-
Technisches Gebiet
-
Die
vorliegende Erfindung ist auf dem medizinischen Gebiet nützlich.
Insbesondere sind neue Imidazolinverbindungen dieser Erfindung als
Neuropeptid-Y-Rezeptor-Antagonisten und als Mittel für die Behandlung
von verschiedenen Arten von cardiovaskulären Störungen, Störungen des zentralen Nervensystems,
metabolischen Erkrankungen oder dergleichen verwendbar.
-
Technischer Hintergrund
-
Neuropeptid
Y (nachstehend als NPY bezeichnet), ein Peptid, das aus 36 Aminosäuren besteht,
wurde zuerst aus Schweinehirn von Tatemoto et al. 1982 (Nature,
296: 659 (1982)) isoliert. NPY ist im zentralen Nervensystem und
dem peripheren Nervensystem breit verteilt und als eines der am
reichlichsten vorhandenen Peptide im Nervensystem spielt es verschiedene
Rollen. Das heißt,
NPY wirkt als orexigene Substanz im zentralen Nervensystem und fördert stark
die Fettakkumulation über
die Sekretion von verschiedenen Hormonen oder die Wirkung des Nervensystems.
Es ist bekannt, dass die kontinuierliche intrazerebroventrikuläre Verabreichung
von NPY Fettsucht und Insulinresistenz, die auf diesen Wirkungen
basieren, induziert (International Journal of Obesity, Band 19:
517 (1995); Endocrinology, Band 133: 1753 (1993)). Es ist ebenfalls
bekannt, dass NPY zentrale Wirkungen, wie Depression, Angstzustand,
Schizophrenie, Schmerz, Demenz oder dergleichen, hat (Drugs, Band
52: 371 (1996)). Weiterhin existiert in der Peripherie NPY gleichzeitig
mit Norepinephrin in sympatischer Nervenendigung und ist in die
Tonizität
des sympatischen Nervensystems einbezogen. Es ist bekannt, dass
periphere Verabreichung von NPY Vasokonstriktion verursacht und
die Wirkungen von anderen vasokonstriktiven Substanzen, wie Norepinephrin,
verstärkt
(British Journal of Pharmacology, Band 95: 419 (1988)). Es wird
auch berichtet, dass NPY in die Erhöhung von Herzhypertrophie im
Ergebnis der Beschleunigung des sympatischen Nervensystems einbezogen
ist (Proceeding National Academic Science USA, Band 97: 1595 (2000)).
-
Weiterhin
wird berichtet, dass NPY auch in die sekretorische Funktion von
Sexualhormonen und Wachstumshormon, sexuelle und reproduktive Funktion,
gastrointestinale Motilität,
Bronchokonstriktion, Entzündung
und Alkoholbevorzugung einbezogen ist (Life Science, Band 55: 551
(1994); The Journal of Allergy and Immunology, Band 101: S345 (1998);
Nature, Band 396: 366 (1998)).
-
NPY
hat eine Vielzahl von pharmakologischen Wirkungen, die sich aus
seinem Binden an gewisse Rezeptoren ergeben, an die Peptid YY und
pankreatisches Polypeptid, welche ähnlich zu NPY sind, ebenfalls
binden. Es ist bekannt, dass diese pharmakologischen Wirkungen durch
die Wirkung von mindestens fünf
Rezeptoren mit oder ohne synergistische Wechselwirkungen verursacht
und vermittelt werden (Trends in Neuroscience, Band 20: 294 (1997)).
-
Es
wird mitgeteilt, dass die zentrale Wirkung, die durch NPY Y1 Rezeptor
vermittelt wird, die starke orexigene Wirkung einschließt (Endocrinology,
Band 137: 3177 (1996); Endocrinology, Band 141: 1011 (2000)). Weiterhin
wird mitgeteilt, dass NPY Y1 Rezeptor in Angstzustand und Schmerz
einbezogen ist (Nature, Band 259: 528 (1993); Brain research, Band
859: 361 (2000)). Außerdem
werden auch Pressorwirkungen, die durch die starke Wirkung von Vasokonstriktion
in der Peripherie vermittelt werden, mitgeteilt. (FEBS Letters,
Band 362: 192 (1995); Nature Medicine, Band 4: 722 (1998)).
-
Es
ist bekannt, dass durch NPY Y2 Rezeptor vermittelte Wirkungen die
inhibitorische Wirkung auf die Freisetzung von verschiedenen Neurotransmittern
in den Nervenendigungen einschließen (British Journal of Pharmacology,
Band 102: 41 (1991); Synapse, Band 2: 299 (1988)). In der Peripherie
verursacht NPY Konstriktion von Blutgefäß oder Vas deferens, direkt
oder durch Steuern dieser Neurotransmitter (The Journal of Pharmacology
and Experimental Therapeutics, Band 261: 863 (1992); British Journal
of Pharmacology, Band 100: 190 (1990)). Zusätzlich ist die Inhibierung
von Lipolyse in adipösen
Geweben bekannt (Endocrinology, Band 131: 1970 (1992)). Weiterhin
wird die Inhibierung von Ionensekretion im Gastrointestinaltrakt
mitgeteilt (British Journal of Pharmacology, Band 101: 247 (1990).
-
Andererseits
wird die Wirkung auf die Funktionen des zentralen Nervensystems,
wie Gedächtnis
und Angstzustand, ebenfalls mitgeteilt (Brain Research, Band 503:
73 (1989); Peptides, Band 19: 359 (1998)).
-
Es
wird berichtet, dass NPY Y3 Rezeptor hauptsächlich im Hirnstamm und im
Herzen exprimiert wird und es wird zur Regulierung von Blutdruck
und Herzfrequenz freigesetzt (The Journal of Pharmacology and Experimental
Therapeutics, Band 258: 633 (1991); Peptides, Band 11: 545 (1990)).
Weiterhin ist bekannt, dass der NPY Y3 Rezeptor in die Steuerung
der Catecholaminsekretion in der adrenalen Drüse einbezogen ist (The Journal
of Pharmacology and Experimental Therapeutics, Band 244: 468 (1988);
Life Science, Band 50: PL7 (1992)).
-
NPY
Y4 Rezeptor hat eine hohe Affinität, insbesondere für pankreatisches
Polypeptid, und hat pharmakologische Wirkungen auf die Inhibierung
von pankreatischer exokriner Sekretion und der gastrointestinalen
Motilität
(Gastroenterology, Band 85: 1411 (1983)). Weiterhin wird berichtet,
dass NPY die Sekretion des Sexualhormons im zentralen Nervensystem
erhöht
(Endocrinology, Band 140: 5171 (1999)).
-
Die
durch NPY Y5 Rezeptor vermittelten Wirkungen schließen die
starke fettakkumulierende Wirkung, einschließlich den orexigenen Wirkung,
ein (Nature, Band 382: 168 (1996); American Journal of Physiology, Band
277: R1428 (1999)). Es wird berichtet, dass der NPY Y5 Rezeptor
auch die Wirkungen des zentralen Nervensystems, wie Anfälle und
Epilepsie, oder Schmerz und die Morphinentzugssymptome vermittelt
(Nature Medicine, Band 3: 761 (1997); Proceeding National Academic
Science, USA, Band 96: 13518 (1999); The Journal of Pharmacology
and Experimental Therapeutics, Band 284: 633 (1998)). Es wird berichtet,
dass in der Peripherie NPY Y5 Rezeptor in diuretische Wirkung und
hypoglycämischen
Effekt einbezogen ist (British Journal of Pharmacology, Band 120:
1335 (1998); Endocrinology, Band 139: 3018 (1998)). Es wird auch
berichtet, dass NPY Herzhypertrophie im Ergebnis der Beschleunigung
des sympatischen Nervensystems verstärkt (Proceeding National Academic
Science USA, Band 97: 1595 (2000)).
-
Die
Funktion von NPY wird durch Binden der NPY-Rezeptoren, die im zentralen oder peripheren
Nervensystem vorliegen, verursacht. Deshalb kann die Expression
der Wirkung von NPY durch Blockieren des Bindens von NPY an NPY-Rezeptoren gedämpft werden.
Substanzen, die dem NPY-Binden an NPY-Rezeptoren entgegenwirken,
können
für die
Prophylaxe oder Behandlung von verschiedenen Erkrankungen, die NPY
betreffen, wie cardiovaskuläre
Störungen,
beispielhaft angegeben durch Bluthochdruck, Nephropathie, Herzerkrankungen
und Vasospasmus, Störungen
des zentralen Nervensystems, beispielhaft angegeben durch Bulimie,
Depression, Angstzustand, Konvulsion, Epilepsie, Demenz, Schmerz,
Alkoholismus und Drogenentzug, metabolische Erkrankungen, beispielhaft
angegeben durch Fettsucht, Diabetes und Hormonanormalität, sexuale
und reproduktive Fehlfunktion, gastrointestinale Motilitätsstörung, Atmungstraktstörung, Entzündung oder
Glaukom oder dergleichen, nützlich
sein (Trends in Pharmacological Science, 15: 153 (1994); Life Science,
55: 551 (1994); Drugs, Band 52: 371 (1996); The Journal of Allergy
and Immunology, Band 101: Seite 345 (1998); Nature, Band 396: 366
(1998); The Journal of Pharmacology and Experimental Therapeutics,
Band 284: 633 (1998); Trends in Pharmacological Science, Band 20: 104
(1999); Proceeding National Academic Science USA, Band 97: 1595
(2000)).
-
Kürzlich hat
die Untersuchung der Autoren der vorliegenden Erfindung ergeben,
dass eine gewisse Art von NPY-Rezeptor-Antagonist
bei der Prophylaxe oder Behandlung von Hypercholesterolämie, Hyperlipidämie und
Arteriosklerose nützlich
ist (Internationale Anmeldungsveröffentlichung WO99/27965).
-
Imidazolonderivate
werden in der Internationalen Anmeldungsveröffentlichung WO99/48888 offenbart,
und es wird beschrieben, dass diese Verbindungen NPY-Rezeptor-Antagonisten-Wirksamkeit
aufweisen. Jedoch werden die erfindungsgemäßen Verbindungen weder offenbart
noch vorgeschlagen.
-
2,4,4-Triphenyl-2-imidazolin
wird in CA 52:17240f von Chemical Abstracts beschrieben; jedoch
werden weder NPY-Rezeptor-Antagonisten-Wirksamkeiten
der Verbindung offenbart, noch vorgeschlagen.
-
Offenbarung der Erfindung
-
Die
Aufgabe der vorliegenden Erfindung ist es, neue Arzneistoffe mit
NPY-antagonistischen Wirksamkeiten bereitzustellen.
-
Die
Erfinder haben gefunden, dass die durch die allgemeine Formel (I)
wiedergegebenen Verbindungen
worin Ar
1,
Ar
2 und Ar
3 unabhängig Aryl
oder Heteroaryl, die substituiert sein können, wobei der Substituent
ausgewählt
ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl,
Halogen(nieder)alkyl, Hydroxy(nieder)alkyl, Cyclo(nieder)alkyl(nieder)alkyl,
Niederalkenyl, Niederalkylamino, Diniederalkylamino, Niederalkanoylamino,
Niederalkylsulfonylamino, Arylsulfonylamino, Hydroxy, Niederalkoxy,
Halogen(nieder)alkoxy, Aryloxy, Heteroaryloxy, Niederalkylthio,
Carboxyl, Formyl, Niederalkanoyl, Niederalkoxycarbonyl, Carbamoyl,
Niederalkylcarbamoyl, Diniederalkylcarbamoyl, Niederalkylsulfonyl,
Arylsulfonyl, Aryl und Heteroaryl, wiedergeben;
n 0, 1 oder
2 wiedergibt;
R
1 Cycloniederalkyl wiedergibt
oder eine Gruppe darstellt, die durch die nachstehende Formel von
-Ar
3 wiedergegeben wird;
R
2 und R
3 unabhängig Wasserstoffatom,
Cyclo(nieder)alkyl, Niederalkenyl oder Niederalkyl, die substituiert sein
können,
wobei der Substituent ausgewählt
ist aus der Gruppe, bestehend aus Halogenatom, Niederalkylamino,
Diniederalkylamino, Niederalkanoylamino, Hydroxy, Niederalkoxy,
Formyl, Niederalkoxycarbonyl, Niederalkylcarbamoyl und Diniederalkylcarbamoyl,
wiedergeben;
sowohl R
10 als auch R
11 Wasserstoffatom wiedergeben oder sie sich
vereinigen, um Oxo wiederzugeben;
R
12 Wasserstoffatom
oder Niederalkyl wiedergibt;
X und Y unabhängig Methylen, Ethenylen, eine
durch die Formel -NR
12- wiedergegebene Gruppe,
Sauerstoffatom oder Schwefelatom wiedergeben;
Z Methin oder
Stickstoffatom wiedergibt (jedoch im Fall, dass sowohl R
2 als auch R
3 Wasserstoffatom
wiedergeben, Ar
1, Ar
2 und
R
1 nicht gleichzeitig eine unsubstituierte
Phenylgruppe wiedergeben); NPY-antagonistische Wirksamkeiten zeigen
und in der Pharmakokinetik ausgezeichnet sind, wie dem Eindringen
im Hirn und die zerebrospinale Flüssigkeit, und haben die vorliegende
Erfindung ausgeführt.
-
Die
erfindungsgemäßen Verbindungen
(I) sind als Mittel für
die Behandlung von verschiedenen Erkrankungen, die NPY betreffen,
das heißt
beispielsweise cardiovaskuläre
Störungen,
beispielhaft angegeben durch Bluthochdruck, Nephropathie, Herzkrankheiten,
Vasospasmus und Arteriosklerose, Störungen des zentralen Nervensystems,
beispielhaft angegeben durch Bulimie, Depression, Angstzustand,
Convulsion, Epilepsie, Demenz, Schmerz, Alkoholismus und Drogenentzug,
metabolische Erkrankungen, beispielhaft angegeben durch Fettsucht,
Diabetes, Hormonanormalität,
Hypercholesterolämie
und Hyperlipidämie,
sexuale und reproduktive Fehlfunktion, gastrointestinale Störung, wie
Inhibierung der Gastrointestinalmotilität, Atmungsstörung, Entzündung oder
Glaukom, oder dergleichen, nützlich,
da sie NPY-antagonistische Wirksamkeiten zeigen und in der inneren
Kinetik, wie intrazerebraler Übergang
oder Übergang
zu zerebrospinaler Flüssigkeit, ausgezeichnet
sind.
-
Insbesondere
sind die erfindungsgemäßen Verbindungen
(I) als Mittel für
die Behandlung von Bulimie, Fettsucht, Diabetes oder dergleichen
verwendbar.
-
Die
vorliegende Erfindung betrifft durch die allgemeine Formel (I) wiedergegebene
Verbindungen, die Salze oder Ester davon, und das Herstellungsverfahren
dafür sowie
deren Verwendung.
-
Weiterhin
betrifft die vorliegende Erfindung die Zwischenprodukte zum Herstellen
der durch die allgemeine Formel (I) wiedergegebenen Verbindungen.
Das heißt,
die vorliegende Erfindung betrifft die durch die allgemeine Formel
(II') wiedergegebenen
Verbindungen:
worin Ar
1p und
Ar
3p unabhängig Aryl oder Heteroaryl,
die substituiert sein können,
wobei der Substituent ausgewählt
ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl,
Halogen(nieder)alkyl, Cyclo(nieder)alkyl(nieder)alkyl, Niederalkenyl,
Di(nieder)alkylamino, Nie deralkoxy, Halogen(nieder)alkoxy, Aryloxy,
Heteroaryloxy, Niederalkylthio, Formyl, Niederalkanoyl, Niederalkoxycarbonyl,
Di(nieder)alkylcarbamoyl, Niederalkylsulfonyl, Arylsulfonyl, Aryl,
Heteroaryl, und einer gegebenenfalls geschützten Hydroxy(nieder)alkyl-,
Niederalkylamino-, Niederalkanoylamino-, Niederalkylsulfonylamino-,
Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder Niederalkylcarbamoylgruppe,
wiedergeben;
n 0, 1 oder 2 wiedergibt;
R
1p Cycloniederalkyl
oder eine Gruppe, die durch die Formel Ar
3p wiedergegeben
wird, wiedergibt; oder
R
2p und R
3p unabhängig Wasserstoffatom,
Cyclo(nieder)alkyl, Niederalkenyl oder Niederalkyl, die substituiert sein
können,
wobei der Substituent ausgewählt
ist aus der Gruppe, bestehend aus Halogenatom, Di(nieder)alkylamino,
Niederalkoxy, Formyl, Niederalkoxycarbonyl, Di(nieder)alkylcarbamoyl,
und einer gegebenenfalls geschützten
Niederalkylamino-, Niederalkanoylamino-, Hydroxy- oder Niederalkylcarbamoylgruppe,
wiedergeben;
sowohl R
10p als auch R
11p Wasserstoffatom wiedergeben oder sie
sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben;
R
12p Wasserstoffatom, Niederalkyl oder eine
Schutzgruppe für
eine Iminogruppe wiedergibt;
X
p und
Y
p unabhängig
Methylen, Ethenylen, eine durch die Formel -NR
12p-
wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben;
Z
Methin oder Stickstoffatom wiedergibt; (jedoch (a) im Fall, dass
sowohl R
2p als auch R
3p Wasserstoffatom wiedergeben,
Ar
1p und R
1p nicht
gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben, und
(b), im Fall, dass R
2p Wasserstoffatom darstellt
und R
3p Methyl, Isopropyl, Isobutyl oder
tert-Butyl darstellt, Ar
1p und R
1p nicht gleichzeitig 4-Methoxyphenyl wiedergeben).
-
Die
Bedeutungen der in der vorliegenden Beschreibung verwendeten Begriffe
werden definiert und die genauere Beschreibung dieser Erfindung
wird nachstehend gezeigt.
-
„Halogenatom" bedeutet Fluoratom,
Chloratom, Bromatom oder Jodatom.
-
„Niederalkyl" bedeutet eine gerad-
oder verzweigtkettige Alkylgruppe von C1 bis
C6 und schließt beispielsweise Methyl, Ethyl,
Propyl, Isopropyl, Butyl, Isobutyl, sec-Butyl, tert-Butyl, Pentyl,
Isopentyl, Hexyl, Isohexyl oder dergleichen ein.
-
„Halogen(nieder)alkyl" bedeutet das vorstehend
angeführte
Niederalkyl, substituiert mit 1 oder nicht weniger als 2, vorzugsweise
1 bis 3 der vorstehend erwähnten
Halogenatome, gleich oder verschieden, an den substituierbaren,
willkürlichen
Positionen, und schließt
beispielsweise Fluormethyl, Difluormethyl, Trifluormethyl, 2-Fluorethyl,
1,2-Difluorethyl, Chlormethyl, 2-Chlorethyl, 1,2-Dichlorethyl, Bromethyl,
Jodmethyl, oder dergleichen ein.
-
„Hydroxy(nieder)alkyl" bedeutet das vorstehend
angeführte
Niederalkyl, substituiert mit 1 oder nicht weniger als 2, vorzugsweise
1 oder 2 Hydroxygruppen an den substituierbaren, willkürlichen
Positionen, und schließt
beispielsweise Hydroxymethyl, 2-Hydroxyethyl, 1-Hydroxy-1-methylethyl,
1,2-Dihydroxyethyl,
3-Hydroxypropyl, oder dergleichen ein.
-
„Cyclo(nieder)alkyl" bedeutet eine Cycloalkylgruppe
von C3 bis C6 und
schließt
Cyclopropyl, Cyclobutyl, Cyclopentyl und Cyclohexyl ein.
-
„Cyclo(nieder)alkyl(nieder)alkyl" bedeutet das vorstehend
angeführte
Niederalkyl, substituiert mit 1 oder nicht weniger als 2 Cyclo(nieder)alkylgruppen,
vorzugsweise 1 vorstehend angeführte
Cyclo(nieder)alkylgruppe an den substituierbaren, willkürlichen
Positionen, und schließt
beispiels weise Cyclopropylmethyl, 2-Cyclopropylethyl, 3-Cyclopropylpropyl,
Cyclobutylmethyl, 2-Cyclobutylethyl, 3-Cyclobutylpropyl, Cyclopentylmethyl,
2-Cyclopentylethyl, 3-Cyclopentylpropyl, Cyclohexylmethyl, 2-Cyclohexylethyl,
3-Cyclohexylpropyl, oder dergleichen ein.
-
„Niederalkenyl" bedeutet eine gerad-
oder verzweigtkettige Alkenylgruppe von C2 bis
C6 und schließt beispielsweise Vinyl, 1-Propenyl,
2-Propenyl, Isopropenyl, 3-Butenyl, 2-Butenyl, 1-Butenyl, 1-Methyl-2-propenyl,
1-Methyl-1-propenyl, 1-Ethyl-1-ethenyl, 2-Methyl-2-propenyl, 2-Methyl-1-propenyl, 3-Methyl-2-butenyl, 4-Pentenyl,
oder dergleichen ein.
-
„Niederalkylamino" bedeutet eine Aminogruppe,
monosubstituiert mit der vorstehend angeführten Niederalkylgruppe, und
schließt
beispielsweise Methylamino, Ethylamino, Propylamino, Isopropylamino,
Butylamino, sec-Butylamino, tert-Butylamino,
oder dergleichen ein.
-
„Diniederalkylamino" bezieht sich auf
eine Aminogruppe, disubstituiert mit gleichem oder verschiedenem,
vorstehend angeführtem
Niederalkyl, und schließt
beispielsweise Dimethylamino, Diethylamino, Ethylmethylamino, Dipropylamino,
Methylpropylamino, Diisopropylamino, oder dergleichen ein.
-
„Niederalkanoyl" bedeutet eine Alkanoylgruppe,
die das vorstehend angeführte
Niederalkyl enthält; das
heißt,
eine Alkanoylgruppe von C2 bis C7, und schließt beispielsweise Acetyl, Propionyl,
Butyryl, Isobutyryl, Valeryl, Isovaleryl, Pivaloyl, oder dergleichen
ein.
-
„Niederalkanoylamino" bedeutet eine Aminogruppe,
monosubstituiert mit dem vorstehend angeführten Niederalkanoyl, und schließt beispielsweise
Acetylamino, Propionylamino, Butyrylamino, Isobutyrylamino, Valerylamino,
Isovalerylamino, Pivaloylamino, oder dergleichen ein.
-
„Niederalkylsulfonyl" bedeutet eine Alkylsulfonylgruppe,
die das vorstehend angeführte
Niederalkyl enthält,
und schließt
beispielsweise Methylsulfonyl, Ethylsulfonyl, Propylsulfonyl, Isopropylsulfonyl,
Butylsulfonyl, sec-Butylsulfonyl, tert-Butylsulfonyl, oder dergleichen
ein.
-
„Niederalkylsulfonylamino" bedeutet eine Aminogruppe,
monosubstituiert mit dem vorstehend erwähnten Niederalkylsulfonyl und
schließt
beispielsweise Methylsulfonylamino, Ethylsulfonylamino, Propylsulfonylamino,
Isopropylsulfonylamino, Butylsulfonylamino, sec-Butylsulfonylamino,
tert-Butylsulfonylamino, oder dergleichen ein.
-
„Aryl" schließt beispielsweise
Phenyl, Naphthyl oder dergleichen ein.
-
„Arylsulfonyl" bedeutet eine Arylsulfonylgruppe,
die das vorstehende Aryl enthält,
und schließt
beispielsweise Phenylsulfonyl, 1-Naphthylsulfonyl, 2-Naphthylsulfonyl
oder dergleichen ein.
-
„Arylsulfonylamino" bedeutet eine Aminogruppe,
monosubstituiert mit dem vorstehend angeführten Arylsulfonyl, und schließt beispielsweise
Phenylsulfonylamino, 1-Naphthylsulfonylamino, 2-Naphthylsulfonylamino
oder dergleichen ein.
-
„Niederalkoxy" bedeutet eine gerad-
oder verzweigtkettige Alkoxygruppe von C1 bis
C6 und schließt beispielsweise Methoxy,
Ethoxy, Propoxy, Isopropoxy, Butoxy, sec-Butoxy, Isobutoxy, tert-Butoxy,
Pentyloxy, Isopentyloxy, Hexyloxy, Isohexyloxy oder dergleichen
ein.
-
„Halogen(nieder)alkoxy" bezieht sich auf
das vorstehend angeführte
Niederalkoxy, substituiert mit 1 oder nicht weniger als 2, vorzugsweise
1 bis 3, der vorstehend angeführten
Halogenatome, gleich oder verschieden, an den substituierbaren,
willkürlichen
Positionen, und schließt
beispielsweise Fluormethoxy, Difluormethoxy, Trifluormethoxy, 2-Fluorethoxy, 1,2-Difluorethoxy,
Chlormethoxy, 2-Chlorethoxy, 1,2-Dichlorethoxy, Brommethoxy, Jodmethoxy
oder dergleichen ein.
-
„Aryloxy" bedeutet eine Aryloxygruppe,
die das vorstehend angeführte
Aryl enthält,
und schließt
beispielsweise Phenoxy, 1-Naphthoxy, 2-Naphthoxy oder dergleichen
ein.
-
„Niederalkylthio" bedeutet eine gerad-
oder verzweigtkettige Alkylthiogruppe von C1 bis
C6 und schließt beispielsweise Methylthio,
Ethylthio, Propylthio, Isopropylthio, Butylthio, sec-Butylthio,
Isobutylthio, tert-Butylthio, Pentylthio, Isopentylthio, Hexylthio,
Isohexylthio oder dergleichen ein.
-
„Niederalkoxycarbonyl" bedeutet eine Alkoxycarbonylgruppe,
die die vorstehend erwähnte
Niederalkoxygruppe enthält;
das heißt,
eine Alkoxycarbonylgruppe von C2 bis C7, und schließt beispielsweise Methoxycarbonyl,
Ethoxycarbonyl, Propoxycarbonyl, Isopropoxycarbonyl, Butoxycarbonyl,
Isobutoxycarbonyl, tert-Butoxycarbonyl, Pentyloxycarbonyl oder dergleichen
ein.
-
„Niederalkylcarbamoyl" bezieht sich auf
eine Carbamoylgruppe, monosubstituiert an dem vorstehend angeführten Niederalkyl,
und schließt
beispielsweise Methylcarbamoyl, Ethylcarbamoyl, Propylcarbamoyl,
Isopropylcarbamoyl, Butylcarbamoyl, sec-Butylcarbamoyl, tert-Butylcarbamoyl
oder dergleichen ein.
-
„Diniederalkylcarbamoyl" bedeutet eine Carbamoylgruppe,
disubstituiert mit dem vorstehend angeführten Niederalkyl, und schließt beispielsweise
Dimethylcarbamoyl, Diethylcarbamoyl, Ethylmethylcarbamoyl, Dipropylcarbamoyl,
Methylpropylcarbamoyl, Diisopropylcarbamoyl oder dergleichen ein.
-
„Heteroaryl" bedeutet eine 5-
oder 6-gliedrige, monocyclische heteroaromatische Gruppe, die 1
oder nicht weniger als 2, vorzugsweise 1 bis 3, Heteroatome, gleich
oder verschieden, ausgewählt
aus der Gruppe von Sauerstoffatom, Stickstoffatom und Schwefelatom,
enthält,
oder „Heteroaryl" bedeutet eine kondensierte, cyclische
heteroaromatische Gruppe, worin die vorstehend angeführte monocyclische
heteroaromatische Gruppe mit der vorstehend angeführten Arylgruppe
kondensiert ist, oder die gleiche oder verschiedene, vorstehend
angeführte,
monocyclische heteroaromatische Gruppe sind miteinander kondensiert,
und schließt
beispielsweise Pyrrolyl, Furyl, Thienyl, Imidazolyl, Pyrazolyl,
Thiazolyl, Iso thiazolyl, Oxazolyl, Isoxazolyl, Triazolyl, Tetrazolyl,
Oxadiazolyl, 1,2,3-Thiadiazolyl, 1,2,4-Thiadiazolyl, 1,2,5-Thiadiazolyl, 1,3,4-Thiadiazolyl,
Pyridyl, Pyrazinyl, Pyrimidinyl, Pyridazinyl, 1,2,4-Triazinyl, 1,3,5-Triazinyl,
Indolyl, Benzofuranyl, Benzothienyl, Benzimidazolyl, Benzoxazolyl,
Benzisoxazolyl, Benzothiazolyl, Benzisothiazolyl, Indazolyl, Purinyl,
Chinolyl, Isochinolyl, Phthalazinyl, Naphthyridinyl, Chinoxalinyl,
Chinazolinyl, Cinnolinyl, Pteridinyl, 1,5-Naphthyridinyl oder dergleichen
ein.
-
„Heteroaryloxy" bedeutet eine Heteroaryloxygruppe,
die die vorstehend angeführte
Heteroarylgruppe enthält,
und schließt
beispielsweise 2-Thienyloxy, 3-Thienyloxy, 2-Pyridyloxy, 3-Pyridyloxy,
4-Pyridyloxy, 3-Indolyloxy, 4-Indolyloxy, 5-Indolyloxy, 6-Indolyloxy
und dergleichen ein.
-
Die
Salze der durch die allgemeine Formel (I) wiedergegebenen Verbindungen
bedeuten die pharmazeutisch verträglichen und üblichen
Salze und schließen
beispielsweise Basenadditionssalz zu der Carboxylgruppe, wenn die
Verbindung eine Carboxylgruppe aufweist, oder Säureadditionssalz zu Amino- oder basischem Heterocyclyl,
wenn die Verbindung eine Amino- oder
eine basische Heterocyclylgruppe aufweist, ein.
-
Die
vorstehend angeführten
Basenadditionssalze schließen
Salze mit Alkalimetallen (beispielsweise Natrium, Kalium, usw.);
Erdalkalimetallen (beispielsweise Calcium, Magnesium, usw.); Ammonium;
organischen Aminen (beispielsweise Trimethylamin, Triethylamin,
Dicyclohexylamin, Ethanolamin, Diethanolamin, Triethanolamin, Procain,
N,N'-Dibenzylethylendiamin,
usw.) oder dergleichen ein.
-
Die
vorstehend erwähnten
Säureadditionssalze
schließen
Salze mit anorganischen Säuren
(beispielsweise Salzsäure,
Schwefelsäure,
Salpetersäure,
Phosphorsäure,
Perchlorsäure,
usw.); organischen Säuren (beispielsweise Äpfelsäure, Fumarsäure, Weinsäure, Zitronensäure, Ascorbinsäure, Trifluoressigsäure, usw.); Sulfonsäuren (beispielsweise
Methansulfonsäure,
Isethionsäure,
Benzolsulfonsäure,
p-Toluolsulfonsäure, usw.);
oder dergleichen ein.
-
Die
Ester der durch die allgemeine Formel (I) wiedergegebenen Verbindungen
beziehen sich auf beispielsweise die pharmazeutisch verträglichen, üblichen
Ester an der Carboxylgruppe, wenn die Verbindung eine Carboxylgruppe
aufweist, und schließen
beispielsweise Ester mit Niederalkylen (beispielsweise Methyl, Ethyl,
Propyl, Isopropyl, Butyl, sec-Butyl,
tert-Butyl, Pentyl, Isopentyl, Neopentyl, Cyclopropyl, Cyclobutyl,
Cyclopentyl, usw.), Aralkylen (beispielsweise Benzyl, Phenethyl,
usw.), Niederalkenylen (beispielsweise Allyl, 2-Butenyl, usw.),
Niederalkoxy(nieder)alkylen (beispielsweise Methoxymethyl, 2-Methoxyethyl,
2-Ethoxyethyl, usw.), Niederalkanoyloxy(nieder)alkylen (beispielsweise
Acetoxymethyl, Pivaloyloxymethyl, 1-Pivaloyloxyethyl, usw.), Niederalkoxycarbonyl(nieder)alkylen
(beispielsweise Methoxycarbonylmethyl, Isopropoxycarbonylmethyl,
usw.), Carboxy(nieder)alkylen (beispielsweise Carboxymethyl, usw.),
Niederalkoxycarbonyloxy(nieder)alkylen (beispielsweise 1-(Ethoxycarbonyloxy)ethyl,
1-(Cyclohexyloxycarbonyloxy)ethyl, usw.), Carbamoyloxy(nieder)alkylen
(beispielsweise Carbamoyloxymethyl, usw.), Phthalidyl, (5-substituiertes
2-Oxo-1,3-dioxol-4-yl)methyl (beispielsweise (5-Methyl-2-oxo-1,3-dioxol-4-yl)methyl, usw.),
oder dergleichen ein.
-
„Ein Mittel
zur Behandlung" bedeutet
einen Arzneistoff, der für
die Behandlung und/oder Prophylaxe von verschiedenen Erkrankungen
angewendet wird.
-
Um
die vorstehend angeführten,
durch die allgemeine Formel (I) wiedergegebenen Verbindungen zu offenbaren,
werden verschiedene, in der Formel (I) angewendete Symbole genauer
durch die Anwendung von bevorzugten Ausführungsformen erläutert.
-
Ar1, Ar2 und Ar3 geben unabhängig Aryl oder Heteroaryl wieder,
das substituiert sein kann, wobei der Substituent ausgewählt ist
aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl,
Halogen(nieder)alkyl, Hydroxy(nieder)alkyl, Cyclo(nieder)alkyl(nieder)alkyl,
Niederalkenyl, Niederalkylamino, Diniederalkylamino, Niederalkanoylamino, Niederalkylsulfonylamino,
Arylsulfonylamino, Hydroxy, Niederalkoxy, Halogen(nieder)alkoxy,
Aryloxy, Heteroaryloxy, Niederalkylthio, Carboxyl, Formyl, Niederalkanoyl,
Niederalkoxycarbonyl, Carbamoyl, Niederalkylcarbamoyl, Diniederalkylcarbamoyl,
Niederalkylsulfonyl, Arylsulfonyl, Aryl und Heteroaryl.
-
Der
Ausdruck „Ar1, Ar2 und Ar3 gibt unabhängig Aryl oder Heteroaryl wieder,
das substituiert sein kann, wobei der Substituent ausgewählt ist
aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl, Halogen(nieder)alkyl,
Hydroxy(nieder)alkyl, Cyclo(nieder)alkyl(nieder)alkyl, Niederalkenyl,
Niederalkylamino, Diniederalkylamino, Niederalkanoylamino, Niederalkylsulfonylamino,
Arylsulfonylamino, Hydroxy, Niederalkoxy, Halogen(nieder)alkoxy,
Aryloxy, Heteroaryloxy, Niederalkylthio, Carboxyl, Formyl, Niederalkanoyl,
Niederalkoxycarbonyl, Carbamoyl, Niederalkylcarbamoyl, Diniederalkylcarbamoyl,
Niederalkylsulfonyl, Arylsulfonyl, Aryl und Heteroaryl", bezieht sich auf
unsubstituiertes, vorstehend angeführtes Aryl, oder das unsubstituierte,
vorstehend angeführte
Heteroaryl, oder das vorstehend angeführte Aryl oder das vorstehend
angeführte Heteroaryl,
das Substituent(en) an der/den substituierbaren, beliebigen Position(en)
aufweist. Der vorstehend angeführte
Substituent kann gleich oder verschieden, 1 oder nicht weniger als
2 Substituenten, vorzugsweise 1 oder 2 Substituenten, ausgewählt aus
der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl, Halogen(nieder)alkyl,
Hydroxy(nieder)alkyl, Cyclo(nieder)alkyl(nieder)alkyl, Niederalkenyl,
Niederalkylamino, Diniederalkylamino, Niederalkanoylamino, Niederalkylsulfonylamino,
Arylsulfonylamino, Hydroxy, Niederalkoxy, Halogen(nieder)alkoxy,
Aryloxy, Heteroaryloxy, Niederalkylthio, Carboxyl, Formyl, Niederalkanoyl,
Niederalkoxycarbonyl, Carbamoyl, Niederalkylcarbamoyl, Diniederalkylcarbamoyl,
Niederalkylsulfonyl, Arylsulfonyl, Aryl und Heteroaryl, sein.
-
Halogenatom,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Fluoratom, Chloratom, Bromatom oder dergleichen
ein.
-
Niederalkyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Methyl, Ethyl, Propyl, Isopropyl oder
dergleichen ein.
-
Halogen(nieder)alkyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Difluormethyl, Trifluormethyl oder dergleichen
ein.
-
Hydroxy(nieder)alkyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Hydroxymethyl, 2-Hydroxyethyl, 1-Hydroxy-1-methylethyl
oder dergleichen ein.
-
Cyclo(nieder)alkyl(nieder)alkyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Cyclopropylmethyl, Cyclobutylmethyl, Cyclopentylmethyl
oder dergleichen ein.
-
Niederalkenyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Vinyl, 1-Propenyl, 2-Methyl-1-propenyl
oder dergleichen ein.
-
Niederalkylamino,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Methylamino, Ethylamino oder dergleichen
ein.
-
Diniederalkylamino,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Dimethylamino, Diethylamino oder dergleichen
ein.
-
Niederalkanoylamino,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Acetylamino, Propionylamino oder dergleichen
ein.
-
Niederalkylsulfonylamino,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Methylsulfonylamino, Ethylsulfonylamino
oder dergleichen ein.
-
Arylsulfonylamino,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Phenylsulfonylamino oder dergleichen ein.
-
Niederalkoxy,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Methoxy, Ethoxy oder dergleichen ein.
-
Halogen(nieder)alkoxy,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Fluormethoxy, Difluormethoxy, Trifluormethoxy
oder dergleichen ein.
-
Aryloxy,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Phenoxy oder dergleichen ein.
-
Heteroaryloxy,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel 2-Pyridyloxy, 3-Pyridyloxy, 4-Pyridyloxy oder dergleichen
ein.
-
Niederalkylthio,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Methylthio, Ethylthio oder dergleichen
ein.
-
Niederalkanoyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Formyl, Acetyl, Propionyl oder dergleichen
ein.
-
Niederalkoxycarbonyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Methoxycarbonyl, Ethoxycarbonyl oder dergleichen
ein.
-
Niederalkylcarbamoyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Methylcarbamoyl, Ethylcarbamoyl oder dergleichen
ein.
-
Diniederalkylcarbamoyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Dimethylcarbamoyl, Diethylcarbamoyl oder
dergleichen ein.
-
Niederalkylsulfonyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Methylsulfonyl, Ethylsulfonyl oder dergleichen
ein.
-
Arylsulfonyl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Phenylsulfonyl oder dergleichen ein.
-
Aryl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Phenyl oder dergleichen ein.
-
Heteroaryl,
als der vorstehend angeführte
Substituent, schließt
vorzugsweise zum Beispiel Thienyl, Thiazolyl, Isothiazolyl, Pyridyl,
Pyrazinyl oder dergleichen ein.
-
Der/Die
Substituent(en) von Ar1 schließt/schließen vorzugsweise
zum Beispiel Halogenatom, Niederalkyl, Halogen(nieder)alkyl, Hydroxy(nieder)alkyl,
Niederalkoxy, Halogen(nieder)alkoxy, Niederalkanoyl oder dergleichen,
bevorzugter Halogenatom, Halogen(nieder)alkyl, Halogen(nieder)alkoxy,
oder dergleichen ein.
-
Aryl
in Ar1 schließt vorzugsweise zum Beispiel
Phenyl oder dergleichen ein, und Heteroaryl in Ar1 schließt vorzugsweise
zum Beispiel Thienyl, Pyridyl oder dergleichen ein.
-
Ar1 wird vorzugsweise beispielhaft durch Phenyl
angegeben, das Substituent(en) aufweist, ausgewählt aus der Gruppe, bestehend
aus Halogenatom, Halogen(nieder)alkyl und Halogen(nieder)alkoxy,
oder durch Thienyl oder Pyridyl, die Substituent(en) aufweisen können, ausgewählt aus
der Gruppe, bestehend aus Halogenatom, Halogen(nieder)alkyl und
Halogen(nieder)alkoxy. Insbesondere ist Ar1 beispielsweise
Phenyl, 2-Fluorphenyl, 3-Fluorphenyl, 4-Fluorphenyl, 2-Chlorphenyl,
3-Chlorphenyl, 4-Chlorphenyl, 2-Bromphenyl, 3-Bromphenyl, 4-Bromphenyl,
2-Methylphenyl, 3-Methylphenyl, 4-Methylphenyl, 2-Trifluormethylphenyl, 3-Trifluormethylphenyl,
4-Trifluormethylphenyl, 2-Hydroxymethylphenyl, 3-Hydroxymethylphenyl,
4-Hydroxymethylphenyl, 2-Methoxyphenyl, 3-Methoxyphenyl, 4-Methoxyphenyl,
2-Difluormethoxyphenyl, 3-Difluormethoxyphenyl, 4-Difluormethoxyphenyl,
2-Trifluormethoxyphenyl, 3-Trifluormethoxyphenyl, 4-Trifluormethoxyphenyl,
2-Formylphenyl,
3-Formylphenyl, 4-Formylphenyl, 2-Thienyl, 4-Chlor-2-thienyl, 5-Chlor-2-thienyl, 4-Brom-2-thienyl,
5-Brom-2-thienyl,
4-Methyl-2-thienyl, 5-Methyl-2-thienyl, 4-Methoxy-2-thienyl, 5-Methoxy-2-thienyl,
3-Thienyl, 5-Chlor-3-thienyl, 5-Methyl-3-thienyl, 5-Methoxy-3-thienyl,
2-Pyridyl, 4-Methyl-2-pyridyl,
5-Methyl-2-pyridyl, 4-Methoxy-2-pyridyl, 5-Methoxy-2-pyridyl, 4-Chlor-2-pyridyl,
5-Chlor-2-pyridyl, 3-Pyridyl, 4-Methyl-3-pyridyl, 5-Methyl-3-pyridyl,
4-Methoxy-3- pyridyl,
5-Methoxy-3-pyridyl, 6-Fluor-3-pyridyl, 4-Chlor-3-pyridyl, 5-Chlor-3-pyridyl,
4-Pyridyl, 2-Fluor-4-pyridyl, 2-Chlor-4-pyridyl,
3-Chlor-4-pyridyl, 2-Methyl-4-pyridyl, 3-Methyl-4-pyridyl, 2-Methoxy-4-pyridyl,
3-Methoxy-4-pyridyl, und dergleichen, vorzugsweise 2-Fluorphenyl,
3-Fluorphenyl, 4-Fluorphenyl, 2-Chlorphenyl, 3-Chlorphenyl, 4-Chlorphenyl,
3-Bromphenyl, 4-Bromphenyl, 3-Methylphenyl, 3-Trifluormethylphenyl,
4-Trifluormethylphenyl, 3-Methoxyphenyl, 4-Methoxyphenyl, 3-Trifluormethoxyphenyl,
4-Trifluormethoxyphenyl, 3-Difluormethoxyphenyl,
4-Difluormethoxyphenyl, 2-Thienyl, 4-Chlor-2-thienyl, 5-Chlor-2-thienyl,
4-Brom-2-thienyl, 5-Brom-2-thienyl,
3-Pyridyl, 4-Methoxy-3-pyridyl, 5-Methoxy-3-pyridyl, 6-Fluor-3-pyridyl,
4-Chlor-3-pyridyl, 5-Chlor-3-pyridyl, 2-Fluor-4-pyridyl und dergleichen,
insbesondere 4-Fluorphenyl, 6-Fluor-3-pyridyl, 2-Fluor-4-pyridyl
oder dergleichen, bevorzugter 4-Fluorphenyl oder dergleichen.
-
Der
Substituent von Ar2 schließt vorzugsweise
zum Beispiel Cyano, Halogenatom, Nitro, Niederalkyl, Halogen(nieder)alkyl,
Niederalkoxy, Halogen(nieder)alkoxy, Formyl, Niederalkanoyl, Niederalkoxycarbonyl, Niederalkylsulfonyl
oder dergleichen, bevorzugter Cyano oder dergleichen, ein.
-
Aryl
in Ar2 schließt beispielsweise Phenyl oder
dergleichen ein, und Heteroaryl in Ar2 schließt beispielsweise
eine 5- oder 6-gliedrige, monocyclische heteroaromatische Gruppe,
die 1 oder nicht weniger als 2, vorzugsweise 1 oder 3, Heteroatome,
gleich oder verschieden, ausgewählt
aus der Gruppe, bestehend aus Sauerstoffatom, Stickstoffatom und
Schwefelatom, insbesondere zum Beispiel Thienyl, Thiazolyl, Isothiazolyl, 1,2,5-Thiadiazolyl,
Pyridyl, Pyrazinyl, Pyrimidinyl, oder dergleichen, vorzugsweise
ein.
-
Ar2 schließt
zum Beispiel Phenyl, das Substituent(en) enthält, ausgewählt aus der Gruppe, bestehend aus
Cyano, Halogenatom, Nitro, Niederalkyl, Halogen(nieder)alkyl, Niederalkoxy,
Halogen(nieder)alkoxy, Formyl, Niederalkanoyl, Niederalkoxycarbonyl
und Niederalkylsulfonyl, oder eine 5- oder 6-gliedrige, monocyclische
heteroaromatische Gruppe, die 1 oder nicht weniger als 2, vorzugsweise
1 oder 3, Heteroatome, gleich oder verschieden, ausgewählt aus
der Gruppe, bestehend aus Sauerstoffatom, Stickstoffatom und Schwefelatom,
enthält,
die Substituent(en) aufweisen können,
ausgewählt
aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl,
Halogen(nieder)alkyl, Niederalkoxy, Halogen(nieder)alkoxy, Formyl,
Niederalkanoyl, Niederalkoxycarbonyl und Niederalkylsulfonyl, ein.
5- oder 6-gliedrige monocyclische heteroaromatische Gruppe schließt vorzugsweise
Thienyl, Thiazolyl, Isothiazolyl, 1,2,5-Thiadiazolyl, Pyridyl, Pyrimidinyl,
Pyrazinyl oder dergleichen ein. Insbesondere schließt Ar2 zum Beispiel Phenyl, 3-Cyanophenyl, 4-Cyanophenyl, 3-Fluorphenyl,
4-Fluorphenyl, 3-Chlorphenyl, 4-Chlorphenyl, 3-Bromphenyl, 4-Bromphenyl, 3-Nitrophenyl, 4-Nitrophenyl,
3-Methylphenyl, 4-Methylphenyl,
3,5-Dimethylphenyl, 3-Trifluormethylphenyl, 4-Trifluormethylphenyl, 3-Hydroxymethylphenyl,
4-Hydroxymethylphenyl, 3-Methoxyphenyl, 4-Methoxyphenyl, 3-Trifluormethoxyphenyl,
4-Trifluormethoxyphenyl, 3-Formylphenyl, 4-Formylphenyl, 3-Acetylphenyl,
3-Methoxycarbonylphenyl, 4-Methoxycarbonylphenyl, 3-Methylsulfonylphenyl,
4-Methylsulfonylphenyl, 2-Thienyl, 4-Cyano-2-thienyl, 5-Cyano-2-thienyl,
4-Chlor-2-thienyl,
5-Chlor-2-thienyl, 3-Thienyl, 2-Thiazolyl, 4-Thiazolyl, 5-Thiazolyl, 3-Isothiazolyl,
4-Isothiazolyl, 1,2,5-Thiadiazol-3-yl, 2-Pyridyl, 4-Cyano-2-pyridyl,
6-Cyano-2-pyridyl, 4-Chlor-2-pyridyl,
6-Chlor-2-pyridyl, 4-Trifluormethyl-2-pyridyl, 6-Trifluormethyl-2-pyridyl,
6-Hydroxy-2-pyridyl, 4-Methoxy-2-pyridyl,
4-Methoxycarbonyl-2-pyridyl, 3-Pyridyl,
5-Cyano-3-pyridyl, 5-Brom-3-pyridyl, 4-Pyridyl, 2-Cyano-4-pyridyl,
2-Chlor-4-pyridyl, Pyrazinyl, 2-Pyrimidinyl, 4-Cyano-2-pyrimidinyl,
4-Chlor-2-pyrimidinyl, 4-Trifluormethyl-2-pyrimidinyl, 4-Carbamoyl-2-pyrimidinyl,
4-Pyrimidinyl, 6-Cyano-4-pyrimidinyl, 6-Chlor-4-pyrimidinyl, 5-Pyrimidinyl,
3-Pyridazinyl, oder dergleichen, vorzugsweise 3-Cyanophenyl, 3-Fluorphenyl,
3-Chlorphenyl, 3-Bromphenyl, 3-Nitrophenyl, 3-Methylphenyl, 3-Trifluormethylphenyl,
3-Trifluormethoxy phenyl, 3-Formylphenyl, 3-Methylsulfonylphenyl,
5-Thiazolyl, 4-Isothiazolyl, 1,2,5-Thiadiazol-3-yl, 4-Cyano-2-pyridyl,
6-Cyano-2-pyridyl,
3-Pyridyl, 5-Cyano-3-pyridyl, 2-Cyano-4-pyridyl, 2-Chlor-4-pyridyl, Pyrazinyl,
4-Cyano-2-pyrimidinyl, 6-Cyano-4-pyrimidinyl und dergleichen, bevorzugter
3-Cyanophenyl, 3-Methylsulfonylphenyl,
4-Cyano-2-pyridyl, 6-Cyano-2-pyridyl,
5-Cyano-3-pyridyl, 2-Chlor-4-pyridyl, 4-Cyano-2-pyrimidinyl oder dergleichen ein.
-
Der/Die
Substituent(en) von Ar3 schließt/schließen vorzugsweise
zum Beispiel Halogenatom, Niederalkyl, Hydroxy(nieder)alkyl, Niederalkenyl,
Di(nieder)alkylamino, Niederalkoxy, Halogen(nieder)alkoxy, Niederalkanoyl,
Aryl oder dergleichen ein.
-
Aryl
in Ar3 schließt vorzugsweise zum Beispiel
Phenyl, Naphthyl oder dergleichen ein, und Heteroaryl in Ar3 schließt
vorzugsweise zum Beispiel Thienyl, Pyridyl, Chinolyl, 1,5-Naphthyridinyl,
oder dergleichen ein.
-
Ar3 ist vorzugsweise Phenyl, Naphthyl, Thienyl,
Pyridyl, Chinolyl, 1,5-Naphthyridinyl oder dergleichen, die Substituent(en)
aufweisen können,
ausgewählt
aus der Gruppe, bestehend aus zum Beispiel Halogenatom, Niederalkyl,
Hydroxy(nieder)alkyl, Niederalkenyl, Di(nieder)alkylamino, Niederalkoxy,
Halogen(nieder)alkoxy, Niederalkanoyl, Aryl und dergleichen, insbesondere
zum Beispiel Phenyl, 4-Cyanophenyl, 2-Fluorphenyl, 3-Fluorphenyl, 4-Fluorphenyl,
2-Chlorphenyl, 3-Chlorphenyl,
4-Chlorphenyl, 2-Methylphenyl, 3-Methylphenyl, 4-Methylphenyl, 3,4-Dimethylphenyl,
2-Trifluormethylphenyl, 3-Trifluormethylphenyl, 4-Trifluormethylphenyl,
3-Hydroxymethylphenyl, 4-Hydroxymethylphenyl, 3-Vinylphenyl, 4-Vinylphenyl,
3-Dimethylaminophenyl, 4-Dimethylaminophenyl, 2-Methoxyphenyl, 3-Methoxyphenyl,
4-Methoxyphenyl, 2-Difluormethoxyphenyl, 3-Difluormethoxyphenyl,
4-Difluormethoxyphenyl, 2-Trifluormethoxyphenyl,
3-Trifluormethoxyphenyl, 4-Trifluormethoxyphenyl, 3-Formylphenyl,
4-Formylphenyl, 4-Biphenylyl, 1-Naphthyl, 2-Naphthyl, 2-Thienyl, 4-Chlor-2-thienyl,
5-Chlor-2-thienyl,
4-Brom-2-thienyl, 5-Brom-2-thienyl, 4-Me thyl-2-thienyl, 5-Methyl-2-thienyl, 4-Methoxy-2-thienyl,
5-Methoxy-2-thienyl,
3-Thienyl, 5-Chlor-3-thienyl, 5-Methyl-3-thienyl, 5-Methoxy-3-thienyl, 2-Pyridyl,
5-Fluor-2-pyridyl, 6-Chlor-2-pyridyl, 5-Chlor-2-pyridyl, 4-Methyl-2-pyridyl,
5-Methyl-2-pyridyl, 4-Methoxy-2-pyridyl,
5-Methoxy-2-pyridyl, 3-Pyridyl, 6-Fluor-3-pyridyl, 4-Chlor-3-pyridyl,
5-Chlor-3-pyridyl, 6-Chlor-3-pyridyl,
4-Methyl-3-pyridyl, 5-Methyl-3-pyridyl,
4-Methoxy-3-pyridyl, 5-Methoxy-3-pyridyl, 6-Methoxy-3-pyridyl, 6-Difluormethoxy-3-pyridyl,
4-Pyridyl, 2-Fluor-4-pyridyl,
2-Chlor-4-pyridyl, 3-Chlor-4-pyridyl, 2-Methyl-4-pyridyl, 3-Methyl-4-pyridyl, 2-Methoxy-4-pyridyl,
3-Methoxy-4-pyridyl,
3-Chinolyl, 6-Chinolyl, 1,5-Naphthyridin-3-yl und dergleichen, vorzugsweise
Phenyl, 3-Fluorphenyl, 4-Fluorphenyl, 3-Chlorphenyl, 4-Chlorphenyl,
2-Methylphenyl, 4-Methylphenyl,
3,4-Dimethylphenyl, 3-Trifluormethylphenyl, 4-Trifluormethylphenyl, 3-Vinylphenyl,
4-Vinylphenyl, 4-Dimethylaminophenyl, 2-Methoxyphenyl, 3-Methoxyphenyl,
4-Methoxyphenyl, 3-Difluormethoxyphenyl, 4-Difluormethoxyphenyl,
3-Trifluormethoxyphenyl,
4-Trifluormethoxyphenyl, 3-Formylphenyl, 4-Biphenylyl, 2-Naphthyl,
2-Thienyl, 5-Chlor-2-thienyl, 5-Methyl-2-thienyl, 5-Methoxy-2-thienyl,
2-Pyridyl, 5-Fluor-2-pyridyl,
3-Pyridyl, 6-Fluor-3-pyridyl, 6-Chlor-3-pyridyl, 6-Methoxy-3-pyridyl, 6-Difluormethoxy-3-pyridyl,
4-Pyridyl, 2-Fluor-4-pyridyl,
3-Chinolyl, 6-Chinolyl, 1,5-Naphthyridin-3-yl,
und dergleichen, insbesondere 6-Fluor-3-pyridyl, 2-Fluor-4-pyridyl und dergleichen,
bevorzugter 6-Fluor-3-pyridyl, oder
dergleichen.
-
R1 gibt Cyclo(nieder)alkyl oder eine durch
die Formel von -Ar3 wiedergegebene Gruppe
wieder;
-
-
Cyclo(nieder)alkyl
in R1 schließt vorzugsweise beispielsweise
Cyclobutyl, Cyclopentyl oder Cyclohexyl oder dergleichen ein.
-
Die
Definition und bevorzugte Beispiele von Ar3 sind
vorstehend angeführt.
-
In
der durch die Formel von (a), (b) oder (c) wiedergegebenen Gruppe
bedeutet n 0, 1 oder 2; sowohl R10 als auch
R11 geben Wasserstoffatom wieder, oder sie
vereinigen sich, um Oxo wiederzugeben; X und Y geben unabhängig Methylen,
Ethenylen, eine durch die Formel von -NR12-
wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wieder;
Z gibt Methin oder Stickstoffatom wieder; und R12 gibt
Wasserstoffatom oder Niederalkyl wieder.
-
Bezüglich der
durch Formel (a), (b) oder (c) wiedergegebenen Gruppe ist es bevorzugt,
dass n 0 oder 1 wiedergibt; sowohl R10 als
auch R11 sich vereinigen, um Oxo wiederzugeben;
X und Y unabhängig
Methylen, Ethenylen, die durch die Formel von -NR12-
wiedergegebene Gruppe oder Sauerstoffatom wiedergeben; Z Methin
wiedergibt; und R12 Wasserstoffatom wiedergibt.
Das heißt,
eine durch die Formel von (a0) oder (b0) wiedergegebene Verbindung, wenn n 0 ist,
oder dergleichen, ist bevorzugt.
-
-
Eine
durch die Formel von (a1), (b1)
oder (c1) wiedergegebene Gruppe, wenn n
1 ist, oder dergleichen, ist bevorzugt.
-
-
Weiterhin
ist es im Fall, n ist 0, bevorzugt, dass die Kombination von X und
Y beispielsweise Sauerstoffatom und Methylen, Sauerstoff und Ethenylen,
Sauerstoffatom und Imino, oder dergleichen, darstellt. Wenn n ist
1, ist es bevorzugt, dass die Kombination von X und Y beispielsweise
Imino und Sauerstoffatom, Sauerstoffatom und Sauerstoffatom und
dergleichen ist.
-
Deshalb
ist insbesondere bezüglich
einer durch die Formel von (a), (b) oder (c) wiedergegebenen Gruppe
eine durch die Formel von (aa) wiedergegebene Gruppe oder dergleichen
bevorzugt.
-
-
Bezüglich R1 ist eine durch die Formel von -Ar3 wiedergegebene Gruppe bevorzugt.
-
R2 und R3 geben unabhängig Wasserstoffatom,
Cyclo(nieder)alkyl, Niederalkenyl oder Niederalkyl wieder, die substituiert
sein können,
wobei der Substituent ausgewählt
ist aus der Gruppe, bestehend aus Halogenatom, Niederalkylamino,
Diniederalkylamino, Niederalkanoylamino, Hydroxy, Niederalkoxy,
Formyl, Niederalkoxycarbonyl, Niederalkylcarbamoyl und Diniederalkylcarbamoyl.
-
Durch
R2 oder R3 wiedergegebenes
Cyclo(nieder)alkyl schließt
vorzugsweise beispielsweise Cyclopentyl, Cyclohexyl oder dergleichen
ein.
-
Durch
R2 oder R3 wiedergegebenes
Niederalkenyl schließt
vorzugsweise beispielsweise Vinyl, 1-Propenyl, 2-Propenyl oder dergleichen ein.
-
„Niederalkyl,
das substituiert sein kann, wobei der Substituent ausgewählt ist
aus der Gruppe, bestehend aus Ha logenatom, Niederalkylamino, Diniederalkylamino,
Niederalkanoylamino, Hydroxy, Niederalkoxy, Formyl, Niederalkoxycarbonyl,
Niederalkylcarbamoyl, und Diniederalkylcarbamoyl" gibt das unsubstituierte, vorstehend
angeführte
Niederalkyl oder das vorstehend angeführte Niederalkyl, das 1 oder
nicht weniger als 2, vorzugsweise 1 oder 2, Substituenten an der/den
substituierbaren, willkürlichen
Position(en), gleich oder verschieden, enthält, wobei der Substituent ausgewählt ist
aus der Gruppe, bestehend aus Halogenatom, Niederalkylamino, Diniederalkylamino,
Niederalkanoylamino, Hydroxy, Niederalkoxy, Formyl, Niederalkoxycarbonyl,
Niederalkylcarbamoyl und Diniederalkylcarbamoyl, wieder.
-
Bezüglich Halogenatom
als Substituenten sind zum Beispiel Fluoratom und dergleichen bevorzugt.
-
Bezüglich Niederalkylamino
als Substituenten sind beispielsweise Methylamino, Ethylamino und
dergleichen bevorzugt.
-
Bezüglich Diniederalkylamino
als Substituenten sind beispielsweise Dimethylamino, Diethylamino
und dergleichen bevorzugt.
-
Bezüglich Niederalkanoylamino
als Substituenten sind beispielsweise Acetylamino, Propionylamino und
dergleichen bevorzugt.
-
Bezüglich Niederalkoxy
als Substituenten sind beispielsweise Methoxy, Ethoxy und dergleichen
bevorzugt. Bezüglich
Niederalkoxycarbonyl als Substituenten sind beispielsweise Methoxycarbonyl,
Ethoxycarbonyl und dergleichen bevorzugt.
-
Bezüglich Niederalkylcarbamoyl
als Substituenten sind beispielsweise Methylcarbamoyl, Ethylcarbamoyl
und dergleichen bevorzugt.
-
Bezüglich Diniederalkylcarbamoyl
als Substituenten sind beispielsweise Dimethylcarbamoyl, Diethylcarbamoyl
und dergleichen bevorzugt.
-
Bezüglich des
Substituenten von Niederalkyl in R2 oder
R3 sind beispielsweise Halogenatom, Niederalkylamino, Diniederalkylamino,
Hydroxyl, Niederalkoxy und dergleichen bevorzugt.
-
Bezüglich Niederalkyl
in R2 oder R3 sind
beispielsweise Methyl, Ethyl, Propyl, Isopropyl, Isobutyl und dergleichen
bevorzugt, und Methyl und dergleichen sind bevorzugter.
-
Insbesondere
schließt
Niederalkyl, das den/die angeführten
Substituent(en) aufweisen kann, und das durch R2 oder
R3 wiedergegeben wird, zum Beispiel Methyl,
Ethyl, Propyl, Isopropyl, Isobutyl, 2-Fluorethyl, 2,2-Difluorethyl,
2,2,2-Trifluorethyl,
Methylaminomethyl, 2-Methylaminoethyl, Dimethylaminomethyl, 2-Dimethylaminoethyl,
2-Diethylaminoethyl, (Acetylamino)methyl, Hydroxymethyl, 2-Hydroxyethyl,
3-Hydroxypropyl, Methoxymethyl, 2-Methoxyethyl, Formylmethyl, Methoxycarbonylmethyl,
Ethoxycarbonylmethyl, (Methylcarbamoyl)methyl, (Ethylcarbamoyl)methyl,
(Dimethylcarbamoyl)methyl, (Diethylcarbamoyl)methyl und dergleichen,
insbesondere Methyl, Ethyl, Propyl, Hydroxymethyl und dergleichen,
bevorzugter Methyl, und dergleichen ein.
-
Bezüglich R2 und R3 ist es bevorzugt,
wenn beide Wasserstoffatom darstellen, oder einer von ihnen Wasserstoffatom
darstellt und der andere Niederalkyl darstellt, das den/die vorstehend
angeführten
Substituent(en) aufweisen kann.
-
Die
erfindungsgemäßen Verbindungen
schließen
vorzugsweise zum Beispiel (a) Verbindungen, worin R1 die
durch die Formel von -Ar3 wiedergegebene
Gruppe darstellt und Ar2 Phenyl darstellt,
das Substituent(en) enthält,
ausgewählt
aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl,
Halogen(nieder)alkyl, Niederalkoxy, Halogen(nieder)alkoxy, Formyl,
Niederalkanoyl, Niederalkoxycarbonyl und Niederalkylsulfonyl; oder
R1 eine durch die Formel von -Ar3 wiedergegebene Gruppe darstellt und Ar2 eine 5- oder 6-gliedrige monocyclische
heteroaromatische Gruppe darstellt, die Substituent(en) aufweisen
kann, ausgewählt
aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl,
Halogen(nieder)alkyl, Niederalkoxy, Halogen(nieder)alkoxy, Formyl,
Niederalkanoyl, Niederalkoxycarbonyl und Niederalkylsul fonyl, die 5-
oder 6-gliedrige monocyclische heteroaromatische Gruppe, die 1 oder
nicht weniger als 2, vorzugsweise 1 bis 3, Heteroatome, gleich oder
verschieden, ausgewählt
aus der Gruppe, bestehend aus Sauerstoffatom, Stickstoffatom und
Schwefelatom, enthält,
ein. Die 5- oder 6-gliedrige monocyclische heteroaromatische Gruppe
ist bevorzugter Thienyl, Thiazolyl, Isothiazolyl, 1,2,5-Thiadiazolyl,
Pyridyl, Pyrimidinyl, Pyrazinyl oder dergleichen.
-
Die
vorstehend in (a) angeführten
Verbindungen schließen
vorzugsweise (b) Verbindungen, worin Ar1 Phenyl,
enthaltend den/die Substituent(en), ausgewählt aus der Gruppe, bestehend
aus Halogenatom, Halogen(nieder)alkyl und Halogen(nieder)alkoxy,
darstellt, oder Ar1 Thienyl oder Pyridyl,
das Substituent(en) aufweisen kann, ausgewählt aus der Gruppe, bestehend
aus Halogenatom, Halogen(nieder)alkyl und Halogen(nieder)alkoxy
und dergleichen, darstellt, ein, und schließt bevorzugter (c) Verbindungen,
worin sowohl R2 als auch R3 Wasserstoff
darstellen; oder entweder R2 oder R3 Wasserstoffatom darstellt und der andere
Niederalkyl, das Substituent(en) aufweisen kann, ausgewählt aus
der Gruppe, bestehend aus Halogenatom, Niederalkylamino, Diniederalkylamino,
Niederalkanoylamino, Hydroxy, Niederalkoxy, Formyl, Niederalkoxycarbonyl,
Niederalkylcarbamoyl und Diniederalkylcarbamoyl und dergleichen,
darstellt, ein.
-
Die
vorstehend in (c) angeführten
Verbindungen schließen
vorzugsweise (d) Verbindungen, worin Ar3 Phenyl,
Naphthyl, Thienyl, Pyridyl, Chinolyl oder 1,5-Napththyridinyl, das
Substituent(en) aufweisen kann, ausgewählt aus der Gruppe, bestehend
aus Halogenatom, Niederalkyl, Hydroxy(nieder)alkyl, Niederalkenyl,
Diniederalkylamino, Niederalkoxy, Halogen(nieder)alkoxy, Niederalkanoyl
und Aryl, darstellt, ein.
-
Weiterhin
schließen
die vorstehend in (a) erwähnten
Verbindungen vorzugsweise (e) Verbindungen ein, worin Ar1 4-Fluorphenyl,
6-Fluor-3-pyridyl bzw. 2-Fluor-4-pyridyl darstellt, Ar3 6-Fluor-3-pyridyl
oder 2-Fluor-4-pyridyl dar stellt, und entweder R2 oder
R3 Wasserstoffatom darstellt und der andere
Methyl darstellt, und bevorzugter insbesondere die Verbindungen,
worin Ar1 4-Fluorphenyl darstellt und Ar3 6-Fluor-3-pyridyl
darstellt; Ar1 4-Fluorphenyl darstellt und
Ar3 2-Fluor-4-pyridyl darstellt; sowohl
Ar1 als auch Ar3 6-Fluor-3-pyridyl darstellen;
oder sowohl Ar1 als auch Ar3 2-Fluor-4-pyridyl darstellen,
oder dergleichen.
-
Jedoch
werden die Verbindungen, worin sowohl R2 als
auch R3 gleichzeitig Wasserstoffatom darstellen und
Ar1, Ar2 und R1 gleichzeitig unsubstituiertes Phenyl darstellen,
aus der vorliegenden Erfindung ausgenommen.
-
Die
erfindungsgemäßen Verbindungen
können
Stereoisomere, wie optische Isomere, Diastereoisomere und geometrische
Isomere, oder Tautomere, in Abhängigkeit
von der Art der Substituenten, einschließen. Somit schließen die
erfindungsgemäßen Verbindungen
alle Stereoisomeren, Tautomeren und ein Gemisch davon ein.
-
Auch
sind die Polymorphe, Hydrate und Solvate der erfindungsgemäßen Verbindungen
innerhalb des Umfangs der Erfindung eingeschlossen.
-
Die
vorliegende Erfindung schließt
Prodrugs der erfindungsgemäßen Verbindungen
in ihren Umfang ein. Im Allgemeinen werden solche Prodrugs funktionelle
Derivate der erfindungsgemäßen Verbindungen
sein, die leicht in vivo in die Verbindungen, die in vivo oder für den lebenden
Körper
erforderlich sind, umwandelbar sind. Somit sollte in den Behandlungsverfahren
der verschiedenen, die vorliegende Erfindung betreffenden Erkrankungen
der Begriff „Verabreichen" Verabreichen einer
Verbindung umfassen, die nicht speziell offenbart sein muss, sich
jedoch in die speziell offenbarte Verbindung in vivo nach Verabreichung
an den Patienten umwandelt, zusätzlich
zum Verabreichen der speziell offenbarten Verbindung. Übliche Verfahren
für die
Auswahl und Herstellung von geeigneten Prodrugderivaten werden beispielsweise
in "Design of Prodrugs" Hrsg. H. Bundgaard,
Elsevier, 1985 oder dergleichen, welches hierin durch Hinweis in
seiner Gesamt heit einbezogen ist, beschrieben. Metaboliten von diesen
Verbindungen schließen
die durch Einführung
der erfindungsgemäßen Verbindungen
in biologisches Milieu erzeugten Wirkstoffe ein und gehören zu der
Kategorie der vorliegenden Erfindung.
-
Das
spezielle Beispiel der durch die allgemeine Formel (I) wiedergegebenen
Verbindung ist zum Beispiel
2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-pyradinyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(1,5-naphthyridin-3-yl)-2-imidazolin,
2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-5-methyl-2-imidazolin,
2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-5-hydroxymethyl-2-imidazolin,
2-(3-Cyanophenyl)-4,4-bis(3-fluorphenyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(3-chinolyl)-4-(2-thienyl)-2-imidazolin,
4-(4-Bromphenyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin,
4,4-Bis(4-chlorphenyl)-2-(3-cyanophenyl)-2-imidazolin,
4-(4-Chlorphenyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-chinolyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-phenyl-4-(4-vinylphenyl)-2-imidazolin,
4-(6-Chlor-3-pyridyl)-2-(3-cyanophenyl)-4-(4-fluorphenyl)-2-imidazolin,
4-(5-Chlor-2-thienyl)-2-(3-cyanophenyl)-4-(4-fluorphenyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-methylphenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(3-methylphenyl)-4-phenyl-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(3-nitrophenyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-phenyl-4-(3-chinolyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-methoxyphenyl)-4-phenyl-2-imidazolin,
4-(3-Chlorphenyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(5-methyl-2-thienyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-phenyl-4-(4-trifluormethylphenyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(3-methoxyphenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(3,4-dimethylphenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-vinylphenyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(2-naphthyl)-4-(3-pyridyl)-2-imidazolin,
4-(3-Bromphenyl)-2-(3-cyanophenyl)-4-(4-fluorphenyl)-2-imidazolin,
4-(2-Chlorphenyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(6-chinolyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-phenyl-4-(2-thienyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-dimethylaminophenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-pyridyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-formylphenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(2-thienyl)-2-imidazolin,
4,4-Diphenyl-2-pyrazinyl-2-imidazolin,
2-(3-Cyanophenyl)-4,4-bis(4-methoxyphenyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-formylphenyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(3-methylsulfonylphenyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(3-formylphenyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(2-methylphenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-hydroxymethylphenyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(1,2,3,4-tetrahydro-1,5-naphthyridin-7-yl)-2-imidazolin,
2-(3-Cyanophenyl)-4-(2-fluorphenyl)-4-(3-pyridyl)-2-imidazolin,
4-(4-Biphenylyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(2-methoxyphenyl)-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-cyclohexyl-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(4-pyridyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(5-pyrimidinyl)-2-imidazolin,
2-(3-Cyanophenyl)-4-cyclopentyl-4-phenyl-2-imidazolin,
2-(3-Cyanophenyl)-4-cyclobutyl-4-phenyl-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(3-pyridazinyl)-2-imidazolin,
4-(4-Fluorphenyl)-5-methyl-2-pyrazinyl-4-(3-chinolyl)-2-imidazolin,
(4S,5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-2-(3-methylsulfonylphenyl)-5-methyl-2-imidazolin,
(4R,5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-2-(3-methylsulfonylphenyl)-5-methyl-2-imidazolin,
(4S,5S)-2-(5-Cyano-3-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(5-Cyano-3-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4S,5S)-2-(2-Cyano-4-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(2-Cyano-4-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(5R)-2-(2-Cyano-4-pyridyl)-4,4-bis(4-fluorphenyl)-5-hydroxymethyl-2-imidazolin,
(5S)-2-(2-Cyano-4-pyridyl)-4,4-bis(4-fluorphenyl)-5-methyl-2-imidazolin,
(4S,5S)-2-(2-Cyano-4-pyridyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(2-Cyano-4-pyridyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin,
(5S)-2-(2-Cyano-4-pyridyl)-4,4-bis(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
4,4-Bis(4-fluorphenyl)-5-methyl-2-pyrazinyl-2-imidazolin,
4,4-Bis(4-fluorphenyl)-5-hydroxymethyl-2-pyrazinyl-2-imidazolin,
5-Ethyl-4,4-bis(4-fluorphenyl)-2-pyrazinyl-2-imidazolin,
4,4-Bis(4-fluorphenyl)-5-propyl-2-pyrazinyl-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(4-isothiazolyl)-2-imidazolin,
2-(3-Pyridyl)-4,4-bis(4-fluorphenyl)-2-imidazolin,
2-(3-Chlorphenyl)-4,4-bis(4-fluorphenyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(3-trifluormethylphenyl)-2-imidazolin,
2-(3-Fluorphenyl)-4,4-bis(4-fluorphenyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(3-trifluormethoxyphenyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(5-thiazolyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(3-methylphenyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(2-thienyl)-2-imidazolin,
2-(5-Brom-3-pyridyl)-4,4-bis(4-fluorphenyl)-2-imidazolin,
2-(3-Bromphenyl)-4,4-bis(4-fluorphenyl)-2-imidazolin,
4,4-Bis(4-fluorphenyl)-2-(3-thienyl)-2-imidazolin,
2-(4-Cyano-2-pyridyl)-9,4-bis(4-fluorphenyl)-2-imidazolin,
(4S,5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-5-(1-hydroxyethyl)-2-imidazolin,
(4S,5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin,
(4S,5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-(1,2,5-thiadiazol-3-yl)-2-imidazolin,
(4R,5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-(1,2,5-thiadiazol-3-yl)-2-imidazolin,
(5R)-4,4-Bis(4-fluorphenyl)-5-hydroxymethyl-2-(6-hydroxy-2-pyridyl)-2-imidazolin,
(5S)-4,4-Bis(4-fluorphenyl)-2-(6-hydroxy-2-pyridyl)-5-methyl-2-imidazolin,
(4S,5S)-2-(2-Chlor-4-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(2-Chlor-4-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4S,5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-2-(6-hydroxy-2-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-2-(6-hydroxy-2-pyridyl)-5-methyl-2-imidazolin,
(5S)-2-(3-Cyanophenyl)-4,4-bis(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4S,5R)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-hydroxymethyl-2-imidazolin,
(4R,5R)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-hydroxymethyl-2-imidazolin,
(4S,5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-5-methyl-4-(1,5-naphthyridin-3-yl)-2-imidazolin,
(4R,5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-5-methyl-4-(1,5-naphthyridin-3-yl)-2-imidazolin,
(4S,5S)-2-(3-Cyanophenyl)-4-(6-fluor-3-pyridyl)-4-(4-fluorphenyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(3-Cyanophenyl)-4-(6-fluor-3-pyridyl)-4-(4-fluorphenyl)-5-methyl-2-imidazolin,
(5S)-2-(5-Cyano-3-pyridyl)-4,4-bis(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4S,5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(5R)-2-(4-Cyano-2-pyridyl)-4,4-bis(4-fluorphenyl)-5-hydroxymethyl-2-imidazolin,
2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-2-imidazolin,
2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-2-imidazolin,
(4S,5R)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-hydroxymethyl-2-imidazolin,
(4R,5R)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-hydroxymethyl-2-imidazolin,
(4S,5S)-2-(6-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(6-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4S,5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin,
(4R,5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin,
(5S)-2-(4-Cyano-2-pyridyl)-4,4-bis(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
(4S,5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-5-methyl-4-(3-pyridyl)-2-imidazolin,
(4R,5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-5-methyl-4-(3-pyridyl)-2-imidazolin,
(5R)-2-(4-Cyano-2-pyrimidyl)-4,4-bis(4-fluorphenyl)-5-hydroxymethyl-2-imidazolin,
(4S,5S)-2-(4-Cyano-2-pyrimidyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin
oder
(4R,5S)-2-(4-Cyano-2-pyrimidyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
oder dergleichen.
-
Das
Verfahren zum Herstellen der erfindungsgemäßen Verbindungen wird wie nachstehend
erläutert.
-
Die
erfindungsgemäßen Verbindungen
(I) können
beispielsweise durch die nachstehenden Verfahren zur Herstellung
oder die in den Beispielen gezeigten Verfahren synthetisiert werden,
jedoch sind diese Ausführungsformen
nicht vorgesehen, das Verfahren zum Herstellen der erfindungsgemäßen Verbindungen
(I) zu begrenzen.
-
Herstellungsverfahren
1
-
Eine
durch die allgemeine Formel (II) wiedergegebene Verbindung:
worin Ar
1p und
Ar
3p unabhängig Aryl oder Heteroaryl,
die substituiert sein können,
wobei der Substituent ausgewählt
ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl,
Halogen(nieder)alkyl, Cyclo(nieder)alkyl(nieder)alkyl, Niederalkenyl,
Di(nieder)alkylamino, Niederalkoxy, Halogen(nieder)alkoxy, Aryloxy,
Heteroaryloxy, Niederalkylthio, Formyl, Niederalkanoyl, Niederalkoxycarbonyl,
Diniederalkylcarbamoyl, Niederalkylsulfonyl, Arylsulfonyl, Aryl,
Heteroaryl, und einer gegebenenfalls geschützten Hydroxy(nieder)alkyl-,
Niederalkylamino-, Niederalkanoylamino-, Niederalkylsulfonylamino-,
Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder Niederalkylcarbamoylgruppe,
wiedergeben;
R
1p Cyclo(nieder)alkyl
wiedergibt oder eine Gruppe, wiedergegeben durch die Formel -Ar
3p, darstellt;
R
2p und R
3p unabhängig Wasserstoffatom,
Cyclo(nieder)alkyl, Niederalkenyl oder Niederalkyl, die substituiert sein
können,
wobei der Substituent ausgewählt
ist aus der Gruppe, bestehend aus Halogenatom, Diniederalkylamino,
Niederalkoxy, Formyl, Niederalkoxycarbonyl, Diniederalkylcarbamoyl,
und einer gegebenenfalls geschützten
Niederalkylamino-, Niederalkanoylamino-, Hydroxy- oder Niederalkylcarbamoylgruppe,
wiedergeben;
sowohl R
10p als auch R
11p Wasserstoffatom wiedergeben oder sie
sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben;
R
12p Wasserstoffatom, Niederalkyl oder eine
Schutzgruppe für
eine Iminogruppe wiedergibt;
X
p und
Y
p unabhängig
Methylen, Ethenylen, eine durch die Formel -NR
12p-
wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben;
n
und Z die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen;
wird
mit einem Säureadditionssalz
der durch die allgemeine Formel (III) wiedergegebenen Verbindung
umgesetzt:
worin Ar
2p Aryl
oder Heteroaryl, die substituiert sein können, wobei der Substituent
ausgewählt
ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, Niederalkyl,
Halogen(nieder)alkyl, Cyclo(nieder)alkyl(nieder)alkyl, Niederalkenyl,
Niederalkylamino, Niederalkoxy, Halogen(nieder)alkoxy, Aryloxy,
Heteroaryloxy, Niederalkylthio, Formyl, Niederalkanoyl, Niederalkoxycarbonyl,
Diniederalkylcarbamoyl, Niederalkylsulfonyl, Arylsulfonyl, Aryl,
Heteroaryl, und ei ner gegebenenfalls geschützten Hydroxy(nieder)alkyl-,
Niederalkylamino-, Niederalkanoylamino-, Niederalkylsulfonylamino-,
Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder Niederalkylcarbamoylgruppe,
wiedergibt;
R
4 Amino oder Niederalkoxy
wiedergibt;
zur Bereitstellung einer Verbindung der allgemeinen
Formel (IV):
worin Ar
1p,
Ar
2p, R
1p, R
2p und R
3p die gleichen
wie vorstehend beschriebenen Bedeutungen aufweisen;
gegebenenfalls
gefolgt von Abspaltung einer Schutzgruppe, unter Gewinnung einer
durch die allgemeine Formel (I) wiedergegebenen Verbindung.
-
Wenn
ein Reaktant eine Amino-, Imino-, Hydroxy-, Carboxyl-, Oxo-, Carbonyl-
oder dergleichen Gruppe aufweist, die nicht an der vorstehenden
Reaktion teilnimmt, kann die Reaktion nach Schützen der Amino-, Imino-, Hydroxy-,
Carboxyl-, Oxo-, Carbonyl- oder dergleichen Gruppe mit einer Schutzgruppe
für Amino
oder Imino, einer Schutzgruppe für
Hydroxyl, einer Schutzgruppe für
Carboxyl oder einer Schutzgruppe für Oxo oder Carbonyl, gefolgt
von Entfernung der Schutzgruppe nach Beendigung der Reaktion, ausgeführt werden.
-
„Schutzgruppe
für Amino
oder Imino" schließt vorzugsweise
Aralkyl (beispielsweise Benzyl, p-Methoxybenzyl, 3,4-Dimethoxybenzyl,
o-Nitrobenzyl, p-Nitrobenzyl, Benzhydryl, Trityl, usw.); Niederalkanoyl
(beispielsweise Formyl, Acetyl, Propionyl, Butyryl, Pivaloyl, usw.);
Benzoyl; Arylalkanoyl (beispielsweise Phenylacetyl, Phenoxyacetyl,
usw.); Niederalkoxycarbonyl (beispielsweise Methoxycarbonyl, Ethoxycarbonyl,
Propyloxycarbonyl, tert-Butoxycarbonyl, usw.); Aralkyloxycarbonyl
(beispielsweise Benzyloxycarbonyl, p-Nitrobenzyloxycarbonyl, Phenethyloxycarbonyl,
usw.); Niederalkylsilyl (beispielsweise Trimethylsilyl, tert-Butyldimethylsilyl, usw.);
oder dergleichen, insbesondere Acetyl, Pivaloyl, Benzoyl, Ethoxycarbonyl,
tert-Butoxycarbonyl oder dergleichen, ein.
-
„Schutzgruppe
für Hydroxy" schließt Niederalkylsilyl
(beispielsweise Trimethylsilyl, tert-Butyldimethylsilyl, usw.);
Niederalkoxymethyl (beispielsweise Methoxymethyl, 2-Methoxyethoxymethyl,
usw.); Tetrahydropyranyl; Trimethylsilylethoxymethyl; Aralkyl (beispielsweise
Benzyl, p-Methoxybenzyl, 2,3-Dimethoxybenzyl, o-Nitrobenzyl, p-Nitrobenzyl,
Trityl, usw.); Acyl (beispielsweise Formyl, Acetyl, usw.) und dergleichen,
insbesondere Methoxymethyl, Tetrahydropyranyl, Trityl, Trimethylsilylethoxymethyl,
tert-Butyldimethylsilyl, Acetyl oder dergleichen, ein.
-
„Schutzgruppe
für Carboxyl" schließt vorzugsweise
Niederalkyl (beispielsweise Methyl, Ethyl, Propyl, Isopropyl, tert-Butyl,
usw.); Halogen(nieder)alkyl (beispielsweise 2,2,2-Trichlorethyl,
usw.); Niederalkenyl (beispielsweise 2-Propenyl, usw.); Aralkyl (beispielsweise
Benzyl, p-Methoxybenzyl, p-Nitrobenzyl, Benzhydryl, Trityl, usw.);
oder dergleichen, insbesondere Methyl, Ethyl, tert-Butyl, 2-Propenyl, Benzyl,
p-Methoxybenzyl, Benzhydryl oder dergleichen ein.
-
„Schutzgruppe
für Oxo
oder Carbonyl" schließt Acetal
oder Ketal (beispielsweise Ethylenketal, Trimethylenketal, Dimethylketal,
usw.) oder dergleichen ein.
-
Die
Reaktion zwischen einer durch die allgemeine Formel (II) wiedergegebenen
Verbindung und einem Säureadditionssalz
einer durch die allgemeine Formel (III) wiedergegebenen Verbindung
wird gewöhnlich durch
Anwenden von 1 bis überschüssigen Mol,
vorzugsweise 1 bis 5 Mol des Säureadditionssalzes
der Verbindung (III) pro Mol der Verbindung (II) ausgeführt.
-
Das
Säureadditionssalz
der Verbindung (III) schließt
vorzugsweise zum Beispiel Hydrochlorid oder dergleichen ein.
-
Die
Reaktion wird gewöhnlich
in einem inerten Lösungsmittel
ausgeführt,
und das inerte Lösungsmittel schließt vorzugsweise
beispielsweise Alkohol (beispielsweise Methanol, Ethanol, usw.),
Dimethylformamid, Dimethylsulfoxid oder ein Gemisch davon oder dergleichen
ein.
-
Die
Reaktionstemperatur ist gewöhnlich –30°C bis 200°C, vorzugsweise
0°C bis
150°C.
-
Die
Reaktionszeit ist gewöhnlich
30 Minuten bis 7 Tage, vorzugsweise 2 Stunden bis 5 Tage.
-
Nach
Beendigung der Reaktion kann das Rohprodukt einer durch die allgemeine
Formel (IV) wiedergegebenen Verbindung durch übliche Behandlung erhalten
werden. Die so durch die allgemeine Formel (IV) wiedergegebene,
erhaltene Verbindung wird durch das übliche Verfahren gereinigt
oder nicht gereinigt, gegebenenfalls gefolgt von Abspaltung der
Schutzgruppen für
Amino, Amino, Hydroxyl, Carboxyl, Oxo und Carbonyl, unter Gewinnung
der durch die allgemeine Formel (I) wiedergegebenen Verbindung.
-
Die
Abspaltung von Schutzgruppen kann beispielsweise durch die in der
Literatur beschriebene Weise ausgeführt werden (siehe Protective
Groups in Organic Synthesis, T. W. Greene, John Wiley & Sons, (1981)), oder
in ähnlicher
Weise, beispielsweise Solvolyse, unter Verwendung von beispielsweise
0,01 Mol eines großen
Säureüberschusses,
vorzugsweise Trifluoressigsäure,
Ameisensäure,
Salzsäure
oder dergleichen, oder 1 Mol eines großen Überschusses an Base, vorzugsweise
Kaliumhydroxid, Calciumhydroxid oder dergleichen; chemische Reduktion,
unter Verwendung von Metallkomplexhydrid oder dergleichen; oder
katalytische Reduktion, unter Anwendung von Palladium-Kohlenstoff-Katalysator,
Raney-Nickel-Katalysator oder dergleichen, in Abhängigkeit
von den Arten der vorstehend angeführten Schutzgruppen, der Stabilität der gewünschten
Verbindung (I) und so weiter.
-
Herstellungsverfahren
2
-
Eine
durch die allgemeine Formel (II) wiedergegebene Verbindung:
worin Ar
1p,
R
1p, R
2p und R
3p die gleichen wie vorstehend beschriebenen
Bedeutungen aufweisen;
wird in Gegenwart von Tri(nieder)alkylaluminium
mit einer durch die allgemeine Formel (V) wiedergegebenen Verbindung:
worin R
5 Niederalkoxy
wiedergibt und Ar
2p die gleiche, wie vorstehend
beschriebene Bedeutung aufweist, umgesetzt;
unter Bereitstellung
einer durch die allgemeine Formel (IV) wiedergegebenen Verbindung:
worin Ar
1p,
Ar
2p, R
1p, R
2p und R
3p die gleichen
wie vorstehend beschriebenen Bedeutungen aufweisen;
gegebenenfalls
gefolgt von Entfernung einer Schutzgruppe, unter Gewinnung einer
durch die allgemeine Formel (I) wiedergegebenen Verbindung.
-
Die
Reaktion in Gegenwart von Tri(nieder)alkylaluminium zwischen einer
durch die allgemeine Formel (II) wiedergegebenen Verbindung und
einer durch die allgemeine Formel (V) wiedergegebenen Verbindung, wird
gewöhnlich
durch Anwenden von 0, 5 bis 5 Mol, vorzugsweise 0, 7 bis 3 Mol,
der Verbindung (V) und 1 bis überschüssige Mol,
vorzugsweise 1 bis 5 Mol, Tri(nieder)alkylaluminium pro Mol der
Verbindung (II), ausgeführt.
-
Für die Reaktion
verwendetes Tri(nieder)alkylaluminium schließt beispielsweise Trimethylaluminium, Triethylalu minium,
Tripropylaluminium, Tributylaluminium und dergleichen ein.
-
Die
Reaktion wird gewöhnlich
in einem inerten Lösungsmittel
ausgeführt
und das inerte Lösungsmittel schließt vorzugsweise
zum Beispiel Benzol, Toluol, Xylol, Methylenchlorid, Chloroform,
Hexan oder ein Gemisch davon oder dergleichen ein.
-
Die
Reaktionstemperatur ist gewöhnlich –20°C bis zu
dem Siedepunkt eines für
die Reaktion verwendeten Lösungsmittels,
vorzugsweise 0°C
bis 110°C.
-
Die
Reaktionszeit ist gewöhnlich
30 Minuten bis 3 Tage, vorzugsweise 3 bis 24 Stunden.
-
Eine
durch die allgemeine Formel (I) wiedergegebene Verbindung kann durch
Behandeln eines Reaktionsgemisches in üblicher Weise nach Entfernung
einer Schutzgruppe, wenn das Produkt eine Schutzgruppe aufweist,
nach Beendigung der Reaktion oder durch Behandeln des Gemisches,
wie es ist, in üblicher
Weise, wenn die Schutzgruppe in dem Reaktionsprodukt nicht vorliegt,
hergestellt werden.
-
Die
Abspaltung der Schutzgruppen und Nachbehandlung und dergleichen
können
gemäß dem wie
in dem vorstehenden Herstellungsverfahren 1 beschriebenen Verfahren
ausgeführt
werden.
-
Herstellungsverfahren
3
-
Eine
durch die allgemeine Formel (II) wiedergegebene Verbindung:
worin Ar
1p,
R
1p, R
2p und R
3p die gleichen wie vorstehend beschriebenen
Bedeutungen aufweisen;
wird mit einer durch die allgemeine
Formel (VI) wiedergegebenen Verbindung:
worin Ar
2 die
gleiche, wie vorstehend beschriebene Bedeutung aufweist, umgesetzt;
unter Bereitstellung einer durch die allgemeine Formel (VII) wiedergegebenen
Verbindung:
worin Ar
1p,
Ar
2p, R
1p, R
2p und R
3p die gleichen,
wie vorstehend beschriebenen Bedeutungen aufweisen.
-
Dann
wird das Produkt (VII) intramolekularer Cyclo-Kondensationsreaktion unterzogen, unter
Bereitstellung einer durch die allgemeine Formel (IV) wiedergegebenen
Verbindung:
worin Ar
1p,
Ar
2p, R
1p, R
2p und R
3p die gleichen
wie vorstehend beschriebenen Bedeutungen aufweisen;
gegebenenfalls
gefolgt von Abspaltung einer Schutzgruppe, unter Gewinnung einer
durch die allgemeine Formel (I) wiedergegebenen Verbindung.
-
Die
Reaktion zwischen einer durch die allgemeine Formel (II) wiedergegebenen
Verbindung und einer durch die allgemeine Formel (VI) wiedergegebenen
Verbindung wird gewöhnlich
durch Anwenden von 0,5 bis überschüssige Mol,
vorzugsweise 1 bis 2 Mol, der Verbindung (VI) pro Mol der Verbindung
(II) ausgeführt.
-
Die
Reaktion wird gewöhnlich
in einem inerten Lösungsmittel
ausgeführt,
und das inerte Lösungsmittel schließt vorzugsweise
zum Beispiel Methylenchlorid, Chloroform, Tetrahydrofuran, Dimethylformamid,
Pyridin oder ein Gemisch davon oder dergleichen ein.
-
Die
vorstehend angeführte
Reaktion wird vorzugsweise in Gegenwart eines Kondensationsmittels ausgeführt, beispielsweise
N,N'-Dicyclohexylcarbodiimid,
N,N'-Diisopropylcarbodiimid,
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid,
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid,
Benzotriazol-1-yloxy-tris-(dimethylamino)phosphoniumhexafluorophosphat,
Benzotriazol-1-yloxy-tris-pyrrolidinophosphoniumhexafluorophosphat,
Diphenylphosphorylazid,
1,1-Carbonyldiimidazol
oder dergleichen.
-
Das
vorstehend angeführte
Kondensationsmittel wird gewöhnlich
bei 1 Mol bis überschüssige Mol, vorzugsweise
1 Mol bis 3 Mol, pro Mol der durch die allgemeine Formel (II) wiedergegebenen
Verbindung, angewendet.
-
Die
Reaktionstemperatur ist gewöhnlich –20°C bis zu
dem Siedepunkt eines für
die Reaktion verwendeten Lösungsmittels,
vorzugsweise 0°C
bis 60°C.
-
Die
Reaktionszeit ist gewöhnlich
30 Minuten bis 3 Tage, vorzugsweise 1 bis 24 Stunden.
-
Eine
durch die allgemeine Formel (VII) wiedergegebene Rohverbindung kann
durch Behandeln des Reaktionsgemisches in der gewöhnlichen
Weise nach Beendigung der Reaktion hergestellt werden. Die durch allgemeine
Formel (VII) wiedergegebene, so erhaltene Rohverbindung wird durch
das übliche
Verfahren gereinigt oder nicht gereinigt, gefolgt von der nächsten intramolekularen
Cyclo-Kondensationsreaktion.
-
Die
intramolekulare Cyclo-Kondensationsreaktion, durch die die Verbindung
(IV) aus der Verbindung (VII) hergestellt wird, wird gewöhnlich in
einem inerten Lösungsmittel
oder ohne Lösungsmittel
ausgeführt.
-
Das
inerte Lösungsmittel
schließt
vorzugsweise zum Beispiel Ethanol, Propanol, Butanol, Pentanol, 1,4-Dioxan, Dimethoxyethan,
Dimethylformamid, Dimethylsulfoxid, Benzol, Toluol, Xylol oder ein
Gemisch davon oder dergleichen ein.
-
Die
Reaktionstemperatur ist gewöhnlich
Raumtemperatur bis zum Siedepunkt eines für die Reaktion verwendeten
Lösungsmittels,
vorzugsweise 80°C
bis 190°C.
-
Die
Reaktionszeit ist gewöhnlich
5 Stunden bis 7 Tage, vorzugsweise 12 Stunden bis 3 Tage.
-
Die
vorstehend beschriebene Cyclisierungsreaktion kann in Gegenwart
eines dehydratisierenden Mittels oder einer katalytischen Menge
Lewis-Säure
ausgeführt
werden. Das dehydratisierende Mittel schließt beispielsweise Phosphoroxychlorid,
Phosphorpentachlorid, Polyphosphorsäure, Thionylchlorid oder dergleichen ein.
Die Lewis-Säure
schließt
beispielsweise Scandiumtrifluormethansulfonat, Yttriumtrifluormethansulfonat, Lanthantrifluormethansulfonat,
Trifluormethansulfonsäurelanthanid
oder dergleichen ein. Die Reaktion wird vorzugsweise ohne Lösungsmittel
oder in einem Lösungsmittel,
beispielsweise Methylenchlorid, Chloroform, Benzol, Toluol, Xylol
oder einem Gemisch davon, oder dergleichen, ausgeführt.
-
Das
vorstehend erwähnte
Dehydratisierungsmittel wird gewöhnlich
bei 1 bis überschüssige Mol,
vorzugsweise 2 bis 10 Mol, pro Mol der durch die allgemeine Formel
(VII) wiedergegebenen Verbindung angewendet. Die Menge an Lewis-Säure ist
1 bis 50 Mol-%, vorzugsweise 5 bis 30 Mol-%.
-
Die
Reaktionstemperatur ist gewöhnlich
Raumtemperatur bis zum Siedepunkt eines für die Reaktion vorzugsweise
angewendeten Lösungsmittels.
-
Die
Reaktionszeit ist gewöhnlich
1 Stunde bis 7 Tage, vorzugsweise 5 Stunden bis 3 Tage.
-
Eine
durch die allgemeine Formel (I) wiedergegebene Verbindung kann durch
Behandeln eines Reaktionsgemisches in üblicher Weise, nach Abspaltung
einer Schutzgruppe, wenn das Produkt eine Schutzgruppe aufweist,
oder durch Behandeln des Gemisches, wie es ist, in üblicher
Weise, wenn die Schutzgruppe nicht vorliegt, hergestellt werden.
-
Die
Abspaltung der Schutzgruppen, Nachbehandlung und dergleichen, können gemäß dem wie
in dem vorstehend angeführten
Herstellungsverfahren 1 beschriebenen Verfahren ausgeführt werden.
-
Die
durch die allgemeine Formel (I) wiedergegebenen Verbindungen können leicht
durch übliche Trenntechniken,
beispielsweise Lösungsmittelextraktion,
Umkristallisation, Säulenchromatographie,
präparative
Dünnschichtchromatographie
oder/und dergleichen, isoliert und gereinigt werden.
-
Diese
Verbindungen können
in die pharmazeutisch verträglichen
Salze oder Ester durch das übliche Verfahren
umgewandelt werden; im Gegensatz kann die Umwandlung der Salze oder
Ester in freie Verbindungen gemäß dem üblichen
Verfahren ausgeführt
werden.
-
Die
durch die allgemeine Formel (II), (III), (V) oder (VI) wiedergegebenen
Verbindungen sind beispielsweise kommerziell erhältlich, oder werden gemäß den bekannten
Verfahren, den Verfahren wie nachstehend oder in den Beispielen
und Bezugsbeispielen beschriebenen Verfahren, dazu analogen Verfahren,
oder gegebenenfalls in Kombination von jenen Verfahren, hergestellt. Herstellungsverfahren
A-1
worin Ar
1p, R
1p,
R
2p und R
3p die
gleichen, wie vorstehend beschriebenen Bedeutungen aufweisen.
-
Dieses
Verfahren bezieht sich auf ein Verfahren zum Herstellen einer durch
die allgemeine Formel (II) wiedergegebenen Verbindung. Gemäß der vorliegenden
Erfindung kann die Verbindung (II) durch Unterziehen dem Verfahren,
dass eine durch die allgemeine Formel (1) wiedergegebene Verbindung
olefiniert, unter Gewinnung einer durch die allgemeine Formel (2)
wiedergegebenen Verbindung, Unterziehen der Verbindung (2) Jodazidierung,
unter Gewinnung einer durch die allgemeine Formel (3) wiedergegebenen
Verbindung, Unterziehen der Verbindung (3) Reduktion, unter Gewinnung
einer durch die allgemeine Formel (4) wiedergegebenen Verbindung,
und Unterziehen der Verbindung (4) Azidierung, gefolgt von Reduktion,
hergestellt werden.
-
Bezüglich der
Olefinierungsreaktion in dem Schritt zum Herstellen der Verbindung
(2) aus der Verbindung (1) kann die Olefinierungsreaktion für Oxo, die
an sich auf dem Fachgebiet der organischen Chemie gut bekannt ist,
beispielsweise die so genannte Horner-Emmons-Reaktion, Wittig-Reaktion,
Peterson-Olefinierung und dergleichen, angewendet werden.
-
Im
Schritt zum Herstellen der Verbindung (3) aus der Verbindung (2)
kann Jodmonochlorid und Jodazid, erzeugt aus Metallazid, beispielsweise
Silberazid, Kaliumazid, Natriumazid oder dergleichen, mit der Verbindung
(2) umgesetzt werden.
-
Die
Reaktion wird gewöhnlich
in einem inerten Lösungsmittel,
beispielsweise Methylenchlorid, Chloroform, Acetonitril, Benzol,
Toluol oder einem Gemisch davon, oder dergleichen, unter Anwendung
von 1 bis überschüssige Mol,
vorzugsweise 1 bis 2 Mol, Jodazid pro Mol der Verbindung (2) ausgeführt.
-
Die
Reaktionstemperatur ist gewöhnlich
0°C bis
zum Siedepunkt des für
die Reaktion verwendeten Lösungsmittels,
vorzugsweise Raumtemperatur bis 80°C. Die Reaktionszeit ist gewöhnlich 30
Minuten bis 7 Tage, vorzugsweise 2 Stunden bis 5 Tage.
-
In
dem Schritt zur Herstellung der Verbindung (4) aus der Verbindung
(3) kann eine Reduktionsreaktion von Azid, die auf dem Fachgebiet
der organischen Chemie an sich bekannt ist, angewendet werden. Insbesondere
kann eine chemische Reduktion, unter Anwendung von beispielsweise
Metall, metallischem Komplexhydrid, Triphenylphosphin oder dergleichen,
oder katalytische Reduktion, unter Anwendung von Palladium-Kohlenstoff-Katalysator,
Raney-Nickel-Katalysator oder dergleichen, angewendet werden.
-
In
dem Verfahren zum Herstellen der Verbindung (II) aus der Verbindung
(4) wird gewöhnlich
die Reaktion mit Azidverbindung (beispielsweise Tetraalkylammoniumazid,
Kaliumazid, Natriumazid oder dergleichen) in Gegenwart von Säure (beispielsweise
Essigsäure,
Ammoniumchlorid), gefolgt von Reduktion der Azidgruppe der gemäß dem an
sich auf dem Fachgebiet der organischen Chemie gut bekannten Verfahren erhaltenen
Verbindung, hergestellt.
-
Das
in dem Schritt für
das Umsetzen der Azidverbindung verwendete Lösungsmittel schließt beispielsweise
Alkohol (beispielsweise Methanol, Ethanol oder dergleichen), Dimethylsulfoxid,
Dimethylformamid, Tetrahydrofuran, Wasser oder dergleichen, oder
ein Gemisch davon oder dergleichen, vorzugsweise ein.
-
Die
Reaktionstemperatur ist gewöhnlich
0°C bis
zum Siedepunkt eines für
die Reaktion angewendeten Lösungsmittels,
vorzugsweise Raumtemperatur bis 100°C. Die Reaktionszeit ist gewöhnlich 3
Stunden bis vorzugsweise 1 Tag.
-
Im
Schritt zur Reduktion von Azid kann die Reduktion insbesondere durch
chemische Reduktion, unter Anwendung von beispielsweise Metall,
Metallkomplexhydrid, Triphenylphosphin oder dergleichen, oder katalytische
Reduktion, unter Anwendung von Palladium-Kohlenstoff-Katalysator,
Raney-Nickel-Katalysator
oder dergleichen, ausgeführt
werden.
-
Die
durch die allgemeine Formel (1) wiedergegebenen Verbindungen und
für die
Olefinierungsreaktion angewendeten Augangsmaterialien können kommerziell
verfügbar
sein, oder können
gemäß den bekannten Verfahren,
oder in Beispielen und Bezugsbeispielen oder analogen Verfahren
dazu, gegebenenfalls in Kombination, hergestellt werden. Herstellungsverfahren
A-2
worin L
1 Wasserstoffatom
oder eine Abgangsgruppe wiedergibt; P eine Schutzgruppe für Amino
wiedergibt und Ar
1p, R
1p,
R
2p und R
3p die
gleichen, wie vorstehend beschriebenen Bedeutungen aufweisen.
-
Dieses
Verfahren ist das alternative Verfahren zum Herstellen einer durch
die allgemeine Formel (II) wiedergegebenen Verbindung. Gemäß diesem
Verfahren kann die Verbindung (II) hergestellt werden durch Unterziehen
einer durch die allgemeine Formel (5) wiedergegebenen Verbindung,
nacheinander Alkylierung oder Arylierung in dieser Reihenfolge an
den aktivierten Carbonylgruppen davon, zur Gewinnung einer durch die
allgemeine Formel (6) wiedergegebenen Verbindung, Unterziehen der
Verbindung (6) der Abspaltung einer Schutzgruppe für die Aminogruppe,
unter Erzeugung einer Hydroxylaminverbindung, gefolgt von Aziridin-Bildung
mit intramolekularer Entwässerung
der erzeugten Hydroxyaminverbindung, unter Gewinnung einer durch die
allgemeine Formel (4) wiedergegebenen Verbindung, und Unterziehen
der Verbindung (4) Azidierung, gefolgt von Reduktion.
-
Eine
Abgangsgruppe für
L1 schließt vorzugsweise zum Beispiel
Halogenatom, Alkoxy, Amid oder dergleichen ein.
-
Eine
Schutzgruppe für
durch P wiedergegebenes Amino schließt eine Schutzgruppe für Amino,
wie in Herstellungsverfahren 1 beschrieben, und bezüglich des
Verfahrens zur Abspaltung einer Schutzgruppe ein, und das in Herstellungsverfahren
1 beschriebene Verfahren kann angewendet werden.
-
Der
Schritt zur Herstellung der Verbindung (6) aus der Verbindung (5)
kann gewöhnlich
durch Unterziehen der Verbindung (5) der Reaktion mit einer Organometallverbindung,
beispielsweise Alkyllithium, Aryllithium, Alkylmagnesium, Arylmagnesium,
Alkylzink, Arylzink, Alkylkupfer, Arylkupfer, oder dergleichen,
die eine durch Ar1p oder R1p wiedergegebene
Gruppe aufweisen, wie Alkyl oder Aryl, ausgeführt werden.
-
Die
Reaktion wird durch die Anwendung von 1 bis überschüssige Mol, vorzugsweise 2 bis
5 Mol, der vorstehend angeführten
Organometallverbindung pro Mol der Verbindung (5) in einem inerten
Lösungsmittel, beispielsweise
ein Etherlösungsmittel
(beispielsweise Diethylether, Tetrahydrofuran, 1,2-Dimethoxyethan), oder
einem Gemisch davon oder dergleichen, ausgeführt.
-
Die
Reaktionstemperatur ist gewöhnlich –130°C bis zum
Siedepunkt eines für
die Reaktion verwendeten Lösungsmittels,
vorzugsweise –100°C bis Raumtemperatur,
und die Reaktionszeit ist gewöhnlich
30 Minuten bis 2 Tage, vorzugsweise 1 Stunde bis 1 Tag.
-
Wenn
L1 Wasserstoffatom darstellt, kann die Verbindung
(6) durch Oxidation einer Alkoholverbindung, die durch Alkylierung
oder Arylierung erhalten wurde, zur Gewinnung der entsprechenden
Ketonverbindung durch die auf dem Gebiet der organischen Chemie
an sich gut bekannte Oxidation und dann wiederum durch Alkylierung
oder Arylierung der erhaltenen Ketonverbindung hergestellt werden.
-
Der
Schritt zum Herstellen der Verbindung (4) aus der Verbindung (6)
kann so durch Umsetzen der nach Entfernung der Schutzgruppe P mit
Halogenid von Trialkyl- oder Triarylphosphin (beispielsweise Dichlortriethylphosphoran,
Dichlortributylphosphoran, Dichlortriphenylphosphoran, Dibromtri ethylphosphoran, Dibromtributylphosphoran,
Dibromtriphenylphosphoran oder dergleichen), gewöhnlich in Gegenwart von Base (beispielsweise
Triethylamin, Diisopropylamin, N,N-Diisopropylethylamin, Pyridin oder dergleichen)
erhaltenen Hydroxyaminoverbindung ausgeführt werden.
-
Die
Reaktion wird gewöhnlich
durch Anwenden von 1 bis überschüssige Mol,
vorzugsweise 2 bis 7 Mol, einer Base und 0,5 bis überschüssige Mol,
vorzugsweise 1 bis 5 Mol, Halogenid von Trialkyl- oder Triarylphosphin,
bezüglich
1 Mol der Verbindung (6), in einem inerten Lösungsmittel, beispielsweise
Diethylether, Tetrahydrofuran, 1,2-Dimethoxyethan, Toluol, Benzol,
Dichlormethan, 1,2-Dichlorethan, Chloroform, Acetonitril oder einem
Gemisch davon und dergleichen, ausgeführt.
-
Die
Reaktionstemperatur ist gewöhnlich –50°C bis zu
dem Siedepunkt eines für
die Reaktion verwendeten Lösungsmittels,
vorzugsweise 0°C
bis 70°C,
und die Reaktionszeit ist gewöhnlich
30 Minuten bis 3 Tage, vorzugsweise 1 Stunde bis 24 Stunden.
-
Das
wie in Herstellungsverfahren A-1 beschriebene Verfahren kann auf
den Schritt zum Herstellen der Verbindung (II) aus der Verbindung
(4) angewendet werden.
-
Durch
eine Reihe von Schritten zum Herstellen der Verbindung (II) aus
der wie vorstehend beschriebenen Verbindung (5) bleibt die Konfiguration
an Kohlenstoffatomen, die an R2p und R3p binden, unverändert. Deshalb kann, wenn Ar1p und R1p von der
gewünschten
Verbindung identisch sind, optisch aktive Verbindung (II) durch
Anwenden eines optisch aktiven Aminosäurederivats, als durch die
allgemeine Formel (5) von dem Ausgangsmaterial wiedergegebene Verbindung,
hergestellt werden. Im Gegensatz dazu kann, wenn Ar1p und R1p von der gewünschten Verbindung verschieden
sind, eine optisch aktive Verbindung (II) durch Umsetzen eines optisch
aktiven Aminosäurederivats,
als durch die allgemeine Formel (5) des Ausgangsmaterials wiedergegebene
Verbindung, gefolgt von Abtrennen des Gemisches von gewöhnlich erzeugten
Diastereoisomeren, die auf der Konfiguration von Kohlenstoffatomen
basie ren, die an Ar1p und R1p an
einer geeigneten Stufe binden, unter Anwendung des an sich bekannten
Verfahrens, und dann gefolgt von Umsetzen entsprechend optisch aktiver
Verbindung in dem anschließenden
Schritt, hergestellt werden.
-
Weiterhin
sind die durch die allgemeine Formel (5) wiedergegebenen Verbindungen
und Organometallverbindungen zur Alkylierung oder Arylierung kommerziell
erhältlich,
oder werden gemäß den in
bekannten Verfahren beschriebenen Verfahren oder in Beispielen und
Bezugsbeispielen oder dazu analogen Verfahren oder möglicher
Kombination von diesen Verfahren hergestellt. Herstellungsverfahren
B
worin Ar
1p und R
1p die gleichen, wie vorstehend beschriebenen
Bedeutungen aufweisen.
-
Dieses
Verfahren bezieht sich auf das Verfahren zum Herstellen einer durch
die allgemeine Formel (II-1) wiedergegebenen Verbindung. Die Verbindung
(II-1) kann aus dem vorliegenden Verfahren, in dem eine durch die
allgemeine Formel (1) wiedergegebene Verbindung Aminocyanierung
der Oxogruppe der durch die allgemeine Formel (1) wiedergegebenen
Verbindung unterzogen wird, unter Gewinnung der durch die allgemeine
Formel (7) wiedergegebenen Verbindung, gefolgt von Reduktion der
Cyanogruppe der Verbindung (7), hergestellt werden.
-
In
den Schritten zum Herstellen der Verbindung (7) aus der Verbindung
(1) kann das so genannte Strecker-Syntheseverfahren, das auf dem Gebiet
der organischen Chemie an sich gut bekannt ist, oder ein verbessertes
Verfahren davon angewendet werden
-
In
dem Schritt zum Herstellen der Verbindung (II-1) aus der Verbindung
(7) kann Reduktionsreaktion einer Cya nogruppe, die auf dem Gebiet
der organischen Chemie an sich gut bekannt ist; das heißt beispielsweise
chemische Reduktion, unter Anwendung von Metall- oder Metallkomplexhydrid
oder dergleichen, oder katalytische Reduktion oder dergleichen,
angewendet werden.
-
Weiterhin
sind die Ausgangsmaterialien für
die Reaktion der Aminocyanierung kommerziell verfügbar, oder
werden gemäß den in
bekannten Verfahren beschriebenen Verfahren, oder in Beispielen
und Bezugsbeispielen oder dazu analogen Verfahren oder einer möglichen
Kombination von diesen Verfahren hergestellt.
-
Das
spezielle Beispiel der durch die allgemeine Formel (II) oder (II') wiedergegebenen
Verbindung ist zum Beispiel
1,1-Bis(4-fluorphenyl)-1,2-ethandiamin,
1-(4-Fluorphenyl)-1-(1,5-naphthyridin-3-yl)-1,2-ethandiamin,
1,1-Bis(4-fluorphenyl)-1,2-propandiamin,
(2R)-1,1-Bis(4-fluorphenyl)-3-(methoxymethoxy)-1,2-propandiamin,
(2S)-1,1-Bis(4-fluorphenyl)-1,2-propandiamin,
(2S)-1,1-Bis(6-fluor-3-pyridyl)-1,2-propandiamin,
(2S)-1,1-Bis(2-fluor-4-pyridyl)-1,2-propandiamin,
(2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamin,
(1S,2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamin,
(1R,2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamin,
(2R)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-3-(methoxymethoxy)-1,2-propandiamin,
(1S,2R)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-3-(methoxymethoxy)-1,2-propandiamin,
(1R,2R)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-3-(methoxymethoxy)-1,2-propandiamin,
(1S,2S)-1-(4-Fluorphenyl)-1-(2-fluor-4-pyridyl)-1,2-propandiamin,
(1R,2S)-1-(4-Fluorphenyl)-1-(2-fluor-4-pyridyl)-1,2-propandiamin,
(1S,2S)-1-(4-Fluorphenyl)-1-(1,5-naphthyridin-3-yl)-1,2-propandiamin,
(1R,2S)-1-(4-Fluorphenyl)-1-(1,5-naphthyridin-3-yl)-1,2-propandiamin,
(1S,2S)-1-(4-Fluorphenyl)-1-(3-pyridyl)-1,2-propandiamin
oder
(1R,2S)-1-(4-Fluorphenyl)-1-(3-pyridyl)-1,2-propandiamin, oder
dergleichen.
-
Die
Anwendbarkeit der erfindungsgemäßen Verbindungen
als Arzneimittel wird durch Aufzeigen von NPY-antagonistischer Wirksamkeit,
beispielsweise in den nachstehenden pharmakologischen Tests, nachgewiesen.
-
Pharmakologischer Test
1 (NPY-Bindungs-Inhibierungs-Test)
-
cDNA-Sequenz,
codierend für
Human-NPY-Y5-Rezeptor (vergleiche Beschreibung der Internationalen
Patent Veröffentlichung
Nummer WO96/16542), wurde in Expressionsvektoren pcDNA3, pRc/RSV
(hergestellt von Invitrogen Inc.) und pCI-neo (hergestellt von Promega
Inc.) geklont. Diese erhaltenen Expressionsvektoren wurden zu Wirtszellen
COS-7, CHO und LM(tk-) (American Type Culture Collection) durch
das kationische Lipidverfahren (vergleiche: Proceedings of the National
Academy of Sciences of the United States of America, Band 84: 7413
(1987)) transfiziert, unter Gewinnung von rekombinanten Zellen,
die NPY-Y5-Rezeptor exprimieren.
-
Eine
Membranprobe, hergestellt aus den Zellen, die NPY Y5-Rezeptor exprimierten,
wurde zusammen mit einer Testverbindung und (125I)Peptid
YY (hergestellt von NEN) (20 000 Cpm) in einem Assaypuffer (25 mM
Tris-Puffer, pH 7,4, enthaltend 10 mM Magnesiumchlorid, 1 mM Phenylmethylsulfonylfluorid,
0,1% Bacitracin und 0,5% Rinderserumalbumin) bei 25°C für 2 Stunden,
dann durch ein Glasfilter GF/C filtriert und mit 5 mM Tris-Puffer
(pH 7,4), enthaltend 0,3% BSA, gewaschen, inkubiert. Die Radioaktivität auf dem
Glasfilter wurde gemessen. Nichtspezifisches Binden wurde in Gegenwart
von 1 μM
Peptid YY und einer 50%igen inhibitorischen Konzentration (IC50) der Testverbindung gegen spezifisches
Peptid YY-Binden bestimmt (vergleiche Endocrinology, Band 131: 2090
(1992)). Die Ergebnisse werden in Tabelle 1 zusammengefasst.
-
Tabelle
1
Inhibitor-Wirkungen auf NPY-Rezeptor-Binden
-
Wie
vorstehend gezeigt, inhibierten die erfindungsgemäßen Verbindungen
stark Peptid YY (NPY-homologes) Binden an NPY Y5-Rezeptoren.
-
Pharmakologischer Test
2 (Antagonistischer Effekt auf das Fressverhalten, induziert durch
D-Trp34 NPY)
-
Eine
chronische Führungskanüle (26 Gauge,
Länge 11
mm) wurde durch stereotaktische Operation in den dritten Ventrikel
von männlichen
SD-Ratten (7–8
Wochen alt, 200–300
g), anästhetisiert
mit Ketamin, eingeschoben. Xylazin (einzelne intraperitoneale Verabreichung
von 74 und 11 mg/kg) und fixiert durch Dentalharz. Die Spitze der
Kanüle
wurde 2,2 mm hinter Scheitelhöhe,
auf der Mittellinie und 8 mm tief von der Oberfläche des Schädelknochens angeordnet. Nach
etwa 1 Woche Erholungszeitraum wurde D-Trp34 NPY
(1 μg/0,4 μl/Schädel, künstliche
cerebrospinale Flüssigkeit,
die 0,05 Rinderserumalbumin enthält)
in den dritten Ventrikel eingespritzt. Die in wässriger 0,5%iger Methylcelluloselösung suspendierte
Testverbindung wurde oral 2 Stunden vor der Verabreichung von D-Trp34 NPY verabreicht und der Nahrungsverbrauch
wurde 2 Stunden nach Verabreichung von D-Trp34 NPY
gemessen.
-
Die
erfindungsgemäßen Verbindungen
inhibierten die Erhöhung
des Nahrungsverbrauchs, der durch D-Trp34 NPY
(homologes von NPY) induziert wurde, welcher zu dem dritten Ventrikel
verabreicht wurde.
-
Pharmakologischer Test
3 (Test für
Pharmakokinetik)
-
Die
getestete Verbindung wurde oral oder intravenös an männliche SD-Ratten (7–10 Wochen
alt, 200–400
g), unter Fastenbedingung über
Nacht, verabreicht, und etwa 100 μl
Blut wurden von der Schwanzvene, unter Anwendung von heparinisierter
Kapillare zu vordefinierten Zeitpunkten genommen. Die Blutproben wurden
durch Zentrifuge (4°C,
6 000 U/min, für
10 Minuten) getrennt, unter Gewinnung von Plasma. Zu dem Plasma
wurde 3-faches Volumen von Ethanol, das die inneren Standardsubstanzen
enthält,
gegeben, und das Gemisch wurde gerührt, dann bei –20°C für 20 Minuten
belassen, gefolgt von Zentrifugierung (4°C, 10 000 U/min für 10 Minuten).
Der Überstand
wurde durch LC/MS/MS analysiert und dann wurde die Konzentration der
Testverbindung in Plasma durch das relative Eichkurvenverfahren
gemessen.
-
Folglich
wurde beispielsweise die Verbindung von Beispiel 7 mit einem biologisch
verfügbaren
Verhältnis
gefunden: 35% und Halbwertszeit im Blut: 5,1 Stunden.
-
Pharmakologischer Test
4 (Hirn- und cerebrospinaler Flüssigkeitseindringtest)
-
Die
getestete Verbindung wurde oral oder intravenös an männliche SD-Ratten (7–10 Wochen
alt, 200–400
g) verabreicht. Die mit Ether anästhetisierten
Ratten und das Vollblut wurden zu vordefinierten Zeitpunkten mit
einer heparinisierten Spritze aus der abdominalen Aorta genommen.
Weiterhin wurde den Ratten die Haut von dem Hinterkopfbereich geschnitten
und eine 30G dentale Nadel zwischen cervicaler Vertebrae in den
subarachnoiden Hohlraum eingeschoben. Und dann wurden 50–100 μl Cerebrospinalflüssigkeit
in einer 1 ml-Spritze durch ein mit der 30G dentalen Nadel verbundenes
Rohr gesammelt, gefolgt von Herausnehmen des Hirns. Zu dem aus der
zentrifugierten Blutprobe (4°C,
6 000 U/min für
10 Minuten) erhaltenen Plasma wurde das 3-fache Volumen von Ethanol,
einschließlich
der inneren Standard-Substanzen, gegeben und das Gemisch wurde gerührt. Zu
der Hirnprobe wurden 2 ml Wasser gegeben und das Gemisch wurde homogenisiert. Zu
einer aliquoten Menge des homogenisierten Gemisches wurde das 3-fache
Ethanolvolumen gegeben, einschließlich der inneren Standardsubstanzen,
gefolgt von Rühren.
Zu der cerebrospinalen Flüssigkeit
wurde das 3-fache Ethanolvolumen, das die inneren Standardsubstanzen
enthält,
gegeben, gefolgt von Rühren.
Die vorstehenden Proben wurden bei –20°C für 20 Minuten belassen und durch
Zentrifuge (4°C,
12 000 G für
10 Minuten) getrennt. Der Überstand
wurde durch LC/MS/MS analysiert und dann jede Konzentration in Plasma, Hirn
und cerebrospinaler Flüssigkeit
durch das relative Eichkurvenverfahren gemessen.
-
Folglich
zeigte beispielsweise die Verbindung von Beispiel 7 cerebrale Konzentration:
2,95 nM/g, cerebrospinale Konzentration: 0,032 μM und Plasmakonzentration: 0,73 μM in 2 Stunden
nach oraler Verabreichung (10 mg/kg).
-
Die
Verbindungen der allgemeinen Formel (I) können oral oder parenteral verabreicht
werden und können
in der Form, die zur Verabreichung geeignet ist, formuliert werden,
unter Bereitstellung eines Mittels zur Behandlung von beispielsweise
cardiovaskulären
Störungen
(zum Beispiel Bluthochdruck, Nephropathie, Herzerkrankung, Vasospasmus,
Arteriosklerose), Störungen
des zentralen Nervensystems (beispielsweise Bulimie, Depression,
Angstzustand, Anfall, Epilepsie, Demenz, Schmerz, Alkoholismus,
Drogenentzug), metabolische Erkrankungen (beispielsweise Fettsucht,
Diabetes, Hormonabnormalität,
Hypercholesterolämie, Hyperlipidämie), sexuelle
und reproduktive Fehlfunktion, gastrische-intestinale Störung, wie
die Inhibierung der gastro-intestinalen Motilität, Atmungsstörung, Entzündung oder
Glaukom, oder derglei chen, bereitstellen. Bei der klinischen Verwendung
können
die erfindungsgemäßen Verbindungen,
nachdem sie formuliert wurden, zusammen mit pharmazeutisch verträglichen
Zusätzen,
in eine geeignete Zubereitung gemäß der Verabreichungsart verabreicht
werden. Für
die Additive, welche die üblicherweise
auf dem Gebiet der pharmazeutischen Formulierung verwendet werden,
können
beispielsweise Gelatine, Lactose, Saccharose, Titanoxid, Stärke, kristalline
Cellulose, Hydroxypropylmethylcellulose, Carboxymethylcellulose,
Maisstärke,
mikrokristallines Wachs, weiße
Vaseline, Magnesiumaluminometasilikat, wasserfreies Calciumphosphat,
Zitronensäure, Trinatriumcitrat,
Hydroxypropylcellulose, Sorbit, Sorbitanfettsäureester, Polysorbat, Saccharosefettsäureester, Polyoxyethylen,
hydriertes Rizinusöl,
Polyvinylpyrrolidon, Magnesiumstearat, leichte wasserfreie Kieselsäure, Talkum,
Pflanzenöl,
Benzylalkohol, Gummi arabicum, Propylenglycol, Polyalkylenglycol,
Cyclodextrin oder Hydroxypropylcyclodextrin, angewendet werden.
-
Ein
Gemisch mit den Additiven kann in Form von festen Zubereitungen
(beispielsweise Tabletten, Kapseln, Granulaten, Pulver, Suppositorien);
oder flüssigen
Zubereitungen (beispielsweise Sirupe, Elixiere, Injektionen) formuliert
werden. Solche Zubereitungen können
gemäß den auf
dem Fachgebiet der pharmazeutischen Formulierung gut bekannten Techniken
formuliert werden. Flüssige
Zubereitungen können
in Form von Zubereitungen vorliegen, die in Wasser oder anderen
geeigneten Medien gelöst
oder suspendiert sind, wenn angewendet, und insbesondere können injizierbare
Zubereitungen in physiologischer Salzlösung oder Glucoselösung, falls
erforderlich, gegebenenfalls zusammen mit einem Puffer und Konservierungsmittel,
gelöst
oder suspendiert werden.
-
Solche
Zubereitungen können
1,0 bis 100 Gewichtsprozent, vorzugsweise 1,0 bis 60 Gewichtsprozent,
der erfindungsgemäßen Verbindungen
enthalten, und können
auch therapeutisch wirksame andere Verbindungen enthalten.
-
Die
erfindungsgemäßen Verbindungen
können
in Kombination mit anderen Mitteln verwendet werden, die zum Behandeln
metabolischer und/oder Essstörungen
verwendbar sind. Die einzelnen Komponenten von solchen Kombinationen
können
getrennt bei verschiedenen Zeiten während des Verlaufs der Therapie
oder gleichberechtigt in geteilten oder einzelnen Dosierungsformen
verabreicht werden. Die vorliegende Erfindung ist deshalb so zu
verstehen, dass alle solchen Regimes von gleichzeitiger oder alternierender
Behandlung umfasst sind, und der Begriff „Verabreichen" ist demgemäß zu interpretieren.
Es ist selbstverständlich,
dass der Umfang von Kombinationen der erfindungsgemäßen Verbindungen
mit anderen Mitteln, die zum Behandeln von metabolischen und/oder
Essstörungen
verwendbar sind, im Prinzip beliebige Kombination mit beliebigen pharmazeutischen
Zusammensetzungen einschließt,
die für
das Behandeln von metabolischen und/oder Essstörungen verwendbar sind.
-
Wenn
die erfindungsgemäßen Verbindungen
klinisch verwendet werden, kann die Dosis oder Häufigkeit der Dosierung in Abhängigkeit
von dem Geschlecht, Alter, Körpergewicht,
dem Grad von Symptomen und der Art und dem Bereich der gewünschten
Behandlungseffekte variiert werden. Eine tägliche Dosis für einen Erwachsenen
ist 0,01 bis 100 mg/kg, vorzugsweise 0,03 bis 1 mg/kg oral in einer
einzelnen oder geteilten Dosis pro Tag, oder 0,001 bis 10 mg/kg,
vorzugsweise 0,001 bis 0,1 mg/kg parenteral, vorzugsweise in einer
einzigen oder geteilten Dosis pro Tag.
-
Ein
Durchschnittsfacharzt, Veterinär
oder Klinikarzt kann die wirksame Menge des zum Inhibieren, Bekämpfen oder
Aufhaltens des Fortschritts des Zustands erforderlichen Arzneistoffs
bestimmen und vorschreiben.
-
Beste Ausführungsform
der Erfindung
-
Die
vorliegende Erfindung wird insbesondere durch die Anwendung von
Beispielen und Bezugsbeispielen erläutert, jedoch ist die Erfindung
nicht auf jene Beispiele begrenzt.
-
Die
Verbindung mit dem Symbol „*" in der Strukturformel
bedeutet die Verbindung einer im Wesentlichen einzelnen Konfiguration
am asymmetrischen Kohlenstoffatom, an das das Symbol angebracht
ist.
-
Der
Schmelzpunkt wurde durch Anwenden von Modell MP-S3 (hergestellt von Yanagimoto Co. Ltd.) bestimmt
und das Ergebnis wurde ohne Korrigieren angegeben. Auch wurde das
Massenspektrum durch das Elektrospray-Ionisationsverfahren (ESI),
unter Verwendung von QuattroII (Micromass Ltd.) bestimmt.
-
Beispiel
1
Herstellung von 2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-2-imidazolin
-
Zu
einer Lösung
von 1,1-Bis(4-fluorphenyl)-1,2-ethandiamin
(500 mg) in Methanol (10 ml) wurde 3-Cyanobenzolimidsäuremethylester
(792 mg) gegeben und das Gemisch wurde 18 h bei Raumtemperatur gerührt. Nach
Auf konzentrieren des Reaktionsgemisches wurde der Rückstand
in Essigsäureethylester
(30 ml) gelöst, mit
gesättigter
wässriger
Natriumhydrogencarbonatlösung
und gesättigter
wässriger
Natriumchloridlösung,
in dieser Reihenfolge, gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und das
organische Lösungsmittel
wurde im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 3 : 1 → 2
: 1) gereinigt, unter Gewinnung der Zielverbindung (370 mg).
Massenspektrum
(ESI): 360,2
1HNMR (300 MHz, CDCl3, δ ppm):
4,15 (1,4H, brs), 4,57 (0,7H, brs), 5,10 (0,7H, brs), 5,38 (0,3H,
brs), 6,94–7,08
(4H, m), 7,22–7,43
(4H, m), 7,54 (1H, t, J = 7,8 Hz), 7,74 (1H, d, J = 7,8 Hz), 8,11
(1H, d, J = 7,6 Hz), 8,19 (1H, s)
-
Beispiel
2
Herstellung von 4,4-Bis(4-fluorphenyl)-2-pyrazinyl-2-imidazolin
-
Zu
einer Lösung
von 1,1-Bis(4-fluorphenyl)-1,2-ethandiamin
(500 mg) in Methanol (10 ml) wurde Pyrazinimidsäuremethylester (385 mg) gegeben
und das Gemisch wurde 18 h bei Raumtemperatur gerührt. Nach
Auf konzentrieren des Reaktionsgemisches wurde der Rückstand
in Essigsäureethylester
(50 ml) gelöst, mit
gesättigter
wässriger
Natriumhydrogencarbonatlösung
und gesättigter
wässriger
Natriumchloridlösung,
in dieser Reihenfolge, gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und das
organische Lösungsmittel
wurde im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie (C-300;
Hexan : Essigsäureethylester
= 1 : 1) gereinigt, unter Gewinnung der Zielverbindung (446 mg).
Schmelzpunkt
144–146°C
1HNMR (300 MHz, CDCl3, δ ppm): 4,17
(1,4H, brs), 4,65 (0,6H, brs), 6,08 (0,7H, brs), 6,51 (0,3H, brs), 6,96–7,04 (4H,
m), 7,26–7,50
(4H, m), 8,54 (1H, s) 8,66 (1H, d, J = 2,4 Hz), 9,45 (0,3H, brs),
9,56 (0,7H, brs)
-
Beispiel
3
Herstellung von 2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(1,5-naphthyridin-3-yl)-2-imidazolin
-
Zu
einer Lösung
von 1-(4-Fluorphenyl)-1-(1,5-naphthyridin-3-yl)-1,2-ethandiamin
(17 mg) in Methanol (1 ml) wurde 3-Cyanobenzolimidsäuremethylester
(23 mg) gegeben und das Gemisch wurde 19 Stunden bei Raumtemperatur
gerührt.
Zu dem Reaktionsgemisch wurde Chloroform gegeben und unlösliche Stoffe
wurden durch Filtration entfernt, gefolgt von Aufkonzentrierung
des Filtrats im Vakuum. Der Rückstand
wurde durch präparative
Dünnschicht-Chromatographie
(100 Essigsäureethylester)
gereinigt, unter Gewinnung der Zielverbindung (8,0 mg).
Massenspektrum
(ESI): 394,0
1HNMR (300 MHz, CDCl3, δ ppm):
4,21–4,26
(1H, m), 4,39–4,44
(1H, m), 5,15 (1H, brs), 7,02–7,09
(2H, m), 7,45–7,51
(1H, m), 7,58 (2H, t, J = 7,8 Hz), 7,60–7,65 (1H, m), 7,78 (1H, dt,
J = 1,4 Hz, 7,7 Hz), 8,13–8,17
(1H, m), 8,22 (1H, brs), 8,36–8,41
(2H, m), 8,95–8,97
(1H, m), 9,06 (1H, brs)
-
Beispiel
4
Herstellung von 2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-5-methyl-2-imidazolin
-
1,1-Bis(4-fluorphenyl)-1,2-propandiamin
(50 mg) wurde in Methanol (1 ml) gelöst und zu der Lösung wurde
3-Cyanobenzolimidsäuremethylester
(75 mg) gegeben. Das Gemisch wurde 24 Stunden bei Raumtemperatur
gerührt.
Nach dem Chloroform (5 ml) zu dem Reaktionsgemisch gegeben wurde,
wurden unlösliche Stoffe
durch Filtration entfernt. Nach Aufkonzentrieren des Filtrats im
Vakuum wurde der erhaltene Rückstand durch
Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 3 : 1 → 1
: 1) gereinigt, unter Gewinnung der Zielverbindung (10 mg).
Massenspektrum
(ESI): 374,1
1HNMR (300 MHz, CDCl3, δ ppm):
0,92 (3H, d, J = 6,6 Hz), 4,60–5,40
(2H, m), 7,40–7,50
(2H, m), 7,52–7,57 (1H,
m), 7,72–7,76
(1H, m), 8,10 (1H, d, J = 8,0 Hz), 8,17 (1H, s)
-
Beispiel
5
Herstellung von 2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-5-hydroxymethyl-2-imidazolin
-
1,1-Bis(4-fluorphenyl)-3-methoxymethoxy-1,2-propandiamin
(218 mg) wurde in Methanol (3 ml) gelöst und zu der Lösung wurde
3-Cyanobenzolimidsäuremethylester
(266 mg) gegeben und das Gemisch wurde 17 Stunden bei Raumtemperatur
gerührt.
Zu dem Reaktionsgemisch wurde eine weitere Menge 3-Cyanobenzolimidsäuremethylester
(266 mg) gegeben und das Reaktionsgemisch wurde bei Raumtemperatur
6 Stunden gerührt.
Zu dem Reaktionsgemisch wurde Essigsäureethylester (50 ml) gegeben
und die organische Schicht wurde mit gesättigter wässriger Natriumhydrogencarbonatlösung und
gesättigter
wässriger
Natriumchloridlösung,
in dieser Reihenfolge, gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und dann
wurde das organische Lösungsmittel
im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 2 : 1) gereinigt, unter Gewinnung von 2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-5-[(methoxymethoxy)methyl]-2-imidazolin
(146 mg) als farbloses Öl.
Die geschützte
Verbindung (115 mg) wurde in der Mischlösung von 2 N Salzsäure (1,5
ml) und Tetrahydrofuran (1,5 ml) gelöst und die Lösung wurde
für 3 Tage
bei 50°C
gerührt.
Nach Kühlen
des Gemisches auf Raumtemperatur durch Stehen desselben bei Raumtemperatur
und Neutralisieren mit gesättigter
wässriger
Natriumhydrogencarbonatlösung
wurde das Gemisch mit Essigsäureethylester
extrahiert. Die organische Schicht wurde mit gesättigter wässriger Natriumchloridlösung gewaschen
und über
wasserfreiem Natriumsulfat getrocknet. Natriumsulfat wurde durch
Filtration entfernt und das organische Lösungsmittel wurde im Vakuum
verdampft, unter Gewinnung der Titelverbindung (105 mg).
Massenspektrum
(ESI): 390,0
1HNMR (300 MHz, CDCl3, δ ppm):
3,09, 3,30 [2H (3,09, brs), (3,30, dd, J = 4,5 Hz, 10,5 Hz)], 4,68
(1H, brs), 5,59 (1H, brs), 7,01 (4H, m), 7,23 (2H, m), 7,48 (2H,
brs), 7,56 (1H, t, J = 7,8 Hz), 7,76 (1H, d, J = 7,8 Hz), 8,11 (1H,
d, J = 7,8 Hz), 8,21 (1H, s)
-
Gemäß einem
der in dem vorstehend erwähnten
Beispiel 1 bis 5 beschriebenen Verfahren wurden das entsprechende
Diamin und Imidsäuremethylester
umgesetzt, unter Gewinnung der nachstehenden Verbindungen.
-
Beispiel 5-1
-
2-(3-Cyanophenyl)-4,4-bis(3-fluorphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 360,1
-
Beispiel 5-2
-
2-(3-Cyanophenyl)-4-(3-chinolyl)-4-(2-thienyl)-2-imidazolin
-
- Massenspektrum (ESI): 381,0
-
Beispiel 5-3
-
4-(4-Bromphenyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 402,0
-
Beispiel 5-4
-
4,4-Bis(4-chlorphenyl)-2-(3-cyanophenyl)-2-imidazolin
-
- Massenspektrum (ESI): 392,0
-
Beispiel 5-5
-
4-(4-Chlorphenyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 358,1
-
Beispiel 5-6
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-chinolyl)-2-imidazolin
-
- Massenspektrum (ESI): 393,1
-
Beispiel 5-7
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 342,2
-
Beispiel 5-8
-
2-(3-Cyanophenyl)-4-phenyl-4-(4-vinylphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 350,1
-
Beispiel 5-9
-
4-(6-Chlor-3-pyridyl)-2-(3-cyanophenyl)-4-(4-fluorphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 377,0
-
Beispiel 5-10
-
4-(5-Chlor-2-thienyl)-2-(3-cyanophenyl)-4-(4-fluorphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 382,1
-
Beispiel 5-11
-
2-(3-Cyanophenyl)-4-(4-methylphenyl)-4-phenyl-2-imidazolin
-
-
Beispiel 5-12
-
2-(3-Cyanophenyl)-4-(3-methylphenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 338,1
-
Beispiel 5-13
-
4,4-Bis(4-fluorphenyl)-2-(3-nitrophenyl)-2-imidazolin
-
-
Beispiel 5-14
-
2-(3-Cyanophenyl)-4-phenyl-4-(3-chinolyl)-2-imidazolin
-
- Massenspektrum (ESI): 375,2
-
Beispiel 5-15
-
2-(3-Cyanophenyl)-4-(4-methoxyphenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 354,0
-
Beispiel 5-16
-
4-(3-Chlorphenyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolinhydrochlorid
-
- Massenspektrum (ESI): 358,1
-
Beispiel 5-17
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(5-methyl-2-thienyl)-2-imidazolin
-
- Massenspektrum (ESI): 362,1
-
Beispiel 5-18
-
2-(3-Cyanophenyl)-4-phenyl-4-(4-trifluormethylphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 392,1
-
Beispiel 5-19
-
2-(3-Cyanophenyl)-4-(3-methoxyphenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 354,1
-
Beispiel 5-20
-
2-(3-Cyanophenyl)-4-(3,4-dimethylphenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 352,1
-
Beispiel 5-21
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-vinylphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 368,1
-
Beispiel 5-22
-
2-(3-Cyanophenyl)-4-(2-naphthyl)-4-(3-pyridyl)-2-imidazolin
-
- Massenspektrum (ESI): 375,1
-
Beispiel 5-23
-
4-(3-Bromphenyl)-2-(3-cyanophenyl)-4-(4-fluorphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 420,1
-
Beispiel 5-24
-
4-(2-Chlorphenyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 358,1
-
Beispiel 5-25
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(6-chinolyl)-2-imidazolin
-
- Massenspektrum (ESI): 393,1
-
Beispiel 5-26
-
2-(3-Cyanophenyl)-4-phenyl-4-(2-thienyl)-2-imidazolin
-
- Massenspektrum (ESI): 330,3
-
Beispiel 5-27
-
2-(3-Cyanophenyl)-4-(4-dimethylaminophenyl)-4-phenyl-2-imidazolin
-
-
Beispiel 5-28
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-pyridyl)-2-imidazolin
-
- Massenspektrum (ESI): 343,0
-
Beispiel 5-29
-
2-(3-Cyanophenyl)-4-(4-formylphenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 352,2
-
Beispiel 5-30
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(2-thienyl)-2-imidazolin
-
- Massenspektrum (ESI): 348,1
-
Beispiel 5-31
-
4,4-Diphenyl-2-pyrazinyl-2-imidazolin
-
-
Beispiel 5-32
-
2-(3-Cyanophenyl)-4,4-bis(4-methoxyphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 384,1
-
Beispiel 5-33
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-formylphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 370,1
-
Beispiel 5-34
-
4,4-Bis(4-fluorphenyl)-2-(3-methylsulfonylphenyl)-2-imidazolin
-
-
Beispiel 5-35
-
4,4-Bis(4-fluorphenyl)-2-(3-formylphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 363,1
-
Beispiel 5-36
-
2-(3-Cyanophenyl)-4-(2-methylphenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 338,1
-
Beispiel 5-37
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(3-hydroxymethylphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 372,1
-
Beispiel 5-38
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(1,2,3,4-tetrahydro-1,5-naphthyridin-7-yl)-2-imidazolin
-
- Massenspektrum (ESI): 398,1
-
Beispiel 5-39
-
2-(3-Cyanophenyl)-4-(2-fluorphenyl)-4-(3-pyridyl)-2-imidazolin
-
- Massenspektrum (ESI): 343,0
-
Beispiel 5-40
-
4-(4-Biphenylyl)-2-(3-cyanophenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 400,3
-
Beispiel 5-41
-
2-(3-Cyanophenyl)-4-(2-methoxyphenyl)-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 354,0
-
Beispiel 5-42
-
2-(3-Cyanophenyl)-4-cyclohexyl-4-phenyl-2-imidazolin
-
-
Beispiel 5-43
-
2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(4-pyridyl)-2-imidazolin
-
- Massenspektrum (ESI): 343,0
-
Beispiel 5-44
-
4,4-Bis(4-fluorphenyl)-2-(5-pyrimidinyl)-2-imidazolin
-
- Massenspektrum (ESI): 337,0
-
Beispiel 5-45
-
2-(3-Cyanophenyl)-4-cyclopentyl-4-phenyl-2-imidazolin
-
- Massenspektrum (ESI): 316,2
-
Beispiel 5-46
-
2-(3-Cyanophenyl)-4-cyclobutyl-4-phenyl-2-imidazolin
-
-
Beispiel 5-47
-
4,4-Bis(4-fluorphenyl)-2-(3-pyridazinyl)-2-imidazolin
-
- Massenspektrum (ESI): 337,0
-
Beispiel 5-48
-
4-(4-Fluorphenyl)-5-methyl-2-pyrazinyl-4-(3-chinolyl)-2-imidazolin
-
- 1HNMR (400 MHz, CD3OD, δ ppm):
0,90 (3H, d, J = 6,4 Hz), 4,97–5,08
(1H, m), 7,06–7,16
(2H, m), 7,32–7,38 und
7,57–7,66
(3H, m), 7,72–7,80
(1H, m), 7,86–8,04
(2H, m), 8,31 (0,8H, s), 8,51 (0,2H, s), 8,64 (0,8H, s), 8,66–8,76 (2H,
m), 8,88 (0,2H, s), 9,34 (0,2H, s), 9,39 (0,8H, s)
-
Beispiel 5-49
-
Das optisch aktive (5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-2-(3-methylsulfonylphenyl)-5-methyl-2-imidazolin,
worin das Ausgangsmaterial das in Bezugsbeispiel 5-1 beschriebene
Diamin ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,90 (3H, d, J = 6,3 Hz), 3,10 (3H, s), 4,72–4,74 (1H, m), 5,10–5,40 (1H, brs),
7,00–8,37
(11H, m) [α]D 25: –263,3° (c0,9, CHCl3)
-
Beispiel 5-50
-
Das optisch aktive (5S)-2-(5-Cyano-3-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 5-1 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,90 (3H, d, J = 6,3 Hz), 4,75 (1H, q, J = 6,3), 5,38 (1H, s), 6,89
(1H, dd, J = 2,9 Hz, 8,6 Hz), 7,03 (2H, t, J = 8,6 Hz), 7,20–7,30 (2H,
m), 7,89 (1H, dt, J = 2,2 Hz, 7,9 Hz), 8,34 (1H, s), 8,50–8,60 (1H,
m), 8,96 (1H, d, J = 2,0 Hz), 9,23 (1H, d, J = 2,0 Hz) [α]D 25: –307,6° (c1,0, MeOH)
-
Beispiel
6
Herstellung des optisch aktiven (5S)-2-(2-Cyano-4-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolinhydrochlorids
-
Zu
einer Lösung
von 4-Cyanopyridin-N-oxid (100 mg) in Methanol (4 ml) wurden Natriummethoxid (13,5
mg) gegeben und das Gemisch wurde 24 Stunden bei Raumtemperatur
gerührt.
Zu dem Reaktionsgemisch wurde Methansulfonsäure (104 mg) gegeben und das
Reaktionsgemisch wurde weitere 10 Minuten bei Raumtemperatur gerührt. Zu
den 3,5 ml dieser Lösung
wurde die Lösung
(2 ml) von (2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamin,
die in Bezugsbeispiel 5-1 beschrieben wurde, gegeben und das Gemisch
wurde 15 Stunden bei Raumtemperatur gerührt. Nach Auf konzentrieren
des Reaktionsgemisches im Vakuum wurde der erhaltene Rückstand
in Essigsäureethylester
gelöst,
mit gesättigter
wässriger
Natriumhydrogencarbonatlösung
und gesättigter
wässriger
Natriumchloridlösung,
in dieser Reihenfolge, gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und das
organische Lösungsmittel
wurde im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchroma tographie
(C-300; Chloroform : Methanol = 9 : 1) gereinigt, unter Gewinnung
von 2-(1-Oxido-pyridin-4-yl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin
(172 mg) als farbloses Öl.
Zu der Lösung (5
ml) der erhaltenen N-Oxidverbindung in Acetonitril wurden Trimethylsilylcyanid
(140 mg) und Triethylamin (142 mg) gegeben und das Gemisch wurde
17 Stunden bei 90°C
gerührt.
Zu dem Gemisch wurden Trimethylsilylcyanid (140 mg) und Triethylamin
(142 mg) gegeben und das Gemisch wurde bei 90°C für 2 Stunden gerührt und
im Vakuum auf konzentriert. Der erhaltene Rückstand wurde in Chloroform
gelöst,
mit gesättigter wässriger
Natriumhydrogencarbonatlösung,
Wasser und gesättigter
wässriger
Natriumchloridlösung,
in dieser Reihenfolge, gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und das
organische Lösungsmittel
wurde im Vakuum eingedampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie (C-300;
Hexan : Essigsäureethylester
= 1 : 1) gereinigt und mit 4 N Chlorwasserstoff-Essigsäureethylester-Lösung behandelt,
unter Gewinnung der Titelverbindung (108 mg).
1HNMR
(300 MHz, DMSO-d6, δ ppm): 0,95 (3H, d, J = 6,6
Hz), 5,40 (1H, brs), 7,23–7,36
(5H, m), 8,22–8,28 (1H,
m), 8,47 (1H, d, J = 4,8 Hz), 8,58 (1H, s), 8,83 (1H, s), 9,12 (1H,
d, J = 5,1 Hz)
[α]D 25: –309,7° (c1,0, MeOH)
-
Durch
Anwenden des in Beispiel 6 beschriebenen Verfahrens wurden das entsprechende
Diamin und 4-Cyanopyridin-N-oxid
Ringkondensationsreaktion unterzogen, gefolgt von Umsetzen mit Trimethylsilylcyanid, gegebenenfalls
wurde die Schutzgruppe durch das Verfahren, ähnlich zu jenem in Beispiel
5 beschriebenen Verfahren entfernt, unter Gewinnung der nachstehenden
Verbindungen.
-
Beispiel 6-1
-
(5R)-2-(2-Cyano-4-pyridyl)-4,4-bis(4-fluorphenyl)-5-hydroxymethyl-2-imidazolin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
3,09 (1H, br), 3,29 (1H, dd, J = 4,4 Hz, 10,6 Hz), 4,74 (0,9H, brs),
5,02 (0,1H, d, J = 5,1 Hz), 5,74 (1H, brs), 6,97–7,05 (4H, m), 7,19–7,23 (2H,
m), 7,42–7,47
(2H, m), 7,92 (1H, d, J = 4,1 Hz), 8,21 (1H, s), 8,81 (1H, d, J
= 5,5 Hz)
-
Beispiel 6-2
-
(5S)-2-(2-Cyano-4-pyridyl)-4,4-bis(4-fluorphenyl)-5-methyl-2-imidazolin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,85–0,97
(3H, m), 4,70–4,78
(1H für
das größere, m),
4,92–5,00
(1H für das
kleinere, m), 5,02 (1H für
das größere, brs),
5,32 (1H für
das kleinere, brs), 6,92–7,07
(4H, m), 7,10–7,53 (4H,
m), 7,87–7,92
(1H, m), 8,10–8,20
(1H, m), 8,77–8,83
(1H, m)
- [α]D 25: –300,0° (c1,0, CHCl3)
-
Beispiel 6-3
-
Das optisch aktive (5S)-2-(2-Cyano-4-pyridyl)-4-(4-fluorphenyl)-4-(2-floor-4-pyridyl)-5-methyl-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 7 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,93 (3H, d, J = 6,6 Hz), 4,71 (1H, q, J = 6,6 Hz), 5,30 (1H, brs),
6,98–7,12 (3H,
m), 7,15–7,24
(3H, m), 7,94 (1H, dd, J = 5,0 Hz, 1,6 Hz), 8,15–8,22 (2H, m), 8,83 (1H, d,
J = 5,0 Hz)
- [α]D 25: –260,0° (c1,0, CHCl3)
-
Beispiel 6-4
-
(5S)-2-(2-Cyano-4-pyridyl)-4,4-bis(6-fluor-3-pyridyl)-5-methyl-2-imidazolinhydrochlorid
-
- 1HNMR (400 MHz, CD3ΟD, δ ppm): 1,15
(3H, d, J = 6,4 Hz), 5,51 (1H, q, J = 6,4 Hz), 7,19 (1H, dd, J =
8,8 Hz, 2,8 Hz), 7,24 (1H, dd, J = 8,8 Hz, 2,8 Hz), 7,84–7,89 (1H,
m), 8,09–8,12
(1H, m), 8,17 (1H, dd, J = 5,2 Hz, 1,6 Hz), 8,23 (1H, d, J = 2,4
Hz), 8,40 (1H, d, J = 1,6 Hz), 8,45 (1H, d, J = 2,4 Hz), 9,07 (1H,
d, J = 5,2 Hz)
- [α]D 25: –239,5° (c0,8, MeΟH)
-
Beispiel
7
Herstellung von 4,4-Bis(4-fluorphenyl)-5-methyl-2-pyrazinyl-2-imidazolin
-
1,1-Bis(4-fluorphenyl)-1,2-propandiamin
(94 mg) und Pyrazincarbonsäure
(53 mg) wurden in Pyridin (1 ml) gelöst und zu der Lösung wurde
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid (103 mg) gegeben. Das Gemisch
wurde 24 Stunden bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde
aufkonzentriert und der erhaltene Rückstand wurde in Chloroform
gelöst,
gefolgt von Waschen mit gesättigter
wässriger
Natriumhydrogencarbonatlösung.
Die wässrige
Schicht wurde zweimal mit Chloroform extrahiert und die vereinigten Chloroformschichten
wurden über
Magnesiumsulfat getrocknet. Nach Verdampfen des Lösungsmittels
im Vakuum wurde der erhaltene Rückstand
durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 1 : 1 → 1
: 3) gereinigt, unter Gewinnung von N-[2-Amino-2,2-bis(4-fluorphenyl)-1-methylethyl]-2-pyrazincarboxamid
(112 mg) als das Gemisch mit Pyrazincarbonsäuremethylester. Das Gemisch
wurde in Methanol (1 ml) gelöst
und zu der Lösung
wurde 2 N wässrige
Natriumhydroxidlösung
(0,5 ml) gegeben und die Lösung
wurde 1 Stunde bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde
mit Chloroform verdünnt, gefolgt
von Waschen mit Wasser. Die wässrige
Schicht wurde zweimal mit Chloroform extrahiert und die vereinigten
Chloroformschichten wurden über
wasserfreiem Magnesiumsulfat getrocknet. Das Lösungsmittel wurde im Vakuum
verdampft, unter Gewinnung der reinen Amidverbindung (95 mg).
-
Zu
einer Lösung
der vorstehend erwähnten
Amidverbindung (157 mg) in Benzol (4 ml) wurde Phosphorpentachlorid
(266 mg) gegeben und das Gemisch wurde 3 Tage bei 50°C gerührt. Nach
Kühlen
auf Raumtemperatur wurde zu dem Reaktionsgemisch 2 N wässrige Natriumhydroxidlösung (4
ml) gegeben und das Reaktionsgemisch wurde für weitere 30 Minuten gerührt. Das
erhaltene Gemisch wurde dreimal mit Chloroform extrahiert und über wasserfreiem
Magnesiumsulfat getrocknet. Das Lösungsmittel wurde im Vakuum
verdampft und der erhaltene Rückstand
wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 1 : 1 → 1
: 2) gereinigt, unter Gewinnung der Zielverbindung (100 mg).
Massenspektrum
(ESI): 351,2
1HNMR (300 MHz, CDCl3, δ ppm):
0,90 (2,1H, d, J = 6,4 Hz), 0,99 (0,9H, d, J = 6,9 Hz), 4,70 (0,7H,
q, J = 6,4 Hz), 5,03 (0,3H, q, J = 6,9 Hz), 5,97–6,00 (0,7H, m), 6,43–6,45 (0,3H,
m), 6,95–7,08
(4H, m), 7,25–7,37
(2,6H, m), 7,51–7,56
(1,4H, m), 8,53–8,55
(1H, m), 8,66 (1H, d, J = 2,5 Hz), 9,42 (0,3H, d, J = 1,7 Hz), 9,54
(0,7H, d, J = 1,5 Hz)
-
Durch
Anwenden des in Beispiel 7 beschriebenen Verfahrens wurden das entsprechende
Diamin und Carbonsäure
cyclisiert und kondensiert, gefolgt von gegebenenfalls Schutzgruppenentfernen
durch das Verfahren, ähnlich
zu dem in Beispiel 5 beschriebenen Verfahren, unter Gewinnung der
nachstehenden Verbindungen.
-
Beispiel 7-1
-
4,4-Bis(4-fluorphenyl)-5-hydroxymethyl-2-pyrazinyl-2-imidazolin
-
- Massenspektrum (ESI): 367,1
- 1HNMR (300 MHz, CDCl3, δ ppm): 3,12,
3,30 [2H (3,12, t, J = 9,6 Hz), (3,30, m)], 4,66, 5,08 [1H (4,66,
dd, J = 3,9, 8,7 Hz), (5,08, m)], 6,55, 6,60 [1H (6,55, s), (6,60,
s)], 7,00 (4H, m), 7,14 (2H, m), 7,35, 7,54 [2H (7,35, dd, J = 4,5
Hz, 5,7 Hz), (3,30, dd, J = 5,4 Hz, 8,1 Hz)], 8,54 (1H, d, J = 2,0
Hz), 8,66 (1H, d, J = 2,0 Hz), 9,43, 9,52 [1H (9,43, s), (9,52,
s)]
-
Beispiel 7-2
-
5-Ethyl-4,4-bis(4-fluorphenyl)-2-pyrazinyl-2-imidazolin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,95–1,35
(3H, m), 4,36–4,45
(1H für
das größere, m),
4,73–4,79
(1H für das
kleinere, m), 6,21–6,29
(1H für
das größere, br),
6,42–6,44
(1H für
das kleinere, br), 6,90–7,18
(4H, m), 7,12–7,21
(2H, m), 7,29–7,60
(2H, m), 8,50–8,58
(1H, m), 8,61–8,68
(1H, m), 9,44 (1H, d, J = 1,5 Hz für das kleinere), 9,55 (1H,
d, J = 1,5 Hz für
das größere)
-
Beispiel 7-3
-
4,4-Bis(4-fluorphenyl)-5-propyl-2-pyrazinyl-2-imidazolinhydrochlorid
-
- 1HNMR (300 MHz, CD3OD, δ ppm):
0,87 (3H, t, J = 7,0 Hz), 1,14–1,66
(4H, m), 5,10–5,20
(1H, m), 7,10–7,30 (6H,
m), 7,54–7,65
(2H, m), 8,87–8,94
(1H, m), 8,95–9,00
(1H, m), 9,36–9,40
(1H, br)
-
Beispiel
8
Herstellung von 4,4-Bis(4-fluorphenyl)-2-(4-isothiazolyl)-2-imidazolin
-
Unter
Rühren
einer Lösung
von 1,1-(4-Fluorphenyl)-1,2-ethandiamin
(330 mg) in wasserfreiem Toluol (8 ml) bei 0°C wurde 0,98 M Trimethylaluminiumhexanlösung (1,36
ml) tropfenweise zu der Lösung
unter Stickstoff gegeben und das Gemisch wurde 5 Minuten bei 0°C gerührt. 4-Isothiazol carbonsäuremethylester
(120 mg) wurde in Toluol (1 ml) gelöst und die Lösung wurde
zu dem vorstehenden Reaktionsgemisch gegeben. Dann wurde das Gemisch
3 Stunden auf 110°C
erhitzt. Nachdem das Reaktionsgemisch gekühlt wurde, wurde Natriumsulfatdecahydrat
zu dem Reaktionsgemisch gegeben und dann wurde das Gemisch 15 Minuten
bei Raumtemperatur gerührt.
Wasserfreies Natriumsulfat wurde zu dem Reaktionsgemisch gegeben
und das Gemisch wurde für
weitere 20 Minuten gerührt.
Das erhaltene Gemisch wurde durch Celite filtriert und das Filtrat wurde
im Vakuum auf konzentriert. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie (C-300;
Hexan : Essigsäureethylester
= 1 : 1) gereinigt, unter Gewinnung der Zielverbindung (116 mg).
Massenspektrum
(ESI): 342,0
1HNMR (300 MHz, CDCl3, δ ppm):
4,26 (2H, brs), 6,97–7,07
(4H, m), 7,32–7,37
(4H, m), 8,93 (1H, s), 9,03 (1H, s)
-
Beispiel
9
Herstellung von 2-(3-Pyridyl)-4,4-bis(4-fluorphenyl)-2-imidazolinhydrochlorid
-
Unter
Rühren
einer Lösung
von 1,1-Bis(4-fluorphenyl)-1,2-ethandiamin
(300 mg) in wasserfreiem Toluol (3 ml) bei 0°C wurde eine Lösung von
0,98 M Trimethylaluminium in Hexan (1,23 ml) tropfenweise zu der Lösung unter
Stickstoff gegeben und das Gemisch wurde 15 Minuten bei Raumtemperatur
gerührt.
Nicotinsäurmethylester
(103 mg) wurde in Toluol (1 ml) gelöst und die Lösung wurde
zu dem vorstehenden Reaktionsgemisch gegeben. Dann wurde das Gemisch
5 Stunden auf 80°C
erhitzt. Nachdem das Reaktionsgemisch gekühlt wurde, wurde Natriumsulfatdecahydrat
zu dem Reaktionsgemisch gegeben und das Gemisch wurde 15 Minuten
bei Raumtemperatur gerührt.
Wasserfreies Natriumsulfat wurde zu dem Reaktionsgemisch gegeben
und das Gemisch wurde für
weitere 20 Minuten gerührt.
Das erhaltene Gemisch wurde durch Celite filtriert und das Filtrat
wurde im Vakuum auf konzentriert. Der erhaltene Rückstand
wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 1 : 2) gereinigt, mit 4 N Chlorwasserstoff-Essigsäureethylester-Lösung behandelt
und im Vakuum getrocknet, unter Gewinnung der Zielverbindung (111
mg).
Schmelzpunkt: 172–176°C
1HNMR (300 MHz, DMSO-d6, δ ppm): 4,70
(2H, s), 7,24–7,33
(4H, m), 7,53–7,60
(4H, m), 7,72–7,76
(1H, m), 8,60–8,65
(1H, m), 8,93–8,95
(1H, m), 9,36 (1H, s), 11,61 (1H, s), 12,29 (1H, s)
-
Durch
Anwenden der in den vorstehend erwähnten Beispielen 8 und 9 beschriebenen
Verfahren wurden das entsprechende Diamin und der Ester umgesetzt,
unter Gewinnung der nachstehenden Verbindungen.
-
Beispiel 9-1
-
2-(3-Chlorphenyl)-4,4-bis(4-fluorphenyl)-2-imidazolinhydrochlorid
-
- Massenspektrum (ESI): 369,0
-
Beispiel 9-2
-
4,4-Bis(4-fluorphenyl)-2-(3-trifluormethylphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 403,1
-
Beispiel 9-3
-
2-(3-Fluorphenyl)-4,4-bis(4-fluorphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 353,1
-
Beispiel 9-4
-
4,4-Bis(4-fluorphenyl)-2-(3-trifluormethoxyphenyl)-2-imidazolin
-
-
Beispiel 9-5
-
4,4-Bis(4-fluorphenyl)-2-(5-thiazolyl)-2-imidazolin
-
- Massenspektrum (ESI): 342,0
-
Beispiel 9-6
-
4,4-Bis(4-fluorphenyl)-2-(3-methylphenyl)-2-imidazolin
-
-
Beispiel 9-7
-
4,4-Bis(4-fluorphenyl)-2-(2-thienyl)-2-imidazolin
-
-
Beispiel 9-8
-
2-(5-Brom-3-pyridyl)-4,4-bis(4-fluorphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 414,0/416,0
-
Beispiel 9-9
-
2-(3-Bromphenyl)-4,4-bis(4-fluorphenyl)-2-imidazolin
-
- Massenspektrum (ESI): 412,9/415,0
-
Beispiel 9-10
-
4,4-Bis(4-fluorphenyl)-2-(3-thienyl)-2-imidazolin
-
-
Beispiel
10
Herstellung von 2-(4-Cyano-2-pyridyl)-4,4-bis(4-fluorphenyl)-2-imidazolin
-
Das
Gemisch von 1,1-Bis(4-fluorphenyl)-1,2-ethandiamin (153 mg) und
4-Cyano-2-ethoxycarbonylpyridin (95 mg) wurde 2 Stunden bei 180°C gerührt. Die
erhaltene ölige
Verbindung wurde durch Kieselgel-Säulenchromatographie (C-300;
Hexan : Essigsäureethylester
= 3 : 2) gereinigt, unter Gewinnung der Zielverbindung (103 mg).
Schmelzpunkt:
206–208°C
-
Beispiel
11
Herstellung des optisch aktiven (5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolins
-
3-Cyanobenzoesäure (84
mg), Triethylamin (159 μl)
und 2-Chlor-1,3-dimethylimidazoliniumchlorid-Dichlormethan 25%ige
Lösung
(212 μl)
wurden zu einer Lösung
von (2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamin
(100 mg), beschrieben in Bezugsbeispiel 5-1, in Chloroform (3 ml),
in dieser Reihenfolge, bei 0°C
gegeben. Dann wurde die Temperatur auf Raumtemperatur erhöht und das
Gemisch wurde 15 Minuten gerührt.
Gesättigte
wässrige
Natriumhydrogencarbonatlösung
wurde zu dem Reaktionsgemisch gegeben und dann wurde das Gemisch
mit Essigsäureethylester
extrahiert. Die organische Schicht wurde mit gesättigter wässriger Natriumchloridlösung gewaschen
und über
wasserfreiem Natriumsulfat getrocknet. Natriumsulfat wurde durch
Filtration entfernt und das organische Lösungsmittel wurde im Vakuum
verdampft. Das erhaltene Rohprodukt wurde für 15 Stunden bei 130°C gerührt. Das
erhaltene Öl
wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 1 : 1) gereinigt, unter Gewinnung der Zielverbindung (140 mg).
1HNMR (300 MHz, CDCl3, δ ppm): 0,89
(3H, d, J = 6,5 Hz), 4,66–4,74
(1H, m), 5,10 (1H, brs), 6,85–6,90
(1H, m), 7,02 (2H, t, J = 8,4 Hz), 7,20–7,30 (2H, m), 7,57 (1H, t,
J = 7,9 Hz), 7,74–7,79
(1H, m), 7,85–7,95
(1H, m), 8,07–8,12
(1H, m), 8,19 (1H, s), 8,37–8,42
(1H, m)
[α]D 25: –295,6° (c1,0, CHCl3)
-
Durch
Anwendung des in dem vorstehend erwähnten Beispiel 11 beschriebenen
Verfahrens wurden das entsprechende Diamin und Carbonsäure cyclisiert
und der Reaktant wurde gegebenenfalls durch das Verfahren, das ähnlich zu
dem in Beispiel 7 beschriebenen Verfahren ist, von den Schutzgruppen
befreit, unter Gewinnung der nachstehenden Verbindungen.
-
Beispiel 11-1
-
2-(3-Cyanophenyl)-4,4-bis(4-fluorphenyl)-5-(1-hydroxyethyl)-2-imidazolin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
1,08 (3H, d, J = 6,3 Hz), 3,54–3,72
(1H, m), 4,47–4,55
(1H für
das größere, m),
4,85–4,90
(1H für
das kleinere, m), 5,17 (1H für
das größere, brs),
5,48 (1H für
das kleinere, brs), 6,93–7,10 (4H,
m), 7,15–7,28
(2H, m), 7,22–7,60
(2H, m), 7,65–7,80
(2H, m), 8,07–8,25
(2H, m)
-
Beispiel 11-2
-
Das optisch aktive (5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin,
worin das Ausgangsmaterial das in Bezugsbeispiel 7 beschriebene
Diamin ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,92 (3H für
das größere, d,
J = 6,6 Hz), 1,02 (3H für
das kleinere, d, J = 6,9 Hz,), 4,66 (1H für das größere, q, J = 6,6 Hz), 4,87
(1H für
das kleinere, q, J = 6,9 Hz), 5,22 (1H für das größere, brs), 5,68 (1H für das kleinere,
brs), 6,97–7,09
(3H, m), 7,12 (1H, s), 7,15–7,35
(3H, m), 7,54–7,61 (1H,
m), 7,75–7,80
(1H, m), 8,08–8,23
(3H, m)
- [α]D 25: –291,6° (c1,0, CHCl3)
-
Beispiel 11-3
-
Das optisch aktive (5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin,
worin das Ausgangsmaterial das in Bezugsbeispiel 8 beschriebene
Diamin ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,98 (3H, d, J = 6,6 Hz), 4,74 (1H, q, J = 6,6 Hz), 4,98–5,08 (1H,
m), 5,07 (1H für
das größere, brs),
5,30 (1H für
das kleinere, brs), 6,91–7,12
(4H, m), 7,47–7,63
(3H, m), 7,78 (1H, d, J = 7,8 Hz), 8,08–8,21 (3H, m)
- [α]D 25: –258,97° (c0,16,
CHCl3)
-
Beispiel
12
Herstellung des optisch aktiven (5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-(1,2,5-thiadiazol-3-yl)-2-imidazolin
-
Triethylamin
(23 mg) und 1,1'-Carbonyldiimidazol
(37 mg) wurden zu einer Lösung
von 1,2,5-Thiadiazol-3-carbonsäure
(30 mg) in Tetrahydrofuran (3 ml) in dieser Reihenfolge gegeben
und das Gemisch wurde 1 Stunde bei Raumtemperatur gerührt. Eine
Lösung
von (2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamin (50 mg), beschrieben
in Bezugsbei spiel 5-1, in Tetrahydrofuran (1 ml) wurde zu dem vorstehenden Reaktionsgemisch
gegeben und das erhaltene Gemisch wurde 17 Stunden bei Raumtemperatur
gerührt.
Das Reaktionsgemisch wurde im Vakuum auf konzentriert und dann wurde
der erhaltene Rückstand
in Essigsäureethylester
gelöst.
Dann wurde die Lösung
mit Wasser und gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und das
organische Lösungsmittel
wurde im Vakuum verdampft. Das erhaltene Rohprodukt wurde in Toluol
(0,5 ml) gelöst.
Ytterbiumtrifluormethansulfonat (12 mg) wurde zu der Lösung gegeben
und dann wurde das Gemisch 10 Stunden bei 100°C gerührt. Das Reaktionsgemisch wurde
mit Essigsäureethylester verdünnt und
die Verdünnung
wurde mit gesättigter
wässriger
Natriumhydrogencarbonatlösung,
Wasser und gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen. Das Gemisch wurde über wasserfreiem
Natriumsulfat getrocknet. Natriumsulfat wurde durch Filtration entfernt
und das organische Lösungsmittel
wurde im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie (C-300;
Hexan : Essigsäureethylester
= 2 : 3) gereinigt, unter Gewinnung der Zielverbindung (37 mg) als
farbloses Öl.
1HNMR (300 MHz, CDCl3, δ ppm): 0,88,
0,98 (3,0H, d, J = 6,6 Hz), 4,70, 4,98 (1,0H, q, J = 6,6 Hz), 5,74,
6,06 (1H, brs), 6,88 (1H, dd, J = 3,0 Hz, 8,7 Hz), 6,99–7,05 (2H,
m), 7,24–7,29
(2H, m), 7,74–7,93
(2H, m), 8,26, 8,40 (1H, d, J = 1,8 Hz), 9,15, 9,19 (1H, s)
[α]D 25: –369,2° (c1,0, MeOH)
-
Durch
Anwenden des in dem vorstehend erwähnten Beispiel 12 beschriebenen
Verfahrens wurden das entsprechende Diamin und Carbonsäure cyclisiert
und kondensiert, und dann wurde der Reaktant gegebenenfalls durch
das Verfahren, das ähnlich
zu dem in Beispiel 7 beschriebenen Verfahren ist, von den Schutzgruppen
befreit, unter Gewinnung der nachstehenden Verbindungen.
-
Beispiel 12-1
-
(5R)-4,4-Bis(4-fluorphenyl)-5-hydroxymethyl-2-(6-hydroxy-2-pyridyl)-2-imidazolin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
3,13 (2H, ds, J = 6,6 Hz), 4,75 (1H, br), 6,68 (1H, d, J = 8,1 Hz),
6,85 (1H, d, J = 6,0 Hz), 6,95–7,00
(4H, m), 7,21–7,25
(4H, m), 7,39–7,53
(3H, m)
- [α]D 25: –269,6° (c0,5, MeOH)
-
Beispiel 12-2
-
(5S)-4,4-Bis(4-fluorphenyl)-2-(6-hydroxy-2-pyridyl)-5-methyl-2-imidazolin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,85–0,95
(3H, m), 4,68–4,95
(1H, m), 5,61 (1H, brs), 6,63–6,75
(2H, m), 6,90–7,07
(4H, m), 7,10–7,20
(2H, m), 7,30–7,51
(3H, m)
- [α]D 25: –305,4° (c0,7, CHCl3)
-
Beispiel 12-3
-
Das optisch aktive (5S)-2-(2-Chlor-4-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin,
worin das Ausgangsmaterial das in Bezugsbeispiel 5-1 beschriebene
Diamin ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,88 (3H, d, J = 6,6 Hz), 4,72–4,84
(1H, m), 5,05–5,15
(1H, brs), 6,90–6,98 (1H,
m), 7,05–7,15
(2H, m), 7,25–7,30
(2H, m), 7,65–7,72
(1H, m), 7,82–7,88
(1H, m), 7,90–7,98
(1H, m), 8,41–8,45
(1H, m), 8,48–8,51
(1H, m)
- [α]D 25: –294,0° (c0,5, MeOH)
-
Beispiel 12-4
-
Das optisch aktive (5S)-4-(4-Fluorphenyl)-4-(6-fluor-3-pyridyl)-2-(6-hydroxy-2-pyridyl)-5-methyl-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 5-1 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,87 (3H, d, J = 6,5 Hz), 4,70–4,80
(1H, m), 5,65–5,85
(1H, br), 6,65–6,80 (2H,
m), 6,86– 6,92
(1H, m), 6,98–7,06
(2H, m), 7,15–7,24
(2H, m), 7,45–7,52
(1H, m), 7,80–7,88
(1H, m), 8,33 (1H, s)
- [α]D 25: –258,8° (c0,7, MeOH)
-
Beispiel 12-5
-
(5S)-2-(3-Cyanophenyl)-4,4-bis(6-fluor-3-pyridyl)-5-methyl-2-imidazolin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,96 (3H, d, J = 6,6 Hz), 4,75 (1H, q, J = 6,6 Hz), 5,16 (1H, s),
6,89–6,93 (2H,
m), 7,55–7,95
(4H, m), 8,08–8,20
(3H, m), 8,41 (1H, s)
- [α]D 25: –256,1° (c0,4, MeOH)
-
Beispiel 12-6
-
Das optisch aktive (5R)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-hydroxymethyl-2-imidazolin,
worin das Ausgangsmaterial das in Bezugsbeispiel 6-1 beschriebene
Diamin ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
3,10 (1H, br), 3,24 (1H, d, J = 4,2 Hz, 10,5 Hz), 4,67 (1H, br),
5,34, 5,83 (1H, br), 6,88 (1H, dd, J = 3,3 Hz, 9,0 Hz), 7,02 (2H,
t, J = 8,7 Hz), 7,28 (2H, t, J = 8,7 Hz), 7,57 (1H, t, J = 8,1 Hz),
7,77 (1H, d, J = 7,8 Hz), 7,88 (1H, td, J = 2,7 Hz, 8,3 Hz), 8,10
(1H, d, J = 8,1 Hz), 8,22 (1H, s), 8,39 (1H, d, J = 0,9 Hz)
- [α]D 25: –268,6° (c1,0, CHCl3)
-
Beispiel 12-7
-
Das optisch aktive (5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-5-methyl-4-(1,5-naphthyridin-3-yl)-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 7-1 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,96 (3H, d, J = 5,9 Hz), 4,80–5,00
(1H, m), 5,42 (1H, brs), 7,03 (2H, t, J = 8,6 Hz), 7,20–7,40 (2H,
m), 7,53 (1H, t, J = 8,6 Hz), 7,63 (1H, dd, J = 4,2 Hz, 8,6 Hz),
7,70–7,80
(1H, m), 8,10–8,20
(1H, m), 8,20 (1H, s), 8,38 (1H, d, J = 8,6 Hz), 8,50 (1H, d, J
= 1,6 Hz), 8,97 (1H, dd, J = 1,6 Hz, 4,2 Hz), 9,08 (1H, s)
- [α]D 25: –363,6° (c0,5, CHCl3)
-
Beispiel 12-8
-
Das optisch aktive (5S)-2-(3-Cyanophenyl)-4-(6-fluor-3-pyridyl)-4-(4-fluorphenyl)-5-methyl-2-imidazolinhydrochlorid,
worin das Ausgangsmaterial das in Bezugsbeispiel 5-2 beschriebene
Diamin ist
-
- 1HNMR (300 MHz, CD3OD, δ ppm):
1,24 (3H, d, J = 7,1 Hz), 5,42 (1H, q, J = 7,1 Hz), 7,15 (1H, dd,
J = 8,5 Hz, 2,6 Hz), 7,27 (2H, dd, J = 8,9 Hz, 8,5 Hz), 7,60 (2H,
dd, J = 8,9 Hz, 4,9 Hz), 7,78–7,92
(2H, m), 8,13–8,29
(3H, m), 8,39–8,40
(1H, m)
- [α]D 25: –283,6° (c0,5, MeOH)
-
Beispiel 12-9
-
Das optisch aktive (5S)-2-(3-Cyanophenyl)-4-(4-fluorphenyl)-5-methyl-4-(1,5-naphthyridin-3-yl)-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 8-1 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
1,02 (3H, d, J = 6,6 Hz), 4,82–4,91
(1H, m), 5,09–5,12
(1H, br), 7,03–7,13 (2H,
m), 7,55–7,70
(4H, m), 7,79 (1H, dt, J = 7,8 Hz, 1,3 Hz), 8,19 (1H, d, J = 7,9
Hz), 8,25 (1H, s), 8,34–8,41 (2H,
m), 8,78 (1H, d, J = 2,5 Hz), 8,96 (1H, dd, J = 4,2 Hz, 1,6 Hz)
-
Beispiel 12-10
-
(5S)-2-(5-Cyano-3-pyridyl)-4,4-bis(6-fluor-3-pyridyl)-5-methyl-2-imidazolinhydrochlorid
-
- 1HNMR (400 MHz, CD3OD, δ ppm):
1,15 (3H, d, J = 6,4 Hz), 5,49 (1H, q, J = 6,4 Hz), 7,19 (1H, dd,
J = 8,8 Hz, 2,8 Hz), 7,24 (1H, dd, J = 8,8 Hz, 2,8 Hz), 7,85–7,90 (1H,
m), 8,09–8,14
(1H, m), 8,24 (1H, d, J = 2,8 Hz), 8,45 (1H, d, J = 2,8 Hz), 8,77–8,78 (1H,
m), 9,29 (1H, d, J = 2,0 Hz), 9,35 (1H, d, J = 2,0 Hz)
- [α]D 25: –252,0° (c0,1, MeOH)
-
Beispiel
13
Herstellung des optisch aktiven (5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin
-
2,4-Dicyanopyridin
(46,5 mg) und Ytterbiumtrifluormethansulfonat (24 mg) wurden zu
einer Lösung des
optisch aktiven (2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamin
(100 mg), beschrieben in Bezugsbeispiel 5-1, in Toluol (0,25 ml)
gegeben und das Gemisch wurde 5 Stunden bei 100°C gerührt. Das Reaktionsgemisch wurde
mit Essigsäureethylester
verdünnt,
mit gesättigter
wässriger
Natriumhydrogencarbonatlösung,
Wasser und gesättigter
wässriger
Natriumchloridlösung,
in dieser Reihenfolge, gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und das
organische Lösungsmittel
wurde im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 2 : 3) gereinigt, unter Gewinnung der Zielverbindung (106 mg)
als einen weißen
Feststoff.
Schmelzpunkt: 177–179°C
1HNMR
(300 MHz, CDCl3, δ ppm): 0,87, 0,98 (3,0H, d,
J = 6,7 Hz), 4,73, 4,98 (1,0H, q, J = 6,6 Hz), 6,85–6,98 (1,0H,
m), 6,99–7,06
(2H, m), 7,18–7,29
(2H, m), 7,61 (1H, d, J = 4,4 Hz), 7,77–7,93 (1H, m), 8,25, 8,39 (1,0H, d,
J = 2,6 Hz), 8,45, 8,48 (1H, s), 8,76 (1H, dd, J = 0,9 Hz, 5,1 Hz)
[α]D 25: –416,9° (c1,0, CHCl3)
-
Beispiel
14
Herstellung von (5R)-2-(4-Cyano-2-pyridyl)-4,4-bis(4-fluorphenyl)-5-hydroxymethyl-2-imidazolin
-
2,4-Dicyanopyridin
(601 mg) und Ytterbiumtrifluormethansulfonat (289 mg) wurden zu
einer Lösung von
(2R)-1,1-Bis(4-fluorphenyl)-3-(methoxymethoxy)-1,2-propandiamin
(1,50 g) in Toluol (30 ml) gegeben und das Gemisch wurde unter Rühren für 2 Tage
unter Rückfluss
erhitzt. Das Reaktionsgemisch wurde im Vakuum auf konzentriert und
dann wurde der Rückstand
in Tetrahydrofuran (20 ml) gelöst.
6 N Salzsäure
(10 ml) wurde zu der Lösung
gegeben und dann wurde das Gemisch 24 Stunden bei Raumtemperatur
gerührt.
Das Reaktionsgemisch wurde im Vakuum auf konzentriert und dann wurde
der Rückstand
durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 2 : 1) gereinigt, unter Gewinnung der Zielverbindung (385 mg).
1HNMR (200 MHz, CD3OD, δ ppm): 3,15
(2H, brs), 4,71 (1H, brs), 7,02 (2H, m), 7,08 (2H, m), 7,23 (2H,
m), 7,57 (2H, m), 7,83 (1H, dd, J = 3,0 Hz, 10,0 Hz), 8,46 (1H,
s), 8,87 (1H, d, J = 10,0 Hz)
[α]D 25: –312,8° (c1,0, CHCl3)
-
Durch
Anwenden der in Beispiel 13 und 14 beschriebenen Verfahren wurden
das entsprechende Diamin und Cyanopyridin cyclisiert, gegebenenfalls
gefolgt von Abspaltung der Schutzgruppen, unter Gewinnung der nachstehenden
Verbindungen.
-
Beispiel 14-1
-
2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-2-imidazolin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
4,16–4,26
(2H, m), 4,54–4,71
(1H, m), 6,20 (0,8H, s), 6,58 (0,2H, s), 6,86–8,78 (10H, m)
-
Beispiel 14-2
-
2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-2-imidazolin
-
-
Beispiel 14-3
-
Das optisch aktive (5R)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-hydroxymethyl-2-imidazolin,
worin das Ausgangsmaterial das in Bezugsbeispiel 6-1 beschriebene
Diamin ist
-
- 1HNMR (400 MHz, CD3OD, δ ppm:)17
(2H, d, J = 6, 2 Hz), 4,76 (1H, t, J = 6,2 Hz), 7,06 (3H, m), 7,28
(2H, m), 7,85 (1H, d, J = 4,8 Hz), 8,12 (1H, ddd, J = 2,8 Hz, 7,6
Hz, 7,6 Hz), 8,43 (1H, d, J = 2,8 Hz), 8,47 (1H, s), 8,88 (1H, d,
J = 4,8 Hz)
- [α]D 25: –304,7° (c0,6, CHCl3)
-
Beispiel 14-4
-
Das optisch aktive (5S)-2-(6-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 5-1 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,88 (2,5H, d, J = 6,5 Hz), 0,98 (0,5H, d, J = 6,8 Hz), 4,68–4,78 (0,8H,
m), 4,92–5,05
(0, 2H, m), 6,16 (0,8H, brs), 6,52 (0,2H, brs), 6,85–7,09 (3H,
m), 7,20–7,25
(2H, m), 7,72–8,00
(3H, m), 8,30–8,55
(2H, m)
- [α]D 25: –335,35° (c0,2, MeOH)
-
Beispiel 14-5
-
Das optisch aktive (5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin (Epimer
in der 4-Position der Verbindung von Beispiel 13), worin das Ausgangsmaterial
das in Bezugsbeispiel 5-2 beschriebene Diamin ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,96 (2,4H, d, J = 6,5 Hz), 1,01 (0,6H, d, J = 6,9 Hz), 4,76 (0,8H,
g, J = 6,5 Hz), 5,07 (0,2H, q, J = 6,9 Hz), 6,14 (0,8H, brs), 6,54
(0,2H, brs), 6,84–6,91
(1H, m), 7,00–7,10
(2H, m), 7,30–7,37
(0,4H, m), 7,48–7,56
(1,6H, m), 7,60–7,71
(2H, m), 8,08 (0,8H, d, J = 2,6 Hz), 8,15 (0,2H, d, J = 2,6 Hz),
8,45 (0,2H, s), 8,57 (0,8H, s), 8,74–8,78 (1H, m)
- [α]D 25: –291,0° (c0,8, MeOH)
-
Beispiel 14-6
-
Das optisch aktive (5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 7 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CD3OD, δ ppm):
0,90 (3H, d, J = 6,3 Hz), 4,80–4,90
(1H, m) 7,05–7,34
(4H, m), 7,23 (1H, d, J = 1,5 Hz), 7,44–7,47 (1H, m), 7,82–7,84 (1H,
m), 8,15 (1H, d, J = 5,1 Hz), 8,44 (1H, s), 8,86 (1H, m)
- [α]D 25: –332,3° (c1,1, CHCl3)
-
Beispiel 14-7
-
(5S)-2-(4-Cyano-2-pyridyl)-4,4-bis(6-fluor-3-pyridyl)-5-methyl-2-imidazolin
-
- Schmelzpunkt: 160–162°C
- [α]D 25: –332,8° (c0,5, MeOH)
-
Beispiel 14-8
-
Das optisch aktive (5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-5-methyl-4-(3-pyridyl)-2-imidazolinhydrochlorid, worin
das Ausgangsmaterial das in Bezugsbeispiel 7-2 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CD3OD, δ ppm):
1,15 (3H, d, J = 6,7 Hz), 5,54 (1H, q, J = 6,7 Hz), 7,16–7,33 (2H,
m), 7,33–7,45
(2H, m), 8,08 (1H, dd, J = 5,5 Hz, 8,3 Hz), 8,16 (1H, dd, J = 1,4
Hz, 4,9 Hz), 8,56 (1H, brs), 8,65 (1H, ddd, J = 1,1 Hz, 2,3 Hz,
8,3 Hz), 8,90 (1H, dd, J = 1,1 Hz, 5,5 Hz), 9,07 (1H, dd, J = 1,1
Hz, 2,3 Hz), 9,10 (1H, dd, J = 1,0 Hz, 4,9 Hz)
- [α]D 25: –292,0° (c0,1, MeOH)
-
Beispiel 14-9
-
Das optisch aktive (5S)-2-(4-Cyano-2-pyridyl)-4-(4-fluorphenyl)-4-(2-fluor-4-pyridyl)-5-methyl-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 8 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,97 (3H, d, J = 6,6 Hz), 4,74–4,90
(1H, m), 6,13 (1H, brs), 6,96 (1H, s), 6,99–7,10 (3H, m), 7,40–7,57 (2H,
m), 7,62 (1H, dd, J = 5,0 Hz, 1,6 Hz), 8,14 (1H, d, J = 5,3 Hz),
8,52–8,61 (1H,
m), 8,76 (1H, d, J = 4,2 Hz)
- [α]D 25: –278,8° (c0,5, CHCl3)
-
Beispiel
15
Herstellung von (5R)-2-(4-Cyano-2-pyrimidyl)-4,4-bis(4-fluorphenyl)-5-hydroxymethyl-2-imidazolin
-
2-Cyano-4-ethoxycarbonylpyrimidin
(207,3 mg) und Scandiumtrifluormethansulfonat (48,8 mg) wurden zu
einer Lösung
von (2R)-1,1-Bis(4-fluorphenyl)-3-methoxymethoxy-1,2-propandiamin (303,5
mg) in Toluol (2,0 ml) gegeben und das Gemisch wurde 4 Stunden bei
110°C gerührt. Das
Reaktions gemisch wurde im Vakuum auf konzentriert und dann wurde
der Rückstand
durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 1 : 1 → 1
: 2) gereinigt, unter Gewinnung von (5R)-2-(4-Ethoxycarbonyl-2-pyrimidyl-4,4-bis(4-fluorphenyl)-5-[(methoxymethoxy)methyl]-2-imidazolin
(279 mg). Die erhaltene Esterverbindung wurde in Methanol (2 ml)
gelöst
und Ammoniakgas wurde durch die Lösung für 20 Minuten bei –78°C geleitet. Dann
wurde die Lösung
20 Stunden bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde
im Vakuum auf konzentriert, und dann wurde der Rückstand durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 1 : 1 → Chloroform
: Methanol = 9 : 1) gereinigt, unter Gewinnung der entsprechenden Amidverbindung
(273 mg). Triethylamin (0,75 ml) und Trifluoressigsäureanhydrid
(0,26 ml) wurden bei –78°C zu einer
Lösung
der erhaltenen Amidverbindung in Tetrahydrofuran (2 ml) gegeben
und die Temperatur des Gemisches wurde auf bis zu Raumtemperatur
erhöht,
gefolgt von Rühren
für 7 Stunden.
Wasser wurde zu dem Reaktionsgemisch gegeben und das Gemisch wurde
mit Essigsäureethylester
extrahiert. Die organische Schicht wurde über wasserfreiem Natriumsulfat
getrocknet und dann wurde Natriumsulfat durch Filtration entfernt.
Dann wurde das organische Lösungsmittel
im Vakuum verdampft. Der Rückstand
wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 1 : 1 → 1
: 2) gereinigt, unter Gewinnung der entsprechenden Cyanoverbindung
(178 mg). Die erhaltene Cyanoverbindung wurde in Tetrahydrofuran
(6 ml) gelöst
und 6 N Salzsäure
(6 ml) wurden zu der Lösung
gegeben. Dann wurde das Gemisch 5 Stunden bei Raumtemperatur gerührt. Gesättigte wässrige Natriumhydrogencarbonatlösung wurde
zu dem Reaktionsgemisch vorsichtig gegeben und das Gemisch wurde
mit Essigsäureethylester
extrahiert. Die organische Schicht wurde über wässerfreiem Natriumsulfat getrocknet
und dann wurde Natriumsulfat durch Filtration entfernt. Das organische
Lösungsmittel
wurde im Vakuum verdampft. Der Rückstand
wurde durch Kieselgel-Säulenchromatographie
(C-300; Chloroform : Essigsäureethylester
= 2 : 1) gereinigt, unter Gewinnung der Titelverbindung (74,7 mg).
1HNMR (300 MHz, CDCl3, δ ppm): 3,10–3,40 (2H,
br), 4,62–4,90
(1H, br), 6,50–6,80
(1H, br), 6,95–7,08
(4H, m), 7,21–7,32
(2H, m), 7,39–7,57
(2H, br), 7,32 (1H, d, J = 4,8 Hz), 9,13 (1H, d, J = 4,9 Hz)
[α]D 25: –295,1° (c0,49,
MeOH)
-
Durch
Anwenden des in Beispiel 15 beschriebenen Verfahrens wurden das
entsprechende Diamin und 2-Cyano-4-ethoxycarbonylpyrimidin cyclisiert und
kondensiert, und die Estergruppe des Produkts wurde in eine Cyanogruppe
umgewandelt, unter Gewinnung der nachstehenden Verbindungen.
-
Beispiel 15-1
-
Das optisch aktive (5S)-2-(4-Cyano-2-pyrimidyl)-4-(4-fluorphenyl)-4-(6-fluor-3-pyridyl)-5-methyl-2-imidazolin, worin
das Ausgangsmaterial das in Bezugsbeispiel 5-1 beschriebene Diamin
ist
-
- 1HNMR (300 MHz, CD3OD, δ ppm):
0,85 (3H, d, J = 6,5 Hz), 4,80–4,93
(1H, m), 6,95–7,13
(3H, m), 7,26–7,35 (2H,
m), 7,88–8,10
(1H, m), 8,0 (1H, d, J = 4,9 Hz), 8,13–8,40 (1H, d, J = 2,0 Hz),
9,18 (1H, d, J = 4,9 Hz)
- [α]D 25: –351,3° (c1,0, MeOH)
-
Bezugsbeispiel
1
Herstellung von 1,1-Bis(4-fluorphenyl)-1,2-ethandiamin
-
4,4'-Difluorbenzophenon
(25 g) wurde in wasserfreiem Toluol (800 ml) gelöst und die Lösung wurde auf –30°C gekühlt. Titantetrachlorid
(21,4 ml) wurde tropfenweise zu der Lösung unter Stickstoff gegeben
und Ammoniakgas wurde für
etwa 20 Minuten durch das Gemisch geleitet. Dann wurde das Gemisch
auf Raumtemperatur erwärmt
und über
Nacht gerührt.
Gesättigte
wässrige
Kaliumcarbonatlösung
(400 ml) wurde zu dem Reaktionsgemisch gegeben und das Gemisch wurde
für 1 weitere
Stunde gerührt.
Die organische Schicht wurde abgetrennt und mit Wasser bzw. gesättigter
Salzlösung
einmal gewaschen. Die organische Schicht wurde über wasserfreiem Natriumsulfat
getrocknet und dann wurde das Lösungsmittel
im Vakuum verdampft, unter Gewinnung von Rohprodukt von 1,1-Bis(4-fluorphenyl)methanimin
(31,7 g). Das Rohprodukt wurde in wasserfreiem Toluol (80 ml) gelöst und Trimethylsilylcyanid
(171,86 ml) und Zinkjodid (1,83 g) wurden zu der Lösung gegeben.
Dann wurde das Gemisch über
Nacht bei Raumtemperatur gerührt.
Das Reaktionsgemisch wurde mit gesättigter wässriger Natriumhydrogencarbonatlösung und
gesättigter
Salzlösung
in dieser Reihenfolge gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Dann wurde das Lösungsmittel
im Vakuum verdampft, unter Gewinnung von Rohprodukt von 2-Amino-2,2-bis(4-fluorphenyl)acetonitril
(30,84 g). Das Rohprodukt wurde in wasserfreiem Toluol (100 ml)
gelöst
und dann wurde tropfenweise 1 M Diisobutylaluminiumhydrid-Toluol-Lösung (500
ml) zu der Lösung
bei –78°C gegeben.
Dann wurde die Temperatur des Gemisches durch Stehen auf Raumtemperatur
erhöht
und das Gemisch wurde über
Nacht gerührt.
Natriumsulfatdecahydrat wurde zu dem Reaktionsgemisch gegeben und
das erhaltene Gemisch wurde 1 Stunde gerührt. Darüber hinaus wurde wasserfreies
Magnesiumsulfat zu dem Reaktionsgemisch gegeben und das Gemisch wurde
eine Stunde gerührt.
Dann wurden unlösliche
Stoffe durch Filtration entfernt. Das Filtrat wurde im Vakuum aufkonzentriert
und dann wurde der erhaltene Rückstand
durch Kieselgel-Säulenchromatographie
(C-300; 100% Essigsäureethylester → 100% Methanol)
gereinigt, unter Gewinnung der Titelverbindung (8,55 g).
1HNMR (300 MHz, CDCl3, δ ppm): 3,32
(2H, s), 6,96–7,00
(4H, m), 7,26–7,33
(4H, m)
-
Bezugsbeispiel
2
Herstellung von 1-(4-Fluorphenyl)-1-(1,5-naphthyridin-3-yl)-1,2-ethandiamin
-
4-Fluorphenyl-1,5-naphthyridin-3-yl-keton
(108 mg) wurde in wasserfreiem Toluol (5 ml) gelöst und die Lösung wurde
auf –30°C gekühlt. Titanchlorid
(0,08 ml) wurde tropfenweise zu der Lösung unter Stickstoff gegeben
und Ammoniakgas wurde durch das Gemisch für etwa 20 Minuten geleitet.
Dann wurde das Gemisch auf Raumtemperatur erwärmt und über Nacht gerührt. Gesättigte wässrige Kaliumcarbonatlösung (1
ml) wurde zu dem Reaktionsgemisch gegeben und das Gemisch wurde
für eine
weitere Stunde gerührt.
Die organische Schicht wurde abgetrennt, mit Wasser bzw. gesättigter
Salzlösung
einmal gewaschen und über
wasserfreiem Natriumsulfat getrocknet. Das Lösungsmittel wurde dann im Vakuum
verdampft, unter Gewinnung von Rohprodukt von (4-Fluorphenyl)(1,5-naphthyridin-3-yl)-methanimin
(103 mg). Das Rohprodukt wurde in wasserfreiem Toluol (2 ml) gelöst und Trimethylsilylcyanid
(0,21 ml) und 1 M Tetrabutylammoniumfluorid-Tetrahydrofuran-Lösung (0,01
ml) wurden zu der Lösung
gegeben. Das Gemisch wurde 6 Tage bei Raumtemperatur gerührt. Das
Reaktionsgemisch wurde mit gesättigter
wässriger
Natriumhydrogencarbonatlösung
bzw. gesättigter
Salzlösung
einmal gewaschen und dann über
wasserfreiem Natriumsulfat getrocknet. Das Lösungsmittel wurde im Vakuum
verdampft, unter Gewinnung von Rohprodukt von 2-Amino-2-(4-fluorphenyl)-2-(1,5-naphthyridin-3-yl)-acetonitril.
Das Rohprodukt wurde in wasserfreiem Toluol (2 ml) gelöst. 1 M
Diisobutylaluminiumhydrid-Toluol-Lösung (1,85 ml) wurde tropfenweise
zu der Lösung
bei –78°C gegeben
und das Gemisch wurde 30 Minuten auf 0°C erwärmt. Natriumsulfatdecahydrat
wurde zu dem Reaktionsgemisch gegeben und das Gemisch wurde eine Stunde
gerührt.
Darüber
hinaus wurde wasserfreies Magnesiumsulfat zu dem Reaktionsgemisch
gegeben und das Gemisch wurde eine Stunde gerührt. Dann wurden unlösliche Stoffe
durch Filtration entfernt. Das Filtrat wurde im Vakuum aufkonzentriert
und dann wurde der erhaltene Rückstand
durch Kieselgel-Säulenchromatographie
(C-300; Chloroform : Methanol = 20 : 1 → 10 : 1 → 2 : 1) gereinigt, unter Gewinnung der
Titelverbindung (17 mg).
1HNMR (200
MHz, CD3OD, δ ppm): 3,52 (2H, s), 6,99–7,10 (2H,
m), 7,34–7,44
(2H, m), 7,63 (1H, dd, J = 4,4 Hz, 8,5 Hz), 8,35–8,40 (1H, m), 8,41–8,43 (1H,
m), 8,92 (1H, d, J = 2,3 Hz), 8,98 (1H, dd, J = 1,7 Hz, 4,4 Hz)
-
Bezugsbeispiel
3
Herstellung von 1,1-Bis(4-fluorphenyl)-1,2-propandiamin
-
Jodmonochlorid
(0,40 g) wurde zu der Suspension von Natriumazid (1,01 g) in Acetonitril
(6 ml) bei 0°C
gegeben. Nachdem die Suspension 10 Minuten gerührt wurde, wurde die Lösung von
1,1-Bis(4-fluorphenyl)-1-propen (1,43 g) in Acetonitril (6 ml) zu
der Suspension gegeben und das Gemisch wurde auf Raumtemperatur
erwärmt
und 38 Stunden gerührt.
Das Reaktionsgemisch wurde in Wasser gegossen und das Gemisch wurde
mit Diethylether extrahiert. Der Extrakt wurde mit 10%iger wässriger
Natriumthiosulfatlösung,
Wasser und gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen und über wasserfreiem Magnesiumsulfat
getrocknet. Das Lösungsmittel
wurde im Vakuum verdampft und dann wurde der Rückstand durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 10 : 1) gereinigt, unter Gewinnung der gewünschten Verbindung, 1,1-Bis(4-fluorphenyl)-2-jodpropylazid
(2,16 g).
-
Die
vorstehend erwähnte
Jodazidverbindung (40 mg) wurde in Diethylether (1 ml) gelöst und Lithiumaluminiumhydrid
(8 mg) wurde zu der Lösung
bei Raumtemperatur gegeben. Dann wurde das Gemisch 2 Stunden bei
Raumtemperatur gerührt.
Natriumsulfatdecahydrat wurde zu dem Reaktionsgemisch gegeben. Das Gemisch
wurde 30 Minuten gerührt.
Darüber
hinaus wurde wasserfreies Magnesiumsulfat zu dem Reaktionsgemisch
gegeben und das Gemisch wurde 30 Minuten gerührt. Unlösliche Stoffe wurden durch
Filtration entfernt und dann wurde das Filtrat im Vakuum auf konzentriert.
Der erhaltene Rückstand
wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 2 : 1) gereinigt, unter Gewinnung der gewünschten Verbindung, 2,2-Bis(4-fluorphenyl)-3-methylaziridin
(9 mg).
-
Die
vorstehend erwähnte
Aziridinverbindung (342 mg) wurde in einem Gemisch von Methanol
(10 ml) und Wasser (2 ml) gelöst
und Natriumazid (439 mg) und Ammoniumchlorid (155 mg) wurden zu
der Lösung gegeben.
Das Gemisch wurde 40 Stunden bei 60°C gerührt. Nachdem das Reaktionsgemisch
durch Stehen gekühlt
wurde, wurde das Reaktionsgemisch mit Essigsäureethylester verdünnt und
die Verdünnung
wurde mit gesättigter
wässriger
Natriumhydrogencarbonatlösung,
Wasser und gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen, über wasserfreiem Natriumsulfat
getrocknet und im Vakuum auf konzentriert. Der erhaltene Rückstand
wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 2 : 1 → 1
: 1) gereinigt, unter Gewinnung der gewünschten Verbindung, 1-Azido-1,1-bis(4-fluorphenyl)-2-propanamin (251 mg).
Die vorstehend erwähnte
Azidverbindung (8 mg) wurde in Gegenwart von 10%igem Palladium-Kohlenstoff-Katalysator (5 mg)
in Methanol unter 1 Atmosphäre
Wasserstoff bei Raumtemperatur reduziert. Der Katalysator wurde
durch Filtration entfernt und dann wurde das Filtrat im Vakuum aufkonzentriert,
unter Gewinnung der Titelverbindung (7 mg).
1HNMR
(200 MHz, CD3OD, δ ppm): 1,14 (3H, d, J = 6,5
Hz), 4,17 (1H, q, J = 6,5 Hz), 7,00–7,13 (4H, m), 7,42–7,55 (4H,
m)
-
Bezugsbeispiel
4
Herstellung von (2R)-1,1-Bis(4-fluorphenyl)-3-(methoxymethoxy)-1,2-propandiamin
-
Unter
Rühren
einer Lösung
von (2S)-2-{[(Benzyloxy)carbonyl]amino}-3-(methoxymethoxy)-propansäuremethylester
(35,70 g) in Tetrahydrofuran (500 ml) unter Stickstoff bei 0°C wurde 1,0
M 4-Fluorphenylmagnesiumbromid-Tetrahydrofuran-Lösung
tropfenweise zu der Lösung
gegeben. Die Temperatur des Gemisches wurde auf Raumtemperatur erhöht und dann
wurde das Gemisch 4 Stunden gerührt.
Gesättigte
wässrige
Ammoniumchloridlösung
(100 ml) wurde zu dem Reaktionsgemisch gegeben und dann wurde das
Reaktionsgemisch im Vakuum auf konzentriert. Gesättigte wässrige Ammoniumchloridlösung wurde
zu dem Rückstand
gegeben und das Gemisch wurde mit Essigsäureethylester extrahiert. Die
organische Schicht wurde mit gesättigter
wässriger
Natriumhydrogencarbonatlösung
und gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und dann
wurde das organische Lösungsmittel
im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 9 : 1 → 5
: 1 → 3
: 1) gereinigt, unter Gewinnung von N-{(1S)-2,2-Bis(4-fluorphenyl)-2-hydroxy-1-[(methoxymethoxy)methyl]ethyl)carbaminsäurebenzylester
(49,97 g) als farbloses Öl.
Das Produkt (49,80 g) wurde in Gegenwart von 10%igem Palladium-Kohlenstoff-Katalysator
(2,00 g) in Methanollösungsmittel
(500 ml) unter 1 atm Wasserstoff für zwei Tage gerührt und
das Reaktionsgemisch wurde auf bis zu 50°C erhitzt und 24 Stunden gerührt. Das
Reaktionsgemisch wurde durch Celite filtriert und dann wurde das
erhaltene Filtrat im Vakuum auf konzentriert. Der Rückstand
wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester =
2 : 1 → Chloroform
: Methanol = 5 : 1) gereinigt, unter Gewinnung von (2S)-2-Amino-1,1-bis(4-fluorphenyl)-3-(methoxymethoxy)-1-propanol (33,30 g)
als gelbes Öl.
Unter Rühren
einer Lösung
von Triphenylphosphin (40,56 g) in Toluol (400 ml) wurde bei 0°C 1,0 M Brom-Benzol-Lösung tropfenweise
zu der Lösung
gegeben und das Gemisch wurde 30 Minuten gerührt. Eine Lösung der vorstehend erwähnten Hydroxyaminverbindung
(20,00 g) in Toluol (50 ml) und Triethylamin (43,11 g) wurde tropfenweise
bei 0°C
in dieser Reihenfolge zugegeben. Anschließend wurde das Reaktionsgemisch
bei 0°C
20 Stunden gerührt,
und gesättigte
wässrige Natriumhydrogencarbonatlösung wurde
zu dem Reaktionsgemisch gegeben. Das erhaltene Gemisch wurde mit
Essigsäureethylester
extrahiert. Die organische Schicht wurde mit gesättigter wässriger Natriumchloridlösung gewaschen
und über
wasserfreiem Natriumsulfat getrocknet. Natriumsulfat wurde durch
Filtration entfernt und dann wurde das organische Lösungsmittel
im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 4 : 1 → 2
: 1) gereinigt, unter Gewinnung von (3R)-2,2-Bis(4-fluorphenyl)-3-[(methoxymethoxy)methyl]aziridin
(7,72 g) als gelbes Öl.
Wasser (40 ml), Natriumazid (7,45 g) und Ammoniumchlorid (2,70 g)
wurden in dieser Reihenfolge zu einer Lösung der Aziridinverbindung
(7,70 g) in N,N-Dimethylformamid
(80 ml) gegeben und das Gemisch wurde 16 Stunden bei 100°C gerührt. Darüber hinaus
wurde Natriumazid (7,45 g) zu dem Reaktionsgemisch gegeben und das Gemisch
wurde 24 Stunden bei 120°C
gerührt.
Anschließend
wurde Wasser zu dem Reaktionsgemisch gegeben und das Gemisch wurde
mit Diethylether extrahiert. Die organische Schicht wurde mit gesättigter
wässriger
Natriumhydrogencarbonatlösung
und gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und dann
wurde das organische Lösungsmittel
im Vakuum verdampft. Der erhal tene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 3 : 1) gereinigt, unter Gewinnung von (2R)-1-Azido-1,1-bis(4-fluorphenyl)-3-(methoxymethoxy)-2-propanamin
(7,68 g) als gelbes Öl.
Die erhaltene Azidverbindung (7,62 g) wurde in Gegenwart von 5%
Palladium-Kohlenstoff-Katalysator
(0,80 g) in Methanollösungsmittel
(80 ml) unter 1 atm Wasserstoff für 17 Stunden gerührt. Das
Reaktionsgemisch wurde durch Celite filtriert und mit Essigsäureethylester
gewaschen. Das erhaltene Filtrat wurde mit gesättigter wässriger Natriumhydrogencarbonatlösung und
gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und dann wurde
das organische Lösungsmittel
im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Essigsäureethylester → Chloroform
: Methanol = 5 : 1) gereinigt, unter Gewinnung des Titeldiamins
(5,70 g) als schwach gelbes Öl.
1HNMR (300 MHz, CDCl3, δ ppm): 3,26–3,33 (1H,
m), 3,32 (3H, s), 3,59 (1H, dd, J = 9,7 Hz, 2,6 Hz), 4,03 (1H, dd,
J = 7,9 Hz, 2,6 Hz), 4,57 (2H, dd, J = 7,8 Hz, 6,5 Hz), 6,92–7,04 (4H,
m), 7,38–7,53
(4H, m)
[α]D 25: –44,3° (c1,0, CHCl3)
-
Die
nachstehenden Verbindungen wurden durch das Verfahren, das ähnlich zu
dem in dem vorstehend erwähnten
Bezugsbeispiel 4 beschriebenen Verfahren ist, hergestellt.
-
Bezugsbeispiel 4-1
-
(2S)-1,1-Bis(4-fluorphenyl)-1,2-propandiamin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
0,99 (3H, d, J = 6,4 Hz), 4,01 (1H, q, J = 6,4 Hz), 6,92–7,03 (4H,
m), 7,38–7,52
(4H, m)
-
Bezugsbeispiel 4-2
-
(2S)-1,1-Bis(6-fluor-3-pyridyl)-1,2-propandiamin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
1,04 (3H, d, J = 6,3 Hz), 4,09 (1H, q, J = 6,3 Hz), 6,88 (2H, dd,
J = 8,5 Hz, 3,1 Hz), 7,88 (1H, ddd, J = 8,5 Hz, 8,0 Hz, 2,5 Hz),
7,96 (1H, ddd, J = 8,5 Hz, 8,0 Hz, 2,5 Hz), 8,35 (1H, d, J = 2,5
Hz), 8,40 (1H, d, J = 2,5 Hz)
-
Bezugsbeispiel 4-3
-
(2S)-1,1-Bis(2-fluor-4-pyridyl)-1,2-propandiamin
-
- 1HNMR (300 MHz, CDCl3, δ ppm):
1,02 (3H, d, J = 6,3 Hz), 4,10 (1H, q, J = 6,3 Hz), 7,12 (1H, s),
7,23 (1H, s), 7,27 (1H, d, J = 5,4 Hz), 7,35 (1H, d, J = 5,7 Hz),
8,16 (1H, d, J = 5,7 Hz), 8,17 (1H, d, J = 5,4 Hz)
-
Bezugsbeispiel
5
Herstellung von (2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamin
-
Die
Lösung
von N-{(1S)-2-(Methoxy(methyl)amino]-1-methyl-2-oxoethyl)-carbaminsäure-t-butylester (50
g) in Tetrahydrofuran (700 ml) wurde tropfenweise langsam bei 0°C zu 2,5
M 4-Fluorphenylmagnesiumbromid-Tetrahydrofuran-Lösung (300 ml), getrennt hergestellt,
gegeben und dann wurde das Reaktionsgemisch 14 Stunden bei Raumtemperatur
gerührt.
Nachdem das Reaktionsgemisch auf 0°C gekühlt wurde, wurde gesättigte wässrige Natriumhydrogencarbonatlösung zu
dem Reaktionsgemisch gegeben und dann wurde das Gemisch zweimal
mit Ether extrahiert. Die organische Schicht wurde mit gesättigter
wässriger
Natriumhydrogencarbonatlösung
und gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und dann
wurde das organische Lösungsmittel
im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 4 : 1) gereinigt, unter Gewinnung von N-[(1S)-2-(4-Fluorphenyl)-1-methyl-2-oxoethyl]carbaminsäure-t-butylester (57,75
g) als schwach gelben Feststoff. Die Lösung der erhaltenen Ketonverbindung
(61,20 g) in Ethylenglycoldimethylether-Lösung (500 ml) wurde tropfenweise zu
der Lösung
von 6-Fluor-3-pyridyllithium in Diethylether (hergestellt durch
die Reaktion von 5-Brom-2-fluorpyridin (59 ml) und 1,6 M Butyllithium-Hexan-Lösung (358
ml) in Diethylether-(1,5 Liter)-Lösungsmittel bei –78°C) bei –78°C gegeben.
Anschließend
wurde das Reaktionsgemisch bei –78°C 30 Minuten
gerührt
und die Temperatur des Reaktionsgemisches wurde auf 0°C erhöht. Gesättigte wässrige Ammoniumchloridlösung wurde
zu dem Reaktionsgemisch gegeben und das Gemisch wurde für weitere
15 Minuten bei Raumtemperatur gerührt. Nachdem die wässrige Schicht
entfernt wurde, wurde die organische Schicht mit Wasser und gesättigter
wässriger
Natriumchloridlösung
in dieser Reihenfolge gewaschen und über wasserfreiem Natriumsulfat
getrocknet. Natriumsulfat wurde durch Filtration entfernt und dann
wurde das organische Lösungsmittel
im Vakuum verdampft. Der Rückstand
wurde in ein wenig Essigsäureethylester
gelöst
und überschüssiger Diisopropylether
wurde zu der Lösung
zum Ausfällen
von N-[(1S)-2-(4-Fluorphenyl)-2-(6-fluor-3-pyridyl)-2-hydroxy-1-methylethyl]carbaminsäure-t-butylester
gegeben. Der Niederschlag wurde filtriert (Ausbeute 74,35 g). Der
Niederschlag wurde mit 4 N Chlorwasserstoff-Essigsäureethylester-Lösung behandelt,
unter Gewinnung von (2S)-2-Amino-1-(4-fluorphenyl)-1-(6-fluor-3-pyridyl)-1-propanol
(32,8 g). Die erhaltene Hydroxyaminoverbindung wurde in Aziridin
durch das Verfahren, das zu dem in Bezugsbeispiel 4 beschriebenen
Verfahren ähnlich
ist, umgewandelt, unter Gewinnung von 2-Fluor-5-[(3S)-2-(4-fluorphenyl)-3-methyl-2-aziridinyl]-pyridin. Die
erhaltene Aziridinverbindung wurde in Azid durch das Verfahren,
das zu dem in Bezugsbeispiel 4 beschriebenen Verfahren ähnlich ist,
umgewandelt, und dann wur de die Azidverbindung durch die Zugabe
von Wasserstoff reduziert, unter Gewinnung des Titel-Diamins. Außerdem ist
das Verhältnis
der Diastereomeren in der zweiten Position des Aziridinrings etwa
3 : 2, und die Diastereomeren können
durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 6 : 4 → 4
: 6) getrennt werden.
-
Bezugsbeispiel
5-1
Herstellung des optisch aktiven (2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamins
-
Das
Gemisch der Diastereomeren von 2-Fluor-5-[(3S)-2-(4-fluorphenyl)-3-methyl-2-aziridinyl]pyridin, erhalten
in Bezugsbeispiel 5, wurde durch Kieselgel-Säulenchromatographie getrennt.
Die Aziridinverbindung, die zuerst eluierte, wurde durch das Verfahren,
das zu dem in Bezugsbeispiel 4 beschriebenen Verfahren ähnlich ist,
behandelt, unter Gewinnung des optisch aktiven Titeldiamins.
1HNMR (300 MHz, CDCl3, δ ppm): 1,00
(3H, d, J = 6,3 Hz), 4,65 (1H, q, J = 6,3 Hz), 6,85 (1H, dd, J =
3,0 Hz, 8,7 Hz), 7,00 (2H, t, J = 8,4 Hz), 7,43 (2H, dd, J = 5,4
Hz, 9,0 Hz), 7,97 (1H, t, J = 8,4 Hz), 8,40 (1H, d, J = 2,7 Hz)
-
Bezugsbeispiel
5-2
Herstellung des optisch aktiven (2S)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-1,2-propandiamins
(Epimer in 1. Position von Bezugsbeispiel 5-1)
-
Das
Gemisch von Diastereomeren von 2-Fluor-5-[(3S)-2-(4-fluorphenyl)-3-methyl-2-aziridinyl]pyridin, erhalten
in Bezugsbeispiel 5, wurde durch Kieselgel-Säulenchromatographie getrennt.
Die als Zweites eluierte Aziridinverbindung wurde durch das Verfahren,
das zu dem in Bezugsbeispiel 4 beschriebenen Verfahren ähnlich ist,
behandelt, unter Gewinnung des optisch aktiven Titeldiamins.
1HNMR (300 MHz, CDCl3, δ ppm): 1,02
(3H, d, J = 6,3 Hz), 4,05 (1H, q, J = 6,3 Hz), 6,84 (1H, dd, J =
8,5 Hz, 3,1 Hz), 7,01 (2H, dd, J = 8,9 Hz, 8,5 Hz), 7,49 (2H, dd,
J = 8,9 Hz, 5,2 Hz), 7,87 (1H, ddd, J = 8,5 Hz, 8,0 Hz, 2,6 Hz),
8,34 (1H, d, J = 2,6 Hz)
-
Bezugsbeispiel
6
Herstellung von (2R)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-3-(methoxymethoxy)-1,2-propandiamin
-
Die
Titelverbindung wurde durch das Verfahren, das zu dem in Bezugsbeispiel
5 beschriebenen Verfahren ähnlich
ist, über
2-Fluor-5-{(3R)-2-(4-fluorphenyl)-3-[(methoxymethoxy)methyl]-2-aziridinyl}pyridin
hergestellt. Das Zwischenprodukt, Aziridin, kann durch Kieselgel-Säulenchromatographie
abgetrennt werden.
-
Bezugsbeispiel
6-1
Herstellung des optisch aktiven (2R)-1-(4-Fluorphenyl)-1-(6-fluor-3-pyridyl)-3-(methoxymethoxy)-1,2-propandiamins
-
Das
Gemisch von Diastereomeren von 2-Fluor-5-{(3R)-2-(4-fluorphenyl)-3-[(methoxymethoxy)methyl]-2-aziridinyl}pyridin,
das in Bezugsbeispiel 6 erhalten wurde, wurde durch Kieselgel-Säulenchromatographie
(eluiert mit gemischtem Lösungsmittel
von Hexan und Essigsäureethylester)
abgetrennt. Die zuerst eluierte Aziridinverbindung wurde durch das
Verfahren, das zu dem in Bezugsbeispiel 4 beschriebenen Verfahren ähnlich ist,
behandelt, unter Gewinnung des optisch aktiven Titeldiamins.
1HNMR (300 MHz, CDCl3, δ ppm): 3,31
(4H, m), 3,56 (1H, dd, J = 2,3 Hz, 9,8 Hz), 4,03 (1H, dd, J = 2,5
Hz, 7,9 Hz), 4,56 (2H, s), 6,85 (1H, dd, J = 3,0 Hz, 6,5 Hz), 7,00
(2H, m), 7,39 (2H, m), 7,94 (1H, dt, J = 2,7 Hz, 7,8 Hz), 8,39 (1H,
s)
-
Bezugsbeispiel
7
Herstellung des optisch aktiven (2S)-1-(4-Fluorphenyl)-1-(2-fluor-4-pyridyl)-1,2-propandiamins
-
Das
Zwischenprodukt von Bezugsbeispiel 5, N-[(1S)-2-(4-Fluorphenyl)-1-methyl-2-oxoethyl]carbaminsäure-t-butylester
und 2-Methyl-2-propansulfinamid, wurden in Gegenwart eines Entwässerungsmittels
kondensiert, unter Gewinnung von N-[(1S)-2-[(t-Butylsulfinyl)imino]-2-(4-fluorphenyl)-1-methylethyl]carbaminsäure-t-butylester.
1,0 M Trimethylaluminium-Hexan-Lösung (0,43
ml) wurde zu der Lösung
der Sulfinyliminverbindung (80 mg) in Toluol (2 ml) bei –78°C gegeben
und das Gemisch wurde 5 Minuten gerührt. Die erhaltene Lösung wurde
tropfenweise langsam zu der 2-Fluor-4-pyridyllithiumlösung (hergestellt
durch die Reaktion von 4-Fluor-2-brompyridin (114 mg) und 1,56 M
Butyllithium-Hexan-Lösung
(0,46 ml) in Diethyletherlösungsmittel (3
ml) bei –78°C) bei –78°C gegeben.
Anschließend
wurde Tetrahydrofuran (3 ml) zu dem Reakti onsgemisch gegeben und
dann wurde das Reaktionsgemisch 2,5 Stunden bei –78°C gerührt. Gesättigte wässrige Natriumchloridlösung wurde
zu dem Reaktionsgemisch gegeben und die Temperatur des Reaktionsgemisches
wurde auf Raumtemperatur erhöht.
Das erhaltene Reaktionsgemisch wurde durch Celite filtriert und
dann wurde die organische Schicht des Filtrats über wasserfreiem Magnesiumsulfat
getrocknet. Magnesiumsulfat wurde durch Filtration entfernt und
dann wurde das organische Lösungsmittel
im Vakuum verdampft. Der erhaltene Rückstand wurde durch Kieselgel-Säulenchromatographie
(C-300; Hexan : Essigsäureethylester
= 2 : 1) gereinigt, unter Gewinnung des optisch aktiven N-[(1S)-2-[(t-Butylsulfinyl)amino]-2-(4-fluorphenyl)-2-(2-fluor-4-pyridyl)-1-methylethyl]carbaminsäure-t-butylesters (49 mg).
Das Produkt wurde mit 4 N Chlorwasserstoff-Dioxan-Lösung behandelt,
unter Gewinnung des optisch aktiven Titeldiamins (31 mg).
1HNMR (300 MHz, CDCl3, δ ppm): 1,03
(3H, d, J = 6,3 Hz), 1,91 (4H, brs), 4,10 (1H, q, J = 6,3 Hz), 6,98–7,48 (6H,
m), 8,12 (1H, d, J = 5,1 Hz)
-
Bezugsbeispiel
7-1
Herstellung des optisch aktiven (2S)-1-(4-Fluorphenyl)-1-(1,5-naphthyridin-3-yl)-1,2-propandiamins
-
1,5-Naphthyridin-3-yllithium
wurde mit N-[(1S)-2-[(t-Butylsulfinyl)imino]-2-(4-fluorphenyl)-1-methylethyl]carbaminsäure-t-butylester
durch das Verfahren, das zu dem in Bezugsbeispiel 7 beschriebenen
Verfahren ähnlich
ist, umgesetzt und die Schutzgruppen des Produkts wurden unter saurer
Bedingung entfernt, unter Gewinnung des optisch aktiven Titeldiamins.
1HNMR (300 MHz, CDCl3, δ ppm): 1,06
(3H, d, J = 6,3 Hz), 4,25 (1H, q, J = 6,3 Hz), 7,00 (2H, t, J =
8,6 Hz), 7,45–7,55
(2H, m), 7,60 (1H, dd, J = 4,2 Hz, 8,5 Hz), 8,35 (1H, d, J = 8,6
Hz), 8,57 (1H, d, J = 1,4 Hz), 8,95 (1H, dd, J = 1,4 Hz, 4,2 Hz),
9,13 (1H, d, J = 2,2 Hz)
-
Bezugsbeispiel
7-2
Herstellung des optisch aktiven (2S)-1-(4-Fluorphenyl)-1-(3-pyridyl)-1,2-propandiamins
-
3-Pyridyllithium
wurde mit N-[(1S)-2-[(t-Butylsulfinyl)imino]-2-(4-fluorphenyl)-1-methylethyl]carbaminsäure-t-butylester durch
das Verfahren, das zu dem in Bezugsbeispiel 7 beschriebenen Verfahren ähnlich ist, umgesetzt
und die Schutzgruppen des Produkts wurden unter saurer Bedingung
entfernt, unter Gewinnung des optisch aktiven Titeldiamins.
1HNMR (300 MHz, CDCl3, δ ppm): 1,01
(3H, d, J = 6,3 Hz), 4,08 (1H, q, J = 6,3 Hz), 6,99 (2H, dd, J =
8,6 Hz, 9,0 Hz), 7,23 (1H, dd, J = 4,7 Hz, 8,1 Hz), 7,45 (2H, dd,
J = 5,3 Hz, 9,0 Hz), 7,86 (1H, ddd, J = 1,6 Hz, 2,5 Hz, 8,1 Hz),
8,44 (1H, dd, J = 1,6 Hz, 4,7 Hz), 8,79 (1H, d, J = 2,5 Hz)
-
Bezugsbeispiel
8
Herstellung des optisch aktiven (2S)-1-(4-Fluorphenyl)-1-(2-fluor-4-pyridyl)-1,2-propandiamins
(Epimer in 1. Position der in Bezugsbeispiel 7 beschriebenen Verbindung)
-
4-Fluorphenyllithium
wurde mit N-[(1S)-2-[(t-Butylsulfinyl)imino]-2-(2-fluor-4-pyridyl)-1-methylethyl]carbamin säure-t-butylester
durch das Verfahren, das zu dem in Bezugsbeispiel 7 beschriebenen
Verfahren ähnlich
ist, umgesetzt und die Schutzgruppen des Produkts wurden unter saurer
Bedingung entfernt, unter Gewinnung des optisch aktiven Titeldiamins.
1HNMR (300 MHz, CDCl3, δ ppm): 0,98
(3H, d, J = 6,4 Hz), 6,98–7,07
(2H, m), 7,10 (1H, s), 4,07 (1H, q, J = 6,4 Hz), 7,24 (1H, dt, J
= 5,4 Hz, 1,7 Hz), 7,48–7,58
(2H, m), 8,11 (1H, d, J = 5,3 Hz)
-
Bezugsbeispiel
8-1
Herstellung des optisch aktiven (2S)-1-(4-Fluorphenyl)-1-(1,5-naphthyridin-3-yl)-1,2-propandiamins
(Epimer in 1. Position der in Bezugsbeispiel 7-1 beschriebenen Verbindung)
-
4-Fluorphenyllithium
wurde mit N-[(1S)-2-[(t-Butylsulfinyl)imino]-2-(1,5-naphthyridin-3-yl)-1-methylethyl]carbaminsäure-t-butylester
durch das Verfahren, das zu dem in Bezugsbeispiel 7 beschriebenen
Verfahren ähnlich
ist, umgesetzt und die Schutzgruppen des Produkts wurden unter saurer
Bedingung entfernt, unter Gewinnung des optisch aktiven Titeldiamins.
1HNMR (300 MHz, CDCl3, δ ppm): 1,09
(1H, d, J = 6,3 Hz), 1,85–2,45
(4H, br), 4,26 (1H, q, J = 6,3 Hz), 7,01 (2H, t, J = 8,6 Hz), 7,46–7,65 (3H,
m), 8,35 (1H, d, J = 8,6 Hz), 8,52 (1H, d, J = 1,8 Hz), 8,96 (1H,
dd, J = 4,1 Hz, 1,5 Hz), 9,07 (1H, d, J = 2,2 Hz)
-
Herstellungsbeispiel 1
-
Die
Verbindung von Beispiel 1, 20,0 g, Lactose, 417 g, kristalline Cellulose,
80 g, und teilweise vorgelatinisierte Stärke, 80 g, wurden unter Anwendung
eines Mischers vom V-Typ vermischt und Magnesiumstearat, 3,0 g,
wurde zu dem Gemisch gegeben. Das erhaltene Gemisch wurde weiter
vermischt. Das Gemisch wurde in einer gewöhnlichen Weise verdichtet,
unter Gewinnung von 3000 Tabletten; der Durchmesser von jeder Tablette
war 7,0 mm und das Gewicht von jeder Tablette war 150 mg.
-
-
Herstellungsbeispiel 2
-
Hydroxypropylcellulose
2910, 10,8 g, und Polyethylenglycol 6000, 2,1 g, wurden in gereinigtem
Wasser, 172,5 g, gelöst
und Titandioxid, 2,1 g, wurde in der Lösung dispergiert, um eine Beschichtungslösung herzustellen.
2500 Tabletten, die getrennt in Herstellungsbeispiel 1 hergestellt
wurden, wurden mit der Beschichtungslösung, unter Verwendung von
HYCOATERMINI sprühbeschichtet,
unter Gewinnung von filmbeschichteten Tabletten, wobei das Gewicht
von jeder Tablette 155 mg war.
-
-
Industrielle Anwendbarkeit
-
Die
erfindungsgemäßen Verbindungen
sind als Mittel für
die Behandlung von verschiedenen Erkrankungen verwendbar, die NPY
betreffen; das heißt
beispielsweise, cardiovaskuläre
Störungen,
beispielhaft angegeben durch Bluthochdruck, Nephropathie, Herzerkrankungen,
Vasospasmus und Arteriosklerose, Störungen des zentralen Nervensystems,
beispielhaft angegeben durch Bulimie, Depression, Angstzustand,
Anfall, Epilepsie, Demenz, Schmerz, Alkoholismus und Drogenentzug,
metabolische Erkrankungen, beispielhaft angegeben durch Fettsucht,
Diabetes, Hormonabnormalität,
Hypercholesterolämie
und Hyperlipidämie,
sexuelle und reproduktive Fehlfunktion, gastrointestinale Störung, wie
die Inhibierung von gastrointestinaler Motilität, Atmungsstörung, Entzündung oder
Glaukom und dergleichen, da sie NPY-antagonistische Wirksamkeiten
zeigen und ausgezeichnet in der Pharmakokinetik sind, wie dem Eindringen
zum Hirn und cerebrospinaler Flüssigkeit.
















 worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen, wie vorstehend beschriebenen Bedeutungen aufweisen.
worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen, wie vorstehend beschriebenen Bedeutungen aufweisen.







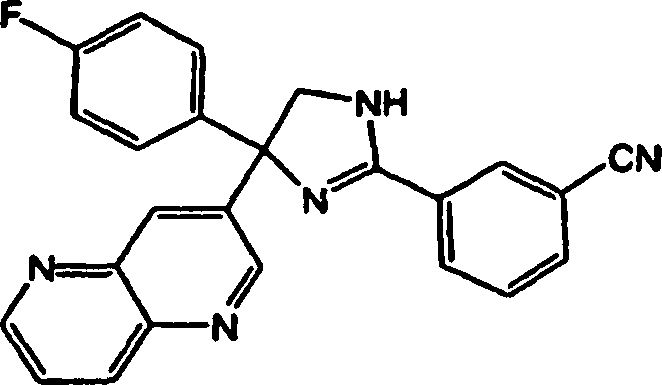






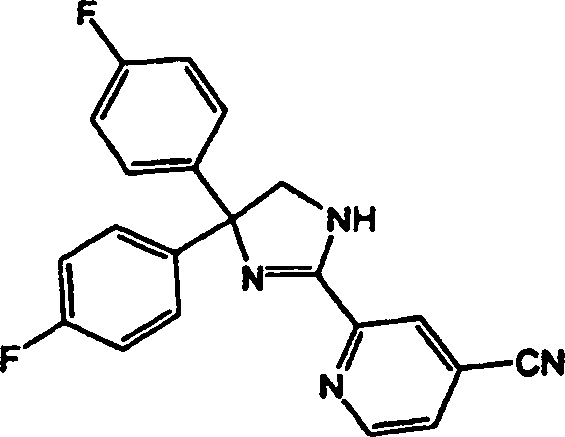





















 R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-al kylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); ein Salz oder Ester davon.
R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-al kylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); ein Salz oder Ester davon.
 R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; n und Z dieselben, wie nachstehend beschriebenen Bedeutungen aufweisen (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); eines Salzes oder Esters davon, umfassend das Umsetzen einer Verbindung der allgemeinen Formel (II):
R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; n und Z dieselben, wie nachstehend beschriebenen Bedeutungen aufweisen (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); eines Salzes oder Esters davon, umfassend das Umsetzen einer Verbindung der allgemeinen Formel (II): worin Ar1p und Ar3p unabhängig Aryl oder Heteroaryl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; n 0, 1 oder 2 wiedergibt; R1p Cyclo-C3-C6-alkyl wiedergibt oder R1p Ar3p darstellt; oder
worin Ar1p und Ar3p unabhängig Aryl oder Heteroaryl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; n 0, 1 oder 2 wiedergibt; R1p Cyclo-C3-C6-alkyl wiedergibt oder R1p Ar3p darstellt; oder R2p und R3p unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, und einer gegebenenfalls geschützten C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, Hydroxy- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; sowohl R10p als auch R11p Wasserstoffatom wiedergeben oder sie sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben; R12p Wasserstoffatom, C1-C6-Alkyl oder eine Schutzgruppe für eine Iminogruppe wiedergibt; Xp und Yp unabhängig Methylen, Ethenylen, eine durch die Formel -NR12p- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder ein Stickstoffatom wiedergibt; mit einem Säureadditionssalz der durch die allgemeine Formel (III) wiedergegebenen Verbindung
R2p und R3p unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, und einer gegebenenfalls geschützten C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, Hydroxy- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; sowohl R10p als auch R11p Wasserstoffatom wiedergeben oder sie sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben; R12p Wasserstoffatom, C1-C6-Alkyl oder eine Schutzgruppe für eine Iminogruppe wiedergibt; Xp und Yp unabhängig Methylen, Ethenylen, eine durch die Formel -NR12p- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder ein Stickstoffatom wiedergibt; mit einem Säureadditionssalz der durch die allgemeine Formel (III) wiedergegebenen Verbindung worin Ar2p Aryl oder Heteroaryl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C1-C6-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carb oxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergibt; R4 Amino oder C1-C6-Alkoxy wiedergibt; zur Bereitstellung einer Verbindung der allgemeinen Formel (IV):
worin Ar2p Aryl oder Heteroaryl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C1-C6-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carb oxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergibt; R4 Amino oder C1-C6-Alkoxy wiedergibt; zur Bereitstellung einer Verbindung der allgemeinen Formel (IV): worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen; gegebenenfalls gefolgt von Abspaltung einer Schutzgruppe.
worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen; gegebenenfalls gefolgt von Abspaltung einer Schutzgruppe.
 R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; n und Z dieselben, wie nachstehend beschriebenen Bedeutungen aufweisen (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); eines Salzes oder Esters davon, umfassend das Umsetzen einer Verbindung der allgemeinen Formel (II):
R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; n und Z dieselben, wie nachstehend beschriebenen Bedeutungen aufweisen (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); eines Salzes oder Esters davon, umfassend das Umsetzen einer Verbindung der allgemeinen Formel (II): worin Ar1p und Ar3p unabhängig Aryl oder Heteroaryl wiedergeben, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; n 0, 1 oder 2 wiedergibt; R1p Cyclo-C3-C6-alkyl wiedergibt oder R1p Ar3p darstellt; oder
worin Ar1p und Ar3p unabhängig Aryl oder Heteroaryl wiedergeben, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; n 0, 1 oder 2 wiedergibt; R1p Cyclo-C3-C6-alkyl wiedergibt oder R1p Ar3p darstellt; oder R2p und R3p unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl und einer gegebenenfalls geschützten C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, Hydroxy- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; sowohl R10p als auch R11p Wasserstoffatom wiedergeben oder sie sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben; R12p Wasserstoffatom, C1-C6-Alkyl oder eine Schutzgruppe für eine Iminogruppe wiedergibt; Xp und Yp unabhängig Methylen, Ethenylen, eine durch die Formel -NR12p- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt; mit einer Verbindung der allgemeinen Formel (V) in Gegenwart von Tri-C1-C6-alkylaluminium:
R2p und R3p unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl und einer gegebenenfalls geschützten C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, Hydroxy- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; sowohl R10p als auch R11p Wasserstoffatom wiedergeben oder sie sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben; R12p Wasserstoffatom, C1-C6-Alkyl oder eine Schutzgruppe für eine Iminogruppe wiedergibt; Xp und Yp unabhängig Methylen, Ethenylen, eine durch die Formel -NR12p- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt; mit einer Verbindung der allgemeinen Formel (V) in Gegenwart von Tri-C1-C6-alkylaluminium: worin Ar2p Aryl oder Heteroaryl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergibt; R5 C1-C6-Alkoxy wiedergibt; zur Bereitstellung einer Verbindung der allgemeinen Formel (IV):
worin Ar2p Aryl oder Heteroaryl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergibt; R5 C1-C6-Alkoxy wiedergibt; zur Bereitstellung einer Verbindung der allgemeinen Formel (IV): worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen; gegebenenfalls gefolgt von Abspaltung einer Schutzgruppe.
worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen; gegebenenfalls gefolgt von Abspaltung einer Schutzgruppe.
 R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; n und Z dieselben, wie nachstehend beschriebenen Bedeutungen aufweisen (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); ei nes Salzes oder Esters davon, umfassend das Umsetzen einer Verbindung der allgemeinen Formel (II):
R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; n und Z dieselben, wie nachstehend beschriebenen Bedeutungen aufweisen (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); ei nes Salzes oder Esters davon, umfassend das Umsetzen einer Verbindung der allgemeinen Formel (II): worin Ar1p und Ar3p unabhängig Aryl oder Heteroaryl wiedergeben, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkyl-amino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; n 0, 1 oder 2 wiedergibt; R1p Cyclo-C3-C6-alkyl wiedergibt oder R1p Ar3p darstellt; oder
worin Ar1p und Ar3p unabhängig Aryl oder Heteroaryl wiedergeben, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkyl-amino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; n 0, 1 oder 2 wiedergibt; R1p Cyclo-C3-C6-alkyl wiedergibt oder R1p Ar3p darstellt; oder R2p und R3p unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, und einer gegebenenfalls geschützten C1-C6-Alkyl amino-, C2-C7-Alkanoylamino-, Hydroxy- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; sowohl R10p als auch R11p Wasserstoffatom wiedergeben oder sie sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben; R12p Wasserstoffatom, C1-C6-Alkyl oder eine Schutzgruppe für die Iminogruppe wiedergibt; Xp und Yp unabhängig Methylen, Ethenylen, eine durch die Formel -NR12p- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt; mit einer Verbindung der allgemeinen Formel (VI):
R2p und R3p unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, und einer gegebenenfalls geschützten C1-C6-Alkyl amino-, C2-C7-Alkanoylamino-, Hydroxy- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; sowohl R10p als auch R11p Wasserstoffatom wiedergeben oder sie sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben; R12p Wasserstoffatom, C1-C6-Alkyl oder eine Schutzgruppe für die Iminogruppe wiedergibt; Xp und Yp unabhängig Methylen, Ethenylen, eine durch die Formel -NR12p- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt; mit einer Verbindung der allgemeinen Formel (VI): worin Ar2p Aryl oder Heteroaryl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergibt; zur Bereitstellung einer Verbindung der allgemeinen Formel (VII):
worin Ar2p Aryl oder Heteroaryl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Cyano, Halogenatom, Nitro, C1-C6-Alkyl, Halogen-C1-C6-alkyl, Cyclo-C3-C6-alkyl-C1-C6-alkyl, C2-C6-Alkenyl, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Halogen-C1-C6-alkoxy, Aryloxy, Heteroaryloxy, C1-C6-Alkylthio, Formyl, C2-C7-Alkanoyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, C1-C6-Alkylsulfonyl, Arylsulfonyl, Aryl, Heteroaryl, und einer gegebenenfalls geschützten Hydroxy-C1-C6-alkyl-, C1-C6-Alkylamino-, C2-C7-Alkanoylamino-, C1-C6-Alkylsulfonylamino-, Arylsulfonylamino-, Hydroxy-, Carboxyl-, Carbamoyl- oder C1-C6-Alkylcarbamoylgruppe, wiedergibt; zur Bereitstellung einer Verbindung der allgemeinen Formel (VII): worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen; gefolgt von Ausführen von intramolekularer Cyclo-Kondensationsreaktion der allgemeinen Formel (VII), zur Bereitstellung einer Verbindung der allgemeinen Formel (IV):
worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen; gefolgt von Ausführen von intramolekularer Cyclo-Kondensationsreaktion der allgemeinen Formel (VII), zur Bereitstellung einer Verbindung der allgemeinen Formel (IV): worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen; gegebenenfalls gefolgt von Abspaltung einer Schutzgruppe.
worin Ar1p, Ar2p, R1p, R2p und R3p die gleichen wie vorstehend beschriebenen Bedeutungen aufweisen; gegebenenfalls gefolgt von Abspaltung einer Schutzgruppe.
 R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); ein Salz oder Ester davon enthält.
R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); ein Salz oder Ester davon enthält.
 R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); ein Salz oder Ester davon enthält.
R2 und R3 unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, C1-C6-Alkylamino, Di-C1-C6-alkylamino, C2-C7-Alkanoylamino, Hydroxy, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, C1-C6-Alkylcarbamoyl und Di-C1-C6-alkylcarbamoyl, wiedergeben; sowohl R10 als auch R11 Wasserstoffatom wiedergeben oder sie sich vereinigen, um Oxo wiederzugeben; R12 Wasserstoffatom oder C1-C6-Alkyl wiedergibt; X und Y unabhängig Methylen, Ethenylen, eine durch die Formel -NR12- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt (jedoch im Fall, dass sowohl R2 als auch R3 Wasserstoffatom wiedergeben, Ar1, Ar2 und R1 nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben); ein Salz oder Ester davon enthält.
 R2p und R3p unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, und einer gegebenenfalls geschützten C1-C6-Alkylamino-, C2- C7-Alkanoylamino-, Hydroxy- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; sowohl R10p als auch R11p Wasserstoffatom wiedergeben oder sie sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben; R12p Wasserstoffatom, C1-C6-Alkyl oder eine Schutzgruppe für eine Iminogruppe wiedergibt; Xp und Yp unabhängig Methylen, Ethenylen, eine durch die Formel -NR12p- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt; (jedoch (a) im Fall, dass sowohl R2p als auch R3p Wasserstoffatom wiedergeben, Ar1p und R1p nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben, und (b), im Fall, dass R2p Wasserstoffatom darstellt und R3p Methyl, Isopropyl, Isobutyl oder tert-Butyl darstellt, Ar1p und R1p nicht gleichzeitig 4-Methoxyphenyl wiedergeben).
R2p und R3p unabhängig Wasserstoffatom, Cyclo-C3-C6-alkyl, C2-C6-Alkenyl oder C1-C6-Alkyl, die substituiert sein können, wobei der Substituent ausgewählt ist aus der Gruppe, bestehend aus Halogenatom, Di-C1-C6-alkylamino, C1-C6-Alkoxy, Formyl, C2-C7-Alkoxycarbonyl, Di-C1-C6-alkylcarbamoyl, und einer gegebenenfalls geschützten C1-C6-Alkylamino-, C2- C7-Alkanoylamino-, Hydroxy- oder C1-C6-Alkylcarbamoylgruppe, wiedergeben; sowohl R10p als auch R11p Wasserstoffatom wiedergeben oder sie sich vereinigen, um eine gegebenenfalls geschützte Oxogruppe wiederzugeben; R12p Wasserstoffatom, C1-C6-Alkyl oder eine Schutzgruppe für eine Iminogruppe wiedergibt; Xp und Yp unabhängig Methylen, Ethenylen, eine durch die Formel -NR12p- wiedergegebene Gruppe, Sauerstoffatom oder Schwefelatom wiedergeben; Z Methin oder Stickstoffatom wiedergibt; (jedoch (a) im Fall, dass sowohl R2p als auch R3p Wasserstoffatom wiedergeben, Ar1p und R1p nicht gleichzeitig eine unsubstituierte Phenylgruppe wiedergeben, und (b), im Fall, dass R2p Wasserstoffatom darstellt und R3p Methyl, Isopropyl, Isobutyl oder tert-Butyl darstellt, Ar1p und R1p nicht gleichzeitig 4-Methoxyphenyl wiedergeben).