US10280229B2 - Linkers and their application towards ADC - Google Patents
Linkers and their application towards ADC Download PDFInfo
- Publication number
- US10280229B2 US10280229B2 US15/326,386 US201515326386A US10280229B2 US 10280229 B2 US10280229 B2 US 10280229B2 US 201515326386 A US201515326386 A US 201515326386A US 10280229 B2 US10280229 B2 US 10280229B2
- Authority
- US
- United States
- Prior art keywords
- group
- linker
- drug
- antibody
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active, expires
Links
- 239000003814 drug Substances 0.000 claims abstract description 46
- 229940079593 drug Drugs 0.000 claims abstract description 40
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 23
- -1 CD11 Proteins 0.000 claims description 128
- 150000001875 compounds Chemical class 0.000 claims description 69
- 210000004027 cell Anatomy 0.000 claims description 61
- 125000001424 substituent group Chemical group 0.000 claims description 54
- 229910052736 halogen Inorganic materials 0.000 claims description 27
- 229910052739 hydrogen Inorganic materials 0.000 claims description 27
- 150000002367 halogens Chemical group 0.000 claims description 25
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 24
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 23
- 238000000034 method Methods 0.000 claims description 21
- 239000000427 antigen Substances 0.000 claims description 19
- 108091007433 antigens Proteins 0.000 claims description 19
- 102000036639 antigens Human genes 0.000 claims description 19
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 17
- 229910052799 carbon Inorganic materials 0.000 claims description 13
- 125000004432 carbon atom Chemical group C* 0.000 claims description 13
- 229910052717 sulfur Inorganic materials 0.000 claims description 13
- 229940022353 herceptin Drugs 0.000 claims description 12
- 102000006495 integrins Human genes 0.000 claims description 12
- 108010044426 integrins Proteins 0.000 claims description 12
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 claims description 11
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 claims description 11
- 239000002254 cytotoxic agent Substances 0.000 claims description 11
- 102000050554 Eph Family Receptors Human genes 0.000 claims description 10
- 108091008815 Eph receptors Proteins 0.000 claims description 10
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 claims description 10
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 claims description 10
- 229940127089 cytotoxic agent Drugs 0.000 claims description 10
- 229910052760 oxygen Inorganic materials 0.000 claims description 10
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 claims description 9
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 claims description 9
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 claims description 9
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 claims description 9
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 claims description 9
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 claims description 9
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 claims description 9
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 claims description 9
- 102100038080 B-cell receptor CD22 Human genes 0.000 claims description 8
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 claims description 8
- 206010006187 Breast cancer Diseases 0.000 claims description 8
- 208000026310 Breast neoplasm Diseases 0.000 claims description 8
- 101150013553 CD40 gene Proteins 0.000 claims description 8
- 102100032912 CD44 antigen Human genes 0.000 claims description 8
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 claims description 8
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 claims description 8
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 claims description 8
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 claims description 8
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 8
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 8
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 claims description 8
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 claims description 8
- 102000001301 EGF receptor Human genes 0.000 claims description 7
- 108060006698 EGF receptor Proteins 0.000 claims description 7
- 101000874179 Homo sapiens Syndecan-1 Proteins 0.000 claims description 7
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 claims description 7
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 claims description 7
- 102100035721 Syndecan-1 Human genes 0.000 claims description 7
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 claims description 7
- 102100025221 CD70 antigen Human genes 0.000 claims description 6
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 claims description 6
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 6
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 claims description 6
- 101000908391 Homo sapiens Dipeptidyl peptidase 4 Proteins 0.000 claims description 6
- 101000610605 Homo sapiens Tumor necrosis factor receptor superfamily member 10A Proteins 0.000 claims description 6
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 claims description 6
- 102100040113 Tumor necrosis factor receptor superfamily member 10A Human genes 0.000 claims description 6
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 claims description 6
- 229940082789 erbitux Drugs 0.000 claims description 6
- 101150029707 ERBB2 gene Proteins 0.000 claims description 5
- 102100037241 Endoglin Human genes 0.000 claims description 5
- 101000881679 Homo sapiens Endoglin Proteins 0.000 claims description 5
- 101000935040 Homo sapiens Integrin beta-2 Proteins 0.000 claims description 5
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 claims description 5
- 102100025390 Integrin beta-2 Human genes 0.000 claims description 5
- 102000003735 Mesothelin Human genes 0.000 claims description 5
- 108090000015 Mesothelin Proteins 0.000 claims description 5
- 101100369076 Mus musculus Tdgf1 gene Proteins 0.000 claims description 5
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 claims description 5
- 229940122803 Vinca alkaloid Drugs 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 229960005532 CC-1065 Drugs 0.000 claims description 4
- 230000004568 DNA-binding Effects 0.000 claims description 4
- 102000051096 EphA2 Receptor Human genes 0.000 claims description 4
- 108010055196 EphA2 Receptor Proteins 0.000 claims description 4
- 108090001061 Insulin Proteins 0.000 claims description 4
- 241000700605 Viruses Species 0.000 claims description 4
- 108010044540 auristatin Proteins 0.000 claims description 4
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 4
- 201000011510 cancer Diseases 0.000 claims description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 4
- 108010087914 epidermal growth factor receptor VIII Proteins 0.000 claims description 4
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 claims description 4
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 claims description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 4
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 claims description 4
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical compound C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 claims description 4
- 238000003786 synthesis reaction Methods 0.000 claims description 4
- 210000004881 tumor cell Anatomy 0.000 claims description 4
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 3
- 230000015572 biosynthetic process Effects 0.000 claims description 3
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 claims description 3
- 229930195731 calicheamicin Natural products 0.000 claims description 3
- 229960004679 doxorubicin Drugs 0.000 claims description 3
- 102000006815 folate receptor Human genes 0.000 claims description 3
- 108020005243 folate receptor Proteins 0.000 claims description 3
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 claims description 3
- 239000008194 pharmaceutical composition Substances 0.000 claims description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 3
- 229960000575 trastuzumab Drugs 0.000 claims description 3
- DLKUYSQUHXBYPB-NSSHGSRYSA-N (2s,4r)-4-[[2-[(1r,3r)-1-acetyloxy-4-methyl-3-[3-methylbutanoyloxymethyl-[(2s,3s)-3-methyl-2-[[(2r)-1-methylpiperidine-2-carbonyl]amino]pentanoyl]amino]pentyl]-1,3-thiazole-4-carbonyl]amino]-2-methyl-5-(4-methylphenyl)pentanoic acid Chemical compound N([C@@H]([C@@H](C)CC)C(=O)N(COC(=O)CC(C)C)[C@H](C[C@@H](OC(C)=O)C=1SC=C(N=1)C(=O)N[C@H](C[C@H](C)C(O)=O)CC=1C=CC(C)=CC=1)C(C)C)C(=O)[C@H]1CCCCN1C DLKUYSQUHXBYPB-NSSHGSRYSA-N 0.000 claims description 2
- 206010009944 Colon cancer Diseases 0.000 claims description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 2
- 229930188224 Cryptophycin Natural products 0.000 claims description 2
- 102000009465 Growth Factor Receptors Human genes 0.000 claims description 2
- 108010009202 Growth Factor Receptors Proteins 0.000 claims description 2
- 206010033128 Ovarian cancer Diseases 0.000 claims description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 2
- 206010060862 Prostate cancer Diseases 0.000 claims description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 2
- 229910006069 SO3H Inorganic materials 0.000 claims description 2
- 206010041067 Small cell lung cancer Diseases 0.000 claims description 2
- 108020004459 Small interfering RNA Proteins 0.000 claims description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 claims description 2
- 206010057644 Testis cancer Diseases 0.000 claims description 2
- 229960002964 adalimumab Drugs 0.000 claims description 2
- 230000001363 autoimmune Effects 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 2
- 239000003153 chemical reaction reagent Substances 0.000 claims description 2
- 108010006226 cryptophycin Proteins 0.000 claims description 2
- PSNOPSMXOBPNNV-VVCTWANISA-N cryptophycin 1 Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 PSNOPSMXOBPNNV-VVCTWANISA-N 0.000 claims description 2
- PSNOPSMXOBPNNV-UHFFFAOYSA-N cryptophycin-327 Natural products C1=C(Cl)C(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 PSNOPSMXOBPNNV-UHFFFAOYSA-N 0.000 claims description 2
- 238000001514 detection method Methods 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 claims description 2
- 229930188854 dolastatin Natural products 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 229930013356 epothilone Natural products 0.000 claims description 2
- 150000003883 epothilone derivatives Chemical class 0.000 claims description 2
- 206010017758 gastric cancer Diseases 0.000 claims description 2
- 229940125396 insulin Drugs 0.000 claims description 2
- 210000002752 melanocyte Anatomy 0.000 claims description 2
- 244000005700 microbiome Species 0.000 claims description 2
- 210000000066 myeloid cell Anatomy 0.000 claims description 2
- 244000045947 parasite Species 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 125000001476 phosphono group Chemical group [H]OP(*)(=O)O[H] 0.000 claims description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 2
- 229960004641 rituximab Drugs 0.000 claims description 2
- 229950008684 sibrotuzumab Drugs 0.000 claims description 2
- 201000011549 stomach cancer Diseases 0.000 claims description 2
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 claims description 2
- 201000003120 testicular cancer Diseases 0.000 claims description 2
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Substances C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 2
- 229930184737 tubulysin Natural products 0.000 claims description 2
- 102100023915 Insulin Human genes 0.000 claims 1
- 229940049595 antibody-drug conjugate Drugs 0.000 abstract description 34
- 239000000611 antibody drug conjugate Substances 0.000 abstract description 18
- 231100000433 cytotoxic Toxicity 0.000 abstract description 10
- 230000001472 cytotoxic effect Effects 0.000 abstract description 10
- 150000001408 amides Chemical class 0.000 abstract description 5
- 230000002132 lysosomal effect Effects 0.000 abstract description 5
- 230000002255 enzymatic effect Effects 0.000 abstract description 4
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 88
- 239000000203 mixture Substances 0.000 description 88
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 85
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 60
- 239000000243 solution Substances 0.000 description 55
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 53
- 239000000543 intermediate Substances 0.000 description 50
- 235000019439 ethyl acetate Nutrition 0.000 description 41
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 40
- 238000006243 chemical reaction Methods 0.000 description 34
- 0 *C(CCC(C)(C)C)CCC(C)(C)C Chemical compound *C(CCC(C)(C)C)CCC(C)(C)C 0.000 description 32
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 28
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 27
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 27
- 239000007787 solid Substances 0.000 description 25
- 125000003118 aryl group Chemical group 0.000 description 22
- 229910052757 nitrogen Inorganic materials 0.000 description 21
- 125000004429 atom Chemical group 0.000 description 20
- 239000011541 reaction mixture Substances 0.000 description 19
- 125000005842 heteroatom Chemical group 0.000 description 18
- 125000005647 linker group Chemical group 0.000 description 17
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 16
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- 125000001072 heteroaryl group Chemical group 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 101150048775 CDI gene Proteins 0.000 description 12
- 239000007832 Na2SO4 Substances 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 12
- 125000000217 alkyl group Chemical group 0.000 description 12
- 125000000623 heterocyclic group Chemical group 0.000 description 12
- 229910052938 sodium sulfate Inorganic materials 0.000 description 12
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 11
- 239000007858 starting material Substances 0.000 description 11
- 125000004433 nitrogen atom Chemical group N* 0.000 description 10
- LOVPHSMOAVXQIH-UHFFFAOYSA-M (4-nitrophenyl) carbonate Chemical compound [O-]C(=O)OC1=CC=C([N+]([O-])=O)C=C1 LOVPHSMOAVXQIH-UHFFFAOYSA-M 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 108010000499 Thromboplastin Proteins 0.000 description 9
- 102000002262 Thromboplastin Human genes 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000012074 organic phase Substances 0.000 description 9
- 102000004169 proteins and genes Human genes 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- 102000005962 receptors Human genes 0.000 description 9
- 108020003175 receptors Proteins 0.000 description 9
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 9
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 8
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 8
- KJJITHAVZJBOHG-NSHDSACASA-N (2s)-2-amino-5-(carbamoylamino)-n-[4-(hydroxymethyl)phenyl]pentanamide Chemical compound NC(=O)NCCC[C@H](N)C(=O)NC1=CC=C(CO)C=C1 KJJITHAVZJBOHG-NSHDSACASA-N 0.000 description 7
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 7
- 108010016626 Dipeptides Proteins 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 125000000753 cycloalkyl group Chemical group 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 150000003254 radicals Chemical class 0.000 description 7
- 238000010898 silica gel chromatography Methods 0.000 description 7
- JDLQOFHTVZPKMT-JRZJBTRGSA-N tert-butyl (2S)-2-[[7-(2,5-dioxopyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl]amino]-3-methylbutanoate Chemical compound O=C1N(C(C=C1)=O)CCCCCC(C(F)(F)F)N[C@H](C(=O)OC(C)(C)C)C(C)C JDLQOFHTVZPKMT-JRZJBTRGSA-N 0.000 description 7
- OYEGZINXBPXCSU-ABLWVSNPSA-N tert-butyl (2S)-3-methyl-2-[(1,1,1-trifluoro-7-hydroxyheptan-2-yl)amino]butanoate Chemical compound CC([C@@H](C(=O)OC(C)(C)C)NC(C(F)(F)F)CCCCCO)C OYEGZINXBPXCSU-ABLWVSNPSA-N 0.000 description 7
- OTHSYBXEYHBLQT-HNNXBMFYSA-N (2S)-2-amino-N-[4-(hydroxymethyl)phenyl]-5-morpholin-4-ylpentanamide Chemical compound N[C@H](C(=O)NC1=CC=C(C=C1)CO)CCCN1CCOCC1 OTHSYBXEYHBLQT-HNNXBMFYSA-N 0.000 description 6
- KPNSJZHOJZHDKV-DTIOYNMSSA-N (2S)-3-methyl-2-(1,1,1-trifluorooct-7-en-2-ylamino)butanoic acid Chemical compound CC([C@@H](C(=O)O)NC(C(F)(F)F)CCCCC=C)C KPNSJZHOJZHDKV-DTIOYNMSSA-N 0.000 description 6
- RXKWVQORPTZUOI-JHJMLUEUSA-N (2s)-3-methyl-2-(1,1,1-trifluorooct-7-en-2-ylamino)butan-1-ol Chemical compound CC(C)[C@@H](CO)NC(C(F)(F)F)CCCCC=C RXKWVQORPTZUOI-JHJMLUEUSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 239000007821 HATU Substances 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 239000000562 conjugate Substances 0.000 description 6
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 6
- JZMSTPGAQVYVOB-KZUDCZAMSA-N tert-butyl (2S)-3-methyl-2-(1,1,1-trifluorooct-7-en-2-ylamino)butanoate Chemical compound CC([C@@H](C(=O)OC(C)(C)C)NC(C(F)(F)F)CCCCC=C)C JZMSTPGAQVYVOB-KZUDCZAMSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 5
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 5
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 5
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 5
- 102000006992 Interferon-alpha Human genes 0.000 description 5
- 108010047761 Interferon-alpha Proteins 0.000 description 5
- SATWKHAVSXUIOT-DBTMUERSSA-N NC(C[C@@H]1CC[C@H](CC1)CNC(OC(C)(C)C)=O)C(=O)NC1=CC=C(C=C1)CO Chemical compound NC(C[C@@H]1CC[C@H](CC1)CNC(OC(C)(C)C)=O)C(=O)NC1=CC=C(C=C1)CO SATWKHAVSXUIOT-DBTMUERSSA-N 0.000 description 5
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 5
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 5
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 5
- 125000002252 acyl group Chemical group 0.000 description 5
- 125000003545 alkoxy group Chemical group 0.000 description 5
- 229940049706 benzodiazepine Drugs 0.000 description 5
- 150000001721 carbon Chemical group 0.000 description 5
- WGLPBDUCMAPZCE-UHFFFAOYSA-N chromium trioxide Inorganic materials O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 description 5
- 238000010828 elution Methods 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 125000001153 fluoro group Chemical group F* 0.000 description 5
- 239000003102 growth factor Substances 0.000 description 5
- 125000004404 heteroalkyl group Chemical group 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 125000001183 hydrocarbyl group Chemical group 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 238000002953 preparative HPLC Methods 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 125000004434 sulfur atom Chemical group 0.000 description 5
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 5
- AXKGIPZJYUNAIW-UHFFFAOYSA-N (4-aminophenyl)methanol Chemical compound NC1=CC=C(CO)C=C1 AXKGIPZJYUNAIW-UHFFFAOYSA-N 0.000 description 4
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 4
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 4
- 102100027207 CD27 antigen Human genes 0.000 description 4
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 4
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 4
- 108010063738 Interleukins Proteins 0.000 description 4
- 102000015696 Interleukins Human genes 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 4
- 102100029268 Neurotrophin-3 Human genes 0.000 description 4
- 208000008589 Obesity Diseases 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 102100025131 T-cell differentiation antigen CD6 Human genes 0.000 description 4
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 4
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 4
- 229940123237 Taxane Drugs 0.000 description 4
- 125000003342 alkenyl group Chemical group 0.000 description 4
- ACBQROXDOHKANW-UHFFFAOYSA-N bis(4-nitrophenyl) carbonate Chemical compound C1=CC([N+](=O)[O-])=CC=C1OC(=O)OC1=CC=C([N+]([O-])=O)C=C1 ACBQROXDOHKANW-UHFFFAOYSA-N 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 229910052796 boron Inorganic materials 0.000 description 4
- 229910052681 coesite Inorganic materials 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 229910052906 cristobalite Inorganic materials 0.000 description 4
- 231100000599 cytotoxic agent Toxicity 0.000 description 4
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 description 4
- IRXSLJNXXZKURP-UHFFFAOYSA-N fluorenylmethyloxycarbonyl chloride Chemical compound C1=CC=C2C(COC(=O)Cl)C3=CC=CC=C3C2=C1 IRXSLJNXXZKURP-UHFFFAOYSA-N 0.000 description 4
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 239000003018 immunosuppressive agent Substances 0.000 description 4
- 235000020824 obesity Nutrition 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 125000004076 pyridyl group Chemical group 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- 229910052682 stishovite Inorganic materials 0.000 description 4
- 229910052905 tridymite Inorganic materials 0.000 description 4
- 230000004614 tumor growth Effects 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 4
- QZWDMBFGQVGQIA-YFKXAPIDSA-N (2S)-2-(2,5-dioxopyrrolidin-1-yl)-2-[[7-(2,5-dioxopyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl]amino]-3-methylbutanoic acid Chemical compound CC(C)[C@](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)(N1C(=O)CCC1=O)C(O)=O QZWDMBFGQVGQIA-YFKXAPIDSA-N 0.000 description 3
- JLMJFFHXSADZTF-LWOQYNTDSA-N (4s)-4-propan-2-yl-2-(trifluoromethyl)-1,3-oxazolidine Chemical compound CC(C)[C@H]1COC(C(F)(F)F)N1 JLMJFFHXSADZTF-LWOQYNTDSA-N 0.000 description 3
- BGMCTGARFXPQML-NSHDSACASA-N (4s)-5-methoxy-5-oxo-4-(phenylmethoxycarbonylamino)pentanoic acid Chemical compound OC(=O)CC[C@@H](C(=O)OC)NC(=O)OCC1=CC=CC=C1 BGMCTGARFXPQML-NSHDSACASA-N 0.000 description 3
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 3
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 3
- ACUIFAAXWDLLTR-UHFFFAOYSA-N 4-(9h-fluoren-9-ylmethoxycarbonylamino)butanoic acid Chemical compound C1=CC=C2C(COC(=O)NCCCC(=O)O)C3=CC=CC=C3C2=C1 ACUIFAAXWDLLTR-UHFFFAOYSA-N 0.000 description 3
- RIMXEJYJXDBLIE-UHFFFAOYSA-N 6-bromohex-1-ene Chemical compound BrCCCCC=C RIMXEJYJXDBLIE-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 101000898454 Bos taurus Cathepsin B Proteins 0.000 description 3
- XOJAIDZTJPEQEG-UHFFFAOYSA-N CC(C)CC1=CNC2=C1C=CC=C2 Chemical compound CC(C)CC1=CNC2=C1C=CC=C2 XOJAIDZTJPEQEG-UHFFFAOYSA-N 0.000 description 3
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 3
- 229940045513 CTLA4 antagonist Drugs 0.000 description 3
- 108020004414 DNA Proteins 0.000 description 3
- 239000012626 DNA minor groove binder Substances 0.000 description 3
- 239000007818 Grignard reagent Substances 0.000 description 3
- 229910004373 HOAc Inorganic materials 0.000 description 3
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 3
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 3
- 102000004877 Insulin Human genes 0.000 description 3
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 3
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 3
- 108010008212 Integrin alpha4beta1 Proteins 0.000 description 3
- 102000008607 Integrin beta3 Human genes 0.000 description 3
- 108010020950 Integrin beta3 Proteins 0.000 description 3
- 102100023123 Mucin-16 Human genes 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 108010025020 Nerve Growth Factor Proteins 0.000 description 3
- 108090000099 Neurotrophin-4 Proteins 0.000 description 3
- 102100033857 Neurotrophin-4 Human genes 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- 101800004937 Protein C Proteins 0.000 description 3
- 102100029981 Receptor tyrosine-protein kinase erbB-4 Human genes 0.000 description 3
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 3
- 102400000827 Saposin-D Human genes 0.000 description 3
- 101800001700 Saposin-D Proteins 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 3
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 150000001414 amino alcohols Chemical class 0.000 description 3
- 230000001028 anti-proliverative effect Effects 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 230000021615 conjugation Effects 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 125000000392 cycloalkenyl group Chemical group 0.000 description 3
- 230000001085 cytostatic effect Effects 0.000 description 3
- 229960003668 docetaxel Drugs 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 150000004795 grignard reagents Chemical class 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 3
- 229940125721 immunosuppressive agent Drugs 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 3
- 125000003386 piperidinyl group Chemical group 0.000 description 3
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 3
- 229960000856 protein c Drugs 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 239000003053 toxin Substances 0.000 description 3
- 231100000765 toxin Toxicity 0.000 description 3
- 108700012359 toxins Proteins 0.000 description 3
- 238000012447 xenograft mouse model Methods 0.000 description 3
- IXSCJZVRYZGHND-RISCZKNCSA-N (2S)-2-[[(2R)-7-(2,5-dioxopyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl]amino]-3-methylbutanoic acid Chemical compound O=C1N(C(C=C1)=O)CCCCC[C@H](C(F)(F)F)N[C@H](C(=O)O)C(C)C IXSCJZVRYZGHND-RISCZKNCSA-N 0.000 description 2
- IXSCJZVRYZGHND-FZMZJTMJSA-N (2S)-2-[[(2S)-7-(2,5-dioxopyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl]amino]-3-methylbutanoic acid Chemical compound O=C1N(C(C=C1)=O)CCCCC[C@@H](C(F)(F)F)N[C@H](C(=O)O)C(C)C IXSCJZVRYZGHND-FZMZJTMJSA-N 0.000 description 2
- FCWYVKWGLXDXRX-HNNXBMFYSA-N (2s)-5-morpholin-4-yl-2-(phenylmethoxycarbonylamino)pentanoic acid Chemical compound C([C@@H](C(=O)O)NC(=O)OCC=1C=CC=CC=1)CCN1CCOCC1 FCWYVKWGLXDXRX-HNNXBMFYSA-N 0.000 description 2
- XSAKVDNHFRWJKS-IIZANFQQSA-N (2s)-n-benzyl-1-[(2s)-1-[(2s)-2-[[(2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methylbutanoyl]pyrrolidine-2-carbonyl]pyrrolidine-2-carboxamide Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC=2C=CC=CC=2)CCC1 XSAKVDNHFRWJKS-IIZANFQQSA-N 0.000 description 2
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 2
- 125000006716 (C1-C6) heteroalkyl group Chemical group 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- GWTBCUWZAVMAQV-UHFFFAOYSA-N 2,2,2-trifluoro-1-methoxyethanol Chemical compound COC(O)C(F)(F)F GWTBCUWZAVMAQV-UHFFFAOYSA-N 0.000 description 2
- AZEKNJGFCSHZID-UHFFFAOYSA-N 4-[[(2-methylpropan-2-yl)oxycarbonylamino]methyl]cyclohexane-1-carboxylic acid Chemical compound CC(C)(C)OC(=O)NCC1CCC(C(O)=O)CC1 AZEKNJGFCSHZID-UHFFFAOYSA-N 0.000 description 2
- WNJNRUDTKVIPOX-UHFFFAOYSA-N 4-amino-N-[4-(hydroxymethyl)phenyl]butanamide Chemical compound NCCCC(=O)NC1=CC=C(C=C1)CO WNJNRUDTKVIPOX-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- DWALWSXWTBOQJR-UHFFFAOYSA-N 9H-fluoren-9-ylmethyl N-[4-[4-(hydroxymethyl)anilino]-4-oxobutyl]carbamate Chemical compound OCC1=CC=C(C=C1)NC(CCCNC(OCC1C2=CC=CC=C2C=2C=CC=CC1=2)=O)=O DWALWSXWTBOQJR-UHFFFAOYSA-N 0.000 description 2
- KBTCNJJDJZRXCX-VWLOTQADSA-N 9h-fluoren-9-ylmethyl n-[(2s)-5-(carbamoylamino)-1-[4-(hydroxymethyl)anilino]-1-oxopentan-2-yl]carbamate Chemical compound O=C([C@@H](NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21)CCCNC(=O)N)NC1=CC=C(CO)C=C1 KBTCNJJDJZRXCX-VWLOTQADSA-N 0.000 description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 2
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 2
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- PIWJENXGYFEJJM-HZCBDIJESA-N C(C)(C)(C)OC(=O)NC[C@@H]1CC[C@H](CC1)C=C(C(=O)NC1=CC=C(C=C1)CO)NC(OCC1=CC=CC=C1)=O Chemical compound C(C)(C)(C)OC(=O)NC[C@@H]1CC[C@H](CC1)C=C(C(=O)NC1=CC=C(C=C1)CO)NC(OCC1=CC=CC=C1)=O PIWJENXGYFEJJM-HZCBDIJESA-N 0.000 description 2
- MHFJJYBVAYOFIU-QAQDUYKDSA-N C(C1=CC=CC=C1)OC(=O)NC(C(=O)O)=C[C@@H]1CC[C@H](CC1)CNC(=O)OC(C)(C)C Chemical compound C(C1=CC=CC=C1)OC(=O)NC(C(=O)O)=C[C@@H]1CC[C@H](CC1)CNC(=O)OC(C)(C)C MHFJJYBVAYOFIU-QAQDUYKDSA-N 0.000 description 2
- TWBCOLHCSOVDRL-IYARVYRRSA-N C(C1=CC=CC=C1)OC(=O)NC(C(=O)OC)=C[C@@H]1CC[C@H](CC1)CNC(=O)OC(C)(C)C Chemical compound C(C1=CC=CC=C1)OC(=O)NC(C(=O)OC)=C[C@@H]1CC[C@H](CC1)CNC(=O)OC(C)(C)C TWBCOLHCSOVDRL-IYARVYRRSA-N 0.000 description 2
- GUSUWBOOGMZWRB-UHFFFAOYSA-N C=C(N)NCCCC(CC(=O)C(NC(C)C)C(C)C)C(=O)C(C)C.CC(C)NC(C(=O)CC(CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CC(C)NC(C(=O)CC(CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CCC1CCC(CC(CC(=O)C(NC(C)C)C(C)C)C(=O)C(C)C)CC1 Chemical compound C=C(N)NCCCC(CC(=O)C(NC(C)C)C(C)C)C(=O)C(C)C.CC(C)NC(C(=O)CC(CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CC(C)NC(C(=O)CC(CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CCC1CCC(CC(CC(=O)C(NC(C)C)C(C)C)C(=O)C(C)C)CC1 GUSUWBOOGMZWRB-UHFFFAOYSA-N 0.000 description 2
- 102100024217 CAMPATH-1 antigen Human genes 0.000 description 2
- PDGRWLSUPDTTRC-GPIUDJANSA-N CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1 Chemical compound CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1 PDGRWLSUPDTTRC-GPIUDJANSA-N 0.000 description 2
- HTGVXBJENLQEDT-DPDHKHMISA-N CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1 Chemical compound CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1 HTGVXBJENLQEDT-DPDHKHMISA-N 0.000 description 2
- RWFQAHSQDFWMEO-FZSLGHSASA-N CC(C)C(CCCCCN1C(=O)C=CC1=O)C(F)(F)F.CC(C)[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F.C[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F Chemical compound CC(C)C(CCCCCN1C(=O)C=CC1=O)C(F)(F)F.CC(C)[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F.C[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F RWFQAHSQDFWMEO-FZSLGHSASA-N 0.000 description 2
- LYLOAMYTLPELEZ-UHFFFAOYSA-N CC(C)C1CC(=O)N(C(C)C)C1=O.CC(C)CC(=O)NC(C)C Chemical compound CC(C)C1CC(=O)N(C(C)C)C1=O.CC(C)CC(=O)NC(C)C LYLOAMYTLPELEZ-UHFFFAOYSA-N 0.000 description 2
- PMXBNHJHDDPDFT-JMQJBWPXSA-N CC(C)C1CC(=O)N(CCCCCC(C(C)C)C(F)(F)F)C1=O.CC(C)C1CC(=O)N(CCCCC[C@@H](C)C(F)(F)F)C1=O.CC(C)C1CC(=O)N(CCCCC[C@H](C(C)C)C(F)(F)F)C1=O Chemical compound CC(C)C1CC(=O)N(CCCCCC(C(C)C)C(F)(F)F)C1=O.CC(C)C1CC(=O)N(CCCCC[C@@H](C)C(F)(F)F)C1=O.CC(C)C1CC(=O)N(CCCCC[C@H](C(C)C)C(F)(F)F)C1=O PMXBNHJHDDPDFT-JMQJBWPXSA-N 0.000 description 2
- YGHNVLKURXFOCG-UHFFFAOYSA-N CC(C)CCC(CCC(C)C)S(=O)(=O)O.CC(C)CCCC(CC(C)C)S(=O)(=O)O.CC(C)CCCC(CC(C)C)S(=O)(=O)O.CC(C)CCCCCC(C)C.CC(C)CCCOCC(C)C.CC(C)CCOCCC(C)C.CC(C)CCOCCOCC(C)C.CC(C)CCOCCOCCOCCC(C)C.CC(C)CCOCCOCCOCCOCC(C)C.CC(C)COCC(CC(C)C)S(=O)(=O)O Chemical compound CC(C)CCC(CCC(C)C)S(=O)(=O)O.CC(C)CCCC(CC(C)C)S(=O)(=O)O.CC(C)CCCC(CC(C)C)S(=O)(=O)O.CC(C)CCCCCC(C)C.CC(C)CCCOCC(C)C.CC(C)CCOCCC(C)C.CC(C)CCOCCOCC(C)C.CC(C)CCOCCOCCOCCC(C)C.CC(C)CCOCCOCCOCCOCC(C)C.CC(C)COCC(CC(C)C)S(=O)(=O)O YGHNVLKURXFOCG-UHFFFAOYSA-N 0.000 description 2
- ALSRABCNEINFPW-UHFFFAOYSA-N CC(C)CCN1CCCCC1.CC(C)CCN1CCCCC1.CCC1CCC(CC(C)C)CC1 Chemical compound CC(C)CCN1CCCCC1.CC(C)CCN1CCCCC1.CCC1CCC(CC(C)C)CC1 ALSRABCNEINFPW-UHFFFAOYSA-N 0.000 description 2
- IPWZGURYZLSCHV-UHFFFAOYSA-N CC(C)N1C(=O)C=CC1=O.CC(C)NC(=O)CBr.CC(C)NC(=O)CCl Chemical compound CC(C)N1C(=O)C=CC1=O.CC(C)NC(=O)CBr.CC(C)NC(=O)CCl IPWZGURYZLSCHV-UHFFFAOYSA-N 0.000 description 2
- VDOTWAHSNFHMDM-UWBLVGDVSA-N CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)ON1C(=O)CCC1=O Chemical compound CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)ON1C(=O)CCC1=O VDOTWAHSNFHMDM-UWBLVGDVSA-N 0.000 description 2
- QLPBPHKTMVXLHW-YAWDEPMISA-N CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O Chemical compound CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O QLPBPHKTMVXLHW-YAWDEPMISA-N 0.000 description 2
- IETDLYPZCCCAKG-RAGYTEFPSA-N CC1=CC=C(C(=O)CC2=CN3C=C(C(=O)N4C[C@@H](CCl)C5=C4C=C(OC(=O)C(C)C)C4=CC=CC(C)=C45)N=C3C=C2)C=C1.CC[C@H](C(=O)N[C@H](C(=O)N(C)[C@@H]([C@@H](C)CC)[C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)C(C)C)OC)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@@H](CC1=CC=CC=C1)C(=O)O)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)C(C)C)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C)C(C)C.[H][C@]12C=NC3=CC(OCCCOC4=CC5=C(C=C4C)C(=O)N4C=C(C6=CC=C(OC)C=C6)C[C@@]4([H])C=N5)=C(OC)C=C3C(=O)N1C=C(C1=CC=C(CC(C)C)C=C1)C2 Chemical compound CC1=CC=C(C(=O)CC2=CN3C=C(C(=O)N4C[C@@H](CCl)C5=C4C=C(OC(=O)C(C)C)C4=CC=CC(C)=C45)N=C3C=C2)C=C1.CC[C@H](C(=O)N[C@H](C(=O)N(C)[C@@H]([C@@H](C)CC)[C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)C(C)C)OC)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@@H](CC1=CC=CC=C1)C(=O)O)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)C(C)C)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C)C(C)C.[H][C@]12C=NC3=CC(OCCCOC4=CC5=C(C=C4C)C(=O)N4C=C(C6=CC=C(OC)C=C6)C[C@@]4([H])C=N5)=C(OC)C=C3C(=O)N1C=C(C1=CC=C(CC(C)C)C=C1)C2 IETDLYPZCCCAKG-RAGYTEFPSA-N 0.000 description 2
- LQFYMYGJJYWYRV-UHFFFAOYSA-N CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C LQFYMYGJJYWYRV-UHFFFAOYSA-N 0.000 description 2
- JCLLMFLSYSRDLE-UHFFFAOYSA-N CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C JCLLMFLSYSRDLE-UHFFFAOYSA-N 0.000 description 2
- HBAUMKJWLJHORE-UHFFFAOYSA-N CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C HBAUMKJWLJHORE-UHFFFAOYSA-N 0.000 description 2
- HLNJIERKCOGZSG-UHFFFAOYSA-N CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)C(CCCNC(N)=O)NC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)C(CCCNC(N)=O)NC(=O)C(NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C HLNJIERKCOGZSG-UHFFFAOYSA-N 0.000 description 2
- FVGGITXPHUMMMP-UHFFFAOYSA-N CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)C(CCCNC(N)=O)NC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)C(CCCNC(N)=O)NC(=O)C(NC(CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C FVGGITXPHUMMMP-UHFFFAOYSA-N 0.000 description 2
- PHZYUUZQUDALPQ-KUAYEOCFSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C PHZYUUZQUDALPQ-KUAYEOCFSA-N 0.000 description 2
- RDYAVCQSWDHWDP-ZIPSUIFHSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C RDYAVCQSWDHWDP-ZIPSUIFHSA-N 0.000 description 2
- YEKWLBXHFSIXKU-HZWPOWGOSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C YEKWLBXHFSIXKU-HZWPOWGOSA-N 0.000 description 2
- NVGGDOMVQIDFSN-OFBDNCCXSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C NVGGDOMVQIDFSN-OFBDNCCXSA-N 0.000 description 2
- OYJNIXJWWBQZBC-FXXIYWSESA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CC(C)C2=O)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CC(C)C2=O)C(C)C)C=C1)C(C)C OYJNIXJWWBQZBC-FXXIYWSESA-N 0.000 description 2
- LWEQCRVDBJNPHA-YQUPJLNNSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C LWEQCRVDBJNPHA-YQUPJLNNSA-N 0.000 description 2
- BPTLFJZMWLEXDJ-WLVAOYTBSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C BPTLFJZMWLEXDJ-WLVAOYTBSA-N 0.000 description 2
- XPZOLLAYPTTXND-AELOKERVSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C XPZOLLAYPTTXND-AELOKERVSA-N 0.000 description 2
- 102000049320 CD36 Human genes 0.000 description 2
- 108010045374 CD36 Antigens Proteins 0.000 description 2
- 108010065524 CD52 Antigen Proteins 0.000 description 2
- 102100027221 CD81 antigen Human genes 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 2
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 2
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 2
- 102100025680 Complement decay-accelerating factor Human genes 0.000 description 2
- 102100032768 Complement receptor type 2 Human genes 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- 108010049207 Death Domain Receptors Proteins 0.000 description 2
- 102000009058 Death Domain Receptors Human genes 0.000 description 2
- LQKSHSFQQRCAFW-UHFFFAOYSA-N Dolastatin 15 Natural products COC1=CC(=O)N(C(=O)C(OC(=O)C2N(CCC2)C(=O)C2N(CCC2)C(=O)C(C(C)C)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C)C(C)C)C(C)C)C1CC1=CC=CC=C1 LQKSHSFQQRCAFW-UHFFFAOYSA-N 0.000 description 2
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 2
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 description 2
- 101000914479 Homo sapiens CD81 antigen Proteins 0.000 description 2
- 101000856022 Homo sapiens Complement decay-accelerating factor Proteins 0.000 description 2
- 101000941929 Homo sapiens Complement receptor type 2 Proteins 0.000 description 2
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 2
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 2
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 2
- 101000835745 Homo sapiens Teratocarcinoma-derived growth factor 1 Proteins 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 2
- 102000048143 Insulin-Like Growth Factor II Human genes 0.000 description 2
- 108010041012 Integrin alpha4 Proteins 0.000 description 2
- 108090001007 Interleukin-8 Proteins 0.000 description 2
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 2
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- AGDQDCWQBCYNIA-INIZCTEOSA-N NC(=O)NCCC[C@H](N)C(=O)NCCCC(=O)CC1=CC=C(CO)C=C1 Chemical compound NC(=O)NCCC[C@H](N)C(=O)NCCCC(=O)CC1=CC=C(CO)C=C1 AGDQDCWQBCYNIA-INIZCTEOSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 108090000742 Neurotrophin 3 Proteins 0.000 description 2
- WYNCHZVNFNFDNH-UHFFFAOYSA-N Oxazolidine Chemical compound C1COCN1 WYNCHZVNFNFDNH-UHFFFAOYSA-N 0.000 description 2
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 2
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 2
- OWPCHSCAPHNHAV-UHFFFAOYSA-N Rhizoxin Natural products C1C(O)C2(C)OC2C=CC(C)C(OC(=O)C2)CC2CC2OC2C(=O)OC1C(C)C(OC)C(C)=CC=CC(C)=CC1=COC(C)=N1 OWPCHSCAPHNHAV-UHFFFAOYSA-N 0.000 description 2
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 2
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 2
- 102100026404 Teratocarcinoma-derived growth factor 1 Human genes 0.000 description 2
- 102100033571 Tissue-type plasminogen activator Human genes 0.000 description 2
- 108010009583 Transforming Growth Factors Proteins 0.000 description 2
- 102000009618 Transforming Growth Factors Human genes 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 125000006242 amine protecting group Chemical group 0.000 description 2
- 230000000340 anti-metabolite Effects 0.000 description 2
- 229940100197 antimetabolite Drugs 0.000 description 2
- 239000002256 antimetabolite Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 125000005002 aryl methyl group Chemical group 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 125000003310 benzodiazepinyl group Chemical class N1N=C(C=CC2=C1C=CC=C2)* 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 230000027455 binding Effects 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 239000003114 blood coagulation factor Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 229940112869 bone morphogenetic protein Drugs 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 230000022534 cell killing Effects 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 238000012054 celltiter-glo Methods 0.000 description 2
- 108010046713 cemadotin Proteins 0.000 description 2
- 229950009017 cemadotin Drugs 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 229960001338 colchicine Drugs 0.000 description 2
- 229940047120 colony stimulating factors Drugs 0.000 description 2
- GLNDAGDHSLMOKX-UHFFFAOYSA-N coumarin 120 Chemical compound C1=C(N)C=CC2=C1OC(=O)C=C2C GLNDAGDHSLMOKX-UHFFFAOYSA-N 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 239000000824 cytostatic agent Substances 0.000 description 2
- CFCUWKMKBJTWLW-UHFFFAOYSA-N deoliosyl-3C-alpha-L-digitoxosyl-MTM Natural products CC=1C(O)=C2C(O)=C3C(=O)C(OC4OC(C)C(O)C(OC5OC(C)C(O)C(OC6OC(C)C(O)C(C)(O)C6)C5)C4)C(C(OC)C(=O)C(O)C(C)O)CC3=CC2=CC=1OC(OC(C)C1O)CC1OC1CC(O)C(O)C(C)O1 CFCUWKMKBJTWLW-UHFFFAOYSA-N 0.000 description 2
- 229960005501 duocarmycin Drugs 0.000 description 2
- 229930184221 duocarmycin Natural products 0.000 description 2
- XOPYFXBZMVTEJF-UHFFFAOYSA-N eleutherobin Natural products C1=CC2(OC)OC1(C)C(OC(=O)C=CC=1N=CN(C)C=1)CC(C(=CCC1C(C)C)C)C1C=C2COC1OCC(O)C(O)C1OC(C)=O XOPYFXBZMVTEJF-UHFFFAOYSA-N 0.000 description 2
- XOPYFXBZMVTEJF-PDACKIITSA-N eleutherobin Chemical compound C(/[C@H]1[C@H](C(=CC[C@@H]1C(C)C)C)C[C@@H]([C@@]1(C)O[C@@]2(C=C1)OC)OC(=O)\C=C\C=1N=CN(C)C=1)=C2\CO[C@@H]1OC[C@@H](O)[C@@H](O)[C@@H]1OC(C)=O XOPYFXBZMVTEJF-PDACKIITSA-N 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical compound C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 2
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 description 2
- 229960001842 estramustine Drugs 0.000 description 2
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 108010021315 integrin beta7 Proteins 0.000 description 2
- 229940047122 interleukins Drugs 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 125000002346 iodo group Chemical group I* 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- XMYQHJDBLRZMLW-UHFFFAOYSA-N methanolamine Chemical compound NCO XMYQHJDBLRZMLW-UHFFFAOYSA-N 0.000 description 2
- QFHHJHQJESUEML-INIZCTEOSA-N methyl (2S)-5-morpholin-4-yl-2-(phenylmethoxycarbonylamino)pentanoate Chemical compound C(C1=CC=CC=C1)OC(=O)N[C@H](C(=O)OC)CCCN1CCOCC1 QFHHJHQJESUEML-INIZCTEOSA-N 0.000 description 2
- YSRDDGAGFTYXDV-HNNXBMFYSA-N methyl (2S)-5-morpholin-4-yl-5-oxo-2-(phenylmethoxycarbonylamino)pentanoate Chemical compound C(C1=CC=CC=C1)OC(=O)N[C@H](C(=O)OC)CCC(=O)N1CCOCC1 YSRDDGAGFTYXDV-HNNXBMFYSA-N 0.000 description 2
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- IDBIFFKSXLYUOT-UHFFFAOYSA-N netropsin Chemical compound C1=C(C(=O)NCCC(N)=N)N(C)C=C1NC(=O)C1=CC(NC(=O)CN=C(N)N)=CN1C IDBIFFKSXLYUOT-UHFFFAOYSA-N 0.000 description 2
- 239000003900 neurotrophic factor Substances 0.000 description 2
- TWLXDPFBEPBAQB-UHFFFAOYSA-N orthoperiodic acid Chemical compound OI(O)(O)(O)(O)=O TWLXDPFBEPBAQB-UHFFFAOYSA-N 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 150000003057 platinum Chemical class 0.000 description 2
- 229960003171 plicamycin Drugs 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- OWPCHSCAPHNHAV-LMONGJCWSA-N rhizoxin Chemical compound C/C([C@H](OC)[C@@H](C)[C@@H]1C[C@H](O)[C@]2(C)O[C@@H]2/C=C/[C@@H](C)[C@]2([H])OC(=O)C[C@@](C2)(C[C@@H]2O[C@H]2C(=O)O1)[H])=C\C=C\C(\C)=C\C1=COC(C)=N1 OWPCHSCAPHNHAV-LMONGJCWSA-N 0.000 description 2
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 2
- WMOVHXAZOJBABW-UHFFFAOYSA-N tert-butyl acetate Chemical compound CC(=O)OC(C)(C)C WMOVHXAZOJBABW-UHFFFAOYSA-N 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- NYVWCWFKYRHGOP-UHFFFAOYSA-N tert-butyl n-[(4-formylcyclohexyl)methyl]carbamate Chemical compound CC(C)(C)OC(=O)NCC1CCC(C=O)CC1 NYVWCWFKYRHGOP-UHFFFAOYSA-N 0.000 description 2
- KSXPGASLEFGROT-UHFFFAOYSA-N tert-butyl n-[[4-(hydroxymethyl)cyclohexyl]methyl]carbamate Chemical compound CC(C)(C)OC(=O)NCC1CCC(CO)CC1 KSXPGASLEFGROT-UHFFFAOYSA-N 0.000 description 2
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical compound C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 2
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 125000005309 thioalkoxy group Chemical group 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 2
- 229960000303 topotecan Drugs 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 2
- 229960003048 vinblastine Drugs 0.000 description 2
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 2
- 229960002066 vinorelbine Drugs 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 description 1
- LOVPHSMOAVXQIH-UHFFFAOYSA-N (4-nitrophenyl) hydrogen carbonate Chemical compound OC(=O)OC1=CC=C([N+]([O-])=O)C=C1 LOVPHSMOAVXQIH-UHFFFAOYSA-N 0.000 description 1
- INAUWOVKEZHHDM-PEDBPRJASA-N (7s,9s)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-7-[(2r,4s,5s,6s)-5-hydroxy-6-methyl-4-morpholin-4-yloxan-2-yl]oxy-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound Cl.N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1 INAUWOVKEZHHDM-PEDBPRJASA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- STDIPSJCFNTMIJ-YJNPBZNESA-N *.CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C(C)C)C=C1)=C2 Chemical compound *.CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C(C)C)C=C1)=C2 STDIPSJCFNTMIJ-YJNPBZNESA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 1
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 description 1
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000005955 1H-indazolyl group Chemical group 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- YZEUHQHUFTYLPH-UHFFFAOYSA-N 2-nitroimidazole Chemical compound [O-][N+](=O)C1=NC=CN1 YZEUHQHUFTYLPH-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical group OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- YIMDLWDNDGKDTJ-QLKYHASDSA-N 3'-deamino-3'-(3-cyanomorpholin-4-yl)doxorubicin Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1C#N YIMDLWDNDGKDTJ-QLKYHASDSA-N 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-M 4-bromobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-M 0.000 description 1
- SPXOTSHWBDUUMT-UHFFFAOYSA-M 4-nitrobenzenesulfonate Chemical compound [O-][N+](=O)C1=CC=C(S([O-])(=O)=O)C=C1 SPXOTSHWBDUUMT-UHFFFAOYSA-M 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- LGZKGOGODCLQHG-CYBMUJFWSA-N 5-[(2r)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol Chemical compound C1=C(O)C(OC)=CC=C1C[C@@H](O)C1=CC(OC)=C(OC)C(OC)=C1 LGZKGOGODCLQHG-CYBMUJFWSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- FJHBVJOVLFPMQE-QFIPXVFZSA-N 7-Ethyl-10-Hydroxy-Camptothecin Chemical compound C1=C(O)C=C2C(CC)=C(CN3C(C4=C([C@@](C(=O)OC4)(O)CC)C=C33)=O)C3=NC2=C1 FJHBVJOVLFPMQE-QFIPXVFZSA-N 0.000 description 1
- VGGWNGWXGFWLRK-UHFFFAOYSA-N 8,9-dihydro-1H-[1,3]oxazolo[4,5-i][1,2]benzodiazepine Chemical class C1=CC=NNC2=C(OCN3)C3=CC=C21 VGGWNGWXGFWLRK-UHFFFAOYSA-N 0.000 description 1
- 108010059616 Activins Proteins 0.000 description 1
- 102000005606 Activins Human genes 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 101800001288 Atrial natriuretic factor Proteins 0.000 description 1
- 102400001282 Atrial natriuretic peptide Human genes 0.000 description 1
- 101800001890 Atrial natriuretic peptide Proteins 0.000 description 1
- 108020003591 B-Form DNA Proteins 0.000 description 1
- 238000011729 BALB/c nude mouse Methods 0.000 description 1
- 229930190007 Baccatin Natural products 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 1
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 1
- 102000013585 Bombesin Human genes 0.000 description 1
- 108010051479 Bombesin Proteins 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 102100031092 C-C motif chemokine 3 Human genes 0.000 description 1
- 101710155856 C-C motif chemokine 3 Proteins 0.000 description 1
- ADEKGEPIBMAOJD-JXINWJLPSA-N C.C.C=C(N)NCCC[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C.CC(C)N[C@H](C(=O)C[C@@H](CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CC(C)N[C@H](C(=O)C[C@@H](CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CCC1CCC(C[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C)CC1.CC[C@H]1CC[C@H](C[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C)CC1 Chemical compound C.C.C=C(N)NCCC[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C.CC(C)N[C@H](C(=O)C[C@@H](CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CC(C)N[C@H](C(=O)C[C@@H](CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CCC1CCC(C[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C)CC1.CC[C@H]1CC[C@H](C[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C)CC1 ADEKGEPIBMAOJD-JXINWJLPSA-N 0.000 description 1
- DGVHDOAFKYDCJG-UHFFFAOYSA-N C.C.CC(=O)O.CC(=O)O.CN.CN.CN.[H]N(C)C(C)=O.[H]N(C)C(C)=O.[H]N(C)C(C)=O.[H]N(C)C(C)=O Chemical compound C.C.CC(=O)O.CC(=O)O.CN.CN.CN.[H]N(C)C(C)=O.[H]N(C)C(C)=O.[H]N(C)C(C)=O.[H]N(C)C(C)=O DGVHDOAFKYDCJG-UHFFFAOYSA-N 0.000 description 1
- XJIIGJFMURQPSO-PJNBODBESA-N C.C=C(N)NCCC[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C.CC(C)N[C@H](C(=O)C[C@@H](CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CC(C)N[C@H](C(=O)C[C@@H](CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CCC1CCC(C[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C)CC1.CC[C@H]1CC[C@H](C[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C)CC1 Chemical compound C.C=C(N)NCCC[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C.CC(C)N[C@H](C(=O)C[C@@H](CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CC(C)N[C@H](C(=O)C[C@@H](CCCN1CCCCC1)C(=O)C(C)C)C(C)C.CCC1CCC(C[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C)CC1.CC[C@H]1CC[C@H](C[C@H](CC(=O)[C@@H](NC(C)C)C(C)C)C(=O)C(C)C)CC1 XJIIGJFMURQPSO-PJNBODBESA-N 0.000 description 1
- UWKWSTIHBRCYJK-IIBDAURMSA-N C/C(=C/[C@H]1CC[C@H](CNC(=O)OC(C)(C)C)CC1)C(=O)NC1=CC=C(CO)C=C1 Chemical compound C/C(=C/[C@H]1CC[C@H](CNC(=O)OC(C)(C)C)CC1)C(=O)NC1=CC=C(CO)C=C1 UWKWSTIHBRCYJK-IIBDAURMSA-N 0.000 description 1
- HPXOCCTUAZDVSW-RLNRARFGSA-N C/C(=C/[C@H]1CC[C@H](CNC(=O)OC(C)(C)C)CC1)C(=O)O Chemical compound C/C(=C/[C@H]1CC[C@H](CNC(=O)OC(C)(C)C)CC1)C(=O)O HPXOCCTUAZDVSW-RLNRARFGSA-N 0.000 description 1
- KPPOPNJCWGRBGM-UHFFFAOYSA-N CC(C)(C)C(=O)C1CC1.CC(C)(C)C1CC(F)C1.CC(C)(C)C1COC1.CC(C)(C)CC1(C)COC1.CC(C)(C)CC1(N)CC1.CC(C)(C)CC1(O)CC1.CC(C)(C)CC1COC1.CN1CC(CC(C)(C)C)C1.[C-]#[N+]C1(CC(C)(C)C)CC1.[C-]#[N+]C1(CC(C)(C)C)CCC1 Chemical compound CC(C)(C)C(=O)C1CC1.CC(C)(C)C1CC(F)C1.CC(C)(C)C1COC1.CC(C)(C)CC1(C)COC1.CC(C)(C)CC1(N)CC1.CC(C)(C)CC1(O)CC1.CC(C)(C)CC1COC1.CN1CC(CC(C)(C)C)C1.[C-]#[N+]C1(CC(C)(C)C)CC1.[C-]#[N+]C1(CC(C)(C)C)CCC1 KPPOPNJCWGRBGM-UHFFFAOYSA-N 0.000 description 1
- DRBHPGLSJWWAJL-UHFFFAOYSA-N CC(C)(C)C1CCC1.CC(C)(C)CC1(C#N)CCC1.CC(C)(C)CC1(C)CC1.CC(C)(C)CC1(C)CC1.CC(C)(C)CC1CC1.CC(C)(C)CC1CC1(C)F Chemical compound CC(C)(C)C1CCC1.CC(C)(C)CC1(C#N)CCC1.CC(C)(C)CC1(C)CC1.CC(C)(C)CC1(C)CC1.CC(C)(C)CC1CC1.CC(C)(C)CC1CC1(C)F DRBHPGLSJWWAJL-UHFFFAOYSA-N 0.000 description 1
- MTLMIEGABHFUSG-LJFHYQCUSA-N CC(C)(C)OC(=O)NCC1CCC(CC(N)C(=O)NC2=CC=C(CO)C=C2)CC1.CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)ON1C(=O)CCC1=O.CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.CCC1=CC=C(CC(=O)CCCNC(=O)[C@@H](N)CCCNC(N)=O)C=C1.N[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(CO)C=C1 Chemical compound CC(C)(C)OC(=O)NCC1CCC(CC(N)C(=O)NC2=CC=C(CO)C=C2)CC1.CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)ON1C(=O)CCC1=O.CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.CCC1=CC=C(CC(=O)CCCNC(=O)[C@@H](N)CCCNC(N)=O)C=C1.N[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(CO)C=C1 MTLMIEGABHFUSG-LJFHYQCUSA-N 0.000 description 1
- GHNJHQKQRFXFON-UHFFFAOYSA-N CC(C)(C)OC(=O)NCC1CCC(OC=O)CC1 Chemical compound CC(C)(C)OC(=O)NCC1CCC(OC=O)CC1 GHNJHQKQRFXFON-UHFFFAOYSA-N 0.000 description 1
- NYVWCWFKYRHGOP-XYPYZODXSA-N CC(C)(C)OC(=O)NC[C@H]1CC[C@H](C=O)CC1 Chemical compound CC(C)(C)OC(=O)NC[C@H]1CC[C@H](C=O)CC1 NYVWCWFKYRHGOP-XYPYZODXSA-N 0.000 description 1
- GYCKGUXZRWLRDG-WNXZPHCCSA-N CC(C)(C)OC(=O)NC[C@H]1CC[C@H](CC(N)C(=O)NC2=CC=C(CO)C=C2)CC1.CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)ON1C(=O)CCC1=O.CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.NC(=O)NCCC[C@H](N)C(=O)NC1=CC=C(CO)C=C1.N[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(CO)C=C1 Chemical compound CC(C)(C)OC(=O)NC[C@H]1CC[C@H](CC(N)C(=O)NC2=CC=C(CO)C=C2)CC1.CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)ON1C(=O)CCC1=O.CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)O.NC(=O)NCCC[C@H](N)C(=O)NC1=CC=C(CO)C=C1.N[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(CO)C=C1 GYCKGUXZRWLRDG-WNXZPHCCSA-N 0.000 description 1
- KSXPGASLEFGROT-XYPYZODXSA-N CC(C)(C)OC(=O)NC[C@H]1CC[C@H](CO)CC1 Chemical compound CC(C)(C)OC(=O)NC[C@H]1CC[C@H](CO)CC1 KSXPGASLEFGROT-XYPYZODXSA-N 0.000 description 1
- NZIZQHPULWMMST-UHFFFAOYSA-N CC(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1 Chemical compound CC(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1 NZIZQHPULWMMST-UHFFFAOYSA-N 0.000 description 1
- GHXPZQFWMNPTAT-UHFFFAOYSA-N CC(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(NC(CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)CC(CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)NCCCC(=O)C(C)(C)C)C=C1.CC(C)C(NC(CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)NC(CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(COC(=O)C(C)(C)C)C=C1 Chemical compound CC(C)C(=O)OCC1=CC=C(NC(=O)C(CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(NC(CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)CC(CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)NCCCC(=O)C(C)(C)C)C=C1.CC(C)C(NC(CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)NC(CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(COC(=O)C(C)(C)C)C=C1 GHXPZQFWMNPTAT-UHFFFAOYSA-N 0.000 description 1
- UJSIZUOWTLOSEQ-UHFFFAOYSA-N CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1 Chemical compound CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCOCC2)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C=C1 UJSIZUOWTLOSEQ-UHFFFAOYSA-N 0.000 description 1
- BKYPIKHCYIDNPF-HAKDTSIYSA-N CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1.CC(C)C1CC(=O)N(CCCCC[C@H](N[C@H](C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC2=CC=C(COC(=O)NCCCC(=O)C(C)(C)C)C=C2)C(C)C)C(F)(F)F)C1=O.CC(C)C1CC(=O)N(CCCCC[C@H](N[C@H](C(=O)N[C@@H](CCCNC(N)=O)C(=O)NCCCC(=O)CC2=CC=C(COC(=O)C(C)(C)C)C=C2)C(C)C)C(F)(F)F)C1=O Chemical compound CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1.CC(C)C1CC(=O)N(CCCCC[C@H](N[C@H](C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC2=CC=C(COC(=O)NCCCC(=O)C(C)(C)C)C=C2)C(C)C)C(F)(F)F)C1=O.CC(C)C1CC(=O)N(CCCCC[C@H](N[C@H](C(=O)N[C@@H](CCCNC(N)=O)C(=O)NCCCC(=O)CC2=CC=C(COC(=O)C(C)(C)C)C=C2)C(C)C)C(F)(F)F)C1=O BKYPIKHCYIDNPF-HAKDTSIYSA-N 0.000 description 1
- PZBBUVFDYOQRLH-HAKDTSIYSA-N CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1.CC(C)[C@H](N[C@@H](CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)NCCCC(=O)C(C)(C)C)C=C1.CC(C)[C@H](N[C@@H](CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)N[C@@H](CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(COC(=O)C(C)(C)C)C=C1 Chemical compound CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1.CC(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C(C)C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1.CC(C)[C@H](N[C@@H](CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)NCCCC(=O)C(C)(C)C)C=C1.CC(C)[C@H](N[C@@H](CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)N[C@@H](CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(COC(=O)C(C)(C)C)C=C1 PZBBUVFDYOQRLH-HAKDTSIYSA-N 0.000 description 1
- WHSRWVPETGBTQX-UHFFFAOYSA-N CC(C)C(CCCCCN1C(=O)C=CC1=O)C(F)(F)F Chemical compound CC(C)C(CCCCCN1C(=O)C=CC1=O)C(F)(F)F WHSRWVPETGBTQX-UHFFFAOYSA-N 0.000 description 1
- UQCCNZXYJJPSTI-UHFFFAOYSA-N CC(C)C(NC(CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)CC(CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)NCCCC(=O)C(C)(C)C)C=C1.CC(C)C(NC(CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)NC(CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(COC(=O)C(C)(C)C)C=C1 Chemical compound CC(C)C(NC(CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)CC(CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)NCCCC(=O)C(C)(C)C)C=C1.CC(C)C(NC(CCCCCN1C(=O)CC(C(C)(C)C)C1=O)C(F)(F)F)C(=O)NC(CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(COC(=O)C(C)(C)C)C=C1 UQCCNZXYJJPSTI-UHFFFAOYSA-N 0.000 description 1
- WJAKBARXIDBTKE-UHFFFAOYSA-N CC(C)CC1=CC=C(COC(=O)C(C)C)C=C1.CC(C)CC1=CC=C(COC(=O)CCCCC(=O)C(C)C)C=C1.CC(C)CC1=CC=C(COC(=O)CCCNC(=O)C(C)C)C=C1.CC(C)CC1=CC=C(COC(=O)CCCOC(=O)C(C)C)C=C1.CC(C)CC1=CC=C(COC(=O)N(C)C2CCCCC2N(C)C(=O)C(C)C)C=C1.CC(C)CCCCC(=O)NC1=CC=C(COC(=O)C(C)C)C=C1.CC(C)CCCNC(=O)NC1=CC=C(COC(=O)C(C)C)C=C1.CC(C)CCCOC(=O)NC1=CC=C(COC(=O)C(C)C)C=C1 Chemical compound CC(C)CC1=CC=C(COC(=O)C(C)C)C=C1.CC(C)CC1=CC=C(COC(=O)CCCCC(=O)C(C)C)C=C1.CC(C)CC1=CC=C(COC(=O)CCCNC(=O)C(C)C)C=C1.CC(C)CC1=CC=C(COC(=O)CCCOC(=O)C(C)C)C=C1.CC(C)CC1=CC=C(COC(=O)N(C)C2CCCCC2N(C)C(=O)C(C)C)C=C1.CC(C)CCCCC(=O)NC1=CC=C(COC(=O)C(C)C)C=C1.CC(C)CCCNC(=O)NC1=CC=C(COC(=O)C(C)C)C=C1.CC(C)CCCOC(=O)NC1=CC=C(COC(=O)C(C)C)C=C1 WJAKBARXIDBTKE-UHFFFAOYSA-N 0.000 description 1
- AMNGVAZCPQMTNM-UHFFFAOYSA-N CC(C)CCCCC(=O)C(C)C Chemical compound CC(C)CCCCC(=O)C(C)C AMNGVAZCPQMTNM-UHFFFAOYSA-N 0.000 description 1
- ULVSDNXGAXITIX-UHFFFAOYSA-N CC(C)NC1=CC=C(COC(=O)C(C)C)C=C1 Chemical compound CC(C)NC1=CC=C(COC(=O)C(C)C)C=C1 ULVSDNXGAXITIX-UHFFFAOYSA-N 0.000 description 1
- WHSRWVPETGBTQX-LLVKDONJSA-N CC(C)[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F Chemical compound CC(C)[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F WHSRWVPETGBTQX-LLVKDONJSA-N 0.000 description 1
- WHSRWVPETGBTQX-NSHDSACASA-N CC(C)[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F Chemical compound CC(C)[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F WHSRWVPETGBTQX-NSHDSACASA-N 0.000 description 1
- FTFAQAHQHVPAGJ-XIAQZKNVSA-N CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(CO)C=C1 Chemical compound CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(CO)C=C1 FTFAQAHQHVPAGJ-XIAQZKNVSA-N 0.000 description 1
- FEAZXHLFPSGJQX-HJMOHWRCSA-N CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)OC2=CC=C([N+](=O)[O-])C=C2)C=C1 Chemical compound CC(C)[C@H](NC(CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)OC2=CC=C([N+](=O)[O-])C=C2)C=C1 FEAZXHLFPSGJQX-HJMOHWRCSA-N 0.000 description 1
- FTFAQAHQHVPAGJ-QURRFSBHSA-N CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(CO)C=C1 Chemical compound CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(CO)C=C1 FTFAQAHQHVPAGJ-QURRFSBHSA-N 0.000 description 1
- PGZCTDMCPLYKMW-IVYMEZCYSA-N CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)NCCCC(=O)O)C=C1 Chemical compound CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)NCCCC(=O)O)C=C1 PGZCTDMCPLYKMW-IVYMEZCYSA-N 0.000 description 1
- FEAZXHLFPSGJQX-LUGQRUKGSA-N CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)OC2=CC=C([N+](=O)[O-])C=C2)C=C1 Chemical compound CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(COC(=O)OC2=CC=C([N+](=O)[O-])C=C2)C=C1 FEAZXHLFPSGJQX-LUGQRUKGSA-N 0.000 description 1
- HRFKFKDBGPZNAW-VWYPKUQYSA-N CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)N[C@@H](CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(CO)C=C1 Chemical compound CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)N[C@@H](CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(CO)C=C1 HRFKFKDBGPZNAW-VWYPKUQYSA-N 0.000 description 1
- LIBOZZPOMNWDFT-UEMWQVMZSA-N CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)N[C@@H](CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(COC(=O)OC2=CC=C([N+](=O)[O-])C=C2)C=C1 Chemical compound CC(C)[C@H](N[C@@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)N[C@@H](CCCNC(N)=O)C(=O)NCCCC(=O)CC1=CC=C(COC(=O)OC2=CC=C([N+](=O)[O-])C=C2)C=C1 LIBOZZPOMNWDFT-UEMWQVMZSA-N 0.000 description 1
- FUAMXYMWLMQALV-DERQMEARSA-N CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(CO)C=C1 Chemical compound CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(CO)C=C1 FUAMXYMWLMQALV-DERQMEARSA-N 0.000 description 1
- AHBIERROMCDMPG-XMUMXOCRSA-N CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(COC(=O)OC2=CC=C([N+](=O)[O-])C=C2)C=C1 Chemical compound CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(COC(=O)OC2=CC=C([N+](=O)[O-])C=C2)C=C1 AHBIERROMCDMPG-XMUMXOCRSA-N 0.000 description 1
- CUUNGTLOUPVIKJ-JBYYAGLTSA-N CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](C[C@H]1CC[C@H](CNC(=O)OC(C)(C)C)CC1)C(=O)NC1=CC=C(CO)C=C1 Chemical compound CC(C)[C@H](N[C@H](CCCCCN1C(=O)C=CC1=O)C(F)(F)F)C(=O)C[C@@H](C[C@H]1CC[C@H](CNC(=O)OC(C)(C)C)CC1)C(=O)NC1=CC=C(CO)C=C1 CUUNGTLOUPVIKJ-JBYYAGLTSA-N 0.000 description 1
- YSPWTTANRUJKCA-UHFFFAOYSA-N CC.CC.[H]N(C)C(C)=O Chemical compound CC.CC.[H]N(C)C(C)=O YSPWTTANRUJKCA-UHFFFAOYSA-N 0.000 description 1
- XPFQOJFIQFZSAT-LIAQTQRMSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)C(CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C(C)C)CC3CCC(CN)CC3)C=C1)=C2.F[S-11] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)C(CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C(C)C)CC3CCC(CN)CC3)C=C1)=C2.F[S-11] XPFQOJFIQFZSAT-LIAQTQRMSA-N 0.000 description 1
- FGEMNCOISSIRKI-QIJKDHMHSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCN3CCCCC3)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C(C)C)C=C1)=C2.F[S-10] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCN3CCCCC3)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C(C)C)C=C1)=C2.F[S-10] FGEMNCOISSIRKI-QIJKDHMHSA-N 0.000 description 1
- JWXKYJQFCPBZQZ-ZEKLFKHASA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCN3CCOCC3)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C(C)C)C=C1)=C2.F[S-9] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCN3CCOCC3)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C(C)C)C=C1)=C2.F[S-9] JWXKYJQFCPBZQZ-ZEKLFKHASA-N 0.000 description 1
- HPLXPSPBLOVJPB-MUMQCMPJSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCCCC3)C=C1)=C2.F[S-] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCCCC3)C=C1)=C2.F[S-] HPLXPSPBLOVJPB-MUMQCMPJSA-N 0.000 description 1
- HLCYOADMIGYLNR-SYFKLQDCSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCCNC3)C=C1)=C2.F[S-8] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCCNC3)C=C1)=C2.F[S-8] HLCYOADMIGYLNR-SYFKLQDCSA-N 0.000 description 1
- HVHZTEUDWOHMRH-MUMQCMPJSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCN(C)CC3)C=C1)=C2.F[SH-2] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCN(C)CC3)C=C1)=C2.F[SH-2] HVHZTEUDWOHMRH-MUMQCMPJSA-N 0.000 description 1
- JPTFVRYHYFCSNV-KKWHCOSTSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCOCC3)C=C1)=C2.F[SH-4] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCOCC3)C=C1)=C2.F[SH-4] JPTFVRYHYFCSNV-KKWHCOSTSA-N 0.000 description 1
- ZYKMWLMNVMGMAP-KKWHCOSTSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCS(=O)(=O)CC3)C=C1)=C2.F[S-7] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCS(=O)(=O)CC3)C=C1)=C2.F[S-7] ZYKMWLMNVMGMAP-KKWHCOSTSA-N 0.000 description 1
- ULKWXPHCGUGDPO-OYIPSJSMSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCS(=O)CC3)C=C1)=C2.F[S-6] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCS(=O)CC3)C=C1)=C2.F[S-6] ULKWXPHCGUGDPO-OYIPSJSMSA-N 0.000 description 1
- VZMPNZMGWSOXKN-KKWHCOSTSA-N CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCSCC3)C=C1)=C2.F[S-5] Chemical compound CC1=CC(=O)OC2=C1C=CC(NC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(=O)OCC3=CC=CC=C3)C3CCSCC3)C=C1)=C2.F[S-5] VZMPNZMGWSOXKN-KKWHCOSTSA-N 0.000 description 1
- KSYNERBUJUEZJQ-COBSHVIPSA-N CC1N[C@@H](C(C)C)CO1 Chemical compound CC1N[C@@H](C(C)C)CO1 KSYNERBUJUEZJQ-COBSHVIPSA-N 0.000 description 1
- GCJRFROPKVKWMC-UHFFFAOYSA-N CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)C(CCCNC(N)=O)NC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)C(CCCNC(N)=O)NC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C GCJRFROPKVKWMC-UHFFFAOYSA-N 0.000 description 1
- SGFAPLFIGQHDEZ-OTWLTCBHSA-N CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)C(CCCNC(N)=O)NC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)C(CCCNC(N)=O)NC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C SGFAPLFIGQHDEZ-OTWLTCBHSA-N 0.000 description 1
- NVJMAGZWENCLPR-QFFDRPPNSA-N CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CCC(C)C(C(CC(=O)N1CCCC1C(OC)C(C)C(=O)CC(C)C(O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)C(C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCNC(N)=O)CC(=O)C(NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C NVJMAGZWENCLPR-QFFDRPPNSA-N 0.000 description 1
- FUCVMRRWHVXWKU-KRWDZBQOSA-N CCC1=CC=C(CC(=O)CCCNC(=O)[C@@H](N)CCCNC(N)=O)C=C1 Chemical compound CCC1=CC=C(CC(=O)CCCNC(=O)[C@@H](N)CCCNC(N)=O)C=C1 FUCVMRRWHVXWKU-KRWDZBQOSA-N 0.000 description 1
- DMDGMEMOEIMLFH-ZEKLFKHASA-N CCOC(=O)N1CCC([C@H](NC(=O)OCC2=CC=CC=C2)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC2=CC=C(COC(=O)NC3=CC4=C(C=C3)C(C)=CC(=O)O4)C=C2)CC1.F[S-3] Chemical compound CCOC(=O)N1CCC([C@H](NC(=O)OCC2=CC=CC=C2)C(=O)C[C@@H](CCCNC(N)=O)C(=O)NC2=CC=C(COC(=O)NC3=CC4=C(C=C3)C(C)=CC(=O)O4)C=C2)CC1.F[S-3] DMDGMEMOEIMLFH-ZEKLFKHASA-N 0.000 description 1
- ZBGKWDKBIQNDKR-WLLVJXJBSA-N CC[C@H](C(=O)N[C@H](C(=O)N(C)[C@@H]([C@@H](C)CC)[C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)C(C)C)OC)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@@H](CC1=CC=CC=C1)C(=O)O)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)C(C)C)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C)C(C)C Chemical compound CC[C@H](C(=O)N[C@H](C(=O)N(C)[C@@H]([C@@H](C)CC)[C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)C(C)C)OC)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@@H](CC1=CC=CC=C1)C(=O)O)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(C)C)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)C(C)C)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C)C(C)C ZBGKWDKBIQNDKR-WLLVJXJBSA-N 0.000 description 1
- DOPHAXSAJSXJOY-KHKDQRCUSA-N CC[C@H](C)C([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C Chemical compound CC[C@H](C)C([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C DOPHAXSAJSXJOY-KHKDQRCUSA-N 0.000 description 1
- LWEQCRVDBJNPHA-MZTQNJRNSA-N CC[C@H](C)[C@@H](C(CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H](C(CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C LWEQCRVDBJNPHA-MZTQNJRNSA-N 0.000 description 1
- RLHWFFWIJWXIAL-ZAAYVOITSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCCC1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCCC1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCCC1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCCC1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C RLHWFFWIJWXIAL-ZAAYVOITSA-N 0.000 description 1
- ZAOFEOSDCMVJOR-QUBXAXNESA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@@H](NC)C(C)C)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@@H](NC)C(C)C)C(C)C ZAOFEOSDCMVJOR-QUBXAXNESA-N 0.000 description 1
- AEVKMFDUKUAKDO-ZRRDAVEKSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)C(CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C AEVKMFDUKUAKDO-ZRRDAVEKSA-N 0.000 description 1
- NMBFCABPBQCXHG-OHCXCDKVSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C NMBFCABPBQCXHG-OHCXCDKVSA-N 0.000 description 1
- AHQGXNAYJWZJDQ-WTMMDOPPSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C AHQGXNAYJWZJDQ-WTMMDOPPSA-N 0.000 description 1
- MVJWYMWYYUKKHW-QWFPMZNFSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C MVJWYMWYYUKKHW-QWFPMZNFSA-N 0.000 description 1
- RXEFHIGJTYHUHS-XUPUEGPHSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C RXEFHIGJTYHUHS-XUPUEGPHSA-N 0.000 description 1
- QZSLENUWNIRUEA-PYGRVAGMSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C QZSLENUWNIRUEA-PYGRVAGMSA-N 0.000 description 1
- OIUOJABWSJIUID-LYZIKBMQSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C OIUOJABWSJIUID-LYZIKBMQSA-N 0.000 description 1
- ZXJXGXLVFNGBAF-KBDPOKNWSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCOCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C ZXJXGXLVFNGBAF-KBDPOKNWSA-N 0.000 description 1
- TWQQQMRWHONJSO-FMBJEFQJSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)CC2CCC(CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C TWQQQMRWHONJSO-FMBJEFQJSA-N 0.000 description 1
- RXEFHIGJTYHUHS-ZZFVWBEHSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C RXEFHIGJTYHUHS-ZZFVWBEHSA-N 0.000 description 1
- QDMSTBKESQJCKE-KYVLRMTKSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C QDMSTBKESQJCKE-KYVLRMTKSA-N 0.000 description 1
- IOJJCVVITOLEHE-ZDEFELDISA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CNC(=O)OC(C)(C)C)CC2)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CNC(=O)OC(C)(C)C)CC2)C=C1)C(C)C IOJJCVVITOLEHE-ZDEFELDISA-N 0.000 description 1
- MVUJOAKFCYYYTP-PHTFLKHTSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C MVUJOAKFCYYYTP-PHTFLKHTSA-N 0.000 description 1
- DOIPQEQGJZUNFH-NXPCEXBVSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@@H](CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C[C@H]2CC[C@H](CN)CC2)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC([Y])C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C DOIPQEQGJZUNFH-NXPCEXBVSA-N 0.000 description 1
- RIYNWMWSUVLTBV-NGHZOXRBSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCN2CCCCC2)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C RIYNWMWSUVLTBV-NGHZOXRBSA-N 0.000 description 1
- LWEQCRVDBJNPHA-MUAOJIETSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C LWEQCRVDBJNPHA-MUAOJIETSA-N 0.000 description 1
- NVJMAGZWENCLPR-AOPWPWSRSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](NC(CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C NVJMAGZWENCLPR-AOPWPWSRSA-N 0.000 description 1
- UOMLTYOTZMGNLL-VAAUVOJASA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C.CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(CC(=O)CCCNC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)CC(C)C2=O)C(F)(F)F)C(C)C)C=C1)C(C)C UOMLTYOTZMGNLL-VAAUVOJASA-N 0.000 description 1
- LWEQCRVDBJNPHA-KEPIUMRXSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](CC(=O)[C@H](C(C)C)N(C)C(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C LWEQCRVDBJNPHA-KEPIUMRXSA-N 0.000 description 1
- WKCJRKGTKPBAFT-ULCFJEJLSA-N CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)C[C@H](C)[C@@H](O)C1=CC=CC=C1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCNC(=O)OCC1=CC=C(NC(=O)[C@H](CCCNC(N)=O)CC(=O)[C@@H](N[C@@H](CCCCCN2C(=O)C=CC2=O)C(F)(F)F)C(C)C)C=C1)C(C)C WKCJRKGTKPBAFT-ULCFJEJLSA-N 0.000 description 1
- 108010009575 CD55 Antigens Proteins 0.000 description 1
- 102100022002 CD59 glycoprotein Human genes 0.000 description 1
- JYGKUWXCLQOUHS-JUKMJGIZSA-N COC(=O)/C(C)=C\[C@H]1CC[C@H](CNC(=O)OC(C)(C)C)CC1 Chemical compound COC(=O)/C(C)=C\[C@H]1CC[C@H](CNC(=O)OC(C)(C)C)CC1 JYGKUWXCLQOUHS-JUKMJGIZSA-N 0.000 description 1
- IOIXKEMJWWZYIB-MRXNPFEDSA-N COC(=O)[C@H](CCC(=O)N1CCOCC1)CC(=O)OCC1=CC=CC=C1 Chemical compound COC(=O)[C@H](CCC(=O)N1CCOCC1)CC(=O)OCC1=CC=CC=C1 IOIXKEMJWWZYIB-MRXNPFEDSA-N 0.000 description 1
- QBEHOKFAFLFWSX-QGZVFWFLSA-N COC(=O)[C@H](CCCN1CCOCC1)CC(=O)OCC1=CC=CC=C1 Chemical compound COC(=O)[C@H](CCCN1CCOCC1)CC(=O)OCC1=CC=CC=C1 QBEHOKFAFLFWSX-QGZVFWFLSA-N 0.000 description 1
- ZLGFHUMXAFINOD-JTQLQIEISA-N C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(CO)C=C1 Chemical compound C[C@@H](CCCNC(N)=O)C(=O)NC1=CC=C(CO)C=C1 ZLGFHUMXAFINOD-JTQLQIEISA-N 0.000 description 1
- UIDQVOONSAXUAS-YFKPBYRVSA-N C[C@@H](CCCNC(N)=O)C(=O)O Chemical compound C[C@@H](CCCNC(N)=O)C(=O)O UIDQVOONSAXUAS-YFKPBYRVSA-N 0.000 description 1
- 102400000113 Calcitonin Human genes 0.000 description 1
- 108060001064 Calcitonin Proteins 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- 108090000565 Capsid Proteins Proteins 0.000 description 1
- 102100036369 Carbonic anhydrase 6 Human genes 0.000 description 1
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102100022641 Coagulation factor IX Human genes 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 102100021906 Cyclin-O Human genes 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 1
- 239000012624 DNA alkylating agent Substances 0.000 description 1
- 102000016911 Deoxyribonucleases Human genes 0.000 description 1
- 108010053770 Deoxyribonucleases Proteins 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 108090000204 Dipeptidase 1 Proteins 0.000 description 1
- 102100020743 Dipeptidase 1 Human genes 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000003951 Erythropoietin Human genes 0.000 description 1
- 108090000394 Erythropoietin Proteins 0.000 description 1
- 108010076282 Factor IX Proteins 0.000 description 1
- 102100031517 Fc receptor-like protein 1 Human genes 0.000 description 1
- 102100031511 Fc receptor-like protein 2 Human genes 0.000 description 1
- 102100031512 Fc receptor-like protein 3 Human genes 0.000 description 1
- 102100031507 Fc receptor-like protein 5 Human genes 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 1
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 1
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 1
- 102100031706 Fibroblast growth factor 1 Human genes 0.000 description 1
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 description 1
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 description 1
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 description 1
- 102400000321 Glucagon Human genes 0.000 description 1
- 108060003199 Glucagon Proteins 0.000 description 1
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 1
- 102000006771 Gonadotropins Human genes 0.000 description 1
- 108010086677 Gonadotropins Proteins 0.000 description 1
- 108010026389 Gramicidin Proteins 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 235000005612 Grewia tenax Nutrition 0.000 description 1
- 244000041633 Grewia tenax Species 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 239000000095 Growth Hormone-Releasing Hormone Substances 0.000 description 1
- 229910004003 H5IO6 Inorganic materials 0.000 description 1
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 1
- 101000897400 Homo sapiens CD59 glycoprotein Proteins 0.000 description 1
- 101000714525 Homo sapiens Carbonic anhydrase 6 Proteins 0.000 description 1
- 101000914324 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 5 Proteins 0.000 description 1
- 101000914321 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 7 Proteins 0.000 description 1
- 101000897441 Homo sapiens Cyclin-O Proteins 0.000 description 1
- 101000846913 Homo sapiens Fc receptor-like protein 1 Proteins 0.000 description 1
- 101000846911 Homo sapiens Fc receptor-like protein 2 Proteins 0.000 description 1
- 101000846910 Homo sapiens Fc receptor-like protein 3 Proteins 0.000 description 1
- 101000846908 Homo sapiens Fc receptor-like protein 5 Proteins 0.000 description 1
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 1
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 1
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 1
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 1
- 101000878605 Homo sapiens Low affinity immunoglobulin epsilon Fc receptor Proteins 0.000 description 1
- 101000961414 Homo sapiens Membrane cofactor protein Proteins 0.000 description 1
- 101000628547 Homo sapiens Metalloreductase STEAP1 Proteins 0.000 description 1
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 1
- 101000617725 Homo sapiens Pregnancy-specific beta-1-glycoprotein 2 Proteins 0.000 description 1
- 101001136592 Homo sapiens Prostate stem cell antigen Proteins 0.000 description 1
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 101000834948 Homo sapiens Tomoregulin-2 Proteins 0.000 description 1
- 108010000521 Human Growth Hormone Proteins 0.000 description 1
- 102000002265 Human Growth Hormone Human genes 0.000 description 1
- 239000000854 Human Growth Hormone Substances 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 108010041014 Integrin alpha5 Proteins 0.000 description 1
- 102000003996 Interferon-beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 description 1
- 239000003810 Jones reagent Substances 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- KJQFBVYMGADDTQ-CVSPRKDYSA-N L-buthionine-(S,R)-sulfoximine Chemical compound CCCCS(=N)(=O)CC[C@H](N)C(O)=O KJQFBVYMGADDTQ-CVSPRKDYSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 229910010084 LiAlH4 Inorganic materials 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 102100038007 Low affinity immunoglobulin epsilon Fc receptor Human genes 0.000 description 1
- 102000009151 Luteinizing Hormone Human genes 0.000 description 1
- 108010073521 Luteinizing Hormone Proteins 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 108090000542 Lymphotoxin-alpha Proteins 0.000 description 1
- 102000004083 Lymphotoxin-alpha Human genes 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102000009571 Macrophage Inflammatory Proteins Human genes 0.000 description 1
- 108010009474 Macrophage Inflammatory Proteins Proteins 0.000 description 1
- 102100028123 Macrophage colony-stimulating factor 1 Human genes 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930126263 Maytansine Natural products 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102100039373 Membrane cofactor protein Human genes 0.000 description 1
- 102100026712 Metalloreductase STEAP1 Human genes 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 102100030173 Muellerian-inhibiting factor Human genes 0.000 description 1
- 101710122877 Muellerian-inhibiting factor Proteins 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- CJUOJDRNZRZLBO-RUZDIDTESA-N NC(=O)NCCC[C@H](CC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)NCCCC(=O)CC1=CC=C(CO)C=C1 Chemical compound NC(=O)NCCC[C@H](CC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)C(=O)NCCCC(=O)CC1=CC=C(CO)C=C1 CJUOJDRNZRZLBO-RUZDIDTESA-N 0.000 description 1
- RJLCLRBDAHBGAT-UHFFFAOYSA-N NCCCC(=O)CC1=CC=C(CO)C=C1 Chemical compound NCCCC(=O)CC1=CC=C(CO)C=C1 RJLCLRBDAHBGAT-UHFFFAOYSA-N 0.000 description 1
- 238000011789 NOD SCID mouse Methods 0.000 description 1
- 108090000028 Neprilysin Proteins 0.000 description 1
- 102000003729 Neprilysin Human genes 0.000 description 1
- 102000015336 Nerve Growth Factor Human genes 0.000 description 1
- 108010042309 Netropsin Proteins 0.000 description 1
- 108090000095 Neurotrophin-6 Proteins 0.000 description 1
- KYRVNWMVYQXFEU-UHFFFAOYSA-N Nocodazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CS1 KYRVNWMVYQXFEU-UHFFFAOYSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- NALXOTGZAZGRJX-UHFFFAOYSA-N O=C(CCCNC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)CC1=CC=C(CO)C=C1 Chemical compound O=C(CCCNC(=O)OCC1C2=C(C=CC=C2)C2=C1C=CC=C2)CC1=CC=C(CO)C=C1 NALXOTGZAZGRJX-UHFFFAOYSA-N 0.000 description 1
- ARNBRXPZSHCIPC-JOCHJYFZSA-N O=C(C[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(CO)C=C1)OCC1=CC=CC=C1 Chemical compound O=C(C[C@@H](CCCN1CCOCC1)C(=O)NC1=CC=C(CO)C=C1)OCC1=CC=CC=C1 ARNBRXPZSHCIPC-JOCHJYFZSA-N 0.000 description 1
- DNFYZSZOVQCJDK-MRXNPFEDSA-N O=C(C[C@@H](CCCN1CCOCC1)C(=O)O)OCC1=CC=CC=C1 Chemical compound O=C(C[C@@H](CCCN1CCOCC1)C(=O)O)OCC1=CC=CC=C1 DNFYZSZOVQCJDK-MRXNPFEDSA-N 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 101100272976 Panax ginseng CYP716A53v2 gene Proteins 0.000 description 1
- 102000003982 Parathyroid hormone Human genes 0.000 description 1
- 108090000445 Parathyroid hormone Proteins 0.000 description 1
- 102000001938 Plasminogen Activators Human genes 0.000 description 1
- 108010001014 Plasminogen Activators Proteins 0.000 description 1
- 108010076181 Proinsulin Proteins 0.000 description 1
- 102100036735 Prostate stem cell antigen Human genes 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- 102400000834 Relaxin A chain Human genes 0.000 description 1
- 101800000074 Relaxin A chain Proteins 0.000 description 1
- 102400000610 Relaxin B chain Human genes 0.000 description 1
- 101710109558 Relaxin B chain Proteins 0.000 description 1
- 102100028255 Renin Human genes 0.000 description 1
- 108090000783 Renin Proteins 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010029180 Sialic Acid Binding Ig-like Lectin 3 Proteins 0.000 description 1
- 102000001555 Sialic Acid Binding Ig-like Lectin 3 Human genes 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 102100022831 Somatoliberin Human genes 0.000 description 1
- 101710142969 Somatoliberin Proteins 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 1
- 102000019197 Superoxide Dismutase Human genes 0.000 description 1
- 108010012715 Superoxide dismutase Proteins 0.000 description 1
- 108091008874 T cell receptors Proteins 0.000 description 1
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 102000011923 Thyrotropin Human genes 0.000 description 1
- 108010061174 Thyrotropin Proteins 0.000 description 1
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 1
- 108050006955 Tissue-type plasminogen activator Proteins 0.000 description 1
- 102100026160 Tomoregulin-2 Human genes 0.000 description 1
- GYDJEQRTZSCIOI-UHFFFAOYSA-N Tranexamic acid Chemical compound NCC1CCC(C(O)=O)CC1 GYDJEQRTZSCIOI-UHFFFAOYSA-N 0.000 description 1
- 102000011117 Transforming Growth Factor beta2 Human genes 0.000 description 1
- 102400001320 Transforming growth factor alpha Human genes 0.000 description 1
- 101800004564 Transforming growth factor alpha Proteins 0.000 description 1
- 101800000304 Transforming growth factor beta-2 Proteins 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- GBOGMAARMMDZGR-UHFFFAOYSA-N UNPD149280 Natural products N1C(=O)C23OC(=O)C=CC(O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 GBOGMAARMMDZGR-UHFFFAOYSA-N 0.000 description 1
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 1
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 241000863480 Vinca Species 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- BBAWTPDTGRXPDG-UHFFFAOYSA-N [1,3]thiazolo[4,5-b]pyridine Chemical compound C1=CC=C2SC=NC2=N1 BBAWTPDTGRXPDG-UHFFFAOYSA-N 0.000 description 1
- LFQRCQSVBIJJTC-UHFFFAOYSA-M [Br-].[Mg+]CCCCC=C Chemical compound [Br-].[Mg+]CCCCC=C LFQRCQSVBIJJTC-UHFFFAOYSA-M 0.000 description 1
- CKUAXEQHGKSLHN-UHFFFAOYSA-N [C].[N] Chemical group [C].[N] CKUAXEQHGKSLHN-UHFFFAOYSA-N 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 229930183665 actinomycin Natural products 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000000488 activin Substances 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 description 1
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 description 1
- 229940024142 alpha 1-antitrypsin Drugs 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001455 anti-clotting effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000005101 aryl methoxy carbonyl group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004935 benzoxazolinyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000005512 benztetrazolyl group Chemical group 0.000 description 1
- MNVHAXXXJWOVLK-QFIPXVFZSA-N benzyl N-[(2S)-1-[4-(hydroxymethyl)anilino]-5-morpholin-4-yl-1-oxopentan-2-yl]carbamate Chemical compound OCC1=CC=C(C=C1)NC([C@H](CCCN1CCOCC1)NC(OCC1=CC=CC=C1)=O)=O MNVHAXXXJWOVLK-QFIPXVFZSA-N 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- DNDCVAGJPBKION-DOPDSADYSA-N bombesin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC=1NC2=CC=CC=C2C=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1NC(=O)CC1)C(C)C)C1=CN=CN1 DNDCVAGJPBKION-DOPDSADYSA-N 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- 108010006025 bovine growth hormone Proteins 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 229960004015 calcitonin Drugs 0.000 description 1
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000004623 carbolinyl group Chemical group 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- NSQLIUXCMFBZME-MPVJKSABSA-N carperitide Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(O)=O)=O)[C@@H](C)CC)C1=CC=CC=C1 NSQLIUXCMFBZME-MPVJKSABSA-N 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- NDAYQJDHGXTBJL-MWWSRJDJSA-N chembl557217 Chemical compound C1=CC=C2C(C[C@H](NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](C(C)C)NC(=O)[C@H](C)NC(=O)[C@H](NC(=O)CNC(=O)[C@@H](NC=O)C(C)C)CC(C)C)C(=O)NCCO)=CNC2=C1 NDAYQJDHGXTBJL-MWWSRJDJSA-N 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 229940117975 chromium trioxide Drugs 0.000 description 1
- GAMDZJFZMJECOS-UHFFFAOYSA-N chromium(6+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Cr+6] GAMDZJFZMJECOS-UHFFFAOYSA-N 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- LGZKGOGODCLQHG-UHFFFAOYSA-N combretastatin Natural products C1=C(O)C(OC)=CC=C1CC(O)C1=CC(OC)=C(OC)C(OC)=C1 LGZKGOGODCLQHG-UHFFFAOYSA-N 0.000 description 1
- 150000004814 combretastatins Chemical class 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- GBOGMAARMMDZGR-JREHFAHYSA-N cytochalasin B Natural products C[C@H]1CCC[C@@H](O)C=CC(=O)O[C@@]23[C@H](C=CC1)[C@H](O)C(=C)[C@@H](C)[C@@H]2[C@H](Cc4ccccc4)NC3=O GBOGMAARMMDZGR-JREHFAHYSA-N 0.000 description 1
- GBOGMAARMMDZGR-TYHYBEHESA-N cytochalasin B Chemical compound C([C@H]1[C@@H]2[C@@H](C([C@@H](O)[C@@H]3/C=C/C[C@H](C)CCC[C@@H](O)/C=C/C(=O)O[C@@]23C(=O)N1)=C)C)C1=CC=CC=C1 GBOGMAARMMDZGR-TYHYBEHESA-N 0.000 description 1
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 229940105423 erythropoietin Drugs 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229960004222 factor ix Drugs 0.000 description 1
- 229940126864 fibroblast growth factor Drugs 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229940028334 follicle stimulating hormone Drugs 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 239000002622 gonadotropin Substances 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 150000002373 hemiacetals Chemical class 0.000 description 1
- 230000002607 hemopoietic effect Effects 0.000 description 1
- 125000004474 heteroalkylene group Chemical group 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 229940124589 immunosuppressive drug Drugs 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000004926 indolenyl group Chemical group 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000000893 inhibin Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 1
- 102000028416 insulin-like growth factor binding Human genes 0.000 description 1
- 108091022911 insulin-like growth factor binding Proteins 0.000 description 1
- 108010021518 integrin beta5 Proteins 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 239000002555 ionophore Substances 0.000 description 1
- 230000000236 ionophoric effect Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000004936 isatinoyl group Chemical group N1(C(=O)C(=O)C2=CC=CC=C12)C(=O)* 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000005438 isoindazolyl group Chemical group 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- NWYYWIJOWOLJNR-RXMQYKEDSA-N l-valinol Chemical compound CC(C)[C@H](N)CO NWYYWIJOWOLJNR-RXMQYKEDSA-N 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- 229940066294 lung surfactant Drugs 0.000 description 1
- 239000003580 lung surfactant Substances 0.000 description 1
- 229940040129 luteinizing hormone Drugs 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical group CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- GSYSFVSGPABNNL-UHFFFAOYSA-N methyl 2-dimethoxyphosphoryl-2-(phenylmethoxycarbonylamino)acetate Chemical compound COC(=O)C(P(=O)(OC)OC)NC(=O)OCC1=CC=CC=C1 GSYSFVSGPABNNL-UHFFFAOYSA-N 0.000 description 1
- 125000006216 methylsulfinyl group Chemical group [H]C([H])([H])S(*)=O 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000006682 monohaloalkyl group Chemical group 0.000 description 1
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 230000007524 negative regulation of DNA replication Effects 0.000 description 1
- 229940053128 nerve growth factor Drugs 0.000 description 1
- 230000000955 neuroendocrine Effects 0.000 description 1
- 229940032018 neurotrophin 3 Drugs 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 229950006344 nocodazole Drugs 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 125000004930 octahydroisoquinolinyl group Chemical group C1(NCCC2CCCC=C12)* 0.000 description 1
- 230000002138 osteoinductive effect Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- QNNHQVPFZIFNFK-UHFFFAOYSA-N oxazolo[4,5-b]pyridine Chemical compound C1=CC=C2OC=NC2=N1 QNNHQVPFZIFNFK-UHFFFAOYSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000004095 oxindolyl group Chemical group N1(C(CC2=CC=CC=C12)=O)* 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- CWODDUGJZSCNGB-HQNRRURTSA-N palytoxin Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CCCCC[C@H](C)C[C@@H]2[C@@]3(C)C[C@H](C)C[C@@](O3)(CCCCCCC[C@H](O)C[C@@H]3[C@@H]([C@@H](O)[C@@H](O)[C@](O)(C[C@H](O)[C@@H](C)\C=C\[C@@H](O)CC[C@@H](O)[C@@H](O)[C@H]4O[C@H](C[C@@H](O)[C@H](O)C[C@@H]5[C@H]([C@H](O)[C@@H](O)[C@H](C[C@H](O)\C=C/C=C/C[C@@H](O)[C@H](O)[C@H](O)C\C=C/C(=C)CC[C@H](O)[C@@H](O)[C@H](O)[C@H](C)C[C@@H]6[C@@H]([C@@H](O)[C@H](O)[C@@H](\C=C/[C@@H](O)[C@H](O)C[C@H]7O[C@H]8C[C@H](O[C@@H]8CC[C@@H]8[C@@H](C[C@@H](CN)O8)O)C7)O6)O)O5)O)[C@@H](O)[C@H](O)C4)O3)O)O2)[C@H](C[C@H](O)[C@H](O)C(\C)=C\[C@H](O)C[C@@H](C)[C@H](O)C(=O)N\C=C\C(=O)NCCCO)[C@H](O)[C@@H](O)[C@@H]1O CWODDUGJZSCNGB-HQNRRURTSA-N 0.000 description 1
- 229960005548 palytoxin Drugs 0.000 description 1
- 239000000199 parathyroid hormone Substances 0.000 description 1
- 229960001319 parathyroid hormone Drugs 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000004932 phenoxathinyl group Chemical group 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 1
- 125000004928 piperidonyl group Chemical group 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 125000004591 piperonyl group Chemical group C(C1=CC=2OCOC2C=C1)* 0.000 description 1
- 229940127126 plasminogen activator Drugs 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 125000006684 polyhaloalkyl group Polymers 0.000 description 1
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 108010087851 prorelaxin Proteins 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- IFOHPTVCEBWEEQ-UHFFFAOYSA-N pyrrolo[2,3-i][1,4]benzodiazepine Chemical class N1=CC=NC2=C3C=CN=C3C=CC2=C1 IFOHPTVCEBWEEQ-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000002534 radiation-sensitizing agent Substances 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 150000003413 spiro compounds Chemical class 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 125000006253 t-butylcarbonyl group Chemical group [H]C([H])([H])C(C(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 1
- RCINICONZNJXQF-XAZOAEDWSA-N taxol® Chemical compound O([C@@H]1[C@@]2(CC(C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3(C21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-XAZOAEDWSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 108010042974 transforming growth factor beta4 Proteins 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 239000003744 tubulin modulator Substances 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 229960005356 urokinase Drugs 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3015—Breast
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
- A61K47/6817—Toxins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
Definitions
- the present invention relates to linkers, containing amide surrogates with a regular or a novel lysosomal enzymatic cleavable dipeptidic unit, to connect cytotoxic drugs to antibodies.
- the present invention also relates to ADCs (antibody-drug conjugates) derived from these amide surrogate linkers for the treatment of cancers.
- ADCs Antibody drug conjugates hold the promise as a target specific and effective treatment for a number of diseases e.g. bacterial infection and cancers, but at the same time with an improved systemic side effect profile.
- the concept to combine a target specific antibody with a cytotoxic compound is very simple and elegant.
- An antibody is well known for its ability to recognize an antigen in specific manner and the often insufficient cell killing activity following an immune response, limited its therapeutic potential.
- cyctotoxic compounds are widely used in the treatment of cancers and their effectiveness are hindered by non-specific systemic adverse events associated with higher dose usage.
- the combination of an antibody with a cyctoxic compound can therefore lead to a therapeutic agent with enhancing efficacy to the antibody and reduced systemic side effects to the cytotoxic drug, attributing to the target specific delivery by the antibody and minimize of normal tissues from drug exposures.
- linker such as the one in Adcertris, which incorporates a lysosomal enzymatic cleavable peptide component.
- the C-terminal amide bond in this part of the linker that can be easily cleaved at target cells by lysosomal enzymes is essential to the drug releasing mechanism.
- Other amide bonds on the linker if undergo prematurely cleavage may lead to undesirable side effects (Formular B-1).
- This application describes the replacement of the N-terminal amide bond of the lysosomal enzymatic cleavable peptide with an amide surrogate (Formullar B-2). This modification can further improved stability of the resulting ADCs in circulation.
- the present invention provides a linker of formula (I) to connect drugs to antibodies
- s 0, 1, 2, 3, or 4;
- the present invention also provides a drug-linker conjugate of formula (II),
- the present invention also provides an ADC of formula (III),
- a is 1, 2, 3, 3.4, 3.5, 4, 4.2, 5, 6, 7, or 8.
- the aforesaid antibody is selected from the group consisting of a resurfaced monoclonal antibody, a resurfaced single chain monoclonal antibody, or a resurfaced monoclonal antibody fragment that preferentially binds to a target cell.
- the aforesaid antibody is selected from the group consisting of a humanized monoclonal antibody, a humanized single chain monoclonal antibody, or a humanized monoclonal antibody fragment.
- the aforesaid antibody is selected from the group consisting of a chimeric antibody, a chimeric antibody fragment, a domain antibody, or a domain antibody fragment thereof.
- the aforesaid antibody is selected from the group consisting of MY9, anti-B4, EpCAM, CD2, CD3, CD4, CD5, CD6, CD11, CD19, CD20, CD22, CD26, CD30, CD33, CD37, CD38, CD40, CD44, CD56, CD79, CD105, CD138, EphA receptors, EphB receptors, EGFR, EGFRvIII, HER2, HER3, mesothelin, cripto, alpha v beta 3 , alpha v beta 5 , alpha v beta 6 integrin or C242.
- the aforesaid antibody is selected from the group consisting of My9-6, B4, C242, N901, DS6, EphA2 receptor, CD38, IGF-IR, CNTO 95, B-B4, Trastuzumab, Tertuzumab, Bevatuzumab, Sibrotuzumab, Rituximab, and Adalimumab.
- the aforesaid antibody is selected from the group consisting of Herceptin and Erbitux.
- the aforesaid antibody binds to target cells selected from tumor cells; virus infected cells, microorganism infected cells, parasite infected cells, autoimmune cells, activated cells, myeloid cells, activated T-cells, B cells, or melanocytes; cells expressing one or more of IGF-IR, CanAg, EGFR, MUCI, MUCI 6, VEGF, TF, MY9, anti-B4, EpCAM, CD2, CD3, CD4, CD5, CD6, CD11, CD11a, CD18, CD19, CD20, CD22, CD26, CD30, CD33, CD37, CD38, CD40, CD44, CD56, CD70, CD79, CD105, CD138, EphA receptors, EphB receptors, EGFRvIII, HER2/neu, HER3, mesothelin, cripto, alpha v beta 3 integrin, alpha v beta 5 integrin, alpha v beta 6 integrin
- the aforesaid tumor cells are selected from breast cancer cells, prostate cancer cells, ovarian cancer cells, colorectal cancer cells, gastric cancer cells, squamous cancer cells, small-cell lung cancer cells, and testicular cancer cells.
- the aforesaid drug is a cytotoxic drug.
- the aforesaid drug is selected from the group consisting of maytansinoid, DNA-binding drug and its analog, calicheamicin, doxorubicin and its analog, vinca alkaloid, cryptophycin, dolastatin, auristatin and analog thereof, tubulysin, epothilone, taxoid and siRNA.
- the aforesaid DNA-binding drug is CC-1065.
- the aforesaid M is selected from the group consisting of:
- the aforesaid drug is a diagnostic or detection reagen.
- the aforesaid drug is a radio-labeled compound.
- the aforesaid drug is labeled with 3 H, 18 F, 11 C, 13 N, 15 O, 201 Tl, 32 P, 51 Cr, 67 Ga, 123 I, 125 I, 131 I, 132 I, 131 Cs, 113 Xe, 133 Xe, 169 Yb, 198 Au, 203 Hg, 99m Tc, 113m In, 133m In, 75 Se, 186 Re, 153 Sm, and 89 Sr.
- the aforesaid L 1 is selected from the group consisting of:
- the aforesaid L 2 is selected from the group consisting of:
- the aforesaid L 2 is selected from the group consisting of:
- the aforesaid ring formed by two geminal R 11 or two adjacent R 11 is cyclohexyl.
- the aforesaid L 3 is selected from the group consisting of:
- the aforesaid linker is selected from the group consisting of:
- the aforesaid linker is selected from the group consisting of:
- the aforesaid conjugate is selected from the group consisting of:
- the aforesaid conjugate is selected from the group consisting of:
- the aforesaid ADC is selected from the group consisting of:
- Y is an antibody, a is an integer or decimal selected from 1-8, concretely a is 1, 2, 3, 3.4, 3.5, 4, 4.2, 5, 6, 7, or 8.
- the aforesaid ADC is selected from the group consisting of:
- Y is an antibody, a is an integer or decimal selected from 1-8, concretely a is 1, 2, 3, 3.4, 3.5, 4, 4.2, 5, 6, 7, or 8.
- the aforesaid ADC is selected from the group consisting of:
- a is an integer or decimal selected from 2-6, concretely a is 2, 3, 3.4, 3.5, 4, 4.2, 5, or 6.
- the aforesaid ADC is selected from the group consisting of:
- a is an integer or decimal selected from 2-6, concretely a is 2, 3, 3.4, 3.5, 4, 4.2, 5, or 6.
- the present invention also provides intermediate compound for the synthesis of the drug-linker conjugate of formula (II) or the ADC of formula (III), comprising:
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising an effective amount of the aforesaid ADC and a pharmaceutically acceptable carrier, diluent or excipient.
- the present invention also provides a use of the aforesaid linker, conjugate, or ADC or the aforesaid composition in the preparation of a medicament for the treatment of cancers.
- the present invention also provides a method for treating or diagnosising cancers, comprising administrating the subject in need thereof an effective amount of the aforesaid ADC or the aforesaid pharmaceutical composition.
- C 1-6 is selected from the group consisting of C 1 , C 2 , C 3 , C 4 , C 5 , and C 6 , the number denotes numbers of carbon atoms;
- C 3-6 is selected from the group consisting of C 3 , C 4 , C 5 , and C 6 .
- C 1-6 alkyl, C 1-6 heteroalkyl, C 3-6 cyclohydrocarbyl, C 3-6 heterocyclohydrocarbyl, C 1-6 alkyl substituted by C 3-6 cyclohydrocarbyl or C 3-6 heterocyclohydrocarbyl, and C 1-6 heteroalkyl substituted by C 3-6 cyclohydrocarbyl or C 3-6 heterocyclohydrocarbyl include, but are not limited to: methyl, ethyl, n-propyl, isopropyl, —CH 2 C(CH 3 )(CH 3 )(OH), cyclopropyl, cyclobutyl, propylmethylene, cycropropylacyl, benzyloxyl, cyclopropylalkylene, trifluoromethyl, aminomethyl, hydroxylmethyl, methyloxyl, methylacyl, methyloxylcarbonyl, methylsulfonyl, methylsulfinyl, eth
- N(CH 3 ) 2 NH(CH 3 ), —CH 2 CF 3 , —CH2CH 2 CF 3 , —CH 2 CH 2 F, —CH 2 CH 2 S( ⁇ O) 2 CH 3 , —CH 2 CH 2 CN,
- substituted means that any one or more hydrogens on the designated atom is replaced with a selection from the indicated group, including deuterium “D” atom, a variant of hydrogen, provided that the designated atom's normal valency is not exceeded, and that the substitution results in a stable compound.
- a substituent is keto (i.e., ⁇ O)
- 2 hydrogens on the atom are replaced.
- Keto substituents are not present on aromatic moieties.
- optionally substituted means that the designated atom can be substituted or unsubstituted by the substituents, and unless otherwise stated, the species and number the substituents are not defined provided that they can be achieved in Chemistry.
- any variable e.g., R
- its definition at each occurrence is independent of its definition at every other occurrence.
- R e.g., R
- said group may optionally be substituted with up to two R groups and R at each occurrence is selected independently from the definition of R.
- substituents and/or variables are permissible only if such combinations result in stable compounds.
- alkyl and heteroalkyl radicals are generically referred to as “alkyl group substituents,” and they can be one or more of a variety of groups selected from, but not limited to: —R′, —OR′, ⁇ O, ⁇ NR′, ⁇ N—OR′, —NR′R′′,
- R′, R′′, R′′′, R′′′′ and R′′′′′ each preferably independently refer to hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl, e.g., aryl substituted with 1-3 halogens, substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups.
- each of the R groups is independently selected as are each R′, R′′, R′′′, R′′′′ and R′′′′′ groups when more than one of these groups is present.
- R′ and R′′ are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6-, or 7-membered ring.
- —NR′R′′ is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl.
- alkyl is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl (e.g., —CF 3 and —CH 2 CF 3 ) and acyl (e.g., —C(O)CH 3 , —C(O)CF 3 , —C(O)CH 2 OCH 3 , and the like).
- haloalkyl e.g., —CF 3 and —CH 2 CF 3
- acyl e.g., —C(O)CH 3 , —C(O)CF 3 , —C(O)CH 2 OCH 3 , and the like.
- substituents for the aryl and heteroaryl groups are generically referred to as “aryl group substituents.”
- the substituents are selected from, for example: —R′, —OR′, —NR′R′′, —SR′, -halogen, —SiR′R′′R′′′, OC(O)R′, —C(O)R′, —CO2R′, —CONR′R′′, —OC(O)NR′R′′, —NR′′C(O)R′, NR′ C(O)NR′′R′′′, —NR′′C(O) 2 R′, —NR′′′′′—C(NR′R′′R′′′) ⁇ NR′′′′, NR′′′′ C(NR′R′′) ⁇ NR′′′, —S(O)R′, —S(O) 2 R′, —S(O) 2 NR′R′′, NR′′SO 2 R′, —CN, —CN, —
- Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)—(CRR′)q-U—, wherein T and U are independently —NR—, —O—, —CRR′— or a single bond, and q is an integer of from 0 to 3.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A(CH 2 )r B—, wherein A and B are independently —CRR′—, —O—, —NR—, —S—, —S(O)—, S(O) 2 —, —S(O) 2 NR′— or a single bond, and r is an integer of from 1 to 4.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula —(CRR′)s-X—(CR′′R′′′)d-, where s and d are independently integers of from 0 to 3, and X is —O—, —NR′—, —S—, —S(O)—, —S(O) 2 —, or —S(O) 2 NR′—.
- the substituents R, R′, R′′ and R′′′ are preferably independently selected from hydrogen or substituted or unsubstituted (C1-C6)alkyl.
- halo or “halogen,” by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl.
- halo(C1-C4)alkyl is mean to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
- haloalkyl examples include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl, and pentachloroethyl.
- Alkoxy represents an alkyl group as defined above with the indicated number of carbon atoms attached through an oxygen bridge.
- C1-6 alkoxy is intended to include C1, C2, C3, C4, C5, and C6 alkoxy groups.
- alkoxy include, but are not limited to, methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, s-butoxy, t-butoxy, n-pentoxy, and s-pentoxy.
- Cycloalkyl is intended to include saturated ring groups, such as cyclopropyl, cyclobutyl, or cyclopentyl. 3-7 cycloalkyl is intended to include C3, C4, C5, C6, and C7 cycloalkyl groups.
- Alkenyl is intended to include hydrocarbon chains of either straight or branched configuration and one or more unsaturated carbon-carbon bonds that may occur in any stable point along the chain, such as ethenyl and propenyl.
- Halo or “halogen” as used herein refers to fluoro, chloro, bromo, and iodo.
- hetero mean, unless otherwise stated, “heteroatom” or “heteroradical”(namely radical containing heteroatom), includeing atoms other than carbon (C) and hydrogen (H), also including the radicals containing these aforesaid heteroatoms. Examples include oxygen (O), nitrogen (N) sulfur (S), silicon (Si), germanium (Ge), aluminum (Al) and boron (B), also include optionally substituted —C( ⁇ O)N(H)—, —N(H)—, —C( ⁇ NH)—, —S( ⁇ O) 2 N(H)—, or —S( ⁇ O) N(H)—.
- Ring as used herein, means a substituted or unsubstituted cycloalkyl, heterocycloalkyl, cycloalkenyl, heterocycloalkenyl, cycloalkynyl, heterocycloalkynyl, aryl, or heteroaryl.
- a ring includes mono, bi, sprio, fused, and bridged ring moieties.
- the number of atoms in a ring is typically defined by the number of members in the ring.
- a “5- to 7-membered ring” means there are 5 to 7 atoms in the encircling arrangement. Unless otherwise specified, the ring optionally includes one to three heteroatoms.
- the term “5- to 7-membered ring” includes, for example phenyl, pyridinyl and piperidinyl.
- the term “ring” further includes a ring system comprising more than one “ring”, wherein each “ring” is independently defined as above.
- heterocycle or “heterocyclic group” is intended to mean a stable monocyclic, bicyclic, or tricyclic ring containing heteroatom or heteroradical, which is saturated, partially unsaturated or unsaturated (aromatic), and which consists of carbon atoms and 1,2, 3, or 4 ring heteroatoms independently selected from the group consisting of N, O and S and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., NO and S (O) p).
- the nitrogen atom may be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined).
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure.
- the heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable.
- a nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and O atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and O atoms in the heterocycle is not more than 1.
- aromatic heterocyclic group or“heteroaryl” is intended to mean a stable 5,6, or 7-membered monocyclic or bicyclic or 7,8, 9, or 10-membered bicyclic heterocyclic aromatic ring which consists of carbon atoms and 1,2, 3, or 4 heterotams independently selected from the group consisting of N, O and S.
- the nitrogen atom may be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined).
- the nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., NO and S (O) p). It is to be noted that total number of S and 0 atoms in the aromatic heterocycle is not more than 1.
- Bridged rings are also included in the definition of heterocycle.
- a bridged ring occurs when one or more atoms (i.e., C, O, N, or S) link two non-adjacent carbon or nitrogen atoms.
- Preferred bridges include, but are not limited to, one carbon atom, two carbon atoms, one nitrogen atom, two nitrogen atoms, and a carbon-nitrogen group. It is noted that a bridge always converts a monocyclic ring into a trycyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge.
- heterocycles include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzoxazolinyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H, 6H-1, 5,2-dithiazinyl, dihydrofuro [2, 3-b] tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1H-indazolyl, indolen
- hydrocarbyl or its lower concept(such as alkyl, alkenyl, alkynyl and phenyl etc.) by itself or as part of another substituent, means, unless otherwise stated, a straight or branched chain, or cyclic hydrocarbon radical, or combination thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals, having the number of carbon atoms designated (i.e. C 1 -C 10 means one to ten carbons).
- “Hydrocarbyl” include, but are not limited to, aliphatic hydrocarbyl and aromatic hydrocarbyl, and the aliphatic hydrocarbyl include linear and cyclic ones, specifically including but not limited to, alkyl, alkenyl, and alkynyl, and the aromatic hydrocarbyl include, but are not limited to, 6-12 membered aromatichydrocarbyl, for example, benzene, and naphthalene.
- the term “alkyl” means a straight or branched chain, or combinations thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals.
- saturated hydrocarbon radicals include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, homologs and isomers of, for example, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like.
- An unsaturated alkyl group is one having one or more double bonds or triple bonds.
- unsaturated alkyl groups include, but are not limited to, vinyl, 2-propenyl, crotyl, 2-isopentenyl, 2-(butadienyl), 2,4-pentadienyl, 3-(1,4-pentadienyl), ethynyl, 1- and 3-propynyl, 3-butynyl, and the higher homologs and isomers.
- heterohydrocarbyl or its lower concept (such as heteroalkyl, heteroalkenyl, teteroalkynyl and heteroaryl etc.) by itself or in combination with another term, means, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom.
- heteroalkyl by itself or in combination with another term, means a stable straight or branched chain, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom.
- the heteroatoms can be selected from the group consisting of B, O, N and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized.
- the heteroatom(s) B, O, N and S may be placed at any interior position of the heterohydrocarbyl group(including the position at which the hydrocarbyl group is attached to the remainder of the molecule).
- Examples include, but are not limited to, —CH2-CH2-O—CH3, —CH2-CH2-NH—CH3, —CH2-CH2-N(CH3)-CH3, —CH2-S—CH2-CH3, —CH2-CH2,—S(O)—CH3, —CH2-CH2-S(O)2-CH3, —CH ⁇ CH—O—CH3, —CH2-CH ⁇ N—OCH3, and —CH ⁇ CH—N(CH3)-CH3.
- Up to two heteroatoms may be consecutive, such as, for example, —CH 2 —NH—OCH 3 .
- alkoxy alkylamino and “alkylthio” (or thioalkoxy) are used in their conventional sense, and refer to those alkyl groups attached to the remainder of the molecule via an oxygen atom, an amino group, or a sulfur atom, respectively.
- cyclohydrocarbyl “heterocyclohydrocarbyl”, or their lower concepts(such as aryl, heteroaryl, cycloalkyl, heterocycloalkyl, cycloalkenyl, heterocycloalkenyl, cycloalkynyl, and heterocycloalkynyl etc.) by themselves or in combination with other terms, represent, unless otherwise stated, cyclic versions of “hydrocarbyl” or “heterohydrocarbyl”, respectively.
- a heteroatom can occupy the position at which the heterocycle is attached to the remainder of the molecule.
- cycloalkyl examples include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like.
- heterocycloalkyl moieties include 1-(1,2,5,6-tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4-morpholinyl, 3-morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1-piperazinyl, 2-piperazinyl.
- aryl means, unless otherwise stated, a polyunsaturated, aromatic, substituent that can be a single ring or multiple rings (preferably from 1 to 3 rings), which are fused together or linked covalently.
- heteroaryl refers to aryl groups (or rings) that contain from one to four heteroatoms.
- the heteroatom is selected from B, N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a heteroaryl group can be attached to the remainder of the molecule through a heteroatom.
- Non-limiting examples of aryl and heteroaryl groups include phenyl, 1-naphthyl, 2-naphthyl, 4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 2-phenyl-4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 5-benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1-isoquinoly
- aryl when used in combination with other terms (e.g., aryloxy, arylthio, arylalkyl) includes both aryl and heteroaryl rings as defined above.
- arylalkyl is meant to include those radicals in which an aryl group is attached to an alkyl group (e.g., benzyl, phenethyl, pyridylmethyl and the like) including those alkyl groups in which a carbon atom (e.g., a methylene group) has been replaced by, for example, an oxygen atom (e.g., phenoxymethyl, 2-pyridyloxymethyl, 3-(1-naphthyloxy)propyl, and the like).
- alkyl group e.g., benzyl, phenethyl, pyridylmethyl and the like
- an oxygen atom e.g., phenoxymethyl, 2-pyridyloxymethyl, 3-(1-naph
- leaving group means a functional group or atom which can be displaced by another functional group or atom in a substitution reaction, such as a nucleophilic substitution reaction.
- representative leaving groups include triflate, chloro, bromo and iodo groups; sulfonic ester groups, such as mesylate, tosylate, brosylate, nosylate and the like; and acyloxy groups, such as acetoxy, trifluoroacetoxy and the like.
- protecting group includes bu not limited to “amino-protecting group”, “hydroxy-protecting group” and “thiol-protecting group”.
- amino-protecting group means a protecting group suitable for preventing undesired reactions at an amino nitrogen.
- Representative amino-protecting groups include, but are not limited to, formyl; acyl groups, for example alkanoyl groups, such as acetyl, trichloroacetyl or trifluoroacetyl; alkoxycarbonyl groups, such as tert-butoxycarbonyl (Boc); arylmethoxycarbonyl groups, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); arylmethyl groups, such as benzyl (Bn), trityl (Tr), and 1,1-di-(4′-methoxyphenyl)methyl; silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
- alkoxycarbonyl groups such as tert-butoxycarbonyl (Boc)
- arylmethoxycarbonyl groups such as benzy
- hydroxy-protecting group means a protecting group suitable for preventing undesired reactions at a hydroxy group.
- Representative hydroxy-protecting groups include, but are not limited to, alkyl groups, such as methyl, ethyl, and tert-butyl; acyl groups, for example alkanoyl groups, such as acetyl; arylmethyl groups, such as benzyl (Bn), p-methoxybenzyl (PMB), 9-fluorenylmethyl (Fm), and diphenylmethyl (benzhydryl, DPM); silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
- alkyl groups such as methyl, ethyl, and tert-butyl
- acyl groups for example alkanoyl groups, such as acetyl
- arylmethyl groups such as benzyl (Bn), p-meth
- the “antibody” is a cell-binding agent, it binds to an antigen that is a polypeptide and may be a transmembrane molecule (e.g. receptor) or a ligand such as a growth factor.
- antigens include molecules such as renin; a growth hormone, including human growth hormone and bovine growth hormone; growth hormone releasing factor; parathyroid hormone; thyroid stimulating hormone; lipoproteins; alpha-1-antitrypsin; insulin A-chain; insulin B-chain; proinsulin; follicle stimulating hormone; calcitonin; luteinizing hormone; glucagon; clotting factors such as factor vmc, factor IX, tissue factor (TF), and von Willebrands factor; anti-clotting factors such as Protein C; atrial natriuretic factor; lung surfactant; a plasminogen activator, such as urokinase or human urine or tissue-type plasminogen activator (t-PA); bombesin; thrombin; hemopoietic growth factor; tumor necrosis factor-alpha and -beta; enkephalinase; RANTES (regulated on activation normally T-cell expressed and secreted); human macrophage inflammatory protein (MIP
- CD proteins such as CD2, CD3, CD4, CD5, CD6, CD8, CD11, CD14, CD18, CD19, CD20, CD21, CD22, CD 25, CD26, CD27, CD28, CD30, CD33, CD36, CD37, CD38, CD40, CD44, CD52, CD55, CD56, CD70, CD79, CD80, CD81, CD103, CD105, CD134, CD137, CD138, and CD152; members of the ErbB receptor family such as the EGF receptor, HER2, HER3 or HER4 receptor; cell adhesion molecules such as LFA-I, MacI, pI50.95, VLA-4, ICAM-I, VCAM, EpCAM, alpha4/beta7 integrin, and alpha5/beta3 integrin including either alpha or beta subunits thereof (e.g.
- anti-CD11a, anti-CD18 or anti-CD11b antibodies growth factors such as VEGF; tissue factor (TF); TGF-beta; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C etc.
- growth factors such as VEGF; tissue factor (TF); TGF-beta; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C etc.
- the most concrete targets herein are IGF-IR, CanAg, EphA2, MUC1, MUC16, VEGF, TF, CD19, CD20, CD22, CD27, CD33, CD37, CD38, CD40, CD44, CD56, CD138, CA6, Her2/neu, EpCAM, CRIPTO (a protein produced at elevated levels in a majority of human breast cancer cells), darpins, alpha v /beta 3 integrin, alpha v /beta 5 integrin, alpha v /beta 6 integrin, TGF-beta, CD11a, CD18, Apo2 and C242 or an antibody which binds to one or more tumor-associated antigens or cell-surface receptors disclosed in US Publication No. 20080171040 or US Publication No. 20080305044 and are incorporated in their entirety by reference.
- CD proteins such as CD3, CD4, CD8, CD19, CD20, CD27, CD34, CD37, CD38, CD46, CD56, CD70 and CD138
- members of the ErbB receptor family such as the EGF receptor, HER2, HER3 or HER4 receptor
- cell adhesion molecules such as LFA-I, Macl, pI50.95, VLA-4, ICAM-I, VCAM, EpCAM, alpha4/beta7 integrin, and alpha v/beta3 integrin including either alpha or beta subunits thereof (e.g.
- anti-CD11a, anti-CD 18 or anti-CD11b antibodies growth factors such as VEGF; tissue factor (TF); TGE- ⁇ ; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C, etc.
- growth factors such as VEGF; tissue factor (TF); TGE- ⁇ ; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C, etc.
- the most concrete targets herein are IGF-IR, CanAg, EGF-R, EGF-RvIII, EphA2, MUCI, MUC16, VEGF, TF, CD19, CD20, CD22, CD27, CD33, CD37, CD38, CD40, CD44, CD56, CD70, CD 138, CA6, Her2/neu, CRIPTO (a protein produced at elevated levels in a majority of human breast cancer cells), alpha v/beta3 integrin, alpha v/beta5 integrin, TGE- ⁇ , CDI Ia, CD18, Apo2, EpCAM and C242.
- Monoclonal antibody techniques allow for the production of specific cell-binding agents in the form of monoclonal antibodies.
- Particularly well known in the art are techniques for creating monoclonal antibodies produced by immunizing mice, rats, hamsters or any other mammal with the antigen of interest such as the intact target cell, antigens isolated from the target cell, whole virus, attenuated whole virus, and viral proteins such as viral coat proteins.
- Sensitized human cells can also be used.
- Another method of creating monoclonal antibodies is the use of phage libraries of sFv (single chain variable region), specifically human sFv (see, e.g., Griffiths et al, U.S. Pat. No. 5,885,793; McCafferty et al, WO 92/01047; Liming et al, WO 99/06587.)
- the monoclonal antibody My9 is a murine IgG2a antibody that is specific for the CD33 antigen found on Acute Myeloid Leukemia (AML) cells (Roy et al. Blood 77:2404-2412 (1991)) and can be used to treat AML patients.
- the monoclonal antibody anti-B4 is a murine IgGi that binds to the CD 19 antigen on B cells (Nadler et al, J Immunol. 131:244-250 (1983)) and can be used if the target cells are B cells or diseased cells that express this antigen such as in non-Hodgkin's lymphoma or chronic lymphoblastic leukemia.
- the antibody N901 is a murine monoclonal IgGi antibody that binds to CD56 found on small cell lung carcinoma cells and on cells of other tumors of neuroendocrine origin (Roy et al. J Nat. Cancer Inst. 88:1136-1145 (1996)); huC242 is an antibody that binds to the CanAg antigen; Trastuzumab is an antibody that binds to HER2/neu; and anti-EGF receptor antibody binds to EGF receptor.
- the drug unit used in this invention can be any cytotoxic, cytostatic or immunosuppressive drug also referred to herein as a cytotoxic, cytostatic or immunosuppressive agent.
- drugs include radioactive isotopes, chemotherapeutic agents, and toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including synthetic analogues and derivatives thereof.
- the Drug unit has an atom that can form a bond with the Linker Unit, in some embodiments, the Drug unit has a nitrogen atom that can form a bond with the Linker unit. In other embodiments, the Drug unit has a carboxylic acid that can form a bond with the Linker unit. In other embodiments, the Drug unit has a sulfhydryl group that can form a bond with the Linker unit . In other embodiments, the Drug unit has a hydroxy! group or ketone that can form a bond with the Linker unit.
- cytotoxic or immunosuppressive agents include, for example, antitubulin agents, auristatins, DNA minor groove binders, DNA replication inhibitors, alkylating agents (e.g. , platinum complexes such as cis-platin, mono(platinum), bis(platinum) and tri-nuelear platinum complexes and carbopiatin), anthracyclin.es, antibiotics, antifolates-, antimetabolites, chemotherapy sensitizers, duocarmycins, etoposides, fluorinated pyrimidines, ionophores, lexitropsins, nitrosoureas, platinois, pre-formirsg compounds, purine antimetabolites, puromycins, radiation sensitizers, steroids, taxanes, topoisornerase inhibitors, vinca alkaloids, or the like.
- alkylating agents e.g. , platinum complexes such as cis-platin, mono(p
- cytotoxic agents include, for example, DNA minor groove binders, DNA alkylating agents, and tubulin inhibitors.
- cytotoxic agents include, for example, auristatins, camptothecins, duocarmycins, etoposides, maytansines and maytansinoids (e.g., DM! and DM4), taxanes, benzodiazepines (e.g., pyrrolo [1,4] benzodiazepines (PBDs). indolinobenzodiazepines, and oxazolidinobenzodiazepines) and vinca alkaloids.
- PBDs pyrrolo [1,4] benzodiazepines
- indolinobenzodiazepines, and oxazolidinobenzodiazepines and vinca alkaloids.
- Select benzodiazepine containing drugs are described in WO 2010/091 150, WO 2012/1 12708, WO 2007/085930, and WO 201 1/
- cytotoxic or immunosuppressive agents include, for example, an androgen, antbramycin (AMC), asparaginase, 5-azacytidine, azathioprine, bleomycin, busuJfan, buthionine sulfoximine, calicheamicin, camptothecin, carbopiatin, carmustine (BSNU), CC-1065, chlorambucil, cisplatin, colchicine, cyclophosphamide, cytarabine, cytidine arabinoside, cytochalasin B, dacarbazine, dactinomycin (formerly actinomycin).
- AMC antbramycin
- asparaginase 5-azacytidine
- azathioprine 5-azacytidine
- azathioprine 5-azacytidine
- azathioprine 5-azacytidine
- azathioprine 5-azacytidine
- azathioprine
- suitable cytotoxic agents include, for example, DNA minor groove binders (e.g., enediynes and exitropsins, a CBI compound; see also U.S. Pat. No. 6,130,237), duocarmycras (see U.S. Publication No.
- taxanes e.g., paclitaxei and docetaxel
- puromycins e.g., vinca alkaloids, CC-1065, SN-38, topotecan, morpholino-doxorubicin, rhizoxin, cyanomorpholino-doxorubicin, echinomvcin, combretastatin, netropsin, epothilone A and B, estramustine, cryptophysins, cemadotin, maytansinoids, discodermoiide, eleutherobin, and mitoxantrone.
- taxanes e.g., paclitaxei and docetaxel
- puromycins e.g., vinca alkaloids, CC-1065, SN-38, topotecan, morpholino-doxorubicin, rhizoxin, cyanomorpholino-doxorubicin, echinomvcin, combret
- the Drug unit is an anti-tubulin agent.
- anti-tubulin agents include, but are not limited to, taxanes (e.g., Taxol® (paclitaxei), Taxotere ⁇ (docetaxel)), T67 (Tularik) and vinca alkyloids (e.g., vincristine, vinblastine, vindesme, and vinorelbine).
- antitubulin agents include, for example, baccatin derivatives, taxane analogs (e.g., epothilone A and B), nocodazole, colchicine and colcimid, estramustine, cryptophysins, cemadotin, maytansinoids, combretastatins, discodermoiide, and eleutherobin.
- the cytotoxic agent is maytansine or a maytansinoid, another group of anti-tubulin agents.
- maytansine or a maytansinoid, another group of anti-tubulin agents.
- the Drug unit is an auristatin.
- Auristatins include, but are not limited to, AE, AFP, AEB, AEVB, MMAF, and MMAE.
- the synthesis and structure of auristatins are described in U.S. Patent Application Publication Nos. 2003-0083263, 2005-0238649 2005-0009751 , 2009-01 1 1756, and 201 1-0020343: International Patent Publication No. WO 04/010957, international Patent Publication No. WO 02/088172, and U.S. Pat. Nos. 7,659,241 and 8,343,928; each of which is incorporated by reference in its entirety and for all purposes.
- Exemplary auristatins of the present invention bind tubulin and exert a cytotoxic or cytostatic effect on the desired cell line.
- Exemplary auristatin Drug units have the following formula or a pharmaceutically acceptable salt thereof wherein the wavy line indicates site of attachment to the linker unit:
- the Drug is a benzodiazepine (including benzodiazepine containing drugs e.g., pyrrolo[l,4]benzodiazepiries (PBDs), indolinobenzodiazepines, and oxazo!idino benzodiazepines).
- PBDs are of the general structure:
- PBDs to form an adduct in the minor groove enables them to interfere with DNA processing, hence their use as antitumour agents.
- the biological activity of these molecules can be potentiated by, for example, joining two PBD units together through their C8/C -hydroxyl functionalities via a flexible alkylene linker.
- the PBD dimers are thought to form sequence-selective DNA lesions such as the palindromic 5′-Pu-GATC-Py-3′ interstrand cross-link which is thought to be mainly responsible for their biological activity.
- An amino alcohol 1 is reacted with a hemiacetal such as trifluoroacetaldehyde methyl hemiacetal in a solvent such as toluene at refluxing temperature with concomitant removal of water to give an oxazolidine intermediate 2.
- Oxazolidine 2 is then reacted with a Grignard or alkali reagent derived from an appropriate precursor to afford an amino alcohol intermediate 3.
- Oxidation under an appropriate condition such as Jones reagent or periodic acid with catalytic amount of chromium trioxide and followed by esterifaction furnishes the ester intermediate 4.
- Appropriate manipulation of functional groups on intermediate 4 affords intermediate 5 with suitable handles for further transformations.
- the amino alcohol 3 in Method 1 can also be prepared in a diastereo-selective manner as described in Gosselin, F., et. al., Org. Lett. 2004, 6, 641 and Roy, A., et. al., J. Org. Chem. 2006, 71, 4320 to give 3a with high distereoselectivity.
- the solution was divided equally into to two tubes (10 mL each), and 1 mL DMSO solution of drug-linker intermediate (25 mg/mL) was added to each tube.
- the ratio of drug-linker intermediate to mAb is 6:1.
- the reaction was carried out at room temperature for ca 12 hour.
- solutions from the two tubes were pooled, centrifuged at 5000 rpm for 5 min and loaded onto a Protein A affinity column to remove the un-reacted drug-Ilinker intermediate.
- the pH of the eluent from the column was adjusted to 6.0 with 1 M Tris buffer.
- the drug antibody ratio (DAR) was determined by hydrophobic interaction chromatography (HIC) HPLC.
- the invention is now is further described by examples.
- the examples given below are for illustration purpose and not meant to limit this invention.
- the compounds of the present invention can be prepared in a number of ways known to one skilled in the art of organic synthesis.
- the examples of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or by variations thereon as appreciated by those skilled in the art. Concrete methods include, but are not limited to, those described below.
- ADC antibody-drug conjugate
- aq is aqueous
- mAb is monoclonal antibody
- MTMH 1-Maleinimidyl-6-trifluoromethylhex-6-yl with the structure of
- FIG. 1 showed the antiproliferative activity of Herceptin and different ADCs and the result suggested that the study ADC001 (EXAMPLE 9) and the control ADC-HA showed comparable activity in the inhibition on the growth of breast cancer cell line HCC1954 in vitro. Herceptin alone is ineffective in this assay.
- FIG. 2 showed the tumor growth curves from each group.
- the result suggested that the study ADC001 (EXAMPLE 9) and the control ADC-HA displayed comparable activity to inhibit tumor growth in the HCC1954 xenograft mice model.
- FIGS. 3 and 4 showed the antiproliferative activity of Herceptin and ADCs (ADC002 to ADC006, EXAMPLE 10 to 14) and the result suggested that the study ADCs and the control ADC-HA showed comparable activity in the inhibition on the growth of breast cancer cell line HCC1954 in vitro. Herceptin alone is ineffective in this assay
- FIGS. 5 to 10 showed the tumor growth curves for control ADC-HA and ADCs (ADC-002 to ADC006, EXAMPLE 10 to 14).
- FIG. 11 showed the comparison of tumor sizes at termination of the study between control ADC-HA and ADCC-002 to ADC006.
- FIG. 12 showed the antiproliferative activity of Erbitux, control ADC-EA, ADC007 (EXAMPLE 15) and ADC008 (EXAMPLE 16).
- FIG. 13 showed the tumor growth curves for control ADC-EA, ADC007 (EXAMPLE 15) and ADC008 (EXAMPLE 16).
- Step 5 (2S)-Tert-butyl 2-((7-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl)amino)-3-methylbutanoate
- Step 6 (2S)-2,5-Dioxopyrrolidin-1-yl-2- ⁇ [7-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-1,1,1-trifluoro-heptan-2-yl]amino ⁇ -3-methylbutanoate
- Step 6 (2S)-Tert-butyl 2-((7-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl)amino)-3-methylbutanoate
- Step 7 (S)-2-(((R)-7-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl)amino)-3-methyl-butanoic acid, (R)-MTMH-Val and (S)-2-(((S)-7-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl)amino)-3-methylbutanoic acid, (S)-MTMH-Val
- Step 4 (S)-Benzyl (1-((4-(hydroxymethyl)phenyl)amino)-5-morpholino-1-oxopentan-2-yl) carbamate
- Step 4 Methyl 2-(((benzyloxy)carbonyl)amino)-3-(trans-4-(((tert-butoxycarbonyl)amino)methyl)cyclohexyl)acrylate
- Step 5 2-(((Benzyloxy)carbonyl)amino)-3-(trans-4-(((tert-butoxycarbonyl)amino)methyl)cyclo-hexyl)acrylic acid
- Step 6 Benzyl (1-(trans-4-(((tert-butoxycarbonyl)amino)methyl)cyclohexyl)-3-((4-(hydroxylmethyl)phenyl)amino)-3-oxoprop-1-en-2-yl)carbamate
- Step 7 tert-Butyl ((trans-4-(2-amino-3-((4-(hydroxymethyl)phenyl)amino)-3-oxopropyl)cyclohexyl)methyl)carbamate
- Step 1 4-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)butanoic acid
- EXAMPLE 1 Herceptin (240 mg) and drug-linker intermediate MTMH-Val-Cit-PAB-OH (11.3 mg), EXAMPLE 1 was coupled according to the general procedure for conjugation of drug-linker intermediate to herceptin to give the title ADC (168 mg, 70%).
- Breast cancer cell line HCC1954 was grown in RPM11640 medium supplemented with 10% fetal bovine serum and maintained in an atmosphere of 5% CO 2 in a humidified 37° C. incubator.
- Epidermoid carcinoma cell line A431 was grown in DMEM medium supplemented with 10% fetal bovine serum and maintained in an atmosphere of 5% CO 2 in a humidified 37° C. incubator.
- mice 7-8 Week old female NOD-SCID mice were injected with HCC1954 cells subcutaneously. Treatment was started 7 days after cell injection once the mean estimated tumor mass reached 153 mm 3 . Mice were grouped randomly according to the tumor volume and body weight (8 animals per group). Animals were treated with ADCs of this invention (15 mpk), Herceptin (15 mpk) as well as vehicle control on day 0 and day 15. Animals were euthanized on day 28 after the start of treatment, when the tumor volume of vehicle control group reached a mass of 1092 mm 3 .
- mice 6-8 Week old female Balb/c nude mice were injected with A431 cells subcutaneously. Treatment was started 10 days after cell injection once the mean estimated tumor mass reached 172 mm 3 . Mice were grouped randomly according to the tumor volume and body weight (8 animals per group). Animals were treated with ADCs of this invention (10 mpk), Erbitux (10 mpk) as well as vehicle control on day 0 and day 15. Animals were euthanized on day 25 after the start of treatment, when the tumor volume of vehicle control group reached a mass of 2000 mm 3 .
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Cell Biology (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Oncology (AREA)
- Toxicology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
The present invention relates to linkers, containing amide surrogates with a regular or a novel lysosomal enzymatic cleavable dipeptidic unit, to connect cytotoxic drugs to antibodies. The present invention also relates to ADCs (antibody-drug conjugates) derived from these amide surrogate linkers for the treatment of cancers.
Description
This application is a National Stage of International Application No. PCT/CN2015/083840, filed on Jul. 13, 2015 and published in English as WO 2016/008392 on Jan. 21, 2016. This application claims the priority to International Application No. PCT/CN2014/082292, filed on Jul. 16, 2014. The entire disclosures of the above applications are incorporated herein by reference.
The present invention relates to linkers, containing amide surrogates with a regular or a novel lysosomal enzymatic cleavable dipeptidic unit, to connect cytotoxic drugs to antibodies. The present invention also relates to ADCs (antibody-drug conjugates) derived from these amide surrogate linkers for the treatment of cancers.
Antibody drug conjugates (ADCs) hold the promise as a target specific and effective treatment for a number of diseases e.g. bacterial infection and cancers, but at the same time with an improved systemic side effect profile.
The concept to combine a target specific antibody with a cytotoxic compound is very simple and elegant. An antibody is well known for its ability to recognize an antigen in specific manner and the often insufficient cell killing activity following an immune response, limited its therapeutic potential. On the other hand, cyctotoxic compounds are widely used in the treatment of cancers and their effectiveness are hindered by non-specific systemic adverse events associated with higher dose usage. The combination of an antibody with a cyctoxic compound can therefore lead to a therapeutic agent with enhancing efficacy to the antibody and reduced systemic side effects to the cytotoxic drug, attributing to the target specific delivery by the antibody and minimize of normal tissues from drug exposures.
One of the keys to the success of ADCs relies on the identification of suitable linkers that are stable during circulation in the blood stream, but rapidly cleave to release the cytotoxic compound at the target for cell killing activity. A demonstrated successful example is the linker, such as the one in Adcertris, which incorporates a lysosomal enzymatic cleavable peptide component. The C-terminal amide bond in this part of the linker that can be easily cleaved at target cells by lysosomal enzymes is essential to the drug releasing mechanism. Other amide bonds on the linker if undergo prematurely cleavage may lead to undesirable side effects (Formular B-1).

This application describes the replacement of the N-terminal amide bond of the lysosomal enzymatic cleavable peptide with an amide surrogate (Formullar B-2). This modification can further improved stability of the resulting ADCs in circulation.

The present invention provides a linker of formula (I) to connect drugs to antibodies,

wherein,
- R is selected from the group consisting of CF2H, CF3, CF2CF3, and PhSO2Me;
- Q is selected from the group consisting of

- L1 is selected from the group consisting of —(CR1R2)m—(CR3R4)—(CR1R2)n—, —(CR1R2)m—(CR3R4)—O—(CR1R2)n—, and —[(CR1R2)(CR1R2)X]p—(CR1R2)q—;
- m is 1, 2, 3, or 4;
- n is 1, 2, 3, or 4;
- p is 1, 2, 3, 4, 5, or 6;
- q is 1 or 2;
- R1 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2.
- R2 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
- R3 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
- R4 is selected from the group consisting of H, C1-3 alkyl, OH, NR5R6, CO2H, P(O)(OH)2, and SO3H, said C1-3 alkyl is optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
- R5 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
- R6 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
- X is selected from the group consisting of NR7, O, S(O)r;
- r is 0, 1, or 2;
- R7 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
- L2 is a di-peptide unit selected from

- R8 is selected from the group consisting of methyl, propyl, isopropyl, sec-butyl, benzyl, and

- R9 is selected from the group consisting of (CH2)4NH2, (CH2)3NHCONH2, (CH2)3NHC(═NH)NH2,

s is 0, 1, 2, 3, or 4;
- L3 is a self-immolative unit selected from the group consisting of

- R10 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
- each R11 is independently selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
- two geminal R11 may be optionally joined together to form a 3-6 membered ring with a carbon atom to which they are attached;
- two adjacent R11 may be optionally joined together to form a 5-7 membered ring with a carbon atom to which they are attached;
- R12 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2; and
- R13 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2.
The present invention also provides a drug-linker conjugate of formula (II),

wherein,
- Q′ is selected from the group consisting of

- M is a drug; and
other variables are as defined above.
The present invention also provides an ADC of formula (III),

wherein,
- Y is an antibody;
- a is an integer or decimal selected from 1-8;
- M is a drug; and
other variables are as defined in above.
In certain embodment of this invention, a is 1, 2, 3, 3.4, 3.5, 4, 4.2, 5, 6, 7, or 8.
In certain embodment of this invention, the aforesaid antibody is selected from the group consisting of a resurfaced monoclonal antibody, a resurfaced single chain monoclonal antibody, or a resurfaced monoclonal antibody fragment that preferentially binds to a target cell.
In certain embodment of this invention, the aforesaid antibody is selected from the group consisting of a humanized monoclonal antibody, a humanized single chain monoclonal antibody, or a humanized monoclonal antibody fragment.
In certain embodment of this invention, the aforesaid antibody is selected from the group consisting of a chimeric antibody, a chimeric antibody fragment, a domain antibody, or a domain antibody fragment thereof.
In certain embodment of this invention, the aforesaid antibody is selected from the group consisting of MY9, anti-B4, EpCAM, CD2, CD3, CD4, CD5, CD6, CD11, CD19, CD20, CD22, CD26, CD30, CD33, CD37, CD38, CD40, CD44, CD56, CD79, CD105, CD138, EphA receptors, EphB receptors, EGFR, EGFRvIII, HER2, HER3, mesothelin, cripto, alphavbeta3, alphavbeta5, alphavbeta6 integrin or C242.
In certain embodment of this invention, the aforesaid antibody is selected from the group consisting of My9-6, B4, C242, N901, DS6, EphA2 receptor, CD38, IGF-IR, CNTO 95, B-B4, Trastuzumab, Tertuzumab, Bevatuzumab, Sibrotuzumab, Rituximab, and Adalimumab.
In certain embodment of this invention, the aforesaid antibody is selected from the group consisting of Herceptin and Erbitux.
In certain embodment of this invention, the aforesaid antibody binds to target cells selected from tumor cells; virus infected cells, microorganism infected cells, parasite infected cells, autoimmune cells, activated cells, myeloid cells, activated T-cells, B cells, or melanocytes; cells expressing one or more of IGF-IR, CanAg, EGFR, MUCI, MUCI 6, VEGF, TF, MY9, anti-B4, EpCAM, CD2, CD3, CD4, CD5, CD6, CD11, CD11a, CD18, CD19, CD20, CD22, CD26, CD30, CD33, CD37, CD38, CD40, CD44, CD56, CD70, CD79, CD105, CD138, EphA receptors, EphB receptors, EGFRvIII, HER2/neu, HER3, mesothelin, cripto, alphavbeta3 integrin, alphavbeta5 integrin, alphavbeta6 integrin, Apo2, and C242 antigens; or cells expressing insulin growth factor receptor, epidermal growth factor receptor, and folate receptor.
In certain embodment of this invention, the aforesaid tumor cells are selected from breast cancer cells, prostate cancer cells, ovarian cancer cells, colorectal cancer cells, gastric cancer cells, squamous cancer cells, small-cell lung cancer cells, and testicular cancer cells.
In certain embodment of this invention, the aforesaid drug is a cytotoxic drug.
In certain embodment of this invention, the aforesaid drug is selected from the group consisting of maytansinoid, DNA-binding drug and its analog, calicheamicin, doxorubicin and its analog, vinca alkaloid, cryptophycin, dolastatin, auristatin and analog thereof, tubulysin, epothilone, taxoid and siRNA.
In certain embodment of this invention, the aforesaid DNA-binding drug is CC-1065.
In certain embodment of this invention, the aforesaid M is selected from the group consisting of:

In certain embodment of this invention, the aforesaid drug is a diagnostic or detection reagen.
In certain embodment of this invention, the aforesaid drug is a radio-labeled compound.
In certain embodment of this invention, the aforesaid drug is labeled with 3H, 18F, 11C, 13N, 15O, 201Tl, 32P, 51Cr, 67Ga, 123I, 125I, 131I, 132I, 131Cs, 113Xe, 133Xe, 169Yb, 198Au, 203Hg, 99mTc, 113mIn, 133mIn, 75Se, 186Re, 153Sm, and 89Sr.
In certain embodment of this invention, the aforesaid L1 is selected from the group consisting of:

In certain embodment of this invention, the aforesaid L2 is selected from the group consisting of:

In certain embodment of this invention, the aforesaid L2 is selected from the group consisting of:

In certain embodment of this invention, the aforesaid ring formed by two geminal R11 or two adjacent R11 is cyclohexyl.
In certain embodment of this invention, the aforesaid L3 is selected from the group consisting of:

In certain embodment of this invention, the aforesaid moiety of

is selected from the group consisting of:

In certain embodment of this invention, the aforesaid moiety of
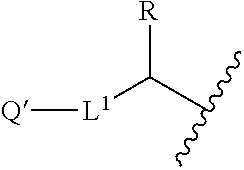
is selected from the group consisting of:

In certain embodment of this invention, the aforesaid linker is selected from the group consisting of:


In certain embodment of this invention, the aforesaid linker is selected from the group consisting of:



In certain embodment of this invention, the aforesaid conjugate is selected from the group consisting of:


In certain embodment of this invention, the aforesaid conjugate is selected from the group consisting of:



In certain embodment of this invention, the aforesaid ADC is selected from the group consisting of:


Y is an antibody, a is an integer or decimal selected from 1-8, concretely a is 1, 2, 3, 3.4, 3.5, 4, 4.2, 5, 6, 7, or 8.
In certain embodment of this invention, the aforesaid ADC is selected from the group consisting of:



Y is an antibody, a is an integer or decimal selected from 1-8, concretely a is 1, 2, 3, 3.4, 3.5, 4, 4.2, 5, 6, 7, or 8.
In certain embodment of this invention, the aforesaid ADC is selected from the group consisting of:



a is an integer or decimal selected from 2-6, concretely a is 2, 3, 3.4, 3.5, 4, 4.2, 5, or 6.
In certain embodment of this invention, the aforesaid ADC is selected from the group consisting of:




a is an integer or decimal selected from 2-6, concretely a is 2, 3, 3.4, 3.5, 4, 4.2, 5, or 6.
The present invention also provides intermediate compound for the synthesis of the drug-linker conjugate of formula (II) or the ADC of formula (III), comprising:


The present invention also provides a pharmaceutical composition comprising an effective amount of the aforesaid ADC and a pharmaceutically acceptable carrier, diluent or excipient.
The present invention also provides a use of the aforesaid linker, conjugate, or ADC or the aforesaid composition in the preparation of a medicament for the treatment of cancers.
The present invention also provides a method for treating or diagnosising cancers, comprising administrating the subject in need thereof an effective amount of the aforesaid ADC or the aforesaid pharmaceutical composition.
Definitions and Explanations
C1-6 is selected from the group consisting of C1, C2, C3, C4, C5, and C6, the number denotes numbers of carbon atoms; C3-6 is selected from the group consisting of C3, C4, C5, and C6.
C1-6 alkyl, C1-6 heteroalkyl, C3-6 cyclohydrocarbyl, C3-6 heterocyclohydrocarbyl, C1-6 alkyl substituted by C3-6 cyclohydrocarbyl or C3-6 heterocyclohydrocarbyl, and C1-6 heteroalkyl substituted by C3-6 cyclohydrocarbyl or C3-6 heterocyclohydrocarbyl include, but are not limited to: methyl, ethyl, n-propyl, isopropyl, —CH2C(CH3)(CH3)(OH), cyclopropyl, cyclobutyl, propylmethylene, cycropropylacyl, benzyloxyl, cyclopropylalkylene, trifluoromethyl, aminomethyl, hydroxylmethyl, methyloxyl, methylacyl, methyloxylcarbonyl, methylsulfonyl, methylsulfinyl, ethyloxyl, ethylacyl, ethylsulfonyl, ethyloxylcarbonyl, dimethylamino, diethylamino, dimethylaminocarbonyl, and diethylaminocarbonyl;
N(CH3)2, NH(CH3), —CH2CF3, —CH2CH2CF3, —CH2CH2F, —CH2CH2S(═O)2CH3, —CH2CH2CN,

—CH2CH(OH)(CH3)2, —CH2CH(F)(CH3)2, —CH2CH2F, —CH2CF3, —CH2CH2CF3, —CH2CH2NH2, —CH2CH2OH, —CH2CH2OCH3, —CH2CH2CH2OCH3, —CH2CH2N(CH3)2, —S(═O)2CH3, —CH2CH2S(═O)2CH3, , , ,

The term “substituted”, as used herein, means that any one or more hydrogens on the designated atom is replaced with a selection from the indicated group, including deuterium “D” atom, a variant of hydrogen, provided that the designated atom's normal valency is not exceeded, and that the substitution results in a stable compound. When a substituent is keto (i.e., ═O), then 2 hydrogens on the atom are replaced. Keto substituents are not present on aromatic moieties. The term “optionally substituted”, as used herein, means that the designated atom can be substituted or unsubstituted by the substituents, and unless otherwise stated, the species and number the substituents are not defined provided that they can be achieved in Chemistry.
When any variable (e.g., R) occurs more than one time in any constituent or formula for a compound, its definition at each occurrence is independent of its definition at every other occurrence. Thus, for example, if a group is shown to be substituted with 0-2 R, then said group may optionally be substituted with up to two R groups and R at each occurrence is selected independently from the definition of R. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
When a bond to a substituent is shown to cross a bond connecting two atoms in a ring, then such substituent may be bonded to any atom on the ring. When a substituent is listed without indicating the atom via which such substituent is bonded to the rest of the compound of a given formula, then such substituent may be bonded via any atom in such substituent. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
Substituents for the alkyl and heteroalkyl radicals (including those groups often referred to as alkylene, alkenyl, heteroalkylene, heteroalkenyl, alkynyl, cycloalkyl, heterocycloalkyl, cycloalkenyl, and heterocycloalkenyl) are generically referred to as “alkyl group substituents,” and they can be one or more of a variety of groups selected from, but not limited to: —R′, —OR′, ═O, ═NR′, ═N—OR′, —NR′R″,
—SR′, -halogen, —SiR′R″R′″, OC(O)R′, —C(O)R′, —CO2R′, —CONR′R″,
—OC(O)NR′R″, —NR″C(O)R′, NR′C(O)NR″R′″, —NR″C(O)2R′,
—NR′″″—C(NR′R″R′″)═NR″″, NR″″ C(NR′R″)═NR′″, —S(O)R′, —S(O)2R′,
—S(O)2NR′R″, NR″SO2R′, —CN, —NO2, —N3, —CH(Ph)2, fluoro(C1-C4)alkoxy, and fluoro(C1-C4)alkyl, in a number ranging from zero to (2m′+1), where m′ is the total number of carbon atoms in such radical. R′, R″, R′″, R″″ and R′″″ each preferably independently refer to hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl, e.g., aryl substituted with 1-3 halogens, substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups. When a compound of the invention includes more than one R group, for example, each of the R groups is independently selected as are each R′, R″, R′″, R″″ and R′″″ groups when more than one of these groups is present. When R′ and R″ are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6-, or 7-membered ring. For example, —NR′R″ is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl. From the above discussion of substituents, one of skill in the art will understand that the term “alkyl” is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl (e.g., —CF3 and —CH2CF3) and acyl (e.g., —C(O)CH3, —C(O)CF3, —C(O)CH2OCH3, and the like).
Similar to the substituents described for the alkyl radical, substituents for the aryl and heteroaryl groups are generically referred to as “aryl group substituents.” The substituents are selected from, for example: —R′, —OR′, —NR′R″, —SR′, -halogen, —SiR′R″R′″, OC(O)R′, —C(O)R′, —CO2R′, —CONR′R″, —OC(O)NR′R″, —NR″C(O)R′, NR′ C(O)NR″R′″, —NR″C(O)2R′, —NR′″″—C(NR′R″R′″)═NR″″, NR″″ C(NR′R″)═NR′″, —S(O)R′, —S(O)2R′, —S(O)2NR′R″, NR″SO2R′, —CN, —NO2, —N3, —CH(Ph)2, fluoro(C1-C4)alkoxy, and fluoro(C1-C4)alkyl, in a number ranging from zero to the total number of open valences on the aromatic ring system; and where R′, R″, R′″, R″″ and R′″″ are preferably independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl and substituted or unsubstituted heteroaryl. When a compound of the invention includes more than one R group, for example, each of the R groups is independently selected as are each R′, R″, R′″, R″″ and R′″″ groups when more than one of these groups is present.
Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)—(CRR′)q-U—, wherein T and U are independently —NR—, —O—, —CRR′— or a single bond, and q is an integer of from 0 to 3. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A(CH2)r B—, wherein A and B are independently —CRR′—, —O—, —NR—, —S—, —S(O)—, S(O)2—, —S(O)2NR′— or a single bond, and r is an integer of from 1 to 4. One of the single bonds of the new ring so formed may optionally be replaced with a double bond. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula —(CRR′)s-X—(CR″R′″)d-, where s and d are independently integers of from 0 to 3, and X is —O—, —NR′—, —S—, —S(O)—, —S(O)2—, or —S(O)2NR′—. The substituents R, R′, R″ and R′″ are preferably independently selected from hydrogen or substituted or unsubstituted (C1-C6)alkyl.
The terms “halo” or “halogen,” by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl. For example, the term “halo(C1-C4)alkyl” is mean to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
Examples of haloalkyl include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl, and pentachloroethyl.“Alkoxy”represents an alkyl group as defined above with the indicated number of carbon atoms attached through an oxygen bridge. C1-6 alkoxy, is intended to include C1, C2, C3, C4, C5, and C6 alkoxy groups. Examples of alkoxy include, but are not limited to, methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, s-butoxy, t-butoxy, n-pentoxy, and s-pentoxy. “Cycloalkyl” is intended to include saturated ring groups, such as cyclopropyl, cyclobutyl, or cyclopentyl. 3-7 cycloalkyl is intended to include C3, C4, C5, C6, and C7 cycloalkyl groups. Alkenyl” is intended to include hydrocarbon chains of either straight or branched configuration and one or more unsaturated carbon-carbon bonds that may occur in any stable point along the chain, such as ethenyl and propenyl.
“Halo” or “halogen” as used herein refers to fluoro, chloro, bromo, and iodo.
As used herein, the term “hetero” , mean, unless otherwise stated, “heteroatom” or “heteroradical”(namely radical containing heteroatom), includeing atoms other than carbon (C) and hydrogen (H), also including the radicals containing these aforesaid heteroatoms. Examples include oxygen (O), nitrogen (N) sulfur (S), silicon (Si), germanium (Ge), aluminum (Al) and boron (B), also include optionally substituted —C(═O)N(H)—, —N(H)—, —C(═NH)—, —S(═O)2 N(H)—, or —S(═O) N(H)—.
“Ring” as used herein, means a substituted or unsubstituted cycloalkyl, heterocycloalkyl, cycloalkenyl, heterocycloalkenyl, cycloalkynyl, heterocycloalkynyl, aryl, or heteroaryl. A ring includes mono, bi, sprio, fused, and bridged ring moieties. The number of atoms in a ring is typically defined by the number of members in the ring. For example, a “5- to 7-membered ring” means there are 5 to 7 atoms in the encircling arrangement. Unless otherwise specified, the ring optionally includes one to three heteroatoms. Thus, the term “5- to 7-membered ring” includes, for example phenyl, pyridinyl and piperidinyl. The term “5- to 7-membered heterocycloalkyl ring”, on the other hand, would include pyridinyl and piperidinyl, but not phenyl. The term “ring” further includes a ring system comprising more than one “ring”, wherein each “ring” is independently defined as above.
As used herein, the term “heterocycle” or “heterocyclic group” is intended to mean a stable monocyclic, bicyclic, or tricyclic ring containing heteroatom or heteroradical, which is saturated, partially unsaturated or unsaturated (aromatic), and which consists of carbon atoms and 1,2, 3, or 4 ring heteroatoms independently selected from the group consisting of N, O and S and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring. The nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., NO and S (O) p). The nitrogen atom may be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined). The heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure. The heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable. A nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and O atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and O atoms in the heterocycle is not more than 1. As used herein, the term “aromatic heterocyclic group”or“heteroaryl”is intended to mean a stable 5,6, or 7-membered monocyclic or bicyclic or 7,8, 9, or 10-membered bicyclic heterocyclic aromatic ring which consists of carbon atoms and 1,2, 3, or 4 heterotams independently selected from the group consisting of N, O and S. The nitrogen atom may be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined). The nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., NO and S (O) p). It is to be noted that total number of S and 0 atoms in the aromatic heterocycle is not more than 1. Bridged rings are also included in the definition of heterocycle. A bridged ring occurs when one or more atoms (i.e., C, O, N, or S) link two non-adjacent carbon or nitrogen atoms. Preferred bridges include, but are not limited to, one carbon atom, two carbon atoms, one nitrogen atom, two nitrogen atoms, and a carbon-nitrogen group. It is noted that a bridge always converts a monocyclic ring into a trycyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge.
Examples of heterocycles include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzoxazolinyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H, 6H-1, 5,2-dithiazinyl, dihydrofuro [2, 3-b] tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1H-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, 3H-indolyl, isatinoyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, methylenedioxyphenyl, morpholinyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-oxadiazolyl, 1,2, 5-oxadiazolyl, 1,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxindolyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, piperidonyl, 4-piperidonyl, piperonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, 2H-pyrrolyl, pyrrolyl, quinazolinyl, quinolinyl, 4H-quinolizinyl, quinoxalinyl, quinuclidinyl, tetrahydrofuranyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, tetrazolyl, 6H-1,2,5-thiadiazinyl, 1,2,3-thiadiazolyl, 1,2, 4-thiadiazolyl, 1,2,5-thiadiazolyl, 1,3,4-thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1,2, 3-triazolyl, 1,2,4-triazolyl, 1,2,5-triazolyl, 1,3,4-triazolyl, and xanthenyl. Also included are fused ring and spiro compounds containing, for example, the above heterocycles.
The term “hydrocarbyl” or its lower concept(such as alkyl, alkenyl, alkynyl and phenyl etc.) by itself or as part of another substituent, means, unless otherwise stated, a straight or branched chain, or cyclic hydrocarbon radical, or combination thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals, having the number of carbon atoms designated (i.e. C1-C10 means one to ten carbons). “Hydrocarbyl” include, but are not limited to, aliphatic hydrocarbyl and aromatic hydrocarbyl, and the aliphatic hydrocarbyl include linear and cyclic ones, specifically including but not limited to, alkyl, alkenyl, and alkynyl, and the aromatic hydrocarbyl include, but are not limited to, 6-12 membered aromatichydrocarbyl, for example, benzene, and naphthalene. In some embodiments, the term “alkyl” means a straight or branched chain, or combinations thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals. Examples of saturated hydrocarbon radicals include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, homologs and isomers of, for example, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like. An unsaturated alkyl group is one having one or more double bonds or triple bonds. Examples of unsaturated alkyl groups include, but are not limited to, vinyl, 2-propenyl, crotyl, 2-isopentenyl, 2-(butadienyl), 2,4-pentadienyl, 3-(1,4-pentadienyl), ethynyl, 1- and 3-propynyl, 3-butynyl, and the higher homologs and isomers.
The term “heterohydrocarbyl” or its lower concept (such as heteroalkyl, heteroalkenyl, teteroalkynyl and heteroaryl etc.) by itself or in combination with another term, means, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom. In some embodiments, the term “heteroalkyl,” by itself or in combination with another term, means a stable straight or branched chain, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom. In an exemplary embodiment, the heteroatoms can be selected from the group consisting of B, O, N and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized. The heteroatom(s) B, O, N and S may be placed at any interior position of the heterohydrocarbyl group(including the position at which the hydrocarbyl group is attached to the remainder of the molecule). Examples include, but are not limited to, —CH2-CH2-O—CH3, —CH2-CH2-NH—CH3, —CH2-CH2-N(CH3)-CH3, —CH2-S—CH2-CH3, —CH2-CH2,—S(O)—CH3, —CH2-CH2-S(O)2-CH3, —CH═CH—O—CH3, —CH2-CH═N—OCH3, and —CH═CH—N(CH3)-CH3. Up to two heteroatoms may be consecutive, such as, for example, —CH2—NH—OCH3.
The terms “alkoxy,” “alkylamino” and “alkylthio” (or thioalkoxy) are used in their conventional sense, and refer to those alkyl groups attached to the remainder of the molecule via an oxygen atom, an amino group, or a sulfur atom, respectively.
The terms “cyclohydrocarbyl”, “heterocyclohydrocarbyl”, or their lower concepts(such as aryl, heteroaryl, cycloalkyl, heterocycloalkyl, cycloalkenyl, heterocycloalkenyl, cycloalkynyl, and heterocycloalkynyl etc.) by themselves or in combination with other terms, represent, unless otherwise stated, cyclic versions of “hydrocarbyl” or “heterohydrocarbyl”, respectively. Additionally, for heterohydrocarbyl or heterocyclohydrocarbyl(such as heteroalkyl and heterocycloalkyl), a heteroatom can occupy the position at which the heterocycle is attached to the remainder of the molecule. Examples of cycloalkyl include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like. Non-limiting examples of heterocycloalkyl moieties include 1-(1,2,5,6-tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4-morpholinyl, 3-morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1-piperazinyl, 2-piperazinyl.
The term “aryl” means, unless otherwise stated, a polyunsaturated, aromatic, substituent that can be a single ring or multiple rings (preferably from 1 to 3 rings), which are fused together or linked covalently. The term “heteroaryl” refers to aryl groups (or rings) that contain from one to four heteroatoms. In an exemplary embodiment, the heteroatom is selected from B, N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. A heteroaryl group can be attached to the remainder of the molecule through a heteroatom. Non-limiting examples of aryl and heteroaryl groups include phenyl, 1-naphthyl, 2-naphthyl, 4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 2-phenyl-4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 5-benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1-isoquinolyl, 5-isoquinolyl, 2-quinoxalinyl, 5-quinoxalinyl, 3-quinolyl, and 6-quinolyl. Substituents for each of the above noted aryl and heteroaryl ring systems are selected from the group of acceptable substituents described below.
For brevity, the term “aryl” when used in combination with other terms (e.g., aryloxy, arylthio, arylalkyl) includes both aryl and heteroaryl rings as defined above. Thus, the term “arylalkyl” is meant to include those radicals in which an aryl group is attached to an alkyl group (e.g., benzyl, phenethyl, pyridylmethyl and the like) including those alkyl groups in which a carbon atom (e.g., a methylene group) has been replaced by, for example, an oxygen atom (e.g., phenoxymethyl, 2-pyridyloxymethyl, 3-(1-naphthyloxy)propyl, and the like).
The term “leaving group” means a functional group or atom which can be displaced by another functional group or atom in a substitution reaction, such as a nucleophilic substitution reaction. By way of example, representative leaving groups include triflate, chloro, bromo and iodo groups; sulfonic ester groups, such as mesylate, tosylate, brosylate, nosylate and the like; and acyloxy groups, such as acetoxy, trifluoroacetoxy and the like.
The term “protecting group” includes bu not limited to “amino-protecting group”, “hydroxy-protecting group” and “thiol-protecting group”. The term “amino-protecting group” means a protecting group suitable for preventing undesired reactions at an amino nitrogen. Representative amino-protecting groups include, but are not limited to, formyl; acyl groups, for example alkanoyl groups, such as acetyl, trichloroacetyl or trifluoroacetyl; alkoxycarbonyl groups, such as tert-butoxycarbonyl (Boc); arylmethoxycarbonyl groups, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); arylmethyl groups, such as benzyl (Bn), trityl (Tr), and 1,1-di-(4′-methoxyphenyl)methyl; silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like. The term “hydroxy-protecting group” means a protecting group suitable for preventing undesired reactions at a hydroxy group. Representative hydroxy-protecting groups include, but are not limited to, alkyl groups, such as methyl, ethyl, and tert-butyl; acyl groups, for example alkanoyl groups, such as acetyl; arylmethyl groups, such as benzyl (Bn), p-methoxybenzyl (PMB), 9-fluorenylmethyl (Fm), and diphenylmethyl (benzhydryl, DPM); silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
Where the “antibody” is a cell-binding agent, it binds to an antigen that is a polypeptide and may be a transmembrane molecule (e.g. receptor) or a ligand such as a growth factor.
Exemplary antigens include molecules such as renin; a growth hormone, including human growth hormone and bovine growth hormone; growth hormone releasing factor; parathyroid hormone; thyroid stimulating hormone; lipoproteins; alpha-1-antitrypsin; insulin A-chain; insulin B-chain; proinsulin; follicle stimulating hormone; calcitonin; luteinizing hormone; glucagon; clotting factors such as factor vmc, factor IX, tissue factor (TF), and von Willebrands factor; anti-clotting factors such as Protein C; atrial natriuretic factor; lung surfactant; a plasminogen activator, such as urokinase or human urine or tissue-type plasminogen activator (t-PA); bombesin; thrombin; hemopoietic growth factor; tumor necrosis factor-alpha and -beta; enkephalinase; RANTES (regulated on activation normally T-cell expressed and secreted); human macrophage inflammatory protein (MIP-1-alpha); a serum albumin, such as human serum albumin; Muellerian-inhibiting substance; relaxin A-chain; relaxin B-chain; prorelaxin; mouse gonadotropin-associated peptide; a microbial protein, such as beta-lactamase; DNase; IgE; a cytotoxic T-lymphocyte associated antigen (CTLA), such as CTLA-4; inhibin; activin; vascular endothelial growth factor (VEGF); receptors for hormones or growth factors; protein A or D; rheumatoid factors; a neurotrophic factor such as bone-derived neurotrophic factor (BDNF), neurotrophin-3, -4, -5, or -6 (NT-3, NT4, NT-5, or NT-6), or a nerve growth factor such as NGF-β; platelet-derived growth factor (PDGF); fibroblast growth factor such as aFGF and bFGF; epidermal growth factor (EGF); transforming growth factor (TGF) such as TGF-alpha and TGF-beta, including TGF-β, TGF-β2, TGF-beta]3, TGF-β4, or TGF-beta5; insulin-like growth factor-I and -II (IGF-I and IGF-II); des(I-3)-IGF-I (brain IGF-I), insulin-like growth factor binding proteins, EpCAM, GD3, FLT3, PSMA, PSCA, MUCI , MUC16, STEAP, CEA, TENB2, EphA receptors, EphB receptors, folate receptor, FOLRI, mesothelin, cripto, alphavbetae, integrins, VEGF, VEGFR, tarnsferrin receptor, IRTAI, IRTA2, IRTA3, IRTA4, IRTA5; CD proteins such as CD2, CD3, CD4, CD5, CD6, CD8, CDI 1 , CD14, CDI 9, CD20, CD21, CD22, CD23, CD25, CD26, CD28, CD30, CD33, CD36, CD37, CD38, CD40, CD44, CD52, CD55, CD56, CD59, CD70, CD79, CD80, CD81 , CDI 03, CDI 05, CDI 34, CDI 37, CDI 38, CDI 52; erythropoietin; osteoinductive factors; immunotoxins; a bone morphogenetic protein (BMP); an interferon, such as interferon-alpha, -beta, and -gamma; colony stimulating factors (CSFs), e.g., M-CSF, GM-CSF, and G-CSF; interleukins (ILs), e.g., IL-I to IL-IO; superoxide dismutase; T-cell receptors; surface membrane proteins; decay accelerating factor; viral antigen such as, for example, a portion of the HIV envelope; transport proteins; homing receptors; addressins; regulatory proteins; integrins, such as CDI Ia, CDI Ib, CDI Ic, CDI 8, an ICAM, VLA-4 and VCAM; a tumor associated antigen such as HER2, HER3 or HER4 receptor; and fragments of any of the above-listed polypeptides, antibody mimics Adnectins (US appl 20070082365), or an antibody which binds to one or more tumor-associated antigens or cell-surface receptors disclosed in US Publication No. 20080171040 or US Publication No. 20080305044 and are incorporated in their entirety by reference.
Concrete antigens for antibodies encompassed by the present invention include CD proteins such as CD2, CD3, CD4, CD5, CD6, CD8, CD11, CD14, CD18, CD19, CD20, CD21, CD22, CD 25, CD26, CD27, CD28, CD30, CD33, CD36, CD37, CD38, CD40, CD44, CD52, CD55, CD56, CD70, CD79, CD80, CD81, CD103, CD105, CD134, CD137, CD138, and CD152; members of the ErbB receptor family such as the EGF receptor, HER2, HER3 or HER4 receptor; cell adhesion molecules such as LFA-I, MacI, pI50.95, VLA-4, ICAM-I, VCAM, EpCAM, alpha4/beta7 integrin, and alpha5/beta3 integrin including either alpha or beta subunits thereof (e.g. anti-CD11a, anti-CD18 or anti-CD11b antibodies); growth factors such as VEGF; tissue factor (TF); TGF-beta; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C etc.
The most concrete targets herein are IGF-IR, CanAg, EphA2, MUC1, MUC16, VEGF, TF, CD19, CD20, CD22, CD27, CD33, CD37, CD38, CD40, CD44, CD56, CD138, CA6, Her2/neu, EpCAM, CRIPTO (a protein produced at elevated levels in a majority of human breast cancer cells), darpins, alphav/beta3 integrin, alphav/beta5 integrin, alphav/beta6 integrin, TGF-beta, CD11a, CD18, Apo2 and C242 or an antibody which binds to one or more tumor-associated antigens or cell-surface receptors disclosed in US Publication No. 20080171040 or US Publication No. 20080305044 and are incorporated in their entirety by reference.
Concrete antigens for antibodies encompassed by the present invention also include CD proteins such as CD3, CD4, CD8, CD19, CD20, CD27, CD34, CD37, CD38, CD46, CD56, CD70 and CD138; members of the ErbB receptor family such as the EGF receptor, HER2, HER3 or HER4 receptor; cell adhesion molecules such as LFA-I, Macl, pI50.95, VLA-4, ICAM-I, VCAM, EpCAM, alpha4/beta7 integrin, and alpha v/beta3 integrin including either alpha or beta subunits thereof (e.g. anti-CD11a, anti-CD 18 or anti-CD11b antibodies); growth factors such as VEGF; tissue factor (TF); TGE-β; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C, etc. The most concrete targets herein are IGF-IR, CanAg, EGF-R, EGF-RvIII, EphA2, MUCI, MUC16, VEGF, TF, CD19, CD20, CD22, CD27, CD33, CD37, CD38, CD40, CD44, CD56, CD70, CD 138, CA6, Her2/neu, CRIPTO (a protein produced at elevated levels in a majority of human breast cancer cells), alpha v/beta3 integrin, alpha v/beta5 integrin, TGE-β, CDI Ia, CD18, Apo2, EpCAM and C242.
Monoclonal antibody techniques allow for the production of specific cell-binding agents in the form of monoclonal antibodies. Particularly well known in the art are techniques for creating monoclonal antibodies produced by immunizing mice, rats, hamsters or any other mammal with the antigen of interest such as the intact target cell, antigens isolated from the target cell, whole virus, attenuated whole virus, and viral proteins such as viral coat proteins. Sensitized human cells can also be used. Another method of creating monoclonal antibodies is the use of phage libraries of sFv (single chain variable region), specifically human sFv (see, e.g., Griffiths et al, U.S. Pat. No. 5,885,793; McCafferty et al, WO 92/01047; Liming et al, WO 99/06587.)
Selection of the antibody is a matter of choice that depends upon the particular cell population that is to be targeted, but in general monoclonal antibodies and epitope binding fragments thereof are preferred, if an appropriate one is available.
For example, the monoclonal antibody My9 is a murine IgG2a antibody that is specific for the CD33 antigen found on Acute Myeloid Leukemia (AML) cells (Roy et al. Blood 77:2404-2412 (1991)) and can be used to treat AML patients. Similarly, the monoclonal antibody anti-B4 is a murine IgGi that binds to the CD 19 antigen on B cells (Nadler et al, J Immunol. 131:244-250 (1983)) and can be used if the target cells are B cells or diseased cells that express this antigen such as in non-Hodgkin's lymphoma or chronic lymphoblastic leukemia. The antibody N901 is a murine monoclonal IgGi antibody that binds to CD56 found on small cell lung carcinoma cells and on cells of other tumors of neuroendocrine origin (Roy et al. J Nat. Cancer Inst. 88:1136-1145 (1996)); huC242 is an antibody that binds to the CanAg antigen; Trastuzumab is an antibody that binds to HER2/neu; and anti-EGF receptor antibody binds to EGF receptor.
The drug unit used in this invention can be any cytotoxic, cytostatic or immunosuppressive drug also referred to herein as a cytotoxic, cytostatic or immunosuppressive agent. Examples of drugs include radioactive isotopes, chemotherapeutic agents, and toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including synthetic analogues and derivatives thereof. The Drug unit has an atom that can form a bond with the Linker Unit, in some embodiments, the Drug unit has a nitrogen atom that can form a bond with the Linker unit. In other embodiments, the Drug unit has a carboxylic acid that can form a bond with the Linker unit. In other embodiments, the Drug unit has a sulfhydryl group that can form a bond with the Linker unit . In other embodiments, the Drug unit has a hydroxy! group or ketone that can form a bond with the Linker unit.
Useful classes of cytotoxic or immunosuppressive agents include, for example, antitubulin agents, auristatins, DNA minor groove binders, DNA replication inhibitors, alkylating agents (e.g. , platinum complexes such as cis-platin, mono(platinum), bis(platinum) and tri-nuelear platinum complexes and carbopiatin), anthracyclin.es, antibiotics, antifolates-, antimetabolites, chemotherapy sensitizers, duocarmycins, etoposides, fluorinated pyrimidines, ionophores, lexitropsins, nitrosoureas, platinois, pre-formirsg compounds, purine antimetabolites, puromycins, radiation sensitizers, steroids, taxanes, topoisornerase inhibitors, vinca alkaloids, or the like. Particularly examples of useful classes of cytotoxic agents include, for example, DNA minor groove binders, DNA alkylating agents, and tubulin inhibitors. Exemplary cytotoxic agents include, for example, auristatins, camptothecins, duocarmycins, etoposides, maytansines and maytansinoids (e.g., DM! and DM4), taxanes, benzodiazepines (e.g., pyrrolo [1,4] benzodiazepines (PBDs). indolinobenzodiazepines, and oxazolidinobenzodiazepines) and vinca alkaloids. Select benzodiazepine containing drugs are described in WO 2010/091 150, WO 2012/1 12708, WO 2007/085930, and WO 201 1/023883.
Individual cytotoxic or immunosuppressive agents include, for example, an androgen, antbramycin (AMC), asparaginase, 5-azacytidine, azathioprine, bleomycin, busuJfan, buthionine sulfoximine, calicheamicin, camptothecin, carbopiatin, carmustine (BSNU), CC-1065, chlorambucil, cisplatin, colchicine, cyclophosphamide, cytarabine, cytidine arabinoside, cytochalasin B, dacarbazine, dactinomycin (formerly actinomycin). daunorubicin, decarbazine, docetaxel, doxorubicin, etoposide, an estrogen, 5-fluordeoxyuridine, 5˜fluorouracil, gemcitabine, gramicidin D, hydroxyurea, idarubicin, ifosfamide, irinoteean, iomustine (CCNU), mayiansine, mechlorethainine, melphalan, 6-mercaptopurine, methotrexate, mithramycin, mitomycin C, mitoxantrone, nitroimidazole, paclitaxei, paly toxin, plicamycin, procarbizine, rhizoxin, streptozotocin, tenoposide, 6-thioguanine, thioTEPA, topotecan, vinblastine, vincristine, vinorelbine, VP-16 and VM-26.
In certain typical embodiments, suitable cytotoxic agents include, for example, DNA minor groove binders (e.g., enediynes and exitropsins, a CBI compound; see also U.S. Pat. No. 6,130,237), duocarmycras (see U.S. Publication No. 20060024317), taxanes (e.g., paclitaxei and docetaxel), puromycins, vinca alkaloids, CC-1065, SN-38, topotecan, morpholino-doxorubicin, rhizoxin, cyanomorpholino-doxorubicin, echinomvcin, combretastatin, netropsin, epothilone A and B, estramustine, cryptophysins, cemadotin, maytansinoids, discodermoiide, eleutherobin, and mitoxantrone.
In certain embodiments, the Drug unit is an anti-tubulin agent. Examples of anti-tubulin agents include, but are not limited to, taxanes (e.g., Taxol® (paclitaxei), Taxotere© (docetaxel)), T67 (Tularik) and vinca alkyloids (e.g., vincristine, vinblastine, vindesme, and vinorelbine). Other antitubulin agents include, for example, baccatin derivatives, taxane analogs (e.g., epothilone A and B), nocodazole, colchicine and colcimid, estramustine, cryptophysins, cemadotin, maytansinoids, combretastatins, discodermoiide, and eleutherobin.
In certain embodiments, the cytotoxic agent is maytansine or a maytansinoid, another group of anti-tubulin agents. (ImmunoGen, Inc.: see also Chari et ah, 1992, Cancer Res. 52:127-131 and U.S. Pat. No. 8,163,888).
In certain embodiments, the Drug unit is an auristatin. Auristatins include, but are not limited to, AE, AFP, AEB, AEVB, MMAF, and MMAE. The synthesis and structure of auristatins are described in U.S. Patent Application Publication Nos. 2003-0083263, 2005-0238649 2005-0009751 , 2009-01 1 1756, and 201 1-0020343: International Patent Publication No. WO 04/010957, international Patent Publication No. WO 02/088172, and U.S. Pat. Nos. 7,659,241 and 8,343,928; each of which is incorporated by reference in its entirety and for all purposes. Exemplary auristatins of the present invention bind tubulin and exert a cytotoxic or cytostatic effect on the desired cell line.
Exemplary auristatin Drug units have the following formula or a pharmaceutically acceptable salt thereof wherein the wavy line indicates site of attachment to the linker unit:

In certain embodiments, the Drug is a benzodiazepine (including benzodiazepine containing drugs e.g., pyrrolo[l,4]benzodiazepiries (PBDs), indolinobenzodiazepines, and oxazo!idino benzodiazepines). PBDs are of the general structure:

but can differ in the number, type and position of substituents, in both their aromatic A rings and pyrrolo C rings, and in the degree of saturation of the C ring. In the B-ring there is either an imine (N═), a carbinolamine(NH—CH(OH))> or a carbinolamine methyl ether (NH—CH(OMe)) at the N10-
General Method to Prepare Linkers, Drug-Linker Conjugates and ADCs:

An amino alcohol 1 is reacted with a hemiacetal such as trifluoroacetaldehyde methyl hemiacetal in a solvent such as toluene at refluxing temperature with concomitant removal of water to give an oxazolidine intermediate 2. Oxazolidine 2 is then reacted with a Grignard or alkali reagent derived from an appropriate precursor to afford an amino alcohol intermediate 3. Oxidation under an appropriate condition such as Jones reagent or periodic acid with catalytic amount of chromium trioxide and followed by esterifaction furnishes the ester intermediate 4. Appropriate manipulation of functional groups on intermediate 4 affords intermediate 5 with suitable handles for further transformations.
Method 2

The amino alcohol 3 in Method 1 can also be prepared in a diastereo-selective manner as described in Gosselin, F., et. al., Org. Lett. 2004, 6, 641 and Roy, A., et. al., J. Org. Chem. 2006, 71, 4320 to give 3a with high distereoselectivity.

Method 4

Intermediate 5 or 6 from Method 1 and intermediate 8 from Method 3 is reacted under an appropriate condition to give intermediate 10. Intermediate 10 is then converted to the 4-nitrophenyl carbonate intermediate 11.

Intermediate 11 from Method 4 is reacted with MMAE under an appropriate condition to give linker-drug intermediate 12.
Method 6

Intermediate 12 is coupled with the desired antibody under an appropriate condition as described in the corresponding experimental section.
General Procedure for Conjugation of Drug-Linker Intermediate to Herceptin:
Step 1: Reduction of Disulfide Bond of Antibody and Purification
To produce 240 mg ADC, 8.955 mL Hereceptin (26.8 mg/mL, around 30 mg·mL) was mixed with 8.95 μL 0.5 M TCEP (final concentration of TCEP in reaction mixture was 0.5 mM), and the reaction was performed at room temperature for 2 hour. Then the reaction solution was loaded on an 1 mL Protein A affinity column to get rid of the remained TCEP and other non-mAb impurities. After the purification, pH of the eluent was adjusted to around 6.0 with 1 M Tris, and the concentration of mAb was measured by nanodrop. The final volume of the solution was about 20 mL, and mAb concentration was 11.6 mg/mL.
Step 2: Conjugation of Drug-Linker and Antibody
The solution was divided equally into to two tubes (10 mL each), and 1 mL DMSO solution of drug-linker intermediate (25 mg/mL) was added to each tube. The ratio of drug-linker intermediate to mAb is 6:1. The reaction was carried out at room temperature for ca 12 hour. When finished, solutions from the two tubes were pooled, centrifuged at 5000 rpm for 5 min and loaded onto a Protein A affinity column to remove the un-reacted drug-Ilinker intermediate. The pH of the eluent from the column was adjusted to 6.0 with 1 M Tris buffer. The drug antibody ratio (DAR) was determined by hydrophobic interaction chromatography (HIC) HPLC.
The invention is now is further described by examples. The examples given below are for illustration purpose and not meant to limit this invention. The compounds of the present invention can be prepared in a number of ways known to one skilled in the art of organic synthesis. The examples of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or by variations thereon as appreciated by those skilled in the art. Concrete methods include, but are not limited to, those described below.
All solvents used are commercially available and are used without further purification. Reactions are typically run using anhydrous solvents under an inert atmosphere of nitrogen. Proton NMR are recorded on Bruker Avance III 400 (400 MHz) spectrometer and chemical shifts are reported as (ppm) down field from tetramethylsilane. Mass spectra are determined on Agilent 1200 series plus 6110 (& 1956A). LC/MS, or Shimadzu MS consisting of a DAD: SPD-M20A (LC) and Shimadzu Micromass 2020 detector. The mass spectrometer is equipped with an electrospray ion source (ESI) operated in a positive or negative mode.
The following abbreviations are used: ADC is antibody-drug conjugate;aq is aqueous; mAb is monoclonal antibody; MTMH is 1-Maleinimidyl-6-trifluoromethylhex-6-yl with the structure of

(S)-MTMH is

(R)-MTMH is

PABO(CO) is

ABA is

MMAE is

Fmoc-Cl is 9-fluorenylmethyl chloroformate; DIAD is diisopropyl azodicarboxylate; Z-Glu-OMe is (S)-4-(((benzyloxy)carbonyl)amino)-5-methoxy-5-oxopentanic acid; HATU is 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate; EDCl is 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide; DMSO is Dimethyl sulfoxide; HOBT is Hydroxybenzotriazole; DCM is dichloromethane; PE is petroleum ether; DMF is N,N-dimethylformamide; DMSO is dimethylsulfoxide; EtOAc is ethyl acetate; EtOH is ethanol; MeOH is methanol; Cbz is benzyloxycarbonyl, a amine protecting group; BOC is tert-butylcarbonyl, amine protecting group; HOAc is acetic acid; NaBH(OAc)3 is sodium triacetoxyborohydride; r.t. is room temperature; THF is tetrahydrofuran; Boc2O is di-tert-butyl dicarbonate; TFA is trifluoroacetic acid; DIPEA is diisopropylethylamine; Pd(dppf)Cl2 is [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II); POCl3 is phosphorus oxychloride; NaH is sodium hydride; LAH is Lithium Aluminium Hydride; Pd(OAc)2 is Palladium (II) acetate; Pd2(dba)3 is tris(dibenzylideneacetone)dipalladium; Pd(PPh3)4 is tetrakis(triphenylphosphine)palladium; Et3SiH is triethylsilane; PPh3 is triphenyl phosphine; Xantphos is 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene; MeSO3H is methanesulfonic acid; Xphos is 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl; Lawesson's reagent is 2,4-Bis(4-methoxylphenyl)-1,3-dithia-2,4-diphosphetane-2,4-disulfide; NBS is N-bromosuccinimide; t-BuOK is potassium tert-butylate;
Compounds are named either manually or by using ChemDraw®, or using vendors catalogue name if commercially available.
To describe the present invention in more detail, the following examples are provided. However, the scope of the present invention is not limited to these.


To a stirred suspension of Mg (10.2 g, 420 mmol) with a few small crystals of iodine in 80 mL of THF was added a small portion of a solution of 6-bromohex-1-ene (22.8 g, 140 mmol) in anhydrous THF (40 mL). After the reaction initiated, the remaining 6-bromohex-1-ene THF solution was added dropwise at a rate to maintain a gentle refluxing. After complete addition, the resulting mixture was heated at 70° C. for 1 h. The Grignard reagent (˜120 mL in THF) obtained was then used directly for the next step.
The Grignard reagent from above was cooled to −78° C. and a solution of (4S)-4-isopropyl-2-(trifluoromethyl)oxazolidine (17 g, 93.4 mmol) in THF (40 mL) was added dropwise. The resulting mixture was warmed up to 0° C. and stirred for 1.5 h. The reaction was quenched with sat. NH4Cl (50 mL), extracted with EtOAc (4×100 mL). The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography elution with Pet. ether/EtOAc (0-4%) to give the title compound (11.5 g, 46%) as a yellow oil.

To a solution of H5IO6 (33.2 g, 145.8 mmol) in anhydrous CH3CN (100.0 mL) was added CrO3 (486 mg, 4.86 mmol) and stirred at room temperature for 1 h. The mixture was then cooled to 0° C. and a solution of (2S)-3-methyl-2-[(1,1,1-trifluorooct-7-en-2-yl)amino]-butan-1-ol (6.5 g, 24.3 mmol, in 20 ml CH3CN) was added dropwise. The resulting mixture was stirred at 0° C. for 1.5 h, quenched with K2HPO4 (16.3 g in 50 mL of H2O) and extracted with EtOAc (3×100 mL). The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography elution with Pet. ether/EtOAc (0-4%-10% EtOAc) to give the title compound (1.04 g, 12.7%) as a yellow oil.

To a solution of (2S)-3-methyl-2-[(1,1,1-trifluorooct-7-en-2-yl)amino]butanoic acid (650 mg, 2.3 mmol) in t-BuOAc (10.0 mL) was added dropwise 70% HClO4 (398 mg, 2.8 mmol) at 0° C. and stirred for 1 h at this temperature. After warming to room temperature and further stirred for 16 h, the reaction mixture was poured into EtOAc (50 mL), washed successively with H2O (30 mL), sat. aq. NaHCO3, (30 ml), and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography elution with Pet. ether/EtOAc (0˜2%˜5%˜10% EtOAc) to give the title compound (0.56 g, 72%) as a colorless oil.

To a solution of (2S)-tert-butyl 3-methyl-2-[(1,1,1-trifluorooct-7-en-2-yl)amino]-butanoate (1.4 g, 4.2 mmol) in anhydrous DCM (6 mL) and MeOH (6 mL) was cooled to −78° C. and charged with O3 for 2 min. The mixture was then warmed to 0° C. and NaBH4 (481 mg, 12.7 mmol) was added. The resulting mixture was stirred at the same temperature for 2 h, quenched with sat. aq NH4Cl (4 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography elution with Pet. ether/EtOAc (4%˜10% EA) to give the title compund 7 (1.05 g, 72%) as a colourless oil.
1H NMR (CDCl3) δ 3.63 (t, J=6.5 Hz, 2H), 3.06 (d, J=6.0 Hz, 1H), 3.01 (d, J=4.6 Hz, 1H), 2.90-2.79 (m, 1H), 1.92-1.80 (m, 1H), 1.67-1.55 (m, 5H), 1.44 (s, 8H), 1.42-1.31 (m, 3H), 0.95-0.83 (m, 6H).

To a solution of triphenyl phosphine (845 mg, 3.2 mmol) in THF (35 mL) at −78° C. under N2 was added DIAD (651 mg, 3.2 mmol) dropwise. The yellow reaction mixture was stirred at −78° C. for 5 min and (2S)-tert-butyl 3-methyl-2-[(1,1,1-trifluoro-7-hydroxyheptan-2-yl)amino]butanoate (1.1 g, 3.22 mmol) in THF (5 mL) was added. After further stirring for 5 min, neopentyl alcohol (142 mg, 1.61 mmol) and 1H-pyrrole-2,5-dione (313 mg, 3.22 mmol) were added to the reaction mixture as solids. The reaction was then stirred overnight at ambient temperature and concentrated. The residue was purified by flash column chromatography elution with Pet. ether/EtOAc (0˜15% EtOAc) to give the title compound (550 mg, 40.7%) as a colorless oil.
1H NMR (CDCl3) δ 6.69 (s, 2 H), 3.52 (t, J=7.2 Hz, 2 H), 3.06-3.01 (m, 1 H), 2.84-2.82 (m, 1 H), 1.89-1.87 (m, 1 H), 1.66-1.52 (m, 8 H), 1.50-1.25 (m, 9 H), 0.95-0.86 (m, 6 H).

To a solution of (2S)-tert-butyl 2-((7-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-1,1,1-trifluoroheptan-2-yl)amino)-3-methylbutanoate (350 mg, 0.84 mmol) in anhydrous DCM (6 mL) under N2 at 0° C. was added TFA (6 mL) dropwise. The resulting mixture was stirred at room temperature for 2 h, concentrated and co-evaporated with toluene (2×10 mL) to remove TFA. The residue was then re-dissloved in anhydrous THF (5 mL) under N2 at 0° C., and triethylamine (251 mg, 2.492 mmol) was added followed by DCC (171 mg, 0.83 mmol). After stirring at room temperature for 5 min, N-hydroxysuccinimide (96 mg, 0.83 mmol) was added. The mixture was stirred at room temperature for 16 h, filtered and concentrated to give the crude title compound (260 mg), which was used for next reaction step without further purification.


To a mixture of (S)-2-amino-3-methylbutan-1-ol (85 g, 824 mmol, 1.0 eq) and 2, 2, 2-trifluoro-1-methoxy-ethanol (75 g, 577 mmol, 0.7 eq) in toluene (1.5 L) was added PPTS (10.4 g, 41.2 mmol, 0.05 Eq) in one portion at room temperature under N2. The mixture was refluxed for 3 h with azeotropic removal of methanol/water (Dean-Stark trap). TLC showed the reaction was complete. The mixture was cooled to room temperature, filtered and concentrated in vacuum to afford the title compound (86 g, crude) as a yellow oil.

To a mixture of magnesium (18.2 g, 750 mmol, 1.5 eq) and I2 (cat) in THF (500 mL) was added dropwise a solution of 6-bromohex-1-ene (81.5 g, 499.8 mmol, 1.0 eq) in THF (500 mL) at 20° C. over a period of 30 min under N2. During the addition, the reaction temperature risen and was maintained at 60° C. After further stirring at 60° C. for another 1.5 h, the mixture was cooled to room temperature and used for the next step without purification.
To Grignard reagent, bromo(hex-5-enyl) magnesium (0.5 M, 500 mL, 1.25 eq) from above at −78° C. under N2 was added dropwise a solution of (4S)-4-isopropyl-2-(trifluoromethyl)oxazolidine (36.6 g, 200 mmol, 1.00 eq) in THF (300 mL). The mixture was stirred at −78° C. for 30 min, then warmed to 20° C. and stirred for 4.5 h. TLC (PE: EA=5:1) showed the reaction was complete. The mixture was quenched with NH4Cl (500 mL) and extracted with EtOAc (1000 mL×3). The combined organic phases were washed with saturated brine (500 mL×2), dried with anhydrous Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (Pet. ether:EtOAc=20:1) to afford the title compound (19 g, 35.5% yield) as a yellow oil.

To a mixture of (2S)-3-methyl-2-[(1,1,1-trifluorooct-7-en-2-yl)amino]butan-1-ol (19 g, 71 mmol, 1 eq) in CH3CN (515 mL) was added pentahydroxy(oxo)-iodane (48.6 g, 213.2 mmol, 3 eq), CrO3 (142 mg, 1.4 mmol, 0.02 eq) and water (4 mL) at WC. The mixture was stirred at 0° C. for 4 h. TLC showed the reaction was complete. The mixture was concentrated in vacuum. The residue was purified by silica gel chromatography (Pet. ether:EtOAc=10:1) to afford the title compound (16 g, 80% yield) as a yellow oil.

To a mixture of (2S)-3-methyl-2-[(1,1,1-trifluorooct-7-en-2-yl)amino]butanoic acid (7 g, 24.9 mmol, 1 eq) in tert-butyl acetate (200 mL) was added 70% HClO4 (7.5 g, 74.7 mmol, 3 eq) in one portion at 30° C. under N2. The mixture was stirred at 30° C. for 3 h. TLC showed the reaction was complete. The mixture was diluted with EtOAc (400 mL) and washed with water (400 mL). The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel chromatography (Pet. ether:EtOAc=60:1) to afford the title compound (7 g, 83.4% yield) as yellow oil.

Ozone was bubbled into a solution of (2S)-tert-butyl 3-methyl-2-[(1,1,1-trifluorooct-7-en-2-yl)amino]butanoate (14.0 g, 41.5 mmol, 1 eq) in DCM (100 mL) and MeOH (10 mL) at −78° C. for 3 min. After excess O3 was purged by N2, NaBH4 (4.7 g, 124.5 mmol, 3 eq) was added at 0° C. The mixture was stirred at 20° C. for 3 h. TLC showed starting material was consumed completely. After quenched by saturated aqueous NH4Cl (50 mL), the mixture was extracted with EtOAc (200 mL). The organic layer was concentrated to give the crude product. The crude product was purified by silica gel chromatography eluted with (Pet. ether:EtOAc=20:1) to give the title compound (11 g, 77.7% yield) as a yellow oil.

To a solution of PPh3 (4.61 g, 17.57 mmol, 1.50 eq) in THF (40 mL) at −78° C. under N2 was added dropwise DIAD (3.55 g, 17.57 mmol, 1.50 eq). After stirred at −78° C. for 5 min, (2S)-tert-butyl 3-methyl-2-[(1,1,1-trifluoro-7-hydroxyheptan-2-yl)amino]butanoate (4.0 g, 11.7 mmol, 1 eq) in THF (5 mL) was added and stirred for 5 min. Followed 2, 2-dimethylpropan-1-ol (774.6 mg, 8.8 mmol, 0.75 eq) and 1H-pyrrole-2,5-dione (1.7 g, 17.6 mmol, 1.5 eq) were added to the reaction mixture as solids. The reaction was stirred for 3 h at 30° C. TLC showed the starting material was consumed. The mixture was concentrated and purified with silica gel column (Pet. ether:EtOAc=10:1) to give the title compound (4.00 g, 9.51 mmol, 81.17% yield) as a white solid.

A mixture of (2S)-tert-butyl 2-((7-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-1,1,1-trifluoro-heptan-2-yl)amino)-3-methylbutanoate (6.0 g, 14.3 mmol, 1 eq) in DCM (40 mL) and TFA (40 mL) was stirred at 30° C. for 12 h. The mixture was then concentrated and purified by prep-HPLC (Instrument: Shimadzu pump LC-20A; Column: SYNERGI 200*50 10 um; Mobile phase: A for H2O (Add 0.75% TFA, v/v) and B for CH3CN; Gradient: B 30-60%; Gradient Time: 30 min; Retention Time: 25-30 min; Flow rate: 80 mL/min) to afford peak 1: INTERMEDIATE 2 (3.20 g, 8.78 mmol, 61.55% yield) as a white solid and peak 2, INTERMEDIATE 3 (1.10 g, 3.02 mmol, 21.16% yield) as a yellow oil.
INTERMEDIATE 2, (R)-MTMH-Val: 1H NMR (CDCl3) δ 6.72 (s, 2H), 3.54 (t, 2H), 3.30 (d, 1H), 3.00 (m, 1H), 2.13 (m, 1H), 1.75-1.25 (m, 8H), 1.03 (d, 3H), 0.98 (d, 3H). LCMS: 365.1 (MO.


To a solution of L-citrulline (20.00 g, 114.16 mmol, 1.00 eq) in 10% Na2CO3 (200 mL) was added dropwise a solution of Fmoc-Cl (35.40 g, 136.84 mmol, 1.20 Eq) in dioxane (100 mL) at 0° C. over a period of 5 min under N2. The reaction mixture was warmed to 20° C. for 4 h. TLC showed the starting material was consumed completely. The reaction mixture was added H2O (50 mL). The aqueous phase was separated and adjusted to PH 2˜3 with 2M HCl (50 mL). The mixture was extracted with EtOAc (150 mL×3). The combined organic phases were washed with saturated brine (100 mL×2), dried over anhydrous Na2SO4, filtered and concentrated to give the title compound (40.00 g, 100.65 mmol, 88.17% yield) as a white solid. LCMS(ESI): 398.1 (MH+).

To a solution of Fmoc-cit (40.00 g, 100.65 mmol, 1.00 Eq) and 4-aminobenzyl alcohol (14.87 g, 120.78 mmol, 1.2 eq) in DCM (1000 mL) was added dropwise a solution of EEDQ (49.78 g, 201.30 mmol, 2.0 eq) in MeOH (500 mL) at 20° C. over a period of 30 min under N2. The reaction mixture was stirred at 20° C. for 16 h. TLC (MeOH:EtOAc=1:10) showed the starting material was consumed completely. The reaction mixture was concentrated and the residue was washed with tert-butyl methyl ether (200 mL×3), filtered and concentrated to give the crude title compound (45.00 g, 89.54 mmol, 88.96% yield) as a white solid.

To a solution of Fmoc-cit-PAB-OH (15.00 g, 29.85 mmol, 1.00 Eq) in DMF (124.5 mL) was added dropwise a solution of piperidine (21.93 g, 257.58 mmol, 8.63 Eq) in DMF (20 mL) at 20° C. over a period of 10 min. The reaction mixture was stirred at 20° C. for 3 h. TLC (DCM:MeOH=5:1) showed the starting material was consumed completely. The reaction was washed with DMF (30 mL×2). The combined organic phases were concentrated to give the residue which was purified by pre-HPLC (formic acid) to give the title compound (7.50 g, 26.76 mmol, 89.63% yield) as a white solid. LCMS(ESI): 218.1 (MH+).


To a solution of Z-Glu-OMe (10 g, 33.87 mmol) in dry DMF (100 mL) was added DIPEA (13.13 g, 101.60 mmol), HATU (15.45 g, 40.64 mmol) and morpholine (4.43 g, 50.8 mmol) and then stirred at 20° C. for 16 h. The mixture was poured into water (200 ml) and EtOAc (500 ml). The organic layer was separated, washed with water (100 mL×2) and brine (100 mL), dried over anhydrous NaSO4 and concentrated to give the title compound (11 g, crude) as an oil. MS: 365 (MH+)

To a solution of (S)-methyl 2-(((benzyloxy)carbonyl)amino)-5-morpholino-5-oxopentanoate (3 g, 8.2 mmol) in dry THF (40 mL) was added a solution of 10M borane methyl sulfide complex in THF (4 mL) at 0° C. and then stirred at 16° C. for 16 h. The mixture was quenched with CH3OH and followed by 1M aqueous HCl (40 mL), stirred at 90° C. for 30 min and evaporated to dryness. The residue was dissolved with water (100 mL), adjusted to pH 10 with aq NaOH and extracted with EtOAc (100 mL×3). The combined organic layers were washed with brine (50 mL×2), dried over anhydrous NaSO4 and concentrated under reduced pressure. The residue from four parallel reactions were pooled and purified by column chromatography (DCM:MeOH=10:1) to give the title compound (5.5 g, yield 47.7%) as an oil. MS: 351 (MH+)

To a solution of (S)-methyl 2-(((benzyloxy)carbonyl)amino)-5-morpholinopentanoate (5.5 g, 15.70 mmol) in THF (50 mL) was added LiOH —H2O (1.32 g, 31.39 mmol). Then the mixture was stirred at 0° C. for 2 h and evaporated to dryness to give the crude title compound (5.0 g, crude).

To a solution of (S)-2-(((benzyloxy)carbonyl)amino)-5-morpholinopentanoic acid (3.00 g, 8.92 mmol) in dry DMF (200 mL) was added HOBt (1.21 g, 8.92 mmol), EDCl (1.71 g, 8.92 mmol) and 4-aminobenzyl alcohol (1.65 g, 13.38 mmol and then stirred at 0° C. for 6 h. The mixture was poured into water (100 mL) and EtOAc (150 mL. The organic layer was separated, washed with water (50 mL×2), brine (50 mL), dried over anhydrous NaSO4 and concentrated under reduced pressure. The residue was purified by column chromatography (DCM:MeOH=10:1) to afford the title compound (1.2 g, yield 30.47%). MS: 442 (MH+)

To a solution of (S)-benzyl (1-((4-(hydroxymethyl)phenyl)amino)-5-morpholino-1-oxopentan-2-yl)carbamate (600.00 mg, 1.36 mmol) in MeOH (50 mL) was added Pd/C (200.00 mg) under N2. The suspension was degassed under vacuum and purged with H2 several times. The mixture was stirred under H2 (15 psi) at 25° C. for 0.5 h. The catalyst was filtered off and the filtrate was concentrated to afford the crude title compound (350.00 mg). MS: 308 (MH+)

To a mixture of trans-4-(aminomethyl)cyclohexanecarboxylic acid (50 g, 318 mmol) and Boc2O (69.4 g, 318 mmol) in H2O (500 mL) was added a solution of NaOH (12.7 g, 318 mmol) in H2O (500 mL) in one portion at 15° C. under N2. The mixture was stirred at 15° C. for 16 h. TLC showed most starting material was consumed. The mixture was adjusted to pH 3 with HOAc (40 mL) and extracted with EtOAc (400 mL×3). The combined organic phases were dried with anhydrous Na2SO4, filtered and concentrated to afford the crude title compound (77 g) as a white solid.
1H NMR (CDCl3) δ 4.62 (m, 1H), 2.98 (m, 2H), 2.25 (m, 1H), 2.04 (d, 2H), 1.83 (d, 2H), 1.438-1.37 (m, 12H), 0.96 (m, 2H).
To a mixture of trans-4-(((tert-Butoxycarbonyl)amino)methyl)cyclohexanecarboxylic acid (37.0 g, 143.8 mmol) in THF (1 L) was added LiAlH4 (8.2 g, 215.7 mmol) at 0° C. under N2. The mixture was stirred at 15° C. for 16 h. TLC showed the reaction was completed. The mixture was quenched with water (8.2 mL), followed by 15% aqueous NaOH solution (8.2 mL), water (24.6 mL) and filtered. The filtrate was concentrated and the residue was purified by silica gel chromatography (Pet.ether:EtOAc=2:1) to afford the title compound (19.0 g, 54.3% yield) as a white solid.
1H NMR (CDCl3) δ 4.61 (br s, 1H), 3.45 (d, 2H), 2.98 (t, 2H), 1.81 (m, 4H), 1.44 (m, 11H), 0.95 (m, 4H).

To a mixture of oxalyl chloride (11.9 g, 93.7 mmol) in DCM (375 mL) was added a solution of DMSO (11.0 g, 140.5 mmol) in DCM (675 mL) at −65° C. under N2. The mixture was stirred at −65° C. for 30 min. A solution of tert-butyl ((trans-4-(hydroxymethyl)cyclohexyl)methyl)carbamate (19.00 g, 78.08 mmol) in DCM (150 mL) was then added at −65° C. The mixture was further stirred at −65° C. for another 30 min. TLC showed most starting material was consumed. A solution of Et3N (19.4 g, 191.3 mmol) in DCM (75 mL) was added at −65° C. and stirred for 30 min. The mixture was warmed to 20° C., quenched with sat.NH4Cl solution (150 mL) and extracted with DCM (300 mL×3). The combined organic phases were dried with anhydrous Na2SO4, filtered and concentrated to afford the crude title compound (19 g) as yellow oil.

To a mixture of tert-butyl ((trans-4-formylcyclohexyl)methyl)carbamate (19.0 g, 78.7 mmol) and methyl 2-(((benzyloxy)carbonyl)amino)-2-(dimethoxyphosphoryl)acetate (28.7 g, 86.6 mmol) in DCM (600 mL) was added DBU (18.0 g, 118.1 mmol) in one portion at 20° C. under N2. The mixture was stirred at 20° C. for 2 h. TLC showed the reaction was completed. The mixture was washed with saturated NaHCO3 (300 mL×2). The organic layer was concentrated and the residue was purified by silica gel chromatography (Pet. ether:EtOAc=3:1) to afford the title compound (20 g, 56.9% yield) as a colorless oil.
1H NMR (CDCl3) δ 7.35 (m, 5H), 6.45 (d, 1H), 6.08 (br s, 1H), 5.15 (s, 2H), 4.60 (br s, 1H), 3.74 (s, 3H), 2.97 (m, 2H), 2.32 (m, 1H), 1.77 (m, 4H), 1.45 (s, 9H), 1.12 (m, 2H), 0.95 (m, 3H).

To a mixture of methyl 2-(((benzyloxy)carbonyl)amino)-3-(trans-4-(((tert-butoxycarbonyl) amino)methyl)cyclohexyl)acrylate (20.0 g, 44.8 mmol) in MeOH (150 mL) was added LiOH.H2O (5.6 g, 134.4 mmol) in H2O (50 mL) at 25° C. under N2. The mixture was stirred at 25° C. for 16 h. TLC showed the reaction was completed. The mixture was adjusted to pH 3 with HOAc and concentrated to remove MeOH. The aqueous phase was extracted with ethyl acetate (50 mL×3). The combined organic phases were dried with anhydrous Na2SO4, filtered and concentrated to afford the crude title compound (18.00 g) as a colorless oil.

A mixture of 2-(((Benzyloxy)carbonyl)amino)-3-(trans-4-(((tert-butoxycarbonyl)amino)methyl)cyclohexyl)acrylic acid (18.0 g, 41.6 mmol), 4-aminobenzy alcohol (7.7 g, 62.4 mmol), HATU (23.7 g, 62.4 mmol) and DIPEA (16.1 g, 124.9 mmol) in DMF (600 mL) was stirred at 25° C. for 16 h under N2. TLC showed the reaction was completed. The mixture was diluted with ethyl acetate (600 mL) and washed with sat.NH4Cl solution (600 mL×3). The organic layer was separated, dried with anhydrous Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (Pet. ether:EtOAc=1:1) to afford the title compound (14.00 g, 26.04 mmol, 62.56% yield) as a brown solid. MS: 538 (MH+).

To a solution of Benzyl (1-(trans-4-(((tert-butoxycarbonyl)amino)methyl)cyclohexyl)-3-((4-(hydroxylmethyl)phenyl)amino)-3-oxoprop-1-en-2-yl)carbamate (7.0 g, 13.0 mmol) in MeOH (100 mL) was added 10% Pd/C (1.0 g) under N2. The suspension was degassed under vacuum and purged with H2 several times. The mixture was stirred under H2 (15psi) at 25° C. for 20 min. TLC showed the starting material was consumed completely. The reaction mixture was filtered and the filtrate was concentrated. The residue was purified by prep-HPLC to give the title compound (1.4 g, 26.3% yield) as a white solid. MS: 406 (MH+)

To a mixture of 4-aminobutanoic acid (5.0 g, 48.5 mmol, 1 eq) in 10% aqueous Na2CO3 solution (16.5 g, 155.7 mmol, 3.2 eq, 150 mL H2O) was added dropwise a solution of (9H-fluoren-9-yl)methyl carbonochloridate (13.8 g, 53.3 mmol, 1.1 eq) in dioxane (100 mL) at 0° C. over a period of 10 min under N2. The mixture was stirred at 20° C. for 4 h. TLC showed the reaction was completed. The mixture was separated and adjusted to PH 2-3 with 2M HCl and extracted with EtOAc (250 mL×3). The combined organic phases were washed with saturated brine (150 mL×2), dried over anhydrous Na2SO4, filtered and concentrated in vacuum to afford the crude title compound (16 g) as a white solid. LCMS: 326.1 (MH+)

To a solution of 4-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)butanoic acid (16.0 g, 49 mmol, 1 eq) in DMF (200 mL) was added then HATU (20.6 g, 54.1 mmol, 1.1 eq) and DIPEA (19.1 g, 147.5 mmol, 3 eq) at 20° C. and stirred for 30 min. Then 4-aminobenzyl alcohol (7.3 g, 59.0 mmol, 1.2 eq) was added in one portion at 20° C. The mixture was stirred at 20° C. for 16 h. TLC showed the reaction was completed. The reaction mixture was concentrated in vacuum. The residue was washed with TBME (300 mL×3), filtered and concentrated to afford the crude title compound (15 g) as a yellow solid. LCMS: 431.2 (MH+)

To a solution of (9H-fluoren-9-yl)methyl (4-((4-(hydroxymethyl)phenyl)amino)-4-oxobutyl)-carbamate (5.0 g, 11.6 mmol, 1 eq) in DMF (40 mL) was added piperidine (7.9 g, 92.9 mmol, 8 eq) in one portion at 20° C. Then the mixture was stirred at 20° C. for 3 h. TLC (DCM: MeOH=5:1) showed starting material was consumed completely. The reaction mixture was concentrated to give the crude title compound (5 g) as a yellow solid. LCMS: 209.1 (MH+)

To a solution of 4-amino-N-(4-(hydroxymethyl)phenyl)butanamide (600 mg, 2.9 mmol, 1 eq) and Fmoc-cit (1.3 g, 3.2 mmol, 1.1 eq) in DCM (8 mL) was added slowly a solution of EEDQ (1.4 g, 5.8 mmol, 2 eq) in MeOH (8 mL) at 20° C. The mixture was stirred at 20° C. for 16 h. TLC (DCM:MeOH=5:1) showed the reaction was completed. The reaction mixture was concentrated and the residue was purified by prep-TLC (SiO2, DCM:MeOH=5:1) to give the title compound (440 mg, 26% yield) as a white solid. LCMS: 588.3 (MH+)

To a solution of Fmoc-Cit-ABA-PAB-OH (440 mg, 749 umol, 1 Eq) in DMF (10 mL) was added piperidine (510 mg, 6 mmol, 8 eq) in one portion at 20° C. The mixture was stirred at 20° C. for 3 h. TLC (DCM:MeOH=5:1) showed the reaction was completed. The reaction mixture was concentrated and the residue was purified by prep-TLC (SiO2, DCM:MeOH=5:1) to give the crude title compound (320 mg) as a white solid. LCMS: 366.2 (MH+)


To a solution of crude (2S)-2,5-dioxopyrrolidin-1-yl-2-{[7-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-1,1,1-trifluoro-heptan-2-yl]amino}-3-methylbutanoate (INTERMEDIATE 1) (0.56 mmol) in DMF (5 mL) was added Cit-PAB-OH (INTERMEDIATE 4) (0.56 mmol) under N2. The resulting mixture was stirred at room temperature for 16 h. Solvent was removed in vacuo. The thick oily residue was washed with tert-butyl methyl ether (10 mL×2) to give the crude title compound, which was used for next step without further purification. LCMS(ESI): 627.3 (MH+)

To a solution of MTMH-Val-Cit-PAB-OH (260 mg crude, 0.56 mmol) in DMF (3 mL) was added bis(4-nitrophenyl) carbonate (340 mg 1.12 mmol) and DIPEA (108 mg, 0.84 mmol). The mixture was stirred at room temperature for about 1 h and concentrated. The residue was washed with tert-butyl methyl ether (10 mL×2) to give the title compound, which was used directly for next step without further purification. LCMS(ESI): 792.3 (MH+)

To a solution of MMAE (0.28 mmol) and MTMH-Val-Cit-PAB (4-nitrophenyl) carbonate (0.56 mmol) in anhydrous DMF (2.5 mL) was added HOBt (23 mg, 0.17 mmol). After stirring at room temperature for 2 min, pyridine (0.5 mL) was added. The mixture was stirred at room temperature for 16 h and then concentrated. The residue was purified by Prep-HPLC to give the title compound (40 mg, 10.4%) as a white solid, LCMS(ESI):=685.9 [(M/2)H+]


To a mixture of (S)-MTMH-Val (INTERMEDIATE 3) (300 mg, 823 umol, 1 eq), HOBt (133.5 mg, 988 umol, 1.20 eq), EDCl (189 mg, 988 umol, 1.20 eq), DIPEA (426 mg, 3.3 mmol, 4 eq) and Cit-PAB-OH (INTERMEDIATE 4) (277 mg, 988 umol, 1.20 eq) in DMF (10 mL) was stirred at 30° C. for 6 h. TLC showed the reaction was completed. The mixture was concentrated in vacuum. The residue was purified by prep-TLC (EtOAc:MeOH=10:1) to afford the title compound (410 mg, 52.3% yield) as a white solid. MS: 627 (MH+)

To a mixture of (S)-MTMH-Val-Cit-PAB-OH (250 mg, 399 umol, 1 eq) in DMF (10 mL) was added DIPEA (77 mg, 598 umol, 1.5 eq) and bis(4-nitrophenyl) carbonate (243 mg, 798 umol, 2.0 eq) in one portion at 25° C. under N2. The mixture was stirred at 25° C. for 15 h. LCMS showed the reaction was completed. The mixture was concentrated in vacuum. The residue was purified by prep-TLC (EtOAc) to afford the title compound (185 mg, 58.6% yield) as a white solid. MS: 792 (MH+)

To a mixture of MMAE (200 mg, 279 umol, 1 eq) and LS)-MTMH-Val-Cit-PAB (4-nitrophenyl) carbonate (250 mg, 316 umol, 1.13 eq) in DMF (10 mL) was added HOBt (25 mg, 185 umol, 0.66 eq) and pyridine (980 mg, 12.4 mmol, 44.5 eq.) in one portion at 25° C. under N2. And then the reaction mixture was allowed to stir at 25° C. for 15 h. LCMS showed the reaction was completed. The mixture was diluted with EtOAc (200 mL) and washed with water (150 ml×3). The organic layer was concentrated to give the residue which was purified by prep-HPLC (FA) to give the title compound (109 mg, 30% yield) as a white solid. LCMS (ESI): 686 [(M/2)H+]

The title compound was prepared in a similar manner as described for (S)-MTMH-Val-Cit-PABO(CO)-MMAE (EXAMPLE 2), step 1 to 3, from (R)-MTMH-Val (INTERMEDIATE 2) and Cit-PAB-OH (INTERMEDIATE 4). LCMS (ESI): 686.1 [(M/2)H+]


To a solution of (S)-MTMH-Val (INTERMEDIATE 2) (350 mg, 0.96 mmol) in DMF (15 mL) was added HOBt (155.75 mg, 1.15 mmol), EDO (220.97 mg, 1.15 mmol), DIPEA (496.59 mg, 3.84 mmol) and (S)-2-amino-N-(4-(hydroxymethyl)phenyl)-5-morpholinopentanamide (INTERMEDIATE 5) (390 mg, 1.1 mmol) under N2 and was then stirred at 25° C. for 6 h. The reaction mixture was evaporated to dryness and the residue was purified by column chromatography (EtOAc:MeOH=10:1) to afford the title compound (400 mg, yield 63.70%). MS: 654 (MO

To a solution of (R)-MTMH-DP1-PAB-OH in DMF (10 mL) was added DIEA (77.10 mg, 0.597 mmol) and bis(4-nitrophenyl) carbonate (349 mg, 1.15 mmol) under N2. The mixture was stirred at 25° C. for 16 h and evaporated to dryness. The residue was purified by prep-TLC (EtOAc) to afford the title compound (300 mg, yield 79.84%). MS: 819 (MO

To a solution of (R)-MTMH-DP1-PAB (4-nitrophenyl) carbonate (200 mg, 0.244 mmol) in DMF (4 mL) was added MMAE (200 mg, 0.278 mmol), HOBt (10 mg, 0.074 mmol) and pyridine (0.8 mL) under N2. The mixture was stirred at 25° C. for 16 h, diluted with EtOAc (50 mL), washed with water (20 mL×2), brine (20 mL) and concentrated. The residue was purified by prep-HPLC (FA) to afford the title compound (160 mg, yield 46.87%). LCMS (ESI): 699.5 [(M/2)H+]

The title compound was prepared in a similar manner as (R)-MTMH-DP1-PAB(CO)-MMAE (EXAMPLE 4) but with piperidine to replace morpholine of INTERMEDIATE 5 for the required corresponding intermediate. LCMS (ESI): 698.6 [(M/2)H+]


To a mixture of (S)-MTMH-Val (INTERMEDIATE 2) (225.0 mg, 617.5 umol) in DMF (12 mL) was added HOBt (108.5 mg, 802.8 umol), EDO (153.9 mg, 802.8 umol) and DIEA (239.4 mg, 1.9 mmol) at 25° C. under N2. After 5 min, tert-butyl ((trans-4-(2-amino-3-((4-(hydroxymethyl)phenyl)amino)-3-oxopropyl)cyclohexyl)methyl)carbamate (INTERMEDIATE 6) (300.5 mg, 741.0 umol) was added and the mixture was further stirred at 25° C. for 4 h. TLC showed that most of starting material was consumed. The mixture was diluted with EtOAc (20 mL), washed with sat.NH4Cl solution (30 mL×3) and concentrated. The residue was purified by prep-TLC and then SFC to give the title compound (peak 1: 140 mg, 30% yield) as a white solid.

The title compound was prepared in a similar manner as described for (S)-MTMH-Val-Cit-PABO(CO)-MMAE (EXAMPLE 2), step 2 to 3, from (R)-MTMH-Boc-DP3-PAB-OH. LCMS [((M−100)/2)H+]: 698.5

To a mixture of (R)-MTMH-Boc-DP3-PABO(CO)-MMAE (200.00 mg, 133.70 umol) in CH3CN (4.25 mL) and H2O (0.25 mL) was added TFA (0.5 mL) in one portion at 25° C. The mixture was stirred at 25-29° C. for 16 h. LCMS showed the reaction was complete. The mixture was concentrated under reduced pressure at 40° C. The residue was purified by pre-HPLC (FA), without further concentration and lyophilized directly to afford the title compound (105.00 mg, 56.3% yield) as a white solid. LCMS: 698.7 [(M/2)H+]


To a mixture of (S)-MTMH-Val-Cit-PAB (4-nitrophenyl) carbonate (420 mg, 531 umol, 1 eq) and 4-aminobutanoic acid (109 mg, 1.1 mmol, 2 eq) in DMF (8 mL) was added DIPEA (205 mg, 1.6 mmol, 3 eq) in one portion at 20° C. under N2. And then the reaction mixture was allowed to stir at 20° C. for 12 h. TLC (Dichloromethane:Methanol=10:1) showed the reaction was complete. The mixture was diluted with EtOAc (50 mL), washed with water (50 ml) and concentrated under vacuum. The residue was purified by pre-TLC (DCM/MeOH=10:1) to give the title compound (236 mg, 46.47% yield, ˜79% purity) as a white solid. MS: 756.3 (MH+)
To a mixture of (S)-MTMH-Val-Cit-PABO(CO)-ABA (50 mg, 66 umol, 1 eq) and DIEA (26 mg, 198 umol, 3 eq) in DMF (3 mL) was added HATU (30 mg, 79 umol, 1.2 eq) in one portion at 10° C. under N2. The mixture was stirred at 10° C. for 30 min, then MMAE (47.5 mg, 66 umol, 1 eq) was added and the mixture was further stirred at 28° C. for 12 h. LCMS showed the reaction was completed. The mixture was concentrated and the residue was purified by pre-HPLC (Column, Phenomenex Synergi C18 150*25*10 um; Condition, 0.225% FA-CAN; Begin B, 45; End B, 75; Gradient Time(min), 10; 100% B Hold Time(min), 2; Flow Rate (ml/min), 25) to give the title compound (25 mg, 26% yield) as a white solid. LCMS: 728.7[(M/2)H+]


To a solution of (S)-MTMH-Val (INTERMEDIATE 3) (383 mg, 1.05 mmol, 1.2 eq) in DMF (8 mL) was added DIPEA (453 mg, 3.50 mmol, 4 eq), HOBt (142 mg, 1.05 mmol, 1.2 eq) and EDCl (202 mg, 1.05 mmol, 1.2 eq) in one portion at 20° C. and stirred for 10 min under N2 Cit-ABA-PAB-OH (INTERMEDIATE 7) (320 mg, 876 umol, 1 eq) was added in one portion at 20° C. and the mixture was further stirred at 20° C. for 16 h under N2. TLC (DCM:MeOH=5:1) showed the reaction was completed. The reaction mixture was concentrated and the residue was purified by prep-TLC (SiO2, DCM:MeOH=5:1) to give the crude title compound (370 mg). LCMS: 712.2 (MH+)

To a solution of (S)-MTMH-Val-Cit-ABA-PAB-OH (370 mg, 520 umol, 1 eq) in DMF (10 mL) was added DIPEA (80.6 mg, 624 umol, 1.2 eq) at 20° C. was added bis(4-nitrophenyl) carbonate (206 mg, 676 umol, 1.3 eq) in one portion at 20° C. under N2. The mixture was stirred at 20° C. for 16 h. TLC (DCM:MeOH=10:1) showed starting material was consumed completely. The mixture was concentrated and the residue was purified by pre-TLC (SiO2, DCM:MeOH=10:1) to give the title compound (70 mg, 79.83 umol, 15.36% yield) as a yellow oil. LCMS: 877.3 (MH+)

To a solution of (S)-MTMH-Val-Cit-ABA-PAB (4-nitrophenyl) carbonate (70 mg, 80 umol, 1 eq) and MMAE (57.3 mg, 79.8 umol, 1 eq) in DMF (6 mL) was added HOBt (5.4 mg, 39.9 umol, 0.5 eq) and pyridine (63.1 mg, 798 umol, 10 eq) at 20° C. The mixture was stirred at 20° C. for 16 h. TLC (DCM:MeOH=10:1) showed the reaction was completed. The mixture was concentrated in vacuum and the residue was purified by Pre-HPLC (FA condition; Column: Phenomenex Synergi Max-RP 250*80 10u; Condition: 0.225% FA-ACN; Begin B: 40; End B: 70) to afford the title compound (40.8 mg, 48.6% yield) as a white solid. LCMS: 728.5 [(M/2)H+].

Herceptin (240 mg) and drug-linker intermediate MTMH-Val-Cit-PAB-OH (11.3 mg), EXAMPLE 1 was coupled according to the general procedure for conjugation of drug-linker intermediate to herceptin to give the title ADC (168 mg, 70%).
Other ADC Examples With Herceptin Were Prepared In A Similar Manner As Example 9

ADC Examples With Erbitux Were Also Prepared In A Similar Manner As Example 9

Screening of Suitable Dipeptide Units for Drug-Linker Intermediates
A convenient method was developed to identify suitable dipeptide units for the use in drug-linker intermediates. Cbz-protected dipeptides incorporated a fluorescent tag (7-amino-4-methylcouramin, 7-AMC) via a para-aminobenzyl carbonyl fragment were prepared by standard peptide chemistry as shown in Scheme 1. These dipeptide substrates were then incubated with bovine cathepsin B under a similar condition as described by Dubowchik (Biorg. Med. Chem. Lett. 1998, 8, 3341-3346). Rate of hydrolysis by bovine cathepsin B was measured by the release of 7-AMC (Table 1). Novel dipeptides with hydrolysis rates slower than the substrate A were rejected. The three dipeptide units in FS-9, FS-10 and FS-11 were selected as additional dipeptide units for drug-linker intermediate preparation.

| TABLE 1 |
| Rate of hydrolysis under the incubation with bovine cathepsin B |
| Rate of Hydrolysis | |||
| Fluorescent Substrate | (nmmol/min/mg) | ||
 | 2.67 | ||
 | 0.84 | ||
 | 0.59 | ||
 | 0.63 | ||
 | 0.84 | ||
 | 0 | ||
 | 0.55 | ||
 | 0 | ||
 | 0 | ||
 | 27.2 | ||
 | 25.1 | ||
 | 20.9 | ||
In Vitro Cytotoxicity Study
- (a) Breast Cancer Cell Line HCC1954
Breast cancer cell line HCC1954 was grown in RPM11640 medium supplemented with 10% fetal bovine serum and maintained in an atmosphere of 5% CO2 in a humidified 37° C. incubator.
The day before treatment, cells were collected and seeded into 96-well plates (2,000 cells per well). On the second day, cells were treated with a dilute concentration of Herceptin and ADCs of this invention (1, 0.3333, 0.1111, 0.037, 0.0123, 0.0041, 0.0014, 0.00046 and 0.00015 μg/ml). Each treatment was performed in triplicate.
After 72 hours treatment, cell viability was assessed by Cell Titer-Glo kit (Promega) according to the manufacturer's instruction.
The study was compared to a control ADC (ADC-HA) with the normal amide bond at the carbon center bearing the trifluoromethyl group.

- (b) A-431 Cell Line
Epidermoid carcinoma cell line A431 was grown in DMEM medium supplemented with 10% fetal bovine serum and maintained in an atmosphere of 5% CO2 in a humidified 37° C. incubator.
The day before treatment, cells were collected and seeded into 96-well plates (2,000 cells per well). On the second day, cells were treated with a dilute concentration of Erbitux and ADCs of this invention (10, 3.33, 1.11, 0.37, 0.123, 0.041, 0.014, 0.0046 and 0.0015 μg/ml). Each treatment was performed in triplicate.
After 72 hours treatment, cell viability was assessed by Cell Titer-Glo kit (Promega) according to the manufacturer's instruction.
The study was compared to a control ADC (ADC-EA) with the normal amide bond at the carbon center bearing the trifluoromethyl group.

In Vivo Efficacy Study
- (a) HCC1954 Xenograft Mice Model
7-8 Week old female NOD-SCID mice were injected with HCC1954 cells subcutaneously. Treatment was started 7 days after cell injection once the mean estimated tumor mass reached 153 mm3. Mice were grouped randomly according to the tumor volume and body weight (8 animals per group). Animals were treated with ADCs of this invention (15 mpk), Herceptin (15 mpk) as well as vehicle control on day 0 and day 15. Animals were euthanized on day 28 after the start of treatment, when the tumor volume of vehicle control group reached a mass of 1092 mm3.
- (b) A-431 Xenograft Mice Model
6-8 Week old female Balb/c nude mice were injected with A431 cells subcutaneously. Treatment was started 10 days after cell injection once the mean estimated tumor mass reached 172 mm3. Mice were grouped randomly according to the tumor volume and body weight (8 animals per group). Animals were treated with ADCs of this invention (10 mpk), Erbitux (10 mpk) as well as vehicle control on day 0 and day 15. Animals were euthanized on day 25 after the start of treatment, when the tumor volume of vehicle control group reached a mass of 2000 mm3.
Claims (29)
1. A linker of formula (I) to connect drugs to antibodies,

wherein,
R is selected from the group consisting of CF2H, CF3, CF2CF3, and PhSO2Me;
Q is selected from the group consisting of

L1 is selected from the group consisting of —(CR1R2)m—(CR3R4)—(CR1R2)n—, —(CR1R2)m—(CR3R4)—O—(CR1R2)n—, and —[(CR1R2)(CR1R2)X]p—(CR1R2)q—;
m is 1, 2, 3, or 4;
n is 1, 2, 3, or 4;
p is 1, 2, 3, 4, 5, or 6;
q is 1 or 2;
R1 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
R2 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
R3 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
R4 is selected from the group consisting of H, C1-3 alkyl, OH, NR5R6, CO2H, P(O)(OH)2, and SO3H, said C1-3 alkyl is optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
R5 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
R6 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
X is selected from the group consisting of NR7, O, S(O)r;
r is 0, 1, or 2;
R7 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
L2 is a di-peptide unit selected from

R8 is selected from the group consisting of methyl, propyl, isopropyl, sec-butyl, benzyl, and

R9 is selected from the group consisting of (CH2)4NH2, (CH2)3NHCONH2, (CH2)3NHC(═NH)NH2,

s is 0, 1, 2, 3, or 4;
L3 is a self-immolative unit selected from the group consisting of

R10 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
each R11 is independently selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2;
two geminal R11 may be optionally joined together to form a 3-6 membered ring with a carbon atom to which they are attached;
two adjacent R11 may be optionally joined together to form a 5-7 membered ring with a carbon atom to which they are attached;
R12 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2; and
R13 is selected from the group consisting of H and C1-3 alkyl optionally substituted with 1, 2 or 3 of substituents independently selected from the group consisting of halogen, OH, NH2, NHMe, and NMe2.
2. A drug-linker conjugate of formula (II),

wherein,
Q′ is selected from the group consisting of

M is a drug; and
other variables are as defined in claim 1 .
3. An anti-drug conjugate (ADC) of formula (III) containing the linker of claim 1 ,

wherein,
Y is an antibody;
a is an integer or decimal selected from 1-8;
M is a drug; and
other variables are as defined in claim 1 .
4. The linker of claim 1 , wherein the antibody is selected from the group consisting of a resurfaced monoclonal antibody, a resurfaced single chain monoclonal antibody, or a resurfaced monoclonal antibody fragment;
or the antibody is selected from the group consisting of a humanized monoclonal antibody, a humanized single chain monoclonal antibody, or a humanized monoclonal antibody fragment;
or the antibody is selected from the group consisting of a chimeric antibody, a chimeric antibody fragment, a domain antibody, or a domain antibody fragment thereof;
or the antibody is selected from the group consisting of MY9, anti-B4, EpCAM, CD2, CD3, CD4, CD5, CD6, CD11, CD19, CD20, CD22, CD26, CD30, CD33, CD37, CD38, CD40, CD44, CD56, CD79, CD105, CD138, EphA receptors, EphB receptors, EGFR, EGFRvIII, HER2, HER3, mesothelin, cripto, alphavbeta3, alphavbeta5, alphavbeta6 integrin or C242;
or the antibody is selected from the group consisting of My9-6, B4, C242, N901, DS6, EphA2 receptor, CD38, IGF-IR, CNTO 95, B-B4, Trastuzumab, Tertuzumab, Bevatuzumab, Sibrotuzumab, Rituximab, and Adalimumab;
or the antibody is selected from the group consisting of Herceptin and Erbitux.
5. The linker of claim 1 , wherein the antibody binds to target cells selected from tumor cells; virus infected cells, microorganism infected cells, parasite infected cells, autoimmune cells, activated cells, myeloid cells, activated T-cells, B cells, or melanocytes; cells expressing one or more of IGF-IR, CanAg, EGFR, MUCI, MUCI 6, VEGF, TF, MY9, anti-B4, EpCAM, CD2, CD3, CD4, CD5, CD6, CD11, CD11a, CD18, CD19, CD20, CD22, CD26, CD30, CD33, CD37, CD38, CD40, CD44, CD56, CD70, CD79, CD105, CD138, EphA receptors, EphB receptors, EGFRvIII, HER2/neu, HER3, mesothelin, cripto, alphavbeta3 integrin, alphavbeta5 integrin, alphavbeta6integrin, Apo2, and C242 antigens; or cells expressing insulin growth factor receptor, epidermal growth factor receptor, and folate receptor.
6. The linker of claim 1 , wherein the drug is a cytotoxic drug, or the drug is a diagnostic or detection reagent.
7. The linker of claim 1 , wherein L1 is selected from the group consisting of:

8. The linker of claim 1 , wherein L2 is selected from the group consisting of

9. The linker of claim 1 , wherein the ring formed by two geminal R11 or two adjacent R11 is cyclohexyl.
10. The linker of claim 1 , wherein L3 is selected from the group consisting of

11. The linker of claim 1 , wherein the moiety of

is selected from the group consisting of

12. The linker of claim 1 , wherein the linker is selected from the group consisting of


13. The drug-linker conjugate of claim 2 , it is selected from the group consisting of


14. The ADC of claim 3 , it is selected from the group consisting of


Y is an antibody, a is an integer or decimal selected from 1-8.
15. A pharmaceutical composition comprising an effective amount of the ADC of claim 3 and a pharmaceutically acceptable carrier, diluent or excipient.
16. A process for treating a patient in need of a medicament for cancers, comprising administering to the patient a medicament comprising an effective amount of the linker in claim 1 .
17. A process for treating or diagnosising a patient in need of a medicament for cancers, comprising administrating to the patient a medicament comprising an effective amount of the ADC of claim 3 .
18. An intermediate compound for the synthesis of the drug-linker conjugate of formula (II) in claim 2 , comprising:

19. The linker of claim 5 , wherein the tumor cells are selected from breast cancer cells, prostate cancer cells, ovarian cancer cells, colorectal cancer cells, gastric cancer cells, squamous cancer cells, small-cell lung cancer cells, and testicular cancer cells.
20. The linker of claim 6 , wherein the drug is selected from the group consisting of maytansinoid, DNA-binding drug and its analog, calicheamicin, doxorubicin and its analog, vinca alkaloid, cryptophycin, dolastatin, auristatin and analog thereof, tubulysin, epothilone, taxoid and siRNA; or the drug is a radio-labeled compound.
21. The linker of claim 20 , wherein the DNA-binding drug is CC-1065; or the drug is labeled with 3H, 18F, 11C, 13N, 15O, 201Tl, 32P, 51Cr, 67Ga, 123I, 125I, 131I, 132I, 131Cs, 113Xe, 133Xe, 169Yb,198Au, 203Hg, 99mTc, 113mIn, 133mIn, 75Se, 186Re, 153Sm, and 89Sr.
22. The drug-linker conjugate of claim 6 , wherein M is selected from the group consisting of

23. The linker of claim 8 , wherein L2 is selected from the group consisting of

24. The drug-linker conjugate of claim 2 , the moiety of

is selected from the group consisting of

25. The linker of claim 12 , wherein the linker is selected from the group consisting of



26. The drug-linker conjugate of claim 13 , it is selected from the group consisting of



27. The ADC of claim 14 , it is selected from the group consisting of



wherein Y is an antibody, a is an integer or decimal selected from 1-8.
28. The ADC of claim 27 , it is selected from the group consisting of


wherein a is an integer or decimal selected from 2-6.
29. The ADC of claim 28 , it is selected from the group consisting of




wherein a is an integer or decimal selected from 2-6.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNPCT/CN2014/082292 | 2014-07-16 | ||
| PCT/CN2014/082292 WO2016008112A1 (en) | 2014-07-16 | 2014-07-16 | Linkers and application towards adc thereof |
| WOPCT/CN2014/082292 | 2014-07-16 | ||
| PCT/CN2015/083840 WO2016008392A1 (en) | 2014-07-16 | 2015-07-13 | Linkers and their application towards adc |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20170202974A1 US20170202974A1 (en) | 2017-07-20 |
| US10280229B2 true US10280229B2 (en) | 2019-05-07 |
Family
ID=55077810
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/326,386 Active 2035-08-13 US10280229B2 (en) | 2014-07-16 | 2015-07-13 | Linkers and their application towards ADC |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10280229B2 (en) |
| EP (1) | EP3169368B1 (en) |
| JP (2) | JP6688790B2 (en) |
| CN (1) | CN107427591B (en) |
| ES (1) | ES2867749T3 (en) |
| TW (1) | TWI579000B (en) |
| WO (2) | WO2016008112A1 (en) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016008112A1 (en) * | 2014-07-16 | 2016-01-21 | Medshine Discovery Inc. | Linkers and application towards adc thereof |
| US10233212B2 (en) | 2015-11-03 | 2019-03-19 | Industrial Technology Research Institute | Compounds, linker-drugs and ligand-drug conjugates |
| CN116672463A (en) * | 2017-02-17 | 2023-09-01 | 浙江特瑞思药业股份有限公司 | Preparation method of targeted CD20 antibody coupling drug, antibody coupling drug and application thereof |
| CN108452319A (en) * | 2017-02-20 | 2018-08-28 | 浙江特瑞思药业股份有限公司 | Target the antibody coupling pharmaceutical preparation of CD20 |
| CN107488231B (en) * | 2017-09-15 | 2020-10-30 | 四川大学 | anti-CD 56 antibodies and uses thereof |
| CN107744592B (en) * | 2017-09-15 | 2020-04-28 | 四川大学 | anti-CD 56 antibody and dolastatin coupling compound and preparation method and application thereof |
| WO2019149116A1 (en) * | 2018-01-30 | 2019-08-08 | 四川科伦博泰生物医药股份有限公司 | Method for preparing conjugate |
| JP2021181405A (en) * | 2018-08-29 | 2021-11-25 | Jsr株式会社 | Polypeptide purification method |
| AU2020204250B2 (en) | 2019-05-20 | 2021-04-08 | Mabplex International Co., Ltd. | One-pot process for preparing intermediate of antibody-drug conjugate |
| CN111670053B (en) * | 2019-05-20 | 2023-08-22 | 烟台迈百瑞国际生物医药股份有限公司 | One-pot method preparation technology of antibody drug conjugate intermediate |
| CN113880731A (en) * | 2021-10-28 | 2022-01-04 | 成都市科隆化学品有限公司 | Fmoc-D-Cit optical purity purification method and obtained product |
Citations (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1999006587A2 (en) | 1997-08-01 | 1999-02-11 | Morphosys Ag | Novel method and phage for the identification of nucleic acid sequences encoding members of a multimeric (poly)peptide complex |
| US5885793A (en) | 1991-12-02 | 1999-03-23 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| US6130237A (en) | 1996-09-12 | 2000-10-10 | Cancer Research Campaign Technology Limited | Condensed N-aclyindoles as antitumor agents |
| WO2002088172A2 (en) | 2001-04-30 | 2002-11-07 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US20030083263A1 (en) | 2001-04-30 | 2003-05-01 | Svetlana Doronina | Pentapeptide compounds and uses related thereto |
| WO2004010957A2 (en) | 2002-07-31 | 2004-02-05 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| WO2004032828A2 (en) | 2002-07-31 | 2004-04-22 | Seattle Genetics, Inc. | Anti-cd20 antibody-drug conjugates for the treatment of cancer and immune disorders |
| US20050238649A1 (en) | 2003-11-06 | 2005-10-27 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| US20060024317A1 (en) | 2004-05-19 | 2006-02-02 | Medarex, Inc | Chemical linkers and conjugates thereof |
| US20070082365A1 (en) | 1998-12-10 | 2007-04-12 | Adnexus Therapeutics, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| WO2007085930A1 (en) | 2006-01-25 | 2007-08-02 | Sanofi-Aventis | Cytotoxic agents comprising new tomaymycin derivatives and their therapeutic use |
| WO2008103693A2 (en) | 2007-02-21 | 2008-08-28 | Medarex, Inc. | Chemical linkers with single amino acids and conjugates thereof |
| WO2008141044A2 (en) | 2007-05-08 | 2008-11-20 | Genentech, Inc. | Cysteine engineered anti-muc16 antibodies and antibody drug conjugates |
| US20090018086A1 (en) | 2005-07-07 | 2009-01-15 | Seattle Genetics, Inc. | Monomethylvaline Compounds Having Phenylalanine Side-Chain Replacements at the C-Terminus |
| US20090111756A1 (en) | 2005-07-07 | 2009-04-30 | Seattle Genectics, Inc. | Monomethylvaline Compounds Having Phenylalanine Carboxy Modifications at the C-Terminus |
| WO2009099741A1 (en) | 2008-02-01 | 2009-08-13 | Genentech, Inc. | Nemorubicin metabolite and analog reagents, antibody-drug conjugates and methods |
| WO2009141240A1 (en) | 2008-05-20 | 2009-11-26 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Novel dual targeting antitumoural conjugates |
| WO2010091150A1 (en) | 2009-02-05 | 2010-08-12 | Immunogen, Inc. | Novel benzodiazepine derivatives |
| US20110020343A1 (en) | 2008-03-18 | 2011-01-27 | Seattle Genetics, Inc. | Auristatin drug linker conjugates |
| WO2011023883A1 (en) | 2009-08-25 | 2011-03-03 | Sanofi-Aventis | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| US8163888B2 (en) | 2003-10-10 | 2012-04-24 | Immunogen, Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates and methods of making said conjugates |
| WO2012112708A1 (en) | 2011-02-15 | 2012-08-23 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives and methods of preparation |
| WO2013163229A1 (en) | 2012-04-24 | 2013-10-31 | Seattle Genetics, Inc. | Dr5 ligand drug conjugates |
| WO2015095227A2 (en) | 2013-12-16 | 2015-06-25 | Genentech, Inc. | Peptidomimetic compounds and antibody-drug conjugates thereof |
| WO2016008112A1 (en) | 2014-07-16 | 2016-01-21 | Medshine Discovery Inc. | Linkers and application towards adc thereof |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102973947A (en) | 2004-06-01 | 2013-03-20 | 健泰科生物技术公司 | Antibody-drug conjugates and methods |
| US20080305044A1 (en) | 2004-11-29 | 2008-12-11 | Seattle Genetics, Inc. | Engineered Antibodies and Immunoconjugates |
| CN108465112B (en) * | 2012-05-15 | 2022-04-29 | 西雅图基因公司 | Self-stabilizing linker conjugates |
-
2014
- 2014-07-16 WO PCT/CN2014/082292 patent/WO2016008112A1/en active Application Filing
-
2015
- 2015-07-13 WO PCT/CN2015/083840 patent/WO2016008392A1/en active Application Filing
- 2015-07-13 EP EP15822852.8A patent/EP3169368B1/en active Active
- 2015-07-13 US US15/326,386 patent/US10280229B2/en active Active
- 2015-07-13 JP JP2017522709A patent/JP6688790B2/en active Active
- 2015-07-13 ES ES15822852T patent/ES2867749T3/en active Active
- 2015-07-13 CN CN201580038671.XA patent/CN107427591B/en active Active
- 2015-07-15 TW TW104122978A patent/TWI579000B/en active
-
2020
- 2020-01-24 JP JP2020009899A patent/JP7042291B2/en active Active
Patent Citations (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5885793A (en) | 1991-12-02 | 1999-03-23 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| US6130237A (en) | 1996-09-12 | 2000-10-10 | Cancer Research Campaign Technology Limited | Condensed N-aclyindoles as antitumor agents |
| WO1999006587A2 (en) | 1997-08-01 | 1999-02-11 | Morphosys Ag | Novel method and phage for the identification of nucleic acid sequences encoding members of a multimeric (poly)peptide complex |
| US20070082365A1 (en) | 1998-12-10 | 2007-04-12 | Adnexus Therapeutics, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| WO2002088172A2 (en) | 2001-04-30 | 2002-11-07 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US20050009751A1 (en) | 2001-04-30 | 2005-01-13 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US20030083263A1 (en) | 2001-04-30 | 2003-05-01 | Svetlana Doronina | Pentapeptide compounds and uses related thereto |
| WO2004010957A2 (en) | 2002-07-31 | 2004-02-05 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| WO2004032828A2 (en) | 2002-07-31 | 2004-04-22 | Seattle Genetics, Inc. | Anti-cd20 antibody-drug conjugates for the treatment of cancer and immune disorders |
| US20060074008A1 (en) | 2002-07-31 | 2006-04-06 | Senter Peter D | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| US8163888B2 (en) | 2003-10-10 | 2012-04-24 | Immunogen, Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates and methods of making said conjugates |
| US20050238649A1 (en) | 2003-11-06 | 2005-10-27 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| US20080226657A1 (en) | 2003-11-06 | 2008-09-18 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| US20060024317A1 (en) | 2004-05-19 | 2006-02-02 | Medarex, Inc | Chemical linkers and conjugates thereof |
| US20090018086A1 (en) | 2005-07-07 | 2009-01-15 | Seattle Genetics, Inc. | Monomethylvaline Compounds Having Phenylalanine Side-Chain Replacements at the C-Terminus |
| US20090111756A1 (en) | 2005-07-07 | 2009-04-30 | Seattle Genectics, Inc. | Monomethylvaline Compounds Having Phenylalanine Carboxy Modifications at the C-Terminus |
| WO2007085930A1 (en) | 2006-01-25 | 2007-08-02 | Sanofi-Aventis | Cytotoxic agents comprising new tomaymycin derivatives and their therapeutic use |
| WO2008103693A2 (en) | 2007-02-21 | 2008-08-28 | Medarex, Inc. | Chemical linkers with single amino acids and conjugates thereof |
| JP2010519310A (en) | 2007-02-21 | 2010-06-03 | メダレックス インコーポレイテッド | Chemical linker having a single amino acid and complex thereof |
| US20080311134A1 (en) | 2007-05-08 | 2008-12-18 | Junutula Jagath R | Cysteine engineered anti-muc16 antibodies and antibody drug conjugates |
| WO2008141044A2 (en) | 2007-05-08 | 2008-11-20 | Genentech, Inc. | Cysteine engineered anti-muc16 antibodies and antibody drug conjugates |
| WO2009099741A1 (en) | 2008-02-01 | 2009-08-13 | Genentech, Inc. | Nemorubicin metabolite and analog reagents, antibody-drug conjugates and methods |
| US20110020343A1 (en) | 2008-03-18 | 2011-01-27 | Seattle Genetics, Inc. | Auristatin drug linker conjugates |
| WO2009141240A1 (en) | 2008-05-20 | 2009-11-26 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Novel dual targeting antitumoural conjugates |
| WO2010091150A1 (en) | 2009-02-05 | 2010-08-12 | Immunogen, Inc. | Novel benzodiazepine derivatives |
| WO2011023883A1 (en) | 2009-08-25 | 2011-03-03 | Sanofi-Aventis | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| WO2012112708A1 (en) | 2011-02-15 | 2012-08-23 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives and methods of preparation |
| WO2013163229A1 (en) | 2012-04-24 | 2013-10-31 | Seattle Genetics, Inc. | Dr5 ligand drug conjugates |
| WO2015095227A2 (en) | 2013-12-16 | 2015-06-25 | Genentech, Inc. | Peptidomimetic compounds and antibody-drug conjugates thereof |
| WO2016008112A1 (en) | 2014-07-16 | 2016-01-21 | Medshine Discovery Inc. | Linkers and application towards adc thereof |
Non-Patent Citations (6)
| Title |
|---|
| Denis C. Roy et al., "Anti-MY9-Blocked-Ricin: An Immunotoxin for Selective Targeting of Acute Myeloid Leukemia Cells", Blood, vol. 77(11), pp. 2404-2412, 1991. |
| Denis C. Roy et al., "Elimination of Neuroblastoma and Small-Cell Lung Cancer Cells With an Anti-Neural Cell Adhesion Molecule Immunotoxin", Journal of the National Cancer Institute, vol. 88(16), pp. 1136-1145, 1996. |
| Jul. 12, 2017, EESR issued in European Patent Application No. 15822852.8. |
| Lee M. Nadler et al., "B4, A Human B Lymphocyte-Associated Antigen Expressed on Normal, Mitogen-Activated, and Malignant B Lymphocytes", The Journal of Immunology, vol. 131(1), pp. 244-250, 1983. |
| Ravi V. J. et al., "Immunoconjugates Containing Novel Maytansinoids: Promising Anticancer Drugs", Cancer Reseach, vol. 52, pp. 127-131, 1992. |
| The First Office Action of Japanese patent application 2017-522709 dated Dec. 4, 2018. |
Also Published As
| Publication number | Publication date |
|---|---|
| ES2867749T3 (en) | 2021-10-20 |
| EP3169368A4 (en) | 2017-08-09 |
| JP7042291B2 (en) | 2022-03-25 |
| US20170202974A1 (en) | 2017-07-20 |
| WO2016008392A1 (en) | 2016-01-21 |
| CN107427591B (en) | 2020-12-29 |
| TW201605481A (en) | 2016-02-16 |
| EP3169368A1 (en) | 2017-05-24 |
| CN107427591A (en) | 2017-12-01 |
| JP2017531027A (en) | 2017-10-19 |
| TWI579000B (en) | 2017-04-21 |
| EP3169368B1 (en) | 2021-02-17 |
| JP2020079251A (en) | 2020-05-28 |
| JP6688790B2 (en) | 2020-05-20 |
| WO2016008112A1 (en) | 2016-01-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10280229B2 (en) | Linkers and their application towards ADC | |
| JP6581630B2 (en) | New benzodiazepine derivatives | |
| US10722588B2 (en) | Cytotoxic benzodiazepine derivatives | |
| US9150649B2 (en) | Potent conjugates and hydrophilic linkers | |
| EP3769787A1 (en) | Conjugates of cysteine engineered antibodies | |
| EP2723389A2 (en) | Novel maytansinoid derivatives with peptide linker and conjugates thereof | |
| JP2023528412A (en) | Anti-BCMA antibody-drug conjugates and methods of use | |
| EP4285937A1 (en) | Conjugate and use thereof | |
| CN118647408A (en) | Conjugate of antibody-eribulin or derivative thereof, intermediate, preparation method, pharmaceutical composition and application | |
| CN115192732A (en) | DNA toxic dimer compound and conjugate thereof | |
| NZ618262B2 (en) | Novel maytansinoid derivatives with peptide linker and conjugates thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: MEDSHINE DISCOVERY INC., CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:LI, CHUN SING;LI, JIAN;CANG, YONG;AND OTHERS;REEL/FRAME:041114/0015 Effective date: 20170112 |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: PUBLICATIONS -- ISSUE FEE PAYMENT VERIFIED |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 4TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1551); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 4 |