CN113527311A - FGFR4 inhibitor, composition and application thereof in preparation of medicines - Google Patents
FGFR4 inhibitor, composition and application thereof in preparation of medicines Download PDFInfo
- Publication number
- CN113527311A CN113527311A CN202110965475.4A CN202110965475A CN113527311A CN 113527311 A CN113527311 A CN 113527311A CN 202110965475 A CN202110965475 A CN 202110965475A CN 113527311 A CN113527311 A CN 113527311A
- Authority
- CN
- China
- Prior art keywords
- cancer
- fgfr4
- compound
- pyrimidin
- dimethoxyphenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 229940125408 FGFR4 inhibitor Drugs 0.000 title claims abstract description 14
- 239000003814 drug Substances 0.000 title claims description 5
- 239000000203 mixture Substances 0.000 title description 21
- 229940079593 drug Drugs 0.000 title description 2
- 238000002360 preparation method Methods 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 89
- -1 LX01 Chemical class 0.000 claims abstract description 79
- 102100027844 Fibroblast growth factor receptor 4 Human genes 0.000 claims abstract description 46
- 101000917134 Homo sapiens Fibroblast growth factor receptor 4 Proteins 0.000 claims abstract description 44
- 230000002401 inhibitory effect Effects 0.000 claims abstract description 16
- 101100334745 Mus musculus Fgfr4 gene Proteins 0.000 claims abstract description 10
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 claims description 44
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 25
- 230000000694 effects Effects 0.000 claims description 22
- 210000004027 cell Anatomy 0.000 claims description 16
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 16
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 13
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 claims description 12
- 150000003839 salts Chemical class 0.000 claims description 10
- 206010028980 Neoplasm Diseases 0.000 claims description 9
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 8
- 201000011510 cancer Diseases 0.000 claims description 7
- 125000003118 aryl group Chemical group 0.000 claims description 6
- 125000000623 heterocyclic group Chemical group 0.000 claims description 6
- 239000003112 inhibitor Substances 0.000 claims description 6
- 201000007270 liver cancer Diseases 0.000 claims description 5
- 208000014018 liver neoplasm Diseases 0.000 claims description 5
- 239000008194 pharmaceutical composition Substances 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 206010006187 Breast cancer Diseases 0.000 claims description 3
- 208000026310 Breast neoplasm Diseases 0.000 claims description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 3
- 208000035475 disorder Diseases 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 2
- 208000009746 Adult T-Cell Leukemia-Lymphoma Diseases 0.000 claims description 2
- 208000016683 Adult T-cell leukemia/lymphoma Diseases 0.000 claims description 2
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 2
- 206010005003 Bladder cancer Diseases 0.000 claims description 2
- 208000011691 Burkitt lymphomas Diseases 0.000 claims description 2
- 208000017604 Hodgkin disease Diseases 0.000 claims description 2
- 208000021519 Hodgkin lymphoma Diseases 0.000 claims description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 2
- 206010052178 Lymphocytic lymphoma Diseases 0.000 claims description 2
- 206010025323 Lymphomas Diseases 0.000 claims description 2
- 206010027406 Mesothelioma Diseases 0.000 claims description 2
- 208000034578 Multiple myelomas Diseases 0.000 claims description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 2
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 2
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 claims description 2
- 125000003647 acryloyl group Chemical group O=C([*])C([H])=C([H])[H] 0.000 claims description 2
- 239000002671 adjuvant Substances 0.000 claims description 2
- 201000006966 adult T-cell leukemia Diseases 0.000 claims description 2
- 230000001684 chronic effect Effects 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 206010017758 gastric cancer Diseases 0.000 claims description 2
- 208000005017 glioblastoma Diseases 0.000 claims description 2
- 201000010536 head and neck cancer Diseases 0.000 claims description 2
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 2
- 208000032839 leukemia Diseases 0.000 claims description 2
- 201000005202 lung cancer Diseases 0.000 claims description 2
- 208000020816 lung neoplasm Diseases 0.000 claims description 2
- 230000001404 mediated effect Effects 0.000 claims description 2
- 201000001441 melanoma Diseases 0.000 claims description 2
- 239000012038 nucleophile Substances 0.000 claims description 2
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 2
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 2
- 201000011549 stomach cancer Diseases 0.000 claims description 2
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 2
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims 2
- 238000004519 manufacturing process Methods 0.000 claims 2
- 206010008342 Cervix carcinoma Diseases 0.000 claims 1
- 206010009944 Colon cancer Diseases 0.000 claims 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims 1
- 206010014733 Endometrial cancer Diseases 0.000 claims 1
- 206010014759 Endometrial neoplasm Diseases 0.000 claims 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims 1
- 208000022072 Gallbladder Neoplasms Diseases 0.000 claims 1
- 208000008839 Kidney Neoplasms Diseases 0.000 claims 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims 1
- 206010033128 Ovarian cancer Diseases 0.000 claims 1
- 206010061535 Ovarian neoplasm Diseases 0.000 claims 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims 1
- 206010060862 Prostate cancer Diseases 0.000 claims 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims 1
- 206010038389 Renal cancer Diseases 0.000 claims 1
- 208000000453 Skin Neoplasms Diseases 0.000 claims 1
- 208000024313 Testicular Neoplasms Diseases 0.000 claims 1
- 206010057644 Testis cancer Diseases 0.000 claims 1
- 208000024770 Thyroid neoplasm Diseases 0.000 claims 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims 1
- 239000012472 biological sample Substances 0.000 claims 1
- 201000010881 cervical cancer Diseases 0.000 claims 1
- 201000004101 esophageal cancer Diseases 0.000 claims 1
- 201000010175 gallbladder cancer Diseases 0.000 claims 1
- 201000010982 kidney cancer Diseases 0.000 claims 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims 1
- 201000002528 pancreatic cancer Diseases 0.000 claims 1
- 208000008443 pancreatic carcinoma Diseases 0.000 claims 1
- 201000000849 skin cancer Diseases 0.000 claims 1
- 201000003120 testicular cancer Diseases 0.000 claims 1
- 201000002510 thyroid cancer Diseases 0.000 claims 1
- 235000018417 cysteine Nutrition 0.000 abstract description 14
- 150000001945 cysteines Chemical class 0.000 abstract description 13
- 108091000080 Phosphotransferase Proteins 0.000 abstract description 11
- 102000020233 phosphotransferase Human genes 0.000 abstract description 11
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 abstract description 3
- DVBPRWJMHURKHP-UHFFFAOYSA-N N-[4-[[3-(3,5-dimethoxyphenyl)-7-[4-(4-methylpiperazin-1-yl)anilino]-2-oxo-4H-pyrimido[4,5-d]pyrimidin-1-yl]methyl]phenyl]prop-2-enamide Chemical compound COC1=CC(=CC(OC)=C1)N1CC2=CN=C(NC3=CC=C(C=C3)N3CCN(C)CC3)N=C2N(CC2=CC=C(NC(=O)C=C)C=C2)C1=O DVBPRWJMHURKHP-UHFFFAOYSA-N 0.000 abstract description 2
- 238000002474 experimental method Methods 0.000 abstract description 2
- 238000012360 testing method Methods 0.000 abstract description 2
- OKGNMRKOGWTADH-UHFFFAOYSA-N 1,4-dihydropyrimidine Chemical compound C1C=CNC=N1 OKGNMRKOGWTADH-UHFFFAOYSA-N 0.000 abstract 1
- HNYOPLTXPVRDBG-UHFFFAOYSA-N barbituric acid Chemical compound O=C1CC(=O)NC(=O)N1 HNYOPLTXPVRDBG-UHFFFAOYSA-N 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 150
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 77
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 73
- 238000006243 chemical reaction Methods 0.000 description 70
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 68
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 63
- 230000015572 biosynthetic process Effects 0.000 description 46
- 238000003786 synthesis reaction Methods 0.000 description 46
- 239000002904 solvent Substances 0.000 description 39
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 38
- 239000003208 petroleum Substances 0.000 description 33
- 239000007787 solid Substances 0.000 description 33
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 32
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 30
- 238000005160 1H NMR spectroscopy Methods 0.000 description 26
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 26
- 239000012074 organic phase Substances 0.000 description 25
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 24
- 239000000243 solution Substances 0.000 description 21
- 238000010898 silica gel chromatography Methods 0.000 description 20
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical compound OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 16
- 239000007858 starting material Substances 0.000 description 16
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 13
- 239000005457 ice water Substances 0.000 description 13
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 239000007788 liquid Substances 0.000 description 12
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 11
- 238000010791 quenching Methods 0.000 description 11
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- 108091008794 FGF receptors Proteins 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 8
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 description 8
- 238000001819 mass spectrum Methods 0.000 description 8
- 238000000034 method Methods 0.000 description 8
- 238000012544 monitoring process Methods 0.000 description 8
- 239000002994 raw material Substances 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 238000001035 drying Methods 0.000 description 7
- QSNVLKZLTDYRKM-UHFFFAOYSA-N ClC1=NC=C(C(=N1)NCC1=CC=C(C=C1)[N+](=O)[O-])CNC1=CC(=CC(=C1)OC)OC Chemical compound ClC1=NC=C(C(=N1)NCC1=CC=C(C=C1)[N+](=O)[O-])CNC1=CC(=CC(=C1)OC)OC QSNVLKZLTDYRKM-UHFFFAOYSA-N 0.000 description 6
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 6
- HFBMWMNUJJDEQZ-UHFFFAOYSA-N acryloyl chloride Chemical compound ClC(=O)C=C HFBMWMNUJJDEQZ-UHFFFAOYSA-N 0.000 description 6
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- 102100031734 Fibroblast growth factor 19 Human genes 0.000 description 5
- 101000846394 Homo sapiens Fibroblast growth factor 19 Proteins 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- METKIMKYRPQLGS-UHFFFAOYSA-N atenolol Chemical compound CC(C)NCC(O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-UHFFFAOYSA-N 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 229940125904 compound 1 Drugs 0.000 description 5
- 125000000753 cycloalkyl group Chemical group 0.000 description 5
- JOXWSDNHLSQKCC-UHFFFAOYSA-N ethenesulfonamide Chemical compound NS(=O)(=O)C=C JOXWSDNHLSQKCC-UHFFFAOYSA-N 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- RFIOZSIHFNEKFF-UHFFFAOYSA-M piperazine-1-carboxylate Chemical compound [O-]C(=O)N1CCNCC1 RFIOZSIHFNEKFF-UHFFFAOYSA-M 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- 239000007868 Raney catalyst Substances 0.000 description 4
- 229910000564 Raney nickel Inorganic materials 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 4
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 4
- 229910000024 caesium carbonate Inorganic materials 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 4
- 238000003828 vacuum filtration Methods 0.000 description 4
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 4
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 3
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 3
- FCMRHMPITHLLLA-UHFFFAOYSA-N 2-methyl-6-nitroaniline Chemical compound CC1=CC=CC([N+]([O-])=O)=C1N FCMRHMPITHLLLA-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 230000001594 aberrant effect Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 229940126142 compound 16 Drugs 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 229940126214 compound 3 Drugs 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 239000012452 mother liquor Substances 0.000 description 3
- 239000010413 mother solution Substances 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- BWZVCCNYKMEVEX-UHFFFAOYSA-N 2,4,6-Trimethylpyridine Chemical compound CC1=CC(C)=NC(C)=C1 BWZVCCNYKMEVEX-UHFFFAOYSA-N 0.000 description 2
- VLOODHWTRPYFIS-UHFFFAOYSA-N 2,4-dichloro-5-(chloromethyl)pyrimidine Chemical compound ClCC1=CN=C(Cl)N=C1Cl VLOODHWTRPYFIS-UHFFFAOYSA-N 0.000 description 2
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 2
- WDHHKNXRNAUJNX-UHFFFAOYSA-N 2-chloro-5-[(3,5-dimethoxyanilino)methyl]-N-[(3-nitrophenyl)methyl]pyrimidin-4-amine Chemical compound COC1=CC(NCC(C(NCC2=CC([N+]([O-])=O)=CC=C2)=N2)=CN=C2Cl)=CC(OC)=C1 WDHHKNXRNAUJNX-UHFFFAOYSA-N 0.000 description 2
- WSQUCDWVLAXPNE-UHFFFAOYSA-N 2-chloro-5-[(3,5-dimethoxyanilino)methyl]-N-[2-(4-nitrophenyl)ethyl]pyrimidin-4-amine Chemical compound COC1=CC(NCC(C(NCCC(C=C2)=CC=C2[N+]([O-])=O)=N2)=CN=C2Cl)=CC(OC)=C1 WSQUCDWVLAXPNE-UHFFFAOYSA-N 0.000 description 2
- IMCBZLGYAYZSAM-UHFFFAOYSA-N 2-cyano-3-methylbut-2-enoic acid Chemical compound CC(C)=C(C#N)C(O)=O IMCBZLGYAYZSAM-UHFFFAOYSA-N 0.000 description 2
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 2
- JDBGXEHEIRGOBU-UHFFFAOYSA-N 5-hydroxymethyluracil Chemical compound OCC1=CNC(=O)NC1=O JDBGXEHEIRGOBU-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000167854 Bourreria succulenta Species 0.000 description 2
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 102000044168 Fibroblast Growth Factor Receptor Human genes 0.000 description 2
- 101710182387 Fibroblast growth factor receptor 4 Proteins 0.000 description 2
- 229930091371 Fructose Natural products 0.000 description 2
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 2
- 239000005715 Fructose Substances 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 108010087230 Sincalide Proteins 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000010609 cell counting kit-8 assay Methods 0.000 description 2
- 239000012295 chemical reaction liquid Substances 0.000 description 2
- 235000019693 cherries Nutrition 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940125810 compound 20 Drugs 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 229940125961 compound 24 Drugs 0.000 description 2
- 229940125846 compound 25 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 229940125808 covalent inhibitor Drugs 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- MLIREBYILWEBDM-UHFFFAOYSA-N cyanoacetic acid Chemical compound OC(=O)CC#N MLIREBYILWEBDM-UHFFFAOYSA-N 0.000 description 2
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 2
- 230000006837 decompression Effects 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- KFOZNPPBKHYHQD-UHFFFAOYSA-N ethenesulfonyl chloride Chemical compound ClS(=O)(=O)C=C KFOZNPPBKHYHQD-UHFFFAOYSA-N 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- MGZKYOAQVGSSGC-DLBZAZTESA-N fisogatinib Chemical compound COc1cc(OC)c(Cl)c(c1Cl)-c1ccc2nc(N[C@@H]3COCC[C@@H]3NC(=O)C=C)ncc2c1 MGZKYOAQVGSSGC-DLBZAZTESA-N 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000004077 genetic alteration Effects 0.000 description 2
- 231100000118 genetic alteration Toxicity 0.000 description 2
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- PCMORTLOPMLEFB-ONEGZZNKSA-N sinapic acid Chemical compound COC1=CC(\C=C\C(O)=O)=CC(OC)=C1O PCMORTLOPMLEFB-ONEGZZNKSA-N 0.000 description 2
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- DQLCYLFCLQPLSY-UHFFFAOYSA-N tert-butyl 4-(3-aminopropyl)piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCN(CCCN)CC1 DQLCYLFCLQPLSY-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- DLZXLCHQWOZGSE-UHFFFAOYSA-N (3-nitrophenyl)methylazanium;chloride Chemical compound Cl.NCC1=CC=CC([N+]([O-])=O)=C1 DLZXLCHQWOZGSE-UHFFFAOYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- KEIPNCCJPRMIAX-HNNXBMFYSA-N 1-[(3s)-3-[4-amino-3-[2-(3,5-dimethoxyphenyl)ethynyl]pyrazolo[3,4-d]pyrimidin-1-yl]pyrrolidin-1-yl]prop-2-en-1-one Chemical compound COC1=CC(OC)=CC(C#CC=2C3=C(N)N=CN=C3N([C@@H]3CN(CC3)C(=O)C=C)N=2)=C1 KEIPNCCJPRMIAX-HNNXBMFYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- BTTNYQZNBZNDOR-UHFFFAOYSA-N 2,4-dichloropyrimidine Chemical compound ClC1=CC=NC(Cl)=N1 BTTNYQZNBZNDOR-UHFFFAOYSA-N 0.000 description 1
- JVMHULJEYUQYSH-UHFFFAOYSA-N 2-(4-nitrophenyl)ethylazanium;chloride Chemical compound Cl.NCCC1=CC=C([N+]([O-])=O)C=C1 JVMHULJEYUQYSH-UHFFFAOYSA-N 0.000 description 1
- WLNLHBMVRFALDZ-UHFFFAOYSA-N 2-N-(3-bromopropyl)benzene-1,2-diamine Chemical compound BrCCCNC1=C(C=CC=C1)N WLNLHBMVRFALDZ-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- VHCSBTPOPKFYIU-UHFFFAOYSA-N 2-chloroethanesulfonyl chloride Chemical compound ClCCS(Cl)(=O)=O VHCSBTPOPKFYIU-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- WNRGWPVJGDABME-UHFFFAOYSA-N 3,5-Dimethoxyaniline Chemical compound COC1=CC(N)=CC(OC)=C1 WNRGWPVJGDABME-UHFFFAOYSA-N 0.000 description 1
- PUIXMSRTTHLNKI-UHFFFAOYSA-N 6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)-8-[3-(4-prop-2-enoylpiperazin-1-yl)propyl]pyrido[2,3-d]pyrimidin-7-one Chemical compound C(C=C)(=O)N1CCN(CC1)CCCN1C(C(=CC2=C1N=C(N=C2)NC)C2=C(C(=CC(=C2Cl)OC)OC)Cl)=O PUIXMSRTTHLNKI-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102100020683 Beta-klotho Human genes 0.000 description 1
- 101710104526 Beta-klotho Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- ZEOWTGPWHLSLOG-UHFFFAOYSA-N Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F Chemical compound Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F ZEOWTGPWHLSLOG-UHFFFAOYSA-N 0.000 description 1
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 1
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 1
- 102100023600 Fibroblast growth factor receptor 2 Human genes 0.000 description 1
- 101710182389 Fibroblast growth factor receptor 2 Proteins 0.000 description 1
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 description 1
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 238000012752 Hepatectomy Methods 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- MBWRLLRCTIYXDW-UHFFFAOYSA-N N-[2-[[6-[(2,6-dichloro-3,5-dimethoxyphenyl)carbamoyl-methylamino]pyrimidin-4-yl]amino]-5-(4-ethylpiperazin-1-yl)phenyl]prop-2-enamide Chemical compound ClC1=C(C(=C(C=C1OC)OC)Cl)NC(N(C)C1=CC(=NC=N1)NC1=C(C=C(C=C1)N1CCN(CC1)CC)NC(C=C)=O)=O MBWRLLRCTIYXDW-UHFFFAOYSA-N 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 229940125828 TAS-120 Drugs 0.000 description 1
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- CIUQDSCDWFSTQR-UHFFFAOYSA-N [C]1=CC=CC=C1 Chemical compound [C]1=CC=CC=C1 CIUQDSCDWFSTQR-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000005236 alkanoylamino group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000009697 arginine Nutrition 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000004658 aryl carbonyl amino group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229910001570 bauxite Inorganic materials 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 239000006189 buccal tablet Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 230000005907 cancer growth Effects 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000007958 cherry flavor Substances 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 210000000232 gallbladder Anatomy 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 238000009650 gentamicin protection assay Methods 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 239000007887 hard shell capsule Substances 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 239000013038 irreversible inhibitor Substances 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000003674 kinase activity assay Methods 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 235000018977 lysine Nutrition 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- XONPDZSGENTBNJ-UHFFFAOYSA-N molecular hydrogen;sodium Chemical compound [Na].[H][H] XONPDZSGENTBNJ-UHFFFAOYSA-N 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- TXEBNKKOLVBTFK-UHFFFAOYSA-N n-[2-[[6-(2,6-dichloro-3,5-dimethoxyphenyl)quinazolin-2-yl]amino]-3-methylphenyl]prop-2-enamide Chemical compound COC1=CC(OC)=C(Cl)C(C=2C=C3C=NC(NC=4C(=CC=CC=4C)NC(=O)C=C)=NC3=CC=2)=C1Cl TXEBNKKOLVBTFK-UHFFFAOYSA-N 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 238000009520 phase I clinical trial Methods 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- PCMORTLOPMLEFB-UHFFFAOYSA-N sinapinic acid Natural products COC1=CC(C=CC(O)=O)=CC(OC)=C1O PCMORTLOPMLEFB-UHFFFAOYSA-N 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007886 soft shell capsule Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000007892 solid unit dosage form Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000025366 tissue development Effects 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000009637 wintergreen oil Substances 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention provides an FGFR4 inhibitor which takes 3, 4-dihydropyrimidine [4,5-d ] pyrimidine-2 (1H) -ketone as a parent nucleus and has a covalent structure. The 9 specific compounds are provided in the example and kinase inhibitory activity tests are carried out on the 9 compounds, wherein the half inhibitory concentration of LX08 on the kinase inhibitory activity of FGFR4 is only 7nM, and the half inhibitory concentration is lower than that of FIIN-2 of a specific control, so that the compound has potential application prospect. In addition, through MALDI-TOF mass spectrometry combination experiments on the synthesized compounds, the compounds such as LX01, LX05, LX06, LX07 and LX08 are found to be only capable of being covalently combined with Cys552 in FGFR4 and not capable of being covalently combined with Cys477 in FGFR4, and the compound such as LX09 is an FGFR4 inhibitor capable of being covalently combined with two Cys552 and Cys477 cysteines in LX FGFR 4.
Description
Technical Field
Described herein are compounds, methods of making the compounds, and methods of using the compounds and compositions to inhibit tyrosine kinase activity.
Background
Fibroblast Growth Factor Receptors (FGFRs) are a family of receptor tyrosine kinases, including FGFR1, FGFR2, FGFR3, FGFR4 and the high affinity receptors for the other 18 different FGF ligands. These ligand-receptor combinations modulate a variety of signaling and endocrine activities during human tissue development. Genetic alterations of FGFR, including mutations, fusions, and gene amplifications, result in aberrant signaling pathways that activate and drive the growth of cancer cells. Researchers have detected genetic alterations in FGFR in a variety of cancer types, including breast cancer, liver cancer, squamous non-small cell lung cancer, squamous head and neck cancer, cholangiocarcinoma, and the like. Clinical validation of FGFR as a therapeutic target has been demonstrated in bladder cancer, liver cancer, lung cancer, breast cancer, and gastric cancer.
In recent years, aberrant fibroblast growth factor receptor 4(FGFR4) signaling has been identified as a major driver of HCC tumorigenesis and progression. FGFR4 is the most highly expressed isoform in hepatocytes, and its ligand FGF19 and co-receptor beta-Klotho bind exclusively to FGFR4 to regulate hepatocyte proliferation, and excessive FGF19 protein increases the probability of proliferation and invasion of HCC cell lines. The liver cancer heterotypic transplanted mouse with high FGFR4 expression can effectively eliminate liver cancer generation in a mouse model by inhibiting the generation of FGF19-FGFR4 or using an FGFR4 antibody. Clinical studies showed that half of all HCC patients had FGFR4 overexpressed, and most of the HCC patients had both FGF19 and FGFR4 upregulated, whereas FGF19 levels were positively correlated with tumor size and post-hepatectomy recurrence, and HCC patients with FGF19 overexpressed survived five years shorter than HCC patients with low expression of FGF 19. Therefore, FGFR4 selective inhibitors can be developed to treat cancer patients driven by aberrant FGFR4 signaling.
The FGFR4 plays an important role in cancer cell metastasis and drug resistance, and the FGFR irreversible inhibitor with good FGFR4 inhibition effect has wide application prospect. Among the covalent inhibitors with high potency and selectivity for inhibition of FGFR4, BLU9931, BLU-554 and H3B-6527 are all covalently bound to Cys552 sulfhydryl group of the hinge region of FGFR4 protein, while PRN1371, FIIN-2 and TAS-120 are covalently bound to Cys477 in the p-loop of FGFR4 protein, all of which are covalently bound to only one cysteine residue, and no inhibitor has been found that can be covalently bound to both cysteine residues in FGFR4 protein. And some of these irreversible inhibitors produced resistance mutations during clinical trials, such as Cys552 mutation in hepatoma cells found in phase I clinical trials of BLU-554. The FGFR4 double-covalent inhibitor capable of being covalently bound with two cysteines in FGFR4 protein simultaneously is developed through strategies of drug combination, group replacement, carbon chain growth, structure simplification and the like.
Disclosure of Invention
The invention aims to provide a structurally optimized FGFR4 inhibitor which has excellent effect of inhibiting fibroblast growth factor receptor 4.
The invention also provides an inhibitor of FGFR4, which contains an FGFR4 double covalent inhibitor capable of being covalently combined with two cysteines (Cys477 and Cys552) in an FGFR4 protein.
The compounds of the present invention have the structure of formula I:
formula I

Wherein R is1Refers to a moiety capable of forming a covalent bond with a nucleophile; r2Is an aryl or heterocyclic group; l is- [ C (R5) (R6)]q-, wherein R5 and R6 are each independently H or C1-C6 alkyl, wherein q is 1-3; wherein A is phenyl, R3Is hydrogen or methyl on the phenyl radical A.
In one embodiment, R1Is an acryloyl group.
In one embodiment, L is independently C1-C3 alkyl.
In one embodiment, R2Is phenyl.
In the synthesis examples of the present invention, the following compounds were synthesized:
n- (4- ((7- ((2-acrylamido-6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-)

Oxy-3, 4-dichloropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) acrylamide;
2-cyano-N- (4- ((7- ((2- (2-cyano-3-methyl-2-butenamido) -6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dihydropyrimidin [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) -3-methyl-2-butenamide;

n- (4- ((3- (3, 5-dimethoxyphenyl) -7- ((2-methyl-6- (ethenesulfonamido) phenyl) amino) -2-oxo-3, 4-dihydropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) ethenesulfonamide;

2-chloro-N- (4- ((7- ((2- (2-chloroacetylamino) -6-methylphenyl) amino) -3- (3, 5-dimethylphenyl) -2-oxo-3, 4-dihydropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) acetamide;

n- (3- ((7- ((2-acrylamido-6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dichloropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) acrylamide;

n- (2- ((8(3- (4-acryloylpiperazin-1-yl) propyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) -3-methylphenyl) acrylamide;

n- (2- ((8- (4-acrylamidobenzyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide;

n- (2- ((8- (4-acrylamidophenethyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide;

n- (2- ((8- (3- (4-acryloylpiperazin-1-yl) propyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide;

the compounds according to the invention are highly potent covalent inhibitors of FGFR4 specificity.
Drawings
Fig. 1 is a comparison of mass spectra of compound LX01 before and after binding to the two cysteines (Cys477 and Cys552) of FGFR 4;
fig. 2 is a comparison of mass spectra of compound LX05 before and after binding to the two cysteines (Cys477 and Cys552) of FGFR 4;
fig. 3 is a comparison of mass spectra of compound LX06 before and after binding to the two cysteines (Cys477 and Cys552) of FGFR 4;
fig. 4 is a comparison of mass spectra of compound LX07 before and after binding to the two cysteines (Cys477 and Cys552) of FGFR 4;
fig. 5 is a comparison of mass spectra of compound LX08 before and after binding to the two cysteines (Cys477 and Cys552) of FGFR 4;
fig. 6 is a mass spectrum comparison of compound LX09 before and after binding to the two cysteines (Cys477 and Cys552) of FGFR 4.
Detailed Description
Unless otherwise specified, the term "cycloalkyl" as used herein alone or as part of another group includes saturated or partially unsaturated (containing 1 or more double bonds) cyclic hydrocarbon groups containing 1 to 2 rings, preferably 3 to 10 carbons, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, and cyclodecyl. "substituted cycloalkyl" includes cycloalkyl optionally substituted with one or more substituents such as halogen, alkyl, alkoxy, hydroxy, aryl, aryloxy, arylalkyl, cycloalkyl, alkylamido, alkanoylamino, oxo, acyl, arylcarbonylamino, amino, nitro, cyano, thiol, and/or alkylthio and/or any of the substituents included in the definition of "substituted alkyl".
Unless otherwise specified, the terms "aryl" or "Ar" as used herein alone or as part of another group refer to monocyclic and polycyclic aromatic groups containing 6 to 10 carbons in the ring portion (e.g., phenyl or naphthyl, including 1-naphthyl and 2-naphthyl) and may optionally include one to three additional rings fused to carbocyclic or heterocyclic rings (e.g., aryl, cycloalkyl, heteroaryl, or cycloheteroalkyl rings).
As used herein, unless otherwise indicated, the term "heterocycle" or "heterocycle" means an unsubstituted or substituted stable 5-to 10-membered monocyclic ring system, which may be saturated or unsaturated, consisting of carbon atoms and 1 to 4 heteroatoms selected from N, O or S, and wherein the nitrogen and sulfur heteroatoms may be optionally oxidized, and the nitrogen heteroatom may be optionally quaternized. Examples of such heterocyclic groups include piperidinyl, piperazinyl, oxopiperazinyl, pyrrolyl, pyrrolidinyl, furanyl, thienyl, pyrazolyl, pyrazolidinyl, imidazolyl.
It is also within the scope of the present invention that the compounds of formula I may exist as pharmaceutically acceptable salts. If the compounds of the formula I have, for example, at least one basic center, they can form acid addition salts. These are formed, for example, using strong inorganic acids, such as mineral acids, for example sulfuric acid, phosphoric acid or hydrohalic acids, strong organic carboxylic acids, such as unsubstituted or substituted (for example by halogen) alkane carboxylic acids of 1 to 4 carbon atoms, for example acetic acid, such as saturated or unsaturated dicarboxylic acids, for example oxalic acid, malonic acid, succinic acid, maleic acid, fumaric acid, phthalic acid or terephthalic acid, such as hydroxycarboxylic acids, for example ascorbic acid, glycolic acid, lactic acid, malic acid, tartaric acid or citric acid, such as amino acids (for example aspartic acid or glutamic acid or lysine or arginine), or benzoic acid, or organic sulfonic acids, such as unsubstituted or substituted (for example by halogen) (C1-C4) alkyl or aryl sulfonic acids, for example methyl or p-toluene-sulfonic acid. If desired, it is also possible to additionally derivatize a basic center to form the corresponding acid addition salts.
The compounds of the invention may be used in the form of a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention as defined herein and a pharmaceutically acceptable carrier or diluent.
The medicaments of the invention are useful for the treatment of FGR4 mediated disorders, in particular cancer. These cancers include hepatocellular, bladder, breast, cervical, colorectal, endometrial, gastric, head and neck, kidney, liver, ovarian, prostate, esophageal, gall bladder, pancreatic, lung, mesothelioma, testicular, squamous cell carcinoma, thyroid, skin, leukemia, multiple myeloma, chronic lymphocytic lymphoma, adult T-cell leukemia, B-cell lymphoma, acute myelogenous leukemia, hodgkin's lymphoma, non-hodgkin's lymphoma, waldenstrom's macroglobulinemia, hairy cell lymphoma, burkitt's lymphoma, glioblastomas, melanoma, and rhabdomyosarcoma.
The form of the pharmaceutical composition according to the invention may be adapted to be administered to a patient in need of treatment, e.g. a mammal such as a human patient, by a variety of routes of administration, e.g. oral, intranasal, intraperitoneal, or parenteral, by intravenous, intramuscular, topical or subcutaneous routes, or by injection into a tissue. Such compositions and formulations should contain at least 0.01% of one or more of the compounds of the invention. The percentage of the compositions and formulations can, of course, vary and can be, for example, from about 0.05% to about 2% by weight of a given unit dosage form. The amount of the compound in such therapeutically useful compositions is such that an effective dosage level is obtained.
The compounds of the invention may be administered systemically, e.g., orally, in combination with a pharmaceutically acceptable carrier, e.g., an inert diluent or an assimilable edible carrier, or by inhalation or insufflation. They may be enclosed in hard or soft shell capsules, may be compressed into tablets, or may be mixed directly with food for consumption by the patient. For oral therapeutic administration, the compounds of the present invention may be combined with one or more excipients and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like. The compounds may be combined with a fine inert powder carrier and inhaled by the patient or insufflated. Such compositions and formulations should contain at least 0.1% of one or more compounds of the invention.
Tablets, troches, pills, capsules and the like may also comprise: binders such as tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; disintegrating agents such as corn starch, potato starch, alginic acid, and the like; lubricants such as magnesium stearate; and a sweetening agent such as sucrose, fructose, lactose or aspartame, or an aromatic agent such as peppermint, oil of wintergreen or cherry flavoring may be added. When the unit dosage form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a vegetable oil or polyethylene glycol. Various other materials may be present as coatings or to otherwise modify the physical form of the solid unit dosage form. For example, tablets, pills, or capsules can be coated with gelatin, wax, shellac, sugar and the like. A syrup or elixir may contain the active compound, sucrose or fructose as a sweetening agent, methyl and propylparabens as preservatives, a dye, and a flavoring such as cherry or orange flavor. Of course, any material used in preparing any unit dosage form should be pharmaceutically acceptable and substantially non-toxic in the amounts employed. In addition, the compounds of the present invention may be incorporated into sustained release formulations and devices. For example, the compounds may be incorporated into time release capsules, time release tablets, and time release pills.
The compounds of the invention may also be administered intravenously or intraperitoneally by infusion or injection. Solutions of the compounds can be prepared in water, optionally mixed with a non-toxic surfactant. Pharmaceutical dosage forms suitable for injection or infusion may include sterile aqueous solutions or dispersions or sterile powders. The liquid carrier can be a solvent or liquid medium including, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, liquid polyethylene glycol, and the like), vegetable oils, non-toxic glycerides, and suitable mixtures thereof.
For topical administration, the compounds of the present invention may be used in pure form. However, it is generally desirable to administer them to the skin as a composition or formulation, together with a dermatologically acceptable carrier, which may be solid or liquid.
Useful solid carriers include finely divided solids such as talc, clay, microcrystalline cellulose, silica, bauxite, and the like. Other solid supports include non-toxic polymeric nanoparticles or microparticles. Useful liquid carriers include water, alcohols or glycols or water/alcohol/glycol blends in which the compounds of the present invention can be dissolved or dispersed at effective levels, optionally with the aid of non-toxic surfactants. Adjuvants such as perfumes and additional antimicrobial agents can be added to optimize performance for a given use. The resulting liquid composition may be applied from an absorbent pad, used to impregnate bandages and other dressings, or sprayed onto the affected area using a pump-type or aerosol sprayer.
Thickeners such as synthetic polymers, fatty acids, fatty acid salts and esters, fatty alcohols, modified celluloses or modified mineral materials can also be used with the liquid carrier to form spreadable pastes, gels, ointments, soaps, and the like, for direct application to the skin of a user.
The concentration of the compound in a liquid composition, such as a lotion, can be from about 0.1 to about 25% by weight, or from about 0.5 to about 10% by weight. The concentration in a semi-solid or solid composition such as a gel or powder may be from about 0.1 to about 5% by weight, or from about 0.5 to about 2.5% by weight.
The amount of a compound of the invention required for use in treatment will vary not only with the particular salt selected, but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will ultimately be at the discretion of the attendant physician or clinician.
The effective dosage and route of administration of the agents of the invention are conventional. The precise amount (effective dose) of an agent will vary from patient to patient, depending upon, for example, the type, age, weight, and general or clinical state of the patient, the severity or mechanism of any condition being treated, the particular agent or carrier employed, the method and extent of administration, and the like. Therapeutically effective dosages can be determined empirically by conventional procedures known to those skilled in the art.
Synthesis example 1 Synthesis of covalent Compound LX01
Step 1: synthesis of 5- (hydroxymethyl) pyrimidine-2, 4-diol

Uracil (20.0g, 178mmol), paraformaldehyde (6.50g, 72.1mmol), potassium hydroxide (6.50g, 116mmol) and 160mL of water were sequentially added to a 100mL single-neck flask, reacted at 60 ℃ for 72 hours, the solvent was removed at 65 ℃ under reduced pressure, 50mL of acetone was added to the residue, stirred for 2 hours, filtered and dried to obtain 26.3g of the objective compound as a white solid with a yield of 100%. mp 290 deg.C
Step 2: synthesis of 2, 4-dichloro-5- (chloromethyl) pyrimidine
5-hydroxymethylpyrimidine-2, 4-diol (5.00g, 35.2mmol) is added into a 100mL single-neck flask,

Phosphorus oxychloride (27.2g, 177mmol) and 10mL of toluene are slowly dropped into a single-mouth flask by a constant pressure dropper under ice bath, DIEA (14.5g, 112mmol) is stirred for 5min, then the temperature is raised to 115 ℃ for reaction for 1h, then the temperature is raised to 125 ℃ for reaction for 5h, TLC is used for monitoring until the reaction is complete (ethyl acetate: petroleum ether is 1:1), the reaction is cooled to room temperature, 50mL of ice water is slowly added for quenching reaction, toluene (50mL × 3) is extracted, organic phases are combined, anhydrous sodium sulfate is dried, the solvent is removed under reduced pressure, and the residue is purified by silica gel column chromatography to obtain 5.2 g of the target compound, colorless oily matter and the yield is 74.9%.1H NMR(500MHz,DMSO-d6):δ8.97 (s,1H),4.86(s,2H).
And step 3: synthesis of N- ((2, 4-dichloropyrimidine) -5-methyl) -3, 5-dimethoxyaniline (1)
2, 4-dichloro-5- (chloromethyl) pyrimidine (4.00g, 20.3 g) was added sequentially to a 50mL single-neck flask

And 4, step 4: synthesis of 2-chloro-5- (((3, 5-dimethoxyphenyl) amino) methyl) -N- (4-nitrobenzyl) pyrimidin-4-amine (2)
Into a 50mL single-neck flask were added compound 1(2.51g, 8.00mmol), 4-nitrobenzylamine salt

Acid salt (2.00g, 10.6mmol), DIEA (3.28g, 25.4mmol) and 20mL dioxane. The reaction was carried out at 60 ℃ for 10h, monitored by TLC (petroleum ether: ethyl acetate: methanol: triethylamine: 20:8:1:1), the solvent was directly removed after completion of the reaction, the residue was purified by silica gel column chromatography to give 2.86g of crude product, which was then slurried with a small amount of ethyl acetate, filtered and dried to give 2.4g of compound 2 as a yellow solid with a yield of 70.0%.1H NMR(500MHz,CDCl3):δ8.12(d,J=9.0Hz,1H),7.95 (s,1H),7.50(m,1H),7.46(d,J=8.4Hz,1H),6.05(s,1H),5.98(s, 2H),4.78(d,J=6.0Hz,2H),4.26(s,2H),3.71(s,6H).
And 5: synthesis of 7-chloro-3- (3, 5-dimethoxyphenyl) -1- (4-nitrobenzyl) -3, 4-dihydropyrimidine [4,5-d ] pyrimidin-2 (1H) -one (3)

Compound 2(2.00g, 4.65mmol), triphosgene (695mg, 2.31mmol) and 15mL of dry THF are added to a 25mL single neck flask, triethylamine (940mg, 9.32mmol) is slowly added dropwise in ice bath and stirred for 1h, the reaction is further heated to 70 ℃ to react for 10h, TLC monitors until the raw material reaction is complete (petroleum ether: ethyl acetate ═ 2:1), 5mL of quench reaction is added, solvent THF is removed under reduced pressure, ethyl acetate (20mL × 3) is extracted, the organic phases are washed successively with saturated sodium bicarbonate and saturated NaCl, combined, dried over anhydrous sodium sulfate, and the residue is purified by silica gel column chromatography to give 1.76g of compound 3, a pale yellow solid, yield 82.9%.1H NMR(500MHz,DMSO-d6):δ8.41(s,1H),8.19(d,J=8.5 Hz,2H),7.61(d,J=8.5Hz,2H),6.61(d,J=2.0Hz,2H),6.47(s, 1H),5.25(s,2H),4.92(s,2H),3.75(s,6H).13C NMR(125MHz,DMSO-d6): δ160.51,158.06,157.60,154.72,151.33,146.56,145.32,143.54, 128.24,123.52,111.86,104.41,98.68,55.42,46.37,43.87.
Step 6: synthesis of 3- (3, 5-dimethoxyphenyl) -7- ((2-methyl-6-nitrophenyl) amino) -1- (4-nitrobenzyl) -3, 4-dihydropyrimidine [4,5-d ] pyrimidin-2 (1H) -one (4)

Compound 3(1.14g, 2.5mmol), 2-methyl-6-nitroaniline (570mg, 3.75mmol), cesium carbonate (2.44g, 7.50mmol), XPhos (238mg, 0.50mmol) and Pd were weighed out in this order2(dba)3(229mg, 0.25mmol) was placed in a 25mL Schlenk tube and 4mL dry DMA was added and reacted at 110 ℃ for 3h under nitrogen. TLC monitored complete reaction of starting material (petroleum ether: ethyl acetate: methanol: 10:1), extracted with ethyl acetate (30mL × 3), washed with saturated sodium bicarbonate, combined organic phases, dried over anhydrous sodium sulfate, and the residue was purified by silica gel column chromatography to give 756mg of compound 4 as a pale yellow solid in 52.9% yield.1H NMR(500MHz,DMSO-d6):δ9.17(s,1H),8.07(s, 1H),8.01(s,2H),7.80(d,J=7.5Hz,1H),7.62(d,J=7.5Hz,1H), 7.40(t,J=15.5Hz,1H),7.26(s,1H),6.60(s,2H),6.45(s,1H),5.04 (s,2H),4.74(s,2H),3.75(s,6H),2.18(s,3H).
And 7: synthesis of N- (4- ((7- ((2-acrylamido-6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dichloropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) acrylamide (LX01)

Into a 50mL single neck flask was added compound 4(500mg, 0.88mmol), 2mL Raney nickel/H2And (3) carrying out hydrogen replacement on O and 20mL of methanol for three times, then carrying out reaction at 25 ℃ for 10h, monitoring the reaction by TLC until the raw materials are completely reacted, filtering the reaction solution by using kieselguhr to obtain a mother solution, removing the solvent by decompression, and carrying out vacuum drying for 12h to obtain 430mg of compound 5 which is a light yellow solid with the yield of 96.1%. Taking compound 5(200mg, 0.39mmol) in a 25mL two-necked flask, adding 10mL dry DCM, triethylamine (87mg, 0.86mmol), nitrogen protection, stirring for 10min under ice salt bath, slowly adding acryloyl chloride (68mg, 0.76mmol) in dichloromethane (1mL) dropwise, TLC monitoring the raw materials until the reaction is complete (petroleum oil)Ether ethyl acetate methanol 10:10:1), 2mL of ice water was added to quench the reaction, ethyl acetate (20mL × 3) was extracted, washed with saturated sodium bicarbonate and saturated NaCl in this order, the organic phases were combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 45mg of compound LX01 as a white solid with a yield of 18.6%.1H NMR(500MHz,DMSO-d6):δ10.05(s,1H),9.49(s,1H),8.28(s,1H), 8.01(s,1H),7.72(s,1H),7.45(s,2H),7.22(t,J=7.6Hz,1H),7.12 (d,J=4.3Hz,1H),6.54(d,J=1.9Hz,2H),6.51(t,J=6.3Hz,1H), 6.42(q,J=4.3Hz,2H),6.23(m,J=7.1Hz,2H),5.72(t,J=11.8 Hz,2H),4.81(s,2H),4.66(s,2H),3.74(s,6H),2.09(s,3H).13C NMR (500MHz,MeOD-d4):δ166.51,166.06,162.73,161.93,157.98,155.03, 145.36,139.08,138.47,136.22,135.37,132.49,132.24,131.77,130.37, 128.74,128.05,128.00,127.70,122.71,121.17,105.62,100.34,55.99, 44.67,18.68.HRMS[M+H]+m/z calculated for C34H33N7O5,620.2577;found, 620.2612.
Synthetic example 2 Synthesis of covalent Compound LX02
Step 1: synthesis of 2-cyano-3-methyl-2-butenoic acid (6)

To a 50mL single-neck flask were added cyanoacetic acid (2.55g, 30.0mmol), acetone (3.48g, 60.0mmol) and 25mL of toluene, reacted at 50 ℃ for 10 hours, TLC monitored until the reaction was complete (ethyl acetate: petroleum ether ═ 2:1), toluene (20mL × 3) extracted, the organic phases combined, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to give 2.25g of compound 6 as white crystals in 60.0% yield.1H NMR(500MHz, CDCl3):δ10.03(s,1H),2.43(s,3H),2.37(s,3H).
Step 2: synthesis of 2-cyano-N- (4- ((7- ((2- (2-cyano-3-methyl-2-butenamido) -6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dihydropyrimidin [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) -3-methyl-2-butenamide (LX02)

A25 mL one-neck flask was charged with 6(500mg, 4.0mmol) and 12mL of thionyl chloride, reacted at 85 ℃ for 4h,TLCthe reaction was monitored until the starting material was completely reacted (petroleum ether: ethyl acetate: methanol: 10:1), and the solvent was removed under reduced pressure to give 550mg of compound 7. A25 mL two-necked flask was charged with 5(200mg, 0.39mmol), 12mL dry dichloromethane, triethylamine (87mg, 0.86mmol), nitrogen blanketed, stirred for 10min in an ice-salt bath, slowly added with a solution of compound 7(108mg, 0.76mmol) in dichloromethane (1mL), TLC monitored for completion of the reaction of the starting materials (petroleum ether: ethyl acetate: methanol 10:10:1), quenched with 2mL ice water, extracted with dichloromethane (20 mL. times.3), washed with saturated sodium bicarbonate, saturated NaCl in that order,the organic phases are combinedAfter drying over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 25mg of compound LX02 as a white solid in 8.8% yield.1H NMR(500MHz,CDCl3): δ8.75(s,1H),7.93(s,1H),7.89(d,J=7.0Hz,2H),7.75(s,1H), 7.30(t,J=7.9Hz,1H),7.19(d,J=7.5Hz,2H),7.12(s,1H),6.88 (s,1H),6.47(d,J=2.2Hz,2H),6.39(t,J=2.1Hz,1H),5.05(s, 2H),4.64(s,2H),3.78(s,6H),2.40(d,J=18.4Hz,6H),2.30(d,J =6.8Hz,6H),2.23(s,3H).13C NMR(500MHz,CDCl3):δ171.23,170.98, 161.30,160.43,159.77,159.44,157.45,153.52,153.09,144.04,135.94, 134.94,130.11,127.65,120.51,117.03,116.80,106.80,106.53,104.37, 102.96,99.46,55.68,47.60,44.01,27.49,18.67.
Synthetic example 3 Synthesis of covalent Compound LX03
Step 1: synthesis of vinyl sulfonyl chloride (8)

A25 mL single-neck flask was charged with 2-chloroethanesulfonyl chloride (2.57g, 15.8mmol) and 10mL of diethyl ether, a solution of 2,4, 6-trimethylpyridine (2.30g, 19.0mmol) in diethyl ether (5mL) was slowly added dropwise at-60 ℃, the reaction was carried out for 10min, then the mixture was moved to 25 ℃ and reacted for 50min, 2mL of 1% sulfuric acid solution was added under ice bath to quench the reaction, extraction was carried out with ethyl acetate (2X 20mL), washing was carried out with saturated NaCl, the organic phases were combined, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to obtain 1.5g of Compound 8 as a colorless oil with a yield of 75.2%.
Step 2: synthesis of N- (4- ((3- (3, 5-dimethoxyphenyl) -7- ((2-methyl-6- (ethenesulfonamido) phenyl) amino) -2-oxo-3, 4-dihydropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) ethenesulfonamide (LX03)

A25 mL two-necked flask was charged with 5(200mg, 0.39mmol), triethylamine (87mg, 0.86mmol) and 12mL dry dichloromethane, stirred for 10min under an ice salt bath under nitrogen blanket, a solution of compound 8(108mg, 0.76mmol) in dichloromethane (1mL) was slowly added for 4h reaction, TLC monitored until the reaction was complete (petroleum ether: ethyl acetate: methanol: 10:1), quenched with 5mL ice water, extracted with ethyl acetate (20 mL. times.3), washed with saturated sodium bicarbonate and saturated NaCl in that order, the organic phases were combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 56mg compound LX03 as a white solid in 21.3% yield.1H NMR(500MHz,DMSO-d6):δ9.90 (s,1H),9.02(s,1H),8.42(s,1H),8.05(s,1H),7.22(t,J=8.9Hz, 2H),7.13(d,J=6.8Hz,1H),6.95(s,3H),6.73(q,J=10Hz,2H),6.57 (d,J=1.9Hz,2H),6.44(s,2H),6.09(d,J=16.4Hz,1H),6.00(d, J=9.9Hz,2H),5.66(s,1H),4.82(s,2H),4.69(s,2H),3.74(s,6H), 2.04(s,3H).HRMS[M+H]+m/z calculated for C32H33N7O7S2,692.1916;found, 692.1954。
Synthesis example 4 Synthesis of covalent Compound LX04
Step 1: synthesis of 2-chloro-N- (4- ((7- ((2- (2-chloroacetylamino) -6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dihydropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) acetamide (LX04)

To a 25mL two-necked flask were added 5(160mg, 0.31mmol), triethylamine (84mg, 0.78mmol) and 10mL dry dichloromethane, stirred under a nitrogen blanket in an ice salt bath for 10min, a solution of chloroacetyl chloride (70mg, 0.62mmol) in dichloromethane (1mL) was slowly added, TLC monitored until the reaction was complete (petroleum ether: ethyl acetate: methanol 10:10:1), quenched with 5mL ice water, extracted with ethyl acetate (20mL × 3), washed successively with saturated sodium bicarbonate, saturated NaCl, combined organic phases, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 36mg compound LX04 as a white solid in 17.3% yield.1H NMR(500MHz,DMSO-d6):δ10.23(s,1H), 9.43(s,1H),8.49(s,1H),8.04(s,1H),7.78(s,1H),7.35(q,J=4.8 Hz,2H),7.23(t,J=7.6Hz,2H),7.12(d,J=6.9Hz,1H),6.81(s, 1H),6.54(d,J=2.0Hz,2H),6.43(s,1H),4.67(s,2H),4.29(s,2H), 4.23(s,2H),3.74(s,6H),2.54(s,2H),2.10(s,3H).HRMS[M+H]+m/z calculated for C32H31Cl2N7O5,664.1797;found,664.1838。
Synthesis example 5 Synthesis of covalent Compound LX05
Step 1: synthesis of 2-chloro-5- (((3, 5-dimethoxyphenyl) amino) methyl) -N- (3-nitrobenzyl) pyrimidin-4-amine (9)
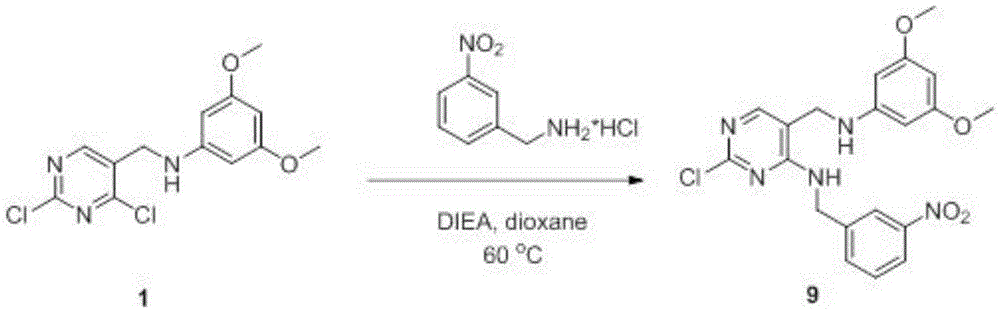
A50 mL single-neck flask was charged with Compound 1(2.51g, 8.0mmol), 3-nitrobenzylamine hydrochloride (2.00g, 10.6mmol), DIEA (3.28g, 25.4mmol), and 20mL dioxane, reacted at 60 ℃ for 10h, TLC monitored until the reaction was complete (petroleum ether: ethyl acetate: methanol: triethylamine: 20:8:1:1), solvent was removed, and the residue was chromatographed on silica gel columnPurification by chromatography gave 2.8g of Compound 9 as a pale yellow oil in 51.0% yield.1H NMR(500MHz,CDCl3):δ8.11(s,1H),8.05(d, J=8.5Hz,1H),7.88(s,1H),7.63(d,J=7.6Hz,1H),7.46(t,J= 7.9Hz,1H),6.91(t,J=5.9Hz,2H),5.93(s,1H),5.87(d,J=1.8 Hz,2H),4.76(d,J=5.9Hz,2H),4.11(d,J=5.2Hz,2H),3.89(t, J=5.2Hz,1H),3.70(s,6H).
Step 2: synthesis of 7-chloro-3- (3, 5-dimethoxyphenyl) -1- (3-nitrobenzyl) -3, 4-dihydropyrimidine [4,5-d ] pyrimidin-2 (1H) -one (10)

Compound 9(1.50g, 3.5mmol), triphosgene (517mg, 2.3mmol) and 15mL of dry THF were added to a 25mL single-neck flask, triethylamine (940mg, 7.0mmol) was slowly added dropwise in an ice bath, after stirring for 1h, the reaction was warmed to 70 ℃ for 10h, TLC monitored until the reaction was complete (petroleum ether: ethyl acetate ═ 2:1), 5mL of ice water was added to quench the reaction, ethyl acetate (20mL × 3) was extracted, washed with saturated sodium bicarbonate and saturated NaCl in that order, the organic phases were combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 1.12g of compound 10 as a pale yellow solid in 70.2% yield.1H NMR(500MHz,CDCl3):δ8.40(s,1H),8.17(s,1H),8.13(d,J=7.3 Hz,1H),7.88(d,J=7.6Hz,1H),7.50(t,J=7.9Hz,1H),6.46(d, J=1.9Hz,2H),6.42(d,J=1.9Hz,1H),5.36(s,2H),4.79(s,2H), 3.79(s,6H).
And step 3: synthesis of 3- (3, 5-dimethoxyphenyl) -7- ((2-methyl-6-nitrophenyl) amino) -1- (3-nitrobenzyl) -3, 4-dihydropyrimidine [4,5-d ] pyrimidin-2 (1H) -one (11)

Compound 10(1.00g, 2.5mmol), 2-methyl-6-nitroaniline (570mg, 3.8mmol), cesium carbonate (2.44g, 7.5mmol), XPhos (238mg, 0.50mmol) and Pd were weighed out in this order2(dba)3(229mg,0.25mmol) was placed in a 25mL Schlenk tube, and 4mL of anhydrous DMA was added, and the mixture was reacted at 110 ℃ for 3 hours under nitrogen. TLC monitored complete reaction of starting material (petroleum ether: ethyl acetate: methanol 10:10:1), extraction with ethyl acetate (30mL × 3), washing with saturated sodium bicarbonate, combining the organic phases, drying over anhydrous sodium sulfate, solvent extraction under reduced pressure, and purification of the residue by silica gel column chromatography afforded 756mg of compound 11 as a pale yellow solid in 52.9% yield.1H NMR(500MHz,CDCl3):δ8.07(d,J=7.2 Hz,2H),8.00(s,1H),7.88(d,J=7.3Hz,1H),7.88(d,J=7.9Hz, 1H),7.61(s,1H),7.54(d,J=7.5Hz,1H),7.50(s,1H),7.37(t,J =8.0Hz,1H),7.32(t,J=7.9Hz,1H),6.50(d,J=2.1Hz,2H),6.43 (t,J=2.1Hz,1H),5.18(s,2H),4.71(s,2H),3.82(s,6H),2.31(s, 3H).
And 4, step 4: synthesis of N- (3- ((7- ((2-acrylamido-6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dichloropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) acrylamide (LX05)

Into a 50mL single neck flask was added compound 11(500mg, 0.87mmol), 2mL Raney nickel/H2O and 20mL of methanol were reacted for 10 hours at 25 ℃ after three times of hydrogen substitution, and the reaction was monitored by TLC until the starting material was completely reacted (petroleum ether: ethyl acetate: methanol: 10:20: 1). The reaction solution was filtered through celite to obtain a mother liquor, the solvent was removed under reduced pressure and dried under vacuum for 12h to obtain 430mg of compound 12 as an off-white solid with a yield of 96.0%. Compound 12(400mg, 0.76mmol) was taken in a 25mL two-necked flask, triethylamine (87mg, 0.86mmol) and 10mL dry dichloromethane were added, stirred for 10min under ice salt bath under nitrogen protection, a solution of acryloyl chloride (68mg, 0.76mmol) in dichloromethane (1mL) was slowly added dropwise, TLC monitored for completion of the reaction of the starting materials (petroleum ether: ethyl acetate: methanol ═ 10:10:1), 2mL ice water was added to quench the reaction, extraction was then performed with ethyl acetate (20mL × 3), washing was performed with saturated sodium bicarbonate, the organic phases were combined, dried over anhydrous sodium sulfate, and purification by column chromatography under reduced pressure of the extraction solvent gave 45mg compound LX05 as a white solid in 18.6% yield.1H NMR(500MHz,DMSO-d6):δ8.27(s,1H),8.03 (s,1H),7.70(d,J=6.4Hz,1H),7.55(d,J=7.7Hz,2H),7.18(t, J=7.8Hz,1H),7.06(d,J=7.5Hz,2H),6.58(d,J=2.2Hz,2H),6.48 (q,J=10.2Hz,1H),6.42(q,J=3.1Hz,2H),6.23(t,J=1.7Hz,1H), 6.20(t,J=1.8Hz,1H),5.71(m,J=1.7Hz,2H),4.86(s,2H),4.70 (s,2H),3.74(s,6H),2.02(s,3H).HRMS[M+H]+m/z calculated for C34H33N7O5,620.2577;found,620.2618.
Synthesis example 6 Synthesis of covalent Compound LX06
Step 1: synthesis of tert-butyl 4- (3-aminopropyl) piperazine-1-carboxylate (14)

To a 50mL single-neck flask were added N- (3-bromopropyl) phenylenediamine (10.00g, 37.3mmol), 1-Boc-piperazine (7.00g, 37.6mmol), potassium iodide (12.40g, 74.6mmol), potassium carbonate (8.88g, 63.4mmol), and 50mL of N, N-dimethylacetamide, reacted at 30 ℃ for 18 hours, TLC monitored that the raw materials reacted completely (ethyl acetate: petroleum ether ═ 1:8), ethyl acetate (50mL × 3) was extracted, washed with saturated sodium bicarbonate and saturated NaCl in this order, the organic phases were combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, 20mL of ethyl acetate was added, slurried, and filtered to obtain 12.2g of intermediate 13 as a white solid with an yield of 87.6%. Taking the intermediate 13(7.40g, 19.8mmol), putting the intermediate into a 50mL single-neck flask, adding 30mL ethanol and 8mL hydrazine hydrate, reacting for 3h at 70 ℃, monitoring raw materials by TLC until the reaction is complete (ethyl acetate: petroleum ether: 1:5), cooling the reaction liquid to normal temperature, carrying out vacuum filtration to obtain a mother liquid, carrying out vacuum filtration to remove the solvent, cooling, adding 20mL diethyl ether and a small amount of anhydrous sodium sulfate, stirring for 10min at 0 ℃, carrying out vacuum filtration to obtain the mother liquid, and carrying out vacuum filtration to obtain 3.9g of a compound 14, a colorless oily substance and a yield of 81.2%.1H NMR(500MHz, CDCl3):δ3.41(t,J=4.8Hz,4H),2.74(t,J=6.8Hz,4H),2.38(q, J=7.2Hz,6H),1.62(m,J=7.0Hz,2H),1.44(s,9H).
Step 2: synthesis of tert-butyl 4- (3- ((2-chloro-5- (((3,5 dimethoxyphenyl) amino) methyl) pyrimidin-4-yl) amino) propyl) piperazine-1-carboxylate (15)

To a 50mL single-neck flask were added compound 1(2.51g, 8.00mmol), compound 14(2.58 g, 10.6mmol), DIEA (3.28g, 25.4mmol) and 20mL dioxane, reacted at 60 ℃ for 10h, monitored by TLC until the starting materials reacted completely (petroleum ether: ethyl acetate: methanol: triethylamine: 16:8:1:1), the solvent was removed under reduced pressure, and the residue was purified by column chromatography to give 3.16g of compound 15 as a pale yellow oil in 76.0% yield.1H NMR(500MHz,CDCl3):δ7.89(d,J=1.4Hz,1H),6.55 (s,1H),5.97(t,J=2.0Hz,1H),5.86(d,J=2.1Hz,2H),4.07(d, J=3.3Hz,2H),3.74(s,6H),3.73(s,2H),3.55(q,J=6.1Hz,2H), 3.37(s,4H),2.35(s,4H),1.45(s,2H),1.44(s,9H).
And step 3: synthesis of tert-butyl 4- (3- (7-chloro-3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dihydropyrimidinyl [4,5-d ] pyrimidin-1 (2H) -yl) propyl) -1-carboxylate (16)
Into a 25mL one-neck flask were added compound 15(1.50g, 2.88mmol), triphosgene (427

And 4, step 4: synthesis of tert-butyl 4- (3- (3, 5-dimethoxyphenyl) -7- ((2-methyl-6-nitrophenyl) amino) -2-oxo-3, 4-dihydropyrimidin [4,5-d ] pyrimidin-1 (2H) -yl) propyl) piperazine-1-carboxylate (17)

Compound 16(850mg, 1.55mmol), 2-methyl-6-nitroaniline (354mg, 2.33mmol), cesium carbonate (1.51g, 4.65mmol), XPhos (151mg, 0.31mmol) and Pd were weighed out in this order2(dba)3(146mg, 0.16mmol) was placed in a 25mL Schlenk tube, and 3mL of dried DMA was added and reacted at 110 ℃ for 3h under nitrogen. TLC monitored the starting material reaction completed (petroleum ether: ethyl acetate: methanol: 10:1), extracted with ethyl acetate (30mL × 3), washed with saturated sodium bicarbonate, washed with saturated NaCl in that order, the organic phases were combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 620mg of compound 17 as a pale yellow solid in 60.4% yield.
And 5: synthesis of N- (2- ((8(3- (4-acryloylpiperazin-1-yl) propyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) -3-methylphenyl) acrylamide (LX06)

Into a 50mL single neck flask was added compound 17(600mg, 0.91mmol), 1mL Raney nickel/H2And (3) carrying out hydrogen replacement on O and 20mL of methanol for three times, then carrying out reaction at 25 ℃ for 10h, monitoring the reaction by TLC until the raw materials are completely reacted, filtering the reaction liquid by using kieselguhr to obtain a mother liquid, removing the solvent by decompression, and drying in vacuum for 12h to obtain 561mg of a compound 18 which is a white solid and has the yield of 97.1%. Compound 18(561mg, 0.89mmol) was taken in a 25mL single vial, 15mL dry dichloromethane, 1mL trifluoroacetic acid was added, stirred overnight at 27 ℃, TLC monitored for completion of the starting material reaction (petroleum ether: ethyl acetate: methanol: 10:1), and the reaction was added to 3 under ice-bath0mL of a saturated sodium bicarbonate solution was stirred for 10min, followed by extraction with dichloromethane (20 mL. times.3), washing with saturated sodium bicarbonate, combining the organic phases, drying over anhydrous sodium sulfate, and removing the solvent under reduced pressure to obtain 450mg of Compound 19. Taking compound 19(200mg, 0.38mmol) into a 25mL double-neck flask, adding 10mL of dried dichloromethane and triethylamine (94mg, 0.86mmol), protecting with nitrogen, stirring for 10min under an ice salt bath, slowly adding a dichloromethane (1mL) solution of acryloyl chloride (68mg, 0.76mmol) dropwise, TLC monitoring that the raw materials are completely reacted, adding 2mL of ice water to quench the reaction, extracting with ethyl acetate (20mL multiplied by 3), washing with saturated sodium bicarbonate and saturated NaCl in sequence, combining organic phases, drying with anhydrous sodium sulfate, removing the solvent under reduced pressure, and purifying the residue by silica gel column chromatography to obtain 32mg of compound LX06 as a white solid with the yield of 13.1%.1H NMR(500MHz,CDCl3):δ8.32(s,1H),7.93 (s,1H),7.91(s,1H),7.21(t,J=7.8Hz,1H),7.07(d,J=7.1Hz, 1H),6.88(s,1H),6.54(q,J=10.6Hz,1H),6.44(d,J=2.1Hz,2H), 6.37(m,J=2.8Hz,2H),6.28(t,J=1.7Hz,1H),6.20(q,J=10.4 Hz,1H),5.69(m,J=8.2Hz,2H),4.61(s,2H),3.86(s,2H),3.77(s, 6H),3.63(s,2H),3.51(s,2H),2.34(s,4H),2.25(s,3H),1.72(s, 3H).13C NMR(500MHz,CDCl3):δ165.46,161.36,161.23,161.02,157.55, 153.14,152.85,143.88,136.23,131.48,128.00,127.73,127.55,127.38, 126.91,104.22,102.83,99.13,55.60,53.05,52.60,47.48,45.76,41.94, 40.12,24.77,18.74.HRMS[M+H]+m/z calculated for C34H40N8O5,641.3155; found,641.3198.
Synthesis example 7 Synthesis of covalent Compound LX07
Step 1: synthesis of 7- ((2-aminophenyl) amino) -3- (3, 5-dimethoxyphenyl) -1- (4-nitrobenzyl) -3, 4-dihydropyrimidinyl [4,5-d ] pyrimidin-2 (1H) -one (20)

Compound 3(500mg, 1.25mmol), o-phenylenediamine (570mg, 1.88mmol), trifluoroacetic acid (214mg, 1.88mmol) and 4mL of sec-butanol were weighed out in that orderThe reaction was carried out in a 25mL Schlenk tube under nitrogen at 100 ℃ for 10 h. The reaction was monitored by TLC until the reaction was complete (ethyl acetate: petroleum ether: TEA ═ 10:10:1), extracted with ethyl acetate (30mL × 3), washed with saturated sodium bicarbonate, the organic phases combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 346mg of compound 20 as a yellow solid in 52.5% yield.1H NMR(500MHz,DMSO-d6):δ8.49(s, 1H),8.09(s,3H),7.46(d,J=7.6Hz,2H),7.10(d,J=7.4Hz,1H), 6.89(t,J=6.9Hz,1H),6.75(d,J=7.0Hz,1H),6.58(s,2H),6.47 (q,J=7.0Hz,2H),5.14(s,2H),4.83(s,2H),4.73(s,2H),3.74(s, 6H),3.63(s,2H),3.51(s,2H),2.34(s,4H),2.25(s,3H),1.72(s, 3H).13C NMR(500MHz,DMSO-d6):δ160.44,160.26,155.73,153.99,152.46, 146.35,146.15,144.16,142.54,128.89,125.69,125.23,124.66,123.25, 116.06,115.56,104.33,101.52,98.34,55.38,46.62,43.28。
Step 2: synthesis of N- (2- ((8- (4-acrylamidobenzyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide (LX07)
Into a 50mL single-neck flask were added compound 20(346mg, 0.66mmol), 2mL Raney

nickel/H2O and 20mL methanol, three times replaced with hydrogen and reacted at 25 ℃ for 10h TLC to monitor the reaction until the starting material was completely reacted (ethyl acetate: petroleum ether: TEA ═ 10:5: 1). The reaction solution was filtered through celite to obtain a mother liquor, the solvent was removed under reduced pressure and dried under vacuum for 12h to obtain 312mg of compound 21 as a white solid with a yield of 97.6%. Taking compound 21(312mg, 0.63mmol) in a 25mL two-port bottle, adding triethylamine (171mg, 1.57mmol) and 10mL dry dichloromethane, stirring for 10min under an ice salt bath under nitrogen protection, slowly adding a dichloromethane (1mL) solution of acryloyl chloride (115mg, 0.76mmol) dropwise, TLC monitoring the raw materials until the reaction is complete (petroleum ether: ethyl acetate: methanol ═ 10:10:1), adding 2mL ice water to quench the reaction, extracting dichloromethane (30mL multiplied by 2), and sequentially using saturated carbonic acidSodium hydrogen, saturated NaCl wash, combine the organic phases, dry over anhydrous sodium sulfate, pump off the solvent under reduced pressure, and purify the residue by silica gel column chromatography to give 56mg of compound LX07 as a white solid in 14.6% yield.1H NMR(500MHz,DMSO-d6):δ10.11 (s,1H),9.88(s,1H),8.60(s,2H),8.11(s,1H),7.55(d,J=7.6Hz, 4H),7.19(d,J=7.7Hz,2H),7.11(s,2H),6.59(s,2H),6.52(q,J =10.3Hz,1H),6.42(q,J=8.9Hz,2H),6.30(d,J=17.0Hz,1H),6.24 (d,J=16.9Hz,1H),5.78(d,J=9.9Hz,1H),6.24(d,J=9.9Hz,1H), 5.04(s,2H),4.74(s,2H),3.75(s,6H).13C NMR(500MHz,DMSO-d6): δ163.78,163.03,160.45,159.28,155.95,153.78,152.50,144.25, 137.71,133.13,132.37,131.90,131.50,129.96,128.16,127.25,126.81, 125.17,124.57,124.44,123.67,119.19,104.32,102.81,98.38,55.40, 46.53,43.03.HRMS[M+H]+m/z calculated for C33H31N7O5,606.2420;found, 606.2615.
Synthesis example 8 Synthesis of covalent Compound LX08
Step 1: synthesis of 2-chloro-5- (((3, 5-dimethoxyphenyl) amino) methyl) -N- (4-nitrophenylethyl) pyrimidin-4-amine (22)

To a 50mL one-neck flask were added compound 1(2.00g, 6.41mmol), 4-nitrophenylethylamine hydrochloride (1.67g, 8.32mmol), DIEA (3.28g, 25.4mmol) and 20mL of dioxane, reacted at 60 ℃ for 10h, monitored by TLC until the starting material was completely reacted (petroleum ether: ethyl acetate: methanol: triethylamine: 20:8:1:1), the solvent was removed under reduced pressure, and the residue was purified by column chromatography to give 2.05g of compound 22 as a yellow oil in 72.3% yield.1H NMR(500MHz,CDCl3):δ8.01(s,1H), 7.99(s,1H),7.83(s,1H),7.28(s,1H),6.35(t,J=5.3Hz,1H),5.99 (t,J=2.0Hz,1H),5.77(d,J=2.1Hz,2H),4.00(s,2H),3.78(q, J=6.1Hz,2H),3.74(s,6H),3.00(t,J=6.7Hz,2H).13C NMR(500MHz, CDCl3):δ162.45,161.81,160.29,154.82,149.00,146.71,146.44, 129.65,123.79,112.72,93.288,91.42,55.256,43.78,41.41,35.00.
Step 2: synthesis of 7-chloro-3- (3, 5-dimethoxyphenyl) -1- (4-nitrophenylethyl) -3, 4-dihydropyrimidine [4,5-d ] pyrimidin-2 (1H) -one (23)

Compound 2(1.00g, 2.25mmol), triphosgene (335m g, 1.13mmol) and 15mL of dry THF were added to a 25mL single-neck flask, triethylamine (4.24g, 5.76mmol) was slowly added dropwise under ice bath, after stirring for 1h, the reaction was warmed to 70 ℃ to react for 10h, TLC monitored until the reaction was complete (petroleum ether: ethyl acetate ═ 2:1), 5mL of ice water was added to quench the reaction, solvent THF was removed under reduced pressure, ethyl acetate (20mL × 3) was extracted, washed with saturated sodium bicarbonate and saturated NaCl in order, the organic phases were combined, dried over anhydrous sodium sulfate, solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 812mg of compound 23 as a pale yellow solid in 76.9% yield.1H NMR(500MHz,DMSO-d6):δ8.35(s,1H), 8.16(d,J=7.9Hz,2H),7.53(d,J=7.9Hz,2H),6.50(s,2H),6.44 (s,1H),4.81(s,2H),4.20(t,J=6.7Hz,2H),3.73(s,6H),3.06(t, J=3.5Hz,2H).13C NMR(500MHz,DMSO-d6):δ160.44,158.17,157.60, 154.48,151.17,147.27,146.16,143.58,130.24,123.45,111.71,104.16, 98.61,55.37,46.09,45.35,33.17.
And step 3: synthesis of 7- ((2-aminophenyl) amino) -3- (3, 5-dimethoxyphenyl) -1- (4-nitrophenyl-ethyl) -3, 4-dihydropyrimidinyl [4,5-d ] pyrimidin-2 (1H) -one (24)

Compound 23(469mg, 1.00mmol), o-phenylenediamine (162mg, 1.50mmol), trifluoroacetic acid (171mg, 1.50mmol) and 3mL of sec-butanol were sequentially weighed out and reacted at 100 ℃ for 10h under nitrogen atmosphere in a 25mL Schlenk tube. TLC monitored the reaction until the starting material was completely reacted (ethyl acetate: petroleum ether: TEA ═ 10:10:1), extracted with ethyl acetate (30mL × 3), and saturated carbonic acidWashed with sodium hydrogen, the organic phases were combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography to give 302mg of compound 24 as a yellow solid in 55.8% yield.1H NMR(500MHz,CDCl3):δ8.07(s,1H), 8.05(s,1H),7.98(s,1H),7.37(d,J=7.8Hz,1H),7.19(t,J=7.45 Hz,1H),7.14(d,J=7.7Hz,2H),6.94(s,2H),6.88(d,J=5.25Hz, 1H),6.84(t,J=7.6Hz,1H),6.46(d,J=1.95Hz,1H),6.41(s,1H), 4.64(s,2H),4.10(t,J=8.25Hz,2H),3.79(s,6H),2.99(t,J=8.0 Hz,2H).
And 4, step 4: synthesis of N- (2- ((8- (4-acrylamidophenethyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide (LX08)

Into a 50mL single neck flask was added compound 24(200mg, 0.37mmol), 1mL Raney nickel/H2O and 20mL of methanol, three times replaced with hydrogen, reacted at 25 ℃ for 10h, and monitored by TLC until the starting material was completely reacted (ethyl acetate: petroleum ether: TEA: 10: 1). The reaction solution was filtered through celite to obtain a mother liquor, the solvent was removed under reduced pressure and dried under vacuum for 12h to obtain 183mg of compound 25 as a white solid. Compound 25(180mg, 0.36mmol) was taken in a 25mL two-necked flask, 10mL of dried dichloromethane, triethylamine (98mg, 0.89mmol) and nitrogen protection were added, stirred for 10min under ice salt bath, a solution of acryloyl chloride (65mg, 0.72mmol) in dichloromethane (1mL) was slowly added dropwise for 2h reaction, TLC monitored until the reaction was complete (petroleum ether: ethyl acetate: methanol ═ 10:10:1), 2mL of ice water was added to quench the reaction, dichloromethane (30mL × 2) was extracted, washed with saturated sodium bicarbonate and saturated NaCl in sequence, the organic phases were combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 36mg of compound LX08 as a white solid with a yield of 16.1%.1H NMR(500MHz,DMSO-d6):δ10.10 (s,1H),9.94(s,1H),8.60(s,1H),8.11(s,1H),7.81(q,J=6.5Hz, 1H),7.58(d,J=8.4Hz,3H),7.18(m,J=7.6Hz,2H),7.05(d,J= 8.3Hz,2H),6.54(m,J=3.9Hz,3H),6.43(m,J=2.2Hz,2H),6.31 (q,J=17.0Hz,1H),6.25(q,J=17.0Hz,1H),5.79(t,J=10.9Hz, 1H),5.75(q,J=10.0Hz,1H),4.68(s,2H),3.99(t,J=7.7Hz,2H), 3.74(s,6H),2.78(t,J=7.9Hz,2H).13C NMR(500MHz,DMSO-d6):δ 164.29,163.45,160.86,159.99,156.57,154.14,152.63,144.68,137.74, 134.41,133.01,132.41,131.95,130.56,129.52,127.74,127.17,125.77, 125.11,124.17,119.80,104.62,103.37,98.83,55.84,46.90,42.87, 33.51.HRMS[M+H]+m/z calculated for C34H33N7O5,620.2577;found, 620.2616.
Synthesis example 9 Synthesis of covalent Compound LX09
Step 1: synthesis of tert-butyl 4- (3- (7- ((2-aminophenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dihydropyrimidin [4,5-d ] pyrimidin-1 (2H) -yl) propyl) piperazine-1-carboxylate (26)

Compound 16(547mg, 1.00mmol), o-phenylenediamine (162mg, 1.50mmol), cesium carbonate (975mg, 3.00mmol), XPhos (98mg, 0.20mmol) and Pd were weighed in this order2(dba)3(92 mg, 0.10mmol) was placed in a 25mL Schlenk tube, followed by 3mL of anhydrous DMA and reacted at 110 ℃ for 3h under nitrogen. TLC monitored the starting material until the reaction was complete (ethyl acetate: petroleum ether: TEA ═ 10:10:1), extracted with ethyl acetate (30mL × 3), washed with saturated sodium bicarbonate, the organic phases combined, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography to give 328mg of compound 26 as a yellow oil in 53.1% yield.1H NMR(500MHz,CDCl3):δ7.93(d,J=7.1Hz,1H), 7.03(d,J=6.0Hz,2H),6.80(s,2H),6.45(s,2H),6.37(s,1H),4.59 (s,2H),3.97(s,2H),3.85(s,2H),3.77(s,6H),3.38(s,1H),2.32 (s,6H),1.84(s,2H),1.44(s,9H).13C NMR(500MHz,CDCl3):δ161.07, 160.677,157.15,154.77,153.16,152.96,143.94,141.37,126.48,125.83, 125.68,119.14,116.98,104.05,102.27,99.01,79.60,55.99,55.50, 52.84,47.41,40.07,28.45,25.06.
Step 2: synthesis of N- (2- ((8- (3- (4-acryloylpiperazin-1-yl) propyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide (LX09)


Compound 26(320mg, 0.52mmol) was taken in a 25mL single neck flask, 15mL of dried dichloromethane and 1mL of trifluoroacetic acid were added, stirred at 27 ℃ overnight, TLC monitored until the starting material reaction was complete (ethyl acetate: petroleum ether: TEA ═ 10:5:1), the reaction was added to 30mL of saturated sodium bicarbonate solution under ice bath, stirred for 10min, dichloromethane (20mL × 3) extracted, saturated sodium bicarbonate washed, the organic phases combined, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to give 254mg of compound 19. Compound 19(200mg, 0.39mmol) was taken in a 25mL two-necked flask, triethylamine (94mg, 0.86mmol) and 10mL dry dichloromethane were added, stirred for 10min under an ice salt bath under nitrogen protection, a solution of acryloyl chloride (70mg, 0.78mmol) in dichloromethane (1mL) was slowly added dropwise, TLC monitored for completion of the reaction of the starting materials (petroleum ether: ethyl acetate: methanol ═ 10:10:1), quenched with 2mL ice water, extracted with dichloromethane (20mL × 3), washed successively with saturated sodium bicarbonate, saturated NaCl, combined organic phases, dried over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to give 36mg compound LX09 as a white solid with a yield of 14.7%.1H NMR(500MHz,CDCl3):δ8.62(s,1H),7.92(s,1H),7.63 (q,J=8.4Hz,3H),7.16(t,J=8.2Hz,3H),6.53(q,J=5.9Hz,1H), 6.45(s,2H),6.39(t,J=12.2Hz,2H),6.25(t,J=9.7Hz,2H),5.69 (q,J=7.3Hz,2H),4.60(s,2H),3.97(s,2H),3.76(s,6H),3.58(s, 2H),3.47(s,2H),2.34(s,4H),2.30(s,2H),1.81(s,2H).HRMS[M+H]+ m/z calculated for C34H40N8O5,627.2999;found,627.3036.
Compound (I)Detection of inhibitory Activity on FGFR kinase
The inhibitory activity of the compounds on FGFR1-4 and FGFR4 mutants was determined using ADP-GloTMKinase Assay (promega, Part No # V9101). The specific experimental process is as follows:
1. the kinase reaction was performed in white opaque 384-well plates and incubated with the laboratory expressing purified kinase (0.1. mu.M), ATP (10. mu.M), substrate (50. mu.g/mL, Part No # ab204877, abcam) and equi-diluted compounds in kinase reaction buffer (40mM Tris-HCl pH 7.5,20mM MgCl2,20mM NaCl,0.1mg/mL BSA,1mM TCEP, and 4% DMSO) for 30min at room temperature;
2. adding reaction termination solution ADP-Glo, incubating at room temperature for 40 minutes, and terminating the reaction;
3. adding a detection solution, incubating for 30-60 minutes at room temperature in a dark place, converting ADP generated by kinase reaction into ATP, and generating cold luminescence;
4. luminescence was measured in a microplate reader and IC50 for the compound was calculated using GraphPad Prism software, the results of which are shown in table 1 below.
TABLE 1 kinase Activity assay results (IC50, nM)

From the aspect of kinase activity, the compounds with the double covalent structures mainly have stronger inhibitory activity on FGFR4, but have weaker activity on FGFR 1-3. Comparing the kinase activity of LX01, LX02, LX03, LX04 on FGFR4, acrylamide groups perform optimally in our chosen electrophilic groups. Tail R2The structure is about 3-6 times less active than the benzene ring for six-membered rings, e.g., LX01, LX05 compared to IC50 for LX06 and LX08 compared to IC50 for LX 09. For the two cysteines (Cys477 and Cys552) in FGFR4, C552A had a greater effect on activity than C477A, compounds LX07, LX08 had at least about a 25-fold reduction in C552A activity over wild-type, LX09 had about a 12-fold reduction in C552A activity, while LX07, LX08, LX09 had only a 4-5-fold reduction in C477A activity over wild-type. Of these 9 compounds, compounds LX01, LX05, LX07, LX08 and LX09 have activity against FGFR4Has higher inhibitory activity and potential medicinal prospect.
Proliferation assay of compounds on FGFR-highly expressed cells
The present invention assesses the inhibitory effect of compounds on the proliferation of cells dependent on the FGFR signaling pathway by survival assays as measured using CCK-8(Vazyme, Part No # A311). The Ba \ F3 cells with high FGFR expression constructed by the experiment are selected, and the specific experimental process is as follows:
1.50 μ L of Ba \ F3 cells were seeded in 96-well plates (about 2000/well) and cultured overnight in a 5% carbon dioxide incubator at 37 ℃;
2. adding 50 μ L of preheated compound diluted with culture medium the next day, mixing well, and culturing for 72 hr;
3. adding 10 mu L of CCK-8 detection reagent into each hole, uniformly mixing, and reacting in an incubator for 1-2 hours;
4. absorbance at 450nm was measured in a microplate reader and IC50 for the compounds was calculated using GraphPad Prism software, the results of which are shown in Table 2 below.
TABLE 2 results of cell Activity assays (IC50, nM)


From the view of cell activity, the inhibitory effect of the compounds on Ba \ F3 cells is consistent with the inhibitory effect of kinase, and the compounds have stronger effect of inhibiting proliferation of Ba \ F3 cells activated by FGFR4 signal channels. The results of kinase and cell activity are combined, and the series of compounds have very good selectivity on FGFR 4.
Covalent binding assay of compounds to FGFR4
To test the covalent binding of the compounds having a significant inhibitory effect on FGFR4 according to the present invention to the two cysteines (Cys477 and Cys552) in FGFR4, changes in molecular weight of kinase mutants FGFR4(C477A) and FGFR4(C552A) before and after binding of the compounds were examined using matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS). The specific experimental process is as follows:
1. protein and compound are mixed according to a molar ratio of 1:5 reacting at room temperature for 1 h;
2. by ddH2Diluting O to 1mL, and concentrating to about 0.5-1mg/mL with 3kD ultrafilter tube;
3. selecting a primary mass spectrum method of linear positive ion mode for detecting large molecular weight, taking sinapic acid as a matrix (20mg/mL), mixing a sample and a saturated matrix solution according to a volume ratio of 1:1, and putting the mixture into a mass spectrometer (AB SCIEX, 5800MADI-TOF) for detection;
4. the data is processed in the data explorer and origin programs, and the results are shown in fig. 1-6.
From the mass spectrometry results, the compounds LX01, LX05, LX06, LX07, LX08 all covalently bind to Cys552 in FGFR4 only and not to Cys477 in FGFR4, while the compound LX09 can covalently bind to Cys552 and Cys477 of two cysteines in FGFR 4.
By integrating kinase activity, cell activity and mass spectrum results, the series of compounds have better selectivity on FGFR4, are covalently bound with single cysteine (Cys552) of FGFR4 or covalently bound with two cysteines (Cys477 and Cys552) of FGFR4 at the same time, are expected to be developed into a new generation of selective FGFR4 inhibitor, and meet the requirements of clinical application.
All documents referred to herein are incorporated by reference into this application as if each were individually incorporated by reference. Furthermore, it will be appreciated that various changes or modifications may be made by those skilled in the art after reading the above teachings of the invention, and such equivalents will fall within the scope of the invention as defined in the appended claims.
Claims (15)
1. An FGFR4 inhibitor having the structure of formula I:

or a pharmaceutically acceptable salt thereof,
wherein R is1Refers to a moiety capable of forming a covalent bond with a nucleophile; r2Is an aryl or heterocyclic group; l is- [ C (R5) (R6)]q-, wherein R5 and R6 are each independently H or C1-C6 alkyl, q is a natural number from 1 to 3, A is phenyl, R is3Is hydrogen or methyl.
2. The FGFR4 inhibitor according to claim 1, wherein R1Is an acryloyl group.
3. The FGFR4 inhibitor according to claim 1, L is independently a C1-C3 alkyl group.
4. The FGFR4 inhibitor according to claim 1, R2Is phenyl.
5. The FGFR4 inhibitor of claim 1 selected from the group consisting of:
n- (4- ((7- ((2-acrylamido-6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dichloropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) acrylamide,
n- (3- ((7- ((2-acrylamido-6-methylphenyl) amino) -3- (3, 5-dimethoxyphenyl) -2-oxo-3, 4-dichloropyrimidine [4,5-d ] pyrimidin-1 (2H) -yl) methyl) phenyl) acrylamide,
n- (2- ((8(3- (4-acryloylpiperazin-1-yl) propyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) -3-methylphenyl) acrylamide,
n- (2- ((8- (4-acrylamidobenzyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide,
LX 08: n- (2- ((8- (3- (4-acryloylpiperazin-1-yl) propyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide,
n- (2- ((8- (4-acrylamidobenzyl) -6- (3, 5-dimethoxyphenyl) -7-oxo-5, 6,7, 8-tetrahydropyrimidinyl [4,5-d ] pyrimidin-2-yl) amino) phenyl) acrylamide.
6. A pharmaceutical composition comprising a therapeutically effective amount of the FGFR4 inhibitor of any of claims 1 to 5, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
7. A pharmaceutical composition comprising a therapeutically effective amount of the FGFR4 inhibitor of any of claims 1 to 5, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable diluent.
8. A pharmaceutical composition comprising a therapeutically effective amount of the FGFR4 inhibitor of any of claims 1 to 5, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable adjuvant.
9. Use of an inhibitor of FGFR4 or a pharmaceutically acceptable salt thereof according to claim 1 in the manufacture of a medicament for the treatment of a FGR 4-mediated disorder.
10. The use according to claim 9, wherein the disorder is cancer.
11. The use of claim 10, wherein the cancer is selected from the group consisting of hepatocellular carcinoma, bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, gastric cancer, head and neck cancer, renal cancer, liver cancer, ovarian cancer, prostate cancer, esophageal cancer, gallbladder cancer, pancreatic cancer, lung cancer, mesothelioma, testicular cancer, squamous cell carcinoma, thyroid cancer, skin cancer, leukemia, multiple myeloma, chronic lymphocytic lymphoma, adult T-cell leukemia, B-cell lymphoma, acute myelogenous leukemia, hodgkin's lymphoma, non-hodgkin's lymphoma, waldenstrom's macroglobulinemia, hairy cell lymphoma, burkitt lymphoma, glioblastoma, melanoma, and rhabdomyosarcoma.
12. The use of claim 11, wherein the cancer is hepatocellular carcinoma.
13. Use of a compound according to claim 1 in the manufacture of a medicament for inhibiting the activity of FGFR4 or a mutant thereof in a patient or a biological sample.
14. The use of claim 11, wherein the activity of FGFR4 or mutants thereof is irreversibly inhibited.
15. The use of claim 11, wherein the activity of FGFR4 or a mutant thereof is irreversibly inhibited by covalently modifying Cys477 and Cys552 of FGFR 4.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110965475.4A CN113527311B (en) | 2021-08-23 | 2021-08-23 | FGFR4 inhibitor, composition and application thereof in preparation of medicines |
| PCT/CN2022/088850 WO2023024545A1 (en) | 2021-08-23 | 2022-04-24 | Fgfr4 inhibitor and composition, and uses thereof in drug preparation |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110965475.4A CN113527311B (en) | 2021-08-23 | 2021-08-23 | FGFR4 inhibitor, composition and application thereof in preparation of medicines |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN113527311A true CN113527311A (en) | 2021-10-22 |
| CN113527311B CN113527311B (en) | 2022-05-06 |
Family
ID=78122728
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110965475.4A Active CN113527311B (en) | 2021-08-23 | 2021-08-23 | FGFR4 inhibitor, composition and application thereof in preparation of medicines |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN113527311B (en) |
| WO (1) | WO2023024545A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023024545A1 (en) * | 2021-08-23 | 2023-03-02 | 中南大学湘雅医院 | Fgfr4 inhibitor and composition, and uses thereof in drug preparation |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105263931A (en) * | 2013-04-19 | 2016-01-20 | 因赛特公司 | Bicyclic heterocycles as FGFR inhibitors |
| CN105307657A (en) * | 2013-03-15 | 2016-02-03 | 西建阿维拉米斯研究公司 | Heteroaryl compounds and uses thereof |
| WO2018113584A1 (en) * | 2016-12-19 | 2018-06-28 | 上海和誉生物医药科技有限公司 | Fgfr4 inhibitor, preparation method therefor and pharmaceutical use thereof |
| CN108948004A (en) * | 2016-05-27 | 2018-12-07 | 杭州英创医药科技有限公司 | Heterocyclic compound as FGFR4 inhibitor |
| CN109384790A (en) * | 2017-08-08 | 2019-02-26 | 南京药捷安康生物科技有限公司 | Fibroblast growth factor acceptor inhibitor and application thereof |
| CN111138459A (en) * | 2018-11-06 | 2020-05-12 | 南京圣和药业股份有限公司 | Optical isomer of FGFR4 inhibitor and application thereof |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113527311B (en) * | 2021-08-23 | 2022-05-06 | 中南大学湘雅医院 | FGFR4 inhibitor, composition and application thereof in preparation of medicines |
-
2021
- 2021-08-23 CN CN202110965475.4A patent/CN113527311B/en active Active
-
2022
- 2022-04-24 WO PCT/CN2022/088850 patent/WO2023024545A1/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105307657A (en) * | 2013-03-15 | 2016-02-03 | 西建阿维拉米斯研究公司 | Heteroaryl compounds and uses thereof |
| CN105263931A (en) * | 2013-04-19 | 2016-01-20 | 因赛特公司 | Bicyclic heterocycles as FGFR inhibitors |
| CN108948004A (en) * | 2016-05-27 | 2018-12-07 | 杭州英创医药科技有限公司 | Heterocyclic compound as FGFR4 inhibitor |
| WO2018113584A1 (en) * | 2016-12-19 | 2018-06-28 | 上海和誉生物医药科技有限公司 | Fgfr4 inhibitor, preparation method therefor and pharmaceutical use thereof |
| CN109384790A (en) * | 2017-08-08 | 2019-02-26 | 南京药捷安康生物科技有限公司 | Fibroblast growth factor acceptor inhibitor and application thereof |
| CN111138459A (en) * | 2018-11-06 | 2020-05-12 | 南京圣和药业股份有限公司 | Optical isomer of FGFR4 inhibitor and application thereof |
Non-Patent Citations (3)
| Title |
|---|
| LI TAN ET AL.: "Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors", 《PNAS》 * |
| RENATA REZENDE MIRANDA ET AL.: "Development of a Potent and Specific FGFR4 Inhibitor for the Treatment of Hepatocellular Carcinoma", 《J. MED. CHEM.》 * |
| WUQING DENG ET AL.: "Investigation of Covalent Warheads in the Design of 2‑Aminopyrimidine-based FGFR4 Inhibitors", 《ACS MED. CHEM. LETT.》 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023024545A1 (en) * | 2021-08-23 | 2023-03-02 | 中南大学湘雅医院 | Fgfr4 inhibitor and composition, and uses thereof in drug preparation |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2023024545A1 (en) | 2023-03-02 |
| CN113527311B (en) | 2022-05-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3584239B1 (en) | O-aminoheteroaryl alkynyl-containing compound, preparation method therefor, and use thereof | |
| CN113544128A (en) | KRAS-G12C inhibitors | |
| CN103237799B (en) | Heterocyclic derivates, preparation processes and medical uses thereof | |
| KR101828187B1 (en) | Novel fused pyrimidine compound or salt thereof | |
| JP2023528903A (en) | KRAS G12C protein inhibitors and uses thereof | |
| EP3173412B1 (en) | 2,4-disubstituted 7h-pyrrolo[2,3-d]pyrimidine derivative, preparation method and medicinal use thereof | |
| CN112552295A (en) | KRAS mutein inhibitors | |
| EP3312180B1 (en) | Use of pteridinone derivative serving as egfr inhibitor | |
| KR20180048635A (en) | Compositions useful for treating disorders related to KIT and PDGFR | |
| WO2014040549A1 (en) | Alkynyl heteroaromatic ring compound and application thereof | |
| CN106187915A (en) | There is inhibitor of ALK Yu EGFR double activity and its preparation method and application | |
| RU2763328C2 (en) | Crystalline fgfr4 inhibitor and its application | |
| WO2018192536A1 (en) | Pyrimido-heterocyclic compound serving as bruton tyrosine kinase inhibitor and applications thereof | |
| WO2022188819A1 (en) | Sos1 proteolysis modulator, preparation method therefor and application thereof | |
| WO2001079184A1 (en) | Substituted piperazine compounds | |
| CN112771049B (en) | FGFR4 inhibitor and application thereof | |
| WO2015058661A1 (en) | Bcr-abl kinase inhibitor and application thereof | |
| EP3865488A1 (en) | Macrocyclic compound as cdk inhibitor, preparation method therefor, and use thereof in medicine | |
| EP3697786B1 (en) | Substituted pyrrolopyridines as inhibitors of activin receptor-like kinase | |
| KR20190021345A (en) | Crystals of aniline pyrimidine compounds provided as EGFR inhibitors | |
| AU2017364720A1 (en) | Novel oxoisoquinoline derivative | |
| CN115322158B (en) | As KRASG12CSubstituted quinazoline compounds of protein inhibitor | |
| WO2014090398A1 (en) | Use of maleimide derivatives for preventing and treating leukemia | |
| CN113527311B (en) | FGFR4 inhibitor, composition and application thereof in preparation of medicines | |
| CN117043163A (en) | Pyrrolopyrimidine or pyrrolopyridine derivative and medical application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |