TWI702954B - Cd73抑制劑 - Google Patents
Cd73抑制劑 Download PDFInfo
- Publication number
- TWI702954B TWI702954B TW108105130A TW108105130A TWI702954B TW I702954 B TWI702954 B TW I702954B TW 108105130 A TW108105130 A TW 108105130A TW 108105130 A TW108105130 A TW 108105130A TW I702954 B TWI702954 B TW I702954B
- Authority
- TW
- Taiwan
- Prior art keywords
- compound
- cancer
- pharmaceutically acceptable
- acceptable salt
- mmol
- Prior art date
Links
- 229940127272 CD73 inhibitor Drugs 0.000 title description 9
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 84
- 150000003839 salts Chemical class 0.000 claims abstract description 80
- 101000678236 Homo sapiens 5'-nucleotidase Proteins 0.000 claims abstract description 74
- 102100022464 5'-nucleotidase Human genes 0.000 claims abstract description 67
- 201000011510 cancer Diseases 0.000 claims abstract description 52
- 230000000694 effects Effects 0.000 claims abstract description 24
- 150000001875 compounds Chemical class 0.000 claims description 189
- 238000000034 method Methods 0.000 claims description 55
- 210000001519 tissue Anatomy 0.000 claims description 18
- 239000003814 drug Substances 0.000 claims description 16
- 238000004519 manufacturing process Methods 0.000 claims description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims description 13
- 210000002966 serum Anatomy 0.000 claims description 11
- 206010006187 Breast cancer Diseases 0.000 claims description 10
- 208000026310 Breast neoplasm Diseases 0.000 claims description 10
- 206010009944 Colon cancer Diseases 0.000 claims description 10
- WHRUIISQCORGKK-KOLCDFICSA-N 5-[6-methyl-5-[(1S,2R)-2-propan-2-ylcyclopropyl]pyridazin-3-yl]-1H-pyrimidine-2,4-dione Chemical compound C(C)(C)[C@@H]1[C@H](C1)C=1C=C(N=NC=1C)C=1C(NC(NC=1)=O)=O WHRUIISQCORGKK-KOLCDFICSA-N 0.000 claims description 9
- SXVLKJCZFKIBAV-WPRPVWTQSA-N 5-[5-[(1S,2S)-2-ethylcyclopropyl]-6-methylpyridazin-3-yl]-1H-pyrimidine-2,4-dione Chemical compound C(C)[C@@H]1[C@H](C1)C=1C=C(N=NC=1C)C=1C(NC(NC=1)=O)=O SXVLKJCZFKIBAV-WPRPVWTQSA-N 0.000 claims description 8
- 201000001441 melanoma Diseases 0.000 claims description 8
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 7
- 206010033128 Ovarian cancer Diseases 0.000 claims description 7
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 7
- 239000003085 diluting agent Substances 0.000 claims description 7
- 201000005202 lung cancer Diseases 0.000 claims description 7
- 208000020816 lung neoplasm Diseases 0.000 claims description 7
- 206010005003 Bladder cancer Diseases 0.000 claims description 6
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 6
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 230000002401 inhibitory effect Effects 0.000 claims description 6
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 6
- 201000002528 pancreatic cancer Diseases 0.000 claims description 6
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 6
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 6
- 208000022072 Gallbladder Neoplasms Diseases 0.000 claims description 5
- 206010025323 Lymphomas Diseases 0.000 claims description 5
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 5
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 5
- 201000010175 gallbladder cancer Diseases 0.000 claims description 5
- 206010017758 gastric cancer Diseases 0.000 claims description 5
- 208000005017 glioblastoma Diseases 0.000 claims description 5
- 201000011549 stomach cancer Diseases 0.000 claims description 5
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 4
- 206010060862 Prostate cancer Diseases 0.000 claims description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 4
- 206010038389 Renal cancer Diseases 0.000 claims description 4
- 125000000217 alkyl group Chemical group 0.000 claims description 4
- 208000029742 colonic neoplasm Diseases 0.000 claims description 4
- 201000010536 head and neck cancer Diseases 0.000 claims description 4
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 4
- 201000010982 kidney cancer Diseases 0.000 claims description 4
- 201000007270 liver cancer Diseases 0.000 claims description 4
- 208000014018 liver neoplasm Diseases 0.000 claims description 4
- 208000015634 Rectal Neoplasms Diseases 0.000 claims 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims 1
- 230000001537 neural effect Effects 0.000 claims 1
- 206010038038 rectal cancer Diseases 0.000 claims 1
- 201000001275 rectum cancer Diseases 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 79
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 75
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 67
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 63
- 238000002360 preparation method Methods 0.000 description 57
- 239000002904 solvent Substances 0.000 description 55
- 239000000203 mixture Substances 0.000 description 47
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 45
- 238000004458 analytical method Methods 0.000 description 40
- 239000000243 solution Substances 0.000 description 40
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 39
- 229960005305 adenosine Drugs 0.000 description 38
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 35
- 235000019439 ethyl acetate Nutrition 0.000 description 33
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 32
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 30
- 239000007787 solid Substances 0.000 description 29
- -1 4,4,5,5-tetramethyl-2-(2-substituted cyclopropyl)-1,3,2-dioxaborolane Chemical class 0.000 description 26
- UDMBCSSLTHHNCD-UHFFFAOYSA-N Coenzym Q(11) Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1O UDMBCSSLTHHNCD-UHFFFAOYSA-N 0.000 description 26
- UDMBCSSLTHHNCD-KQYNXXCUSA-N adenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O UDMBCSSLTHHNCD-KQYNXXCUSA-N 0.000 description 26
- 238000006243 chemical reaction Methods 0.000 description 25
- LNQVTSROQXJCDD-UHFFFAOYSA-N adenosine monophosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(CO)C(OP(O)(O)=O)C1O LNQVTSROQXJCDD-UHFFFAOYSA-N 0.000 description 24
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 23
- 210000004027 cell Anatomy 0.000 description 23
- 230000002829 reductive effect Effects 0.000 description 23
- 238000003756 stirring Methods 0.000 description 23
- 238000004587 chromatography analysis Methods 0.000 description 22
- 239000010410 layer Substances 0.000 description 22
- 239000003795 chemical substances by application Substances 0.000 description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 20
- 239000003921 oil Substances 0.000 description 20
- 229920006395 saturated elastomer Polymers 0.000 description 20
- 238000005481 NMR spectroscopy Methods 0.000 description 16
- 229940079593 drug Drugs 0.000 description 15
- 239000003112 inhibitor Substances 0.000 description 15
- 239000007864 aqueous solution Substances 0.000 description 14
- 239000011780 sodium chloride Substances 0.000 description 14
- 238000005160 1H NMR spectroscopy Methods 0.000 description 13
- 239000000460 chlorine Substances 0.000 description 13
- 239000011541 reaction mixture Substances 0.000 description 13
- 239000011734 sodium Substances 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 239000003480 eluent Substances 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 11
- 230000005764 inhibitory process Effects 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 229950006790 adenosine phosphate Drugs 0.000 description 10
- 210000004970 cd4 cell Anatomy 0.000 description 10
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- 238000012360 testing method Methods 0.000 description 10
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 9
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 9
- 239000012074 organic phase Substances 0.000 description 9
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 8
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- 102000045309 human NT5E Human genes 0.000 description 8
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 8
- 238000004949 mass spectrometry Methods 0.000 description 8
- 239000002609 medium Substances 0.000 description 8
- 238000010898 silica gel chromatography Methods 0.000 description 8
- NEZWQTAIJWQNHI-UHFFFAOYSA-N 2-ethylcyclopropane-1-carboxylic acid Chemical compound CCC1CC1C(O)=O NEZWQTAIJWQNHI-UHFFFAOYSA-N 0.000 description 7
- ZJJNMXUSBPHRDY-UHFFFAOYSA-N 4-chloro-6-(2,4-dimethoxypyrimidin-5-yl)-3-methylpyridazine Chemical compound ClC1=C(N=NC(=C1)C=1C(=NC(=NC=1)OC)OC)C ZJJNMXUSBPHRDY-UHFFFAOYSA-N 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 210000001744 T-lymphocyte Anatomy 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 238000010790 dilution Methods 0.000 description 7
- 239000012895 dilution Substances 0.000 description 7
- 239000000284 extract Substances 0.000 description 7
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 229960003786 inosine Drugs 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- 239000000377 silicon dioxide Substances 0.000 description 7
- 238000012546 transfer Methods 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 6
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 6
- JGPOSNWWINVNFV-UHFFFAOYSA-N carboxyfluorescein diacetate succinimidyl ester Chemical compound C=1C(OC(=O)C)=CC=C2C=1OC1=CC(OC(C)=O)=CC=C1C2(C1=C2)OC(=O)C1=CC=C2C(=O)ON1C(=O)CCC1=O JGPOSNWWINVNFV-UHFFFAOYSA-N 0.000 description 6
- 239000012230 colorless oil Substances 0.000 description 6
- 230000008878 coupling Effects 0.000 description 6
- 238000010168 coupling process Methods 0.000 description 6
- 238000005859 coupling reaction Methods 0.000 description 6
- 230000017858 demethylation Effects 0.000 description 6
- 238000010520 demethylation reaction Methods 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 239000012091 fetal bovine serum Substances 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 239000001963 growth medium Substances 0.000 description 6
- 229940045207 immuno-oncology agent Drugs 0.000 description 6
- 239000002584 immunological anticancer agent Substances 0.000 description 6
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 6
- 238000012799 strong cation exchange Methods 0.000 description 6
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 6
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 6
- CFMZSMGAMPBRBE-UHFFFAOYSA-N 2-hydroxyisoindole-1,3-dione Chemical class C1=CC=C2C(=O)N(O)C(=O)C2=C1 CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- 239000012981 Hank's balanced salt solution Substances 0.000 description 5
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 5
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 235000019253 formic acid Nutrition 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- LKGKUACPLXCVOF-UHFFFAOYSA-N (2,4-dimethoxypyrimidin-5-yl)boronic acid Chemical compound COC1=NC=C(B(O)O)C(OC)=N1 LKGKUACPLXCVOF-UHFFFAOYSA-N 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 4
- DWOZNANUEDYIOF-UHFFFAOYSA-L 4-ditert-butylphosphanyl-n,n-dimethylaniline;dichloropalladium Chemical compound Cl[Pd]Cl.CN(C)C1=CC=C(P(C(C)(C)C)C(C)(C)C)C=C1.CN(C)C1=CC=C(P(C(C)(C)C)C(C)(C)C)C=C1 DWOZNANUEDYIOF-UHFFFAOYSA-L 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 238000002965 ELISA Methods 0.000 description 4
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 4
- 229930010555 Inosine Natural products 0.000 description 4
- 206010027476 Metastases Diseases 0.000 description 4
- 229940024606 amino acid Drugs 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 238000012742 biochemical analysis Methods 0.000 description 4
- 235000011089 carbon dioxide Nutrition 0.000 description 4
- 229960005395 cetuximab Drugs 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000000132 electrospray ionisation Methods 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 238000000684 flow cytometry Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 230000009401 metastasis Effects 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 4
- 238000004808 supercritical fluid chromatography Methods 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 229910052723 transition metal Inorganic materials 0.000 description 4
- 150000003624 transition metals Chemical class 0.000 description 4
- KOKIOKRQOCRQHK-IUCAKERBSA-N 2-[(1S,2S)-2-ethylcyclopropyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound CC[C@H]1C[C@@H]1B1OC(C)(C)C(C)(C)O1 KOKIOKRQOCRQHK-IUCAKERBSA-N 0.000 description 3
- HBYCKISMJZSUDJ-UHFFFAOYSA-N 2-propan-2-ylcyclopropane-1-carboxylic acid Chemical compound CC(C)C1CC1C(O)=O HBYCKISMJZSUDJ-UHFFFAOYSA-N 0.000 description 3
- NGLRCSLJXJGKHV-ZJUUUORDSA-N 4,4,5,5-tetramethyl-2-[(1S,2S)-2-propan-2-ylcyclopropyl]-1,3,2-dioxaborolane Chemical compound CC(C)[C@H]1C[C@@H]1B1OC(C)(C)C(C)(C)O1 NGLRCSLJXJGKHV-ZJUUUORDSA-N 0.000 description 3
- SWLRTVHQASOVRZ-UHFFFAOYSA-N 4,6-dichloro-3-methylpyridazine Chemical compound CC1=NN=C(Cl)C=C1Cl SWLRTVHQASOVRZ-UHFFFAOYSA-N 0.000 description 3
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 3
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 150000004703 alkoxides Chemical class 0.000 description 3
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 3
- 239000001099 ammonium carbonate Substances 0.000 description 3
- 150000001500 aryl chlorides Chemical class 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 238000005888 cyclopropanation reaction Methods 0.000 description 3
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 3
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 3
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 3
- 102000015694 estrogen receptors Human genes 0.000 description 3
- 108010038795 estrogen receptors Proteins 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 3
- 229910052737 gold Inorganic materials 0.000 description 3
- 239000010931 gold Substances 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000002372 labelling Methods 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 3
- VYNDHICBIRRPFP-UHFFFAOYSA-N pacific blue Chemical compound FC1=C(O)C(F)=C2OC(=O)C(C(=O)O)=CC2=C1 VYNDHICBIRRPFP-UHFFFAOYSA-N 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 229920001296 polysiloxane Polymers 0.000 description 3
- 238000010186 staining Methods 0.000 description 3
- 230000004614 tumor growth Effects 0.000 description 3
- 238000010792 warming Methods 0.000 description 3
- HBYCKISMJZSUDJ-WDSKDSINSA-N (1s,2s)-2-propan-2-ylcyclopropane-1-carboxylic acid Chemical compound CC(C)[C@@H]1C[C@@H]1C(O)=O HBYCKISMJZSUDJ-WDSKDSINSA-N 0.000 description 2
- GMPHCAJAEHBRPV-UFOMNOBLSA-N (2R,3R,4S,5R)-2-(6-amino-4,5-dihydropurin-9-yl)-5-(hydroxy(113C)methyl)(2,3,4,5-13C4)oxolane-3,4-diol Chemical compound NC1=NC=NC2C1N=CN2[13C@@H]1O[13C@H]([13CH2]O)[13C@@H](O)[13C@H]1O GMPHCAJAEHBRPV-UFOMNOBLSA-N 0.000 description 2
- REYZXWIIUPKFTI-YFKPBYRVSA-N (2r)-2-propan-2-yloxirane Chemical compound CC(C)[C@@H]1CO1 REYZXWIIUPKFTI-YFKPBYRVSA-N 0.000 description 2
- OJRHUICOVVSGSY-RXMQYKEDSA-N (2s)-2-chloro-3-methylbutan-1-ol Chemical compound CC(C)[C@H](Cl)CO OJRHUICOVVSGSY-RXMQYKEDSA-N 0.000 description 2
- DDTJFSPKEIAZAM-BYPYZUCNSA-N (2s)-2-chloro-3-methylbutanoic acid Chemical compound CC(C)[C@H](Cl)C(O)=O DDTJFSPKEIAZAM-BYPYZUCNSA-N 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- KILDQAGHULQJNI-BQYQJAHWSA-N 2-[(e)-but-1-enyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound CC\C=C\B1OC(C)(C)C(C)(C)O1 KILDQAGHULQJNI-BQYQJAHWSA-N 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- BSKZXOSKFBUCNU-UHFFFAOYSA-N 3,6-dichloro-4-cyclopropylpyridazine Chemical compound N1=NC(Cl)=CC(C2CC2)=C1Cl BSKZXOSKFBUCNU-UHFFFAOYSA-N 0.000 description 2
- XBTQAFYIRNRNMN-UHFFFAOYSA-N 3-chloro-4-cyclobutyl-6-(2,4-dimethoxypyrimidin-5-yl)pyridazine Chemical compound ClC=1N=NC(=CC=1C1CCC1)C=1C(=NC(=NC=1)OC)OC XBTQAFYIRNRNMN-UHFFFAOYSA-N 0.000 description 2
- QYTURVMFLCIJNG-UHFFFAOYSA-N 3-chloro-4-cyclopropyl-6-(2,4-dimethoxypyrimidin-5-yl)pyridazine Chemical compound ClC=1N=NC(=CC=1C1CC1)C=1C(=NC(=NC=1)OC)OC QYTURVMFLCIJNG-UHFFFAOYSA-N 0.000 description 2
- XTJQCYPNKDSUIZ-UHFFFAOYSA-N 4-cyclobutyl-6-(2,4-dimethoxypyrimidin-5-yl)-3-methoxypyridazine Chemical compound C1(CCC1)C1=C(N=NC(=C1)C=1C(=NC(=NC=1)OC)OC)OC XTJQCYPNKDSUIZ-UHFFFAOYSA-N 0.000 description 2
- XWJHOJKPWMVOCN-UHFFFAOYSA-N 5-(6-chloro-5-cyclopropylpyridazin-3-yl)-1H-pyrimidine-2,4-dione Chemical compound ClC1=C(C=C(N=N1)C=1C(NC(NC=1)=O)=O)C1CC1 XWJHOJKPWMVOCN-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 229940125431 BRAF inhibitor Drugs 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- NGLRCSLJXJGKHV-VHSXEESVSA-N C(C)(C)[C@H]1[C@@H](C1)B1OC(C(O1)(C)C)(C)C Chemical compound C(C)(C)[C@H]1[C@@H](C1)B1OC(C(O1)(C)C)(C)C NGLRCSLJXJGKHV-VHSXEESVSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000984753 Homo sapiens Serine/threonine-protein kinase B-raf Proteins 0.000 description 2
- 238000006546 Horner-Wadsworth-Emmons reaction Methods 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 2
- 208000000172 Medulloblastoma Diseases 0.000 description 2
- XUMBMVFBXHLACL-UHFFFAOYSA-N Melanin Chemical compound O=C1C(=O)C(C2=CNC3=C(C(C(=O)C4=C32)=O)C)=C2C4=CNC2=C1C XUMBMVFBXHLACL-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- ZRKWMRDKSOPRRS-UHFFFAOYSA-N N-Methyl-N-nitrosourea Chemical compound O=NN(C)C(N)=O ZRKWMRDKSOPRRS-UHFFFAOYSA-N 0.000 description 2
- 206010029260 Neuroblastoma Diseases 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 238000010240 RT-PCR analysis Methods 0.000 description 2
- 102100027103 Serine/threonine-protein kinase B-raf Human genes 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 2
- 229950001573 abemaciclib Drugs 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- RWMOFKJKPRCTKI-NWDGAFQWSA-N benzyl (1R,2R)-2-acetylcyclopropane-1-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)[C@H]1[C@@H](C1)C(C)=O RWMOFKJKPRCTKI-NWDGAFQWSA-N 0.000 description 2
- 239000012455 biphasic mixture Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- FUSUHKVFWTUUBE-UHFFFAOYSA-N buten-2-one Chemical compound CC(=O)C=C FUSUHKVFWTUUBE-UHFFFAOYSA-N 0.000 description 2
- PGIRZIYDZWBFCD-KRWDZBQOSA-N butyl-[[(2S)-oxiran-2-yl]methoxy]-diphenylsilane Chemical group C(CCC)[Si](C1=CC=CC=C1)(C1=CC=CC=C1)OC[C@H]1OC1 PGIRZIYDZWBFCD-KRWDZBQOSA-N 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 230000007969 cellular immunity Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 229960002465 dabrafenib Drugs 0.000 description 2
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 2
- 238000003113 dilution method Methods 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- YQPYKYQROJHYBV-SFYZADRCSA-N ethyl (1S,2R)-2-propan-2-ylcyclopropane-1-carboxylate Chemical compound CCOC(=O)[C@H]1C[C@@H]1C(C)C YQPYKYQROJHYBV-SFYZADRCSA-N 0.000 description 2
- LUUSYESYLFJQHB-WHFBIAKZSA-N ethyl (1S,2S)-2-(difluoromethyl)cyclopropane-1-carboxylate Chemical compound CCOC(=O)[C@H]1C[C@@H]1C(F)F LUUSYESYLFJQHB-WHFBIAKZSA-N 0.000 description 2
- MCRPKBUFXAKDKI-UHFFFAOYSA-N ethyl pyridine-4-carboxylate Chemical compound CCOC(=O)C1=CC=NC=C1 MCRPKBUFXAKDKI-UHFFFAOYSA-N 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 229960002584 gefitinib Drugs 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000002013 hydrophilic interaction chromatography Methods 0.000 description 2
- 238000013388 immunohistochemistry analysis Methods 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 238000009169 immunotherapy Methods 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- UZWDCWONPYILKI-UHFFFAOYSA-N n-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine Chemical compound C1CN(CC)CCN1CC(C=N1)=CC=C1NC1=NC=C(F)C(C=2C=C3N(C(C)C)C(C)=NC3=C(F)C=2)=N1 UZWDCWONPYILKI-UHFFFAOYSA-N 0.000 description 2
- 230000036963 noncompetitive effect Effects 0.000 description 2
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 239000002777 nucleoside Substances 0.000 description 2
- 229960003278 osimertinib Drugs 0.000 description 2
- DUYJMQONPNNFPI-UHFFFAOYSA-N osimertinib Chemical group COC1=CC(N(C)CCN(C)C)=C(NC(=O)C=C)C=C1NC1=NC=CC(C=2C3=CC=CC=C3N(C)C=2)=N1 DUYJMQONPNNFPI-UHFFFAOYSA-N 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- 150000002924 oxiranes Chemical class 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 2
- 238000010837 poor prognosis Methods 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000002553 single reaction monitoring Methods 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 239000012089 stop solution Substances 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- YUFLXVKNWNTRNT-NVXWUHKLSA-N tert-butyl-[2-[(1S,2R)-2-[6-(2,4-dimethoxypyrimidin-5-yl)-3-methylpyridazin-4-yl]cyclopropyl]ethoxy]-dimethylsilane Chemical compound C(C)(C)(C)[Si](OCC[C@H]1[C@@H](C1)C1=C(N=NC(=C1)C=1C(=NC(=NC=1)OC)OC)C)(C)C YUFLXVKNWNTRNT-NVXWUHKLSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 229960004066 trametinib Drugs 0.000 description 2
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 2
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 238000005292 vacuum distillation Methods 0.000 description 2
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 2
- 229960003862 vemurafenib Drugs 0.000 description 2
- ONTVBKAFCGRFDD-QWWZWVQMSA-N (1R,2R)-2-(1,1-difluoroethyl)cyclopropane-1-carboxylic acid Chemical compound CC(F)(F)[C@@H]1C[C@H]1C(O)=O ONTVBKAFCGRFDD-QWWZWVQMSA-N 0.000 description 1
- AQPYFAYLGMEWCD-HRFVKAFMSA-N (1S,2S)-2-(difluoromethyl)cyclopropane-1-carboxylic acid Chemical compound OC(=O)[C@H]1C[C@@H]1C(F)F AQPYFAYLGMEWCD-HRFVKAFMSA-N 0.000 description 1
- NEZWQTAIJWQNHI-WHFBIAKZSA-N (1S,2S)-2-ethylcyclopropane-1-carboxylic acid Chemical compound C(C)[C@@H]1[C@H](C1)C(=O)O NEZWQTAIJWQNHI-WHFBIAKZSA-N 0.000 description 1
- QAKGVPAQOZSDOZ-DMTCNVIQSA-N (1s,2s)-2-(hydroxymethyl)cyclopropane-1-carboxylic acid Chemical compound OC[C@H]1C[C@@H]1C(O)=O QAKGVPAQOZSDOZ-DMTCNVIQSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- RBACIKXCRWGCBB-SCSAIBSYSA-N (2r)-2-ethyloxirane Chemical compound CC[C@@H]1CO1 RBACIKXCRWGCBB-SCSAIBSYSA-N 0.000 description 1
- YXTKHLHCVFUPPT-YYFJYKOTSA-N (2s)-2-[[4-[(2-amino-5-formyl-4-oxo-1,6,7,8-tetrahydropteridin-6-yl)methylamino]benzoyl]amino]pentanedioic acid;(1r,2r)-1,2-dimethanidylcyclohexane;5-fluoro-1h-pyrimidine-2,4-dione;oxalic acid;platinum(2+) Chemical compound [Pt+2].OC(=O)C(O)=O.[CH2-][C@@H]1CCCC[C@H]1[CH2-].FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 YXTKHLHCVFUPPT-YYFJYKOTSA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- CTKINSOISVBQLD-GSVOUGTGSA-N (R)-Glycidol Chemical compound OC[C@@H]1CO1 CTKINSOISVBQLD-GSVOUGTGSA-N 0.000 description 1
- HBXRPXYBMZDIQL-UHFFFAOYSA-N 1$l^{2}-borolane Chemical compound [B]1CCCC1 HBXRPXYBMZDIQL-UHFFFAOYSA-N 0.000 description 1
- IBXMKLPFLZYRQZ-UHFFFAOYSA-N 1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1C=CC(=O)C=CC1=CC=CC=C1 IBXMKLPFLZYRQZ-UHFFFAOYSA-N 0.000 description 1
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 description 1
- KLNQXUGBWGMCRP-WCQYABFASA-N 2,4-dimethoxy-5-[6-methyl-5-[(1R,2S)-2-propan-2-ylcyclopropyl]pyridazin-3-yl]pyrimidine Chemical class C(C)(C)[C@H]1[C@@H](C1)C=1C=C(N=NC=1C)C=1C(=NC(=NC=1)OC)OC KLNQXUGBWGMCRP-WCQYABFASA-N 0.000 description 1
- KLNQXUGBWGMCRP-YPMHNXCESA-N 2,4-dimethoxy-5-[6-methyl-5-[(1S,2R)-2-propan-2-ylcyclopropyl]pyridazin-3-yl]pyrimidine Chemical compound C(C)(C)[C@@H]1[C@H](C1)C=1C=C(N=NC=1C)C=1C(=NC(=NC=1)OC)OC KLNQXUGBWGMCRP-YPMHNXCESA-N 0.000 description 1
- KEVRHVMWBKFGLO-UHFFFAOYSA-N 2,4-dimethoxypyrimidine Chemical compound COC1=CC=NC(OC)=N1 KEVRHVMWBKFGLO-UHFFFAOYSA-N 0.000 description 1
- YFTHTJAPODJVSL-UHFFFAOYSA-N 2-(1-benzothiophen-5-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(SC=C2)C2=C1 YFTHTJAPODJVSL-UHFFFAOYSA-N 0.000 description 1
- ARRZLLPGBZPUEG-ZYHUDNBSSA-N 2-[(1S,2R)-2-[6-(2,4-dimethoxypyrimidin-5-yl)-3-methylpyridazin-4-yl]cyclopropyl]ethanol Chemical compound COC1=NC=C(C(=N1)OC)C1=CC(=C(N=N1)C)[C@H]1[C@@H](C1)CCO ARRZLLPGBZPUEG-ZYHUDNBSSA-N 0.000 description 1
- LFHMRRMWPATNIL-DGCLKSJQSA-N 2-[(1S,2R)-2-[6-(2,4-dimethoxypyrimidin-5-yl)-3-methylpyridazin-4-yl]cyclopropyl]ethyl methanesulfonate Chemical compound CS(=O)(=O)OCC[C@H]1[C@@H](C1)C1=C(N=NC(=C1)C=1C(=NC(=NC=1)OC)OC)C LFHMRRMWPATNIL-DGCLKSJQSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- WAKFRZBXTKUFIW-UHFFFAOYSA-M 2-bromo-2-phenylacetate Chemical compound [O-]C(=O)C(Br)C1=CC=CC=C1 WAKFRZBXTKUFIW-UHFFFAOYSA-M 0.000 description 1
- AXAQZUPPTUBENW-UHFFFAOYSA-N 2-chloro-1h-pyridazine Chemical compound ClN1NC=CC=C1 AXAQZUPPTUBENW-UHFFFAOYSA-N 0.000 description 1
- APOYTRAZFJURPB-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)-n-(trifluoro-$l^{4}-sulfanyl)ethanamine Chemical compound COCCN(S(F)(F)F)CCOC APOYTRAZFJURPB-UHFFFAOYSA-N 0.000 description 1
- IBZKBSXREAQDTO-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)ethanamine Chemical class COCCNCCOC IBZKBSXREAQDTO-UHFFFAOYSA-N 0.000 description 1
- USMRYMUZKSMTRL-UHFFFAOYSA-N 2-methyl-1,3,2-dioxaborolane Chemical compound CB1OCCO1 USMRYMUZKSMTRL-UHFFFAOYSA-N 0.000 description 1
- AFXKCBFBGDUFAM-UHFFFAOYSA-N 2-methylpropan-2-amine;hydrofluoride Chemical compound [F-].CC(C)(C)[NH3+] AFXKCBFBGDUFAM-UHFFFAOYSA-N 0.000 description 1
- JADVVTZXHQUFLS-UHFFFAOYSA-N 3,4-dichloropyridazine Chemical class ClC1=CC=NN=C1Cl JADVVTZXHQUFLS-UHFFFAOYSA-N 0.000 description 1
- GUSWJGOYDXFJSI-UHFFFAOYSA-N 3,6-dichloropyridazine Chemical compound ClC1=CC=C(Cl)N=N1 GUSWJGOYDXFJSI-UHFFFAOYSA-N 0.000 description 1
- NQUQLGGUKAKWMS-SVRRBLITSA-N 3-chloro-4-[(1S,2S)-2-(difluoromethyl)cyclopropyl]-6-(2,4-dimethoxypyrimidin-5-yl)pyridazine Chemical compound ClC=1N=NC(=CC=1[C@@H]1[C@H](C1)C(F)F)C=1C(=NC(=NC=1)OC)OC NQUQLGGUKAKWMS-SVRRBLITSA-N 0.000 description 1
- ASFHDLDAWYTMJS-UHFFFAOYSA-N 3-methoxypyridazine Chemical compound COC1=CC=CN=N1 ASFHDLDAWYTMJS-UHFFFAOYSA-N 0.000 description 1
- USCSRAJGJYMJFZ-UHFFFAOYSA-N 3-methyl-1-butyne Chemical compound CC(C)C#C USCSRAJGJYMJFZ-UHFFFAOYSA-N 0.000 description 1
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 1
- PDMWYFJWQXSWRH-ZJUUUORDSA-N 4-[(1S,2S)-2-(difluoromethyl)cyclopropyl]-6-(2,4-dimethoxypyrimidin-5-yl)-3-methylpyridazine Chemical compound FC([C@@H]1[C@H](C1)C1=C(N=NC(=C1)C=1C(=NC(=NC=1)OC)OC)C)F PDMWYFJWQXSWRH-ZJUUUORDSA-N 0.000 description 1
- PQQCEIMCLFXJLN-UHFFFAOYSA-N 4-butylphosphanyl-n,n-dimethylaniline Chemical compound CCCCPC1=CC=C(N(C)C)C=C1 PQQCEIMCLFXJLN-UHFFFAOYSA-N 0.000 description 1
- 108700004024 5'-Nucleotidase Proteins 0.000 description 1
- UHCYLLWRLWNSMG-UHFFFAOYSA-N 5-(5-cyclobutyl-6-methoxypyridazin-3-yl)-1H-pyrimidine-2,4-dione Chemical compound C1(CCC1)C=1C=C(N=NC=1OC)C=1C(NC(NC=1)=O)=O UHCYLLWRLWNSMG-UHFFFAOYSA-N 0.000 description 1
- RPPTWNXASIKORE-ZYHUDNBSSA-N 5-[5-[(1R,2S)-2-(2-fluoroethyl)cyclopropyl]-6-methylpyridazin-3-yl]-2,4-dimethoxypyrimidine Chemical compound COC1=NC=C(C(=N1)OC)C=1N=NC(=C(C=1)[C@H]1[C@@H](C1)CCF)C RPPTWNXASIKORE-ZYHUDNBSSA-N 0.000 description 1
- GBFONUQMSKQSQQ-JQWIXIFHSA-N 5-[5-[(1S,2S)-2-ethylcyclopropyl]-6-methylpyridazin-3-yl]-2,4-dimethoxypyrimidine Chemical compound C(C)[C@@H]1[C@H](C1)C=1C=C(N=NC=1C)C=1C(=NC(=NC=1)OC)OC GBFONUQMSKQSQQ-JQWIXIFHSA-N 0.000 description 1
- IQOAJMSCDHFOGX-UHFFFAOYSA-N 5-[5-[2-(difluoromethyl)cyclopropyl]-6-methylpyridazin-3-yl]-1H-pyrimidine-2,4-dione Chemical compound FC(C1C(C1)C=1C=C(N=NC=1C)C=1C(NC(NC=1)=O)=O)F IQOAJMSCDHFOGX-UHFFFAOYSA-N 0.000 description 1
- RGNFIBMZIVGHTM-OCCSQVGLSA-N 6-(1,2-dihydropyrimidin-5-yl)-3-methyl-4-[(1S,2R)-2-propan-2-ylcyclopropyl]pyridazine Chemical compound C(C)(C)[C@@H]1[C@H](C1)C=1C=C(N=NC1C)C=1C=NCNC1 RGNFIBMZIVGHTM-OCCSQVGLSA-N 0.000 description 1
- 102000009346 Adenosine receptors Human genes 0.000 description 1
- 108050000203 Adenosine receptors Proteins 0.000 description 1
- 101710134784 Agnoprotein Proteins 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 241000208340 Araliaceae Species 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- WHRUIISQCORGKK-ONGXEEELSA-N C(C)(C)[C@H]1[C@H](C1)C=1C=C(N=NC1C)C=1C(NC(NC1)=O)=O Chemical compound C(C)(C)[C@H]1[C@H](C1)C=1C=C(N=NC1C)C=1C(NC(NC1)=O)=O WHRUIISQCORGKK-ONGXEEELSA-N 0.000 description 1
- KOKIOKRQOCRQHK-RKDXNWHRSA-N C(C)[C@H]1[C@@H](C1)B1OC(C(O1)(C)C)(C)C Chemical compound C(C)[C@H]1[C@@H](C1)B1OC(C(O1)(C)C)(C)C KOKIOKRQOCRQHK-RKDXNWHRSA-N 0.000 description 1
- 0 C*(C)(C=C(C1=CC(C2C(*)C2)=C(C)**1)C(*1)=O)C1=O Chemical compound C*(C)(C=C(C1=CC(C2C(*)C2)=C(C)**1)C(*1)=O)C1=O 0.000 description 1
- 229940116741 CD137 agonist Drugs 0.000 description 1
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 1
- 101100463133 Caenorhabditis elegans pdl-1 gene Proteins 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- DYCYMKHGOOBDNF-UHFFFAOYSA-N Cl.C1(C=CC=C1)[Zr+2]C1C=CC=C1 Chemical compound Cl.C1(C=CC=C1)[Zr+2]C1C=CC=C1 DYCYMKHGOOBDNF-UHFFFAOYSA-N 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- 239000012824 ERK inhibitor Substances 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 101000599940 Homo sapiens Interferon gamma Proteins 0.000 description 1
- 101001033249 Homo sapiens Interleukin-1 beta Proteins 0.000 description 1
- 101001002657 Homo sapiens Interleukin-2 Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- 101000611183 Homo sapiens Tumor necrosis factor Proteins 0.000 description 1
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- 229940113303 Indoleamine 2,3-dioxygenase inhibitor Drugs 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 1
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 1
- IVRXNBXKWIJUQB-UHFFFAOYSA-N LY-2157299 Chemical compound CC1=CC=CC(C=2C(=C3CCCN3N=2)C=2C3=CC(=CC=C3N=CC=2)C(N)=O)=N1 IVRXNBXKWIJUQB-UHFFFAOYSA-N 0.000 description 1
- HLFSDGLLUJUHTE-SNVBAGLBSA-N Levamisole Chemical compound C1([C@H]2CN3CCSC3=N2)=CC=CC=C1 HLFSDGLLUJUHTE-SNVBAGLBSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 238000006050 Minisci radical substitution reaction Methods 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100381978 Mus musculus Braf gene Proteins 0.000 description 1
- 101000591390 Mus musculus Neurotensin receptor type 2 Proteins 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- 206010029113 Neovascularisation Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 235000005035 Panax pseudoginseng ssp. pseudoginseng Nutrition 0.000 description 1
- 235000003140 Panax quinquefolius Nutrition 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- 238000006932 Simmons-Smith cyclopropanation reaction Methods 0.000 description 1
- 208000033749 Small cell carcinoma of the bladder Diseases 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- 239000012505 Superdex™ Substances 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 238000006161 Suzuki-Miyaura coupling reaction Methods 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 230000006052 T cell proliferation Effects 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- 108091005735 TGF-beta receptors Proteins 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- OLCWZBFDIYXLAA-IOSLPCCCSA-N adenosine 5'-methylenediphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)CP(O)(O)=O)[C@@H](O)[C@H]1O OLCWZBFDIYXLAA-IOSLPCCCSA-N 0.000 description 1
- 229940121359 adenosine receptor antagonist Drugs 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical compound [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 238000009175 antibody therapy Methods 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 229960003852 atezolizumab Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- MSEKITBTLPJWOI-GHMZBOCLSA-N benzyl (1R,2R)-2-(1,1-difluoroethyl)cyclopropane-1-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)[C@H]1[C@@H](C1)C(C)(F)F MSEKITBTLPJWOI-GHMZBOCLSA-N 0.000 description 1
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- PJGJQVRXEUVAFT-UHFFFAOYSA-N chloroiodomethane Chemical compound ClCI PJGJQVRXEUVAFT-UHFFFAOYSA-N 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 238000007333 cyanation reaction Methods 0.000 description 1
- TXWOGHSRPAYOML-UHFFFAOYSA-N cyclobutanecarboxylic acid Chemical compound OC(=O)C1CCC1 TXWOGHSRPAYOML-UHFFFAOYSA-N 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- YMGUBTXCNDTFJI-UHFFFAOYSA-M cyclopropanecarboxylate Chemical compound [O-]C(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-M 0.000 description 1
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 150000001989 diazonium salts Chemical class 0.000 description 1
- 238000006193 diazotization reaction Methods 0.000 description 1
- MIILMDFFARLWKZ-UHFFFAOYSA-L dichlorozirconium;1,2,3,4,5-pentamethylcyclopentane Chemical compound [Cl-].[Cl-].CC1=C(C)C(C)=C(C)C1(C)[Zr+2]C1(C)C(C)=C(C)C(C)=C1C MIILMDFFARLWKZ-UHFFFAOYSA-L 0.000 description 1
- HQWPLXHWEZZGKY-UHFFFAOYSA-N diethylzinc Chemical compound CC[Zn]CC HQWPLXHWEZZGKY-UHFFFAOYSA-N 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 125000005594 diketone group Chemical group 0.000 description 1
- AXAZMDOAUQTMOW-UHFFFAOYSA-N dimethylzinc Chemical compound C[Zn]C AXAZMDOAUQTMOW-UHFFFAOYSA-N 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000012154 double-distilled water Substances 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 229950009791 durvalumab Drugs 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 210000004696 endometrium Anatomy 0.000 description 1
- 208000023965 endometrium neoplasm Diseases 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 1
- UFNVPOGXISZXJD-XJPMSQCNSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-XJPMSQCNSA-N 0.000 description 1
- 229960003649 eribulin Drugs 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- YHHXIIBFDYCARD-BQBZGAKWSA-N ethyl (1S,2S)-2-ethylcyclopropane-1-carboxylate Chemical compound CCOC(=O)[C@H]1C[C@@H]1CC YHHXIIBFDYCARD-BQBZGAKWSA-N 0.000 description 1
- AOQBVPPEXCRXAW-RITPCOANSA-N ethyl (1s,2s)-2-(hydroxymethyl)cyclopropane-1-carboxylate Chemical compound CCOC(=O)[C@H]1C[C@@H]1CO AOQBVPPEXCRXAW-RITPCOANSA-N 0.000 description 1
- JWYSLVLBKXDZCW-WDSKDSINSA-N ethyl (1s,2s)-2-methylcyclopropane-1-carboxylate Chemical compound CCOC(=O)[C@H]1C[C@@H]1C JWYSLVLBKXDZCW-WDSKDSINSA-N 0.000 description 1
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 1
- LDDOSDVZPSGLFZ-UHFFFAOYSA-N ethyl cyclopropanecarboxylate Chemical compound CCOC(=O)C1CC1 LDDOSDVZPSGLFZ-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000017188 evasion or tolerance of host immune response Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- JYEFSHLLTQIXIO-SMNQTINBSA-N folfiri regimen Chemical compound FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 JYEFSHLLTQIXIO-SMNQTINBSA-N 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 210000000232 gallbladder Anatomy 0.000 description 1
- 229950000456 galunisertib Drugs 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 235000008434 ginseng Nutrition 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 102000057041 human TNF Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 238000006197 hydroboration reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000012135 ice-cold extraction buffer Substances 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 238000000126 in silico method Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- FABUFPQFXZVHFB-CFWQTKTJSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@H](C)C(=O)C(C)(C)[C@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-CFWQTKTJSA-N 0.000 description 1
- 229960002014 ixabepilone Drugs 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229960001614 levamisole Drugs 0.000 description 1
- 229950011263 lirilumab Drugs 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 108010082117 matrigel Proteins 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 229910052987 metal hydride Inorganic materials 0.000 description 1
- 150000004681 metal hydrides Chemical class 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 239000002032 methanolic fraction Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- 210000002200 mouth mucosa Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- 229960000513 necitumumab Drugs 0.000 description 1
- 230000025020 negative regulation of T cell proliferation Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229960003301 nivolumab Drugs 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000007339 nucleophilic aromatic substitution reaction Methods 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- HEGSGKPQLMEBJL-RKQHYHRCSA-N octyl beta-D-glucopyranoside Chemical compound CCCCCCCCO[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O HEGSGKPQLMEBJL-RKQHYHRCSA-N 0.000 description 1
- 229950008516 olaratumab Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 201000008129 pancreatic ductal adenocarcinoma Diseases 0.000 description 1
- 210000004923 pancreatic tissue Anatomy 0.000 description 1
- WYMSBXTXOHUIGT-UHFFFAOYSA-N paraoxon Chemical compound CCOP(=O)(OCC)OC1=CC=C([N+]([O-])=O)C=C1 WYMSBXTXOHUIGT-UHFFFAOYSA-N 0.000 description 1
- 230000007119 pathological manifestation Effects 0.000 description 1
- 229960002621 pembrolizumab Drugs 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 229950010773 pidilizumab Drugs 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 208000023958 prostate neoplasm Diseases 0.000 description 1
- 108010043671 prostatic acid phosphatase Proteins 0.000 description 1
- 239000000296 purinergic P1 receptor antagonist Substances 0.000 description 1
- PJESVVYWPFAJCS-UHFFFAOYSA-N pyridazine-3-carbonitrile Chemical compound N#CC1=CC=CN=N1 PJESVVYWPFAJCS-UHFFFAOYSA-N 0.000 description 1
- 150000004892 pyridazines Chemical class 0.000 description 1
- 229960002633 ramucirumab Drugs 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 230000027756 respiratory electron transport chain Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000010414 supernatant solution Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- MHYGQXWCZAYSLJ-UHFFFAOYSA-N tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1 MHYGQXWCZAYSLJ-UHFFFAOYSA-N 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- UQCWXKSHRQJGPH-UHFFFAOYSA-M tetrabutylazanium;fluoride;hydrate Chemical compound O.[F-].CCCC[N+](CCCC)(CCCC)CCCC UQCWXKSHRQJGPH-UHFFFAOYSA-M 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 238000006276 transfer reaction Methods 0.000 description 1
- 229950007217 tremelimumab Drugs 0.000 description 1
- GGUBFICZYGKNTD-UHFFFAOYSA-N triethyl phosphonoacetate Chemical compound CCOC(=O)CP(=O)(OCC)OCC GGUBFICZYGKNTD-UHFFFAOYSA-N 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 229950005972 urelumab Drugs 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 201000007710 urinary bladder small cell neuroendocrine carcinoma Diseases 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 229960004295 valine Drugs 0.000 description 1
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本發明提供5-[5-[2-環烷基]-6-噠嗪-3-基]-1H-嘧啶-2,4-二酮化合物或其醫藥學上可接受之鹽,其抑制CD73之活性且可用於治療癌症。
Description
本發明係關於5-[5-[2-環烷基]-6-噠嗪-3-基]-1H-嘧啶-2,4-二酮化合物或其醫藥學上可接受之鹽以及包含該等化合物之醫藥組合物,其等抑制CD73之活性及可適用於治療癌症。
CD73 (亦稱為5'-核苷酸酶或胞外-5'核苷酸酶(EC 3.1.3.5))為將5'單核苷酸轉化為核苷之酶。CD73在許多組織中表現且在癌組織中上調,且CD73路徑藉由限制抗腫瘤T細胞免疫性經由腺苷受體信號傳導促進腫瘤生長(Zhang B,Cancer Research 2010
; 70: 6407-6411;Antonioli L等人,Drug Discovery Today 2017
; 22: 1686-1696)。
CD73缺陷型小鼠增加了抗腫瘤免疫性且對實驗性轉移具有抗性(Stagg J等人,Cancer Research 2011
; 71: 2892-2900)。由腫瘤CD73產生之細胞外腺苷積聚於腫瘤微環境中,損害抗腫瘤T細胞免疫性(Zhang B,Cancer Research 2010
; 70: 6407-6411; Antonioli L等人,Drug Discovery Today 2017
; 22: 1686-1696),且與腫瘤免疫逃逸、增殖、遷移、新血管生成、轉移及腫瘤細胞之化學抗性有關(Inoue Y等人,Oncotarget 2017
; 8: 8738-8751)。
亦已報導升高的CD73表現與增加的免疫抑止相關聯(Jin D等人,Cancer Res. 70
: 2245-2255 (2010);Beavis PA等人,Trends Immunol 33
: 231-7 (2012);Beavis, PA等人,Cancer Immunol Res. 3
: 506-17 (2015);Loi S等人,Proc. Natl. Acad. Sci. USA 110
: 11091-11096 (2013))。
因此,CD73路徑發揮免疫抑止作用,並且已表明阻斷CD73路徑可適用於治療癌症(Antonioli L等人,Oncoimmunol. 2016
; 5: e1216292, doi: 10.1080/2162402X.2016.1216292;Antonioli L等人,Trends in Cancer 2016
; 2: 95-109;Allard D等人,Immunotherapy 2016
; 8: 145-163)。CD73抑制劑在臨床試驗中用於治療癌症(Hay CM等人,Oncoimmunol. 2016
; 5(8): e1208875;Allard D等人,Immunotherapy 2016
; 8: 145-163)。
仍需要提供替代性CD73抑制劑,更特定言之,以用於治療癌症。特定言之,仍需要口服可用小分子CD73抑制劑,且其在腫瘤微環境中呈現更全面的標靶抑制。
因此,本發明提供可適用於治療癌症之CD73抑制劑化合物。本發明提供式I化合物: 其中n為0-3;
其中R1
為-H、-F、-偕-二氟、-偕-二甲基、-C1-4
烷基、-CHF2
、-CHF2
CH3
或-CH2
CH2
F;
且
R2
選自-H、-CH3
、-F、-Cl、-CN或-OCH3
;
或其醫藥學上可接受之鹽。
其中n為0-3;
其中R1
為-H、-F、-偕-二氟、-偕-二甲基、-C1-4
烷基、-CHF2
、-CHF2
CH3
或-CH2
CH2
F;
且
R2
選自-H、-CH3
、-F、-Cl、-CN或-OCH3
;
或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供式I化合物,其中n為0,R1
為-C1-4
烷基,且R2
為-CH3
;或其醫藥學上可接受之鹽。
在另一實施例中,化合物為

 或其醫藥學上可接受之鹽。
或其醫藥學上可接受之鹽。
在另一實施例中,化合物為

 ,或其醫藥學上可接受之鹽。
,或其醫藥學上可接受之鹽。
在另一實施例中,化合物為
 ,或其醫藥學上可接受之鹽。
,或其醫藥學上可接受之鹽。
在另一實施例中,化合物為
 ,
或其醫藥學上可接受之鹽。
,
或其醫藥學上可接受之鹽。
在另一實施例中,化合物為
 、
、 、
、 或
或 ,
或其醫藥學上可接受之鹽。
,
或其醫藥學上可接受之鹽。
在另一實施例中,化合物為 ,
或其醫藥學上可接受之鹽。
,
或其醫藥學上可接受之鹽。
在一個實施例中,式I化合物具有下式: 其中R1
為-CH2
CH3
或-CH(CH3
)2
,或其醫藥學上可接受之鹽。在另一實施例中,R1
為-CH2
CH3
,或其醫藥學上可接受之鹽。在另一實施例中,R1
為-CH(CH3
)2
,或其醫藥學上可接受之鹽。
其中R1
為-CH2
CH3
或-CH(CH3
)2
,或其醫藥學上可接受之鹽。在另一實施例中,R1
為-CH2
CH3
,或其醫藥學上可接受之鹽。在另一實施例中,R1
為-CH(CH3
)2
,或其醫藥學上可接受之鹽。
在另一實施例中,式I化合物具有下式: 或
或 其中R1
為CH2
CH3
或CH(CH3
)2
,或其醫藥學上可接受之鹽。
其中R1
為CH2
CH3
或CH(CH3
)2
,或其醫藥學上可接受之鹽。
在另一實施例中,式I化合物具有下式: 其中R1
為-CH2
CH3
或-CH(CH3
)2
,或其醫藥學上可接受之鹽。
其中R1
為-CH2
CH3
或-CH(CH3
)2
,或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供以下化合物: ,
或其醫藥學上可接受之鹽。
,
或其醫藥學上可接受之鹽。
在一個實施例中,本發明提供以下化合物: ,
或其醫藥學上可接受之鹽。
,
或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供以下化合物: ,
或其醫藥學上可接受之鹽。
,
或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供以下化合物: ,
或其醫藥學上可接受之鹽。
,
或其醫藥學上可接受之鹽。
在一個實施例中,本發明提供一種化合物5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供一種化合物5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在一個實施例中,本發明提供一種醫藥組合物,其包含5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽,以及一或多種醫藥學上可接受之載劑、稀釋劑或賦形劑。
在另一實施例中,本發明提供一種醫藥組合物,其包含5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,以及一或多種醫藥學上可接受之載劑、稀釋劑或賦形劑。
在一個實施例中,本發明提供一種用於治療癌症之方法,其包含向有需要之患者投與有效量之5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供一種用於治療癌症之方法,其包含向有需要之患者投與有效量之5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在一個實施例中,本發明提供用於療法中之5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供用於療法中之5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在一個實施例中,本發明提供用於治療癌症之5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供用於治療癌症之5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在一個實施例中,本發明提供一種用於治療癌症之醫藥組合物,該醫藥組合物包含5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供一種用於治療癌症之醫藥組合物,該醫藥組合物包含5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
在一個實施例中,本發明提供5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽在製造用於治療癌症之藥物中的用途。
在另一實施例中,本發明提供5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽在製造用於治療癌症之藥物中的用途。
在一個實施例中,本發明提供4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪之製備及用途,其可在結構上表示為: ,
其適用於製造用於治療癌症之化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪。
,
其適用於製造用於治療癌症之化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪。
在一個實施例中,本發明提供4,4,5,5-四甲基-2-(2位取代的環丙基)-1,3,2-二氧雜硼雜環戊烷之製備及用途,其可在結構上表示為: ,
其中R1
= CH2
CH3
或CH(CH3
)2
,其適用於製造用於治療癌症之藥物。在一個實施例中,R = CH2
CH3
。在另一實施例中,R = CH(CH3
)2
。在另一實施例中,本發明提供用於製造化合物或藥物之4,4,5,5-四甲基-2-(2位取代的環丙基)-1,3,2-二氧雜硼雜環戊烷。
,
其中R1
= CH2
CH3
或CH(CH3
)2
,其適用於製造用於治療癌症之藥物。在一個實施例中,R = CH2
CH3
。在另一實施例中,R = CH(CH3
)2
。在另一實施例中,本發明提供用於製造化合物或藥物之4,4,5,5-四甲基-2-(2位取代的環丙基)-1,3,2-二氧雜硼雜環戊烷。
在一個實施例中,本發明提供2-[(1S,2S)-2-乙基-環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷之製備及用途,其可在結構上表示為: ,
其適用於製造用於治療之癌症化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之2-[(1S,2S)-2-乙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷。
,
其適用於製造用於治療之癌症化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之2-[(1S,2S)-2-乙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷。
在一個實施例中,本發明提供2-[(1S,2S)-2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷之製備及用途,其可在結構上表示為: ,
其適用於製造用於治療癌症之化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之2-[(1S,2S)-2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷。
,
其適用於製造用於治療癌症之化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之2-[(1S,2S)-2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷。
在一個實施例中,本發明提供使用(1S,2S)-2-乙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯)對掌性合成2-[(1S,2S)-2-乙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷,其可在結構上表示為: ,
其中R1
= CH2
CH3
或CH(CH3
)2
,其適用於製造用於治療癌症之化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之(1S,2S)-2-乙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯)。
,
其中R1
= CH2
CH3
或CH(CH3
)2
,其適用於製造用於治療癌症之化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之(1S,2S)-2-乙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯)。
在一個實施例中,本發明提供使用(1S,2S)-2-乙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯)對掌性合成2-[(1S,2S)-2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷,其可在結構上表示為: ,
其適用於製造用於治療癌症之化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之(1S,2S)-2-異丙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯)。
,
其適用於製造用於治療癌症之化合物或藥物。在另一實施例中,本發明提供用於製造化合物或藥物之(1S,2S)-2-異丙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯)。
CD73蛋白在許多組織中表現,包括大腦、甲狀腺、骨髓、淋巴結、扁桃體、脾臟、心肌、平滑肌、肺、鼻咽、肝、膽囊、胰腺、唾液腺、口腔黏膜、食道、胃、十二指腸、小腸、結腸、直腸、腎臟、膀胱、睾丸、前列腺、輸卵管、陰道、乳房、子宮頸、子宮、子宮內膜、卵巢、軟組織及皮膚(proteinatlas.org/ENSG00000135318-NT5E/tissue)。CD73在多種類型之腫瘤中過度表現(Antonioli L等人,Nat. Rev. Cancer 13
, 842-857 (2013);Antonioli L等人,Trends Cancer 2
: 95-109 (2016)。CD73亦在正常及病理性人類肝膽胰臟組織中表現,包括肝細胞、胰管腺癌及肝外膽管細胞癌(Sciarra, A.等人,Cancer Immunol Immunother 2019
; doi.org/10.1007/s00262-018-2290-1)。
升高的CD73表現及活性與腫瘤侵襲性及轉移性相關聯,且與較短的患者存活時間相關聯(Jin D等人,Cancer Research 2010
; 70: 2245-2255)。增加的CD73表現與不良預後相關聯(Inoue K等人,Oncotarget
8: 8738-8751 (2017);Turcotte K等人,Cancer Res.
5: 4494-4503 (2015);Lu YT等人,World J. Gastroenterol. 19
: 1912-1918 (2013);Wu XR等人,J. Surg. Oncol. 106
: 130-137 (2012);Buisseret S等人,Ann. Oncol.
29: 1056-1062 (2017);Leclerc R等人,Clin. Cancer Res. 22
: 158-66 (2016))。
三陰性乳癌中之CD73表現與較糟的臨床結果及增加的對蒽環黴素化療之抗性相關聯(Allard B等人,Expert Opin. Ther. Targets 2014
; 18: 863-881)。CD73與頭頸部鱗狀細胞癌之不良預後相關聯(Ren Z-H等人,Oncotargets 2016
; 7: 61690-61702)。CD73表現亦與腎細胞癌之進展相關聯(Yu Y等人,Oncol. Lett. 2015
; 9: 2485-2494)。
CD73在子宮內膜腫瘤(Aliagas E等人,Mediators of Inflammation 2014
; doi: 10.1155/2014/509027)、前列腺腫瘤組織(Leclerc BG等人,Clin. Cancer Res. 2016
; 22: 158-166)、非小細胞肺癌組織(Inoue Y等人,Oncotarget 2017
; 8: 8738-8751)中過度表現。已報導CD73在腫瘤中過度表現,該等腫瘤包括乳癌、結腸直腸癌、卵巢癌、胃癌及膽囊癌(Gao Z-W及Zhang H-Z,Biomed Res. Int. 2014
; doi: 10.1155/2014/460654)。已報導CD73在癌細胞株中過度表現,該等癌細胞株包括神經膠母細胞瘤、黑素瘤、乳癌、卵巢癌、神經管母細胞瘤及膀胱癌細胞株(Gao Z-W及Zhang H-Z,Biomed Res.Int. 2014
; doi: 10.1155/2014/460654)。
已報導CD73-腺苷減小卵巢癌患者之免疫反應及存活率(Gaudreau P-O等人,Oncoimmunology 2016
; 5(5): e1127496)。
已報導用選擇性CD73抑制劑α,β-亞甲基腺苷5'-二磷酸酯在活體內進行之CD73阻斷在皮下注射有EG7 (淋巴瘤)、MC38 (結腸)、AT-3 (乳房)及B16F10 (黑素瘤)腫瘤細胞之同基因型小鼠中減少腫瘤生長(Stagg J等人,Cancer Research 2011
; 71: 2892-2900;Forte G等人,J. Immunol. 2012
; 189: 2226-2233)。已報導抗CD73抗體療法抑制乳房腫瘤生長及轉移(Stagg J等人,PNAS 2010
; 107: 1547-1552;Terp MG,J. Immunol. 2013
; 191: 4165-4173)。
另外,已報導腺苷及CD73酶活性在癌症患者中增加(Huang N等人,Cancer Res.75
:1538 (2015),且已報導對免疫檢查點抑制劑無反應之患者比有反應之患者具有更高的腺苷含量(Giannakis HX, et al.,J. Clin. Oncol.
35: 15 Suppl. 3036-3036 (2017))。
已報導致癌活化及雌激素受體喪失與增加的CD73表現相關聯(Reinhardt J等人,Cancer Res. 77
: 4697-4709 (2017);Spychala J等人,Clin. Cancer Res. 10
:708-17 (2004);Ascierto及McArthur J.,Transl. Med. 15
: 173 (2017);Young A等人,Cancer Res. 77
:4684-4696 (2017))。
可受益於CD73抑制劑治療之個體包括:具有對抗PD1/PDL-1抑制劑具有抗性或難以用該等抑制劑治療之腫瘤的個體,該等腫瘤諸如非小細胞肺癌、膀胱癌及黑素瘤;患有EGFR/BRAF/Kras突變型癌症之個體,該等癌症諸如非小細胞肺癌、膀胱癌、黑素瘤、結腸及胰腺;患有雌激素受體(-)癌症之個體,該等癌症諸如三陰性乳癌;具有高表現量之CD73之個體,諸如胰臟癌及結腸直腸癌。視需要,此類個體可經選擇以供基於如藉由IHC分析所量測到的其腫瘤中高CD73表現量之存在;或基於如藉由RT-PCR分析所偵測到的其腫瘤中EGFR及BRAF突變之存在;或基於如藉由IHC或RT-PCR所分析所偵測到的其腫瘤中雌激素受體之損失;或基於如藉由LC-MS分析所偵測到的其腫瘤或血漿中高含量腺苷及AMP之存在而用本文中所揭示之化合物或其醫藥學上可接受之鹽進行治療。對於藥效動力學評估,本文中所描述之基於LC-MS之離體分析可用於量測CD73抑制劑對血液中將AMP轉化為腺苷的影響。
本發明亦提供一種治療患者之癌症的方法,其包含向有需要之患者投與有效量的式I化合物或其醫藥學上可接受之鹽。
本發明亦提供一種治療癌症之方法,其包含向有需要之個體投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽,其中該癌症為膀胱癌、乳癌、膽管癌、結腸直腸癌、結腸癌、胃癌、膽囊癌、神經膠母細胞瘤、頭頸癌、肝癌、肺癌、淋巴瘤、神經管母細胞瘤、黑素瘤、卵巢癌、胰臟癌、前列腺癌或腎癌。在一個實施例中,乳癌為三陰性乳癌。在另一實施例中,肺癌為非小細胞肺癌。在一個較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮。
在一個實施例中,患者為已測定血清CD73活性之患者。在一個較佳實施例中,「測定CD73活性」意謂測定是否存在CD73活性。用於測定CD73表現量或CD73活性之方法為一般熟習此項技術者已知,例如參見S Morello等人,J. Trans. Med. 2017
; 15:244。在另一較佳實施例中,「測定CD73活性」意謂定量AMP藉由CD73轉化為腺苷之量,且有助於定量CD73活性之量的基於LC-MS之分析提供於本文中。
在另一實施例中,患者為已測定組織中之CD73表現之患者。在另一實施例中,組織為腫瘤組織。用於例如使用西方墨點法或免疫組織化學測定組織中之CD73表現量的方法為一般熟習此項技術者已知(X-R Wu等人,J. Surg. Oncol. 2012
; 106: 130-137)。
本發明亦提供一種方法,其包含測定來自已投與如技術方案1至12中任一項之化合物或其醫藥學上可接受之鹽的患者之組織中之CD73表現。在一較佳實施例中,組織為腫瘤組織。
本發明亦提供一種方法,其包含測定來自已投與根據本文中之式I化合物的化合物或其醫藥學上可接受之鹽的患者之血清中之CD73活性。此方法係靈敏的,不涉及腫瘤活體組織切片,不涉及抗體或免疫組織化學,有助於定量CD73活性(例如,藉由有助於計算EC50
)。在一個較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮。在另一較佳實施例中,該方法進一步包含使用放射性標記之單磷酸腺苷(AMP)分析CD73活性且使用質譜法測定放射性標記之AMP濃度。在另一較佳實施例中,放射性標記之AMP為13C5_15N5-AMP。
在另一較佳實施例中,該方法包含(a)將來自患者之血清提供於複數個容器(例如,微量滴定培養盤)中之每一者中,該等容器經組態以含有向患者投與的各種濃度之化合物,(b)將13C10_15N5_AMP提供於複數個容器中之各容器中,(c)在有助於混合(例如,震盪)的條件下培育複數個容器,(d)使複數個容器離心,(e)將上澄液自各容器轉移至新的各別容器,(f)將含有內部標準物之萃取溶液提供於新的各別容器中之每一者中,(g)使新的各別容器離心,(h)將上澄液自各新的各別容器轉移至各別分析容器,(i)如本文中所描述藉由LC/MS (「用於腺苷及腺苷純化之質譜法(Mass Spectroscopy for Adenosine and Adenosine Purification)」)針對13C10_15N5_adenosine、13C10_15N4_inosine及15N4次黃嘌呤分析各各別分析容器中之上澄液,以及(j)計算EC50
。在一個較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮。在一較佳實施例中,內部標準物為13C5-AMP、13C5-腺苷、15N4-肌苷、及13C5-次黃嘌呤。
本發明亦提供本文中之式I化合物或其醫藥學上可接受之鹽在療法中的用途。在一個實施例中,療法為癌症治療。在另一實施例中,該癌症為膀胱癌、乳癌、膽管癌、結腸直腸癌、結腸癌、胃癌、膽囊癌、神經膠母細胞瘤、頭頸癌、肝癌、肺癌、淋巴瘤、神經管母細胞瘤、黑素瘤、卵巢癌、胰臟癌、前列腺癌或腎癌。在一個實施例中,乳癌為三陰性乳癌。在另一實施例中,肺癌為非小細胞肺癌。在一個較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮。
本發明亦提供本文中之式I化合物或其醫藥學上可接受之鹽用於製造用於治療癌症之藥物的用途。在一個實施例中,該癌症為膀胱癌、乳癌、膽管癌、結腸直腸癌、結腸癌、胃癌、膽囊癌、神經膠母細胞瘤、頭頸癌、肝癌、肺癌、淋巴瘤、神經管母細胞瘤、黑素瘤、卵巢癌、胰臟癌、前列腺癌或腎癌。在一個實施例中,乳癌為三陰性乳癌。在另一實施例中,肺癌為非小細胞肺癌。在一個較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮。
在一個實施例中,本發明提供一種治療癌症之方法,其包含與一或多種抗腫瘤劑組合同時、分開或依序投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽。抗腫瘤劑之非限制性實例包括雷莫蘆單抗(ramucirumab)、萊西單抗(necitumumab)、奧拉單抗(olaratumab)、吉西他濱(gemcitabine)、培美曲塞(pemetrexed)、高倫替布(galunisertib)、阿貝馬昔布(abemaciclib)、吉非替尼(gefitinib)、維羅非尼(vemurafenib)、達拉非尼(dabrafenib)、曲美替尼(trametinib)、順鉑(cisplatin)、卡鉑(carboplatin)、達卡巴嗪(dacarbazine)、脂質多柔比星(doxorubicin)、多西他賽(docetaxel)、環磷醯胺(cyclophosphamide)及多柔比星、溫諾平(navelbine)、艾瑞布林(eribulin)、太平洋紫杉醇(paclitaxel)、可注射懸浮液之太平洋紫杉醇蛋白結合顆粒、伊沙匹隆(ixabepilone)、卡培他濱(capecitabine)、FOLFOX (甲醯四氫葉酸、氟脲嘧啶及奧沙利鉑(oxaliplatin))、FOLFIRI (甲醯四氫葉酸、氟脲嘧啶及伊立替康(irinotecan))、西妥昔單抗(cetuximab)、EGFR抑制劑、Raf抑制劑、B-Raf抑制劑、ERK抑制劑、CDK4/6抑制劑、吲哚胺2,3-雙加氧酶抑制劑、TGFβ抑制劑及TGFβ受體抑制劑。
在另一實施例中,本發明提供一種治療癌症之方法,其包含與一或多種免疫腫瘤學藥劑組合同時、分開或依序投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽。在一個較佳實施例中,免疫腫瘤學藥劑為抗PD-1抗體、抗PD-L1抗體、抗CD137促效劑抗體或抗CTLA4抗體。免疫腫瘤學藥劑之非限制性實例包括納武單抗(nivolumab)、伊派利單抗(ipilimumab)、皮立珠單抗(pidilizumab)、派立珠單抗(pembrolizumab)、曲美木單抗(tremelimumab)、烏瑞魯單抗(urelumab)、利瑞路單抗(lirilumab)、阿特珠單抗(atezolizumab)、德瓦魯單抗(durvalumab)及抗PD-L1抗體LY3300054 (分別於WO 2017/034916及US 2017/0058033中闡述為SEQ ID NO: 10及SEQ ID NO: 11之重鏈及輕鏈序列)。在一個較佳實施例中,免疫腫瘤學藥劑為抗PD-1抗體。在另一較佳實施例中,免疫腫瘤學藥劑為抗PD-L1抗體。在另一較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,且免疫腫瘤學藥劑為LY3300054。
在一個實施例中,本發明提供一種治療非小細胞肺癌之方法,其包含與另一種藥劑組合同時、分開或依序投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽。在一個較佳實施例中,另一種藥劑為奧希替尼(osimertinib)、西妥昔單抗或阿貝馬昔布。在另一較佳實施例中,另一種藥劑為奧希替尼。在另一較佳實施例中,另一種藥劑為西妥昔單抗。在另一較佳實施例中,另一種藥劑為阿貝馬昔布。
在另一實施例中,本發明提供一種治療黑素瘤之方法,其包含與另一種藥劑組合同時、分開或依序投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽。在一個較佳實施例中,另一種藥劑為BRAF抑制劑、抗PD-1抗體或抗PD-L1抗體。在另一較佳實施例中,另一種藥劑為BRAF抑制劑。在另一較佳實施例中,另一種藥劑為抗PD-1抗體。在另一較佳實施例中,另一種藥劑為抗PD-L1抗體。在另一較佳實施例中,抗PD-L1抗體為LY3300054。在另一較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,且另一種藥劑為抗PD-L1抗體LY3300054。
在另一實施例中,本發明提供一種治療結腸直腸癌之方法,其包含與另一種藥劑組合同時、分開或依序投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽。在一個較佳實施例中,另一種藥劑為阿貝馬昔布。在另一較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,且另一種藥劑為阿貝馬昔布。
在另一實施例中,本發明提供一種治療胰臟癌之方法,其包含與另一種藥劑組合同時、分開或依序投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽。在一個較佳實施例中,另一種藥劑為阿貝馬昔布。在另一較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,且另一種藥劑為阿貝馬昔布。
在另一實施例中,本發明提供一種治療三陰性乳癌之方法,其包含與另一種藥劑組合同時、分開或依序投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽。在一個較佳實施例中,化合物為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮。
本發明亦提供一種抑制患者體內之CD73酶活性的方法,其包含向有需要之患者投與有效量的式I化合物或其醫藥學上可接受之鹽。本發明亦提供一種抑制AMP在患者體內轉化為腺苷之方法,其包含向有需要之患者投與有效量的式I化合物或其醫藥學上可接受之鹽。
在另一實施例中,本發明提供一種治療癌症之方法,其包含與一或多種調節腺苷之藥劑組合同時、分開或依序投與有效量的本文中之式I化合物或其醫藥學上可接受之鹽。調節腺苷之藥劑之非限制性實例包括抗CD73抗體及腺苷受體拮抗劑。
本發明亦提供一種醫藥組合物,其包含式I化合物或其醫藥學上可接受之鹽,以及一或多種醫藥學上可接受之載劑、稀釋劑或賦形劑。本發明進一步提供一種用於製備醫藥組合物之方法,其包含摻合式I化合物或其醫藥學上可接受之鹽與一或多種醫藥學上可接受之載劑、稀釋劑或賦形劑。本發明亦涵蓋合成式I化合物之新穎中間產物及方法。
本申請案根據35 U.S.C. §119(e)規定主張2018年3月1日申請之美國臨時申請案第62/636,978號及2018年12月5日申請之美國臨時申請案第62/775,553號之權益;該等申請案之揭示內容以引用之方式併入本文中。
如上文使用且貫穿本發明之描述,除非另有指示,否則以下術語應理解為具有如下含義:
「醫藥學上可接受之載劑、稀釋劑或賦形劑」為此項技術中大體上認可之用於將生物活性劑遞送至哺乳動物(例如,人類)的介質。
術語「治療(treatment/treat/treating)」及其類似者意欲包括減緩或逆轉病症之進展。此等術語亦包括緩解、改善、衰減、消除或減輕病症或病況之一或多個症狀(即使病症或病況未實際上消除及即使病症或病況之進展本身未減緩或逆轉)。
「有效量」意謂將引發治療臨床醫師對患者之生物學或醫學反應或所期望治療效果的本發明之化合物或其醫藥學上可接受之鹽或含有本發明之化合物或其醫藥學上可接受之鹽的醫藥組合物的量。在一個實施例中,化合物或其醫藥學上可接受之鹽在活體外或離體CD73酶分析抑制AMP轉化為腺苷。在另一實施例中,化合物或其醫藥學上可接受之鹽在來自經不同劑量之化合物治療之動物的小鼠全血抑制AMP轉化為腺苷。
「偕-二氟」係指鍵結至同一碳之兩個氟原子。
「偕-二甲基」係指鍵結至同一碳之兩個甲基。
如本文中所使用,術語「患者」係指人類。
有效量可易於由作為熟習此項技術者之主治診斷醫師藉由使用已知技術且藉由觀察在類似情況下得到之結果來測定。在測定用於患者之有效量時,主治診斷醫師考慮多個因素,包括但不限於:患者之物種;其體型、年齡及一般健康狀況;所涉及之特定疾病或病症;涉及程度或疾病或病症之嚴重程度;個別患者之反應;所投與之特定化合物;投與模式;所投與之製劑之生物可用性特徵;所選擇之劑量方案;伴隨藥療之使用;及其他相關情況。
本發明化合物可在廣泛劑量範圍內使用。舉例而言,每日劑量通常在約0.01至約50 mg/kg體重之範圍內。
本發明化合物較佳調配為藉由使得化合物具有生物可用性之任何途徑投與的醫藥組合物,該途徑包括經口、靜脈內及經皮途徑。最佳地,此類組合物用於經口投與。此類醫藥組合物及其製備方法為此項技術中所熟知的。參見例如Remington: The Science and Practice of Pharmacy (D.B. Troy編, 第21版, Lippincott, Williams & Wilkins, 2006)。
熟習此項技術者應理解,本發明化合物能夠形成鹽。本發明化合物含有鹼性雜環,且因此與多種無機酸及有機酸中之任一者反應,以形成醫藥學上可接受之酸加成鹽。此類醫藥學上可接受之酸加成鹽及其常見製備方法為此項技術中所熟知的。參見例如,P. Stahl等人, PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE, (VCHA/Wiley-VCH, 2008)。
「一或多種醫藥學上可接受之鹽」係指相對無毒的無機鹽及有機鹽或本發明化合物之鹽(S.M. Berge等人, 「Pharmaceutical Salts」,Journal of Pharmaceutical Sciences
, 第66卷, 第1期, 1977年1月)。
化合物名中「異構體1」之名稱表示,當一對對映異構體之混合物在下文「製備及實例」中所描述的條件下藉由對掌性層析分離時,本發明對應中間產物或化合物為兩個溶離對映異構體中之第一個。化合物名中「異構體2」之名稱表示,當一對對映異構體之混合物在下文「製備及實例」中所描述的條件下藉由對掌性層析分離時,本發明對應中間產物或化合物為兩個溶離對映異構體中之第二個。
可根據此項技術中熟知且瞭解之合成方法來製備本發明化合物。此等反應步驟之適合反應條件係此項技術中熟知的,且合適的替代溶劑及輔試劑係在技術領域內。熟習此項技術者亦應瞭解,藉由熟知技術視需要或期望可分離及/或純化合成中間產物(且此舉頻繁),在後續合成步驟中直接使用具有少量或不純化之各種中間產物係有可能的。此外,熟習此項技術者將瞭解,在某些情況下所介紹之部分次序並非至關重要的。如熟練的化學家所充分理解,產生本發明化合物所必需之具體步驟順序取決於具體的合成複合物、起始複合物及經取代部分的相對不利條件。所有取代物(除非另有指示)係皆如先前所定義且所有反應劑皆為此項技術中所熟知及理解。
如本文中所使用,以下術語具有指示以下之含義:「ACN」係指乙腈;「DAST」係指三氟化二乙基胺基硫;「DCM」係指二氯甲烷;「DMAP」係指4-二甲胺基吡啶;「dmso」或「DMSO」係指二甲亞碸;「ee」係指對映異構體過量;「ES/MS」係指電噴質譜分析;「EtOAc」係指乙酸乙酯;「Et2
O」係指乙醚;「FBS」係指胎牛血清;「GC-MS」係指氣相層析-質譜分析;「HBSS」係指漢克氏平衡鹽溶液(Hank's Balanced Salt Solution);「IC50
」係指半數最大抑制濃度;「LAH」係指氫化鋰鋁;「LC-ES/MS」係指液相層析電噴質譜分析;「MS」係指質譜分析;「MeOH」係指甲醇;「MTBE」係指甲基第三丁基醚;「nBuLi」係指正丁基鋰;「nm」係指奈米;「NMR」係指核磁共振;「OAc」係指乙酸酯;「psi」係指磅/平方吋;「RT」係指室溫或環境溫度;「SCX」係指StrongCation Exchange;「SFC」係指超臨界流體層析;「SN
Ar」係指親核芳族取代;「TEA」係指三乙胺;「THF」係指四氫呋喃;「tR
」係指滯留時間;且「w/w」係指溶液中之重量/重量比例。
本發明化合物可如以下流程所說明來合成。流程 1 
流程1描繪式Ia之化合物之製備。使用熟知的Suzuki類型條件,市售(2,4-二甲氧基嘧啶-5-基)硼酸1可偶合至4,6-二氯-3-甲基-噠嗪。可實現3的氯部分與經適當取代之反式環丙基硼酸酯的後續Suzuki偶合以獲得4。熟習此項技術者將認識到化合物4之對映異構體可使用此項技術中所熟知的對掌性分離技術來分離。可藉由去保護4中之甲氧基在此項技術中充分描述之一系列去甲基化條件下來製備式Ia之化合物。流程 2 
流程2描繪製備式II化合物所需的必備環丙基硼酸酯。熟習此項技術者將認識到可在一系列條件下,尤其在存在諸如Cu或Zr之過渡金屬催化劑的情況下實現經適當取代之炔烴5之硼氫化,以獲得硼酸烯酯6。硼酸烯酯可在此項技術中所熟知的穩定類碳烯型條件下環丙烷化,諸如西蒙斯-史密斯環丙烷化(Simmons-Smith cyclopropanation)、科里-柴可夫斯基反應(Corey-Chaykovsky reaction)及用具有(例如,Cu、Pd或Ni)及不具有(例如,熱或光化學)過渡金屬催化劑的重氮甲烷(或重氮化合物)進行環丙烷化之方法,以獲得經適當取代之反式-環丙基硼酸酯7。熟習此項技術者將認識到熱力學上有利的環丙烷化產物將為反式對映異構體於7中之混合物。流程 3 
流程3描繪式Ib化合物之對掌性合成。經適當取代之胺基酸8在經修改山德邁耳條件(基團SN
Ar)下重氮化以得到9為此項技術中所熟知。可利用此項技術中所熟知的一系列還原劑(包括氫化鋁及二硼烷作為還原劑)實現至醇10的後續還原。在此項技術中充分描述對掌性環氧化物11在鹼性條件下之環化。可藉由用適合膦酸酯(例如,2-二乙氧基磷醯基乙酸乙酯或2-三乙基磷醯基乙酸乙酯)及適合鹼基(例如,烷基鋰、金屬醇鹽或金屬氫化物)處理對掌性環氧化物11來使用立體選擇性霍納-沃茲沃茨-埃蒙斯反應(Horner-Wadsworth-Emmons reaction),以獲得反式-環丙烷衍生物12 (參見例如,L. Delhaye; A. Merschaert; P. Delbeke;W. Brione.Org. Proc. Res. & Dev. 2007
,11
, 689-692)。可在此項技術中充分描述之各種鹼性條件下實現化合物12至對應酸13之水解。在此項技術中充分描述了利用適合酸活化劑(例如,羰基二咪唑)在存在溫和非親核鹼的情況下用經適當取代之N-羥基鄰苯二甲醯亞胺進行之酸13的後續偶合,以製備化合物14。可在此項技術中已知之許多條件下,包括在存在過渡金屬催化劑(例如,鈴木-宮浦反應(Suzuki-Miyaura reaction))的情況下,在光解條件下,或藉由單電子轉移反應完成N-羥基鄰苯二甲醯亞胺酯14之去羧硼醯化,包括例如N-羥基鄰苯二甲醯亞胺酯與二硼在由吡啶-硼自由基促進之自由基偶合中的錯合,以獲得反式環丙烷硼酸酯15。(參見例如,W.-M. Cheng; S. Rui; B. Zhao; W.-L. Xing; Y. Fu.Org. Lett. 2017
,19
, 4291-4294。) 可與流程1中所描述之流程類似地進行硼酸酯15與芳基氯4之偶合及後續去甲基化,以獲得式Ib之對掌性化合物類型。流程 4 
流程4描繪式Ic化合物之合成。可在此項技術中所熟知之自由基條件(例如,Minisci反應)下進行二氯噠嗪17之親核取代,以獲得經取代之噠嗪19。可與流程1中所描述之流程類似地進行硼酸酯20與芳基氯19之偶合及後續去甲基化,以獲得式Ic之化合物類型。流程 5 
流程5描繪式Id化合物之合成。可在此項技術中通常已知之親核條件下進行2-氯噠嗪21之置換以提供甲氧基噠嗪22。可與流程1中所描述之流程類似地進行22之後續去甲基化以獲得式Id之化合物類型。流程 6 
流程6描繪式Ie化合物之合成。可在此項技術中已知之各種條件下,包括過渡金屬(例如,Pd、Cu、Rh)催化之反應實現式Ic之氰化,以提供式Ie化合物。流程 7 
流程7描繪If化合物之合成。硼酸烯酯可與流程2中所描述之流程類似地環丙烷化。可與流程1中所描述之流程類似地進行硼酸酯25與芳基氯3之偶合以獲得對掌性化合物26。可藉由用適合酸處理來實現將二乙醇縮乙醛26解蔽成醛27。用各種氟化試劑(例如,DAST、XtalFluor®
、Fluolead™或Deoxo-Fluor®
)處理醛27可提供二氟甲基28。可與流程1中所描述之流程類似地進行28之後續去甲基化以獲得式If之化合物類型。流程 8 
流程8描繪Ig化合物之合成。可使用熟習此項技術者已知之取代技術用諸如氟化鉀或氟化第三丁基銨之試劑將甲磺酸酯29轉化為氟化物30。可與流程1中所描述之流程類似地進行後續去甲基化以獲得式Ig之對掌性化合物。
製備及實例
以下製備及實例進一步說明本發明且表示本發明化合物之典型合成,但不應理解為以任何方式限制本發明之範疇。試劑及起始材料易於利用或可易於由一般熟習此項技術者合成。應理解,製備及實例以說明而非限制之方式闡述,且一般熟習此項技術者可進行各種修改。
在Agilent HP1100液相層析系統上進行LC-ES/MS。在介接至HP1100 HPLC之質量選擇性偵測器四極質譜儀上進行電噴質譜分析量測(在正及/或負模式中獲得)。LC-MS條件(低pH):管柱:PHENOMENEX®
GEMINI®
NX C-18 2.1 × 50 mm 3.0 μm;梯度:5%-100% B,3分鐘,接著100% B,0.75分鐘;管柱溫度:50℃ +/- 10℃;流動速率:1.2 mL/min;溶劑A:具有0.1% HCOOH之去離子水;溶劑B:具有0.1%甲酸之ACN;波長214 nm。替代LC-MS條件(高pH):管柱:WATERSTM
XTERRA®
MSC-18管柱2.1 × 50 mm,3.5 μm;梯度: 5% 溶劑A,0.25分鐘,5%至100%溶劑B,3分鐘及100%溶劑B,0.5分鐘或10%至100%溶劑B,3分鐘及100%溶劑B,0.75分鐘之梯度;管柱溫度:50℃ +/- 10℃;流動速率:1.2 mL/min;溶劑A:10 mM NH4
HCO3
pH 9;溶劑B:ACN;波長:214 nm。
製備型逆相層析在配備有質量選擇性偵測器質譜儀及Leap自動進樣器/溶離份收集器之Agilent1200 LC-ES/MS上進行。高pH方法在具有10 × 20 mm保護件之75 × 30 mm PHENOMENEX®
GEMINI®
-NX,5 µm粒徑管柱上執行。流動速率為85 mL/min。溶離劑為含10 mM碳酸氫銨(pH 10)之ACN。
NMR光譜在Bruker AVIII HD 400 MHz NMR質譜儀上進行,使用殘餘溶劑[CDCl3
,7.26 ppm;(CD3
)2
SO,2.50 ppm]作為參考標準,獲得如以ppm為單位報導之CDCl3
或(CD3
)2
SO溶液。當報告峰多峰性時,可使用以下縮寫:s (單峰)、d (雙重峰)、t (三重峰)、q (四重峰)、m (多重峰)、br-s (寬單峰)、dd (雙重峰之雙重峰)、dt (三重峰之雙重峰)。在報導時,偶合常數(J)以赫茲(Hz)為單位報導。
製備1
外消旋-反-2-(2-乙基環丙基)-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷 向2-[(E
)-丁-1-烯基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(1.65 g,9.06 mmol)於Et2
O (45.30 mL)中之溶液中添加Pd(OAc)2
(0.20 g,0.90 mmol)。音波處理5分鐘且冷卻至-5℃。
向2-[(E
)-丁-1-烯基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(1.65 g,9.06 mmol)於Et2
O (45.30 mL)中之溶液中添加Pd(OAc)2
(0.20 g,0.90 mmol)。音波處理5分鐘且冷卻至-5℃。
在250 mL錐形瓶中,添加40% KOH水溶液(162 mL),隨後添加Et2
O (245 mL)。在冰/飽和NaCl水浴中將雙相混合物冷卻至-5℃。歷經10分鐘逐份添加N-亞硝基-N-甲基脲(36.1 g,350 mmol)。攪拌15分鐘且置放於乾冰/丙酮浴中。將醚層傾析至量筒中且在-5℃將Et2
O混合物(71 mL,約10當量CH2
N2
)添加至上文所描述之溶液中。2小時後,經由矽藻土過濾反應內容物,經MgSO4
乾燥,且在真空中濃縮,獲得標題化合物(1.08 g,58%)。1
H NMR (d6
-DMSO) δ: -0.51 (dt, J = 9.3, 5.7 Hz, 1H), 0.33-0.37 (m, 1H), 0.50-0.54 (m, 1H), 0.76-0.84 (m, 1H), 0.91 (t, J = 7.4 Hz, 3H), 1.14 (s, 12H), 1.21-1.30 (m, 2H)。
製備1之替代程序
向在氮下於0℃冷卻之雙(五甲基環戊二烯基)二氯化鋯(11.1 g,25.5 mmol)於THF (3.4 L)中之溶液中添加三乙基硼氫化鋰於THF中之1 M溶液(41.3 mL,41.3 mmol)。升溫至RT且攪拌1小時,同時用鋁箔保護燒瓶避光。
在氮下在-78℃冷卻的另一燒瓶中,冷凝1-丁-1-炔(50.6 g,936 mmol)且添加頻哪醇硼烷(pinacolborane) (55.0 g,425 mmol)。在-78℃攪拌所得混合物30分鐘且經由套管添加第一個燒瓶之內容物。添加TEA (5.93 mL,42.6 mmol)且攪拌反應混合物,同時升溫至RT歷20小時。藉由經由套管添加冰/水混合物(1.5 L)淬滅反應,添加EtOAc (300 mL),且在RT攪拌30分鐘。分離所得層並用EtOAc (2 × 200 mL)再萃取水相。合併有機萃取物,用Na2
SO4
乾燥,過濾,且在真空中濃縮,得到呈黃色油狀之2-[(E)-丁-1-烯基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(53 g,65%)。GC-MS (m/z): 182 (M+)。
在-20℃歷經10分鐘將二乙基鋅於甲苯中之溶液(15% w/w,550 mL,611 mmol)添加至氯碘甲烷(55 mL,755 mmol)中,且攪拌35分鐘。歷經20分鐘逐滴添加2-[(E)-丁-1-烯基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(53 g,277 mmol)於甲苯(100 mL)中之溶液,同時將反應之內部溫度保持於-20℃。攪拌混合物2小時同時升溫至0℃。藉由緩慢添飽和NH4
Cl水溶液(750 mL)淬滅反應。分離有機層且用EtOAc (2 × 250 mL)再萃取水相。合併有機萃取物,經MgSO4
乾燥,過濾,且蒸發,得到呈油狀之標題化合物(48 g,84%),適合不經額外純化即使用。GC-MS (m/z): 180 (M-16)。
製備2
4,4,5,5-四甲基-2-[(E)-3-甲基丁-1-烯基]-1,3,2-二氧雜硼雜環戊烷 歷經5分鐘將頻哪醇硼烷(9.5 mL,64 mmol)逐滴添加至冰冷的3-甲基丁-1-炔(4.91 g,72.0 mmol)中。密封壓力容器,升溫至RT且攪拌18小時。添加氫氯化雙(環戊二烯基)鋯(IV) (2.0 g,7.4 mmol)及TEA (1.1 mL,7.9 mmol)。密封壓力容器,置放於60℃油浴中,且攪拌10分鐘。將所得紅色溶液冷卻至RT持續2.5小時。用DCM (200 mL)稀釋反應混合物,用飽和NaHCO3
水溶液(100 mL)、飽和NaCl水溶液(50 mL)洗滌,經MgSO4
乾燥,經由矽膠墊(150 mL)過濾,用DCM (700 mL)沖洗矽膠,且在真空中濃縮以得到標題化合物(11.5 g,82%)。ES/MS (m/z): 196 (M+H)。1
H NMR (CDCl3
) δ: 1.03 (d, J= 6.7 Hz, 6H), 1.29 (s, 12H), 2.37 (m, 1H), 5.40 (dd, 1H), 6.64 (dd, 1H)。
歷經5分鐘將頻哪醇硼烷(9.5 mL,64 mmol)逐滴添加至冰冷的3-甲基丁-1-炔(4.91 g,72.0 mmol)中。密封壓力容器,升溫至RT且攪拌18小時。添加氫氯化雙(環戊二烯基)鋯(IV) (2.0 g,7.4 mmol)及TEA (1.1 mL,7.9 mmol)。密封壓力容器,置放於60℃油浴中,且攪拌10分鐘。將所得紅色溶液冷卻至RT持續2.5小時。用DCM (200 mL)稀釋反應混合物,用飽和NaHCO3
水溶液(100 mL)、飽和NaCl水溶液(50 mL)洗滌,經MgSO4
乾燥,經由矽膠墊(150 mL)過濾,用DCM (700 mL)沖洗矽膠,且在真空中濃縮以得到標題化合物(11.5 g,82%)。ES/MS (m/z): 196 (M+H)。1
H NMR (CDCl3
) δ: 1.03 (d, J= 6.7 Hz, 6H), 1.29 (s, 12H), 2.37 (m, 1H), 5.40 (dd, 1H), 6.64 (dd, 1H)。
製備3
外消旋-反式-2-[2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷 向將N-亞硝基-N-甲基脲逐份添加至Et2
O (70 mL)及KOH水溶液(30.5 g,435 mmol,70 mL H2
O)之冰冷雙相混合物中。攪拌直至固體溶解為止(< 5分鐘)。將所得重氮甲烷溶液吸取至快速攪拌之Pd(OAc)2
(237 mg,1.05 mmol)及4,4,5,5-四甲基-2-[(E)-3-甲基丁-1-烯基]-1,3,2-二氧雜硼雜環戊烷(4.00 g,20.4 mmol)於Et2
O (70 mL)中之冰冷懸浮液中。在完成添加之後,將反應混合物升溫至RT,經由矽藻土過濾,且在真空中濃縮濾液。將所得殘餘物溶解於DCM中,經由矽膠塞(25 g)過濾,且在真空中濃縮以獲得標題化合物(4.32 g,> 99%)。ES/MS (m/z): 210 (M+H)。1
H NMR (CDCl3
) δ: -0.35 (m, 1H), 0.45 (m, 1H), 0.65 (m, 1H), 0.78 (m, 1H), 0.98 (m, 7H), 1.23 (s, 12H)。
向將N-亞硝基-N-甲基脲逐份添加至Et2
O (70 mL)及KOH水溶液(30.5 g,435 mmol,70 mL H2
O)之冰冷雙相混合物中。攪拌直至固體溶解為止(< 5分鐘)。將所得重氮甲烷溶液吸取至快速攪拌之Pd(OAc)2
(237 mg,1.05 mmol)及4,4,5,5-四甲基-2-[(E)-3-甲基丁-1-烯基]-1,3,2-二氧雜硼雜環戊烷(4.00 g,20.4 mmol)於Et2
O (70 mL)中之冰冷懸浮液中。在完成添加之後,將反應混合物升溫至RT,經由矽藻土過濾,且在真空中濃縮濾液。將所得殘餘物溶解於DCM中,經由矽膠塞(25 g)過濾,且在真空中濃縮以獲得標題化合物(4.32 g,> 99%)。ES/MS (m/z): 210 (M+H)。1
H NMR (CDCl3
) δ: -0.35 (m, 1H), 0.45 (m, 1H), 0.65 (m, 1H), 0.78 (m, 1H), 0.98 (m, 7H), 1.23 (s, 12H)。
製備4
4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪 在壓力瓶中,將2,4-二甲氧基-5-嘧啶基硼酸(6.75 g,36.7 mmol)、4,6-二氯-3-甲基-噠嗪(5.98 g,36.7 mmol)、[1,1'-雙(二苯基膦基)二茂鐵]二氯鈀(II) (0.55 g,0.73 mmol)、Cs2
CO3
(29.9 g,91.8 mmol)合併於1,4-二噁烷/H2
O之4:1混合物(151 mL)中。抽空空氣且用N2
回填。密封容器且在70℃下加熱3小時。經由矽藻土過濾殘餘物且用EtOAc沖洗。用水,隨後飽和N aCl水溶液洗滌有機混合物,經MgSO4
乾燥,且蒸乾。藉由層析經由矽膠,使用具有0-30% DCM/(33% MeOH/DCM)之梯度的330 g REDISEP®
管柱歷經15分鐘以200毫升/分鐘之流動速率純化所得黑色殘餘物,以在蒸發層析溶離份之後得到標題化合物(6.2 g,63%)。ES/MS (m/z) (35
Cl/37
Cl) 267/269 [M+1]+
H NMR (d6
-DMSO) δ: 2.74 (s, 3H), 4.00 (s, 3H), 4.03 (s, 3H), 8.20 (s, 1H), 8.87 (s, 1H)。
在壓力瓶中,將2,4-二甲氧基-5-嘧啶基硼酸(6.75 g,36.7 mmol)、4,6-二氯-3-甲基-噠嗪(5.98 g,36.7 mmol)、[1,1'-雙(二苯基膦基)二茂鐵]二氯鈀(II) (0.55 g,0.73 mmol)、Cs2
CO3
(29.9 g,91.8 mmol)合併於1,4-二噁烷/H2
O之4:1混合物(151 mL)中。抽空空氣且用N2
回填。密封容器且在70℃下加熱3小時。經由矽藻土過濾殘餘物且用EtOAc沖洗。用水,隨後飽和N aCl水溶液洗滌有機混合物,經MgSO4
乾燥,且蒸乾。藉由層析經由矽膠,使用具有0-30% DCM/(33% MeOH/DCM)之梯度的330 g REDISEP®
管柱歷經15分鐘以200毫升/分鐘之流動速率純化所得黑色殘餘物,以在蒸發層析溶離份之後得到標題化合物(6.2 g,63%)。ES/MS (m/z) (35
Cl/37
Cl) 267/269 [M+1]+
H NMR (d6
-DMSO) δ: 2.74 (s, 3H), 4.00 (s, 3H), 4.03 (s, 3H), 8.20 (s, 1H), 8.87 (s, 1H)。
製備4之替代程序
使氮氣流通過(2,4-二甲氧基嘧啶-5-基)硼酸(85 g,439 mmol)、4,6-二氯-3-甲基-噠嗪(75 g,437 mmol)及Cs2
CO3
(358 g,1099 mmol)於1,4-二噁烷(1175 mL)及H2
O (340 mL)中之混合物5分鐘。添加[1,1'-雙(二苯基膦基)二茂鐵]二氯鈀(II) (6.6 g,8.7 mmol)且在75℃下攪拌所得混合物16小時。冷卻反應至RT,經由矽藻土過濾混合物,且用EtOAc沖洗濾餅。分離所得層並用飽和NaCl水溶液洗滌有機相兩次,經MgSO4
乾燥,過濾,且在真空中濃縮。將水(500 mL)添加至所得殘餘物中,在RT下攪拌16小時,且過濾所得固體。用H2
O洗滌所收集固體且在真空下乾燥16小時以獲得呈褐色固體狀之所要化合物(70 g,54%)。ES/MS (m/z) (35
Cl/37
Cl) 267/269 [M+1]+
以下實例可基本上如製備4中所描述來製備。 
製備6
反式-5-[2-乙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶 在20 mL壓力瓶中,添加4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪(0.6 g,2.0 mmol)、雙(二第三丁基(4-二甲胺基苯基)膦)二氯鈀(II) (114 mg,0.16 mmol)、K2
CO3
(0.69 g,4.9 mmol)、H2
O (2.25 mL)及外消旋反式-2-[2-乙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(0.66 g,3.37 mmol)。抽空燒瓶且用N2
回填。密封容器且在90℃下加熱隔夜。經由矽藻土過濾反應混合物,用水,隨後飽和NaCl水溶液洗滌有機層,經MgSO4
乾燥,且在真空中濃縮。藉由逆相層析,使用275 g REDISEP®
Gold C18管柱純化所得殘餘物,歷經20分鐘以150毫升/分鐘之流動速率用30-50% 10mM NH4
HCO3
/ACN之梯度溶離,以在蒸發層析溶離份之後得到呈白色固體狀之標題化合物(異構體之基本上外消旋混合物) (475 mg,70%)。
在20 mL壓力瓶中,添加4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪(0.6 g,2.0 mmol)、雙(二第三丁基(4-二甲胺基苯基)膦)二氯鈀(II) (114 mg,0.16 mmol)、K2
CO3
(0.69 g,4.9 mmol)、H2
O (2.25 mL)及外消旋反式-2-[2-乙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(0.66 g,3.37 mmol)。抽空燒瓶且用N2
回填。密封容器且在90℃下加熱隔夜。經由矽藻土過濾反應混合物,用水,隨後飽和NaCl水溶液洗滌有機層,經MgSO4
乾燥,且在真空中濃縮。藉由逆相層析,使用275 g REDISEP®
Gold C18管柱純化所得殘餘物,歷經20分鐘以150毫升/分鐘之流動速率用30-50% 10mM NH4
HCO3
/ACN之梯度溶離,以在蒸發層析溶離份之後得到呈白色固體狀之標題化合物(異構體之基本上外消旋混合物) (475 mg,70%)。
藉由SFC對所得異構體進行純化(管柱:PHENOMENEX®
LUX®
纖維素-4,4.6 × 150 mm;40% MeOH/CO2
等度;流動速率:5毫升/分鐘,UV 250 nm)以得到0.197 g異構體1:tR
= 2.71分鐘(UV);及0.195 g異構體2:tR
3.55分鐘(UV),兩者均> 98% ee。ES/MS (m/z): 301 (M+H)。
製備7
反式-5-[2-異丙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶 組合4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪(1.97 g,7.39 mmol)、外消旋-反式-2-[2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(3.32 g,15.8 mmol)、雙(二第三丁基(4-二甲胺基苯基)膦)二氯鈀(II) (1.35 g,1.85 mmol)、1,4-二噁烷(37 mL)及1 M Na2
CO3
水溶液(18 mL,18 mmol)。用N2
吹掃反應容器且加熱至90℃持續18小時。冷卻至RT,用EtOAc (150 mL)稀釋,且分離各層。用1 M Na2
CO3
水溶液、飽和NaCl水溶液連續洗滌有機層,經MgSO4
乾燥,過濾,且在真空中濃縮濾液。藉由二氧化矽層析純化所得殘餘物,用60-100% EtOAc/DCM之梯度溶離,以在蒸發層析溶離份之後得到呈異構體之基本上外消旋混合物形式之標題化合物(1.48 g,64%)。
組合4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪(1.97 g,7.39 mmol)、外消旋-反式-2-[2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(3.32 g,15.8 mmol)、雙(二第三丁基(4-二甲胺基苯基)膦)二氯鈀(II) (1.35 g,1.85 mmol)、1,4-二噁烷(37 mL)及1 M Na2
CO3
水溶液(18 mL,18 mmol)。用N2
吹掃反應容器且加熱至90℃持續18小時。冷卻至RT,用EtOAc (150 mL)稀釋,且分離各層。用1 M Na2
CO3
水溶液、飽和NaCl水溶液連續洗滌有機層,經MgSO4
乾燥,過濾,且在真空中濃縮濾液。藉由二氧化矽層析純化所得殘餘物,用60-100% EtOAc/DCM之梯度溶離,以在蒸發層析溶離份之後得到呈異構體之基本上外消旋混合物形式之標題化合物(1.48 g,64%)。
藉由SFC對異構體進行純化(管柱:PHENOMENEX®
LUX®
纖維素-4,4.6 × 150 mm;40% MeOH/CO2
等度;流動速率:5 mL/分鐘,UV 250 nm)以得到647 mg異構體1:tR
= 2.56 min及647 mg異構體2:tR
= 3.75 min。ES/MS (m/z): 315 (M+H)。
製備8
(1S,2S)-2-乙基環丙烷羧酸乙酯 歷經10分鐘向在冰/水浴(內部溫度:8℃)中冷卻的2-二乙氧基磷醯基乙酸乙酯(62.2 g,277 mmol)於1,4-二噁烷(400 mL)中之溶液逐滴添加nBuLi於己烷中之2.5 M溶液(110 mL,280 mmol)。移除冷卻浴液且在RT下攪拌30分鐘。經由套管將溶液轉移至1 L壓力容器且添加(2R)-2-乙基環氧乙烷(20 g,280 mmol)。在150℃ (50 psi壓力)下攪拌所得混合物17小時。冷卻反應至RT且添加水(250 mL)。分離有機相且用MTBE (2 × 200 mL)再萃取水相。組合有機萃取物,用飽和NaCl水溶液(2 × 150 mL)洗滌,經MgSO4
乾燥,且在真空中濃縮,以得到適合於未經額外純化即使用的呈黃色油狀之粗標題化合物(49.1 g,定量)。GC-MS (m/z): 142 (M+), 97 (M-45)。
歷經10分鐘向在冰/水浴(內部溫度:8℃)中冷卻的2-二乙氧基磷醯基乙酸乙酯(62.2 g,277 mmol)於1,4-二噁烷(400 mL)中之溶液逐滴添加nBuLi於己烷中之2.5 M溶液(110 mL,280 mmol)。移除冷卻浴液且在RT下攪拌30分鐘。經由套管將溶液轉移至1 L壓力容器且添加(2R)-2-乙基環氧乙烷(20 g,280 mmol)。在150℃ (50 psi壓力)下攪拌所得混合物17小時。冷卻反應至RT且添加水(250 mL)。分離有機相且用MTBE (2 × 200 mL)再萃取水相。組合有機萃取物,用飽和NaCl水溶液(2 × 150 mL)洗滌,經MgSO4
乾燥,且在真空中濃縮,以得到適合於未經額外純化即使用的呈黃色油狀之粗標題化合物(49.1 g,定量)。GC-MS (m/z): 142 (M+), 97 (M-45)。
製備9
(1S,2S)-2-乙基環丙烷羧酸 在100℃下攪拌(1S,2S)-2-乙基環丙烷羧酸乙酯(39.44 g,277.4 mmol)、1,4-二噁烷(315 mL)及25%氫氧化鈉水溶液(315 mL)之混合物16小時。冷卻混合物至室溫,用MTBE (2 × 300 mL)萃取,且丟棄有機相。用37% HCl水溶液酸化水相直至pH約為1-2為止,用MTBE (3 × 300 mL)萃取,分離各層,用飽和NaCl水溶液洗滌有機層,經MgSO4
乾燥,且在真空中濃縮以得到呈琥珀色油狀之標題化合物(25.1 g,75%)。1
H NMR (400 MHz, CDCl3
) δ: 0.79-0.85 (m, 1H), 1.00 (t, J=7.5 Hz, 3H), 1.22-1.26 (m, 1H), 1.32-1.42 (m, 3 H), 1.43-1.48 (m, 1H), 9.0-12.0 (br-s, 1H)。
在100℃下攪拌(1S,2S)-2-乙基環丙烷羧酸乙酯(39.44 g,277.4 mmol)、1,4-二噁烷(315 mL)及25%氫氧化鈉水溶液(315 mL)之混合物16小時。冷卻混合物至室溫,用MTBE (2 × 300 mL)萃取,且丟棄有機相。用37% HCl水溶液酸化水相直至pH約為1-2為止,用MTBE (3 × 300 mL)萃取,分離各層,用飽和NaCl水溶液洗滌有機層,經MgSO4
乾燥,且在真空中濃縮以得到呈琥珀色油狀之標題化合物(25.1 g,75%)。1
H NMR (400 MHz, CDCl3
) δ: 0.79-0.85 (m, 1H), 1.00 (t, J=7.5 Hz, 3H), 1.22-1.26 (m, 1H), 1.32-1.42 (m, 3 H), 1.43-1.48 (m, 1H), 9.0-12.0 (br-s, 1H)。
製備10
(1S,2S)-2-乙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯) 在0℃下攪拌(1S,2S)-2-乙基環丙烷羧酸(25.1 g,209 mmol)、2-羥基異吲哚啉-1,3-二酮(34.8 g,209 mmol)及DMAP (2.58 g,20.9 mmol)於DCM (360 mL)中之懸浮液,且逐滴添加N,N'-二異丙基碳化二亞胺(29.3 g,230 mmol)。移除冷卻浴液且在RT下攪拌反應物2小時。經由矽膠栓塞過濾懸浮液,用DCM溶離。蒸發溶劑且藉由層析經由矽膠純化所得殘餘物,用15%己烷/丙酮溶離,以在自層析溶離份移除溶劑之後得到呈淡黃色固體狀之標題化合物(51.56 g,84%)。ES/MS (m/z): 260 (M+1)。
在0℃下攪拌(1S,2S)-2-乙基環丙烷羧酸(25.1 g,209 mmol)、2-羥基異吲哚啉-1,3-二酮(34.8 g,209 mmol)及DMAP (2.58 g,20.9 mmol)於DCM (360 mL)中之懸浮液,且逐滴添加N,N'-二異丙基碳化二亞胺(29.3 g,230 mmol)。移除冷卻浴液且在RT下攪拌反應物2小時。經由矽膠栓塞過濾懸浮液,用DCM溶離。蒸發溶劑且藉由層析經由矽膠純化所得殘餘物,用15%己烷/丙酮溶離,以在自層析溶離份移除溶劑之後得到呈淡黃色固體狀之標題化合物(51.56 g,84%)。ES/MS (m/z): 260 (M+1)。
製備11
2-[(1S,2S)-2-乙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷 使N2
流通過(1S,2S)-2-乙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯) (51 g,173.1 mmol)及雙(頻哪醇根基)二硼(87.9 g,346 mmol)於EtOAc (1 L)中之溶液5分鐘。添加異菸鹼酸乙酯(5.34 g,35 mmol)且在85℃下攪拌混合物24小時。冷卻所得懸浮液,過濾且丟棄固體,並在減壓下濃縮棕色濾液。經由矽膠栓塞過濾所得粗殘餘物,用2% EtOAc/己烷溶離。自濾液移除溶劑且使用層析經由矽膠再純化所得殘餘物,用3% EtOAc/己烷溶離,以在自層析溶離份移除溶劑之後獲得呈無色油狀之標題化合物(17.1 g,49%)。GC-MS (m/z): 180 (M-16)。
製備12
(2S)-2-氯-3-甲基-丁酸
使N2
流通過(1S,2S)-2-乙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯) (51 g,173.1 mmol)及雙(頻哪醇根基)二硼(87.9 g,346 mmol)於EtOAc (1 L)中之溶液5分鐘。添加異菸鹼酸乙酯(5.34 g,35 mmol)且在85℃下攪拌混合物24小時。冷卻所得懸浮液,過濾且丟棄固體,並在減壓下濃縮棕色濾液。經由矽膠栓塞過濾所得粗殘餘物,用2% EtOAc/己烷溶離。自濾液移除溶劑且使用層析經由矽膠再純化所得殘餘物,用3% EtOAc/己烷溶離,以在自層析溶離份移除溶劑之後獲得呈無色油狀之標題化合物(17.1 g,49%)。GC-MS (m/z): 180 (M-16)。
製備12
(2S)-2-氯-3-甲基-丁酸 在0℃下冷卻L-纈胺酸溶液(286 g,2.44 mol)及5 M HCl水溶液(3.25 L,16.3 mol)。歷經2小時逐滴添加4 M NaNO2
水溶液(1 L,4 mol),保持內部溫度低於5℃。攪拌反應物2小時同時升溫至RT,並在RT下攪拌額外16小時。歷經30分鐘逐份添加Na2
CO3
(242 g,2.28 mol)。用MTBE (3 × 1000 mL)萃取所得溶液,用飽和NaCl水溶液(500 mL)洗滌經合併之有機萃取物,經MgSO4
使有機萃取物乾燥,且在真空中濃縮。藉由真空蒸餾(15 mbar/140℃)純化所得殘餘物以得到呈油狀之標題化合物(248 g,68%)。1
H NMR (400 MHz, CDCl3
) δ: 1.1 (dt, J=6.6, 2.6 Hz, 6H), 2.34-2.42 (m, 1H), 4.19-4.23 (m, 1H), 10.0-12.0 (br-s, 1H)。
在0℃下冷卻L-纈胺酸溶液(286 g,2.44 mol)及5 M HCl水溶液(3.25 L,16.3 mol)。歷經2小時逐滴添加4 M NaNO2
水溶液(1 L,4 mol),保持內部溫度低於5℃。攪拌反應物2小時同時升溫至RT,並在RT下攪拌額外16小時。歷經30分鐘逐份添加Na2
CO3
(242 g,2.28 mol)。用MTBE (3 × 1000 mL)萃取所得溶液,用飽和NaCl水溶液(500 mL)洗滌經合併之有機萃取物,經MgSO4
使有機萃取物乾燥,且在真空中濃縮。藉由真空蒸餾(15 mbar/140℃)純化所得殘餘物以得到呈油狀之標題化合物(248 g,68%)。1
H NMR (400 MHz, CDCl3
) δ: 1.1 (dt, J=6.6, 2.6 Hz, 6H), 2.34-2.42 (m, 1H), 4.19-4.23 (m, 1H), 10.0-12.0 (br-s, 1H)。
製備13
(2S)-2-氯-3-甲基-丁-1-醇 將(2S)-2-氯-3-甲基-丁酸(137 g,1 mol)於2-甲基四氫呋喃(500 mL)中之溶液冷卻至0℃。歷經2.5小時逐滴添加LAH於甲基四氫呋喃(480 mL,1.1 mol)中之2.3 M溶液,保持內部溫度低於10℃。升溫至RT且在RT下攪拌混合物1小時且在50℃下攪拌混合物1小時。冷卻反應至0℃且連續及緩慢地添加H2
O (1.48 mL)、15% NaOH水溶液(1.48 mL)及H2
O (4.46 mL)。使混合物升溫至RT,經由矽藻土之床層過濾,且在真空中蒸發溶劑以得到呈無色油狀之標題化合物(110 g,81%)。1
H NMR (400 MHz, CDCl3
) δ: 0.97- 1.1 (m, 6H), 1.94-2.18 (m, 1H), 2.60-3.09 (br-s, 1H), 3.71-3.78 (m, 1H), 3.80-3.85 (m, 1H), 3.90-3.96 (m, 1H)。
將(2S)-2-氯-3-甲基-丁酸(137 g,1 mol)於2-甲基四氫呋喃(500 mL)中之溶液冷卻至0℃。歷經2.5小時逐滴添加LAH於甲基四氫呋喃(480 mL,1.1 mol)中之2.3 M溶液,保持內部溫度低於10℃。升溫至RT且在RT下攪拌混合物1小時且在50℃下攪拌混合物1小時。冷卻反應至0℃且連續及緩慢地添加H2
O (1.48 mL)、15% NaOH水溶液(1.48 mL)及H2
O (4.46 mL)。使混合物升溫至RT,經由矽藻土之床層過濾,且在真空中蒸發溶劑以得到呈無色油狀之標題化合物(110 g,81%)。1
H NMR (400 MHz, CDCl3
) δ: 0.97- 1.1 (m, 6H), 1.94-2.18 (m, 1H), 2.60-3.09 (br-s, 1H), 3.71-3.78 (m, 1H), 3.80-3.85 (m, 1H), 3.90-3.96 (m, 1H)。
製備14
(2R)-2-異丙基環氧乙烷 將KOH (195 g,3.48 mol)於H2
O (195 mL)中之溶液冷卻至0℃且歷經20分鐘添加純(2S)-2-氯-3-甲基-丁-1-醇(110 g,801 mmol),同時保持內部溫度低於5℃。使反應混合物升溫至RT。藉由在100 mbar下真空蒸餾純化反應混合物,自23℃升溫至50℃,以得到呈無色油狀之標題化合物(47 g,64%)。1
H NMR (400 MHz, CDCl3
) δ: 0.98 (d, J=6.9 Hz, 3H), 1.05 (d, J=6.9 Hz, 3H), 1.51 (o, J=6.9 Hz, 1H), 2.52-2.54 (m, 1H), 2.70-2.75 (m, 2H)。
將KOH (195 g,3.48 mol)於H2
O (195 mL)中之溶液冷卻至0℃且歷經20分鐘添加純(2S)-2-氯-3-甲基-丁-1-醇(110 g,801 mmol),同時保持內部溫度低於5℃。使反應混合物升溫至RT。藉由在100 mbar下真空蒸餾純化反應混合物,自23℃升溫至50℃,以得到呈無色油狀之標題化合物(47 g,64%)。1
H NMR (400 MHz, CDCl3
) δ: 0.98 (d, J=6.9 Hz, 3H), 1.05 (d, J=6.9 Hz, 3H), 1.51 (o, J=6.9 Hz, 1H), 2.52-2.54 (m, 1H), 2.70-2.75 (m, 2H)。
製備15
(1S,2R)-2-異丙基環丙烷羧酸乙酯 歷經25分鐘將nBuLi於己烷中之溶液(310 mL,780 mmol)逐滴添加至在冰/水浴(內部溫度:8℃)中冷卻的2-二乙氧基磷醯基乙酸乙酯(153 mL,772 mmol)於1,4-二噁烷(870 mL)中之溶液中。升溫至RT且攪拌40分鐘。經由套管將溶液轉移至3 L壓力容器且將(2R)-2-異丙基環氧乙烷(70 g,772 mmol)添加於1,4-二噁烷(180 mL)中。在150℃下於50 psi壓力下攪拌反應混合物14小時。冷卻反應混合物至RT且添加H2
O (700 mL)。分離各層,且用MTBE (2 × 500 mL)再萃取水相。合併有機相,用NaCl飽和水溶液 (2 × 350 mL)洗滌,經MgSO4
乾燥,且在真空中濃縮,以獲得適合於隨後未經進一步純化即使用的呈黃色油狀之粗標題化合物(107.7 g,> 99%)。GC-MS (m/z): 156 (M+)。
歷經25分鐘將nBuLi於己烷中之溶液(310 mL,780 mmol)逐滴添加至在冰/水浴(內部溫度:8℃)中冷卻的2-二乙氧基磷醯基乙酸乙酯(153 mL,772 mmol)於1,4-二噁烷(870 mL)中之溶液中。升溫至RT且攪拌40分鐘。經由套管將溶液轉移至3 L壓力容器且將(2R)-2-異丙基環氧乙烷(70 g,772 mmol)添加於1,4-二噁烷(180 mL)中。在150℃下於50 psi壓力下攪拌反應混合物14小時。冷卻反應混合物至RT且添加H2
O (700 mL)。分離各層,且用MTBE (2 × 500 mL)再萃取水相。合併有機相,用NaCl飽和水溶液 (2 × 350 mL)洗滌,經MgSO4
乾燥,且在真空中濃縮,以獲得適合於隨後未經進一步純化即使用的呈黃色油狀之粗標題化合物(107.7 g,> 99%)。GC-MS (m/z): 156 (M+)。
製備16 (1S,2R)-2-異丙基環丙烷羧酸 在100℃下攪拌含有25% NaOH水溶液(800 mL)的(1S,2R)-2-異丙基環丙烷羧酸乙酯(107.7 g,482.6 mmol)於1,4-二噁烷(800 mL)中之混合物7小時。冷卻混合物至RT,添加水(300 mL),用MTBE (2 × 500 mL)萃取,且丟棄有機相。用37% HCl水溶液(約500 mL)酸化水相直至pH約為1-2為止。
在100℃下攪拌含有25% NaOH水溶液(800 mL)的(1S,2R)-2-異丙基環丙烷羧酸乙酯(107.7 g,482.6 mmol)於1,4-二噁烷(800 mL)中之混合物7小時。冷卻混合物至RT,添加水(300 mL),用MTBE (2 × 500 mL)萃取,且丟棄有機相。用37% HCl水溶液(約500 mL)酸化水相直至pH約為1-2為止。
用MTBE (2 × 600 mL)萃取經酸化水性混合物,有機層用飽和NaCl水溶液洗滌,經MgSO4
乾燥,且在真空中濃縮,以得到呈琥珀色油狀之標題化合物(54.2 g,75%)。1
H NMR (400 MHz, CDCl3
) δ: 0.81-0.86 (m, 1H), 1.01 (dd, J=5.9, 3.7 Hz, 6H), 1.04-1.13 (m, 1H), 1.20-1.24 (m, 1H), 1.28-1.37 (m, 1H), 1.39-1.44 (m, 1H)。
製備17
(1S,2R)-2-異丙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯) 在0℃下攪拌(1S,2R)-2-異丙基環丙烷羧酸(54.2 g,359 mmol)、2-羥基異吲哚啉-1,3-二酮(59.8 g,359 mmol)及DMAP (4.44 g,35.9 mmol)於DCM (690 mL)中之懸浮液。逐滴添加N,N'-二異丙基碳化二亞胺(50.4 g,395 mmol),升溫至RT,且在RT下攪拌所得反應混合物2小時。添加H2
O (600 mL)且分離各相。用DCM (2 × 300 mL)萃取水相,合併有機相,經MgSO4
乾燥,過濾,且蒸發。藉由二氧化矽層析純化所得固體殘餘物,用100% DCM溶離,以在蒸發層析溶離份之後得到呈淡黃色固體狀之標題化合物(96 g,88%)。ES/MS (m/z): 274 (M+1)。
在0℃下攪拌(1S,2R)-2-異丙基環丙烷羧酸(54.2 g,359 mmol)、2-羥基異吲哚啉-1,3-二酮(59.8 g,359 mmol)及DMAP (4.44 g,35.9 mmol)於DCM (690 mL)中之懸浮液。逐滴添加N,N'-二異丙基碳化二亞胺(50.4 g,395 mmol),升溫至RT,且在RT下攪拌所得反應混合物2小時。添加H2
O (600 mL)且分離各相。用DCM (2 × 300 mL)萃取水相,合併有機相,經MgSO4
乾燥,過濾,且蒸發。藉由二氧化矽層析純化所得固體殘餘物,用100% DCM溶離,以在蒸發層析溶離份之後得到呈淡黃色固體狀之標題化合物(96 g,88%)。ES/MS (m/z): 274 (M+1)。
製備18
2-[(1S,2S)-2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷 使氮氣流通過(1S,2S)-2-異丙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯) (96 g,316 mmol)及雙(頻哪醇根基)二硼(160 g,630 mmol)於EtOAc (480 mL)中之溶液15分鐘。在85℃下攪拌混合物且歷經10分鐘逐滴添加異菸鹼酸乙酯(24.38 g,157 mmol)。在85℃下攪拌所得混合物16小時。冷卻所得懸浮液,過濾及丟棄固體,且在減壓下蒸發棕色濾液。經由矽膠栓塞過濾粗殘餘物,用2% EtOAc/己烷溶離。自濾液移除溶劑且使用層析經由矽膠再純化所得殘餘物,用3% EtOAc/己烷溶離,以在自層析溶離份移除溶劑之後獲得呈無色油狀之標題化合物(25.3 g,38%)。1
H NMR (400 MHz, CDCl3
): -0.33_
-0.38 (m, 1H), 0.42-0.47 (m, 1H), 0.63-0.67 (m, 1H), 0.75-0.82 (m, 1H), 0.89-1.02 (m, 1H), 0.98 (m, 6H), 1.24 (s, 12H)。
使氮氣流通過(1S,2S)-2-異丙基環丙烷羧酸(1,3-二側氧基異吲哚啉-2-基酯) (96 g,316 mmol)及雙(頻哪醇根基)二硼(160 g,630 mmol)於EtOAc (480 mL)中之溶液15分鐘。在85℃下攪拌混合物且歷經10分鐘逐滴添加異菸鹼酸乙酯(24.38 g,157 mmol)。在85℃下攪拌所得混合物16小時。冷卻所得懸浮液,過濾及丟棄固體,且在減壓下蒸發棕色濾液。經由矽膠栓塞過濾粗殘餘物,用2% EtOAc/己烷溶離。自濾液移除溶劑且使用層析經由矽膠再純化所得殘餘物,用3% EtOAc/己烷溶離,以在自層析溶離份移除溶劑之後獲得呈無色油狀之標題化合物(25.3 g,38%)。1
H NMR (400 MHz, CDCl3
): -0.33_
-0.38 (m, 1H), 0.42-0.47 (m, 1H), 0.63-0.67 (m, 1H), 0.75-0.82 (m, 1H), 0.89-1.02 (m, 1H), 0.98 (m, 6H), 1.24 (s, 12H)。
製備19
4,4,5,5-四甲基-2-[(E)-4-甲基戊-1-烯基]-1,3,2-二氧雜硼雜環戊烷 將[(E)-4-甲基戊-1-烯基]硼酸(0.975 g,7.62 mmol)及頻哪醇(1.11 g,9.14 mmol)溶解於乾燥DCM (9.7 mL)中。添加MgSO4
(0.7313 g,6.05 mmol)且在室溫下攪拌3天。過濾不可溶固體且在減壓下移除溶劑以得到呈透明油狀之標題化合物(1.6 g,100%)。1H NMR (400.13 MHz, DMSO): 0.86 (d, J=6.6 Hz, 6H), 1.08 (s, 6H), 1.19 (s, 12H), 1.62-1.72 (m, 1H), 2.01 (td, J=6.9, 1.4 Hz, 2H), 3.92 (s, 1H), 5.31 (dt, J=17.9, 1.3 Hz, 1H), 6.47 (dt, J=17.9, 6.9 Hz, 1H)。
將[(E)-4-甲基戊-1-烯基]硼酸(0.975 g,7.62 mmol)及頻哪醇(1.11 g,9.14 mmol)溶解於乾燥DCM (9.7 mL)中。添加MgSO4
(0.7313 g,6.05 mmol)且在室溫下攪拌3天。過濾不可溶固體且在減壓下移除溶劑以得到呈透明油狀之標題化合物(1.6 g,100%)。1H NMR (400.13 MHz, DMSO): 0.86 (d, J=6.6 Hz, 6H), 1.08 (s, 6H), 1.19 (s, 12H), 1.62-1.72 (m, 1H), 2.01 (td, J=6.9, 1.4 Hz, 2H), 3.92 (s, 1H), 5.31 (dt, J=17.9, 1.3 Hz, 1H), 6.47 (dt, J=17.9, 6.9 Hz, 1H)。
製備20
第三丁基-[[(2S)-環氧乙烷-2-基]甲氧基]-二苯基-矽烷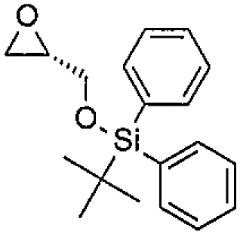 向[(2R)-環氧乙烷-2-基]甲醇(15.0 g,202.6 mmol)於DMF (135 mL)中之冰冷溶液中添加咪唑(33.34 g,489.8 mmol)且攪拌15分鐘。歷經30分鐘逐滴添加第三丁基-氯-二苯基-矽烷(77 mL,302.5 mmol)且攪拌隔夜至室溫。用己烷(1 L)、醚(500 mL)及水(1 L)稀釋。分離各層且用醚(2 x 1 L)萃取水層。合併有機物且用水(3 × 750 mL)、飽和NaHCO3
、飽和NaCl水溶液洗滌,且經MgSO4
乾燥。在減壓下蒸發溶劑。藉由矽膠層析純化:溶離劑 0-10% EtOAc/己烷,以在自層析溶離份移除溶劑之後得到呈油狀物之標題化合物(11.77 g,18%)。1H NMR (400.13 MHz, CDCl3
): 1.08 (s, 9H), 2.64 (dd, J=2.6, 5.1 Hz, 1H), 2.77 (t, J=4.6 Hz, 1H), 3.15 (五重峰, J=3.5 Hz, 1H), 3.73 (dd, J=4.7, 11.8 Hz, 1H), 3.88 (dd, J=3.2, 11.9 Hz, 1H), 7.40-7.48 (m, 6H), 7.71 (m, 4H)。
向[(2R)-環氧乙烷-2-基]甲醇(15.0 g,202.6 mmol)於DMF (135 mL)中之冰冷溶液中添加咪唑(33.34 g,489.8 mmol)且攪拌15分鐘。歷經30分鐘逐滴添加第三丁基-氯-二苯基-矽烷(77 mL,302.5 mmol)且攪拌隔夜至室溫。用己烷(1 L)、醚(500 mL)及水(1 L)稀釋。分離各層且用醚(2 x 1 L)萃取水層。合併有機物且用水(3 × 750 mL)、飽和NaHCO3
、飽和NaCl水溶液洗滌,且經MgSO4
乾燥。在減壓下蒸發溶劑。藉由矽膠層析純化:溶離劑 0-10% EtOAc/己烷,以在自層析溶離份移除溶劑之後得到呈油狀物之標題化合物(11.77 g,18%)。1H NMR (400.13 MHz, CDCl3
): 1.08 (s, 9H), 2.64 (dd, J=2.6, 5.1 Hz, 1H), 2.77 (t, J=4.6 Hz, 1H), 3.15 (五重峰, J=3.5 Hz, 1H), 3.73 (dd, J=4.7, 11.8 Hz, 1H), 3.88 (dd, J=3.2, 11.9 Hz, 1H), 7.40-7.48 (m, 6H), 7.71 (m, 4H)。
製備21
(1S,2S)-2-(羥基甲基)環丙烷羧酸乙酯 在室溫下將膦醯基乙酸三乙酯(7.5 mL,38 mmol)緩慢添加至第三丁醇鈉(3.67 g,37.82 mmol)於1,4-二噁烷(40 mL)中之懸浮液中。1小時後,添加第三丁基-[[(2S)-環氧乙烷-2-基]甲氧基]-二苯基-矽烷(11.77 g,33.76 mmol)且在密封壓力容器中於140℃下加熱隔夜。添加額外膦醯基乙酸三乙酯(2 mL)且在160℃下加熱一小時。使混合物冷卻且用DCM (300 ml)及水(150 mL)稀釋。分離各層,用DCM (3 × 150 mL)萃取水層。合併有機物並用飽和NaHCO3
水溶液、飽和NaCl水溶液洗滌,且經MgSO4
乾燥。經由矽藻土過濾且在減壓下蒸發溶劑,以得到呈油狀物之氧基甲基]環丙烷羧酸乙酯(1S,2S)-2-[[第三丁基(二苯基)矽烷基酯](18.45 g)。在室溫下溶解於THF(75 mL)中且添加氟化四丁銨(1 M THF,67 mL,67 mmol,1.0 M)。在130 mbar及30℃下攪拌48小時且移除溶劑。藉由矽膠層析純化,溶離劑:0-50% EtOAc/己烷,以在自層析溶離份移除溶劑之後分離呈油狀物之標題化合物(3.35 g,62%)。ES/MS (m/z): 142 (M+1)。1H NMR (400.13 MHz, CDCl3
): 0.90-0.86 (m, 1H), 1.23 (dd, J=4.5, 8.9 Hz, 1H), 1.28 (t, J=7.2 Hz, 4H), 1.56-1.60 (m, 1H), 1.69-1.77 (m, 2H), 2.06 (s, 2H), 3.50 (dd, J=6.9, 11.5 Hz, 1H), 3.64 (dd, J=6.0, 11.4 Hz, 1H), 4.14 (qd, J=7.1, 2.9 Hz, 3H)。
在室溫下將膦醯基乙酸三乙酯(7.5 mL,38 mmol)緩慢添加至第三丁醇鈉(3.67 g,37.82 mmol)於1,4-二噁烷(40 mL)中之懸浮液中。1小時後,添加第三丁基-[[(2S)-環氧乙烷-2-基]甲氧基]-二苯基-矽烷(11.77 g,33.76 mmol)且在密封壓力容器中於140℃下加熱隔夜。添加額外膦醯基乙酸三乙酯(2 mL)且在160℃下加熱一小時。使混合物冷卻且用DCM (300 ml)及水(150 mL)稀釋。分離各層,用DCM (3 × 150 mL)萃取水層。合併有機物並用飽和NaHCO3
水溶液、飽和NaCl水溶液洗滌,且經MgSO4
乾燥。經由矽藻土過濾且在減壓下蒸發溶劑,以得到呈油狀物之氧基甲基]環丙烷羧酸乙酯(1S,2S)-2-[[第三丁基(二苯基)矽烷基酯](18.45 g)。在室溫下溶解於THF(75 mL)中且添加氟化四丁銨(1 M THF,67 mL,67 mmol,1.0 M)。在130 mbar及30℃下攪拌48小時且移除溶劑。藉由矽膠層析純化,溶離劑:0-50% EtOAc/己烷,以在自層析溶離份移除溶劑之後分離呈油狀物之標題化合物(3.35 g,62%)。ES/MS (m/z): 142 (M+1)。1H NMR (400.13 MHz, CDCl3
): 0.90-0.86 (m, 1H), 1.23 (dd, J=4.5, 8.9 Hz, 1H), 1.28 (t, J=7.2 Hz, 4H), 1.56-1.60 (m, 1H), 1.69-1.77 (m, 2H), 2.06 (s, 2H), 3.50 (dd, J=6.9, 11.5 Hz, 1H), 3.64 (dd, J=6.0, 11.4 Hz, 1H), 4.14 (qd, J=7.1, 2.9 Hz, 3H)。
製備22
(1S,2S)-2-甲醯基環丙烷羧酸乙酯 向(1S,2S)-2-(羥基甲基)環丙烷羧酸乙酯(12.25 g,84.9 mmol)於DCM (425 mL)中之冰冷溶液中一次性添加氯鉻酸吡啶(26.09 g,118.6 mmol)。30分鐘後,移除冰浴且在室溫下攪拌4小時。添加水(200 mL)且經由矽藻土過濾。用額外的水(100 mL)及DCM (800 mL)沖洗。使矽藻土濾餅懸浮於DCM中且過濾。重複此操作兩次。合併有機物,分離水層,且經由二氧化矽墊過濾有機物。用額外DCM沖洗且在減壓下蒸發溶劑以得到呈油狀物之標題化合物(11.0 g,91%)。1H NMR (400.13 MHz, CDCl3
): 1.30 (t, J=7.3 Hz, 3H), 1.57-1.50 (m, 1H), 1.60-1.65 (m, 2H), 2.26-2.30 (m, 1H), 2.45 (td, J=9.0, 4.2 Hz, 1H), 4.13-4.22 (m, 3H), 9.32 (d, J=4.2 Hz, 1H)。
向(1S,2S)-2-(羥基甲基)環丙烷羧酸乙酯(12.25 g,84.9 mmol)於DCM (425 mL)中之冰冷溶液中一次性添加氯鉻酸吡啶(26.09 g,118.6 mmol)。30分鐘後,移除冰浴且在室溫下攪拌4小時。添加水(200 mL)且經由矽藻土過濾。用額外的水(100 mL)及DCM (800 mL)沖洗。使矽藻土濾餅懸浮於DCM中且過濾。重複此操作兩次。合併有機物,分離水層,且經由二氧化矽墊過濾有機物。用額外DCM沖洗且在減壓下蒸發溶劑以得到呈油狀物之標題化合物(11.0 g,91%)。1H NMR (400.13 MHz, CDCl3
): 1.30 (t, J=7.3 Hz, 3H), 1.57-1.50 (m, 1H), 1.60-1.65 (m, 2H), 2.26-2.30 (m, 1H), 2.45 (td, J=9.0, 4.2 Hz, 1H), 4.13-4.22 (m, 3H), 9.32 (d, J=4.2 Hz, 1H)。
製備23
(1S,2S)-2-(二氟甲基)環丙烷羧酸乙酯 歷經3分鐘將三氟化二乙基胺基硫(24 mL,172.3 mmol)添加至(1S,2S)-2-甲醯基環丙烷羧酸乙酯(11.00 g,77.38 mmol)於DCM (220 mL)中之冰冷溶液中。1小時後,自冰浴移除且在室溫下攪拌2小時。添加三氟化二乙基胺基硫(3.5 mL)且攪拌額外一小時。在冰浴中冷卻混合物且小心地倒入飽和NaHCO3
水溶液中。分離各層且用DCM (2 × 50 mL)萃取水層。
歷經3分鐘將三氟化二乙基胺基硫(24 mL,172.3 mmol)添加至(1S,2S)-2-甲醯基環丙烷羧酸乙酯(11.00 g,77.38 mmol)於DCM (220 mL)中之冰冷溶液中。1小時後,自冰浴移除且在室溫下攪拌2小時。添加三氟化二乙基胺基硫(3.5 mL)且攪拌額外一小時。在冰浴中冷卻混合物且小心地倒入飽和NaHCO3
水溶液中。分離各層且用DCM (2 × 50 mL)萃取水層。
合併有機物且經MgSO4
乾燥。經由小二氧化矽墊過濾樣品且在減壓(250 mbar及30℃)下蒸發溶劑以得到呈油狀物之標題化合物(11.10 g,87%)。1H NMR (400.13 MHz, CDCl3
): 1.14-1.19 (m, 1H), 1.29 (m, 1H), 1.30 (t, J=7.2 Hz, 3H), 1.97-1.89 (m, 2H), 4.22 (q, J=7.2 Hz, 3H), 5.79 (td, JH-F=56.8 Hz, J=3.6 Hz, 1H)。
製備24
(1S,2S)-2-(二氟甲基)環丙烷羧酸 將1 N NaOH (75 mL)添加至(1S,2S)-2-(二氟甲基)環丙烷羧酸乙酯(11.10 g,67.62 mmol)於MeOH (75 mL)中之溶液中且在RT下攪拌隔夜。添加DCM (70 mL)且分離各層。用DCM (70 mL)萃取水層。在冰浴中冷卻水層且藉由添加35% HCl將pH調節至1。添加DCM (50 mL)且分離兩個層。用DCM萃取水層。合併有機物,經MgSO4
乾燥,且在減壓(200 mbar,30℃)下蒸發溶劑以得到呈油狀物之標題化合物(6.25 g,68%)。1H NMR (400.13 MHz, CDCl3
): 1.26 (dt, J= 8.4, 5.9 Hz, 1H), 1.34-1.39 (m, 1H), 1.92-1.96 (m, 1H), 2.01-2.07 (m, 1H), 5.82 (td, JH-F=59Hz, J=3.2 Hz, 1H)。
將1 N NaOH (75 mL)添加至(1S,2S)-2-(二氟甲基)環丙烷羧酸乙酯(11.10 g,67.62 mmol)於MeOH (75 mL)中之溶液中且在RT下攪拌隔夜。添加DCM (70 mL)且分離各層。用DCM (70 mL)萃取水層。在冰浴中冷卻水層且藉由添加35% HCl將pH調節至1。添加DCM (50 mL)且分離兩個層。用DCM萃取水層。合併有機物,經MgSO4
乾燥,且在減壓(200 mbar,30℃)下蒸發溶劑以得到呈油狀物之標題化合物(6.25 g,68%)。1H NMR (400.13 MHz, CDCl3
): 1.26 (dt, J= 8.4, 5.9 Hz, 1H), 1.34-1.39 (m, 1H), 1.92-1.96 (m, 1H), 2.01-2.07 (m, 1H), 5.82 (td, JH-F=59Hz, J=3.2 Hz, 1H)。
製備25
反式-苄基-2-乙醯基環丙烷羧酸酯 將K2
CO3
(36.2 g,261.93 mmol) 添加至苯基溴乙酸酯(40 g,174.618 mmol)、甲基乙烯基酮(43 mL,523.85 mmol)、1,4-二氮雜雙環[2.2.2]辛烷(2.3 g,20.9 mmol)於乙腈(400 mL)中之混合物中。在N2
下於80℃下攪拌隔夜。使其冷卻,過濾且在減壓下蒸發溶劑。藉由矽膠層析純化,溶離劑0-50% EtOAc/己烷,以在自層析溶離份移除溶劑之後得到標題化合物(13.4 g,35%)。1H NMR (400.13 MHz, d6
-DMSO): 1.33-1.39 (m, 2H), 2.08-2.13 (m, 1H), 2.24 (s, 3H), 2.54-2.59 (m, 2H), 5.13 (s, 2H), 7.39-7.37(m, 6H)。
將K2
CO3
(36.2 g,261.93 mmol) 添加至苯基溴乙酸酯(40 g,174.618 mmol)、甲基乙烯基酮(43 mL,523.85 mmol)、1,4-二氮雜雙環[2.2.2]辛烷(2.3 g,20.9 mmol)於乙腈(400 mL)中之混合物中。在N2
下於80℃下攪拌隔夜。使其冷卻,過濾且在減壓下蒸發溶劑。藉由矽膠層析純化,溶離劑0-50% EtOAc/己烷,以在自層析溶離份移除溶劑之後得到標題化合物(13.4 g,35%)。1H NMR (400.13 MHz, d6
-DMSO): 1.33-1.39 (m, 2H), 2.08-2.13 (m, 1H), 2.24 (s, 3H), 2.54-2.59 (m, 2H), 5.13 (s, 2H), 7.39-7.37(m, 6H)。
製備26
反式-苄基-2-(1,1-二氟乙基)環丙烷羧酸酯 在0℃下將EtOH (0.05 eq)添加至反式-苄基-2-乙醯基環丙烷羧酸酯(2.76 g,12.6 mmol)及三氟化雙(2-甲氧基乙基)胺基硫(23.2 mL,126 mmol)之混合物中。使其升溫至室溫且在50℃下加熱40小時。用DCM稀釋且在冰浴中冷卻混合物,以緩慢添加飽和NaHCO3
水溶液。分離各層且用DCM (2 × 100 mL)萃取水層。合併有機物且經無水Na2
SO4
乾燥。在減壓下蒸發溶劑。藉由矽膠層析純化,溶離劑:10% MTBE/己烷,以在自層析溶離份移除溶劑之後得到呈無色油狀之標題化合物(2.30 g,76%)。1H NMR (400.13 MHz, d6
-DMSO): 1.19 (t, J=7.5 Hz, 2H), 1.67 (t, J H-F=16 Hz, 3H), 1.93-1.98 (m, 1H), 2.00-2.08 (m, 1H), 5.13 (s, 2H), 7.38-7.40 (m, 5H)。
在0℃下將EtOH (0.05 eq)添加至反式-苄基-2-乙醯基環丙烷羧酸酯(2.76 g,12.6 mmol)及三氟化雙(2-甲氧基乙基)胺基硫(23.2 mL,126 mmol)之混合物中。使其升溫至室溫且在50℃下加熱40小時。用DCM稀釋且在冰浴中冷卻混合物,以緩慢添加飽和NaHCO3
水溶液。分離各層且用DCM (2 × 100 mL)萃取水層。合併有機物且經無水Na2
SO4
乾燥。在減壓下蒸發溶劑。藉由矽膠層析純化,溶離劑:10% MTBE/己烷,以在自層析溶離份移除溶劑之後得到呈無色油狀之標題化合物(2.30 g,76%)。1H NMR (400.13 MHz, d6
-DMSO): 1.19 (t, J=7.5 Hz, 2H), 1.67 (t, J H-F=16 Hz, 3H), 1.93-1.98 (m, 1H), 2.00-2.08 (m, 1H), 5.13 (s, 2H), 7.38-7.40 (m, 5H)。
製備27
反式-2-(1,1-二氟乙基)環丙烷羧酸 將10% Pd/C (1.53 g,14.4 mmol)添加至反式-苄基-2-(1,1-二氟乙基)環丙烷羧酸酯(6.56 g,27.3 mmol)於EtOAc (136 mL,0.2 M)中之溶液中。使用氣囊在RT下於H2
下攪拌3小時。經由矽藻土過濾且用EtOAc沖洗。在減壓下蒸發溶劑以得到呈無色油狀之標題化合物(4.03 g,98%)。1H NMR (400.13 MHz, d6
-DMSO): 1.10 (t, J=7.4 Hz, 2H), 1.66 (t, J H-F= 18.4 Hz, 3H), 1.72-1.77 (m, 1H), 1.96-1.99 (m, 1H), 12.49 (s, 1H)。
將10% Pd/C (1.53 g,14.4 mmol)添加至反式-苄基-2-(1,1-二氟乙基)環丙烷羧酸酯(6.56 g,27.3 mmol)於EtOAc (136 mL,0.2 M)中之溶液中。使用氣囊在RT下於H2
下攪拌3小時。經由矽藻土過濾且用EtOAc沖洗。在減壓下蒸發溶劑以得到呈無色油狀之標題化合物(4.03 g,98%)。1H NMR (400.13 MHz, d6
-DMSO): 1.10 (t, J=7.4 Hz, 2H), 1.66 (t, J H-F= 18.4 Hz, 3H), 1.72-1.77 (m, 1H), 1.96-1.99 (m, 1H), 12.49 (s, 1H)。
製備28
5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶 藉由使N2
鼓泡經由混合物使4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪(16 g,54.0 mmol)、2-[(1S,2S)-2-乙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(17 g,83.22 mmol)、2 M Na2
CO3
水溶液(70 mL,140 mmol)及1,4-二噁烷(290 mL)之混合物脫氣10分鐘。添加雙(二第三丁基(4-二甲胺基苯基)膦)二氯鈀(II) (2.0 g,2.74 mmol)且在90℃下攪拌所得混合物16小時。冷卻反應混合物至PT,用H2
O稀釋,且用EtOAc萃取。分離所得相,經無水MgSO4
使有機相乾燥,且在真空中濃縮。藉由層析經由二氧化矽純化所得殘餘物,用60-100% 己烷/EtOAc之梯度溶離,以在自層析溶離份移除溶劑之後獲得呈琥珀色油狀之標題化合物(12.95 g,77%)。在靜置時在RT下油固化成灰白色固體。ES/MS (m/z): 301 (M+1)。
藉由使N2
鼓泡經由混合物使4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪(16 g,54.0 mmol)、2-[(1S,2S)-2-乙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(17 g,83.22 mmol)、2 M Na2
CO3
水溶液(70 mL,140 mmol)及1,4-二噁烷(290 mL)之混合物脫氣10分鐘。添加雙(二第三丁基(4-二甲胺基苯基)膦)二氯鈀(II) (2.0 g,2.74 mmol)且在90℃下攪拌所得混合物16小時。冷卻反應混合物至PT,用H2
O稀釋,且用EtOAc萃取。分離所得相,經無水MgSO4
使有機相乾燥,且在真空中濃縮。藉由層析經由二氧化矽純化所得殘餘物,用60-100% 己烷/EtOAc之梯度溶離,以在自層析溶離份移除溶劑之後獲得呈琥珀色油狀之標題化合物(12.95 g,77%)。在靜置時在RT下油固化成灰白色固體。ES/MS (m/z): 301 (M+1)。
製備29
5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶 混合4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪(19.1 g,70.6 mmol)、K3
PO4
(45.9 g,212 mmol)、1,4-二噁烷(300 mL)及H2
O (75 mL)且用N2
使混合物脫氣10分鐘。添加[1,1'-雙(二苯基膦基)二茂鐵]二氯鈀(II) (7.98 g 10.6 mmol)。用氮氣鼓泡額外2分鐘且一次性添加2-[(1S,2S)-2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(22.2 g,106 mmol)。在80℃下攪拌所得混合物16小時。冷卻反應至RT,添加H2
O,且用EtOAc萃取。分離有機層,經無水MgSO4
乾燥,且在減壓下濃縮。藉由層析經由二氧化矽純化所得殘餘物,用85%己烷/EtOAc溶離,以在自層析溶離份移除溶劑之後分離呈琥珀色油狀之標題化合物(21.5 g,92%)。在靜置時在RT下油固化成灰白色固體。ES/MS (m/z): 315 (M+1)。
混合4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪(19.1 g,70.6 mmol)、K3
PO4
(45.9 g,212 mmol)、1,4-二噁烷(300 mL)及H2
O (75 mL)且用N2
使混合物脫氣10分鐘。添加[1,1'-雙(二苯基膦基)二茂鐵]二氯鈀(II) (7.98 g 10.6 mmol)。用氮氣鼓泡額外2分鐘且一次性添加2-[(1S,2S)-2-異丙基環丙基]-4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷(22.2 g,106 mmol)。在80℃下攪拌所得混合物16小時。冷卻反應至RT,添加H2
O,且用EtOAc萃取。分離有機層,經無水MgSO4
乾燥,且在減壓下濃縮。藉由層析經由二氧化矽純化所得殘餘物,用85%己烷/EtOAc溶離,以在自層析溶離份移除溶劑之後分離呈琥珀色油狀之標題化合物(21.5 g,92%)。在靜置時在RT下油固化成灰白色固體。ES/MS (m/z): 315 (M+1)。
製備30
4-[(1S,2S)-2-(二氟甲基)環丙基]-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪 將二甲基鋅(2 M於甲苯中,1.5 mL,3.0 mmol)添加至3-氯-4-[(1S,2S)-2-(二氟甲基)環丙基]-6-(2,4-二甲氧基嘧啶-5-基)噠嗪(502 mg,1.46 mmol)及1,1'-雙(二苯基膦基)二茂鐵-二氯化鈀(II) (53 mg,0.07 mmol)於THF (6 mL)中之脫氣懸浮液中。在密封瓶中在60℃下加熱2小時。使其冷卻至室溫。添加飽和NH4
Cl水溶液且用DCM (3×)萃取。合併有機物,經無水MgSO4
乾燥且在減壓下移除溶劑。藉由矽膠層析純化,溶離劑:EtOAc,以在自層析溶離份移除溶劑之後得到呈黃色殘餘物之標題化合物(433 mg,91.8%)。ES/MS (m/z): 323 (M+1)。
將二甲基鋅(2 M於甲苯中,1.5 mL,3.0 mmol)添加至3-氯-4-[(1S,2S)-2-(二氟甲基)環丙基]-6-(2,4-二甲氧基嘧啶-5-基)噠嗪(502 mg,1.46 mmol)及1,1'-雙(二苯基膦基)二茂鐵-二氯化鈀(II) (53 mg,0.07 mmol)於THF (6 mL)中之脫氣懸浮液中。在密封瓶中在60℃下加熱2小時。使其冷卻至室溫。添加飽和NH4
Cl水溶液且用DCM (3×)萃取。合併有機物,經無水MgSO4
乾燥且在減壓下移除溶劑。藉由矽膠層析純化,溶離劑:EtOAc,以在自層析溶離份移除溶劑之後得到呈黃色殘餘物之標題化合物(433 mg,91.8%)。ES/MS (m/z): 323 (M+1)。
製備31
3,6-二氯-4-環丙基-噠嗪 向3,6-二氯噠嗪(10.00 g,67.12 mmol)於水(300 mL)中之懸浮液中添加10 mL濃縮H2
SO4
及環丙烷羧酸(5.85 mL,73.7 mmol),在70℃下加熱且用N2
脫氣。歷經30秒添加AgNO3
(2.28 g,13.4 mmol)於10 mL H2
O中之溶液,隨後歷經30分鐘逐滴添加(NH4
)2
S2
O8
(46 g,201.577 mmol)於150 mL H2
O中之溶液。一小時後使混合物冷卻至RT且倒在冰上,藉由添加濃縮NH4
OH將pH調節至9。用EtOAc稀釋且分離有機相。用額外EtOAc萃取水層。合併有機物,經無水Na2
SO4
乾燥,且在減壓下蒸發溶劑。藉由逆相層析(C18 Gold 415 g,梯度25-100% ACN於10 mM碳酸氫銨中;150 mL/min,30分鐘)純化,以在自層析溶離份移除溶劑之後分離呈白色固體狀之標題化合物(6.83 g,54%)。ES/MS (m/z): (35
Cl/37
Cl) 189/191。
向3,6-二氯噠嗪(10.00 g,67.12 mmol)於水(300 mL)中之懸浮液中添加10 mL濃縮H2
SO4
及環丙烷羧酸(5.85 mL,73.7 mmol),在70℃下加熱且用N2
脫氣。歷經30秒添加AgNO3
(2.28 g,13.4 mmol)於10 mL H2
O中之溶液,隨後歷經30分鐘逐滴添加(NH4
)2
S2
O8
(46 g,201.577 mmol)於150 mL H2
O中之溶液。一小時後使混合物冷卻至RT且倒在冰上,藉由添加濃縮NH4
OH將pH調節至9。用EtOAc稀釋且分離有機相。用額外EtOAc萃取水層。合併有機物,經無水Na2
SO4
乾燥,且在減壓下蒸發溶劑。藉由逆相層析(C18 Gold 415 g,梯度25-100% ACN於10 mM碳酸氫銨中;150 mL/min,30分鐘)純化,以在自層析溶離份移除溶劑之後分離呈白色固體狀之標題化合物(6.83 g,54%)。ES/MS (m/z): (35
Cl/37
Cl) 189/191。
可基本上如製備22中所描述來製備以下實例。 
製備43
3-氯-4-環丙基-6-(2,4-二甲氧基嘧啶-5-基)噠嗪 混合(2,4-二甲氧基嘧啶-5-基)硼酸(4.60 g,25.0 mmol)、3,6-二氯-4-環丙基-噠嗪(5.2 g,28 mmol)及K2
CO3
(4.4 g,32 mmol)、1,4-二噁烷(125 mL)以及H2
O (42 mL)且用N2
使混合物脫氣10分鐘。添加[1,1'-雙(二苯基膦基)二茂鐵]二氯鈀(II)二氯甲烷錯合物(1.5 g,1.8 mmol)且用N2
進一步脫氣。在60℃下攪拌所得混合物1小時。冷卻反應至RT,添加H2
O,且用EtOAc萃取。分離有機層。用額外EtOAc萃取水層。合併有機物,經無水Na2
SO4
乾燥,且在減壓下蒸發溶劑。藉由層析經由二氧化矽純化所得殘餘物,用70%己烷/(3:2丙酮:DCM)溶離,以在自層析溶離份移除溶劑之後分離呈淡黃色固體狀之標題化合物(2.4 g,33%)。ES/MS (m/z): (35
Cl/37
Cl) 293/295 [M+1]+
混合(2,4-二甲氧基嘧啶-5-基)硼酸(4.60 g,25.0 mmol)、3,6-二氯-4-環丙基-噠嗪(5.2 g,28 mmol)及K2
CO3
(4.4 g,32 mmol)、1,4-二噁烷(125 mL)以及H2
O (42 mL)且用N2
使混合物脫氣10分鐘。添加[1,1'-雙(二苯基膦基)二茂鐵]二氯鈀(II)二氯甲烷錯合物(1.5 g,1.8 mmol)且用N2
進一步脫氣。在60℃下攪拌所得混合物1小時。冷卻反應至RT,添加H2
O,且用EtOAc萃取。分離有機層。用額外EtOAc萃取水層。合併有機物,經無水Na2
SO4
乾燥,且在減壓下蒸發溶劑。藉由層析經由二氧化矽純化所得殘餘物,用70%己烷/(3:2丙酮:DCM)溶離,以在自層析溶離份移除溶劑之後分離呈淡黃色固體狀之標題化合物(2.4 g,33%)。ES/MS (m/z): (35
Cl/37
Cl) 293/295 [M+1]+
以下實例可基本上如製備6中所描述來製備。 
製備52
4-環丁基-6-(2,4-二甲氧基嘧啶-5-基)-3-甲氧基-噠嗪 3-根據製備21及33自環丁基羧酸獲得氯-4-環丁基-6-(2,4-二甲氧基嘧啶-5-基)噠嗪。
3-根據製備21及33自環丁基羧酸獲得氯-4-環丁基-6-(2,4-二甲氧基嘧啶-5-基)噠嗪。
將3-氯-4-環丁基-6-(2,4-二甲氧基嘧啶-5-基)噠嗪(0.42 g,1.4 mmol)添加至藉由將Na (0.16 g,6.8 mmol)溶解於MeOH (12 mL)中所製備之NaOMe溶液中。在封蓋瓶中於60℃下加熱混合物隔夜。使混合物冷卻至RT,添加50%飽和NaCl水溶液且用DCM (3×)萃取。合併有機物且經無水MgSO4
乾燥。在減壓下移除溶劑以得到呈白色固體狀之標題化合物(0.38 g,93%)。ES/MS (m/z): 303 (M+1)。
以下實例可基本上如製備52中所描述來製備。 
製備59
反式-第三丁基-二甲基-[2-[2-(4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷-2-基)環丙基]乙氧基]矽烷 可基本上如製備3中所描述由市售第三丁基-二甲基-[(E)-4-(4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷-2-基)丁-3-烯氧基]矽烷來製備。1H NMR (400.13 MHz, CDCl3): 3.70-3.66 (m, 2H), 1.51-1.38 (m, 2H), 1.23 (s, 12H), 1.04-0.96 (m, 1H), 0.91 (s, 9H), 0.71-0.67 (m, 1H), 0.46-0.41 (m, 1H), 0.07 (s, 6H), -0.38 (dt, J= 9.3, 5.8 Hz, 1H)。
可基本上如製備3中所描述由市售第三丁基-二甲基-[(E)-4-(4,4,5,5-四甲基-1,3,2-二氧雜硼雜環戊烷-2-基)丁-3-烯氧基]矽烷來製備。1H NMR (400.13 MHz, CDCl3): 3.70-3.66 (m, 2H), 1.51-1.38 (m, 2H), 1.23 (s, 12H), 1.04-0.96 (m, 1H), 0.91 (s, 9H), 0.71-0.67 (m, 1H), 0.46-0.41 (m, 1H), 0.07 (s, 6H), -0.38 (dt, J= 9.3, 5.8 Hz, 1H)。
製備60
反式-第三丁基-二甲基-[2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙氧基]矽烷 可基本上如製備4中所描述來製備。ES/MS (m/z): 417 (M+1)。
可基本上如製備4中所描述來製備。ES/MS (m/z): 417 (M+1)。
製備61
反式-2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙醇 添加含氟化四丁銨(5 mL,5 mmol,1 M於THF中)及反式-第三丁基-二甲基-[2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙氧基]矽烷(1.15 g,8.9 mmol)之DCM (3 mL)中且在60℃下攪拌1小時。冷卻至室溫。用DCM (80 mL)稀釋且用飽和NH4
Cl (3 × 30 mL)洗滌。合併有機物,經無水Na2
SO4
乾燥,且在減壓下移除溶劑。藉由矽膠層析純化,溶離劑0-30% MeOH/EtOAc,以在自層析溶離份移除溶劑之後得到呈白色固體狀之標題化合物(0.53 g,53%)。ES/MS (m/z): 317 (M+1)。1H NMR (399.80 MHz, CDCl3): 8.92 (s, 1H), 7.29 (m, 1H), 4.03 (s, 3H), 4.02 (s, 3H), 3.81 (t, J= 6.2 Hz, 2H), 2.76 (s, 3H), 1.73-(m, 3H), 1.26 (m, 2H), 1.03(m, 2H)。
添加含氟化四丁銨(5 mL,5 mmol,1 M於THF中)及反式-第三丁基-二甲基-[2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙氧基]矽烷(1.15 g,8.9 mmol)之DCM (3 mL)中且在60℃下攪拌1小時。冷卻至室溫。用DCM (80 mL)稀釋且用飽和NH4
Cl (3 × 30 mL)洗滌。合併有機物,經無水Na2
SO4
乾燥,且在減壓下移除溶劑。藉由矽膠層析純化,溶離劑0-30% MeOH/EtOAc,以在自層析溶離份移除溶劑之後得到呈白色固體狀之標題化合物(0.53 g,53%)。ES/MS (m/z): 317 (M+1)。1H NMR (399.80 MHz, CDCl3): 8.92 (s, 1H), 7.29 (m, 1H), 4.03 (s, 3H), 4.02 (s, 3H), 3.81 (t, J= 6.2 Hz, 2H), 2.76 (s, 3H), 1.73-(m, 3H), 1.26 (m, 2H), 1.03(m, 2H)。
製備62
反式-2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙基甲磺酸酯 在0℃下在N2
下將甲磺醯氯(0.2 mL,3 mmol)添加至反式-2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙醇(0.390 g,1.22 mmol)及N,N-二異丙基乙胺(0.4 mL,2 mmol)於DCM (8 mL)中之溶液中。在0℃下攪拌40分鐘。用50 mL DCM稀釋且用5% NaHCO3
(2 × 30 mL)及水(30 mL)洗滌。合併有機物,經無水Na2
SO4
乾燥,且在減壓下蒸發溶劑。藉由矽膠層析純化,溶離劑:0-20% MeOH/EtOAc,以在自層析溶離份移除溶劑之後得到呈褐色固體狀之標題化合物(0.280 g,58%)。ES/MS (m/z): 395 (M+1)。1H 1H NMR (399.80 MHz, CDCl3): 8.98 (s, 1H), 7.38 (s, 1H), 4.37 (t, J= 6.2 Hz, 2H), 4.07 (s, 3H), 4.05 (s, 3H), 3.01 (s, 3H), 2.81 (s, 3H), 2.04 (m, 1H), 1.90-1.78 (m, 2H), 1.27 (m, 1H), 1.06 (t, J= 7.1 Hz, 2H)。
在0℃下在N2
下將甲磺醯氯(0.2 mL,3 mmol)添加至反式-2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙醇(0.390 g,1.22 mmol)及N,N-二異丙基乙胺(0.4 mL,2 mmol)於DCM (8 mL)中之溶液中。在0℃下攪拌40分鐘。用50 mL DCM稀釋且用5% NaHCO3
(2 × 30 mL)及水(30 mL)洗滌。合併有機物,經無水Na2
SO4
乾燥,且在減壓下蒸發溶劑。藉由矽膠層析純化,溶離劑:0-20% MeOH/EtOAc,以在自層析溶離份移除溶劑之後得到呈褐色固體狀之標題化合物(0.280 g,58%)。ES/MS (m/z): 395 (M+1)。1H 1H NMR (399.80 MHz, CDCl3): 8.98 (s, 1H), 7.38 (s, 1H), 4.37 (t, J= 6.2 Hz, 2H), 4.07 (s, 3H), 4.05 (s, 3H), 3.01 (s, 3H), 2.81 (s, 3H), 2.04 (m, 1H), 1.90-1.78 (m, 2H), 1.27 (m, 1H), 1.06 (t, J= 7.1 Hz, 2H)。
製備63
反2,4-二甲氧基-5-[6-甲基-5-[2-(2-氟乙基)環丙基]噠嗪-3-基]嘧啶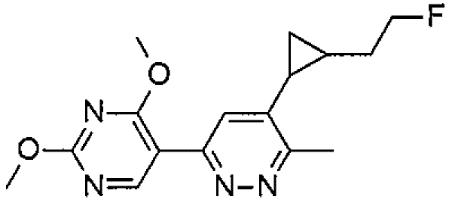 將氟化四丁銨水合物(3 mL,3 mmol,1 M於THF中)添加至反式-2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙基甲磺酸酯(0.280 g,0.71 mmol)於THF (3 mL)中之溶液中。在70℃下加熱2小時。冷卻至RT。用EtOAc稀釋,用NaCl飽和水溶液洗滌,經無水Na2
SO4
乾燥,且在減壓下移除溶劑。藉由矽膠層析純化,溶離劑:0-20% MeOH/EtOAc,以在自層析溶離份移除溶劑之後得到呈淡黃色油狀之標題化合物(0.170 g,71%)。ES/MS (m/z): 319 (M+1)。1H NMR (399.80 MHz, CDCl3): 9.01 (m, 1H), 7.39 (s, 1H), 4.67-4.52 (dt, J H-F 48 Hz, J= 6.7 Hz, 2H), 4.08 (s, 3H), 4.07 (s, 3H), 2.83 (s, 3H), 2.00-1.94 (m, 3H), 1.27 (m, 1H), 1.06 (m, 2H)。
將氟化四丁銨水合物(3 mL,3 mmol,1 M於THF中)添加至反式-2-[2-[6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-噠嗪-4-基]環丙基]乙基甲磺酸酯(0.280 g,0.71 mmol)於THF (3 mL)中之溶液中。在70℃下加熱2小時。冷卻至RT。用EtOAc稀釋,用NaCl飽和水溶液洗滌,經無水Na2
SO4
乾燥,且在減壓下移除溶劑。藉由矽膠層析純化,溶離劑:0-20% MeOH/EtOAc,以在自層析溶離份移除溶劑之後得到呈淡黃色油狀之標題化合物(0.170 g,71%)。ES/MS (m/z): 319 (M+1)。1H NMR (399.80 MHz, CDCl3): 9.01 (m, 1H), 7.39 (s, 1H), 4.67-4.52 (dt, J H-F 48 Hz, J= 6.7 Hz, 2H), 4.08 (s, 3H), 4.07 (s, 3H), 2.83 (s, 3H), 2.00-1.94 (m, 3H), 1.27 (m, 1H), 1.06 (m, 2H)。
對掌性分離:管柱Lux Cellulose-4,250 × 21 mm,流動速率70 g/min,溶離劑:40% MeOH/CO2
。
對映異構體1 > 99% ee,rt 2.59 min (Lux Cellulose-4,4.6 × 150 mm,40% MeOH/CO2
,5 mL/min,225 nm)。對映異構體2 > 99% ee,rt 3.34 min (Lux Cellulose-4,4.6 × 150 mm,40% MeOH/CO2
,5 mL/min,225 nm)。
實例1
5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮 將5-[5-[(-2-乙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶異構體1 (197 mg,0.66 mmol)溶解於1 M HCl水溶液(4 mL)中且將所得混合物加熱至70℃隔夜。冷卻反應混合物至RT,在-78℃下在丙酮/乾冰浴中冷凍,且藉由凍乾移除溶劑,以得到呈淡黃色固體狀之標題化合物(0.176 g,97%)。ES/MS (m/z): 273 (M+H)。1
H NMR (d6
-DMSO) δ: 1.00 (t, J= 7.3 Hz, 3H), 1.11-1.16 (m, 1H), 1.27-1.33 (m, 2H), 1.47-1.52 (m, 2H), 1.92-1.96 (m, 1H), 2.80 (s, 3H), 8.00 (s, 1H), 8.41 (d, J= 5.5 Hz, 1H), 11.73 (s, 1H), 11.99-11.91 (m, 1H)。
將5-[5-[(-2-乙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶異構體1 (197 mg,0.66 mmol)溶解於1 M HCl水溶液(4 mL)中且將所得混合物加熱至70℃隔夜。冷卻反應混合物至RT,在-78℃下在丙酮/乾冰浴中冷凍,且藉由凍乾移除溶劑,以得到呈淡黃色固體狀之標題化合物(0.176 g,97%)。ES/MS (m/z): 273 (M+H)。1
H NMR (d6
-DMSO) δ: 1.00 (t, J= 7.3 Hz, 3H), 1.11-1.16 (m, 1H), 1.27-1.33 (m, 2H), 1.47-1.52 (m, 2H), 1.92-1.96 (m, 1H), 2.80 (s, 3H), 8.00 (s, 1H), 8.41 (d, J= 5.5 Hz, 1H), 11.73 (s, 1H), 11.99-11.91 (m, 1H)。
實例1-5之替代程序
在45℃下攪拌5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶(16.2 g,51.5 mmol)及1 M HCl水溶液(135 mL,135 mmol)之懸浮液16小時。冷卻至RT,添加2 M K2
HPO4
水溶液至pH為約6 (約150 mL),且在RT下攪拌16小時。過濾及收集所得固體,用水洗滌,且在45℃下在真空烘箱中乾燥16小時,以得到呈白色固體狀之標題化合物(13.8 g,93%)。ES/MS (m/z): 273 (M+1)。使5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮結晶
將5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-溶解於甲醇中,在50℃下攪拌1小時且使其冷卻至環境溫度,其中其自溶液中結晶。藉由真空過濾分離固體且在70℃下在真空下短暫乾燥。
實例2
5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮 將反式-5-[5-[2-異丙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶異構體1 (628 mg,2.00 mmol)溶解於MeOH (3 mL)中。添加1 M HCl水溶液(5 mL)且加熱至70℃持續3小時。冷卻至RT,將反應混合物負載於經MeOH洗滌之SCX管柱(20 g,Silicycle SILIABOND對甲苯磺酸)上,用MeOH (140 mL)洗滌SCX管柱且用2 M NH3
/MeOH (140 ml)溶離所要產物。濃縮NH3
/MeOH溶離份以獲得呈奶黃色固體狀之標題化合物(546 mg,95%)。ES/MS (m/z): 287 (M+H)。1
H NMR (d6
-DMSO) δ: 0.99 (m, 9H), 1.24 (m, 1H), 1.81 (m, 1H), 2.72 (s, 3H), 7.67 (s, 1H), 8.23 (s, 1H), 11.43 (bs, 2H)。
將反式-5-[5-[2-異丙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶異構體1 (628 mg,2.00 mmol)溶解於MeOH (3 mL)中。添加1 M HCl水溶液(5 mL)且加熱至70℃持續3小時。冷卻至RT,將反應混合物負載於經MeOH洗滌之SCX管柱(20 g,Silicycle SILIABOND對甲苯磺酸)上,用MeOH (140 mL)洗滌SCX管柱且用2 M NH3
/MeOH (140 ml)溶離所要產物。濃縮NH3
/MeOH溶離份以獲得呈奶黃色固體狀之標題化合物(546 mg,95%)。ES/MS (m/z): 287 (M+H)。1
H NMR (d6
-DMSO) δ: 0.99 (m, 9H), 1.24 (m, 1H), 1.81 (m, 1H), 2.72 (s, 3H), 7.67 (s, 1H), 8.23 (s, 1H), 11.43 (bs, 2H)。
實例1-5之替代程序
在45℃下攪拌5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-2,4-二甲氧基-嘧啶(21.5 g,65.6 mmol)及1 M HCl水溶液(165 mL,135 mmol)之懸浮液16小時。冷卻至RT且用MTBE萃取。丟棄有機相且向水相添加2 M K2
HPO4
水溶液直至pH為約6 (約150 mL)為止。在RT下攪拌所得混合物16小時。過濾及收集所得固體,用水洗滌且在45℃下在真空烘箱中乾燥16小時,以得到呈白色固體狀之標題化合物(14.3 g,76%)。ES/MS (m/z): 287 (M+1)。
實例3
5-[5-[2-(二氟甲基)環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮 可基本上如實例1中所描述來製備5-[5-[2-(二氟甲基)環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮。
可基本上如實例1中所描述來製備5-[5-[2-(二氟甲基)環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮。
以下實例可基本上如實例1中所描述來製備。 
實例7
5-(6-氯-5-環丙基-噠嗪-3-基)-1H-嘧啶-2,4-二酮 將1 M HCl (14 mL,14 mmol)添加至3-氯-4-環丙基-6-(2,4-二甲氧基嘧啶-5-基)噠嗪(1.0 g,3.4 mmol)於MeOH (17 mL)中之溶液中且在50℃下攪拌隔夜。在減壓下移除溶劑以得到呈白色固體狀之標題化合物(0.9 g,100%)。ES/MS (m/z): (35
Cl/37
Cl) 265/267
將1 M HCl (14 mL,14 mmol)添加至3-氯-4-環丙基-6-(2,4-二甲氧基嘧啶-5-基)噠嗪(1.0 g,3.4 mmol)於MeOH (17 mL)中之溶液中且在50℃下攪拌隔夜。在減壓下移除溶劑以得到呈白色固體狀之標題化合物(0.9 g,100%)。ES/MS (m/z): (35
Cl/37
Cl) 265/267
以下實例可基本上如實例7中所描述來製備。 
實例16
4-環丙基-6-(2,4-二側氧基-1H-嘧啶-5-基)噠嗪-3-甲腈 用N2
使5-(6-氯-5-環丙基-噠嗪-3-基)-1H-嘧啶-2,4-二酮(A,57 mg,0.22 mmol,100質量%)於DMF (1 mL,12.9 mmol)中之溶液脫氣。添加Zn(CN)2
(20 mg,0.22 mmol)、參(二亞苄基丙酮)二鈀(0) (5 mg,0.0054 mmol)及1,1'-雙(二苯基膦基)二茂鐵(6 mg,0.011 mmol)且進一步用N2
脫氣。緊密封蓋且在120℃下加熱隔夜。冷卻至RT且經由矽藻土過濾。藉由反相層析(C18 Gold 15.5 g,梯度5-20% ACN/10 mM碳酸氫銨;20 CV)純化以分離呈淡黃色固體狀之標題化合物(0.032 g,58%)。ES/MS (m/z): 256 (M+1)。
用N2
使5-(6-氯-5-環丙基-噠嗪-3-基)-1H-嘧啶-2,4-二酮(A,57 mg,0.22 mmol,100質量%)於DMF (1 mL,12.9 mmol)中之溶液脫氣。添加Zn(CN)2
(20 mg,0.22 mmol)、參(二亞苄基丙酮)二鈀(0) (5 mg,0.0054 mmol)及1,1'-雙(二苯基膦基)二茂鐵(6 mg,0.011 mmol)且進一步用N2
脫氣。緊密封蓋且在120℃下加熱隔夜。冷卻至RT且經由矽藻土過濾。藉由反相層析(C18 Gold 15.5 g,梯度5-20% ACN/10 mM碳酸氫銨;20 CV)純化以分離呈淡黃色固體狀之標題化合物(0.032 g,58%)。ES/MS (m/z): 256 (M+1)。
以下實例可基本上如實例16中所描述來製備。 
實例23
5-(5-環丁基-6-甲氧基-噠嗪-3-基)-1H-嘧啶-2,4-二酮 將1 M HCl (5.2 mL,5.2 mmol)添加至4-環丁基-6-(2,4-二甲氧基嘧啶-5-基)-3-甲氧基-噠嗪(0.39 g,1.3 mmol)中且在50℃下攪拌6小時,接著在RT下攪拌隔夜。在減壓下移除溶劑。經由SCX筒(10 g,溶離劑60 mL DCM,60 mL50% MeOH/DCM及120 mL 50% 7 M NH3
/MeOH/DCM)純化以得到呈白色固體狀之標題化合物(0.337 g,96%)。ES/MS (m/z): 275 (M+1)。
將1 M HCl (5.2 mL,5.2 mmol)添加至4-環丁基-6-(2,4-二甲氧基嘧啶-5-基)-3-甲氧基-噠嗪(0.39 g,1.3 mmol)中且在50℃下攪拌6小時,接著在RT下攪拌隔夜。在減壓下移除溶劑。經由SCX筒(10 g,溶離劑60 mL DCM,60 mL50% MeOH/DCM及120 mL 50% 7 M NH3
/MeOH/DCM)純化以得到呈白色固體狀之標題化合物(0.337 g,96%)。ES/MS (m/z): 275 (M+1)。
以下實例可基本上如實例23中所描述來製備。 
實例30
5-[5-[(1S,2R)-2-(2-氟乙基)環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮
將HCl (1 M於H2
O中,2 mL,2 mmol)添加至反式-2,4-二甲氧基-5-[6-甲基-5-[2-(2-氟乙基)環丙基]噠嗪-3-基]嘧啶之對映異構體1 (0.043 g,0.14 mmol)中且在50℃下加熱隔夜。在減壓下移除溶劑。經由SCX筒(12 g,溶離劑70 mL MeOH,70 mL 2 M NH3
/MeOH)純化以得到呈白色固體狀之標題化合物(0.037 g,89%)。ES/MS (m/z): 291 (M+1)。
生物分析
以下分析顯示,本發明之例示性化合物為CD73活性之抑制劑,且適用於治療癌症。
CD73 蛋白 表現及純化
C端6-HIS標記之人類CD73 (胺基酸1-547)係藉由用CD73基因瞬時轉染HEK293F哺乳動物細胞而在該細胞中表現,且使用Ni2+
親和力及Superdex 200尺寸排阻層析純化。如上文所描述來表現及純化C端6-HIS標記之小鼠CD73 (胺基酸1-549)。如先前所描述來表現及純化C端6-HIS標記之大鼠CD73 (胺基酸1-549)。
用於腺苷及腺苷純化之質譜法
Agilent 300 RapidFire自動化萃取系統(Agilent, Santa Clara, CA)與三個HPLC四元泵(quaternary pumps)一起使用,耦合至具有電噴霧電離(ESI)介面源之Sciex 6500三重四極(triple quadrupole)質譜儀(AB Sciex, Framingham, MA)。RapidFire質譜系統負載可再用的RapidFire HILIC (H1)固相萃取(SPE)筒(G9203-80109)。
用於樣品負載及洗滌之溶劑A為含有5% (v/v) ACN之50 mM甲酸銨(pH 4.0)。用於樣品溶離之溶劑B為含0.3%甲酸 + 2%氫氧化銨之70% ACN/30% MeOH。藉由在真空下直接自多孔培養盤抽吸10 μL至收集環來連續分析樣品。使用溶劑A以1.25毫升/分鐘之流動速率藉由四元泵1將10 μL樣品負載至HILIC筒上且洗3000 ms。使用溶劑B以1.25毫升/分鐘之流動速率藉由四元泵3使殘留分析物溶離至質譜儀持續3000 ms。使用溶劑A以1.25 mL/min之流動速率藉由四元泵1使系統再平衡3000 ms。
三重四極質譜儀裝備電噴霧電離(ESI)源且使用所選反應監測(SRM)正模式(M+H)+監測分析物。在m/z
268.05/136.0監測腺苷且在m/z
348.1/136.0監測單磷酸腺苷。使用13C5腺苷及15N5 AMP作為內部標準物分別計算腺苷及單磷酸腺苷之面積比值。
人類 CD73 生物化學分析
此分析之目的應為鑑別及表徵CD73酶活性之抑制劑。將含有2 μM單磷酸腺苷(Sigma #01930)、10 mM Tris pH 7.5、100 mM NaCl、0.01% BSA、0.2 mM辛基葡糖苷及50 pMCD73蛋白之反應混合物(20 μL)添加至384孔培養盤(Nunc #264573)。在RT下培育30分鐘後,藉由添加含有2%甲酸及10 μM13
C5-腺苷(用13
C5核糖標記)之20 μL停止溶液(Cambridge Isotope Laboratories - #CLM-3678-0),隨後添加40 μL dH2
O來終止反應。利用如上文所描述之質譜法來測定腺苷-13
C5及腺苷核糖-13
C5 (內部標準物)含量。使用信號比(腺苷峰整合/腺苷內部標準物峰整合)來定量各反應。藉由使用方程式{抑制% = 100 × [1 - (X - MIN)/(MAX - MIN)]}來計算抑制百分比,其中在存在> 10× IC50
之已知競爭性抑制劑的情況下,X等於孔信號比,MAX等於DMSO對照組之中位值信號比,且MIN等於酶活性之信號比。出於篩選之目的,在1% DMSO中以50 μM測試各化合物。藉由在0.0025至50 μM之10個濃度下測試各化合物來測定各化合物之IC50
(使用1:3稀釋流程)。
5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮之IC50
為0.028 μM。
5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮之IC50
為0.043 μM。
本文中所揭示之實例之所有化合物呈現小於0.062 μM之IC50
。
人類 CD73 機制分析
此分析之目的為測定所關注化合物之作用機制。如上所指示對人類CD73生物化學分析進行分析。使用1:3稀釋流程在0.023至50 μM之8個不同濃度下,但使用3倍稀釋流程在0.023至50 μM之AMP之8個不同濃度下來測試各化合物。使用GraphPad Prism 7.00標繪且使用專用混合模型抑制擬合不同抑制劑之面積比及底物濃度以測定抑制之Vmax、Km、Ki及α值{(Vmaxapp
= Vmax / (1+[I] /(Alpha * Ki));Kmapp
= Km * (1 + [I] / Ki) / (1 + [I] / (Alpha * Ki));Y=Vmaxapp
*X / (Kmapp
+ X),其中α、Vmax、Km及Ki共用於各化合物}。
5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮及5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮中之每一者為結合至酶-磷酸酯錯合物,但並未結合至酶蛋白自身的無競爭性抑制劑。
隨著無競爭性抑制劑,例如5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮之IC50值降低,增加AMP之底物濃度增強了該等抑制劑之效能,如表1中所示。 
小鼠 CD73 機制分析
此分析之目的為關於小鼠CD73酶活性之抑制來評定抑制劑。如上文所描述對人類CD73生物化學分析進行此分析,不同之處在於使用3 μM AMP及50 pM小鼠CD73酶。
5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮之IC50
為0.175 μM。
Calu6 人類細胞分析
此分析之目的為在基於細胞之分析中針對CD73測試化合物。使Calu6細胞(1500個細胞/孔)在96孔經聚-D-賴胺酸塗佈之培養盤(BD #356640)中生長,該培養盤含有100 μL介質MEM (Gibco #11095-072) + 1%丙酮酸鈉(Gibco #11360-070) + 1% NEAA (Gibco #11140-050) + 10% FBS (Hyclone #SH30071)。在RT下培育培養盤30分鐘,隨後在37℃/5% CO2
下培育隔夜。細胞用分析緩衝液(10 mM Tris-HCl pH 7.2,10 mM D-葡萄糖,1 mM KCl,125 mM NaCl,2 mM MgCl2
) (90微升/孔)洗滌兩次。隨後,將90 μL分析緩衝液添加至各孔中,隨後添加10微升/孔之AMP及化合物預混合物(50 μM AMP,化合物於1% DMSO中之不同濃度)。在室溫下培育培養盤60分鐘。接著,移除10 μL上澄液/孔且添加至新的培養盤,隨後添加20 μL停止溶液(2%甲酸,1.2 μM腺苷核糖-13
C5 (Cambridge Isotope Laboratories - #CLM-3678-0))及90 μL ddH2O以用於質譜分析。藉由利用如上文所描述之質譜法(Agilent RapidFire)來測定腺苷-13
C5及腺苷核糖-13
C5 (內部標準物)含量以用於人類CD73生物化學分析。亦如上文所描述來計算抑制百分比。
5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮之IC50
為0.0073 uM (實例2之化合物)。
5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮之IC50
為0.028 uM。
離體標靶抑制 分析
此分析之目的為在基於離體分析中針對小鼠血液中之鼠類CD73來測試化合物。在腫瘤達至約400 mm3
之後動物(6隻/組)經口給藥有在20% HPBCD (2-羥丙基-β-環糊精) (pH 2)中調配之各化合物。在治療之後,將血液收集至肝素管中且用於將13
C10-15
N5-AMP轉化為經標記腺苷、肌苷及次黃嘌呤的離體分析,如針對使用自經由經口給藥經受化合物治療之動物收集之全血的離體分析所描述。
在來自用不同劑量之化合物治療之動物的小鼠全血中,5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮抑制AMP轉化為腺苷、肌苷及次黃嘌呤,如表2中所示。 
*:統計顯著性。
5-[5-[(1S,2S)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮之IC50
為0.0073 uM。
人類 CD73 分析之基於 Calu6 腫瘤之活體內標靶抑制
此分析之目的為在活體內標靶抑制分析中針對來源於人類癌症Calu6細胞之異種移植腫瘤中的人類CD73來測試化合物。使Calu6細胞(ATCC)在補充有10%胎牛血清之HBSS介質中生長。收集具有胰蛋白酶之亞匯合細胞且用無血清之生長介質沖洗兩次。藉由在裸鼠(The Harlon Laboratory)之後側腹中注射5 × 106
之HBSS及MATRIGEL® (BD Biosciences, Franklin Lakes, NJ)之1:1混合物來引發皮下腫瘤之生長。當平均腫瘤體積達至約400-500 mm3
時,藉由腫瘤大小及體重隨機分配動物且如所指示將其置放至各別治療組。治療後,在含有如下文所描述之內部標準物的1 mL冰冷萃取緩衝液中收集且處理腫瘤樣本(各自50-80 mg)。
將箔條及液體N2
添加至砂漿中以預冷。將腫瘤組織滴至箔條上,且添加液體N2
。將另一箔條經放於腫瘤組織頂部上且用研杵錘擊直至腫瘤徹底磨碎為止。將50至100 mg之腫瘤組織置放於導管(Fishers Scientific,目錄號02-681-302)中且置放於乾冰上。將一種金屬珠粒(Qiagen目錄號69989)及含有內部標準物13
C5
-腺苷、13
C5
-AMP、15
N5
-GTP、15
N4
-肌苷5'-單磷酸及13
C-15
N-次黃嘌呤(Cambridge Isotope Lab及Cayman Chemical)的1 mL 80%甲醇添加至導管中,且在-80℃下儲存樣本直至用於LC/MS分析為止。
亦將血液收集至肝素管中且用於將13
C5
-15
N5-AMP轉化為經標記腺苷、肌苷及次黃嘌呤的離體分析,如針對使用自經由經口給藥經受化合物治療之動物收集之全血的離體分析所描述。
如表3中所示,在用不同劑量之化合物治療之Calu6腫瘤中,5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮抑制AMP轉化為腺苷。 
*:統計顯著性。
T 細胞抑制分析
將羧基螢光素二乙酸丁二醯亞胺基酯(CFSE)用作標記藥劑人類PBMC,或經分離CD4細胞(0.5 × 106
-1 × 108
個細胞)用含有5% HI FBS (Gibco # 10082)之標記緩衝液(RPMI 1640 w/L-麩醯胺酸,GIBCO目錄號11875)洗滌且懸浮於1 ml標記緩衝液中。將細胞與含有50 µM CFSE (Biolegend目錄號423801)之110 µl PBS混合,且在室溫下培育5 min。經標記細胞用含有5% HI FBS (Gibco # 10082)之PBS洗滌一次且用含有1×青黴素/鏈黴素之T細胞正常生長介質(X-Vivo 15,Lonza,目錄號04-744Q)洗滌一次,且懸浮於用於PMBC及CD4細胞之生長介質中。
T 細胞活化及處理
將經CFSE標記之人類PBMC (600,000個細胞/毫升)或CD4細胞(500,000個細胞/毫升)與Dynabeads®人類T-活化子CD3/CD28 (Gibco,目錄號11131D)以細胞/珠粒及人類IL-2 (60 IU/ml,Roche,目錄號:11011456001)之1:1比率混合。將細胞置放於37℃水浴中10分鐘以預活化T細胞。將PBMC (125微升/孔)或CD4細胞(100微升/孔)轉移至96孔培養盤(Costar 3799, Corning Inc.)。為使用PBMC測試化合物,在含有400-600 µM AMP之細胞正常生長介質(125 µL)中製備呈各種濃度之測試化合物。為使用CD4細胞測試化合物,在含有200-250 µM AMP之細胞正常生長介質(100 µL)中製備呈各種濃度之測試化合物。在37℃、5% CO2
下培養經處理細胞68-70小時。
將含有經處理PBMC之介質(160 µl)轉移至培養盤且將用於細胞介素分析之剩餘介質轉移至AcroPrep 96培養盤(AcroPrep 96過濾器板,Omega 3K NTRL 350 ul孔,Pall Life Science目錄號8033)以用於製備用於代謝物之LC-MS分析的樣本。
將含有CD4細胞之介質(100 µl)轉移至另一培養盤且經由以1500 g離心2小時經由AcroPrep培養盤過濾至另一培養盤。將經過濾介質(50 µl/孔)與相同體積之80%甲醇、1% NH4
OH及內部標準物(250 ng/mL13
C5-AMP及250 ng/mL13
C5-腺苷)混合以用於代謝物之LC-MS分析。
經處理PBMC或CD4細胞經收集且針對流式細胞測量術染色以定量T細胞增殖。
流式細胞測量術
PBMC或CD4細胞在DPBS中以1:200稀釋用Zombie Aqua (Biolegend,目錄號423102,批號B195875)染色15 min,且隨後在冰上在流式染色緩衝液(DPBS-2%HIFBS-0.5%BSA)中以1:5稀釋用人類Fc受體結合抑制劑(eBioscience目錄號14-9161-73)阻斷於15分鐘。用APC/Cy7抗人類CD3 (Biolegend目錄號300426,1:20稀釋)、APC抗人類CD4 (Biolegend,目錄號317416,1:20稀釋)及Pacific Blue抗人類CD8a (Biolegend,目錄號300927,1:20稀釋)之混合液染色PBMC,且在冰上用含APC抗人類CD4 (Biolegend,目錄號317416,1:20稀釋)之流式染色緩衝液中染色CD4細胞30分鐘。
藉由BD FACS Verse獲得流式資料。適當地補償各螢光通道。用以下選通策略在FloJo版本7.6.5上處理資料:在FSC相對於SSC點陣圖上選通的淋巴球;在SSC相對於Zombie Aqua點陣圖上選通的淋巴球之活細胞;在APC-Cy7相對於Zombie Aqua點陣圖上選通的可存活淋巴球之CD3細胞;在APC相對於Pacific Blue點陣圖上選通的CD4細胞及CD8細胞;在APC相對於CFSE (FITC)及Pacific Blue相對於CFSE (FITC)點陣圖上進一步選通的CD4及CD8細胞。藉由增殖工具分析CD4及CD8細胞之增殖。增殖指數(PI)如總細胞數量/起始細胞數量所限定,且化合物拯救%經計算為(PIw/o AMP w/o cpd
- PIsample
)/(PIw/o AMP w/o cpd
- PIw AMP w/o cpd
)*100。
如表4中所示,藉由5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮來抑制CD73拯救了腺苷介導之T細胞增殖之抑制,其與生長介質中腺苷含量之劑量依賴性降低相關。 
細胞介素分析
來自經處理PBMC之介質用來自細胞介素ELISA套組之1× ELISA/ELISPOT稀釋劑以1:7.5、1:2.5及1:50稀釋,並且基於製造商之手冊使用人類TNF α ELISA Ready-SET-GO!套組(eBioscience目錄號88-7346-22)分析TNFα,使用人類IL-1β ELISA ready-SET-GO!套組(第2代) (eBioscience,目錄號88-7261-22)分析IL-1β,且使用人類IFN γ ELISA ready-SET-GO!套組(eBioscience,目錄號88-7316-22)分析IFNγ。
如表5中所示,藉由5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮來抑制CD73增加了CD4+
T細胞中之TNF-α含量。 
如表6中所示,藉由5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮抑制CD73增加CD4+
T細胞中之TNF-γ含量。 
活體內模型
視需要,可例如根據此項技術中所闡述之方法進行本發明化合物或其醫藥學上可接受之鹽之CD73抑制作用之臨床前模型化,該等方法例如闡述於Rongvaux A等人,Annual Rev. Immunology 2013
; 31: 635-74及其中所引用之文獻;Sanmamed MF等人,Annals of Oncology 2016
; 27: 1190-1198及其中所引用之文獻中。
用於量測人類血清中之 CD73 活性的 基於 LC/MS 之 分析
在室溫下以1,500g
使新鮮正常人類血液離心15分鐘,且收集含有血清之上層溶離份。將所收集血清(25微升/孔)轉移至含有各種濃度之實例2之化合物及固定濃度之左旋咪唑(1,500 uM)的96深孔培養盤(DWP,Analytical Sales & Services Inc.目錄號968820)。在冰上培育60 min後,將13
C5-15
N5-AMP (50 uM)添加至培養盤中之各孔中,且在室溫下培育培養盤15 min。接著隨著添加200微升/孔之17.3 TCA而將培養盤置放於乾冰上,隨後用培養盤震盪機(Qiagen)以26 fps震盪3分鐘。隨後在4℃下以2940g
使培養盤離心20 min。離心後,將來自各孔之100微升/孔之上澄液轉移至新的96深孔培養盤且在冰上與18.4微升/孔之2.5M Na2
CO3
混合,隨後添加含有內部標準物(為:13
C5-AMP、13
C5-腺苷、13
C5-次黃嘌呤及15
N4-肌苷)之200 ul萃取溶液。在4℃下以2940g
進一步離心20 min後,200微升/孔之上澄液用於藉由如上文所描述之LC/MS進行13
C10-15
N5-腺苷、13
C10-15
N5-肌苷及15
N5-次黃嘌呤之分析。對於EC50計算,用來源於電腦(in silico)模型之未結合溶離份(%)或以實驗方式校正血清中之CD73抑制劑濃度。
表7含有藉由5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮(實例2之化合物)來抑制人類血清中之CD73活性的資料。 
表8含有藉由本文中所揭示之某些化合物來抑制人類血清中之CD73活性的資料。 

Claims (19)
- 一種下式化合物:

- 如請求項1之化合物,其為:

- 如請求項2之化合物,其為:

- 如請求項2之化合物,其為:

- 如請求項2之化合物,其為:

- 如請求項1之化合物,其中n為0,R1為-C1-4烷基,且R2為-CH3,或其醫藥學上可接受之鹽。
- 如請求項6中之化合物,其中R1為異丙基,或其醫藥學上可接受之鹽。
- 如請求項1之化合物,其為



- 如請求項1至8中任一項之化合物,其為:

- 如請求項1至8中任一項之化合物,其為5-[5-[(1S,2R)-2-異丙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
- 如請求項1至8中任一項之化合物,其為:

- 如請求項1至8中任一項之化合物,其為5-[5-[(1S,2S)-2-乙基環丙基]-6-甲基-噠嗪-3-基]-1H-嘧啶-2,4-二酮,或其醫藥學上可接受之鹽。
- 一種醫藥組合物,其包含如請求項1至12中任一項之化合物,或其醫藥學上可接受之鹽,以及一或多種醫藥學上可接受之載劑、稀釋劑或賦形劑。
- 一種用於評定化合物之CD73抑制活性的方法,其包含測定已投與如請求項1至12中任一項之化合物或其醫藥學上可接受之鹽的患者血清中之CD73活性。
- 一種用於評定化合物之CD73抑制活性的方法,其包含測定已投與如請求項1至12中任一項之化合物或其醫藥學上可接受之鹽的患者組織中之CD73表現。
- 一種如請求項1至12中任一項之化合物或其醫藥學上可接受之鹽的用途,其用於製造用於治療患者之癌症的藥物。
- 如請求項16之用途,其中該癌症為膀胱癌、乳癌、膽管癌、直腸癌、結腸癌、胃癌、膽囊癌、神經膠母細胞瘤、頭頸癌、肝癌、肺癌、淋巴瘤、神經管母細胞瘤、黑素瘤、卵巢癌、胰臟癌、前列腺癌或腎癌。
- 如請求項16之用途,其中該患者為已測定血清CD73活性之患者。
- 如請求項16之用途,其中該患者為已測定組織CD73表現之患者。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862636978P | 2018-03-01 | 2018-03-01 | |
| US62/636,978 | 2018-03-01 | ||
| US201862775553P | 2018-12-05 | 2018-12-05 | |
| US62/775,553 | 2018-12-05 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW201945002A TW201945002A (zh) | 2019-12-01 |
| TWI702954B true TWI702954B (zh) | 2020-09-01 |
Family
ID=65686120
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW108105130A TWI702954B (zh) | 2018-03-01 | 2019-02-15 | Cd73抑制劑 |
Country Status (33)
| Country | Link |
|---|---|
| US (1) | US11028074B2 (zh) |
| EP (1) | EP3759096B1 (zh) |
| JP (1) | JP6794560B2 (zh) |
| KR (1) | KR20200116965A (zh) |
| CN (1) | CN111819173B (zh) |
| AU (1) | AU2019228473B2 (zh) |
| BR (1) | BR112020016066A2 (zh) |
| CA (1) | CA3092661C (zh) |
| CL (1) | CL2020002158A1 (zh) |
| CO (1) | CO2020010191A2 (zh) |
| CR (1) | CR20200376A (zh) |
| CY (1) | CY1125145T1 (zh) |
| DK (1) | DK3759096T3 (zh) |
| DO (1) | DOP2020000148A (zh) |
| EC (1) | ECSP20052897A (zh) |
| ES (1) | ES2909701T3 (zh) |
| HR (1) | HRP20220459T1 (zh) |
| IL (1) | IL277006B2 (zh) |
| JO (1) | JOP20200209A1 (zh) |
| LT (1) | LT3759096T (zh) |
| MA (1) | MA52413A (zh) |
| MX (1) | MX2020009115A (zh) |
| PE (1) | PE20210177A1 (zh) |
| PH (1) | PH12020551464A1 (zh) |
| PL (1) | PL3759096T3 (zh) |
| PT (1) | PT3759096T (zh) |
| RS (1) | RS63073B1 (zh) |
| SG (1) | SG11202008366RA (zh) |
| SI (1) | SI3759096T1 (zh) |
| TW (1) | TWI702954B (zh) |
| UA (1) | UA123890C2 (zh) |
| WO (1) | WO2019168744A1 (zh) |
| ZA (1) | ZA202004805B (zh) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20220038453A (ko) * | 2019-08-29 | 2022-03-28 | 일라이 릴리 앤드 캄파니 | Cd73 억제제의 결정질 형태 |
| US20210198239A1 (en) * | 2019-12-06 | 2021-07-01 | Plexxikon Inc. | Compounds and methods for cd73 modulation and indications therefor |
| JP2023522949A (ja) * | 2020-04-23 | 2023-06-01 | オプナ バイオ ソシエテ アノニム | Cd73調節のための化合物及び方法並びにそれらの表示 |
| BR112022020769A2 (pt) | 2020-05-01 | 2022-12-20 | Gilead Sciences Inc | Compostos de 2,4-dioxopirimidina de inibição de cd73 |
| WO2022007677A1 (zh) * | 2020-07-07 | 2022-01-13 | 贝达药业股份有限公司 | Cd73抑制剂及其在医药上的应用 |
| CN116348118A (zh) | 2020-09-08 | 2023-06-27 | 贝达药业股份有限公司 | Cd73抑制剂及其在医药上的应用 |
| CN114315839A (zh) * | 2020-09-30 | 2022-04-12 | 武汉人福创新药物研发中心有限公司 | 嘧啶二酮类化合物及其用途 |
| WO2022090711A1 (en) | 2020-10-26 | 2022-05-05 | AdoRx Therapeutics Limited | Compounds as cd73 inhibitors |
| CN114437038A (zh) * | 2020-11-05 | 2022-05-06 | 武汉人福创新药物研发中心有限公司 | 哒嗪炔烃类化合物及其用途 |
| CN114437039A (zh) | 2020-11-05 | 2022-05-06 | 武汉人福创新药物研发中心有限公司 | Cd73抑制剂及其应用 |
| WO2022121914A1 (zh) * | 2020-12-10 | 2022-06-16 | 上海翰森生物医药科技有限公司 | 氧代氮环类衍生物调节剂、其制备方法和应用 |
| WO2022195499A1 (en) * | 2021-03-19 | 2022-09-22 | Aurigene Discovery Technologies Limited | Substituted pyridazine compounds as cd73 inhibitors |
| CA3235986A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Science, Inc. | Cd73 compounds |
| CN118829639A (zh) * | 2022-03-07 | 2024-10-22 | 贝达药业股份有限公司 | 一种哒嗪类衍生物的晶型、制备方法及其应用 |
| WO2023201267A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| US20240116928A1 (en) * | 2022-07-01 | 2024-04-11 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024013206A1 (en) * | 2022-07-14 | 2024-01-18 | F. Hoffmann-La Roche Ag | Heterocycle compounds for the treatment of cancer |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017120508A1 (en) * | 2016-01-08 | 2017-07-13 | Arcus Biosciences, Inc. | Modulators of 5'-nucleotidase, ecto and the use thereof |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2434337T3 (es) | 2006-04-03 | 2013-12-16 | Glaxo Group Limited | Derivados de azabiciclo[3.1.0]hexilo como moduladores de los receptores D3 de la dopamina |
| EP1860113A1 (en) | 2006-05-24 | 2007-11-28 | Rheinische Friedrich-Wilhelms-Universität Bonn | Ectonucleotidase inhibitors |
| AR105654A1 (es) | 2015-08-24 | 2017-10-25 | Lilly Co Eli | Anticuerpos pd-l1 (ligando 1 de muerte celular programada) |
| WO2017153952A1 (en) * | 2016-03-10 | 2017-09-14 | Glaxosmithkline Intellectual Property Development Limited | 5-sulfamoyl-2-hydroxybenzamide derivatives |
-
2019
- 2019-02-15 TW TW108105130A patent/TWI702954B/zh not_active IP Right Cessation
- 2019-02-22 CN CN201980016287.8A patent/CN111819173B/zh active Active
- 2019-02-22 UA UAA202005110A patent/UA123890C2/uk unknown
- 2019-02-22 CR CR20200376A patent/CR20200376A/es unknown
- 2019-02-22 MX MX2020009115A patent/MX2020009115A/es unknown
- 2019-02-22 PL PL19709335T patent/PL3759096T3/pl unknown
- 2019-02-22 RS RS20220310A patent/RS63073B1/sr unknown
- 2019-02-22 US US16/481,146 patent/US11028074B2/en active Active
- 2019-02-22 BR BR112020016066-0A patent/BR112020016066A2/pt not_active IP Right Cessation
- 2019-02-22 JO JOP/2020/0209A patent/JOP20200209A1/ar unknown
- 2019-02-22 AU AU2019228473A patent/AU2019228473B2/en not_active Ceased
- 2019-02-22 ES ES19709335T patent/ES2909701T3/es active Active
- 2019-02-22 SI SI201930184T patent/SI3759096T1/sl unknown
- 2019-02-22 IL IL277006A patent/IL277006B2/en unknown
- 2019-02-22 PE PE2020001302A patent/PE20210177A1/es unknown
- 2019-02-22 KR KR1020207025071A patent/KR20200116965A/ko active IP Right Grant
- 2019-02-22 CA CA3092661A patent/CA3092661C/en active Active
- 2019-02-22 DK DK19709335.4T patent/DK3759096T3/da active
- 2019-02-22 EP EP19709335.4A patent/EP3759096B1/en active Active
- 2019-02-22 MA MA052413A patent/MA52413A/fr unknown
- 2019-02-22 JP JP2019557361A patent/JP6794560B2/ja active Active
- 2019-02-22 WO PCT/US2019/019074 patent/WO2019168744A1/en active Application Filing
- 2019-02-22 PT PT197093354T patent/PT3759096T/pt unknown
- 2019-02-22 LT LTEPPCT/US2019/019074T patent/LT3759096T/lt unknown
- 2019-02-22 HR HRP20220459TT patent/HRP20220459T1/hr unknown
- 2019-02-22 SG SG11202008366RA patent/SG11202008366RA/en unknown
-
2020
- 2020-07-28 DO DO2020000148A patent/DOP2020000148A/es unknown
- 2020-08-03 ZA ZA2020/04805A patent/ZA202004805B/en unknown
- 2020-08-19 CO CONC2020/0010191A patent/CO2020010191A2/es unknown
- 2020-08-21 CL CL2020002158A patent/CL2020002158A1/es unknown
- 2020-08-31 EC ECSENADI202052897A patent/ECSP20052897A/es unknown
- 2020-09-01 PH PH12020551464A patent/PH12020551464A1/en unknown
-
2022
- 2022-04-12 CY CY20221100276T patent/CY1125145T1/el unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017120508A1 (en) * | 2016-01-08 | 2017-07-13 | Arcus Biosciences, Inc. | Modulators of 5'-nucleotidase, ecto and the use thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI702954B (zh) | Cd73抑制劑 | |
| JP6820254B2 (ja) | 治療用化合物およびその使用 | |
| JP6911031B2 (ja) | 免疫調節剤としての複素環化合物 | |
| EP3468965B1 (en) | 1-tetrahydropyranylcarbonyl-2,3-dihydro-1h-indole compounds for treating cancer | |
| JP6995623B2 (ja) | Lsd1阻害剤としてのヘテロ環式化合物 | |
| JP7402167B2 (ja) | アルギナーゼ阻害剤 | |
| AU2020340299B2 (en) | Crystalline forms of a CD73 inhibitor | |
| EP3697786B1 (en) | Substituted pyrrolopyridines as inhibitors of activin receptor-like kinase | |
| TW202313602A (zh) | Axl化合物 | |
| JP7117029B2 (ja) | 2-置換ピラゾールアミノ-4-置換アミノ-5-ピリミジンホルムアミド系化合物、組成物およびその使用 | |
| KR20210135521A (ko) | 피롤로피라졸 유도체 | |
| WO2020221209A1 (zh) | 一种cd73抑制剂,其制备方法和应用 | |
| EA041789B1 (ru) | Ингибиторы cd73 | |
| WO2022002243A1 (zh) | 咪唑并嘧啶类衍生物、其制备方法及其在医药上的应用 | |
| WO2022152140A1 (zh) | 桥杂环基取代的嘧啶类化合物及其制备方法和医药用途 | |
| RU2788628C2 (ru) | Ингибиторы активин-рецептороподобной киназы | |
| EA045742B1 (ru) | Кристаллические формы ингибитора cd73 | |
| EA044500B1 (ru) | Нафтиридиноновые соединения для применения в качестве активаторов t-клеток |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | Annulment or lapse of patent due to non-payment of fees |