JP2019506861A - マイクロ流体装置における腸細胞の成長のためのシステム及び方法 - Google Patents
マイクロ流体装置における腸細胞の成長のためのシステム及び方法 Download PDFInfo
- Publication number
- JP2019506861A JP2019506861A JP2018540028A JP2018540028A JP2019506861A JP 2019506861 A JP2019506861 A JP 2019506861A JP 2018540028 A JP2018540028 A JP 2018540028A JP 2018540028 A JP2018540028 A JP 2018540028A JP 2019506861 A JP2019506861 A JP 2019506861A
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- intestinal
- ipsc
- chip
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 210000001842 enterocyte Anatomy 0.000 title claims abstract description 85
- 238000000034 method Methods 0.000 title claims description 145
- 230000012010 growth Effects 0.000 title description 21
- 210000004027 cell Anatomy 0.000 claims abstract description 694
- 230000000968 intestinal effect Effects 0.000 claims abstract description 192
- 238000012258 culturing Methods 0.000 claims abstract description 80
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims abstract description 52
- 210000000981 epithelium Anatomy 0.000 claims abstract description 48
- 210000004263 induced pluripotent stem cell Anatomy 0.000 claims description 227
- 210000002220 organoid Anatomy 0.000 claims description 145
- 210000002438 upper gastrointestinal tract Anatomy 0.000 claims description 97
- 210000002569 neuron Anatomy 0.000 claims description 65
- 230000002267 hypothalamic effect Effects 0.000 claims description 58
- 230000014509 gene expression Effects 0.000 claims description 52
- 230000004069 differentiation Effects 0.000 claims description 48
- 150000001875 compounds Chemical class 0.000 claims description 42
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 claims description 42
- 210000001900 endoderm Anatomy 0.000 claims description 41
- AQGNHMOJWBZFQQ-UHFFFAOYSA-N CT 99021 Chemical compound CC1=CNC(C=2C(=NC(NCCNC=3N=CC(=CC=3)C#N)=NC=2)C=2C(=CC(Cl)=CC=2)Cl)=N1 AQGNHMOJWBZFQQ-UHFFFAOYSA-N 0.000 claims description 36
- 101800003838 Epidermal growth factor Proteins 0.000 claims description 35
- 102000003945 NF-kappa B Human genes 0.000 claims description 35
- 108010057466 NF-kappa B Proteins 0.000 claims description 35
- 229940116977 epidermal growth factor Drugs 0.000 claims description 35
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 claims description 34
- 229930002330 retinoic acid Natural products 0.000 claims description 34
- 229960001727 tretinoin Drugs 0.000 claims description 34
- 238000005259 measurement Methods 0.000 claims description 24
- 230000006540 mitochondrial respiration Effects 0.000 claims description 24
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 claims description 23
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 claims description 23
- 108010023082 activin A Proteins 0.000 claims description 23
- 210000003750 lower gastrointestinal tract Anatomy 0.000 claims description 22
- 238000004519 manufacturing process Methods 0.000 claims description 21
- 238000012216 screening Methods 0.000 claims description 21
- 239000000203 mixture Substances 0.000 claims description 20
- 108010082117 matrigel Proteins 0.000 claims description 17
- 102000045246 noggin Human genes 0.000 claims description 17
- 108700007229 noggin Proteins 0.000 claims description 17
- 230000007423 decrease Effects 0.000 claims description 16
- VFSUUTYAEQOIMW-YHBQERECSA-N 3-chloro-N-[trans-4-(methylamino)cyclohexyl]-N-[3-(pyridin-4-yl)benzyl]-1-benzothiophene-2-carboxamide Chemical compound C1C[C@@H](NC)CC[C@@H]1N(C(=O)C1=C(C2=CC=CC=C2S1)Cl)CC1=CC=CC(C=2C=CN=CC=2)=C1 VFSUUTYAEQOIMW-YHBQERECSA-N 0.000 claims description 14
- 230000026731 phosphorylation Effects 0.000 claims description 14
- 238000006366 phosphorylation reaction Methods 0.000 claims description 14
- FYBHCRQFSFYWPY-UHFFFAOYSA-N purmorphamine Chemical compound C1CCCCC1N1C2=NC(OC=3C4=CC=CC=C4C=CC=3)=NC(NC=3C=CC(=CC=3)N3CCOCC3)=C2N=C1 FYBHCRQFSFYWPY-UHFFFAOYSA-N 0.000 claims description 14
- 239000003112 inhibitor Substances 0.000 claims description 13
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 12
- 210000002451 diencephalon Anatomy 0.000 claims description 12
- 239000011435 rock Substances 0.000 claims description 12
- HIJMSZGHKQPPJS-UHFFFAOYSA-N 3-(6-methylpyridin-2-yl)-n-phenyl-4-quinolin-4-ylpyrazole-1-carbothioamide Chemical compound CC1=CC=CC(C=2C(=CN(N=2)C(=S)NC=2C=CC=CC=2)C=2C3=CC=CC=C3N=CC=2)=N1 HIJMSZGHKQPPJS-UHFFFAOYSA-N 0.000 claims description 10
- CDOVNWNANFFLFJ-UHFFFAOYSA-N 4-[6-[4-(1-piperazinyl)phenyl]-3-pyrazolo[1,5-a]pyrimidinyl]quinoline Chemical compound C1CNCCN1C1=CC=C(C2=CN3N=CC(=C3N=C2)C=2C3=CC=CC=C3N=CC=2)C=C1 CDOVNWNANFFLFJ-UHFFFAOYSA-N 0.000 claims description 7
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 claims description 7
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 claims description 7
- DWJXYEABWRJFSP-XOBRGWDASA-N DAPT Chemical compound N([C@@H](C)C(=O)N[C@H](C(=O)OC(C)(C)C)C=1C=CC=CC=1)C(=O)CC1=CC(F)=CC(F)=C1 DWJXYEABWRJFSP-XOBRGWDASA-N 0.000 claims description 7
- 238000011977 dual antiplatelet therapy Methods 0.000 claims description 7
- 231100000507 endocrine disrupting Toxicity 0.000 claims description 7
- 230000001939 inductive effect Effects 0.000 claims description 7
- 238000000926 separation method Methods 0.000 claims description 7
- 210000002966 serum Anatomy 0.000 claims description 6
- 239000000725 suspension Substances 0.000 claims description 6
- IDDDVXIUIXWAGJ-DDSAHXNVSA-N 4-[(1r)-1-aminoethyl]-n-pyridin-4-ylcyclohexane-1-carboxamide;dihydrochloride Chemical compound Cl.Cl.C1CC([C@H](N)C)CCC1C(=O)NC1=CC=NC=C1 IDDDVXIUIXWAGJ-DDSAHXNVSA-N 0.000 claims description 5
- 230000002757 inflammatory effect Effects 0.000 claims description 5
- 210000001020 neural plate Anatomy 0.000 claims description 3
- 108700013515 Wnt3A Proteins 0.000 claims 6
- 102000044880 Wnt3A Human genes 0.000 claims 6
- 102000009024 Epidermal Growth Factor Human genes 0.000 claims 2
- 101710177179 Cytochrome b5 Proteins 0.000 claims 1
- 102100024745 DNA-directed RNA polymerase, mitochondrial Human genes 0.000 claims 1
- 101000686765 Homo sapiens DNA-directed RNA polymerase, mitochondrial Proteins 0.000 claims 1
- 101000874364 Homo sapiens Protein SCO2 homolog, mitochondrial Proteins 0.000 claims 1
- 101000835023 Homo sapiens Transcription factor A, mitochondrial Proteins 0.000 claims 1
- 102100035546 Protein SCO2 homolog, mitochondrial Human genes 0.000 claims 1
- 102100026155 Transcription factor A, mitochondrial Human genes 0.000 claims 1
- 210000001519 tissue Anatomy 0.000 abstract description 87
- 210000000056 organ Anatomy 0.000 abstract description 56
- 230000001965 increasing effect Effects 0.000 abstract description 44
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 32
- 201000010099 disease Diseases 0.000 abstract description 24
- 239000012530 fluid Substances 0.000 abstract description 24
- 241000894006 Bacteria Species 0.000 abstract description 14
- 230000003833 cell viability Effects 0.000 abstract description 13
- 210000002865 immune cell Anatomy 0.000 abstract description 12
- 230000001413 cellular effect Effects 0.000 abstract description 7
- 230000007614 genetic variation Effects 0.000 abstract description 4
- 208000002551 irritable bowel syndrome Diseases 0.000 abstract description 4
- 230000035790 physiological processes and functions Effects 0.000 abstract description 4
- 230000002459 sustained effect Effects 0.000 abstract description 3
- 238000000059 patterning Methods 0.000 abstract description 2
- 239000000598 endocrine disruptor Substances 0.000 description 164
- 108090000623 proteins and genes Proteins 0.000 description 115
- 238000011282 treatment Methods 0.000 description 101
- 239000002609 medium Substances 0.000 description 77
- 230000008672 reprogramming Effects 0.000 description 61
- 230000000694 effects Effects 0.000 description 56
- 210000002919 epithelial cell Anatomy 0.000 description 52
- 102000000905 Cadherin Human genes 0.000 description 51
- 108050007957 Cadherin Proteins 0.000 description 51
- 210000004379 membrane Anatomy 0.000 description 50
- 239000012528 membrane Substances 0.000 description 50
- 210000000130 stem cell Anatomy 0.000 description 48
- 102100023050 Nuclear factor NF-kappa-B p105 subunit Human genes 0.000 description 44
- 102000004169 proteins and genes Human genes 0.000 description 44
- 230000028327 secretion Effects 0.000 description 41
- 102000004874 Synaptophysin Human genes 0.000 description 36
- 108090001076 Synaptophysin Proteins 0.000 description 36
- 238000002474 experimental method Methods 0.000 description 36
- -1 phospho Chemical class 0.000 description 36
- 102100035100 Transcription factor p65 Human genes 0.000 description 34
- 238000010899 nucleation Methods 0.000 description 34
- 102400001368 Epidermal growth factor Human genes 0.000 description 33
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 32
- 210000002490 intestinal epithelial cell Anatomy 0.000 description 32
- 230000002124 endocrine Effects 0.000 description 31
- 230000037361 pathway Effects 0.000 description 31
- 101800001586 Ghrelin Proteins 0.000 description 30
- 102400000442 Ghrelin-28 Human genes 0.000 description 30
- 101000954805 Homo sapiens Protein Wnt-3a Proteins 0.000 description 30
- 102100037051 Protein Wnt-3a Human genes 0.000 description 30
- GNKDKYIHGQKHHM-RJKLHVOGSA-N ghrelin Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)CN)COC(=O)CCCCCCC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CC=CC=C1 GNKDKYIHGQKHHM-RJKLHVOGSA-N 0.000 description 30
- 238000003365 immunocytochemistry Methods 0.000 description 28
- 230000002829 reductive effect Effects 0.000 description 28
- 230000004044 response Effects 0.000 description 28
- 238000012360 testing method Methods 0.000 description 28
- 239000003981 vehicle Substances 0.000 description 28
- 238000003119 immunoblot Methods 0.000 description 27
- 230000002438 mitochondrial effect Effects 0.000 description 27
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 26
- 230000002496 gastric effect Effects 0.000 description 26
- 102000005747 Transcription Factor RelA Human genes 0.000 description 25
- 108010031154 Transcription Factor RelA Proteins 0.000 description 25
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 24
- 239000010410 layer Substances 0.000 description 24
- 230000035800 maturation Effects 0.000 description 24
- 230000000241 respiratory effect Effects 0.000 description 24
- 239000013598 vector Substances 0.000 description 24
- 102100031655 Cytochrome b5 Human genes 0.000 description 23
- 239000003153 chemical reaction reagent Substances 0.000 description 23
- 238000001000 micrograph Methods 0.000 description 23
- 208000008589 Obesity Diseases 0.000 description 22
- SNGREZUHAYWORS-UHFFFAOYSA-N perfluorooctanoic acid Chemical compound OC(=O)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F SNGREZUHAYWORS-UHFFFAOYSA-N 0.000 description 22
- PIILXFBHQILWPS-UHFFFAOYSA-N tributyltin Chemical compound CCCC[Sn](CCCC)CCCC PIILXFBHQILWPS-UHFFFAOYSA-N 0.000 description 22
- 210000003890 endocrine cell Anatomy 0.000 description 21
- 235000020824 obesity Nutrition 0.000 description 21
- CDEURGJCGCHYFH-DJLDLDEBSA-N 5-ethynyl-2'-deoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(C#C)=C1 CDEURGJCGCHYFH-DJLDLDEBSA-N 0.000 description 20
- 230000001684 chronic effect Effects 0.000 description 20
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 20
- 102100023059 Nuclear factor NF-kappa-B p100 subunit Human genes 0.000 description 19
- 238000001994 activation Methods 0.000 description 19
- 230000015572 biosynthetic process Effects 0.000 description 19
- 238000011161 development Methods 0.000 description 19
- 230000018109 developmental process Effects 0.000 description 19
- 238000010586 diagram Methods 0.000 description 19
- 239000003550 marker Substances 0.000 description 19
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 18
- 230000004913 activation Effects 0.000 description 18
- 238000011284 combination treatment Methods 0.000 description 18
- 230000003247 decreasing effect Effects 0.000 description 18
- 239000003814 drug Substances 0.000 description 18
- 210000003158 enteroendocrine cell Anatomy 0.000 description 18
- 210000002175 goblet cell Anatomy 0.000 description 18
- 230000001404 mediated effect Effects 0.000 description 18
- 230000000394 mitotic effect Effects 0.000 description 18
- 241000283973 Oryctolagus cuniculus Species 0.000 description 17
- 239000006285 cell suspension Substances 0.000 description 17
- 230000002068 genetic effect Effects 0.000 description 16
- 230000006698 induction Effects 0.000 description 16
- 210000000936 intestine Anatomy 0.000 description 16
- 210000001778 pluripotent stem cell Anatomy 0.000 description 16
- 108010007167 Cytochromes b5 Proteins 0.000 description 15
- 238000003556 assay Methods 0.000 description 15
- 238000012512 characterization method Methods 0.000 description 15
- 210000004347 intestinal mucosa Anatomy 0.000 description 15
- 230000011664 signaling Effects 0.000 description 15
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 14
- 241001559542 Hippocampus hippocampus Species 0.000 description 14
- 101000979342 Homo sapiens Nuclear factor NF-kappa-B p105 subunit Proteins 0.000 description 14
- 229940079593 drug Drugs 0.000 description 14
- 239000012091 fetal bovine serum Substances 0.000 description 14
- 238000003384 imaging method Methods 0.000 description 14
- 238000010166 immunofluorescence Methods 0.000 description 14
- 238000011534 incubation Methods 0.000 description 14
- 238000001543 one-way ANOVA Methods 0.000 description 14
- YNXLOPYTAAFMTN-SBUIBGKBSA-N C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 Chemical compound C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 YNXLOPYTAAFMTN-SBUIBGKBSA-N 0.000 description 13
- 102000004127 Cytokines Human genes 0.000 description 13
- 108090000695 Cytokines Proteins 0.000 description 13
- 108010088847 Peptide YY Proteins 0.000 description 13
- 102100029909 Peptide YY Human genes 0.000 description 13
- 238000011529 RT qPCR Methods 0.000 description 13
- 230000027455 binding Effects 0.000 description 13
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 13
- 210000004698 lymphocyte Anatomy 0.000 description 13
- 210000003470 mitochondria Anatomy 0.000 description 13
- 230000001105 regulatory effect Effects 0.000 description 13
- 238000010186 staining Methods 0.000 description 13
- 230000003068 static effect Effects 0.000 description 13
- 108020004414 DNA Proteins 0.000 description 12
- 238000002965 ELISA Methods 0.000 description 12
- 208000018522 Gastrointestinal disease Diseases 0.000 description 12
- 108010078814 Tumor Suppressor Protein p53 Proteins 0.000 description 12
- 210000001035 gastrointestinal tract Anatomy 0.000 description 12
- 239000001963 growth medium Substances 0.000 description 12
- 108091070501 miRNA Proteins 0.000 description 12
- 206010028980 Neoplasm Diseases 0.000 description 11
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 11
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 11
- 208000010643 digestive system disease Diseases 0.000 description 11
- 238000005516 engineering process Methods 0.000 description 11
- 208000018685 gastrointestinal system disease Diseases 0.000 description 11
- 230000007246 mechanism Effects 0.000 description 11
- 210000003097 mucus Anatomy 0.000 description 11
- 230000035882 stress Effects 0.000 description 11
- 238000001262 western blot Methods 0.000 description 11
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 10
- 101000687905 Homo sapiens Transcription factor SOX-2 Proteins 0.000 description 10
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 10
- 102100024270 Transcription factor SOX-2 Human genes 0.000 description 10
- 238000004458 analytical method Methods 0.000 description 10
- 230000004888 barrier function Effects 0.000 description 10
- 230000008859 change Effects 0.000 description 10
- 238000007877 drug screening Methods 0.000 description 10
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 10
- 238000002073 fluorescence micrograph Methods 0.000 description 10
- 230000001976 improved effect Effects 0.000 description 10
- 230000005764 inhibitory process Effects 0.000 description 10
- 239000000463 material Substances 0.000 description 10
- 230000003278 mimic effect Effects 0.000 description 10
- 230000004898 mitochondrial function Effects 0.000 description 10
- 239000002243 precursor Substances 0.000 description 10
- 238000003762 quantitative reverse transcription PCR Methods 0.000 description 10
- FAWLNURBQMTKEB-URDPEVQOSA-N 213546-53-3 Chemical compound N([C@@H](C)C(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N1[C@@H](CCC1)C(O)=O)C(C)C)C(C)C)C(=O)[C@@H]1CCCN1C(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)N)C(C)C FAWLNURBQMTKEB-URDPEVQOSA-N 0.000 description 9
- 108050008832 Fatty acid-binding protein, intestinal Proteins 0.000 description 9
- 102100026748 Fatty acid-binding protein, intestinal Human genes 0.000 description 9
- 102400000921 Gastrin Human genes 0.000 description 9
- 108010052343 Gastrins Proteins 0.000 description 9
- 101710151321 Melanostatin Proteins 0.000 description 9
- 102400000064 Neuropeptide Y Human genes 0.000 description 9
- 108090000189 Neuropeptides Proteins 0.000 description 9
- 102000005157 Somatostatin Human genes 0.000 description 9
- 108010056088 Somatostatin Proteins 0.000 description 9
- 230000005754 cellular signaling Effects 0.000 description 9
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 9
- 238000004891 communication Methods 0.000 description 9
- 238000013461 design Methods 0.000 description 9
- 238000000684 flow cytometry Methods 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 238000002825 functional assay Methods 0.000 description 9
- 239000003102 growth factor Substances 0.000 description 9
- 229940088597 hormone Drugs 0.000 description 9
- 239000005556 hormone Substances 0.000 description 9
- 238000003125 immunofluorescent labeling Methods 0.000 description 9
- 238000000338 in vitro Methods 0.000 description 9
- 210000000265 leukocyte Anatomy 0.000 description 9
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 description 9
- 239000000523 sample Substances 0.000 description 9
- 229940076279 serotonin Drugs 0.000 description 9
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 9
- 229960000553 somatostatin Drugs 0.000 description 9
- 230000025366 tissue development Effects 0.000 description 9
- WHNFPRLDDSXQCL-UAZQEYIDSA-N α-msh Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(N)=O)NC(=O)[C@H](CO)NC(C)=O)C1=CC=C(O)C=C1 WHNFPRLDDSXQCL-UAZQEYIDSA-N 0.000 description 9
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 8
- 102000003849 Cytochrome P450 Human genes 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 8
- 241000283074 Equus asinus Species 0.000 description 8
- 108700005087 Homeobox Genes Proteins 0.000 description 8
- 108020005196 Mitochondrial DNA Proteins 0.000 description 8
- 101710126211 POU domain, class 5, transcription factor 1 Proteins 0.000 description 8
- 102000040945 Transcription factor Human genes 0.000 description 8
- 108091023040 Transcription factor Proteins 0.000 description 8
- NIJJYAXOARWZEE-UHFFFAOYSA-N Valproic acid Chemical compound CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 8
- 230000002411 adverse Effects 0.000 description 8
- 210000000601 blood cell Anatomy 0.000 description 8
- 238000012217 deletion Methods 0.000 description 8
- 230000037430 deletion Effects 0.000 description 8
- 230000001627 detrimental effect Effects 0.000 description 8
- 230000008482 dysregulation Effects 0.000 description 8
- 230000003993 interaction Effects 0.000 description 8
- 230000007774 longterm Effects 0.000 description 8
- 230000006609 metabolic stress Effects 0.000 description 8
- 244000005700 microbiome Species 0.000 description 8
- 210000000110 microvilli Anatomy 0.000 description 8
- 230000004065 mitochondrial dysfunction Effects 0.000 description 8
- 230000035772 mutation Effects 0.000 description 8
- 230000035699 permeability Effects 0.000 description 8
- 230000009467 reduction Effects 0.000 description 8
- 239000002356 single layer Substances 0.000 description 8
- 210000002784 stomach Anatomy 0.000 description 8
- 101100257359 Caenorhabditis elegans sox-2 gene Proteins 0.000 description 7
- 101150047030 ERO1 gene Proteins 0.000 description 7
- 101710128765 Enhancer of filamentation 1 Proteins 0.000 description 7
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 7
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 7
- 102100028538 Guanylate-binding protein 4 Human genes 0.000 description 7
- 101710110797 Guanylate-binding protein 4 Proteins 0.000 description 7
- 101000617805 Homo sapiens Staphylococcal nuclease domain-containing protein 1 Proteins 0.000 description 7
- 101000830894 Homo sapiens Targeting protein for Xklp2 Proteins 0.000 description 7
- 102100040061 Indoleamine 2,3-dioxygenase 1 Human genes 0.000 description 7
- 101710120843 Indoleamine 2,3-dioxygenase 1 Proteins 0.000 description 7
- 102400000740 Melanocyte-stimulating hormone alpha Human genes 0.000 description 7
- 101710200814 Melanotropin alpha Proteins 0.000 description 7
- 101100257363 Mus musculus Sox2 gene Proteins 0.000 description 7
- 101100494726 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pep-4 gene Proteins 0.000 description 7
- 101000942603 Schizosaccharomyces pombe (strain 972 / ATCC 24843) Condensin complex subunit 3 Proteins 0.000 description 7
- 102100021996 Staphylococcal nuclease domain-containing protein 1 Human genes 0.000 description 7
- 102100035071 Vimentin Human genes 0.000 description 7
- 108010065472 Vimentin Proteins 0.000 description 7
- 102000013814 Wnt Human genes 0.000 description 7
- 108050003627 Wnt Proteins 0.000 description 7
- 101000963191 Xenopus laevis Maternal DNA replication licensing factor mcm3 Proteins 0.000 description 7
- 230000002159 abnormal effect Effects 0.000 description 7
- 210000003295 arcuate nucleus Anatomy 0.000 description 7
- 230000033228 biological regulation Effects 0.000 description 7
- 201000011510 cancer Diseases 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 230000000052 comparative effect Effects 0.000 description 7
- 230000006378 damage Effects 0.000 description 7
- 210000002744 extracellular matrix Anatomy 0.000 description 7
- 230000033001 locomotion Effects 0.000 description 7
- 108020004999 messenger RNA Proteins 0.000 description 7
- 230000002503 metabolic effect Effects 0.000 description 7
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 7
- 108090000765 processed proteins & peptides Proteins 0.000 description 7
- 238000011002 quantification Methods 0.000 description 7
- 238000011084 recovery Methods 0.000 description 7
- 230000029058 respiratory gaseous exchange Effects 0.000 description 7
- 150000003384 small molecules Chemical class 0.000 description 7
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 7
- 239000000758 substrate Substances 0.000 description 7
- 210000005048 vimentin Anatomy 0.000 description 7
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 6
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 6
- 230000004568 DNA-binding Effects 0.000 description 6
- 101000922386 Homo sapiens Cytochrome b5 Proteins 0.000 description 6
- 231100000002 MTT assay Toxicity 0.000 description 6
- 238000000134 MTT assay Methods 0.000 description 6
- 102000016943 Muramidase Human genes 0.000 description 6
- 108010014251 Muramidase Proteins 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 6
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 6
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 6
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 6
- 210000003892 absorptive cell Anatomy 0.000 description 6
- 230000003321 amplification Effects 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 238000001574 biopsy Methods 0.000 description 6
- BJQHLKABXJIVAM-UHFFFAOYSA-N bis(2-ethylhexyl) phthalate Chemical compound CCCCC(CC)COC(=O)C1=CC=CC=C1C(=O)OCC(CC)CCCC BJQHLKABXJIVAM-UHFFFAOYSA-N 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- 206010009887 colitis Diseases 0.000 description 6
- 230000001276 controlling effect Effects 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 230000009931 harmful effect Effects 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 210000004692 intercellular junction Anatomy 0.000 description 6
- 101150111214 lin-28 gene Proteins 0.000 description 6
- 229960000274 lysozyme Drugs 0.000 description 6
- 239000004325 lysozyme Substances 0.000 description 6
- 235000010335 lysozyme Nutrition 0.000 description 6
- 208000030159 metabolic disease Diseases 0.000 description 6
- 230000001537 neural effect Effects 0.000 description 6
- 238000003199 nucleic acid amplification method Methods 0.000 description 6
- 238000005457 optimization Methods 0.000 description 6
- 230000002269 spontaneous effect Effects 0.000 description 6
- 239000013589 supplement Substances 0.000 description 6
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 5
- 231100000716 Acceptable daily intake Toxicity 0.000 description 5
- 229920002307 Dextran Polymers 0.000 description 5
- 239000004375 Dextrin Substances 0.000 description 5
- 229920001353 Dextrin Polymers 0.000 description 5
- 101150059079 EBNA1 gene Proteins 0.000 description 5
- 102100030013 Endoribonuclease Human genes 0.000 description 5
- 208000031448 Genomic Instability Diseases 0.000 description 5
- 101001010783 Homo sapiens Endoribonuclease Proteins 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 101100310648 Mus musculus Sox17 gene Proteins 0.000 description 5
- 102000003797 Neuropeptides Human genes 0.000 description 5
- 208000005718 Stomach Neoplasms Diseases 0.000 description 5
- 238000010171 animal model Methods 0.000 description 5
- 239000000090 biomarker Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 210000004556 brain Anatomy 0.000 description 5
- 238000004113 cell culture Methods 0.000 description 5
- 230000010261 cell growth Effects 0.000 description 5
- 238000003501 co-culture Methods 0.000 description 5
- 235000019425 dextrin Nutrition 0.000 description 5
- 239000004205 dimethyl polysiloxane Substances 0.000 description 5
- 230000008175 fetal development Effects 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 206010017758 gastric cancer Diseases 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- 235000003642 hunger Nutrition 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 238000011068 loading method Methods 0.000 description 5
- 230000000955 neuroendocrine Effects 0.000 description 5
- 230000010627 oxidative phosphorylation Effects 0.000 description 5
- 210000003134 paneth cell Anatomy 0.000 description 5
- 239000013612 plasmid Substances 0.000 description 5
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 230000037351 starvation Effects 0.000 description 5
- 201000011549 stomach cancer Diseases 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- UPTYCYWTFGTCCG-UHFFFAOYSA-N 5-(1-piperazinylsulfonyl)isoquinoline Chemical compound C=1C=CC2=CN=CC=C2C=1S(=O)(=O)N1CCNCC1 UPTYCYWTFGTCCG-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 241000283707 Capra Species 0.000 description 4
- 206010009900 Colitis ulcerative Diseases 0.000 description 4
- 208000011231 Crohn disease Diseases 0.000 description 4
- 102000000634 Cytochrome c oxidase subunit IV Human genes 0.000 description 4
- 108090000365 Cytochrome-c oxidases Proteins 0.000 description 4
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 description 4
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 4
- 102100035688 Guanylate-binding protein 1 Human genes 0.000 description 4
- 101710110781 Guanylate-binding protein 1 Proteins 0.000 description 4
- 102100030641 Homeobox protein orthopedia Human genes 0.000 description 4
- 101001002634 Homo sapiens Interleukin-1 alpha Proteins 0.000 description 4
- 101000983161 Homo sapiens Phospholipase A2, membrane associated Proteins 0.000 description 4
- 102000004877 Insulin Human genes 0.000 description 4
- 108090001061 Insulin Proteins 0.000 description 4
- 102100020881 Interleukin-1 alpha Human genes 0.000 description 4
- 102100031036 Leucine-rich repeat-containing G-protein coupled receptor 5 Human genes 0.000 description 4
- 101710174256 Leucine-rich repeat-containing G-protein coupled receptor 5 Proteins 0.000 description 4
- 102000001796 Melanocortin 4 receptors Human genes 0.000 description 4
- 108700011259 MicroRNAs Proteins 0.000 description 4
- 102000007296 Mucin-2 Human genes 0.000 description 4
- 108010008705 Mucin-2 Proteins 0.000 description 4
- 208000012868 Overgrowth Diseases 0.000 description 4
- 102100026831 Phospholipase A2, membrane associated Human genes 0.000 description 4
- 108091027967 Small hairpin RNA Proteins 0.000 description 4
- 108010021436 Type 4 Melanocortin Receptor Proteins 0.000 description 4
- 201000006704 Ulcerative Colitis Diseases 0.000 description 4
- 241000700605 Viruses Species 0.000 description 4
- 238000009825 accumulation Methods 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 230000000845 anti-microbial effect Effects 0.000 description 4
- 239000004599 antimicrobial Substances 0.000 description 4
- 230000011712 cell development Effects 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- 239000000356 contaminant Substances 0.000 description 4
- 238000005520 cutting process Methods 0.000 description 4
- 230000001086 cytosolic effect Effects 0.000 description 4
- 230000006735 deficit Effects 0.000 description 4
- 238000000326 densiometry Methods 0.000 description 4
- 238000003255 drug test Methods 0.000 description 4
- 230000004064 dysfunction Effects 0.000 description 4
- 210000001671 embryonic stem cell Anatomy 0.000 description 4
- 230000007613 environmental effect Effects 0.000 description 4
- 230000004634 feeding behavior Effects 0.000 description 4
- 230000008014 freezing Effects 0.000 description 4
- 238000007710 freezing Methods 0.000 description 4
- 230000003054 hormonal effect Effects 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 230000004060 metabolic process Effects 0.000 description 4
- 108091031881 miR-290 stem-loop Proteins 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 239000002773 nucleotide Substances 0.000 description 4
- 125000003729 nucleotide group Chemical group 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 239000002994 raw material Substances 0.000 description 4
- 238000003860 storage Methods 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- 238000010257 thawing Methods 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 238000013518 transcription Methods 0.000 description 4
- 230000035897 transcription Effects 0.000 description 4
- 230000004906 unfolded protein response Effects 0.000 description 4
- 230000003827 upregulation Effects 0.000 description 4
- 229960000604 valproic acid Drugs 0.000 description 4
- 230000002407 ATP formation Effects 0.000 description 3
- 102000015735 Beta-catenin Human genes 0.000 description 3
- 108060000903 Beta-catenin Proteins 0.000 description 3
- 101150087009 CYB5A gene Proteins 0.000 description 3
- 241000766026 Coregonus nasus Species 0.000 description 3
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 3
- 102100035965 Gastrokine-1 Human genes 0.000 description 3
- 102100028539 Guanylate-binding protein 5 Human genes 0.000 description 3
- 101001058850 Homo sapiens Guanylate-binding protein 5 Proteins 0.000 description 3
- 102000008070 Interferon-gamma Human genes 0.000 description 3
- 108010074328 Interferon-gamma Proteins 0.000 description 3
- 108010085895 Laminin Proteins 0.000 description 3
- 101710128836 Large T antigen Proteins 0.000 description 3
- 108090001090 Lectins Proteins 0.000 description 3
- 102000004856 Lectins Human genes 0.000 description 3
- 101150059949 MUC4 gene Proteins 0.000 description 3
- 101150111110 NKX2-1 gene Proteins 0.000 description 3
- 208000007683 Pediatric Obesity Diseases 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 3
- 102100026918 Phospholipase A2 Human genes 0.000 description 3
- 108010058864 Phospholipases A2 Proteins 0.000 description 3
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 3
- 102100037886 Regenerating islet-derived protein 3-gamma Human genes 0.000 description 3
- 101710086144 Regenerating islet-derived protein 3-gamma Proteins 0.000 description 3
- 108010044012 STAT1 Transcription Factor Proteins 0.000 description 3
- 102100029904 Signal transducer and activator of transcription 1-alpha/beta Human genes 0.000 description 3
- 102100021796 Sonic hedgehog protein Human genes 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 102000008816 Trefoil Factor-2 Human genes 0.000 description 3
- 108010088411 Trefoil Factor-2 Proteins 0.000 description 3
- 101710158352 Type III intermediate filament Proteins 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 230000001464 adherent effect Effects 0.000 description 3
- 230000000844 anti-bacterial effect Effects 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 230000002354 daily effect Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000006471 dimerization reaction Methods 0.000 description 3
- 230000003828 downregulation Effects 0.000 description 3
- 239000003344 environmental pollutant Substances 0.000 description 3
- 239000003256 environmental substance Substances 0.000 description 3
- 210000005081 epithelial layer Anatomy 0.000 description 3
- 238000013401 experimental design Methods 0.000 description 3
- 210000003754 fetus Anatomy 0.000 description 3
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 3
- 235000013305 food Nutrition 0.000 description 3
- 235000013373 food additive Nutrition 0.000 description 3
- 239000002778 food additive Substances 0.000 description 3
- 102000006602 glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 3
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 3
- 230000013632 homeostatic process Effects 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 238000012760 immunocytochemical staining Methods 0.000 description 3
- 230000001771 impaired effect Effects 0.000 description 3
- 208000027866 inflammatory disease Diseases 0.000 description 3
- 229960003130 interferon gamma Drugs 0.000 description 3
- 210000003963 intermediate filament Anatomy 0.000 description 3
- 230000003870 intestinal permeability Effects 0.000 description 3
- 239000002523 lectin Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000002826 magnetic-activated cell sorting Methods 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 230000004769 mitochondrial stress Effects 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- 230000017128 negative regulation of NF-kappaB transcription factor activity Effects 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 238000007427 paired t-test Methods 0.000 description 3
- 230000007310 pathophysiology Effects 0.000 description 3
- 229940049954 penicillin Drugs 0.000 description 3
- 230000000737 periodic effect Effects 0.000 description 3
- 230000008855 peristalsis Effects 0.000 description 3
- 230000003094 perturbing effect Effects 0.000 description 3
- 231100000614 poison Toxicity 0.000 description 3
- 239000013641 positive control Substances 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 238000009331 sowing Methods 0.000 description 3
- 229960005322 streptomycin Drugs 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 210000001578 tight junction Anatomy 0.000 description 3
- 239000003440 toxic substance Substances 0.000 description 3
- 210000005042 type III intermediate filament Anatomy 0.000 description 3
- 210000004291 uterus Anatomy 0.000 description 3
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 2
- 208000037068 Abnormal Karyotype Diseases 0.000 description 2
- 108700021677 Agouti-Related Proteins 0.000 description 2
- IGAZHQIYONOHQN-UHFFFAOYSA-N Alexa Fluor 555 Chemical compound C=12C=CC(=N)C(S(O)(=O)=O)=C2OC2=C(S(O)(=O)=O)C(N)=CC=C2C=1C1=CC=C(C(O)=O)C=C1C(O)=O IGAZHQIYONOHQN-UHFFFAOYSA-N 0.000 description 2
- 102100027308 Apoptosis regulator BAX Human genes 0.000 description 2
- 108050006685 Apoptosis regulator BAX Proteins 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 2
- 102100028914 Catenin beta-1 Human genes 0.000 description 2
- 108010016788 Cyclin-Dependent Kinase Inhibitor p21 Proteins 0.000 description 2
- 102100033270 Cyclin-dependent kinase inhibitor 1 Human genes 0.000 description 2
- 239000004803 Di-2ethylhexylphthalate Substances 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 108010067306 Fibronectins Proteins 0.000 description 2
- 102000016359 Fibronectins Human genes 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- 102000054184 GADD45 Human genes 0.000 description 2
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 2
- 102100031150 Growth arrest and DNA damage-inducible protein GADD45 alpha Human genes 0.000 description 2
- 102100031153 Growth arrest and DNA damage-inducible protein GADD45 beta Human genes 0.000 description 2
- 101150033506 HOX gene Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000916173 Homo sapiens Catenin beta-1 Proteins 0.000 description 2
- 101001075218 Homo sapiens Gastrokine-1 Proteins 0.000 description 2
- 101001066158 Homo sapiens Growth arrest and DNA damage-inducible protein GADD45 alpha Proteins 0.000 description 2
- 101001066164 Homo sapiens Growth arrest and DNA damage-inducible protein GADD45 beta Proteins 0.000 description 2
- 101001066163 Homo sapiens Growth arrest and DNA damage-inducible protein GADD45 gamma Proteins 0.000 description 2
- 101001033249 Homo sapiens Interleukin-1 beta Proteins 0.000 description 2
- 101000972286 Homo sapiens Mucin-4 Proteins 0.000 description 2
- 101001094700 Homo sapiens POU domain, class 5, transcription factor 1 Proteins 0.000 description 2
- 101000606748 Homo sapiens Pepsin A-5 Proteins 0.000 description 2
- 101000713275 Homo sapiens Solute carrier family 22 member 3 Proteins 0.000 description 2
- 101000659774 Homo sapiens Taste receptor type 1 member 3 Proteins 0.000 description 2
- 101000611183 Homo sapiens Tumor necrosis factor Proteins 0.000 description 2
- 101001127470 Homo sapiens p53 apoptosis effector related to PMP-22 Proteins 0.000 description 2
- 208000026350 Inborn Genetic disease Diseases 0.000 description 2
- 102100039065 Interleukin-1 beta Human genes 0.000 description 2
- 102000004889 Interleukin-6 Human genes 0.000 description 2
- 108090001005 Interleukin-6 Proteins 0.000 description 2
- 101710172072 Kexin Proteins 0.000 description 2
- 239000002211 L-ascorbic acid Substances 0.000 description 2
- 235000000069 L-ascorbic acid Nutrition 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- 229930182816 L-glutamine Natural products 0.000 description 2
- 102000016267 Leptin Human genes 0.000 description 2
- 108010092277 Leptin Proteins 0.000 description 2
- 101800001751 Melanocyte-stimulating hormone alpha Proteins 0.000 description 2
- 108010008699 Mucin-4 Proteins 0.000 description 2
- 102000007270 Mucin-4 Human genes 0.000 description 2
- 102100022693 Mucin-4 Human genes 0.000 description 2
- 102100021878 Neuronal pentraxin-2 Human genes 0.000 description 2
- 101710155147 Neuronal pentraxin-2 Proteins 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- 102100039652 Pepsin A-5 Human genes 0.000 description 2
- 101150071480 Polrmt gene Proteins 0.000 description 2
- 101710183548 Pyridoxal 5'-phosphate synthase subunit PdxS Proteins 0.000 description 2
- 102100035459 Pyruvate dehydrogenase protein X component, mitochondrial Human genes 0.000 description 2
- 108700008625 Reporter Genes Proteins 0.000 description 2
- 101150064954 SCO2 gene Proteins 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 108090000787 Subtilisin Proteins 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- 101150080074 TP53 gene Proteins 0.000 description 2
- 102100035942 Taste receptor type 1 member 3 Human genes 0.000 description 2
- 101150080431 Tfam gene Proteins 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical class O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 108050001370 Tight junction protein ZO-1 Proteins 0.000 description 2
- 108700009124 Transcription Initiation Site Proteins 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- 108090000901 Transferrin Proteins 0.000 description 2
- 102100040247 Tumor necrosis factor Human genes 0.000 description 2
- 102000044820 Zonula Occludens-1 Human genes 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000000853 adhesive Substances 0.000 description 2
- 230000001070 adhesive effect Effects 0.000 description 2
- 210000001789 adipocyte Anatomy 0.000 description 2
- 210000000577 adipose tissue Anatomy 0.000 description 2
- 230000009118 appropriate response Effects 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 210000000270 basal cell Anatomy 0.000 description 2
- 238000003766 bioinformatics method Methods 0.000 description 2
- 239000003181 biological factor Substances 0.000 description 2
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 2
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 230000007910 cell fusion Effects 0.000 description 2
- 230000011748 cell maturation Effects 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 235000013339 cereals Nutrition 0.000 description 2
- 208000037976 chronic inflammation Diseases 0.000 description 2
- 230000006020 chronic inflammation Effects 0.000 description 2
- 230000005757 colony formation Effects 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 210000004748 cultured cell Anatomy 0.000 description 2
- 206010061428 decreased appetite Diseases 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 230000002939 deleterious effect Effects 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 230000009547 development abnormality Effects 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- BVTBRVFYZUCAKH-UHFFFAOYSA-L disodium selenite Chemical compound [Na+].[Na+].[O-][Se]([O-])=O BVTBRVFYZUCAKH-UHFFFAOYSA-L 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000000428 dust Substances 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 230000002900 effect on cell Effects 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 230000007368 endocrine function Effects 0.000 description 2
- 210000004039 endoderm cell Anatomy 0.000 description 2
- 230000001973 epigenetic effect Effects 0.000 description 2
- 230000003090 exacerbative effect Effects 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 230000001605 fetal effect Effects 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 239000012595 freezing medium Substances 0.000 description 2
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 2
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 2
- 238000012239 gene modification Methods 0.000 description 2
- 238000001415 gene therapy Methods 0.000 description 2
- 208000016361 genetic disease Diseases 0.000 description 2
- 230000005017 genetic modification Effects 0.000 description 2
- 235000013617 genetically modified food Nutrition 0.000 description 2
- 238000010362 genome editing Methods 0.000 description 2
- 230000034659 glycolysis Effects 0.000 description 2
- 210000003128 head Anatomy 0.000 description 2
- 238000012744 immunostaining Methods 0.000 description 2
- 238000007901 in situ hybridization Methods 0.000 description 2
- 238000010874 in vitro model Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 238000013383 initial experiment Methods 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 229940039781 leptin Drugs 0.000 description 2
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 2
- 230000003902 lesion Effects 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 239000002679 microRNA Substances 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 238000000520 microinjection Methods 0.000 description 2
- 108091064355 mitochondrial RNA Proteins 0.000 description 2
- 210000005087 mononuclear cell Anatomy 0.000 description 2
- AFRBPRLOPBAGCM-UHFFFAOYSA-N n-[2-(1h-indol-4-yl)ethyl]-n-propylpropan-1-amine Chemical compound CCCN(CCC)CCC1=CC=CC2=C1C=CN2 AFRBPRLOPBAGCM-UHFFFAOYSA-N 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 208000015122 neurodegenerative disease Diseases 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 210000000440 neutrophil Anatomy 0.000 description 2
- 238000012758 nuclear staining Methods 0.000 description 2
- 230000006911 nucleation Effects 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 238000000879 optical micrograph Methods 0.000 description 2
- 108700025694 p53 Genes Proteins 0.000 description 2
- 102100030898 p53 apoptosis effector related to PMP-22 Human genes 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 230000000849 parathyroid Effects 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 230000010412 perfusion Effects 0.000 description 2
- 230000002572 peristaltic effect Effects 0.000 description 2
- 238000011338 personalized therapy Methods 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000008741 proinflammatory signaling process Effects 0.000 description 2
- 210000004129 prosencephalon Anatomy 0.000 description 2
- 238000011552 rat model Methods 0.000 description 2
- 101150085542 relA gene Proteins 0.000 description 2
- 230000003252 repetitive effect Effects 0.000 description 2
- 230000004043 responsiveness Effects 0.000 description 2
- 230000036186 satiety Effects 0.000 description 2
- 235000019627 satiety Nutrition 0.000 description 2
- 210000004739 secretory vesicle Anatomy 0.000 description 2
- 238000010187 selection method Methods 0.000 description 2
- 230000009131 signaling function Effects 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 239000004055 small Interfering RNA Substances 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- 239000011781 sodium selenite Substances 0.000 description 2
- 229960001471 sodium selenite Drugs 0.000 description 2
- 235000015921 sodium selenite Nutrition 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 210000001082 somatic cell Anatomy 0.000 description 2
- 230000009211 stress pathway Effects 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 230000005945 translocation Effects 0.000 description 2
- 238000007492 two-way ANOVA Methods 0.000 description 2
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 230000035899 viability Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 description 1
- 108020004465 16S ribosomal RNA Proteins 0.000 description 1
- RCEBHKDQZCVBTC-UHFFFAOYSA-N 2-acetamidothiophene-3-carboxylic acid Chemical compound CC(=O)NC=1SC=CC=1C(O)=O RCEBHKDQZCVBTC-UHFFFAOYSA-N 0.000 description 1
- 102100022289 60S ribosomal protein L13a Human genes 0.000 description 1
- 229940126565 ATP-synthase inhibitor Drugs 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- 239000012103 Alexa Fluor 488 Substances 0.000 description 1
- 239000012109 Alexa Fluor 568 Substances 0.000 description 1
- 239000012110 Alexa Fluor 594 Substances 0.000 description 1
- 239000012114 Alexa Fluor 647 Substances 0.000 description 1
- 239000012099 Alexa Fluor family Substances 0.000 description 1
- 229930182536 Antimycin Natural products 0.000 description 1
- 102400000059 Arg-vasopressin Human genes 0.000 description 1
- 101800001144 Arg-vasopressin Proteins 0.000 description 1
- 241001436672 Bhatia Species 0.000 description 1
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 description 1
- BMZRVOVNUMQTIN-UHFFFAOYSA-N Carbonyl Cyanide para-Trifluoromethoxyphenylhydrazone Chemical compound FC(F)(F)OC1=CC=C(NN=C(C#N)C#N)C=C1 BMZRVOVNUMQTIN-UHFFFAOYSA-N 0.000 description 1
- 102100032378 Carboxypeptidase E Human genes 0.000 description 1
- 108010058255 Carboxypeptidase H Proteins 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102000020313 Cell-Penetrating Peptides Human genes 0.000 description 1
- 108010051109 Cell-Penetrating Peptides Proteins 0.000 description 1
- 102100024297 Cilia- and flagella-associated protein 410 Human genes 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 102100027476 Cytochrome c oxidase assembly protein COX19 Human genes 0.000 description 1
- 102000018832 Cytochromes Human genes 0.000 description 1
- 108010052832 Cytochromes Proteins 0.000 description 1
- 230000005778 DNA damage Effects 0.000 description 1
- 231100000277 DNA damage Toxicity 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- 102000016911 Deoxyribonucleases Human genes 0.000 description 1
- 108010053770 Deoxyribonucleases Proteins 0.000 description 1
- 208000012239 Developmental disease Diseases 0.000 description 1
- 208000002699 Digestive System Neoplasms Diseases 0.000 description 1
- 108010031111 EBV-encoded nuclear antigen 1 Proteins 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 208000030814 Eating disease Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 208000019454 Feeding and Eating disease Diseases 0.000 description 1
- 210000000712 G cell Anatomy 0.000 description 1
- 101710205777 Gastrokine-1 Proteins 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003693 Hedgehog Proteins Human genes 0.000 description 1
- 108090000031 Hedgehog Proteins Proteins 0.000 description 1
- 238000010867 Hoechst staining Methods 0.000 description 1
- 101710178632 Homeobox protein orthopedia Proteins 0.000 description 1
- 101000681240 Homo sapiens 60S ribosomal protein L13a Proteins 0.000 description 1
- 101000980066 Homo sapiens Cilia- and flagella-associated protein 410 Proteins 0.000 description 1
- 101000770637 Homo sapiens Cytochrome c oxidase assembly protein COX15 homolog Proteins 0.000 description 1
- 101000725442 Homo sapiens Cytochrome c oxidase assembly protein COX19 Proteins 0.000 description 1
- 101001139134 Homo sapiens Krueppel-like factor 4 Proteins 0.000 description 1
- 101000984042 Homo sapiens Protein lin-28 homolog A Proteins 0.000 description 1
- 101000652324 Homo sapiens Transcription factor SOX-17 Proteins 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 1
- 102100029098 Hypoxanthine-guanine phosphoribosyltransferase Human genes 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 108091006081 Inositol-requiring enzyme-1 Proteins 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 102100020677 Krueppel-like factor 4 Human genes 0.000 description 1
- 208000025972 Maternal Obesity Diseases 0.000 description 1
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 1
- 241001599018 Melanogaster Species 0.000 description 1
- 241000736262 Microbiota Species 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 102000015728 Mucins Human genes 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 108010052419 NF-KappaB Inhibitor alpha Proteins 0.000 description 1
- 102000008125 NF-kappa B p52 Subunit Human genes 0.000 description 1
- 108010074852 NF-kappa B p52 Subunit Proteins 0.000 description 1
- 102100039337 NF-kappa-B inhibitor alpha Human genes 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- 108010018674 Neurophysins Proteins 0.000 description 1
- 102000002710 Neurophysins Human genes 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 101800001442 Peptide pr Proteins 0.000 description 1
- 241000233805 Phoenix Species 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 208000001280 Prediabetic State Diseases 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 102100025460 Protein lin-28 homolog A Human genes 0.000 description 1
- 108010026552 Proteome Proteins 0.000 description 1
- 102100022762 R-spondin-1 Human genes 0.000 description 1
- 101710110302 R-spondin-1 Proteins 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 238000011530 RNeasy Mini Kit Methods 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 101000713578 Rattus norvegicus Tubulin beta-3 chain Proteins 0.000 description 1
- 229940123934 Reductase inhibitor Drugs 0.000 description 1
- 108700005075 Regulator Genes Proteins 0.000 description 1
- 230000018199 S phase Effects 0.000 description 1
- 108010028723 SN50 peptide Proteins 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 102100030243 Transcription factor SOX-17 Human genes 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 108091000117 Tyrosine 3-Monooxygenase Proteins 0.000 description 1
- 102000048218 Tyrosine 3-monooxygenases Human genes 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000036982 action potential Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 238000000246 agarose gel electrophoresis Methods 0.000 description 1
- 239000003905 agrochemical Substances 0.000 description 1
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 1
- 150000005005 aminopyrimidines Chemical class 0.000 description 1
- 229940025084 amphetamine Drugs 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 230000000578 anorexic effect Effects 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000002519 antifouling agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- CQIUKKVOEOPUDV-IYSWYEEDSA-N antimycin Chemical compound OC1=C(C(O)=O)C(=O)C(C)=C2[C@H](C)[C@@H](C)OC=C21 CQIUKKVOEOPUDV-IYSWYEEDSA-N 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 210000004082 barrier epithelial cell Anatomy 0.000 description 1
- 239000007640 basal medium Substances 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 230000007698 birth defect Effects 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000004641 brain development Effects 0.000 description 1
- 235000015496 breakfast cereal Nutrition 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 238000011072 cell harvest Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000013553 cell monolayer Substances 0.000 description 1
- 238000003654 cell permeability assay Methods 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 230000008614 cellular interaction Effects 0.000 description 1
- 201000010897 colon adenocarcinoma Diseases 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 238000010219 correlation analysis Methods 0.000 description 1
- 239000008406 cosmetic ingredient Substances 0.000 description 1
- 230000002559 cytogenic effect Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 238000009795 derivation Methods 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 208000016097 disease of metabolism Diseases 0.000 description 1
- 235000014632 disordered eating Nutrition 0.000 description 1
- 238000012137 double-staining Methods 0.000 description 1
- 239000003651 drinking water Substances 0.000 description 1
- 235000020188 drinking water Nutrition 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000000857 drug effect Effects 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000002828 effect on organs or tissue Effects 0.000 description 1
- 238000002001 electrophysiology Methods 0.000 description 1
- 230000007831 electrophysiology Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000019439 energy homeostasis Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 230000004890 epithelial barrier function Effects 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000010195 expression analysis Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- 238000012921 fluorescence analysis Methods 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 239000004811 fluoropolymer Substances 0.000 description 1
- 229920002313 fluoropolymer Polymers 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 210000001156 gastric mucosa Anatomy 0.000 description 1
- 210000005095 gastrointestinal system Anatomy 0.000 description 1
- 238000003209 gene knockout Methods 0.000 description 1
- 238000003208 gene overexpression Methods 0.000 description 1
- 230000030279 gene silencing Effects 0.000 description 1
- 210000001654 germ layer Anatomy 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 230000002414 glycolytic effect Effects 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 108091005708 gustatory receptors Proteins 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 239000000383 hazardous chemical Substances 0.000 description 1
- 231100000206 health hazard Toxicity 0.000 description 1
- 231100000508 hormonal effect Toxicity 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 210000003016 hypothalamus Anatomy 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 231100000986 in utero exposure Toxicity 0.000 description 1
- 239000002440 industrial waste Substances 0.000 description 1
- 239000012678 infectious agent Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 210000005027 intestinal barrier Anatomy 0.000 description 1
- 230000007358 intestinal barrier function Effects 0.000 description 1
- 208000028774 intestinal disease Diseases 0.000 description 1
- 230000003871 intestinal function Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 238000013332 literature search Methods 0.000 description 1
- 238000010859 live-cell imaging Methods 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000010946 mechanistic model Methods 0.000 description 1
- 239000013028 medium composition Substances 0.000 description 1
- 210000002901 mesenchymal stem cell Anatomy 0.000 description 1
- 210000003716 mesoderm Anatomy 0.000 description 1
- 230000004066 metabolic change Effects 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- 238000002493 microarray Methods 0.000 description 1
- 244000005706 microflora Species 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 230000006676 mitochondrial damage Effects 0.000 description 1
- 210000001700 mitochondrial membrane Anatomy 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 238000004264 monolayer culture Methods 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 230000003843 mucus production Effects 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- AEMBWNDIEFEPTH-UHFFFAOYSA-N n-tert-butyl-n-ethylnitrous amide Chemical compound CCN(N=O)C(C)(C)C AEMBWNDIEFEPTH-UHFFFAOYSA-N 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 210000003061 neural cell Anatomy 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000009907 neuroendocrine response Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000008062 neuronal firing Effects 0.000 description 1
- BPGXUIVWLQTVLZ-OFGSCBOVSA-N neuropeptide y(npy) Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 BPGXUIVWLQTVLZ-OFGSCBOVSA-N 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 230000005937 nuclear translocation Effects 0.000 description 1
- 235000015816 nutrient absorption Nutrition 0.000 description 1
- 238000012803 optimization experiment Methods 0.000 description 1
- 230000005305 organ development Effects 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- JSGHQDAEHDRLOI-UHFFFAOYSA-N oxomalononitrile Chemical compound N#CC(=O)C#N JSGHQDAEHDRLOI-UHFFFAOYSA-N 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 230000036284 oxygen consumption Effects 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 210000004923 pancreatic tissue Anatomy 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 238000003068 pathway analysis Methods 0.000 description 1
- 239000013610 patient sample Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000034190 positive regulation of NF-kappaB transcription factor activity Effects 0.000 description 1
- 230000023603 positive regulation of transcription initiation, DNA-dependent Effects 0.000 description 1
- 230000003334 potential effect Effects 0.000 description 1
- 201000009104 prediabetes syndrome Diseases 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 239000000955 prescription drug Substances 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 210000002243 primary neuron Anatomy 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 238000000575 proteomic method Methods 0.000 description 1
- 230000004063 proteosomal degradation Effects 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000007261 regionalization Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 230000008521 reorganization Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 230000004202 respiratory function Effects 0.000 description 1
- 230000036387 respiratory rate Effects 0.000 description 1
- 230000001020 rhythmical effect Effects 0.000 description 1
- 229940080817 rotenone Drugs 0.000 description 1
- JUVIOZPCNVVQFO-UHFFFAOYSA-N rotenone Natural products O1C2=C3CC(C(C)=C)OC3=CC=C2C(=O)C2C1COC1=C2C=C(OC)C(OC)=C1 JUVIOZPCNVVQFO-UHFFFAOYSA-N 0.000 description 1
- 239000005060 rubber Substances 0.000 description 1
- 239000013535 sea water Substances 0.000 description 1
- 210000002955 secretory cell Anatomy 0.000 description 1
- MFBOGIVSZKQAPD-UHFFFAOYSA-M sodium butyrate Chemical compound [Na+].CCCC([O-])=O MFBOGIVSZKQAPD-UHFFFAOYSA-M 0.000 description 1
- 238000002174 soft lithography Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 210000002536 stromal cell Anatomy 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000820 toxicity test Toxicity 0.000 description 1
- 231100000041 toxicology testing Toxicity 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 230000034512 ubiquitination Effects 0.000 description 1
- 238000010798 ubiquitination Methods 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 239000012130 whole-cell lysate Substances 0.000 description 1
- 239000002676 xenobiotic agent Substances 0.000 description 1
- 230000022814 xenobiotic metabolic process Effects 0.000 description 1
Images












































































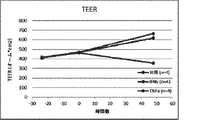
































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0679—Cells of the gastro-intestinal tract
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12M—APPARATUS FOR ENZYMOLOGY OR MICROBIOLOGY; APPARATUS FOR CULTURING MICROORGANISMS FOR PRODUCING BIOMASS, FOR GROWING CELLS OR FOR OBTAINING FERMENTATION OR METABOLIC PRODUCTS, i.e. BIOREACTORS OR FERMENTERS
- C12M21/00—Bioreactors or fermenters specially adapted for specific uses
- C12M21/08—Bioreactors or fermenters specially adapted for specific uses for producing artificial tissue or for ex-vivo cultivation of tissue
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12M—APPARATUS FOR ENZYMOLOGY OR MICROBIOLOGY; APPARATUS FOR CULTURING MICROORGANISMS FOR PRODUCING BIOMASS, FOR GROWING CELLS OR FOR OBTAINING FERMENTATION OR METABOLIC PRODUCTS, i.e. BIOREACTORS OR FERMENTERS
- C12M23/00—Constructional details, e.g. recesses, hinges
- C12M23/02—Form or structure of the vessel
- C12M23/16—Microfluidic devices; Capillary tubes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/11—Epidermal growth factor [EGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/119—Other fibroblast growth factors, e.g. FGF-4, FGF-8, FGF-10
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/13—Nerve growth factor [NGF]; Brain-derived neurotrophic factor [BDNF]; Cilliary neurotrophic factor [CNTF]; Glial-derived neurotrophic factor [GDNF]; Neurotrophins [NT]; Neuregulins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/155—Bone morphogenic proteins [BMP]; Osteogenins; Osteogenic factor; Bone inducing factor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/16—Activin; Inhibin; Mullerian inhibiting substance
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/24—Interferons [IFN]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/25—Tumour necrosing factors [TNF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/415—Wnt; Frizzeled
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/998—Proteins not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/999—Small molecules not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/45—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from artificially induced pluripotent stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2535/00—Supports or coatings for cell culture characterised by topography
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Cell Biology (AREA)
- Sustainable Development (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Clinical Laboratory Science (AREA)
- Dispersion Chemistry (AREA)
- Transplantation (AREA)
- Developmental Biology & Embryology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Physics & Mathematics (AREA)
- Tropical Medicine & Parasitology (AREA)
- Neurology (AREA)
- Food Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Neurosurgery (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
Abstract
Description
本発明の目的のために、次の用語を以下に定義する。
胎児発育の重大な時期における高レベルの人工内分泌攪乱化学物質(EDC)へのヒトの持続的な曝露は、内分泌組織前駆細胞における代謝ホメオスタシスの長期的な撹乱をもたらし得るものであり、したがって、小児期の肥満に寄与し得る。ヒトの腸及び脳の内分泌組織におけるEDC誘発性発達異常を試験するための実行可能なプラットフォームは存在しない。したがって、本発明者らは、パーフルオロオクタン酸(PFOA)、トリブチルスズ(TBT)、及び、ブチルヒドロキシトルエン(BHT)を含む、本発明者らの食料及び上水道を汚染する一般的なEDCに対する低用量慢性曝露の効果を決定するためのプラットフォームを、2つのヒト誘導多能性幹細胞(hiPSC)に由来する内分泌組織、すなわち前腸上皮細胞(iFGE)及び神経ペプチド視床下部ニューロン(iHTN)を用いて開発した。
本明細書には、3つの異なるEDCのそれぞれ、パーフルオロオクタン酸(PFOA)、トリブチルスズ(TBT)、及びブチルヒドロキシトルエン(BHT)と、それらの組み合わせによる効果が記載される。PFOAは、フルオロポリマーにおける界面活性剤であることが知られており、いつまでも環境中に残留することが知られている。2007年の調査では、米国の人口の約98%が、血液中に検出可能なレベルのPFOAを保有しており、産業廃棄物、耐汚染性カーペット、ハウスダスト、水、及び、調理器具のコーティングを介して、それらの曝露を受ける可能性がある。有機スズであるTBTは、生物の付着から船舶を保護するための塗料に用いられる防汚剤として使用されている。しかしながら、ハウスダストに存在することが、ヒトに対する曝露への主要な原因となっている。BHTは、一般的な食品添加物、パーソナルケア製品成分、及び、化粧品成分、農薬、プラスチック、及び、ゴムの成分である。しかしながら、これは、一般的に消費される朝食シリアル商品での抗酸化剤としても利用されている。ヒト誘導多能性幹細胞(hiPSC)の使用は、腸及び視床下部神経肥大ニューロンの発達に対する慢性低用量EDC曝露の有害作用と機序を解明し、そして、EDCへの子宮曝露を模倣するためのプラットフォームとして役立つ。
本明細書には、ある量の誘導多能性幹細胞(iPSC)を提供すること、及び、1つ以上の因子の存在下で培養することを含み、当該1つ以上の因子が、iPSCを分化させることができる、誘導多能性幹細胞を分化させる方法がさらに記載される。
一実施形態において、本発明は、細胞を培養する方法であって、a)上面と底面を含む膜を含む流体装置を提供すること、b)当該底面上に細胞を播種すること、及び、c)当該播種された細胞を、腸オルガノイドの増殖を助ける条件下で培養することを含む、方法を企図する。一実施形態において、当該細胞は、胃腸系の障害と診断された患者の腸組織生検試料に由来する。一実施形態において、当該細胞は、胃腸系の障害と診断された患者由来の誘導多能性幹細胞に由来する。一実施形態において、当該患者は、ヒト患者である。一実施形態において、当該胃腸障害は、過敏性腸疾患である。一実施形態において、この方法は、当該細胞を当該上面に播種すること、及び、当該上面に播種された細胞を、当該少なくとも1つの腸管絨毛構造の成熟を助ける条件下で培養することを、さらに含む。一実施形態において、当該少なくとも1つの腸管絨毛構造は、腸オルガノイドルーメンに向かって分極化する。一実施形態において、当該少なくとも1つの腸管絨毛は、インビボの腸管絨毛と形態的に類似している。一実施形態において、当該腸管絨毛は、腸細胞型を含み、当該腸細胞型として、パネート細胞、杯細胞、腸内分泌細胞、及び、腸細胞などがあるが、これらに限定されない。一実施形態において、腸細胞型は免疫細胞化学によって確認される。一実施形態において、腸細胞型は、Igr5+を含む。一実施形態において、パネート細胞は、抗菌剤を分泌する。一実施形態において、この方法は、STAT1がリン酸化されるような条件下でIFNγを腸オルガノイドに投与することを、さらに含む。一実施形態において、当該方法は、IFNγ応答遺伝子がアップレギュレートされるような条件下でIFNγを腸オルガノイドに投与することを、さらに含む。一実施形態において、当該IFNγ応答遺伝子として、IDO1、GBP4及び/またはGBP5などがあるが、これらに限定されない。一実施形態において、IFNγ投与は、腸管上皮サブタイプ特異的遺伝子をさらにアップレギュレートする。一実施形態において、腸管上皮サブタイプ特異的遺伝子として、ホスホリパーゼA2群2A及び/またはMuc4があるが、これらに限定されない。一実施形態において、当該方法は、当該腸オルガノイドにおける遺伝子発現を測定することをさらに含む。一実施形態において、当該方法は、当該腸オルガノイドにおける抗菌分泌を測定することをさらに含む。一実施形態において、本方法は、腸のオルガノイド機能に関する作用因子の影響を評価することをさらに含み、当該作用因子として、管腔微生物、免疫細胞、及び/または、サイトカインなどがあるが、これらに限定されない。
本明細書では、腸細胞の集団を含むマイクロ流体装置を製造するための方法を記載する。様々な実施形態において、当該方法は、誘導多能性幹細胞(iPSC)からヒト腸オルガノイド(HIO)の生成、及び、腸管上皮細胞をマイクロ流体装置へ播種することを含む。様々な実施形態において、当該マイクロ流体装置は、組織化した構造を有する腸細胞の集団を含み、HIOを単細胞に分離させすること、および当該単細胞を装置に加えることを含む。様々な実施形態において、当該単細胞は、装置に添加する前に、CD326+発現に基づいて精製される。様々な実施形態において、当該単細胞を当該装置に添加することは、ROCK阻害剤、SB202190、及び、A83−01の1つ以上を含む培地での再懸濁を含む。様々な実施形態において、当該HIOは、分離の前に、ROCK阻害剤の存在下で培養する。様々な実施形態において、HIOは、iPSCに由来する。様々な実施形態において、当該iPSCは、リプログラミングされたリンパ芽球様B細胞由来の誘導多能性幹細胞(LCL−iPSC)である。様々な実施形態において、当該iPSCは、炎症性腸疾患及び/または病態に罹患した対象から得られたリプログラミングされた細胞である。様々な実施形態において、iPSCからのHIOの誘導は、アクチビンA及びWnt3Aの存在下でのiPSCの培養による胚体内胚葉の生成、FGFとWnt3AまたはCHIR99021のいずれかとの存在下での胚体内胚葉の培養による後腸への分化、上皮スフィアまたは上皮チューブの回収、マトリゲルでの上皮スフィアまたは上皮チューブの懸濁、ならびにCHIR99021、ノギン、及びEGFの存在下での培養を含む。様々な実施形態において、当該組織化した構造は、絨毛を含む。様々な実施形態において、当該絨毛は、吸収細胞、杯細胞、腸内分泌細胞、及びパネート細胞からなる群から選択される1つ以上の上皮細胞株により裏打ちされている。様々な実施形態において、当該組織化した構造は、バリア機能、シトクロムP450活性、及び/または頂端粘液分泌を有する。
様々な実施形態において、この方法は、iPSCからHIOの生成を含み、当該生成は、iPSCから胚体内胚葉、上皮構造、及びオルガノイドへの分化を含む。様々な実施形態において、胚体内胚葉の誘導は、1日、2日、3日、4日、またはそれ以上の日数にわたって、アクチビンA及びWnt3AとともにiPSCを培養すること、及び、FBSの濃度を時間とともに増加させることを含む。様々な実施形態において、胚体内胚葉の誘導は、アクチビンA(例えば、100ng/ml)、Wnt3A(25ng/ml)とともに、1日、2日、3日、4日、または、それ以上の日数にわたってiPSCの培養すること、及びFBSの濃度を時間とともに(1日目、2日目、及び、3日目に、それぞれ、0%、0.2%、及び2%)増加させることを含む。例えば、胚体内胚葉の誘導は、アクチビンA(例えば、100ng/ml)、Wnt3A(25ng/ml)とともに、1日、2日、3日、4日、または、それ以上の日数にわたってiPSCを培養すること、及びFBSの濃度を時間とともに(1日目、2日目、及び、3日目に、それぞれ、0%、0.2%、及び2%)増加させることを含む。様々な実施形態において、アクチビンAの濃度は、約0〜25ng/ml、約25〜50ng/ml、約50〜75ng/ml、約100〜125ng/ml、約125〜150ng/mlを含む。様々な実施形態において、Wnt3Aの濃度は、〜約25ng/ml、約25〜50ng/ml、約50〜75ng/ml、約100〜125ng/ml、約125〜150ng/mlを含む。様々な実施形態において、FBSの経時的な濃度は、第1日目、第2日目、及び、第3日目に、それぞれ、約0%〜0.2%、約0.2%〜0.5%、約0.5%〜1%、約1%〜2%、及び、2%超である。様々な実施形態において、後腸の形成は、FBS及びFGF4とともに改良DMEM/F12などの培地で、1日間、2日間、3日間、4日間、または、それ以上の日数にわたって胚体内胚葉細胞を培養することを含む。様々な実施形態において、後腸の形成は、1日間、2日間、3日間、4日間、または、それ以上の日数にわたって、胚体内胚葉細胞を、0%〜0.2%、約0.2%〜0.5%、約0.5%〜1%、約1%〜約2%、及び、2%以上の濃度のFBS、及び約50〜100ng/ml、約100〜250ng/ml、約250〜500ng/ml、及び500ng/ml以上の濃度のFGF4を含む培地において培養することを含む。例えば、後腸の形成は、2%FBS及びFGF4(500ng/ml)とともに改良DMEM/F12などの培地で、1日間、2日間、3日間、4日間、または、それ以上の日数にわたる胚体内胚葉細胞の培養を含むことができる。様々な実施形態において、Wnt3A、CHIR99021、またはその双方が加えられる。様々な実施形態において、Wnt3Aの濃度は、約100〜250ng/ml、約250〜500ng/ml、及び500ng/mlを含み、CHIR99021の濃度は、約0.5〜1μM、約1〜1.5μM、約1.5〜2μM、または2μM、またはそれ以上が添加される。例えば、Wnt3A(500ng/ml)、CHIR99021(2μM)、または、その双方が添加される。様々な実施形態において、約3〜4日後に、この方法は、遊離懸濁上皮スフィア、及び、緩慢に付着した上皮チューブを含むオルガノイドの単離を含む。様々な実施形態において、当該単離されたオルガノイドは、マトリゲルに懸濁され、次いで、CHIR99021、ノギン、EGF、及び、B27を含有する腸培地に重層される。様々な実施形態において、当該単離されたオルガノイドは、マトリゲルに懸濁され、次いで、CHIR99021、ノギン、EGF、及び、B27を含有する腸培地に重層される。様々な実施形態において、CHIR99021の濃度は、約0.5〜1μM、約1〜1.5μM、約1.5〜2μM、または2μMであり、ノギンの濃度は、約50〜100ng/ml、約100〜250ng/ml、約250〜500ng/ml、及び500ng/ml以上であり、EGFの濃度は、約50〜100ng/ml、約100〜250ng/ml、約250〜500ng/ml、及び500ng/mlであり、ならびにB27の濃度は、約0.25X〜0.5X、約0.5〜1X、約1X〜2X、または2X以上である。例えば、当該培地は、CHIR99021(2μM)、ノギン(100ng/ml)、及びEGF(100ng/ml)、及びB27(1X)を含有する。様々な実施形態において、HIOは、その後、7〜10日毎に継代される。様々な実施形態において、腸組織の集団は、腸臓器の特徴を含んだ組織化された集団である。様々な実施形態において、腸細胞は、絨毛へと組織化される。様々な実施形態において、当該絨毛は、小腸の4つすべての上皮細胞株(吸収、粘液分泌、腸内分泌、及び、パネート)で裏打ちされている。様々な実施形態において、当該腸細胞の集団は、バリア機能、シトクロムP450活性、及び/または、頂端粘液分泌を有する。
本明細書では、腸細胞の集団を含むマイクロ流体装置であって、当該集団が、組織化した構造を含む当該装置を記載する。様々な実施形態において、当該組織化した構造は、絨毛を含む。様々な実施形態において、当該絨毛は、吸収細胞、杯細胞、腸内分泌細胞、及びパネート細胞からなる群から選択される1つ以上の上皮細胞株で裏打ちされている。様々な実施形態において、当該組織化した構造は、バリア機能、シトクロムP450活性、及び/または頂端粘液分泌を有する。様々な実施形態において、当該腸細胞は、ヒト腸オルガノイド(HIO)に由来しており、単細胞に分離され、及び、CD326+発現に基づいて精製される。様々な実施形態において、当該HIOは、アクチビンA及びWnt3Aの存在下でのiPSCの培養による胚体内胚葉の生成、FGFと、Wnt3AまたはCHIR99021のいずれかとの存在下での胚体内胚葉の培養による後腸への分化、上皮スフィアまたは上皮チューブの回収、マトリゲルにおける上皮スフィアまたは上皮チューブの懸濁、ならびに、CHIR99021、ノギン、及びEGFの存在下での培養を含む方法によって、iPSCから誘導される。
A節には、体細胞源由来の誘導多能性幹細胞(iPSC)を生成することに関する方法の実施形態が記載されており、当該体細胞源として、白血球源があるがこれに限定されず、また、B節には、リンパ芽球B細胞の形態の例示的な白血球源に由来するiPSCを生成するためのかような使用の例が記載されている。リンパ芽球B細胞は、iPSCを作製するための当初の供給源物質として使用するのに望ましい白血球の一種であり、以下のA節を含む本明細書に記載する方法によって、その後にリプログラミングされる。これらの白血球由来iPSCは、後に、他の細胞型に分化し、当該細胞型として、腸細胞、視床下部ニューロン、内皮細胞などがあるが、これらに限定されない。したがって、以下のB節及び本明細書に記載の例示的な原材料を用いて、以下のA節及び本明細書に記載の原材料を操作する技術は、本明細書に記載のマイクロ流体チップと共に使用するために記載された様々な分化細胞を幅広く生成することができる。
また、本明細書では、ある量の細胞を提供すること、ある量のリプログラミング因子を細胞に送達すること、リプログラミング培地で、少なくとも4日間、細胞を培養することを含む、誘導多能性幹細胞を生成するための効率的な方法が記載されており、当該リプログラミング因子を送達及び培養することで、誘導多能性幹細胞が生成される。ある特定の実施形態において、当該細胞は、初代培養細胞である。他の実施形態において、当該細胞は、血液細胞(BC)である。ある特定の実施形態において、当該血液細胞は、T細胞である。他の実施形態において、当該血液細胞は、非T細胞である。他の実施形態において、当該細胞は、例えば、末梢血単核細胞(PBMC)を含む単核細胞(MNC)である。他の実施形態において、当該細胞は、原発性顆粒球、単球、及びBリンパ球である。
「LCL−iPSC」は、上記A節に記載の技術を用いて生成される。
本明細書において引用する全ての文献は、あたかも完全に記載が盛り込まれているかのように、その全内容を参照により援用する。特に断りがない限り、本明細書において使用する技術用語、及び、科学用語は、本発明が属する技術分野における当業者が通常理解するのと同じ意味を有する。Allen et al.,Remington:The Science and Practice of Pharmacy 22nd ed.,Pharmaceutical Press(September 15,2012)、Hornyak et al.,Introduction to Nanoscience and Nanotechnology,CRC Press(2008)、Singleton and Sainsbury,Dictionary of Microbiology and Molecular Biology 3rd ed.,revised ed.,J.Wiley & Sons(New York,NY 2006)、Smith,March‘s Advanced Organic Chemistry Reactions,Mechanisms and Structure,7th ed.,J.Wiley & Sons(New York,NY 2013)、Singleton,Dictionary of DNA and Genome Technology 3rd ed.,Wiley−Blackwell(November 28,2012)、及びGreen and Sambrook、Molecular Cloning:A Laboratory Manual 4th ed.,Cold Spring Harbor Laboratory Press(Cold Spring Harbor,NY 2012)は、本出願において使用した数多くの用語に関する一般的な説明を、当業者に対して提供する。抗体を調製する方法に関する参考文献については、Greenfield,Antibodies A Laboratory Manual 2nd ed.,Cold Spring Harbor Press(Cold Spring Harbor NY,2013)、Kohler and Milstein,Derivation of specific antibody−producing tissue culture and tumor lines by cell fusion,Eur.J.Immunol.1976 Jul,6(7):511−9、Queen and Selick,Humanized immunoglobulins,米国特許第5,585,089号(1996年12月)、及びRiechmann et al.,Reshaping human antibodies for therapy,Nature 1988 Mar 24,332(6162):323−7を参照されたい。
研究計画
発達中の腸管及び視床下部神経肥大ニューロンに対する慢性低用量EDC曝露の有害効果と機序とを解明するためのヒト誘導多能性幹細胞(hiPSC)の使用が本明細書において記載され、これは、EDCに対する子宮内曝露を模倣するためのプラットフォームとして役立つ。このようなスクリーニングプラットフォームは、ヒトの発達モデルを忠実に模倣するだけでなく、スクリーニングされた化合物によって撹乱させる可能性のある発達の手がかりに関する貴重な考察も提供することができる。
前腸スフェロイド細胞分化(iFGE)
分化のために、iPSCを、アクターゼで処理し、及び、ROCK阻害剤Y27632(10μM、Stemgent)を含むE8培地中、1ウェルあたり100万個の密度で6ウェルのマトリゲル被覆ディッシュに播種した。翌日に、アクチビンA(100ng/ml、R&D)及びWnt3A(1日目のみ25ng/ml、Peprotech)を含むRPMI 1640(Gibco)に、3日間、曝露することで、iPSCを胚体内胚葉へと分化させた。この3日間、当該細胞を、0%、0.2%、及び、2%の漸増濃度の規定FBS(dFBS、Hyclone)に曝露した。胚体内胚葉誘導の後に、2%dFBS、2μM CHIR99021(2μM、Cayman)、FGF4(500ng/ml、Peprotech)、LDN(2μM、Cayman)、及びレチノイン酸(2μM、Cayman)を含む改良DMEM/F12培地(Gibco)で、次の3日間、培養することで、それら細胞を、前腸スフェロイドを形成するように指示した。その結果、半懸濁スフェロイドとなり、次いで、それらを選択的に採取し、そして、さらなる成熟及び実験のためにマトリゲル被覆実験プレートに移した。採取した前腸スフェロイドを成熟させるために、N2(Invitrogen)、B27(Invitrogen)、グルタマックス、ペニシリン/ストレプトマイシン/抗真菌薬、及び、EGF(100ng/ml、Peprotech)を有する改良DMEM/F12を含む培地で、それらを培養した。必要に応じて、培地を2〜3日毎に交換し、及び、スフェロイドを、20日目まで上皮単層に発生させた。
視床下部ニューロン分化(iHTN)
iHTNへの分化のために、iPSCをアクターゼで処理し、単一細胞として、ROCK阻害剤Y27632(10μM、Stemgent)を含むE8培地中、1ウェルあたり約100万個の細胞密度で、6ウェルのマトリゲル被覆ディッシュに播種した。翌日に、iHTN分化を、LDN193189(1μM、Cayman)、及び、SB431542(10μM、Cayman)を用いた二重SMAD阻害による神経外胚葉分化によって開始し、及び、この処理を、48時間行う。これに続き、3日目から8日目で、スムーズンドアゴニストSAG(1μM、Tocris)とプルモルファミン(PMN、1μM、Tocris)とによるソニックヘッジホッグ活性化及びIWR−endo(10μM、Cayman)を用いたWntシグナル伝達阻害によって、腹側間脳へ細胞を向かわせ、2日毎に定期的に培地を交換した。9日目から13日目に、当該細胞を、腹側化剤レチノイン酸(0.1μM、Cayman)の存在下で、DAPT(10μM、Cayman)を用いて、徐々に細胞周期から逸脱させる。14日目に、細胞を、アクターゼで処理し、脳由来神経栄養因子BDNF(10ng/ml、Miltenyi)を含有する成熟培地の存在下で、ラミニン被覆プレート上に再移植し、そして、40日目まで維持した。
EDC処置
本発明者らは、3つの異なるEDC、すなわち、パーフルオロオクタン酸(PFOA)(2.5μM、Sigma−Aldrich)、トリブチルスズ(TBT)(10nM、Sigma−Aldrich)、及びブチル化ヒドロキシトルエン(BHT)(10nM、Cayman)を、個別に、及び、組み合わせて使用した。したがって、本発明者らは、6つの処置群、すなわち、ビヒクル対照(媒体)、PFOA、TBT、BHT、及び、併用処置を行った。EDCのiFGE処置は、上記のように分化を行い、最後の12日間の分化の間、すなわち、8日目〜20日目に、EDC処置を加えて実施した。同様に、上で詳述したプロトコール、及び最後の12日間の分化、すなわち、28日目〜40日目のEDC処置を行ってiHTNを分化させた。NFκBi(SN50)を用いる救済実験のために、EDC処置をする前に、当該細胞を、最初に、NFκBiに対して24時間曝露した。続いて、当該細胞を、NFκBiとともに併用処置で処置を行った。NFκBi処置は、併用EDC処置条件と組み合わせただけであることに留意すべきである。
免疫蛍光
免疫蛍光に供された細胞を、最初に、4%パラホルムアルデヒド(PFA)を用いて、20分間、固定し、及び、続いて、PBSで洗浄した。5%ロバ血清(Millipore)と0.2%トリトンX−100(Bio−rad)とを含むPBSで、短くとも2時間、細胞をブロッキングした後に、当該細胞を、適切な濃度の関連する一次抗体の組み合わせ(1:250)により、4℃で、一晩、インキュベートした。0.1%Tween−20を含むPBSを用いて十分に洗浄をした後に、当該細胞を、適切な種特異的アレクサフルオル結合二次抗体の組み合わせで、45分間(1:500)処理する。ヘキスト染色を用いて核を標識し、及び、当該細胞を、次いで、ImageXpress Micro XLS(Molecular devices)を利用する適切な蛍光フィルターを用いて細胞を可視化した。
免疫ブロット
細胞ペレットを回収し、そして、溶解し(哺乳動物PER、Thermo scientific+1xプロテアーゼ阻害剤カクテル、Thermo Scientific)、タンパク質を定量した後に、試料を調製した。本発明者らは、ポリアクリルアミドゲル(NuPAGE(商標)Novex(商標)4〜12%ビス−トリスタンパク質ゲル)のレーンあたり約15μgのタンパク質を負荷した。ゲルが一旦溶解したら、それらを、ニトロセルロース膜上に移し、続いて、5%乳溶液で、短くとも2時間かけてブロックした。この後に、一次抗体で膜を処理する一段階i−Bindプロセス、洗浄、そして、二次抗体ステップ(Life technologies)が続いた。本発明者らは、LiCor(登録商標)IRDye二次抗体(680及び800波長の赤外染料)を使用し、LiCor ODyssey CLx撮像装置(Li−Cor)において、バンドの検出を行った。
定量的PCR
RNeasy Mini Kit(Qiagen)を用いて、全RNAを単離し、まず、RNA(2μg)をDNase処理し、Promega Reverse Transcriptase System(Promega)を用いて、オリゴ(dT)でcDNAに逆転写した。各遺伝子に特異的なプライマー配列を用いて、SYBR Green master mix(Applied Biosystems)を用いて、3連で、反応を行った。各PCRサイクルは、95℃で10分間、95℃で30秒間→58℃で60秒間を、50サイクル行い、及び、72℃で5分間であった。対象となる遺伝子を、ミトコンドリア遺伝子についてのRPL13Aまたは16srRNAのいずれかに対して正規化した。
MTTアッセイ
MTTアッセイによって、細胞生存率を評価した。1ウェルあたり100μlの培地に10,000個の細胞の密度で、96ウェルプレートに細胞を播種した。アッセイ当日に、新鮮な培地(100μl)を加え、10μlのMTT溶液を、培地(12mMのストックMTT溶液)に加えて、37℃で、4時間インキュベートした。各ウェルに50μlのDMSOを添加して反応を停止させた。バックグラウンドを差し引くために、細胞を含まない陰性対照を含めた。自動マルチウェル分光光度計(Perkin Elmer)を用いて、吸光度値を、540nmで読み取った。
代謝表現型解析及びシーホース呼吸計測アッセイ
シーホースXFe24細胞外フラックスアナライザー(Seahorse Biosciences)を用いて、ミトコンドリアストレス試験を行い、細胞内の酸素消費率(OCR)のリアルタイム測定値を得た。EDCで処置をした、または処置していないiFGE及びiHTNを、24ウェルシーホース培養プレートに、10,000〜15,000個の細胞/ウェルの密度で播種した。OCRの分析のために、細胞を、シーホース基本培地で再構成し、測定を行う前に、非CO2インキュベーターで、37℃で、1時間、静置した。化学試薬(Sigma)は、以下の最終濃度のものを用いた。1μMオリゴマイシン−ATP合成酵素阻害剤、1μM(FCCP)シアン化カルボニル4−(トリフルオロメトキシ)フェニルヒドラゾン−脱共役剤、及び、0.5μMアンチマイシンA−シトクロムC還元酵素阻害剤と0.5μMロテノンの混合物−複合体I阻害剤。結果を、BCAアッセイ(Thermo Scientific)によって決定されたタンパク質濃度に対して正規化した。
統計分析
すべてのデータを、平均±標準偏差または標準誤差として表している。p<0.05を、有意とみなした。すべての統計解析は、スチューデントの対応のあるt検定、または、一元配置分散分析(ANOVA)、及び、多重比較のためのニューマン−コイルス事後検定を用いて、グラフパッドプリズムで行った。
一次及び二次抗体
免疫細胞化学染色。一次。α−MSH、ウサギ、Phoenix Pharmaceuticals、H−43−01、1:250。β−カテニン、ウサギ、Santa Cruz、sc7199、1:500。CART、ヤギ、Santa Cruz、sc18068、1:250。CPE、ヤギ、R&D Systems、AF3587、1:250。E−カドヘリン、ヤギ、R&D Systems、AF648、1:250。GABA、ウサギ、Sigma−Aldrich、A2025、1:250。ガストリン、ウサギ、Dako、A056801−2、1:250。グレリン、ヤギ、Santa Cruz、sc10368、1:250。NF−κB(ホスホ Ser−311)、マウス、Santa Cruz、sc166748、1:250。NP−II、ヤギ、Santa Cruz、sc27093、1:250。NPY、ウサギ、MerckMillipore、AB9608、1:250。OTP、ウサギ、Genetex、GTX119601、1:250。ペプチドYY、ウサギ、Abcam、ab22663、1:250。セロトニン、ウサギ、Immunostar、20080、1:250。ソマトスタチン、ウサギ、Santa Cruz、sc13099、1:250。Sox17、マウス、Novus、47996,1:250。Sox2、ウサギ、Stemgent、09−0024、1:500。シナプトフィジン、マウス、Santa Cruz、sc17750、1:250。TH、マウス、Immunostar、22941、1:250。二次(1:200)。AlexaFluor 488 ロバ抗ウサギ、AlexaFluor 555 ロバ抗マウス、AlexaFluor 594 ロバ抗マウス、AlexaFluor 568 ロバ抗ヤギ、AlexaFluor 647 ロバ抗ヤギ。
プライマー配列:

末梢血単核細胞を多能性に対してエピソーム的にリプログラミングする。
iPSCに対する末梢血単核細胞(PBMC)の非取り込みリプログラミングを、本発明者らの研究室で公表したプロトコールと同様のエピソーム(OriP/EBNA1)プラスミドに基づいた方法を用いて行った。これには、エピソームに発現する7つのリプログラミング因子である、OCT3/4、SOX2、KLF4、LIN28、非形質転換L−MYC、SV40ラージT抗原(SV40LT)、及びp53に対するshRNA(図S1A)の核トランスフェクションを含んでいた。このプロトコールは、27〜32日後に機械的に単離して増殖することができる血液由来の非取り込みiPSCクローンを首尾よく生成した(図S1A)。この研究で使用した独立したドナー由来のiPSC株(80i対照 Tn2、及び、201i対照 NTn4)の代表的な画像は、図S1Bの明視野画像で示されるように、核と細胞質の比が高い密着したコロニーなど、多能性幹細胞の典型的な特徴を示した。また、堅固なアルカリホスファターゼ活性を示し、核(OCT3/4、NANOG、SOX2)及び表面(SSEA−4、TRA−1−81、TRA−1−60)多能性タンパク質の強い発現を示した(図S1B)。生成したPBMC−iPSCは、高い多能性と低い新規性スコアとを示して(図S1C)PluriTestアッセイにも合格し、また、G−バンド核型スプレッド(図S1D及びE)に示したように、正常な細胞遺伝的状態を維持している。
ヒトiPSCは、WNT、FGF、BMP、及び、レチノイン酸シグナル伝達の調節によって内分泌的に活性な前腸上皮(iFGE)へと分化する
胃のオルガノイドを成熟させるために3Dマトリゲルバブルを使用したWellsのグループによって以前に発表された3D胃オルガノイド分化に基づいて、本発明者らは、内分泌機能を有する胃上皮の2D単層を生成するために彼らのプロトコールの変法を用いた。内分泌細胞タイプを含む前腸腔上皮へのiPSCの特定化は、(1)アクチビンA及びWnt3Aが媒介した胚体内胚葉の特定化、(2)BMPシグナル伝達の抑制と、WNT(CHIR)、FGF(FGF4)、及び、レチノイン酸(RA)シグナル伝達の活性化とを同時に行うこと、及び、(3)高濃度の表皮成長因子(EGF)での前腸上皮を含有する内分泌細胞の最終生成(図1A)、による段階的方法で首尾よく達成できた。胚体内胚葉を誘導した後、iPSCの6日後に、腸管様オルガノイド構造が、内胚葉単層から出現する。腸管オルガノイドを再播種すると、接着性の上皮様細胞層が、iPSCの7日後から20日後まで一貫して出現する(図1B)。関連する胃/前腸特異的遺伝子の発現をモニターすることによって、20日目のiPSC由来の前腸上皮(iFGE)の特徴付けを確認した。SOX2(前腸前駆細胞)、PDX1(前腸腔)、GKN1(ガストロカイン1、胃粘膜)、PGA5(消化酵素)、TAS1R3(前腸における味覚受容体)、及び、TFF2(トレフォイル因子2、胃粘膜の安定分泌タンパク質)遺伝子が、20日目の接着性iFGEにおいて観察された(図1C)。iFGEは後腸特異的CDX2の発現を示さなかったことに留意することが重要である(図1C)。上皮細胞表面に特異的なタンパク質を評価すると、CDH1(E−カドヘリン)とCTNNB(β−カテニン)とが形成されたiFGEのシートとして、多角形の石畳の形をした細胞の表面に規則的に観察された(図1D)。内胚葉及び前腸前駆細胞特異的転写因子であるSox17及びSox2は、それぞれ、iFGEの前腸の識別を確認する(図1D)。重要なことに、シナプトフィジン(SYP)、ソマトスタチン、及び、セロトニンなどの内分泌的に活性な前腸に存在することが知られている神経内分泌マーカーが、20日目に、iFGEによって発現された(図1E)。とりわけ、iFGEは、ガストリン(G細胞)、グレリン(壁細胞)、及び、幾つかのペプチドYY(粘膜)細胞などの胃特異的ホルモン発現腸内分泌細胞に対しても免疫陽性であった(図1F)。
機能的ニューロペプチダーゼ性視床下部ニューロン(iHTN)は、SHHの活性化及びWNTシグナル伝達の阻害によって、hiPSC−神経上皮から得ることができる。
指定したパターン形成と、iPSCの二重SMAD阻害(SMADi)小分子処置で神経上皮を特定化した後に、当該iHTNを生成した。続いて、早期WNT阻害、及び、SHH活性化は、視床下部及び弓形核が存在する、腹側間脳が同一の前脳細胞タイプを特定化した(図2A)。40日までに前脳前駆細胞と分化ニューロンの終末成熟を同期させると、AgRP(アグーチ関連ペプチド、食欲促進神経ペプチド)、MC4R(メラノコルチン4受容体、摂食及び代謝の調節)、Nkx2.1(腹側間脳マーカー)、NPY(神経ペプチドY、AgRPで同時発現した食欲促進神経ペプチド)、及びPCSK2(前駆タンパク質転換酵素スブチリシン/ケキシン2型、神経内分泌遺伝子)などの視床下部及び神経肥大遺伝子の発現を増加した(図2B)。不可欠な視床下部神経ペプチドNPY及びα−メラノサイト刺激ホルモン(α−MSH)の分泌は、ELISAを用いて確認され、そして、その結果は、40日目のiHTNにおいて両神経ペプチドが有意に高いレベルにあることを明らかにした(図2C及びD)。免疫蛍光染色は、OTP(ホメオボックスタンパク質オーソペディア、図2E)、α−MSH(図2F)、NPY(図2G)、SST(ソマトスタチン、図2H)、GABA(図2I)、CPE(カルボキシペプチダーゼE、図2J)、CART(コカイン−、及び、アンフェタミン−調節転写産物、図2K)、NP−II(ニューロフィジンII/アルギニンバソプレシン、図2L)、5−HT(セロトニン、図2M)、及び、TH(チロシンヒドロキシラーゼ、図2N)などの幾つかの神経内分泌及び視床下部弓状核特異的タンパク質を発現するニューロンを示した。多電極アレイ(MEA)プラットフォームを用いた電気生理学的測定は、0日目の無活動と比較して、40日目のニューロンでは自発的活動電位と反復的発光との規則的な一連の流れを示し、したがって、真正なニューロンの識別とiHTNの電気的成熟とを確認している(図2O)。
慢性低用量EDC処置は、細胞の生存率に影響を与えずに、iFGE及びiHTNでのNF−κBシグナル伝達を攪乱する。
本発明者らは、iPSC−内分泌細胞培養の分化を首尾よく終えた後に、12日間の処置パラダイムにわたって、低用量のEDCを用いて、これらの組織を攪乱することにした。EDC処置の最適濃度は、分化したiPSC−内分泌培養物(図S2)において、10%の細胞生存率の喪失さえも観察された用量に満たない対数濃度または半対数濃度として決定した。加えて、ヒトの許容可能な1日摂取量(TDI)を把握するために文献検索を利用し、細胞生存率に対する化合物の各々の範囲の効果を実施し、そして、ペルフルオロオクタン酸(PFOA、2.5μM)、トリブチルスズ(TBT、10nM)、及び、ブチルヒドロキシトルエン(BHT、10nM)を、複数のEDCに対して同時に環境曝露を行うのと類似の併用処置パラダイムとともに与えた(図S2)。発達中のiPSC由来の内分泌組織のEDCで処理すると、iFGE細胞においてリン酸化NF−κB p65免疫陽性細胞数の有意な増加が、iHTNにおいて、1.35〜1.5倍(図3C)、及び、1.2〜1.3倍(図3D)(p<0.001)で認められた。これらの培養物の免疫ブロットは、NF−κB p65リン酸化レベルが、EDC処置iFGE(図3E)(p<0.001)、及びiHTN(図3F)(p<0.01)において有意に上昇することを確認した。EDCの添加と、その結果として表れるNF−κBホスホ−p65の増加が、細胞生存率または一般的な細胞傷害性におけるEDC誘導性喪失の結果ではないことを確認するために、EDC処置、及び、ビヒクル処置iFGE及びiHTNに対していMTT細胞生存率アッセイを実施した。これらの処置ではEDCは、双方のiPSC由来組織型において、細胞生存率に対して有意の影響を及ぼさないことが認められた(図3G及びH)。
ミトコンドリア呼吸を撹乱することによって、EDCは代謝活性に作用する。
本発明者らの1つの目的は、慢性EDC撹乱がヒト内分泌組織における代謝活性及び呼吸に影響するかを決定することを目的としていたので、本発明者らは、NF−κBのリン酸化が、この現象にどのように寄与もし得るかをを調べた。興味深いことに、関連する核及びミトコンドリアにコードされた呼吸遺伝子の転写を直接的及び間接的に制御することによってNF−κBシグナル伝達がミトコンドリア機能に影響を及ぼすという、癌生物学における幾つかの証拠がある。まず、本発明者らは、XFe24 Seahorse Extracellular Flux Analyzerで、ミトコンドリアストレス試験を行うことで、ミトコンドリア呼吸機能に対するEDCの効果を決定した。本発明者らは、iFGEにおいて、BHT(p<0.05)の添加、及び、PFOA、TBT、及び、BHTの併用処置(p<0.01)を行うことで、最大呼吸が減少し、また、予備呼吸能の40〜50%が減少する結果となったことを確認した(図5A)。iHTNにおいて同様の効果を示され、TBT、BHT、及び、併用処置で処置すると、最大呼吸、及び、予備呼吸能において40〜50%の減少を示した(図5B)。ミトコンドリア質量に対する処置の効果は、全ての処置期間においてCOX IV(内部ミトコンドリア膜酵素)レベルが変化しなかったので、除外した(図S5)。
NF−κB阻害は、経路活性化及びミトコンドリア障害から細胞を救済する。
iPSC−内分泌細胞の発達において、EDC曝露に起因する有害なNF−κB経路撹乱とミトコンドリア機能不全効果とが顕著であることを考慮して、本発明者らは、これらの表現型が、NF−κB経路の活性化を単に遮断することによって、ことによると救済できるかを調べた。したがって、本発明者らは、細胞透過性阻害ペプチドであるNF−κB阻害剤(NFκBi)SN50を使用して、これが有害な併用EDC処置で処置されたiPSC−内分泌培養における従前の表現型を救済し得るかどうかを決定した。SN50ペプチドは、NF−κB調節遺伝子の核誘導を阻害することが知られている。本発明者らは、iFGEにおけるEDCとNF−κBiとの同時処置が行われ次第に、ホスホ−p65、古典的(p50/p105)、及び、非古典的(p52/p100)経路の全体的な減少が、ビヒクル対照のレベルにまでほぼ回帰すことを見出した(p<0.001)。NF−κBiは、p50だけに特異的な阻害効果を示すのではなく、併用処置だけの場合と比較して、活性化されたp65、p50、及び、p52レベルに関する一般的な阻害効果よりも大きい効果を示した(図6A)。免疫陽性pNF−κB細胞が、ビヒクル対照レベルに近づくように減少した時にも、SN50 NFκBi処置のこの救済効果は確認された(図6B)。特に、NF−κBi処置は、併用EDC処置した細胞のミトコンドリア予備呼吸能も有意に改善した(図6C)。最も興味深い発見は、SCO2、POLRMT、TFAM、及び、CYTB5などのミトコンドリア機能に関与するタンパク質の転写調節が、EDC併用処置と比較して、NFκBi処置ですべて回復したことである(図6D)。これらの結果は、NF−κBi処置が併用EDC処置媒介効果を有意に逆転させたiHTNにおいて再現している(図7)。NF−κB経路撹乱と重篤なミトコンドリア機能障害とを関連付けるこの新規な発見は、特に、内分泌攪乱の状況において、いずれの系においても示されていない。
考察
「環境オビーソゲン」仮説によれば、内分泌攪乱化学物質(EDC)として知られている広範囲の環境汚染物質のサブセットは、ホルモンシグナル伝達経路を標的とし、正常組織の発達を妨げ、そして、身体の恒常性の制御を妨害する。子宮内または幼少期の幹細胞発生の重要な期間に、主にヒトの食物を介して、有機スズ、ペルフルオロケミカル、及び、食品添加物などの遍在する「オビーソゲン」EDCを繰り返し曝露すると、遺伝的に素因がある個体の正常な代謝制御が永久に改変され、人生の後半に肥満になる。注目すべきは、これらのEDCが、本発明者らの日常環境に存在し続けており、健康被害を引き起こし続けているという事実である。最近の文献は、EPA(環境保護庁)の勧告に従ってPFOAの使用を段階的に廃止する努力をしているにもかかわらず、テネシー川から供給された飲料水にPFOAが含まれていたことを報告していた。同様に、シリアルの添加物としてBHTを排除する努力がなされており、主要なブランドは、BHTをシリアル用添加物から首尾よく除去した。これらEDCへの日々の曝露が、我々に対して内分泌撹乱とそれに関連する影響を与え続けているとすれば、これらEDCの影響は、もっと詳細に研究される必要がある。
さらなる研究
様々な薬物/化合物/汚染物質の胎児発達に関する影響は、先天性欠損や発達障害を回避し、次世代の生活の質を向上させるために、速やかに対処する必要がある、手段となっている。地球規模の汚染レベルが絶えず増加しているため、有害な環境汚染物質や有害物質への曝露の必然的危険度も高まっている。系内でのこれらの有毒物質の影響に関する明確な示唆を提供する一貫した薬物スクリーニングプラットフォームが必要である。
追加の研究
本実施例は、EDC媒介調節不全に関連する例示的な結果を記載する。
実施例4の一部に記載したような、EDC効果に関する、追加の実験。
バイオインフォマティクス
NFκB−p65(RELA)及びTP53の推定上のDNA結合部位のバイオインフォマティクス決定を図20に示す。(a)SCO2、POLRMT、TFAM、CYB5Aなどの関心のある遺伝子上の、NFκB−p65及びTP53結合モチーフの推定結合部位の数、及びIL1A、IL1B、TNF、IL6などのNFκB−p65(RELA)によって調節される既知の各遺伝子、あるいは、GADD45A、GADD45B、GADD45G、PERP、BAXなどのTP53によって調節される既知の各遺伝子の特定を示すチャート。(b)関心のある遺伝子上のNFκB−p65及びTP53のDNA結合モチーフの転写開始部位の上流の塩基対における最短距離の特定。HOX遺伝子を、中性遺伝子、あるいは、文献であまり報告されていない、NFκB−p65及びTP53のいずれかによって制御される遺伝子として用いた。ヌクレオチドの配列保存の配列ロゴグラフィカル表現として表したDNA結合モチーフ。そこではヌクレオチド文字の大きさが、バイオインフォマティクス分析で使用した(c)NFκB−p65及び(d)TP53についての配列でのその位置での当該文字の頻度を表す。
胃(前腸)マイクロ流体チップの開発
本実施例では、部分的に、試験薬剤及び薬剤の発生効果のためのヒトモデルとして使用する、前腸/胃チップを開発するための例示的な材料、細胞、及び、方法を記載する。他の実施形態において、前腸細胞由来の誘導胃細胞が、薬剤及び薬物を試験するために使用される。
成熟した前腸細胞とホルモン作用
幾つかの実施形態において、成熟に対するEGFレベルの変化の影響について成熟前腸細胞を試験したが、その理由の一部は、流動下のSYP+細胞の数が比較的少ないためである。特に、10μlの流動(前出の実施例を参照されたい)とともに、EGFレベルは、100ng/mlから、当初は、2ng/mlまで徐々に減少し、中間のある段階では、10ng/mlに減少した。図30を参照されたい。部分的に、これが内分泌細胞種への成熟を、より良好にしたかを確認するために、これを行った。この条件は、実際に、流動条件下では、より少数のSox2+細胞と、より多数のSYP+細胞を示した。しかし、発明者らは、上皮の完全な被覆を得ておらず、むしろ斑点状のものでしかなかった。
ホルモン効果とがん
発明者らは、発明者らのオルガノイド(O)の分泌能を比較するための陽性対照として、ヒト胃癌細胞株を使用した。発明者らは、染色(SYP)及びELISA(グレリン)の双方で認められた内分泌細胞を得た。発明者らは、EGFレベルを制御することによって、増殖を制御することができた。また、発明者らは、EGFレベルを制御することによって、内分泌細胞の成熟を増加させることもできた。iFG−SRのグレリン分泌レベルは、HGC分泌レベルに匹敵することが観察された。
例示的実験フローチャート及び設定
本実施例は、例示的なチップの設定と、無流動条件下と流動条件下例えば、(10μl/時間の流速)との間の比較を説明する。チップの設定:iPSC由来の胃オルガノイド及びiFG−MOを、頂端チャネルに播種した。機能的アッセイ及びイメージングのために、基底チャネルにiHTNを播種した。モニターされる条件として、チップにおけるiHTNの成長、ならびにSox2、E−カドヘリン、及びTuJ1などの前腸マーカー及び神経マーカーのためのイメージングがあるが、これらに限定されない。図37、例示的な実験フローチャート及び設定、を参照されたい。例示的なチップ、実験条件、及びアッセイの例を示す概略的な時系列図。iPSC由来胃オルガノイド及びiFG−MOを、頂端チャネルに播種した。iHTNを、機能的アッセイ、及び、イメージングのために基底チャネルに播種した。チップにおけるiHTNの成長、ならびにSox2、E−カドヘリン、及びTuJ1などの前腸マーカー及び神経マーカーのイメージング。無流動及び流動(10μl/時間の流量)条件で、2連で培養した。
リンパ芽球細胞株の一般的なiPSCリプログラミングプロトコール
疾患モデリングは、患者に特異的な幹細胞を使用して疾患の特徴を再現し、発生の特徴を観察し得るので、大きな利益を得ることができる。iPSC生成のための重要な資源として、世界中に様々な保存施設が存在しているリンパ芽球細胞株がある。これらの供給源からリプログラミングのために改良がされた方法は、最初に、プラスミドの組み合わせを用いた標的宿主細胞の核形成、それに続く2日間のインキュベーション、3〜5日目に、毎日、リプログラミング培地(古い培地は吸引しない)の添加、6日目に、リプログラミング培地の(吸引による)交換、7日目〜9日目の各日に、毎日、リプログラミング培地の(古い培地は吸引しない)添加、10日目に、リプログラミング培地の(吸引による)交換、10日目〜16日目の隔日での、リプログラミング培地の(古い培地は吸引しない)添加として記載することができる。早ければ11日目にも小さなコロニーが出現し、17日目までには、相当数のコロニーが視認できるようになる。漸進的に増量する無血清完全培地、mTeSR1、への培地交換は、18〜20日目に行われる。24日目までに、リプログラムされたコロニーは容易に明らとなり、確認のための生存細胞イメージングのために抗体染色し得る。25〜29日目を通して、追加のコロニーを、サブクローニングのために単離し得る。30日目までに、先に単離したコロニーが付着し始め、正常なiPSC形態を示し、及び、保存するか、または、連続して細胞株として継代することができる。記載した技術を用いて、本発明者らは、少なくとも10%の変換効率を達成することができ、このことは、既存のリプログラミング研究と比較して、少なくとも3〜8倍の改善を示す。さらなる詳細は、PCT出願第PCT/US2015/034532を参照されたい。当該出願は、本明細書にその全内容を参照することにより援用する
iPSCからの三次元腸オルガノイド及び腸管上皮細胞
胚体内胚葉の形成を誘導するために、すべてのiPSCを、高用量のアクチビンA(100ng/ml、R&D Systems)とともに、FBSの濃度を経時的に高めながら(1日目、2日目、及び、3日目に、それぞれ、0%、0.2%、及び、2%で)培養をした。内胚葉分化の初日に、Wnt3A(25ng/ml、R&D Systems)も加えた。後腸形成を誘導するために、細胞を、Wnt3A及びFGF4(500ng/ml、R&D Systems)と併せて、2%FBSを含む改良DMEM/F12で培養をした。3〜4日後、浮遊性の上皮スフィア、及び、緩やかに付着した上皮チューブが見えてくるようになり、そして、回収を行った。その後、これらの上皮構造を、R−スポンジン−1、ノギン、EGF(それぞれ、500ng/ml、100ng/ml、及び、100ng/ml、すべてR&D Systems)を含有するマトリゲルに懸濁し、次に、R−スポンジン−1、ノギン、EGF(それぞれ、500ng/ml、100ng/ml、及び、100ng/ml、すべてR&D Systems)及び、B27(1×、Invitrogen)を含有する腸用培地に重層した。その後、オルガノイドを、7〜10日ごとに継代した。
腸管上皮細胞のマイクロ流体装置への播種
腸管上皮細胞をマイクロ流体装置に播種するために、HIOをまず解離させ、次に、蛍光活性化細胞選別を用いて腸管上皮細胞を得た。選別の24時間前に、ROCK阻害剤(10μM、Tocris)を、HIO培養培地に添加した。その翌日に、HIOをマトリゲルから除去し、続いて、オルガノイドが、単細胞懸濁液に完全に解離するまで、TrypLE Express(Life Technologies)において、20〜40分間、インキュベートした。次いで、これらの細胞を、30マイクロメートルのフィルターに通し、そして、CD326(Biolegend)で、30分間、染色した。次いで、細胞をCD326について、陽性選別をした。細胞を回収し、そして、ROCK阻害剤(10μM、Tocris)、SB202190(10μM、Tocris)、及び、A83−01(500nM、Tocris)を含有する腸の培地で、5×106/mlの密度に再懸濁した。死滅した/付着していない細胞を、3〜6時間後に、装置に通して培地を洗い流すことで除去し、及び、流動を、8〜24時間後に、60μl/時間の速度で開始した。
腸管上皮細胞のマイクロ流体装置への播種
iPSC由来の腸管上皮を、マイクロ流体装置に播種し、続いて、腸管上皮サブタイプの特徴付けを行う。経上皮抵抗性及びデキストランFITC流出による透過性の検査を含む機能的アッセイは、基礎的条件下、または、インターフェロン−γ(IFNγ)及び/または腫瘍壊死因子−α(TNFα)などの炎症性サイトカインへの応答のいずれかで評価する。また、様々な腸管上皮サブタイプを調節し得る薬物候補を調べて、そのようなサブタイプが、実際に調節され得るかどうかを評価する。このようなアッセイを確立した後、遺伝的に定義された炎症性腸疾患(IBD)患者由来のIPSCが生成され、腸オルガノイドに分化させ、解離し、その後にマイクロ流体装置に播種され、IBDに関連する遺伝的変異の機能的結果を評価する。
肥満モデル
本実施例(及び、次の実施例)は、肥満モデルに関連する細胞に関する。非統合iPSC株は、正常肥満度指数(BMI<25)、及び、BMI>50の超肥満(SO)を示す個体から生成した。内分泌組織−胃腸(GI)オルガノイド及び視床下部(HT)ニューロペプチダーゼニューロンへのiPSC再分化の実現可能性を示した。iPSC−内分泌細胞から、異なる基準の全細胞プロテオームプロファイルを生成した。iPSCの胃腸オルガノイド(iGIO)及び視床下部ニューロン(iHTN)への分化は、「臓器チップ」マイクロ流体装置に細胞を播種する前に行った。使用を企図する例示的なマイクロ流体装置を図38に示し、チップでiGIO及びiHTNを使用する例示的な結果を図39に示す。
EDCを用いたマイクロ流体「臓器チップ」装置の慢性低用量処置
発明者らは、内分泌攪乱化学物質(EDC)に対する慢性の低用量曝露は、初期のヒト内分泌組織発生期間において有害であり、その結果、永続的なミトコンドリア機能障害を伴った過反応性のNF−κB及びHMGタンパク質前炎症性シグナル伝達をもたらすとの仮説をたてた。このことを試験するために、「臓器チップ」マイクロ流体装置(実施例31)に播種した胃腸オルガノイド(iGIO)及び視床下部ニューロン(iHTN)を、EDC汚染物質/混合物(トリブチルスズ(TBT)、パーフルオロオクタン酸(PFOA)、ブチル化ヒドロキシトルエン(BHT)、及び、ビス(2−エチルヘキシル)フタレート(DEHP)の慢性低用量処置(TDI範囲))に曝露する。調節不全分泌タンパク質群を、定量的プロテオミクスによって特定する。
単細胞懸濁液が播種されたマイクロ流体「臓器チップ」装置
一実施形態において、iPSCは、HIOを形成するように指示され、続いて、単細胞懸濁液に解離された。次いで、これらの細胞を、多孔質可撓性膜によって分離された2つのチャンバからなる、小型マイクロ流体装置(SMD)に播種した。図40を、参照されたい。
トランスウェル培養装置と比較したマイクロ流体「臓器チップ」装置
iGIO及びiHTNを、ダイナミックフローマイクロ流体装置、図38A、及び、静的トランスウェル装置、図38Bの双方に播種した。例示的な図38は、ダイナミックフロー臓器チップ(ダイナミックフローOoC)、及び、静的トランスウェル培養として、iGIO(頂端)、及び、iHTN(基底)のEDC撹乱のための例示的な播種を示している。これらのシステムは、例示的化合物も用いて(+/−(対照))を試験し、当該化合物として、TNF−αやEDCが含まれるが、これらに限定されない。ダイナミックフローOoC:PDMS膜は、7μmの孔を有する。頂端チャネルの高さは、1mmであり、基底チャネルの高さは、0.2mmである。
マイクロ流体培養における上皮細胞
IPSC由来のヒト腸管上皮細胞を、10ng/mlのIFNγで処置をした。IFNγの基礎投与は、IPSC由来のヒト腸管上皮細胞における経上皮抵抗性の低下と、デキストランFITCの流出の増加とをもたらす。基本的に、このことは、このサイトカインに応答して腸管上皮がより透過性であることを意味している。TNFαの添加は、腸の透過性に変化を誘発しない。
マイクロ流体「臓器チップ」装置における使用のために開発された三次元オルガノイドシステム
発明者らは、例えば、図41に示すように、腸内で一般的に認められる全ての細胞型を有する腸オルガノイドを成長させた。例として、個々の細胞を、図42A〜Dの顕微鏡写真に蛍光染色して示している。これらは、栄養吸収に関与する腸細胞、粘液産生に関与する杯細胞、抗菌剤産生に関与するパネート細胞、及び、ホルモン産生に関与する腸内分泌細胞を含む。
マイクロ流体「臓器チップ」装置
例示的な概略図及びマイクロ流体チップで増殖する細胞を、図44A〜Eに示す。A)チップの概略図を示す。B及びC)は、「腸管チップ」の部分及びサイズを識別するオーバレイの写真を示す。C)は、さらに、膜の顕微鏡写真を示す。D)は、機械的歪みを持たないチップ、及び、機械的歪みを有するチップの、それぞれの表記の下に結果として生じる細胞の顕微鏡写真を伴った概略図を示す。及びE)は、印加圧力(kPa)に対する、基板の歪み(%)と細胞の歪み(%)とのグラフを示す。
Caco−2上皮細胞は、マイクロ流体「臓器チップ」装置で増殖した場合、腸管様組織とは異なる。
チップで成長したCaco−2上皮細胞は、チップで成長した腸管様組織が示すのと同じIFNγに対する応答を示さない。実際、IFNγで処置した腸管様組織細胞と、IFNγを有する及びIFNγを有さないCaco−2上皮細胞との相対発現を比較するマーカーのパネルは、試験した各遺伝子マーカーに対して異なる応答を示した。図48は、IFNγ処置をした、及びIFNγ処置をしないグリセルアルデヒド3−リン酸デヒドロゲナーゼ(GADPH)に対して正規化した遺伝子マーカーの相対的な例示的発現のグラフを示す。A)IDO1(インドールアミン2,3−ジオキシゲナーゼ1)。B)GBP1(グアニル酸結合タンパク質1)。C)GBP4(グアニル酸結合タンパク質4)。D)LYZ(リゾチーム)。E)PLA2G2A(ホスホリパーゼA2グループIIA)。F)分泌された抗菌レクチン(RegIIIγ)。G)LRG5(Gタンパク質結合受容体5を含むロイシンリッチリピート)。H)OLM4(オルファクトメディン4)。及びI)MUC4(ムチン4)。
マイクロ流体「臓器チップ」装置における分極した腸の絨毛様構造の自発的形成
ヒト腸オルガノイド由来の腸管上皮細胞を、本明細書に記載するようにマイクロ流体チップで増殖させた。チップに播種してから12日後、細胞は、チップの上部チャネル端部の屈曲部を越えて延在する連続層とコンフルエントになった。図49を、参照されたい。
マイクロ流体チップの細胞播種密度
本実施例は、腸内腸管様組織の単細胞懸濁液中の異なる量の細胞を使用するチップの播種の例示的な結果を示す。少なくとも5つの異なるチップに、40μlの液体あたり様々な細胞の量を播種した。インキュベーションの6日目である図55D及びEは、濃度3.75×106細胞/ml(40μlに150K)、及び、E)2.5×106細胞/ml(40μlに100K)を播種したマイクロ流体チップで増殖した腸細胞の画像であり、インキュベーションの7日目である図56Cでは、十分な細胞を播種しなかった。また、マイクロ流体チップの膜の頂部で成長する細胞の高拡大画像も、これらの細胞数で、コンフルエントな被覆率に至らないことを裏付けた。例えば、図56は、3.75×106細胞/ml(40μlに150K)が、コンフルエントな被覆率を提供するには不十分であることを示している。赤で示した例示的な被覆されていない領域を参照されたい。図57は、2.5×106細胞/ml(40μlに100K)が、コンフルエントな被覆率を提供するには不十分であることを示している、赤で示した幾つかの例示的な被覆されていない領域を参照されたい。対照的に、7.5×106細胞/ml(40μlに300K)、6.25×106細胞/ml(40μlに250K)、5.0×106細胞/ml(40μlに200K)は、コンフルエントな細胞の層を提供するのに十分な細胞であった。
マイクロ流体腸オルガノイドチップの頂端及び基底チャネルに使用するための培地製剤を特定する。
チップに播種するために使用する前のオルガノイドの最適な培養時間、例えば、チップに播種するオルガノイドの齢を特定し、上部チャネルのオルガノイド単細胞懸濁播種密度の範囲を確立し、30μl/時間の流速(両方のチャネルにおける)が、絨毛様構造の自発的形成を誘導することを発見した後に、マイクロ流体チップの上部チャネルの生存可能な細胞被覆率をもたらす培地処方を特定するために培地組成を試験した。一部には、所望の細胞増殖及び特徴付けのために、増殖因子を含有する培地が、上部及び下部の双方のチャネルに必要であるかどうかを評価することが一つの目標であった。例示的な培地処方物を、本実施例において後述する。
マイクロ流体腸オルガノイドチップにおいて成長する腸細胞のフローサイトメトリー分析
本実施例は、本明細書に記載するマイクロ流体チップで増殖する、iPSC腸管様組織由来の腸細胞集団のパーセンテージの例示的な結果を示す。マイクロ流体チップで増殖する細胞の大部分は、上皮細胞である(例示的には、83.4%及び72%の細胞集団)。さらに、非上皮細胞集団は、例示的な集団において、腸細胞の15.6%及び28.6%として特定された。さらに、パネート細胞(5.03%)、腸内分泌細胞(0.153%)、杯細胞(0.131%)、及び、腸細胞(1.06%)などの腸小細胞集団においても、特異的な非上皮細胞タイプが検出された。
分裂細胞は、腸の絨毛の下部に位置する−パルスチェイス実験。
本実施例は、分裂細胞が、マイクロ流体腸管チップにおいて、主に、腸の絨毛の下部に位置していることを実証する。
経時的多重実験での使用のためにiPS細胞を凍結する。
ヒトiPS細胞株由来の腸内腸管様組織細胞の使用に関する一つの制約は、これらの細胞を、生存可能で再現性のあるマイクロ流体腸管チップを作製するために、ある特定の期間に使用する必要があることである。しかしながら、本発明の開発の際に、これらの細胞を用いた最初の実験から長時間離間した実験において、同じヒト腸内腸管様組織細胞の複数のアリコート(すなわち、複製試料)を使用するための方法及び条件が開発された。あるいは、ヒトiPS細胞株に由来する腸内腸管様組織細胞は、マイクロ流体チップで使用する前に長期間貯蔵し得る。
臓器チップ設計の変形
マイクロ流体臓器チップの設計の追加の実施形態を、図65〜図67に示しており、そこでは、複数の臓器のためのマイクロ流体チップが、流体的に取り付けられている。
超肥満iPSC由来視床下部ニューロンは、調節不全の神経内分泌応答経路を有する。
視床下部の弓状核(ARC)でのニューロンは、飢餓や満腹などの摂食行動を調節する。それらは、胃腸管、膵臓、及び、脂肪組織からのホルモンシグナルに応答した神経ペプチドの分泌によって、生理学的代謝プロセスを制御する。この応答の調節不全は、肥満などの代謝性疾患における細胞病態生理学の中心にある。本研究において、発明者らは、多能性幹細胞からヒトの視床下部ニューロン(iHTN)を分化させるためのロバスト法を記載し、(a)摂食行動を制御する重要な外因性ホルモンを有するiHTNを撹乱させ、(b)超肥満個体(BMI>50)に由来するiHTNと、健常な正常対照(BMI<25)に由来するiHTNとを比較することにより、代謝性疾患をモデル化することにおいて、それらの機能的有用性を実証する。
Claims (40)
- 腸細胞を生成する方法であって、
アクチビンA及びWnt3Aの存在下でのiPSCの培養による胚体内胚葉の生成、
FGF4、及びWnt3AまたはCHIR99021のいずれかの存在下での胚体内胚葉の培養による後腸への分化、
上皮スフィアまたは上皮チューブの回収、
上皮スフィアまたは上皮チューブのマトリゲルでの懸濁、ならびに
CHIR99021、ノギン、及びEGFの存在下で培養して腸細胞を生成すること
を含む、前記方法。 - 前記iPSCが、リプログラミングされたリンパ芽球様B細胞由来の誘導多能性幹細胞(LCL−iPSC)である、請求項1に記載の方法。
- 前記iPSCが、炎症性腸疾患及び/または炎症性腸病態に罹患している対象から得たリプログラミングされた細胞である、請求項2に記載の方法。
- アクチビンA及びWnt3Aの存在下でのiPSCの培養が、約3〜4日間である、請求項1に記載の方法。
- FGF4、及びWnt3AまたはCHIR99021のいずれかの存在下での胚体内胚葉の培養が、約1〜4日間である、請求項1に記載の方法。
- CHIR99021、ノギン、及びEGFの存在下での培養が、約18〜21日間の期間である、請求項1に記載の方法。
- 前記腸細胞を単細胞に分離させ、CD326+発現に基づいて精製する、請求項1に記載の方法。
- 前記腸細胞を、分離の前に、ROCK阻害剤の存在下で培養する、請求項7に記載の方法。
- 前記単細胞が、ROCK阻害剤、SB202190、及びA83−01の1つ以上を含む培地で再懸濁される、請求項7に記載の方法。
- 前記腸細胞が、装置に播種される、請求項1に記載の方法。
- 請求項1に記載の方法によって製造された腸細胞を含む組成物。
- 請求項1に記載の方法によって製造された腸細胞を含むオルガノイド。
- 絨毛を含む組織化した構造を含む、請求項12に記載のオルガノイド。
- 誘導多能性幹細胞を分化させる方法であって、
ある量の誘導多能性幹細胞(iPSC)を提供すること、及び
1つ以上の因子の存在下で培養すること
を含み、
該1つ以上の因子が、iPSCを分化させることができる、前記方法。 - アクチビンA及びWnt3Aを含む1つ以上の因子の存在下で培養することにより、前記iPSCを胚体内胚葉へと分化させる、請求項14に記載の方法。
- アクチビンA及びWnt3Aを含む1つ以上の因子の存在下での培養が、約3日間である、請求項15に記載の方法。
- 前記分化するiPSCが、最初に、血清を含まない条件下で培養され、続いて、血清が添加される、請求項16に記載の方法。
- CHIR99021、FGF(FGF4)、LDN、及びレチノイン酸(RA)を含む1つ以上の因子の存在下でさらに培養することにより、胚体内胚葉を前腸スフェロイドへと分化させる、請求項15に記載の方法。
- CHIR99021、FGF(FGF4)、LDN、及びレチノイン酸(RA)を含む1つ以上の因子の存在下での培養が、約3日間である、請求項18に記載の方法。
- コーティングされた表面上で培養することにより、前記前腸スフェロイドを前腸上皮へと分化させる、請求項18に記載の方法。
- 表皮成長因子(EGF)を含む1つ以上の因子の存在下でのさらなる培養によって、前記前腸スフェロイドを前腸上皮へと分化させる、請求項18に記載の方法。
- 表皮成長因子(EGF)を含む1つ以上の因子の存在下での前記さらなる培養が、約20日間である、請求項21に記載の方法。
- 前記iPSCを、最初に、ROCK阻害剤Y27632の存在下で培養する、請求項14に記載の方法。
- LDN193189及びSB431542を含む1つ以上の因子の存在下で培養することにより、前記iPSCを神経外胚葉へと分化させる、請求項23に記載の方法。
- LDN193189及びSB431542を含む1つ以上の因子の存在下での培養が、約2日間である、請求項24に記載の方法。
- スムーズンドアゴニストSAG、プルモルファミン(PMN)及びIWR−endoを含む1つ以上の因子の存在下で培養することにより、前記神経外胚葉を腹側間脳へと分化させる、請求項24に記載の方法。
- スムーズンドアゴニストSAG、プルモルファミン(PMN)、及びIWR−endoを含む1つ以上の因子の存在下での培養が、約5〜6日間である、請求項26に記載の方法。
- DAPT、レチノイン酸(RA)を含む1つ以上の因子の存在下で培養することにより、腹側間脳を成熟させる、請求項26に記載の方法。
- DAPT、レチノイン酸(RA)を含む1つ以上の因子の存在下での培養が、約4〜5日間である、請求項28に記載の方法。
- BDNFを含む1つ以上の因子の存在下で培養することにより、前記成熟腹側間脳をさらに成熟させる、請求項29に記載の方法。
- BDNFを含む1つ以上の因子の存在下での培養が、約20〜27日間である、請求項30に記載の方法。
- 請求項14に記載の方法によって製造された前腸細胞を含む、分化したiPSCの組成物。
- 請求項14に記載の方法によって製造された視床下部ニューロンを含む、分化したiPSCの組成物。
- 化合物スクリーニングの方法であって、
ある量の分化した誘導多能性幹細胞(iPSC)を提供すること、
該分化したiPSCを、1つ以上の化合物と接触させること、
該分化したiPSCの1つ以上の特性を測定すること
を含み、
該分化したiPSCの該1つ以上の特性の測定が、該1つ以上の化合物の特徴を特定する、前記方法。 - 化合物スクリーニングが、内分泌攪乱についてのスクリーニングを含む、請求項34に記載の方法。
- 前記1つ以上の化合物の特徴が、NF−kBのリン酸化を誘導することを含む、請求項35に記載の方法。
- 前記1つ以上の化合物の特徴が、ミトコンドリア呼吸の減少を含む、請求項35に記載の方法。
- 前記1つ以上の化合物の特徴が、SCO2、POLRMT、TFAM、及びCYTB5の1つ以上の発現の減少を含む、請求項35に記載の方法。
- 前記分化したiPSCが、前腸上皮である、請求項34に記載の方法。
- 前記分化したiPSCが、視床下部ニューロンである、請求項34に記載の方法。
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662289521P | 2016-02-01 | 2016-02-01 | |
| US62/289,521 | 2016-02-01 | ||
| US201662332849P | 2016-05-06 | 2016-05-06 | |
| US62/332,849 | 2016-05-06 | ||
| US201662354040P | 2016-06-23 | 2016-06-23 | |
| US62/354,040 | 2016-06-23 | ||
| US201662437314P | 2016-12-21 | 2016-12-21 | |
| US62/437,314 | 2016-12-21 | ||
| PCT/US2017/016098 WO2017136479A1 (en) | 2016-02-01 | 2017-02-01 | Systems and methods for growth of intestinal cells in microfluidic devices |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2019506861A true JP2019506861A (ja) | 2019-03-14 |
| JP2019506861A5 JP2019506861A5 (ja) | 2019-04-25 |
Family
ID=59500254
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018540028A Pending JP2019506861A (ja) | 2016-02-01 | 2017-02-01 | マイクロ流体装置における腸細胞の成長のためのシステム及び方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (4) | US11473061B2 (ja) |
| EP (2) | EP3411470A4 (ja) |
| JP (1) | JP2019506861A (ja) |
| AU (3) | AU2017214468B2 (ja) |
| CA (2) | CA3013357A1 (ja) |
| GB (1) | GB2564582B (ja) |
| WO (2) | WO2017136479A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022025269A1 (ja) * | 2020-07-30 | 2022-02-03 | 国立研究開発法人理化学研究所 | 膀胱オルガノイド及びその製造方法 |
| US11414648B2 (en) | 2017-03-24 | 2022-08-16 | Cedars-Sinai Medical Center | Methods and compositions for production of fallopian tube epithelium |
| US11473061B2 (en) | 2016-02-01 | 2022-10-18 | Cedars-Sinai Medical Center | Systems and methods for growth of intestinal cells in microfluidic devices |
| US11767513B2 (en) | 2017-03-14 | 2023-09-26 | Cedars-Sinai Medical Center | Neuromuscular junction |
| US11913022B2 (en) | 2017-01-25 | 2024-02-27 | Cedars-Sinai Medical Center | In vitro induction of mammary-like differentiation from human pluripotent stem cells |
| WO2024053406A1 (ja) * | 2022-09-09 | 2024-03-14 | 国立大学法人大阪大学 | 小腸上皮様細胞及びその作製方法 |
| US11981918B2 (en) | 2018-04-06 | 2024-05-14 | Cedars-Sinai Medical Center | Differentiation technique to generate dopaminergic neurons from induced pluripotent stem cells |
| US12042791B2 (en) | 2016-01-12 | 2024-07-23 | Cedars-Sinai Medical Center | Method of osteogenic differentiation in microfluidic tissue culture systems |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011140441A2 (en) | 2010-05-06 | 2011-11-10 | Children's Hospital Medical Center | Methods and systems for converting precursor cells into intestinal tissues through directed differentiation |
| US10174289B2 (en) | 2014-05-28 | 2019-01-08 | Children's Hospital Medical Center | Methods and systems for converting precursor cells into gastric tissues through directed differentiation |
| CA2963704A1 (en) | 2014-10-17 | 2016-04-21 | Children's Hospital Medical Center | In vivo model of human small intestine using pluripotent stem cells and methods of making and using same |
| ES2929758T3 (es) | 2016-05-05 | 2022-12-01 | Childrens Hospital Med Ct | Métodos para la fabricación in vitro de tejido del fondo gástrico y composiciones relacionadas con el mismo |
| EP3512935B1 (en) | 2016-09-13 | 2022-02-23 | President and Fellows of Harvard College | Methods relating to intestinal organ-on-a-chip |
| CN118909916A (zh) | 2016-12-05 | 2024-11-08 | 儿童医院医学中心 | 结肠类器官及其制备和使用方法 |
| EP3625331B1 (en) * | 2017-05-19 | 2024-07-10 | Cedars-Sinai Medical Center | Systems and methods for growth of intestinal cells |
| WO2019060735A1 (en) | 2017-09-21 | 2019-03-28 | EMULATE, Inc. | PHYSIOLOGY AND PATHOPHYSIOLOGY OF HUMAN INTESTINES: INTESTIN-ON-CHIP |
| CN107828655A (zh) * | 2017-11-15 | 2018-03-23 | 大连理工大学 | 一种微流控芯片及其应用 |
| CN111670245B (zh) * | 2017-12-22 | 2024-03-12 | 蒂斯由斯股份有限公司 | 建立将iPSC衍生的细胞分化为器官等效物的新型多器官芯片 |
| US20190293634A1 (en) * | 2018-02-28 | 2019-09-26 | Albert Li | Isolated intestinal mucosa and uses thereof |
| GB2612702B (en) * | 2018-05-09 | 2024-04-10 | Emulate Inc | Host-biome interactions |
| WO2019222870A1 (zh) * | 2018-05-21 | 2019-11-28 | 深圳华大生命科学研究院 | 一种仿生肠道器官芯片及其制备方法和应用 |
| KR20210040107A (ko) * | 2018-07-26 | 2021-04-12 | 칠드런즈 호스피탈 메디칼 센터 | 간-담도-췌장 조직 및 이를 제조하는 방법 |
| US11859214B1 (en) | 2018-08-17 | 2024-01-02 | The Government Of The United States, As Represented By The Secretary Of The Army | Automated system for simulating the human lower gastrointestinal tract |
| WO2020232086A1 (en) * | 2019-05-15 | 2020-11-19 | The Cleveland Clinic Foundation | Induced human colitic organoids |
| JP2022534778A (ja) | 2019-05-28 | 2022-08-03 | シリン・シェン | 患者由来ミクロオルガノスフェアのための方法及び装置 |
| EP3976070A4 (en) * | 2019-05-31 | 2023-06-14 | Children's Hospital Medical Center | SHAPED ORGANOID COMPOSITIONS AND PROCESSES FOR THEIR PRODUCTION |
| US20210003559A1 (en) * | 2019-07-02 | 2021-01-07 | Hoffmann-La Roche Inc. | Method for Assessing a Compound Interacting with a Target on Epithelial Cells |
| US20230314412A1 (en) * | 2020-03-21 | 2023-10-05 | Florica Therapeutics, Inc. | Neuronal diencephalon stem cells, preparation and uses for treatment and prevention of hormonal disorders and other diseases |
| WO2022009142A2 (en) * | 2020-07-09 | 2022-01-13 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | Methods of determining cancer therapy |
| GB202109913D0 (en) * | 2021-07-09 | 2021-08-25 | Stichting Hubrecht Organoid Tech | Organoid-derived monolayers and uses thereof |
| WO2024107345A1 (en) * | 2022-11-15 | 2024-05-23 | Corning Incorporated | Systems and methods of differentiating stem cells within bioreactor |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014171434A (ja) * | 2013-03-08 | 2014-09-22 | National Institute Of Advanced Industrial & Technology | 胃組織細胞の作製方法 |
| WO2014159356A1 (en) * | 2013-03-14 | 2014-10-02 | The Brigham And Women's Hospital, Inc. | Compositions and methods for epithelial stem cell expansion and culture |
| JP2015504676A (ja) * | 2012-01-13 | 2015-02-16 | ザ ジェネラル ホスピタル コーポレイション | 単離ヒト肺前駆細胞およびその使用 |
| WO2015188131A1 (en) * | 2014-06-05 | 2015-12-10 | Cedars-Sinai Medical Center | A novel and efficient method for reprogramming immortalized lymphoblastoid cell lines to induced pluripotent stem cells |
Family Cites Families (87)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5610281A (en) * | 1994-05-03 | 1997-03-11 | Brigham & Women's Hospital, Inc. | Antibodies for modulating heterotypic E-cadherin interactions with human T lymphocytes |
| AU2002236683A1 (en) | 2000-10-27 | 2002-05-21 | Beth Israel Deaconess Medical Center | Non-isotopic detection of osteoblastic activity in vivo using modified bisphosphonates |
| US20040247571A1 (en) | 2001-04-23 | 2004-12-09 | Xia Meijer | Neural cells expressing tyrosine hydroxylase |
| US20080004332A1 (en) | 2002-03-07 | 2008-01-03 | Alkon Daniel L | Methods for alzheimer's disease treatment and cognitive enhancement |
| US7960166B2 (en) | 2003-05-21 | 2011-06-14 | The General Hospital Corporation | Microfabricated compositions and processes for engineering tissues containing multiple cell types |
| KR20080109943A (ko) | 2003-08-29 | 2008-12-17 | 위스콘신 얼럼나이 리서어치 화운데이션 | 영장류 배아 줄기 세포 기원의 신경 줄기 세포, 운동 뉴런 및 도파민 뉴런의 시험관내 분화 방법 |
| DE102004061947A1 (de) * | 2004-12-22 | 2006-07-06 | Ccs Cell Culture Service Gmbh | Verfahren zum Einfrieren und Lagern von Zellen |
| US20070077649A1 (en) | 2005-09-06 | 2007-04-05 | Sammak Paul J | Transplantable cell growth niche and related compositions and methods |
| CA2641446C (en) | 2006-02-03 | 2016-06-07 | President And Fellows Of Harvard College | Engineered cell growth on polymeric films and biotechnological applications thereof |
| US20080044847A1 (en) | 2006-06-23 | 2008-02-21 | Shusta Eric V | Blood-Brain Barrier Model |
| KR100790880B1 (ko) * | 2006-07-05 | 2008-01-02 | 삼성전자주식회사 | 자성 비드가 결합되어 있는 소수성 다공성 중합체가 벽면에결합되어 있는 마이크로채널 또는 마이크로챔버를포함하는 미세유동장치 및 그를 이용하는 방법 |
| EP2139990A1 (en) | 2007-03-23 | 2010-01-06 | California Stem Cell, Inc. | Human late stage motor neuron progenitor cells and methods of making and using same |
| JP2008307007A (ja) | 2007-06-15 | 2008-12-25 | Bayer Schering Pharma Ag | 出生後のヒト組織由来未分化幹細胞から誘導したヒト多能性幹細胞 |
| JP2009247276A (ja) * | 2008-04-04 | 2009-10-29 | Erii Kk | 生体供給用、生体移植用、生体添加用、生体投与用の解凍器官若しくは組織または解凍細胞群とその製造方法及びそれらに用いる過冷却液並びに製造装置 |
| AU2015204375A1 (en) | 2008-07-16 | 2015-08-06 | Children's Medical Center Corporation | Organ mimic device with microchannels and methods of use and manufacturing thereof |
| CN104328050B (zh) | 2008-07-16 | 2017-12-15 | 儿童医疗中心有限公司 | 具有微通道的器官模仿装置及其使用和制造方法 |
| US8642334B2 (en) * | 2009-02-17 | 2014-02-04 | Memorial Sloan Kettering Cancer Center | Methods of neural conversion of human embryonic stem cells |
| WO2010108005A2 (en) | 2009-03-18 | 2010-09-23 | University Of Georgia Research Foundation | Novel neural progenitors from pluripotent stem cells, methods of producing same and use to produce neural cells |
| KR20110134939A (ko) | 2009-04-03 | 2011-12-15 | 더 맥클린 하스피털 코퍼레이션 | 유도된 다능성 줄기 세포 |
| AU2010281524A1 (en) | 2009-08-06 | 2012-02-23 | Neuraltus Pharmaceuticals, Inc. | Treatment of macrophage-related disorders |
| US9708582B2 (en) * | 2009-10-13 | 2017-07-18 | Stemcell Technologies Inc. | Method of differentiating stem cells |
| JP5808349B2 (ja) | 2010-03-01 | 2015-11-10 | カリス ライフ サイエンシズ スウィッツァーランド ホールディングスゲーエムベーハー | セラノーシスのためのバイオマーカー |
| WO2011140441A2 (en) * | 2010-05-06 | 2011-11-10 | Children's Hospital Medical Center | Methods and systems for converting precursor cells into intestinal tissues through directed differentiation |
| CN103459591B (zh) * | 2010-11-02 | 2016-05-04 | 国立大学法人熊本大学 | 肠细胞的制造方法 |
| EP3162386B1 (en) | 2010-12-13 | 2019-10-16 | TheraDep Technologies, Inc. | Implantable medical devices |
| US20120211373A1 (en) | 2011-02-22 | 2012-08-23 | The Regents Of The University Of Michigan | Microfluidic system for measuring cell barrier function |
| RU2612915C2 (ru) | 2011-02-28 | 2017-03-13 | Президент Энд Феллоуз Ов Харвард Колледж | Система культивирования клеток |
| WO2012122405A2 (en) | 2011-03-08 | 2012-09-13 | The Trustees Of Columbia University In The City Of New York | Screening assays using stem cells and stem cell-derived neurons from mouse models of alzheimer's disease |
| CA2836843A1 (en) | 2011-06-09 | 2012-12-13 | F. Hoffmann-La Roche Ag | Method for differentiation of pluripotent stem cells into vascular bed cells |
| CN103764166B (zh) | 2011-06-22 | 2017-10-24 | 通用医疗公司 | 蛋白质病的治疗 |
| ES2928319T3 (es) * | 2011-10-31 | 2022-11-17 | Riken | Método para el cultivo de células madre |
| CA2855932A1 (en) | 2011-11-13 | 2013-05-16 | Blanchette Rockefeller Neurosciences Institute | Pkc activators and combinations thereof |
| US9725687B2 (en) | 2011-12-09 | 2017-08-08 | President And Fellows Of Harvard College | Integrated human organ-on-chip microphysiological systems |
| US9149806B2 (en) | 2012-01-10 | 2015-10-06 | Biopico Systems Inc | Microfluidic devices and methods for cell sorting, cell culture and cells based diagnostics and therapeutics |
| WO2013152120A2 (en) | 2012-04-03 | 2013-10-10 | Whitehead Institute For Biomedical Research | Compositions and methods for promoting intestinal stem cell function |
| KR101200049B1 (ko) | 2012-05-14 | 2012-11-13 | 한국원자력연구원 | 테크네튬-99엠 트리카보닐 표지 글리신 단량체 또는 올리고머 활용 생리활성분자를 이용한 프로브의 제조방법 및 이를 포함하는 영상제 조성물 |
| EP2861612A4 (en) | 2012-06-13 | 2016-02-17 | Stemgent Inc | METHODS FOR PREPARING PLURIPOTENT STEM CELLS |
| US9513280B2 (en) | 2012-08-28 | 2016-12-06 | University Of Utah Research Foundation | Microfluidic biological barrier model and associated method |
| GB201216796D0 (en) | 2012-09-20 | 2012-11-07 | Cambridge Entpr Ltd | In vitro pancreatic differentiation |
| US20140134732A1 (en) | 2012-11-14 | 2014-05-15 | Wisconsin Alumni Research Foundation | Simplified Compositions and Methods for Generating Neural Stem Cells From Human Pluripotent Stem Cells |
| US9932559B2 (en) | 2012-11-16 | 2018-04-03 | The Johns Hopkins University | Platform for creating an artificial blood brain barrier |
| US10160950B2 (en) | 2013-03-01 | 2018-12-25 | Wisconsin Alumni Research Foundation | Methods of maintaining, expanding and differentiating neuronal subtype specific progenitors |
| US10266807B2 (en) | 2013-04-03 | 2019-04-23 | FUJIFILM Cellular Dynamics, Inc. | Methods and compositions for culturing endoderm progenitor cells in suspension |
| US10638948B2 (en) | 2013-04-19 | 2020-05-05 | Cedars-Sinai Medical Center | Imaging biomarkers for the diagnosis and prognosis of back pain and related conditions |
| EP3498824A1 (en) * | 2013-04-26 | 2019-06-19 | Memorial Sloan-Kettering Cancer Center | Cortical interneurons and other neuronal cells produced by the directed differentiation of pluripotent and multipotent cells |
| RU2016100219A (ru) | 2013-06-11 | 2017-07-17 | Президент Энд Феллоус Оф Гарвард Колледж | КЛЕТКИ SC-β И КОМПОЗИЦИИ, И СПОСОБЫ ДЛЯ ИХ СОЗДАНИЯ |
| US10017734B2 (en) | 2013-08-06 | 2018-07-10 | Takeda Pharmaceutical Company Limited | Method for producing dopaminergic neurons |
| GB201317869D0 (en) * | 2013-10-09 | 2013-11-20 | Cambridge Entpr Ltd | In vitro production of foregut stem cells |
| WO2015057261A1 (en) * | 2013-10-16 | 2015-04-23 | The Brigham And Women's Hospital, Inc. | Methods of generating intermediate mesoderm cells from human pluripotent stem cells |
| US11119093B2 (en) | 2013-12-20 | 2021-09-14 | President And Fellows Of Harvard College | Low shear microfluidic devices and methods of use and manufacturing thereof |
| WO2015138032A2 (en) | 2013-12-20 | 2015-09-17 | President And Fellows Of Harvard College | Organomimetic devices and methods of use and manufacturing thereof |
| EP3107995B1 (en) | 2014-02-18 | 2019-10-30 | Massachusetts Institute Of Technology | Biophysically sorted osteoprogenitors from culture expanded bone marrow derived mesenchymal stromal cells (mscs) |
| ES2939807T3 (es) | 2014-03-21 | 2023-04-27 | Fujifilm Cellular Dynamics Inc | Producción de neuronas dopaminérgicas del mesencéfalo y métodos para su utilización |
| EP3126484B1 (en) | 2014-03-31 | 2020-09-23 | Brigham and Women's Hospital, Inc. | Systems and methods for biomimetic fluid processing |
| CN107075466A (zh) | 2014-04-23 | 2017-08-18 | 新加坡科技研究局 | 细胞重编程的方法 |
| JP6588969B2 (ja) | 2014-05-16 | 2019-10-09 | ヤンセン バイオテツク,インコーポレーテツド | 膵内分泌細胞内のmafa発現を強化するための小分子の使用 |
| WO2015181253A1 (en) | 2014-05-27 | 2015-12-03 | Fundación Pública Andaluza Progreso Y Salud | Neural progenitor cell population |
| US10174289B2 (en) * | 2014-05-28 | 2019-01-08 | Children's Hospital Medical Center | Methods and systems for converting precursor cells into gastric tissues through directed differentiation |
| CA2963704A1 (en) | 2014-10-17 | 2016-04-21 | Children's Hospital Medical Center | In vivo model of human small intestine using pluripotent stem cells and methods of making and using same |
| AU2015336453B2 (en) | 2014-10-24 | 2021-05-20 | Riken | Production method for nerve tissue |
| WO2016086040A1 (en) | 2014-11-25 | 2016-06-02 | Los Alamos National Security, Llc | Patterned neuromuscular junctions and methods of use |
| JP5777127B1 (ja) | 2014-12-09 | 2015-09-09 | 公立大学法人横浜市立大学 | 原始腸内胚葉細胞及びその作製方法 |
| US10344259B2 (en) | 2015-04-09 | 2019-07-09 | Biolamina Ab | Methods and compositions for producing stem cell derived dopaminergic cells for use in treatment of neurodegenerative diseases |
| JP2018521113A (ja) | 2015-05-11 | 2018-08-02 | アルコン、ダニエル・エル. | ApoE4対立遺伝子の存在を決定した後のPKC活性化因子を使用した神経変性状態の治療 |
| WO2017035119A1 (en) | 2015-08-24 | 2017-03-02 | National University Of Singapore | Blood brain barrier model in a 3d co-culture microfluidic system |
| US20180057788A1 (en) | 2016-08-29 | 2018-03-01 | EMULATE, Inc. | Development of spinal cord on a microfluidic chip |
| WO2017070224A1 (en) | 2015-10-19 | 2017-04-27 | EMULATE, Inc. | Microfluidic model of the blood brain barrier |
| US20170226478A1 (en) | 2015-10-19 | 2017-08-10 | EMULATE, Inc. | Neuromuscular Junction: NMJ-ON-CHIP |
| US20180306780A1 (en) | 2015-10-29 | 2018-10-25 | The Regents Of The University Of California | Astrocyte differentiation protocol |
| WO2017078807A1 (en) | 2015-11-04 | 2017-05-11 | Fate Therapeutics, Inc. | Methods and compositions for inducing hematopoietic cell differentiation |
| GB2569058B (en) | 2016-01-12 | 2021-04-14 | Cedars Sinai Medical Center | A method of osteogenic differentiation in microfluidic tissue culture systems |
| JP2019506861A (ja) | 2016-02-01 | 2019-03-14 | シーダーズ−サイナイ メディカル センター | マイクロ流体装置における腸細胞の成長のためのシステム及び方法 |
| WO2017143049A1 (en) | 2016-02-16 | 2017-08-24 | President And Fellows Of Harvard College | Improved blood-brain barrier endothelial cells derived from pluripotent stem cells for blood-brain barrier models |
| EP3423564A4 (en) | 2016-03-03 | 2019-08-21 | New York Stem Cell Foundation, Inc. | MICROGLIAL CELLS DERIVED FROM PLURIPOTENT STEM CELLS AND METHODS OF PREPARATION AND USE THEREOF |
| US10214724B2 (en) | 2016-04-05 | 2019-02-26 | Wisconsin Alumni Research Foundation | Methods for differentiation of human pluripotent stem cells to brain microvascular endothelial cells |
| EP3458572A4 (en) | 2016-05-17 | 2020-03-04 | Agency for Science, Technology and Research | ENDOTHEL CELLS FROM HUMAN STEM CELLS, ENDOTHEL-HEPATOCYTE COCULTIVATION SYSTEM AND USES THEREOF |
| US10221395B2 (en) | 2016-06-16 | 2019-03-05 | Cedars-Sinai Medical Center | Efficient method for reprogramming blood to induced pluripotent stem cells |
| US12091650B2 (en) | 2016-08-29 | 2024-09-17 | EMULATE, Inc. | Development of spinal cord on a microfluidic chip |
| AU2018235950B2 (en) | 2017-03-14 | 2022-11-17 | Cedars-Sinai Medical Center | Neuromuscular junction: NMJ-on-chip |
| US11767513B2 (en) | 2017-03-14 | 2023-09-26 | Cedars-Sinai Medical Center | Neuromuscular junction |
| GB2575574B (en) | 2017-03-14 | 2022-08-17 | Cedars Sinai Medical Center | Neuromuscular junction |
| EP3625331B1 (en) | 2017-05-19 | 2024-07-10 | Cedars-Sinai Medical Center | Systems and methods for growth of intestinal cells |
| WO2019183597A1 (en) | 2018-03-23 | 2019-09-26 | Cedars-Sinai Medical Center | Methods of use of islet cells |
| EP3775161A4 (en) | 2018-04-06 | 2022-04-06 | Cedars-Sinai Medical Center | NEURODEGENERATIVE DISEASE MODELS DERIVED FROM HUMAN PLURIPOTENTIC STEM CELLS ON A MICROFLUIDIC CHIP |
| WO2019195800A1 (en) | 2018-04-06 | 2019-10-10 | Cedars-Sinai Medical Center | Novel differentiation technique to generate dopaminergic neurons from induced pluripotent stem cells |
| WO2019212690A1 (en) | 2018-04-30 | 2019-11-07 | Cedars-Sinai Medical Center | Pkc pathway in parkinson's disease |
-
2017
- 2017-02-01 JP JP2018540028A patent/JP2019506861A/ja active Pending
- 2017-02-01 GB GB1814294.3A patent/GB2564582B/en active Active
- 2017-02-01 US US16/074,747 patent/US11473061B2/en active Active
- 2017-02-01 WO PCT/US2017/016098 patent/WO2017136479A1/en active Application Filing
- 2017-02-01 WO PCT/US2017/016079 patent/WO2017136462A2/en active Application Filing
- 2017-02-01 AU AU2017214468A patent/AU2017214468B2/en active Active
- 2017-02-01 EP EP17748084.5A patent/EP3411470A4/en active Pending
- 2017-02-01 EP EP17748100.9A patent/EP3411472A4/en active Pending
- 2017-02-01 CA CA3013357A patent/CA3013357A1/en active Pending
- 2017-02-01 CA CA3013337A patent/CA3013337C/en active Active
- 2017-02-01 AU AU2017213795A patent/AU2017213795A1/en not_active Abandoned
-
2018
- 2018-07-31 US US16/051,004 patent/US11326149B2/en active Active
-
2020
- 2020-12-16 AU AU2020289787A patent/AU2020289787A1/en not_active Abandoned
-
2022
- 2022-02-23 US US17/678,485 patent/US11952592B2/en active Active
-
2024
- 2024-04-04 US US18/626,855 patent/US20240254449A1/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015504676A (ja) * | 2012-01-13 | 2015-02-16 | ザ ジェネラル ホスピタル コーポレイション | 単離ヒト肺前駆細胞およびその使用 |
| JP2014171434A (ja) * | 2013-03-08 | 2014-09-22 | National Institute Of Advanced Industrial & Technology | 胃組織細胞の作製方法 |
| WO2014159356A1 (en) * | 2013-03-14 | 2014-10-02 | The Brigham And Women's Hospital, Inc. | Compositions and methods for epithelial stem cell expansion and culture |
| WO2015188131A1 (en) * | 2014-06-05 | 2015-12-10 | Cedars-Sinai Medical Center | A novel and efficient method for reprogramming immortalized lymphoblastoid cell lines to induced pluripotent stem cells |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12042791B2 (en) | 2016-01-12 | 2024-07-23 | Cedars-Sinai Medical Center | Method of osteogenic differentiation in microfluidic tissue culture systems |
| US11473061B2 (en) | 2016-02-01 | 2022-10-18 | Cedars-Sinai Medical Center | Systems and methods for growth of intestinal cells in microfluidic devices |
| US11913022B2 (en) | 2017-01-25 | 2024-02-27 | Cedars-Sinai Medical Center | In vitro induction of mammary-like differentiation from human pluripotent stem cells |
| US11767513B2 (en) | 2017-03-14 | 2023-09-26 | Cedars-Sinai Medical Center | Neuromuscular junction |
| US11414648B2 (en) | 2017-03-24 | 2022-08-16 | Cedars-Sinai Medical Center | Methods and compositions for production of fallopian tube epithelium |
| US11981918B2 (en) | 2018-04-06 | 2024-05-14 | Cedars-Sinai Medical Center | Differentiation technique to generate dopaminergic neurons from induced pluripotent stem cells |
| WO2022025269A1 (ja) * | 2020-07-30 | 2022-02-03 | 国立研究開発法人理化学研究所 | 膀胱オルガノイド及びその製造方法 |
| WO2024053406A1 (ja) * | 2022-09-09 | 2024-03-14 | 国立大学法人大阪大学 | 小腸上皮様細胞及びその作製方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| GB2564582A (en) | 2019-01-16 |
| AU2017214468B2 (en) | 2020-09-17 |
| CA3013337A1 (en) | 2017-08-10 |
| EP3411470A4 (en) | 2019-10-09 |
| EP3411472A4 (en) | 2020-01-22 |
| AU2020289787A1 (en) | 2021-01-21 |
| US20190031992A1 (en) | 2019-01-31 |
| GB201814294D0 (en) | 2018-10-17 |
| CA3013337C (en) | 2023-05-09 |
| US11952592B2 (en) | 2024-04-09 |
| AU2017214468A1 (en) | 2018-09-13 |
| US20190153395A1 (en) | 2019-05-23 |
| US20220282221A1 (en) | 2022-09-08 |
| AU2017213795A1 (en) | 2018-08-16 |
| US11326149B2 (en) | 2022-05-10 |
| EP3411470A2 (en) | 2018-12-12 |
| WO2017136462A2 (en) | 2017-08-10 |
| US11473061B2 (en) | 2022-10-18 |
| GB2564582B (en) | 2021-09-22 |
| WO2017136479A1 (en) | 2017-08-10 |
| US20240254449A1 (en) | 2024-08-01 |
| WO2017136462A3 (en) | 2017-09-14 |
| CA3013357A1 (en) | 2017-08-10 |
| EP3411472A1 (en) | 2018-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11952592B2 (en) | Systems and methods for growth of intestinal cells in microfluidic devices | |
| CN110382012B (zh) | 肝类器官疾病模型以及其制备和使用方法 | |
| Barker | Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration | |
| US20210208131A1 (en) | Immune cell organoid co-cultures | |
| JP2022095796A (ja) | ヒト腸神経堤系統由来多能性幹細胞によって可能にされるヒルシュスプルング病における細胞ベースの処置および薬物発見 | |
| Clarke et al. | The adult mouse dentate gyrus contains populations of committed progenitor cells that are distinct from subependymal zone neural stem cells | |
| Seaberg et al. | Intrinsic differences distinguish transiently neurogenic progenitors from neural stem cells in the early postnatal brain | |
| JP2018518938A (ja) | 神経変性疾患の処置における使用のための幹細胞由来のドパミン作用性細胞を生成するための方法および組成物 | |
| US20220401493A1 (en) | Isolation of fusion-competent myoblasts and therapeutic applications thereof related to muscular dystrophy | |
| JP2022132249A (ja) | 多能性幹細胞の中腸内胚葉細胞への分化 | |
| Torreggiani et al. | Protocol for the long‐term culture of human primary keratinocytes from the normal colorectal mucosa | |
| Nishikawa et al. | Migration and differentiation of transplanted enteric neural crest-derived cells in murine model of Hirschsprung’s disease | |
| Martín-Ibáñez et al. | Insights in spatio-temporal characterization of human fetal neural stem cells | |
| Pérez Millán et al. | Pituitary stem cells: past, present and future perspectives | |
| Loret et al. | Preliminary characterization of jejunocyte and colonocyte cell lines isolated by enzymatic digestion from adult and young cattle | |
| US20240287430A1 (en) | Systems and Methods for Cellular Lumen Formation and Cellular Differentiation | |
| WO2024059208A2 (en) | Compositions and methods for obtaining suspension human intestinal organoids having a serosal mesothelial layer | |
| Wiegering et al. | A differential requirement for ciliary transition zone proteins in human and mouse neural progenitor fate specification | |
| Xiong et al. | SALL2 regulates neural differentiation of mouse embryonic stem cells through Tuba1a | |
| Sharma | Lineage commitment and specification of progenitors of the caudal mesoderm | |
| Frith | Delineating the signals in anterior-posterior patterning of hPSC derived neural crest cells | |
| Gracz | Intrinsic and extrinsic regulation of potency in the intestinal stem cell niche | |
| Seaberg | Mammalian brain development: The role of distinct neural stem and progenitor cells from embryonic neural induction to adult neurogenesis. | |
| Gracz | The role of SRY-box (Sox) transcription factors in epithelial stem cell biology of the gastrointestinal tract | |
| Surette et al. | Rohan R. Nadkarni, Soumeya Abed, Brian J. Cox, 4, 8 Sonam Bhatia, Jennifer T. Lau, 3, 6 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180829 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190212 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200128 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210301 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20210308 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20210601 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210830 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20220131 |