EP3777979B1 - Anti-inflammatory compound and preparation and use thereof - Google Patents
Anti-inflammatory compound and preparation and use thereof Download PDFInfo
- Publication number
- EP3777979B1 EP3777979B1 EP19791850.1A EP19791850A EP3777979B1 EP 3777979 B1 EP3777979 B1 EP 3777979B1 EP 19791850 A EP19791850 A EP 19791850A EP 3777979 B1 EP3777979 B1 EP 3777979B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- dimethylpyridin
- methoxyphenyl
- compound
- cyclopropylmethoxy
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims description 246
- 230000003110 anti-inflammatory effect Effects 0.000 title description 29
- 238000002360 preparation method Methods 0.000 title description 6
- 239000000047 product Substances 0.000 claims description 93
- 239000013067 intermediate product Substances 0.000 claims description 78
- 125000000217 alkyl group Chemical group 0.000 claims description 71
- WEVYAHXRMPXWCK-UHFFFAOYSA-N acetonitrile Substances CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 52
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 45
- 125000003118 aryl group Chemical group 0.000 claims description 44
- 229910052799 carbon Inorganic materials 0.000 claims description 41
- 229910052736 halogen Inorganic materials 0.000 claims description 39
- 150000002367 halogens Chemical class 0.000 claims description 39
- 125000004432 carbon atom Chemical group C* 0.000 claims description 37
- -1 hydroxy, thio Chemical group 0.000 claims description 34
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 31
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 31
- 125000000304 alkynyl group Chemical group 0.000 claims description 30
- 125000003342 alkenyl group Chemical group 0.000 claims description 28
- 238000000034 method Methods 0.000 claims description 28
- 239000001257 hydrogen Substances 0.000 claims description 26
- 229910052739 hydrogen Inorganic materials 0.000 claims description 26
- GLELJCWVINJXQJ-VOTSOKGWSA-N 1-[(E)-2-(3-hydroxy-4-methoxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC GLELJCWVINJXQJ-VOTSOKGWSA-N 0.000 claims description 25
- VAQJMHQHWIMLKW-UHFFFAOYSA-N 1-[2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O VAQJMHQHWIMLKW-UHFFFAOYSA-N 0.000 claims description 22
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 20
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 19
- 125000004185 ester group Chemical group 0.000 claims description 18
- 229910052760 oxygen Inorganic materials 0.000 claims description 18
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 17
- 239000001301 oxygen Substances 0.000 claims description 17
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 claims description 16
- 201000004624 Dermatitis Diseases 0.000 claims description 15
- 150000002431 hydrogen Chemical class 0.000 claims description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 14
- 125000004001 thioalkyl group Chemical group 0.000 claims description 12
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 11
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 10
- 238000006467 substitution reaction Methods 0.000 claims description 10
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- 150000001721 carbon Chemical group 0.000 claims description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 9
- 229910052717 sulfur Inorganic materials 0.000 claims description 9
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 claims description 8
- 238000007259 addition reaction Methods 0.000 claims description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- 150000001923 cyclic compounds Chemical class 0.000 claims description 8
- 125000001033 ether group Chemical group 0.000 claims description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 claims description 8
- 239000007858 starting material Substances 0.000 claims description 8
- IGWBDMBNLBOHGE-ODLFYWEKSA-N C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)N)=C/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)N)=C/N1C(=CC(C=C1C)=C=O)C IGWBDMBNLBOHGE-ODLFYWEKSA-N 0.000 claims description 7
- 229910052757 nitrogen Inorganic materials 0.000 claims description 7
- LUJMEECXHPYQOF-UHFFFAOYSA-N 3-hydroxyacetophenone Chemical class CC(=O)C1=CC=CC(O)=C1 LUJMEECXHPYQOF-UHFFFAOYSA-N 0.000 claims description 6
- IAVREABSGIHHMO-UHFFFAOYSA-N 3-hydroxybenzaldehyde Chemical class OC1=CC=CC(C=O)=C1 IAVREABSGIHHMO-UHFFFAOYSA-N 0.000 claims description 6
- 125000003277 amino group Chemical group 0.000 claims description 6
- 150000003457 sulfones Chemical class 0.000 claims description 6
- 150000003462 sulfoxides Chemical class 0.000 claims description 6
- OSYBYWBWSUYNBA-QQOXCAACSA-N 1-[(E)-2-[4-(difluoromethoxy)-3-[(3R)-thiolan-3-yl]oxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound S1C[C@@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F OSYBYWBWSUYNBA-QQOXCAACSA-N 0.000 claims description 5
- UNCZQGKVTALYQE-IWHGQQBYSA-N 1-[(E)-2-[4-methoxy-3-[(3S)-thiolan-3-yl]oxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound S1C[C@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC UNCZQGKVTALYQE-IWHGQQBYSA-N 0.000 claims description 5
- SJWCNQKBINEVPB-ODLFYWEKSA-N C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)O)=C/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)O)=C/N1C(=CC(C=C1C)=C=O)C SJWCNQKBINEVPB-ODLFYWEKSA-N 0.000 claims description 5
- SOBPGDAPYQIKIW-KEBDBYFISA-N C1(CC1)COC=1C=C(C=CC=1OC)/C=C(\C#N)/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C=C(\C#N)/N1C(=CC(C=C1C)=C=O)C SOBPGDAPYQIKIW-KEBDBYFISA-N 0.000 claims description 5
- SVRONUHDCMFEDC-UHFFFAOYSA-N C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1)=O)C)=O Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1)=O)C)=O SVRONUHDCMFEDC-UHFFFAOYSA-N 0.000 claims description 5
- AJFUNEJIJGCVJZ-BQYQJAHWSA-N CSCCOC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC Chemical compound CSCCOC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC AJFUNEJIJGCVJZ-BQYQJAHWSA-N 0.000 claims description 5
- 150000001851 cinnamic acid derivatives Chemical class 0.000 claims description 5
- KWEAWRKKPUPNMT-CMDGGOBGSA-N 1-[(E)-2-(3-cyclopropyloxy-4-methoxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC KWEAWRKKPUPNMT-CMDGGOBGSA-N 0.000 claims description 4
- XCIYAYJHUICTOS-LFVJCYFKSA-N 1-[(E)-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-phenylethenyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(=C/N1C(=CC(C=C1C)=O)C)/C1=CC=CC=C1 XCIYAYJHUICTOS-LFVJCYFKSA-N 0.000 claims description 4
- XJOHOHQZRJOKOE-VQHVLOKHSA-N 1-[(E)-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]ethenyl]-2-methylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(/C=C/N2C(=CC(C=C2)=O)C)C=CC=1OC XJOHOHQZRJOKOE-VQHVLOKHSA-N 0.000 claims description 4
- JDPWCRFUSURYTN-VOTSOKGWSA-N 1-[(E)-2-[4-(difluoromethoxy)-3-(2-methylsulfanylethoxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound CSCCOC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F JDPWCRFUSURYTN-VOTSOKGWSA-N 0.000 claims description 4
- RSDXJFQXJIAKPC-VOTSOKGWSA-N 1-[(E)-2-[4-(difluoromethoxy)-3-(methylsulfanylmethoxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound CSCOC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F RSDXJFQXJIAKPC-VOTSOKGWSA-N 0.000 claims description 4
- KEUTTWBHDUMSMY-BQYQJAHWSA-N 1-[(E)-2-[4-methoxy-3-(methylsulfanylmethoxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound CSCOC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC KEUTTWBHDUMSMY-BQYQJAHWSA-N 0.000 claims description 4
- VLUHOGMJTVRTQT-UHFFFAOYSA-N 1-[2-[4-methoxy-3-(2-methylsulfanylethoxy)phenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound CSCCOC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O VLUHOGMJTVRTQT-UHFFFAOYSA-N 0.000 claims description 4
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 claims description 4
- LIOSRQRMAMJIHF-HAHDFKILSA-N C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)N(CC)CC)=C/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)N(CC)CC)=C/N1C(=CC(C=C1C)=C=O)C LIOSRQRMAMJIHF-HAHDFKILSA-N 0.000 claims description 4
- 150000008062 acetophenones Chemical class 0.000 claims description 4
- 239000012752 auxiliary agent Substances 0.000 claims description 4
- 150000003935 benzaldehydes Chemical class 0.000 claims description 4
- 230000002140 halogenating effect Effects 0.000 claims description 4
- 125000004434 sulfur atom Chemical group 0.000 claims description 4
- 239000004094 surface-active agent Substances 0.000 claims description 4
- URKMASJKHXXJHZ-PDGQHHTCSA-N (E)-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-3-(2,6-dimethyl-4-oxopyridin-1-yl)prop-2-enenitrile Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)\C(=C/N1C(=CC(C=C1C)=O)C)\C#N URKMASJKHXXJHZ-PDGQHHTCSA-N 0.000 claims description 3
- FNRMOLNTSRVAAS-MDZDMXLPSA-N 1-[(E)-2-(3-but-2-ynoxy-4-methoxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C(C#CC)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC FNRMOLNTSRVAAS-MDZDMXLPSA-N 0.000 claims description 3
- WXCHZLDQLHKNOC-ZHACJKMWSA-N 1-[(E)-2-(3-cyclopent-3-en-1-yloxy-4-methoxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC=CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC WXCHZLDQLHKNOC-ZHACJKMWSA-N 0.000 claims description 3
- YDXHWGWNKKUOHC-CMDGGOBGSA-N 1-[(E)-2-(4-methoxy-3-propan-2-yloxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C(C)(C)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC YDXHWGWNKKUOHC-CMDGGOBGSA-N 0.000 claims description 3
- SAPUWAJVKDOCMX-CMDGGOBGSA-N 1-[(E)-2-(4-methoxy-3-propoxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C(CC)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC SAPUWAJVKDOCMX-CMDGGOBGSA-N 0.000 claims description 3
- JBCIABNKIWRAPE-BQYQJAHWSA-N 1-[(E)-2-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F JBCIABNKIWRAPE-BQYQJAHWSA-N 0.000 claims description 3
- BDBVJOFIFBIYES-RMKNXTFCSA-N 1-[(E)-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]ethenyl]pyridin-4-one Chemical compound C1(CC1)COC=1C=C(/C=C/N2C=CC(C=C2)=O)C=CC=1OC BDBVJOFIFBIYES-RMKNXTFCSA-N 0.000 claims description 3
- OSYBYWBWSUYNBA-UABRLCRWSA-N 1-[(E)-2-[4-(difluoromethoxy)-3-[(3S)-thiolan-3-yl]oxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound S1C[C@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F OSYBYWBWSUYNBA-UABRLCRWSA-N 0.000 claims description 3
- HTXZMGUDUNFWKD-CMDGGOBGSA-N 1-[(E)-2-[4-methoxy-3-(2-methylpropoxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C(C(C)C)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC HTXZMGUDUNFWKD-CMDGGOBGSA-N 0.000 claims description 3
- YNRCRVRZLNSWNT-HBLYWKAGSA-N 1-[(E)-2-[4-methoxy-3-[(3R)-1-oxothiolan-3-yl]oxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound COC1=C(C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=C1)O[C@H]1CS(CC1)=O YNRCRVRZLNSWNT-HBLYWKAGSA-N 0.000 claims description 3
- UNCZQGKVTALYQE-JHTBKMLMSA-N 1-[(E)-2-[4-methoxy-3-[(3R)-thiolan-3-yl]oxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound S1C[C@@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC UNCZQGKVTALYQE-JHTBKMLMSA-N 0.000 claims description 3
- YNRCRVRZLNSWNT-FASULOODSA-N 1-[(E)-2-[4-methoxy-3-[(3S)-1-oxothiolan-3-yl]oxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound COC1=C(C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=C1)O[C@@H]1CS(CC1)=O YNRCRVRZLNSWNT-FASULOODSA-N 0.000 claims description 3
- IAPUNTIPGIHIRU-UHFFFAOYSA-N 1-[2-(3,4-dimethoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O IAPUNTIPGIHIRU-UHFFFAOYSA-N 0.000 claims description 3
- VHVHSTUVZDNFLG-UHFFFAOYSA-N 1-[2-(3-but-2-ynoxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(C#CC)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O VHVHSTUVZDNFLG-UHFFFAOYSA-N 0.000 claims description 3
- YUBBNFRIDGVYML-UHFFFAOYSA-N 1-[2-(3-butoxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(CCC)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O YUBBNFRIDGVYML-UHFFFAOYSA-N 0.000 claims description 3
- DHTQXPQLWALFEF-UHFFFAOYSA-N 1-[2-(3-cyclohexyloxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CCCCC1)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O DHTQXPQLWALFEF-UHFFFAOYSA-N 0.000 claims description 3
- TYSVOKCKYHTHKE-UHFFFAOYSA-N 1-[2-(3-cyclopent-3-en-1-yloxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC=CC1)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O TYSVOKCKYHTHKE-UHFFFAOYSA-N 0.000 claims description 3
- KGBYJWURIWGDCF-UHFFFAOYSA-N 1-[2-(3-ethoxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(C)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O KGBYJWURIWGDCF-UHFFFAOYSA-N 0.000 claims description 3
- QXFMSLCLIXLVQF-UHFFFAOYSA-N 1-[2-(3-hexoxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(CCCCC)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O QXFMSLCLIXLVQF-UHFFFAOYSA-N 0.000 claims description 3
- TXKXGFRMCHUWPF-UHFFFAOYSA-N 1-[2-(4-methoxy-3-pentoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(CCCC)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O TXKXGFRMCHUWPF-UHFFFAOYSA-N 0.000 claims description 3
- MDOAHKROLZSOJP-UHFFFAOYSA-N 1-[2-(4-methoxy-3-prop-2-ynoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(C#C)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O MDOAHKROLZSOJP-UHFFFAOYSA-N 0.000 claims description 3
- MHFDDAZZPJPZIL-UHFFFAOYSA-N 1-[2-(4-methoxy-3-propan-2-yloxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(C)(C)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O MHFDDAZZPJPZIL-UHFFFAOYSA-N 0.000 claims description 3
- UEASXNDFPJOQRZ-UHFFFAOYSA-N 1-[2-(4-methoxy-3-propoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(CC)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O UEASXNDFPJOQRZ-UHFFFAOYSA-N 0.000 claims description 3
- FZNZUUJCFXMHCJ-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC(F)F)C(CN1C(=CC(C=C1C)=O)C)=O FZNZUUJCFXMHCJ-UHFFFAOYSA-N 0.000 claims description 3
- KMUSGAQSENXGDY-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]cyclopropyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C1C(C1)N1C(=CC(C=C1C)=O)C KMUSGAQSENXGDY-UHFFFAOYSA-N 0.000 claims description 3
- PZWYBRCUKZISBR-UHFFFAOYSA-N 1-[2-[4-methoxy-3-(2-methylpropoxy)phenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C(C(C)C)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O PZWYBRCUKZISBR-UHFFFAOYSA-N 0.000 claims description 3
- HHVSTLMXFNACEG-UHFFFAOYSA-N 1-[2-[4-methoxy-3-(3-methylbut-2-enoxy)phenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound CC(=CCOC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O)C HHVSTLMXFNACEG-UHFFFAOYSA-N 0.000 claims description 3
- ITDRKYXOJTUMKQ-UHFFFAOYSA-N 1-[2-[4-methoxy-3-(oxetan-3-yloxy)phenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound O1CC(C1)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O ITDRKYXOJTUMKQ-UHFFFAOYSA-N 0.000 claims description 3
- YMEMUGKVZZHYNY-UHFFFAOYSA-N 2-chloro-1-[2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-oxoethyl]pyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1)=O)Cl)=O YMEMUGKVZZHYNY-UHFFFAOYSA-N 0.000 claims description 3
- XMEYKMOWXJJKME-UHFFFAOYSA-N C(C#CC)(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 Chemical compound C(C#CC)(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 XMEYKMOWXJJKME-UHFFFAOYSA-N 0.000 claims description 3
- MZJOAEDADZLQLJ-UHFFFAOYSA-N C(C)(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 Chemical compound C(C)(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 MZJOAEDADZLQLJ-UHFFFAOYSA-N 0.000 claims description 3
- WWIAOZZDAXCDRZ-UHFFFAOYSA-N C(C1=CC=CC=C1)(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 Chemical compound C(C1=CC=CC=C1)(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 WWIAOZZDAXCDRZ-UHFFFAOYSA-N 0.000 claims description 3
- JWLYUSBDWXPMRD-UHFFFAOYSA-N C(CC)(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 Chemical compound C(CC)(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 JWLYUSBDWXPMRD-UHFFFAOYSA-N 0.000 claims description 3
- JXRCWEFWTUVTRV-UHFFFAOYSA-N C1(CC1)C(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 Chemical compound C1(CC1)C(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1 JXRCWEFWTUVTRV-UHFFFAOYSA-N 0.000 claims description 3
- PVODUDNMGIMXRY-BKUYFWCQSA-N C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)N(C)C)=C/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)N(C)C)=C/N1C(=CC(C=C1C)=C=O)C PVODUDNMGIMXRY-BKUYFWCQSA-N 0.000 claims description 3
- FHLNDJUZOFXTBJ-NDENLUEZSA-N C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)NC)=C/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)NC)=C/N1C(=CC(C=C1C)=C=O)C FHLNDJUZOFXTBJ-NDENLUEZSA-N 0.000 claims description 3
- WRDRPRGSBLREAZ-NDENLUEZSA-N C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)OC)=C/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)OC)=C/N1C(=CC(C=C1C)=C=O)C WRDRPRGSBLREAZ-NDENLUEZSA-N 0.000 claims description 3
- CDNZTFPMLDAPMF-BKUYFWCQSA-N C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)OCC)=C/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)OCC)=C/N1C(=CC(C=C1C)=C=O)C CDNZTFPMLDAPMF-BKUYFWCQSA-N 0.000 claims description 3
- YAJHOSVQWHMBQH-VOTSOKGWSA-N CC1=CC(=C=O)C=C(N1/C=C/C2=CC(=C(C=C2)OC)CS(=O)(=O)O)C Chemical compound CC1=CC(=C=O)C=C(N1/C=C/C2=CC(=C(C=C2)OC)CS(=O)(=O)O)C YAJHOSVQWHMBQH-VOTSOKGWSA-N 0.000 claims description 3
- YXFURKGNZCQRSS-BQYQJAHWSA-N CC=1N(C(=CC(C=1)=C=O)C)/C=C/C=1C=CC(=C(OCC(=O)NC)C=1)OC Chemical compound CC=1N(C(=CC(C=1)=C=O)C)/C=C/C=1C=CC(=C(OCC(=O)NC)C=1)OC YXFURKGNZCQRSS-BQYQJAHWSA-N 0.000 claims description 3
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 claims description 3
- DUDOVLKRJONDFE-QXDHYYDYSA-N FC(OC1=C(C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=C1)O[C@H]1CS(CC1)=O)F Chemical compound FC(OC1=C(C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=C1)O[C@H]1CS(CC1)=O)F DUDOVLKRJONDFE-QXDHYYDYSA-N 0.000 claims description 3
- ADNWDIWPVODWDG-UHFFFAOYSA-N O1C(CCC1)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O Chemical compound O1C(CCC1)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O ADNWDIWPVODWDG-UHFFFAOYSA-N 0.000 claims description 3
- ULZAMULVPPXUEN-QQOXCAACSA-N O=S1(C[C@@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F)=O Chemical compound O=S1(C[C@@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F)=O ULZAMULVPPXUEN-QQOXCAACSA-N 0.000 claims description 3
- ULZAMULVPPXUEN-UABRLCRWSA-N O=S1(C[C@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F)=O Chemical compound O=S1(C[C@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F)=O ULZAMULVPPXUEN-UABRLCRWSA-N 0.000 claims description 3
- 241000534944 Thia Species 0.000 claims description 3
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 claims description 3
- 229940088644 n,n-dimethylacrylamide Drugs 0.000 claims description 3
- 150000005299 pyridinones Chemical group 0.000 claims description 3
- 230000009467 reduction Effects 0.000 claims description 3
- QVSHTQSGTWVZJZ-MDZDMXLPSA-N 1-[(E)-2-(3-cyclobutyloxy-4-methoxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CCC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC QVSHTQSGTWVZJZ-MDZDMXLPSA-N 0.000 claims description 2
- RKMLXIIMYQOPBU-WYMLVPIESA-N 1-[(E)-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]prop-1-enyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(=C/N1C(=CC(C=C1C)=O)C)/C RKMLXIIMYQOPBU-WYMLVPIESA-N 0.000 claims description 2
- FOFKSLFAOPJUGC-JHTBKMLMSA-N 1-[(E)-2-[3-[(3R)-1,1-dioxothiolan-3-yl]oxy-4-methoxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound O=S1(C[C@@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC)=O FOFKSLFAOPJUGC-JHTBKMLMSA-N 0.000 claims description 2
- FUTCUJFBGWRZML-VOTSOKGWSA-N 1-[(E)-2-[4-(difluoromethoxy)-3-methoxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound COC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F FUTCUJFBGWRZML-VOTSOKGWSA-N 0.000 claims description 2
- APDMKMQJEIFKFB-CPNJWEJPSA-N 1-[(Z)-2-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]pent-1-en-3-ynyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC(F)F)/C(=C/N1C(=CC(C=C1C)=O)C)/C#CC APDMKMQJEIFKFB-CPNJWEJPSA-N 0.000 claims description 2
- DJCACNJIRCKHPF-XSFVSMFZSA-N 1-[(Z)-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]pent-1-en-3-ynyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(=C/N1C(=CC(C=C1C)=O)C)/C#CC DJCACNJIRCKHPF-XSFVSMFZSA-N 0.000 claims description 2
- IWVWTBJSSVEDKS-UHFFFAOYSA-N 1-[2-(3-cyclobutyloxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CCC1)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O IWVWTBJSSVEDKS-UHFFFAOYSA-N 0.000 claims description 2
- DFZHRXLDVFDLKC-UHFFFAOYSA-N 1-[2-(3-cyclopentyloxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CCCC1)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O DFZHRXLDVFDLKC-UHFFFAOYSA-N 0.000 claims description 2
- ROYLPVKZPREXBB-UHFFFAOYSA-N 1-[2-(3-cyclopropyloxy-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O ROYLPVKZPREXBB-UHFFFAOYSA-N 0.000 claims description 2
- PAPXGHYPWZCGBO-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-oxoethyl]pyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C=CC(C=C1)=O)=O PAPXGHYPWZCGBO-UHFFFAOYSA-N 0.000 claims description 2
- PUXURQAFSDDGDQ-UHFFFAOYSA-N 1-[2-[4-methoxy-3-(methylsulfanylmethoxy)phenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound CSCOC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O PUXURQAFSDDGDQ-UHFFFAOYSA-N 0.000 claims description 2
- RYVZGIMMTKABME-CMDGGOBGSA-N C1(CC1)COC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC Chemical compound C1(CC1)COC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC RYVZGIMMTKABME-CMDGGOBGSA-N 0.000 claims description 2
- AOEIBGVVKFWZTE-UHFFFAOYSA-N CC(=CC(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1)C Chemical compound CC(=CC(=O)OC(CN1C(=CC(C=C1C)=C=O)C)C1=CC(=C(C=C1)OC)OCC1CC1)C AOEIBGVVKFWZTE-UHFFFAOYSA-N 0.000 claims description 2
- FOFKSLFAOPJUGC-IWHGQQBYSA-N O=S1(C[C@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC)=O Chemical compound O=S1(C[C@H](CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC)=O FOFKSLFAOPJUGC-IWHGQQBYSA-N 0.000 claims description 2
- 239000003153 chemical reaction reagent Substances 0.000 claims description 2
- 239000003795 chemical substances by application Substances 0.000 claims description 2
- 230000008030 elimination Effects 0.000 claims description 2
- 238000003379 elimination reaction Methods 0.000 claims description 2
- 230000003301 hydrolyzing effect Effects 0.000 claims description 2
- 230000001590 oxidative effect Effects 0.000 claims description 2
- 150000002923 oximes Chemical class 0.000 claims description 2
- KHHWWVUYCPMGEL-UHFFFAOYSA-N 1,2-dimethylpyridin-4-one Chemical compound CC1=CC(=O)C=CN1C KHHWWVUYCPMGEL-UHFFFAOYSA-N 0.000 claims 1
- LTBQUOJQQAPFTQ-BQYQJAHWSA-N 1-[(E)-2-[4-methoxy-3-(methylsulfinylmethoxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound COC1=C(OCS(C)=O)C=C(\C=C\N2C(C)=CC(=O)C=C2C)C=C1 LTBQUOJQQAPFTQ-BQYQJAHWSA-N 0.000 claims 1
- XMKQXTJLFJRPJO-BQYQJAHWSA-N 1-[(E)-2-[4-methoxy-3-(methylsulfonylmethoxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound COC1=C(OCS(C)(=O)=O)C=C(\C=C\N2C(C)=CC(=O)C=C2C)C=C1 XMKQXTJLFJRPJO-BQYQJAHWSA-N 0.000 claims 1
- MKZCGQFBSQHZOF-UHFFFAOYSA-N 1-[2-[4-methoxy-3-(2-methylsulfonylethoxy)phenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound COC1=C(OCCS(C)(=O)=O)C=C(C=C1)C(=O)CN1C(C)=CC(=O)C=C1C MKZCGQFBSQHZOF-UHFFFAOYSA-N 0.000 claims 1
- KZHGTOHYLCRUCQ-UHFFFAOYSA-N 1-[2-[4-methoxy-3-(methylsulfinylmethoxy)phenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound COC1=C(OCS(C)=O)C=C(C=C1)C(=O)CN1C(C)=CC(=O)C=C1C KZHGTOHYLCRUCQ-UHFFFAOYSA-N 0.000 claims 1
- CAZNGAIEHMJDAI-VXLYETTFSA-N CC(N(/C(\C(N)=O)=C/C(C=C1)=CC(OCC2CC2)=C1OC)C(C)=C1)=CC1=C=O Chemical compound CC(N(/C(\C(N)=O)=C/C(C=C1)=CC(OCC2CC2)=C1OC)C(C)=C1)=CC1=C=O CAZNGAIEHMJDAI-VXLYETTFSA-N 0.000 claims 1
- MHSYPTOKJQAQHW-VOTSOKGWSA-N CC1=CC(=O)C=C(C)N1\C=C\C1=CC(OCCS(C)(=O)=O)=C(OC(F)F)C=C1 Chemical compound CC1=CC(=O)C=C(C)N1\C=C\C1=CC(OCCS(C)(=O)=O)=C(OC(F)F)C=C1 MHSYPTOKJQAQHW-VOTSOKGWSA-N 0.000 claims 1
- SRHAGQZXDLAGGK-VOTSOKGWSA-N CC1=CC(=O)C=C(C)N1\C=C\C1=CC(OCCS(C)=O)=C(OC(F)F)C=C1 Chemical compound CC1=CC(=O)C=C(C)N1\C=C\C1=CC(OCCS(C)=O)=C(OC(F)F)C=C1 SRHAGQZXDLAGGK-VOTSOKGWSA-N 0.000 claims 1
- DHULWNUBEIVOAG-VOTSOKGWSA-N CC1=CC(=O)C=C(C)N1\C=C\C1=CC(OCS(C)(=O)=O)=C(OC(F)F)C=C1 Chemical compound CC1=CC(=O)C=C(C)N1\C=C\C1=CC(OCS(C)(=O)=O)=C(OC(F)F)C=C1 DHULWNUBEIVOAG-VOTSOKGWSA-N 0.000 claims 1
- XFVMQHZVSAODCD-VOTSOKGWSA-N CC1=CC(=O)C=C(C)N1\C=C\C1=CC(OCS(C)=O)=C(OC(F)F)C=C1 Chemical compound CC1=CC(=O)C=C(C)N1\C=C\C1=CC(OCS(C)=O)=C(OC(F)F)C=C1 XFVMQHZVSAODCD-VOTSOKGWSA-N 0.000 claims 1
- JHBLMRZYURZCEB-BQYQJAHWSA-N COC1=C(OCCS(C)(=O)=O)C=C(\C=C\N2C(C)=CC(=O)C=C2C)C=C1 Chemical compound COC1=C(OCCS(C)(=O)=O)C=C(\C=C\N2C(C)=CC(=O)C=C2C)C=C1 JHBLMRZYURZCEB-BQYQJAHWSA-N 0.000 claims 1
- QVBSROJRIUPCKH-UHFFFAOYSA-N COC1=C(OCCS(C)=O)C=C(C=C1)C(=O)CN1C(C)=CC(=O)C=C1C Chemical compound COC1=C(OCCS(C)=O)C=C(C=C1)C(=O)CN1C(C)=CC(=O)C=C1C QVBSROJRIUPCKH-UHFFFAOYSA-N 0.000 claims 1
- SZQRMMRRVXGPTQ-BQYQJAHWSA-N COC1=C(OCCS(C)=O)C=C(\C=C\N2C(C)=CC(=O)C=C2C)C=C1 Chemical compound COC1=C(OCCS(C)=O)C=C(\C=C\N2C(C)=CC(=O)C=C2C)C=C1 SZQRMMRRVXGPTQ-BQYQJAHWSA-N 0.000 claims 1
- UNGZJCPBGYAGRJ-UHFFFAOYSA-N COC1=C(OCS(C)(=O)=O)C=C(C=C1)C(=O)CN1C(C)=CC(=O)C=C1C Chemical compound COC1=C(OCS(C)(=O)=O)C=C(C=C1)C(=O)CN1C(C)=CC(=O)C=C1C UNGZJCPBGYAGRJ-UHFFFAOYSA-N 0.000 claims 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 396
- 238000006243 chemical reaction Methods 0.000 description 337
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 177
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 146
- 239000000243 solution Substances 0.000 description 135
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 122
- 238000005160 1H NMR spectroscopy Methods 0.000 description 116
- 239000007787 solid Substances 0.000 description 106
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 100
- 238000004007 reversed phase HPLC Methods 0.000 description 98
- 239000012074 organic phase Substances 0.000 description 93
- 238000001035 drying Methods 0.000 description 79
- 238000001914 filtration Methods 0.000 description 78
- 238000000746 purification Methods 0.000 description 78
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 76
- 239000012299 nitrogen atmosphere Substances 0.000 description 69
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 61
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 60
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 51
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 46
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 39
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 36
- 229920006395 saturated elastomer Polymers 0.000 description 33
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 30
- 229910000027 potassium carbonate Inorganic materials 0.000 description 30
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 26
- 229910000024 caesium carbonate Inorganic materials 0.000 description 26
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 238000004440 column chromatography Methods 0.000 description 24
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 23
- 235000010265 sodium sulphite Nutrition 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- 230000000694 effects Effects 0.000 description 19
- 239000003921 oil Substances 0.000 description 19
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 18
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 18
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 17
- 239000012043 crude product Substances 0.000 description 17
- 229940086542 triethylamine Drugs 0.000 description 17
- 102000004127 Cytokines Human genes 0.000 description 15
- 108090000695 Cytokines Proteins 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 208000010668 atopic eczema Diseases 0.000 description 13
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 12
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 12
- WQAATLMNJKIDRL-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-hydroxyethyl]pyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C=CC(C=C1)=O)O WQAATLMNJKIDRL-UHFFFAOYSA-N 0.000 description 10
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 229940079593 drug Drugs 0.000 description 10
- 230000005764 inhibitory process Effects 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- 101100296720 Dictyostelium discoideum Pde4 gene Proteins 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 206010061218 Inflammation Diseases 0.000 description 8
- 101100082610 Plasmodium falciparum (isolate 3D7) PDEdelta gene Proteins 0.000 description 8
- 201000010099 disease Diseases 0.000 description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 8
- 230000004054 inflammatory process Effects 0.000 description 8
- JVTZFYYHCGSXJV-UHFFFAOYSA-N isovanillin Chemical compound COC1=CC=C(C=O)C=C1O JVTZFYYHCGSXJV-UHFFFAOYSA-N 0.000 description 8
- 239000012280 lithium aluminium hydride Substances 0.000 description 8
- 108090000790 Enzymes Proteins 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 7
- 201000004681 Psoriasis Diseases 0.000 description 7
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 7
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 7
- 235000019270 ammonium chloride Nutrition 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- USZAGAREISWJDP-UHFFFAOYSA-N crisaborole Chemical compound C=1C=C2B(O)OCC2=CC=1OC1=CC=C(C#N)C=C1 USZAGAREISWJDP-UHFFFAOYSA-N 0.000 description 7
- 229950008199 crisaborole Drugs 0.000 description 7
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 7
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 7
- 230000000770 proinflammatory effect Effects 0.000 description 7
- 239000012312 sodium hydride Substances 0.000 description 7
- 229910000104 sodium hydride Inorganic materials 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 201000008937 atopic dermatitis Diseases 0.000 description 6
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- GRDGKQILTBTXSJ-UHFFFAOYSA-N 3-(cyclopropylmethoxy)-4-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1OCC1CC1 GRDGKQILTBTXSJ-UHFFFAOYSA-N 0.000 description 5
- JWMLCCRPDOIBAV-UHFFFAOYSA-N chloro(methylsulfanyl)methane Chemical compound CSCCl JWMLCCRPDOIBAV-UHFFFAOYSA-N 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- BCLXBTMKOCKJCN-AATRIKPKSA-N 1-[(E)-2-[4-(difluoromethoxy)-3-hydroxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F BCLXBTMKOCKJCN-AATRIKPKSA-N 0.000 description 4
- CTSQBUOHTFZQHF-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O CTSQBUOHTFZQHF-UHFFFAOYSA-N 0.000 description 4
- MYPRJISTEPVEAA-UHFFFAOYSA-N 2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-trimethylsilyloxyacetonitrile Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(C#N)O[Si](C)(C)C MYPRJISTEPVEAA-UHFFFAOYSA-N 0.000 description 4
- AHVVCELVGCPYGI-UHFFFAOYSA-N 3-(cyclopropylmethoxy)-4-(difluoromethoxy)benzaldehyde Chemical compound FC(F)OC1=CC=C(C=O)C=C1OCC1CC1 AHVVCELVGCPYGI-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 206010012438 Dermatitis atopic Diseases 0.000 description 4
- 102000000589 Interleukin-1 Human genes 0.000 description 4
- 108010002352 Interleukin-1 Proteins 0.000 description 4
- 102000003814 Interleukin-10 Human genes 0.000 description 4
- 108090000174 Interleukin-10 Proteins 0.000 description 4
- 108090001005 Interleukin-6 Proteins 0.000 description 4
- 102000004889 Interleukin-6 Human genes 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- 230000001363 autoimmune Effects 0.000 description 4
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 125000003375 sulfoxide group Chemical group 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- XCIYAYJHUICTOS-JLPGSUDCSA-N 1-[(Z)-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-phenylethenyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)\C(=C/N1C(=CC(C=C1C)=O)C)\C1=CC=CC=C1 XCIYAYJHUICTOS-JLPGSUDCSA-N 0.000 description 3
- NDBBMUYPBWFJSE-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-hydroxyethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)O NDBBMUYPBWFJSE-UHFFFAOYSA-N 0.000 description 3
- DUDISXARBBWPJY-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-hydroxyethyl]-2-methylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1)=O)C)O DUDISXARBBWPJY-UHFFFAOYSA-N 0.000 description 3
- LNNXOEHOXSYWLD-UHFFFAOYSA-N 1-bromobut-2-yne Chemical compound CC#CCBr LNNXOEHOXSYWLD-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N 1-butanol Substances CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- MYFKLQFBFSHBPA-UHFFFAOYSA-N 1-chloro-2-methylsulfanylethane Chemical compound CSCCCl MYFKLQFBFSHBPA-UHFFFAOYSA-N 0.000 description 3
- SUHRRMYAMHCKCS-UHFFFAOYSA-N 2-amino-1-[3-(cyclopropylmethoxy)-4-methoxyphenyl]ethanol Chemical compound COC1=CC=C(C(O)CN)C=C1OCC1CC1 SUHRRMYAMHCKCS-UHFFFAOYSA-N 0.000 description 3
- GZJWXXSAKWZSOE-UHFFFAOYSA-N 2-bromo-1-[3-(cyclopropylmethoxy)-4-methoxyphenyl]ethanone Chemical compound BrCC(=O)C1=CC(=C(C=C1)OC)OCC1CC1 GZJWXXSAKWZSOE-UHFFFAOYSA-N 0.000 description 3
- ZLIKNROJGXXNJG-UHFFFAOYSA-N 4-(difluoromethoxy)-3-hydroxybenzaldehyde Chemical compound OC1=CC(C=O)=CC=C1OC(F)F ZLIKNROJGXXNJG-UHFFFAOYSA-N 0.000 description 3
- SOBPGDAPYQIKIW-JMIUGGIZSA-N C1(CC1)COC=1C=C(C=CC=1OC)\C=C(\C#N)/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)\C=C(\C#N)/N1C(=CC(C=C1C)=C=O)C SOBPGDAPYQIKIW-JMIUGGIZSA-N 0.000 description 3
- 102000013462 Interleukin-12 Human genes 0.000 description 3
- 108010065805 Interleukin-12 Proteins 0.000 description 3
- 108010044467 Isoenzymes Proteins 0.000 description 3
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 3
- 125000003368 amide group Chemical group 0.000 description 3
- 230000007012 clinical effect Effects 0.000 description 3
- 239000000411 inducer Substances 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- BGULGZGECGLSCE-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-hydroxyethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC(F)F)C(CN1C(=CC(C=C1C)=O)C)O BGULGZGECGLSCE-UHFFFAOYSA-N 0.000 description 2
- LPVUPTPNWKMUTE-UHFFFAOYSA-N 1-[2-bromo-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(=CN1C(=CC(C=C1C)=O)C)Br LPVUPTPNWKMUTE-UHFFFAOYSA-N 0.000 description 2
- SIAJDYMIOROCGU-UHFFFAOYSA-N 1-[2-chloro-2-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]ethyl]-2,6-dimethylpyridin-4-one Chemical compound ClC(CN1C(=CC(C=C1C)=O)C)C1=CC(=C(C=C1)OC(F)F)OCC1CC1 SIAJDYMIOROCGU-UHFFFAOYSA-N 0.000 description 2
- PGTNZBDEBUNRJY-UHFFFAOYSA-N 1-[2-hydroxy-2-[4-methoxy-3-(methylsulfanylmethoxy)phenyl]ethyl]-2,6-dimethylpyridin-4-one Chemical compound CSCOC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)O PGTNZBDEBUNRJY-UHFFFAOYSA-N 0.000 description 2
- VRZMABCEAJKNLL-UHFFFAOYSA-N 1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]but-2-yn-1-ol Chemical compound C1(CC1)COC=1C=C(C=CC=1OC(F)F)C(C#CC)O VRZMABCEAJKNLL-UHFFFAOYSA-N 0.000 description 2
- IIJYTNAWSAAUSQ-UHFFFAOYSA-N 1-[3-(cyclopropylmethoxy)-4-methoxyphenyl]ethanone Chemical compound COC1=CC=C(C(C)=O)C=C1OCC1CC1 IIJYTNAWSAAUSQ-UHFFFAOYSA-N 0.000 description 2
- GZFQJUDCOLHDCR-UHFFFAOYSA-N 1-amino-2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]propan-2-ol Chemical compound NCC(C)(O)C1=CC(=C(C=C1)OC)OCC1CC1 GZFQJUDCOLHDCR-UHFFFAOYSA-N 0.000 description 2
- HLVFKOKELQSXIQ-UHFFFAOYSA-N 1-bromo-2-methylpropane Chemical compound CC(C)CBr HLVFKOKELQSXIQ-UHFFFAOYSA-N 0.000 description 2
- LOYZVRIHVZEDMW-UHFFFAOYSA-N 1-bromo-3-methylbut-2-ene Chemical compound CC(C)=CCBr LOYZVRIHVZEDMW-UHFFFAOYSA-N 0.000 description 2
- CYNYIHKIEHGYOZ-UHFFFAOYSA-N 1-bromopropane Chemical compound CCCBr CYNYIHKIEHGYOZ-UHFFFAOYSA-N 0.000 description 2
- JKTCBAGSMQIFNL-UHFFFAOYSA-N 2,3-dihydrofuran Chemical compound C1CC=CO1 JKTCBAGSMQIFNL-UHFFFAOYSA-N 0.000 description 2
- YBDLZOQQPWYTIK-UHFFFAOYSA-N 2-(3-cyclopropyloxy-4-methoxyphenyl)-2-trimethylsilyloxyacetonitrile Chemical compound C1(CC1)OC=1C=C(C=CC=1OC)C(C#N)O[Si](C)(C)C YBDLZOQQPWYTIK-UHFFFAOYSA-N 0.000 description 2
- ZCDRHPBFASHMRN-UHFFFAOYSA-N 2-(4-methoxy-3-phenylmethoxyphenyl)-2-trimethylsilyloxyacetonitrile Chemical compound COC1=CC=C(C(O[Si](C)(C)C)C#N)C=C1OCC1=CC=CC=C1 ZCDRHPBFASHMRN-UHFFFAOYSA-N 0.000 description 2
- PWIQYFPCYRZDMD-UHFFFAOYSA-N 2-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-trimethylsilyloxyacetonitrile Chemical compound C1(CC1)COC=1C=C(C=CC=1OC(F)F)C(C#N)O[Si](C)(C)C PWIQYFPCYRZDMD-UHFFFAOYSA-N 0.000 description 2
- XUUSEAUYAOEXBW-UHFFFAOYSA-N 2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-trimethylsilyloxypropanenitrile Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(C#N)(C)O[Si](C)(C)C XUUSEAUYAOEXBW-UHFFFAOYSA-N 0.000 description 2
- RNNMJZMPHTVUFM-UHFFFAOYSA-N 2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-3-(2,6-dimethyl-4-oxopyridin-1-yl)-2-trimethylsilyloxypropanenitrile Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)(O[Si](C)(C)C)C#N RNNMJZMPHTVUFM-UHFFFAOYSA-N 0.000 description 2
- YFJSRGHISLWTOZ-UHFFFAOYSA-N 2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]cyclopropan-1-amine Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C1C(C1)N YFJSRGHISLWTOZ-UHFFFAOYSA-N 0.000 description 2
- YTINPEPHNGQOSR-UHFFFAOYSA-N 2-[4-(difluoromethoxy)-3-phenylmethoxyphenyl]-2-trimethylsilyloxyacetonitrile Chemical compound C(C1=CC=CC=C1)OC=1C=C(C=CC=1OC(F)F)C(C#N)O[Si](C)(C)C YTINPEPHNGQOSR-UHFFFAOYSA-N 0.000 description 2
- WNDOPVHLPKTKBO-UHFFFAOYSA-N 2-[4-methoxy-3-(methylsulfanylmethoxy)phenyl]-2-trimethylsilyloxyacetonitrile Chemical compound CSCOC=1C=C(C=CC=1OC)C(C#N)O[Si](C)(C)C WNDOPVHLPKTKBO-UHFFFAOYSA-N 0.000 description 2
- APIXJSLKIYYUKG-UHFFFAOYSA-N 3 Isobutyl 1 methylxanthine Chemical compound O=C1N(C)C(=O)N(CC(C)C)C2=C1N=CN2 APIXJSLKIYYUKG-UHFFFAOYSA-N 0.000 description 2
- QURCVMIEKCOAJU-UHFFFAOYSA-N 3-Hydroxy 4-Methoxy Cinnamic acid Chemical compound COC1=CC=C(C=CC(O)=O)C=C1O QURCVMIEKCOAJU-UHFFFAOYSA-N 0.000 description 2
- NUPJLUSVFMBCOM-UHFFFAOYSA-N 3-cyclopropyloxy-4-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1OC1CC1 NUPJLUSVFMBCOM-UHFFFAOYSA-N 0.000 description 2
- JBVTWVXASGBUGN-UHFFFAOYSA-N 4-(difluoromethoxy)-3-phenylmethoxybenzaldehyde Chemical compound FC(F)OC1=CC=C(C=O)C=C1OCC1=CC=CC=C1 JBVTWVXASGBUGN-UHFFFAOYSA-N 0.000 description 2
- DCVWCCVNXLMTBB-UHFFFAOYSA-N 4-methoxy-3-(methylsulfanylmethoxy)benzaldehyde Chemical compound COC1=CC=C(C=O)C=C1OCSC DCVWCCVNXLMTBB-UHFFFAOYSA-N 0.000 description 2
- VQVQZFHUXRSRBZ-UHFFFAOYSA-N 4-methoxy-3-phenylmethoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1OCC1=CC=CC=C1 VQVQZFHUXRSRBZ-UHFFFAOYSA-N 0.000 description 2
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 2
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 2
- 102100032367 C-C motif chemokine 5 Human genes 0.000 description 2
- ZREPXIQAWSPEAH-UQQQWYQISA-N C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)=C/N1C(=CC(C=C1C)=C=O)C Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)/C(/C(=O)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)=C/N1C(=CC(C=C1C)=C=O)C ZREPXIQAWSPEAH-UQQQWYQISA-N 0.000 description 2
- MTWQHOPAUVXLFB-UHFFFAOYSA-N CC=1N(C(=CC(C=1)=C=O)C)CC#N Chemical compound CC=1N(C(=CC(C=1)=C=O)C)CC#N MTWQHOPAUVXLFB-UHFFFAOYSA-N 0.000 description 2
- 108010055166 Chemokine CCL5 Proteins 0.000 description 2
- 108010012236 Chemokines Proteins 0.000 description 2
- 102000019034 Chemokines Human genes 0.000 description 2
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 2
- 206010012434 Dermatitis allergic Diseases 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 208000004232 Enteritis Diseases 0.000 description 2
- 108010008165 Etanercept Proteins 0.000 description 2
- 239000012981 Hank's balanced salt solution Substances 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- 102000051628 Interleukin-1 receptor antagonist Human genes 0.000 description 2
- 108700021006 Interleukin-1 receptor antagonist Proteins 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 102000000588 Interleukin-2 Human genes 0.000 description 2
- 102000013264 Interleukin-23 Human genes 0.000 description 2
- 108010065637 Interleukin-23 Proteins 0.000 description 2
- 102000004388 Interleukin-4 Human genes 0.000 description 2
- 108090000978 Interleukin-4 Proteins 0.000 description 2
- 108090001007 Interleukin-8 Proteins 0.000 description 2
- 102000004890 Interleukin-8 Human genes 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- CMCJGROFOLESTP-YFKPBYRVSA-N [(3S)-thiolan-3-yl] methanesulfonate Chemical compound CS(=O)(=O)O[C@@H]1CSCC1 CMCJGROFOLESTP-YFKPBYRVSA-N 0.000 description 2
- CMCJGROFOLESTP-RXMQYKEDSA-N [(3r)-thiolan-3-yl] methanesulfonate Chemical compound CS(=O)(=O)O[C@@H]1CCSC1 CMCJGROFOLESTP-RXMQYKEDSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- UDMBCSSLTHHNCD-KQYNXXCUSA-N adenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O UDMBCSSLTHHNCD-KQYNXXCUSA-N 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- KXVUSQIDCZRUKF-UHFFFAOYSA-N bromocyclobutane Chemical compound BrC1CCC1 KXVUSQIDCZRUKF-UHFFFAOYSA-N 0.000 description 2
- BRTFVKHPEHKBQF-UHFFFAOYSA-N bromocyclopentane Chemical compound BrC1CCCC1 BRTFVKHPEHKBQF-UHFFFAOYSA-N 0.000 description 2
- FJWFZRXGXZLAGU-UHFFFAOYSA-N cyclopent-3-en-1-yl methanesulfonate Chemical compound CS(=O)(=O)OC1CC=CC1 FJWFZRXGXZLAGU-UHFFFAOYSA-N 0.000 description 2
- LPIQUOYDBNQMRZ-UHFFFAOYSA-N cyclopentene Chemical compound C1CC=CC1 LPIQUOYDBNQMRZ-UHFFFAOYSA-N 0.000 description 2
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 2
- BNETVPCXYPZHQU-UHFFFAOYSA-N cyclopropylmethyl 2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]cyclopropane-1-carboxylate Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C1C(C1)C(=O)OCC1CC1 BNETVPCXYPZHQU-UHFFFAOYSA-N 0.000 description 2
- 229950003468 dupilumab Drugs 0.000 description 2
- 210000003979 eosinophil Anatomy 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 210000004969 inflammatory cell Anatomy 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 2
- YLTGFGDODHXMFB-UHFFFAOYSA-N isoacetovanillone Chemical compound COC1=CC=C(C(C)=O)C=C1O YLTGFGDODHXMFB-UHFFFAOYSA-N 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 210000001616 monocyte Anatomy 0.000 description 2
- QMLWSAXEQSBAAQ-UHFFFAOYSA-N oxetan-3-ol Chemical compound OC1COC1 QMLWSAXEQSBAAQ-UHFFFAOYSA-N 0.000 description 2
- 230000008506 pathogenesis Effects 0.000 description 2
- 230000037368 penetrate the skin Effects 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 2
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 208000017520 skin disease Diseases 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- CMCJGROFOLESTP-UHFFFAOYSA-N thiolan-3-yl methanesulfonate Chemical compound CS(=O)(=O)OC1CCSC1 CMCJGROFOLESTP-UHFFFAOYSA-N 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- HMLGSIZOMSVISS-ONJSNURVSA-N (7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-(2,2-dimethylpropanoyloxymethoxyimino)acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@@H]1C(N2C(=C(C=C)CSC21)C(O)=O)=O)C(=O)\C(=N/OCOC(=O)C(C)(C)C)C1=CSC(N)=N1 HMLGSIZOMSVISS-ONJSNURVSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- JESMDBFDDFQXCU-MDZDMXLPSA-N 1-[(E)-2-(3-butoxy-4-methoxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C(CCC)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC JESMDBFDDFQXCU-MDZDMXLPSA-N 0.000 description 1
- YUCDCQKJIMDCOR-ZHACJKMWSA-N 1-[(E)-2-(3-cyclopentyloxy-4-methoxyphenyl)ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CCCC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC YUCDCQKJIMDCOR-ZHACJKMWSA-N 0.000 description 1
- XDZNPFVKFKKABY-FNORWQNLSA-N 1-[(E)-2-(3-hydroxy-4-methoxyphenyl)ethenyl]-2-methylpyridin-4-one Chemical compound OC=1C=C(/C=C/N2C(=CC(C=C2)=O)C)C=CC=1OC XDZNPFVKFKKABY-FNORWQNLSA-N 0.000 description 1
- HDJKOKGCXKFGDQ-MDZDMXLPSA-N 1-[(E)-2-[3-(2,2-dimethylpropoxy)-4-methoxyphenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound C(C(C)(C)C)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC HDJKOKGCXKFGDQ-MDZDMXLPSA-N 0.000 description 1
- OSYBYWBWSUYNBA-FNORWQNLSA-N 1-[(E)-2-[4-(difluoromethoxy)-3-(thiolan-3-yloxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound S1CC(CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC(F)F OSYBYWBWSUYNBA-FNORWQNLSA-N 0.000 description 1
- UNCZQGKVTALYQE-SOFGYWHQSA-N 1-[(E)-2-[4-methoxy-3-(thiolan-3-yloxy)phenyl]ethenyl]-2,6-dimethylpyridin-4-one Chemical compound S1CC(CC1)OC=1C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=CC=1OC UNCZQGKVTALYQE-SOFGYWHQSA-N 0.000 description 1
- REVMYAPVZNCMGX-UHFFFAOYSA-N 1-[2-(3-cyclopentyl-4-methoxyphenyl)-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CCCC1)C=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O REVMYAPVZNCMGX-UHFFFAOYSA-N 0.000 description 1
- PZPLXBAGSDRVOD-UHFFFAOYSA-N 1-[2-[3-(cyclobutylmethoxy)-4-methoxyphenyl]-2-oxoethyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CCC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)=O PZPLXBAGSDRVOD-UHFFFAOYSA-N 0.000 description 1
- ZOUYPXQRLBOVFM-UHFFFAOYSA-N 1-[2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-hydroxypropyl]-2,6-dimethylpyridin-4-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)(C)O ZOUYPXQRLBOVFM-UHFFFAOYSA-N 0.000 description 1
- VNFNAFFPPLDHAY-UHFFFAOYSA-N 1-[2-hydroxy-2-(3-hydroxy-4-methoxyphenyl)ethyl]-2,6-dimethylpyridin-4-one Chemical compound OC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)O VNFNAFFPPLDHAY-UHFFFAOYSA-N 0.000 description 1
- MNQQSCHYJUQBCQ-UHFFFAOYSA-N 1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]but-2-yn-1-one Chemical compound C1(CC1)COC=1C=C(C=CC=1OC(F)F)C(C#CC)=O MNQQSCHYJUQBCQ-UHFFFAOYSA-N 0.000 description 1
- CQWYAXCOVZKLHY-UHFFFAOYSA-N 1-bromo-2,2-dimethylpropane Chemical compound CC(C)(C)CBr CQWYAXCOVZKLHY-UHFFFAOYSA-N 0.000 description 1
- MPPPKRYCTPRNTB-UHFFFAOYSA-N 1-bromobutane Chemical compound CCCCBr MPPPKRYCTPRNTB-UHFFFAOYSA-N 0.000 description 1
- MNDIARAMWBIKFW-UHFFFAOYSA-N 1-bromohexane Chemical compound CCCCCCBr MNDIARAMWBIKFW-UHFFFAOYSA-N 0.000 description 1
- YZWKKMVJZFACSU-UHFFFAOYSA-N 1-bromopentane Chemical compound CCCCCBr YZWKKMVJZFACSU-UHFFFAOYSA-N 0.000 description 1
- VSYFZULSKMFUJJ-UHFFFAOYSA-N 2,6-dimethylpyran-4-one Chemical compound CC1=CC(=O)C=C(C)O1 VSYFZULSKMFUJJ-UHFFFAOYSA-N 0.000 description 1
- ZISARVXKZDUBKD-UHFFFAOYSA-N 2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-1-phenylethanone Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)CC(=O)C1=CC=CC=C1 ZISARVXKZDUBKD-UHFFFAOYSA-N 0.000 description 1
- KFCSJWGGHVRWRC-UHFFFAOYSA-N 2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-3-(2,6-dimethyl-4-oxopyridin-1-yl)-2-hydroxypropanenitrile Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C(CN1C(=CC(C=C1C)=O)C)(O)C#N KFCSJWGGHVRWRC-UHFFFAOYSA-N 0.000 description 1
- RVGPBGMZHQLHAD-UHFFFAOYSA-N 2-[3-(cyclopropylmethoxy)-4-methoxyphenyl]cyclopropane-1-carboxylic acid Chemical compound C1(CC1)COC=1C=C(C=CC=1OC)C1C(C1)C(=O)O RVGPBGMZHQLHAD-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- NRQIRHDKBIIAKP-UHFFFAOYSA-N 2-amino-1-(3-cyclopropyloxy-4-methoxyphenyl)ethanol Chemical compound COC1=CC=C(C(O)CN)C=C1OC1CC1 NRQIRHDKBIIAKP-UHFFFAOYSA-N 0.000 description 1
- JJBPZTLYONVMMO-UHFFFAOYSA-N 2-amino-1-(4-methoxy-3-phenylmethoxyphenyl)ethanol Chemical compound COC1=CC=C(C(O)CN)C=C1OCC1=CC=CC=C1 JJBPZTLYONVMMO-UHFFFAOYSA-N 0.000 description 1
- LFLWMKBYZNHAAN-UHFFFAOYSA-N 2-amino-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]ethanol Chemical compound NCC(O)C1=CC(=C(C=C1)OC(F)F)OCC1CC1 LFLWMKBYZNHAAN-UHFFFAOYSA-N 0.000 description 1
- NWUCQESEGOSJFM-UHFFFAOYSA-N 2-amino-1-[4-(difluoromethoxy)-3-phenylmethoxyphenyl]ethanol Chemical compound NCC(O)C1=CC(=C(C=C1)OC(F)F)OCC1=CC=CC=C1 NWUCQESEGOSJFM-UHFFFAOYSA-N 0.000 description 1
- AURNAHLNVYDOGZ-UHFFFAOYSA-N 2-amino-1-[4-methoxy-3-(methylsulfanylmethoxy)phenyl]ethanol Chemical compound NCC(O)C1=CC(=C(C=C1)OC)OCSC AURNAHLNVYDOGZ-UHFFFAOYSA-N 0.000 description 1
- XFKYKTBPRBZDFG-UHFFFAOYSA-N 2-aminoacetonitrile;hydrochloride Chemical compound Cl.NCC#N XFKYKTBPRBZDFG-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- PMTPFBWHUOWTNN-UHFFFAOYSA-N 2-chloro-4-methoxypyridine Chemical compound COC1=CC=NC(Cl)=C1 PMTPFBWHUOWTNN-UHFFFAOYSA-N 0.000 description 1
- HOZLOOPIXHWKCI-UHFFFAOYSA-N 2-chloro-n-methylacetamide Chemical compound CNC(=O)CCl HOZLOOPIXHWKCI-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- ZPSJGADGUYYRKE-UHFFFAOYSA-N 2H-pyran-2-one Chemical compound O=C1C=CC=CO1 ZPSJGADGUYYRKE-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- YYPNJNDODFVZLE-UHFFFAOYSA-N 3-methylbut-2-enoic acid Chemical compound CC(C)=CC(O)=O YYPNJNDODFVZLE-UHFFFAOYSA-N 0.000 description 1
- BDUBTLFQHNYXPC-UHFFFAOYSA-N 3-methylbut-2-enoyl chloride Chemical compound CC(C)=CC(Cl)=O BDUBTLFQHNYXPC-UHFFFAOYSA-N 0.000 description 1
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- MWVSSYOXUWYVTJ-UHFFFAOYSA-N 4-methoxy-2-methylpyridine Chemical compound COC1=CC=NC(C)=C1 MWVSSYOXUWYVTJ-UHFFFAOYSA-N 0.000 description 1
- UPJKSWLLCONYMW-UHFFFAOYSA-N 5'-Adenosine monophosphate Natural products COc1cc(O)c(C(=O)C)c(OC2OC(COC3OC(C)C(O)C(O)C3O)C(O)C(O)C2O)c1 UPJKSWLLCONYMW-UHFFFAOYSA-N 0.000 description 1
- MJZJYWCQPMNPRM-UHFFFAOYSA-N 6,6-dimethyl-1-[3-(2,4,5-trichlorophenoxy)propoxy]-1,6-dihydro-1,3,5-triazine-2,4-diamine Chemical compound CC1(C)N=C(N)N=C(N)N1OCCCOC1=CC(Cl)=C(Cl)C=C1Cl MJZJYWCQPMNPRM-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- XASOHFCUIQARJT-UHFFFAOYSA-N 8-methoxy-6-[7-(2-morpholin-4-ylethoxy)imidazo[1,2-a]pyridin-3-yl]-2-(2,2,2-trifluoroethyl)-3,4-dihydroisoquinolin-1-one Chemical compound C(N1C(=O)C2=C(OC)C=C(C=3N4C(=NC=3)C=C(C=C4)OCCN3CCOCC3)C=C2CC1)C(F)(F)F XASOHFCUIQARJT-UHFFFAOYSA-N 0.000 description 1
- AEOBEOJCBAYXBA-UHFFFAOYSA-N A2P5P Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1OP(O)(O)=O AEOBEOJCBAYXBA-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- 208000023328 Basedow disease Diseases 0.000 description 1
- HUGJUYPSXULVQQ-UHFFFAOYSA-M Br[Mg]C#C Chemical compound Br[Mg]C#C HUGJUYPSXULVQQ-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- ARXFGTLWCZVXIH-XDHOZWIPSA-N C(CC)C=1C(=C(C=CC=1OC)/C=C(\C(=O)N)/N1C(=CC(C=C1C)=C=O)C)OC Chemical compound C(CC)C=1C(=C(C=CC=1OC)/C=C(\C(=O)N)/N1C(=CC(C=C1C)=C=O)C)OC ARXFGTLWCZVXIH-XDHOZWIPSA-N 0.000 description 1
- 102100036150 C-X-C motif chemokine 5 Human genes 0.000 description 1
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 1
- 208000006344 Churg-Strauss Syndrome Diseases 0.000 description 1
- UDMBCSSLTHHNCD-UHFFFAOYSA-N Coenzym Q(11) Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1O UDMBCSSLTHHNCD-UHFFFAOYSA-N 0.000 description 1
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 1
- 206010013774 Dry eye Diseases 0.000 description 1
- 206010014025 Ear swelling Diseases 0.000 description 1
- 208000018428 Eosinophilic granulomatosis with polyangiitis Diseases 0.000 description 1
- DUDOVLKRJONDFE-WXRGWGKQSA-N FC(OC1=C(C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=C1)O[C@@H]1CS(CC1)=O)F Chemical compound FC(OC1=C(C=C(/C=C/N2C(=CC(C=C2C)=O)C)C=C1)O[C@@H]1CS(CC1)=O)F DUDOVLKRJONDFE-WXRGWGKQSA-N 0.000 description 1
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 208000001204 Hashimoto Disease Diseases 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 101000947186 Homo sapiens C-X-C motif chemokine 5 Proteins 0.000 description 1
- 101000959820 Homo sapiens Interferon alpha-1/13 Proteins 0.000 description 1
- 101000988424 Homo sapiens cAMP-specific 3',5'-cyclic phosphodiesterase 4B Proteins 0.000 description 1
- 206010020649 Hyperkeratosis Diseases 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 102100040019 Interferon alpha-1/13 Human genes 0.000 description 1
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 1
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 1
- 102000003815 Interleukin-11 Human genes 0.000 description 1
- 108090000177 Interleukin-11 Proteins 0.000 description 1
- 102000003816 Interleukin-13 Human genes 0.000 description 1
- 108090000176 Interleukin-13 Proteins 0.000 description 1
- 108050003558 Interleukin-17 Proteins 0.000 description 1
- 102000013691 Interleukin-17 Human genes 0.000 description 1
- 102000003810 Interleukin-18 Human genes 0.000 description 1
- 108090000171 Interleukin-18 Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 102000000646 Interleukin-3 Human genes 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 102000000704 Interleukin-7 Human genes 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 102000000585 Interleukin-9 Human genes 0.000 description 1
- 208000001126 Keratosis Diseases 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- 208000032514 Leukocytoclastic vasculitis Diseases 0.000 description 1
- 101710151805 Mitochondrial intermediate peptidase 1 Proteins 0.000 description 1
- 240000001307 Myosotis scorpioides Species 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- 229940099471 Phosphodiesterase inhibitor Drugs 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- 208000003251 Pruritus Diseases 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 206010040844 Skin exfoliation Diseases 0.000 description 1
- NWGKJDSIEKMTRX-AAZCQSIUSA-N Sorbitan monooleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O NWGKJDSIEKMTRX-AAZCQSIUSA-N 0.000 description 1
- 108700012920 TNF Proteins 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- 208000027515 Tracheal disease Diseases 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- CESGKXMBHGUQTB-VONOSFMSSA-N [(1S,2S,6R,10S,11R,13S,14R,15R)-1,6,14-trihydroxy-8-(hydroxymethyl)-4,12,12,15-tetramethyl-5-oxo-13-tetracyclo[8.5.0.02,6.011,13]pentadeca-3,8-dienyl] tetradecanoate Chemical compound C1=C(CO)C[C@]2(O)C(=O)C(C)=C[C@H]2[C@@]2(O)[C@H](C)[C@@H](O)[C@@]3(OC(=O)CCCCCCCCCCCCC)C(C)(C)[C@H]3[C@@H]21 CESGKXMBHGUQTB-VONOSFMSSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 229940119059 actemra Drugs 0.000 description 1
- 229960002964 adalimumab Drugs 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 229960002459 alefacept Drugs 0.000 description 1
- 208000002205 allergic conjunctivitis Diseases 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229960004238 anakinra Drugs 0.000 description 1
- 238000003975 animal breeding Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 208000024998 atopic conjunctivitis Diseases 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- LKXYJYDRLBPHRS-UHFFFAOYSA-N bromocyclopropane Chemical compound BrC1CC1 LKXYJYDRLBPHRS-UHFFFAOYSA-N 0.000 description 1
- FLHFTXCMKFVKRP-UHFFFAOYSA-N bromomethylcyclobutane Chemical compound BrCC1CCC1 FLHFTXCMKFVKRP-UHFFFAOYSA-N 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- LUEHNHVFDCZTGL-UHFFFAOYSA-N but-2-ynoic acid Chemical compound CC#CC(O)=O LUEHNHVFDCZTGL-UHFFFAOYSA-N 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 229960003115 certolizumab pegol Drugs 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229960001701 chloroform Drugs 0.000 description 1
- 229940090100 cimzia Drugs 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- GDTNRAWOOVYBER-UHFFFAOYSA-N cyclopropylmethyl 3-[3-(cyclopropylmethoxy)-4-methoxyphenyl]prop-2-enoate Chemical compound C1(CC1)COC=1C=C(C=CC(=O)OCC2CC2)C=CC=1OC GDTNRAWOOVYBER-UHFFFAOYSA-N 0.000 description 1
- JJZCWPPVJPGNIV-UHFFFAOYSA-N cyclopropylmethyl 3-[3-(cyclopropylmethyl)-4-methoxyphenyl]prop-2-enoate Chemical compound C1(CC1)CC=1C=C(C=CC(=O)OCC2CC2)C=CC=1OC JJZCWPPVJPGNIV-UHFFFAOYSA-N 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000035618 desquamation Effects 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000003651 drinking water Substances 0.000 description 1
- 235000020188 drinking water Nutrition 0.000 description 1
- 229940073621 enbrel Drugs 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Natural products C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- GWQVMPWSEVRGPY-UHFFFAOYSA-N europium cryptate Chemical compound [Eu+3].N=1C2=CC=CC=1CN(CC=1N=C(C=CC=1)C=1N=C(C3)C=CC=1)CC(N=1)=CC(C(=O)NCCN)=CC=1C(N=1)=CC(C(=O)NCCN)=CC=1CN3CC1=CC=CC2=N1 GWQVMPWSEVRGPY-UHFFFAOYSA-N 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 102000048114 human PDE4B Human genes 0.000 description 1
- 229940048921 humira Drugs 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 208000030603 inherited susceptibility to asthma Diseases 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 239000002085 irritant Substances 0.000 description 1
- 231100000021 irritant Toxicity 0.000 description 1
- 230000007803 itching Effects 0.000 description 1
- 210000002510 keratinocyte Anatomy 0.000 description 1
- 229940054136 kineret Drugs 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- QBNOPZJAURRQCE-UHFFFAOYSA-M magnesium;prop-1-yne;bromide Chemical compound [Mg+2].[Br-].CC#[C-] QBNOPZJAURRQCE-UHFFFAOYSA-M 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 239000012514 monoclonal antibody product Substances 0.000 description 1
- 210000004980 monocyte derived macrophage Anatomy 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 108091008819 oncoproteins Proteins 0.000 description 1
- 102000027450 oncoproteins Human genes 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 1
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000009522 phase III clinical trial Methods 0.000 description 1
- PHEDXBVPIONUQT-RGYGYFBISA-N phorbol 13-acetate 12-myristate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCCCCCCCCCCCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(C)=O)C1(C)C PHEDXBVPIONUQT-RGYGYFBISA-N 0.000 description 1
- 239000002644 phorbol ester Substances 0.000 description 1
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 1
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical compound OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 1
- 229940124606 potential therapeutic agent Drugs 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- RZWZRACFZGVKFM-UHFFFAOYSA-N propanoyl chloride Chemical compound CCC(Cl)=O RZWZRACFZGVKFM-UHFFFAOYSA-N 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 230000016515 regulation of signal transduction Effects 0.000 description 1
- 229940116176 remicade Drugs 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 201000003068 rheumatic fever Diseases 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229960003323 siltuximab Drugs 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- RSIJVJUOQBWMIM-UHFFFAOYSA-L sodium sulfate decahydrate Chemical compound O.O.O.O.O.O.O.O.O.O.[Na+].[Na+].[O-]S([O-])(=O)=O RSIJVJUOQBWMIM-UHFFFAOYSA-L 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 229940071598 stelara Drugs 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940053017 sylvant Drugs 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- YCGJWFCBFZPGJK-UHFFFAOYSA-N thietan-3-ol Chemical compound OC1CSC1 YCGJWFCBFZPGJK-UHFFFAOYSA-N 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- BPLKQGGAXWRFOE-UHFFFAOYSA-M trimethylsulfoxonium iodide Chemical compound [I-].C[S+](C)(C)=O BPLKQGGAXWRFOE-UHFFFAOYSA-M 0.000 description 1
- 229940046728 tumor necrosis factor alpha inhibitor Drugs 0.000 description 1
- 239000002452 tumor necrosis factor alpha inhibitor Substances 0.000 description 1
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 1
- 229960003824 ustekinumab Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 229940045860 white wax Drugs 0.000 description 1
- WCJYTPVNMWIZCG-UHFFFAOYSA-N xylylcarb Chemical compound CNC(=O)OC1=CC=C(C)C(C)=C1 WCJYTPVNMWIZCG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4412—Non condensed pyridines; Hydrogenated derivatives thereof having oxo groups directly attached to the heterocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/68—One oxygen atom attached in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/443—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4436—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to the field of pharmaceutical chemistry, and in particular to an anti-inflammatory compound, a preparation method, and use thereof in the treatment of dry eye syndrome.
- Cytokines include pro-inflammatory cytokines (including IL-1, IL-1 ⁇ , IL-2, IL-3, IL-6, IL-7, IL-9, IL-12, IL-17, IL-18, IL-23, TNF-, LT, LIF, oncoproteins and IFN-); anti-inflammatory cytokines (IL-4, IL-10, IL-11, IL-13 and TGF- ⁇ ); and chemokines (IL-8, Gro-a, MIP-1, MCP-1, ENA-78 and RANTES).
- pro-inflammatory cytokines including IL-1, IL-1 ⁇ , IL-2, IL-3, IL-6, IL-7, IL-9, IL-12, IL-17, IL-18, IL-23, TNF-, LT, LIF, oncoproteins and IFN-
- anti-inflammatory cytokines IL-4, IL-10, IL-11, IL-13 and TGF- ⁇
- chemokines IL-8, Gro-a,
- pro-inflammatory cytokines especially TNF-, IL-1 ⁇ and IL-6, and anti-inflammatory cytokine IL-10, show important roles in the pathogenesis of various inflammation-related diseases and can thus be used as potential therapeutic agents.
- pro-inflammatory cytokines TNF-, IFN, IL-1, IL-2, IL-6, and IL-12
- chemokines IL-8, MCP-1, and RANTES
- inflammation-related diseases such as eczema, psoriasis, enteritis, Graves' disease, and Hashimoto thyroiditis
- IL-10 inhibits the increase in production of pro-inflammatory cytokines in LPMC culture in vitro and in patients.
- Phosphodiesterase (PDE) isozymes are involved in the regulation of signal transduction cascade in cells by regulating the cyclic nucleotide level. So far, a family of 11 PDE isozyme genes have been identified. The difference between these isozymes lies in their cell distribution and biochemical functions. High PDE4 activity was found in leukocytes of patients with atopic dermatitis, particularly children ( Butle, JM, et al., J. Allergy Clin. Immunol.1983, 71: 490-497 ). PDE4 is a main isozyme in inflammatory cells, such as monocytes and monocyte-derived macrophages ( Gantner et al., Br. J.
- PDE4 inhibitors show a highly potent anti-inflammatory effect by increasing the intracellular cAMP level. By inhibiting cAMP degradation, the PDE4 inhibitors can regulate intracellular functions (e.g., reduce peroxide production) and gene transcription (e.g., inhibit the synthesis and/or release of inflammatory cytokines). Because PDE4 is also expressed in keratinocytes, these cells can be used as other possible pharmacological targets for controlling inflammatory skin diseases using PDE4 inhibitors.
- PDE4 inhibitors are useful in diseases related to eosinophil activity, particularly inflammatory tracheal diseases, such as bronchial asthma, allergic rhinitis, allergic conjunctivitis, atopic dermatitis, eczema, allergic angiitis, inflammation and proliferative skin diseases, such as psoriasis or keratosis.
- inflammatory tracheal diseases such as bronchial asthma, allergic rhinitis, allergic conjunctivitis, atopic dermatitis, eczema, allergic angiitis, inflammation and proliferative skin diseases, such as psoriasis or keratosis.
- TNF- ⁇ inhibitors such as Etanercept (Enbrel), infliximab (Remicade) and adalimumab (Humira), certolizumab pegol (Cimzia) can be used for treating immune system-related diseases such as rheumatic arthritis, psoriasis and irritant enteritis; and several drugs targeting IL-1 (Anakinra (Kineret)), IL-4 (dupilumab (Dupixent)), IL-6 (tocilizumab (Actemra), and siltuximab (Sylvant)), or IL-12/IL-23 (Ustekinumab (Stelara)) and Alefacept (Amevive) inhibiting T immune cells can be used for treating various immune diseases.
- IL-1 Adakinra (Kineret)
- IL-4 diupilumab (Dupixent)
- IL-6 tocilizumab (Actemra
- Crisaborole a PDE4 inhibitor, has proved its efficacy and safety in the treatment of mild to moderate eczema in children and adults in Phase III clinical trials (research project Nos.: AD-301 and AD-302) of eczema (also known as allergic dermatitis, atopic dermatitis, or atopic dermatitis) ( Paller AS et al., J Am Acad Dermatol. 2016; 75(3): 494-503 ).
- PDE4 is a key regulatory point for the production of inflammatory cytokines in eczema, which mainly acts by degrading cyclic adenosine monophosphate. In circulating inflammatory cells in patients with eczema, the PDE4 activity increases, and in-vitro tests show that when PDE4 in monocytes is inhibited, the release of cytokines that promote inflammatory response decreases.
- Crisaborole has been proved to have good safety, but limited clinical effect, as indicated by a clinical effect on eczema that is only about 10% higher than that of the blank control. Also, a large proportion (about 50%) of patients cannot benefit from Crisaborole. Crisaborole has an insignificant clinical effect in the treatment of psoriasis due to the low activity (where the enzyme inhibition activity is about 100 nM, and the cell activity inhibiting cytokine release is about 500 nM) (not approved for use).
- the present invention provides a novel anti-inflammatory drug that has a potent inhibition effect on PDE4, an important target for autoimmune activation, and is easy to penetrate the skin, and readily degradable.
- the present disclosure describes an anti-inflammatory compound, which is a compound having a structure shown below:
- R A is hydrogen, alkyl, or aryl, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, cycloalkyl, aryl, or halogen,
- R B is hydrogen, alkyl, cycloalkyl, heterocycloalkyl, aryl, alkenyl, or alkynyl, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, cycloalkyl, heterocycloalkyl, aryl, or halogen, and one or more carbon atom(s) in the groups is/are optionally substituted with sulfur, sulfoxide, sulfone, or sulfonyl; or R B is a hydroxyl protecting group,
- R C is hydrogen, alkyl, halogen, alkoxy, or cyano
- R E is a group where one or more hydrogen atom(s) attached to carbon on the pyridinone ring of G1 is/are substituted with alkyl, aryl, cyano, or halogen, where the alkyl, aryl and halogen are as defined in the scenario of R A .
- the anti-inflammatory compound provided in the present invention is further characterized by G1 that is a three-membered ring, and having a specific structure shown below: in which X is carbon.
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises: using 3-hydroxybenzaldehyde derivative A as a starting material, and substituting the hydrogen in the of the 3-hydroxybenzaldehyde derivative A with R B to obtain an intermediate product B;
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises subjecting the hydroxyl group on the Middle bridge of the type-A target product to an addition/substitution reaction to obtain a type-A-1 target product,
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises oxidizing the hydroxyl group on the middle bridge of the type-A target product to obtain a type-B target product, where the type-B target product is a compound having a structure shown below:
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises deprotecting the group R B that is a hydroxyl protecting group on the type-B target product to obtain a type-B-1 target product, where the type-B-1 target product is a compound having a structure shown below:
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises subjecting the hydroxyl group on the phenyl ring of the type B-1 target product to an addition/substitution reaction to obtain a type B-2 target product,
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises oximating the carbonyl group on the middle bridge of the type-B target product to obtain a type B-3 target product, where the type-B-3 target product is a compound having a structure shown below:
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises subjecting the hydroxyl group on the oxime of the type-B-3 target product to an addition/substitution reaction to obtain a type-B-4 target product,
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises: using a 3-hydroxyacetophenone derivative I as a starting material, and substituting the hydrogen in the of the 3-hydroxyacetophenone derivative I with R B , to obtain an intermediate product II;
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises removing the hydroxyl group on the middle bridge of the type-A target product to obtain a type-C target product, or
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises reducing and then eliminating the carbonyl group on the middle bridge of the type-B-2 target product to obtain a type-C-1 target product, where the type-C-1 target product is a compound having a structure shown below:
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises subjecting the hydroxyl group on the phenyl ring of the type C-1 target product to an addition/substitution reaction to obtain a type C-2 target product,
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises reacting the carbonyl group on the middle bridge of the type-B target product with a halogenating reagent to obtain an intermediate product X1;
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises: reacting an acetophenone derivative 1 as a starting material with trimethylsilyl cyanide, to obtain an intermediate product 2;
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises removing the hydroxyl group on the middle bridge of the type-A' target product to obtain a type-C' target product, where the type-C' target product is a compound having a structure shown below:
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises reacting the carbonyl group on the middle bridge of the type-B target product with trimethylsilyl cyanide to obtain an intermediate product Y1;
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises reacting a six-membered N-acetonitrile compound Z1 with a benzaldehyde derivative Z2 to obtain a type-C" target product,
- the present invention also provides a method for preparing the anti-inflammatory compound, which comprises: esterifying a cinnamic acid derivative 1 as a starting material, to obtain an intermediate product 2;
- the present invention provides a compound, wherein the compound has a structure shown below:
- the present invention also provides use of the anti-inflammatory compound as a PDE4 inhibitor.
- the present invention also provides use of the anti-inflammatory compound in the treatment of inflammatory skin diseases.
- the present invention also provides a pharmaceutical composition comprising 0.01-10% of the compound mentioned above.
- the present invention provides a novel anti-inflammatory compound that has a potent inhibition effect on PDE4, an important target for autoimmune activation, and is easy to penetrate the skin, and readily degradable. It is more effective than existing drugs (such as Eucrisa) or has fewer side effects than existing drugs (such as hormones, and tacrolimus), thus being a topical drug for treating eczema having good effects and no toxic side effects.
- 3-hydroxy-4-methoxybenzaldehyde (3.04 g, 20 mmol) was dissolved in acetonitrile (80 mL), and then potassium carbonate (5.52 g, 40 mmol) and bromomethylcyclopropane (4.05 g, 30 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, a saturated aqueous sodium chloride solution (60 mL) was added, and extracted with dichloromethane (3 x 100 mL).
- 3-cyclopropylmethoxy-4-methoxybenzaldehyde (3.09 g, 15 mmol) was dissolved in dichloromethane (30 mL); and under nitrogen atmosphere, triethyl amine (4.16 mL, 30 mmol) and trimethylsilyl cyanide (3.75 g, 30 mmol) were added, and stirred for 6 hours at room temperature.
- the reaction solution was concentrated and rotary dried to obtain a crude product of 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4 H )-yl)ethyl) acetate (116 mg, yield 30%, pale yellow oil).
- reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4 H )-yl)ethyl) propionate (140 mg, yield 35%, pale yellow oil).
- reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4 H )-yl)ethyl) cyclopropylcarboxylate (103 mg, yield 25%, pale yellow oil).
- reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4 H )-yl)ethyl) benzoate (94 mg, yield 21%, white solid).
- reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4 H )-yl)ethyl) crotonate (127 mg, yield 31%, white solid).
- reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4 H )-yl)ethyl)3-methyl crotonate (137 mg, yield 32%, white solid).
- reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4 H )-yl)ethyl)but-2-ynoate (37 mg, 9%, white solid).
- 3-hydroxy-4-methoxybenzaldehyde (1.52 g, 10 mmol) was dissolved in acetonitrile (40 mL), and then potassium carbonate (2.76 g, 20 mmol) and benzyl bromide (2.56 g, 15 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, a saturated aqueous sodium chloride solution (30 mL) was added, and extracted with dichloromethane (3 x 50 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by column chromatography to obtain 3-benzoxy-4-methoxybenzaldehyde (2.30 g, 95%, white solid).
- reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain 1-(2-(3-(tetrahydrofuran-2-yl)oxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1 H )-one (28 mg, yield 44%, white solid).
- 3-methylthiomethoxy-4-methoxybenzaldehyde (1.7 g, 8 mmol) was dissolved in dichloromethane (20 mL); and under nitrogen atmosphere, triethyl amine (1.6 g, 16 mmol) and trimethylsilyl cyanide (1.6 g, 16 mmol) were added, and stirred for 6 hours at room temperature.
- the reaction solution was concentrated and rotary dried to obtain 2-(3-methylthiomethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- 3-cyclopropylmethoxy-4-methoxybenzaldehyde (3.09 g, 15 mmol) was dissolved in dichloromethane (30 mL); and under nitrogen atmosphere, triethyl amine (4.16 mL, 30 mmol) and trimethylsilyl cyanide (3.75 g, 30 mmol) were added, and stirred for 6 hours at room temperature.
- the reaction solution was concentrated and rotary dried to obtain 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- 3-hydroxy-4-methoxyacetophenone (1.66 g, 10 mmol) was dissolved in acetonitrile (30 mL); and under nitrogen atmosphere, potassium carbonate (2.76 g, 20 mmol) and bromomethylcyclopropane (2.0 g, 15mmol) were added, and stirred for 6 hours at 80°C.
- the reaction solution was filtered.
- the filtrate was concentrated, rotary dried, and purified by column chromatography to obtain 1-(3-cyclopropylmethoxy-4-methoxyphenyl)ethanone (2.1 g, yield 95%, white solid).
- 3-hydroxy-4-difluoromethoxybenzaldehyde (1.88 g, 10.0 mmol) was dissolved in acetonitrile (10 mL), and then potassium carbonate (2.07 g, 15.0 mmol) and bromomethylcyclopropane (1.76 g, 13.0 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, saturated saline (30 mL) was added, and extracted with ethyl acetate (3 x 60 mL).
- 3-hydroxy-4-difluoromethoxybenzaldehyde (2.36 g, 12.5 mmol) was dissolved in acetonitrile (40 mL), and then potassium carbonate (3.46 g, 25.1 mmol) and benzyl bromide (2.79 g, 16.3 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, a saturated aqueous sodium chloride solution (30 mL) was added, and extracted with dichloromethane (3 x 120 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by column chromatography to obtain 3-benzoxy-4-difluoromethoxybenzaldehyde (3.30 g, 95%).
- 3-benzoxy-4-difluoromethoxybenzaldehyde (3.30 g, 11.9 mmol) was dissolved in dichloromethane (30 mL), and then triethyl amine (2.40 g, 23.7 mmol) and trimethylsilyl cyanide (3.53 g, 35.6 mmol) were added in sequence in an ice bath and stirred for 16 hours under nitrogen atmosphere at room temperature. After the reaction was completed, the reaction solution was directly rotary dried to obtain 2-(3-benzoxy-4-difluoromethoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- reaction was diluted with anhydrous tetrahydrofuran (200 mL), and then water (1.35 mL), an aqueous sodium hydroxide solution (1.35 mL, 15%), and water (4.05 mL) were added in sequence, stirred for half an hour, dried over anhydrous sodium sulfate, filtered, and rotary dried to obtain a crude product of 2-amino-1-(3-benzoxy-4-difluoromethoxyphenyl)ethanol, which was directly used in the next reaction.
- reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain ( E )-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-methylethenyl)-2,6-dimethylpyridin-4(1 H )-one (27 mg, yield 80%, white solid).
- Trisphenyl phosphite (1.49 g, 4.80 mmol) was dissolved in anhydrous tetrahydrofuran (15 mL), and then triethyl amine (0.53 mg, 5.20 mmol) and the compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)pyridin-2,6-dimethyl-4(1 H )-one (1.36 mg, 4.00 mmol) was slowly added to the reaction solution at -60°C and stirred for 15 min. Then, bromine (0.77 mg, 4.80 mmol) was slowly added to the reaction solution.
- 3-(cyclopropylmethoxy)-4-methoxybenzaldehyde (1.03 g, 5 mmol) was dissolved in anhydrous tetrahydrofuran (5 mL); and under nitrogen atmosphere, 1-ethynylmagnesium bromide (0.5 M, 12 mL, 6 mmol) was added dropwise at 0°C, warmed to room temperature, and stirred for 3 hours at room temperature. The reaction was quenched with a saturated aqueous ammonium chloride solution, and extracted with dichloromethane (3 x 30 mL).
- the compound 1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-alkynyl-1-butanol (1.50 g, 5 mmol) was dissolved in dichloromethane (20 mL); and Dess-Martin Periodinane (3.18g, 7.5 mmol) and solid sodium bicarbonate (6.30 g, 75 mmol) were added at 35°C, and stirred for 1 hr. After the reaction was completed, a saturated sodium bicarbonate solution was added, and extracted with dichloromethane (3 x 30 mL).
- reaction solution was rotary dried, and purified to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxypent-3-yn-1-yl)-2,6-dimethylpyridin-4(1 H )-one (20 mg, yield 3%).
- reaction solution was directly rotary dried, and purified by column chromatography to obtain ( Z )-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4 H )-yl)-acrylamide (140 mg, 27%, white solid).
- reaction solution was quenched with a saturated aqueous ammonium chloride solution, and the reaction solution was directly rotary dried, and purified by column chromatography to obtain ( Z )-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2, 6-dimethyl-4-carbonylpyridin-1(4 H )-yl)- N -methylacrylamide (5 mg, 17%, white solid).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Dermatology (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
- The present invention relates to the field of pharmaceutical chemistry, and in particular to an anti-inflammatory compound, a preparation method, and use thereof in the treatment of dry eye syndrome.
- Human autoimmune inflammation is a main factor causing many human diseases. People with degenerative diseases usually exhibit excessive levels of pro-inflammatory modulators in their blood. A class of such pro-inflammatory modulators are cytokines. Cytokines include pro-inflammatory cytokines (including IL-1, IL-1β, IL-2, IL-3, IL-6, IL-7, IL-9, IL-12, IL-17, IL-18, IL-23, TNF-, LT, LIF, oncoproteins and IFN-); anti-inflammatory cytokines (IL-4, IL-10, IL-11, IL-13 and TGF-β); and chemokines (IL-8, Gro-a, MIP-1, MCP-1, ENA-78 and RANTES).
- In many situations of inflammation, pro-inflammatory cytokines, especially TNF-, IL-1β and IL-6, and anti-inflammatory cytokine IL-10, show important roles in the pathogenesis of various inflammation-related diseases and can thus be used as potential therapeutic agents. For example, elevated levels of pro-inflammatory cytokines (TNF-, IFN, IL-1, IL-2, IL-6, and IL-12) and chemokines (IL-8, MCP-1, and RANTES) have been observed in inflammation-related diseases, such as eczema, psoriasis, enteritis, Graves' disease, and Hashimoto thyroiditis, with simultaneous increases in their soluble TNF receptors, IL-1 receptor antagonists, and anti-inflammatory cytokine IL-10. It has been confirmed that IL-10 inhibits the increase in production of pro-inflammatory cytokines in LPMC culture in vitro and in patients.
- Phosphodiesterase (PDE) isozymes are involved in the regulation of signal transduction cascade in cells by regulating the cyclic nucleotide level. So far, a family of 11 PDE isozyme genes have been identified. The difference between these isozymes lies in their cell distribution and biochemical functions. High PDE4 activity was found in leukocytes of patients with atopic dermatitis, particularly children (Butle, JM, et al., J. Allergy Clin. Immunol.1983, 71: 490-497). PDE4 is a main isozyme in inflammatory cells, such as monocytes and monocyte-derived macrophages (Gantner et al., Br. J. Pharmacol.,1997, 121: 221-2317), eosinophils (Dent et al., J. Pharmacol. Exp. Ther., 1994, 271: 1167-1174) and B lymphocytes (Cooper et al., J. Invest. Dermatol., 2985, 84: 477-482). PDE4 inhibitors show a highly potent anti-inflammatory effect by increasing the intracellular cAMP level. By inhibiting cAMP degradation, the PDE4 inhibitors can regulate intracellular functions (e.g., reduce peroxide production) and gene transcription (e.g., inhibit the synthesis and/or release of inflammatory cytokines). Because PDE4 is also expressed in keratinocytes, these cells can be used as other possible pharmacological targets for controlling inflammatory skin diseases using PDE4 inhibitors.
- PDE4 inhibitors are useful in diseases related to eosinophil activity, particularly inflammatory tracheal diseases, such as bronchial asthma, allergic rhinitis, allergic conjunctivitis, atopic dermatitis, eczema, allergic angiitis, inflammation and proliferative skin diseases, such as psoriasis or keratosis. These compounds can be administered possibly in the form of oral, transdermal, topical, inhalable and intranasal preparations.
- In recent years, great changes have taken place in the treatment of inflammation-related diseases. Due to the more and more attention paid to the severity of these diseases and the insight into the important role of cytokines in their immune pathogenesis by the patients and physicians, many drugs targeting cytokines are available in the clinic and some are available in the market to treat these diseases related to the autoimmune system. Most of the efforts are focused on targeting TNF-α and IL-1. At present, many drugs are available on the market. For example, TNF-α inhibitors such as Etanercept (Enbrel), infliximab (Remicade) and adalimumab (Humira), certolizumab pegol (Cimzia) can be used for treating immune system-related diseases such as rheumatic arthritis, psoriasis and irritant enteritis; and several drugs targeting IL-1 (Anakinra (Kineret)), IL-4 (dupilumab (Dupixent)), IL-6 (tocilizumab (Actemra), and siltuximab (Sylvant)), or IL-12/IL-23 (Ustekinumab (Stelara)) and Alefacept (Amevive) inhibiting T immune cells can be used for treating various immune diseases.
- However, when these monoclonal antibody products are systemically administered, the immunity of patients is inhibited, resulting in infection or other systemic side effects. Therefore, these drugs are clinically approved for treating patients with moderate to severe inflammatory diseases. Most patients with psoriasis and eczema are mild to moderate, so these drugs cannot help them.
- Crisaborole, a PDE4 inhibitor, has proved its efficacy and safety in the treatment of mild to moderate eczema in children and adults in Phase III clinical trials (research project Nos.: AD-301 and AD-302) of eczema (also known as allergic dermatitis, atopic dermatitis, or atopic dermatitis) (Paller AS et al., J Am Acad Dermatol. 2016; 75(3): 494-503). PDE4 is a key regulatory point for the production of inflammatory cytokines in eczema, which mainly acts by degrading cyclic adenosine monophosphate. In circulating inflammatory cells in patients with eczema, the PDE4 activity increases, and in-vitro tests show that when PDE4 in monocytes is inhibited, the release of cytokines that promote inflammatory response decreases.
- Crisaborole has been proved to have good safety, but limited clinical effect, as indicated by a clinical effect on eczema that is only about 10% higher than that of the blank control. Also, a large proportion (about 50%) of patients cannot benefit from Crisaborole. Crisaborole has an insignificant clinical effect in the treatment of psoriasis due to the low activity (where the enzyme inhibition activity is about 100 nM, and the cell activity inhibiting cytokine release is about 500 nM) (not approved for use).
- In view of this, it can be found that the existing drugs for eczema and psoriasis cannot meet the needs of patients, particularly the majority of mild-moderate patients, and more effective and safe local drugs are still needed to be developed to treat most mild-moderate inflammatory skin diseases (such as psoriasis, eczema, etc.).
- The present invention provides a novel anti-inflammatory drug that has a potent inhibition effect on PDE4, an important target for autoimmune activation, and is easy to penetrate the skin, and readily degradable.
- The invention is set out in the appended set of Claims.
- The present disclosure describes an anti-inflammatory compound, which is a compound having a structure shown below:

- In the structural formula, RA is hydrogen, alkyl, or aryl, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, cycloalkyl, aryl, or halogen,
- the alkyl is an alkyl group having 1 to 15 carbon atoms, generally a group having the general formula CnH2n+1-, and preferably a linear or branched alkyl group where n is not greater than 6;
- the aryl group is selected from any aromatic groups consisting of pure carbocyclic or heterocyclic ring(s) with 5-24 carbon atoms, such as cyclopentadienyl, phenyl, naphthyl, quinolinyl, pyrrolyl, pyridinyl, furyl, and the like; and
- the "one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl (which generally refers to an alkyl group with no more than 10 carbon atoms), cycloalkyl (three-, four-, five-, six-, or seven-membered), heterocycloalkyl (aza/thia/oxa-three/four/five/six/seven-membered), aryl (which generally refers to an aryl group having 5 to 20 carbon atoms), or halogen (fluoro, chloro, bromo, iodo)" means the forms of substitution of hydrogen on an alkyl group, for example, -CH2Cl, -CH2CH2Cl, -CH2(Br)CH3, -CH2CH2F, -CHF2, -CH(CH3)CHF2, -CH(Ph)CH3,

- or the forms of substitution of hydrogen on an aromatic group, for example,


- RB is hydrogen, alkyl, cycloalkyl, heterocycloalkyl, aryl, alkenyl, or alkynyl, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, cycloalkyl, heterocycloalkyl, aryl, or halogen, and one or more carbon atom(s) in the groups is/are optionally substituted with sulfur, sulfoxide, sulfone, or sulfonyl; or RB is a hydroxyl protecting group,
- where the alkyl is as defined in the scenario of RA;
- the aryl is as defined in the scenario of RA;
- the cycloalkyl refers to a three-membered, four-membered, five-membered, or six-membered ring;
- the heterocycloalkyl refers to an oxalazalthia three-membered, four-membered, five-membered, or six-membered ring;
- the alkenyl may be selected from linear or branched alkenyl with 2-10 carbon atoms, such as ethenyl (-=), 2-propenyl (-CH2-=), 2-butenyl (-CH2-=-CH3), 3-butenyl (-CH2-CH2-=), cyclopentenyl

- the alkynyl may be selected from linear or branched alkynyl with 2-10 carbon atoms, such as ethynyl


- the "one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, cycloalkyl, heterocycloalkyl, aryl, or halogen" has the same meaning as in the scenario of RA;
- the "one or more carbon atom(s) in the groups is/are optionally substituted with sulfur, sulfoxide, sulfone, or sulfonyl refers to a situation in which one or more carbon atom(s) such as C1 and C2 on the structure



- the hydroxyl protecting group means that a protectant reacts with a hydroxyl group to converts the hydroxyl group into silicon ether, benzyl ether, methyl ether, allyl ether and other forms, so that it is not affected by oxidation, reduction and other reactions.
- RC is hydrogen, alkyl, halogen, alkoxy, or cyano,
- where the alkyl is selected from a linear or branched alkyl group with 1-20 carbon atoms; and
- the alkoxy refers to the group -O-R, in which R is a branched or linear alkyl group with 1-20 carbon atoms;
- RD1 is hydrogen, oxygen, nitrogen, hydroxy, cyano, amino, amido, alkyl, aryl, an ester group, carboxyl, alkynyl, or alkenyl, and in the above groups one or more hydrogen atom(s) attached to carbon/oxygen (that is, hydrogen in -OH)/nitrogen (that is, hydrogen in -NH2, -NH(R), or =NH) in the groups is/are optionally substituted with alkyl, cycloalkyl, alkynyl, alkenyl, aryl, halogen, sulfonyl, a sulfoxide group, or an ether group, and one or more carbon atom(s) in the groups is/are optionally substituted with sulfur, sulfoxide, sulfone, or sulfonyl,
- where the alkyl is as defined in the scenario of RA;
- the amino refers to -NH2;
- the amido refers to a group denoted by

- the aryl is as defined in the scenario of RA;
- the alkenyl is as defined in the scenario of RB;
- the alkynyl is as defined in the scenario of RB;
- the ester group refers to a group denoted by

- where the alkyl is as defined in the scenario of RB, and
- the aryl is as defined in the scenario of RB;
- in the "one or more hydrogen atom(s) attached to carbon/oxygen/nitrogen in the groups is/are optionally substituted with alkyl, cycloalkyl, alkynyl, alkenyl, aryl, halogen, sulfonyl, a sulfoxide group, or an ether group", the alkyl, cycloalkyl, aryl, and halogen are as defined in the scenario of RA; the alkynyl generally refers to an alkynyl substituent with no more than 20 carbon atoms, and may be



- the "one or more carbon atom(s) in the groups is/are optionally substituted with sulfur, sulfoxide, sulfone, or sulfonyl" has the same meaning as in the scenario of RB.
- C1-RD1 bond is a single bond, or a double bond (e.g. C=O, C=N, and the like);
- RD2 is hydrogen, cyano, alkyl, cycloalkyl, aryl, an ester group, or carboxyl, and in the above groups one or more hydrogen atom(s) attached to carbon/oxygen/nitrogen in the groups is/are optionally substituted with alkyl, cycloalkyl, alkynyl, alkenyl, aryl, halogen, sulfonyl, a sulfoxide group, or an ether group,
- where the amino, amido, alkyl, cycloalkyl, aryl, ester group, "one or more hydrogen atom(s) attached to carbon/oxygen/nitrogen in the groups is/are optionally substituted with alkyl, cycloalkyl, alkynyl, alkenyl, aryl, halogen, sulfonyl, a sulfoxide group, or an ether group" have the same meaning as those in the scenario of RD1.
- G1 is a single bond, a double bond or a ring comprising C1 and C2,
where the ring may be a ring-opening product of C1 and C2 double bonds, for example, cyclopropane, and propylene oxide; or a cyclization reaction product of a compound having a double bond or a triple bond such as cis-butadiene, cyclopentene and the like with C1=C2 double bond. - RE is a group where one or more hydrogen atom(s) attached to carbon on the pyridinone ring of G1 is/are substituted with alkyl, aryl, cyano, or halogen,
where the alkyl, aryl and halogen are as defined in the scenario of RA. - Further, the anti-inflammatory compound provided in the present invention is further characterized by G1 that is a three-membered ring, and having a specific structure shown below:

- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises: using 3-hydroxybenzaldehyde derivative A as a starting material, and substituting the hydrogen in the of the 3-hydroxybenzaldehyde derivative A with RB to obtain an intermediate product B;
- reacting the intermediate product B in the presence of trimethylsilyl cyanide to obtain an intermediate product C;
- reducing the intermediate product C to obtain an intermediate product D having an amino group; and
- reacting the amino group in the intermediate product D with a six-membered oxygen-containing cyclic compound to obtain a type-A target product,
- where the 3-hydroxybenzaldehyde derivative A is a compound having a structure shown below:

- the intermediate product B is a compound having a structure shown below:

- the intermediate product C is a compound having a structure shown below:
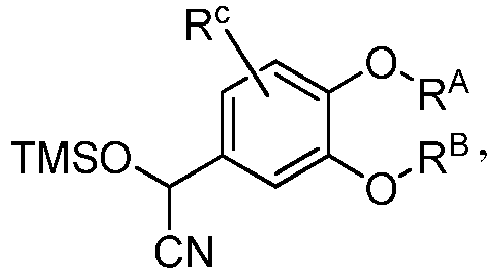
- the intermediate product D is a compound having a structure shown below:

- the type-A target product is a compound having a structure shown below:

- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises subjecting the hydroxyl group on the Middle bridge of the type-A target product to an addition/substitution reaction to obtain a type-A-1 target product,
- where the type-A-1 target product is a compound having a structure shown below:

- in which Rd is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl.
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises oxidizing the hydroxyl group on the middle bridge of the type-A target product to obtain a type-B target product,
where the type-B target product is a compound having a structure shown below:
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises deprotecting the group RB that is a hydroxyl protecting group on the type-B target product to obtain a type-B-1 target product,
where the type-B-1 target product is a compound having a structure shown below:
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises subjecting the hydroxyl group on the phenyl ring of the type B-1 target product to an addition/substitution reaction to obtain a type B-2 target product,
- where the type-B-2 target product is a compound having a structure shown below:

- in which Rd is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl.
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises oximating the carbonyl group on the middle bridge of the type-B target product to obtain a type B-3 target product,
where the type-B-3 target product is a compound having a structure shown below:
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises subjecting the hydroxyl group on the oxime of the type-B-3 target product to an addition/substitution reaction to obtain a type-B-4 target product,
- where the type-B-4 target product is a compound having a structure shown below:

- in which Rd-1 is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the group is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, heterocycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl.
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises: using a 3-hydroxyacetophenone derivative I as a starting material, and substituting the hydrogen in the of the 3-hydroxyacetophenone derivative I with RB, to obtain an intermediate product II;
- reacting the intermediate product II in the presence of a halogenating agent to obtain an intermediate product III; and
- reacting the halogen in the intermediate product III with a six-membered oxygen-containing cyclic compound to obtain a type-B target product,
- where the 3-hydroxyacetophenone derivative I is a compound having a structure shown below:

- the intermediate product II is a compound having a structure shown below:

- the intermediate product III is a compound having a structure shown below:

- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises removing the hydroxyl group on the middle bridge of the type-A target product to obtain a type-C target product,
or - reducing and then eliminating the carbonyl group on the middle bridge of the type-B target product to obtain a type-C target product,
- where the type-C target product is a compound having a structure shown below:

- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises reducing and then eliminating the carbonyl group on the middle bridge of the type-B-2 target product to obtain a type-C-1 target product,
where the type-C-1 target product is a compound having a structure shown below:
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises subjecting the hydroxyl group on the phenyl ring of the type C-1 target product to an addition/substitution reaction to obtain a type C-2 target product,
- where the type-C-2 target product is a compound having a structure shown below:

- in which Rb-1 is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, heterocycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl.
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises reacting the carbonyl group on the middle bridge of the type-B target product with a halogenating reagent to obtain an intermediate product X1; and
- replacing the halogen in the intermediate product X1 to obtain a type-C-3 target product,
- where the intermediate product X1 is a compound having a structure shown below:

- where the type-C-3 target product is a compound having a structure shown below:

- in which Rd-2 is aryl, alkyl, cycloalkyl, an ether group, or an ester group where one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl.
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises: reacting an acetophenone derivative 1 as a starting material with trimethylsilyl cyanide, to obtain an intermediate product 2;
- reducing the intermediate product 2 to obtain an intermediate product 3; and
- reacting the amino group in the intermediate product 3 with a six-membered oxygen-containing cyclic compound to obtain a type-A' target product,
- where the acetophenone derivative 1 is a compound having a structure shown below:

- the intermediate product 2 is a compound having a structure shown below:

- the intermediate product 3 is a compound having a structure shown below:

- the type-A' target product is a compound having a structure shown below:

- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises removing the hydroxyl group on the middle bridge of the type-A' target product to obtain a type-C' target product,
where the type-C' target product is a compound having a structure shown below:
- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises reacting the carbonyl group on the middle bridge of the type-B target product with trimethylsilyl cyanide to obtain an intermediate product Y1; and
- subjecting the intermediate product Y1 to reduction and elimination to obtain a type-C" target product,
- where the intermediate product Y1 is a compound having a structure shown below:

- the type-C" target product is a compound having a structure shown below:

- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises reacting a six-membered N-acetonitrile compound Z1 with a benzaldehyde derivative Z2 to obtain a type-C" target product,
- where the six-membered N-acetonitrile compound Z1 is a compound having a structure shown below:

- the benzaldehyde derivative Z2 is a compound having a structure shown below:

- In addition, the present invention also provides a method for preparing the anti-inflammatory compound, which comprises: esterifying a cinnamic acid derivative 1 as a starting material, to obtain an intermediate product 2;
- forming a ring from the double bond on the intermediate product 2 to obtain an intermediate product 3;
- hydrolyzing the terminal ester group of the intermediate product 3 to give an intermediate product 4 having a carboxyl group;
- aminating the terminal carboxyl group of the intermediate product 4 to obtain an intermediate product 5; and
- reacting the intermediate product 5 with a six-membered oxygen-containing cyclic compound to obtain a type-D target product,
- where the cinnamic acid derivative 1 is a compound having a structure shown below:

- the intermediate product 2 is a compound having a structure shown below:

- the intermediate product 3 is a compound having a structure shown below:

- the intermediate product 4 is a compound having a structure shown below:

- the intermediate product 5 is a compound having a structure shown below:

- The present invention provides a compound, wherein the compound has a structure shown below:

- wherein RA is hydrogen, alkyl comprising from 1 to 6 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is three-membered, four-membered, five-membered or six-membered cycle, or halogen;
- RB is hydrogen, alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is a three-membered, four-membered, five-membered or six-membered cycle, heterocycloalkyl which is an oxo/thia three-membered, four-membered, five-membered or six-membered cycle, alkenyl comprising from 2 to 4 carbon atoms, or alkynyl comprising from 2 to 4 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is a three-membered, four-membered, five-membered or six-membered cycle, or halogen, and one or more carbon atom(s) in the groups is/are optionally substituted with a sulfur atom, sulfoxide, sulfone, or sulfonyl;
- RC is hydrogen;
- RD1 is hydrogen, oxygen, nitrogen, hydroxy, cyano, alkyl comprising from 1 to 6 carbon atoms, phenyl, an ester group, carboxyl, or alkynyl comprising from 2 to 4 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon/oxygen in the groups is/are optionally substituted with alkyl comprising 1 to 6 carbon atoms, cycloalkyl which is a three-membered, a four-membered, five-membered or six-membered ring, alkynyl comprising from 2 to 4 carbon atoms, alkenyl comprising from 2 to 4 carbon atoms, or phenyl, and one or more carbon atom(s) in the groups is/are optionally substituted with a sulfur atom;
- C1-RD1 bond is a single bond or a double bond;
- RD2 is hydrogen or cyano;
- G1 is a single bond, a double bond or cyclopropane comprising C1 and C2; and
- RE is a group where one or more hydrogen atom(s) attached to carbon on the pyridinone ring is/are substituted with alkyl comprising from 1 to 6 carbon atoms or halogen.
- In addition, the present invention also provides use of the anti-inflammatory compound as a PDE4 inhibitor.
- In addition, the present invention also provides use of the anti-inflammatory compound in the treatment of inflammatory skin diseases.
- In addition, the present invention also provides a pharmaceutical composition comprising 0.01-10% of the compound mentioned above; and
- other components selected from a surfactant, a lipid compound, and an auxiliary agent;
- where the amount of the surfactant accounts for 10-30% of the total weight of the pharmaceutical composition;
- the amount of the lipid compound accounts for 50-85% of the total weight of the pharmaceutical composition; and
- the amount of the auxiliary agent accounts for 10-30% of the total weight of the pharmaceutical composition.
- The present invention provides a novel anti-inflammatory compound that has a potent inhibition effect on PDE4, an important target for autoimmune activation, and is easy to penetrate the skin, and readily degradable. It is more effective than existing drugs (such as Eucrisa) or has fewer side effects than existing drugs (such as hormones, and tacrolimus), thus being a topical drug for treating eczema having good effects and no toxic side effects.
-

- The specific reaction scheme is as shown below:

- At room temperature, 3-hydroxy-4-methoxybenzaldehyde (3.04 g, 20 mmol) was dissolved in acetonitrile (80 mL), and then potassium carbonate (5.52 g, 40 mmol) and bromomethylcyclopropane (4.05 g, 30 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, a saturated aqueous sodium chloride solution (60 mL) was added, and extracted with dichloromethane (3 x 100 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by column chromatography to obtain 3-cyclopropylmethoxy-4-methoxybenzaldehyde (3.50 g, 85%).
- 3-cyclopropylmethoxy-4-methoxybenzaldehyde (3.09 g, 15 mmol) was dissolved in dichloromethane (30 mL); and under nitrogen atmosphere, triethyl amine (4.16 mL, 30 mmol) and trimethylsilyl cyanide (3.75 g, 30 mmol) were added, and stirred for 6 hours at room temperature. The reaction solution was concentrated and rotary dried to obtain a crude product of 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile (4.57 g, 15 mmol) obtained in the above step was dissolved in anhydrous tetrahydrofuran (50 mL), and then lithium aluminum hydride (1.14 g, 30 mmol) was added portion-wise in an ice bath, and stirred overnight at room temperature. After the reaction was completed, water (1.2 mL), an aqueous sodium hydroxide solution (1.2 mL, 15%), and water (3.6 mL) were added in sequence, stirred for half an hour, dried over anhydrous sodium sulfate, filtered, and rotary dried to obtain a crude product of 2-amino-1-(3-cyclopropylmethoxy-4-methoxyphenyl)ethanol.
- 2-amino-1-(3-(cyclopropylmethoxy)-4-methoxyphenyl)ethanol obtained in the above step was dissolved in ethanol (20 mL), and then 2,6-dimethyl-4H-pyran-4-one (1 g, 10.41 mmol) and an aqueous sodium hydroxide solution (2 M, 20 mL) were added, and stirred overnight at 60°C. After the reaction was completed, the reaction solution was rotary dried, and purified by column chromatography to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6, dimethylpyridin-4(1H)-one (2.01 g, yield 58%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.03 (d, J = 1.6 Hz, 2H), 6.90 (dd, J = 8.0, 1.6 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H), 6.05 (s, 2H), 5.03 (dd, J = 9.6, 3.2 Hz, 1H), 4.03 (dd, J = 15.2, 10.0 Hz, 1H), 3.93-3.84 (m, 6H), 2.47 (s, 6H), 1.39-1.26 (m, 1H), 0.69-0.64 (m, 2H), 0.40-0.36 (m, 2H); LC-MS: m/z 344.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (343 mg, 1.0 mmol) was dissolved in dichloromethane (8 mL), and then acetyl chloride (118 mg, 1.5 mmol) and triethyl amine (202 mg, 2.0 mmol) were added in sequence, and stirred overnight at room temperature. After the reaction was completed, the reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) acetate (116 mg, yield 30%, pale yellow oil). 1H NMR (400 MHz, CD3OD) δ 7.05-6.96 (m, 5H), 6.12-6.08 (m, 1H), 4.82-4.75 (m, 1H), 4.62-4.56 (m, 1H), 3.86-3.83 (m, 5H), 2.75 (s, 6H), 2.03 (s, 3H) 1.29-1.21 (m, 1H), 0.64-0.60 (m, 2H), 0.37-0.33 (m, 2H); LC-MS: m/z 386.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (343 mg, 1.0 mmol) was dissolved in dichloromethane (8 mL), and then propionyl chloride (139 mg, 1.5 mmol) and triethyl amine (202 mg, 2.0 mmol) were added in sequence, and stirred overnight at room temperature. After the reaction was completed, the reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) propionate (140 mg, yield 35%, pale yellow oil). 1H NMR (400 MHz, CD3OD) δ 7.05-6.97 (m, 5H), 6.15-6.12 (m, 1H), 4.82-4.76 (m, 1H), 4.64-4.59 (m, 1H), 3.86-3.83 (m, 5H), 2.77 (s, 6H), 2.38-2.29 (m, 2H), 1.29-1.24 (m, 1H), 1.05-1.01 (m, 3H), 0.64-0.60 (m, 2H), 0.36-0.33 (m, 2H); LC-MS: m/z 399.9 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (343 mg, 1.0 mmol) was dissolved in dichloromethane (8 mL), and then cyclopropanecarbonyl chloride (157 mg, 1.5 mmol) and triethyl amine (202 mg, 2.0 mmol) were added in sequence, and stirred overnight at room temperature. After the reaction was completed, the reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) cyclopropylcarboxylate (103 mg, yield 25%, pale yellow oil). 1H NMR (400 MHz, CD3OD) δ 7.04-7.01 (m, 2H), 6.95 (s, 1H), 6.35 (s, 2H), 6.11-6.06 (m, 1H), 4.56-4.50 (m, 1H), 4.37-4.32 (m, 1H), 3.89-3.81 (m, 5H), 2.53 (s, 6H), 0.92-0.81 (m, 6H), 0.66-0.62 (m, 2H), 0.39-0.36 (m, 2H); LC-MS: m/z 412.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (343 mg, 1.0 mmol) was dissolved in dichloromethane (8 mL), and then benzoyl chloride (211 mg, 1.5 mmol) and triethyl amine (202 mg, 2.0 mmol) were added in sequence and stirred overnight at room temperature. After the reaction was completed, the reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) benzoate (94 mg, yield 21%, white solid). 1H NMR (400 MHz, CD3OD) δ 8.04-8.00 (m, 2H), 7.70-7.65 (m, 1H), 7.56-7.51 (m, 2H), 7.22 (dd, J = 8.4, 2.0 Hz, 1H), 7.12 (d, J = 2.0 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 7.00 (s, 2H), 6.48-6.44 (m, 1H), 5.00-4.93 (m, 1H), 4.81-4.75 (m, 1H), 3.91-3.87 (m, 5H), 2.82 (s, 6H), 2.03 (s, 3H) 1.32-1.25 (m, 1H), 0.66-0.61 (m, 2H), 0.39-0.35 (m, 2H); LC-MS: m/z 448.2 [M + H]+.
-

- The specific reaction scheme is as shown below:
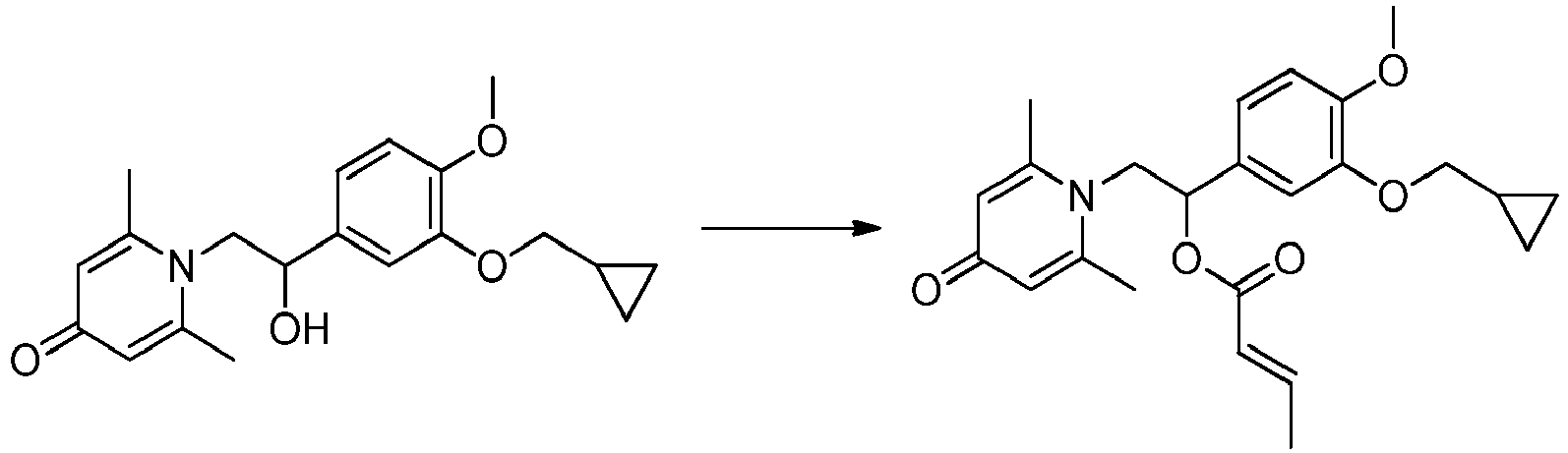
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (343 mg, 1.0 mmol) was dissolved in dichloromethane (8 mL), and then crotonyl chloride (157 mg, 1.5 mmol) and triethyl amine (202 mg, 2.0 mmol) were added in sequence, and stirred overnight at room temperature. After the reaction was completed, the reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) crotonate (127 mg, yield 31%, white solid). 1H NMR (400 MHz, CDCl3) δ 6.87 (d, J = 8.4 Hz, 1H), 6.82 (dd, J = 8.4, 2.0 Hz, 1H), 6.77 (d, J = 2.0 Hz, 1H), 6.33 (s, 2H), 5.98-5.89 (m, 1H), 5.86-5.76 (m, 1H), 4.37-4.28 (m, 1H), 4.13-4.04 (m, 1H), 3.88 (s, 3H), 3.84 (d, J = 6.8 Hz, 2H), 2.37 (s, 6H), 1.33-1.28 (m, 1H), 0.69-0.64 (m, 2H), 0.39-0.35 (m, 2H); LC-MS: m/z 412.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (343 mg, 1.0 mmol) was dissolved in dichloromethane (8 mL), and then 3-methylcrotonyl chloride (178 mg, 1.5 mmol) and triethyl amine (202 mg, 2.0 mmol) were added in sequence, and stirred overnight at room temperature. After the reaction was completed, the reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl)3-methyl crotonate (137 mg, yield 32%, white solid). 1H NMR (400 MHz, CDCl3) δ 6.87 (d, J = 8.4 Hz, 1H), 6.83 (dd, J = 8.4,2.0 Hz, 1H), 6.78 (d, J = 2.0 Hz, 1H), 6.28 (s, 2H), 5.90 (dd, J = 9.2, 4.8 Hz, 1H), 4.91 (s, 1H), 4.30 (dd, J = 15.2, 9.2 Hz, 1H), 4.05 (dd, J = 15.2, 4.8 Hz, 1H), 3.88 (s, 3H), 3.83 (d, J = 7.2 Hz, 2H), 3.02 (s, 2H), 2.37 (s, 6H), 1.69 (s, 3H), 1.35-1.27 (m, 1H), 0.69-0.64 (m, 2H), 0.39-0.35 (m, 2H); LC-MS: m/z 426.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (197 mg, 0.69 mmol) and 2-butynoic acid (72 mg, 0.86 mmol) were dissolved in dichloromethane (10 mL), and then 4-dimethylaminopyridine (105 mg, 0.86 mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (165 mg, 0.86 mmol) were added in sequence, and stirred overnight at 30°C. After the reaction was completed, the reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl)but-2-ynoate (37 mg, 9%, white solid). 1H NMR (400 MHz, CDCl3) δ 6.88-6.82 (m, 2H), 6.77 (d, J = 1.6 Hz, 1H), 6.22 (s, 2H), 5.90 (dd, J = 8.4, 5.2 Hz, 1H), 4.31 (dd, J = 15.2, 8.4 Hz, 1H), 4.04 (dd, J = 15.2, 5.2 Hz, 1H), 3.88 (s, 3H), 3.84 (d, J = 7.2 Hz, 2H), 2.33 (s, 6H), 2.01 (s, 3H), 1.32-1.28 (m, 1H), 0.69-0.64 (m, 2H), 0.39-0.35 (m, 2H). LC-MS: m/z 410.4 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (50 mg, 0.15 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then 1-bromo-2-butyne (40 mg, 0.30 mmol) and cesium carbonate (98 mg, 0.30 mmol) were added and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(but-2-yn-1-yloxy)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)ethyl)-2,26-dimethylpyridin-4-(1H)-one (19 mg, yield 32%, pale yellow oil). 1H NMR (400 MHz, CDCl3) δ 6.89 (d, J = 7.6 Hz, 1H), 6.84-6.81 (m, 2H), 6.58 (s, 2H), 4.70-4.66 (m, 1H), 4.30-4.24 (m, 1H), 4.14-4.08 (m, 1H), 4.05-4.00 (m, 1H), 3.91-3.86 (m, 5H), 3.82-3.77 (m, 1H), 2.45 (s, 6H), 1.37-1.31 (m, 1H), 1.27 (s, 3H), 0.72-0.66 (m, 2H), 0.42-0.38 (m, 2H); LC-MS: m/z 396.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- At room temperature, 3-hydroxy-4-methoxybenzaldehyde (1.52 g, 10 mmol) was dissolved in acetonitrile (40 mL), and then potassium carbonate (2.76 g, 20 mmol) and benzyl bromide (2.56 g, 15 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, a saturated aqueous sodium chloride solution (30 mL) was added, and extracted with dichloromethane (3 x 50 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by column chromatography to obtain 3-benzoxy-4-methoxybenzaldehyde (2.30 g, 95%, white solid).
- 3-benzoxy-4-methoxybenzaldehyde (2.30 g, 9.5 mmol) was dissolved in dichloromethane (30 mL), and then triethyl amine (1.92 g, 19 mmol) and trimethylsilyl cyanide (2.82 g, 28.5 mmol) were added in sequence in an ice bath and stirred for 16 hours under nitrogen atmosphere at room temperature. After the reaction was completed, direct rotary drying gave 2-(3-benzoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- 2-(3-benzoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile obtained in the above step was dissolved in anhydrous tetrahydrofuran (40 mL), and lithium aluminum hydride (1.08 g, 28.5 mmol) was added portion-wise in an ice bath and stirred overnight at room temperature. After the reaction was completed, water (1.1 mL), an aqueous sodium hydroxide solution (1.1 mL, 15%), and water (3.3 mL) were added in sequence, stirred for half an hour, dried over anhydrous sodium sulfate, filtered, and rotary dried to obtain a crude product of 2-amino-1-(3-benzoxy-4-methoxyphenyl)ethanol, which was directly used in the next reaction.
- 2-Amino-1-(3-benzoxy-4-methoxyphenyl)ethanol obtained in the above step was dissolved in ethanol (60 mL), and then 2,6-dimethyl-4H-pyran-4-one (1.24 g, 10 mmol), sodium hydroxide (800 mg, 20 mmol) and water (10 mL) were added in sequence, heated to 60°C, and stirred overnight under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried, and purified by column chromatography to obtain 1-(2-(3-benzoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (1.62 g, 45%, white solid). LC-MS m/z 380.2 [M + H]+.
- 1-(2-(3-benzoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (1.62 g, 4.26 mmol) was dissolved in dichloromethane (30 mL), and then the Dess-Martin Periodinane (2.16 g, 5.11 mmol) was added and stirred at room temperature for 2 hours. The reaction solution was filtered, and washed with saturated brine. The organic phase was dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by column chromatography to obtain 1-(2-(3-benzoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (1.37 g, yield 85%, white solid). LC-MS m/z 378.2 [M + H]+.
- The compound 1-(2-(3-benzoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (1.37 g, 3.61 mmol) was dissolved in methanol (50 mL), then Pd/C (137 mg) and triethyl amine (1 mL) were added, and hydrogen was introduced. The system was stirred overnight at room temperature. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (660 mg, yield 64%, gray solid). 1H NMR (400 MHz, CD3OD) δ 7.73 (dd, J = 8.4, 2.0 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.04 (s, 2H), 5.97 (s, 2H), 3.98 (s, 3H), 2.51 (s, 6H); LC-MS: m/z 288.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (28 mg, 0.1 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then iodomethane (19 mg, 0.13 mmol) and potassium carbonate (21 mg, 0.15 mmol) were added and stirred for 1 hr at room temperature under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3,4-dimethoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (10 mg, yield 30%, pale yellow oil). 1H NMR (400 MHz, CD3OD) δ 7.85 (dd, J = 8.4, 2.0 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 7.05 (s, 2H), 6.02 (s, 2H), 4.07 (s, 3H), 3.96 (s, 3H), 2.52 (s, 6H); LC-MS: m/z 302.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (28 mg, 0.1 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then iodoethane (21 mg, 0.13 mmol) and potassium carbonate (21 mg, 0.15 mmol) were added and stirred for 1 hr at room temperature under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-ethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (15 mg, yield 48%, pale yellow oil). 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.02 (s, 2H), 6.91 (d, J = 8.0 Hz, 1H), 6.15 (s, 2H), 4.32 (q, J = 6.0 Hz, 2H), 3.92 (s, 3H), 2.62 (s, 6H), 1.51 (t, J = 6.0 Hz, 1H); LC-MS m/z 316.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (20 mg, 0.07 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromopropane (10 mg, 0.08 mmol) and potassium carbonate (15 mg, 0.11 mmol) were added and stirred for 2 hours at room temperature under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-propoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (8 mg, yield 34%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.22 (d, J = 2.0 Hz, 1H), 7.14 (dd, J = 8.4, 2.0 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 6.38 (s, 2H), 6.02 (s, 2H), 4.03 (t, J = 6.4 Hz, 2H), 3.89 (s, 3H), 2.37 (s, 6H), 1.90-1.80 (m, 2H), 1.08 (t, J = 7.2 Hz, 3H); LC-MS: m/z 330.2 [M+H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (35 mg, 0.12 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromo-isopropane (17 mg, 0.14 mmol) and potassium carbonate (25 mg, 0.18 mmol) were added, and stirred for 2 hours at room temperature under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-isopropoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (10 mg, yield 63%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.02 (dd, J = 8.4, 2.0 Hz, 1H), 6.98 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 14.4 Hz, 1H), 6.60 (d, J = 14.4 Hz, 1H), 6.30 (s, 2H), 4.69-4.60 (m, 1H), 3.90 (s, 3H), 2.57 (s, 6H), 1.35 (d, J = 6.0 Hz, 6H); LC-MS: m/z 330.2 [M+H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromo-isopropane (95 mg, 0.7 mmol) and potassium carbonate (37 mg, 0.7mmol) were added, and stirred for 2 hours at room temperature under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-n-butoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (26 mg, yield 42%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.85 (dd, J = 8.4, 2.0 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 7.05 (s, 2H), 6.02 (s, 2H), 4.07 (t, J = 6.4 Hz, 2H), 3.96 (s, 3H), 2.52 (s, 6H), 1.84-1.77 (m, 2H), 1.58-1.48 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); LC-MS: m/z 344.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromo-isobutane (27 mg, 0.2 mmol) and potassium carbonate (37 mg, 0.27 mmol) were added, and stirred for 2 hours at room temperature under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-iso-butoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (25 mg, yield 40%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.02 (dd, J = 8.4, 2.0 Hz, 1H), 6.98 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 14.4 Hz, 1H), 6.60 (d, J = 14.4 Hz, 1H), 6.30 (s, 2H), 3.92 (s, 3H), 3.81 (d, J = 6.8 Hz, 2H), 2.27 (s, 6H), 2.25-2.17 (m, 1H), 1.08 (d, J = 6.8 Hz, 6H); LC-MS: m/z 344.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then 1-bromopentane (105 mg, 0.7 mmol) and potassium carbonate (97 mg, 0.7mmol) were added, and stirred for 5 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-n-pentyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 32%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.85 (dd, J = 8.4, 2.0 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 7.07 (s, 2H), 6.03 (s, 2H), 4.06 (t, J = 6.4 Hz, 2H), 3.96 (s, 3H), 2.53 (s, 6H), 1.86-1.79 (m, 2H), 1.52-1.36 (m, 4H), 0.95 (t, J = 7.2 Hz, 3H); LC-MS: m/z 358.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then 1-bromon-hexane (115 mg, 0.7 mmol) and potassium carbonate (97 mg, 0.7 mmol) were added and stirred for 5 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-n-hexyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (43 mg, yield 66%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.85 (dd, J = 8.4, 2.0 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 7.08 (s, 2H), 6.04 (s, 2H), 4.07 (t, J = 6.4 Hz, 2H), 3.96 (s, 3H), 2.53 (s, 6H), 1.86-1.78 (m, 2H), 1.54-1.46 (m, 2H), 1.40-1.34 (m, 4H), 0.93 (t, J = 7.2 Hz, 3H); LC-MS: m/z 372.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 3-hydroxy-4-methoxybenzaldehyde (1 g, 6.6 mmol), bromo-cyclopropane (2.4 mg, 19.8 mmol), cesium carbonate (6.5 g, 19.8 mmol), and potassium iodide (168 mg,0.15 mmol) were dissolved in N, N-dimethylformamide (10 mL), stirred for 1 hr in a sealed tube at 180°Cunder nitrogen atmosphere, then heated to 220°C, and stirred for another 1 hr. After the reaction was completed, saturated saline (20 mL) was added, and extracted with dichloromethane (3 x 20 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by column chromatography gave 3-cyclopropyloxy-4-methoxybenzaldehyde (170 mg, yield 13%, pale yellow oil). LC-MS: m/z 193.4 [M + H]+.
- 3-cyclopropyloxy-4-methoxybenzaldehyde (170 mg, 0.88 mmol) was dissolved in dichloromethane (4 mL); and under nitrogen atmosphere, triethyl amine (356 mg, 3.52 mmol) and trimethylsilyl cyanide (349 mg, 3.52 mmol) were added, and stirred for 6 hours at room temperature. After the reaction was completed, the reaction solution was concentrated and rotary dried to obtain 2-(3-cyclopropyloxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- 2-(3-cyclopropyloxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile obtained in the above step was dissolved in anhydrous tetrahydrofuran (20 mL), and then lithium aluminum hydride (100 mg, 2.6 mmol) was added portion-wise in an ice bath and stirred overnight at room temperature. After the reaction was completed, water (0.1 mL), an aqueous sodium hydroxide solution (0.1 mL, 15%), and water (0.3 mL) were added in sequence, stirred for half an hour, dried over anhydrous sodium sulfate, and filtered. The filtrate was rotary dried to obtain a crude product of 2-amino-1-(3-cyclopropyloxy-4-methoxyphenyl)ethanol (300 mg).
- 2-Amino-1-(3-(cyclopropyloxy)-4-methoxyphenyl)ethanol obtained in the above step was dissolved in ethanol (5 mL), and then 2,6-dimethyl-4H-pyran-4-one (124 mg, 1 mmol) and aqueous sodium hydroxide solution (2 M, 2 mL) were added, and stirred overnight at 60°C. The reaction was complete as indicated by LCMS. The reaction solution was rotary dried, and purified by column chromatography to obtain 1-(2-(3-cyclopropyloxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (100 mg, 34%, white solid). LC-MS: m/z 330.1 [M + H]+.
- 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (30 mg, 0.1 mmol) was dissolved in dichloromethane (5 mL), and stirred for 2 hours at normal temperature. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain 1-(2-(3-cyclopropyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (15 mg, yield 45%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.97 (dd, J = 8.4, 2.0 Hz, 1H), 7.96 (d, J = 2.0 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H), 6.44 (s, 2H), 5.75 (s, 2H), 4.02 (s, 3H), 2.32 (s, 6H), 2.23-2.19 (m, 1H), 0.94-0.90 (m, 4H); LC-MS: m/z 328.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromomethylcyclopropane (27 mg, 0.2 mmol) and potassium carbonate (37 mg, 0.27mmol) were added, and stirred for 2 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (36 mg, yield 62%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.79 (dd, J = 8.4, 2.0 Hz, 1H), 7.50 (d, J = 2.0 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 5.99 (s, 2H), 5.62 (s, 2H), 3.90 (s, 3H), 3.53 (d, J = 6.8 Hz, 2H), 2.11 (s, 6H), 1.26-1.21 (m, 1H), 0.62-0.56 (m, 2H), 0.36-0.31 (m, 2H); LC-MS: m/z 342.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then cyclobutyl bromide (69 mg, 0.51 mmol) and potassium carbonate (71 mg, 0.51 mmol) were added and stirred for 5 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-cyclobutyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (16 mg, yield 26%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.85 (d, J = 8.4 Hz, 1H), 7.45 (s, 1H), 7.14 (d, J = 8.4 Hz, 1H), 7.04 (s, 2H), 5.99 (s, 2H), 4.79-4.73 (m, 1H), 3.96 (s, 3H), 2.51 (s, 6H), 2.51-2.45 (m, 1H), 2.23-2.13 (m, 1H), 1.91-1.83 (m, 1H), 1.79-1.70 (m, 1H); LC-MS: m/z 342.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromomethylcyclobutane (76 mg, 0.51 mmol) and potassium carbonate (71 mg, 0.51mmol) were added, and stirred for 4 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-cyclobutylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (16 mg, yield 24%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.84 (d, J = 8.4 Hz, 1H), 7.61 (s, 1H), 7.14 (d, J = 8.4 Hz, 1H), 6.91 (s, 2H), 5.95 (s, 2H), 4.04 (d, J = 6.8 Hz, 2H), 3.95 (s, 3H), 2.84-2.78 (m, 1H), 2.47 (s, 6H), 2.20-2.10 (m, 2H), 2.02-1.86 (m, 4H); LC-MS: m/z 356.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromocyclopentane (76 mg, 0.51 mmol) and potassium carbonate (71 mg, 0.51mmol) were added, and stirred for 4 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-cyclopentyl-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 33%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.84 (dd, J = 8.4, 2.0 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 6.39 (s, 2H), 5.71 (s, 2H), 4.95-4.91 (m, 1H), 3.96 (s, 3H), 2.31 (s, 6H), 2.01-1.80 (m, 6H), 1.71-1.61 (m, 2H); LC-MS m/z 356.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromocycloohexane (83 mg, 0.51 mmol) and potassium carbonate (71 mg, 0.51mmol) were added, and stirred for 4 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-cyclohexyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (8 mg, yield 13%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.86 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.16 (d, J = 8.4 Hz, 1H), 7.04 (s, 2H), 6.00 (s, 2H), 4.39-4.32 (m, 1H), 3.95 (s, 3H), 2.52 (s, 6H), 2.02-1.93 (m, 2H), 1.86-1.77 (m, 2H), 1.62-1.52 (m, 2H), 1.45-1.28 (m, 4H); LC-MS m/z 370.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (30 mg, 0.11 mmol) was dissolved in acetonitrile (5 mL), and then cyclopent-3-en-1-yl methanesulfonate (68 mg, 0.42 mmol) and potassium carbonate (58 mg, 0.42 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain 1-(2-(3-(cyclopent-3-en-1-yloxy)-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (12 mg, yield 32%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.4 Hz, 1H), 7.55 (s, 1H), 7.18 (s, 2H), 7.01 (d, J = 8.4 Hz, 1H), 6.04-5.91 (m, 2H), 5.77 (s, 2H), 5.17-5.11 (m, 1H), 3.96 (s, 3H), 2.95-2.89 (m, 2H), 2.63-2.60 (m, 2H), 2.53 (s, 6H); LC-MS m/z 354.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.18 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then 3-bromopropene (26 mg, 0.21 mmol) and potassium carbonate (37 mg, 0.27 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-allyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (25 mg, yield 42%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.83 (d, J = 8.4 Hz, 1H), 7.61 (s, 1H), 7.13 (d, J = 8.4 Hz, 1H), 6.34 (s, 2H), 6.14-6.03(m, 1H), 5.68 (s, 2H), 5.43 (d, J = 17.2 Hz, 1H), 5.27 (d, J = 10.4 Hz, 1H), 4.64 (d, J = 4.8 Hz, 2H), 3.95 (s, 3H), 2.26 (s, 6H). LC-MS m/z 328.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.18 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then 1-bromo-3-methyl-2-butene (31 mg, 0.21 mmol) and potassium carbonate (37 mg, 0.27 mmol) were added, and stirred for 1 hr 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-((3-methylbut-2-en-1-yl)oxy)-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (25 mg, yield 40%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.4 Hz, 1H), 7.55 (d, J = 2.0 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 6.32 (s, 2H), 5.51 (d, J = 6.8 Hz, 1H), 5.32 (s, 2H), 4.65 (d, J = 6.8 Hz, 2H), 3.98 (s, 3H), 2.21 (s, 6H), 1.79 (s, 3H), 1.77(s, 3H); LC-MS m/z 356.2 [M + H]+.
-

- The specific reaction scheme is as shown below:
- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.18 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then propargyl bromide (25 mg, 0.21 mmol) and potassium carbonate (37 mg, 0.27 mmol) were added, and stirred for 4 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-propargyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (21 mg, yield 35%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.0 Hz, 1H), 7.70 (s, 1H), 7.04 (d, J = 8.0 Hz, 1H), 7.03 (s, 2H), 5.78 (s, 2H), 4.85 (d, J = 2.4 Hz, 2H), 4.00 (s, 3H), 2.56 (t, J = 2.4 Hz, 3H), 2.46 (s, 6H); LC-MS: m/z 326.3 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.18 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then 1-bromo-2-butyne (83 mg, 0.70 mmol) and potassium carbonate (97 mg, 0.70 mmol) were added, and stirred for 2 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-(but-2-yn-1-yloxy)-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (14 mg, yield 23%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.90 (dd, J = 8.4, 2.0 Hz, 1H), 7.72 (d, J = 2.0 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.09 (s, 2H), 6.03 (s, 2H), 4.78 (q, J = 2.4 Hz, 2H), 3.96 (s, 3H), 2.54 (s, 6H), 1.82 (t, J = 2.4 Hz, 3H); LC-MS: m/z 340.0 [M + H]+.
-

-

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (158 mg, 0.55 mmol), oxacyclobutan-3-ol (81.4 mg, 1.1 mmol) and triphenylphosphine (288 mg, 1.1 mmol) were dissolved in anhydrous tetrahydrofuran (10 mL), the reaction solution was cooled in an ice bath, diisopropyl azodicarboxylate (222 mg, 1.1 mmol) was added dropwise over 5 min under nitrogen atmosphere, and then reacted for 24 hours at room temperature. After the reaction was completed, saturated saline (20 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-(oxacyclobutan-3-yl-oxy)-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (45 mg, yield 24%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.84 (dd, J = 8.4, 2.0 Hz, 1H), 7.31 (d, J = 2.0 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.13 (s, 2H), 6.06 (s, 2H), 5.38-5.32 (m, 1H), 5.06-5.02 (m, 2H), 4.78-4.75 (m, 2H), 4.00 (s, 3H), 2.56 (s, 6H); LC-MS: m/z 344.4 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in dichloromethane (5 mL), and then 2,3-dihydrofuran (68 mg, 0.42 mmol) and pyridinium toluene-4-sulphonate (4.6 mg, 0.018 mmol) were added, and stirred overnight at room temperature. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain 1-(2-(3-(tetrahydrofuran-2-yl)oxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (28 mg, yield 44%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.87 (dd, J = 8.4, 2.0 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 6.40 (s, 2H), 5.87 (d, J = 4.4 Hz, 1H), 5.70 (s, 2H), 4.07-4.02 (m, 1H), 3.97-3.92 (m, 1H), 3.94 (s, 3H), 2.31-2.11 (m, 4H), 2.29 (s, 6H); LC-MS m/z 358.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- Under nitrogen atmosphere, 3-hydroxy-4-methoxybenzaldehyde (2 g, 13 mmol) was dissolved in N,N-dimethylformamide (20 mL), and then chloromethyl methyl sulfide (1.5 g, 15.6 mmol) and cesium carbonate (6 g, 19.5 mmol) were added and stirred overnight at room temperature under nitrogen atmosphere. After the reaction was completed, saturated saline (40 mL) was added, and extracted with dichloromethane (3 x 30 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by column chromatography gave 3-methylthiomethoxy-4-methoxybenzaldehyde (1.7 g, yield 61%, yellow oil). LC-MS: m/z 213.1 [M + H]+.
- 3-methylthiomethoxy-4-methoxybenzaldehyde (1.7 g, 8 mmol) was dissolved in dichloromethane (20 mL); and under nitrogen atmosphere, triethyl amine (1.6 g, 16 mmol) and trimethylsilyl cyanide (1.6 g, 16 mmol) were added, and stirred for 6 hours at room temperature. The reaction solution was concentrated and rotary dried to obtain 2-(3-methylthiomethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- 2-(3-Methylthiomethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile obtained in the above step was dissolved in anhydrous tetrahydrofuran (50 mL), and then lithium aluminum hydride (608 mg, 16 mmol) was added portion-wise in an ice bath and stirred overnight at room temperature. After the reaction was completed, water (0.6 mL), an aqueous sodium hydroxide solution (0.6 mL, 15%), and water (1.8 mL) were added in sequence, stirred for half an hour, dried over anhydrous sodium sulfate, and filtered. The filtrate was rotary dried to obtain a crude product of 2-amino-1-(3-methylthiomethoxy-4-methoxyphenyl)ethanol.
- 2-Amino-1-(3-(methylthiomethoxy)-4-methoxyphenyl)ethanol obtained in the above step was dissolved in ethanol (10 mL), and then 2,6-dimethyl-4-pyranone (1.24 g, 10 mmol) and an aqueous sodium hydroxide solution (2 M, 10 mL) were added, and stirred overnight at 60°C. The reaction was complete as indicated by LCMS. The reaction solution was rotary dried, and purified by column chromatography to obtain 1-(2-(3-methylthiomethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (1 g, 35%). LC-MS: m/z 350.1 [M + H]+.
- 1-(2-(3-methylthiomethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.14 mmol) was dissolved in dimethylsulfoxide (5 mL), and then sulfur trioxide pyridine complex (111 mg, 0.7 mmol) in DMSO (2.5ml) was slowly added and stirred overnight at normal temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-methylthiomethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (25 mg, yield 51%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.67 (dd, J = 8.4, 2.0 Hz, 1H), 7.55 (d, J = 2.0 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.28 (s, 2H), 5.31 (s, 2H), 5.23 (s, 2H), 3.93 (s, 3H), 3.68 (s, 3H), 2.12 (s, 6H); LC-MS: m/z 348.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- 1-(2-(3-methylthiomethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (100 mg, 0.29 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (56 mg, 0.28 mmol) was added and stirred at room temperature for 2 hours. Then, a saturated aqueous sodium sulfite solution (5 mL) was added and stirred for 10 min. The reaction solution was extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by column chromatography gave 1-(2-(3-methylsulfoxidemethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (38 mg, yield 36%, white solid). LC-MS: m/z 366.2 [M + H]+.
- 1-(2-(3-methylsulfoxidemethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (38 mg, 0.1 mmol) was dissolved in dichloromethane (10 mL), and then the Dess-Martin Periodinane (85 mg, 0.2 mmol) was added and stirred at room temperature for 2 hours. The reaction solution was filtered, and washed with saturated brine. The organic phase was dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by reverse-phase HPLC to obtain 1-(2-(3-methylsulfoxidemethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (10 mg, yield 37%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 2.0 Hz, 1H), 7.74 (dd, J = 8.4, 2.0 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.26 (s, 2H), 5.25 (s, 2H), 5.23 (s, 2H), 3.93 (s, 3H), 3.68 (s, 3H), 2.12 (s, 6H); LC-MS: m/z 364.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- 1-(2-(3-methylthiomethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (100 mg, 0.29 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (176 mg, 0.87 mmol) was added and stirred overnight at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added and stirred for 10 min. The reaction solution was extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by column chromatography gave 1-(2-(3-methylsulfonemethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, yield 55%, white solid). LC-MS: m/z 382.2 [M + H]+.
- 1-(2-(3-methylsulfonemethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.13 mmol) was dissolved in dichloromethane (10 mL), and then the Dess-Martin Periodinane (110 mg, 0.26 mmol) was added and stirred at room temperature for 2 hours. The reaction solution was filtered, and washed with saturated brine. The organic phase was dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by reverse-phase HPLC to obtain 1-(2-(3-methylsulfoxidemethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (30 mg, yield 61%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.84 (dd, J = 8.4, 2.0 Hz, 1H), 7.70 (d, J = 2.0 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 6.26 (s, 2H), 5.35 (s, 2H), 5.23 (s, 2H), 3.98 (s, 3H), 3.78 (s, 3H), 2.25 (s, 6H); LC-MS: m/z 380.1 [M + H]+.
-

- The specific reaction scheme is as shown below:
- The compound 1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (287 mg, 1 mmol) was dissolved in N, N-dimethylformamide (5 mL), and then chloroethyl methyl sulfide (166 mg, 1.5 mmol) and potassium carbonate (276 mg, 2 mmol) were added and stirred for 2 hours at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave 1-(2-(3-methylthioethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one200 mg, yield 55%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.66 (dd, J = 8.4, 2.0 Hz, 1H), 7.56 (d, J = 2.0 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.28 (s, 2H), 5.31 (s, 2H), 4.27 (t, J = 6.8 Hz, 2H), 3.98 (s, 3H), 2.95 (t, J = 6.8 Hz, 2H), 2.24 (s, 3H), 2.19 (s, 6H); LC-MS: m/z 362.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-methylthioethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.15 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (28 mg, 0.14 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfoxideethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 40%, colorless oil). 1H NMR (400 MHz, CDCl3) δ 7.71 (dd, J = 8.4, 2.0 Hz, 1H), 7.66 (d, J = 2.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.26 (s, 2H), 5.32 (s, 2H), 4.57-4.54 (m, 2H), 3.97 (s, 3H), 3.35-3.31 (m, 1H), 3.15-3.09 (m, 1H), 2.75 (s, 3H), 2.18 (s, 6H); LC-MS: m/z 378.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-methylthioethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.15 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (71 mg, 0.35 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfoneethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 36%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.74 (dd, J = 8.4, 2.0 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.26 (s, 2H), 5.30 (s, 2H), 4.53 (t, J = 4.2 Hz, 2H), 3.95 (s, 3H), 3.51 (t, J = 4.2 Hz, 2H), 3.18 (s, 3H), 2.18 (s, 6H); LC-MS: m/z 394.2 [M + H]+.
-

-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (341 mg, 1 mmol) and hydroxylamine hydrochloride (139 mg, 2 mmol) were dissolved in pyridine (10 mL), and refluxed for 24 hours under nitrogen atmosphere. After the reaction was completed, pyridine was rotary dried, and and the reaction solution was purified by reverse-phase HPLC to obtain (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (96 mg, yield 27%, white solid) and (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one(43 mg, yield 12%, white solid).
- (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one. 1H NMR (400 MHz, CDCl3) δ 7.45 (d, J = 2.0 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 6.93 (dd, J = 8.4, 2.0 Hz, 1H), 6.48 (s, 2H), 5.00 (s, 2H), 3.92 (s, 3H), 3.89 (d, J = 6.8 Hz, 2H), 2.41 (s, 6H), 1.42-1.32 (m, 1H), 0.68-0.62 (m, 2H), 0.40-0.35 (m, 2H); LC-MS: m/z 357.4 [M + H]+.
- (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 2.0 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 6.93 (dd, J = 8.4, 2.0 Hz, 1H), 6.49 (s, 2H), 5.00 (s, 2H), 3.92 (s, 3H), 3.89 (d, J = 6.8 Hz, 2H), 2.41 (s, 6H), 1.42-1.32 (m, 1H), 0.68-0.62 (m, 2H), 0.40-0.35 (m, 2H); LC-MS: m/z 357.4 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (36 mg, 0.1 mmol) was dissolved in N,N-dimethylformamide (2 mL), and then 60% sodium hydride (12 mg, 0.3 mmol) was added at 0°C, and stirred for half an hour. Then, iodomethane (28.4 mg, 0.2 mmol) was added, and stirred at room temperature for 4 hours. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 55%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 2.0 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.02 (dd, J = 8.4, 2.0 Hz, 1H), 6.52 (s, 2H), 5.02 (s, 2H), 3.93 (s, 3H), 3.92 (s, 3H), 3.89 (d, J = 6.8 Hz, 2H), 2.45 (s, 6H), 1.32-1.23 (m, 1H), 0.70-0.65 (m, 2H), 0.50-0.38 (m, 2H); LC-MS: m/z 371.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (36 mg, 0.1 mmol) was dissolved in N,N-dimethylformamide (2 mL), and then 60% sodium hydride (12 mg, 0.3 mmol) was added at 0°C, and stirred for half an hour. Then, iodomethane (28.4 mg, 0.2 mmol) was added, and stirred at room temperature for 4 hours. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (18 mg, yield 49%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 2.0 Hz, 1H), 7.11 (d, J = 8.4 Hz, 1H), 7.00 (dd, J = 8.4, 2.0 Hz, 1H), 6.46 (s, 2H), 5.00 (s, 2H), 3.95 (s, 3H), 3.93 (s, 3H), 3.88 (d, J = 6.8 Hz, 2H), 2.49 (s, 6H), 1.42-1.32 (m, 1H), 0.68-0.62 (m, 2H), 0.40-0.35 (m, 2H); LC-MS: m/z 371.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (300 mg, 0.84 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then cesium carbonate (548 mg, 1.68 mmol) and chloromethylmethyl sulfide (122 mg, 1.26 mmol) were added, and stirred overnight at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methylthiomethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (200 mg, yield 57%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.23 (d, J = 8.4 Hz, 1H), 7.18 (s, 1H), 7.08 (d, J = 8.4 Hz, 1H), 6.96 (s, 2H), 6.35 (s, 2H), 5.18 (s, 2H), 3.91 (s, 2H), 3.85 (d, J = 6.8 Hz, 2H), 2.47 (s, 6H), 2.40 (s, 3H), 1.36-1.28 (m, 1H), 0.69-0.64 (m, 2H), 0.35-0.32 (m, 2H); LC-MS: m/z 417.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (36 mg, 0.1 mmol) was dissolved in N,N-dimethylformamide (2 mL), and then cesium carbonate (65 mg, 0.2 mmol) and chloromethylmethyl sulfide (15 mg, 0.15 mmol) were added, and stirred overnight at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methylthiomethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 48%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.36 (d, J = 8.4 Hz, 1H), 7.25 (s, 1H), 6.88 (d, J = 8.4 Hz, 1H), 6.86 (s, 2H), 6.38 (s, 2H), 5.10 (s, 2H), 3.96 (s, 2H), 3.90 (d, J = 6.8 Hz, 2H), 2.47 (s, 6H), 2.40 (s, 3H), 1.36-1.28 (m, 1H), 0.69-0.64 (m, 2H), 0.35-0.32 (m, 2H); LC-MS: m/z 417.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methylthiomethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (42 mg, 0.1 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (18 mg, 0.09 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methylsulfoxidemethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 46%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.28 (d, J = 8.4 Hz, 1H), 7.21 (s, 1H), 6.96 (d, J = 8.4 Hz, 1H), 6.90 (s, 2H), 6.36 (s, 2H), 5.15 (s, 2H), 3.94 (s, 2H), 3.88 (d, J = 6.8 Hz, 2H), 2.46 (s, 6H), 2.42 (s, 3H), 1.38-1.30 (m, 1H), 0.69-0.65 (m, 2H), 0.38-0.33 (m, 2H); LC-MS: m/z 433.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methylthiomethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (42 mg, 0.1 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (61 mg, 0.3 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methylsulfonemethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one (26 mg, yield 58%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.23 (d, J = 8.4 Hz, 1H), 7.18 (s, 1H), 7.08 (d, J = 8.4 Hz, 1H), 6.96 (s, 2H), 6.35 (s, 2H), 5.18 (s, 2H), 3.91 (s, 2H), 3.85 (d, J = 6.8 Hz, 2H), 2.47 (s, 6H), 2.40 (s, 3H), 1.36-1.28 (m, 1H), 0.69-0.64 (m, 2H), 0.35-0.32 (m, 2H); LC-MS: m/z 449.2 [M + H]+.
-

- The specific reaction scheme is as shown below:
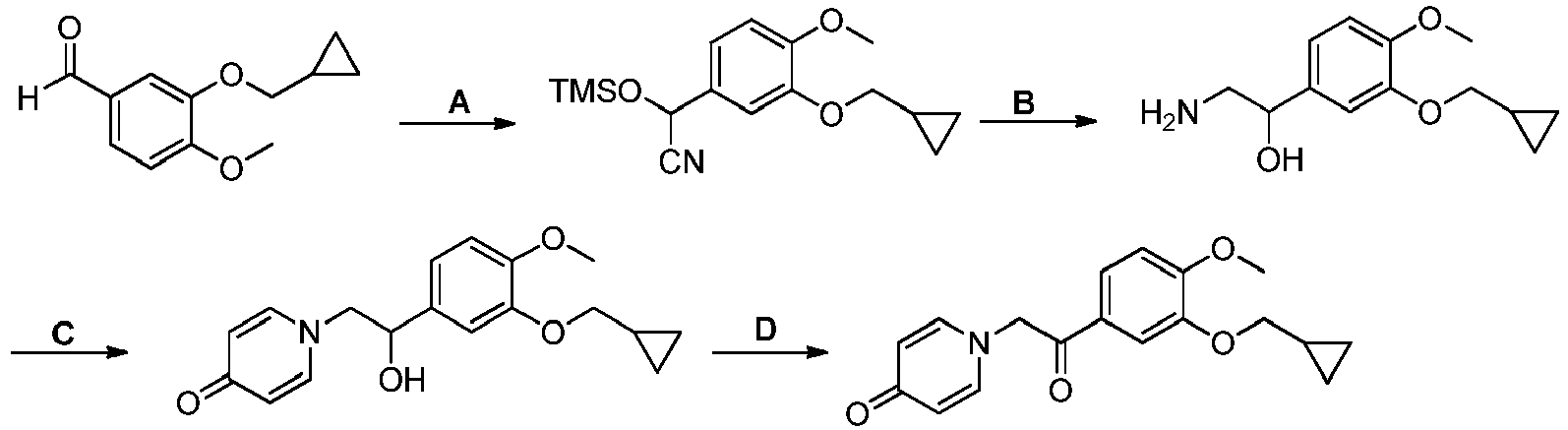
- 3-cyclopropylmethoxy-4-methoxybenzaldehyde (3.09 g, 15 mmol) was dissolved in dichloromethane (30 mL); and under nitrogen atmosphere, triethyl amine (4.16 mL, 30 mmol) and trimethylsilyl cyanide (3.75 g, 30 mmol) were added, and stirred for 6 hours at room temperature. The reaction solution was concentrated and rotary dried to obtain 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- The compound 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-trimethylsiloxyacetonitrile (4.57 g, 15 mmol) was dissolved in anhydrous tetrahydrofuran (50 mL), and then lithium aluminum hydride (1.14 g, 30 mmol) was added portion-wise in an ice bath, and stirred overnight at room temperature. After the reaction was completed, water (1.2 mL), an aqueous sodium hydroxide solution (1.2 mL, 15%), and water (3.6 mL) were added in sequence, stirred for half an hour, dried over anhydrous sodium sulfate, filtered, and rotary dried to obtain a crude product of 2-amino-1-(3-cyclopropylmethoxy-4-methoxyphenyl)ethanol.
- The compound 2-amino-1-(3-(cyclopropylmethoxy)-4-methoxyphenyl)ethanol was dissolved in ethanol (20 mL), and then pyrone (1 g, 10.41 mmol) and aqueous sodium hydroxide solution (2M, 20 mL) were added, and stirred overnight at 60°C. The reaction was complete as indicated by LCMS. The reaction solution was rotary dried, and purified by column chromatography to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (2 g, 63%). LC-MS: m/z 316.2 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (280 mg, 0.88 mmol) was dissolved in dichloromethane (10 mL); and under nitrogen atmosphere, Dess-Martin Periodinane (746 mg, 1.76 mmol) was added, and stirred for 3 hours at room temperature. After the reaction was completed, the reaction solution was concentrated, rotary dried, and purified by column chromatography to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)pyridin-4(1H)-one (50 mg, yield 18%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.83 (d, J = 7.6 Hz, 2H), 7.68 (dd, J = 8.4, 1.2 Hz, 1H), 7.45 (d, J = 1.2 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 6.47 (d, J = 7.6 Hz, 2H), 5.72 (s, 2H), 3.89 (s, 3H), 3.87 (d, J = 6.8 Hz, 2H), 1.26-1.21 (m, 1H), 0.62-0.56 (m, 2H), 0.36-0.31 (m, 2H); LC-MS: m/z 314.1 [M + H]+.
-

- The specific reaction scheme is as shown below.

- 3-hydroxy-4-methoxyacetophenone (1.66 g, 10 mmol) was dissolved in acetonitrile (30 mL); and under nitrogen atmosphere, potassium carbonate (2.76 g, 20 mmol) and bromomethylcyclopropane (2.0 g, 15mmol) were added, and stirred for 6 hours at 80°C. The reaction solution was filtered. The filtrate was concentrated, rotary dried, and purified by column chromatography to obtain 1-(3-cyclopropylmethoxy-4-methoxyphenyl)ethanone (2.1 g, yield 95%, white solid).
- The compound 1-(3-cyclopropylmethoxy-4-methoxyphenyl)ethanone (2.2 g, 10 mmol) was dissolved in methanol (30 mL), and then N-bromosuccinimide (2.14g, 12 mmol) and p-toluenesulfonic acid (1.7 g, 10 mmol) were added to the solution and reacted at 65°C for 12 hours. After the reaction was completed, the reaction solution was concentrated, rotary dried, and purified by column chromatography to obtain 2-bromo-1-(3-cyclopropylmethoxy-4-methoxyphenyl)ethanone (800 mg, yield 27%, white solid).
- The compound 2-bromo-1-(3-cyclopropylmethoxy-4-methoxyphenyl)ethanone (185 mg, 1.5 mmol) and 4-methoxy-2methylpyridine (500 mg, 1.67 mmol) were dissolved in acetonitrile (10 mL), and stirred at 80°C for 48 hours. After the reaction was completed, the reaction solution was concentrated, rotary dried, and purified by column chromatography to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2-methylpyridin-4(1H)-one (300 mg, yield 61%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.76 (dd, J = 8.4, 2.0 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.55 (d, J = 1.2 Hz, 1H), 7.12 (d, J = 8.8 Hz, 2H), 6.42-6.38 (m, 2H), 5.72 (s, 2H), 3.95 (s, 3H), 3.91 (d, J = 6.8 Hz, 2H), 2.24 (s, 3H), 1.34-1.24 (m, 1H), 0.66-0.60 (m, 2H), 0.39-0.34 (m, 2H); LC-MS: m/z 328.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- 2-bromo-1-(3-cyclopropylmethoxy-4-methoxyphenyl)ethanone (594 mg, 2 mmol) and 4-methoxy-2chloropyridine (288 mg, 2 mmol) were dissolved in acetonitrile (10 mL), and stirred at 80°C for 48 hours. After the reaction was completed, the reaction solution was concentrated, rotary dried, and purified by column chromatography to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2-chloropyridin-4(1H)-one (117 mg, yield 17%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.73 (d, J = 7.6 Hz, 2H), 7.47 (d, J = 2.0 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 6.38 (d, J = 2.4 Hz, 1H), 6.18 (dd, J = 8.0, 2.4 Hz, 1H), 5.77 (s, 2H), 3.89 (s, 3H), 3.87 (d, J = 6.8 Hz, 2H), 1.27-1.22 (m, 1H), 0.62-0.56 (m, 2H), 0.36-0.32 (m, 2H); LC-MS: m/z 348.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- 3-hydroxy-4-methoxycinnamic acid (194 mg, 1 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromomethylcyclopropane (338 mg, 2.5 mmol) and potassium carbonate (414 mg, 3 mmol) were added. The reaction solution was stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with ethyl acetate (3 x 10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by column chromatography to obtain cyclopropylmethyl 3-cyclopropylmethoxy-4-methoxycinnamate (200 mg, yield 61%, yellow liquid). LC-MS: m/z 303.3 [M + H]+.
- Under nitrogen atmosphere, dimethylsulfoxide (4 mL), 60% sodium hydride (28.8 mg, 0.36 mmol) and trimethylsulfoxonium iodide (160 mg, 0.36 mmol) were stirred for 20 min at room temperature. A solution (2 mL) of cyclopropylmethyl 3-cyclopropylmethyl-4-methoxycinnamate (200 mg, 0.66 mmol) in tetrahydrofuran was slowly added. After the dropwise addition, the reaction solution was stirred at room temperature for 1 hr, then heated to 50°C and continuously stirred for 1 hr. After the reaction was completed, the reaction solution was poured into iced water, and extracted with ethyl acetate (3 x 10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by column chromatography to obtain cyclopropylmethyl 2-(3-cyclopropylmethoxy-4-methoxyphenyl)cyclopropylcarboxylate (80 mg, yield 56%, white solid). LC-MS: m/z 317.3 [M + H]+.
- Cyclopropylmethyl 2-(3-cyclopropylmethoxy-4-methoxyphenyl)cyclopropylcarboxylate (80 mg, 0.25 mmol) was dissolved in methanol (2 mL), and then a sodium hydroxide solution (2N, 1 mL) was added, and stirred for 2 hr under reflux. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification gave 2-(3-cyclopropylmethoxy-4-methoxyphenyl)cyclopropylcarboxylic acid (50 mg, yield 76%, white solid).
-

- 2-(3-cyclopropylmethoxy-4-methoxyphenyl)cyclopropylcarboxylic acid (50 mg, 0.19 mmol) was dissolved in 1,4-dioxane (2 mL), and then triethyl amine (26 mg, 0.26 mmol) and diphenylphosphoryl azide(64 mg, 0.26 mmol) were added. This reaction solution was stirred for 16 hours at room temperature, then heated to 80°C and stirred for 1 hr. A mixture of 10% hydrochloric acid in 1,4-dioxane was added and continuously stirred at room temperature for 18 hours. After the reaction was completed, the reaction was quenched by adding a sodium hydroxide solution (1 mL, 3N), and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification gave 2-(3-cyclopropylmethoxy-4-methoxyphenyl)cyclopropylamine (12 mg, yield: 25%, yellow oil).
-

- The compound 2-(3-cyclopropylmethoxy-4-methoxyphenyl)cyclopropylamine (60 mg, 0.24 mmol) was dissolved in ethanol (3 mL), and then 2.6-dimethyl-4H-pyran-4-one (45 mg, 0.36 mmol) and sodium hydroxide (20 mg, 0.48 mmol) were added, and stirred overnight at 60°C. The reaction was complete as indicated by LCMS. The reaction solution was rotary dried, and purified to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)cyclopropyl)-2,6-dimethylpyridin-4(1H)-one (10 mg, yield 11%, yellow oil). 1H NMR (400 MHz, CD3OD) δ 6.83 (d, J = 8.8 Hz, 1H), 6.75-6.58 (m, 2H), 6.18 (s, 2H), 3.76 (d, J = 6.8 Hz, 2H), 3.73 (s, 3H), 2.40 (s, 6H), 1.63-1.54 (m, 2H), 1.29-1.12 (m, 2H), 0.54-0.48 (m, 2H), 0.26-0.22 (m, 2H); LC-MS: m/z 340.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- At room temperature, 3-hydroxy-4-difluoromethoxybenzaldehyde (1.88 g, 10.0 mmol) was dissolved in acetonitrile (10 mL), and then potassium carbonate (2.07 g, 15.0 mmol) and bromomethylcyclopropane (1.76 g, 13.0 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, saturated saline (30 mL) was added, and extracted with ethyl acetate (3 x 60 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by reverse-phase HPLC to obtain 3-cyclopropylmethoxy-4-difluoromethoxybenzaldehyde (2.20 g, 91%). LC-MS: m/z 243.2 [M + H]+.
- The compound 3-cyclopropylmethoxy-4-difluoromethoxybenzaldehyde (2.20 g, 9.1 mmol) was dissolved in dichloromethane (30 mL), and then triethyl amine (1.84 g, 18.2 mmol) and trimethylsilyl cyanide (2.7 g, 27.3 mmol) were added in sequence in an ice bath and stirred for 16 hours under nitrogen atmosphere at room temperature. After the reaction was completed, the reaction solution was directly rotary dried to obtain the product 2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-trimethylsiloxyacetonitrile (3.1 g, 100%), which was directly used in the next reaction.
- The compound 2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-trimethylsiloxyacetonitrile (3.1 g, 9.1 mmol) was dissolved in anhydrous tetrahydrofuran (50 mL), and then lithium aluminum hydride (1.04 g, 27.3 mmol) was added portion-wise in an ice bath, and stirred overnight at room temperature. After the reaction was completed, the reaction solution was diluted with anhydrous tetrahydrofuran (200 mL), and extracted with water (1 mL). An aqueous sodium hydroxide solution (3 mL, 1M) and then water (3 mL) were added, stirred for half an hour, dried over anhydrous sodium sulfate, filtered, and rotary dried to obtain a crude product of 2-amino-1-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)ethanol (2.70 g, 100%). LC-MS: m/z 258.1 [M-18+1]+.
- The compound 2-amino-1-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)ethanol (2.70 g, 9.1 mmol) was dissolved in ethanol (30 mL), and then 2,6-dimethyl-4H-pyran-4-one (1.86 g, 15.0 mmol), sodium hydroxide (0.60 g, 15.0 mmol) and water (10 mL) were added in sequence, heated to 60°C, and stirred for 16 hours under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried, and purified to obtain 1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (2.4 g, 69%). LC-MS: m/z 380.2 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.13 mmol) was dissolved in dichloromethane (10 mL), and then the Dess-Martin Periodinane (110 mg, 0.26 mmol) was added and stirred at room temperature for 2 hours. The reaction solution was filtered, and washed with saturated brine. The organic phase was dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by reverse-phase HPLC to obtain 1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one (35 mg, yield 72%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.66 (s, 2H), 7.64 (d, J = 8.4 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H), 6.79 (t, JH-F = 74.4 Hz, 1H), 6.45 (s, 2H), 5.50 (s, 2H), 3.99 (d, J = 7.2 Hz, 2H), 2.26 (s, 6H) 1.37-1.28 (m, 1H), 0.72-0.66 (m, 2H), 0.43-0.37 (m, 2H); LC-MS: m/z 378.3 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)pyridin-4(1H)-one (270 mg, 0.86 mmol) and p-toluenesulfonic acid (443 mg, 2.58 mmol) were dissolved in toluene (5 mL), and stirred overnight under reflux. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(3-cyclopropylmethoxy-4-methoxystyryl)pyridin-4(1H)-one (50 mg, yield 20%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 8.02 (d, J = 7.6 Hz, 2H), 7.52 (d, J = 14.4 Hz, 1H), 7.10 (s, 1H), 6.97-6.93 (m, 2H), 6.85 (d, J = 14.4 Hz, 1H), 6.21 (d, J = 7.6 Hz, 2H), 3.83 (d, J = 7.2 Hz, 2H), 3.77 (s, 3H), 1.26-1.21 (m, 1H), 0.62-0.56 (m, 2H), 0.36-0.31 (m, 2H); LC-MS: m/z 298.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2-methylpyridin-4(1H)-one (654 mg, 2 mmol) was dissolved in methanol (10 mL), and then sodium borohydride (114 mg, 3 mmol) was added portion-wise and stirred at room temperature for 3 hours. After the reaction was completed, a saturated ammonium chloride solution (20 mL) was added, and the reaction solution was filtered, concentrated, and purified by column chromatography to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2-methylpyridin-4(1H)-one (620 mg, yield 95%, white solid). LC-MS: m/z 330.4 [M + H]+
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2-methylpyridin-4(1H)-one (620 mg, 1.88 mmol) and p-toluenesulfonic acid (970 mg, 5.64 mmol) were dissolved in toluene (15 mL), and stirred overnight under reflux. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(3-cyclopropylmethoxy-4-methoxystyryl)-2-methylpyridin-4(1H)-one (520 mg, yield 84%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 8.32 (d, J = 7.2 Hz, 1H), 7.65 (d, J = 14.0 Hz, 1H), 7.22 (d, J = 1.6 Hz, 1H), 7.12 (dd, J = 8.4, 1.6 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 14.0 Hz, 1H), 6.66-6.62 (m, 2H), 3.86 (d, J = 7.2 Hz, 2H), 3.81 (s, 3H), 2.51(s, 3H), 1.28-1.22 (m, 1H), 0.62-0.57 (m, 2H), 0.36-0.31 (m, 2H); LC-MS: m/z 312.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2-methylpyridin-4(1H)-one (5.74 g, 20 mmol) was dissolved in methanol (100 mL), and then sodium borohydride (1.14 g, 30 mmol) was added portion-wise and stirred at room temperature for 3 hours. After the reaction was completed, a saturated ammonium chloride solution (20 mL) was added, and the reaction solution was filtered, concentrated, and purified by column chromatography to obtain 1-(2-(3-hydroxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (5.09 g, yield 88%, colorless oil). LC-MS: m/z 290.4 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2-methylpyridin-4(1H)-one (5.09 g, 17.6 mmol) and p-toluenesulfonic acid (9.08 g, 52.8 mmol) were dissolved in toluene (100 mL), and stirred overnight under reflux. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(3-cyclopropylmethoxy-4-methoxystyryl)-2-methylpyridin-4(1H)-one (3.05 g, yield 64%, gray solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.34 (s, 1H), 7.16 (s, 2H), 7.05 (d, J = 14.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.62 (d, J = 14.0 Hz, 1H), 3.92 (s, 2H), 2.48(s, 6H); LC-MS: m/z 272.1 [M + H]+.
-

- The specific reaction scheme is as shown below:
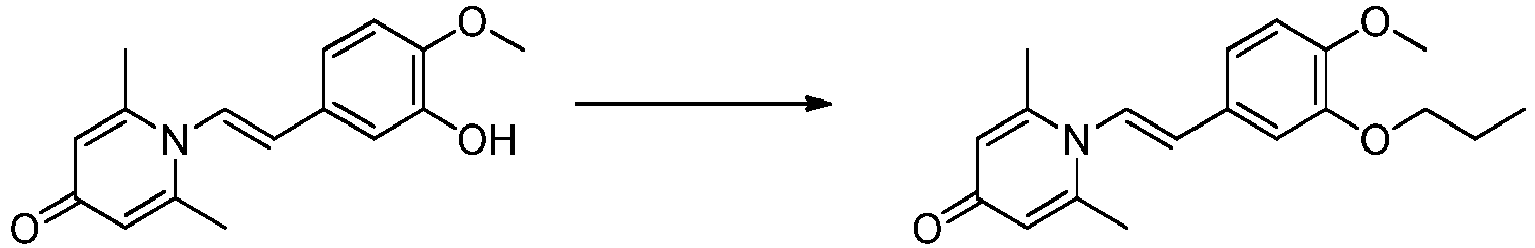
- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromopropane (34.5 mg, 0.28 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 2 hours at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-propoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (25 mg, yield 44%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.22 (d, J = 2.0 Hz, 1H), 7.14 (dd, J = 8.4, 2.0 Hz, 1H), 7.09 (d, J = 14.2 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 14.2 Hz, 1H), 6.38 (s, 2H), 4.03 (t, J = 6.4 Hz, 2H), 3.89 (s, 3H), 2.37 (s, 6H), 1.90-1.80 (m, 2H), 1.08 (t, J = 7.2 Hz, 3H); LC-MS: m/z 314.2 [M+H]+.
-
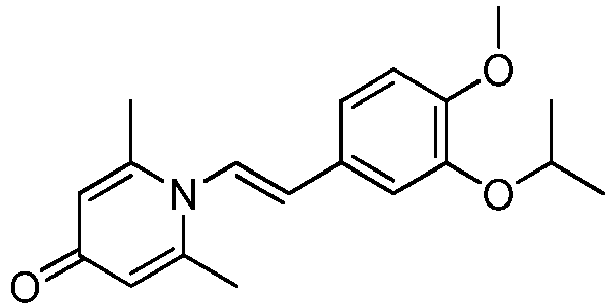
- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromoisopropane (34.5 mg, 0.28 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 2 hours at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-isopropoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (8 mg, yield 14%, colorless oil). 1H NMR (400 MHz, CD3OD) δ 7.26 (d, J = 2.0 Hz, 1H), 7.22 (dd, J = 8.4, 2.0 Hz, 1H), 7.21 (d, J = 14.4 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 6.97 (s, 2H), 6.89 (d, J = 14.4 Hz, 1H), 4.69-4.60 (m, 1H), 3.90 (s, 3H), 2.57 (s, 6H), 1.35 (d, J = 6.0 Hz, 6H). LC-MS: m/z 314.2 [M+H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then n-butyl bromide (38 mg, 0.28 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 2 hours at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-n-butoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 34%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.57 (s, 2H), 7.27 (d, J = 14.4 Hz, 1H), 7.10 (d, J = 1.6 Hz, 1H), 7.04-6.98 (m, 2H), 6.95 (d, J = 14.4 Hz, 1H), 4.34 (t, J = 6.4 Hz, 2H), 3.82 (s, 3H), 2.60 (s, 6H), 1.82-1.75 (m, 2H), 1.50-1.40 (m, 2H), 0.96 (t, J =7.2 Hz, 1H); LC-MS: m/z 328.2 [M + H]+.
-
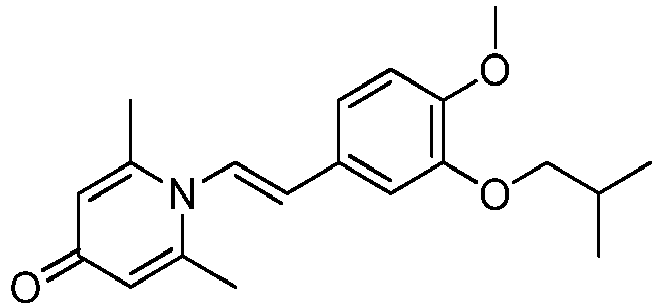
- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromoisobutane (38 mg, 0.28 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 2 hours at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-iso-butoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (25 mg, yield 42%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.02 (dd, J = 8.4, 2.0 Hz, 1H), 6.98 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 14.4 Hz, 1H), 6.60 (d, J = 14.4 Hz, 1H), 6.30 (s, 2H), 3.92 (s, 3H), 3.81 (d, J = 6.8 Hz, 2H), 2.27 (s, 6H), 2.24-2.17 (m, 1H), 1.08 (d, J = 6.8 Hz, 6H); LC-MS: m/z 328.2 [M+H]+.
-

- The specific reaction scheme is as shown below:
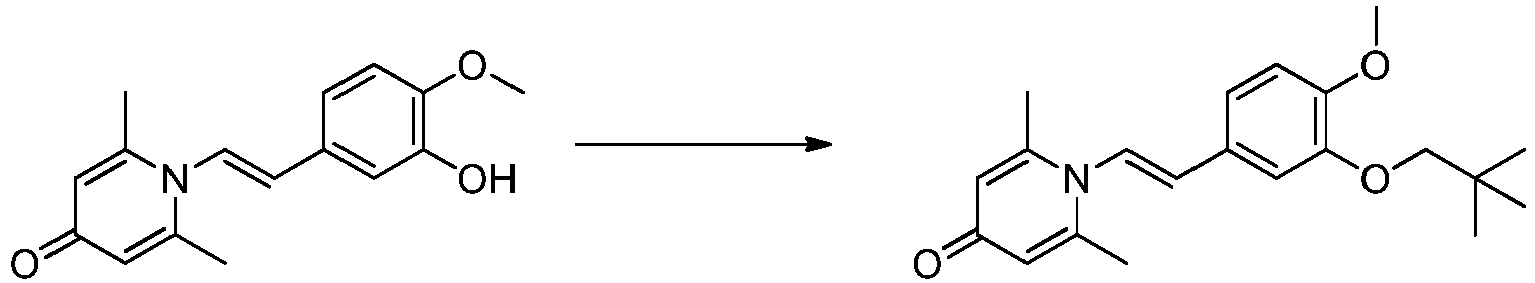
- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (200 mg, 0.74 mmol) was dissolved in N,N -dimethylformamide (5 mL), and then bromo-neopentane (224 mg, 1.48 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 24 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-neopentyloxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 8%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.25 (s, 2H), 7.05-7.00 (m, 2H), 6.97 (d, J = 14.4 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.70 (d, J = 14.4 Hz, 1H), 3.91 (s, 3H), 3.68 (s, 2H), 2.54 (s, 6H), 1.08 (s, 9H); LC-MS m/z 342.3 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (100 mg, 0.3 mmol) and p-toluenesulfonic acid (68.4 mg, 0.6 mmol) were dissolved in toluene (5 mL), and then N,N-dimethylformamide (1 mL) was added and stirred overnight under reflux. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(3-cyclopropyloxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, yield 53%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.15 (s, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.06 (d, J = 14.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 14.4 Hz, 1H), 6.32 (s, 2H), 4.02 (s, 3H), 2.32 (s, 6H), 2.23-2.19 (m, 1H), 0.94-0.90 (m, 4H); LC-MS m/z 312.3 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (186 mg, 0.54 mmol) and p-toluenesulfonic acid (140 mg, 0.81 mmol) were dissolved in toluene (5 mL), and then N,N-dimethylformamide (1 mL) was added and stirred overnight under reflux. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(3-cyclopropylmethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one(80 mg, yield 45%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.13 (dd, J = 8.4, 2.0 Hz, 1H), 7.19 (d, J = 2.0 Hz, 1H), 7.07 (d, J = 14.4 Hz, 1H), 7.11 (s, 2H), 6.99 (d, J = 8.4 Hz, 1H), 6.75 (d, J = 14.4 Hz, 1H), 6.41 (s, 2H), 3.89 (d, J = 7.2 Hz, 2H), 3.87 (s, 3H), 2.36 (s, 6H), 1.33-1.24 (m, 1H), 0.65-0.60 (m, 2H), 0.38-0.34 (m, 2H); LC-MS: m/z 326.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-cyclopropyloxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (10 mg, 0.03 mmol) was dissolved in ethanol (3 mL), and then Pd/C(50 mg) was added, and stirred for 2 hours under hydrogen atmosphere at room temperature. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(3-cyclopropylmethoxy-4-methoxyphenethyl)-2,6-dimethylpyridin-4(1H)-one (2.9 mg, yield 30%, colorless oil). 1H NMR (400 MHz, CD3OD) δ 7.13 (dd, J = 8.4, 2.0 Hz, 1H), 7.19 (d, J = 2.0 Hz, 1H), 7.11 (s, 2H), 6.99 (d, J = 8.4 Hz, 1H), 6.41 (s, 2H), 3.89 (d, J = 7.2 Hz, 2H), 3.87 (s, 3H), 2.89-2.83 (m, 2H), 2.83-2.77 (m, 2H), 2.36 (s, 6H), 1.33-1.24 (m, 1H), 0.65-0.60 (m, 2H), 0.38-0.34 (m, 2H); LC-MS: m/z 328.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (100 mg, 0.37 mmol) was dissolved in N,N -dimethylformamide (5 mL), and then bromocyclobutane (100 mg, 0.74 mmol) and cesium carbonate (241 mg, 0.74 mmol) were added, and stirred for 4 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-cyclobutyloxy-4-methoxy-styryl)-2,6-dimethylpyridin-4(1H)-one(18 mg, yield 15%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.13 (dd, J = 8.4, 2.0 Hz, 1H), 7.05 (d, J = 14.4 Hz, 1H), 7.03 (d, J = 2.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.75 (d, J = 14.4 Hz, 1H), 6.36 (s, 2H), 4.78-4.71 (m, 1H), 3.86 (s, 3H), 2.52-2.44 (m, 2H), 2.34 (s, 6H), 2.22-2.12 (m, 2H), 1.89-1.81 (m, 1H), 1.77-1.65 (m, 1H); LC-MS m/z 326.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (100 mg, 0.37 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then bromo-cyclopentane (110 mg, 0.74 mmol) and cesium carbonate (241 mg, 0.74 mmol) were added, and stirred for 4 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-cyclopentyloxy-4-methoxy-styryl)-2,6-dimethylpyridin-4(1H)-one (25 mg, yield 20%, pale yellow solid). 1H NMR (400 MHz, CD3OD) δ 7.16 (s, 1H), 7.13 (d, J = 8.4 Hz, 1H), 7.06 (d, J = 14.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 14.4 Hz, 1H), 6.36 (s, 2H), 3.35-3.83 (m, 1H), 3.86 (s, 3H), 2.52-2.44 (m, 2H), 2.35 (s, 6H), 1.93-1.79 (m, 6H), 1.69-1.58 (m, 2H); LC-MS m/z 340.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (100 mg, 0.37 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then cyclopent-3-en-1-yl methanesulfonate (110 mg, 0.74 mmol) and cesium carbonate (241 mg, 0.74 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-(cyclopent-3-en-1-yloxy)-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (22 mg, yield 15%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.45 (s, 2H), 7.08 (d, J = 8.0 Hz, 1H), 7.02-6.99 (m, 2H), 6.92 (d, J = 8.0 Hz, 1H), 6.74 (d, J = 14.4 Hz, 1H), 5.77 (s, 2H), 5.12-5.05 (m, 1H), 3.90 (s, 3H), 2.89-2.82 (m, 2H), 2.69-2.63 (m, 2H), 2.58 (s, 6H); LC-MS m/z 338.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.18 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then 3-bromopropene (44 mg, 0.36 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 2 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-allyloxy-4-methoxy-styryl)-2,6-dimethylpyridin-4(1H)-one (12 mg, yield 20%, pale yellow oil). 1H NMR (400 MHz, CDCl3) δ 7.23 (s, 2H), 7.09-6.92 (m, 4H), 6.73-6.69 (m, 1H), 6.16-6.05 (m, 1H), 5.45 (d, J = 17.2 Hz, 1H), 5.34 (d, J = 10.4 Hz, 1H), 4.67 (d, J= 4.0 Hz, 2H), 3.94 (s, 3H), 2.54 (s, 6H); LC-MS m/z 312.2 [M + H]+.
-
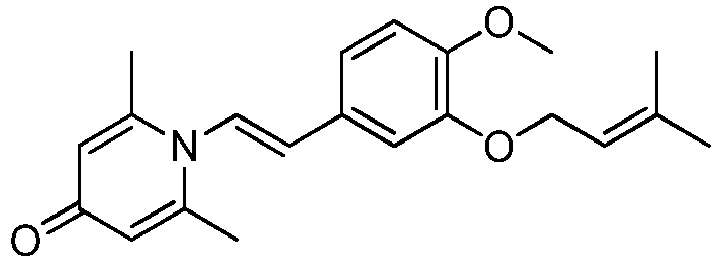
- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.18 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then 1-bromo-3-methyl-2-butene (55 mg, 0.36 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 1 hr at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-((3-(3-methylbut-2-en-1-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (15 mg, yield 18%, yellow solid). 1H NMR (400 MHz, CDCl3) δ 7.08 (s, 2H), 7.08-7.04 (m, 2H), 6.92-6.90 (m, 1H), 6.91 (d, J = 14.4 Hz, 1H), 6.69 (d, J = 14.4 Hz, 1H), 5.54 (t, J = 6.0 Hz, 1H), 4.62 (d, J = 6.0 Hz, 2H), 3.92 (s, 3H), 2.51 (s, 6H), 1.80 (s, 3H), 1.72 (s, 3H); LC-MS m/z 340.0 [M + H]+.
-
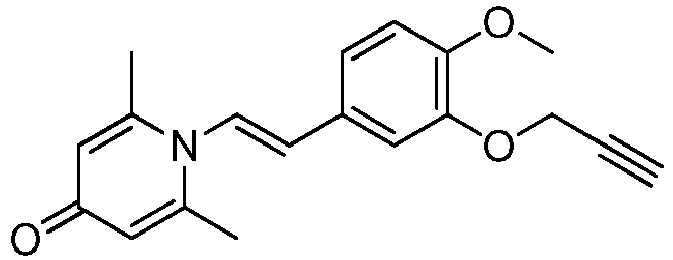
- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.18 mmol) was dissolved in N,N -dimethylformamide (5 mL), and then 3-bromo-propyne (44 mg, 0.36 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 3 hours at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-(prop-2-yn-1-yloxy)-4-methoxy)styryl)-2,6-dimethylpyridin-4(1H)-one (10 mg, yield 15%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.26-7.14 (m, 3H), 7.13 (s, 2H), 6.96 (d, J = 8.4 Hz, 1H), 6.73 (d, J = 14.4 Hz, 1H), 4.83 (s, 2H), 3.94 (s, 3H), 2.56 (s, 1H), 2.52 (s, 6H); LC-MS m/z 310.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.18 mmol) was dissolved in N,N -dimethylformamide (5 mL), and then 1-bromo-2-butyne (49 mg, 0.36 mmol) and cesium carbonate (117 mg, 0.36 mmol) were added, and stirred for 1 hr at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-(but-2-yn-1-yloxy)-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (15 mg, yield 25%, yellow solid). 1H NMR (400 MHz, CDCl3) δ 7.17 (s, 1H), 7.13 (d, J = 8.4 Hz, 1H), 6.09 (s, 2H), 6.98 (d, J = 14.4 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 14.4 Hz, 1H), 4.78 (s, 2H), 3.93 (s, 3H), 2.52 (s, 6H), 1.86 (s, 3H); LC-MS m/z 324.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (150 mg, 0.55 mmol), oxacyclobutan-3-ol (81.4 mg, 1.1 mmol) and triphenylphosphine (288 mg, 1.1 mmol) were dissolved in anhydrous tetrahydrofuran (10 mL), the reaction solution was cooled in an ice bath, diisopropyl azodicarboxylate(222 mg, 1.1 mmol) was added dropwise over 5 min under nitrogen atmosphere, and then reacted for 24 hours at room temperature. After the reaction was completed, saturated saline (20 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-((3-(oxacyclobutan-3-yl-oxy)-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (40 mg, yield 22%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 8.4 Hz, 1H), 7.07 (s, 2H), 6.96 (d, J = 8.4 Hz, 1H), 6.94 (d, J = 13.2 Hz, 1H), 6.70 (s, 1H), 6.69 (d, J = 13.2 Hz, 1H), 5.27 (m, 1H), 5.01 (m, 1H), 4.88 (m, 1H), 3.94 (s, 3H), 2.53 (s, 6H); LC-MS: m/z 328.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (150 mg, 0.55 mmol), thiacyclobutan-3-ol (99 mg, 1.1 mmol) and triphenylphosphine (288 mg, 1.1 mmol) were dissolved in anhydrous tetrahydrofuran (10 mL), the reaction solution was cooled in an ice bath, diisopropyl azodicarboxylate(222 mg, 1.1 mmol) was added dropwise over 5 min under nitrogen atmosphere, and then reacted for 24 hours at room temperature. After the reaction was completed, saturated saline (20 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-((3-(thiacyclobutan-3-yl-oxy)-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, yield 16%, pale yellow solid). 1H NMR (400 MHz, CD3OD) δ 7.37 (s, 2H), 7.19 (d, J = 14.4 Hz, 1H), 7.13 (d, J = 2.0 Hz, 1H), 7.05 (dd, J = 8.4, 2.0 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 14.4 Hz, 1H), 5.77-5.69 (m, 1H), 3.90 (s, 3H), 3.65-3.61 (m, 2H), 3.57-3.53 (m, 2H), 2.66 (s, 6H); LC-MS: m/z 344.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.15 mmol) was dissolved in dichloromethane (5 mL), and then 2,3-dihydrofuran (68 mg, 0.42 mmol) and pyridinium toluene-4-sulphonate (4.6 mg, 0.018 mmol) were added, and stirred for 3 hours at room temperature. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-((3-(tetrahydrofuran-2-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (16 mg, yield 24%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 2.0 Hz, 1H), 7.26 (s, 2H), 7.09 (dd, J = 8.4, 2.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 14.4 Hz, 1H), 6.75 (d, J = 14.4 Hz, 1H), 5.82 (d, J = 4.4 Hz, 1H), 5.70 (s, 2H), 4.13-4.08 (m, 1H), 4.01-3.95 (m, 1H), 3.90 (s, 3H), 2.35 (s, 6H), 2.24 -2.14 (m, 2H), 2.02 -1.95 (m, 2H); LC-MS m/z 342.3 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (200 mg, 0.74 mmol) was dissolved in N,N -dimethylformamide (5 mL), and then 2-chloro-N-methylacetamide (119 mg, 1.11 mmol) and potassium carbonate (153 mg, 1.11 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-2-(5-(2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethenyl)-2methoxyphenoxy)-N-methylacetamide (100 mg, yield 40%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.28 (d, J = 2.0 Hz, 1H), 7.23 (dd, J = 8.4, 2.0 Hz, 1H), 7.08 (d, J = 14.2 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 14.2 Hz, 1H), 6.35 (s, 2H), 4.55 (s, 2H), 3.92 (s, 3H), 2.84 (s, 3H), 2.34 (s, 6H); LC-MS m/z 343.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.17 mmol) was dissolved in dichloromethane (5 mL), and then cyclopropanecarbonyl chloride (27 mg, 0.26 mmol) and potassium carbonate (36 mg, 0.26 mmol) were added and stirred for 2 hours at room temperature. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-((3-cyclopropylformyloxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 35%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.37 (dd, J = 8.4, 2.0 Hz, 1H), 7.27 (d, J = 2.0 Hz, 1H), 7.11 (s, 2H), 7.04 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 14.4 Hz, 1H), 6.72 (d, J = 14.4 Hz, 1H), 3.92 (s, 3H), 2.52 (s, 6H), 1.96-1.89 (m, 1H), 1.25-1.21 (m, 2H), 1.12-1.06 (m, 2H); LC-MS: m/z 340.2 [M+H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (271 mg, 1.0 mmol) was dissolved in N, N-dimethylformamide (10 mL), and then triethyl amine (202 mg, 2.0mmol), and methylsulfonyl chloride (172 mg, 1.5 mmol) were added, and stirred overnight at room temperature. After the reaction was completed, saturated saline (20 mL) was added, and extracted with dichloromethane (3 x 20 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-5-(2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethenyl)-2-methoxyphenylmethyl sulfonate (130 mg, yield 40%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.61 (d, J = 2.4 Hz, 1H), 7.58 (dd, J = 8.8, 2.4 Hz, 1H), 7.28 (d, J = 14.4 Hz, 1H), 7.25 (d, J = 8.8 Hz, 1H), 7.05 (s, 2H), 6.96 (d, J = 14.4 Hz, 1H), 3.96 (s, 3H), 2.58 (s, 6H). LC-MS: m/z 350.1 [M+H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (70 mg, 0.26 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then chloromethylmethyl sulfide(50 mg, 0.52 mmol) and cesium carbonate (170 mg, 0.52 mmol) were added, and stirred overnight at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylthiomethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one(50 mg, yield 58%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 2.0 Hz, 1H), 7.25(dd, J = 8.4, 2.0 Hz, 1H), 7.08 (d, J = 14.4 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 14.4 Hz, 1H), 6.77 (s, 2H), 5.03 (s, 2H), 3.90 (s, 3H), 2.36 (s, 6H), 2.26 (s, 3H); LC-MS: m/z 332.5 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-methylthiomethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.15 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (28 mg, 0.14 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfoxidemethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (36 mg, yield 69%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 2.0 Hz, 1H), 7.28(dd, J = 8.4, 2.0 Hz, 1H), 7.09 (d, J = 14.4 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 7.03 (s, 2H), 6.68 (d, J = 14.4 Hz, 1H), 5.26 (d, J = 11.2 Hz, 1H), 5.11 (d, J = 11.2 Hz, 1H), 3.96 (s, 3H), 2.36 (s, 6H), 2.26 (s, 3H); LC-MS: m/z 348.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-methylthiomethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, 0.09 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (54 mg, 0.27 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfonemethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (26 mg, yield 79%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.50 (d, J = 2.0 Hz, 1H), 7.32 (dd, J = 8.4, 2.0 Hz, 1H), 7.11 (d, J = 8.4, 1H), 7.10 (d, J = 14.0 Hz, 1H), 6.79 (d, J = 14.0 Hz, 1H), 6.37 (s, 2H), 5.20 (s, 2H), 3.93 (s, 3H), 3.11 (s, 3H), 2.35 (s, 6H); LC-MS m/z 364.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (200 mg, 0.73 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then chloroethyl methyl sulfide (165 mg, 1.5 mmol) and cesium carbonate (489 mg, 1.5 mmol) were added, and stirred overnight at room temperature. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylthioethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (220 mg, yield 87%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.05 (dd, J = 8.4, 2.0 Hz, 1H), 7.01 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 14.4 Hz, 1H), 6.59 (d, J = 14.4 Hz, 1H), 6.26 (s, 2H), 4.24 (t, J = 7.2 Hz, 2H), 3.91 (s, 3H), 2.96 (t, J = 7.2 Hz, 2H), 2.26 (s, 6H), 2.24 (s, 3H); LC-MS: m/z 346.5 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-methylthioethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.15 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (28 mg, 0.14 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfoxideethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, yield 53%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.07 (d, J = 2.0 Hz, 1H), 6.99 (dd, J = 8.4, 2.0 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.72 (d, J = 14.4 Hz, 1H), 6.51 (d, J = 14.4 Hz, 1H), 6.17 (s, 2H), 4.50-4.41 (m, 2H), 3.83 (s, 3H), 3.27-3.20 (m, 1H), 3.07-3.01 (m, 1H), 2.68 (s, 3H), 2.17 (s, 6H); LC-MS: m/z 362.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-methylthioethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, 0.09 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (54 mg, 0.54 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfoneethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one(35 mg, yield 61%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.08 (dd, J = 8.4, 2.0 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 14.4 Hz, 1H), 6.58 (d, J = 14.4 Hz, 1H), 6.31 (s, 2H), 4.50 (t, J = 4.2 Hz, 2H), 3.89 (s, 3H), 3.50 (t, J = 4.2 Hz, 2H), 3.19 (s, 3H), 2.26 (s, 6H); LC-MS: m/z 378.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (45 mg, 0.15 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then tetrahydrothiophen-3-yl methanesulfonate (53 mg, 0.29 mmol) and cesium carbonate (95 mg, 0.29 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (35 mg, yield 65%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.10 (dd, J = 8.4, 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 14.4 Hz, 1H), 6.58 (d, J = 14.4 Hz, 1H), 6.31 (s, 2H), 5.16-5.13 (m, 1H), 3.90 (s, 3H), 3.18-3.08 (m, 1H), 3.09 (d, J = 3.6 Hz, 2H), 2.98-2.93 (m, 1H), 2.49-2.42 (m, 1H), 2.27 (s, 6H), 2.08-1.99 (m, 1H); LC-MS: m/z 358.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-((3-(tetrahydrothiophen-3-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (68 mg, 0.19 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (116 mg, 0.57 mmol) was added and stirred for 3 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-((1,1, -dioxotetrahydrothiophen-3-yl)oxy)-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (62 mg, yield 86%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.28 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 7.13 (d, J = 14.4 Hz, 1H), 7.10 (s, 2H), 6.97 (d, J = 14.4 Hz, 1H), 5.34-5.27 (m, 1H), 3.91 (s, 3H), 3.52-3.44 (m, 1H), 3.41-3.32 (m, 2H), 3.23-3.17 (m, 1H), 2.70-2.61 (m, 1H), 2.58-2.47 (m, 1H), 2.54 (s, 6H); LC-MS: m/z 390.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (45 mg, 0.15 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then (R)-tetrahydrothiophen-3-yl methanesulfonate (53 mg, 0.29 mmol) and cesium carbonate (95 mg, 0.29 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (S,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, yield 56%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.10 (dd, J = 8.4, 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 14.4 Hz, 1H), 6.58 (d, J = 14.4 Hz, 1H), 6.31 (s, 2H), 5.16-5.13 (m, 1H), 3.90 (s, 3H), 3.18-3.08 (m, 1H), 3.09 (d, J = 3.6 Hz, 2H), 2.98-2.93 (m, 1H), 2.49-2.42 (m, 1H), 2.27 (s, 6H), 2.08-1.99 (m, 1H); LC-MS: m/z 358.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (S,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (81 mg, 0.23 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (41 mg, 0.20 mmol) was added and stirred for 3 hours at 0°C. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave a diastereoisomer of 1-((E)-4-methoxy-3-(((3S)-1-oxotetrahydrothiophen-3-yl)oxy)styryl)-2,6-dimethylpyridin-4(1H)-one show below:

- (25 mg, yield 29%, pale yellow oil). 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 1.6 Hz, 1H), 7.12 (dd, J = 8.4, 1.6 Hz, 1H), 7.07 (d, J = 14.4 Hz, 1H), 7.01 (s, 2H), 6.94 (d, J = 8.4 Hz, 1H), 6.68 (d, J = 14.4 Hz, 1H), 5.28-5.24 (m, 1H), 3.91 (s, 3H), 3.34-3.14 (m, 4H), 2.93-2.85 (m, 1H), 2.51 (s, 6H), 2.34-2.25 (m, 1H); LC-MS: m/z 374.1 [M + H]+.

- (20 mg, yield 23%, pale yellow oil)1H NMR (400 MHz, CDCl3) δ 7.17 (d, J = 8.4 Hz, 1H), 7.15 (s, 1H), 7.07 (d, J = 14.4 Hz, 1H), 7.03 (s, 2H), 6.95 (d, J = 8.4 Hz, 1H), 6.71 (d, J = 14.4 Hz, 1H), 5.48-5.43 (m, 1H), 3.89 (s, 3H), 3.64 (d, J = 15.2 Hz, 1H), 3.21-3.07 (m, 3H), 2.83-2.74 (m, 1H), 2.65-2.59 (m, 1H), 2.51 (s, 6H); LC-MS: m/z 374.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-((3-(tetrahydrothiophen-3-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (81 mg, 0.23 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (140 mg, 0.69 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-((1,1, -dioxotetrahydrothiophen-3-yl)oxy)-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (58 mg, yield 66%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 14.4 Hz, 1H), 7.05 (s, 2H), 6.97 (d, J = 8.4 Hz, 1H), 6.71 (d, J = 14.4 Hz, 1H), 5.32-5.27 (m, 1H), 3.91 (s, 3H), 3.52-3.43 (m, 1H), 3.41-3.31 (m, 2H), 3.22-3.16 (m, 1H), 2.69-2.62 (m, 1H), 2.57-2.47 (m, 1H), 2.52 (s, 6H); LC-MS m/z 390.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (45 mg, 0.15 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then (S)-tetrahydrothiophen-3-yl methanesulfonate (53 mg, 0.29 mmol) and cesium carbonate (95 mg, 0.29 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (S,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, yield 56%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.10 (dd, J = 8.4, 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 14.4 Hz, 1H), 6.58 (d, J = 14.4 Hz, 1H), 6.31 (s, 2H), 5.16-5.13 (m, 1H), 3.90 (s, 3H), 3.18-3.08 (m, 1H), 3.09 (d, J = 3.6 Hz, 2H), 2.98-2.93 (m, 1H), 2.49-2.42 (m, 1H), 2.27 (s, 6H), 2.08-1.99 (m, 1H); LC-MS: m/z 358.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (R,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (57 mg, 0.16 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (29 mg, 0.15 mmol) was added and stirred for 3 hours at 0°C. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave a diastereoisomer of 1-((E)-4-methoxy-3-(((3R)-1-oxotetrahydrothiophen-3-yl)oxy)styryl)-2,6-dimethylpyridin-4(1H)-one shown below:

- (15 mg, yield 25%, pale yellow oil). 1H NMR (400 MHz, CD3OD) δ 7.26 (d, J = 2.0 Hz, 1H), 7.20 (dd, J = 8.4, 2.0 Hz, 1H), 7.12 (d, J = 14.4 Hz, 1H), 6.99 (d, J = 7.6, 1H), 6.96 (s, 2H), 6.80 (d, J = 14.4, 1H), 5.18-5.13 (m, 1H), 3.80 (s, 3H), 3.36-3.25(m, 2H), 3.09-3.00 (m, 2H), 2.69-2.61 (m, 1H), 2.49 (s, 6H), 2.27-2.19(m, 1H); LC-MS: m/z 374.1 [M + H]+.

- (10 mg, yield 17%, pale yellow oil)1H NMR (400 MHz, CD3OD) δ 7.33 (d, J = 2.0 Hz, 1H), 7.30 (dd, J = 8.4, 2.0 Hz, 1H), 7.26 (d, J = 14.4 Hz, 1H), 7.10 (d, J = 7.6, 1H), 7.09 (s, 2H), 6.93 (d, J = 14.4, 1H), 5.52-5.49 (m, 1H), 3.89 (s, 3H), 3.76-3.71 (m, 1H), 3.30-3.22 (m, 2H), 3.76-3.71 (m, 1H), 2.77-2.67 (m, 1H), 2.65-2.63 (m, 1H), 2.61 (s, 6H); LC-MS: m/z 374.1 [M + H]+.
-
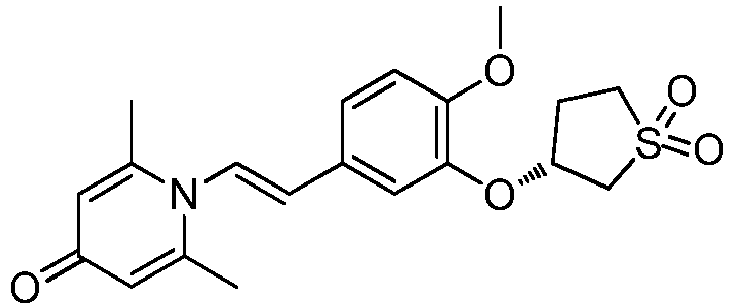
- The specific reaction scheme is as shown below:

- The compound (E)-1-((3-(tetrahydrothiophen-3-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, 0.08 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (49 mg, 0.24 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-((1,1, -dioxotetrahydrothiophen-3-yl)oxy)-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (12 mg, yield 40%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.23 (s, 1H), 7.22 (dd, J = 8.4, 2.0 Hz, 1H), 7.12 (d, J = 14.4 Hz, 1H), 7.10 (dd, J = 8.4, 2.0 Hz, 1H), 6.96 (s, 2H), 6.81 (d, J = 14.4, 1H), 5.20-5.16 (m, 1H), 3.80 (s, 3H), 3.38-3.28 (m, 2H), 3.25-3.24 (m, 1H), 3.13-3.07 (m, 1H), 2.49 (s, 6H), 2.47-2.34 (m, 2H); LC-MS: m/z 390.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (80 mg, 0.21 mmol) was dissolved in trichloromethane (10 mL), and heated to 80°C. Then thionyl chloride (0.5 mL) was added and stirred at 80°C for 15 min. After the reaction was completed, the reaction solution was directly rotary dried to obtain a crude product of 1-(2-chloro-2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)ethyl)-2,6-dimethylpyridin-4(1H)-one (85 mg, 100%). LC-MS: m/z 398.2 [M + H]+.
- The compound 1-(2-chloro-2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)ethyl)-2,6-dimethylpyridin-4(1H)-one (85 mg, 0.21 mmol) was dissolved in ethanol (10 mL), and then sodium hydroxide (64 mg, 1.60 mmol) and water (2 mL) were added in sequence, heated to 100°C, and stirred for 16 hours under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried, and purified to obtain (E)-1-(3-(cyclopropylmethoxy)-4-(difluoromethoxy)styryl)-2,6-dimethylpyridin-4(1H)-one (20 mg, 26%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.41 (d, J = 2.0 Hz, 1H), 7.34 (d, J = 14.4 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.17 (dd, J = 8.4, 2.0 Hz, 1H), 7.12 (t, JH-F = 74.4 Hz, 1H), 6.91 (d, J = 14.4 Hz, 1H), 6.49 (s, 2H), 3.95 (d, J = 7.2 Hz, 2H), 2.34 (s, 6H), 1.31-1.21 (m, 1H), 0.62-0.57 (m, 2H), 0.37-0.33 (m, 2H); LC-MS: m/z 362.2 [M + H]+.
-
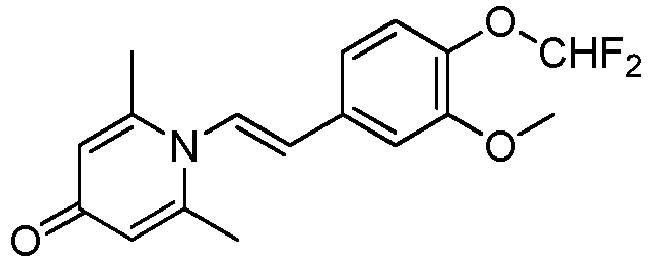
- The specific reaction scheme is as shown below:

- At room temperature, 3-hydroxy-4-difluoromethoxybenzaldehyde (2.36 g, 12.5 mmol) was dissolved in acetonitrile (40 mL), and then potassium carbonate (3.46 g, 25.1 mmol) and benzyl bromide (2.79 g, 16.3 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, a saturated aqueous sodium chloride solution (30 mL) was added, and extracted with dichloromethane (3 x 120 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified by column chromatography to obtain 3-benzoxy-4-difluoromethoxybenzaldehyde (3.30 g, 95%).
- 3-benzoxy-4-difluoromethoxybenzaldehyde (3.30 g, 11.9 mmol) was dissolved in dichloromethane (30 mL), and then triethyl amine (2.40 g, 23.7 mmol) and trimethylsilyl cyanide (3.53 g, 35.6 mmol) were added in sequence in an ice bath and stirred for 16 hours under nitrogen atmosphere at room temperature. After the reaction was completed, the reaction solution was directly rotary dried to obtain 2-(3-benzoxy-4-difluoromethoxyphenyl)-2-trimethylsiloxyacetonitrile, which was directly used in the next reaction.
- The compound 2-(3-benzoxy-4-difluoromethoxyphenyl)-2-trimethylsiloxyacetonitrile (4.47 g, 11.9 mmol) was dissolved in anhydrous tetrahydrofuran (40 mL), and then lithium aluminum hydride (1.35 g, 35.6 mmol) was added portion-wise in an ice bath, and stirred overnight at room temperature. After the reaction was completed, the reaction was diluted with anhydrous tetrahydrofuran (200 mL), and then water (1.35 mL), an aqueous sodium hydroxide solution (1.35 mL, 15%), and water (4.05 mL) were added in sequence, stirred for half an hour, dried over anhydrous sodium sulfate, filtered, and rotary dried to obtain a crude product of 2-amino-1-(3-benzoxy-4-difluoromethoxyphenyl)ethanol, which was directly used in the next reaction.
- The compound 2-amino-1-(3-benzoxy-4-difluoromethoxyphenyl)ethanol (3.67 g, 11.9 mmol) was dissolved in ethanol (60 mL), and then 2,6-dimethyl-4H-pyran-4-one (2.19 g, 17.7 mmol), sodium hydroxide (708 mg, 17.7 mmol) and water (10 mL) were added in sequence, heated to 60°C, and stirred for 16 hours under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried, and purified by column chromatography to obtain 1-(2-(3-benzoxy-4-difluoromethoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (2.3 g, 47%). LC-MS m/z 416.2 [M + H]+.
- The compound 1-(2-(3-benzoxy-4-difluoromethoxyphenyl)-2-hydroxyethyl)-2,6-dimethylpyridin-4(1H)-one (2.3 g, 5.3 mmol) was dissolved in trichloromethane (80 mL), and then heated to 90°C. Thionyl chloride (3 mL) was added and stirred at 90°C for 15 min. After the reaction was completed, the reaction solution was directly rotary dried to obtain a crude product of 1-(2-(3-benzoxy-4-difluoromethoxyphenyl)-2-chloroethyl)-2,6-dimethylpyridin-4(1H)-one (2.15 g, 93%). LC-MS m/z 434.2 [M + H]+.
- The compound 1-(2-(3-benzoxy-4-difluoromethoxyphenyl)-2-chloroethyl)-2,6-dimethylpyridin-4(1H)-one (2.15 g, 5.0 mmol) was dissolved in ethanol (40 mL), and then sodium hydroxide (1.2 g, 30 mmol) and water (15 mL) were added in sequence, heated to 90°C, and stirred for 16 hours under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried, and purified to obtain (E)-1-(3-benzoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one(1.5 g, 75%). LC-MS: m/z 398.2 [M + H]+.
- The compound (E)-1-(3-benzoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (400 mg, 1.0 mmol) was dissolved in anhydrous dichloromethane (100 mL), and titanium tetrachloride (1 M, 2 mL, 2.0 mmol) was slowly added dropwise in an ice bath, and stirred overnight at room temperature. After the reaction was completed, the reaction solution was rotary dried, and purified to obtain (E)-1-(3-hydroxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 16%). LC-MS: m/z 308.2 [M + H]+.
- The compound (E)-1-(3-hydroxy-4-difluoromethoxy-styryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.16 mmol) was dissolved in acetonitrile (5 mL), and then iodomethane (30 mg, 0.21 mmol) and potassium carbonate (90 mg, 0.65 mmol) were added, and stirred for 2 hours at 80°C under nitrogen atmosphere. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(3-methoxy-4-difluoromethoxy-styryl)-2,6-dimethylpyridin-4(1H)-one (38 mg, yield 71%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.0 Hz, 1H), 7.08-7.05 (m, 2H), 6.96 (d, J = 14.0 Hz, 1H), 6.69 (d, J = 14.0 Hz, 1H), 6.59 (t, JH-F = 74.8 Hz, 1H), 6.41 (s, 2H), 3.95 (s, 3H), 2.31 (s, 6H); LC-MS: m/z 322.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-difluoromethoxy-styryl)-2,6-dimethylpyridin-4(1H)-one (35 mg, 0.11 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then chloromethylmethyl sulfide (22 mg, 0.23 mmol) and cesium carbonate (150 mg, 0.46 mmol) were added, and stirred for 1 hr at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylthiomethoxy-4-difluoromethoxy-styryl)-2,6-dimethylpyridin-4(1H)-one(30 mg, yield 71%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.24-7.22 (m, 2H), 7.16 (dd, J = 8.4, 2.0 Hz, 1H), 7.08 (d, J = 14.4 Hz, 1H), 6.77 (s, 2H), 6.76 (d, J = 14.4 Hz, 1H), 6.59 (t, JH-F = 74.0 Hz, 1H), 5.29 (s, 2H), 2.41 (s, 6H), 2.29 (s, 3H); LC-MS: m/z 368.4 [M + H]+.
-
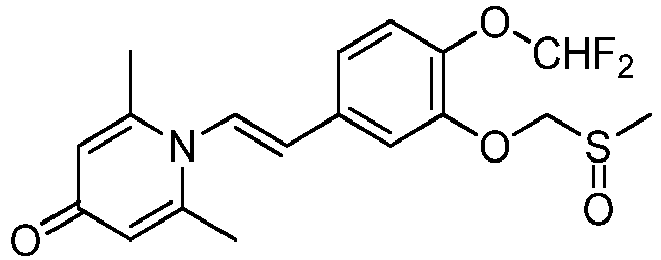
- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-methylthiomethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (78 mg, 0.21 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (39 mg, 0.19 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfoxidemethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (64 mg, yield 78%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.72 (s, 1H), 7.27 (d, J = 14.8 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.03 (s, 2H), 6.80 (d, J = 14.8 Hz, 1H), 6.64 (t, JH-F = 73.6 Hz, 1H), 5.26 (d, J = 11.2 Hz, 1H), 5.11 (d, J = 11.2 Hz, 1H), 2.77 (s, 3H), 2.52 (s, 6H); LC-MS: m/z 384.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-methylthiomethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (78 mg, 0.21 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (128 mg, 0.63 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfonemethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one(35 mg, yield 41%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.56 (d, J = 2.0 Hz, 1H), 7.27 (d, J = 8.4, 2.0 Hz, 1H), 7.26 (d, J = 14.4 Hz, 1H), 7.21 (d, J = 8.4 Hz, 1H), 6.97 (s, 2H), 6.91 (d, J = 14.4 Hz, 1H), 6.79 (t, JH-F = 73.6 Hz, 1H), 5.23 (s, 2H), 3.01 (s, 3H), 2.49 (s, 6H); LC-MS: m/z 399.9 [M + H]+.
-

- The specific reaction scheme is as shown below:

- At room temperature, (E)-1-(4-(difluoromethoxy)-3-hydroxystyryl)-2,6-dimethylpyridin-4(1H)- one (50 mg, 0.16 mmol) was dissolved in N,N -dimethylformamide (2 mL), and then potassium carbonate (34 mg, 0.24 mmol) and chloroethyl methyl sulfide (36 mg, 0.33 mmol) were added in sequence, and heated to 80°C for 16 hours with stirring under nitrogen atmosphere. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(3-methylthioethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (8 mg, 14%). 1H NMR (400 MHz, CDCl3) δ 7.18 (d, J = 8.4 Hz, 1H), 7.06-6.99 (m, 2H), 6.84 (d, J = 14.0 Hz, 1H), 6.59 (t, J = 14.0 Hz, 1H), 6.58 (d, JH-F = 75.2 Hz, 1H), 6.19 (s, 2H), 4.20 (t, J = 6.4 Hz, 2H), 2.87 (t, J = 6.4 Hz, 2H), 2.19 (s, 6H), 2.16 (s, 3H); LC-MS: m/z 382.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-methylthioethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (55 mg, 0.14 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (27 mg, 0.13 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfoxideethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (16 mg, yield 28%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.38 (s, 1H), 7.26 (d, J = 14.4 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 6.97 (s, 2H), 6.79 (d, J = 14.4 Hz, 1H), 6.58 (t, JH-F = 74.0 Hz, 1H), 4.60 (t, J = 8.8 Hz, 1H), 3.33-3.26 (m, 1H), 3.16-3.10 (m, 1H), 2.75 (s, 3H), 2.51 (s, 6H); LC-MS: m/z 398.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-methylthioethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (55 mg, 0.14 mmol) was dissolved in dichloromethane (5 mL), and then m-chloroperoxybenzoic acid (85 mg, 0.42 mmol) was added and stirred for 2 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-methylsulfoneethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (33 mg, yield 75%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.45 (s, 1H), 7.36 (d, J = 14.4 Hz, 1H), 7.30-7.25 (m, 2H), 7.04 (s, 2H), 7.00 (d, J = 14.4 Hz, 1H), 6.58 (t, JH-F = 74.4 Hz, 1H), 4.55 (t, J = 5.2 Hz, 1H), 3.66 (t, J = 5.2 Hz, 1H), 3.15 (s, 3H), 2.59 (s, 6H); LC-MS: m/z 414.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one (42 mg, 0.14 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then tetrahydrothiophen-3-yl methanesulfonate (53 mg, 0.29 mmol) and cesium carbonate (95 mg, 0.29 mmol) were added, and stirred for 1 hr at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, yield 54%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.41 (d, J = 2.0 Hz, 1H), 7.27 (d, J = 14.4 Hz, 1H), 7.26 (dd, J = 8.4, 2.0 Hz, 1H), 7.21 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 14.4 Hz, 1H), 6.79 (t, JH-F = 75.0 Hz, 1H), 6.65 (s, 2H), 5.36-5.32 (m, 1H), 3.16 (dd, J = 12.0, 4.4 Hz, 1H), 3.11-3.02 (m, 2H), 2.99-2.94 (m, 1H), 2.49-2.41 (m, 1H), 2.45 (s, 6H), 2.10-2.01 (m, 1H); LC-MS: m/z 394.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (50 mg, 0.15 mmol) was dissolved in N,N-dimethylformamide (5 mL), and then (R)-tetrahydrothiophen-3-yl methanesulfonate (53 mg, 0.29 mmol) and cesium carbonate (95 mg, 0.29 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (S,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (10 mg, yield 17%, pale yellow solid). 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 8.4 Hz, 1H), 7.11 (dd, J= 8.4, 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H), 6.87 (d, J = 14.4 Hz, 1H), 6.77 (d, J = 14.4 Hz, 1H), 6.64 (d, J = 14.4 Hz, 1H), 6.61 (t, JH-F = 74.8 Hz, 1H), 6.29 (s, 2H), 5.21-5.16 (m, 1H), 3.16-3.09 (m, 3H), 3.03-2.97 (m, 1H), 2.52-2.47 (m, 1H), 2.26 (s, 6H), 2.11-2.02 (m, 1H); LC-MS: m/z 394.2 [M + H]+.
-

- The specific reaction scheme is as shown below:
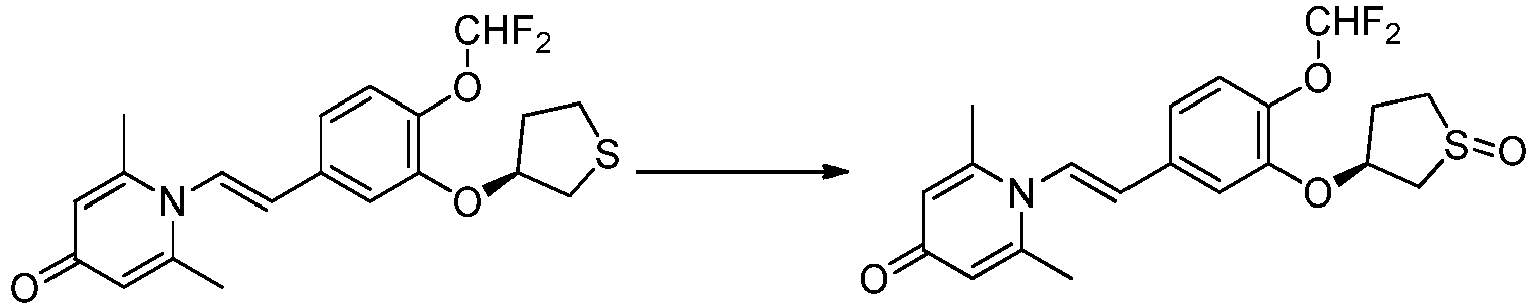
- The compound (E)-1-((3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (103 mg, 0.26 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (48 mg, 0.24 mmol) was added and stirred for 2 hours at 0°C. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave a diastereoisomer of 1-((E)-4-difluoromethoxy-3-(((3S)-1-oxotetrahydrothiophen-3-yl)oxy)styryl)-2,6-dimethylpyridin-4(1H)-one shown below:

- (42 mg, yield 39%, pale yellow oil). 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1H), 7.37 (d, J = 14.4 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 6.94 (s, 2H), 6.81 (d, J = 14.4 Hz, 1H), 6.80 (t, JH-F = 72.4 Hz, 1H), 5.46-5.38 (m, 1H), 3.46-3.38 (m, 1H), 3.33-3.17 (m, 3H), 2.94-2.85 (m, 1H), 2.54 (s, 6H), 2.41-2.32 (m, 1H); LC-MS: m/z 410.1 [M + H]+.

- (30 mg, yield 28%, pale yellow oil). 1H NMR (400 MHz, CDCl3) δ 7.35 (s, 1H), 7.30 (d, J = 14.4 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.16 (dd, J = 8.4, 1.2 Hz, 1H), 6.95 (s, 2H), 6.82 (d, J = 14.4 Hz, 1H), 6.48 (t, JH-F = 73.6 Hz, 1H), 5.58-5.53 (m, 1H), 3.60 (d, J = 15.2 Hz, 1H), 3.17 (dd, J = 15.2, 4.8 Hz, 1H), 3.11-3.07 (m, 2H), 2.90-2.80 (m, 1H), 2.65-2.61 (m, 1H), 2.51 (s, 6H); LC-MS: m/z 410.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-((3-(tetrahydrothiophen-3-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one (55 mg, 0.14 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (85 mg, 0.42 mmol) was added and stirred for 3 hours at room temperature. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (S,E)-1-(3-(1,1-dioxotetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (52 mg, yield 86%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.44 (s, 1H), 7.36 (d, J = 14.0, Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 6.96 (s, 2H), 6.81 (d, J = 14.0 Hz, 1H), 6.60 (t, JH-F = 73.6 Hz, 1H), 5.45-5.40 (m, 1H), 3.50-3.35 (m, 3H), 3.27-2.21(m, 1H), 2.70-2.58 (m, 2H), 2.53 (s, 6H); LC-MS m/z 426.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-1-(3-hydroxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (200 mg, 0.58 mmol) was dissolved in N,N-dimethylformamide (10 mL), and then (S)-tetrahydrothiophen-3-yl methanesulfonate (212 mg, 1.16 mmol) and cesium carbonate (758 mg, 2.3 mmol) were added, and stirred overnight at 80°C under nitrogen atmosphere. After the reaction was completed, saturated saline (20 mL) was added, and extracted with dichloromethane (3 x 20 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (R,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (30 mg, yield 13%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 8.4 Hz, 1H), 7.11 (dd, J = 8.4 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H), 6.89 (d, J = 14.4 Hz, 1H), 6.65 (d, J = 14 Hz, 1H), 6.60 (m, J = 14.4 Hz, 1H), 6.63 (t, J = 74.8 Hz, 1H), 6.31 (s, 2H), 5.21-5.16 (m, 1H), 3.16-3.09 (m, 3H), 3.03-2.97 (m, 1H), 2.52-2.47 (m, 1H), 2.26 (s, 6H), 2.11-2.02 (m, 1H); LC-MS m/z 394.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (R,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (55 mg, 0.14 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (26 mg, 0.13 mmol) was added and stirred for 3 hours at 0°C. Then, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave a diastereoisomer of 1-((E)-4-difluoromethoxy-3-(((3R)-1-oxotetrahydrothiophen-3-yl)oxy)styryl)-2,6-dimethylpyridin-4(1H)-one shown below:

- (20 mg, yield 35%, pale yellow oil). 1H NMR (400 MHz, CD3Cl) δ 7.50 (s, 1H), 7.20 (d, J = 12.8 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.95 (s, 2H), 6.82 (s, 1H), 6.80 (m, J = 76.4 Hz, 1H), 5.43 (m, 1H), 3.44-3.38 (m, 2H), 3.29-3.16 (m, 2H), 2.92-2.89 (m, 1H), 2.54 (s, 6H), 2.41-2.33 (m, 1H); LC-MS: m/z 410.0 [M + H]+.

- (10 mg, yield 17%, pale yellow oil)1H NMR (400 MHz, CD3Cl) δ 7.34 (s, 1H), 7.30 (d, J = 14.4 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 6.94 (s, 2H), 6.82 (d, J = 14.4 Hz, 1H), 6.48 (t, J = 73.6 Hz, 1H), 5.55 (m, 1H), 3.59 (d, J = 14.8 Hz, 1H), 3.19-3.14 (m, 1H), 3.11-3.08 (m, 2H), 2.90-2.81 (m, 1H), 2.64-2.61 (m, 1H), 2.54 (s, 6H); LC-MS: m/z 410.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (R,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (40 mg, 0.1 mmol) was dissolved in dichloromethane (5 mL), and then 85% m-chloroperoxybenzoic acid (61 mg, 0.3 mmol) was added and stirred for 2 hours at room temperature. After the reaction was completed, a saturated aqueous sodium sulfite solution (10 mL) was added, stirred for 10 min, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (R,E)-1-(3-(1,1-dioxotetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one (33 mg, yield 75%, white solid). 1H NMR (400 MHz, CD3OD) δ 7.43 (d, J = 1.6 Hz, 1H), 7.38 (dd, J = 8.4, 2.0 Hz, 1H), 7.36 (d, J = 2.0 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 7.06 (s, 2H), 7.01 (d, J = 14.4 Hz, 1H), 6.87 (t, J = 75.2 Hz, 1H), 5.43-5.39 (m, 1H), 3.56-8.51 (m, 1H), 3.44-3.36 (m, 2H), 3.31-3.25 (m, 1H), 2.66-2.62 (m, 2H), 2.61 (s, 6H); LC-MS: m/z 426.0 [M + H]+.
-

- The specific reaction scheme is as shown below:

- 3-Cyclopropylmethoxy-4-methoxyphenylacetophenone (220 mg, 1.0 mmol) was dissolved in acetonitrile (2 mL); and under nitrogen atmosphere, cesium fluoride (76 mg, 0.50 mmol) was added, trimethylsilyl cyanide (149 mg, 1.50 mmol) was slowly added dropwise at 0°C, and stirred for 5 hours. The reaction solution was filtered, and rotary dried to obtain a crude product of 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-trimethylsiloxy-propionitrile. LC-MS: m/z 320.2 [M + H]+.
- The compound 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-trimethylsiloxy-propionitrile (319 mg, 1.0 mmol) was dissolved in anhydrous tetrahydrofuran (3 mL); and under nitrogen atmosphere, lithium aluminum hydride (96 mg, 2.0 mmol) was slowly added dropwise at 0°C, and the solution was stirred at room temperature for 1 hr. After the reaction was completed, sodium sulfate decahydrate was added to quench the reaction. Then, the reaction solution was filtered, and the filtrate was rotary dried to obtain a crude product of 1-amino-2-(3-(cyclopropylmethoxy)-4-methoxyphenyl)-2-propanol. LC-MS: m/z 252.2 [M + H]+.
- The compound 1-amino-2-(3-(cyclopropylmethoxy)-4-methoxyphenyl)-2-propanol (251 mg, 1.0 mmol) was dissolved in ethanol (3 mL), and then 2.6-dimethyl-4H-pyran-4-one (186 mg, 1.5 mmol) and sodium hydroxide (80 mg, 2.0 mmol) were added, and stirred overnight at 60°C. The reaction was complete as indicated by LCMS. The reaction solution was rotary dried, and purified to obtain 1-(2-(3-(cyclopropylmethoxy)-4-methoxyphenyl)-2-hydroxypropyl)-2,6-dimethylpyridin-4(1H)-one (40 mg, yield 11%). LC-MS: m/z 358.0 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxypropyl)-2,6-dimethylpyridin-4(1H)-one (36 mg, 0.1 mmol) was dissolved in toluene (3 mL), and then p-toluenesulfonic acid (29 mg, 0.15 mmol) was added and stirred overnight at 110°C. The reaction was complete as indicated by TLC. The reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-methylethenyl)-2,6-dimethylpyridin-4(1H)-one (27 mg, yield 80%, white solid). 1H NMR (400MHz, CDCl3) δ 7.05-7.03 (m, 1H), 6.92-6.90 (m, 2H), 6.68(s, 2H), 6.10 (s, 1H), 3.92 (s, 3H), 3.88 (d, J = 6.8 Hz, 2H), 2.42 (s, 3H), 2.52 (s, 6H), 1.12-1.06 (m, 1H), 0.72-0.68 (m, 2H), 0.40-0.36 (m, 2H); LC-MS: m/z 340.2 [M + H]+.
-

-

-

- Trisphenyl phosphite (1.49 g, 4.80 mmol) was dissolved in anhydrous tetrahydrofuran (15 mL), and then triethyl amine (0.53 mg, 5.20 mmol) and the compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)pyridin-2,6-dimethyl-4(1H)-one (1.36 mg, 4.00 mmol) was slowly added to the reaction solution at -60°C and stirred for 15 min. Then, bromine (0.77 mg, 4.80 mmol) was slowly added to the reaction solution. After the reaction was completed, the reaction was quenched with a saturated aqueous ammonium chloride solution, and the reaction solution was directly rotary dried, and purified by column chromatography to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-bromoethenyl)-2,6-dimethylpyridin-4(1H)-one (708 mg, yield 44%). LC-MS: m/z 403.9 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-bromoethenyl)-2,6-dimethylpyridin-4(1H)-one (200 mg, 0.50 mmol) was dissolved in tetrahydrofuran (2 mL) and water (1 mL). Under nitrogen atmosphere, palladium acetate (5.5 mg, 5 mol%), sodium carbonate (5 mg, 0.50 mmol) and phenylboronic acid (73.0 mg, 0.60 mmol) were added to the reaction solution, and stirred at 80°C for 1 hr. After the reaction was completed, a saturated ammonium chloride solution (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. The reaction solution was filtered, rotary dried, and purified by reverse-phase HPLC to obtain (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-phenylethenyl)-2,6-dimethylpyridin-4(1H)-one (69 mg, yield 34%, white solid) and (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-phenylethenyl)-2,6-dimethylpyridin-4(1H)-one (30.4 mg, yield 15%, white solid).
- (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-phenylethenyl)-2,6-dimethylpyridin-4(1H)-one: 1H NMR(400 MHz, CDCl3) δ 8.40 (s, 1H), 8.35 (s, 1H), 8.06-8.04 (m, 2H), 7.68-7.66 (m, 3H), 7.26-7.20 (m, 2H), 7.04 (d, J = 8.4 Hz, 1H), 6.33 (m, 1H), 3.85 (d, J = 6.8Hz, 2H), 3.81 (s, 3H), 2.83 (s, 6H), 0.89-0.82 (m, 1H), 0.62-0.58 (m, 2H), 0.34-0.30 (m, 2H); LC-MS: m/z 401.9 [M + H]+.
- (Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-phenylethenyl)-2,6-dimethylpyridin-4(1H)-one: 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 8.39 (s, 1H), 8.12-8.08 (m, 2H), 7.65-7.62 (m, 3H), 7.32 (d, J = 8.4 Hz, 1H), 7.10-7.00 (m, 2H), 6.13 (m, 1H), 3.86(d, J = 6.8 Hz, 2H), 3.81 (s, 3H), 2.83 (s, 6H), 0.89-0.82 (m, 1H), 0.62-0.58 (m, 2H), 0.34-0.30 (m, 2H); LC-MS: m/z 401.9 [M + H]+.
-

- The specific reaction scheme is as shown below:
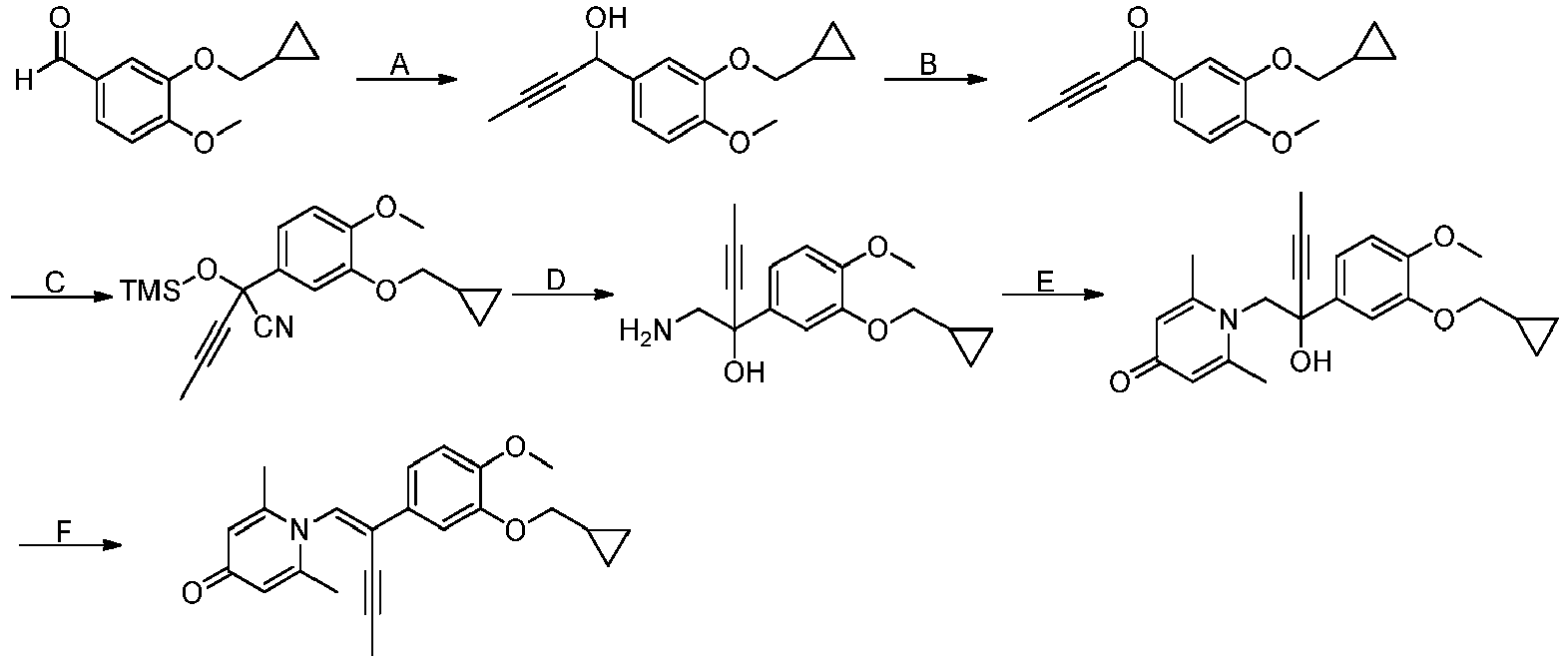
- 3-(cyclopropylmethoxy)-4-methoxybenzaldehyde (1.03 g, 5 mmol) was dissolved in anhydrous tetrahydrofuran (5 mL); and under nitrogen atmosphere, 1-ethynylmagnesium bromide (0.5 M, 12 mL, 6 mmol) was added dropwise at 0°C, warmed to room temperature, and stirred for 3 hours at room temperature. The reaction was quenched with a saturated aqueous ammonium chloride solution, and extracted with dichloromethane (3 x 30 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, and rotary dried to obtain a crude product of 1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-alkynyl-1-butanol (1.50 g). LC-MS: m/z 228.9 [M - 17]+.
- The compound 1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-alkynyl-1-butanol (1.50 g, 5 mmol) was dissolved in dichloromethane (20 mL); and Dess-Martin Periodinane (3.18g, 7.5 mmol) and solid sodium bicarbonate (6.30 g, 75 mmol) were added at 35°C, and stirred for 1 hr. After the reaction was completed, a saturated sodium bicarbonate solution was added, and extracted with dichloromethane (3 x 30 mL). The organic phases were combined, and dried over anhydrous sodium sulfate, filtered, rotary dried, and purified to obtain a crude product of 1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-alkynyl-1-butanol, which was directly used in the next reaction. LC-MS: m/z 249.9 [M + H]+.
- The compound 1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-alkynyl-1-butanone (336 mg, 1.5 mmol) was dissolved in acetonitrile (5 mL); and under nitrogen atmosphere, cesium fluoride (114 mg, 0.75 mmol) was added, trimethylsilyl cyanide (225 mg, 2.25 mmol) was slowly added dropwise at 0°C, and stirred for 1.5 hours. After the reaction was completed, the reaction solution was filtered, and rotary dried to obtain a crude product of 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(trimethylsiloxy)pent-3-ynyl nitrile. LC-MS: m/z 344.2 [M + H]+.
- The compound 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(trimethylsiloxy)-3-alkynylpentyl nitrile (413 mg, 1.5 mmol) was dissolved in anhydrous tetrahydrofuran (4 mL); and under nitrogen atmosphere, lithium aluminum hydride (114 mg, 3.0 mmol) was slowly added dropwise at 0°C, and the solution was stirred at room temperature for 1 hr. After the reaction was completed, sodium sulfate decahydrate was added to quench the reaction. The reaction solution was filtered, and the filtrate was rotary dried to obtain a crude product of 1-amino-2-(3-cyclopropylmethoxy)4-methoxyphenyl)-3-alkynyl-2-pentanol. LC-MS: m/z 276.2 [M + H]+.
- The compound 1-amino-2-(3-(cyclopropylmethoxy)-4-methoxyphenyl)-3-alkynyl-2-pentanol (413 mg, 1.5 mmol) was dissolved in ethanol (5 mL), and then 2.6-dimethylpyran-4-one (279 mg, 2.25 mmol) and sodium hydroxide (120 mg, 3.0 mmol) were added, and stirred overnight at 60°C. The reaction was complete as indicated by LCMS. The reaction solution was rotary dried, and purified to obtain 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxypent-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one (20 mg, yield 3%). 1H NMR(400 MHz, CDCl3) δ 7.12-7.07 (m, 2H), 6.86 (d, J = 8.4 Hz, 1H), 6.20 (s, 1H), 6.10 (s, 1H), 4.22-4.12 (m, 2H), 3.88 (s, 3H), 3.83 (d, J = 7.2 Hz, 2H), 2.42 (s, 3H), 2.20 (s, 3H), 1.88 (s, 3H), 1.34-1.28 (m, 1H), 0.67-0.62 (m, 2H), 0.39-0.35 (m, 2H); LC-MS: m/z 381.9 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxypent-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one (20 mg, 0.05 mmol) was dissolved in toluene (3 mL), and then p-toluenesulfonic acid (15 mg, 0.08 mmol) was added and stirred overnight at 60°C. The reaction was complete as indicated by TLC. The reaction solution was rotary dried, and purified to obtain (Z)-1-(2-(3-cyc1opropylmethoxy-4-methoxyphenyl)pent-1-en-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one (18 mg, yield 90%, white solid). 1H NMR(400 MHz, CDCl3) δ 7.27 (dd, J = 8.4, 2.0 Hz, 1H), 7.16 (J = 8.4 Hz, 1H), 6.92-6.90 (m, 2H), 6.31 (s, 2H), 3.92-3.90 (m, 5H), 2.25 (s, 6H), 1.98 (s, 3H), 1.40-1.31 (m, 1H), 0.70-0.65 (m, 2H), 0.41-0.37 (m, 2H). LC-MS: m/z 363.9 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2, 6-dimethylpyridin-4(1H)-one (2.50 g, 7.3 mmol) was dissolved in dichloromethane (50 mL); and Dess-Martin Periodinane (4.80 g, 11.0 mmol) and sodium bicarbonate (9.00 g, 110 mmol) were slowly added at 35°C, and stirred for 1 hr. After the reaction was completed, a saturated sodium bicarbonate solution (50 mL) was added, and extracted with dichloroethane (3 x 30 mL). The organic phases were combined and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification gave 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2, 6-dimethylpyridin-4(1H)-one (1.98 g, yield 80%, white solid). LC-MS: m/z 342.4 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2, 6-dimethylpyridin-4(1H)-one (500 mg, 1.5 mmol) was dissolved in acetonitrile (25 mL); and under nitrogen atmosphere, triethyl amine (456 mg, 4.5 mmol) was added, trimethylsilyl cyanide (600 mg, 6.0 mmol) was slowly added dropwise at 0°C, and stirred for 4 hours. The reaction solution was rotary dried to obtain a crude product of 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-cyano-2-(trimethylsilyloxy)ethyl)-2, 6-dimethylpyridin-4(1H)-one. LC-MS: m/z 441.4 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-cyano-2-(trimethylsilyloxy)ethyl)-2, 6-dimethylpyridin-4(1H)-one (660 mg, 1.5 mmol) was dissolved in acetonitrile (25 mL), and aqueous hydrochloric acid (3M, 1.5 mL) was added and stirred at 80°C for 0.5 hours. The reaction was complete as indicated by TLC. The reaction solution was rotary dried to obtain a crude product of 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-cyano-2-hydroxyethyl)-2, 6-dimethylpyridin-4(1H)-one. LC-MS: m/z 369.4 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-cyano-2-hydroxyethyl)-2, 6-dimethylpyridin-4(1H)-one (552 mg, 1.5 mmol) was dissolved in chloroform (25 mL), and then thionyl chloride (1.0 mL) was added and stirred for 0.5 hours at 80°C. The reaction was complete as indicated by TLC. The reaction solution was rotary dried, and purified to obtain (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-cyanoethenyl)-2,6-dimethylpyridin-4(1H)-one (283 mg, 54%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.44 (s, 1H), 7.29 (d, J = 2.0 Hz, 1H), 7.14 (d, J =1.2 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 6.36 (s, 2H), 3.95-3.92 (m, 5H), 2.30(s, 6H), 1.38-1.30 (m, 1H), 0.71-0.67 (m, 2H), 0.42-0.38 (m, 2H). LC-MS: m/z 351.4 [M + H]+.
-

- The specific reaction scheme is as shown below:

- (E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-cyanoethenyl)-2,6-dimethylpyridin-4(1H)-one (500 mg, 1.4 mmol) was dissolved in acetonitrile (10 mL), and then potassium hydroxide (10 wt%, 10 mL) was added at room temperature and stirred at 80°C for 2 hours. The reaction was complete as indicated by TLC. The reaction solution was directly rotary dried, and purified by column chromatography to obtain (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylamide (140 mg, 27%, white solid). 1H NMR(400 MHz, DMSO-d6 ) δ 7.73 (s, 1H), 7.56 (s, 1H), 7.13 (s, 1H), 7.04 (s, 2H), 6.99 (s, 1H), 5.96 (s, 2H), 3.85 (d, J = 7.2 Hz, 2H), 3.80 (s, 3H), 2.27 (m, 6H), 0.87-0.84 (m, 1H), 0.61-0.56 (m, 2H), 0.35-0.31 (m, 2H); LC-MS: m/z 369.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylamide (30 mg, 0.08 mmol) was dissolved in N, N-dimethylformamide (0.5 mL), and then 60% sodium hydride (16 mg, 0.4 mmol) was added, and stirred at 70°C for 0.5 hours. Iodomethane (12 mg, 0.08 mmol) was added to the reaction solution, and stirred at 70°C for another 1 hr. The reaction solution was quenched with a saturated aqueous ammonium chloride solution, and the reaction solution was directly rotary dried, and purified by column chromatography to obtain (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2, 6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N-methylacrylamide (5 mg, 17%, white solid). 1H NMR(400 MHz, CDCl3) δ 7.04-7.02 (m, 1H), 6.93-6.91 (m, 2H), 6.63(s, 1H), 6.28 (s, 2H), 6.02 (s, 1H), 3.92 (s, 3H), 3.88 (d, J = 6.8 Hz, 2H), 2.81 (t, J = 4.8 Hz, 3H), 2.34 (s, 6H), 0.97-0.83 (m, 1H), 0.76-0.62 (m, 2H), 0.40-0.36 (m, 2H); LC-MS: m/z 383.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylamide (30 mg,0.08 mmol) was dissolved in N, N-dimethylformamide (0.5 mL), and then 60% sodium hydride (16 mg,0.4 mmol) was added, and stirred at 70°C for 0.5 hours. Iodomethane (24 mg, 0.16 mmol) was added to the reaction solution, and stirred at 70°C for another 1 hr. The reaction was quenched with a saturated aqueous ammonium chloride solution, and the reaction solution was directly rotary dried and purified by column chromatography to obtain (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-dimethylacrylamide (10 mg, 50%, white solid). 1H NMR(400MHz,CDCl3) δ 6.70-6.97 (m, 1H), 6.93-6.91 (m, 2H), 6.70(s, 1H), 6.27 (s, 2H), 3.92 (s, 3H), 3.88 (d, J = 6.8 Hz, 2H), 2.92 (s, 3H), 2.78 (s, 3H), 2.37 (s, 6H), 1.37-1.29 (m, 1H), 0.69-0.65 (m, 2H), 0.40-0.36 (m, 2H). LC-MS: m/z 397.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylamide (20 mg, 0.06 mmol) was dissolved in N, N-dimethylformamide (0.5 mL), and then 60% sodium hydride (12 mg, 0.3 mmol) was added, and stirred at 70°C for 0.5 hours. Iodoethane (19 mg, 0.12mmol) was added to the reaction solution, and stirred at 70°C for another 1 hr. The reaction was quenched with a saturated aqueous ammonium chloride solution, and the reaction solution was directly rotary dried and purified by column chromatography to obtain (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-diethylacrylamide (6 mg, 30%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.04-7.01 (m, 1H), 6.95-6.91 (m, 2H), 6.72(s, 1H), 6.41 (s, 2H), 3.92 (s, 3H), 3.88 (d, J = 7.2 Hz, 2H), 3.34-3.31 (m, 2H), 3.18 (q, J = 6.8 Hz, 2H), 1.01 (t, J = 7.2 Hz, 3H), 0.90-0.82 (m, 1H), 0.81 (t, J = 7.2 Hz, 3H), 0.69-0.65 (m, 2H), 0.40-0.36 (m, 2H); LC-MS: m/z 425.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-diethylacrylamide (60.0 mg, 0.16 mmol) was dissolved in N,N-dimethylformamide (5 mL). 4-dimethylaminopyridine (4.0 mg, 0.03 mmol) and triethyl amine (36 mg, 0.36 mmol) were added at 35°C, and then di-tert butyl dicarbonate (71 mg, 0.33 mmol) was slowly added to the reaction solution and stirred for 2 hours. After the reaction was completed, the reaction solution was rotary dried to obtain a crude product of (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-di-tert butyloxycarbonylacrylamide. LC-MS: m/z 569.2 [M + H]+.
- The compound (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-di-tert butyloxycarbonylacrylamide (90 mg, 0.16 mmol) was dissolved in methanol (2 mL), and a sodium hydroxide solution (2N, 1 mL) was added to the reaction solution at 35°C and stirred for 2 hours. After the reaction was completed, water (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified to obtain (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylic acid (23.1mg, yield 39%, white solid). 1H NMR (400MHz, CDCl3) δ 7.18-7.16 (d, J = 8.0 Hz, 1H), 7.11(s, 2H), 6.94 (d, J = 8.0 Hz, 1H), 6.82 (s, 1H), 3.92 (s, 3H), 3.90(d, J = 6.4 Hz, 2H), 2.6 (s, 6H), 1.33-1.27 (m, 1H), 0.68-0.63 (m, 2H), 0.41-0.35 (m, 2H). LC-MS: m/z 370.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylic acid (10 mg, 0.03 mmol) was dissolved in dichloromethane (0.5 mL), and one drop of N,N-dimethylformamide was added to the reaction solution. Oxalyl chloride (8 mg, 0.03 mmol) was slowly added to the reaction solution, and stirred for 1 hr. Then, methanol (0.5 mL) was added to the reaction solution. After the reaction was completed, the reaction was quenched with a saturated aqueous ammonium chloride solution, and the reaction solution was directly rotary dried and purified by column chromatography to obtain (Z)-methyl 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylate (3 mg, yield 26%, white solid). 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H),7.95 (s, 2H), 7.15 (s, 1H), 7.09(d, J = 8.4Hz, 1H), 6.94(d, J = 8.4Hz, 1H), 3.94-3.93 (m, 5H), 3.70 (s, 3H), 2.83 (s, 6H), 1.70-1.59 (m, 1H), 0.68-0.63 (m, 2H), 0.40-0.39(m, 2H); LC-MS: m/z 384.3 [M + H]+.
-

- The specific reaction scheme is as shown below:

- (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylic acid (10 mg, 0.03 mmol) was dissolved in dichloromethane (0.5 mL), and one drop of N,N-dimethylformamide was added to the reaction solution. Oxalyl chloride (8 mg, 0.03 mmol) was slowly added to the reaction solution, and stirred for 1 hr. Then, ethanol (0.5 mL) was added to the reaction solution. After the reaction was completed, the reaction was quenched with a saturated aqueous ammonium chloride solution, and the reaction solution was directly rotary dried and purified by column chromatography to obtain (Z)-ethyl 2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylate (4 mg, yield 34%, white solid). 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 7.65 (s, 2H), 7.28 (s, 1H), 7.16 (d, J = 8.0 Hz, 1H), 6.92(d, J = 8.4 Hz, 1H), 4.18 (q, J = 7.2 Hz, 2H), 4.00(d, J = 6.8 Hz, 2H), 3.92(m, 3H), 2.92 (s, 6H), 1.45-1.32 (m, 1H), 1.15(t, J = 14.4 Hz, 3H), 0.67-0.63 (m, 2H), 0.44-0.40 (m, 2H); LC-MS: m/z 398.3 [M + H]+.
-

- The specific reaction scheme is as shown below:

- At room temperature, 4-(difluoromethoxy)-3-hydroxybenzaldehyde (0.964 g, 5.13 mmol) was dissolved in acetonitrile (10 mL), and then potassium carbonate (1.06 g, 7.69 mmol) and bromomethylcyclopropane (0.9 g, 6.67 mmol) were added in sequence, and heated to 80°C for 3 hours with stirring under nitrogen atmosphere. After the reaction was completed, water (30 mL) was added, and extracted with dichloromethane (3 x 30 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified to obtain 3-cyclopropylmethoxy-4-difluoromethoxybenzaldehyde (1.16 g, yield 93%). LC-MS: m/z 343.2 [M + H]+.
- The compound 3-cyclopropylmethoxy-4-difluoromethoxybenzaldehyde (1.16 g, 4.79 mmol) was dissolved in anhydrous tetrahydrofuran (30 mL), and then prop-1-yn-1-ylmagnesium bromide (0.5 N, 19.2 mL, 9.6 mmol) was slowly added in an ice bath, and stirred for 16 hours at room temperature under nitrogen atmosphere. After the reaction was completed, the reaction solution was quenched with water (30 mL), and extracted with ethyl acetate (3 x 60 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, rotary dried, and purified to obtain 1-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)but-2-yn-1-ol (1.01 g, 74%).
- The compound 1-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)but-2-yn-1-ol (1.01 g, 3.58 mmol) was dissolved in dichloromethane (50 mL), and then Dess-Martin periodinane (3.04 g, 7.16 mmol) were added and stirred overnight at room temperature. After the reaction was completed, the reaction solution was filtered, rotary dried, and purified to obtain 1-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)but-2-yn-1-one (0.96 g, yield 96%).
- The compound 1-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)but-2-yn-1-one (0.96 g, 3.43 mmol) was dissolved in dichloromethane (10 mL), and then triethyl amine (0.693 g, 6.86 mmol) and trimethylsilyl cyanide (1.02 g, 10.29 mmol) were added in sequence in an ice bath and stirred for 16 hours under nitrogen atmosphere at room temperature. After the reaction was completed, the reaction solution was directly rotary dried to obtain a crude product of 2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-(trimethylsiloxy)pent-3-ynyl nitrile, which was directly used in the next reaction.
- The compound 2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-(trimethylsiloxy)pent-3-ynyl nitrile (1.30 g, 3.43 mmol) was dissolved in anhydrous tetrahydrofuran (40 mL); and then lithium aluminum hydride (0.391 g, 10.29 mmol) was added portion-wise in an ice bath, and stirred overnight at room temperature. After the reaction was completed, water (0.4 mL), a 15% aqueous sodium hydroxide solution (0.4 mL), and water (1.2 mL) were added in sequence, stirred for half an hour, dried over anhydrous sodium sulfate, filtered, and rotary dried to obtain a crude product of 1-amino-2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)pent-3-yn-2-ol, which was directly used in the next reaction.
- The compound 1-amino-2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)pent-3-yn-2-ol (1.07 g, 3.43 mmol) was dissolved in ethanol (40 mL), and then 2,6-dimethyl-4H-pyran-4-one (0.553 g, 4.46 mmol), sodium hydroxide (206 mg, 5.15 mmol) and water (6 mL) were added in sequence, heated to 60°C, and stirred for 72 hours under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried, and purified by column chromatography to obtain 1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-hydroxypent-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one (120 mg, yield 8%). LC-MS: m/z 418.2 [M + H]+.
- The compound 1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-hydroxypent-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one (120 mg, 0.29 mmol) was dissolved in trichloromethane (10 mL), and heated to 90°C. Then thionyl chloride (0.5 mL) was added and stirred at 90°C for 15 min. After the reaction was completed, the reaction solution was directly rotary dried to obtain a crude product of 1-(2-chloro-2-(3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl)pent-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one. LC-MS: m/z 436.2 [M + H]+.
- The compound 1-(2-chloro-2-(3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl)pent-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one (125 mg, 0.29 mmol) was dissolved in ethanol (10 mL), and then sodium hydroxide (72 mg, 1.8 mmol) and water (2 mL) were added in sequence, heated to 90°C, and stirred for 16 hours under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried, and purified by reverse-phase HPLC to obtain the product (Z)-1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)pent-1-en-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one (2.3 mg, yield 2%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.38-7.28 (m, 3H), 7.21 (d, J = 8.0 Hz, 1H), 7.01 (s, 2H), 6.69 (t, J = 75.2 Hz, 1H), 3.97 (d, J = 6.4 Hz, 2H), 2.48 (s, 6H), 1.95 (s, 3H), 1.35-1.25 (m, 1H), 0.70-0.65 (m, 2H), 0.42-0.36 (m, 2H); LC-MS: m/z 400.1 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (Z)-3-(2,6dimethyl-4-carbonylpyridine)-1(4H)-yl)-2-(3-hydroxy4-methoxyphenyl)acrylonitrile (100 mg, 0.34 mmol) was dissolved in acetonitrile (10 mL), and then iodomethane (96 mg, 0.68 mmol) and potassium carbonate (187 mg, 1.35 mmol) were added and stirred for 5 hours at 85°C under nitrogen atmosphere. After the reaction was completed, the reaction solution was filtered, and the filtrate was rotary dried, and purified by reverse-phase HPLC to obtain (Z)-2-(3,4-dimethoxyphenyl)-3-(2,6dimethyl-4-carbonylpyridine)-1(4H)-yl)acrylonitrile (21 mg, yield 33%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.65 (s, 1H), 7.29 (dd, J = 8.4, 2.0 Hz, 1H), 7.18 (d, J = 2.0 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 6.51 (s, 2H), 3.98 (s, 3H), 3.96 (s, 3H), 2.37 (s, 6H); LC-MS m/z 311.2 [M + H]+.
-

-

- The specific reaction scheme is as shown below:

- 2,6-dimethyl-4H-pyran-4-one (620 mg, 5 mmol) and aminoacetonitrile hydrochloride (463 mg, 6 mmol) were dissolved in pyridine (10 mL), and stirred for 46 hours at 80°C under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried, and purified by column chromatography to obtain 2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acetonitrile (583 mg, yield 72%, white solid). LC-MS: m/z 163.1 [M + H]+.
- 2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acetonitrile (162 mg, 1 mmol) and 3-cyclopropylmethoxy-4-methoxybenzaldehyde (206 mg, 1mmol) were dissolved in pyridine (10 mL), and stirred overnight at 100°C under nitrogen atmosphere. After the reaction was completed, the reaction solution was rotary dried to remove pyridine, and purified by reverse-phase HPLC to obtain (E)-3-(3-cyclopropylmethoxy-4methoxyphenyl)-2(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylonitrile (126 mg, yield 36%, white solid) and (Z)-3-(3-cyclopropylmethoxy-4methoxyphenyl)-2(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylonitrile (20 mg, yield 6%, white solid).
- (E)-3-(3-cyclopropylmethoxy-4-methoxyphenyl)-2(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylonitrile. 1H NMR (400 MHz, DMSO-d6 ) δ 8.85 (s, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.07 (dd, J = 8.4, 2.0 Hz, 1H), 6.65 (d, J = 2.0 Hz, 1H), 6.38 (s, 2H), 3.92 (s, 3H), 3.88 (d, J = 6.8 Hz, 2H), 2.22 (s, 6H), 1.26-1.17 (m, 1H), 0.65-0.58 (m, 2H), 0.32-0.26 (m, 2H); LC-MS: m/z 351.2 [M + H]+.
- (Z)-3-(3-cyclopropylmethoxy-4methoxyphenyl)-2(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylonitrile. 1H NMR (400 MHz, DMSO-d6 ) δ 7.58 (s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 6.97 (dd, J = 8.4, 1.6 Hz, 1H), 6.69 (d, J = 1.6 Hz, 1H), 6.09 (s, 2H), 3.80 (s, 3H), 3.53 (d, J = 6.8 Hz, 2H), 3.34 (s, 6H), 2.02 (s, 6H), 1.16-1.11 (m, 1H), 0.56-0.52 (m, 2H), 0.27-0.23 (m, 2H); LC-MS: m/z 351.2 [M + H]+.
-

- The specific reaction scheme is as shown below:

- (E)-3-(3-cyclopropylmethoxy-4methoxyphenyl)-2(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylonitrile (350 mg, 1 mmol) was dissolved in acetonitrile (10 mL), and then potassium hydroxide (10 wt%, 10 mL) was added at room temperature and stirred at 80°C for 2 hours. After the reaction was completed, the reaction solution was directly rotary dried, and purified by reverse-phase HPLC to obtain (Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylamide (247 mg, 67%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.82 (s, 1H), 7.75 (s, 1H), 7.54 (s, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.87 (dd, J = 8.4, 2.0 Hz, 1H), 6.38 (d, J = 2.0 Hz, 1H), 6.14 (s, 2H), 3.80 (s, 3H), 3.51 (d, J = 6.8 Hz, 2H), 1.94 (s, 6H), 1.16-1.07 (m, 1H), 0.56-0.51 (m, 2H), 0.27-0.24 (m, 2H); LC-MS: m/z 369.4 [M + H]+.
-

- The specific reaction scheme is as shown below:

- The compound (E)-3-(3-(cyclopropylmethoxy)-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridine)-1(4H)acrylamide (37 mg, 0.1 mmol) was dissolved in N,N-dimethylformamide (2 mL), and then 60% sodium hydride (24 mg, 0.6 mmol) was added at 0°C, and stirred for half an hour. Then, iodomethane (43 mg, 0.3 mmol) was added, and stirred at room temperature for 4 hours. After the reaction was completed, saturated saline (10 mL) was added, and extracted with dichloromethane (3 x 10 mL). The organic phases were combined, and dried over anhydrous sodium sulfate. Filtration, rotary drying, and purification by reverse-phase HPLC gave (E)-3-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridine)-1(4H)-yl)-N, N-dimethylacrylamide (22 mg, yield 55%, white solid). 1H NMR (400 MHz, DMSO-d6 ) δ 7.28 (s, 1H), 7.03 (d, J = 8.4 Hz, 1H), 6.87 (dd, J = 8.4, 1.6 Hz, 1H), 6.49 (d, J = 1.6 Hz, 1H), 6.09 (s, 2H), 3.80 (s, 3H), 3.53 (d, J = 6.8 Hz, 2H), 3.34 (s, 6H), 2.02 (s, 6H), 1.16-1.11 (m, 1H), 0.56-0.52 (m, 2H), 0.27-0.23 (m, 2H); LC-MS: m/z 397.2 [M + H]+.
- The inhibitory effect of the compounds on human PDE4B 1 enzyme activity was determined by quantifying the 5'-adenosine monophosphate (5'-AMP) formed from 3',5'-cyclic adenosine monophosphate (cAMP).
- The test compound or water (control) and human recombinant PDE4B1 enzyme (4.8 U) were mixed in a buffer solution (pH 7.4) consisting of 1x Hanks' balanced salt solution (HBSS), 5 mM HEPES, 3 mM MgCl2, and 0.1% BSA and incubated for 10 min. A cAMP enzyme substrate (final concentration 40 nM) was added, and the mixture was incubated at room temperature for 60 min. Then a fluorescent acceptor (Dye2 labeled with cAMP), a fluorescent donor (anti-cAMP antibody labeled with europium cryptate) and a non-specific phosphodiesterase inhibitor IBMX (3-isobutyl-1-methyl xanthine; final concentration 1 mM) were added. After 60 min, the fluorescence transfer related to the amount of remaining cAMP was measured on a microplate reader (Rubystar, BMG) at λex = 337 nm, λem = 620 nm and λem = 665 nm. The enzyme activity is calculated from the ratio of the signals measured at 665 nm and 620 nm. The results are expressed as percent inhibition of the enzyme activity of the control (without PDE4 inhibitor). The enzyme was omitted for measurement of the basic control. The IC50 value (IC50 = the concentration that caused the half-maximum inhibition of the specific activity of the control) is derived from dose-response measurements with eight different concentrations (n=2; repeated 2 times)
- The experimental results obtained are listed in Table 1.
Example IC50 (nM) Example IC50 (nM) Example IC50 (nM) 1 760 42 8.2 83-2 106 2 125 43 4.9 84 230 3 39 44 30 85 3.8 4 36 45 170 86-1 310 5 5 46 25 86-2 81 6 19 47 7.1 87 78 7 5.7 48 420 88 2.8 8 23 49 2.9 89 6.9 9 25 50 180 90 3.8 10 3300 51 57 91 75 11 44 52 N.A. 92 94 12 N.A. 53 15 93 12.9 13 4.9 54 26 94 137 14 21 55 54 95 260 15 33 56 16 96 2.4 16 10.3 57 100 97 3.7 17 16 58 5.5 98-1 97 18 4.5 59 5.1 98-2 12.5 19 4.6 60 1200 99 59 20 13 61 6.2 100 1.5 21 4.6 62 10.2 101-1 99 22 20 63 11.1 101-2 26 23 5 64 43 102 50 24 23 65 520 103 14 25 11.5 66 270 104-1 1700 26 39 67 170 104-2 8400 27 65 68 27 105 3 28 92 69 5200 106 2.7 29 93 70 8.1 107 450 30 116 71 1420 108 760 31 26 72 3900 109 180 32 13.9 73 2000 110 460 33 830 74 38 111 6600 34 420 75 290 112 138 35 32 76 105 113 170 36 1250 77 26.4 114 1.6 37 510 78 36 115 9.9 38-1 1090 79 160 116-1 460 38-2 71 80 7 116-2 35 39 1070 81 1.15 117 3600 40 6.8 82 7.5 118 320 41 1.22 83-1 550 - The effect of the compounds on the release characteristics of TNFa cytokines in frozen human peripheral blood mononuclear cells (PBMCs) was determined.
- Fresh human PBMCs were isolated, and suspended in a fresh medium. The PBMC medium (CM) is RPMI1640 containing 10% FBS, 1% PBS, and 2 mM L-glutamine. The cells were plated at a density of 2*10^5 cells/well (100ul). The test compound was dissolved in DMSO to form a 10 mM sample (DMSO, 100%). The compound was diluted with PBMC medium to the required concentrations. The sample (50 uL) was incubated for 1 hr with cells at 37°C, 5% CO2, after which the inducer (LPS, 1ug/mL) was added. The inducer+vehicle (LPS+0.05%DMS0) was used as a control in this experiment. The vehicle without inducer was used as a negative control. Crisaborole was used as a positive control. After 24 hours of incubation, the supernatant was extracted and stored at -80°C. The supernatant was thawed and the TNFα level in the supernatant was measured by Luminex4-plex assay.
- Using this experimental method, it is determined that the EC50 of Example 77 is = 11 nM, the EC50 of Example 80 is <5 nM; and the EC50 of Example 88 is <5 nM.
- Test of Inhibition on allergic dermatitis on mouse ear induced by locally administered phorbol ester
- The test compounds are used to treat various skin diseases such as itching, redness, dryness, crusting, desquamation, inflammation and discomfort.
- The test compound was topically applied to the right ear of the test animal 30 minutes before and 15 minutes after the administration of (12-)phorbol myristate (-13-) acetate (PMA). The dosing volume was 20 ul/ear for a solvent vehicle or 20 mg/ear for a cream.
- Male CD-1 mice weighing 24±2g were used. All animals were maintained in an environment at a controlled temperature (22°C-23°C) and humidity (70%-80%), with a 12-h-light and 12-h-dark photoperiod. The animals were bred in the animal room for at least 1 week before they are used for experimental test. The mice were allowed to access food and drinking water freely. Generally, all aspects of this work were carried out in accordance with the guidelines of Care and Use of Laboratory Animals (National Academy Press, Washington, D.C., 1996), including animal breeding, experimentation, and handling.
- Each group had 5 mice. PMA (4 pg in 20 uL acetone) was applied locally to the front and back surfaces of the right ear of each animal. The vehicle (ethanol: acetone/1:1, 20 µL/ear) or test compound (specified dose in 20 uL of 1:1 acetone: ethanol, for each ear) was applied 30 minutes before and 15 minutes after PMA administration, and crisaborole was administered as a positive control. Then, 6 hours after PMA administration, ear swelling was measured as an indicator of inflammation using a staining micrometer. The percentage of inhibition is calculated according to the following formula: ([IC-IT]/IC)X100%, where IC and IT means the increase in ear thickness (mm) of mice in the control and treatment groups, respectively. 30% or more inhibition is considered to have significant anti-inflammatory activity.
- Compounds of the present invention, such as compound 88, can be administered as a gel, lotion, ointment and solution, and the route of administration includes, but is not limited to, local administration, instillation, aerosol, transdermal patch, via insertion, or oral administration.
- The following is a preparation method of 1% ointment preparation (weight percentage):
1% compound, 15% PEG400, 0.02% butylated hydroxytoluene, 2% span 80, 10% white wax, and 71.98% white petrolatum were mechanically stirred until an ointment was formed.
Claims (16)
- A compound, wherein the compound has a structure shown below:
 wherein RA is hydrogen, alkyl comprising from 1 to 6 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is three-membered, four-membered, five-membered or six-membered cycle, or halogen;RB is hydrogen, alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is a three-membered, four-membered, five-membered or six-membered cycle, heterocycloalkyl which is an oxo/thia three-membered, four-membered, five-membered or six-membered cycle, alkenyl comprising from 2 to 4 carbon atoms, or alkynyl comprising from 2 to 4 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is a three-membered, four-membered, five-membered or six-membered cycle, or halogen, and one or more carbon atom(s) in the groups is/are optionally substituted with a sulfur atom, sulfoxide, sulfone, or sulfonyl;RC is hydrogen;RD1 is hydrogen, oxygen, nitrogen, hydroxy, cyano, alkyl comprising from 1 to 6 carbon atoms, phenyl, an ester group, carboxyl, or alkynyl comprising from 2 to 4 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon/oxygen in the groups is/are optionally substituted with alkyl comprising 1 to 6 carbon atoms, cycloalkyl which is a three-membered, a four-membered, five-membered or six-membered ring, alkynyl comprising from 2 to 4 carbon atoms, alkenyl comprising from 2 to 4 carbon atoms, or phenyl, and one or more carbon atom(s) in the groups is/are optionally substituted with a sulfur atom;C1-RD1 bond is a single bond or a double bond;RD2 is hydrogen or cyano;G1 is a single bond, a double bond or cyclopropane comprising Ci and C2; andRE is a group where one or more hydrogen atom(s) attached to carbon on the pyridinone ring is/are substituted with alkyl comprising from 1 to 6 carbon atoms or halogen.
wherein RA is hydrogen, alkyl comprising from 1 to 6 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is three-membered, four-membered, five-membered or six-membered cycle, or halogen;RB is hydrogen, alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is a three-membered, four-membered, five-membered or six-membered cycle, heterocycloalkyl which is an oxo/thia three-membered, four-membered, five-membered or six-membered cycle, alkenyl comprising from 2 to 4 carbon atoms, or alkynyl comprising from 2 to 4 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl comprising from 1 to 6 carbon atoms, cycloalkyl which is a three-membered, four-membered, five-membered or six-membered cycle, or halogen, and one or more carbon atom(s) in the groups is/are optionally substituted with a sulfur atom, sulfoxide, sulfone, or sulfonyl;RC is hydrogen;RD1 is hydrogen, oxygen, nitrogen, hydroxy, cyano, alkyl comprising from 1 to 6 carbon atoms, phenyl, an ester group, carboxyl, or alkynyl comprising from 2 to 4 carbon atoms, and in the above groups one or more hydrogen atom(s) attached to carbon/oxygen in the groups is/are optionally substituted with alkyl comprising 1 to 6 carbon atoms, cycloalkyl which is a three-membered, a four-membered, five-membered or six-membered ring, alkynyl comprising from 2 to 4 carbon atoms, alkenyl comprising from 2 to 4 carbon atoms, or phenyl, and one or more carbon atom(s) in the groups is/are optionally substituted with a sulfur atom;C1-RD1 bond is a single bond or a double bond;RD2 is hydrogen or cyano;G1 is a single bond, a double bond or cyclopropane comprising Ci and C2; andRE is a group where one or more hydrogen atom(s) attached to carbon on the pyridinone ring is/are substituted with alkyl comprising from 1 to 6 carbon atoms or halogen. - The compound according to claim 1, wherein
G1 is a three-membered ring having a specific structure shown below:
- A method for preparing the compound according to claim 1, comprising:using a 3-hydroxybenzaldehyde derivative A as a starting material, and substituting the hydrogen in the of the 3-hydroxybenzaldehyde derivative A with RB to obtain an intermediate product B;reacting the intermediate product B with trimethylsilyl cyanide to obtain an intermediate product C;reducing the intermediate product C to obtain an intermediate product D having an amino group; andreacting the amino group in the intermediate product D with a six-membered oxygen-containing cyclic compound to obtain a type-A target product,wherein the 3-hydroxybenzaldehyde derivative A is a compound having a structure shown below:
 the intermediate product B is a compound having a structure shown below:
the intermediate product B is a compound having a structure shown below: the intermediate product C is a compound having a structure shown below:
the intermediate product C is a compound having a structure shown below: the intermediate product D is a compound having a structure shown below:
the intermediate product D is a compound having a structure shown below: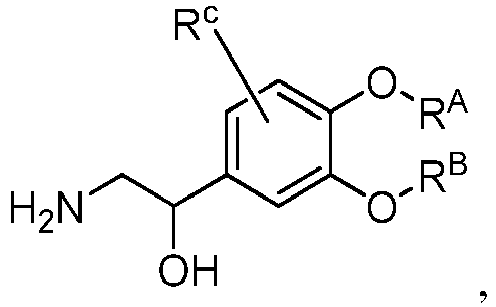 the type-A target product is a compound having a structure shown below:
the type-A target product is a compound having a structure shown below:
- A method for preparing the compound according to claim 3, comprising:i) subjecting the hydroxyl group on the middle bridge of the type-A target product to an addition/substitution reaction to obtain a type-A-1 target product,wherein the type-A-1 target product is a compound having a structure shown below:
 in which Rd is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl;orii) oxidizing the hydroxyl group on the middle bridge of the type-A target product to obtain a type-B target product,
in which Rd is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl;orii) oxidizing the hydroxyl group on the middle bridge of the type-A target product to obtain a type-B target product,
wherein the type-B target product is a compound having a structure shown below: iii) removing the hydroxyl group on the middle bridge of the type-A target product to obtain a type-C target product,
iii) removing the hydroxyl group on the middle bridge of the type-A target product to obtain a type-C target product,
wherein the type-C target product is a compound having a structure shown below:
- The method for preparing the compound according to claim 4, comprising:iv) deprotecting the group RB on the type-B targetproduct to obtain a type-B-1 target product,wherein the type-B-1 target product is a compound having a structure shown below:
 v) oximating the carbonyl group on the middle bridge of the type-B target product to obtain a type-B-3 target product,wherein the type-B-3 target product is a compound having a structure shown below:
v) oximating the carbonyl group on the middle bridge of the type-B target product to obtain a type-B-3 target product,wherein the type-B-3 target product is a compound having a structure shown below: vi) reducing and then eliminating the carbonyl group on the middle bridge of the type-B target product to obtain a type-C target product,wherein the type-C target product is a compound having a structure shown below:
vi) reducing and then eliminating the carbonyl group on the middle bridge of the type-B target product to obtain a type-C target product,wherein the type-C target product is a compound having a structure shown below: vii) reacting the carbonyl group on the middle bridge of the type-B target product with a halogenating reagent to obtain an intermediate product X1, andreplacing the halogen in the intermediate product X1 to obtain a type-C-3 target product,wherein the intermediate product X1 is a compound having a structure shown below:
vii) reacting the carbonyl group on the middle bridge of the type-B target product with a halogenating reagent to obtain an intermediate product X1, andreplacing the halogen in the intermediate product X1 to obtain a type-C-3 target product,wherein the intermediate product X1 is a compound having a structure shown below: the type-C-3 target product is a compound having a structure shown below:
the type-C-3 target product is a compound having a structure shown below: in which Rd-2 is aryl, alkyl, cycloalkyl, an ether group, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the group is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl;
in which Rd-2 is aryl, alkyl, cycloalkyl, an ether group, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the group is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl;
orviii) reacting the carbonyl group on the middle bridge of the type-B target product with trimethylsilyl cyanide to obtain an intermediate product Yl, andsubjecting the intermediate product Y1 to reduction and elimination to obtain a type-C" target product,wherein the intermediate product Y1 is a compound having a structure shown below: the type-C" target product is a compound having a structure shown below:
the type-C" target product is a compound having a structure shown below:
- A method for preparing the compound according to claim 5, comprising:subjecting the hydroxyl group on the benzene ring of the type-B-1 target product to an addition/substitution reaction to obtain a type-B-2 target product,wherein the type-B-2 target product is a compound having a structure shown below:
 in which Rb is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl;
in which Rb is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl;
orsubjecting the hydroxyl group on the oxime of the type-B-3 target product to an addition/substitution reaction to obtain a type-B-4 target product,wherein the type-B-4 target product is a compound having a structure shown below: in which Rd-1 is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl.
in which Rd-1 is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl. - The method for preparing the compound according to claim 4, comprising:using a 3-hydroxyacetophenone derivative I as a starting material, and substituting the hydrogen in the of the 3-hydroxyacetophenone derivative I with RB to obtain an intermediate product II;reacting the intermediate product II in the presence of a halogenating agent to obtain an intermediate product III; andreacting the halogen of the intermediate product III with a six-membered oxygen-containing cyclic compound to obtain the type-B target product,wherein the 3-hydroxyacetophenone derivative I is a compound having a structure shown below:
 the intermediate product II is a compound having a structure shown below:
the intermediate product II is a compound having a structure shown below: the intermediate product III is a compound having a structure shown below:
the intermediate product III is a compound having a structure shown below: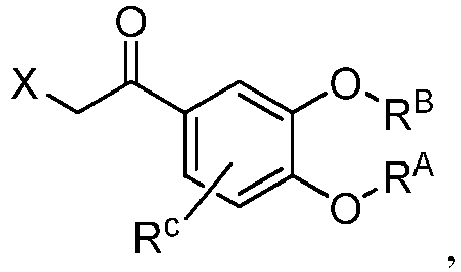
- A method for preparing the compound according to claim 6, comprising:reducing and then eliminating the carbonyl group on the middle bridge of the type-B-2 target product to obtain a type-C-1 target product,wherein the type-C-1 target product is a compound having a structure shown below:

- A method for preparing the compound according to claim 8, comprising:subjecting the hydroxyl group on the phenyl ring of the type-C-1 target product to an addition/substitution reaction to obtain a type-C-2 target product,wherein the type-C-2 target product is a compound having a structure shown below:
 in which Rb-1 is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl.
in which Rb-1 is alkyl, cycloalkyl, or an ester group, and in the above groups one or more hydrogen atom(s) attached to carbon in the groups is/are optionally substituted with alkyl, alkynyl, alkenyl, cycloalkyl, aryl, halogen, hydroxy, thio, cyano, or thioalkyl. - A method for preparing the compound according to claim 1, comprising:reacting an acetophenone derivative 1 as a starting material with trimethylsilyl cyanide, to obtain an intermediate product 2;reducing the intermediate product 2 to obtain an intermediate product 3; andreacting the amino group in the intermediate product 3 with a six-membered oxygen-containing cyclic compound to obtain a type-A' target product,wherein the acetophenone derivative 1 is a compound having a structure shown below:
 the intermediate product 2 is a compound having a structure shown below:
the intermediate product 2 is a compound having a structure shown below: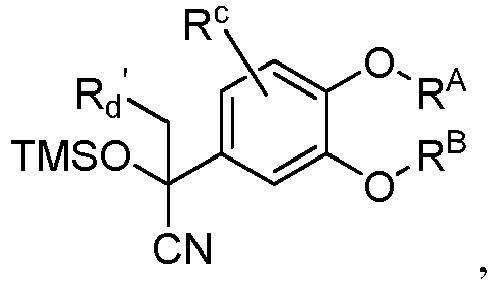 the intermediate product 3 is a compound having a structure shown below:
the intermediate product 3 is a compound having a structure shown below: the type-A' target product is a compound having a structure shown below:
the type-A' target product is a compound having a structure shown below:
- A method for preparing the compound according to claim 10, comprising:removing the hydroxyl group on the middle bridge of the type-A' target product to obtain a type-C' target product,wherein the type-C' target product is a compound having a structure shown below:

- The method for preparing the compound according to claim 1, comprising:reacting a six-membered N-acetonitrile compound Z1 with a benzaldehyde derivative Z2 to obtain a type-C" target product,wherein the six-membered N-acetonitrile compound Z1 is a compound having a structure shown below:
 the benzaldehyde derivative Z2 is a compound having a structure shown below:
the benzaldehyde derivative Z2 is a compound having a structure shown below:
- A method for preparing a compound according to claim 1, comprising:using a cinnamic acid derivative 1 as a starting material, and esterifying the cinnamic acid derivative 1 to obtain an intermediate product 2;forming a ring from the double bond on the intermediate product 2 to obtain an intermediate product 3;hydrolyzing the terminal ester group on the intermediate product 3 to give an intermediate product 4 having a carboxyl group;aminating the terminal carboxyl group of the intermediate product 4 to obtain an intermediate product 5; andreacting the intermediate product 5 with a six-membered oxygen-containing cyclic compound to obtain a type-D target product,wherein the cinnamic acid derivative 1 is a compound having a structure shown below:
 the intermediate product 2 is a compound having a structure shown below:
the intermediate product 2 is a compound having a structure shown below: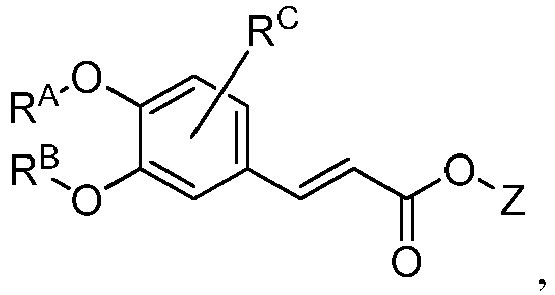 the intermediate product 3 is a compound having a structure shown below:
the intermediate product 3 is a compound having a structure shown below: the intermediate product 4 is a compound having a structure shown below:
the intermediate product 4 is a compound having a structure shown below: the intermediate product 5 is a compound having a structure shown below:
the intermediate product 5 is a compound having a structure shown below:
- The compound according to claim 1, wherein the compound is selected from the group consisting of:1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-hydroxyethyl)-2,6, dimethylpyridin-4(1H)-one;(1-(3-cyclopropylmethoxy-4methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) acetate;(1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) propionate;(1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) cyclopropylcarboxylate;(1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) benzoate;(1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) crotonate;(1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) 3-methylcrotonate;(1-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethyl) but-2-ynoate;1-(2-(but-2-yn-1-yloxy)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)ethyl)-2,6-dimethylpyridin-4-(1H)-one;1-(2-(3-hydroxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3,4-dimethoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-ethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-propoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-isopropoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-n-butoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)- one;1-(2-(3-iso-butoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-n-pentyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-n-hexyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-cyclopropyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxo)ethyl-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-cyclobutyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-cyclobutylmethoxy-4-methoxyphenyl)-2-oxo)ethyl-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-cyclopentyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-cyclohexyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-(cyclopent-3-en-1-yloxy)-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-allyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-((3-methylbut-2-en-1-yl)oxy)-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-propargyloxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-(but-2-yn-1-yloxy)-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-(oxacyclobutan-3-yl-oxy)-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-(tetrahydrofuran-2-yl)oxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-methylthiomethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-methylsulfinylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-methylsulfonylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-methylthioethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-methylsulfinylethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-methylsulfonylethoxy-4-methoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(hydroxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(methoxylimido)ethyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2- (methylthiomethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2- (methylthiomethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2- (methylsulfinylmethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2- (methylsulfonylmethoxyimido)ethyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)pyridin-4(1H)-one;1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2-methylpyridin-4(1H)-one;1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-oxoethyl)-2-chloropyridin-4(1H)-one;1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)cyclopropyl)-2,6-dimethylpyridin-4(1H)-one;1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)-2-oxoethyl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-cyclopropylmethoxy-4-methoxystyryl)pyridin-4(1H)-one;(E)-1-(3-cyclopropylmethoxy-4-methoxystyryl)-2-methylpyridin-4(1H)-one;(E)-1-(3-hydroxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-propoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-isopropoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-n-butoxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-iso-butoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-neopentyloxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-cyclopropyloxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-cyclopropylmethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-cyclopropylmethoxy-4-methoxyphenethyl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-cyclobutyloxy-4-methoxy-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-cyclopentyloxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-(cyclopent-3-en-1-yloxy)-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-allyloxy-4-methoxy-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-(3-methylbut-2-en-1-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-(prop-2-yn-1-yloxy)-4-methoxy)styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-(but-2-yn-1-yloxy)-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-(oxacyclobutan-3-yl-oxy)-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-(thiacyclobutan-3-yl-oxy)-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-(tetrahydrofuran-2-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-2-(5-(2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethenyl)-2-methoxyphenoxy)-N-methylacetamide;(E)-1-((3-cyclopropylformyloxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-5-(2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)ethenyl)-2-methoxyphenylmethyl sulfonate;(E)-1-(3-methylthiomethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylsulfinylmethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylsulfonylmethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylthioethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylsulfinylethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylsulfonylethoxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-(tetrahydrothiophen-3-yl)oxy-4-methoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-((1,1,-dioxotetrahydrothiophen-3-yl)oxy)-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(S,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;1-((E)-4-methoxy-3-(((3S)-1-oxotetrahydrothiophen-3-yl)oxy)styryl)-2,6-dimethylpyridin-4(1H)-one;(S,E)-1-(3-((1,1-dioxotetrahydrothiophen-3-yl)oxy)-4-methoxy-styryl)-2,6-dimethylpyridin-4(1H)-one;(R,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-methoxystyryl)-2,6-dimethylpyridin-4(1H)-one;1-((E)-4-methoxy-3-(((3R)-1-oxotetrahydrothiophen-3-yl)oxy)styryl)-2,6-dimethylpyridin-4(1H)-one;(R,E)-1-(3-((1,1-dioxotetrahydrothiophen-3-yl)oxy)-4-methoxy-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-(cyclopropylmethoxy)-4-(difluoromethoxy)styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methoxy-4-difluoromethoxy-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylthiomethoxy-4-difluoromethoxy-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)- 1-(3-methylsulfinylmethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylsulfonylmethoxy-4-difluoromethoxy-styryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylthioethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylsulfinylethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(3-methylsulfonylethoxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-((3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxy)-styryl)-2,6-dimethylpyridin-4(1H)-one;(S,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;

 (S,E)-1-(3-(1,1-dioxotetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(R,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;1-((E)-4-difluoromethoxy-3-(((3R)-1-oxotetrahydrothiophen-3-yl)oxy)styryl)-2,6-dimethylpyridin-4(1H)-one;(R,E)-1-(3-(1,1-dioxotetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-methylethenyl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-phenylethenyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)pent-1-en-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-cyanoethenyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylamide;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N-methylacrylamide;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-dimethylacrylamide;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-diethylacrylamide;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylic acid;(Z)-methyl-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylate;(Z)-ethyl-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylate;(Z)-1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)pent-1-en-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one;(Z)-2-(3,4-dimethoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridine)-1(4H)-yl)acrylonitrile;(E)-3-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylonitrile;(E)-3-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylamide; and(E)-3-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridine)-1(4H)-yl)-N,N-dimethylacrylamide.
(S,E)-1-(3-(1,1-dioxotetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(R,E)-1-(3-(tetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;1-((E)-4-difluoromethoxy-3-(((3R)-1-oxotetrahydrothiophen-3-yl)oxy)styryl)-2,6-dimethylpyridin-4(1H)-one;(R,E)-1-(3-(1,1-dioxotetrahydrothiophen-3-yl)oxy-4-difluoromethoxystyryl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-methylethenyl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-phenylethenyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)pent-1-en-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one;(E)-1-(2-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-cyanoethenyl)-2,6-dimethylpyridin-4(1H)-one;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylamide;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N-methylacrylamide;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-dimethylacrylamide;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-N,N-diethylacrylamide;(Z)-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylic acid;(Z)-methyl-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylate;(Z)-ethyl-2-(3-cyclopropylmethoxy-4-methoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)-acrylate;(Z)-1-(2-(3-cyclopropylmethoxy-4-difluoromethoxyphenyl)pent-1-en-3-yn-1-yl)-2,6-dimethylpyridin-4(1H)-one;(Z)-2-(3,4-dimethoxyphenyl)-3-(2,6-dimethyl-4-carbonylpyridine)-1(4H)-yl)acrylonitrile;(E)-3-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylonitrile;(E)-3-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridin-1(4H)-yl)acrylamide; and(E)-3-(3-cyclopropylmethoxy-4-methoxyphenyl)-2-(2,6-dimethyl-4-carbonylpyridine)-1(4H)-yl)-N,N-dimethylacrylamide. - The compound according to claim 1 for use as a PDE4 inhibitor or in the treatment of inflammatory skin diseases.
- A pharmaceutical composition comprising 0.01-10% of the compound according to claim 1; and other components selected from a surfactant, a lipid compound, and an auxiliary agent;wherein the amount of the surfactant accounts for 10-30% of the total weight of the pharmaceutical composition;the amount of the lipid compound accounts for 50-85% of the total weight of the pharmaceutical composition; andthe amount of the auxiliary agent accounts for 10-30% of the total weight of the pharmaceutical composition.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810386572.6A CN110407741B (en) | 2018-04-26 | 2018-04-26 | Anti-inflammatory compound and preparation and application thereof |
| PCT/CN2019/078964 WO2019205843A1 (en) | 2018-04-26 | 2019-03-21 | Anti-inflammatory compound and preparation and use thereof |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP3777979A1 EP3777979A1 (en) | 2021-02-17 |
| EP3777979A4 EP3777979A4 (en) | 2021-08-04 |
| EP3777979B1 true EP3777979B1 (en) | 2024-01-17 |
Family
ID=68294751
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP19791850.1A Active EP3777979B1 (en) | 2018-04-26 | 2019-03-21 | Anti-inflammatory compound and preparation and use thereof |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US11440886B2 (en) |
| EP (1) | EP3777979B1 (en) |
| JP (1) | JP7364142B2 (en) |
| KR (1) | KR20210004982A (en) |
| CN (1) | CN110407741B (en) |
| AU (1) | AU2019258242B2 (en) |
| CA (1) | CA3096905C (en) |
| ES (1) | ES2975791T3 (en) |
| IL (1) | IL278165B2 (en) |
| RU (1) | RU2759626C1 (en) |
| SG (1) | SG11202009982PA (en) |
| TW (1) | TWI701239B (en) |
| WO (1) | WO2019205843A1 (en) |
| ZA (1) | ZA202006347B (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024174931A1 (en) * | 2023-02-23 | 2024-08-29 | 启元生物(杭州)有限公司 | Novel crystal form of compound as well as preparation method therefor and use thereof |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0228185A (en) * | 1988-04-14 | 1990-01-30 | Tanabe Seiyaku Co Ltd | Cephalosporin compound and synthetic intermediate thereof |
| CA2005787A1 (en) * | 1988-12-29 | 1990-06-29 | Masao Wada | Cephalosporin compounds |
| GB9723078D0 (en) * | 1997-10-31 | 1998-01-07 | Cerebrus Ltd | Chemical compounds |
| US20060004056A1 (en) * | 2002-07-02 | 2006-01-05 | Bernard Cote | Di-aryl-substituted-ethan pyridone pde4 inhibitors |
| US8263648B2 (en) * | 2006-09-11 | 2012-09-11 | Mylan Laboratories Ltd. | Diebenzofuran derivatives as inhibitors of PDE-4 and PDE-10 |
| UA96783C2 (en) * | 2006-12-22 | 2011-12-12 | Лео Фарма А/С | Substituted methylphenylketones for use as pde4 inhibitors |
| GB201113689D0 (en) * | 2011-08-09 | 2011-09-21 | Amakem Nv | Novel PDE4 inhibitors |
| CN102603544A (en) * | 2012-04-06 | 2012-07-25 | 南京大学 | Anti-inflammatory compound, preparation and use thereof |
-
2018
- 2018-04-26 CN CN201810386572.6A patent/CN110407741B/en active Active
-
2019
- 2019-03-21 IL IL278165A patent/IL278165B2/en unknown
- 2019-03-21 RU RU2020129300A patent/RU2759626C1/en active
- 2019-03-21 KR KR1020207029585A patent/KR20210004982A/en not_active Application Discontinuation
- 2019-03-21 SG SG11202009982PA patent/SG11202009982PA/en unknown
- 2019-03-21 JP JP2020542547A patent/JP7364142B2/en active Active
- 2019-03-21 CA CA3096905A patent/CA3096905C/en active Active
- 2019-03-21 ES ES19791850T patent/ES2975791T3/en active Active
- 2019-03-21 EP EP19791850.1A patent/EP3777979B1/en active Active
- 2019-03-21 WO PCT/CN2019/078964 patent/WO2019205843A1/en active Application Filing
- 2019-03-21 AU AU2019258242A patent/AU2019258242B2/en active Active
- 2019-03-22 TW TW108109957A patent/TWI701239B/en active
-
2020
- 2020-08-14 US US16/993,289 patent/US11440886B2/en active Active
- 2020-10-13 ZA ZA2020/06347A patent/ZA202006347B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA3096905A1 (en) | 2019-10-31 |
| NZ767619A (en) | 2023-12-22 |
| TW201945341A (en) | 2019-12-01 |
| AU2019258242A1 (en) | 2020-09-24 |
| EP3777979A4 (en) | 2021-08-04 |
| JP7364142B2 (en) | 2023-10-18 |
| ES2975791T3 (en) | 2024-07-15 |
| ZA202006347B (en) | 2022-01-26 |
| KR20210004982A (en) | 2021-01-13 |
| AU2019258242B2 (en) | 2022-01-27 |
| US20200377460A1 (en) | 2020-12-03 |
| IL278165A (en) | 2020-12-31 |
| EP3777979A1 (en) | 2021-02-17 |
| IL278165B2 (en) | 2024-10-01 |
| US11440886B2 (en) | 2022-09-13 |
| RU2759626C1 (en) | 2021-11-16 |
| TWI701239B (en) | 2020-08-11 |
| WO2019205843A1 (en) | 2019-10-31 |
| CN110407741A (en) | 2019-11-05 |
| JP2021518838A (en) | 2021-08-05 |
| CN110407741B (en) | 2023-03-21 |
| CA3096905C (en) | 2024-01-09 |
| SG11202009982PA (en) | 2020-11-27 |
| IL278165B1 (en) | 2024-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3212189B1 (en) | Substituted chromanes and method of use | |
| EP1305026B1 (en) | Barbituric acid analogs as therapeutic agents | |
| FR2552760A1 (en) | Process for obtaining carbostyril derivatives and synthesis intermediates. | |
| KR20050040814A (en) | Amide derivatives as glycogen synthase kinase 3-beta inhibitors | |
| EP2513102B1 (en) | Ethynyl derivatives | |
| KR960014871B1 (en) | Pharmaceutical compositions useful as antiproliferative and antimetastatic agents containing 5-amino or substituted amino 1,2,3-triazoles | |
| EP4186896A1 (en) | Shp2 inhibitor and composition and application thereof | |
| Johnson et al. | Synthesis and biological activity of a new class of cytotoxic agents: N-(3-oxoprop-1-enyl)-substituted pyrimidines and purines | |
| EP2036905A1 (en) | Pyridylisoxazole derivative | |
| Akhtar et al. | Synthesis of hybrids of dihydropyrimidine and pyridazinone as potential anti-breast cancer agents | |
| EP3777979B1 (en) | Anti-inflammatory compound and preparation and use thereof | |
| EP2044957A1 (en) | Treatment agent for inflammatory bowel disease | |
| EP2225245B1 (en) | N-phenyl-imidazo(1,2-a)pyridine-2-carboxamide compounds, preparation thereof and therapeutic application thereof | |
| EP0432209B1 (en) | Phenylpyrrolic compounds used as drugs, their preparation and application | |
| EP2225241B1 (en) | Derivatives of N-phenyl-imidazo-(1,2-A)-pyridine-2-carboxamides, preparation thereof and therapeutic application thereof | |
| EP1814879B1 (en) | Derivatives of 4,7-dioxobenzothiazole-2-carboxamides, preparation method thereof and therapeutic uses of same | |
| CN107459491A (en) | Benzamide compound containing 1,2,3 triazole structures and application thereof | |
| EP3613735B1 (en) | Novel sirt 1 activator and medicinal use thereof | |
| EP3184525A1 (en) | 3-phenyl-5-ureidoisothiazole-4-caboximide and 3-amino-5-phenylisothiazole derivatives as kinase inhibitors | |
| EP1832584A1 (en) | Pyrimidinylisoxazol derivative | |
| EP3394053B1 (en) | (4-hydroxy-2-phenyl-1,3-thiazol-5-yl) methanone derivatives as trpm8 antagonists | |
| Lin et al. | Cytotoxicities and Topoisomerase I Inhibitory Activities of 2‐[2‐(2‐Alkynylphenyl) ethynyl] benzonitriles, 1‐Aryldec‐3‐ene‐1, 5‐diynes, and Related Bis (enediynyl) arene Compounds | |
| Sharma et al. | Conventional and Microwave Induced Synthesis of 4-Oxo-Thiazolidine Derivatives and Their Biological Activities |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE INTERNATIONAL PUBLICATION HAS BEEN MADE |
|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: REQUEST FOR EXAMINATION WAS MADE |
|
| 17P | Request for examination filed |
Effective date: 20201113 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| AX | Request for extension of the european patent |
Extension state: BA ME |
|
| A4 | Supplementary search report drawn up and despatched |
Effective date: 20210705 |
|
| DAV | Request for validation of the european patent (deleted) | ||
| DAX | Request for extension of the european patent (deleted) | ||
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: E-NITIATE BIOPHARMACEUTICALS (HANGZHOU) CO., LTD |
|
| RIC1 | Information provided on ipc code assigned before grant |
Ipc: A61P 37/00 20060101AFI20210629BHEP Ipc: A61P 17/00 20060101ALI20210629BHEP Ipc: C07D 401/04 20060101ALI20210629BHEP |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: EXAMINATION IS IN PROGRESS |
|
| 17Q | First examination report despatched |
Effective date: 20220818 |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: GRANT OF PATENT IS INTENDED |
|
| INTG | Intention to grant announced |
Effective date: 20230413 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAJ | Information related to disapproval of communication of intention to grant by the applicant or resumption of examination proceedings by the epo deleted |
Free format text: ORIGINAL CODE: EPIDOSDIGR1 |
|
| GRAL | Information related to payment of fee for publishing/printing deleted |
Free format text: ORIGINAL CODE: EPIDOSDIGR3 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: EXAMINATION IS IN PROGRESS |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: GRANT OF PATENT IS INTENDED |
|
| INTC | Intention to grant announced (deleted) | ||
| INTG | Intention to grant announced |
Effective date: 20230822 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE PATENT HAS BEEN GRANTED |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602019045271 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: FP |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG9D |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20240424 Year of fee payment: 6 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 1650176 Country of ref document: AT Kind code of ref document: T Effective date: 20240117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240517 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20240410 Year of fee payment: 6 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20240513 Year of fee payment: 6 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240418 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2975791 Country of ref document: ES Kind code of ref document: T3 Effective date: 20240715 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240417 Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 20240528 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 20240507 Year of fee payment: 6 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240417 Ref country code: NO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240417 Ref country code: LT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240517 Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240418 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20240430 Year of fee payment: 6 Ref country code: IT Payment date: 20240509 Year of fee payment: 6 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240517 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240517 Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20240117 |