KR20190016516A - 페닐 프로판아미드 유도체와 이를 제조하는 방법, 및 이를 약학적으로 사용하는 방법 - Google Patents
페닐 프로판아미드 유도체와 이를 제조하는 방법, 및 이를 약학적으로 사용하는 방법 Download PDFInfo
- Publication number
- KR20190016516A KR20190016516A KR1020187037174A KR20187037174A KR20190016516A KR 20190016516 A KR20190016516 A KR 20190016516A KR 1020187037174 A KR1020187037174 A KR 1020187037174A KR 20187037174 A KR20187037174 A KR 20187037174A KR 20190016516 A KR20190016516 A KR 20190016516A
- Authority
- KR
- South Korea
- Prior art keywords
- cycloalkyl
- alkyl
- amino
- compound
- heterocyclyl
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1016—Tetrapeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/07—Tetrapeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/06—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/06—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents
- C07K1/061—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/06—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents
- C07K1/061—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups
- C07K1/062—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups for alpha- or omega-carboxy functions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/1008—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atoms, i.e. Gly, Ala
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/101—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms, e.g. Val, Ile, Leu
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Analytical Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
본 발명은, 식(I)으로 표시된 페닐 프로판아미드 유도체, 이 유도체의 제조 방법, κ-오피오이드 수용체(KOR) 작용제로서 유도체의 사용 방법, 및 통증 및 통증 관련 질병을 치료 및/또는 예방하기 위한 의약품을 제조하기 위해 유도체를 사용하는 방법을 제공한다.

Description
본 발명은 의약 분야에 속하고, 페닐 프로판아미드 유도체, 그 제조 방법, 및 의약에서 이를 사용하는 방법에 관한 것이다. 특히, 본 발명은, 식(I)으로 표시된 페닐 프로판아미드 유도체, 그 제조 방법, 및 이를 포함하는 약학 조성물, κ-오피오이드(opioid) 수용체(KOR) 작용제(agonist)로서 이를 사용하는 방법, 및 통증 및 통증 관련 질병을 치료 및/또는 예방하기 위한 약제의 제조에 이를 사용하는 방법에 관한 것이다.
오피오이드 수용체는 G-단백질-결합 수용체의 중요한 종류이고, 내인성 오피오이드 펩티드와 오피오이드의 조합의 표적이다. 활성화된 오피오이드 수용체는 신경계 면역 및 내분비계에서 조절 역할을 한다. 오피오이드는 현재 가장 강력하고 가장 일반적으로 사용되는 중추 진통제이다. 내인성 오피오이드 펩티드는 포유동물에서 자연적으로 발생하는 오피오이드 활성 물질이다. 현재 알려져 있는 내인성 오피오이드 펩티드는 엔케팔린(enkephalins), 엔돌핀, 다이놀핀(dynorphins) 및 네오모르핀으로 대략 분류된다(Pharmacol Rev 2007; 59: 88-123). 중추 신경계에는 상응하는 오피오이드 수용체, 즉, μ, δ, κ 수용체 등이 있다.
κ-오피오이드 수용체(KOR)는 380개의 아미노산으로 이루어지고, 다이놀핀이 그 내인성 리간드이다. 이것은 감각 뉴런, 후근 신경절 세포(dorsal root ganglion cells) 및 주요 구심성 뉴런(primary afferent neurons)에서 발현되고, 통증, 신경 내분비, 정서적 행동 및 인지와 같은 중요한 생리적 활성에 관여한다. 인간 KOR은 OPRK1 유전자에 의해 코딩되고 염색체 8q11-12에 위치하는 것으로 현재 알려져 있다(Simonin F, Gaveriaux Ruff C, Kieffer BL, 등의, Proc Natl Acad Sci USA 1995, 92(15): 7006-10). KOR 활성화는 G 단백질 Gi/G0와 결부되고, 이는 포스포디에스테라아제(phosphodiesterase) 활성을 증가시키고, 아데닐레이트 시클라아제(adenylate cyclase)의 활성을 억제하며, 세포내 cAMP 수준을 감소시킴으로써, 신경 억제를 생성한다. KOR 작용제는 수용체에 반복적으로 작용하여 탈감각화(desensitization)를 일으키고, 아데닐레이트 시클라아제 활성의 억제를 감소시킨다(Raynor K, Kong H, Hines J, et al. J Pharmacol Exp Ther, 1994, 270:1381-6). KOR은 또한 내향성 정류기(inward rectifier) 칼륨 채널 및 N-형 칼슘 이온 채널과 결부된다{Henry DJ, Grandy DK, Lester HA, Davidson N, Chavkin C (Mar 1995) Molecular Pharmacology 47 (3): 551-7}. KOR 작용제는 말초 감각 신경 종말로부터 전-손상(pre-hurt) 및 전-염증(pre-inflammatory) 물질(P)의 (칼슘-의존성) 방출을 억제할 수 있고, 이것은 이들의 통각억제 및 항염증 효과를 일으킬 수 있다. 다이놀핀 이외에도, 다양한 천연 알칼로이드와 합성 리간드가 KOR에 결합할 수 있다. KOR은 자연적인 중독 통제 메커니즘을 제공하므로, 수용체 작용제로서의 약물은 약물 중독 치료에 대한 잠재력을 갖는다.
이러한 관찰, 예를 들어, 설치류 당뇨병성 신경병증에서 KOR 작용제인 아시마돌린(asimadoline)의 효과(Jolivalt 등의, Diabetologia 2006, 49(11): 2775-85; Epub 8월 19일) 및 신경병성 통증을 가진 쥐의 만성 압축 손상(CCI) 모델에서 KOR 작용제인 U-50488 효과와 이 효과에 대한 오피오이드 길항제인 날록손(naloxone)의 차단의 영향(Bileviciute-Ljungar 등의, Eur. J. Pharm 2004. 494 :139-46)은, 당뇨병, 바이러스 및 화학요법으로 인해 발생하는 신경병성 통증의 치료에 KOR 작용제를 사용하는 것을 지지한다. 월경불순 및 자궁내막증과 같은 부인과 질환을 포함하는, 내장 통증의 치료 또는 예방에 KOR 작용제를 사용하는 것이 또한 평가되었다(Riviere, Br. J. Pharmacol 2004. 141: 1331-4).
κ-오피오이드 작용제는 물의 신장 배설을 증가시키고 소변의 나트륨 배설을 감소시킨다{즉, 수분 촉진(water-promoting)으로도 알려진, 선택적 수분 이뇨(water diuresis)를 일으킴}. 많은 연구자들은 이 효과가 바소프레신의 뇌하수체 분비의 억제 때문인 것으로 믿는다. 중추적으로 작용하고 주장된 말초 선택적 κ 오피오이드를 비교하는 연구는, 혈액 뇌 장벽 내의 KOR이 이러한 효과를 매개하고 있는 것으로 결론을 내렸다. 일부 연구자들은, 노시셉틴(nociceptin) 펩티드 또는 주변(periphery)의 노시셉틴 수용체에 작용하는 하전된 펩티드 컨쥬게이트로 저나트륨 혈증을 치료할 것을 제안하였고, 이 노시셉틴 수용체는 KOR과 관련이 있지만 상이하다(DR Kapusta, Life Sci., 60:15-21, 1997).
현재 KOR 작용제를 개시하고 있는 특허 출원은, WO20071398, WO2008060552, WO09932510, WO2013184794, WO2014089019, WO2014184356 및 WO2015065867을 포함한다.
κ-오피오이드 수용체(KOR 수용체) 작용제는 제약 산업에서 양호한 적용 가능성을 갖는다. 더 나은 치료 효과를 이루고 시장의 요구를 더 잘 충족시키기 위해, 본 발명자는 높은 효과와 낮은 독성을 갖는 새로운 세대의 KOR 수용체 작용제를 개발하기 희망한다. 본 발명은, 놀랍게도 우수한 효과와 작용을 나타내는, 신규한 κ 오피오이드 수용체(KOR 수용체) 작용제 화합물(코어 구조에서 글리신의 아미노기의 추가 변형을 갖는)을 제공할 것이다. 특히, 글리신의 아미노기 상의 치환기가 치환 또는 비치환 에틸렌기일 때, 화합물은 예상치 못한 효과를 갖는다.
본 발명은 식(I)의 화합물:
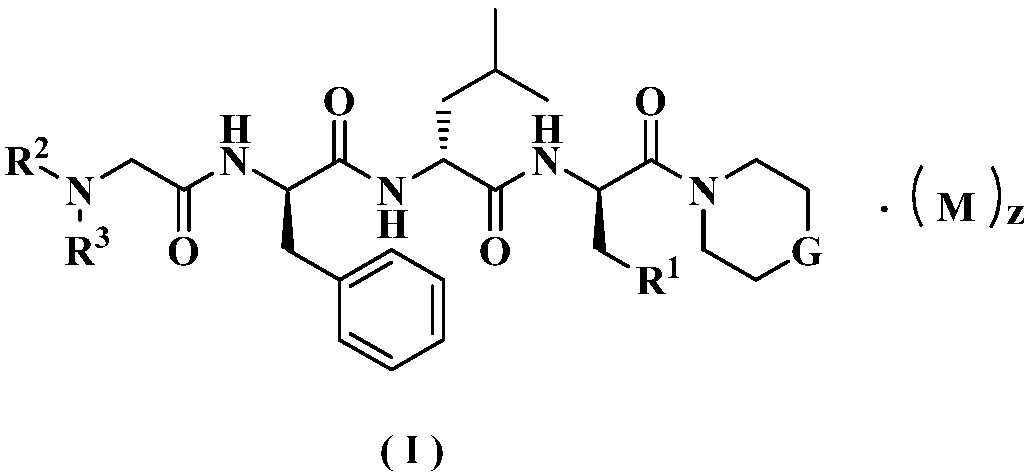
또는 그 토토머(tautomer), 메소머(mesomer), 라세미체(racemate), 거울상 이성질체(enantiomer), 부분 입체 이성질체(diastereomer), 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염에 관한 것이고,
상기 식에서:
M은 무기산 또는 유기산, 바람직하게는 유기산, 더 바람직하게는 트리플루오로아세트산이고;
G는 O, -NR4 및 -CR5R6으로 이루어진 군으로부터 선택되며;
R1은 수소, 알킬, 알콕시, 할로알킬, 할로겐, 아미노, 니트로, 히드록시, 시아노, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7 및 -NR8R9로 이루어진 군으로부터 선택되고, 여기에서, 알킬, 할로알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R2는 수소, 알킬, 알콕시, 할로알킬, 시클로알킬, 시클로알킬알킬, 헤테로시클릴, 헤테로시클릴알킬, 아릴, 아릴알킬, 헤테로아릴, 헤테로아릴알킬, -OR7, -C(O)R7 및 -C(O)OR7로 이루어진 군으로부터 선택되고, 여기에서, 알킬, 할로알킬, 시클로알킬, 시클로알킬알킬, 헤테로시클릴, 헤테로시클릴알킬, 아릴, 아릴알킬, 헤테로아릴 및 헤테로아릴알킬은 각각 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R3은 수소, 알킬, 알콕시, 할로알킬, 시클로알킬, 시클로알킬알킬, 헤테로시클릴, 헤테로시클릴알킬, 아릴, 아릴알킬, 헤테로아릴, 헤테로아릴알킬, -OR7, -C(O)R7 및 -C(O)OR7로 이루어진 군으로부터 선택되고, 여기에서, 알킬, 할로알킬, 시클로알킬, 시클로알킬알킬, 헤테로시클릴, 헤테로시클릴알킬, 아릴, 아릴알킬, 헤테로아릴 및 헤테로아릴알킬은 각각 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R4는 수소, 알킬, 할로알킬, 시클로알킬, 알콕시, 히드록시알킬, 히드록시, 아미노, 알콕시카르보닐, 헤테로시클릴, 아릴, 헤테로아릴, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7, -NR8R9 및 -NHC(O)NR8R9로 이루어진 군으로부터 선택되고, 여기 에서, 알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로겐, 히드록시, 아미노, 알콕시카르보닐, 니트로, 시아노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R5와 R6은 수소, 알킬, 알콕시, 히드록시알킬, 히드록시, 아미노, 알콕시카르보닐, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7, -NR8R9 및 -NHC(O)NR8R9로 이루어진 군으로부터 각각 독립적으로 선택되고, 여기에서, 알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로겐, 히드록시, 아미노, 알콕시카르보닐, 니트로, 시아노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R7은 수소, 알킬, 아미노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고, 여기에서, 알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로겐, 히드록시, 아미노, 니트로, 시아노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R8과 R9는 수소, 알킬, 알콕시, 히드록시알킬, 히드록시, 아미노, 알콕시카르보닐, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 각각 독립적으로 선택되고, 여기에서, 알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로겐, 히드록시, 아미노, 알콕시카르보닐, 니트로, 시아노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
z는 0, 1, 2, 3 또는 4이고;
m은 0, 1 또는 2이다.
본 발명의 바람직한 구현예에서, 식(I)의 화합물은 또한 다음 식(II)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
M, G, R2, R3 및 z는 식(I)에서 정의된 바와 같다.
본 발명의 바람직한 구현예에서, 식 (I) 또는 (II)의 화합물은 또한 식(III)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
M, G, R2 및 z는 식(I)에서 정의된 바와 같다.
본 발명의 바람직한 구현예에서, 식 (I), (II) 또는 (III)의 화합물은 또한 식(IV)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
M, R2 및 z는 식(I)에서 정의된 바와 같다.
본 발명의 바람직한 구현예에서, 식 (I), (II), (III) 또는 (IV)의 화합물에서, R2는 아릴알킬, 시클로알킬알킬 및 시클로알킬로 이루어진 군으로부터 선택되고, 여기에서, 아릴알킬, 시클로알킬알킬 및 시클로알킬은 각각 알킬, 시클로알킬 및 아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환된다.
본 발명의 바람직한 구현예에서, 식 (I), (II) 또는 (III)의 화합물은 또한 식(III-A)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
G는 O 또는 CR5R6; 바람직하게는 CR5R6이고;
R10은 수소, 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되며;
R11과 R12는 동일하거나 또는 상이하고, 각각 수소, 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 독립적으로 선택되거나;
또는 R10과 R11은 함께 시클로알킬을 형성하거나;
또는 R11과 R12는 함께 시클로알킬을 형성하고;
R13은 수소, 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되며;
s는 0, 1 또는 2이고;
R5 내지 R6, M 및 z는 식(I)에서 정의된 바와 같다.
본 발명의 바람직한 구현예에서, 식 (I), (II), (III), (IV) 또는 (III-A)의 화합물은 또한 식(IV-A)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
R10 내지 R13, M, z 및 s는 식(III-A)에서 정의된 바와 같다.
본 발명의 바람직한 구현예에서, 식 (I), (II), (III), (IV), (III-A) 또는 (IV-A)의 화합물은 또한 식(IV-B)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
R10 내지 R11, R13, M, z 및 s는 식(III-A)에서 정의된 바와 같다.
본 발명의 바람직한 구현예에서, 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물에서, z는 0 또는 1이다.
식(I)의 전형적인 화합물은 다음 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염을 포함하지만, 이에 제한되지 않는다:




다른 양상에서, 본 발명은 또한 식(II)의 화합물을 제조하기 위한 중간체인 식(V)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염에 관한 것이고,
상기 식에서:
Ra는 아미노-보호기, 바람직하게는 t-부톡시카르보닐, 9-플루오레닐메톡시카르보닐, 알릴옥시카르보닐, 트리클로로에톡시카르보닐, 트리메틸실릴에톡시카르보닐, 벤질옥시카르보닐, p-메틸벤젠설포닐, p-니트로벤젠설포닐 또는 tert-부틸(즉, Boc, Fmoc, Alloc, Teoc, CBz, 토실(Tosyl), 노실(Nosyl) 및 t-Bu)이고;
G, R2 및 R3은 식(II)에서 정의된 바와 같다.
다른 양상에서, 본 발명은 또한 식(II)의 화합물을 제조하는 공정에 관한 것으로, 다음의 단계:

산성 조건 하에서 식(V)의 화합물 상의 보호기 Ra를 제거하여 식(II)의 화합물을 수득하는 단계를 포함하고;
상기 식에서:
M, G, z, R2 및 R3은 식(II)에서 정의된 바와 같고, Ra는 식(V)에서 정의된 바와 같다.
다른 양상에서, 본 발명은 또한 식(III)의 화합물을 제조하기 위한 중간체인 식(VI)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염에 관한 것이고,
상기 식에서:
Ra는 아미노-보호기, 바람직하게는 t-부톡시카르보닐, 9-플루오레닐메톡시카르보닐, 알릴옥시카르보닐, 트리클로로에톡시카르보닐, 트리메틸실릴에톡시카르보닐, 벤질옥시카르보닐, p-메틸벤젠설포닐, p-니트로벤젠설포닐 또는 tert-부틸이고;
G와 R2는 식(III)에서 정의된 바와 같다.
다른 양상에서, 본 발명은 또한 식(III)의 화합물을 제조하는 공정에 관한 것으로, 다음의 단계:

산성 조건 하에서 식(VI)의 화합물 상의 보호기 Ra를 제거하여 식(III)의 화합물을 수득하는 단계를 포함하고;
상기 식에서:
M, G, z, 및 R2는 식(III)에서 정의된 바와 같고, Ra는 식(VI)에서 정의된 바와 같다.
산성 조건을 제공하는 산성 시약은 바람직하게는 트리플루오로아세트산 또는 1,4-디옥산 중의 염화수소의 용액이다.
또한, 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물에서 z가 0이 아닌 경우, 선택적으로, 유리 상태 생성물, 즉, 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물을 수득하기 위해 유리 반응(free reaction)을 실행하도록 약 염기가 첨가된다.
다른 양상에서, 본 발명은 또한 상기 언급된 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염의 치료 유효량, 및 하나 이상의 약학적으로 허용 가능한 담체(carrier), 희석제 또는 부형제를 포함하는 약학 조성물에 관한 것이다.
본 발명은 또한 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염을, 하나 이상의 약학적으로 허용 가능한 담체, 희석제 또는 부형제와 혼합하는 단계를 포함하는, 상기 언급된 조성물을 제조하는 공정에 관한 것이다.
일 구현예에서, 본 발명의 약학 조성물은, 오피오이드, 카나비노이드(cannabinoids), 항우울제, 항경련제(anticonvulsants), 신경 안정제(tranquilizers), 코르티코스테로이드(corticosteroids), 이온 채널 차단제 또는 비스테로이드성 항염증제(NSAID) 중 하나 이상을 추가로 포함한다.
본 발명은 또한 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염, 또는 이를 포함하는 약학 조성물을, κ 오피오이드 수용체(KOR 수용체)를 작용(agonizing) 또는 길항(antagonizing)시키기 위한 약제의 제조에 사용하는 방법에 관한 것이다.
본 발명은 또한 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염, 또는 이를 포함하는 약학 조성물을, κ 오피오이드 수용체(KOR 수용체) 작용제 매개 및 관련 질병을 예방 및/또는 치료하기 위한 약제의 제조에 사용하는 방법에 관한 것으로, 여기에서 κ 오피오이드 수용체(KOR 수용체) 작용제 매개 및 관련 장애는 바람직하게는 통증, 염증, 가려움증, 부종(edema), 저나트륨 혈증(hyponatremia), 저칼륨 혈증(hypokalemia), 장 폐쇄증, 기침 및 녹내장으로 이루어진 군으로부터 선택되고, 더 바람직하게는 통증이다.
본 발명은 또한 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염, 또는 이를 포함하는 약학 조성물을, 포유동물(예를 들어, 인간)의 통증 및 통증 관련 질병을 예방 및/또는 치료하기 위한 약제의 제조에 사용하는 방법에 관한 것으로, 여기에서 통증은 수술후 통증, 암에 의해 발생한 통증, 신경병성 통증(neuropathic pain), 외상성 통증(traumatic pain) 및 염증에 의해 발생한 통증 등일 수 있다.
본 발명은 또한 κ 오피오이드 수용체(KOR 수용체)를 작용 또는 길항시키기 위한 방법에 관한 것으로, 본 발명의 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염의 치료 유효량을 이를 필요로 하는 환자에게 투여하는 단계를 포함한다.
본 발명은 또한 KOR 수용체 작용제 매개 및 관련 질병을 예방 및/또는 치료하기 위한 방법에 관한 것으로, 본 발명의 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염의 치료 유효량을 이를 필요로 하는 환자에게 투여하는 단계를 포함한다. 이 방법은 현저한 효능을 나타내고 부작용은 더 적게 나타낸다. 여기에서, κ 오피오이드 수용체(KOR 수용체) 작용제 매개 및 관련 장애는 통증, 염증, 가려움증, 부종, 저나트륨 혈증, 저칼륨 혈증, 장 폐쇄증, 기침 및 녹내장으로 이루어진 군으로부터 선택되고, 바람직하게는 통증이다.
본 발명은 또한 약제로서 사용하기 위한 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B), 특히 식 (I)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염에 관한 것이다.
본 발명은 또한 κ 오피오이드 수용체(KOR 수용체)를 작용 또는 길항시키는 데 사용하기 위한 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B), 특히 식 (I)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염, 또는 이를 포함하는 약학 조성물에 관한 것이다.
본 발명은 또한 KOR 수용체 작용제 매개 및 관련 질병을 예방 및/또는 치료하는 데 사용하기 위한 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염에 관한 것이다.
본 발명은 또한 포유동물(예를 들어, 인간)의 통증 및 통증 관련 질병을 예방 및/또는 치료하는 데 사용하기 위한 식 (I), (II), (III), (IV), (III-A), (IV-A) 또는 (IV-B)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염, 또는 이를 포함하는 약학 조성물에 관한 것이다. 여기에서, κ 오피오이드 수용체(KOR 수용체) 작용제 매개 및 관련 질병, 장애, 또는 상태는, 급성 또는 만성 통증, 염증, 가려움증, 저나트륨 혈증, 부종, 장 폐쇄증, 기침 및 녹내장을 포함하지만, 이에 제한되지 않는 임의의 κ 오피오이드 수용체(KOR 수용체) 작용제 매개 상태일 수 있다. 예를 들어, κ 오피오이드 수용체(KOR 수용체) 관련 통증은 신경병성 통증, 체성 통증(somatic pain), 내장 통증(visceral pain) 또는 피부 통증일 수 있다. 일부 질병, 장애 또는 상태는 하나를 초과하는 형태의 통증과 관련이 있다. 예를 들어, 수술 후 통증은 사용된 수술의 유형과 범위에 따라 신경병성 통증, 체성 통증, 내장 통증, 또는 피부 통증 중 일부 또는 전부일 수 있다.
본 발명에서 수반된 κ 오피오이드 수용체(KOR 수용체) 관련 염증은, 축농증, 류마티스성 관절염, 건초염, 점액낭염(bursitis), 힘줄염(tendonitis), 상완골 상과염(humeral epicondylitis), 유착성 관절낭염(adhesive capsulitis), 골수염, 골관절염, 염증성 장 질환(IBD), 과민성 대장 증후군(IBS), 안구 염증, 귀 염증, 또는 자가면역성 염증을 포함하지만, 이에 제한되지 않는 임의의 염증성 질병 또는 상태일 수 있다.
본 발명에서 수반된 κ 오피오이드 수용체(KOR 수용체) 관련 소양증(pruritus)은, 임의의 소양증 질병 및 상태, 예를 들어, 결막염 안구 가려움증과 같은 안구 가려움증, 가려움증, 및 말기 신장 질병(많은 환자들이 신장 투석을 받음)과 관련된 가려움증 및 다른 형태의 담즙정체{원발성 담관 간경화, 임신중 간내 담즙정체, 만성 콜레스테롤 간 질병, 요독증, 악성 담즙정체, 황달, 및 아토피성 피부염 또는 접촉성 피부염을 포함하는 습진(피부염), 피부 잡티, 적혈구 증가증(polycythemia), 편평태선(lichen planus), 만성 단순 이끼(chronic simple moss), 이 기생증(pediculosis), 갑상선 중독증, 무좀, 두드러기, 옴(scabies), 질염, 여드름 관련 항문 가려움증, 곤충 물림 가려움증, 및 약물에 의해 일어난 가려움증(μ 오피오이드에 의해 일어난 가려움증과 같은)과 같은 피부 상태를 포함하는}일 수 있다.
본 발명에서 수반된 κ 오피오이드 수용체(KOR 수용체) 관련 부종은, 울혈성 심장 질병으로 인해 일어난 부종이나 또는 항이뇨 호르몬(ADH)의 부적절한 분비 증후군에 의해 일어난 부종과 같은 임의의 부종성 질병 또는 상태일 수 있다.
본 발명에서 수반된 κ 오피오이드 수용체(KOR 수용체) 관련 장 폐쇄증은, 수술 후 장 폐쇄증 또는 오피오이드 유발 장 기능장애를 포함하지만, 이에 제한되지 않는 임의의 장 폐쇄성 질병 또는 상태일 수 있다.
본 발명에서 수반된 κ 오피오이드 수용체(KOR 수용체) 관련 신경병성 통증은, 임의의 신경병성 통증, 예를 들어, 삼차 신경통(trigeminal neuralgia), 당뇨병성 통증, 대상 포진 관련 통증과 같은 바이러스 유발성 통증, 화학 요법 유발 통증, 침습성 신경 전이 암 통증, 외상 및 수술 관련 신경병성 통증, 및 편두통과 같이 신경병리학적 요인을 갖는 다양한 두통 변종일 수 있다.
본 발명에서 수반된 κ 오피오이드 수용체(KOR 수용체) 관련 통증은, 안구 통증, 예를 들어, 굴절 각막 절제술(PRK), 안구 파열, 안저 골절(fundus fracture), 화학적 화상, 각막 상피 마모 또는 자극 후 눈 통증, 또는 결막염, 각막 궤양, 공막염(scleritis), 공막 염증, 공막 각막염, 안구 대상 포진, 간질성 각막염, 급성 홍채염, 건성 각결막염, 안와 봉와직염, 안와 의사종양, 천포창(pemphigus), 트라코마, 또는 포도막염과 관련된 안구 통증을 포함한다.
본 발명에서 수반된 κ 오피오이드 수용체(KOR 수용체) 관련 통증은 또한 인후통, 특히, 알레르기성 비염, 급성 기관지염, 감기, 접촉 궤양, 단순 헤르페스 바이러스 손상, 전염성 단핵증, 인플루엔자, 후두암, 급성 후두염, 급성 괴사성 궤양 치은염, 편도선 농양, 인두 작열감(pharyngeal burning), 인두염, 역류성 인두염, 급성 부비강염(acute sinusitis) 및 편도선염(tonsillitis)과 같은 염증 상태와 관련된 인후통을 포함한다.
κ 오피오이드 수용체(KOR 수용체) 관련 통증은, 관절염 통증, 신장 결석, 요결석 및 담관 결석 통증, 자궁경련, 월경불순, 자궁 내막증, 유방염, 소화불량, 수술 후 통증{예를 들어, 맹장 수술, 개방 대장 수술, 탈장 봉합술, 전립선 절제술, 대장 절제술, 위 절제술, 비장 절제술, 결장 절제술, 인공항문성형술(colostomy), 골반내 복강경 검사(pelvic laparoscopy), 난관 결찰술(tubal ligation), 자궁 적출술, 정관 절제술 또는 담낭 절제술에 의해 발생한 수술후 통증}, 의학적 치료 후 통증(예를 들어, 결장 내시경 검사, 방광경 검사, 자궁경 검사 또는 자궁 경부 또는 자궁 내막 생검 후 통증), 이염 통증(otitis pain), 전격성 암 통증, 및 IBD 또는 IBS와 같은 GI 장애 또는 기타 염증성 상태와 관련된 통증, 특히 내장 염증과 관련된 통증{예를 들어, 위식도 역류성 질병, 췌장염, 급성 신우신염, 궤양성 대장염, 담낭염, 간경변, 간 낭충증(hepatic cyst), 간염, 십이지장 궤양 또는 위궤양, 식도염, 위염, 위장염, 대장염, 게실염(diverticulitis), 장 폐쇄증, 난소 낭종, 골반 염증성 질병, 궤양 천공, 복막염, 전립선염, 간질성 방광염}, 또는 독과의 접촉에 의해 발생한 통증{예를 들어, 곤충 독소, 또는 살리실레이트(염) 또는 NSAIDs와 같은 약물}일 수 있다.
κ 오피오이드 수용체(KOR 수용체) 관련 저나트륨 혈증은, 저나트륨 혈증(저나트륨 상태)이 존재하는 임의의 질병 또는 상태일 수 있고, 예를 들어, 인간에서 혈장의 나트륨 농도가 135 mmol/ℓ 미만으로 존재하면, 이상(abnormality)이 단독으로 일어날 수 있거나, 또는 다른 의학적 상태의 합병증으로서 또는 나트륨 결핍을 일으키는 약물 사용의 결과로서 더 흔하게 보여지고, 여기에서, 저나트륨 혈증 관련 질병은, 폐, 십이지장, 췌장, 난소, 방광 및 요관의 암, 흉선종, 중피종, 기관지 선종, 유암종 종양, 신경절 신경종(ganglioneuroma) 및 유잉 육종(ewing's sarcoma)을 포함하는, 과도한 ADH 분비를 일으키는 종양 요인; 감염, 예를 들어, 폐렴(세균성 또는 바이러스성), 농양(폐 또는 뇌), 공포 형성(vacuolation)(아스페르길루스증), 결핵(폐 또는 뇌), 수막염(세균성 또는 바이러스성), 뇌염 및 AIDS; 혈관 요인, 예를 들어: 뇌혈관 경색 또는 출혈 및 해면 정맥동 색전증(cavernous sinus embolism); 신경학적 요인, 예를 들어, 길리안 바르 증후군(Guillan-Barre syndrome), 다발성 경화증, 진전 섬망증(delirium tremen), 근육 측부 경화증, 뇌수종, 정신병, 말초 신경병증, 두부 외상(폐쇄성 및 관통성), CNS 종양 또는 감염, 및 사상하부 삼투 수용체(hypothalamic osmoreceptor)에 영향을 미치는 CNS 손상; 뇌량의 무발생증(agenesis of the corpus callosum), 구순 구개열(cleft lip and palate) 및 기타 중간선 결함을 포함하는 선천성 기형; 대사성 요인, 예를 들어, 급성 간헐성 포르피린증(acute intermittent porphyria), 천식, 기흉 및 양압 호흡; 약물, 예를 들어, 티아지드 이뇨제, 파라세타몰, 바르비투레이트(barbiturates), 콜린, 에스트로겐, 경구 혈당 강하제(oral hypoglycemic agents), 바소프레신 또는 데스모프레신, 고용량 옥시토신 클로로프로파미드, 빈크리스틴, 카르바마제핀, 니코틴, 페노티아진, 시클로포스파미드, 트리시클릭 항우울제, 모노아민 옥시다아제 억제제 및 세로토닌 재흡수 억제제; 예를 들어, 입원 중, 수술 중 또는 신체 활동 중 또는 신체 활동 후(즉, 운동 관련 저나트륨 혈증) 과도한 저장액(hypotonic fluid)의 투여, 및 고령자에서 저나트륨 영양 보충제의 적용을 포함하지만, 이에 제한되지 않고, 저나트륨 혈증과 관련된 다른 상태는, 신부전, 신증후군(nephrotic syndrome)(모델 신장병과 최소 병변 질병), 악성 성질, 영양 실조, 횡문근융해(rhabdomyolysis), 외과적 치료, 선택적 심장 카테터 삽입, 실혈(blood loss), 및 삼투성 이뇨를 일으킬 수 있는 당뇨(glycosuria)의 고칼슘 혈증(hypercalcemia), 저칼륨 혈증, 및 고혈당증(hyperglycemia)을 포함한다.
본 발명은 또한 κ 오피오이드 수용체(KOR 수용체) 매개 및 관련 질병, 장애, 또는 상태를 예방 및/또는 치료하기 위한 방법에 관한 것으로, 각 식의 화합물, 특히 식(I)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염의 치료 유효량을 이를 필요로 하는 환자에게 투여하는 단계를 포함한다. 이 방법은 현저한 효능을 나타내고 부작용은 더 적게 나타낸다. 여기에서, κ 오피오이드 수용체(KOR 수용체) 매개 및 관련 질병은 급성 또는 만성 통증, 염증, 가려움증, 저나트륨 혈증, 부종, 장 폐쇄증, 기침 및 녹내장을 포함하지만, 이에 제한되지 않는다.
본 발명은 또한 포유동물의 통증 및 통증 관련 질병을 예방 및/또는 치료하기 위한 방법에 관한 것으로, 식 (I), (II), (III)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염의 치료 유효량을 이를 필요로 하는 포유동물에게 투여하는 단계를 포함한다. 이 방법은 현저한 효능을 나타내고 부작용은 더 적게 나타낸다. 여기에서, 통증은 수술후 통증, 암에 의해 발생한 통증, 신경병성 통증, 외상성 통증, 체성 통증, 내장 통증, 피부 통증 및 염증에 의해 발생한 통증일 수 있고, 예를 들어, 수술 후 통증은 사용된 수술의 유형과 범위에 따라 신경병성 통증, 체성 통증, 내장 통증, 또는 피부 통증 중 어느 하나 또는 모든 요인일 수 있으며; 암은 유방암, 자궁내막암, 자궁경부암, 피부암, 전립선암, 난소암, 나팔관 종양, 난소 종양, 혈우병 및 백혈병으로 이루어진 군으로부터 선택될 수 있다.
활성 성분을 함유하는 약학 조성물은 경구 투여에 적합한 형태, 예를 들어, 정제, 트로키제(troche), 마름모꼴 정제(lozenge), 수성 또는 유성 현탁액, 분산성 분말 또는 과립, 에멀션, 경질 또는 연질 캡슐, 또는 시럽 또는 일릭서(elixir)일 수 있다. 경구 조성물은 약학 조성물을 제조하기 위한 이 기술분야에 공지된 임의의 방법에 따라 제조될 수 있다. 이러한 조성물은 만족스럽고 입에 맞는 약학 제제를 제공하기 위해, 감미료, 착향료, 착색제 및 보존제로 이루어진 군으로부터 선택된 하나 이상의 약품을 함유할 수 있다. 정제는 정제를 제조하기에 적합한 무독성의 약학적으로 허용 가능한 부형제와 혼합된 활성 성분을 함유한다. 이들 부형제는 불활성 부형제, 과립화제(granulating agents), 붕해제(disintegrating agents) 및 윤활제일 수 있다. 정제는 코팅되지 않거나 약물의 맛을 감추거나 또는 위장관에서 활성 성분의 붕해 및 흡수를 지연시키도록 공지된 기술에 의해 코팅되어, 연장된 기간 동안 지속적인 방출을 제공할 수 있다.
경구 제제는, 활성 성분이 불활성 고체 희석제와 혼합되거나, 또는 활성 성분이 수용성 담체 또는 오일 매질 또는 올리브 오일과 혼합된 연질 젤라틴 캡슐로 제공될 수 있다.
수성 현탁액은 수성 현탁액의 제조에 적합한 부형제와 혼합된 활성 성분을 함유한다. 이러한 부형제는 현탁화제, 분산제 또는 습윤제이다. 수성 현탁액은 또한 에틸파라벤 또는 n-프로필파라벤과 같은 하나 이상의 보존제, 하나 이상의 착색제, 하나 이상의 착향료, 및 하나 이상의 감미료를 함유할 수 있다.
오일 현탁액은 식물성 오일에 활성 성분을 현탁시킴으로써 제제화될 수 있다. 오일 현탁액은 증점제(thickener)를 함유할 수 있다. 상기 언급한 감미료와 착향료는 입에 맞는 조제물을 제공하기 위해 첨가될 수 있다. 이들 조성물은 산화방지제를 첨가함으로써 보존될 수 있다.
분산제 또는 습윤제, 현탁화제 또는 하나 이상의 보존제와 혼합된 활성 성분은 물을 첨가함으로써 수성 현탁액의 제조에 적합한 분산성 분말 또는 과립으로 제조될 수 있다. 적합한 분산제 또는 습윤제 및 현탁화제는 이미 앞에서 명시된 것들로 예시된다. 감미료, 착향료 및 착색제와 같은 추가 부형제가 또한 첨가될 수 있다.
본 발명의 약학 조성물은 또한 수중유(oil-in-water) 에멀션의 형태일 수 있다. 오일상은 식물성 오일, 또는 유동 파라핀(liquid paraffin)과 같은 광물유, 또는 이들의 혼합물일 수 있다. 적합한 에멀션화제(emulsifying agent)는 천연 발생하는 인지질 또는 부분 에스테르일 수 있다. 에멀션은 또한 감미료, 착향료, 보존제 및 산화방지제를 함유할 수 있다.
본 발명의 약학 조성물은 멸균 수용액의 형태일 수 있다. 사용될 수 있는 허용 가능한 비히클 또는 용매는 물, 링거 용액 및 등장성 염화나트륨 용액이다. 멸균 주사 가능 조제물은 또한 활성 성분이 오일 상에 용해되는 주사 가능한 멸균 수중유 마이크로 에멀션(microemulsion)일 수 있다. 주사 가능 용액 또는 마이크로 에멀션은 국소 볼러스 주사(local bolus injection)에 의해 개인의 혈류 안으로 도입될 수 있다.
본 발명의 약학 조성물은 근육내 및 피하 투여를 위한 주사 가능한 멸균 수성 또는 유성 현탁액의 형태일 수 있다. 이러한 현탁액은 공지된 기술에 따라 상기 기술된 바와 같은 적합한 분산제 또는 습윤제 및 현탁화제로 제제화될 수 있다. 멸균 주사 가능 조제물은 또한 무독성의 비경구적으로 허용 가능한 희석제 또는 용매에 제조된 멸균 주사 가능 용액 또는 현탁액일 수 있다. 또한, 멸균 고정유(fixed oil)는 용매 또는 현탁화 매질로서 용이하게 사용될 수 있다.
본 발명의 화합물은 직장 투여용 좌약의 형태로 투여될 수 있다. 이들 약학 조성물은 상온에서는 고체이지만 직장에서는 액체인 적절한 비자극성 부형제와 약물을 혼합함으로써 제조될 수 있어서, 직장에서 용해되어 약물을 방출한다.
이 기술분야의 당업자에게는 약물의 투여량이 다음 요인: 특정 화합물의 활성, 환자의 나이, 환자의 체중, 환자의 일반적인 건강, 환자의 행동, 환자의 식이요법(diet), 투여 시간, 투여 경로, 배설 비율(excretion rate), 약물 조합 등을 포함하지만, 이에 제한되지 않는 다양한 요인에 의존하는 것으로 잘 알려져 있다. 또한, 치료 모드, 식(I)의 화합물의 1일 투여량 또는 약학적으로 허용 가능한 그 염의 유형과 같은 최상의 치료는 종래의 치료 요법에 의해 입증될 수 있다.
도 1은 쥐의 카라기난에 의해 유발된 카라기난 염증성 통증에 대한 본 출원의 화합물의 효과를 도시한다.
본 발명의 상세한 설명
달리 명시되지 않는 한, 상세한 설명과 청구항에서 사용된 용어는 아래에 기술된 의미를 갖는다.
"알킬"은 C1 내지 C20 직쇄(straight chain) 및 분지쇄(branched chain) 기(group)를 포함하는 포화 지방족 탄화수소기, 바람직하게는 1 내지 12개의 탄소 원자를 갖는 알킬, 및 더 바람직하게는 1 내지 6개의 탄소 원자를 갖는 알킬을 나타낸다. 비제한적인 예는, 메틸, 에틸, n-프로필, 이소프로필, n-부틸, 이소부틸, tert-부틸, sec-부틸, n-펜틸, 1,1-디메틸프로필, 1,2-디메틸프로필, 2,2-디메틸프로필, 1-에틸프로필, 2-메틸부틸, 3-메틸부틸, n-헥실, 1-에틸-2-메틸프로필, 1,1,2-트리메틸프로필, 1,1-디메틸부틸, 1,2-디메틸부틸, 2,2-디메틸부틸, 1,3-디메틸부틸, 2-에틸부틸, 2-메틸펜틸, 3-메틸펜틸, 4-메틸펜틸, 2,3-디메틸부틸, n-헵틸, 2-메틸헥실, 3-메틸헥실, 4-메틸헥실, 5-메틸헥실, 2,3-디메틸펜틸, 2,4-디메틸펜틸, 2,2-디메틸펜틸, 3,3-디메틸펜틸, 2-에틸펜틸, 3-에틸펜틸, n-옥틸, 2,3-디메틸헥실, 2,4-디메틸헥실, 2,5-디메틸헥실, 2,2-디메틸헥실, 3,3-디메틸헥실, 4,4-디메틸헥실, 2-에틸헥실, 3-에틸헥실, 4-에틸헥실, 2-메틸-2-에틸펜틸, 2-메틸-3-에틸펜틸, n-노닐, 2-메틸-2-에틸헥실, 2-메틸-3-에틸헥실, 2,2-디에틸펜틸, n-데실, 3,3-디에틸헥실, 2,2-디에틸헥실, 및 그 분지형 이성질체를 포함한다. 더 바람직하게는, 알킬기는 1 내지 6개의 탄소 원자를 갖는 저급 알킬(lower alkyl)이고, 비제한적인 예는, 메틸, 에틸, n-프로필, 이소프로필, n-부틸, 이소부틸, tert-부틸, sec-부틸, n-펜틸, 1,1-디메틸프로필, 1,2-디메틸프로필, 2,2-디메틸프로필, 1-에틸프로필, 2-메틸부틸, 3-메틸부틸, n-헥실, 1-에틸-2-메틸프로필, 1,1,2-트리메틸프로필, 1,1-디메틸부틸, 1,2-디메틸부틸, 2,2-디메틸부틸, 1,3-디메틸부틸, 2-에틸부틸, 2-메틸펜틸, 3-메틸펜틸, 4-메틸펜틸, 2,3-디메틸부틸, 등을 포함한다. 알킬기는 치환되거나 또는 치환되지 않을 수 있다. 치환시, 치환기(들)는 임의의 이용 가능한 연결 지점에서 치환될 수 있다. 치환기(들)는 바람직하게는 알킬, 알케닐, 알키닐, 알콕시, 알킬티오, 알킬아미노, 할로겐, 티올, 히드록시, 니트로, 시아노, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, 시클로알콕시, 헤테로고리 알콕시, 시클로알킬티오, 헤테로고리 알킬티오, 옥소, 카르복시 및 알콕시카르보닐로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 기이다.
"시클로알킬"은 3 내지 20개의 탄소 원자, 바람직하게는 3 내지 12개의 탄소 원자, 더 바람직하게는 3 내지 6개의 탄소 원자, 및 가장 바람직하게는 5 내지 6개의 탄소 원자를 갖는 포화 또는 부분적으로 불포화된 단환식(monocyclic) 또는 다환식(polycyclic) 탄화수소기를 나타낸다. 단환식 시클로알킬의 비제한적인 예는, 시클로프로필, 시클로부틸, 시클로펜틸, 시클로펜테닐, 시클로헥실, 시클로헥세닐, 시클로헥사디에닐, 시클로헵틸, 시클로헵타트리에닐, 시클로옥틸 등을 포함한다. 다환식 시클로알킬은 스피로 고리(spiro ring), 축합된 고리(fused ring) 또는 가교 고리(bridged ring)를 갖는 시클로알킬을 포함한다.
"스피로 시클로알킬"은 하나의 공통 탄소 원자(스피로 원자라 불림)를 통해 연결된 고리를 갖는 5 내지 20 원(membered) 다환식 기를 나타내고, 여기에서, 하나 이상의 고리는 하나 이상의 이중 결합을 함유할 수 있지만, 고리 중 어느 것도 완전하게 컨쥬게이트된 파이-전자계를 갖지 않는다. 바람직하게는 6 내지 14 원 스피로 시클로알킬, 및 더 바람직하게는 7 내지 10 원 스피로 시클로알킬이다. 고리 사이에서 공유된 스피로 원자의 수에 따라, 스피로 시클로알킬은 모노-스피로 시클로알킬, 디-스피로 시클로알킬, 또는 폴리-스피로 시클로알킬, 및 바람직하게는 모노-스피로 시클로알킬 또는 디-스피로 시클로알킬, 및 더 바람직하게는 4-원/4-원, 4-원/5-원, 4-원/6-원, 5-원/5-원, 또는 5-원/6-원 모노-스피로 시클로알킬로 나누어질 수 있다. 스피로 시클로알킬의 비제한적인 예는 다음을 포함한다:

"축합된 시클로알킬"은 5 내지 20 원의 모든 탄소 다환식 기(all-carbon polycyclic group)를 나타내고, 여기에서 계(system) 안의 각 고리는 탄소 원자의 인접한 쌍을 다른 고리와 공유하고, 여기에서 하나 이상의 고리는 하나 이상의 이중 결합을 함유할 수 있지만, 고리 중 어느 것도 완전하게 컨쥬게이트된 파이-전자계를 갖지 않는다. 바람직하게는 6 내지 14 원 축합 시클로알킬, 및 더 바람직하게는 7 내지 10 원 축합 시클로알킬이다. 구성된 고리(membered rings)의 수에 따라, 축합된 시클로알킬은 이환식(bicyclic), 삼환식(tricyclic), 사환식(tetracyclic) 또는 다환식 축합 시클로알킬, 바람직하게는 이환식 또는 삼환식 축합 시클로알킬, 및 더 바람직하게는 5-원/5-원, 또는 5-원/6-원 이환식 축합 시클로알킬로 나누어질 수 있다. 축합 시클로알킬의 비제한적인 예는 다음을 포함한다:

"가교 시클로알킬(Bridged cycloalkyl)"은 5 내지 20 원의 모든 탄소 다환식 기를 나타내고, 여기에서 계 안의 모든 2개 고리는 2개의 연결되지 않은 탄소 원자를 공유하고, 여기에서 고리는 하나 이상의 이중 결합을 가질 수 있지만, 고리 중 어느 것도 완전하게 컨쥬게이트된 파이-전자계를 갖지 않는다. 바람직하게는 6 내지 14 원 가교 시클로알킬, 및 더 바람직하게는 7 내지 10 원 가교 시클로알킬이다. 구성된 고리의 수에 따라, 가교 시클로알킬은 이환식, 삼환식, 사환식 또는 다환식 가교 시클로알킬, 및 바람직하게는 이환식, 삼환식 또는 사환식 가교 시클로알킬, 및 더 바람직하게는 이환식 또는 삼환식 가교 시클로알킬로 나누어질 수 있다. 가교 시클로알킬의 비제한적인 예는 다음을 포함한다:

시클로알킬의 고리는 아릴, 헤테로아릴 또는 헤테로시클릴의 고리에 축합될 수 있고, 여기에서 모 구조(parent structure)에 결합된 고리는 시클로알킬이다. 비제한적인 예는, 인다닐, 테트라하이드로나프틸, 벤조시클로헵틸 등, 바람직하게는 벤조시클로펜틸, 테트라하이드로나프틸을 포함한다. 시클로알킬은 선택적으로 치환되거나 또는 비치환될 수 있다. 치환시, 치환기(들)는 바람직하게는 알킬, 알케닐, 알키닐, 알콕시, 알킬티오, 알킬아미노, 할로겐, 티올, 히드록시, 니트로, 시아노, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, 시클로알콕시, 헤테로고리 알콕시, 시클로알킬티오, 헤테로고리 알킬티오, 옥소, 카르복시 및 알콕시카르보닐로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 기이다.
"헤테로시클릴"은, 고리 원자로서 N, O 및 S(O)m(여기에서, m은 0 내지 2의 정수임)으로 이루어진 군으로부터 선택된 하나 이상의 헤테로 원자를 갖지만, 고리에서 -O-O-, -O-S- 또는 -S-S-를 제외하고, 나머지 고리 원자는 탄소 원자인, 3 내지 20 원 포화 또는 부분적으로 불포화된 단환식 또는 다환식 탄화수소기를 나타낸다. 바람직하게는, 헤테로시클릴은 3 내지 12개의 원자를 갖고, 여기에서 1 내지 4개의 원자가 헤테로 원자이며, 더 바람직하게는 3 내지 8개의 원자를 갖고, 여기에서 1 내지 3개의 원자가 헤테로 원자이며, 가장 바람직하게는 5 내지 6개의 원자를 갖고, 여기에서 1 내지 2개 또는 1 내지 3개의 원자가 헤테로 원자이다. 단환식 헤테로시클릴의 비제한적인 예는, 피롤리디닐, 이미다졸리디닐, 테트라히드로퓨라닐, 테트라히드로피라닐, 테트라히드로티에닐, 디히드로이미다졸릴, 디히드로퓨라닐, 디히드로피라졸릴, 디히드로피롤릴, 피페리딜, 피페라지닐, 모르폴리닐, 티오모르폴리닐, 호모피페라지닐 등을 포함하고, 바람직하게는 테트라히드로피라닐, 피페리딜 또는 피롤리디닐을 포함한다. 다환식 헤테로시클릴은 스피로 고리, 축합 고리 또는 가교 고리를 갖는 헤테로시클릴을 포함한다.
"스피로 헤테로시클릴"은 하나의 공통 원자(스피로 원자라 불림)를 통해 연결된 고리를 갖는 5 내지 20 원 다환식 헤테로시클릴을 나타내고, 여기에서 고리는 고리 원자로서 N, O 및 S(O)m(여기에서, m은 0 내지 2의 정수임)으로 이루어진 군으로부터 선택된 하나 이상의 헤테로 원자를 갖고, 나머지 고리 원자는 탄소 원자이며, 여기에서 하나 이상의 고리는 하나 이상의 이중 결합을 함유할 수 있지만, 고리 중 어느 것도 완전하게 컨쥬게이트된 파이-전자계를 갖지 않는다. 바람직하게는 6 내지 14 원 스피로 헤테로시클릴, 및 더 바람직하게는 7 내지 10 원 스피로 헤테로시클릴이다. 고리 사이에서 공유된 스피로 원자의 수에 따라, 스피로 헤테로시클릴은 모노-스피로 헤테로시클릴, 디-스피로 헤테로시클릴, 또는 폴리-스피로 헤테로시클릴, 바람직하게는 모노-스피로 헤테로시클릴 또는 디-스피로 헤테로시클릴, 및 더 바람직하게는 4-원/4-원, 4-원/5-원, 4-원/6-원, 5-원/5-원, 또는 5-원/6-원 모노-스피로 헤테로시클릴로 나누어질 수 있다. 스피로 헤테로시클릴의 비제한적인 예는 다음을 포함한다:

"축합된 헤테로시클릴"은 5 내지 20 원의 다환식 헤테로시클릴 기를 나타내고, 여기에서 계 안의 각 고리는 원자의 인접한 쌍을 다른 고리와 공유하고, 여기에서 하나 이상의 고리는 하나 이상의 이중 결합을 함유할 수 있지만, 고리 중 어느 것도 완전하게 컨쥬게이트된 파이-전자계를 갖지 않고, 여기에서 고리는 고리 원자로서 N, O 및 S(O)m(여기에서, m은 0 내지 2의 정수임)으로 이루어진 군으로부터 선택된 하나 이상의 헤테로 원자를 갖고, 나머지 고리 원자는 탄소 원자이다. 바람직하게는 6 내지 14 원 축합 헤테로시클릴, 및 더 바람직하게는 7 내지 10 원 축합 헤테로시클릴이다. 구성된 고리의 수에 따라, 축합된 헤테로시클릴은 이환식, 삼환식, 사환식 또는 다환식 축합 헤테로시클릴, 바람직하게는 이환식 또는 삼환식 축합 헤테로시클릴, 및 더 바람직하게는 5-원/5-원, 또는 5-원/6-원 이환식 축합 헤테로시클릴로 나누어질 수 있다. 축합 헤테로시클릴의 비제한적인 예는 다음을 포함한다:


"가교 헤테로시클릴"은 5 내지 14 원의 다환식 헤테로시클릴기를 나타내고, 여기에서 계 안의 모든 2개 고리는 2개의 연결되지 않은 원자를 공유하고, 여기에서 고리는 하나 이상의 이중 결합을 가질 수 있지만, 고리 중 어느 것도 완전하게 컨쥬게이트된 파이-전자계를 갖지 않으며, 여기에서 고리는 고리 원자로서 N, O 및 S(O)m(여기에서, m은 0 내지 2의 정수임)으로 이루어진 군으로부터 선택된 하나 이상의 헤테로 원자를 갖고, 나머지 고리 원자는 탄소 원자이다. 바람직하게는 6 내지 14 원 가교 헤테로시클릴, 및 더 바람직하게는 7 내지 10 원 가교 헤테로시클릴이다. 구성된 고리의 수에 따라, 가교된 헤테로시클릴은 이환식, 삼환식, 사환식 또는 다환식 가교 헤테로시클릴, 및 바람직하게는 이환식, 삼환식 또는 사환식 가교 헤테로시클릴, 및 더 바람직하게는 이환식 또는 삼환식 가교 헤테로시클릴로 나누어질 수 있다. 가교 헤테로시클릴의 비제한적인 예는 다음을 포함한다:

헤테로시클릴 고리는 아릴, 헤테로아릴 또는 시클로알킬의 고리에 축합될 수 있고, 여기에서 모 구조에 결합된 고리는 헤테로시클릴이다. 비제한적인 예는 다음을 포함한다:

헤테로시클릴은 선택적으로 치환되거나 또는 비치환될 수 있다. 치환시, 치환기(들)는 바람직하게는 알킬, 알케닐, 알키닐, 알콕시, 알킬티오, 알킬아미노, 할로겐, 티올, 히드록시, 니트로, 시아노, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, 시클로알콕시, 헤테로고리 알콕시, 시클로알킬티오, 헤테로고리 알킬티오, 옥소, 카르복시 및 알콕시카르보닐로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 기이다.
"아릴"은, 완전하게 컨쥬게이트된 파이-전자계를 가지는 6 내지 14 원의 모든 탄소 단환식 고리 또는 다환식 축합 고리(즉, 계 안의 각 고리는 계 안의 다른 고리와 인접한 쌍의 탄소 원자를 공유함)를 나타낸다. 바람직하게는 6 내지 10 원 아릴, 더 바람직하게는 5 내지 6 원 아릴, 예를 들어, 페닐과 나프틸이다. 아릴 고리는 헤테로아릴, 헤테로시클릴 또는 시클로알킬의 고리에 축합될 수 있고, 여기에서 모 구조에 결합된 고리는 아릴 고리이다. 비제한적인 예는 다음을 포함한다:

아릴은 선택적으로 치환되거나 또는 비치환될 수 있다. 치환시, 치환기(들)는 바람직하게는 알킬, 알케닐, 알키닐, 알콕시, 알킬티오, 알킬아미노, 할로겐, 티올, 히드록시, 니트로, 시아노, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, 시클로알콕시, 헤테로고리 알콕시, 시클로알킬티오, 헤테로고리 알킬티오, 카르복시, 및 알콕시카르보닐로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 기이다.
"헤테로아릴"은, 고리 원자로서 O, S 및 N으로 이루어진 군으로부터 선택된 1 내지 4개의 헤테로 원자를 갖는 5 내지 14 원의 헤테로 방향족계, 바람직하게는 1 내지 3개의 헤테로 원자를 갖는 5 내지 10 원 헤테로아릴, 및 더 바람직하게는 1 내지 2개의 헤테로 원자를 갖는 5 또는 6 원 헤테로아릴, 예를 들어, 이미다졸릴, 푸릴, 티에닐, 티아졸릴, 피라졸릴, 옥사졸릴, 피롤릴, 테트라졸릴, 피리딜, 피리미디닐, 티아디아졸릴, 피라지닐 등, 바람직하게는 이미다졸릴, 피라졸릴, 피리미디닐 또는 티아졸릴, 및 더 바람직하게는 피라졸릴을 나타낸다. 헤테로아릴 고리는 아릴, 헤테로시클릴 또는 시클로알킬의 고리에 축합될 수 있고, 여기에서 모 구조에 결합된 고리는 헤테로아릴 고리이다. 비제한적인 예는 다음을 포함한다:


헤테로아릴은 선택적으로 치환되거나 또는 비치환될 수 있다. 치환시, 치환기(들)는 바람직하게는 알킬, 알케닐, 알키닐, 알콕시, 알킬티오, 알킬아미노, 할로겐, 티올, 히드록시, 니트로, 시아노, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, 시클로알콕시, 헤테로고리 알콕시, 시클로알킬티오, 헤테로고리 알킬티오, 카르복시, 및 알콕시카르보닐로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 기이다.
"알콕시"는 -O-(알킬) 또는 -O-(비치환된 시클로알킬) 기를 나타내고, 여기에서 알킬은 상기 정의된 바와 같다. 비제한적인 예는, 메톡시, 에톡시, 프로폭시, 부톡시, 시클로프로필옥시, 시클로부틸옥시, 시클로펜틸옥시, 시클로헥실옥시 등을 포함한다. 알콕시는 선택적으로 치환되거나 또는 비치환될 수 있다. 치환시, 치환기는 바람직하게는 알킬, 알케닐, 알키닐, 알콕시, 알킬티오, 알킬아미노, 할로겐, 티올, 히드록시, 니트로, 시아노, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, 시클로알콕시, 헤테로고리 알콕시, 시클로알킬티오, 헤테로고리 알킬티오, 카르복시, 및 알콕시카르보닐로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 기이다.
"히드록시알킬"은 히드록시(들)에 의해 치환된 알킬을 나타내고, 여기에서 알킬은 상기 정의된 바와 같다.
"할로알킬"은 하나 이상의 할로겐에 의해 치환된 알킬을 나타내고, 여기에서 알킬은 상기 정의된 바와 같다.
"시클로알킬알킬"은 하나 이상의 시클로알킬에 의해 치환된 알킬을 나타내고, 여기에서 시클로알킬과 알킬은 상기 정의된 바와 같다.
"헤테로시클릴알킬"은 하나 이상의 헤테로시클릴에 의해 치환된 알킬을 나타내고, 여기에서 헤테로시클릴과 알킬은 상기 정의된 바와 같다.
"아릴알킬"은 하나 이상의 아릴에 의해 치환된 알킬을 나타내고, 여기에서 아릴과 알킬은 상기 정의된 바와 같다.
"히드록시"는 -OH 기를 나타낸다.
"할로겐"은 플루오르, 염소, 브롬 또는 요오드를 나타낸다.
"아미노"는 -NH2 기를 나타낸다.
"시아노"는 -CN 기를 나타낸다.
"니트로"는 -NO2 기를 나타낸다.
"카르복시"는 -C(O)OH 기를 나타낸다.
"알콕시카르보닐"은 -C(O)O(알킬) 또는 -C(O)O(시클로알킬) 기를 나타내고, 여기에서 알킬과 시클로알킬은 상기 정의된 바와 같다.
"아실 할라이드"는 -C(O)-할로겐 기를 포함하는 화합물을 나타낸다.
"X는 A, B, 또는 C로 이루어진 군으로부터 선택된다", "X는 A, B 및 C로 이루어진 군으로부터 선택된다", "X는 A, B 또는 C이다", "X는 A, B 및 C이다" 등의 모두는 같은 의미이다. 이는, X가 A, B, 및 C 중 어느 하나 이상일 수 있음을 의미한다.
"선택적인(optional)" 또는 "선택적으로(optionally)"는, 이후에 기술된 사건 또는 상황이 일어날 수 있지만, 반드시 일어날 필요는 없음을 의미하고, 이 설명은 사건 또는 상황이 일어나거나 또는 일어나지 않는 상황을 포함한다. 예를 들어, "알킬에 의해 선택적으로 치환된 헤테로고리 기"는 알킬기가 존재할 수 있지만 반드시 존재할 필요는 없음을 의미하고, 이 설명은 헤테로고리 기가 알킬에 의해 치환 및 헤테로고리 기가 알킬에 의해 치환되지 않는 상황을 포함한다.
"치환된"은, 상응하는 수의 치환기에 의해 독립적으로 치환된, 기에서 하나 이상의 수소 원자, 바람직하게는 최대 5개, 더 바람직하게는 1 내지 3개의 수소 원자를 나타낸다. 치환체가 이들의 가능한 화학적 위치에만 존재하는 것은 말할 필요도 없다. 이 기술분야의 당업자는 과도한 노력을 기울이지 않고도 실험 또는 이론에 의해 치환이 가능한지 또는 불가능한지 결정할 수 있다. 예를 들어, 유리 수소를 갖는 아미노 또는 히드록시와 불포화 결합(올레핀과 같은)을 갖는 탄소 원자의 조합은 불안정할 수 있다.
"약학 조성물"은 본 발명에 따른 화합물 중 하나 이상 또는 생리학적으로/약학적으로 허용 가능한 그 염 또는 전구약물(prodrug)과, 다른 화학 성분, 및 생리학적으로/약학적으로 허용 가능한 담체 및 부형제와 같은 다른 성분의 혼합물을 나타낸다. 약학 조성물의 목적은 유기체에 대한 화합물의 투여를 촉진시키는 것으로서, 활성 성분의 흡수에 도움이 되고 따라서 생물학적 활성을 나타낸다.
"약학적으로 허용 가능한 염"은 포유 동물에서 안전하고 효과적이며, 원하는 생물학적 활성을 갖는 본 발명의 화합물의 염을 나타낸다.
약어표:

본 발명의 화합물의 합성 방법
본 발명의 목적을 이루기 위해, 본 발명은 다음의 기술적 해결 방법을 적용한다.
본 발명의 식(II)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염을 제조하는 공정은 다음 단계를 포함한다:



식(II-A)의 화합물은 알칼리성 조건 하에서 식(II-B)의 화합물과 반응하여 식(II-C)의 화합물을 수득하고, 여기에서 이 조건 하의 알칼리성 시약은 바람직하게는 트리에틸아민이다. 생성된 식(II-C)의 화합물은 가열 및 알칼리성 조건 하에서 요오드화 칼륨의 존재 하에 식(II-D)의 화합물과 반응하여 식(II-E)의 화합물을 수득하고, 여기에서 이 조건 하의 알칼리성 시약은 바람직하게는 탄산 칼륨이다. 생성된 식(II-E)의 화합물의 탈보호를 거쳐서 식(II-F)의 화합물을 수득한다. 생성된 식(II-F)의 화합물은 축합제의 조건 하에 식(II-J)의 화합물과 반응하여 식(V)의 화합물을 수득하고, 여기에서 이 조건 하의 축합 시약은 바람직하게는 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트이다. 생성된 식(V)의 화합물은 산성 조건 하에서 아미노기 상의 보호기의 제거를 거쳐서 식(II)의 화합물을 수득하고, 여기에서 이 조건 하의 산성 시약은 바람직하게는 트리플루오로아세트산 또는 1,4-디옥산 중의 염산의 용액이다.
또한, 식(II)의 화합물에서 z가 0이 아닌 경우, 선택적으로, 유리 반응을 실행하기 위해 약 염기가 첨가되어 유리 상태 생성물, 즉, 식(II)의 화합물을 수득한다.
알칼리성 조건을 제공하는 시약은 유기 염기와 무기 염기를 포함하고, 여기에서 상기 유기 염기는 피리딘, 피페리딘, 트리에틸아민, N,N-디이소프로필에틸아민, n-부틸리튬, 리튬 디이소프로필아미드, 아세트산 칼륨, tert-부톡시화 나트륨 및 tert-부톡시화 칼륨을 포함하지만, 이에 제한되지 않고, 여기에서 무기 염기는 수소화 나트륨, 인산 칼륨, 탄산 나트륨, 탄산 칼륨, 탄산 세슘, 수산화 나트륨 및 수산화 리튬을 포함하지만, 이에 제한되지 않는다.
산성 조건을 제공하는 시약은 염화수소, 트리플루오로아세트산, 포름산, 아세트산, 염산, 황산, 및 메탄설폰산을 포함하지만, 이에 제한되지 않는다.
축합제는, 1-(3-디메틸아미노프로필)-3-에틸카르보디이미드 염산염, N,N '-디시클로헥실카르보디이미드, N,N '-디이소프로필카르보디이미드, O-벤조트리아졸-N,N,N',N'-테트라메틸우로늄 테트라플루오로보레이트, 1-히드록시벤조트리아졸, 1-히드록시-7-아자벤조트리아졸, O-벤조트리아졸-N,N,N ',N'-테트라메틸우론 헥사플루오로포스페이트, 2-(7-아자벤조트리아졸-1-일)-N,N,N ',N'-테트라메틸우레아 헥사플루오로포스페이트, 벤조트리아졸-1-일옥시트리스(디메틸아미노)포스포늄 헥사플루오로포스페이트 및 벤조트리아졸-1-일-옥시트리피롤리디노-포스포늄 헥사플루오로포스페이트로 이루어진 군으로부터 선택되고, 바람직하게는 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우론 헥사플루오로포스페이트이다.
사용된 용매는 아세트산, 메탄올, 에탄올, 톨루엔, 테트라하이드로퓨란, 디클로로메탄, 디메틸 설폭시드, 1,4-디옥산, 물 및 N,N-디메틸포름아미드를 포함하지만, 이에 제한되지 않는다.
상기 식에서:
Ra는 아미노 보호기, 바람직하게는 t-부톡시카르보닐, 9-플루오레닐메톡시카르보닐, 알릴옥시카르보닐, 트리클로로에톡시카르보닐, 트리메틸실릴옥시카르보닐, 벤질옥시카르보닐, p-메틸벤젠설포닐, p-니트로벤젠설포닐 및 tert-부틸이고;
Rb는 카르복실 보호기, 바람직하게는 DMB, Bn, 알릴, Pfp, Me, PMB, MEM 및 t-Bu이며;
M, G, z, R2 및 R3는 식(II)에서 정의된 바와 같다.
본 발명의 식(III)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염을 제조하는 공정은 다음 단계를 포함한다:



식(II-C)의 화합물은 가열 및 알칼리성 조건 하에서 요오드화 칼륨의 존재 하에 식(III-1)의 화합물과 반응하여 식(III-2)의 화합물을 수득하고, 여기에서 이 조건 하의 알칼리성 시약은 바람직하게는 탄산 칼륨이다. 생성된 식(III-2)의 화합물에 아미노 보호기가 첨가되어 식(III-3)의 화합물을 수득한다. 생성된 식(III-3)의 화합물은 축합제의 존재 하에 식(II-J)의 화합물과 반응하여 식(VI)의 화합물을 수득하고, 여기에서 이 조건 하의 축합 시약은 바람직하게는 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트이다. 생성된 식(VI)의 화합물은 산성 조건 하에서 아미노기 상의 보호기의 제거를 거쳐서 식(III)의 화합물을 수득하고, 여기에서 이 조건 하의 산성 시약은 바람직하게는 트리플루오로아세트산 또는 1,4-디옥산 중의 염산의 용액이다.
또한, 식(III)의 화합물에서 z가 0이 아닌 경우, 선택적으로, 유리 반응을 실행하기 위해 약 염기가 첨가되어 유리 상태 생성물, 즉, 식(III)의 화합물을 수득한다.
알칼리성 조건을 제공하는 시약은 유기 염기와 무기 염기를 포함하고, 여기에서 상기 유기 염기는 피리딘, 피페리딘, 트리에틸아민, N,N-디이소프로필에틸아민, n-부틸리튬, 리튬 디이소프로필아미드, 아세트산 칼륨, tert-부톡시화 나트륨 및 tert-부톡시화 칼륨을 포함하지만, 이에 제한되지 않고, 여기에서 무기 염기는 수소화 나트륨, 인산 칼륨, 탄산 나트륨, 탄산 칼륨, 탄산 세슘, 수산화 나트륨 및 수산화 리튬을 포함하지만, 이에 제한되지 않는다.
산성 조건을 제공하는 시약은 염화수소, 트리플루오로아세트산, 포름산, 아세트산, 염산, 황산, 및 메탄설폰산을 포함하지만, 이에 제한되지 않는다.
축합제는, 1-(3-디메틸아미노프로필)-3-에틸카르보디이미드 염산염, N,N '-디시클로헥실카르보디이미드, N,N '-디이소프로필카르보디이미드, O-벤조트리아졸-N,N,N',N'-테트라메틸우로늄 테트라플루오로보레이트, 1-히드록시벤조트리아졸, 1-히드록시-7-아자벤조트리아졸, O-벤조트리아졸-N,N,N ',N'-테트라메틸우론 헥사플루오로포스페이트, 2-(7-아자벤조트리아졸-1-일)-N,N,N ',N'-테트라메틸우레아 헥사플루오로포스페이트, 벤조트리아졸-1-일옥시트리스(디메틸아미노)포스포늄 헥사플루오로포스페이트 및 벤조트리아졸-1-일-옥시트리피롤리디노-포스포늄 헥사플루오로포스페이트로 이루어진 군으로부터 선택되고, 바람직하게는 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우론 헥사플루오로포스페이트이다.
사용된 용매는 아세트산, 메탄올, 에탄올, 톨루엔, 테트라하이드로퓨란, 디클로로메탄, 디메틸 설폭시드, 1,4-디옥산, 물 및 N,N-디메틸포름아미드를 포함하지만, 이에 제한되지 않는다.
상기 식에서:
Ra는 아미노 보호기, 바람직하게는 t-부톡시카르보닐, 9-플루오레닐메톡시카르보닐, 알릴옥시카르보닐, 트리클로로에톡시카르보닐, 트리메틸실릴옥시카르보닐, 벤질옥시카르보닐, p-메틸벤젠설포닐, p-니트로벤젠설포닐 및 tert-부틸이고;
Rb는 카르복실 보호기, 바람직하게는 DMB, Bn, 알릴, Pfp, Me, PMB, MEM 및 t-Bu이며;
M, G, z 및 R2는 식(III)에서 정의된 바와 같다.
본 발명의 식(III-A)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염을 제조하는 공정은 다음 단계를 포함한다:



식(II-C)의 화합물은 가열 및 알칼리성 조건 하에서 요오드화 칼륨의 존재 하에 식(III-1-1)의 화합물과 반응하여 식(III-2-1)의 화합물을 수득하고, 여기에서 이 조건 하의 알칼리성 시약은 바람직하게는 탄산 칼륨이다. 생성된 식(III-2-1)의 화합물에 아미노 보호기가 첨가되어 식(III-3-1)의 화합물을 수득한다. 생성된 식(III-3-1)의 화합물은 축합제의 존재 하에 식(II-J)의 화합물과 반응하여 식(III-5)의 화합물을 수득하고, 여기에서 이 조건 하의 축합 시약은 바람직하게는 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트이다. 생성된 식(III-5)의 화합물은 산성 조건 하에서 아미노기 상의 보호기의 제거를 거쳐서 식(III-A)의 화합물을 수득하고, 여기에서 이 조건 하의 산성 시약은 바람직하게는 트리플루오로아세트산 또는 1,4-디옥산 중의 염산의 용액이다.
또한, 식(III-A)의 화합물에서 z가 0이 아닌 경우, 선택적으로, 유리 반응을 실행하기 위해 약 염기가 첨가되어 유리 상태 생성물, 즉, 식(III-A)의 화합물을 수득한다.
알칼리성 조건을 제공하는 시약은 유기 염기와 무기 염기를 포함하고, 여기에서 상기 유기 염기는 피리딘, 피페리딘, 트리에틸아민, N,N-디이소프로필에틸아민, n-부틸리튬, 리튬 디이소프로필아미드, 아세트산 칼륨, tert-부톡시화 나트륨 및 tert-부톡시화 칼륨을 포함하지만, 이에 제한되지 않고, 여기에서 무기 염기는 수소화 나트륨, 인산 칼륨, 탄산 나트륨, 탄산 칼륨, 탄산 세슘, 수산화 나트륨 및 수산화 리튬을 포함하지만, 이에 제한되지 않는다.
산성 조건을 제공하는 시약은 염화수소, 트리플루오로아세트산, 포름산, 아세트산, 염산, 황산, 및 메탄설폰산을 포함하지만, 이에 제한되지 않는다.
축합제는, 1-(3-디메틸아미노프로필)-3-에틸카르보디이미드 염산염, N,N '-디시클로헥실카르보디이미드, N,N '-디이소프로필카르보디이미드, O-벤조트리아졸-N,N,N',N'-테트라메틸우로늄 테트라플루오로보레이트, 1-히드록시벤조트리아졸, 1-히드록시-7-아자벤조트리아졸, O-벤조트리아졸-N,N,N ',N'-테트라메틸우론 헥사플루오로포스페이트, 2-(7-아자벤조트리아졸-1-일)-N,N,N ',N'-테트라메틸우레아 헥사플루오로포스페이트, 벤조트리아졸-1-일옥시트리스(디메틸아미노)포스포늄 헥사플루오로포스페이트 및 벤조트리아졸-1-일-옥시트리피롤리디노-포스포늄 헥사플루오로포스페이트로 이루어진 군으로부터 선택되고, 바람직하게는 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우론 헥사플루오로포스페이트이다.
사용된 용매는 아세트산, 메탄올, 에탄올, 톨루엔, 테트라하이드로퓨란, 디클로로메탄, 디메틸 설폭시드, 1,4-디옥산, 물 및 N,N-디메틸포름아미드를 포함하지만, 이에 제한되지 않는다.
상기 식에서:
Ra는 아미노 보호기, 바람직하게는 t-부톡시카르보닐, 9-플루오레닐메톡시카르보닐, 알릴옥시카르보닐, 트리클로로에톡시카르보닐, 트리메틸실릴옥시카르보닐, 벤질옥시카르보닐, p-메틸벤젠설포닐, p-니트로벤젠설포닐 및 tert-부틸이고;
Rb는 카르복실 보호기, 바람직하게는 DMB, Bn, 알릴, Pfp, Me, PMB, MEM 및 t-Bu이며;
R10 내지 R13, M, G 및 z는 식(III-A)에서 정의된 바와 같다.
본 발명의 식(IV-A)의 화합물, 또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염을 제조하는 공정은 다음 단계를 포함한다:


식(II-C)의 화합물은 가열 및 알칼리성 조건 하에서 요오드화 칼륨의 존재 하에 식(III-1-1)의 화합물과 반응하여 식(III-2-1)의 화합물을 수득하고, 여기에서 이 조건 하의 알칼리성 시약은 바람직하게는 탄산 칼륨이다. 생성된 식(III-2-1)의 화합물에 아미노 보호기가 첨가되어 식(III-3-1)의 화합물을 수득한다. 생성된 식(III-3-1)의 화합물은 축합제의 존재 하에 식(IV-1)의 화합물과 반응하여 식(IV-2)의 화합물을 수득하고, 여기에서 이 조건 하의 축합 시약은 바람직하게는 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트이다. 생성된 식(IV-2)의 화합물은 산성 조건 하에서 보호기 Rc와 Rb의 제거를 거쳐서 식(IV-3)의 화합물을 수득한다. 생성된 식(IV-3)의 화합물은 보호기 Ra의 제거를 추가로 거쳐서 식(IV-A)의 화합물을 수득하고, 여기에서 이 조건 하의 산성 시약은 바람직하게는 트리플루오로아세트산 또는 1,4-디옥산 중의 염산의 용액이다.
또한, 식(IV-A)의 화합물에서 z가 0이 아닌 경우, 선택적으로, 유리 반응을 실행하기 위해 약 염기가 첨가되어 유리 상태 생성물, 즉, 식(IV-A)의 화합물을 수득한다.
알칼리성 조건을 제공하는 시약은 유기 염기와 무기 염기를 포함하고, 여기에서 상기 유기 염기는 피리딘, 피페리딘, 트리에틸아민, N,N-디이소프로필에틸아민, n-부틸리튬, 리튬 디이소프로필아미드, 아세트산 칼륨, tert-부톡시화 나트륨 및 tert-부톡시화 칼륨을 포함하지만, 이에 제한되지 않고, 여기에서 무기 염기는 수소화 나트륨, 인산 칼륨, 탄산 나트륨, 탄산 칼륨, 탄산 세슘, 수산화 나트륨 및 수산화 리튬을 포함하지만, 이에 제한되지 않는다.
산성 조건을 제공하는 시약은 염화수소, 트리플루오로아세트산, 포름산, 아세트산, 염산, 황산, 및 메탄설폰산을 포함하지만, 이에 제한되지 않는다.
축합제는, 1-(3-디메틸아미노프로필)-3-에틸카르보디이미드 염산염, N,N '-디시클로헥실카르보디이미드, N,N '-디이소프로필카르보디이미드, O-벤조트리아졸-N,N,N',N'-테트라메틸우로늄 테트라플루오로보레이트, 1-히드록시벤조트리아졸, 1-히드록시-7-아자벤조트리아졸, O-벤조트리아졸-N,N,N ',N'-테트라메틸우론 헥사플루오로포스페이트, 2-(7-아자벤조트리아졸-1-일)-N,N,N ',N'-테트라메틸우레아 헥사플루오로포스페이트, 벤조트리아졸-1-일옥시트리스(디메틸아미노)포스포늄 헥사플루오로포스페이트 및 벤조트리아졸-1-일-옥시트리피롤리디노-포스포늄 헥사플루오로포스페이트로 이루어진 군으로부터 선택되고, 바람직하게는 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우론 헥사플루오로포스페이트이다.
사용된 용매는 아세트산, 메탄올, 에탄올, 톨루엔, 테트라하이드로퓨란, 디클로로메탄, 디메틸 설폭시드, 1,4-디옥산, 물 및 N,N-디메틸포름아미드를 포함하지만, 이에 제한되지 않는다.
상기 식에서:
Ra는 아미노 보호기, 바람직하게는 t-부톡시카르보닐, 9-플루오레닐메톡시카르보닐, 알릴옥시카르보닐, 트리클로로에톡시카르보닐, 트리메틸실릴옥시카르보닐, 벤질옥시카르보닐, p-메틸벤젠설포닐, p-니트로벤젠설포닐 및 tert-부틸이고;
Rb는 카르복실 보호기, 바람직하게는 DMB, Bn, 알릴, Pfp, Me, PMB, MEM 및 t-Bu이며;
Rc는 아미노 보호기, 바람직하게는 벤질옥시카르보닐, t-부톡시카르보닐, 9-플루오레닐메톡시카르보닐, 알릴옥시카르보닐, 트리클로로에톡시카르보닐, 트리메틸실릴옥시카르보닐, p-메틸벤젠설포닐, p-니트로벤젠설포닐 또는 tert-부틸이고;
R10 내지 R13, M 및 z는 식(IV-A)에서 정의된 바와 같다.
바람직한
구현예
본 발명은 다음 실시예를 참조하여 추가 기술될 것이지만, 실시예는 본 발명의 범위를 제한하는 것으로 간주되지 않아야 한다.
실시예
화합물의 구조는 핵 자기 공명(NMR) 및/또는 질량 분광법(MS)에 의해 확인된다. NMR 화학 시프트(chemical shift)(δ)는 10-6(ppm) 단위로 주어진다. NMR은 Bruker AVANCE-400 NMR 기기에 의해 결정된다. 측정용 용매는 중수소화(deuterated)-디메틸 설폭시드(DMSO-d6), 중수소화-클로로포름(CDCl3) 및 중수소화-메탄올(CD3OD)이고, 내부 표준물질(internal standard)은 테트라메틸실란(TMS)이다.
MS는 FINNIGAN LCQAd (ESI) 질량 분석기에 의해 결정된다(제조업체: Thermo, 유형: Finnigan LCQ 어드밴티지 MAX).
키랄 고성능 액체 크로마토그래피(HPLC) 분석은 LC-10A vp(Shimadzu) 또는 SFC-애너리티컬(Berger Instruments Inc.) 상에서 결정된다.
옌타이 황해(Yantai Huanghai) HSGF254 또는 칭다오(Qingdao) GF254 실리카겔 플레이트가 박층 실리카겔 크로마토그래피(TLC)를 위해 사용된다. TLC에 사용된 실리카겔 플레이트의 치수는 0.15 mm 내지 0.2 mm이고, 생성물 정제에 사용된 실리카겔 플레이트의 치수는 0.4 mm 내지 0.5 mm이다.
옌타이 황해 200 내지 300 메시 실리카겔이 컬럼 크로마토그래피를 위한 담체로서 사용된다.
Prep Star SD-1(Varian Instruments Inc.) 또는 SFC-멀티그램(Berger Instruments Inc.)가 키랄 분취용 컬럼 크로마토그래피를 위해 사용된다.
평균 키나아제 억제율(kinase inhibition rate)과 IC50 값은 NovoStar ELISA(BMG Co., Germany)에 의해 결정된다.
본 발명의 알려진 원료는 이 기술분야에 알려진 종래의 합성 방법에 의해 제조될 수 있거나, 또는 ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc., 또는 Dari chemical Company, 등으로부터 구입될 수 있다.
달리 명시되지 않는 한, 반응은 질소 대기 또는 아르곤 대기 하에서 실행된다.
"질소 대기" 또는 "아르곤 대기"라는 용어는 반응 플라스크가 1 L 질소 또는 아르곤 벌룬(balloon)이 장착된 것을 의미한다.
"수소 대기"라는 용어는 반응 플라스크가 1 L 수소 벌룬이 장착된 것을 의미한다.
가압 수소화 반응은 Parr 3916EKX 수소화 기기와 QL-500 수소 발생기 또는 HC2-SS 수소화 기기로 실행된다.
수소화 반응에서, 반응계는 일반적으로 진공이 되고 수소로 충전되며, 상기 작업이 3회 반복된다.
마이크로파 반응에서 CEM 디스커버-S 908860 유형의 마이크로파 반응기가 사용된다.
달리 명시되지 않는 한, 용액은 수용액을 나타낸다.
달리 명시되지 않는 한, 반응에서 반응 온도는 20℃ 내지 30℃ 범위의 실온을 나타낸다.
반응 공정은 박층 크로마토그래피(TLC)에 의해 모니터링되고, 전개 용매(developing solvent)의 계는 다음을 포함한다: A: 디클로로메탄과 메탄올 계, B: n-헥산과 아세트산 에틸 계, C: 석유 에테르와 아세트산 에틸 계, D: 아세톤. 용매의 부피의 비는 화합물의 극성에 따라 조절될 수 있다.
컬럼 크로마토그래피와 박층 크로마토그래피로 화합물을 정제하기 위한 용리계는 다음을 포함한다: A: 디클로로메탄과 메탄올 계, B: n-헥산과 아세트산 에틸 계, C: 디클로로메탄과 아세톤 계. 용매의 부피의 비는 화합물의 극성에 따라 조절될 수 있고, 때로 트리에틸아민과 같은 알칼리성 시약 또는 아세트산과 같은 산성 시약이 약간 첨가될 수 있다.
실시예에서 고성능 액체 크로마토그래피에 사용된 고압 액체 크로마토그래피 기기는 Gilson-281이고, 크로마토그래피 컬럼은 Shimadzu의 Shim-pack PREP-ODS이며, 사용된 이동상은 트리플루오로아세트산 완충계, 즉, 물(0.05% 트리플루오로아세테이트 함유)-아세토니트릴이다.
실시예에서 트리플루오로아세테이트 염 형태인 각각의 화합물은 다음의 일반적인 방법에 의해 유리 상태로 수득될 수 있다: 그 트리플루오로아세테이트 염을 적절한 용매(예를 들어, 메탄올, 에탄올, 테트라하이드로퓨란, 아세톤 등)에 용해시키고, 약 염기(중탄산 나트륨, 탄산 나트륨, 탄산 칼륨 등과 같은)를 첨가하여 pH를 중성으로 조절하고, 혼합물을 감압 하에 농축시키며, 잔류물을 정제하여 유리 상태를 수득하였다.
실시예 1
4-아미노-1-((6R,9R,12R)-12-(4-아미노부틸)-6-벤질-9-이소부틸-4,7,10-트리옥소-1-(1-페닐시클로프로필)-2,5,8,11-테트라아자트리데칸-13-오일)피페리딘-4-카르복시산 1




제1 단계
4-벤질 1-tert-부틸 4-(((벤질옥시)카르보닐)아미노)피페리딘-1,4-디카르복실레이트 1b
4-(((벤질옥시)카르보닐)아미노)-1-(tert-부톡시카르보닐)피페리딘-4-카르복시산 1a(1.2 g, 0.0032 mol, "Bioorganic Medicinal Chemistry Letters, 2007, 7(9), 2448-2451"에 개시된 공지된 방법에 의해 제조), 브롬화 벤질(0.65 g, 0.0038 mol) 및 탄산 세슘(2.1 g, 0.0064 mol)을 20 mL의 N,N-디메틸포름아미드에 용해시키고, 실온에서 12시간 동안 교반하였다. 반응 용액을 물에 붓고 아세트산 에틸(30 mL×3)로 추출하였다. 유기 상을 합하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시키고, 생성된 잔류물을 용리계 B를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 1b(800 mg, 수득률: 53%)를 수득하였다.
제2 단계
벤질 4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 염산염 1c
1b(800 mg, 1.71 mmol)를 2 mL의 디클로로메탄에 용해시키고, 1,4-디옥산 중의 4M 염산 용액 2 mL를 첨가하였다. 실온에서 4시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켜 미정제 표제 화합물 1c(800 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제3 단계
(R)-벤질 1-(2-((((9H-플루오렌-9-일)메톡시)카르보닐)아미노)-6-((tert- 부톡시카르보닐)아미노)헥사노일)-4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 1e
미정제 화합물 1c(800 mg, 1.97 mmol)와 (R)-2-((((9H-플루오렌-9-일)메톡시)카르보닐)아미노)-6-((tert-부톡시카르보닐)아미노)헥사노익산 1d(926 mg, 1.97 mmol, "ChemMedChem, 2015, 10(7), 1232-1239"에 개시된 공지된 방법에 따라 제조)를 20 mL의 N,N-디메틸포름아미드에 용해시켰다. 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(1.12 g, 3.0 mmol)와 N,N-디이소프로필에틸아민(0.7 mL, 3.94 mmol)을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 2N 시트르산 용액에 붓고 아세트산 에틸(30 mL×3)로 추출하였다. 유기 상을 합하고, 포화 중탄산 나트륨 용액으로 세척하여, 무수 황산나트륨으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 1e(1.6 g)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제4 단계
(R)-벤질 1-(2-아미노-6-((tert-부톡시카르보닐)아미노)헥사노일)-4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 1f
미정제 화합물 1e(1.6 g, 0.002 mol)를 10 mL의 디클로로메탄에 용해시킨 다음, 10 mL의 피페리딘을 첨가하였다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 1f(900 mg, 수득률: 77%)를 수득하였다.
제5 단계
(R)-벤질 2-((R)-2-(2-클로로아세트아미도)-3-페닐프로피온아미도)-4-메틸펜타노에이트 1i
(R)-벤질 2-((R)-2-아미노-3-페닐프로판아미도)-4-메틸펜타노에이트 1g(500 mg, 1.36 mmol, 특허 출원 "US20110212882A1"에 개시된 방법에 의해 제조)와 트리에틸아민(275 mg, 2.72 mmol)을 10 mL의 디클로로메탄에 용해시킨 다음, 염화 클로로아세틸(230 mg, 2 mmol)을 적가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 물에 붓고 포화 염화 암모늄 용액으로 세척하였다. 유기 상을 무수 황산나트륨으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 생성물 1i(500 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제6 단계
(R)-벤질 4-메틸-2-((R)-3-페닐-2-(2-(((1-페닐시클로프로필)메틸)아미노)아세트아미도)프로판아미도)펜타노에이트 1k
미정제 화합물 1i(150 mg, 0.33 mmol)와 (1-페닐시클로프로필)메탄아민 염산염 1j(74 mg, 0.4 mmol, "Journal of American Chemical Society, 2015, 137(5), 2042- 2046"에 개시된 공지된 방법에 의해 제조)를 10 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 요오드화 칼륨(110 mg, 0.67 mmol)과 탄산 칼륨(139 mg, 1 mmol)을 첨가하였다. 반응 용액을 60℃까지 가온하고 5시간 동안 교반한 다음, 감압 하에 농축시켰다. 생성된 잔류물에 물을 첨가하고 디클로로메탄(30 mL×3)으로 추출하였다. 유기 상을 합하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 1k(187 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제7 단계
(9R,12R)-벤질 9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-((1-페닐시클로프로필)메틸)-3-옥사-5,8,11-트리아자트리데칸-13-오에이트 1l
미정제 화합물 1k(187 mg, 0.337 mmol)를 디클로로메탄에 용해시킨 다음, 디-tert-부틸 디카르보네이트(147 mg, 0.67 mmol)와 N,N-디이소프로필에틸아민(130 mg, 1.01 mmol)을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 실리카겔 컬럼 크로마토그래피로 정제하여 표제 화합물 1l(100 mg, 수득률: 45.5%)을 수득하였다.
제8 단계
(9R,12R)-9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-((1-페닐시클로프로필)메틸)-3-옥사-5,8,11-트리아자트리데칸-13-오익산 1m
1l(100 ㎎, 0.152 mmol)을 10 mL의 에탄올에 용해시킨 다음, 팔라듐-탄소(100 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 5시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 1m(86 mg)을 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제9 단계
벤질 1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-((1-페닐시클로프로필)메틸)-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)-4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 1n
미정제 화합물 1m(86 mg, 0.152 mmol), 1f(91 mg, 0.152 mmol) 및 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(115 mg, 0.3 mmol)를 10 mL의 N,N-디메틸포름아미드에 용해시켰다. 실온에서 5시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 실리카겔 컬럼 크로마토그래피로 정제하여 표제 화합물 1n(100 mg, 수득률: 57.5%)을 수득하였다.
제10 단계
4-아미노-1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-((1-페닐시클로프로필)메틸)-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)피페리딘-4-카르복시산 1o
1n(100 mg, 0.087 mmol)을 10 mL의 에탄올에 용해시킨 다음, 팔라듐-탄소(100 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 1o(80 mg)을 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제11 단계
4-아미노-1-((6R,9R,12R)-12-(4-아미노부틸)-6-벤질-9-이소부틸-4,7,10-트리옥소-1-(1-페닐시클로프로필)-2,5,8,11-테트라아자트리데칸-13-오일)피페리딘-4-카르복시산 트리플루오로아세테이트 1p
미정제 화합물 1o(80 mg, 0.087 mmol)을 10 mL의 디클로로메탄에 용해시킨 다음, 2 mL의 트리플루오로아세트산을 첨가하였다. 실온에서 5시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 고성능 액체 크로마토그래피로 정제하여 표제 화합물 1p(10 mg, 수득률: 15.9%)를 수득하였다.
MS m/z (ESI): 720.4 [M+1]
1H NMR (400 MHz, CD3OD) δ 8.47-8.40 (m, 2H), 7.42-7.27 (m, 11H), 4.84-4.81 (m, 1H), 4.70-4.67 (m, 1H), 4.40-4.38 (m, 1H), 4.25-4.10 (m, 1H), 3.95-3.85 (m, 2H), 3.78-3.70 (m, 2H), 3.61-3.52 (m, 1H), 3.5-3.41 (m, 1H), 3.25-3.10 (m, 3H), 3.10-2.95 (m, 2H), 2.95-2.89 (m, 2H), 2.89-2.75 (m, 2H), 2.31-2.24 (m, 2H), 1.95-1.45 (m, 13H), 1.1-0.9 (m, 6H), 0.9-0.86 (m, 4H).
제12 단계
4-아미노-1-((6R,9R,12R)-12-(4-아미노부틸)-6-벤질-9-이소부틸-4,7,10-트리옥소-1-(1-페닐시클로프로필)-2,5,8,11-테트라아자트리데칸-13-오일)피페리딘-4-카르복시산 1
1p(10 mg, 0.012 mmol)를 1 mL의 디클로로메탄과 메탄올의 혼합 용액(V/V = 10:1)에 용해시킨 다음, 포화 탄산나트륨 수용액을 적가하여 pH를 약 7로 조절하였다. 반응 용액을 실온에서 30분 동안 교반하고 방치하여 분리되도록 하였다. 유기 상을 수거하고, 무수 황산 마그네슘으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 1(8.6 mg, 수득률: 100%)을 수득하였다.
MS m/z (ESI): 720.4 [M+1]
실시예 2
4-아미노-1-((2R,5R,8R,14S)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 2



제1 단계
(R)-벤질 4-메틸-2-((R)-3-페닐-2-(2-(((S)-2-페닐프로필)아미노)아세트아미도)프로판아미드)펜타노에이트 2b
1i(500 mg, 1.12 mmol)와 (S)-2-페닐프로판-1-아민 2a(228 mg, 1.68 mmol, "Advanced Synthesis & Catalysis, 2015, 357(18), 3875-3879"에 개시된 공지된 방법에 의해 제조)를 10 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 요오드화 칼륨(372 mg, 2.24 mmol)과 탄산 칼륨(309 mg, 2.24 mmol)을 첨가하였다. 반응 용액을 60℃까지 가온하고 12시간 동안 교반하였다. 반응 용액을 실온으로 냉각하고, 물을 첨가하고, 디클로로메탄(30 mL×3)으로 추출하였다. 유기 상을 합하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 2b(600 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제2 단계
(9R,12R)-벤질 9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-((S)-2-페닐프로필)-3-옥사-5,8,11-트리아자트리데칸-13-오에이트 2c
미정제 화합물 2b(600 mg, 1.1 mmol)를 10 mL의 디클로로메탄에 용해시킨 다음, 디-tert-부틸 디카보네이트(360 mg, 1.65 mmol)와 트리에틸아민(222 mg, 2.2 mmol)을 첨가하였다. 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 2c(380 mg, 수득률: 54%)를 수득하였다.
제3 단계
(9R,12R)-9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-((S)-2-페닐프로필)-3-옥사-5,8,11-트리아자트리데칸-13-오익산 2d
2c(380 mg, 0.59 mmol)를 15 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(40 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 2d(300 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제4 단계
벤질 1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-((S)-2-페닐프로필)-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)-4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 2e
미정제 화합물 2d(300 mg, 0.54 mmol), 1f(356 mg, 0.6 mmol), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(308 mg, 0.81 mmol) 및 N,N-디이소프로필에틸아민(104 mg, 0.81 mmol)을 10 mL의 N,N-디메틸포름아미드에 용해시켰다. 실온에서 1.5시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 2e(500 mg, 수득률: 81.8%)를 수득하였다.
제5 단계
4-아미노-1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-((S)-2-페닐프로필)-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)피페리딘-4-카르복시산 2f
2e(500 mg, 0.44 mmol)를 10 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(50 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 2f(400 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제6 단계
4-아미노-1-((2R,5R,8R,14S)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 트리플루오로아세테이트 2g
미정제 화합물 2f(400 mg, 0.44 mmol)를 5 mL의 디클로로메탄에 용해시킨 다음, 1,4-디옥산 중의 4M 염산 용액 2 mL를 첨가하였다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 고성능 액체 크로마토그래피로 정제하여 표제 화합물 2g(150 mg, 수득률: 48%)를 수득하였다.
MS m/z (ESI): 708.6 [M+1]
1H NMR (400 MHz, CD3OD) δ 7.40-7.17 (m, 11H), 4.89-4.82 (m, 1H), 4.79-4.74 (m, 1H), 4.40-4.39 (m, 1H), 4.22-4.15 (m, 1H), 4.01-3.95 (m, 1H), 3.85-3.60 (m, 5H), 3.49-3.36 (m, 1H), 3.21-3.09 (m, 5H), 2.96-2.92 (m, 4H), 2.27-2.25 (m, 3H), 1.83-1.45 (m, 14H), 1.34-1.33 (m, 3H), 1.01-0.92 (m, 6H)
제7 단계
4-아미노-1-((2R,5R,8R,14S)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 2
2g(150 mg, 0.182 mmol)를 1 mL의 디클로로메탄과 메탄올의 혼합 용액(V/V = 10:1)에 용해시킨 다음, 포화 탄산나트륨 수용액을 적가하여 pH를 약 7로 조절하였다. 반응 용액을 실온에서 30분 동안 교반하고 방치하여 분리되도록 하였다. 유기 상을 수거하고, 무수 황산 마그네슘으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 2(129 mg, 수득률: 100%)를 수득하였다.
MS m/z (ESI): 708.6 [M+1]
실시예 3
4-아미노-1-((2R,5R,8R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-14-메틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 3



제1 단계
(9R,12R)-벤질 9-벤질-12-이소부틸-2,2-디메틸-5-(2-메틸-2-페닐프로필)-4,7,10-트리옥소-3-옥사-5,8,11-트리아자트리데칸-13-오에이트 3b
미정제 화합물 1i(130 mg, 0.293 mmol)와 2-메틸-2-페닐프로판-1-아민 3a (130mg, 0.878 mmol, 특허 출원 "WO2007030582"에 개시된 방법에 의해 제조)를 2 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 요오드화 칼륨(73 mg, 0.44 mmol)과 탄산 칼륨(121 mg, 0.878 mmol)을 첨가하였다. 반응 용액을 80℃까지 가온하고 12시간 동안 교반하였다. 반응 용액을 실온으로 냉각한 다음, 1 mL의 테트라하이드로퓨란과 1 mL의 물을 첨가하였다. 균일하게 교반한 후, 디-tert-부틸 디카보네이트(96 mg, 0.439 mmol)를 첨가하였다. 반응 용액을 실온에서 2시간 동안 교반한 다음, 감압 하에 농축시켰다. 잔류물에 물을 첨가하고 아세트산 에틸(30 mL×3)로 추출하였다. 유기 상을 합하고, 포화 염화나트륨 용액으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 B를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 3b(110 mg, 수득률: 57%)를 수득하였다.
제2 단계
(9R,12R)-9-벤질-12-이소부틸-2,2-디메틸-5-(2-메틸-2-페닐프로필)-4,7,10-트리옥소-3-옥사-5,8,11-트리아자트리데칸-13-오익산 3c
3b(110 mg, 0.162 mmol)를 메탄올에 용해시킨 다음, 팔라듐-탄소(20 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 30℃까지 가온하며 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 3c(74 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제3 단계
메틸 1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-5-(2-메틸-2-페닐프로필)-4,7,10,13-테트라옥소-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)-4-((tert-부톡시카르보닐)아미노)피페리딘-4-카르복시레이트 3e
미정제 화합물 3c(74 mg, 0.13 mmol), (R)-메틸 1-(2-아미노-6-((tert-부톡시카르보닐)아미노)헥사노일)-4-((tert-부톡시카르보닐)아미노)피페리딘-4-카르복실레이트 3d(70 mg, 0.143 mmol, 특허 출원 "JP5807140B1"에 개시된 방법에 의해 제조), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(74 mg,0.195 mmol) 및 N,N-디이소프로필에틸아민(50 mg, 0.39 mmol)을 2 mL의 N,N-디메틸포름아미드에 용해시키고, 0℃에서 2시간 동안 교반하였다. 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물에 물을 첨가하고, 아세트산 에틸(30 mL×3)로 추출하였다. 유기 상을 합하고, 포화 염화나트륨 용액으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 3e(134 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제4 단계
4-아미노-1-((2R,5R,8R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-14-메틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 트리플루오로아세테이트 3f
미정제 화합물 3e(134 mg, 0.13 mmol)를 2 mL의 테트라하이드로퓨란과 메탄올의 혼합 용매(V/V = 3:1)에 용해시킨 다음, 0.65 mL의 1M 수산화리튬을 첨가하였다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 고성능 액체 크로마토그래피로 정제하여 표제 화합물 3f(40 mg, 수득률: 30%)를 수득하였다.
MS m/z (ESI): 722.6 [M+1]
1H NMR (400 MHz, CD3OD) δ 7.90-7.759 (m, 2H), 7.45-7.15 (m, 10H), 4.80-4.71 (m, 1H), 4.45-4.37 (m, 1H), 4.17-4.10 (m, 1H), 4.02-3.85 (m, 2H), 3.80-3.72 (m, 3H), 3.65-3.50 (m, 1H), 3.48-3.40 (m, 1H), 3.25-3.15 (m, 3H), 3.05-2.80 (m, 5H), 2.38-2.20 (m, 3H), 2.02-1.40 (m, 20H), 1.05-0.92 (m, 6H).
제5 단계
4-아미노-1-((2R,5R,8R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-14-메틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 3
3f(40 mg, 0.048 mmol)를 1 mL의 디클로로메탄과 메탄올의 혼합 용액(V/V = 10:1)에 용해시켰다. 포화 탄산나트륨 수용액을 적가하여 pH를 약 7로 조절하였다. 반응 용액을 실온에서 30분 동안 교반하고 방치하여 분리되도록 하였다. 유기 상을 수거하고, 무수 황산 마그네슘으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 3(34.6 mg, 수득률: 100%)을 수득하였다.
MS m/z (ESI): 722.6 [M+1]
실시예 4
4-아미노-1-((2R,5R,8R,14R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-15-메틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자헥사데칸-1-오일)피페리딘-4-카르복시산 4


제1 단계
(R)-벤질 4-메틸-2-((R)-2-(2-(((R)-3-메틸-2-페닐부틸)아미노)아세트아미도)-3-페닐프로판아미도)펜타노에이트 4b
미정제 화합물 1i(1 g, 2.45 mmol)와 (R)-3-메틸-2-페닐부탄-1-아민 4a(500 mg, 3 mmol, "Tetrahedron:Asymmetry, 2003, 14(16), 2401-2406"에 개시된 공지된 방법에 의해 제조)를 10 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 요오드화 칼륨(1 g, 6 mmol)과 탄산 칼륨(1.3 g, 9.2 mmol)을 첨가하였다. 반응 용액을 60℃까지 가온하고 12시간 동안 교반한 다음, 감압 하에 농축시켰다. 생성된 잔류물에 물을 첨가하고 디클로로메탄(30 mL×3)으로 추출하였다. 유기 상을 합하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 4b(500 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제2 단계
(9R,12R)-벤질 9-벤질-12-이소부틸-2,2-디메틸-5-((R)-3-메틸-2-페닐부틸) -4,7,10-트리옥소-3-옥사-5,8,11-트리아자트리데칸-13-오에이트 4c
미정제 화합물 4b(500 mg, 0.875 mmol)를 디클로로메탄에 용해시킨 다음, 디-tert-부틸 디카보네이트(380 mg, 1.75 mmol)와 N,N-디이소프로필에틸아민(340 mg, 2.62 mmol)을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 실리카겔 컬럼 크로마토그래피로 정제하여 표제 화합물 4c(300 mg, 수득률: 51.1%)를 수득하였다.
제3 단계
(9R,12R)-9-벤질-12-이소부틸-2,2-디메틸-5-((R)-3-메틸-2-페닐부틸)-4,7,10-트리옥소-3-옥사-5,8,11-트리아자트리데칸-13-오익산 4d
4c(300 mg, 0.447 mmol)을 10 mL의 에탄올에 용해시킨 다음, 팔라듐-탄소(100 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 4d(260 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제4 단계
벤질 1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-5-((R)-3-메틸-2-페닐부틸)-4,7,10,13-테트라옥소-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)-4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 4e
미정제 화합물 4d(260 mg, 0.447 mmol), 1f(270 mg, 0.447 mmol), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(500 mg, 1.34 mmol) 및 3 mL의 트리에틸아민을 10 mL의 N,N-디메틸포름아미드에 용해시켰다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 4e(100 mg, 수득률: 19.2%)를 수득하였다.
제5 단계
4-아미노-1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-5-((R)-3-메틸-2-페닐부틸)-4,7,10,13-테트라옥소-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)피페리딘-4-카르복시산 4f
4e(100 mg, 0.086 mmol)을 20 mL의 에탄올에 용해시키고, 팔라듐-탄소(100 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 4f(81 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제6 단계
4-아미노-1-((2R,5R,8R,14R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-15-메틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자헥사데칸-1-오일)피페리딘-4-카르복시산 트리플루오로아세테이트 4g
미정제 화합물 4f(81 mg, 0.086 mmol)를 10 mL의 디클로로메탄에 용해시킨 다음, 3 mL의 트리플루오로아세트산을 첨가하였다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 고성능 액체 크로마토그래피로 정제하여 표제 화합물 4g(8 mg, 수득률: 12.7%)를 수득하였다.
MS m/z (ESI): 736.4 [M+1]
1H NMR (400 MHz, CD3OD) δ 7.45-7.15 (m, 10H), 4.9-4.8 (m, 1H), 4.8-4.62 (m, 1H), 4.41-4.28 (m, 1H), 4.1-4.0 (m, 1H), 3.98-3.85 (m, 1H), 3.85-3.65 (m, 3H), 3.65-3.55 (m, 1H), 3.55-3.45 (m, 1H) 3.38-3.28 (m, 2H), 3.25-3.08 (m, 2H), 3.25-3.05 (m, 2H), 2.95-2.87 (m, 2H), 2.85-2.75 (m, 1H), 2.75-2.65 (m, 2H), 2.4-2.15 (m, 3H), 2-1.35 (m, 14H), 1.2-0.83 (m, 9H), 0.71-0.62 (d, 3H).
제7 단계
4-아미노-1-((2R,5R,8R,14R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-15-메틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자헥사데칸-1-오일)피페리딘-4-카르복시산 4
4g(8 mg, 0.009 mmol)를 1 mL의 디클로로메탄과 메탄올의 혼합 용매(V/V = 10:1)에 용해시켰다. 포화 탄산나트륨 수용액을 적가하여 pH를 약 7로 조절하였다. 반응 용액을 실온에서 30분 동안 교반하고 방치하여 분리되도록 하였다. 유기 상을 수거하고, 무수 황산 마그네슘으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 4(6.9 mg, 수득률: 100%)를 수득하였다.
MS m/z (ESI): 736.4 [M+1]
실시예 5
4-아미노-1-((2R,5R,8R,14R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 5


제1 단계
(R)-벤질 4-메틸-2-((R)-3-페닐-2-(2-(((R)-2-페닐프로필)아미노)아세트아미도)프로판아미도)펜타노에이트 5b
1i(500 mg, 1.12 mmol)와 (R)-2-페닐프로판-1-아민 5a(228mg, 1.68 mmol, "Angewandte Chemie,International Edition, 2003, 42(39), 4793-4795"에 개시된 공지된 방법에 의해 제조)를 10 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 요오드화 칼륨(372 mg, 2.24 mmol)과 탄산 칼륨(309 mg, 2.24 mmol)을 첨가하였다. 반응 용액을 60℃까지 가온하고 12시간 동안 교반하였다. 반응 용액을 실온으로 냉각하고, 물을 첨가하고, 디클로로메탄(30 mL×3)으로 추출하였다. 유기 상을 합하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 5b(600 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제2 단계
(9R,12R)-벤질 9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-((R)-2-페닐프로필)-3-옥사-5,8,11-트리아자트리데칸-13-오에이트 5c
미정제 화합물 5b(600 mg, 1.1 mmol)를 20 mL의 디클로로메탄에 용해시킨 다음, 디-tert-부틸 디카보네이트(361 mg, 1.66 mmol)와 트리에틸아민(222 mg, 2.2 mmol)을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 5c(580 mg, 수득률: 82%)를 수득하였다.
제3 단계
(9R,12R)-9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-((R)-2-페닐프로필)-3-옥사-5,8,11-트리아자트리데칸-13-오익산 5d
5c(580 mg, 0.9 mmol)를 10 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(60 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 5d(500 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제4 단계
벤질 1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-((R)-2-페닐프로필)-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)-4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 5e
미정제 화합물 5d(365 mg, 0.66 mmol), 1f(393 mg, 0.66 mmol), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(376 mg, 0.99 mmol) 및 N,N-디이소프로필에틸아민(0.16 mL, 0.99 mmol)을 10 mL의 N,N-디메틸포름아미드에 용해시켰다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 5e(170 mg, 수득률: 23%)를 수득하였다.
제5 단계
4-아미노-1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-((R)-2-페닐프로필)-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)피페리딘-4-카르복시산 5f
5e(80 mg, 0.0706 mmol)를 10 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(10 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 5f(60 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제6 단계
4-아미노-1-((2R,5R,8R,14R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 트리플루오로아세테이트 5g
미정제 생성물 5f(60 mg, 0.066 mmol)를 2 mL의 디클로로메탄에 용해시킨 다음, 1,4-디옥산 중의 4M 염산 용액 1 mL를 첨가하였다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 고성능 액체 크로마토그래피로 정제하여 표제 화합물 5g(30 mg, 수득률: 55.6%)를 수득하였다.
MS m/z (ESI): 708.6 [M+1]
제7 단계
4-아미노-1-((2R,5R,8R,14R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자펜타데칸-1-오일)피페리딘-4-카르복시산 5
5g(30 mg, 0.028 mmol)를 5 mL의 메탄올/물의 혼합 용매에 용해시킨 다음, 중탄산나트륨 고체(10 mg)를 첨가하여 pH를 7로 조절하였다. 반응 용액을 30분 동안 교반한 다음, 감압 하에서 농축시켰다. 생성된 잔류물에 10 mL의 디클로로메탄을 첨가하고, 30분 동안 교반하고, 여과하였다. 여과 케이크(filter cake)를 10 mL의 디클로로메탄으로 헹구고, 여과액을 감압 하에 농축시켜 표제 화합물 5(17 mg, 수득률: 85.9%)를 수득하였다.
MS m/z (ESI): 708.6 [M+1]
1H NMR (400 MHz, CD3OD): δ 7.33-7.19 (m, 10H), 4.90-4.84 (m, 2H), 4.64-4.61 (m, 2H), 4.42-4.39 (m, 1H), 3.86-3.74 (m, 5H), 3.20-3.12 (m, 4H), 2.94-2.84 (m, 4H), 2.61-2.54 (m, 2H), 2.20-2.15 (m, 3H), 1.79-1.70 (m, 2H), 1.68-1.60 (m, 8H), 1.45-1.40 (m, 3H), 1.30-1.20 (m, 5H), 0.99-0.76 (m, 6H).
실시예 6
(R)-N-((R)-6-아미노-1-모르폴리노-1-옥소헥산-2-일)-4-메틸-2-((R)-3-페닐-2-(2-(((1S,2R)-2-페닐시클로프로필)아미노)아세트아미도)프로판아미도)펜탄아미드 6


제1 단계
(R)-벤질 tert-부틸(6-모르폴리노-6-옥소헥산-1,5-디일)디카바메이트 6b
(R)-2-(((벤질옥시)카르보닐)아미노)-6-((tert-부톡시카르보닐)아미노)헥사노익산 6a(1.14 g, 3 mmol, "African Journal of pure and Applied Chemistry, 2009, 3(6), 108-115"에 개시된 공지된 방법에 의해 제조), 모르폴리노(0.31 mL, 3.6 mmol), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(1.73 g, 4.5 mmol) 및 N,N-디이소프로필에틸아민(0.8 mL, 4.5 mmol)을 10 mL의 N,N-디메틸포름아미드에 용해시키고, 실온에서 2시간 동안 교반하였다. 반응 용액에 50 mL의 아세트산 에틸을 첨가하고, 포화 염화암모늄 용액, 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 무수 황산나트륨으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 6b(1.3 g)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제2 단계
(R)-tert-부틸 (5-아미노-6-모르폴리노-6-옥소헥실)카바메이트 6c
미정제 화합물 6b(1.3 g, 2.9 mmol)를 15 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(350 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 6c(914 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제3 단계
(R)-벤질 4-메틸-2-((R)-3-페닐-2-(2-(((1S,2R)-2-페닐시클로프로필)아미노)아세트아미도)프로판아미도)펜타노에이트 6e
미정제 화합물 1i(300 mg, 0.675 mmol)와 (1S,2S)-2-페닐시클로프로판아민 6d(120 mg, 0.68 mmol, 특허 출원 "US20060116370A1"에 개시된 방법에 의해 제조)를 10 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 요오드화 칼륨(560 mg, 3.375 mmol)과 탄산 칼륨(465 mg, 3.375 mmol)을 첨가하였다. 반응 용액을 60℃까지 가온하고 5시간 동안 교반한 다음, 감압 하에 농축시켰다. 생성된 잔류물에 물을 첨가하고 디클로로메탄(30 mL×3)으로 추출하였다. 유기 상을 합하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 6e(200 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제4 단계
(9R,12R)-벤질 9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-((1S,2R)-2-페닐시클로프로필)-3-옥사-5,8,11-트리아자트리데칸-13-오에이트 6f
미정제 화합물 6e(200 mg, 0.35 mmol)를 디클로로메탄에 용해시킨 다음, 디-tert-부틸 디카르보네이트(100 mg, 0.525 mmol)와 트리에틸아민(110 mg, 1.05 mmol)을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 실리카겔 컬럼 크로마토그래피로 정제하여 표제 화합물 6f(140 mg, 수득률: 62.5%)를 수득하였다.
제5 단계
(9R,12R)-9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-((1S,2R)-2-페닐시클로프로필)-3-옥사-5,8,11-트리아자트리데칸-13-오익산 6g
6f(140 mg, 0.218 mmol)를 4.5 mL의 테트라하이드로퓨란, 메탄올 및 물의 혼합 용매(V/V/V = 4:4:1)에 용해시킨 다음, 수산화리튬 일수화물(55 mg, 1.31 mmol)을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켜 메탄올과 테트라하이드로퓨란 용매를 제거하였다. 물을 첨가하고, 1M의 염산을 적가하여 pH를 6으로 조절하였다. 반응 용액을 디클로로메탄(30 mL×3)으로 추출하였다. 유기 상을 합하고, 포화 염화나트륨 용액으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 6g(130 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제6 단계
tert-부틸 ((10R,13R,16R)-16-페닐-13-이소부틸-2,2-디메틸-10-(모르폴린-4-카르보닐)-4,12,15,18-테트라옥소-3-옥사-5,11,14,17-테트라아자노나데칸-19-일)((1S,2R)-2-페닐시클로프로필)카바메이트 6h
미정제 화합물 6g(130 mg, 0.218 mmol), 미정제 화합물 6c(85 mg, 0.26 mmol), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(125 mg, 0.327 mmol) 및 N,N-디이소프로필에틸아민(85mg, 0.654 mmol)을 5 mL의 N,N-디메틸포름아미드에 용해시키고, 실온에서 12시간 동안 교반하였다. 반응 용액에 30 mL의 아세트산 에틸을 첨가하고, 포화 염화암모늄 용액, 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 6h(130 mg, 수득률: 70%)를 수득하였다.
제7 단계
(R)-N-((R)-6-아미노-1-모르폴리노-1-옥소헥산-2-일)-4-메틸-2-((R)-3-페닐-2-(2-(((1S,2R)-2-페닐시클로프로필)아미노)아세트아미도)프로판아미도)펜탄아미드 6
6h(60 mg, 0.071 mmol)를 3 mL의 디클로로메탄에 용해시킨 다음, 1,4-디옥산 중의 4M 염산 용액 0.8 mL를 첨가하였다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 메탄올과 물의 혼합 용매(V/V = 20:1)에 용해시켰다. 탄산나트륨을 첨가하여 pH를 8보다 크게 조절하였다. 용액을 감압 하에 농축시켰다. 생성된 잔류물에 10 mL의 디클로로메탄을 첨가하고, 10분 동안 교반하고, 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 6(19 mg, 수득률: 43.5%)을 수득하였다.
MS m/z (ESI): 649.3 [M+1]
1H NMR (400 MHz, CD3OD) δ 7.26-7.23 (m, 10H), 4.95-4.93 (m, 1H), 4.80-4.78 (m, 1H), 4.70-4.68 (m, 1H), 3.66-3.60 (m, 8H), 3.32-3.30 (m, 6H), 3.28-3.26 (m, 1H), 3.18-3.16(m, 1H), 2.94-2.91 (m, 1H), 2.65-2.63 (m, 1H), 2.26-2.23 (m, 1H), 1.71-1.68 (m, 5H), 1.48-1.42 (m, 6H), 0.95-0.93 (dd, 6H).
실시예 7
4-아미노-1-((2R,5R,8R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자헥사데칸-1-오일)피페리딘-4-카르복시산 7


제1 단계
(9R,12R)-9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-(2-페닐부틸)-3-옥사-5,8,11-트리아자트리데칸-13-오익산 7a
1l(300 mg, 0.458 mmol)를 10 mL의 메탄올에 용해시킨 다음, 촉매량의 팔라듐-탄소를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 7a(189 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제2 단계
벤질 1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-(2-페닐부틸)-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)-4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 7b
미정제 화합물 7a(189 mg, 0.34mmol), 1f(200 mg, 0.34 mmol), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(194 mg, 0.51 mmol) 및 트리에틸아민(67 mg, 0.68 mmol)을 10 mL의 N,N-디메틸포름아미드에 용해시켰다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 7b(80 mg, 수득률: 20%)를 수득하였다.
제3 단계
4-아미노-1-((9R,12R,15R)-9-벤질-15-(4-((tert-부톡시카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-(2-페닐부틸)-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)피페리딘-4-카르복시산 7c
7b(80 mg, 0.07 mmol)를 10 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(10 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 7c(50 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제4 단계
4-아미노-1-((2R,5R,8R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자헥사데칸-1-오일)피페리딘-4-카르복시산 트리플루오로아세테이트 7d
미정제 화합물 7c(50 mg, 0.054 mmol)를 10 mL의 디클로로메탄에 용해시킨 다음, 1,4-디옥산 중의 4M 염산 용액 2 mL를 첨가하였다. 실온에서 1시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 고성능 액체 컬럼 크로마토그래피로 정제하여 표제 화합물 7d(10 mg, 수득률: 25.6%)를 수득하였다.
MS m/z (ESI): 722.6 [M+1]
1H NMR (400 MHz, CD3OD) δ 7.42-7.25 (m, 10H), 4.84-4.69 (m, 3H), 4.39-4.38 (m, 2H), 3.90-3.73 (m, 8H), 3.22-3.19 (m, 4H), 2.94-2.67 (m, 5H), 2.24-2.19 (m, 3H), 1.79-1.58 (m, 15H), 0.99-0.93 (m, 6H), 0.78 (t, 3H)
제5 단계
4-아미노-1-((2R,5R,8R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자헥사데칸-1-오일)피페리딘-4-카르복시산 7
7d(10 mg, 0.012 mmol)를 1 mL의 디클로로메탄과 메탄올의 혼합 용매(V/V = 10:1)에 용해시킨 다음, 포화 탄산나트륨 수용액을 첨가하여 pH를 약 7로 조절하였다. 반응 용액을 실온에서 30분 동안 교반하고 방치하여 분리되도록 하였다. 유기 상을 수거하고, 무수 황산 마그네슘으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 7(8.6 mg, 수득률: 100%)을 수득하였다.
MS m/z (ESI): 722.6 [M+1]
실시예 8
4-아미노-1-((2R,5R,8R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자테트라데칸-1-오일)피페리딘-4-카르복시산 8



제1 단계
(R)-메틸 2-아미노-6-(((벤질옥시)카르보닐)아미노)헥사노에이트 염산염 8b
1.3 mL의 디클로로설폭시드를 20 mL의 메탄올에 용해시키고, 아이스 배스(ice bath)에서 0℃로 냉각시켰다. (R)-2-아미노-6-(((벤질옥시)카르보닐)아미노)헥사노익산 8a(2 g, 7.1 mmol)을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켜 미정제 표제 화합물 8b(2.09 g)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제2 단계
(9R,12R)-벤질 9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-페닐에틸-3-옥사-5,8,11-트리아자트리데칸-13-오에이트 8d
2-((tert-부톡시카르보닐)(페닐에틸)아미노)아세트산 8c(332 mg, 1.19 mmol, 특허 출원 "US6245746B1"에 개시된 방법에 의해 제조)와 1g(439 mg, 1.19 mmol)를 6.6 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(679 mg, 1.785 mmol)와 N,N-디이소프로필에틸아민(615 mg, 4.76 mmol)을 첨가하였다. 용액을 0℃에서 2시간 동안 교반한 다음, 감압 하에 농축시켰다. 생성된 잔류물에 물을 첨가하고 아세트산 에틸(30 mL×3)로 추출하였다. 유기 상을 합하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 B를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 8d(410 mg, 수득률: 55%)를 수득하였다.
제3 단계
(9R,12R)-9-벤질-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-5-페닐에틸-3-옥사-5,8,11-트리아자트리데칸-13-오익산 8e
8d(410 mg, 0.65 mmol)를 20 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(60 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 8e(313 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제4 단계
(9R,12R,15R)-메틸 9-벤질-15-(4-(((벤질옥시)카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-페닐에틸-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오에이트 8f
미정제 화합물 8e(100 mg, 0.125 mmol)와 미정제 화합물 8b(85 mg, 0.231 mmol)를 5 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 2-(7-아자벤조트리아졸-1-일)-N,N,N ',N'-테트라메틸우로늄 헥사플루오로포스페이트(105 mg, 0.277 mmol)와 N,N-디이소프로필에틸아민(72 mg, 0.555 mmol)을 첨가하였다. 실온에서 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물에 물을 첨가하고 아세트산 에틸(30 mL×3)로 추출하였다. 유기 상을 합하고, 포화 염화암모늄 용액(30 mL×3), 포화 중탄산나트륨 용액(30 mL×3) 및 포화 염화나트륨 용액(30 mL×3)으로 연속적으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 8f(140 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제5 단계
(9R,12R,15R)-9-벤질-15-(4-(((벤질옥시)카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-페닐에틸-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오익산 8g
미정제 화합물 8f(140 mg, 0.17 mmol)를 7 mL의 테트라하이드로퓨란, 메탄올 및 물의 혼합 용매(V/V/V = 3:3:1)에 용해시킨 다음, 수산화리튬 일수화물(40 mg, 0.85 mmol)을 첨가하였다. 실온에서 0.5시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켜 메탄올과 테트라하이드로퓨란 용매를 제거하였다. 물을 첨가한 다음, 1M의 염산을 적가하여 pH를 6으로 조절하였다. 반응 용액을 디클로로메탄(30 mL×3)으로 추출하였다. 유기 상을 합하고, 포화 염화나트륨 용액으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 8g(220 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제6 단계
벤질 1-((9R,12R,15R)-9-벤질-15-(4-(((벤질옥시)카르보닐)아미노)부틸)-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-페닐에틸-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)-4-(((벤질옥시)카르보닐)아미노)피페리딘-4-카르복실레이트 8h
미정제 화합물 8g(220 mg, 0.17 mmol)와 미정제 화합물 1c(130 mg, 0.26 mmol)를 5 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(100 mg, 0.26 mmol)와 N,N-디이소프로필에틸아민(66 mg, 0.51 mmol)을 첨가하였다. 실온에서 3시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물에 물을 첨가하고 아세트산 에틸(30 mL×3)로 추출하였다. 유기 상을 합하고, 포화 염화암모늄 용액(30 mL×3), 포화 중탄산나트륨 용액(30 mL×3) 및 포화 염화나트륨 용액(30 mL×3)으로 연속적으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 8h(170 mg, 수득률: 87%)를 수득하였다.
제7 단계
1-((9R,12R,15R)-15-(4-아미노부틸)-9-벤질-12-이소부틸-2,2-디메틸-4,7,10,13-테트라옥소-5-페닐에틸-3-옥사-5,8,11,14-테트라아자헥사데칸-16-오일)-4-(카르복시아미노)피페리딘-4-카르복시산 8i
8h(170 mg, 0.147 mmol)를 5 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(50 mg, 촉매량)를 첨가하였다. 첨가 완료 후, 반응계를 수소로 3회 퍼징하고, 실온에서 12시간 동안 교반하였다. 반응 용액을 셀라이트를 통해 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 8i(140 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제8 단계
4-아미노-1-((2R,5R,8R)-2-(4-아미노부틸)-8-벤질-5-이소부틸-4,7,10-트리옥소-14-페닐-3,6,9,12-테트라아자테트라데칸-1-오일)피페리딘-4-카르복시산 8
미정제 화합물 8i(140 mg, 0.167 mmol)를 3 mL의 디클로로메탄에 용해시킨 다음, 1,4-디옥산 중의 4M 염산 용액 0.5 mL를 첨가하였다. 실온에서 1시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 메탄올과 물의 혼합 용매(V/V = 20:1)에 용해시킨 다음, 탄산나트륨을 첨가하여 pH를 8보다 크게 조절하였다. 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물에 10 mL의 디클로로메탄을 첨가하고, 10분 동안 교반하고 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 8(19 mg, 수득률: 17.4%)을 수득하였다.
MS m/z (ESI): 694.4 [M+1]
1H NMR (400 MHz, CD3OD) δ 7.32-7.01 (m, 10H), 4.68-4.66 (m, 1H), 4.44-4.42 (m, 1H), 3.75-3.65 (m, 6H), 3.35 (s, 2H), 3.32-3.28 (m, 6H), 3.20-3.18 (m, 2H), 3.00 (s, 2H), 2.70-2.58 (m, 5H), 2.10-1.98 (m, 3H), 1.55-1.50 (m, 6H), 0.95-0.93 (dd, 6H).
실시예 9
(R)-N-((R)-6-아미노-1-(4-(3-메틸우레이도)피페리딘-1-일)-1-옥소헥산-2-일)-4-메틸-2-((R)-3-페닐-2-(2-(((R)-2-페닐프로필)아미노)아세트아미도)프로판아미도)펜탄아미드 9


제1 단계
tert-부틸 4-(3-메틸우레이도)피페리딘-1-카르복실레이트 9c
9a(8.11 g, 40 mmol, 특허 출원 "WO2006115353"에 개시된 방법에 의해 제조)를 130 mL의 디클로로메탄에 용해시킨 다음, N,N-디이소프로필에틸아민(15.51 g, 120 mmol)을 첨가하였다. 0℃로 냉각시킨 후, 반응 용액에 9b(3.74 g, 40 mmol)를 첨가하고 2시간 동안 실온에서 교반하였다. 반응 용액에 200 mL의 포화 중탄산나트륨 용액을 첨가하고 디클로로메탄(200 mL×3)으로 추출하였다. 유기 상을 합하고, 포화 염화나트륨 용액(200 mL)으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 9c(9.3 g)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제2 단계
1-메틸-3-(피페리딘-4-일)우레아 염산염 9d
미정제 화합물 9c(1 g, 4 mmol)를 10 mL의 디클로로메탄에 용해시킨 다음, 1,4-디옥산 중의 4M 염산 용액 2 mL를 첨가하였다. 2시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켜 미정제 표제 화합물 9d(1 g, 백색 고형물)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제3 단계
(R)-(9H-플루오렌-9-일)메틸 tert-부틸(6-(4-(3-메틸우레이도)피페리딘-1-일)-6-옥소헥산-1,5-디일)디카바메이트 9e
미정제 화합물 9d(1 g, 5.16 mmol)와 1d(2.42 g, 5.16 mmol)를 20 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(2.94 g, 7.74 mmol)와 트리에틸아민(1.03 g, 10.32 mmol)을 첨가하였다. 실온에서 4시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 9e(1 g, 수득률: 32%)를 수득하였다.
제4 단계
(R)-tert-부틸(5-아미노-6-(4-(3-메틸우레이도)피페리딘-1-일)-6-옥소헥실)카바메이트 9f
9e(304 mg, 0.5 mmol)를 2 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 6 mL의 트리에틸아민을 첨가하였다. 반응 용액을 12시간 동안 교반한 다음, 정제 없이 바로 다음 단계에서 사용하였다.
제5 단계
tert-부틸((10R,13R,16R)-16-벤질-13-이소부틸-2,2-디메틸-10-(4-(3-메틸우레이도)피페리딘-1-카르보닐)-4,12,15,18-테트라옥소-3-옥사-5,11,14,17-테트라아자노나데칸-19-일)((R)-2-페닐프로필)카바메이트 9g
미정제 화합물 9f(193 mg, 0.5 mmol), 미정제 화합물 5d(277 mg, 0.5 mmol), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(380 mg, 1 mmol) 및 N,N-디이소프로필에틸아민(129 mg, 1 mmol)을 30 mL의 N,N-디메틸포름아미드에 용해시켰고, 실온에서 12시간 동안 교반하였다. 반응 용액에 10 mL의 포화 시트르산 용액과 20 mL의 물을 첨가하고, 아세트산 에틸(30 mL×3)로 추출하였다. 유기 상을 합하고, 포화 중탄산나트륨 용액(20 mL)과 포화 염화나트륨 용액(20 mL)으로 연속적으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 9g(500 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제6 단계
(R)-N-((R)-6-아미노-1-(4-(3-메틸우레이도)피페리딘-1-일)-1-옥소헥산-2-일)-4-메틸-2-((R)-3-페닐-2-(2-(((R)-2-페닐프로필)아미노)아세트아미도)프로판아미도)펜탄아미드 트리플루오로아세테이트 9
미정제 화합물 9g(184 mg, 0.2 mmol)를 10 mL의 디클로로메탄에 용해시킨 다음, 1,4-디옥산 중의 4M 염산 용액 2 mL를 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 고성능 액체 크로마토그래피로 정제하여 표제 화합물 9(20 mg, 수득률: 14%)를 수득하였다.
MS m/z (ESI): 721.6 [M+1]
1H NMR (400 MHz, CD3OD) δ 8.37-8.25 (m, 1H), 7.73-7.65 (m, 1H), 7.41-7.37 (m, 2H), 7.39-7.29 (m, 7H), 7.18-7.16 (m, 2H), 4.93-4.90 (m, 1H), 4.83-4.82 (m, 1H), 4.54-4.50 (m, 2H), 4.00-3.92 (m, 1H), 3.88-3.60 (m, 6H), 3.15-3.08 (m, 5H), 3.05-2.98 (m, 4H), 2.70 (s, 3H), 2.15-1.88 (m, 3H), 1.79-1.61 (m, 15H), 1.01-0.96 (m, 6H).
제7 단계
(R)-N-((R)-6-아미노-1-(4-(3-메틸우레이도)피페리딘-1-일)-1-옥소헥산-2-일)-4-메틸-2-((R)-3-페닐-2-(2-(((R)-2-페닐프로필)아미노)아세트아미도)프로판아미도)펜탄아미드 9
9h(20 mg, 0.024 mmol)를 1 mL의 디클로로메탄과 메탄올의 혼합 용매(V/V = 10:1)에 용해시킨 다음, 포화 탄산나트륨 수용액을 적가하여 pH를 약 7로 조절하였다. 반응 용액을 실온에서 30분 동안 교반하고 방치하여 분리되도록 하였다. 유기 상을 수거하고, 무수 황산 마그네슘으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 9(17 mg, 수득률: 100%)을 수득하였다.
MS m/z (ESI): 721.6 [M+1]
실시예 10
(R)-N-((R)-6-아미노-1-모르폴리노-1-옥소헥산-2-일)-2-((R)-2-(2-((2,3-디하이드로-1H-인덴-2-일)아미노)아세트아미도)-3-페닐프로판아미도)-4-메틸펜탄아미드 10



제1 단계
(R)-(9H-플루오렌-9-일)메틸 (6-((1-(4,4-디메틸-2,6-디옥소시클로헥실리덴)에틸)아미노)-1-모르폴리노-1-옥소헥산-2-일)카바메이트 10b
(R)-2-((((9H-플루오렌-9-일)메톡시)카르보닐)아미노)-6-((1-(4,4-디메틸-2,6-디옥소시클로헥실리덴)에틸)아미노)헥사노익산 10a(1.06 g, 2 mmol, Accela ChemBio Inc.에서 구입)와 모르폴린(200 mg, 2.4 mmol)을 10 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(1.51 g, 4 mmol)와 트리에틸아민(400 mg, 4 mmol)을 첨가하였다. 실온에서 3시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물에 5 mL의 포화 시트르산 용액과 50 mL의 물을 첨가하고, 아세트산 에틸(100 mL×3)로 추출하였다. 유기 상을 합하고, 물(100 mL)로 세척하며, 무수 황산나트륨으로 건조시키고, 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 10b(1.3 g)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제2 단계
(R)-2-(1-((5-아미노-6-모르폴리노-6-옥소헥실)아미노)에틸리덴)-5,5-디메틸시클로헥산-1,3-디온 10c
미정제 화합물 10b(1.3 g, 2 mmol)를 5 mL의 디클로로메탄에 용해시킨 다음, 2 mL의 피페리딘을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 10c(500 mg, 수득률: 75%)를 수득하였다.
제3 단계
(R)-벤질 2-((R)-2-(2-((2,3-디하이드로-1H-인덴-2-일)아미노)아세트아미도)-3-페닐프로판아미도)-4-메틸펜타노에이트 10e
미정제 화합물 1i(222 mg, 0.5 mmol)와 2,3-디하이드로-1H-인덴-2-아민 염산염 10d(127 mg, 0.4 mmol, "Tetrahedron, 2005, 61(28), 6801-6807"에 개시된 공지된 방법에 의해 제조)를 5 mL의 N,N-디메틸포름아미드에 용해시킨 다음, 요오드화 칼륨(415 mg, 2.5 mmol)과 탄산 칼륨(345 mg, 2.5 mmol)을 첨가하였다. 반응 용액을 60℃까지 가온하고 12시간 동안 교반한 다음, 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 10e(300 mg, 수득률: 100%)를 수득하였다.
제4 단계
(9R,12R)-벤질 9-벤질-5-(2,3-디하이드로-1H-인덴-2-일)-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-3-옥사-5,8,11-트리아자트리데칸-13-오에이트 10f
10e(300 mg, 0.55 mmol)를 10 mL의 디클로로메탄에 용해시킨 다음, 디-tert-부틸 디카보네이트(181 mg, 0.83 mmol)와 N,N-디이소프로필에틸아민(0.3 mL, 1.65 mmol)을 첨가하였다. 실온에서 12시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 용리계 A를 갖는 박층 크로마토그래피로 정제하여 표제 화합물 10f(170 mg, 수득률: 48%)를 수득하였다.
제5 단계
(9R,12R)-9-벤질-5-(2,3-디하이드로-1H-인덴-2-일)-12-이소부틸-2,2-디메틸-4,7,10-트리옥소-3-옥사-5,8,11-트리아자트리데칸-13-오익산 10g
10f(170 mg, 0.26 mmol)를 10 mL의 메탄올에 용해시킨 다음, 팔라듐-탄소(50 mg, 촉매량)를 첨가하였다. 반응계를 수소로 3회 퍼징하고 실온에서 12시간 동안 교반하였다. 반응 용액을 여과하고, 여과액을 감압 하에 농축시켜 미정제 표제 화합물 10g(112 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제6 단계
tert-부틸 ((4R,7R,10R)-4-벤질-16-(4,4-디메틸-2,6-디옥소시클로헥실리덴)-7-이소부틸-10-(모르폴린-4-카르보닐)-2,5,8-트리옥소-3,6,9,15-테트라아자헵타데실)(2,3-디하이드로-1H-인덴-2-일)카바메이트 10h
미정제 화합물 10g(112 mg, 0.2 mmol), 10c(78 mg, 0.2 mmol), 2-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트(114 mg, 0.3 mmol) 및 N,N-디이소프로필에틸아민(0.1 mL, 0.6 mmol)을 15 mL의 N,N-디메틸포름아미드에 용해시켰고, 실온에서 2시간 동안 교반하였다. 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물에 100 mL의 아세트산 에틸을 첨가하고, 물(50 mL×3)과 포화 염화암모늄 용액(50 mL×3)으로 연속적으로 세척하고, 무수 황산나트륨으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 미정제 표제 화합물 10h(183 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제7 단계
tert-부틸 (2-(((R)-1-(((R)-1-(((R)-6-아미노-1-모르폴리노-1-옥소헥산-2-일)아미노)-4-메틸-1-옥소펜탄-2-일)아미노)-1-옥소-3-페닐프로판-2-일)아미노)-2-옥소에틸)(2,3-디하이드로-1H-인덴-2-일)카바메이트 10i
미정제 화합물 10h(183 mg, 0.2 mmol)를 10 mL의 메탄올에 용해시킨 다음, 0.5 mL의 히드라진 수화물(hydrazine hydrate)을 첨가하였다. 실온에서 1시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켜 미정제 표제 화합물 10i(150 mg)를 수득하였고, 이것을 정제 없이 바로 다음 단계에서 사용하였다.
제8 단계
(R)-N-((R)-6-아미노-1-모르폴리노-1-옥소헥산-2-일)-2-((R)-2-(2-((2,3-디하이드로-1H-인덴-2-일)아미노)아세트아미도)-3-페닐프로판아미도)-4-메틸펜탄아미드 트리플루오로아세테이트 10j
미정제 화합물 10i(150 mg, 1.31 mmol)를 5 mL의 디클로로메탄에 용해시킨 다음, 1 mL의 트리플루오로아세트산을 첨가하였다. 실온에서 1시간 동안 교반한 후, 반응 용액을 감압 하에 농축시켰다. 생성된 잔류물을 고성능 액체 크로마토그래피로 정제하여 표제 화합물 10j(5 mg, 수득률: 4%)를 수득하였다.
MS m/z (ESI): 649.4 [M+1]
1H NMR (400 MHz, DMSO-d 6 ) d 8.36 (d, 1H), 7.88 (d, 2H), 7.21-7.33 (m, 9H), 4.61-4.64 (m, 1H), 4.36-4.47 (m, 1H), 4.02-4.15 (m, 2H), 3.60-3.70 (m, 3H), 3.45-3.59 (m, 5H), 3.35-3.44 (m, 3H), 2.90-3.25 (m, 9H), 1.55-1.80 (m, 6H), 1.30-1.45 (m, 4H), 0.98 (d, 3H), 0.92 (d, 3H).
제9 단계
(R)-N-((R)-6-아미노-1-모르폴리노-1-옥소헥산-2-일)-2-((R)-2-(2-((2,3-디하이드로-1H-인덴-2-일)아미노)아세트아미도)-3-페닐프로판아미도)-4-메틸펜탄아미드 10
10j(5 mg, 0.0057 mmol)를 2 mL의 디클로로메탄과 메탄올의 혼합 용매(V/V = 10:1)에 용해시킨 다음, 포화 탄산나트륨을 적가하여 pH를 약 7로 조절하였다. 반응 용액을 실온에서 30분 동안 교반하고 방치하여 분리되도록 하였다. 유기 상을 수거하고, 무수 황산 마그네슘으로 건조시키고 여과하였다. 여과액을 감압 하에 농축시켜 표제 화합물 10(4 mg, 수득률: 100%)을 수득하였다.
MS m/z (ESI): 649.4 [M+1]
생물학적 분석(BIOLOGICAL ASSAY)
본 발명은 다음 시험 실시예를 참조하여 추가로 기술될 것이지만, 실시예는 본 발명의 범위를 제한하는 것으로 간주되지 않아야 한다.
시험 실시예 1
1. 실험 목적
이 실험의 목적은 인간 KOR(h-KOR) 수용체에 대한 본 발명의 화합물의 작용 효과(agonistic effect)를 결정하고, EC50의 값에 따라 화합물의 시험관 내 활성을 평가하는 것이다.
2. h-KOR 활성 시험
2.1 실험 목적
본 발명의 화합물은 h-KOR 수용체를 활성화시켜, 세포내 cAMP 수준(level)을 감소시킬 수 있다. 2차 전달자(second messenger) cAMP는 핵에 들어가고 DNA의 CRE에 결합하여, 다운스트림 루시페라아제(downstream luciferase)의 발현을 개시한다. 루시페라아제는 그 기질과 반응하여 형광을 방출하고, 측정된 형광 신호는 화합물의 작용 활성(agonistic activity)을 반영한다.
2.2 실험 방법
h-KOR에 작용하고 다운스트림 cAMP 수준에 영향을 미치는 시험 실시예 화합물의 활성은 다음 방법으로 시험하였다.
2.1.1 실험 재료 및 기기

2.2.2 실험 절차
1) HEK293/KOR/CRE 단일 클론성 세포주(monoclonal cell lines) 수득
KOR/pcDNA3.1(+) 및 CRE/pGL4.29를 HEK293 세포로 전이하였다. G418 및 하이그로마이신(hygromycin)을 배양 배지에 첨가하고, HEK293/KOR/CRE 단일 클론성 세포주를 96-웰(well) 세포 배양 평판(cell culture plate)에서 스크리닝하였다.
2) h-KOR에 대한 실시예 화합물의 작용 효과
HEK293/h-KOR/CRE 단일 클론성 세포를 DMEM/고 글루코오스 배지(10% FBS, 1 mg/ml G418, 200 ㎍/㎖ 하이그로마이신, 균일하게 혼합됨)에서 배양하고, 3일마다 계대배양(passage)하였다. 실험 당일, 새로운 세포 배지로 세포 현탁액을 제조하고, 20,000 세포/웰을 갖는 96 웰 평판(BD, #356692)에 첨가하고, 37℃에서 5% CO2에서 배양하였다. 2일째에, 화합물을 20 mM 농도의 순수한 DMSO에 용해시킨 다음, DMSO로 제제화하여 200 nM의 제1 농도로 만들고, 3배 농도 기울기(concentration gradient)로 8가지 농도로 희석시켰다. 90 ㎕의 DMSO를 블랭크(blank) 및 대조(control) 웰에 첨가하였다. 화합물 용액을 10 μM의 포스콜린(Forskolin)을 함유하는 DMEM/하이퍼글루코오스(hyperglucose){SH30243.01B, 하이클론(Hyclone)} 배지로 20배 희석시켰다. 1일째 접종한 세포 배양 평판을 꺼내고, 희석된 약물 또는 대조군(0.5% DMSO) 10 ㎕를 각각의 웰에 첨가하였다. 평판을 부드럽게 흔들고, 37℃에서 4시간 두었다. 96-웰 세포 배양 평판에서, 100 ㎕의 루시페라아제 분석 용액(Promega, # E6110)을 각 웰에 첨가하였다. 평판을 실온에서 5분 동안 두었다. 빅터(Victor) 3.0을 사용하여 화학 발광 값(chemiluminescence value)을 측정하였다. 화합물의 각 농도 및 상응하는 신호 값을 기초로 그래프패드 프리즘(Graphpad Prism) 소프트웨어를 사용하여 화합물의 EC50 값을 계산하였다.
2.3 시험 결과
h-KOR에 작용하고 다운스트림 cAMP 수준에 영향을 미치는 본 발명의 화합물의 활성은 상기 시험에 의해 측정하였고, EC50 값이 표 1.1에 나타나 있다.
표 1.1: h-KOR 수용체에 작용하고 cAMP 수준에 영향을 미치는 본 발명의 화합물의 EC50

결론: 본 발명의 화합물은 h-KOR 수용체에 상당한 작용 효과를 갖는다. 특히, 글리신의 아미노기 상의 치환기가 치환 또는 비치환된 에틸렌 기인 경우, 화합물은 예상치 못한 효과를 갖는다.
약물 동력학 평가
시험 실시예 2. 쥐에서 본 발명의 실시예 2, 5 및 8의 화합물의 약물 동력학 분석
1. 요약
쥐(rat)를 시험 동물로 사용하였다. 상이한 시점에서 혈장 내의 약물 농도는 실시예 2, 5 및 8의 화합물을 쥐에게 정맥내 투여한 후, LC/MS/MS에 의해 측정되었다. 본 발명의 화합물의 약물 동력학적 거동은 SD 쥐에서 연구 및 평가되었다.
2. 프로토콜(Protocol)
2.1 시험 화합물
실시예 2, 5 및 8의 화합물
2.2 시험 동물
절반이 수컷이고 절반이 암컷인 12 마리의 스프래그-다우리(Sprague-Dawley, SD) 쥐는 SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., CO{라이센스 번호: SCXK (상하이) 2008-0016}에서 구입하였다.
2.3 시험 화합물의 제조
적당량의 시험 화합물의 중량을 측정하고, 5% DMSO + 5% PEG400 + 90% 생리 식염수(normal saline)를 연속적으로 첨가하였다.
2.4 투여
밤새 단식 후, 절반이 수컷이고 절반이 암컷인 12 마리의 SD 쥐를 똑같이 3개의 그룹으로 나누고, 5 mL/kg의 투여 부피의 투여량으로 정맥내 투여하였다.
3. 공정
정맥 군(venous group)에서, 투여 전 및 투여 후 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, 11.0 및 24.0 시간에 안와 부비강(orbital sinus)으로부터 혈액(0.2 mL)을 채취하였다. 샘플을 헤파린 첨가 튜브(heparinized tube)에 보관하고, 10분 동안 3,500 rpm으로 원심 분리하여 혈장을 분리하였다. 혈장 샘플을 -20℃에서 보관하였다.
정맥내 투여 후 SD 쥐 혈장에서 시험 화합물의 농도를 LC-MS/MS로 측정하였다.
4. SD 쥐에서 약물 동력학적 파라미터의 결과
본 발명의 실시예 2, 5 및 8의 화합물의 약물 동력학적 파라미터는 아래에 나타나 있다.

결론: 본 발명의 화합물은 쥐에서 양호한 약물 동력학적 특성을 갖는다.
시험 실시예 3. 개에서 본 발명의 실시예 5의 화합물의 약물
동력학
분석
1. 요약
비글 개(Beagle dog)를 시험 동물로 사용하였다. 상이한 시점에서 혈장 내의 약물 농도가 실시예 5의 화합물을 비글 개에게 정맥내 투여한 후, LC/MS/MS에 의해 측정되었다. 본 발명의 화합물의 약물 동력학적 거동이 비글 개에서 연구 및 평가되었다.
2. 프로토콜
2.1 시험 화합물
실시예 5의 화합물
2.2 시험 동물
한 그룹의 3 마리의 수컷 비글 개를 Medicilon Pharmaceutical Technology (상하이) Co., Ltd에서 구입하였다.
2.3 시험 화합물의 제조
적당량의 시험 화합물의 중량을 측정하고, 100% 생리 식염수를 첨가하였다.
2.4 투여
밤새 단식 후, 수컷인, 한 그룹의 3 마리의 비글 개에게 2 mL/kg의 투여 부피의 투여량으로 정맥내 투여하였다.
3. 공정
정맥 군에서, 투여 전 및 투여 후 5분, 15분, 0.5, 1.0, 2.0, 4.0, 8.0, 12.0 및 24.0 시간에 경정맥(jugular vein)으로부터 혈액(1 mL)을 채취하였다. 샘플을 헤파린 첨가 튜브에 보관하고, 10분 동안 3,500 rpm으로 원심 분리하여 혈장을 분리하였다. 혈장 샘플을 -80℃에서 보관하였다.
정맥내 투여 후 비글 개 혈장에서 시험 화합물의 농도를 LC-MS/MS로 측정하였다.
4. 비글 개에서 약물 동력학적 파라미터의 결과
본 발명의 실시예 5의 화합물의 약물 동력학적 파라미터는 아래에 나타나 있다.

결론: 본 발명의 화합물은 비글 개에서 양호한 약물 동력학적 특성을 갖는다.
시험 실시예 4. 쥐에서 카라기난(carrageenan) 유도 염증성 통증의 치료에서 KOR 작용제의 실험 보고서
1. 실험 목적
쥐에서 염증성 통증에 대한 KOR 작용제의 치료 효과를 평가하기 위해, 쥐에서 카라기난 염증성 통증 모델이 확립되었다.
2. 실험 방법과 실험 재료
2.1. 시험 동물과 사육(feeding) 조건
수컷 위스타 쥐(Male Wistar rat)는 상하이 Slac Laboratory Animal Co., Ltd.{상하이, 중국, 증명서 번호(Certificate No.) 2015000513408, 라이센스 번호 SCXK (상하이) 2012-0002}에서 구입하였다. 쥐는 150 ~ 180 g이었고, 12/12 시간 명/암 주기 조절, 23±1℃의 일정한 온도, 50 ~ 60%의 습도, 및 음식과 물에 자유롭게 접근하는 조건에서, 우리(cage)당 5 마리가 사육되었다. 구입 후, 동물들은 실험이 시작되기 전 7일 동안 이 조건에 적응되었다.
2.2. 시험 화합물
실시예 5의 화합물;
λ-카라기난, 배치 번호(Batch No.) BCBP8978V, sigma에서 구입.
0.9% 염화나트륨 용액(500 mL, 4.5 g)
1% λ-카라기난을 생리 식염수에 넣고, 밤새 교반하여 젤리와 같은 현탁액을 형성하였다.
화합물 투여량은 염기를 기초로 계산되었다.
2.3. 실험 설계와 실험 방법
2.3.1 동물 그룹화(grouping)
적응을 돕는 사육 후에, 쥐를 다음과 같이 그룹으로 나누었다:

주: NS: 카라기난 용액을 준비하는 데 사용된 생리 식염수(normal saline); i.v.: 정맥내 주사; s.c.: 피하 주사.
2.3.2. 실험 방법
[1][2]
:
실험 방법은 문헌 1(Kazunari Nakao 등)의 방법에 따라 수정되었다. 염증성 통증 실험 전에, 쥐를 체중에 따라 다음의 그룹으로 무작위로 나누었다: 블랭크 대조 그룹, 모델 그룹, 실시예 5 ~ 0.1 mg/kg 그룹, 및 실시예 5 ~ 0.3 mg/kg 그룹. 각 그룹에는 8 마리의 쥐가 있었다. 염증성 통증 모델은 1% 카라기난(100 ㎕)으로 피하 주사된 위스타 쥐 발바닥(footpad)에서 이루어졌다. 4시간 후, 쥐는 기계적 통증 역치(pain threshold)를 평가하기 위해 발바닥의 압통 시험(plantar tenderness test)을 거쳤다. 검출 30분 전에 약물의 단일 꼬리 정맥(tail vein) 투여(1 ml/kg)를 실시하고, 대조 그룹과 모델 그룹은 상응하는 용매를 투여 받았다.
주: 문헌 1, CJ-023,423, a Novel, Potent and Selective Prostaglandin KOR Receptor Antagonist with Anti-hyperalgesic Properties[J]. The Journal of Pharmacology and Experimental Therapeutics, 2007, 322(2):686-694.
2.4 실험 장치
전자 폰 프레이(Electronic Von Frey): UGO BASILE, 타입 38450.
2.5 데이터 표현과 통계 처리
실험 데이터는 평균 ± 표준 편차(S.D.)로 표현되었다. 통계적인 비교는 엑셀 소프트웨어 시험을 사용하여 수행되었다. 모델 그룹과 대조 그룹 사이의 데이터를 분석하고 비교하여 유의한 통계적 유의도가 있는지 결정하였다. *P < 0.05는 모델 그룹과 대조 그룹 사이에 유의미한 차이가 있음을 나타내고, ** P < 0.01는 모델 그룹과 대조 그룹 사이에 높은 유의미한 차이가 있음을 나타내며, #P < 0.05는 모델 그룹과 투여 그룹 사이에 유의미한 차이가 있음을 나타내고, ##P < 0.01는 모델 그룹과 투여 그룹 사이에 높은 유의미한 차이가 있음을 나타낸다.
3. 결과: 쥐에서 카라기난 유도 카라기난 염증성 통증에 대한 본 발명의 화합물의 효과
쥐의 실험 결과는 블랭크 대조 그룹에서 압통의 역치(threshold of tenderness)가 약 20 g이고, 모델 그룹에서 압통의 역치가 7.6 g임을 보여주었다. 블랭크 대조 그룹과 비교해서, 모델 그룹에서 압통의 역치가 유의미하게 감소하였다(P < 0.01). 모델 그룹과 비교해서, 모든 약물은 염증성 쥐의 압통 역치를 유의미하게 증가시킬 수 있었다(P < 0.01). 실시예 5 ~ 0.1 mg/kg 및 실시예 5 ~ 0.3 mg/kg의 압통의 역치는 각각 13.7 g과 23.2 g이었다. 증가는 각각 79.5%와 204.5%였고, 유의미한 투여량 의존성을 가졌다. (도 1 참조).
4. 토론
λ-카라기난은 수생 식물에서 추출된 콜로이드 물질로, 알레르기성 자극 효과를 갖는다. 카라기난 단독으로 염증을 유발하고 통증을 일으킬 수 있다. 이 실험에서, 쥐에서 KOR 작용제 투여 후 압통의 역치의 변화를 관찰하고, 아급성(subacute) 염증성 통증 및 그 강도에 대한 약물의 진통 효과를 평가하기 위해, 카라기난 염증성 통증의 모델을 설정하였다. 이 실험은 압통에 대한 쥐의 반응을 측정하기 위해 전자 촉각 측정 기기를 사용하였다. 전자 촉각 측정 기기(e-VF)는 쥐와 마우스의 알레르기 및 이질통증(allodynia)을 평가하기 위해 Ugo Basile 오리지널 디자인을 사용하여 설계되었다. 기기는 동물의 자극 시간과 자극 강도를 자동으로 기록한다. 독특한 프리즘 디자인은 실험 중 시험 동물의 발바닥 영역을 쉽게 관찰할 수 있도록 한다. 감지하는 동안, 기기는 시험 동물이 시험 발톱(test claw)을 수축시키는 것을 감지할 수 있거나, 또는 발 스위치(foot switch)에 의해 판단될 수 있다. 보다 집중된 포지셔닝은 국소 통증 및 신경병성 통증 측정에 더욱 적합하다.
5. 결론
시험 화합물은 쥐의 염증성 통증을 투여량 의존 방식으로 개선할 수 있다.
Claims (18)
- 식(I)의 화합물:

또는 그 토토머(tautomer), 메소머(mesomer), 라세미체(racemate), 거울상 이성질체(enantiomer), 부분 입체 이성질체(diastereomer), 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염에 있어서,
상기 식에서:
M은 무기산 또는 유기산, 바람직하게는 유기산, 더 바람직하게는 트리플루오로아세트산이고;
G는 O, -NR4 및 -CR5R6으로 이루어진 군으로부터 선택되며;
R1은 수소, 알킬, 알콕시, 할로알킬, 할로겐, 아미노, 니트로, 히드록시, 시아노, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7 및 -NR8R9로 이루어진 군으로부터 선택되고, 여기에서, 알킬, 할로알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R2는 수소, 알킬, 알콕시, 할로알킬, 시클로알킬, 시클로알킬알킬, 헤테로시클릴, 헤테로시클릴알킬, 아릴, 아릴알킬, 헤테로아릴, 헤테로아릴알킬, -OR7, -C(O)R7 및 -C(O)OR7로 이루어진 군으로부터 선택되고, 여기에서, 알킬, 할로알킬, 시클로알킬, 시클로알킬알킬, 헤테로시클릴, 헤테로시클릴알킬, 아릴, 아릴알킬, 헤테로아릴 및 헤테로아릴알킬은 각각 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R3은 수소, 알킬, 알콕시, 할로알킬, 시클로알킬, 시클로알킬알킬, 헤테로시클릴, 헤테로시클릴알킬, 아릴, 아릴알킬, 헤테로아릴, 헤테로아릴알킬, -OR7, -C(O)R7 및 -C(O)OR7로 이루어진 군으로부터 선택되고, 여기에서, 알킬, 할로알킬, 시클로알킬, 시클로알킬알킬, 헤테로시클릴, 헤테로시클릴알킬, 아릴, 아릴알킬, 헤테로아릴 및 헤테로아릴알킬은 각각 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R4는 수소, 알킬, 할로알킬, 시클로알킬, 알콕시, 히드록시알킬, 히드록시, 아미노, 알콕시카르보닐, 헤테로시클릴, 아릴, 헤테로아릴, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7, -NR8R9 및 -NHC(O)NR8R9로 이루어진 군으로부터 선택되고, 여기 에서, 알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로겐, 히드록시, 아미노, 알콕시카르보닐, 니트로, 시아노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R5와 R6은 수소, 알킬, 알콕시, 히드록시알킬, 히드록시, 아미노, 알콕시카르보닐, 시클로알킬, 헤테로시클릴, 아릴, 헤테로아릴, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7, -NR8R9 및 -NHC(O)NR8R9로 이루어진 군으로부터 각각 독립적으로 선택되고, 여기에서, 알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로겐, 히드록시, 아미노, 알콕시카르보닐, 니트로, 시아노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R7은 수소, 알킬, 아미노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고, 여기에서, 알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로겐, 히드록시, 아미노, 니트로, 시아노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
R8과 R9는 수소, 알킬, 알콕시, 히드록시알킬, 히드록시, 아미노, 알콕시카르보닐, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 각각 독립적으로 선택되고, 여기에서, 알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴은 각각 알킬, 할로겐, 히드록시, 아미노, 알콕시카르보닐, 니트로, 시아노, 알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되며;
z는 0, 1, 2, 3 또는 4이고;
m은 0, 1 또는 2인, 식(I)의 화합물. - 제1항에 있어서,
식(II)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
M, G, R2, R3 및 z는 제1항에서 정의된 바와 같은, 식(I)의 화합물. - 제1항 또는 제2항에 있어서,
식(III)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
M, G, R2 및 z는 제1항에서 정의된 바와 같은, 식(I)의 화합물. - 제1항 내지 제3항 중 어느 한 항에 있어서,
식(IV)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
M, R2 및 z는 제1항에서 정의된 바와 같은, 식(I)의 화합물. - 제1항 내지 제4항 중 어느 한 항에 있어서,
R2는 아릴알킬, 시클로알킬알킬 및 시클로알킬로 이루어진 군으로부터 선택되고, 여기에서, 아릴알킬, 시클로알킬알킬 및 시클로알킬은 각각 알킬, 시클로알킬 및 아릴로 이루어진 군으로부터 선택된 하나 이상의 기에 의해 선택적으로 치환되는, 식(I)의 화합물. - 제1항 내지 제3항 중 어느 한 항에 있어서,
식(III-A)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
G는 O 또는 CR5R6; 바람직하게는 CR5R6이고;
R10은 수소, 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되며;
R11과 R12는 동일하거나 또는 상이하고, 각각 수소, 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 독립적으로 선택되거나;
또는 R11과 R12는 함께 시클로알킬을 형성하고;
R13은 수소, 알킬, 할로알킬, 할로겐, 아미노, 니트로, 시아노, 히드록시, 알콕시, 할로알콕시, 히드록시알킬, 시클로알킬, 헤테로시클릴, 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되며;
s는 0, 1 또는 2이고;
R5 내지 R6, M 및 z는 제1항에서 정의된 바와 같은, 식(I)의 화합물. - 제1항 내지 제6항 중 어느 한 항에 있어서,
식(IV-A)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
R10 내지 R13, M, z 및 s는 제6항에서 정의된 바와 같은, 식(I)의 화합물. - 제1항 내지 제7항 중 어느 한 항에 있어서,
식(IV-B)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염이고,
상기 식에서:
R10 내지 R11, R13, M, z 및 s는 제6항에서 정의된 바와 같은, 식(I)의 화합물. - 제1항 내지 제8항 중 어느 한 항에 있어서,
z는 0 또는 1인, 식(I)의 화합물. - 제1항 내지 제9항 중 어느 한 항에 있어서,
화합물은 다음으로 이루어진 군:




으로부터 선택되는, 식(I)의 화합물. - 식(VI)의 화합물:

또는 그 토토머, 메소머, 라세미체, 거울상 이성질체, 부분 입체 이성질체, 또는 이들의 혼합물, 또는 약학적으로 허용 가능한 그 염에 있어서,
상기 식에서:
Ra는 아미노-보호기, 바람직하게는 t-부톡시카르보닐, 9-플루오레닐메톡시카르보닐, 알릴옥시카르보닐, 트리클로로에톡시카르보닐, 트리메틸실릴에톡시카르보닐, 벤질옥시카르보닐, p-메틸벤젠설포닐, p-니트로벤젠설포닐 또는 tert-부틸이고;
G와 R2는 제3항에서 정의된 바와 같은, 식(VI)의 화합물. - 제3항에 따른 식(III)의 화합물을 제조하는 방법에 있어서,
다음의 단계:

산성 조건 하에서 식(VI)의 화합물 상의 보호기 Ra를 제거하여 식(III)의 화합물을 수득하는 단계를 포함하고;
상기 식에서:
M, G, z, 및 R2는 제3항에서 정의된 바와 같고, Ra는 제11항에서 정의된 바와 같은, 식(III)의 화합물을 제조하는 방법. - 제1항 내지 제10항 중 어느 한 항에 따른 식(I)의 화합물의 치료 유효량, 및 하나 이상의 약학적으로 허용 가능한 담체(carrier), 희석제 또는 부형제를 포함하는, 약학 조성물.
- 제1항 내지 제10항 중 어느 한 항에 따른 식(I)의 화합물, 또는 제13항에 따른 약학 조성물을, κ 오피오이드 수용체 작용제 매개 및 관련 질병을 예방 및/또는 치료하기 위한 약제의 제조에 사용하는 방법.
- 제14항에 있어서,
κ 오피오이드 수용체 작용제 매개 및 관련 질병은, 통증, 염증, 가려움증, 부종(edema), 저나트륨 혈증(hyponatremia), 저칼륨 혈증(hypokalemia), 장 폐쇄증, 기침 및 녹내장으로 이루어진 군으로부터 선택되고, 바람직하게는 통증인, 사용 방법. - 제1항 내지 제10항 중 어느 한 항에 따른 식(I)의 화합물, 또는 제13항에 따른 약학 조성물을, 통증 및 통증 관련 질병을 예방 및/또는 치료하기 위한 약제의 제조에 사용하는 방법.
- 제15항 또는 제16항에 있어서,
통증은, 신경병성 통증, 체성 통증(trunk pain), 내장 통증(visceral pain), 피부 통증, 관절염 통증, 신장 결석 통증, 자궁 경련, 월경 불순, 자궁 내막증, 소화불량, 수술 후 통증, 의학적 치료 후 통증, 눈 통증, 이염 통증(otitis pain), 전격성 암 통증(fulminant cancer pain), 및 GI 장애 관련 통증으로 이루어진 군으로부터 선택되는, 사용 방법. - 제1항 내지 제10항 중 어느 한 항에 따른 식(I)의 화합물, 또는 제13항에 따른 약학 조성물을, κ 오피오이드 수용체를 작용(agonizing)시키기 위한 약제의 제조에 사용하는 방법.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610397516 | 2016-06-07 | ||
| CN201610397516.3 | 2016-06-07 | ||
| PCT/CN2017/087328 WO2017211272A1 (zh) | 2016-06-07 | 2017-06-06 | 苯基丙酰胺类衍生物、其制备方法及其在医药上的应用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20190016516A true KR20190016516A (ko) | 2019-02-18 |
| KR102401743B1 KR102401743B1 (ko) | 2022-05-24 |
Family
ID=60578399
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020187037174A KR102401743B1 (ko) | 2016-06-07 | 2017-06-06 | 페닐 프로판아미드 유도체와 이를 제조하는 방법, 및 이를 약학적으로 사용하는 방법 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20190144499A1 (ko) |
| EP (1) | EP3466962A4 (ko) |
| JP (1) | JP7120927B2 (ko) |
| KR (1) | KR102401743B1 (ko) |
| CN (3) | CN108290926B (ko) |
| AU (1) | AU2017277003B2 (ko) |
| BR (1) | BR112018074431A2 (ko) |
| CA (1) | CA3025710A1 (ko) |
| HK (1) | HK1255567A1 (ko) |
| MX (1) | MX2018015082A (ko) |
| RU (1) | RU2738207C2 (ko) |
| TW (1) | TWI781938B (ko) |
| UA (1) | UA123737C2 (ko) |
| WO (1) | WO2017211272A1 (ko) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108290926B (zh) * | 2016-06-07 | 2022-02-08 | 江苏恒瑞医药股份有限公司 | 苯基丙酰胺类衍生物、其制备方法及其在医药上的应用 |
| US11084847B2 (en) | 2016-09-27 | 2021-08-10 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Polyamide compound and use thereof |
| CA3084201A1 (en) | 2017-12-06 | 2019-06-13 | Jiangsu Hengrul Medicine Co., Ltd. | Salt of phenylpropionamide derivative and preparation method therefor |
| CN111065390B (zh) * | 2017-12-06 | 2023-01-24 | 江苏恒瑞医药股份有限公司 | Kor激动剂与mor激动剂联合在制备治疗疼痛的药物中的用途 |
| CN109879934B (zh) * | 2017-12-06 | 2020-12-08 | 江苏恒瑞医药股份有限公司 | 一种苯基丙酰胺类衍生物的盐及其制备方法 |
| TW202015716A (zh) | 2018-05-16 | 2020-05-01 | 大陸商江蘇恆瑞醫藥股份有限公司 | 一種kor受體激動劑醫藥組成物 |
| CN110790817A (zh) | 2019-11-12 | 2020-02-14 | 成都诺和晟泰生物科技有限公司 | 一种多肽类化合物、制剂、药物组合物及制备方法、应用 |
| CN113493490B (zh) * | 2020-04-03 | 2024-03-12 | 成都诺和晟泰生物科技有限公司 | 一种合成肽酰胺类化合物及其在医药领域的用途 |
| WO2021219134A1 (zh) * | 2020-04-30 | 2021-11-04 | 南京明德新药研发有限公司 | 苯基丙酰胺类化合物及其应用 |
| AU2020454871A1 (en) | 2020-06-25 | 2023-01-19 | Humanwell Pharmaceutical US | Peptides for treatment of medical disorders |
| CN115925699B (zh) * | 2022-02-25 | 2023-10-03 | 南京知和医药科技有限公司 | 具有镇痛活性的稠环化合物及其制备方法与用途 |
| WO2023179659A1 (zh) | 2022-03-23 | 2023-09-28 | 江苏恩华药业股份有限公司 | 多酰胺类化合物、其制备方法及其医药用途 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010509343A (ja) * | 2006-11-10 | 2010-03-25 | カラ セラピューティクス インコーポレイテッド | 合成ペプチドアミド |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8236766B2 (en) * | 2006-11-10 | 2012-08-07 | Cara Therapeutics, Inc. | Uses of synthetic peptide amides |
| US8906859B2 (en) * | 2006-11-10 | 2014-12-09 | Cera Therapeutics, Inc. | Uses of kappa opioid synthetic peptide amides |
| WO2009156889A1 (en) * | 2008-06-25 | 2009-12-30 | Pfizer Inc. | Diaryl compounds and uses thereof |
| WO2010006267A2 (en) * | 2008-07-11 | 2010-01-14 | University Of Kansas | Cyclic tetrapeptides |
| WO2010105636A1 (en) * | 2009-03-17 | 2010-09-23 | United Technologies Ut Ag | Topical compositions comprising interleukin-1 alpha and peptides and cosmetic methods thereof |
| CA2866299C (en) * | 2012-03-05 | 2017-12-05 | Dr. Reddy's Laboratories Ltd. | Substituted heterocyclic acetamides as kappa opioid receptor (kor) agonists |
| WO2013184794A2 (en) * | 2012-06-05 | 2013-12-12 | Cara Therapeutics, Inc. | Peripheral kappa receptor agonists for reducing pain and inflammation |
| US20150272927A1 (en) * | 2012-12-03 | 2015-10-01 | University Of Washington Through Its Center For Commercialization | Methods and compositions for treating vasomotor symptoms |
| WO2014184356A1 (en) * | 2013-05-17 | 2014-11-20 | Dr. August Wolff Gmbh & Co. Kg Arzneimittel | Perhydroquinoxaline derivatives useful as analgesics |
| WO2015065867A2 (en) * | 2013-10-28 | 2015-05-07 | Cara Therapeutics, Inc. | Peripheral kappa opioid receptor agonists for preventing, inhibiting or treating nausea and vomiting |
| CN105523996B (zh) * | 2014-10-17 | 2020-03-06 | 江苏恒瑞医药股份有限公司 | 酰胺类衍生物、其制备方法及其在医药上的应用 |
| CN108290926B (zh) * | 2016-06-07 | 2022-02-08 | 江苏恒瑞医药股份有限公司 | 苯基丙酰胺类衍生物、其制备方法及其在医药上的应用 |
-
2017
- 2017-06-06 CN CN201780004217.1A patent/CN108290926B/zh active Active
- 2017-06-06 US US16/306,950 patent/US20190144499A1/en not_active Abandoned
- 2017-06-06 TW TW106118675A patent/TWI781938B/zh not_active IP Right Cessation
- 2017-06-06 AU AU2017277003A patent/AU2017277003B2/en not_active Ceased
- 2017-06-06 WO PCT/CN2017/087328 patent/WO2017211272A1/zh unknown
- 2017-06-06 RU RU2018145808A patent/RU2738207C2/ru active
- 2017-06-06 JP JP2018563586A patent/JP7120927B2/ja active Active
- 2017-06-06 CA CA3025710A patent/CA3025710A1/en active Pending
- 2017-06-06 UA UAA201812607A patent/UA123737C2/uk unknown
- 2017-06-06 EP EP17809711.9A patent/EP3466962A4/en not_active Withdrawn
- 2017-06-06 CN CN202210043910.2A patent/CN114349820A/zh active Pending
- 2017-06-06 BR BR112018074431-0A patent/BR112018074431A2/pt not_active IP Right Cessation
- 2017-06-06 CN CN202210044042.XA patent/CN114940700A/zh active Pending
- 2017-06-06 MX MX2018015082A patent/MX2018015082A/es unknown
- 2017-06-06 KR KR1020187037174A patent/KR102401743B1/ko active IP Right Grant
-
2018
- 2018-11-15 HK HK18114605.8A patent/HK1255567A1/zh unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010509343A (ja) * | 2006-11-10 | 2010-03-25 | カラ セラピューティクス インコーポレイテッド | 合成ペプチドアミド |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2018145808A (ru) | 2020-07-09 |
| CN114349820A (zh) | 2022-04-15 |
| US20190144499A1 (en) | 2019-05-16 |
| CA3025710A1 (en) | 2017-12-14 |
| MX2018015082A (es) | 2019-04-22 |
| CN108290926B (zh) | 2022-02-08 |
| RU2738207C2 (ru) | 2020-12-09 |
| AU2017277003B2 (en) | 2021-01-07 |
| KR102401743B1 (ko) | 2022-05-24 |
| RU2018145808A3 (ko) | 2020-07-09 |
| CN108290926A (zh) | 2018-07-17 |
| JP7120927B2 (ja) | 2022-08-17 |
| EP3466962A1 (en) | 2019-04-10 |
| WO2017211272A1 (zh) | 2017-12-14 |
| EP3466962A4 (en) | 2020-01-08 |
| UA123737C2 (uk) | 2021-05-26 |
| BR112018074431A2 (pt) | 2019-03-06 |
| TWI781938B (zh) | 2022-11-01 |
| JP2019517530A (ja) | 2019-06-24 |
| CN114940700A (zh) | 2022-08-26 |
| AU2017277003A1 (en) | 2018-12-13 |
| TW201742858A (zh) | 2017-12-16 |
| HK1255567A1 (zh) | 2019-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102401743B1 (ko) | 페닐 프로판아미드 유도체와 이를 제조하는 방법, 및 이를 약학적으로 사용하는 방법 | |
| JP6927999B2 (ja) | サイクリン依存性キナーゼ9(cdk9)阻害剤のe3リガーゼリガンドとのコンジュゲーションによるcdk9の分解および使用法 | |
| JP2756742B2 (ja) | N−アシル−2,3−ベンゾジアゼピン誘導体、その製造法、それを含有する医薬組成物、およびその製造法 | |
| US8653127B2 (en) | Bicyclic pyrazolo-heterocycles | |
| CN107098871B (zh) | 苯基丙酰胺类衍生物、其制备方法及其在医药上的应用 | |
| CN112566916B (zh) | 作为pad4抑制剂的经取代的噻吩并吡咯 | |
| US6794510B2 (en) | Processes for the preparation of peripheral opioid antagonist compounds and intermediates thereto | |
| DE69417235T2 (de) | IMIDAZO[1,2-a]PYRAZIN-4-ON-DERIVATE ANTAGONISTEN DER AMPA UND NMDA REZEPTOREN | |
| KR20160018538A (ko) | 베타-세크레타제(bace) 저해제로서의 4-아미노-6-페닐-5,6-디하이드로이미다조[1,5-a]피라진-3(2h)-온 유도체 | |
| KR19980702735A (ko) | Eaa 길항제로서의 인돌 유도체 | |
| KR20160018537A (ko) | 베타-세크레타제(bace) 저해제로서의 4-아미노-6-페닐-5,6-디하이드로이미다조[1,5-a]피라진 유도체 | |
| KR20160019893A (ko) | 베타-세크레타제(bace) 저해제로서의 4-아미노-6-페닐-6,7-디하이드로[1,2,3]트리아졸로[1,5-a]피라진 유도체 | |
| CN107098876B (zh) | 苯基丙酰胺类衍生物、其制备方法及其在医药上的应用 | |
| US8211926B2 (en) | Bicyclic pyrazolo-heterocycles | |
| CN114181277A (zh) | 一种用于雄激素受体蛋白靶向降解的嵌合体化合物、其制备方法及其在医药上的应用 | |
| CN113754635A (zh) | 稠环类化合物及其制备方法和用途 | |
| WO2024006403A2 (en) | Selective histone deacetylase 8 (hdac8) degraders and methods of use thereof | |
| FI102276B (fi) | Menetelmä uusien terapeuttisesti käyttökelpoisten 5-N,N-dialkyylisulfa moyyli-1H-6,7,8,9-tetrahydro£g|indoli-2,3-dioni-3-oksiimien valmistami seksi | |
| WO2023230612A1 (en) | Heterocyclic pad4 inhibitors | |
| CA3169225A1 (en) | Adamts inhibitors, preparation methods and medicinal uses thereof | |
| WO2023116763A1 (zh) | 一种哒嗪类化合物、其药物组合物及应用 | |
| CN116693446A (zh) | 苯基脲类衍生物及其医药用途 | |
| WO2023230609A1 (en) | Heterocyclic pad4 inhibitors | |
| CN116693470A (zh) | 组胺h3受体抑制剂及其医药用途 | |
| NZ738078A (en) | Mct4 inhibitors for treating disease |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant |