RU2440978C2 - Способ получения 1-(3, 4-дихлорбензил)-5-октилбигуанида или его соли - Google Patents
Способ получения 1-(3, 4-дихлорбензил)-5-октилбигуанида или его соли Download PDFInfo
- Publication number
- RU2440978C2 RU2440978C2 RU2009111276/04A RU2009111276A RU2440978C2 RU 2440978 C2 RU2440978 C2 RU 2440978C2 RU 2009111276/04 A RU2009111276/04 A RU 2009111276/04A RU 2009111276 A RU2009111276 A RU 2009111276A RU 2440978 C2 RU2440978 C2 RU 2440978C2
- Authority
- RU
- Russia
- Prior art keywords
- salt
- compound
- acid
- formula
- represented
- Prior art date
Links
- 0 *c(cc1)cc(Cl)c1Cl Chemical compound *c(cc1)cc(Cl)c1Cl 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C277/00—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C277/02—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups of guanidine from cyanamide, calcium cyanamide or dicyandiamides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C277/00—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C277/08—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups of substituted guanidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C279/00—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C279/20—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups containing any of the groups, X being a hetero atom, Y being any atom, e.g. acylguanidines
- C07C279/24—Y being a hetero atom
- C07C279/26—X and Y being nitrogen atoms, i.e. biguanides
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Изобретение относится к способу получения 1-(3,4-дихлорбензил)-5-октилбигуанида, представленного формулой (1), или его соли, заключающемуся во взаимодействии 1-циано-3-октилгуанидина или его соли с 3,4-дихлорбензиламином или его солью в сложноэфирном органическом растворителе. Технический результат предложенного изобретения состоит в получении целевого продукта с высоким выходом безопасным и несложным способом. 3 н. и 4 з.п. ф-лы.
Description
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу получения 1-(3,4-дихлорбензил)-5-октилбигуанида или его соли
УРОВЕНЬ ТЕХНИКИ
1-(3,4-дихлорбензил)-5-октилбигуанид, представленный формулой (1),

(далее по тексту в некоторых случаях именуемый просто «соединение A») и его соли являются соединениями с высокой противомикробной активностью. Поэтому исследуются бактерицидные препараты, содержащие эти соединения в качестве действующих ингредиентов.
До настоящего времени соединение A производили по способу, раскрытому в не прошедшей экспертизу японской патентной публикации № H5-194361.
Конкретно в этой публикации раскрыт способ, где соединение, представленное формулой (2)

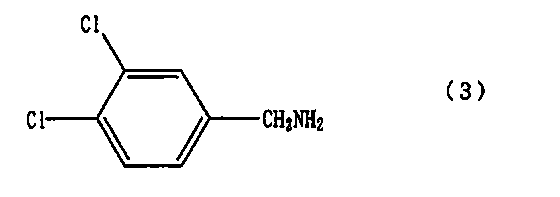
вводят во взаимодействие с соединением, представленным формулой (3)

с получением соединения A.
В способе, раскрытом в упомянутой публикации, взаимодействие соединения (2) с соединением (3) проводят в присутствии или отсутствии растворителя, такого как 2-этоксиэтанол, 2-метоксиэтанол, o-дихлорбензол, мезитилен или подобных растворителей.
Однако упомянутые выше растворители, а именно 2-этоксиэтанол, 2-метоксиэтанол и o-дихлорбензол, обладают нежелательными свойствами, такими как положительный результат в хромосомных тестах, мутагенность, тератогенность и т.д., и не подходят в качестве растворителей для применения в производстве лекарственных препаратов. Кроме того, проблемой является высокая температура кипения о-дихлорбензола. Следовательно, 2-этоксиэтанол, 2-метоксиэтанол и о-дихлорбензол не могут применяться при промышленном производстве соединения A. Таким образом в указанной публикации из числа перечисленных растворителей только мезитилен конкретно применяется в примерах.
В примере 1 упомянутой публикации конкретно описан способ, где соединение (2) и гидрохлорид соединения (3) нагревали до кипения с обратным холодильником с применением мезитилена в качестве растворителя, получая таким образом моногидрохлорид соединения A. Однако выход по данному способу составлял лишь 53,1% в пересчете на соединение (2) и лишь 56,9% в пересчете на гидрохлорид соединения (3), что далеко от удовлетворительного результата. Далее, поскольку реакцию проводили при температуре кипения мезитилена, т.е. при высокой температуре 162-164°C, был необходим специальный нагреватель, и осуществление этой реакции в промышленных масштабах с применением оборудования общего назначения является весьма затруднительным.
Таким образом, упомянутый выше известный способ не подходит для получения соединения А в промышленном масштабе.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Проблемы, которые предполагалось решить настоящим изобретением
Цель настоящего изобретения заключается в разработке безопасного и легко реализуемого способа получения соединения А или его солей с высокой чистотой и высоким выходом, с тем чтобы этот способ обеспечил возможность проводить реакцию при низкой температуре в течение непродолжительного времени с применением оборудования общего назначения.
Средства для решения указанных проблем
Для достижения указанной выше цели авторы настоящего изобретения осуществили обширные исследования для выявления безопасного и легко реализуемого способа получения соединения А или его солей. Одна из идей, направленных на исключение необходимости в специальном нагревателе, заключалась в применении растворителя с низкой температурой кипения, но использование различных растворителей с низкой температурой кипения позволило установить, что они не способны улучшить выход соединения A или его солей. Далее авторы настоящего изобретения исследовали различные растворители с низкой температурой кипения и обнаружили, что указанная выше цель может быть достигнута только при применении определенных органических растворителей, представляющих собой сложные эфиры. Работа над настоящим изобретением была завершена на основе этих данных.
В настоящем изобретении разработан способ получения 1-(3,4-дихлорбензил)-5-октилбигуанида или его солей, который изложен в следующих пунктах 1-7.
Пункт 1. Способ получения 1-(3,4-дихлорбензил)-5-октилбигуанида, представленного формулой (1)
или его соли, причем способ включает взаимодействие 1-циано-3-октилгуанидина, представленного формулой (2)
или его соли с 3,4-дихлорбензиламином, представленным формулой (3)
или его солью в сложноэфирном органическом растворителе.
Пункт 2. Способ по п.1, где сложноэфирный органический растворитель представляет собой по меньшей мере один растворитель, выбранный из группы, состоящей из н-бутилацетата, изобутилацетата, втор-бутилацетата, трет-бутилацетата, н-пентилацетата, изопентилацетата и н-пропилпропионата.
Пункт 3. Способ по п.2, где сложноэфирный органический растворитель представляет собой н-бутилацетат.
Пункт 4. Способ по любому из пп. 3, где реакцию проводят в присутствии кислоты.
Пункт 5. Способ по п.4, где кислота представляет собой по меньшей мере одну кислоту, выбранную из группы, состоящей из хлористоводородной кислоты, серной кислоты, фосфорной кислоты и бромистоводородной кислоты.
Пункт 6. Способ получения 1-(3,4-дихлорбензил)-5-октилбигуанида, представленного формулой (1)
или его соли, где способ включает стадии взаимодействия н-октиламина, представленного формулой (4)
H
2
N-(CH
2
)
7
-CH
3
(4)
или его соли, с соединением, представленным формулой (5)
M-N(CN)
2
(5)
где M представляет собой щелочной металл, или его соль, в сложноэфирном органическом растворителе с получением 1-циано-3-октилгуанидина, представленного формулой (2)
или его соли;
и взаимодействия 1-циано-3-октилгуанидина или его соли, полученных на предыдущей стадии, с 3,4-дихлорбензиламином, представленным формулой (3)
или его солью в сложноэфирном органическом растворителе.
Пункт 7. Способ получения 1-(3,4-дихлорбензил)-5-октилбигуанида, представленного формулой (1)
или его соли, где способ включает взаимодействие н-октиламина, представленного формулой (4)
H
2
N-(CH
2
)
7
-CH
3
(4)
или его соли, с соединением, представленным формулой (5)
M-N(CN)
2
(5)
где M представляет собой щелочной металл, или с его солью, в сложноэфирном органическом растворителе с получением 1-циано-3-октилгуанидина, представленного формулой (2)
или его соли;
и затем, без выделения 1-циано-3-октилгуанидина или его соли, добавление 3,4-дихлорбензиламина, представленного формулой (3)
или его соли к реакционной смеси для осуществления взаимодействия 1-циано-3-октилгуанидина или его соли с 3,4-дихлорбензиламином или его солью.
Способ получения 1-(3,4-дихлорбензил)-5-октилбигуанида или его соли по настоящему изобретению описан ниже.
Схема реакции-1

Соединение (1) или его соль могут быть получены взаимодействием соединения (2) или его соли с соединением (3) или его солью в сложноэфирном органическом растворителе.
Например, можно упомянуть применяемые в этой реакции в качестве сложноэфирного растворителя пропилацетат, н-бутилацетат, изобутилацетат, втор-бутилацетат, трет-бутилацетат, н-пентилацетат, изопентилацетат, н-пропилпропионат, диэтилкарбонат и т.д.
Среди перечисленных выше сложноэфирных органических растворителей предпочтительными являются растворители с температурой кипения в районе 100-140°C.
Кроме того, эфиры карбоновых кислот являются предпочтительным типом сложноэфирных органических растворителей.
В качестве предпочтительных примеров сложных эфиров карбоновых кислот для применения в способе по настоящему изобретению могут быть упомянуты сложные эфиры карбоновых кислот, представленные формулой (6)
R
1
COOR
2
(6)
где R1 представляет собой C1-3 алкильную группу и R2 представляет собой C4-6 алкильную группу. Конкретные примеры сложных эфиров карбоновых кислот, представленных формулой (6), включают н-бутилацетат, изобутилацетат, втор-бутилацетат, трет-бутилацетат, н-пентилацетат, изопентилацетат, н-пропилпропионат, и т.д., причем н-бутилацетат является более предпочтительным. При применении указанных сложных эфиров карбоновых кислот соединение (1) может быть получено со значительно более высоким выходом.
В показанной выше реакции соединение (3) обычно применяют в количестве от 0,5 до 1,5 моль, предпочтительно 0,8-1,1 моль и наиболее предпочтительно 0,9-1,1 моль на один моль соединения (2).
Количество сложноэфирного органического растворителя обычно составляет от 2 до 20 мл, предпочтительно от 3 до 10 мл и более предпочтительно от 5 до 8 мл на один грамм соединения (2).
Если соединение (3) взято в форме соли (соли кислоты), предпочтительно не допускать присутствия кислоты в реакционной системе.
Если соединение (3) взято в свободной форме, предпочтительно обеспечить наличие кислоты в реакционной системе.
Количество кислоты составляет от 0,5 до 1,5 эквивалентов и предпочтительно от 0,8 до 1 эквивалента на эквивалент соединения (3).
Примеры кислот включают неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.д.; органические кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, глутаминовая кислота и т.д.; или смесь упомянутых выше органических кислот и упомянутых выше неорганических кислот. Среди упомянутых выше кислот предпочтительными являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., причем более предпочтительной является хлористоводородная кислота.
Реакцию проводят при нагревании, обычно при температуре приблизительно от 50 до 150°C, и предпочтительно при температуре приблизительно от 100 до 140°C. Обычно реакция завершается в течение примерно 0,5-15 часов.
Соединение, которое предполагается получить по приведенной выше схеме реакции (1), может быть выделено и очищено путем, например, охлаждения реакционной смеси, выделения неочищенного продукта реакции при помощи одного из способов выделения, например фильтрования, концентрирования, экстракции и т.д., и последующей очистки выделенного продукта с помощью обычной методики очистки, например, колоночной хроматографии, перекристаллизации и т.д.
Если соль соединения (1) образовалась в виде кристаллов, предпочтительно очищать эту соль соединения по следующей методике перекристаллизации, повышая тем самым чистоту целевого продукта. Перекристаллизацию выполняют с применением растворителя для перекристаллизации. В качестве растворителя для перекристаллизации можно применять, например воду, органический растворитель или смесь воды и органического растворителя. Подходят органические растворители, смешивающиеся с водой. В качестве конкретных примеров таких органических растворителей могут быть упомянуты ацетон, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, ацетонитрил, диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФА), а также спирты (например, метанол, этанол, 1-пропанол, изопропанол, 1-бутанол, изобутанол и т.д.).
Конкретно перекристаллизацию проводят, добавляя соль соединения (1) в растворитель, нагревая полученную смесь при перемешивании до достижения температуры кипения применяемого растворителя, чтобы таким образом добиться растворения соли соединения (1), и охлаждая полученный раствор до заранее установленной температуры для выпадения кристаллов.
Осажденные таким образом кристаллы отделяют фильтрованием, центрифугированием и т.д., при необходимости промывают полученный осадок незначительным количеством холодного растворителя и затем высушивают, получая целевое соединение в очищенном состоянии.
В описанном выше способе перекристаллизации заранее определенная температура для выделения кристаллов в процессе охлаждения не ограничена. В качестве примера, при использовании 15% этанола, раствор соли соединения (1) в 15% этаноле охлаждают предпочтительно до температуры около 40°C или ниже, более предпочтительно от 0 до 35°C и еще более предпочтительно от 10 до 30°C. При охлаждении до температуры в указанном диапазоне целевое соединение может быть получено с высокой чистотой.
Кроме того, если соль соединения (1) представляет собой гидрохлорид, описанную выше методику перекристаллизации предпочтительно выполняют с применением в качестве растворителя для перекристаллизации смеси воды со спиртом, таким как этанол, изопропанол и т.д.
Помимо этого, если соль соединения (1) полученная по способу, изображенному выше на схеме реакции-1, образовалась в виде кристаллов дигидрохлорида, эти кристаллы легко превращаются в моногидрохлорид при обработке, например при перемешивании или т.п. этих кристаллов в воде или смешанном растворителе, состоящем из воды и органического растворителя.
Более конкретно дигидрохлорид соединения (1) в растворителе нагревают при перемешивании до достижения температуры кипения использованного растворителя. После этого полученный раствор охлаждают для выпадения кристаллов и кристаллы отделяют фильтрованием, центрифугированием и т.д. с последующим высушиванием, получая моногидрохлорид соединения (1).
Не существует ограничения на температуру, до которой охлаждают раствор в описанной выше методике, и, например, раствор дигидрохлорида соединения (1), полученный после описанной тепловой обработки, предпочтительно охлаждают до температуры около 40°C, более предпочтительно от 0 до 35°C и еще более предпочтительно от 10 до 30°C.
Кроме того, моногидрохлорид соединения (1) может быть получен с помощью такой обработки, как перемешивание или т.п. дигидрохлорида соединения (1) в растворителе в суспендированном состоянии без растворения дигидрохлорида соединения (1) при нагревании, и отделение кристаллов с помощью фильтрования, центрифугирование и т.д. с последующим высушиванием.
В этом случае суспензию дигидрохлорида соединения (1) перемешивают обычно при температуре около 40°C или ниже, предпочтительно примерно 0-35°C и еще более предпочтительно 10-35°C, однако приведенные цифры не ограничивают температурный режим.
Предпочтительный способ получения моногидрохлорида соединения (1) из дигидрохлорида соединения (1) заключается в перемешивании дигидрохлорида соединения (1) в воде при температуре 10-35°C, не добиваясь растворения дигидрохлорида соединения (1) за счет нагревания.
В этом способе предпочтительно применяются органические растворители, смешиваемые с водой. Конкретные примеры таких органических растворителей включают ацетон, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, ацетонитрил, диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФА) и т.д., а также спирты (например, метанол, этанол, 1-пропанол, изопропанол, 1-бутанол, изобутанол и т.д.).
Если моногидрохлорид соединения (1), полученный по описанному выше способу, содержит примесь гидрата, полученное соединение легко превратить в безводное такой обработкой, как, например, перемешивание или т.п. продукта при температуре от 0 до 60°C и предпочтительно от 30 до 50°C в течение 0,5-4 часов и предпочтительно 1-2 часов в водно-спиртовом растворе, включающем спирт, например этанол, изопропанол и т.п. в количестве 15% или более, предпочтительно 30% или более. Более конкретно моногидрохлорид·1/2 гидрат соединения (1) подвергают такой обработке, как перемешивание или т.п. при 30-50°C в водно-этанольном растворе, содержащем 30% или более этанола в течение примерно 2 часов с получением безводного моногидрохлорида соединения (1). В случае водно-этанольного раствора, содержащего 15% этанола, безводный моногидрохлорид соединения (1) может быть получен такой обработкой, как перемешивание или т.п. при температуре 40-60°C или выше в течение примерно 2 часов.
В настоящем изобретении соединение, представленное формулой (2) и применяемое в качестве исходного вещества, получают, например, показанным ниже способом.
Схема реакции-2

где M представляет собой щелочной металл.
Соединение (2) или его соль могут быть получены взаимодействием соединения, представленного формулой (4) (далее по тексту в некоторых случаях именуемого просто «соединением (4)»), или его соли, с соединением, представленным формулой (5) (далее по тексту в некоторых случаях именуемого просто «соединением (5)»), или его солью.
В качестве подходящего щелочного металла в соединении (5) могут быть упомянуты литий, натрий, калий и т.д.
Это взаимодействие проводят в инертном растворителе или без растворителя. Примеры инертного растворителя для применения в этой реакции включают: воду; ароматические углеводородные органические растворители, такие как бензол, толуол, ксилол и т.д.; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан, моноглим, диглим и т.д.; галогенированные углеводороды, такие как дихлорметан, дихлорэтан, хлороформ, четыреххлористый углерод и т.д.; низшие спирты, такие как метанол, этанол, изопропанол, бутанол, трет-бутанол, этиленгликоль и т.д.; жирные кислоты, такие как уксусная кислота и т.д.; сложные эфиры, такие как этилацетат, пропилацетат, н-бутилацетат, изобутилацетат, втор-бутилацетат, трет-бутилацетат, н-пентилацетат, изопентилацетат, н-пропилпропионат, диэтилкарбонат и т.д.; кетоны, такие как ацетон, метилэтилкетон и т.д.; ацетонитрил, пиридин, ДМФА, ДМСО, гексаметилтриамид фосфорной кислоты или их смесь и т.д.
Если соединение (4) взято в форме соли (кислой соли), предпочтительно не допускать присутствия кислоты в реакционной смеси.
Если соединение (4) взято в свободной форме, предпочтительно обеспечить присутствие кислоты в реакционной смеси.
Количество кислоты составляет от 0,5 до 1,5 эквивалентов, и предпочтительно от 0,8 до 1 эквивалента на эквивалент соединения (4).
Примеры кислот включают: неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.д.; органические кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, глутаминовая кислота и т.д.; или смесь упомянутых выше неорганических кислот и упомянутых выше органических кислот. Среди упомянутых выше кислот предпочтительными являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., причем более предпочтительной является хлористоводородная кислота.
При применении упомянутого выше растворителя его количество обычно составляет от 5 до 20 мл, предпочтительно от 8 до 12 мл и более предпочтительно от 9 до 11 мл на эквивалент 1 г соединения (4).
Эту реакцию обычно проводят при температуре приблизительно от 50 до 150°C и предпочтительно примерно от 70 до 130°C. Реакция, как правило, завершается в течение примерно от 0,5 до 40 часов.
При проведении реакции вышеупомянутого соединения (4) или его соли с соединением (5) или его солью в сложноэфирном органическом растворителе реакцию синтеза соединения (2), представленную приведенной выше схемой реакции-2, и реакцию по приведенной выше схеме реакции-1 с использованием соединения (2) в качестве исходного вещества, можно проводить в одну стадию. Соответственно последовательность этих реакций может быть проведена без выделения соединения (2) или его соли в качестве промежуточного соединения, что приводит к значительному улучшению выхода.
Исходные соединения, применяемые в приведенных выше схемах реакций, могут представлять собой подходящие соли, и целевые соединения, полученные по приведенным выше схемам, могут быть в форме подходящих солей.
Подходящие соли являются фармацевтически приемлемыми солями, включая, например, соли металлов, например, соли щелочных металлов (например, соли натрия, соли калия и т.д.), соли щелочноземельных металлов (например, соли кальция, соли магния и т.д.) и т.д., соли аммония, карбонаты щелочных металлов (например, карбонат лития, карбонат калия, карбонат натрия, карбонат цезия и т.д.), гидрокарбонаты щелочных металлов (например, гидрокарбонат лития, гидрокарбонат натрия, гидрокарбонат калия и т.д.), гидроксиды щелочных металлов (например, гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид цезия и т.д.) и другие соли неорганических оснований; три(низший алкил)аминов (например, триметиламина, триэтиламина, N-этилдиизопропиламина и т.д.), пиридина, хинолина, пиперидина, имидазола, пиколина, диметиламинопиридина, диметиланилина, N-(низший алкил)морфолинов (например, N-метилморфолина и т.п.), 1,5-диазабицикло[4.3.0]нонена-5(DBN), 1,8-диазабицикло[5.4.0]ундецена-7 (DBU), 1,4-диазабицикло[2.2.2]октана (DABCO) и другие соли органических оснований; гидрохлориды, гидробромиды, гидройодиды, сульфаты, нитраты, фосфаты и другие соли неорганических кислот; формиаты, ацетаты, пропионаты, оксалаты, малонаты, сукцинаты, фумараты, малеаты, лактаты, малаты, цитраты, тартраты, карбонаты, пикраты, метансульфонаты, этансульфонаты, п-толуолсульфонаты, глутаматы, а также другие соли органических кислот; и т.д.
Исходные вещества и целевые соединения, показанные на каждой схеме реакции, включают их сольваты (например, гидраты, этанолаты и т.д.). Предпочтительные сольваты включают гидраты. Целевые соединения, полученные по приведенной выше схеме реакции (2), могут быть выделены из реакционной смеси и очищены с помощью, например охлаждения реакционной смеси, выделения неочищенного продукта реакции из реакционной смеси при помощи одного из способов выделения, например, фильтрования, концентрирования, экстракции и/или других способов выделения и последующей очистки неочищенного продукта с помощью колоночной хроматографии, перекристаллизации и/или другой традиционной методики очистки.
ЭФФЕКТЫ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению, хотя реакцию проводят при низкой температуре с применением оборудования общего назначения без необходимости использования специального нагревателя, желаемое соединение, т.е. 1-(3,4-дихлорбензил)-5-октилбигуанид или его соль, могут быть получены за короткое время безопасным, простым способом и с высоким выходом. Настоящее изобретение обладает также тем преимуществом, что применение сложноэфирного органического растворителя дает возможность проведения реакции соединения (4) с соединением (5) с получением соединения (2), и взаимодействия соединения (2) с соединением (3) с получением соединения (1) в одном и том же растворителе и в одном и том же реакторе. Далее, поскольку способ по настоящему изобретению в основном не приводит к получению побочных продуктов, 1-(3,4-дихлорбензил)-5-октилбигуанид или его соли могут быть получены с более высокой чистотой с применением несложной методики очистки.
НАИЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Справочные примеры, примеры и сравнительные примеры приведены ниже для более подробной иллюстрации настоящего изобретения.
Справочный пример 1: 1-циано-3-н-октилгуанидин
Соединение (4) в количестве 7,00 кг (54,16 моль) растворяли в 105 литрах этилацетата и полученную смесь охлаждали до 5°C или ниже. К полученному раствору при перемешивании и температуре 40°C или ниже по каплям добавляли 2,66 кг концентрированной серной кислоты (27,12 моль). К полученной таким образом суспензии 1/2 сульфата соединения (4) добавляли 5,06 кг дицианамида натрия (56,83 моль) и полученную суспензию нагревали до кипения с обратным холодильником в течение 7 часов. Реакционный раствор охлаждали до 40°C или ниже и добавляли к нему 70 литров воды. Затем полученный раствор нагревали до 80-90°C (внутренняя температура) с целью отгонки этилацетата. Оставшуюся жидкость охлаждали до 40°C или ниже и добавляли 70 литров толуола с последующей экстракцией 1-циано-3-н-октилгуанидина при температуре примерно 50°C. Толуоловый слой с экстрагированным продуктом промывали 35 литрами воды при температуре примерно 50°C и охлаждали до 10°C или ниже, после чего перемешивали в течение примерно 30 минут. Отделяли образовавшийся кристаллический осадок и промывали его 7 литрами толуола. Полученные кристаллы высушивали при 40°C в течение 7,5 часов, получая 1-циано-3-н-октилгуанидин.
Выход: 9,11 кг (выход составил 85,7% в пересчете на соединение (4)).
Получены белые кристаллы с температурой плавления от 69 до 74°C (явно выраженной температуры плавления не наблюдалось).
ИК-спектр (KBr): 3439, 3296, 2916, 2164, 1659, 1556, 1160, 718 и 572 см-1.
Термогравиметрия/дифференциальный термический анализ: 73,5°C (слабый) и эндотермический пик при 77,5°C.
1H-ЯМР (CDCl3) спектр: 0,88 ppm (т, J=6,6 Гц, 3Н), 1,20-1,38 ppm (м, 10Н), 1,43-1,62 ppm (м, 2Н), 3,17 ppm (дд, J=6,9 Гц, J=6,0 Гц, 2Н), 5,60-5,70 ppm (ушир.с, 2Н), 5,80-5,95 ppm (ушир.с, 1Н).
Справочный пример 2: Ацидолиз дигидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида
Дигидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида в количестве 1 г растворяли в 15 мл 10% этанола и затем кипятили с обратным холодильником в течение 5 часов. Проводили ВЭЖХ-анализ в описанных ниже условиях.
Выход 1-[N-(3,4-дихлорбензил)карбамоил-3-октил]гуанидина (время удерживания: 9,84 минуты) составлял 0,91% и выход 1-(N-октилкарбамоил)-3-(3,4-дихлорбензил)гуанидина (время удерживания: 10,54 минут) составлял 0,22%.
Условия ВЭЖХ-анализа:
Колонка: YMC AM302 4,6 мм внутр.диаметр × 150 мм;
Элюент: MeCN/0,05M водный раствор 1-октансульфоната натрия/уксусная кислота = 700/300/1;
Детектор: УФ 254 нм.
Полученный 1-[N-(3,4-дихлорбензил)карбамоил-3-октил]гуанидин обладал следующими физическими свойствами:
ЯМР (ДМСО-d6) δ: 0,86 (3Н, т, J=6,0 Гц,), 1,07-1,35 (10Н, м), 1,35-1,49 (2Н, м), 2,95-3,15 (2Н, м), 4,12 (2Н, д, J=6,3 Гц), 6,78-7,40 (4Н, м), 7,23 (1Н, дд, J=2,1 Гц, J=8,4 Гц), 7,46 (1Н, д, J=2,1 Гц), 7,54 (1Н, д, J=8,4 Гц).
Полученный 1-(N-октилкарбамоил)-3-(3,4-дихлорбензил)гуанидин обладал следующими физическими свойствами:
ЯМР (ДМСО-d6) δ: 0,85 (3Н, т, J=6,6 Гц,), 1,02-1,40 (12Н, м), 2,89-2,95 (2Н, м), 4,33 (2Н, ушир.с), 5,76-7,00 (4Н, м), 7,28 (1Н, дд, J=2,1 Гц, J=8,1 Гц), 7,52 (1Н, д, J=2,1 Гц), 7,58 (1Н, д, J=8,1 Гц).
Пример 1: 1/2 гидрат моногидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида
Соединение (2) в количестве 9,82 г (0,05 моль) и 10,63 г 3,4-дихлорбензиламина (0,05 моль) добавляли к 49 мл бутилацетата и затем кипятили с обратным холодильником в течение 6 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли смесь 12 мл воды и 47 мл изопропилового спирта и растворяли в ней остаток. К полученному таким образом раствору по каплям добавляли 10,13 г концентрированной соляной кислоты. Полученную смесь перемешивали при 28-30°C в течение 30 минут и отфильтровывали выпавшие кристаллы. Полученные описанным способом кристаллы промывали небольшим количеством изопропилового спирта, получая 23,42 г (невысушенного) дигидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида. Эти кристаллы без высушивания суспендировали в 167 мл воды, затем перемешивали суспензию при 25-27°C в течение 2 часов с последующим отделением кристаллов фильтрованием. Полученные таким образом кристаллы промывали незначительным количеством воды и высушивали при 40°C в течение 20 часов, получая 17,05 г 1/2 гидрата моногидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида, имеющего чистоту 99,9%, с выходом 81,6%.
Пример 2: дигидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида
Соединение (4) в количестве 100 г (0,774 моль) растворяли в 1 литре н-бутилацетата и к этому раствору при перемешивании добавляли 37,6 г концентрированной серной кислоты (0,383 моль). К полученной таким способом суспензии 1/2 сульфата соединения (4) добавляли 68,9 г дицианамида натрия (0,774 моль) и образовавшуюся суспензию нагревали до кипения с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали примерно до 20°C и ее органический слой последовательно промывали примерно 500 мл каждого из следующих составов: (i) 5% хлористоводородной кислотой, (ii) 5% водным раствором едкого натра, (iii) 5% водным раствором бикарбоната натрия и (iv) водой.
К полученному описанным способом раствору соединения (2) в н-бутилацетате при перемешивании добавляли 118,5 г соединения (3) (0,673 моль) и затем 58,4 мл концентрированной хлористоводородной кислоты. Реакционную смесь нагревали и отгоняли примерно 800 мл н-бутилацетата при атмосферном давлении (нормальном давлении), после чего нагревали реакционную смесь до кипения с обратным холодильником в течение 3,5 часов. После этого реакционную смесь охлаждали примерно до 40°C и к ней добавляли 900 мл изопропанола, 100 мл воды и 134 мл концентрированной хлористоводородной кислоты. Эту смесь перемешивали при 60-70°C в течение 1 часа, охлаждали до 10°C или ниже и затем отделяли выпавшие кристаллы. Полученные кристаллы промывали 200 мл изопропанола и высушивали при 60°C, получая дигидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида.
Выход: 243,8 г (выход составлял 81,3% в пересчете на соединение (3)).
Температура плавления: 228,9°C.
ИК-спектр (KBr): 2920, 1682, 1634, 1337, 1035, 820 и 640 см-1.
Пример 3: Моногидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида 1/2 H
2
O
Дигидрохлорид соединения A в количестве 100 г (0,225 моль) добавляли к 1 л 15% водного раствора изопропанола и полученную смесь нагревали до растворения дигидрохлорида соединения A. Полученный раствор охлаждали примерно до 35°C, добавляли в него 0,2 г кристаллической затравки и затем перемешивали в течение 1 часа при температуре 25-35°C. После перемешивания раствор охлаждали до 10°C или ниже и затем отделяли выпавшие кристаллы. Кристаллический осадок промывали 200 мл воды, получая влажные кристаллы.
После перекристаллизации этих влажных кристаллов из 1 литра 15% водного раствора изопропанола полученные кристаллы высушивали при 40°C, получая неочищенные кристаллы моногидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида 1/2 H2O.
Выход: 90,54 г (выход составлял 96,5% в пересчете на дигидрохлорид соединения A).
Чистота (ВЭЖХ) 99,9% или выше.
Размер частиц: цель может быть достигнута, если кристаллы могут проходить через сито с ячейками 870 мкм.
Температура плавления: 173-174°C.
1H-ЯМР (ДМСО-d6) спектр: 0,85 ppm (т, J=6,8 Гц, 3Н), 1,10-1,50 ppm (м, 12Н), 2,92-3,08 ppm (м, 2Н), 4,33 ppm (д, J=6,3 Гц, 2Н), 6,80-7,20 ppm (ушир.с, 3Н), 7,30 ppm (д, J=8,4 Гц, 1Н), 7,48-7,62 ppm (м, 3Н), 7,70-7,90 ppm (ушир.с, 0,5Н).
ИК-спектр (KBr): 3316, 3190, 2928, 1584, 1549, 1152, 1032 и 723 см-1.
Термогравиметрия/дифференциальный термический анализ:
Наблюдались три эндотермических пика в районе 40±10°C, 90±10°C и 170±5°C. Температуры эндотермических пиков незначительно различались для разных партий вещества; однако наблюдались три хорошо различимых пика.
Спектр дифракции рентгеновских лучей на порошке (2θ): 3,6°, 7,2°, 10,9°, 18,1° и 25,5°.
Пример 4: Моногидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида 1/2 H
2
O
Неочищенные кристаллы дигидрохлорида соединения A в количестве 8,92 кг добавляли в 15% водный раствор этанола (смесь 114 литров очищенной воды и 20 литров этанола), полученную смесь нагревали до растворения неочищенных кристаллов. Полученный раствор охлаждали примерно до 40°C, добавляли к нему 90 г кристаллической затравки и затем перемешивали при 30-40°C в течение примерно 2 часов. После перемешивания раствор охлаждали примерно до 10°C и отделяли выпавшие кристаллы центрифугированием. Полученные кристаллы высушивали при 40°C, получая моногидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида 1/2 H2O.
Выход: 8,15 кг (выход составлял 97,4% в пересчете на неочищенные кристаллы дигидрохлорида соединения A).
Чистота (ВЭЖХ) 99,6% или выше.
Температура плавления: 173-174°C.
1H-ЯМР (ДМСО-d6) спектр: 0,85 ppm (т, J=6,8 Гц, 3Н), 1,10-1,50 ppm (м, 12Н), 2,92-3,08 ppm (м, 2Н), 4,33 ppm (д, J=6,3 Гц, 2Н), 6,80-7,20 ppm (ушир.с, 3Н), 7,30 ppm (д, J=8,4 Гц, 1Н), 7,48-7,62 ppm (м, 3Н), 7,70-7,90 ppm (ушир.с, 0,5Н).
ИК-спектр (KBr): 3316, 3190, 2928, 1584, 1549, 1152, 1032 и 723 см-1.
Термогравиметрия/дифференциальный термический анализ: Наблюдалось три эндотермических пика в районе 40±10°C, 90±10°C и 170±5°C. Температуры эндотермических пиков незначительно различались для разных партий вещества; однако наблюдались три хорошо различимых пика.
Спектр дифракции рентгеновских лучей на порошке (2θ): 3,6°, 7,2°, 10,9°, 18,1° и 25,5°.
Пример 5: Моногидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида 1/2 H
2
O
Соединение (4) в количестве 9 кг (69,64 моль) растворяли в 90 литрах н-бутилацетата и затем охлаждали до 10°C или ниже. К полученному раствору при перемешивании и температуре, не превышающей 40°C, добавляли 3,35 кг концентрированной серной кислоты (34,16 моль) (в течение примерно 4 минут).
К полученной таким образом суспензии 1/2 сульфата соединения (4) добавляли 6,2 кг (69,64 моль) дицианамида натрия и затем нагревали до кипения с обратным холодильником в течение примерно 3 часов. Полученную реакционную смесь охлаждали примерно до 20-40°C и ее органический слой последовательно промывали примерно 45 литрами каждого из следующих составов: (i) 5% водный раствор едкого натра, (ii) 5% хлористоводородная кислота, (iii) 5% водный раствор бикарбоната натрия и (iv) 5% водный раствор соли.
Полученный описанным способом раствор соединения (2) в н-бутилацетате охлаждали примерно до 15°C и к нему при перемешивании и температуре, не превышающей 35°C, последовательно добавляли 11,6 кг соединения (3) (65,89 моль) и 6,7 кг концентрированной хлористоводородной кислоты. При кипячении полученного раствора с обратным холодильником отгоняли примерно 72 литра растворителя в течение примерно 6,5 часов. Реакционную смесь охлаждали примерно до 80°C и добавляли к ней 72 литра изопропанола и 18 кг очищенной воды. Затем реакционную смесь нагревали при перемешивании при температуре примерно 60°C до повторного растворения выпавших кристаллов. К полученному раствору при температуре примерно 44-55°C добавляли концентрированную хлористоводородную кислоту в количестве 14,1 кг, и смесь перемешивали в течение 1 часа без нагревания. Полученную смесь охлаждали до 10°C или ниже и затем отделяли выпавшие кристаллы. Полученные кристаллы суспендировали в 54 литрах изопропанола и полученную суспензию перемешивали при 10°C или ниже в течение примерно 1 часа, после чего отделяли кристаллы. Полученные таким образом кристаллы (дигидрохлорид) без высушивания добавляли к 225 литрам очищенной воды и смесь перемешивали при 25-35°C в течение примерно 2 часов и затем отделяли кристаллы. Полученные кристаллы промывали 45 литрами очищенной воды и подвергали вакуумной сушке (15 мм.Hg) примерно при 40°C, применяя конический сушильный аппарат и получая неочищенные кристаллы моногидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида 1/2 H2O. Выход: 21,26 кг (выход составлял 77,2% в пересчете на соединение (3)).
Пример 6: (Способ очистки)
Неочищенные кристаллы моногидрохлорида соединения A·1/2 H2O добавляли к 15% водному раствору этанола (смесь 179 литров очищенной воды и 32 литров этанола) и полученную смесь нагревали (при температуре 80°C или ниже) до растворения неочищенных кристаллов с последующим фильтрованием в горячем состоянии. Полученный фильтрат вновь кипятили с обратным холодильником для подтверждения полного растворения неочищенных кристаллов и затем охлаждали примерно до 35°C. После этого к раствору добавляли 76 г кристаллической затравки и смесь охлаждали до 20°C на 1 час и затем до 10°C на 30 минут. Выпавшие кристаллы отделяли центрифугированием и промывали 42 литрами очищенной воды в центрифуге. Полученные кристаллы высушивали при 40°C в течение 22 часов, получая моногидрохлорид соединения A·1/2 H2O.
Выход: 18,34 кг (выход составлял 96,5% в пересчете на неочищенные кристаллы моногидрохлорида соединения A·1/2 H2O);
Чистота (ВЭЖХ) 99,9% или выше.
Размер частиц: цель может быть достигнута, если кристаллы могут проходить через сито с ячейками 870 мкм.
Температура плавления: 173-174°C.
1H-ЯМР (ДМСО-d6) спектр: 0,85 ppm (т, J=6,8 Гц, 3Н), 1,10-1,50 ppm (м, 12Н), 2,92-3,08 ppm (м, 2Н), 4,33 ppm (д, J=6,3 Гц, 2Н), 6,80-7,20 ppm (ушир.с, 3Н), 7,30 ppm (д, J=8,4 Гц, 1Н), 7,48-7,62 ppm (м, 3Н), 7,70-7,90 ppm (ушир.с, 0,5Н).
ИК-спектр (KBr): 3316, 3190, 2928, 1584, 1549, 1152, 1032 и 723 см-1.
Термогравиметрия/дифференциальный термический анализ: Наблюдались три эндотермических пика в районе 40±10°C, 90±10°C и 170±5°C. Температуры эндотермических пиков незначительно различались для разных партий вещества; однако наблюдались три хорошо различимых пика.
Спектр дифракции рентгеновских лучей на порошке (2θ): 3,6°, 7,2°, 10,9°, 18,1° и 25,5°.
Пример 7: моногидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида
Проводили исследование с целью определения того, каким образом концентрация и температура водного раствора этанола влияют на превращение моногидрохлорида соединения A·1/2 H2O в безводную форму (кристаллическую форму I). Моногидрохлорид соединения A 1/2 H2O суспендировали в водном растворе этанола с концентрацией 30% или выше, и его превращение в кристаллическую форму I завершалось в течение 2 часов после инициирования реакции при 30°C. При 25°C образование кристаллов формы I наблюдалось в течение 2 часов после инициирования реакции, и превращение в кристаллическую форму I завершалось через 4 часа. При 20°C образование кристаллической формы I наблюдалось в течение 2 часов после инициирования реакции, но превращение в кристаллическую форму I не завершалось даже через 6 часов.
Такое же исследование проводили с использованием 15% водного раствора этанола. При 35°C образование кристаллической формы I наблюдали через 4 часа после инициирования реакции при 35°C, и при 40°C превращение в кристаллическую форму I завершалось в течение 2 часов. Если использовали 10% водный раствор этанола, превращение в кристаллическую форму I не завершалось даже через 6 часов даже после повышения температуры до 57°C.
Из описанного выше исследования становится ясно, что моногидрохлорид соединения A·1/2 H2O может быть полностью превращен в кристаллическую форму I при суспендировании в водном растворе этанола, имеющем концентрацию 30% или выше и перемешивании полученной суспензии при температуре в диапазоне от 30 до 45°C в течение 2 часов или более. Кроме того, становится ясно, что если моногидрохлорид соединения A·1/2 H2O суспендирован в 15% водном растворе этанола, трансформация в кристаллическую форму I может быть проведена путем перемешивания полученной суспензии при 40°C в течение 2 часов или более.
(кристаллическая форма I):
Температура плавления: 177-179°C;
Спектр дифракции рентгеновских лучей на порошке (2θ): 3,9°, 17,5°, 21,9° и 22,5°.
Сравнительный пример 1: моногидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида
Мезитилен в количестве 200 мл добавляли к 20 г соединения (2) и 20,2 г гидрохлорида соединения (3) и затем нагревали до кипения с обратным холодильником в течение 1,5 часов. После завершения реакции температуру реакционной смеси понижали до комнатной и из нее удаляли мезитилен. К остатку добавляли 10% водный раствор этанола в количестве 200 мл. Полученную смесь нагревали и последовательно промывали 10% раствором этанола, водой и изопропиловым эфиром, получая 28,1 г неочищенного продукта. Полученный неочищенный продукт подвергали перекристаллизации с применением этилацетата, получая 22,1 г моногидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида в форме белых призматических кристаллов.
Выход составлял 53,1% в пересчете на соединение (2) и 56,9% в пересчете на соединение (3).
Сравнительный пример 2: моногидрохлорид 1-(3,4-дихлорбензил)-5-октилбигуанида
Неочищенный продукт (4 г), полученный в сравнительном примере 1, растворяли в 60 мл 15% EtOH при нагревании и затем охлаждали до 40°C с последующим перемешиванием при этой температуре в течение 4 часов. Выпавшие кристаллы отделяли фильтрованием, получая 3,6 г моногидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида. Выход составлял 90% в пересчете на неочищенный продукт.
Температура плавления: белые кристаллы, температура плавления 169-170°C.
ИК-спектр (KBr): 3314, 3176, 2920, 1595, 1545, 1146, 1027 и 723 см-1.
Термогравиметрия/дифференциальный термический анализ: Наблюдались два сильных эндотермических пика в районе 110±5°C и 170±5°C. Температуры эндотермических пиков незначительно различались для разных партий вещества; однако наблюдались два хорошо различимых пика.
Сравнительный пример 3: применение толуола в качестве реакционного растворителя
К 15 мл толуола добавляли 1 г гидрохлорида октиламина (6,0 ммоль) и 0,56 г NaN(CN)2 (6,3 ммоль) и затем кипятили с обратным холодильником в течение 2 часов. К полученной смеси добавляли 1,2 г 3,4-дихлорбензиламина (5,7 ммоль), после чего дополнительно кипятили с обратным холодильником в течение 2,5 часов. Полученную реакционную смесь исследовали с помощью ВЭЖХ-анализа. Полученная смесь помимо моногидрохлорида 1-(3,4-дихлорбензил)-5-октилбигуанида (5,85 минут, 55%), содержала 1,5-диоктилбигуанид (11,40 минут, 9%), 1,5-бис(3,4-дихлорбензил)бигуанид (3,46 минут, 6%), 1-(3,4-дихлорбензил)-3-цианогуанидин (2,1 минуты, 4%) и 1-октил-3-цианогуанидин (2,60 минут, 11%).
Условия ВЭЖХ-анализа:
Колонка YMCAM302 4,6 мм внутр.диаметр × 150 мм №188.
Элюент: MeCN/0,05 М водный раствор 1-октансульфоната натрия/уксусная кислота = 700/300/1.
Claims (7)
1. Способ получения 1-(3,4-дихлорбензил)-5-октилбигуанида, представленного формулой (1)

или его соли, причем способ включает взаимодействие 1-циано-3-октилгуанидина, представленного формулой (2)

или его соли с 3,4-дихлорбензиламином, представленным формулой (3)

или его солью, в сложноэфирном органическом растворителе.
или его соли, причем способ включает взаимодействие 1-циано-3-октилгуанидина, представленного формулой (2)
или его соли с 3,4-дихлорбензиламином, представленным формулой (3)
или его солью, в сложноэфирном органическом растворителе.
2. Способ по п.1, где сложноэфирный органический растворитель представляет собой, по меньшей мере, один растворитель, выбранный из группы, состоящей из н-бутилацетата, изобутилацетата, втор-бутилацетата, трет-бутилацетата, н-пентилацетата, изопентилацетата и н-пропилпропионата.
3. Способ по п.2, где сложноэфирный органический растворитель представляет собой н-бутилацетат.
4. Способ по любому из пп.1-3, где реакцию проводят в присутствии кислоты.
5. Способ по п.4, где кислота представляет собой по меньшей мере одну кислоту, выбранную из группы, состоящей из хлористоводородной кислоты, серной кислоты, фосфорной кислоты и бромистоводородной кислоты.
6. Способ получения 1-(3,4-дихлорбензил)-5-октилбигуанида, представленного формулой (1)

или его соли, где способ включает стадии взаимодействия н-октиламина, представленного формулой (4)
H2N-(CH2)7-CH3 (4)
или его соли с соединением, представленным формулой (5)
M-N(CN)2 (5)
где М представляет собой щелочной металл, или его солью в сложноэфирном органическом растворителе с получением 1-циано-3-октилгуанидина, представленного формулой (2)

или его соли;
и взаимодействия 1-циано-3-октилгуанидина или его соли, полученных на предыдущей стадии, с 3,4-дихлорбензил амином, представленным формулой (3)
или его солью в сложноэфирном органическом растворителе.
или его соли, где способ включает стадии взаимодействия н-октиламина, представленного формулой (4)
H2N-(CH2)7-CH3 (4)
или его соли с соединением, представленным формулой (5)
M-N(CN)2 (5)
где М представляет собой щелочной металл, или его солью в сложноэфирном органическом растворителе с получением 1-циано-3-октилгуанидина, представленного формулой (2)
или его соли;
и взаимодействия 1-циано-3-октилгуанидина или его соли, полученных на предыдущей стадии, с 3,4-дихлорбензил амином, представленным формулой (3)
или его солью в сложноэфирном органическом растворителе.
7. Способ получения 1-(3,4-дихлорбензил)-5-октилбигуанида, представленного формулой (1)

или его соли, где способ включает взаимодействие н-октиламина, представленного формулой (4)
H2N-(CH2)7-CH3 (4)
или его соли с соединением, представленным формулой (5)
M-N(CN)2 (5)
где М представляет собой щелочной металл, или его солью в сложноэфирном органическом растворителе с получением 1-циано-3-октилгуанидина, представленного формулой (2)

или его соли;
и последующее без выделения 1-циано-3-октилгуанидина или его соли добавление 3,4-дихлорбензиламина, представленного формулой (3)

или его соли в реакционную смесь для осуществления взаимодействия 1-циано-3-октилгуанидина или его соли с 3,4-дихлорбензиламином или его солью.
или его соли, где способ включает взаимодействие н-октиламина, представленного формулой (4)
H2N-(CH2)7-CH3 (4)
или его соли с соединением, представленным формулой (5)
M-N(CN)2 (5)
где М представляет собой щелочной металл, или его солью в сложноэфирном органическом растворителе с получением 1-циано-3-октилгуанидина, представленного формулой (2)
или его соли;
и последующее без выделения 1-циано-3-октилгуанидина или его соли добавление 3,4-дихлорбензиламина, представленного формулой (3)
или его соли в реакционную смесь для осуществления взаимодействия 1-циано-3-октилгуанидина или его соли с 3,4-дихлорбензиламином или его солью.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006-232922 | 2006-08-30 | ||
| JP2006232922 | 2006-08-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2009111276A RU2009111276A (ru) | 2010-10-10 |
| RU2440978C2 true RU2440978C2 (ru) | 2012-01-27 |
Family
ID=38871529
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2009111276/04A RU2440978C2 (ru) | 2006-08-30 | 2007-08-28 | Способ получения 1-(3, 4-дихлорбензил)-5-октилбигуанида или его соли |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US7868207B2 (ru) |
| EP (1) | EP2066623B1 (ru) |
| JP (1) | JP5078993B2 (ru) |
| KR (1) | KR101087454B1 (ru) |
| CN (1) | CN101506152B (ru) |
| AR (1) | AR062576A1 (ru) |
| AU (1) | AU2007289597B2 (ru) |
| BR (1) | BRPI0715831A2 (ru) |
| CA (1) | CA2661272C (ru) |
| ES (1) | ES2422161T3 (ru) |
| HK (1) | HK1130468A1 (ru) |
| IL (1) | IL196982A (ru) |
| MX (1) | MX2009002258A (ru) |
| MY (1) | MY146360A (ru) |
| RU (1) | RU2440978C2 (ru) |
| TW (1) | TWI385143B (ru) |
| WO (1) | WO2008026757A1 (ru) |
| ZA (1) | ZA200900838B (ru) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007062222A2 (en) | 2005-11-22 | 2007-05-31 | University Of South Florida | Inhibition of cell proliferation |
| US20100184851A1 (en) * | 2008-08-29 | 2010-07-22 | University Of South Florida | Inhibition of cell proliferation |
| ES2734299T3 (es) * | 2015-09-11 | 2019-12-05 | Bayer Cropscience Ag | Procedimiento para producir sales de biguanida y s-triazinas |
| JPWO2021132365A1 (ru) * | 2019-12-26 | 2021-07-01 | ||
| US20220251026A1 (en) * | 2019-12-26 | 2022-08-11 | Showa Denko K.K. | Method for producing highly polymerizable n-vinyl carboxlyic acid amide monomer |
| US11718579B2 (en) | 2019-12-26 | 2023-08-08 | Showa Denko K.K. | Method for producing highly polymerizable N-vinyl carboxylic acid amide monomer |
| JPWO2021132367A1 (ru) * | 2019-12-26 | 2021-07-01 | ||
| JPWO2021132363A1 (ru) * | 2019-12-26 | 2021-07-01 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5286905A (en) * | 1990-12-28 | 1994-02-15 | Idemitsu Kosan Company Limited | Process for producing biguanide derivative |
| CA2064664C (en) * | 1991-04-05 | 2000-01-11 | Hiroshi Ishikawa | Biguanide derivatives, manufacturing method thereof, and disinfectants containing the derivatives |
| JPH04308562A (ja) * | 1991-04-05 | 1992-10-30 | Otsuka Pharmaceut Co Ltd | ビスビグアナイド誘導体とこの誘導体を含有した消毒薬 |
| JP2662343B2 (ja) * | 1991-06-19 | 1997-10-08 | 大塚製薬株式会社 | モノビグアナイド誘導体とこの誘導体を含有した消毒薬 |
| JP3493692B2 (ja) * | 1993-09-09 | 2004-02-03 | 三菱マテリアル株式会社 | ビスビグアナイド系化合物およびそれを含む殺菌剤 |
| DK1634589T3 (da) * | 2003-05-28 | 2011-04-18 | Otsuka Pharma Co Ltd | Vandig olanexidinopløsning, fremgangsmåde til fremstilling heraf og desinfektionsmiddel |
-
2007
- 2007-08-28 JP JP2009509802A patent/JP5078993B2/ja not_active Expired - Fee Related
- 2007-08-28 EP EP07806580.2A patent/EP2066623B1/en not_active Not-in-force
- 2007-08-28 RU RU2009111276/04A patent/RU2440978C2/ru not_active IP Right Cessation
- 2007-08-28 MX MX2009002258A patent/MX2009002258A/es active IP Right Grant
- 2007-08-28 ZA ZA200900838A patent/ZA200900838B/xx unknown
- 2007-08-28 AU AU2007289597A patent/AU2007289597B2/en not_active Ceased
- 2007-08-28 US US12/439,481 patent/US7868207B2/en not_active Expired - Fee Related
- 2007-08-28 CA CA2661272A patent/CA2661272C/en not_active Expired - Fee Related
- 2007-08-28 ES ES07806580T patent/ES2422161T3/es active Active
- 2007-08-28 WO PCT/JP2007/067107 patent/WO2008026757A1/en active Application Filing
- 2007-08-28 KR KR1020097006445A patent/KR101087454B1/ko not_active IP Right Cessation
- 2007-08-28 BR BRPI0715831-9A patent/BRPI0715831A2/pt not_active IP Right Cessation
- 2007-08-28 MY MYPI20090486A patent/MY146360A/en unknown
- 2007-08-28 TW TW096131817A patent/TWI385143B/zh not_active IP Right Cessation
- 2007-08-28 CN CN2007800317486A patent/CN101506152B/zh not_active Expired - Fee Related
- 2007-08-29 AR ARP070103824A patent/AR062576A1/es unknown
-
2009
- 2009-02-09 IL IL196982A patent/IL196982A/en active IP Right Grant
- 2009-11-09 HK HK09110398.8A patent/HK1130468A1/xx not_active IP Right Cessation
Non-Patent Citations (1)
| Title |
|---|
| Ken Umehara et al. "In vitro characterization of the oxidative cleavage of the octyl side chain of olanexidine, a novel antimicrobial agent, in dog liver microsomes" DRUG METABOLISM AND DISPOSITION 200, vol.28, №12, p.1417-1424. Tsubouchi, H., Ohguro, K. "Synthesis and structure-activity relationships of novel antiseptics" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS 1997, vol.7, №13, p.1721-1724. * |
Also Published As
| Publication number | Publication date |
|---|---|
| IL196982A (en) | 2013-03-24 |
| CN101506152B (zh) | 2012-11-21 |
| KR20090045409A (ko) | 2009-05-07 |
| KR101087454B1 (ko) | 2011-11-25 |
| IL196982A0 (en) | 2009-11-18 |
| CA2661272C (en) | 2014-03-18 |
| MY146360A (en) | 2012-08-15 |
| US7868207B2 (en) | 2011-01-11 |
| TWI385143B (zh) | 2013-02-11 |
| RU2009111276A (ru) | 2010-10-10 |
| AU2007289597A1 (en) | 2008-03-06 |
| WO2008026757A1 (en) | 2008-03-06 |
| JP2010502567A (ja) | 2010-01-28 |
| MX2009002258A (es) | 2009-03-20 |
| CA2661272A1 (en) | 2008-03-06 |
| US20090270653A1 (en) | 2009-10-29 |
| TW200817316A (en) | 2008-04-16 |
| EP2066623B1 (en) | 2013-06-19 |
| AU2007289597B2 (en) | 2011-03-03 |
| EP2066623A1 (en) | 2009-06-10 |
| ZA200900838B (en) | 2010-05-26 |
| HK1130468A1 (ru) | 2009-12-31 |
| JP5078993B2 (ja) | 2012-11-21 |
| ES2422161T3 (es) | 2013-09-09 |
| BRPI0715831A2 (pt) | 2013-07-23 |
| AR062576A1 (es) | 2008-11-19 |
| CN101506152A (zh) | 2009-08-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2440978C2 (ru) | Способ получения 1-(3, 4-дихлорбензил)-5-октилбигуанида или его соли | |
| JP6061158B2 (ja) | 6−(7−((1−アミノシクロプロピル)メトキシ)−6−メトキシキノリン−4−イルオキシ)−n−メチル−1−ナフトアミド、またはそれの薬学的に許容される塩の合成中間体およびその使用 | |
| CA3040720C (en) | Method for producing substituted 5-fluoro-1h-pyrazolopyridines | |
| WO2013049605A1 (en) | Processes for the preparation of an intermediate in the synthesis of eltrombopag | |
| CN112047888B (zh) | 一种合成恩杂鲁胺的方法 | |
| JP6985179B2 (ja) | プロリンアミド化合物の製造方法 | |
| MXPA03003459A (es) | Base venlafaxina cristalina y polimorfos novedosos de clorhidrato de venlafaxina, procesos para su preparacion. | |
| EP4375282A1 (en) | Preparation method for hepatitis b virus nucleocapsid inhibitor | |
| WO2011124638A1 (en) | Pimobendan manufacturing process | |
| NZ724215B2 (en) | Method for producing substituted 5-fluoro-1h-pyrazolopyridines | |
| JPS62132870A (ja) | N−(2−ヒドロキシエチル)−2−メチル−3−カルボン酸アミド−キノキサリン−1,4−ジ−n−オキシドの精製法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20180829 |