EP0009227B1 - Fluorination process and fluoro analogs of hydrocodone and oxycodone - Google Patents
Fluorination process and fluoro analogs of hydrocodone and oxycodone Download PDFInfo
- Publication number
- EP0009227B1 EP0009227B1 EP19790103475 EP79103475A EP0009227B1 EP 0009227 B1 EP0009227 B1 EP 0009227B1 EP 19790103475 EP19790103475 EP 19790103475 EP 79103475 A EP79103475 A EP 79103475A EP 0009227 B1 EP0009227 B1 EP 0009227B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- carbon atoms
- epoxy
- formula
- fluoro
- methoxymorphinan
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
- 238000000034 method Methods 0.000 title claims description 28
- 230000008569 process Effects 0.000 title claims description 13
- 125000001153 fluoro group Chemical group F* 0.000 title description 8
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 title description 5
- 238000003682 fluorination reaction Methods 0.000 title description 4
- BRUQQQPBMZOVGD-XFKAJCMBSA-N Oxycodone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C BRUQQQPBMZOVGD-XFKAJCMBSA-N 0.000 title description 2
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 title description 2
- LLPOLZWFYMWNKH-CMKMFDCUSA-N hydrocodone Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)CC(=O)[C@@H]1OC1=C2C3=CC=C1OC LLPOLZWFYMWNKH-CMKMFDCUSA-N 0.000 title description 2
- 229960000240 hydrocodone Drugs 0.000 title description 2
- 229960002085 oxycodone Drugs 0.000 title description 2
- LLPOLZWFYMWNKH-UHFFFAOYSA-N trans-dihydrocodeinone Natural products C1C(N(CCC234)C)C2CCC(=O)C3OC2=C4C1=CC=C2OC LLPOLZWFYMWNKH-UHFFFAOYSA-N 0.000 title description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 65
- 150000001875 compounds Chemical class 0.000 claims description 63
- -1 methylallyl Chemical group 0.000 claims description 44
- 125000004432 carbon atom Chemical group C* 0.000 claims description 37
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical group CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 claims description 34
- 239000000203 mixture Substances 0.000 claims description 19
- 125000000217 alkyl group Chemical group 0.000 claims description 18
- 150000002576 ketones Chemical class 0.000 claims description 15
- 239000002253 acid Substances 0.000 claims description 14
- 239000001257 hydrogen Substances 0.000 claims description 11
- 229910052739 hydrogen Inorganic materials 0.000 claims description 11
- 150000003839 salts Chemical class 0.000 claims description 10
- DPUCMWXOSSWNDZ-DCXHWPDHSA-N (4r,4ar,12bs)-7-fluoro-9-methoxy-3-methyl-2,4,4a,5,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical compound O1C2C(F)=CC[C@H]3[C@]4([H])N(C)CC[C@]23C2=C1C(OC)=CC=C2C4 DPUCMWXOSSWNDZ-DCXHWPDHSA-N 0.000 claims description 9
- 239000002904 solvent Substances 0.000 claims description 9
- LFOMGBBERKOUPQ-UHFFFAOYSA-N NS(F)(F)F Chemical class NS(F)(F)F LFOMGBBERKOUPQ-UHFFFAOYSA-N 0.000 claims description 8
- 125000004423 acyloxy group Chemical group 0.000 claims description 8
- 239000002798 polar solvent Substances 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 239000012025 fluorinating agent Substances 0.000 claims description 7
- 229910052731 fluorine Inorganic materials 0.000 claims description 7
- 239000011737 fluorine Substances 0.000 claims description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 7
- 229910052500 inorganic mineral Inorganic materials 0.000 claims description 7
- 239000011707 mineral Substances 0.000 claims description 7
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 claims description 6
- 125000001589 carboacyl group Chemical group 0.000 claims description 6
- 239000012454 non-polar solvent Substances 0.000 claims description 6
- YEUDLIJXGQPHDY-BZOQIEQVSA-N (4r,4ar,12bs)-3-(cyclopropylmethyl)-7-fluoro-9-methoxy-2,4,4a,5,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical compound C([C@@]12[C@H]3CC=C(F)C1OC=1C(OC)=CC=C(C2=1)C[C@]31[H])CN1CC1CC1 YEUDLIJXGQPHDY-BZOQIEQVSA-N 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 5
- 125000001844 prenyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 5
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 4
- 125000006224 2-tetrahydrofuranylmethyl group Chemical group [H]C([H])(*)C1([H])OC([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 4
- 125000004981 cycloalkylmethyl group Chemical group 0.000 claims description 4
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical group FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 claims description 4
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 4
- 229920002554 vinyl polymer Polymers 0.000 claims description 4
- 125000006017 1-propenyl group Chemical group 0.000 claims description 3
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 claims description 3
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 claims description 3
- MRQYTJXVULSNIS-UHFFFAOYSA-N 4-bromo-3-fluorophenol Chemical group OC1=CC=C(Br)C(F)=C1 MRQYTJXVULSNIS-UHFFFAOYSA-N 0.000 claims description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 3
- 239000003795 chemical substances by application Substances 0.000 claims description 3
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 3
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 claims description 3
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 230000009467 reduction Effects 0.000 claims description 3
- ZASWJVJRLOHFCK-GJZMFAHQSA-N (4r,4ar,12bs)-7,7-difluoro-9-methoxy-3-methyl-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical compound C1C(C2=3)=CC=C(OC)C=3OC3[C@]22CCN(C)[C@@]1([H])[C@@H]2CCC3(F)F ZASWJVJRLOHFCK-GJZMFAHQSA-N 0.000 claims description 2
- 239000004593 Epoxy Substances 0.000 claims 2
- XJQPAFFHNPXEDF-HKCCTKGBSA-N (4r,4ar,12bs)-3-(cyclobutylmethyl)-7,7-difluoro-9-methoxy-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical compound C([C@@]12C3OC=4C(OC)=CC=C(C1=4)C[C@@]1([C@@H]2CCC3(F)F)[H])CN1CC1CCC1 XJQPAFFHNPXEDF-HKCCTKGBSA-N 0.000 claims 1
- HNCCTNVZWNRQMS-LDXROWBESA-N (4r,4ar,12bs)-6-(cyclopropylmethyl)-7,7-difluoro-9-methoxy-2,3,4,4a,5,6,7a,13-octahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical compound C1([C@]23CCN[C@@H]([C@@H]3C3)CC4=CC=C(C(O1)=C42)OC)C(F)(F)C3CC1CC1 HNCCTNVZWNRQMS-LDXROWBESA-N 0.000 claims 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 claims 1
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 37
- 239000007787 solid Substances 0.000 description 16
- 239000000243 solution Substances 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 14
- 239000004480 active ingredient Substances 0.000 description 14
- 239000003921 oil Substances 0.000 description 14
- 235000019198 oils Nutrition 0.000 description 14
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 150000001408 amides Chemical class 0.000 description 9
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 241000699670 Mus sp. Species 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 8
- 230000000202 analgesic effect Effects 0.000 description 8
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 239000000730 antalgic agent Substances 0.000 description 7
- 239000003887 narcotic antagonist Substances 0.000 description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 7
- HXSPOSPPXZJZHZ-YIZMJLDNSA-N (4r,4ar,12bs)-7-fluoro-3-methyl-2,4,4a,5,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-9-ol;hydrochloride Chemical compound Cl.O1C2C(F)=CC[C@H]3[C@]4([H])N(C)CC[C@]23C2=C1C(O)=CC=C2C4 HXSPOSPPXZJZHZ-YIZMJLDNSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 238000010521 absorption reaction Methods 0.000 description 6
- 229940035676 analgesics Drugs 0.000 description 6
- 239000003814 drug Substances 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 6
- 150000007513 acids Chemical class 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 238000002329 infrared spectrum Methods 0.000 description 5
- 230000008018 melting Effects 0.000 description 5
- 238000002844 melting Methods 0.000 description 5
- 235000010755 mineral Nutrition 0.000 description 5
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- MMGUCDDWAUUIOB-JSAQVEJNSA-N (4r,4ar,12bs)-3-(cyclopropylmethyl)-7,7-difluoro-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline-9-ol Chemical compound C([C@@]12C3OC=4C(O)=CC=C(C1=4)C[C@@]1([C@@H]2CCC3(F)F)[H])CN1CC1CC1 MMGUCDDWAUUIOB-JSAQVEJNSA-N 0.000 description 4
- RLQZIECDMISZHS-UHFFFAOYSA-N 2-phenylcyclohexa-2,5-diene-1,4-dione Chemical compound O=C1C=CC(=O)C(C=2C=CC=CC=2)=C1 RLQZIECDMISZHS-UHFFFAOYSA-N 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- BSVWENIBPSVBCC-VCTRGTQWSA-N [(4r,4ar,12bs)-7,7-difluoro-9-methoxy-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline-3-yl]-cyclobutylmethanone Chemical compound C([C@@]12C3OC=4C(OC)=CC=C(C1=4)C[C@@]1([C@@H]2CCC3(F)F)[H])CN1C(=O)C1CCC1 BSVWENIBPSVBCC-VCTRGTQWSA-N 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 150000003840 hydrochlorides Chemical class 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- GJLBNVWSAGGJCA-YPTPCZDSSA-N (1S,9R,10R)-13-fluoro-17-azatetracyclo[7.5.3.01,10.02,7]heptadeca-2,4,6-triene Chemical compound C1C(F)CC[C@H]2[C@]3([H])NCC[C@@]21C1=CC=CC=C1C3 GJLBNVWSAGGJCA-YPTPCZDSSA-N 0.000 description 3
- KZLOUAQROWASPE-NPDDNSPTSA-N (4r,4ar,12bs)-3-(cyclobutylmethyl)-7,7-difluoro-9-methoxy-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline;hydrochloride Chemical compound Cl.C([C@@]12C3OC=4C(OC)=CC=C(C1=4)C[C@@]1([C@@H]2CCC3(F)F)[H])CN1CC1CCC1 KZLOUAQROWASPE-NPDDNSPTSA-N 0.000 description 3
- OFLMEEWFNOKPDN-UQQIXKAISA-N (4r,4ar,12bs)-3-(cyclopropylmethyl)-7,7-difluoro-9-methoxy-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical compound C([C@@]12C3OC=4C(OC)=CC=C(C1=4)C[C@@]1([C@@H]2CCC3(F)F)[H])CN1CC1CC1 OFLMEEWFNOKPDN-UQQIXKAISA-N 0.000 description 3
- PRTXRUZFMVBJSJ-RUALPGEESA-N (4r,4ar,12bs)-7,7-difluoro-9-methoxy-3-methyl-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline;hydrochloride Chemical compound Cl.C1C(C2=3)=CC=C(OC)C=3OC3[C@]22CCN(C)[C@@]1([H])[C@@H]2CCC3(F)F PRTXRUZFMVBJSJ-RUALPGEESA-N 0.000 description 3
- ZRXLINBCJRKJDY-CVHNIZFNSA-N (4r,4ar,12bs)-7-fluoro-9-methoxy-3-methyl-2,4,4a,5,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline;hydrochloride Chemical compound Cl.O1C2C(F)=CC[C@H]3[C@]4([H])N(C)CC[C@]23C2=C1C(OC)=CC=C2C4 ZRXLINBCJRKJDY-CVHNIZFNSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- KVCCEYMOXHCPKJ-WOFHWGLUSA-N [(4r,4ar,12bs)-7-fluoro-9-methoxy-2,4,4a,5,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-3-yl]-cyclobutylmethanone Chemical compound C([C@@]12[C@H]3CC=C(F)C1OC=1C(OC)=CC=C(C2=1)C[C@]31[H])CN1C(=O)C1CCC1 KVCCEYMOXHCPKJ-WOFHWGLUSA-N 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- ULFHSQLFQYTZLS-UHFFFAOYSA-N difluoroamine Chemical compound FNF ULFHSQLFQYTZLS-UHFFFAOYSA-N 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- ZKQFHRVKCYFVCN-UHFFFAOYSA-N ethoxyethane;hexane Chemical compound CCOCC.CCCCCC ZKQFHRVKCYFVCN-UHFFFAOYSA-N 0.000 description 3
- 150000002431 hydrogen Chemical group 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 229920000609 methyl cellulose Polymers 0.000 description 3
- 239000001923 methylcellulose Substances 0.000 description 3
- 235000010981 methylcellulose Nutrition 0.000 description 3
- 229960005181 morphine Drugs 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- JACGNUSQQWYLGR-BJJXKVORSA-N (1R,9R,10R)-5,6-difluoro-17-azatetracyclo[7.5.3.01,10.02,7]heptadeca-2(7),3,5-triene Chemical compound C1CCC[C@H]2[C@]3([H])NCC[C@@]21C1=CC=C(F)C(F)=C1C3 JACGNUSQQWYLGR-BJJXKVORSA-N 0.000 description 2
- HPRRQMGZRLNYEC-AFSSITGASA-N (4R,4aR,7S,7aR,12bS)-7-fluoro-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical class F[C@@H]1[C@H]2[C@]34C=5C(=C(C=CC=5C[C@H]([C@@H]3C=C1)N(C)CC4)OC)O2 HPRRQMGZRLNYEC-AFSSITGASA-N 0.000 description 2
- NBSGIQCKUQWGAU-TWIAOXOGSA-N (4r,4ar,12bs)-3-(cyclopropylmethyl)-7-fluoro-2,4,4a,5,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-9-ol;hydrochloride Chemical compound Cl.C([C@@]12[C@H]3CC=C(F)C1OC=1C(O)=CC=C(C2=1)C[C@]31[H])CN1CC1CC1 NBSGIQCKUQWGAU-TWIAOXOGSA-N 0.000 description 2
- WTUSIWWHFUVKSW-ZFDIKFDDSA-N (4r,4ar,7s,7ar,12bs)-7-fluoro-3-methyl-2,4,4a,7,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-9-ol Chemical class O([C@H]1[C@H](C=C[C@H]23)F)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O WTUSIWWHFUVKSW-ZFDIKFDDSA-N 0.000 description 2
- ZQXCQTAELHSNAT-UHFFFAOYSA-N 1-chloro-3-nitro-5-(trifluoromethyl)benzene Chemical compound [O-][N+](=O)C1=CC(Cl)=CC(C(F)(F)F)=C1 ZQXCQTAELHSNAT-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- FZRKAZHKEDOPNN-UHFFFAOYSA-N Nitric oxide anion Chemical compound O=[N-] FZRKAZHKEDOPNN-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000003610 charcoal Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 150000002222 fluorine compounds Chemical class 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 229910052740 iodine Chemical group 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 229960004715 morphine sulfate Drugs 0.000 description 2
- GRVOTVYEFDAHCL-RTSZDRIGSA-N morphine sulfate pentahydrate Chemical compound O.O.O.O.O.OS(O)(=O)=O.O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O.O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O GRVOTVYEFDAHCL-RTSZDRIGSA-N 0.000 description 2
- 230000003533 narcotic effect Effects 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N p-hydroxybenzoic acid methyl ester Natural products COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000004237 preparative chromatography Methods 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- HIFJUMGIHIZEPX-UHFFFAOYSA-N sulfuric acid;sulfur trioxide Chemical compound O=S(=O)=O.OS(O)(=O)=O HIFJUMGIHIZEPX-UHFFFAOYSA-N 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GTKOZGJHIBCFME-NXCFDTQHSA-N (1R,9R,10R)-17-decyl-17-azatetracyclo[7.5.3.01,10.02,7]heptadeca-2,4,6-triene Chemical compound C1CCC[C@H]2[C@]3([H])N(CCCCCCCCCC)CC[C@@]21C1=CC=CC=C1C3 GTKOZGJHIBCFME-NXCFDTQHSA-N 0.000 description 1
- KDOQCCVLGFVOIW-LUZMIZLMSA-N (4r,4ar,7ar,12bs)-7,7-difluoro-9-methoxy-3-(2-phenylethyl)-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical compound C([C@@]12C3=C4O[C@H]2C(F)(F)CC[C@H]1[C@H]1CC3=CC=C4OC)CN1CCC1=CC=CC=C1 KDOQCCVLGFVOIW-LUZMIZLMSA-N 0.000 description 1
- VLJROMNXPZLYLX-OCOKVPCDSA-N (4r,4ar,7ar,12bs)-7-fluoro-9-methoxy-3-(2-phenylethyl)-2,4,4a,5,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline Chemical compound C([C@@]12C3=C4O[C@H]2C(F)=CC[C@H]1[C@H]1CC3=CC=C4OC)CN1CCC1=CC=CC=C1 VLJROMNXPZLYLX-OCOKVPCDSA-N 0.000 description 1
- WFLOTYSKFUPZQB-UHFFFAOYSA-N 1,2-difluoroethene Chemical class FC=CF WFLOTYSKFUPZQB-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- OXHPTABOQVHKLN-UHFFFAOYSA-N 1-(2-bromoethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCBr)C=C1 OXHPTABOQVHKLN-UHFFFAOYSA-N 0.000 description 1
- NAMDIHYPBYVYAP-UHFFFAOYSA-N 1-methoxy-2-(2-methoxyethoxy)ethane Chemical compound COCCOCCOC.COCCOCCOC NAMDIHYPBYVYAP-UHFFFAOYSA-N 0.000 description 1
- FUOHKPSBGLXIRL-UHFFFAOYSA-N 2-(chloromethyl)thiophene Chemical compound ClCC1=CC=CS1 FUOHKPSBGLXIRL-UHFFFAOYSA-N 0.000 description 1
- BDZHKUAKSMWSAJ-UHFFFAOYSA-N 2-chloro-n,n-diethyl-1,1,2-trifluoroethanamine Chemical compound CCN(CC)C(F)(F)C(F)Cl BDZHKUAKSMWSAJ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Natural products OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 244000290333 Vanilla fragrans Species 0.000 description 1
- 235000009499 Vanilla fragrans Nutrition 0.000 description 1
- 235000012036 Vanilla tahitensis Nutrition 0.000 description 1
- PSDYQSWHANEKRV-UHFFFAOYSA-N [S]N Chemical class [S]N PSDYQSWHANEKRV-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000000397 acetylating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229910000102 alkali metal hydride Inorganic materials 0.000 description 1
- 150000008046 alkali metal hydrides Chemical class 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 239000002269 analeptic agent Substances 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 230000003064 anti-oxidating effect Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- UUWSLBWDFJMSFP-UHFFFAOYSA-N bromomethylcyclohexane Chemical compound BrCC1CCCCC1 UUWSLBWDFJMSFP-UHFFFAOYSA-N 0.000 description 1
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- ZVTQWXCKQTUVPY-UHFFFAOYSA-N chloromethylcyclopropane Chemical compound ClCC1CC1 ZVTQWXCKQTUVPY-UHFFFAOYSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 229960004126 codeine Drugs 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- JFWMYCVMQSLLOO-UHFFFAOYSA-N cyclobutanecarbonyl chloride Chemical compound ClC(=O)C1CCC1 JFWMYCVMQSLLOO-UHFFFAOYSA-N 0.000 description 1
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexyloxide Natural products O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 1
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 1
- 239000012649 demethylating agent Substances 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- XUCNUKMRBVNAPB-UHFFFAOYSA-N fluoroethene Chemical class FC=C XUCNUKMRBVNAPB-UHFFFAOYSA-N 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 239000012433 hydrogen halide Substances 0.000 description 1
- 229910000039 hydrogen halide Inorganic materials 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 239000011630 iodine Chemical group 0.000 description 1
- TYQCGQRIZGCHNB-JLAZNSOCSA-N l-ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(O)=C(O)C1=O TYQCGQRIZGCHNB-JLAZNSOCSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- PQIOSYKVBBWRRI-UHFFFAOYSA-N methylphosphonyl difluoride Chemical group CP(F)(F)=O PQIOSYKVBBWRRI-UHFFFAOYSA-N 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- INAXVFBXDYWQFN-XHSDSOJGSA-N morphinan Chemical compound C1C2=CC=CC=C2[C@]23CCCC[C@H]3[C@@H]1NCC2 INAXVFBXDYWQFN-XHSDSOJGSA-N 0.000 description 1
- BGEHHDVYQRNMJB-UHFFFAOYSA-N morpholine;sulfuric acid Chemical compound OS([O-])(=O)=O.C1COCC[NH2+]1 BGEHHDVYQRNMJB-UHFFFAOYSA-N 0.000 description 1
- CZHYJTHTVKIGAJ-UHFFFAOYSA-N n,n-diethylethanamine;hexane;propan-2-one Chemical compound CC(C)=O.CCCCCC.CCN(CC)CC CZHYJTHTVKIGAJ-UHFFFAOYSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 235000019629 palatability Nutrition 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- VOKSWYLNZZRQPF-GDIGMMSISA-N pentazocine Chemical compound C1C2=CC=C(O)C=C2[C@@]2(C)[C@@H](C)[C@@H]1N(CC=C(C)C)CC2 VOKSWYLNZZRQPF-GDIGMMSISA-N 0.000 description 1
- 229960005301 pentazocine Drugs 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 239000008354 sodium chloride injection Substances 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000002195 soluble material Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- CBXCPBUEXACCNR-UHFFFAOYSA-N tetraethylammonium Chemical compound CC[N+](CC)(CC)CC CBXCPBUEXACCNR-UHFFFAOYSA-N 0.000 description 1
- 125000005301 thienylmethyl group Chemical group [H]C1=C([H])C([H])=C(S1)C([H])([H])* 0.000 description 1
- 229940098465 tincture Drugs 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000563 toxic property Toxicity 0.000 description 1
- UFXIRMVZNARBDL-UHFFFAOYSA-N trifluoro(morpholin-4-yl)-$l^{4}-sulfane Chemical compound FS(F)(F)N1CCOCC1 UFXIRMVZNARBDL-UHFFFAOYSA-N 0.000 description 1
- KRVIQSHPVVXYIT-UHFFFAOYSA-N trifluoro(piperidin-1-yl)-$l^{4}-sulfane Chemical compound FS(F)(F)N1CCCCC1 KRVIQSHPVVXYIT-UHFFFAOYSA-N 0.000 description 1
- CCKAYTBOIIPZGA-UHFFFAOYSA-N trifluoro(pyrrolidin-1-yl)-$l^{4}-sulfane Chemical compound FS(F)(F)N1CCCC1 CCKAYTBOIIPZGA-UHFFFAOYSA-N 0.000 description 1
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/22—Bridged ring systems
- C07D221/28—Morphinans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D489/00—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula:
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D489/00—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula:
- C07D489/02—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula: with oxygen atoms attached in positions 3 and 6, e.g. morphine, morphinone
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D489/00—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula:
- C07D489/06—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula: with a hetero atom directly attached in position 14
- C07D489/08—Oxygen atom
Definitions
- This invention relates to a fluorination process for production of compounds which are useful as intermediates in syntheses of novel fluorine analogs of hydrocodone and oxycodone, the fluorine analogs exhibiting analgesic and/or narcotic antagonist properties in mammals.
- a fluorinating agent such as N-(2-chloro-1,1,2-trifluoroethyl)diethylamine.
- 6-deoxy-6-fluoromorphines and -codeines are said to be "active analgesics and central nervous system stimulants like the parent alkaloids, but in addition they possess pharmacologic. advantages. Illustratively, 6-deoxy-6-fluorocodeine is a more active analgesic than codein when administered orally, and side effects are reduced. Similarly, 6-deoxy-6-fluoromorphine is an analgesic as active as morphine, but it elicits only comparatively mild side effects.” No actual biological data are disclosed.
- the compounds of the instant invention are vinylic fluorides and in addition encompass variations of the nitrogen substituent, providing compounds with mixed agonist-antagonist profiles (see below, page 3, line 30-32 whereat cyclopropylmethyl and cyclobutylmethyl derivatives are disclosed).
- the compound of US-A 3 137 701 shown on page 1 of the instant description is relatively inactive as an analgesic (the PQW column) and has no antagonist component (the anti-straub tail column, a measure of the narcotic tendency of the compound), in marked contrast to most of the compounds of the present invention.
- Diethylaminosulfur trifluoride is a convenient reagent for replacing the carbonyl oxygen of aldehydes and ketones with two fluorine atoms.
- DAST Diethylaminosulfur trifluoride
- the compounds of Examples 5-7, 11 and 12 illustrate the best mode of the invention of which Examples 5 and 11 are preferred.
- MA mineral acid
- R in formula I is H (obtained as shown in Example 13)
- the corresponding 6-fluoride is converted to a pharmaceutically superior compound by alkylation of the HN group with RX, usually under basic conditions, wherein R is as previously defined except that it is not H and X is generally halogen (Cl, Br or I), for example, cyclopropylmethyl bromide.
- the substituent in the 3-position is generally the 3-methoxy substituent.
- a demethylating agent such as a hydrogen halide
- the 3-hydroxy compound from which can be prepared the corresponding alkyl ether wherein the alkyl group contains 1-4 carbon atoms, for example, the propoxy or butoxy derivative, or the corresponding ester of a C 1 ⁇ C 12 alkanoic acid, for example, the formate, butyrate or dodecanoate, using general synthetic methods.
- the esters can exhibit greater potency as pharmaceuticals.
- the more active compounds are preferably prepared by reacting under substantially anhydrous conditions the ketone of the formula wherein
- the reduction can be accomplished using conventional chemical techniques, for example, by subjecting the intermediate of formula IV to reaction with LiAIH 4 , as exemplified in the examples below.
- Other reducing agents which can be used include other alkali metal hydrides, such as sodium or potassium borohydride.
- R 2 when R 2 is to be hydroxyl in the final product, the compound subjected to fluorination should have R 2 as the ester (or equivalent protective group) which is subsequently converted to the hydroxyl.
- R 2 is OH in the starting compound, the use of excess fluorinating agent, disubstituted aminosulfur trifluoride, results in the formation of a product wherein R 2 is F.
- the starting ketone is the mineral acid salt, for example, the hydrogen sulfate of formula III, a good yield of the desired fluorinated compound is obtained.
- the starting ketone which is the amide of formula II. It is more preferred to use such amide wherein R 3 has 3-8 carbon atoms..
- the disubstituted aminosulfur trifluorides are known compounds. Particularly useful are diethylaminosulfur trifluoride (bAST), pyrrolidinosulfur trifluoride, morpholinosulfur trifluoride and piperidylsulfur trifluoride.
- the reaction is normally carried out in a solvent medium, preferably catalytically using a highly polar catalyst.
- the solvent can be polar or nonpolar but it must be nonreactive with the disubstituted aminosulfur trifluoride.
- the use of a polar solvent tends to give more of the 6-fluoro-6,7-unsaturated codeine (n is 1) whereas the use of a nonpolar solvent generally gives more of the 6,6-difluoro compound (n is 2).
- polar solvent is meant a solvent that has a high dielectric constant.
- Solvents which favor conversion of ketones to vinylene fluorides are polar and include dioxane, diethyl ether, tetrahydrofuran, ethylene glycol dimethyl ether (glyme), diethylene glycol dimethyl ether (diglyme) and triethylene glycol dimethyl ether.
- Nonpolar solvents such as hydrocarbons and halogenated hydrocarbons, for example, CCl 3 F, generally increase the amount of 6,6-difluoro (gem-) compound formed.
- a strong acid such as fuming sulfuric acid
- a highly polar catalyst increases the rate of formation of the 6,7-unsaturated-6-fluoride.
- Other useful catalysts include strong mineral acids that, in the quantities used, do not react with the carbonyl group, with the carbon- carbon double bond or with other groups present in the codeine-type compounds, for example, perchloric, polyphosphoric and fluorosulfonic acids.
- the useful acids generally have a log K a of greater than -2.
- the amount of catalyst used is generally 0.001 to 1% by weight of the ketone starting material. The catalyst is believed to function by increasing the polarity of the reaction medium and by increasing the rate of the reaction.
- the reaction is conducted, under substantially anhydrous conditions, in a vessel which is suitably glass, but a metal or ceramic vessel can be used.
- the reaction is conducted at -40°C to +80°C with the range 0-30°C being generally preferred.
- the reaction time is dependent upon the reactants and the temperature, with times of less than one hour to one week or more being useful. Pressure is not critical but ambient or autogeneous pressure is preferred.
- the desired fluoro compound can be separated from the reaction mixture by conventional procedures.
- Preparative chromatography is a particularly useful procedure for separation and purification but other means, for example, crystaliation or extraction can be used.
- the reaction mixture also contained minor amounts of the monofluoro- ⁇ , ⁇ -unsaturated compound (5) or (2).
- the N-cyclopropylcarbonylcodone (12) was obtained by reacting 18 g of cyclopropane carbonyl chloride with 30.7 g of the amine (11) at room temperature in methylene chloride in the presence of 20 g of tetraethylamine; yield 33 g as a white solid; identical with that of Gates, J.Med.Chem. 7, 127 (1964).
- the amide (13) (7 g) was dissolved in 100 ml of tetrahydrofuran and mixed with 3 g of LiAIH 4 in 100 ml of tetrahydrofuran. After refluxing for 16 hours, 3 ml of water was added followed by 3 ml of 15% aqueous NaOH and 9 ml of water to give 6.13 g of product which was chromatographed using 1:1 hexane:acetone containing 1% triethylamine to give 3.6 g of (15) in 96% purity by 19 F nmr or 91% by HPLC as a white solid m.p. 69-73°. The hydrochloride of the latter softened at 100° and had a mp of 123-133°. HMRS Calcd for C 2 ,H 25 NO 2 F 2 : 361.1852; Found: 361.1846.
- Example 5 The 0-methyl ether (16) of Example 5 (2.9 g) was reacted with 7 g of pyridine hydrochloride as in Example 6; there was obtained a tan solid which was taken up in ether and converted to the hydrochloride salt. The salt was recrystallized from ethanol to give 0.6 g of white solid (18) mp 285-290°. HPLC showed one major peak (94.9%).
- Example 5 To 10 g of amine (11) of Example 5 in 150 ml of dimethylformamide was added 4 g of allyl bromide and 2.8 g of NaHC0 3 . The mixture was heated at 80-85° for 5 hours, diluted with water, extracted with ether and converted to 12.7 g of the amine salt (19). The salt was reacted with diethylaminosulfur trifluoride according to the procedure of Example 2 and the hydrochloride salts (20) and (21) thus formed were separated by chromatography as described in Example 5. 19 F nmr (CDCl 3 ): ⁇ -19.6, -94.2, -104.1 and 106.7 ppm.
- Example 11 The procedure of Example 11 was used to prepare the acetoxy derivative of the corresponding difluoride.
- the difluoride (30) was obtained as a white solid melting at 128-130°.
- the fluoronorcodeinones (32) and (33) of this example are valuable intermediates in synthesis of N-substituted compounds of formula I.
- they can be alkylated with RX, wherein X is CI or Br, in a solvent in the presence of a base.
- Suitable alkylating agents include cyclohexylmethyl bromide, dimethylallyl, chloride, 2-thienylmethyl chloride, p-methoxyphenylethyl bromide and cyclopropylmethyl chloride; these give, respectively, the corresponding N-cyclohexylmethyl, dimethylallylic, thienylmethyl and phenylethyl compounds.
- N-butyl- or N-dimethylallylcodeine Starting with N-butyl- or N-dimethylallylcodeine and using the procedures of Examples 1 and 2, the corresponding N-butyl and N-dimethylallyl substituted compounds are obtained. Similarly, N-(2-furanylmethyl), N-(2-thienylmethyl) and N-(2-thienylethyl) substituted compounds corresponding to those of Example 5 can be prepared and used as described above.
- compositions of this invention include those made with physiologically acceptable acids that are known in the art; such salts include hydrochloride, sulfate, phosphate, nitrate, citrate, and maleate.
- the process of this invention provides compounds which are useful intermediates in the synthesis of the N-substituted compounds, of this invention, which are active pharmaceutically as analgesics and/or narcotic antagonists.
- the compounds of this invention can be administered as analgesic agents to alleviate pain or as narcotic antagonists by any means that produces contact of the active agent with the agent's site of action in the body of a mammal. They can be administered either as individual therapeutic agents or in a combination of therapeutic agents, generally with a pharmaceutical carrier.
- the dosage depends upon: the pharmacodyrramic characteristics of the particular agent, and its mode and route of administration; the age, health, and weight of the recipient; the nature and extent of the symptoms; the kind of concurrent treatment; the frequency of treatment; and the effect desired.
- a daily dosage of active ingredient can be about 0.05 to 100 milligrams per kilogram of body weight in divided doses 2 to 4 times a day or in sustained release form.
- Dosage forms (compositions) suitable for internal administration can contain about 25 milligrams to about 75 milligrams of active ingredient per dosage unit.
- the active ingredient will ordinarily be present in an amount of about 0.5-95% by weight based on the total weight of the composition.
- the active ingredient can be administered orally in solid dosage forms such as capsules, tablets, and powders, or in liquid dosage forms such as elixirs, syrups and suspensions. It can also be administered parenterally in sterile liquid dosage forms or rectally in the form of suppositories.
- Capsules or tablets contain the active ingredient and powdered carriers, such as lactose, sucrose, mannitol, starch, cellulose derivatives, magnesium stearate and stearic acid; they can be manufactured as sustained release products.
- powdered carriers such as lactose, sucrose, mannitol, starch, cellulose derivatives, magnesium stearate and stearic acid; they can be manufactured as sustained release products.
- suitable carriers for parenteral solutions include water, a suitable oil, saline, aqueous dextrose (glucose), and related sugar solutions, and glycols such as propylene glycol or polyethylene glycols.
- Solutions for parenteral administration contain, preferably: a water-soluble salt-of the active ingredient, for example, the hydrochloride; suitable stabilizing agents; buffer substances; anti-oxidizing agents such as ascorbic acid; and preservatives such as benzalkonium chloride, methyl- or propylparaben and chlorobutanol.
- Suppositories contain the active ingredient in a suitable oleaginous or water-soluble base.
- the oleaginous class includes cocoa butter and fats having similar properties; the water-soluble class includes polyethylene glycols.
- Suitable pharmaceutical carriers are described in Remington's Pharmaceutical Sciences, E. W.
- Useful pharmaceutical dosage-forms for administration of the compounds of this invention can be illustrated by the following:
- a large number of unit capsules are prepared by filling standard two-piece, hard gelatin capsules, each with 50 milligrams of powdered active ingredients, 110 milligrams of lactose, 32 milligrams of talc and 8 milligrams of magnesium stearate.
- a mixture of active ingredient in soybean oil is prepared and injected by means of a positive displacement pump into gelatin to form soft gelatin capsules containing 50 milligrams of the active ingredient.
- the capsules are washed in petroleum ether and dried.
- a large number of tablets are prepared by conventional procedures so that the dosage unit is 50 milligrams of active ingredient, 7 milligrams of ethyl cellulose, 0.2 milligram of colloidal silicon dioxide, 7 milligrams of magnesium stearate, 11 milligrams of microcrystalline cellulose, 11 milligrams of comstarch and 98.8 milligrams of lactose.
- Appropriate coatings may be applied to increase palatability or delay absorption.
- a parenteral composition suitable for administration by injection is prepared by stirring 1.5% by weight of active ingredient in 10% by volume of propylene glycol and water. The solution is sterilized by filtration.
- composition suitable for administration by injection is prepared by dissolving 1% by weight of active ingredient in sodium chloride injection U.S.P. XV and adjusting the pH of the solution to between 6 and 7. The solution is sterilized by filtration.
- An aqueous suspension is prepared for oral administration so that each 5 milliliters contain 10 milligrams of finely divided active ingredient, 500 milligrams of acacia, 5 milligrams of sodium benzoate, 1.0 gram of sorbitol solution, U.S.P., 5 milligrams of sodium saccharin and 0.025 millilitre of vanilla tincture.
- a standard procedure for detecting and comparing the analgesic activity of compounds in this series for which there is good correlation with human efficacy is the standard phenylquinone writhing test modified from Siegmund, et. al., Proc. Soc. Exp. Biol. Med 95, 729 (1957).
- a test compound suspended in 1% methylcellulose was given orally to fasted (17-21 hours) female white mice, 5 ⁇ 20 animals per double blind test.
- Aqueous (0.01% phenyl-p-benzoquinone) phenylquinone was injected intraperitoneally at 24 minutes later using 0.20 ml per mouse.
- mice were observed for 10 minutes for a characteristic stretching or writhing syndrome which is indicative of pain induced by phenylquinone.
- the effective analgesic dose for 50% of the mice was calculated by the moving average method of Thompson, U.R., Bact. Rev. 11 115-145 (1947). This is reported as PQW ED 50 in the table below.
- Some of the compounds of this invention are particularly useful as analgesics by subcutaneous injection; for example, the compound (29) of Example 11 had an ED so of 0.0016 fifteen minutes after injection.
- Narcotic analgesics produce in mice an erection and arching of the tail (90° or more) which is referable to spinal cord stimulation. This Straub tail reaction is not produced by other analgesics, including the narcotic antagonists.
- the method used was modified from Shemano (Shemano, I., and Wendel, H., Tox. Appl. Pharm. 6, 334-9 (1964)).
- CF,S female mice (18-21 g), 10-20 mice per dose were injected subcutaneously with log scaled doses of analgesic in 0.9% saline or 0.45% saline containing 0.5% methylcellulose.
- a positive Straub tail response was recorded if a tail was erected for 3 seconds at any time within 25 minutes after dosing.
- a quantal Straub tail ED 50 was calculated by the moving average method and is an indication of physical dependence.
- the narcotic antagonist (anti-Straub tail) property of the compounds is estimated by their ability to prevent morphine-induced Straub tail in mice. In this test the compound is injected intraperitoneally into mice and 10 minutes later 53 mg/kg of morphine sulfate is given subcutaneously. Prevention of the induction of a 90° Straub tail for minutes after the morphine sulfate injection is considered to indicate narcotic antagonism in the compound tested.
- the following table shows the activity in mg/kg exhibited by various compounds including those of this invention (in the hydrochloride salt form to provide solubility), when tested by the procedures given above. A value of 135 mg/kg or higher is considered to indicate an inactive compound.
- the Straub Tail Antagonism test was modified by giving test drugs (in 1% methyl cellulose with .1.25% "Tween 80") subcutaneously, followed by intraperitoneal injection of morpholine sulfate (at 35 mg/kg) 5 minutes after the test drug. After 15 minutes, the animals were observed for 5 minutes for evidence of Straub tail.
- ED 50's were calculated as above with a 95%confidence limit as described by Litchfield, J. Pharmacol. Exp. Ther. 96, 99 (1949). When compound (17) of Example 6 was tested, the ED SO was 0.0031 mg/kg; the value for Pentazocine was 1.9 mg/kg.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
- This invention relates to a fluorination process for production of compounds which are useful as intermediates in syntheses of novel fluorine analogs of hydrocodone and oxycodone, the fluorine analogs exhibiting analgesic and/or narcotic antagonist properties in mammals.
- Morphine and codeine analgesics exhibit toxic properties or have addictive action. Considerable effort has been made to find derivatives that are free from these qualities and still have analgesic effects. Compounds which are narcotic antagonists are also useful in medicine, such as in the treatment of addicts. Fluorine derivatives of codein, i.e. 6-deoxy-6-fluorcodeines and -morphines and their dihydro derivatives have been reported by Ayer in US-A-3 137 701. They are prepared by the reaction of a 6-hydroxyalkaloid having the codeine ring structure with a fluorinating agent such as N-(2-chloro-1,1,2-trifluoroethyl)diethylamine. One such compound obtained by Ayer, and confirmed by Bognar et al., Acta Chimica Academiae Scientiarum Hungaricae 67, 63-69 (1971), is of the formula

- The 6-deoxy-6-fluoromorphines and -codeines are said to be "active analgesics and central nervous system stimulants like the parent alkaloids, but in addition they possess pharmacologic. advantages. Illustratively, 6-deoxy-6-fluorocodeine is a more active analgesic than codein when administered orally, and side effects are reduced. Similarly, 6-deoxy-6-fluoromorphine is an analgesic as active as morphine, but it elicits only comparatively mild side effects." No actual biological data are disclosed.
- The compounds with the 7,8-double bond are allylic fluorides. Only N-methyl derivatives are contemplated in US-A-3 137 701.
- Contrary to the fact that the disclosed compounds are allylic fluorides, the compounds of the instant invention are vinylic fluorides and in addition encompass variations of the nitrogen substituent, providing compounds with mixed agonist-antagonist profiles (see below, page 3, line 30-32 whereat cyclopropylmethyl and cyclobutylmethyl derivatives are disclosed). It should be noted from the last entry in the table on page 32 of the instant application that the compound of US-A 3 137 701 shown on page 1 of the instant description is relatively inactive as an analgesic (the PQW column) and has no antagonist component (the anti-straub tail column, a measure of the narcotic tendency of the compound), in marked contrast to most of the compounds of the present invention.
- Diethylaminosulfur trifluoride (DAST) is a convenient reagent for replacing the carbonyl oxygen of aldehydes and ketones with two fluorine atoms. W. J. Middleton, J. Org. Chem., 40, 574 (1975), "New · Fluorinating Reagents, Dialkylaminosulfur Fluorides" and L. N. Markovskij, V. E. Pashinnik and A. V. Kirsanov, Synthesis, 787 (1973). Middleton discloses CClaF, diglyme, methylene dichloride and benzene as reaction solvents and temperatures of 25-85°C. Markovskij et al. disclose that the carbonyl compound and dialkylaminosulfur trifluoride are simply mixed in equal molar amounts and heated.
- One aspect of the invention resides in compounds of the formula

- R is hydrogen, alkyl of 1-10 carbon atoms, allyl, methylallyl, dimethylallyl, cycloalkylmethyl of 4-7 carbon atoms, 2-furanylmethyl, 2-tetrahydrofuranylmethyl, 2-thienylmethyl, 2-thienylethyl or phenylethyl which may be ring substituted with chloro, bromo, fluoro, methoxy or 1-3 carbon atom alkyl substituents;
- Y is H or R1;
- R1 is alkyl of 1--4 carbon atoms or alkanoyl of 1-12 carbon atoms;
- R2 is hydrogen, hydroxy or alkanoyloxy of 1-4 carbon atoms;
- n is 1 or 2; and
- a is a 6,7 double bond when n is 1 and a single bond when n is 2;
- Particularly preferred are compounds of formula I wherein R is cyclopropylmethyl or cyclobutylmethyl. Those compounds wherein Y is H or acetyl are generally more active, as are those wherein R2 is H. For reasons of availability and pharmacological activity, generally, n is 1 and, therefore, a is a double bond. The compounds of Examples 5-7, 11 and 12 illustrate the best mode of the invention of which Examples 5 and 11 are preferred.
- Compounds of this invention can be prepared by reacting a disubstituted aminosulfur trifluoride with a mineral acid (MA), for example, HCl or H2SO4 salt of a 6-keto compound corresponding to that of formula I wherein Y is R1, R2 is H or alkanoyloxy of 1―4 carbon atoms, a is a single bond and (F)n is replaced by carbonyl oxygen (=0). In such process the ketone of the formula

- R is hydrogen, alkyl of 1-10 carbon atoms, allyl, methylallyl, dimethylallyl, cycloalkylmethyl of 4-7 carbon atoms, 2-furanylmethyl, 2-tetrahydrofuranylmethyl, 2-thienylmethyl, 2-thienylethyl or phenylethyl which may be ring substituted with chloro, bromo, fluoro, methoxy or 1-3 carbon atom alkyl substituents;
- R1 is alkyl of 1-4 carbon atoms or alkanoyl of 1-12 carbon atoms;
- RZ' is hydrogen or alkanoyloxy of 1-4 carbon atoms; and
- MA is a mineral acid.
is reacted with a disubstituted aminosulfur trifluoride fluorinating agent of the formula R4R5NSF wherein each R4 and R5, alike or different, is a primary alkyl group of 1-4 carbon atoms; or R4 and R when taken together are ―(CH2)4―, ―(CH2)5― or ―CH2CH2OCH2CH2―; at a temperature of―40°C to +80°C; in the presence of a polar or nonpolar solvent; and recovering the fluorine-containing compound of the formula
- R, R1 and R2' are as defined above;
- n is 1 or 2; and
- a is a 6,7 double bond when n is 1 and a single bond when n is 2.
- When R in formula I is H (obtained as shown in Example 13), the corresponding 6-fluoride is converted to a pharmaceutically superior compound by alkylation of the HN group with RX, usually under basic conditions, wherein R is as previously defined except that it is not H and X is generally halogen (Cl, Br or I), for example, cyclopropylmethyl bromide.
- In the compounds obtainable by the procedures herein, the substituent in the 3-position is generally the 3-methoxy substituent. Treatment of such compunds with a demethylating agent, such as a hydrogen halide, gives the 3-hydroxy compound from which can be prepared the corresponding alkyl ether wherein the alkyl group contains 1-4 carbon atoms, for example, the propoxy or butoxy derivative, or the corresponding ester of a C1―C12 alkanoic acid, for example, the formate, butyrate or dodecanoate, using general synthetic methods. The esters can exhibit greater potency as pharmaceuticals.
- The more active compounds are preferably prepared by reacting under substantially anhydrous conditions the ketone of the formula

- R3 is alkyl of 1-9 carbon atoms, vinyl, 1-propenyl, isobutenyl, cycloalkyl of 3-6 carbon atoms, 2-furanyl, 2-tetrahydrofuranyl, 2-thienyl, 2-thienylmethyl or phenylmethyl which may be ring substituted with chloro, bromo, fluoro, methoxy or 1-3 carbon atom alkyl substituents;
- R' is as defined above;
- R2' is hydrogen or alkanoyloxy of 1-4 carbon atoms; and
- MA represents a mineral acid,
with a disubstituted aminosulfur trifluoride of the formula R4R5NSF3 wherein each of R4 and R5, alike or different, is a primary alkyl group of 1-4 carbon atoms, or R4 and R5 taken together are ―(CH2)4―, ―(CH2)5― or―CH2CH2OCH2CH2―, at a temperature of ―40°C to +80°C, in the presence of a polar or nonpolar solvent, and recovering the fluorine-containing compound of formula described below. - When the amide starting material of formula II is used, the aforesaid fluorination step produces an intermediate compound of the formula


- This intermediate is subjected to reduction whereupon the


- The reduction can be accomplished using conventional chemical techniques, for example, by subjecting the intermediate of formula IV to reaction with LiAIH4, as exemplified in the examples below. Other reducing agents which can be used include other alkali metal hydrides, such as sodium or potassium borohydride.
- When the compound of formula III is used as the starting material in the aforesaid reaction, the product will be of the formula


- Regarding formula I, when R2 is to be hydroxyl in the final product, the compound subjected to fluorination should have R2 as the ester (or equivalent protective group) which is subsequently converted to the hydroxyl. When R2 is OH in the starting compound, the use of excess fluorinating agent, disubstituted aminosulfur trifluoride, results in the formation of a product wherein R2 is F. When the starting ketone is the mineral acid salt, for example, the hydrogen sulfate of formula III, a good yield of the desired fluorinated compound is obtained. For better yields it is preferred to use the starting ketone which is the amide of formula II. It is more preferred to use such amide wherein R3 has 3-8 carbon atoms..
- The disubstituted aminosulfur trifluorides are known compounds. Particularly useful are diethylaminosulfur trifluoride (bAST), pyrrolidinosulfur trifluoride, morpholinosulfur trifluoride and piperidylsulfur trifluoride.
- The reaction is normally carried out ina solvent medium, preferably catalytically using a highly polar catalyst. The solvent can be polar or nonpolar but it must be nonreactive with the disubstituted aminosulfur trifluoride. The use of a polar solvent tends to give more of the 6-fluoro-6,7-unsaturated codeine (n is 1) whereas the use of a nonpolar solvent generally gives more of the 6,6-difluoro compound (n is 2). By polar solvent is meant a solvent that has a high dielectric constant. Solvents which favor conversion of ketones to vinylene fluorides

- The addition of a small amount of a strong acid, such as fuming sulfuric acid, as a highly polar catalyst increases the rate of formation of the 6,7-unsaturated-6-fluoride. Other useful catalysts include strong mineral acids that, in the quantities used, do not react with the carbonyl group, with the carbon- carbon double bond or with other groups present in the codeine-type compounds, for example, perchloric, polyphosphoric and fluorosulfonic acids. The useful acids generally have a log Ka of greater than -2. The amount of catalyst used is generally 0.001 to 1% by weight of the ketone starting material. The catalyst is believed to function by increasing the polarity of the reaction medium and by increasing the rate of the reaction.
- The reaction is conducted, under substantially anhydrous conditions, in a vessel which is suitably glass, but a metal or ceramic vessel can be used. The reaction is conducted at -40°C to +80°C with the range 0-30°C being generally preferred. The reaction time is dependent upon the reactants and the temperature, with times of less than one hour to one week or more being useful. Pressure is not critical but ambient or autogeneous pressure is preferred.
- The desired fluoro compound can be separated from the reaction mixture by conventional procedures. Preparative chromatography is a particularly useful procedure for separation and purification but other means, for example, crystaliation or extraction can be used.
- All temperatures reported herein are in °C.
-

- A solution of 40 ml of diethylaminosulfur trifluoride (DAST) in 160 ml of glyme was added dropwise to a suspension of 8.0 g of hydrocodone hydrochloride and 1.6 ml of fuming sulfuric acid in 240 ml of glyme cooled to -78°. The reaction mixture was warmed to room temperature and stirred for 8 days. The mixture was poured over ice, made basic with sodium bicarbonate and extracted with methylene chloride. Evaporation of the methylene chloride extracts gave a viscous oil which was taken in ether and filtered. Concentration of the ether filtrate gave 4.87 g of a white solid which was chromatographed on silica. Elution with 1:1 hexane:acetone containing 1% of diethylamine gave 0.58 g of white solid 17-methyl-6,7-didehydro-4,5-epoxy-6-fluoro-3-methoxymorphinan melting at 167-169° (HCI salt 214-217°) and 2.09 g of white solid 17-methyl-6,7-dihydro-4-hydroxy-5-chloro-6-fluoro-3-methoxymorphinan melting at 169-173° (HCI salt% 178-182°). The infrared spectrum showed =CF absorption at 5.91 µ with no indication of C=O. 19F nmr (CDCl3) for (3) δ -116.6 ppm; for (2) δ -116.2 ppm.


-

- A mixture of 10.0 g (0.029 mole) of hydrocodone hydrochloride in 200 ml of CCl3F (Freon®11) was cooled to -78°. To this was added dropwise a solution of 50 ml of diethylaminosulfur trifluoride (DAST) in 50 ml of CCl3F. The mixture was warmed to room temperature, stirred for 5 days, then poured over crushed ice. The solution was made basic with sodium bicarbonate and the layers were separated. The aqueous layer was extracted with methylene chloride and the combined organic extracts were washed with water, then brine and dried (MgS04). This gave 8.65 g of crude product as a viscous oil. A 1.6 g sample was fractionated by preparative chromatography to give 800 mg of the pure difluoro derivative 17-methyl-4,5-epoxy-6,6-difluoro-3-methoxymorphinan. A sample converted to the hydrochloride salt melted at 270―275°. 19F nmr (CDCl3) δ -91.6, -94.2, -104.1 and -106.7 ppm.
- The reaction mixture also contained minor amounts of the monofluoro-α,β-unsaturated compound (5) or (2).
-

- A mixture of 800 mg of the methoxy derivative 2 of Example 1 and 2.4 g of pyridine hydrochloride was heated at 190° for 3 hours, cooled and diluted with water. The aqueous solution was made basic with sodium hydroxide and extracted with methylene chloride. The methylene chloride extracts were dried (MgS04) and concentrated. The resulting oil was taken in ether and converted to the hydrochloride salt to give 200 mg of the 0-demethylated derivative (6) as a white crystalline solid. High performance liquid chromatography (HPLC) showed one major component (95%).

-
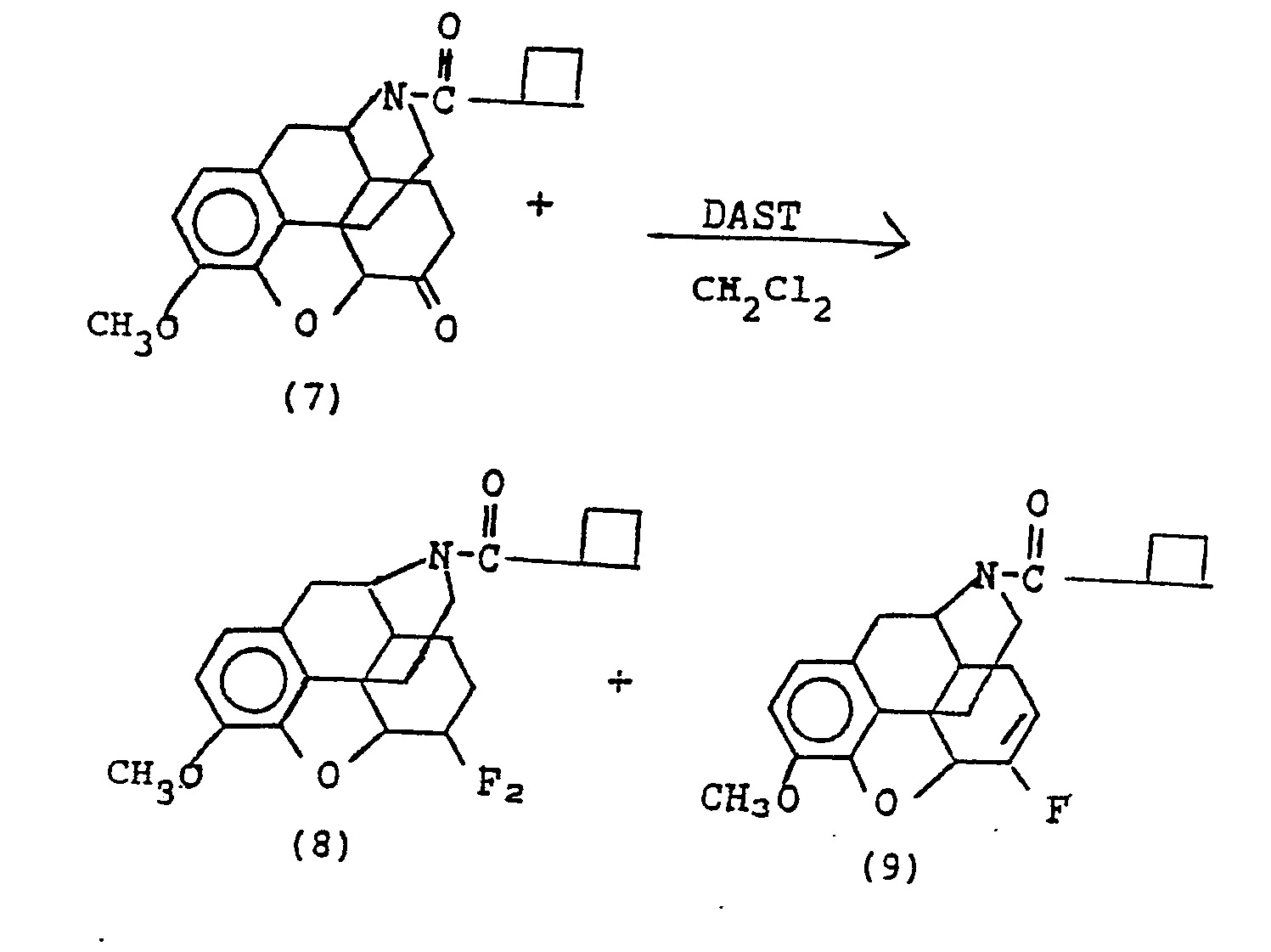
- A solution of 25.0 g (68 mmoles) of the keto amide (7), prepared by reaction of cyclobutyl carbonyl chloride with the amine (11) of Example 5, in 200 ml of dry methylene chloride was cooled to -78°. To this was added dropwise a solution of 25 ml (200 mmoles) of DAST in 100 ml of dry methylene chloride. The mixture was stirred at 25° for 6 days, poured onto crushed ice and neutralized, separated, washed with water, then brine and dried over MgS04. The resulting oil was collected with ether to give 14.32 g of white solid mixture of (8) and (9), mostly (8). Concentration of the ether filtrate gave an additional 10.0 g of solid. 19F nmr (CDCl3): δ -91.6, -94.2, -104.3 and -106.9 ppm (―CF2) and -116.4 ppm (=CF).
-

- To a stirred suspension of 4.5 g of LiAIH4 in 200 ml of anhydrous tetrahydrofuran was added dropwise a mixture of 12.2 g of the amides (8) and (9) from part I in 100 ml of anhydrous tetrahydrofuran. The mixture was heated at reflux for 24 hours, cooled and then hydrolyzed using: 4.5 ml of water, 4.5 ml of 15% aqueous sodium hydroxide and 13.5 ml of water. This gave 6.1 g of crude amines. A 2.2 g sample was chromatographed on silica gel. Elution with methylene chloride containing 2% methanol and 1% diethylamine gave 1.5 g of 90% pure difluoride as a viscous oil. This was dissolved in ether and converted to the hydrochoride salt ( 10).19F nmr (CDDl3) δ -91.7, -94.3, -104.2 and -106.8 ppm (S). Infrared spectrum showed


-

- The N-cyclopropylcarbonylcodone (12) was obtained by reacting 18 g of cyclopropane carbonyl chloride with 30.7 g of the amine (11) at room temperature in methylene chloride in the presence of 20 g of tetraethylamine; yield 33 g as a white solid; identical with that of Gates, J.Med.Chem. 7, 127 (1964).
- When 18.5 g of the amide (12) was dissolved in 150 ml of methylene chloride and cooled and 18 ml of DAST was added, after 3 days at room temperature 19.2 g of brown oil resulted. This was dissolved in ether and filtered and the oil was chromatographed twice on silica using 90% ether- cyclohexanone with 1% methylene chloride and 0.001 % water. There was obtained 4.2 g of 87% pure difluoride (13) and 6.8 g of about a 50:50 mixture of difluoride (13) and monofluoro compound (14).
- The amide (13) (7 g) was dissolved in 100 ml of tetrahydrofuran and mixed with 3 g of LiAIH4 in 100 ml of tetrahydrofuran. After refluxing for 16 hours, 3 ml of water was added followed by 3 ml of 15% aqueous NaOH and 9 ml of water to give 6.13 g of product which was chromatographed using 1:1 hexane:acetone containing 1% triethylamine to give 3.6 g of (15) in 96% purity by 19F nmr or 91% by HPLC as a white solid m.p. 69-73°. The hydrochloride of the latter softened at 100° and had a mp of 123-133°. HMRS Calcd for C2,H25NO2F2: 361.1852; Found: 361.1846.
- Ten g of codone amide (12) was dissolved in 120 ml of glyme and 0.8 ml of sulfuric acid and 10 ml of DAST in 80 ml of glyme were added at -78°. After stirring at room temperature for 6 days the mixture was poured on ice, neutralized and extracted with methylene chloride. The extracts were evaporated and the residue was dissolved in ether to give 10.2 g of the 6-fluoromorphinan (14) which was chromatographed on silica using 90% ether-hexane containing 1% methylene chloride and 0.1% water. About 2.3 g of this product was dissolved in 50 ml of tetrahydrofuran and reduced with 1.1 g of LiAIH4 in 50 ml of tetrahydrofuran by refluxing for 16 hours. There was obtained 1.4 g of white product (16) as the HCI salt, mp 93-97°.
-

- The O-methyl ether (15) of Example 5 (2.8 g) was heated at 190-195° for 3 hours with 7 g of pyridine hydrochloride, diluted with water and extracted with methylene chloride; the extracted material was dissolved in toluene, filtered, concentrated, dissolved in ether and crystallized from ether-hexane by adding hexane; the resultant white powder (1 g) mp 78-84° was converted to the HCI salt (17) and purified by HPLC (96% pure), mp 312―317°. HRMS Calcd for C20H23NO2F2: 347.1695; Found: 347.1708.
-

- The 0-methyl ether (16) of Example 5 (2.9 g) was reacted with 7 g of pyridine hydrochloride as in Example 6; there was obtained a tan solid which was taken up in ether and converted to the hydrochloride salt. The salt was recrystallized from ethanol to give 0.6 g of white solid (18) mp 285-290°. HPLC showed one major peak (94.9%).

-

- To 10 g of amine (11) of Example 5 in 150 ml of dimethylformamide was added 4 g of allyl bromide and 2.8 g of NaHC03. The mixture was heated at 80-85° for 5 hours, diluted with water, extracted with ether and converted to 12.7 g of the amine salt (19). The salt was reacted with diethylaminosulfur trifluoride according to the procedure of Example 2 and the hydrochloride salts (20) and (21) thus formed were separated by chromatography as described in Example 5. 19F nmr (CDCl3):δ -19.6, -94.2, -104.1 and 106.7 ppm.
-

- A solution of 23.0 g of the morphinan (22) in 100 ml of acetic anhydride was heated at reflux for 0.5 hour cooled and then concentrated. The residue was dissolved in 200 ml of water, cooled to 0° and adjusted to pH 9 with ammonium hydroxide. A viscous oil precipitated. The water layer was decanted and the oil was dissolved in methylene chloride and dried over Na2S04. Concentration of the methylene chloride gave 22.3 g of viscous oil. The infrared spectrum was substantially free of OH- absorption.
-

- A solution of 18.0 g of the ketoamide (23) in 150 ml of dry methylene chloride was cooled to -780. A solution of 18 ml of DAST in 150 ml of methylene chloride was added dropwise. The reaction mixture was stirred at room temperature for three days, poured over ice and neutralized with sodium bicarbonate. The organic layer was separated, washed with brine and dried (K2C03). This gave 16.6 g of the crude fluorides as a viscous oil.

- The above fluoro derivatives (24) and (25) were separated using HPLC. Elution with hexane- acetone-triethylamine gave 8 g of the difluoride (24) as a colorless viscous oil. 19F nmr (CDCl3) δ -91.3, -93.9 and 102.3, 104.4 ppm (―CF2).
-

- An 8 g sample of the difluoro amide (24) was reduced as in Example 4, II. This gave 4.5 g of tan solid which was dissolved in ether; some insoluble solid was filtered off. Concentration of the ether- soluble material gave 2.6 g of pale yellow solid which was taken in ether and decolorized by use of charcoal to give a white solid melting at 162-164°. When converted to the hydrochloride salt (in ether) the difluoromorphinan (26) melted at 266-269°. Its infrared spectrum showed hydroxyl absorption at 3400 cm-1 but no amide or carbonyl. absorption.
-

- Using the procedures described hereinabove, starting with (27) and acetylating, fluorinating with DAST, recovering the difluoride and hydrolyzing and reducing, as in Example 9, gave the hydrochloride salt (28) which melted at 248-252°.

-

- A solution of 3.0 g of the crude phenol (18) of Example 7 in 25 ml of acetic anhydride was heated at reflux for 0.5 hour and then concentrated. The residue was treated with ice water and made basic with ammonia. The aqueous base was extracted with methylene chloride, treated with decolorizing charcoal and dried (K2CO3). Removal of the solvent gave 3.6 g of tan oil which was recrystallized from hexane-ether to give 0.84 g of the acetoxy derivative (29) as a white solid melting at 86-88°. The infrared spectrum showed strong carbonyl absorption at 1730 cm-1.

- A sample of the base was dissolved in ether and converted to the hydrochloride salt which melted at 139° (dec.).
-

- The procedure of Example 11 was used to prepare the acetoxy derivative of the corresponding difluoride. The difluoride (30) was obtained as a white solid melting at 128-130°.

-

- A mixture of 13.5 g of the nordihydrocodeinone hydrochloride (31) in 250 ml of methylene chloride was treated with 13 ml of DAST using the procedures described hereinabove. After work-up the residue was taken in ether and filtered. This gave 6.7 g of a mixture of the fluoro derivatives (32) and (33) as a tan solid. These compounds were separated by chromatography as described in Example 1. 19F nmr -93.13, -105.57 ppm (difluoride) and -116.12 ppm (vinyl).

- The fluoronorcodeinones (32) and (33) of this example are valuable intermediates in synthesis of N-substituted compounds of formula I. For example, they can be alkylated with RX, wherein X is CI or Br, in a solvent in the presence of a base. Suitable alkylating agents include cyclohexylmethyl bromide, dimethylallyl, chloride, 2-thienylmethyl chloride, p-methoxyphenylethyl bromide and cyclopropylmethyl chloride; these give, respectively, the corresponding N-cyclohexylmethyl, dimethylallylic, thienylmethyl and phenylethyl compounds.
- When the general procedures of Examples 4 and 5 are carried out using compounds corresponding to (7) and (12) but where R3 is CH2C6H5, C6H11 or (CH2)9H, there are obtained the corresponding compounds N-phenylethyl-6,7-didehydro-4,5a-epoxy-6-fluoro-3-methoxymorphinan, N-phenylethyl-4,5a-epoxy-6,6-difluoro-3-methoxymorphinan and the N-cyclohexylmethyl- or N-n-decylmorphinan.
- Starting with N-butyl- or N-dimethylallylcodeine and using the procedures of Examples 1 and 2, the corresponding N-butyl and N-dimethylallyl substituted compounds are obtained. Similarly, N-(2-furanylmethyl), N-(2-thienylmethyl) and N-(2-thienylethyl) substituted compounds corresponding to those of Example 5 can be prepared and used as described above.
- When the N-cyclobutanoylnoroxycodone-14-acetate is used in the general procedures of Example 9, the 6,6-difluoro- and 6-fluoro-Δ6,7-cyclobutylmethylnoroxycodone-14-acetate are obtained. Hydrolysis of the 14-acetate compounds gives the 14-hydroxy compounds. The corresponding 14-butanoyl, 14-propionyl and 14-formyl compounds can be prepared by suitable acylation of the 14-hydroxy compounds with appropriate alkanoyl halides.
- Pharmaceutically suitable acid addition salts of the compounds of this invention include those made with physiologically acceptable acids that are known in the art; such salts include hydrochloride, sulfate, phosphate, nitrate, citrate, and maleate.
- The process of this invention provides compounds which are useful intermediates in the synthesis of the N-substituted compounds, of this invention, which are active pharmaceutically as analgesics and/or narcotic antagonists.
- The compounds of this invention can be administered as analgesic agents to alleviate pain or as narcotic antagonists by any means that produces contact of the active agent with the agent's site of action in the body of a mammal. They can be administered either as individual therapeutic agents or in a combination of therapeutic agents, generally with a pharmaceutical carrier. The dosage depends upon: the pharmacodyrramic characteristics of the particular agent, and its mode and route of administration; the age, health, and weight of the recipient; the nature and extent of the symptoms; the kind of concurrent treatment; the frequency of treatment; and the effect desired. Usually a daily dosage of active ingredient can be about 0.05 to 100 milligrams per kilogram of body weight in divided doses 2 to 4 times a day or in sustained release form.
- Dosage forms (compositions) suitable for internal administration can contain about 25 milligrams to about 75 milligrams of active ingredient per dosage unit. In the pharmaceutical compositions herein the active ingredient will ordinarily be present in an amount of about 0.5-95% by weight based on the total weight of the composition.
- The active ingredient can be administered orally in solid dosage forms such as capsules, tablets, and powders, or in liquid dosage forms such as elixirs, syrups and suspensions. It can also be administered parenterally in sterile liquid dosage forms or rectally in the form of suppositories.
- Capsules or tablets contain the active ingredient and powdered carriers, such as lactose, sucrose, mannitol, starch, cellulose derivatives, magnesium stearate and stearic acid; they can be manufactured as sustained release products.
- In general, suitable carriers for parenteral solutions include water, a suitable oil, saline, aqueous dextrose (glucose), and related sugar solutions, and glycols such as propylene glycol or polyethylene glycols. Solutions for parenteral administration contain, preferably: a water-soluble salt-of the active ingredient, for example, the hydrochloride; suitable stabilizing agents; buffer substances; anti-oxidizing agents such as ascorbic acid; and preservatives such as benzalkonium chloride, methyl- or propylparaben and chlorobutanol.
- Suppositories contain the active ingredient in a suitable oleaginous or water-soluble base. The oleaginous class includes cocoa butter and fats having similar properties; the water-soluble class includes polyethylene glycols.
- Suitable pharmaceutical carriers are described in Remington's Pharmaceutical Sciences, E. W.
- Useful pharmaceutical dosage-forms for administration of the compounds of this invention can be illustrated by the following:
- A large number of unit capsules are prepared by filling standard two-piece, hard gelatin capsules, each with 50 milligrams of powdered active ingredients, 110 milligrams of lactose, 32 milligrams of talc and 8 milligrams of magnesium stearate.
- A mixture of active ingredient in soybean oil is prepared and injected by means of a positive displacement pump into gelatin to form soft gelatin capsules containing 50 milligrams of the active ingredient. The capsules are washed in petroleum ether and dried.
- A large number of tablets are prepared by conventional procedures so that the dosage unit is 50 milligrams of active ingredient, 7 milligrams of ethyl cellulose, 0.2 milligram of colloidal silicon dioxide, 7 milligrams of magnesium stearate, 11 milligrams of microcrystalline cellulose, 11 milligrams of comstarch and 98.8 milligrams of lactose. Appropriate coatings may be applied to increase palatability or delay absorption.
- A parenteral composition suitable for administration by injection is prepared by stirring 1.5% by weight of active ingredient in 10% by volume of propylene glycol and water. The solution is sterilized by filtration.
- Another parenteral composition suitable for administration by injection is prepared by dissolving 1% by weight of active ingredient in sodium chloride injection U.S.P. XV and adjusting the pH of the solution to between 6 and 7. The solution is sterilized by filtration.
- An aqueous suspension is prepared for oral administration so that each 5 milliliters contain 10 milligrams of finely divided active ingredient, 500 milligrams of acacia, 5 milligrams of sodium benzoate, 1.0 gram of sorbitol solution, U.S.P., 5 milligrams of sodium saccharin and 0.025 millilitre of vanilla tincture.
- A standard procedure for detecting and comparing the analgesic activity of compounds in this series for which there is good correlation with human efficacy is the standard phenylquinone writhing test modified from Siegmund, et. al., Proc. Soc. Exp. Biol. Med 95, 729 (1957). A test compound suspended in 1% methylcellulose was given orally to fasted (17-21 hours) female white mice, 5―20 animals per double blind test. Aqueous (0.01% phenyl-p-benzoquinone) phenylquinone was injected intraperitoneally at 24 minutes later using 0.20 ml per mouse. Commencing at 30 minutes after the oral administration of the test compound, the mice were observed for 10 minutes for a characteristic stretching or writhing syndrome which is indicative of pain induced by phenylquinone. The effective analgesic dose for 50% of the mice (ED 50) was calculated by the moving average method of Thompson, U.R., Bact. Rev. 11 115-145 (1947). This is reported as PQW ED50 in the table below.
- Some of the compounds of this invention are particularly useful as analgesics by subcutaneous injection; for example, the compound (29) of Example 11 had an EDso of 0.0016 fifteen minutes after injection.
- Narcotic analgesics produce in mice an erection and arching of the tail (90° or more) which is referable to spinal cord stimulation. This Straub tail reaction is not produced by other analgesics, including the narcotic antagonists.
- The method used was modified from Shemano (Shemano, I., and Wendel, H., Tox. Appl. Pharm. 6, 334-9 (1964)). CF,S female mice (18-21 g), 10-20 mice per dose were injected subcutaneously with log scaled doses of analgesic in 0.9% saline or 0.45% saline containing 0.5% methylcellulose. A positive Straub tail response was recorded if a tail was erected for 3 seconds at any time within 25 minutes after dosing. A quantal Straub tail ED50 was calculated by the moving average method and is an indication of physical dependence.
- . The narcotic antagonist (anti-Straub tail) property of the compounds is estimated by their ability to prevent morphine-induced Straub tail in mice. In this test the compound is injected intraperitoneally into mice and 10 minutes later 53 mg/kg of morphine sulfate is given subcutaneously. Prevention of the induction of a 90° Straub tail for minutes after the morphine sulfate injection is considered to indicate narcotic antagonism in the compound tested.
- The following table shows the activity in mg/kg exhibited by various compounds including those of this invention (in the hydrochloride salt form to provide solubility), when tested by the procedures given above. A value of 135 mg/kg or higher is considered to indicate an inactive compound.

- The Straub Tail Antagonism test was modified by giving test drugs (in 1% methyl cellulose with .1.25% "Tween 80") subcutaneously, followed by intraperitoneal injection of morpholine sulfate (at 35 mg/kg) 5 minutes after the test drug. After 15 minutes, the animals were observed for 5 minutes for evidence of Straub tail. ED 50's were calculated as above with a 95%confidence limit as described by Litchfield, J. Pharmacol. Exp. Ther. 96, 99 (1949). When compound (17) of Example 6 was tested, the EDSO was 0.0031 mg/kg; the value for Pentazocine was 1.9 mg/kg.
and pharmaceutically acceptable salts of the compounds. Formula I and all formulas shown hereinafter are depicted by planar representations. One skilled in the art will readily understand the nonplanar aspects of these representations, for example, that the carbon-oxygen bond in the 5-position is exo or alpha. Pharmaceutically acceptable salts are those made with physiologically acceptable acids that are known in the art; such salts include the hydrochloride, sulfate, phosphate, nitrate, citrate and maleate. The invention also resides in pharmaceutically active compositions which include these compounds.
In accordance with the process of the invention, many cyclic and acylic ketones are transformed by the disubstituted aminosulfur.trifluoride into vinyl fluorides as well as gem-difluorides.
Claims (28)

and pharmaceutically acceptable salts of the compound.






with a disubstituted aminosulfur trifluoride fluorinating agent of the formula R4R5NSF3 wherein each R4 and R5, alike or different, is a primary alkyl group of 1-4 carbon atoms; or R4 and R5 when taken together are ―(CH2)4―, ―(CH2)5― or―CH2CH2OCH2CH2―; at a temperature of -40°C to +80°C; in the presence of a polar or nonpolar solvent; and recovering the fluorine-containing compound of the formula


Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US94419778A | 1978-09-19 | 1978-09-19 | |
| US944197 | 1978-09-19 | ||
| US05/944,198 US4236008A (en) | 1978-09-19 | 1978-09-19 | Fluorination of precursors for fluorine analogs of hydrocodone and oxycodone |
| US944198 | 1978-09-19 | ||
| US06/054,225 US4241065A (en) | 1979-07-02 | 1979-07-02 | Fluoro analogs of hydrocodone and oxycodone useful as analgesics, narcotic antagonists or both |
| US54225 | 1979-07-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0009227A1 EP0009227A1 (en) | 1980-04-02 |
| EP0009227B1 true EP0009227B1 (en) | 1982-09-01 |
Family
ID=27368595
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP19790103475 Expired EP0009227B1 (en) | 1978-09-19 | 1979-09-17 | Fluorination process and fluoro analogs of hydrocodone and oxycodone |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP0009227B1 (en) |
| CA (1) | CA1143376A (en) |
| DE (1) | DE2963614D1 (en) |
| DK (1) | DK388679A (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4613668A (en) * | 1983-12-22 | 1986-09-23 | The United States Of America As Represented By The Department Of Health And Human Services | Short total synthesis or morphinan compounds which uses cyclization of a cycloalkylcarbonyl compound selected from cyclopropylcarbonyl and cyclobutylcarbonyl |
| WO2011154827A2 (en) * | 2010-06-11 | 2011-12-15 | Rhodes Technologies | Transition metal-catalyzed processes for the preparation of n-allyl compounds and use thereof |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3137701A (en) * | 1962-04-19 | 1964-06-16 | Upjohn Co | Process for the preparation of 6-deoxy-6-fluoromorphines, 6-deoxy-6-fluorocodeines, and related compounds |
-
1979
- 1979-09-17 EP EP19790103475 patent/EP0009227B1/en not_active Expired
- 1979-09-17 CA CA000335799A patent/CA1143376A/en not_active Expired
- 1979-09-17 DE DE7979103475T patent/DE2963614D1/en not_active Expired
- 1979-09-18 DK DK388679A patent/DK388679A/en not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| EP0009227A1 (en) | 1980-04-02 |
| DE2963614D1 (en) | 1982-10-28 |
| CA1143376A (en) | 1983-03-22 |
| DK388679A (en) | 1980-03-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4272541A (en) | 7,8 and 7-8 Substituted 4,5α-epoxymorphinan-6-one compounds, and methods of treating pain and drug dependence with them | |
| EP1562953B1 (en) | Process for the preparation of quaternary n-alkyl morphinan alkaloid salts | |
| US4795813A (en) | Synthesis of derivatives of codeine and other 3-O-alkylmorphines | |
| EP2222678B1 (en) | Processes for the production of (+)-'nal' morphinan compounds | |
| EP0485636B1 (en) | Indole derivative | |
| EP0039066A2 (en) | 17-Substituted 6-desoxy-7,8-dihydro-6-alpha-methylnoroxymorphone narcotic antagonists | |
| CA1195324A (en) | Morphinane derivatives | |
| US4241065A (en) | Fluoro analogs of hydrocodone and oxycodone useful as analgesics, narcotic antagonists or both | |
| US8106201B2 (en) | Process for the synthesis of morphinane compounds and intermediates thereof | |
| JPS626554B2 (en) | ||
| US4272540A (en) | 14-Methoxymorphinan-6-one compounds and therapeutic methods of treating pain and drug dependence with them | |
| WO2012102360A1 (en) | Morphinan derivative | |
| US4236008A (en) | Fluorination of precursors for fluorine analogs of hydrocodone and oxycodone | |
| US4230712A (en) | 7-Methyl-8β-lower alkyl or 7-methyl-8-lower alkyl B/C cis or trans morphinan-6-one compounds and therapeutic method of treating pain employing them | |
| US3686167A (en) | Benzazocine derivatives | |
| EP0009227B1 (en) | Fluorination process and fluoro analogs of hydrocodone and oxycodone | |
| US4243668A (en) | Octahydro-1H-benzo[4,5]furo[3,2-e]-isoquinoline analgesic and narcotic antagonistic compounds | |
| AU2002224086B8 (en) | Indole derivatives and use thereof in medicines | |
| CA1100136A (en) | 4a-aryl octahydro-1h-2-pyrindines | |
| US3766188A (en) | Carbonyldioxymorphinan derivatives | |
| US4194044A (en) | Process for preparing 3-phenoxy morphinans | |
| EP0070427B1 (en) | Octahydro-4a,7-ethano- and -etheno-benzofuro(3,2-e)isoquinoline derivatives | |
| US4423221A (en) | 7-Carboethoxy-morphinan-6-one compounds | |
| US4260761A (en) | Intermediates for the preparation of octahydro-1H-benzo[4,5]furo[3,2,-e]-isoquinoline analgesic and narcotic antagonistic compounds | |
| US4242514A (en) | Method for the introduction of a methyl group into the 7 position of the morphinan nucleus |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB IT LU NL |
|
| 17P | Request for examination filed | ||
| ITF | It: translation for a ep patent filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB IT LU NL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19820930 |
|
| REF | Corresponds to: |
Ref document number: 2963614 Country of ref document: DE Date of ref document: 19821028 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19830623 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19830624 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19830627 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19830630 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19830701 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 19830801 Year of fee payment: 5 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19840930 Ref country code: BE Effective date: 19840930 |
|
| BERE | Be: lapsed |
Owner name: E.I. DU PONT DE NEMOURS AND CY Effective date: 19840917 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19850401 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19850531 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19850601 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19881118 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |