag nuclear localization signal sequence (from amino to carboxy ends:
- APKKKRKVG (SEQ ID NO: 8)
- CMX- ⁇ gal contains the E. coli ⁇ -galactosidase coding sequence derived from plasmid pCH110 (accession number U 02445) inserted downstream of the CMV promoter in plasmid pCMX.
- RXRm contains a single point mutation changing Asp-322 to Pro in the LBD of human RXR ⁇ .
- Luciferase reporter plasmids (termed TK-Luc) were constructed by placing the cDNA encoding firefly luciferase immediately downstream from the herpes virus thymidine kinase promoter (located at nucleotide residues ⁇ 105 to +51) of the thymidine kinase nucleotide sequence), which is linked in turn to the various response elements.
- Response elements were inserted in plasmid TK-Luc at the unique Hind III site.
- the yeast GAL4 UASG response element has the nucleotide sequence, and was inserted in 4 direct repeats to yield UASG ⁇ 4:
- hsp EcRE ecdysone response element
- CV-1 African Green Monkey cells were grown in Dulbecco's Modified Eagle's medium supplemented with 10% resin-charcoal stripped fetal bovine serum (FBS), 50 U/ml penicillin G and 50 ⁇ g/ml streptomycin sulfate (DMEM-FBS) at 37° C. in 5% CO 2 .
- FBS fetal bovine serum
- DMEM-FBS 50 U/ml penicillin G
- Liposomes (N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-ammonium methyl sulfate, sold by Boehringer Mannheim under the name DOTAP) were formed according to the manufacturer's instructions.
- the liposomes contained reporter gene constructs (300 ng/10 5 cells); cytomegalovirus driven expression vectors (25 ng/10 5 cells) were added as indicated along with CMX-Bgal (500 ng/10 5 cells) as an internal control. After 2 hours the liposomes were removed and replaced with fresh media. Cells were incubated for approximately 40 hours with phenol red-free DMEM-FBS containing the indicated compounds. After exposure to the specified ligand, the cells were harvested.
- the harvested cells were assayed for the presence of luciferase activity.
- Cells were lysed in 0.1 M KPO4 (pH 7.8), 1.0% TRITON® X-100, 1.0 mM dithiolthreitol (DTT) and 2 mM ethylenediamine tetracetic acid (EDTA).
- DTT dithiolthreitol
- EDTA ethylenediamine tetracetic acid
- Luciferase activity was measured by reaction of the cell lysates with luciferin in a reaction buffer comprising: 20 mM tricine, 1.07 mM Mg(CO 3 ) 4 -Mg(OH) 2 -5H 2 O, 2.67 mM MgSO 4 .7H 2 O, 0.1 mM EDTA, 0.5 mM Sodium luciferin, 0.15 mg/ml Coenzyme A, 5 mM DTT, and 0.5 mM adenosine triphosphate (ATP). Resulting chemiluminescence was measured in a luminometer. See de Wet et al., Mol. Cell Biol. 7:725 (1987) (hereby incorporated by reference herein). All points were assayed in triplicate and varied by less than 15%. Each experiment was repeated three or more times with similar results. No cytotoxicity was observed with any of the compounds when used at the indicated concentrations and treatment times.
- HepG2 cells (a hepatoma cell line used as a hepatocyte model) were maintained in Eagle's minimal essential medium supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM L-glutamine, non-essential amino acids, 50 U/ml penicillin G and 50 ⁇ g/ml streptomycin sulfate.
- Caco-2 cells (a cell line derived from a colon carcinoma capable of spontaneously differentiating into cells sharing characteristics with small intestinal cells) were maintained in DMEM supplemented with 20% FBS, 50 U/ml penicillin G and 50 ⁇ g/ml streptomycin sulfate. Caco-2 cells were maintained for 20 days post-confluence to allow differentiation. They were fed twice a week with their regular media during this period.
- Northern blots were prepared from polyA + RNA using the Oligotex method (Qiagen) and analyzed with the following probes: human CYP7A (accession #M93133) nucleotides 1617-2576 (Karam and Chiang, 185 Biochem. Biophys. Res. Commun. 588 (1992)), human SHP (accession #L76571) nucleotides 888-1355, human IBABP (accession # A1311734), an EST containing the entire coding sequence of IBABP.
- Coactivators which bind agonist-activated nuclear receptors including, without limitation, RAR and RXR, function to assist the activated nuclear receptor exert its activity as a transcription factor. Characterized co-activators include CBP, p300, RIP140, SRC-1, ACTR, TIF2 (also called GRIP 1) and TIFI. In most cases coactivator will not bind the receptor in the absence of an agonist, moreover, receptor antagonists will often block coactivator binding, either directly or through the recruitment of a corepressor. Coactivator recruitment assays are therefore a valuable method for directly visualizing associative changes to a receptor caused by the addition of a prospective ligand.
- GRIP 1 was expressed as a fusion protein with glutathione-S-transferase, an enzyme which selectively binds glutathione and can thus be used as an affinity reagent. See e.g., U.S. Pat. No. 5,654,176.
- the GST-GRIP1 fusion protein was expressed in E. coli and purified on glutathione-Sepharose columns. In vitro translated BAR/FXR ⁇ RXR (0.6-1.2 ⁇ l each) and GST-GRIP 1 (5 ⁇ g) were incubated for 30 min at room temperature with 100,000 cpm of the E.
- Oligonucleotide probes were made to identify specific receptor dimers; an optimized DR-1 (direct repeat 1; a nucleotide sequence motif to which RXR will bind) probe (5′-GCTACC AGGTCA A AGGTCA CGTAGCT) (SEQ ID NO: 12) was used for RXR homodimers and the IR-1 sequence of the IBABP promoter, to which the BAR/RXR heterodimer binds, was used for BAR/RXR heterodimers.
- DR-1 direct repeat 1; a nucleotide sequence motif to which RXR will bind
- the transfected cells were then treated with the indicated ligands for 40 hours. Activity was measured using luciferase expression in the cotransfection assay described above.
- AGN 29 and AGN 31 robustly activated BAR/FXR (91- and 85-fold respectively) when used at a concentration of 5 ⁇ M, whereas CDCA resulted in a nearly 200-fold activation at 100 ⁇ M (FIG. 1A).
- TTNPB as previously demonstrated, also induced significant BAR/FXR activity (65-fold).
- the RXR-specific ligand LG268 also activated the BAR-RXR heterodimer via the RXR subunit as previously demonstrated (FIG. 1A).
- TTNPB is a strong RAR activator
- AGN 29 and AGN 31 we tested the ability of AGN 29 and AGN 31 to activate both RAR and RXR.
- DBD Gal 4 DNA binding domain
- LBD ligand binding domain
- Gal-L-RAR or Gal-L-RXR fusions were transfected into CV-1 cells with a Gal 4 reporter (MH100 ⁇ 4) and the effect of different ligands was evaluated.
- FIG. 1B TTNPB was able to strongly activate RAR, but treatment with AGN 29 and AGN 31 resulted in a loss of specificity for RAR.
- RXR RXR heterodimers containing nuclear receptors including AR, mPXR, hPXR, ER ⁇ , CAR ⁇ , LXR ⁇ , PPAR ⁇ , PPAR ⁇ , PPAR ⁇ , VDR and TR ⁇ .
- CV-1 cells transfected with full length BAR/FXR were treated with increasing doses of AGN 29 and AGN 31.
- the EC 50 for AGN29 and AGN31 is approximately 2 ⁇ M compared with an EC 50 of approximately 50 ⁇ M for CDCA.
- EC 50 is meant the concentration at which the response is 50% of maximal.
- Co-activator recruitment assays were performed by mixing AGN 29 and AGN 31 with BAR, RXRm, a 32 P-labeled BAR/FXR response element (IBABP IR1) and the receptor interaction domain of GRIP 1.
- RXRm is an RXR mutant impaired in its ability to bind ligand and used to determine whether AGN 29 and AGN 31 bind to the BAR/FXR subunit and not to RXR. After the binding reactions, the resulting complexes were separated on polyacrylamide gels. As expected, BAR-RXRm heterodimers failed to recruit co-activator in the absence of ligand (FIG. 2B).
- AGN 34 did not affect the basal activity of luciferase gene expression, but repressed CDCA-activated BAR-mediated transcription nearly 10-fold (FIG. 4A).
- Dose-response analysis increasing concentration of AGN 34; constant concentration of CDCA) in the same assay system indicated that AGN34 is a very strong repressor, displaying approximately 85% repression at 0.03 ⁇ M and showing maximal activity at 1 ⁇ M (FIG. 4B).
- AGN34 is a selective BAR/FXR modulator (BARM); that is, a BAR/FXR partial antagonist, that regulates different BAR/FXR target genes differentially.
- BARM selective BAR/FXR modulator
- Antagonism of IBABP expression by AGN 34 is useful as a therapeutic method for the treatment of conditions such as colorectal cancer characterized by the presence of excessive levels of bile acid.
- Cyp 7A means that such therapeutic use is quite specific and will therefore have a minimum of undesired side effects.
- Luciferase reporter constructs containing the Herpes virus thymidine kinase promoter ( ⁇ 105/+51) linked to the indicated number of copies of the following response elements: hsp27 EcRE ⁇ 6 and MH100 ⁇ 4 (UASG ⁇ 4) were used in a co-transfection assay conjunction with the following expression plasmids: GAL-L-hRXR ⁇ , hFXR, rFXRop, rnFXR and hRXRop. As shown below, in most cases hRXRop was recombinantly coexpressed with one other of the indicated receptor constructs, so as to permit the formation of hetero-or homodimers containing hRXRop.
- the data indicate that AGN 34 antagonizes the CDCA-mediated stimulation of BAR/FXR transcriptional activation, and that both the stimulation of transactivational activity by CDCA and the antagonism of this activity by AGN 34 is selective for the BAR/FXR receptor or the BAR:RXR heterodimer; neither agent shows activity in a system containing only RXR (and thus presumably only RXR homodimers.
- LG 268 stimulates RXR-mediated reporter gene transcription when used as the sole ligand in this experiment, and when RXR is the only receptor recombinantly expressed by the cell. LG 268 also stimulates transctivation of the reporter gene which RXR is co-expressed with rat or mouse BAR, but not with human recombinant BAR/FXR This result suggests that LG 268 may have a measure of BAR/FXR (or BAR:RXR) agonist activity on the rat and mouse receptors as well as on RXR itself.
- Example 4 An experiment conducted in a manner similar to that of Example 4 was carried out using either CDCA (50 ⁇ M) alone or in combination with AGN 34 (1 ⁇ M) or LG 754(1 ⁇ M) (an RXR homodimer antagonist but activator of PPAR-RXR) as test agents.
- Ethyl 4-Hex-1-ynylbenzoate (3) A solution of 1-hexyne (1.72 mL, 15 mmol), ethyl 4-iodobenzoate (1.38 g, 5 mmol), triethylamine (1.05 mL, 7.5 mmol), and THF (20 mL) was degassed with argon for ten minutes. The solution was treated with bis(triphenylphosphine)palladium (II) chloride (17.5 mg, 0.25 mmol) and copper iodide (11.4 mg, 0.06 mg) and it was stirred at room temperature for 24 h. The solution was concentrated under reduced pressure, and the residue was purified by silica gel chromatography (hexane) to give the title compound.
- the residue was purified by silica gel chromatography using 9:1 hexane:ethyl acetate as the eluent to give an inseparable mixture of isomeric esters.
- the esters were hydrolyzed with 2N aqueous KOH (1 mL) in ethanol (4 mL), acidified with 1N HCl, and the products were extracted with ethyl acetate.
- the organic extracts were washed with brine and dried (MgSO4).
- the filtered solvent was removed in vacuo and the resulting solid recrystalized from a solution of hexane and ethyl acetate to give the title compound.
- Ethyl 2-Hexyloxy-3,5-diisopropylbenzoate (10). A solution of ethyl 2-hydroxy-3,5-diisopropylbenzoate (19.1 g, 76.4 mmol) and 10 mL DMF was added slowly to a suspension of sodium hydride (4 g, 99.3 mmol) and DMF (90 mL) at O ° C. After 10 minutes, 1-iodohexane (17.0 mL, 114.6 mmol) was added, and the solution was stirred at room temperature for 18 hours. The reaction was quenched by the addition of water and the products extracted with ethyl acetate. The organic layers were combined, and washed with brine, and dried over MgSO4. The filtered solvents were removed by rotary evaporation to give the title compound as a yellow oil that was used in the next step without further purification.
- (2E, 4E, 6Z)-7-(2-Hexyloxy-3,5-diisopropylphenyl)-3-methylocta-2,4,6-trienoic Acid AGN 34.
- a solution of ethyl (2E, 4E, 6Z)-7-(2-hexyloxy-3,5-diisopropylphenyl)-3-methylocta-2,4,6-trienoate (1.2 g, 2.73 mmol) and ethanol (40 mL) was treated with 2N aqueous NaOH (30 mL, 60 mmol). The solution was stirred at 60° C.
- the layers were separated and the aqueous layer was extracted with 50 mL of dichloromethane.
- the combined organic extracts were washed with brine, and dried (MgSO4), and filtered, and the solvents were removed in vacuo.
- the residue was filtered through a plug (6@ ⁇ 2@) of silica gel using a solution of 97% hexane/ethyl acetate. After removal of the solvent the residue was heated under vacuum (3 torr) to 170° C. for 1 hour to remove a low-boiling impurity. The remaining material is the title compound.
- the solution was purged with argon for 15 minutes and bis(triphenylphosphine)palladium (II) chloride (83 mg, 0.12 mmol) and copper (I) iodide (22 mg, 0.12 mmol) were added and the solution stirred at ambient temperature for 3 days.
- PNMR 300 MHz, CDCl 3 ) ⁇ 0.10 (s, 9H), 1.29 (s, 12H), 1.68 (s, 4H), 2.24 (s, 3H), 4.72 (s, 2H), 6.87 (s, 1H), 7.07 (s, 1H), 7.17 (s, 1H), 7.35 (s, 4H).
- the solution was stirred at room temperature for 16 hours, the manganese dioxide filtered off, and the hexane removed in vacuo.
- the residue was dissolved in 2 mL of ethanol and treated with sodium cyanide (37.5 mg, 0.77 mmol) and acetic acid (13.7 mg, 0.23 mmol). After 15 minutes, the solution was treated with 265 mg (3.0 mmol) of manganese dioxide.
- the suspension was stirred at room temperature for 6 hours and the manganese dioxide removed by filtration.
- the solution was poured into a separatory funnel containing water and ether. The layers were separated and the aqueous layer was extracted 3 times with ether.
- Cerevisiae TL-lvc promoter 10 cgacggagta ctgtcctcg agct 24 11 22 DNA Artificial Sequence hsp EcRe 11 tggacaagtg cattgaaccc tt 22 12 26 DNA Artificial Sequence TK-luc reporter 12 gctaccaggt caaaggtcac gtagct 26 13 26 DNA Artificial Sequence TK-luc reporter 13 gctaccaggt caaaggtcac gtagct 26 14 27 DNA Artificial Sequence TIC-lvc repater 14 ccttaaggtg aataaccttg gggctcc 27
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
- This application is a continuation-in-part of Ser. No. 10/006,450, filed on Nov. 19, 2001.
- The present invention is relevant to the fields of human and veterinary medicine, physiology and biochemistry, particularly in the regulation of lipid metabolism and catabolism and cholesterol synthesis and breakdown.
- A vast array of specific metabolic, developmental, and catabolic processes appear to be directly or indirectly regulated in vivo by comparatively small molecules such as steroids, retinoids and thyroid hormones. The mechanism whereby a single such compound can contribute to the regulation of numerous different cellular events was the subject of much speculation until relatively recently, when it was discovered that these compounds each share the ability to bind to transcriptionally active proteinaceous receptors. These protein receptors, in turn, are able to bind specific cis-acting nucleic acid regulatory sequence regions, termed response elements or RE's, located upstream of the coding sequence of certain genes and to activate the transcription of these genes. Thus, the proteinaceous receptors can serve as specific, ligand-dependent regulators of gene transcription and expression.
- The amino acid sequences of these various receptors were quickly found to share regions of homology, thus making each such receptor a member of a family of ligand-modulated receptor molecules. This family has been termed the steroid superfamily of nuclear hormone receptors; nuclear, because the receptors are usually found in high concentration in the nucleus of the cell.
- Further study of the structural and functional relationship between the nuclear hormone receptors has shown certain characteristics in common between them in addition to sequence homology. See e.g., Evans et al. Science 240:889-895 (1988). As stated above, the nuclear hormone receptors are able to bind to cis-acting regulatory elements present in the promoters of the target genes. The glucocorticoid, estrogen, androgen, progestin, and mineralcorticoid receptors have been found to bind as homodimers to specific response elements organized as inverted repeats.
- Another class of nuclear hormone receptors, which includes the retinoid receptor RAR (retinoic acid receptor), the thyroid receptor, the vitamin D receptor, the peroxisome proliferator receptor, and the insect ecdysone receptor bind their response element as a heterodimer in conjunction with the retinoid X receptor (RXR), which in turn is positively activated by 9-cis retinoic acid. See Mangelsdorf, et al., The Retinoid Receptors in The Retinoids: Biology, Chemistry and Medicine Ch.8 (Sporn et al., eds. 2d ed., Raven Press Ltd. 1994); Nagpal and Chandraratna, Current Pharm. Design 2:295-316 (1996), which are both incorporated by reference herein. The retinoid receptors RAR and RXR, like many nuclear receptors, exist as a number of subtypes (RARα, RARβ, RARγ, and RXRα, RXRβ, and RXRγ). Additionally, each subtype may exist in different isoforms.
- While the nuclear hormone receptors referenced above have all been shown to have specific ligand partners, nucleic acid and amino acid sequencing experiments and sequence alignment and comparison have revealed a class of protein molecules retaining significant sequence homology and structural similarity to the nuclear hormone receptor superfamily, but for which no corresponding ligand has yet been discovered. In fact, some of these “receptors” have been discovered to require no ligand binding to exhibit transcriptional activity. Collectively, these unassigned receptors have been collectively termed “orphan” receptors.
- Products of intermediate metabolism are known transcriptional regulators in prokaryotes and lower eukaryotes such as yeast; thus there has been speculation that such metabolites may also serve this function in higher organisms, perhaps through interaction with the nuclear hormone receptors.
- Farnesol is an isoprenoid involved in the mevalonate biosynthetic pathway, which leads to the synthesis of cholesterol, bile acids, porphyrin, dolichol, ubiquinone, carotenoids, retinoids, vitamin D, steroid hormones, and farnesylated proteins. Farnesyl pyrophosphate, a derivative of farnesol, is the last common intermediate in the mevalonate biosynthetic pathway.
- Forman et al., Cell 81:687-693 (1995) have demonstrated that an orphan receptor, now termed farnesoid X-activated receptor (FXR) or Bile Acid Receptor (BAR), is activated by farnesol and related molecules. This reference is hereby incorporated by reference herein. BAR/FXR is expressed in the liver, gut, adrenal gland, and kidney.
- The amino acid sequence of BAR/FXR reveals a conserved DNA-binding domain (DBD) and ligand-binding domain (LBD). The LBD comprises subdomains responsible for ligand binding, receptor dimerization, and transactivation. Additionally, cells expressing chimeric proteins that contain the LBD of BAR/FXR fused to the DBD of the yeast GAL4 transcription activator did not transcribe a reporter gene containing a GAL4 response element unless the BAR/FXR construct was coexpressed with another protein comprising the dimerization and ligand binding subdomains of RXR. These data suggested that BAR/FXR and RXR interact to form a transcriptionally active dimer. No interaction was seen between BAR/FXR and any other nuclear hormone receptors that were tested. Id.
- Among the nuclear hormone receptors amino acid sequence homology to BAR/FXR is high in the insect ecdysone receptor (EcR), which dimerizes with an RXR homolog. When dimerized with RXRα, BAR/FXR was shown to specifically bind hsp27, an EcR response element, however, binding was not seen when BAR/FXR was expressed alone. BAR/FXR and RXR bind to certain sequences as a heterodimer.
- The BAR-RXRα complex was found to be activated by juvenile hormone III (JH III) incubation of cells transfected with RXR and BAR. The cells were also transfected with a reporter plasmid containing 5 copies of the hsp27 response element within a portion of the mouse mammary tumor virus (MTV) promoter; the promoter was positioned upstream of the firefly luciferase gene. Activation of this gene results in the expression of luciferase, which is easily quantifiable as a measure of transactivation activity. Other potential ligands, including selected steroids, and eicosanoids were found to have no effect in this system. JH III failed to activate other nuclear hormone receptors, and does not activate either BAR/FXR or RXR alone. Forman et al., Cell 81:687-693 (1995).
- JH III is a derivative of farnesyl pyrophosphate. Other farnesyl derivatives have been tested for the ability to activate the BAR-RXR complex. Farnesol was demonstrated to strongly activate the heterodimer. Other derivatives such as farnesal, farnesyl acetate, farnesoic acid and geranylgeraniol activated the BAR-RXR complex somewhat less strongly; the farnesyl metabolites geraniol, squalene and cholesterol did not activate BAR-RXR. Id.
- Cholesterol synthesis is closely regulated by modulation of the levels of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA), which regulates the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A to mevalonate. Through a series of phosphorylations and a decarboxylation reaction, mevalonate is converted into 3-isopentenyl pyrophosphoric acid, which isomerizes to 3,3-dimethylallyl pyrophosphoric acid. An enzyme-mediated condensation reaction between the 5 carbon isoprenyl compounds 3-isopentenyl pyrophosphoric acid and 3,3-dimethylallyl pyrophosphoric acid results in the formation of the 10 carbon diisoprenyl compound geranyl pyrophosphoric acid. This, in turn, reacts with another molecule of 3-isopentenyl pyrophosphoric acid to form the 15 carbon compound farnesyl pyrophosphate. Two molecules of this latter compound react to form the 30 carbon molecule presqualine pyrophosphate, which is dephosphorylated to form squaline. Squaline is then cyclized to form cholesterol. Thus, HMG-CoA reductase mediates the initial formation of the isoprene units that are subsequently assembled in series and cyclized to form cholesterol.
- The levels of HMG-CoA reductase are governed in part by controlling the gene transcription, translation, and by degradation of the enzyme. Farnesol has been shown to be involved in the regulation of HMG-CoA reductase degradation. Evidence exists for the synergistic promotion of HMG-CoA reductase degradation by farnesol and a sterol component, such as 25-hydroxycholesterol. See e.g., Meigs et al., J. Biol. Chem. 271:7916-7922 (1996), hereby incorporated by reference herein.
- Cholesterol is the precursor of various compounds such as sterols, bile acids such as cholic acid, and the steroid hormones such as testosterone and progesterone. All these compounds retain the basic cholesterol nucleus. The more polar bile acids are formed in the liver and secreted into the small intestine, where they aid in the absorption of lipids. The formation of bile acids from cholesterol is therefore an important degradation pathway for cholesterol, and is a key determinant of the steady-state concentration of cholesterol in the body.
- The rate-limiting enzyme in the formation of bile acids from cholesterol is cholesterol 7α-hydroxylase (Cyp7a). For some time it has been known that bile acids act in a negative feedback loop to limit their own production via this pathway, but the means by which this is accomplished has remained elusive. Recently, there has been evidence that Cyp7a synthesis and expression is inhibited by bile acids. Chiang, Front. Biosci. 3:D176-93 (1998) hereby incorporated by reference herein.
- Despite the fact that cholesterol is essential for the synthesis of cell membranes and various hormones and other small molecules, raised levels of cholesterol, particularly in the form of low density lipoprotein (LDL), have been strongly linked to arteriosclerosis and other cardiovascular diseases. Additionally, maintenance of appropriate bile acid concentrations is important in regulating lipid metabolism, and may be useful in the prevention of colon cancer and gallstone formation.
- Specifically, bile acids such as CDCA and DCA activate cyclooxygenase-2 (COX-2) transcription. COX-2 is overexpressed in many cancer cell lines and results in the production of prostaglandins capable of inhibiting apoptosis (an important element in the body's defense against cancers) and which have been implicated in the stimulation of angiogenesis and invasiveness. Inhibitors of COX-2 expression are known to decrease the size and occurrence of intestinal polyps. Thus, the maintenance of bile acid concentrations within the body may be very important.
- Among currently available drugs for the treatment of hypercholesterolemia are ion exchange media such as colestipol and cholestyramine. These drugs function by sequestering bile acids in the gut; the bile acids are then excreted in the feces. Because the intestine does not reabsorb the sequestered bile acids, the bile acids are no longer available to inhibit the formation of bile acids by cholesterol degradation. As a result, bile acid synthesis is “derepressed” with the result that the steady-state concentration of cholesterol is lowered.
- Unfortunately, these ion exchange drugs have been associated with an increased incidence of intestinal tumors in rodents. Additionally, since the drugs are highly charged, they are capable of adsorbing other compounds, such as ingested drugs, naturally occurring hormones, regulatory factors and the like.
- A poster displayed by Neisor, Flach, Weinberger & Bentzen at an AACR conference on Nuclear Receptors in Palm Springs, Calif. held on Jan. 8-11, 1999 discussed the ability of certain 1,1-biphosphonate esters to activate BAR/FXR and to lower plasma cholesterol levels in mammals. This poster abstract is incorporated by reference herein.
- A patent application was filed by certain of the present inventors and was published Dec. 21, 2000. The invention claimed in this patent application, publication number WO00/76523, was under an obligation of assignment to the same assignees as the present invention. The invention claimed therein is drawn to methods of modulating BAR/FXR receptor activity using certain compounds.
- Additionally, an International Patent Application, WO00/40965 describing the role of BAR/FXR in bile acid synthesis was published on Jul. 13, 2000. No synthetic compounds for modulating cholesterol or bile acid synthesis were disclosed therein.
- It is now known that BAR/FXR is integrally involved in bile acid biology. Bile acid-activated BAR/FXR induces expression of SHP (small heterodimer partner), a molecule lacking a DNA binding site which can bind many nuclear receptors. Additionally, BAR/FXR regulates the expression of ileal bile acid binding protein (IBABP), a protein which helps to prevent bile acids from exerting cytotoxic activity when they are shuttled within cells. Specific bile acid transporters are required in order for bile acids to enter cells; the expression of at least one such transporter, BSEP, is increased by bile acid-activated BAR. BAR/FXR is also involved in the bile acid mediated control of triglyceride levels; CDCA reduces triglyceride levels in humans, and BAR/FXR mediates the synthesis of CDCA.
- Numerous epidemiologic studies have demonstrated an association between high dietary cholesterol and the development of colon cancer. See e.g., Potter, J. D. Colorectal Cancer: Molecules And Populations J Natl Cancer Inst 91: 916-32, 1999. However, the precise mechanism by which cholesterol metabolites contribute to colon cancer remains unclear. Cholesterol is converted to bile acids in the liver which are then excreted into the gastrointestinal tract. Thus, diets high in cholesterol result in high concentrations of bile acid in intestinal contents. The link between dietary cholesterol and elevated bile acids is worrisome as fecal bile acid concentration is significantly higher in patients with colorectal cancer than in normal individuals. See Hill, M. J., Drasar, B. S., Williams, R. E., Meade, T. W., Cox, A. G., Simpson, J. E. and Morson, B. C. Faecal Bile-Acids And Clostridia In Patients With Cancer Of The Large Bowel Lancet 1: 535-9, 1975.
- In addition to epidemiological studies in humans, bile acids have been shown to promote the growth of colon cancer in various rodent models. For example, oral or intrarectal administration of bile acids induced significantly greater numbers of colon adenomas and adenocarcinomas in a carcinogen-induced model of colon cancer. In humans, mutational inactivation of the APC gene initiates most colon carcinomas. Another useful rodent model of colon carcinogenesis is the APC/Min (multiple intestinal neoplasia) mouse, which develops intestinal polyps due to a mutation the APC gene. Administration of bile acids to these mice result in increased numbers of ampullary tumors. Thus, experimental results from these rodent models support the epidemiological studies linking elevated bile acid levels to an increased risk of developing colon cancer.
- What mechanisms underlie the link between bile acids and colon cancer? Several studies have shown that endogenous bile acids can alter the balance between cell proliferation and apoptosis. Depletion of endogenous bile acids has been reported to decrease the rate of intestinal epithelial cell proliferation in rodents Roy, C. C., Laurendeau, G., Doyon, G., Chartrand, L. and Rivest, M. R. The Effect Of Bile And Of Sodium Taurocholate On The Epithelial Cell Dynamics Of The Rat Small Intestine Proc Soc Exp Biol Med 149: 1000-4, 1975.
- Conversely, acute exposure (1-2 days) to bile acids, now known to activate FXR, has been shown to lead to an increase in intestinal epithelial proliferation of as much as 3-fold. Clearly, it would not be possible to chronically sustain this acute increase in proliferation without compensatory measures that would maintain a constant epithelial cell mass. Indeed, after chronic exposure (>2-4 weeks) to bile acids, this increase in proliferation is not observed or is dramatically reduced.
- In addition to cell proliferation, apoptosis is also critical to the development of colon cancer as the progression from colon adenomas to adenocarcinomas is associated with an inhibition of apoptosis. Several bile acids have been shown to induce apoptosis in colon cancer cell lines. In contrast, bile acids decreased apoptosis in a cell line derived from a benign colon adenoma. While the role of bile acids in regulating apoptosis in vivo remains to be studied, these studies suggest that bile acids may have different effects on benign adenomas as compared to the less differentiated adenocarcinomas. The precise mechanisms governing how bile acids transduce signals that ultimately regulate cell proliferation and apoptosis remain to be elucidated.
- Bile acids have also been shown to regulate transcriptional events critical to colon carcinogenesis. In particular, chenodeoxycholic acid (CDCA) and DCA activate cyclooxygenase-2 (COX-2) transcription in gastrointestinal cell lines. This is significant as COX-2, which is over expressed in many colon cancers, produces prostaglandins that inhibit apoptosis and stimulate angiogenesis and invasiveness. Moreover, selective COX-2 inhibitors decrease the number and size of polyps in APC/Min mice and are currently being evaluated in clinical trials. Thus, the ability of bile acids derived from dietary cholesterol to regulate transcription may have important implications for the development of colon cancer. However, it is unknown how bile acids transduce a signal to stimulate COX-2 gene transcription.
- Taken together, the above studies demonstrate a significant epidemiological and experimental association between bile acids and colon cancer. The ability of bile acids to regulate transcription, particularly combined with recent evidence that bile acids bind to BAR, indicates that BAR, as a ligand-regulated transcription factor, may mediate the activities of bile acids in colon cancer.
- The identification of a nuclear receptor such as BAR/FXR as a potential mediator of colon cell growth has significant clinical value. First, this provides a molecular basis by which to refine dietary guidelines regarding cholesterol intake and the risk of developing colon cancer. Second, such findings provide a specific molecular target for future therapies aimed at treating and/or preventing colon cancer. “Hormonal” therapies could be highly effective, particularly if they are designed to be specific, relatively non-toxic (i.e. with a minimum of unwanted activities).
- Thus, there remains a need in the art for methods of modulating the steady-state concentration of cholesterol and/or bile acids in vivo. Preferably such modulation would be at least somewhat selective for the bile acid synthesis pathway and would be dissociated from BAR's effect on the expression of genes not directly involved in bile acid synthesis. Additionally, it would be useful if compounds were found that selectively inhibit the expression of certain BAR/FXR while not inhibiting, or actually increasing, the expression of other BAR/FXR target genes.
- FIG. 1A Shows the activation of CV-1 cells transfected with CMX-BAR, CMX-RXR and the EcRE×6 TK luc reporter. Reporter activation was measured in transfectant cells alone and upon treatment with 100 gM CDCA, 5 μM AGN 29, 5 μM AGN 31, 5 μM TTNPB and 100 nM LG268.
- FIG. 1B shows the activation of CV-1 cells transfected with CMX-BAR, CMX-RXR and the EcRE×6 TK luc reporter. Cells were also transfected with the RAR fusion vector Gal-L-RAR. Reporter activation was measured in transfectant cells alone and upon treatment with 100 μM CDCA, 5 μM AGN 29, 5 μM AGN 31, 5 μM TTNPB and 100 nM LG268.
- FIG. 1C shows the activation of CV-1 cells transfected with CMX-BAR, CMX-RXR and the EcRE×6 TK luc reporter. Cells were also transfected with the RXRA fusion vector Gal-L-RXR. Reporter activation was measured in transfectant cells alone and upon treatment with 100 μM CDCA, 5 μM AGN 29, 5 μM AGN 31, 5 μM TTNPB and 100 nM LG268.
- FIG. 2A shows the activation of CV-1 cells transfected with full length BAR/FXR upon treatment with increasing doses of CDCA, AGN 29 and AGN 31.
- FIG. 2B shows the change in polyacrylamide gel electrophoresis migration of BAR:RXRM heterodimers upon incubation with the receptor interaction domain of the
co-activator GRIP 1 and differing amounts of either AGN 29 or AGN 31. - FIG. 3A shows a Northern blot analysis of IBABP expression in Caco-2 cells upon incubation alone or in the presence of CCDA, AGN 29, or AGN 31.
- FIG. 3B shows a Northern blot analysis of CYP7A, SHP and GAPDH expression in HepG2 cells upon incubation alone or in the presence of CCDA, AGN 29, or AGN 31.
- FIG. 4A shows the results of a co-transfection assay conducted in which the combined effects of AGN 34 and CDCA on Bar/FXR activity were determined.
- FIG. 4B shows a dose-response curve of transactivation activity when cells are incubated in the presence of a constant amount of CDCA and increasing amounts of AGN 34.
- FIG. 4C shows the effect upon the transactivational activity of a variety of nuclear receptors of incubation with AGN 34.
- FIG. 4D shows a “gel shift” co-activator (GRIP) recruitment assay of AGN 34 and AGN 29, alone and together, in which the concentration of AGN 34 is increased.
- FIG. 5A is a Western blot of IBABP, SHP and GAPDH RNA expression upon treatment of Caco-2 cells with CDCA, AGN34 and CDCA and AGN 34.
- FIG. 5B is a Western blot of IBABP, SHP and GAPDH RNA expression upon treatment of HepG2 cells with CDCA, AGN34 and CDCA and AGN 34.
- The present invention is directed to methods for modulating the transcriptional activity of BAR/FXR through the use of synthetic ligands of the BAR:RXR heterodimer. Such ligands are able to cause BAR, preferably in combination with another nuclear hormone receptor such as RXR, to suppress, inhibit, or stimulate the transcription of a given target gene. In one preferred embodiment, the invention is directed to methods for stimulating BAR/FXR activity comprising administering an effective dose of a synthetic agonist of BAR/FXR activity. Preferred synthetic agonists are identified as AGN 29 and AGN31.
- In another preferred embodiment the invention is directed to methods for inhibiting the BAR-mediated stimulation of Intestinal Bile Acid Binding Protein (IBABP) gene expression, comprising administering an effective dose of AGN 34.
- Contemplated by the present invention are methods for regulating the concentration of bile acids in a mammal. A heightened concentration of bile acids in mammals has been associated with an increased occurrence of colon cancer; thus, the use of BAR/FXR ligands which do not significantly increase, or which decrease Cyp7a expression may effectively lower abnormally high bile acid concentrations therefore providing a therapeutic and/or prophylactic effect for this indication. The transcription of proteins other than Cyp7a are regulated by bile acids; these include Intestinal Bile Acid Binding Protein and Cyclooxygenase 2 (both up-regulated by CDCA), and sterol-27-hydroxylase, Intestinal Bile Acid Transporter, and Liver Bile Acid Transporter (these proteins are down regulated by CDCA). The methods of the present invention are therefore useful in modulating the expression of these proteins as well.
- Thus, in another preferred embodiment the present invention is directed to a method of treating colorectal cancer through the administration to a patient in need thereof of a pharmaceutically effective dose of a BARIFXR ligand which causes a decrease or inhibition of the formation of a IBABP:bile acid complex. Preferably the BAR/FXR ligand decreases the expression of IBABP without antagonizing at least one other activity characteristic of agonism of the BAR/FXR receptor. Particularly preferred as a BAR/FXR ligand is AGN 34.
- By “BAR/FXR ligand” is meant that the ligand binds either to the BAR/FXR receptor or to a complex intermolecular complex or multimer which comprises the BAR/FXR receptor and which modulates an activity associated with the BAR/FXR receptor.
- The BAR/FXR ligands of the present invention may be BAR/FXR antagonists, BAR/FXR inverse agonists, or have attributes of more than one of these. By “agonist” is meant that the ligand stimulates a ligand-dependent BAR/FXR activity above any baseline levels present in the absence of ligand. By “antagonist” is meant that the ligand binds to BAR, and functions as a competitive or non-competitive inhibitor of BAR/FXR agonist activity. By “inverse agonist” is meant that the ligand will bind to BAR/FXR and cause the suppression of an BAR/FXR activity to a level lower than seen in the absence of any BAR/FXR ligand.
- By modulating an activity associated with the BAR/FXR receptor is meant that the ligand affects an activity associated primarily with the BAR/FXR receptor alone or in combination with another factor, with the BAR:RXR heterodimer, but not with the RXR homodimer. A ligand may exert its activity by binding the BARIFXR subunit, by binding the RXR subunit, or by binding both the BAR/FXR and RXR subunit. The mechanism of modulation is irrelevant to this invention.
- In another aspect the present invention pertains to methods of stimulating or inhibiting an activity, or stimulating one activity and inhibiting another activity associated with a BARIFXR receptor of a mammal by treating such a mammal with a pharmaceutically acceptable composition comprising a compound selected from the group consisting of: AGN 29, AGN 31 and AGN 34.
- AGN 29 has the following structure:

- AGN 31 has the following structure:

- AGN 34 has the following structure:

- Other aspects and embodiments of the invention are contained in the disclosure that follows and the claims that conclude this specification.
- The present invention is directed to methods for modulating the activity of a mammalian BAR/FXR receptor, preferably the human BAR/FXR protein.
- Such methods involve the use of a BAR/FXR ligand which will bind the BAR/FXR receptor or a complex containing the BAR/FXR receptor, thereby affecting the ability of BAR/FXR to exert its biological effects, either directly or by blocking the ability of a naturally occurring ligand to exert its affects. The BAR/FXR ligands of the present invention may be BAR/FXR antagonists, BAR/FXR agonists, or BAR/FXR inverse agonists. Preferably, although not necessarily, the BAR/FXR ligands have substantially no activity at the retinoid nuclear receptors, RAR and RXR. In another embodiment, the BAR/FXR ligand may be a bi-specific compound able to bind and modulate both RXR and BAR.
- Also included are aspects of the invention directed to methods for increasing the plasma concentration of cholesterol in a mammal pathologically deficient in cholesterol through the use of an BAR/FXR agonist.
- While not wishing to be bound by theory, the Applicants currently believe that the BAR/FXR receptor, when bound by an BAR/FXR agonist, may inhibit the transcription of the oxysterol receptor LXRα, which in turn activates transcription of Cyp7a. Repression of transcription of this key enzyme in the biosynthesis of bile acids therefore results in a lower concentration of bile acids within the body; high bile acid concentrations have been associated with a heightened risk of colon cancer.
- As an aid in the further understanding of this invention, Applicants offer the following Examples, which are intended to illustrate the invention, but not to limit the scope of the claims.
- Materials and Methods
- All mammalian expression vectors were derivatives of the bacterial/mammalian shuttle vector pCMX, an expression vector containing the cytomegalovirus (CMV) promoter/enhancer, followed by a bacteriophage T7 promoter for transcription of the cloned gene in vitro. Plasmid pCMX also contains the SV40 small t intron/poly adenylation signal sequence, polyoma virus enhancer/origin and the SV40 enhancer/origin of plasmid CDM8 (see Seed, Nature 329:840-842 (1987), hereby incorporated by reference herein) cloned into the large Pvu II fragment of pUC 19. PUC 19 is a commonly used cloning vector available from New England Biolabs, Inc. This Pvu II fragment contains a Col El origin of replication and an ampicillin resistance gene for plasmid selection, but lacks the pUCl 9 polylinker cloning site. To create a new polylinker site, a synthetic polylinker comprising the following restriction sites: 5′-KpnI, EcoRV, BamHI, MscI, NheI-3′ followed by a translational termination sequence inserted in all three reading frames. See Umesono et al., Cell 65:1255-1266 (1991), hereby incorporated by reference herein.
- The nucleic acid regions encoding the following full-length proteins were cloned into a CMV expression vector. The sequences of these genes and/or their corresponding polypeptides have the indicated GenBank accession numbers: rat BARIFXR (accession #U18374), human RXRA (accession #X52773), human TRβ (accession #X04707), human LXRα (accession #U22662), mouse CARP (accession #AF009327), mouse PPARa (accession #X57638), mouse PPARδ (accession #U10375), mouse PPARγ(accession #U10374), VDR (accession #NM —000376). Gal4 fusions containing the indicated protein fragments were fused to the C-terminal end of the yeast Gal4 DNA binding domain (amino acids 1-147, accession #X85976): Gal-RAR (human RAR ligand binding domain, Glu 156-Ser 463, accession #X06614), Gal-RXR (human RXRa igand binding domain, Glu 203-Thr 462, accession #X52773). The βgal contains the E. coli P-galactosidase coding sequences derived from pCH110 (accession #U02445). RXRm contains a single point mutation (Asp-322-Pro) in the LBD of human RXRα. Luciferase reporter constructs (TK-luc) contain the Herpes virus thymidine kinase promoter (−105/+51) linked to the indicated number of copies of the following response elements: hsp27 EcRE×6 (see Yao et al., 71 Cell 63-72 (1992)); IBABP IR-1×3 (CCTTAAGGTGAATAACCTTGGGGCTCC) (SEQ ID NO: 13); UASG×4 (MH100×4); PPRE×3 (see Forman et al., 81 Cell 687 (1995)); βRE2×3 (see Forman et al., 395 Nature 612 (1998); LXREx3 (see Willy et al., 9 Genes Dev. 1033 (1995)); SPP×3 (see Umesono et al, 65 Cell 1255 (1991)) and T3RE (MLV)×3 (see Perlmann et al., 7 Genes Dev. 1411 (1993). All references, including each of those above, cited in this application are hereby incorporated by reference herein unless specifically indicated otherwise. The GenBank information corresponding to these accession numbers is hereby incorporated by reference herein in its entirety. The rat BAR/FXR amino acid sequence, mouse BAR/FXR amino acid sequence, and human BAR/FXR amino acid sequence are provided herein as SEQ ID NO: 1, SEQ ID NO: 2, and SEQ ID NO: 3, respectively. The human RXRα amino acid sequence is provided herein as SEQ ID NO: 4.
- GAL4 fusion proteins were constructed using standard molecular biological methods (see e.g., Sambrook et al., Molecular Cloning: A Laboratory Manual (2d ed. Cold Spring Harbor Laboratory Press 1989), incorporated by reference herein in its entirety) by inserting a nucleotide sequence encoding the indicated polypeptide immediately downstream of the yeast GAL4 DNA-binding domain in plasmid pSG424, described in Sadowski et al., Nucleic Acids Research 17:7539, hereby incorporated by reference herein. The amino acid sequence of the yeast GAL4 DBD, hereby designated SEQ ID NO: 5, is as follows:
NH2-MKLLSSIEQA CDICRLKKLK CSKEKPKCAK CLKNNWECRY SPKTKRSPLT RAHLTEVESR LERLEQLFLL IFPREDLDM ILKMDSLQD IKALLTGLF VQDNVNKDAV TDRLASVETD MPLTLRQHRI SATSSSEESS NKGQRQLTVS-COOH - Fusion proteins were made, as indicated above, using common molecular biological techniques by creation of open nucleic acid reading frames encoding the indicated polypeptides, and cloning into the polylinker portion of pCMX.
- For GAL-L-RXR, the plasmid nucleic acids encoded amino acids Glu 203 to Thr462 of human RXRα (SEQ ID NO: 4) fused to the GAL4 sequences. The junction between the carboxyl terminal section of GAL4 and the amino terminal portion of the RXR LBD had the following structure:
EcoRI Asp718 Sal/Xho GTA-TCG-CCG-GAA-TTC-GGT-ACC-GTC-GAG-GCC-GTG-CAG- GAG-.. Val-Ser Glu-Ala-Val-Gln- GLu-.. GAL4 -> 203 --> hRXRa LBD - This junction nucleotide sequence
- 5′GTATCGCCGGAATTCGGTACCGTCGAGGCCGTGCAGGAG3′ is hereby designated SEQ ID NO: 6.
- For GAL-L-BAR, the plasmid nucleic acids encoded amino acids Leu 181 to Gln469 of rat BAR/FXR (SEQ ID NO: 1) fused to the GAL4 sequences. The junction between the carboxyl terminal section of GAL4 and the amino terminal portion of the BAR/FXR LBD had the following structure:
EcoRI former | KpnI/NaeI GTATCGCCGGAATTCGGGCTAAGGAAGTGCAGAGAGATGGGAATG TTGGCTGAATG ValSerProGluPheGlyLeuArgLysCysArgGluMetGlyMet LeuAlaGlu GAL4>| |<---rBARa AA 181 |<---LBD - This junction nucleotide sequence (from 5′ to 3′)
GTATCGCCGCAATTCGGGCTAAGGAAGTGCAGAGAGATGGGAATGTTG GCTGAATG - is hereby designated SEQ ID NO: 7.
- RXR ligand binding domain (LBD) expression construct L-RXR contains nucleotide residues encoding the SV40 Tag nuclear localization signal sequence (from amino to carboxy ends:
- APKKKRKVG (SEQ ID NO: 8)
- located immediately upstream (i.e., to the 5′ side on the coding strand) of a nucleotide sequence encoding the human RXRα LBD (Glu 203 to Thr462). CMX-βgal contains the E. coli β-galactosidase coding sequence derived from plasmid pCH110 (accession number U 02445) inserted downstream of the CMV promoter in plasmid pCMX. RXRm contains a single point mutation changing Asp-322 to Pro in the LBD of human RXRα.
- Luciferase reporter plasmids (termed TK-Luc) were constructed by placing the cDNA encoding firefly luciferase immediately downstream from the herpes virus thymidine kinase promoter (located at nucleotide residues −105 to +51) of the thymidine kinase nucleotide sequence), which is linked in turn to the various response elements. The promoter region of the TK-Luc plasmids has the following structure:
SalI HindIII SphI PstI | | | | | GGTTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGTGCCAAGCTTGCATGCCTGCAGG 61 ---------+---------+---------+---------+---------+---------+ 120 BaiCI BstBI XbaI BamI EcoRI | | | | | pUC18 —|— HSV-TK TCGACTCTAGAGGATCCGGCCCCGCCCAGCGTCTTGTCATTGGCGAATTCGAACACGCAG 121 ---------+---------+---------+---------+---------+---------+ 180 AvaII AvaII* | AvaII | | MluI | | | | ATGCAGTCGGGGCGGCGCGGTCCCAGGTCCACTTCGCATATTAAGGTGACGCGTGTGGCC 181 ---------+---------+---------+---------+---------+---------+ 240 PstI BglII | | |-- transcription start — HSV-TK —| TCGAACACCGAGCGACCCTGCAGCGACCCGCTTAACAGCGTCAACAGCGTGCCGCAGATC 241 ---------+---------+---------+---------+---------+---------+ 300 EheI PaeR7I NarI| XhoI KasI|| | ||| TCTCGAGTCCGGTACTGTTGGTAAAATGGAAGACGCCAAAAACATAAAGAAAGGCCCGGC 301 ---------+---------+---------+---------+---------+---------+ 360 MetGluAspAlaLysAsnIleLysLysGlyProAla - |— luciferase XbaI FokI | | GCCATTCTATCCTCTAGAGGATGGAACCGCTGGAGAGCAACTGCATAAGGCTATGAAGAG 361 ---------+---------+---------+---------+---------+---------+ 420 ProPheTyrProLeuGluAspGlyThrAlaGlyGluGlnLeuHisLysAlaMetLysArg - This nucleotide sequence (continuous from 5′ to 3′)
5′-GGTTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGTGCCAAGCTT GCATGCCTGCAGGTCGACTCTAGAGGATCCGGCCCCGCCCAGCGTCTTGT CATTGGCGAATTCGAACACGCAGATGCAGTCGGGGCGGCGCGGTCCCAGG TCCACTTCGCATATTAAGGTGACGCGTGTGGCCTCGAACACCGAGCGACC CTGCAGCGACCCGCTTAACAGCGTCAACAGCGTGCCGCAGATCTCTCGAG TCCGGTACTGTTGGTAAAATGGAAGACGCCAAAAACATAAAGAAAGGCCC GGCGCCATTCTATCCTCTAGAGGATGGAACCGCTGGAGAGCAACTGCATA AGGCTATGAAGAG-3′ - is designated SEQ ID NO: 9.
- Response elements were inserted in plasmid TK-Luc at the unique Hind III site. As an example, the yeast GAL4 UASG response element has the nucleotide sequence, and was inserted in 4 direct repeats to yield UASG×4:
- 5-CGACGGAGTACTGTCCTCCGAGCT-3′ (SEQ ID NO: 10)
- As another example, the hsp EcRE (ecdysone response element) was inserted into the Hind III site of plasmid TK-Luc as six direct repeats of the following sequence:
- 5′-TGGACAAGTGCATTGAACCCTT-3′ (SEQ ID NO: 11)
- to yield hsp EcRE×6.
- The person of ordinary skill in the art will recognize that the sequences of other response elements discussed herein are disclosed in the cited references, and are readily available e.g., from the National Institutes of Health's National Center for Biotechnology Information (NCBI) website (https://www.ncbi.nlm.nih.gov/) on the Worldwide Web, which is hereby incorporated by reference herein in its entirety.
- Transient Transfection Assay
- CV-1 African Green Monkey cells were grown in Dulbecco's Modified Eagle's medium supplemented with 10% resin-charcoal stripped fetal bovine serum (FBS), 50 U/ml penicillin G and 50 μg/ml streptomycin sulfate (DMEM-FBS) at 37° C. in 5% CO 2. One day prior to transfection, cells were plated to 50-80% confluence using phenol red-free DMEM-FBS. Cells were transiently transfected by lipofection as described in Forman et al., 81 Cell 687 (1995). Liposomes (N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-ammonium methyl sulfate, sold by Boehringer Mannheim under the name DOTAP) were formed according to the manufacturer's instructions. The liposomes contained reporter gene constructs (300 ng/105 cells); cytomegalovirus driven expression vectors (25 ng/105 cells) were added as indicated along with CMX-Bgal (500 ng/105 cells) as an internal control. After 2 hours the liposomes were removed and replaced with fresh media. Cells were incubated for approximately 40 hours with phenol red-free DMEM-FBS containing the indicated compounds. After exposure to the specified ligand, the cells were harvested.
- The harvested cells were assayed for the presence of luciferase activity. Cells were lysed in 0.1 M KPO4 (pH 7.8), 1.0% TRITON® X-100, 1.0 mM dithiolthreitol (DTT) and 2 mM ethylenediamine tetracetic acid (EDTA). Luciferase activity was measured by reaction of the cell lysates with luciferin in a reaction buffer comprising: 20 mM tricine, 1.07 mM Mg(CO 3)4-Mg(OH)2-5H2O, 2.67 mM MgSO4.7H2O, 0.1 mM EDTA, 0.5 mM Sodium luciferin, 0.15 mg/ml Coenzyme A, 5 mM DTT, and 0.5 mM adenosine triphosphate (ATP). Resulting chemiluminescence was measured in a luminometer. See de Wet et al., Mol. Cell Biol. 7:725 (1987) (hereby incorporated by reference herein). All points were assayed in triplicate and varied by less than 15%. Each experiment was repeated three or more times with similar results. No cytotoxicity was observed with any of the compounds when used at the indicated concentrations and treatment times.
- Northern Analysis
- HepG2 cells (a hepatoma cell line used as a hepatocyte model) were maintained in Eagle's minimal essential medium supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM L-glutamine, non-essential amino acids, 50 U/ml penicillin G and 50 μg/ml streptomycin sulfate. Caco-2 cells (a cell line derived from a colon carcinoma capable of spontaneously differentiating into cells sharing characteristics with small intestinal cells) were maintained in DMEM supplemented with 20% FBS, 50 U/ml penicillin G and 50 μg/ml streptomycin sulfate. Caco-2 cells were maintained for 20 days post-confluence to allow differentiation. They were fed twice a week with their regular media during this period.
- One day prior to treatment, confluent HepG2 and differentiated Caco-2 cells were switched to phenol red-free media containing resin-charcoal stripped FBS and then treated for and additional 24 hours with the indicated compounds. Total RNA was isolated using the Trizol reagent. Northern blots were prepared from polyA + RNA using the Oligotex method (Qiagen) and analyzed with the following probes: human CYP7A (accession #M93133) nucleotides 1617-2576 (Karam and Chiang, 185 Biochem. Biophys. Res. Commun. 588 (1992)), human SHP (accession #L76571) nucleotides 888-1355, human IBABP (accession # A1311734), an EST containing the entire coding sequence of IBABP.
- Coactivator Recruitment Assay
- Coactivators, which bind agonist-activated nuclear receptors including, without limitation, RAR and RXR, function to assist the activated nuclear receptor exert its activity as a transcription factor. Characterized co-activators include CBP, p300, RIP140, SRC-1, ACTR, TIF2 (also called GRIP 1) and TIFI. In most cases coactivator will not bind the receptor in the absence of an agonist, moreover, receptor antagonists will often block coactivator binding, either directly or through the recruitment of a corepressor. Coactivator recruitment assays are therefore a valuable method for directly visualizing associative changes to a receptor caused by the addition of a prospective ligand.
-
GRIP 1 was expressed as a fusion protein with glutathione-S-transferase, an enzyme which selectively binds glutathione and can thus be used as an affinity reagent. See e.g., U.S. Pat. No. 5,654,176. The GST-GRIP1 fusion protein was expressed in E. coli and purified on glutathione-Sepharose columns. In vitro translated BAR/FXR\RXR (0.6-1.2 μl each) and GST-GRIP 1 (5 μg) were incubated for 30 min at room temperature with 100,000 cpm of the E. coli DNA polymerase Klenow fragment-labeled probes in 10 mM Tris (pH 8.0), 50 mM KCl, 6% glycerol, 0.05% NP-40, 1 mM DTT, 12.5 ng/μl poly dI-dC, and the indicated ligands. Complexes were electrophoresed through 5% polyacrylamide gel in 0.5% TBE (45 mM Tris-base, 45 mM boric acid, 1 mM EDTA) at room temperature. Oligonucleotide probes were made to identify specific receptor dimers; an optimized DR-1 (direct repeat 1; a nucleotide sequence motif to which RXR will bind) probe (5′-GCTACCAGGTCAAAGGTCACGTAGCT) (SEQ ID NO: 12) was used for RXR homodimers and the IR-1 sequence of the IBABP promoter, to which the BAR/RXR heterodimer binds, was used for BAR/RXR heterodimers. - Identification of Synthetic BAR/FXR Agonists
- Because of the central role played by BAR/FXR in cholesterol degradation/bile acid formation, we proposed that effective BAR/FXR activators and repressors could have beneficial pharmacological effects in a variety of bile acid-related diseases. As our starting point in the identification of BAR/FXR modulators, we made use of the observation that the synthetic retinoid TTNPB, [E]-4-[2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl) propen-1-yl]benzoic acid, activates BAR. TTNPB has the structure:

- We used a sensitive, high throughput, cell-based transient transfection assay, described above, to screen for BAR/FXR activators. Using this method, we identified 2 potent agonists which we designated AGN29 and AGN31. Full-length BAR/FXR and RXR expression plasmids were cotransfected in CV-1 cells with a luciferase reporter construct containing BAR/FXR binding sites derived from the IBABP promoter (IBBABP IR-1).
- The transfected cells were then treated with the indicated ligands for 40 hours. Activity was measured using luciferase expression in the cotransfection assay described above. AGN 29 and AGN 31 robustly activated BAR/FXR (91- and 85-fold respectively) when used at a concentration of 5 μM, whereas CDCA resulted in a nearly 200-fold activation at 100 μM (FIG. 1A). TTNPB, as previously demonstrated, also induced significant BAR/FXR activity (65-fold). The RXR-specific ligand LG268 also activated the BAR-RXR heterodimer via the RXR subunit as previously demonstrated (FIG. 1A). Since TTNPB is a strong RAR activator, we tested the ability of AGN 29 and AGN 31 to activate both RAR and RXR. To eliminate the effect of ligands on endogenous receptors from our assay, we used fusion proteins between the
Gal 4 DNA binding domain (DBD) and the ligand binding domain (LBD) of the receptor of interest. Thus, Gal-L-RAR or Gal-L-RXR fusions were transfected into CV-1 cells with aGal 4 reporter (MH100×4) and the effect of different ligands was evaluated. As seen in FIG. 1B, TTNPB was able to strongly activate RAR, but treatment with AGN 29 and AGN 31 resulted in a loss of specificity for RAR. These compounds on the other hand retained some ability to activate RXR (FIG. 1C), but had no effect on other RXR heterodimers containing nuclear receptors including AR, mPXR, hPXR, ERα, CARβ, LXRα, PPARα, PPARβ, PPARδ, VDR and TRβ. - To test the relative potency of the compounds, CV-1 cells transfected with full length BAR/FXR were treated with increasing doses of AGN 29 and AGN 31. As shown in FIG. 2A, the EC 50 for AGN29 and AGN31 is approximately 2 μM compared with an EC50 of approximately 50 μM for CDCA. By EC50 is meant the concentration at which the response is 50% of maximal.
- To address the question of whether the two identified compounds are ligands for BAR, we used an in vitro co-activator recruitment assay. Most nuclear receptors, including BAR, are inactive in the absence of an agonist because they are unable to interact with co-activators. In the presence of agonist, LBD appears to undergo conformational changes that enable recruitment of co-activators and subsequent transcription. By recruitment is meant the formation of a reversible non-covalent association between a receptor LBD and a co-activator or co-repressor, usually in response to the addition of such a receptor co-modulator. Because the association between co-activator and receptor results in the formation of a new higher molecular weight species, the ligand-dependent recruitment of co-activator can be detected in gel shift assays and is commonly used as an indicator for ligand binding. Blumberg et al., 12 Genes Dev. 1269 (1998); Forman et al., 395 Nature 612 (1998); Kliewer et al., 92 Cell 73 (1998); Krey et al., 11 Mol. Endocrinol. 779 (1997).
- Co-activator recruitment assays were performed by mixing AGN 29 and AGN 31 with BAR, RXRm, a 32P-labeled BAR/FXR response element (IBABP IR1) and the receptor interaction domain of
GRIP 1. RXRm is an RXR mutant impaired in its ability to bind ligand and used to determine whether AGN 29 and AGN 31 bind to the BAR/FXR subunit and not to RXR. After the binding reactions, the resulting complexes were separated on polyacrylamide gels. As expected, BAR-RXRm heterodimers failed to recruit co-activator in the absence of ligand (FIG. 2B). However, addition of AGN 29 or AGN 31 shifted the majority of the complexes with the co-activator GRIP1 suggesting that AGN 29 and AGN 31 bind BAR/FXR and not RXR. These experiments were performed with a range of doses for AGN 29 and AGN 31 and co-activator recruitment was seen at doses that parallel the potency of these compounds in transient transfection assays (FIG. 2B). - We next tested the ability of AGN 29 and AGN 31 to regulate BAR/FXR target genes. We used the hepatoma cell line HepG2 as a hepatocyte model as this cell line has been used extensively to study cholesterol metabolism and bile acid-mediated gene regulation. We monitored the expression of two well known BAR/FXR target genes: CYP7A, the rate-limiting enzyme for bile acid synthesis, is down regulated by bile acids in HepG2 cells. It is now clear from gene-targeting studies that bile acids exert their inhibitory role on this gene through BAR/FXR (Sinal et al., 102 Cell 731 (2000). We also monitored the expression of the nuclear receptor SHP, a recently recognized target gene for BAR. Caco-2 cells were used as a model for ileal enterocytes. This cell line was used to monitor the levels of SHP and IBABP. Both cell lines were treated for 24 hours with AGN 29 or AGN 31 (10 μM), or with CDCA (100 μM) as a positive control.
- As assayed by Northern blot analysis, in differentiated Caco-2 cells AGN 29 and AGN 31 strongly induced IBABP and SHP expression to the same level as CDCA (FIG. 3A). In HepG2 cells, AGN29 and AGN31 were able to repress CYP7A expression and induce the transcription of SHP (FIG. 3B). These ligands had no effect on BAR/FXR expression or on GAPDH These results complement the in vitro data and confirm that AGN 29 and AGN 31 are agonist ligands for BAR.
- We next asked whether compounds previously identified by us in these assays as incapable of activating BAR/FXR might possess antagonist activity. We identified one such compound, AGN34, whose structure is shown above. To test the ability of AGN34 to repress BAR/FXR activity, we transiently transfected CV-1 cells with full length BAR/FXR and RXR and with the IR-1-containing luciferase reporter gene. Cells were then treated with suboptimal levels of CDCA (50 μM) with or without AGN 34 (1 μM). Addition of 50 μM CDCA resulted in nearly 100-fold activation of BAR.
- In the cotransfection assay AGN 34 did not affect the basal activity of luciferase gene expression, but repressed CDCA-activated BAR-mediated transcription nearly 10-fold (FIG. 4A). Dose-response analysis (increasing concentration of AGN 34; constant concentration of CDCA) in the same assay system indicated that AGN34 is a very strong repressor, displaying approximately 85% repression at 0.03 μM and showing maximal activity at 1 μM (FIG. 4B).
- Various other nuclear receptor/RXR heterodimers were incubated with the cognate ligand of the non-RXR component of the heterodimer. These were: human AR (androgen receptor), mouse PXR (10 μM pregnenolone-16-carbonitrile), human PXR (10 μM rifampicin), ERα (100 nM 17β-estradiol), human LXRα (30 μM hyodeoxycholic acid methyl ester), mouse PPARα (5 μM Wy 14,643), mouse PPARγ (1 μM rosiglitazone), mouse PPARδ (1 μM carbaprostacyclin), human VDR (100
nM 1,25-dihydroxyvitamin D3) and human TRB (100 nM triiodothyronine). No ligand was added to mouse CARβ which is constitutively active. Each compound was incubated in the presence or absence of 1 mM AGN 34, then assayed for luciferase reporter activity. As indicated in FIG. 4C, AGN34 was unable to repress the specific ligand-induced activity of these nuclear receptors, while AGN 34 remained able to repress the BAR/RXR heterodimer almost 10-fold. - We next tested the ability of AGN34 to displace the GRIP-1 co-activator from BAR. The results are shown in FIG. 4D. BAR:RXR:GRIP1 form a complex only in the presence of agonist; in this case 0.75 μM AGN 29 (lane 2). The addition of increasing concentrations of AGN 34 resulted in the progressive dissociation of that complex, suggesting that AGN 34 competes with agonists for binding to BAR. The experiments were repeated with RXRm, a mutant RXR in which asp322 within the RXR LBD has been replaced with a proline, rendering the RXRm with over 100-fold reduced Kd for ligand. Competition experiments using the BAR/RXRm heterodimer binds yielded the same results as obtained using the BAR/RXR heterodimer. (FIG. 4D).
- To verify that AGN34 represses BAR/FXR in vivo, we looked at its effect on BAR/FXR target genes in cultured cells by Northern blot analysis. Differentiated Caco-2 cells and HepG2 cells were treated for 24 hours with 100 μM CDCA alone or in combination with 1 μM AGN34, then the RNA extracted and Northern blot performed as indicated above.
- As expected, CDCA increased IBABP expression sharply in Caco-2 cells and the addition of AGN34 reduced the induced IBABP levels close to basal levels (FIG. 5A). To further confirm the role of AGN34, we looked at its effect on SHP expression both in Caco-2 and HepG2 cells. In both cell lines, AGN 34 did not affect basal SHP levels nor did it reduce CDCA-elevated expression. The effect of AGN 34 was also tested on CYP7A expression. In contrast to what would be expected from a BAR/FXR full antagonist, AGN 34 repressed rather than stimulated CYP7A expression, and this activity was repressed even further by the combination of AGN 34 and CDCA (FIG. 5B).
- These results clearly indicate that AGN34 is a selective BAR/FXR modulator (BARM); that is, a BAR/FXR partial antagonist, that regulates different BAR/FXR target genes differentially. Antagonism of IBABP expression by AGN 34 is useful as a therapeutic method for the treatment of conditions such as colorectal cancer characterized by the presence of excessive levels of bile acid. Additionally, the fact that AGN 34 does not antagonize other BAR-regulated genes, such as Cyp 7A, means that such therapeutic use is quite specific and will therefore have a minimum of undesired side effects.
- To verify that AGN 34 modulates an activity that is characteristic of the BAR/FXR receptor, and that it does not function through the RXR homodimer, the following experiment was performed.
- Luciferase reporter constructs (TK-luc) containing the Herpes virus thymidine kinase promoter (−105/+51) linked to the indicated number of copies of the following response elements: hsp27 EcRE×6 and MH100×4 (UASG×4) were used in a co-transfection assay conjunction with the following expression plasmids: GAL-L-hRXRα, hFXR, rFXRop, rnFXR and hRXRop. As shown below, in most cases hRXRop was recombinantly coexpressed with one other of the indicated receptor constructs, so as to permit the formation of hetero-or homodimers containing hRXRop.
- These constructs were tested in combination with the following test agents: CDCA, AGN 34, and various concentrations of these two agents. Methods are the same as for all transfections. Concentrations are noted in the second row of the table in 1 μM. Except for the “none” column, all columns contained 50 μM CDCA+/− the indicated amount of AGN 34 in μM—e.g. last column is 50 μM CDCA+10 μM AGN34. Expression of the luciferase reporter gene product was detected and quantified as described above. The results are shown in the following table:
TABLE 1 Transfected Transfected AGN CDCA + CDCA + CDCA + CDCA + CDCA + Reporter Receptor Receptor None CDCA 34 AGN 34 AGN 34 AGN 34 AGN 34 AGN 34 Ligand 50 10 0.01 0.1 1 3 10 Amount MH100 × 4 GAL-L- 0.33 0.38 0.38 0.34 0.31 0.37 0.35 0.37 hRXRa EcRE × 6 HFXR HRXRop 0.34 21.00 0.41 5.14 4.02 2.42 1.98 2.25 EcRE × 6 RFXRop HRXRop 0.32 19.29 0.27 2.92 2.05 1.28 0.98 1.28 EcRE × 6 MFXR HRXRop 0.22 17.71 0.26 4.37 2.45 1.38 1.29 1.36 Fold Activation MH100 × 4 GAL-L- 1.00 1.14 1.14 1.03 0.92 1.12 1.05 1.10 hRXRa EcRE × 6 HFXR HRXRop 1.00 61.32 1.21 15.00 11.73 7.08 5.78 6.58 EcRE × 6 RFXRop HRXRop 1.00 59.59 0.84 9.03 6.32 3.94 3.04 3.94 EcRE × 6 MFXR HRXRop 1.00 79.50 1.18 19.61 10.99 6.19 5.77 6.11 - These data indicate that when recombinant RXR is expressed alone (thus promoting the formation of RXR homodimers), the addition of CDCA, AGN 34 and the indicated concentrations of both ingredients does not result in the stimulation of cis-transactivation of the reporter gene. However, when human RXR is co-expressed with human, rat or mouse FXR (BAR) in transfected CV-1 cells, a resulting stimulation of gene expression is detected when the bile acid CDCA is added. AGN 34 alone does not cause a stimulation of transcription of the reporter gene. Increasing concentrations of AGN 34 added to a constant amount of CDCA in this system reduces the CDCA-mediated stimulation of reporter gene transcription in a dose-dependent manner. Thus, the data indicate that AGN 34 antagonizes the CDCA-mediated stimulation of BAR/FXR transcriptional activation, and that both the stimulation of transactivational activity by CDCA and the antagonism of this activity by AGN 34 is selective for the BAR/FXR receptor or the BAR:RXR heterodimer; neither agent shows activity in a system containing only RXR (and thus presumably only RXR homodimers.
- The same reporter and expression constructs were then tested in the same manner in combination with the following test agents: AGN 34, LG 268, and various concentrations of AGN 34 and at a constant concentration of LG 268. LG 268 is an RXR agonist. The results are shown in the following table:
TABLE 2 AGN LG268 + LG268 + LG268 + LG268 + LG268 + None LG268 34 AGN 34 AGN 34 AGN 34 AGN 34 AGN 34 Reporter 0.1 10 0.01 0.1 1 3 10 Activity MH100 × 4 GAL-L- 0.33 17.16 0.37 11.17 2.86 0.36 0.35 0.33 hRXRa EcRE × 6 HFXR HRXRop 0.32 1.47 0.44 0.76 0.47 0.40 0.41 0.39 EcRE × 6 RFXRop HRXRop 0.28 11.94 0.29 5.24 0.44 0.25 0.26 0.25 EcRE × 6 MFXR HRXRop 0.25 12.95 0.35 6.73 0.39 0.27 0.26 0.29 Fold Activation MH100 × 4 GAL-L- 1.00 51.55 1.12 33.56 8.61 1.07 1.05 0.98 hRXRa EcRE × 6 HFXR HRXRop 1.00 4.54 1.35 2.36 1.45 1.23 1.26 1.22 EcRE × 6 RFXRop HRXRop 1.00 42.42 1.02 18.59 1.55 0.88 0.92 0.90 EcRE × 6 MFXR HRXRop 1.00 52.10 1.39 27.08 1.56 1.07 1.04 1.18 - As can be seen from the data, LG 268 stimulates RXR-mediated reporter gene transcription when used as the sole ligand in this experiment, and when RXR is the only receptor recombinantly expressed by the cell. LG 268 also stimulates transctivation of the reporter gene which RXR is co-expressed with rat or mouse BAR, but not with human recombinant BAR/FXR This result suggests that LG 268 may have a measure of BAR/FXR (or BAR:RXR) agonist activity on the rat and mouse receptors as well as on RXR itself.
- The addition of increasing concentrations of AGN 34 at a constant LG 268 concentration results in an attenuation of the LG 268-mediated transactivation activity in a manner consistent with antagonism of LG 268 activity. AGN 34 alone, does not stimulate receptor-mediated transactivation.
- Subsequent gel shift experiments indicate that AGN 34 fully displaces associated co-activator (GRIP1) from BAR:RXR heterodimers in which LG 268 was added. Much lower doses of AGN 34 are necessary to cause full displacement than for the displacement of co-activator from RXR:BAR-CDCA complexes. Similar results are seen in a mammalian two hybrid assay detecting recruitment of CoA; AGN 34 inhibits recruitment of CoA, to the BAR:RXR heterodimer.
- An experiment conducted in a manner similar to that of Example 4 was carried out using either CDCA (50 μM) alone or in combination with AGN 34 (1 μM) or LG 754(1 μM) (an RXR homodimer antagonist but activator of PPAR-RXR) as test agents. The results are shown in the following table:
TABLE 3 CDCA + CDCA + None CDCA AGN 34 LG 754 Fold Activation MH100 × 4 GAL-L-hRXRa 1.00 0.96 0.38 0.79 EcRE × 6 HFXR HRXRop 1.00 31.41 5.51 16.67 EcRE × 6 RFXRop HRXRop 1.00 71.94 4.99 16.40 EcRE × 6 MFXR HRXRop 1.00 58.61 5.26 26.02 - These results confirm, as above, that CDCA is able to stimulate BAR-mediated transactivation of the reporter gene. LG 754 antagonizes the CDCA-mediated activity, but to a much lesser degree than AGN 34.
- The following example provides a detailed description of compounds having BAR/FXR modulating activity, as well as methods of making such compounds.
-

- Ethyl 4-Hex-1-ynylbenzoate (3). A solution of 1-hexyne (1.72 mL, 15 mmol), ethyl 4-iodobenzoate (1.38 g, 5 mmol), triethylamine (1.05 mL, 7.5 mmol), and THF (20 mL) was degassed with argon for ten minutes. The solution was treated with bis(triphenylphosphine)palladium (II) chloride (17.5 mg, 0.25 mmol) and copper iodide (11.4 mg, 0.06 mg) and it was stirred at room temperature for 24 h. The solution was concentrated under reduced pressure, and the residue was purified by silica gel chromatography (hexane) to give the title compound.
- 1HNMR (CDCl3, 300 MHz): δ 0.95 (t, 3H, J=7.3 Hz), 1.39 (t, 3H, J=7.1 Hz), 1.49 (m, 2H), 1.59 (m, 2H), 2.43 (t, 2H, J=7.1 Hz), 4.36 (q, 2H, J=7.3 Hz), 7.44 (d, 2H, J=8.3 Hz), 7.95 (d, 2H, J=8.3 Hz).
- 6-Iodo-1,1,4,4-tetramethyl-1,2,3,4-tetrahydronaphthalene (6). Aluminum trichloride was added very slowly to an ice-cold solution of iodobenzene (6.1 mL, 54.6 mmol) and 2,5-dichloro-2,5-dimethylhexane (5 g, 27.3 mmol). After 20 minutes, the solution was diluted with hexane and poured over ice water. The layers were separated and the aqueous layer extracted two times with hexane. The combined organic layers were washed with water and brine, dried over MgSO 4, and the filtered solvents were removed under reduced pressure. The excess iodobenzene was removed under high vacuum to give the title compound as a colorless solid.
- 1HNMR (CDCl3, 300 MHz): δ 0.1.25 (s, 6H), 1.26 (s, 6H), 1.65 (s, 2H), 1.66 (s, 2H), 7.03 (d, 1H, J=8.3 Hz), 7.42 (dd, H, J=2.0, 8.3 Hz), 7.59 (d, 1H, J=2.0 Hz).
- 4-[2-(5,5,8,8-Tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)hex-1-enyl]benzoic Acid (AGN 31). Ethyl 4-hex-1-ynylbenzoate (192 mg, 0.83 mmol) and 6-iodo-1,1,4,4-tetramethyl-1,2,3,4-tetrahydronaphthalene (628 mg, 2 mmol) were mixed with triethylamine (0.4 mL). Bis(triphenylphosphine)palladium (II) diacetate (16 mg, 0.021 mmol) and acetonitrile (0.3 mL) were added, and the solution was purged with argon for a few minutes. Acetic acid (0.083 mL, 2.20 mmol) was added and the solution was stirred overnight at 80° C. The reaction was diluted with water and ether, the layers were separated, and the aqueous layer extracted twice with ether. The combined ether layers were washed with brine, and dried (MgSO4), and filtered, and the solvent was removed under reduced pressure. The residue was purified by silica gel chromatography using 9:1 hexane:ethyl acetate as the eluent to give an inseparable mixture of isomeric esters. The esters were hydrolyzed with 2N aqueous KOH (1 mL) in ethanol (4 mL), acidified with 1N HCl, and the products were extracted with ethyl acetate. The organic extracts were washed with brine and dried (MgSO4). The filtered solvent was removed in vacuo and the resulting solid recrystalized from a solution of hexane and ethyl acetate to give the title compound.
- 1HNMR (CDCl3, 300 MHz): δ 0.882 (t, 3H, J=7.1 Hz), 1.22-1.55 (m, 4H), 1.31(s, 6H), 1.32 (s, 6H), 1.71 (s, 4H), 2.69 (t, 2H, J=8.3 Hz), 6.72 (s, 1H), 7.24 (d, 1H, J=8.2 Hz), 7.30 (d, 1H, J=8.3 Hz), 7.40 (s, 1H), 7.42 (d, 1H, J=8.4 Hz), 8.11 (d, 1H, J=8.4 Hz).
-


- Ethyl 2-Hydroxy-3,5-diisopropylbenzoate (9). Thionyl chloride (48 mL, 675 mmol) was added to a solution of 2-hydroxy-3,5-diisopropylbenzoic acid (15 g, 67.5 mmol) and dichloromethane (60 mL), and the resulting solution was heated to reflux for 18 hours. The solution was cooled to room temperature and the solvents were removed under vacuum. Ethanol was added and the solution was stirred at room temperature for 4 hours. The solvent was evaporated, and the residue was purified by silica gel chromatography (hexane:ethyl acetate::4:1).
- 1HNMR (CDCl3, 300 MHz): δ 1.24 (d, 6H, J=3.6 Hz), 1.27 (d, 6H, J=3.6 Hz), 1.43 (t, 3H, J=7.0 Hz), 2.87 (m, 1H, J=3.6 Hz), 3.37 (m, 1H, J=3.6 Hz), 4.42 (q, 2H, J=7.0 Hz), 7.27 (s, 1H), 7.55 (s, 1H), 11.1 (s, 1H).
- Ethyl 2-Hexyloxy-3,5-diisopropylbenzoate (10). A solution of ethyl 2-hydroxy-3,5-diisopropylbenzoate (19.1 g, 76.4 mmol) and 10 mL DMF was added slowly to a suspension of sodium hydride (4 g, 99.3 mmol) and DMF (90 mL) at O ° C. After 10 minutes, 1-iodohexane (17.0 mL, 114.6 mmol) was added, and the solution was stirred at room temperature for 18 hours. The reaction was quenched by the addition of water and the products extracted with ethyl acetate. The organic layers were combined, and washed with brine, and dried over MgSO4. The filtered solvents were removed by rotary evaporation to give the title compound as a yellow oil that was used in the next step without further purification.
- 1HNMR (CDCl3, 300 MHz): δ 0.91 (t, 3H, J=7.2 Hz), 1.15-1.53 (m, 6H), 1.23 (d, 12H, J=6.9 Hz), 1.41 (t, 3H, J=7.2 Hz), 1.81 (m, 3H, J=7.2 Hz), 2.89 (m, 1H, J=7.2 Hz), 3.39 (m, 1H, J=7.2 Hz), 3.85 (t, 2H, J=6.9 Hz), 4.39 (q, 2H, J=6.9 Hz), 7.25 (d, 1H, J=2.4 Hz), 7.45 (d, 1H, J=2.4 Hz).
- 2-Hexyloxy-3,5-diisopropylbenzoic Acid (11). A solution of ethyl 2-hydroxy-3,5-diisopropylbenzoate (25.5 g, 76.4 mmol) and ethanol (200 mL) was treated with 2N aqueous NaOH (50 mL, 100 mmol). The solution was stirred at room temperature for 3 days, and acidified with 2N HCl, and the products were extracted with ethyl acetate. The combined organic extracts were washed with brine, and dried over MgSO4. The filtered solvent was concentrated under reduced pressure, and the residue was purified by silica gel chromatography (ethyl acetate:hexane::3:2) to give the title compound as a yellow solid.
- 1HNMR (CDCl3, 300 MHz): δ 0.91 (t, 3H, J=7.2 Hz), 1.18-1.42 (m, 4H), 1.24 (d, 6H, J=6.9 Hz), 1.26 (d, 6H, J=6.9 Hz), 1.48 (m, 2H), 1.88 (m, 3H, J=7.2 Hz), 2.92 (m, 1H, J=6.9 Hz), 3.28 (m, 1H, J=6.9 Hz), 3.92 (t, 2H, J=6.9 Hz), 7.34 (d, 1H, J=2.4 Hz), 7.82 (d, 1H, J=2.4 Hz).
- 2-Hexyloxy-3,5-diisopropylbenzophenone (12). A solution of CH 3Li in ether (1.4 M, 48 mL, 67.2 mmol) was added to a solution of 2-hexyloxy-3,5-diisopropylbenzoic acid (5.2 g, 16.9 mmol) and THF (100 mL) at 0° C. under argon. The solution was stirred at 0° C. for 3.5 hours and treated with trimethylsilyl chloride (50 mL). The resulting cloudy solution was stirred for 20 minutes at room temperature and then the reaction was quenched by the addition of 2N HCl. After stirring the solution for an hour at room temperature, the product was extracted with ethyl acetate three times. The extracts were combined and washed with brine, and dried (MgSO4), and filtered, and the solvents were concentrated in vacuo to give the title compound as a brown oil, which was not further purified.
- 1HNMR (CDCl3, 300 MHz): δ 0.91 (t, 3H, J=6.9 Hz), 1.16-1.42 (m, 4H), 1.24 (d, 12H, J=7.2 Hz), 1.45 (m, 2H), 1.79 (m, 3H, J=7.2 Hz), 2.63 (s, 3H), 2.89 (m, 1H, J=7.2 Hz), 3.35 (m, 1H, J=6.9 Hz), 3.72 (t, 2H, J=7.2 Hz), 7.21 (d, 1H, J=2.1 Hz), 7.24 (d, 1H, J=2.1 Hz).
- 1-Ethynyl-2-hexyloxy-3,5-diisopropylbenzene (13). To a solution of diisopropylamine (3.4 mL, 24.2 mmol) in THF (20 mL) at 0° C. was added a solution of n-BuLi in hexane (1.6 M, 17 mL, 27.4 mmol). The solution was stirred at 0° C. for 30 minutes and then cooled to −78° C. A solution of 2-Hexyloxy-3,5-diisoprpoylbenzophenone (4.9 g, 16.1 mmol) and THF (20 mL) was added, and the solution was stirred for 1 hour at −78° C. Diethyl chlorophosphate (3.1 mL, 20.9 mmol) was added to the solution and the reaction was allowed to slowly warm up to room temperature over three hours. A second solution of lithium diisopropylamine (LDA) was prepared as described above by adding n-BuLi (33 mL, 82.2 mmol) to a solution of diisopropylamine (10.2 mL, 72.6 mmol) and THF (20 mL) at 0° C. and then cooling this solution to −78° C. The first solution was added to the LDA at −78° C., and the resulting solution was allowed to warm to room temperature over three hours. Adding water and ethyl acetate quenched the reaction. The layers were separated and the aqueous layer was extracted two times with ethyl acetate. The combined extracts were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography using hexane and ethyl acetate in a 4:1 ratio to give the title compound as a yellow oil.
- 1HNMR (CDCl3, 300 MHz): δ 0.92 (t, 3H, J=6.9 Hz), 1.13-1.45 (m, 4H), 1.22 (d, 6H, J=7.2 Hz), 1.23 (d, 6H, J=7.2 Hz), 1.50 (m, 2H), 1.82 (m, 3H, J=7.2 Hz), 2.84 (m, 1H, J=6.9 Hz), 3.22 (s, 1H), 3.33 (m, 1H, J=6.9 Hz), 4.03 (t, 2H, J=6.6 Hz), 7.08 (d, 1H, J=2.1 Hz), 7.17 (d, 1H, J=2.1 Hz).
- Cyanatobenzene. A solution of cyanogen bromide (24.3 g, 230 mmol) and water (75 mL) was added to a solution of phenol (20.7 g, 220 mmol) and carbon tetrachloride (75 mL) at 0° C. The solution was treated with triethylamine (31 mL, 220 mmol.) over 30 minutes, and then it was allowed to warm to room temperature and stirred for 18 hours. The mixture was diluted with ethyl acetate, and washed with brine, and dried (MgSO4), and filtered, and the solvent was removed by rotary evaporation. The residue was purified by flash chromatography on silica gel (4:1/hexane:ethyl acetate) to give the title compound as a clear oil.
- 1HNMR (CDCl3, 300 MHz): δ 7.34 (br s, 3H), 7.45 (br s, 2H).
- (2-Hexyloxy-3,5-diisopropylphenyl)propynenitrile (14). A solution of n-BuLi in hexanes (1.6 M, 3.2 mL, 8.06 mmol) was added slowly to a solution of 1-ethynyl-2-hexyloxy-3,5-diisopropylbenzene (2.1 g, 7.33 mmol) and THF (20 mL) at −78° C. The solution was stirred at −78° C. for ten minutes and treated with a solution of cyanatobenzene (1.0 g, 8.06 mmol) and THF (3 mL). The solution was warmed to room temperature over two hours, and then 2N NaOH was added. The products were extracted with ethyl acetate (3×), the organic layers were combined and washed with brine and dried over MgSO 4. The filtered solvents were concentrated under reduced pressure and the residue was purified by flash chromatography (SiO2, 4:1/hexane:ethyl acetate) to give the title compound as a yellow oil.
- 1HNMR (CDCl3, 300 MHz): δ 0.94 (t, 3H, J=6.9 Hz), 1.13-1.40 (m, 4H), 1.22 (d, 6H, J=6.9 Hz), 1.23 (d, 6H, J=6.9 Hz), 1.53 (m, 2H), 1.83 (m, 3H), 2.85 (m, 1H, J=6.9 Hz), 3.30 (m, 1H, J=6.9 Hz), 3.99 (t, 2H, J=6.6 Hz), 7.24 (s, 1H), 7.24 (s, 1H).
- (Z)-3-(2-Hexyloxy-3,5-diisopropylphenyl)but-2-enenitrile (15). A solution of MeLi and ether (1.4 M, 27.5 mL, 38.5 mmol) was added to a stirring solution of copper iodide (3.70 g, 19.2 mL) in THF (50 mL) at 0° C. The solution was cooled to −78° C. and a solution of (2-hexyloxy-3,5-diisopropylohenyl)propynenitrile (2.99 g, 9.62 mmol) in THF (5 mL) was added slowly. The solution was stirred at −78° C. for 1 hour and then quenched by the addition of 15 mL of methanol. The products were extracted with ethyl acetate and saturated aqueous NH 4Cl. The combined organic layers were washed with brine and dried over MgSO4. The filtered solvent was concentrated under reduced pressure and the concentrate was purified by silica gel chromatography using a 95:5 mixture of hexane:ethyl acetate to produce the title compound as a yellow oil.
- 1HNMR (CDCl3, 300 MHz): δ 0.92 (t, 3H, J=6.6 Hz), 1.13-1.40 (m, 4H), 1.24 (d, 6H, J=6.6 Hz), 1.25 (d, 6H, J=6.9 Hz), 1.44 (m, 2H), 1.72 (m, 3H, J=6.9 Hz), 2.30 (d, 3H, J=1.5 Hz), 2.89 (m, 1H, J=6.9 Hz), 3.32 (m, 1H, J=6.9 Hz), 3.70 (t, 2H, J=6.6 Hz), 5.42 (d, 1H, J=1.5 Hz), 6.92 (d, 1H, J=2.1 Hz), 7.12 (d, 1H, J=2.1 Hz).
- (Z)-3-(2-Hexyloxy-3,5-diisopropylphenyl)but-2-enal (16). A solution of DEBAL-H in dichloromethane (1.0 M, 4.50 mL, 4.50 mmol) was added to a solution of (Z)-3-(2-hexyloxy-3,5-diisopropylphenyl)but-2-enenitrile (1.02 g, 3.12 mmol) and hexane (30 mL) at −78° C. The solution was stirred at −78° C. for six hours, and a solution of 20% sodium potassium tartrate was added at −78° C. The solution was warmed to room temperature and the products extracted with ethyl acetate (3×). The combined organic layers were washed with brine, and dried (MgSO 4), and filtered. The solvent was removed by rotary evaporation and the residue purified by flash chromatography on silica gel using a 95:5 mixture of hexane and ethyl acetate to give the title compound as yellow oil.
- 1HNMR (CDCl3, 300 MHz): δ 0.91 (t, 3H, J=6.3 Hz), 1.13-1.40 (m, 4H), 1.23 (d, 6H, J=6.6 Hz), 1.25 (d, 6H, J=6.6 Hz), 1.44 (m, 2H), 1.72 (m, 3H, J=6.9 Hz), 2.30 (s, 3H), 2.89 (m, 1H, J=6.6 Hz), 3.32 (m, 1H, J=6.6 Hz), 3.64 (t, 2H, J=6.6 Hz), 6.21 (d, 1H, J=8.4 Hz), 6.81 (d, 1H, J=2.4 Hz), 7.12 (d, 1H, J=2.4 Hz), 9.44 (d, 1H, J=8.4 Hz).
- Ethyl (2E, 4E, 6Z)-7-(2-Hexyloxy-3,5-diisopropylphenyl)-3-methylocta-2,4,6-trienoate (18). A solution of n-BuLi in hexanes (2.5 M, 6.4 mL, 15.9 mmol) was added to a solution of DMPU (4.0 mL) and Ethyl (E)-4-(diethoxyphosphoryl)-3-methylbut-2-enoatel (4.2 g, 15.9 mmol) and THF (20 mL) at −78° C. After 15 minutes, a solution of (Z)-3-(2-hexyloxy-3,5-diisopropylphenyl)but-2-enal (1.05 g, 3.18 mmol) and THF (5 mL) was added dropwise over 10 to 15 minutes, and the solution was warmed to 0° C. and stirred for one hour. The reaction was quenched at 0° C. by the addition of aqueous NH 4Cl. The products were extracted with ethyl acetate and the combined organic layers were washed with brine and dried over MgSO4. The filtered solvent was concentrated under reduced pressure and the concentrate was purified by silica gel chromatography using a 90:10 mixture of hexane:ethyl acetate to produce the title compound as a yellow oil.
- HNMR (CDCl 3, 300 MHz): δ 0.89 (t, 3H, J=6.6 Hz), 1.13-1.45 (m, 4H), 1.23 (d, 6H, J=6.9 Hz), 1.25 (d, 6H, J=6.9 Hz), 1.28 (t, 3H, J=7.1 Hz), 1.39 (m, 2H), 1.65 (m, 3H, J=6.6 Hz), 2.15 (s, 3H), 2.21 (s, 3H), 2.86 (m, 1H, J=6.9 Hz), 3.34 (m, 1H, J=6.9 Hz), 3.63 (t, 2H, J=6.3 Hz), 4.15 (q, 2H, J=7.2 Hz), 5.74 (s, 1H), 6.21 (d, 1H, J=15.4 Hz), 6.22 (d, 1H, J=10.5 Hz), 6.50 (dd, 1H, J=10.5, 15.4 Hz), 6.75 (d, 1H, J=2.3 Hz), 7.04 (d, 1H, J=2.3 Hz).
- (2E, 4E, 6Z)-7-(2-Hexyloxy-3,5-diisopropylphenyl)-3-methylocta-2,4,6-trienoic Acid (AGN 34). A solution of ethyl (2E, 4E, 6Z)-7-(2-hexyloxy-3,5-diisopropylphenyl)-3-methylocta-2,4,6-trienoate (1.2 g, 2.73 mmol) and ethanol (40 mL) was treated with 2N aqueous NaOH (30 mL, 60 mmol). The solution was stirred at 60° C. for 18 hours, acidified with 2N HCl, and the products were extracted with ethyl acetate. The combined organic extracts were washed with brine, and dried over MgSO 4. The filtered solvent was concentrated under reduced pressure, and the residue was purified by recrystalization from ethanol to give the title compound as a yellow solid.
- 1HNMR (CDCl3, 300 MHz): δ 0.89 (t, 3H, J=6.3 Hz), 1.13-1.45 (m, 4H), 1.22 (d, 6H, J=6.9 Hz), 1.24 (d, 6H, J=6.9 Hz), 1.29 (m, 2H), 1.65 (m, 3H, J=6.3 Hz), 2.15 (s, 3H), 2.21 (s, 3H), 2.86 (m, 1H,J=6.9 Hz), 3.33 (m, 1H,J=6.9 Hz), 3.62 (t, 2H, J=6.3 Hz), 5.75 (s, 1H), 6.23 (d, 1H, J=15.3 Hz), 6.25 (d, 1H, J=10.8 Hz), 6.62 (dd, 1H, J=10.8, 15.3 Hz), 6.74 (d, 1H, J=2.1 Hz), 7.03 (d, 1H,J=2.1 Hz).
- Synthesis of AGN 29
- 4-Bromobenzyl tert-butyldiphenylsilyl ether Tert-butyldiphenylsilyl chloride (10.4 mL, 40.1 mmol) was added to a solution of 4-bromobenzyl alcohol (5.0 g, 26.7 mmol) and 50 mL of dichloromethane. The solution was treated with triethylamine (3.72 mL, 26.7 mmol) and (dimethylamino)pyridine (163 mg, 1.34 mmol) and stirred overnight at room temperature. The solution was diluted with 300 mL of dichloromethane and washed with 50 mL of 10% aqueous HCl. The layers were separated and the aqueous layer was extracted with 50 mL of dichloromethane. The combined organic extracts were washed with brine, and dried (MgSO4), and filtered, and the solvents were removed in vacuo. The residue was filtered through a plug (6@×2@) of silica gel using a solution of 97% hexane/ethyl acetate. After removal of the solvent the residue was heated under vacuum (3 torr) to 170° C. for 1 hour to remove a low-boiling impurity. The remaining material is the title compound.
- PNMR (300 MHz, CDCl 3) δ 1.09 (s, 9H), 4.70 (s, 2H), 7.20 (d, 2H, J=7.9 Hz), 7.35-7.45 (m, 8H), 7.65 (overlapping ds, 4H).
- 4-[(trimethylsilyl)ethynyl]benzyl tert-butyldiphenylsilyl ether
- A 25 mL round bottom flask was flame-dried under high vacuum. The vacuum was broken by the addition of dry argon, and the flask was allowed to cool to room temperature. The flask was charged with 2.0 g (4.70 mmol) of 4-bromobenzyl tert-butyldiphenylsilyl ether (Compound 1), 2.0 mL (14.1 mmol) of (trimethylsilyl)acetylene, and 16.5 mL of triethylamine. The solution was purged with argon for 15 minutes and bis(triphenylphosphine)palladium (II) chloride (83 mg, 0.12 mmol) and copper (I) iodide (22 mg, 0.12 mmol) were added and the solution stirred at ambient temperature for 3 days. The solution was poured into a separatory funnel containing water and ether. The layers were separated and the aqueous layer was extracted 3 times with ether. The combined ether layers were washed once with brine, and dried over magnesium sulfate, and the solvents were removed under reduced pressure. The residue was purified by distillation (bp=180° B 185° C., 1 torr) to give the title compound.
- PNMR (300 MHz, CDCl 3) δ 0.23 (s, 9H), 1.09 (s, 9H), 4.73 (s, 2H), 7.23 (d, 2H, J=7.9 Hz), 7.31-7.45 (m, 8H), 7.65 (overlapping ds, 4H).
- (Z)-4-[2-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-2-(trimethylsilyl)vinyl]benzyl alcohol A 3-
neck 25 mL round bottom flask was fitted with a reflux condenser, and flame-dried under high vacuum. The vacuum was broken by the addition of dry argon (3×), and the flask was allowed to cool to room temperature. The flask was charged with 0.5 mL (1.0 mmol) of borane-methyl sulfide and THF (0.3 mL) and cooled to 0° C. The solution was treated with 0.20 mL (2 mmol) of cyclohexene and stirred at 0° C. for 1 hour. Neat 4-[(trimethylsilyl)ethynyl]benzyl tert-butyldiphenylsilyl ether (443 mg, 1 mmol) was added and, after 15 minutes the solution was warmed to room temperature and stirred for 2.25 hours. In a second flask was prepared a solution of tetrakis(triphenylphosphine)palladium (0) (58 mg, 0.05 mmol) and 2-bromo-3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalene (1.26 g, 4.5 mmol) in 5 mL of THF, which was purged with argon for 10 minutes. The solvents in the first flask were removed under high vacuum, and the residue dissolved in 1 mL of THF and 1 mL of 2 M aqueous NaOH, and the resulting solution was purged with argon for 10 minutes. A 1 mL aliquot of the solution from the second flask was added to the first flask, and the reaction was protected from light and refluxed for 5 hours. The reaction was cooled to room temperature and treated with 2 M NaOH (1 mL) and 30% hydrogen peroxide (0.4 mL). The solution was poured into a separatory funnel containing water and pentane. The layers were separated and the aqueous layer was extracted 3 times with pentane. The combined organic layers were washed once with brine, and dried over magnesium sulfate, and the solvents were removed under reduced pressure. The residue was partially purified by silica gel chromatography (99:1, hexane:ethyl acetate). The later fractions were combined and concentrated under reduced pressure. The residue (203 mg) was dissolved in 3.2 mL of THF and treated with 313 mg of tetrabutylammonium fluoride (Tbaf) adsorbed onto silica gel (1.6 mmol fluoride per gram). The suspension was stirred for 5 hours at room temperature and then the silica gel was washed with ether, and the separated ether extracts were dried over magnesium sulfate. The filtered solvents were removed under reduced pressure and the residue purified by silica gel chromatography (4:1, hexane:ethyl acetate) to give the title compound. - PNMR (300 MHz, CDCl 3)−0.10 (s, 9H), 1.29 (s, 12H), 1.68 (s, 4H), 2.24 (s, 3H), 4.72 (s, 2H), 6.87 (s, 1H), 7.07 (s, 1H), 7.17 (s, 1H), 7.35 (s, 4H).
- Ethyl (Z)-4-[2-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-2-(trimethylsilyl)vinyl]benzoate Manganese dioxide (265 mg, 2.96 mmol) was added to a solution of (Z)-4-[2-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-2-(trimethylsilyl)vinyl]benzyl alcohol (60 mg, 0.15 mmol) and 3.65 mL of hexane. The solution was stirred at room temperature for 16 hours, the manganese dioxide filtered off, and the hexane removed in vacuo. The residue was dissolved in 2 mL of ethanol and treated with sodium cyanide (37.5 mg, 0.77 mmol) and acetic acid (13.7 mg, 0.23 mmol). After 15 minutes, the solution was treated with 265 mg (3.0 mmol) of manganese dioxide. The suspension was stirred at room temperature for 6 hours and the manganese dioxide removed by filtration. The solution was poured into a separatory funnel containing water and ether. The layers were separated and the aqueous layer was extracted 3 times with ether. The combined organic layers were washed once with brine, and dried over magnesium sulfate, and the solvents were removed under reduced pressure. The residue was purified by silica gel chromatograhy (97:3, hexane:ethyl acetate) to give the title compound.
- PNMR (300 MHz, CDCl 3)−0.11 (s, 9H), 1.28 (s, 12H), 1.41 (t, 3H, J=7.1 Hz), 1.68 (s, 4H), 2.23 (s, 3H), 4.39 (q, 2H, J=7.1 Hz), 6.86 (s, 1H), 7.08 (s, 1H), 7.17 (s, 1H), 7.41 (d, 2H, J=8.5 Hz), 8.03 (d, 2H, J=8.5 Hz).
- (Z)-4-[2-(3,5,5,8,8-Pentamethy1-5,6,7,8-tetrahydronaphthalen-2-yl)-2-(trimethylsilyl) vinyl]benzoic Acid (AGN 29) To a solution of ethyl (Z)-4-[2-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-2-(trimethylsilyl)vinyl]benzoate (0.034 g, 0.076 mmol) and 2 mL of ethyl alcohol was added aqueous 1 N KOH (0.5 mL). The resulting solution was heated in an 50° C. bath until the hydrolysis reaction was completed, as judged by thin layer chromatography. The solution was cooled to room temperature, diluted with water and washed once with 1:1 ether:hexane solution, and the layers were separated. The aqueous layer was acidified with 1 N aqueous HCl and the product extracted 3 times with ethyl acetate. The combined organic extracts were washed with brine, and dried over MgSO 4, and filtered, and the solvents were removed in vacuo to give AGN 29 as a white solid.
- PNMR (300 MHz, CDCl 3) δ−0.09 (s, 9H), 1.28 (s, 12H), 1.68 (s, 4H), 2.24 (s, 3H), 6.86 (s, 1H), 7.08 (s, 1H), 7.18 (s, 1H), 7.46 (d, 2H,J=8.1 Hz), 8.11 (d,2H,J=8.1 Hz).
- Further disclosure can be found in U.S. Pat. No. 5,675,033 and in International Patent Application No. WO00/77011, both of which are hereby incorporated by reference herein. The latter publication also discloses the synthesis of AGN 29.
- The examples set forth herein are meant to be illustrative only, and are not intended to limit the scope of the invention, which should be defined solely with reference to the claims that conclude this specification.
-
1 14 1 469 PRT Rattus norvegicus 1 Met Asn Leu Ile Gly Pro Ser His Leu Gln Ala Thr Asp Glu Phe Ala 1 5 10 15 Leu Ser Glu Asn Leu Phe Gly Val Leu Thr Glu His Ala Ala Gly Pro 20 25 30 Leu Gly Gln Asn Leu Asp Leu Glu Ser Tyr Ser Pro Tyr Asn Asn Val 35 40 45 Gln Phe Pro Gln Val Gln Pro Gln Ile Ser Ser Ser Ser Tyr Tyr Ser 50 55 60 Asn Leu Gly Phe Tyr Pro Gln Gln Pro Glu Asp Trp Tyr Ser Pro Gly 65 70 75 80 Leu Tyr Glu Leu Arg Arg Met Pro Thr Glu Ser Val Tyr Gln Gly Glu 85 90 95 Thr Glu Val Ser Glu Met Pro Val Thr Lys Lys Pro Arg Met Ala Ala 100 105 110 Ser Ser Ala Gly Arg Ile Lys Gly Asp Glu Leu Cys Val Val Cys Gly 115 120 125 Asp Arg Ala Ser Gly Tyr His Tyr Asn Ala Leu Thr Cys Glu Gly Cys 130 135 140 Lys Gly Phe Phe Arg Arg Ser Ile Thr Lys Asn Ala Val Tyr Lys Cys 145 150 155 160 Lys Asn Gly Gly Asn Cys Val Met Asp Met Tyr Met Arg Arg Lys Cys 165 170 175 Gln Asp Cys Arg Leu Arg Lys Cys Arg Glu Met Gly Met Leu Ala Glu 180 185 190 Cys Leu Leu Thr Glu Ile Gln Cys Lys Ser Lys Arg Leu Arg Lys Asn 195 200 205 Val Lys Gln His Ala Asp Gln Thr Val Asn Glu Asp Ser Glu Gly Arg 210 215 220 Asp Leu Arg Gln Val Thr Ser Thr Thr Lys Leu Cys Arg Glu Lys Thr 225 230 235 240 Glu Leu Thr Val Asp Gln Gln Thr Leu Leu Asp Tyr Ile Met Asp Ser 245 250 255 Tyr Ser Lys Gln Arg Met Pro Gln Glu Ile Thr Asn Lys Ile Leu Lys 260 265 270 Glu Glu Phe Ser Ala Glu Glu Asn Phe Leu Ile Leu Thr Glu Met Ala 275 280 285 Thr Ser His Val Gln Ile Leu Val Glu Phe Thr Lys Arg Leu Pro Gly 290 295 300 Phe Gln Thr Leu Asp His Glu Asp Gln Ile Ala Leu Leu Lys Gly Ser 305 310 315 320 Ala Val Glu Ala Met Phe Leu Arg Ser Ala Glu Ile Phe Asn Lys Lys 325 330 335 Leu Pro Ala Gly His Ala Asp Leu Leu Glu Glu Arg Ile Arg Lys Ser 340 345 350 Gly Ile Ser Asp Glu Tyr Ile Thr Pro Met Phe Ser Phe Tyr Lys Ser 355 360 365 Val Gly Glu Leu Lys Met Thr Gln Glu Glu Tyr Ala Leu Leu Thr Ala 370 375 380 Ile Val Ile Leu Ser Pro Asp Arg Gln Tyr Ile Lys Asp Arg Glu Ala 385 390 395 400 Val Glu Lys Leu Gln Glu Pro Leu Leu Asp Val Leu Gln Lys Leu Cys 405 410 415 Lys Ile Tyr Gln Pro Glu Asn Pro Gln His Phe Ala Cys Leu Leu Gly 420 425 430 Arg Leu Thr Glu Leu Arg Thr Phe Asn His His His Ala Glu Met Leu 435 440 445 Met Ser Trp Arg Val Asn Asp His Lys Phe Thr Pro Leu Leu Cys Glu 450 455 460 Ile Trp Asp Val Gln 465 2 484 PRT Mus musculus 2 Met Val Met Gln Phe Gln Gly Leu Glu Asn Pro Ile Gln Ile Ser Leu 1 5 10 15 His His Ser His Arg Leu Ser Gly Phe Val Pro Asp Gly Met Ser Val 20 25 30 Lys Pro Ala Lys Gly Met Leu Thr Glu His Ala Ala Gly Pro Leu Gly 35 40 45 Gln Asn Leu Asp Leu Glu Ser Tyr Ser Pro Tyr Asn Asn Val Pro Phe 50 55 60 Pro Gln Val Gln Pro Gln Ile Ser Ser Ser Ser Tyr Tyr Ser Asn Leu 65 70 75 80 Gly Phe Tyr Pro Gln Gln Pro Glu Asp Trp Tyr Ser Pro Gly Ile Tyr 85 90 95 Glu Leu Arg Arg Met Pro Ala Glu Thr Gly Tyr Gln Gly Glu Thr Glu 100 105 110 Val Ser Glu Met Pro Val Thr Lys Lys Pro Arg Met Ala Ala Ala Ser 115 120 125 Ala Gly Arg Ile Lys Gly Asp Glu Leu Cys Val Val Cys Gly Asp Arg 130 135 140 Ala Ser Gly Tyr His Tyr Asn Ala Leu Thr Cys Glu Gly Cys Lys Gly 145 150 155 160 Phe Phe Arg Arg Ser Ile Thr Lys Asn Ala Val Tyr Lys Cys Lys Asn 165 170 175 Gly Gly Asn Cys Val Met Asp Met Tyr Met Arg Arg Lys Cys Gln Glu 180 185 190 Cys Arg Leu Arg Lys Cys Arg Glu Met Gly Met Leu Ala Glu Cys Leu 195 200 205 Leu Thr Glu Ile Gln Cys Lys Ser Lys Arg Leu Arg Lys Asn Val Lys 210 215 220 Gln His Ala Asp Gln Thr Val Asn Glu Asp Asp Ser Glu Gly Arg Asp 225 230 235 240 Leu Arg Gln Val Thr Ser Thr Thr Lys Phe Cys Arg Glu Lys Thr Glu 245 250 255 Leu Thr Ala Asp Gln Gln Thr Leu Leu Asp Tyr Ile Met Asp Ser Tyr 260 265 270 Asn Lys Gln Arg Met Pro Gln Glu Ile Thr Asn Lys Ile Leu Lys Glu 275 280 285 Glu Phe Ser Ala Glu Glu Asn Phe Leu Ile Leu Thr Glu Met Ala Thr 290 295 300 Ser His Val Gln Ile Leu Val Glu Phe Thr Lys Lys Leu Pro Gly Phe 305 310 315 320 Gln Thr Leu Asp His Glu Asp Gln Ile Ala Leu Leu Lys Gly Ser Ala 325 330 335 Val Glu Ala Met Phe Leu Arg Ser Ala Glu Ile Phe Asn Lys Lys Leu 340 345 350 Pro Ala Gly His Ala Asp Leu Leu Glu Glu Arg Ile Arg Lys Ser Gly 355 360 365 Ile Ser Asp Glu Tyr Ile Thr Pro Met Phe Ser Phe Tyr Lys Ser Val 370 375 380 Gly Glu Leu Lys Met Thr Gln Glu Glu Tyr Ala Leu Leu Thr Ala Ile 385 390 395 400 Val Ile Leu Ser Pro Asp Arg Gln Tyr Ile Lys Asp Arg Glu Ala Val 405 410 415 Glu Lys Leu Gln Glu Pro Leu Leu Asp Val Leu Gln Lys Leu Cys Lys 420 425 430 Met Tyr Gln Pro Glu Asn Pro Gln His Phe Ala Cys Leu Leu Gly Arg 435 440 445 Leu Thr Glu Leu Arg Thr Phe Asn His His His Ala Glu Met Leu Met 450 455 460 Ser Trp Arg Val Asn Asp His Lys Phe Thr Pro Leu Leu Cys Glu Ile 465 470 475 480 Trp Asp Val Gln 3 476 PRT Homo Sapiens 3 Pro Arg Thr His Met Gly Ser Lys Met Asn Leu Ile Glu His Ser His 1 5 10 15 Leu Pro Thr Thr Asp Glu Phe Ser Phe Ser Glu Asn Leu Phe Gly Val 20 25 30 Leu Thr Glu Gln Val Ala Gly Pro Leu Gly Gln Asn Leu Glu Val Glu 35 40 45 Pro Tyr Ser Gln Tyr Ser Asn Val Gln Phe Pro Gln Val Gln Pro Gln 50 55 60 Ile Ser Ser Ser Ser Tyr Tyr Ser Asn Leu Gly Phe Tyr Pro Gln Gln 65 70 75 80 Pro Glu Glu Trp Tyr Ser Pro Gly Ile Tyr Glu Leu Arg Arg Met Pro 85 90 95 Ala Glu Thr Leu Tyr Gln Gly Glu Thr Glu Val Ala Glu Met Pro Val 100 105 110 Thr Lys Lys Pro Arg Met Gly Ala Ser Ala Gly Arg Ile Lys Gly Asp 115 120 125 Glu Leu Cys Val Val Cys Gly Asp Arg Ala Ser Gly Tyr His Tyr Asn 130 135 140 Ala Leu Thr Cys Glu Gly Cys Lys Gly Phe Phe Arg Arg Ser Ile Thr 145 150 155 160 Lys Asn Ala Val Tyr Lys Cys Lys Asn Gly Gly Asn Cys Val Met Asp 165 170 175 Met Tyr Met Arg Arg Lys Cys Gln Glu Cys Arg Leu Arg Lys Cys Lys 180 185 190 Glu Met Gly Met Leu Ala Glu Cys Leu Leu Thr Glu Ile Gln Cys Lys 195 200 205 Ser Lys Arg Leu Arg Lys Asn Val Lys Gln His Ala Asp Gln Thr Val 210 215 220 Asn Glu Asp Ser Glu Gly Arg Asp Leu Arg Gln Val Thr Ser Thr Thr 225 230 235 240 Lys Ser Cys Arg Glu Lys Thr Glu Leu Thr Pro Asp Gln Gln Thr Leu 245 250 255 Leu His Phe Ile Met Asp Ser Tyr Asn Lys Gln Arg Met Pro Gln Glu 260 265 270 Ile Thr Asn Lys Ile Leu Lys Glu Glu Phe Ser Ala Glu Glu Asn Phe 275 280 285 Leu Ile Leu Thr Glu Met Ala Thr Asn His Val Gln Val Leu Val Glu 290 295 300 Phe Thr Lys Lys Leu Pro Gly Phe Gln Thr Leu Asp His Glu Asp Gln 305 310 315 320 Ile Ala Leu Leu Lys Gly Ser Ala Val Glu Ala Met Phe Leu Arg Ser 325 330 335 Ala Glu Ile Phe Asn Lys Lys Leu Pro Ser Gly His Ser Asp Leu Leu 340 345 350 Glu Glu Arg Ile Arg Asn Ser Gly Ile Ser Asp Glu Tyr Ile Thr Pro 355 360 365 Met Phe Ser Phe Tyr Lys Ser Ile Gly Glu Leu Lys Met Thr Gln Glu 370 375 380 Glu Tyr Ala Leu Leu Thr Ala Ile Val Ile Leu Ser Pro Asp Arg Gln 385 390 395 400 Tyr Ile Lys Asp Arg Glu Ala Val Glu Lys Leu Gln Glu Pro Leu Leu 405 410 415 Asp Val Leu Gln Lys Leu Cys Lys Ile His Gln Pro Glu Asn Pro Gln 420 425 430 His Phe Ala Cys Leu Leu Gly Arg Leu Thr Glu Leu Arg Thr Phe Asn 435 440 445 His His His Ala Glu Met Leu Met Ser Trp Arg Val Asn Asp His Lys 450 455 460 Phe Thr Pro Leu Leu Cys Glu Ile Trp Asp Val Gln 465 470 475 4 462 PRT Homo Sapiens 4 Met Asp Thr Lys His Phe Leu Pro Leu Asp Phe Ser Thr Gln Val Asn 1 5 10 15 Ser Ser Leu Thr Ser Pro Thr Gly Arg Gly Ser Met Ala Ala Pro Ser 20 25 30 Leu His Pro Ser Leu Gly Pro Gly Ile Gly Ser Pro Gly Gln Leu His 35 40 45 Ser Pro Ile Ser Thr Leu Ser Ser Pro Ile Asn Gly Met Gly Pro Pro 50 55 60 Phe Ser Val Ile Ser Ser Pro Met Gly Pro His Ser Met Ser Val Pro 65 70 75 80 Thr Thr Pro Thr Leu Gly Phe Ser Thr Gly Ser Pro Gln Leu Ser Ser 85 90 95 Pro Met Asn Pro Val Ser Ser Ser Glu Asp Ile Lys Pro Pro Leu Gly 100 105 110 Leu Asn Gly Val Leu Lys Val Pro Ala His Pro Ser Gly Asn Met Ala 115 120 125 Ser Phe Thr Lys His Ile Cys Ala Ile Cys Gly Asp Arg Ser Ser Gly 130 135 140 Lys His Tyr Gly Val Tyr Ser Cys Glu Gly Cys Lys Gly Phe Phe Lys 145 150 155 160 Arg Thr Val Arg Lys Asp Leu Thr Tyr Thr Cys Arg Asp Asn Lys Asp 165 170 175 Cys Leu Ile Asp Lys Arg Gln Arg Asn Arg Cys Gln Tyr Cys Arg Tyr 180 185 190 Gln Lys Cys Leu Ala Met Gly Met Lys Arg Glu Ala Val Gln Glu Glu 195 200 205 Arg Gln Arg Gly Lys Asp Arg Asn Glu Asn Glu Val Glu Ser Thr Ser 210 215 220 Ser Ala Asn Glu Asp Met Pro Val Glu Arg Ile Leu Glu Ala Glu Leu 225 230 235 240 Ala Val Glu Pro Lys Thr Glu Thr Tyr Val Glu Ala Asn Met Gly Leu 245 250 255 Asn Pro Ser Ser Pro Asn Asp Pro Val Thr Asn Ile Cys Gln Ala Ala 260 265 270 Asp Lys Gln Leu Phe Thr Leu Val Glu Trp Ala Lys Arg Ile Pro His 275 280 285 Phe Ser Glu Leu Pro Leu Asp Asp Gln Val Ile Leu Leu Arg Ala Gly 290 295 300 Trp Asn Glu Leu Leu Ile Ala Ser Phe Ser His Arg Ser Ile Ala Val 305 310 315 320 Lys Asp Gly Ile Leu Leu Ala Thr Gly Leu His Val His Arg Asn Ser 325 330 335 Ala His Ser Ala Gly Val Gly Ala Ile Phe Asp Arg Val Leu Thr Glu 340 345 350 Leu Val Ser Lys Met Arg Asp Met Gln Met Asp Lys Thr Glu Leu Gly 355 360 365 Cys Leu Arg Ala Ile Val Leu Phe Asn Pro Asp Ser Lys Gly Leu Ser 370 375 380 Asn Pro Ala Glu Val Glu Ala Leu Arg Glu Lys Val Tyr Ala Ser Leu 385 390 395 400 Glu Ala Tyr Cys Lys His Lys Tyr Pro Glu Gln Pro Gly Arg Phe Ala 405 410 415 Lys Leu Leu Leu Arg Leu Pro Ala Leu Arg Ser Ile Gly Leu Lys Cys 420 425 430 Leu Glu His Leu Phe Phe Phe Lys Leu Ile Gly Asp Thr Pro Ile Asp 435 440 445 Thr Phe Leu Met Glu Met Leu Glu Ala Pro His Gln Met Thr 450 455 460 5 147 PRT S. Cerevisiae GAL4 DNA Binding Domain 5 Met Lys Leu Leu Ser Ser Ile Glu Gln Ala Cys Asp Ile Cys Arg Leu 1 5 10 15 Lys Lys Leu Lys Cys Ser Lys Glu Lys Pro Lys Cys Ala Lys Cys Leu 20 25 30 Lys Asn Asn Trp Glu Cys Arg Tyr Ser Pro Lys Thr Lys Arg Ser Pro 35 40 45 Leu Thr Arg Ala His Leu Thr Glu Val Glu Ser Arg Leu Glu Arg Leu 50 55 60 Glu Gln Leu Phe Leu Leu Ile Phe Pro Arg Glu Asp Leu Asp Met Ile 65 70 75 80 Leu Lys Met Asp Ser Leu Gln Asp Ile Lys Ala Leu Leu Thr Gly Leu 85 90 95 Phe Val Gln Asp Asn Val Asn Lys Asp Ala Val Thr Asp Arg Leu Ala 100 105 110 Ser Val Glu Thr Asp Met Pro Leu Thr Leu Arg Gln His Arg Ile Ser 115 120 125 Ala Thr Ser Ser Ser Glu Glu Ser Ser Asn Lys Gly Gln Arg Gln Leu 130 135 140 Thr Val Ser 145 6 39 DNA Artificial Sequence Linker 6 gtatcgccgg aattcggtac cgtcgaggcc gtgcaggag 39 7 56 DNA Artificial Sequence Linker 7 gtatcgccgg aattcgggct aaggaagtgc agagagatgg gaatgttggc tgaatg 56 8 9 PRT SV 40 Tag 8 Ala Pro Lys Lys Lys Arg Lys Val Gly 1 5 9 360 DNA Artificial Sequence TK-luc promoter 9 ggttttccca gtcacgacgt tgtaaaacga cggccagtgc caagcttgca tgcctgcagg 60 tcgactctag aggatccggc cccgcccagc gtcttgtcat tggcgaattc gaacacgcag 120 atgcagtcgg ggcggcgcgg tcccaggtcc acttcgcata ttaaggtgac gcgtgtggcc 180 tcgaacaccg agcgaccctg cagcgacccg cttaacagcg tcaacagcgt gccgcagatc 240 tctcgagtcc ggtactgttg gtaaaatgga agacgccaaa aacataaaga aaggcccggc 300 gccattctat cctctagagg atggaaccgc tggagagcaa ctgcataagg ctatgaagag 360 10 24 DNA S. Cerevisiae TL-lvc promoter 10 cgacggagta ctgtcctccg agct 24 11 22 DNA Artificial Sequence hsp EcRe 11 tggacaagtg cattgaaccc tt 22 12 26 DNA Artificial Sequence TK-luc reporter 12 gctaccaggt caaaggtcac gtagct 26 13 26 DNA Artificial Sequence TK-luc reporter 13 gctaccaggt caaaggtcac gtagct 26 14 27 DNA Artificial Sequence TIC-lvc repater 14 ccttaaggtg aataaccttg gggctcc 27
Claims (10)
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/104,385 US20030144360A1 (en) | 2001-11-19 | 2002-03-22 | Composition and method for modulating BAR/FXR receptor activity |
| US10/910,688 US20050004165A1 (en) | 2001-11-19 | 2004-08-03 | Composition and method for modulating BAR/FXR receptor activity |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US645001A | 2001-11-19 | 2001-11-19 | |
| US10/104,385 US20030144360A1 (en) | 2001-11-19 | 2002-03-22 | Composition and method for modulating BAR/FXR receptor activity |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US645001A Continuation-In-Part | 2001-11-19 | 2001-11-19 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/910,688 Continuation US20050004165A1 (en) | 2001-11-19 | 2004-08-03 | Composition and method for modulating BAR/FXR receptor activity |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20030144360A1 true US20030144360A1 (en) | 2003-07-31 |
Family
ID=21720945
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/104,385 Abandoned US20030144360A1 (en) | 2001-11-19 | 2002-03-22 | Composition and method for modulating BAR/FXR receptor activity |
| US10/910,688 Abandoned US20050004165A1 (en) | 2001-11-19 | 2004-08-03 | Composition and method for modulating BAR/FXR receptor activity |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/910,688 Abandoned US20050004165A1 (en) | 2001-11-19 | 2004-08-03 | Composition and method for modulating BAR/FXR receptor activity |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US20030144360A1 (en) |
| EP (1) | EP1453496A4 (en) |
| JP (1) | JP2005523241A (en) |
| AU (1) | AU2002366094A1 (en) |
| WO (1) | WO2003043581A2 (en) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1392714B1 (en) | 2001-03-12 | 2005-08-31 | Intercept Pharmaceuticals, Inc. | Steroids as agonists for fxr |
| US9498484B2 (en) | 2004-03-12 | 2016-11-22 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using FXR ligands |
| US10987362B2 (en) | 2004-03-12 | 2021-04-27 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using FXR ligands |
| ITMI20050912A1 (en) | 2005-05-19 | 2006-11-20 | Erregierre Spa | PROCESS OF PREPARATION OF ACIDS 3-A-YA (B) -DIDROSSI-6-A (B) -ALCHIL-5B-COLANICI |
| US8607281B2 (en) * | 2006-09-07 | 2013-12-10 | Porto Vinci Ltd. Limited Liability Company | Control of data presentation in multiple zones using a wireless home entertainment hub |
| MX2009007728A (en) | 2007-01-19 | 2009-12-15 | Intercept Pharmaceuticals Inc | 23-substituted bile acids as tgr5 modulators and methods of use thereof. |
| CA2732323C (en) | 2008-07-30 | 2017-06-27 | Intercept Pharmaceuticals, Inc. | Tgr5 modulators and methods of use thereof |
| JP5535233B2 (en) | 2008-11-19 | 2014-07-02 | インターセプト ファーマシューティカルズ, インコーポレイテッド | TGR5 modulator and method of using the same |
| AU2013277429B2 (en) | 2012-06-19 | 2016-01-14 | Intercept Pharmaceuticals, Inc. | Preparation, uses and solid forms of obeticholic acid |
| US9982008B2 (en) | 2012-06-19 | 2018-05-29 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| IT201600068742A1 (en) * | 2016-07-01 | 2018-01-01 | Bar Pharmaceuticals Soc A Responsabilita Limitata | DERIVATIVES OF IODESEXICOLIC ACID AND THEIR USE |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU774538B2 (en) * | 1999-06-11 | 2004-07-01 | Allergan, Inc. | Organosilyl compounds having nuclear hormone receptor modulating activity |
| CA2377320A1 (en) * | 1999-06-11 | 2000-12-21 | Allergan Sales, Inc. | Methods for modulating fxr receptor activity |
-
2002
- 2002-03-22 US US10/104,385 patent/US20030144360A1/en not_active Abandoned
- 2002-11-19 AU AU2002366094A patent/AU2002366094A1/en not_active Abandoned
- 2002-11-19 EP EP02803683A patent/EP1453496A4/en not_active Withdrawn
- 2002-11-19 JP JP2003545262A patent/JP2005523241A/en active Pending
- 2002-11-19 WO PCT/US2002/037192 patent/WO2003043581A2/en active Application Filing
-
2004
- 2004-08-03 US US10/910,688 patent/US20050004165A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| US20050004165A1 (en) | 2005-01-06 |
| WO2003043581A3 (en) | 2003-12-04 |
| EP1453496A2 (en) | 2004-09-08 |
| AU2002366094A1 (en) | 2003-06-10 |
| AU2002366094A8 (en) | 2003-06-10 |
| WO2003043581A2 (en) | 2003-05-30 |
| EP1453496A4 (en) | 2008-10-08 |
| JP2005523241A (en) | 2005-08-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Forman et al. | Identification of a nuclear receptor that is activated by farnesol metabolites | |
| EP1185277B1 (en) | Use of FXR synthetic ligands for the treatment of diseseas linked to cholesterol imbalance and colon cancer | |
| Khan et al. | Reviews: current topicsrole of nuclear receptors in the regulation of gene expression by dietary fatty acids | |
| Baxter et al. | Selective modulation of thyroid hormone receptor action | |
| US20030144360A1 (en) | Composition and method for modulating BAR/FXR receptor activity | |
| Hashimoto et al. | Nuclear receptor antagonists designed based on the helix-folding inhibition hypothesis | |
| EP0808374B1 (en) | A farnesoid activated receptor (FAR) and method for screening for modulators of processes mediated by the FAR | |
| US8022052B2 (en) | Inhibition of PPAR gamma expression by specific osteogenic oxysterols | |
| Greschik et al. | Structure-activity relationship of nuclear receptor-ligand interactions | |
| US20020132223A1 (en) | Methods for modulating activity of the FXR nuclear receptor | |
| Miyata et al. | Receptor-interacting protein 140 interacts with and inhibits transactivation by, peroxisome proliferator-activated receptor α and liver-X-receptor α | |
| JP2004510682A (en) | Methods for influencing cholesterol catabolism using nuclear bile acid receptors | |
| Kamenecka et al. | Synthetic modulators of the retinoic acid receptor-related orphan receptors | |
| Carlberg | Lipid soluble vitamins in gene regulation | |
| Berkenstam et al. | Nuclear receptors and their relevance to diseases related to lipid metabolism | |
| US6906057B1 (en) | Methods for modulating FXR receptor activity | |
| Traves et al. | Selective activation of liver X receptors by acanthoic acid-related diterpenes | |
| US6893830B1 (en) | Method of screening oxysterol activation of LXRα | |
| JP2002526405A5 (en) | ||
| EP0879223B1 (en) | Aromatic polycyclic retinoid-type derivatives, method for preparing same, and use thereof for making pharmaceutical and cosmetic compositions | |
| Vodička et al. | The effects of RARα and RXRα proteins on growth, viability, and differentiation of v-myb-transformed monoblasts | |
| Cidlowski et al. | CPF Redfern, Department of Endocrinology, Medical Molecular Biology Group, Faculty of Medicine, University of Newcastle upon | |
| Chintharlapalli | New mechanism-based anticancer drugs that act as orphan nuclear receptor agonists | |
| JP2004528316A (en) | Synergistic combinations of retinoid receptor ligands and selected cytotoxic agents for cancer treatment | |
| Yoshihara | Studies of antagonists and selective agonists of the thyroid hormone receptors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: ALLERGAN SALES, INC., CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:BEARD, RICHARD L.;CHANDRARATNA, ROSHANTHA A.;REEL/FRAME:012918/0425 Effective date: 20020416 |
|
| AS | Assignment |
Owner name: ALLERGAN, INC., CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:ALLERGAN SALES, INC. (MERGED INTO ALLERGAN SALES, LLC 6/3/2002);REEL/FRAME:013898/0170 Effective date: 20030331 |
|
| AS | Assignment |
Owner name: CITY OF HOPE, CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:FORMAN, BARRY M.;DUSSAULT, ISABELLE;REEL/FRAME:015564/0718;SIGNING DATES FROM 20040607 TO 20040614 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |