CN1310718A - 用作旋转异构酶抑制剂的杂环化合物 - Google Patents
用作旋转异构酶抑制剂的杂环化合物 Download PDFInfo
- Publication number
- CN1310718A CN1310718A CN99808997A CN99808997A CN1310718A CN 1310718 A CN1310718 A CN 1310718A CN 99808997 A CN99808997 A CN 99808997A CN 99808997 A CN99808997 A CN 99808997A CN 1310718 A CN1310718 A CN 1310718A
- Authority
- CN
- China
- Prior art keywords
- compound
- alkyl
- formula
- benzoxazole
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Physical Education & Sports Medicine (AREA)
- Psychology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Immunology (AREA)
- Ophthalmology & Optometry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Enzymes And Modification Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Hydrogenated Pyridines (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines Containing Plant Substances (AREA)
Abstract
式(Ⅰ)化合物,其中A是任意选择性地被C1-C6烷基取代的直链C3-C5亚烷基;X是O、S、NH或N(C1-C6烷基);Y是O、S、NH或N(C1-C6烷基);R是通过碳连接的4至6元非芳香性的含有1个氮杂原子的杂环基团,所述基团任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C2-C6链烯基、C3-C7环烷基、芳基、het、-CO2(C1-C6烷基)、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代,所述烷基和链烯基任意选择性地被1或2个彼此独立地选自C3-C7环烷基、芳基、het、-O(芳基)、-O(C1-C2亚烷基)芳基、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代;该化合物是旋转异构酶、特别是FKBP-12和FKBP-52的抑制剂。因此,该化合物可调节神经元的再生和分枝,可用于治疗由于神经变性疾病引起的神经病学疾病或其它涉及神经损伤的疾病。
Description
本发明涉及1-杂芳基-吡咯烷、-哌啶和-高哌啶衍生物及其制备方法、其制备中所用的中间体、含有该衍生物的组合物以及所述衍生物的用途。
据报道,免疫抑制剂FK-506可以在体外促进神经细胞系和培养模型中的轴突分枝(参见Lyons等,《美国国家科学院院报》(Pro.Natl.Acad.Sci.),1994,91,3191-95和Snyder等,《天然药物》(NatureMedicine),1995,1,32-37)。WO-A-96/40140、WO-A-96/40633和WO-A-97/16190公开了具有神经营养活性但对蛋白磷酸酶钙调磷酸酶缺乏抑制作用从而没有免疫抑制作用的化合物。US-A-5,721,256公开了磺胺类化合物,WO-A-98/13343和WO-A-98/13355公开了杂环类化合物,这些化合物均具有神经营养活性而不显示任何明显的免疫抑制活性。WO-A-92/21313公开了具有免疫抑制活性的磺胺类化合物。
WO-A-96/40140和WO-A-96/40633中提出,这些化合物的神经营养作用至少部分是由与FK-506结合蛋白例如FKBP-12或FKBP-52的高亲和性相互作用所介导的。但是,由这种与FKBP-型亲免素的相互作用引起神经营养作用的机制目前还不清楚。对可以通过该神经营养/非免疫抑制剂类型的化合物实现的神经营养活性的范围进行了研究,结果发现,可以在大鼠中在面部神经压碎和坐骨神经压碎后促进轴突的再生。还观察到了可以通过文中所公开的化合物在小鼠中促进用毒素MPTP损伤的多巴胺神经元功能的恢复。此外,还报道了在用6-羟基多巴胺损伤多巴胺能神经元后,可以用文中所公开的化合物促进大鼠纹状体神经分布的恢复。(参见Hamilton&Steiner,《当代药物设计》(Current Pharmaceutical Design),1997,3,405-428)。
现已发现,本发明的化合物是对FKBP-型亲免素具有亲和性的神经营养剂。具体地讲,它们是酶活性、尤其是FKBP-型亲免素、特别是亲免素FKBP-12的顺-反脯氨酰异构酶(旋转异构酶)活性的强效抑制剂。本发明的化合物对蛋白磷酸酶钙调磷酸酶没有明显的抑制作用,因此没有免疫抑制活性。
因此,本发明的化合物减轻神经元变性并促进神经元的再生和分枝,因此可用于治疗由于神经变性疾病引起的神经病学疾病或其它涉及神经损伤的疾病。可以治疗的神经病学疾病包括老年性痴呆(早老性痴呆)和其它痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、影响中枢或外周神经系统的各种形式的变性疾病(例如小脑-脑干萎缩、进行性共济失调综合征)、各种形式的肌营养不良、进行性肌萎缩、进行性延髓肌萎缩、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、突出、破裂或脱出的椎间盘综合征、颈椎关节强硬、神经丛疾病、胸腔出口综合征、各种形式的外周神经病(糖尿病性的和非糖尿病性的)、三叉神经痛、舌咽神经痛、面神经麻痹、导致中枢或外周神经系统损伤的各种形式的自身免疫相关性疾病(例如多发性硬化、重症肌无力、格-巴二氏综合征)、神经系统的AIDS相关性疾病、氨苯砜抽搐(dapsone ticks)、眼神经的眼球和眼球后病变(例如视网膜病和眼球后神经炎)、听力障碍例如耳鸣和朊病毒疾病。
优选本发明的化合物可用于治疗老年性痴呆(早老性痴呆)或其它的痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、外周神经痛(糖尿病性的或非糖尿病性的)、多发性硬化或听力障碍例如耳鸣。
本发明提供下式的化合物或其可药用盐:其中
A是任意选择性地被C1-C6烷基取代的直链C3-C5亚烷基;
X是O、S、NH或N(C1-C6烷基);
Y是O、S、NH或N(C1-C6烷基);
R是通过碳连接的4至6元非芳香性的含有1个氮杂原子的杂环基团,所述基团任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C2-C6链烯基、C3-C7环烷基、芳基、het、-CO2(C1-C6烷基)、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代,所述烷基和链烯基任意选择性地被1或2个彼此独立地选自C3-C7环烷基、芳基、het、-O(芳基)、-O(C1-C2亚烷基)芳基、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代;
R1、R2、R3和R4彼此独立地选自H、卤素、C1-C6烷基、C3-C7环烷基、卤素(C1-C6)烷基、C1-C6烷氧基、-CONR5R6、C3-C7环烷氧基、C3-C7环烷基-(C2-C4)亚烷基、C3-C7环烷基(C2-C4)烷氧基和-CO2(C1-C6烷基);
R5和R6彼此独立地选自H和C1-C6烷基,或者当它们合在一起时,表示直链的C3-C5亚烷基;
“芳基”是指任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C1-C6烷氧基、卤素、-CONR5R6、卤素(C1-C6烷基)和-NR5R6的取代基取代的苯基;
“het”是指含有1至3个彼此独立地选自N、O和S的杂原子的5或6元单环或8、9或10元二环的杂环基团,所述基团任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C1-C6烷氧基、卤素、卤素(C1-C6烷基)、苯基和-NR5R6的取代基所取代。
在上述定义中,“卤素”是指氟、氯、溴或碘;含有必要碳原子数的烷基、烷氧基、链烯基和亚烷基除非指明可以是直链或支链的。
式(Ⅰ)化合物的可药用盐包括其酸加成盐和碱盐。
适宜的酸加成盐是由形成无毒盐的酸形成,所述无毒盐的例子是盐酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、硫酸氢盐、硝酸盐、磷酸盐、磷酸氢盐、乙酸盐、马来酸盐、富马酸盐、乳酸盐、酒石酸盐、柠檬酸盐、葡萄糖酸盐、琥珀酸盐、蔗糖盐、苯甲酸盐、甲磺酸盐、乙磺酸盐、苯磺酸盐、对甲苯磺酸盐和双羟萘酸盐(pamoate)。
适宜的碱盐是由形成无毒盐的碱形成,所述无毒盐的例子是钠盐、钾盐、铝盐、钙盐、镁盐、锌盐和二乙醇胺盐。
适宜的盐可以参见Berge等,《药学杂志》(J.Pharm.Sci.),1977,66,1-19。
式(Ⅰ)化合物的可药用溶剂化物包括其水合物。
本发明还包括式(Ⅰ)化合物的多晶型物及其放射标记的衍生物。
式(Ⅰ)化合物含有一个或多个不对称碳原子,因此以两种或多种立体异构体形式存在。当式(Ⅰ)化合物含有链烯基或亚链烯基(alkenylene)基团时,还可以出现顺式(E)和反式(Z)异构。本发明包括式(Ⅰ)化合物的各种立体异构体及其(如果有的话)各种互变异构体形式以及它们的混合物。
非对映体或顺式和反式异构体的分离可以通过常规方法来完成,例如,通过将式(Ⅰ)化合物或其适宜的盐或衍生物的立体异构体混合物进行分级结晶、色谱分离或HPLC分离。如果合适,式(Ⅰ)化合物的单个对映体还可以从相应的光学纯的中间体制备或通过拆分、例如将相应的外消旋体用适宜的手性载体进行HPLC分离或将由相应的外消旋体与适宜的光学活性的酸或碱反应形成的非对映体盐进行分级结晶制得。
特别优选下式的化合物:
其中R、R1、R2、R3、R4、A、X和Y分别如以上式(Ⅰ)化合物中所定义。
在以上式(Ⅰ)和(ⅠA)化合物的定义中,优选如下定义:
优选A是1,4-亚丁基。
优选X是O、S或NH。
优选X是O或NH。
优选Y是O或NH。
优选Y是NH。
优选R是氮杂环丁烷基、吡咯烷基或哌啶基,它们均任意选择性地按照以上式(Ⅰ)化合物中R的定义被取代。
优选R是3-氮杂环丁烷基、3-吡咯烷基、3-哌啶基或4-哌啶基,它们均任意选择性地按照以上式(Ⅰ)化合物中R的定义被取代。
优选R是氮杂环丁烷基、吡咯烷基或哌啶基,它们均任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、het、-CO2(C1-C6烷基)和-CO(het)的取代基所取代,所述烷基任意选择性地被1或2个彼此独立地选自C3-C7环烷基、芳基、het、-O(芳基)、-O(C1-C2亚烷基)芳基和-CONR5R6的取代基所取代。
优选R是3-氮杂环丁烷基、3-吡咯烷基、3-哌啶基或4-哌啶基,它们均任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、het、-CO2(C1-C6烷基)和-CO(het)的取代基所取代,所述烷基任意选择性地被1或2个彼此独立地选自C3-C7环烷基、芳基、het、-O(芳基)、-O(C1-C2亚烷基)芳基和-CONR5R6的取代基所取代。
优选R是氮杂环丁烷基、吡咯烷基或哌啶基,它们均任意选择性地被1、2或3个彼此独立地选自乙基、2-吡啶基、叔丁氧羰基、喹啉-2-基羰基、2-苯基喹啉-4-基羰基、4-甲氧基喹啉-2-基羰基、6-甲氧基-2-苯基喹啉-4-基羰基、2-哌啶子基喹啉-4-基羰基、2-氯喹啉-4-基羰基、1H-苯并吡唑-6-基羰基、环丙基甲基、苯基甲基、二苯基甲基、2-吡啶基甲基、3-吡啶基甲基、4-吡啶基甲基、2-(2-吡啶基)乙基、2-(2-甲基咪唑-1-基)乙基、(1H-1,2,4-三唑-3-基)甲基、(2-氯喹啉-3-基)甲基、喹啉-4-基甲基、喹啉-2-基甲基、喹啉-3-基甲基、1-(喹啉-4-基)乙基、(2-氟吡啶-4-基)甲基、苯氧基甲基、苄氧基甲基、氨基羰基甲基、2-(氨基羰基)乙基和3-(氨基羰基)苯基甲基的取代基所取代。
优选R是3-氮杂环丁烷基、3-吡咯烷基、3-哌啶基或4-哌啶基,它们均任意选择性地被1、2或3个彼此独立地选自乙基、2-吡啶基、叔丁氧羰基、喹啉-2-基羰基、2-苯基喹啉-4-基羰基、4-甲氧基喹啉-2-基羰基、6-甲氧基-2-苯基喹啉-4-基羰基、2-哌啶子基喹啉-4-基羰基、2-氯喹啉-4-基羰基、1H-苯并吡唑-6-基羰基、环丙基甲基、苯基甲基、二苯基甲基、2-吡啶基甲基、3-吡啶基甲基、4-吡啶基甲基、2-(2-吡啶基)乙基、2-(2-甲基咪唑-1-基)乙基、(1H-1,2,4-三唑-3-基)甲基、(2-氯喹啉-3-基)甲基、喹啉-4-基甲基、喹啉-2-基甲基、喹啉-3-基甲基、1-(喹啉-4-基)乙基、(2-氟吡啶-4-基)甲基、苯氧基甲基、苄氧基甲基、氨基羰基甲基、2-(氨基羰基)乙基和3-(氨基羰基)苯基甲基的取代基所取代。
优选R1、R2、R3和R4彼此独立地选自H、卤素(C1-C6)烷基和卤素。
优选R1、R2、R3和R4彼此独立地选自H、氟、氯、溴和三氟甲基。
优选R5和R6分别是H,或者,当合在一起时,是1,5-亚戊基。
优选“芳基”是指任意选择性地被CONR5R6取代的苯基,其中R5和R6优选均是H。
优选“het”是指吡啶基、咪唑基、三唑基、喹啉基或苯并吡唑基,它们均任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C1-C6烷氧基、卤素、苯基和-NR5R6的取代基所取代。
优选“bet”是指吡啶基、咪唑基、三唑基、喹啉基或苯并吡唑基,它们均任意选择性地被1、2或3个彼此独立地选自甲基、甲氧基、氟、氯、苯基和哌啶子基的取代基所取代。
优选“het”是指2-吡啶基、3-吡啶基、4-吡啶基、咪唑-1-基、1H-1,2,4-三唑-3-基、喹啉-2-基、喹啉-3-基、喹啉-4-基或1H-苯并吡唑-6-基,它们均任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C1-C6烷氧基、卤素、苯基和-NR5R6的取代基所取代。
优选“het”是指2-吡啶基、3-吡啶基、4-吡啶基、咪唑-1-基、1H-1,2,4-三唑-3-基、喹啉-2-基、喹啉-3-基、喹啉-4-基或1H-苯并吡唑-6-基,它们均任意选择性地被1、2或3个彼此独立地选自甲基、甲氧基、氟、氯、苯基和哌啶子基的取代基所取代。
优选“het”是指2-吡啶基、3-吡啶基、4-吡啶基、1H-1,2,4-三唑-3-基、喹啉-2-基、喹啉-3-基、喹啉-4-基、1H-苯并吡唑-6-基、2-甲基咪唑-1-基、2-氯喹啉-3-基、2-苯基喹啉-4-基、4-甲氧基喹啉-2-基、6-甲氧基-2-苯基喹啉-4-基、2-哌啶子基喹啉-4-基、2-氯喹啉-4-基或2-氟吡啶-4-基。
优选式(Ⅰ)化合物中下式的基团
是1,3-苯并噁唑-2-基、1,3-苯并噻唑-2-基、1H-苯并咪唑-2-基、6-溴-1,3-苯并噁唑-2-基或6-氯-1,3-苯并噻唑-2-基。
在以下实施例部分中描述的特别优选的式(Ⅰ)化合物的例子是:(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(2-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(3-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(4-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[3-(氨基羰基)苯基甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[(2-氯喹啉-3-基)甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[(喹啉-3-基)甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[(喹啉-4-基)甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[(喹啉-2-基)甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[1-(喹啉-4-基)乙基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[喹啉-2-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[2-苯基喹啉-4-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[4-甲氧基喹啉-2-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[6-甲氧基-2-苯基喹啉-4-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[2-哌啶子基喹啉-4-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[2-氯喹啉-4-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[1H-苯并吡唑-6-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-[(3S)-1-苄基吡咯烷-3-基]-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-(1-苄基-3-哌啶基)-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-(3R,5S)-5-[(苄氧基)甲基]吡咯烷-3-基-2-哌啶甲酰胺盐酸盐;和1-(1H-1,3-苯并咪唑-2-基)-N2-[(3S)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺。
式(Ⅰ)化合物可以用常规方法例如以下描述的方法进行制备,若无另外说明,其中的R、R1、R2、R3、R4、A、X和Y如以上式(Ⅰ)化合物中所定义。1)其中X是O或S的式(Ⅰ)化合物可以通过将下式化合物 其中X是O或S,与下式化合物进行脱水偶联制得:
其中X是O或S,与下式化合物进行脱水偶联制得:
H-Y-R (Ⅲ)。
式(Ⅱ)化合物可以按照本文的制备例3中所描述的方法进行制备。采用常规方法进行该制备的适宜条件是本领域技术人员熟知的,例如标准教材如《高等有机化学》(Advanced Organic Chemistry),第3版,Jerry March,0-56,371-4页中所描述的条件。
适宜条件的例子如下:(a)可以首先将式(Ⅱ)化合物用1-羟基苯并三唑水合物和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐在适宜的酸接受体例如三乙胺的存在下转变成活泼酯,然后就地用式(Ⅲ)化合物处理。反应可以在适宜的溶剂例如二氯甲烷中进行。还可以使用催化量的适宜催化剂,例如4-二乙基氨基吡啶。(b)可以将式(Ⅱ)和(Ⅲ)的化合物与三苯膦和偶氮二甲酸二乙酯在适宜溶剂例如四氢呋喃的存在下混合。(c)可以将式(Ⅱ)和(Ⅲ)的化合物与1,1’-羰基二咪唑在适宜溶剂例如四氢呋喃或二氯甲烷的存在下混合。(d)直接将式(Ⅱ)和(Ⅲ)的化合物一起加热,反应在任意选择性存在适宜溶剂如N,N-二甲基乙酰胺、环戊醇或二苯基醚的条件下进行,当Y是O时,在任意选择性存在适宜的酸性催化剂的条件下进行。
式(Ⅱ)的中间体化合物可以通过常规方法制备,例如,通过反应路线1中所示的路线制备。
反应路线1其中R7是C1-C4烷基(优选甲基)或苄基L1是适宜的离去基,例如卤素(优选氯)、-SCH3、-SH、-SO2CH3、-SO2CF3、-OSO2CH3或-OSO2CF3。
式(Ⅲ)、(Ⅳ)和(Ⅵ)的化合物可以通过常规方法制备。2)其中X=NH的式(Ⅰ)化合物(即式(ⅠB)的化合物)可以通过反应方案2中所示的路线进行制备,即通过将式(ⅩⅢA)或(ⅩⅢB)的化合物与式(Ⅲ)化合物反应进行制备。
反应方案2
 其中L2是适宜的离去基,例如以上在L1中所定义的那些,R8是C1-C4烷基(优选叔丁基)或苄基,R9是C1-C4烷基或苄基。
其中L2是适宜的离去基,例如以上在L1中所定义的那些,R8是C1-C4烷基(优选叔丁基)或苄基,R9是C1-C4烷基或苄基。
式(Ⅷ)和(Ⅹ)的化合物可以通过常规的方法制备。在制备例46和47中描述了使用市售的式(Ⅷ)和(Ⅹ)化合物的反应。3)其中X是O或S的式(Ⅰ)化合物可以通过将式(ⅩⅣ)化合物其中X是O或S,与式(Ⅲ)化合物反应制得:
H-Y-R(Ⅲ)其中L3是适宜的离去基,例如(ⅰ)卤素,优选氯或溴,(ⅱ)可以提供活泼酯的基团,例如通过将式(Ⅱ)化合物与1-羟基苯并三唑、苯并三唑-1-基氧基三(二甲基氨基)-鏻六氟磷酸盐、O-(1H-苯并三唑-1-基)-N,N,N’,N’-四甲基脲四氟硼酸盐(tetramethyluronium tetrafluoroborate)或五氟苯酚反应得到的基团,(ⅲ)可以提供混合酸酐的基团,例如通过将式(Ⅱ)化合物与氯甲酸异丁酯反应得到的基团,或(ⅳ)可以提供咪唑酰胺的基团,例如通过将式(Ⅱ)化合物与1,1’-羰基二咪唑反应得到的基团。
反应可以用常规的方法进行。
式(ⅩⅣ)化合物可以通过常规方法例如从式(Ⅱ)化合物制备。4)所有式(Ⅰ)化合物均可以通过将式(ⅩⅤ)化合物与式(ⅩⅥ)化合物反应制得:其中L3如以上式(Ⅵ)化合物中的L1所定义,优选是氯。
在一个优选的方法中,其中L3是氯,反应可以在适宜的酸接受剂例如乙基二异丙基胺的存在下、在适宜的溶剂例如乙腈或N,N-二甲基乙酰胺中在加热的条件下进行。
当X=S时,反应可以方便地用铜粉、三乙胺盐酸盐和二甲苯在加热条件下进行。式(ⅩⅤ)化合物可以通过与本文中的制备例47所述类似的常规方法制备。式(ⅩⅥ)化合物可以通过例如本文中的制备例47所述的常规方法制备。
可以理解,某些式(Ⅰ)化合物可以通过常规方法,例如采用常规的相互转变技术转变成其它的式(Ⅰ)化合物。所有上述反应以及前述方法中所用的新原料的制备均是常规的,通过参考所引用的文献以及本文中的实施例和制备例,进行这些反应所用的适宜试剂和反应条件或所需产物的制备及分离方法对于本领域技术人员是显而易见的。本领域技术人员可以理解,在所述的某些方法中,所采用的合成步骤的顺序可以改变,这特别取决于具体反应物中所存在的其它功能基的性质、关键中间体的可得性以及准备采用的保护基策略(如果有的话)等因素。很明显,这些因素将会影响所述合成步骤中所用试剂的选择。
如果合适,式(Ⅰ)化合物的可药用盐可以很方便地通过将式(Ⅰ)化合物与所需的酸或碱的溶液混合在一起制得。可使盐从溶液中析出沉淀并通过过滤进行收集,或通过蒸除溶剂进行回收。
式(Ⅰ)化合物对FKBP-12的亲和性可以采用与文献方法(例如,参见Kofron,J.L.等,《生物化学》(Biochemistry),1991,30,6127-6134,Zarnt,T.等,《生物化学杂志》(Biochem.J.)1995,305,159-164,Holt,D.A.等,《美国化学会志》(J.Am.Chem.Soc.),1993,115,9925-9938)类似的操作在偶联比色PPI酶试验中进行体外测定。在这些方法中,四肽底物中的疏水性氨基酸-脯氨酸键(例如N-琥珀酰-ala-phe-pro-phe-对硝基酰苯胺[琥珀酰-AFPF-pNA]中的苯丙氨酸-脯氨酸键)的顺-反异构化可以通过监测由过量的胰凝乳蛋白酶从含有反Pro的肽中裂解出pNA来测定。
IC50(产生50%抑制的式(Ⅰ)化合物的浓度)值用如下分析方法测定。将分析缓冲液(2.175ml)(50mM 4-(2-羟基乙基)-1-哌嗪乙磺酸(HEPES)、100mM NaCl、1mM二硫苏糖醇(DTT),pH8.0)在比色杯中平衡至10℃。依次向分析缓冲液中加入12.5μl本发明化合物的DMSO溶液、250μl 60mg/mlα-胰凝乳蛋白酶在1mM盐酸中的溶液和50μl人重组FKBP-12(4.5μM)的溶液并混合。通过加入12.5μl 20mM琥珀酰-AFPF-pNA的DMSO溶液引发反应。于390nM监测吸收值1分钟,每0.25秒收集一次数据。将数据用带有偏移量(offset)的一级速率方程拟合,用得到的速率常数校正未催化的底物异构化速率。将在不同抑制剂浓度(10nM至100μM)下测定的速率常数用对照速率常数的%抑制表示。用符合S形剂量响应数据的非线性最小二乘方曲线评估IC50。
用以下描述的试验方法测定本发明化合物的Ki,app(表观抑制常数)。将分析缓冲液(2.175ml)(50mM(HEPES)、100mMNaCl、1mM DTT,pH8.0)在比色杯中平衡至10℃。依次向分析缓冲液中加入12.5μl本发明化合物的DMSO溶液、250μl 60mg/mlα-胰凝乳蛋白酶在1mM盐酸中的溶液和50μl人重组FKBP-12(1.5μM)的溶液并混合。通过加入12.5μl无水琥珀酰-ALPF-pNA(100μM最终浓度)在400mM LiCl的三氟乙醇溶液中的溶液引发反应。于390nM监测吸收值3分钟,每0.5秒收集一次数据。将数据用带有偏移量的一级速率方程拟合,从t0时的顺(re leu-pro键)-琥珀酰-ALPF-pNA的浓度和不同抑制剂浓度(Ⅰ)下的一级速率常数计算初始速率(v)。将v抑制剂/v对照v.[Ⅰ]形式的数据用可逆紧密结合抑制的方程拟合以生成Ki,app值(参见Morrison,J.F.等,《分子细胞生物物理学评论》(Comments Mol.Cell Biophys.),1985,2,347-368)。当Ki,app接近试验中FKBP-12的浓度时(30nM)采用该分析。用Dixon分析(参见Dixon,M.,《生物化学杂志》(Biochem.J.),1953,55,170-171)生成效力较弱的化合物的Ki,app值。用同样的方法生成FKBP52的Ki,app值,其中进行了如下改变:用40μl人重组FKBP52(5.2μM)代替FKBP12并在试验中使用2.185ml分析缓冲液。
本发明化合物对FKBP-12酶具有抑制活性。早期的实验表明,本发明的化合物还对FKPB-52酶具有抑制活性。
FKBP-52酶可以通过Peattie,D.A.等,《美国国家科学院院报》(Proc.Natl.Acad.Sci.USA)1992 Nov.15;89(22):10974-8中描述的方法进行表达和鉴定。在以下参考文献中对FKPB-52酶进行了讨论:Miyata,Y.等,《美国国家科学院院报》(Proc.Natl.Acad.Sci.USA)1997 Dec.23;94(26):14500-5;Tai,P.K.等,《生物化学》(Biochemistry)1993 Aug.31;32(34):8842-7;Bose,S.等,《科学》(Science),274,1715-5,1996和Czar,M.J.等,《分子内分泌学》(Molecular Endocrinology)9,1549-1560,1995.
式(Ⅰ)化合物促进轴突分枝的活性可以在鸡胚胎背根神经节的外植块培养物中进行测定。背根神经节(DRG)按照Bray的方法(参见“神经细胞培养(Culturing Nerve Cells)”G.Banker和K.Goslin编,MITPress,Cambridge,MA,1991,119页)无菌分离。将单个的神经节保存在置于冰上的不含Ca2+/Mg2+的Tyrodes缓冲液中直至收集了许多的神经节。然后将单个的神经节转移至含有Neurobasal培养基+B27添充物的胶原涂层的24孔培养板中并在37℃于5%CO2气氛下保温。保温4小时使神经节附着,然后加入本发明的化合物。培养24或48小时后将外植块固定并用考马斯蓝染色。对于每次的处理,分析4至6个神经节并通过用成像分析评估相对于外植块直径的轴突分枝程度进行计分。将本发明的化合物在含或不合10ng/ml神经生长因子(NGF)的条件下进行测试并与仅含10ng/ml神经生长因子时的分枝相比较。
用于测定FKBP-12PPI酶抑制剂的促进轴突分枝活性的另一种系统是Gold,B.G.等,《实验神经病学》(Exp.Neurol.),1997,147(2),269-278中描述的SH-SY-5Y成神经细胞瘤模型。将细胞在37℃和7%CO2气氛下保持在补充有10%胎牛血清(FCS)、50U/ml青霉素、50μg/ml链霉素的Dulbecco’s改良的Eagle’s培养基(DMEM)中。将细胞以1×106细胞/孔置于平板中并用400nM阿非迪霉素处理5天。然后将细胞洗涤并用10ng/ml NGF±各种化合物浓度处理7天以测定化合物是否可以在存在最适度以下的NGF浓度(和/或不含NGF)下促进轴突的分枝。通过用成像分析在20个随机的区域内测量轴突的长度来确定轴突的分枝。
本发明化合物的神经营养活性可以用大鼠坐骨神经压碎模型作为外周神经再生模型(参见Bridge,P.M.等,《实验神经病学》(Experimental Neurology),1994,127,284-290,Medinaceli,L.等,《实验神经病学》(Expl.Neurology),1982,77,634-643,Gold,B.G.等,《恢复神经病学和神经学》(Restorative Neurology andNeuroscience),1994,6,287-296)、用在各种动物中的1-甲基-4-苯基-1,2,3,6-四氢吡啶(MPTP)和6-羟基多巴胺模型作为帕金森氏症中的再生模型(参见Mokry,J.,《生理学研究》(Physiol.Res.),1995,44(3),143-150)以及用伞穹窿损伤作为早老性痴呆中的再生模型(参见Cassel,J.C.,Duconseille,E.,Jeltsch,H.和Will,B.,《神经病学进展》(Prog.Neurol.),1997,51,663-716)进行体内评估。
式(Ⅰ)化合物可以单独给药,但通常是以其与根据预定的给药途径和常规药物实践选择的适宜药物赋形剂、稀释剂或载体的混合物的形式给药。
例如,可将式(Ⅰ)化合物以可以含有矫味剂或着色剂的片剂、胶囊、卵形片、酏剂、溶液剂或混悬剂的形式口服或舌下给药,用于立即或控制释放应用。
适宜的片剂可以含有赋形剂如微晶纤维素、乳糖、柠檬酸钠、碳酸钙、磷酸二钙和甘氨酸,崩解剂如淀粉(优选玉米淀粉、土豆淀粉或木薯淀粉)、藻酸和某些复合硅酸盐,以及制粒粘合剂如聚乙烯吡咯烷酮、蔗糖、明胶和阿拉伯胶。此外,还可以含有润滑剂如硬脂酸镁、十二烷基硫酸钠和滑石。
相似类型的固体组合物还可用作明胶胶囊的填料。就此而言,优选的赋形剂包括乳糖以及高分子量的聚乙二醇。对于含水混悬液和/或酏剂,可将式(Ⅰ)化合物与各种甜味剂或矫味剂、着色剂或染料、乳化剂和/或助悬剂以及稀释剂如水、乙醇、丙二醇、甘油及其混合物相混合。
式(Ⅰ)化合物还可以进行胃肠外注射,例如静脉内、腹膜内、鞘内、心室内、胸骨内、颅内、肌肉内或皮下注射,或者可以将其通过输注的方法给药。最好是将它们以无菌含水溶液的形式给药,该溶液还可含有其它物质,例如可以使溶液与血液等渗的足够量的盐或葡萄糖。如需要,应将含水溶液进行适当的缓冲(优选缓冲至pH3到9)。在无菌条件下制备适宜的胃肠外制剂很容易通过本领域技术人员熟知的常规制药技术来完成。
对于人类患者的口服和胃肠外给药,式(Ⅰ)化合物的每日剂量水平通常为1mg/kg至25mg/kg(单次给药或分多次给药)。
因此,式(Ⅰ)化合物的胶囊或片剂可以含有0.05mg至1.0g活性化合物用于每次给药1粒、两粒或多粒。不论怎样,医生均可以确定最适于具体患者的实际剂量,该剂量将随着具体患者的年龄、体重和反应而改变。以上剂量是一般情况的例子。当然,有些个体可能需要更高或更低的剂量范围,这些也包括在本发明的范围内。
式(Ⅰ)化合物还可以鼻内或通过吸入给药,它通常以干粉吸入剂或气雾剂的形式从加压的容器或喷雾器中释放,该制剂使用适宜的抛射剂例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、氢氟烷烃如1,1,1,2-四氟乙烷(HFA 134A[商标]或1,1,1,2,3,3,3-七氟丙烷(HFA227EA[商标])、二氧化碳或其它适宜的气体。对于加压气雾剂,剂量单位可以通过装配能够释放计量量的阀门来确定。加压容器或喷雾器可以含有活性化合物的溶液或混悬液,例如,用乙醇和抛射剂的混合物作为溶剂,其中还可含有润滑剂如脱水山梨醇三油酸酯。用于吸入或吹入剂的胶囊和药筒(例如由明胶制成的)可以含有式(Ⅰ)化合物和适宜的粉末基质如乳糖或淀粉的粉末混合物。
优选对气雾剂或干粉制剂进行设置,从而使每一计量剂量或每一“喷”向患者传递20μg至20mg式(Ⅰ)化合物。气雾剂的每日总剂量在20μg至20mg的范围内,可以单次给药,或者,更常用的是在一天内分成多次给药。
或者,可以将式(Ⅰ)化合物以栓剂或阴道栓的形式给药,或将其以洗剂、溶液剂、霜剂、软膏或扑粉的形式局部应用。还可将式(Ⅰ)化合物用皮肤贴剂进行经皮给药。还可将其通过眼的途径进行给药,特别是用于治疗眼的神经病学疾病。
对于眼科应用,可将化合物配制成在等渗的、调节过pH值的无菌盐水中的微粉化混悬液的形式,或者,优选配制成在等渗的、调节过pH值的无菌盐水中的溶液的形式,其中可任意选择性地含有防腐剂如氯化苄烷氧
盐(benzylalkonium chloride)。或者,可将其在软膏如凡士林中配制。
对于皮肤的局部应用,可将式(Ⅰ)化合物配制成适宜的软膏形式,其含有悬浮或溶解在例如一种或多种如下物质的混合物中的活性化合物:矿物油、液体矿脂、白凡士林、丙二醇、聚氧乙烯聚氧丙烯化合物、乳化蜡和水。或者,可将其配制成悬浮或溶解在例如一种或多种如下物质的混合物中的适宜的洗剂或霜剂:矿物油、脱水山梨醇一硬脂酸酯、聚乙二醇、液体石蜡、吐温60(polysorbate 60)、鲸蜡酯醇(cetearylalcohol)、鲸蜡基硬脂醇、2-辛基十二烷醇、苄醇和水。
还可将式(Ⅰ)化合物与其它神经营养剂例如神经营养生长因子(NGF)、神经胶质衍生的生长因子、脑衍生的生长因子、睫状神经营养因子和/或神经营养蛋白-3一起给药。神经营养剂的剂量水平取决于联合用药的神经营养效果和采用的给药途径。
应当理解,本文所提到的治疗包括治愈、减轻和预防性治疗。
因此,本发明还提供:(ⅰ)含有式(Ⅰ)化合物或其可药用盐或溶剂化物以及可药用赋形剂、稀
释剂或载体的药物组合物:(ⅱ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物用作药物:(ⅲ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物在生产用于治疗
神经元变性的药物中的用途:(ⅳ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物在生产用于促进
神经元再生和分枝的药物中的用途:(ⅴ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物在生产用于治疗神
经病学疾病或障碍如神经变性疾病的药物中的用途:(ⅵ)如(ⅴ)中所述的用途,其中的神经病学疾病或障碍选自老年性痴呆(早老性痴呆)和其它痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、影响中枢或外周神经系统的各种形式的变性疾病(例如小脑-脑干萎缩、进行性共济失调综合征)、各种形式的肌营养不良、进行性肌萎缩、进行性延髓肌萎缩、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、突出、破裂或脱出的椎间盘综合征、颈椎关节强硬、神经丛疾病、胸腔出口综合征、各种形式的外周神经病(糖尿病性的和非糖尿病性的)、三叉神经痛、舌咽神经痛、面神经麻痹、导致中枢或外周神经系统损伤的各种形式的自身免疫相关性疾病(例如多发性硬化、重症肌无力、格-巴二氏综合征)、神经系统的AIDS相关性疾病、氨苯砜抽搐、眼神经的眼球和眼球后病变(例如视网膜病和眼球后神经炎)、听力障碍例如耳鸣和朊病毒疾病;
(ⅶ)如(ⅵ)中所述的用途,其中的神经病学疾病或障碍是老年性痴呆(早老性痴呆)或其它的痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、外周神经病(糖尿病性的或非糖尿病性的)、多发性硬化或听力障碍例如耳鸣;
(ⅷ)治疗人类患者神经元变性的方法,该方法包括,将所述患者用有效量的式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物进行治疗;
(ⅸ)促进人类患者神经元再生和分枝的方法,该方法包括,将所述患者用有效量的式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物进行治疗;
(ⅹ)治疗人类患者神经病学疾病或障碍如神经变性疾病的方法,该方法包括,将所述患者用有效量的式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物进行治疗;
(ⅹⅰ)如(ⅹ)中所述的方法,其中的神经病学疾病或障碍选自老年性痴呆(早老性痴呆)和其它痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、影响中枢或外周神经系统的各种形式的变性疾病(例如小脑-脑干萎缩、进行性共济失调综合征)、各种形式的肌营养不良、进行性肌萎缩、进行性廷髓肌萎缩、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、突出、破裂或脱出的椎间盘综合征、颈椎关节强硬、神经丛疾病、胸腔出口综合征、各种形式的外周神经病(糖尿病性的和非糖尿病性的)、三叉神经痛、舌咽神经痛、面神经麻痹、导致中枢或外周神经系统损伤的各种形式的自身免疫相关性疾病(例如多发性硬化、重症肌无力、格-巴二氏综合征)、神经系统的AIDS相关性疾病、氨苯砜抽搐、眼神经的眼球和眼球后病变(例如视网膜病和眼球后神经炎)、听力障碍例如耳鸣和朊病毒疾病;
(ⅹⅱ)如(ⅹⅰ)中所述的方法,其中的神经病学疾病或障碍是老年性痴呆(早老性痴呆)或其它的痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、外周神经病(糖尿病性的或非糖尿病性的)、多发性硬化或听力障碍例如耳鸣;和
(ⅹⅲ)文中所描述的所有新中间体。
(ⅹⅳ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物在生产用于治疗由于FKBP-12或FKBP-52缺乏或过度生成所引起之疾病的药物中的用途。
以下实施例说明了式(Ⅰ)化合物的制备。用ACD/IUPAC Pro软件程序作为所制备化合物命名的基础。
实施例1(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3R)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺
将三乙胺(0.167ml)加入到(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸(100mg)[参见制备例3]、1-羟基苯并三唑水合物(60.4mg)、(3R)-1-苄基吡咯烷-3-基胺(78.7mg)和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(85.4mg)的二氯甲烷(30ml)溶液中。将反应混合物室温搅拌18小时,然后将混合物用水稀释并分出有机相,用无水硫酸钠干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用50∶50∶0→25∶75∶0→20∶80∶1(体积比)己烷∶乙酸乙酯:0.88氨水的溶剂梯度洗脱得到黄色油状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3R)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺(94mg)。1H-NMR(CDCl3)δ:7.40(1H,d),7.30-7.20(7H,m),7.10(1H,m),6.70(1H,d),4.90(1H,s),4.45(1H,m),4.25(1H,d),3.70-3.50(2H,m),3.20(1H,t),2.80(1H,m),2.50(2H,m),2.40-2.20(3H,m),1.80-1.50(6H,m).分析:实测值C,69.70;H,7.15;N,13.16;C24H28N4O2·0.5H2O的理论值C,69.71;H,7.07;N,13.09%。旋光:[◇]25 D=-40.9°(c=0.09,甲醇)。
实施例2(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺
标题化合物按照与实施例1类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]和(3S)-1-苄基吡咯烷-3-基胺制备。将粗产物通过硅胶柱色谱纯化,用50∶50∶1→20∶80∶1(体积比)己烷∶乙酸乙酯∶0.88氨水的溶剂梯度以10%的增量洗脱。将产物用乙酸乙酯∶己烷重结晶进-步纯化得到白色固体状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺。1H-NMR(CDCl3)δ:7.40(1H,d),7.30(1H,m),7.20(6H,s),7.05(1H,t),6.60(1H,d),4.85(1H,s),4.45(1H,bs),4.25(1H,d),3.60(2H,s),3.20(1H,t),2.75(1H,m),2.55(2H,m),2.35(1H,m),2.25(2H,m),1.80-1.50(4H,m),1.30(2H,m).分析:实测值C,71.00;H,7.00;N,13.80;C24H28N4O2的理论值C,71.26;H,6.98;N,13.85%。旋光:[◇]25 D=-102.0°(c=0.1,甲醇)。
实施例3
(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3R)-吡咯烷-3-基]-2-哌啶甲酰胺
将20%w/w吸附在碳上的氢氧化钯(12.5mg)加入到(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3R)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺(62.5mg)[参见实施例1]的乙醇(10ml)溶液中。将反应混合物在414kPa(60p.s.i.)下氢化56小时,然后滤除催化剂并减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用95∶5→90∶10(体积比)乙酸乙酯:二乙胺的溶剂梯度洗脱得到油状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3R)-吡咯烷-3-基]-2-哌啶甲酰胺(1mg)。1H-NMR(CDCl3)δ:7.35(1H,d),7.25(1H,m),7.20(1H,t),7.00(1H,m),6.65(1H,bs),4.85(1H,bs),4.40(1H,bs),4.25(1H,d),3.20(1H,t),2.80(1H,bs),2.65(1H,m),2.55(1H,m),2.40-2.20(4H,m),1.80-1.60(6H,m).MS:314(MH+).
实施例4(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺
标题化合物通过与实施例3类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-苄基吡咯烷-3-基]-2-哌啶甲酰胺[参见实施例2]和20%吸附在碳的氢氧化钯制备,得到棕色泡沫状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3R)-吡咯烷-3-基]-2-哌啶甲酰胺。1H-NMR(CDCl3)δ:7.30(1H,d),7.25(1H,m),7.20(1H,m),7.00(1H,m),6.60(1H,d),4.80(1H,s),4.40(1H,m),4.20(1H,d),3.25(1H,t),3.20(1H,m),3.00-2.80(2H,m),2.70(1H,d),2.40(1H,m),2.15(1H,m),1.80-1.50(7H,m).分析:实测值C,60.85;H,7.14;N,15.79;C17H22N4O2·0.25CH2Cl20.4H2O的理论值C,60.43;H,6.85;N,16.34%.
实施例5(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(2-吡啶基)吡咯烷-3-基]-2-哌啶甲酰胺
将碳酸氢钠(28.6mg)加入到(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺(104.9mg)[参见实施例4]和2-溴吡啶(52.7mg)的乙腈(5ml)溶液中。将反应混合物于75℃加热48小时,然后补加碳酸氢钠(28.6mg)并将混合物继续加热回流24小时。减压蒸除溶剂,将残余物在乙酸乙酯和水之间进行分配,分出有机层,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统93∶7∶1(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱,然后用第二根柱子进一步纯化,用95∶5(体积比)乙酸乙酯∶二乙胺洗脱得到棕色油状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(2-吡啶基)吡咯烷-3-基]-2-哌啶甲酰胺(11mg)。1H-NMR(CDCl3)δ:8.15(1H,d),7.40(1H,m),7.25(2H,m),7.20(1H,m),7.05(1H,m),6.80(1H,d),6.60(1H,t),6.30(1H,t),4.90(1H,m),4.60(1H,m),4.25(1H,d),3.75(1H,m),3.50(2H,m),3.45(1H,d),3.20(1H,t),2.40-2.20(2H,m),2.05(1H,m),1.80-1.60(5H,m).精确质量:实测值392.2073(MH+),C22H25N5O2理论值392.2086(MH+)。
实施例6(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(2-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺
将碳酸钾(0.052g)于0℃下加入到(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺(108.2mg)[参见实施例4]和2-(氯甲基)吡啶的乙腈(6.8ml,0.055M)溶液中。[2-(氯甲基)吡啶通过将2-(氯甲基)吡啶盐酸盐在乙醚和饱和碳酸氢钠水溶液之间进行分配制得。将分出的有机相用盐水洗涤,用硫酸镁干燥然后减压蒸除溶剂。将残余的游离碱立即溶于乙腈并使用]。将反应混合物室温搅拌18小时,然后减压蒸除溶剂并将残余物在乙酸乙酯和水之间进行分配。分出有机层,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统93∶7∶1(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱得到油状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(2-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺(63.1mg)。1H-NMR(CDCl3)δ:8.45(1H,d),7.50(1H,t),7.40(1H,d),7.30-7.00(5H,m),6.80(1H,d),4.85(1H,s),4.50(1H,m),4.25(1H,d),3.70(2H,t),3.20(1H,t),2.80(1H,m),2.65(1H,m),2.55(1H,d),2.40-2.00(4H,m),1.80-1.60(5H,m).分析:实测值C,62.01;H,6.38;N,15.39;C23H27N5O2·1.25 H2O·0.25CH2Cl2理论值C,62.16;H,6.73;N,15.59%。
实施例7-9
以下列表的实施例(表1)中的如下通式的化合物通过与实施例6类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺[参见实施例4]和相应的卤化物制得:
表1脚注1.盐酸盐通过与实施例11类似的方法制备。
实施例10(2S)-1-(1,3-苯并噁唑-2-基)-N2-(3S)-1-[2-(2-吡啶基)乙基]吡咯烷-3-基-2-哌啶甲酰胺
将2-乙烯基吡啶(0.02ml)加入到(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺(53.6mg)[参见实施例4]的乙醚(5ml)溶液中。加入苄基三甲基氢氧化铵(40%w/w水溶液)(1ml)并将反应混合物回流搅拌48小时,然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用93∶7∶1(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱,然后将产物在第二根柱子上进一步纯化,用95∶5(体积比)乙酸乙酯∶二乙胺洗脱得到油状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-(3S)-1-[2-(2-吡啶基)乙基]吡咯烷-3-基-2-哌啶甲酰胺(19.8mg)。1H-NMR(CDCl3)δ:8.50(1H,m),7.60(1H,m),7.40(1H,d),7.25(1H,m),7.20-7.00(4H,m),6.65(1H,bs),4.90(1H,s),4.50(1H,m),4.25(1H,d),3.20(1H,m),3.00-2.80(5H,m),2.65(2H,s),2.40-2.20(4H,m),1.80-1.60(5H,m).MS:420(MH+).
实施例11(2S)-1-(1,3-苯并噁唑-2-基)-N2-(3S)-1-[2-(2-甲基-1H-咪唑-1-基)乙基]吡咯烷-3-基-2-哌啶甲酰胺盐酸盐
标题化合物通过与实施例5类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺[参见实施例4]和1-(2-氯乙基)-2-甲基-1H-咪唑[参见美国专利3962274,CAN 85:177416]制备。将粗产物通过硅胶柱色谱纯化,用溶剂系统93∶7∶1(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱得到(2S)-1-(1,3-苯并噁唑-2-基)-N2-(3S)-1-[2-(2-甲基-1H-咪唑-1-基)乙基]吡咯烷-3-基-2-哌啶甲酰胺。将残余物的胶状物溶于甲醇并用1N氯化氢的乙醚溶液处理。将形成的悬浮液蒸发然后干燥得到红色固体状的产物的盐酸盐。1H-NMR(DMSO-d6)δ:8.55(1H,m),7.60(1H,d),7.50(1H,d),7.40(1H,d),7.25(1H,m),7.15(1H,m),7.00(1H,m),4.75(1H,m),4.45(2H,t),4.40(1H,m),4.00(1H,m),3.80-3.20(7H,m),2.70-2.25(2H,m),2.60(3H,s),2.20(1H,d),1.80-1.60(3H,m),1.50-1.10(2H,m).精确质量:实测值423.2516(MH+),C23H30N6O2理论值423.2509(MH+)。
实施例12(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(1H-1,2,4-三唑-3-基甲基)吡咯烷-3-基]-2-哌啶甲酰胺 将3-(氯甲基)-1H-1,2,4-三唑(70.8mg)[参见Bazhenov D.N.等,Zh.Org.Khim,(1994),30(5),791-792和其中引用的文献]于0℃下加入到(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺(91.8mg)[参见实施例4]、碳酸钾(91mg)和碘化钠(10mg)的乙腈(10ml)溶液中。将反应混合物室温搅拌18小时,然后减压蒸除溶剂并将残余物在乙酸乙酯和水之间进行分配。分出有机层,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统93∶7∶1(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱得到固体状(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(1H-1,2,4-三唑-3-基甲基)吡咯烷-3-基]-2-哌啶甲酰胺(12.6mg)。1H-NMR(CDCl3)δ:8.00(1H,s),7.60(1H,bs),7.40(1H,d),7.25(1H,m),7.15(1H,m),7.00(1H,m),4.85(1H,d),4.45(1H,d),4.20(1H,m),3.95(2H,m),3.25(1H,t),3.00-2.20(7H,m),1.80-1.25(5H,m).精确质量:实测值396.2138(MH+),C20H25N7O2理论值396.2148(MH+)。
将3-(氯甲基)-1H-1,2,4-三唑(70.8mg)[参见Bazhenov D.N.等,Zh.Org.Khim,(1994),30(5),791-792和其中引用的文献]于0℃下加入到(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺(91.8mg)[参见实施例4]、碳酸钾(91mg)和碘化钠(10mg)的乙腈(10ml)溶液中。将反应混合物室温搅拌18小时,然后减压蒸除溶剂并将残余物在乙酸乙酯和水之间进行分配。分出有机层,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统93∶7∶1(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱得到固体状(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(1H-1,2,4-三唑-3-基甲基)吡咯烷-3-基]-2-哌啶甲酰胺(12.6mg)。1H-NMR(CDCl3)δ:8.00(1H,s),7.60(1H,bs),7.40(1H,d),7.25(1H,m),7.15(1H,m),7.00(1H,m),4.85(1H,d),4.45(1H,d),4.20(1H,m),3.95(2H,m),3.25(1H,t),3.00-2.20(7H,m),1.80-1.25(5H,m).精确质量:实测值396.2138(MH+),C20H25N7O2理论值396.2148(MH+)。
实施例13(2S)-N2-[(3S)-1-((氨基羰基)甲基)吡咯烷-3-基]-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺
标题化合物按照与实施例5类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺[参见实施例4]和2-溴乙酰胺制备。将粗产物通过硅胶柱色谱纯化,用93∶7∶1→90∶10∶1(体积比)二氯甲烷∶甲醇∶0.88氨水的溶剂梯度洗脱得到油状的(2S)-N2-[(3S)-1-((氨基羰基)甲基)吡咯烷-3-基]-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺。1H-NMR(CDCl3)δ:7.40(1H,d),7.30(1H,m),7.20(1H,t),7.10(1H,t),6.80(1H,d),6.60(1H,bs),5.20(1H,bs),4.85(1H,s),4.45(1H,m),4.25(1H,d),3.25(1H,t),3.10(2H,s),2.85(1H,m),2.75(1H,m),2.65(1H,m),2.50-2.20(3H,m),1.80-1.60(6H,m),精确质量:实测值372.2046(MH+),C19H25N5O3理论值372.2036(MH+)。
实施例13A(2S)-N2-{(3S)-1-[3-(氨基羰基)苯基甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺
标题化合物通过与实施例5类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺[参见实施例4]和3-氯甲基苯甲酰胺[《生物有机药物化学》(Biorg.Med.Chem.)1998,6,721-734]制备。将粗产物通过硅胶柱色谱纯化,用98∶1.75∶0.25→93∶7∶1→90∶10∶1(体积比)二氯甲烷∶甲醇∶0.88氨水的溶剂梯度洗脱得到灰白色固体状(2S)-N2-{(3S)-1-[3-(氨基羰基)苯基甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺。NMR(CDCl3)d:7.80(2H,m),7.30(4H,m),7.20(1H,m),7.05(1H,m),6.70(2H,bm),5.50(1H,bs),4.85(1H,s),4.45(1H,s),4.25(1H,d),3.60(2H,s),3.20(1H,t),2.75(1H,m),2.50(2H,m),2.40(1H,d),2.25(2H,m),1.50-1.85(6H,m).分析:实测值C,66.43,H,6.51,N,15.42,C25H29N5O3·0.25 H2O理论值C,66.43,H,6.58,N,15.49%.MS:448(MH+)。
实施例14(2S)-N2-[(3S)-1-(氨基羰基乙基)吡咯烷-3-基]-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺
将丙烯酰胺(11.6mg)加入到(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-吡咯烷-3-基]-2-哌啶甲酰胺(51.6mg)[参见实施例4]的乙醚(3ml)溶液中。将反应混合物回流18小时,然后将乙醚层用水稀释。分出水层然后用乙酸乙酯萃取。将合并的有机层用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用93∶7∶1→90∶10∶1二氯甲烷∶甲醇∶0.88氨水的溶剂梯度洗脱得到灰白色泡沫状的(2S)-N2-[(3S)-1-(氨基羰基乙基)吡咯烷-3-基]-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺(26.4mg)。1H-NMR(CDCl3)δ:7.40(1H,d),7.30(1H,m),7.20(1H,m),7.10(1H,t),6.70(1H,bs),5.10(1H,bs),4.90(1H,s),4.45(1H,m),4.25(1H,d),3.20(1H,t),2.85(1H,m),2.75-2.60(4H,m),2.40-2.20(5H,m),1.80-1.55(6H,m).MS:386(MH+).
实施例15(2S)-N2-[(3S)-1-苄基吡咯烷-3-基]-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酰胺
标题化合物通过与实施例1类似的方法从(2S)-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例6]和(3R)-1-苄基吡咯烷-3-基胺制备。将粗产物通过硅胶柱色谱纯化,用50∶50∶1→40∶60∶1(体积比)己烷∶乙酸乙酯∶三乙胺的溶剂梯度洗脱。将产物用乙酸异丙酯重结晶进行进一步纯化得到油状的(2S)-N2-[(3S)-1-苄基吡咯烷-3-基]-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酰胺。1H-NMR(CDCl3)δ:7.40(1H,s),7.30(1H,d),7.20(6H,m),6.60(1H,d),4.80(1H,s),4.40(1H,bs),4.20(1H,d),3.55(2H,s),3.20(1H,t),2.80(1H,t),2.55(2H,m),2.40-2.20(3H,m),1.80-1.50(6H,m).分析:实测值C,59.54;H,5.58;N,11.54;C24H27N4O2Br理论值C,59.63;H,5.63;N,11.59%。
实施例16-20
以下列表的实施例(表2)中的如下通式的化合物通过与实施例1类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]和相应的胺[参见制备例8和13至16(表2b)]制得,所不同的是在酰胺偶联中还使用催化量的4-二乙基氨基吡啶:
表2

实施例21-26
以下列表的实施例(表3)中的如下通式的化合物通过与实施例1类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]和相应的胺[参见制备例24至30(表3b)]制得,所不同的是在酰胺偶联中还使用催化量的4-二乙基氨基吡啶:
表3 
实施例28(2S)-N2-(1-二苯基甲基-3-氮杂环丁基)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺
标题化合物通过与实施例1类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]和1-二苯甲基-3-氮杂环丁烷胺[参见《药物化学杂志》(J.Med.Chem.)(1977),21(1),78-82]制备。将粗产物通过硅胶柱色谱纯化,用80∶20→60∶40→50∶50(体积比)己烷∶乙酸乙酯的溶剂梯度洗脱得到白色泡沫状(2S)-N2-(1-二苯基甲基-3-氮杂环丁基)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺。1H-NMR(CDCl3)δ:7.40(1H,d),7.30(5H,m),7.20(5H,m),7.15(2H,m),7.10(1H,t),6.80(1H,d),4.90(1H,s),4.55(1H,m),4.25(1H,s),4.20(1H,s),3.50(2H,t),3.20(1H,t),2.85(1H,m),2.75(1H,m),2.35(1H,m),1.80-1.50(5H,m).分析:实测值C,73.84;H,6.48;N,11.79,C29H30N4O2·0.33 H2O理论值C,73.71;H,6.54;N,11.86%。
实施例29
(2S)-1-(1,3-苯并噁唑-2-基)-N2-(1-乙基-3-氮杂环丁基)-2-哌啶甲酰胺
将20%w/w吸附在碳上的氢氧化钯(31mg)加入到(2S)-N2-(1-二苯甲基-3-氮杂环丁烷基)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺(120mg)[参见实施例28]的乙醇(5ml)溶液中。将反应混合物在414kPa(60p.s.i.)下氢化18小时,然后补加20%w/w吸附在碳上的氢氧化钯(32mg)并将混合物继续氢化72小时。滤除催化剂并用乙醇洗涤,然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用100∶0→90∶10(体积比)二氯甲烷∶甲醇的溶剂梯度以2%的增量洗脱得到泡沫状(2S)-1-(1,3-苯并噁唑-2-基)-N2-(1-乙基-3-氮杂环丁基)-2-哌啶甲酰胺(37.1mg)。1H-NMR(CDCl3)δ:7.40(1H,d),7.30(1H,m),7.20(1H,t),7.05(1H,t),4.95(1H,s),4.60(1H,m),4.25(1H,d),3.75(2H,t),3.30-3.10(3H,m),2.60(2H,q),2.40(1H,m),1.80-1.60(5H,m),1.00(3H,t).分析:实测值C,59.71;H,7.17;N,14.45;C18H24N4O2·0.55 CH2Cl2理论值C,59.39;H,6.74;N,14.94%。MS:329(MH+)。
实施例30(2S)-1-(1,3-苯并噁唑-2-基)-N2-(1-苄基-3-哌啶基)-2-哌啶甲酰胺
标题化合物通过与实施例1类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]和1-苄基-3-哌啶基胺[参见《药物化学杂志》(J.Med.Chem.)(1980),23(8),848-851]制备。将粗产物通过硅胶柱色谱纯化,用90∶10→40∶60(体积比)己烷∶乙酸乙酯的溶剂梯度以10%的增量洗脱得到黄色胶状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-(1-苄基-3-哌啶基)六氢-2-哌啶甲酰胺。1H-NMR(CDCl3)δ:7.40-7.00(9H,m),6.95(1H,bs),4.90(1H,m),4.30-4.20(2H,2×d),4.10(1H,m),3.40(1H,t),3.25(1H,t),2.60(1H,m),2.40-2.20(3H,m),2.10(1H,m),1.90(1H,m),1.80-1.40(8H,m).MS:419(MH+).
实施例31(2S)-1-(1,3-苯并噁唑-2-基)-N2-(4-哌啶基)-2-哌啶甲酰胺
将三氟乙酸(10ml)于0℃下加入到4-([(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶基]羰基氨基)哌啶-1-甲酸叔丁酯(1.631g)[参见制备例31]的二氯甲烷(10ml)溶液中。将反应混合物升温至室温并搅拌2小时,然后减压蒸除溶剂并将残余物溶于水。加入碳酸氢钠直至溶液达到pH 8,然后将产物用乙酸乙酯萃取。分出有机层,用硫酸镁干燥然后减压蒸除溶剂得到白色泡沫状(2S)-1-(1,3-苯并噁唑-2-基)-N2-(4-哌啶基)-2-哌啶甲酰胺(1.48g)。1H-NMR(CDCl3)δ:7.40(1H,d),7.30(1H,m),7.20(1H,m),7.10(1H,t),6.80(1H,d),4.85(1H,s),4.30(1H,d),4.00(1H,m),3.30(2H,t),3.20(1H,t),2.90(2H,m),2.35(1H,d),2.05(2H,m),1.80-1.60(6H,m),1.30(2H,m).MS:329(MH+).
实施例32(2S)-1-(1,3-苯并噁唑-2-基)-N2-(1-苄基-4-哌啶基)-2-哌啶甲酰胺
标题化合物通过与实施例6类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-N2-(4-哌啶基)-2-哌啶甲酰胺[参见实施例31]和苄基溴制备。将粗产物通过硅胶柱色谱纯化,用溶剂系统93∶7∶1(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱得到油状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-(1-苄基-4-哌啶基)-2-哌啶甲酰胺。1H-NMR(CDCl3)δ:7.40(1H,d),7.30-7.20(7H,m),7.10(1H,t),6.45(1H,d),4.90(1H,s),4.25(1H,d),3.85(1H,m),3.40(2H,s),3.20(1H,t),2.65(2H,m),2.40(1H,d),2.15(2H,m),1.90(3H,m),1.80-1.60(4H,m),1.30(2H,m).分析:实测值C,69.87;H,7.39;N,12.79,C25H30N4O2·0.15CH2Cl2理论值C,70.04;H,7.08;N,12.99%。
实施例33(2S)-1-(1,3-苯并噁唑-2-基)-N2-[1-(4-吡啶基甲基)-4-哌啶基]-2-哌啶甲酰胺
将4-(氯甲基)吡啶(88.1mg)于0℃加入到(2S)-1-(1,3-苯并噁唑-2-基)-N2-(4-哌啶基)-2-哌啶甲酰胺(118mg)[参见实施例31]、碳酸钾(56.6mg)和碘化钠(6.7mg)的乙腈(10ml)溶液中。将反应混合物室温搅拌18小时,然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统93∶7∶1(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱得到红色胶状(2S)-1-(1,3-苯并噁唑-2-基)-N2-[1-(4-吡啶基甲基)-4-哌啶基]-2-哌啶甲酰胺(14.8mg)。1H-NMR(CDCl3)δ:8.55(2H,d),7.40(1H,m),7.30(2H,m),7.25(2H,m),7.10(1H,t),6.50(1H,d),4.90(1H,s),4.25(1H,d),3.90(1H,m),3.40(2H,s),3.20(1H,t),2.75-2.60(2H,m),2.40(1H,d),2.20(2H,m),1.90(2H,m),1.80-1.60(5H,m),1.50(2H,m).MS:420(MH+).
实施例34(2S,4R)-4-([(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶基]羰基氨基)-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯
标题化合物通过与实施例1类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]和(2S,4R)-4-氨基-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯[参见制备例34]制备。将粗产物通过硅胶柱色谱纯化,用70∶30→50∶50(体积比)己烷∶乙酸乙酯的溶剂梯度洗脱得到泡沫状(2S,4R)-4-([(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶基]羰基氨基)-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯。1H-NMR(CDCl3)δ:7.40-7.25(7H,m),7.20(1H,t),7.05(1H,t),6.55(1H,d),4.90(1H,s),4.55(1H,m),4.50(2H,s),4.25(1H,d),4.05-3.90(1H,m),3.65-3.50(3H,m),3.30-3.10(2H,m),2.35(2H,m),1.85-1.30(14H,m),0.90(1H,m).分析:实测值,C,66.66;H,7.22;N,10.31;C30H38N4O5·0.25 H2O理论值C,66.83;H,7.20;N,10.39%。旋光:[◇]25 D=-51.0°(c=0.1,甲醇)。
实施例35(2S)-1-(1,3-苯并噁唑-2-基)-N2-(3R,5S)-5-[(苄氧基)甲基]吡咯烷-3-基-2-哌啶甲酰胺盐酸盐
标题化合物通过与制备例8类似的方法从(2S,4R)-4-([(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶基]羰基氨基)-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯[参见实施例34]和氯化氢气体制备,得到白色固体状的(2S)-1-(1,3-苯并噁唑-2-基)-N2-(3R,5S)-5-[(苄氧基)甲基]吡咯烷-3-基-2-哌啶甲酰胺盐酸盐。1H-NMR(DMSO-d6)δ:9.60(1H,bs),8.95(1H,bs),8.45(1H,d),7.40-7.25(7H,m),7.15(1H,t),7.00(1H,t),4.80(1H,d),4.55(2H,s),4.40(1H,m),4.15-4.05(1H,m),4.00(1H,m),3.70-3.60(2H,m),3.40(2H,m),3.10(1H,m),2.25(1H,d),2.00(2H,m),1.80-1.60(3H,m),1.55(1H,m),1.35(1H,m).分析:实测值C,55.60;H,6.39;N,10.14;C25H30N4O3·2HCl·2H2O理论值C,55.25;H,6.67;N,10.30%。旋光:[◇]25 D=-19.0°(c=0.1,甲醇)。
实施例36(3S)-1-(2-吡啶基甲基)-3-哌啶基(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸酯盐酸盐
将(3R)-1-(2-吡啶基甲基)-3-哌啶醇(117mg)[参见制备例35]加入到搅拌中的(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸(150mg)[参见制备例3]、三苯膦(192mg)和偶氮二甲酸二乙酯(0.115ml)的干燥四氢呋喃(6ml)溶液中。将反应混合物回流搅拌16小时,然后减压蒸除溶剂并将残余物在乙酸乙酯和0.5M盐酸之间进行分配。将水层用15%氢氧化钠调至碱性然后将产物用乙酸乙酯萃取。分出有机层,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用98∶2(体积比)乙酸乙酯∶甲醇洗脱得到黄色油状的(3S)-1-(2-吡啶基甲基)-3-哌啶基(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸酯(140mg)。
通过将化合物溶于乙酸乙酯然后向溶液中通入氯化氢气体制备盐酸盐,得到白色固体状的标题化合物。1H-NMR(CDCl3)(游离碱)δ:8.30-8.20(1H,m),7.60-7.30(1H,m),7.20-7.15(1H,m),7.15-7.00(2H,m),7.00-6.90(1H,m),5.00-4.85(2H,m),4.20-4.00(3H,m),3.55(1H,s),3.50-3.45(1H,d),3.40-3.25(1H,m),2.90-2.75(2H,m),2.75-2.60(1H,m),2.50-2.40(1H,m),2.35-2.15(2H,m),1.90-1.40(4H,m),1.35-1.25(1H,m),1.25-1.10(1H,m).MS:421(MH+).旋光:[◇]25 D=-40.70°(c=1.0,甲醇)。
实施例37(3S,5S)-5-(苯氧基甲基)-1-(4-吡啶基甲基)吡咯烷-3-基(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸酯盐酸盐
标题化合物通过与实施例36类似的方法从(3R,5S)-5-(苯氧基甲基)-1-(4-吡啶基甲基)吡咯烷-3-醇[参见制备例38]和(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]制备。将粗产物通过硅胶柱色谱纯化,用3∶1→10∶1(体积比)乙酸乙酯∶己烷的溶剂梯度洗脱得到标题化合物。盐酸盐通过将饱和氯化氢气体的乙醚溶液加入到产物的乙酸乙酯溶液中制得,分离得到白色固体。1H-NMR(CDCl3)(游离碱)δ:8.54-8.50(1H,d),8.39-8.37(1H,d),7.34-7.27(1H,m),7.26-7.18(4H,m),7.18-7.09(2H,m),7.02-6.95(1H,m),6.95-6.87(1H,m),6.84-6.80(1H,d),6.78-6.72(1H,d),5.08-5.00(0.5H,m),5.00-4.94(1H,m),4.70-4.64(0.5H,m),4.42-4.32(1H,m),4.30-4.20(0.5H,m),4.20-4.13(1H,m),4.10-4.02(0.5H,d),3.57(1H,s),3.38-3.28(1H,m),3.26-3.20(0.5H,d),3.11-3.05(0.5H,d),3.05-2.97(1h,m),2.92-2.83(0.5H,m),2.54-2.48(0.5H,m),2.46-2.41(0.5H,dd),2.39-2.31(0.5H,m),2.30-2.24(1H,d),2.16-2.06(1H,m),1.90-1.68(3H,m),1.64-1.50(1H,m),1.35-1.22(2H,m).MS:513(MH+).旋光:[◇]25 D=-18.00°(c=1.0,甲醇)。
实施例38(3S)-1-[(2-氟-4-吡啶基)甲基]-3-哌啶基(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸酯盐酸盐
标题化合物通过与实施例36类似的方法从(3R)-1-[(2-氟-4-吡啶基)甲基]-3-哌啶醇[参见制备例39]和(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]制备。将粗产物通过硅胶柱色谱部分纯化,用3∶1→1∶1(体积比)乙酸乙酯∶己烷的溶剂梯度洗脱,将产物通过用95∶5(体积比)热的己烷∶乙酸乙酯研制,然后用热的石油醚∶乙醚95∶5研制进一步纯化得到(3S)-1-[(2-氟-4-吡啶基)甲基]-3-哌啶基(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸酯。通过将饱和的氯化氢气体的乙醚溶液加入到产物的乙酸乙酯溶液中制备盐酸盐,分离得到白色固体。1H-NMR(CDCl3)游离碱δ:7.93-7.92(1H,d),7.29-6.85(5H,m),6.72(1H,s),5.00-4.80(2H,m),4.20-4.10(2H,dd),4.08-3.99(1H,dd),3.95-3.86(1H,d),3.35-3.21(2H,m),3.20-3.15(1H,d),2.79-2.65(2H,m),2.38-2.10(2H,m),2.10-1.95(1H,dd),1.90-1.60(3H,m),1.60-1.40(2H,m),1.40-1.20(1H,m).MS:439(MH+).旋光:[◇]25 D=-52.50°(c=1.0,甲醇)。
实施例39(2S)-1-(1,3-苯并噻唑-2-基)-N-[(3S)-1-(3-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺
标题化合物通过与实施例1类似的方法从(2S)-1-(1,3-苯并噻唑-2-基)-2-哌啶甲酸[参见制备例43]和(3S)-1-(3-吡啶基甲基)吡咯烷-3-胺[参见制备例41]制备,所不同的是还使用催化量的4-二甲基氨基吡啶,制得(2S)-1-(1,3-苯并噻唑-2-基)-N-[(3S)-1-(3-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺,为白色固体。1H-NMR(CDCl3)δ:8.40(1H,m),7.60(1H,m),7.55-7.40(2H,m),7.22(1H,m),7.10(2H,m),6.80(1H,m),4.95(1H,d),4.40(1H,bs),3.80(1H,m),3.60-3.40(2H,m),3.25(1H,m),2.70(1H,m),2.60-2.30(2H,m),2.30-2.10(3H,m),1.80-1.40(6H,m).MS:422(MH+).
实施例40(2S)-N2-[(3S)-1-苄基吡咯烷-3-基]-1-(6-氯-1,3-苯并噻唑-2-基)-2-哌啶甲酰胺
标题化合物通过与实施例1类似的方法从(2S)-1-(6-氯-1,3-苯并噻唑-2-基)-2-哌啶甲酸[参见制备例45]和(3S)-1-苄基吡咯烷-3-基胺[参见《药物化学杂志》(J.Med.Chem.)(1989),31(8),1586-1590]制备,得到固体状的(2S)-N2-[(3S)-1-苄基吡咯烷-3-基]-1-(6-氯-1,3-苯并噻唑-2-基)-2-哌啶甲酰胺。1H-NMR(CDCl3)δ:7.55(1H,s),7.40(1H,m),7.20(5H,m),6.75(1H,m),4.90(1H,bs),4.40(1H,bs),3.75(1H,m),3.60-3.20(3H,m),2.80(1H,m),2.55(2H,m),2.20(2H,m),1.80(1H,m),1.80-1.40(6H,m).MS:455(MH+).
实施例411-(1H-1,3-苯并咪唑-2-基)-N2-[(3S)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺
将1,3,4,12a-四氢吡啶并[1’,2’:3,4]咪唑并[1,2-a][1,3]苯并咪唑-12(2H)-酮(73mg)[参见制备例49]和(3S)-1-苄基吡咯烷-3-基胺(62mg)在1,4-二氧六环(0.5ml)中混合。将反应混合物加热至90℃并搅拌4小时,然后减压蒸除溶剂,将残余物溶于二氯甲烷∶甲醇然后预吸附在硅胶上。然后将粗产物通过硅胶柱色谱纯化,用溶剂系统99∶1∶0.1→97∶3∶0.3(体积比)二氯甲烷∶甲醇∶0.88氨水洗脱得到固体状1-(1H-1,3-苯并咪唑-2-基)-N2-[(3S)-1-苄基-1-吡咯烷-3-基]-2-哌啶甲酰胺(69mg)。1H-NMR(CDCl3)δ:8.90-8.70(1H,d),7.50-7.00(9H,m),4.80(1H,d),4.40(1H,bs),3.80(2H,d),3.60(2H,d),3.35(1H,m),2.75(1H,m),2.60-2.40(2H,m),2.20(3H,m),1.80-1.50(5H,m).分析:实测值C,71.33,H,7.33;N,17.26,C24H29N5O理论值C,71.44;H,7.24;N,17.36%。旋光:[◇]25 D=+6.00°(c=0.1,甲醇)。
以下制备例描述了前述实施例中所用的某些原料的制备。
制备例1
(2S)-2-(甲氧羰基)哌啶鎓氯化物
将(2S)-哌啶甲酸L-酒石酸盐(20.0g)[参见WO-A-96/11185]于0℃下滴加到亚硫酰氯(54ml)的甲醇(270ml)溶液中。然后将反应混合物室温搅拌18小时,然后减压蒸除溶剂并将残余物与甲苯(3×100ml)共沸。将粗产物通过用甲醇(15ml,加入乙醚使其混浊)重结晶进行纯化,得到白色结晶状(2S)-2-(甲氧基羰基)哌啶鎓氯化物(11.06g)。1H-NMR(D2O)δ:3.95(1H,d),3.70(3H,m),3.40(1H,d),3.00(1H,t),2.20(1H,d),1.80(2H,m),1.70-1.40(3H,m).旋光:[◇]25 D=-8.40°(c=0.1,甲醇)。MS:144(MH+)。
制备例2
(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸甲酯
将乙基二异丙基胺(6.52ml)加入到(2S)-2-(甲氧基羰基)哌啶鎓氯化物(3.057g)[参见制备例1]和2-氯苯并噁唑(2.13ml)的乙腈(50ml)溶液中。将反应混合物室温搅拌18小时,然后于50℃继续搅拌2小时。减压蒸除溶剂并将残余物在乙酸乙酯和水之间进行分配,分出有机层,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用80∶10∶0→0∶100∶0→0∶95∶5(体积比)己烷∶乙酸乙酯∶甲醇的溶剂梯度洗脱得到固体状(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸甲酯(3.18g)。1H-NMR(CDCl3)δ:7.35(1H,d),7.25(1H,d),7.15(1H,m),7.00(1H,m),5.00(1H,d),4.20(1H,m),3.70(3H,s),3.35(1H,t),2.30(1H,d),1.80(3H,m),1.60(1H,m),1.35(1H,m).MS:261(MH+).
制备例3(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸
将氢氧化锂水溶液(1N,51ml)于0℃下加入到(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸甲酯(8.987g)[参见制备例2]的甲醇(306ml)溶液中。将反应混合物室温搅拌18小时,然后减压蒸除溶剂并将残余物在乙酸乙酯和水之间进行分配。分出水层并用2N盐酸酸化至pH2,将产物用乙酸乙酯萃取,用硫酸镁干燥然后减压蒸除溶剂得到白色固体状(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸(8.17g)。1H-NMR(CDCl3)δ:7.40(1H,d),7.25(1H,m),7.15(1H,t),7.00(1H,t),5.80(1H,bs),4.95(1H,bs),4.15(1H,d),3.40(1H,t),2.40(1H,d),1.80(3H,m),1.60-1.40(2H,m).旋光:[◇]25 D=-116.2°(c=0.1,甲醇)。MS:247(MH+)。
制备例41-(碘甲基)环丙烷
将碘化钠(6.07g)加入到1-(溴甲基)环丙烷(1.09g)的丙酮(10ml)溶液中。将反应混合物加热至回流然后搅拌18小时,然后滤出白色固体并减压蒸除溶剂。将残余物在乙醚和水之间进行分配,分出有机层并用硫代硫酸钠洗涤,用硫酸镁于燥然后减压蒸除溶剂得到无色液体状1-(碘甲基)环丙烷(0.269g)。1H-NMR(CDCl3)δ:3.15(2H,d),1.30(1H,m),0.80(2H,m),0.30(2H,m).
制备例5(2S)-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酸甲酯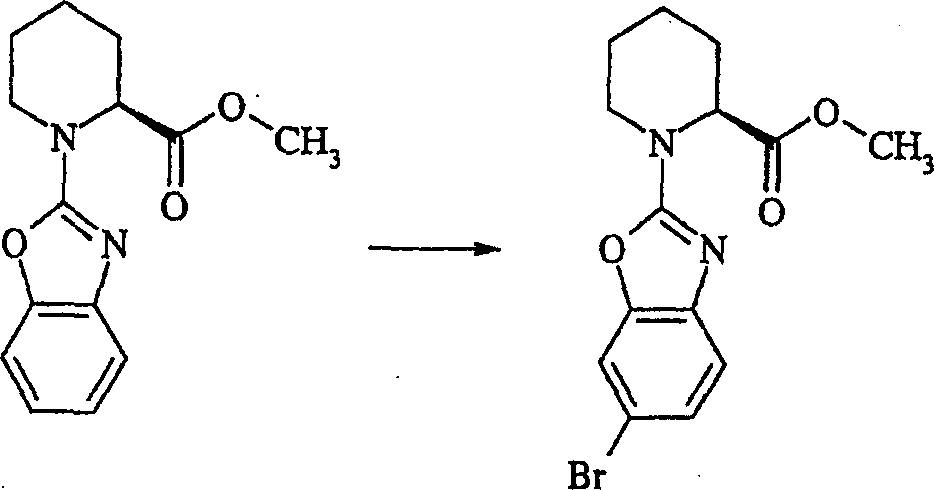
将2,4,4,6-四溴-2,5-环己二烯-1-酮(4.7g)于-10℃下用10分钟的时间加入到(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸甲酯(3.0g)[参见制备例2]的二氯甲烷(60ml)溶液中。然后将反应混合物升温至室温并用二氯甲烷稀释。将有机层用饱和碳酸氢钠洗涤,然后用1N氢氧化钠溶液洗涤,用硫酸钠干燥然后减压蒸除溶剂得到紫色油状(2S)-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酸甲酯(3.7g)。1H-NMR(CDCl3)δ:7.40(1H,s),7.25(1H,d),7.20(1H,d),5.00(1H,d),4.20(1H,d),3.80(3H,s),3.40(1H,t),2.40(1H,d),1.80(3H,m),1.70(2H,m),1.40(1H,m).
制备例6(2S)-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酸
标题化合物通过与制备例3类似的方法从(2S)-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酸甲酯[参见制备例5]和1N氢氧化锂水溶液制备,得到粉红色泡沫状的(2S)-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酸。1H-NMR(CDCl3)δ:7.40(1H,s),7.25-7.20(2H,m),5.00(1H,d),4.80(1H,bs),4.20(1H,d),3.40(1H,t),2.40(1H,d),1.90(3H,m),1.70-1.40(2H,m).MS:325(MH+).
制备例7
N-(3S)-1-[(2-氯-3-喹啉基)甲基]吡咯烷-3-基氨基甲酸叔丁酯
将三乙酰氧基硼氢化钠(2.3g)加入到N-[(3S)-吡咯烷-3-基]氨基甲酸叔丁酯(10g)[参见《杂环化学杂志》(J.Het.Chem.)(1990),27,1527-1536]和2-氯-3-喹啉甲醛(1.1g)[参见《印度化学会志》(Ind.J.Chem.Soc.)(1985),24,1286-1287]的二氯甲烷(20ml)溶液中。将反应混合物室温搅拌18小时,然后加入水(20ml)并将混合物继续搅拌1小时。分出有机层,用饱和碳酸氢钠水溶液洗涤,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统1∶1(体积比)乙酸乙酯∶氯仿洗脱得到油状的N-(3S)-1-[(2-氯-3-喹啉基)甲基]-1-吡咯烷-3-基氨基甲酸叔丁酯(1.6g)。1H-NMR(CDCl3)δ:8.20(1H,s),7.98(1H,d),7.80(1H,d),7.68(1H,m),7.55(1H,m),5.85(1H,bs),4.22(1H,bs),3.82(2H,s),2.85(1H,bs),2.78(1H,m),2.65(1H,m),2.50(1H,m),2.28(1H,m),1.65(1H,m),1.40(9H,s).MS:262(MH+).
制备例8
(3S)-1-[(2-氯-3-喹啉基)甲基]吡咯烷-3-胺盐酸盐
将氯化氢气体通入到N-(3S)-1-[(2-氯-3-喹啉基)甲基]吡咯烷-3-基氨基甲酸叔丁酯(1.6g)[参见制备例7]的氯仿(20ml)溶液中直至饱和。将反应混合物室温搅拌18小时,然后减压蒸除溶剂得到白色固体状(3S)-1-[(2-氯-3-喹啉基)甲基]吡咯烷-3-胺盐酸盐(1.5g)。MS:262(MH+)。Rf∶0.1(10∶1(体积比)乙酸乙酯∶氯仿)。
制备例9至12
以下列表的制备例(表2a)中的如下通式的化合物通过与制备例7类似的方法从N-[(3S)-吡咯烷-3-基]氨基甲酸叔丁酯[参见《杂环化学杂志》(J.Het.Chem.)(1990),27,1527-1536]和相应的醛或酮制备。
表2a
 脚注1.原料制备
脚注1.原料制备
制备例13至16
以下列表的制备例(表2b)中化合物通过与制备例8类似的方法从相应的氨基甲酸叔丁酯[参见表2a]制备。
表2b
制备例17至23
以下列表的制备例(表3a)中的如下通式的化合物通过与实施例1类似的方法从N-[(3S)-吡咯烷-3-基]氨基甲酸叔丁酯[参见《杂环化学杂志》(J.Het.Chem.)(1990),27,1527-1536]和相应的羧酸制备
表3a 
 脚注1.原料制备
脚注1.原料制备
制备例24至30
以下列表的制备例(表3b)中的化合物通过与制备例8类似的方法从相应的氨基甲酸叔丁酯[参见表3a]制备。
表3b 
制备例314-([(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶基]羰基氨基)哌啶甲酸叔丁酯
标题化合物通过与实施例1类似的方法从(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酸[参见制备例3]和4-氨基-1(2H)-哌啶甲酸叔丁酯[参见Takatani,Muneo等,WO9740051]制备。将粗产物通过硅胶柱色谱纯化,用2∶1∶0→0∶95∶5(体积比)己烷∶乙酸乙酯∶甲醇的溶剂梯度洗脱得到白色泡沫状4-([(2S)-1-(1,3-苯并噁唑-2-基)-2-哌啶基]羰基氨基)哌啶甲酸叔丁酯。1H-NMR(CDCl3)δ:7.40(1H,m),7.30(1H,m),7.20(1H,m),7.10(1H,m),6.40(1H,d),4.90(1H,s),4.30(1H,d),4.00(3H,m),3.20(1H,t),2.90(2H,m),2.40(1H,d),1.90(2H,m),1.80-1.60(5H,m),1.40(9H,s),1.30(2H,m).MS:429(MH+).
制备例32
(2S,4S)-2-[(苄氧基)甲基]-4-[(4-甲基苯基)磺酰基]氧基吡咯烷-1-甲酸叔丁酯
将4-甲基-1-苯磺酸甲酯(0.8g)、三苯膦(1.12g)和偶氮二甲酸二乙酯(0.68ml)在10℃及氮气氛下依次加入到(2S,4R)-2-[(苄氧基)甲基]-4-羟基-1-吡咯烷-1-甲酸叔丁酯(1.1g)[参见Takado,Seiichi等,《化学会志化学通讯》(J.Chem.Soc.Chem Commun.)(1988),23,1527-1528]的干燥四氢呋喃(10ml)溶液中。将反应混合物室温搅拌48小时,然后减压蒸除溶剂并将残余物在乙酸乙酯和水之间进行分配。分出有机层,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统85∶15(体积比)己烷∶乙酸乙酯洗脱得到无色胶状标题化合物。1H-NMR(CDCl3)δ:7.80(2H,d),7.30(7H,m),5.05(1H,m),4.50(2H,m),4.00(1H,m),3.70(2H,m),3.50(2H,m),2.50(3H,s),2.40-2.20(2H,m),1.45(9H,s).MS:462(MH+).
制备例33
(2S,4R)-4-叠氮基-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯
将叠氮化钠(0.32g)加入到(2S,4S)-2-[(苄氧基)甲基]-4-[(4-甲基苯基)磺酰基]氧基吡咯烷-1-甲酸叔丁酯(1.15g)[参见制备例32]的乙醇(20ml)和二甲基甲酰胺(5ml)溶液中。将反应混合物加热至80℃4小时,然后将冷却的反应混合物在乙醚和水之间进行分配。分出有机层并将水层用乙醚萃取两次,将合并的有机层用硫酸镁干燥然后减压蒸除溶剂。将残余物与二氯甲烷共沸得到油状的(2S,4R)-4-叠氮基-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯(820mg)。1H-NMR(CDCl3)δ:7.30(5H,m),4.50(2H,s),4.20-4.00(2H,m),3.70-3.40(4H,m),2.25(1H,m),2.15(1H,m),1.45(9H,m).MS:333(MH+).
制备例34(2S,4R)-4-氨基-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯
将三苯膦(421mg)加入到(2S,4R)-4-叠氮基-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯(455mg)[参见制备例33]的干燥四氢呋喃(10ml)溶液中。将反应混合物搅拌至不再有氮气溢出,加入水(0.036ml),然后将混合物继续搅拌72小时。减压蒸除溶剂,将残余物溶于乙醚然后加入己烷直至混合物变为混浊。分出上清液然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统90∶10(体积比)二氯甲烷∶甲醇洗脱得到无色油状(2S,4R)-4-氨基-2-[(苄氧基)甲基]吡咯烷-1-甲酸叔丁酯(225mg)。1H-NMR(CDCl3)δ:7.30(5H,m),4.55(2H,s),4.10(1H,m),3.70-3.40(4H,m),3.10(1H,m),2.25(1H,m),1.80(1H,m),1.50(9H,m).MS:307(MH+).
制备例35(3R)-1-(2-吡啶基甲基)-3-哌啶醇
将(3R)-3-哌啶醇盐酸盐(10.0g)和三乙胺(10.13ml)的干燥1,2-二氯乙烷(350ml)溶液于50℃搅拌15分钟。加入2-吡啶甲醛(7.63ml)和冰乙酸(4.16ml)并将反应混合物回流搅拌1.5小时。然后分批加入三乙酰氧基硼氢化钠(34.65g),将形成的混合物冷却至室温并继续搅拌1小时。然后加入水(350ml)和1M氢氧化钠水溶液直至混合物达到pH12。分出有机层,将水层用氯仿萃取,将合并的有机层用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统95∶5(体积比)氯仿∶甲醇洗脱得到棕色油状(3R)-1-(2-吡啶基甲基)-3-哌啶醇(8.10g)。1H-NMR(CDCl3)δ:8.55-8.50(1H,m),7.65-7.60(1H,m),7.30-7.25(1H,m),7.15-7.10(1H,m),3.85-3.75(1H,m),3.60(2H,s),2.80-2.70(1H,bs),2.60-2.50(1H,m),2.50-2.40(2H,bs),2.40-2.25(1H,m),1.80-1.70(1H,m),1.70-1.60(1H.,m),1.60-1.40(2H,m).Rf∶0.26(95∶5(体积比),氯仿∶甲醇)。
制备例36(2S,4R)-4-羟基-2-(苯氧基甲基)吡咯烷-1-甲酸苄酯
将(2S,4R)-4-羟基-2-(羟基甲基)吡咯烷-1-甲酸苄酯(2.0g)[参见Ceulemans等,《欧洲化学杂志》(Chem.Eur.J.)(1997),3(12),1997-2010]加入到苯酚(1.12g)、三苯膦(2.51g)和偶氮二甲酸二乙酯(1.51ml)的干燥四氢呋喃(40ml)溶液中。将反应混合物室温搅拌20小时,然后减压蒸除溶剂并将残余物溶于氯仿。将有机溶液依次用15%氢氧化钠水溶液和盐水洗涤,用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统1∶1(体积比)己烷∶乙酸乙酯洗脱得到透明油状(2S,4R)-4-羟基-2-(苯氧基甲基)吡咯烷-1-甲酸苄酯(0.69g)。1H-NMR(CDCl3)δ:7.40-7.20(7H,m),7.0-6.90(1H,m),6.85-6.75(2H,d),5.20-5.00(3H,m),4.80(1H,s),4.30-3.75(2H,m),3.75-3.60(3H,m),2.30-2.15(1H,m),2.10-2.00(1H,m).Rf∶0.4(1∶1(体积比)己烷∶乙酸乙酯)。
制备例37(3R,5S)-5-(苯氧基甲基)吡咯烷-3-醇
将10%w/w钯碳(0.05g)加入到(2S,4R)-4-羟基-2-(苯氧基甲基)吡咯烷-1-甲酸苄酯(0.25g)[参见制备例36]和5M甲酸铵水溶液(1.45ml)的甲醇(20ml)溶液中。将反应混合物加热回流3天,然后将反应液用硅藻土过滤并减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统90∶10∶0.5(体积比)氯仿∶甲醇∶0.88氨水洗脱得到棕色油状(3R,5S)-5-(苯氧基甲基)吡咯烷-3-醇(0.25g)。1H-NMR(CDCl3)δ:7.24-7.20(2H,t),6.91-6.85(1H,t),6.80-6.75(2H,d),4.78-4.71(1H,m),3.80-3.68(2H,bs),3.64-3.59(1H,dd),3.56-3.49(1H,dd),3.26-3.19(1H,m),3.19-3.16(1H,d),3.02-2.94(1H,dd),2.24-2.14(1H,ddd),1.70-1.60(1H,ddd).MS:194(MH+).
制备例38(3R,5S)-5-(苯氧基甲基)-1-(4-吡啶基甲基)吡咯烷-3-醇
标题化合物通过与制备例35类似的方法从(3R,5S)-5-(苯氧基甲基)吡咯烷-3-醇[参见制备例37]和4-吡啶甲醛制备。将粗产物通过硅胶柱色谱纯化,用溶剂系统95∶5(体积比)氯仿∶甲醇洗脱得到白色固体状(3R,5S)-5-(苯氧基甲基)-1-(4-吡啶基甲基)吡咯烷-3-醇。1H-NMR(CDCl3)δ:8.45-8.40(2H,m),7.25-7.10(4H,m),6.90-6.80(1H,m),6.80-6.70(2H,m),4.73-4.66(1H,m),4.08-4.02(1H,d),3.73-3.66(1H,dd),3.56-3.50(1H,dd),3.36-3.28(1H,d),3.20-3.13(2H,d),2.87-2.80(1H,m),2.53-2.46(1H,dd),2.43-2.35(1H,m),2.12-2.03(1H,m).旋光:[◇]25 D=-12.50°(c=1.0,氯仿)。MS:285(MH+)。
制备例39(3R)-1-[(2-氟-4-吡啶基)甲基]-3-哌啶醇
将(3R)-3-羟基哌啶盐酸盐(0.188g)加入到4-(溴甲基)-2-氟吡啶(0.26g)[参见Porter等,WO9622978]和碳酸钾(0.189g)的乙腈(15ml)溶液中。将反应混合物回流5天,然后减压蒸除溶剂并将残余物在二氯甲烷和水之间进行分配。分出有机层,将水层调至pH 12然后将产物用二氯甲烷萃取。将合并的有机层用硫酸镁干燥然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用溶剂系统95∶5(体积比)氯仿∶甲醇洗脱得到浅黄色油状的(3R)-1-[(2-氟-4-吡啶基)甲基]-3-哌啶醇(0.255g)。1H-NMR(CDCl3)δ:7.97-7.96(1H,d),7.03-7.01(1H,d),6.80(1H,s),3.70-3.60(1H,m),3.44-3.36(2H,m),3.32(1H,s),2.55-2.45(1H,d),2.35-2.20(1H,bs),2.20-2.05(2H,d),1.70-1.60(2H,d),1.50-1.35(1H,m),1.35-1.25(1H,m).MS:211(MH+).
制备例40
N-[(3S)-1-(3-吡啶基甲基)吡咯烷-3-基]氨基甲酸叔丁酯
标题化合物通过与制备例7类似的方法从N-[(3S)-1-吡咯烷-3-基]氨基甲酸叔丁酯(参见《杂环化学杂志》(J.Het.Chem.),1990,27,1286-1287)和3-吡啶甲醛制得,得到的N-[(3S)-1-(3-吡啶基甲基)吡咯烷-3-基]氨基甲酸叔丁酯立即用于制备例41。
制备例41
(3S)-1-(3-吡啶基甲基)吡咯烷-3-胺
标题化合物通过与制备例8类似的方法从N-[(3S)-1-(3-吡啶基甲基)-1-吡咯烷-3-基]氨基甲酸叔丁酯[参见制备例40]和氯化氢制备,得到白色固体状的(3S)-1-(3-吡啶基甲基)吡咯烷-3-胺。MS:178(MH+)。
制备例42
(2S)-1-(1,3-苯并噻唑-2-基)-2-哌啶甲酸乙酯
将2-氯-1,3-苯并噻唑(503mg)加入到(2S)-2-哌啶甲酸乙酯(471mg)[J.A.C.S.(1993),115(22),9925-9938]、三乙胺盐酸盐(414mg)和铜粉(38mg)的二甲苯(5ml)溶液中。将反应混合物回流28小时,然后将乙酸乙酯(20ml)加入到冷却的混合物中并滤出固体。将有机层用水洗涤,用硫酸镁干燥然后减压蒸除溶剂得到棕色固体状(2S)-1-(1,3-苯并噻唑-2-基)-2-哌啶甲酸乙酯(705mg)。1H-NMR(CDCl3)δ:7.58(1H,d),7.50(1H,d),7.20(1H,t),7.00(1H,t),5.10(1H,d),4.18(2H,q),3.80(1H,m),3.42(1H,m),2.25(1H,d),1.95-1.80(3H,m),1.60(1H,m),1.40(1H,m),1.20(3H,t).MS:291(MH+).
制备例43
(2S)-1-(1,3-苯并噻唑-2-基)-2-哌啶甲酸
标题化合物通过与制备例3类似的方法从(2S)-1-(1,3-苯并噻唑-2-基)-2-哌啶甲酸乙酯[参见制备例42]和1N氢氧化锂水溶液制备。将粗产物通过硅胶柱色谱纯化,用溶剂系统10∶1(体积比)二氯甲烷∶甲醇洗脱得到固体状的(2S)-1-(1,3-苯并噻唑-2-基)-2-哌啶甲酸。1H-NMR(CDCl3)δ:7.42(2H,m),7.10(1H,m),6.95(1H,m),4.80(1H,m),3.50(2H,m),2.10(1H,m),1.50(5H,m).MS:261(MH+).
制备例44
(2S)-1-(6-氯-1,3-苯并噻唑-2-基)-2-哌啶甲酸乙酯标题化合物通过与制备例42类似的方法从2,6-二氯-1,3-苯并噻唑[参见《印度化学会志》(J.Ind.Chem.Soc.),(1993),10,565-569]和(2S)-2-哌啶甲酸乙酯(471mg)[参见J.A.C.S.(1993),115(22),9925-9938]制备,得到固体状的(2S)-1-(6-氯-1,3-苯并噻唑-2-基)-2-哌啶甲酸乙酯。1H-NMR(CDCl3)δ:7.55(1H,s),7.40(1H,d),7.15(1H,d), 5.05(1H,d),4.15(2H,q),3.65(1H,m),3.45(1H,m),2.22(1H,m),1.80(2H,m),1.60(1H,m),1.35(1H,m),1.15(3H,t).MS:325(MH+).
制备例45
(2S)-1-(6-氯-1,3-苯并噻唑-2-基)-2-哌啶甲酸
标题化合物通过与制备例3类似的方法从(2S)-1-(6-氯-1,3-苯并噻唑-2-基-2-哌啶甲酸乙酯[参见制备例44]和1N氢氧化锂水溶液制备,得到固体状的(2S)-1-(6-氯-1,3-苯并噻唑-2-基)-2-哌啶甲酸。1H-NMR(DMSO-d6)δ:7.80(1H,s),7.70(1H,d),7.30(1H,d),5.60(1H,bs),4.75(1H,bs),3.40(2H,m),2.20(1H,m),1.80-1.60(3H,m),1.50(1H,m),1.30(1H,m).MS:295(MH+).
制备例46
2-氯-1H-1,3-苯并咪唑-1-甲酸叔丁酯
将2-氯-1H-1,3-苯并咪唑(1.07g)加入到二碳酸二叔丁酯(1.83g)和4-二甲氨基吡啶(86mg)的乙腈(15ml)溶液中。将反应混合物室温搅拌30分钟,然后减压蒸除溶剂。将粗产物通过硅胶柱色谱纯化,用100∶0→90∶10(体积比)己烷∶乙酸乙酯的溶剂梯度洗脱得到白色固体状2-氯-1H-1,3-苯并咪唑-1-甲酸叔丁酯(1.68g)。1H-NMR(CDCl3)δ:7.90(1H,m),7.35(1H,m),7.40(2H,m),1.80(9H,s).MS:253(MH+).
制备例47
2-[(2S)-2-[(苄氧基)羰基]-1-哌啶基]-1H-1,3-苯并咪唑-1-甲酸叔丁酯
标题化合物通过与制备例2类似的方法从2-氯-1H-1,3-苯并咪唑-1-甲酸叔丁酯[参见制备例46]和(2S)-2-哌啶甲酸苄酯[参见J.A.C.S.(1996),118(7),1629-1644]制备。将粗产物通过硅胶柱色谱纯化,用90∶10→80∶20(体积比)己烷∶乙酸乙酯的溶剂梯度以5%的增量洗脱得到油状的2-[(2S)-2-[(苄氧基)羰基]-1-哌啶基]-1H-1,3-苯并咪唑-1-甲酸叔丁酯。1H-NMR(CDCl3)δ:7.65(1H,d),7.45(1H,d),7.40(2H,s),7.25(5H,m),5.20(2H,s),4.70(1H,m),4.65(1H,m),3.60(1H,m),2.20(1H,m),2.05(1H,m),1.80-1.50(13H,m).MS:436(MH+).
制备例48
(2S)-1-[1-(叔丁氧基羰基)-1H-1,3-苯并咪唑-2-基]-2-哌啶甲酸
将10%w/w钯碳(300mg)加入到2-[(2S)-2-[(苄氧基)羰基]-1-哌啶基]-1H-1,3-苯并咪唑-1-甲酸叔丁酯(900mg)[参见制备例47]的乙醇(30ml)溶液中。将反应混合物在103.5kPa(15psi)下室温氢化18小时。然后滤出催化剂并减压蒸除溶剂得到白色泡沫状(2S)-1-[1-(叔丁氧基羰基)-1H-1,3-苯并咪唑-2-基]-2-哌啶甲酸(700mg)。1H-NMR(DMSO-d6)δ:7.65(1H,d),7.35(1H,d),7.20(1H,t),7.10(1H,t),4.40(1H,m),3.50(2H,m),2.05(1H,d),1.90(1H,m),1.70-1.40(13H,m).
制备例49
1,3,4,12a-四氢吡啶并[1’,2’:3,4]咪唑并[1,2-a][1,3]苯并咪唑-12(2H)-酮
标题化合物通过与实施例1类似的方法从(2S)-1-[1-(叔丁氧基羰基)-1H-1,3-苯并咪唑-2-基]-2-哌啶甲酸[参见制备例48]和(3S)-1-苄基吡咯烷-3-基胺制备。将粗产物通过硅胶柱色谱纯化,用70∶30→50∶50(体积比)己烷∶乙酸乙酯、然后是90∶10∶1二氯甲烷∶甲醇∶0.88氨水的溶剂梯度洗脱得到白色固体状1,3,4,12a-四氢吡啶并[1’,2’:3,4]咪唑并[1,2-a][1,3]苯并咪唑-12(2H)-酮。1H-NMR(CDCl3)δ:7.60(1H,d),7.40(1H,d),7.30(1H,m),7.10(1H,t),4.20(2H,m),3.20(1H,m),2.35(1H,d),2.05(1H,m),1.80(1H,d),1.70-1.50(3H,m).MS:227(MH+).
可以理解,本发明将要求保护如下内容:(ⅰ)式(Ⅰ)化合物或其可药用盐或溶剂化物;(ⅱ)式(Ⅰ)化合物或其可药用盐或溶剂化物的制备方法;(ⅲ)含有式(Ⅰ)化合物或其可药用盐或溶剂化物以及可药用赋形剂、稀释剂或载体的药物组合物;(ⅳ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物用作药物;(ⅴ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物在生产用于治疗神经元变性的药物中的用途;(ⅵ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物在生产用于促进神经元再生和分枝的药物中的用途;(ⅶ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物在生产用于治疗神经病学疾病或障碍如神经变性疾病的药物中的用途;(ⅷ)如(ⅶ)中所述的用途,其中的神经病学疾病或障碍选自老年性痴呆(早老性痴呆)和其它痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、影响中枢或外周神经系统的各种形式的变性疾病(例如小脑-脑干萎缩、进行性共济失调综合征)、各种形式的肌营养不良、进行性肌萎缩、进行性延髓肌萎缩、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、突出、破裂或脱出的椎间盘综合征、颈椎关节强硬、神经丛疾病、胸腔出口综合征、各种形式的外周神经病(糖尿病性的和非糖尿病性的)、三叉神经痛、舌咽神经痛、面神经麻痹、导致中枢或外周神经系统损伤的各种形式的自身免疫相关性疾病(例如多发性硬化、重症肌无力、格-巴二氏综合征)、神经系统的AIDS相关性疾病、氨苯砜抽搐、眼神经的眼球和眼球后病变(例如视网膜病和眼球后神经炎)、听力障碍例如耳鸣和朊病毒疾病;(ⅸ)如(ⅷ)中所述的用途,其中的神经病学疾病或障碍是老年性痴呆(早老性痴呆)或其它的痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、外周神经病(糖尿病性的或非糖尿病性的)、多发性硬化或听力障碍例如耳鸣;(ⅹ)治疗人类患者神经元变性的方法,该方法包括,将所述患者用有效量的式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物进行治疗;(ⅹⅰ)促进人类患者神经元再生和分枝的方法,该方法包括,将所述患者用有效量的式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物进行治疗;(ⅹⅱ)治疗人类患者神经病学疾病或障碍如神经变性疾病的方法,该方法包括,将所述患者用有效量的式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物进行治疗;(ⅹⅲ)如(ⅹⅱ)中所述的方法,其中的神经病学疾病或障碍选自老年性痴呆(早老性痴呆)和其它痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、影响中枢或外周神经系统的各种形式的变性疾病(例如小脑-脑干萎缩、进行性共济失调综合征)、各种形式的肌营养不良、进行性肌萎缩、进行性延髓肌萎缩、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、突出、破裂或脱出的椎间盘综合征、颈椎关节强硬、神经丛疾病、胸腔出口综合征、各种形式的外周神经病(糖尿病性的和非糖尿病性的)、三叉神经痛、舌咽神经痛、面神经麻痹、导致中枢或外周神经系统损伤的各种形式的自身免疫相关性疾病(例如多发性硬化、重症肌无力、格-巴二氏综合征)、神经系统的AIDS相关性疾病、氨苯砜抽搐、眼神经的眼球和眼球后病变(例如视网膜病和眼球后神经炎)、听力障碍例如耳鸣和朊病毒疾病;(ⅹⅳ)如(ⅹⅲ)中所述的方法,其中的神经病学疾病或障碍是老年性痴呆(早老性痴呆)或其它的痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、外周神经病(糖尿病性的或非糖尿病性的)、多发性硬化或听力障碍例如耳鸣;和(ⅹⅴ)文中所描述的所有新中间体。(ⅹⅵ)式(Ⅰ)化合物或其可药用盐、溶剂化物或组合物在生产用于治疗由于FKBP-12或FKBP-52缺乏或过度生成所引起之疾病的药物中的用途。
本发明的化合物对旋转异构酶FKBP-12显示出抑制活性。特别是某些优选的化合物,即实施例2、6、7、8、13a、15、16、17、18、19、20、21、22、23、24、25、26、27、30、35和41的化合物抑制FKBP-12酶的IC50值低于1200nM。实施例2的化合物抑制FKBP-52酶的IC50值为2790nM。
Claims (37)
1.式(Ⅰ)化合物或其可药用盐:其中
A是任意选择性地被C1-C6烷基取代的直链C3-C5亚烷基;
X是O、S、NH或N(C1-C6烷基);
Y是O、S、NH或N(C1-C6烷基);
R是通过碳连接的4至6元非芳香性的含有1个氮杂原子的杂环基团,所述基团任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C2-C6链烯基、C3-C7环烷基、芳基、het、-CO2(C1-C6烷基)、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代,所述烷基和链烯基任意选择性地被1或2个彼此独立地选自C3-C7环烷基、芳基、het、-O(芳基)、-O(C1-C2亚烷基)芳基、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代;
R1、R2、R3和R4彼此独立地选自H、卤素、C1-C6烷基、C3-C7环烷基、卤素(C1-C6)烷基、C1-C6烷氧基、-CONR5R6、C3-C7环烷氧基、C3-C7环烷基-(C2-C4)亚烷基、C3-C7环烷基(C2-C4)烷氧基和-CO2(C1-C6烷基);
R5和R6彼此独立地选自H和C1-C6烷基,或者当它们合在一起时,表示直链的C3-C5亚烷基;
“芳基”是指任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C1-C6烷氧基、卤素、-CONR5R6、卤素(C1-C6烷基)和-NR5R6的取代基取代的苯基;
“het”是指含有1至3个彼此独立地选自N、O和S的杂原子的5或6元单环或8、9或10元二环的杂环基团,所述基团任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C1-C6烷氧基、卤素、卤素(C1-C6烷基)、苯基和-NR5R6的取代基所取代。
2.权利要求1所述的化合物,其具有式(ⅠA)的立体化学结构: 其中R、R1、R2、R3、R4、A、X和Y如权利要求1中的式(Ⅰ)化合物所定义。
其中R、R1、R2、R3、R4、A、X和Y如权利要求1中的式(Ⅰ)化合物所定义。
3.权利要求1或2的化合物,其中A是1,4-亚丁基。
4.权利要求1至3中任意一项所述的化合物,其中X是O、S或NH。
5.权利要求4的化合物,其中X是O或NH。
6.权利要求1至5中任意一项所述的化合物,其中Y是O或NH。
7.权利要求6的化合物,其中Y是NH。
8.权利要求1至7中任意一项所述的化合物,其中R是氮杂环丁烷基、吡咯烷基或哌啶基,它们均选择性地被1、2或3个彼此独立地选自C1-C6烷基、C2-C6链烯基、C3-C7环烷基、芳基、het、-CO2(C1-C6烷基)、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代,所述烷基和链烯基任意选择性地被1或2个彼此独立地选自G3-C7环烷基、芳基、bet、-O(芳基)、-O(C1-C2亚烷基)芳基、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代。
9.权利要求1至8中任意一项所述的化合物,其中R是氮杂环丁烷基、吡咯烷基或哌啶基,它们均任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、het、-CO2(C1-C6烷基)和-CO(het)的取代基所取代,所述烷基任意选择性地被1或2个彼此独立地选自C3-C7环烷基、芳基、het、-O(芳基)、-O(C1-C2亚烷基)芳基和-CONR5R6的取代基所取代。
10.权利要求1至9中任意一项所述的化合物,其中R是氮杂环丁烷基、吡咯烷基或哌啶基,它们均任意选择性地被1、2或3个彼此独立地选自乙基、2-吡啶基、叔丁氧羰基、喹啉-2-基羰基、2-苯基喹啉-4-基羰基、4-甲氧基喹啉-2-基羰基、6-甲氧基-2-苯基喹啉-4-基羰基、2-哌啶子基喹啉-4-基羰基、2-氯喹啉-4-基羰基、1H-苯并吡唑-6-基羰基、环丙基甲基、苯基甲基、二苯基甲基、2-吡啶基甲基、3-吡啶基甲基、4-吡啶基甲基、2-(2-吡啶基)乙基、2-(2-甲基咪唑-1-基)乙基、(1H-1,2,4-三唑-3-基)甲基、(2-氯喹啉-3-基)甲基、喹啉-4-基甲基、喹啉-2-基甲基、喹啉-3-基甲基、1-(喹啉-4-基)乙基、(2-氟吡啶-4-基)甲基、苯氧基甲基、苄氧基甲基、氨基羰基甲基、2-(氨基羰基)乙基和3-(氨基羰基)苯基甲基的取代基所取代。
11.权利要求1至10中任意一项所述的化合物,其中R是任意选择性取代的3-氮杂环丁烷基、3-吡咯烷基、3-哌啶基或4-哌啶基。
12.权利要求1至11中任意一项所述的化合物,其中的芳基是任意选择性地被CONR5R6取代的苯基。
13.权利要求1至12中任意一项所述的化合物,其中的“het”基团是吡啶基、咪唑基、三唑基、喹啉基或苯并吡唑基,它们均任意选择性地被1、2或3个彼此独立地选自甲基、甲氧基、氟、氯、苯基和哌啶子基的取代基所取代。
14.权利要求1至13中任意一项所述的化合物,其中R1、R2、R3和R4彼此独立地选自H、卤素(C1-C6)烷基和卤素。
15.权利要求1至14中任意一项所述的化合物,其中R1、R2、R3和R4彼此独立地选自H、氟、氯、溴和三氟甲基。
16.权利要求1至15中任意一项所述的化合物,其中,权利要求1的式(Ⅰ)化合物中的下式基团 是1,3-苯并噁唑-2-基、1,3-苯并噻唑-2-基、1H-苯并咪唑-2-基、6-溴-1,3-苯并噁唑-2-基或6-氯-1,3-苯并噻唑-2-基。
是1,3-苯并噁唑-2-基、1,3-苯并噻唑-2-基、1H-苯并咪唑-2-基、6-溴-1,3-苯并噁唑-2-基或6-氯-1,3-苯并噻唑-2-基。
17.权利要求1至16中任意一项所述的化合物,其中,A是1,4-亚丁基;X是O或NH;Y是NH;R是3-氮杂环丁烷基、3-吡咯烷基、3-哌啶基或4-哌啶基,它们均任意选择性地被1、2或3个彼此独立地选自乙基、2-吡啶基、叔丁氧羰基、喹啉-2-基羰基、2-苯基喹啉-4-基羰基、4-甲氧基喹啉-2-基羰基、6-甲氧基-2-苯基喹啉-4-基羰基、2-哌啶子基喹啉-4-基羰基、2-氯喹啉-4-基羰基、1H-苯并吡唑-6-基羰基、环丙基甲基、苯基甲基、二苯基甲基、2-吡啶基甲基、3-吡啶基甲基、4-吡啶基甲基、2-(2-吡啶基)乙基、2-(2-甲基咪唑-1-基)乙基、(1H-1,2,4-三唑-3-基)甲基、(2-氯喹啉-3-基)甲基、喹啉-4-基甲基、喹啉-2-基甲基、喹啉-3-基甲基、1-(喹啉-4-基)乙基、(2-氟吡啶-4-基)甲基、苯氧基甲基、苄氧基甲基、氨基羰基甲基、2-(氨基羰基)乙基和3-(氨基羰基)苯基甲基的取代基所取代。
18.前述权利要求中任意一项所述的化合物,其选自:(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(2-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(3-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-[(3S)-1-(4-吡啶基甲基)吡咯烷-3-基]-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[3-(氨基羰基)苯基甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[(2-氯喹啉-3-基)甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[(喹啉-3-基)甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[(喹啉-4-基)甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[(喹啉-2-基)甲基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[1-(喹啉-4-基)乙基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[喹啉-2-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[2-苯基喹啉-4-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[4-甲氧基喹啉-2-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[6-甲氧基-2-苯基喹啉-4-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[2-哌啶子基喹啉-4-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[2-氯喹啉-4-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-{(3S)-1-[1H-苯并吡唑-6-基羰基]吡咯烷-3-基}-1-(1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-N2-[(3S)-1-苄基吡咯烷-3-基]-1-(6-溴-1,3-苯并噁唑-2-基)-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-(1-苄基-3-哌啶基)-2-哌啶甲酰胺;(2S)-1-(1,3-苯并噁唑-2-基)-N2-(3R,5S)-5-[(苄氧基)甲基]吡咯烷-3-基-2-哌啶甲酰胺盐酸盐;和1-(1H-1,3-苯并咪唑-2-基)-N2-[(3S)-1-苄基吡咯烷-3-基]-2-哌啶甲酰胺。
19.含有权利要求1至18中任意一项所述的化合物或其可药用盐或溶剂化物以及可药用赋形剂、稀释剂或载体的药物组合物。
20.权利要求1至18中任意一项所述的化合物或其可药用盐、溶剂化物或组合物用作药物。
21.权利要求1至18中任意一项所述的化合物或其可药用盐、溶剂化物或组合物在生产用于治疗神经元变性的药物中的用途。
22.权利要求1至18中任意一项所述的化合物或其可药用盐、溶剂化物或组合物在生产用于促进神经元再生和分枝的药物中的用途。
23.权利要求1至18中任意一项所述的化合物或其可药用盐、溶剂化物或组合物在生产用于治疗神经病学疾病或障碍如神经变性疾病的药物中的用途。
24.权利要求23所述的用途,其中的神经病学疾病或障碍选自老年性痴呆(早老性痴呆)和其它痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、影响中枢或外周神经系统的各种形式的变性疾病(例如小脑-脑干萎缩、进行性共济失调综合征)、各种形式的肌营养不良、进行性肌萎缩、进行性延髓肌萎缩、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、突出、破裂或脱出的椎间盘综合征、颈椎关节强硬、神经丛疾病、胸腔出口综合征、各种形式的外周神经病(糖尿病性的和非糖尿病性的)、三叉神经痛、舌咽神经痛、面神经麻痹、导致中枢或外周神经系统损伤的各种形式的自身免疫相关性疾病(例如多发性硬化、重症肌无力、格-巴二氏综合征)、神经系统的AIDS相关性疾病、氨苯砜抽搐、眼神经的眼球和眼球后病变(例如视网膜病和眼球后神经炎)、听力障碍例如耳鸣和朊病毒疾病。
25.权利要求23所述的用途,其中的神经病学疾病或障碍是老年性痴呆(早老性痴呆)或其它的痴呆症、肌萎缩性侧索硬化和其它形式的运动神经元疾病、帕金森氏症、杭廷顿氏舞蹈病、与中风有关的神经病学缺陷、对中枢或外周神经系统(例如脊髓)的物理或创伤性损伤、外周神经病(糖尿病性的或非糖尿病性的)、多发性硬化或听力障碍例如耳鸣。
26.治疗人类患者神经元变性的方法,该方法包括,将所述患者用有效量的权利要求1至18中任意一项所述的化合物或其可药用盐、溶剂化物或组合物进行治疗。
27.促进人类患者神经元再生和分枝的方法,该方法包括,将所述患者用有效量的权利要求1至18中任意一项所述的化合物或其可药用盐、溶剂化物或组合物进行治疗。
28.治疗人类患者神经病学疾病或障碍如神经变性疾病的方法,该方法包括,将所述患者用有效量的权利要求1至18中任意一项所述的化合物或其可药用盐、溶剂化物或组合物进行治疗。
29.权利要求1至18中任意一项所述的式(Ⅰ)化合物的制备方法:其中
X是O、S、NH或N(C1-C6烷基);
A是任意选择性地被C1-C6烷基取代的直链C3-C5亚烷基;
Y是O、S、NH或N(C1-C6烷基);
R是通过碳连接的4至6元非芳香性的含有1个氮杂原子的杂环基团,所述基团任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C2-C6链烯基、C3-C7环烷基、芳基、het、-CO2(C1-C6烷基)、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代,所述烷基和链烯基任意选择性地被1或2个彼此独立地选自C3-C7环烷基、芳基、het、-O(芳基)、-O(C1-C2亚烷基)芳基、-CO(het)、-CONR5R6和-CO(芳基)的取代基所取代;
R1、R2、R3和R4彼此独立地选自H、卤素、C1-C6烷基、C3-C7环烷基、卤素(C1-C6)烷基、C1-C6烷氧基、-CONR5R6、C3-C7环烷氧基、C3-C7环烷基-(C2-C4)亚烷基、C3-C7环烷基(C2-C4)烷氧基和-CO2(C1-C6烷基);
R5和R6彼此独立地选自H和C1-C6烷基,或者当它们合在一起时,表示直链的C3-C5亚烷基;
“芳基”是指任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C1-C6烷氧基、卤素、-CONR5R6、卤素(C1-C6烷基)和-NR5R6的取代基取代的苯基;和
“het”是指含有1至3个彼此独立地选自N、O和S的杂原子的5或6元单环或8、9或10元二环的杂环基团,所述基团任意选择性地被1、2或3个彼此独立地选自C1-C6烷基、C1-C6烷氧基、卤素、卤素(C1-C6烷基)、苯基和-NR5R6的取代基所取代,
该方法包括:(a)将通式(Ⅱ)的化合物进行脱水偶联:其中X是O或S,R1、R2、R3和R4如该权利要求中前面所定义;或者(b)将通式(ⅩⅢA)或(ⅩⅢB)的化合物 其中X是NH或N(C1-C6烷基),R1、R2、R3、R4和A如该权利要求中前面所定义,与式(Ⅲ)的化合物进行加成反应:
其中X是NH或N(C1-C6烷基),R1、R2、R3、R4和A如该权利要求中前面所定义,与式(Ⅲ)的化合物进行加成反应:
H-Y-R(Ⅲ),其中R和Y如该权利要求中前面所定义,其中,式(Ⅰ)的化合物可任意选择性地形成所需化合物的可药用或可兽药用的盐或其可药用或可兽药用的溶剂化物。
30.权利要求29所述的式(Ⅰ)化合物的制备方法,该方法包括,将式(Ⅴ)化合物: 其中A如权利要求29中的式(Ⅰ)化合物所定义,R7是C1-C4烷基或苄基,与通式(Ⅵ)的化合物:其中X是O或S,R1、R2、R3和R4如权利要求29中的式(Ⅰ)化合物所定义,L’是适宜的离去基,进行N-取代反应以形成式(Ⅶ)的化合物:
其中A如权利要求29中的式(Ⅰ)化合物所定义,R7是C1-C4烷基或苄基,与通式(Ⅵ)的化合物:其中X是O或S,R1、R2、R3和R4如权利要求29中的式(Ⅰ)化合物所定义,L’是适宜的离去基,进行N-取代反应以形成式(Ⅶ)的化合物: 其中A、X、R1、R2、R3和R4如权利要求29中的式(Ⅰ)化合物所定义,然后可将其通过碱水解转变成式(Ⅱ)的化合物:其中X、R1、R2、R3和R4如该权利要求中前面所定义,然后可将所述式(Ⅱ)的化合物通过权利要求29的方法转变成式(Ⅰ)的化合物。
其中A、X、R1、R2、R3和R4如权利要求29中的式(Ⅰ)化合物所定义,然后可将其通过碱水解转变成式(Ⅱ)的化合物:其中X、R1、R2、R3和R4如该权利要求中前面所定义,然后可将所述式(Ⅱ)的化合物通过权利要求29的方法转变成式(Ⅰ)的化合物。
31.通式(Ⅱ)的化合物:其中X是O或S,R1、R2、R3和R4如权利要求29中所定义。
32.通式(Ⅶ)的化合物: 其中R1、R2、R3、R4和A如权利要求29中的式(Ⅰ)化合物所定义,X是O或S,其中的R7是C1-C4烷基或苄基。
其中R1、R2、R3、R4和A如权利要求29中的式(Ⅰ)化合物所定义,X是O或S,其中的R7是C1-C4烷基或苄基。
33.权利要求1至18中任意一项所述的通式(Ⅰ)化合物的制备方法,该方法包括,将通式(Ⅹ)的化合物: 其中A如权利要求29中的式(Ⅰ)化合物所定义,R9是C1-C4烷基或苄基,与通式(Ⅸ)的化合物:
其中A如权利要求29中的式(Ⅰ)化合物所定义,R9是C1-C4烷基或苄基,与通式(Ⅸ)的化合物: 其中R1、R2、R3和R4如权利要求29中的式(Ⅰ)化合物所定义,R8是C1-C4烷基或苄基,L2是适宜的离去基,进行N-取代反应以形成式(Ⅺ)的化合物:
其中R1、R2、R3和R4如权利要求29中的式(Ⅰ)化合物所定义,R8是C1-C4烷基或苄基,L2是适宜的离去基,进行N-取代反应以形成式(Ⅺ)的化合物: 其中A、R1、R2、R3、R4、R8和R9如该权利要求中前面所定义,然后可以通过酯裂解将其转变成式(Ⅻ)的化合物:
其中A、R1、R2、R3、R4、R8和R9如该权利要求中前面所定义,然后可以通过酯裂解将其转变成式(Ⅻ)的化合物: 其中A、R1、R2、R3、R4和R8如该权利要求中前面所定义,然后将其通过环化反应转变成式(ⅩⅢA)或(ⅩⅢB)的化合物:其中R1、R2、R3、R4和A如该权利要求中前面所定义,所述式(ⅩⅢA)或(ⅩⅢB)的化合物可以通过权利要求29中所述的方法转变成式(Ⅰ)化合物。
其中A、R1、R2、R3、R4和R8如该权利要求中前面所定义,然后将其通过环化反应转变成式(ⅩⅢA)或(ⅩⅢB)的化合物:其中R1、R2、R3、R4和A如该权利要求中前面所定义,所述式(ⅩⅢA)或(ⅩⅢB)的化合物可以通过权利要求29中所述的方法转变成式(Ⅰ)化合物。
34.通式(Ⅸ)的化合物: 其中R1、R2、R3和R4如权利要求29中的式(Ⅰ)化合物所定义,R8是C1-C4烷基或苄基,L2是适宜的离去基。
其中R1、R2、R3和R4如权利要求29中的式(Ⅰ)化合物所定义,R8是C1-C4烷基或苄基,L2是适宜的离去基。
35.式(Ⅺ)的化合物: 其中A、R1、R2、R3、R4、R8和R9如权利要求33所定义。
其中A、R1、R2、R3、R4、R8和R9如权利要求33所定义。
36.式(Ⅻ)的化合物:其中A、R1、R2、R3、R4和R8如权利要求33所定义。
37.式(ⅩⅢA)或(ⅩⅢB)化合物:其中R1、R2、R3、R4和A如权利要求33所定义。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9815880.1 | 1998-07-21 | ||
| GBGB9815880.1A GB9815880D0 (en) | 1998-07-21 | 1998-07-21 | Heterocycles |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2004100399747A Division CN1611499A (zh) | 1998-07-21 | 1999-06-28 | 用作旋转异构酶抑制剂的杂环化合物 |
| CNA2003101239079A Division CN1511837A (zh) | 1998-07-21 | 1999-06-28 | 制备作为旋转异构酶抑制剂的杂环化合物所用的中间体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1310718A true CN1310718A (zh) | 2001-08-29 |
| CN1174978C CN1174978C (zh) | 2004-11-10 |
Family
ID=10835920
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2003101239079A Pending CN1511837A (zh) | 1998-07-21 | 1999-06-28 | 制备作为旋转异构酶抑制剂的杂环化合物所用的中间体 |
| CNA2004100399747A Pending CN1611499A (zh) | 1998-07-21 | 1999-06-28 | 用作旋转异构酶抑制剂的杂环化合物 |
| CNB998089974A Expired - Fee Related CN1174978C (zh) | 1998-07-21 | 1999-06-28 | 用作旋转异构酶抑制剂的杂环化合物 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2003101239079A Pending CN1511837A (zh) | 1998-07-21 | 1999-06-28 | 制备作为旋转异构酶抑制剂的杂环化合物所用的中间体 |
| CNA2004100399747A Pending CN1611499A (zh) | 1998-07-21 | 1999-06-28 | 用作旋转异构酶抑制剂的杂环化合物 |
Country Status (45)
Families Citing this family (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9815696D0 (en) * | 1998-07-20 | 1998-09-16 | Pfizer Ltd | Heterocyclics |
| GB9815880D0 (en) * | 1998-07-21 | 1998-09-16 | Pfizer Ltd | Heterocycles |
| CA2349227C (en) | 1998-11-03 | 2008-02-05 | Basf Aktiengesellschaft | Substituted 2-phenylbenzimidazoles, the production thereof and their use |
| DOP2000000107A (es) | 1999-12-01 | 2002-09-16 | Agouron Pharmaceutical Inc | Compuestos, composiciones y metodos para estimular el crecimiento y alongamiento de las neuronas |
| FR2805818B1 (fr) * | 2000-03-03 | 2002-04-26 | Aventis Pharma Sa | Derives d'azetidine, leur preparation et les compositions pharmaceutiques les contenant |
| DE10124953A1 (de) * | 2001-05-21 | 2002-12-12 | Marlies Knipper | Substanz für die therapeutische Behandlung von Tinnitus |
| EP1778637B1 (en) * | 2004-06-29 | 2012-02-22 | Aventis Pharmaceuticals Inc. | FKBP-binding composition and pharmaceutical use thereof |
| TWI375673B (en) * | 2005-04-11 | 2012-11-01 | Abbott Lab | 1h-benzimidazole-4-carboxamides substituted with a quaternary carbon at the 2-position are potent parp inhibitors |
| US7728026B2 (en) * | 2005-04-11 | 2010-06-01 | Abbott Laboratories, Inc. | 2-substituted-1 h-benzimidazile-4-carboxamides are PARP inhibitors |
| ATE536349T1 (de) * | 2005-09-29 | 2011-12-15 | Abbott Lab | In der 2-stellung durch phenyl substituierte 1h- benzimidazol-4-carbonsäureamide sind wirksame parp-inhibitoren |
| CA2628461C (en) * | 2005-11-15 | 2013-09-24 | Abbott Laboratories | Substituted 1h-benzimidazole-4-carboxamides are potent parp inhibitors |
| SI1976828T1 (sl) | 2005-12-29 | 2017-07-31 | Celtaxsys, Inc. | Diaminski derivati kot inhibitorji levkotrien A4 hidrolaze |
| RU2462458C2 (ru) * | 2006-02-10 | 2012-09-27 | Байомарин Ига Лимитед | Лечение миодистрофии дюшена |
| AU2007213451B2 (en) * | 2006-02-10 | 2013-02-07 | Summit (Oxford) Limited | Treatment of Duchenne muscular dystrophy |
| GB0602768D0 (en) * | 2006-02-10 | 2006-03-22 | Vastox Plc | Treatment of muscular dystrophy |
| US20070259937A1 (en) * | 2006-05-02 | 2007-11-08 | Giranda Vincent L | Substituted 1h-benzimidazole-4-carboxamides are potent parp inhibitors |
| JP5528812B2 (ja) * | 2006-12-20 | 2014-06-25 | アムジエン・インコーポレーテツド | 複素環式化合物ならびに炎症、血管形成および癌の治療におけるこれらの使用 |
| CL2008000666A1 (es) * | 2007-03-07 | 2008-06-13 | Xenon Pharmaceuticals Inc | Compuestos derivados de triciclos sustituidos, inhibidores del transportador de metales divalentes-1; y uso para tratar una enfermedad asociada con un trastorno del hierro. |
| US8067613B2 (en) * | 2007-07-16 | 2011-11-29 | Abbott Laboratories | Benzimidazole poly(ADP ribose)polymerase inhibitors |
| EP3251694A1 (en) * | 2007-08-03 | 2017-12-06 | Summit (Oxford) Limited | Drug combinations for the treatment of duchenne muscular dystrophy |
| GB0715937D0 (en) * | 2007-08-15 | 2007-09-26 | Vastox Plc | Method of treatment og duchenne muscular dystrophy |
| GB0715939D0 (en) * | 2007-08-15 | 2007-09-26 | Vastox Plc | Method of treatment of duchenne muscular dystrophy |
| DE102008060549A1 (de) | 2008-12-04 | 2010-06-10 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Wirkstoff-Peptid-Konstrukt zur extrazellulären Anreicherung |
| WO2010083199A1 (en) * | 2009-01-19 | 2010-07-22 | Abbott Laboratories | Benzthiazole inhibitors of poly(adp-ribose)polymerase |
| GB0905664D0 (en) * | 2009-04-02 | 2009-05-13 | Summit Corp Plc | Compounds for treatment of duchenne muscular dystrophy |
| AU2014249168B2 (en) | 2013-03-12 | 2018-07-12 | Celltaxis, Llc | Methods of inhibiting leukotriene A4 hydrolase |
| RU2690489C2 (ru) | 2013-03-14 | 2019-06-04 | Селтакссис, Инк. | Ингибиторы лейкотриен а4-гидролазы |
| JP6534650B2 (ja) | 2013-03-14 | 2019-06-26 | セルタクシス,インコーポレイテッド | ロイコトリエンa4加水分解酵素の阻害剤 |
| CA2906035A1 (en) | 2013-03-14 | 2014-09-25 | Celtaxsys, Inc. | Inhibitors of leukotriene a4 hydrolase |
| EP3145925B1 (en) * | 2014-05-19 | 2020-11-04 | Boehringer Ingelheim Animal Health USA Inc. | Anthelmintic compounds |
| AU2019278935A1 (en) | 2018-05-31 | 2020-12-10 | Celltaxis, Llc | Method of reducing pulmonary exacerbations in respiratory disease patients |
| CN109111376B (zh) * | 2018-09-18 | 2021-09-14 | 四川医立特生物医药有限公司 | 一种2,5-双脱氧链霉胺衍生物及其应用 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5612332A (en) * | 1984-03-19 | 1997-03-18 | Alteon Inc. | Di- and triaminoguanidines, and methods of use |
| JPS6428245A (en) | 1987-07-22 | 1989-01-30 | Sumitomo Electric Industries | Production of fluoride glass |
| MX9202466A (es) | 1991-05-24 | 1994-06-30 | Vertex Pharma | Compuestos inmunosupresores novedosos. |
| CO4520280A1 (es) * | 1993-10-26 | 1997-10-15 | Boehringer Ingelheim Pharma | Derivados de pirrolidina, inhibidores de la bioscientesis de leucotrienos |
| DE19514313A1 (de) | 1994-08-03 | 1996-02-08 | Bayer Ag | Benzoxazolyl- und Benzothiazolyloxazolidinone |
| US5696135A (en) * | 1995-06-07 | 1997-12-09 | Gpi Nil Holdings, Inc. | Inhibitors of rotamase enzyme activity effective at stimulating neuronal growth |
| US5859031A (en) | 1995-06-07 | 1999-01-12 | Gpi Nil Holdings, Inc. | Small molecule inhibitors of rotamase enzyme activity |
| US5801197A (en) * | 1995-10-31 | 1998-09-01 | Gpi Nil Holdings, Inc. | Rotamase enzyme activity inhibitors |
| US5801187A (en) * | 1996-09-25 | 1998-09-01 | Gpi-Nil Holdings, Inc. | Heterocyclic esters and amides |
| US5786378A (en) * | 1996-09-25 | 1998-07-28 | Gpi Nil Holdings, Inc. | Heterocyclic thioesters |
| US5780484A (en) | 1996-11-13 | 1998-07-14 | Vertex Pharmaceuticals Incorporated | Methods for stimulating neurite growth with piperidine compounds |
| US5840736A (en) | 1996-11-13 | 1998-11-24 | Vertex Pharmaceuticals Incorporated | Methods and compositions for stimulating neurite growth |
| US5811434A (en) | 1996-11-13 | 1998-09-22 | Vertex Pharmacueticals Incorporated | Methods and compositions for stimulating neurite growth |
| US5721256A (en) | 1997-02-12 | 1998-02-24 | Gpi Nil Holdings, Inc. | Method of using neurotrophic sulfonamide compounds |
| GB9815696D0 (en) * | 1998-07-20 | 1998-09-16 | Pfizer Ltd | Heterocyclics |
| GB9815880D0 (en) * | 1998-07-21 | 1998-09-16 | Pfizer Ltd | Heterocycles |
-
1998
- 1998-07-21 GB GBGB9815880.1A patent/GB9815880D0/en not_active Ceased
-
1999
- 1999-06-28 ES ES99963123T patent/ES2191484T3/es not_active Expired - Lifetime
- 1999-06-28 CA CA002338214A patent/CA2338214C/en not_active Expired - Fee Related
- 1999-06-28 CN CNA2003101239079A patent/CN1511837A/zh active Pending
- 1999-06-28 KR KR10-2001-7000927A patent/KR100450008B1/ko not_active IP Right Cessation
- 1999-06-28 BR BR9912330-4A patent/BR9912330A/pt not_active Application Discontinuation
- 1999-06-28 AT AT99963123T patent/ATE233261T1/de not_active IP Right Cessation
- 1999-06-28 EP EP99963123A patent/EP1100797B1/en not_active Expired - Lifetime
- 1999-06-28 HU HU0103413A patent/HUP0103413A3/hu unknown
- 1999-06-28 SK SK77-2001A patent/SK772001A3/sk unknown
- 1999-06-28 CN CNA2004100399747A patent/CN1611499A/zh active Pending
- 1999-06-28 NZ NZ522270A patent/NZ522270A/en unknown
- 1999-06-28 CN CNB998089974A patent/CN1174978C/zh not_active Expired - Fee Related
- 1999-06-28 WO PCT/IB1999/001211 patent/WO2000005232A1/en not_active Application Discontinuation
- 1999-06-28 DE DE69905582T patent/DE69905582T2/de not_active Expired - Fee Related
- 1999-06-28 GE GEAP19995715A patent/GEP20033028B/en unknown
- 1999-06-28 ID IDW20010155A patent/ID26991A/id unknown
- 1999-06-28 PL PL99345734A patent/PL345734A1/xx not_active Application Discontinuation
- 1999-06-28 AU AU42858/99A patent/AU765925B2/en not_active Ceased
- 1999-06-28 AP APAP/P/2001/002046A patent/AP2001002046A0/en unknown
- 1999-06-28 EA EA200100052A patent/EA003513B1/ru not_active IP Right Cessation
- 1999-06-28 KR KR10-2004-7007852A patent/KR100468185B1/ko not_active IP Right Cessation
- 1999-06-28 NZ NZ508838A patent/NZ508838A/xx unknown
- 1999-06-28 OA OA1200100023A patent/OA11585A/en unknown
- 1999-06-28 JP JP2000561188A patent/JP3795329B2/ja not_active Expired - Fee Related
- 1999-06-28 TR TR2001/00135T patent/TR200100135T2/xx unknown
- 1999-06-28 SI SI9930246T patent/SI1100797T1/xx unknown
- 1999-06-28 DK DK99963123T patent/DK1100797T3/da active
- 1999-06-28 EE EEP200100044A patent/EE200100044A/xx unknown
- 1999-06-28 IL IL14024499A patent/IL140244A0/xx unknown
- 1999-07-07 HN HN1999000106A patent/HN1999000106A/es unknown
- 1999-07-13 TW TW088111868A patent/TWI229672B/zh not_active IP Right Cessation
- 1999-07-19 AR ARP990103544A patent/AR019427A1/es unknown
- 1999-07-19 CO CO99045677A patent/CO5080782A1/es unknown
- 1999-07-20 MY MYPI99003048A patent/MY118222A/en unknown
- 1999-07-20 SV SV1999000102A patent/SV1999000102A/es not_active Application Discontinuation
- 1999-07-20 DZ DZ990151A patent/DZ2851A1/xx active
- 1999-07-20 PE PE1999000727A patent/PE20001037A1/es not_active Application Discontinuation
- 1999-07-20 PA PA19998478501A patent/PA8478501A1/es unknown
- 1999-07-20 TN TNTNSN99147A patent/TNSN99147A1/fr unknown
- 1999-07-20 CR CR6073A patent/CR6073A/es not_active Application Discontinuation
- 1999-07-20 GT GT199900115A patent/GT199900115A/es unknown
- 1999-07-20 MA MA25691A patent/MA24936A1/fr unknown
- 1999-07-21 US US09/358,107 patent/US6372736B1/en not_active Expired - Fee Related
-
2000
- 2000-12-22 IS IS5790A patent/IS5790A/is unknown
-
2001
- 2001-01-19 HR HR20010052A patent/HRP20010052A2/hr not_active Application Discontinuation
- 2001-01-19 NO NO20010322A patent/NO20010322L/no not_active Application Discontinuation
- 2001-02-14 BG BG105254A patent/BG105254A/xx unknown
-
2002
- 2002-01-23 US US10/056,901 patent/US6562964B1/en not_active Expired - Fee Related
- 2002-02-06 HK HK02100933.8A patent/HK1039779A1/zh unknown
-
2003
- 2003-04-09 JP JP2003105099A patent/JP2004002374A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1310718A (zh) | 用作旋转异构酶抑制剂的杂环化合物 | |
| CN1061654C (zh) | 四唑衍生物的制备方法 | |
| CN1304390C (zh) | 新颖的γ—分泌酶抑制剂 | |
| JP3668133B2 (ja) | ロータマーゼ酵素の阻害剤としての複素環式化合物 | |
| CN1756740A (zh) | 具有2,6-二取代苯乙烯基的含氮杂环衍生物 | |
| JP3545341B2 (ja) | Fkbp阻害剤 | |
| CN1285828A (zh) | 被含有一个氮原子的五、六或七元杂环取代的咪唑基烷基 | |
| US6509464B1 (en) | FKBP inhibitors | |
| JP3091844B2 (ja) | 複素環式化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1039779 Country of ref document: HK |