CN114555830A - 靶核酸的检测方法、核酸结合分子的检测方法、及核酸结合能力的评价方法 - Google Patents
靶核酸的检测方法、核酸结合分子的检测方法、及核酸结合能力的评价方法 Download PDFInfo
- Publication number
- CN114555830A CN114555830A CN202080072133.3A CN202080072133A CN114555830A CN 114555830 A CN114555830 A CN 114555830A CN 202080072133 A CN202080072133 A CN 202080072133A CN 114555830 A CN114555830 A CN 114555830A
- Authority
- CN
- China
- Prior art keywords
- nucleic acid
- target nucleic
- target
- detecting
- region
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images




















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/12—Transferases (2.) transferring phosphorus containing groups, e.g. kinases (2.7)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/111—General methods applicable to biologically active non-coding nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6809—Methods for determination or identification of nucleic acids involving differential detection
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6858—Allele-specific amplification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y102/00—Oxidoreductases acting on the aldehyde or oxo group of donors (1.2)
- C12Y102/01—Oxidoreductases acting on the aldehyde or oxo group of donors (1.2) with NAD+ or NADP+ as acceptor (1.2.1)
- C12Y102/01012—Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating) (1.2.1.12)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y207/00—Transferases transferring phosphorus-containing groups (2.7)
- C12Y207/11—Protein-serine/threonine kinases (2.7.11)
- C12Y207/11022—Cyclin-dependent kinase (2.7.11.22)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2523/00—Culture process characterised by temperature
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Plant Pathology (AREA)
- Pathology (AREA)
- Oncology (AREA)
- Hospice & Palliative Care (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
本发明为一种靶核酸的检测方法,是从碱基序列或修饰状态与靶核酸部分不同的非靶核酸中识别并检测靶核酸的方法,将所述非靶核酸中与所述靶核酸不同的区域作为靶区域,将所述靶核酸中与所述非靶核酸不同的区域作为对应靶区域,以供试核酸样本为模板,在与所述非靶核酸中的所述靶区域特异性结合的分子存在下,使用与所述靶核酸及非靶核双方杂交的引物,在所述分子能够与所述非靶核酸结合的温度条件下进行核酸扩增反应,基于有无扩增产物来检测靶核酸。
Description
技术领域
本发明涉及一种识别基因突变、基因多态性、核酸的修饰等碱基序列或结构近似的多个核酸以检测目标核酸的方法、检测核酸结合分子或评价其核酸结合能力的方法、及用于这些方法的试剂盒。
本申请基于2019年10月18日在日本申请的日本特愿2019-191409号主张优先权,其内容引用于本文。
背景技术
基因的碱基序列变化导致的基因突变多是产生了仅1~数个碱基的插入、缺失、向其它碱基转化等。因而,基因突变的检测中,需要识别并检测除1~数个碱基的突变位点以外由相同的碱基序列构成的野生型核酸和突变型核酸。
作为检测DNA或RNA的1~数个碱基的突变的方法,具有多种技术。作为利用PCR(polymerase chain reaction)检测突变的方法,可列举例如使用基于荧光物质和猝灭剂的修饰寡核苷酸探针的PCR法。在该方法中,在与预测产生了突变的突变型DNA区域杂交并经荧光物质和猝灭剂修饰的寡核苷酸探针的存在下,对包含预测产生了突变的DNA区域的DNA片段进行PCR扩增反应。对于突变型DNA,该探针进行退火,通过用于PCR的DNA聚合酶的核酸外切酶活性切割探针,荧光物质和猝灭剂解离,结果反应系统中产生荧光。而在野生型DNA的情况下,该探针不进行退火,因此不会产生荧光。但是,在野生型和突变型的DNA混合存在的样品中,在进行定量评价时需要繁琐的操作,因此难以用于检测杂合突变。
另外,还有利用SURVEYOR核酸酶的SURVEYOR测定(参见非专利文献1)。该测定将经PCR扩增的对照DNA和测试DNA在试管内混合,进行热变性及重新双链化,使用SURVEYOR核酸酶切割错配碱基的3’侧,从而检测出测试DNA包含与对照DNA不同的碱基。该方法较为简便,主要用于从经基因组编辑的细胞群中提取DNA并检测基因组编辑的效率。通常,基因组编辑效率达不到100%,基因组编辑的方法也多种多样,因此无需加入对照DNA。但是,在各种基因组编辑细胞的检测中,为了检测纯合的突变,需要混合来自野生型细胞的DNA。另外,在分析各种细胞来源的情况下,通过桑格法等确定碱基序列的方法更加直接,因此通常不使用SURVEYOR测定。
此外,还有ORNi-PCR法,该方法向反应系统中添加与PCR反应中扩增的DNA区域互补的17~29碱基左右的短链RNA(寡核苷酸、ORN)(例如,参见非专利文献2)。该方法中,特异性抑制ORN与之杂交的DNA区域的PCR扩增。即,仅ORN不与之杂交的DNA被PCR扩增。
现有技术文献
专利文献
专利文献1:日本特开2011-103900号公报
非专利文献
非专利文献1:Zhu,et al,Scientific Reports,2014,4:6420,DOI:10.1038/srep06420
非专利文献2:Fujita,et al,DNA Research,2018,vol.25,p.395-407.
非专利文献3:Notomi,et al,Nucleic Acids Research,2000,vol.28(12),e63.
非专利文献4:Dean,et al,Proceedings of the National Academy ofSciences of the United States of America,2002,vol.99(8),p.5261-5266.
非专利文献5:Fujita,et al.Scientific Reports,2016,6:30485.
非专利文献6:Fujita,et al.PLoS One,2015,10(6),e0116579.
非专利文献7:Abudayyeh,et al.Science,2016,353(6299),aaf5573.
非专利文献8:Gootenberg,et al.Science,2017,356(6336),p.438-442.
非专利文献9:Rascle and Lees,Nucleic Acids Research,2003,vol.31(23),p.6882-6890.
非专利文献10:Abudayyeh,et al.Nature,2017,vol.550(7675),p.280-284.
发明内容
本发明的主要目的在于,提供一种利用与作为模板的核酸结合的分子对核酸扩增反应的抑制来检测核酸的差异、修饰、检测具有DNA结合能力的分子、或评价DNA结合能力的方法。
本发明人等经过潜心研究,结果发现,在诸如RPA(重组酶聚合酶扩增)法那样可以在等温条件下扩增具有靶碱基序列的核酸片段的方法中,通过使反应系统中存在与具有特定的碱基序列或处于修饰状态下的核酸特异性结合的分子,能够特异性地仅检测出不与该分子结合的核酸片段,从而完成了本发明。
即,本发明的靶核酸的检测方法、核酸结合分子的检测方法、及核酸结合能力的评价方法、以及用于这些方法的试剂盒如下所述。
[1]一种靶核酸的检测方法,从碱基序列或修饰状态与所述靶核酸部分不同的非靶核酸中识别并检测靶核酸,
将所述非靶核酸中碱基序列或修饰状态与所述靶核酸不同的区域作为靶区域,将所述靶核酸中碱基序列或修饰状态与所述非靶核酸不同的区域作为对应靶区域,以供试核酸样本为模板,在与所述非靶核酸中的所述靶区域特异性结合的分子(其中,仅由核酸构成的分子除外)存在下,使用与所述靶核酸及所述非靶核酸双方杂交的引物,在所述分子能够与所述非靶核酸结合的温度条件下进行核酸扩增反应,基于有无扩增产物来检测所述靶核酸。
[2]根据所述[1]所述的靶核酸的检测方法,其中,在通过所述核酸扩增反应得到了扩增产物的情况下,所述供试核酸样本中包含所述靶核酸。
[3]根据所述[1]或[2]所述的靶核酸的检测方法,其中,在65℃以下的温度条件下进行所述核酸扩增反应。
[4]根据所述[1]~[3]中任一项所述的靶核酸的检测方法,其中,所述核酸扩增反应为等温核酸扩增反应。
[5]根据所述[1]~[4]中任一项所述的靶核酸的检测方法,其中,所述核酸扩增反应为重组酶聚合酶扩增法。
[6]根据所述[1]~[5]中任一项所述的靶核酸的检测方法,其中,所述靶核酸中的所述对应靶区域和所述非靶核酸中的所述靶区域的碱基序列不同。
[7]根据所述[6]所述的靶核酸的检测方法,其中,所述靶区域为基因突变的突变位点或基因多态性的多态性位点。
[8]根据所述[1]~[7]中任一项所述的靶核酸的检测方法,其中,所述分子为DNA链断裂活性缺失型Cas9蛋白质和gRNA的复合物。
[9]根据所述[8]所述的靶核酸的检测方法,其中,所述gRNA特异性识别并结合由与所述非靶核酸中的所述靶区域互补的碱基序列构成的DNA。
[10]根据所述[1]~[9]中任一项所述的靶核酸的检测方法,其中,所述供试核酸样本经亚硫酸氢盐处理,
所述对应靶区域和所述靶区域的甲基化状态存在差别,包含通过亚硫酸氢盐处理而在所述对应靶区域和所述靶区域之间产生差别的碱基。
[11]根据所述[1]~[7]中任一项所述的靶核酸的检测方法,其中,所述分子为RNA链断裂活性缺失型Cas13a蛋白质和gRNA的复合物。
[12]根据所述[11]所述的靶核酸的检测方法,其中,所述gRNA特异性识别并结合由与所述非靶核酸中的所述靶区域互补的碱基序列构成的RNA。
[13]根据所述[1]~[5]中任一项所述的靶核酸的检测方法,其中,所述靶核酸中的所述对应靶区域和所述非靶核酸中的所述靶区域的修饰状态不同。
[14]根据所述[13]所述的靶核酸的检测方法,其中,所述靶核酸中的所述对应靶区域为未经CpG甲基化修饰的区域,
所述非靶核酸中的所述靶区域为经CpG甲基化修饰的区域,
所述分子为CpG甲基化DNA结合蛋白质。
[15]一种用于检测靶核酸的试剂盒,用于所述[8]~[10]中任一项所述的靶核酸的检测方法,具有:
与所述靶核酸及所述非靶核酸双方杂交的引物、DNA链断裂活性缺失型Cas9蛋白质、及gRNA。
[16]一种用于检测靶核酸的试剂盒,用于所述[14]所述的靶核酸的检测方法,具有:
与所述靶核酸及所述非靶核酸双方杂交的引物、及CpG甲基化DNA结合蛋白质。
[17]根据所述[15]或[16]所述的用于检测靶核酸的试剂盒,其中,进一步包含重组酶、单链DNA结合蛋白质、及DNA聚合酶。
[18]一种用于检测靶核酸的试剂盒,用于所述[11]或[12]所述的靶核酸的检测方法,具有:
与所述靶核酸及所述非靶核酸双方杂交的引物、RNA链断裂活性缺失型Cas13a蛋白质、及gRNA。
[19]一种核酸结合分子的检测方法,是检测核酸结合分子的方法,
使用供试样本、核酸及与所述核酸杂交的引物进行核酸扩增反应,
在所述供试样本中包含所述核酸结合分子的情况下,通过所述核酸扩增反应不能得到扩增产物,在所述供试样本中不包含所述核酸结合分子的情况下,通过所述核酸扩增反应得到扩增产物。
[20]根据所述[19]所述的核酸结合分子的检测方法,其中,所述核酸结合分子为碱基序列特异性地与核酸结合的分子。
[21]根据所述[19]所述的核酸结合分子的检测方法,其中,所述核酸结合分子为CpG甲基化DNA结合蛋白质。
[22]一种用于检测核酸结合分子的试剂盒,用于所述[19]~[21]所述任一项的核酸结合分子的检测方法,具有:
核酸、及与所述核酸杂交的引物。
[23]一种核酸结合能力的评价方法,是评价供试物质对于核酸的结合能力的方法,
使用供试物质、作为评价所述供试物质的核酸结合能力的对象的核酸、及与所述核酸杂交的引物,在所述供试物质能够与所述核酸结合的温度条件下进行核酸扩增反应,在得到扩增产物的情况下,评价所述供试物质不具有对于所述核酸的结合能力,在未得到扩增产物的情况下,评价所述供试物质具有对于所述核酸的结合能力。
[24]根据所述[23]所述的核酸结合能力的评价方法,其中,所述供试物质为DNA链断裂活性缺失型Cas9蛋白质和gRNA的复合物。
[25]根据所述[23]所述的核酸结合能力的评价方法,其中,所述核酸为CpG甲基化DNA。
[26]一种用于评价核酸结合能力的试剂盒,用于所述[23]~[25]中任一项所述的核酸结合能力的评价方法,具有:
作为评价所述供试物质的核酸结合能力的对象的核酸、及与所述核酸杂交的引物。
通过本发明的靶核酸的检测方法,即使不需要PCR中所需的用于进行温度控制的热循环仪,也能够作为核酸扩增产物的阳性信号检测到靶核酸。
另外,通过本发明的用于检测靶核酸的试剂盒,能够更简便地实施所述靶核酸的检测方法,以检测靶核酸。
通过本发明的核酸结合分子的检测方法及核酸结合能力的评价方法,能够以核酸扩增产物的减少量为指标来检测并评价核酸结合能力。
另外,根据本发明的用于检测核酸结合分子的试剂盒及用于评价核酸结合能力的试剂盒,能够更简便地实施所述核酸结合分子的检测方法及核酸结合能力的评价方法。
附图说明
图1是示意性示出本发明的靶核酸的检测方法的原理中使用DNA链断裂活性缺失型Cas9蛋白质(dCas9)和gRNA作为靶区域特异性结合分子来检测基因突变的方案的图。
图2是示意性示出本发明的靶核酸的检测方法的原理中将RNA作为靶核酸通过RT-PCR来检测基因突变时的方法的图。使用RNA链断裂活性缺失型Cas13a蛋白质(dCas13a)和gRNA的复合物作为靶区域特异性结合分子。
图3是示出实施例1中所使用的293T细胞的KRAS基因的部分区域、和所使用的两种gRNA的碱基序列的图。
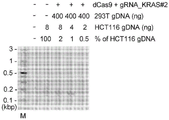
图4是实施例1中各种gRNA与dCas9共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图5是实施例2中各种gRNA与dCas9共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图6是示意性示出实施例2中在添加有gRNA_KRAS#2的反应液中所进行的RPA反应的图。
图7是示出实施例2中向gRNA_KRAS导入单碱基置换的突变而得到的gRNA_KRAS_mut的碱基序列的图。
图8是实施例2中gRNA_KRAS#2与dCas9共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图9是示出实施例3中所使用的HCT116细胞的CDKN2A(p16)基因的部分区域、和所使用的gRNA_p16_Gx5#2的碱基序列的图。
图10是实施例3中gRNA_KRAS或gRNA_p16_Gx5#2与dCas9共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图11是示意性示出实施例4中对CDKN2A(p16)基因进行的基因组编辑的图、及在添加有gRNA_mid2的反应液中进行的RPA反应的图。
图12是实施例4中gRNA_mid2与dCas9共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图13是实施例5中gRNA_KRAS#2与dCas9共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图14是实施例5中gRNA_KRAS#2与dCas9共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图15是实施例6中与MBD2蛋白质共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图16是实施例6中与MBD2蛋白质共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图17是实施例6中与MBD2蛋白质共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图18是实施例7中与LexA蛋白质或者dCas9共同进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图19是实施例8中将人NEAT1基因的mRNA(NEAT1-RNA)作为靶核酸,gRNA_NEAT1与dCas13a共同进行RT-PCR,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图20是实施例8中将人NEAT1基因的mRNA(NEAT1-RNA)作为靶核酸,gRNA_NEAT1_2与dCas13a共同进行RT-PCR,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
图21是实施例9中将Cis质粒或CisM质粒作为模板,在未IL-3处理或处理完毕的核提取液存在下进行RPA反应,并将得到的反应液进行琼脂糖电泳而分离出的条带的染色图像。
具体实施方式
<靶核酸的检测方法>
在本发明及本申请说明书中,“靶核酸”是指作为检测对象的核酸。作为靶核酸,只要其中规定了碱基序列,以足以能够设计供利用引物和聚合酶进行的核酸扩增反应使用的引物,则不受特别限定。具体而言,能够将单链DNA、双链DNA、单链RNA、RNA-DNA杂合体(单链RNA和单链DNA形成了双链的核酸)等作为靶核酸。另外,靶核酸也包括具有特定的修饰状态的核酸。该修饰状态可列举例如:甲基化、氟化、硫代磷酸酯化、二硫代磷酸酯化、糖基化、PEG(聚乙二醇)加成、肽加成等。
在本发明及本申请说明书中,“非靶核酸”表示具有与靶核酸相同的结构,但部分碱基序列或修饰状态与靶核酸不同的核酸。作为非靶核酸,只要其的碱基序列及修饰状态等结构与靶核酸的差异明显,且可以与靶核酸区分,则不受特别限定。例如,能够将经CpG甲基化修饰的核酸作为靶核酸,将由与靶核酸同源的碱基序列构成,但未经CpG甲基化修饰的核酸作为非靶核酸。
在本发明及本申请说明书中,“对应靶区域”表示靶核酸中与非靶核酸的结构不同的区域,“靶区域”表示非靶核酸中与靶核酸的结构不同的区域。靶核酸中的对应靶区域的存在位置相当于非靶核酸中的靶区域的存在位置。例如,在靶核酸和非靶核酸的结构的差异为碱基序列的差异的情况下,靶核酸中碱基序列与非靶核酸不同的位点(差异位点)为对应靶区域,非靶核酸中碱基序列与靶核酸不同的位点(差异位点)为靶区域。同理,在靶核酸和非靶核酸的结构的差异为修饰状态的差异的情况下,靶核酸中修饰状态与非靶核酸不同的位点(差异位点)为对应靶区域,非靶核酸中修饰状态与靶核酸不同的位点(差异位点)为靶区域。靶核酸中所含的对应靶区域可以为一个,也可以为两个以上。
在本发明及本申请说明书中,“靶碱基序列”表示靶核酸中包含对应靶区域的区域的碱基序列。靶碱基序列可以为仅由对应靶区域构成的碱基序列,也可以为包含对应靶区域的靶核酸的部分区域的碱基序列,还可以为靶核酸的全长的碱基序列。
需要指出,在本发明及本申请说明书中,“碱基序列相同”表示“碱基序列同源”,“碱基序列互补”表示“碱基序列彼此互补”。
在检测包含特定的碱基序列的核酸时,通过核酸扩增反应扩增包含该碱基序列的区域,并检测扩增产物,由此提高检测灵敏度。在利用这样的核酸扩增反应的检测方法中,不仅是由作为检测对象的碱基序列构成的核酸片段被扩增,有时与该碱基序列类似的碱基序列的核酸片段也被扩增。在核酸扩增反应中,通过抑制与作为检测对象的碱基序列类似的碱基序列的核酸片段的扩增来提高由作为检测对象的碱基序列构成的核酸片段的检测精度。
本发明的靶核酸的检测方法以供试核酸样本为模板,使用引物和聚合酶进行核酸扩增反应,检测作为核酸扩增产物的该供试核酸样本中的靶核酸。在本发明的靶核酸的检测方法中,在与非靶核酸中的靶区域结合、且不与靶核酸中的对应靶区域结合的分子(其中,仅由核酸构成的分子除外)存在下,进行该核酸扩增反应。下面,有时将“与非靶核酸中的靶区域结合、且不与靶核酸中的对应靶区域结合的分子(其中,仅由核酸构成的分子除外)”称为“靶区域特异性结合分子”。以靶区域特异性结合分子所结合的非靶核酸为模板的核酸扩增反应,通过与靶区域结合的靶区域特异性结合分子来抑制引物对于模板核酸的退火、及基于聚合酶的核酸延伸反应。由此,抑制非靶核酸的核酸扩增,增加靶核酸的扩增产物在扩增产物中所占的比例,提高靶核酸的检测精度。
具体而言,本发明的靶核酸的检测方法从非靶核酸中识别并检测靶核酸,以供试核酸样本为模板,在靶区域特异性结合分子存在下,使用与靶核酸及非靶核酸双方杂交的引物进行核酸扩增反应,基于有无扩增产物来检测靶核酸。靶核酸通过该核酸扩增反应扩增,因此,在供试核酸样本中包含靶核酸的情况下,通过该核酸扩增反应得到扩增产物。即,供试核酸样本中的靶核酸能够作为该核酸扩增反应的扩增产物被检测到。
在本发明中,靶核酸和非靶核酸只要在相同的结构中具有彼此结构不同的区域,则不受特别限定。本发明的靶核酸的检测方法甚至可以检测核酸结构的微小的差异,因此,在靶核酸和非靶核酸的结构的差异为碱基序列的差异的情况下,靶核酸中的对应靶区域优选为20碱基以下的区域,更优选为10碱基以下的区域,进一步优选为1~5碱基的区域,更进一步优选为1或2碱基的区域。
在本发明及本申请说明书中,“供试核酸样本”是指包含核酸的样本,其供靶核酸的检测使用。在本发明中,针对供试核酸样本中的核酸,检测是否包含靶核酸。供试核酸样本只要为包含核酸的样本,则不受特别限定。例如,可以将从动物、植物、微生物、病毒、培养细胞等中提取的核酸作为供试核酸样本。另外,化学合成的核酸及有意经修饰处理的核酸也可以作为供试核酸样本。可以通过苯酚/氯仿法等公知的方法从细胞等中提取核酸。
供试核酸样本中所含的核酸只要为能够作为使用该样本的核酸扩增反应的模板的核酸,则不受特别限定。由于能够利用以通用的DNA为模板的核酸扩增反应,因此,供本发明的靶核酸的检测方法使用的供试核酸样本中的核酸优选DNA,更优选双链DNA。供本发明的靶核酸的检测方法使用的供试核酸样本中的核酸还可以为RNA。在供试核酸样本中的核酸为RNA的情况下,可以将以该RNA为模板的逆转录反应作为本发明的靶核酸的检测方法中的核酸扩增反应,另外,也可以预先通过逆转录反应合成cDNA之后,再供核酸扩增反应使用。
在本发明中,用于扩增核酸扩增反应中使用的靶核酸的引物与靶核酸及非靶核酸双方杂交。因而,在不存在靶区域特异性结合分子时,供试核酸样本中所含的非靶核酸也会被使用该引物的核酸扩增反应扩增。在本发明中,通过在靶区域特异性结合分子存在下进行核酸扩增反应,抑制了非靶核酸的核酸扩增。需要指出,在本发明中,用于扩增核酸扩增反应中使用的靶核酸的引物只要在进行核酸扩增反应的条件下与靶核酸及非靶核酸双方杂交即可,可以为与靶核酸中碱基序列及修饰状态与非靶核酸相同的部分区域进行杂交的引物,也可以为与靶核酸中碱基序列或修饰状态与非靶核酸不同的部分区域进行杂交的引物。
在本发明中,用于扩增核酸扩增反应中使用的靶核酸的引物是核苷酸通过磷酸二酯结合的寡核苷酸或其修饰体。这些引物可以仅由DNA及RNA等天然型核苷酸(天然存在的核苷酸)构成;也可以仅由人工核苷酸构成,该人工核苷酸是通过改造天然型核苷酸而可以与天然型核苷酸进行磷酸二酯结合的核苷酸;还可以为包含天然型核苷酸和人工核苷酸两者的嵌合分子。作为人工核苷酸,可列举:天然型核苷酸的侧链等被氨基等官能团修饰的核苷酸、核糖骨架的2’位的羟基被甲氧基、氟基、甲氧基乙基等取代的核苷酸、硫代磷酸酯型核苷酸(磷酸基的氧原子被硫原子取代)、吗啉基(Morpholino)型核苷酸(核糖、脱氧核糖被吗啉环取代)、BNA(Bridged nucleic acid,桥接核酸)、HNA(Hexitol NucleicAcid,己醇核酸)、LNA(Locked Nucleic Acid,锁核酸)、PNA(Peptide Nucleic Acid,肽核酸)、TNA(Threose nucleic acid,苏糖核酸)、GNA(Glycerol nucleic acid,甘油核酸)、CeNA(Cyclohexenyl nucleic acid,环己烯基核酸)等。另外,作为寡核苷酸的修饰体,可列举例如被有助于扩增产物检测的标记物质修饰后的修饰体。作为该标记物质,可列举:荧光物质、放射性同位素、化学发光体、酶、抗体等。以不会抑制引物的核酸扩链反应的方式结合标记物质。
在本发明中,核酸扩增反应中使用的引物可以为一种,也可以为两种,还可以为三种以上。能够根据所实施的核酸扩增反应适当确定。
例如,能够使用由正向引物和反向引物构成的引物组。例如,靶碱基序列为靶核酸的全长或其部分区域的碱基序列,可以设置为在对应靶区域的5’侧和3’侧均存在结构与非靶核酸相同的区域,具体而言,存在碱基序列及修饰状态与非靶核酸相同的区域。能够使用和位于靶碱基序列的5’端且结构与非靶核酸相同的区域进行杂交的正向引物、及和位于靶碱基序列的3’端且结构与非靶核酸相同的区域进行杂交的、用于扩增该靶碱基序列的核酸片段的引物组。
在本发明的靶核酸的检测方法中,核酸扩增反应在靶区域特异性结合分子能够与非靶核酸结合的温度条件下进行。该核酸扩增反应只要在整个反应工序中、在靶区域特异性结合分子能够与非靶核酸结合的温度条件下进行即可,也可以具备PCR法等的温度循环。
在本发明的靶核酸的检测方法中,该核酸扩增反应优选在65℃以下的温度条件下进行。通过在65℃以下的温度条件下进行,能够将蛋白质等耐热性较低的分子用作靶区域特异性结合分子。需要指出,通过使用由耐热蛋白质构成的靶区域特异性结合分子,并使用耐热性聚合酶等,也可以在超过65℃、例如超过65℃且小于等于95℃左右的温度范围内进行核酸扩增反应。
由于不需要PCR中所需的用于进行温度控制的热循环仪,因此本发明的靶核酸的检测方法中的核酸扩增反应优选在等温条件下进行。需要指出,“等温条件下”表示在反应期间相对于所设置的温度保持在±3℃或±1℃的温度范围内。
作为等温条件,只要能够进行核酸扩增反应,则没有特别限制,例如DNA聚合酶的最佳温度中包含的一定的温度。作为等温条件,可列举例如:10℃以上、15℃以上、20℃以上、25℃以上、或30℃以上的一定的温度,及65℃以下、60℃以下、50℃以下、45℃以下、或40℃以下的一定的温度。另外,等温条件可以为例如10℃~65℃的范围中包含的一定的温度、15℃~50℃的范围中包含的一定的温度、20℃~45℃的范围中包含的一定的温度、或30℃~45℃的范围中包含的一定的温度。在本说明书中,“等温下孵育”、“等温条件下保温”、“等温下反应”等术语表示反应期间相对于设置的温度保持在±7℃、±5℃、±3℃或±1℃的温度范围内。
在本发明的靶核酸的检测方法中,在等温条件下进行的核酸扩增反应不受特别限定,能够进行公知的等温核酸扩增反应或其变形反应。作为公知的等温核酸扩增反应,可列举:RPA法(专利文献1)、LAMP(环介导等温扩增)法(非专利文献3)、MDA(多重置换扩增)法(非专利文献4)、使用逆转录酶的逆转录反应、NASBA(酸序列依赖性扩增)法等。
RPA法是指如下方法:将与靶碱基序列的一个末端区域杂交的引物、和与另一个末端区域杂交的引物分别与重组酶形成复合物,使该复合物与模板核酸接触,与单链DNA结合蛋白质(single-strand binding protein(SSB))形成复制叉后,通过DNA聚合酶合成双链核酸。所使用的引物可以与PCR用引物相同地进行设计。所使用的SSB及DNA聚合酶、制备反应液的缓冲液等可以从RPA法中常用的物质及其变体中适当选用。另外,也可以利用“TwistAmp(注册商标)”(TwistDx公司制)等市售的RPA用试剂盒。作为反应条件,可以在RPA法中常用的条件或对其进行改变后的条件下适当进行。
LAMP法是指如下方法:从靶碱基序列中选择六个区域,使用组合的四种引物(FIP引物、F3引物、BIP引物、及B3引物),利用链置换反应进行扩增。F3引物相当于本发明中的“与靶碱基序列的5’端区域杂交的正向引物”,B3引物相当于本发明中的“与靶碱基序列的3’端区域杂交的反向引物”。这些引物能够使用例如LAMP法引物设计辅助软件“PrimerExplorer”(荣研化学公司制)来设计。所使用的链置换型DNA聚合酶及制备反应液的缓冲液等能够从LAMP法中常用的物质及其变体中适当选用,作为反应条件,也可以在LAMP法中常用的条件或对其进行改变后的条件下适当进行。
MDA法通常是指使用随机引物,通过Phi29 DNA聚合酶从引物所结合的位置来合成单链核酸的方法。Phi29 DNA聚合酶自身具有类解旋酶活性,即使在核酸合成过程中遇到双链核酸(例:模板DNA和引物的缔合体)的情况下,也能够解开该双链,同时进行DNA合成反应。已知通过30℃、12小时的反应合成70,000碱基长以上。通过使用随机引物及靶碱基序列附近的引物,能够扩增靶碱基序列。所使用的引物及DNA聚合酶、制备反应液的缓冲液等能够从MDA法中常用的物质及对其进行改变后的变体中适当选用,作为反应条件,能够在MDA法中常用的条件或将对其进行改变后的条件下适当进行。
NASBA法是指以RNA为模板并使用AMV逆转录酶、RNase H、及T7 RNA聚合酶和两种引物(F引物和5’侧具有T7启动子序列的R引物)所进行的等温核酸扩增反应。首先,R引物(相当于本发明中“与靶碱基序列的3’端区域杂交的反向引物”)对模板RNA进行退火,通过逆转录酶合成cDNA,得到的RNA/DNA链的RNA链部分被RNase H消化。F引物(相当于本发明中“与靶碱基序列的5’端区域杂交的正向引物”)对剩余的cDNA进行退火,通过逆转录酶合成cDNA,以得到的双链cDNA为模板,通过T7 RNA聚合酶的转录反应合成反义的单链RNA。F引物对该单链RNA进行退火,通过逆转录酶得到RNA/DNA链之后,RNA链部分被RNase H消化。R引物对剩余的cDNA进行退火,通过逆转录酶得到双链cDNA,将其作为模板,通过T7 RNA聚合酶的转录反应合成反义的单链RNA。如上反复操作,对作为靶核酸的RNA的反义单链RNA进行扩增。所使用的引物及逆转录酶、RNase H、及T7 RNA聚合酶、制备反应液的缓冲液等从ASBA法中常用的物质及其变体中适当选用,作为反应条件,也可以在NASBA法中常用的条件或对其进行改变后的条件下适当进行。
逆转录反应是指以RNA为模板,通过逆转录酶来合成模板RNA的反义cDNA链的反应。通过RT-PCR法,能够高灵敏度地检测靶RNA,RT-PCR法与以得到的cDNA为模板的PCR进行组合。所使用的引物及逆转录酶、DNA聚合酶、制备反应液的缓冲液等能够从逆转录反应中常用的物质及其变体中适当选用。
RPA法、LAMP法、MDA法、NASBA法、及逆转录反应中的反应温度只要在所使用的酶显示酶活性的温度范围内,则不受特别限定。另外,在等温核酸扩增反应中,得到的扩增产物依赖于反应时间。因而,反应时间能够根据目标扩增产物的量来适当设置。例如,在RPA法和LAMP法中,能够设置为5分钟~6小时,优选设置为5分钟~1小时,更优选设置为5~30分钟。在MDA法中,能够设置为5分钟~32小时,优选设置为5分钟~24小时,更优选设置为5分钟~16小时。在NASBA法中,能够设置为5分钟~6小时,优选设置为5分钟~3小时,更优选设置为5分钟~1.5小时。在逆转录反应中,能够设置为5分钟~3小时,优选设置为5分钟~1小时,更优选设置为5~30分钟。
在本发明的靶核酸的检测方法中,核酸扩增反应的反应系统中含有的靶区域特异性结合分子只要为能够与非靶核酸特异性结合以抑制核酸扩增的分子,则不受特别限定,其中,非靶核酸为可以通过核酸扩增反应扩增的除靶核酸以外的核酸,该核酸扩增反应使用靶核酸的扩增中使用的引物。
在靶核酸和非靶核酸的结构的差异为碱基序列的差异的情况下,作为靶区域特异性结合分子,能够使用特异性识别并结合非靶核酸的靶区域的碱基序列的分子。靶核酸为DNA时,该分子可列举例如DNA链断裂活性缺失型Cas9(dCas9)蛋白质和gRNA(guide RNA)的复合物(CRISPR复合物)。靶核酸为RNA时,该分子可列举例如构成与RNA结合的CRISPR复合物的Cas13a(Cas13a)(非专利文献7)的RNA链断裂活性缺失型(dCas13a)蛋白质和gRNA的复合物(CRISPR复合物)。特别是报道了Cas13a/gRNA复合物能够识别RNA的1个碱基的差别(非专利文献8)。因而,在本发明的靶核酸的检测方法中,通过使用Cas13a/gRNA复合物作为靶区域特异性结合分子,能够将仅1个碱基不同的靶核酸和非靶核酸进行区分,从而检测靶核酸。
作为dCas9蛋白质,可列举例如:向Cas9蛋白质中的核酸酶结构域导入突变,保持DNA结合能力的同时,使核酸酶活性灭活而得到的蛋白质。作为该蛋白质,可列举例如:至少导入了来自化脓性链球菌(Streptococcus pyogenes)的野生型Cas9蛋白质中的RuvC核酸酶结构域内的D10A(将第10个天冬氨酸置换为丙氨酸的点突变)和HNH核酸酶结构域内的H840A(将第840个组氨酸置换为丙氨酸的点突变)这两个点突变中至少一个点突变而得到的突变体。另外,也可以使用向各种Cas9蛋白质中导入相当于来自化脓性链球菌的Cas9蛋白质的D10A及H840A的两个点突变而得到的突变体。作为dCas9蛋白质,还可以使用“EnGenSpy dCas9(SNAP-tag)”(New England Biolabs公司制)等市售的蛋白质。
Cas13a也被称为C2c2,其是构成靶向RNA的CRISPR复合物的蛋白质,具有两个HEPN结构域(Higher Eukaryotes and Prokaryotes Nucleotide-binding domain)和一个核酸酶结构域。作为dCas13a,可列举例如:向Cas13a蛋白质中的核酸酶结构域导入突变,保持RNA结合能力的同时,使核酸酶活性灭活而得到的蛋白质。作为该蛋白质,可列举例如:导入来自韦德纤毛菌(Leptotrichia wadei)的野生型Cas13a蛋白质中的核酸酶结构域内的R474A(将第474个精氨酸置换为丙氨酸的点突变)和R1046A(将第1046个精氨酸置换为丙氨酸的点突变)这两个点突变中的至少一个点突变而得到的突变体(非专利文献10)。另外,也可以使用向各种Cas13a蛋白质中导入相当于来自韦德纤毛菌的Cas13a蛋白质的R474A及R1046A的两个点突变而得到的突变体。
在使用Cas9的情况,gRNA包含来自细菌的crRNA(CRISPR RNA)及tracrRNA(trans-activating CRISPR RNA)。crRNA是包含由与tracrRNA的一部分互补的碱基序列构成的区域(与tracrRNA结合的区域)、和由与特异性识别并结合的碱基序列互补的碱基序列构成的区域(靶核酸结合区域)的单链RNA。靶核酸为DNA时,靶核酸结合区域为DNA结合区域,靶核酸为RNA时,靶核酸结合区域为RNA结合区域。tracrRNA是具有由与crRNA的一部分互补的碱基序列构成的区域(与crRNA结合的区域)、并在该区域内与crRNA杂交而形成发夹结构的单链RNA。crRNA和tracrRNA可以分别为独立的单链RNA,也可以为两者通过适当的RNA接头连接后的单链RNA(sgRNA)。它们可以通过与基因组编辑常用的方法相同的方法来设计。另一方面,Cas13a不需要tracrRNA,仅将crRNA用作gRNA。因此,也可以将单链RNA作为gRNA发挥功能。
本发明中使用的gRNA中的靶核酸结合区域(DNA结合区域或RNA结合区域)是与非靶核酸中的靶区域结合的区域,是由与该靶区域的碱基序列互补的碱基序列构成的区域。通过在非靶核酸的靶区域上结合CRISPR复合物,即,当靶核酸为DNA时结合dCas9蛋白质和gRNA的复合物,当靶核酸为RNA时结合dCas13a蛋白质和gRNA的复合物,从而抑制引物对于模板核酸的退火、及基于聚合酶的核酸扩链反应,进而抑制非靶核酸的扩增。
在使用Cas9的情况下,gRNA中的靶核酸结合区域(DNA结合区域)通常选择其后的紧邻PAM序列的区域的碱基序列。PAM序列是被dCas9蛋白质识别的序列,依赖于所用的Cas9蛋白质等来确定。dCas9蛋白质所结合的DNA结合区域的碱基长不受特别限定,例如可以制成15~30碱基长左右,优选18~23碱基长。
在使用Cas13a的情况下,gRNA中的靶核酸结合区域(RNA结合区域)通常选择其后紧邻PFS序列的区域的碱基序列。PFS序列是被dCas13a蛋白质识别的序列,依赖于所用的Cas13a蛋白质等来确定。dCas13a蛋白质所结合的RNA结合区域的碱基长不受特别限定,例如可以制成15~40碱基长左右,优选20~30碱基长。
反应液中的dCas9蛋白质或Cas13a蛋白质的量不受特别限定,例如可以设为每20μL反应液200ng以下,优选10~80ng。另外,反应液中的gRNA的量不受特别限定,例如可以设为50nM以下,优选2.5~20nM。
作为靶区域特异性结合分子,除CRISPR复合物以外,还可以使用不与靶核酸中的对应靶区域结合、但与非靶核酸中的靶区域特异性结合的、具备DNA序列特异性结合能力的蛋白质、及具备RNA序列特异性结合能力的蛋白质等分子。作为这样的蛋白质,可列举例如:识别并特异性结合特定的DNA序列的锌指(zinc finger)蛋白质、TAL效应子(transcription-activator like effector:TALE)、LexA蛋白质等转录控制因子等。另外,还可列举例如:识别并特异性结合特定的RNA序列的PPR(pentatricopeptide repeat)蛋白质等。
在非靶核酸为修饰状态与靶核酸不同的核酸的情况下,当非靶核酸中的靶区域为经过特定的修饰的状态、且靶核酸中的对应靶区域为未经过该修饰的状态时,可以将与经过该修饰的核酸特异性结合的分子用作靶区域特异性结合分子。例如,当非靶核酸中的靶区域经CpG甲基化、且靶核酸中的对应靶区域未经CpG甲基化时,可以将CpG甲基化DNA结合蛋白质用作靶区域特异性结合分子。作为CpG甲基化DNA结合蛋白质,能够适当使用具有MBD(Methyl-CpG-binding domain)的蛋白质(MBD蛋白质)及识别经甲基化的CpG的抗体等公知的蛋白质。反应液中的MBD2蛋白质的量不受特别限定,例如能够采用每20μL反应液10μg以下,优选0.05~2μg。
在非靶核酸为修饰状态和靶核酸不同的核酸的情况下,当非靶核酸中的靶区域为未经过特定的修饰的状态、且靶核酸中的对应靶区域为经过该修饰的状态时,可以将与未经过该修饰的核酸特异性结合、且不与经过该修饰的核酸结合的分子用作靶区域特异性结合分子。例如,当非靶核酸中的靶区域未经过CpG甲基化、且靶核酸中的对应靶区域经过CpG甲基化时,可以将与未经过CpG甲基化的DNA特异性结合、不与经过CpG甲基化的核酸结合的蛋白质用作靶区域特异性结合分子。作为与未经过CpG甲基化的DNA特异性结合的蛋白质,可列举例如使酶活性灭活而得到的CpG甲基转移酶突变体。另外,如果反应液中不存在能够作为甲基供体的物质,则CpG甲基转移酶的野生型蛋白质也能够用作该蛋白质。反应液中的与未经过CpG甲基化的DNA特异性结合的蛋白质的量不受特别限定,例如,能够采用每20μL反应液10μg以下,优选0.05~2μg。
在非靶核酸为两种以上的情况下,可以向核酸扩增反应的反应系统中添加针对每种非靶核酸的靶区域特异性结合分子。
在本发明的靶核酸的检测方法中,在通过核酸扩增反应得到了扩增产物的情况下,检测到靶核酸,判断供试核酸样本中包含靶核酸。另一方面,在未得到扩增产物的情况下,未检测到靶核酸,判断供试核酸样本中不包含靶核酸。由于靶核酸被作为扩增产物这一阳性信号来检测,因此,能够以足够的检测灵敏度检测靶核酸。
本发明的靶核酸的检测方法适合用于例如基因突变的检测等。将对应靶区域作为所检测的目标突变位点,以将靶核酸或非靶核酸作为模板时得到包含该突变位点的区域的核酸扩增产物的方式,设计用于核酸扩增反应的引物。例如,将突变位点为突变型的核酸(突变型核酸)作为靶核酸,将突变位点为野生型的核酸(野生型核酸)作为非靶核酸,将与突变位点为野生型的碱基序列的核酸特异性结合、但不与突变位点为突变型的碱基序列的核酸结合的分子(其中,仅由核酸构成的分子除外)作为靶区域特异性结合分子。在该靶区域特异性结合分子存在下,且在该靶区域特异性结合分子不会丧失与野生型核酸的结合活性的温度条件下,进行核酸扩增反应。在供试的供试核酸样本中包含突变型核酸的情况下,得到扩增产物,在供试核酸样本中不包含突变型核酸的情况下,不能得到扩增产物。可以根据有无扩增产物来检测作为靶核酸的突变型核酸。因而,在该方法中,不仅能够检测纯合突变,也能够检测杂合突变。
图1是示意性示出使用dCas9蛋白质和gRNA作为靶区域特异性结合分子来检测基因突变时的方法的图。图中,左侧是以野生型核酸为模板的扩增反应,右侧是以突变型核酸为模板的扩增反应。
图2是示意性示出将RNA作为靶核酸并通过RT-PCR来检测基因突变时的方法的图。作为靶区域特异性结合分子,使用与野生型RNA结合的dCas13a蛋白质/gRNA复合物。图中,右侧是以突变型RNA为模板的扩增反应,左侧是以野生型RNA为模板的扩增反应。由于野生型RNA上结合dCas13a蛋白质/gRNA复合物,因此,抑制基于逆转录酶的cDNA扩链,从而不能得到PCR的扩增产物。突变型RNA通过RT-PCR得到扩增产物。
本发明的靶核酸的检测方法也适合例如基因多态性的检测。将靶区域作为所检测的目标多态性位点,以将靶核酸或非靶核酸作为模板时得到包含该多态性位点的区域的核酸扩增产物的方式,设计核酸扩增反应中使用的引物。例如,将多态性位点为靶基因型的核酸作为靶核酸,将多态性位点为靶基因型以外的基因型的核酸作为非靶核酸,将与多态性位点为靶基因型以外的基因型的核酸特异性结合的分子(其中,仅由核酸构成的分子除外)作为靶区域特异性结合分子。在该靶区域特异性结合分子存在下,且在该靶区域特异性结合分子不会丧失与非靶核酸的结合活性的温度条件下,进行核酸扩增反应。在供试的供试核酸样本中包含所检测的目标基因型核酸的情况下,得到扩增产物,在供试核酸样本中不包含该基因型核酸的情况下,不能得到扩增产物。能够通过有无扩增产物来检测作为靶核酸的特定的基因型的基因多态性。该方法同样也不仅能够检测纯合多态性,也能够检测杂合多态性。
DNA的甲基化分析具有利用亚硫酸氢盐处理的方法。在甲基化碱基不脱氨基、非甲基化碱基脱氨基的适当的反应时间,对作为分析对象的DNA进行亚硫酸氢盐处理,然后进行基于聚合酶的核酸扩增反应,得到扩增产物,扩增产物中,甲基化碱基是原来的碱基,非甲基化碱基被脱氨基而转化为其它碱基。例如,胞嘧啶通过脱氨基转化为尿嘧啶,因此,在扩增产物中,甲基化胞嘧啶为胞嘧啶,非甲基化胞嘧啶为胸腺嘧啶。
在对应靶区域和靶区域的甲基化状态存在差别的情况下,通过亚硫酸氢盐处理,使对应靶区域和靶区域之间碱基产生差别。所以,通过使用经亚硫酸氢盐处理的核酸作为供试核酸样本,能够通过本发明的靶核酸的检测方法来分析核酸中的特定的碱基有无甲基化。可以将作为分析对象的碱基经甲基化的核酸作为靶核酸,也可以将作为分析对象的碱基未经过甲基化的核酸作为靶核酸。
例如,在分析特定的胞嘧啶有无甲基化的情况下,当将作为分析对象的胞嘧啶经甲基化的核酸进行了亚硫酸氢盐处理而得到的核酸作为靶核酸时,将靶核酸中包含作为分析对象的胞嘧啶(甲基化胞嘧啶)的部分区域作为对应靶区域,将该作为分析对象的胞嘧啶未经过甲基化的核酸进行了亚硫酸氢盐处理而得到的核酸作为非靶核酸。在该情况下,非靶核酸中的作为分析对象的胞嘧啶通过亚硫酸氢盐处理转化为尿嘧啶。将包含该尿嘧啶的部分区域作为靶区域。在供试核酸样本中包含作为分析对象的胞嘧啶为甲基化胞嘧啶的核酸的情况下,通过在靶区域特异性结合分子存在下进行核酸扩增反应,得到该碱基为胞嘧啶的核酸扩增产物。在供试核酸样本中所含的核酸全部的作为分析对象的胞嘧啶均为非甲基化胞嘧啶的情况下,通过在靶区域特异性结合分子存在下进行核酸扩增反应,不能得到扩增产物。
另外,在将作为分析对象的胞嘧啶未经过甲基化的核酸进行了亚硫酸氢盐处理而得到的核酸作为靶核酸的情况下,将靶核酸中的包含作为分析对象的胞嘧啶(非甲基化胞嘧啶)的部分区域作为对应靶区域,将该作为分析对象的胞嘧啶经甲基化的核酸进行了亚硫酸氢盐处理而得到的核酸作为非靶核酸。在该情况下,非靶核酸中的作为分析对象的胞嘧啶经过了甲基化,因此,即使亚硫酸氢盐处理后,其仍保持为甲基化胞嘧啶。将包含该甲基化胞嘧啶的部分区域作为靶区域。在供试核酸样本中包含作为分析对象的胞嘧啶为非甲基化胞嘧啶的核酸的情况下,通过在靶区域特异性结合分子存在下进行核酸扩增反应,得到该碱基为胸腺嘧啶的核酸扩增产物。在供试核酸样本中包含的核酸全部的作为分析对象的胞嘧啶均为甲基化胞嘧啶的情况下,通过在靶区域特异性结合分子存在下进行核酸扩增反应,不能得到扩增产物。
需要指出,在上述各种方法中,作为与靶区域特异性结合、且不与对应靶区域结合的靶区域特异性结合分子,能够使用例如CRISPR复合物等,在CRISPR复合物中,gRNA的靶核酸结合区域(DNA结合区域或RNA结合区域)为与靶区域互补的碱基序列。
通过核酸扩增反应得到的靶核酸的扩增产物能够通过检测PCR等核酸扩增反应的扩增产物时常用的各种方法来检测。作为该检测方法,可列举例如:通过溴化乙锭等对利用琼脂糖凝胶电泳分级的条带进行染色的方法、嵌入剂法、熔融曲线分析法。嵌入剂法是指利用Cybergreen等荧光性嵌入剂与所生成的双链DNA结合而发出荧光的方法,荧光强度随着扩增产物的增加而提高。通过向反应液照射激发光并测定荧光强度,能够检测扩增产物的生成量。另外,在使用被标记物质修饰的引物的情况下,能够以该标记物质为指标进行检测。在使用被荧光物质标记的引物的情况下,通过柱层析等从核酸扩增反应后的反应液中除去未反应的引物之后,测定荧光强度,由此能够检测扩增产物。
通过将本发明的靶核酸的检测方法中使用的试剂等制成试剂盒,能够更简便地进行该方法。在本发明的靶核酸的检测方法中,作为用于使用dCas9蛋白质及gRNA作为靶区域特异性结合分子的方法的试剂盒,优选其具有与靶核酸及非靶核酸杂交的引物、dCas9蛋白质、及gRNA。在本发明的靶核酸的检测方法中,作为用于使用dCas13a蛋白质及gRNA作为靶区域特异性结合分子的方法的试剂盒,优选其具有与靶核酸及非靶核酸杂交的引物、dCas13a蛋白质、及gRNA。另外,作为用于使用CpG甲基化DNA结合蛋白质作为靶区域特异性结合分子的方法的试剂盒,优选其具有与靶核酸及非靶核酸双方杂交的引物、及CpG甲基化DNA结合蛋白质。另外,作为用于使用转录控制因子作为靶区域特异性结合分子的方法的试剂盒,优选其具有与靶核酸及非靶核酸双方杂交的引物、及转录控制因子。
优选本发明的用于检测靶核酸的试剂盒中还包含用于核酸扩增反应的酶、用于制备反应液的浓缩缓冲液、dNTP等。例如,通过RPA法进行核酸扩增反应的方法中使用的试剂盒优选进一步包含重组酶、SSB、及DNA聚合酶。
本发明的用于检测靶核酸的试剂盒优选进一步包括使用说明书,该使用说明书中记录了用于使用该试剂盒来进行本发明的靶核酸的检测方法的文字。该使用说明书也可以记载于收纳该试剂盒的容器的表面。
<核酸结合分子的检测方法>
通过在作为模板的核酸上结合一些物质,抑制引物对于模板核酸的退火及基于聚合酶的核酸扩链反应,利用该反应,能够检测核酸结合分子(具有核酸结合能力的分子)。
即,本发明的核酸结合分子的检测方法使用供试样本、核酸、及与所述核酸杂交的引物来进行核酸扩增反应。在供试样本中包含与用作模板的核酸结合的核酸结合分子的情况下,不能通过核酸扩增反应得到扩增产物。另一方面,在供试样本中不包含与该核酸结合的核酸结合分子的情况下,通过核酸扩增反应得到扩增产物。即,基于核酸扩增反应后的核酸扩增产物的量,能够检测该供试样本中所存在的核酸结合分子。
作为供本发明的核酸结合分子的检测方法使用的供试样本,只要其为有望存在核酸结合分子的样本,则不受特别限定。例如,能够使用动物及植物的组织或细胞的提取液(裂解液)、培养细胞的细胞提取液、土壌等从自然界采集的样本、及其粗精制物等。需要指出,组织及细胞的提取液能够通过常规方法来制备。
本发明的核酸结合分子的检测方法中所检测的核酸结合分子不受特别限定,优选为肽、蛋白质、或低分子化合物,也可以为核酸和蛋白质等其它分子的复合物。另外,该核酸结合分子可以为未经鉴定的物质。例如,通过将如各种细胞的细胞提取液及核提取液那样含有各种物质的物品作为供试样本,能够考察该供试样本中是否包含有核酸结合分子。另外,本发明的核酸结合分子的检测方法中所检测的核酸结合分子的核酸结合能力不受特别限定,可以为识别并特异性结合特定的碱基序列的碱基序列特异性核酸结合分子,也可以为识别并结合特定的修饰状态的核酸结合分子,还可以为不受碱基序列或修饰状态的影响,广泛与全部核酸结合的核酸结合分子。
本发明的核酸结合分子的检测方法中使用的核酸被用作核酸扩增反应的模板,其只要能够作为核酸扩增反应的模板,则不受特别限定,优选DNA或RNA。该核酸可以规定碱基序列,也可以不规定碱基序列。另外,该核酸可以仅由一种核酸构成,也可以如由细胞中提取及精制的DNA那样为多种核酸的混合物。例如,在检测不识别碱基序列则与核酸结合的核酸结合分子的情况下,作为模板的核酸可以为碱基序列不确定的核酸或多种核酸的混合物。
例如,在检测识别并结合特定的碱基序列的序列特异性核酸结合分子的情况下,将具有该特定的碱基序列的核酸作为模板,使用以与模板核酸中的该特定的碱基序列的上游(5’侧)杂交方式设计的引物,以在核酸扩增反应中对模板核酸中包含该特定的碱基序列的区域进行扩增。引物能够基于作为模板的核酸的碱基序列通过常规方法来设计合成。
在检测与处于特定的修饰状态的核酸特异性结合的核酸结合分子的情况下,将处于该特定的修饰状态的核酸作为模板。该修饰状态可列举例如:甲基化、氟化、硫代磷酸酯化、二硫代磷酸酯化、糖基化、PEG(聚乙二醇)加成、肽加成等。
作为本发明的核酸结合分子的检测方法中进行的核酸扩增反应,可列举与本发明的靶核酸的检测方法中进行的所述核酸扩增反应相同的反应。在所检测的目标核酸结合分子为蛋白质等耐热性较低的分子的情况下,核酸扩增反应优选在65℃以下的温度范围进行。相反地,在想要检测耐热性的核酸结合分子的情况下,使用耐热性聚合酶,在超过65℃且小于等于100℃的温度范围内进行核酸扩增反应。另外,作为本发明的核酸结合分子的检测方法中进行的核酸扩增反应,不需要PCR中所需的用于进行温度控制的热循环仪,因此,优选RPA法、LAMP法、MDA法、NASBA法等在等温条件下进行的核酸扩增反应。
例如,使用供试样本、核酸、及与所述核酸杂交的引物进行核酸扩增反应,将得到的核酸扩增产物量与除不添加供试样本之外在相同的条件下进行核酸扩增反应而得到的核酸扩增产物量进行比较。在供试样本中包含与用作模板的核酸结合的核酸结合分子的情况下,在该供试样本存在下得到的核酸扩增产物量比不存在该供试样本时得到的核酸扩增产物量少。相反地,在供试样本存在下得到的核酸扩增产物量大于等于不存在供试样本时得到的核酸扩增产物量的情况下,该供试样本中不存在与用作模板的核酸结合的核酸结合分子。
通过将本发明的核酸结合分子的检测方法中使用的试剂等制成试剂盒,能够更简便地进行该方法。例如,作为使用该方法的用于检测核酸结合分子的试剂盒,优选其含有作为模板的核酸、及与该核酸杂交的引物。此外,优选还包含用于核酸扩增反应的酶、用于制备反应液的浓缩缓冲液、及dNTP等。例如,通过RPA法进行核酸扩增反应的方法中使用的试剂盒优选进一步包含重组酶、SSB及DNA聚合酶。
本发明的用于检测核酸结合分子的试剂盒还优选进一步包含使用说明书,使用说明书上记载了用于使用该试剂盒进行本发明的核酸结合分子的检测方法的文字。该使用说明书也可以记载于收纳该试剂盒的容器的表面。
<核酸结合能力的评价方法>
通过在作为模板的核酸上结合一些物质,抑制引物对于模板核酸的退火及基于聚合酶的核酸扩链反应,通过该反应,能够检测核酸结合分子的核酸结合能力。
即,本发明的核酸结合能力的评价方法使用供试物质、作为评价该供试物质的核酸结合能力的对象的核酸、及与该核酸杂交的引物来进行核酸扩增反应。在供试物质具备对于用作模板的核酸的结合能力的情况下,不能通过核酸扩增反应得到扩增产物。另一方面,在供试物质不具备对于该核酸的结合能力的情况下,通过核酸扩增反应得到扩增产物。即,基于核酸扩增反应后的核酸扩增产物的量,能够评价该供试物质对于用作模板的核酸的结合能力。
本发明的核酸结合能力的评价方法中作为评价的对象的供试物质不受特别限定,优选为肽、蛋白质或低分子化合物。此外,也可以为核酸和蛋白质等其它分子的复合物。另外,该供试物质除精制物之外,也可以为包含供试物质以外的物质的组合物。并且,该供试物质还可以为未鉴定的物质。例如,如各种细胞的细胞提取液及核提取液那样含有各种物质的物品也可以用作供试物质。
本发明的核酸结合能力的评价方法中评价的核酸结合能力不受特别限定,可以为识别并结合特定的碱基序列的序列特异性核酸结合能力,也可以为识别并结合特定的修饰状态的核酸结合能力,还可以为不受碱基序列及修饰状态影响,广泛与全部核酸结合的核酸结合能力。
本发明的核酸结合能力的评价方法中使用的核酸是作为评价供试物质是否与之结合的对象的核酸,其只要能够作为核酸扩增反应的模板,则不受特别限定,优选DNA或RNA。该核酸可以规定碱基序列,也可以不规定碱基序列。另外,该核酸可以仅由一种核酸构成,也可以为多种核酸的混合物。
例如,在评价识别并结合特定的碱基序列的序列特异性核酸结合能力的情况下,将具有该特定的碱基序列的核酸作为模板,使用以与模板核酸中的该特定的碱基序列的上游(5’侧)的杂交的方式设计的引物,以在核酸扩增反应中对模板核酸中包含该特定的碱基序列的区域进行扩增。引物能够基于作为模板的核酸的碱基序列通过常规方法来设计合成。
在评价与处于特定的修饰状态的核酸特异性结合的核酸结合能力的情况下,将处于该特定的修饰状态的核酸作为模板。该修饰状态可列举例如:甲基化、氟化、硫代磷酸酯化、二硫代磷酸酯化、糖基化、PEG(聚乙二醇)加成、肽加成等。
作为本发明的核酸结合能力的评价方法中进行的核酸扩增反应,可列举与本发明的靶核酸的检测方法中进行的所述核酸扩增反应相同的反应。在供试物质为蛋白质等耐热性较低的分子的情况下,核酸扩增反应优选在65℃以下的温度范围进行。相反地,在供试物质为耐热性高的分子、并评价高温环境下的核酸结合能力的情况下,使用耐热性聚合酶,在超过65℃且小于等于100℃的温度范围内进行核酸扩增反应。另外,作为本发明的核酸结合能力的评价方法中进行的核酸扩增反应,不需要PCR中所需的用于温度控制的热循环仪,因此,优选RPA法、LAMP法、MDA法、NASBA法等在等温条件下进行的核酸扩增反应。
例如,使用供试物质、核酸、及与所述核酸杂交的引物进行核酸扩增反应,将得到的核酸扩增产物量与除不添加供试物质之外在相同条件下进行核酸扩增反应得到的核酸扩增产物量进行比较。在供试物质在进行核酸扩增反应的温度条件等下具有对于模板核酸的核酸结合能力的情况下,在该供试物质存在下得到的核酸扩增产物量比不存在该供试物质时得到的核酸扩增产物量少。即,在供试物质存在下得到的核酸扩增产物量比在不存在供试物质时得到的核酸扩增产物量少的情况下,评价该供试物质在进行核酸扩增反应的温度条件等下具有对于用作模板的核酸的核酸结合能力。相反地,在供试物质存在下得到的核酸扩增产物量大于等于不存在供试物质时得到的核酸扩增产物量的情况下,评价该供试物质不具有对于用作模板的核酸的核酸结合能力。
通过将本发明的核酸结合能力的评价方法中使用的试剂等制成试剂盒,能够更简便地进行该方法。例如,作为使用该方法的用于评价核酸结合能力的试剂盒,优选其含有作为评价供试物质的核酸结合能力的对象的核酸、及与该核酸杂交的引物。此外,优选也包含用于核酸扩增反应的酶、用于制备反应液的浓缩缓冲液、dNTP等。例如,通过RPA法进行核酸扩增反应的方法中使用的试剂盒优选进一步包含重组酶、SSB、及DNA聚合酶。
本发明的用于评价核酸结合能力的试剂盒也优选进一步包含使用说明书,使用说明书记载了用于使用该试剂盒进行本发明的核酸结合能力的评价方法的文字。该使用说明书也可以记载于收纳该试剂盒的容器的表面。
实施例
接着,通过实施例等对本发明进行更详细说明,但本发明并不受这些例子限定。
[RNA及引物]
下面的实验中使用的RNA使用委托FASMAC公司化学合成的RNA。将所使用的RNA示于表1。
[表1]

下面的实验中使用的引物使用委托eurofin公司化学合成的引物。将所使用的引物示于表2。
[表2]
| 引物 | 序列(5′→3′) | 序列号 |
| KRAS-RPA-G12-F | tagtgtattaaccttatgtgtgacatgttctaat | 7 |
| KRAS-RPA-G12-R | aaacaagatttacctctattgttggatcatattc | 8 |
| p16-RPA-F2 | ggcggcggggagcagcatggagccttcggctgac | 9 |
| p16-RPA-R2 | ctacccacctggatcggcctccgaccgtaactat | 10 |
| p14-RPA-F | gtcccagtctgcagttaagggggcaggagt | 11 |
| p14-RPA-R | gggcctttcctacctggtcttctaggaa | 12 |
| p16-RPA-F | gaggaagaaagaggaggggctggctggtcacc | 13 |
| p16-RPA-R | ctgcagaccctctacccacctggatcggcctc | 14 |
| p14ARF-CpG_island-F | gtgggtcccagtctgcagttaag | 15 |
| p14ARF-CpG_island-R | acttttcgagggcctttcctac | 16 |
| Pax5-LexA-RPA-F | gcatcagtcgcccttcgcctccttctctcg | 17 |
| Pax5-LexA-RPA-R | gcgagggcggaacgtgactttgccctgcgg | 18 |
[gRNA的制作]
将1μL 10μM crRNA、1μL 10μM tracrRNA、及2μL无核酸酶水混合,在98℃下孵育2分钟,由此形成gRNA(crRNA/tracrRNA复合物)。
[RPA反应]
如下进行RPA反应。首先,向一管(装入冻干试剂)RPA试剂(产品名“TwistAmp(注册商标)Basic kit”,TwistDx公司制)中加入29.5μL rehydration buffer、2.5μL 10μM正向引物、2.5μL 10μM反向引物并进行混合。接着,从所制备的溶液中取13.6μL到另外的管中,添加20ng模板DNA和无核酸酶水,由此制备19μL RPA反应准备溶液。向该RPA反应准备溶液中添加1μL 280mM MgOAc溶液之后,在37℃下孵育30分钟,进行RPA反应。
如下进行使用dCas9蛋白质和gRNA的RPA反应。需要指出,作为dCas9,使用向来自化脓性链球菌的Cas9中导入D10A及H840A的点突变而得到的蛋白质,该蛋白质委托Sysmex公司(ProCube,制造编号14M_029)合成。首先,将0.8μL gRNA、0.4μg dCas9蛋白质及无核酸酶水混合,制成10μL,将其作为CRISPR溶液。RPA反应时,向所述制备的RPA反应准备溶液中添加1μL CRISPR溶液(添加后的溶液中,dCas9蛋白质为40ng,gRNA为10nM),在37℃下孵育5分钟。然后,向反应液中添加1μL 280mM MgOAc溶液,在37℃下孵育30分钟,进行RPA反应。
反应结束后,使用“PCR/Gel DNApurification kit”(日本genetics公司制)从各反应液中精制核酸。使用包含“SYBR(注册商标)Safe DNA Gel Stain”(ThermoFisherScientific公司制)的琼脂糖凝胶对精制产物进行电泳。
[实施例1]
针对培养细胞株293T细胞的KRAS基因,制作两种gRNA,将其与dCas9一起添加在反应系统中,进行RPA反应。
使用从293T细胞提取并精制的基因组DNA(293T gDNA)作为模板,使用gRNA_KRAS(序列号1及6)及gRNA_KRAS#2(序列号2及6)作为gRNA。作为引物组,使用正向引物(序列号7)和反向引物(序列号8),该正向引物(序列号7)和反向引物(序列号8)对KRAS基因的基因组DNA中的包含gRNA_KRAS及gRNA_KRAS#2的靶区域的区域进行扩增。将293T细胞的KRAS基因的部分区域和与所使用的两种gRNA的基因组DNA互补的序列部分示于图3。并且,作为对照,使用针对CDKN2A(p16)基因的gRNA(gRNA_p16_Gx5#2、序列号3及6)。
将电泳的结果示于图4。在添加了gRNA_p16_Gx5#2的反应液中,与未添加gRNA和dCas9的反应液相同地,检测到了扩增产物的条带。另一方面,添加有gRNA_KRAS及gRNA_KRAS#2的反应液均未检测到扩增产物的条带,抑制了正向引物和反向引物对于KRAS基因的核酸扩增。由以上结果确认,通过使反应液中含有针对想要抑制其核酸扩增的DNA的gRNA和dCas9,能够抑制通过RPA反应产生的核酸扩增。
[实施例2]
在培养细胞株HCT116细胞的KRAS基因中,仅单侧等位基因具有单碱基置换的突变(GGC→GAC),第13个甘氨酸被置换为冬氨酸。通过该单碱基置换,图3中所示的gRNA_KRAS#2中的PAM序列(TGG)变为TGA,不再是PAM序列(NGG)。
使用从HCT116细胞中提取并精制的基因组DNA(HCT116 gDNA)作为模板核酸,除此之外,与实施例1相同地进行RPA反应,精制后进行电泳。
将电泳的结果示于图5。与仅将具有野生型KRAS基因的293T gDNA作为模板的实施例1相同地,在添加有gRNA_p16_Gx5#2的反应液中,也确认到了核酸扩增产物,在添加有gRNA_KRAS的反应液中,未确认到扩增产物。即,即使gRNA和模板核酸有一个碱基的错配,gRNA_KRAS也能够作为gRNA发挥功能。另一方面,与实施例1不同,添加有gRNA_KRAS#2的反应液中,确认到了核酸扩增产物。
通过序列分析对各反应液中得到的扩增产物的碱基序列进行确认,结果未添加gRNA的反应液和添加有gRNA_p16_Gx5#2的反应液的扩增产物中,包含HCT116 gDNA中原本包含的野生型KRAS和突变型KRAS(G13D)两者。另一方面,添加有gRNA_KRAS#2的反应液的扩增产物中,仅包含突变型KRAS(G13D),未能确认到野生型KRAS的核酸扩增。图6中示意性示出在添加有gRNA_KRAS#2的反应液中进行的RPA反应。在野生型KRAS中,gRNA_KRAS#2与dCas9一同结合于PAM序列附近,抑制基于DNA聚合酶的扩增反应,在突变型KRAS中,gRNA_KRAS#2所结合的预定区域附近没有PAM序列,因此不会重排dCas9,扩增反应正常进行。即确认,通过以PAM序列紧随突变位点之后的方式设计gRNA,可以非常高精度地检测1个碱基突变。
接着,如图7所示,在gRNA_KRAS上导入单碱基置换的突变。该gRNA_KRAS_mut(序列号4及6)与野生型KRAS有一个碱基不同,与突变型KRAS(G13D)有两个碱基不同。使用HCT116gDNA或293T gDNA作为模板核酸,使用gRNA_KRAS_mut作为gRNA,除此之外,与实施例1相同地进行RPA反应,精制后进行电泳。
应液中,通过gRNA_KRAS_mut抑制了核酸扩增。在添加有HCT116gDNA的反应液中,即使在添加有gRNA_KRAS_mut的情况下,也检测到了扩增产物的条带。对添加有HCT116gDNA的反应液中的扩增产物的碱基序列进行确认,结果在未添加gRNA_KRAS_mut的情况下,包含野生型KRAS和突变型KRAS(G13D)两者。另一方面,在添加有gRNA_KRAS_mut的情况下,仅包含突变型KRAS(G13D)。即,即使gRNA和模板核酸有一个碱基的错配,也能够作为gRNA发挥功能,从而能够抑制RPA反应中的核酸扩增,若gRNA和模板核酸有两个碱基的错配,则不能作为gRNA发挥功能,RPA反应产生的核酸扩增正常进行。由以上结果确认,即使在PAM序列上不存在突变的情况下,通过人为向gRNA导入突变,并增加与靶碱基序列的错配,也能够区分1个碱基突变。
[实施例3]
如图9所示,在HCT116细胞的CDKN2A(p16)基因中,仅单侧等位基因插入有一个G(Gx5)。因而,gRNA_p16_Gx5#2的碱基序列与Gx5等位基因同源,但与其它等位基因(Gx4)不一致。
使用从HCT116细胞中提取并精制的基因组DNA(HCT116 gDNA)作为模板核酸,作为引物组,使用正向引物(序列号9)和反向引物(序列号10),该正向引物(序列号9)和反向引物(序列号10)用于对CDKN2A(p16)基因的基因组DNA中的包含gRNA_p16_Gx5#2的靶区域的区域进行扩增,作为gRNA,使用gRNA_p16_Gx5#2或gRNA_KRAS,除此之外,与实施例1相同地进行RPA反应,精制后进行电泳。
将电泳的结果示于图10。即使在添加有gRNA_p16_Gx5#2的反应液中,扩增产物也比添加有gRNA_KRAS的反应液及未添加gRNA的反应液的扩增产物更少,但确认到了扩增产物。
通过序列分析对各反应液中得到的扩增产物的碱基序列进行确认,结果未添加gRNA的反应液和添加有gRNA_KRAS的反应液的扩增产物中,包含HCT116 gDNA中原本包含的Gx5型p16和Gx4型p16两者。另一方面,添加有gRNA_p16_Gx5#2的反应液的扩增产物中,仅包含Gx4型p16,不能确认到Gx5型p16的核酸扩增。gRNA_p16_Gx5#2与dCas9共同被重排至Gx5型p16序列,抑制基于DNA聚合酶的扩增反应,但Gx4型p16序列上未重排dCas9,扩增反应正常进行。即确认,通过本发明的靶核酸的检测方法,能够非常高精度地检测插入或缺失1个碱基的突变。
[实施例4]
对293T细胞的CDKN2A(p16)基因进行基因组编辑,在靶向野生型核酸的gRNA(gRNA_mid2,序列号5及6)和dCas9存在下对编辑后的基因组进行RPA反应,仅扩增并检测到经基因组编辑的核酸。图11中示意性示出对CDKN2A(p16)基因进行的基因组编辑。
在未经过基因组编辑的情况下,保持野生型的碱基序列,因此,通过gRNA_mid2和dCas9抑制RPA扩增。另一方面,在经过基因组编辑而导致碱基序列改变的情况下,未产生gRNA_mid2和dCas9带来的抑制扩增的效果,可见RPA扩增。
首先,如下进行基因组编辑。使用转染试剂“Lipofectamine 3000”(ThermoFisherScienticif公司制)将2μg Cas9表达质粒(Addgene公司制,#41815)及2μg靶向人CDKN2A(p16)基因的sgRNA表达质粒(sgRNA-mid2)(非专利文献5)转染至293T细胞(4×105细胞)。培养2天后,回收细胞,并提取及精制基因组DNA。
接着,作为模板,使得到的基因组DNA的总量为100ng,并使用正向引物(序列号9)和反向引物(序列号10),该正向引物(序列号9)和反向引物(序列号10)用于对293T细胞的CDKN2A(p16)基因的基因组DNA中包含gRNA_mid2所靶向的碱基序列的区域进行RPA扩增,除此之外,与实施例1相同地进行RPA反应,精制后进行电泳。需要指出,通过向未经过基因组编辑的293T gDNA中少量混合经过基因组编辑的293T gDNA,降低了经基因组编辑的基因组DNA的比率。
将电泳的结果示于图12。在仅以未经过基因组编辑的293T gDNA为模板的反应液中,未确认到扩增产物,通过gRNA_mid2抑制了核酸扩增反应。另一方面,在以未经过基因组编辑的293T gDNA(80ng)和经过基因组编辑的293T gDNA(20ng)的混合物为模板的反应液中,确认到扩增产物。对各扩增产物的碱基序列进行确认,结果以未经过基因组编辑的293TgDNA为模板的反应液的扩增产物与CDKN2A(p16)基因的野生型核酸序列相同。以未经过基因组编辑的293T gDNA和经过基因组编辑的293T gDNA的混合物为模板的反应液的扩增产物大部分是野生型核酸序列,但也存在一些考虑为经过基因组编辑的序列的、多样性丰富的碱基序列。另一方面,在以未经过基因组编辑的293T gDNA和经过基因组编辑的293TgDNA的混合物为模板,且加入了dCas9和gRNA_mid2的反应液的扩增产物中,仅检测到多样性丰富的碱基序列,野生型核酸序列未被扩增。
[实施例5]
向293T gDNA中混合少量的HCT116 gDNA,将降低了突变型KRAS(G13D)的存在比率([来自具有突变型KRAS(G13D)的细胞的gDNA的含量]/([来自具有突变型KRAS(G13D)的细胞的gDNA的含量]+[来自仅具有野生型KRAS的细胞的gDNA的含量])×100%)的核酸作为模板,考察是否能够通过gRNA_KRAS#2和dCas9抑制野生型KRAS的RPA扩增。
将HCT116 gDNA固定为20ng,将293T gDNA和HCT116 gDNA混合,并使突变型KRAS(G13D)的存在比率如图13所示,将混合物作为模板,并使用gRNA_KRAS#2作为gRNA,除此之外,与实施例1相同地进行RPA反应,精制后进行电泳。
将电泳的结果示于图13。即使在来自具有突变型KRAS(G13D)的细胞的gDNA的存在比率仅为2%的反应液中,也确认到核酸扩增产物。对各扩增产物的碱基序列进行确认,结果在来自具有突变型KRAS(G13D)的细胞的gDNA的存在比率为5~100%的反应液的扩增产物中,仅检测到突变型KRAS(G13D),但在来自具有突变型KRAS(G13D)的细胞的gDNA的存在比率为2%的反应液的扩增产物中,确认到突变型KRAS(G13D)和野生型KRAS两者。
接着,将293T gDNA固定为400ng,将293T gDNA和HCT116 gDNA混合,并使突变型KRAS(G13D)的存在比率如图14所示,将混合物作为模板,并使用gRNA_KRAS#2作为gRNA,除此之外,与实施例1相同地进行RPA反应,精制后进行电泳。
将电泳的结果示于图14。即使在来自具有突变型KRAS(G13D)的细胞的gDNA的存在比率仅为1%的反应液中,也确认到核酸扩增产物。对各扩增产物的碱基序列进行确认,结果来自具有突变型KRAS(G13D)的细胞的gDNA的存在比率为1~100%的反应液的扩增产物中,仅检测到突变型KRAS(G13D)。以上结果表明,即使靶核酸(本实验中为突变型KRAS(G13D))的存在比率仅为1%,也能够检测靶核酸。
[实施例6]
在HCT116细胞的CDKN2A(p14ARF)基因中,仅在单侧等位基因缺失一个G(Gx4)。另外,HCT116细胞的CDKN2A(p14ARF)基因中,Gx4等位基因中的Gx4位置周围的胞嘧啶不经甲基化修饰,但Gx5等位基因中的Gx5位置周围的胞嘧啶经过甲基化修饰。因而,将CpG甲基化DNA结合蛋白质添加于RPA反应的反应液,抑制以Gx5等位基因为模板的RPA扩增,进而检测Gx4等位基因。作为CpG甲基化DNA结合蛋白质,使用MBD2蛋白质(“EpiXplore(注册商标)Methylated DNA Enrichment Kit”,Takara Bio公司制)。
使用0.25μg MBD2蛋白质来代替gRNA和dCas9,除此之外,与实施例1相同地进行RPA反应,精制后进行电泳。需要指出,作为引物组,使用用于对CDKN2A(p14ARF)基因的Gx4·Gx5位置进行扩增的正向引物(序列号11)和反向引物(序列号12)。两个引物均不包含CpG。
将电泳的结果示于图15。在未添加MBD2的反应液中,确认到核酸扩增产物。即使在添加了MBD2的情况下,也可见扩增产物。对各扩增产物的碱基序列进行确认,结果在未添加MBD2的反应液的扩增产物中,包含Gx4型p14ARF和Gx5型p14ARF两者。另一方面,在添加有MBD2的反应液的扩增产物中,仅包含Gx4型p14ARF,未能确认Gx5型p14ARF的核酸扩增。由以上结果表明,通过使RPA反应的反应液中存在CpG甲基化DNA结合蛋白质,能够抑制以经CpG甲基化的核酸为模板的核酸扩增,进而能够检测未经CpG甲基化的靶核酸。
在HCT116细胞的CDKN2A(p16)基因中,Gx5等位基因中的Gx5位置周围的胞嘧啶不经甲基化修饰,但Gx4等位基因中的Gx4位置周围的胞嘧啶经过甲基化修饰。因而,将CpG甲基化DNA结合蛋白质添加于RPA反应的反应液,抑制以Gx4等位基因为模板的RPA扩增,进而检测Gx5等位基因。作为CpG甲基化DNA结合蛋白质,使用MBD2。
使用0.5μg MBD2蛋白质来代替gRNA和dCas9,除此之外,与实施例3相同地进行RPA反应,精制后进行电泳。需要指出,作为引物组,使用用于对CDKN2A(p16)基因的Gx4·Gx5位置进行扩增的正向引物(序列号13)和反向引物(序列号14)。该反向引物(序列号14)包含一处CpG。
将电泳的结果示于图16。在未添加MBD2的反应液中,确认到核酸扩增产物。即使在添加了MBD2的情况下,也可见扩增产物。对各扩增产物的碱基序列进行确认,结果在未添加MBD2的反应液的扩增产物中,包含Gx5型p16和Gx4型p16两者。另一方面,在添加有MBD2的反应液的扩增产物中,仅包含Gx5型p16,未能确认Gx4型p16的核酸扩增。以上结果也表明,通过使RPA反应的反应液中存在CpG甲基化DNA结合蛋白质,抑制以经CpG甲基化的核酸为模板的核酸扩增,进而能够检测未经CpG甲基化的靶核酸。
接着,针对在试管内人工CpG甲基化的DNA,也考察是否通过本方法抑制核酸扩增。首先,使用夹持CDKN2A(p14ARF)基因的Gx4·Gx5位置的正向引物(序列号15)和反向引物(序列号16),通过PCR对被两个引物夹持的碱基序列进行扩增。需要指出,PCR使用“AmpliTaq Gold(注册商标)360Master Mix”(ThermoFisher Scientific公司制),制备PCR反应液,10μL PCR反应液中包含10ng HCT116基因组DNA和0.5μM的各引物。反应时,首先,在95℃下变性10分钟,接着,按照95℃、15秒;60℃、30秒;及72℃、30秒进行30循环,然后,在72℃下处理1分钟。扩增产物通过“PCR/Gel DNA purification kit”(日本genetics公司制)精制后,克隆至T-Vector pMD20(Takara Bio公司制),并通过Competent Quick DH5a(东洋纺公司制)扩增。扩增后的质粒使用“NucleoBond(注册商标)Xtra Midi Plus”(Takara Bio公司制)精制。作为精制后的质粒,准备包含Gx4的质粒(p14_Gx4质粒)和包含Gx5的质粒(p14_Gx5质粒)两种。
在HCT116细胞内,CDKN2A(p14ARF)基因的Gx4位置周围的胞嘧啶不经甲基化修饰,Gx5位置周围的胞嘧啶经过甲基化修饰。但是,由大肠杆菌精制的质粒不经过这些修饰。因而,有意将Gx4位置周围而不是Gx5位置周围的胞嘧啶在试管内进行甲基化修饰。具体而言,使精制后的1μg p14_Gx4质粒与6单位的CpG甲基转移酶M.SssI(New England BioLabs公司制)及160μM S-腺苷甲硫氨酸一同在37℃下反应1小时,由此进行甲基化处理。甲基化后的p14_Gx4质粒、及未经甲基化处理的p14_Gx5质粒使用“PCR/Gel DNA purification kit”(日本genetics公司制)精制。
使用1pg质粒DNA代替基因组DNA,除此之外,与实施例1相同地制备RPA反应准备溶液。将0.5μg MBD2蛋白质添加于RPA反应准备溶液之后,在37℃下孵育10分钟,然后,加入1μL 280mM的MgOAc溶液,并在37℃下孵育10分钟,从而进行RPA反应。需要指出,作为引物组,使用用于对CDKN2A(p14ARF)基因的Gx4·Gx5位置进行扩增的正向引物(序列号11)和反向引物(序列号12)。反应后,反应液不进行精制,取2μL进行电泳。
将电泳的结果示于图17。在未添加MBD2的反应液中,确认到核酸扩增产物。在添加有MBD2的情况下,可见来自未经甲基化的p14_Gx5质粒的核酸扩增,但抑制了来自经甲基化的p14_Gx4质粒的核酸扩增。以上结果表明,也可以利用人工CpG甲基化的DNA作为模板;另外,CpG甲基化直接参与对核酸扩增的抑制。
[实施例7]
探讨了除dCas9/gRNA及MBD2以外的其它DNA结合蛋白质是否能抑制RPA反应。鸡DT40#205-2细胞株在Pax5基因启动子区域具有LexA结合序列,作为细菌的DNA结合蛋白质的LexA蛋白质结合于LexA结合序列(非专利文献6)。因而,考察了在以DT40#205-2细胞基因组DNA为模板,并向RPA反应的反应液添加了LexA蛋白质的情况下,是否抑制LexA结合序列的RPA扩增。
使用20ng LexA蛋白质来代替gRNA和dCas9,除此之外,与实施例1相同地进行RPA反应,精制后进行电泳。需要指出,作为LexA蛋白质,使用委托Sysmex公司(ProCube,制造编号13T_0170)合成LexA蛋白质的DNA结合结构域而得到的蛋白质。阴性对象蛋白质使用20ngdCas9蛋白质。作为引物组,使用用于对包含LexA结合序列的区域进行扩增的正向引物(序列号17)和反向引物(序列号18)。
将电泳的结果示于图18。在未添加LexA蛋白质的反应液中,可见核酸扩增。在添加有LexA蛋白质的情况下,抑制来自基因组DNA的核酸扩增。在添加了dCas9作为阴性对象蛋白质的情况下,未抑制来自基因组DNA的核酸扩增。这些结果表明,即使不是CRISPR复合物或MBD2蛋白质,只要为DNA结合蛋白质,则能够抑制靶核酸的扩增。通过利用本技术,能够评价特定的DNA结合分子对于特定的碱基序列是否具有结合能力。另外,也能够用于检测分子群体中是否存在对特定的碱基序列具有结合能力的分子。
[实施例8]
通过RT-PCR,作为靶核酸来检测供试核酸样本中的单链RNA。将人NEAT1基因的mRNA(NEAT1-RNA)作为靶核酸。
[gRNA及引物]
本实验中,将由表3中记载的碱基序列构成的单链RNA用作gRNA。表3的gRNA的碱基序列中,大写字母的区域是与靶核酸NEAT1-RNA的部分区域互补的区域(RNA结合区域)。gRNA使用委托Gene Design公司化学合成的物质。作为gRNA,预先将1μL 10μM gRNA及3μL无核酸酶水混合,在100℃下孵育2分钟后,在室温下冷却,然后用于实验。
[表3]

本实验中,使用由表4中记载的碱基序列构成的引物。正向引物(Human NEAT1-F2)和反向引物(Human NEAT1-R2)是以夹持与dCas13a/gRNA_NEAT1所靶向的RNA序列互补的cDNA序列的方式设计的引物。正向引物(MY-0119)和反向引物(MY-0129)是以夹持与dCas13a/gRNA_NEAT1_2所靶向的靶RNA序列互补的cDNA序列的方式设计的引物。作为对照,使用正向引物(hGAPDH-dCas13a-F3)和反向引物(hGAPDH-dCas13a-R3),以将人GAPDH基因的mRNA的cDNA作为模板进行扩增。引物使用委托eurofin公司化学合成的引物。
[表4]
| 引物 | 序列(5′→3′) | 序列号 |
| Human NEAT1-F2 | ctaaattgagcctccggtca | 21 |
| Human NEAT1-R2 | acaagaaggcaggcaaacag | 22 |
| MY-0119 | cactggtactgggagggatg | 23 |
| MY-0129 | cccttcaacctgcatttcctac | 24 |
| hGAPDH-dCas13a-F3 | gagccaaaagggtcatcatctct | 25 |
| hGAPDH-dCas13a-R3 | cacgataccaaagttgtcatgga | 26 |
[供试核酸样本的制备]
将从人源培养细胞MRC-5细胞中提取的总RNA作为供试核酸样本。MRC-5细胞在包含10%FBS(胎牛血清)、青霉素-链霉素(西格玛公司制)的E-MEM(和光公司制)培养基中培养。MRC-5细胞的RNA使用市售的RNA提取用试剂“Isogen II”(NIPPON GENE公司制)提取及精制。
[dCas13a]
作为dCas13a蛋白质,使用向来自韦德纤毛菌的野生型Cas13a中导入R474A及R1046A的点突变而得到的突变型蛋白质,该蛋白质委托Sysmex公司(ProCube,制造编号17T_042)合成。
[使用gRNA_NEAT1的RT-PCR]
将0.46μL gRNA、0.24μg dCas13a蛋白质、及无核酸酶水混合,制成5μL,将其作为dCas13a/gRNA溶液。
首先,使用“ReverTraAce qPCR RT Master Mix with gDNA Remover”(TOYOBO公司制)进行逆转录反应(RT)。首先,将2μL“4×DN Master Mix”(已添加gDNA Remover)、1ngRNA、5μL dCas13a/gRNA溶液、及无核酸酶水混合,制成8μL,在37℃下孵育5分钟。然后,向该反应液中添加2μL“5×RT Master Mix II”,在37℃下孵育30分钟,接着,在50℃下孵育5分钟,接着,在98℃下孵育5分钟,从而进行逆转录反应。向逆转录反应后的溶液中混合10μL无核酸酶水,制得20μL cDNA溶液。
接着,使用“EmeraldAmp(注册商标)MAX PCR Master Mix”(Takara Bio公司制)进行PCR。制备PCR反应液,10μL PCR反应液中包含1μL cDNA、0.5μM的Human NEAT1-F2引物及Human NEAT1-R2引物。反应时,首先,在94℃下变性1分钟,接着,按照94℃、15秒;60℃、15秒;及72℃、1分钟进行35循环,然后,在72℃下处理1分钟。
[使用gRNA_NEAT1_2的RT-PCR]
dCas13a/gRNA溶液的制备及RT与使用gRNA_NEAT1的RT-PCR相同地进行。
接着,使用“AmpliTaq Gold(注册商标)360Master Mix”(ThermoFisherScientific公司制)进行PCR。制备PCR反应液,10μL PCR反应液中包含1μL cDNA、0.5μM的MY-0119引物及MY-0129引物。反应时,首先,在95℃下变性10分钟,接着,按照95℃、15秒;55℃、30秒;及72℃、30秒进行38循环,然后,在72℃下处理1分钟。
[GAPDH的RT-PCR]
GAPDH的扩增使用“EmeraldAmp(注册商标)MAX PCR Master Mix”(Takara Bio公司制)进行。制备PCR反应液,10μL PCR反应液中包含1μL cDNA、0.5μM的hGAPDH-dCas13a-F3引物及hGAPDH-dCas13a-R3引物。反应时,首先,在94℃下变性1分钟,接着,按照94℃、15秒;60℃、15秒;及72℃、1分钟进行28或32循环,然后,在72℃下处理1分钟。
[电泳]
使用包含“SYBR(注册商标)Safe DNAGel Stain”(ThermoFisher Scientific公司制)的琼脂糖凝胶对各扩增反应中得到的扩增产物进行电泳。
将使用gRNA_NEAT1的RT-PCR的扩增产物的结果示于图19,将使用gRNA_NEAT1_2的RT-PCR的扩增产物的结果示于图20。如图19所示,与未添加dCas13a及仅添加dCas13a的反应液相比,添加了dCas13a/gRNA_NEAT1复合物溶液的反应液中,PCR对NEAT1的扩增减弱。另一方面,在扩增了dCas13a/gRNA_NEAT1复合物未靶向的GAPDH的情况下,未见这种DNA扩增减弱。图20中也确认到同样的情况。以上结果表明,dCas13a/gRNA_NEAT1复合物特异性抑制NEAT1的逆转录反应,从而确认,通过使反应液中存在dCas13a/gRNA复合物,能够抑制序列特异性逆转录反应。
[实施例9]
已有报道指出,在Ba/F3细胞内,Stat5在IL-3刺激(10ng/mL)30分钟后与Cis启动子结合(非专利文献9)。因而,使用具有Stat5结合位点(TTCNNNGAA)的双链DNA、及用于对包含该双链DNA的Stat5结合位点的区域进行RPA扩增的引物组,检测Ba/F3细胞的核提取液中的Stat5。
[模板核酸]
将夹持小鼠Cis基因启动子内的Stat5结合位点的区域(表5中的Cis区域:序列号27)作为模板核酸,制备插入有该模板核酸的质粒(Cis质粒)。该区域内包含四个Stat5结合位点。表5的序列号27中的白心部位是Stat5结合位点。另外,作为阴性对照,向Stat5结合位点加入突变,以使其缺失Stat5结合能力,将得到的突变序列(表5中的CisM区域:序列号28)作为模板核酸,制备插入有该模板核酸的质粒(CisM质粒)。表5的序列号28中的白心部位为加入了突变的Stat5结合位点。
[表5]

首先,将Ba/F3细胞的基因组DNA作为模板,使用夹持小鼠Cis基因启动子内的所述Cis区域的正向引物(mCis_-259/-199_F)和反向引物(mCis_-188/-104_R),通过PCR扩增被两个引物夹持的碱基序列。PCR中,使用“EmeraldAmp(注册商标)MAX PCR Master Mix”(Takara Bio公司制),制备PCR反应液,10μL PCR反应液中包含10ng Ba/F3基因组DNA、0.5μM各引物。反应时,首先,在94℃下变性1分钟,接着,按照94℃、15秒;60℃、15秒;及72℃、1分钟进行35循环,然后,在72℃下处理1分钟。
通过“PCR/Gel DNApurification kit”(日本genetics公司制)精制得到的扩增产物后,将其克隆至T-Vector pMD20(Takara Bio公司制),通过“Competent Quick DH5a”(东洋纺公司制)扩增。扩增后的质粒使用“NucleoSpin(注册商标)Plasmid QuickPure”(Takara Bio公司制)精制(Cis质粒)。
如下制作CisM质粒。将Cis质粒作为模板,使用夹持Stat5结合位点的正向引物(mCis_-259/-199_mut_F)和反向引物(mCis_-188/-104_mut_R),通过PCR扩增被两个引物夹持的碱基序列。需要指出,PCR中,使用“EmeraldAmp(注册商标)MAX PCR Master Mix”(Takara Bio公司制),制备PCR反应液,10μL PCR反应液中包含1pg Cis质粒、0.5μM的各引物。反应时,首先,在94℃下变性1分钟,接着,按照94℃、15秒;55℃、15秒;及72℃、1分钟进行30循环,然后,在72℃下处理1分钟。与Cis区域的扩增产物相同地,将扩增产物精制后整合至T-Vector pMD20,扩增后再进行精制,由此得到CisM质粒。进行DNA序列分析,结果确认,CisM质粒中,虽然在反向引物侧发现预想之外的突变,但Stat5结合位点上导入了表5所示的突变。
[表6]

[Ba/F3细胞的核提取液的制备]
如下制备无IL-3刺激的Ba/F3细胞及IL-3刺激处理后的Ba/F3细胞的核提取液。
用RPMI-1640(和光公司制)培养基培养Ba/F3细胞,培养基中包含10%FBS、10mMHEPES缓冲液(pH7.2)(NACALAI TESQUE公司制)、1×非必需氨基酸、1mM丙酮酸钠(NACALAITESQUE公司制)、5μM 2-巯基乙醇(Sigma-Aldrich公司制)、1ng/mL IL-3(ThermoFisherScientific公司制)、及青霉素-链霉素(NACALAI TESQUE公司制)。
制备无IL-3刺激及IL-3刺激处理后的核提取液时,首先,将Ba/F3细胞用PBS清洗3次,然后,利用除去IL-3的培养基培养6小时(无IL-3刺激)。然后,向培养基中添加IL-3至0ng/mL,在37℃下培养30分钟(IL-3刺激处理)。回收无IL-3刺激及IL-3刺激处理后的Ba/F3细胞,使用“NE-PER(注册商标)Nuclear and Cytoplasmic Extraction Reagents”(ThermoFisher Scientific公司制)制备核提取液。
[RPA反应]
如下进行RPA反应。首先,向一管(装入冻干试剂)RPA试剂(产品名“TwistAmp(注册商标)Basic kit”,TwistDx公司制)中加入29.5μL rehydration buffer、2.5μL 10μM正向引物(M13Primer RV)、2.5μL 10μM反向引物(M13 Primer M4)、及无核酸酶水,制成50μL,并进行混合。接着,将所制备的溶液中的0μL取至另外的管中,添加1pg质粒DNA(Cis质粒或CisM质粒)和3ng核提取液之后,添加10mM Tris(pH8.0),从而制备11.3μL反应液。将该反应液在37℃下孵育5分钟,然后,加入1μL 280mM的MgOAc溶液,在37℃下孵育30分钟,从而进行RPA反应。反应后,该反应液不精制,取2μL进行电泳。
[表7]
| ·引物 | 序列(5′→3′) | 序列号 |
| M13 Primer RV | CAGGAAACAGCTATGAC | 33 |
| M13 Primer M4 | GTTTTCCCAGTCACGAC | 34 |
将电泳的结果示于图21。在将Cis质粒作为模板的RPA反应中,作为添加了IL-3刺激处理后的核提取液的反应液,与添加了无IL-3刺激核提取液的反应液相比,RPA产生的DNA扩增减弱。这是因为,IL-3刺激处理后的核提取液中存在的Stat5与Cis质粒中的Stat5结合位点结合,从而抑制了DNA扩增,而无IL-3刺激核提取液中不存在Stat5,因此未抑制RPA反应。另一方面,在将CisM质粒作为模板的RPA反应中,无论有无IL-3刺激处理,均发现同等程度的DNA扩增。根据以上结果,可以通过RPA产生的核酸扩增来评价细胞溶解液等溶液中是否存在与靶DNA结合的蛋白质。
[实施例10]
将基因组DNA库作为模板,并在CpG甲基化DNA结合蛋白质存在下进行RPA反应,考察未经过CpG甲基化的DNA是否被扩增。
[基因组DNA库]
使用从HCT116细胞中提取及精制的基因组DNA,委托Promega公司制作DNA库(产品号:KK0500)。制作DNA库时所追加的适配子序列中包含用于illumina公司的新一代序列的index15(ATGTCA)。追加适配子时不使用DNA扩增操作。
[RPA反应]
如下使用与适配子序列互补的引物来进行RPA反应。首先,向一管(装入冻干试剂)RPA试剂(产品名“TwistAmp(注册商标)Basic kit”,TwistDx公司制)中添加29.5μLrehydration buffer、2.5μL 10μM正向引物(P5-NGS-Lib-Promega-RPA-F)、2.5μL 10μM反向引物(P7-NGS-Lib-Promega-RPA-R)、及9.5μL无核酸酶水,并进行混合。接着,将所制备的溶液中的17.5μL取至其它管中,添加0.5μL DNA库、0.5μg MBD2蛋白质(“EpiXplore(注册商标)Methylated DNA Enrichment Kit”,Takara Bio公司制)、无核酸酶水,由此制备19μLRPA反应准备溶液。将该RPA反应准备溶液在37℃下孵育10分钟,然后,加入1μL 280mMMgOAc溶液,在37℃下孵育10分钟,从而进行RPA反应。反应结束后,使用“PCR/Gel DNApurification kit”(日本genetics公司制),从反应液中精制核酸。
[对RPA反应的扩增产物进行分析]
在HCT116细胞的CDKN2A(p14ARF)基因中,仅单侧等位基因缺失一个G(Gx4)。另外,在HCT116细胞的CDKN2A(p14ARF)基因中,Gx4等位基因中的Gx4位置周围的胞嘧啶未经甲基化修饰,Gx5等位基因中的Gx5位置周围的胞嘧啶经过甲基化修饰。因而,在向RPA反应的反应液中添加MBD2蛋白质的情况下,通过PCR确认是否抑制了以Gx5等位基因为模板的RPA扩增。具体而言,为了扩增CDKN2A(p14ARF)基因,使用正向引物(hp14ARF-Ex1-F)和反向引物(hp14ARF-Ex1-R),用PCR扩增被两个引物夹持的碱基序列。使用“AmpliTaq Gold(注册商标)360Master Mix”(ThermoFisher Scientific公司制)进行PCR。制备PCR反应液,10μLPCR反应液中包含0.5μL RPA反应后DNA、0.5μM hp14ARF-Ex1-F引物及HP14ARF-Ex1-R引物。PCR反应时,首先,在95℃下变性10分钟,接着,按照95℃、15秒;60℃、30秒;及72℃、30秒进行30循环,然后,在72℃下处理1分钟。
[表8]

PCR反应后,该反应液不进行精制,取2μL进行电泳。结果无论RPA反应中是否添加MBD2蛋白质,均确认到PCR扩增产物。对各扩增产物的碱基序列进行确认,结果在将未添加MBD2蛋白质的RPA反应的扩增产物DNA作为模板进行PCR的情况下,Gx4型p14ARF和Gx5型p14ARF均被扩增。另一方面,在添加MBD2蛋白质进行RPA反应并将得到的RPA反应的扩增产物DNA作为模板进行PCR的情况下,Gx4型p14ARF优先被扩增,Gx5型p14ARF的扩增减少。以上结果表明,在以基因组DNA库为模板,并使用使整个库扩增的引物组的情况下,通过使RPA反应的反应液中存在CpG甲基化DNA结合蛋白质,抑制将经CpG甲基化的DNA作为模板的核酸扩增,仅能够扩增未经CpG甲基化的DNA。
虽然以RPA反应后的扩增产物为模板,通过PCR扩增及DNA序列分析(桑格序列)确认了有无CpG甲基化DNA的扩增,但也可以使用新一代序列分析来代替DNA序列分析。即,通过新一代序列对以基因组DNA库为模板、在MBD2蛋白质存在下所进行的RPA反应的扩增产物的碱基序列进行全面分析,并与以该基因组DNA库为模板、在不存在MBD2蛋白质时所进行的RPA反应的扩增产物的碱基序列进行比较,从而能够全面鉴定基因组上的CpG甲基化位点。
序列表
<110> 株式会社Epigeneron
<120> PC-30396
<130> 靶核酸的检测方法、核酸结合分子的检测方法、及核酸结合能力的评价方法
<160> 38
<210> 1
<211> 42
<212> RNA
<213> 人工序列
<220>
<223> crRNA_KRAS
<400> 1
guaguuggag cugguggcgu guuuuagagc uaugcuguuu ug 42
<210> 2
<211> 42
<212> RNA
<213> 人工序列
<220>
<223> crRNA_KRAS#2
<400> 2
cuugugguag uuggagcugg guuuuagagc uaugcuguuu ug 42
<210> 3
<211> 42
<212> RNA
<213> 人工序列
<220>
<223> crRNA_hp16_Gx5#2
<400> 3
acggccgcgg cccggggguc guuuuagagc uaugcuguuu ug 42
<210> 4
<211> 42
<212> RNA
<213> 人工序列
<220>
<223> crRNA_KRAS_mut
<400> 4
guaguuggag cuggaggcgu guuuuagagc uaugcuguuu ug 42
<210> 5
<211> 40
<212> RNA
<213> 人工序列
<220>
<223> crRNA_mid2
<400> 5
caccuccucu acccgacccc guuuuagagc uaugcuguuu 40
<210> 6
<211> 69
<212> RNA
<213> 人工序列
<220>
<223> tracrRNA
<400> 6
aaacagcaua gcaaguuaaa auaaggcuag uccguuauca acuugaaaaa guggcaccga 60
gucggugcu 69
<210> 7
<211> 34
<212> DNA
<213> 人工序列
<220>
<223> KRAS-RPA-G12-F 引物
<400> 7
tagtgtatta accttatgtg tgacatgttc taat 34
<210> 8
<211> 34
<212> DNA
<213> 人工序列
<220>
<223> KRAS-RPA-G12-R 引物
<400> 8
aaacaagatt tacctctatt gttggatcat attc 34
<210> 9
<211> 34
<212> DNA
<213> 人工序列
<220>
<223> p16-RPA-F2 引物
<400> 9
ggcggcgggg agcagcatgg agccttcggc tgac 34
<210> 10
<211> 34
<212> DNA
<213> 人工序列
<220>
<223> p16-RPA-R2 引物
<400> 10
ctacccacct ggatcggcct ccgaccgtaa ctat 34
<210> 11
<211> 30
<212> DNA
<213> 人工序列
<220>
<223> p14-RPA-F 引物
<400> 11
gtcccagtct gcagttaagg gggcaggagt 30
<210> 12
<211> 28
<212> DNA
<213> 人工序列
<220>
<223> p14-RPA-R 引物
<400> 12
gggcctttcc tacctggtct tctaggaa 28
<210> 13
<211> 32
<212> DNA
<213> 人工序列
<220>
<223> p16-RPA-F 引物
<400> 13
gaggaagaaa gaggaggggc tggctggtca cc 32
<210> 14
<211> 32
<212> DNA
<213> 人工序列
<220>
<223> p16-RPA-R 引物
<400> 14
ctgcagaccc tctacccacc tggatcggcc tc 32
<210> 15
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> p14ARF-CpG_island-F 引物
<400> 15
gtgggtccca gtctgcagtt aag 23
<210> 16
<211> 22
<212> DNA
<213> 人工序列
<220>
<223> p14ARF-CpG_island-R 引物
<400> 16
acttttcgag ggcctttcct ac 22
<210> 17
<211> 30
<212> DNA
<213> 人工序列
<220>
<223> Pax5-LexA-RPA-F 引物
<400> 17
gcatcagtcg cccttcgcct ccttctctcg 30
<210> 18
<211> 30
<212> DNA
<213> 人工序列
<220>
<223> Pax5-LexA-RPA-R 引物
<400> 18
gcgagggcgg aacgtgactt tgccctgcgg 30
<210> 19
<211> 64
<212> RNA
<213> 人工序列
<220>
<223> gRNA_NEAT1
<400> 19
gauuuagacu accccaaaaa cgaaggggac uaaaaccauc aaucugcguu guggcaucaa 60
cguu 64
<210> 20
<211> 65
<212> RNA
<213> 人工序列
<220>
<223> gRNA_NEAT1_2
<400> 20
gauuuagacu accccaaaaa cgaaggggac uaaaacuauc ucuaaccaac ccucuccccu 60
ucuuc 65
<210> 21
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 人 NEAT1-F2 引物
<400> 21
ctaaattgag cctccggtca 20
<210> 22
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 人NEAT1-R2 引物
<400> 22
acaagaaggc aggcaaacag 20
<210> 23
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> MY-0119 引物
<400> 23
cactggtact gggagggatg 20
<210> 24
<211> 22
<212> DNA
<213> 人工序列
<220>
<223> MY-0129 引物
<400> 24
cccttcaacc tgcatttcct ac 22
<210> 25
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> hGAPDH-dCas13a-F3 引物
<400> 25
gagccaaaag ggtcatcatc tct 23
<210> 26
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> hGAPDH-dCas13a-R3 引物
<400> 26
cacgatacca aagttgtcat gga 23
<210> 27
<211> 156
<212> DNA
<213> 人工序列
<220>
<223> 小鼠Cis基因启动子中的Cis区
<400> 27
caactctagg agctcccgcc cagttttcct ggaaagttct tggaaatctg tcaaaggtgt 60
ttcctttctc ggtccaaagc actagacgcc tgcacccccg ttcccctccg ggccgccgca 120
aagcccgcgg ttctaggaag atgaggcttc cgggaa 156
<210> 28
<211> 153
<212> DNA
<213> 人工序列
<220>
<223> CisM区
<400> 28
caactctagg agctcccgcc cagttggcct ggccagggct tggccatctg tcaaaggtgt 60
ttcctttctc ggtccaaagc actagacgcc tgcacccccg ttcccctccg ggccgccgca 120
aagcccgcgg ggctaggccg atgaggcggc caa 153
<210> 29
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> mCis_-259/-199_F 引物
<400> 29
caactctagg agctcccgcc 20
<210> 30
<211> 19
<212> DNA
<213> 人工序列
<220>
<223> mCis_-188/-104_R 引物
<400> 30
ttcccggaag cctcatctt 19
<210> 31
<211> 52
<212> DNA
<213> 人工序列
<220>
<223> mCis_-259/-199_mut_F 引物
<400> 31
caactctagg agctcccgcc cagttggcct ggccagggct tggccatctg tc 52
<210> 32
<211> 32
<212> DNA
<213> 人工序列
<220>
<223> mCis_-188/-104_mut_R 引物
<400> 32
ggcccggccg cctcatcggc ctagccccgc gg 32
<210> 33
<211> 17
<212> DNA
<213> 人工序列
<220>
<223> M13 Primer RV 引物
<400> 33
caggaaacag ctatgac 17
<210> 34
<211> 17
<212> DNA
<213> 人工序列
<220>
<223> M13 Primer M4 引物
<400> 34
gttttcccag tcacgac 17
<210> 35
<211> 33
<212> DNA
<213> 人工序列
<220>
<223> P5-NGS-Lib-Promega-RPA-F 引物
<400> 35
atacggcgac caccgagatc tacactcttt ccc 33
<210> 36
<211> 33
<212> DNA
<213> 人工序列
<220>
<223> P7-NGS-Lib-Promega-RPA-R 引物
<400> 36
caagcagaag acggcatacg agattctgac atg 33
<210> 37
<211> 22
<212> DNA
<213> 人工序列
<220>
<223> hp14ARF-Ex1-F 引物
<400> 37
agtgagggtt ttcgtggttc ac 22
<210> 38
<211> 22
<212> DNA
<213> 人工序列
<220>
<223> hp14ARF-Ex1-R 引物
<400> 38
cctagacgct ggctcctcag ta 22
Claims (26)
1.一种靶核酸的检测方法,从碱基序列或修饰状态与所述靶核酸部分不同的非靶核酸中识别并检测靶核酸,
将所述非靶核酸中碱基序列或修饰状态与所述靶核酸不同的区域作为靶区域,将所述靶核酸中碱基序列或修饰状态与所述非靶核酸不同的区域作为对应靶区域,以供试核酸样本为模板,在与所述非靶核酸中的所述靶区域特异性结合的分子存在下,使用与所述靶核酸及所述非靶核酸双方杂交的引物,在所述分子能够与所述非靶核酸结合的温度条件下进行核酸扩增反应,基于有无扩增产物来检测所述靶核酸,其中,与所述非靶核酸中的所述靶区域特异性结合的分子不包括仅由核酸构成的分子。
2.根据权利要求1所述的靶核酸的检测方法,其中,
在通过所述核酸扩增反应得到了扩增产物的情况下,所述供试核酸样本中包含所述靶核酸。
3.根据权利要求1或2所述的靶核酸的检测方法,其中,在65℃以下的温度条件下进行所述核酸扩增反应。
4.根据权利要求1~3中任一项所述的靶核酸的检测方法,其中,所述核酸扩增反应为等温核酸扩增反应。
5.根据权利要求1~4中任一项所述的靶核酸的检测方法,其中,所述核酸扩增反应为重组酶聚合酶扩增法。
6.根据权利要求1~5中任一项所述的靶核酸的检测方法,其中,所述靶核酸中的所述对应靶区域和所述非靶核酸中的所述靶区域的碱基序列不同。
7.根据权利要求6所述的靶核酸的检测方法,其中,所述靶区域为基因突变的突变位点或基因多态性的多态性位点。
8.根据权利要求1~7中任一项所述的靶核酸的检测方法,其中,所述分子为DNA链断裂活性缺失型Cas9蛋白质和gRNA的复合物。
9.根据权利要求8所述的靶核酸的检测方法,其中,所述gRNA特异性识别并结合由与所述非靶核酸中的所述靶区域互补的碱基序列构成的DNA。
10.根据权利要求1~9中任一项所述的靶核酸的检测方法,其中,所述供试核酸样本经亚硫酸氢盐处理,
所述对应靶区域和所述靶区域的甲基化状态存在差别,包含通过亚硫酸氢盐处理而在所述对应靶区域和所述靶区域之间产生差别的碱基。
11.根据权利要求1~7中任一项所述的靶核酸的检测方法,其中,所述分子为RNA链断裂活性缺失型Cas13a蛋白质和gRNA的复合物。
12.根据权利要求11所述的靶核酸的检测方法,其中,所述gRNA特异性识别并结合由与所述非靶核酸中的所述靶区域互补的碱基序列构成的RNA。
13.根据权利要求1~5中任一项所述的靶核酸的检测方法,其中,所述靶核酸中的所述对应靶区域和所述非靶核酸中的所述靶区域的修饰状态不同。
14.根据权利要求13所述的靶核酸的检测方法,其中,所述靶核酸中的所述对应靶区域为未经CpG甲基化修饰的区域,
所述非靶核酸中的所述靶区域为经CpG甲基化修饰的区域,
所述分子为CpG甲基化DNA结合蛋白质。
15.一种用于检测靶核酸的试剂盒,用于权利要求8~10中任一项所述的靶核酸的检测方法,具有:
与所述靶核酸及所述非靶核酸双方杂交的引物、
DNA链断裂活性缺失型Cas9蛋白质、及
gRNA。
16.一种用于检测靶核酸的试剂盒,用于权利要求14所述的靶核酸的检测方法,具有:
与所述靶核酸及所述非靶核酸双方杂交的引物、及
CpG甲基化DNA结合蛋白质。
17.根据权利要求15或16所述的用于检测靶核酸的试剂盒,进一步包含重组酶、单链DNA结合蛋白质、及DNA聚合酶。
18.一种用于检测靶核酸的试剂盒,用于权利要求11或12所述的靶核酸的检测方法,具有:
与所述靶核酸及所述非靶核酸双方杂交的引物、
RNA链断裂活性缺失型Cas13a蛋白质、及
gRNA。
19.一种核酸结合分子的检测方法,是检测核酸结合分子的方法,
使用供试样本、核酸及与所述核酸杂交的引物进行核酸扩增反应,
在所述供试样本中包含所述核酸结合分子的情况下,通过所述核酸扩增反应不能得到扩增产物,在所述供试样本中不包含所述核酸结合分子的情况下,通过所述核酸扩增反应得到扩增产物。
20.根据权利要求19所述的核酸结合分子的检测方法,其中,所述核酸结合分子为碱基序列特异性地与核酸结合的分子。
21.根据权利要求19所述的核酸结合分子的检测方法,其中,所述核酸结合分子为CpG甲基化DNA结合蛋白质。
22.一种用于检测核酸结合分子的试剂盒,用于权利要求19~21中任一项所述的核酸结合分子的检测方法,具有:
核酸、及
与所述核酸杂交的引物。
23.一种核酸结合能力的评价方法,是评价供试物质对核酸的结合能力的方法,
使用供试物质、作为评价所述供试物质的核酸结合能力的对象的核酸、及与所述核酸杂交的引物,在所述供试物质能够与所述核酸结合的温度条件下进行核酸扩增反应,在得到了扩增产物的情况下,评价所述供试物质不具有对于所述核酸的结合能力,在未得到扩增产物的情况下,评价所述供试物质具有对于所述核酸的结合能力。
24.根据权利要求23所述的核酸结合能力的评价方法,其中,所述供试物质为DNA链断裂活性缺失型Cas9蛋白质和gRNA的复合物。
25.根据权利要求23所述的核酸结合能力的评价方法,其中,所述核酸为CpG甲基化DNA。
26.一种用于评价核酸结合能力的试剂盒,用于权利要求23~25中任一项所述的核酸结合能力的评价方法,具有:
作为评价所述供试物质的核酸结合能力的对象的核酸、及
与所述核酸杂交的引物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-191409 | 2019-10-18 | ||
| JP2019191409 | 2019-10-18 | ||
| PCT/JP2020/039128 WO2021075555A1 (ja) | 2019-10-18 | 2020-10-16 | 標的核酸の検出方法、核酸結合分子の検出方法、及び核酸結合能の評価方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN114555830A true CN114555830A (zh) | 2022-05-27 |
Family
ID=75538256
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202080072133.3A Pending CN114555830A (zh) | 2019-10-18 | 2020-10-16 | 靶核酸的检测方法、核酸结合分子的检测方法、及核酸结合能力的评价方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20230031001A1 (zh) |
| EP (1) | EP4047091A4 (zh) |
| JP (1) | JPWO2021075555A1 (zh) |
| KR (1) | KR20220083700A (zh) |
| CN (1) | CN114555830A (zh) |
| CA (1) | CA3156337A1 (zh) |
| TW (1) | TW202126817A (zh) |
| WO (1) | WO2021075555A1 (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11814689B2 (en) | 2021-07-21 | 2023-11-14 | Montana State University | Nucleic acid detection using type III CRISPR complex |
| CN116240200A (zh) * | 2022-07-01 | 2023-06-09 | 中国科学院基础医学与肿瘤研究所(筹) | 一种基于可编程核酸酶的超灵敏目标核酸富集检测方法 |
| GB202214125D0 (en) | 2022-09-27 | 2022-11-09 | Genomic Labs Ltd | Nucleic acid amplification; improved methods |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2718482T3 (es) | 2004-06-01 | 2019-07-02 | Alere San Diego Inc | Kit para amplificación de recombinasa polimerasa |
| JP5322471B2 (ja) * | 2008-03-27 | 2013-10-23 | シスメックス株式会社 | メチル化dnaの解析方法及びプライマーセット |
| WO2010022098A2 (en) * | 2008-08-18 | 2010-02-25 | Life Technologies Corporation | Dna methylation detection methods |
| WO2010065626A1 (en) * | 2008-12-02 | 2010-06-10 | Globeimmune, Inc. | Genotyping tools, methods and kits |
| WO2015058008A2 (en) * | 2013-10-18 | 2015-04-23 | California Institute Of Technology | Enhanced nucleic acid identification and detection |
| EP3795697B1 (en) * | 2014-02-04 | 2024-09-18 | Jumpcode Genomics, Inc. | Genome fractioning |
| JP6731942B2 (ja) * | 2014-12-19 | 2020-07-29 | エピゲノミクスアーゲー | CpGメチル化の検出方法及び癌の診断方法 |
| GB201604261D0 (en) * | 2016-03-11 | 2016-04-27 | Ucl Business Plc | Allele-specific gene suppression |
| KR101964746B1 (ko) * | 2016-09-07 | 2019-04-03 | 울산대학교 산학협력단 | dCas9 단백질 및 표적 핵산 서열에 결합하는 gRNA를 이용한 핵산 검출의 민감도 및 특이도 향상용 조성물 및 방법 |
| CN107034188B (zh) * | 2017-05-24 | 2018-07-24 | 中山大学附属口腔医院 | 一种靶向骨的外泌体载体、CRISPR/Cas9基因编辑系统及应用 |
| WO2019178346A1 (en) * | 2018-03-14 | 2019-09-19 | The Trustees Of The University Of Pennsylvania | Enrichment of nucleic acids |
| JP7086698B2 (ja) | 2018-04-26 | 2022-06-20 | キヤノン株式会社 | 画像形成装置、画像形成システム、及び画像形成方法 |
-
2020
- 2020-10-16 KR KR1020227012384A patent/KR20220083700A/ko unknown
- 2020-10-16 TW TW109136034A patent/TW202126817A/zh unknown
- 2020-10-16 EP EP20875877.1A patent/EP4047091A4/en not_active Withdrawn
- 2020-10-16 US US17/769,721 patent/US20230031001A1/en active Pending
- 2020-10-16 CA CA3156337A patent/CA3156337A1/en active Pending
- 2020-10-16 WO PCT/JP2020/039128 patent/WO2021075555A1/ja unknown
- 2020-10-16 JP JP2021552470A patent/JPWO2021075555A1/ja active Pending
- 2020-10-16 CN CN202080072133.3A patent/CN114555830A/zh active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| EP4047091A4 (en) | 2023-11-15 |
| CA3156337A1 (en) | 2021-04-22 |
| US20230031001A1 (en) | 2023-02-02 |
| KR20220083700A (ko) | 2022-06-20 |
| WO2021075555A1 (ja) | 2021-04-22 |
| TW202126817A (zh) | 2021-07-16 |
| EP4047091A1 (en) | 2022-08-24 |
| JPWO2021075555A1 (zh) | 2021-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230392191A1 (en) | Selective degradation of wild-type dna and enrichment of mutant alleles using nuclease | |
| EP2906715B1 (en) | Compositions, methods, systems and kits for target nucleic acid enrichment | |
| CN113166797A (zh) | 基于核酸酶的rna耗尽 | |
| EP1130113A1 (en) | Multiplex ligation dependent amplification assay | |
| KR20230116944A (ko) | 고온 내성 Cas 단백질의 용도, 표적 핵산 분자의 검출방법 및 시약 키트 | |
| US20130059734A1 (en) | Epigenetic analysis | |
| WO2013192292A1 (en) | Massively-parallel multiplex locus-specific nucleic acid sequence analysis | |
| CN114555830A (zh) | 靶核酸的检测方法、核酸结合分子的检测方法、及核酸结合能力的评价方法 | |
| WO2011139920A2 (en) | Methylation-specific competitive allele-specific taqman polymerase chain reaction (cast-pcr) | |
| KR101600039B1 (ko) | 염기 특이 반응성 프라이머를 이용한 핵산 증폭방법 | |
| EP3378948B1 (en) | Method for quantifying target nucleic acid and kit therefor | |
| CN109680044B (zh) | 一种基于选择性消除野生链背景干扰的基因突变检测方法 | |
| US20180051330A1 (en) | Methods of amplifying nucleic acids and compositions and kits for practicing the same | |
| CN111989408A (zh) | 检测靶核酸区域内的与参考序列的差异的方法 | |
| US6316192B1 (en) | Method for enrichment of unique DNA fragments through cyclical removal of PCR adapter attached to DNA fragments whose sequences are shared between two DNA pools | |
| CN117242190A (zh) | 单链dna的扩增 | |
| KR20230124636A (ko) | 멀티플렉스 반응에서 표적 서열의 고 감응성 검출을위한 조성물 및 방법 | |
| JP2003510011A (ja) | カップル性ポリメラーゼ連鎖反応−制限エンドヌクレアーゼ消化−リガーゼ検出反応法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |