JP2006510630A - 糖尿病を治療又は予防するためのジペプチジルペプチダーゼ阻害剤としてのフェニルアラニン誘導体 - Google Patents
糖尿病を治療又は予防するためのジペプチジルペプチダーゼ阻害剤としてのフェニルアラニン誘導体 Download PDFInfo
- Publication number
- JP2006510630A JP2006510630A JP2004557341A JP2004557341A JP2006510630A JP 2006510630 A JP2006510630 A JP 2006510630A JP 2004557341 A JP2004557341 A JP 2004557341A JP 2004557341 A JP2004557341 A JP 2004557341A JP 2006510630 A JP2006510630 A JP 2006510630A
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- unsubstituted
- substituted
- alkoxy
- halogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 206010012601 diabetes mellitus Diseases 0.000 title claims abstract description 19
- 150000002993 phenylalanine derivatives Chemical class 0.000 title abstract description 4
- 102000003779 Dipeptidyl-peptidases and tripeptidyl-peptidases Human genes 0.000 title 1
- 108090000194 Dipeptidyl-peptidases and tripeptidyl-peptidases Proteins 0.000 title 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 169
- 238000011282 treatment Methods 0.000 claims abstract description 54
- 239000003112 inhibitor Substances 0.000 claims abstract description 36
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 30
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims abstract description 25
- 201000010099 disease Diseases 0.000 claims abstract description 21
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 21
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 claims abstract description 19
- 102000016622 Dipeptidyl Peptidase 4 Human genes 0.000 claims abstract 5
- 125000000217 alkyl group Chemical group 0.000 claims description 185
- 229910052736 halogen Inorganic materials 0.000 claims description 157
- 150000002367 halogens Chemical class 0.000 claims description 157
- 125000003545 alkoxy group Chemical group 0.000 claims description 122
- -1 Cyano, Hydroxy Chemical group 0.000 claims description 79
- 125000001424 substituent group Chemical group 0.000 claims description 64
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Substances N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 60
- 229910052799 carbon Inorganic materials 0.000 claims description 46
- 238000000034 method Methods 0.000 claims description 44
- 229910052739 hydrogen Inorganic materials 0.000 claims description 39
- 229940125396 insulin Drugs 0.000 claims description 31
- 102000004877 Insulin Human genes 0.000 claims description 29
- 108090001061 Insulin Proteins 0.000 claims description 29
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 29
- 125000000623 heterocyclic group Chemical group 0.000 claims description 29
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 27
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 24
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 24
- 230000000694 effects Effects 0.000 claims description 24
- 230000005764 inhibitory process Effects 0.000 claims description 24
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 24
- 150000003839 salts Chemical class 0.000 claims description 24
- 102100040918 Pro-glucagon Human genes 0.000 claims description 23
- 239000001257 hydrogen Substances 0.000 claims description 23
- 229910052757 nitrogen Inorganic materials 0.000 claims description 23
- 241000124008 Mammalia Species 0.000 claims description 22
- 125000004432 carbon atom Chemical group C* 0.000 claims description 21
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 claims description 20
- 239000004480 active ingredient Substances 0.000 claims description 20
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 20
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 claims description 17
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 17
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 claims description 15
- 239000000556 agonist Substances 0.000 claims description 14
- 125000003118 aryl group Chemical group 0.000 claims description 14
- 125000003831 tetrazolyl group Chemical group 0.000 claims description 13
- 125000000335 thiazolyl group Chemical group 0.000 claims description 13
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 12
- 150000002431 hydrogen Chemical class 0.000 claims description 12
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 11
- 208000008589 Obesity Diseases 0.000 claims description 11
- 239000008103 glucose Substances 0.000 claims description 11
- 208000024891 symptom Diseases 0.000 claims description 11
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 10
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 10
- 235000020824 obesity Nutrition 0.000 claims description 10
- 206010022489 Insulin Resistance Diseases 0.000 claims description 9
- 102000023984 PPAR alpha Human genes 0.000 claims description 9
- 125000001072 heteroaryl group Chemical group 0.000 claims description 9
- 201000001421 hyperglycemia Diseases 0.000 claims description 9
- 230000002401 inhibitory effect Effects 0.000 claims description 9
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 claims description 9
- 208000035475 disorder Diseases 0.000 claims description 8
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims description 8
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 7
- 229940100389 Sulfonylurea Drugs 0.000 claims description 6
- 150000002632 lipids Chemical class 0.000 claims description 6
- 229940123208 Biguanide Drugs 0.000 claims description 5
- 108010004684 Pituitary adenylate cyclase-activating polypeptide Proteins 0.000 claims description 5
- 102000002808 Pituitary adenylate cyclase-activating polypeptide Human genes 0.000 claims description 5
- 125000002252 acyl group Chemical group 0.000 claims description 5
- 150000004283 biguanides Chemical class 0.000 claims description 5
- 208000006575 hypertriglyceridemia Diseases 0.000 claims description 5
- NFFXEUUOMTXWCX-UHFFFAOYSA-N 5-[(2,4-dioxo-1,3-thiazolidin-5-yl)methyl]-2-methoxy-n-[[4-(trifluoromethyl)phenyl]methyl]benzamide Chemical group C1=C(C(=O)NCC=2C=CC(=CC=2)C(F)(F)F)C(OC)=CC=C1CC1SC(=O)NC1=O NFFXEUUOMTXWCX-UHFFFAOYSA-N 0.000 claims description 4
- 208000032928 Dyslipidaemia Diseases 0.000 claims description 4
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 claims description 4
- 208000035150 Hypercholesterolemia Diseases 0.000 claims description 4
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 4
- 229940122199 Insulin secretagogue Drugs 0.000 claims description 4
- 208000017170 Lipid metabolism disease Diseases 0.000 claims description 4
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 claims description 4
- 108010016731 PPAR gamma Proteins 0.000 claims description 4
- 208000017442 Retinal disease Diseases 0.000 claims description 4
- 206010038923 Retinopathy Diseases 0.000 claims description 4
- 125000003342 alkenyl group Chemical group 0.000 claims description 4
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 claims description 4
- 125000006260 ethylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])C([H])([H])[H] 0.000 claims description 4
- 125000001153 fluoro group Chemical group F* 0.000 claims description 4
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 claims description 4
- 208000017169 kidney disease Diseases 0.000 claims description 4
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 claims description 4
- 125000004458 methylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])[H] 0.000 claims description 4
- 201000001119 neuropathy Diseases 0.000 claims description 4
- 230000007823 neuropathy Effects 0.000 claims description 4
- 125000004043 oxo group Chemical group O=* 0.000 claims description 4
- 208000033808 peripheral neuropathy Diseases 0.000 claims description 4
- 229940044601 receptor agonist Drugs 0.000 claims description 4
- 239000000018 receptor agonist Substances 0.000 claims description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 4
- 208000004611 Abdominal Obesity Diseases 0.000 claims description 3
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 claims description 3
- 201000001320 Atherosclerosis Diseases 0.000 claims description 3
- 206010065941 Central obesity Diseases 0.000 claims description 3
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 3
- 208000011231 Crohn disease Diseases 0.000 claims description 3
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 3
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 claims description 3
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 3
- 239000004472 Lysine Substances 0.000 claims description 3
- 206010033645 Pancreatitis Diseases 0.000 claims description 3
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 3
- 239000003888 alpha glucosidase inhibitor Substances 0.000 claims description 3
- 239000003098 androgen Substances 0.000 claims description 3
- 239000003963 antioxidant agent Substances 0.000 claims description 3
- 230000009977 dual effect Effects 0.000 claims description 3
- 108010036598 gastric inhibitory polypeptide receptor Proteins 0.000 claims description 3
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 3
- 230000004770 neurodegeneration Effects 0.000 claims description 3
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 3
- 230000002611 ovarian Effects 0.000 claims description 3
- 201000010065 polycystic ovary syndrome Diseases 0.000 claims description 3
- 208000037803 restenosis Diseases 0.000 claims description 3
- 208000011580 syndromic disease Diseases 0.000 claims description 3
- 230000002792 vascular Effects 0.000 claims description 3
- MVQVNTPHUGQQHK-UHFFFAOYSA-N 3-pyridinemethanol Chemical compound OCC1=CC=CN=C1 MVQVNTPHUGQQHK-UHFFFAOYSA-N 0.000 claims description 2
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 claims description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 2
- 229940124213 Dipeptidyl peptidase 4 (DPP IV) inhibitor Drugs 0.000 claims description 2
- 229940122904 Glucagon receptor antagonist Drugs 0.000 claims description 2
- 229940089838 Glucagon-like peptide 1 receptor agonist Drugs 0.000 claims description 2
- 102000014743 Pituitary Adenylate Cyclase-Activating Polypeptide Receptors Human genes 0.000 claims description 2
- 108010064032 Pituitary Adenylate Cyclase-Activating Polypeptide Receptors Proteins 0.000 claims description 2
- 102000001494 Sterol O-Acyltransferase Human genes 0.000 claims description 2
- 108010054082 Sterol O-acyltransferase Proteins 0.000 claims description 2
- 239000002404 acyltransferase inhibitor Substances 0.000 claims description 2
- 239000003529 anticholesteremic agent Substances 0.000 claims description 2
- 229940127226 anticholesterol agent Drugs 0.000 claims description 2
- 239000002220 antihypertensive agent Substances 0.000 claims description 2
- 229940030600 antihypertensive agent Drugs 0.000 claims description 2
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 claims description 2
- 125000001246 bromo group Chemical group Br* 0.000 claims description 2
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 2
- 230000001906 cholesterol absorption Effects 0.000 claims description 2
- 239000003603 dipeptidyl peptidase IV inhibitor Substances 0.000 claims description 2
- 229940121380 ileal bile acid transporter inhibitor Drugs 0.000 claims description 2
- 229960003512 nicotinic acid Drugs 0.000 claims description 2
- 235000001968 nicotinic acid Nutrition 0.000 claims description 2
- 239000011664 nicotinic acid Substances 0.000 claims description 2
- 229960004738 nicotinyl alcohol Drugs 0.000 claims description 2
- 239000003801 protein tyrosine phosphatase 1B inhibitor Substances 0.000 claims description 2
- 239000003352 sequestering agent Substances 0.000 claims description 2
- YROXIXLRRCOBKF-UHFFFAOYSA-N sulfonylurea Chemical class OC(=N)N=S(=O)=O YROXIXLRRCOBKF-UHFFFAOYSA-N 0.000 claims description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 36
- 229940125542 dual agonist Drugs 0.000 claims 3
- 102100039994 Gastric inhibitory polypeptide Human genes 0.000 claims 2
- 230000001419 dependent effect Effects 0.000 claims 2
- 230000003278 mimic effect Effects 0.000 claims 2
- 206010061218 Inflammation Diseases 0.000 claims 1
- 229940122355 Insulin sensitizer Drugs 0.000 claims 1
- 102000000536 PPAR gamma Human genes 0.000 claims 1
- 229940122054 Peroxisome proliferator-activated receptor delta agonist Drugs 0.000 claims 1
- 229940121363 anti-inflammatory agent Drugs 0.000 claims 1
- 239000002260 anti-inflammatory agent Substances 0.000 claims 1
- 239000003877 glucagon like peptide 1 receptor agonist Substances 0.000 claims 1
- 230000004054 inflammatory process Effects 0.000 claims 1
- 230000001568 sexual effect Effects 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 84
- 230000002265 prevention Effects 0.000 abstract description 11
- 230000006806 disease prevention Effects 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 172
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 84
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 76
- 239000000243 solution Substances 0.000 description 70
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical group C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 50
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 45
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 43
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 43
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 40
- 238000006243 chemical reaction Methods 0.000 description 40
- 239000000047 product Substances 0.000 description 37
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 35
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 30
- 239000000543 intermediate Substances 0.000 description 30
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 29
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 28
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 27
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 26
- 229920006395 saturated elastomer Polymers 0.000 description 25
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 25
- 239000003814 drug Substances 0.000 description 24
- 229940079593 drug Drugs 0.000 description 23
- 238000003818 flash chromatography Methods 0.000 description 21
- 239000007787 solid Substances 0.000 description 21
- 239000000741 silica gel Substances 0.000 description 20
- 229910002027 silica gel Inorganic materials 0.000 description 20
- 235000017557 sodium bicarbonate Nutrition 0.000 description 20
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- 239000012267 brine Substances 0.000 description 19
- 239000003921 oil Substances 0.000 description 19
- 235000019198 oils Nutrition 0.000 description 19
- 239000012074 organic phase Substances 0.000 description 19
- 238000000746 purification Methods 0.000 description 19
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 18
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 17
- 150000001721 carbon Chemical group 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 16
- 238000007792 addition Methods 0.000 description 16
- 239000011541 reaction mixture Substances 0.000 description 16
- 239000002253 acid Substances 0.000 description 15
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 15
- 235000019341 magnesium sulphate Nutrition 0.000 description 15
- 238000003756 stirring Methods 0.000 description 13
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 150000002148 esters Chemical class 0.000 description 12
- 229910052938 sodium sulfate Inorganic materials 0.000 description 12
- 235000011152 sodium sulphate Nutrition 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 11
- 239000002904 solvent Substances 0.000 description 10
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 102000004190 Enzymes Human genes 0.000 description 9
- 108090000790 Enzymes Proteins 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000008346 aqueous phase Substances 0.000 description 9
- 239000012044 organic layer Substances 0.000 description 9
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 8
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 description 8
- 239000000095 Growth Hormone-Releasing Hormone Substances 0.000 description 8
- 101710142969 Somatoliberin Proteins 0.000 description 8
- 102100022831 Somatoliberin Human genes 0.000 description 8
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 239000013058 crude material Substances 0.000 description 7
- 238000009472 formulation Methods 0.000 description 7
- 239000000546 pharmaceutical excipient Substances 0.000 description 7
- 108090000765 processed proteins & peptides Proteins 0.000 description 7
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 6
- 241000282326 Felis catus Species 0.000 description 6
- 101800000221 Glucagon-like peptide 2 Proteins 0.000 description 6
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- 238000005859 coupling reaction Methods 0.000 description 6
- 235000014113 dietary fatty acids Nutrition 0.000 description 6
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 6
- 239000000194 fatty acid Substances 0.000 description 6
- 229930195729 fatty acid Natural products 0.000 description 6
- 150000004665 fatty acids Chemical class 0.000 description 6
- TWSALRJGPBVBQU-PKQQPRCHSA-N glucagon-like peptide 2 Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(O)=O)[C@@H](C)CC)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)CC)C1=CC=CC=C1 TWSALRJGPBVBQU-PKQQPRCHSA-N 0.000 description 6
- 239000000859 incretin Substances 0.000 description 6
- 239000004615 ingredient Substances 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 231100000252 nontoxic Toxicity 0.000 description 6
- 230000003000 nontoxic effect Effects 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 239000003765 sweetening agent Substances 0.000 description 6
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- 0 C*(C*CN(C(*)C1*)C(C(*)C(*)[Al])=O)*C1I(C)* Chemical compound C*(C*CN(C(*)C1*)C(C(*)C(*)[Al])=O)*C1I(C)* 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- 101000908391 Homo sapiens Dipeptidyl peptidase 4 Proteins 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 description 5
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 150000001540 azides Chemical class 0.000 description 5
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 5
- 239000007859 condensation product Substances 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 235000012631 food intake Nutrition 0.000 description 5
- 235000003599 food sweetener Nutrition 0.000 description 5
- 238000006460 hydrolysis reaction Methods 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 150000002576 ketones Chemical class 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- SWLAMJPTOQZTAE-UHFFFAOYSA-N 4-[2-[(5-chloro-2-methoxybenzoyl)amino]ethyl]benzoic acid Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(C(O)=O)C=C1 SWLAMJPTOQZTAE-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 102000019034 Chemokines Human genes 0.000 description 4
- 108010012236 Chemokines Proteins 0.000 description 4
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 4
- 208000013016 Hypoglycemia Diseases 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- 102000002072 Non-Receptor Type 1 Protein Tyrosine Phosphatase Human genes 0.000 description 4
- 108010015847 Non-Receptor Type 1 Protein Tyrosine Phosphatase Proteins 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 235000019270 ammonium chloride Nutrition 0.000 description 4
- 239000007900 aqueous suspension Substances 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 238000010494 dissociation reaction Methods 0.000 description 4
- 230000005593 dissociations Effects 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 239000012065 filter cake Substances 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- 230000037406 food intake Effects 0.000 description 4
- 230000011132 hemopoiesis Effects 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- MGXWVYUBJRZYPE-YUGYIWNOSA-N incretin Chemical class C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)[C@@H](C)O)[C@@H](C)CC)C1=CC=C(O)C=C1 MGXWVYUBJRZYPE-YUGYIWNOSA-N 0.000 description 4
- 229950004994 meglitinide Drugs 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 238000012746 preparative thin layer chromatography Methods 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 239000002002 slurry Substances 0.000 description 4
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- LENYOXXELREKGZ-WCCKRBBISA-N (3s)-3-fluoropyrrolidin-1-ium;chloride Chemical compound Cl.F[C@H]1CCNC1 LENYOXXELREKGZ-WCCKRBBISA-N 0.000 description 3
- AEMWUHCKKDPRSK-UHFFFAOYSA-N (ne)-n-diazo-2,4,6-tri(propan-2-yl)benzenesulfonamide Chemical compound CC(C)C1=CC(C(C)C)=C(S(=O)(=O)N=[N+]=[N-])C(C(C)C)=C1 AEMWUHCKKDPRSK-UHFFFAOYSA-N 0.000 description 3
- UTDVHCQTKWTQEA-UHFFFAOYSA-N 1-(2-aminoacetyl)-n-(4-methyl-2-oxochromen-7-yl)pyrrolidine-2-carboxamide Chemical compound C1=CC=2C(C)=CC(=O)OC=2C=C1NC(=O)C1CCCN1C(=O)CN UTDVHCQTKWTQEA-UHFFFAOYSA-N 0.000 description 3
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 3
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical compound O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 3
- 244000215068 Acacia senegal Species 0.000 description 3
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 3
- 241000283690 Bos taurus Species 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 102000006573 Chemokine CXCL12 Human genes 0.000 description 3
- 108010008951 Chemokine CXCL12 Proteins 0.000 description 3
- 241000283086 Equidae Species 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 229920000084 Gum arabic Polymers 0.000 description 3
- 206010020772 Hypertension Diseases 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 3
- 208000001132 Osteoporosis Diseases 0.000 description 3
- 241001494479 Pecora Species 0.000 description 3
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 3
- 230000006044 T cell activation Effects 0.000 description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 3
- 235000010489 acacia gum Nutrition 0.000 description 3
- 239000000205 acacia gum Substances 0.000 description 3
- 229960000583 acetic acid Drugs 0.000 description 3
- 210000000577 adipose tissue Anatomy 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 235000013305 food Nutrition 0.000 description 3
- 208000007565 gingivitis Diseases 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 230000002218 hypoglycaemic effect Effects 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 229940057995 liquid paraffin Drugs 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000009401 metastasis Effects 0.000 description 3
- 229960003105 metformin Drugs 0.000 description 3
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 210000003205 muscle Anatomy 0.000 description 3
- 230000001537 neural effect Effects 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 210000000496 pancreas Anatomy 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 229960003243 phenformin Drugs 0.000 description 3
- ICFJFFQQTFMIBG-UHFFFAOYSA-N phenformin Chemical compound NC(=N)NC(=N)NCCC1=CC=CC=C1 ICFJFFQQTFMIBG-UHFFFAOYSA-N 0.000 description 3
- DHHVAGZRUROJKS-UHFFFAOYSA-N phentermine Chemical compound CC(C)(N)CC1=CC=CC=C1 DHHVAGZRUROJKS-UHFFFAOYSA-N 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 230000019100 sperm motility Effects 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 2
- OHVNGWREPWMVFE-BPNCWPANSA-N (2s)-1-[(2s,3s)-2-amino-3-(4-fluorophenyl)butanoyl]pyrrolidine-2-carbonitrile Chemical compound O=C([C@@H](N)[C@@H](C)C=1C=CC(F)=CC=1)N1CCC[C@H]1C#N OHVNGWREPWMVFE-BPNCWPANSA-N 0.000 description 2
- ZVZRWSMWJQAIPJ-CABZTGNLSA-N (2s,3s)-2-amino-3-(4-fluorophenyl)-1-(1,3-thiazolidin-3-yl)butan-1-one Chemical compound O=C([C@@H](N)[C@@H](C)C=1C=CC(F)=CC=1)N1CCSC1 ZVZRWSMWJQAIPJ-CABZTGNLSA-N 0.000 description 2
- KLLUAXNPIZSSBO-XDTLVQLUSA-N (2s,3s)-2-amino-3-(4-fluorophenyl)-1-[(3s)-3-fluoropyrrolidin-1-yl]butan-1-one Chemical compound O=C([C@@H](N)[C@@H](C)C=1C=CC(F)=CC=1)N1CC[C@H](F)C1 KLLUAXNPIZSSBO-XDTLVQLUSA-N 0.000 description 2
- WQADWIOXOXRPLN-UHFFFAOYSA-N 1,3-dithiane Chemical compound C1CSCSC1 WQADWIOXOXRPLN-UHFFFAOYSA-N 0.000 description 2
- LOZWAPSEEHRYPG-UHFFFAOYSA-N 1,4-dithiane Chemical compound C1CSCCS1 LOZWAPSEEHRYPG-UHFFFAOYSA-N 0.000 description 2
- PLRACCBDVIHHLZ-UHFFFAOYSA-N 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Chemical compound C1N(C)CCC(C=2C=CC=CC=2)=C1 PLRACCBDVIHHLZ-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- VRPJIFMKZZEXLR-UHFFFAOYSA-N 2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid Chemical compound CC(C)(C)OC(=O)NCC(O)=O VRPJIFMKZZEXLR-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- APOYTRAZFJURPB-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)-n-(trifluoro-$l^{4}-sulfanyl)ethanamine Chemical compound COCCN(S(F)(F)F)CCOC APOYTRAZFJURPB-UHFFFAOYSA-N 0.000 description 2
- YYVPZQADFREIFR-UHFFFAOYSA-N 3,3-difluoropyrrolidine;hydrochloride Chemical compound [Cl-].FC1(F)CC[NH2+]C1 YYVPZQADFREIFR-UHFFFAOYSA-N 0.000 description 2
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 2
- NKOHRVBBQISBSB-UHFFFAOYSA-N 5-[(4-hydroxyphenyl)methyl]-1,3-thiazolidine-2,4-dione Chemical compound C1=CC(O)=CC=C1CC1C(=O)NC(=O)S1 NKOHRVBBQISBSB-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 235000003911 Arachis Nutrition 0.000 description 2
- 244000105624 Arachis hypogaea Species 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 2
- 102100032367 C-C motif chemokine 5 Human genes 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 108010055166 Chemokine CCL5 Proteins 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 229920002785 Croscarmellose sodium Polymers 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- HEMJJKBWTPKOJG-UHFFFAOYSA-N Gemfibrozil Chemical compound CC1=CC=C(C)C(OCCCC(C)(C)C(O)=O)=C1 HEMJJKBWTPKOJG-UHFFFAOYSA-N 0.000 description 2
- 102000051325 Glucagon Human genes 0.000 description 2
- 108060003199 Glucagon Proteins 0.000 description 2
- 108010086246 Glucagon-Like Peptide-1 Receptor Proteins 0.000 description 2
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 2
- 102100032882 Glucagon-like peptide 1 receptor Human genes 0.000 description 2
- 239000007818 Grignard reagent Substances 0.000 description 2
- 206010056438 Growth hormone deficiency Diseases 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- 208000031886 HIV Infections Diseases 0.000 description 2
- 208000037357 HIV infectious disease Diseases 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- 101001135571 Mus musculus Tyrosine-protein phosphatase non-receptor type 2 Proteins 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 206010070834 Sensitisation Diseases 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 206010064390 Tumour invasion Diseases 0.000 description 2
- 229960002632 acarbose Drugs 0.000 description 2
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 150000004703 alkoxides Chemical class 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 2
- 235000008206 alpha-amino acids Nutrition 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 150000007514 bases Chemical class 0.000 description 2
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- MBLJFGOKYTZKMH-LLVKDONJSA-N benzyl (3r)-3-hydroxypyrrolidine-1-carboxylate Chemical compound C1[C@H](O)CCN1C(=O)OCC1=CC=CC=C1 MBLJFGOKYTZKMH-LLVKDONJSA-N 0.000 description 2
- MBLJFGOKYTZKMH-NSHDSACASA-N benzyl (3s)-3-hydroxypyrrolidine-1-carboxylate Chemical compound C1[C@@H](O)CCN1C(=O)OCC1=CC=CC=C1 MBLJFGOKYTZKMH-NSHDSACASA-N 0.000 description 2
- XZMBERXRQLASNY-UHFFFAOYSA-N benzyl 3,3-difluoropyrrolidine-1-carboxylate Chemical compound C1C(F)(F)CCN1C(=O)OCC1=CC=CC=C1 XZMBERXRQLASNY-UHFFFAOYSA-N 0.000 description 2
- 229960000516 bezafibrate Drugs 0.000 description 2
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 230000009400 cancer invasion Effects 0.000 description 2
- ZDQWVKDDJDIVAL-UHFFFAOYSA-N catecholborane Chemical compound C1=CC=C2O[B]OC2=C1 ZDQWVKDDJDIVAL-UHFFFAOYSA-N 0.000 description 2
- 239000000460 chlorine Chemical group 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 229960001214 clofibrate Drugs 0.000 description 2
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- UWCHSDIUMBNDLT-UHFFFAOYSA-L copper;methylsulfanylmethane;dibromide Chemical compound CSC.Br[Cu]Br UWCHSDIUMBNDLT-UHFFFAOYSA-L 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 108010015198 endomorphin 2 Proteins 0.000 description 2
- XIJHWXXXIMEHKW-LJWNLINESA-N endomorphin-2 Chemical compound C([C@H](N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)C1=CC=C(O)C=C1 XIJHWXXXIMEHKW-LJWNLINESA-N 0.000 description 2
- 239000002024 ethyl acetate extract Substances 0.000 description 2
- 229960002297 fenofibrate Drugs 0.000 description 2
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 2
- MQOBSOSZFYZQOK-UHFFFAOYSA-N fenofibric acid Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1C(=O)C1=CC=C(Cl)C=C1 MQOBSOSZFYZQOK-UHFFFAOYSA-N 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 230000030136 gastric emptying Effects 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 229960003627 gemfibrozil Drugs 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 2
- 229960001381 glipizide Drugs 0.000 description 2
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 2
- 229960004666 glucagon Drugs 0.000 description 2
- 230000014101 glucose homeostasis Effects 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 2
- 206010020718 hyperplasia Diseases 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 230000002779 inactivation Effects 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000000185 intracerebroventricular administration Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 230000037356 lipid metabolism Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- MLTFKRDFAJSIEX-UHFFFAOYSA-N lithium;1,3-oxazolidin-2-one Chemical compound [Li].O=C1NCCO1 MLTFKRDFAJSIEX-UHFFFAOYSA-N 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 210000005228 liver tissue Anatomy 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 2
- 229940049920 malate Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 239000002808 molecular sieve Substances 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 230000000324 neuroprotective effect Effects 0.000 description 2
- 208000004235 neutropenia Diseases 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 2
- 238000005897 peptide coupling reaction Methods 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 230000004043 responsiveness Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 230000008313 sensitization Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 235000010265 sodium sulphite Nutrition 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- 125000006633 tert-butoxycarbonylamino group Chemical group 0.000 description 2
- QCCUDVIZRVMXBM-TUSQITKMSA-N tert-butyl n-[(2s,3s)-4-(dimethylamino)-1-[(3s)-3-fluoropyrrolidin-1-yl]-3-[4-[(4-methoxybenzoyl)-methylamino]phenyl]-1,4-dioxobutan-2-yl]carbamate Chemical compound C1=CC(OC)=CC=C1C(=O)N(C)C1=CC=C([C@@H]([C@H](NC(=O)OC(C)(C)C)C(=O)N2C[C@@H](F)CC2)C(=O)N(C)C)C=C1 QCCUDVIZRVMXBM-TUSQITKMSA-N 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 229960005371 tolbutamide Drugs 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- GXPHKUHSUJUWKP-UHFFFAOYSA-N troglitazone Chemical compound C1CC=2C(C)=C(O)C(C)=C(C)C=2OC1(C)COC(C=C1)=CC=C1CC1SC(=O)NC1=O GXPHKUHSUJUWKP-UHFFFAOYSA-N 0.000 description 2
- 229960001641 troglitazone Drugs 0.000 description 2
- GXPHKUHSUJUWKP-NTKDMRAZSA-N troglitazone Natural products C([C@@]1(OC=2C(C)=C(C(=C(C)C=2CC1)O)C)C)OC(C=C1)=CC=C1C[C@H]1SC(=O)NC1=O GXPHKUHSUJUWKP-NTKDMRAZSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- DBGIVFWFUFKIQN-VIFPVBQESA-N (+)-Fenfluramine Chemical compound CCN[C@@H](C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-VIFPVBQESA-N 0.000 description 1
- DBGIVFWFUFKIQN-UHFFFAOYSA-N (+-)-Fenfluramine Chemical compound CCNC(C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-UHFFFAOYSA-N 0.000 description 1
- QDZOEBFLNHCSSF-PFFBOGFISA-N (2S)-2-[[(2R)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-1-[(2R)-2-amino-5-carbamimidamidopentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-N-[(2R)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]pentanediamide Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CCCNC(N)=N)C1=CC=CC=C1 QDZOEBFLNHCSSF-PFFBOGFISA-N 0.000 description 1
- JNTMAZFVYNDPLB-PEDHHIEDSA-N (2S,3S)-2-[[[(2S)-1-[(2S,3S)-2-amino-3-methyl-1-oxopentyl]-2-pyrrolidinyl]-oxomethyl]amino]-3-methylpentanoic acid Chemical compound CC[C@H](C)[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(O)=O JNTMAZFVYNDPLB-PEDHHIEDSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- ZHNBEQRHEJAAIE-XDTLVQLUSA-N (2s,3s)-2-amino-3-(4-bromophenyl)-1-[(3s)-3-fluoropyrrolidin-1-yl]butan-1-one Chemical compound O=C([C@@H](N)[C@@H](C)C=1C=CC(Br)=CC=1)N1CC[C@H](F)C1 ZHNBEQRHEJAAIE-XDTLVQLUSA-N 0.000 description 1
- XXMXYMMFWJVZLC-CABZTGNLSA-N (2s,3s)-3-(4-fluorophenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]butanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)[C@@H](C)C1=CC=C(F)C=C1 XXMXYMMFWJVZLC-CABZTGNLSA-N 0.000 description 1
- TWNOWKXKGBUSMW-YYWHXJBOSA-N (2s,3s)-3-amino-2-[4-(4-cyanophenoxy)phenyl]-4-[(3s)-3-fluoropyrrolidin-1-yl]-n,n-dimethyl-4-oxobutanamide Chemical compound O=C([C@@H](N)[C@@H](C(=O)N(C)C)C=1C=CC(OC=2C=CC(=CC=2)C#N)=CC=1)N1CC[C@H](F)C1 TWNOWKXKGBUSMW-YYWHXJBOSA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- LENYOXXELREKGZ-PGMHMLKASA-N (3r)-3-fluoropyrrolidine;hydrochloride Chemical compound Cl.F[C@@H]1CCNC1 LENYOXXELREKGZ-PGMHMLKASA-N 0.000 description 1
- JHHZLHWJQPUNKB-SCSAIBSYSA-N (3r)-pyrrolidin-3-ol Chemical compound O[C@@H]1CCNC1 JHHZLHWJQPUNKB-SCSAIBSYSA-N 0.000 description 1
- FIPZWVLCZIYEMW-UHFFFAOYSA-N (4-cyanophenoxy)boronic acid Chemical compound OB(O)OC1=CC=C(C#N)C=C1 FIPZWVLCZIYEMW-UHFFFAOYSA-N 0.000 description 1
- PRYJZYXVMVOEEG-AFAVFJNCSA-N (4r)-3-[(2r,3s)-2-bromo-3-(4-bromophenyl)butanoyl]-4-phenyl-1,3-oxazolidin-2-one Chemical compound C1([C@H]2N(C(OC2)=O)C(=O)[C@H](Br)[C@@H](C)C=2C=CC(Br)=CC=2)=CC=CC=C1 PRYJZYXVMVOEEG-AFAVFJNCSA-N 0.000 description 1
- PXOVZNUACNXJJU-DYVFJYSZSA-N (4r)-3-[(3r)-3-(4-bromophenyl)butanoyl]-4-phenyl-1,3-oxazolidin-2-one Chemical compound C1([C@H]2N(C(OC2)=O)C(=O)C[C@@H](C)C=2C=CC(Br)=CC=2)=CC=CC=C1 PXOVZNUACNXJJU-DYVFJYSZSA-N 0.000 description 1
- QDMNNMIOWVJVLY-QMMMGPOBSA-N (4r)-4-phenyl-1,3-oxazolidin-2-one Chemical compound C1OC(=O)N[C@@H]1C1=CC=CC=C1 QDMNNMIOWVJVLY-QMMMGPOBSA-N 0.000 description 1
- DCVIMWRFFDRWJM-SZHMKSFOSA-N (4s)-3-[(e)-but-2-enoyl]-4-phenyl-1,3-oxazolidin-2-one Chemical compound C1OC(=O)N(C(=O)/C=C/C)[C@H]1C1=CC=CC=C1 DCVIMWRFFDRWJM-SZHMKSFOSA-N 0.000 description 1
- QDMNNMIOWVJVLY-MRVPVSSYSA-N (4s)-4-phenyl-1,3-oxazolidin-2-one Chemical compound C1OC(=O)N[C@H]1C1=CC=CC=C1 QDMNNMIOWVJVLY-MRVPVSSYSA-N 0.000 description 1
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 description 1
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 description 1
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 1
- ZQNVTVOTCKZKHJ-NSCUHMNNSA-N (e)-4-(4-bromophenyl)but-3-en-2-one Chemical compound CC(=O)\C=C\C1=CC=C(Br)C=C1 ZQNVTVOTCKZKHJ-NSCUHMNNSA-N 0.000 description 1
- QVAMPRCBAQIQBI-SGJFDWMWSA-N (e,2s)-4-(4-bromophenyl)but-3-en-2-ol Chemical compound C[C@H](O)\C=C\C1=CC=C(Br)C=C1 QVAMPRCBAQIQBI-SGJFDWMWSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- ZMMRLWFGWQVVJD-UHFFFAOYSA-O (n,n-dimethylcarbamimidoyl)-dimethylazanium;azide Chemical compound [N-]=[N+]=[N-].CN(C)C(N)=[N+](C)C ZMMRLWFGWQVVJD-UHFFFAOYSA-O 0.000 description 1
- VMKAFJQFKBASMU-QGZVFWFLSA-N (r)-2-methyl-cbs-oxazaborolidine Chemical compound C([C@@H]12)CCN1B(C)OC2(C=1C=CC=CC=1)C1=CC=CC=C1 VMKAFJQFKBASMU-QGZVFWFLSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- VDFVNEFVBPFDSB-UHFFFAOYSA-N 1,3-dioxane Chemical compound C1COCOC1 VDFVNEFVBPFDSB-UHFFFAOYSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- IMLSAISZLJGWPP-UHFFFAOYSA-N 1,3-dithiolane Chemical compound C1CSCS1 IMLSAISZLJGWPP-UHFFFAOYSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- ZOBPZXTWZATXDG-UHFFFAOYSA-N 1,3-thiazolidine-2,4-dione Chemical compound O=C1CSC(=O)N1 ZOBPZXTWZATXDG-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- ZYKWABRWFBVPIJ-UHFFFAOYSA-N 1-benzhydryl-3-fluoroazetidine Chemical compound C1C(F)CN1C(C=1C=CC=CC=1)C1=CC=CC=C1 ZYKWABRWFBVPIJ-UHFFFAOYSA-N 0.000 description 1
- MMAJXKGUZYDTHV-UHFFFAOYSA-N 1-benzhydrylazetidin-3-ol Chemical compound C1C(O)CN1C(C=1C=CC=CC=1)C1=CC=CC=C1 MMAJXKGUZYDTHV-UHFFFAOYSA-N 0.000 description 1
- ZQXCQTAELHSNAT-UHFFFAOYSA-N 1-chloro-3-nitro-5-(trifluoromethyl)benzene Chemical compound [O-][N+](=O)C1=CC(Cl)=CC(C(F)(F)F)=C1 ZQXCQTAELHSNAT-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- JKTCBAGSMQIFNL-UHFFFAOYSA-N 2,3-dihydrofuran Chemical compound C1CC=CO1 JKTCBAGSMQIFNL-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- RGWUPQHXIQNHFN-UHFFFAOYSA-N 3-(4-fluorophenyl)butanoic acid Chemical compound OC(=O)CC(C)C1=CC=C(F)C=C1 RGWUPQHXIQNHFN-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- KRVWLMWHONJSCH-UHFFFAOYSA-N 3-fluoroazetidine;2,2,2-trifluoroacetic acid Chemical compound FC1CNC1.OC(=O)C(F)(F)F KRVWLMWHONJSCH-UHFFFAOYSA-N 0.000 description 1
- HRASFKASEOUOPD-UHFFFAOYSA-N 3-oxopyrrolidine-1-carboxylic acid Chemical compound OC(=O)N1CCC(=O)C1 HRASFKASEOUOPD-UHFFFAOYSA-N 0.000 description 1
- NCTCGHLIHJJIBK-UHFFFAOYSA-N 3-phenyl-1,3-oxazolidin-2-one Chemical compound O=C1OCCN1C1=CC=CC=C1 NCTCGHLIHJJIBK-UHFFFAOYSA-N 0.000 description 1
- CFOOTBBXHJHHMT-UHFFFAOYSA-N 4,4-diphenyl-1-propan-2-ylpiperidine Chemical compound C1CN(C(C)C)CCC1(C=1C=CC=CC=1)C1=CC=CC=C1 CFOOTBBXHJHHMT-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- IXENWFQXVCOHAZ-UHFFFAOYSA-N 4-fluoropiperidine;hydrochloride Chemical compound Cl.FC1CCNCC1 IXENWFQXVCOHAZ-UHFFFAOYSA-N 0.000 description 1
- SIOAPROKUUGJAQ-UHFFFAOYSA-N 4-methoxy-n-methylbenzamide Chemical compound CNC(=O)C1=CC=C(OC)C=C1 SIOAPROKUUGJAQ-UHFFFAOYSA-N 0.000 description 1
- MVDXXGIBARMXSA-PYUWXLGESA-N 5-[[(2r)-2-benzyl-3,4-dihydro-2h-chromen-6-yl]methyl]-1,3-thiazolidine-2,4-dione Chemical compound S1C(=O)NC(=O)C1CC1=CC=C(O[C@@H](CC=2C=CC=CC=2)CC2)C2=C1 MVDXXGIBARMXSA-PYUWXLGESA-N 0.000 description 1
- STDKZZIKAJFATG-UHFFFAOYSA-N 5-benzyl-1,3-thiazolidine-2,4-dione Chemical compound S1C(=O)NC(=O)C1CC1=CC=CC=C1 STDKZZIKAJFATG-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 208000011732 Abnormal glucose homeostasis Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-M Aminoacetate Chemical compound NCC([O-])=O DHMQDGOQFOQNFH-UHFFFAOYSA-M 0.000 description 1
- 102000004400 Aminopeptidases Human genes 0.000 description 1
- 108090000915 Aminopeptidases Proteins 0.000 description 1
- 102000007592 Apolipoproteins Human genes 0.000 description 1
- 108010071619 Apolipoproteins Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 208000023328 Basedow disease Diseases 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-N Betaine Natural products C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000002080 C09CA02 - Eprosartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- 229940124802 CB1 antagonist Drugs 0.000 description 1
- 108090000312 Calcium Channels Proteins 0.000 description 1
- 102000003922 Calcium Channels Human genes 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 206010070901 Diabetic dyslipidaemia Diseases 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- 108010067722 Dipeptidyl Peptidase 4 Proteins 0.000 description 1
- 102100031107 Disintegrin and metalloproteinase domain-containing protein 11 Human genes 0.000 description 1
- 101710121366 Disintegrin and metalloproteinase domain-containing protein 11 Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 208000004232 Enteritis Diseases 0.000 description 1
- 102100023688 Eotaxin Human genes 0.000 description 1
- 101710139422 Eotaxin Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- LUTBBSKDGQJPJM-UHFFFAOYSA-N FC1=CCNC1 Chemical compound FC1=CCNC1 LUTBBSKDGQJPJM-UHFFFAOYSA-N 0.000 description 1
- PSCXEUSWZWRCMQ-UHFFFAOYSA-N F[S](F)F Chemical compound F[S](F)F PSCXEUSWZWRCMQ-UHFFFAOYSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 101000825742 Homo sapiens Somatoliberin Proteins 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- 108010001127 Insulin Receptor Proteins 0.000 description 1
- 102000003746 Insulin Receptor Human genes 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- VLJNHYLEOZPXFW-BYPYZUCNSA-N L-prolinamide Chemical compound NC(=O)[C@@H]1CCCN1 VLJNHYLEOZPXFW-BYPYZUCNSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- IBAQFPQHRJAVAV-ULAWRXDQSA-N Miglitol Chemical compound OCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO IBAQFPQHRJAVAV-ULAWRXDQSA-N 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-O N,N,N-trimethylglycinium Chemical compound C[N+](C)(C)CC(O)=O KWIUHFFTVRNATP-UHFFFAOYSA-O 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 102000003797 Neuropeptides Human genes 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108010015181 PPAR delta Proteins 0.000 description 1
- 108010050095 PT-100 dipeptide Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 208000018262 Peripheral vascular disease Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 238000006619 Stille reaction Methods 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 102400000096 Substance P Human genes 0.000 description 1
- 101800003906 Substance P Proteins 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 208000029052 T-cell acute lymphoblastic leukemia Diseases 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- FPPMQVOJFIYTFU-RCOVLWMOSA-N [(1s,2s)-1-carboxy-2-(4-fluorophenyl)propyl]-diazonioazanide Chemical compound [N-]=[N+]=N[C@H](C(O)=O)[C@@H](C)C1=CC=C(F)C=C1 FPPMQVOJFIYTFU-RCOVLWMOSA-N 0.000 description 1
- SVUIMOOZXQQLJS-RCOVLWMOSA-N [(1s,2s)-2-(4-bromophenyl)-1-carboxypropyl]-diazonioazanide Chemical compound [N-]=[N+]=N[C@H](C(O)=O)[C@@H](C)C1=CC=C(Br)C=C1 SVUIMOOZXQQLJS-RCOVLWMOSA-N 0.000 description 1
- FKCMADOPPWWGNZ-YUMQZZPRSA-N [(2r)-1-[(2s)-2-amino-3-methylbutanoyl]pyrrolidin-2-yl]boronic acid Chemical compound CC(C)[C@H](N)C(=O)N1CCC[C@H]1B(O)O FKCMADOPPWWGNZ-YUMQZZPRSA-N 0.000 description 1
- WERKSKAQRVDLDW-ANOHMWSOSA-N [(2s,3r,4r,5r)-2,3,4,5,6-pentahydroxyhexyl] (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO WERKSKAQRVDLDW-ANOHMWSOSA-N 0.000 description 1
- 201000010390 abdominal obesity-metabolic syndrome 1 Diseases 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- VJHCJDRQFCCTHL-UHFFFAOYSA-N acetic acid 2,3,4,5,6-pentahydroxyhexanal Chemical compound CC(O)=O.OCC(O)C(O)C(O)C(O)C=O VJHCJDRQFCCTHL-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000008484 agonism Effects 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000036592 analgesia Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 210000004198 anterior pituitary gland Anatomy 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 150000001503 aryl iodides Chemical class 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- OISFUZRUIGGTSD-LJTMIZJLSA-N azane;(2r,3r,4r,5s)-6-(methylamino)hexane-1,2,3,4,5-pentol Chemical compound N.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO OISFUZRUIGGTSD-LJTMIZJLSA-N 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- DGQYMNWQXVFIBE-LLVKDONJSA-N benzyl (3r)-3-fluoropyrrolidine-1-carboxylate Chemical compound C1[C@H](F)CCN1C(=O)OCC1=CC=CC=C1 DGQYMNWQXVFIBE-LLVKDONJSA-N 0.000 description 1
- RQFCHCFLVLLDPD-ZDUSSCGKSA-N benzyl (3s)-3-acetyloxypyrrolidine-1-carboxylate Chemical compound C1[C@@H](OC(=O)C)CCN1C(=O)OCC1=CC=CC=C1 RQFCHCFLVLLDPD-ZDUSSCGKSA-N 0.000 description 1
- MBLJFGOKYTZKMH-UHFFFAOYSA-N benzyl 3-hydroxypyrrolidine-1-carboxylate Chemical compound C1C(O)CCN1C(=O)OCC1=CC=CC=C1 MBLJFGOKYTZKMH-UHFFFAOYSA-N 0.000 description 1
- LMHWEUQNJRXMCD-UHFFFAOYSA-N benzyl 3-oxopyrrolidine-1-carboxylate Chemical compound C1CC(=O)CN1C(=O)OCC1=CC=CC=C1 LMHWEUQNJRXMCD-UHFFFAOYSA-N 0.000 description 1
- ZYMOCSXYVNFNNT-UHFFFAOYSA-N benzyl 4-fluoropiperidine-1-carboxylate Chemical compound C1CC(F)CCN1C(=O)OCC1=CC=CC=C1 ZYMOCSXYVNFNNT-UHFFFAOYSA-N 0.000 description 1
- YSVYYENQIFSZTO-UHFFFAOYSA-N benzyl 4-fluoropiperidine-1-carboxylate;4-fluoropiperidine;hydrochloride Chemical compound Cl.FC1CCNCC1.C1CC(F)CCN1C(=O)OCC1=CC=CC=C1 YSVYYENQIFSZTO-UHFFFAOYSA-N 0.000 description 1
- VZOVOHRDLOYBJX-UHFFFAOYSA-N benzyl 4-oxopiperidine-1-carboxylate Chemical compound C1CC(=O)CCN1C(=O)OCC1=CC=CC=C1 VZOVOHRDLOYBJX-UHFFFAOYSA-N 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- 229940076810 beta sitosterol Drugs 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- LGJMUZUPVCAVPU-UHFFFAOYSA-N beta-Sitostanol Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(C)CCC(CC)C(C)C)C1(C)CC2 LGJMUZUPVCAVPU-UHFFFAOYSA-N 0.000 description 1
- 108010065875 beta-casomorphins Proteins 0.000 description 1
- NJKOMDUNNDKEAI-UHFFFAOYSA-N beta-sitosterol Natural products CCC(CCC(C)C1CCC2(C)C3CC=C4CC(O)CCC4C3CCC12C)C(C)C NJKOMDUNNDKEAI-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000002306 biochemical method Methods 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- SUAVNZMIOODRBR-UHFFFAOYSA-N bis(trifluoromethylsulfonyloxy)boranyl trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OB(OS(=O)(=O)C(F)(F)F)OS(=O)(=O)C(F)(F)F SUAVNZMIOODRBR-UHFFFAOYSA-N 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 150000001649 bromium compounds Chemical group 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 235000019577 caloric intake Nutrition 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 239000002458 cell surface marker Substances 0.000 description 1
- 229960005110 cerivastatin Drugs 0.000 description 1
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 230000035605 chemotaxis Effects 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229940090805 clavulanate Drugs 0.000 description 1
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000003271 compound fluorescence assay Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 1
- 239000012004 corey–bakshi–shibata catalyst Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000000139 costimulatory effect Effects 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229960005168 croscarmellose Drugs 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- MGNCLNQXLYJVJD-UHFFFAOYSA-N cyanuric chloride Chemical compound ClC1=NC(Cl)=NC(Cl)=N1 MGNCLNQXLYJVJD-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960004597 dexfenfluramine Drugs 0.000 description 1
- QYMPJVZAQCBGRM-VUCTXSBTSA-N diazonio-[(2s,3s)-3-(4-fluorophenyl)-1-oxo-1-[(4s)-2-oxo-4-phenyl-1,3-oxazolidin-3-yl]butan-2-yl]azanide Chemical compound C1([C@@H]2N(C(OC2)=O)C(=O)[C@@H](N=[N+]=[N-])[C@@H](C)C=2C=CC(F)=CC=2)=CC=CC=C1 QYMPJVZAQCBGRM-VUCTXSBTSA-N 0.000 description 1
- FAVAVMFXAKZTMV-UHFFFAOYSA-N dibutylboranyl trifluoromethanesulfonate Chemical compound CCCCB(CCCC)OS(=O)(=O)C(F)(F)F FAVAVMFXAKZTMV-UHFFFAOYSA-N 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000004582 dihydrobenzothienyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 description 1
- SPCNPOWOBZQWJK-UHFFFAOYSA-N dimethoxy-(2-propan-2-ylsulfanylethylsulfanyl)-sulfanylidene-$l^{5}-phosphane Chemical compound COP(=S)(OC)SCCSC(C)C SPCNPOWOBZQWJK-UHFFFAOYSA-N 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 108010054812 diprotin A Proteins 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- WLGSIWNFEGRXDF-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O.CCCCCCCCCCCC(O)=O WLGSIWNFEGRXDF-UHFFFAOYSA-N 0.000 description 1
- 210000005064 dopaminergic neuron Anatomy 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 229950005627 embonate Drugs 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 210000003989 endothelium vascular Anatomy 0.000 description 1
- 229950002375 englitazone Drugs 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 229960004563 eprosartan Drugs 0.000 description 1
- OROAFUQRIXKEMV-LDADJPATSA-N eprosartan Chemical compound C=1C=C(C(O)=O)C=CC=1CN1C(CCCC)=NC=C1\C=C(C(O)=O)/CC1=CC=CS1 OROAFUQRIXKEMV-LDADJPATSA-N 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- 229950000206 estolate Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 231100000318 excitotoxic Toxicity 0.000 description 1
- 230000003492 excitotoxic effect Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 1
- 229960000815 ezetimibe Drugs 0.000 description 1
- 229960001582 fenfluramine Drugs 0.000 description 1
- 229960000701 fenofibric acid Drugs 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 239000006261 foam material Substances 0.000 description 1
- 238000010575 fractional recrystallization Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 210000003731 gingival crevicular fluid Anatomy 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 230000009229 glucose formation Effects 0.000 description 1
- 230000004153 glucose metabolism Effects 0.000 description 1
- 230000004190 glucose uptake Effects 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 150000004795 grignard reagents Chemical class 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 239000003324 growth hormone secretagogue Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 208000013210 hematogenous Diseases 0.000 description 1
- 231100000304 hepatotoxicity Toxicity 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- FBPFZTCFMRRESA-UHFFFAOYSA-N hexane-1,2,3,4,5,6-hexol Chemical compound OCC(O)C(O)C(O)C(O)CO FBPFZTCFMRRESA-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000004957 immunoregulator effect Effects 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000004968 inflammatory condition Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000030214 innervation Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 210000004347 intestinal mucosa Anatomy 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000011630 iodine Chemical group 0.000 description 1
- 229910052740 iodine Chemical group 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 239000013038 irreversible inhibitor Substances 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000004898 kneading Methods 0.000 description 1
- 208000006443 lactic acidosis Diseases 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000004130 lipolysis Effects 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- AHCNXVCAVUYIOU-UHFFFAOYSA-M lithium hydroperoxide Chemical compound [Li+].[O-]O AHCNXVCAVUYIOU-UHFFFAOYSA-M 0.000 description 1
- 230000007056 liver toxicity Effects 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- BRKADVNLTRCLOW-UHFFFAOYSA-M magnesium;fluorobenzene;bromide Chemical compound [Mg+2].[Br-].FC1=CC=[C-]C=C1 BRKADVNLTRCLOW-UHFFFAOYSA-M 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 208000011661 metabolic syndrome X Diseases 0.000 description 1
- 229940102396 methyl bromide Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical compound CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- PQIOSYKVBBWRRI-UHFFFAOYSA-N methylphosphonyl difluoride Chemical group CP(F)(F)=O PQIOSYKVBBWRRI-UHFFFAOYSA-N 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 229960001110 miglitol Drugs 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 210000002161 motor neuron Anatomy 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- KVKFRMCSXWQSNT-UHFFFAOYSA-N n,n'-dimethylethane-1,2-diamine Chemical compound CNCCNC KVKFRMCSXWQSNT-UHFFFAOYSA-N 0.000 description 1
- KRKPYFLIYNGWTE-UHFFFAOYSA-N n,o-dimethylhydroxylamine Chemical compound CNOC KRKPYFLIYNGWTE-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 210000001577 neostriatum Anatomy 0.000 description 1
- PKWDZWYVIHVNKS-UHFFFAOYSA-N netoglitazone Chemical compound FC1=CC=CC=C1COC1=CC=C(C=C(CC2C(NC(=O)S2)=O)C=C2)C2=C1 PKWDZWYVIHVNKS-UHFFFAOYSA-N 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000005015 neuronal process Effects 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- GYCKQBWUSACYIF-UHFFFAOYSA-N o-hydroxybenzoic acid ethyl ester Natural products CCOC(=O)C1=CC=CC=C1O GYCKQBWUSACYIF-UHFFFAOYSA-N 0.000 description 1
- 239000007764 o/w emulsion Substances 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 238000007410 oral glucose tolerance test Methods 0.000 description 1
- 239000008203 oral pharmaceutical composition Substances 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 1
- 229960001243 orlistat Drugs 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 210000000963 osteoblast Anatomy 0.000 description 1
- IVMHDOBGNQOUHO-UHFFFAOYSA-N oxathiane Chemical compound C1CCSOC1 IVMHDOBGNQOUHO-UHFFFAOYSA-N 0.000 description 1
- OOFGXDQWDNJDIS-UHFFFAOYSA-N oxathiolane Chemical compound C1COSC1 OOFGXDQWDNJDIS-UHFFFAOYSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 238000005949 ozonolysis reaction Methods 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 208000028169 periodontal disease Diseases 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003562 phentermine Drugs 0.000 description 1
- 229960005190 phenylalanine Drugs 0.000 description 1
- DJFBJKSMACBYBD-UHFFFAOYSA-N phosphane;hydrate Chemical compound O.P DJFBJKSMACBYBD-UHFFFAOYSA-N 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 229960003912 probucol Drugs 0.000 description 1
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical compound C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 229950004405 prodipine Drugs 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- ZVJHJDDKYZXRJI-UHFFFAOYSA-N pyrroline Natural products C1CC=NC1 ZVJHJDDKYZXRJI-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- JSDRRTOADPPCHY-HSQYWUDLSA-N quinapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC=CC=C2C1)C(O)=O)CC1=CC=CC=C1 JSDRRTOADPPCHY-HSQYWUDLSA-N 0.000 description 1
- 229960001455 quinapril Drugs 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 238000011808 rodent model Methods 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 235000021003 saturated fats Nutrition 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 210000000582 semen Anatomy 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- KZJWDPNRJALLNS-VJSFXXLFSA-N sitosterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CC[C@@H](CC)C(C)C)[C@@]1(C)CC2 KZJWDPNRJALLNS-VJSFXXLFSA-N 0.000 description 1
- 229950005143 sitosterol Drugs 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- KKCBUQHMOMHUOY-UHFFFAOYSA-N sodium oxide Chemical compound [O-2].[Na+].[Na+] KKCBUQHMOMHUOY-UHFFFAOYSA-N 0.000 description 1
- 229910001948 sodium oxide Inorganic materials 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- XWQMFFMAJDXTJN-RCBQFDQVSA-N tert-butyl n-[(2s,3s)-3-(4-fluorophenyl)-1-[(3s)-3-fluoropyrrolidin-1-yl]-1-oxobutan-2-yl]carbamate Chemical compound O=C([C@@H](NC(=O)OC(C)(C)C)[C@@H](C)C=1C=CC(F)=CC=1)N1CC[C@H](F)C1 XWQMFFMAJDXTJN-RCBQFDQVSA-N 0.000 description 1
- GHCLQCWJCIQQQY-ACRUOGEOSA-N tert-butyl n-[(2s,3s)-4-(dimethylamino)-1-[(3s)-3-fluoropyrrolidin-1-yl]-1,4-dioxo-3-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]butan-2-yl]carbamate Chemical compound O=C([C@@H](NC(=O)OC(C)(C)C)[C@@H](C(=O)N(C)C)C=1C=CC(=CC=1)B1OC(C)(C)C(C)(C)O1)N1CC[C@H](F)C1 GHCLQCWJCIQQQY-ACRUOGEOSA-N 0.000 description 1
- KNBQRLAGPJMELA-XIRDDKMYSA-N tert-butyl n-[(2s,3s)-4-(dimethylamino)-1-[(3s)-3-fluoropyrrolidin-1-yl]-3-(4-hydroxyphenyl)-1,4-dioxobutan-2-yl]carbamate Chemical compound O=C([C@@H](NC(=O)OC(C)(C)C)[C@@H](C(=O)N(C)C)C=1C=CC(O)=CC=1)N1CC[C@H](F)C1 KNBQRLAGPJMELA-XIRDDKMYSA-N 0.000 description 1
- RNBRBEMOHXZFNB-OQNFGNQXSA-N tert-butyl n-[(e,2s,3s)-3-(4-bromophenyl)-1-[(3s)-3-fluoropyrrolidin-1-yl]-1-oxohex-4-en-2-yl]carbamate Chemical compound O=C([C@@H](NC(=O)OC(C)(C)C)[C@@H](/C=C/C)C=1C=CC(Br)=CC=1)N1CC[C@H](F)C1 RNBRBEMOHXZFNB-OQNFGNQXSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- OSBSFAARYOCBHB-UHFFFAOYSA-N tetrapropylammonium Chemical compound CCC[N+](CCC)(CCC)CCC OSBSFAARYOCBHB-UHFFFAOYSA-N 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000001228 trophic effect Effects 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- WCJYTPVNMWIZCG-UHFFFAOYSA-N xylylcarb Chemical compound CNC(=O)OC1=CC=C(C)C(C)=C1 WCJYTPVNMWIZCG-UHFFFAOYSA-N 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- 229940126158 β3 adrenergic receptor agonist Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/02—Drugs for disorders of the endocrine system of the hypothalamic hormones, e.g. TRH, GnRH, CRH, GRH, somatostatin
- A61P5/04—Drugs for disorders of the endocrine system of the hypothalamic hormones, e.g. TRH, GnRH, CRH, GRH, somatostatin for decreasing, blocking or antagonising the activity of the hypothalamic hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/28—Antiandrogens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/04—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Physical Education & Sports Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Rheumatology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurology (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Vascular Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Emergency Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
Description
構造式
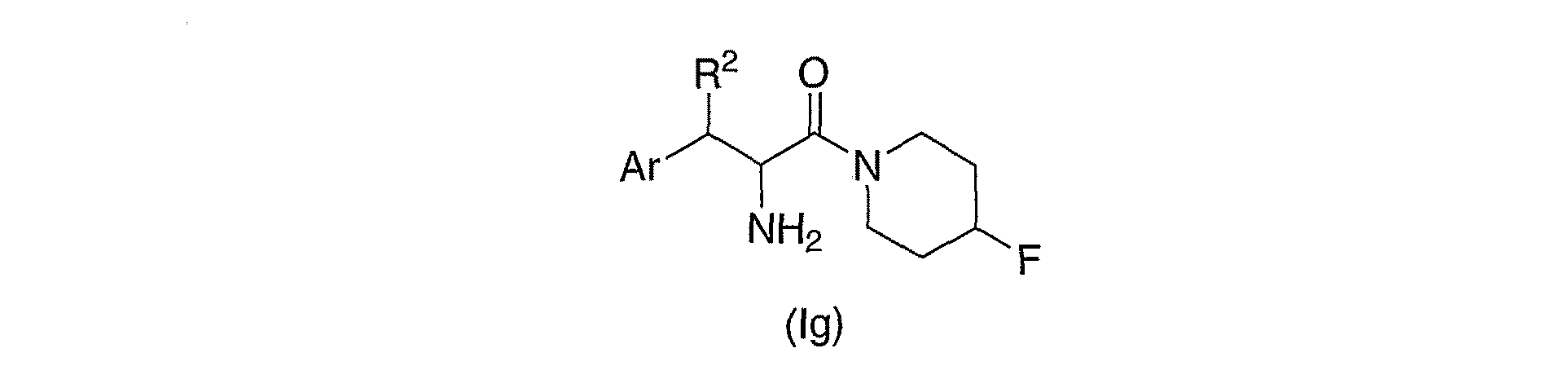
各nは、独立に、0、1又は2であり;
m及びpは、独立に、0又は1であり;
qは、1又は2であり;
Xは、CH2、S、CHF又はCF2であり;
Arは、置換されていないフェニル又は1ないし5個のR3置換基で置換されたフェニルであり;
R1は、水素又はシアノであり;
R2は、
C1−10アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
C2−10アルケニル(アルケニルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−アリール(アリールは、置換されていないか、又はヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−ヘテロアリール(ヘテロアリールは、置換されていないか、又はヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)n−ヘテロシクリル(ヘテロシクリルは、置換されていないか、又はオキソ、ヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル(シクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)nCOOH、
(CH2)nCOOC1−6アルキル、
(CH2)nCONR4R5(式中、R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキルからなる群から独立に選択されるか(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、又は、
R4及びR5は、両者で、それらが結合している窒素原子とともに、アゼチジン、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択され;
R2中の任意のメチレン(CH2)炭素原子は、置換されていないか、又はハロゲン、ヒドロキシ、C1−4アルキル及びC1−4アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし2個の基で置換されており;
各R3は、
ハロゲン、
シアノ、
ヒドロキシ、
フェニルオキシ(置換されていないか、又はハロゲン、ヒドロキシ、CO2H、シアノ、C1−6アルキルオキシカルボニル、C1−6アルキル、C3−6シクロアルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、
C1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
C1−6アルコキシ(アルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
(CH2)n−NR4R5、
(CH2)n−CONR4R5、
(CH2)n−OCONR4R5、
(CH2)n−SO2NR4R5、
(CH2)n−SO2R6、
(CH2)n−NR7SO2R6、
(CH2)n−NR7CONR4R5、
(CH2)n−NR7COR7、
(CH2)n−NR7CO2R6、
(CH2)n−COOH、
(CH2)n−COOC1−6アルキル、
(CH2)q−アリール(アリールは、置換されていないか、又はハロゲン、ヒドロキシ、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル、C3−6シクロアルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)q−ヘテロアリール(ヘテロアリールは、置換されているか、又はヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル、C3−6シクロアルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)q−ヘテロシクリル(ヘテロシクリルは、置換されていないか、又はオキソ、ヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル、C3−6シクロアルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル(シクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
からなる群から独立に選択され、
R3中の任意のメチレン(CH2)炭素原子は、置換されていないか、又はハロゲン、ヒドロキシ、C1−4アルキル及びC1−4アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし2個の基で置換されており;
R6は、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)からなる群から独立に選択され、R6中の任意のメチレン(CH2)炭素原子は、置換されていないか、又はハロゲン、ヒドロキシ、C1−4アルキル及びC1−4アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし2個の基で置換されており;
各R7は、水素又はR6である。)
本発明の化合物の一実施形態において、*の印が付された炭素原子は、式Ia
に図示された立体化学配置を有する。
に図示された立体化学配置を有する。
に図示されているように、mは1であり、pは0である。
に図示されている立体化学配置を有する。
式Ie:
に図示されているように、R1は水素であり、XはCHFであり、m及びpは0である。
に図示されている立体化学配置を有する。
に図示されている立体化学配置を有する。
C1−6アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
C2−6アルケニル(アルケニルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル(シクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)nCOOH、
(CH2)nCOOC1−6アルキル、及び
(CH2)nCONR4R5(R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキルからなる群から独立に選択されるか(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、又は
R4及びR5は、両者で、それらが結合している窒素原子とともに、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択され、
R2中の任意のメチレン(CH2)炭素原子は、置換されていないか、又はハロゲン、ヒドロキシ、C1−4アルキル及びC1−4アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし2個の基で置換されている。
C1−3アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル、
COOH、
COOC1−6アルキル、及び
CONR4R5(R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキルからなる群から独立に選択されるか(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、又は
R4とR5は、両者で、それらが結合している窒素原子とともに、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択される。
メチル、
エチル、
CH2−シクロプロピル、
COOH、
COOMe、
COOEt、
CONMe2、
CONH2、
CONHMe、
CONHEt、
ピロリジン−1−イルカルボニル、
アゼチジン−1−イルカルボニル、及び
[(テトラゾール−5−イル)アミノ]カルボニル
からなる群から選択される。
に図示された立体化学配置を有し;
R2は、
C1−6アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル、
COOH、
COOC1−6アルキル、及び
CONR4R5(R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキルからなる群から独立に選択されるか(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、又は
R4及びR5は、両者で、それらが結合している窒素原子とともに、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択され;
各R3は
ハロゲン、
ヒドロキシ、
C1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
C1−6アルコキシ(アルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
フェニルオキシ(置換されていないか、又はハロゲン、ヒドロキシ、シアノ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル(シクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
からなる群から独立に選択される。
メチル、
エチル、
CH2−シクロプロピル、
COOH、
COOMe、
COOEt、
CONMe2、
CONH2、
CONHMe、
CONHEt、
ピロリジン−1−イルカルボニル、
アゼチジン−1−イルカルボニル、及び
[(テトラゾール−5−イル)アミノ]カルボニル、
からなる群から選択される。
フルオロ、
クロロ、
ブロモ、
トリフルオロメチル、
トリフルオロメトキシ、及び
メトキシ、
からなる群から選択される。
Xは、CH2、S、CHF又はCF2であり;
Arは、置換されていないフェニル又は1ないし5個のR3置換基で置換されたフェニルであり;
R1は、水素又はシアノであり;
R2は、
C1−6アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル、
COOH、
COOC1−6アルキル、及び
CONR4R5(R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキルからなる群から独立に選択されるか(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、又は
R4とR5は、両者で、それらが結合している窒素原子とともに、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択され;
各R3は、
ハロゲン、
C1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
C1−6アルコキシ(アルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
フェニルオキシ(置換されていないか、又はハロゲン及びシアノから独立に選択される1ないし3個の置換基で置換されている。)、及び
フェニル(CH2)nCON(Me)−(フェニルは、置換されていないか、又はハロゲン、トリフルオロメチル及びC1−4アルキルから独立に選択される1ないし3個の置換基で置換されている。)、
からなる群から独立に選択される。)
の化合物を包含する。
「アルキルチオ」という用語は、指定された炭素原子の数の直鎖若しくは分枝鎖アルキルスルフィド(例えば、C1−6アルキルチオ)、又はこの範囲に属する任意の数の直鎖若しくは分枝鎖アルキルスルフィド(すなわち、メチルチオ(MeS−)、エチルチオ、イソプロピルチオなど)を表す。
(b)(i)グリタゾン(例えば、トログリタゾン、ピオグリタゾン、エングリタゾン、MCC−555、ロシグリタゾンなど)などのPPARγアゴニスト及びKRP−297などのPPARα/γデュアルアゴニストなどの他のPPARリガンド及びフェノフィブリン酸誘導体(ゲムフィブロジル、クロフィブレート、フェノフィブレート及びベザフィブレート)などのPPARαアゴニスト、(ii)メトホルミン及びフェンホルミンなどのビグアニド、並びに(iii)タンパク質チロシンホスファターゼ1B(PTP−1B)阻害剤などのインシュリン増感剤;
(c)インシュリン又はインシュリン模倣物;
(d)スルホニル尿素、並びにトルブタミド、グリブリド、グリピジド、グリメピリド及びメグリチニド(レパグリニドなど)などの他のインシュリン分泌促進物質;
(e)α−グルコシダーゼ阻害剤(アカルボース及びミグリトールなど);
(f)WO 98/04528号、WO 99/01423号、WO 00/39088号、及びWO 00/69810号に開示されているものなどのグルカゴン受容体アンタゴニスト;
(g)WO 00/42026号及びWO 00/59887号に開示されているものなどのGLP−1、GLP−1模倣物及びGLP−1受容体アゴニスト;
(h)GIP及びWO 00/58360号に開示されているものなどのGLP模倣物、並びにGIP受容体アゴニスト:
(i)PACAP、PACAP模倣物及びWO 01/23420号に開示されているものなどのPACAP受容体アゴニスト;
(j)(i)HMG−CoA還元酵素阻害剤(ロバスタチン、シンバスタチン、プラバスタチン、セリバスタチン、フルバスタチン、アトルバスタチン、イタバスタチン及びロスバスタチン、並びに他のスタチン)、(ii)金属イオン封鎖剤(コレスチラミン、コレスチポール、及び架橋されたデキストランのジアルキルアミノアルキル誘導体)、(iii)ニコチニルアルコール、ニコチン酸又はそれらの塩、(iv)フェノフィブリン酸誘導体(ゲムフィブロジル、クロフィブレート、フェノフィブレート及びベザフィブレレート)などのPPARαアゴニスト、(v)KRP−297などのPPARα/γデュアルアゴニスト、(vi)β−シトステロール及びエゼチミブなどのコレステロール吸収の阻害剤、(vii)アバシミブ(avasimibe)などのアシルCoA:コレステロールアシルトランスフェラーゼ阻害剤、並びに(viii)プロブコールなどの抗酸化剤などの、コレステロール降下剤;
(k)WO 97/28149号に開示されているものなどの、PPARδアゴニスト;
(l)フェンフルラミン、デクスフェンフルラミン、フェンテルミン、シブトラミン、オーリスタット、ニューロペプチドY5阻害剤、CB−1アンタゴニスト、MC−4Rアゴニスト、β3アドレナリン作動性受容体アゴニストなどの肥満抑制化合物;
(m)回腸の胆汁酸輸送体阻害剤;
(n)アスピリン、非ステロイド性抗炎症剤、グルココルチコイド、アザルフィジン、及び選択的シクロオキシゲナーゼ−2阻害剤などの炎症症状に使用するための薬剤;
(o)ACE阻害剤(エナラプリル、リシノプリル、カプトプリル、キナプリル、タンドラプリル(tandolapril))、A−II受容体遮断薬(ロサルタン、カンデサルタン、イルベサルタン、バルサルタン、テルミサルタン、エプロサルタン)、β遮断薬及びカルシウムチャンネル遮断薬などの降圧剤。
本発明の薬学的組成物及び方法は、上記の病態の治療に一般的に使用される、本明細書に記載されている治療的に活性な他の化合物をさらに含むことができる。
ステップA:(3R)−ヒドロキシピロリジン−1−カルボン酸ベンジル
機械的攪拌器、熱電対、添加漏斗(addition funnel)及び窒素気泡器を備えた22Lの三つ口丸底フラスコに、(3R)−3−ヒドロキシピロリジン425g(4.88mol)、ジクロロメタン8L、トリエチルアミン1L(7.17mol)を加えた。この溶液を氷浴で5ないし10℃まで冷やした後、反応温度を20℃未満に保ちながら、約1.5時間にわたって、クロロギ酸ベンジル1000g(5.86mol)を滴下して添加した。この反応混合物を氷浴中でさらに1時間攪拌した後、氷浴を取り除き、反応混合物を室温まで一晩暖めた。飽和炭酸水素ナトリウム水溶液約15Lを含有する巨大な抽出機の中にこの混合物を注いだ。2つに分けた2Lのジクロロメタンを用いて、水相を逆抽出した。合わせた有機物を硫酸マグネシウム上で乾燥させ、濃縮し、オレンジ色の油を得た。この粗精製物をジクロロメタン中に取り込み、50%酢酸エチル/ヘキサン中の予め充填されたシリカゲルの5kgカラムに適用し、8Lの50%酢酸エチル/ヘキサン、16Lの75%酢酸エチル/ヘキサン、次いで100%酢酸エチル/ヘキサンで順次溶出し、静置したときに結晶化する黄色の油として表題化合物を得た。
機械的攪拌器、熱電対、添加漏斗及び窒素気泡器を備えた5Lの三つ口丸底フラスコに、(ジエチルアミノ)三フッ化硫黄375mL(2.84mol)及びジクロロメタン400mLを加えた。この溶液を−78℃まで冷却した。反応温度を−70℃未満に保ちながら、ジクロロメタン400mL中の(3R)−3−ヒドロキシピロリジン−1−カルボン酸ベンジル304g(1.37mol)の溶液を、2時間にわたり、添加漏斗を介して、この溶液に添加した。この反応混合物を攪拌し、室温まで一晩ゆっくりと暖めた。氷、水及び飽和炭酸水素ナトリウム水溶液を含有する巨大な抽出機に、少しずつ、注意しながらこの混合物を加えた。この混合物を酢酸エチル8Lで抽出した。有機層を塩水で洗浄し、硫酸マグネシウム上で乾燥させ、濃縮し、茶色の油を得た。フラッシュクロマトグラフィー(シリカゲル、10%から30%への酢酸エチル/ヘキサンのグラジエントで溶出)による精製によって、表題化合物が茶色の油として得られた。
(3S)−3−フルオロピロリジン−1−カルボン酸ベンジル(249g、1.11mmol)をエタノール2.3L中に溶かし、次いで、水115mLを加えた後、10%の炭素担持パラジウム30gを加えた。この混合物を、40psiの水素下で、約24時間揺動させた。さらに10g、次いで5gの触媒を加えた。40psiの水素下で、この混合物を反応が完結するまで攪拌した。この混合物をろ過し、ろ塊をエタノールで洗浄した。ろ液と洗浄液を合わせたものを、濃塩酸185mLで処理し、濃縮して無色の油を得た。残留物にトルエンを加えて共沸混合物とした後、ジエチルエーテル2Lを加えた。この油が結晶化するまで、イソプロピルアルコールを加えた。この混合物を、週末まで、室温で寝かした。結晶を集めて、ジエチルエーテルで洗浄し、真空中で乾燥させて、表題化合物を得た。母液と洗浄液を合わせて、濃縮し、トルエンを加えて共沸混合物とし、ジエチルエーテル/イソプロピルアルコールで練和した。この第二集団を集めて、真空中で乾燥させて、さらなる表題化合物を得た。[α]D=+8.64°(c=4、メタノール)。
ステップA:(3S)−3−アセトキシピロリジン−1−カルボン酸ベンジル
機械的攪拌器、熱電対、添加漏斗及び窒素気泡器を備えた22Lの三つ口丸底フラスコに、(3R)−3−ヒドロキシピロリジン−1−カルボン酸ベンジル(中間体1、ステップA)422g(1.91mol)、トルエン12L、トリフェニルホスフィン751g(2.86mol)、氷酢酸164mL(2.86mol)を加えた。得られた混合物を室温で攪拌した後、冷たい水浴で内部温度を28℃未満に保ちながら、約30分の期間にわたって、添加漏斗を介して、アゾジカルボン酸ジエチル500g(2.87mol)を添加した。この反応物を室温で一晩攪拌した。真空中で溶媒を除去し、ジエチルエーテル6Lを用いて残留物を練和した。固体をろ過して除去し、ジエチルエーテルでよく洗浄した。ろ液とエーテル洗浄液を合わせて、濃縮すると、濃黄色の油が固体とともに得られた。フラッシュクロマトグラフィー(シリカゲル、5%の酢酸エチル/ヘキサン及び10%から30%への酢酸エチル/ヘキサンのグラジエントで順次溶出)による精製によって、表題化合物を淡黄色の油として得た。
(3S)−3−アセトキシピロリジン−1−カルボン酸ベンジル427g(1.62mol)を含有する20Lの三ツ口丸底フラスコに、無水エタノール4Lを加えた後、約400mLの水中の水酸化カリウム101g(1.57mol)を加えた。約15分後、この反応混合物を水8L中に注ぎ、酢酸エチル8Lで抽出した。次いで、さらに4Lの酢酸エチルで水層を抽出した。合わせた有機物を塩水で洗浄し、硫酸マグネシウム上で乾燥させ、濃縮し、濃い油及び固体を得た。
中間体1、ステップBに概説した操作に実質的に従って、(3S)−3−ヒドロキシピロリジン−1−カルボン酸ベンジルの366g(1.62mol)部を表題化合物に変換した。
中間体1、ステップCに概説した操作に実質的に従って、(3R)−3−フルオロピロリジン−1−カルボン酸ベンジルの222g(1.0mol)部を表題化合物に変換した。[α]D=−8.61(c=4、メタノール)。
ステップA:3−オキソピロリジン−1−カルボン酸ベンジル
機械的攪拌器、熱電対、コンデンサー及び窒素気泡器を備えた12Lの三つ口丸底フラスコに、(3R)−3−ヒドロキシピロリジン−1−カルボン酸ベンジル(中間体1、ステップA)351g(1.61mol)、ジクロロメタン6L、粉末化された分子篩500g及びN−メチルモルホリン−N−オキシド400g(3.41mol)を加えた。得られた懸濁液を室温で攪拌し、これに、テトラプロピルアンモニウム過ルテニウム酸塩12.9g(0.0367mol)を添加した。冷たい水浴を用いて、反応温度を30℃以下に保った。この混合物を室温で2時間攪拌した。この混合物を、5kgのシリカゲルのプラグ上に注いで、10%酢酸エチル/ジクロロメタンで溶出し、オレンジ色の油として表題化合物を得た。
機械的攪拌器、熱電対、添加漏斗及び窒素気泡器を備えた12Lの三つ口丸底フラスコに、3−オキソピロリジン−1−カルボン酸ベンジル292g(1.33mol)及びジクロロメタン3Lを加えた。室温で攪拌している前記溶液に、冷たい水浴を用いて内部温度を25℃未満に保ちながら、約3時間にわたって、(ジエチルアミノ)三フッ化硫黄530mL(4.0mol)を滴下しながら添加した。この混合物を室温で一晩攪拌した。氷及び固体炭酸水素ナトリウムを含有する巨大な抽出機の中にこの混合物を注いだ。次いで、酢酸エチル8Lを加え、炭酸水素ナトリウムを用いてこの混合物を塩基性にした。層を分離した。有機物を硫酸マグネシウム上で乾燥させ、濃縮し、茶色の油309gを得た。フラッシュクロマトグラフィー(シリカゲル10%から20%酢酸エチル/ヘキサンへのグラジエント)による精製によって、表題化合物を得た。
中間体1、ステップCに概説した操作に実質的に従って、3,3−ジフルオロピロリジン−1−カルボン酸ベンジルの242g(1.00mol)部を表題化合物に変換した。1H NMR(500MHz、CD3OD):δ3.7(t,2H)、3.6(t,2H)、2.55(m,2H)。
ステップA:4−フルオロ−1−ピペリジンカルボン酸ベンジル
1Lの丸底フラスコに、4−オキソ−1−ピペリジンカルボン酸ベンジル12.64g(51.4mmol)及びジクロロメタン300mLを加えた。−78℃で攪拌している前記溶液に、約1時間にわたり、添加漏斗を介して、[ビス(2−メトキシエチル)アミノ]三フッ化硫黄19mL(102.8mmol)を添加した。この反応混合物を、室温になるまで、ゆっくりと一晩暖めた。水及び飽和炭酸水素ナトリウム水溶液を含有する巨大な抽出機の中に、少しずつ、注意しながらこの混合物を注いだ。この混合物をジクロロメタン(3x300mL)で抽出した。合わせた有機層を、飽和炭酸ナトリウム水溶液で1度、10%塩酸水溶液及び塩水で2度洗浄し、硫酸ナトリウムで乾燥させ、真空中で濃縮した。Biotage(R)システム上でのフラッシュクロマトグラフィー(グラジエント、ヘキサンから65%酢酸エチル/ヘキサン)による精製によって、所望の生成物を得た。LC/MS 242.1(M+1)。
エタノール80mL中に、4−フルオロ−1−ピペリジンカルボン酸ベンジル(5.5g、23.2mmol)を溶かし、20%の炭素担持水酸化パラジウム(無水ベース)1.0gを前記混合物に添加した。40psiの水素下で約12時間、混合物を揺動させた後、セライトパッドを通してろ過し、メタノール100mLで洗浄した。ろ液と洗浄液を合わせたものを、ジエチルエーテル中の1M塩酸60mLで処理し、濃縮して白色の蝋状固体を得た。この固体を真空中で乾燥させて、正体不明の微少な不純物とともに、表題化合物を固体として得た。さらなる精製を行わずに、この物質を用いた。1H NMR(CDCl3):δ4.95(d,J=47.4Hz,1H),3.70(br s,1H),3.34−3.27(m,4H),2.29(dt,J=37.1,12.3Hz,2H),2.16(br s,2H)。
ステップA:1−ベンズヒドリル−3−フルオロアゼチジン
250mLの丸底フラスコに、1−ベンズヒドリル−3−ヒドロキシアゼチジン3.0g(12.5mmol)及びジクロロメタン80mLを加えた。−78℃で攪拌している前記溶液に、約3時間にわたり、添加漏斗を介して、[ビス(2−メトキシエチル)アミノ]三フッ化硫黄4.6mL(25mmol)を添加した。この反応混合物を、室温になるまで、ゆっくりと一晩暖めた。水及び飽和炭酸水素ナトリウム水溶液を含有する巨大な抽出機の中に、少しずつ、(注意しながら)この反応混合物を添加した。この混合物をジクロロメタン80mLで3回抽出した。合わせた有機層を、飽和炭酸水素ナトリウム水溶液、水及び塩水で順次洗浄し、硫酸ナトリウムで乾燥させ、真空中で濃縮した。Biotage(R)システムを用いたフラッシュクロマトグラフィー(グラジエント、ヘキサンから80%酢酸エチル/ヘキサン)による精製によって、所望の生成物を得た。LC/MS 242.1(M+1)。
エタノール60mL及び20%の炭素担持水酸化パラジウム(無水ベース)500mg中に、1−ベンズヒドリル−3−フルオロアゼジチン(1.7g、7.04mmol)を溶かした。40psiの水素気体下で約12時間、混合物を揺動させた。セライトパッドを通してこの混合物をろ過し、メタノール100mLでろ塊を洗浄した。合わせた洗浄液をトリフルオロ酢酸10mLで処理し、濃縮して2つの油を得、このうち密度が高いものが所望のフルオロアゼチジン塩である。この混合物は、さらに精製しなかった。1H NMR(CDCl3) δ5.45−4.30(dm,J=56.7Hz,1H),4.46−4.38(m,2H),4.24−2.17(m,2H)。
ステップA:(4S)−3−[(2E)−ブタ−2−エノイル]−4−フェニル−1,3−オキサゾリジン−2−オン
テトラヒドロフラン(THF、100mL)中の攪拌された4(S)−4−フェニル−2−オキサゾリジノン(97.0g、43.0mmol)の溶液に、−78℃でn−ブチルリチウム(30.0mL、1.6M ヘキサン中、48.0mmol)を添加した。この混合物を20分間攪拌した後、塩化クロトニル(5.0g、43.0mmol)を加えた。得られた溶液を−78℃で30分間、室温で1.5時間攪拌した。飽和塩化アンモニウム水溶液を添加することによって、反応を停止させた。得られた水性スラリーを水で希釈し、3部の酢酸エチルで抽出した。合わせた有機層を、飽和炭酸水素ナトリウム水溶液及び塩水で洗浄し、硫酸ナトリウムで乾燥させ、真空中で濃縮した。フラッシュクロマトグラフィー(シリカゲル、83:17のヘキサン/酢酸エチル)による精製によって、所望の生成物を得た。
無水THF(60mL)及びジメチルスルフィド(30mL)中の攪拌された臭化銅(II)ジメチルスルフィド錯体(6.85g、33.3mmol)の溶液に、−40℃で4−フルオロフェニル臭化マグネシウム(30.0mL、1.0M THF中、30.0mmol)を添加した。得られた混合物を−40℃で30分間攪拌した後、−10℃まで加温した。 THF(30mL)中のステップAから得られた生成物(1.18g、5.10mmol)を、1時間にわたって、−10℃で、上記反応混合物に添加した。得られた混合物を−10℃で2時間攪拌した後、室温までゆっくり加温し、室温で12時間攪拌した。飽和塩化アンモニウム水溶液をゆっくり添加することによって、反応を停止させた。有機相を分離させ、2部の酢酸エチルを用いて水相を抽出した。合わせた有機相を塩水で洗浄し、真空中で濃縮した。フラッシュクロマトグラフィー(シリカゲル、80:20のヘキサン/酢酸エチル)による精製によって、所望の生成物を得た。
無水THF(40mL)中のステップBで得た生成物(1.18g、3.61mmol)の攪拌された溶液に、−78℃でヘキサメチルジシラジドカリウム(8.7mL、0.5M トルエン中、4.35mmol)を添加した。この反応混合物を−78℃で30分間攪拌した。予め冷却したTHF(30mL)中のトリシルアジド(1.56g、5.04mmol)の溶液を、カニューラを通じて加えた。1.5分後に、氷酢酸(0.95mL、16.6mmol)及び酢酸カリウム(1.77g、18.1mmol)で反応を停止させた。冷却槽を取り除き、この反応物を室温で12時間攪拌した。この溶液を、酢酸エチル及び塩水間に分配した。水相を3部の酢酸エチルで抽出した。有機相を合わせて、飽和炭酸水素ナトリウム水溶液で洗浄し、硫酸ナトリウム上で乾燥させ、真空中で蒸発させた。フラッシュクロマトグラフィー(シリカゲル、80:20のヘキサン/酢酸エチル)による精製によって、所望の生成物を得た。
THF(30mL)中のステップCで得た生成物(0.88g、2.39mmol)の攪拌された溶液に、水(10mL)を加えた。0℃で15分間、この溶液を攪拌し、次いで30%の過酸化水素(1.60mL、14.3mmol)を添加した後、水酸化リチウム(0.17g、7.23mmol)をゆっくりと添加した。得られた混合物を0℃で4時間攪拌した。飽和亜硫酸ナトリウム水溶液を添加して、反応を停止させ、30分間室温で攪拌した。水相を分離し、3部のジクロロメタンで抽出した。次いで、3Nの塩酸で水相をpH1の酸性にし、3部の酢酸エチルで抽出した。この酢酸エチル抽出物を合わせて、硫酸ナトリウム上で乾燥させ、真空中で蒸発させて生成物を得、これを次のステップで直接使用した。LC/MS 224.1(M+1)。
10:8:1のメタノール/テトラヒドロフラン/トリフルオロ酢酸15mL中に溶かしたステップDで得たアジド500mg(2.24mmol)に、10%の炭素担持パラジウム50mgを添加した。反応フラスコに窒素を流した後、水素雰囲気下(1atm)で12時間攪拌した。この反応物をセライトのパッドに通した。次いで、メタノールでろ塊を洗浄し、真空中で溶媒を除去して、黄色の泡状物質を得た。この泡状物質に、ジオキサン5mLと飽和炭酸水素ナトリウム水溶液5mLを添加した後、ジ−tert−ブチルジカルボネート1.47g(6.70mmol)を添加した。この反応液を室温で12時間攪拌した。酢酸エチルで反応混合物を希釈し、1Nの塩酸水溶液を用いてpH1の酸性にした。相を分離させ、2部の酢酸エチルで水層を抽出した。有機抽出液を合わせて、塩水で洗浄し、硫酸ナトリウム上で乾燥させ、真空中で濃縮した。フラッシュクロマトグラフィー(シリカゲル、95:5のジクロロメタン/メタノール)によって、残留物を精製し、所望の生成物を得た。LC/MS 298.0(M+1)。
無水ジメチルホルムアミド(DMF、2mL)中のステップDで得られた生成物(0.19g、0.65mmol)の攪拌された溶液に、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(EDC、0.19g、0.99mmol)、ヒドロキシベンゾトリアゾール(HOBT、0.13g、0.96mmol)、チアゾリジン(0.26mL、3.3mmol)及びN,N’−ジイソプロピルエチルアミン(0.33mL、1.9mmol)を加えた。室温で12時間攪拌した後に、この反応液を酢酸エチルで希釈した。塩水、1N塩酸及び1N水酸化ナトリウム水溶液で、有機相を順次洗浄し、硫酸ナトリウム上で乾燥させ、真空中で蒸発させて、未精製の結合された生成物を得、次のステップで直接使用した。
ジクロロメタン(5mL)中のステップE由来の生成物の撹拌溶液に、トリフルオロ酢酸(1mL)を室温で添加した。室温で1時間撹拌した後、真空中で溶媒を除去し、残留物をHPLC(YMC Pro−C18カラム、グラジエント溶出、10−90% アセトニトリル/0.1%TFAを含有する水)で精製し、表題化合物を得た。LC/MS 269.1(M+1)。
ステップA:(βS)−N−(tert−ブトキシカルボニル)−4−フルオロ−β−メチル−L−フェニルアラニル−L−プロリンアミド
無水N,N−ジメチルホルムアミド(DMF、1.5mL)中の(2S,3S)−2−アジド−3−(4−フルオロフェニル)ブタン酸(実施例1、ステップD、91.8mg、0.309mmol)の撹拌溶液に、[O−(7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート](HATU、141mg、0.371mmol)、1−ヒドロキシ−7−アザベンゾトリアゾール(HOAT、50.5mg、0.371mmol)、L−プロリンアミド(38.8mg、0.340mmol)及びN,N’−ジイソプロピルエチルアミン(0.135mL、0.773mmol)を添加した。室温にて16時間撹拌した後、反応物を酢酸エチルで希釈した。水、飽和炭酸水素ナトリウム水溶液及び塩水で有機相を順次洗浄し、乾燥させ(硫酸マグネシウム)、減圧下で濃縮した。分取用薄層クロマトグラフィー(シリカ、酢酸エチル溶出液)で残留物を精製し、表題化合物を得た。
無水DMF(1mL)中のステップA由来の生成物(91.7mg、0.233mmol)の撹拌溶液に、塩化シアヌル(86mg、0.466mmol)を添加した。室温にて16時間撹拌した後、反応物を酢酸エチルで希釈した。水、飽和炭酸水素ナトリウム水溶液及び塩水で有機相を順次洗浄し、乾燥させ(硫酸マグネシウム)、減圧下で濃縮した。分取用薄層クロマトグラフィー(シリカ、酢酸エチル溶出液)で精製し、表題化合物を得た。
ジクロロメタン(1mL)中のステップB由来の生成物75.0mgの撹拌溶液に、トリフルオロ酢酸(1mL)を室温で添加した。1時間撹拌した後、真空中で溶媒を除去し、表題化合物を得た。LC/MS 275.1(M+1)。
ステップA:(3S)−1−[(2S,3S)−2−[(tert−ブトキシルカルボニル)アミノ]3−(4−フルオロフェニル)−1−オキソブタニル]−3−フルオロピロリジン
無水DMF(2mL)中の(βS)−N−(tert−ブトキシカルボニル)−4−フルオロ−β−メチル−L−フェニルアラニン(実施例1、ステップE、0.15g、0.51mmol)の撹拌溶液に、EDC(0.25g、1.33mmol)、HOBT(0.18g、1.33mmol)、(3S)−3−フルオロピロリジン塩酸塩(0.18g、1.45mmol)及びN,N’−ジイソプロピルエチルアミン(0.23mL、1.31mmol)を添加した。室温にて12時間撹拌した後、反応物を酢酸エチルで希釈した。塩水、1N 塩酸及び1N 水酸化ナトリウム水溶液で有機相を順次洗浄し、硫酸ナトリウムで乾燥させ、真空中で蒸発させて、未精製のカップリング生成物を得て、次のステップで直接用いた。
ジクロロメタン(5mL)中のステップA由来の生成物の撹拌溶液に、室温でトリフルオロ酢酸(1mL)を添加した。室温で1時間撹拌した後、真空中で溶媒を除去し、残留物をHPLC(YMC Pro−C18カラム、グラジエント溶出、10−90% アセトニトリル/0.1%TFAを含有する水)で精製し、表題化合物を得た。LC/MS 269.0(M+1)。
ステップA:(4R)−3−[(2E)−3−(4−ブロモフェニル)プロパ−2−エノイル]−4−フェニル−1,3−オキサゾリジン−2−オン
−78℃にて、無水THF(250mL)中の4−ブロモケイ皮酸(5.79g、22.5mmol)の撹拌溶液に、トリエチルアミン(4.60mL、34.6mmol)を添加し、続いて塩化トリメチルアセチル(3.54mL、24.7mmol)を添加した。得られた懸濁液を−78℃にて15分間、0℃にて1時間、続いて−78℃にて15分間撹拌し、その後、0℃にて、4(R)−4−フェニル−2−オキサゾリジノンリチウムのスラリー(−78℃で無水THF(150mL)中の4(R)−4−フェニル−2−オキサゾリジノン(5.0g、30.6mmol)の溶液に、n−ブチルリチウム(19.1mL、30.5mmol)を添加することにより、−78℃にて15分前に調製した。)の中に、カニューレを用いて移した。得られたスラリーを−78℃で1時間、室温にて12時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させた。有機相を分離し、真空中で濃縮し、次のステップでこの未精製生成物を直接用いた。LC/MS 372.0(M+1)。
THF(60mL)及びジメチルスルフィド(30mL)中の臭化銅(II)ジメチルスルフィド錯体(8.78g、42.7mmol)の撹拌溶液に、−40℃で臭化メチルマグネシウム(12.7mL、ジエチルエーテル中3.0M、38.1mmol)を添加した。得られた混合物を−40℃で30分間撹拌し、その後、−20℃まで温めた。THF(30mL)中のステップA由来の生成物(3.53g、9.48mmol)を、上記反応混合物に対して、−20℃で1時間にわたり添加した。得られた混合物を−20℃で2時間撹拌し、次に、室温までゆっくりと温め、室温で12時間撹拌した。飽和塩化アンモニウム水溶液をゆっくりと添加してその反応を停止させた。有機相を分離し、水相を酢酸エチルで2回に分けて抽出した。合わせた有機層を食塩水で洗浄し、真空中で濃縮した。フラッシュクロマトグラフィーによる精製を行い(シリカゲル、83:17 ヘキサン/酢酸エチル)、所望の生成物を得た。
ジクロロメタン(40mL)中のステップB由来の生成物(2.87g、7.39mmol)の撹拌溶液に、−78℃にて、ジイソプロピルエチルアミン(1.93mL、11.1mmol)及びジブチルボロントリフレート(9.6mL、ジクロロメタン中1M溶液、9.60mmol)を添加した。その淡黄色の溶液を−78℃で15分間、0℃で1時間撹拌し、15分間、−78℃に再冷却した。予め冷却しておいたジクロロメタン(40mL)中のN−ブロモスクシンイミド(3.93g、22.2mmol)の懸濁液へ、上記溶液を、カニューレを用いて移した。得られた混合物を−78℃で1時間、0℃で3時間撹拌した。0.5N 亜硫酸水素ナトリウム水溶液によりその反応を停止させた。有機相を分離し、水相を酢酸エチルで2回に分けて抽出した。合わせた有機層を塩水で洗浄し、真空中で濃縮した。フラッシュクロマトグラフィーによる精製を行い(シリカゲル、83:17 ヘキサン/酢酸エチル)、所望の生成物を得た。
アセトニトリル(40mL)中のステップC由来の生成物(2.71g、6.39mmol)の撹拌溶液に、テトラメチルグアニジニウムアジド(3.51g、22.2mmol)を添加した。反応物を室温で12時間撹拌した。固形物を濾取し、ろ液を蒸発させた。未精製生成物をフラッシュクロマトグラフィーで精製し(83:17 ヘキサン/酢酸エチル)、所望の生成物を得た。
THF(60mL)中のステップD由来の生成物(2.77g、6.23mmol)の撹拌溶液に、水(20mL)を添加した。この溶液を0℃で15分間撹拌し、次に30%過酸化水素(6.0mL、52.9mmol)を添加し、続いて水酸化リチウム(0.50g、21.2mmol)をゆっくりと添加した。得られた混合物を0℃で4時間撹拌した。亜硫酸ナトリウム飽和水溶液を添加して反応を停止させ、室温で30分間撹拌した。水相を分離し、ジクロロメタンで3回洗浄した。次に、水相を3N 塩酸でpH1まで酸性化して、酢酸エチルで3回に分けて抽出した。酢酸エチル抽出物を合わせ、硫酸ナトリウムで乾燥させ、真空中で蒸発させ、生成物を得て、次のステップで直接用いた。
無水DMF(10mL)に溶解させた酸1.20g(4.22mmol)に、EDC(2.29g、11.9mmol)、HOBT(1.62g、11.9mmol)、(3S)−3−フルオロピロリジン塩酸塩(1.50g、11.9mmol)及びN,N’−ジイソプロピルエチルアミン(4.2mL、23.6mmol)を添加した。室温にて12時間撹拌した後、反応物を酢酸エチルで希釈した。塩水、1N 塩酸及び1N 水酸化ナトリウム水溶液で有機相を洗浄し、硫酸ナトリウムで乾燥させ、真空中で蒸発させて、黄色の泡状物質を回収した。この泡状物質に、ジオキサン40mL、水4mL及びトリフェニルホスフィン(4.70g、17.9mmol)を添加した。反応物を90℃で12時間加熱し、その後、室温まで冷却した。真空中で溶媒を除去し、ジオキサン20mL及び飽和炭酸水素ナトリウム水溶液20mL中に残留物を溶解した。得られた混合物に、ジ−tert−ブチルジカルボネート7.8g(35.8mmol)を添加した。反応物を室温で12時間撹拌した。この反応混合物を酢酸エチルで希釈し、1N 塩酸でpH1に酸性化した。層を分離し、水層を酢酸エチルで2回に分けて抽出した。有機抽出物を合わせ、食塩水で洗浄し、硫酸ナトリウムで乾燥させ、真空中で濃縮した。フラッシュクロマトグラフィーによる精製を行い(シリカゲル、66:34 ヘキサン/酢酸エチル)、所望の生成物を得た。LC/MS 429.1(M+1)。
室温にて、ジクロロメタン(5mL)中のステップF由来の生成物(100mg、0.23mmol)の撹拌溶液に、トリフルオロ酢酸(1mL)を添加した。室温で1時間撹拌した後、真空中で溶媒を除去し、残留物をHPLC(YMC Pro−C18カラム、グラジエント溶出、10−90% アセトニトリル/0.1%TFAを含有する水)で精製し、表題化合物を得た。LC/MS 329.0(M+1)。
ステップA:トランス−4−(4−ブロモフェニル)−3−ブテン−2−オン
無水ジクロロメタン(500mL)中に溶解させた4−ブロモケイ皮酸25.0g(110mmol)に、EDC(28.8g、150mmol)、HOBT(20.3g、150mmol)、N,O−ジメチルヒドロキシルアミン塩酸塩(14.6g、150mmol)及びN,N’−ジイソプロピルエチルアミン(23mL、150mmol)を添加した。室温で24時間撹拌した後、反応物を濃縮し、次いで、10%塩酸水溶液400mLで希釈した。次に、得られた混合物を300mLのジエチルエーテルで3回抽出し、有機相を合わせ、10%塩酸、飽和炭酸水素ナトリウム水溶液及び飽和塩水(各100mL)で順次洗浄した。次に、有機相を硫酸マグネシウムで乾燥させ、ろ過し、真空中で蒸発させて、粘性のある油状物質としてWeinrebアミドを得、さらに精製せずに使用した。この油状物質に対して、無水テトラヒドロフラン300mLを添加し、得られた溶液を−78℃に冷却した。この溶液に対して、臭化メチルマグネシウム60mL(180mmol、ジエチルエーテル中3N)を添加した。この撹拌混合物を1時間にわたり、ゆっくりと0℃まで温めた。次に、水及び5% 塩酸水溶液(各100mL)を用いて、慎重にこの混合物を停止させ、次に、濃縮してテトラヒドロフランを除去した。得られた混合物を、300mLのジエチルエーテルで3回に分けて抽出し、有機相を合わせ、5%塩酸、飽和炭酸水素ナトリウム水溶液及び飽和塩水(各100mL)で順次洗浄した。次に、有機相を硫酸マグネシウムで乾燥させ、ろ過し、真空中で蒸発させて、粘性のある油を得た。Biotage(R)システム(シリカゲル、0−15%酢酸エチル/ヘキサングラジエント)を用いたフラッシュクロマトグラフィーによってこの未精製物質を精製し、淡黄色の結晶性固体として表題化合物を得た。LC/MS 225.0(M+1),227.0(M+3)。
トルエン100mLに溶解させたステップA由来のケトン、5.55g(24.7mmol)に、(R)−2−メチル−CBS−オキサザボロリジン触媒3.7mL(3.7mmol、トルエン中1M)を添加し、得られた混合物を室温で15分間撹拌した。この混合物を−78℃まで冷却し、トルエン30mL中のカテコールボラン4.0mL(37.1mmol)を、30分間にわたり滴下添加した。添加後、そのスラリーを−78℃で60分間撹拌し、ゆっくりと均一化した。次に、この溶液を−78℃でさらに4時間(反応時間は4ないし24時間と異なる。)、TLCで出発物質が完全に消失したことが分かるまで撹拌した。次に、反応混合物を水100mLで希釈し、各100mLのジエチルエーテルで3回に分けて、得られた混合物を抽出した。次に、有機相を合わせ、各100mLの1N NaOH水溶液で2回、各100mLの5%塩酸溶液で2回、飽和塩水100mLで洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させ、未精製のろう状固体を得た。次に、Biotage(R)システム(シリカゲル、0−20% 酢酸エチル/ヘキサン勾配)を用いたフラッシュクロマトグラフィーによってこの未精製物質を精製して、無色の結晶性固体としてアルコールを得た。この化合物をヘキサン中で再結晶化させ、無色結晶としてアルコールを得た(Mosherのエステル分析により、96% ee)。LC/MS 209.0(M−H2O+1),211.0(M−H2O+3)。
無水ジクロロメタン(300mL)中に溶解させたステップB由来のアルコール12.6g(55mmol)に、EDC(23g、120mmol)、HOBT(16g、120mmol)、N−(tert−ブトキシカルボニル)グリシン(21g、120mmol)及びN,N’−ジイソプロピルエチルアミン(19mL、120mmol)を添加した。5時間後、この混合物を濃縮し、10%塩酸水溶液200mLで希釈した。次に、得られた混合物を各300mLのジエチルエーテルで3回に分けて抽出し、有機相を合わせ、5%塩酸、飽和炭酸水素ナトリウム水溶液及び飽和塩水(各100mL)で順次洗浄した。次に、有機相を硫酸マグネシウムで乾燥させ、ろ過し、真空中で蒸発させて、粘性のある油状物質として未精製物質を回収した。Biotage(R)システム(シリカゲル、0−20% 酢酸エチル/ヘキサングラジエント)を用いたフラッシュクロマトグラフィーによってこの未精製物質を精製して、無色の結晶性固体として表題化合物を得た。LC/MS 328.1(M−tBu+1),330.1(M−tBu+3)。
無水テトラヒドロフラン(50mL)中のステップC由来のエステル(18.1g、47mmol)を、−78℃に予め冷却しておいたリチウムヘキサメチルジシラジド溶液105mL(105mmol、テトラヒドロフラン中1M)に、カニューレを用いて添加した。その温度で10分間撹拌した後、−78℃にて、塩化亜鉛溶液55mL(55mmol、ジエチルエーテル中1M)を添加した。得られた混合物を−78℃で5時間撹拌し、室温までゆっくりと3時間にわたり温めた。室温でさらに2時間撹拌した後、水及び5%塩酸(各100mL)でこの混合物の反応を停止させた。次に、得られた混合物を各300mLの酢酸エチルで3回に分けて抽出し、有機相を合わせ、5%塩酸、飽和炭酸水素ナトリウム水溶液及び飽和塩水(各200mL)で順次洗浄した。次に、有機相を硫酸マグネシウムで乾燥させ、ろ過し、真空中で蒸発させて、黄色の泡状物質として未精製物質を得た。LC/MS 384.1(M+1),386.1(M+3)。1:1 ジエチルエーテル/メタノール500mL中に、この未精製物質を溶解し、0℃に冷却した。黄色が持続するようになるまで、トリメチルシリルジアゾメタン溶液(75mL、150mmol、ヘキサン中2M)を分割して添加した。室温に温めた後、この溶液をさらに8時間撹拌し、次に真空中で濃縮した。Biotage(R)システム(シリカゲル、0−15% 酢酸エチル/ヘキサン勾配)を用いたフラッシュクロマトグラフィーによってこの未精製物質を精製して、無色の油として表題化合物を得た。LC/MS 298.0(M−Boc+1),300.0(M−Boc+3)。
テトラヒドロフラン(THF)600mL中のメチル(βS)−4−ブロモ−N−(tert−ブトキシカルボニル)−β−[(1E)−プロパ−1−エニル]−L−フェニルアラニナート(ステップD)25g(62.8mmol)の溶液に、メタノール200mL及び1N 水酸化ナトリウム水溶液200mL(200mmol)を連続して添加した。その反応混合物を室温で3時間撹拌し、次に、メタノール及びTHFを減圧下で除去した。その水性混合物に、1N 塩酸 250mLを添加し、この混合物を酢酸エチル(3x300mL)で抽出した。合わせた有機抽出物を塩水(300mL)で洗浄し、次に硫酸ナトリウムで乾燥させ、濾過し、真空中で濃縮して、カルボン酸を得て、さらに精製せずに用いた。
DMF50mL中の、ステップEで得た(βS)−4−ブロモ−N−(tert−ブトキシカルボニル)−β−[(1E)−プロパ−1−エニル]−L−フェニルアラニン2.00g(5.21mmol)及び(3S)−3−フルオロピロリジン塩酸塩(中間体1)0.781g(6.25mmol)の溶液に、N、N’−ジイソプロピルエチルアミン2.71mL(15.6mmol)、0.774g(5.73mmol)のHOBT及び1.10g(5.73mmol)のEDCを添加した。室温で16時間後に、0.5Mの炭酸水素ナトリウム水溶液200mLを添加して反応を停止させた。この混合物を各300mLの酢酸エチルで2回抽出した。各200mLの0.5M 炭酸水素ナトリウム水溶液、塩水で2回さらに有機層を洗浄し、乾燥させ(硫酸マグネシウム)、真空中で濃縮して、透明な油を得た。シリカゲルクロマトグラフィー(20%から50%酢酸エチル/ヘキサンへのグラジエント)による精製によって、無色の油として表題化合物を得た。LC/MS 355.3(M+1)及び357.3(M+3)。
水30mL中の、過ヨウ素酸ナトリウム2.37g(11.1mmol)、炭酸カリウム0.306g(2.21mmol)及び過マンガン酸カリウム0.087g(0.550mmol)の溶液に、室温で、tert−ブタノール70mLを添加した。tert−ブタノール20mL中の、ステップFで得た(3S)−1−[(2S,3S,4E)−3−(4−ブロモフェニル)−2−(tert−ブトキシカルボニルアミノ)ヘキサ−4−エノイル]−3−フルオロピロリジン2.21g(1.01mol)の溶液に、得られた懸濁液を添加した。次いで、得られた混合物を40℃まで9時間温めた。次いで、この混合物を室温まで冷却し、1Mの亜硫酸水素ナトリウム水溶液200mL上に注いだ。得られた混合物を各250mLの酢酸エチルで2回抽出した。有機層を塩水で洗浄し、乾燥させ(硫酸マグネシウム)、真空中で濃縮し、黄色の固体を得た。この物質を26mLのDMF中に溶かした。この溶液に、THF中のジメチルアミンの2.0M溶液1.33mL(2.65mmol)を添加した後、N、N’−ジイソプロピルエチルアミン1.15mL(6.63mmol)、0.328g(2.43mmol)のHOBT及び0.466g(2.43mmol)のEDCを添加した。室温で16時間攪拌した後に、0.5Mの炭酸水素ナトリウム水溶液100mLでこの反応物を希釈した。この混合物を各250mLの酢酸エチルで2回抽出した。各100mLの0.5M 炭酸水素ナトリウム水溶液、塩水で有機層を2回さらに洗浄し、乾燥させ(硫酸マグネシウム)、真空中で濃縮して、黄色の油を得た。シリカゲルクロマトグラフィー(0%から10%MeOH/酢酸エチルへのグラジエント)による精製によって、黄色の固体として表題化合物を得た。LC/MS 385.8(M+1)及び387.8(M+3)。
密封可能なチューブ中のトルエン1mL中の、ステップGで得た(3S)−1−[(2S,3S)−3−(4−ブロモフェニル)−2−(tert−ブトキシカルボニルアミノ)−3−(ジメチルアミノカルボニル)−1−オキソプロパニル]−3−ジフルオロピロリジン56.9mg(0.117mmol)の溶液に、N−メチル−4−メトキシベンゾアミド48.5mg(0.293mmol)、固体の炭酸カリウム48.5mg(0.351mmol)及び固体のヨウ化銅(I)22.3mg(0.117mmol)を添加した。この混合物に、トルエン1mL中のN,N’−ジメチルエチレンジアミン0.0378mLの溶液を添加した。10分間窒素を通気させて、この混合物を脱気した後、密封し、48時間110℃まで温めた。この混合物を室温まで冷却し、1:1:1の酢酸エチル/ジクロロメタン/メタノールの溶液で、この固体を練和し、ろ過した。この溶液を真空中で濃縮し、分取用薄層クロマトグラフィー(10%メタノール/酢酸エチル)によって、得られた残留物を精製して、白色の固体として表題化合物を得た。LC/MS 509.2(M+1)。
ジクロロメタン1mL中の、ステップHで得たN−(4−{(1S,2S)−2−[(tert−ブトキシカルボニル)アミノ]−1−[(ジメチルアミノ)カルボニル]−3−[(3S)−3−フルオロピロリジン−1−イル]−3−オキソプロピル}フェニル)−N−メチル−4−メトキシベンゾアミド17.6mg(0.0346mmol)の溶液に、トリフルオロ酢酸1mLを添加した。室温で1時間後に、この溶液を真空中で濃縮した。逆相分取用HPLC(10%から80%へのアセトニトリル水溶液)による精製によって、泡状の白色固体として表題化合物を得た。LC/MS 471.2(M+1)。
ステップA:(3S)−1−[(2S,3S)−2−(tert−ブトキシカルボニルアミノ)−3−(ジメチルアミノカルボニル)−1−オキソ−3−[4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)フェニル]プロパニル]−3−フルオロピロリジン
実施例5のステップGで得られた化合物5.48g(11.27mmol)、ビス(ピナコラート)二ホウ素(5.72g、22.54mmol)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(1.84g、2.25mmol)及び酢酸カリウム(3.32g、33.81mmol)の混合物に窒素を吹き込んだ。次いで、メチルスルホキシド(60mL)を添加し、この溶液を脱気し、窒素を再度充填した後、この反応混合物を90℃で一晩温めた。得られた黒色混合物を室温まで冷却し、セライトのパッドを通してろ過し、酢酸エチルでろ塊を洗浄した。合わせたろ液を、さらに酢酸エチル(500mL)で希釈し、飽和炭酸水素ナトリウム水溶液、水及び飽和塩水で順次洗浄し、乾燥させ(硫酸ナトリウム)、減圧下で濃縮した。フラッシュクロマトグラフィー(シリカゲル、1メタノール/酢酸エチル溶出液)によって、粘性のある暗色の残留物を精製し、表題化合物を得た。LC−MS 534.3(M+1)。
0℃のテトラヒドロフラン75mL中の、上記ステップAで得られた物質4.0g(7.50mmol)の溶液に、30%過酸化水素の水溶液1.25mLを添加した。1時間後に、この溶液を室温まで温め、攪拌を2時間継続した。この反応混合物を1Nの塩酸でpH4に酸性化した。さらに水を添加し、酢酸エチルで水層を抽出した(3回)。合わせた有機抽出物を、飽和塩水で洗浄し、乾燥させ(硫酸マグネシウム)、減圧下で濃縮した。フラッシュクロマトグラフィー(シリカゲル、メタノール/酢酸エチル)によって、得られた油を精製し、表題化合物を得た。LC−MS 424.2(M+1)。
8mLのジクロロメタン中の、ステップBで得られた化合物324mg(0.766mmol)の懸濁液に、4−シアノフェニルホウ酸(225mg、1.532mmol)、酢酸銅(139mg、0.766mmol)、ピリジン(0.31mL、3.830mmol)及び4Å分子篩を添加した。室温で、空気のバルーン下にて、この混合物を40時間攪拌した後、セライトのパッドを通して濾過した。減圧下でろ液を濃縮して油を得、フラッシュクロマトグラフィー(シリカゲル、メタノール/ジクロロメタン溶出液)によって精製し、表題化合物を得た。LC−MS 525.3(M+1)。
ジクロロメタン(1mL)中の、上記ステップCで得られた化合物(4.0mg)の溶液を、トリフルオロ酢酸(1mL)で処理した。1時間後に、減圧下で溶液を濃縮し、表題化合物を得た。LC−MS 425.3(M+1)。
経口用薬学的組成物の具体的な実施形態として、100mgの効力錠剤は、本発明の何れかの化合物100mg、微結晶性セルロース268mg、クロスカルメロースナトリウム20mg及びステアリン酸マグネシウム4mgから構成される。まず、活性な、微結晶性セルロースとクロスカルメロースを混合する。次いで、ステアリン酸マグネシウムによってこの混合物を潤滑化し、圧縮して錠剤にする。
Claims (28)
- 構造式Iの化合物:
(式中、
各nは、独立に、0、1又は2であり;
m及びpは、独立に、0又は1であり;
qは、1又は2であり;
Xは、CH2、S、CHF又はCF2であり;
Arは、置換されていないフェニル又は1ないし5個のR3置換基で置換されたフェニルであり;
R1は、水素又はシアノであり;
R2は、
C1−10アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
C2−10アルケニル(アルケニルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−アリール(アリールは、置換されていないか、又はヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−ヘテロアリール(ヘテロアリールは、置換されていないか、又はヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)n−ヘテロシクリル(ヘテロシクリルは、置換されていないか、又はオキソ、ヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル(シクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)nCOOH、
(CH2)nCOOC1−6アルキル、
(CH2)nCONR4R5(式中、R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキルからなる群から独立に選択されるか(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、又は、
R4及びR5は、両者で、それらが結合している窒素原子とともに、アゼチジン、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択され;
R2中の任意のメチレン(CH2)炭素原子は、置換されていないか、又はハロゲン、ヒドロキシ、C1−4アルキル及びC1−4アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし2個の基で置換されており;
各R3は、
ハロゲン、
シアノ、
ヒドロキシ、
C1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
C1−6アルコキシ(アルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
フェニルオキシ(置換されていないか、又はハロゲン、ヒドロキシ、CO2H、シアノ、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−NR4R5、
(CH2)n−CONR4R5、
(CH2)n−OCONR4R5、
(CH2)n−SO2NR4R5、
(CH2)n−SO2R6、
(CH2)n−NR7SO2R6、
(CH2)n−NR7CONR4R5、
(CH2)n−NR7COR7、
(CH2)n−NR7CO2R6、
(CH2)n−COOH、
(CH2)n−COOC1−6アルキル、
(CH2)q−アリール(アリールは、置換されていないか、又はハロゲン、ヒドロキシ、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル、C3−6シクロアルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)q−ヘテロアリール(ヘテロアリールは、置換されているか、又はヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル、C3−6シクロアルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)q−ヘテロシクリル(ヘテロシクリルは、置換されていないか、又はオキソ、ヒドロキシ、ハロゲン、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル、C3−6シクロアルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル(シクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
からなる群から独立に選択され、
R3中の任意のメチレン(CH2)炭素原子は、置換されていないか、又はハロゲン、ヒドロキシ、C1−4アルキル及びC1−4アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし2個の基で置換されており;
R6は、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)からなる群から独立に選択され、R6中の任意のメチレン(CH2)炭素原子は、置換されていないか、又はハロゲン、ヒドロキシ、及びC1−4アルキル(置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし2個の基で置換されており;
各R7は、水素又はR6である。) - *の印が付された炭素原子が、式Ia:
- R1に結合されている**の印が付された炭素原子が、式Ib:
- 構造式Ic:
- *の印が付された炭素原子及び**の印が付された炭素原子が、式Id:
- 構造式Ie
- *の印が付された炭素原子が、式If:
- 構造式Ig
- *の印が付された炭素原子が、式Ih:
- R2は、
C1−6アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
C2−6アルケニル(アルケニルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル(シクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、CO2H、C1−6アルキルオキシカルボニル、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)nCOOH、
(CH2)nCOOC1−6アルキル、及び
(CH2)nCONR4R5(R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキルからなる群から独立に選択されるか(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)、又は
R4及びR5は、両者で、それらが結合している窒素原子とともに、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択され、
R2中の任意のメチレン(CH2)炭素原子は、置換されていないか、又はハロゲン、ヒドロキシ、C1−4アルキル及びC1−4アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし2個の基で置換されている、請求項1に記載の化合物。 - R2が、
C1−3アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル、
COOH、
COOC1−6アルキル、
CONR4R5(R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキル(式中、アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)からなる群から独立に選択されるか、又は
R4とR5は、両者で、それらが結合している窒素原子とともに、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル、及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択される、請求項10に記載の化合物。 - R2が、
メチル、
エチル、
CH2−シクロプロピル、
COOH、
COOMe、
COOEt、
CONMe2、
CONH2、
CONHMe、
CONHEt、
ピロリジン−1−イルカルボニル、
アゼチジン−1−イルカルボニル、及び
[(テトラゾール−5−イル)アミノ]カルボニル
からなる群から選択される、請求項11に記載の化合物。 - *の印が付された炭素原子、R1に結合されている**の印が付された炭素原子、及びR2に結合されている***の印が付された炭素原子が、式Ii:
R2が、
C1−6アルキル(アルキルは、置換されていないか、又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている)、
(CH2)n−C3−6シクロアルキル、
COOH、
COOC1−6アルキル、及び
CONR4R5(R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)からなる群から独立に選択されるか、又は、
R4とR5は、両者で、それらが結合している窒素原子とともに、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、該複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択され;
各R3が、
ハロゲン、
ヒドロキシ、
C1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
C1−6アルコキシ(アルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
フェニルオキシ(置換されていないか、又はハロゲン、ヒドロキシ、シアノ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
(CH2)n−C3−6シクロアルキル(シクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし3個の置換基で置換されている。)、
からなる群から独立に選択される、請求項1に記載の化合物。 - R2が、
メチル、
エチル、
CH2−シクロプロピル、
COOH、
COOMe、
COOEt、
CONMe2、
CONH2、
CONHMe、
CONHEt、
ピロリジン−1−イルカルボニル、
アゼチジン−1−イルカルボニル、及び
[(テトラゾール−5−イル)アミノ]カルボニル
からなる群から選択される、請求項13に記載の化合物。 - R3が、
フルオロ、
クロロ、
ブロモ、
トリフルオロメチル、
トリフルオロメトキシ、及び
メトキシ
からなる群から選択される、請求項14に記載の化合物。 - 構造式Ijを有する請求項1に記載の化合物。
Xは、CH2、S、CHF、又はCF2であり;
Arは、置換されていないフェニル又は1ないし5個のR3置換基で置換されたフェニルであり;
R1は、水素又はシアノであり;
R2は、
C1−6アルキル(アルキルは、置換されていないか又はハロゲン若しくはヒドロキシから独立に選択される1ないし5個の置換基で置換されている)、
(CH2)n−C3−6シクロアルキル、
COOH、
COOC1−6アルキル、及び
CONR4R5(R4及びR5は、水素、テトラゾリル、チアゾリル、(CH2)n−フェニル、(CH2)n−C3−6シクロアルキル及びC1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されており、フェニル及びシクロアルキルは、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)からなる群から独立に選択されるか、又は、
R4とR5は、両者で、それらが結合している窒素原子とともに、ピロリジン、ピペリジン、ピペラジン及びモルホリンから選択される複素環を形成しており、前記複素環は、置換されていないか、又はハロゲン、ヒドロキシ、C1−6アルキル及びC1−6アルコキシ(アルキル及びアルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)から独立に選択される1ないし5個の置換基で置換されている。)
からなる群から選択され;
各R3は、
ハロゲン、
ヒドロキシ、
C1−6アルキル(アルキルは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
C1−6アルコキシ(アルコキシは、置換されていないか、又は1ないし5個のハロゲンで置換されている。)、
フェニルオキシ(置換されていないか、又はハロゲン及びシアノから独立に選択される1ないし3個の置換基で置換されている。)、及び
フェニル(CH2)nCON(Me)−(フェニルは、置換されていないか、又はハロゲン、トリフルオロメチル及びC1−4アルキルから独立に選択される1ないし3個の置換基で置換されている。)、
からなる群から独立に選択される。) - からなる群から選択される構造式を有する請求項16の化合物又は薬学的に許容されるそれらの塩。
- 請求項1に記載の化合物と薬学的に許容される担体とを含む薬学的組成物。
- 請求項1に記載の化合物の有効量を哺乳動物に投与することを含む、ジペプチジルペプチダーゼ−IV酵素活性の阻害を必要とする哺乳動物においてジペプチジルペプチダーゼ−IV酵素活性を阻害する方法。
- 請求項1に記載の化合物の治療的有効量を哺乳動物に投与することを含む、糖尿病の治療を必要とする哺乳動物において糖尿病を治療する方法。
- 請求項1に記載の化合物の治療的有効量を哺乳動物に投与することを含む、非インシュリン依存性(2型)糖尿病の治療を必要としている哺乳動物において非インシュリン依存性(2型)糖尿病を治療する方法。
- 請求項1に記載の化合物の治療的有効量を哺乳動物に投与することを含む、高血糖の治療を必要としている哺乳動物において高血糖を治療する方法。
- 請求項1に記載の化合物の治療的有効量を哺乳動物に投与することを含む、肥満の治療を必要としている哺乳動物において肥満を治療する方法。
- 請求項1に記載の化合物の治療的有効量を哺乳動物に投与することを含む、異脂肪血症、高脂血症、高トリグリセリド血症、高コレステロール血症、低HDL及び高LDLの群から選択される一又は複数の脂質障害の治療を必要とする哺乳動物において前記脂質障害を治療する方法。
- 請求項1に記載の化合物の治療的有効量を哺乳動物に投与することを含む、(1)高血糖、(2)低グルコース耐性、(3)インシュリン抵抗性、(4)肥満、(5)脂質障害、(6)異脂肪血症、(7)高脂血症、(8)高トリグリセリド血症、(9)高コレステロール血症、(10)低HDLレベル、(11)高LDLレベル、(12)アテローム性動脈硬化症及びその続発症、(13)血管再狭窄、(14)過敏性腸症候群、(15)クローン病及び潰瘍性大腸炎を含む炎症性腸疾患、(16)その他の炎症性症状、(17)膵炎、(18)腹部肥満、(19)神経変性疾患、(20)網膜症、(21)腎症、(22)神経障害、(23)シンドロームX、(24)卵巣アンドロゲン過剰症(多嚢胞性卵巣症候群)、並びにインシュリン抵抗性を要素とする他の疾患、からなる群から選択される一又は複数の症状の治療を必要としている哺乳動物を治療する方法。
- (a)第二のジペプチジルペプチダーゼIV阻害剤;
(b)PPARγアゴニスト、PPARα/γデュアルアゴニスト、PPARαアゴニスト、ビグアニド、及びタンパク質チロシンホスファターゼ−1B阻害剤からなる群から選択されるインシュリン増感剤;
(c)インシュリン又はインシュリン模倣物;
(d)スルホニル尿素又はその他のインシュリン分泌促進物質;
(e)α−グルコシダーゼ阻害剤;
(f)グルカゴン受容体アンタゴニスト;
(g)GLP−1、GLP−1模倣物又はGLP−1受容体アゴニスト;
(h)GIP、GIP模倣物又はGIP受容体アゴニスト;
(i)PACAP、PACAP模倣物、又はPACAP受容体アゴニスト;
(j)(i)HMG−CoA還元酵素阻害剤、(ii)金属イオン封鎖剤、(iii)ニコチニルアルコール、ニコチン酸又はその塩、(iv)PPARαアゴニスト、(v)PPARα/γデュアルアゴニスト、(vi)コレステロール吸収阻害剤、(vii)アシルCoA:コレステロールアシルトランスフェラーゼ阻害剤、及び(viii)抗酸化剤などの、コレステロール降下剤;
(k)PPARδアゴニスト;
(1)肥満抑制化合物;
(m)回腸の胆汁酸輸送体阻害剤;
(n)抗炎症剤;並びに
(o)降圧剤、
からなる群から選択される一又は複数の活性成分がさらに追加された、請求項18に記載の薬学的組成物。 - PPARα/γデュアルアゴニストがKRP−297である、請求項26に記載の薬学的組成物。
- PPARα/γデュアルアゴニストKRP−297とともに、請求項1に記載の化合物の治療的有効量を哺乳動物に投与することを含む、糖尿病の治療を必要としている哺乳動物において糖尿病を治療する方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US43083602P | 2002-12-04 | 2002-12-04 | |
| PCT/US2003/037825 WO2004050022A2 (en) | 2002-12-04 | 2003-11-26 | Phenylalanine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2006510630A true JP2006510630A (ja) | 2006-03-30 |
| JP2006510630A5 JP2006510630A5 (ja) | 2006-09-28 |
Family
ID=32469540
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004557341A Pending JP2006510630A (ja) | 2002-12-04 | 2003-11-26 | 糖尿病を治療又は予防するためのジペプチジルペプチダーゼ阻害剤としてのフェニルアラニン誘導体 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7309714B2 (ja) |
| EP (1) | EP1578414A4 (ja) |
| JP (1) | JP2006510630A (ja) |
| AU (1) | AU2003297564A1 (ja) |
| CA (1) | CA2508487A1 (ja) |
| WO (1) | WO2004050022A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011509916A (ja) * | 2007-01-16 | 2011-03-31 | カイノス・メディシン・インコーポレイテッド | チアゾリジン誘導体およびその製造方法 |
Families Citing this family (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE380175T1 (de) * | 2001-06-27 | 2007-12-15 | Smithkline Beecham Corp | Pyrrolidine als dipeptidyl peptidase inhibitoren |
| EP1554256B1 (en) | 2002-07-15 | 2009-12-09 | Merck & Co., Inc. | Piperidino pyrimidine dipeptidyl peptidase inhibitors for the treatment of diabetes |
| AU2003275404A1 (en) | 2002-10-07 | 2004-05-04 | Merck & Co., Inc. | Antidiabetic beta-amino heterocyclic dipeptidyl peptidase inhibitors |
| EP1556362B1 (en) | 2002-10-18 | 2008-03-26 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| AU2004206812A1 (en) | 2003-01-17 | 2004-08-05 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7388019B2 (en) | 2003-01-31 | 2008-06-17 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7560455B2 (en) | 2003-05-14 | 2009-07-14 | Merck & Co., Inc. | 3-Amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| CA2526770A1 (en) | 2003-06-06 | 2004-12-23 | Merck & Co., Inc. | Fused indoles as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| EP1638950A4 (en) | 2003-06-17 | 2010-06-30 | Merck Sharp & Dohme | CYCLOHEXYLGLYCIN DERIVATIVES AS INHIBITORS OF DIPEPTIDYL PEPTIDASE FOR THE TREATMENT OR PREVENTION OF DIABETES |
| ATE417832T1 (de) | 2003-07-31 | 2009-01-15 | Merck & Co Inc | Hexahydrodiazepinone als inhibitoren des dipeptidylpeptidase-iv zur behandlung bzw. prävention von diabetes |
| JP2007510651A (ja) * | 2003-11-04 | 2007-04-26 | メルク エンド カムパニー インコーポレーテッド | 糖尿病の治療又は予防のためのジペプチジルペプチダーゼ−iv阻害剤としての縮合フェニルアラニン誘導体 |
| WO2005108382A1 (en) | 2004-05-04 | 2005-11-17 | Merck & Co., Inc. | 1,2,4-oxadiazole derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| DK1753748T3 (da) | 2004-05-12 | 2009-10-05 | Pfizer Prod Inc | Prolinderivater og deres anvendelse som dipeptidyl-peptidase-IV-inhibitorer |
| EP1756106A4 (en) * | 2004-05-18 | 2008-12-17 | Merck & Co Inc | CYCLOHEXYLALANINE DERIVATIVES AS INHIBITORS OF DIPEPTIDYL PEPTIDASE-IV FOR THE TREATMENT OR PREVENTION OF DIABETES |
| EP1794120B1 (en) | 2004-07-23 | 2012-04-11 | Nuada, LLC | Peptidase inhibitors |
| ATE473742T1 (de) | 2004-08-23 | 2010-07-15 | Merck Sharp & Dohme | Kondensierte triazolderivate als dipeptidylpeptidase-iv-hemmer zur behandlung bzw. prävention von diabetes |
| CN101031300A (zh) * | 2004-10-01 | 2007-09-05 | 默克公司 | 用于治疗或预防糖尿病的作为二肽基肽酶-ⅳ抑制剂的氨基哌啶化合物 |
| DOP2006000008A (es) * | 2005-01-10 | 2006-08-31 | Arena Pharm Inc | Terapia combinada para el tratamiento de la diabetes y afecciones relacionadas y para el tratamiento de afecciones que mejoran mediante un incremento de la concentración sanguínea de glp-1 |
| NZ556614A (en) | 2005-01-26 | 2010-10-29 | Allergan Inc | 3-aryl-3-hydroxy-2-amino-propionic acid amides, 3-heteroayryl-3-hydroxy-2-amino-propionic acid amides and related compounds having analgesic and/or immunostimulant activity |
| CN101374523B (zh) | 2005-09-14 | 2012-04-11 | 武田药品工业株式会社 | 用于治疗糖尿病的二肽基肽酶抑制剂 |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| CA2633167A1 (en) * | 2005-12-16 | 2007-07-12 | Merck & Co., Inc. | Pharmaceutical compositions of combinations of dipeptidyl peptidase-4 inhibitors with metformin |
| GB0526291D0 (en) | 2005-12-23 | 2006-02-01 | Prosidion Ltd | Therapeutic method |
| US20090156465A1 (en) | 2005-12-30 | 2009-06-18 | Sattigeri Jitendra A | Derivatives of beta-amino acid as dipeptidyl peptidase-iv inhibitors |
| SG10201404220TA (en) | 2006-04-11 | 2014-10-30 | Arena Pharm Inc | Methods of using gpr119 receptor to identify compounds useful for increasing bone mass in an individual |
| PE20071221A1 (es) | 2006-04-11 | 2007-12-14 | Arena Pharm Inc | Agonistas del receptor gpr119 en metodos para aumentar la masa osea y para tratar la osteoporosis y otras afecciones caracterizadas por masa osea baja, y la terapia combinada relacionada a estos agonistas |
| MX2008013130A (es) | 2006-04-12 | 2008-11-19 | Probiodrug Ag | Inhibidores de enzima. |
| US8324383B2 (en) | 2006-09-13 | 2012-12-04 | Takeda Pharmaceutical Company Limited | Methods of making polymorphs of benzoate salt of 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-benzonitrile |
| JP5379692B2 (ja) | 2006-11-09 | 2013-12-25 | プロビオドルグ エージー | 潰瘍、癌及び他の疾患の治療のためのグルタミニルシクラーゼの阻害薬としての3−ヒドロキシ−1,5−ジヒドロ−ピロール−2−オン誘導体 |
| TW200838536A (en) | 2006-11-29 | 2008-10-01 | Takeda Pharmaceutical | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| SI2091948T1 (sl) | 2006-11-30 | 2012-07-31 | Probiodrug Ag | Novi inhibitorji glutaminil ciklaze |
| US8093236B2 (en) | 2007-03-13 | 2012-01-10 | Takeda Pharmaceuticals Company Limited | Weekly administration of dipeptidyl peptidase inhibitors |
| EP2142514B1 (en) | 2007-04-18 | 2014-12-24 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| EP2108960A1 (en) * | 2008-04-07 | 2009-10-14 | Arena Pharmaceuticals, Inc. | Methods of using A G protein-coupled receptor to identify peptide YY (PYY) secretagogues and compounds useful in the treatment of conditons modulated by PYY |
| AR077642A1 (es) | 2009-07-09 | 2011-09-14 | Arena Pharm Inc | Moduladores del metabolismo y el tratamiento de trastornos relacionados con el mismo |
| MX2012002993A (es) | 2009-09-11 | 2012-04-19 | Probiodrug Ag | Derivados heterociclicos como inhibidores de ciclasa glutaminilo. |
| US9181233B2 (en) | 2010-03-03 | 2015-11-10 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| SG183229A1 (en) | 2010-03-10 | 2012-09-27 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011127051A1 (en) | 2010-04-06 | 2011-10-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| EP2560953B1 (en) | 2010-04-21 | 2016-01-06 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
| EP2571876B1 (en) | 2010-05-21 | 2016-09-07 | Merck Sharp & Dohme Corp. | Substituted seven-membered heterocyclic compounds as dipeptidyl peptidase-iv inhibitors for the treatment of diabetes |
| CA2812061A1 (en) | 2010-09-22 | 2012-03-29 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| EP2648517B1 (en) | 2010-12-06 | 2015-08-05 | Merck Sharp & Dohme Corp. | Tricyclic heterocycles useful as dipeptidyl peptidase-iv inhibitors |
| DK2686313T3 (en) | 2011-03-16 | 2016-05-02 | Probiodrug Ag | Benzimidazole derivatives as inhibitors of glutaminyl cyclase |
| US20140018371A1 (en) | 2011-04-01 | 2014-01-16 | Arena Pharmaceuticals, Inc. | Modulators Of The GPR119 Receptor And The Treatment Of Disorders Related Thereto |
| WO2012145361A1 (en) | 2011-04-19 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012145604A1 (en) | 2011-04-22 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| US20140038889A1 (en) | 2011-04-22 | 2014-02-06 | Arena Pharmaceuticals, Inc. | Modulators Of The GPR119 Receptor And The Treatment Of Disorders Related Thereto |
| WO2012170702A1 (en) | 2011-06-08 | 2012-12-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| CN103702562A (zh) | 2011-06-29 | 2014-04-02 | 默沙东公司 | 用于制备手性二肽基肽酶-iv抑制剂的方法 |
| US9051329B2 (en) | 2011-07-05 | 2015-06-09 | Merck Sharp & Dohme Corp. | Tricyclic heterocycles useful as dipeptidyl peptidase-IV inhibitors |
| WO2013055910A1 (en) | 2011-10-12 | 2013-04-18 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2013122920A1 (en) | 2012-02-17 | 2013-08-22 | Merck Sharp & Dohme Corp. | Dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2014018355A1 (en) | 2012-07-23 | 2014-01-30 | Merck Sharp & Dohme Corp. | Treating diabetes with dipeptidyl peptidase-iv inhibitors |
| US9315508B2 (en) | 2012-07-23 | 2016-04-19 | Merck Sharp & Dohme Corp. | Treating diabetes with dipeptidyl peptidase-IV inhibitors |
| TWI500613B (zh) | 2012-10-17 | 2015-09-21 | Cadila Healthcare Ltd | 新穎之雜環化合物 |
| WO2014074668A1 (en) | 2012-11-08 | 2014-05-15 | Arena Pharmaceuticals, Inc. | Modulators of gpr119 and the treatment of disorders related thereto |
| CN112521369A (zh) | 2013-03-13 | 2021-03-19 | 福马治疗股份有限公司 | 用于抑制fasn的化合物及组合物 |
| WO2016014324A1 (en) | 2014-07-21 | 2016-01-28 | Merck Sharp & Dohme Corp. | Process for preparing chiral dipeptidyl peptidase-iv inhibitors |
| GB201415598D0 (en) | 2014-09-03 | 2014-10-15 | Univ Birmingham | Elavated Itercranial Pressure Treatment |
| MX2017011586A (es) | 2015-03-09 | 2017-10-26 | Intekrin Therapeutics Inc | Metodos para el tratamiento de enfermedad de higado graso no alcoholico y/o lipodistrofia. |
| CN110996951A (zh) | 2017-04-03 | 2020-04-10 | 科赫罗斯生物科学股份有限公司 | 治疗进行性核上性麻痹的PPARγ激动剂 |
| EP3461819B1 (en) | 2017-09-29 | 2020-05-27 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
| CN110483432A (zh) * | 2018-05-14 | 2019-11-22 | 中国科学院上海药物研究所 | 一类丙烯酸类化合物及其制备方法、药物组合物和用途 |
| TWI767148B (zh) | 2018-10-10 | 2022-06-11 | 美商弗瑪治療公司 | 抑制脂肪酸合成酶(fasn) |
| US10793554B2 (en) | 2018-10-29 | 2020-10-06 | Forma Therapeutics, Inc. | Solid forms of 4-(2-fluoro-4-(1-methyl-1H-benzo[d]imidazol-5-yl)benzoyl)piperazin-1-yl)(1-hydroxycyclopropyl)methanone |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09509921A (ja) * | 1993-12-03 | 1997-10-07 | フェーリング ベスローテン フェンノートシャップ | 酵素インヒビター |
| WO2001040180A2 (en) * | 1999-11-30 | 2001-06-07 | Ferring Bv | N-substituted 2-cyanopyrrolidines and their use as antidiabetic agents |
| JP3234236B2 (ja) * | 1996-08-28 | 2001-12-04 | 塩野義製薬株式会社 | チアゾリル―アラニン残基を有する新規ペプチド誘導体 |
| JP2002265439A (ja) * | 2001-03-08 | 2002-09-18 | Mitsubishi Pharma Corp | シアノピロリジン誘導体およびその医薬用途 |
| WO2002076450A1 (en) * | 2001-03-27 | 2002-10-03 | Merck & Co., Inc. | Dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| JP2002539244A (ja) * | 1999-03-23 | 2002-11-19 | フェリング ベスローテン フェンノートシャップ | 成長促進用組成物 |
| JP2002363167A (ja) * | 2001-03-28 | 2002-12-18 | Lab Servier | 新規α−アミノ酸スルホニル化合物、その製造法、およびそれらを含む医薬組成物 |
| JP2002363157A (ja) * | 2001-05-15 | 2002-12-18 | Lab Servier | 新規α−アミノ酸化合物、その調製方法及びそれを含有する医薬組成物 |
| WO2003002530A2 (en) * | 2001-06-27 | 2003-01-09 | Smithkline Beecham Corporation | Pyrrolidines as dipeptidyl peptidase inhibitors |
| JP4413863B2 (ja) * | 2002-11-07 | 2010-02-10 | メルク エンド カムパニー インコーポレーテッド | 糖尿病の治療又は予防のためのジペチジルペプチダーゼ阻害剤としてのフェニルアラニン誘導体 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4386090A (en) | 1980-12-22 | 1983-05-31 | Delalande S.A. | Nitrogen containing 2,3-dihydro naphthalenes, compositions and use |
| DE122010000020I1 (de) | 1996-04-25 | 2010-07-08 | Prosidion Ltd | Verfahren zur Senkung des Blutglukosespiegels in Säugern |
| US6011155A (en) | 1996-11-07 | 2000-01-04 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| CO5150173A1 (es) | 1998-12-10 | 2002-04-29 | Novartis Ag | Compuestos n-(glicilo sustituido)-2-cianopirrolidinas inhibidores de peptidasa de dipeptidilo-iv (dpp-iv) los cuales son efectivos en el tratamiento de condiciones mediadas por la inhibicion de dpp-iv |
| US6432969B1 (en) | 2000-06-13 | 2002-08-13 | Novartis Ag | N-(substituted glycyl)-2 cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| UA74912C2 (en) | 2001-07-06 | 2006-02-15 | Merck & Co Inc | Beta-aminotetrahydroimidazo-(1,2-a)-pyrazines and tetratriazolo-(4,3-a)-pyrazines as inhibitors of dipeptylpeptidase for the treatment or prevention of diabetes |
-
2003
- 2003-11-26 CA CA002508487A patent/CA2508487A1/en not_active Abandoned
- 2003-11-26 WO PCT/US2003/037825 patent/WO2004050022A2/en active Application Filing
- 2003-11-26 AU AU2003297564A patent/AU2003297564A1/en not_active Abandoned
- 2003-11-26 EP EP03812453A patent/EP1578414A4/en not_active Withdrawn
- 2003-11-26 US US10/537,476 patent/US7309714B2/en active Active
- 2003-11-26 JP JP2004557341A patent/JP2006510630A/ja active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09509921A (ja) * | 1993-12-03 | 1997-10-07 | フェーリング ベスローテン フェンノートシャップ | 酵素インヒビター |
| JP3234236B2 (ja) * | 1996-08-28 | 2001-12-04 | 塩野義製薬株式会社 | チアゾリル―アラニン残基を有する新規ペプチド誘導体 |
| JP2002539244A (ja) * | 1999-03-23 | 2002-11-19 | フェリング ベスローテン フェンノートシャップ | 成長促進用組成物 |
| WO2001040180A2 (en) * | 1999-11-30 | 2001-06-07 | Ferring Bv | N-substituted 2-cyanopyrrolidines and their use as antidiabetic agents |
| JP2002265439A (ja) * | 2001-03-08 | 2002-09-18 | Mitsubishi Pharma Corp | シアノピロリジン誘導体およびその医薬用途 |
| WO2002076450A1 (en) * | 2001-03-27 | 2002-10-03 | Merck & Co., Inc. | Dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| JP2002363167A (ja) * | 2001-03-28 | 2002-12-18 | Lab Servier | 新規α−アミノ酸スルホニル化合物、その製造法、およびそれらを含む医薬組成物 |
| JP2002363157A (ja) * | 2001-05-15 | 2002-12-18 | Lab Servier | 新規α−アミノ酸化合物、その調製方法及びそれを含有する医薬組成物 |
| WO2003002530A2 (en) * | 2001-06-27 | 2003-01-09 | Smithkline Beecham Corporation | Pyrrolidines as dipeptidyl peptidase inhibitors |
| JP4413863B2 (ja) * | 2002-11-07 | 2010-02-10 | メルク エンド カムパニー インコーポレーテッド | 糖尿病の治療又は予防のためのジペチジルペプチダーゼ阻害剤としてのフェニルアラニン誘導体 |
Non-Patent Citations (1)
| Title |
|---|
| JPN5005010657, STOCKEL−MASCHEK A A, BIOCHIMICA ET BIOPHYSICA ACTA. PROTEIN STRUCTURE AND MOLECULAR ENZYMOLOGY, 20000615, V1479 N1−2, P15−31, NL, ELSEVIEER * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011509916A (ja) * | 2007-01-16 | 2011-03-31 | カイノス・メディシン・インコーポレイテッド | チアゾリジン誘導体およびその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2004050022A3 (en) | 2004-08-05 |
| WO2004050022A2 (en) | 2004-06-17 |
| WO2004050022B1 (en) | 2004-09-30 |
| EP1578414A2 (en) | 2005-09-28 |
| CA2508487A1 (en) | 2004-06-17 |
| AU2003297564A1 (en) | 2004-06-23 |
| EP1578414A4 (en) | 2007-10-24 |
| US7309714B2 (en) | 2007-12-18 |
| WO2004050022A8 (en) | 2004-12-16 |
| US20060111336A1 (en) | 2006-05-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7309714B2 (en) | Phenylalanine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes | |
| JP4579239B2 (ja) | 糖尿病の治療または予防するためのジペプチジルペプチダーゼ阻害剤としてのシクロヘキシルグリシン誘導体 | |
| JP4413863B2 (ja) | 糖尿病の治療又は予防のためのジペチジルペプチダーゼ阻害剤としてのフェニルアラニン誘導体 | |
| JP4352001B2 (ja) | 糖尿病の治療または予防のためのベータ−アミノ複素環式ジペプチジルペプチダーゼ阻害剤 | |
| US7238683B2 (en) | Fused phenylalanine derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes | |
| EP1635818B1 (en) | Fused indoles as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes | |
| US7671073B2 (en) | Cyclohexylalanine derivatives as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes | |
| US7687492B2 (en) | 1,2,4-Oxadiazole derivatives as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes | |
| JP2007500704A (ja) | 糖尿病の治療又は予防のためのジペプチジルペプチダーゼiv−阻害剤としてのヘキサヒドロジアゼピノン | |
| JP2006528693A (ja) | 糖尿病を治療又は予防するためのジペプチジルペプチダーゼ阻害剤としての3−アミノ−4−フェニルブタン酸誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060808 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060808 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100209 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100507 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100514 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100608 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100615 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100706 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20110118 |