EP1495128B1 - Ehanced protein expression in bacillus - Google Patents
Ehanced protein expression in bacillus Download PDFInfo
- Publication number
- EP1495128B1 EP1495128B1 EP03726142.7A EP03726142A EP1495128B1 EP 1495128 B1 EP1495128 B1 EP 1495128B1 EP 03726142 A EP03726142 A EP 03726142A EP 1495128 B1 EP1495128 B1 EP 1495128B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- bacillus
- subtilisin
- gene
- strain
- sequence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 108090000623 proteins and genes Proteins 0.000 title claims description 194
- 241000193830 Bacillus <bacterium> Species 0.000 title claims description 149
- 102000004169 proteins and genes Human genes 0.000 title description 38
- 230000014509 gene expression Effects 0.000 title description 27
- 108020004414 DNA Proteins 0.000 claims description 115
- 238000000034 method Methods 0.000 claims description 102
- 239000003550 marker Substances 0.000 claims description 62
- 238000012217 deletion Methods 0.000 claims description 59
- 230000037430 deletion Effects 0.000 claims description 59
- 230000002759 chromosomal effect Effects 0.000 claims description 49
- 235000014469 Bacillus subtilis Nutrition 0.000 claims description 42
- 108090000787 Subtilisin Proteins 0.000 claims description 34
- 244000063299 Bacillus subtilis Species 0.000 claims description 31
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 26
- 230000028327 secretion Effects 0.000 claims description 20
- 108091033319 polynucleotide Proteins 0.000 claims description 17
- 239000002157 polynucleotide Substances 0.000 claims description 17
- 102000040430 polynucleotide Human genes 0.000 claims description 17
- 230000035772 mutation Effects 0.000 claims description 15
- 230000001131 transforming effect Effects 0.000 claims description 14
- 230000002779 inactivation Effects 0.000 claims description 13
- 230000000415 inactivating effect Effects 0.000 claims description 12
- 108010056079 Subtilisins Proteins 0.000 claims description 10
- 102000005158 Subtilisins Human genes 0.000 claims description 10
- 238000002744 homologous recombination Methods 0.000 claims description 7
- 230000006801 homologous recombination Effects 0.000 claims description 7
- 241000194108 Bacillus licheniformis Species 0.000 claims description 6
- 241000193385 Geobacillus stearothermophilus Species 0.000 claims description 6
- 101150089588 degU gene Proteins 0.000 claims description 6
- 241000193744 Bacillus amyloliquefaciens Species 0.000 claims description 5
- 241000193422 Bacillus lentus Species 0.000 claims description 5
- 241000194107 Bacillus megaterium Species 0.000 claims description 5
- 241000193388 Bacillus thuringiensis Species 0.000 claims description 5
- 241000700124 Octodon degus Species 0.000 claims description 5
- 230000002708 enhancing effect Effects 0.000 claims description 5
- 101150077915 oppA gene Proteins 0.000 claims description 5
- 241000193752 Bacillus circulans Species 0.000 claims description 4
- 241001328122 Bacillus clausii Species 0.000 claims description 4
- 241000193749 Bacillus coagulans Species 0.000 claims description 4
- 241000193764 Brevibacillus brevis Species 0.000 claims description 4
- 101150085919 degQ gene Proteins 0.000 claims description 4
- 101150083941 degS gene Proteins 0.000 claims description 4
- 241000194103 Bacillus pumilus Species 0.000 claims description 3
- 108010020132 microbial serine proteinases Proteins 0.000 claims description 3
- 241000194109 Paenibacillus lautus Species 0.000 claims description 2
- 125000003275 alpha amino acid group Chemical group 0.000 claims 3
- 229940054340 bacillus coagulans Drugs 0.000 claims 1
- 229940097012 bacillus thuringiensis Drugs 0.000 claims 1
- 239000013615 primer Substances 0.000 description 108
- 210000004027 cell Anatomy 0.000 description 87
- 150000007523 nucleic acids Chemical class 0.000 description 53
- 239000012634 fragment Substances 0.000 description 51
- 210000000349 chromosome Anatomy 0.000 description 48
- 238000003752 polymerase chain reaction Methods 0.000 description 48
- 102000039446 nucleic acids Human genes 0.000 description 37
- 108020004707 nucleic acids Proteins 0.000 description 37
- 239000013612 plasmid Substances 0.000 description 36
- 235000018102 proteins Nutrition 0.000 description 35
- 238000006243 chemical reaction Methods 0.000 description 29
- 238000003199 nucleic acid amplification method Methods 0.000 description 25
- 230000003321 amplification Effects 0.000 description 24
- 102000035195 Peptidases Human genes 0.000 description 23
- 108091005804 Peptidases Proteins 0.000 description 23
- 230000000845 anti-microbial effect Effects 0.000 description 21
- 108091008146 restriction endonucleases Proteins 0.000 description 21
- 239000004365 Protease Substances 0.000 description 20
- 125000003729 nucleotide group Chemical group 0.000 description 20
- 108091026890 Coding region Proteins 0.000 description 19
- 239000000523 sample Substances 0.000 description 19
- 230000009466 transformation Effects 0.000 description 19
- 239000002773 nucleotide Substances 0.000 description 18
- 238000011144 upstream manufacturing Methods 0.000 description 18
- 239000013598 vector Substances 0.000 description 17
- 229940088598 enzyme Drugs 0.000 description 16
- 108090000765 processed proteins & peptides Proteins 0.000 description 16
- 102000004190 Enzymes Human genes 0.000 description 15
- 108090000790 Enzymes Proteins 0.000 description 15
- 238000004519 manufacturing process Methods 0.000 description 15
- 230000006870 function Effects 0.000 description 14
- 230000004927 fusion Effects 0.000 description 14
- 102000004196 processed proteins & peptides Human genes 0.000 description 14
- 235000019419 proteases Nutrition 0.000 description 14
- 230000000295 complement effect Effects 0.000 description 13
- 238000003780 insertion Methods 0.000 description 13
- 230000037431 insertion Effects 0.000 description 13
- 239000000203 mixture Substances 0.000 description 13
- 229920001184 polypeptide Polymers 0.000 description 13
- 150000001413 amino acids Chemical group 0.000 description 12
- 238000009396 hybridization Methods 0.000 description 12
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 11
- 241000588724 Escherichia coli Species 0.000 description 11
- 238000003556 assay Methods 0.000 description 11
- 239000013611 chromosomal DNA Substances 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 230000012010 growth Effects 0.000 description 11
- 101150088738 pckA gene Proteins 0.000 description 11
- 101150067708 pckG gene Proteins 0.000 description 11
- 238000013518 transcription Methods 0.000 description 11
- 230000035897 transcription Effects 0.000 description 11
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- 230000008569 process Effects 0.000 description 10
- 108091034117 Oligonucleotide Proteins 0.000 description 9
- 239000002299 complementary DNA Substances 0.000 description 9
- 239000000872 buffer Substances 0.000 description 8
- 238000010367 cloning Methods 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 229910001868 water Inorganic materials 0.000 description 8
- 239000000499 gel Substances 0.000 description 7
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 6
- 238000010276 construction Methods 0.000 description 6
- 239000013604 expression vector Substances 0.000 description 6
- 244000005700 microbiome Species 0.000 description 6
- 101150029104 prpC gene Proteins 0.000 description 6
- 101150065786 sigD gene Proteins 0.000 description 6
- 101150079130 sopB gene Proteins 0.000 description 6
- 241000894007 species Species 0.000 description 6
- 241000194110 Bacillus sp. (in: Bacteria) Species 0.000 description 5
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 5
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 5
- 239000004473 Threonine Substances 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 229960002685 biotin Drugs 0.000 description 5
- 235000020958 biotin Nutrition 0.000 description 5
- 239000011616 biotin Substances 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 238000000855 fermentation Methods 0.000 description 5
- 230000004151 fermentation Effects 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 230000010354 integration Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 230000010076 replication Effects 0.000 description 5
- -1 scoC4 Proteins 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 229960000268 spectinomycin Drugs 0.000 description 5
- UNFWWIHTNXNPBV-WXKVUWSESA-N spectinomycin Chemical compound O([C@@H]1[C@@H](NC)[C@@H](O)[C@H]([C@@H]([C@H]1O1)O)NC)[C@]2(O)[C@H]1O[C@H](C)CC2=O UNFWWIHTNXNPBV-WXKVUWSESA-N 0.000 description 5
- QRBLKGHRWFGINE-UGWAGOLRSA-N 2-[2-[2-[[2-[[4-[[2-[[6-amino-2-[3-amino-1-[(2,3-diamino-3-oxopropyl)amino]-3-oxopropyl]-5-methylpyrimidine-4-carbonyl]amino]-3-[(2r,3s,4s,5s,6s)-3-[(2s,3r,4r,5s)-4-carbamoyl-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-4,5-dihydroxy-6-(hydroxymethyl)- Chemical compound N=1C(C=2SC=C(N=2)C(N)=O)CSC=1CCNC(=O)C(C(C)=O)NC(=O)C(C)C(O)C(C)NC(=O)C(C(O[C@H]1[C@@]([C@@H](O)[C@H](O)[C@H](CO)O1)(C)O[C@H]1[C@@H]([C@](O)([C@@H](O)C(CO)O1)C(N)=O)O)C=1NC=NC=1)NC(=O)C1=NC(C(CC(N)=O)NCC(N)C(N)=O)=NC(N)=C1C QRBLKGHRWFGINE-UGWAGOLRSA-N 0.000 description 4
- 241000276408 Bacillus subtilis subsp. subtilis str. 168 Species 0.000 description 4
- 102000012410 DNA Ligases Human genes 0.000 description 4
- 108010061982 DNA Ligases Proteins 0.000 description 4
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 239000006137 Luria-Bertani broth Substances 0.000 description 4
- LTQCLFMNABRKSH-UHFFFAOYSA-N Phleomycin Natural products N=1C(C=2SC=C(N=2)C(N)=O)CSC=1CCNC(=O)C(C(O)C)NC(=O)C(C)C(O)C(C)NC(=O)C(C(OC1C(C(O)C(O)C(CO)O1)OC1C(C(OC(N)=O)C(O)C(CO)O1)O)C=1NC=NC=1)NC(=O)C1=NC(C(CC(N)=O)NCC(N)C(N)=O)=NC(N)=C1C LTQCLFMNABRKSH-UHFFFAOYSA-N 0.000 description 4
- 108010035235 Phleomycins Proteins 0.000 description 4
- 108010022999 Serine Proteases Proteins 0.000 description 4
- 102000012479 Serine Proteases Human genes 0.000 description 4
- 238000000137 annealing Methods 0.000 description 4
- 238000003491 array Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000005520 cutting process Methods 0.000 description 4
- 238000001502 gel electrophoresis Methods 0.000 description 4
- 239000002777 nucleoside Substances 0.000 description 4
- 125000003835 nucleoside group Chemical group 0.000 description 4
- 238000003757 reverse transcription PCR Methods 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 230000014616 translation Effects 0.000 description 4
- 230000035899 viability Effects 0.000 description 4
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 3
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 3
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- 101100010747 Escherichia coli (strain K12) epd gene Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 101150098454 GAPA2 gene Proteins 0.000 description 3
- 101150036652 GAPB gene Proteins 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 101100335749 Halobacterium salinarum (strain ATCC 700922 / JCM 11081 / NRC-1) gap gene Proteins 0.000 description 3
- 108091092195 Intron Proteins 0.000 description 3
- 239000007987 MES buffer Substances 0.000 description 3
- 229920001213 Polysorbate 20 Polymers 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000004599 antimicrobial Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 101150036876 cre gene Proteins 0.000 description 3
- 230000003413 degradative effect Effects 0.000 description 3
- 238000004925 denaturation Methods 0.000 description 3
- 230000036425 denaturation Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 230000001939 inductive effect Effects 0.000 description 3
- 229910001629 magnesium chloride Inorganic materials 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 239000013600 plasmid vector Substances 0.000 description 3
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 3
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 3
- 235000019833 protease Nutrition 0.000 description 3
- 210000001938 protoplast Anatomy 0.000 description 3
- 238000012807 shake-flask culturing Methods 0.000 description 3
- 101150105742 spoIIE gene Proteins 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 230000002103 transcriptional effect Effects 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 101100222170 Bacillus subtilis (strain 168) cssS gene Proteins 0.000 description 2
- 101000583086 Bunodosoma granuliferum Delta-actitoxin-Bgr2b Proteins 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 108020004705 Codon Proteins 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 102100031780 Endonuclease Human genes 0.000 description 2
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- 238000012408 PCR amplification Methods 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 108010076504 Protein Sorting Signals Proteins 0.000 description 2
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 241001659629 Virgibacillus Species 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 239000011543 agarose gel Substances 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- 101150009206 aprE gene Proteins 0.000 description 2
- 238000010170 biological method Methods 0.000 description 2
- 230000001851 biosynthetic effect Effects 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 229960005091 chloramphenicol Drugs 0.000 description 2
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 2
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 2
- SUYVUBYJARFZHO-UHFFFAOYSA-N dATP Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-UHFFFAOYSA-N 0.000 description 2
- RGWHQCVHVJXOKC-SHYZEUOFSA-J dCTP(4-) Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)C1 RGWHQCVHVJXOKC-SHYZEUOFSA-J 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000011033 desalting Methods 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 238000001962 electrophoresis Methods 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 238000012224 gene deletion Methods 0.000 description 2
- 235000021472 generally recognized as safe Nutrition 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 239000011121 hardwood Substances 0.000 description 2
- 230000014726 immortalization of host cell Effects 0.000 description 2
- 238000003018 immunoassay Methods 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 239000012092 media component Substances 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 238000003127 radioimmunoassay Methods 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000003248 secreting effect Effects 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 239000001226 triphosphate Substances 0.000 description 2
- 235000011178 triphosphate Nutrition 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 239000011534 wash buffer Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- NHJVRSWLHSJWIN-UHFFFAOYSA-N 2,4,6-trinitrobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O NHJVRSWLHSJWIN-UHFFFAOYSA-N 0.000 description 1
- WTLKTXIHIHFSGU-UHFFFAOYSA-N 2-nitrosoguanidine Chemical compound NC(N)=NN=O WTLKTXIHIHFSGU-UHFFFAOYSA-N 0.000 description 1
- 108020005345 3' Untranslated Regions Proteins 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 108020003589 5' Untranslated Regions Proteins 0.000 description 1
- OPIFSICVWOWJMJ-AEOCFKNESA-N 5-bromo-4-chloro-3-indolyl beta-D-galactoside Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OC1=CNC2=CC=C(Br)C(Cl)=C12 OPIFSICVWOWJMJ-AEOCFKNESA-N 0.000 description 1
- VHTUHGNVVZPWGO-UHFFFAOYSA-N 7-(2-hydroxyethyl)-1,3-dimethyl-8-(pyridin-3-ylmethyl)purine-2,6-dione Chemical compound OCCN1C=2C(=O)N(C)C(=O)N(C)C=2N=C1CC1=CC=CN=C1 VHTUHGNVVZPWGO-UHFFFAOYSA-N 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 241001147780 Alicyclobacillus Species 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 241001147782 Amphibacillus Species 0.000 description 1
- 241000555286 Aneurinibacillus Species 0.000 description 1
- 241001626813 Anoxybacillus Species 0.000 description 1
- 241000972773 Aulopiformes Species 0.000 description 1
- 241000304886 Bacilli Species 0.000 description 1
- 241000006382 Bacillus halodurans Species 0.000 description 1
- 101100038071 Bacillus subtilis (strain 168) putC gene Proteins 0.000 description 1
- 108700016166 Bacillus subtilis sboA Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 108091005658 Basic proteases Proteins 0.000 description 1
- 241000555281 Brevibacillus Species 0.000 description 1
- 101150111062 C gene Proteins 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 101710123154 Chloramphenicol resistance protein Proteins 0.000 description 1
- 108090000317 Chymotrypsin Proteins 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 238000007399 DNA isolation Methods 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- 241000721047 Danaus plexippus Species 0.000 description 1
- 241000238557 Decapoda Species 0.000 description 1
- 102000016911 Deoxyribonucleases Human genes 0.000 description 1
- 108010053770 Deoxyribonucleases Proteins 0.000 description 1
- 101100364969 Dictyostelium discoideum scai gene Proteins 0.000 description 1
- 108090000204 Dipeptidase 1 Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 101100100608 Escherichia coli (strain K12) trpC gene Proteins 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 108091029865 Exogenous DNA Proteins 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- 241000321606 Filobacillus Species 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- 241001261512 Gracilibacillus Species 0.000 description 1
- 241000193004 Halobacillus Species 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- 101710192606 Latent membrane protein 2 Proteins 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 239000006391 Luria-Bertani Medium Substances 0.000 description 1
- 241000193386 Lysinibacillus sphaericus Species 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 101100261636 Methanothermobacter marburgensis (strain ATCC BAA-927 / DSM 2133 / JCM 14651 / NBRC 100331 / OCM 82 / Marburg) trpB2 gene Proteins 0.000 description 1
- 229910017621 MgSO4-7H2O Inorganic materials 0.000 description 1
- 108020005196 Mitochondrial DNA Proteins 0.000 description 1
- 101100364971 Mus musculus Scai gene Proteins 0.000 description 1
- 108091061960 Naked DNA Proteins 0.000 description 1
- 108091005507 Neutral proteases Proteins 0.000 description 1
- 102000035092 Neutral proteases Human genes 0.000 description 1
- 238000002944 PCR assay Methods 0.000 description 1
- 229920010824 PPS UR Polymers 0.000 description 1
- 241000179039 Paenibacillus Species 0.000 description 1
- 108010067372 Pancreatic elastase Proteins 0.000 description 1
- 102000016387 Pancreatic elastase Human genes 0.000 description 1
- 239000001888 Peptone Substances 0.000 description 1
- 108010080698 Peptones Proteins 0.000 description 1
- 108010002747 Pfu DNA polymerase Proteins 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 101100408135 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) phnA gene Proteins 0.000 description 1
- 108010066717 Q beta Replicase Proteins 0.000 description 1
- 238000012181 QIAquick gel extraction kit Methods 0.000 description 1
- 108700005075 Regulator Genes Proteins 0.000 description 1
- 101100109408 Staphylococcus aureus (strain COL) arg gene Proteins 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 208000037065 Subacute sclerosing leukoencephalitis Diseases 0.000 description 1
- 206010042297 Subacute sclerosing panencephalitis Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 101710137500 T7 RNA polymerase Proteins 0.000 description 1
- 108010006785 Taq Polymerase Proteins 0.000 description 1
- 101710109576 Terminal protein Proteins 0.000 description 1
- 241001291204 Thermobacillus Species 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 241000321595 Ureibacillus Species 0.000 description 1
- 101100071188 Vibrio parahaemolyticus serotype O3:K6 (strain RIMD 2210633) tdh1 gene Proteins 0.000 description 1
- 101100071190 Vibrio parahaemolyticus serotype O3:K6 (strain RIMD 2210633) tdh2 gene Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 102000004139 alpha-Amylases Human genes 0.000 description 1
- 108090000637 alpha-Amylases Proteins 0.000 description 1
- 101150050280 alsD gene Proteins 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- 101150032882 arcB gene Proteins 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- 102000005936 beta-Galactosidase Human genes 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 102000006635 beta-lactamase Human genes 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000006696 biosynthetic metabolic pathway Effects 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 229940041514 candida albicans extract Drugs 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003593 chromogenic compound Substances 0.000 description 1
- 229960002376 chymotrypsin Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000013599 cloning vector Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- HAAZLUGHYHWQIW-KVQBGUIXSA-N dGTP Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 HAAZLUGHYHWQIW-KVQBGUIXSA-N 0.000 description 1
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 1
- 101150023726 degR gene Proteins 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 229960000633 dextran sulfate Drugs 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 235000020774 essential nutrients Nutrition 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000010195 expression analysis Methods 0.000 description 1
- 239000010200 folin Substances 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 101150023858 frpC gene Proteins 0.000 description 1
- 238000003500 gene array Methods 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-O guanidinium Chemical compound NC(N)=[NH2+] ZRALSGWEFCBTJO-UHFFFAOYSA-O 0.000 description 1
- XLYOFNOQVPJJNP-ZSJDYOACSA-N heavy water Substances [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 1
- 229940094991 herring sperm dna Drugs 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 230000001744 histochemical effect Effects 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000009630 liquid culture Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 108010026228 mRNA guanylyltransferase Proteins 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 238000002887 multiple sequence alignment Methods 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- VBEGHXKAFSLLGE-UHFFFAOYSA-N n-phenylnitramide Chemical compound [O-][N+](=O)NC1=CC=CC=C1 VBEGHXKAFSLLGE-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 229940124276 oligodeoxyribonucleotide Drugs 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 235000019319 peptone Nutrition 0.000 description 1
- 210000002706 plastid Anatomy 0.000 description 1
- 230000000379 polymerizing effect Effects 0.000 description 1
- 229920000069 polyphenylene sulfide Polymers 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000003614 protease activity assay Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000008521 reorganization Effects 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 239000011369 resultant mixture Substances 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 101150062862 rocD gene Proteins 0.000 description 1
- 101150112902 rocF gene Proteins 0.000 description 1
- 235000019515 salmon Nutrition 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 239000006152 selective media Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 230000000405 serological effect Effects 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000012064 sodium phosphate buffer Substances 0.000 description 1
- NCGJACBPALRHNG-UHFFFAOYSA-M sodium;2,4,6-trinitrobenzenesulfonate Chemical compound [Na+].[O-][N+](=O)C1=CC([N+]([O-])=O)=C(S([O-])(=O)=O)C([N+]([O-])=O)=C1 NCGJACBPALRHNG-UHFFFAOYSA-M 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 101150058720 tdh gene Proteins 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 238000005382 thermal cycling Methods 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- 125000002264 triphosphate group Chemical class [H]OP(=O)(O[H])OP(=O)(O[H])OP(=O)(O[H])O* 0.000 description 1
- HRXKRNGNAMMEHJ-UHFFFAOYSA-K trisodium citrate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O HRXKRNGNAMMEHJ-UHFFFAOYSA-K 0.000 description 1
- 229940038773 trisodium citrate Drugs 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 101150019416 trpA gene Proteins 0.000 description 1
- 101150081616 trpB gene Proteins 0.000 description 1
- 101150111232 trpB-1 gene Proteins 0.000 description 1
- 101150100816 trpD gene Proteins 0.000 description 1
- 101150044170 trpE gene Proteins 0.000 description 1
- 101150099081 trpF gene Proteins 0.000 description 1
- 101150079930 trpGD gene Proteins 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 239000012137 tryptone Substances 0.000 description 1
- 108010087967 type I signal peptidase Proteins 0.000 description 1
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 101150028950 ycgN gene Proteins 0.000 description 1
- 239000012138 yeast extract Substances 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/74—Vectors or expression systems specially adapted for prokaryotic hosts other than E. coli, e.g. Lactobacillus, Micromonospora
- C12N15/75—Vectors or expression systems specially adapted for prokaryotic hosts other than E. coli, e.g. Lactobacillus, Micromonospora for Bacillus
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/195—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- C07K14/32—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria from Bacillus (G)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
- C12N1/20—Bacteria; Culture media therefor
- C12N1/205—Bacterial isolates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/52—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from bacteria or Archaea
- C12N9/54—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from bacteria or Archaea bacteria being Bacillus
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/02—Preparation of peptides or proteins having a known sequence of two or more amino acids, e.g. glutathione
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12R—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES C12C - C12Q, RELATING TO MICROORGANISMS
- C12R2001/00—Microorganisms ; Processes using microorganisms
- C12R2001/01—Bacteria or Actinomycetales ; using bacteria or Actinomycetales
- C12R2001/07—Bacillus
- C12R2001/125—Bacillus subtilis ; Hay bacillus; Grass bacillus
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
Definitions
- the present invention provides cells that have been genetically manipulated to have an altered capacity to produce expressed proteins.
- the present invention relates to Gram-positive microorganisms, such as Bacillus species having enhanced secretion of a subtilisin, wherein one or more chromosomal genes have been inactivated, and preferably wherein one or more chromosomal genes have been deleted from the Bacillus chromosome.
- Important production enzymes include ⁇ -amylases, neutral proteases, and alkaline (or serine) proteases.
- the present invention provides means for improved secretion of a subtilisin in Bacillus. More particularly, in these embodiments, the present invention involves inactivation of one or more chromosomal genes in a Bacillus host strain, wherein the inactivated genes are not necessary for strain viability.
- the present invention provides methods and compositions for enhancing secretion of a subtilisin from a Bacillus cell.
- the methods comprise inactivating sbo, in a Bacillus host strain to produce an altered Bacillus strain; growing the altered Bacillus strain under suitable growth conditions; and allowing a subtilisin to be secreted in the altered Bacillus, wherein the secretion of the subtilisin is enhanced, compared to the corresponding unaltered Bacillus host strain.
- Inactivation of a chromosomal gene can comprise the deletion of a gene to produce the altered Bacillus strain.
- inactivation of a chromosomal gene comprises insertional inactivation.
- the present invention provides altered Bacillus strains comprising the deletion of sbo .
- the altered Bacillus strain is a subtilisin producing strain.
- the altered Bacillus strain further comprises a mutation In a gene selected from the group consisting of degU, degQ, degS, scoC4, spoIIE, and oppA.
- the present invention provides DNA constructs comprising an incoming sequence.
- the incoming sequence includes a selective marker and a gene or gene fragment of sbo .
- the selective marker is located in between two fragments of the gene.
- the incoming sequence comprises a selective marker and a homology box, wherein the homology box flanks the 5' and/or 3' end of the marker.
- a host cell is transformed with the DNA construct.
- the host cell is a Bacillus cell. The DNA construct is chromosomally integrated into the host cell.
- Disclosed are methods for obtaining an altered Bacillus strain expressing a protein of interest which comprises transforming a Bacillus host cell with the DNA construct of the present, wherein the DNA construct is integrated into the chromosome of the Bacillus host cell; producing an altered Bacillus strain, wherein one or more chromosomal genes have been inactivated; and growing the altered Bacillus strain under suitable growth conditions for the expression of a protein of interest
- the Bacillus host strain is selected from the group consisting of B . licheniformis, B. lentus, B. subtilis, B. amyloliquefaciens B . brevis, B. stearothermophilus, B. alkalophilus, B. coagulans, B.
- Bacillus host strain is a recombinant host.
- the protein of interest is recovered.
- the selective marker is excised from the altered Bacillus.
- the method comprises transforming a Bacillus host cell with a DNA construct comprising an incoming sequence wherein the incoming sequence comprises a selective marker and of sbo, wherein the DNA construct is integrated into the chromosome of the Bacillus host cell and results in the deletion of one or more gene(s); obtaining an altered Bacillus strain, and growing the altered Bacillus strain under suitable growth conditions for the secretion of the protein of interest.
- the methods comprise the steps of transforming a Bacillus subtilis host cell with a DNA construct according to the invention; allowing homologous recombination of the DNA construct and a homologous region of the Bacillus chromosome wherein sbo, is deleted from the Bacillus chromosome; obtaining an altered Bacillus subtilis strain; and growing the altered Bacillus strain under conditions suitable for the expression of a protease.
- the protease producing Bacillus is a subtilisin producing strain.
- the protease producing strain further includes a mutation in a gene selected from the group consisting of degU, degQ, degS, scoC4, spoIIE, and oppA.
- the inactivation comprises the insertional inactivation of the gene.
- the present invention provides methods for enhancing the secretion of a subtilisin in Bacillus comprising: obtaining an altered Bacillus strain produced by introducing a DNA construct including a selective marker and an inactivating chromosomal segment into a Bacillus host strain, wherein the DNA construct is integrated into the Bacillus chromosome resulting in the deletion of an indigenous chromosomal region or fragment thereof from the Bacillus host cell; and growing the altered Bacillus strain under suitable growth conditions, wherein expression of a protein of interest is greater in the altered Bacillus strain compared to the expression of the protein of interest is the corresponding unaltered Bacillus host cell.
- Zheng et al. J. Bacteriol. Vol 181, No. 23, pp 7346-7355 discloses the construction of a Bacillus strain lacking the sbo gene. According to Zheng et al ., genes of the abo-alb locus are required for production of the antilisteral bateriocin subtilosin.
- Van der laan et al. (Appl. Environ. Microbiol. Vol. 57, No. 4, pp 901-909 ) discloses the cloning, characterisation and multiple chromosomal integration of the Bacillus PB92 alkaline protease gene.
- nucleic acids are written left to right in 5' to 3' orientation; amino acid sequences are written left to right in amino to carboxy orientation, respectively.
- the headings provided herein are not limitations of the various aspects or embodiments of the invention that can be had by reference to the specification as a whole. Accordingly, the terms defined immediately below are more fully defined by reference to the Specification as a whole.
- host cell refers to a cell that has the capacity to act as a host or expression vehicle for a newly introduced DNA sequence.
- the host cells are Bacillus sp. or E . coli cells.
- the genus Bacillus includes all species within the genus " Bacillus ,” as known to those of skill in the art, including but not limited to B . subtilis, B. licheniformis, B. lentus, B. brevis, B. stearothermophilus, B. alkalophilus, B. amyloliquefaciens, B. clausii, B. halodurans, B. megaterium, B. coagulans, B. circulans, B. lautus, and B. thuringiensis. It is recognized that the genus Bacillus continues to undergo taxonomical reorganization.
- the genus include species that have been reclassified, including but not limited to such organisms as B . stearothermophilus, which is now named " Geobacillus stearothermophilus .”
- Geobacillus stearothermophilus The production of resistant endospores in the presence of oxygen is considered the defining feature of the genus Bacillus, although this characteristic also applies to the recently named Alicyclobacillus , Amphibacillus, Aneurinibacillus, Anoxybacillus, Brevibacillus, Filobacillus, Gracilibacillus, Halobacillus, Paenibacillus, Salibacillus, Thermobacillus, Ureibacillus, and Virgibacillus.
- nucleic acid refers to a nucleotide or polynucleotide sequence, and fragments or portions thereof, as well as to DNA, cDNA, and RNA of genomic or synthetic origin which may be double-stranded or single-stranded, whether representing the sense or antisense strand. It will be understood that as a result of the degeneracy of the genetic code, a multitude of nucleotide sequences may encode a given protein.
- the term "gene” means a chromosomal segment of DNA involved in producing a polypeptide chain that may or may not include regions preceding and following the coding regions (e.g. 5' untranslated (5' UTR) or leader sequences and 3' untranslated (3' UTR) or trailer sequences, as well as intervening sequence (introns) between individual coding segments (exons)).
- 5' untranslated (5' UTR) or leader sequences and 3' untranslated (3' UTR) or trailer sequences as well as intervening sequence (introns) between individual coding segments (exons)
- vector refers to any nucleic acid that can be replicated in cells and can carry new genes or DNA segments into cells. Thus, the term refers to a nucleic acid construct designed for transfer between different host cells.
- An "expression vector” refers to a vector that has the ability to incorporate and express heterologous DNA fragments in a foreign cell. Many prokaryotic and eukaryotic expression vectors are commercially available. Selection of appropriate expression vectors is within the knowledge of those having skill in the art.
- DNA construct As used herein, the terms "DNA construct,” “expression cassette,” and “expression vector,” refer to a nucleic acid construct generated recombinantly or synthetically, with a series of specified nucleic acid elements that permit transcription of a particular nucleic acid in a target cell (i.e ., these are vectors or vector elements, as described above).
- the recombinant expression cassette can be incorporated into a plasmid, chromosome, mitochondrial DNA, plastid DNA, virus, or nucleic acid fragment.
- the recombinant expression cassette portion of an expression vector includes, among other sequences, a nucleic acid sequence to be transcribed and a promoter.
- transforming DNA As used herein, "transforming DNA,” “transforming sequence,” and “DNA construct” refer to DNA that is used to introduce sequences into a host cell or organism. Transforming DNA is DNA used to introduce sequences into a host cell or organism. The DNA may be generated in vitro by PCR or any other suitable techniques. The transforming DNA comprises an incoming sequence, flanked by homology boxes. The transforming DNA can comprise other non-homologous sequences, added to the ends ( i.e ., stuffer sequences or flanks). The ends can be closed such that the transforming DNA forms a closed circle, such as, for example, insertion into a vector.
- plasmid refers to a circular double-stranded (ds) DNA construct used as a cloning vector, and which forms an extrachromosomal self-replicating genetic element in many bacteria and some eukaryotes. In some embodiments, plasmids become incorporated into the genome of the host cell.
- isolated and purified refer to a nucleic acid or amino acid (or other component) that is removed from at least one component with which it is naturally associated.
- the present invention provides means for enhancing subtilisin secretion, such that the enhanced strains produced a greater quantity and/or quality of a protein of interest than the parental strain (e.g ., the wild-type and/or originating strain).
- expression refers to a process by which a polypeptide is produced based on the nucleic acid sequence of a gene.
- the process includes both transcription and translation.
- the term "introduced” refers to any method suitable for transferring the nucleic acid sequence into the cell. Such methods for introduction include but are not limited to protoplast fusion, transfection, transformation, conjugation, and transduction (See e.g ., Ferrari et al., “Genetics,” in Hardwood et al, (eds.), Bacillus, Plenum Publishing Corp., pages 57-72, [1989 ]).
- the terms “transformed” and “stably transformed” refers to a cell that has a non-native (heterologous) polynucleotide sequence integrated into its genome or as an episomal plasmid that is maintained for at least two generations.
- an incoming sequence refers to a DNA sequence that is introduced into the Bacillus chromosome.
- the incoming sequence is part of a DNA construct.
- the incoming sequence encodes one or more proteins of interest.
- the incoming sequence comprises a sequence that may or may not already be present in the genome of the cell to be transformed ( i.e ., it may be either a homologous or heterologous sequence).
- the incoming sequence encodes a sequence that is already present in the chromosome of the host cell to be transformed.
- the incoming sequence comprises of sbo, in yet another embodiment, the incoming sequence includes a selective marker. In a further embodiment the incoming sequence includes two homology boxes.
- homology box refers to a nucleic acid sequence, which is homologous to a sequence in the Bacillus chromosome. More specifically, a homology box is an upstream or downstream region having between about 80 and 100% sequence identity, between about 90 and 100% sequence identity, or between about 95 and 100% sequence identity with the immediate flanking coding region of a gene or part of a gene to be inactivated according to the invention. These sequences direct where in the Bacillus chromosome a DNA construct is integrated and directs what part of the Bacillus chromosome is replaced by the incoming sequence. While not meant to limit the invention, a homology box may include about between 1 base pair (bp) to 200 kilobases (kb).
- a homology box includes about between 1 bp and 10.0 kb; between 1 bp and 5.0 kb; between 1 bp and 2.5 kb; between 1 bp and 1.0 kb, and between 0.25 kb and 2.5 kb.
- a homology box may also include about 10.0 kb, 5.0 kb, 2.5 kb, 2.0 kb, 1.5 kb, 1.0 kb, 0.5 kb, 0.25 kb and 0.1 kb.
- the 5' and 3' ends of a selective marker are flanked by a homology box wherein the homology box comprises nucleic acid sequences immediately flanking the coding region of the gene.
- selectable marker-encoding nucleotide sequence refers to a nucleotide sequence which is capable of expression in the host cells and where expression of the selectable marker confers to cells containing the expressed gene the ability to grow in the presence of a corresponding selective agent or lack of an essential nutrient.
- selectable marker refers to a nucleic acid (e.g ., a gene) capable of expression in host cell which allows for ease of selection of those hosts containing the vector.
- selectable markers include but are not limited to antimicrobials.
- selectable marker refers to genes that provide an indication that a host cell has taken up an incoming DNA of interest or some other reaction has occurred.
- selectable markers are genes that confer antimicrobial resistance or a metabolic advantage on the host cell to allow cells containing the exogenous DNA to be distinguished from cells that have not received any exogenous sequence during the transformation.
- a "residing selectable marker” is one that is located on the chromosome of the microorganism to be transformed.
- a residing selectable marker encodes a gene that is different from the selectable marker on the transforming DNA construct. Selective markers are well known to those of skill in the art.
- the marker is an antimicrobial resistant marker (e.g ., amp R ; phleo R ; spec R ; kan R ; ery R ; tet R ; cmp R ; and neo R ; See e.g ., Guerot-Fleury, Gene, 167:335-337 [1995 ]; Palmeros et al., Gene 247:255-264 [2000 ]; and Trieu-Cuot et al., Gene, 23:331-341 [1983 ]).
- an antimicrobial resistant marker e.g ., amp R ; phleo R ; spec R ; kan R ; ery R ; tet R ; cmp R ; and neo R ; See e.g ., Guerot-Fleury, Gene, 167:335-337 [1995 ]; Palmeros et al., Gene 247:255-264 [
- chloramphenicol resistance gene e.g ., the gene present on pC194, as well as the resistance gene present in the Bacillus licheniformis genome.
- This resistance gene is particularly useful in the present invention, as well as in embodiments involving chromosomal amplification of chromosomally integrated cassettes and integrative plasmids ( See e.g ., Albertini and Galizzi, Bacteriol., 162:1203-1211 [1985 ]; and Stahl and Ferrari, J. Bacteriol., 158:411-418 [1984 ]).
- the DNA sequence of this naturally-occurring chloramphenicol resistance gene is shown below:
- markers useful in accordance with the invention include, but are not limited to auxotrophic markers, such as tryptophan; and detection markers, such as ⁇ -galactosidase.
- promoter refers to a nucleic acid sequence that functions to direct transcription of a downstream gene.
- the promoter is appropriate to the host cell in which the target gene is being expressed.
- the promoter, together with other transcriptional and translational regulatory nucleic acid sequences (also termed “control sequences") is necessary to express a given gene.
- control sequences also termed “control sequences”
- the transcriptional and translational regulatory sequences include, but are not limited to, promoter sequences, ribosomal binding sites, transcriptional start and stop sequences, translational start and stop sequences, and enhancer or activator sequences.
- a nucleic acid is "operably linked" when it is placed into a functional relationship with another nucleic acid sequence.
- DNA encoding a secretory leader i.e ., a signal peptide
- a promoter or enhancer is operably linked to a coding sequence if it affects the transcription of the sequence
- a ribosome binding site is operably linked to a coding sequence if it is positioned so as to facilitate translation.
- operably linked means that the DNA sequences being linked are contiguous, and, in the case of a secretory leader, contiguous and in reading phase. However, enhancers do not have to be contiguous. Linking is accomplished by ligation at convenient restriction sites. If such sites do not exist, the synthetic oligonucleotide adaptors or linkers are used in accordance with conventional practice.
- inactivation includes any method that prevents the functional expression of sbo, wherein the gene or gene product is unable to exert its known function. Inactivation or enhancement occurs via any suitable means, including deletions, substitutions ( e.g ., mutations), interruptions, and/or insertions in the nucleic acid gene sequence.
- inactivation is achieved by deletion.
- the gene is deleted by homologous recombination.
- a DNA construct comprising an incoming sequence having a selective marker flanked on each side by a homology box is used.
- the homology box comprises nucleotide sequences homologous to nucleic acids flanking regions of the chromosomal sbo gene.
- the DNA construct aligns with the homologous sequences of the Bacillus host chromosome and in a double crossover event the sbo gene is excised out of the host chromosome.
- deletion of a gene refers to deletion of the entire coding sequence, deletion of part of the coding sequence, or deletion of the coding sequence including flanking regions.
- the deletion may be partial as long as the sequences left in the chromosome provides the desired biological activity of the gene.
- the flanking regions of the coding sequence may include from about 1 bp to about 500 bp at the 5' and 3' ends.
- the flanking region may be larger than 500 bp but will preferably not include other genes in the region which may be inactivated or deleted according to the invention. The end result is that the deleted gene is effectively non-functional.
- a “deletion” is defined as a change in either nucleotide or amino acid sequence in which one or more nucleotides or amino acid residues, respectively, have been removed ( i.e. , are absent).
- a “deletion mutant” has fewer nucleotides or amino acids than the respective wild-type organism.
- an "insertion” or “addition” is a change in a nucleotide or amino acid sequence which has resulted in the addition of one or more nucleotides or amino acid residues, respectively, as compared to the naturally occurring sequence.
- activation is by insertion in a single crossover event with a plasmid as the vector.
- a sbo chromosomal gene is aligned with a plasmid comprising the gene or part of the gene coding sequence and a selective marker.
- the selective marker is located within the gene coding sequence or on a part of the plasmid separate from the gene.
- the vector is integrated into the Bacillus chromosome, and the gene is inactivated by the insertion of the vector in the coding sequence.
- substitution results from the replacement of one or more nucleotides or amino acids by different nucleotides or amino acids, respectively.
- homologous genes refers to a pair of genes from different, but usually related species, which correspond to each other and which are identical or very similar to each other.
- the term encompasses genes that are separated by speciation (i.e ., the development of new species) (e.g ., orthologous genes), as well as genes that have been separated by genetic duplication (e.g. , paralogous genes).
- ortholog and “orthologous genes” refer to genes in different species that have evolved from a common ancestral gene (i.e ., a homologous gene) by speciation. Typically, orthologs retain the same function in during the course of evolution. Identification of orthologs finds use in the reliable prediction of gene function in newly sequenced genomes.
- paralogous genes refer to genes that are related by duplication within a genome. While orthologs retain the same function through the course of evolution, paralogs evolve new functions, even though some functions are often related to the original one. Examples of paralogous genes include, but are not limited to genes encoding trypsin, chymotrypsin, elastase, and thrombin, which are all serine proteinases and occur together within the same species.
- homology refers to sequence similarity or identity, with identity being preferred. This homology is determined using standard techniques known in the art (see e.g. , Smith and Waterman, Adv. Appl. Math., 2:482 [1981 ]; Needleman and Wunsch, J. Mol. Biol., 48:443 [1970 ]; Pearson and Lipman, Proc. Natl. Acad. Sci. USA 85:2444 [1988 ]; programs such as GAP, BESTFIT, FASTA, and TFASTA in the Wisconsin Genetics Software Package (Genetics Computer Group, Madison, WI); and Devereux et al., Nucl. Acid Res., 12:387-395 [1984 ]).
- an "analogous sequence” is one wherein the function of the gene is essentially the same as the gene designated from Bacillus subtilis strain 168. Additionally, analogous genes include at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% sequence identity with the sequence of the Bacillus subtilis strain 168 gene. Alternately, analogous sequences have an alignment of between 70 to 100% of the genes found in the B . subtilis 168 region and/or have at least between 5 - 10 genes found in the region aligned with the genes in the B . subtilis 168 chromosome. In additional embodiments more than one of the above properties applies to the sequence. Analogous sequences are determined by known methods of sequence alignment. A commonly used alignment method is BLAST, although as indicated above and below, there are other methods that also find use in aligning sequences.
- PILEUP creates a multiple sequence alignment from a group of related sequences using progressive, pairwise alignments. It can also plot a tree showing the clustering relationships used to create the alignment. PILEUP uses a simplification of the progressive alignment method of Feng and Doolittle ( Feng and Doolittle, J. Mol. Evol., 35:351-360 [1987 ]). The method is similar to that described by Higgins and Sharp ( Higgins and Sharp, CABIOS 5:151-153 [1989 ]).
- Useful PILEUP parameters including a default gap weight of 3.00, a default gap length weight of 0.10, and weighted end gaps.
- BLAST BLAST algorithm
- Altschul et al. J. Mol. Biol., 215:403-410, [1990 ]; and Karlin et al., Proc. Natl. Acad. Sci. USA 90:5873-5787 [1993 ]
- the HSP S and HSP S2 parameters are dynamic values and are established by the program itself depending upon the composition of the particular sequence and composition of the particular database against which the sequence of interest is being searched. However, the values may be adjusted to increase sensitivity.
- a % amino acid sequence identity value is determined by the number of matching identical residues divided by the total number of residues of the "longer" sequence in the aligned region. The "longer" sequence is the one having the most actual residues in the aligned region (gaps introduced by WU-Blast-2 to maximize the alignment score are ignored).
- percent (%) nucleic acid sequence identity is defined as the percentage of nucleotide residues in a candidate sequence that are identical with the nucleotide residues of the sequence shown in the nucleic acid figures.
- a preferred method utilizes the BLASTN module of WU-BLAST-2 set to the default parameters, with overlap span and overlap fraction set to 1 and 0.125, respectively.
- the alignment may include the introduction of gaps in the sequences to be aligned.
- sequences which contain either more or fewer nucleosides than those of the nucleic acid figures it is understood that the percentage of homology will be determined based on the number of homologous nucleosides in relation to the total number of nucleosides. Thus, for example, homology of sequences shorter than those of the sequences identified herein and as discussed below, will be determined using the number of nucleosides in the shorter sequence.
- hybridization refers to the process by which a strand of nucleic acid joins with a complementary strand through base pairing, as known in the art.
- a nucleic acid sequence is considered to be "selectively hybridizable" to a reference nucleic acid sequence if the two sequences specifically hybridize to one another under moderate to high stringency hybridization and wash conditions.
- Hybridization conditions are based on the melting temperature (Tm) of the nucleic acid binding complex or probe.
- Tm melting temperature
- maximum stringency typically occurs at about Tm-5°C (5° below the Tm of the probe); “high stringency” at about 5-10°C below the Tm; “intermediate stringency” at about 10-20°C below the Tm of the probe; and “low stringency” at about 20-25°C below the Tm.
- maximum stringency conditions may be used to identify sequences having strict identity or near strict identity with the hybridization probe; while an intermediate or low stringency hybridization can be used to identify or detect polynucleotide sequence homologs.
- Moderate and high stringency hybridization conditions are well known in the art.
- An example of high stringency conditions includes hybridization at about 42°C in 50% formamide, 5X SSC, 5X Denhardt's solution, 0.5% SDS and 100 ⁇ g/ml denatured carrier DNA followed by washing two times in 2X SSC and 0.5% SDS at room temperature and two additional times in 0.1X SSC and 0.5% SDS at 42°C.
- recombinant includes reference to a cell or vector, that has been modified by the introduction of a heterologous nucleic acid sequence or that the cell is derived from a cell so modified.
- recombinant cells express genes that are not found in identical form within the native (non-recombinant) form of the cell or express native genes that are otherwise abnormally expressed, under expressed or not expressed at all as a result of deliberate human intervention.
- “Recombination, "recombining,” or generating a “recombined” nucleic acid is generally the assembly of two or more nucleic acid fragments wherein the assembly gives rise to a chimeric gene.
- library of mutants refers to a population of cells which are identical in most of their genome but include different homologues of one or more genes. Such libraries find use for example, in methods to identify genes or operons with improved traits.
- amplification and “gene amplification” refer to a process by which specific DNA sequences are disproportionately replicated such that the amplified gene becomes present in a higher copy number than was initially present in the genome.
- selection of cells by growth in the presence of a drug results in the amplification of either the endogenous gene encoding the gene product required for growth in the presence of the drug or by amplification of exogenous ( i.e ., input) sequences encoding this gene product, or both.
- amplification reagents refers to those reagents (deoxyribonucleotide triphosphates, buffer, etc.), needed for amplification except for primers, nucleic acid template and the amplification enzyme.
- amplification reagents along with other reaction components are placed and contained in a reaction vessel (test tube, microwell, etc.).
- RT-PCR refers to the replication and amplification of RNA sequences.
- reverse transcription is coupled to PCR, most often using a one enzyme procedure in which a thermostable polymerase is employed, as described in U.S. Patent No. 5,322,770 , herein incorporated by reference.
- the RNA template is converted to cDNA due to the reverse transcriptase activity of the polymerase, and then amplified using the polymerizing activity of the polymerase ( i.e ., as in other PCR methods).
- restriction site refers to a nucleotide sequence recognized and cleaved by a given restriction endonuclease and is frequently the site for insertion of DNA fragments.
- restriction sites are engineered into the selective marker and into 5' and 3' ends of the DNA construct.
- ingenous chromosomal region and "a fragment of an indigenous chromosomal region” refer to a segment of the Bacillus chromosome which is deleted from a Bacillus host cell in some embodiments of the present invention.
- deleted segments include one or more genes with known functions, while in other embodiments, deleted segments include one or more genes with unknown functions, and in other embodiments, the deleted segments include a combination of genes with known and unknown functions.
- indigenous chromosomal regions or fragments thereof include as many as 200 genes or more.
- An indigenous chromosomal region or fragment thereof can have a necessary function under certain conditions, but the region is not necessary for Bacillus strain viability under laboratory conditions.
- Preferred laboratory conditions include but are not limited to conditions such as growth in a fermenter, in a shake flask on plated media, etc., at standard temperatures and atmospheric conditions ( e.g ., aerobic).
- the chromosome of the altered Bacillus strain may include 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 85%, 80%, 75% or 70% of the corresponding unaltered Bacillus host chromosome.
- the chromosome of an altered Bacillus strain according to the invention will include about 99 to 90%; 99 to 92%; and 98 to 94% of the corresponding unaltered Bacillus host strain chromosome genome.
- chromosomal integration refers to the process whereby the incoming sequence is introduced into the chromosome of a host cell (e.g., Bacillus ).
- the homologous regions of the transforming DNA align with homologous regions of the chromosome.
- the sequence between the homology boxes is replaced by the incoming sequence in a double crossover (i.e ., homologous recombination).
- Homologous sections of an inactivating chromosomal segment of a DNA construct align with the flanking homologous regions of the indigenous chromosomal region of the Bacillus chromosome.
- the indigenous chromosomal region is deleted by the DNA construct in a double crossover (i.e., homologous recombination).
- a "multi-contiguous single gene region” is a region wherein at least the coding regions of two genes occur in tandem and in some embodiments, include intervening sequences preceding and following the coding regions.
- the present invention includes embodiments that involve singe gene deletions.
- the incoming sequence comprises a selective marker flanked by a homology box on both ends.
- the incoming sequence includes a sequence that interrupts the transcription and/or translation of the coding sequence.
- the DNA construct includes restriction sites engineered at the upstream and downstream ends of the construct.
- the DNA construct is incorporated into a vector or used without the presence of plasmid DNA, it is used to transform microorganisms. It is contemplated that any suitable method for transformation will find use with the present invention.
- at least one copy of the DNA construct is integrated into the host Bacillus chromosome.
- one or more DNA constructs of the invention are used to transform host cells.
- the incoming sequence comprises a selective marker located between two Iox P sites ( See , Kuhn and Torres, Meth. Mol. Biol., 180:175-204 [2002 ]), and the antimicrobial is then deleted by the action of Cre protein. In some embodiments, this results in the insertion of a single loxP site, as well as a deletion of native DNA, as determined by the primers used to construct homologous flanking DNA and antimicrobial-containing incoming DNA.
- host cells are transformed with one or more DNA constructs according to the present invention to produce an altered Bacillus strain wherein two or more genes have been inactivated in the host cell.
- two or more genes are deleted from the host cell chromosome.
- two or more genes are inactivated by insertion of a DNA construct.
- the inactivated genes are contiguous (whether inactivated by deletion and/or insertion), while in other embodiments, they are not contiguous genes.
- secretion of a protein of interest is higher in the altered strain obtained using the present invention than in a corresponding unaltered host.
- the altered Bacillus cells produced using the present invention are maintained and grown under conditions suitable for the expression and recovery of a polypeptide of interest from cell culture ( See e.g ., Hardwood and Cutting (eds.) Molecular Biological Methods for Bacillus, John Wiley & Sons [1990 ]).
- Primers for the antimicrobial markers contained BamHI sites at both ends of the fragment. Where possible, PCR primers were designed to remove promoters of deleted indigenous chromosomal regions, but to leave all terminators in the immediate area.
- the primary source of chromosome sequence, gene localization, and promoter and terminator information was obtained from Kunststoff et al., (1997) supra and also obtainable from the SubtiList World Wide Web Server known to those in the art ( See e.g ., Moszer et al ., supra ). Numerous deletions have been made using the present invention. A list of primer sequences from deletions created by this method is provided in Table 1. Reference is also made to Figure 2 for an explanation of the primer naming system. Table 1.
- PCR reactions carried out in 150 ⁇ L Eppendorf tubes containing 84 ⁇ L water, 10 ⁇ L PCR buffer, 1 ⁇ L of each primer (i.e. , PKS-UF and PKS-UR), 2 ⁇ L of dNTPs, 1 ⁇ L of wild type Bacillus chromosomal DNA template, and 1 ⁇ L of polymerase.
- DNA polymerases used included Taq Plus Precision polymerase and Herculase (Stratagene). Reactions were carried out in a Hybaid PCRExpress thermocycler using the following program. The samples were first heated at 94°C for 5 minutes, then cooled to a 50° hold. Polymerase was added at this point. Twenty-five cycles of amplification consisted of 1 minute at 95°C, 1 minute at 50°C and 1 minute at 72°C. A final 10 minutes at 72°C ensured complete elongation. Samples were held at 4°C for analysis.
- Digests were performed at 37°C for 1 hour as a 20 ⁇ L reaction consisting of 9 ⁇ L of water, 2 ⁇ L of 10xBSA, 2 ⁇ L of an appropriate NEB restriction buffer (according to the 2000-01 NEB Catalog and Technical Reference), 5 ⁇ L of template, and 1 ⁇ L of each restriction enzyme.
- NEB New England BioLabs
- the digested fragments were purified by gel electrophoresis and extraction using the Qiagen Qiaquick gel extraction kit following the manufacturer's instructions.
- Figures 5 and 6 provide gels showing the results for various deletions.
- the pJM102 plasmid was cut with the unique restriction enzyme sites appropriate for each deletion ( See , Table 2; for cssS , Xba I and Sac I were used) and purified as described above prior to ligation. This recircularized plasmid was transformed into Invitrogen's "Top Ten" E. coli cells, using the manufacturers One Shot transformation protocol.
- Transformants were selected on Luria-Bertani broth solidified with1.5% agar (LA) plus 50 ppm carbanicillin containing X-gal for blue-white screening. Clones were picked and grown overnight at 37°C in 5mL of Luria Bertani broth (LB) plus 50 ppm carbanicillin and plasmids were isolated using Qiagen's Qiaquick Mini-Prep kit. Restriction analysis confirmed the presence of the insert by cutting with the restriction sites at each end of the insert to drop an approximately 2 kb band out of the plasmid.
- Method 2 Upstream and downstream fragments were amplified as in Method 1, except the primers were designed with 25 bp "tails" complementary to the antimicrobial marker's primer sequences.
- a “tail” is defined herein as base pairs on the 5' end of a primer that are not homologous to the sequence being directly amplified, but are complementary to another sequence of DNA.
- the primers for amplifying the antimicrobial contain "tails" that are complementary to the fragments' primers.
- the DeletionX-UFfus and DeletionX-URfus are direct complements of one another. This is also true for the DFfus and DR-fus primer sets.
- these primers contain restriction enzyme sites similar to those used in Method 1 for use in creating a plasmid vector (See, Table 3 and U.S. Patent No. 5,023,171 ).
- Table 3 provides a list of primers useful for creation of deletion constructs by PCR fusion.
- Table 4 provides an additional list of primers useful for creation of deletion constructs by PCR fusion. However, in this Table, all deletion constructs would include the phleo R marker.
- Primers Primer name Restriction enzyme engineered into primer Sequence SEQ ID. NO.
- the fragments listed in Tables 3 and 4 were size-verified by gel electrophoresis as described above. If correct, 1 ⁇ L each of the upstream, downstream, and antimicrobial resistance marker fragments were placed in a single reaction tube with the DeletionX-UF and DeletionX-DR primers or nested primers where listed. Nested primers are 25 base pairs of DNA homologous to an internal portion of the upstream or downstream fragment, usually about 100 base pairs from the outside end of the fragment (See, Figure 2 ). The use of nested primers frequently enhances the success of fusion.
- the PCR reaction components were similar to those described above, except 82 ⁇ L of water was used to compensate for additional template volume.
- the PCR reaction conditions were similar to those described above, except the 72°C extension was lengthened to 3 minutes. During extension, the antimicrobial resistance gene was fused in between the upstream and downstream pieces. This fusion fragment can be directly transformed into Bacillus without any purification steps or with a simple Qiagen Qiaquick PRC purification done according to manufacturer's instructions.
- Method 3 a method for creating DNA constructs using ligation of PCR fragments and direct transformation of Bacillus are described.
- modification of prpC, sigD and tdh / kbl are provided to demonstrate the method of ligation.
- sigD and tdh / kbl were constructed by one method and prpC by an alternate method.
- the upstream and downstream fragments adjacent to the tdh/kbl region of the Bacillus subtilis chromosome were amplified by PCR similar to as described in Method 1, except that the inside primer of the flanking DNA was designed to contain type II s restriction sites.
- Primers for the loxP-spectinomycin-loxP cassette were designed with the same type II s restriction site as the flanks and complementary overhangs.
- Unique overhangs for the left flank and the right flank allowed directional ligation of the antimicrobial cassette between the upstream and downstream flanking DNA. All DNA fragments were digested with the appropriate restriction enzymes, and the fragments were purified with a Qiagen Qiaquick PCR purification kit using the manufacturer's instructions.
- An additional example of creating a DNA molecule by ligation of PCR amplified DNA fragments for direct transformation of Bacillus involved a partial in-frame deletion of the gene prpC.
- a 3953 bp fragment of Bacillus subtilis chromosomal DNA containing the prpC gene was amplified by PCR using primers p95 and p96. The fragment was cleaved at unique restriction sites PfIM I and Bst XI. This yielded three fragments, an upstream, a downstream, and a central fragment. The latter is the fragment deleted and consists of 170 bp located internal to the prpC gene.
- the digestion mixture was purified with a Qiagen Qiaquick PCR purification kit, followed by desalting in a 1 mL spin column containing BioRad P-6 gel and equilibrated with 2 mM Tris-HCl, pH 7.5.
- the antimicrobial cassette, IoxP-spectinomycin-loxP was amplified with the primer containing a Bst XI site and the downstream primer containing a Pfl MI site both with cleavage sites complementary to the sites in the genomic DNA fragment.
- the fragment was digested with Pfl MI and Bst XI and purified as described for the chromosomal fragment above.
- pckA was also modified.
- the PCR primers pckA UF, pckA-2Urfus, spc ffus, spc rfus, pckA Dffus and pckA DR were used for PCR and PCR fusion reactions using the chromosomal DNA of a Bacillus subtilis I168 derivative and pDG1726 ( See , Guerout-Fleury et al., Gene 167(1-2):335-6 [1995 ]) as template.
- the primers are shown in Table 8.
- the method used in constructing these deletion mutants was the same as Method 1, described above. Table 8.
- Cells of a host strain Bacillus subtilis with partial genotype xylRcomK were rendered competent by growth for 2 hours in Luria-Bertani medium containing 1% xylose, as described in U.S. Patent Appln. Ser. No. 09/927,161, filed August 10, 2001 , herein incorporated by reference, to an OD 550 of 1.
- This culture was seeded from a 6 hour culture. All cultures were grown at 37°C, with shaking at 300 rpm. Aliquots of 0.3 mL of were frozen as 1:1 mixtures of culture and 30% glycerol in round bottom 2 mL tubes and stored in liquid nitrogen for future use.
- transformants were picked into Luria-Bertani (100 ppm spectinomycin) and grown at 37 °C for genomic DNA isolation performed as known in the art ( See e.g ., Harwood and Cuttings, Molecular Biological Methods for Bacillus, John Wiley and Son, New York, N.Y. [1990], at p. 23 ). Typically 400 to 1400 transformants were obtained from 100 uL transformation mix, when 5 uL of ligation reaction mix was used in the transformation.
- the marker could be removed by transforming the strain with a plasmid containing the cre gene capable of expression the Cre protein.
- Cells were transformed with pCRM-TS-pleo ( See below) cultured at 37 °C to 42 °C, plated onto LA and after colonies formed patched onto LA containing 100 ppm spectinomycin. Patches which did not grow after overnight incubation were deemed to have lost the antimicrobial maker. Loss of maker was verified by PCR assay with primers appropriate for the given gene.
- pCRM-TS-pleo has the following sequence (SEQ ID NO:205):
- transcriptome DNA array methods were used in the development of mutants of the present invention.
- target RNA was harvested from a Bacillus strain by guanidinium acid phenol extraction as known in the art ( See e.g ., Farrell, RNA Methodologies, (2nd Ed.). Academic Press, San Diego, at pp. 81 ] and time-point was reverse-transcribed into biotin-labeled cDNA by a method adopted from deSaizieu et al. ( deSaizieu et a/., J. Bacteriol., 182: 4696-4703 [2000 ]) and described herein.
- RNA 25 mg was incubated 37°C overnight in a 100-mL reaction: 1x GIBCO first-strand buffer (50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl 2 ); 10 mM DTT; 40 mM random hexamer; 0.3 mM each dCTP, dGTP and dTTP; 0.12 mM dATP; 0.3 mM biotin-dATP (NENO; 2500 units SuperScript II reverse-transcriptase (Roche). To remove RNA, the reaction was brought to 0.25 M NaOH and incubated at 65°C for 30 minutes.
- 1x GIBCO first-strand buffer 50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl 2 ); 10 mM DTT; 40 mM random hexamer; 0.3 mM each dCTP, dGTP and dTTP; 0.12
- reaction yield was approximately 20-25 mg biotin-labeled cDNA.
- Hybridizations were performed as described in the Affymetrix Expression Analysis Technical Manual (Affymetrix) using reagent suppliers as suggested. Briefly, 10 mg of fragmented biotin-labeled cDNA were added to a 220-mL hybridization cocktail containing: 100 mM MES (N-morpholinoethanesufonic acid), 1M Na + , 20 mM EDTA, 0.01% Tween 20; 5 mg/mL total yeast RNA; 0.5 mg/mL BSA; 0.1 mg/mL herring-sperm DNA; 50 pM control oligonucleotide (AFFX-B1).
- MES N-morpholinoethanesufonic acid
- the cocktails were heated to 95°C for 5 minutes, cooled to 40°C for 5 minutes, briefly centrifuged to remove particulates, and 200 mL was injected into each pre-warmed pre-rinsed (1x MES buffer + 5 mg/ml yeast RNA) GeneChip cartridge. The arrays were rotated at 40°C overnight.
- the signals in the arrays were detected with the Hewlett-Packard Gene Array Scanner using 570 nm laser light with 3-mm pixel resolution.
- the signal intensities of the 4351 ORF probe sets were scaled and normalized across all time points comprising a time course experiment. These signals were then compared to deduce the relative expression levels of genes under investigation.
- the threonine biosynthetic and degradative genes were simultaneous transcribed, indicating inefficient threonine utilization. Deletion of the degradative threonine pathway improved expression of the desired product ( See , Figure 7 ).
- the present invention provides means to modify pathways with transcription profiles that are similar to threonine biosynthetic and degradative profiles.
- the present invention also finds use in the modification of pathways with transcription profiles similar to threonine in order to optimize Bacillus strains.
- at least one gene selected from the group consisting of rocA, ycgN, ycgm rocF and rocD is deleted or otherwise modified.
- the present invention provides means to improve strain protein production through the combination of pckA deletion or modification and deletion or modification of gapB and/or fbp.
- the tryptophan biosynthetic pathway genes showed unbalanced transcription.
- the present invention will find use in producing strains that exhibit increased transcription of genes such as those selected from the group consisting of trpA, trpB, frpC, trpD, trpE, and/or trpF, such that the improved strains provide improved expression of the desired product, as compared to the parental ( i.e ., wild-type and/or originating strain).
- modifications of these genes in any combination will lead to improved expression of the desired product.
- a mutant strain was compared to parent strain to judge improvements. Although this mutant pckA strain did not show improvement under these conditions, it is contemplated that improvements will be produced under modified culture conditions ( i.e ., as known to those in the art), and/or incorporation of additional genes.
- these additional genes are selected from the group consisting of gapB, alsD, and/or fbp
- the DNA construct was created by Method 1 or 2 as described above, it was transformed into a suitable Bacillus subtilis lab strain (e.g ., BG2036 or BG2097; any competent Bacillus immediate host cell may be used in the methods of the present invention).
- the cells were plated on a selective media of 0.5 ppm phleomycin or 100 ppm spectinomycin as appropriate (Ferrari and Miller, Bacillus Expression: A Gram-Positive Model in Gene Expression Systems: Using Nature for the Art of Expression, pgs 65-94 [1999 ]).
- the laboratory strains were used as a source of chromosomal DNA carrying the deletion that was transformed into a Bacillus subtilis production host strain twice or BG3594 and then MDT 98-113 once. Transformants were streaked to isolate a single colony, picked and grown overnight in 5 mL of LB plus the appropriate antimicrobial. Chromosomal DNA was isolated as known in the art ( See e.g. , Hardwood et al ., supra ).
- the presence of the integrated DNA construct was confirmed by three PCR reactions, with components and conditions as described above. For example, two reactions were designed to amplify a region from outside the deletion cassette into the antimicrobial gene in one case (primers 1 and 11) and through the entire insert in another (primers 1 and 12). A third check amplified a region from outside the deletion cassette into the deleted region (primers 1 and 4). Figure 4 shows that a correct clone showed a band in the first two cases but not the third. Wild-type Bacillus subtilis chromosomal DNA was used as a negative control in all reactions, and should only amplify a band with the third primer set.
- subtilisin activity was measured by shake flask assays and the activity was compared to wild type levels. Assays were performed in 250 ml baffled flasks containing 50 mL of growth media suitable for subtilisin production as known in the art ( See , Christianson et al., Anal. Biochem., 223:119-129 [1994 ]; and Hsia et al., Anal. Biochem. 242:221 - 227 [1996 ]). The media were inoculated with 50 ⁇ L of an 8 hour 5mL culture and grown for 40 hours at 37°C with shaking at 250 RPM.
- Figure 8 provides a graph showing improved protease secretion as measured from shake flask cultures in Bacillus subtilis wild-type strain (unaltered) and corresponding altered deletion strains ( -sbo ) and (- slr ). Protease activity (g/L) was measured after 17, 24 and 40 hours.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Microbiology (AREA)
- Medicinal Chemistry (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- General Chemical & Material Sciences (AREA)
- Gastroenterology & Hepatology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Tropical Medicine & Parasitology (AREA)
- Virology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Description
- The present invention provides cells that have been genetically manipulated to have an altered capacity to produce expressed proteins. In particular, the present invention relates to Gram-positive microorganisms, such as Bacillus species having enhanced secretion of a subtilisin, wherein one or more chromosomal genes have been inactivated, and preferably wherein one or more chromosomal genes have been deleted from the Bacillus chromosome.
- Genetic engineering has allowed the improvement of microorganisms used as industrial bioreactors, cell factories and in food fermentations. In particular, Bacillus species produce and secrete a large number of useful proteins and metabolites (Zukowski, "Production of commercially valuable products," In: Doi and McGlouglin (eds.) Biology of Bacilli: Applications to Industry, Butterworth-Heinemann, Stoneham. Mass pp 311-337 [1992]). The most common Bacillus species used in industry are B. licheniformis, B. amyloliquefaciens and B. subtilis. Because of their GRAS (generally recognized as safe) status, strains of these Bacillus species are natural candidates for the production of proteins utilized in the food and pharmaceutical industries. Important production enzymes include α-amylases, neutral proteases, and alkaline (or serine) proteases. However, in spite of advances in the understanding of production of proteins in Bacillus host cells, there remains a need for methods to increase expression of these proteins.
- The present invention relates to Gram-positive microorganisms, such as Bacillus species having enhanced secretion of a subtilisin, wherein one or more chromosomal genes have been inactivated, and preferably wherein one or more chromosomal genes have been deleted from the Bacillus chromosome. The present invention provides methods and compositions for the improved secretion of a subtilisin in Bacillus.
- The present invention provides means for improved secretion of a subtilisin in Bacillus. More particularly, in these embodiments, the present invention involves inactivation of one or more chromosomal genes in a Bacillus host strain, wherein the inactivated genes are not necessary for strain viability.
- The present invention provides methods and compositions for enhancing secretion of a subtilisin from a Bacillus cell. The methods comprise inactivating sbo, in a Bacillus host strain to produce an altered Bacillus strain; growing the altered Bacillus strain under suitable growth conditions; and allowing a subtilisin to be secreted in the altered Bacillus, wherein the secretion of the subtilisin is enhanced, compared to the corresponding unaltered Bacillus host strain. Inactivation of a chromosomal gene can comprise the deletion of a gene to produce the altered Bacillus strain. In additional embodiments, inactivation of a chromosomal gene comprises insertional inactivation.
- The present invention provides altered Bacillus strains comprising the deletion of sbo. The altered Bacillus strain is a subtilisin producing strain. In yet other embodiments, the altered Bacillus strain further comprises a mutation In a gene selected from the group consisting of degU, degQ, degS, scoC4, spoIIE, and oppA.
- In further embodiments, the present invention provides DNA constructs comprising an incoming sequence. In some embodiments, the incoming sequence includes a selective marker and a gene or gene fragment of sbo. In alternative embodiments, the selective marker is located in between two fragments of the gene. In other embodiments, the incoming sequence comprises a selective marker and a homology box, wherein the homology box flanks the 5' and/or 3' end of the marker. In additional embodiments, a host cell is transformed with the DNA construct. In further embodiments, the host cell is a Bacillus cell. The DNA construct is chromosomally integrated into the host cell.
- Disclosed are methods for obtaining an altered Bacillus strain expressing a protein of interest which comprises transforming a Bacillus host cell with the DNA construct of the present, wherein the DNA construct is integrated into the chromosome of the Bacillus host cell; producing an altered Bacillus strain, wherein one or more chromosomal genes have been inactivated; and growing the altered Bacillus strain under suitable growth conditions for the expression of a protein of interest In yet additional embodiments, the Bacillus host strain is selected from the group consisting of B. licheniformis, B. lentus, B. subtilis, B. amyloliquefaciens B. brevis, B. stearothermophilus, B. alkalophilus, B. coagulans, B. circulans, B. pumilus, B. thuringiensis, B. clausii, B. megaterium, and preferably, B. subtilis. In some embodiments, the Bacillus host strain is a recombinant host. In yet additional embodiments, the protein of interest is recovered. In further embodiments, the selective marker is excised from the altered Bacillus.
- Disclosed are methods for obtaining an altered Bacillus strain expressing a protein of interest. The method comprises transforming a Bacillus host cell with a DNA construct comprising an incoming sequence wherein the incoming sequence comprises a selective marker and of sbo, wherein the DNA construct is integrated into the chromosome of the Bacillus host cell and results in the deletion of one or more gene(s); obtaining an altered Bacillus strain, and growing the altered Bacillus strain under suitable growth conditions for the secretion of the protein of interest.
- Disclosed are methods for obtaining Bacillus subtilis strains that demonstrate enhanced protease production. The methods comprise the steps of transforming a Bacillus subtilis host cell with a DNA construct according to the invention; allowing homologous recombination of the DNA construct and a homologous region of the Bacillus chromosome wherein sbo, is deleted from the Bacillus chromosome; obtaining an altered Bacillus subtilis strain; and growing the altered Bacillus strain under conditions suitable for the expression of a protease. The protease producing Bacillus is a subtilisin producing strain. In additional embodiments, the protease producing strain further includes a mutation in a gene selected from the group consisting of degU, degQ, degS, scoC4, spoIIE, and oppA. In some embodiments, the inactivation comprises the insertional inactivation of the gene.
- The present invention provides methods for enhancing the secretion of a subtilisin in Bacillus comprising: obtaining an altered Bacillus strain produced by introducing a DNA construct including a selective marker and an inactivating chromosomal segment into a Bacillus host strain, wherein the DNA construct is integrated into the Bacillus chromosome resulting in the deletion of an indigenous chromosomal region or fragment thereof from the Bacillus host cell; and growing the altered Bacillus strain under suitable growth conditions, wherein expression of a protein of interest is greater in the altered Bacillus strain compared to the expression of the protein of interest is the corresponding unaltered Bacillus host cell.
- Zheng et al. (J. Bacteriol. Vol 181, No. 23, pp 7346-7355) discloses the construction of a Bacillus strain lacking the sbo gene. According to Zheng et al., genes of the abo-alb locus are required for production of the antilisteral bateriocin subtilosin.
- Van der laan et al. (Appl. Environ. Microbiol. Vol. 57, No. 4, pp 901-909) discloses the cloning, characterisation and multiple chromosomal integration of the Bacillus PB92 alkaline protease gene.
-
-
Figure 1 , Panels A and B illustrate a general schematic diagram of one method ("Method 1"' See, Example 1) provided. In this method, flanking regions of a gene and/or an indigenous chromosomal region are amplified out of a wild-type Bacillus chromosome, cut with restriction enzymes (including at least BamHI) and ligated into pJM102. The construct is cloned through E. coli and the plasmid is isolated, linearized with BamHI and ligated to an antimicrobial marker with complementary ends. After cloning again in E. coli, a liquid culture is grown and used to isolate plasmid DNA for use in transforming a Bacillus host strain (preferably, a competent Bacillus host strain). -
Figure 2 illustrates the location of primers used in the construction of a DNA cassette. The diagram provides an explanation of the primer naming system used herein.Primers 1 and 4 are used for checking the presence of the deletion. These primers are referred to as "DeletionX-UF-chk" and "DeletionX-UR-chk-del." DeletionX-UF-chk is also used in a PCR reaction with a reverse primer inside the antimicrobial marker (Primer 11: called for example PBSX-UR-chk-Del) for a positive check of the cassette's presence in the chromosome.Primers 2 and 6 are used to amplify the upstream flanking region. These primers are referred to as "DeletionX-UF" and "DeletionX-UR," and contain engineered restriction sites at the black vertical bars.Primers Primers -
Figure 3 is a general schematic diagram of one method ("Method 2"; See Example 2). Flanking regions are engineered to include 25 bp of sequence complementary to a selective marker sequence. The selective marker sequence also includes 25 bp tails that complement DNA of one flanking region. Primers near the ends of the flanking regions are used to amplify all three templates in a single reaction tube, thereby creating a fusion fragment. This fusion fragment or DNA construct is directly transformed into a competent Bacillus host strain. -
Figure 4 provides an electrophoresis gel of Bacillus DHB deletion clones.Lanes primers Lane 3 depicts the wild-type control for this reaction. Only non-specific amplification is observed.Lanes 4 and 5 depict the DHB deleted strains amplified withprimers 9 and 12. This 2 kb band amplifies through the antibiotic region to below the downstream section of the inactivated chromosomal segment. Lane 6 is the negative control for this reaction and a band is not illustrated.Lanes primers 1 and 4 and the illustration confirms that the DHB region is missing. Lane 9 is the wild-type control. -
Figure 5 illustrates gel electrophoresis of two clones of a production strain of Bacillus subtilis (wild-type) wherein slr is replaced with a phleomycin (phleo) marker which results in a deletion of the slr gene.Lanes locations Lane 3 is the wild-type chromosomal DNA amplified with the same primers. A 1.2 kb band is observed for the insert.Lanes 4 and 5 represent the clones amplified with primers atlocations 9 and 12. Lane 6 is the wild-type chromosomal DNA amplified with the same primers. Correct transformants include a 2 kb band.Lanes locations 2 and 4. Lane 9 is the wild-type chromosomal DNA amplified with the same primers. No band is observed for the deletion strains, but a band around 1 kb is observed in the wild-type. Reference is made toFigure 2 for an explanation of primer locations. -
Figure 6 provides an electrophoresis gel of a clone of a production strain of Bacillus subtilis (wild-type) wherein cssS is inactivated by the integration of a spc marker into the chromosome.Lane 1 is a control without the integration and is approximately 1.5kb smaller. -
Figure 7 provides a bar graph showing improved subtilisin secretion measured from shake flask cultures with Bacillus subtilis wild-type strain (unaltered) and corresponding altered Bacillus subtilis strains having various deletions. Protease activity (g/L) was measured after 17, 24 and 40 hours or was measured at 24 and 40 hours. -
Figure 8 provides a bar graph showing improved protease secretion as measured from shake flask cultures in Bacillus subtilis wild-type strain (unaltered) and corresponding altered deletion strains (-sbo) and (-slr). Protease activity (g/L) was measured after 17, 24 and 40 hours. - Unless defined otherwise herein, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs (See e.g., Singleton et al., DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOGY, 2D ED., John Wiley and Sons, New York [1994]; and Hale and Marham, THE HARPER COLLINS DICTIONARY OF BIOLOGY, Harper Perennial, NY [1991], both of which provide one of skill with a general dictionary of many of the terms used herein). Although any methods and materials similar or equivalent to those described herein can be used in the practice or testing of the present invention, the preferred methods and materials are described. Numeric ranges are inclusive of the numbers defining the range. As used herein and in the appended claims, the singular "a", "an" and "the" includes the plural reference unless the context clearly dictates otherwise. Thus, for example, reference to a "host cell" includes a plurality of such host cells.
- Unless otherwise indicated, nucleic acids are written left to right in 5' to 3' orientation; amino acid sequences are written left to right in amino to carboxy orientation, respectively. The headings provided herein are not limitations of the various aspects or embodiments of the invention that can be had by reference to the specification as a whole. Accordingly, the terms defined immediately below are more fully defined by reference to the Specification as a whole.
- As used herein, "host cell" refers to a cell that has the capacity to act as a host or expression vehicle for a newly introduced DNA sequence. In preferred embodiments of the present invention, the host cells are Bacillus sp. or E. coli cells.
- As used herein, "the genus Bacillus" includes all species within the genus "Bacillus," as known to those of skill in the art, including but not limited to B. subtilis, B. licheniformis, B. lentus, B. brevis, B. stearothermophilus, B. alkalophilus, B. amyloliquefaciens, B. clausii, B. halodurans, B. megaterium, B. coagulans, B. circulans, B. lautus, and B. thuringiensis. It is recognized that the genus Bacillus continues to undergo taxonomical reorganization. Thus, it is intended that the genus include species that have been reclassified, including but not limited to such organisms as B. stearothermophilus, which is now named "Geobacillus stearothermophilus." The production of resistant endospores in the presence of oxygen is considered the defining feature of the genus Bacillus, although this characteristic also applies to the recently named Alicyclobacillus, Amphibacillus, Aneurinibacillus, Anoxybacillus, Brevibacillus, Filobacillus, Gracilibacillus, Halobacillus, Paenibacillus, Salibacillus, Thermobacillus, Ureibacillus, and Virgibacillus.
- As used herein, "nucleic acid" refers to a nucleotide or polynucleotide sequence, and fragments or portions thereof, as well as to DNA, cDNA, and RNA of genomic or synthetic origin which may be double-stranded or single-stranded, whether representing the sense or antisense strand. It will be understood that as a result of the degeneracy of the genetic code, a multitude of nucleotide sequences may encode a given protein.
- As used herein the term "gene" means a chromosomal segment of DNA involved in producing a polypeptide chain that may or may not include regions preceding and following the coding regions (e.g. 5' untranslated (5' UTR) or leader sequences and 3' untranslated (3' UTR) or trailer sequences, as well as intervening sequence (introns) between individual coding segments (exons)).
- As used herein, the term "vector" refers to any nucleic acid that can be replicated in cells and can carry new genes or DNA segments into cells. Thus, the term refers to a nucleic acid construct designed for transfer between different host cells. An "expression vector" refers to a vector that has the ability to incorporate and express heterologous DNA fragments in a foreign cell. Many prokaryotic and eukaryotic expression vectors are commercially available. Selection of appropriate expression vectors is within the knowledge of those having skill in the art.
- As used herein, the terms "DNA construct," "expression cassette," and "expression vector," refer to a nucleic acid construct generated recombinantly or synthetically, with a series of specified nucleic acid elements that permit transcription of a particular nucleic acid in a target cell (i.e., these are vectors or vector elements, as described above). The recombinant expression cassette can be incorporated into a plasmid, chromosome, mitochondrial DNA, plastid DNA, virus, or nucleic acid fragment. Typically, the recombinant expression cassette portion of an expression vector includes, among other sequences, a nucleic acid sequence to be transcribed and a promoter.
- As used herein, "transforming DNA," "transforming sequence," and "DNA construct" refer to DNA that is used to introduce sequences into a host cell or organism. Transforming DNA is DNA used to introduce sequences into a host cell or organism. The DNA may be generated in vitro by PCR or any other suitable techniques. The transforming DNA comprises an incoming sequence, flanked by homology boxes. The transforming DNA can comprise other non-homologous sequences, added to the ends (i.e., stuffer sequences or flanks). The ends can be closed such that the transforming DNA forms a closed circle, such as, for example, insertion into a vector.
- As used herein, the term "plasmid" refers to a circular double-stranded (ds) DNA construct used as a cloning vector, and which forms an extrachromosomal self-replicating genetic element in many bacteria and some eukaryotes. In some embodiments, plasmids become incorporated into the genome of the host cell.
- As used herein, the terms "isolated" and "purified" refer to a nucleic acid or amino acid (or other component) that is removed from at least one component with which it is naturally associated.
- The present invention provides means for enhancing subtilisin secretion, such that the enhanced strains produced a greater quantity and/or quality of a protein of interest than the parental strain (e.g., the wild-type and/or originating strain).
- As used herein the term "expression" refers to a process by which a polypeptide is produced based on the nucleic acid sequence of a gene. The process includes both transcription and translation.
- As used herein in the context of introducing a nucleic acid sequence into a cell, the term "introduced" refers to any method suitable for transferring the nucleic acid sequence into the cell. Such methods for introduction include but are not limited to protoplast fusion, transfection, transformation, conjugation, and transduction (See e.g., Ferrari et al., "Genetics," in Hardwood et al, (eds.), Bacillus, Plenum Publishing Corp., pages 57-72, [1989]).
- As used herein, the terms "transformed" and "stably transformed" refers to a cell that has a non-native (heterologous) polynucleotide sequence integrated into its genome or as an episomal plasmid that is maintained for at least two generations.
- As used herein "an incoming sequence" refers to a DNA sequence that is introduced into the Bacillus chromosome. The incoming sequence is part of a DNA construct. The incoming sequence encodes one or more proteins of interest. In some embodiments, the incoming sequence comprises a sequence that may or may not already be present in the genome of the cell to be transformed (i.e., it may be either a homologous or heterologous sequence). The incoming sequence encodes a sequence that is already present in the chromosome of the host cell to be transformed. The incoming sequence comprises of sbo, in yet another embodiment, the incoming sequence includes a selective marker. In a further embodiment the incoming sequence includes two homology boxes.
- As used herein, "homology box" refers to a nucleic acid sequence, which is homologous to a sequence in the Bacillus chromosome. More specifically, a homology box is an upstream or downstream region having between about 80 and 100% sequence identity, between about 90 and 100% sequence identity, or between about 95 and 100% sequence identity with the immediate flanking coding region of a gene or part of a gene to be inactivated according to the invention. These sequences direct where in the Bacillus chromosome a DNA construct is integrated and directs what part of the Bacillus chromosome is replaced by the incoming sequence. While not meant to limit the invention, a homology box may include about between 1 base pair (bp) to 200 kilobases (kb). Preferably, a homology box includes about between 1 bp and 10.0 kb; between 1 bp and 5.0 kb; between 1 bp and 2.5 kb; between 1 bp and 1.0 kb, and between 0.25 kb and 2.5 kb. A homology box may also include about 10.0 kb, 5.0 kb, 2.5 kb, 2.0 kb, 1.5 kb, 1.0 kb, 0.5 kb, 0.25 kb and 0.1 kb. In some embodiments, the 5' and 3' ends of a selective marker are flanked by a homology box wherein the homology box comprises nucleic acid sequences immediately flanking the coding region of the gene.
- As used herein, the term "selectable marker-encoding nucleotide sequence" refers to a nucleotide sequence which is capable of expression in the host cells and where expression of the selectable marker confers to cells containing the expressed gene the ability to grow in the presence of a corresponding selective agent or lack of an essential nutrient.
- As used herein, the terms "selectable marker" and "selective marker" refer to a nucleic acid (e.g., a gene) capable of expression in host cell which allows for ease of selection of those hosts containing the vector. Examples of such selectable markers include but are not limited to antimicrobials. Thus, the term "selectable marker" refers to genes that provide an indication that a host cell has taken up an incoming DNA of interest or some other reaction has occurred. Typically, selectable markers are genes that confer antimicrobial resistance or a metabolic advantage on the host cell to allow cells containing the exogenous DNA to be distinguished from cells that have not received any exogenous sequence during the transformation. A "residing selectable marker" is one that is located on the chromosome of the microorganism to be transformed. A residing selectable marker encodes a gene that is different from the selectable marker on the transforming DNA construct. Selective markers are well known to those of skill in the art. As indicated above, preferably the marker is an antimicrobial resistant marker (e.g., ampR; phleoR; specR; kanR; eryR; tetR; cmpR; and neoR; See e.g., Guerot-Fleury, Gene, 167:335-337 [1995]; Palmeros et al., Gene 247:255-264 [2000]; and Trieu-Cuot et al., Gene, 23:331-341 [1983]). Provided is a chloramphenicol resistance gene (e.g., the gene present on pC194, as well as the resistance gene present in the Bacillus licheniformis genome). This resistance gene is particularly useful in the present invention, as well as in embodiments involving chromosomal amplification of chromosomally integrated cassettes and integrative plasmids (See e.g., Albertini and Galizzi, Bacteriol., 162:1203-1211 [1985]; and Stahl and Ferrari, J. Bacteriol., 158:411-418 [1984]). The DNA sequence of this naturally-occurring chloramphenicol resistance gene is shown below:


- The deduced amino acid sequence of this chloramphenicol resistance protein is:

- Other markers useful in accordance with the invention include, but are not limited to auxotrophic markers, such as tryptophan; and detection markers, such as β-galactosidase.
- As used herein, the term "promoter" refers to a nucleic acid sequence that functions to direct transcription of a downstream gene. In preferred embodiments, the promoter is appropriate to the host cell in which the target gene is being expressed. The promoter, together with other transcriptional and translational regulatory nucleic acid sequences (also termed "control sequences") is necessary to express a given gene. In general, the transcriptional and translational regulatory sequences include, but are not limited to, promoter sequences, ribosomal binding sites, transcriptional start and stop sequences, translational start and stop sequences, and enhancer or activator sequences.
- A nucleic acid is "operably linked" when it is placed into a functional relationship with another nucleic acid sequence. For example, DNA encoding a secretory leader (i.e., a signal peptide), is operably linked to DNA for a polypeptide if it is expressed as a preprotein that participates in the secretion of the polypeptide; a promoter or enhancer is operably linked to a coding sequence if it affects the transcription of the sequence; or a ribosome binding site is operably linked to a coding sequence if it is positioned so as to facilitate translation. Generally, "operably linked" means that the DNA sequences being linked are contiguous, and, in the case of a secretory leader, contiguous and in reading phase. However, enhancers do not have to be contiguous. Linking is accomplished by ligation at convenient restriction sites. If such sites do not exist, the synthetic oligonucleotide adaptors or linkers are used in accordance with conventional practice.
- The term "inactivation" includes any method that prevents the functional expression of sbo, wherein the gene or gene product is unable to exert its known function. Inactivation or enhancement occurs via any suitable means, including deletions, substitutions (e.g., mutations), interruptions, and/or insertions in the nucleic acid gene sequence.
- In some preferred embodiments, inactivation is achieved by deletion. The gene is deleted by homologous recombination. For example, when sbo is the gene to be deleted, a DNA construct comprising an incoming sequence having a selective marker flanked on each side by a homology box is used. The homology box comprises nucleotide sequences homologous to nucleic acids flanking regions of the chromosomal sbo gene. The DNA construct aligns with the homologous sequences of the Bacillus host chromosome and in a double crossover event the sbo gene is excised out of the host chromosome.
- As used herein, "deletion" of a gene refers to deletion of the entire coding sequence, deletion of part of the coding sequence, or deletion of the coding sequence including flanking regions. The deletion may be partial as long as the sequences left in the chromosome provides the desired biological activity of the gene. The flanking regions of the coding sequence may include from about 1 bp to about 500 bp at the 5' and 3' ends. The flanking region may be larger than 500 bp but will preferably not include other genes in the region which may be inactivated or deleted according to the invention. The end result is that the deleted gene is effectively non-functional. In simple terms, a "deletion" is defined as a change in either nucleotide or amino acid sequence in which one or more nucleotides or amino acid residues, respectively, have been removed (i.e., are absent). Thus, a "deletion mutant" has fewer nucleotides or amino acids than the respective wild-type organism.
- In another preferred embodiment, inactivation is by insertion. For example, in some embodiments, when sbo is the gene to be inactivated, a DNA construct comprises an incoming sequence having the sbo gene interrupted by a selective marker. The selective marker will be flanked on each side by sections of the sbo coding sequence. The DNA construct aligns with essentially identical sequences of the sbo gene in the host chromosome and in a double crossover event the sbo gene is inactivated by the insertion of the selective marker. In simple terms, an "insertion" or "addition" is a change in a nucleotide or amino acid sequence which has resulted in the addition of one or more nucleotides or amino acid residues, respectively, as compared to the naturally occurring sequence.
- In another embodiment, activation is by insertion in a single crossover event with a plasmid as the vector. For example, a sbo chromosomal gene is aligned with a plasmid comprising the gene or part of the gene coding sequence and a selective marker. In some embodiments, the selective marker is located within the gene coding sequence or on a part of the plasmid separate from the gene. The vector is integrated into the Bacillus chromosome, and the gene is inactivated by the insertion of the vector in the coding sequence.
- As used herein, a "substitution" results from the replacement of one or more nucleotides or amino acids by different nucleotides or amino acids, respectively.
- As used herein, "homologous genes" refers to a pair of genes from different, but usually related species, which correspond to each other and which are identical or very similar to each other. The term encompasses genes that are separated by speciation (i.e., the development of new species) (e.g., orthologous genes), as well as genes that have been separated by genetic duplication (e.g., paralogous genes).
- As used herein, "ortholog" and "orthologous genes" refer to genes in different species that have evolved from a common ancestral gene (i.e., a homologous gene) by speciation. Typically, orthologs retain the same function in during the course of evolution. Identification of orthologs finds use in the reliable prediction of gene function in newly sequenced genomes.
- As used herein, "paralog" and "paralogous genes" refer to genes that are related by duplication within a genome. While orthologs retain the same function through the course of evolution, paralogs evolve new functions, even though some functions are often related to the original one. Examples of paralogous genes include, but are not limited to genes encoding trypsin, chymotrypsin, elastase, and thrombin, which are all serine proteinases and occur together within the same species.
- As used herein, "homology" refers to sequence similarity or identity, with identity being preferred. This homology is determined using standard techniques known in the art (See e.g., Smith and Waterman, Adv. Appl. Math., 2:482 [1981]; Needleman and Wunsch, J. Mol. Biol., 48:443 [1970]; Pearson and Lipman, Proc. Natl. Acad. Sci. USA 85:2444 [1988]; programs such as GAP, BESTFIT, FASTA, and TFASTA in the Wisconsin Genetics Software Package (Genetics Computer Group, Madison, WI); and Devereux et al., Nucl. Acid Res., 12:387-395 [1984]).
- As used herein, an "analogous sequence" is one wherein the function of the gene is essentially the same as the gene designated from Bacillus subtilis strain 168. Additionally, analogous genes include at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% sequence identity with the sequence of the Bacillus subtilis strain 168 gene. Alternately, analogous sequences have an alignment of between 70 to 100% of the genes found in the B. subtilis 168 region and/or have at least between 5 - 10 genes found in the region aligned with the genes in the B. subtilis 168 chromosome. In additional embodiments more than one of the above properties applies to the sequence. Analogous sequences are determined by known methods of sequence alignment. A commonly used alignment method is BLAST, although as indicated above and below, there are other methods that also find use in aligning sequences.
- One example of a useful algorithm is PILEUP. PILEUP creates a multiple sequence alignment from a group of related sequences using progressive, pairwise alignments. It can also plot a tree showing the clustering relationships used to create the alignment. PILEUP uses a simplification of the progressive alignment method of Feng and Doolittle (Feng and Doolittle, J. Mol. Evol., 35:351-360 [1987]). The method is similar to that described by Higgins and Sharp (Higgins and Sharp, CABIOS 5:151-153 [1989]). Useful PILEUP parameters including a default gap weight of 3.00, a default gap length weight of 0.10, and weighted end gaps.
- Another example of a useful algorithm is the BLAST algorithm, described by Altschul et al., (Altschul et al., J. Mol. Biol., 215:403-410, [1990]; and Karlin et al., Proc. Natl. Acad. Sci. USA 90:5873-5787 [1993]). A particularly useful BLAST program is the WU-BLAST-2 program (See, Altschul et al., Meth. Enzymol." 266:460-480 [1996]). WU-BLAST-2 uses several search parameters, most of which are set to the default values. The adjustable parameters are set with the following values: overlap span =1, overlap fraction = 0.125, word threshold (T) = 11. The HSP S and HSP S2 parameters are dynamic values and are established by the program itself depending upon the composition of the particular sequence and composition of the particular database against which the sequence of interest is being searched. However, the values may be adjusted to increase sensitivity. A % amino acid sequence identity value is determined by the number of matching identical residues divided by the total number of residues of the "longer" sequence in the aligned region. The "longer" sequence is the one having the most actual residues in the aligned region (gaps introduced by WU-Blast-2 to maximize the alignment score are ignored).
- Thus, "percent (%) nucleic acid sequence identity" is defined as the percentage of nucleotide residues in a candidate sequence that are identical with the nucleotide residues of the sequence shown in the nucleic acid figures. A preferred method utilizes the BLASTN module of WU-BLAST-2 set to the default parameters, with overlap span and overlap fraction set to 1 and 0.125, respectively.
- The alignment may include the introduction of gaps in the sequences to be aligned. In addition, for sequences which contain either more or fewer nucleosides than those of the nucleic acid figures, it is understood that the percentage of homology will be determined based on the number of homologous nucleosides in relation to the total number of nucleosides. Thus, for example, homology of sequences shorter than those of the sequences identified herein and as discussed below, will be determined using the number of nucleosides in the shorter sequence.
- As used herein, the term "hybridization" refers to the process by which a strand of nucleic acid joins with a complementary strand through base pairing, as known in the art.
- A nucleic acid sequence is considered to be "selectively hybridizable" to a reference nucleic acid sequence if the two sequences specifically hybridize to one another under moderate to high stringency hybridization and wash conditions. Hybridization conditions are based on the melting temperature (Tm) of the nucleic acid binding complex or probe. For example, "maximum stringency" typically occurs at about Tm-5°C (5° below the Tm of the probe); "high stringency" at about 5-10°C below the Tm; "intermediate stringency" at about 10-20°C below the Tm of the probe; and "low stringency" at about 20-25°C below the Tm. Functionally, maximum stringency conditions may be used to identify sequences having strict identity or near strict identity with the hybridization probe; while an intermediate or low stringency hybridization can be used to identify or detect polynucleotide sequence homologs.
- Moderate and high stringency hybridization conditions are well known in the art. An example of high stringency conditions includes hybridization at about 42°C in 50% formamide, 5X SSC, 5X Denhardt's solution, 0.5% SDS and 100 µg/ml denatured carrier DNA followed by washing two times in 2X SSC and 0.5% SDS at room temperature and two additional times in 0.1X SSC and 0.5% SDS at 42°C. An example of moderate stringent conditions include an overnight incubation at 37°C in a solution comprising 20% formamide, 5 x SSC (150mM NaCl, 15 mM trisodium citrate), 50 mM sodium phosphate (pH 7.6), 5 x Denhardt's solution, 10% dextran sulfate and 20 mg/ml denaturated sheared salmon sperm DNA, followed by washing the filters in 1 x SSC at about 37 - 50°C. Those of skill in the art know how to adjust the temperature, ionic strength, etc. as necessary to accommodate factors such as probe length and the like.
- As used herein, "recombinant" includes reference to a cell or vector, that has been modified by the introduction of a heterologous nucleic acid sequence or that the cell is derived from a cell so modified. Thus, for example, recombinant cells express genes that are not found in identical form within the native (non-recombinant) form of the cell or express native genes that are otherwise abnormally expressed, under expressed or not expressed at all as a result of deliberate human intervention. "Recombination, "recombining," or generating a "recombined" nucleic acid is generally the assembly of two or more nucleic acid fragments wherein the assembly gives rise to a chimeric gene.
- Mutant DNA sequences can be generated with site saturation mutagenesis in at least one codon. In another preferred embodiment, site saturation mutagenesis is performed for two or more codons. Mutant DNA sequences can have more than 40%, more than 45%, more than 50%, more than 55%, more than 60%, more than 65%, more than 70%, more than 75%, more than 80%, more than 85%, more than 90%, more than 95%, or more than 98% homology with the wild-type sequence. In alternative embodiments, mutant DNA is generated in vivo using any known mutagenic procedure such as, for example, radiation, nitrosoguanidine and the like. The desired DNA sequence is then isolated and used in the methods provided herein.
- As used herein, the term "target sequence" refers to a DNA sequence in the host cell that encodes the sequence where it is desired for the incoming sequence to be inserted into the host cell genome. In some embodiments, the target sequence encodes a functional wild-type gene or operon, while in other embodiments the target sequence encodes a functional mutant gene or operon, or a non-functional gene or operon.
- As used herein, a "flanking sequence" refers to any sequence that is either upstream or downstream of the sequence being discussed (e.g., for genes A-B-C, gene B is flanked by the A and C gene sequences). In a preferred embodiment, the incoming sequence is flanked by a homology box on each side. In another embodiment, the incoming sequence and the homology boxes comprise a unit that is flanked by stuffer sequence on each side. In some embodiments, a flanking sequence is present on only a single side (either 3' or 5'), but in preferred embodiments, it is on each side of the sequence being flanked. The sequence of each homology box is homologous to a sequence in the Bacillus chromosome. These sequences direct where in the Bacillus chromosome the new construct gets integrated and what part of the Bacillus chromosome will be replaced by the incoming sequence. In a preferred embodiment, the 5' and 3' ends of a selective marker are flanked by a polynucleotide sequence comprising a section of the inactivating chromosomal segment. It is present on each side of the sequence being flanked.
- As used herein, the term "stuffer sequence" refers to any extra DNA that flanks homology boxes (typically vector sequences). However, the term encompasses any non-homologous DNA sequence. Not to be limited by any theory, a stuffer sequence provides a noncritical target for a cell to initiate DNA uptake.
- As used herein, the term "library of mutants" refers to a population of cells which are identical in most of their genome but include different homologues of one or more genes. Such libraries find use for example, in methods to identify genes or operons with improved traits.
- As used herein, the terms "hypercompetent" and "super competent" mean that greater than 1% of a cell population is transformable with chromosomal DNA (e.g., Bacillus DNA). Alternatively, the terms are used in reference to cell populations in which greater than10% of a cell population is transformable with a self-replicating plasmid (e.g., a Bacillus plasmid). Preferably, the super competent cells are transformed at a rate greater than observed for the wild-type or parental cell population. Super competent and hypercompetent are used interchangeably herein.
- As used herein, the terms "amplification" and "gene amplification" refer to a process by which specific DNA sequences are disproportionately replicated such that the amplified gene becomes present in a higher copy number than was initially present in the genome. In some embodiments, selection of cells by growth in the presence of a drug (e.g., an inhibitor of an inhibitable enzyme) results in the amplification of either the endogenous gene encoding the gene product required for growth in the presence of the drug or by amplification of exogenous (i.e., input) sequences encoding this gene product, or both.
- "Amplification" is a special case of nucleic acid replication involving template specificity. It is to be contrasted with non-specific template replication (i.e., replication that is template-dependent but not dependent on a specific template). Template specificity is here distinguished from fidelity of replication (i.e., synthesis of the proper polynucleotide sequence) and nucleotide (ribo- or deoxyribo-) specificity. Template specificity is frequently described in terms of "target" specificity. Target sequences are "targets" in the sense that they are sought to be sorted out from other nucleic acid. Amplification techniques have been designed primarily for this sorting out.
- As used herein, the term "co-amplification" refers to the introduction into a single cell of an amplifiable marker in conjunction with other gene sequences (i.e., comprising one or more non-selectable genes such as those contained within an expression vector) and the application of appropriate selective pressure such that the cell amplifies both the amplifiable marker and the other, non-selectable gene sequences. The amplifiable marker may be physically linked to the other gene sequences or alternatively two separate pieces of DNA, one containing the amplifiable marker and the other containing the non-selectable marker, may be introduced into the same cell.
- As used herein, the terms "amplifiable marker," "amplifiable gene," and "amplification vector" refer to a gene or a vector encoding a gene which permits the amplification of that gene under appropriate growth conditions.
- "Template specificity" is achieved in most amplification techniques by the choice of enzyme. Amplification enzymes are enzymes that, under conditions they are used, will process only specific sequences of nucleic acid in a heterogeneous mixture of nucleic acid. For example, in the case of Qβ replicase, MDV-1 RNA is the specific template for the replicase (See e.g., Kacian et al., Proc. Natl. Acad. Sci. USA 69:3038 [1972]). Other nucleic acids are not replicated by this amplification enzyme. Similarly, in the case of T7 RNA polymerase, this amplification enzyme has a stringent specificity for its own promoters (See, Chamberlin et al., Nature 228:227 [1970]). In the case of T4 DNA ligase, the enzyme will not ligate the two oligonucleotides or polynucleotides, where there is a mismatch between the oligonucleotide or polynucleotide substrate and the template at the ligation junction (See, Wu and Wallace, Genomics 4:560 [1989]). Finally, Taq and Pfu polymerases, by virtue of their ability to function at high temperature, are found to display high specificity for the sequences bounded and thus defined by the primers; the high temperature results in thermodynamic conditions that favor primer hybridization with the target sequences and not hybridization with non-target sequences.
- As used herein, the term "amplifiable nucleic acid" refers to nucleic acids which may be amplified by any amplification method. It is contemplated that "amplifiable nucleic acid" will usually comprise "sample template."
- As used herein, the term "sample template" refers to nucleic acid originating from a sample which is analyzed for the presence of "target" (defined below). In contrast, "background template" is used in reference to nucleic acid other than sample template which may or may not be present in a sample. Background template is most often inadvertent. It may be the result of carryover, or it may be due to the presence of nucleic acid contaminants sought to be purified away from the sample. For example, nucleic acids from organisms other than those to be detected may be present as background in a test sample.
- As used herein, the term "primer" refers to an oligonucleotide, whether occurring naturally as in a purified restriction digest or produced synthetically, which is capable of acting as a point of initiation of synthesis when placed under conditions in which synthesis of a primer extension product which is complementary to a nucleic acid strand is induced, (i.e., in the presence of nucleotides and an inducing agent such as DNA polymerase and at a suitable temperature and pH). The primer is preferably single stranded for maximum efficiency in amplification, but may alternatively be double stranded. If double stranded, the primer is first treated to separate its strands before being used to prepare extension products. Preferably, the primer is an oligodeoxyribonucleotide. The primer must be sufficiently long to prime the synthesis of extension products in the presence of the inducing agent. The exact lengths of the primers will depend on many factors, including temperature, source of primer and the use of the method.
- As used herein, the term "probe" refers to an oligonucleotide (i.e., a sequence of nucleotides), whether occurring naturally as in a purified restriction digest or produced synthetically, recombinantly or by PCR amplification, which is capable of hybridizing to another oligonucleotide of interest. A probe may be single-stranded or double-stranded. Probes are useful in the detection, identification and isolation of particular gene sequences. It is contemplated that any probe used in the present invention will be labeled with any "reporter molecule," so that is detectable in any detection system, including, but not limited to enzyme (e.g., ELISA, as well as enzyme-based histochemical assays), fluorescent, radioactive, and luminescent systems. It is not intended that the present invention be limited to any particular detection system or label.
- As used herein, the term "target," when used in reference to the polymerase chain reaction, refers to the region of nucleic acid bounded by the primers used for polymerase chain reaction. Thus, the "target" is sought to be sorted out from other nucleic acid sequences. A "segment" is defined as a region of nucleic acid within the target sequence.
- As used herein, the term "polymerase chain reaction" ("PCR") refers to the methods of
U.S. Patent Nos. 4,683,195 4,683,202 , and4,965,188 , which include methods for increasing the concentration of a segment of a target sequence in a mixture of genomic DNA without cloning or purification. This process for amplifying the target sequence consists of introducing a large excess of two oligonucleotide primers to the DNA mixture containing the desired target sequence, followed by a precise sequence of thermal cycling in the presence of a DNA polymerase. The two primers are complementary to their respective strands of the double stranded target sequence. To: effect amplification, the mixture is denatured and the primers then annealed to their complementary sequences within the target molecule. Following annealing, the primers are extended with a polymerase so as to form a new pair of complementary strands. The steps of denaturation, primer annealing and polymerase extension can be repeated many times (i.e., denaturation, annealing and extension constitute one "cycle"; there can be numerous "cycles") to obtain a high concentration of an amplified segment of the desired target sequence. The length of the amplified segment of the desired target sequence is determined by the relative positions of the primers with respect to each other, and therefore, this length is a controllable parameter. By virtue of the repeating aspect of the process, the method is referred to as the "polymerase chain reaction" (hereinafter "PCR"). Because the desired amplified segments of the target sequence become the predominant sequences (in terms of concentration) in the mixture, they are said to be "PCR amplified". - As used herein, the term "amplification reagents" refers to those reagents (deoxyribonucleotide triphosphates, buffer, etc.), needed for amplification except for primers, nucleic acid template and the amplification enzyme. Typically, amplification reagents along with other reaction components are placed and contained in a reaction vessel (test tube, microwell, etc.).
- With PCR, it is possible to amplify a single copy of a specific target sequence in genomic DNA to a level detectable by several different methodologies (e.g., hybridization with a labeled probe; incorporation of biotinylated primers followed by avidin-enzyme conjugate detection; incorporation of 32P-labeled deoxynucleotide triphosphates, such as dCTP or dATP, into the amplified segment). In addition to genomic DNA, any oligonucleotide or polynucleotide sequence can be amplified with the appropriate set of primer molecules. In particular, the amplified segments created by the PCR process itself are, themselves, efficient templates for subsequent PCR amplifications.
- As used herein, the terms "PCR product," "PCR fragment," and "amplification product" refer to the resultant mixture of compounds after two or more cycles of the PCR steps of denaturation, annealing and extension are complete. These terms encompass the case where there has been amplification of one or more segments of one or more target sequences.
- As used herein, the term "RT-PCR" refers to the replication and amplification of RNA sequences. In this method, reverse transcription is coupled to PCR, most often using a one enzyme procedure in which a thermostable polymerase is employed, as described in
U.S. Patent No. 5,322,770 , herein incorporated by reference. In RT-PCR, the RNA template is converted to cDNA due to the reverse transcriptase activity of the polymerase, and then amplified using the polymerizing activity of the polymerase (i.e., as in other PCR methods). - As used herein, the terms "restriction endonucleases" and "restriction enzymes" refer to bacterial enzymes, each of which cut double-stranded DNA at or near a specific nucleotide sequence.
- A "restriction site" refers to a nucleotide sequence recognized and cleaved by a given restriction endonuclease and is frequently the site for insertion of DNA fragments. In certain embodiments of the invention restriction sites are engineered into the selective marker and into 5' and 3' ends of the DNA construct.
- As used herein "an inactivating chromosomal segment" comprises two sections. Each section comprises polynucleotides that are homologous with the upstream or downstream genomic chromosomal DNA that immediately flanks an indigenous chromosome region as defined herein. "Immediately flanks" means the nucleotides comprising the inactivating chromosomal segment do not include the nucleotides defining the indigenous chromosomal region. The inactivating chromosomal segment directs where in the Bacillus chromosome the DNA construct gets integrated and what part of the Bacillus chromosome will be replaced.
- As used herein, "indigenous chromosomal region" and "a fragment of an indigenous chromosomal region" refer to a segment of the Bacillus chromosome which is deleted from a Bacillus host cell in some embodiments of the present invention. In general, the terms "segment," "region," "section," and "element" are used interchangeably herein. In some embodiments, deleted segments include one or more genes with known functions, while in other embodiments, deleted segments include one or more genes with unknown functions, and in other embodiments, the deleted segments include a combination of genes with known and unknown functions. In some embodiments, indigenous chromosomal regions or fragments thereof include as many as 200 genes or more.
- An indigenous chromosomal region or fragment thereof can have a necessary function under certain conditions, but the region is not necessary for Bacillus strain viability under laboratory conditions. Preferred laboratory conditions include but are not limited to conditions such as growth in a fermenter, in a shake flask on plated media, etc., at standard temperatures and atmospheric conditions (e.g., aerobic).
- An indigenous chromosomal region or fragment thereof may encompass a range of about 0.5kb to 500 kb; about 1.0 kb to 500 kb; about 5 kb to 500 kb; about 10 kb to 500kb; about 10 kb to 200kb; about 10kb to 100kb; about 10kb to 50kb; about 100kb to 500kb; and about 200kb to 500 kb of the Bacillus chromosome. When an indigenous chromosomal region or fragment thereof has been deleted, the chromosome of the altered Bacillus strain may include 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 85%, 80%, 75% or 70% of the corresponding unaltered Bacillus host chromosome. Preferably, the chromosome of an altered Bacillus strain according to the invention will include about 99 to 90%; 99 to 92%; and 98 to 94% of the corresponding unaltered Bacillus host strain chromosome genome.
- As used herein, "strain viability" refers to reproductive viability. The deletion of an indigenous chromosomal region or fragment thereof, does not deleteriously affect division and survival of the altered Bacillus strain under laboratory conditions.
- As used herein, "altered Bacillus strain" refers to a genetically engineered Bacillus sp. wherein a protein of Interest has an enhanced level of production as compared to the production of the same protein of interest in a corresponding unaltered Bacillus host strain grown under essentially the same growth conditions. The enhanced level of expression results from the inactivation of one or more chromosomal genes. In one embodiment, the enhanced level of expression results from the deletion of one or more chromosomal genes. The enhanced level of production results from the insertional inactivation of one or more chromosomal genes. The inactivated gene is sbo.
- The altered Bacillus strain can comprise two inactivated genes, while in other embodiments, there are three inactivated genes, four inactivated genes, five inactivated genes, six inactivated genes, or more.
- As used herein, a "corresponding unaltered Bacillus strain" is the host strain (e.g., the originating and/or wild-type strain) from which the indigenous chromosomal region or fragment thereof is deleted or modified and from which the altered strain is derived.
- As used herein, the term "chromosomal integration" refers to the process whereby the incoming sequence is introduced into the chromosome of a host cell (e.g., Bacillus). The homologous regions of the transforming DNA align with homologous regions of the chromosome. Subsequently, the sequence between the homology boxes is replaced by the incoming sequence in a double crossover (i.e., homologous recombination). Homologous sections of an inactivating chromosomal segment of a DNA construct align with the flanking homologous regions of the indigenous chromosomal region of the Bacillus chromosome. Subsequently, the indigenous chromosomal region is deleted by the DNA construct in a double crossover (i.e., homologous recombination).
- "Homologous recombination" means the exchange of DNA fragments between two DNA molecules or paired chromosomes at the site of identical or nearly identical nucleotide sequences. Chromosomal integration is homologous recombination.
- "Homologous sequences" as used herein means a nucleic acid or polypeptide sequence having 100%, 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 88%, 85%, 80%, 75%, or 70% sequence identity to another nucleic acid or polypeptide sequence when optimally aligned for comparison. Homologous sequences can have between 85% and 100% sequence identity, between 90% and 100% sequence identity, or there is 95% and 100% sequence identity.
- As used herein "amino acid" refers to peptide or protein sequences or portions thereof. The terms "protein", "peptide" and "polypeptide" are used interchangeably.
- As used herein, "protein of interest" and "polypeptide of interest" refer to a subtilisin
- The protein of interest is a secreted polypeptide which is fused to a signal peptide (i.e., an amino-terminal extension on a protein to be secreted). Nearly all secreted proteins use an amino- terminal protein extension which plays a crucial role in the targeting to and translocation of precursor proteins across the membrane. This extension is proteolytically removed by a signal peptidase during or immediately following membrane transfer.
- As used herein, the term "heterologous protein" refers to a protein or polypeptide that does not naturally occur in the host cell.
- As used herein, "homologous protein" refers to a protein or polypeptide native or naturally occurring in a cell.
- As used herein, an "operon region" comprises a group of contiguous genes that are transcribed as a single transcription unit from a common promoter, and are thereby subject to co-regulation. In some embodiments, the operon includes a regulator gene. In most preferred embodiments, operons that are highly expressed as measured by RNA levels, but have an unknown or unnecessary function are used.
- As used herein, a "multi-contiguous single gene region" is a region wherein at least the coding regions of two genes occur in tandem and in some embodiments, include intervening sequences preceding and following the coding regions.
- As indicated above, the present invention includes embodiments that involve singe gene deletions.
- The present invention includes a DNA construct comprising an incoming sequence. The DNA construct is assembled in vitro, followed by direct cloning of the construct into a competent Bacillus host, such that the DNA construct becomes integrated into the Bacillus chromosome. For example, PCR fusion and/or ligation can be employed to assemble a DNA construct in vitro. In some embodiments, the DNA construct is a non-plasmid construct, while in other embodiments it is incorporated into a vector (e.g., a plasmid). In some embodiments, circular plasmids are used. In preferred embodiments, circular plasmids are designed to use an appropriate restriction enzyme (i.e., one that does not disrupt the DNA construct). Thus, linear plasmids find use in the present invention (See,
Figure 1 ). However, other methods are suitable for use in the present invention, as known to those in the art (See e.g., Perego, "Integrational Vectors for Genetic Manipulation in Bacillus subtilis," in (Sonenshein et al. (eds.), Bacillus subtilis and Other Gram-Positive Bacteria, American Society for Microbiology, Washington, DC [1993]). - In some embodiments, the incoming sequence includes a selective marker. The incoming sequence includes sbo. In yet other embodiments, the incoming sequence comprises a selective marker flanked on the 5' and 3' ends with a fragment of the gene sequence. In some embodiments, when the DNA construct comprising the selective marker and gene, gene fragment or homologous sequence thereto is transformed into a host cell, the location of the selective marker renders the gene non-functional for its intended purpose. In some embodiments, the incoming sequence comprises the selective marker located in the promoter region of the gene. In other embodiments, the incoming sequence comprises the selective marker located after the promoter region of gene. In yet other embodiments, the incoming sequence comprises the selective marker located in the coding region of the gene. In further embodiments, the incoming sequence comprises a selective marker flanked by a homology box on both ends. In still further embodiments, the incoming sequence includes a sequence that interrupts the transcription and/or translation of the coding sequence. In yet additional embodiments, the DNA construct includes restriction sites engineered at the upstream and downstream ends of the construct.
- Whether the DNA construct is incorporated into a vector or used without the presence of plasmid DNA, it is used to transform microorganisms. It is contemplated that any suitable method for transformation will find use with the present invention. In preferred embodiments, at least one copy of the DNA construct is integrated into the host Bacillus chromosome. In some embodiments, one or more DNA constructs of the invention are used to transform host cells.
- The DNA construct also includes a polynucleotide encoding a protein of interest. In some of these preferred embodiments, the DNA construct also includes a constitutive or inducible promoter that is operably linked to the sequence encoding the protein of interest. The protein of interest is a subtilisin, the promoter is selected from the group consisting of a tac promoter, a β-lactamase promoter, or an aprE promoter (DeBoer et al., Proc. Natl. Acad. Sci. USA 80:21-25 [1983]). However, any suitable promoter known to those in the art finds use with the present invention. Nonetheless, in particularly preferred embodiments, the promoter is the B. subtilis aprE promoter.
- Various methods are known for the transformation of Bacillus species. Indeed, methods for altering the chromosome of Bacillus involving plasmid constructs and transformation of the plasmids into E. coli are well known. In most methods, plasmids are subsequently isolated from E. coli and transformed into Bacillus. However, it is not essential to use such intervening microorganisms such as E. coli, and in some preferred embodiments, the DNA construct is directly transformed into a competent Bacillus host.
- The well-known Bacillus subtilis strain 168 finds use in the present invention. Indeed, the genome of this strain has been well-characterized (See, Kunst et al., Nature 390:249-256 [1997]; and Henner et al., Microbiol. Rev., 44:57-82 [1980]). The genome is comprised of one 4215 kb chromosome. While the coordinates used herein refer to the 168 strain, the invention encompasses analogous sequences from Bacillus strains. The incoming chromosomal sequence includes sbo. The DNA coding sequences of this gene from B. subtilis 168 is provided in SEQ ID NO: 1.
- The incoming sequence which comprises a sbo, may further include immediate chromosomal coding region flanking sequences.
- Sequences of these genes and gene products are provided below. The numbering used herein is that used in subtilist (See e.g., Moszer et al., Microbiol., 141:261-268 [1995]).
- The sbo coding sequence of B. subtilis 168 is shown below:
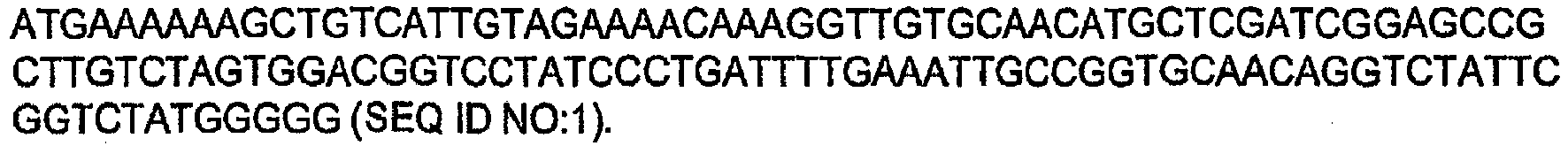
- The deduced amino acid sequence for Sbo is:
- MKKAVIVENKGCATCSIGAACLVDGPIPDFEIAGATGLFGLWG (SEQ ID NO: 2).
- As indicated above, it is contemplated that inactivated analogous genes found in other Bacillus hosts will find use in the present invention.
- The host cell is a member of the genus Bacillus, while in some embodiments, the Bacillus strain of interest is alkalophilic. Numerous alkalophilic Bacillus strains are known (See e.g.,
U.S. Pat. 5,217,878 ; and Aunstrup et al., Proc IV IFS: Ferment. Technol. Today, 299-305 [1972]). In some preferred embodiments, the Bacillus strain of interest is an industrial Bacillus strain. Examples of industrial Bacillus strains include, but are not limited to B. licheniformis, B. lentus, B. subtilis, and B. amyloliquefaciens. In additional embodiments, the Bacillus host strain is selected from the group consisting of B. lentus, B. brevis, B. stearothermophilus, B. alkalophilus, B. coagulans, B. circulans, B. pumilus, B. thuringiensis, B. clausii, and B. megaterium, as well as other organisms within the genus Bacillus, as discussed above. In some particularly preferred embodiments, B. subtilis is used. For example,U.S. Patents 5,264,366 and4,760,025 (RE 34,606 ) describe various Bacillus host strains that find use in the present invention, although other suitable strains are contemplated for use in the present invention. - An industrial strain may be a non-recombinant strain of a Bacillus sp., a mutant of a naturally occurring strain or a recombinant strain. Preferably, the host strain is a recombinant host strain wherein a polynucleotide encoding a subtilisin has been introduced into the host. A further preferred host strain is a Bacillus subtilis host strain and particularly a recombinant Bacillus subtilis host strain. Numerous B. subtilis strains are known, including but not limited to 1A6 (ATCC 39085), 168 (1A01), SB19, W23, Ts85, B637, PB1753 through PB1758, PB3360, JH642, 1A243 (ATCC 39,087), ATCC 21332, ATCC 6051, MI113, DE100 (ATCC 39,094), GX4931, PBT 110, and PEP 211strain (See e.g., Hoch et al., Genetics, 73:215-228 [1973];
U.S. Patent No. 4,450,235 ;U.S. Patent No. 4,302,544 ; andEP 0134048 ). The use of B. subtilis as an expression host is further described by Palva et al. and others (See, Palva et al., Gene 19:81-87 [1982]; also see Fahnestock and Fischer, J. Bacteriol., 165:796-804 [1986]; and Wang et al., Gene 69:39-47 [1988]).. - Industrial protease producing Bacillus strains provide particularly preferred expression hosts. In some preferred embodiments, use of these strains in the present invention provides further enhancements in efficiency and protease production. Two general types of proteases are typically secreted by Bacillus sp., namely neutral (or "metalloproteases") and alkaline (or "serine") proteases. Serine proteases are enzymes which catalyze the hydrolysis of peptide bonds in which there is an essential serine residue at the active site. Serine proteases have molecular weights in the 25,000 to 30,000 range (See, Priest, Bacteriol. Rev., 41:711-753 [1977]). Subtilisin is a serine protease for use in the present invention. A wide variety of Bacillus subtilisins have been identified and sequenced, for example, subtilisin 168, subtilisin BPN', subtilisin Carlsberg, subtilisin DY, subtilisin 147 and subtilisin 309 (See e.g.,
EP 414279 B WO 89/06279 WO 99/20770 WO 99/20726 WO 99/20769 WO 89/06279 RE 34,606 U.S. Patent No. 4,914,031 ;U.S. Patent No. 4,980,288 ;U.S. Patent No. 5,208,158 ;U.S. Patent No. 5,310,675 ;U.S. Patent No. 5,336,611 ;U.S. Patent No. 5,399,283 ;U.S. Patent No. 5,441,882 ;U.S. Patent No. 5,482,849 ;U.S. Patent No. 5,631,217 ;U.S. Patent No. 5,665,587 ;U.S. Patent No. 5,700,676 ;U.S. Patent No. 5,741,694 ;U.S. Patent No. 5,858,757 ;U.S. Patent No. 5,880,080 ;U.S. Patent No. 6,197,567 ; andU.S. Patent No. 6,218,165 ). - In yet another embodiment, a preferred Bacillus host is a Bacillus sp. that includes a mutation or deletion in at least one of the following genes, degU, degS, degR and degQ. Preferably the mutation is in a degU gene, and more preferably the mutation is degU(Hy)32. (See, Msadek et al., J. Bacteriol., 172:824-834 [1990]; and Olmos et al., Mol. Gen. Genet., 253:562-567 [1997]). A most preferred host strain is a Bacillus subtilis carrying a degU32(Hy) mutation. In a further embodiment, the Bacillus host comprises a mutation or deletion in scoC4, (See, Caldwell et al., J. Bacteriol., 183:7329-7340 [2001]); spoIIE (See, Arigoni et al., Mol. Microbiol., 31:1407-1415 [1999]); oppA or other genes of the opp operon (See, Perego et al., Mol. Microbiol., 5:173-185 [1991]). Indeed, it is contemplated that any mutation in the opp operon that causes the same phenotype as a mutation in the oppA gene will find use in some embodiments of the altered Bacillus strain of the present invention. In some embodiments, these mutations occur alone, while in other embodiments, combinations of mutations are present. In some embodiments, an altered Bacillus of the invention is obtained from a Bacillus host strain that already includes a mutation to one or more of the above-mentioned genes. In alternate embodiments, an altered Bacillus of the invention is further engineered to include mutation of one or more of the above-mentioned genes.
- In yet another embodiment, the incoming sequence comprises a selective marker located between two IoxP sites (See, Kuhn and Torres, Meth. Mol. Biol., 180:175-204 [2002]), and the antimicrobial is then deleted by the action of Cre protein. In some embodiments, this results in the insertion of a single loxP site, as well as a deletion of native DNA, as determined by the primers used to construct homologous flanking DNA and antimicrobial-containing incoming DNA.
- Those of skill in the art are well aware of suitable methods for introducing polynucleotide sequences into Bacillus cells (See e.g., Ferrari et al., "Genetics," in Harwood et al. (ed.), Bacillus, Plenum Publishing Corp. [1989], pages 57-72; See also, Saunders et al., J. Bacteriol., 157:718-726 [1984]; Hoch et al., J. Bacteriol., 93:1925 -1937 [1967]; Mann et al., Current Microbiol., 13:131-135 [1986]; and Holubova, Folia Microbiol., 30:97 [1985]; for B. subtilis, Chang et al., Mol. Gen. Genet., 168:11-115 [1979]; for B. megaterium, Vorobjeva et al., FEMS Microbiol. Lett., 7:261-263 [1980]; for B amyloliquefaciens, Smith et al., Appl. Env. Microbiol., 51:634 (1986); for B. thuringiensis, Fisher et al., Arch. Microbiol., 139:213-217 [1981]; and for B. sphaericus, McDonald, J. Gen. Microbiol., 130:203 [1984]). Indeed, such methods as transformation including protoplast transformation and congression, transduction, and protoplast fusion are known and suited for use in the present invention. Methods of transformation are particularly preferred to introduce a DNA construct provided by the present invention into a host cell.
- In addition to commonly used methods, in some embodiments, host cells are directly transformed (i.e., an intermediate cell is not used to amplify, or otherwise process, the DNA construct prior to introduction into the host cell). Introduction of the DNA construct into the host cell includes those physical and chemical methods known in the art to introduce DNA into a host cell without insertion into a plasmid or vector. Such methods include, but are not limited to calcium chloride precipitation, electroporation, naked DNA, liposomes and the like. In additional embodiments, DNA constructs are co-transformed with a plasmid, without being inserted into the plasmid. In further embodiments, a selective marker is deleted from the altered Bacillus strain by methods known in the art (See, Stahl et al., J. Bacteriol., 158:411-418 [1984]; and Palmeros et al., Gene 247:255 - 264 [2000]).
- In some embodiments, host cells are transformed with one or more DNA constructs according to the present invention to produce an altered Bacillus strain wherein two or more genes have been inactivated in the host cell. In some embodiments, two or more genes are deleted from the host cell chromosome. In alternative embodiments, two or more genes are inactivated by insertion of a DNA construct. In some embodiments, the inactivated genes are contiguous (whether inactivated by deletion and/or insertion), while in other embodiments, they are not contiguous genes.
- There are various assays known to those of ordinary skill in the art for detecting and measuring activity of intracellularly and extracellularly expressed polypeptides. In particular, for proteases, there are assays based on the release of acid-soluble peptides from casein or hemoglobin measured as absorbance at 280 nm or colorimetrically using the Folin method (See e.g., Bergmeyer et al., "Methods of Enzymatic Analysis" vol. 5, Peptidases, Proteinases and their Inhibitors, Verlag Chemie, Weinheim [1984]). Other assays involve the solubilization of chromogenic substrates (See e.g., Ward, "Proteinases," in Fogarty (ed.)., Microbial Enzymes and Biotechnology, Applied Science, London, [1983], pp 251-317). Other exemplary assays include succinyl-Ala-Ala-Pro-Phe-para nitroanilide assay (SAAPFpNA) and the 2,4,6-trinitrobenzene sulfonate sodium salt assay (TNBS assay). Numerous additional references known to those in the art provide suitable methods (See e.g., Wells et al., Nucleic Acids Res. 11:7911-7925 [1983]; Christianson et al., Anal. Biochem., 223:119 -129 [1994]; and Hsia et al., Anal Biochem.,242:221-227 [1999]).
- Means for determining the levels of secretion of a protein of interest in a host cell and detecting expressed proteins include the use of immunoassays with either polyclonal or monoclonal antibodies specific for the protein. Examples include enzyme-linked immunosorbent assay (ELISA), radioimmunoassay (RIA), fluorescence immunoassay (FIA), and fluorescent activated cell sorting (FACS). However, other methods are known to those in the art and find use in assessing the protein of interest (See e.g., Hampton et al., Serological Methods, A Laboratory Manual, APS Press, St. Paul, MN [1990]; and Maddox et al., J. Exp. Med., 158:1211 [1983]). In some preferred embodiments, secretion of a protein of interest is higher in the altered strain obtained using the present invention than in a corresponding unaltered host. As known in the art, the altered Bacillus cells produced using the present invention are maintained and grown under conditions suitable for the expression and recovery of a polypeptide of interest from cell culture (See e.g., Hardwood and Cutting (eds.) Molecular Biological Methods for Bacillus, John Wiley & Sons [1990]).
- The manner and method of carrying out the present invention may be more fully understood by those of skill in the art by reference to the following examples.
- The following Examples are provided in order to demonstrate and further illustrate certain preferred embodiments and aspects of the present invention.
- In the experimental disclosure which follows, the following abbreviations apply: °C (degrees Centigrade); rpm (revolutions per minute); H2O (water); dH2O (deionized water); (HCl (hydrochloric acid); aa (amino acid); bp (base pair); kb (kilobase pair); kD (kilodaltons); gm (grams); µg (micrograms); mg (milligrams); ng (nanograms); µl (microliters); ml (milliliters); mm (millimeters); nm (nanometers); µm (micrometer); M (molar); mM (millimolar); µM (micromolar); U (units); V (volts); MW (molecular weight); sec (seconds); min(s) (minute/minutes); hr(s) (hour/hours); MgCl2 (magnesium chloride); NaCl (sodium chloride); OD280 (optical density at 280 nm); OD600 (optical density at 600 nm); PAGE (polyacrylamide gel electrophoresis); PBS (phosphate buffered saline [150 mM NaCl, 10 mM sodium phosphate buffer, pH 7.2]); PEG (polyethylene glycol); PCR (polymerase chain reaction); RT-PCR (reverse transcription PCR); SDS (sodium dodecyl sulfate); Tris (tris(hydroxymethyl)aminomethane); w/v (weight to volume); v/v (volume to volume); LA medium (per liter: Difco Tryptone Peptone 20g, Difco Yeast Extract 10g, EM Science NaCl 1g, EM Science Agar 17.5g, dH20 to 1L); ATCC (American Type Culture Collection, Rockville, MD); Clontech (CLONTECH Laboratories, Palo Alto, CA); Difco (Difco Laboratories, Detroit, MI); GIBCO BRL or Gibco BRL (Life Technologies, Inc., Gaithersburg, MD); Invitrogen (Invitrogen Corp., San Diego, CA); NEB (New England Biolabs, Beverly, MA); Sigma (Sigma Chemical Co., St. Louis, MO); Takara (Takara Bio Inc. Otsu, Japan); Roche Diagnostics and Roche (Roche Diagnostics, a division of F. Hoffmann La Roche, Ltd., Basel, Switzerland); EM Science (EM Science, Gibbstown, NJ); Qiagen (Qiagen, Inc., Valencia, CA); Stratagene (Stratagene Cloning Systems, La Jolla, CA); Affymetrix (Affymetrix, Santa Clara, California).
- This Example describes "
Method 1," which is also depicted inFigure 1 . In this method, E. coli was used to produce a pJM102 plasmid vector carrying the DNA construct to be transformed into Bacillus strains. (See, Perego, supra). Regions immediately flanking the 5' and 3' ends of the deletion site were PCR amplified. PCR primers were designed to be approximately 37 base pairs in length, including 31 base pairs homologous to the Bacillus subtilis chromosome and a 6 base pair restriction enzyme site located 6 base pairs from the 5' end of the primer. Primers were designed to engineer unique restriction sites at the upstream and downstream ends of the construct and a BamHI site between the two fragments for use in cloning. Primers for the antimicrobial markers contained BamHI sites at both ends of the fragment. Where possible, PCR primers were designed to remove promoters of deleted indigenous chromosomal regions, but to leave all terminators in the immediate area. The primary source of chromosome sequence, gene localization, and promoter and terminator information was obtained from Kunst et al., (1997) supra and also obtainable from the SubtiList World Wide Web Server known to those in the art (See e.g., Moszer et al., supra). Numerous deletions have been made using the present invention. A list of primer sequences from deletions created by this method is provided in Table 1. Reference is also made toFigure 2 for an explanation of the primer naming system.Table 1. Primers Primer Name Restriction Enzyme Engineered Into Primer Primer Sequence SEQ ID NO PBSX-UF XbaI CTACATTCTAGACGATTTGTTTGATCGATATGTGGAAGC 60 PBSX-UR BamHI GGCTGAGGATCCATTCCTCAGCCCAGAAGAGAACCTA 61 PBSX-DF BamHI TCCCTCGGATCCGAAATAGGTTCTGCTTATTGTATTCG 62 PBSX-DR SacI AGCGTTGAGCTCGCGCCATGCCATTATATTGGCTGCTG 63 Pphage 1- EcoRI GTGACGGAATTCCACGTGCGTCTTATATTGCTGAGCTT 64 Pphage 1- BamHI CGTTTTGGATCCAAAAACACCCCTTTAGATAATCTTAT 65 Pphage 1- BamHI ATCAAAGGATCCGCTATGCTCCAAATGTACACCTTTCCGT 66 Pphage 1- PstI ATATTTCTGCAGGCTGATATAAATAATACTGTGTGTTCC 67 Pphage 2- SacI CATCTTGAATTCAAAGGGTACAAGCACAGAGACAGAG 68 Pphage 2- BamHI TGACTTGGATCCGGTAAGTGGGCAGTTTGTGGGCAGT 69 Pphage 2- BamHI TAGATAGGATCCTATTGAAAACTGTTTAAGAAGAGGA 70 Pphage 2- PstI CTGATTCTGCAGGAGTGTTTTTGAAGGAAGCTTCATT 71 Pphage 4- KpnI CTCCGCGGTACCGTCACGAATGCGCCTCTTATTCTAT 72 Pphage 4- BamHI TCGCTGGGATCCTTGGCGCCGTGGAATCGATTTTGTCC 73 Pphage 4- BamHI GCAATGGGATCCTATATCAACGGTTATGAATTCACAA 74 Pphage 4- PstI CCAGAACTGCAGGAGCGAGGCGTCTCGCTGCCTGAAA 75 PPS-UF SacI GACAAGGAGCTCATGAAAAAAAGCATAAAGCTTTATGTTGC 76 PPS-UR BamHI GACAAGGGATCCCGGCATGTCCGTTATTACTTAATTTC 77 PPS-DF BamHI GACAAGGGATCCTGCCGCTTACCGGAAACGGA 78 PPS-DR XbaI GACAAGTCTAGATTATCGTTTGTGCAGTATTACTTG 79 SPβ-F SacI ACTGATGAGCTCTGCCTAAACAGCAAACAGCAGAAC 80 SPβ-UR BamHI ACGAATGGATCCATCATAAAGCCGCAGCAGATTAAATAT 81 SPβ-DF BamHI ACTGATGGATCCATCTTCGATAAATATGAAAGTGGC 82 SPβ-DR XbaI ACTGATTCTAGAGCCTTTTTCTCTTGATGCAATTCTTC 83 PKS-UF XbaI GAGCCTCTAGAGCCCATTGAATCATTTGTTT 84 PKS-UR BamHI GAGCCGGATCCTTAAGGATGTCGTTTTTGTGTCT 85 PKS-DF BamHI GAGCCGGATCCATTTCGGGGTTCTCAAAAAAA 86 PKS-DR SacI GAGCCGAGCTCATGCAAATGGAAAAATTGAT 87 Skin-UF XbaI GAAGTTCTAGAGATTGTAATTACAAAAGGGGGGTG 88 Skin-UR BamHI GAAGTGGATCCTTTCACCGATCATAAAAGCCC 89 Skin-DF BamHI TGAAAGGATCCATTTTTCATTGATTGTTAAGTC 90 Skin-DR SacI GAAGTTAGAGCTCGGGGGGGCATAAATTTCCCG 91 Phleo-UF BamHI GCTTATGGATCCGATACAAGAGAGGTCTCTCG 92 Phleo-DR BamHI GCTTATGGATCCCTGTCATGGCGCATTAACG 93 Spec-UF BamHI ACTGATGGATCCATCGATTTTCGTTCGTGAATACATG 94 Spec-DR BamHI ACTGATGGATCCCATATGCAAGGGTTTATTGTTTTC 95 CssS-UF XbaI GCACGTTCTAGACCACCGTCCCCTGTGTTGTATCCAC 96 CssS-UR BamHI AGGAAGGGATCCAGAGCGAGGAAGATGTAGGATGATC 97 CssS-DF BamHI TGACAAGGATCCTGTATCATACCGCATAGCAGTGCC 98 CssS-DR SacI TTCCGCGAGCTCGGCGAGAGCTTCAGACTCCGTCAGA 99 SBO- Xbal GAGCCTCTAGATCAGCGATTTGACGCGGCGC 100 SBO- BamHl TTATCTGGATCCCTGATGAGCAATGATGGTAAGATAGA 101 SBO- BamHl GGGTAA GGATCC CCCAAAAGGGCATAGTCATTCTACT 102 SBO- Asp718 GAGATCGGTACC CTTTTGGGCCATATCGTGGATTTC 103 PhrC-UF HindIII GAGCC AAGCTT CATTGACAGCAACCAGGCAGATCTC 104 PhrC-DF PstI GCTTATAAGCTTGATACAAGAGAGGTCTCTCG 105 PhrC-UR PstI GCTTATAAGCTTCTGTCATGGCGCATTAACG 106 PhrC-DR SacI GAGCCGAGCTC CATGCCGATGAAGTCATCGTCGAGC 107 PhrC-UF- Hindlll CGTGAA AAGCTT TCGCGGGATGTATGAATTTGATAAG 108 PhrC-DR- SacI TGTAGGGAGCTC GATGCGCCACAATGTCGGTACAACG 109 The restriction sites are designated as follows: XbaI is TCTAGA; BamHI is GGATCC; SacI is GAGCTC; Asp718 is GGTACC; PstI is CTGCAG and HindIII is AAGCTT. Also prophage is designated as "Pphage." - In this method, 100 µL PCR reactions carried out in 150µL Eppendorf tubes containing 84µL water, 10µL PCR buffer, 1µL of each primer (i.e., PKS-UF and PKS-UR), 2µL of dNTPs, 1 µL of wild type Bacillus chromosomal DNA template, and 1 µL of polymerase. DNA polymerases used included Taq Plus Precision polymerase and Herculase (Stratagene). Reactions were carried out in a Hybaid PCRExpress thermocycler using the following program. The samples were first heated at 94°C for 5 minutes, then cooled to a 50° hold. Polymerase was added at this point. Twenty-five cycles of amplification consisted of 1 minute at 95°C, 1 minute at 50°C and 1 minute at 72°C. A final 10 minutes at 72°C ensured complete elongation. Samples were held at 4°C for analysis.
- After completion of the PCR, 10µL of each reaction were run on an Invitrogen 1.2% agarose E-gel at 60 volts for 30 minutes to check for the presence of a band at the correct size. All the gel electrophoresis methods described herein used these conditions. If a band was present, the remainder of the reaction tube was purified using the Qiagen Qiaquick® PCR purification kit according to the manufacturer's instructions, then cut with the appropriate restriction enzyme pair. Digests were performed at 37°C for 1 hour as a 20 µL reaction consisting of 9µL of water, 2µL of 10xBSA, 2µL of an appropriate NEB restriction buffer (according to the 2000-01 NEB Catalog and Technical Reference), 5 µL of template, and 1µL of each restriction enzyme. For example, the PBSX upstream fragment and CssS upstream fragments were cut with XbaI and BamHI in NEB (New England BioLabs) restriction buffer B. The digested fragments were purified by gel electrophoresis and extraction using the Qiagen Qiaquick gel extraction kit following the manufacturer's instructions.
Figures 5 and 6 provide gels showing the results for various deletions. - Ligation of the fragments into a plasmid vector was done in two steps, using either the Takara ligation kit following the manufacturer's instructions or T4 DNA ligase (Reaction contents: 5 µL each insert fragment, 1µL cut pJM102 plasmid, 3 µL T4 DNA ligase buffer, and 1 µL T4 DNA ligase). First, the cut upstream and downstream fragments were ligated overnight at 15°C into unique restriction sites in the pJM102 plasmid polylinker, connecting at the common BamHI site to re-form a circular plasmid. The pJM102 plasmid was cut with the unique restriction enzyme sites appropriate for each deletion (See, Table 2; for cssS, XbaI and SacI were used) and purified as described above prior to ligation. This recircularized plasmid was transformed into Invitrogen's "Top Ten" E. coli cells, using the manufacturers One Shot transformation protocol.
- Transformants were selected on Luria-Bertani broth solidified with1.5% agar (LA) plus 50 ppm carbanicillin containing X-gal for blue-white screening. Clones were picked and grown overnight at 37°C in 5mL of Luria Bertani broth (LB) plus 50 ppm carbanicillin and plasmids were isolated using Qiagen's Qiaquick Mini-Prep kit. Restriction analysis confirmed the presence of the insert by cutting with the restriction sites at each end of the insert to drop an approximately 2 kb band out of the plasmid. Confirmed plasmids with the insert were cut with BamHI to linearize them in digestion reactions as described above (with an additional 1 µL of water in place of a second restriction enzyme), treated with 1 µL calf intestinal and shrimp phosphatases for 1 hour at 37°C to prevent re-circularization, and ligated to the antimicrobial resistance marker as listed in Table 2. Antimicrobial markers were cut with BamHI and cleaned using the Qiagen Gel Extraction Kit following manufacturer's instructions prior to ligation. This plasmid was cloned into E. coli as before, using 5 ppm phleomycin (phl) or 100 ppm spectinomycin (spc) as appropriate for selection. Confirmation of marker insertion in isolated plasmids was done as described above by restriction analysis with BamHI. Prior to transformation into B. subtilis, the plasmid was linearized with ScaI to ensure a double crossover event.
Table 2. Unique Restriction Enzyme Pairs Used in Deletion Constructs Deletion Name Unique Restriction Enzyme Pair Antimicrobial Marker Sbo XbaI-Asp718 spc Slr XbaI - SacI phleo YbcO XbaI - SacI spc Csn XbaI - Sall phleo PBSX XbaI-SacI phl PKS XbaI-SacI phl SPβ XbaI-SacI spec PPS XbaI-SacI spec Skin XbaI-SacI phl - This Example describes "
Method 2," which is also depicted inFigure 3 . Upstream and downstream fragments were amplified as inMethod 1, except the primers were designed with 25 bp "tails" complementary to the antimicrobial marker's primer sequences. A "tail" is defined herein as base pairs on the 5' end of a primer that are not homologous to the sequence being directly amplified, but are complementary to another sequence of DNA. Similarly, the primers for amplifying the antimicrobial contain "tails" that are complementary to the fragments' primers. For any given deletion, the DeletionX-UFfus and DeletionX-URfus are direct complements of one another. This is also true for the DFfus and DR-fus primer sets. In addition, in some embodiments, these primers contain restriction enzyme sites similar to those used inMethod 1 for use in creating a plasmid vector (See, Table 3 andU.S. Patent No. 5,023,171 ). Table 3 provides a list of primers useful for creation of deletion constructs by PCR fusion. Table 4 provides an additional list of primers useful for creation of deletion constructs by PCR fusion. However, in this Table, all deletion constructs would include the phleoR marker.Table 3. Primers Primer name Restriction enzyme engineered into primer Sequence SEQ ID. NO. DHB-UF XbaI CGAGAATCTAGAACAGGATGAATCATCTGTGGCGGG 110 DHB-UFfus-phleo BamHI 
111 DHB-URfus-phleo BamHI 
112 DHB-DFfus-phleo BamHI 
113 DHB-DRfus-phleo BamHI 
114 DHB-DR SacI GACTTCGTCGACGAGTGCGGACGGCCAGCATCACCA 115 DHB-UF-nested Xbal GGCATATCTAGAGACATGAAGCGGGAAACAGATG 116 DHB-DR-nested SacI GGTGCGGAGCTCGACAGTATCACAGCCAGCGCTG 117 YvfF-yveK-UF XbaI AAGCGTTCTAGACTGCGGATGCAGATCGATCTCGGG 118 YvfF-yveK-UF-phleo BamHI 
119 YvfF-yveK-UR-phleo BamHI 
120 YvfF-yveK-DF-phleo BamHI 
121 YvfF-yveK-DR-phleo BamHI 
122 YvfF-yveK-DR PstI CAAGCACTGCAGCCCACACTTCAGGCGGCTCAGGTC 123 YvfF-yveK-UF- XbaI GAGATATCTAGAATGGTATGAAGCGGAATTCCCG 124 YvfF-yveK-DR- KpnI ATAAACGGTACCCCCCTATAGATGCGAACGTTAGCCC 125 Prophage7-UF EcoRI AAGGAGGAATTCCATCTTGAGGTATACAAACAGTCAT 126 Prophage 7-UF- BamHI TCTCCGAGAAAGACAGGCAGGATCGGGATCC 127 Prophage 7-UR- BamHI TTGCTGAGTCTGGCTTTCGGTAAGCGGATCC 128 Skin+prophage7- Asp718 AAGGACGGTACCGGCTCATTACCCTCTTTTCAAGGGT 129 Skin+pro7-UF-phleo BamHI 
130 Skin+pro7-UR-phleo BamHI 
131 Skin+pro7-DF-phleo BamHI 
132 Skin+pro7-DR-phleo BamHI 
133 Skin+pro7-DR PstI GTCAACCTGCAGAGCGGCCCAGGTACAAGTTGGGGA 134 Skin+pro7-UF- SacI GGATCAGAGCTCGCTTGTCCTCCTGGGAACAGCCGG 135 Skin+pro7-DR- PstI TATATGCTGCAGGGCTCAGACGGTACCGGTTGTTCCT 136 The restriction sites are designated as follows: XbaI is TCTAGA; BamHI is GGATCC; SacI is GAGCTC; Asp718 is GGTACC; PstI is CTGCAG and HindIII is AAGCTT. Table 4. Additional Primers Used to Create Deletion Constructs by PCR Fusion*. Primer Name Restriction Enzyme Engineered Into Primer Sequence SEQ ID NO: Slr-UF XbaI CTGAACTCTAGACCTTCACCAGGCACAGAGGAGGTGA 137 Sir-Uffus BamHI 
138 Slr-Urfus BamHI 
139 Slr-Dffus BamHI 
140 Slr-Drfus BamHI 
141 Slr-DR SacI TGAGACGAGCTCGATGCATAGGCGACGGCAGGGCGCC 142 Slr-UF- nested XbaI CGAAATTCTAGATCCCGCGATTCCGCCCTTTGTGG 143 Slr-DR-nested SacI TTCCAAGAGCTCGCGGAATACCGGAAGCAGCCCC 144 YbcO-UF XbaI CAATTCTCTAGAGCGGTCGGCGCAGGTATAGGAGGGG 145 YbcO-UF BamHI 
146 YbcO-UR BamHI 
147 YbcO-DF BamHI 
148 YbcO-DR BamHI 
149 YbcO-DR SacI CAGGCGGAGCTCCCATTTATGACGTGCTTCCCTAAGC 150 Csn-UF XbaI TACGAATCTAGAGATCATTGCGGAAGTAGAAGTGGAA 151 Csn-UF BamHI 
152 Csn-UR BamHI 
153 Csn-DF BamHI 
154 Csn-DR BamHI 
155 Csn-DR SalI ATACTCGTCGACATACGTTGAATTGCCGAGAAGCCGC 156 Csn-UF- NA CTGGAGTACCTGGATCTGGATCTCC 157 Csn-DR- NA GCTCGGCTTGTTTCAGCTCATTTCC 158 SigB-UF SacI CGGTTTGAGCTCGCGTCCTGATCTGCAGAAGCTCATT 159 SigB-UF BamHI 
160 SigB-UR BamHI 
161 SigB-DF BamHI 
162 SigB-DR BamHI 
163 SigB-DR SalI GCTTCGGTCGACTTTGCCGTCTGGATATGCGTCTCTCG 164 SigB-UF- SacI GTCAAAGAGCTCTATGACAGCCTCCTCAAATTGCAGG 165 SigB-DR- SalI TTCCATGTCGACGCTGTGCAAAACCGCCGGCAGCGCC 166 SpoIISA-UF EcoRI ACATTCGAATTCAGCAGGTCAATCAGCTCGCTGACGC 167 SpoIISA-UF BamHI 
168 SpoIISA-UR BamHI 
169 SpoIISA-DF BamHI 
170 SpoIISA-DR BamHI 
171 SpoIISA-DR HindIII AGTCAT AAGCTTTCTGGCGTTTGATTTCATCAACGGG 172 SpoIISA-UF- NA CAGCGCGACTTGTTAAGGGACAATA 173 SpoIISA-DR- NA GGCTGCTGTGATGAACTTTGTCGGA 174 *All deletion constructs include the phleoR marker - The fragments listed in Tables 3 and 4 were size-verified by gel electrophoresis as described above. If correct, 1 µL each of the upstream, downstream, and antimicrobial resistance marker fragments were placed in a single reaction tube with the DeletionX-UF and DeletionX-DR primers or nested primers where listed. Nested primers are 25 base pairs of DNA homologous to an internal portion of the upstream or downstream fragment, usually about 100 base pairs from the outside end of the fragment (See,
Figure 2 ). The use of nested primers frequently enhances the success of fusion. The PCR reaction components were similar to those described above, except 82 µL of water was used to compensate for additional template volume. The PCR reaction conditions were similar to those described above, except the 72°C extension was lengthened to 3 minutes. During extension, the antimicrobial resistance gene was fused in between the upstream and downstream pieces. This fusion fragment can be directly transformed into Bacillus without any purification steps or with a simple Qiagen Qiaquick PRC purification done according to manufacturer's instructions. - In this Example, a method ("
Method 3") for creating DNA constructs using ligation of PCR fragments and direct transformation of Bacillus are described. By way of example, modification of prpC, sigD and tdh/kbl are provided to demonstrate the method of ligation. Indeed, sigD and tdh/kbl were constructed by one method and prpC by an alternate method. - The upstream and downstream fragments adjacent to the tdh/kbl region of the Bacillus subtilis chromosome were amplified by PCR similar to as described in
Method 1, except that the inside primer of the flanking DNA was designed to contain type II s restriction sites. Primers for the loxP-spectinomycin-loxP cassette were designed with the same type II s restriction site as the flanks and complementary overhangs. Unique overhangs for the left flank and the right flank allowed directional ligation of the antimicrobial cassette between the upstream and downstream flanking DNA. All DNA fragments were digested with the appropriate restriction enzymes, and the fragments were purified with a Qiagen Qiaquick PCR purification kit using the manufacturer's instructions. This purification was followed by desalting in a 1 mL spin column containing BioRad P-6 gel and equilibrated with 2 mM Tris-HCl, pH 7.5. Fragments were concentrated to 124 to 250 ng/µL using a Savant Speed Vac SC110 system. Three piece ligations of 0.8 to 1 µg of each fragment were performed with 12U T4 ligase (Roche) in a 15 to 25 µL reaction volume at 14 to 16°C for 16 hours. The total yield of the desired ligation product was >100 ng per reaction, as estimated by comparison to a standard DNA ladder on an agarose gel. The ligation mixture was used without purification for transformation reactions. Primers for this construction are shown in Table 5, belowTable 5. Primers for tdh/kbl Deletion Primer Name Restriction Enzyme Engineered Into Primer Primer Sequence SEQ ID NO: p70 DR none CTCAGTTCATCCATCAAATCACCAAGTCCG 175 P82 DF Bbsl 
176 p71 UF none AACCTTCCAGTCCGGTTTACTGTCGC 177 P83 UR BbsI 
178 p98spc F BbsI CCTTGTCTTGAAGACGGAGCTGGATCCATAACTTCGTATAATG 179 p106 spc R BbsI GTACCATAAGAAGACGGCTAGAGGATGCATATGGCGGCCGC 180 p112 UF* none CATATGCTCCGGCTCTTCAAGCAAG (analytical primer) 181 p113 DR* none CCTGAGATTGATAAACATGAAGTCCTC (analytical primer) 182 *primers for analytical PCR - The construct for the sigD deletion closely followed construction of tdh/kbl. The primers used for the sigD construction are provided in Table 6.
Table 6. Primers for sigD Construction Primer Name Restriction Enzyme Engineered Into Primer Primer Sequence SEQ ID NO: SigD UF none ATATTGAAGTCGGCTGGATTGTGG 183 SigD UR BgIII GCGGCAGATCTCGGCGCATTAAGTCGTCA 184 SigD DF EcoRI GCGGCGAATTCTCTGCTGGAAAAAGTGATACA 185 SigD DR none TTCGCTGGGATAACAACAT 186 Loxspc UF BgIII GCGGCAGATCTTAAGCTGGATCCATAACTTCG 187 Loxspc DR EcoRI GCGGCGAATTCATATGGCGGCCGCATAACTTC 188 SigD UO none CAATTTACGCGGGGTGGTG 189 SigD DO none GAATAGGTTACGCAGTTGTTG 190 Spc UR none CTCCTGATCCAAACATGTAAG 191 Spc DF none AACCCTTGCATATGTCTAG 192 - An additional example of creating a DNA molecule by ligation of PCR amplified DNA fragments for direct transformation of Bacillus involved a partial in-frame deletion of the gene prpC. A 3953 bp fragment of Bacillus subtilis chromosomal DNA containing the prpC gene was amplified by PCR using primers p95 and p96. The fragment was cleaved at unique restriction sites PfIMI and BstXI. This yielded three fragments, an upstream, a downstream, and a central fragment. The latter is the fragment deleted and consists of 170 bp located internal to the prpC gene. The digestion mixture was purified with a Qiagen Qiaquick PCR purification kit, followed by desalting in a 1 mL spin column containing BioRad P-6 gel and equilibrated with 2 mM Tris-HCl, pH 7.5. In a second PCR reaction, the antimicrobial cassette, IoxP-spectinomycin-loxP, was amplified with the primer containing a BstXI site and the downstream primer containing a PflMI site both with cleavage sites complementary to the sites in the genomic DNA fragment. The fragment was digested with PflMI and BstXI and purified as described for the chromosomal fragment above. A three piece ligation of the upstream, antimicrobial cassette, and the downstream fragments was carried out as for tdh/kbl, described above. The yield of desired ligation product was similar and the ligation product was used without further treatment for the transformation of xylRcomK competent Bacillus subtilis, as described in greater detail below.
Table 7. Primers for prpC Deletion Primer Name Restriction Enzyme Engineered Into Primer Primer Sequence SEQ ID NO: p95 DF none GCGCCCTTGATCCTAAGTCAGATGAAAC 193 p96 UR none CGGGTCCGATACTGACTGTAAGTTTGAC 194 p100 spc R PflMI GTACCATAACCATGCCTT'GGTTAGGATGCATATGGCGGCCGC 195 p101 spc F BstXI CCTTGTCTTCCATCTTGCTGGAGCTGGATCCATAACTTCGTATAATG 196 p114 anal. none GAGAGCAAGGACATGACATTGACGC 197 p115 anal.,* none GATCTTCACCCTCTTCAACTTGTAAAG 198 *anal., analytical PCR primer - In addition to the above deletions, pckA was also modified. The PCR primers pckA UF, pckA-2Urfus, spc ffus, spc rfus, pckA Dffus and pckA DR, were used for PCR and PCR fusion reactions using the chromosomal DNA of a Bacillus subtilis I168 derivative and pDG1726 (See, Guerout-Fleury et al., Gene 167(1-2):335-6 [1995]) as template. The primers are shown in Table 8. The method used in constructing these deletion mutants was the same as
Method 1, described above.Table 8. Primers Used for PckA Deletion Primer Name Restriction Enzyme Engineered Into Primer Primer Sequence Seq ID NO: pckA UF none TTTGCTTCCTCCTGCACAAGGCCTC 199 pckA-2URfus none CGTTATTGTGTGTGCATTTCCATTGT 200 spc ffus none 
201 pckA DFfus none GTAATGGCCCTCTCGTATAAAAAAC 202 spc rfus none 
203 pckA DR none GACCAAAATGTTTCGATTCAGCATTCCT 204 - Cells of a host strain Bacillus subtilis with partial genotype xylRcomK, were rendered competent by growth for 2 hours in Luria-Bertani medium containing 1% xylose, as described in
U.S. Patent Appln. Ser. No. 09/927,161, filed August 10, 2001 round bottom 2 mL tubes and stored in liquid nitrogen for future use. - For transformation, frozen competent cells were thawed at 37 °C and immediately after thawing was completed, DNA from ligation reaction mixtures was added at a level of 5 to 15 µL per tube. Tubes were then shaken at 1400 rpm (Tekmar VXR S-10) for 60 min at 37 °C. The transformation mixture was plated without dilution in 100 uL aliquots on 8 cm LA plates containing 100 ppm of spectinomycin. After growth over night, transformants were picked into Luria-Bertani (100 ppm spectinomycin) and grown at 37 °C for genomic DNA isolation performed as known in the art (See e.g., Harwood and Cuttings, Molecular Biological Methods for Bacillus, John Wiley and Son, New York, N.Y. [1990], at p. 23). Typically 400 to 1400 transformants were obtained from 100 uL transformation mix, when 5 uL of ligation reaction mix was used in the transformation.
- When the antimicrobial marker was located between two IoxP sites in the incoming DNA, the marker could be removed by transforming the strain with a plasmid containing the cre gene capable of expression the Cre protein. Cells were transformed with pCRM-TS-pleo (See below) cultured at 37 °C to 42 °C, plated onto LA and after colonies formed patched onto LA containing 100 ppm spectinomycin. Patches which did not grow after overnight incubation were deemed to have lost the antimicrobial maker. Loss of maker was verified by PCR assay with primers appropriate for the given gene.
- pCRM-TS-pleo has the following sequence (SEQ ID NO:205):



- In addition to the above methods, transcriptome DNA array methods were used in the development of mutants of the present invention. First, target RNA was harvested from a Bacillus strain by guanidinium acid phenol extraction as known in the art (See e.g., Farrell, RNA Methodologies, (2nd Ed.). Academic Press, San Diego, at pp. 81] and time-point was reverse-transcribed into biotin-labeled cDNA by a method adopted from deSaizieu et al. (deSaizieu et a/., J. Bacteriol., 182: 4696-4703 [2000]) and described herein. Total RNA (25 mg) was incubated 37°C overnight in a 100-mL reaction: 1x GIBCO first-strand buffer (50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl2); 10 mM DTT; 40 mM random hexamer; 0.3 mM each dCTP, dGTP and dTTP; 0.12 mM dATP; 0.3 mM biotin-dATP (NENO; 2500 units SuperScript II reverse-transcriptase (Roche). To remove RNA, the reaction was brought to 0.25 M NaOH and incubated at 65°C for 30 minutes. The reaction was neutralized with HCl and the nucleic acid precipitated at -20°C in ethanol with 2.5 M ammonium-acetate. The pellet was washed, air-dried, resuspended in water, and quantitated by UV spectroscopy. The reaction yield was approximately 20-25 mg biotin-labeled cDNA.
- Twelve mg of this cDNA were fragmented in 33 mL 1x One-Phor-All buffer (Amersham-Pharmacia #27-0901-02) with 3.75 milliunits of DNasel I at 37°C for 10 minutes. After heat-killing the DNase, fragmentation was validated by running 2 mg of the fragmented cDNA on a 3% agarose gel. Biotin-containing cDNA routinely ranged in size from 25 to 125 nucleotides. The remaining 10 mg of cDNA were hybridized to an Affymetrix Bacillus GeneChip array.
- Hybridizations were performed as described in the Affymetrix Expression Analysis Technical Manual (Affymetrix) using reagent suppliers as suggested. Briefly, 10 mg of fragmented biotin-labeled cDNA were added to a 220-mL hybridization cocktail containing: 100 mM MES (N-morpholinoethanesufonic acid), 1M Na+, 20 mM EDTA, 0.01% Tween 20; 5 mg/mL total yeast RNA; 0.5 mg/mL BSA; 0.1 mg/mL herring-sperm DNA; 50 pM control oligonucleotide (AFFX-B1). The cocktails were heated to 95°C for 5 minutes, cooled to 40°C for 5 minutes, briefly centrifuged to remove particulates, and 200 mL was injected into each pre-warmed pre-rinsed (1x MES buffer + 5 mg/ml yeast RNA) GeneChip cartridge. The arrays were rotated at 40°C overnight.
- The samples were removed and the arrays were filled with non-stringent wash buffer (6x SSPE, 0.01 % Tween 20) and washed on the Affymetrix fluidics station with protocol Euk-GE-WS2, using non-stringent and stringent (0.1 M MES, 0.1 M [Na+], 0.01% Tween 20) wash buffers. Arrays were stained in three steps: (1) streptavidin; (2) anti-streptavidin antibody tagged with biotin; (3) streptavidin-phycoerythrin conjugate.
- The signals in the arrays were detected with the Hewlett-Packard Gene Array Scanner using 570 nm laser light with 3-mm pixel resolution. The signal intensities of the 4351 ORF probe sets were scaled and normalized across all time points comprising a time course experiment. These signals were then compared to deduce the relative expression levels of genes under investigation. The threonine biosynthetic and degradative genes were simultaneous transcribed, indicating inefficient threonine utilization. Deletion of the degradative threonine pathway improved expression of the desired product (See,
Figure 7 ). The present invention provides means to modify pathways with transcription profiles that are similar to threonine biosynthetic and degradative profiles. Thus, the present invention also finds use in the modification of pathways with transcription profiles similar to threonine in order to optimize Bacillus strains. In some preferred embodiments, at least one gene selected from the group consisting of rocA, ycgN, ycgm rocF and rocD is deleted or otherwise modified. Using the present invention as described herein resulted in the surprising discovery that the sigD regulon was transcribed. Deletion of this gene resulted in better expression of the desired product (See,Figure 7 ). It was also surprising to find the transcription of gapB and pckA. Deletion of pckA did not result in improvement or detriment. However, the present invention provides means to improve strain protein production through the combination of pckA deletion or modification and deletion or modification of gapB and/or fbp. In addition, during the development of the present invention, it was observed that the tryptophan biosynthetic pathway genes showed unbalanced transcription. Thus, it is contemplated that the present invention will find use in producing strains that exhibit increased transcription of genes such as those selected from the group consisting of trpA, trpB, frpC, trpD, trpE, and/or trpF, such that the improved strains provide improved expression of the desired product, as compared to the parental (i.e., wild-type and/or originating strain). Indeed, it is contemplated that modifications of these genes in any combination will lead to improved expression of the desired product. - Analysis of the strains produced using the above constructs were conducted following fermentation. Cultures at 14 L scale were conducted in Biolafitte® fermenters. Media components per 7 liters are listed in Table 9.
Table 9. Media Components per 7L Fermentation NaH2PO4-H2O 0.8% 56g KH2PO4 0.8% 56g MgSO4-7H2O 0.28% 19.6g antifoam 0.1% 7g CaCl2-2H2O 0.01% 0.7g ferrous sulfate-7H2O 0.03% 2.1g MnCl2-4H2O 0.02% 1.4g trace metals 100 x stock* 1% 70g H2SO4 0.16% 11.2g 60% glucose 1.29% 90 *See, Harwood and Cutting, supra, at p. 549 - The tanks were stirred at 750 rpm and airflow was adjusted to 11 Liters per minute, the temperature was 37°C, and the pH was maintained at 6.8 using NH4OH. A 60% glucose solution was fed starting at about 14 hours in a linear ramp from 0.5 to 2.1 grams per minute to the end of the fermentation. Off-gasses were monitored by mass spectrometry. Carbon balance and efficiency were calculated from glucose fed, yield of protein product, cell mass yield, other carbon in broth, and CO2 evolved. A mutant strain was compared to parent strain to judge improvements. Although this mutant pckA strain did not show improvement under these conditions, it is contemplated that improvements will be produced under modified culture conditions (i.e., as known to those in the art), and/or incorporation of additional genes. In some preferred embodiments, these additional genes are selected from the group consisting of gapB, alsD, and/or fbp
- Once the DNA construct was created by
Method - The presence of the integrated DNA construct was confirmed by three PCR reactions, with components and conditions as described above. For example, two reactions were designed to amplify a region from outside the deletion cassette into the antimicrobial gene in one case (
primers 1 and 11) and through the entire insert in another (primers 1 and 12). A third check amplified a region from outside the deletion cassette into the deleted region (primers 1 and 4).Figure 4 shows that a correct clone showed a band in the first two cases but not the third. Wild-type Bacillus subtilis chromosomal DNA was used as a negative control in all reactions, and should only amplify a band with the third primer set. - Once the DNA construct was stably integrated into a competent Bacillus subtilis strain, the subtilisin activity was measured by shake flask assays and the activity was compared to wild type levels. Assays were performed in 250 ml baffled flasks containing 50 mL of growth media suitable for subtilisin production as known in the art (See, Christianson et al., Anal. Biochem., 223:119-129 [1994]; and Hsia et al., Anal. Biochem. 242:221 - 227 [1996]). The media were inoculated with 50 µL of an 8 hour 5mL culture and grown for 40 hours at 37°C with shaking at 250 RPM. Then, 1 mL samples were taken at 17, 24 and 40 hours for protease activity assays. Protease activity was measured at 405 nM using the Monarch Automatic Analyser. Samples in duplicate were diluted 1:11 (3.131 g/L) in buffer. As a control to ensure correct machine calibration one sample was diluted 1:6 (5.585 g/L), 1:12 (2.793 g/L and 1:18 (1.862 g/L).
Figure 7 illustrates the protease activity in various altered Bacillus subtilis clones.Figure 8 provides a graph showing improved protease secretion as measured from shake flask cultures in Bacillus subtilis wild-type strain (unaltered) and corresponding altered deletion strains (-sbo) and (-slr). Protease activity (g/L) was measured after 17, 24 and 40 hours. - Cell density was also determined using spectrophotometric measurement at an OD of 600. No significant differences were observed for the samples at the measured time (data not shown).
Claims (15)
- A method for providing an altered Bacillus strain having enhanced secretion of a subtilisin comprising: transforming a Bacillus strain with a DNA construct comprising a polynucleotide sequence flanked on each side by nucleotide sequences of the flanking regions of the chromosomal sbo gene, wherein the chromosomal sbo gene encodes the amino acid sequence of SEQ ID No. 2, for inactivating said sbo gene by homologous recombination between the DNA construct and the genome of said Bacillus strain.
- The method according to claim 1, wherein the polynucleotide sequence further comprises a selective marker.
- The method according to claim 2, wherein the selective marker is flanked by loxP sites.
- The method according to claim 1, wherein said inactivating comprises deletion of said sbo gene.
- The method according to claim 1, wherein said inactivating comprises insertional inactivation of said sbo gene.
- The method according to claim 1, wherein said subtilisin is selected from the group consisting of subtilisin 168, subtilisin BPN', subtilisin Carlsberg, subtilisin DY, subtilisin 147, subtilisin 309.
- The method according to any of the claims 1 to 6, wherein said Bacillus strain is selected from the group consisting of Bacillus licheniformis, Bacillus lentus, Bacillus subtilis, Bacillus amyloliquefaciens, Bacillus brevis, Bacillus stearothermophilus, Bacillus alkalophilus, Bacillus coagulans, Bacillus circulans, Bacillus pumilus, Bacillus lautus, Bacillus clausii, Bacillus megaterium, and Bacillus thuringiensis.
- The method according to any of the claims 1 to 7, wherein said Bacillus strain further comprises a mutation in a gene selected from the group consisting of degU, degQ, degS, scoC4, spollE, and oppA.
- A DNA construct comprising a polynucleotide sequence flanked on each side by nucleotide sequences of the flanking regions of the chromosomal sbo gene, wherein the chromosomal sbo gene encodes the amino acid sequence of SEQ ID No. 2, said construct further comprising a polynucleotide sequence encoding a subtilisin.
- The DNA construct according to claim 9, wherein the polynucleotide sequence comprises a selective marker.
- The DNA construct according to claim 10, wherein the selective marker is flanked by loxP sites.
- The DNA construct according to claim 9, wherein said subtilisin is selected from the group consisting of subtilisin 168, subtilisin BPN', subtilisin Carlsberg, subtilisin DY, subtilisin 147, subtilisin 309.
- A Bacillus strain comprising the DNA construct according to any of the claims 9 to 14.
- A method for enhancing secretion of a subtilisin from a Bacillus strain comprising growing the Bacillus strain according to claim 15, under conditions such that the subtilisin is produced by said Bacillus strain.
- A method for enhancing secretion of a subtilisin from a Bacillus strain comprising:inactivating the chromosomal sbo gene in a Bacillus strain to produce an altered Bacillus strain, wherein the chromosomal sbo gene encodes the amino acid sequence of SEQ ID No. 2; andgrowing said altered Bacillus strain under suitable conditions such that a subtilisin is produced by said altered Bacillus strain, wherein the secretion of said subtilisin is enhanced as compared to the secretion of said subtilisin in the corresponding unaltered Bacillus strain.
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11150881A EP2341070A3 (en) | 2002-03-29 | 2003-03-28 | Ehanced protein expression in bacillus |
| EP11150879A EP2339017A3 (en) | 2002-03-29 | 2003-03-28 | Enhanced protein expression in bacillus |
| DK11150877.6T DK2339016T3 (en) | 2002-03-29 | 2003-03-28 | Increased production of subtilisins |
| EP11150877.6A EP2339016B8 (en) | 2002-03-29 | 2003-03-28 | Enhanced production of subtilisins in Bacillus |
| EP15196066.3A EP3023501A1 (en) | 2002-03-29 | 2003-03-28 | Enhanced protein expression in bacillus |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US36885802P | 2002-03-29 | 2002-03-29 | |
| US36894902P | 2002-03-29 | 2002-03-29 | |
| US368949P | 2002-03-29 | ||
| US368858P | 2002-03-29 | ||
| US37634302P | 2002-04-29 | 2002-04-29 | |
| US376343P | 2002-04-29 | ||
| PCT/US2003/009585 WO2003083125A1 (en) | 2002-03-29 | 2003-03-28 | Ehanced protein expression in bacillus |
Related Child Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP11150877.6A Division EP2339016B8 (en) | 2002-03-29 | 2003-03-28 | Enhanced production of subtilisins in Bacillus |
| EP11150877.6A Division-Into EP2339016B8 (en) | 2002-03-29 | 2003-03-28 | Enhanced production of subtilisins in Bacillus |
| EP11150879A Division-Into EP2339017A3 (en) | 2002-03-29 | 2003-03-28 | Enhanced protein expression in bacillus |
| EP15196066.3A Division EP3023501A1 (en) | 2002-03-29 | 2003-03-28 | Enhanced protein expression in bacillus |
| EP11150881A Division-Into EP2341070A3 (en) | 2002-03-29 | 2003-03-28 | Ehanced protein expression in bacillus |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP1495128A1 EP1495128A1 (en) | 2005-01-12 |
| EP1495128A4 EP1495128A4 (en) | 2006-11-02 |
| EP1495128B1 true EP1495128B1 (en) | 2014-05-07 |
Family
ID=28678930
Family Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP03726142.7A Expired - Lifetime EP1495128B1 (en) | 2002-03-29 | 2003-03-28 | Ehanced protein expression in bacillus |
| EP11150881A Withdrawn EP2341070A3 (en) | 2002-03-29 | 2003-03-28 | Ehanced protein expression in bacillus |
| EP11150879A Withdrawn EP2339017A3 (en) | 2002-03-29 | 2003-03-28 | Enhanced protein expression in bacillus |
| EP15196066.3A Withdrawn EP3023501A1 (en) | 2002-03-29 | 2003-03-28 | Enhanced protein expression in bacillus |
| EP11150877.6A Expired - Lifetime EP2339016B8 (en) | 2002-03-29 | 2003-03-28 | Enhanced production of subtilisins in Bacillus |
Family Applications After (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP11150881A Withdrawn EP2341070A3 (en) | 2002-03-29 | 2003-03-28 | Ehanced protein expression in bacillus |
| EP11150879A Withdrawn EP2339017A3 (en) | 2002-03-29 | 2003-03-28 | Enhanced protein expression in bacillus |
| EP15196066.3A Withdrawn EP3023501A1 (en) | 2002-03-29 | 2003-03-28 | Enhanced protein expression in bacillus |
| EP11150877.6A Expired - Lifetime EP2339016B8 (en) | 2002-03-29 | 2003-03-28 | Enhanced production of subtilisins in Bacillus |
Country Status (7)
| Country | Link |
|---|---|
| US (4) | US8124399B2 (en) |
| EP (5) | EP1495128B1 (en) |
| JP (3) | JP4680512B2 (en) |
| AU (1) | AU2003228393A1 (en) |
| CA (2) | CA2915148C (en) |
| DK (2) | DK2339016T3 (en) |
| WO (1) | WO2003083125A1 (en) |
Families Citing this family (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK2339016T3 (en) * | 2002-03-29 | 2017-01-23 | Danisco Us Inc | Increased production of subtilisins |
| JP4336184B2 (en) * | 2003-11-07 | 2009-09-30 | 花王株式会社 | Recombinant microorganism |
| DK2664670T3 (en) | 2003-12-03 | 2015-07-27 | Danisco Us Inc | perhydrolase |
| EP1735339A2 (en) * | 2004-04-09 | 2006-12-27 | Genencor International, Inc. | Pcka modifications and enhanced protein expression in bacillus |
| JP2005348641A (en) * | 2004-06-09 | 2005-12-22 | Kao Corp | Host microorganism |
| US7981659B2 (en) | 2005-10-13 | 2011-07-19 | Kao Corporation | Bacillus subtilis mutant strain |
| DE102006004871A1 (en) * | 2006-02-02 | 2007-08-09 | Wacker Chemie Ag | Microbial strain useful for producing recombinant proteins comprises a gene coding for a recombinant protein and a gene that codes for a host protein and is mutated to reduce expression of the host protein |
| KR20130006529A (en) * | 2006-04-24 | 2013-01-16 | 아지노모토 가부시키가이샤 | Bacterium capable of producing purine substance, and process for production of purine substance |
| DK2029732T3 (en) | 2006-05-31 | 2010-02-01 | Novozymes As | Chloramphenicol resistance selection in Bacillus licheniformis |
| CN101641438B (en) * | 2007-03-12 | 2013-12-25 | 丹尼斯科美国公司 | Modified proteases |
| ES2533874T3 (en) * | 2007-10-31 | 2015-04-15 | Medimmune, Llc | Protein frame |
| BRPI0909373A2 (en) * | 2008-03-28 | 2015-08-04 | Danisco Us Inc | Method for locus amplification in bacterial cells |
| CN102906112B (en) | 2010-04-13 | 2016-12-07 | 米迪缪尼有限公司 | TRAIL R2-specificity polymer support |
| CN108948196B (en) | 2011-10-11 | 2022-09-23 | 维埃拉生物股份有限公司 | CD 40L-specific TN 3-derived scaffolds and methods of use thereof |
| WO2013138351A1 (en) | 2012-03-13 | 2013-09-19 | Board Of Trustees Of The University Of Arkansas | Separatome-based protein expression and purification platform |
| US8945837B2 (en) | 2013-03-15 | 2015-02-03 | American Sterilizer Company | Sterilization indicator including a simplified genetically engineered biological indicator |
| EP2796556A1 (en) | 2013-04-25 | 2014-10-29 | Rijksuniversiteit Groningen | Improved means and methods for expressing recombinant proteins |
| CN105378079B (en) | 2013-07-12 | 2019-09-27 | 诺维信公司 | The direct transfer of polynucleotides between genome |
| CA2924650C (en) | 2013-09-17 | 2019-10-22 | Board Of Trustees Of The University Of Arkansas | E. coli separatome-based protein expression and purification platform |
| JP2015156844A (en) * | 2014-02-25 | 2015-09-03 | 花王株式会社 | Bacillus subtilis variant and method for producing of dipicolinic acid using the same |
| JP6437737B2 (en) * | 2014-05-09 | 2018-12-12 | 花王株式会社 | Bacillus subtilis mutant and method for producing poly-γ-glutamic acid using the same |
| JP6527669B2 (en) * | 2014-05-12 | 2019-06-05 | 花王株式会社 | Bacillus subtilis mutant strain and method for producing dipicolinic acid using the same |
| UA126325C2 (en) | 2014-09-17 | 2022-09-21 | Споуджен Байотек Інк. | Fusion proteins, recombinant bacteria, and methods for using recombinant bacteria |
| US10683528B2 (en) | 2014-12-16 | 2020-06-16 | Danisco Us Inc | Enhanced protein expression |
| WO2018053058A1 (en) | 2016-09-14 | 2018-03-22 | Danisco Us Inc. | Lignocellulosic biomass fermentation-based processes |
| DK3585910T3 (en) | 2017-02-24 | 2024-06-17 | Danisco Us Inc | COMPOSITIONS AND METHODS FOR INCREASED PROTEIN PRODUCTION IN BACILLUS LICHENIFORMIS |
| KR102533803B1 (en) * | 2017-04-07 | 2023-05-18 | 듀폰 뉴트리션 바이오사이언시즈 에이피에스 | Bacillus host cells producing β-galactosidase and lactase in the absence of p-nitrobenzylesterase activator |
| US11879127B2 (en) | 2017-08-23 | 2024-01-23 | Danisco Us Inc. | Methods and compositions for efficient genetic modifications of Bacillus licheniformis strains |
| EP3682010A1 (en) | 2017-09-13 | 2020-07-22 | Danisco US Inc. | Modified 5'-untranslated region (utr) sequences for increased protein production in bacillus |
| NZ763625A (en) | 2017-09-20 | 2024-07-26 | Spogen Biotech Inc | Fusion proteins, recombinant bacteria, and exosporium fragments for plant health |
| EP3735478B1 (en) | 2018-01-03 | 2023-08-16 | Danisco US Inc. | Mutant and genetically modified bacillus cells and methods thereof for increased protein production |
| CN109355303B (en) * | 2018-11-14 | 2022-05-06 | 天津大学 | Application of inhibition and/or knockout gene in improving expression quantity of monoclonal antibody |
| WO2020112609A1 (en) | 2018-11-28 | 2020-06-04 | Danisco Us Inc | Novel promoter sequences and methods thereof for enhanced protein production in bacillus cells |
| CN109868253B (en) * | 2019-01-11 | 2020-03-13 | 齐鲁工业大学 | Bacillus licheniformis engineering bacteria for inhibiting bacterial autolysis and construction method and application thereof |
| EP4031560A1 (en) | 2019-08-14 | 2022-07-27 | Danisco US Inc | Compositions and methods for increased protein production in bacillus licheniformis |
| CA3155372A1 (en) | 2019-10-28 | 2021-05-06 | Sharief Barends | Microbial host cells for the production of heterologous cyanuric acid hydrolases and biuret hydrolases |
| US20220389372A1 (en) | 2019-11-11 | 2022-12-08 | Danisco Us Inc. | Compositions and methods for enhanced protein production in bacillus cells |
| JP2023524334A (en) | 2020-01-15 | 2023-06-12 | ダニスコ・ユーエス・インク | Compositions and methods for enhanced protein production in Bacillus licheniformis |
| CN116897160A (en) | 2021-02-22 | 2023-10-17 | 丹尼斯科美国公司 | Methods and compositions for producing a protein of interest in a pigment-deficient bacillus cell |
| US20240263185A1 (en) | 2021-05-24 | 2024-08-08 | Danisco Us Inc. | Compositions and methods for enhanced protein production in bacillus cells |
| MX2023015455A (en) | 2021-06-24 | 2024-02-23 | Basf Se | Improved bacillus production host. |
| MX2023015460A (en) | 2021-06-24 | 2024-01-18 | Basf Se | Improved bacillus host cell with altered rema/remb protein. |
| WO2023023642A2 (en) | 2021-08-20 | 2023-02-23 | Danisco Us Inc. | Methods and compositions for enhanced protein production in bacillus cells |
| EP4433588A1 (en) | 2021-11-16 | 2024-09-25 | Danisco US Inc. | Compositions and methods for enhanced protein production in bacillus cells |
| WO2023183865A1 (en) | 2022-03-24 | 2023-09-28 | Danisco Us Inc. | Compositions and methods for industrial scale production and recovery of lectins |
| WO2023192953A1 (en) | 2022-04-01 | 2023-10-05 | Danisco Us Inc. | Pro-region mutations enhancing protein production in gram-positive bacterial cells |
| WO2023245191A2 (en) * | 2022-06-17 | 2023-12-21 | Motif Foodworks, Inc. | Inducible cell lysis system |
| WO2024040043A1 (en) | 2022-08-16 | 2024-02-22 | International N&H Denmark Aps | Expression systems for phosphatases |
| WO2024050503A1 (en) | 2022-09-02 | 2024-03-07 | Danisco Us Inc. | Novel promoter and 5'-untranslated region mutations enhancing protein production in gram-positive cells |
| WO2024064653A1 (en) | 2022-09-20 | 2024-03-28 | Dupont Nutrition Biosciences Aps | Variant anti-viral polypeptides |
| WO2024091804A1 (en) | 2022-10-24 | 2024-05-02 | Danisco Us Inc. | Compositions and methods for enhanced protein production in bacillus cells |
Family Cites Families (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4302544A (en) | 1979-10-15 | 1981-11-24 | University Of Rochester | Asporogenous mutant of B. subtilis for use as host component of HV1 system |
| US4450235A (en) | 1982-04-21 | 1984-05-22 | Cpc International Inc. | Asporogenic mutant of bacillus subtilis useful as a host in a host-vector system |
| US4760025A (en) | 1984-05-29 | 1988-07-26 | Genencor, Inc. | Modified enzymes and methods for making same |
| NZ208612A (en) | 1983-06-24 | 1991-09-25 | Genentech Inc | Method of producing "procaryotic carbonyl hydrolases" containing predetermined, site specific mutations |
| US5310675A (en) * | 1983-06-24 | 1994-05-10 | Genencor, Inc. | Procaryotic carbonyl hydrolases |
| WO1985000382A1 (en) | 1983-07-06 | 1985-01-31 | Gist-Brocades N.V. | Molecular cloning and expression in industrial microorganism species |
| US5264366A (en) * | 1984-05-29 | 1993-11-23 | Genencor, Inc. | Protease deficient bacillus |
| US5700676A (en) | 1984-05-29 | 1997-12-23 | Genencor International Inc. | Modified subtilisins having amino acid alterations |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4965188A (en) | 1986-08-22 | 1990-10-23 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences using a thermostable enzyme |
| WO1987004461A1 (en) | 1986-01-15 | 1987-07-30 | Amgen | THERMALLY STABLE AND pH STABLE SUBTILISIN ANALOGS AND METHOD FOR PRODUCTION THEREOF |
| US4980288A (en) | 1986-02-12 | 1990-12-25 | Genex Corporation | Subtilisin with increased thermal stability |
| GB8608194D0 (en) | 1986-04-03 | 1986-05-08 | Massey Ferguson Services Nv | Valve control system |
| US4863768A (en) * | 1986-06-19 | 1989-09-05 | Asahi Kasei Kogyo Kabushiki Kaisha | Heat shrinkable cylindrical laminated film |
| US5322770A (en) | 1989-12-22 | 1994-06-21 | Hoffman-Laroche Inc. | Reverse transcription with thermostable DNA polymerases - high temperature reverse transcription |
| PT86864B (en) | 1987-02-27 | 1992-05-29 | Gist Brocades Nv | PROCESS FOR MOLECULAR CLONING AND EXPRESSION OF GENES ENCODING FOR PROTEOLITIC ENZYMES |
| US4914031A (en) | 1987-04-10 | 1990-04-03 | Amgen, Inc. | Subtilisin analogs |
| DK6488D0 (en) | 1988-01-07 | 1988-01-07 | Novo Industri As | ENZYMES |
| CN1056187C (en) | 1988-02-11 | 2000-09-06 | 金克克国际有限公司 | Proteolytic enzymes and their use in detergents |
| CA2003078A1 (en) * | 1988-11-18 | 1990-05-18 | Alan Sloma | Protease deletion |
| US5665587A (en) | 1989-06-26 | 1997-09-09 | Novo Nordisk A/S | Modified subtilisins and detergent compositions containing same |
| US5023171A (en) | 1989-08-10 | 1991-06-11 | Mayo Foundation For Medical Education And Research | Method for gene splicing by overlap extension using the polymerase chain reaction |
| ES2095233T3 (en) | 1989-08-11 | 1997-02-16 | Gist Brocades Nv | EFFECTIVE PRODUCTION OF MUTANT PROTEASE. |
| US5585253A (en) | 1989-11-27 | 1996-12-17 | The Regents Of The University Of California | Extracellular serine protease and a Bacillus subtilis alkaline neutral an serine protease mutant strain |
| IE892893A1 (en) | 1990-03-11 | 1991-09-25 | Charles O Kane | Phage-like bacteriocins |
| DK97190D0 (en) | 1990-04-19 | 1990-04-19 | Novo Nordisk As | OXIDATION STABLE DETERGENT ENZYMER |
| US5482849A (en) | 1990-12-21 | 1996-01-09 | Novo Nordisk A/S | Subtilisin mutants |
| WO1992019721A1 (en) | 1991-04-26 | 1992-11-12 | Genencor International, Inc. | Gene expression in bacilli |
| WO1992019729A1 (en) | 1991-05-01 | 1992-11-12 | Novo Nordisk A/S | Stabilized enzymes and detergent compositions |
| DE4411223A1 (en) | 1994-03-31 | 1995-10-05 | Solvay Enzymes Gmbh & Co Kg | Use of alkaline proteases in commercial textile washing processes |
| WO1999003984A2 (en) | 1997-07-15 | 1999-01-28 | Genencor International, Inc. | Proteases from gram-positive organisms |
| MA24811A1 (en) | 1997-10-23 | 1999-12-31 | Procter & Gamble | WASHING COMPOSITIONS CONTAINING MULTISUBSTITUTED PROTEASE VARIANTS |
| US6528255B1 (en) | 1997-12-30 | 2003-03-04 | Genencor International, Inc. | Proteases from gram positive organisms |
| US6835550B1 (en) | 1998-04-15 | 2004-12-28 | Genencor International, Inc. | Mutant proteins having lower allergenic response in humans and methods for constructing, identifying and producing such proteins |
| US6509185B1 (en) | 2000-01-07 | 2003-01-21 | Genencor International, Inc. | Mutant aprE promotor |
| JP4824257B2 (en) * | 2000-06-14 | 2011-11-30 | ダニスコ・ユーエス・インコーポレーテッド | Production of secreted polypeptides |
| EP1309677B2 (en) | 2000-08-11 | 2012-04-11 | Genencor International, Inc. | Bacillus transformation, transformants and mutant libraries |
| DE10043331A1 (en) * | 2000-09-02 | 2002-03-14 | Degussa | New polynucleotide from coryneform bacteria, useful when overexpressed for increasing fermentative amino acid production, encodes sigma factor D |
| WO2002055717A2 (en) * | 2000-10-10 | 2002-07-18 | Genencor Int | Enhanced secretion of a polypeptide by a microorganism |
| DK2339016T3 (en) * | 2002-03-29 | 2017-01-23 | Danisco Us Inc | Increased production of subtilisins |
-
2003
- 2003-03-28 DK DK11150877.6T patent/DK2339016T3/en active
- 2003-03-28 DK DK03726142.7T patent/DK1495128T3/en active
- 2003-03-28 US US10/507,720 patent/US8124399B2/en not_active Expired - Fee Related
- 2003-03-28 WO PCT/US2003/009585 patent/WO2003083125A1/en active Application Filing
- 2003-03-28 JP JP2003580558A patent/JP4680512B2/en not_active Expired - Fee Related
- 2003-03-28 CA CA2915148A patent/CA2915148C/en not_active Expired - Fee Related
- 2003-03-28 CA CA2480277A patent/CA2480277C/en not_active Expired - Fee Related
- 2003-03-28 EP EP03726142.7A patent/EP1495128B1/en not_active Expired - Lifetime
- 2003-03-28 EP EP11150881A patent/EP2341070A3/en not_active Withdrawn
- 2003-03-28 AU AU2003228393A patent/AU2003228393A1/en not_active Abandoned
- 2003-03-28 EP EP11150879A patent/EP2339017A3/en not_active Withdrawn
- 2003-03-28 EP EP15196066.3A patent/EP3023501A1/en not_active Withdrawn
- 2003-03-28 EP EP11150877.6A patent/EP2339016B8/en not_active Expired - Lifetime
-
2009
- 2009-06-01 JP JP2009132313A patent/JP5007319B2/en not_active Expired - Fee Related
-
2010
- 2010-08-19 JP JP2010183904A patent/JP5170793B2/en not_active Expired - Fee Related
-
2012
- 2012-01-23 US US13/356,572 patent/US8383366B2/en not_active Expired - Fee Related
-
2013
- 2013-01-18 US US13/745,630 patent/US9175294B2/en not_active Expired - Fee Related
-
2015
- 2015-09-17 US US14/857,343 patent/US9617549B2/en not_active Expired - Fee Related
Non-Patent Citations (1)
| Title |
|---|
| LAAN VAN DER J C ET AL: "CLONING, CHARACTERIZATION, AND MULTIPLE CHROMOSOMAL INTEGRATION OF A BACILLUS ALKALINE PROTEASE GENE", APPLIED AND ENVIRONMENTAL MICROBIOLOGY, AMERICAN SOCIETY FOR MICROBIOLOGY, US, vol. 57, no. 4, 1 April 1991 (1991-04-01), pages 901 - 909, XP001007884, ISSN: 0099-2240 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2339016B8 (en) | 2016-01-27 |
| EP2339017A3 (en) | 2011-10-12 |
| CA2480277C (en) | 2016-02-09 |
| US8383366B2 (en) | 2013-02-26 |
| JP4680512B2 (en) | 2011-05-11 |
| JP5170793B2 (en) | 2013-03-27 |
| WO2003083125A1 (en) | 2003-10-09 |
| AU2003228393A1 (en) | 2003-10-13 |
| US20160168582A1 (en) | 2016-06-16 |
| EP2339016A3 (en) | 2011-11-16 |
| US9175294B2 (en) | 2015-11-03 |
| JP2011004759A (en) | 2011-01-13 |
| US20120183995A1 (en) | 2012-07-19 |
| US9617549B2 (en) | 2017-04-11 |
| EP2339016B1 (en) | 2015-11-25 |
| EP1495128A4 (en) | 2006-11-02 |
| EP2339017A2 (en) | 2011-06-29 |
| EP2339016A2 (en) | 2011-06-29 |
| JP2009225804A (en) | 2009-10-08 |
| CA2915148C (en) | 2018-03-13 |
| EP2341070A3 (en) | 2011-10-19 |
| JP2005521407A (en) | 2005-07-21 |
| EP1495128A1 (en) | 2005-01-12 |
| DK1495128T3 (en) | 2014-08-11 |
| DK2339016T3 (en) | 2017-01-23 |
| CA2480277A1 (en) | 2003-10-09 |
| JP5007319B2 (en) | 2012-08-22 |
| EP3023501A1 (en) | 2016-05-25 |
| US8124399B2 (en) | 2012-02-28 |
| EP2341070A2 (en) | 2011-07-06 |
| CA2915148A1 (en) | 2003-10-09 |
| US20140011234A1 (en) | 2014-01-09 |
| US20060073559A1 (en) | 2006-04-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1495128B1 (en) | Ehanced protein expression in bacillus | |
| EP2421973B1 (en) | Proteases with modified pro regions | |
| EP2129779B2 (en) | Modified proteases | |
| US20080009033A1 (en) | Secretion, transcription and sporulation genes in Bacillus clausii | |
| US20090011463A1 (en) | Pcka Modifications and Enhanced Protein Expression in Bacillus | |
| EP3089990B1 (en) | Enhanced protein expression | |
| CA2935066C (en) | Enhanced protein expression |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20041025 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LI LU MC NL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL LT LV MK |
|
| A4 | Supplementary search report drawn up and despatched |
Effective date: 20060929 |
|
| 17Q | First examination report despatched |
Effective date: 20090113 |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| INTG | Intention to grant announced |
Effective date: 20131125 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LI LU MC NL PT RO SE SI SK TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: REF Ref document number: 666752 Country of ref document: AT Kind code of ref document: T Effective date: 20140515 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 60346137 Country of ref document: DE Effective date: 20140618 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 Effective date: 20140804 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: T3 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 666752 Country of ref document: AT Kind code of ref document: T Effective date: 20140507 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140808 Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140908 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 60346137 Country of ref document: DE |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20150210 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 60346137 Country of ref document: DE Effective date: 20150210 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20150328 Ref country code: MC Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: MM4A |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20150328 Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20150331 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20150331 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 14 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 15 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HU Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT; INVALID AB INITIO Effective date: 20030328 Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140507 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 16 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20190312 Year of fee payment: 17 Ref country code: FI Payment date: 20190311 Year of fee payment: 17 Ref country code: GB Payment date: 20190327 Year of fee payment: 17 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DK Payment date: 20190322 Year of fee payment: 3 Ref country code: NL Payment date: 20190313 Year of fee payment: 17 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20200217 Year of fee payment: 18 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20200214 Year of fee payment: 18 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R119 Ref document number: 60346137 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: FI Ref legal event code: MAE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200328 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: EBP Effective date: 20200331 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: MM Effective date: 20200401 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200401 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20201001 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20200328 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200328 Ref country code: DK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200331 |
|
| REG | Reference to a national code |
Ref country code: BE Ref legal event code: MM Effective date: 20210331 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210331 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210331 |