JP2004170323A - Screening method for cutaneous disease therapeutic agent - Google Patents
Screening method for cutaneous disease therapeutic agent Download PDFInfo
- Publication number
- JP2004170323A JP2004170323A JP2002338738A JP2002338738A JP2004170323A JP 2004170323 A JP2004170323 A JP 2004170323A JP 2002338738 A JP2002338738 A JP 2002338738A JP 2002338738 A JP2002338738 A JP 2002338738A JP 2004170323 A JP2004170323 A JP 2004170323A
- Authority
- JP
- Japan
- Prior art keywords
- par2
- group
- therapeutic agent
- test substance
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- BCTPSHLVPKFXTF-UHFFFAOYSA-N CC(CCCC1)N1C(c1n[n]2c(C(F)(F)F)cc(-c3ccccc3)nc2c1)=O Chemical compound CC(CCCC1)N1C(c1n[n]2c(C(F)(F)F)cc(-c3ccccc3)nc2c1)=O BCTPSHLVPKFXTF-UHFFFAOYSA-N 0.000 description 1
- ATBJQBRIAPZDIC-UHFFFAOYSA-N OC(c1n[n]2c(C(F)(F)F)cc(-c3ccccc3)nc2c1)=O Chemical compound OC(c1n[n]2c(C(F)(F)F)cc(-c3ccccc3)nc2c1)=O ATBJQBRIAPZDIC-UHFFFAOYSA-N 0.000 description 1
Landscapes
- Investigating Or Analysing Biological Materials (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
【0001】
【発明の属する技術分野】
本発明は、皮膚疾患治療剤のスクリーニング方法、詳しくは、痒みを抑制する皮膚疾患治療剤及びそのスクリーニング方法に関する。
【0002】
【従来の技術】
プロテアーゼ受容体PAR (proteinase−activated receptor) はGタンパク共役7回膜貫通型受容体の一種であり、現在までに4つのPAR;PAR1, PAR2, PAR3, PAR4 がクローニングされている。
PAR2はトリプシン、トリプターゼ、または、血液凝固第VIIa因子もしくは第Xa因子によって、N末端の部分ペプチドが切断され、新しいN末端が生成することによって活性化される。例えば、ヒトPAR2の場合、N末端36残基が切断されて、新たにSLIGKVの配列ではじまるN末端を有するポリペプチドとなって活性化される。また、活性化されて生成するN末端のペプチドの配列に基づく、5〜6個のアミノ酸から成る合成ペプチド(アゴニストペプチド)によっても、活性化されることが知られている(非特許文献1および非特許文献2)。該アゴニストペプチドとしては、SLIGKVの配列及びSLIGRLの配列で表される部分ペプチドやその改変ペプチドtrans−cinnamoyl−LIGRLO等が挙げられる(非特許文献3)。
【0003】
PAR2は、消化液の分泌、神経性炎症、疼痛、アレルギー反応(非特許文献4及び非特許文献5)などの種々の生理活性に関与している。
一方、痒みは皮膚固有の感覚である痒みをひきおこす生理活性物質としては、疾患により異なるが、例えば、ヒスタミン、セロトニン、ロイコトリエンB4、サブスタンスP、VIP、トリプシン(非特許文献6)等が報告されている。痒みを知覚するメカニズムについては不明な点も多いが、痒み刺激に対してヒスタミン等の生理活性物質が肥満細胞等から分泌され、知覚神経の一種であるC繊維を通して脳へと伝達されると考えられている。
上記に関連して、PAR2は、C線維(非特許文献7)及び肥満細胞に発現していること、並びに、PAR2の作用により脱顆粒が生じること(非特許文献8)等が報告されているが、PAR2と皮膚の痒み症状との関係は不明であった。
【非特許文献1】
Dery, Oら、「American Journal of Physiology」1998 第274巻, p.C1429−52
【非特許文献2】
Macfarlane, S.R.ら、「Pharmacological Reviews」2001 第53巻,p 245−282
【非特許文献3】
Al−Ani, B.ら、「Journal of Pharmacology and Experimental Therapeutics」1999 第290巻, p753−760
【非特許文献4】
Sun, G.ら、「Journal of Immunology」2001 第167巻, p1014−1021
【非特許文献5】
Miike, S.ら、「Journal of Immunology」2001 第167巻, p6615−6622
【非特許文献6】
Hagermark, O.ら、 「Seminars in Dermatol」1995 第14巻,p271−276
【非特許文献7】
Steinhoff, M.ら、「Nature Medicine」2000 第6巻, p151−158
【非特許文献8】
Grant, R.S.ら、「Journal of Pharmacology and Experimental Therapeutics」2002 第302巻, p466−474
【0004】
【発明が解決しようとする課題】
本発明の目的は、新規な作用機序に基づく、痒み症状を改善する皮膚疾患治療剤、およびそのスクリーニング方法を提供することにある。
【0005】
【課題を解決するための手段】
本発明者らは、鋭意検討を重ねた結果、PAR2アゴニストペプチドが、マウスにおいて痒み症状を誘発することを見出した。本発明は以上の知見に基づき、完成するに至ったものである。
【0006】
すなわち、本発明は、
[1] 以下の工程(1)及び(2):
(1)被験物質のPAR2阻害活性を評価する工程、
(2)(1)の結果に基づいて、PAR2阻害活性を有する被験物質を皮膚疾患治療剤の有効成分として選別する工程、
を含む、皮膚疾患治療剤のスクリーニング方法、
[2] (1)の工程が、下記の工程(a)〜(d):
(a)被験物質とPAR2を発現可能な細胞とを接触させる工程、
(b)(a)におけるPAR2の発現量を測定する工程、
(c)(b)の値を、被験物質を接触させない対照におけるPAR2の発現量と比較する工程、
(d)(c)の結果に基づいて、被験物質のPAR2阻害活性を評価する工程
を含むことを特徴とする、[1]記載の皮膚疾患治療剤のスクリーニング方法、
[3] (1)の工程が、下記の工程(a)〜(d):
(a)被験物質と、PAR2及びPAR2活性化因子を含む細胞、または該細胞から調製した細胞画分とを接触させる工程、
(b)(a)におけるPAR2由来の機能を測定する工程、
(c)(b)の値を、被験物質を接触させない対照におけるPAR2由来の機能と比較する工程、
(d)(c)の結果に基づいて、被験物質のPAR2阻害活性を評価する工程、
を含むことを特徴とする、[1]記載の皮膚疾患治療剤のスクリーニング方法、
[4] (d)の工程が、以下の工程:
(d−1)被験物質のPAR2阻害活性と、式:
【化2】
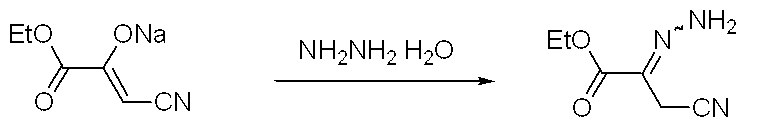
(d−2)前記化合物と同等以上のPAR2阻害活性を示す被験物質を、皮膚疾患治療剤候補化合物として選択する工程、
を含むことを特徴とする、[3]記載のスクリーニング方法。
[5] (1)の工程が、下記の工程(a)〜(d):
(a)被験物質と、PAR2、及びPAR2リガンドを含む水溶液、細胞、または該細胞から調製した細胞膜画分とを接触させる工程、
(b)(a)におけるPAR2に結合したPAR2リガンド量を測定する工程、
(c)(b)の値を、被験物質を接触させない対照における、PAR2に結合したPAR2アゴニスト量と比較する工程、
(d)(c)の結果に基づいて、被験物質のPAR2阻害活性を評価する工程、
を含むことを特徴とする、[1]記載の皮膚疾患治療剤のスクリーニング方法、
[6] 皮膚疾患が、痒みを伴う疾患であることを特徴とする、[1]〜[5]のいずれか記載のスクリーニング方法、
[7] 皮膚疾患治療剤が、痒み症状を改善することを特徴とする治療剤である、[1]〜[6]のいずれか記載のスクリーニング方法、
【0007】
[8] PAR2阻害活性を有する化合物を有効成分として含有する、皮膚疾患治療剤、
[9] PAR2阻害活性を有する化合物が、一般式(1):
【化3】
R3は、水素原子、アルキル基、ハロアルキル基、または置換もしくは無置換のアリール基を表し、
R4およびR5は、独立して、水素原子、ハロゲン原子、アルキル基、アルコキシ基、メチレンジオキシ基、またはハロアルキル基を表す。)
で表される化合物、又はその薬学上許容される塩である、[8]記載の治療剤、
[10] R3がハロアルキル基で表されることを特徴とする、[9]記載の治療剤、
[11] ハロアルキル基がトリフルオロメチル基であることを特徴とする、[9]記載の治療剤、
[12] R1とR2が隣接する炭素原子とともに結合して、アルキル基、アルキルカルボニル基、アリールカルボニル基、アルコキシカルボニル基、フェニル基、またはベンジル基で置換されていてもよい、ピペリジン、パーヒドロアゼピン、モルホリン、チオモルホリン、またはピペラジンを形成していることを特徴とする、[8]〜[10]のいずれか記載の治療剤、
[13] R1とR2が隣接する炭素原子とともに結合して、アルキル基、または、アルコキシカルボニル基で置換されていてもよい、ピペリジン、またはパーヒドロアゼピンを形成していることを特徴とする、[11]記載の治療剤、
[14] 一般式(2):
【化4】
【化5】
【化6】
から選択される基を表し、R3’は、水素原子、アルキル基、ハロアルキル基、またはフェニル基を表し、
R4 ’およびR5’は、独立して、水素原子、ハロゲン原子、アルキル基、またはハロアルキル基を表す。]
で表される化合物、またはその薬学上許容される塩を有効成分として含有する皮膚疾患治療剤、
【0008】
[15] 皮膚疾患が、痒みを伴う疾患であることを特徴とする、[8]〜[14]のいずれか記載の治療剤、
[16] 皮膚疾患治療剤が、痒み症状を改善することを特徴とする治療剤である、[8]〜[14]のいずれか記載の治療剤、
[17] PAR2又はPAR2活性化因子を有効成分として含有する、痒み症状を改善する薬剤を評価するための動物モデルにおける痒み惹起用試薬、
に関するものである。
【0009】
【本発明の実施の形態】
本明細書において、アミノ酸、(ポリ)ペプチド、(ポリ)ヌクレオチドなどの略号による表示は、IUPAC−IUBの規定〔IUPAC−IUB Communication onBiological Nomenclature, Eur. J. Biochem., 138: 9 (1984)〕、「塩基配列又はアミノ酸配列を含む明細書等の作成のためのガイドライン」(日本国特許庁編)、および当該分野における慣用記号に従う。
【0010】
本明細書において、「PAR2」とは、Gタンパク共役7回膜貫通型受容体に属するトリプシンやトリプターゼ等のプロテアーゼによって活性化される受容体(proteinase−activated receptor)蛋白質を表し、そのアミノ酸配列は、ヒトについては、Bohm, S. K. et al. Biochem.l J., 314, 1009−16, 1996 (Genbank Ac. No.U34038) に記載されており、他の動物種のホモログについては、マウス: Nystedt S. et al., J. Biol. Chem., 270, 5950−55, 1995(Genbank Ac. No. Z48043)、及びラット:Saifeddine M. et al., Br. J. Pharmacol., 118, 521−30, 1996(Genbank Ac. No. U61373)についてそれぞれ公知である。
【0011】
本明細書において「PAR2」には、上記のアミノ酸配列を有するものだけでなく、これらと生物学的機能が同等であることを限度として、その同族体(ホモログやスプライスバリアント)、変異体、誘導体、成熟体及びアミノ酸修飾体などが包含される。ここで上記以外の動物種のホモログは、HomoloGene(http://www.ncbi.nlm.nih.gov/HomoloGene/)により同定された遺伝子の塩基配列から演繹的に同定することができる。また変異体には、天然に存在するアレル変異体、天然に存在しない変異体、及び人為的に欠失、置換、付加および挿入されることによって改変されたアミノ酸配列を有する変異体が包含される。なお、上記変異体としては、変異のないタンパク質または(ポリ)ペプチドと、少なくとも70%、好ましくは80%、より好ましくは95%、さらにより好ましくは97%相同なものを挙げることができる。またアミノ酸修飾体には、天然に存在するアミノ酸修飾体、天然に存在しないアミノ酸修飾体が包含され、具体的にはアミノ酸のリン酸化体が挙げられる。
【0012】
本明細書において、「PAR2遺伝子」とは、前記PAR2をコードする遺伝子を表し、その塩基配列は、ヒトについてはBohm, S. K. et al. Biochem.l J., 314, 1009−16, 1996 (Genbank Ac. No. U34038)に記載されており、他の動物種のホモログについては、マウス:Nystedt S. et al., J. Biol. Chem., 270, 5950−55,1995(Genbank Ac. No. Z48043)、ラット:Saifeddine M. et al., Br. J. Pharmacol., 118, 521−30, 1996(Genbank Ac. No. U61373)等についてそれぞれ公知である。
また、本明細書において「PAR2遺伝子」には、上記の塩基配列を有するものだけでなく、これらと生物学的機能が同等であることを限度として、その同族体(ホモログやスプライスバリアント)、変異体等が包含される。
ここで上記以外の動物種のホモログは、HomoloGene(http://www.ncbi.nlm.nih.gov/HomoloGene/)により同定された遺伝子の塩基配列から演繹的に同定することができる。また変異体には、天然に存在するアレル変異体、天然に存在しない変異体、及び人為的に欠失、置換、付加および挿入されることによって改変された塩基配列を有する変異体が包含される。なお、上記変異体としては、変異のないポリヌクレオチドと、少なくとも70%、好ましくは80%、より好ましくは95%、さらにより好ましくは97%相同なものを挙げることができる。
【0013】
「生物学的機能が同等である」とは、具体的には、既知のPAR2アゴニストとの結合活性を保持しているか、又は以下に述べるPAR2の機能を保持してことを表す。
【0014】
本明細書において、「PAR2活性化因子」とは、▲1▼PAR2のN末端の部分ペプチドを加水分解させることによってPAR2を活性化する酵素蛋白質、及び▲2▼▲1▼の酵素蛋白質非存在下にPAR2を活性化することができるアゴニスト化合物を表し、PAR2活性化とは▲1▼▲2▼により生ずる作用を共に含む概念である。
すなわち、PAR2の天然における活性化機構は、PAR2活性化因子によりN末端のペプチド(ヒトPAR2の場合N末端36残基に相当する。)が加水分解されて新たに生成するN末端部分ペプチド(以下、活性型N末端部分ペプチドと称する場合がある。)が、リガンドとしてPAR2に結合し、これによりPAR2由来の生理活性が生じるものであり、具体的な前記▲1▼のPAR2活性化因子(酵素蛋白質)としては、トリプシン、トリプタ−ゼ、血液凝固第Xa因子、又は血液凝固第VIIa因子等を例示することができる。
一方、前記▲2▼のPAR活性化因子(アゴニスト)としては、活性化されたPAR2のN末端部分ペプチド配列に基づくペプチド化合物及びその誘導体が挙げられる。具体的には、SLIGKV(配列番号:1)及びSLIGRL(配列番号:2)で表される部分ペプチドやその改変ペプチドtrans−cinnamoyl−LIGRLO(配列番号:3)等が挙げられる。また、膜結合型セリンプロテアーゼ 1(Takeuchi, T. et al., J. Biol. Chem., 275, 26333−26342 (2000))、及び膜結合型セリンプロテアーゼ 3 (Wallrapp, C. et. al., Cancer Res., 60, 2602−2606 (2000))等もアゴニストとして例示することができる。
【0015】
本明細書において、「PAR2阻害活性を有する化合物」とは、PAR2の機能を阻害する方向へ作用する化合物であれば何ら限定されるものではなく、PAR2の発現量を減少させる活性を有する化合物、PAR2アンタゴニスト活性を有する化合物、PAR2の機能を抑制する化合物等を含む概念である。
ここで「PAR2の発現量を減少させる」とは、PAR2をコードするmRNAの発現量を減少させること、またはPAR2の蛋白質量を減少させることを表す。
「PAR2のアンタゴニスト活性」とは、N末端が切断されて活性化されたPAR2の活性型N末端部分ペプチドとPAR2の結合や、アゴニストとPAR2の結合を阻害する活性を言う。
また、「PAR2の機能」としては、細胞内カルシウムの濃度上昇や細胞内イノシトール3リン酸の産生増加等のGPCRに特有の機能、プロテインキナーゼCもしくはMAP キナーゼによる細胞内タンパク質のリン酸化増加、又は細胞からのプロスタグランジンE2(PGE2)放出促進、又は、哺乳動物における唾液分泌促進等の機能が挙げられる。
【0016】
以下、本発明の実施の形態を詳細に説明する。
本発明の第1の態様は、以下の工程(1)被験物質のPAR2阻害活性を評価する工程、及び(2)(1)の結果に基づいて、PAR2阻害活性を有する被験物質を皮膚疾患治療剤の有効成分として選別する工程を含む、皮膚疾患治療剤のスクリーニング方法である。
上記(1)において、PAR2阻害活性を評価する方法としては、I.PAR2の発現量を測定する方法、II.PAR2の機能を測定する方法、III.PAR2結合阻害活性を測定する方法等が挙げられる。以下それぞれについて、具体的に述べる。
【0017】
I.PAR2の発現量を測定する方法
PAR2の発現量に基づき、PAR阻害活性を評価する方法は、以下の(a)〜(d)の工程:
(a)被験物質とPAR2を発現可能な細胞とを接触させる工程、
(b)(a)におけるPAR2の発現量を測定する工程、
(c)(b)の値を、被験物質を接触させない対照におけるPAR2の発現量と比較する工程、
(d)(c)の結果に基づいて、被験物質のPAR2阻害活性を評価する工程
を含む。
【0018】
ここで、PAR2を発現可能な細胞としては、内在性および外来性を問わず、PAR2を発現する培養細胞全般を挙げることができる。培養細胞においてこれら本発明遺伝子が発現しているか否かは、公知のノーザンブロット法やRT−PCR法にてこれらの遺伝子発現を検出することにより、容易に確認することができる。
当該スクリーニングに用いられる細胞の具体例としては、例えば以下の(1)〜(3):
(1) PAR2を発現している哺乳動物由来細胞
(2)上記PAR2活性化因子で処理した細胞
(3)PAR2をコードする遺伝子を導入した細胞、
等を挙げることができる。
ここで、前記(1)の細胞としては、膵臓組織由来細胞、ケラチノサイト由来細胞、肥満細胞、血管内皮細胞、Tリンパ球、血小板、骨芽細胞、肝細胞、又は各種の株化された細胞が挙げられる。具体的には、MIA PaCa−2細胞(ヒト膵臓癌由来細胞株)、HEK293(ヒト胎児腎臓由来細胞株)、A549(ヒト肺ガン由来細胞株)、HFL1(ヒト胎児肺由来細胞株)、SW982(ヒト滑膜様細胞株)、又はSW1353(ヒト軟骨様細胞株)を例示することができる。
前記(2)において、PAR2活性化因子としては、前記と同じものが挙げられる。処理する細胞としては、前記(1)に記載された細胞が挙げられる。
【0019】
前記(3)の遺伝子を導入した細胞としては、前記(1)及び(2)に挙げた細胞の他、通常遺伝子導入に用いられる宿主細胞、すなわちCOP、L、C127、Sp2/0、NS−1、NIH3T3、ST2等のマウス由来細胞、ラット由来細胞、BHK、CHO等のハムスター由来細胞、COS1、COS3、COS7、CV1、Vero等のサル由来細胞、HeLa、293等のヒト由来細胞、およびSf9、Sf21、High Five等の昆虫由来細胞などが例示される。
【0020】
さらに、本発明のスクリーニング方法に用いられる細胞には、細胞の集合体である組織なども含まれる。
また本発明スクリーニングに際して、被験物質と細胞とを接触させる条件は、特に制限されないが、該細胞が死滅せず且つ本発明遺伝子を発現できる培養条件(温度、pH、培地組成など)を選択するのが好ましい。
【0021】
PAR2の発現量を測定する方法としては、試料として工程(a)の細胞から調製されるRNAを用いるノーザンブロット法、RT−PCR法、DNAチップ解析法、もしくはin situハイブリダイゼーション解析法、又は、試料として工程(a)の細胞から調製される蛋白質を用いるウェスタンブロッティング法などの公知の方法が挙げられる。測定試料は、通常細胞を破砕、希釈、又は遠心分離する等の必要な処理を行い、RNAもしくは蛋白質を含む画分として調製する。
【0022】
ノーザンブロット法を利用する場合は、PAR2遺伝子の塩基配列において連続する少なくとも15塩基を有するポリヌクレオチド及び/又はその相補的なポリヌクレオチドをプローブとして用いることによって、RNA中のPAR2の発現の有無やその発現レベルを検出、測定することができる。具体的には、前記プローブを放射性同位元素(32P、33Pなど:RI)や蛍光物質などで標識し、これを、常法に従ってナイロンメンブレン等にトランスファーした、工程(a)の細胞由来RNAとハイブリダイズさせた後、形成されたプローブ(DNA)とRNAとの二重鎖を、疾患マーカーの標識物(RI若しくは蛍光物質)に由来するシグナルを放射線検出器(BAS−1800II、富士フィルム社製)または蛍光検出器で検出、測定する方法を例示することができる。また、AlkPhos Direct Labelling and Detection System (Amersham Pharmacia Biotech社製)を用いて、該プロトコールに従ってプローブDNAを標識し、前記RNAとハイブリダイズさせた後、前記プローブの標識物に由来するシグナルをマルチバイオイメージャーSTORM860(Amersham Pharmacia Biotech社製)で検出、測定する方法を使用することもできる。
【0023】
RT−PCR法を利用する場合は、本発明の上記プローブと同様の塩基配列をプライマーとして用いることによって、RNA中のPAR2の発現の有無や発現レベルを検出、測定することができる。具体的には、工程(a)の細胞由来RNAから常法に従ってcDNAを調製して、これを鋳型として標的のPAR2の遺伝子の領域が増幅できるように、前記プライマー(上記cDNA(−鎖)に結合する正鎖、+鎖に結合する逆鎖)をこれとハイブリダイズさせて、常法に従ってPCR法を行い、得られた増幅二本鎖DNAを検出する方法を例示することができる。なお、増幅された二本鎖DNAの検出は、上記PCRを予めRIや蛍光物質で標識しておいたプライマーを用いて行うことによって産生される標識二本鎖DNAを検出する方法、産生された二本鎖DNAを常法に従ってナイロンメンブレン等にトランスファーさせて、標識されたプライマーとハイブリダイズさせて検出する方法などを用いることができる。なお、生成された標識二本鎖DNA産物はアジレント2100バイオアナライザ(横河アナリティカルシステムズ社製)などで測定することができる。また、SYBR Green RT−PCR Reagents (Applied Biosystems 社製)で該プロトコールに従ってRT−PCR反応液を調製し、ABI PRISM 7700 Sequence Detection System (Applied Biosystems 社製)で反応させて、該反応物を検出することもできる。
DNAチップ解析を利用する場合は、上記ノーザンハイブリダイゼーション法でプローブとして用いたDNAプローブ(1本鎖または2本鎖)を樹脂上のチップに結合させたものを用意し、これに工程(a)の細胞由来RNAから常法によって調製されたcRNAをハイブリダイズさせて、形成されたDNAとcRNAとの二本鎖を、前記プライマーから調製される標識プライマーと結合させて検出する方法を挙げることができる。
【0024】
また、PAR2を認識する抗体を用いて、ウエスタンブロット法などの公知方法で、PAR2を検出、定量する方法を挙げることができる。
ウエスタンブロット法は、一次抗体を試料と反応後、二次抗体として125Iなどの放射性同位元素、蛍光物質、ホースラディッシュペルオキシターゼ(HRP)などの酵素等で標識した標識抗体(一次抗体に結合する抗体)を用い、得られる標識化合物の放射性同位元素、蛍光物質などに由来するシグナルを放射線測定器(BAS−1800II:富士フィルム社製など)、蛍光検出器などで検出し、測定することによって実施できる。また、一次抗体として本発明疾患マーカーを用いた後、ECL Plus Western Blotting Detection System (アマシャム ファルマシアバイオテク社製)を用いて、該プロトコールに従って検出し、マルチバイオイメージャーSTORM860(アマシャム ファルマシアバイオテク社製)で測定することもできる。
【0025】
ここで前記抗体は、ポリクローナル抗体であっても、またそのモノクローナル抗体であってもよく、その製造方法は、すでに周知であり、本発明の抗体もこれらの常法に従って製造することができる(Current Protocol in Molecular Biology, Chapter 11.12〜11.13(2000))。具体的には、本発明の抗体がポリクローナル抗体の場合には、常法に従って大腸菌等で発現し精製したPAR2を用いて、あるいは常法に従ってこれら本発明タンパク質の部分アミノ酸配列を有するオリゴペプチドを合成して、家兎等の非ヒト動物に免疫し、該免疫動物の血清から常法に従って得ることが可能である。一方、モノクローナル抗体の場合には、常法に従って大腸菌等で発現し精製したPAR2、またはこれらタンパク質の部分アミノ酸配列を有するオリゴペプチドをマウス等の非ヒト動物に免疫し、得られた脾臓細胞と骨髄腫細胞とを細胞融合させて調製したハイブリドーマ細胞の中から得ることができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley and Sons. Section 11.4〜11.11)。
【0026】
抗体の作製に免疫抗原として使用されるPAR2は、前記公知文献に記載された配列情報に基づいて、DNAクローニング、各プラスミドの構築、宿主へのトランスフェクション、形質転換体の培養および培養物からのタンパク質の回収の操作により得ることができる。これらの操作は、当業者に既知の方法、あるいは文献記載の方法(Molecular Cloning, T.Maniatis et al., CSH Laboratory (1983),DNA Cloning, DM. Glover, IRL PRESS (1985))などに準じて行うことができる。
【0027】
上記のように測定した被験物質存在下におけるPAR2発現量(工程(b)の測定値)を、被験物質を接触させない対照におけるPAR2の発現量と比較することによって、被験物質のPAR2阻害活性を評価することができる。
具体的には、被験物質を添加した上記(1)〜(3)のいずれかの細胞におけるPAR2の発現レベルが、被験物質を添加しない細胞(対照)のそのレベルに比して低くなることをもって、行うことができる。また、PAR2の発現に発現誘導物質を必要とする細胞を用いる場合は、発現誘導物質としてPAR2活性化因子を用いることによって誘導される発現が被験物質の存在によって制御されること、すなわち発現誘導物質の存在下で被験物質を接触させた細胞の遺伝子発現が、発現誘導物質存在下で被験物質を接触させなかった対照細胞(正のコントロール)に比して低くなることを指標として、当該被験物質をPAR2阻害活性を有する化合物として評価することができる。
【0028】
上記のように評価した被験物質のPAR2阻害活性を指標として、皮膚疾患治療剤の候補物質を選別することができる。例えば、被験物質存在下におけるPAR2の発現量が、被験物質を接触させない対照細胞における発現量と比較して、10%、好ましくは30%、特に好ましくは50%以上減少する被験物質を候補物質として選別することができる。
【0029】
またPAR2の発現レベルの検出及び定量は、これを制御する遺伝子領域(発現制御領域)に、例えばルシフェラーゼ遺伝子などのマーカー遺伝子をつないだ融合遺伝子を導入した細胞株を用いて、マーカー遺伝子由来のタンパク質の活性を測定することによっても実施できる。従って、PAR2の発現制御物質のスクリーニング方法には、かかるマーカー遺伝子の発現量を指標として標的物質を探索する方法も包含されるものであり、この意味において、PAR2の発現制御領域とマーカー遺伝子との融合遺伝子を用いたスクリーニングも本発明の範疇である。
【0030】
なお、上記マーカー遺伝子としては、発光反応や呈色反応を触媒する酵素の構造遺伝子が好ましい。具体的には、上記のルシフェラーゼ遺伝子のほか、分泌型アルカリフォスファターゼ遺伝子、クロラムフェニコール・アセチルトランスフェラーゼ遺伝子、βグルクロニダーゼ遺伝子、βガラクトシダーゼ遺伝子、及びエクオリン遺伝子などのレポーター遺伝子が例示できる。
また、ここで前記本発明遺伝子の発現制御領域は、例えば該遺伝子の転写開始部位上流約1kb、好ましくは約2kbを用いることができる。
【0031】
本発明遺伝子の発現制御領域は、例えば(i)5’−RACE法(例えば、5’full Race Core Kit(宝酒造社製)等を用いて実施される)、オリゴキャップ法、S1プライマーマッピング等の通常の方法により、5’末端を決定するステップ;(ii)Genome Walker Kit(クローンテック社製)等を用いて5’−上流領域を取得し、得られた上流領域について、プロモーター活性を測定するステップ;を含む手法等により同定することができる。また融合遺伝子の作成、およびマーカー遺伝子由来の活性測定は公知の方法で行うことができる。
【0032】
II.PAR2の機能を測定する方法
PAR2の機能に基づき、PAR阻害活性を評価する方法は、以下の(a)〜(d)の工程:
(a)被験物質と、PAR2及びPAR2活性化因子を含む細胞、または該細胞から調製した細胞画分とを接触させる工程、
(b)(a)におけるPAR2由来の機能を測定する工程、
(c)(b)の値を、被験物質を接触させない対照におけるPAR2由来の機能と比較する工程、
(d)(c)の結果に基づいて、被験物質のPAR2阻害活性を評価する工程、
を含む。
【0033】
ここで、PAR2の公知の機能に基づく如何なる機能測定方法をも利用することができる。すなわち、PAR2の公知の機能・活性測定系に被験物質を添加し、当該PAR2の公知の機能を抑制する被験物質を、PAR2阻害活性を有する化合物として評価することができる。
【0034】
本発明のスクリーニングは、PAR2を含む細胞または該細胞から調製した細胞画分と、被験物質とを接触させることにより行うことができる。
【0035】
また、本発明のスクリーニング方法に用いられる細胞としては、内在性及び外来性を問わず、PAR2を発現し得る細胞を挙げることができる。該細胞としては、具体的には、前記Iにおいて(1)〜(3)に記載した細胞などを用いることができる。
また本発明のスクリーニング方法に用いられる細胞画分とは、上記細胞に由来する各種の画分を意味し、これには、例えば細胞膜画分などが含まれる。
【0036】
前記スクリーニングにおいて用いられるPAR2は、公知のタンパク質であり、DNAクローニング、各プラスミドの構築、宿主へのトランスフェクション、形質転換細胞の培養、および必要に応じて培養物からのタンパク質の回収の操作により得ることができる。これらの操作は、当業者に既知の方法、あるいは文献記載の方法(Molecular Cloning, T.Maniatis et al., CSH Laboratory (1983), DNACloning, DM. Glover, IRL PRESS (1985))などに準じて行うことができる。
【0037】
PAR2の機能を測定する方法としては、具体的には、細胞内カルシウム濃度の上昇活性、アラキドン酸遊離(Kong, W. et. al., Proc. Nat. Acad. Sci. USA, 94, 8884−8889 (1997); McHowat, J., et al., J. Urology, 165, 2063−2067(2001))、細胞内イノシトール3リン酸の産生促進、細胞膜電位変動、c−fos活性化(Hoogerwerf, W. A., et al., J. Neurosci., 21, 9036−9042 (2001); Coelho, A. M., et. al., Gastroenterology, 122, 1035−1047 (2002))、又はプロテインキナーゼCもしくはMAPキナーゼによる細胞内蛋白質のリン酸化促進活性等のGPCR特有の機能を測定する方法、または細胞からのプロスタグランジンE2(PGE2)放出促進活性、哺乳動物における唾液分泌促進活性等が挙げられる。
該生理活性は、公知の方法、または市販の測定用キットを用いて測定することができる。すなわち、PAR2を含有する細胞をマルチウェルプレート等に培養する。新鮮な培地、あるいは細胞毒性を示さない適当なバッファーに交換し、被験化合物、および/またはリガンド等を添加して一定時間インキュベートした後、細胞を抽出、あるいは上清液を回収して生成した産物をそれぞれ該当する方法に従って定量する。生理活性の指標とする物質(例えばアラキドン酸など)の生成が、細胞が含有する分解酵素によって測定不能な場合は、該分解酵素に対する阻害剤を併用することもできる。具体的な測定方法を以下に例示する。
【0038】
▲1▼Fluorescent imaging plate reader(FLIPR)法
細胞内カルシウム濃度上昇活性の測定方法としては、ヒト胎児腎臓由来細胞株HEK293細胞、ヒト肺ガン由来細胞株A549、またはヒト胎児肺由来細胞株HFL1等のPAR2を発現し得る培養細胞株に、Fluo−3 AM(Molecular Probe社)等のカルシウム結合性蛍光プローブ、及びprobenecid(色素を細胞内に保持するために、multiple drug−resistance pumpの阻害剤として用いる。)を含む培地で培養し、PAR2アゴニスト(NH2−SLIGRL−CONH2等の合成ペプチド)もしくはトリプシン等のPAR2活性化因子を添加して、更に一定時間(例えば30分〜2時間)培養した後、PAR2を活性化することによって変動する細胞内カルシウム濃度を、蛍光シグナルの変化としてFLIPR(Molecular Devices社)にて検出する。検出は、488nmのアルゴンレーザーで細胞を励起し、カルシウムイオンが結合したFluo−3の蛍光を500−560nmの波長でとらえることによって行う。例えば、リガンド添加直後から60秒後までの蛍光強度から、添加前の値をバックグラウンドとして差し引いた値を活性とすることができる。被験物質を添加する場合はPAR2活性化因子を添加する前に添加することが好ましい。被験物質のPAR2機能阻害活性は、被験物質存在下におけるカルシウムイオン量が、被験物質非存在下におけるカルシウムイオン量に比して減少するか否かを指標にして評価することができる。
尚、上記において、PAR2の発現ベクターと、Gq蛋白、もしくはGqs5蛋白(Gqs5とは、C末端に、Gs蛋白のC末端アミノ酸5残基を置換したGq蛋白を意味する。)、もしくはGqi5蛋白(Gqi5とは、C末端に、Gi蛋白のC末端アミノ酸5残基を置換したGq蛋白を意味する。)の発現ベクターを作製し、CHO−K1細胞等に遺伝子導入試薬Lipofectoamine (Invitrogen社)等を用いて共導入し、形質転換細胞を調製して用いることもできる。
【0039】
▲2▼イノシトール3リン酸の産生量を測定する方法
ヒト膵臓癌由来細胞株 MIA PaCa−2細胞等の培養細胞株に、PAR2アゴニスト(NH2−SLIGRL−CONH2等の合成ペプチド)もしくはトリプシン等のPAR2活性化因子を添加して、PAR2を活性化する。一定時間後にトリクロロ酢酸等の酸を加えて反応を停止させた後、細胞を回収し、ジエチルエーテル等の有機溶媒を含む水溶液等を用いて細胞内のイノシトール3リン酸を抽出し、イノシトール3リン酸の量を定量することができる。
定量方法としては、[3H]アッセイシステム(Amersham、TRK1000;IP3抗体及び[3H]IP3を用いて生体内IP3量を測定するシステム)で定量する方法が挙げられる。
【0040】
▲3▼プロテインキナーゼC活性化測定方法
ヒト膵臓癌由来細胞株、MIA PaCa−2細胞等の培養細胞株に、[32P]ATP存在下で、PAR2アゴニスト(H−SLIGRL−NH2等の合成ペプチド)もしくはトリプシン等のPAR2活性化因子を添加して、PAR2を活性化する。一定時間後に0℃に冷却することによって反応を停止させた後、細胞をホモゲナイズし、バッファー(例えば、0.25mM sucrose、20mM triethanolamine、2mM EDTA、0.5mM EGTA、1mM DTE、10ug/ml Leupeptin、1mM PMSF溶液)を加えて超音波処理後に遠心分離を行い、上清▲1▼と沈渣を共に回収する。沈渣を更にバッファー(例えば、TritonX−100含有バッファー)中でホモゲナイズし、遠心分離を行って上清▲2▼を回収する。前記上清▲1▼及び▲2▼をDEAEイオン交換カラムなどを用いてPKC画分を分離精製し、PKCの基質である[32P]ATP由来の[32P]リン酸量を測定することにより、PKCの量を定量することができる。
【0041】
▲4▼PGE2量の測定
ヒト滑膜様細胞株SW982細胞を被験物質存在下に、炭酸ガス培養装置内で一定時間培養した後、アゴニストペプチドを添加し、更に一定時間(例えば24時間)培養する。培養上清中のプロスタグランジンE2(PGE2)の量を、プロスタグランジンE2 EIA (Cayman chemical 514010) を用いて測定することができる。候補物質の選択は、被験物質の存在下におけるPGE2の量が、被験物質非存在下におけるPGE2に比して減少するか否かを指標にして行うことができる。
【0042】
上記のように測定した被験物質存在下におけるPAR2の機能(工程(b)の測定値)を、被験物質を接触させない対照におけるPAR2の機能と比較することによって、被験物質のPAR2阻害活性を評価することができる。
具体的には、被験物質を添加した上記(1)〜(3)のいずれかの細胞におけるPAR2の機能レベルが、被験物質を添加しない細胞(対照)のそのレベルに比して低くなることをもって、行うことができる。
【0043】
上記のように評価した被験物質のPAR2阻害活性を指標として、皮膚疾患治療剤の候補物質を選別することができる。例えば、被験物質存在下におけるPAR2の機能レベルが、被験物質を接触させない対照細胞における機能レベルと比較して、10%、好ましくは30%、特に好ましくは50%以上低下する被験物質を候補物質として選別することができる。
【0044】
III. PAR2結合阻害活性を測定する方法
PAR2は受容体であるので、受容体結合阻害活性に基づき、PAR2阻害活性を評価する方法は、以下の工程(a)〜(d):
(a)被験物質と、PAR2、及びPAR2リガンドを含む水溶液、細胞、または該細胞から調製した細胞膜画分とを接触させる工程、
(b)(a)におけるPAR2に結合したPAR2アゴニスト量を測定する工程、
(c)(b)の値を、被験物質を接触させない対照における、PAR2に結合したPAR2アゴニスト量と比較する工程、
(d)(c)の結果に基づいて、被験物質のPAR2阻害活性を評価する工程、
を含む。
ここで、PAR2のリガンド(受容体結合物質)としては、PAR2アゴニスト、PAR2アンタゴニスト、及びそれらを標識化した標識リガンドが挙げられる。また、公知アゴニスト化合物の誘導体をペプチド合成等により調製したものを用いることもできる。
【0045】
具体的には、被験物質存在下におけるPAR2に対するリガンド(アゴニスト化合物等)の結合量を測定し、当該結合量を被験物質非存在下でのPAR2に対するリガンド(アゴニスト化合物等)の結合量(対照結合量)と比較する。
【0046】
この場合、前記PAR2とリガンドとの結合阻害は、▲1▼受容体に結合することによって、リガンドのPAR2への結合を阻害する態様のものであってもよいし、▲2▼リガンドに結合することによって、リガンドのPAR2への結合を阻害する態様のものであってもよい。但し、とりわけ▲1▼の態様の場合は、PAR2に結合してもリガンドと同じ作用を発揮しない被験物質を選択することが必要である。
このため、上記のスクリーニング方法で選別された被験物質は更に、選択された被験物質の中から、更にリガンドと同じ作用を有する被験物質、又はリガンドの作用を拮抗する被験物質を選択する工程に供することが好ましい。具体的には、上記II.に記載された方法を挙げることができる。
【0047】
ここで、PAR2は、精製物(単離物)又はその水溶液であっても良いし、当該PAR2を含有する細胞またはその細胞画分の形態であっても良い。 ここでPAR2を含む水溶液としては、例えばPAR2を含む水溶液の他、これらのタンパク質を含む細胞溶解液、細胞破砕液、核抽出液あるいは細胞の培養上清などを例示することができる。
本発明スクリーニングで用いるPAR2を含有する細胞は、当該受容体を天然に発現している細胞を用いても良いし、またPAR2をコードする遺伝子を細胞に導入して作製した形質転換細胞を用いても良い。
【0048】
前記形質転換細胞は、Molecular Cloning 2nd Edt., Cold Spring Harbor Laboratory Press (1989)等の基本書に従い、当業者にとって公知の方法で調製することができる。例えば、前記受容体のcDNAをpCAGGS(Gene 108,193−199(1991))、pcDNA1.1、pcDNA3.1誘導体(インビトロジェン社)などの公知の発現ベクターに挿入する。その後、適当な宿主に導入し、培養することにより、導入した受容体のDNAに対応するタンパク質を発現させた形質転換細胞を作製することができる。宿主としては、一般的に広く普及している、CHO細胞、C127細胞、BHK21細胞、COS細胞などを用いることができるが、これに限定されることなく、酵母、細菌、昆虫細胞などを用いることもできる。
【0049】
PAR2のcDNAを有する発現ベクターの宿主細胞への導入方法としては、公知の発現ベクターの宿主細胞への導入方法であれば、どのような方法でもよく、例えばリン酸カルシウム法(J. Virol., 52, 456−467(1973))、LT−1(Panvera社製)を用いる方法、遺伝子導入用リピッド(Lipofectamine、Lipofectin;Gibco−BRL社製)を用いる方法などが挙げられる。
前記PAR2を天然に発現する細胞、および前記PAR2を発現する形質転換細胞は、そのままスクリーニングに用いることができるが、細胞膜をスクリーニングに用いる場合は、例えば以下のようにして細胞膜画分を得ることができる。すなわちまず細胞に低張バッファーを添加し、細胞を低張破壊した後、ホモジナイズし、遠心分離することにより細胞膜画分の沈殿物を得る。そしてこの沈殿物をバッファーに懸濁することにより、PAR2を含有する細胞膜画分を得ることができる。得られた細胞膜画分は、抗体を結合させたカラム等により常法で精製することもできる。
【0050】
リガンドとしては、アゴニスト化合物をそのままで用いても良いし、任意の標識物質で標識された標識化アゴニスト化合物を用いることもできる。ここで標識物質としては、放射性同位体(3H、14C、35S、125I等)、蛍光物質(Molecular Probes社Alexa Protein Labeling Kits等)、化学発光物質(Assay Designs社Chemiluminescence Labeling Kit等)などを例示することができる。
【0051】
受容体結合阻害活性の測定は、具体的には、以下の方法が例示される。すなわち、受容体(PAR2)を含有する水溶液(通常緩衝液が用いられる)、前記細胞若しくは細胞膜画分に、10−3〜10−10Mの適当な濃度に調製した被験化合物溶液(通常溶媒には水もしくは緩衝液が用いられるが、溶解度に応じてエタノールやDMSOを添加することもできる)を加えた後、リガンドを加え、一定時間(通常、10分〜2時間)反応させる。その後遠心分離等により上清を分離してリガンド量を計測する。あるいは、形質転換細胞もしくは細胞膜画分を含む沈殿物を界面活性剤、塩基を含む溶液に溶解した後溶液に含まれるリガンド量を計測することもできる。ここでリガンドが放射性同位元素で標識された標識化アゴニスト化合物であれば、放射活性を測定すればよい。また、リガンド量は、HPLCなどで試料を精製して計測してもよい。例えば、標識化されていないリガンドを用いる場合は、HPLCを用いてリガンドを含む画分を分離してUV(例えば220nmや254nm)で検出することもできる。
【0052】
上記の数値を、被験化合物の代わりに溶媒をブランクとして用いて実施した場合の値(対照結合量)と比較することにより、被験化合物が、受容体と標識リガンドの結合を阻害するか否かを評価することができる。すなわち候補物質のスクリーニングは、被験物質存在下での結合量が、被験物質非存在下での結合量に比して、減少するか否かを指標にして行うことができる。
具体的な阻害率(%)については、以下の式:
{1−(被験物質を添加した場合にPAR2と結合した標識アゴニストリガンド量)/(被験物質非添加時におけるPAR2と結合した標識アゴニストリガンド量)}X100
で算出することによって求めることができる。
【0053】
上記のように測定した被験物質の阻害率に基づき、被験物質のPAR2阻害活性を評価することができる。例えば、阻害率が、10%以上、好ましくは30%以上、特に好ましくは50%以上である被験物質を候補物質として選別することができる。
【0054】
本発明スクリーニング方法によってスクリーニングされる被験物質(候補物質)は、制限されないが、核酸(本発明遺伝子のアンチセンスヌクレオチドを含む)、ペプチド、タンパク質、有機化合物、無機化合物などであり、本発明スクリーニングは、具体的にはこれらの被験物質またはこれらを含む試料(被験試料)を上記細胞および/または組織と接触させることにより行われる。かかる被験試料としては、被験物質を含む細胞抽出液、遺伝子ライブラリーの発現産物、合成低分子化合物、合成ペプチド、天然化合物などが挙げられるが、これらに制限されない。
【0055】
本発明の第2の態様は、PAR2阻害活性を有する化合物を有効成分として含有する、皮膚疾患治療剤に関する。PAR2阻害活性を有する化合物、又はその薬学上許容される塩は、皮膚の痒み症状を抑える効果を有し、皮膚疾患治療剤、詳しくは痒み症状を伴う皮膚疾患治療剤として有効である。
ここで、PAR2阻害活性を有する化合物は、上記スクリーニング方法で述べたPAR2の発現や機能を抑制する化合物であれば、特に限定は無く、合成低分子化合物、合成ペプチド、天然化合物、蛋白質、核酸、糖化合物等が挙げられる。好ましくは、分子量200〜2000、更に好ましくは分子量300〜1000の化合物であり、例えば、実施例18に記載された方法において、10μg/mlにおいて30%以上のPAR2阻害活性を示す化合物である。
【0056】
PAR2阻害活性を有する化合物として、具体的には、前記式(1)で表される化合物又はその薬学上許容される塩が挙げられる。
式(1)において、アルキル基とは炭素数1〜10のアルキル基を表し、好ましくは炭素数1〜6のアルキル基を、更に好ましくは炭素数1〜4のアルキル基を表す。具体的にはメチル基、エチル基、プロピル基、1−メチルエチル基、またはブチル基等が挙げられる。
式(1)において、アルコキシ基とは炭素数1〜10のアルコキシ基を表し、好ましくは炭素数1〜6のアルコキシ基を、更に好ましくは炭素数1〜4のアルコキシ基を表す。具体的にはメトキシ基、エトキシ基、プロポキシ基、1−メチルエトキシ基、またはブトキシ基等が挙げられる。
式(1)において、ハロゲン原子とは、フッ素原子、塩素原子、臭素原子、またはヨウ素原子を表し、好ましくは、フッ素原子または塩素原子を表す。
本明細書において、ハロアルキル基とは、同一または異なる、1〜3個のハロゲン原子を含む炭素数1〜2のハロアルキル基を表し、具体的には、トリフルオロメチル基、ジフルオロメチル基、1,1,1−トリフルオロエチル基、または2,2−ジフルオロエチル基等が挙げられる。好ましくは、トリフルオロメチル基が挙げられる。
式(1)において、ヘテロシクロアルカンとしては、ピロリジン、ピペリジン、ピペラジン、モルホリン、チオモルホリン、チオモルホリンオキシド、チオモルホリンジオキシド、パーヒドロアゼピン、または、パーヒドロジアゼピン等が挙げられる。好ましくは、ピロリジン、ピペリジン、ピペラジン、または、パーヒドロアゼピンが挙げられる。
前記ヘテロシクロアルカンが置換されている場合の置換基としては、アルキル基、置換もしくは無置換のアリールカルボニル基、アルキルカルボニル基、アルコキシカルボニル基、置換もしくは無置換のアリール基、または置換もしくは無置換のアリールアルキル基が挙げられる。
好ましくは、炭素原子上で置換されている場合の置換基としては、炭素数1〜4のアルキル基、または炭素数2〜4のアルコキシカルボニル基が挙げられる。
また、前記ヘテロシクロアルカンが窒素原子上で置換されている場合の置換基としては、炭素数1〜4のアルキル基、炭素数2〜4のアルコキシカルボニル基、炭素数1〜4のアルキルカルボニル基、置換もしくは無置換のアリールカルボニル基、置換もしくは無置換のアリール基、または置換もしくは無置換のアリールアルキル基が挙げられる。
前記置換もしくは無置換のアリールカルボニル基,置換もしくは無置換のアリール基、または置換もしくは無置換のアリールアルキル基におけるアリールとしては、フェニル、1−ナフチル、または2−ナフチルが挙げられ、特に好ましくはフェニルが挙げられる。
また、前記置換アリールカルボニル基、置換アリール基、または置換アリールアルキル基における置換基としては、ハロゲン原子、炭素数1〜4のアルキル基、炭素数1〜4のアルコキシ基、またはメチレンジオキシ基等が挙げられる。
【0057】
一般式(1)で表される化合物において、R1およびR2は好ましくは、隣接する炭素原子とともに結合して、アルキル基、アリールカルボニル基、アルキルカルボニル基、またはアルコキシカルボニル基で置換されていてもよい1〜2個の窒素原子、0〜1個の酸素原子、および0〜1個の硫黄原子を含むヘテロシクロアルカンを形成しており、該へテロシクロアルカンは、好ましくは、ピペリジン、ピペラジン、またはパーヒドロアゼピンである。
あるいは、一般式(1)で表される化合物において、R1が炭素数1〜4のアルキル基を表し、R2が、水酸基、アルコキシ基、ニトリル基、置換もしくは無置換のアリール基、または置換もしくは無置換のヘテロアリール基で置換されていてもよいアルキル基を表すのもまた、好ましい態様の一つである。前記アリール基としてはフェニル基が挙げられる。また、前記へテロアリール基としては、ピリジル基が挙げられる。R2におけるアルキル基の置換基が置換アリール基もしくは置換ヘテロアリール基を表す場合の、アリール基もしくはヘテロアリール基の置換基としては、ハロゲン原子、炭素数1〜4のアルキル基、炭素数1〜4のハロアルキル基、または炭素数1〜4のアルコキシ基が挙げられる。
一般式(1)で表される化合物において、R3におけるアリール基としては、フェニル基が挙げられ、該アリール基が置換されている場合の置換基としては、ハロゲン原子、炭素数1〜4のアルキル基、炭素数1〜2のハロアルキル基、または炭素数1〜4のアルコキシ基が挙げられる。R3は好ましくは炭素数1〜3のアルキル基、フェニル基、または炭素数1〜2のハロアルキル基を表す。特に好ましい例として、トリフルオロメチル基が挙げられる。
一般式(1)で表される化合物において、R4およびR5は、好ましくは、同一または異なって、水素原子、または、ハロゲン原子を表す。
【0058】
一般式(2)において、R6は、好ましくは、水素原子、炭素数2〜4のアルキルカルボニル基、またはベンゾイル基を表す。
一般式(2)において、R7は、好ましくは、水素原子、炭素数1〜3のアルキル基を表し、更に好ましくは、水素原子、メチル基、を表す。
一般式(2)において、R8は、好ましくは、水素原子、炭素数1〜3のアルキル基、または炭素数2〜4のアルコキシカルボニル基を表し、更に好ましくは、水素原子、メチル基、メトキシカルボニル基、またはエトキシカルボニル基を表す。ここでR8がアルコキシカルボニル基を表す場合、R7は好ましくは水素原子を表す。
一般式(2)において、R9は、好ましくは、水素原子、炭素数1〜3のアルキル基、または炭素数2〜4のアルコキシカルボニル基を表し、更に好ましくは、水素原子、メトキシカルボニル基、またはエトキシカルボニル基を表す。
一般式(2)において、R10は、好ましくは、メチル基もしくはエチル基を表し、R11は、好ましくは水酸基、もしくはピリジル基で置換されていてもよいエチル基、もしくはプロピル基を表す。
【0059】
式(1)で表される化合物は、その多くが公知であり、市販品として得ることができる。また、以下の方法で製造することもできる。
製造法:
【化7】
式(1−2)のヒドラゾンは、公知化合物である式(1−1)の化合物とヒドラジン1水和物を、酸性条件下、0〜100℃で反応させることで製造できる。溶媒としては、メタノール、エタノールなどのアルコール系溶媒、クロロホルム、ジクロロメタンなどのハロゲン系溶媒、ジメチルホルムアミド、ジメチルスルフォキシト゛、アセトン、アセトニトリル、ジオキサンやテトラヒドロフランなどのエーテル系溶媒、水またはこれらの混合溶媒等が用いられる。メタノール、エタノールなどのアルコール系溶媒がより好ましい。酸としては、酢酸、塩酸、硫酸、トリフルオロ酢酸などを用いることができる。この反応では、クロロホルム中、酸として硫酸を用いる等の方法により、式(1−2)の化合物を単離することなく、式(1−3)の化合物を製造することができる。
式(1−2)の化合物を、メタノール、エタノールなどのアルコール系溶媒中、塩基を加えて加熱することにより、式(1−3)の化合物を得ることができる。塩基としては、水酸化ナトリウム、水酸化カリウム、炭酸カリウム、炭酸ナトリウム、もしくは炭酸水素ナトリウムなどの無機塩基や、トリエチルアミン、もしくはピリジンなどの有機塩基が用いられる。好ましくは、炭酸カリウム、または炭酸ナトリウムを用いることができる。
式(1−5)の化合物は、式(1−3)の化合物と式(1−4)で表される1,3−ジケトンを、塩基もしくは酸の存在、または非存在下に、室温から100℃で反応させることにより製造できる。溶媒としては、メタノール、エタノールなどのアルコール系溶媒、クロロホルム、ジクロロメタンなどのハロゲン系溶媒、ジメチルホルムアミド、ジメチルスルフォキシト゛、アセトン、アセトニトリル、ジオキサンやテトラヒドロフランなどのエーテル系溶媒、水、またはこれらの混合溶媒等が用いられる。このうち、メタノール、エタノールなどのアルコール系溶媒がより好ましい。酸としては、酢酸、塩酸、または、塩化亜鉛などのルイス酸などを用いることができる。塩基として、ピロリジン、またはピペリジンなどの有機塩基を用いることができる。
式(1−4)の化合物は、公知化合物を用いるか、あるいは本明細書実施例に記載された方法で製造することができる。また、式(1−5)の化合物は、式(1−2)の化合物から、式(1−3)の化合物を単離することなく製造することもできる。
式(1−6)の化合物は、式(1−5)の化合物を加水分解してカルボン酸とした後、公知の方法に従い市販のアミン化合物と縮合させることにより製造できる。縮合方法としては、本明細書記載の水溶性カルボジイミド(WSCI)を用いる方法、混合酸無水物法、または、酸ハライドを用いる方法等が挙げられる。該縮合方法については「Comprehensive Organic Transformation(Larock, R.C., VCH Publishers, Inc. 1989)」等に記載されている。
【0060】
一般式(1)で表される化合物の特に好ましいものとして、以下の式51〜64の化合物が例示される。
【化8】
薬学上許容される塩としては、酸付加塩及び塩基付加塩が挙げられる。酸付加塩としては、例えば塩酸塩、臭化水素酸塩、硫酸塩、ヨウ化水素酸塩、硝酸塩、リン酸塩等の無機酸塩、クエン酸塩、シュウ酸塩、酢酸塩、ギ酸塩、プロピオン酸塩、安息香酸塩、トリフルオロ酢酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、ベンゼンスルホン酸、パラトルエンスルホン酸塩等の有機酸塩が挙げられ、塩基付加塩としては、ナトリウム塩、カリウム塩、カルシウム塩、マグネシウム塩、アンモニウム塩等の無機塩基塩、トリエチルアンモニウム塩、トリエタノールアンモニウム塩、ピリジニウム塩、ジイソプロピルアンモニウム塩等の有機塩基塩等が挙げられ、アルギニン、アスパラギン酸、グルタミン酸などの塩基性あるいは酸性アミノ酸といったアミノ酸塩が挙げられる。また、本発明のPAR阻害活性を有する化合物は、水和物、又はエタノール和物等の溶媒和物であってもよい。
【0062】
一般式(1)で表される化合物、またはその薬学上許容される塩は、皮膚疾患治療剤、詳しくは、痒み症状を伴う皮膚疾患治療剤として有効である。
【0063】
痒み症状を伴う皮膚疾患としては、アトピー性皮膚炎、接触性皮膚炎、乾癬、湿疹、蕁麻疹、乾皮症、虫刺され、しもやけ、あせも、疥癬等が挙げられる。好ましくは、乾癬、アトピー性皮膚炎が挙げられる
【0064】
以下、本発明のPAR2阻害活性を有する化合物を有効成分として含有する、上記疾患の治療剤を医薬品として用いる場合の投与量および投与形態などにつき記述する。
本発明の化合物又はその薬学上許容される塩は、経口または非経口投与、好ましくは経口投与することができ、薬剤としてそれぞれ経口または非経口投与に適した種々の剤型で、ヒトおよびヒト以外の動物に使用される。例えば、経口的に投与する場合、通常用いられる投与形態、例えば錠剤、カプセル剤、シロップ剤、懸濁液などで投与することができる。非経口的に投与する場合、溶液、乳剤、懸濁液などの液剤を注射剤として投与すること、坐剤の型で直腸投与すること、軟膏、ローション剤、クリーム剤等の塗布剤の型で経皮投与すること、スプレー剤、エアゾル剤等を噴霧剤の型で吸入により投与すること、点滴液(点眼薬など)として投与すること等ができる。このような投与剤型は、通常の担体、賦型剤、結合剤、安定化剤などの助剤と有効成分を配合することにより一般的方法に従って製造することができる。注射剤型で用いる場合は、緩衝剤、溶解補助剤、等張剤などを添加することもできる。投与量、投与回数は、対象とする疾患、患者の症状、年齢、体重等、および投与形態などによって異なるが、経口投与する場合、有効成分は通常は成人に対し1日あたり約1〜1000mgの範囲、好ましくは、約10〜500mgの範囲を1回または数回に分けて投与することができる。注射剤として投与する場合、有効成分は約0.1〜約500mgの範囲、好ましくは約3〜約100mgの範囲を1回または数回に分けて投与することができる。
【0065】
【実施例】
以下に実施例及び参考例を挙げて本発明を詳細に説明するが、本発明はもとよりこれに限定されるものではない。
実施例1
(2E)−3−シアノ−2−ヒドラゾノプロパン酸エチル
【化9】
1H−NMR (400MHz, CDCl3)δppm : 1.34−1.41(m, 3H), 3.46(s), 3.63(s), 4.26−4.38(m, 2H), 6.52(brs), 8.50(br)
【0066】
実施例2
エチル 5−フェニル−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン−2−カルボキシレート
【化10】
【0067】
実施例3
5−フェニル−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン−2−カルボン酸カリウム
【化11】
1H−NMR (400MHz, DMSO−d6)δppm : 6.87(s, 1H), 7.53−7.59(m, 3H), 8.02(s, 1H), 8.24−8.31(m, 2H)
【0068】
実施例4
5−フェニル−2−(ピペリジン−1−イルカルボニル)−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン
【化12】
1H−NMR (400MHz, CDCl3)δppm : 1.62−1.78(m, 6H), 3.75−3.83(m, 4H), 7.17(s, 1H), 7.54−7.59(m, 3H), 7.68(s, 1H), 8.09−8.16(m, 2H)
【0069】
実施例5
2−[(2−メチルピペリジン−1−イル)カルボニル]−5−フェニル−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン
【化13】
1H−NMR (400MHz, CDCl3)δppm : 1.30−1.40(m, 3H), 1.49−1.90(m, 6H), 2.90−3.30(m, 1H), 4.20−4.74(m, 1H), 4.55−5.14(m, 1H), 7.15(s, 1H), 7.54−7.59(m, 3H), 7.67(s, 1H), 8.09−8.15(m, 2H)
【0070】
実施例6
2−(アゼパン−1−イルカルボニル)−5−フェニル−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン
【化14】
1H−NMR (400MHz, CDCl3)δppm : 1.60−1.73(m, 4H), 1.81−1.93(m, 4H), 3.73−3.85(m, 4H), 7.23(s, 1H), 7.54−7.59(m, 3H), 7.68(s, 1H), 8.09−8.15(m, 2H)
【0071】
実施例7
2−[(4−ベンゾイルピペラジン−1−イル)カルボニル]−5−フェニル−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン
【化15】
1H−NMR (400MHz, CDCl3)δppm : 3.50−4.25(m, 8H), 7.28(s, 1H), 7.55−7.60(m, 3H), 7.72(brs, 1H), 8.10−8.15(m, 2H)
【0072】
実施例8
エチル 1−{[5−フェニル−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン−2−イル]カルボニル}ピペリジン−2−カルボキシレート
【化16】
1H−NMR (400MHz, CDCl3)δppm : 1.26−1.35(m, 3H), 1.40−1.90(m, 5H), 2.28−2.43(m, 1H), 3.00−3.40(m, 1H), 4.20−4.32(m, 2H), 4.45−4.80(m, 1H), 5.45−5.60(m, 1H), 7.21−7.28(m, 1H), 7.53−7.60(m, 3H), 7.67−7.71(m, 1H), 8.10−8.06(m, 2H)
【0073】
実施例9
エチル 1−{[5−フェニル−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン−2−イル]カルボニル}ピペリジン−4−カルボキシレート
【化17】
1H−NMR (400MHz, CDCl3)δppm : 1.28(t, 3H, J=7.1Hz), 1.80−2.12(m, 4H), 2.64(m, 1H), 3.13(m, 1H), 3.37(m, 1H), 4.18(q, 1H, J=7.1Hz), 4.46−4.62(m, 2H), 7.21(s, 1H), 7.54−7.60(m, 3H), 7.69(s, 1H), 8.10−8.16(m, 2H)
実施例10
1−(4−クロロフェニル)−4,4,4−トリフルオロブタン−1,3−ジオン
【化18】
1H−NMR (400MHz, DMSO−d6)δppm : 7.02(s, 1H), 7.66(d, 2H, J=8.7Hz), 8.14(d, 2H, J=8.7Hz)
【0074】
実施例11
エチル 5−(4−クロロフェニル)−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン−2−カルボキシレート
【化19】
1H−NMR (400MHz, CDCl3) δppm : 1.47(t, 3H, J=7.1Hz), 4.51(q, 2H, J=7.1Hz), 7.38(s, 1H), 7.52−7.57(m, 2H), 7.71(s, 1H), 8.07−8.12(m, 2H)
【0075】
実施例12
5−(4−クロロフェニル)−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン−2−カルボン酸カリウム
【化20】
【0076】
実施例13
5−(4−クロロフェニル)−2−(ピペリジン−1−イルカルボニル)−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン
【化21】
1H−NMR (400MHz, CDCl3)δppm : 1.61−1.77(m, 6H), 3.76−3.83(m, 2H), 7.18(s, 1H), 7.51−7.56(m, 2H), 7.63(s, 1H), 8.06−8.11(m, 2H)
【0077】
実施例14
5−(4−クロロフェニル)−2−[(2−メチルピペリジン−1−イル)カルボニル]−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン
【化22】
1H−NMR (400MHz, CDCl3)δppm : 1.31−1.40(m, 3H), 1.50−1.90(m, 6H), 2.90−3.30(m, 1H), 4.20−4.73(m, 1H), 4.53−5.15(m, 1H), 7.14(s, 1H), 7.51−7.56(m, 2H), 7.63(s, 1H), 8.06−8.11(m, 2H)
【0078】
実施例15
2−(アゼパン−1−イルカルボニル)−5−(4−クロロフェニル)−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン
【化23】
1H−NMR (400MHz, CDCl3) δppm : 1.60−1.71(m, 4H), 1.80−1.92(m, 4H), 3.73−3.84(m, 4H), 7.23(s, 1H), 7.51−7.56(m, 2H), 7.63(s, 1H), 8.06−8.11(m, 2H)
【0079】
実施例16
エチル 1−{[5−(4−クロロフェニル)−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン−2−イル]カルボニル}ピペリジン−2−カルボキシレート
【化24】
実施例17
5−(4−クロロフェニル)−N−(2−ヒドロキシエチル)−N−メチル−7−(トリフルオロメチル)ピラゾロ[1,5−a]ピリミジン−2−カルボキシアミド
【化25】
1H−NMR (400MHz, CDCl3)δppm : 3.05−3.24(m, 1H), 3.24−3.47(m, 3H), 3.78−4.00(m, 4H), 7.27−7.33(m, 1H), 7.51−7.58(m, 2H), 7.65−7.70(m, 1H), 8.06−8.12(m, 2H) 。
【0080】
実施例18: Fluorometric Imaging Plate Reader (FLIPR) を用いた細胞内カルシウム濃度測定
[材料と方法]
牛胎児血清10%(10%FBS)添加DMEM培養液(インビトロジェン社)を用いて培養したヒト胎児腎臓由来細胞株HEK293細胞を 2.5X105cells/mlに調製し、ポリDリジンでコートした96穴黒色プレート(透明底)(ファルコン社)に80μl/well播いて、炭酸ガス培養装置で一晩培養した。Hanks−20mM Hepes 緩衝液(pH7.4)で調製したFLIPR Calcium Assay Kit(Molecular Devices社)を細胞に80μl/well添加して、1時間炭酸ガス培養装置にて培養した。Hanks−20mM Hepes緩衝液(pH7.4)で、PAR2活性化因子として0.0005%に調製したtrypsin (SIGMA社 T8642)を、96穴V底ポリプロピレンプレート(ヌンク社)に添加して試薬プレートとした。FLIPR(Molecular Devices社)で測定する直前に、Hanks−20mM Hepes緩衝液(pH7.4)で調製した被験物質を、細胞に40μl/well添加し、プレートシェーカーで攪拌した。被験物質を添加した細胞プレートと試薬プレートをFLIPRにセットし、細胞内カルシウム濃度の変化をCCDカメラにて検出した。測定は室温で70秒間行い、試薬プレートから細胞プレートへの試薬の添加(50μl/well)は、FLIPR本体内蔵の96well自動分注機で行った。
被験物質の細胞内カルシウム上昇抑制率は、以下の式:
(被験物質存在下における細胞内カルシウム濃度の変化−Trypsin非存在下における細胞内カルシウム濃度の変化)/(被験物質非存在下における細胞内カルシウム濃度の変化−被験物質及びTrypsin非存在下における細胞内カルシウム濃度の変化)
で求めることができる。
[結果]被験物質の細胞内カルシウム上昇抑制率(%)を、表1に示した。
【表1】
【0081】
実施例19: PGE2量の測定を利用したPAR2阻害活性の評価
SW982細胞を2.5x105細胞/mlになるように各培地で希釈し、96穴プレートに100μl/wellの細胞を播きこんだ。炭酸ガス培養装置内で一晩前培養した後、培地を除去し、5μg/ml 化合物入り培地を 80μl /well添加した(コントロール群、コントロールペプチド群、アゴニストペプチド単独群、IL−1β群には0.1%DMSO含有培地を添加した)。化合物を入れたウェルには 500uM アゴニストペプチド入り培地を 20μl /well 添加した(終濃度は 4μg/ml 化合物・100uM アゴニストペプチト)。コントロール群のウェルには各培地を20ul /well、コントロールペプチド群のウェルには500μM コントロールペプチド入り培地を 20μl /well (終濃度100μM)、アゴニストペプチド単独群のウェルには500uM アゴニストペプチド入り培地を 20μl /well (終濃度100μM)、IL−1β群のウェルには50ng/mlIL−1β入り培地を 20μl /well (終濃度10ng/ml)の割合で添加した。24時間後 (N=3) に各上清を回収し−80℃で測定時まで保管した。各上清中のPGE2量の測定は、プロスタグランジンE2 EIA (Cayman chemical 514010) を用いて行った。コントロールペプチドのPGE2量の値を 阻害率100%の値、アゴニストペプチドのPGE2量を阻害率0%の値として、実施例の化合物のPGE2産生阻害率を計算した。すなわち、被験物質のPAR2阻害率は以下の式:
(被験物質におけるアゴニスト存在下でのPGE2量−被験物質におけるコントロールペプチド存在下でのPGE2量)/(対照におけるアゴニスト存在下でのPGE2量−対照におけるコントロールペプチド存在下でのPGE2量)
で求めた。実施例の化合物のPGE2産生抑制率を表2に示した。
【表2】
【0082】
実施例20:PAR2の痒み惹起活性
7週齢の雄ICRマウスの肩背部皮内にアゴニストペプチドまたはコントロールペプチドを50pmol/20ul投与した。投与後10分間、後肢で投与部付近を引っかく行動を計測した(後肢が地面から離れて、再度戻るまでを1回と計測した)。結果を表3に示した。この結果から、アゴニストペプチド(配列番号:2)を投与した群は、コントロールペプチド(配列番号:4)を投与した群と比較して、著しく投与部位付近を引っかく回数が多いことがわかった。
更に、実施例17の化合物(200mg/kg)を経口で投与した後、投与化合物の血中濃度を考慮して30分後にアゴニストペプチドを投与した。上記と同様にアゴニストペプチド投与後10分間、後肢で投与部付近を引っかく行動を計測した。結果を表4に示した。この結果から、実施例1の化合物により、アゴニストペプチドにより惹起された引っかき行動が抑制されることがわかった。
【表3】
投与液 引っかき回数
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
生理食塩水(ペプチド溶解溶媒) 2回
SLIGRL(アゴニストペプチド) 25回
LSIGRL(コントロールペプチド) 7回
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
【表4】
投与液 引っかき回数
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
実施例17の化合物+SLIGRL 12回
SLIGRL 24回
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
【0083】
【発明の効果】
本発明により、痒みを伴う皮膚疾患治療剤をスクリーニングする方法を提供することが可能になった。また、PAR2阻害活性を有する化合物を有効成分として含有する痒みを伴う皮膚疾患治療剤を提供することが可能になった。[0001]
TECHNICAL FIELD OF THE INVENTION
The present invention relates to a method for screening a therapeutic agent for skin diseases, and more particularly, to a therapeutic agent for skin diseases that suppresses itch and a method for screening the same.
[0002]
[Prior art]
The protease receptor PAR (proteinase-activated receptor) is a kind of G protein-coupled seven-transmembrane receptor, and four PARs; PAR1, PAR2, PAR3, and PAR4 have been cloned to date.
PAR2 is activated by cleavage of the N-terminal partial peptide by trypsin, tryptase, or blood coagulation factor VIIa or factor Xa to generate a new N-terminal. For example, in the case of human PAR2, the N-terminal 36 residues are truncated to form a polypeptide having an N-terminal newly starting with the sequence of SLIGKV and activated. It is also known that the peptide is activated by a synthetic peptide (agonist peptide) consisting of 5 to 6 amino acids based on the sequence of the N-terminal peptide generated by activation (Non-Patent Document 1 and Non-patent document 2). Examples of the agonist peptide include a partial peptide represented by the sequence of SLIGKV and the sequence of SLIGRL, a modified peptide thereof, trans-cinnamoyl-LIGRLO, and the like (Non-Patent Document 3).
[0003]
PAR2 is involved in various physiological activities such as secretion of digestive juice, neurogenic inflammation, pain, allergic reaction (Non-Patent Documents 4 and 5).
On the other hand, although itching varies depending on the disease as a physiologically active substance that causes itching, which is an intrinsic sensation of the skin, for example, histamine, serotonin, leukotriene B4, substance P, VIP, trypsin (Non-patent Document 6) and the like have been reported. I have. There are many unclear points about the mechanism of perception of itch, but it is thought that bioactive substances such as histamine are secreted from mast cells etc. in response to itch stimulation and transmitted to the brain through C fibers, a type of sensory nerve. Have been.
In connection with the above, it has been reported that PAR2 is expressed in C fibers (Non-Patent Document 7) and mast cells, and that degranulation occurs by the action of PAR2 (Non-Patent Document 8). However, the relationship between PAR2 and skin itch symptoms was unknown.
[Non-patent document 1]
Derry, O. et al., "American Journal of Physiology" 1998, vol. 274, p. C1429-52
[Non-patent document 2]
Macfarlane, S.M. R. Et al., "Pharmalogical Reviews" 2001, Vol. 53, p 245-282.
[Non-Patent Document 3]
Al-Ani, B .; Et al., "Journal of Pharmacology and Experimental Therapeutics", Vol. 290, p. 753-760.
[Non-patent document 4]
Sun, G .; Et al., "Journal of Immunology", 2001, Vol. 167, p1014-1021.
[Non-Patent Document 5]
Mike, S.M. Et al., "Journal of Immunology", 2001, Vol. 167, pp. 6615-6622.
[Non-Patent Document 6]
Hagermark, O .; Et al., "Seminars in Dermatol," 1995, Vol. 14, p271-276.
[Non-Patent Document 7]
Steinhoff, M .; Et al., "Nature Medicine" 2000, Vol. 6, p. 151-158.
[Non-Patent Document 8]
Grant, R.A. S. Et al., "Journal of Pharmacology and Experimental Therapeutics", 2002, Vol. 302, p. 466-474.
[0004]
[Problems to be solved by the invention]
An object of the present invention is to provide a therapeutic agent for skin diseases which improves itch symptoms based on a novel mechanism of action, and a method for screening the same.
[0005]
[Means for Solving the Problems]
The present inventors have conducted extensive studies and found that a PAR2 agonist peptide induces itching symptoms in mice. The present invention has been completed based on the above findings.
[0006]
That is, the present invention
[1] The following steps (1) and (2):
(1) a step of evaluating the PAR2 inhibitory activity of the test substance,
(2) a step of selecting a test substance having PAR2 inhibitory activity as an active ingredient of a therapeutic agent for skin diseases based on the result of (1);
A method for screening a therapeutic agent for skin diseases, comprising:
[2] The step (1) includes the following steps (a) to (d):
(A) contacting a test substance with a cell capable of expressing PAR2,
(B) measuring the expression level of PAR2 in (a),
(C) comparing the value of (b) with the expression level of PAR2 in a control not contacted with the test substance;
(D) a step of evaluating the PAR2 inhibitory activity of the test substance based on the results of (c)
[1] The method for screening a therapeutic agent for skin diseases according to [1], which comprises:
[3] The step (1) includes the following steps (a) to (d):
(A) contacting a test substance with a cell containing PAR2 and a PAR2 activator, or a cell fraction prepared from the cell;
(B) measuring the PAR2-derived function in (a),
(C) comparing the value of (b) with a PAR2-derived function in a control not contacted with the test substance;
(D) evaluating the PAR2 inhibitory activity of the test substance based on the results of (c),
[1] The method for screening a therapeutic agent for skin diseases according to [1], which comprises:
[4] The step (d) comprises the following steps:
(D-1) PAR2 inhibitory activity of test substance and formula:
Embedded image
(D-2) selecting a test substance exhibiting a PAR2 inhibitory activity equal to or higher than that of the compound as a candidate compound for a therapeutic agent for skin disease;
The screening method according to [3], comprising:
[5] The step (1) includes the following steps (a) to (d):
(A) contacting the test substance with an aqueous solution containing PAR2 and PAR2 ligand, a cell, or a cell membrane fraction prepared from the cell;
(B) measuring the amount of PAR2 ligand bound to PAR2 in (a),
(C) comparing the value of (b) with the amount of PAR2 agonist bound to PAR2 in a control not contacted with the test substance;
(D) evaluating the PAR2 inhibitory activity of the test substance based on the results of (c),
[1] The method for screening a therapeutic agent for skin diseases according to [1], which comprises:
[6] the screening method of any one of [1] to [5], wherein the skin disease is a disease accompanied by itch;
[7] The screening method according to any one of [1] to [6], wherein the therapeutic agent for a skin disease is a therapeutic agent characterized by improving itch symptoms.
[0007]
[8] A therapeutic agent for skin diseases, comprising a compound having PAR2 inhibitory activity as an active ingredient;
[9] The compound having PAR2 inhibitory activity is represented by the general formula (1):
Embedded image
R3Represents a hydrogen atom, an alkyl group, a haloalkyl group, or a substituted or unsubstituted aryl group,
R4And R5Independently represents a hydrogen atom, a halogen atom, an alkyl group, an alkoxy group, a methylenedioxy group, or a haloalkyl group. )
Or a pharmaceutically acceptable salt thereof, wherein the therapeutic agent according to [8],
[10] R3Is a haloalkyl group, the therapeutic agent according to [9],
[11] the therapeutic agent of [9], wherein the haloalkyl group is a trifluoromethyl group;
[12] R1And R2Is bonded with an adjacent carbon atom, and may be substituted with an alkyl group, an alkylcarbonyl group, an arylcarbonyl group, an alkoxycarbonyl group, a phenyl group, or a benzyl group, piperidine, perhydroazepine, morpholine, thiomorpholine, Or a therapeutic agent according to any one of [8] to [10], wherein the therapeutic agent forms piperazine;
[13] R1And R2Is combined with an adjacent carbon atom to form piperidine or perhydroazepine, which may be substituted with an alkyl group or an alkoxycarbonyl group, and the therapeutic agent according to [11]. ,
[14] General formula (2):
Embedded image
Embedded image
Embedded image
Represents a group selected from3 'Represents a hydrogen atom, an alkyl group, a haloalkyl group, or a phenyl group,
R4 'And R5 'Independently represents a hydrogen atom, a halogen atom, an alkyl group, or a haloalkyl group. ]
A compound represented by the formula, or a therapeutic agent for skin diseases containing a pharmaceutically acceptable salt thereof as an active ingredient,
[0008]
[15] The therapeutic agent according to any of [8] to [14], wherein the skin disease is a disease accompanied by itch.
[16] The therapeutic agent according to any one of [8] to [14], wherein the therapeutic agent for skin disease is a therapeutic agent characterized by improving itch symptoms.
[17] a reagent for inducing itch in an animal model for evaluating a drug for improving itch symptoms, which comprises PAR2 or a PAR2 activator as an active ingredient;
It is about.
[0009]
[Embodiment of the present invention]
In the present specification, the abbreviations of amino acids, (poly) peptides, (poly) nucleotides, and the like are referred to by the IUPAC-IUB [IUPAC-IUB Communication on Biological Nomenclature, Eur. J. Biochem. , 138: 9 (1984)], "Guidelines for Preparation of Specifications and the like Containing Base Sequences or Amino Acid Sequences" (edited by the Japan Patent Office) and conventional symbols in the art.
[0010]
In the present specification, "PAR2" refers to a receptor (proteinase-activated receptor) protein that is activated by a protease such as trypsin or tryptase, which belongs to a G protein-coupled seven-transmembrane receptor, and its amino acid sequence is And for humans, Bohm, S.M. K. et al. Biochem. lJ. , 314, 1009-16, 1996 (Genbank Ac. No. U34038), and homologs of other animal species can be found in mice: Nysteadt S. et al. et al. , J. et al. Biol. Chem. , 270, 5950-55, 1995 (Genbank Ac. No. Z48043), and rats: Saifedine M., et al. et al. , Br. J. Pharmacol. , 118, 521-30, 1996 (Genbank Ac. No. U61373), respectively.
[0011]
In the present specification, “PAR2” includes not only those having the above-mentioned amino acid sequences but also homologs (homologs and splice variants), variants and derivatives thereof as long as they have the same biological function. , Mature forms and amino acid modifications. Here, homologs of animal species other than the above can be deduced from the nucleotide sequence of the gene identified by HomoloGene (https://www.ncbi.nlm.nih.gov/HomoloGene/). Variants also include naturally occurring allelic variants, non-naturally occurring variants, and variants having amino acid sequences altered by artificial deletion, substitution, addition and insertion. . In addition, examples of the above mutants include those that are at least 70%, preferably 80%, more preferably 95%, and still more preferably 97% homologous to a protein or (poly) peptide having no mutation. Amino acid modifications include naturally occurring amino acids and non-naturally occurring amino acids, and specifically include phosphorylated amino acids.
[0012]
As used herein, the term "PAR2 gene" refers to the gene encoding PAR2, and its nucleotide sequence is the same as that of humans in Bohm, S.M. K. et al. Biochem. lJ. , 314, 1009-16, 1996 (Genbank Ac. No. U34038), and homologs of other animal species can be found in mice: Nysteadt S. et al. et al. , J. et al. Biol. Chem. , 270, 5950-55, 1995 (Genbank Ac. No. Z48043), rat: Saiddine M. et al. et al. , Br. J. Pharmacol. , 118, 521-30, 1996 (Genbank Ac. No. U61373) and the like.
As used herein, the term “PAR2 gene” includes not only those having the above-mentioned nucleotide sequences, but also homologs (homologs and splice variants) and mutations thereof, as long as they have the same biological function. Body etc. are included.
Here, homologs of animal species other than the above can be deduced from the nucleotide sequence of the gene identified by HomoloGene (https://www.ncbi.nlm.nih.gov/HomoloGene/). Mutants also include naturally occurring allelic variants, non-naturally occurring variants, and variants having base sequences modified by artificial deletion, substitution, addition and insertion. . In addition, examples of the mutant include those that are at least 70%, preferably 80%, more preferably 95%, and even more preferably 97% homologous to a polynucleotide having no mutation.
[0013]
The phrase “equivalent biological functions” specifically means that the compound has a binding activity with a known PAR2 agonist or has the function of PAR2 described below.
[0014]
As used herein, "PAR2 activator" refers to (1) an enzyme protein that activates PAR2 by hydrolyzing a partial peptide at the N-terminus of PAR2, and (2) the absence of an enzyme protein of (1). An agonist compound capable of activating PAR2 is shown below, and PAR2 activation is a concept including both the actions caused by (1) and (2).
In other words, the natural activation mechanism of PAR2 is that the N-terminal peptide (corresponding to 36 residues at the N-terminal in the case of human PAR2) is hydrolyzed by the PAR2 activator to newly generate an N-terminal partial peptide (hereinafter referred to as “N-terminal peptide”). , Which may be referred to as an active N-terminal partial peptide) binds to PAR2 as a ligand, thereby producing a PAR2-derived physiological activity. Specifically, the PAR2 activator (enzyme) Examples of the protein include trypsin, tryptase, blood coagulation factor Xa, and blood coagulation factor VIIa.
On the other hand, examples of the PAR activator (agonist) of the above (2) include peptide compounds based on the activated N-terminal partial peptide sequence of PAR2 and derivatives thereof. Specific examples thereof include partial peptides represented by SLIGKV (SEQ ID NO: 1) and SLIGRL (SEQ ID NO: 2), and modified peptides trans-cinnamoyl-LIGRLO (SEQ ID NO: 3). In addition, membrane-bound serine protease 1 (Takeuchi, T. et al., J. Biol. Chem., 275, 26333-26342 (2000)), and membrane-bound serine protease 3 (Wallrapp, C. et. , Cancer Res., 60, 2602-2606 (2000)), and the like.
[0015]
In the present specification, the “compound having PAR2 inhibitory activity” is not particularly limited as long as it is a compound that acts in the direction of inhibiting the function of PAR2, and a compound having an activity of reducing the expression level of PAR2, The concept includes a compound having PAR2 antagonist activity, a compound that suppresses the function of PAR2, and the like.
Here, “reducing the expression level of PAR2” means reducing the expression level of mRNA encoding PAR2 or reducing the amount of PAR2 protein.
The term “PAR2 antagonist activity” refers to the activity of inhibiting the binding between PAR2 and the activated N-terminal partial peptide of PAR2, the N-terminal of which has been cleaved, and the binding between agonist and PAR2.
The “function of PAR2” includes functions specific to GPCRs such as increased intracellular calcium concentration and increased production of intracellular inositol triphosphate, increased phosphorylation of intracellular proteins by protein kinase C or MAP kinase, or Functions include promotion of prostaglandin E2 (PGE2) release from cells and promotion of salivary secretion in mammals.
[0016]
Hereinafter, embodiments of the present invention will be described in detail.
The first aspect of the present invention comprises the following steps (1) a step of evaluating the PAR2 inhibitory activity of a test substance, and (2) treating a test substance having a PAR2 inhibitory activity on the basis of the results of (1) for treating a skin disease. A method for screening a therapeutic agent for skin diseases, comprising a step of selecting as an active ingredient of an agent.
In the above (1), the method for evaluating the PAR2 inhibitory activity includes the method described in Method for measuring the expression level of PAR2, II. Method of measuring PAR2 function, III. A method for measuring PAR2 binding inhibitory activity and the like can be mentioned. Each of these will be specifically described below.
[0017]
I. Method for measuring PAR2 expression level
The method for evaluating the PAR inhibitory activity based on the expression level of PAR2 includes the following steps (a) to (d):
(A) contacting a test substance with a cell capable of expressing PAR2,
(B) measuring the expression level of PAR2 in (a),
(C) comparing the value of (b) with the expression level of PAR2 in a control not contacted with the test substance;
(D) a step of evaluating the PAR2 inhibitory activity of the test substance based on the results of (c)
including.
[0018]
Here, examples of cells that can express PAR2 include all cultured cells that express PAR2 regardless of whether they are endogenous or exogenous. Whether or not these genes of the present invention are expressed in cultured cells can be easily confirmed by detecting the expression of these genes by a known Northern blot method or RT-PCR method.
Specific examples of the cells used for the screening include, for example, the following (1) to (3):
(1) PAR2-expressing mammalian cells
(2) Cells treated with the PAR2 activator
(3) a cell into which a gene encoding PAR2 has been introduced,
And the like.
Here, the cells of the above (1) include pancreatic tissue-derived cells, keratinocyte-derived cells, mast cells, vascular endothelial cells, T lymphocytes, platelets, osteoblasts, hepatocytes, or various established cells. No. Specifically, MIA PaCa-2 cells (human pancreatic cancer-derived cell line), HEK293 (human fetal kidney-derived cell line), A549 (human lung cancer-derived cell line), HFL1 (human fetal lung-derived cell line), SW982 (Human synovial cell line) or SW1353 (human cartilage-like cell line).
In the above (2), the same PAR2 activator as described above can be used. The cells to be treated include the cells described in (1) above.
[0019]
Examples of the cells into which the gene of (3) has been introduced include, in addition to the cells described in (1) and (2), host cells usually used for gene transfer, namely, COP, L, C127, Sp2 / 0, NS- 1. Mouse-derived cells such as NIH3T3 and ST2, rat-derived cells, hamster-derived cells such as BHK and CHO, monkey-derived cells such as COS1, COS3, COS7, CV1, and Vero; human-derived cells such as HeLa and 293; and Sf9. , Sf21, High Five, and other insect-derived cells.
[0020]
Furthermore, the cells used in the screening method of the present invention also include tissues that are aggregates of cells.
In the screening of the present invention, the conditions under which the test substance is brought into contact with the cells are not particularly limited, and culture conditions (temperature, pH, medium composition, etc.) that do not kill the cells and can express the gene of the present invention are selected. Is preferred.
[0021]
As a method for measuring the expression level of PAR2, Northern blotting, RT-PCR, DNA chip analysis, or the like using RNA prepared from cells in step (a) as a sample, orin situKnown methods such as a hybridization analysis method and a Western blotting method using a protein prepared from the cells in step (a) as a sample are exemplified. The measurement sample is usually prepared by performing necessary treatment such as crushing, diluting, or centrifuging the cells to prepare a fraction containing RNA or protein.
[0022]
When the Northern blot method is used, the presence or absence of PAR2 expression in RNA can be determined by using as a probe a polynucleotide having at least 15 consecutive nucleotides in the base sequence of the PAR2 gene and / or its complementary polynucleotide. The expression level can be detected and measured. Specifically, the probe is replaced with a radioisotope (32P,33P or the like: labeled with RI) or a fluorescent substance, and then hybridized with the cell-derived RNA of step (a) transferred to a nylon membrane or the like according to a conventional method. A method for detecting and measuring a signal derived from a label (RI or fluorescent substance) of a disease marker using a radiation detector (BAS-1800II, manufactured by Fuji Film Co., Ltd.) or a fluorescence detector for the double strand of Can be. Further, using AlkPhos Direct Labeling and Detection System (manufactured by Amersham Pharmacia Biotech), the probe DNA is labeled according to the protocol, and the probe is hybridized with the RNA. A method of detecting and measuring with STORM860 (manufactured by Amersham Pharmacia Biotech) can also be used.
[0023]
When the RT-PCR method is used, the presence or absence and expression level of PAR2 in RNA can be detected and measured by using the same nucleotide sequence as the above-described probe of the present invention as a primer. Specifically, a cDNA is prepared from the cell-derived RNA in step (a) according to a conventional method, and the primer (the above cDNA (-strand) is used as a template to amplify the target PAR2 gene region. The positive and negative chains that bind to each other are hybridized with this, a PCR method is performed according to a conventional method, and the resulting amplified double-stranded DNA is detected. The amplified double-stranded DNA was detected by a method for detecting labeled double-stranded DNA produced by performing the PCR using primers previously labeled with RI or a fluorescent substance. A method in which double-stranded DNA is transferred to a nylon membrane or the like according to a conventional method, hybridized with a labeled primer, and detected can be used. The generated labeled double-stranded DNA product can be measured using an Agilent 2100 Bioanalyzer (manufactured by Yokogawa Analytical Systems). In addition, an RT-PCR reaction solution is prepared according to the protocol using SYBR Green RT-PCR Reagents (manufactured by Applied Biosystems), and ABI PRISM 7700 Sequence Detection System (a reaction of Applied Biosystems is performed by Applied Biosystems). You can also.
When using DNA chip analysis, a DNA probe (single-stranded or double-stranded) used as a probe in the above Northern hybridization method is attached to a chip on a resin. CRNA prepared by a conventional method from cell-derived RNA is hybridized, and a double-stranded DNA and cRNA formed is bound to a labeled primer prepared from the primer to detect the double-stranded DNA. it can.
[0024]
In addition, a method of detecting and quantifying PAR2 by a known method such as Western blotting using an antibody that recognizes PAR2 can be mentioned.
In Western blotting, the primary antibody is reacted with the sample and then125Using a labeled antibody (an antibody that binds to the primary antibody) labeled with a radioisotope such as I, a fluorescent substance, an enzyme such as horseradish peroxidase (HRP), etc., and derived from the radioisotope, the fluorescent substance, etc. of the obtained labeled compound This signal can be detected and detected by a radiation meter (BAS-1800II: manufactured by Fuji Film Co., Ltd.), a fluorescence detector, or the like. Further, after using the disease marker of the present invention as a primary antibody, detection is performed using ECL Plus Western Blotting Detection System (manufactured by Amersham Pharmacia Biotech) according to the protocol, and the multi-bio imager STORM860 (manufactured by Amersham Pharmacia Biotech) is used. You can also.
[0025]
Here, the antibody may be a polyclonal antibody or a monoclonal antibody thereof. The production method thereof is already well known, and the antibody of the present invention can be produced according to these conventional methods (Current). Protocol in Molecular Biology, Chapter 11.12-11.13 (2000)). Specifically, when the antibody of the present invention is a polyclonal antibody, an oligopeptide having a partial amino acid sequence of the protein of the present invention is synthesized using PAR2 expressed and purified in E. coli or the like according to a conventional method, or according to a conventional method. Then, a non-human animal such as a rabbit can be immunized and obtained from the serum of the immunized animal according to a conventional method. On the other hand, in the case of a monoclonal antibody, a non-human animal such as a mouse is immunized with PAR2 expressed and purified in Escherichia coli or the like according to a conventional method, or an oligopeptide having a partial amino acid sequence of these proteins, and the obtained spleen cells and bone marrow. Can be obtained from hybridoma cells prepared by cell fusion with tumor cells (Current Protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley and Sons 1st 1st. .
[0026]
PAR2 used as an immunizing antigen for the production of an antibody can be prepared by DNA cloning, construction of each plasmid, transfection into a host, culture of a transformant, and culturing of a transformant based on the sequence information described in the above-mentioned known literature. It can be obtained by the operation of recovering the protein. These operations are performed according to methods known to those skilled in the art, or methods described in the literature (Molecular Cloning, T. Maniatis et al., CSH Laboratory (1983), DNA Cloning, DM. Glover, IRL PRESS (1985)) and the like. Can be done.
[0027]
The PAR2 inhibitory activity of the test substance was evaluated by comparing the PAR2 expression level measured in the presence of the test substance (measured value in step (b)) as described above with the PAR2 expression level in a control not contacted with the test substance. can do.
Specifically, the expression level of PAR2 in the cells of any of the above (1) to (3) to which the test substance was added was lower than that of the cells to which the test substance was not added (control). ,It can be carried out. When cells that require an expression inducer for PAR2 expression are used, expression induced by using a PAR2 activator as an expression inducer is controlled by the presence of the test substance. The gene expression of cells contacted with the test substance in the presence of the test substance was determined to be lower than that of control cells not contacted with the test substance in the presence of the expression inducing substance (positive control). Can be evaluated as a compound having PAR2 inhibitory activity.
[0028]
Using the PAR2 inhibitory activity of the test substance evaluated as described above as an index, a candidate substance for a therapeutic agent for skin diseases can be selected. For example, a test substance whose PAR2 expression level in the presence of the test substance is reduced by 10%, preferably 30%, particularly preferably 50% or more compared to the expression level in control cells not contacted with the test substance is considered as a candidate substance. Can be sorted out.
[0029]
Further, the detection and quantification of the expression level of PAR2 can be performed by using a cell line in which a fusion gene in which a marker gene such as a luciferase gene is linked is introduced into a gene region (expression control region) that regulates PAR2, using a marker gene-derived protein. The activity can also be measured by measuring the activity of Therefore, the method of screening for a PAR2 expression control substance also includes a method of searching for a target substance using the expression level of such a marker gene as an index. In this sense, the method of screening for a PAR2 expression control region and a marker gene is used. Screening using the fusion gene is also within the scope of the present invention.
[0030]
The marker gene is preferably a structural gene of an enzyme that catalyzes a luminescence reaction or a color reaction. Specifically, in addition to the above-mentioned luciferase gene, reporter genes such as secreted alkaline phosphatase gene, chloramphenicol acetyltransferase gene, β-glucuronidase gene, β-galactosidase gene, and aequorin gene can be exemplified.
The expression control region of the gene of the present invention may be, for example, about 1 kb, preferably about 2 kb upstream of the transcription start site of the gene.
[0031]
The expression control region of the gene of the present invention may be prepared by, for example, (i) 5′-RACE method (for example, using 5 ′ full Race Core Kit (manufactured by Takara Shuzo)), oligocap method, S1 primer mapping, etc. A step of determining the 5 'end by a usual method; (ii) obtaining a 5'-upstream region using Genome Walker Kit (manufactured by Clonetech) and measuring the promoter activity of the obtained upstream region; And the like. Preparation of the fusion gene and measurement of the activity derived from the marker gene can be performed by known methods.
[0032]
II. How to measure PAR2 function
The method for evaluating the PAR inhibitory activity based on the function of PAR2 includes the following steps (a) to (d):
(A) contacting a test substance with a cell containing PAR2 and a PAR2 activator, or a cell fraction prepared from the cell;
(B) measuring the PAR2-derived function in (a),
(C) comparing the value of (b) with a PAR2-derived function in a control not contacted with the test substance;
(D) evaluating the PAR2 inhibitory activity of the test substance based on the results of (c),
including.
[0033]
Here, any function measurement method based on the known function of PAR2 can be used. That is, a test substance is added to a system for measuring the known function and activity of PAR2, and the test substance that suppresses the known function of PAR2 can be evaluated as a compound having PAR2 inhibitory activity.
[0034]
The screening of the present invention can be performed by bringing a cell containing PAR2 or a cell fraction prepared from the cell into contact with a test substance.
[0035]
In addition, examples of the cells used in the screening method of the present invention include cells that can express PAR2 regardless of whether they are endogenous or exogenous. As the cells, specifically, the cells described in (1) to (3) in I above can be used.
The cell fraction used in the screening method of the present invention means various fractions derived from the above-mentioned cells, and includes, for example, a cell membrane fraction and the like.
[0036]
PAR2 used in the screening is a known protein, and is obtained by operations such as DNA cloning, construction of each plasmid, transfection into a host, culture of transformed cells, and, if necessary, recovery of the protein from the culture. be able to. These operations are performed according to a method known to those skilled in the art, or a method described in the literature (Molecular Cloning, T. Maniatis et al., CSH Laboratory (1983), DNA Cloning, DM. Glover, IRL PRESS (1985)). It can be carried out.
[0037]
As a method for measuring the function of PAR2, specifically, the activity of increasing the intracellular calcium concentration, arachidonic acid release (Kong, W. et. Al., Proc. Nat. Acad. Sci. USA, 94, 8884- 8889 (1997); McHowat, J., et al., J. Urology, 165, 2063-2067 (2001)), promotion of production of intracellular inositol triphosphate, fluctuation of cell membrane potential, activation of c-fos (Hogerwerf, W. A., et al., J. Neurosci., 21, 9036-9042 (2001); Coelho, AM, et. Al., Gastroenterology, 122, 1035-1047 (2002)), or protein inkers. A method for measuring a GPCR-specific function such as an activity of promoting the phosphorylation of an intracellular protein by ZeC or MAP kinase, or a prostaglandin E2 (PGE2) release promoting activity from a cell, a salivary secretion promoting activity in a mammal, and the like. Can be
The physiological activity can be measured using a known method or a commercially available measurement kit. That is, cells containing PAR2 are cultured in a multiwell plate or the like. Replace with a fresh medium or an appropriate buffer that does not show cytotoxicity, add the test compound and / or ligand, etc., incubate for a certain period of time, and then extract the cells or collect the supernatant to produce the product Is quantified according to the respective method. When the production of a substance (for example, arachidonic acid) as an indicator of a physiological activity cannot be measured by a degrading enzyme contained in a cell, an inhibitor for the degrading enzyme can be used in combination. A specific measuring method will be described below.
[0038]
(1) Fluorescent imaging plate reader (FLIPR) method
As a method for measuring the intracellular calcium concentration increasing activity, a human embryonic kidney-derived cell line HEK293 cell, a human lung cancer-derived cell line A549, or a human fetal lung-derived cell line HFL1, such as a cultured cell line that can express PAR2, Cultivation in a medium containing a calcium-binding fluorescent probe such as -3 AM (Molecular Probe) and probenecid (used as an inhibitor of multiple drug-resistance pump to retain the dye in the cell), and a PAR2 agonist (Synthetic peptide such as NH2-SLIGRL-CONH2) or a PAR2 activator such as trypsin is added, and the cells are further cultured for a certain period of time (for example, 30 minutes to 2 hours). Calcium concentration Detecting at FLIPR (Molecular Devices, Inc.) as a change in fluorescence signal. The detection is carried out by exciting the cells with a 488 nm argon laser and capturing the fluorescence of Fluo-3 bound to calcium ions at a wavelength of 500-560 nm. For example, the value obtained by subtracting the value before the addition as the background from the fluorescence intensity from immediately after the addition of the ligand to 60 seconds after can be used as the activity. When a test substance is added, it is preferable to add it before adding the PAR2 activator. The PAR2 function inhibitory activity of the test substance can be evaluated by using as an index whether or not the amount of calcium ions in the presence of the test substance decreases as compared to the amount of calcium ions in the absence of the test substance.
In the above, a PAR2 expression vector, a Gq protein or a Gqs5 protein (Gqs5 means a Gq protein in which the C-terminal is substituted with 5 C-terminal amino acid residues of the Gs protein) or a Gqi5 protein ( Gqi5 means a Gq protein in which the C-terminal is replaced with the C-terminal 5 amino acid residues of Gi protein.) An expression vector is prepared, and a gene transfer reagent Lipofectamine (Invitrogen) or the like is introduced into CHO-K1 cells or the like. And co-introduced to prepare transformed cells for use.
[0039]
(2) Method for measuring the amount of inositol triphosphate produced
Human pancreatic cancer-derived cell line PAR2 is activated by adding a PAR2 agonist (a synthetic peptide such as NH2-SLIGRL-CONH2) or a PAR2 activator such as trypsin to a cultured cell line such as MIA PaCa-2 cells. After a certain time, such as trichloroacetic acidacidAfter stopping the reaction by adding, the inositol triphosphate in the cells is extracted using an aqueous solution containing an organic solvent such as diethyl ether or the like, and the amount of inositol triphosphate can be quantified. it can.
As a quantification method,3H] Assay system (Amersham, TRK1000; IP3 antibody and [3H] A system for measuring the amount of IP3 in vivo using IP3).
[0040]
(3) Method for measuring protein kinase C activation
Cultured cell lines such as human pancreatic cancer-derived cell lines, MIA PaCa-2 cells,32[P] In the presence of ATP, a PAR2 agonist (a synthetic peptide such as H-SLIGRL-NH2) or a PAR2 activator such as trypsin is added to activate PAR2. After stopping the reaction by cooling to 0 ° C. after a certain period of time, the cells were homogenized and buffered (eg, 0.25 mM sucrose, 20 mM triethanolamine, 2 mM EDTA, 0.5 mM EGTA, 1 mM DTE, 10 ug / ml Leuptin, (1 mM PMSF solution), and after sonication, centrifugation is performed to collect both the supernatant (1) and the sediment. The precipitate is further homogenized in a buffer (for example, a buffer containing Triton X-100), and centrifuged to collect the supernatant (2). The supernatants (1) and (2) were separated and purified from a PKC fraction using a DEAE ion exchange column or the like, and were used as PKC substrates [32[P] ATP-derived [32By measuring the amount of [P] phosphoric acid, the amount of PKC can be quantified.
[0041]
(4) Measurement of PGE2 amount
After the human synovial cell line SW982 cells are cultured for a certain period of time in a carbon dioxide gas culturing device in the presence of a test substance, an agonist peptide is added, and the cells are further cultured for a certain period of time (for example, 24 hours). The amount of prostaglandin E2 (PGE2) in the culture supernatant can be measured using prostaglandin E2 EIA (Cayman chemical 514010). The selection of the candidate substance can be performed by using as an index whether or not the amount of PGE2 in the presence of the test substance decreases as compared to PGE2 in the absence of the test substance.
[0042]
The PAR2 inhibitory activity of the test substance is evaluated by comparing the PAR2 function (measured value in step (b)) in the presence of the test substance measured as described above with the PAR2 function in a control not contacted with the test substance. be able to.
Specifically, the function level of PAR2 in the cells of any of the above (1) to (3) to which the test substance was added was lower than that of the cells to which the test substance was not added (control). ,It can be carried out.
[0043]
Using the PAR2 inhibitory activity of the test substance evaluated as described above as an index, a candidate substance for a therapeutic agent for skin diseases can be selected. For example, a test substance in which the functional level of PAR2 in the presence of the test substance is reduced by 10%, preferably 30%, particularly preferably 50% or more as compared with the functional level in control cells not contacted with the test substance is considered as a candidate substance. Can be sorted out.
[0044]
III. Method for measuring PAR2 binding inhibitory activity
Since PAR2 is a receptor, a method for evaluating PAR2 inhibitory activity based on receptor binding inhibitory activity comprises the following steps (a) to (d):
(A) contacting the test substance with an aqueous solution containing PAR2 and PAR2 ligand, a cell, or a cell membrane fraction prepared from the cell;
(B) measuring the amount of PAR2 agonist bound to PAR2 in (a),
(C) comparing the value of (b) with the amount of PAR2 agonist bound to PAR2 in a control not contacted with the test substance;
(D) evaluating the PAR2 inhibitory activity of the test substance based on the results of (c),
including.
Here, the PAR2 ligand (receptor binding substance) includes a PAR2 agonist, a PAR2 antagonist, and a labeled ligand obtained by labeling them. In addition, a derivative of a known agonist compound prepared by peptide synthesis or the like can also be used.
[0045]
Specifically, the amount of binding of a ligand (agonist compound, etc.) to PAR2 in the presence of the test substance is measured, and the amount of binding is determined by the amount of binding of the ligand (agonist compound, etc.) to PAR2 in the absence of the test substance (control binding). Amount).
[0046]
In this case, the binding inhibition between the PAR2 and the ligand may be (1) a mode of inhibiting the binding of the ligand to PAR2 by binding to the receptor, or (2) a binding to the ligand. Accordingly, the embodiment may inhibit the binding of the ligand to PAR2. However, particularly in the case of the mode (1), it is necessary to select a test substance which does not exert the same effect as the ligand even when bound to PAR2.
Therefore, the test substance selected by the above screening method is further subjected to a step of selecting a test substance having the same action as the ligand or a test substance that antagonizes the action of the ligand from the selected test substances. Is preferred. Specifically, the above II. Can be mentioned.
[0047]
Here, PAR2 may be a purified product (isolate) or an aqueous solution thereof, or may be a cell containing the PAR2 or a cell fraction thereof. Here, examples of the aqueous solution containing PAR2 include, for example, an aqueous solution containing PAR2, a cell lysate, a cell lysate, a nuclear extract, or a cell culture supernatant containing these proteins.
As the cell containing PAR2 used in the screening of the present invention, a cell which naturally expresses the receptor may be used, or a PAR2-encoding gene may be introduced into a cell to obtain a transformed cell. Is also good.
[0048]
The transformed cells were obtained from Molecular Cloning 2nd Edt. , Cold Spring Harbor Laboratory Press (1989), etc., according to a method known to those skilled in the art. For example, the cDNA of the receptor is inserted into a known expression vector such as pCAGGS (Gene 108, 193-199 (1991)), pcDNA1.1, pcDNA3.1 derivative (Invitrogen). Thereafter, the cells are introduced into an appropriate host and cultured, whereby a transformed cell expressing a protein corresponding to the DNA of the introduced receptor can be prepared. As the host, CHO cells, C127 cells, BHK21 cells, COS cells, and the like, which are generally widely used, can be used, but are not limited thereto, and yeast, bacteria, insect cells, and the like can be used. You can also.
[0049]
As a method for introducing an expression vector having PAR2 cDNA into a host cell, any method may be used as long as it is a method for introducing a known expression vector into a host cell. For example, the calcium phosphate method (J. Virol., 52, 456-467 (1973)), a method using LT-1 (manufactured by Panvera), a method using a lipid for gene transfer (Lipofectamine, Lipofectin; manufactured by Gibco-BRL), and the like.
The cells that naturally express PAR2 and the transformed cells that express PAR2 can be used for screening as they are. When cell membranes are used for screening, for example, a cell membrane fraction can be obtained as follows. it can. That is, first, a hypotonic buffer is added to the cells, the cells are hypotonically disrupted, homogenized, and centrifuged to obtain a precipitate of the cell membrane fraction. By suspending this precipitate in a buffer, a cell membrane fraction containing PAR2 can be obtained. The obtained cell membrane fraction can be purified by a conventional method using a column to which an antibody is bound.
[0050]
As the ligand, an agonist compound may be used as it is, or a labeled agonist compound labeled with an arbitrary labeling substance may be used. Examples of the labeling substance include radioisotopes (3H, 14C, 35S, 125I, etc.), fluorescent substances (Molecular Probes, Alexa Protein Labeling Kits, etc.), and chemiluminescent substances (Assay Designs, Chemiluminescence Labeling Kit, etc.). be able to.
[0051]
The measurement of the receptor binding inhibitory activity is specifically exemplified by the following method. That is, an aqueous solution containing a receptor (PAR2) (usually a buffer solution is used), a test compound solution prepared at an appropriate concentration of 10 −3 to 10 −10 M in the cell or cell membrane fraction (usually, a solvent is used). Water or a buffer is used, but ethanol or DMSO may be added depending on the solubility), and then a ligand is added, followed by reacting for a certain time (usually 10 minutes to 2 hours). Thereafter, the supernatant is separated by centrifugation or the like, and the amount of ligand is measured. Alternatively, the precipitate containing the transformed cell or cell membrane fraction can be dissolved in a solution containing a surfactant and a base, and then the amount of ligand contained in the solution can be measured. Here, if the ligand is a labeled agonist compound labeled with a radioisotope, the radioactivity may be measured. The amount of ligand may be measured by purifying a sample by HPLC or the like. For example, when an unlabeled ligand is used, a fraction containing the ligand can be separated using HPLC and detected by UV (for example, 220 nm or 254 nm).
[0052]
By comparing the above values with the values obtained when a solvent was used as a blank in place of the test compound (control binding amount), it was determined whether or not the test compound inhibited the binding between the receptor and the labeled ligand. Can be evaluated. That is, screening of a candidate substance can be performed by using as an index whether or not the amount of binding in the presence of the test substance is smaller than the amount of binding in the absence of the test substance.
For the specific inhibition rate (%), the following formula:
{1- (amount of labeled agonist ligand bound to PAR2 when test substance was added) / (amount of labeled agonist ligand bound to PAR2 when no test substance was added)} X100
Can be obtained by calculating
[0053]
The PAR2 inhibitory activity of the test substance can be evaluated based on the test substance inhibition rate measured as described above. For example, a test substance having an inhibition rate of 10% or more, preferably 30% or more, particularly preferably 50% or more can be selected as a candidate substance.
[0054]
The test substance (candidate substance) screened by the screening method of the present invention is not limited, but includes nucleic acids (including antisense nucleotides of the gene of the present invention), peptides, proteins, organic compounds, inorganic compounds, and the like. Specifically, the test is carried out by bringing these test substances or a sample containing them (test sample) into contact with the cells and / or tissues. Such test samples include, but are not limited to, cell extracts containing test substances, expression products of gene libraries, synthetic low molecular weight compounds, synthetic peptides, natural compounds, and the like.
[0055]
A second aspect of the present invention relates to a therapeutic agent for skin diseases, comprising a compound having PAR2 inhibitory activity as an active ingredient. The compound having a PAR2 inhibitory activity or a pharmaceutically acceptable salt thereof has an effect of suppressing skin itch symptoms, and is effective as a therapeutic agent for skin diseases, specifically, a therapeutic agent for skin diseases accompanied by itch symptoms.
Here, the compound having PAR2 inhibitory activity is not particularly limited as long as it is a compound that suppresses the expression and function of PAR2 described in the above-mentioned screening method, and is not particularly limited, and may be a synthetic low-molecular compound, a synthetic peptide, a natural compound, a protein, a nucleic acid, Sugar compounds and the like. Preferably, it is a compound having a molecular weight of 200 to 2,000, more preferably 300 to 1,000, and for example, a compound having a PAR2 inhibitory activity of 30% or more at 10 μg / ml in the method described in Example 18.
[0056]
Specific examples of the compound having the PAR2 inhibitory activity include the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof.
In the formula (1), the alkyl group represents an alkyl group having 1 to 10 carbon atoms, preferably an alkyl group having 1 to 6 carbon atoms, and more preferably an alkyl group having 1 to 4 carbon atoms. Specific examples include a methyl group, an ethyl group, a propyl group, a 1-methylethyl group, and a butyl group.
In the formula (1), the alkoxy group represents an alkoxy group having 1 to 10 carbon atoms, preferably an alkoxy group having 1 to 6 carbon atoms, and more preferably an alkoxy group having 1 to 4 carbon atoms. Specific examples include a methoxy group, an ethoxy group, a propoxy group, a 1-methylethoxy group, and a butoxy group.
In the formula (1), the halogen atom represents a fluorine atom, a chlorine atom, a bromine atom, or an iodine atom, and preferably represents a fluorine atom or a chlorine atom.
In the present specification, the haloalkyl group represents the same or different haloalkyl group having 1 to 2 carbon atoms containing 1 to 3 halogen atoms, and specifically, a trifluoromethyl group, a difluoromethyl group, Examples thereof include a 1,1-trifluoroethyl group and a 2,2-difluoroethyl group. Preferably, a trifluoromethyl group is used.
In Formula (1), examples of the heterocycloalkane include pyrrolidine, piperidine, piperazine, morpholine, thiomorpholine, thiomorpholine oxide, thiomorpholine dioxide, perhydroazepine, and perhydrodiazepine. Preferably, pyrrolidine, piperidine, piperazine or perhydroazepine is used.
Examples of the substituent when the heterocycloalkane is substituted include an alkyl group, a substituted or unsubstituted arylcarbonyl group, an alkylcarbonyl group, an alkoxycarbonyl group, a substituted or unsubstituted aryl group, or a substituted or unsubstituted group. Arylalkyl groups.
Preferably, the substituent when substituted on a carbon atom includes an alkyl group having 1 to 4 carbon atoms or an alkoxycarbonyl group having 2 to 4 carbon atoms.
When the heterocycloalkane is substituted on a nitrogen atom, examples of the substituent include an alkyl group having 1 to 4 carbon atoms, an alkoxycarbonyl group having 2 to 4 carbon atoms, and an alkylcarbonyl group having 1 to 4 carbon atoms. A substituted or unsubstituted arylcarbonyl group, a substituted or unsubstituted aryl group, or a substituted or unsubstituted arylalkyl group.
Examples of the aryl in the substituted or unsubstituted arylcarbonyl group, the substituted or unsubstituted aryl group, or the substituted or unsubstituted arylalkyl group include phenyl, 1-naphthyl, and 2-naphthyl, and particularly preferably phenyl. Is mentioned.
Examples of the substituent in the substituted arylcarbonyl group, substituted aryl group, or substituted arylalkyl group include a halogen atom, an alkyl group having 1 to 4 carbon atoms, an alkoxy group having 1 to 4 carbon atoms, and a methylenedioxy group. Is mentioned.
[0057]
In the compound represented by the general formula (1), R1And R2Is preferably 1 to 2 nitrogen atoms or 0 to 1 oxygen atom which may be substituted with an alkyl group, an arylcarbonyl group, an alkylcarbonyl group, or an alkoxycarbonyl group by bonding together with an adjacent carbon atom. , And a heterocycloalkane containing 0 to 1 sulfur atom, wherein the heterocycloalkane is preferably piperidine, piperazine, or perhydroazepine.
Alternatively, in the compound represented by the general formula (1),1Represents an alkyl group having 1 to 4 carbon atoms;2Is also a preferred embodiment in which represents a hydroxyl group, an alkoxy group, a nitrile group, a substituted or unsubstituted aryl group, or an alkyl group which may be substituted with a substituted or unsubstituted heteroaryl group. The aryl group includes a phenyl group. The heteroaryl group includes a pyridyl group. R2When the substituent of the alkyl group in the above represents a substituted aryl group or a substituted heteroaryl group, examples of the substituent of the aryl group or the heteroaryl group include a halogen atom, an alkyl group having 1 to 4 carbon atoms, and an alkyl group having 1 to 4 carbon atoms. Examples thereof include a haloalkyl group and an alkoxy group having 1 to 4 carbon atoms.
In the compound represented by the general formula (1), R3Examples of the aryl group include a phenyl group. When the aryl group is substituted, the substituent may be a halogen atom, an alkyl group having 1 to 4 carbon atoms, a haloalkyl group having 1 to 2 carbon atoms, or a carbon atom. And alkoxy groups of formulas 1 to 4. R3Preferably represents an alkyl group having 1 to 3 carbon atoms, a phenyl group, or a haloalkyl group having 1 to 2 carbon atoms. A particularly preferred example is a trifluoromethyl group.
In the compound represented by the general formula (1), R4And R5Preferably represents the same or different and represents a hydrogen atom or a halogen atom.
[0058]
In the general formula (2), R6Preferably represents a hydrogen atom, an alkylcarbonyl group having 2 to 4 carbon atoms, or a benzoyl group.
In the general formula (2), R7Preferably represents a hydrogen atom or an alkyl group having 1 to 3 carbon atoms, and more preferably represents a hydrogen atom or a methyl group.
In the general formula (2), R8Preferably represents a hydrogen atom, an alkyl group having 1 to 3 carbon atoms, or an alkoxycarbonyl group having 2 to 4 carbon atoms, and more preferably represents a hydrogen atom, a methyl group, a methoxycarbonyl group, or an ethoxycarbonyl group. . Where R8Represents an alkoxycarbonyl group;7Preferably represents a hydrogen atom.
In the general formula (2), R9Preferably represents a hydrogen atom, an alkyl group having 1 to 3 carbon atoms, or an alkoxycarbonyl group having 2 to 4 carbon atoms, and more preferably represents a hydrogen atom, a methoxycarbonyl group, or an ethoxycarbonyl group.
In the general formula (2), R10Preferably represents a methyl group or an ethyl group;11Represents preferably an ethyl group or a propyl group which may be substituted with a hydroxyl group or a pyridyl group.
[0059]
Many of the compounds represented by the formula (1) are known and can be obtained as commercial products. Moreover, it can also be manufactured by the following method.
Manufacturing method:
Embedded image
The hydrazone of the formula (1-2) can be produced by reacting a compound of the formula (1-1), which is a known compound, with hydrazine monohydrate at 0 to 100 ° C under acidic conditions. Examples of the solvent include alcohol solvents such as methanol and ethanol, halogen solvents such as chloroform and dichloromethane, ether solvents such as dimethylformamide, dimethyl sulfoxide, acetone, acetonitrile, dioxane and tetrahydrofuran, water, and a mixed solvent thereof. Is used. Alcohol solvents such as methanol and ethanol are more preferred. Acetic acid, hydrochloric acid, sulfuric acid, trifluoroacetic acid and the like can be used as the acid. In this reaction, the compound of the formula (1-3) can be produced by a method such as using sulfuric acid as an acid in chloroform without isolating the compound of the formula (1-2).
The compound of formula (1-3) can be obtained by adding a base to an alcoholic solvent such as methanol or ethanol and heating the compound of formula (1-2). As the base, an inorganic base such as sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, or sodium hydrogen carbonate, or an organic base such as triethylamine or pyridine is used. Preferably, potassium carbonate or sodium carbonate can be used.
The compound of the formula (1-5) is prepared by converting a compound of the formula (1-3) and a 1,3-diketone of the formula (1-4) from room temperature in the presence or absence of a base or an acid. It can be produced by reacting at 100 ° C. Examples of the solvent include alcohol solvents such as methanol and ethanol, halogen solvents such as chloroform and dichloromethane, ether solvents such as dimethylformamide, dimethyl sulfoxide, acetone, acetonitrile, dioxane and tetrahydrofuran, water, and a mixed solvent thereof. Are used. Among them, alcohol solvents such as methanol and ethanol are more preferable. As the acid, acetic acid, hydrochloric acid, or a Lewis acid such as zinc chloride can be used. As the base, an organic base such as pyrrolidine or piperidine can be used.
The compound of the formula (1-4) can be prepared by using a known compound or by a method described in Examples in the present specification. Further, the compound of the formula (1-5) can also be produced from the compound of the formula (1-2) without isolating the compound of the formula (1-3).
The compound of the formula (1-6) can be produced by hydrolyzing the compound of the formula (1-5) to a carboxylic acid and condensing it with a commercially available amine compound according to a known method. Examples of the condensation method include a method using a water-soluble carbodiimide (WSCI) described in this specification, a mixed acid anhydride method, a method using an acid halide, and the like. The condensation method is described in "Comprehensive Organic Transformation (Larock, RC, VCH Publishers, Inc. 1989)" and the like.
[0060]
Particularly preferred examples of the compound represented by the general formula (1) include compounds represented by the following formulas 51 to 64.
Embedded image
Pharmaceutically acceptable salts include acid addition salts and base addition salts. Examples of the acid addition salt include inorganic salts such as hydrochloride, hydrobromide, sulfate, hydroiodide, nitrate and phosphate, citrate, oxalate, acetate, formate, and the like. Organic acid salts such as propionate, benzoate, trifluoroacetate, maleate, tartrate, methanesulfonate, benzenesulfonic acid, and paratoluenesulfonate are included. Inorganic base salts such as salts, potassium salts, calcium salts, magnesium salts, and ammonium salts; and organic base salts such as triethylammonium salts, triethanolammonium salts, pyridinium salts, diisopropylammonium salts, and the like.Arginine, aspartic acid, and glutamic acid And amino acid salts such as basic or acidic amino acids. Further, the compound having a PAR inhibitory activity of the present invention may be a hydrate or a solvate such as ethanolate.
[0062]
The compound represented by the general formula (1) or a pharmaceutically acceptable salt thereof is effective as a therapeutic agent for skin diseases, specifically, a therapeutic agent for skin diseases accompanied by itch symptoms.
[0063]
Examples of skin diseases accompanied by itching symptoms include atopic dermatitis, contact dermatitis, psoriasis, eczema, hives, xerosis, insect bites, soybeans, burning, scabies and the like. Preferable are psoriasis and atopic dermatitis
[0064]
Hereinafter, the dose and the dosage form when the therapeutic agent for the above-mentioned diseases is used as a pharmaceutical containing the compound having the PAR2 inhibitory activity of the present invention as an active ingredient will be described.
The compound of the present invention or a pharmaceutically acceptable salt thereof can be administered orally or parenterally, preferably orally, and in various dosage forms suitable for oral or parenteral administration, respectively, as a medicament, human and non-human. Used for animals. For example, when administered orally, it can be administered in a commonly used dosage form, for example, a tablet, capsule, syrup, suspension or the like. For parenteral administration, liquids such as solutions, emulsions and suspensions are administered as injections, rectally administered in the form of suppositories, and ointments, lotions, creams, and other application forms. It can be administered transdermally, a spray, an aerosol, or the like can be administered by inhalation in the form of a spray, or can be administered as a drip (eg, eye drops). Such a dosage form can be manufactured according to a general method by blending an active ingredient with auxiliaries such as a usual carrier, excipient, binder and stabilizer. When used in the form of an injection, a buffer, a solubilizing agent, an isotonic agent and the like can be added. The dose and frequency of administration vary depending on the disease to be treated, the condition of the patient, age, body weight, etc., and the form of administration. However, when administered orally, the active ingredient is usually administered to an adult in an amount of about 1 to 1000 mg per day. A range, preferably, a range of about 10-500 mg, can be administered once or in several divided doses. When administered as an injection, the active ingredient can be administered in a range of about 0.1 to about 500 mg, preferably in a range of about 3 to about 100 mg, once or in several divided doses.
[0065]
【Example】
Hereinafter, the present invention will be described in detail with reference to Examples and Reference Examples, but the present invention is not limited thereto.
Example 1
(2E) -3-Cyano-2-hydrazonopropanoic acid ethyl
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.34-1.41 (m, 3H), 3.46 (s), 3.63 (s), 4.26-4.38 (m, 2H), 6.52 (brs), 8.50 (br)
[0066]
Example 2
Ethyl 5-phenyl-7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine-2-carboxylate
Embedded image
[0067]
Example 3
Potassium 5-phenyl-7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine-2-carboxylate
Embedded image

1H-NMR (400 MHz, DMSO-d6) Δ ppm: 6.87 (s, 1H), 7.53-7.59 (m, 3H), 8.02 (s, 1H), 8.24-8.31 (m, 2H)
[0068]
Example 4
5-phenyl-2- (piperidin-1-ylcarbonyl) -7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.62-1.78 (m, 6H), 3.75-3.83 (m, 4H), 7.17 (s, 1H), 7.54-7.59 (m, 3H). , 7.68 (s, 1H), 8.09-8.16 (m, 2H)
[0069]
Example 5
2-[(2-methylpiperidin-1-yl) carbonyl] -5-phenyl-7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.30-1.40 (m, 3H), 1.49-1.90 (m, 6H), 2.90-3.30 (m, 1H), 4.20-4.74 ( m, 1H), 4.55-5.14 (m, 1H), 7.15 (s, 1H), 7.54-7.59 (m, 3H), 7.67 (s, 1H), 8 .09-8.15 (m, 2H)
[0070]
Example 6
2- (azepan-1-ylcarbonyl) -5-phenyl-7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.60-1.73 (m, 4H), 1.81-1.93 (m, 4H), 3.73-3.85 (m, 4H), 7.23 (s, 1H). , 7.54-7.59 (m, 3H), 7.68 (s, 1H), 8.09-8.15 (m, 2H).
[0071]
Example 7
2-[(4-benzoylpiperazin-1-yl) carbonyl] -5-phenyl-7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 3.50-4.25 (m, 8H), 7.28 (s, 1H), 7.55-7.60 (m, 3H), 7.72 (brs, 1H), 8.10 -8.15 (m, 2H)
[0072]
Example 8
Ethyl 1-{[5-phenyl-7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidin-2-yl] carbonyl} piperidine-2-carboxylate
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.26-1.35 (m, 3H), 1.40-1.90 (m, 5H), 2.28-2.43 (m, 1H), 3.00-3.40 ( m, 1H), 4.20-4.32 (m, 2H), 4.45-4.80 (m, 1H), 5.45-5.60 (m, 1H), 7.21-7. 28 (m, 1H), 7.53-7.60 (m, 3H), 7.67-7.71 (m, 1H), 8.10-8.06 (m, 2H)
[0073]
Example 9
Ethyl 1-{[5-phenyl-7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidin-2-yl] carbonyl} piperidine-4-carboxylate
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.28 (t, 3H, J = 7.1 Hz), 1.80-2.12 (m, 4H), 2.64 (m, 1H), 3.13 (m, 1H), 3 .37 (m, 1H), 4.18 (q, 1H, J = 7.1 Hz), 4.46-4.62 (m, 2H), 7.21 (s, 1H), 7.54-7 .60 (m, 3H), 7.69 (s, 1H), 8.10-8.16 (m, 2H)
Example 10
1- (4-chlorophenyl) -4,4,4-trifluorobutane-1,3-dione
Embedded image
1H-NMR (400 MHz, DMSO-d6) Δ ppm: 7.02 (s, 1H), 7.66 (d, 2H, J = 8.7 Hz), 8.14 (d, 2H, J = 8.7 Hz)
[0074]
Example 11
Ethyl 5- (4-chlorophenyl) -7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine-2-carboxylate
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.47 (t, 3H, J = 7.1 Hz), 4.51 (q, 2H, J = 7.1 Hz), 7.38 (s, 1H), 7.52-7.57 ( m, 2H), 7.71 (s, 1H), 8.07-8.12 (m, 2H)
[0075]
Example 12
Potassium 5- (4-chlorophenyl) -7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine-2-carboxylate
Embedded image
[0076]
Example 13
5- (4-chlorophenyl) -2- (piperidin-1-ylcarbonyl) -7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.61-1.77 (m, 6H), 3.76-3.83 (m, 2H), 7.18 (s, 1H), 7.51-7.56 (m, 2H). , 7.63 (s, 1H), 8.06-8.11 (m, 2H).
[0077]
Example 14
5- (4-chlorophenyl) -2-[(2-methylpiperidin-1-yl) carbonyl] -7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.31-1.40 (m, 3H), 1.50-1.90 (m, 6H), 2.90-3.30 (m, 1H), 4.20-4.73 ( m, 1H), 4.53-5.15 (m, 1H), 7.14 (s, 1H), 7.51-7.56 (m, 2H), 7.63 (s, 1H), 8 .06-8.11 (m, 2H)
[0078]
Example 15
2- (azepan-1-ylcarbonyl) -5- (4-chlorophenyl) -7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 1.60-1.71 (m, 4H), 1.80-1.92 (m, 4H), 3.73-3.84 (m, 4H), 7.23 (s, 1H) , 7.51-7.56 (m, 2H), 7.63 (s, 1H), 8.06-8.11 (m, 2H).
[0079]
Example 16
Ethyl 1-{[5- (4-chlorophenyl) -7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidin-2-yl] carbonyl} piperidine-2-carboxylate
Embedded image
Example 17
5- (4-chlorophenyl) -N- (2-hydroxyethyl) -N-methyl-7- (trifluoromethyl) pyrazolo [1,5-a] pyrimidine-2-carboxamide
Embedded image
1H-NMR (400 MHz, CDCl3) Δ ppm: 3.05-3.24 (m, 1H), 3.24-3.47 (m, 3H), 3.78-4.00 (m, 4H), 7.27-7.33 ( m, 1H), 7.51-7.58 (m, 2H), 7.65-7.70 (m, 1H), 8.06-8.12 (m, 2H).
[0080]
Example 18: Measurement of intracellular calcium concentration using Fluorometric Imaging Plate Reader (FLIPR)
[Materials and methods]
A human embryonic kidney-derived cell line HEK293 cell cultured in a DMEM culture solution (Invitrogen) supplemented with 10% fetal bovine serum (10% FBS) was 2.5 × 10 55The cells were adjusted to cells / ml, and seeded at 80 μl / well on a 96-well black plate (clear bottom) (Falcon) coated with poly-D-lysine, and cultured overnight in a carbon dioxide gas culture apparatus. A FLIPR Calcium Assay Kit (Molecular Devices) prepared with Hanks-20 mM Hepes buffer (pH 7.4) was added to the cells at 80 μl / well, and the cells were cultured for 1 hour in a carbon dioxide gas culture device. Trypsin (SIGMA T8642) adjusted to 0.0005% as a PAR2 activator with Hanks-20 mM Hepes buffer (pH 7.4) was added to a 96-well V-bottom polypropylene plate (Nunc) to prepare a reagent plate. did. Immediately before measurement with FLIPR (Molecular Devices), a test substance prepared with Hanks-20 mM Hepes buffer (pH 7.4) was added to the cells at 40 μl / well, followed by stirring with a plate shaker. The cell plate and the reagent plate to which the test substance was added were set on the FLIPR, and changes in intracellular calcium concentration were detected with a CCD camera. The measurement was performed at room temperature for 70 seconds, and the addition of the reagent from the reagent plate to the cell plate (50 μl / well) was performed with a 96-well automatic dispenser built in the FLIPR main body.
The inhibition rate of increase in intracellular calcium of a test substance is calculated by the following formula:
(Change in intracellular calcium concentration in the presence of test substance-change in intracellular calcium concentration in the absence of Trypsin) / (Change in intracellular calcium concentration in the absence of test substance-intracellular in the absence of test substance and Trypsin) Change in calcium concentration)
Can be obtained by
[Results] Table 1 shows the inhibition rate (%) of the intracellular calcium increase of the test substance.
[Table 1]
[0081]
Example 19: Evaluation of PAR2 inhibitory activity using measurement of PGE2 amount
SW982 cells 2.5 × 105The cells were diluted to each cell / ml with each medium, and 100 μl / well of cells were seeded on a 96-well plate. After pre-incubation overnight in a carbon dioxide gas culture apparatus, the medium was removed, and a medium containing 5 μg / ml compound was added at 80 μl / well (0 μl for the control group, control peptide group, agonist peptide alone group, and IL-1β group). Medium containing 1% DMSO was added). A medium containing 500 uM agonist peptide was added at 20 μl / well to the well containing the compound (final concentration: 4 μg / ml compound · 100 uM agonist peptide). 20 μl / well of each medium in the wells of the control group, 20 μl / well (final concentration 100 μM) of the medium containing the control peptide in the wells of the control peptide group, and 20 μl / well of the medium containing the agonist peptide in the wells of the agonist peptide alone group. / Well (final concentration: 100 μM) and a medium containing 50 ng / ml IL-1β were added to the wells of the IL-1β group at a ratio of 20 μl / well (final concentration: 10 ng / ml). Twenty-four hours later (N = 3), each supernatant was collected and stored at −80 ° C. until measurement. The amount of PGE2 in each supernatant was measured using prostaglandin E2 EIA (Cayman chemical 514010). The PGE2 production inhibition rate of the compound of the example was calculated by setting the value of the PGE2 amount of the control peptide to the value of the inhibition rate of 100% and the amount of the PGE2 of the agonist peptide to the value of the inhibition rate of 0%. That is, the PAR2 inhibition rate of the test substance is expressed by the following formula:
(Amount of PGE2 in the presence of an agonist in the test substance-Amount of PGE2 in the presence of a control peptide in the test substance) / (Amount of PGE2 in the presence of the agonist in the control-Amount of PGE2 in the presence of the control peptide in the control)
I asked for it. Table 2 shows the PGE2 production inhibitory rates of the compounds of the examples.
[Table 2]
[0082]
Example 20: Itching-inducing activity of PAR2
A 7-week-old male ICR mouse was intradermally injected with an agonist peptide or a control peptide at 50 pmol / 20 ul into the back of the shoulder. Ten minutes after the administration, the behavior of scratching the vicinity of the administration part with the hind limb was measured (one measurement was taken until the hind limb left the ground and returned again). The results are shown in Table 3. From this result, it was found that the group to which the agonist peptide (SEQ ID NO: 2) was administered was significantly more frequently scratched near the administration site than the group to which the control peptide (SEQ ID NO: 4) was administered.
Further, after the compound of Example 17 (200 mg / kg) was orally administered, the agonist peptide was administered 30 minutes later in consideration of the blood concentration of the administered compound. In the same manner as described above, the behavior of scratching the vicinity of the administration site on the hind limb for 10 minutes after administration of the agonist peptide was measured. The results are shown in Table 4. From these results, it was found that the compound of Example 1 suppressed the scratching behavior induced by the agonist peptide.
[Table 3]
Dosing solution Number of scratches
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
Physiological saline (solvent for peptide dissolution) twice
SLIGRL (agonist peptide) 25 times
LSIGRL (control peptide) 7 times
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
[Table 4]
Dosing solution Number of scratches
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
Compound of Example 17 + SLIGRL 12 times
SLIGRL 24 times
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
[0083]
【The invention's effect】
ADVANTAGE OF THE INVENTION By this invention, it became possible to provide the method of screening the therapeutic agent for the skin disease accompanied by itch. Further, it has become possible to provide a therapeutic agent for a skin disease accompanied by itch, which comprises a compound having PAR2 inhibitory activity as an active ingredient.
Claims (10)
(1)被験物質のPAR2阻害活性を評価する工程、
(2)(1)の結果に基づいて、PAR2阻害活性を有する被験物質を皮膚疾患治療剤の有効成分として選別する工程、
を含む、皮膚疾患治療剤のスクリーニング方法。The following steps (1) and (2):
(1) a step of evaluating the PAR2 inhibitory activity of the test substance,
(2) selecting a test substance having PAR2 inhibitory activity as an active ingredient of a therapeutic agent for skin diseases based on the result of (1);
A screening method for a therapeutic agent for skin diseases, comprising:
(a)被験物質とPAR2を発現可能な細胞とを接触させる工程、
(b)(a)におけるPAR2の発現量を測定する工程、
(c)(b)の値を、被験物質を接触させない対照におけるPAR2の発現量と比較する工程、
(d)(c)の結果に基づいて、被験物質のPAR2阻害活性を評価する工程
を含むことを特徴とする、請求項1記載の皮膚疾患治療剤のスクリーニング方法。The step (1) includes the following steps (a) to (d):
(A) contacting a test substance with a cell capable of expressing PAR2,
(B) measuring the expression level of PAR2 in (a),
(C) comparing the value of (b) with the expression level of PAR2 in a control not contacted with the test substance;
(D) The method for screening a therapeutic agent for skin diseases according to claim 1, comprising a step of evaluating the PAR2 inhibitory activity of the test substance based on the results of (c).
(a)被験物質と、PAR2及びPAR2活性化因子を含む細胞、または該細胞から調製した細胞画分とを接触させる工程、
(b)(a)におけるPAR2由来の機能を測定する工程、
(c)(b)の値を、被験物質を接触させない対照におけるPAR2由来の機能と比較する工程、
(d)(c)の結果に基づいて、被験物質のPAR2阻害活性を評価する工程、
を含むことを特徴とする、請求項1記載の皮膚疾患治療剤のスクリーニング方法。The step (1) includes the following steps (a) to (d):
(A) contacting a test substance with a cell containing PAR2 and a PAR2 activator, or a cell fraction prepared from the cell;
(B) measuring the PAR2-derived function in (a),
(C) comparing the value of (b) with a PAR2-derived function in a control not contacted with the test substance;
(D) evaluating the PAR2 inhibitory activity of the test substance based on the results of (c),
The method for screening a therapeutic agent for skin diseases according to claim 1, which comprises:
R3は、水素原子、アルキル基、ハロアルキル基、または置換もしくは無置換のアリール基を表し、
R4およびR5は、独立して、水素原子、ハロゲン原子、アルキル基、アルコキシ基、メチレンジオキシ基、またはハロアルキル基を表す。)
で表される化合物、又はその薬学上許容される塩である、請求項6記載の治療剤。The compound having PAR2 inhibitory activity is represented by the general formula (1):
R 3 represents a hydrogen atom, an alkyl group, a haloalkyl group, or a substituted or unsubstituted aryl group;
R 4 and R 5 independently represent a hydrogen atom, a halogen atom, an alkyl group, an alkoxy group, a methylenedioxy group, or a haloalkyl group. )
The therapeutic agent according to claim 6, which is a compound represented by the formula: or a pharmaceutically acceptable salt thereof.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002338738A JP2004170323A (en) | 2002-11-22 | 2002-11-22 | Screening method for cutaneous disease therapeutic agent |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002338738A JP2004170323A (en) | 2002-11-22 | 2002-11-22 | Screening method for cutaneous disease therapeutic agent |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004170323A true JP2004170323A (en) | 2004-06-17 |
Family
ID=32701867
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002338738A Pending JP2004170323A (en) | 2002-11-22 | 2002-11-22 | Screening method for cutaneous disease therapeutic agent |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004170323A (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005030773A1 (en) * | 2003-09-26 | 2005-04-07 | Dainippon Sumitomo Pharma Co., Ltd. | Novel pyrazolopyrimidine derivatives |
| WO2006127379A3 (en) * | 2005-05-26 | 2007-02-22 | Acadia Pharm Inc | Par2-modulating compounds and their use |
| WO2012026766A2 (en) | 2010-08-25 | 2012-03-01 | (주)네오팜 | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| JP2012531457A (en) * | 2009-07-01 | 2012-12-10 | ピエール、ファブレ、デルモ‐コスメティーク | L-serine for use as a medicament for preventing and / or treating an inflammatory response of the skin |
| JP2013504759A (en) * | 2009-09-18 | 2013-02-07 | ザ プロクター アンド ギャンブル カンパニー | A non-invasive method for measuring histamine from the skin as an objective measure of itching |
| WO2015048245A1 (en) * | 2013-09-25 | 2015-04-02 | Vertex Pharmaceuticals Incorporated | Imidazopyridazines useful as inhibitors of the par-2 signaling pathway |
| WO2017129801A1 (en) * | 2016-01-27 | 2017-08-03 | Universität Zürich | Use of gabaa receptor modulators for treatment of itch |
| WO2018043461A1 (en) | 2016-08-31 | 2018-03-08 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | PYRAZOLO[1,5-a]PYRIMIDINE COMPOUND |
| WO2019163956A1 (en) | 2018-02-26 | 2019-08-29 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | SALT OF PYRAZOLO[1,5-a]PYRIMIDINE COMPOUND AND CRYSTALS THEREOF |
| JPWO2021084760A1 (en) * | 2019-11-01 | 2021-05-06 | ||
| RU2781636C2 (en) * | 2016-01-27 | 2022-10-17 | Университет Цюрих | Use of gabaa-receptor modulators for treatment of itch |
| WO2023067388A1 (en) * | 2021-10-22 | 2023-04-27 | Ildong Pharmaceutical Co., Ltd. | Uses of cftr modulator and/or pde4 inhibitor compounds |
| US11787812B2 (en) | 2020-12-11 | 2023-10-17 | Ildong Pharmaceutical Co., Ltd. | Substituted pyrazolo[4,3-d]pyrimidines and imidazo[5,1 -f][1,2,4]triazines as androgen receptor and phosphodiesterase dual inhibitors |
| US11827640B2 (en) | 2020-10-23 | 2023-11-28 | Ildong Pharmaceutical Co., Ltd. | Substituted pyrazolo[1,5-a]pyrimidines as CFTR modulators |
| KR20240017855A (en) | 2021-06-02 | 2024-02-08 | 오츠카 세이야쿠 가부시키가이샤 | Pyrazolo[1,5-a]pyrimidine compounds for the treatment of skin disorders |
| WO2024096629A1 (en) * | 2022-11-02 | 2024-05-10 | 일동제약 주식회사 | Eye drop composition comprising cftr modulator compound |
| US12134619B2 (en) | 2018-04-18 | 2024-11-05 | Neurocycle Therapeutics, Inc. | Crystalline salts or co-crystals of 2′,6-difluoro-5′-[3-(1-hydroxy-1-methylethyl)-imidazo[1,2-b][1,2,4]triazin-7-yl]biphenyl-2-carbonitrile with p-toluenesulofonic acid as GABAA positive allosteric modulators |
-
2002
- 2002-11-22 JP JP2002338738A patent/JP2004170323A/en active Pending
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005030773A1 (en) * | 2003-09-26 | 2005-04-07 | Dainippon Sumitomo Pharma Co., Ltd. | Novel pyrazolopyrimidine derivatives |
| WO2006127379A3 (en) * | 2005-05-26 | 2007-02-22 | Acadia Pharm Inc | Par2-modulating compounds and their use |
| JP2012531457A (en) * | 2009-07-01 | 2012-12-10 | ピエール、ファブレ、デルモ‐コスメティーク | L-serine for use as a medicament for preventing and / or treating an inflammatory response of the skin |
| JP2013504759A (en) * | 2009-09-18 | 2013-02-07 | ザ プロクター アンド ギャンブル カンパニー | A non-invasive method for measuring histamine from the skin as an objective measure of itching |
| WO2012026766A2 (en) | 2010-08-25 | 2012-03-01 | (주)네오팜 | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| WO2012026765A2 (en) | 2010-08-25 | 2012-03-01 | (주)네오팜 | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| EP2617721A2 (en) * | 2010-08-25 | 2013-07-24 | Neopharm Co., Ltd. | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| EP2617721A4 (en) * | 2010-08-25 | 2014-01-08 | Neopharm Co Ltd | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| US10030024B2 (en) | 2013-09-25 | 2018-07-24 | Vertex Pharmaceuticals Incorporated | Imidazopyridazines useful as inhibitors of the PAR-2 signaling pathway |
| WO2015048245A1 (en) * | 2013-09-25 | 2015-04-02 | Vertex Pharmaceuticals Incorporated | Imidazopyridazines useful as inhibitors of the par-2 signaling pathway |
| CN105683192A (en) * | 2013-09-25 | 2016-06-15 | 沃泰克斯药物股份有限公司 | Imidazopyridazines useful as inhibitors of the par-2 signaling pathway |
| WO2017129801A1 (en) * | 2016-01-27 | 2017-08-03 | Universität Zürich | Use of gabaa receptor modulators for treatment of itch |
| US11529359B2 (en) | 2016-01-27 | 2022-12-20 | Universitat Zurich | Use of GABAA receptor modulators for treatment of itch |
| US10786513B2 (en) | 2016-01-27 | 2020-09-29 | Universitat Zurich | Use of GABAA receptor modulators for treatment of itch |
| RU2781636C2 (en) * | 2016-01-27 | 2022-10-17 | Университет Цюрих | Use of gabaa-receptor modulators for treatment of itch |
| WO2018043461A1 (en) | 2016-08-31 | 2018-03-08 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | PYRAZOLO[1,5-a]PYRIMIDINE COMPOUND |
| CN109071554A (en) * | 2016-08-31 | 2018-12-21 | 卫材R&D管理有限公司 | Pyrazolo [1,5-a] pyrimidine compound |
| US10227349B2 (en) | 2016-08-31 | 2019-03-12 | Eisai R&D Management Co., Ltd. | Pyrazolo[1,5-a]pyrimidine compound |
| KR20190039884A (en) | 2016-08-31 | 2019-04-16 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | Pyrazolo [1,5-a] pyrimidine compound |
| JPWO2018043461A1 (en) * | 2016-08-31 | 2019-06-24 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Pyrazolo [1,5-a] pyrimidine compounds |
| TWI732031B (en) * | 2016-08-31 | 2021-07-01 | 日商衛材R&D企管股份有限公司 | PYRAZOLO[1,5-a]PYRIMDINE COMPOUND |
| CN109071554B (en) * | 2016-08-31 | 2021-03-16 | 卫材R&D管理有限公司 | Pyrazolo [1,5-a ] pyrimidine compounds |
| KR102388621B1 (en) | 2016-08-31 | 2022-04-21 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | Pyrazolo[1,5-a]pyrimidine compound |
| CN111655694A (en) * | 2018-02-26 | 2020-09-11 | 卫材R&D管理有限公司 | Salt of pyrazolo [1,5-a ] pyrimidine compound and crystal thereof |
| CN111655694B (en) * | 2018-02-26 | 2023-03-28 | 卫材R&D管理有限公司 | Salt of pyrazolo [1,5-a ] pyrimidine compound and crystal thereof |
| EP3760631A4 (en) * | 2018-02-26 | 2021-10-20 | Eisai R&D Management Co., Ltd. | Salt of pyrazolo[1,5-a]pyrimidine compound and crystals thereof |
| KR102723164B1 (en) | 2018-02-26 | 2024-10-30 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | Salts of pyrazolo[1,5-a]pyrimidine compounds and their crystals |
| US11312720B2 (en) | 2018-02-26 | 2022-04-26 | Eisai R&D Management Co., Ltd. | Hydrochloride salt of 2-((3R,4S)-1-(5-(4-chloro-3,5-difluorophenyl)-7-((2-fluoro-6-methylphenyl)(methyl)amino)pyrazolo[1,5-a]pyrimidine-2-carbonyl)-3-methoxypiperidin-4-yl) acetic acid and crystals thereof |
| KR20200124655A (en) | 2018-02-26 | 2020-11-03 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | Salts of pyrazolo[1,5-a]pyrimidine compounds and crystals thereof |
| WO2019163956A1 (en) | 2018-02-26 | 2019-08-29 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | SALT OF PYRAZOLO[1,5-a]PYRIMIDINE COMPOUND AND CRYSTALS THEREOF |
| US12134619B2 (en) | 2018-04-18 | 2024-11-05 | Neurocycle Therapeutics, Inc. | Crystalline salts or co-crystals of 2′,6-difluoro-5′-[3-(1-hydroxy-1-methylethyl)-imidazo[1,2-b][1,2,4]triazin-7-yl]biphenyl-2-carbonitrile with p-toluenesulofonic acid as GABAA positive allosteric modulators |
| WO2021084760A1 (en) * | 2019-11-01 | 2021-05-06 | 株式会社 資生堂 | Method for skin state evaluation using activated par-2 in skin as index, method for screening par-2 activation promoter or inhibitor, and par-2 activation inhibitor |
| JP7263538B2 (en) | 2019-11-01 | 2023-04-24 | 株式会社 資生堂 | Skin condition evaluation method using active PAR-2 in skin as index, screening method for PAR-2 activation promoter or inhibitor, and PAR-2 activation inhibitor |
| JPWO2021084760A1 (en) * | 2019-11-01 | 2021-05-06 | ||
| US11827640B2 (en) | 2020-10-23 | 2023-11-28 | Ildong Pharmaceutical Co., Ltd. | Substituted pyrazolo[1,5-a]pyrimidines as CFTR modulators |
| US11787812B2 (en) | 2020-12-11 | 2023-10-17 | Ildong Pharmaceutical Co., Ltd. | Substituted pyrazolo[4,3-d]pyrimidines and imidazo[5,1 -f][1,2,4]triazines as androgen receptor and phosphodiesterase dual inhibitors |
| KR20240017855A (en) | 2021-06-02 | 2024-02-08 | 오츠카 세이야쿠 가부시키가이샤 | Pyrazolo[1,5-a]pyrimidine compounds for the treatment of skin disorders |
| WO2023067388A1 (en) * | 2021-10-22 | 2023-04-27 | Ildong Pharmaceutical Co., Ltd. | Uses of cftr modulator and/or pde4 inhibitor compounds |
| WO2024096629A1 (en) * | 2022-11-02 | 2024-05-10 | 일동제약 주식회사 | Eye drop composition comprising cftr modulator compound |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2004170323A (en) | Screening method for cutaneous disease therapeutic agent | |
| Kim et al. | Melastatin-type transient receptor potential channel 7 is required for intestinal pacemaking activity | |
| EP1536799B1 (en) | Novel 2-amino-4-oxoquinazolones as lxr nuclear receptor binding compounds with partial agonistic properties | |
| Capra | Molecular and functional aspects of human cysteinyl leukotriene receptors | |
| US7220723B2 (en) | Inhibitors of the interaction between HMGB polypeptides and toll-like receptor 2 as anti-inflammatory agents | |
| JP5613671B2 (en) | Novel compound II | |
| US8188041B2 (en) | Inhibitors of the interaction between HMGB polypeptides and toll-like receptor 2 as anti-inflammatory agents | |
| US7531320B2 (en) | Modulation of β-catenin/TCF-activated transcription | |
| JP6804605B2 (en) | Organic compounds | |
| Svelander et al. | Inhibition of cathepsin K reduces bone erosion, cartilage degradation and inflammation evoked by collagen-induced arthritis in mice | |
| JP2001213802A (en) | Compound for treating female sexual dysfunction | |
| US20240307537A1 (en) | Methods and Compositions for Treating Cancer | |
| JP2005536187A (en) | Diagnosis and treatment of diseases associated with N-formyl peptide receptor-like 1 (FPRL1) | |
| KR20080034877A (en) | Pharmacological chaperones for treating obesity | |
| US10385027B2 (en) | Triazole derivatives and their use as PDE4 activators | |
| JP2011516486A (en) | Diagnosis, prevention and treatment method of bone mass disease | |
| US9206115B2 (en) | ATGListatin and pharmaceutical composition comprising the same | |
| US20130338374A1 (en) | Gpr 17 agonists and screening assay | |
| JP2007523166A (en) | Furosemide derivatives as modulators of HM74 and their use to treat inflammation | |
| Jiang et al. | Phosphorylation-Regulated Dynamic Phase Separation of HIP-55 Protects Against Heart Failure | |
| EP2948433B1 (en) | Atglistatin as lipase inhibitor | |
| US20220273751A1 (en) | Gpcr heteromer inhibitors and uses thereof | |
| JP4153693B2 (en) | Novel CaMKK inhibitor | |
| JP2007526917A (en) | HM74 Oxydecahydronaphthalene Modulator | |
| JP2005534311A (en) | Screening for suitable agents for the treatment of leukocyte-related inflammatory diseases |