-
Technisches Gebiet
-
Die
vorliegende Erfindung bezieht sich auf eine Tricyclo[5.2.1.0
2,6]decan-Verbindung oder Tricyclo[5.2.1.0
2,6]dece-3-en-Verbindung, wie sie in Formel
[1] unten gezeigt ist, oder auf die härtbare Harzzusammensetzung,
enthaltend die oben beschriebene Verbindung
worin A
1 und
A
2 unabhängig
Wasserstoff oder eine Acryloyl-Gruppe
oder Methacryloyl-Gruppe gezeigt in Formel [2] unten:
worin R
1 Wasserstoff
oder C
1-4-Alkyl ist; und R
2 und
R
3 unabhängig
Wasserstoff oder C
1-10-Alkyl sind; oder
eine 2-Vinyloxyethyl-Gruppe gezeigt in Formel (3) unten sind:
-CH2CH2OCH=CH2 [3]worin
die gepunktete Linie eine Einfachbindung oder eine Doppelbindung
ist, mit der Maßgabe,
daß A
1 und A
2 nicht gleichzeitig
Wasserstoff sind.
-
Die
Verbindung in der vorliegenden Erfindung bezieht sich auf das Monomer,
das auf dem Gebiet von Muster-Style-Materialien verwendet wird,
die in der Lithographie mit aktiver Lichtstrahlung, zum Beispiel
Ultraviolettstrahlen, Strahlen des fernen Ultraviolett, Elektronenstrahlen,
Ionenstrahlen und Röntgenstrahlen
geeignet sind, und auf die härtbare
Harzzusammensetzung, die das genannte Monomer enthält. Außerdem bezieht sich
die Verbindung in der vorliegenden Erfindung auf die monomeren Ausgangsmaterialien,
optischen Materialien und Klebemittel, da sie reaktive ethylenisch
ungesättigte
Gruppen innerhalb des Moleküls
hat sowie eine hochwärmebeständige Tricyclodecan-Struktur
hat, und auf die härtbare
Harzzusammensetzung, die das Monomer enthält.
-
Stand der Technik
-
Bis
jetzt ist die in der unten stehenden Formel gezeigte Verbindung
als die Verbindung bekannt, die mehr als eine ungesättigte Gruppe
im Molekül
einer alicyclischen Verbindung hat, oder als die Hauptkomponente
von monomeren Ausgangsmaterialien für die Deckschichtzusammensetzung
für optische
Platten bekannt (japanische Offenlegungsschrift Hei 1-121370).
-
-
Die
Harzzusammensetzung für
optische Rohmaterialien, die aus der in der Formel unten gezeigten Verbindung
als der Hauptkomponente der Zusammensetzung besteht, ist ebenfalls
bekannt (JP Offenlegungsschrift Hei 2-115205).
-
-
Zusätzlich hat
der Erfinder der vorliegenden Erfindung bereits die Patentanmeldung
eingereicht, die sich auf Tricyclo[5.2.1.02,6]dece-3-en-8,9-dicarbonsäurediallyl
bezieht (als TDEA abgekürzt),
das in der Formel unten gezeigt ist (JP-Offenlegungsschrift Sho
60-156683).
-
-
Allerdings
haben Polyester, die TDEA verwenden eine niedrige Polymerisationsreaktivität der Allyl-Gruppe
gezeigt und es wurde kein zufriedenstellendes Resultat bei der Verwendung
der genannten Harzzusammensetzung für optische Acryl-Rohmaterialien erzielt.
-
Copolymer
mit einer Adamantylmethacrylat-Einheit von alicyclischem Polymer
als Polymerverbindung mit der Klarheit bei 193 nm Wellenlänge und
noch mit Trockenätzbeständigkeit
[Takechi, S. et al., Journal of Photopolymer Science and Technology,
5(3): 439–446
(1992); und JP-Offenlegung Hei 5-265212],
Poly(norbornylmethacrylat) [Endo M. et al., Proceedings of IEDM],
CA 14-18, San Francisco (1992)] oder Copolymer mit Poly(isobornylmethacrylat)-Einheit
(Wallraff, G. M. et al., Journal of Vacuum Science and Technology,
B11 (6): 2783–2788
(1993)] und Copolymer mit Poly(menthylmethacrylat)-Einheit [JP-Offenlegung
Hei 8-82925] wurde vorgeschlagen.
-
Andererseits
sind aromatische Derivate, zum Beispiel 2,2-Bis-(4-(2-vinyloxy)ethoxy)phenyl)propan, gezeigt
in der Formel unten
oder aliphatische Vinylether-Derivate,
zum Beispiel Bis(2-vinyloxyethyl)ether,
1,2-Bis[(2-vinyloxy)ethoxy]ethan und dgl. als Vinylether-Derivate
bisher bekannt [Chemistry of Materials, 6(10): 1854–1860 (1994)].
-
Allerdings
waren alicyclische Derivate, die eine Vinyloxyethyloxymethyl-Gruppe
an Position 8 und 9 von Tricyclo[5.2.1.02,6]dece-3-en
oder Tricyclo[5.2.1.o2,6]decan haben, überhaupt
nicht bekannt.
-
Der
existierende Stand der Technik, der oben beschrieben wurde, enthält keine
Reste, die den Unterschied in der Löslichkeit vor und nach der
Belichtung innerhalb der Resteinheit einer alicyclischen Gruppe (Adamantyl,
Norbornyl, Isobornyl oder Menthyl) manifestieren können, was
in einer Trockenätzbeständigkeit resultiert.
Daher wurde die Verwendung als Harzkomponente für das Resist durch Herstellen
des Copolymers mit Comonomer, zum Beispiel t-Butylmethacrylat oder
Tetrahydropyranylmethacrylat, welche die Differenz bei der Löslichkeit
zeigen können,
im einschlägigen
Stand der Technik durchgeführt.
Allerdings wurde die Musterbildung bisher im einschlägigen Stand
der Technik verwendet. Allerdings erfordert die Musterbildung etwa
30 bis 50% Comonomergehalt und als Resultat war die Wirkung der
Trockenätzbeständigkeit
infolge einer alicyclischen Gruppenstruktur drastisch verringert
und ihre Nützlichkeit
wurde fraglich.
-
Der
existierende Stand der Technik, der bisher in Verwendung ist, hat
keine polare Stelle innerhalb der Moleküle und daher ist eine Adhäsion am
Siliciumsubstrat schlecht. Außerdem
ist die Empfindlichkeit wegen der geringen Löslichkeit in alkalischer wäßriger Entwicklungslösung niedrig,
und daher gibt es die Unzulänglichkeit
von Bodensatz, der bei der Entwicklungslösung aller Wahrscheinlichkeit
zurückbleibt.
-
Offenbarung der Erfindung
-
Die
Aufgabe der vorliegenden Erfindung besteht darin, die neue alicyclische
Verbindung herzustellen, die als Photo-Monomer für die Resist-Ausgangsmaterialien
dadurch geeignet ist, daß es
hohe Klarheit bei kurzwelligem ultraviolettem Licht hat und ausgezeichnet
bezüglich
der Trockenätzbeständigkeit
infolge einer Tricycloringstruktur ist und Adhäsion für das Substrat und hohe Löslichkeit
in einer alkalischen Entwicklerlösung
zusätzlich
zu hoher Empfindlichkeit und größerer Auflösung zeigt,
als Monomer für
optische Materialien und Disk-Überzugsmaterialien
dadurch geeignet ist, daß es
hohe Klarheit, niedrige Doppelbrechung und niedrige Wasserabsorptionsrate
hat, und als Monomer für
die Beschichtungsausgangsmaterialien, zum Beispiel EB-härtbare Beschichtung, dadurch
geeignet ist, daß es
eine hohe Reaktivität,
hohe Verschleißmerkmale
und Wasserbeständigkeit
hat, sie besteht auch in der Bereitstellung der härtbaren
Harzzusammensetzung, die die Verbindung enthält.
-
Die
Erfinder der vorliegenden Erfindung haben äußerste Anstrengungen bei der
Lösung
der oben beschriebenen Probleme unternommen. Die vorliegende Erfindung
wurde nach der Auswahl einer Tricyclodecanyl-Gruppe als alicyclische
Gruppe, um hohe Klarheit und hohe Trockenätzbeständigkeit zu erhalten, und Einführung einer
Hydroxymethyl-Gruppe als ein polarer Substituent, um eine gute Adhäsion für das Substrat
und hohe Löslichkeit
in alkalischer Entwicklerlösung
zusätzlich
zu hoher Empfindlichkeit und größerer Auflösung zu
erreichen, vollendet.
-
Demnach
bezieht sich die vorliegende Erfindung auf eine Tricyclo[5.2.1.0
2,6]decan-Verbindung oder Tricyclo[5.2.1.0
2,6]dece-3-en-Verbindung, gezeigt in Formel
[1] unten
worin A
1 und
A
2 unabhängig
Wasserstoff oder eine Acryloyl-Gruppe
oder Methacryloyl-Gruppe gezeigt in Formel [2] unten:
worin R
1 Wasserstoff
oder C
1-4-Alkyl ist; und R
2 und
R
3 unabhängig
Wasserstoff oder C
1-10-Alkyl sind; oder
eine 2-Vinyloxyethyl-Gruppe gezeigt in Formel [3] unten sind:
-CH2CH2OCH=CH2 [3]worin
die gepunktete Linie eine Einfachbindung oder eine Doppelbindung
ist, mit der Maßgabe,
daß A
1 und A
2 nicht gleichzeitig
Wasserstoff sind; die Erfindung bezieht sich auch auf die härtbare Harzzusammensetzung, die
die Verbindung darin enthält.
-
C1-4-Alkyl-Gruppe in der Definition von R1
ist eine geradkettige oder verzweigte Alkyl-Gruppe, zum Beispiel
Methyl, Ethyl, Propyl, Isopropyl, n-Butyl, Isobutyl, s-Butyl, t-Butyl
und dgl.
-
C1-10-Alkyl-Gruppe in der Definition von R2 und R3 ist auch
eine geradkettige oder verzweigte Alkyl-Gruppe, zum Beispiel 2-Ethylbutyl,
n-Pentyl, Isopentyl, 1-Methylpentyl, 1,3-Dimethylbutyl, n-Hexyl,
1-Methylhexyl, n-Heptyl, Isoheptyl, 1-Methylheptyl, 3-Methylheptyl,
n-Octyl, 2-Ethylhexyl, Nonyl, Decyl und dgl. zusätzlich zu Gruppen, die oben
beschrieben wurden.
-
Folgende
Verbindungen, die unten beschrieben werden, können abgekürzt werden, wie es auf der
linken Seite angegeben ist.
- DOL:
- 8,9-Bis(hydroxymethyl)tricyclo[5.2.1.02,6]dece-3-en
- DH-DOL:
- 8,9-Bis(hydroxymethyl)tricyclo[5.2.1.02,6]decan
- DH-DOLMA:
- Acryloyloxymethyl-9-hydroxymethyltricyclo
[5.2.1.02,6]decan
- DH-DOLMM:
- 8-Methacryloyloxymethyl-9-hydroxymethyltricyclo[5.2.1.02,6]decan
- DH-DOLDA:
- 8,9-Bis(acryloxymethyl)tricyclo[5.2.1.02,6]decan
- DH-DOLDM:
- 8,9-Bis(methacryloyloxymethyl)tricyclo[5.2.1.02,6]decan
- DOLMV:
- 8(oder 9)-Hydroxymethyl-9-(oder
8)-(2-vinyloxyethyl)oxymethyl-tricyclo[5.2.1.02,6]dece-3-en
- DH-DOLMV:
- 8-Hydroxymethyl-9-(2-vinyloxyethyl)oxymethyl-tricyclo[5.2.1.02,6]decan
- DOLVE:
- 8,9-Bis[(2-vinyloxyethyl)oxymethyl]tricyclo[5.2.1.02,6]dece-3-en
- DH-DOLVE:
- 8,9-Bis[(2-vinyloxyethyl)oxymethyl]tricyclo[5.2.1.02,6]decan
- HVE:
- 2-Halogenethylvinylether
- VEC:
- 2-Chlorethylvinylether
-
Alicyclische
Verbindungen, gezeigt in der Formel [1] oben, der vorliegenden Erfindung
können
wie unten gezeigt in A)–D)
gruppiert werden.
- A) 8-(Meth)acryloyloxymethyl-9-hydroxymethyltricyclo[5.2.1.02,6]decan-Verbindung oder 9-(Meth)acryloyloxymethyl- 8-hydroxymethyltricyclo[5.2.1.02,6]dece-3-en-Verbindung oder 8-(Meth)acryloyloxymethyl-9-hydroxymethyltricyclo[5.2.1.02,6]dece-3-en-Verbindung, gezeigt der Formel
[4a] oder [4b] unten worin
R1 Wasserstoff oder C1-4-Alkyl
ist; R2 und R3 unabhängig Wasserstoff
oder C1-10-Alkyl sind und die gepunktete
Linie eine Einfachbindung oder eine Doppelbindung ist.
- B) 8,9-Bis((meth)acryloyloxymethyl)tricyclo[5.2.1.02,6]decan-Verbindung
oder 8,9-Bis((meth)acryloyloxymethyl)tricyclo[5.2.1.02,6]dece-3-en-Verbindung,
gezeigt in der Formel [5] unten worin R1 Wasserstoff
oder C1-4-Alkyl ist; R2 und
R3 unabhängig
Wasserstoff oder C1-10-Alkyl sind und die
gepunktete Linie eine Einfachbindung oder eine Doppelbindung ist.
- C) 8-Hydroxymethyl-9-(2-vinyloxyethyl)oxymethyl-tricyclo[5.2.1.02,6]decan oder 8-Hydroxymethyl-9-(2-vinyloxyethyl)oxymethyltricyclo[5.2.1.02,6]dece-3-en oder 9-Hydroxymethyl-8-(2-vinyloxyethyl)oxymethyl-tricyclo[5.2.1.02,6]dece-3-en, gezeigt in der Formel [6a]
oder [6b] unten worin
die gepunktete Linie eine Einfachbindung oder eine Doppelbindung
ist
- D) 8,9-Bis[(2-vinyloxyethyl)oxymethyl]tricyclo[5.2.1.02,6]decan oder 8,9-Bis[(2-vinyloxyethyl)oxymethyl]tricyclo[5.2.1.02,6]dece-3-en, gezeigt in der Formel [7]
unten worin
die gepunktete Linie eine Einfachbindung oder eine Doppelbindung
ist.
-
Das
Verfahren zur Herstellung von (Meth)acryl-Verbindungen, gezeigt
in der Formel [4a] und [4b] und [5], wird in (1)–(3) des Reaktionsschemas I
unten beschrieben Reaktionsschema
I
worin
R
1, R
2, R
3 und die gepunktete Linie dieselbe Bedeutung
wie oben haben; R
4 Wasserstoff oder C
1-10-Alkyl ist und X Halogen ist.
-
8,9-Bis(hydroxymethyl)tricyclo[5.2.1.02,6]dece-3-en (DOL) gezeigt in der Formel
[8], als Ausgangsrohmaterial kann durch das Verfahren erhalten werden,
das in der JP-Offenlegungsschrift
Hei 8-206741 offenbart ist, das unten beschrieben ist.
-
-
Das
heißt,
8,9-Bis(alkoxycarbonyl)tricyclo[5.2.1.02,6]dece-3-en (TCDE) kann aus
der Reaktion mit Dicyclopentadien (DCPD), Kohlenmonoxid und Alkoholverbindung
unter Vorliegen Eisen(III)-chlorid mit Palladium-Katalysator erhalten
werden. DOL kann außerdem
durch die Reduktion dieses TCDE erhalten werden.
-
Außerdem kann
8,9-Bis(hydroxymethyl)tricyclo[5.2.1.02,6]decan
(DH-DOL) durch das Verfahren, das in der JP-Offenlegungsschrift Hei 7-206740 von
den Erfindern der vorliegenden Erfindung offenbart wurde und das
unten beschrieben ist, erhalten werden.
-
-
Die
genannte Verbindung kann nämlich
entweder durch das Verfahren, in dem zuerst die Doppelbindung in
TCDE reduziert wird, bevor die Reduktion der Carbonyl-Gruppe erfolgt,
oder durch das Verfahren, in welchem zunächst die Carbonyl-Gruppe in
TCDE reduziert wird und dann die Doppelbindung in dem so erhaltenen
DOL reduziert wird, erhalten werden.
-
Außerdem ist
R1 in einer (Meth)acryl-Verbindung, gezeigt
in der Formel [9] oder der Formel [10], als anderes Ausgangsmaterial
Wasserstoff oder eine C1-4-Alkyl-Gruppe
und vorzugsweise Wasserstoff oder eine C1-2-Alkyl-Gruppe.
Darüber
hinaus sind R2, R3 und R4 Wasserstoff oder eine C1-10-Alkyl-Gruppe und vorzugsweise
Wasserstoff oder eine C1-5-Alkyl-Gruppe. Eine Alkyl-Gruppe
in diesen genannten Beispielen hat die Anzahl von Kohlenstoffatomen,
die den Beispielen der Alkyl-Gruppe in den oben beschriebenen Ausführungsformen
entspricht.
-
Genaue
Beispiele für
diese (Meth)acryl-Verbindungen sind zum Beispiel Acrylsäure, Methacrylsäure, Tiglinsäure, 3,3-Dimethylacrylsäure, 2-Methyl-2-pentansäure, 2-Ethyl-2-hexansäure und
2-Octansäure
und dgl. und C1-10-Acrylsäureester-Verbindungen davon,
zum Beispiel Acrylsäuremethylester,
Acrylsäureethylester,
Methacrylsäuremethylester,
Methacrylsäureethylester,
Tiglinsäuremethylester,
Tiglinsäureethylester, 3,3-Dimethylacrylsäuremethylester,
3,3-Dimethylacrylsäureethylester,
2-Methyl-2-pentansäuremethylester, 2-Methyl-2-pentansäureethylester,
2-Ethyl-2-hexansäuremethylester,
2-Ethyl-2-hexansäureethylester,
2-Octansäuremethylester
und 2-Octansäureethylester
und dgl.
-
Die
Reaktion (1) und (2) in Schema I erfordern einen Katalysator und
als solcher kann eine Mineralsäure,
zum Beispiel Schwefelsäure,
Salzsäure
und Salpetersäure
verwendet werden; Schwefelsäure
ist speziell bevorzugt. Außerdem
kann eine organische Säure,
zum Beispiel Benzolsulfonsäure,
p-Toluolsulfonsäure, Methansulfonsäure, Ethansulfonsäure und
Trifluoressigsäure
verwendet werden, und p-Toluolsulfonsäure ist bevorzugt.
-
Darüber hinaus
können
Wolframsäure,
Molybdänsäure und
ihre Heteropolysäure
als Katalysator genannt werden. Typische Beispiele für Heteropolysäure sind
H3PW12O40,
H4SiW12O40, H4TiW12O40, H5CoW12O40, H5FeW12O40, H6P2W18O62,
H7PW11O33,
H4TiMO12O40, H3PMo12O40, H7PMo11O39, H6P2Mo18O62,
H4PMoW11O40, H4PVMo11O40, H4SiMo12O40, H5PV2Mo10O40,
H3PMo6W6O40, H0,5Cs2,5PW12O40 und
ihre entsprechenden Hydrate. Zusätzlich
kann ein Katalysator mit Kohlenstoff oder Siliciumdioxid als Träger genannt
werden. H3PW12O40, H3PMo12O40 und deren Hydrate
sind unter diesen Heteropolysäuren
am bevorzugtesten.
-
Darüber hinaus
kann auch ein Kationenaustauschharz, zum Beispiel Amberlite IR120
(Marke), und ein Zeolith H-Typ, zum Beispiel H-ZSM-5, verwendet
werden.
-
Als
Beispiele für
einen Katalysator für
ein Umesterungsverfahren können
speziell Fettsäuresalze
der Verbindung aus der zweiten Gruppe des Periodensystem, dargestellt
durch 3ZnO-2B2O3,
Cadmiumacetat, Zinkacetat und Calciumacetat, zusätzlich zu Mineralsäuren, Heteropolysäuren, organischen
Säuren,
Kationenaustauschharz und Zeolith vom H-Typ, die oben beschrieben
wurden, eingesetzt werden.
-
Die
verwendete Menge dieser Katalysatoren kann 0,1–100 Gew.-%, bezogen auf DOL
oder DH-DOL, sein, und unter wirtschaftlichen Gesichtspunkten sind
1–20 Gew.-%
bevorzugt.
-
In
den Reaktionen (1) und (2) in Schema I kann eine Überschußmenge an
Acrylsäure-Verbindung
oder Acrylsäureester-Verbindung eingesetzt
werden, allerdings ist die Verwendung von Lösungsmitteln normalerweise
bevorzugter, da die Reaktion im Lösungsmittel durchgeführt werden
kann, nachdem die Menge an Allylalkohol, die verwendet wird, nahezu
auf einen theoretischen Wert gesenkt war. Die Arten an Lösungsmittel, die
verwendet werden, sind zum Beispiel halogenierter Kohlenwasserstoff,
zum Beispiel 1,2-Dichlorethan (EDC) oder 1,1,1-Trichlorethan, aromatischer
Kohlenwasserstoff, zum Beispiel Benzol, Toluol oder Xylol, und Ether,
zum Beispiel 1,2-Dimethoxyethanether oder Diethylenglykoldimethylether.
-
Die
verwendete Lebensmittelmenge ist die 1- bis 20-fache Gewichtsmenge,
bevorzugter die 1- bis 6-fache Gewichtsmenge, bezogen auf DOL oder
DH-DOL. Die Reaktionstemperatur kann 50–200°C, bevorzugter 70–150°C sein. Die
Reaktion kann unter Normaldruck oder unter erhöhtem Druck durchgeführt werden. Die
Reaktionszeit kann 1–50
Stunden betragen, allerdings ist es unter normalen Umständen praktisch,
die Reaktion in 2–12
Stunden durchzuführen.
-
Das
Herstellungsverfahren für
die Reaktion (3) in Schema I wird im folgenden beschrieben.
-
(Meth)acrylsäurehalogenid,
gezeigt in der allgemeine Formel [11], kann erhalten werden, indem
die (Meth)acrylsäure-Verbindung mit halogeniertem
Thionyl in Säurehalogenid
umgewandelt wird. Halogenatome, gezeigt als Rest X, können F,
Cl, Br und I sein, allerdings wird normalerweise das billigste Cl
gewählt.
Typische Beispiele für
diese Verbindungen sind zum Beispiel Acryloylchlorid, Methacryloylchlorid,
Tiglyloylchlorid, 3,3-Dimethylacryloylchlorid,
2-Methyl-2-pentenoylchlorid, 2-Ethyl-2-hexanoylchlorid und 2-Octyloylchlorid.
Die verwendete Menge ist vorzugsweise 2,0–2,5 Äquivalente DOL oder DH-DOL.
-
Basen,
zum Beispiel verzweigte Alkylamin-Verbindungen wie Trimethylamin,
Triethylamin und Tripropylamin, aromatische Amin-Verbindung, zum
Beispiel Pyridin, Anilin und N-Methylanilin, cyclische Alkylamin-Verbindung,
zum Beispiel 1,5-Diazabicyclo[4,3,0]-5-nonan (DBN), 1,4-Diazabicyclo[2,2,2]octan
(DBO) und 1,8-Diazabicyclo[5,4,0]undec-7-en (DBU) und Metallcarbonat,
zum Beispiel Natriumcarbonat, Natriumhydrogencarbonat und Kaliumcarbonat,
sind in dem in der Reaktionsformel (3) in Schema I gezeigten Verfahren unentbehrlich.
Die bevorzugte Base unter diesen ist Triethylamin oder Tripropylamin.
Die verwendete Menge ist vorzugsweise 2,0–2,5 Äquivalente (dasselbe Äquivalent
wie Säurechlorid)
bezüglich
DOL oder DH-DOL.
-
Das
bevorzugte Verfahren zu der Reaktion (3) in Schema I ist es, Lösungsmittel
zu verwenden. Typische Beispiele für ein bevorzugtes Lösungsmittel
sind eine Ether-Verbindung, zum Beispiel Tetrahydrofuran (THF),
1,2-Dimethoxyethan, Dioxan und 2-Methoxyethylether, N,N-Dimethylformamid
(DMF), N,N-Dimethylacetamid (DMAc), N-Methylpyrrolidon und 1,3-Dimethyl-2-imidazolidinon
(DMI). Unter diesen ist das billigere 1,2-Dimethoxyethan oder DMF
bevorzugt. Die bevorzugte Menge, die zu verwenden ist, ist die 1-
bis 10-fache Gewichtsmenge
und speziell die 2- bis 5-fache Gewichtsmenge DOL oder DH-DOL.
-
Die
Reaktionstemperatur ist vorzugsweise 0–100°C und am bevorzugtesten 0–50°C. Wasser
wird zugesetzt, um das nach der Reaktion zurückgebliebene Säurechlorid
zu hydrolysieren, Lösungsmittel
wird abdestilliert, es wird mit einem in Wasser unlöslichen
Lösungsmittel
(Ethersystem oder Estersystem) extrahiert und dann wird durch Destillation
oder Säulenchromatographie
unter Erhalt der gewünschten
Verbindung gereinigt.
-
Tricyclo[5.2.1.0
2,6]decan-Verbindung oder Tricyclo[5.2.1.0
2,6]dece-3-en-Verbindung, worin beide, A
1 und A
2, eine 2-Vinyloxyethyl-Gruppe
sind oder entweder A
1 oder A
2 2-Vinyloxyethyl
ist, während
das andere Wasserstoff ist, kann außerdem durch das unten beschriebene
Reaktionsschema II synthetisiert werden. Reaktionsschema
II
worin
X ein Halogenatom ist.
- (1) 8 (oder 9)-Hydroxymethyl-9
(oder 8)-(2-vinyloxyethyl)oxymethyl-tricyclo[5.2.1.02,6]dece-3-en
(DOLMV) kann nämlich
erhalten werden, indem 8,9-Bis(hydroxymethyl)tricyclo[5.2.1.02,6]dece-3-en (DOL) und 2-Halogenethylvinylether
(HVE) in Gegenwart von Basen umgesetzt werden.
- (2) 8-Hydroxymethyl-9-(2-vinyloxyethyl)oxymethyltricyclo[5.2.1.02,6]decan (DH-DOLMV) kann erhalten werden,
indem 8,9-Bis(hydroxymethyl)tricyclo[5.2.1.02,6]decan
(DH-DOL) und HVE
unter Vorliegen von Basen umgesetzt werden.
- (3) 8,9-Bis[(2-vinyloxyethyl)oxymethyl]tricyclo[5.2.1.02,6]dece-3-en (DOLVE) kann erhalten werden,
indem DOL und die 2-fache
Molmenge an HVE unter Vorliegen von Basen umgesetzt werden.
- (4) 8,9-Bis-[(2-vinyloxyethyl)oxymethyl]tricyclo[5.2.1.02,6]decan (DH-DOLVE) kann erhalten werden,
indem DH-DOL und die 2-fache Molmenge an HVE bei Vorliegen von Basen
umgesetzt werden.
- (5) DOLVE kann erhalten werden, indem DOLMV und HVE unter Vorliegen
von Basen umgesetzt werden.
- (6) DH-DOLVE kann erhalten werden, indem DH-DOLMV und HVE bei
Vorliegen von Basen umgesetzt werden.
-
Dabei
sind typische Beispiele für
2-Halogenethylvinylether, einem anderen Ausgangsmaterial zusätzlich zu
DOL und DH-DOL, das in der Formel [12] unten gezeigt wird XCN2CH2OCH=CH2 [12] worin
X ein Halogenatom ist, 2-Fluorethylvinylether, 2-Chlorethylvinylether,
2-Bromethylvinylether und 2-Iodethylvinylether, und 2-Chlorethylvinylether
wird unter wirtschaftlichen Gesichtspunkt eingesetzt. Die bevorzugteste
Verwendung der Menge ist die 1- bis 10-fache Molmenge, speziell
die 2- bis 5-fache Molmenge bezüglich DOL
oder DH-DOL.
-
Das
Vorliegen von Basen ist für
die Reaktion (1)–(6)
im Schema II oben unerläßlich. Die
Art der Basen, die verwendet werden können, können Hydroxid oder Hydrid von
Alkalimetall und Erdalkalimetall sein. Konkrete Beispiele sind zum
Beispiel LiOH, NaOH, KOH, RbOH, CsOH, Mg(OH)2,
Ca(OH)2, Sr(OH)2,
Ba(OH)2, LiH, NaH, KH, MgH2,
CaH2, SrH2 und Ba(OH)2. NaOH, KOH, Ca(OH)2,
Ba(OH)2, NaH und KH sind bevorzugt. Die
zu verwendende Menge ist die 1- bis 10-fache Molmenge bezüglich des
Reaktionssubstrats und speziell die 2- bis 5-fache Molmenge ist bevorzugt.
-
Die
Verwendung von organischen Lösungsmitteln
ist für
die Reaktion (1) bis (6) in Schema II bevorzugt. Die Art des bevorzugten
organischen Lösungsmittels
sind Lösungsmittel
der Amid-Familie, zum Beispiel N,N-Dimethylformamid (DMF), N,N-Dimethylacetamid
(DMAc) oder N-Methylpryrrolidon, Lösungsmittel der Sulfoxid-Familie,
zum Beispiel Dimethylsulfoxid (DMSO) oder Sulfolan, Lösungsmittel
der aliphatischen Kohlenwasserstoff-Familie, zum Beispiel Hexan,
Heptan oder Cyclohexan, Lösungsmittel
der aromatischen Kohlenwasserstoff-Familie, zum Beispiel Toluol,
Xylol oder Ethylbenzol, Lösungsmittel
der Ether-Familie, zum Beispiel Dioxan, Tetrahydrofuran (THF), 1,2-Dimethoxyethan
(DME) oder Diethylglykoldimethylether und Wasser, wobei eine oder
mehr als zwei Arten in einem homogenen System oder einem Zweiphasensystem
verwendet werden kann/können.
Die zu verwendende Menge kann die 1- bis 20-fache Gewichtsmenge bezüglich des
Reaktionssubstrats sein, und wirtschaftlich ist speziell die 2-
bis 5-fache Gewichtsmenge.
-
Darüber hinaus
ist auch die Verwendung eines Phasentransferkatalysators für die Reaktion
(1)–(6)
in Schema II wirksam. Die Verwendung eines Phasentransferkatalysators
ist produktiv, zum Beispiel wenn ein Alkalimetall oder Erdalkalimetall
in einem Lösungsmittel
der Amid-Familie verwendet werden soll oder wenn ein Alkalimetall
oder Erdalkalimetall in einem Zweiphasen-Lösungsmittelsystem
mit Kohlenwasserstoff-Lösungsmitteln
und Wasser verwendet werden soll. Beispiele für Phasentransferkatalysatoren
sind zum Beispiel Tetrabutylammoniumchlorid, Tetrabutylammoniumbromid,
Trimethylbenzylammoniumchlorid, Triethylbenzylammoniumchlorid, Tetramethylammoniumchlorid,
Tetraethylammoniumbromid, Tetra-n-butylammoniumhydrogensulfat, Trimethylbenzylammoniumhydroxid-Methanol-Lösung und
Tetra-n-butylammoniumhydroxid-Lösung.
-
Die
zu verwendende Menge ist 0,1–20
Gew.-%, und vorzugsweise sind 0,5–10 Gew.-%, bezogen auf das
Reaktionssubstrat, geeignet.
-
Die
Reaktionstemperatur beträgt üblicherweise
0–150°C, jedoch
ist eine Reaktionstemperatur von 40–120°C bevorzugt, so daß die Ausbeute
der gewünschten
Verbindung erhöht
wird. Die Reaktion kann unter Normaldruck oder unter erhöhtem Druck
durchgeführt
werden.
-
Die
Reaktionszeit variiert in Abhängigkeit
von der Art der Basen, der Menge, der Reaktionstemperatur und des
Lösungsmittels
unter anderen Faktoren, aber üblicherweise
wird die Reaktion in 1–50
Stunden durchgeführt,
und die Bedingungen, bei denen sie in 2–20 Stunden durchgeführt werden
kann, sind bevorzugt. Die Reaktion kann durch das Chargenverfahren
oder durch das kontinuierliche Verfahren durchgeführt werden.
-
Auf
die Reaktion kann eine Probennahme in der Reaktionslösung und
ein Analysieren durch Gaschromatographie folgen. Die Isolierung
des Produkts kann durch Zugabe von Wasser und Isopropylether, Ethylacetat
oder Methylethylketon (MEK) unter anderen, Dehydratisierung und
Konzentrierung der organischen Phase erfolgen und der Rest kann
durch Destillieren und/oder Anwenden von Säulenchromatographie auf das Endprodukt
gereinigt werden.
-
Eine
so in der vorliegenden Erfindung erhaltene Acryloyl-Gruppe oder Methacryloyl-Gruppe
oder alicyclische Verbindung mit einer 2-Vinyloxyethyl-Gruppe kann
als eine Monomerkomponente eines härtbaren Harzes verwendet werden.
In diesem Fall kann das Monomer der vorliegenden Erfindung alleine
verwendet werden, allerdings ist die Verwendung in Kombination mit
einem anderen Monomer der Acryl-Familie im allgemeinen die Norm.
Auf jeden Fall wird eine Polymerisation mit Katalysator (Polymerisationsinitiator)
und Lösungsmittel,
falls erforderlich, durchgeführt.
-
Monomere,
die in Kombination verwendet werden können, sind ohne Beschränkung zum
Beispiel Trimethylpropantriacrylat, 1,6-Hexandioldiacrylat, Neopentylglykoldiacrylat,
Trimethylolpropandipropoxytriacrylat, Polyethylenglykoldiacrylat,
Tetrahydrofurfurylacrylat, 4·5,8-Bis(acryloyloxymethyl)tricyclodecan,
Oxyethylenbisphenol A-diacrylat,
(Meth)acrylsäure
von Bisphenol A-glycidylether, (Meth)acrylat von Phenol-Novolak-Polyepoxy-Verbindung,
Methyl(meth)acrylat, Carboxytricyclodecanylmethyl(meth)acrylat,
2-Methyladamantyl(meth)acrylat, Trimethylnorbornyl(meth)acrylat,
Hydroxymethyl(meth)acrylat und Hydroxypivalinsäureneopentylglykol.
-
Die
zu verwendende Menge an anderem Monomer, zum Beispiel (Meth)acrylat,
als die oben beschriebenen ist weniger als 80 Gew.-% und vorzugsweise
weniger 70 Gew.-%, bezogen auf die Gesamtmenge der Zusammensetzung,
einschließlich
der Verbindung der vorliegenden Erfindung. Die Leistungsfähigkeit,
zum Beispiel die Adhäsion
von härtbarem
Produkt kann nicht erreicht werden, wenn die zu verwendende Menge über 80 Gew.-%
liegt.
-
Der
Initiator wird normalerweise der Verbindung der vorliegenden Erfindung
oder der härtbaren
Harzzusammensetzung, die die Verbindung enthält, zugesetzt, um die Härtungsreaktion
zu beschleunigen. Beispielsweise wird ein Radikalpolymerisationsinitiator
für die
Härtungsreaktion
mit Wärmeenergie
verwendet. Typische Beispiele für
den Radikalpolymerisationsinitiator sind ohne Beschränkung organisches
Peroxid, zum Beispiel Benzoylperoxid, Acetylperoxid, Di-t-butylperoxid,
t-Butylperoxylaurat, Dicumylperoxid, α,α'-Bis-t-butylperoxy-p-diisopropylbenzol,
2,5-Dimethyl,2,5-di-t-butylperoxyhexan, 2,5-Di-t-butylperoxyhexen, t-Butylperoxybenzoat,
n-Butyl-4,4-bis-t-butylperoxyvalerat,
p-Menthanhydroperoxid und t-Butylcumylperoxid, oder eine Azobis-Verbindung,
zum Beispiel 2,2'-Azobis(isobutyronitril),
2,2'-Azobis(2,4,4-trimethylvaleronitril)
und 2,2'-Azobis(2-cyclopropylpropionitril).
-
Ein
Lichtpolymerisationsinitiator wird für eine Härtungsreaktion mit Energiestrahlen
verwendet. Ein bekannter Lichtpolymerisationsinitiator kann zu diesem
Zweck verwendet werden, und Beispiele dafür sind ohne Beschränkung Benzoin,
Benzoinmethylether, Benzoinethylether, Benzoinisopropylether, Benzoinbutylether, Benzoinphenylether,
1-Hydroxycyclohexylphenylketon, 3-Methylacetophenon, 2,2-Dimethoxyacetophenon, 2,2-Dimethoxy-2-phenylacetophenon,
4-Dialkylaminoacetophenon, 2-Phenylthioacetophenon, Benzyl, Benzyldimethylketal,
Benzoylbenzoat, Anthrachinon, 2-Ethylanthrachinon, Naphthochinon,
2,4-Diisopropylthioxanthon, Azobisisobutyronitril, 2,2'-Azobis-2,4-dimethylvaleronitril,
Benzoylperoxid, Di-t-butylperoxid,
Diphenyldisulfid, Tetramethylthiurammonosulfid, Tetraethylthiuramdisulfid,
Benzophenon, Pivaloinethylether, Dibenzylsulfid, Cinnamoylchlorid,
Dimethyldiphenylendisulfid und Dibenzothiazoldisulfid.
-
Darüber hinaus
können
Triborfluoridether, Titansäuretetraisopropyl
und dgl. auch als Kationenpolymerisationsinitiator eingesetzt werden.
-
Die
Menge dieser Initiatoren, Radikalpolymerisationsinitiator, Lichtpolymerisationsinitiator
und Kationenpolymerisationsinitiator, die zu verwenden ist, ist
mehr als 0,01 Gew.-%, aber weniger als 5 Gew.-% und vorzugsweise
mehr als 0,05 Gew.-%, aber weniger als 3 Gew.-%, bezogen auf die
Verbindung der vorliegenden Erfindung oder die härtbare Harzzusammensetzung,
die die Verbindung enthält.
Die Härtungsrate
ist nicht für
die Praxis geeignet, wenn sie weniger als 0,01 Gew.-% ist. Wenn
sie 5 Gew.-% übersteigt,
kann kein Verbesserungseffekt für
die Härtungsrate
erwartet werden und die Leistungsfähigkeit des gehärteten Films
nimmt ab, was nicht bevorzugt ist.
-
Der
Sensibilisator kann zur Verstärkung
des Effekt eines Lichtpolymerisationsinitiators in die Verbindung
der vorliegenden Erfindung, welche die Härtungsreaktion mit Energiestrahlen
initiiert, oder in die härtbare Harzzusammensetzung,
die die Verbindung enthält,
gegeben werden. Der Sensibilisator kann mit der Bestrahlung durch
Energiestrahlen nicht selbst aktiviert werden, allerdings erhöht die Kombination
die Wirksamkeit, wenn sie mit dem Lichtinitiator verwendet wird,
verglichen mit dem Lichtinitiator alleine. Typische Beispiele sind,
jedoch ohne Beschränkung,
zum Beispiel Triethylamin, Triethyltetramin, n-Butylamin, D-n-butylamin, Tri-n-butylphosphin,
Allylthioharnstoff, S-Benzylisothiuranium-p-toluolsulfinat und Diethylaminoethylmethacrylat.
-
Die
zu verwendende Menge des Sensibilisators ist mehr als 0,01 Gew.-%,
aber weniger als 5 Gew.-% und bevorzugter mehr als 0,05 Gew.-%,
aber weniger als 3 Gew.-%, bezogen auf die Verbindung der vorliegenden
Erfindung oder die härtbare
Harzzusammensetzung, die die Verbindung darin hat. Wenn sie weniger als
0,01 Gew.-% ist, ist die Härtungsrate
für die
Praxis zu langsam. Wenn sie jedoch 5 Gew.-% übersteigt, kann kein weiterer
Verbesserungseffekt durch den Sensibilisator erwartet werden und
die Leistungsfähigkeit
des gehärteten
Films nimmt ab, was nicht bevorzugt ist.
-
Beispiele
für das
zur Polymerisation verwendete Lösungsmittel
sind die cyclische Ether-Gruppe, zum Beispiel Tetrahydrofuran und
Dioxan, die Glykolether-Gruppe, zum Beispiel Ethylenglykolmonomethylether, Ethylenglykolmonoethylether,
Diethylenglykolmonomethylether, Diethylenglykolmonoethylether und
Diethylenglykoldimethylether, die Propylenglykolalkyletheracetat-Gruppe,
zum Beispiel Propylenglykol(mono)methyletheracetat und Propylenglykolpropyletheracetat,
die Keton-Gruppe zum Beispiel Methylethylketon, Cyclohexanon und
4-Hydroxy-4-methyl-2-pentanon,
die Ester-Gruppe, zum Beispiel 2-Hydroxypropionsäureethyl, 2-Hydroxy-2-methylpropionsäureethyl,
2-Hydroxy-2-methylpropionsäureethyl,
Ethoxyethylacetat, Hydroxysäureethyl,
2-Hydroxy-3-methylbutansäuremethyl,
3-Methoxypropionsäuremethyl,
3-Methoxypropionsäureethyl,
3-Ethoxypropionsäureethyl,
3-Ethoxypropionsäuremethyl,
Ethylacetat, Butylacetat, Methyllactat und Ethyllactat, die Cellosolveester-Gruppe
zum Beispiel Methylcellosolveacetat und Ethylcellosolveacetat, die aromatische
Kohlenwasserstoff-Gruppe, zum Beispiel Benzol, Toluol und Xylol,
und aprotische polare Lösungsmittel,
zum Beispiel DMF, und zwar unter normalen Umständen in einer Menge von 20
bis 1000 Gew.-Teilen, bezogen auf 100 Gew.-Teile der polymeren Komponente.
-
In
der vorliegenden Erfindung können
Komponenten, zum Beispiel anorganische Füllstoffmaterialien, Egalisiermittel,
Färbemittel,
zum Beispiel Pigment oder Farbstoff, Klebeeigenschaften verleihende
Mittel, Weichmacher, Lösungsmittel
und Lagerungsstabilisatoren je nach Bedarf der Verbindung der vorliegenden
Erfindung oder der härtbaren
Harzzusammensetzung, die die Verbindung darin enthält, zugesetzt
werden.
-
Für die Verbindung
der vorliegenden Erfindung, bei der Energiestrahlen zu einer Härtungsreaktion
führen,
oder für
die härtbare
Harzzusammensetzung, die die Verbindung darin hat, sind typische
Beispiele für
aktives Licht zur Durchführung
der Härtungsreaktion
mit Bestrahlung der Verbindung oder der Zusammensetzung ultraviolette
Strahlung, Elektronenstrahlen oder Röntgenstrahlen. Sonnenlicht,
eine chemische Lampe, eine Niederdruckquecksilberdampflampe, eine
Hochdruckquecksilberdampflampe, eine Metallhalogenidlampe oder eine
Xenonlampe können
u.a. als Lichtquelle für
ultraviolette Strahlen verwendet werden.
-
Das
Anwendungsgebiet für
die härtbare
Harzzusammensetzung, die die alicyclische Verbindung der vorliegenden
Erfindung hat, sind zum Beispiel Kleber, Anstrichmittel, Tinte,
Baumaterialien, Beleuchtung, glasfaserverstärktes Harz, Rost- und Korrosionsprävention,
optische Linsen, Beschichtung für
optische Fasern, UV·EB-härtbares
Harz und Resist.
-
Die
alicyclische Verbindung der vorliegenden Erfindung zeichnet sich
speziell als lichtempfindliches Monomer aus, das für Resist-Ausgangsmaterial
geeignet ist, und zwar wegen der hohen Klarheit für ultraviolette
Strahlen kurzer Wellenlänge,
wegen seiner Trockenätzbeständigkeit
infolge der tricyclischen Ringstruktur und der starken Klebereigenschaften
gegenüber
Siliconsubstrat, ist als optisches Ausgangsmaterial und Scheibenüberzugsausgangsmaterial
geeignet, und zwar wegen der hohen Klarheit, der niedrigen Doppelbrechung
und der niedrigen Wasserabsorptionsrate, und ist als Überzugsausgangsmaterial,
zum Beispiel UV·EB-härtbares
Beschichtungsmaterial geeignet, und zwar wegen der hohen Reaktivität, der hohen
Verschleißeigenschaften
und der hohen Wasserbeständigkeit.
-
Darüber hinaus
kann die Verbindung der vorliegenden Erfindung auch als Ausgangsmaterial
zur Zahnwiederherstellung eingesetzt werden. Es ist bezüglich der
Klebeeigenschaften gegenüber
Dentin und der blockierenden Eigenschaften für die Nachbarschaft von Dentin
als Wiederherstellungsmaterial hervorragend. Außerdem hat der dentale Kleber
mit Mono(meth)acrylat-Verbindung mit Hydroxy-Gruppe im Molekül gemäß der vorliegenden
Erfindung eine gute Ausgewogenheit unter anderem für Klebeeigenschaft,
Blockierungseigenschaft und hydrophile Natur.
-
Bester Modus zur Durchführung der
Erfindung
-
Die
vorliegende Erfindung wird detaillierter beschrieben, indem die
Beispiele gezeigt werden, allerdings wird die Erfindung nicht auf
die besonderen hierin beschriebenen Ausführungsformen beschränkt.
-
-
3,92
g (20 mmol) DH-DOL, 40 ml Tetrahydrofuran (THF) und 2,02 g (20 mmol)
Triethylamin wurden in einen 100 ml-Vierhals-Reaktionskolben gefüllt und
auf 5°C
gekühlt.
Unter Rühren
wurde die gemischte Lösung von
1,82 g (20 mmol) Acryloylchlorid und 10 m THF tropfenweise zugesetzt.
Die Reaktionslösung
wurde für
1 Stunde bei 5°C
gerührt
und dann für
6 Stunden bei 25°C
weiter umgesetzt. Die Reaktionslösung
wurde filtriert und das Filtrat wurde konzentriert. Der Rückstand
wurde in Chloroform gelöst,
mit 0,5 N Salzsäure-Lösung, gesättigter Salzlösung, 3%iger
Natriumcarbonat-Lösung
und gesättigter
Salzlösung
der Reihe nach gewaschen und unter reduziertem Druck nach Dehydratisierung
mit wasserfreiem Magnesiumsulfat konzentriert. Der konzentrierte
Rückstand
wurde durch Silicagel-Säulenchromatographie
mit n-Hexan/Ethylacetat = 4/1 gereinigt, wodurch 1,14 g (Ausbeute
23%) 8-Acryloyloxymethyl-9-hydroxymethyltricyclo[5.2.1.02,6]decan (DH-DOLMA) und 1,25 g (Ausbeute
21%) 8,9-Bis(acryloyloxymethyl)tricyclo[5.2.1.02,6]decan (DH-DOLDA) erhalten wurden.
-
Die
Struktur von DH-DOLMA wurde durch unten beschriebenen analytischen
Resultate bestätigt.
Massenspektrum
(FAB+, m/e (%)): 251 (M+1,
16), 220 (25), 179 (27), 177 (47), 161 (92), 133 (35), 119 (44), 101
(67), 94 (100), 84 (63), 71 (40).
1H-NMR
(CDCl3, δ ppm):
1,37 (dt, J1 = 10,25 Hz, J2 =
1,47 Hz, 1H), 1,48–1,69
(m, 7H), 2,06 (d, J = 12,0 Hz, 2H), 2,12 (d, J = 7,7 Hz, 1H), 2,15
(s, 1H), 2,24 (dd, J1 = 15,29 Hz, J2 = 8,33 Hz, 1H), 2,44 (d, J = 2,56 Hz, 2H), 3,49
(dd, J1 = 10,62 Hz, J2 =
7,69 Hz, 1H), 3,69 (dd, J1 = 10,53 Hz, J2 = 6,68 Hz, 1H), 4,06 (dd, J1 =
11,08 Hz, J2 = 8,33 Hz, 1H), 4,25 (dd, J1 = 10,99 Hz, J2 =
6,59 Hz, 1H), 5,83 (dd, J1 = 8,61 Hz, J2 = 1,47 Hz, 1H), 6,12 (dd, J1 =
17,30 Hz, J2 = 10,34 Hz, 1H), 6,40 (dd,
J1 = 17,39 Hz, J2 =
1,47, 1H).
13C-NMR (CDCl3, δ ppm): 26,78
(2), 29,01, 37,11, 38,31, 40,71 (2), 44,68, 45,02, 45,53, 45,59,
62,92, 65,16, 128,50, 130,8, 166,29.
IR (KBr, cm–1):
3420, 2950, 1720, 1410, 1300, 1200, 1050, 1020, 980, 810.
-
-
7,84
g (40 mmol) DH-DOL, 3,24 g (45 mmol) Acrylsäure, 0,23 g p-Toluolsulfonsäuremonohydrat,
0,20 g Hydrochinon und 20 ml Benzol wurden in einen 100 ml-Vierhals-Reaktionskolben
eingefüllt,
das Gemisch wurde für
4 Stunden gerührt,
während
das Produkt Wasser mit gleichzeitiger Erhöhung der Temperatur von 90 auf
104°C abdestilliert
wurde. Das Gemisch wurde nach Beendigung der Reaktion abgekühlt und
dann wurden 1,2-Dichlorethan (EDC) und Wasser zugesetzt und die
EDC-Schicht wurde
mit gesättigter
Salzlösung,
5%iger Natriumhydrogencarbonat-Lösung
und erneut gesättigter
Salzlösung
in Folge gewaschen. Diese EDC-Schicht wurde unter reduziertem Druck
konzentriert und der Rückstand
wurde durch Silicagel-Säulenchromatographie gereinigt,
wodurch 2,08 g (Ausbeute 21%) DH-DOLMA als Zielprodukt erhalten
wurden.
-
-
3,92
g (20 mmol) DH-DOL, 30 g THF und 2,02 g (20 mmol) Triethylamin wurden
in einen 100 ml-Vierhals-Reaktionskolben gefüllt und auf 5°C gekühlt. Die
gemischte Lösung
von 2,09 g (20 mmol) Methacryloylchlorid und 10 g THF wurde unter
Rühren
tropfenweise in das Reaktionsgemisch gegeben. Das Rühren wurde für 1 Stunde
bei 5°C
fortgesetzt und dann wurde das Gemisch zusätzlich für 6 Stunden bei 25°C umgesetzt. Das
Gemisch wurde unter reduziertem Druck bei 25°C konzentriert und dann wurden
1,2-Dichlorethan (EDC) und Wasser zu dem Rückstand gegeben und die EDC-Schicht
wurde abgetrennt. Diese EDC-Schicht wurde mit 0,5%iger Salzsäure-Lösung, gesättigter
Salzlösung,
3%iger Natriumhydrogencarbonat-Lösung
und erneut gesättigter
Salzlösung
nacheinander gewaschen und die Schicht wurde mit wasserfreiem Natriumsulfat
getrocknet und unter reduziertem Druck konzentriert. Dieser konzentrierte
Rückstand
wurde durch Silicagel-Säulenchromatographie
mit n-Hexan/Ethylacetat = 4/1 gereinigt, wodurch 1,48 g (Ausbeute
28%) 8-Methacryloyloxymethyl-9-hydroxymethyltricyclo[5.2.1.02,6]decan (DH-DOLMM) und 1,39 g (Ausbeute
21%) 8,9-Bis(methacryloyloxymethyl)tricyclo[5.2.1.02,6]decan
(DH-DOLDM) erhalten wurden.
-
Die
Struktur von DH-DOLMM wurde mit den unten beschriebenen Analysendaten
bestätigt.
Massenspektrum
(FAB+, m/e (%)): 265 (M+1,
55), 178 (100).
1H-NMR (CDCl3, δ ppm):
1,27 (s, 2H), 1,48–1,68
(m, 9H), 1,95 (s, 3H), 2,07 (d, J = 15,5 Hz, 2H), 2,45 (s, 2H), 3,48
(dd, J1 = 8,05 Hz, J2 =
10,25 Hz, 1H), 3,68 (dd, J1 = 6,77 Hz, J2 = 10,44 Hz, 1H), 4,04 (dd, J1 =
8,51 Hz, J2 = 10,89 Hz, 1H), 4,24 (dd, J1 = 6,23 Hz, J2 =
10,99 Hz, 1H), 5,56 (dd, J1 = 1,47 Hz, J2 = 3,11 Hz, 1H), 6,09 (s, 1H).
13C-NMR (CDCl3, δ ppm): 14,13,
18,30, 22,71, 26,82, 29,04, 31,90, 37,13, 38,26, 40,67, 44,69, 45,62,
62,7, 65,24, 125,45, 136,38, 167,50.
-
-
7,84
g (40 mmol) DH-DOL, 3,87 g (45 mmol) Methacrylsäure, 0,23 g p-Toluolsulfonsäuremonohydrat, 0,20
g Hydroxychinon und 20 ml Benzol wurden in einen 100 ml-Vierhals-Reaktionskolben gefüllt und
das Gemisch wurde für
4 Stunden gerührt,
während
das Produkt Wasser bei gleichzeitiger Erhöhung der Temperatur von 90–106°C abdestilliert
wurde. Das Gemisch wurde nach Beendigung der Reaktion gekühlt und
dann wurden 1,2-Dichlorethan (EDC) und Wasser zugesetzt und die
EDC-Schicht wurde mit gesättigter
Salzlösung, 5%iger
Natriumhydrogencarbonat-Lösung
und erneut mit gesättigter
Salzlösung
der Reihe nach gewaschen. Nachdem diese EDC-Schicht unter reduziertem Druck konzentriert
worden war, wurde der Rückstand
durch Silicagel-Säulenchromatographie
gereinigt, wodurch 1,19 g (Ausbeute 18%) an DH-DOLMM als Zielprodukt erhalten
wurden.
-
-
422
g (2,15 mol) DH-DOL wurden in 6 1 Tetrahydrofuran (THF) gelöst und es
wurden 438 g (4,84 mol) Acryloylchlorid zugesetzt. 480 g (4,84 mol)
Triethylamin wurden tropfenweise über 30 Minuten nach Kühlung auf
0°C in das
Gemisch gegeben. Nach Beendigung der tropfenweisen Zugabe wurde
die Reaktion 2 Stunden nach allmählichem
Erhöhen
der Temperatur auf Raumtemperatur (25°C) beendet. Das Gemisch wurde
unter reduziertem Druck nach Zusatz von 3 l Wasser konzentriert
und THF wurde abdestilliert. Der pH der Wasserschicht wurde mit
Triethylamin auf pH 8 eingestellt und dann dreimal mit Diisopropylether
extrahiert. Die extrahierten Lösungen
wurden kombiniert und unter reduziertem Druck konzentriert, und
dann wurde der Rückstand durch
eine Silicagel-Säule
(SiO2: 3 kg × 2, Eluat:n-Hexan/Ethylacetat
= 10/1) gereinigt. 510 g (Ausbeute 77,9%) öliges Produkt wurden bei Raumtemperatur
als die Hauptfraktion erhalten. Dieses ölige Produkt wurde fest, nachdem
es in einem Kühlschrank
mit –40°C über Nacht
gehalten worden war und dann auf Raumtemperatur zurückgebracht
worden war. Die Analysendaten dieses Kristalls waren wie folgt.
Schmelzpunkt
(°C): 33–34°C
Massenspektrum
(FD+, m/e (%)): 304 (M+,
100), 160 (45).
1H-NMR (CDCl3, δ ppm):
1,41 (d, J = 10,3 Hz, 2H), 2,08 (s, 2H), 2,26–2,29 (m, 2H), 2,46 (s, 2H),
4,06–4,10
(m, 2H), 4,20 (dd, J1 = 5,86 Hz, J2 = 11,0 Hz, 2H), 5,83 (dd, J1 =
1,47 Hz, J2 = 10,4 Hz, 2H), 6,13 (c, J1 = 10,4 Hz, J2 =
17,2 Hz, 2H), 6,41 (dd, J1 = 1,47 Hz, J2 = 1,72 Hz, 2H).
-
Durch
die oben beschriebenen Resultate wurde das Produkt als 8,9-Bis(acryloyloxymethyl)tricyclo[5.2.1.02,6]decan bestätigt.
-
Beispiel 6
-
7,84
g (40 mmol) DH-DOL, 6,48 g (90 mmol) Acrylsäure, 0,23 g p-Toluolsulfonsäure, 0,20
g Hydroxychinon und 4 ml Benzol wurden gemischt und unter Entfernung
von Wasser gerührt,
während
die Temperatur auf 90–106°C erhöht wurde.
Nachdem die Reaktion beendet war, wurde das Gemisch nach Zugabe
von 1,4 ml Wasser unter reduziertem Druck konzentriert. Das Gemisch
wurde unter Zusatz von 20 ml Diisopropylether extrahiert und dann
mit 5% NaCl-Lösung,
2% NaOH-Lösung
und außerdem
5% NaCl-Lösung
gewaschen. Nach Konzentrierung unter verringertem Druck wurde der
Rückstand
durch das Silicagel gereinigt, wodurch 3,5 g (Ausbeute 28,7%) 8,9-Bis(acryloyloxymethyl)tricyclo[5.2.1.02,6]decan als Zielprodukt erhalten wurden.
-
Beispiel 7
-
340
g (1,73 mol) DH-DOL wurden in 5,1 l Tetrahydrofuran (THF) gelöst und dann
wurden 526 g (5,19 mol) Triethylamin zugesetzt. 408 g (5,19 mol)
Methacryloyloxychlorid wurden nach Kühlen des Gemisches auf 0°C zugesetzt.
Nach der Beendigung der tropfenweise Zugabe wurde das Gemisch kontinuierlich
für 3 Stunden
gerührt,
während
die Temperatur allmählich
auf Raumtemperatur (25°C)
erhöht
wurde, um die Reaktion zu vervollständigen. Dann wurde das Gemisch
nach Zugabe von Wasser unter reduziertem Druck konzentriert, und THF
wurde abdestilliert. Isopropylether (IPE) wurde dem Rückstand
zugesetzt und das Produkt wurde extrahiert.
-
Diese
IPE-Lösung
wurde unter reduziertem Druck konzentriert, wodurch 550 g weiße Kristalle
erhalten wurden. Diese unreinen Kristalle wurden mit n-Hexan/Ethylacetat
= 20/1 umkristallisiert, wodurch 110,3 g weiße Kristalle mit einer Reinheit
durch Flüssigchromatographie
mit 99,1% erhalten wurden. Das Filtrat wurde nach Konzentrierung
durch die Silicagel-Säule
(SiO2: 5 kg) weiter gereinigt, wodurch 206,6
g weiße
Kristalle mit einer Reinheit von 99,3% mit Flüssigchromatographie erhalten
wurden. Dasselbe Produkt wurde nach Umkristallisieren erhalten.
Die Resultate sind unten beschrieben.
Schmelzpunkt (°C): 72–73
Massenspektrum
(FD+, m/e (%)): 332 (M+,
100), 279 (35).
1H-NMR (CDCl3, δ ppm):
1,41 (d, J = 10,3 Hz, 1H), 1,41 (d, J = 10,3 Hz, 1H), 1,52–1,56 (m,
6H), 1,70 (d, J = 10,3 Hz, 1H), 1,95 (s, 6H), 2,09 (s, 2H), 2,27
(t, J = 3,9 Hz, 2H), 2,46 (s, 2H), 4,04–4,09 (m, 2H), 4,19–4,22 (m, 2H),
5,56 (s, 2H), 6,11 (s, 2H).
-
Durch
die oben beschriebenen Resultate wurde das Produkt als 8,9-Bis(methacryloyloxymethyl)tricyclo[5.2.1.02,6]decan bestätigt.
-
-
20
g (102 mmol) 8,9-Bis(hydroxymethyl)tricyclo[5.2.1.02,6]decan
(DH-DOL), 13,47 g (204 mmol) 85%iges Kaliumhydroxid und 90 ml DMSO
wurden in einen 300 ml Vierhals-Reaktionskolben gefüllt und
auf 60°C
erhitzt. Als nächstes
wurden 43,47 g (408 mmol) 2-Chlorethylvinylether (VEC) bei 70°C tropfenweise
zugesetzt. Das Gemisch wurde dann für 3 Stunden bei 75–80°C gerührt. 6,74
g (102 mmol) 85%iges Kaliumhydroxid und 21,74 g (204 mmol) VEC wurden
außerdem
zugesetzt. Das Gemisch wurde für
eine weitere Stunde unter Erwärmen
gerührt
und das Reaktionsgemisch wurde durch Dünnschichtchromatographie untersucht, und
es wurde nur das Produkt gleichzeitig mit dem Verschwinden von Ausgangsmaterialien
identifiziert.
-
Als
nächstes
wurden Wasser und Ethylacetat zu dem Reaktionsgemisch gegeben und
die organische Schicht wurde mit Wasser gewaschen, über wasserfreiem
Magnesiumsulfat getrocknet und unter reduziertem Druck konzentriert.
Der konzentrierte Rückstand
wurde durch Silicagel-Säulenchromatographie
mit n-Hexan/Ethylacetat = 6/1–2/1
gereinigt, wodurch 27,8 g (Ausbeute 100%) an öligem Produkt erhalten wurden
(wurde später
bei Lagerung bei 5°C
fest). Die Analysenresultate dieses Produkts waren wie folgt:
Schmelzpunkt
(°C): 33–34.
Massenspektrum
(FD+, m/e (%)): 266 (M+, 100), 249 (18),
194 (13).
1H-NMR (CDCl3, δ ppm): 1,32
(d, J = 8,4 Hz, 1H), 1,49 (d, J = 9,8 Hz, 3H), 1,55–1,64 (m,
4H), 1,88 (d, J = 12,4 Hz, 2H), 2,17 (s, 1H), 2,31 (s, 1H), 2,39
(s, 2H), 3,40–3,45
(m, 2H), 3,64–3,75
(m, 5H), 3,83 (t, J = 4,0 Hz, 2H), 4,01–4,04 (m, 1H), 4,16–4,20 (m,
1H), 6,47–6,52
(m, 1H).
13C-NMR (CDCl3, δ ppm): 26,71,
26,76, 29,00, 38,09, 39,68, 41,66, 45,62, 45,72, 45,85, 45,94, 64,04,
66,79, 69,30, 73,52, 86,85, 151,56.
-
Aus
den beschriebenen Resultaten wurde das Produkt als 8-Hydroxymethyl-9-(2-vinyloxyethyl)oxymethyltricyclo[5.2.1.02,6]decan bestätigt.
-
Beispiel 9
-
1,96
g (10 mmol) DH-DOL und 20 ml DMF wurden in einen 50 ml-Vierhals-Reaktionskolben
gefüllt
und 1 g (25 mmol) 60%iges NaH wurde unter Rühren in einem Eisbad zugesetzt.
Die Temperatur dieses Gemisches wurde dann allmählich von Raumtemperatur auf
50°C erhöht und für 2 Stunden
gerührt.
2,67 g (25 mmol) VEC wurden tropfenweise zugesetzt, nachdem das
Gemisch wieder auf 0°C
wieder gekühlt
worden war. Die Temperatur wurde erneut auf Raumtemperatur eingestellt
und die Reaktion wurde dann für
3 Stunden durchgeführt.
Wasser und Ethylacetat wurden zu dem Reaktionsgemisch gegeben und
dann wurde die organische Schicht mit Wasser gewaschen und konzentriert
und der konzentrierte Rückstand
wurde durch Silicagel-Säulenchromatographie
gereinigt, wodurch 1,14 g (Ausbeute 42,9%) DH-DOLMV und 0,71 g (Rückgewinnung
36,2%) DH-DOL, das Ausgangsmaterial, isoliert wurden.
-
Beispiel 10
-
3,92
g (20 mmol) DH-DOL und 60 ml DMF wurden in einen 100 ml-Vierhals-Reaktionskolben
gefüllt und
4,8 g (120 mmol) 60%iges NaH wurden unter Rühren in einem Eisbad über 30 Minuten
zugesetzt. Die Temperatur wurde allmählich von Raumtemperatur auf
100°C erhöht und es
wurde für
18 Stunden gerührt. 8,53
g (80 mmol) VEC wurden nach erneuten Kühlen des Gemisches auf 0°C tropfenweise
zugegeben. Die Temperatur des Gemisches wurde erneut auf Raumtemperatur
eingestellt und die Reaktion wurde für 3 Stunden durchgeführt. Wasser
und Ethylacetat wurden zugegeben und die organische Schicht wurde
mit Wasser gewaschen und konzentriert und der konzentrierte Rückstand
wurde durch Silicagel-Säulenchromatographie gereinigt,
wodurch 3,68 g (Ausbeute 69,1%) DH-DOLMV und 0,82 g (Rückgewinnung
21,0%) DH-DOLO, das Ausgangsmaterial, isoliert wurden.
-
Beispiel 11
-
1,96
g (10 mmol) DH-DOL, 10 ml Toluol, 3,96 g (60 mmol) 85%iges KOH,
10 ml Wasser und 1,29 g (4 mmol) Tetrabutylammoniumbromid wurden
in einen 50 ml-Vierhals-Reaktionskolben
eingefüllt
und 4,26 g (40 mmol) VEC wurden tropfenweise unter Rühren zugesetzt.
Nachdem die Temperatur auf 70°C
erhöht
worden war und die Reaktion für
67 Stunden durchgeführt
worden war, wurden außerdem
2,13 g (20 mmol) VEC zugesetzt und es wurde für 24 Stunden umgesetzt. Nachdem
das Gemisch abgekühlt
worden war und unter Zusatz von Wasser getrennt worden war, wurde
die Wasserschicht mit Ethylacetat extrahiert, die kombinierten organischen
Schichten wurde mit Wasser gewaschen, dann mit wasserfreiem Magnesiumsulfat
dehydratisiert und unter reduziertem Druck konzentriert. Der konzentrierte
Rückstand
wurde durch Silicagel-Säulenchromatographie
mit n-Hexan/Ethylacetat = 6/1 bis 2/1 gereinigt, wodurch 1,85 g
(Ausbeute 70%) DH-DOLMV und 1,07 g (Ausbeute 28%) einer neuen Komponente
erhalten wurden. Die Analysenresultate für dieses neue ölige Produkt
sind wie unten beschrieben.
Massenspektrum (FD+, m/e (%)):
336 (M+, 100), 269 (28), 249 (19), 195 (23).
1H-NMR (CDCl3, δ ppm): 1,30
(d, J = 9,9 Hz, 1H), 1,48 (d, J = 9,0 Hz, 1H), 1,59 (dd, J1 = 12,8 Hz, J2 =
10,3 Hz, 6H), 2,05 (s, 2H), 2,16 (d, J = 11,5 Hz, 2H), 2,40 (s,
2H), 3,33 (dd, J1 = 1,5 Hz, J2 =
7,6 Hz, 2H), 3,51–3,54
(m, 2H), 3,59–3,66
(m, 4H), 3,79–3,83
(m, 4H), 3,99–4,01
(m, 2H), 4,18 (dd, J1 = 12,2 Hz, J2 = 2,1 Hz, 2H), 6,50 (dd, J1 =
7,47 Hz, J2 = 5,87 Hz, 2H).
13C-NMR (CDCl3, δ ppm): 26,78,
29,06, 37,83, 38,56, 44,98, 45,73, 67,44, 69,13, 71,96, 76,82, 86,61,
151,89.
-
Aus
den oben erhaltenen Resultaten wurde das Produkt als 8,9-Bis[(2-vinyloxyethyl)oxymethyl]tricyclo[5.2.1.02,6]decan (DH-DOLVE) bestätigt.
-
Beispiel 12
-
145
g (0,74 mmol) DH-DOL, 40 ml Toluol, 95,4 g (0,3 mmol) Tetrabutylammoniumbromid,
293 g (4,44 mmol) 85%iges KOH und 740 ml Wasser wurden in einen
2 l-Vierhalt-Reaktionskolben gefüllt
und tropfenweise wurden 315,5 g (2,96 mmol) VEC tropfenweise unter
Rühren
bei Raumtemperatur zugegeben. Die Temperatur wurde auf 70°C erhöht und es
wurde kontinuierlich für
95 Stunden gerührt.
Nach der weiteren Zugabe von 139,5 g (1,31 mmol) VEC wurde die Reaktion
für 48
Stunden durchgeführt.
Nach dem Abkühlen
wurde Wasser zugesetzt, die Toluolschicht wurde abgetrennt, die
Wasserschicht wurde mit Ethylacetat extrahiert und der Extrakt wurde
mit der Toluolschicht vermischt, dann wurde mit wasserfreiem Magnesiumsulfat
dehydratisiert, um das ölige
Produkt bei Konzentrierung zu erhalten. Nach Zugabe von 0,5 g Hydrochinon
zu diesem öligen
Produkt wurden Verunreinigungen mit niedrigem Siedepunkt durch Destillation
unter reduziertem Druck (Ölbad 106–135°C/1,6 mmHg)
entfernt, und der Rückstand
wurde durch Silicagel-Säulenchromatographie
mit n-Hexan/Ethylacetat = 6/1–2/1
gereinigt. Das erhaltene unreine DH-DOLVE und das unreine DH-DOLMV
wurden jeweils in Aceton gelöst,
mit Aktivkohle (5% Zugabe) entfärbt,
filtriert und dann wurde Aceton abdestilliert, wodurch 41 g (Ausbeute
16%) DH-DOLVE und 123 g (Ausbeute 63%) DH-DOLMV erhalten wurden.
(Stabilisator: Zusatz von etwa 100 ppm Hydrochinonmonomethylether).
-
Beispiel 13
-
1,96
g (10 mmol) DH-DOL, 3,5 ml DMSO und 1,32 g (20 mmol) 85%iges KOH
wurden in einen 50 ml-Vierhals-Reaktionskolben gegeben und es wurde
für 1 Stunde
bei 60°C
gerührt.
Nach der tropfenweisen Zugabe von 4,26 g (40 mmol) VEC mit 70°C wurde das
Gemisch für
3 Stunden bei 75–80°C gerührt. 0,32
g (1 mmol) Tetrabutylammoniumbromid, 0,66 g (10 mmol) 85%iges KOH
und 2,13 g (20 mmol) VEC wurden außerdem zugegeben und das Gemisch
wurde für
1 Stunde unter Erwärmen
gerührt.
Obgleich DH-DOL, das Ausgangsmaterial, verschwand, wurden außerdem 0,66
g (10 mmol) 85%iges KOH und 2,13 g (20 mmol) VEC zugegeben und es
wurde für
5 Stunden unter Erwärmen
gerührt.
Nach dem Kühlen
wurden Wasser und Ethylacetat zugesetzt, die organische Schicht
wurde mit Wasser gewaschen, mit wasserfreiem Magnesiumsulfat getrocknet
und konzentriert. Der konzentrierte Rückstand wurde durch Silicagel-Säulenchromatographie gereinigt,
wodurch 0,58 g (Ausbeute 17%) DH-DOLVE und 1,89 g (Ausbeute 71%)
DH-DOLMV erhalten wurden.
-
Beispiel 14
-
2,66
g (10 mmol) DH-DOLMV, 10 ml Toluol, 0,32 g (1 mmol) Tetrabutylammoniumbromid
und 0,8 g (20 mmol) NaOH, gelöst
in 5 ml Wasser, wurden in einen 50 ml-Vierhals-Reaktionskolben gegeben
und das Gemisch wurde für
1 Stunde bei 60°C
gerührt.
Als nächstes
wurden 4,26 g (40 mmol) VEC tropfenweise bei 70°C zugegeben. Die Temperatur
des Gemisches wurde auf 75–80°C erhöht und es
wurde für
3 Stunden gerührt. Nach
der weiteren Zugabe von 2,0 g (50 mmol) NaOH, wurden 4,26 g (40
mmol) VEC zugesetzt und es wurde für 3 Stunden gerührt.
-
Nachdem
die Reaktion beendet war, wurde die Toluol-Schicht abgetrennt und
mit dem Ethylacetat-Extrakt der Wasserschicht kombiniert, mit wasserfreiem
Magnesiumsulfat getrocknet, um nach Konzentrierung das ölige Produkt
zu erhalten. Das ölige
Produkt wurde durch Silicagel-Säulenchromatographie
gereinigt, wodurch 1,25 g (Ausbeute 37%) DH-DOLVE mit 95,8% Reinheit
nach Gaschromatographie erhalten wurden.
-
Beispiel 15
-
2,66
g (10 mmol) DH-DOLMV, erhalten in Beispiel 12, 10 ml DMSO und 1,32
g (20 mmol) 85%iges KOH wurden in einen 50 ml-Vierhals-Reaktionskolben gefüllt und
für eine
Stunde bei 60°C
gerührt.
Nach der weiteren Zugabe von 4,26 g (40 mmol) VEC tropfenweise bei
70°C wurde
das Gemisch für
3 Stunden bei 75–80°C gerührt. Nach
der Zugabe von 1,32 g (20 mmol) 85%iges KOH wurden weitere 4,26
g (40 mmol) VEC zugesetzt und es wurde kontinuierlich für 3 Stunden
gerührt.
Nachdem die Reaktionslösung
auf Raumtemperatur abgekühlt
war, wurde Wasser und Ethylacetat zugesetzt, und die organische
Schicht wurde mit wasserfreiem Magnesiumsulfat dehydratisiert, dann
konzentriert und der Rückstand
wurde durch Silicagel-Säulenchromatographie
gereinigt, wodurch 0,98 g (Ausbeute 29%) DH-DOLVE erhalten wurden.
-
Beispiel 16
-
(DG-DOLDA-Lichthärtung und Merkmale)
-
Eine
Probe mit der folgenden Zusammensetzung wurde hergestellt und zur
Evaluierung verwendet. 5 Gew.-Teile 1-Hydroxycyclohexylphenylketon
(produziert von Ciba Speciality Chemicals Co., Irgcure-184®)
als Lichtpolymerisationsinitiator wurden zu der Zusammensetzung
gegeben, umfassend 45 Gew.-Teile 8,9-Bis(acryloyloxymethyl)tricyclo[5.2.1.02,6]decan (DH- DOLDA), erhalten in Beispiel 5, 20 Gew.-Teile
Trimethylolpropandipropoxytriacrylat (produziert von Nihon Kayaku
K. K., TPA-320®),
30 Gew.-Teile Hydroxypivalinsäureneopentylglykol
(produziert von Nihon Kayakaku K. K., KYARAD-MANDA®) und
5 Gew.-Teile Tetrahydrofurfurylacrylat (produziert von Osak Yuki
Xo., THFA®).
Das erhaltene Gemisch wurde durch Schleuderbeschichtung auf das
Glassubstrat aufgetragen und dann mit einer Hochdruckquecksilberlampe
lichtgehärtet,
bis die Klebrigkeit vollständig
verschwunden war. Die erhaltene Beschichtung wurden auf die folgenden
Merkmale untersucht.
- 1) Klarheit; die Durchlässigkeit
bei 400 nm durch den Film mit einer Dicke von 3 μm auf dem Glassubstrat nach
Härtung
war 98%. Die Trübung
war kleiner als 0,1% und er war transparent;
- 2) Brechungsindex; 1,5097 (633 nm), Doppelbrechung 0,0000;
- 3) Bleistifthärte:
HB;
- 4) Wasserabsorptionsrate: 0,62%;
- 5) Wärmebeständigkeit:
Temperatur bei 10%iger Gewichtsreduktion: 241°C;
- 6) Elektrische Merkmale: dielektrische Konstante 3,25 (1 KHz),
dielektrischer Dissipationsfaktor 0,0342 (1 kHz), spezifischer Durchgangswiderstand
5 × 10
15 Ω·cm;
- 7) Klebeeigenschaften: Cellophanband-Abziehtest wurde nach einem
Gitterschnitt auf einem bloßen
Silicium-Substrat
durchgeführt,
es wurde aber kein Ablösen
beobachtet.
-
Beispiel 17
-
(Synthese von DH-DOLMV-Homopolymer)
-
Unter
Stickstoffgasatmosphäre
wurden 2 g 8-Hydroxymethyl-9-(2-vinyloxyethyl)oxymethyl-tricyclo[5.2.1.02,6]decan (DH-DOLMV) zu 10 Gew.-% in Diglyme (Diethylenglykoldimethylether)
gelöst.
Drei Tropfen Bortrifluoridether-Lösung als Kationenpolymerisationsinitiator
wurden mit einer Pipette in diese Lösung gegeben und es wurde eine
Kationenpolymerisation bei 70°C
für 24
Stunden unter Erwärmen
und konstantem Rühren
durchgeführt.
Die Molekulargewichtsverteilung des erhaltenen Polymers mit Gelfiltrationschromatographie (Polystyrol-Umwandlung) wurde
bestätigt
und das zahlenmittlere Molekulargewicht (Mn) war 8543, das gewichtsmittlere
Molekulargewicht (Mw) war 27 337 und das Verhältnis Ms/Mn war 3,2.
-
Beispiel 18
-
(Synthese von DH-DOLMA und Acrylsäuremethyl-Copolymer)
-
Unter
Stickstoffgasatmosphäre
wurden 2,5 g 8-Acryloyloxymethyl-9-hydroxymethyltricyclo[5.2.1.02,6]decan (DH-DOLMA) und 2,5 g Acrylsäuremethyl
in 20 g Cyclohexanon gelöst.
0,1 g 2,2'-Azobis(isobutyronitril)
wurden als Polymerisationsinitiator in die Lösung gegeben und es wurde eine
Radikalpolymerisation bei 70°C
für 24
Stunden unter Erwärmen
und konstantem Rühren
durchgeführt.
Die Molekulargewichtsverteilung des erhaltenen Polymers wurde mit
Gelfiltrationschromatographie (Polystyrol-Umwandlung) bestätigt, und
Mn war 7841, Mw war 29 012 und das Verhältnis Mw/Mn war 3,7. Die Umwandlungsrate
Monomer zu Polymer wurde durch Gaschromatgraphie verfolgt, allerdings
bleibt die Umwandlungsrate nahezu dieselbe und es wurde ein statistisches
Copolymer erhalten.
-
Beispiel 19
-
(Synthese von DH-DOLMA-Homopolymer und
seine Eigenschaften)
-
Unter
Stickstoffgasatmosphäre
wurde 2 g 8-Acryloyloxymethyl-9-hydroxymethyltricyclo[5.2.1.02,6]decan (DH-DOLMA) einer Lösung, bestehend
aus 2-Heptanon und Propylglykolmonomethyletheracetat im Gewichtsverhältnis 1:1,
gelöst.
0,16 g 2,2'-Azobis(isobutyronitril)
wurden als Radikalpolymerisationsinitiator in die Lösung gegeben
und es wurde eine Radikalpolymerisation bei 70°C für 24 Stunden unter Erwärmen und
konstanten Rühren
durchgeführt.
Die Molekulargewichtsverteilung des erhaltenen Polymers wurde mit
Gelfiltrationschromatographie (Polystyrolumwandlung) bestätigt und
Mn war 2434, Mw war 5841 und das Verhältnis Mw/Mn war 2,4. Spezielle
Eigenschaften, wie sie unten beschrieben sind, wurden für das erhaltenen
Polymer untersucht.
- 1) Brechungsindex: 1,536
(633 nm);
- 2) Wasserabsorptionsrate: 0,61%;
- 3) Wärmebeständigkeit:
Temperatur bei 10%iger Gewichtsreduktion: 251°C;
- 4) Glasübergangstemperatur:
bis zu 150°C
mit Differentialscanningkalorimetriemessung nicht detektiert;
- 5) Klebeeigenschaften: Cellophanband-Abziehtest wurde nach einem
Gitterschnitt auf dem reinen Siliciumsubstrat durchgeführt, allerdings
wurde kein Ablösen
beobachtet.
-
[Industrielle Anwendbarkeit]
-
Die
alicyclische Verbindung der vorliegenden Erfindung ist eine industriell
nützliche
Verbindung als ein Monomer für
ein härtbares
Harz, und die härtbare
Harzzusammensetzung, die die Verbindung darin hat, ist geeignet
als Resist-Ausgangsmaterialien,
und zwar wegen der Trockenätzbeständigkeit
und der Verbesserung bei den Klebeeigenschaften, als optische Ausgangsmaterialien
und Disk-Überzugsausgangsmaterialien
wegen der Verbesserung der Klarheit, der niedrigen Doppelbrechung
und der niedrigen Wasserabsorptionsrate und auch als Beschichtungausgangsmaterialien,
zum Beispiel UV·EB-Härten des Überzugs,
und zwar wegen der Reaktivität,
der Verschleißbeständigkeit
und Wasserbeständigkeit.
 worin R1 Wasserstoff oder C1-4-Alkyl ist; und R2 und R3 unabhängig Wasserstoff oder C1-10-Alkyl sind; oder eine 2-Vinyloxyethyl-Gruppe gezeigt in Formel (3) unten sind:
worin R1 Wasserstoff oder C1-4-Alkyl ist; und R2 und R3 unabhängig Wasserstoff oder C1-10-Alkyl sind; oder eine 2-Vinyloxyethyl-Gruppe gezeigt in Formel (3) unten sind:




 worin R1 Wasserstoff oder C1-4-Alkyl ist; und R2 und R3 unabhängig Wasserstoff oder C1-10-Alkyl sind; oder eine 2-Vinyloxyethyl-Gruppe gezeigt in Formel [3] unten sind:
worin R1 Wasserstoff oder C1-4-Alkyl ist; und R2 und R3 unabhängig Wasserstoff oder C1-10-Alkyl sind; oder eine 2-Vinyloxyethyl-Gruppe gezeigt in Formel [3] unten sind:




 worin R1, R2, R3 und die gepunktete Linie dieselbe Bedeutung wie oben haben; R4 Wasserstoff oder C1-10-Alkyl ist und X Halogen ist.
worin R1, R2, R3 und die gepunktete Linie dieselbe Bedeutung wie oben haben; R4 Wasserstoff oder C1-10-Alkyl ist und X Halogen ist.


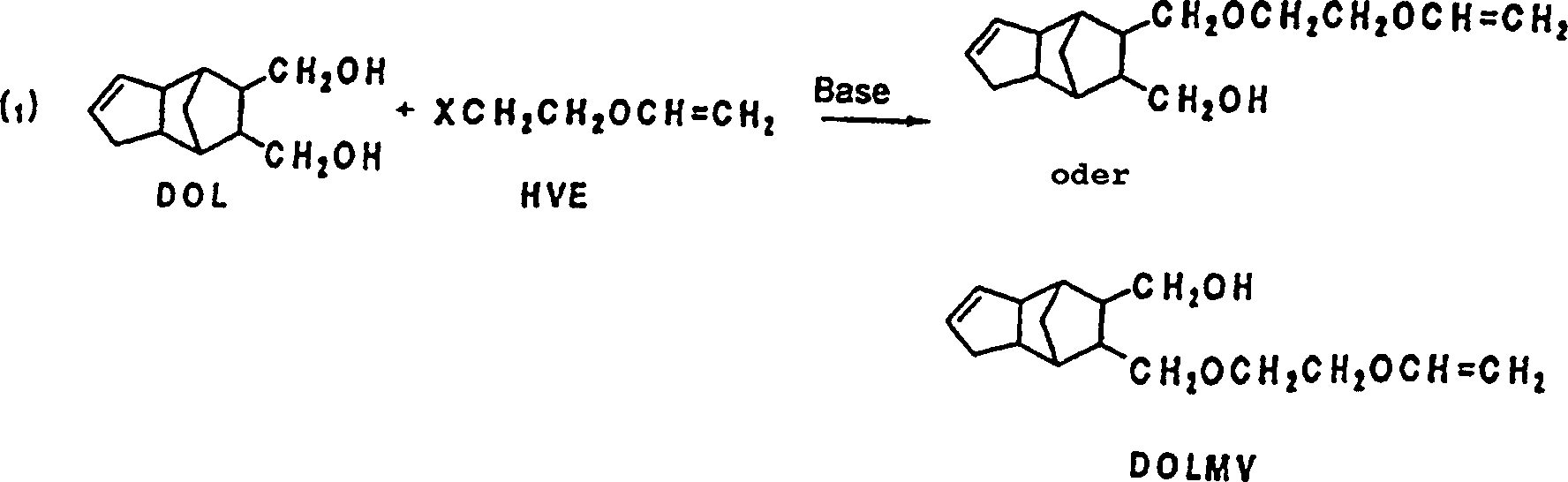 worin X ein Halogenatom ist.
worin X ein Halogenatom ist.






 worin R1 Wasserstoff oder C1-C4-Alkyl ist; und R2 und R3 unabhängig Wasserstoff oder C1-C10-Alkyl sind; oder eine 2-Vinyloxyethylgruppe gezeigt in Formel (3) unten sind:
worin R1 Wasserstoff oder C1-C4-Alkyl ist; und R2 und R3 unabhängig Wasserstoff oder C1-C10-Alkyl sind; oder eine 2-Vinyloxyethylgruppe gezeigt in Formel (3) unten sind:
 worin R1 Wasserstoff oder C1-C4-Alkyl ist; R2 und R3 unabhängig Wasserstoff oder C1-C10-Alkyl sind; und die gepunktete Linie eine Einfachbindung oder eine Doppelbindung ist.
worin R1 Wasserstoff oder C1-C4-Alkyl ist; R2 und R3 unabhängig Wasserstoff oder C1-C10-Alkyl sind; und die gepunktete Linie eine Einfachbindung oder eine Doppelbindung ist.
 worin R1 Wasserstoff oder C1-C4-Alkyl ist; R2 und R3 unabhängig Wasserstoff oder C1-C10-Alkyl sind; und die gepunktete Linie eine Einfachbindung oder eine Doppelbindung ist.
worin R1 Wasserstoff oder C1-C4-Alkyl ist; R2 und R3 unabhängig Wasserstoff oder C1-C10-Alkyl sind; und die gepunktete Linie eine Einfachbindung oder eine Doppelbindung ist.


 worin R1 Wasserstoff oder C1-C4-Alkyl ist; R2 und R3 unabhängig Wasserstoff oder C1-C10-Alkyl sind; oder eine 2-Vinyloxyethylgruppe gezeigt in Formel (3) unten sind:
worin R1 Wasserstoff oder C1-C4-Alkyl ist; R2 und R3 unabhängig Wasserstoff oder C1-C10-Alkyl sind; oder eine 2-Vinyloxyethylgruppe gezeigt in Formel (3) unten sind: