TWI690521B - 作為隱花色素調節劑之含有咔唑之醯胺類、胺基甲酸酯類及脲類 - Google Patents
作為隱花色素調節劑之含有咔唑之醯胺類、胺基甲酸酯類及脲類 Download PDFInfo
- Publication number
- TWI690521B TWI690521B TW104111004A TW104111004A TWI690521B TW I690521 B TWI690521 B TW I690521B TW 104111004 A TW104111004 A TW 104111004A TW 104111004 A TW104111004 A TW 104111004A TW I690521 B TWI690521 B TW I690521B
- Authority
- TW
- Taiwan
- Prior art keywords
- carbazol
- hydroxypropyl
- difluoro
- compound
- aryl
- Prior art date
Links
- 0 CC(C)COC(NC(*)C(O)=O)=O Chemical compound CC(C)COC(NC(*)C(O)=O)=O 0.000 description 9
- GSRASLAJJIMZQV-UHFFFAOYSA-N O=C(c1ccccc1)N(CCC1(F)F)C1=O Chemical compound O=C(c1ccccc1)N(CCC1(F)F)C1=O GSRASLAJJIMZQV-UHFFFAOYSA-N 0.000 description 2
- RBIJPMMJKZSNPP-UHFFFAOYSA-N C=C(CC1)NC1=O Chemical compound C=C(CC1)NC1=O RBIJPMMJKZSNPP-UHFFFAOYSA-N 0.000 description 1
- JTYYWQRSEPVEHP-UHFFFAOYSA-N CC(CCN)NC Chemical compound CC(CCN)NC JTYYWQRSEPVEHP-UHFFFAOYSA-N 0.000 description 1
- JQXZBJAAOLPTKP-LURJTMIESA-N C[C@@H](CN)NC(OC(C)(C)C)=O Chemical compound C[C@@H](CN)NC(OC(C)(C)C)=O JQXZBJAAOLPTKP-LURJTMIESA-N 0.000 description 1
- YVIVRJLWYJGJTJ-SCSAIBSYSA-N C[C@H](CC1)NC1=O Chemical compound C[C@H](CC1)NC1=O YVIVRJLWYJGJTJ-SCSAIBSYSA-N 0.000 description 1
- AZRBAHRGYUSOJN-UHFFFAOYSA-N NCc(cccc1)c1NCc1ccccc1 Chemical compound NCc(cccc1)c1NCc1ccccc1 AZRBAHRGYUSOJN-UHFFFAOYSA-N 0.000 description 1
- JPHLZZHHKOBKQH-GFCCVEGCSA-N O=C(CCCBr)N([C@H](Cc1ccccc1)CO1)C1=O Chemical compound O=C(CCCBr)N([C@H](Cc1ccccc1)CO1)C1=O JPHLZZHHKOBKQH-GFCCVEGCSA-N 0.000 description 1
- RICNFGUUMAJPAW-UHFFFAOYSA-N O=C1NCCCC1F Chemical compound O=C1NCCCC1F RICNFGUUMAJPAW-UHFFFAOYSA-N 0.000 description 1
- BEERPRMSTNISRY-KCXAZCMYSA-N O[C@@H](CN([C@@H]1C[C@H]2CC1)C2=O)C[n]1c(ccc(F)c2)c2c2cc(F)ccc12 Chemical compound O[C@@H](CN([C@@H]1C[C@H]2CC1)C2=O)C[n]1c(ccc(F)c2)c2c2cc(F)ccc12 BEERPRMSTNISRY-KCXAZCMYSA-N 0.000 description 1
Images






















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/88—Carbazoles; Hydrogenated carbazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/118—Prognosis of disease development
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T436/00—Chemistry: analytical and immunological testing
- Y10T436/14—Heterocyclic carbon compound [i.e., O, S, N, Se, Te, as only ring hetero atom]
- Y10T436/142222—Hetero-O [e.g., ascorbic acid, etc.]
- Y10T436/143333—Saccharide [e.g., DNA, etc.]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Neurology (AREA)
- Genetics & Genomics (AREA)
- Analytical Chemistry (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Molecular Biology (AREA)
- Physics & Mathematics (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Immunology (AREA)
- Psychiatry (AREA)
- Physical Education & Sports Medicine (AREA)
- Pathology (AREA)
- Obesity (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
Abstract
本文之標的為結構式I之含有咔唑之醯胺、胺基甲酸脂、及脲衍生物及其醫藥上可接受之鹽或水合物,其中變數R1、R2、R3、R4、R5、R6、R7、A、D、E、G、J、L、M、Q、a、及b係相應地說明。亦提供含有式I化合物之醫藥組成物以治療Cry媒介性疾病或失調,諸如糖尿病、與糖尿病有關之併發症、庫欣氏症候群(Cushing’s syndrome)、NASH、NAFLD、氣喘、及COPD。
Description
本申請案主張2014年4月7日提出之美國暫時專利申請案案號61/976,350(U.S.Provisional Application No.61/976,350)之優先權,該暫時申請案乃整體併入本文中以供參考。
本文揭示之標的尤其係關於含有咔唑之醯胺、胺基甲酸脂、及脲衍生物,含這些化合物之醫藥組成物,彼等用於治療隱花色素媒介性疾病或失調之方法,及彼等之製備方法。亦提供診斷、偵檢、或監測接受本文所揭示之化合物及組成物之個體的隱花色素依賴性疾病之進展的方法。
日夜生理時鐘為內在之計時機轉,其控制許多生理過程之每日節律,諸如腄眠/覺醒行為、體溫、荷爾蒙分
泌、及代謝(Takahashi,J.S.et al.Nat.Rev.Genet.2008,9,764;Green,C.B.et al.Cell,2008,134,728;Zhang,E.E.et al.Nat.Rev.Mol.Cell.Biol.2010,11,764)。日夜節律係以細胞自主方式經由時鐘基因之轉錄調控網路而產生。於核心回饋迴路中,轉錄因子CLOCK及BMAL1活化Period(Per1及Per2)及隱花色素(Cry1及Cry2)基因。轉譯及核定位後,PER及CRY蛋白抑制CLOCK-BMAL1的功能,導致持續之節律基因表現。許多生理路徑係在日夜生理時鐘的控制之下(Panda,S.et al.Cell,2002,109,307),包括許多肝過程的直接調控(Rey,G.et al.PLoS Biol.2011,9,e1000595;Bugge,A.et al.Genes Dev.2012,26,657)。
日夜節律紊亂係與胰島素敏感性的受損有關(Spiegel,K.et al.J.Appl.Physiol.2005,99,2008;Spiegel,K.et al.Lancet,1999,354,1435),降低瘦素濃度且導致可與糖尿病前期相比之高血糖症、高胰島素血症及飯後葡萄糖反應(Scheer,F.A.et al.Proc.Natl.Acad.Sci.USA,2009,106,4453)。一些全基因組關聯研究發現Cry2在哺乳動物葡萄糖濃度的調控中可能是重要的(Dupuis,J.et al.Nat.Genet.2010,42,105;Liu,C.et al.PLoS One,2011,6,e21464;Barker,A.et al.Diabetes,2011,60,1805)。
血液中葡萄糖濃度因為內分泌胰腺之胰島素敏感性及胰島素分泌能力的變化而為高度節律性的(Polonsky,K.S.et al.N.Engl.J.Med.1988,318,1231)。Clock△19突變小
鼠發展成年齡依賴性高血糖症且這些動物亦發展對飲食誘導性肥胖症的易感性,具有不適當低濃度的胰島素(Turek,F.W.et al.Science,2005,308,1043)且因應胰島素治療而顯現更急速之血糖下降,表示這些動物的胰島素敏感性增強,因而掩蔽彼等之β細胞缺乏(Marcheva,B.et al.Nature,2010,466,627)。小鼠Bmal1之肝特異性刪除導致葡萄糖耐受性受損及胰島素敏感性提高(Lamia,K.A.et al.Proc.Natl.Acad.Sci.USA,2008,105,15172)。第2型糖尿病的個體且甚至其尚未被疾病影響的一等親顯現葡萄糖耐受性節律性的改變(Boden,G.et al.Diabetes,1999,48,2182)。又,與無疾病之人類相比,第2型糖尿病病人之Per2、Per3、及Cry2的表現顯著較低(Stamenkovich,J.A.et al.Metabolism,2012,61,978)。糖質新生基因磷酸烯醇丙酮酸羧化激酶(Pck1)及葡萄糖6-磷酸酶(G6pc)係被CRY及Bmal1基因調控子REV-ERB所控制(Zhang,E.E.et al.Nat.Med.2010,16,1152;Lamia,K.A.et al.Nature,2011,480,552;Yin,L.et al.Science,2007,318,1786)。糖質新生作用係被多重信號機轉所牢牢控制,而且於小鼠之研究顯示,Cry1及Cry2之調節可擾亂糖質新生作用及調控血糖濃度(Zhang,E.E.et al.Nat.Med.2010,16,1152)。
於單一療法或組合療法之上下文中,新穎且確立之口服抗糖尿病劑具有不均勻且受限之有效性。口服抗糖尿病療法受不良且受限之血糖控制所苦,或者因為不可接受之
副作用諸如水腫、體重增加、或者甚至嚴重併發症如低血糖症而導致病患順從性不佳。二甲雙胍(metformin,一種經取代雙胍)可導致腹瀉及胃腸不適。最後,水腫、體重增加、且一些情況下,肝毒性及心臟毒性已與一些噻二唑-2,4-二酮抗糖尿病劑(例如羅格列酮(Rosiglitazone)及吡格列酮(Pioglitazone))之投予有關。使用二或多種上述藥劑的組合療法是常見的,但通常僅漸增地改善血糖之控制。
Cry1及Cry2亦與葡萄糖皮質素受體(GR)交互作用以全面地改變對葡萄糖皮質素的轉錄反應(Lamia,K.A.et al.Nature,2011,480,552)。Cry1及/或Cry2的喪失導致葡萄糖耐受不良及本質地高濃度之循環皮質酮,顯示下視丘-腦下垂體-腎上腺軸線的抑制作用降低連同肝內葡萄糖皮質素轉活化作用提高。在基因方面,Cry1及Cry2以荷爾蒙依賴性方式與Pck1啟動子中的葡萄糖反應元件有關,而Pck1基因之地塞米松(dexamethasone)誘導性轉錄作用於缺乏隱花色素的肝中顯著地提高。此顯示用於抑制發炎之葡萄糖皮質素的不期望代謝副作用(例如迥血糖症、胰島素抗性及腎上腺功能之抑制)可藉由將彼等與可使Cry1及/或Cry2安定化之藥劑組合而減輕。
本文之標的係關於隱花色素(Cry)調節性化合物,含Cry調節性化合物之醫藥組成物及藉投予Cry調節性化合物以治療Cry相關性疾病或失調,諸如糖尿病、肥胖症、
代謝症候群、庫欣氏症候群(Cushing’s syndrome)及青光眼之方法。
一方面,本文揭示之標的係關於式I化合物:

或其醫藥上可接受之鹽或水合物,其中A、D、E、G、J、L、M、及Q各自獨立地為N或C;當A、D、E、G、J、L、M、或Q為C時,R1及R2各自獨立地選自H、鹵基、氰基、硝基、-CF3、-CHF2、-CH2F、三氟甲氧基、疊氮基、羥基、(C1-C6)烷氧基、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R8、-(C=O)-O-R8、-O-(C=O)-R8、-NR8(C=O)-R10、-(C=O)-NR8R9、-NR8R9、-NR8OR9、-S(O)cNR8R9、-S(O)d(C1-C8)烷基、-O-SO2-R8、NR8-S(O)c、-(CR8R9)d(3-10)員環烷基、-(CR8R9)e(C6-C10)芳基、-(CR8R9)e(4-10)員雜環基、-(CR8R9)f(C=O)(CR8R9)e(C6-C10)芳基、-(CR8R9)f(C=O)(CR8R9)e(4-10)員雜環基、-(CR8R9)eO(CR8R9)f(C6-C10)芳基、
-(CR8R9)eO(CR8R9)f(4-10)員雜環基、-(CR8R9)fS(O)d(CR8R9)e(C6-C10)芳基、及-(CR8R9)fS(O)d(CR8R9)e(4-10)員雜環基;R3及R5各自獨立地選自H、氰基、-CF3、-CHF2、-CH2F、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R8、-(C=O)-O-R8、-(C=O)-NR8R9、-S(O)cNR8R9、-S(O)d(C1-C8)烷基、-(CR8R9)d(3-10)員環烷基、-(CR8R9)e(C6-C10)芳基、-(CR8R9)e(4-10)員雜環基、-(CR8R9)f(C=O)(CR8R9)e(C6-C10)芳基、-(CR8R9)f(C=O)(CR8R9)e(4-10)員雜環基、-(CR8R9)eO(CR8R9)f(C6-C10)芳基、-(CR8R9)eO(CR8R9)f(4-10)員雜環基、-(CR8R9)fS(O)d(CR8R9)e(C6-C10)芳基、及-(CR8R9)fS(O)d(CR8R9)e(4-10)員雜環基;其中R3基各自隨意地彼此鍵聯成4-12員單環或雙環;其中R5基各自隨意地彼此鍵聯成4-12員單環或雙環;R4為H、-CF3、-CHF2、-CH2F、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R8、-(C=O)-O-R8、-(C=O)-NR8R9、-(CR8R9)d(3-10)員環烷基、-(CR8R9)e(C6-C10)芳基、-(CR8R9)e(4-10)員雜環基、-(CR8R9)f(C=O)(CR8R9)e(C6-C10)芳基、-(CR8R9)f(C=O)(CR8R9)e(4-10)員雜環基、
-(CR8R9)eO(CR8R9)f(C6-C10)芳基、-(CR8R9)eO(CR8R9)f(4-10)員雜環基、-CR8R9)fS(O)d(CR8R9)e(C6-C10)芳基、及-(CR8R9)fS(O)d(CR8R9)e(4-10)員雜環基;其中R6及R7彼此鍵聯成4-12員單環或雙環;R8、R9及R10各自獨立地選自H、(C1-C6)烷基、-(CR11R12)e(3-10)員環烷基、-(CR11R12)g(C6-C10)芳基、及-(CR11R12)g(4-10)員雜環基;前述R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、R15、及R16之(C1-C6)烷基、(3-10)員環烷基、(C6-C10)芳基及(4-10)員雜環基之任何碳原子獨立地隨意地經1至3個R14取代基取代,該R14取代基各自獨立地選自鹵基、氰基、硝基、-CF3、-CHF2,-CH2F、三氟甲氧基、疊氮基、羥基、-O-R15,(C1-C6)烷氧基、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R11、-(C=O)-R15、-(C=O)-O-R11、-(C=O)-O-R15、-O-(C=O)-R11、-O-(C=O)-R15、-NR11(C=O)-R13、-(C=O)-NR11R12、-(C=O)-NR11R15、-NR11R12、-NR11R15、-NR11OR12、-NR11OR15、-S(O)cNR11R12、-S(O)cNR11R15、-S(O)d(C1-C6)烷基、-S(O)dR15、-O-SO2-R11、-O-SO2-R15、-NR11-S(O)c、-NR15-S(O)c、-(CR11R12)e(3-10)員環烷基、-(CR11R12)e(C6-C10)芳基、-(CR11R12)e(4-10)員雜環基、-(CR11R12)f(C=O)(CR11R12)e(C6-C10)芳基、
-(CR11R12)f(C=O)(CR11R12)e(4-10)員雜環基、-(CR11R12)eO(CR11R12)f(C6-C10)芳基、-(CR11R12)eO(CR11R12)f(4-10)員雜環基、-(CR11R12)fS(O)d(CR11R12)e(C6-C10)芳基、及-(CR11R12)fS(O)d(CR11R12)e(4-10)員雜環基;前述R14之(C1-C6)烷基、(3-10)員環烷基、(C6-C10)芳基及(4-10)員雜環基之任何碳原子獨立地隨意地經1至3個R16取代基取代,該R16取代基各自獨立地選自鹵基、氰基、硝基、-CF3、-CHF2、-CH2F、三氟甲氧基、疊氮基、(CH2)eOH、(C1-C6)烷氧基、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R11、-(C=O)-R15、-(C=O)-O-R11、-(C=O)-O-R15、-O-(C=O)-R11、-O-(C=O)-R15、-NR11(C=O)-R13、-(C=O)-NR11R12、-NR11R12、及-NR11R15;前述R1、R2、R3、R4、R5、R6、R7,、R8、R9、R10、R14、及R15之(4-10)員雜環基之任何氮原子獨立地隨意地經(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R11、-(C=O)-O-R11、-(C=O)-NR11R12、-(CR11R12)e(3-10)員環烷基、-(CR11R12)e(C6-C10)芳基、-(CR11R12)e(4-10)員雜環基、-(CR11R12)f(C=O)(CR11R12)e(C6-C10)芳基、或-(CR11R12)f(C=O)(CR11R12)e(4-10)員雜環基取代;R11、R12、及R13各自獨立地為H或(C1-C6)烷基;R15為-(CR11R12)e(3-10)員環烷基、-(CR11R12)e(C6-C10)芳基、或-(CR11R12)e(4-10)員雜環基;
a及b各自獨立地為1、2、3、或4;c為1或2;d為0、1、或2;且e、f、及g各自獨立地為0、1、2、3、4、或5。
一些實施例中,A、D、E、G、J、L、M、及Q各自為C;R1及R2各自獨立地選自H或鹵基;R4為H或(C1-C6)烷基,R3及R5為H;R6及R7彼此鍵聯成4-12員單環或雙環狀醯胺環;R8、R9、R10、R11、R12、R13、R14、R15、R16、a、b、c、d、e、及f為如同本文中所定義者。
其他實施例中,A、D、E、G、J、L、M、及Q各自為C;R1及R2各自獨立地選自H或鹵基;R4為H或(C1-C6)烷基,R3及R5為H;R6及R7彼此鍵聯成4-12員單環或雙環狀脲環;R8、R9、R10、R11、R12、R13、R14、R15、R16、a、b、c、d、e、及f為如同本文中所定義者。
一些實施例中,式I化合物為在C-3帶有(R)-組態之單一鏡像異構物,其中A、D、E、G、J、L、M、及Q各自為C;R1及R2各自獨立地選自H或鹵基;R4為H或(C1-C6)烷基,R3及R5為H;R6及R7彼此鍵聯成4-12員單環或雙環狀醯胺環;R8、R9、R10、R11、R12、R13、R14、R15、R16、a、b、c、d、e、及f為如同本文中所定義者。
本文揭示之標的的其他實施例中,式I化合物為在C-3帶有(R)-組態之單一鏡像異構物,其中A、D、E、G、J、L、M、及Q各自為C;R1及R2各自獨立地選自H或
鹵基;R4為H或(C1-C6)烷基,R3及R5為H;R6及R7彼此鍵聯成4-12員單環或雙環狀脲環;R8、R9、R10、R11、R12、R13、R14、R15、R16、a、b、c、d、e、及f為如同本文中所定義者。
本文揭示之標的的其他實施例為選自由以下所組成之群組的化合物:1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;2-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;(1R,4S)-2-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;(R)-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;(R)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;(S)-1-((S)-3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-4-甲基咪唑啶-2-酮;或其醫藥上可接受之鹽或水合物。
另一方面,本文所述之化合物可調節Cry1或Cry2。
Cry1或Cry2之調節包括下列之任一者:結合至Cry1或Cry2;抑制Cry1或Cry2之修飾;改變Cry1或Cry2之局部化;提高或降低Cry1或Cry2之安定化;提高或降低Cry1或Cry2至標靶間的結合;提高或降低Cry1或Cry2之活性;及提高或降低Cry1或Cry2標靶之活性。Cry1及/或Cry2之標靶包括(但不限定於)Per1、Per2、葡萄糖皮質素受體(GR)、CLOCK、BMAL1、或CLOCK-BMAL1啟動子序列。
另一方面,本文所述之標的係提供醫藥組成物,其包含式I化合物、或其醫藥上可接受之鹽或水合物,及醫藥上可接受之載體、佐劑、或稀釋劑。一些實施例中,醫藥組成物進一步包含一或多種額外之治療劑。額外治療劑之實例包括(但不限定於)DPP-IV抑制劑諸如西他列汀(sitagliptin)、阿格列汀(alogliptin)、維格列汀(vildagliptin)、沙克列汀(saxagliptin)及利拉列汀(linagliptin);GLP-1激動劑諸如艾塞那肽(exenatide)、利拉魯肽(liraglutide)及阿必魯肽(albiglutide);SGLT2抑制劑諸如卡那利福心(canagliflozin)、爾土利福心(ertugliflozin)及多百利福心(dapagliflozin);二甲雙胍(metformin);及磺醯脲類諸如格列本脲(glyburide)。其他額外治療劑之實例包括Signifor®、酮康唑(ketoconazole)、美替拉酮(metyrapone)、米托坦(mitotane)、依托咪酯(etomidate)、Korlym®、表皮生長因子抑制劑、醛固酮合成酶/11β-羥化酶抑制劑LCI699、及
左旋酮康唑(levoketoconazole,COR-003)。
另一方面係提供治療個體之Cry媒介性疾病或失調的方法,係將治療有效量之本文所述之醫藥組成物投予該個體。另一方面,本發明提供減輕個體之Cry媒介性疾病或失調的症狀之方法,係藉將治療有效量之本文所述之醫藥組成物投予該個體。疾病或失調可選自由以下所組成之群組:糖尿病、糖尿病併發症諸如糖尿病神經病變、糖尿病視網膜病變、糖尿病腎病變、白內障形成、青光眼、糖尿病血管病變、動脈粥狀硬化;非酒精性脂肪肝炎(NASH);非酒精性脂肪肝疾病(NAFLD);氣喘;慢性阻塞性肺病(COPD);代謝症候群;胰島素抗性症候群;肥胖症、青光眼、庫欣氏症候群(Cushing’s syndrome);精神病性憂鬱症;阿滋海默氏症;神經病性疼痛;藥物濫用;骨質疏鬆症;癌症;黃斑部病變;及肌病變。
本文所述之任何方法亦可包含將一或多種額外之治療劑投予該個體。額外治療劑之實例包括(但不限定於)DPP-IV抑制劑諸如西他列汀(sitagliptin)、阿格列汀(alogliptin)、維格列汀(vildagliptin)、沙克列汀(saxagliptin)及利拉列汀(linagliptin);GLP-1激動劑諸如艾塞那肽(exenatide)、利拉魯肽(liraglutide)及阿必魯肽(albiglutide);SGLT2抑制劑諸如卡那利福心(canagliflozin)、爾土利福心(ertugliflozin)及多百利福心(dapagliflozin);二甲雙胍(metformin);磺醯脲類諸如格列本脲(glyburide);Signifor®、酮康唑(ketoconazole)、美
替拉酮(metyrapone)、米托坦(mitotane)、依托咪酯(etomidate)、Korlym®、表皮生長因子抑制劑、醛固酮合成酶/11β-羥化酶抑制劑LCI699、及左旋酮康唑(levoketoconazole,COR-003)。
另一方面提供監測個體之Cry媒介性疾病或失調的進展或預後之方法,其包含量測第一段時間取自個體之第一樣品中的一或多種隱花色素或隱花色素調節基因之有效量;量測第二段時間取自個體之第二樣品中的一或多種隱花色素或隱花色素調節基因之有效量;將第一樣品中偵檢出之一或多種隱花色素或隱花色素調節基因的量與第二樣品中偵檢出之一或多種隱花色素或隱花色素調節基因的量,或與參考值相比較。隱花色素調節基因之實例包括於彼等之啟動子中含有E-box序列之基因。此些基因包括(但不限定於)Dbp、Rev-erb α、Rev-erb β、Ror α、Ror β、Ror γ、Per1、Per2、Per3、Cry1、Cry2、Pck1、G6Pc、Avp、Vip、Cck、SP(物質P)、AA-Nat、PK2(前動力蛋白2,Prokinectin 2)、c-Myc、MyoD及Nampt。
一些實施例中,該監測包含評估在該個體發展成該Cry媒介性疾病或失調之風險的變化。
用於給藥予人類之最適時間預期為傍晚,相當於人類Cry表現之高峰及活性(白天)期的結束。
該個體可包含先前已治療該Cry媒介性疾病或失調者,先前尚未治療該Cry媒介性疾病或失調者,或先前尚未被診斷為患有該Cry媒介性疾病或失調者。樣品可為全
血、血清、血漿、血球、內皮細胞、活體組織、淋巴液、腹水、組織間液、骨髓、腦脊髓液(CSF)、精液、唾液、黏液、痰、汗、或尿。
一些實施例中,該第一樣品係於對個體治療該Cry媒介性疾病或失調之前取自該個體且該第二樣品係於對個體治療該Cry媒介性疾病或失調之後取自該個體。其他實施例中,該個體係以含本文揭示之式I化合物的醫藥組成物治療。某些實施例中,該監測進一步包含選擇對該個體之治療法及/或監測對該Cry媒介性疾病或失調之治療法的有效性,其中該對該Cry媒介性疾病或失調之治療法包含進行手術、單獨地投予本文所定義之醫藥組成物或與一或多種額外治療劑組合投予、於單獨地投予本文所定義之醫藥組成物或與一或多種額外治療劑組合投予之後或之前進行手術、或未採取進一步行動。
其他實施例中,該參考值包含指標值、衍生自一或多種Cry媒介性疾病或失調風險預測演算法之值、衍生自未患有Cry媒介性疾病或失調之個體的值、或衍生自被診斷為患有Cry媒介性疾病或失調之個體的值。一些實施例中,該量測包含偵檢是否有該一或多種隱花色素,定量該一或多種隱花色素之量,定性該一或多種隱花色素之類型,及評估一或多種隱花色素結合至標靶之能力。該標靶可為Per1、Per2、或CLOCK-BMAL1啟動子序列。
如同本文所揭示地,Cry媒介性疾病或失調選自由以下所組成之群組:糖尿病、糖尿病併發症諸如糖尿病神經
病變、糖尿病視網膜病變、糖尿病腎病變、白內障形成、青光眼、糖尿病血管病變、動脈粥狀硬化;非酒精性脂肪肝炎(NASH);非酒精性脂肪肝疾病(NAFLD);氣喘;慢性阻塞性肺病(COPD);代謝症候群;胰島素抗性症候群;肥胖症、青光眼、庫欣氏症候群(Cushing’s syndrome);精神病性憂鬱症;阿滋海默氏症;神經病性疼痛;藥物濫用;骨質疏鬆症;癌症;黃斑部病變;及肌病變。
一實施例中,於本文揭示之式I化合物中,A、D、E、G、J、L、M、及Q為碳。另一實施例中,於式I化合物中,R1及R2為氫。又另一實施例中,於式I化合物中,R1及R2為氟,且a及b為1。其他實施例中,於式I化合物中,R3及R5為氫。另一實施例中,於式I化合物中,R3、R4、及R5為氫。一些實施例中,於式I化合物中,R6及R7鍵聯形成隨意經取代之單環。
其他實施例中,於式I化合物中,R6及R7鍵聯形成隨意經取代之稠合雙環、隨意經取代之橋聯雙環、隨意經取代之螺雙環、隨意經取代之吡咯啶酮環、隨意經取代之咪唑啶酮環、隨意經取代之哌啶酮環、及/或隨意經取代之嘧啶酮環。同樣地,這些實施例之任一者中,該藉由R6及R7所形成之環只經氟、甲基、乙基、異丙基、C3-6環烷或苯基取代。
除非另有定義,否則本文所用之所有技術及科學術語均具有與本發明有關之熟知技藝者一般理解者相同之定義。雖然與本文所述者類似或同等之方法及材料可用於本
發明之操作中,但適當之方法及材料乃述於下。所有公開、專利申請案、專利、及本文所述之其他參考資料乃整體明確地併入以供參考。如有抵觸,則將由本專利說明書(包括定義)來控制。此外,本文所述之材料、方法、及實例僅供闡述而非意在限制。
本發明之其他特性及優點將經由下列詳述之說明書及申請專利範圍而顯見。
圖1A-H為將化合物72投予小鼠後,顯現核心時鐘基因表現的一系列圖。核心時鐘基因Per2(A及B)、Bmal1(C及D)、Cry1(E及F)、及Cry2(G及H)之mRNA表現係於24小時期間以六小時為間隔地於以載劑(水)或化合物72處理之C57Bl/6J DIO(A、C、E、G)或Balb/c(B、D、F、H)小鼠的肝內量測。轉錄量係藉RT-qPCR測定且與ZT8時之載劑相比較,伴隨著將暗期繪成陰影。將每一時點之得自經化合物72處理之mRNA量與載劑藉T-檢定相比較:*<0.05,**<0.01,***<0.001,****<0.0001。
圖2A-D為將化合物72投予小鼠後,顯現糖質新生基因表現的一系列圖。糖質新生基因Pck1(PEPCK;A及B)、G6Pc(葡萄糖6-磷酸酶催化亞單位;C及D)係於24小時期間以六小時為間隔地於以載劑(水)或化合物72處理之C57Bl/6J DIO(A及C)或Balb/c(B及D)小鼠的肝內
量測。轉錄量係藉RT-qPCR測定且與ZT8時之載劑相比較,伴隨著將暗期繪成陰影。將每一時點之得自經化合物72處理之mRNA量與載劑藉T-檢定相比較:*<0.05,**<0.01,***<0.001,****<0.0001。
圖3A-C為將化合物72、化合物48、化合物9、或化合物57投予ICR小鼠後,於肝內顯現核心時鐘基因表現的一系列圖。核心時鐘基因Per2(A),Bmal1(B),and Cry2(C)之mRNA表現係於以化合物72、化合物48、化合物9、化合物57、或載劑處理4天BID之ICR小鼠之肝內量測。mRNA量係藉將最終ZT0劑量後於ZT6取出之樣品進行RT-qPCR測定。將每一時點之得自經化合物72處理之mRNA量與載劑藉T-檢定相比較:*<0.05,**<0.01,***<0.001,****<0.0001。
圖4為於Cry1表現之高峰時刻投予化合物72之三次每日劑量後顯現Dbp基因表現的圖。Dbp mRNA的表現係於三次100毫克/公斤化合物72之每日劑量後、於ZT7.5、於db/db小鼠之血液中量測。轉錄量係藉RT-qPCR測定且與取自ZT7.5之經載劑(10% kolliphor)處理之小鼠的血液相比較。將每一種化合物處理之mRNA量與載劑藉T-檢定相比較:(***;p 0.001)。
0.001)。
圖5A-D為於Cry1表現之高峰或最底點時刻投予單次劑量之化合物72後顯現核心時鐘基因表現的一系列圖。核心時鐘基因Per2(A)、Bmal1(B)、Cry1(C)及Cry2(D)之mRNA表現係於100毫克/公斤化合物72之單次劑
量後、於ZT7.5(Cry1表現之高峰)或ZT17.5(Cry1表現之最底點)於C57Bl/6J DIO小鼠之肝內量測。轉錄量係藉RT-qPCR測定且與經載劑(10% kolliphor)處理之肝相比較。將每一時點之得自經化合物72處理之mRNA量與載劑藉T-檢定相比較:*<0.05,**<0.01,***<0.001,****<0.0001。
圖6為顯現以db/db小鼠進行之化合物72對葡萄糖耐受試驗(OGTT)之效應的一系列圖。化合物72(50毫克/公斤,口服)或10% kolliphor(對照組)係以單次劑量方式於Cry1及Bmal1基因表現之高峰(ZT0)(A)或最底點(ZT10)(B)時刻投予。
圖7為顯現以db/db小鼠進行之化合物72對葡萄糖曲線下面積(AUC)之效應的圖。化合物72(50毫克/公斤,口服)或10% kolliphor(對照組)係以單次劑量方式於Cry1及Bmal1基因表現之高峰(ZT0)時刻投予。
圖8A-C為顯現以db/db小鼠進行之化合物72投予7天對葡萄糖代謝之效應的一系列圖。化合物72(50毫克/公斤,口服(PO))或10% kolliphor(對照組)係投予7天。A)空腹血液葡萄糖濃度,B)口服葡萄糖耐受試驗(OGTT);C)葡萄糖AUC。
圖9A-B為顯現以db/db小鼠進行之化合物72投予7天對胰島素濃度之效應的一系列圖。化合物72(50毫克/公斤,口服(PO))或10% kolliphor(對照組)經投予。A)葡萄糖負荷之前(於0小時)及之後(於2小時)之胰島素濃
度;B)經穩態模型評估法評估之胰島素抗性(HOMA-IR)。
圖10為顯現投予最後一次劑量(50毫克/公斤,口服(PO))後約8小時於血漿及肝中量測之化合物72的化合物濃度之圖。化合物72於Per2分析法中之EC50濃度在圖上以虛線標示。
圖11A-C為顯現以db/db小鼠進行之提高劑量之化合物72(10毫克/公斤,50毫克/公斤,及100毫克/公斤)對葡萄糖代謝之效應的一系列圖。10% kolliphor係用於作為載劑對照組。A)空腹血液葡萄糖濃度;B)OGTT;C)葡萄糖AUC。
圖12A-B為顯現以db/db小鼠進行之提高劑量之化合物72(10毫克/公斤,50毫克/公斤,及100毫克/公斤)對胰島素濃度之效應的一系列圖。10% kolliphor係用於作為載劑對照組。A)葡萄糖負荷之前(於0小時)及之後(於2小時)之胰島素濃度;B)經穩態模型評估法評估之胰島素抗性(HOMA-IR)。
圖13為顯現於以提高之劑量投予最後一次劑量(10毫克/公斤,50毫克/公斤,及100毫克/公斤))後約8小時於血漿及肝中量測之化合物72的化合物濃度之圖。化合物72於Per2分析法中之EC50濃度在圖上以虛線標示。
圖14A-C為顯現以db/db小鼠進行之提高劑量之化合物9(30毫克/公斤,100毫克/公斤,及300毫克/公斤)對葡萄糖代謝之效應的一系列圖。10% kolliphor係用於作為對照組。A)空腹血液葡萄糖濃度;B)OGTT;C)葡萄糖
AUC。
圖15A-B為顯現以db/db小鼠進行之各種劑量之化合物9(30毫克/公斤,100毫克/公斤,及300毫克/公斤)對胰島素濃度之效應的一系列圖。10% kolliphor係用於作為對照組。A)葡萄糖負荷之前(於0小時)及之後(於2小時)之胰島素濃度;B)經穩態模型評估法評估之胰島素抗性(HOMA-IR)。
圖16為顯現於以提高之劑量投予最後一次劑量(30毫克/公斤,100毫克/公斤,及300毫克/公斤)後約8小時於血漿及肝中之化合物9的化合物濃度之圖。化合物9於Per2分析法中之EC50濃度在圖上以虛線標示。
圖17A-C為顯現以C57/B16J DIO小鼠進行之化合物72對葡萄糖代謝之效應的圖。化合物72(100毫克/公斤,口服(PO))、10% kolliphor(對照組)、或羅格列酮(rosiglitazone,30毫克/公斤)係投予7天。A)空腹血液葡萄糖濃度,B)OGTT;C)葡萄糖AUC。
圖18A-B為顯現以C57/B16J DIO小鼠進行之化合物72對胰島素濃度之效應的一系列圖。化合物72(100毫克/公斤,口服(PO))、10% kolliphor(對照組)、或羅格列酮(rosiglitazone,30毫克/公斤)係投予7天。A)葡萄糖負荷之前(於0小時)及之後(於2小時)之胰島素濃度;B)經穩態模型評估法評估之胰島素抗性(HOMA-IR)。
圖19為顯現化合物72對皮質酮誘導性胰島素抗性之大鼠模型的效應之一系列圖。皮質酮(30毫克/公斤,皮下
(SC))係連同載劑、化合物72(50毫克/公斤,口服(PO))或美服培酮(mifepristone,30毫克/公斤,口服(PO))一起投服7天。(A)空腹血漿葡萄糖濃度及(B)空腹血漿胰島素濃度。
圖20為顯現以皮質酮誘導性胰島素抗性之大鼠模型進行之化合物72(50毫克/公斤,口服(PO))投予7天對HOMA-IR之效應的圖。皮質酮(30毫克/公斤,皮下(SC))係連同載劑、化合物72(50毫克/公斤,口服(PO))或美服培酮(mifepristone,30毫克/公斤,口服(PO))一起投服7天。
圖21為顯現化合物72對CRY1 FAD-結合性結構域之體外熱安定性之效應的圖。如同藉差示掃描螢光(“熱漂移”)分析法所測得地,將純化之CRY1 FAD-結合性結構域以化合物72處理可導致蛋白質熔化溫度之劑量依賴性提高。
圖22A-C為顯現以DIO小鼠進行之提高劑量之化合物72(10毫克/公斤,30毫克/公斤,及100毫克/公斤)對葡萄糖代謝之效應的一系列圖。化合物72(100毫克/公斤,口服(PO))、10% kolliphor(對照組)、或美服培酮(mifepristone,30毫克/公斤,口服(PO))係投服7天。A)空腹血液葡萄糖濃度;B)OGTT;C)葡萄糖AUC。
本文本中引述之每一申請案及專利,以及每一申請案
及專利中引用之每一文件或參考資料(包括在每一公告專利的審查期間;“申請案引用文件”),及相對應這些申請案及專利之任一者或由這些申請案及專利之任一者中請求優先權的每一美國及外國申請案或專利,及每一申請案引用文件中引用或提及之每一文件,均明確地併入本文中以供參考。更通常地,本文本所引用之文件或參考資料,無論是申請專利範圍之前的參考資料列表,或是文本本身;以及這些文件或參考資料之每一者(“本文引用之參考資料”)、以及每一本文引用參考資料所引用之每一文件或參考資料(包括任何製造商的規格、教示等等)均明確地併入本文中以供參考。併入本文本中以供參考的文件可用於本發明之操作中。整個本專利說明書中所述之特性、結構、或特徵可以任何適當方式組合於一或多個實施例中。例如,整個專利說明書中之詞組“例示實施例”、“實例實施例”、“一些實施例”或其他類似語言文字所意指的事實為,所述之與實施例有關之特定特性、結構、或特徵可包括於本文中所述之至少一個實施例中。故,整個專利說明書中之詞組“例示實施例”、“實例實施例”、“一些實施例中”、“其他實施例中”或其他類似語言文字的出現並不必然地全意指同一實施例群組,且所述之特性、結構、或特徵可以任何方式組合於一或多個實施例中。
欲促進對本說明書的了解,一些術語乃定義於下。本文所定義之術語具有熟諳本文所述標的領域者一般已理解的定義。術語諸如“a”、“an”及“the”不意在僅意指單一實
體,而係包括可使用具體實例闡述之一般類別者。本文之專門術語係用於說明本文所述標的之具體實施例,但其用法並未界定於標的,除非於申請專利範圍中概述。
本文中所用之述語“包含”、“包括”、或“具有”係以其開放、非限制性意義被使用。
除非另有指定,否則本文中所用之述語“鹵基”意指氟基、氯基、溴基、或碘基。
除非另有指定,否則本文中所用之述語“烷基”包括具有直或支鏈部分之飽和單價烴基。
除非另有指定,否則本文中所用之述語“烯基”表示含一或多個碳-碳雙鍵之具2至6個碳的單價直或支鏈基,其實例為乙烯基、1-丙烯基、2-丙烯基、2-甲基-1-丙烯基、1-丁烯基、2-丁烯基等等。
除非另有指定,否則本文中所用之述語“炔基”表示含碳-碳三鍵之具二至六個碳原子的單價直或支鏈基,其實例為乙炔基、1-丙炔基等等。
除非另有指定,否則本文中所用之述語“烷氧基”表示O-烷基,其中烷基為如上所定義者。
術語“Me”意指甲基,且“Et”意指乙基。
除非另有指定,否則本文中所用之述語“環烷基”意指含有共3至10個碳原子之非芳族、飽和或部分飽和、單環或稠合、螺或未稠合雙環或三環烴。環烷基之實例衍生自(但不限定於)下列者:

除非另有指定,否則本文中所用之述語“芳基”包括衍生自芳族烴藉由移除一個氫而得之有機基團,諸如苯基或萘基。
除非另有指定,否則本文中所用之述語“(4-12)員雜環基”包括含有一至四個各自選自O、S、及N之雜原子的芳族及非芳族雜環基,其中每一雜環基於其環中具有4-12個原子,且前提是該基團之環並不含有兩個相鄰之O或S原子。非芳族雜環基包括於其環系統中僅具有3個原子之基團,但芳族雜環基必需於其環系統中具有至少5個原子。雜環基包括苯並稠合環系統。3員雜環基之實例為吖環丙烷,4員環雜環基之實例為四氫吖唉基(衍生自四氫吖唉)。5員環雜環基之實例為噻唑基,7員環之實例為氮雜環庚三烯基且10員雜環基之實例為喹啉基。非芳族雜環基之實例為吡咯啶基、四氫呋喃基、二氫呋喃基、四氫噻吩基、四氫哌喃基、二氫哌喃基、四氫噻喃釷、1-哌啶基、N- 啉基、N-硫代
啉基、N-硫代 啉基、噻
啉基、噻 烷基、哌
烷基、哌 基、四氫吖唉基、氧雜環丁烷基、硫雜環丁烷基、高哌啶基、氧雜環庚烷基、硫雜環庚烷基、氧氮雜環庚三烯基、二氮雜環庚三烯基、硫氮雜環庚三烯基、1,2,3,6-四氫吡啶基、2-吡咯啉基、3-吡咯啉基、吲哚啉基、2H-哌喃基、4H-哌喃
基、二
基、四氫吖唉基、氧雜環丁烷基、硫雜環丁烷基、高哌啶基、氧雜環庚烷基、硫雜環庚烷基、氧氮雜環庚三烯基、二氮雜環庚三烯基、硫氮雜環庚三烯基、1,2,3,6-四氫吡啶基、2-吡咯啉基、3-吡咯啉基、吲哚啉基、2H-哌喃基、4H-哌喃
基、二 烷基、1,3-二
烷基、1,3-二 基、吡唑啉基、二噻烷基、二噻
基、吡唑啉基、二噻烷基、二噻 基、二氫哌喃基、二氫噻吩基、二氫呋喃基、吡唑啶基、咪唑啉基、咪唑啶基、3-氮雜雙環[3.1.0]已烷基、3-氮雜雙環[4.1.0]庚烷基、3H-吲哚基及喹
基、二氫哌喃基、二氫噻吩基、二氫呋喃基、吡唑啶基、咪唑啉基、咪唑啶基、3-氮雜雙環[3.1.0]已烷基、3-氮雜雙環[4.1.0]庚烷基、3H-吲哚基及喹 基。芳族雜環基之實例為吡啶基、咪唑基、嘧啶基、吡唑基、三唑基、吡
基。芳族雜環基之實例為吡啶基、咪唑基、嘧啶基、吡唑基、三唑基、吡 基、四唑基、呋喃基、噻吩基、異
基、四唑基、呋喃基、噻吩基、異 唑基、噻唑基、
唑基、噻唑基、 唑基、異噻唑基、吡咯基、喹啉基、異喹啉基、吲哚基、苯並咪唑基、苯並呋喃基、
唑基、異噻唑基、吡咯基、喹啉基、異喹啉基、吲哚基、苯並咪唑基、苯並呋喃基、 啉基、吲唑基、吲哚
啉基、吲唑基、吲哚 基、酞
基、酞 基、嗒
基、嗒 基、三
基、三 基、異吲哚基、喋啶基、嘌呤基、
基、異吲哚基、喋啶基、嘌呤基、 二唑基、噻二唑基、呋咕基、苯並呋咕基、苯並噻吩基、苯並噻唑基、苯並
二唑基、噻二唑基、呋咕基、苯並呋咕基、苯並噻吩基、苯並噻唑基、苯並 唑基、喹唑啉基、喹
唑基、喹唑啉基、喹 啉基、萘啶基及呋喃並吡啶基。前述衍生自以上列表之基團當有可能時,可為C-連接或N-連接。例如,衍生自吡咯之基團可為吡咯-1-基(N-連接)或吡咯-3-基(C-連接)。此外,衍生自咪唑之基團可為咪唑-1-基(N-連接)或咪唑-3-基(C-連接)。4-12員雜環可於任一環碳、硫或氮原子上隨意地每環經一或兩個側氧基取代。其中2個環原子經側氧基部分所取代之雜環基的實例為1,1-二側氧基-硫代
啉基、萘啶基及呋喃並吡啶基。前述衍生自以上列表之基團當有可能時,可為C-連接或N-連接。例如,衍生自吡咯之基團可為吡咯-1-基(N-連接)或吡咯-3-基(C-連接)。此外,衍生自咪唑之基團可為咪唑-1-基(N-連接)或咪唑-3-基(C-連接)。4-12員雜環可於任一環碳、硫或氮原子上隨意地每環經一或兩個側氧基取代。其中2個環原子經側氧基部分所取代之雜環基的實例為1,1-二側氧基-硫代 啉基。4-12員雜環之其他實例衍生自(但不限定於)下列者:
啉基。4-12員雜環之其他實例衍生自(但不限定於)下列者:

本文所用之述語“經取代”意指指定原子上之任一或多個氫原子經選定之指定基團替代,前提是不超過該指定原子之常價,且該取代導致安定化合物之產生。當取代基為酮基(亦即=O)時,則原子上的2個氫原子被替代。酮基取代基不存在於芳族部分上。本文所用之環雙鍵為在兩個相鄰環原子間形成的雙鍵(例如C=C、C=N或N=N)。此基團之非限制性實例包括(但非限制)H、CH3、NO2、SO2N(CH3)2、SO2N((CH3)SO2)、COOH、COOCH3、CO(N(CH3))、烷基、烯基、炔基、芳基、芳烷基、環烷基、雜環基、烷芳基、雜芳基、雜環烷基、烷氧基(亦即甲氧基、乙氧基等等)、烷羰氧基、芳羰氧基、烷氧基羰氧基、芳氧基羰氧基、羧酸基、烷羰基、烷胺羰基、芳烷胺羰基、烯胺羰基、烷羰基、芳羰基、芳烷羰基、烯羰基、烷氧羰基、胺羰基、烷硫羰基、三氟甲基、五氟乙基、鹵素(亦即氯基、氟基、溴基、碘基)、氰基、硫基、醯胺基、醚、酯、羥基、羥烷基、飽和或不飽和脂肪酸、疊氮基、磷醯胺基、磺醯胺基、內醯胺、磷酸基、膦酸
基、亞膦酸基、胺基(包括烷胺基、二烷胺基、芳胺基、二芳胺基及烷芳胺基)、醯胺基(包括烷羰胺基、芳羰胺基、胺基甲醯基及脲基)、甲脒基、亞胺基、胍基、氫硫基、烷硫基、芳硫基、硫代羧酸基、硫酸基、烷亞磺醯基、磺酸基、胺磺醯基、磺醯胺基、硝基、氰基、疊氮基等等。
本文揭示之標的係提供可調節一或多種隱花色素分子之含咔唑的磺醯胺化合物。這些化合物具有式I所示之一般結構:

或其醫藥上可接受之鹽或水合物,其中A、D、E、G、J、L、M、及Q各自獨立地為N或C;當A、D、E、G、J、L、M、或Q為C時,R1及R2各自獨立地選自H、鹵基、氰基、硝基、-CF3、-CHF2、-CH2F、三氟甲氧基、疊氮基、羥基、(C1-C6)烷氧基、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R8、-(C=O)-O-R8、-O-(C=O)-R8、-NR8(C=O)-R10、
-(C=O)-NR8R9、-NR8R9、-NR8OR9、-S(O)cNR8R9、-S(O)d(C1-C8)烷基、-O-SO2-R8、NR8-S(O)c、-(CR8R9)d(3-10)員環烷基、-(CR8R9)e(C6-C10)芳基、-(CR8R9)e(4-10)員雜環基、-(CR8R9)f(C=O)(CR8R9)e(C6-C10)芳基、-(CR8R9)f(C=O)(CR8R9)e(4-10)員雜環基、-(CR8R9)eO(CR8R9)f(C6-C10)芳基、-(CR8R9)eO(CR8R9)f(4-10)員雜環基、-(CR8R9)fS(O)d(CR8R9)e(C6-C10)芳基、及-(CR8R9)fS(O)d(CR8R9)e(4-10)員雜環基;R3及R5各自獨立地選自H、氰基、-CF3、-CHF2、-CH2F、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R8、-(C=O)-O-R8、-(C=O)-NR8R9、-S(O)cNR8R9、-S(O)d(C1-C8)烷基、-(CR8R9)d(3-10)員環烷基、-(CR8R9)e(C6-C10)芳基、-(CR8R9)e(4-10)員雜環基、-(CR8R9)f(C=O)(CR8R9)e(C6-C10)芳基、-(CR8R9)f(C=O)(CR8R9)e(4-10)員雜環基、-(CR8R9)eO(CR8R9)f(C6-C10)芳基、-(CR8R9)eO(CR8R9)f(4-10)員雜環基、-(CR8R9)fS(O)d(CR8R9)e(C6-C10)芳基、及-(CR8R9)fS(O)d(CR8R9)e(4-10)員雜環基;其中R3基各自隨意地彼此鍵聯成4-12員單環或雙環;其中R5基各自隨意地彼此鍵聯成4-12員單環或雙
環;R4為H、-CF3、-CHF2、-CH2F、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R8、-(C=O)-O-R8、-(C=O)-NR8R9、-(CR8R9)d(3-10)員環烷基、-(CR8R9)e(C6-C10)芳基、-(CR8R9)e(4-10)員雜環基、-(CR8R9)f(C=O)(CR8R9)e(C6-C10)芳基、-(CR8R9)f(C=O)(CR8R9)e(4-10)員雜環基、-(CR8R9)eO(CR8R9)f(C6-C10)芳基、-(CR8R9)eO(CR8R9)f(4-10)員雜環基、-CR8R9)fS(O)d(CR8R9)e(C6-C10)芳基、及-(CR8R9)fS(O)d(CR8R9)e(4-10)員雜環基;其中R6及R7彼此鍵聯成4-12員單環或雙環;R8、R9及R10各自獨立地選自H、(C1-C6)烷基、-(CR11R12)e(3-10)員環烷基、-(CR11R12)g(C6-C10)芳基、及-(CR11R12)g(4-10)員雜環基;前述R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、R15、及R16之(C1-C6)烷基、(3-10)員環烷基、(C6-C10)芳基及(4-10)員雜環基之任何碳原子獨立地隨意地經1至3個R14取代基取代,該R14取代基各自獨立地選自鹵基、氰基、硝基、-CF3、-CHF2,-CH2F、三氟甲氧基、疊氮基、羥基、-O-R15,(C1-C6)烷氧基、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R11、-(C=O)-R15、-(C=O)-O-R11、-(C=O)-O-R15、-O-(C=O)-R11、-O-(C=O)-R15、-NR11(C=O)-R13、
-(C=O)-NR11R12、-(C=O)-NR11R15、-NR11R12、-NR11R15、-NR11OR12、-NR11OR15、-S(O)cNR11R12、-S(O)cNR11R15、-S(O)d(C1-C6)烷基、-S(O)dR15、-O-SO2-R11、-O-SO2-R15、-NR11-S(O)c、-NR15-S(O)c、-(CR11R12)e(3-10)員環烷基、-(CR11R12)e(C6-C10)芳基、-(CR11R12)e(4-10)員雜環基、-(CR11R12)f(C=O)(CR11R12)e(C6-C10)芳基、-(CR11R12)f(C=O)(CR11R12)e(4-10)員雜環基、-(CR11R12)eO(CR11R12)f(C6-C10)芳基、-(CR11R12)eO(CR11R12)f(4-10)員雜環基、-(CR11R12)fS(O)d(CR11R12)e(C6-C10)芳基、及-(CR11R12)fS(O)d(CR11R12)e(4-10)員雜環基;前述R14之(C1-C6)烷基、(3-10)員環烷基、(C6-C10)芳基及(4-10)員雜環基之任何碳原子獨立地隨意地經1至3個R16取代基取代,該R16取代基各自獨立地選自鹵基、氰基、硝基、-CF3、-CHF2、-CH2F、三氟甲氧基、疊氮基、(CH2)eOH、(C1-C6)烷氧基、(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R11、-(C=O)-R15、-(C=O)-O-R11、-(C=O)-O-R15、-O-(C=O)-R11、-O-(C=O)-R15、-NR11(C=O)-R13、-(C=O)-NR11R12、-NR11R12、及-NR11R15;前述R1、R2、R3、R4、R5、R6、R7,、R8、R9、R10、R14、及R15之(4-10)員雜環基之任何氮原子獨立地隨意地經(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(C=O)-R11、
-(C=O)-O-R11、-(C=O)-NR11R12、-(CR11R12)e(3-10)員環烷基、-(CR11R12)e(C6-C10)芳基、-(CR11R12)e(4-10)員雜環基、-(CR11R12)f(C=O)(CR11R12)e(C6-C10)芳基、或-(CR11R12)f(C=O)(CR11R12)e(4-10)員雜環基取代;R11、R12、及R13各自獨立地為H或(C1-C6)烷基;R15為-(CR11R12)e(3-10)員環烷基、-(CR11R12)e(C6-C10)芳基、或-(CR11R12)e(4-10)員雜環基;a及b各自獨立地為1、2、3、或4;c為1或2;d為0、1、或2;且e、f、及g各自獨立地為0、1、2、3、4、或5。
式I化合物之例示實施例中,A、D、E、G、J、L、M、及Q各自為C;R1及R2各自獨立地選自H或鹵基;R4為H或(C1-C6)烷基,R3及R5為H;R6及R7彼此鍵聯成4-12員單環或雙環狀醯胺環;R8、R9、R10、R11、R12、R13、R14、R15、R16、a、b、c、d、e、及f為如同本文中所定義者。
一些實施例中,A、D、E、G、J、L、M、及Q各自為C;R1及R2各自獨立地選自H或鹵基;R4為H或(C1-C6)烷基,R3及R5為H;R6及R7彼此鍵聯成4-12員單環或雙環狀脲環;R8、R9、R10、R11、R12、R13、R14、R15、R16、a、b、c、d、e、及f為如同本文中所定義者。
本文所揭示之標的的一些實施例中,式I化合物為在C-3帶有(R)-組態之單一鏡像異構物,其中A、D、E、
G、J、L、M、及Q各自為C;R1及R2各自獨立地選自H或鹵基;R4為H或(C1-C6)烷基,R3及R5為H;R6及R7彼此鍵聯成4-12員單環或雙環狀醯胺環;R8、R9、R10、R11、R12、R13、R14、R15、R16、a、b、c、d、e、及f為如同本文中所定義者。
其他實施例中,式I化合物為在C-3帶有(S)-組態之單一鏡像異構物,其中A、D、E、G、J、L、M、及Q各自為C;R1及R2各自獨立地選自H或鹵基;R4為H或(C1-C6)烷基,R3及R5為H;R6及R7彼此鍵聯成4-12員單環或雙環狀脲環;R8、R9、R10、R11、R12、R13、R14、R15、R16、a、b、c、d、e、及f為如同本文中所定義者。
某些實施例中,化合物可選自由以下所組成之群組:1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;2-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;(1R,4S)-2-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;(R)-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;(R)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;
(S)-1-((S)-3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-4-甲基咪唑啶-2-酮;或其醫藥上可接受之鹽或水合物。
本文所用之術語“醫藥上可接受”意指材料諸如載體或稀釋劑,其不會使本文所述化合物之生物活性或性質消失,且為相對無毒性的,亦即該材料可投予個體而不會導致不期望的生物效應或以有害方式與組成物內所含之任何組份交互作用。
本文所用之術語“醫藥上可接受之鹽”意指鹽,其保留特定化合物之游離酸及鹼之生物有效性且並非生物學或其他方面不期望者。式I化合物之醫藥上可接受之鹽包括其酸加成及鹼鹽。適當之酸加成鹽係由可形成無毒性鹽之酸中形成。實例包括乙酸鹽、己二酸鹽、阿拉伯半乳聚糖磺酸鹽、抗壞血酸鹽、天冬胺酸鹽、苯甲酸鹽、苯磺酸鹽、碳酸氫鹽/碳酸鹽、硫酸氫鹽/硫酸鹽、硼酸鹽、樟腦磺酸鹽、膽酸鹽、檸檬酸鹽、乙二磺酸鹽、依托酸鹽(estolate)、乙磺酸鹽、甲酸鹽、富馬酸鹽、半乳糖醛酸鹽、葡庚糖酸鹽、葡萄糖酸鹽、葡萄糖醛酸鹽、麩胺酸鹽、六氟磷酸鹽、海苯酸鹽、馬尿酸鹽、氫氯酸鹽/氯化物、氫溴酸鹽/溴化物、氫碘酸鹽/碘化物、3-羥基-2-萘甲酸鹽、1-羥基-2-萘甲酸鹽、羥乙磺酸鹽、乳酸鹽、乳糖醛酸鹽、蘋果酸鹽、馬來酸鹽、丙二酸鹽、苦杏仁酸鹽、甲
磺酸鹽、甲基硫酸鹽、黏酸鹽、萘二磺酸鹽、萘甲酸鹽、2-萘磺酸鹽、烟酸鹽、硝酸鹽、油酸鹽、乳清酸鹽、草酸鹽、棕櫚酸鹽、巴莫酸鹽、磷酸鹽/磷酸氫鹽/磷酸二氫鹽、醣二酸鹽、柳酸鹽、硬脂酸鹽、琥珀酸鹽、磺柳酸鹽、酒石酸鹽、甲苯磺酸鹽、三氟乙酸鹽及色胺酸鹽。
適當之鹼鹽係由可形成無毒性鹽之鹼中形成。實例包括腺嘌呤、鋁、2-胺基-2-甲基丙-1-醇、精胺酸、苯乙苄胺、苄星、鈣、膽鹼、胞嘧啶、二乙胺、二醇胺、伊泊胺(epolamine)、三級丁胺(erbumine)、乙二胺、葡萄糖胺、甘胺酸、胍、鳥嘌呤、海巴明(hydrabamine)、離胺酸、鎂、甲葡胺、 啉、菸鹼醯胺、乙醇胺、鳥胺酸、哌
啉、菸鹼醯胺、乙醇胺、鳥胺酸、哌 、鉀、普羅卡因(procaine)、脯胺酸、吡哆醇、絲胺酸、銀、鈉、三乙醇胺、緩血酸胺、酪胺酸、纈胺酸及鋅鹽。欲回顧適當之鹽,請參見“Handbook of Pharmaceutical Salts:Properties,Selection,and Use”by Stahl and Wermuth(Wiley-VCH,Weinheim,Germany,2002)。
、鉀、普羅卡因(procaine)、脯胺酸、吡哆醇、絲胺酸、銀、鈉、三乙醇胺、緩血酸胺、酪胺酸、纈胺酸及鋅鹽。欲回顧適當之鹽,請參見“Handbook of Pharmaceutical Salts:Properties,Selection,and Use”by Stahl and Wermuth(Wiley-VCH,Weinheim,Germany,2002)。
式I化合物之醫藥上可接受之鹽可藉將式I化合物溶液視情況與期望之酸或鹼一起混合而輕易製得。鹽可由溶液中沈澱出且藉過濾法收集或者可藉將溶劑蒸發而復收。鹽中之離子化程度可由完全離子化至幾乎未離子化間變化。
式I化合物亦可以各種結晶形式存在,稱之為多形體。多形體包括化合物之相同元素組成物的不同結晶充填排列。多形體可具有不同之X光繞射圖式、紅外線光譜、
熔點、密度、硬度、結晶形狀、光學及電性質、安定性、溶劑合物及溶解度。各種因素諸如再結晶溶劑、結晶速率、及貯存溫度可導致單一結晶形式佔有優勢。
“溶劑合物”欲意指特定化合物之醫藥上可接受之溶劑合物形式,其保留此化合物之生物有效性。溶劑合物之實例包括本發明化合物與水、異丙醇、乙醇、甲醇、二甲亞碸、乙酸乙酯、乙酸、或乙醇胺組合。術語“水合物”意指其中溶劑為水之溶劑合物。術語“醇化物”其中溶劑為醇之溶劑合物。水合物係藉將一或多分子之水與一分子之物質組合而形成,其中水保留其為H2O之分子狀態。水合物之非限制性實例包括單水合物、二水合物等等。
本發明化合物包括本文所定義之式I化合物、其多形體、前藥、及異構物(包括光學、幾何、及互變異構物)以及同位素標記之式I化合物。
本發明化合物可以前藥形式投予。故式I化合物之某些衍生物(其本身具有鮮少或無藥理活性)當投予體內或投予體上時,可轉化成具有期望活性之式I化合物,例如藉水解裂解作用轉化。此衍生物稱為‘前藥’。前藥使用方面的進一步訊息可見於“Pro-drugs as Novel Delivery Systems,Vol.14,ACS Symposium Series(T.Higuchi and W.Stella)及“Bioreversible Carriers in Drug Design”,Pergamon Press,1987(Ed.E.B.Roche,American Pharmaceutical Association)中。前藥可(例如)藉將式I化合物中之存在之適當官能性以熟諳此藝者已知之某些部分
替代而製得,該某些部分為例如如同於H.Bundgaard之“Design of Prodrugs”(Elsevier,1985)中所述之‘前部分,。
此前藥之一些實例包括當式I化合物含有羧酸官能性(-CO2H)時,為其酯,例如氫以(C1-C8)烷基替代;當式I化合物含有醇官能性(-OH)時,為其醚,例如氫以(C1-C8)烷醯氧基甲基替代;且當式I化合物含有二級胺基官能性(-NHR,其中R不為H)時,為其醯胺,例如一個氫以(C1-C10)烷醯基替代。根據前述實例之替代基的進一步實例及其他前藥型式之實例為熟諳此藝者已知。
式I化合物含有一或多個不對稱碳原子。應該理解的是,對應於式I化合物之所有鏡像異構物及/或非鏡像異構物可藉類似之方法製得。式I化合物之所有光學異構物及立體異構物及其混合物被視為在本發明之範圍內。有關於式I化合物方面,本發明包括消旋物、一或多種鏡像異構物形式、一或多種非鏡像異構物形式、或其混合物之用途。式I化合物亦可以互變異構物形式存在。本發明關於所有此些互變異構物及其混合物之用途。
本發明化合物內所含之某些官能基可取代生物電子等排基團,亦即具有類似於母基團之空間或電子要求,但顯現不同或改善之物理化學或其他性質之基團。適當實例為熟諳此藝者已知且包括(但不限定於)Patini,et al.Chem Rev.1996,96,3147-3176及其中引用之參考資料中所述之部分。
包括在所請求式I化合物範圍內者為醫藥上可接受之
酸加成鹽或鹼鹽,其中該相對離子為光學活性,例如D-乳酸鹽或L-離胺酸,或消旋性例如DL-酒石酸鹽或DL-精胺酸。順式/反式異構物可藉熟諳此藝者詳知之慣用技術例如層析法及分段結晶法分離。用於製備/離析出個別鏡像異構物之慣用技術包括由適當光學活性先質中進行手性合成或將消旋物(或鹽或衍生物之消旋物)使用(例如)手性高壓液相層析(HPLC)進行離析。
另外,消旋物(或消旋物先質)可與適當之光學活性化合物例如醇或者當式I化合物含有酸性或鹼性部分,則與酸或鹼諸如酒石酸或1-苯基乙胺反應。所得之非鏡像異構物混合物可藉層析法及/或分段結晶法分離且藉熟諳此藝者詳知之方法將非鏡像異構物轉化成相應之純鏡像異構物及/或非鏡像異構物。本發明之手性化合物(及其手性先質)可使用層析法典型地HPLC法以鏡像異構物及/或非鏡像異構物富集形式得到,該層析係於不對稱樹脂上使用由烴組成之流動相,典型地庚烷或己烷,含0至50%、典型地2至20%異丙醇及含0至5%烷胺典型地0.1%二乙胺進行。將洗提液濃縮以得富集混合物。鏡像異構物及/或非鏡像異構物之混合物可藉熟諳此藝者慣用之技術分離。例如參見“Stereochemistry of Organic Compounds”by E.L.Eliel(Wiley,New York,1994)。
式I化合物可為同位素標記的,其中一或多個原子被具有相同原子數但原子質量或質量數不同於自然界通常發現之原子質量或質量數的原子所替代。適於包括在本發明
化合物中的同位素實例包括氫之同位素,諸如2H及3H;碳之同位素,諸如11C、13C及14C;氯之同位素,諸如36Cl;氟之同位素,諸如18F;碘之同位素,諸如123I及125I;氮之同位素,諸如13N及15N;氧之同位素,諸如15O、17O及18O;磷之同位素,諸如32P;及硫之同位素,諸如35S。某些式I同位素標記化合物,例如併入放射活性同位素者,可用於藥物及/或基質組織分佈研究中。放射活性同位素氚(亦即3H)及碳-14(亦即14C)特別可用於供此目的,因其易於併入且檢測方法方便之故。以較重之同位素諸如氘(亦即2H)取代則可能因為較大之代謝安定性,例如提高於體內之半生期或降低劑量需求而得到某些醫療優勢,因此在一些狀況下可能較佳。以正子發射同位素諸如11C、18F、15O及13N取代則可用於正子斷層造影(PET)研究中以檢查基質受體之佔有率。式I之同位素標記化合物通常可藉熟諳技藝者已知之慣用技術或藉類似於附加之實例及製備例中所述之方法,使用適當之同位素標記試劑取代先前使用之未標記試劑製得。
本發明化合物調節Cry1及/或Cry2。本文所用之“調節”意指提高、降低、或改變Cry1及Cry2之功能、活性或內在特徵。Cry1或Cry2之調節包括下列之任一者:結合至Cry1或Cry2;抑制Cry1或Cry2之修飾;改變Cry1或Cry2之局部化;提高或降低Cry1或Cry2之安定化;提高或降低Cry1或Cry2至標靶間的結合;提高或降低Cry1或Cry2之活性;及提高或降低Cry1或Cry2標靶之
活性。
Cry1及Cry2之調節包括本發明化合物經由直接交互作用亦或經由間接交互作用結合至Cry1及/或Cry2。一些方面,本發明化合物可結合至含Cry1及/或Cry2之錯合物。偵檢小分子與蛋白質間之交互作用的方法於技藝中已知,例如免疫沈澱技術、層析法、及各種陣列格式。
Cry1及Cry2之內在特徵諸如轉譯後修飾、安定性、或局部化,可藉由本發明化合物而改變。Cry1及Cry2之轉譯後修飾在決定Cry1及Cry2之活性、安定性、或細胞局部化中扮演關鍵性角色。一些研究已顯示,磷酸化可改變Cry1及Cry2之安定性。本發明化合物可防止或提高Cry1及Cry2之轉譯後修飾,例如磷酸化、泛素化、乙醯化、醣化、核糖基化、或小泛素化。
偵檢Cry1或Cry2之轉譯後修飾的方法可由熟諳此藝者輕易進行。此些偵檢方法包括西方墨點法及放射免疫分析法。Cry1及Cry2於特定條件下,例如於與Per1及Per2異二聚化之後,局部化至核。一旦在核內,Cry1及Cry2在由起始轉錄中破壞核CLOCK-BMAL1複合物扮演關鍵性角色,因此反向調控負回饋迴路中之日夜節律基因,這在保持日夜擺動是重要的。蛋白質之局部化可由熟諳此藝者例如藉免疫螢光法、次細胞分群法及西方墨點法輕易測定。Cry1及Cry2之反向調控對日夜擺動亦具關鍵性,且於轉錄及蛋白質層級媒介。Cry1及Cry2之安定性可藉技藝中已知之方法以及實例5-8呈現者測量。
本文所用之Cry1及Cry2之活性包括Cry1或Cry2至標靶間的結合及下游Cry1或Cry2標靶之活性。本發明化合物可提高或降低Cry1或Cry2至標靶間的結合。結合至Cry1及/或Cry2之標靶為技藝中已知,且包括Per1、Per2、葡萄糖皮質素受體、CLOCK-BMAL1啟動子序列、及VEGF啟動子序列。其他標靶包括其表現被Cry1或Cry2所調節之基因,包括於彼等之啟動子中含有E-box序列之基因。此些基因包括(但未限制)Dbp、Rev-erb α、Rev-erb β、Ror α、Ror β、Ror γ、Per1、Per2、Per3、Cry1、Cry2、Pck1、G6Pc、Avp、Vip、Cck、SP(物質P)、AA-Nat、PK2(前動力蛋白2,Prokinectin 2)、c-Myc、MyoD及Nampt。本文中提及之Cry1及Cry2標靶亦包括彼些尚待鑑定之標靶。
Cry1或Cry2與標靶間的結合可藉(例如)免疫沈澱法、酵母菌雙雜合法、親和層析法測定。Cry1或Cry2標靶之下游活性包括CLOCK-BMAL1媒介性轉錄、Cry1或Cry2結合至CLOCK-BMAL-1啟動子、Cry1或Cry2結合至VEGF啟動子、Per1或Per2局部化或安定性、CLOCK-BMAL1二聚化、CLOCK-BMAL1標靶基因Cry1,Cry2,Per1,Per2,Rev-erb α and β,Rora、TIM蛋白、及VEGF之表現。偵檢啟動子活性之方法可藉如同實例3及4中所述之染色質免疫沈澱法、電泳遷移率變動分析法、或啟動子-螢光素酶分析法測定。測定標靶基因表現之方法包括基因表現分析法及微陣列,其可由熟諳此藝者輕易進行。
一些實施例中,測定推定效力之方法或分析法可用於鑑定適於治療或減輕Cry媒介性疾病或失調症狀之本文所述的特定化合物。一方面,於特定暴露時間後,誘導基線反應與最大反應之間的半程之化合物濃度(本文稱之為EC50值或濃度)可藉體外分析法測量化合物對核心時鐘基因表現的效應而測知。將可操作地連接至核心時鐘基因啟動子序列(亦即Per1、Per2、Cry1、Cry2或Bmal1)之螢光素酶報導子引入以本發明化合物處理之細胞中(亦即轉染、轉導、傳染),再隨著時間測量發光(或時鐘基因傳動表現)。尤其,測定發光之期間、幅度、及相,且與測量與日夜節律有關之預期表現相比。化合物之EC50值可使用熟諳此藝者輕易可得之方法計算出。此分析法之實例述於本文實例3中。EC50值可用於評估本發明化合物之效能。
其他方面,具有提高效力之本發明化合物可藉體內分析法測定。將化合物投予個體(亦即小鼠模型)一段時間。將生物樣品由個體中分離出來,再測量生物樣品中存在之化合物濃度。生物樣品為(例如)全血或其任何部分(亦即血清或血漿)、或組織諸如受Cry-媒介性疾病或失調影響之組織。如同於相關之體外分析法(如上文及實例3中所述地)檢測地,將經處理個體之樣品中偵檢出的濃度與相同化合物之EC50濃度值相比較。所量測於體內之化合物濃度大於所測EC50值的化合物為較佳之本發明化合物。這些較佳化合物可證實具有提高之效力以治療或減輕Cry-媒
介性疾病或失調之症狀。
本文所揭示標的之其他方面,提供醫藥組成物,其包含根據式I之化合物及醫藥上可接受之載體、佐劑或稀釋劑。製備具有特定量活性化合物之各種醫藥組成物的方法為熟諳此藝者已知或將顯而易見。此外,熟諳此藝者熟悉調配及投予技術。此標題將例如於Goodman and Gilman’s“The Pharmaceutical Basis of Therapeutics”,current edition,Pergamon Press;and“Remington’s Pharmaceutical Sciences”,current edition,Mack Publishing,Co.,Easton,PA中討論。這些技術可用於本文所述方法及組成物之適當方面及實施例中。醫藥組成物較佳地於GMP條件下製造。下列實例係提供以僅供闡述之目的而不意在用於限制本發明。
因為本文所述之化合物意在用於醫藥組成物中,應輕易理解的是,彼等各自較佳地以實質地純形式提供,例如至少50%純、至少55%純、至少60%純、至少65%純、至少70%純、至少75%純、至少80%純、至少85%純、至少90%純、至少95%純、至少96%純、至少97%純、至少98%純、或至少99%純度。本文中提供之百分比係以重量計。化合物之不純製劑可用於製備醫藥組成物中所用之更純形式;這些較不純的化合物製劑應含有至少1%、更適當地至少5%,例如10至49%之式I化合物。
式I化合物可以適當之局部、經口、鼻部、眼部、黏膜部、直腸部、陰道部、及非經腸部醫藥配方形式提供以
用於治療Cry媒介性疾病。本發明化合物可以片劑或膠囊、以油性或水性懸浮液、菱形含錠、錠劑、粉末、顆粒、乳膠、糖漿或酏劑形式經口投予。經口使用之組成物包括一或多種用於調味、甜化、著色及防腐之化學劑以求產生醫藥上優雅且可口之製劑。片劑可含有醫藥上可接受之賦形劑、載體、稀釋劑、及佐劑作為此片劑製造中之助劑。如同技藝中慣用的,這些片劑可包以醫藥上可接受之腸溶包衣,諸如甘油單硬脂酸酯或甘油二硬脂酸酯以延緩於胃腸道中之崩解及吸收以提供更長期之持續作用。水難溶性化合物之溶解速率可藉由使用噴霧乾燥分散液諸如述於Takeuchi,H.et al.J.Pharm.Pharmacol.1987,39,769-773者而增強。
經口使用之配方可為硬明膠膠囊形式,其中活性成分係與惰性固體稀釋劑例如碳酸鈣、磷酸鈣或高嶺土混合。彼等亦可為軟明膠膠囊形式,其中活性成分係與水或油介質諸如花生油、液體石蠟或橄欖油混合。
水性懸浮液一般含有活性成分且與適於製造水性懸浮液之賦形劑混合。此賦形劑可為懸浮劑,諸如Kolliphor、羧甲基纖維素鈉、甲基纖維素、羥丙基甲基纖維素、藻酸鈉、聚乙烯基吡咯啶酮、黃蓍膠及金合歡膠;分散或潤濕劑,其可為天然存在之磷脂諸如卵磷脂、環氧乙烷與長鏈脂肪酸之縮合產物諸如聚氧乙烯硬脂酸酯、環氧乙烷與長鏈脂肪醇之縮合產物諸如十七碳乙烯氧鯨蠟醇(heptadecaethyleneoxycetanol)、環氧乙烷與衍生自脂肪酸
及己糖醇之偏酯的縮合產物諸如聚氧乙烯山梨糖醇單油酸酯、或環氧乙烷與衍生自脂肪酸己糖醇酐之偏酯的縮合產物諸如聚氧乙烯山梨糖醇酐單油酸酯。
醫藥組成物可為無菌注射水性或油性懸浮液形式。此懸浮液可根據已知之方法調配成水性等張溶液或懸浮液,而栓劑可由脂肪乳膠或懸浮液中製得。組成物可為無菌及/或含有佐劑諸如防腐劑、安定劑、潤濕劑或乳化劑、溶解促進劑、調控滲透壓之鹽及/或緩衝劑。此外,彼等亦可含有其他有治療價值之物質。無菌注射製劑亦可於無毒素非經腸部可接受之稀釋劑或溶劑中調配成懸浮液形式,例如為溶於1,3-丁二醇中之溶液形式。可接受之載劑及溶劑當中,可使用的為水、林格氏(Ringers)溶液及等張氯化鈉溶液。欲供此目的,任何溫和之固定油可予使用包括合成單或二甘油酯。此外,脂肪酸諸如油酸發現使用於注射製劑中。
式I化合物亦可以栓劑形式投予以供藥物之直腸部投予。這些組成物可藉將藥物與適當無刺激性賦形劑混合而製得,該賦形劑於約25℃為固體但在直腸溫度時為液體,因此會於直腸中溶化而釋出藥物。此材料包括可可油及其他甘油酯。
在局部或經皮使用之製劑方面,例如乃使用含有本發明化合物之乳油、軟膏、膠凍溶液或懸浮液。用於經皮應用之適當配方包括有效量之本發明化合物與載體。載體可包括可吸收之藥理學可接受之溶劑以協助通過主人的皮
膚。例如,經皮裝置為繃帶形式,其包含底撐件、含有本化合物隨意地與載體之儲槽、隨意之速率控制隔層以將化合物以受控制且預定之速率長時間地將化合物遞送至主人的皮膚、及將該裝置固定至皮膚的工具。基質經皮配方及離子電滲裝置亦可使用。用於局部應用例如應用至皮膚及眼睛之適當配方較佳地為技藝中詳知之水性溶液、軟膏、乳油或凝膠。此可含有溶解劑、安定劑、張力增強劑、緩衝劑及防腐劑。
活性化合物可以醫藥上可接受之載體製備,該載體可保護化合物免於由身體快速排除,諸如控釋型配方包括植入物及微型膠囊遞送系統。生物可降解、生物可相容性聚合物可予使用,諸如乙烯乙酸乙烯酯、聚酐、聚乙醇酸、膠原、聚原酸酯、及聚乳酸。製備此些配方之方法對熟諳此藝者是顯而易見的。
式I化合物亦可製備於微脂粒遞送系統形式諸如小單層泡、大單層泡及多層泡中。微脂粒亦可由各種磷脂諸如膽固醇、硬脂醯胺或磷脂醯膽鹼中形成。
活性醫藥成分或二種活性醫藥成分之適當緩釋形式可為基質片劑或膠囊組成物。適當之基質形成材料包括(例如)蠟(例如棕櫚蠟、蜂蠟、石蠟、純地蠟、蟲膠蠟、脂肪酸、及脂肪醇),油、硬化油或脂肪(例如硬化菜籽油、蓖麻油、牛油、棕櫚油、及大豆油),及聚合物(例如羥丙基纖維素、聚乙烯基吡咯啶酮、羥丙基甲基纖維素、及聚乙二醇)。其他適當之基質製片材料為微晶纖維素、粉狀纖
維素、羥丙基纖維素、乙基纖維素、與其他載體、及填料。片劑亦可含有顆粒、包衣粉末、或丸粒。片劑亦可多層化。當諸活性成分具有顯著不同之藥物動力學概況時,則多層化片劑尤佳。完成之片劑可隨意地予以包衣或未包衣。
包衣組成物典型地含有不可溶之基質聚合物(約包衣組成物之15-85重量%)及水溶性材料(約包衣組成物之15-85重量%)。腸溶聚合物(約包衣組成物之1-99重量%)可隨意地使用或包括。適當之水溶性材料包括聚合物諸如聚乙二醇、羥丙基纖維素、羥丙基甲基纖維素、聚乙烯基吡咯啶酮、聚乙烯醇、及單體材料諸如糖類(例如乳糖、蔗糖、果糖、甘露糖醇等等)、鹽類(例如氯化鈉、氯化鉀等等)、有機酸(例如富馬酸、琥珀酸、乳酸、及酒石酸)、及其混合物。適當之腸溶聚合物包括羥丙基甲基纖維素、乙酸琥珀酸酯、羥丙基甲基纖維素、鄰苯二甲酸酯、聚乙酸乙烯鄰苯二甲酸酯、鄰苯二甲酸乙酸纖維素、偏苯三甲酸乙酸纖維素、蟲膠、玉米蛋白、及含羧基之聚甲基丙烯酸酯。
包衣組成物可根據包衣摻合物之性質予以塑化,諸如主要組份或組份混合物之玻璃轉移溫度或用於施用包衣組成物之溶劑。適當之塑化劑可以包衣組成物之0至50重量%加入且包括(例如)鄰苯二甲酸二乙酯、檸檬酸酯、聚乙二醇、甘油、乙醯化甘油酯、乙醯化檸檬酸酯、癸二酸二丁酯、及蓖麻油。如有需要,包衣組成物可包括填料。
填料的量可佔包衣組成物總重之1重量%至約99重量%且可為不可溶材料諸如二氧化矽、二氧化鈦、滑石、高嶺土、氧化鋁、澱粉、粉狀纖維素、MCC、或波拉克林鉀(polacrilin potassium)。包衣組成物可以於有機溶劑或水性溶劑或其混合液中之溶液或乳膠形式施用。如果施用溶液,則溶劑可以佔已溶解固體總重之約25-99重量%之量存在。適當之溶劑為水、低級醇、低級氯化烴類、酮類、或其混合物。如果施用乳膠,則溶劑以佔乳膠中聚合材料量之約25-97重量%之量存在。溶劑主要地可為水。
本發明化合物之劑量為約0.5毫克/公斤體重至約100毫克/公斤體重,或者在其間的任何遞增。較佳劑量率在約30毫克/公斤體重至約100毫克/公斤體重之間。總每日劑量可以單劑量或分劑量投予。式I化合物之適當治療劑量可在每天每公斤接受者體重1微克(μg)至1000毫克(mg)之範圍內,且在其間的任何遞增諸如1、2、3、5、10、25、50、75、100、200、300、400、500、600、700、800、900、或1000微克(1毫克);2、3、5、10、25、50、75、100、200、300、400、500、600、700、800、900或1000毫克。然而,應該理解的是,任何特定患者之特定劑量將依一些因素包括所投予之特定化合物之活性、年齡、體重、一般健康狀況、姓別、飲食、投予時間、投予路徑、排泄速率、藥物組合及接受療法之特定疾病的嚴重性而定。
劑量方案可予調整以提供最適之期望反應。例如,可
投予單次劑量,可隨著時間投予數次分劑量或者可依治療情況之迫切性所指示地將劑量成比例地降低或提高。特別有利地係將非經腸部組成物調配於劑量單位形式中以易於投予及提供劑量均勻性。本文所用之劑量單位形式意指供待治療哺乳動物個體用之適於作為單一式劑量之物理離散單位;每一單位含有經計算可產生期望療效之預定量的活性化合物且聯合所需之醫藥載體。本發明劑量單位形式之規格乃藉由下列者所指定且直接依下列者而定:(a)治療劑之獨特特徵及待達成之特定治療或預防效應,及(b)對個體敏感性治療之此活性化合物調技術中所固有之限制。因此,熟知技藝者應了解,以本文所提供之說明為基準,將劑量及投藥方案根據治療技藝中詳知之方法調整。亦即,最大可耐受劑量可輕易確立,且提供患者可察覺之治療利益的有效量亦可測定,就如同可為用於投予每一藥劑以提供患者可察覺之治療利益的短暫要求。因此,雖然某些劑量及投予方案在本文中例示,但這些實例絕非限制在操作本發明中可提供予患者之劑量及投予方案。
一些實施例中,本文所述之化合物及組成物係於晚上約就寢時間或睡眠時間投予個體。較佳地,本文所述之化合物及組成物係於就寢時間之前或之後6小時、5小時、4小時、3小時、120分鐘、90分鐘、60分鐘、45分鐘、30分鐘、25分鐘、20分鐘、15分鐘、10分鐘、5分鐘內、或立即地投予個體。較佳地,本發明之化合物或組成物係於就寢時間之前2小時內或立即地投予個體。本文所
用之“就寢時間”意指個體有休息意圖而上床或睡著的晚上時間。
其他實施例中,本文所述之化合物及組成物可隨著或不隨著食物投予。當化合物或組成物隨著食物投予時,較佳地可在進餐諸如早餐、千餐、晚餐或點心之前或之後4小時內投予。例如,本發明之化合物或組成物係在進餐之前或之後的6小時內、5小時內、4小時內、3小時內、120分鐘內、90分鐘內、60分鐘內、45分鐘內、30分鐘內、25分鐘內、20分鐘內、15分鐘內、10分鐘內、5分鐘內、或立即投予。較佳地,本發明之化合物或組成物係在晚餐之後4小時內、3小時內、120分鐘內、90分鐘內、或60分鐘內投予。
本文所揭示標的之另一方面,提供治療Cry媒介性疾病或失調之方法,其包含投予治療有效量之如同本文先前任一實施例中所述之根據式I之化合物。本發明之較佳實施例為治療Cry媒介性疾病或失調之方法,其中該以Cry之異常濃度為特徵之疾病或失調選自由以下所組成之群組:糖尿病、與糖尿病有關之併發症、代謝症候群、胰島素抗性症候群、肥胖症、青光眼、庫欣氏症候群(Cushing’s syndrome)、炎性失調、粒腺體失調、弗里德利希共濟失調(Friedrich’s ataxia)、精神病性憂鬱症、阿滋海默氏症、神經病性疼痛、藥物濫用、骨質疏鬆症、癌症、黃斑部病變、及肌病變。藉本文所揭示化合物治療之特佳Cry媒介性疾病或失調包括糖尿病、糖尿病併發症諸
如糖尿病神經病變、糖尿病視網膜病變、糖尿病腎病變、白內障形成、青光眼、糖尿病血管病變、動脈粥狀硬化;非酒精性脂肪肝炎(NASH);非酒精性脂肪肝疾病(NAFLD);氣喘;及慢性阻塞性肺病(COPD)。
本文所用之術語“投予”等等意指可使用以能夠將化合物或組成物遞送至期望之生物作用位的方法。這些方法包括(但不限定於)經口、非經腸部、局部、黏膜部、眼部、眼睛、陰道、及直腸部投予。熟諳此藝者熟悉本文所述之化合物及方法可使用之投予技術,例如如同於Goodman and Gilman,The Pharmacological Basis of Therapeutics,current ed.;Pergamon;and Remington’s,Pharmaceutical Sciences(current edition),Mack Publishing Co.,Easton,Pa中所述地。本文中所用之醫藥組成物的“非經腸部投予”包括以個體組織之物理破壞及將醫藥組成物經由組織破壞而投予為特徵之任何投予路徑。故非經腸部投予包括(但不限定於)醫藥組成物藉由組成物之注射、藉由組成物經由手術切口之施予、藉由組成物經由組織穿透非手術傷口之施予等等投予。尤其、非經腸部投予欲包括(但不限定於)皮下、腹膜內、肌內、及胸骨內注射,及腎臟透析輸注技術。
本發明上下文中之“個體”較佳地為哺乳動物。哺乳動物可為人類、非人類之靈長類、小鼠、大鼠、狗、貓、馬、或母牛,但不限定為這些實例。除人類以外之哺乳動物可有利地作為代表Cry媒介性疾病或失調的動物模型,
諸如ob/ob小鼠。個體可為雄性或雌性。個體可為先前已診斷或鑑定為患有Cry媒介性疾病或失調,且隨意地已接受或正接受該疾病或失調之治療干預或治療法者。另外,個體亦可為先前尚未被診斷為患有Cry媒介性疾病或失調者。例如,個體可為顯現Cry媒介性疾病或失調之一或多種風險因子者,或為並未顯現Cry媒介性疾病或失調之風險因子之個體,或為無Cry媒介性疾病或失調症狀之個體。個體亦可為罹患或處於發展Cry媒介性疾病或失調之風險者,或為罹患或處於發展Cry媒介性疾病或失調復發之風險者。個體亦可為先前已治療Cry媒介性疾病或失調者,無論是藉由單獨地投予本文所揭示之化合物及組成物或將本文所揭示之化合物及組成物與其他治療劑、手術、或先前之任何組合法組合投予。本文所用之術語“個體”及“患者”可互換使用。
“Cry媒介性疾病或失調”可包括(未限制)糖尿病(包括(未限制)胰島素依賴性“第I型”糖尿病、非胰島素依賴性“第II型”糖尿病、妊娠型糖尿病及糖尿病相關併發症如糖尿病神經病變、糖尿病視網膜病變、糖尿病心肌病變、糖尿病腎病變、牙周病、及糖尿病酮酸中毒)、代謝症候群、胰島素抗性症候群、肥胖症、青光眼、庫欣氏症候群(Cushing’s syndrome);精神病性憂鬱症;阿滋海默氏症;神經病性疼痛;藥物濫用;骨質疏鬆症;癌症;黃斑部病變;及肌病變。
本文所用之術語“治療(treating)”、“治療(treat)”、或
“治療(treatment)”包括預防性(例如預防疾病性)、暫時性、輔助性、及治癒性治療。例如本文所用之第2型糖尿病之治療意指具有第2型糖尿病或處於具第2型糖尿病風險之患者可根據本文所述之方法治療。對接受預防性治療的患者而言,預防性治療所致之疾病狀態發生率的降低為預防性治療的可預見結果。
本文所用之術語“減輕(alleviating)”或“減輕(alleviate)”說明失調之現象或症狀的嚴重度被減少、降低、或抑制之過程。重要地,症狀可被減輕而未被消除。較佳實施例中,本發明醫藥組成物之投予導致症狀之消除,然而消除是没有必要的。治療有效量之本文所述化合物或醫藥組成物預期可減少症狀之嚴重度。
本文所用之術語“症狀”經定義為疾病、患病、損傷、或身體一些不對勁的指示。症狀被經歷症狀的個體所感覺或注意到,但可能不被其他者輕易注意到。其他者係由健康照護或臨床專業人員定義。
除非另有指定,否則本文所用之術語“代謝症候群”意指牛皮癬、糖尿病、傷口癒合、發炎、神經退化性疾病、半乳糖血症、楓糖尿症、苯酮尿症、高肌胺酸血症、胸腺嘧啶尿嘧啶尿症、四價硫化物血症(sulfinuria)、異戊酸血症、酵母胺酸尿症、4-羥基丁酸尿症、葡萄糖-6-磷酸脫氫酶缺乏症、及丙酮酸脫氫酶缺乏症。
本文所用之術語“肥胖症”或“肥胖”通常意指以他/她的年齡、姓別及身高而言,起過平均體重之至少約20-30%
的個體。技術上,“肥胖”的定義對男性而言為身體質量指數大於27.8公斤/平方公尺的個體,且對女性而言,為身體質量指數大於27.3公斤/平方公尺的個體。熟諳此藝者輕易知道,本發明方法不限制於落在上述準則內者。實際上,本發明方法亦可有利地藉由落在這些傳統準則外側的個體,例如藉由可能有肥胖症傾向者進行操作。
本文所用之術語“炎性失調”意指失調諸如氣喘、慢性阻塞性肺病(COPD)、類風濕性關節炎、僵直性脊椎炎、牛皮癬性關節炎、牛皮癬、軟骨鈣質沈著病、痛風、炎性腸病、潰瘍性結腸炎,克隆氏病、纖維肌痛、及惡病質。
本文所用之術語“庫欣氏症候群(Cushing’s syndrome)”意指因為增加濃度之皮質醇的長期暴露所致之現象及症狀之群集。庫欣氏症候群(Cushing’s syndrome)可因內源性或外源性原因所致。內源性庫欣氏症候群的原因包括腦下垂體腫瘤(亦稱為庫辛氏病)、腎上腺腫瘤、及促腎上腺皮質激素(ACTH)及/或促腎上腺皮質激素釋放激素(CRH)由其他腫瘤(包括但不限定於小細胞肺癌)之異位分泌。外源性(或醫原性)庫欣氏症候群係因為使用用於治療各種失調包括炎性失調包括(但不限定於)氣喘、牛皮癬及類風濕性關節炎之皮質類固醇所致。
本文所用之術語“粒線體疾病”意指疾病諸如粒線體腦肌肉病變、乳酸中毒、及類中風症候群(MELAS)、肌抽躍癲癇合併紅色襤褸肌纖維症(MERRF)、凱恩塞瑞氏症候群(Kearns-Sayre syndrome)、慢性進行性外眼肌麻痺症、
Leber氏遺傳性視神經病變、萊氏症候群(Leigh syndrome)、糖尿病、聾、神經性肌肉無力、運動失調、及色素沉積性視網膜炎(NARP)、及肌神經源性胃腸性腦病變。
本文所用之術語“癌症”意指以不受控之細胞生長及/或增殖為特徵之失調及疾病,且包括良性及惡性癌症,高度增生失調及疾病,及轉移。特佳之癌症實例包括實質固態瘤癌症或上皮癌,包括但不限定於:肺癌;腦癌;胰臟癌;頭頸部癌症(例如鱗狀細胞癌);乳癌;結腸直腸癌;肝癌;胃癌;腎癌;卵巢癌;攝護腺癌;或腺癌。其他癌症為具有VEGF的表現提高、血管新生提高、或缺氧性腫瘤者。
本文所用之詞組“治療有效量”意指研究者、獸醫、醫師或其他者尋求之引發組織、系統、動物、或人類之生物或醫學反應的藥物或醫藥劑之量。
本文所用之詞組“有效降低血液葡萄糖濃度的量”意指充分提供循環濃度高到足以達成期望效應之化合物的量。此濃度典型地落在約10nM上至2μM之範圍內;以約100nM上至500nM範圍內之濃度較佳。如同先前所述,既然落在上述式I定義內之不同化合物的活性可有相當大的變化,且既然個別個體之症狀嚴重度可呈現廣泛的差異,故由醫師決定個體對治療之反應且因此修改劑量。
本文所用之詞組“胰島素抗性”意指全身或個別組織諸如骨骼肌組織、心肌組織、脂肪組織或肝組織對胰島素作
用之敏感性降低。胰島素抗性發生在許多有或無糖尿病的個體。
本文所用之詞“胰島素抗性症候群”意指集群表現,其包括胰島素抗性、高胰島素血症、非胰島素依賴性糖尿病(NIDDM)、動脈高血壓、中央型(內臟型)肥胖症、及血脂異常。
本發明化合物亦可用於治療其他與葡萄糖之利用受損及胰島素抗性有關之代謝失調包括NIDDM之主要晚期併發症,諸如糖尿病血管病變、動脈粥狀硬化、非酒精性脂肪肝炎(NASH)、非酒精性脂肪肝疾病(NAFLD)、糖尿病腎病變、糖尿病神經病變、及糖尿病眼部併發症諸如視網膜病變、白內障形成及青光眼,及與NIDDM有關聯之許多其他併發症,包括血脂異常、萄萄糖皮質素誘導性胰島素抗性、多囊性卵巢症候群、肥胖症、高血糖症、高血脂症、高膽固醇血症、高三酸甘油酯血症、高胰島素血症、及高血壓。這些病症之簡略說明可於任何醫學字典例如“Stedman’s Medical Dictionary”(Xth Ed.)中得到。
本文所揭示化合物及組成物可以治療有效量地與一或多種如本文所定義之額外治療療劑組合投予。例如,協同效應可與其他用於治療Cry媒介性疾病或失調的物質發生。當本發明化合物與其他療法一起投予時,共同投予之化合物的劑量當然將依所用共同藥物、所用特定藥物、待治療之病症等等而變。
本文所用之可交替使用之術語“組合治療(combination
treatment)”、“組合療法(combination therapy)”、“組合治療(combined treatment)”或“組合治療(combinatorial treatment)”意指個體以至少兩種不同治療劑的治療。本文所用之術語“共同投予”或“組合投予”等等意欲包含將選定之治療劑投予單一個體,且欲包括其中各藥劑不必要同一投予路徑或同時地投予之治療方案。本文所述之組合治療意在提供由本文揭示之化合物及組成物及一或多種額外治療劑之共同作用所得之有利效應。組合之有利效應包括(但不限定於)由本文所揭示化合及治療劑之組合所致之藥物動力學或藥效學共同作用。這些治療劑之組合投予典型地於定義之時限間進行(通常依所選定之組合而為分鐘、小時、天或星期)。一些實施例中,本發明化合物係與一或多種額外治療劑係以同步或相繼方式組合投予。當同步投予時,本發明化合物可(例如)與額外治療劑於同一膠囊中投予。另外地,本發明化合物及額外治療劑可涵蓋於個別組成物(亦即膠囊)中而於同時間投予。當相繼投予時,本發明化合物可在投予額外治療劑之前或之後投予。術語“醫藥組成物”意指由將一種以上之活性成分混合或組合所得之產物且包括活性成分之固定及非固定組合。“固定組合”意指活性成分例如本文揭示之化合物及額外治療劑均以單一實體或劑量形式同步投予患者。“非固定組合”意指活性成分例如本文揭示之化合物及額外治療劑均以個別之實體形式同步地、同時地或無時間限制地相繼投予患者,其中此投予提供治療有效濃度之2種化合物於患者體內。
後者亦應用雞尾酒療法,例如3或更多種有效成分之投予。
用於治療糖尿病、代謝症候群、肥胖症、胰島素抗性症候群、糖尿病併發症或癌症之治療劑包括但不限定於下列者:胰島素、降向糖劑、抗糖尿病劑、抗發炎劑、降脂劑、抗高血壓劑諸如鈣通道阻斷劑、β-腎上腺素能受體阻斷劑、環加氧酶-2抑制劑、血管緊張素系統抑制劑、ACE抑制劑、腎素抑制劑、化療劑、放射療法、荷爾蒙調節劑、免疫調節劑、抗血管生成劑,連同常見之風險因子改良劑。
胰島素包括速效形式諸如Insulin lispro rDNA來源:HUMALOG(1.5毫升,10毫升,Eli Lilly and Company,Indianapolis,Ind.),取自牛肉及豬肉之胰島素注射液(常規胰島素)(常規ILETIN I,Eli Lilly],人類:rDNA:HUMULIN R(Eli Lilly),NOVOLIN R(Novo Nordisk),半合成:VELOSULIN人類(Novo Nordisk),rDNA人類,緩衝化:VELOSULIN BR,豬肉:常規胰島素(Novo Nordisk),純化豬肉:豬肉常規ILETIN II(Eli Lilly),常規純化豬肉胰島素(Novo Nordisk),及常規(濃縮)ILETIN II U-500(500單位/毫升,Eli Lilly);中效形式諸如胰島素鋅懸浮液,牛肉及豬肉:LENTE ILETIN G I(Eli Lilly),人類,rDNA:HUMULIN L(Eli Lilly),NOVOLIN L(Novo Nordisk),純化豬肉:LENTE ILETIN II(Eli Lilly),等項胰島素懸浮液(NPH):牛肉及豬肉:NPH ILETIN I(Eli
Lilly),人類,rDNA:HUMULIN N(Eli Lilly),Novolin N(Novo Nordisk),純化豬肉:Pork NPH Iletin II(Eli Lilly),NPH-N(Novo Nordisk);及長效形式諸如胰島素鋅懸浮液,長效(ULTRALENTE,Eli Lilly),人類,rDNA:HUMULIN U(Eli Lilly)。
降血脂劑包括(未限制)磺醯脲類:乙醯苯磺醯環己脲(Acetohexamide(Dymelor))、氯磺丙尿(Chlorpropamide(Diabinese))、甲苯磺丁脲(Tolbutamide(Orinase));二代磺醯脲類:吡磺環己脲(Glipizide(Glucotrol、Glucotrol XL))、格列本脲(Glyburide(Diabeta;Micronase;Glynase))、格列美脲(Glimepiride(Amaryl));雙胍類:二甲雙胍(Glucophage);α-葡萄糖苷酶抑制劑:阿卡波糖(Acarbose(Precose))、米格列醇(Miglitol(Glyset)),噻唑啶二酮類:羅格列酮(Rosiglitazone(Avandia))、吡咯列酮(Pioglitazone(Actos))、曲格列酮(Troglitazone(Rezulin));美格替奈類(Meglitinides):瑞格列奈(Repaglinide(Prandin));及其他降血糖劑諸如阿卡波糖(Acarbose);丁二胍(Buformin);丁氧胺氫氯酸鹽(Butoxamine Hydrochloride);卡格列波糖(Camiglibose);環格列酮(Ciglitazone);恩格列酮鈉(Englitazone Sodium);達格列酮鈉(Darglitazone Sodium);氫氯酸依托雙胍(Etoformin Hydrochloride);格列胺脲(Gliamilide);格列波脲(Glibomuride);格列他尼格列齊特鈉(Glicetanile Gliclazide Sodium);氟嘧醯胺(Gliflumide);升糖素
(Glucagon);格列己脲(Glyhexamide);格列嘧啶鈉(Glymidine Sodium);格列辛脲(Glyoctamide);格列帕脲(Glyparamide);利諾格列(Linogliride);富馬酸利諾格列(Linogliride Fumarate);帕莫酸甲酯(Methyl Palmoxirate);帕莫酸鈉(Palmoxirate Sodium);酒石酸吡咯格列(Pirogliride Tartrate);人類胰島素原(Proinsulin Human);乙酸司格列肽(Seglitide Acetate);甲磺氮草脲(Tolazamide);甲苯磺吡胺(Tolpyrramide);唑泊司他(Zopolrestat)。
抗糖尿病劑包括(未限制)二肽基肽酶抑制劑諸如西他列汀(sitagliptin)、阿格列汀(alogliptin)、維格列汀(vildagliptin)、沙克列汀(saxagliptin)、利拉列汀(linagilptin)、阿拉格列汀(anagliptin)、替格列汀(teneligliptin)、吉格列汀(gemigliptin)、杜拓格列汀(dutogliptin)或現今發展中之任何格列汀類(gliptins)、小蘗鹼及羽扇豆醇;及GLP-1激動劑諸如艾塞那肽(exenatide)、利拉魯肽(liraglutide)、阿必魯肽(albiglutide)、他司魯泰(taspogenitde)、及AVE0010。
抗發炎劑包括阿氯芬酸(Alclofenac);二丙酸阿氯米松(Alclometasone Dipropionate);丙縮阿爾孕酮(Algestone Acetonide);α-澱粉酶;阿西法爾(Amcinafal);安西非特(Amcinafide);胺芬酸鈉(Amfenac Sodium);氫氯酸胺普立糖(Amiprilose Hydrochloride);阿那白滯素(Anakinra);阿尼羅酸(Anirolac);阿尼扎芬(Anitrazafen);阿扎丙宗
(Apazone);巴柳氮二鈉(Balsalazide Disodium);苄達酸(Bendazac);苯惡洛芬(Benoxaprofen);氫氯酸苄達明(Benzydamine Hydrochloride);菠蘿蛋白酶(Bromelains);溴派莫(Broperamole);布地奈德(Budesonide);卡洛芬(Carprofen);環洛芬(Cicloprofen);辛噴他宗(Cintazone);克利洛芬(Cliprofen);丙酸氯倍他索(Clobetasol Propionate);丁酸氯倍他索(Clobetasone Butyrate);氯吡酸(Clopirac);丙酸氯硫卡松(Cloticasone Propionate);乙酸三氟米松(Cormethasone Acetate);去氧可的松(Cortodoxone);地夫可特(Deflazacort);地奈德(Desonide);去羥米松(Desoximetasone);二丙酸地塞米松(Dexamethasone Dipropionate);雙氯芬酸鉀(Diclofenac Potassium);雙氯芬酸鈉(Diclofenac Sodium);二乙酸二氟拉松(Diflorasone Diacetate);二氟米酮鈉(Diflumidone Sodium);二氟尼柳(Diflunisal);二氟潑尼酯(Difluprednate);雙酞榛酮(Diftalone);二甲亞碸;羥西奈德(Drocinonide);因甲羥松(Endrysone);恩莫單抗(Enlimomab);伊諾利康鈉(Enolicam Sodium);依匹唑(Epirizole);依托度酸(Etodolac);依托芬那酯(Etofenamate);聯苯乙畯(Felbinac);非那莫(Fenamole);芬存芬(Fenbufen);芬氯酸(Fenclofenac);苯克洛酸(Fenclorac);芬度柳(Fendosal);苯吡惡二酮(Fenpipalone);芬替酸(Fentiazac);夫拉扎酮(Flazalone);氟惡米松(Fluazacort);氟芬那酸(Flufenamic
Acid);氟咪唑(Flumizole);乙酸氟尼縮松(Flunisolide Acetate);氟尼辛(Flunixin);氟尼辛葡甲胺(Flunixin Meglumine);氟可丁丁酯(Fluocortin Butyl);氟甲孕酮乙酸酯(Fluorometholone Acetate);氟喹宗(Fluquazone);氟比洛芬(Flurbiprofen);氟瑞托芬(Fluretofen);丙酸氟替卡松(Fluticasone Propionate);呋喃洛芬(Furaprofen);呋羅布芬(Furobufen);哈西縮松(Halcinonide);鹵貝他索丙酸酯(Halobetasol Propionate);乙酸鹵潑尼松(Halopredone Acetate);異丁芬酸(Ibufenac);布洛芬(Ibuprofen);布洛芬鋁(Ibuprofen Aluminum);布洛芬吡啶甲醇(Ibuprofen Piconol);伊洛達普(Ilonidap);吲哚美辛(Indomethacin);吲哚美辛鈉(Indomethacin Sodium);吲哚洛芬(Indoprofen);吲哚克索(Indoxole);吲四唑(Intrazole);乙酸異氟潑尼松(Isoflupredone Acetate);伊索克酸(Isoxepac);伊索昔康(Isoxicam);酮洛芬(Ketoprofen);氫氯酸洛非咪唑(Lofemizole Hydrochloride);氯諾昔康(Lornoxicam);伊碳酸氯替潑諾(Loteprednol Etabonate);美克芬那梅鈉(Meclofenamate Sodium);甲氯芬那酸(Meclofenamic Acid);二丁酸甲氯松(Meclorisone Dibutyrate);甲芬那酸(Mefenamic Acid);美沙胺(Mesalamine);美西拉宗(Meseclazone);磺庚甲潑尼龍(Methylprednisolone Suleptanate);莫尼氟酯(Morniflumate);萘丁美酮(Nabumetone);萘普生(Naproxen);萘普生鈉(Naproxen Sodium);萘普索
(Naproxol);尼馬宗(Nimazone);奧柳氮鈉(Olsalazine Sodium);奧古蛋白(Orgotein);奧帕諾辛(Orpanoxin);奧沙普秦(Oxaprozin);羥保松(Oxyphenbutazone);氫氯酸瑞尼托林(Paranyline Hydrochloride);木聚硫鈉(Pentosan Polysulfate Sodium);甘油保泰松鈉(Phenbutazone Sodium Glycerate);吡非尼酮(Pirfenidone);吡羅昔康(Piroxicam);肉桂酸吡羅昔康(Piroxicam Cinnamate);吡羅昔康乙醇胺(Piroxicam Olamine);吡咯洛(Pirprofen);潑那扎特(Prednazate);普立非酮(Prifelone);普羅度酸(Prodolic Acid);普羅喹宗(Proquazone);普羅沙唑(Proxazole);檸檬酸普羅沙唑(Proxazole Citrate);利美索龍(Rimexolone);氯馬扎利(Romazarit);柳膽來司(Salcolex);沙那西定(Salnacedin);雙柳酯(Salsalate);柳酸酯;血根氯銨(Sanguinarium Chloride);司克拉宗(Seclazone);絲美辛(Sermetacin);舒多昔康(Sudoxicam);舒林達(Sulindac);舒洛芬(Suprofen);他美辛(Talmetacin);氯煙酞酯(Talniflumate);他洛柳酯(Talosalate);特丁非隆(Tebufelone);替尼達普(Tenidap);替尼達普鈉(Tenidap Sodium);替諾昔康(Tenoxicam);替昔康(Tesicam);替西米德(Tesimide);四氫甲吲胺(Tetrydamine);硫平酸(Tiopinac);替可的松匹伐酯(Tixocortol Pivalate);妥美汀(Tolmetin);妥美汀鈉(Tolmetin Sodium);三氯氟松(Triclonide);三氟胺酯(Triflumidate);齊多美辛(Zidometacin);葡萄糖皮質素;
苯醯吡酸鈉(Zomepirac Sodium)。重要的抗發炎劑為阿斯匹靈。
其他抗發炎劑為細胞激素抑制劑包括細胞激素拮抗劑(例如IL-6受體拮抗劑)、氮雜-烷基溶血磷脂(AALP)、及腫瘤壞死因子-α(TNF-α)抑制劑,諸如抗-TNF-α抗體、可溶性TNF受體、TNF-α、反義核酸分子、多價丙脒腙(CNI-1493)、N-乙醯半胱胺酸、配妥西菲林(pentoxiphylline)、己酮可可鹼(oxpentifylline)、碳環核苷類似物、小分子S9a、RP 55778(a TNF-α合成抑制劑)、地塞比諾(Dexanabinol(HU-211))、MDL 201,449A(9-[(1R,3R)-反-環戊-3-醇]腺嘌呤、及曲可達莫(trichodimerol(BMS-182123)。)。其他TNF-α抑制劑包括恩博(Etanercept(ENBREL、Immunex、Seattle))及英夫利昔單抗(Infliximab(REMICADE,Centocor,Malvern,Pa.))。
降脂劑包括吉非羅齊(gemfibrozil)、考來烯胺(cholystyramine)、膽利泊(colestipol)、菸鹼酸、及HMG-CoA還原酶抑制劑。可用於投予或與根據本發明之其他藥劑共同投予之HMG-CoA還原酶抑制劑包括(但不限定於)辛伐他汀(simvastatin(美國專利號碼4,444,784))、洛伐他汀(lovastatin(美國專利號碼4,231,938))、普伐他汀鈉(pravastatin sodium(美國專利號碼4,346,227))、氟伐他汀(fluvastatin(美國專利號碼4,739,073))、阿托伐他汀(atorvastatin(美國專利號碼5,273,995))、及西利伐他丁(cerivastatin)。
鈣通道阻斷劑包括二氫吡啶類諸如尼非待平(nifedipine),苯基烷基胺類諸如維拉帕米(verapamil)、及苯並硫氮平類諸如地爾硫卓(diltiazem)。其他鈣通道阻斷劑包括(但不限定於)胺利酮(amrinone)、胺氯地平(amlodipine)、苄環烷(bencyclane)、非洛地平(felodipine)、芬地林(fendiline)、氟苯桂 (flunarizine)、依拉地平(isradipine)、尼卡地平(nicardipine)、尼莫地平(nimodipine)、哌克昔林(perhexilene)、加洛帕米(gallopamil)、噻帕米(tiapamil)及噻帕米類似物(諸如1993RO-11-2933)、苯妥英(phenytoin)、巴比妥類(barbiturates)、及肽類強啡肽(dynorphin)、ω-芋螺毒素(omega-conotoxin)、及ω-美洲蜘蛛毒素(omega-agatoxin)等等及/或其醫藥上可接受之鹽。
(flunarizine)、依拉地平(isradipine)、尼卡地平(nicardipine)、尼莫地平(nimodipine)、哌克昔林(perhexilene)、加洛帕米(gallopamil)、噻帕米(tiapamil)及噻帕米類似物(諸如1993RO-11-2933)、苯妥英(phenytoin)、巴比妥類(barbiturates)、及肽類強啡肽(dynorphin)、ω-芋螺毒素(omega-conotoxin)、及ω-美洲蜘蛛毒素(omega-agatoxin)等等及/或其醫藥上可接受之鹽。
β-腎上腺素能受體阻斷劑包括(但不限定於)阿替洛爾(atenolol)、醋丁洛爾(acebutolol)、烯丙心安(alprenolol)、苯呋洛爾(befunolol)、倍他洛爾(betaxolol)、布尼洛爾(bunitrolol)、卡替洛爾(carteolol)、塞利洛爾(celiprolol)、海卓洛爾(hedroxalol)、茚諾洛爾(indenolol)、拉貝洛爾(labetalol)、左布諾洛爾(levobunolol)、甲吲洛爾(mepindolol)、美替洛爾(methypranol)、吲哚美辛(metindol)、美托洛爾(metoprolol)、美卓拉洛爾(metrizoranolol)、氧烯洛爾(oxprenolol)、品多洛爾(pindolol)、普萘洛爾(propranolol)、普拉洛爾(practolol)、普拉洛爾
(practolol)、索他洛爾那多洛爾(sotalolnadolol)、替普洛爾(tiprenolol)、托馬洛爾(tomalolol)、噻馬洛爾(timolol)、布拉洛爾(bupranolol)、噴布洛爾(penbutolol)、三甲苯心安(trimepranol)、2-(3-(1,1-二甲基乙基)-胺基-2-羥基丙氧基)-3-吡啶腈氫氯酸鹽、1-丁胺基-3-(2,5-二氯苯氧基-)-2-丙醇、1-異丙胺基-3-(4-(2-環丙基甲氧基乙基)苯氧基)-2-丙醇、3-異丙胺基-1-(7-二基二氫茚-4-基氧基)-2-丁醇、2-(3-三級丁胺基-2-羥基-苯硫基)-4-(5-胺基甲醯基-2-噻吩基)噻唑、7-(2-羥基-3-三級丁胺基丙氧基)酞內酯。以上鑑定之化合物可以其異構物混合物形式、或以其個別之左旋或右旋形式使用。
選擇性COX-2抑制劑為技藝中已知且包括(但不限定於)述於美國專利號碼5,474,995;美國專利號碼5,521,213;美國專利號碼5,536,752;美國專利號碼5,550,142;美國專利號碼5,552,422;美國專利號碼5,604,253;美國專利號碼5,604,260;美國專利號碼5,639,780;美國專利號碼5,677,318;美國專利號碼5,691,374;美國專利號碼5,698,584;美國專利號碼5,710,140;美國專利號碼5,733,909;美國專利號碼5,789,413;美國專利號碼5,817,700;美國專利號碼5,849,943;美國專利號碼5,861,419;美國專利號碼5,922,742;美國專利號碼5,925,631;及美國專利號碼5,643,933中之COX-2抑制劑。一些以上鑑定之COX-2抑制劑為選擇性COX-2抑制劑之前藥,且包括述於WO
95/00501、WO 95/18799、及美國專利號碼5,474,995(1995年12月12日公告)中者。
血管緊張素II拮抗劑之實例包括:肽化合物(例如沙拉辛(saralasin)、[(San1)(Val5)(Ala8)]血管緊張素-(1-8)八肽及相關類似物);N-取代之咪唑-2-酮(美國專利號碼5,087,634);咪唑乙酸鹽衍生物包括2-N-丁基-4-氯-1-(2-氯苄基)咪唑-5-乙酸(參見Long et al.,J.Pharmacol.Exp.Ther.247(1),1-7(1988));4,5,6,7-四氫-1H-咪唑並[4,5-c]吡啶-6-羧酸及類似物衍生物(美國專利號碼4,816,463);N2-四唑β-葡糖苷酸類似物(美國專利號碼5,085,992);經取代之吡咯、吡唑、及三唑(tryazoles)(美國專利號碼5,081,127);酚及雜環衍生物諸如1,3-咪唑(美國專利號碼5,073,566);咪唑並-稠合之7員環雜環(美國專利號碼5,064,825);肽(例如美國專利號碼4,772,684);針對血管緊張素II之抗體(例如美國專利號碼4,302,386);及芳烷基咪唑化合物諸如聯苯-甲基取代之咪唑(例如1988年1月20日之EP 253,310);ES8891(N- 啉基乙醯基-(-1-萘基)-L-丙胺醯基-1-(4,噻唑基)-L-丙胺醯基(35,45)-4-胺基-3-羥基-5-環-己戊醯基--N-己醯胺日本東京三共株式會社(Sankyo Company,Ltd.,Tokyo,Japan);SKF108566(E-α-2-[2-丁基-1-(羧基苯基)甲基]1H-咪唑-5-基[甲基-e]-2-噻吩丙酸,Smith Kline Beecham Pharmaceuticals,Pa.);氯沙坦(Losartan(DUP753/MK954,DuPont Merck Pharmaceutical Company));瑞米吉崙(Remikirin(RO42-
5892,F.Hoffman LaRoche AG));A2激動劑(Marion Merrill Dow)及某些非肽雜環(G.D.Searle and Company)。
啉基乙醯基-(-1-萘基)-L-丙胺醯基-1-(4,噻唑基)-L-丙胺醯基(35,45)-4-胺基-3-羥基-5-環-己戊醯基--N-己醯胺日本東京三共株式會社(Sankyo Company,Ltd.,Tokyo,Japan);SKF108566(E-α-2-[2-丁基-1-(羧基苯基)甲基]1H-咪唑-5-基[甲基-e]-2-噻吩丙酸,Smith Kline Beecham Pharmaceuticals,Pa.);氯沙坦(Losartan(DUP753/MK954,DuPont Merck Pharmaceutical Company));瑞米吉崙(Remikirin(RO42-
5892,F.Hoffman LaRoche AG));A2激動劑(Marion Merrill Dow)及某些非肽雜環(G.D.Searle and Company)。
血管緊張素轉化酶(ACE)抑制劑包括胺基酸及其衍生物、肽類包括二肽及三肽及針對ACE之抗體,其藉由抑制ACE活性因此降低或消除加壓物質血管緊張素II之形成而干擾腎素-血管緊張素系統,。其他ACE抑制劑包括醯基巰基及巰基烷醯基脯胺酸類諸如卡托普利(captopril(美國專利號碼4,105,776))及佐芬普利(zofenopril(美國專利號碼4,316,906)),羧基烷基二肽類諸如伊那普利(enalapril(美國專利號碼4,374,829))、賴諾普利(lisinopril(美國專利號碼4,374,829))、喹那普利(quinapril(美國專利號碼4,344,949))、雷米普利(ramipril(美國專利號碼4,587,258))、及培哚普利(perindopril(美國專利號碼4,508,729)),羧基烷基二肽模擬物諸如西拉普利(cilazapril(美國專利號碼4,512,924))及貝那普利(benazapril(美國專利號碼4,410,520)),氧膦基烷醯基脯胺酸類諸如福辛普利(fosinopril(美國專利號碼4,337,201)及群多普利(trandolopril)。
腎素抑制劑包括胺基酸及其衍生物、肽類及其衍生物、及針對腎素之抗體。其他腎素抑制劑包括肽之尿素衍生物(美國專利號碼5,116,835);藉由非肽鍵連接之胺基酸(美國專利號碼5,114,937);二肽及三肽衍生物(美國專利號碼5,106,835);胺基酸及其衍生物(美國專利號碼
5,104,869及5,095,119);二醇磺醯胺及亞磺醯基類(美國專利號碼5,098,924);修飾肽類(美國專利號碼5,095,006);肽基β-胺基醯基胺基二醇胺基甲酸酯(美國專利號碼5,089,471);吡咯咪唑酮類(美國專利號碼5,075,451);含氟及氯司特丁或司特酮(fluorine and chlorine statine or statone)之肽類(美國專利號碼5,066,643);肽基胺基二醇類(美國專利號碼5,063,208及4,845,079);N- 啉基衍生物(美國專利號碼5,055,466);胃酶抑素衍生物(美國專利號碼4,980,283);N-雜環型醇類(美國專利號碼4,885,292);針對腎素之單株抗體(美國專利號碼4,780,401);及各種其他肽類及其類似物(美國專利號碼5,071,837、5,064,965、5,063,207、5,036,054、5,036,053、5,034,512、及4,894,437)。
啉基衍生物(美國專利號碼5,055,466);胃酶抑素衍生物(美國專利號碼4,980,283);N-雜環型醇類(美國專利號碼4,885,292);針對腎素之單株抗體(美國專利號碼4,780,401);及各種其他肽類及其類似物(美國專利號碼5,071,837、5,064,965、5,063,207、5,036,054、5,036,053、5,034,512、及4,894,437)。
可用於治療糖尿病及相關失調之其他治療劑包括(但不限定於)脂肪酶抑制劑諸如西替利司他(cetilistat(ATL-962));合成澱粉不溶素類似物諸如含或不含重組瘦素之普蘭林肽(Symlin pramlintide);鈉-葡萄糖共同轉運體2(SGLT2)抑制劑如舍格列淨(sergliflozin(869682;KGT-1251))、YM543、卡那利福心(canagliflozin)、爾土利福心(ertugliflozin)、多百利福心(dapagliflozin)、GlaxoSmithKline分子189075、及Sanofi-Aventis分子AVE2268;雙重脂肪三酸甘油酯脂肪酶及PI3激酶活化劑如Adyvia(ID 1101);神經肽Y2、Y4、Y5受體之拮抗劑如Nastech分子PYY3-36、人類荷爾蒙PYY3-36之合
成類以物及胰臟多肽(7TM分子TM30338);Shionogi分子S-2367;大麻素CB1受體拮抗劑諸如利莫那斑(rimonabant(Acomplia))、泰倫那斑(taranabant)、CP-945,598、Solvay分子SLV319、Vernalis分子V24343;荷爾蒙樣油醯雌酮;血清素、多巴胺、及降腎上腺素之抑制劑(亦於技藝中稱之為三單胺再吸收抑制劑)如特索芬辛(tesofensine(Neurosearch分子NS2330));降腎上腺素及多巴胺再吸收抑制劑如Contrave(安非他酮(bupropion)加上類雅片拮抗劑那曲酮(naltrexone))及Excalia(安非他酮(bupropion)加上抗痙劑唑尼沙胺(zonisaminde));11β-羥基固醇脫氫酶第1型(11b-HSD1)之抑制劑如Incyte分子INCB13739;皮質醇合成抑制劑諸如酮康唑(ketoconazole(DiObex分子DIO-902));糖質新生抑制劑諸如Metabasis/Daiichi分子CS-917;葡萄糖激酶活化劑如Roche分子R1440;蛋白質酪胺酸磷酸酶-1B之反義抑制劑諸如ISIS 113715;以及其他藥劑如NicOx分子NCX 4016;胃泌激素及上皮生長因子(EGF)類似物之注射諸如胰島再生療法(E1-I.N.T.);貝他司汀(betahistine(Obecure分子OBE101));膽酸螯合劑(例如考來烯胺(cholestyramine)及膽利泊(colestipol))、維他命B3(亦稱為菸鹼酸(nicotinic acid或niacin))、維他命B6(吡哆醇)、維化命B12(氰鈷胺素)、纖維酸衍生物(例如吉非羅齊(gemfibrozil)、安妥明(clofibrate)、非諾貝特(fenofibrate)及苯扎貝特(benzafibrate))、普羅布考
(probucol)、硝基甘油(nitroglycerin)、及膽固醇吸收抑制劑(例如β-谷固醇(β-sitosterol)及醯基CoA-膽固醇醯基轉移酶(ACAT)抑制劑諸如甲亞油醯胺(melinamide))、HMG-CoA合成酶抑制劑、角鯊烯環氧酶抑制劑及角鯊烯合成酶抑制劑。
用於治療庫欣氏症候群(Cushing’s syndrome)之藥劑的實例包括(未限制)神經調節劑(Signifor®(pasireotide)、卡麥角林(cabergoline));腎上腺類固醇生成抑制劑(酮康唑(ketoconazole)、美替拉酮(metyrapone)、米托坦(mitotane)、依托咪酯(etomidate));及核受體調節劑(Korlym®(mifepristone)、視網酸)。其他藥劑包括(未限制)上皮生長因子受體抑制劑(例如吉非替尼(gefitinib))、醛固酮合成酶/11β-羥化酶抑制劑LCI699、及左旋酮康唑levoketoconazole(COR-003))。
經常用於治療疼痛包括神經病性疼痛之止痛劑實例包括(未限制)類雅片或非類雅片止痛劑。適當之類鴉片止痛劑包括但不限定於 啡、海洛因、氫
啡、海洛因、氫 啡酮(hydromorphone)、氫可酮(hydrocodone)、氧
啡酮(hydromorphone)、氫可酮(hydrocodone)、氧 啡酮(oxymorphone)、氧可酮(oxycodone)、美託酮(metopon)、變
啡酮(oxymorphone)、氧可酮(oxycodone)、美託酮(metopon)、變 啡鹼(apomorphine)、去甲
啡鹼(apomorphine)、去甲 啡(normorphine)、羥戊甲
啡(normorphine)、羥戊甲 啡(etorphine)、丁丙諾啡(buprenorphine)、麥佩裡定(meperidine)、洛派丁胺(lopermide)、阿尼利定(anileridine)、依索庚
啡(etorphine)、丁丙諾啡(buprenorphine)、麥佩裡定(meperidine)、洛派丁胺(lopermide)、阿尼利定(anileridine)、依索庚 (ethoheptazine)、匹米諾定(piminidine)、倍他羅定(betaprodine)、地芬諾酯
(diphenoxylate)、芬太尼(fentanyl)、舒芬太尼(sufentanil)、阿芬太尼(alfentanil)、雷米芬太尼(remifentanil)、左啡諾(levorphanol)、右美沙芬(dextromethorphan)、非那左辛(phenazocine)、潘他唑新(pentazocine)、環佐辛(cyclazocine)、美沙酮(methadone)、異美沙酮(isomethadone)及丙氧芬(propoxyphene)。適當之非類鴉片止痛劑包括(但不限定於)阿斯匹靈、塞來昔布(celecoxib)、羅非昔布(rofecoxib)、雙氯芬酸(diclofinac)、二氟尼柳(diflusinal)、依托度酸(etodolac)、非諾洛芬(fenoprofen)、氟比洛芬(flurbiprofen)、布洛芬(ibuprofen)、酮洛酚(ketoprofen)、吲哚美辛(indomethacin)、酮咯酸(ketorolac)、美克芬那梅(meclofenamate)、甲芬那酸(mefanamic acid)、萘丁美酮(nabumetone)、萘普生(naproxen)、吡羅昔康(piroxicam)及舒林達(Sulindac)。
(ethoheptazine)、匹米諾定(piminidine)、倍他羅定(betaprodine)、地芬諾酯
(diphenoxylate)、芬太尼(fentanyl)、舒芬太尼(sufentanil)、阿芬太尼(alfentanil)、雷米芬太尼(remifentanil)、左啡諾(levorphanol)、右美沙芬(dextromethorphan)、非那左辛(phenazocine)、潘他唑新(pentazocine)、環佐辛(cyclazocine)、美沙酮(methadone)、異美沙酮(isomethadone)及丙氧芬(propoxyphene)。適當之非類鴉片止痛劑包括(但不限定於)阿斯匹靈、塞來昔布(celecoxib)、羅非昔布(rofecoxib)、雙氯芬酸(diclofinac)、二氟尼柳(diflusinal)、依托度酸(etodolac)、非諾洛芬(fenoprofen)、氟比洛芬(flurbiprofen)、布洛芬(ibuprofen)、酮洛酚(ketoprofen)、吲哚美辛(indomethacin)、酮咯酸(ketorolac)、美克芬那梅(meclofenamate)、甲芬那酸(mefanamic acid)、萘丁美酮(nabumetone)、萘普生(naproxen)、吡羅昔康(piroxicam)及舒林達(Sulindac)。
經常用於治療青光眼之治療劑實例包括膽鹼能激動劑(例如匹魯卡品(pilocarpine)及卡巴可(carbachol))、膽鹼酯酶抑制劑(例如毒扁豆鹼(physostigmine)、新斯狄明(neostigmine)、德美卡靈(demacarium)、碘磷靈(echothiophate iodide)及異氟磷(isofluorophate))、碳酸酐抑制劑(例如乙醯唑胺(acetazolamide)、二氯磺胺(dichlorphenamide)、甲醋唑胺(methazolamide)、乙氧苯唑胺(ethoxzolamide)及杜塞醯胺(dorzolamide))、非選擇性腎
上腺素能激動劑(例如腎上腺素及地匹福林(dipivefrin))、α2-選擇性腎上腺素能激動劑(例如阿可樂定(apraclonidine)及溴莫尼定(brimonidine))、β-阻斷劑(例如噻馬洛爾(timolol)、貝他左羅(betazolol)、左布諾洛爾(levobunolol)、卡替洛爾(carteolol)及美替洛爾(metipranolol))、前列腺素類似物(例如舒而坦(latanoprost))及滲透性利尿劑(例如甘油、甘露糖醇及異山梨酯(isosorbide));皮質素類固醇類諸如倍氯米松(beclomethasone)、甲潑尼龍(Methylprednisolone)、倍他米松(betamethasone)、潑尼龍(prednisone)、潑尼松龍(prenisolone)、地塞米松(dexamethasone)、氟替卡松(fluticasone)及氫化可的松(hydrocortisone)、及皮質類固醇類似物諸如布地奈德(Budesonide)。
經常用於治療阿滋海默氏症之治療劑實例包括β-分泌酶抑制劑或γ-分泌酶抑制劑;甘胺酸轉運抑制劑、tau磷酸化抑制劑;Aβ寡聚物形成之阻斷劑;p25/CDK5抑制劑;HMG-CoA還原酶抑制劑;PPARγ激動劑諸如吡咯列酮(pioglitazone)及羅格列酮(rosiglitazone);NK1/NK3受體拮抗劑;非類固醇抗發炎藥(NSAID)包括布洛芬(ibuprofen);維他命E;抗類澱粉蛋白抗體包括抗類澱粉蛋白人源化單株抗體;COX-2抑制劑;抗發炎化合物諸如(R)-氟比洛芬((R)-flurbiprofen);CB-1受體拮抗劑或CB-1受體反向激動劑;抗體諸如多西環素(doxycycline)及利福平(rifampin);N-甲基-D-天冬胺酸(NMDA)受體拮抗劑諸
如二甲金剛胺(memantine)及奈拉美生(neramexane);NR2B拮抗劑;雄激素受體調節劑;乙醯膽鹼酯酶抑制劑諸如雪花蓮胺(galantamine)、利凡斯的明(rivastigmine)、多奈哌齊(donepezil)、及他克林(tacrine);mGluR5調節劑;生長激素促分泌劑諸如伊布莫侖(ibutamoren)、伊布莫侖甲磺酸鹽(ibutamoren mesylate)、及卡普瑞林(capromorelin);組織胺H3拮抗劑;AMPA激動劑;PDE IV抑制劑;GABAA反向激動劑;GABAA α5受體配位體;GABAB受體配位體;鉀通道阻斷劑;神經元菸鹼激動劑;P-450抑制劑諸如利托那韋(ritonavir)。
經常用於治療情感失調諸如憂鬱症之治療劑實例包括(未限制)阿米替林(amitriptyline)、氧化阿米替林(amitriptyline oxide)、地昔帕明(desipramine)、二苯西平(dibenzepin)、度硫平(dosulepin)、多塞平(doxepin)、氯米帕明(clomipramine)、丙咪 (imipramine)、去甲替林(nortriptyline)、米塞林(mianserin)、馬普替林(maprotiline)、曲米帕明(trimipramine)、CP-122721、艾扎索南(elzasonan)、PD-171729、MK-869、DOV-216303、DOV-21947、利卡西平(licarbazepine)、安非布他酮(amfebutamone)、雷達伐辛(radafaxine)、維拉佐酮(vilazodone)、GSK-679769、GW-597599、NS-2359、GSK-876008、普拉克索(pramipexole)、度洛西汀(duloxetine)、阿托莫西汀(atomoxetine)、LY-628535、去甲文拉法辛(desvenlafaxine)、依他普侖(escitalopram)、
LU-AA21004、沙瑞度坦(saredutant)、SR-58611、SSR-149415、SSR-146977、馬氯貝胺(moclobemide)、R-673、R-1204、BMS-469458、DPC-368、Org-34517、Org-34850、CRH受體之抑制劑、ONO-2333Ms、NBI-876008、AAG-561、NBI-34041、DPC-368、PD-171729、SSR-125543、維洛沙秦(viloxazine)、曲唑酮(trazodone)、奈法唑酮(nefazodone)、米氮平(mirtazapine)、萬拉法辛(venlafaxine)、瑞波西汀(reboxetine)、強內心百樂明(tranylcypromine)、溴法羅明(brofaromine)、馬氯貝胺(moclobemide)、西酞普蘭(citalopram)、艾司西酞普蘭(escitalopram)、帕羅西汀(paroxetine)、氟西汀(fluoxetine)、氟伏沙明(fluvoxamine)、舍曲林(sertraline)、Hypericum(St.John’s Wort)、阿普唑侖(alprazolam)、氯硝西泮(clonazepam)、地西泮(diazepam)、勞拉西泮(lorazepam)、哈拉西泮(halazepam)、氯二氮平(chlordiazepoxide),及其他藥物諸如丁螺環酮(buspirone)、可樂定(clonidine)、帕戈隆(pagoclone)、利培酮(risperidone)、奧氮平(olanzapine)、喹硫平(quetiapine)、齊拉西酮(ziprasidone)、塞來昔布(celecoxib)、吡羅昔康(piroxicam)、帕瑞昔布(parecoxib)、伐地考昔(valdecoxib)、PMI-001、PH-686464、SC-58236、依托昔布(etoricoxib)、羅菲可西保(rofecoxib)、L-776967、羅美昔布(lumiracoxib)、GW-406381、GW-644784、美洛昔康(meloxicam)、SVT-
2016、PAC-10649、CS-706、LAS-34475、西米考昔(cimicoxib)、A-183827.0、或尼美舒利(nimesulide)。
(imipramine)、去甲替林(nortriptyline)、米塞林(mianserin)、馬普替林(maprotiline)、曲米帕明(trimipramine)、CP-122721、艾扎索南(elzasonan)、PD-171729、MK-869、DOV-216303、DOV-21947、利卡西平(licarbazepine)、安非布他酮(amfebutamone)、雷達伐辛(radafaxine)、維拉佐酮(vilazodone)、GSK-679769、GW-597599、NS-2359、GSK-876008、普拉克索(pramipexole)、度洛西汀(duloxetine)、阿托莫西汀(atomoxetine)、LY-628535、去甲文拉法辛(desvenlafaxine)、依他普侖(escitalopram)、
LU-AA21004、沙瑞度坦(saredutant)、SR-58611、SSR-149415、SSR-146977、馬氯貝胺(moclobemide)、R-673、R-1204、BMS-469458、DPC-368、Org-34517、Org-34850、CRH受體之抑制劑、ONO-2333Ms、NBI-876008、AAG-561、NBI-34041、DPC-368、PD-171729、SSR-125543、維洛沙秦(viloxazine)、曲唑酮(trazodone)、奈法唑酮(nefazodone)、米氮平(mirtazapine)、萬拉法辛(venlafaxine)、瑞波西汀(reboxetine)、強內心百樂明(tranylcypromine)、溴法羅明(brofaromine)、馬氯貝胺(moclobemide)、西酞普蘭(citalopram)、艾司西酞普蘭(escitalopram)、帕羅西汀(paroxetine)、氟西汀(fluoxetine)、氟伏沙明(fluvoxamine)、舍曲林(sertraline)、Hypericum(St.John’s Wort)、阿普唑侖(alprazolam)、氯硝西泮(clonazepam)、地西泮(diazepam)、勞拉西泮(lorazepam)、哈拉西泮(halazepam)、氯二氮平(chlordiazepoxide),及其他藥物諸如丁螺環酮(buspirone)、可樂定(clonidine)、帕戈隆(pagoclone)、利培酮(risperidone)、奧氮平(olanzapine)、喹硫平(quetiapine)、齊拉西酮(ziprasidone)、塞來昔布(celecoxib)、吡羅昔康(piroxicam)、帕瑞昔布(parecoxib)、伐地考昔(valdecoxib)、PMI-001、PH-686464、SC-58236、依托昔布(etoricoxib)、羅菲可西保(rofecoxib)、L-776967、羅美昔布(lumiracoxib)、GW-406381、GW-644784、美洛昔康(meloxicam)、SVT-
2016、PAC-10649、CS-706、LAS-34475、西米考昔(cimicoxib)、A-183827.0、或尼美舒利(nimesulide)。
經常用於治療成癮症及藥物濫用之治療劑實例包括(未限制)苯乙肼(phenelzine)、苯烷肼(phenylalkylhydrazine(美國專利號碼4,786,653))、二硫龍(disulfiram(“Antabuse”))、2-亞胺基-5-苯基-4- 唑啶酮、α-甲基-對-酪胺酸或芙莎酸、哌
唑啶酮、α-甲基-對-酪胺酸或芙莎酸、哌 衍生物(美國專利號碼4,325,952)、可樂定(clonidine)連同三環抗憂鬱藥(美國專利號碼4,788,189)、γ-哌哢類諸如麥芽醇或乙基麥芽醇(美國專利號碼4,276,890)、阿坎酸(acamprosate)、加巴噴丁(gabapentin)、胺己烯酸(vigabatrin)、巴氯芬(baclofen)、N-乙醯半胱胺酸、右旋安非他命(nocaine)、莫達非尼(modanafil)、帕羅西汀(paroxetine)、安非他酮(bupropion)、米氮平(mirtazapine)、妥品美(topiramate)、恩丹西酮(ondansetron)、伐尼克蘭(varenicline)、類鴉片受體拮抗劑諸如那曲酮(naltrexone)、納洛酮(naloxone)、納美芬(nalmephine)、恩他酮(antaxone)、L-α-乙醯美沙醇(L-α-acetyl methadol)、潘他唑新(pentazocine)、美妥芬諾(butorphanol)、納布芬(nalbuphine)、丁丙諾非(buprenorphine)、及美沙酮(methadone)。
衍生物(美國專利號碼4,325,952)、可樂定(clonidine)連同三環抗憂鬱藥(美國專利號碼4,788,189)、γ-哌哢類諸如麥芽醇或乙基麥芽醇(美國專利號碼4,276,890)、阿坎酸(acamprosate)、加巴噴丁(gabapentin)、胺己烯酸(vigabatrin)、巴氯芬(baclofen)、N-乙醯半胱胺酸、右旋安非他命(nocaine)、莫達非尼(modanafil)、帕羅西汀(paroxetine)、安非他酮(bupropion)、米氮平(mirtazapine)、妥品美(topiramate)、恩丹西酮(ondansetron)、伐尼克蘭(varenicline)、類鴉片受體拮抗劑諸如那曲酮(naltrexone)、納洛酮(naloxone)、納美芬(nalmephine)、恩他酮(antaxone)、L-α-乙醯美沙醇(L-α-acetyl methadol)、潘他唑新(pentazocine)、美妥芬諾(butorphanol)、納布芬(nalbuphine)、丁丙諾非(buprenorphine)、及美沙酮(methadone)。
經常用於骨質疏鬆症治療且可調節骨礦物質含量之治療劑實例包括(但不限定於)雙膦酸鹽類(bisphosphonates)諸如阿侖膦酸鹽(alendronate(Fosamax®))、利塞膦酸鹽(risedronate(Actonel®))、依替膦酸鹽(etidronate
(Didronel®))、帕米膦酸鹽(pamidronate)、替魯膦酸鹽(tiludronate(Skelid®))、氯屈膦酸鹽(clodronate(Bonefos®;Loron®))、奈立膦酸鹽(neridronate)、奧帕膦酸鹽(olpadronate)、唑來膦酸鹽(zoledronate(Zometa®))、及伊斑膦酸鹽(ibandronate(Boniva®)),選擇性雌激素受體調節劑(SERM)諸如雷洛昔芬(raloxifene(Evista®))、阿佐昔芬(arzoxifene)、氯米芬(clomifene)、巴多昔芬(bazedoxifene)、拉索昔芬(lasofoxifene)、奧美昔芬(ormeloxifene)、它莫昔芬(tamoxifen)、及托瑞米芬(toremifine),合成代謝療法諸如特立帕肽(teriparatide(Forteo®;重組副甲狀腺荷爾蒙)、及雷奈酸鍶(strontium ranelate)、及副甲狀腺荷爾蒙之重組肽片段,雌激素/黃體酮替代療法,單株抗體、核因子kB配位體受體活化劑(RANKL)之抑制劑諸如保髂麗(denosumab)、骨保護素(osteoprotegerin)及胃酶抑素A(Pepstatin A),組織蛋白酶K抑制劑諸如但不限定於OST-4077(呋喃-2-羧酸-(1-{1-[4-氟-2-(2-側氧基-吡咯啶-1-基)-苯基]-3-側氧基-哌啶-3-基胺基甲醯基}-環己基)-醯胺)、亮肽素、Cbz-Phe-Ala-CHN2、Cbz-Leu-Leu-Leu-醛、血清胱蛋白,不可逆半胱胺酸蛋白酶抑制劑如肽鹵甲基酮、肽重氮甲基酮、及環氧化物,靜態不可逆半胱胺酸蛋白酶抑制劑諸如醯氧基甲基酮、氮雜肽、麥克受質(Michael acceptor)如肽乙烯酯、碸及磺酸鹽,可逆半胱胺酸蛋白酶抑制劑諸如肽醛、a-酮基酯及a-酮基醯胺,肽甲基酮及其羥基、烷氧基、芳氧基、
烷硫基、及芳硫基衍生物,1,3-雙-(醯基胺基)-2-丙酮、1,3-雙-(醯基肼基)-羰基、醯基胺基-吡唑酮、哌啶酮、及噻唑-羰基-醯肼,整合素Avb3(技藝中亦稱為玻璃黏連蛋白(vitronectin))拮抗劑,鈣離子阻斷化合物(Ca2+受體拮抗劑,其提高PTH之分泌),降鈣素(MiacalcinÒ),硝酸鹽包括但不限定於單硝酸異山梨酯(ISMO)或硝酸甘油軟膏(NTG),及膳食增補劑諸如鈣及維他命D,及其組合。
本發明之另一實施例為以由採自個體之樣品中量測時鐘基因(例如Cry1及Cry2)表現程度為基準以鑑定需要治療之患者的方法(Bjarnason,G.A.et al.Am.J.Pathol.2001,158,1793;Akashi,M.et.al.Proc.Natl.Acad.Sci.USA,2010,107,15643)。由取自個體之樣品中量測之人類時鐘基因包括Cry1及Cry2之節律性mRNA表現概況顯示日夜生理時鐘存在於周邊組織中(Mohawk,J.A.et al.Ann.Rev.Neurosci.2012,Epub ahead of print)。這些樣品中之日夜生理時鐘相關基因已證實在一天當中有不同變化。而且,人類周邊血液單核細胞中之時鐘基因(例如Cry1及Cry2)表現樣式藉由疾病諸如肥胖症而改變(Tahira,K.et al.Arch.Med.Sci.2011,7,933)。周邊血液單核細胞中之時鐘基因(例如Cry1及Cry2)表現的變化可與血清瘦素、脂聯素、胰島素及hsCRP濃度、血漿脂肪、葡萄糖、褪黑激素及皮質醇濃度有關,而於動物中,與組織包括肝、脂肪、胰臟及骨髂肌中之時鐘基因(例如Cry1及Cry2)表現有關。藉由將取自個體之樣品以式I化
合物接觸處理,再量測節律mRNA或蛋白質表現概況,可鑑定出需要治療的患者及可評估藥理學活性。其他實施例中,一或多種隱花色素之活性可評估(例如)隱花色素結合至標靶諸如Per1、Per2、葡萄糖皮質素受體(GR)、或含Cry識別部位之啟動子序列諸如CLOCK-BMAL1啟動子之能力。
因此,本文所揭示標的之一方面係關於監測個體之Cry媒介性疾病或失調的進展或預後之方法,其包含量測第一段時間取自個體之第一樣品中的一或多種隱花色素之有效量;量測第二段時間取自個體之第二樣品中的一或多種隱花色素之有效量;再將第一樣品中偵檢出之一或多種隱花色素的量與第二樣品中偵檢出之一或多種隱花色素的量,或與參考值相比較。
“診斷(Diagnosis)”、“診斷(diagnose)”、“預後(prognose)”或“預後(prognosis)”並未限定於確定性或幾乎確定性判定個體具有疾病,而係亦包括與健康個體或一般族群相比,判定個體具有提高之具有或發展疾病的可能性。
本文所用之“表現”及“表現程度”包括但不限定於一或多種下列者:基因轉錄成先質mRNA;先質mRNA剪接及經過其他過程以產生成熟mRNA;mRNA安定性;成熟mRNA轉譯成蛋白質(包括密碼子使用及tRNA可利用性);及轉譯產物之糖化及/或其他修飾,如需要用於適當表現及功能。
“公式”、“演算法”或“模型”為任何數學方程式、演算、分析或程式化過程,或統計技術,其採用一或多種連續性或類別性輸入(本文稱為“參數”),再計算出輸出值,偶爾稱之為“指標”或“指標值”。“演算法”之非限制性實例包括總和、比率、及回歸算子諸如係數或指數、值變換及值正規化(包括但未限定為以臨床參數諸如姓別、年齡、身體質量指數、或種族為基準之正規化組合)、規則及指引、統計分類模型、及於歷史族群上訓練之神經網路。特別有用於量測本文所定義之Cry者為線性及非線性方程式及統計分類分析法以將個體樣品中偵檢出之Cry濃度與發展Cry媒介性疾病或失調之個體風險之間的闗係產生“關聯”。
“量測(Measuring)”或“量測(measurement)”意指評估給定物質於臨床或個體衍生樣品中之存在、不存在、或量(quantity或amount)(其可為有效量),包括此物質之定性或定量濃度之衍生,或者另評估個體臨床參數之值或分類。量測(Measurement或measuring)亦可包含型式之定性及物質之鑑別。量測亦包含一或多種Cry結合至標靶之能力,其中該標靶可為period基因或蛋白Per1及Per2、葡萄糖皮質素受體(GR)、或CLOCK-BMAL1基因之啟動子區。Cry之量測型用於診斷、偵檢、或鑑定個體之Cry媒介性疾病或失調,監測個體之Cry媒介性疾病或失調的進展或預後,預測個體之Cry媒介性疾病或失調之再發,或將個體分類成具有發展Cry媒介性疾病或失調之低風險或
高風險者或Cry媒介性疾病或失調之再發者。
本發明上下文中之“風險”係關於事件會在特定期限發生之可能性,例如Cry媒介性疾病或失調之發展,且可意指個體之“絕對”風險或“相對”風險。絕對風險可經由相關時間群組之量測後的實際觀察量測,或者以由已追蹤相關期間之統計有效歷史群組中發展出的指標值進行量測。相對風險意指個體之絕對風險相比於低風險群組之絕對風險或平均族群風險的比值,其可藉由如何評估臨床風險因子而變化。勝算比(給定測試結果之正向事件與負向事件的比例)亦常使用(勝算係根據式p/(1-p),其中p為事件之可能性且(1-p)為無事件之可能性)以無需換算。另外,本發明上下文中可評估之另外之連續量測包括Cry媒介性疾病或失調之發展的時間,或Cry媒介性疾病或失調之不同階段之進展的時間,包括Cry媒介性疾病或失調之進展或發展及治療轉換風險降低比。
本文所揭示標的上下文中之“風險評估(Risk evaluation)”或“風險評估(evaluation of risk)”包含對於事件或疾病狀態發生之可能性(probability)、勝算、或可能性(likelihood),由一種疾病狀態轉換成另一種,亦即由“正常”狀況轉換成處於發展Cry媒介性疾病或失調風險狀況、或由處於風險狀況轉換成Cry媒介性疾病或失調之事件或轉換發生率,或再發性疾病或失調之發展進行預測。風險評估亦可包含Cry媒介性疾病或失調之其他指標的預測,以有關於先前量測族群之絕對或相對方式表達。本發
明方法可用於對轉換成Cry媒介性疾病或失調之風險進行連續性或分類性量測,故可診斷及界定經定義為發展疾病或失調風險類別之個體的風險譜。在分類方案中,本發明可用於區分出正常與處於風險之群體。其他實施例中,本發明可予使用以便區分出處於風險狀況和疾病狀況,或區分出疾病狀況和正常。
本文所用之“樣品”為由個體分離出來之生物樣品且可包括(例示但未限制)全血、血清、血漿、血液細胞、內皮細胞、活體組織、淋巴液、腹水、組織間液(亦稱為“細胞外液”,且涵蓋細胞間的空間中發現的流體,尤其包括齦溝液)、骨髓、精液、腦脊髓液(CSF)、唾液、黏液、痰、汗、尿、或任何其他分泌物、排泄物、或其他體液。
“統計上顯著的”意指該改變大於預期僅只偶然發生(其可為“偽陽性”)的。統計顯著性可藉技藝中已知之任何方法測定。顯著性的常用量測法包括p值,其呈現得到至少如同給定數據點般地極端的結果之可能性,假定該數據點為單只偶然之結果。p值於0.05或更低時,結果通常被視為具高度顯著性。
Cry媒介性疾病或失調之風險可藉量測樣品(例如衍生自個體之樣品)中一或多種隱花色素之“有效量”,再將有效量與參考值相比較,通常使用數學演算法或公式以將得自多個個體之結果的資訊結合成單一量測而偵檢出。經鑑定具有提高Cry媒介性疾病或失調風險之個體可隨意地選出以接受治療方案或治療干預,諸如將本文所定義之式I
化合物以單一藥物療法之方式或與一或多種額外治療劑組合地投予,或者執行手術(其可在單獨地投予式I化合物或與額外治療劑或其他療法組合投予之後或之前執行)。
偵檢樣品中這些隱花色素的方法具有許多之應用。例如,一或多種隱花色素可予量測以協助Cry媒介性疾病或失調之診斷或預後。另一實例中,偵檢隱花色素之方法可用以監測個體對Cry媒介性疾病或失調之治療的反應。另一實例中,這些方法可用以分析或鑑定可於體內或體外調節隱花色素表現之化合物。
本發明可用於對轉換成Cry媒介性疾病或失調之風險進行連續性或分類性量測,故可診斷及界定經定義為處於發展疾病或失調風險之個體類別的風險譜。在分類方案中,本發明可用於區分出正常與處於風險之群體。其他實施例中,本發明可予使用以便區分出處於風險和疾病,或區分出疾病和正常。此不同之使用可能需要個體小組或概況、數學演算法、及/或截止點之不同組合,但進行相同之精確量測法以供所欲之用途。
鑑定處於風險之個體使能夠選擇及開始各種治療干預或治療方案以求延緩、降低、或預防個體之轉換成Cry媒介性疾病或失調。隱花色素蛋白質、核酸、多型體、代謝物、或其他分析物之有效量濃度亦容許在治療過程期間待監測。此方法中,生物樣品可由接受Cry媒介性疾病或失調之治療方案例如治療性處置之個體提供。此治療方案可包括(但不限定於)進行手術及以用於經診斷或鑑定患有
Cry媒介性疾病或失調之個體中的治療劑例如本文所述之式I化合物治療。如有需要,生物樣品得自個體治療之前、期間、或之後之各種不同時間點。例如,藉將個體之隱花色素概況與參考組之隱花色素概況相比較以判定疾病狀態可重覆進行一次以上,其中該個體概況可得自取自每一時間之個別樣品,方法重覆。樣品可取自界定之時間間隔,諸如4小時、8小時、12小時、24小時、48小時、72小時、或熟諳此藝者進行之任何適當時間間隔。
個體基因組成之差異可導致其代謝各種藥物的相對能力不同,該些藥物可調節Cry媒介性疾病或失調之症狀或風險因子。患有Cry媒介性疾病或失調之個體或處於Cry媒介性疾病或失調風險之個體可因年齡、種族、及其他參數而變化。因此,測量本文所定義之一或多種隱花色素之有效量(單獨地或與用於藥物代謝之已知遺傳因子一起組合地)容許預定可預測性濃度,所選定個體中待測試之推定治療劑或預防劑將適於治療或預防個體之Cry媒介性疾病或失調。
欲鑑定適於特定個體之治療劑或藥物,得自個體之試驗樣品亦可暴露至治療劑或藥物,且可測定一或多種隱花色素蛋白質、核酸、多型體、剪接變異體、代謝物或其他分析物之濃度或活性。其他被影響或直接或間接結合至一或多種隱花色素之基因或蛋白質(例如Per1、Per2、GR、CLOCK-BMAL1啟動子等等)亦可量測。一或多種隱花色素之濃度可將衍生自處理Cry媒介性疾病或失調例如治療
或暴露至治療劑或藥物之前或之後的個體之樣品相比較,或者可將一或多個因為此治療或暴露而顯現風險因子改善之個體的樣品相比較。
核酸可以熟諳此藝者已知之許多方法得自樣品,例如萃取法包括(例如)溶劑萃取法、親和純化法及離心法。選擇性沈澱法亦可純化核酸。層析法亦可使用包括凝膠過濾、離子交換、選擇性吸附、或親和力結合層析法。核酸可為(例如)RNA、DNA或可合成為cDNA。核酸可使用技藝中已知之微陣列技術,例如Affymetrix陣列其後多維標度技術進行偵檢。參見R.Ekins,R.and Chu,F.W.(1999)Trends Biotechnol.17:217-218;D.D.Shoemaker,et al.,(2001)Nature 409(6822):922-927及美國專利號碼5,750,015。
如有需要,樣品可藉(例如)預分離法製備以增強一或多種隱花色素之可偵檢性。預分離法包括(例如)汽巴龍藍瓊脂糖層析法(Cibacron blue agarose chromatography)、尺寸排阻層析法、離子交換層析法、肝素層析法、凝集素層析法、親和層析法、單股DNA親和層析法、序列萃取法、凝膠電泳法、及液相層析法。分析物亦可在偵檢之前修飾。樣品可藉將高量存在或者可能干擾樣品中受闗注分子之偵檢的蛋白質移出而予以預分離。例如,於血液血清樣品中,血清白蛋白以高量存在且可能混淆一或多種隱花色素之分析。故,血液血清樣品可藉使用(例如)包含可特異地結合血清白蛋白之吸附劑的基質將血清白蛋白移出而
予以預分離,親和柱或抗血清白蛋白抗體可予使用。
其他實施例中,樣品中有關之分子可藉高解析電泳法例如一或二維凝膠電泳法分離。分離份可分離出來,再藉氣相離子光譜術進一步分析。較佳地,二維凝膠電泳法係用於產生二維陣列之斑點,包括一或多種隱花色素。例如參見Jungblut and Thiede,(1997)Mass Spectr.Rev.16:145-162。二維凝膠電泳法可使用技藝中已知之方法進行。例如參見Deutscher ed.,Methods in Enzymology vol.182。典型地,樣品可藉(例如)等電聚焦法分離,此期間樣品中之一或多種隱花色素於pH梯度中分離直至彼等到達彼等之淨電荷為零之斑點處為止(亦即等電點)。此第一分離步驟導致一維陣列。一維陣列中之分子進一步使用通常可與第一步驟中所用者區分之技術分離。例如,於第二維中,藉等電聚焦法分離出之有關分子使用聚丙烯醯胺凝膠諸如於十二烷基硫酸鈉之存在下進行聚丙烯醯胺凝膠電泳法(SDS-PAGE)進一步分離。SDS-PAGE凝膠容許以分子質量為基準進行進一步分離。典型地,二維凝膠電泳法可化學地在複雜混合物當中分離出分子質量範圍在1000-200,000Da之不同的有關分子。
二維陣列中之有關分子可使用技藝中已知之任何適當方法偵檢。例如,凝膠中有關之分子可予標記或染色(例如考馬斯藍(Coomassie Blue)或銀染色)。如果凝膠電泳產生相當於本發明一或多種隱花色素分子量之斑點,該斑點可被切除且藉(例如)氣相離子光譜術、質譜術、或高效能
液相層析法進一步分析。另外,含有關分子之凝膠可藉施加電場而轉移至惰性膜。然後,約相當於有關分子之分子量的膜上斑點可藉(例如)氣相離子光譜術、質譜術、或HPLC分析。
隨意地,有關分子可在分析前進行修飾以改善其解析度或測知其本體。例如,樣品可在分析前接受蛋白分解消化作用。任何蛋白酶均可使用。以很可能地將蛋白質裂解成不連續數目之片段的蛋白酶諸如胰蛋白酶特別有用。因消化作用所致之片段可充作有關分子之指紋,藉以能夠進行彼等之間接偵檢。此在當有具有類似分子質量之有關分子可能混淆討論中之較佳分子亦即隱花色素時特別有用。而且,蛋白分解片段化可用於高分子量分子,因為較小分子較易於藉質譜術解析之故。另一實例中,分子可予修飾以改善偵檢之解析度。例如,神經胺酸苷酶可用以由糖蛋白中移出終端唾液酸殘基以改善結合至陰離子吸附劑(例如陽離子交換陣列)及改善偵檢之解析度。另一實例中,分子可藉由可特異地結合至另一分子實體的特定分子量標籤之連接,進一步區分彼等而修飾。隨意地,偵檢此有關之修飾分子後,分子本體可進一步藉將修飾版之物理及化學特徵於蛋白質資料庫(例如SwissProt)中匹配而測定。
一旦捕獲於基質例如生物晶片或抗體上,則任何適當方法諸如本文所述者以及技藝中已知之其他方法可用以量測樣品中之一或多種隱花色素。此分子之濃度或量的實際量測可使用技藝中已知之任何方法測定。這些方法包括
(未限制)質譜術(例如雷射脫附/離子化質譜術)、螢光(例如三明治免疫分析法)、表面電漿子共振、橢圓偏光術及原子力顯微鏡。方法可進一步包括藉一或多種微陣列、PCR法、質譜術(例如包括且未限制為ESI-MS、ESI-MS/MS、ESI-MS/(MS)n、基質輔助雷射脫附/離子化飛行時間質譜術(MALDI-TOF-MS)、表面增強雷射脫附/離子化飛行時間質譜術(SELDI-TOF-MS)、矽上之脫附/離子化(DIOS)、二次離子質譜術(SIMS)、四極飛行時間(Q-TOF)、大氣壓化學離子化質譜術(APCI-MS)、APCI-MS/MS、APCI-(MS)n、大氣壓光離子化質譜術(APPI-MS)、APPI-MS/MS、及APPI-(MS)n、四極質譜術、傅里葉(Fourier)變換質譜術(FTMS)、及離子阱質譜術)、核酸晶片、北方墨點雜交法、TMA、SDA、NASBA、PCR、即時PCR、反轉錄酶PCR、即時反轉錄酶PCR、原位PCR、層析分離連結質譜術、使用固定抗體進行之蛋白質補獲或藉傳統免疫分析法。例如參見美國專利5,723,591;5,801,155及6,084,102及Higuchi,1992 and 1993。PCR分析法可(例如)於多孔式血結構中或於晶片諸如BioTrove OPEN ARRAY晶片(BioTrove,Woburn,MA)中進行。
例如,相當於隱花色素之序列資料庫入口內的序列可用於建構探子,該探子於(例如)北方墨點雜交分析或可特異地且較佳地定量地擴增核酸序列之方法中用於偵檢RNA序列。另一實例,序列可用以建構引子,該引子可特異地或選擇性地雜交至隱花色素序列且其於(例如)以擴
增為基底之偵檢法諸如以反轉錄為基底之聚合酶鏈鎖反應(RT-PCR)例如定量即時RT-PCR中用於擴增此序列。當基因表現之改變與基因擴增、刪除、多型現象、及突變有關時,則試驗及參考族群中的序列比較可藉將個體及參考細胞族群中之相對量之經檢查DNA序列相比較來進行。本文所用之術語“特異地(或選擇性地)雜交”當關連到核酸時,意指結合反應,其為核酸異源族群中之核酸存在的決定因素。故於指定之分析條件下,特定之核酸探子(包括抑制性核酸)可結合或雜交至特定之有關核酸,該結合或雜交至少兩倍於背景值且並未以顯著之量實質地結合或雜交至樣品中存在的其他核酸。
隱花色素的濃度亦可藉免疫分析法測定。抗體可為單株、多株、嵌合、或前述抗體之片段,如同本文詳細討論地,且偵檢反應產物之步驟可以任何適當之免疫分析法進行。詞組“特異地(或選擇性地)結合”至抗體或“特異地(或選擇性地)與...產生免疫反應”當關聯到蛋白質或肽時,意指結合反應,其為蛋白質或其他生物學之異源族群中之蛋白質存在的決定因素。故於指定之免疫分析法條件下,該特定抗體結合至特定蛋白質之能力至少兩倍於背景值且並未以顯著之量實質地結合至樣品中存在的其他蛋白質。於此條件下特異結合至抗體可能需要選出對特定蛋白質具特異性的抗體。例如可選擇針對來自特定物種諸如大鼠、小鼠、或人類之隱花色素的多株抗體以僅得到特異地與彼隱花色素產生免疫反應而不與其他蛋白質產生免疫反應的多
株抗體,惟隱花色素之多型變體及對偶基因除外。此選擇可藉由扣除掉可與來自其他物種之隱花色素交互反應的抗體而達成。
根據本發明進行之免疫分析法可為均相分析法或非均相刀析法。均相分析法中,免疫反應通常包含特異性抗體(例如抗隱花色素蛋白質抗體)、標記分析物、及有關之樣品。於將抗體結合至標記分析物後,將標記所產生的記號直接或間接地修飾。免疫反應及其程度之偵檢均可於均相溶液中進行。可使用之免疫化學標記包括自由基、放射性同位素、螢光染料、酵素、噬菌體、或輔酶。
非均相分析法中,試劑通常為樣品、抗體、及用於產生可偵檢信號的工具。可使用如上所述的樣品。抗體可固定於載體諸如珠(諸如蛋白A及蛋白G瓊脂糖珠)、盤或載片上,再與懷疑含有抗原的試樣於液相中接觸。然後將載體由液相中分離出,再使用可產生此信號的工具以檢查載體相或液相。信號係於樣品中分析物的存在有關。用於產生可偵檢信號的工具包括可偵檢標記之使用。可偵檢標記之實例包括磁珠(例如DYNABEADSTM)、螢光染料、酵素(例如辣根過氧化物、鹼性磷酸酶及其他常用於ELISA中者)、放射標記(例如35S、125I、131I)、及螢光標記(例如螢光素、Alexa、綠色螢光蛋白、玫瑰紅)及比色標記諸如膠體金或著色玻璃或根據已知技術之塑膠珠。
此外,樣品中有關之分子可使用間接分析法偵檢,其中(例如)係使用第二標記抗體以偵檢結合之隱花色素特異
性抗體,及/或於競爭或抑制分析法中,其中(例如)可結合至隱花色素明顯之抗原決定位的單株抗體係與混合物同時保溫。例如,如果待偵檢之抗原含有第二結合位,則結合至彼位之抗體可被軛合至可偵檢基團上,再於分離步驟之前,加至液相反應溶液中。固體載體上可偵檢標記之存在顯示測試樣品中之抗原的存在。量測抗體-抗原複合物之量或存在的方法包括(例如)螢光、冷光、化學冷光、吸光度、反射率、透射率、雙折射或折射率(例如表面表面電漿子共振、橢圓偏光術、共振鏡法、光柵偶合器波導法或干涉量度法)。光學方法包括顯微鏡術(共焦及非共焦二者)、成像法及非成像法。電化學法包括電位測定法及電流測定法。射頻法包括多極共振光譜術。適當免疫分析法實例包括(但不限定於)免疫墨點法(例如西方墨點法、狹縫墨點分析法)、免疫沈澱法、免疫螢光法、化學冷光法、電化學冷光法(ECL)或酵素連結免疫分析法例如酵素連結免疫吸附分析法(ELISA)及放射免疫分析法(RIA)。通常參見E.Maggio,Enzyme-Immunoassay,(1980)(CRC Press,Inc.,Boca Raton,Fla.);亦參見美國專利號碼4,727,022;4,659,678;4,376,110;4,275,149;4,233,402;及4,230,767。這些方法亦述於(例如)Methods in Cell Biology:Antibodies in Cell Biology,volume 37(Asai,ed.1993);Basic and Clinical Immunology(Stites & Terr,eds.,7th ed.1991);及Harlow & Lane,同上。所有均併入本文中以供參考。
免疫分析法可用以測定樣品中是否有一或多種隱花色素以及於樣品中的量。抗體-標誌複合物的量可藉與標準品比較而測知。標準品可為(例如)已知化合物或已知存在於樣品中的另一蛋白質。如上所提,一或多種隱花色素的測試量不必以絕對單位量測,只要該量測單位可與對照組比較即可。
蛋白質經常以複數個以可偵檢地不同質量為特徵的不同形式存在於樣品中。這些形式可由轉譯前及轉譯後修飾其中一者或二者所致。轉譯前修飾的形式包括對偶基因變種、剪切變種及RNA編輯形式。轉譯後修飾形式包括因蛋白分解裂解(例如母蛋白質之片段)、糖化、磷酸化、脂質化、氧化、甲基化、胱胺酸基化、磺酸化及乙醯化所致的形式。抗體亦可用於偵檢蛋白質、多肽、突變、及多型體之轉譯後修飾,諸如酪胺酸磷酸化、蘇胺酸磷酸化、絲胺酸磷酸化、糖化(例如O-GlcNAc)。此抗體特別地偵檢有關之蛋白質或諸蛋白質中的磷酸化胺基酸,且可用於本文所述之免疫墨點法、免疫螢光法、及ELISA分析法中。這些抗體為熟諳此藝者詳知,且為市面可得。轉譯後修飾亦可使用介穩離子於反射模式基質輔助雷射脫附/離子化飛行時間質譜術(MALDI-TOF)(Wirth,U.et al.(2002)Proteomics 2(10):1445-51)中測定。蛋白質包括特定蛋白質及其所有修飾形式之總體在本文中稱之為“蛋白質集群”。特定蛋白質之所有修飾形式之總體(排除該特定蛋白質本身)在本文中稱之為“修飾蛋白質集群”。任何隱花色
素之修飾形式本身亦可使用於本文所揭示之方法中。某些情況下,修飾形式在診斷上顯現比本文所提及之特定形式更佳的識別能力。修飾形式最初可藉技藝中已知之任何方法偵檢。
另外,可量測隱花色素蛋白質及核酸。術語“代謝物”包括代謝過程之任何化學或生化產物,諸如藉由生物分子(例如蛋白質、核酸、醣類、或脂肪)之製程、裂解或消耗所產生之任何化合物。代謝物可以熟諳此藝者已知之各種方法偵檢,包括折射率光譜術(RI)、紫外線光譜術(UV)、螢光分析法、放射化學分析、近紅外線光譜術(near-IR)、核磁共振光譜術(NMR)、光散射分析(LS)、質譜術、熱解質譜術、濁度測定法、色散拉曼光譜術(dispersive Raman spectroscopy)、氣相層析結合質譜術、液相層析(包括高效能液相層析(HPLC)),其可與質譜術結合,基質輔助雷射脫附離子化-飛行時間(MALDI-TOF)結合質譜術、離子噴霧光譜術結合質譜術、毛細管電泳、離子遷移率光譜術、表面增強雷射脫附/離子化(SELDI)、光學方法、電化學法、原子力顯微鏡術、射頻法、表面電漿子共振、橢圓偏光術、NMR及IR偵檢法。(參見國際專利申請案公開號WO 04/056456及WO 04/088309,每一者均整體併入本文中以供參考)。就此方面,其他分析物可使用上述偵檢法、或熟知技藝者已知之其他方法量測。例如,可使用螢光染料諸如尤其使用Fluo系列、Fura-2A、Rhod-2來偵檢樣品中之循環鈣離子(Ca2+)。其他代謝物可使用特別設計
或量身訂做來偵檢此些代謝物的試劑進行類似的偵檢。
Cry媒介性疾病或失調可包含一或多種隱花色素活性之改變,或一或多種隱花色素結合至標靶之能力。無需被理論所束縛,隱花色素蛋白質咸信可結合至Period蛋白Per 1及/或Per2成雜二聚物,然後結合至CLOCK-BMAL1之啟動子區以促進回饋迴路中的轉錄阻抑,其可影響許多的代謝過程。故量測根據本發明之一或多種隱花色素的有效量可包括評估Cry蛋白結合至Per1及/或Per2、至葡萄糖皮質素受體(GR)、或熟諳此藝者已知之任何其他Cry結合標靶之能力的提高或降低。蛋白質-蛋白質交互作用之量測可藉技藝中已知之方法促進,包括免疫共沈澱法、酵母菌雙雜合分析法、表面表面電漿子共振、雙分子螢光互補法、串聯親和純化法、噬菌體展示法、螢光偏振極化/各向異性、雙偏振極化干涉測量法、螢光關聯光譜術、螢光共振能量轉移等等。
一或多種隱花色素之活性亦可藉結合至DNA序列亦即CLOCK-BMAL1基因之啟動子區、或含有被一或多種隱花色素所識別之結合位的基因之啟動子區之能力的提高或降低來量測。“啟動子”、“啟動子序列”、或“啟動子區”可結合細胞中的RNA聚合酶、引發下游(3’方向)編碼序列之轉錄,藉此控制其表現。欲達定義本發明之目的,啟動子序列於其3’藉轉錄引發位結合且延伸上游(5’方向)至包括以超過背景值之可偵檢程度引發轉錄所需之最小數目之鹼基或元件。啟動子內,將發現轉錄引發位(方便地例如
藉以核酸酶S1作圖來定義)序列、以及負責RNA聚合酶結合之蛋白質結合結構域(共同序列)序列。啟動子可全體地衍生自天然基因,或由自然界發現之不同啟動子所衍生之不同元件所組成,或者甚至包含合成DNA片段。大多數情況下,調控序列的精確邊界尚未被完全定義,不同長度之DNA片段可能具有相同的啟動子活性。
CLOCK-BMAL1啟動子(或含有Cry結合或識別位之任何其他啟動子區)可以“可操作地連接”至報導子基因。術語“可操作地連接”意指單一核酸片段上之核酸序列的聯合,故一者的功能被其他者所影響。例如,當啟動子可影響編碼序列之表現時(亦即編碼序列係在啟動子之轉錄控制之下),則啟動子與彼編碼序列可操作地連接。編碼序列可以有義或反義方向可操作地連接至調控序列。術語“報導子基因”意指編碼鑑定因子的核酸,該鑑定因子可以報導子基因之效應為基底被鑑定出,其中該效應係用於追蹤核酸之繼承性,以鑑定出已繼承之有關核酸的細胞或生物體,及/或量測基因表現誘導作用或轉錄。已知且用於技藝中之報導子基因實例包括:螢光素酶(Luc)、綠色螢光蛋白(GFP)、鹼性磷酸酶(ALP)、氯霉素乙醯基轉移酶(CAT)、β-半乳糖苷酶(LacZ)、β-葡萄糖醛酸酶(Gus)等等。選擇性標誌基因亦可視為是報導子基因。啟動子-報導子基因架構可含於質體或表現載體中,質體或表現載體再轉移或轉染至細胞中。報導子基因之表現可藉測定基因產物之活性,例如如果使用以上舉例之報導子基因則藉測
定酵素活性而偵檢。
術語“質體”意指帶有基因之染色體外的元件,其並非細胞中央代謝的一部分,且通常為環狀雙股DNA分子形式。此元件可為自主複製序列、基因組整合序列、噬菌體或核苷酸序列、線性、環狀、或超螺旋之單或雙股DNA或RNA,衍生自任何來源,其中一些核苷酸序列已連結或重組至唯一架構中,該架構可將供選定基因產物用之啟動子片段及DNA序列連同適當3’未轉譯序列一起引入細胞中。術語“表現載體”意指載體、質體或載劑,其經設計使在轉形至宿主後能夠使插入核酸序列表現。載體可藉技藝中已知之方法引入期望之宿主細胞中,諸如轉染法、電穿孔法、顯微注射法、轉導法、細胞融合、DEAE聚葡糖、磷酸鈣沈澱法、脂質轉染法(溶體融合)、基因槍之使用、或DNA載體運體。任何細胞均可用以進行報導子分析法,諸如原核細胞或真核細胞。較佳地,細胞可為細菌細胞、真菌細胞、酵母菌細純、線蟲細胞、昆蟲細胞、魚細胞、植物細胞、鳥類細胞、動物細胞、及哺乳動物細胞。細胞可為初代細胞或可以細胞系方式連續傳代。細胞及細胞系實例為熟諳此藝者已知。
量測一或多種隱花色素結合至DNA序列之活性或能力的其他方法包括染色質免疫沈澱分析法、電泳遷移率變動分析法、DNA拉下分析法(DNA pull-down assay)、微盤捕獲及偵檢法(microplate capture and detection)等等。
隱花色素蛋白質、核酸、多型體、代謝物、或其他分
析物的有效量濃度、或隱花色素蛋白質或直接或間接地結合至隱花色素蛋白質之標靶的活性可繼而測定並與參考值(例如疾病狀態已知之對照組個體或族群、或指標值或基線值)相比。參考樣品或指標值或基線值可取自或衍生自一或多個已暴露至該治療之個體,或可取自或衍生自一或多個處於發展Cry媒介性疾病或失調之低風險的個體,或可取自或衍生自因暴露至治療而顯現疾病風險因子改善之個體。另外,參考樣品或指標值或基線值可取自或衍生自一或多個尚未暴露至該治療之個體。例如,樣品可收集自已接受Cry媒介性疾病或失調之起始治療及監測該治療進展之接續疾病或失調治療的個體。一些實施例中,第一樣品可於第一段時間取自個體,例如在以本文所定義式I化合物單獨地或與一或多種額外治療劑組合地治療之前,其後量測或偵檢一或多種如本文所述之隱花色素(或隱花色素標靶)。其後,第二樣品可於第二段時間取自個體,例如在以本文所定義式I化合物單獨地或與一或多種額外治療劑組合地治療之後,再量測一或多種隱花色素或隱花色素標靶。任何數目的樣品可以任何時間間隔在整個治療期間採取以評估其有效性。
參考值亦可包含由族群研究中之風險預測演算法或計算指數諸如本文所揭示者所衍出之值。本文中之類似術語為“對照組”,其可為(例如)正常個體或非疾病個體諸如未能偵檢出Cry媒介性疾病或失調之個體的正常個體可比較血清中存在的隱花色素之平均量或中位量。對照組的量係
於與量測測試量相同或類似的實驗條件下量測。相關性可考慮到測試樣品中隱花色素的存在與否以及對照組中相同分子的偵檢頻率。相關性可將這些因素均考慮以幫助疾病狀態之測定。
未患有Cry媒介性疾病或失調、或不預期會發展Cry媒介性疾病或失調之彼些個體的參考組概況亦可根據本文所揭示之方法準備。一或多種隱花色素的量測亦可用於產生取自患有Cry媒介性疾病或失調之個體的“個體概況”。個體概況可與參考組概況相比以診斷或鑑定出處於發展Cry媒介性疾病或失調之風險的個體、以監測疾病之進展、以及疾病之進展速率、以及監測治療方式或個體處置之有效性。
本發明之參考組及個體概況可含於機器可讀取之媒介物中諸如(但不限定於)類比或數位磁帶例如尤其可藉VCR、CD-ROM、DVD-ROM、USB flash media讀取者。此機器可讀取之媒介物亦可含有額外之測試結果,諸如(未限制)臨床參數及傳統實驗室風險因子之量測。另外或額外地,機器可讀取之媒介物亦可包含個體資訊諸如病史或相關家族史。機器可讀取之媒介物亦可含有與其他風險演算法及計算指數諸如本文所述者有關之資訊。
本文揭示之任何方法中,得自樣品的數據可由偵檢工具直接送至含診斷演算法的電腦中。另外,所得數據可手工地或經由自動化工具送至含有診斷演算法的個別電腦中。因此,本發明之實施例包括將隱花色素之偵檢與Cry
媒介性疾病或失調的可能診斷產生關聯之方法。該相關性可考慮到一或多種隱花色素於樣品中的量且與對照組(例如於未能偵檢出Cry媒介性疾病或失調之正常個體)之量相比較(隱花色素之向上或向下調節)。相關性可考慮到測試樣品中隱花色素的存在與否以及對照組中相同分子的偵檢頻率。相關性可將這些因素均考慮以幫助測知個體是否患有Cry媒介性疾病或失調。
數據分析可包括測定所偵檢標誌之信號強度(例如峰高度)及移出“離群值”(偏離預定統計分佈之數據)之步驟。觀察峰可予正規化,再計算每一峰高度相對於一些參考組的過程。例如,參考組可為因儀器及化學品(例如能量吸收分子)所產生之背景雜音,將其級別設定為零。每一有關分子偵檢出之信號強度可以期望等級(例如100)之相對強度顯示。另外,標準品(例如血清蛋白質)可加入樣品中以使得自標準品之峰可用於作為參考組而計算出所偵檢之每一有關分子的相對強度。
所得數據可轉變或轉換成各種顯示格式。一格式中,稱之為“光譜視圖或滯留物圖”,可顯示標準之光譜視圖,其中該視圖描述分子以每一特定分子量到達偵檢器的量。另一格式稱之為“峰圖”,僅有峰高度及質量資訊由譜視圖中保留,得到更乾淨的影像且使有關之分子能夠具有更易見到之幾近相同之分子量。又另一格式稱為“凝膠視圖”,得自峰視圖的每一質量可以每一峰高度為基底而轉化成灰階影像,產生類似於電泳凝膠帶狀條紋的外觀。又另一格
式稱為“3-D疊圖(3-D overlay)”,可將某些光譜重疊以研究相對峰高度之細微變化。又另一格式稱為“差異圖視圖”,可將二或多個光譜相比,便利地突出惟一的有關分子,其在樣品間被向上及向下調節。得自任二樣品之概況(光譜)可目視比較。又另一格式中,可使用Spotfir散佈圖,其中受偵檢之有關分子以點狀繪於圖中,其中圖的一個軸代表所偵檢隱花色素的表觀分子量且另一個軸代表所偵檢隱花色素的信號強度。對每一樣品而言,所偵檢出之有關分子及樣品中存在之分子的量可存於電腦可讀取之媒介物中。此數據可繼而與對照組或參考組概況或參考值(例如對照組(例如未能偵檢出Cry媒介性疾病或失調的個體)偵檢出之分子概況或量)相比較。
本文所揭示之方法中所產生的數據可使用利用分類模型進行之圖型識別法分類。一些實施例中,使用樣品諸如“已知樣品”產生的數據可繼而用以“訓練”分類模型。“已知樣品”為已預分類之樣品(例如疾病或無疾病)。使用已知樣品所產生的數據可繼而用以“訓練”分類模型。“已知樣品”為已預分類之樣品。可用以形成分類模型之數據可稱之為“訓練數據集”。一旦受訓練,該分類模型可辨識使用未知樣品所產生之數據中的圖型。分類模型可繼而用以將未知樣品分類。此可用於(例如)預測特定生物樣品是否與某些生物病症有關(例如致病或未致病的)。用以形成分類模型之訓練數據集可包含原始數據或預處理數據。一些實施例中,原始數據可直接由飛行時間光譜或質譜中獲得,
且繼而可隨意地以任何適當方式“預處理”。預處理步驟諸如這些可用以降低用於訓類分類模型之數據的量。
分類模型可使用任何適當的統計分類(或“學習”)法形成,該統計分類(或“學習”)法試圖以數據中存在之目標參數為基底將數據體分離成類。分類方法可為監督或未監督。監督或未監督分類法的實例乃述於Jain,“Statistical Pattern Recognition:A Review”,IEEE Transactions on Pattern Analysis and Machine Intelligence,Vol.22,No.1,January 2000中,其乃整體併入本文中以供參考。監督分類中,含有已知類別之實例的數據經提交至學習機轉,該學習機轉學習一或多種界定每一已知類別的關聯組集。新數據可繼而應用至學習機轉,該學習機轉繼而使用所學習之關聯將新數據分類。
監督分類法之實例包括線性回歸法(例如多重線性回歸(MLR)、淨最小平方(PLS)回歸法及主成分回歸法(PCR))、二元決策樹(例如遞迴分割法諸如CART-分類與迴歸樹)、類神經網路諸如反向傳播網路、區別分析法(例如貝葉斯分類器(Bayesian classifier)或費雪分析法(Fischer analysis))、邏輯分類器、及支援向量分類器(支援向量機)。較佳之監督分類法為遞迴分割法(美國專利申請案公開號20020138208)。未監督分類法試圖以訓練數據集之類似性為基底學習分類,而未將衍生訓練數據集的光譜預先分類。未監督學習法包括集群分析。集群分析試圖將數據分成“群集”或群組,該“群集”或群組理想地應具有
彼此極為類似的成員,且與其他群集極為不同。類似性乃繼而使用一些距離尺度量測,其量測數據項之間的距離,且將彼此較密切的數據項群集一起。群集技術包括MacQueen’s K-means演算法及Kohonen氏自我組織映射圖演算法(Kohonen’s Self-Organizing Map algorithm)。主張用於分類生物資訊的學習演算法乃述於(例如)國際專利申請案公開號WO 01/31580及美國專利申請案公開號20020193950、20030004402、及20030055615中。其他分類法包括使用單一最大分離分析(Unified Maximum Separability Analysis(“USMA”))分類器非線性版之多變量預測模型。USMA分類器之細節乃述於美國專利申請案公開號20030055615中。
其他分類演算法及公式尤其包括(但不限定於)主成分分析法(PCA)、互關聯、因素軸旋轉、邏輯回歸(LogReg)、線性區別分析法(LDA)、Eigengene線性區別分析(ELDA)、隨機森林法(RF)、遞迴分割樹(RPART)、以及其他相關決策樹分類技術、縮小重心法(Shrunken Centroids(SC))、StepAIC、Kth-最近鄰居法(Kth-Nearest Neighbor)、Boosting法、決策樹、神經網路、貝葉斯網路(Bayesian Networks)、支援向量機、留一法(LOO)、10倍交叉驗證法(10-Fold CV)、及隱馬科夫模型(Hidden Markov Model)。
一或多種隱花色素之偵檢及相關性亦可使用任何適當方法,包括軟體包,例如Applied Maths,GenExploreTM、
雙向集群分析法、主成分分析法、區別分析法、自我組織映射圖;BioDiscovery,Inc.,Los Angeles,California(ImaGeneTM,特殊影像處理及數據提取軟體,MatLab®動力支持;GeneSight:階層分群法、人工神經網路(SOM)、主成分分析法、時間系列;AutoGeneTM;CloneTrackerTM);GeneData AG(Basel,Switzerland);分子圖型識別網站於MIT之Whitehead Genome Center;Rosetta Inpharmatics,Kirkland,Washington。ResolverTM Expression Data Analysis System;Scanalytics,Inc.,Fairfax,VA。其微陣列分析套件使研究者能夠取得、看見、處理、及分析基因表現微陣列數據;TIGR(基因組研究所The Institute for Genome Research)提供陣列分析的軟體工具。例如亦參見Eisen and Brown,(1999)Methods Enzymol.303:179-205。
定性疾病狀態之方法的某些實施例中,該些方法進一步包含以疾病或失調狀態為基底進行臨床治療之處置及改善。例如,如果本發明方法之結果不確定或者有需要證實疾病或失調狀態的原因,則醫師可開立更多的試驗(例如CT掃描、PET掃描、MRI掃描、PET-CT掃描、X光、生檢、血液試驗。另外,如果狀態顯示治療適當,則醫師可安排個體之治療計劃。其他情況下,替代地或除了手術以外,個體可接受治療性治療(諸如將本文所定義之式I化合物單獨地或與一或多種額外治療劑組合地投予)。不需要採取進一步行動。又,如果結果顯示治療成功,則可能需要維持療法或者不需更進一步處置。
本文揭示之標的亦提供於個體之臨床治療後再度量測隱花色素之方法。這些情況下,該些方法係用以監測Cry媒介性疾病或失調之狀態,例如對治療之反應,疾病之緩解或疾病之進展。該些方法亦可於個體接受每次治療後重覆進行,得以令醫師追蹤治療之有效性。如果結果顯示治療無效,則可因此改變治療過程。
本發明提供用於定性疾病狀態及/或偵檢或診斷疾病之套組,其中該套組可用以偵檢一或多種隱花色素。例如,套組可用以偵檢本文所述之任一或多種隱花色素,其一或多種隱花色素差別地存在於疾病個體及正常個體之樣品中。本發明之套組有許多之應用。例如,套組可用於本文所述之本發明任一方法中,尤其諸如用以區分個體是否患有Cry媒介性疾病或失調或具有陰性診斷,故幫助診斷。另一實例中,套組可用以藉使用體外或體內之Cry媒介性疾病或失調之動物模型鑑定出可調節一或多種隱花色素表現之化合物,可調節一或多種隱花色素活性之化合物(亦即影響一或多種隱花色素結合至標靶諸如Per1、Per2、葡萄糖皮質素受體(GR)、或被隱花色素所識別之啟動子序列諸如CLOCK-BMAL1啟動子或任何其他啟動子序列之能力)。另一實例中,套組可用以鑑定出如本文所定義之一或多種隱花色素蛋白之結合標靶。
本發明之套組可包括偵檢試劑,例如可藉具有同源性核酸序列特異地鑑定一或多種隱花色素核酸之核酸,諸如寡核苷酸序列、引子、或適配體,與一部分核酸互補或針
對以包裝一起之核酸編碼之蛋白質的抗體。寡核苷酸可為基因片段。寡核苷酸可為單股或雙股。例如寡核苷酸可為200、150、100、50、25、10或更少核苷酸長度。另外,偵檢試劑可為一或多種抗體,其可特異地或選擇性地結合至一或多種隱花色素蛋白或標靶。套組可將核酸或抗體(已結合至固體基質或與供將彼等結合至基質之試劑分別包裝)、對照組配方(陽性及/或陰性)、及/或可偵檢標記尤其諸如螢光素、綠色螢光蛋白、玫瑰紅、花青染料、Alexa染料、螢光素酶、放射標記含於個別容器中。用於進行分析法及用於疾病關聯性之教示(例如書面、磁帶、VCR、CD-ROM等等)可包括於套組中。
例如,偵檢試劑可固定於固體基質諸如多孔帶條上以形成至少一個偵檢部位。多孔帶條之偵檢區的量測可包括複數個含有核酸的部位。測試帶條亦可含有供陰性及/或陽性對照組用之部位。另外,對照組部位可座落在有別於測試帶條之個別帶條上。隨意地,不同偵檢部位可含有不同量的固定化核酸,例如第一偵檢部位含有較高量且接續部位含有較低量。加入測試樣品後,顯示可偵檢信號之部位數提供定量顯示之存在於樣品中的隱花色素量。偵檢部位可以任何適當可偵檢形狀配置且典型地為跨度測試帶條寬度的棒或點之形狀。基板陣列可在(例如)固體基板例如如同於美國專利號碼5,744,305所述之“晶片”上。另外,基板陣列可為溶液陣列例如xMAP(Luminex,Austin,TX)、Cyvera(Illumina,San Diego,CA)、CellCard(Vitra
Bioscience,Mountain View,CA)及Quantum Dots’Mosaic(Invitrogen,Carlsbad,CA)。套組亦可含有試劑、及/或酵素以用於擴增或分離出樣品DNA。套組可包括用於即時PCR之試劑,例如TaqMan探子及/或引子、及酵素。
一些實施例中,套組包含:(a)基板,其上含有吸附劑,其中該吸附劑保留或以另外方式適用於結合隱花色素,及b)藉將樣品與吸附劑接觸再偵檢吸附劑所保留的隱花色素之教示。一些實施例中,套組可包含洗提液(作為替代品或與教示組合)或製造洗提液之教示,其中該吸附劑與該洗提液之組合得以使用氣相離子光譜術進行隱花色素之偵檢。
其他實施例中,套組可包含第一基板,該第一基板於其上包含吸附劑(例如以吸附劑官能化之微粒),及第二基板,第一基板可放置在該第二基板上以形成探針,其可被移出再插至機器諸如氣相離子光譜儀中。其他實施例中,套組可包含單一基板,其為伴隨著吸附劑於基板上之探針形式,該基板可移出並插至機器中。另一實施例中,套組可進一步包含預分離旋轉柱(例如汽巴龍藍瓊脂糖管柱(Cibacron blue agarose column)、抗-HSA瓊脂糖管柱、K-30尺寸排阻管柱、Q-陰離子交換旋轉管柱、單股DNA管柱、凝集素等等)。另一實施例中,套組包含(a)抗體,其特異地結合至一或多種隱花色素;及(b)偵檢試劑。抗體可(例如)為針對隱花色素基因之基因產物的抗體。
隨意地,套組可進一步包含標準品或對照組資訊使得
測試樣品可與對照組資訊標準相比較以測知樣品中偵檢出之一或多種隱花色素的測試量是否為與Cry媒介性疾病或失調之診斷一致的診斷量。
雖然一些變化已於上文詳細說明,但亦可能有其他的修飾或添加。尤其,除了本文所提及者之外,亦可提供進一步的特性及/或變化。例如,上述之執行可導向所揭示特性之各種組合及次組合,及/或以上所揭示之一些進一步特性的組合及次組合。此外,本文所述之邏輯流程並不需要所示的特定順序、或接續順序來達到期望結果。其他實施例可在申請專利範圍之範圍內。
下列反應圖,反應圖I、II、III、IV、V、及VI描述合成式I化合物之方法。製備式I化合物之一般方法中,除非另有指定,否則變數R1、R2、R3、R4、R5、R6、R7、a、及b為如同先前式I化合物所定義者。本文所述之反應圖意在提供用於製備給定之許多化合物所用之方法的一般說明。然而,從詳細之說明來看,很明顯地所用之製備模式比本文所述之一般步驟進一步地更為廣泛。尤其,值得注意的是,根據反應圖所製備之化合物可進一步修飾以提供在本發明範圍內之新穎化合物。下列化合物中所用之試劑及中間體為市售或可根據熟知有機合成技藝者之標準文獻步驟製備。
以下反應圖I描述式I化合物之合成。將式IV之經適當取代之溴化物衍生物以式V之適當咔唑、於適當溶劑諸如N,N-二甲基甲醯胺或N,N-二甲基乙醯胺、於約0℃至150℃之溫度範圍內處理約5分鐘至24小時以得式III之相應環氧乙烷化合物。用於將式IV之溴化物化合物與式V之咔唑化合物反應以得式III化合物的較佳條件包括將反應於N,N-二甲基甲醯胺中、於0℃至室溫、於氫氧化鉀之存在下進行20至24小時,其後進行萃取處理。將式III化合物以式II之適當醯胺或脲、於適當溶劑諸如N,N-二甲基甲醯胺、二甲亞碸或N,N-二甲基乙醯胺中、於約室溫至150℃之溫度範圍內處理約5分鐘至3天以得式I之相應醯胺或脲化合物。用於將式III之環氧乙烷化合物反應以得式I化合物之較佳條件包括將反應於N,N-二甲基甲醯胺中與氫化鈉於室溫進行20至24小時,其後進行萃取處理。另外,式III之環氧乙烷化合物可與式II之醯胺或脲於適當溶劑諸如二甲亞碸中,與適當鹼諸如三級丁醇鉀於室溫反應3天,即得式I化合物。
反應圖I

以下反應圖II描述式I化合物之另一合成法。將式III之經適當取代之環氧乙烷衍生物以式VII之適當二胺、於適當溶劑諸如乙醇中、於0℃至150℃之溫度範圍內處理約5分鐘至24小時以得式VI之相應二胺化合物。用於將式III之環氧乙烷化合物與式VII之二胺反應以得式VI化合物的較佳條件包括將反應於乙醇中、於40℃進行20至24小時。將式VI之化合物以適當之羰基化劑諸如1,1’-羰基二咪唑、於適當溶劑諸如四氫呋喃中,於室溫處理5分鐘至24小時,即得式I之相應化合物。
反應圖II

以下反應圖III描述式I化合物之另一合成法。將式II之醯胺或脲化合物以式IX之經適當取代之溴化物衍生物、於適當溶劑諸如四氫呋喃中、於約0℃至65℃之溫度範圍內處理約5分鐘至24小時以得式VIII之相應環氧乙烷化合物。用於將式IX之溴化物化合物與式II之醯胺或脲反應以得式VIII化合物的較佳條件包括將反應於四氫呋喃中、於0℃至室溫、於氫化鈉之存在下進行20至24小時,其後進行萃取處理。將式VIII化合物以式V之適當咔唑於適當溶劑諸如N,N-二甲基甲醯胺中、於0℃至70℃之溫度範圍內處理約5分鐘至24小時,即得式I之相應化合物。用於將式VIII之環氧乙烷化合物與式V之咔唑反應以得式I化合物的較佳條件包括將反應於N,N-二甲基甲醯胺中、於室溫至70℃、於氫化鈉之存在下進行20至24小時,即得式I化合物。

以下反應圖IV描述式I化合物之另一合成法。將式XIII之經Boc保護之胺基酸化合物以氨、適當偶合試劑諸如N,N,N',N'-四甲基-O-(1H-苯並三唑-1-基)脲鎓六氟磷酸鹽、適當鹼諸如N,N-二異丙基乙胺及適當溶劑諸如二甲基甲醯胺、於約0℃至65℃之溫度範圍內處理約5分鐘至24小時以得式XII之相應醯胺化合物。將式XII之經Boc保護之胺基醯胺化合物以適當還原劑諸如甲硼烷、於適當溶劑諸如四氫呋喃中、於約0℃至100℃之溫度範圍內處理約5分鐘至24小時以得式XI之相應經Boc保護之二胺化合物。用於將式III之環氧乙烷化合物與式XI之經Boc保護之二胺化合物反應以得式X化合物的較佳條件包括將反應於乙醇中、於70℃進行16至24小時。將式X化合物以適當鹼諸如三級丁醇鉀、於適當溶劑諸如四氫呋喃
中、於約0℃至100℃之溫度範圍內處理約5分鐘至24小時,即得式I化合物。

以下反應圖V描述式I化合物之另一合成法。將式XVI之經苄基保護之二胺化合物以適當之羰基化劑諸如1,1’-羰基二咪唑、於適當溶劑諸如四氫呋喃中、於室溫處理5分鐘至24小時以得式XV之相應化合物。用於將式III之環氧乙烷化合物與式XV之經苄基保護之脲反應以得式XIV化合物的較佳條件包括將反應於N,N-二甲基甲醯胺中、於室溫至70℃、於氫化鈉之存在下進行16至24小時。將式XIV之經苄基保護之脲化合物以1至50psi之氫、於適當催化劑諸如氫氧化鈀/碳之存在下、以適當之酸諸如乙酸、於適當溶劑諸如四氫呋喃中、於約室溫至
100℃之溫度範圍內處理約5分鐘至5天,即得式I之相應化合物。

以下反應圖VI描述式I化合物之另一合成法。將式XVII或XVIII之經適當取代之手性環氧乙烷衍生物以式II之適當醯胺或脲化合物、以適當鹼諸如氫化鈉、於適當溶劑諸如N,N-二甲基甲醯胺或四氫呋喃中、於0℃至150℃之溫度範圍內處理約5分鐘至24小時,即得式I之相應手性醯胺或脲化合物。
反應圖VI

以下反應圖VII描述式I化合物之另一合成法。將式XVII或XVIII之經適當取代之手性環氧乙烷衍生物以式VII之適當二胺、於適當溶劑諸如乙醇中、於0℃至150℃之溫度範圍內處理約5分鐘至24小時以得式VI之相應手性二胺化合物。用於將式XVII或XVIII之環氧乙烷化合物與式VII之二胺反應以得式VI化合物的較佳條件包括將反應於乙醇中、於55℃進行5至24小時。將式VI化合物以適當羰基化劑諸如1,1’-羰基二咪唑、於適當溶劑諸如四氫呋喃中、於室溫處理5分鐘至24小時,即得式I之相應化合物。
反應圖VII

本文所述之反應圖中,應該理解的是,用於製備式I化合物之中間體中的羥基可依所需地被熟諳此藝者已知之慣用基團保護。例如,含有羥基之中間體可以相應之三級丁基二甲基矽基醚形式保護,其後藉以四正丁基氟化銨處理予以脫保護以得自由態羥基衍生物。適當之保護基及其移除方法闡述於“Protective Groups in Organic Synthesis”,3rd Ed.,T.W.Greene and P.G.M.Wuts(Wiley & Sons,1999)中。
所有情況下之1H核磁共振(NMR)光譜均符合所提出的結構。特徵化學位移(δ)係以在四甲基矽烷低場的百萬分點表示,且主峰的指定使用慣用之縮寫:例如s,單峰;d,二重峰;t,三重峰;q,四重峰;m,多重峰;br,寬峰。質譜(m/z)使用電噴霧離子化(ESI)或大氣壓力化學離子化(APCI)記錄。當薄層層析(TLC)被使用時,其稱之為矽膠TLC,使用矽膠60 F254板,Rf為TLC板上之化合物移動的距離除以溶劑前沿移動的距離。HPLC意指高效能液相層析。
下列具體實例係載明以用於闡述之目的而不應解釋為對本說明書之限制。

於圓底燒瓶內裝入1-溴-4-氟苯(13.0克,74.3毫莫耳)、2-氯-4-氟苯胺(11.354克,78.0毫莫耳)、無水甲苯(200毫升)及三級丁醇鉀(10.003克,89.1毫莫耳)。將混合物脫氣,再反填充氮,然後將參(二亞苄基丙酮)二鈀(0)(2.041克,2.2毫莫耳)及三-三級丁膦(0.902克,4.5毫莫耳)加入,再將反應於氮下、於100℃攪拌16小時。冷卻後,將混合物以6M水性氫氯酸處理成酸性pH,然後以固體碳酸鈉反調節回鹼性pH。將混合物乾燥(無水硫酸鎂),通過矽藻土(Celite)中過濾,再將濾塊以乙酸乙酯清洗。將濾液濃縮,再將殘留物藉矽膠層析(0-20%乙酸乙酯/己烷)予以純化,即得黃色油狀物(14克,79%)。1H NMR(300MHz,CDCl3):δ 7.14(dd,1H,J=8.4,3.0Hz),7.12-6.98(m,5H),6.88(td,1H,J=8.7,3.0Hz),5.80(br s,1H)。

將碳酸鉀(26.528克,191.9毫莫耳)、2-氯-4-氟-N-(4-氟苯基)苯胺(23.0克,96.0毫莫耳)、三環己基鏻四氟硼酸鹽(3.534克,9.6毫莫耳)、二乙酸鈀(1.077克,4.8毫莫耳)、及無水N,N-二甲基乙醯胺(200毫升)之混合物於氮下、於130℃攪拌16小時。冷卻後,將混合物濃縮,再將殘留物以乙酸乙酯處理,通過矽藻土(Celite)中過濾,再將濾塊以乙酸乙酯清洗。將濾液濃縮,再將殘留物藉短矽膠管柱(20-50%二氯甲烷/己烷)予以純化,以得粗產物,將其由己烷-二氯甲烷中予以再結晶,即得白色粉狀之純產物(17.2克,88%)。1H NMR(300MHz,CDCl3):δ 8.00(br s,1H),7.67(dd,2H,J=8.7,2.7Hz),7.36(dd,2H,J=8.7,4.2Hz),7.19(td,2H,J=9.0,2.7Hz)。

將粉狀氫氧化鉀(3.36克,60毫莫耳)加至咔唑(8.36
克,50毫莫耳)之無水N,N-二甲基甲醯胺(50毫升)溶液中,再於周圍溫度攪拌1小時。將反應混合物於冰浴中冷卻,再將表溴醇(10.3毫升,125毫莫耳)加入。將冰浴移出,再將反應於室溫攪拌20小時。將混合物分配於乙酸乙酯與水之間。將有機層接續地以水及飽和水性氯化鈉溶液清洗,乾燥(無水硫酸鈉),過濾,再濃縮。將生料以己烷研磨,再由乙酸乙酯/己烷中予以再結晶,即得白色針狀之期望產物(6.41克,58%產率)。第二份收成之結晶係由母液中結晶出,即得附加產物(1.2克,11%)。1H NMR(300MHz,CDCl3)δ 8.11-8.08(m,2H),7.46-7.44(m,4H),7.28-7.25(m,2H),4.68-4.62(dd,1H,J=3.1,15.8Hz)4.45-4.38(dd,1H,J=4.8,15.9Hz),3.37(m,1H),2.84-2.81(dd,1H,J=4.2,4.3Hz),2.60-2.57(dd,1H,J=2.5,5.0Hz);HPLC分析:(C18,20分鐘期間5-95%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):7.83分,98.7%。
下列化合物係以類似之方法製得:


將4-二甲胺基吡啶(0.075克)及苯甲醯氯(6.9毫升,59.5毫莫耳,1.1當量)加至冷0℃ 2-吡咯啶酮(4.4克,51.7毫莫耳,1.0當量)及三乙胺(15.4毫升,111.2毫莫耳,2.1當量)之無水四氫呋喃(120毫升)溶液中。將所得混合物於室溫攪拌16小時。將混合物倒至水中,再以乙酸乙酯萃取。將有機部分以0.1M水性氫氯酸、飽和水性碳酸氫鈉、及飽和水性氯化鈉溶液清洗,於無水硫酸鈉上乾燥,過濾再於真空中濃縮以得紅色油狀物。將粗產物藉矽膠管柱層析以20-65%梯度之乙酸乙酯之己烷液洗提予以純化,即得灰白色固狀物(5.63克,58%)。1H NMR(300MHz,CDCl3):δ 7.61-7.57(m,2H),7.53-7.47(tt,1H,J=1.5,7.5Hz),7.42-7.37(m,2H),3.98-3.94(t,2H,J=7.1Hz),2.61-2.58(t,2H,J=8.0Hz),2.20-2.10(quint,2H,J=7.5Hz)。ESI(m/z):190.1(M+H)。

將二異丙基胺鋰(3.382毫升2M之四氫呋喃溶液,6.8毫莫耳,1.3當量)加至-78℃之1-苯甲醯基吡咯啶-2-酮(1克,5.3毫莫耳,1.0當量)之無水四氫呋喃(26毫升)溶液中,再將混合物於-78℃攪拌30分鐘。將N-氟苯磺醯亞胺(2.5克,7.9毫莫耳,1.5當量)之無水四氫呋喃
(5毫升)溶液於-78℃徐緩加入,再將反應於-40℃攪拌1小時。將飽和水性碳酸氫鈉加入,將溶液加溫至室溫,再以乙酸乙酯萃取。將有機層以飽和水性氯化鈉清洗,於無水硫酸鈉上乾燥,過濾,再於真空中濃縮以得黃色固狀物。將粗產物藉矽膠管柱層析以15-60%梯度乙酸乙酯之己烷液洗提予以純化,即得白色固狀物(0.595克,54%)。1H NMR(300MHz,CDCl3):δ 7.64-7.61(m,2H),7.57-7.52(tt,1H,J=1.5,7.5Hz),7.45-7.39(m,2H),5.28-5.06(dt,1H,J=7.8,51Hz),4.15-4.07(m,1H),3.87-3.78(m,1H),2.68-2.56(m,1H),2.45-2.27(m,1H)。19F NMR(282MHz,CDCl3):δ -188.9至-189.2(ddd,J=12.1,24.2,51.8Hz)。

將辛胺(0.259毫升,1.6毫莫耳,1.1當量)加至1-苯甲醯基-3-氟吡咯啶-2-酮(0.282克,1.4毫莫耳,1.0當量)之無水四氫呋喃(5毫升)溶液中,再將反應於室溫攪拌16小時。將反應混合物於減壓下濃縮以得黃色油狀物。將粗產物藉矽膠管柱層析以70-100%梯度乙酸乙酯之己烷液洗提予以純化,即得白色固狀物。(0.104克,74%產率)。1H NMR(300MHz,CDCl3):δ 7.81(br s,1H),5.11-4.89(ddd,1H,J=6.3,7.8,52.8Hz),3.49-3.42(m,1H),
3.36-3.27(m,1H),2.57-2.41(m,1H),2.34-2.13(m,1H)。13C NMR(75MHz,CDCl3):δ 173.5-173.3(d,J=20Hz),89.9-87.4(d,J=182Hz),39.1(d,J=4Hz),28.6-28.4(d,J=20Hz)。19F NMR(282MHz,CDCl3):δ -190.1至-190.4(ddd,J=15,27,52Hz)。

將二碳酸二三級丁酯(16.512克,75.7毫莫耳,1.5當量)加至於0℃之已攪拌之哌啶-2-酮(5克,50.4毫莫耳,1.0當量)、三乙胺(14.022毫升,100.9毫莫耳,2.0當量)及N,N-4-二甲胺基吡啶(0.123克,1.0毫莫耳)之二氯甲烷(100毫升)溶液中。將混合物徐緩加溫至室溫,再攪拌48小時。以水令反應中止,再將有機層接續地以1N水性氫氯酸、飽和水性碳酸弳鈉及飽和水性氯化鈉清洗,再於無水硫酸鈉上乾燥,過濾及於真空中濃縮。將殘留物藉矽膠管柱(0-100%乙酸乙酯/己烷)予以純化,即得黃色油狀之期望產物(8.5克,85%)。1H NMR(300MHz,CDCl3):δ 3.72-3.62(m,2H),2.58-2.48(m,2H),1.90-1.78(m,4H),1.55(s,9H)。

將雙(三甲基矽烷基)胺鈉(22.586毫升1M之四氫呋喃溶液,22.6毫莫耳,1.5當量)於30分鐘期間逐滴加至於氮下、於-78℃之已攪拌之2-側氧基哌啶-1-羧酸三級丁酯(3克,15.1毫莫耳,1.0當量)之無水四氫呋喃(70毫升)溶液中。將所得溶液於-78℃攪拌45分鐘,然後將N-氟苯磺醯亞胺(7.122克,22.6毫莫耳,1.5當量)之無水四氫呋喃(30毫升)溶液於30分鐘期間逐滴加入。將反應於-78℃攪拌1小時,然後令其於2小時期間徐緩加溫至室溫,再於室溫攪拌1小時。以飽和水性氯化銨令反應中止,再以乙酸乙酯萃取。將有機相以飽和水性氯化鈉清洗,再於無水硫酸鎂上乾燥,過濾及於真空中濃縮。將殘留物以乙醚處理,再將固狀物丟棄。將溶液濃縮,再將殘留物藉矽膠管柱層析(0-100%乙酸乙酯/己烷)予以純化以得白色固狀之粗產物流份及二氟基副產物(1.5克)。將粗產物流份藉第二回合之矽膠層析予以進一步純化,即得濃稠油狀之期望產物(0.46克,14%)。1H NMR(300MHz,CDCl3):δ 4.92(ddd,1H,J=47.4,8.7,6.3Hz),3.78-3.60(m,2H),2.35(m,1H),2.15-1.80(m,3H),1.55(s,9H)。19F NMR(282MHz,CDCl3):δ -185.2(dt,J=45.7,15.5Hz)。

將三氟乙酸(1毫升,13.5毫莫耳,6.5當量)加至0℃之3-氟-2-側氧基哌啶-1-羧酸三級丁酯(0.450克,2.1毫莫耳,1.0當量)之二氯甲烷(5毫升)溶液中,再將所得溶液攪拌3小時。將反應於減壓下濃縮,再將殘留物藉矽膠管柱(0-100%乙酸乙酯/己烷,然後0-20%甲醇/乙酸乙酯)予以純化,即得白色粉狀之期望產物(0.23克,95%)。1H NMR(300MHz,CDCl3):δ 6.36(br s,1H),4.85(ddd,1H,J=46.8,8.1,5.4Hz),3.50-3.20(m,2H),2.40-1.70(m,4H)。19F NMR(282MHz,CDCl3):δ -186.5(dt,J=46.5,15.5Hz)。

3,3-二氟哌啶-2-酮係根據已報告之步驟(Kim,B.C.et al.Synthesis 2012,44,3165-3170)製得。

將二異丙基胺鋰(0.905毫升2M之四氫呋喃溶液,1.8毫莫耳,1.3當量)加至-78℃之得自製備例21B之1-苯甲醯基-3-氟吡咯啶-2-酮(0.3克,1.4毫莫耳,1.0當量)及N-氟苯磺醯亞胺(0.639克,2.0毫莫耳,1.4當量)之無水四氫呋喃(10毫升)溶液中,再將混合物於-78℃攪拌30分鐘。將另外份之二異丙基胺鋰溶液(0.5當量)及N-氟苯磺醯亞胺(0.5當量之0.5毫升無水四氫呋喃液)之加入,再將反應於-78℃攪拌1小時。將飽和水性碳酸氫鈉加入,將溶液加溫至室溫,再以乙酸乙酯萃取。將有機層以飽和水性氯化鈉清洗,於無水硫酸鈉上乾燥,過濾,再於真空中濃縮。將粗產物藉矽膠管柱層析以15-50%梯度乙酸乙酯之己烷液洗提予以純化,即得白色固狀物(0.09克,23%)。1H NMR(300MHz,CDCl3):δ 7.66-7.61(m,2H),7.59-7.55(m,1H),7.47-7.44(m,2H),4.02-3.97(m,2H),2.70-2.56(tt,2H,J=6.6,14.7Hz)。19F NMR(282MHz,CDCl3):δ -106.0 to-106.1(t,J=15Hz)。

將辛胺(0.075毫升,0.5毫莫耳,1.1當量)加至1-苯甲醯基-3,3-二氟吡咯啶-2-酮(0.085克,0.4毫莫耳,1.0當量)之無水四氫呋喃(1毫升)溶液中,再將反應於室溫攪拌16小時。將混合物於減壓下濃縮以得黃色油狀物。將粗製殘留物藉矽膠管柱層析以50-100%梯度乙酸乙酯之己烷液洗提予以純化,即得白色固狀物(0.024克,52%產率)。1H NMR(300MHz,CDCl3):δ 7.93(br s,1H),3.50-3.46(br t,2H,J=6.0Hz),2.63-2.48(tt,2H,J=6.6,15.2Hz)。19F NMR(282MHz,CDCl3):δ -107.33 to -107.44(t,J=15.2Hz)。13C NMR(75MHz,CDCl3):δ 167.5-166.7(t,J=31Hz),121.1-114.4(t,J=248Hz),37.1(t,J=3.3Hz),31.2-30.6(t,J=23.1Hz)。

將二異丙基胺鋰(2.4毫升2M溶液,4.8毫莫耳,2.0當量)加至冷-78℃之1-苄基-2-吡咯啶酮(0.422克,2.4毫莫耳,1.0當量)之無水四氫呋喃(15毫升)溶液中,再將所得紅色溶液於-78℃攪拌30分鐘,再將碘甲烷(0.6毫升,9.6毫莫耳,4.0當量)加入。將溶液於-78℃攪拌1小時,再令其徐緩加溫至室溫16小時。將飽和水性氯化銨加入,再將混合物以乙酸乙酯萃取。將有機部分
以飽和水性氯化鈉清洗,於無水硫酸鈉上乾燥,過濾及於真空中濃縮。將粗產物藉矽膠管柱層析以35-80%梯度乙酸乙酯之己烷液洗提予以純化,即得棕褐色液狀產物(0.374克,82%)。1H NMR(300MHz,CDCl3):δ 7.37-7.34(m,5H),4.52-4.41及4.46-4.41(ABq,2H,J=14.6Hz),3.27-3.15(m,2H),2.60-2.46(m,1H),2.28-2.15(m,1H),1.68-1.58(m,1H),1.28-1.25(d,3H,J=7.2Hz)。
下列化合物係以類似之方法製得:



於氮氣氛下,將10%鈀/碳(0.09克)加至1-苄基-3-(環己-2-烯-1-基)哌啶-2-酮(0.6克,2.3毫莫耳)之乙醇(10毫升)溶液中。將混合物置於氫氣氛下,再攪拌2天。將懸浮液通過矽藻土(Celite)中過濾,再於減壓下濃縮以得清澈液狀之期望產物(0.578克,98%)。1H NMR(300MHz,CDCl3):δ 7.33-7.22(m,5H),4.66-4.61及4.59-4.54(ABq,
2H,J=14.7Hz),3.19-3.14(m,2H),2.34-2.21(m,2H),1.86-1.52(m,9H),1.39-1.04(m,5H);ESI(m/z):272.2(M+H)。

根據de Filippis,A.et al.Tetrahedron,2004,60,9757之步驟合成。將雙(三甲基矽烷基)胺鋰(6.4毫升1M之無水四氫呋喃溶液,6.4毫莫耳,2.0當量)加至冷(-20℃)己攪拌之N-苄基-2-哌啶酮(1.326克,7.0毫莫耳,2.2當量)之無水四氫呋喃(14毫升,0.5M)溶液中,再將混合物於-20℃攪拌20分鐘。將氯化鋅(0.955克,7.0毫莫耳,2.2當量)之無水四氫呋喃(8毫升)溶液加入,再將溶液於-20℃攪拌20分鐘。將所得溶液經由套管送至2-二環己膦基-2’-(N,N-二甲胺基)聯苯(0.094克)、參(二亞苄基丙酮)二鈀(0)(0.092克)、及溴苯(0.335毫升,3.2毫莫耳,1.0當量)之無水四氫呋喃(6毫升)溶液中,再將所得溶液於70℃加熱6小時。以水性氯化銨令反應中止,再以乙酸乙酯萃取。將有機部分以飽和水性氯化鈉清洗,於無水硫酸鈉上乾燥,過濾及於真空中濃縮。將粗產物藉矽膠管柱層析以15-60%梯度乙酸乙酯之己烷液洗提予以純化,即得黃色液狀物(0.629克,74%)。1H NMR(300MHz,
CDCl3):δ 7.38-7.21(m,10H),4.74-4.69及4.66-4.61(AB,2H,J=14.4Hz),3.77-3.72(dd,1H,J=6.0,8.1Hz),3.41-3.28(m,2H),2.23-2.13(m,1H),2.05-1.69(m,3H)。

將三氟甲烷磺酸(0.604毫升,6.8毫莫耳,4.0當量)加至1-苄基-3-甲基吡咯啶-2-酮(0.323克,1.7毫莫耳,1.0當量)之甲苯(2毫升,1M)溶液中。將混合物於195℃、於微波反應器中加熱25分鐘。將混合物倒至小量飽和水性碳酸氫鈉中,以乙酸乙酯萃取,以飽和水性氯化鈉清洗,再將結合之水性層以乙酸乙酯再度萃取。將結合之有機部分於無水硫酸鈉上乾燥,過濾,再於真空中濃縮。將粗製殘留物藉矽膠管柱層析以0-10%甲醇之二氯甲烷液洗提予以純化,即得期望產物(0.087克)。1H NMR(300MHz,CDCl3):δ 6.49(br s),3.37-3.26(m,2H),2.53-2.28(m,2H),1.80-1.65(m,1H),1.21-1.19(d,3H,J=6.6Hz)。
下列化合物係以類似之方法製得:



將二異丙基胺鋰(3.15毫升2M之四氫呋喃溶液,1.1當量)加至冷(-78℃)1-苄基-2-吡咯啶酮(1.0克,5.7毫莫耳,1.0當量)之無水四氫呋喃(19毫升)溶液中,再將混合物於-78℃攪拌1小時。將環丁酮(0.426毫升,5.7毫莫耳,1.0當量)及三氟化硼乙醚(0.704毫升,5.7毫莫耳,1.0當量)加入,再將反應混合物於-78℃攪拌4小時。以飽和水性氯化銨令反應中止,再以乙酸乙酯萃取。將有機部分以飽和水性氯化鈉清洗,於無水硫酸鈉上乾燥,過濾及於減壓下濃縮。將粗產物藉矽膠管柱層析由矽膠中以50-100%梯度乙酸乙酯之己烷液洗提予以純化,即得白色固狀物(0.714克,51%)。1H NMR(300MHz,CDCl3):δ 7.36-7.20(m,5H),4.56-4.51及4.42-4.37(AB,2H,J=14.7Hz),4.24(s,1H),3.28-3.21(m,2H),2.78-2.72(t,1H,J=2.7Hz),2.34-1.89(m,7H),1.66-1.52(m,1H);ESI(m/z):246.0(M+H)。
下列化合物係以類似之方法製得:


將N,N-二異丙基乙胺(2.485毫升,14.3毫莫耳,5.0當量)、N,N-4-二甲胺基吡啶(0.07克,0.6毫莫耳,0.2當量)、及甲磺醯氯(0.331毫升,4.3毫莫耳,1.5當量)加至冷(0℃)1-苄基-3-(1-羥基環丁基)吡咯啶-2-酮(0.7克,2.9毫莫耳,1.0當量)之無水二氯甲烷(12毫升)溶液中。將混合物於0℃攪拌2小時,於室溫攪拌16小時,再迴流攪拌3小時。將混合物冷卻至室溫再分配於乙酸乙酯與飽和水性氯化銨之間。將有機層以飽和水性氯化鈉清洗,於無水硫酸鈉上乾燥,過濾及於真空中濃縮以得橙色油狀物。將粗產物藉矽膠管柱層析以20-60%梯度乙酸乙酯之己烷液洗提予以純化,即得黃色油狀物(0.25克,38%)。1H NMR(300MHz,CDCl3):δ 7.33-7.23(m,5H),4.47(s,2H),3.27-3.19(m,4H),2.75-2.69(m,2H),2.52-2.46(m,
2H),2.18-2.08(quint,2H,J=7.8Hz);ESI(m/z):228.2(M+H)。
下列化合物係以類似之方法製得:


將10%鈀/碳(0.05克)加至1-苄基-3-亞環丁基吡咯啶-2-酮(0.25克,1.0毫莫耳)之乙醇(11毫升)溶液中,再將反應混合物於氫氣氛下攪拌72小時。將混合物通過矽藻土(Celite)中過濾,再於減壓下濃縮,即得清澈油狀物(0.242克,100%)。1H NMR(300MHz,CDCl3):δ 7.34-7.20(m,5H),4.49-4.47及4.40-4.35(ABq,2H,J=14.4Hz),3.18-3.13(m,2H),2.69-2.47(m,2H),2.20-1.64(m,8H);ESI(m/z):230.2(M+H)。
下列化合物係以類似之方法製得:


以類似於製備例18之方法由1-苄基-3-環丁基吡咯啶-2-酮中製得。1H NMR(300MHz,CDCl3):δ 5.45(br s,1H),3.34-3.27(m,2H),2.65-2.54(m,1H),2.43-2.35(m,1H),2.28-1.79(m,8H)。
下列化合物係以類似之方法製得:


將氰基氫硼化鈉(8.017克,127.6毫莫耳,2.0當量)
加至環己酮(6.26克,63.8毫莫耳)、乙二胺(42.64毫升,637.8毫莫耳,10.0當量)、乙酸(36.515毫升,637.8毫莫耳,10.0當量)、及4Å分子篩(25克)之無水甲醇(250毫升)混合液中。將混合物攪拌48小時,過濾以移除固狀物,再濃縮成半固狀物。將生料溶於3N水性氫氧化鈉(150毫升)中,再以二氯甲烷萃取三次。將結合之有機部分以微鹼性之飽和水性氯化鈉溶液清洗,於無水硫酸鈉上乾燥,過濾,再於真空中濃縮以得淺黃色液狀物,將其藉真空蒸餾法予以純化,即得清澈液狀物(4.1克,45%)。1H NMR(300MHz,CDCl3):δ 2.80-2.76(td,2H,J=0.9,6.0Hz),2.68-2.64(td,2H,J=0.9,6.0Hz),2.43-2.34(m,1H),1.89-1.83(m,2H),1.74-1.70(m,2H),1.62-1.57(m,1H),1.32-0.98(m,8H)。

於室溫將環丁胺(5.90毫升,59.8毫莫耳,1.0當量)於15分鐘期間逐滴加至丙烯腈(4.76克,89.7毫莫耳,1.5當量)之甲醇(7毫升)溶液中。將混合物於室溫攪拌30分鐘,再於迴流攪拌1小時,冷卻至室溫,於減壓下濃縮,再將期望產物於真空下蒸餾,即得清澈液狀物(7.7克,98%)。1H NMR(300MHz,CDCl3):δ 3.29-3.21(m,1H),2.88-2.83(t,2H,J=6.6Hz),2.50-2.46(t,2H,J=6.6
Hz),2.26-2.20(m,2H),1.76-1.63(m,4H),1.30(br s,1H)。

將3-(環丁胺基)丙腈(5.0克,40.3毫莫耳,1.0當量)之無水乙醚(40毫升)溶液於45分鐘期間逐滴加至已冷卻(0℃)之氫化鋁鋰(3.056克,80.5毫莫耳,2.0當量)之無水乙醚(120毫升)懸浮液中。將反應混合物於室溫攪拌15分鐘,再於迴流攪拌4小時,冷卻至室溫,再攪拌1小時。將混合物冷卻至0℃,再強烈攪拌同時將水(3.1毫升)逐滴加入,其後將15%水性氫氧化鈉(3.1毫升),最後將水(9.3毫升)加入。將所得漿液加溫至室溫,攪拌15分鐘,再將硫酸鎂加入,同時攪拌另15分鐘。將固體材料藉通過玻璃多孔濾器中以溫二氯甲烷清洗多次地過濾移出,再將有機溶液於減壓下濃縮,即得淺黃色液狀之期望產物(3.44克,66%)。1H NMR(300MHz,CDCl3):δ 3.14(m,1H),2.69-2.62(m,2H),2.53-2.45(m,2H),2.13-2.10(m,2H),1.56-1.48(m,6H),1.33(br s,3H)。

於室溫將環戊胺(5.794毫升,58.7毫莫耳,1.0當量)逐滴加至丙烯腈(5.79毫升,88.1毫莫耳,1.5當量)之甲醇(7毫升)溶液中。將溶液於室溫攪拌30分鐘,再於迴流攪拌1小時,冷卻至室溫,於減壓下濃縮,再將期望產物於真空下蒸餾,即得清澈液狀物(7.4克,91%)。1H NMR(300MHz,CDCl3):δ 3.14-3.04(quin,1H,J=6.3Hz),2.91-2.87(t,2H,J=6.9Hz),2.53-2.48(td,2H,J=0.9,6.9Hz),1.88-1.78(m,2H),1.73-1.49(m,4H),1.36-1.24(m,2H),1.19(br s,1H)。

將3-(環戊胺基)丙腈(6.0克,43.4毫莫耳,1.0當量)之無水乙醚(40毫升)溶液於45分鐘期間逐滴加至已冷卻(0℃)之氫化鋁鋰(3.295克,86.8毫莫耳,2.0當量)之無水乙醚(150毫升)懸浮液中。將反應混合物於室溫攪拌15分鐘,再於迴流攪拌4小時,冷卻至室溫,再攪拌1小時。將混合物冷卻至0℃,再強烈攪拌同時將水(3.4毫升)逐滴加入,其後將15%水性氫氧化鈉(3.4毫升),最後將水(10.2毫升)加入。將所得漿液加溫至室溫,攪拌15分鐘,再將硫酸鎂加入,同時攪拌另15分鐘。將固體材料藉通過玻璃多孔濾器中以溫二氯甲烷清洗多次地過濾移
出,再將有機溶液於減壓下濃縮,即得清澈油狀之期望產物(4.5克,73%)。1H NMR(300MHz,CDCl3):δ 3.05-2.96(quint,1H,J=6.6Hz),2.74-2.71(t,2H,J=6.6Hz),2.68-2.58(t,2H,J=6.9Hz),1.85-1.68(m,2H),1.62-1.42(m,6H),1.34(br s,3H),1.30-1.21(m,2H)。

將3,6-二氟-9-((2-甲基環氧乙烷-2-基)甲基)-9H-咔唑(1.0當量,或者3,6-二氟-9-(環氧乙烷-2-基甲基)-9H-咔唑)加至N-環己基-1,3-丙烷二胺(或其他N-官能化1,3-丙烷二胺,8.0當量)之乙醇(1M)溶液中,再將反應混合物於70℃攪拌16小時或者直至藉LCMS測知反應完成為止,冷卻至室溫,於真空中濃縮以得粗製殘留物,將其藉管柱層析由HP矽膠中以適當梯度甲醇之二氯甲烷及0.1%三乙胺液洗提予以純化,即得期望產物。




將Boc-DL-丙胺酸(5.0克,26.4毫莫耳,1.0當量)、N,N,N',N'-四甲基-O-(1H-苯並三唑-1-基)脲鎓六氟磷酸鹽(15.033克,39.6毫莫耳,1.5當量)、N,N-二異丙基乙胺(8.735毫升,52.9毫莫耳,2.0當量)及無水二甲基甲醯胺(50毫升)之混合物於室溫攪拌20分鐘,然後以冰水冷卻,再將氨(2.250克,132.1毫莫耳,5.0當量)徐緩地成泡吹至混合物中。將反應於室溫於密封容器中攪拌3小時。將反應以水稀釋,再以乙酸乙酯萃取。將有機層以飽和水性氯化鈉清洗,於無水硫酸鈉上乾燥,過濾及於真空中濃縮。將所得固狀物以冷乙酸乙酯及乙醚清洗,再乾燥,即得白色粉狀產物(2.9克,58%)。1H NMR(300MHz,CDCl3):δ 6.20(br s,1H),5.50(br s,1H),5.00(br s,1H),4.20(m,1H),1.47(s,9H),1.40(d,3H,J=7.2Hz)。
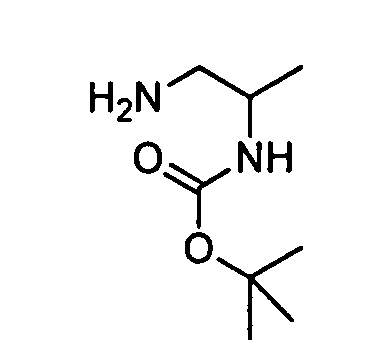
將(1-胺基-1-側氧基丙-2-基)胺基甲酸三級丁酯(2.2
克)溶於無水四氫呋喃(100毫升)中,再將甲硼烷(40毫升1M之四氫呋喃溶液)加入。將混合物於室溫攪拌2小時,然後於90℃加熱2小時。冷卻至室溫後,以甲醇令反應中止直至無氣泡產生為止。將混合物於90℃加熱1小時,然後濃縮至乾,即得漿狀粗產物(2.2克),其係直接使用於下一步驟反應。1H NMR(300MHz,CDCl3):δ 4.60(br s,1H),3.65(m,1H),2.76(dd,1H,J=12.9,5.1Hz),2.64(dd,1H,J=12.9,6.3Hz),1.47(s,9H),1.14(d,3H,J=6.9Hz)。

於氮氣氛下,將3,6-二氟-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.7克)及(1-胺基丙-2-基)胺基甲酸三級丁酯(1.5克)之乙醇(50毫升)溶液於70℃攪拌16小時。將混合物於減壓下濃縮,再藉矽膠管柱層析以0-20%梯度甲醇之二氯甲烷液洗提予以純化,即得灰白色泡沫狀物(1.28克)。產物係直接用於下一步驟中而不必額外純化:1H NMR(300MHz,CDCl3):δ 7.67(dd,2H,J=8.7,2.7Hz),7.42-7.37
(m,2H),7.22(td,2H,J=9.0,2.7Hz),4.55-4.30(m,3H),4.13(m,1H),3.78(br s,1H),2.88及2.82(dd,1H,J=12.0,3.6Hz),2.70-2.50(m,3H),1.45(s,9H),1.13及1.11(d,3H,J=6.6Hz);ESI(m/z):434.0(M+H)。

將環丙胺(4.214毫升,60.8毫莫耳,1.0當量)徐緩加至於室溫之丙烯腈(4.840克,91.2毫莫耳,1.5當量)之甲醇(7毫升)溶液中,再攪拌30分鐘。將反應加熱至迴流,再攪拌1小時,冷卻,濃縮,再於真空下蒸餾,即得5.5克清澈液狀物(5.5克,82%)。1H NMR(300MHz;CDCl3):δ 2.99(t,2H,J=6.3Hz),2.51(t,2H,J=6.3Hz),2.12(m,1H),1.78(br s,1H),0.49-0.32(m,4H);ESI(m/z):111.5(M+H)。

將3-(環丙胺基)丙腈(5.000克,45.4毫莫耳,1.0當量)之無水四氫呋喃(20毫升)溶液於十分鐘期間徐緩地加
至已冷卻(0℃)之氫化鋁鋰(3.445克,90.8毫莫耳,2.0當量)之無水四氫呋喃(120毫升)懸浮液中。將反應於室溫攪拌15分鐘,然後加熱至迴流,再攪拌3小時。將混合物冷卻至室溫,再將十水合硫酸鈉加入直至停止起泡為止。將懸浮液攪拌10分鐘,再將固狀物濾出(邊以四氫呋喃清洗)。將溶液於減壓下濃縮,以得粗產物,其係直接用於下一步驟中。1H NMR(300MHz,CDCl3):δ 2.74-2.69(m,4H),2.10-2.04(m,1H),1.65-1.56(m,2H),0.43-0.27(m,4H);ESI(m/z):115.4(M+H)。

邊強烈攪拌邊將適當N-官能化1,3-丙烷二胺或1,2-乙二胺(10.0毫莫耳,1.0當量)加至1,1’-羰基二咪唑(1.622克,10.0毫莫耳,1.0當量)之無水四氫呋喃(0.05M)溶液中,將其以外部冰浴保持於0℃。令溶液徐緩加溫至室溫,再攪拌16小時。將混合物以下列步驟操作:i)將混合物於減壓下濃縮,再藉管柱層析由矽膠中以梯度甲醇之二氯甲烷液洗提予以純化,即得期望產物;或ii)將混合物以乙酸乙酯稀釋,再接續地以1N水性氫氯酸清洗兩次及以飽和水性氯化鈉清洗一次,將有機層以乙酸乙酯反萃
取一次,將結合之有機部分於無水硫酸鈉上乾燥,過濾,再於真空中濃縮,即得期望產物,其係使用而不必進一步純化。



將60%氫化鈉之礦油液(1.1當量)加至1-乙基四氫嘧
啶-2(1H)-酮,或另外製備例6所產生之環狀脲(1.0當量),之無水四氫呋喃(0.2M)溶液中,再將所得懸浮液於室溫攪拌1小時。將表溴醇(3.0當量)加入,再將混合物於室溫或於35℃攪拌24小時。將矽膠加入,再將懸浮液於減壓下濃縮,再藉矽膠管柱層析且將期望產物以適當梯度甲醇之二氯甲烷液洗提出予以直接純化。



將60%氫化鈉之礦油液(1.1當量)加至製備例7所產生之環狀脲(1.0當量)之無水四氫呋喃(0.2M)溶液中,再將所得懸浮液於室溫攪拌1小時。將2-(氯甲基)-2-甲基環氧乙烷(4.0當量)於室溫加入,再將混合物於70-90℃於密封管中攪拌過夜。將矽膠加入,再將懸浮液於減壓下濃縮,再藉矽膠管柱層析且將期望產物以適當梯度甲醇之二氯甲烷液洗提出予以直接純化。


將甲胺(158.584毫升40% w/w水性溶液,1842.3毫莫耳,10.0當量)及巴豆腈(15毫升,184.2毫莫耳,1.0當量)之混合物於室溫攪拌16小時。將反應以二氯甲烷(3 x 100毫升)萃取,再將結合之有機層於無水硫酸鈉上乾燥,過濾及於真空中濃縮,即得無色油狀之期望產物(18克,100%)。1H NMR(300MHz,CDCl3):δ 2.96(m,1H),2.47(d,2H,J=6.0Hz),2.46(s,3H),1.36(d,3H,J=6.6Hz)。

於Parr搖動燒瓶內裝入甲醇(100毫升),冷卻至0℃,再以氨起泡。將3-(甲胺基)丁腈(4.8克,48.9毫莫耳,1.0當量)加入,再將一匙量之雷氏鎳(Raney Ni)加入。將反應於氫下、於50psi搖動10小時。將反應通過矽藻土(Celite)中過濾,再將濾液於真空中濃縮,即得清澈油狀物5.0克,100%)。1H NMR(300MHz,CDCl3):δ 2.90-2.60(m,3H),2.43(s,3H),1.70-1.40(m,2H),1.08(d,3H,J=6.0Hz)。
將1,1’-羰基二咪唑(7.935克,48.9毫莫耳,1.0當量)加至於0℃之已攪拌之N3-甲基丁烷-1,3-二胺(5.0克,48.9毫莫耳,1.0當量)之無水四氫呋喃(100毫升)溶液中。將混合物徐緩加溫至室溫,再攪拌16小時。將反應於減壓下濃縮,再將殘留物以飽和水性氯化銨處理,再以二氯甲烷萃取兩次。將結合之有機層以飽和水性氯化鈉清洗,於無水硫酸鈉上乾燥,過濾及於真空中濃縮。將殘留物藉矽膠管柱(0-20%乙醇/二氯甲烷)予以純化,即得白色固狀之期望產物(1.8克,29%)。1H NMR(300MHz,CDCl3):δ 4.66(br s,1H),3.53-3.35(m,2H),3.25(m,1H),2.95(s,3H),2.05(m,1H),1.70(m,1H),1.24(d,3H,J=6.3Hz)。

於氮氣氛下,將2-(苄胺基)苯甲腈(4.0克,19.2毫莫耳,1.0當量)之無水四氫呋喃(25毫升)溶液徐緩加至冷(0℃)氫化鋁鋰(2.187克,57.6毫莫耳,3.0當量)之無水四氫呋喃(60毫升)懸浮液中。將混合物冷卻,再將十水合硫
酸鈉加入直至停止起泡為止(使用外部水/冰浴)。將混合物過濾以移除固狀物,再將溶液於真空中濃縮,即得清澈液狀物(3.2克,79%)。1H NMR(300MHz,CDCl3):δ 7.42-7.23(m,5H),7.17-7.11(td,1H,J=1.8,7.5Hz),7.07-7.04(m,1H),6.68-6.61(m,2H),6.29(br s,1H),4.40(s,2H),3.99(s,2H),1.31(br s,2H)。

將1,1’-羰基二咪唑(1.318克,8.1毫莫耳,1.1當量)加至2-(胺甲基)-N-苄基苯胺(1.5克,7.1毫莫耳,1.0當量)之無水四氫呋喃(142毫升)溶液中,再將溶液於室溫攪拌24小時。將1N水性氫氯酸(20毫升)加入,再將混合物以乙酸乙酯萃取三次。將有機部分以無水硫酸鈉乾燥,過濾及真空中濃縮,即得白色固狀物(1.68克,99%)。1H NMR(300MHz,CDCl3):δ 7.33-7.20(m,5H),7.11-7.03(m,2H),6.95-6.91(m,1H),6.74-6.71(d,1H,J=8.1Hz),5.13(s,2H),4.54(s,2H),1.60(br s,2H)。ESI(m/z):239.2(M+H)。

將60%氫化鈉之礦油液(0.01克,0.3毫莫耳,1.0當量)加至己攪拌之1-苄基-3,4-二氫喹唑啉-2(1H)-酮(0.105克,0.4毫莫耳,1.8當量)之無水二甲基甲醯胺(1毫升)溶液中,再將混合物攪拌30分鐘。將3,6-二氟-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.065克,0.3毫莫耳,1.0當量)加入,再將混合物於55℃攪拌16小時。將混合物倒至飽和水性氯化銨中,再以乙酸乙酯萃取三次。將有機部分於無水硫酸鈉上乾燥,過濾及濃縮以得棕褐色油狀物。將粗製殘留物藉矽膠管柱層析以20-60%梯度乙酸乙酯/己烷洗提予以純化,即得產物,其係直接用於下一步驟中而不必額外純化(35%)。ESI(m/z):498.2(M+H);HPLC分析:(C18,20分鐘期間5-95%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):15.2分,51%。

將60%氫化鈉之礦油液(0.01克,0.3毫莫耳,1.0當量)加至已攪拌之1-苄基-3,4-二氫喹唑啉-2(1H)-酮(0.099克,0.4毫莫耳,1.8當量)之無水二甲基甲醯胺(1毫升)溶液中,再將混合物攪拌30分鐘。將3,6-二氟-9-((2-甲基環氧乙烷-2-基)甲基)-9H-咔唑(0.065克,0.2毫莫耳,1.0當量)加入,再將混合物於55℃攪拌16小時。將混合物倒至飽和水性氯化銨中,再以乙酸乙酯萃取三次。將有機部分於硫酸鈉上乾燥,過濾及濃縮以得棕褐色油狀物。將粗製殘留物藉矽膠管柱層析以20-60%梯度乙酸乙酯/己烷洗提予以純化,即得灰白色固狀物(0.086克,65%)。1H NMR(300MHz,CDCl3):δ 7.66-7.62(dd,2H,J=2.4,8.7Hz),7.45-7.40(dd,2H,J=4.1,8.9Hz),7.30-7.09(m,8H),7.04-6.93(m,2H),6.75-6.72(d,1H,J=8.1Hz),5.20-5.15及5.08-5.03(ABq,2H,J=16.6Hz),4.74-4.69及4.59-4.54(ABq,2H,J=14.3Hz),4.39-4.34及4.32-4.26(ABq,2H,J=15.3Hz),4.09(s,1H),3.97-3.93及3.51-3.47(ABq,2H,J=14.4Hz),1.36(s,3H);ESI(m/z):512.3(M+H)。

將碎4Å活化分子篩(15.0克)及二氯甲烷(300毫升)之混合物冷卻至-10℃,將異丙醇鈦(IV)(2.072毫升,7.0
毫莫耳,0.05當量)及D-酒石酸(-)-二乙酯(2.126克,10.3毫莫耳,0.07當量)藉由注射管加入,其後將80%氫過氧化異丙苯(50.000克,262.8毫莫耳,1.8當量)加入。將混合物於-10℃攪拌30分鐘。將混合物冷卻至-35℃,再將2-甲基-2-丙烯-1-醇(10.620克,147.3毫莫耳,1.0當量)之二氯甲烷(10毫升)溶液藉由注射管於30分鐘期間以1-2毫升為一份地分次加入。將反應混合物於-35℃攪拌1小時,然後置於-20℃冷凍器中3天。將混合物加溫至0℃,將水加入,再於室溫攪拌30分鐘。將混合物冷卻至0℃,再將30%氫氧化鈉之飽和水性氯化鈉(10毫升)溶液加入,再於此溫攪拌1小時。將混合物通過矽藻土(Celite)墊中過濾,再以二氯甲烷(50毫升)清洗。將水性部分分離出,再以二氯甲烷萃取(3 x 30毫升)。將有機層結合,再乾燥(無水硫酸鎂),過濾及於真空中濃縮以得無色液狀物。將粗製殘留物藉矽膠管柱層析以20-100%梯度乙醚/己烷洗提予以純化,即得無色油狀物(6.2349克,48%)。1H NMR(300MHz,CDCl3):δ 3.76(dd,1H,J=4.5,12.3Hz),3.6(dd,1H,J=8.1,12.3Hz),2.92(d,1H,J=4.5Hz),2.66(d,1H,J=4.8Hz),1.79(br m,1H),1.36(s,3H)。
(S)鏡像異構物係以類似之方法由酒石酸(+)-二乙酯中製得。

將3-硝基苯磺醯氯(15.092克,68.1毫莫耳,1.2當量)分成小份地於15分鐘期間徐緩加至於-20℃之已攪拌之(R)-(2-甲基環氧乙烷-2-基)甲醇(5.000克,56.8毫莫耳,1.0當量)、N,N-4-二甲胺基吡啶(0.100克,0.8毫莫耳,1.4莫耳%)及N,N-二異丙基乙胺(15.776毫升,88.9毫莫耳,2.0當量)之無水二氯甲烷(100毫升)溶液中。令混合物達0℃,再攪拌3小時。以水令反應中止,再以二氯甲烷萃取。將結合之有機層接續地以水、1N水性氫氯酸、飽和水性碳酸氫鈉、及飽和水性氯化鈉清洗。將有機層乾燥(無水硫酸鈉),過濾及於真空中濃縮。將粗製殘留物藉矽膠管柱(0-80%乙酸乙酯/己烷)予以純化,即得黃色油狀之期望產物(6.73克,43.4%)。1H NMR(300MHz,CDCl3):δ 8.77(t,1H,J=1.8Hz),8.54(m,1H),8.27(m,1H),7.84(t,1H,J=7.95Hz),4.29(d,1H,J=11.1Hz),4.05(d,1H,J=11.1Hz),2.73(dd,2H,J=17.7,4.8Hz),1.37(s,3H)。
(R)鏡像異構物係以類似之方法合成。

將60%氫化鈉之礦油液(1.355克,33.9毫莫耳,1.1當量)加至於0℃之已攪拌之咔唑(5.15克,30.8毫莫耳,1.0當量)之無水二甲基甲醯胺(100毫升)溶液中,再將混合物於0℃攪拌1小時。將間硝基苯磺酸(R)-(-)-環氧丙酯(9.981克,38.5毫莫耳,1.3當量)加入,再將反應混合物攪拌1小時,然後徐緩加溫至室溫,再攪拌16小時。將混合物分配於水與乙酸乙酯之間。將有機層以飽和水性氯化鈉清洗,乾燥(無水硫酸鈉),過濾及於減壓下濃縮以得紅色油狀物。將粗製殘留物藉矽膠管柱(15-80%二氯甲烷/己烷)予以純化,即得白色固狀之期望產物(4.95克,72%)。1H NMR(300MHz,CDCl3):δ 8.11-8.08(dt,2H,J=0.9,7.5Hz),7.48-7.46(m,4H),7.28-7.23(m,2H),4.67-4.61(dd,1H,J=2.4,15.9Hz),4.45-4.38(dd,1H,J=5.0,15.9Hz),3.39-3.34(m,1H),2.83-2.80(t,1H,J=4.2Hz),2.60-2.57(dd,1H,J=2.4,4.8Hz)。HPLC分析:(C18,20分鐘期間5-95%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):13.2分,99.5%。手性HPLC分析:(Chiralcel AD-H,20分鐘期間5-15%異丙醇之己烷液;滯留時間,於254nm之面積%):8.12分,3.8%;8.54分,96.1%(92.2% ee)。
下列化合物係以類似之方法製得:





將(1R)-(-)-2-氮雜雙環[2.2.1]庚-5-烯-3-酮(1.0克,9.2毫莫耳)及10%鈀/碳(0.4克)之甲醇(50毫升)混合液於氫氣氛下攪拌3小時。將混合物通過矽藻土(Celite)中過濾,再將濾塊以甲醇清洗。將結合之有機相於真空中濃縮,即得所需產物(1克,98%)。1H NMR(300MHz,CDCl3):δ 5.91(br s,1H),3.89(s,1H),2.74(s,1H),1.96-1.59(m,5H),1.42-1.37(m,1H)。

將(1S)-(+)-2-氮雜雙環[2.2.1]庚-5-烯-酮(1.0克,9.2毫莫耳,1.0當量)及10%鈀/碳(0.4克)之甲醇(40毫升)混合液於氫氣氛下、於周圍溫度攪拌2小時。將混合物通過矽藻土(Celite)中過濾,再將濾塊以甲醇清洗。將結合之有機相於真空中濃縮,即得所需產物(1克,98%)。1H NMR(300MHz,CDCl3):δ 5.83(br s,1H),3.89(s,1H),2.74(s,1H),1.96-1.59(m,5H),1.42-1.38(m,1H)。

(R)-(-)-5-(羥甲基)-2-吡咯啶酮(1.0克,8.7毫莫耳,1.0當量)溶於二氯甲烷(40毫升)中。將三乙胺(1.569毫升,11.3毫莫耳,1.3當量)、對甲苯磺醯氯(1.904克,10.0毫莫耳,1.1當量)及4-(二甲胺基)吡啶(0.12克)於冰冷卻下加入,再將混合物於室溫攪拌18小時。將反應混合物於減壓下濃縮,將0.5N水性氫氯酸加入,再將混合物以乙酸乙酯萃取。將有機層以0.5N水性氫氯酸、水、飽和水性碳酸氫鈉、及飽和水性氯化鈉清洗。將有機部分乾燥(無水硫酸鈉),過濾及於減壓下濃縮,即得白色固狀
物(1.91克,81%)。1H NMR(300MHz,CDCl3):δ 7.79-7.76(m,2H),7.38-7.35(m,2H),5.73(br s,1H),4.08-4.04(dd,1H,J=3.5,9.5Hz),3.98-3.93(m,1H),3.85-3.82(dd,1H,J=7.5,9.5Hz),2.47(s,3H),2.36-2.22(m,3H),1.81-1.73(m,1H)。

4-甲苯磺酸(S)-(5-側氧基吡咯啶-2-基)甲酯(4.5克,16.7毫莫耳,1.0當量)溶於無水乙腈(140毫升)中,將碘化鈉(5.009克,33.4毫莫耳,2.0當量)加入,再將混合物加熱至迴流8小時。將反應混合物冷卻至室溫,於減壓下濃縮,將水加入,再將混合物以乙酸乙酯萃取。將有機層以飽和水性硫代硫酸鈉、水、及飽和水性氯化鈉清洗。將有機部分乾燥(無水硫酸鈉),過濾及於真空中濃縮,即得白色固狀之標題化合物(2.0克,53%)。1H NMR(300MHz,CDCl3):δ 6.42(br s,1H),3.90-3.82(s,1H),3.27-3.16(m,2H),2.53-2.29(m,3H),1.88-1.17(m,1H)。

將對甲苯磺酸(R)-5-(羥甲基)-2-吡咯啶酮(1.9克,7.1毫莫耳,1.0當量)溶於無水乙腈(60毫升)中,將碘化鈉(2.09克)加入,再將混合物於迴流加熱8小時。將反應混合物冷卻至室溫,於減壓下濃縮,將水加入,再將混合物以乙酸乙酯萃取。將有機層以飽和水性硫代硫酸鈉、水、及飽和水性氯化鈉清洗。將有機部分乾燥(無水硫酸鈉),過濾及於真空中濃縮,即得灰白色固狀之標題化合物(0.97克,61%)。1H NMR(300MHz,CDCl3):δ 5.92(br s,1H),3.89-3.85(m,1H),3.28-3.15(m,2H),2.53-2.30(m,3H),1.88-1.78(m,1H)。

(S)-5-(碘甲基)吡咯啶-2-酮(2.0克,8.9毫莫耳,1.0當量)溶於乙醇(60毫升)中,將碳酸鈉(1.036克,9.8毫莫耳,1.1當量)及10%鈀/碳(0.4克)加入,再將混合物於氫氣氛下攪拌16小時。將混合物通過矽藻土(Celite)中過濾,再將濾液於減壓下濃縮以得橙色半固狀物。將5%水性硫代硫酸鈉加至所得殘留物中,再將混合物以乙酸乙酯萃取。將有機層乾燥(無水硫酸鈉),過濾及於真空中濃
縮,即得淺黃色油狀之標題化合物(0.564克,64%)。1H NMR(300MHz,CDCl3):δ 6.61(br s,1H),3.82-3.72(sext,1H,J=6.6Hz),2.45-2.22(m,3H),1.71-1.59(m,1H),1.23-1.21(d,3H,J=6.6Hz)。

將(S)-5-碘甲基吡咯啶-2-酮(1.12克)溶於乙醇(30毫升)中,將碳酸鈉(0.53克)及10%鈀/碳(0.22克)加入,再將混合物於氫氣氛下攪拌8小時。將混合物通過矽藻土(Celite)中過濾,再將濾液於減壓下濃縮。將5%水性硫代硫酸鈉加至所得殘留物中,再將混合物以乙酸乙酯萃取。將有機層乾燥(無水硫酸鈉),過濾及於真空中濃縮,即得標題化合物(0.263克,61%)。1H NMR(300MHz,CDCl3):δ 6.34(br s,1H),3.83-3.72(sext,1H,J=6.3Hz),2.46-2.21(m,3H),1.72-1.60(m,1H),1.23-1.21(d,3H,J=6.0Hz)。

將(R)-3,6-二氟-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.1克,0.4毫莫耳,1.0當量)及乙二胺(0.232克,3.9毫莫耳,10.0當量)之乙醇(2毫升)混合液於55℃攪拌5小時。將反應混合物冷卻至室溫,再於真空中濃縮以得淡黃色漿狀物,其徐緩固化以形成灰白色固狀物(0.122克,96%)。1H NMR(300MHz,CDCl3):δ 7.68(dd,2H,J=8.7,2.7Hz),7.42(dd,2H,J=8.7,4.2Hz),7.22(td,2H,J=9.0,2.7Hz),4.33-4.31(d,2H,J=5.4Hz),4.15(m,1H),2.85-2.55(m,6H)1.84(br s,4H)。ESI(m/z):320.2(M+H)。


於圓底燒瓶內裝入4-溴丁酸(19.226克,115.1毫莫耳,1.0當量)及無水乙醚(500毫升)。將所得溶液冷卻至-78℃,再將三乙胺(16.473毫升,118.5毫莫耳,1.1當量)其後將三甲基乙醯氯(14.596毫升,118.5毫莫耳,1.1當量)加入。有白色沈澱物形成,其後將冷浴移出,再令反應混合物加溫至0℃,再攪拌2小時,將漿液再冷卻至
-78℃。於另一反應容器中,於-78℃將2.5M正丁基鋰之己烷溶液(45.146毫升,112.9毫莫耳,1.0當量)逐滴加至正於無水四氫呋喃(170毫升)中攪拌的(R)-4-苄基-2- 唑啶酮(20.000克,112.9毫莫耳,1.0當量)溶液中。10分鐘後,將所得漿液藉由大孔徑套管加至混合酐(已以約100毫升無水四氫呋喃清洗)中。於-78℃ 15分鐘後,令混合物於30分鐘期間加溫至0℃,然後保持於此溫1小時。然後以水(180毫升)小心地令漿液之反應中止,攪拌另5分鐘,然後加溫至室溫。將所得溶液以乙酸乙酯(2 x 500毫升)萃取。將結合之有機萃取物以水、飽和水性碳酸氫鈉(500毫升)、飽和水性氯化鈉(400毫升)清洗,乾燥(無水硫酸鈉),過濾,再於真空中濃縮。將粗製殘留物藉急驟層析(矽膠,10-50%乙酸乙酯/己烷)予以純化,即得清澈油狀物(26.0克,71%)。1H NMR(300MHz,CDCl3):δ 7.19-7.37(m,5H),4.63-4.71(m,1H),4.16-4.25(m,2H),3.52(t,2H,J=6.5Hz),3.29(dd,1H,J=3.3,13.5Hz),3.08-3.18(m,2H),2.78(dd,1H,J=9.6,13.5Hz),2.21-2.30(m,2H)。
唑啶酮(20.000克,112.9毫莫耳,1.0當量)溶液中。10分鐘後,將所得漿液藉由大孔徑套管加至混合酐(已以約100毫升無水四氫呋喃清洗)中。於-78℃ 15分鐘後,令混合物於30分鐘期間加溫至0℃,然後保持於此溫1小時。然後以水(180毫升)小心地令漿液之反應中止,攪拌另5分鐘,然後加溫至室溫。將所得溶液以乙酸乙酯(2 x 500毫升)萃取。將結合之有機萃取物以水、飽和水性碳酸氫鈉(500毫升)、飽和水性氯化鈉(400毫升)清洗,乾燥(無水硫酸鈉),過濾,再於真空中濃縮。將粗製殘留物藉急驟層析(矽膠,10-50%乙酸乙酯/己烷)予以純化,即得清澈油狀物(26.0克,71%)。1H NMR(300MHz,CDCl3):δ 7.19-7.37(m,5H),4.63-4.71(m,1H),4.16-4.25(m,2H),3.52(t,2H,J=6.5Hz),3.29(dd,1H,J=3.3,13.5Hz),3.08-3.18(m,2H),2.78(dd,1H,J=9.6,13.5Hz),2.21-2.30(m,2H)。
(S)鏡像異構物係以類似之方法合成。


將疊氮化鈉(7.623克,117.3毫莫耳,1.5當量)加至己攪拌之(R)-4-苄基-3-(4-溴丁醯基) 唑啶-2-酮(25.500克,78.2毫莫耳,1.0當量)之無水N,N-二甲基甲醯胺(225毫升)溶液中。將所得溶液於55℃加熱15分鐘及於70℃加熱2小時。冷卻至室溫後,將溶液倒至飽和水性氯化鈉/乙酸乙酯(1.4升)中,再以水及飽和水性氯化鈉清洗,乾燥(無水硫酸鈉),過濾,再於真空中濃縮以得油狀物(22.0克,98%),其係使用而不必進一步純化。1H NMR(300MHz,CDCl3):δ 7.36-7.19(m,5H),4.62-4.72(m,1H),4.10-4.25(m,2H),3.41(t,2H,J=6.6Hz),3.30(dd,1H,J=3.3,13.2Hz),2.99-3.10(m,2H),2.79(dd,2H,J=9.9,13.5Hz),1.93-2.02(m,2H);HPLC分析:(C18,20分鐘期間10-90%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):12.3分,95%。
唑啶-2-酮(25.500克,78.2毫莫耳,1.0當量)之無水N,N-二甲基甲醯胺(225毫升)溶液中。將所得溶液於55℃加熱15分鐘及於70℃加熱2小時。冷卻至室溫後,將溶液倒至飽和水性氯化鈉/乙酸乙酯(1.4升)中,再以水及飽和水性氯化鈉清洗,乾燥(無水硫酸鈉),過濾,再於真空中濃縮以得油狀物(22.0克,98%),其係使用而不必進一步純化。1H NMR(300MHz,CDCl3):δ 7.36-7.19(m,5H),4.62-4.72(m,1H),4.10-4.25(m,2H),3.41(t,2H,J=6.6Hz),3.30(dd,1H,J=3.3,13.2Hz),2.99-3.10(m,2H),2.79(dd,2H,J=9.9,13.5Hz),1.93-2.02(m,2H);HPLC分析:(C18,20分鐘期間10-90%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):12.3分,95%。
(S)鏡像異構物係以類似之方法合成。


將1.0M雙(三甲基矽烷基)胺鈉之四氫呋喃溶液(119.088毫升,71.5毫莫耳,1.0當量)徐緩地加至正於-78℃無水四氫呋喃(125毫升)中攪拌之(R)-3-(4-疊氮基丁
醯基)-4-苄基 唑啶-2-酮(20.600克,71.5毫莫耳,1.0當量)溶液中。15分鐘後,將所得溶液以碘甲烷(4.671毫升,75.0毫莫耳,1.0當量)處理。將冷卻浴移出,且於5分鐘後將反應容器置於冰水浴中,再攪拌另15分鐘。以飽和水性碳酸氫鈉(300毫升)令反應中止,再以乙酸乙酯(2 x 300毫升)萃取。將結合之有機萃取物以飽和水性氯化鈉(2 x 200毫升)清洗,乾燥(無水硫酸鎂),過濾,再於真空中濃縮。藉急驟層析(400克矽膠,10-50%乙酸乙酯/己烷)予以純化,即得清澈油狀之期望產物(9.7克,45%)。1H NMR(300MHz,CDCl3):δ 7.20-7.38(m,5H),4.65-4.72(m,1H),4.15-4.26(m,2H),3.85-3.79(m,1H),3.35(app.t,2H,J=6.0Hz),3.25(dd,1H,J=13.5,3.6Hz),2.78(dd,1H,J=13.0Hz,9.0Hz),2.04-2.18(m,1H),1.67-1.78(m,1H),1.27(d,3H,J=6.9Hz);HPLC分析:(C18,20分鐘期間10-90%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):13.0分,99.4%。
唑啶-2-酮(20.600克,71.5毫莫耳,1.0當量)溶液中。15分鐘後,將所得溶液以碘甲烷(4.671毫升,75.0毫莫耳,1.0當量)處理。將冷卻浴移出,且於5分鐘後將反應容器置於冰水浴中,再攪拌另15分鐘。以飽和水性碳酸氫鈉(300毫升)令反應中止,再以乙酸乙酯(2 x 300毫升)萃取。將結合之有機萃取物以飽和水性氯化鈉(2 x 200毫升)清洗,乾燥(無水硫酸鎂),過濾,再於真空中濃縮。藉急驟層析(400克矽膠,10-50%乙酸乙酯/己烷)予以純化,即得清澈油狀之期望產物(9.7克,45%)。1H NMR(300MHz,CDCl3):δ 7.20-7.38(m,5H),4.65-4.72(m,1H),4.15-4.26(m,2H),3.85-3.79(m,1H),3.35(app.t,2H,J=6.0Hz),3.25(dd,1H,J=13.5,3.6Hz),2.78(dd,1H,J=13.0Hz,9.0Hz),2.04-2.18(m,1H),1.67-1.78(m,1H),1.27(d,3H,J=6.9Hz);HPLC分析:(C18,20分鐘期間10-90%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):13.0分,99.4%。
(S)鏡像異構物係以類似之方法合成。

將三苯膦(16.744克,63.8毫莫耳,2.0當量)加至於室溫之正於無水四氫呋喃(125毫升)中攪拌的(R)-3-((R)-4-
疊氮基-2-甲基丁醯基)-4-苄基 唑啶-2-酮(9.650克,31.9毫莫耳,1.0當量)溶液中。5分鐘後(溶液轉為黃色且有氣泡),將水(0.575毫升,31.9毫莫耳,1.0當量)加入,再將反應混合物於25℃攪拌18小時。將混合物濃縮,再將所得殘留物藉急驟層析(矽膠,75-100%乙酸乙酯/己烷,然後100%至90%乙酸乙酯/甲醇梯度)予以純化,即得期望產物(2.98克,95%)。1H NMR(300MHz,CDCl3):δ 6.10(br.s,1H),3.25-3.35(m,2H),2.27-2.50(m,2H),1.68-1.80(m,1H),1.20(d,3H,J=7.5Hz)。
唑啶-2-酮(9.650克,31.9毫莫耳,1.0當量)溶液中。5分鐘後(溶液轉為黃色且有氣泡),將水(0.575毫升,31.9毫莫耳,1.0當量)加入,再將反應混合物於25℃攪拌18小時。將混合物濃縮,再將所得殘留物藉急驟層析(矽膠,75-100%乙酸乙酯/己烷,然後100%至90%乙酸乙酯/甲醇梯度)予以純化,即得期望產物(2.98克,95%)。1H NMR(300MHz,CDCl3):δ 6.10(br.s,1H),3.25-3.35(m,2H),2.27-2.50(m,2H),1.68-1.80(m,1H),1.20(d,3H,J=7.5Hz)。
(S)鏡像異構物係以類似之方法合成。

遵循Harris,B.D.et al.Synth.Commun.1986,16,1815之步驟。將三甲基矽烷基氯(0.805毫升,6.3毫莫耳,0.1當量)加至於室溫之已攪拌之(S)-(-)-4-胺基-2-羥基丁酸(15.000克,125.9毫莫耳,1.0當量)之二甲苯(1升)及六甲基二矽氮烷(184.757毫升,881.5毫莫耳,7.0當量)溶液中。將反應混合物加熱至迴流4小時,冷卻至室溫,再以純乙醇/甲醇(1升)稀釋。將溶劑於減壓下移出以得粗產物,將其藉管柱層析使用乙酸乙酯/二氯甲烷(30-80%梯度)作為洗提液予以純化,即得白色固狀物(17.9克,82%)。1H NMR(300MHz,CDCl3):δ 6.49(br s,1H),
4.28-4.23(t,1H,J=7.7Hz),3.40-3.21(m,2H),2.42-2.32(m,2H),2.08-1.95(m,2H),0.18(s,9H)。

遵循DiRocco,D.A.et al.J.Am.Chem.Soc.2009,131,10872 & DiRocco,D.A.et al.WO 2012/009372之步驟。將二碳酸二三級丁酯(15.11克,69.24毫莫耳,2.0當量)、三乙胺(4.82毫升,34.62毫莫耳,1.0當量)、及4-二甲胺基吡啶(4.23克,34.62毫莫耳,1.0當量)加至(S)-3-(三甲基矽烷氧基)吡咯啶-2-酮(6.00克,34.62毫莫耳,1.0當量)之無水二氯甲烷(150毫升)溶液中。將混合物於室溫攪拌過夜,然後將1N水性氫氯酸(100毫升)加入,再分層。將有機層以1N水性氫氯酸(2×50毫升)、及飽和水性氯化鈉(1×50毫升)清洗,乾燥(無水硫酸鈉)及過濾。將溶液於真空中濃縮以得粗製油狀物,將其藉矽膠層析(以0-15%乙酸乙酯/己烷洗提)予以純化,即得清澈黏滯油狀物,其於冷凍器中固化(3.93克,42%)。1H NMR(300MHz,CDCl3):δ 4.32-4.26(dd,1H,J=8.4,9.3Hz),3.82-3.74(ddd,1H,J=2.1,9.0,11.1Hz),3.50-3.41(m,1H),2.28-2.25(m,1H),1.94-1.87(m,1H),1.51(s,9H),0.18(s,9H)。

將2-側氧基-3-((三甲基矽烷基)氧基)吡咯啶-1-羧酸(S)-三級丁酯(3克,11毫莫耳)之無水二氯甲烷(55毫升)溶液冷卻至-78℃,於此時點將二乙胺基三氟化硫(2.9毫升,22毫莫耳,2.0當量)逐滴加入。然後令溶液徐緩地加溫至室溫,繼而將飽和水性碳酸氫鈉(50毫升)加入以令反應中止。分層,然後將有機層以飽和水性氯化銨(2 x 25毫升)清洗,乾燥(無水硫酸鈉),過濾,再於真空中濃縮以得粗製固狀物。然後將此生料溶於二氯甲烷(40毫升)中,再將三氟乙酸(2.5毫升,33毫莫耳,3.0當量)加入。將溶液攪拌3小時,此時點氣體的放出已消退。於真空中濃縮以得棕褐色液狀物,將其藉矽膠層析(以0-10%甲醇/二氯甲烷洗提)予以純化,即得白色固狀之期望產物(0.77克,68%)。1H NMR(300MHz,CDCl3):δ 7.35(br s,1H),5.15-4.93(dt,1H,J=7.6,53.4Hz),3.50-3.34(m,2H),2.53(m,1H),2.39-2.21(m,1H)。

將三苯膦(26.461克,100.9毫莫耳,2.0當量)加至於氮氣氛下之正攪拌之4-硝基苯甲酸(9.273克,55.5毫莫耳,1.1當量)及(S)-(-)-3-羥基-2-吡咯啶酮(5.100克,50.4毫莫耳,1.0當量)之無水四氫呋喃(175毫升)混合液中。將偶氮二羧酸二異丙酯(14.898毫升,75.7毫莫耳,1.5當量)逐滴加至此反應混合物中(邊以冷水浴進行外部冷卻)。將反應於室溫攪拌過夜。將反應混合物於真空中濃縮以得粗製殘留物。將甲醇(130毫升)加至殘留物中,其後將碳酸鉀(0.38克)於室溫加入。將反應混合物於室溫攪拌8小時。將反應混合物以二氯甲烷稀釋,再通過矽藻土(Celite)中過濾。將矽藻土(Celite)床以1%甲醇之二氯甲烷液清洗。將濾液結合,再濃縮至乾。將殘留物分配於乙酸乙酯:稀水性氫氯酸(20毫升,9:1)之間,再攪拌15分鐘。分層,再將水性層以乙酸乙酯清洗三次。將水性層濃縮至乾而得固狀殘留物。將粗製殘留物以1-2%甲醇之二氯甲烷液(3 x 50毫升)清洗,乾燥(無水硫酸鈉),過濾,再濃縮,即得棕褐色油狀物(3.38克,60%)。1H NMR(300MHz,CDCl3):δ 4.32-4.27(t,1H,J=8.5Hz),3.36-3.19(m,2H),2.48-2.40(m,1H),2.07-1.93(m,1H),1.16-1.14(d,1H,J=6.3Hz)。

三甲基矽烷基氯(0.405毫升,3.2毫莫耳,0.1當量)加至於室溫之已攪拌之(R)-3-羥基-2-吡咯啶酮(3.200克,31.7毫莫耳,1.0當量)、二甲苯(45毫升)、及六甲基二矽氮烷(39.8毫升,189毫莫耳,6.0當量)之懸浮液中。將反應混合物於迴流溫度加熱5小時,再以純乙醇(50毫升)稀釋。將溶劑於減壓下移出。將粗產物藉管柱層析使用乙酸乙酯之二氯甲烷液(30-80%梯度)作為洗提液予以純化以得清澈油狀物,其於靜置後固化成灰白色固狀物(2.57克,47%)。1H NMR(300MHz,CDCl3):δ 6.89(br s,1H),4.28-4.23(t,1H,J=8.1Hz),3.40-3.21(m,2H),2.42-2.32(m,1H),2.08-1.95(m,1H),0.18(s,9H)。

將二碳酸二三級丁酯(6.971克,31.9毫莫耳,2.0當量)、三乙胺(2.222毫升,16.0毫莫耳,1.0當量)、及N,N-4-二甲胺基吡啶(1.953克,16.0毫莫耳,1.0當量)加至(R)-3-((三甲基矽烷基)氧基)吡咯啶-2-酮(2.770克,16.0毫莫耳,1.0當量)之無水二氯甲烷(75毫升)溶液中。將混合物於室溫攪拌過夜,然後以二氯甲烷稀釋,再以0.1N水性氫氯酸(100毫升)清洗。將有機層以0.1N水性
氫氯酸(2×100毫升)、及飽和水性氯化鈉(1×100毫升)清洗,乾燥(無水硫酸鈉),再過濾。將溶液於真空中濃縮,再藉矽膠層析(以0-15%乙酸乙酯/己烷洗提)予以純化,即得清澈黏滯油狀物(2.1克,48%)。1H NMR(300MHz,CDCl3):δ 4.32-4.26(dd,1H,J=8.1,9.3Hz),3.82-3.74(ddd,1H,J=2.1,9.0,11.1Hz),3.50-3.41(m,1H),2.32-2.23(m,1H),1.97-1.87(m,1H),1.54(s,9H),0.18(s,9H)。

將2-側氧基-3-((三甲基矽烷基)氧基)吡咯啶-1-羧酸(R)-三級丁酯(2.100克,7.7毫莫耳,1.0當量)之無水二氯甲烷(37毫升)溶液冷卻至-78℃,於此時點將二乙胺基三氟化硫(2.030毫升,15.4毫莫耳,2.0當量)逐滴加入。然後令溶液徐緩加溫至室溫,然後將飽和水性碳酸氫鈉(33毫升)加入以令反應中止。分層,再將有機層以飽和水性氯化銨(2 x16毫升)清洗,乾燥(無水硫酸鈉),過濾,再於真空中濃縮以得粗製固狀物。然後將此生料溶於二氯甲烷(30毫升)中,再將三氟乙酸(1.7毫升,33毫莫耳,3.0當量)加入。將溶液攪拌3小時,此時點氣體的放出已消退。於真空中濃縮,再藉矽膠層析(以0-10%甲醇/二氯甲烷洗提)予以純化,即得灰白色固狀之期望產物(0.71克,
90%)。1H NMR(300MHz,CDCl3):δ 7.35(br s,1H),5.15-4.93(dt,1H,J=7.6,53.4Hz),3.50-3.34(m,2H),2.53(m,1H),2.39-2.21(m,1H)。

遵循Oikawa,M.et al.Tetrahedron,1995,51,62377之步驟。將含有哌啶(54.425毫升,551.0毫莫耳,2.0當量)及碳酸鉀(13.774克,99.7毫莫耳,0.4當量)之燒瓶浸於水浴中,再邊強烈攪拌邊將丙醛(16.000克,275.5毫莫耳,1.0當量)於20分鐘期間加入。攪拌18小時後,藉通過矽藻土(Celite)墊(墊以乙醚清洗)過濾以將不可溶之材料移出。將濾液乾燥(無水硫酸鈉),過濾及於真空中濃縮。將所得之粗製烯胺溶於乙腈(150毫升)中,再將丙烯酸甲酯(47.433克,551.0毫莫耳,2.0當量)逐滴加入。將反應混合物於迴流攪拌24小時,其後將乙酸(31.541毫升,551.0毫莫耳,2.0當量)及水(150毫升)加入。於迴流攪拌24小時後,將其以氯化鈉飽和,再以乙醚(3 x 100毫升)萃取。將結合之有機萃取物乾燥(無水硫酸鎂),過濾,再於真空中濃縮。藉矽膠急驟層析(0-20%乙酸乙酯/己烷)予以純化,即得無色油狀之加成物(22克,55%)。1H NMR(300MHz,CDCl3):δ 9.64(d,1H,J=1.8Hz),3.69(s,3H),2.50-2.35(m,3H),2.07(sext,1H,J=7.2Hz),1.71
(sext,1H,J=7.2Hz),1.14(d,3H,J=7.2Hz)。



遵循Amat,M.et al.J.Org.Chem.2014,79,2792之步驟。將(R)-(-)-苯基甘胺醇(5.500克,40.1毫莫耳,1.0當量)加至已攪拌之4-甲基-5-側氧基戊酸甲酯(5.780克,40.1毫莫耳,1.0當量)之甲苯(100毫升)溶液中。將反應混合物於迴流加熱25小時,同時以迪胺-斯塔克裝置(Dean-Stark apparatus)將所產生之水共沸移出。冷卻後,將混合物於真空中濃縮,再將殘留物藉矽膠管柱(柱以TEA預處理,然後以0-70%乙酸乙酯/己烷洗提)予以純化,即得淡黃色固狀之期望產物:(3R,8S,8aS)-8-甲基-3-苯基四氫-2H- 唑並[3,2-a]吡啶-5(3H)-酮(1.3克,14%)及無色漿狀之(3R,8S,8aR)-8-甲基-3-苯基四氫-2H-
唑並[3,2-a]吡啶-5(3H)-酮(1.3克,14%)及無色漿狀之(3R,8S,8aR)-8-甲基-3-苯基四氫-2H- 唑並[3,2-a]吡啶-5(3H)-酮(6.5克,70%)。(3R,8S,8aS)-8-甲基-3-苯基四氫-2H-
唑並[3,2-a]吡啶-5(3H)-酮(6.5克,70%)。(3R,8S,8aS)-8-甲基-3-苯基四氫-2H- 唑並[3,2-a]吡啶-5(3H)-酮:1H NMR(300MHz,CDCl3):δ 7.40-7.20(m,5H),5.25(t,1H,J=7.5Hz),4.61(d,1H,J=8.4Hz),4.48(t,1H,J=8.7Hz),3.75(dd,1H,J=9.0,7.8Hz),2.55(dd,1H,J=18.0,6.0Hz),
2.46-2.33(m,1H),1.88-1.51(m,3H),1.19(d,3H,J=5.7Hz);ESI(m/z):232.7(M+H)。(3R,8S,8aR)-8-甲基-3-苯基四氫-2H-
唑並[3,2-a]吡啶-5(3H)-酮:1H NMR(300MHz,CDCl3):δ 7.40-7.20(m,5H),5.25(t,1H,J=7.5Hz),4.61(d,1H,J=8.4Hz),4.48(t,1H,J=8.7Hz),3.75(dd,1H,J=9.0,7.8Hz),2.55(dd,1H,J=18.0,6.0Hz),
2.46-2.33(m,1H),1.88-1.51(m,3H),1.19(d,3H,J=5.7Hz);ESI(m/z):232.7(M+H)。(3R,8S,8aR)-8-甲基-3-苯基四氫-2H- 唑並[3,2-a]吡啶-5(3H)-酮:1H NMR(300MHz,CDCl3):δ 7.40-7.20(m,5H),4.93(d,1H,J=6.3Hz),4.44(d,1H,J=9.3Hz),4.14(dd,1H,J=8.7,6.3Hz),4.01(dd,1H,J=8.7,1.2Hz),2.47-2.25(m,2H),2.05-1.87(m,2H),1.60-1.43(m,1H),1.20(d,3H,J=6.6Hz);ESI(m/z):232.7(M+H)。
唑並[3,2-a]吡啶-5(3H)-酮:1H NMR(300MHz,CDCl3):δ 7.40-7.20(m,5H),4.93(d,1H,J=6.3Hz),4.44(d,1H,J=9.3Hz),4.14(dd,1H,J=8.7,6.3Hz),4.01(dd,1H,J=8.7,1.2Hz),2.47-2.25(m,2H),2.05-1.87(m,2H),1.60-1.43(m,1H),1.20(d,3H,J=6.6Hz);ESI(m/z):232.7(M+H)。

遵循Amat,M.et al.J.Org.Chem.2014,79,2792之步驟。將三乙基矽烷(7.458毫升,46.7毫莫耳,3.0當量)及氯化鈦(IV)(7.697毫升,70.0毫莫耳,4.5當量)加至己攪拌之(3R,8S,8aR)-8-甲基-3-苯基四氫-2H- 唑並[3,2-a]吡啶-5(3H)-酮(3.600克,15.6毫莫耳,1.0當量)之無水二氯甲烷(100毫升)溶液中,再將混合物於50℃攪拌24小時。然後將另外之氯化鈦(IV)(7.7毫升)及三乙基矽烷(7.5毫升)加入,再於50℃持續攪拌24小時。將混合物倒至飽和水性碳酸氫鈉(100毫升)中。將水性相於矽藻土(Celite)上過濾再以二氯甲烷萃取。將結合之有機萃取物乾燥(無水硫酸鈉),過濾,再於真空中濃縮以得殘留物,
將其於矽膠上進行層析(0-100%乙酸乙酯/己烷,然後純乙酸乙酯),即得無色油狀之期望產物(1.6克,44%)且回收起始材料(0.6克)。1H NMR(300MHz,CDCl3):δ 7.40-7.20(m,5H),5.80(dd,1H,J=9.6,4.8Hz),4.20-4.00(m,2H),2.97(ddd,1H,J=11.7,4.8,2.4Hz),2.84(dd,1H,J=11.7,9.9Hz),2.70(br s,1H),2.59(ddd,1H,J=17.7,6.3,3.0Hz),2.47(ddd,1H,J=17.7,11.1,6.6Hz),1.90-1.75(m,2H),1.50(m,1H),0.93(d,3H,J=6.3Hz)。
唑並[3,2-a]吡啶-5(3H)-酮(3.600克,15.6毫莫耳,1.0當量)之無水二氯甲烷(100毫升)溶液中,再將混合物於50℃攪拌24小時。然後將另外之氯化鈦(IV)(7.7毫升)及三乙基矽烷(7.5毫升)加入,再於50℃持續攪拌24小時。將混合物倒至飽和水性碳酸氫鈉(100毫升)中。將水性相於矽藻土(Celite)上過濾再以二氯甲烷萃取。將結合之有機萃取物乾燥(無水硫酸鈉),過濾,再於真空中濃縮以得殘留物,
將其於矽膠上進行層析(0-100%乙酸乙酯/己烷,然後純乙酸乙酯),即得無色油狀之期望產物(1.6克,44%)且回收起始材料(0.6克)。1H NMR(300MHz,CDCl3):δ 7.40-7.20(m,5H),5.80(dd,1H,J=9.6,4.8Hz),4.20-4.00(m,2H),2.97(ddd,1H,J=11.7,4.8,2.4Hz),2.84(dd,1H,J=11.7,9.9Hz),2.70(br s,1H),2.59(ddd,1H,J=17.7,6.3,3.0Hz),2.47(ddd,1H,J=17.7,11.1,6.6Hz),1.90-1.75(m,2H),1.50(m,1H),0.93(d,3H,J=6.3Hz)。

於備有乾冰冷凝器之二頸式燒瓶內裝入(S)-1-((R)-2-羥基-1-苯乙基)-5-甲基哌啶-2-酮(1.600克,6.9毫莫耳,1.0當量)及無水四氫呋喃(20毫升)。將混合物於氮氣氛下冷卻至-78℃,然後將氨(50毫升)冷凝。將反應溫度升至-33℃。將小碎片金屬鈉加入直至持續藍色為止,再將混合物於-33℃攪拌5分鐘。藉加入固體氯化銨令反應中止直至藍色消失為止。將混合物於室溫攪拌5小時,再將二氯甲烷加入。將混合物通過矽藻土(Celite)中過濾,再將濾液於真空中濃縮。將殘留物藉矽膠管柱(0-100%乙酸乙酯/己烷,然後20%甲醇/乙酸乙酯)予以純化,即得無色油狀之期望產物(0.480克,62%)。1H NMR(300MHz,CDCl3):
δ 5.84(br s,1H),3.10(m,1H),2.94(t,1H,J=10.8Hz),2.50-2.28(m,2H),2.05-1.80(m,2H),1.50(m,1H),1.03(d,3H,J=6.9Hz)。

遵循Amat,M.et al.J.Org.Chem.2014,79,2792之步驟。將三乙基矽烷(2.072毫升,13.0毫莫耳,3.0當量)及氯化鈦(IV)(2.138毫升,19.5毫莫耳,4.5當量)加至己攪拌之(3R,8R,8aS)-8-甲基-3-苯基四氫-2H- 唑並[3,2-a]吡啶-5(3H)-酮(1.000克,4.3毫莫耳,1.0當量)之無水二氯甲烷(50毫升)溶液中,再將混合物於50℃攪拌6小時。將混合物倒至飽和水性碳酸氫鈉(50毫升)中。將水性相於矽藻土(Celite)上過濾,再以二氯甲烷萃取。將結合之有機萃取物乾燥(無水硫酸鈉)、過濾,及於真空中濃縮以得殘留物,將其進行層析(0-100%乙酸乙酯/己烷,然後乙酸乙酯),即得無色漿狀之期望產物(0.200克,20%)。1H NMR(300MHz,CDCl3):δ 7.40-7.20(m,5H),5.67(dd,1H,J=7.8,6.3Hz),4.20-4.05(m,2H),3.15-3.00(m,2H),2.65-2.40(m,3H),2.00-1.75(m,2H),1.40(m,1H),0.89(d,3H,J=6.9Hz)。
唑並[3,2-a]吡啶-5(3H)-酮(1.000克,4.3毫莫耳,1.0當量)之無水二氯甲烷(50毫升)溶液中,再將混合物於50℃攪拌6小時。將混合物倒至飽和水性碳酸氫鈉(50毫升)中。將水性相於矽藻土(Celite)上過濾,再以二氯甲烷萃取。將結合之有機萃取物乾燥(無水硫酸鈉)、過濾,及於真空中濃縮以得殘留物,將其進行層析(0-100%乙酸乙酯/己烷,然後乙酸乙酯),即得無色漿狀之期望產物(0.200克,20%)。1H NMR(300MHz,CDCl3):δ 7.40-7.20(m,5H),5.67(dd,1H,J=7.8,6.3Hz),4.20-4.05(m,2H),3.15-3.00(m,2H),2.65-2.40(m,3H),2.00-1.75(m,2H),1.40(m,1H),0.89(d,3H,J=6.9Hz)。

於備有乾冰冷凝器之二頸式燒瓶內裝入(R)-1-((S)-2-羥基-1-苯乙基)-5-甲基哌啶-2-酮(0.200克,0.9毫莫耳,1.0當量)及無水四氫呋喃(5毫升)。將混合物於氮氣氛下冷卻至-78℃,然後將氨(15毫升)冷凝。將反應溫度升至-33℃。將小碎片金屬鈉加入直至持續藍色為止,再將混合物於-33℃攪拌5分鐘。藉加入固體氯化銨令反應中止直至藍色消失為止。將混合物於室溫攪拌5小時,再將二氯甲烷加入。將混合物通過矽藻土(Celite)中過濾,再將濾液於真空中濃縮。將殘留物藉矽膠管柱(0-100%乙酸乙酯/己烷,然後20%甲醇/乙酸乙酯)予以純化,即得黃色油狀之期望產物(0.080克,82%)。1H NMR(300MHz,CDCl3):δ 5.85(br s,1H),3.10(m,1H),2.93(t,1H,J=10.8Hz),2.50-2.28(m,2H),2.05-1.80(m,2H),1.50(m,1H),1.03(d,3H,J=6.6Hz)。

將Boc-Ala-OMe(5.000克,24.6毫莫耳,1.0當量)及28%水性氫氧化銨(100.00毫升,1479.7毫莫耳,60.1當量)之甲醇(100毫升)混合液於室溫攪拌16小時。將反應混合物於真空中濃縮,即得白色固狀之期望產物(4.65克,100%)。1H NMR(300MHz,CDCl3):δ 6.15(br,1H),5.55(br,1H),4.95(br,1H),4.20(br,1H),1.46(s,9H),1.39(d,3H,J=7.2Hz)。

將1.0M甲硼烷-四氫呋喃錯合物之四氫呋喃液(85.536毫升,85.5毫莫耳,3.5當量)加至於氮下之已攪拌之Boc-Ala-NH2(4.600克,24.4毫莫耳,1.0當量)之無水四氫呋喃(100毫升)溶液中。將混合物於室溫攪拌16小時,然後於70℃加熱2小時。冷卻後,以甲醇令反應中止直至無氣泡產生為止。將混合物於70℃加熱2小時,然後於真空中濃縮。將殘留物藉矽膠管柱(0-100%乙酸乙酯/己烷,然後0-30%甲醇/二氯甲烷)予以純化,即得半固狀之期望產物(1.4克,33%)。1H NMR(300MHz,CDCl3):δ 4.60(br s,1H),3.65(m,1H),2.76(dd,1H,J=12.9,4.8Hz),2.64(dd,1H,J=12.9,6.6Hz),1.45(s,9H),1.13(d,3H,J=6.9Hz)。

將(R)-3,6-二氟-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.600克,2.3毫莫耳,1.0當量)及(1-胺基丙-2-基)胺基甲酸(S)-三級丁酯(1.210克,6.9毫莫耳,3.0當量)之乙醇(10毫升)混合液於70℃攪拌。15小時後,將反應混合物於真空中濃縮,再藉矽膠管柱(0-100%乙酸乙酯/己烷,然後0-30%甲醇/二氯甲烷)予以純化,即得白色泡沫狀之期望產物(0.750克,75%)及回收胺起始材料(0.2克)。1H NMR(300MHz,CDCl3):δ 7.67(dd,2H,J=8.7,2.4Hz),7.42(dd,2H,J=8.7,3.9Hz),7.22(td,2H,J=9.0,2.7Hz),4.50-4.25(m,3H),4.13(m,1H),3.78(br s,1H),2.86(dd,1H,J=12.0,3.6Hz),2.70-2.50(m,3H),1.43(s,9H),1.10(d,3H,J=6.3Hz);ESI(m/z):434.0(M+H)。

將(R)-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.150克,0.7毫莫耳,1.0當量)及(1-胺基丙-2-基)胺基甲酸(S)-三級丁酯(0.200克,1.1毫莫耳,1.7當量)之乙醇(5毫升)混合液於70℃攪拌。15小時後,將反應混合物於真空中濃縮,再藉矽膠管柱(0-100%乙酸乙酯/己烷)予以純化,即得白色泡沫狀之期望產物(0.120克,45%)。1H NMR(300MHz,CDCl3):δ 8.09(d,2H,J=7.8Hz),7.52-7.41(m,4H),7.25-7.20(m,2H),4.50-4.35(m,3H),4.17(m,1H),3.73(m,1H),2.83(dd,1H,J=11.7,3.6Hz),2.66-2.47(m,3H),1.43(s,9H),1.08(d,3H,J=6.6Hz);ESI(m/z):398.1(M+H)。

將Boc-D-Ala-OMe(5.000克,24.6毫莫耳,1.0當量)及28%水性氫氧化銨(100毫升,1479.7毫莫耳,60.1
當量)之甲醇(100毫升)混合液於室溫攪拌16小時。將反應混合物於真空中濃縮,即得白色固狀之期望產物(4.65克,100%)。1H NMR(300MHz,CDCl3):δ 6.15(br,1H),5.60(br,1H),5.00(br,1H),4.20(br,1H),1.46(s,9H),1.39(d,3H,J=6.9Hz)。

將1.0M甲硼烷-四氫呋喃錯合物之四氫呋喃液(97.756毫升,97.8毫莫耳,4.0當量)加至於氮下之已攪拌之Boc-D-Ala-NH2(4.600克,24.4毫莫耳,1.0當量)之無水四氫呋喃(100毫升)溶液中。將混合物於室溫攪拌16小時,然後於70℃加熱2小時。冷卻後,以甲醇令反應中止直至無氣泡產生為止。將混合物於70℃加熱2小時,然後於真空中濃縮。將殘留物藉矽膠管柱(0-30%甲醇/二氯甲烷)予以純化,即得無色油狀之期望產物(1.5克,35%)。1H NMR(300MHz,CDCl3):δ 4.60(br s,1H),3.65(m,1H),2.75(dd,1H,J=12.9,5.1Hz),2.63(dd,1H,J=12.9,6.6Hz),1.45(s,9H),1.41(s,2H),1.12(d,3H,J=6.3Hz)。

將(R)-3,6-二氟-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.200克,0.8毫莫耳,1.0當量)及(1-胺基丙-2-基)胺基甲酸(R)-三級丁酯(0.403克,2.3毫莫耳,3.0當量)之乙醇(10毫升)混合液於70℃攪拌。15小時後,將反應混合物於真空中濃縮,再藉矽膠管柱(0-30%甲醇/二氯甲烷)予以純化,即分離出白色粉狀之期望產物(0.250克,75%)。1H NMR(300MHz,CDCl3):δ 7.68(dd,2H,J=8.7,2.7Hz),7.41(dd,2H,J=9.3,4.2Hz),7.22(td,2H,J=9.3,2.4Hz),4.43(br s,1H),4.35(d,2H,J=5.7Hz),4.12(m,1H),3.78(br s,1H),2.80(dd,1H,J=12.3,3.6Hz),2.63(dd,1H,J=12.3,5.7Hz),2.57(d,2H,J=7.2Hz),1.44(s,9H),1.11(d,3H,J=6.6Hz);ESI(m/z):434.0(M+H)。

將(R)-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.150克,0.7毫莫耳,1.0當量)及(1-胺基丙-2-基)胺基甲酸(R)-三級丁酯(0.351克,2.0毫莫耳,3.0當量)之乙醇(10毫升)混合液於70℃攪拌。15小時後,將反應混合物於真空中濃縮,再藉矽膠管柱(0-30%甲醇/二氯甲烷)予以純化,即分離出白色泡沫狀之期望產物(0.210克,78%)。1H NMR(300MHz,CDCl3):δ 8.10(d,2H,J=7.5Hz),7.55-7.40(m,4H),7.27-7.20(m,2H),4.60-4.30(m,3H),4.16(m,1H),3.78(m,1H),2.80(dd,1H,J=12.3,3.6Hz),2.67(dd,1H,J=12.0,8.4Hz),2.60-2.50(m,2H),1.44(s,9H),1.10(d,3H,J=6.6Hz);ESI(m/z):398.1(M+H)。

將三級丁醇鉀(0.151克,1.3毫莫耳)加至已攪拌之哌啶-2-酮(0.133克,1.3毫莫耳)之二甲亞碸(5毫升)溶液
中,再將混合物於室溫攪拌1小時。將9-(環氧乙烷-2-基甲基)-9H-咔唑(0.150克,0.7毫莫耳)加入,再將混合物於室溫攪拌16小時。將混合物以水稀釋,再以乙酸乙酯萃取。將有機層以飽和水性氯化鈉清洗,乾燥(無水硫酸鈉),過濾及於真空中濃縮。將殘留物藉矽膠管柱(50-100%乙酸乙酯/己烷)然後藉製備HPLC(C18,於0-8分鐘期間含0.1%甲酸之30-80%乙腈/水)予以純化,即得白色泡沫狀產物(0.098克,45%)。1H NMR(300MHz,CDCl3):δ 8.08(d,2H,J=7.8Hz),7.50-7.40(m,4H),7.28-7.18(m,2H),4.60-4.25(m,4H),3.87(dd,1H,J=14.1,7.8Hz),3.15-2.95(m,3H),2.36(t,2H,J=5.7Hz),1.85-1.55(m,4H);ESI(m/z):323.2(M+H)。
化合物2至72係藉類似於用於化合物1之步驟或藉使用氫化鈉(0.4當量)取代三級丁醇鉀製得。另外,亦可使用磷腈鹼P4-t-Bu取代三級丁醇鉀。在當用於下列標靶之起始材料非市售之情況下,合成法乃述於先前之製備實例中。




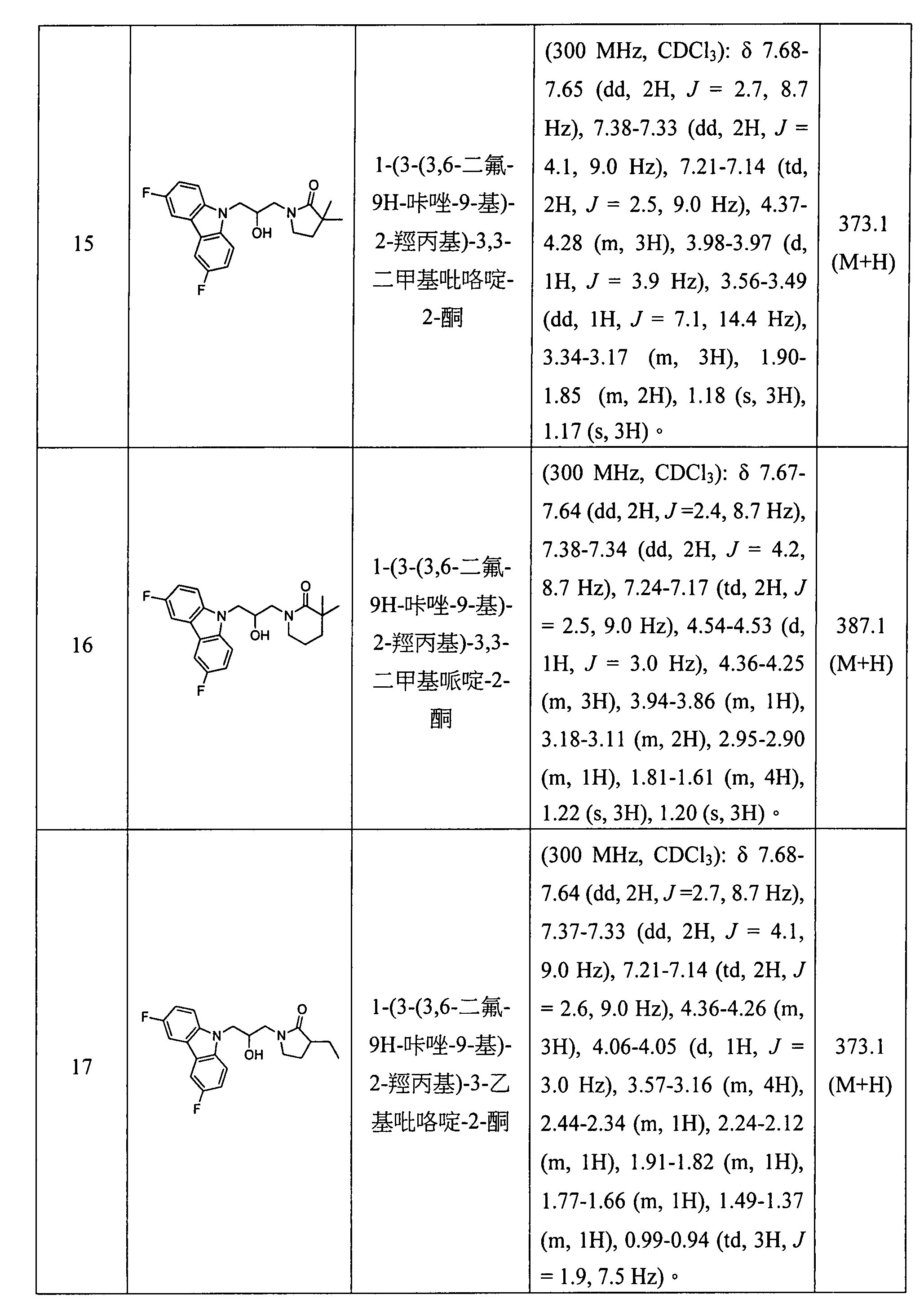

























將3,6-二氟-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.06克,0.2毫莫耳)及1,3-二胺基丙烷(0.195毫升,2.3毫莫耳)之乙醇(2毫升)混合液於40℃攪拌5小時。將反應混合物於真空中濃縮以得淡黃色油狀物。ESI(m/z):334.2(M+H)。於0℃將殘留物溶於二氯甲烷中,將4-二甲胺基吡啶(0.005克)及1,1’-羰基二咪唑(0.056克,0.3毫莫耳)
加入。將混合物加溫至室溫,再攪拌16小時。將反應於真空中濃縮,再將殘留物藉矽膠層析(0-10%甲醇/二氯甲烷)予以純化,即得白色粉狀之期望產物(0.060克,72%)。1H NMR(300MHz,d6-DMSO):δ 7.99(dd,2H,J=9.6,2.7Hz),7.56(dd,2H,J=9.0,4.2Hz),7.30(td,2H,J=9.0,2.7Hz),6.29(s,1H),5.24(d,1H,J=5.4Hz),4.35(dd,1H,J=15.0,3.6Hz),4.24(dd,1H,J=15.0,8.1Hz),4.07(m,1H),3.55-3.05(m,6H),1.90-1.70(m,2H);ESI(m/z):360.1(M+H)。
化合物72至84係藉類似於用於化合物71之步驟使用適當二胺諸如N-甲基-1,3-二胺基丙烷、乙二胺、N-乙基乙二胺、N1-環己基丙烷-1,3-二胺,N1-環丁基丙烷-1,3-二胺,N1-環戊基丙烷-1,3-二胺或丙烷-1,2-二胺取代1,3-二胺基丙烷製得。所用之非市售二胺之製備乃述於中間體之製備中。







將(1-((3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)胺基)丙-2-基)胺基甲酸三級丁酯(1.400克,80%純度,2.6毫莫耳,1.0當量)及三級丁醇鉀(0.290克,2.6毫莫耳,1.0當量)之無水四氫呋喃(270毫升)混合液於迴流攪拌2小時。將反應混合物冷卻至室溫,將乙酸(0.1毫升)及矽膠加入,再將混合物於減壓下濃縮成漿液。將粗製殘留物藉矽
膠管柱層析藉以0-10%甲醇/二氯甲烷之梯度洗提予以純化以得固狀物。將固狀物由乙酸乙酯中結晶,即得白色固狀物(0.673克,72%)。1H NMR(300MHz,CDCl3;非鏡像異構物之混合物):δ 7.69-7.65(dd,2H,J=2.7,8.7Hz),7.40-7.35(dd,2H,J=4.1,9.0Hz),7.24-7.18(td,2H,J=2.7,9.0Hz),4.47-4.19(m,5H),3.82(br m,1H),3.58-2.93(m,4H),1.26-1.24(d,3H,J=6.3Hz);ESI(m/z):360.9(M+H)。

60%氫化鈉之礦油液(0.009克,0.2毫莫耳)加至3,6-二氟-9H-咔唑(0.065克,0.3毫莫耳)之無水N,N-二甲基甲醯胺(0.3毫升)溶液中,再將混合物於室溫攪拌30分鐘。將1-乙基-3-(環氧乙烷-2-基甲基)四氫嘧啶-2(1H)-酮(0.055克,0.3毫莫耳)之無水N,N-二甲基甲醯胺(0.3毫升)溶液加入,再將混合物於70℃加熱8小時。將反應混合物以甲醇稀釋,過濾,再藉製備HPLC(C18,30-95%乙腈之水液)予以純化,即得白色固狀物(0.072克,62%)。1H NMR(300MHz,CDCl3):δ 7.67-7.63(dd,2H,J=2.6,9.0Hz),7.39-7.35(dd,2H,J=4.2,8.7Hz),7.23-7.16
(td,2H,J=2.4,9.0Hz),5.52(d,1H,J=2.7Hz),4.37-4.21(m,3H),3.86-3.79(m,1H),3.43-2.98(m,6H),2.88-2.83(dd,1H,J=1.8,14.7Hz),1.91-1.83(quin,2H,J=5.9Hz),1.12-1.07(t,3H,J=7.4Hz);ESI(m/z):388.1(M+H);HPLC分析:(C18,20分鐘期間10-90%乙腈之水液;滯留時間,於254nm之面積%):12.8分,100%。
化合物87-100係藉類似於用於化合物88之步驟製得。








將60%氫化鈉之礦油液(0.075克,1.9毫莫耳,1.2當量)加至已攪拌之3,6-二氟-9H-咔唑(0.38克,1.9毫莫耳,1.2當量)之無水N,N-二甲基乙醯胺(2毫升)溶液中。於室溫攪拌2小時後,所得咔唑鈉溶液隨時備用。
將60%氫化鈉之礦油液(0.075克,1.9毫莫耳,1.2當
量)加至於0℃之已攪拌之1,6-二甲基四氫嘧啶-2(1H)-酮(0.2克,1.6毫莫耳,1.0當量)之無水N,N-二甲基乙醯胺(3毫升)溶液中,再將混合物於室溫攪拌1小時。將表溴醇(0.155毫升,1.9毫莫耳,1.2當量)於0℃加入,再將混合物徐緩加溫至室溫,再攪拌16小時。將咔唑鈉溶液加入,再將混合物於70℃加熱5小時。將反應以水稀釋,再以乙酸乙酯萃取。將有機層以飽和水性氯化鈉清洗,於硫酸鈉上乾燥,過濾及於真空中濃縮。將殘留物藉矽膠管柱(0-100%乙酸乙酯/己烷)予以純化,即得白色泡沫狀之純產物(0.116克,19%)。1H NMR(300MHz,CDCl3):(兩種非鏡像異構物之混合物)δ 7.68(dd,2H,J=8.7,2.7Hz),7.40(dd,2H,J=9.0,4.2Hz),7.27-7.18(m,2H),5.51 and 5.06(d,1H,J=2.7Hz),4.50-4.20(m,3H),3.90-3.74(m,1H),3.50-2.75(m,7H),2.05(m,1H),1.60(m,1H),1.21 and 1.17(d,3H,J=6.6Hz);ESI(m/z):388.2(M+H)。

1-苄基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3,4-二氫喹唑啉-2(1H)-酮(0.102克,0.2毫莫耳,1.0當量)及20%氫氧化鈀/碳(0.035克)之乙酸(4毫升)及四
氫呋喃(2毫升)混合液於50psi氫下攪拌48小時。將20%氫氧化鈀/碳(0.02克)及10%鈀/碳(0.01克)加入,再將反應混合物於50psi氫下攪拌72小時。將混合物過濾,濃縮,再藉製備HPLC(C18,40-80%乙腈之水液)予以純化,即得白色固狀物(0.013克,15%)。1H NMR(300MHz,CD3OD):δ 7.73-7.70(dd,2H,J=2.6,8.6Hz),7.55-7.51(dd,2H,J=4.1,9.2Hz),7.27-7.05(m,3H),7.07-7.05(d,1H,J=7.2Hz),6.96-6.91(td,1H,J=1.1,7.5Hz),6.81-6.78(d,1H,J=8.1Hz),4.71(s,2H),4.33(s,2H)3.75-3.70,3.59-3.54(ABq,2H,J=14.4Hz),1.22(s,3H);ESI(m/z):422.1(M+H);HPLC分析:(C18,20分鐘期間5-95%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):13.5分,91.5%。
下列化合物係藉類似於用於化合物102之步驟製得。


將60%氫化鈉之礦油液(0.009克,0.2毫莫耳,1.0當量)加至已攪拌之(1S,4R)-2-氮雜雙環[2.2.1]庚-3-酮(0.051克,0.5毫莫耳,2.0當量)之無水四氫呋喃(1.5毫升)溶液中,再將混合物於室溫攪拌60分鐘。將(R)-3,6-二氟-9-(環氧乙烷-2-基甲基)-9H-咔唑(0.060克,0.2毫莫耳,1.0當量)加入,再將混合物於80℃於密封管中攪拌16小時。將混合物冷卻至室溫,再將1N乙酸(0.02毫升)之甲醇液加入,然後於減壓下濃縮。將粗製殘留物藉製備HPLC(C18,30-95%乙腈之水液)予以純化,即得白色固狀物(0.053克,61%)。1H NMR(300MHz,CDCl3):δ 7.68-7.65(dd,2H,J=2.6,8.6Hz),7.38-7.33(dd,2H,J=4.1,8.9Hz),7.24-7.17(td,2H,J=2.5,9.0Hz),4.38-4.28(m,4H),3.64(br s,1H),3.43-3.38(dd,1H,J=2.7,14.1Hz),3.02-2.96(dd,1H,J=2.7,14.4Hz),2.90-2.89(m,1H),1.96-1.40(m,6H);ESI(m/z):371.1(M+H);HPLC分析:(C18,20分鐘期間5-95%乙腈之水液+0.1%三氟乙酸:滯留時間,於254nm之面積%):12.2分,100%;
手性HPLC分析(Chiralcel AD-H,42分鐘期間15%乙醇之己烷液:滯留時間,於254nm之面積%):7.7分,98.5%;12.6分,0.7%(98.5% de)。
化合物105-138係藉類似於用於化合物104之步驟製得。




















用於評估式I化合物之特定分析法包括如下所述之用於評估測試化合物效力之Per2分析法及用於評估測試化合物標靶之Cry1分析法。
化合物係使用如前先前述於Zhang,E.E.et al.Cell,2009,139,199-210之高通量日夜分析系統進行篩選。簡單地說,將攜帶Per2-dLuc之安定U2OS報導子細胞以30,000個細胞/孔之密度塗佈於Corning 96孔式固狀白色平底之經TC處理之微孔盤(Corning®)中,再於37℃、於5%二氧化碳之存在下、於補充有10%胎牛血清(FBS)及盤尼西林(100單位/毫升)-鏈黴素(100微克/毫升)之杜伯爾科
改良式伊格爾培養基(DMEM)中培育48小時。將式I化合物典型地以2毫克/毫升之濃度溶於二甲亞碸(DMSO)中。然後將DMSO儲液於DMSO中系列稀釋,典型地每次稀釋步驟稀釋3倍。48小時期限後,將細胞培養基由塗佈之細胞中移出,再將細胞以200微升/孔之補充有5μM毛喉素(forskolin(Tocris®))及1mM甲蟲螢光素(beetle luciferin(Promega®))的完全細胞培養基(上述)同步化。同步化之後立即地將1微升化合物稀釋液加至每一孔中。將盤密封,簡短地搖動,再藉於35℃量測冷光(Tecan® Infinite M200 or Tecan® Infinite M200 Pro)連續最少3天以監測基因表現。原始冷光數據(計數)首先使用MulticycleTM軟體(Actimetrics公司)分析以測出每一化合物濃度之幅度(amp)、周期及相(phz)。對照組孔(亦即無化合物,僅二甲亞碸(DMSO))之周期長度應為26-30小時。然後將幅度數據相對於對數化合物濃度(M)繪圖,再藉非線性回歸分析法分析以測出EC50。
下表提供特定化合物之Per2 EC50數據。EC50值係以微莫耳濃度表示。


熟諳此藝者可輕易地將此分析法最適化以測得本文所述任何化合物之Per2 EC50數據。
化合物結合至分離出來之人類CRY1蛋白(hCRY1)之FAD結合結構域之作用係使用差示掃描螢光(‘熱漂移’)分
析法(Pantoliano et al.(2001)J Biomol Screening 6,429;Niesen et al.(2007)Nature Protocols 2,2212)測定。hCRY1之FAD結合結構域(胺基酸殘基1-494)連同C端Myc-DDK標籤(FADBD)係藉HEK293T細胞(型號# CRL-3216,美國菌種中心(American Type Culture Collection))之瞬時轉染製造且藉抗-FLAG親和層析法(型號# A2220,Sigma-Aldrich)純化。將FADBD(每孔0.5微克)與稀釋於DMSO中之化合物稀釋液(反應中為5% DMSO最終濃度)於17.5微升Tris緩衝鹽水(TBS)中、於冰上培育10分鐘,然後將2.5微升8X Sypro澄染料(Sypro-Orange Dye)(Life Technologies)加至每一孔中。每一化合物濃度一式三份地進行孔之分析。熔化溫度係於ABI7500定量PCR儀器中、使用熔化曲線模式以於25℃ 2分鐘之熱概況,其後1℃/分鐘之斜坡率上升至99℃進行量測。每一孔之熔化溫度係由熔化曲線之第一導函數中測出。熔化溫度之改變(△Tm)藉由扣除單獨地FADBD之5% DMSO液而得。如圖21所示,化合物72可觀察到△Tm之劑量依賴性提高,顯示化合物與hCRY1 FADBD蛋白密切相關。
此實例中,係檢測含有咔唑之醯胺類、胺基甲酸酯類及脲類對各種小鼠模型之時鐘及糖質新生基因表現之效應。尤其使用四種不同的小鼠模型:ICR小鼠、Balb/c小鼠、C57B1/6J DIO(飲食誘導性肥胖症)小鼠、及db/db小
鼠。C57B1/6J DIO及db/db小鼠均為技藝中已知之糖尿病、肥胖症、及血脂異常模型。飲食誘導性肥胖(DIO)小鼠,其因應於高脂餵食而顯現第II型糖尿病表型,發展出肥胖症、高胰島素血症、胰島素抗性及葡萄糖不耐性(Srinivasan and Ramarao,2007)。db/db小鼠(leprdb小鼠)具有db基因之突變,該db基因編碼瘦素受體。Db/db小鼠自然地攝食過度且變得肥胖、高血糖、高胰島素血及胰島素抗性。
檢測Cry調節劑化合物對基因表現之效應的體內研究係使用下述之各種實驗模型進行。
四天研究. 雄ICR小鼠(重30-35克),得自Charles River實驗室(加州霍利斯特),係在至少3天之環境適應後用於進行實驗。將小鼠投予載劑(WFI或10% Kolliphor)或化合物(50毫克/公斤,劑量體積5毫升/公斤,口服)4天BID(一日兩次),由第一天的下午開始。化合物或載劑的最後一次劑量係在採檢日早上投予。於採檢當日(最終劑量之後6小時),以二氧化碳窒息法使小鼠安樂死,再切出50毫克肝組統,再置於含500微升RNAlater之管中。
24小時基因表現研究. 購買17個星期大之C57B1/6J DIO小鼠(The Jackson Laboratory,加州沙加緬度),且於用於實驗前先適應環境2星期。整體之群組大小為每次處理為15隻,分成5個時間點而得最終之群組大小為3隻小鼠。將小鼠以5毫升/公斤BID之劑量體積經由胃管經
口餵食法投予載劑(注射用水(WFI))或Cry調節劑化合物化合物72(50毫克/公斤於WFI中)2天,最終之第五次投予劑量係在採檢前12小時。
每隻小鼠於實驗期間總計接受5次之化合物劑量。在研究開始之前,將小鼠稱重,再以晚上的體重為基準隨機分配至每一處理組。於第3天之下午3:00開始,使用二氧化碳窒息法使經載劑或化合物72處理之動物群組安樂死,分別切出50毫克肝、副睪脂肪及骨骼肌,再置於含500微升RNALater之管中。此步驟亦對剩餘之時間點群組於其給定之採檢時間進行。血漿樣品亦取自每隻動物再冰凍以待稍後用於化合物級別之量測。
小鼠肝之總RNA之製備. 使用E.Z.N.A.® HP總RNA分離套組(E.Z.N.A.® HP Total RNA Isolation Kits)(R6812-01且擬案述於手冊中,2010年修訂)以由肝樣品中製備及分離出RNA。欲製備RNA樣品,乃將10-30毫克樣品由RNA-Later中移出,再置於1.5毫升微離心管(microfuge tube)中。將GTC溶解緩衝液(700微升)加至組織中,再以轉子-定子均質機(例如Tissue-Tearor,模型# 985370 BioSpec產品含4.5毫米探針,型號# 985370-04)均質化,然後全速離心( 13,000 x g)5分鐘。將清澈之上層液藉吸移法轉移至已預插於2毫升收集管(Collection Tube)之DNA清除管柱(DNA Clearance Column)中。將組裝之管柱以13,000 x g之速離心1分鐘,再將流經物存起來。將等量(700微升)70%乙醇加至
溶解產物中,再混合。然後將樣品施至已置於2毫升收集管之HiBind RNA旋轉離心管柱,將其以10,000 x g之速於室溫離心60秒。將RNA清洗緩衝液I(RNA Wash Buffer I)(250微升)藉吸移法直接加至已插於2毫升收集管中之新HiBind RNA管柱上。將組裝之管柱以10,000 x g之速離心60秒。將RNA管柱置於新2毫升收集管中。將DNA酶I(DNase I)儲液直接吸移(75微升)至每一管柱之HiBind RNA樹脂上(使用不含RNA酶之DNA酶組(E1091)進行DNA酶消化:用於每一HiBind RNA管柱,DNA酶I(DNase I)儲液製備如下:E.Z.N.A.® DNase I消化緩衝液73.5微升,不含RNA酶之DNA酶I(RNase Free DNase I)(20Kunitz/微升)1.5微升=總體積75微升)。將管柱與結合之RNA於室溫(25-30℃)培育15分鐘。將RNA清洗緩衝液I(RNA Wash Buffer I)(500微升)加至管柱中,再置於工作台2分鐘。以10,000 x g之速離心60秒後,丟棄流經物,將500微升RNA清洗緩衝液II(RNA Wash Buffer II)加入,再以10,000 x g之速離心60秒。將另500微升RNA清洗緩衝液II(RNA Wash Buffer II)加入,再將管柱組裝體以10,000 x g之速離心60秒。將管柱以最大速離心2分鐘至完全將HiBind基質乾燥。將管柱置於乾淨之1.5毫升微離心管中,再將40-70微升分子生物學級水加入。靜置1分鐘後,將管柱以最大速離心2分鐘以洗提出RNA。將分離出之RNA收集至收集管中。
13,000 x g)5分鐘。將清澈之上層液藉吸移法轉移至已預插於2毫升收集管(Collection Tube)之DNA清除管柱(DNA Clearance Column)中。將組裝之管柱以13,000 x g之速離心1分鐘,再將流經物存起來。將等量(700微升)70%乙醇加至
溶解產物中,再混合。然後將樣品施至已置於2毫升收集管之HiBind RNA旋轉離心管柱,將其以10,000 x g之速於室溫離心60秒。將RNA清洗緩衝液I(RNA Wash Buffer I)(250微升)藉吸移法直接加至已插於2毫升收集管中之新HiBind RNA管柱上。將組裝之管柱以10,000 x g之速離心60秒。將RNA管柱置於新2毫升收集管中。將DNA酶I(DNase I)儲液直接吸移(75微升)至每一管柱之HiBind RNA樹脂上(使用不含RNA酶之DNA酶組(E1091)進行DNA酶消化:用於每一HiBind RNA管柱,DNA酶I(DNase I)儲液製備如下:E.Z.N.A.® DNase I消化緩衝液73.5微升,不含RNA酶之DNA酶I(RNase Free DNase I)(20Kunitz/微升)1.5微升=總體積75微升)。將管柱與結合之RNA於室溫(25-30℃)培育15分鐘。將RNA清洗緩衝液I(RNA Wash Buffer I)(500微升)加至管柱中,再置於工作台2分鐘。以10,000 x g之速離心60秒後,丟棄流經物,將500微升RNA清洗緩衝液II(RNA Wash Buffer II)加入,再以10,000 x g之速離心60秒。將另500微升RNA清洗緩衝液II(RNA Wash Buffer II)加入,再將管柱組裝體以10,000 x g之速離心60秒。將管柱以最大速離心2分鐘至完全將HiBind基質乾燥。將管柱置於乾淨之1.5毫升微離心管中,再將40-70微升分子生物學級水加入。靜置1分鐘後,將管柱以最大速離心2分鐘以洗提出RNA。將分離出之RNA收集至收集管中。
小鼠血液之總RNA之製備. 欲進行全血之RNA研
究,乃使用db/db小鼠(9星期大,The Jackson Laboratory,緬因州巴港)於實驗,且每個實驗群組之n=8隻小鼠。將小鼠於ZT0(早上7:00)(ZT意指授時時間(Zeitgeber Time),表示開啟光照以刺激小鼠設施中的白天之時間)投予化合物72(100毫克/公斤,口服;劑量體積5毫升/公斤,於10% Kolliphor中)、或10% Kolliphor每日一次地達三天。於最終日之ZT7.5(下午2:30),使用二氧化碳窒息法使動物安樂死,再經由心臟穿刺收集來自心臟的血液。將血液置於RNALater溶液中達1.5毫升總量。總RNA係使用Ambion小鼠RiboPure血液RNA分離套組AM1951(Ambion Mouse RiboPure Blood RNA Isolation Kit AM1951)如下地製備。將樣品離心3分鐘,丟棄上層液。將二毫升之溶解溶液加入,再渦動,轉移至15毫升管,其後將微升3M乙酸鈉加入。將溶解緩衝液加至3.8毫升總量,再渦動。將樣品混合物以1.5毫升酸酚(Acid Phenol):氯仿萃取,再將水性相復收。將0.5份體積之100%乙醇加入且渦動後,令樣品通過套組提供之過濾管柱,再以750微升清洗緩衝液1清洗。將濾器以2次通過之750微升清洗緩衝液2/3清洗,再乾燥。將RNA於200微升分子生物學級水(無RNA酶)中洗提出。
總RNA之定量. RediPlate 96 RiboGreen RNA定量套組(RediPlate 96 RiboGreen RNA Quantification Kit)(Invitrogen)RediPlate標準曲線係藉將20微升再構成之RNA標準劑轉移至已於180微升RediPlate TE緩衝
液中再構成之RediPlate孔中製得。有關由30-100毫克組織中使用類似於Omega Bio-Tek HP總RNA套組之套組製備且於無RNA酶之水中以50微升之量洗提出的肝RNA方面,乃將5微升的總RNA於195微升RediPlate TE緩衝液中稀釋(RNA之1:40稀釋)、混合。轉移5微升至已於195微升TE緩衝液中再構成之RediPlate孔且於室溫培育10分鐘後,於Tecan M200中以480nm之激發設定及520nm之放射設定以及增益設定至約70%以讀取標準劑及樣品之螢光強度。另外,可使用FlexStation 3以激發波長488nm、放射波長525nm且截止波長515nm進行。標準曲線於GraphPad Prism中產生且樣品之讀取(未知)經由線性回歸分析法內插,再計算出RNA樣品濃度。
cDNA之製備. 高性能cDNA反轉錄套組(High-Capacity cDNA Reverse Transcription Kit)(Invitrogen)將10X RT緩衝液(10X RT Buffer)(40-70微升)、dNTP及隨機引子於冰上解凍。每一樣品使用相同量之投入RNA(通常0.5-4.0微克),反應設定於40微升之總量。將適量之總RNA及無核酸酶之水(Nuclease-Free H2O)混合以得20微升之總量。主混合(master mix)係以4.2微升無RNA酶之水(RNAse-Free H2O)、2微升10X RT緩衝液(10X RT Buffer)、0.8微升25X dNTP、及2微升隨機引子(Random Primer)產生以用於每一反應(隨意地,可多加10%至全數待進行之反應中以確保足夠的量)。每一反應將反轉錄酶
(Reverse Transcriptase)(1微升)加入,再無需渦動地小心混合。一些一式二份之樣品(總數之約10%)經設計為RT(-)組,而足量之缺乏反轉錄酶(Reverse Transcriptase)的主混合則製備以包括這些對照組。將主混合(Master mix)(20微升)(或RT(-)主混合)加至20微升固定化投入之RNA樣品中。將反應於37℃培育2小時,加熱至85℃ 5分鐘,再置於冰上。如果隔天使用,將cDNA樣品貯存於4℃,或者貯存於-20℃更長之時間。
RT-PCR. 欲進行PCR分析,所用之套組為TaqMan® Fast Universal主混合(2x),無AmpErase® UNG(TaqMan® Fast Universal Master Mix(2x),No AmpErase® UNG)。欲於ABI 7500上進行反應,乃將2微升cDNA模板或RT(-)對照組模板置於每一孔中。主混合(master mix)乃製備以包括1.0微升TaqMan表現分析試劑盒(TaqMan Expression Assay)(引子/探子)及7微升無核酸酶之水(Nuclease-Free H2O)以供每一進行之樣品使用,再將18微升主混合(master mix)+表現分析試劑盒(Expression Assay)+無核酸酶之水(Nuclease-Free H2O)加至每一2微升樣品中,藉吸移法混合。將盤密封,旋轉下降,再裝載於ABI7500中。至少一種管家mRNA表現分析試劑盒(例如GAPDH;Mm03302249_g1或Hs02758991_g1)包括在待評估之每一組RNA樣品中。
Cry調節劑化合物72對核心時鐘功能之效應乃於糖尿病及非糖尿病小鼠中檢測。將17個星期大之雄
C57B1/6J DIO小鼠(The Jackson Laboratory,加州沙加緬度)保持高脂飲食(HFD),於治療前適應環境2星期以重現糖尿病及肥胖症。將三隻小鼠以化合物或載劑處理各5個時間點。將載劑(注射用水(WFI))或化合物72(50毫克/公斤於注射用水中)以5毫升/公斤之劑量體積BID(每日兩次)經由胃管經口餵食法投予小鼠。總計,每隻小鼠於整個實驗期間接受5次劑量之化合物。在研究開始之前,將小鼠稱重,再以晚上的體重為基準隨機分配至每一處理組。於第3天之下午3:00開始,使用二氧化碳窒息法使經載劑或化合物72處理之動物群組安樂死,分別切出50毫克肝、副睪白脂肪及骨骼肌以用於RNA之製備。
化合物72對日夜mRNA之效應亦於正常Balb/c小鼠中、於24小時期間檢測。小鼠為8個星期大,得自Charles River,且令其適應環境2個星期。將鼠投予BID達3天,總計每隻動物投予7次劑量,最後一次劑量在取得組織之前12小時。將載劑(注射用水(WFI))或化合物72(50毫克/公斤於注射用水中)以5毫升/公斤之劑量體積BID(每日兩次)經由胃管經口餵食法投予小鼠。第3天之下午3:00開始,將載劑動物群組及或化合物72群組宰殺,切出約50毫克肝、肺、腎、腎上腺、脾臟、副睪脂肪、及棕色脂肪組織,再置於RNALater中。
經載劑處理之C57B1/6J DIO小鼠之核心時鐘mRNA顯現特有之日夜表現樣式(圖1)。然而化合物72處理組之Per2 mRNA在C57B1/6J DIO及Balb/c小鼠中、在24小時
期間均受到抑制,且這些在ZT8時顯現最大降低,且在24小時後的ZT8再度顯現最大降低(圖1 A & B)。兩種品種之小鼠之Bmal1 mRNA於最初ZT0之後於ZT8及32小時均藉由化合物72之處理而實質地提高。Bmal1轉錄本於C57B1/6J DIO小鼠亦顯現顯著之相位延遲,而於Balb/c小鼠顯現較小程度(圖1C & D)。Cry1之mRNA在其表現峰期間、在黑暗期間受到抑制(圖1E & F,陰影區)。於DIO小鼠所觀察到之經載劑處理之Cry1轉錄本相對於經載劑處理之Balb/c小鼠者之相位超前可能一部分係因為高脂飲食對許多一日間樣式之已知效應所致(Eckel-Mahan et al.(2013)Cell)。與Cry1 mRNA對照之下,Cry2 mRNA於經載劑處理之小鼠中係於白天的小時中達到高峰,但此現象在經化合物72處理之C57B1/6J DIO及Balb/c小鼠中均強烈地減弱(圖1G & H)。
於與上述相同之24小時研究中,相對於載劑,C57B1/6J DIO小鼠之糖質新生基因Pck1(PEPCK)及G6Pc(葡萄糖6-磷酸酶催化單位)之mRNA的日夜樣式被化合物72實質地改變(圖2A & C)。於其他研究中可觀察到經載劑處理之小鼠之比正常餵養之C57B1/6J小鼠者顯現平坦化及相位超前的樣式(Hughes et al.(2009))。經化合物72處理之C57B1/6J DIO小鼠之這兩種糖質新生基因於傍晚(ZT14)均顯現表現峰,其較靠近於彼等於正常餵養之小鼠中所觀察到之表現高峰時間(Hughes et al.(2009))。Pck1及G6Pc之一日間表現樣式於C57B1/6J DIO小鼠中藉化合
物72相對於載劑而改變的程度比於Balb/c小鼠中者為大(圖2B & D)。
檢測多重Cry調節劑化合物,化合物72、化合物48、化合物9及化合物57於ICR小鼠的效應。將小鼠以50毫克/公斤之每一種化合物(化合物72、化合物48、化合物9、及化合物57)PO(經口)以5毫升/公斤之劑量體積BID或載劑對照組投予4天。每一種化合物均導致肝Per2表現之抑制(圖3A)。化合物48處理導致Bmal1 mRNA於ZT6呈8倍提高,經化合物72及化合物9處理之小鼠顯現4倍提高,而經化合物57處理之小鼠顯現至少2倍提高(圖3B)。化合物72、化合物48及化合物9亦導致Cry2轉錄本的降低(圖3C)。
核心時鐘基因mRNA於全血中的表現量可提供測定化合物於經處理個體中之效應的非侵入性方法。使用雄db/db小鼠(9星期大,The Jackson Laboratory,緬因州巴港)且每一實驗群組之n=8。將小鼠於ZT0(早上7:00)(ZT意指授時時間(Zeitgeber Time),表示開啟光照以刺激小鼠設施中的白天之時間)投予化合物72(100毫克/公斤,口服;劑量體積5毫升/公斤,於10% Kolliphor中)、或10% Kolliphor每日一次地達三天。於最終日之ZT7.5(下午2:30),使用二氧化碳窒息法使動物安樂死,再經由心臟穿刺收集來自心臟的血液。將全血轉移至含RNALater之管以供藉RT-qPCR分析。
D-box結合蛋白Dbp係以強烈日夜方式受到調控。化
合物72於此研究中可導致Dbp基因表現統計上顯著的抑制(圖4),證實經此化合物處理之小鼠的白血細胞可提供有關化合物對整個生物體中之核心時鐘機轉之效應的訊息。此訊息可用作為診斷標誌,或作為評估Cry調節劑及其他衝擊核心日夜機轉之治療劑的效應的生物標誌。
對直接與核心時鐘機轉直接交互作用的化合物而言,投予時間可能對使其效應最大化是緊要的。將Db/db小鼠以單一劑量之化合物72(50毫克/公斤)於ZT0或ZT10給予。前者(ZT0)與小鼠肝中之Cry1及Bmal1蛋白的高峰一致,而後者(ZT10)相當於Cry1及Bmal1之約略最底點。於7.5小時後取出每一者之肝組織,再藉RT-qPCR檢測樣品之核心時鐘基因mRNA。與最底點相比,既然化合物72於高峰時對時鐘mRNA具有較大之相對衝擊(圖5),故在約前者之時間投藥可提供對時鐘機轉之較大效應,且此可進一步導致對時鐘之代謝輸出的較大效應。
於第II型糖尿病db/db小鼠模型中,評估當將化合物72以單一劑量型式於核心時鐘基因Cryl/Bmal1表現之高峰或最低點時刻投予時對葡萄糖代謝之效應。
Leprdb為同合子之雄db/db小鼠(6星期大),得自The Jackson Laboratory(緬因州巴港)。令小鼠群居於正常之亮/暗循環(開啟光照:07:00-19:00小時)且隨意地接近標準
九粒狀小鼠飲食及水。實驗前使動物習慣於這些條件2星期。將小鼠分成兩個研究臂,於Cry1及Bmal1基因表現之高峰或最底點時刻給藥。將小鼠於ZT0(高峰,早上7:00)或ZT10(最底點,下午5:00)投予載劑(10% Kolliphor,Sigma-Aldrich)或化合物72(50毫克/公斤於10% Kolliphor之水液中)以5毫升/公斤之劑量體積每日一次(QD)地經由胃管經口餵食法投予一次。將動物於第0天稱重,再隨意地分配成處理群組使得每一群組具有類似的平均起始重量。當將小鼠轉移至乾淨籠中且自由給予水但無食物12小時時,令於ZT0投藥之小鼠於下午10:30開始禁食過夜。於ZT10投藥之小鼠以相同方式於早上8:30開始禁食。於研究當天,投藥後,在量測禁食血液葡萄糖之前,小鼠先接受切尾創傷2小時,以令其由步驟可能導致之任何應力中恢復。於早上10:30或下午8:30使用AlphaTRAK血糖儀(Abbott Laboratories,美國)估測動物之禁食血液葡萄糖(FBG)。於早上11:30(高峰時刻給藥之小鼠)或下午9:30(最低點時刻給藥之小鼠),每隻動物投予0.5克/公斤葡萄糖,然後於葡萄糖負載之後之t=15、30、60、90及120分鐘量測血液葡萄糖。於最後一次血液收集及採收組織及血液以供其他端點測定後,將動物處死。
將OGTT期間取得之禁食血液葡萄糖值及葡萄糖量測平均,再繪圖(GraphPad Prism,GraphPad軟體,加州拉霍雅)。計算出每一個別動物之曲線下面積(AUC)。統計
分析係使用單因子變異數分析(one-way ANOVA)進行,其後進行適當事後檢定(post-test)。當p<0.05時,則顯著性被接受。數據以平均值及S.E.M呈現。
相比於載劑對照組,將化合物72(50毫克/公斤,口服)以單一劑量於Cry1及Bmal1基因表現之高峰時刻投予db/db小鼠可導致對OGTT量測之明顯效應(圖6A),但當於最底點時刻投予時則無效應(圖6B)。由在基因表現高峰時刻投藥之動物之OGTT所計算出之AUC顯示,化合物72之處理可導致葡萄糖的波動降低14%(74098 +/- 4194對63842 +/- 4318;圖7)。
當將化合物72以單一劑量形式於7天期間投予第II型糖尿病db/db小鼠模型時,化合物72對葡萄糖代謝及胰島素濃度之效應。
Leprdb為同合子之雄db/db小鼠(6星期大),得自The Jackson Laboratory(緬因州巴港)。令小鼠群居於正常之亮/暗循環(開啟光照:07:00-19:00小時)且隨意地接近標準丸粒狀小鼠飲食及水。實驗前使動物習慣於這些條件2星期。將小鼠於ZT0(早上7:00)投予載劑(10% Kolliphor,Sigma-Aldrich)或化合物72(50毫克/公斤於10% Kolliphor之水液中)以5毫升/公斤之劑量體積每日一次(QD)地經由胃管經口餵食法投予七天。將動物於第0天稱
重,再隨意地分配至任一處理群組使得每一群組具有類似的平均起始重量。於端點量測前的晚上10:30,將小鼠置於乾淨籠中且自由給予水但無食物12小時,其後進行禁食血液葡萄糖之量測。於研究最後一天,將動物如常地投藥,然後在量測禁食血液葡萄糖之前接受切尾創傷2小時,以令其由步驟可能導致之任何應力中恢復。於早上10:30使用AlphaTRAK血糖儀(Abbott Laboratories,美國)估測動物之禁食血液葡萄糖(FBG)。FBG量測後,使用尾部壓擠技術將每隻小鼠之血液收集至毛細管中。將毛細管於血球離心機(BD Triac 0200)中離心,再將所得血漿轉移至eppendorff小離心管。將此樣品(標記為t=0小時)於-80℃冷凍以令稍後之胰島素量測。於早上11:30,每隻動物投予0.5克/公斤葡萄糖,然後於葡萄糖負載之後之t=15、30、60、90及120分鐘量測血液葡萄糖。在OGTT結束時,收集血液以進行t=2小時之如上所述之胰島素測定。於最後一次血液收集及採收組織及血液以供其他端點測定(其他處詳述)後,將動物處死。將經化合物72處理之動物的血漿及肝組織提交委外研究機構(CRO)以使用LC/MS/MS且與已知化合物量之標準曲線相比較以量測化合物濃度。
將OGTT期間取得之禁食血液葡萄糖值及葡萄糖量測平均,再繪圖(GraphPad Prism,GraphPad軟體,加州拉霍雅)。計算出每一個別動物之曲線下面積(AUC)。血漿胰島素濃度係使用超敏感性胰島素酵素連結性免疫吸附分析
法(Ultrasensitive Insulin ELISA(ALPCO,新罕布夏州沙連))測定。HOMA-IR(體內平衡模型評估-胰島素抗性)係使用下列公式計算出:(FPI(微單位/升) x FPG(毫莫耳/升))/22.5,其中FPI及FPG分別代表禁食血漿胰島素及禁食血漿葡萄糖。胰島素數據亦以GraphPad Prism格式表示。統計分析係使用單因子變異數分析(one-way ANOVA)進行,其後進行適當事後檢定(post-test)。當p<0.05時,則顯著性被接受。數據以平均值及S.E.M呈現。
與載劑對照組相比,將化合物72(50毫克/公斤,口服)投予db/db小鼠7天導致FBG顯著降低(484.9 +/- 34.37毫克/分升對385.0 +/- 29.69毫克/分升;圖8A)。OGTT量測過程期間,經化合物72處理之動物低於載劑對照群組(圖8B)。由OGTT計算出之AUC顯示化合物72之投予導致萄糖波動之顯著降低(54845 +/- 4112對35942 +/- 3192;圖8C)。
血漿胰島素之量測由採自t=0及t=2小時之樣品中獲得且示於圖9A中。胰島素藉以化合物72處理於t=0及t=2小時之時點分別降低20%及21%(分別為4.70 +/- 0.76對3.78 +/- 0.69奈克/毫升及3.17 +/- 0.67對2.53 +/- 0.50奈克/毫升)。HOMA-IR(胰島素再敏化之指標)降低33%,由139.91 +/- 26.57至93.69 +/- 23.60單位(圖9B)。
化合物72之化合物濃度係於研究終止時採取之樣品中估測且示於圖10中,圖10亦顯示由如同實例3所述之
Per2分析法測得之EC50值以供比較之目的。化合物72於投予最後一次劑量後之約8小時發現存在於血漿亦存在於肝中(血漿:0.53 +/- 0.03μM;肝:0.67 +/- 0.05μM,以平均值及S.E.M表示)。兩個案例中,化合物濃度均略高於化合物72測出之Per2 EC50值(0.4μM;分別高於血漿及肝中之EC50值的1.3倍及1.7倍)。
於第II型糖尿病db/db小鼠模型中評估以漸增劑量投予化合物72達7天對葡萄糖代謝及胰島素濃度之效應。
Leprdb為同合子之雄db/db小鼠(5星期大),得自The Jackson Laboratory(緬因州巴港)。令小鼠群居於正常之亮/暗循環(開啟光照:07:00-19:00小時)且隨意地接近標準九粒狀小鼠飲食及水。實驗前使動物習慣於這些條件2星期。將小鼠於ZT0(早上7:00)投予載劑(10% Kolliphor,Sigma-Aldrich)或10、50或100毫克/公斤之化合物72(於10% Kolliphor之水液中)以5毫升/公斤之劑量體積每日一次(QD)地經由胃管經口餵食法投予七天。進行之實驗方法與實例11詳述者相同。經化合物72處理之動物的血漿及肝組織中之化合物濃度係使用LC/MS/MS量測且與已知化合物量之標準曲線相比較。
與載劑對照組相比,將化合物72以上升劑量投予db/db小鼠7天導致於50及100毫克/公斤時之禁食血液
葡萄糖濃度的降低,然而其並未達到統計顯著性(載劑對照組:491.0 +/- 51.30毫克/分升,50毫克/公斤:402.8 +/- 25.25毫克/分升,100毫克/公斤:420.7 +/- 26.44毫克/分升;圖11A)。以10毫克/公斤劑量投予之化合物72證實禁食血液葡萄糖濃度未降低(503.5 +/- 49.68毫克/分升)。OGTT量測過程期間,經化合物72處理之動物證實對葡萄糖負荷後之動物的葡萄糖波動產生劑量依賴性效應(圖11B)。由OGTT計算出之曲線下面積顯示,化合物72之投予可以劑量依賴方式降低葡萄糖AUC,其於100毫克/公斤具顯著性(載劑:46088 +/- 3303,10毫克/公斤:39771 +/- 4244,50毫克/公斤:35527 +/- 3215,100毫克/公斤:28499 +/- 3079;圖11C)。
血漿胰島素之量測由採自t=0及t=2小時之樣品中獲得且示於圖12A中。當以100毫克/公斤投予時,藉以化合物72處理之胰島素於t=0時降低(由5.68 +/- 0.43至4.63 +/- 0.17奈克/毫升(分別為載劑及100毫克/公斤群組))。HOMA-IR於化合物72投予後,以劑量依賴性方式降低(載劑:169.3 +/- 18.41,10毫克/公斤:172.7 +/- 16.61,50毫克/公斤:149.2 +/- 16.49,100毫克/公斤:111.9 +/- 9.02單位;圖12B)。化合物72於100毫克/公斤之效應為顯著的(與載劑對照群組相比降低34%)。
化合物72之化合物濃度係於研究終止時採取之樣品中估測且示於圖13中,圖13亦顯示Per2 EC50值(如同實例3所述)以供比較之目的。化合物72於投予最後一次劑
量後之約8小時發現存在於血漿亦存在於肝中,且暴露濃度相對於投予劑量之提高而提高(血漿,10毫克/公斤:0.09 +/- 0.01μM;50毫克/公斤:0.57 +/- 0.03μM;100毫克/公斤:1.22 +/- 0.17μM;肝,10毫克/公斤:0.12 +/- 0.01μM;50毫克/公斤:0.78 +/- 0.06μM;100毫克/公斤:1.81 +/- 0.22μM)。血漿及肝之暴露濃度於50毫克/公斤時分別高於血漿及肝中之Per2 EC50值的1.4倍及1.95倍,於100毫克/公斤時則分別高3倍及4.5倍。
於第II型糖尿病db/db小鼠模型中評估以漸增劑量投予化合物9達7天對葡萄糖代謝及胰島素濃度之效應。
Leprdb為同合子之雄db/db小鼠(5星期大),得自The Jackson Laboratory(緬因州巴港)。令小鼠群居於正常之亮/暗循環(開啟光照:07:00-19:00小時)且隨意地接近標準九粒狀小鼠飲食及水。實驗前使動物習慣於這些條件2星期。將小鼠於ZT0(早上7:00)投予載劑(10% Kolliphor,Sigma-Aldrich)或30、100或300毫克/公斤之化合物9、或30毫克/公斤之羅格列酮(Rosiglitazone)(於10% Kolliphor之水液中)以5毫升/公斤之劑量體積每日一次(QD)地經由胃管經口餵食法投予七天。羅格列酮(Rosiglitazone)為用於陽性對照組之抗糖尿病治療劑。進行之實驗方法與實例11詳述者相同。經化合物9處理之動物的血漿及肝組織中之化合物濃度係使用LC/MS/MS量
測且與已知化合物量之標準曲線相比較。
與載劑對照群組相比,將化合物9以上升劑量投予db/db小鼠7天導致100毫克/公斤時之禁食血液葡萄糖濃度的降低(由492.8 +/- 48.07至403.1 +/- 39.73毫克/分升;圖14A),但整體無統計的顯著效應。以30及100毫克/公斤劑量投予之化合物9證實OGTT期間葡萄糖濃度的量測為劑量依賴性降低,而於300毫克/公斤之最高測試劑量未觀察到提高的效應(圖14B)。由OGTT計算出之曲線下面積顯示,化合物9之投予可以劑量依賴方式降低葡萄糖AUC,其於100及300毫克/公斤均具顯著性(載劑:56046 +/- 3204,30毫克/公斤:44442 +/- 3895,100毫克/公斤:33643 +/- 4822,300毫克/公斤:33650 +/- 4688;圖8C)。比起載劑對照群組,葡萄糖AUC於30、100及300毫克/公斤分別降低21%、40%及40%。用於作為動物模型陽性對照組之羅格列酮(Rosiglitazone)可顯著地抑制禁食血液葡萄糖(由492.8 +/- 48.07至280.4 +/- 13.66毫克/分升;圖14A)、及葡萄糖AUC(由56046 +/- 3204至11502 +/- 2118單位;圖14C)。
血漿胰島素之量測由採自t=0及t=2小時之樣品中獲得且示於圖15A中。藉以化合物9處理後之胰島素於t=0(載劑:14.89 +/- 2.93,30毫克/公斤:10.94 +/- 1.62,100毫克/公斤:7.71 +/- 1.26,300毫克/公斤:10.54 +/- 1.6奈克/毫升)及t=2小時(載劑:7.44 +/- 0.92,30毫克/公斤:5.76 +/- 0.11,100毫克/公斤:3.70
+/- 0.29,300毫克/公斤:4.01 +/- 0.44奈克/毫升)均降低。HOMA-IR於投予化合物9之後以劑量依賴性方式降低,然而於300毫克/公斤之化合物對此端點之效應較低(載劑:438.8 +/- 87.88、30毫克/公斤:289.9 +/- 24.40,100毫克/公斤:175.3 +/- 27.52,300毫克/公斤:301.4 +/- 52.66單位;圖15B)。於100毫克/公斤,化合物9之效應具顯著性(與載劑對照組相比降低60%)。羅格列酮(Rosiglitazone)於t=0及t=2小時均降低胰島素濃度(分別至3.05 +/- 0.14及2.28 +/- 0.08奈克/毫升;圖15A),以及顯著地降低HOMA-IR(至50.58 +/- 3.52單位,圖15B)。
化合物9之組織濃度係於研究終止時採取之肝樣品中估測且示於圖16中,圖16亦顯示Per2 EC50值以供比較之目的。化合物9於投予最後一次劑量後之約8小時發現存在於血漿肝中,且投予30毫克/公斤至100毫克/公斤間的劑量後之暴露濃度乃相對於劑量之提高而提高。於300毫克/公斤之暴露濃度顯示藥物之累積(7.1倍提高而非預期的3倍)。投予30、100或300毫克/公斤化合物9之動物肝樣品中之化合物濃度分別為0.19 +/- 0.02μM、0.67 +/- 0.05μM及4.77 +/- 1.06μM。投予30毫克/公斤化合物9後之肝暴露濃度低於Per2 EC50 value(0.3μM)值之約1.6倍,而於100毫克/公斤及300毫克/公斤則高2.2倍及15.9倍。
檢測化合物72於第II型糖尿病之飲食誘導性肥胖症(DIO)小鼠模型中的效應。
雄C57/B16J DIO小鼠得自The Jackson Laboratory(加州沙加緬度)。令小鼠群居於正常之亮/暗循環(開啟光照:07:00-19:00小時)且隨意地接近高脂飲食(D12492(60千卡%脂肪),Research Diets,Inc.)及水。實驗前使動物習慣於這些條件至少2星期且於約24星期大時使用。將小鼠於ZT0(早上7:00)投予載劑(10% Kolliphor,Sigma-Aldrich)、化合物72(100毫克/公斤於10% Kolliphor之水液中)或羅格列酮(Rosiglitazone)(30毫克/公斤於10% Kolliphor之水液中)以5毫升/公斤之劑量體積每日一次(QD)地經由胃管經口餵食法投予七天。羅格列酮(Rosiglitazone)為用於陽性對照組之抗糖尿病治療劑。將小鼠於第0天稱重,再隨意地分配至任一處理群組使得每一群組具有類似的平均起始重量。於端點量測前的晚上10:30,將小鼠置於乾淨籠中且自由給予水但無食物12小時,其後進行禁食血液葡萄糖之量測。於研究最後一天,將動物如常地投藥,然後在量測禁食血液葡萄糖之前接受切尾創傷2小時,以令其由步驟可能導致之任何應力中恢復。於早上10:30使用AlphaTRAK血糖儀(Abbott Laboratories,美國)估測動物之禁食血液葡萄糖(FBG)。FBG量測後,使用尾部壓擠技術將每隻小鼠之血液收集至
毛細管中。將毛細管於血球離心機(BD Triac 0200)中離心,再將所得血漿轉移至eppendorff小離心管。將此樣品(標記為t=0小時)於-80℃冷凍以令稍後之胰島素量測。於早上11:30,每隻動物投予1.5克/公斤葡萄糖,然後於葡萄糖負載之後之t=15、30、60、90及120分鐘量測血液葡萄糖。在OGTT結束時,收集血液以進行t=2小時之如上所述之胰島素測定。於最後一次血液收集及採收組織及血液以供其他端點測定後,將動物處死。
將OGTT期間取得之禁食血液葡萄糖值及葡萄糖量測平均,再繪圖(GraphPad Prism,GraphPad軟體,加州拉霍雅)。計算出每一個別動物之曲線下面積(AUC)。血漿胰島素濃度係使用超敏感性胰島素酵素連結性免疫吸附分析法(Ultrasensitive Insulin ELISA(ALPCO,新罕布夏州沙連))測定。HOMA-IR(體內平衡模型評估-胰島素抗性)係使用下列公式計算出:(FPI(微單位/升) x FPG(毫莫耳/升))/22.5,其中FPI及FPG分別代表禁食血漿胰島素及禁食血漿葡萄糖。胰島素數據亦以GraphPad Prism格式表示。統計分析係使用單因子變異數分析(one-way ANOVA)進行,其後進行適當事後檢定(post-test)。當p<0.05時,則顯著性被接受。數據以平均值及S.E.M呈現。
與載劑對照組相比,將化合物72(100毫克/公斤,口服)投予C57/B16J DIO小鼠7天導致禁食血液葡萄糖濃度顯著降低(237.2 +/- 15.29毫克/分升對177.1 +/- 8.28毫克/分升;圖17A)。OGTT量測過程期間,經化合物72
處理之動物遠低於載劑對照群組(圖17B)。由OGTT計算出之AUC顯示化合物72之投予導致萄糖波動之顯著降低(31511 +/- 1670對17055 +/- 769.1;圖17C)。用於作為動物模型陽性對照組之羅格列酮(Rosiglitazone)可降低禁食血液葡萄糖至153.9 +/- 5.05毫克/分升及葡萄糖AUC至11500 +/- 1104單位。
血漿胰島素之量測由採自t=0及t=2小時之樣品中獲得且示於圖18A中。藉以化合物72處理後之胰島素於t=0(載劑:5.00 +/- 0.92,100毫克/公斤:3.12 +/- 0.24,奈克/毫升)及t=2小時(載劑:4.82 +/- 0.60,100毫克/公斤:2.88 +/- 0.21奈克/毫升)均降低。HOMA-IR於投予化合物72之後顯著降低(載劑:70.76 +/- 11.30,100毫克/公斤:32.54 +/-3.37單位;圖18B)。羅格列酮(Rosiglitazone)(30毫克/公斤)於t=0及t=2小時可降低胰島素(分別至2.70 +/-0.12及2.30 +/- 0.06奈克/毫升)及顯著地降低HOMA-IR(至24.60 +/- 1.42單位)。
重覆將皮質酮投予大鼠6天可誘導與血漿胰島素及葡萄糖之顯著提高有關之體重的顯著降低。這些效應經由11-βHSD1活性所產生之皮質醇所媒介。葡萄糖皮質素受體拮抗劑諸如美服培酮(mifepristone)可改善皮質醇對胰島素抗性之效應。這些實驗的目標係用於測定化合物72對
大鼠之皮質酮誘導性胰島素抗性之發展的效應。
將皮質酮(30毫克/公斤,皮下(sc),每日一次(qd))與測試化合物組合地投予動物6天,且於最後一次皮質酮劑量後之27小時將動物處死。參考標準劑美服培酮(Mifepristone)亦包括。21-乙酸皮質酮(Sigma C-3130)由RenaSci供應且使用5毫克/公斤之劑量體積經由皮下路徑以於1%甲基纖維素中之微細懸浮液形式投予。化合物72(50毫克/公斤於10% Kolliphor之水液中)每日一次地(QD)使用5毫克/公斤之劑量體積經由胃管經口餵食法投予。美服培酮(Mifepristone(Sigma M8046))由RenaSci提供。
葡萄糖及胰島素之測定係於得自尾靜脈血之血漿樣品進行,該血漿樣品於禁食12小時後,於最後一次皮質酮劑量後之約27小時取得。然後將動物處死,再取出終端(心臟)血液以從中製得血漿。
三十四隻Sprague Dawley大鼠(重量範圍200-250克)訂購自Charles River,Margate,Kent,英國。令大鼠群居於正常之亮/暗循環(開啟光照:07:00-19:00小時)且隨意地接近標準九粒狀大鼠飲食及自來水。實驗前使動物習慣於這些條件2星期。接著,動物接受3日之基線期,此期間彼等以載劑於t=0小時(07:00)每日投予一次。此步驟經發現可降低研究中之應力相關性效應的發生率。所有藥物均如同以下表2所示地投予6天。體重於07:00(t=0小時)開始投藥之前立即記錄。皮質酮經由皮下路徑(sc)投予,
而化合物72及美服培酮(mifepristone)經由胃管經口餵食法於t=0小時之皮質酮投予之後立即投予。

投藥之第6天,將大鼠於22:30開始禁食12小時(安排在第7天終止之際)。於第7天07:00將7隻大鼠如常地投予載劑但無皮質酮(皮下(sc)),其後經口投予載劑/化合物72/美服培酮(mifepristone)。於第7天10:30,最後一次皮質酮劑量後之27小時,採取側尾靜脈之血液樣品(300微升)至含EDTA之管(Sarstedt 16.444)中。將血液離心,再將所得血漿整除份貯存於-75℃。藉二氧化碳窒息法將動物安樂死,其後將頸部脫位。藉心臟穿刺法將終端血液(約10毫升)收集至含EDTA之管(Sarstedt 5毫升32.332)中,然後離心,再將血漿貯存於-75℃。使用市售之臨床試劑(Thermoelectron Infinity葡萄糖試劑(TR15421))分析尾靜脈血漿之葡萄糖及使用Mercodia超敏感大鼠胰島素大鼠酵素連結性免疫吸附分析法(ultrasensitive rat insulin rat ELISA)(10-1251-10)分析胰島素(n=1)。
血漿葡萄糖及胰島素藉由穩健回歸法或一般之線性模型以處理作為因子及以放血次序及基線體重作為共變量而分析。如適當,亦使用對數變換。使用多重比較檢定(雙尾)以測定載劑群組與皮質酮群組間的顯著差異。P<0.05被視為具統計顯著性。
化合物72之投予大鼠顯著地降低因皮質酮之投予處理而導致之血漿葡萄糖及胰島素提高。血漿葡萄糖濃度在以皮質酮處理後由6.28 +/- 0.30Mm提高至10.17 +/- 0.51mM,此可藉由化合物72(50毫克/公斤)而顯著地降低至8.55 +/- 0.3mM(p<0.01;平均值及S.E.M)。血漿胰島素濃度於皮質酮處理後由0.70 +/- 0.11奈克/毫升增至8.19 +/- 0.91奈克/毫升,其藉由化合物72(50毫克/公斤)而降至5.24 +/- 1.11奈克/毫升(p<0.05;數據以平均值及S.E.M表示;圖19)。用於作為動物模型陽性對照組之美服培酮(Mifepristone)顯著地降低血漿葡萄糖及血漿胰島素分別至7.43 +/- 0.27奈克/毫升及3.62 +/- 0.29奈克/毫升。
HOMA-IR值乃如同化合物9中所述地計算,且數據示於圖20中。與載劑:載劑對照群組(5.27 +/- 1.04單位)相比,皮質酮處理提高HOMA-IR至95.57 +/- 11.4單位。化合72(50毫克/公斤)及美服培酮(mifepristone)之投予顯著降低胰島素抗性大鼠之HOMA-IR值分別至56.94 +/- 11.18單位及29.99 +/- 2.54單位。
使用雄ICR小鼠(重量30-40克,Charles River Laboratories)於實驗且每個實驗群組之n=3隻小鼠(總共27隻小鼠用於研究)。將小鼠投予Cry調節劑化合物9或化合物72(50毫克/公斤,口服(P.O);劑量體積5毫升/公斤,於10% Kolliphor中)。投服後於下列之時間點收集血液及肝組織:15、30、60、90分鐘,3、6、12及24小時。對照群組之動物(T0)亦予採樣。使用二氧化碳將動物安樂死,再使用心臟穿刺法收集來自心臟之血液,轉移至EDTA管,然後以5400rpm之速於4℃離心5分鐘。將所得血漿使用乾冰冷凍,然後則存於-80℃直至準備好用於分析為止。將肝組織由每隻動物中移出,收集0.5克至eppendorff小離心管,冷凍再接受藥物動力學量測。將每隻動物之血漿及肝組織提交委外研究機構(CRO)以使用LC/MS/MS且與血漿及肝中已知化合物量之標準曲線相比較以量測化合物濃度。原始數據使用用於PK參數的WinNonLin(Cmax、Tmax、消除半衰期(elimination t1/2)、MRT(平均滯留時間)、AUC(曲線下面積)-(0-最後及外推%)分析。
使用雄SD大鼠(重量250-300克,Charles River Laboratories)於實驗且每個實驗群組之n=4隻大鼠。將大鼠投予化合物72(50毫克/公斤,口服(P.O);劑量體積5毫升/公斤,於10% Kolliphor中)。投予後於下列之時間點收集血液:15、30、60、90分鐘,3、6、12及24小
時。亦收集投藥前之樣品。在重置(Reset)之遞送前,由Charles River之技術人員將動物插管。於每一時點由右總頸靜脈之套管收集全血(0.3毫升)。將全血轉移至EDTA管,然後以5400rpm之速於4℃離心5分鐘。將所得血漿使用乾冰冷凍,然後則存於-80℃直至準備好用於分析為止。於每次抽血後將0.9%氯化鈉(0.3毫升)投予以供流體之補充。12小時時點後,使用0.1毫升肝素鈉(500IU/毫升)作為封管溶液。樣品如上述地分析。表3及4總結分析之結果。


估測化合物72隨著劑量之提高於第II型糖尿病之飲食誘導性肥胖症(DIO)小鼠模型中的效應。
雄C57/B16J DIO小鼠得自The Jackson Laboratory(加州沙加緬度)。令小鼠群居於正常之亮/暗循環(開啟光照:07:00-19:00小時)且隨意地接近高脂飲食(D12492(60千卡%脂肪),Research Diets,Inc.)及水。實驗前使動物習慣於這些條件至少2星期且於約26星期大時使用。將小鼠於ZT0(早上7:00)投予載劑(10% Kolliphor,Sigma-Aldrich)、化合物72(10、30或100毫克/公斤於10% Kolliphor之水液中)或羅格列酮(Rosiglitazone)(30毫克/公斤於10% Kolliphor之水液中)以5毫升/公斤之劑量體積每日一次(QD)地經由胃管經口餵食法投予七天。進行
之實驗方法與實例10詳述者相同。
與載劑對照組相比,將化合物72以上升劑量投予C57/B16J DIO小鼠7天導致禁食血液葡萄糖濃度的降低,其於100毫克/公斤達到顯著性(載劑對照組:226.9 +/- 13.11毫克/分升,10毫克/公斤:206.8 +/- 8.36毫克/分升,30毫克/公斤:197.5 +/- 12.06毫克/分升,100毫克/公斤:176.3 +/- 7.83毫克/分升,圖22A)。OGTT量測過程期間,經化合物72處理之動物證實葡萄糖負荷後之葡萄糖波動降低(圖22B)。由OGTT計算出之曲線下面積顯示,化合物72之投予可降低葡萄糖AUC,於30及100毫克/公斤證實具有顯著性(載劑:26090 +/- 1917,10毫克/公斤:22563 +/- 1224,30毫克/公斤:19033 +/- 1934,100毫克/公斤:19502 +/- 2404單位;圖22C)。用於作為動物模型陽性對照組之羅格列酮(Rosiglitazone)可顯著地抑制禁食血液葡萄糖(由226.9 +/- 13.11至161.1 +/- 8.06毫克/分升;圖22A)、及葡萄糖AUC(由26090 +/- 1917至9858 +/- 1281單位;圖22C)。

Claims (44)
- 一種式I化合物,

- 根據申請專利範圍第1項之化合物,其中A、D、E、G、J、L、M、及Q各自為碳;R1及R2各自獨立地選自氫或鹵基;R4為氫或(C1-C6)烷基;R3及R5為氫;且R6及R7彼此鍵聯成吡咯啶酮環。
- 根據申請專利範圍第1項之化合物,其中A、D、E、G、J、L、M、及Q各自為碳;R1及R2各自獨立地選自氫或鹵基;R4為氫或(C1-C6)烷基;R3及R5為氫;且R6及R7彼此鍵聯成咪唑啶酮環。
- 根據申請專利範圍第1項之化合物,其中A、D、E、G、J、L、M、及Q各自為碳;R1及R2各自獨立地選自氫或鹵基;R4為氫或(C1-C6)烷基;R3及R5為氫;且R6及R7彼此鍵聯成7員單環或雙環。
- 根據申請專利範圍第1項之化合物,其中A、D、E、G、J、L、M、及Q各自為碳;R1及R2各自獨立地選自氫或鹵基;R4為氫或(C1-C6)烷基;R3及R5為氫;且R6及R7彼此鍵聯成10員單環或雙環。
- 根據申請專利範圍第1項之化合物,其選自由以下所組成之群組:1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮; 2-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;(1R,4S)-2-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;(R)-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;(R)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;(S)-1-((S)-3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-4-甲基咪唑啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3,3-二甲基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-乙基吡咯啶-2-酮; 3-環戊基-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-異丙基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-甲基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-甲基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4,4-二甲基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-5-乙基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-5-乙基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-5-甲基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-5-甲基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4-異丙基吡咯啶-2-酮;4-環丙基-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4-甲基吡咯啶-2-酮; 1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-4-甲基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4-乙基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-4-乙基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4,5-二甲基吡咯啶-2-酮;3-環丁基-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-苯基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-苯基吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3,3-二氟吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3,3-二氟吡咯啶-2-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)-4-環丙基吡咯啶-2-酮;1-(3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)吡咯啶-2-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)吡咯啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-乙基咪唑 啶-2-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4-甲基咪唑啶-2-酮;1-環己基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-苯基咪唑啶-2-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-異丙基咪唑啶-2-酮;1-環戊基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;1-環丙基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;1-環丁基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-環丁基咪唑啶-2-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-環丙基咪唑啶-2-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-異丙基咪唑啶-2-酮;3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3,4-二氫喹唑啉-2(1H)-酮; 3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3,4-二氫喹唑啉-2(1H)-酮;(1S,4R)-2-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;(1S,4R)-2-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;(1R,4S)-2-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;(1R,4S)-2-((S)-3-(9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;(R)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-5-甲基吡咯啶-2-酮;(S)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-5-甲基吡咯啶-2-酮;(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-5-甲基吡咯啶-2-酮;(S)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-5-甲基吡咯啶-2-酮;(R)-1-((S)-3-(9H-咔唑-9-基)-2-羥丙基)-5-甲基吡咯啶-2-酮;(S)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-甲基吡咯啶-2-酮;(R)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-甲基吡咯啶-2-酮; (S)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-3-甲基吡咯啶-2-酮;(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-3-甲基吡咯啶-2-酮;(S)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;(S)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;(R)-1-((S)-3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;(S)-1-((S)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;(R)-1-((S)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;(S)-1-((R)-3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;(S)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4-甲基咪唑啶-2-酮;(R)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4-甲基咪唑啶-2-酮;及 (S)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-4-甲基咪唑啶-2-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第6項之化合物,其為1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第6項之化合物,其為2-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氫雜雙環[2.2.1]庚-3-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第6項之化合物,其為1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第6項之化合物,其為(1R,4S)-2-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-2-氮雜雙環[2.2.1]庚-3-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第6項之化合物,其為(R)-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)咪唑啶-2-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第6項之化合物,其為(R)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟吡咯啶-2-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第6項之化合物,其為(S)-1-((S)-3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟吡咯啶-2-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第6項之化合物,其為(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-4-甲基咪唑啶-2-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第1項之化合物,其中A、D、E、G、J、L、M、及Q為碳。
- 根據申請專利範圍第1項之化合物,其中R1及R2為氫。
- 根據申請專利範圍第1項之化合物,其中R1及R2為氟,且a及b為1。
- 根據申請專利範圍第1項之化合物,其中R3及R5為氫。
- 根據申請專利範圍第1項之化合物,其中R3、R4及R5為氫。
- 根據申請專利範圍第1項之化合物,其中R6及R7係鍵聯形成隨意地經取代之吡咯啶酮環。
- 根據申請專利範圍第1項之化合物,其中R6及R7係鍵聯形成隨意地經取代之咪唑啶酮環。
- 根據申請專利範圍第1項之化合物,其中R6及R7係鍵聯形成隨意地經取代之7員單環或雙環。
- 根據申請專利範圍第1項之化合物,其中R6及R7係鍵聯形成隨意地經取代之10員單環或雙環。
- 根據申請專利範圍第1項之化合物,其中藉由R6及R7所形成的環係經一或多個氟基、甲基、乙基、異丙基、C3-6環烷基、或苯基取代。
- 一種式I化合物,

- 根據申請專利範圍第25項之化合物,其中R1及R2各自為氫或鹵基。
- 根據申請專利範圍第25項之化合物,其中R3、 R4、及R5各自獨立地選自氫及甲基。
- 根據申請專利範圍第25項之化合物,其中該化合物係選自:1-(3-(9H-咔唑-9-基)-2-羥丙基)哌啶-2-酮(化合物1);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)哌啶-2-酮(化合物4);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)哌啶-2-酮(化合物5);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-氟哌啶-2-酮(化合物11);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-氟哌啶-2-酮(化合物12);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3,3-二氟哌啶-2-酮(化合物13);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3,3-二氟哌啶-2-酮(化合物14);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3,3-二甲基哌啶-2-酮(化合物16);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-苯基哌啶-2-酮(化合物19);3-環己基-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)哌啶-2-酮(化合物20);3-環己基-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲 基丙基)哌啶-2-酮(化合物21);3-環戊基-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)哌啶-2-酮(化合物23);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-乙基哌啶-2-酮(化合物24);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-異丙基哌啶-2-酮(化合物25);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-異丙基哌啶-2-酮(化合物26);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-甲基哌啶-2-酮(化合物27);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-甲基哌啶-2-酮(化合物28);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-4-甲基哌啶-2-酮(化合物36);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-5-甲基哌啶-2-酮(化合物37);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-5-甲基哌啶-2-酮(化合物38);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-6-甲基哌啶-2-酮(化合物39);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-6-甲基哌啶-2-酮(化合物40);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-4-甲基哌啶- 2-酮(化合物49);3-環丁基-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)哌啶-2-酮(化合物51);3-環丁基-1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)哌啶-2-酮(化合物52);1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-甲氧基哌啶-2-酮(化合物57);1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-甲氧基哌啶-2-酮(化合物60);1-(3-(9H-咔唑-9-基)-2-羥丙基)-4-甲基哌啶-2-酮(化合物61);1-(3-(9H-咔唑-9-基)-2-羥丙基)-5-甲基哌啶-2-酮(化合物62);1-(3-(9H-咔唑-9-基)-2-羥丙基)-6-甲基哌啶-2-酮(化合物63);1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-甲基哌啶-2-酮(化合物64);1-(3-(9H-咔唑-9-基)-2-羥基-2-甲基丙基)哌啶-2-酮(化合物66);1-(3-(9H-咔唑-9-基)-2-羥丙基)-3,3-二氟哌啶-2-酮(化合物69);1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-氟哌啶-2-酮(化合物70);(S)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-5-甲基哌啶-2- 酮(化合物129);(S)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-5-甲基哌啶-2-酮(化合物130);(R)-1-((R)-3-(9H-咔唑-9-基)-2-羥丙基)-5-甲基哌啶-2-酮(化合物131);(S)-1-((S)-3-(9H-咔唑-9-基)-2-羥丙基)-5-甲基啶啶-2-酮(化合物132);(R)-1-((S)-3-(9H-咔唑-9-基)-2-羥丙基)-5-甲基哌啶-2-酮(化合物133);及(R)-1-((R)-3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-5-甲基哌啶-2-酮(化合物134);或其醫藥上可接受之鹽或水合物。
- 一種式I化合物,

- 根據申請專利範圍第29項之化合物,其中該化合物係選自:1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-甲基四氫嘧 啶-2(1H)-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)四氫嘧啶-2(1H)-酮;1-環己基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)四氫嘧啶-2(1H)-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-苯基四氫嘧啶-2(1H)-酮;1-環戊基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)四氫嘧啶-2(1H)-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-異丙基四氫嘧啶-2(1H)-酮;1-環丁基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)四氫嘧啶-2(1H)-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-苯基四氫嘧啶-2(1H)-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-異丙基四氫嘧啶-2(1H)-酮;1-環丁基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)四氫嘧啶-2(1H)-酮;1-環己基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)四氫嘧啶-2(1H)-酮;1-環丙基-3-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)四氫嘧啶-2(1H)-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3-乙基四氫嘧 啶-2(1H)-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)-3-乙基四氫嘧啶-2(1H)-酮;1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3,5-二甲基四氫嘧啶-2(1H)-酮;及1-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)-3,4-二甲基四氫嘧啶-2(1H)-酮;或其醫藥上可接受之鹽或水合物。
- 一種化合物,選自:1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-甲基四氫嘧啶-2(1H)-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)四氫嘧啶-2(1H)-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-乙基四氫嘧啶-2(1H)-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-環丙基四氫嘧啶-2(1H)-酮;1-(3-(9H-咔唑-9-基)-2-羥丙基)-3-環丁基四氫嘧啶-2(1H)-酮;4-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥丙基)啉-3-酮;及4-(3-(3,6-二氟-9H-咔唑-9-基)-2-羥基-2-甲基丙基)
 啉-3-酮;或其醫藥上可接受之鹽或水合物。
啉-3-酮;或其醫藥上可接受之鹽或水合物。
- 根據申請專利範圍第1至31項中任一項之化合物, 其中該化合物調節Cry1或Cry2。
- 根據申請專利範圍第32項之化合物,其中該調節包含下列之任一者:(i)結合至Cry1或Cry2;(ii)抑制Cry1或Cry2之修飾;(iii)改變Cry1或Cry2之局部化;(iv)提高或降低Cry1或Cry2之安定化;(v)提高或降低Cry1或Cry2至標靶間的結合;(vi)提高或降低Cry1或Cry2之活性;及(vii)提高或降低Cry1或Cry2標靶之活性。
- 根據申請專利範圍第33項之化合物,其中該標靶為Per1、Per2、葡萄糖皮質素受體(GR)、CLOCK、BMAL1、或CLOCK-BMAL1啟動子序列。
- 一種醫藥組成物,其包含根據申請專利範圍第1至31項中任一項之化合物、或其醫藥上可接受之鹽或水合物,及醫藥上可接受之載體、佐劑、或稀釋劑。
- 根據申請專利範圍第35項之醫藥組成物,其進一步包含一或多種額外之治療劑。
- 根據申請專利範圍第36項之醫藥組成物,其中該一或多種額外之治療劑選自由以下所組成之群組:DPP-IV抑制劑、SGLT2抑制劑、二甲雙胍(metformin)、及磺醯脲類。
- 根據申請專利範圍第36項之醫藥組成物,其中該一或多種額外之治療劑選自由以下所組成之群組: Signifor®、酮康唑(ketoconazole)、美替拉酮(metyrapone)、米托坦(mitotane)、依托咪酯(etomidate)、Korlym®、表皮生長因子受體抑制劑、醛固酮合成酶/11β-羥化酶抑制劑LCI699、及左旋酮康唑(levoketoconazole,COR-003)。
- 一種根據申請專利範圍第35至38項中任一項之醫藥組成物於製造藥品之用途,該藥品用於治療個體之Cry媒介性疾病或失調或用於減輕Cry媒介性疾病或失調的症狀。
- 根據申請專利範圍第39項之用途,其中該Cry媒介性疾病或失調係選自由以下所組成之群組:糖尿病、糖尿病併發症、非酒精性脂肪肝炎(NASH)、非酒精性脂肪肝疾病(NAFLD)、氣喘、慢性阻塞性肺病(COPD)、代謝症候群、胰島素抗性症候群、肥胖症、青光眼、庫欣氏症候群(Cushing’s syndrome)、精神病性憂鬱症、阿滋海默氏症、神經病性疼痛、藥物濫用、骨質疏鬆症、癌症、黃斑部病變、及肌病變。
- 根據申請專利範圍第40項之用途,其中該糖尿病併發症係選自由以下所組成之群組:糖尿病神經病變、糖尿病視網膜病變、糖尿病腎病變、白內障形成、青光眼、糖尿病血管病變、及動脈粥狀硬化。
- 根據申請專利範圍第35至38項中任一項之醫藥組成物,其用於治療個體之Cry媒介性疾病或失調或用於減輕個體之Cry媒介性疾病或失調的症狀。
- 根據申請專利範圍第42項之醫藥組成物,其中該Cry媒介性疾病或失調選自由以下所組成之群組:糖尿病、糖尿病併發症、非酒精性脂肪肝炎(NASH)、非酒精性脂肪肝疾病(NAFLD)、氣喘、慢性阻塞性肺病(COPD)、代謝症候群、胰島素抗性症候群、肥胖症、青光眼、庫欣氏症候群(Cushing’s syndrome)、精神病性憂鬱症、阿滋海默氏症、神經病性疼痛、藥物濫用、骨質疏鬆症、癌症、黃斑部病變、及肌病變。
- 根據申請專利範圍第43項之醫藥組成物,其中該糖尿病併發症係選自由以下所組成之群組糖尿病神經病變、糖尿病視網膜病變、糖尿病腎病變、白內障形成、青光眼、糖尿病血管病變、及動脈粥狀硬化。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461976350P | 2014-04-07 | 2014-04-07 | |
| US61/976,350 | 2014-04-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW201605835A TW201605835A (zh) | 2016-02-16 |
| TWI690521B true TWI690521B (zh) | 2020-04-11 |
Family
ID=53008857
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW104111004A TWI690521B (zh) | 2014-04-07 | 2015-04-02 | 作為隱花色素調節劑之含有咔唑之醯胺類、胺基甲酸酯類及脲類 |
Country Status (22)
| Country | Link |
|---|---|
| US (6) | US10005759B2 (zh) |
| EP (2) | EP3939971A1 (zh) |
| JP (4) | JP6601916B2 (zh) |
| KR (1) | KR102224423B1 (zh) |
| CN (2) | CN106458891B (zh) |
| AR (1) | AR099970A1 (zh) |
| AU (1) | AU2015244060B2 (zh) |
| CA (1) | CA2944074C (zh) |
| DK (1) | DK3129366T3 (zh) |
| ES (1) | ES2886469T3 (zh) |
| HR (1) | HRP20211372T1 (zh) |
| HU (1) | HUE055547T2 (zh) |
| IL (1) | IL248240B (zh) |
| LT (1) | LT3129366T (zh) |
| MX (1) | MX371096B (zh) |
| NZ (1) | NZ724683A (zh) |
| PL (1) | PL3129366T3 (zh) |
| RU (1) | RU2705094C2 (zh) |
| SG (1) | SG11201608020YA (zh) |
| SI (1) | SI3129366T1 (zh) |
| TW (1) | TWI690521B (zh) |
| WO (1) | WO2015157182A1 (zh) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013170186A1 (en) | 2012-05-11 | 2013-11-14 | Reset Therapeutics, Inc. | Carbazole-containing sulfonamides as cryptochrome modulators |
| TWI690521B (zh) | 2014-04-07 | 2020-04-11 | 美商同步製藥公司 | 作為隱花色素調節劑之含有咔唑之醯胺類、胺基甲酸酯類及脲類 |
| WO2018013546A1 (en) * | 2016-07-11 | 2018-01-18 | The Regents Of The University Of California | Bic inhibitor of cry-cry and cry-cib oligomerization/clustering |
| JP2019534266A (ja) | 2016-10-14 | 2019-11-28 | 江蘇恒瑞医薬股▲ふん▼有限公司 | 5員ヘテロアリール環の架橋した環誘導体、その製造方法およびその医学的使用 |
| JP6835308B2 (ja) * | 2016-10-19 | 2021-02-24 | 国立研究開発法人理化学研究所 | クリプトクロムの機能を抑制するための組成物 |
| WO2018132383A1 (en) * | 2017-01-10 | 2018-07-19 | The Regents Of The University Of California | Cry1-clock-bmal1 complex-disrupting agents and methods of identifying and using same |
| US11713470B2 (en) * | 2017-03-20 | 2023-08-01 | University of Pittsburgh—of the Commonwealth System of Higher Education | Targeted gene therapies for pain and other neuro-related disorders |
| JP7254094B2 (ja) * | 2018-03-22 | 2023-04-07 | オーリジーン ディスカバリー テクノロジーズ リミテッド | Prmt5阻害剤としての置換イミダゾリジン-2-オン誘導体 |
| US20210267939A1 (en) * | 2018-06-18 | 2021-09-02 | Duke University | Compositions and methods for treating nafld/nash and related disease phenotypes |
| EP3886854A4 (en) | 2018-11-30 | 2022-07-06 | Nuvation Bio Inc. | PYRROLE AND PYRAZOLE COMPOUNDS AND METHODS OF USE THERE |
| MA54395A (fr) | 2018-12-07 | 2022-03-16 | Neurocrine Biosciences Inc | Antagoniste du récepteur crf1, formulations pharmaceutiques et formes solides correspondantes pour le traitement de l'hyperplasie surrénalienne congénitale |
| CN111285793B (zh) * | 2019-03-01 | 2022-04-22 | 广州中医药大学(广州中医药研究院) | 氟取代双分子咔唑衍生物、其制备方法及应用 |
| CN111217741B (zh) * | 2019-03-01 | 2022-03-15 | 广东省中医院(广州中医药大学第二附属医院、广州中医药大学第二临床医学院、广东省中医药科学院) | 氟取代单咔唑类衍生物、其制备方法及应用 |
| CA3132319A1 (en) | 2019-03-04 | 2020-09-10 | Strongbridge Dublin Limited | Methods of treating disease with levoketoconazole |
| WO2021026185A1 (en) * | 2019-08-08 | 2021-02-11 | New York University | Indole compounds as modulators of rage activity and uses thereof |
| CN112162028A (zh) * | 2020-09-29 | 2021-01-01 | 中国农业科学院农业质量标准与检测技术研究所 | 一种用于草莓组织中维生素c的质谱成像方法 |
| GB202106143D0 (en) * | 2021-04-29 | 2021-06-16 | Adaptive Diagnostics Ltd | Determination of the presence of a target species |
| CN112986451B (zh) * | 2021-05-06 | 2022-05-17 | 湖南师范大学 | 一种基于风味特征的杂交鱼类肉品质评价方法 |
| CN113461674B (zh) * | 2021-08-09 | 2022-05-13 | 山东农业大学 | 一种促进植物根系生长的酰胺类化合物及其制备方法和应用 |
| EP4382529A1 (en) | 2022-12-07 | 2024-06-12 | Bayer Consumer Care AG | A process for preparing pure (3s)-pyrrolidin-3-ol and pure (3s)-pyrrolidin-3-ol hydrochloride |
| CN115974763A (zh) * | 2022-12-26 | 2023-04-18 | 常州大学 | 一种咔唑衍生物及其应用 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130303524A1 (en) * | 2012-05-11 | 2013-11-14 | Reset Therapeutics, Inc. | Carbazole-Containing Sulfonamides as Cryptochrome Modulators |
Family Cites Families (105)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4302386A (en) | 1978-08-25 | 1981-11-24 | The Ohio State University | Antigenic modification of polypeptides |
| US4105776A (en) | 1976-06-21 | 1978-08-08 | E. R. Squibb & Sons, Inc. | Proline derivatives and related compounds |
| US4230767A (en) | 1978-02-08 | 1980-10-28 | Toyo Boseki Kabushiki Kaisha | Heat sealable laminated propylene polymer packaging material |
| US4275149A (en) | 1978-11-24 | 1981-06-23 | Syva Company | Macromolecular environment control in specific receptor assays |
| US4233402A (en) | 1978-04-05 | 1980-11-11 | Syva Company | Reagents and method employing channeling |
| US4325952A (en) | 1978-04-18 | 1982-04-20 | Aziende Chimiche Riunite Angelini Francesco | Method of treating abstinence syndrome with cycloaklyltriazoles |
| US4316906A (en) | 1978-08-11 | 1982-02-23 | E. R. Squibb & Sons, Inc. | Mercaptoacyl derivatives of substituted prolines |
| IL58849A (en) | 1978-12-11 | 1983-03-31 | Merck & Co Inc | Carboxyalkyl dipeptides and derivatives thereof,their preparation and pharmaceutical compositions containing them |
| US4276890A (en) | 1979-04-11 | 1981-07-07 | Fichera Anthony T | Tobacco smoking inhibitor |
| US4231938A (en) | 1979-06-15 | 1980-11-04 | Merck & Co., Inc. | Hypocholesteremic fermentation products and process of preparation |
| US4508729A (en) | 1979-12-07 | 1985-04-02 | Adir | Substituted iminodiacids, their preparation and pharmaceutical compositions containing them |
| US4444784A (en) | 1980-08-05 | 1984-04-24 | Merck & Co., Inc. | Antihypercholesterolemic compounds |
| MX7065E (es) | 1980-06-06 | 1987-04-10 | Sankyo Co | Un procedimiento microbiologico para preparar derivados de ml-236b |
| US4376110A (en) | 1980-08-04 | 1983-03-08 | Hybritech, Incorporated | Immunometric assays using monoclonal antibodies |
| US4344949A (en) | 1980-10-03 | 1982-08-17 | Warner-Lambert Company | Substituted acyl derivatives of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acids |
| ZA817261B (en) | 1980-10-23 | 1982-09-29 | Schering Corp | Carboxyalkyl dipeptides,processes for their production and pharmaceutical compositions containing them |
| US4337201A (en) | 1980-12-04 | 1982-06-29 | E. R. Squibb & Sons, Inc. | Phosphinylalkanoyl substituted prolines |
| US4410520A (en) | 1981-11-09 | 1983-10-18 | Ciba-Geigy Corporation | 3-Amino-[1]-benzazepin-2-one-1-alkanoic acids |
| GB2128984B (en) | 1982-05-12 | 1985-05-22 | Hoffmann La Roche | Diaza-bicyclic compounds |
| US4659678A (en) | 1982-09-29 | 1987-04-21 | Serono Diagnostics Limited | Immunoassay of antigens |
| US4739073A (en) | 1983-11-04 | 1988-04-19 | Sandoz Pharmaceuticals Corp. | Intermediates in the synthesis of indole analogs of mevalonolactone and derivatives thereof |
| US4727022A (en) | 1984-03-14 | 1988-02-23 | Syntex (U.S.A.) Inc. | Methods for modulating ligand-receptor interactions and their application |
| US4780401A (en) | 1984-04-09 | 1988-10-25 | Ciba-Geigy Corporation | Novel monoclonal antibodies to human renin and hybridoma cells, processes for their preparation and their applications |
| US4845079A (en) | 1985-01-23 | 1989-07-04 | Luly Jay R | Peptidylaminodiols |
| US5066643A (en) | 1985-02-19 | 1991-11-19 | Sandoz Ltd. | Fluorine and chlorine statine or statone containing peptides and method of use |
| US4894437A (en) | 1985-11-15 | 1990-01-16 | The Upjohn Company | Novel renin inhibiting polypeptide analogs containing S-aryl-D- or L- or DL-cysteinyl, 3-(arylthio)lactic acid or 3-(arylthio)alkyl moieties |
| US4885292A (en) | 1986-02-03 | 1989-12-05 | E. R. Squibb & Sons, Inc. | N-heterocyclic alcohol renin inhibitors |
| US4816463A (en) | 1986-04-01 | 1989-03-28 | Warner-Lambert Company | Substituted diimidazo [1,5-a: 4',5'-d]pyridines having antihypertensive activity |
| CA1334092C (en) | 1986-07-11 | 1995-01-24 | David John Carini | Angiotensin ii receptor blocking imidazoles |
| US4772684A (en) | 1987-01-20 | 1988-09-20 | Triton Biosciences, Inc. | Peptides affecting blood pressure regulation |
| US4786653A (en) | 1987-08-24 | 1988-11-22 | Golwyn Daniel H | Treatment of neurotransmitter-linked drug abuse |
| US4980283A (en) | 1987-10-01 | 1990-12-25 | Merck & Co., Inc. | Renin-inhibitory pepstatin phenyl derivatives |
| US5089471A (en) | 1987-10-01 | 1992-02-18 | G. D. Searle & Co. | Peptidyl beta-aminoacyl aminodiol carbamates as anti-hypertensive agents |
| US5034512A (en) | 1987-10-22 | 1991-07-23 | Warner-Lambert Company | Branched backbone renin inhibitors |
| US5063207A (en) | 1987-10-26 | 1991-11-05 | Warner-Lambert Company | Renin inhibitors, method for using them, and compositions containing them |
| US5055466A (en) | 1987-11-23 | 1991-10-08 | E. R. Squibb & Sons, Inc. | N-morpholino derivatives and their use as anti-hypertensive agents |
| US5081127A (en) | 1988-01-07 | 1992-01-14 | E. I. Du Pont De Nemours And Company | Substituted 1,2,3-triazole angiotensin II antagonists |
| US5036054A (en) | 1988-02-11 | 1991-07-30 | Warner-Lambert Company | Renin inhibitors containing alpha-heteroatom amino acids |
| US4788189A (en) | 1988-02-29 | 1988-11-29 | Glazer Howard I | Method to treat smoking withdrawal symptoms by potentiated central noradrenergic blocking |
| US5036053A (en) | 1988-05-27 | 1991-07-30 | Warner-Lambert Company | Diol-containing renin inhibitors |
| DE3841520A1 (de) | 1988-12-09 | 1990-06-13 | Hoechst Ag | Enzymhemmende harnstoffderivate von dipeptiden, verfahren zu ihrer herstellung, diese enthaltende mittel und ihre verwendung |
| US5106835A (en) | 1988-12-27 | 1992-04-21 | American Cyanamid Company | Renin inhibitors |
| DE4004820A1 (de) | 1989-08-05 | 1991-04-25 | Bayer Ag | Renininhibitoren, verfahren zur herstellung und ihre verwendung in arzneimitteln |
| US5064825A (en) | 1989-06-01 | 1991-11-12 | Merck & Co., Inc. | Angiotensin ii antagonists |
| US5744101A (en) | 1989-06-07 | 1998-04-28 | Affymax Technologies N.V. | Photolabile nucleoside protecting groups |
| FI94339C (fi) | 1989-07-21 | 1995-08-25 | Warner Lambert Co | Menetelmä farmaseuttisesti käyttökelpoisen /R-(R*,R*)/-2-(4-fluorifenyyli)- , -dihydroksi-5-(1-metyylietyyli)-3-fenyyli-4-/(fenyyliamino)karbonyyli/-1H-pyrroli-1-heptaanihapon ja sen farmaseuttisesti hyväksyttävien suolojen valmistamiseksi |
| US5063208A (en) | 1989-07-26 | 1991-11-05 | Abbott Laboratories | Peptidyl aminodiol renin inhibitors |
| US5098924A (en) | 1989-09-15 | 1992-03-24 | E. R. Squibb & Sons, Inc. | Diol sulfonamide and sulfinyl renin inhibitors |
| US5104869A (en) | 1989-10-11 | 1992-04-14 | American Cyanamid Company | Renin inhibitors |
| US5114937A (en) | 1989-11-28 | 1992-05-19 | Warner-Lambert Company | Renin inhibiting nonpeptides |
| US5073566A (en) | 1989-11-30 | 1991-12-17 | Eli Lilly And Company | Angiotensin ii antagonist 1,3-imidazoles and use thereas |
| US5750015A (en) | 1990-02-28 | 1998-05-12 | Soane Biosciences | Method and device for moving molecules by the application of a plurality of electrical fields |
| US5095119A (en) | 1990-03-08 | 1992-03-10 | American Home Products Corporation | Renin inhibitors |
| US5075451A (en) | 1990-03-08 | 1991-12-24 | American Home Products Corporation | Pyrrolimidazolones useful as renin inhibitors |
| US5064965A (en) | 1990-03-08 | 1991-11-12 | American Home Products Corporation | Renin inhibitors |
| US5085992A (en) | 1990-07-19 | 1992-02-04 | Merck & Co., Inc. | Microbial transformation process for antihypertensive products |
| US5087634A (en) | 1990-10-31 | 1992-02-11 | G. D. Searle & Co. | N-substituted imidazol-2-one compounds for treatment of circulatory disorders |
| US5071837A (en) | 1990-11-28 | 1991-12-10 | Warner-Lambert Company | Novel renin inhibiting peptides |
| US5604260A (en) | 1992-12-11 | 1997-02-18 | Merck Frosst Canada Inc. | 5-methanesulfonamido-1-indanones as an inhibitor of cyclooxygenase-2 |
| US5474995A (en) | 1993-06-24 | 1995-12-12 | Merck Frosst Canada, Inc. | Phenyl heterocycles as cox-2 inhibitors |
| GB9602877D0 (en) | 1996-02-13 | 1996-04-10 | Merck Frosst Canada Inc | 3,4-Diaryl-2-hydroxy-2,5- dihydrofurans as prodrugs to cox-2 inhibitors |
| US5538848A (en) | 1994-11-16 | 1996-07-23 | Applied Biosystems Division, Perkin-Elmer Corp. | Method for detecting nucleic acid amplification using self-quenching fluorescence probe |
| BR9408478A (pt) | 1994-01-10 | 1997-08-26 | Merck Frosst Canada Inc | Composto composição farmacéutica processos de tratmento de uma doença suscetível ao tratamento com agente anti-inflamatório não esteroidal e de tratamento de doenças medidas pela ciclo-oxigenase, sal farmaceuticamente eficaz e uso de um composto |
| US5521213A (en) | 1994-08-29 | 1996-05-28 | Merck Frosst Canada, Inc. | Diaryl bicyclic heterocycles as inhibitors of cyclooxygenase-2 |
| CA2200462A1 (en) | 1994-10-27 | 1996-05-09 | Merck Frosst Canada Inc. | Stilbene derivatives useful as cyclooxygenase-2 inhibitors |
| US5552422A (en) | 1995-01-11 | 1996-09-03 | Merck Frosst Canada, Inc. | Aryl substituted 5,5 fused aromatic nitrogen compounds as anti-inflammatory agents |
| US5801155A (en) | 1995-04-03 | 1998-09-01 | Epoch Pharmaceuticals, Inc. | Covalently linked oligonucleotide minor grove binder conjugates |
| US5691374A (en) | 1995-05-18 | 1997-11-25 | Merck Frosst Canada Inc. | Diaryl-5-oxygenated-2-(5H) -furanones as COX-2 inhibitors |
| US5604253A (en) | 1995-05-22 | 1997-02-18 | Merck Frosst Canada, Inc. | N-benzylindol-3-yl propanoic acid derivatives as cyclooxygenase inhibitors |
| US5639780A (en) | 1995-05-22 | 1997-06-17 | Merck Frosst Canada, Inc. | N-benzyl indol-3-yl butanoic acid derivatives as cyclooxygenase inhibitors |
| US5643933A (en) | 1995-06-02 | 1997-07-01 | G. D. Searle & Co. | Substituted sulfonylphenylheterocycles as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US5733909A (en) | 1996-02-01 | 1998-03-31 | Merck Frosst Canada, Inc. | Diphenyl stilbenes as prodrugs to COX-2 inhibitors |
| US5789413A (en) | 1996-02-01 | 1998-08-04 | Merck Frosst Canada, Inc. | Alkylated styrenes as prodrugs to COX-2 inhibitors |
| GB9607503D0 (en) | 1996-04-11 | 1996-06-12 | Merck Frosst Canada Inc | Bisaryl cyclobutenes derivatives as cyclooxygenase inhibitors |
| US5922742A (en) | 1996-04-23 | 1999-07-13 | Merck Frosst Canada | Pyridinyl-2-cyclopenten-1-ones as selective cyclooxygenase-2 inhibitors |
| US5677318A (en) | 1996-07-11 | 1997-10-14 | Merck Frosst Canada, Inc. | Diphenyl-1,2-3-thiadiazoles as anti-inflammatory agents |
| US5861419A (en) | 1996-07-18 | 1999-01-19 | Merck Frosst Canad, Inc. | Substituted pyridines as selective cyclooxygenase-2 inhibitors |
| AR008331A1 (es) | 1997-01-23 | 1999-12-29 | Smithkline Beecham Corp | Compuestos antagonistas de un receptor de il-8, uso de los mismos para la fabricacion de medicamentos, procedimiento para su obtencion, composicionesfarmaceuticas que los contienen |
| US6475744B1 (en) | 1999-07-22 | 2002-11-05 | The General Hospital Corporation | Methods for identifying compounds which modulate circadian rhythm |
| US6399631B1 (en) | 1999-07-23 | 2002-06-04 | Pfizer Inc. | Carbazole neuropeptide Y5 antagonists |
| EP1236173A2 (en) | 1999-10-27 | 2002-09-04 | Biowulf Technologies, LLC | Methods and devices for identifying patterns in biological systems |
| AU2001280581A1 (en) | 2000-07-18 | 2002-01-30 | Correlogic Systems, Inc. | A process for discriminating between biological states based on hidden patterns from biological data |
| CN1262337C (zh) | 2000-11-16 | 2006-07-05 | 赛弗根生物系统股份有限公司 | 质谱分析方法 |
| US7113896B2 (en) | 2001-05-11 | 2006-09-26 | Zhen Zhang | System and methods for processing biological expression data |
| US20020193950A1 (en) | 2002-02-25 | 2002-12-19 | Gavin Edward J. | Method for analyzing mass spectra |
| WO2003105759A2 (en) * | 2002-06-12 | 2003-12-24 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Pharmacologic inhibition of myc function |
| US20040121305A1 (en) | 2002-12-18 | 2004-06-24 | Wiegand Roger Charles | Generation of efficacy, toxicity and disease signatures and methods of use thereof |
| US20050101023A1 (en) | 2003-03-28 | 2005-05-12 | Rogers James A. | Methods for diagnosing urinary tract and prostatic disorders |
| DE602005009811D1 (de) | 2004-03-26 | 2008-10-30 | Hoffmann La Roche | Tetrahydrocarbazole und derivate |
| BRPI0514397A (pt) | 2004-08-19 | 2008-06-10 | Aventis Pharma Inc | derivados de 3-ariltioindol-2-carboxamida e análogos destes como inibidores de caseìna cinase iépsilon |
| IN2014KN02423A (zh) | 2006-04-07 | 2015-05-01 | Vertex Pharma | |
| CL2007003591A1 (es) | 2006-12-12 | 2008-02-29 | Wyeth Corp | Compuestos derivados de sulfonamida ciclicos, inhibidores de retoma de monoamina; procedimiento de preparacion; composicion farmaceutica; y uso para el tratamiento o prevencion de disfuncion sexual, trastorno gastrointestinal, trastorno genitourinari |
| WO2009086303A2 (en) * | 2007-12-21 | 2009-07-09 | University Of Rochester | Method for altering the lifespan of eukaryotic organisms |
| UA107652C2 (en) * | 2008-10-06 | 2015-02-10 | Incuron Llc | Carbazole compounds and therapeutic uses of the compounds |
| US9162980B2 (en) * | 2009-01-09 | 2015-10-20 | Board Of Regents Of The University Of Texas System | Anti-depression compounds |
| US8362277B2 (en) | 2009-01-09 | 2013-01-29 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| EP2385829B1 (en) * | 2009-01-09 | 2018-08-01 | Board of Regents of the University of Texas System | Pro-neurogenic compounds |
| US9962368B2 (en) | 2009-01-09 | 2018-05-08 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| AU2011274787B2 (en) * | 2010-07-07 | 2016-06-16 | Board Of Regents Of The University Of Texas System | Pro-neurogenic compounds |
| WO2012009372A2 (en) | 2010-07-12 | 2012-01-19 | Colorado State University Research Foundation | Triazolium carbene catalysts and processes for asymmetric carbon-carbon bond formation |
| JP6231566B2 (ja) * | 2012-08-24 | 2017-11-15 | ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム | 神経新生促進化合物 |
| US9648880B2 (en) | 2012-09-04 | 2017-05-16 | University Of Massachusetts | Antifungal agents and uses thereof |
| US9357781B2 (en) * | 2013-05-03 | 2016-06-07 | Inscent, Inc. | Honeybee repellents and uses thereof |
| US9353078B2 (en) * | 2013-10-01 | 2016-05-31 | New York University | Amino, amido and heterocyclic compounds as modulators of rage activity and uses thereof |
| TWI690521B (zh) | 2014-04-07 | 2020-04-11 | 美商同步製藥公司 | 作為隱花色素調節劑之含有咔唑之醯胺類、胺基甲酸酯類及脲類 |
-
2015
- 2015-04-02 TW TW104111004A patent/TWI690521B/zh active
- 2015-04-06 CA CA2944074A patent/CA2944074C/en active Active
- 2015-04-06 PL PL15719041T patent/PL3129366T3/pl unknown
- 2015-04-06 HU HUE15719041A patent/HUE055547T2/hu unknown
- 2015-04-06 MX MX2016013105A patent/MX371096B/es active IP Right Grant
- 2015-04-06 NZ NZ724683A patent/NZ724683A/en unknown
- 2015-04-06 LT LTEPPCT/US2015/024537T patent/LT3129366T/lt unknown
- 2015-04-06 SG SG11201608020YA patent/SG11201608020YA/en unknown
- 2015-04-06 US US14/679,846 patent/US10005759B2/en active Active
- 2015-04-06 EP EP21178088.7A patent/EP3939971A1/en active Pending
- 2015-04-06 RU RU2016143304A patent/RU2705094C2/ru active
- 2015-04-06 EP EP15719041.4A patent/EP3129366B1/en active Active
- 2015-04-06 JP JP2016561780A patent/JP6601916B2/ja active Active
- 2015-04-06 AU AU2015244060A patent/AU2015244060B2/en active Active
- 2015-04-06 SI SI201531691T patent/SI3129366T1/sl unknown
- 2015-04-06 WO PCT/US2015/024537 patent/WO2015157182A1/en active Application Filing
- 2015-04-06 DK DK15719041.4T patent/DK3129366T3/da active
- 2015-04-06 CN CN201580030117.7A patent/CN106458891B/zh active Active
- 2015-04-06 CN CN201910308138.0A patent/CN110003172B/zh active Active
- 2015-04-06 ES ES15719041T patent/ES2886469T3/es active Active
- 2015-04-06 KR KR1020167030709A patent/KR102224423B1/ko active IP Right Grant
- 2015-04-07 AR ARP150101040A patent/AR099970A1/es active IP Right Grant
-
2016
- 2016-10-06 IL IL248240A patent/IL248240B/en active IP Right Grant
-
2018
- 2018-05-21 US US15/985,168 patent/US10214507B2/en active Active
- 2018-12-27 US US16/233,761 patent/US10759777B2/en active Active
-
2019
- 2019-05-07 JP JP2019087865A patent/JP2019142959A/ja not_active Withdrawn
-
2020
- 2020-07-17 US US16/931,948 patent/US20210171495A1/en not_active Abandoned
-
2021
- 2021-06-30 JP JP2021108890A patent/JP2021155449A/ja not_active Withdrawn
- 2021-08-30 HR HRP20211372TT patent/HRP20211372T1/hr unknown
-
2022
- 2022-01-10 US US17/571,839 patent/US20220411399A1/en not_active Abandoned
-
2023
- 2023-06-27 US US18/341,950 patent/US20240190837A1/en active Pending
- 2023-08-21 JP JP2023134051A patent/JP2023154093A/ja active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130303524A1 (en) * | 2012-05-11 | 2013-11-14 | Reset Therapeutics, Inc. | Carbazole-Containing Sulfonamides as Cryptochrome Modulators |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI690521B (zh) | 作為隱花色素調節劑之含有咔唑之醯胺類、胺基甲酸酯類及脲類 | |
| US10383880B2 (en) | Carbazole-containing sulfonamides as cryptochrome modulators | |
| BR112016023446B1 (pt) | Composto modulador de criptocromo (cry), composição farmacêutica que compreende o mesmo e uso da dita composição para o tratamento ou alívio de um sintoma de uma doença ou transtorno mediado por cry |