CN113274507A - Preparation and use of immunostimulatory conjugate complexes for targeted delivery and activation - Google Patents
Preparation and use of immunostimulatory conjugate complexes for targeted delivery and activation Download PDFInfo
- Publication number
- CN113274507A CN113274507A CN202010106067.9A CN202010106067A CN113274507A CN 113274507 A CN113274507 A CN 113274507A CN 202010106067 A CN202010106067 A CN 202010106067A CN 113274507 A CN113274507 A CN 113274507A
- Authority
- CN
- China
- Prior art keywords
- asn
- qhl
- compound
- dox
- ala
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 28
- 230000004913 activation Effects 0.000 title abstract description 36
- 230000003308 immunostimulating effect Effects 0.000 title abstract description 6
- 150000001875 compounds Chemical class 0.000 claims abstract description 165
- 239000003814 drug Substances 0.000 claims abstract description 115
- 229940079593 drug Drugs 0.000 claims abstract description 100
- 230000002829 reductive effect Effects 0.000 claims abstract description 59
- 206010028980 Neoplasm Diseases 0.000 claims description 118
- 150000003839 salts Chemical class 0.000 claims description 61
- 239000007787 solid Substances 0.000 claims description 49
- 238000011282 treatment Methods 0.000 claims description 46
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 32
- 102000009027 Albumins Human genes 0.000 claims description 28
- 108010088751 Albumins Proteins 0.000 claims description 28
- 102000030431 Asparaginyl endopeptidase Human genes 0.000 claims description 25
- 108010055066 asparaginylendopeptidase Proteins 0.000 claims description 25
- 239000008194 pharmaceutical composition Substances 0.000 claims description 22
- 201000011510 cancer Diseases 0.000 claims description 20
- 230000002401 inhibitory effect Effects 0.000 claims description 17
- 210000004185 liver Anatomy 0.000 claims description 17
- 208000014018 liver neoplasm Diseases 0.000 claims description 16
- 150000003057 platinum Chemical class 0.000 claims description 15
- 201000007270 liver cancer Diseases 0.000 claims description 14
- 229960004679 doxorubicin Drugs 0.000 claims description 12
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 claims description 10
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 9
- 208000010706 fatty liver disease Diseases 0.000 claims description 9
- 210000004981 tumor-associated macrophage Anatomy 0.000 claims description 9
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 claims description 8
- 238000004519 manufacturing process Methods 0.000 claims description 8
- 229930192392 Mitomycin Natural products 0.000 claims description 7
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 claims description 7
- 229940024606 amino acid Drugs 0.000 claims description 7
- 235000001014 amino acid Nutrition 0.000 claims description 7
- 125000003277 amino group Chemical group 0.000 claims description 7
- 210000004072 lung Anatomy 0.000 claims description 7
- 229960004857 mitomycin Drugs 0.000 claims description 7
- 230000001737 promoting effect Effects 0.000 claims description 7
- 208000004930 Fatty Liver Diseases 0.000 claims description 6
- 125000000539 amino acid group Chemical group 0.000 claims description 6
- 230000001093 anti-cancer Effects 0.000 claims description 6
- 229960000390 fludarabine Drugs 0.000 claims description 6
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 claims description 6
- 210000002865 immune cell Anatomy 0.000 claims description 6
- 150000001413 amino acids Chemical class 0.000 claims description 5
- 210000000481 breast Anatomy 0.000 claims description 5
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 claims description 5
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 4
- HAWSQZCWOQZXHI-FQEVSTJZSA-N 10-Hydroxycamptothecin Chemical compound C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-FQEVSTJZSA-N 0.000 claims description 4
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 claims description 4
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 4
- HAWSQZCWOQZXHI-UHFFFAOYSA-N CPT-OH Natural products C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-UHFFFAOYSA-N 0.000 claims description 4
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 claims description 4
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 claims description 4
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 claims description 4
- 206010019708 Hepatic steatosis Diseases 0.000 claims description 4
- 229930012538 Paclitaxel Natural products 0.000 claims description 4
- 206010070863 Toxicity to various agents Diseases 0.000 claims description 4
- 230000033115 angiogenesis Effects 0.000 claims description 4
- 230000005809 anti-tumor immunity Effects 0.000 claims description 4
- 229940127093 camptothecin Drugs 0.000 claims description 4
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 claims description 4
- 229910052799 carbon Inorganic materials 0.000 claims description 4
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 4
- 229960000975 daunorubicin Drugs 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 229960001904 epirubicin Drugs 0.000 claims description 4
- 210000003734 kidney Anatomy 0.000 claims description 4
- 229960001592 paclitaxel Drugs 0.000 claims description 4
- 210000000496 pancreas Anatomy 0.000 claims description 4
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 claims description 4
- 229960004618 prednisone Drugs 0.000 claims description 4
- 230000002265 prevention Effects 0.000 claims description 4
- 231100000240 steatosis hepatitis Toxicity 0.000 claims description 4
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims description 4
- AAQGRPOPTAUUBM-ZLUOBGJFSA-N Ala-Ala-Asn Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(O)=O AAQGRPOPTAUUBM-ZLUOBGJFSA-N 0.000 claims description 3
- 208000007082 Alcoholic Fatty Liver Diseases 0.000 claims description 3
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 claims description 3
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 claims description 3
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 3
- DCXYFEDJOCDNAF-UWTATZPHSA-N D-Asparagine Chemical compound OC(=O)[C@H](N)CC(N)=O DCXYFEDJOCDNAF-UWTATZPHSA-N 0.000 claims description 3
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 claims description 3
- 206010016262 Fatty liver alcoholic Diseases 0.000 claims description 3
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 claims description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 claims description 3
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 claims description 3
- 102000035195 Peptidases Human genes 0.000 claims description 3
- 108091005804 Peptidases Proteins 0.000 claims description 3
- DFTCYYILCSQGIZ-GCJQMDKQSA-N Thr-Ala-Asn Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(O)=O DFTCYYILCSQGIZ-GCJQMDKQSA-N 0.000 claims description 3
- UEOOXDLMQZBPFR-ZKWXMUAHSA-N Val-Ala-Asn Chemical compound C[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](C(C)C)N UEOOXDLMQZBPFR-ZKWXMUAHSA-N 0.000 claims description 3
- 230000001476 alcoholic effect Effects 0.000 claims description 3
- 208000026594 alcoholic fatty liver disease Diseases 0.000 claims description 3
- 210000004556 brain Anatomy 0.000 claims description 3
- 229960004117 capecitabine Drugs 0.000 claims description 3
- 210000003679 cervix uteri Anatomy 0.000 claims description 3
- 229960000684 cytarabine Drugs 0.000 claims description 3
- 230000006378 damage Effects 0.000 claims description 3
- 229960003668 docetaxel Drugs 0.000 claims description 3
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 claims description 3
- 229950005454 doxifluridine Drugs 0.000 claims description 3
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 claims description 3
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 claims description 3
- 210000003238 esophagus Anatomy 0.000 claims description 3
- 229960005420 etoposide Drugs 0.000 claims description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 claims description 3
- 229960002949 fluorouracil Drugs 0.000 claims description 3
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 claims description 3
- 229960005277 gemcitabine Drugs 0.000 claims description 3
- 210000003494 hepatocyte Anatomy 0.000 claims description 3
- 210000005008 immunosuppressive cell Anatomy 0.000 claims description 3
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 claims description 3
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 claims description 3
- 229960001924 melphalan Drugs 0.000 claims description 3
- 229960000485 methotrexate Drugs 0.000 claims description 3
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 claims description 3
- 229960001156 mitoxantrone Drugs 0.000 claims description 3
- 210000004985 myeloid-derived suppressor cell Anatomy 0.000 claims description 3
- 210000001989 nasopharynx Anatomy 0.000 claims description 3
- 229960001420 nimustine Drugs 0.000 claims description 3
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 claims description 3
- 210000001672 ovary Anatomy 0.000 claims description 3
- 230000035755 proliferation Effects 0.000 claims description 3
- 210000002307 prostate Anatomy 0.000 claims description 3
- 210000003491 skin Anatomy 0.000 claims description 3
- 210000002784 stomach Anatomy 0.000 claims description 3
- 210000001550 testis Anatomy 0.000 claims description 3
- 229960000303 topotecan Drugs 0.000 claims description 3
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 claims description 3
- 210000003932 urinary bladder Anatomy 0.000 claims description 3
- 210000004291 uterus Anatomy 0.000 claims description 3
- 229960004528 vincristine Drugs 0.000 claims description 3
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 claims description 3
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 claims description 3
- IFKQPMZRDQZSHI-GHCJXIJMSA-N Ala-Ile-Asn Chemical compound [H]N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(O)=O IFKQPMZRDQZSHI-GHCJXIJMSA-N 0.000 claims description 2
- XQNRANMFRPCFFW-GCJQMDKQSA-N Ala-Thr-Asn Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](C)N)O XQNRANMFRPCFFW-GCJQMDKQSA-N 0.000 claims description 2
- XSLGWYYNOSUMRM-ZKWXMUAHSA-N Ala-Val-Asn Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O XSLGWYYNOSUMRM-ZKWXMUAHSA-N 0.000 claims description 2
- SWLOHUMCUDRTCL-ZLUOBGJFSA-N Asn-Ala-Asn Chemical compound C[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](CC(=O)N)N SWLOHUMCUDRTCL-ZLUOBGJFSA-N 0.000 claims description 2
- PTSDPWIHOYMRGR-UGYAYLCHSA-N Asn-Ile-Asn Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(O)=O PTSDPWIHOYMRGR-UGYAYLCHSA-N 0.000 claims description 2
- QYRMBFWDSFGSFC-OLHMAJIHSA-N Asn-Thr-Asn Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](CC(=O)N)N)O QYRMBFWDSFGSFC-OLHMAJIHSA-N 0.000 claims description 2
- MJIJBEYEHBKTIM-BYULHYEWSA-N Asn-Val-Asn Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](CC(=O)N)N MJIJBEYEHBKTIM-BYULHYEWSA-N 0.000 claims description 2
- PAXANSWUSVPFNK-IUKAMOBKSA-N Thr-Ile-Asn Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H]([C@@H](C)O)N PAXANSWUSVPFNK-IUKAMOBKSA-N 0.000 claims description 2
- AAZOYLQUEQRUMZ-GSSVUCPTSA-N Thr-Thr-Asn Chemical compound C[C@@H](O)[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@H](C(O)=O)CC(N)=O AAZOYLQUEQRUMZ-GSSVUCPTSA-N 0.000 claims description 2
- BKIOKSLLAAZYTC-KKHAAJSZSA-N Thr-Val-Asn Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O BKIOKSLLAAZYTC-KKHAAJSZSA-N 0.000 claims description 2
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical compound IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 claims description 2
- BZMIYHIJVVJPCK-QSFUFRPTSA-N Val-Ile-Asn Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](C(C)C)N BZMIYHIJVVJPCK-QSFUFRPTSA-N 0.000 claims description 2
- PQSNETRGCRUOGP-KKHAAJSZSA-N Val-Thr-Asn Chemical compound CC(C)[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@H](C(O)=O)CC(N)=O PQSNETRGCRUOGP-KKHAAJSZSA-N 0.000 claims description 2
- 208000027418 Wounds and injury Diseases 0.000 claims description 2
- 125000002947 alkylene group Chemical group 0.000 claims description 2
- 208000019425 cirrhosis of liver Diseases 0.000 claims description 2
- 210000001072 colon Anatomy 0.000 claims description 2
- 238000001976 enzyme digestion Methods 0.000 claims description 2
- 230000002489 hematologic effect Effects 0.000 claims description 2
- 208000019691 hematopoietic and lymphoid cell neoplasm Diseases 0.000 claims description 2
- 208000006454 hepatitis Diseases 0.000 claims description 2
- 208000014674 injury Diseases 0.000 claims description 2
- 208000018191 liver inflammation Diseases 0.000 claims description 2
- 210000000664 rectum Anatomy 0.000 claims description 2
- 230000021615 conjugation Effects 0.000 claims 1
- 230000007863 steatosis Effects 0.000 claims 1
- 229940035722 triiodothyronine Drugs 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 34
- 231100000135 cytotoxicity Toxicity 0.000 abstract description 5
- 230000003013 cytotoxicity Effects 0.000 abstract description 5
- 239000000543 intermediate Substances 0.000 description 145
- 238000006243 chemical reaction Methods 0.000 description 117
- 210000004027 cell Anatomy 0.000 description 96
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 87
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 83
- 239000000243 solution Substances 0.000 description 79
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 76
- 230000015572 biosynthetic process Effects 0.000 description 75
- 238000003786 synthesis reaction Methods 0.000 description 75
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 70
- 239000002904 solvent Substances 0.000 description 68
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 53
- 235000002639 sodium chloride Nutrition 0.000 description 52
- 238000003756 stirring Methods 0.000 description 49
- 238000012360 testing method Methods 0.000 description 46
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 45
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 45
- 238000004128 high performance liquid chromatography Methods 0.000 description 37
- 239000012043 crude product Substances 0.000 description 35
- 239000000047 product Substances 0.000 description 32
- 229910052757 nitrogen Inorganic materials 0.000 description 29
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 28
- 241000699670 Mus sp. Species 0.000 description 27
- 238000000034 method Methods 0.000 description 27
- 239000012071 phase Substances 0.000 description 26
- 239000002609 medium Substances 0.000 description 25
- 239000000203 mixture Substances 0.000 description 25
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 23
- 238000001704 evaporation Methods 0.000 description 22
- 238000001816 cooling Methods 0.000 description 21
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 21
- 238000012544 monitoring process Methods 0.000 description 20
- 230000002441 reversible effect Effects 0.000 description 20
- 125000005647 linker group Chemical group 0.000 description 19
- 241001465754 Metazoa Species 0.000 description 18
- 238000010438 heat treatment Methods 0.000 description 18
- -1 Ala-Ala amino acids Chemical class 0.000 description 17
- 239000002504 physiological saline solution Substances 0.000 description 17
- 239000002994 raw material Substances 0.000 description 17
- 229910001873 dinitrogen Inorganic materials 0.000 description 16
- 238000011580 nude mouse model Methods 0.000 description 16
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 16
- 238000000967 suction filtration Methods 0.000 description 16
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- 229910002092 carbon dioxide Inorganic materials 0.000 description 15
- 238000004809 thin layer chromatography Methods 0.000 description 15
- 210000001519 tissue Anatomy 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 14
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 14
- 238000004090 dissolution Methods 0.000 description 14
- 239000000523 sample Substances 0.000 description 14
- 238000005406 washing Methods 0.000 description 14
- 206010027476 Metastases Diseases 0.000 description 13
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 13
- 239000007864 aqueous solution Substances 0.000 description 13
- 238000001914 filtration Methods 0.000 description 13
- 230000009401 metastasis Effects 0.000 description 13
- 238000004537 pulping Methods 0.000 description 13
- 210000004322 M2 macrophage Anatomy 0.000 description 12
- 241000699660 Mus musculus Species 0.000 description 12
- 238000002474 experimental method Methods 0.000 description 12
- 239000012091 fetal bovine serum Substances 0.000 description 12
- 230000001965 increasing effect Effects 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 11
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 11
- 108091006905 Human Serum Albumin Proteins 0.000 description 11
- 102000008100 Human Serum Albumin Human genes 0.000 description 11
- 241000699666 Mus <mouse, genus> Species 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 238000001514 detection method Methods 0.000 description 11
- AJDPNPAGZMZOMN-UHFFFAOYSA-N diethyl (4-oxo-1,2,3-benzotriazin-3-yl) phosphate Chemical compound C1=CC=C2C(=O)N(OP(=O)(OCC)OCC)N=NC2=C1 AJDPNPAGZMZOMN-UHFFFAOYSA-N 0.000 description 11
- 238000011065 in-situ storage Methods 0.000 description 11
- 239000006228 supernatant Substances 0.000 description 11
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- 239000011324 bead Substances 0.000 description 10
- 238000012258 culturing Methods 0.000 description 10
- 239000007924 injection Substances 0.000 description 10
- 238000002347 injection Methods 0.000 description 10
- 230000001225 therapeutic effect Effects 0.000 description 10
- 210000004881 tumor cell Anatomy 0.000 description 10
- 238000011725 BALB/c mouse Methods 0.000 description 9
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000001963 growth medium Substances 0.000 description 9
- 210000002540 macrophage Anatomy 0.000 description 9
- 238000002156 mixing Methods 0.000 description 9
- 108090000765 processed proteins & peptides Proteins 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- 230000008878 coupling Effects 0.000 description 8
- 238000010168 coupling process Methods 0.000 description 8
- 238000005859 coupling reaction Methods 0.000 description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 238000004108 freeze drying Methods 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 229910001961 silver nitrate Inorganic materials 0.000 description 8
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 description 8
- 230000003442 weekly effect Effects 0.000 description 8
- 206010006187 Breast cancer Diseases 0.000 description 7
- 208000026310 Breast neoplasm Diseases 0.000 description 7
- 239000006285 cell suspension Substances 0.000 description 7
- 239000012295 chemical reaction liquid Substances 0.000 description 7
- 239000007795 chemical reaction product Substances 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 229940090044 injection Drugs 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- 239000000758 substrate Substances 0.000 description 7
- 239000008215 water for injection Substances 0.000 description 7
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 230000000259 anti-tumor effect Effects 0.000 description 6
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 6
- 229960004562 carboplatin Drugs 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000004069 differentiation Effects 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 239000012065 filter cake Substances 0.000 description 6
- 238000009169 immunotherapy Methods 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 238000009987 spinning Methods 0.000 description 6
- 238000005303 weighing Methods 0.000 description 6
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 description 5
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 5
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- 230000003213 activating effect Effects 0.000 description 5
- 235000009582 asparagine Nutrition 0.000 description 5
- 238000004821 distillation Methods 0.000 description 5
- 238000009826 distribution Methods 0.000 description 5
- 239000011259 mixed solution Substances 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 238000012216 screening Methods 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- KJYAFJQCGPUXJY-UMSFTDKQSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-4-oxo-4-(tritylamino)butanoic acid Chemical compound C([C@@H](C(=O)O)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21)C(=O)NC(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 KJYAFJQCGPUXJY-UMSFTDKQSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 4
- 229920000858 Cyclodextrin Polymers 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- MWWSFMDVAYGXBV-RUELKSSGSA-N Doxorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-RUELKSSGSA-N 0.000 description 4
- 102000005593 Endopeptidases Human genes 0.000 description 4
- 108010059378 Endopeptidases Proteins 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 108010023302 HDL Cholesterol Proteins 0.000 description 4
- 206010033128 Ovarian cancer Diseases 0.000 description 4
- 206010061535 Ovarian neoplasm Diseases 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 239000002246 antineoplastic agent Substances 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 239000001569 carbon dioxide Substances 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- 238000002648 combination therapy Methods 0.000 description 4
- 238000005520 cutting process Methods 0.000 description 4
- 238000002784 cytotoxicity assay Methods 0.000 description 4
- 231100000263 cytotoxicity test Toxicity 0.000 description 4
- 229960002918 doxorubicin hydrochloride Drugs 0.000 description 4
- 239000003684 drug solvent Substances 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 230000008595 infiltration Effects 0.000 description 4
- 238000001764 infiltration Methods 0.000 description 4
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 4
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 239000012488 sample solution Substances 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 230000004083 survival effect Effects 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 238000002054 transplantation Methods 0.000 description 4
- 229910021642 ultra pure water Inorganic materials 0.000 description 4
- 239000012498 ultrapure water Substances 0.000 description 4
- 239000011534 wash buffer Substances 0.000 description 4
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 206010009944 Colon cancer Diseases 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- 239000001116 FEMA 4028 Substances 0.000 description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 3
- 206010025323 Lymphomas Diseases 0.000 description 3
- 239000004472 Lysine Substances 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 229940009456 adriamycin Drugs 0.000 description 3
- 229940041181 antineoplastic drug Drugs 0.000 description 3
- 229960001230 asparagine Drugs 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 235000011175 beta-cyclodextrine Nutrition 0.000 description 3
- 229960004853 betadex Drugs 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 210000001185 bone marrow Anatomy 0.000 description 3
- 208000024207 chronic leukemia Diseases 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 231100000433 cytotoxic Toxicity 0.000 description 3
- 230000001472 cytotoxic effect Effects 0.000 description 3
- 229960002465 dabrafenib Drugs 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 206010017758 gastric cancer Diseases 0.000 description 3
- 208000014829 head and neck neoplasm Diseases 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 208000019423 liver disease Diseases 0.000 description 3
- 201000005202 lung cancer Diseases 0.000 description 3
- 208000020816 lung neoplasm Diseases 0.000 description 3
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 3
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 3
- 210000001616 monocyte Anatomy 0.000 description 3
- 210000005087 mononuclear cell Anatomy 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 description 3
- 239000003880 polar aprotic solvent Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 201000011549 stomach cancer Diseases 0.000 description 3
- 230000002195 synergetic effect Effects 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 3
- BQEUNUXHHDRAHJ-QMMMGPOBSA-N (2s)-2-(tert-butylamino)-4-methylpentanoic acid Chemical compound CC(C)C[C@@H](C(O)=O)NC(C)(C)C BQEUNUXHHDRAHJ-QMMMGPOBSA-N 0.000 description 2
- BZNDDHWTEVCBAD-BQBZGAKWSA-N (2s)-2-[[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoyl]amino]propanoic acid Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)OC(C)(C)C BZNDDHWTEVCBAD-BQBZGAKWSA-N 0.000 description 2
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 2
- PUAQLLVFLMYYJJ-UHFFFAOYSA-N 2-aminopropiophenone Chemical compound CC(N)C(=O)C1=CC=CC=C1 PUAQLLVFLMYYJJ-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- 206010000830 Acute leukaemia Diseases 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- 241001456553 Chanodichthys dabryi Species 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 206010019695 Hepatic neoplasm Diseases 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- 206010024264 Lethargy Diseases 0.000 description 2
- 108010013709 Leukocyte Common Antigens Proteins 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 101100519207 Mus musculus Pdcd1 gene Proteins 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 2
- 208000015634 Rectal Neoplasms Diseases 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 125000003275 alpha amino acid group Chemical group 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 150000001508 asparagines Chemical class 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 238000007664 blowing Methods 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 239000006143 cell culture medium Substances 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 229940044683 chemotherapy drug Drugs 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 238000011284 combination treatment Methods 0.000 description 2
- 238000005336 cracking Methods 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 2
- 230000002354 daily effect Effects 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- 201000004101 esophageal cancer Diseases 0.000 description 2
- VBMSIFVVWOBOHI-NTEUORMPSA-N ethyl (e)-3-[[3-hydroxy-2-methyl-6-[[3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6,11-dioxo-2,4-dihydro-1h-tetracen-1-yl]oxy]oxan-4-yl]amino]but-2-enoate Chemical class O1C(C)C(O)C(NC(/C)=C/C(=O)OCC)CC1OC1C2=C(O)C(C(=O)C3=C(OC)C=CC=C3C3=O)=C3C(O)=C2CC(O)(C(=O)CO)C1 VBMSIFVVWOBOHI-NTEUORMPSA-N 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 201000010536 head and neck cancer Diseases 0.000 description 2
- 235000009200 high fat diet Nutrition 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 230000002434 immunopotentiative effect Effects 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 201000010982 kidney cancer Diseases 0.000 description 2
- DWKPPFQULDPWHX-VKHMYHEASA-N l-alanyl ester Chemical compound COC(=O)[C@H](C)N DWKPPFQULDPWHX-VKHMYHEASA-N 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000010172 mouse model Methods 0.000 description 2
- TYRGLVWXHJRKMT-QMMMGPOBSA-N n-benzyloxycarbonyl-l-serine-betalactone Chemical compound OC(=O)[C@H](C)NC(=O)OCC1=CC=CC=C1 TYRGLVWXHJRKMT-QMMMGPOBSA-N 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- 229940108949 paclitaxel injection Drugs 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 230000003285 pharmacodynamic effect Effects 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 206010038038 rectal cancer Diseases 0.000 description 2
- 201000001275 rectum cancer Diseases 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000011265 semifinished product Substances 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000017550 sodium carbonate Nutrition 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 210000004989 spleen cell Anatomy 0.000 description 2
- 235000000891 standard diet Nutrition 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 201000002510 thyroid cancer Diseases 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000001291 vacuum drying Methods 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- XULIXFLCVXWHRF-UHFFFAOYSA-N 1,2,2,6,6-pentamethylpiperidine Chemical compound CN1C(C)(C)CCCC1(C)C XULIXFLCVXWHRF-UHFFFAOYSA-N 0.000 description 1
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- KZMAWJRXKGLWGS-UHFFFAOYSA-N 2-chloro-n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]-n-(3-methoxypropyl)acetamide Chemical compound S1C(N(C(=O)CCl)CCCOC)=NC(C=2C=CC(OC)=CC=2)=C1 KZMAWJRXKGLWGS-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 102000009422 Aspartic endopeptidases Human genes 0.000 description 1
- 108030004804 Aspartic endopeptidases Proteins 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 229920001824 Barex® Polymers 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 239000004412 Bulk moulding compound Substances 0.000 description 1
- HJGMBZLWVIFLFH-UHFFFAOYSA-N C(CCCCCCCCCCCCCCCCC)(=O)OCCCCCCCCCCCC.[Li] Chemical compound C(CCCCCCCCCCCCCCCCC)(=O)OCCCCCCCCCCCC.[Li] HJGMBZLWVIFLFH-UHFFFAOYSA-N 0.000 description 1
- 238000011740 C57BL/6 mouse Methods 0.000 description 1
- 208000005590 Choroidal Neovascularization Diseases 0.000 description 1
- 206010060823 Choroidal neovascularisation Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 102000005927 Cysteine Proteases Human genes 0.000 description 1
- 108010005843 Cysteine Proteases Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 102100031547 HLA class II histocompatibility antigen, DO alpha chain Human genes 0.000 description 1
- 206010019668 Hepatic fibrosis Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000866278 Homo sapiens HLA class II histocompatibility antigen, DO alpha chain Proteins 0.000 description 1
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 241000521257 Hydrops Species 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- 108010007622 LDL Lipoproteins Proteins 0.000 description 1
- 102000007330 LDL Lipoproteins Human genes 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 102000017095 Leukocyte Common Antigens Human genes 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100028123 Macrophage colony-stimulating factor 1 Human genes 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- FEWJPZIEWOKRBE-XIXRPRMCSA-N Mesotartaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-XIXRPRMCSA-N 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- NSGDYZCDUPSTQT-UHFFFAOYSA-N N-[5-bromo-1-[(4-fluorophenyl)methyl]-4-methyl-2-oxopyridin-3-yl]cycloheptanecarboxamide Chemical compound Cc1c(Br)cn(Cc2ccc(F)cc2)c(=O)c1NC(=O)C1CCCCCC1 NSGDYZCDUPSTQT-UHFFFAOYSA-N 0.000 description 1
- 238000013232 NAFLD rodent model Methods 0.000 description 1
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- HHAIXNHKPAADOF-UHFFFAOYSA-N ONC(CO)(CO)CO.[Na] Chemical compound ONC(CO)(CO)CO.[Na] HHAIXNHKPAADOF-UHFFFAOYSA-N 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 201000002154 Pterygium Diseases 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- 208000000260 Warts Diseases 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 230000007059 acute toxicity Effects 0.000 description 1
- 231100000403 acute toxicity Toxicity 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 108010056243 alanylalanine Proteins 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940045985 antineoplastic platinum compound Drugs 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- FAOSYNUKPVJLNZ-UHFFFAOYSA-N butylstannane Chemical compound CCCC[SnH3] FAOSYNUKPVJLNZ-UHFFFAOYSA-N 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 229960004106 citric acid Drugs 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 201000010989 colorectal carcinoma Diseases 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 229960001270 d- tartaric acid Drugs 0.000 description 1
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 229940048879 dl tartaric acid Drugs 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000000857 drug effect Effects 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003759 ester based solvent Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229960004756 ethanol Drugs 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- IRXSLJNXXZKURP-UHFFFAOYSA-N fluorenylmethyloxycarbonyl chloride Chemical compound C1=CC=C2C(COC(=O)Cl)C3=CC=CC=C3C2=C1 IRXSLJNXXZKURP-UHFFFAOYSA-N 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 102000034238 globular proteins Human genes 0.000 description 1
- 108091005896 globular proteins Proteins 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 238000007490 hematoxylin and eosin (H&E) staining Methods 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 206010019847 hepatosplenomegaly Diseases 0.000 description 1
- CTRFEEQNDJFNNL-UHFFFAOYSA-N hexanoic acid;pyrrole-2,5-dione Chemical compound O=C1NC(=O)C=C1.CCCCCC(O)=O CTRFEEQNDJFNNL-UHFFFAOYSA-N 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229910052588 hydroxylapatite Inorganic materials 0.000 description 1
- 230000005934 immune activation Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000001024 immunotherapeutic effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 239000002085 irritant Substances 0.000 description 1
- 231100000021 irritant Toxicity 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- FEWJPZIEWOKRBE-LWMBPPNESA-N levotartaric acid Chemical compound OC(=O)[C@@H](O)[C@H](O)C(O)=O FEWJPZIEWOKRBE-LWMBPPNESA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000002932 luster Substances 0.000 description 1
- 230000000527 lymphocytic effect Effects 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 239000010413 mother solution Substances 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229940074355 nitric acid Drugs 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 230000031787 nutrient reservoir activity Effects 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 150000003058 platinum compounds Chemical class 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 238000005096 rolling process Methods 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 231100000241 scar Toxicity 0.000 description 1
- 201000004409 schistosomiasis Diseases 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 230000007226 seed germination Effects 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical group C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 201000010153 skin papilloma Diseases 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 210000004988 splenocyte Anatomy 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 239000008227 sterile water for injection Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229960001367 tartaric acid Drugs 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 210000002303 tibia Anatomy 0.000 description 1
- 238000002723 toxicity assay Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 230000005760 tumorsuppression Effects 0.000 description 1
- 230000002100 tumorsuppressive effect Effects 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 210000000689 upper leg Anatomy 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 201000009825 uterine corpus cancer Diseases 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 238000009423 ventilation Methods 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table compounds of the platinum group
- C07F15/0086—Platinum compounds
- C07F15/0093—Platinum compounds without a metal-carbon linkage
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/555—Heterocyclic compounds containing heavy metals, e.g. hemin, hematin, melarsoprol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39558—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against tumor tissues, cells, antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/545—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/643—Albumins, e.g. HSA, BSA, ovalbumin or a Keyhole Limpet Hemocyanin [KHL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table compounds of the platinum group
- C07F15/0086—Platinum compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/0806—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atoms, i.e. Gly, Ala
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/0808—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms, e.g. Val, Ile, Leu
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/1008—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atoms, i.e. Gly, Ala
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Endocrinology (AREA)
- Oncology (AREA)
- Biomedical Technology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
The invention provides the preparation and use of immunostimulatory conjugate complexes for targeted delivery and activation. In particular, the invention provides compounds of formula MI-S-C-A, useful as linker arms, and drug-linked pharmaceutical compounds of formula MI-S-C-A-D. The pharmaceutical compounds of the present invention have improved aqueous solubility, reduced cytotoxicity, enhanced pharmaceutical activity.
Description
Technical Field
The invention relates to an anti-tumor drug compound, in particular to preparation and application of an immunostimulatory coupling compound for targeted delivery and activation
Background
Legumain was first identified in leguminous seeds as an asparaginyl endopeptidase, a member of the cysteine protease C13 family. Legumain can treat storage proteins during seed germination. Legumain's finding in parasites and mammals including humans later proved that the protease was highly conserved. Pig-derived Legumain was first cloned and identified in 1997. Legumain is highly expressed in most solid tumors. Differential expression of Legumain in normal and tumor tissues makes Legumain an ideal target for tumor therapy. Legumain is a peptide chain endopeptidase capable of specifically cleaving the peptide bond at the C-terminal of asparagine in a peptide chain under weakly acidic conditions. CN201210573744.3 discloses aspartase-targeted activated polypeptide doxorubicin derivatives that release Leu-doxorubicin compounds in tumors by Legumain cleavage of the tetrapeptide group (linker).
Disclosure of Invention
According to the invention, through further compound screening and biological system research, a chemically modified activated linker is developed, which can further enhance the activation efficiency. In addition, the chemically modified activating linker arms of the invention can enhance the selectivity of the conjugate drug to immune cells, produce immunotherapeutic enhancing properties in therapy, and enhance the synergistic efficacy of the combination with PD-1 antibodies.
The technical problem to be solved by the invention is to create a coupling arm with high efficiency and specificity selection. Previous studies found that asparagine endopeptidases preferentially recognize the substrate peptide sequence of tetrapeptides and cleave the amide bond between Asn and other residues. The idea of improving the activation efficiency is as follows: a large number of structurally different compounds are synthesized at both ends of tripeptides (such as AAN), and then the working mechanism of asparagine endopeptidase is further researched by using the compounds. Based on the mapping of the crystal structure of asparagine endopeptidase (FIG. 1), since the active center of asparagine endopeptidase is located at the bottom of the invagination, activation of the substrate peptide requires the active center of enzyme to be close to the bottom; in the figure, the C189 attack at position S1 of the asparaginyl endopeptidase cleaves the Asn peptide chain, whereas the adjacent positions S2, S3, S4 and S1, which bind to the substrate, determine the efficiency of the cleavage, and S2, S3 correspond to the Ala-Ala amino acids of the substrate peptide. In view of the possible interaction or steric hindrance of linking compounds such as doxorubicin to the MI group, the assay further synthesized and screened a large number of chemically modified structures at both ends of the substrate tripeptide. After connecting the synthesized compounds with different groups with MI and adriamycin, screening the activation efficiency under the condition of tumor tissues or asparaginyl endopeptidase to optimize and obtain a novel compound couplet with mutual structure-activity relationship, wherein the schematic diagram of the structure is shown in figure 2 and comprises an MI group, a selective group S, a tripeptide group C cut by the asparaginyl endopeptidase, an auxiliary group A and a coupled drug. The added groups bring new functions: in addition to enhancing the activation activity of asparaginyl endopeptidase on the conjugated drug compound (D), the physical properties and biological functions of the drug compound are improved. The pharmaceutical compound provided by the invention is hydrophilic, and the cell membrane permeability of the pharmaceutical compound is changed, so that the pharmaceutical compound is the most suitable for developing a patent drug. In addition, the invention also discovers that the pharmaceutical compound shown as the formula (II) has cell selectivity, can be specifically phagocytized by tumor-related macrophages, attacks or inhibits the tumor-related macrophages and MDSC cells, thereby relieving the immune inhibition effect of the tumor-related macrophages and promoting the immunotherapy. The invention also discovers that when the adriamycin is coupled, the activation efficiency is influenced by the length of the S group, and the longer the chain length of the S is, the more the steric hindrance is, the adverse effect is caused on the combination of the compound and the enzyme, and the activation efficiency is reduced.
Human Serum Albumin (HSA) is a small globular protein consisting of 585 amino acids (66-69kd), many charged residues (e.g. lysine, aspartic acid and groups without prosthetic groups or carbohydrates), and a small number of tryptophan or methionine residues. The compound of formula (II) is coupled into a macromolecular drug at the 34 th cysteine coupled with human serum albumin; experiments show that the albumin covalently coupled compound of formula (II) or EMC-AANL-DOX of the invention has reduced toxicity, improved drug stability and greatly improved therapeutic efficacy.
In conclusion, the connecting arm of the formula (I) and the drug compound of the formula (II) have enhanced activation efficiency, enhanced selectivity on immune cells, enhanced tissue selectivity, proper water solubility and lipid solubility and drug stability.
Accordingly, the present invention provides compounds of formula (I) (linker arms) and pharmaceutical compounds of formula (II) (conjugates) as described herein, and pharmaceutically acceptable salts thereof.
The present invention also provides platinum derivatives having the structure:

the invention also provides a pharmaceutical compound represented by formula (II) of the invention or a pharmaceutically acceptable salt thereof, covalently linked to albumin, and EMC-AANL-DOX covalently linked to albumin; preferably, albumin is linked to the MI or EMC moiety of formula (II) through its cysteine residue at position 36.
The invention also provides a pharmaceutical composition, which comprises the compound shown in the formula (II) or the pharmaceutically acceptable salt thereof, the platinum derivative or the pharmaceutically acceptable salt thereof, the pharmaceutical compound shown in the formula (II) or the pharmaceutically acceptable salt thereof, which is covalently linked with albumin, or EMC-AANL-DOX or the pharmaceutically acceptable salt thereof, which is covalently linked with albumin, and a pharmaceutically acceptable carrier.
The invention also provides an application of the compound of the formula (II) or a pharmaceutically acceptable salt thereof, the platinum derivative or a pharmaceutically acceptable salt thereof, the pharmaceutical compound of the formula (II) or a pharmaceutically acceptable salt thereof, which is covalently linked with albumin, or EMC-AANL-DOX or a pharmaceutically acceptable salt thereof, which is covalently linked with albumin, in preparing a medicament for treating or preventing cancer, fatty liver (including alcoholic and non-alcoholic fatty liver), steatohepatitis, fatty liver disease, hepatic fibrosis, hepatic inflammation and fatty degeneration phenomena caused by hepatic cell injury; preferably, the cancer is a solid cancer or a hematological tumor, preferably of the bladder, brain, breast/breast, cervix, colon, rectum, esophagus, kidney, liver, lung, nasopharynx, pancreas, prostate, skin, stomach, uterus, ovary, testis, and hematological location.
The invention also provides the use of a compound of formula (I) to enhance the drug's aqueous solubility, reduce drug toxicity, improve drug efficacy, and/or improve drug selectivity for immune cells, or to prepare a drug molecule for delivering a drug to the liver.
The invention also provides an EMC-AANL-DOX compound with the structure shown in the formula or an albumin-coupled drug thereof (preferably, the 36 th cysteine residue of the albumin is covalently linked with the EMC part) in the preparation of a drug for treating liver cancer, and an anti-PD-1 antibody and/or an anti-PD-L1 antibody in the preparation of a drug for combined treatment of tumors:

the invention also provides application of the compound shown in the formula (II) or a pharmaceutically acceptable salt thereof, the platinum derivative or a pharmaceutically acceptable salt thereof, the pharmaceutical compound shown in the formula (II) or a pharmaceutically acceptable salt thereof covalently linked with albumin, or EMC-AANL-DOX or a pharmaceutically acceptable salt thereof covalently linked with albumin in preparation of medicines for inhibiting immunosuppressive cells, inhibiting tumor-related macrophages, inhibiting MDSC cells, inhibiting angiogenesis, promoting antitumor immunity and/or promoting T lymphocyte proliferation.
The invention also provides application of the compound shown in the formula (II) or a pharmaceutically acceptable salt thereof, the platinum derivative or a pharmaceutically acceptable salt thereof, the pharmaceutical compound shown in the formula (II) or a pharmaceutically acceptable salt thereof, which is covalently connected with albumin, or EMC-AANL-DOX or a pharmaceutically acceptable salt thereof, which is covalently connected with albumin, and an anti-PD-1 antibody in preparation of medicines for combined treatment of tumors.
Drawings
FIG. 1: crystal structure and action substrate map of asparaginyl endopeptidase.
FIG. 2: schematic representation of immunostimulatory conjugate complexes for targeted delivery and activation.
FIG. 3: comparison of cleavage kinetics of preferred compounds.
FIG. 4: mouse bone marrow mononuclear cell isolation and induced differentiation of M2 macrophages.
FIG. 5: cytotoxicity assay of compounds on CD8+ T cells.
FIG. 6: cytotoxicity assay of compounds on M2 macrophages.
FIG. 7: the compound is used for the curative effect experiment of HT1080 tumors.
FIG. 8: EMC-AANL-DOX has a high distribution profile in liver and liver cancer tissues.
FIG. 9: QHL-087-DOX has a high distribution profile in liver and liver cancer tissues.
FIG. 10: QHL-087-DOX in combination with an anti-PD-1 antibody and for the treatment of hepatoma in situ.
FIG. 11: the curative effect of EMC-AANL-DOX and anti-PD-1 antibody combined therapy on the hepatoma in situ is better than that of the Ranvatinib and anti-PD-1 antibody combined therapy.
FIG. 12: combined therapeutic effects of HSA-EMC-AANL-DOX, QHL-087-DOX and anti-PD-1 antibody
FIG. 13: in vitro cytotoxicity assay of N-CBP.
FIG. 14: HSA-QHL-095-N-CBP cytotoxicity assay.
FIG. 15: single agents of HSA-QHL-095-N-CBP and combination therapy effects with anti-PD-1 antibodies.
Detailed Description
The technical solution of the present invention is further described with reference to the following specific examples.
Linker arm Compounds
The present invention provides compounds having the structure shown in formula (I) below, which can be used as linkers to enhance the drug solubility of the compounds, reduce the drug toxicity, enhance the drug efficacy, and/or increase the selectivity of the drugs for immune cells when linked to a drug of interest (e.g., an anticancer compound):
MI-S-C-A (I)
wherein MI is a maleimido group; s is a group for improving the enzyme digestion efficiency or the selectivity; c is an amino acid connecting arm which can be broken by proteolytic enzyme; and A is an auxiliary connecting arm.
An exemplary MI is a maleimide group represented by the formula:

wherein the wavy line indicates the position of connection to S.
In some embodiments, S in formula (I) may be represented as S1-S2-S3, wherein S1 is selected from:

wherein Rx is absent or selected from: c1-6Alkylene radical, C1-6Alkylene amino group, C1-6Alkylene carboxyl and C1-6An alkylenecarbonylamino group, the wavy line indicating the position of attachment to the adjacent moiety; s2 is absent or is- [ (CH)2)pO]q-, wherein p is an integer of 1 to 4, preferably 2, q is an integer of 0 to 15, preferably 1 to 15, more preferably 2 to 6; s3 is absent or selected from polar amino acid residues such as: glu, Asp, Gly, Ala, Val, Leu, Ile, Met, Phe, Trp, Ser, Thr, Cys, Tyr, Asn, Gln, Lys, Arg and His, preferably Glu and Asp.
It is understood that at least one of S1, S2, and S3 is present.
Preferably, MI, S1, S2, S3, C and a are connected to each other by any one of:

wherein, the wavy line indicates the connecting portion with the adjacent portion; preferably, S is linked to C by a group selected from:

in some embodimentsS is-R1-[(CH2)pO]q-R2-R3-, in which R1Is linked to MI, is absent or selected from C1-6Alkylene or C1-6An alkylenecarbonylamino group; r2Is selected from C1-6An alkylene group; r3Selected from-C (O) O-, -NH-, -O-or-C (O) -R4Wherein R is4Is an amino acid residue selected from Glu, Asp, Gly, Ala, Val, Leu, Ile, Met, Phe, Trp, Ser, Thr, Cys, Tyr, Asn, Gln, Lys, Arg and His, and preferably Glu and Asp, and R4Forming an amide bond with the-c (o) -through an amino group thereof; p is an integer of 1 to 4; q is an integer of 0 to 15, preferably 1 to 15, more preferably 2 to 6. Preferably, R1Absent, p is 2 or 3, q is 1 to 15, preferably 2 to 6, R2Is C1-4Alkylene radical, R3Selected from the group consisting of-C (O) O-, -NH-and-O-. In some embodiments, preferably, R1Is absent, q is 0, R2Is C1-6Alkylene radical, R3is-C (O) -R4,R4Preferably Glu and Asp, and R4An amide bond is formed with the-C (O) -through the amino group thereof. In some embodiments, preferably, R1 is C1-6An alkylenecarbonylamino group, p is 2 or 3, q is 1 to 15, preferably 2 to 6, R2Is C1-4Alkylene radical, R3is-C (O) -R4,R4Preferably Glu and Asp, and R4An amide bond is formed with the-C (O) -through the amino group thereof.
Exemplary MI-S are selected from:



preferably, the C attached to any of the MI-S described above is AAN and A is any of the structures described below.
Preferably, in the compound of formula (I) according to the present invention, C is selected from the group consisting of a group that cleaves asparagine endopeptidase when expressed in the tumor microenvironment, and the group comprises an Asn residue. In some embodiments, C is X1X2X3Wherein X is1Selected from L or D form Ala, Thr, Val and Asn; x2Selected from L or D form Ala, Thr, Val and Ile; x3Is Asn, preferably not D-Asn. Exemplary C is selected from: Ala-Ala-Asn, Thr-Ala-Asn, Val-Ala-Asn, Asn-Ala-Asn, Thr-Thr-Asn, Val-Thr-Asn, Asn-Thr-Asn, Ala-Val-Asn, Thr-Val-Asn, Asn-Val-Asn, Ala-Ile-Asn, Thr-Ile-Asn, Val-Ile-Asn, Asn-Ile-Asn, Ala-Thr-Asn, D-Thr-L-Val-L-Asn, D-Thr-L-Ala-L-Asn, D-Ala-L-Val-L-Asn, L-Thr-D-Val-L-Asn, L-Thr-D-Ala-L-Asn, L-Ala-D-Val-L-Asn, D-Thr-D-Val-L-Asn, D-Thr-D-Ala-L-Asn, D-Ala-D-Val-L-Asn. In some particularly preferred embodiments, C is AAN.
In the compounds of formula (I) according to the invention, A is preferably selected from Leu, PABC-OH and PABC-NH2The structures are respectively shown as the following formulas:



wherein the wavy line indicates the position of the connection to C.
In some embodiments, the compounds of formula (I) of the present invention wherein S and A are selected from any one of the following groups 1-137 [ wherein "2 peg" represents- (CH)2CH2O)2-, 3peg represents- (CH)2CH2O)3-, 4peg represents- (CH)2CH2O)4-, 6peg represents- (CH)2CH2O)6-) by analogy]:






Preferably, in the compound of formula (I) of the present invention, MI is a maleimido group, S and A are any one of groups QHL-001 to QHL-162, and C is AAN.
Particularly preferred compounds of formula (I) according to the invention (linker arms) are selected from: QHL-005, QHL-006, QHL-008, QHL-086, QHL-087, QHL-089, QHL-090, QHL-092, QHL-093, QHL-095, QHL-096, QHL-098, QHL-099, QHL-101, QHL-102, QHL-104, QHL-105, QHL-107, QHL-108 and QHL-116, QHL-119, QHL-138, QHL-140, QHL-141, QHL-143, QHL-144, QHL-146, QHL-147, QHL-150, QHL-153, QHL-154, QHL-155, QHL-156, QHL-157, QHL-158, QHL-159, QHL-160, QHL-161 and QHL-162, more preferably QHL-086, QHL-087, QHL-089 and QHL-090.
Pharmaceutical compounds
The present invention provides a compound (conjugate) represented by the following formula (II) or a pharmaceutically acceptable salt thereof:
MI-S-C-A-D (II)
wherein MI, S, C and A form a linker arm compound according to any embodiment of the invention; d is a drug, preferably an anticancer compound.
In formula II, when a is the linking group, it is selected from:



wherein the wavy line indicates the connecting position with C and D. Preferably, C is attached via-NH-.
Preferably, D is selected from the group consisting of ranimustine, prednisone, triiodothyronine (T3), doxorubicin, daunorubicin, epirubicin, methotrexate, fludarabine, gemcitabine, cytarabine, melphalan, nimustine, mitoxantrone, mitomycin, camptothecin, 10-hydroxycamptothecin, topotecan, fluorouracil, doxifluridine, etoposide, fludarabine, capecitabine, vincristine, epothilone B, paclitaxel, docetaxel, darafenib, doxifyllib, motexenib, compound a, compound B, and platinum derivatives of the formula:

wherein the structures of the compound a and the compound b are as follows:


more preferably, D is selected from daunorubicin, polyvirginib, epirubicin, compound a, compound b, mitomycin, dabrafenib, motexenib, ranisimetride, prednisone, and T3. Preferably, the compounds of formula (I) of the invention (linker arms) used for linking to these drugs (D) are selected from: QHL-005, QHL-006, QHL-008, QHL-086, QHL-087, QHL-089, QHL-090, QHL-092, QHL-093, QHL-095, QHL-096, QHL-098, QHL-099, QHL-101, QHL-102, QHL-104, QHL-105, QHL-107, QHL-108 and QHL-116, QHL-119, QHL-138, QHL-140, QHL-141, QHL-143, QHL-144, QHL-146, QHL-147, QHL-150, QHL-153, QHL-154, QHL-155, QHL-156, QHL-157, QHL-158, QHL-159, QHL-160, QHL-161 and QHL-162, more preferably QHL-086, QHL-087, QHL-089 and QHL-090.
Preferably, A and D are connected by any one of the following means:

wherein the wavy line indicates the connecting portion with the adjacent portion.
More preferably, A and D are linked by a-CO-NH-linkage, wherein the carbonyl group is linked to or is part of A (e.g., when A is Leu) and the amino group is linked to or is part of D. Typically, the drug compound is attached to a at a location that does not affect the biological activity of the drug, e.g., at a location that is remote from the active center of the drug compound.
Preferably, the pharmaceutical compound of formula (II) according to the present invention is selected from:









in some embodiments of the present invention, the present invention also provides platinum derivatives, prodrugs thereof, or pharmaceutically acceptable salts thereof, having the structure:

the pharmaceutical compositions of the present invention may be covalently coupled to albumin to form novel pharmaceutical compounds. Accordingly, the present invention also includes pharmaceutical compounds of formula (II) of the present invention covalently linked to albumin. Typically, albumin is linked to the MI of the linker arm. In some embodiments, the invention also includes EMC-AANL-DOX linked to albumin, pharmaceutical compositions thereof, and uses thereof. The present invention also relates to pharmaceutical compounds of formula (II) of the present invention, or a pharmaceutically acceptable salt thereof, covalently linked to albumin.
In the present invention, the pharmaceutically acceptable salt may be various pharmaceutically acceptable salts known in the art, including inorganic and organic acid salts such as hydrochloride, hydrobromide, phosphate, sulfate, citrate, lactate, tartrate, maleate, fumarate, mandelate and oxalate; and inorganic and organic base salts formed with bases such as sodium hydroxy, TRIS (hydroxymethyl) aminomethane (TRIS, tromethamine) and N-methylglucamine.
Third, preparation method
An exemplary process for the preparation of the compounds of formulae (I) and (II) of the present invention comprises:
step 1: preparation of tripeptide-PABC or tetrapeptide: coupling amino acid residues and separating to obtain the formed tripeptide-PABC or tetrapeptide, namely C-A;
step 2: preparation of MI-S: selecting a compound suitable for an MI-S group, and carrying out condensation or cyclization to obtain MI-S with a carboxyl at one end;
and step 3: preparation of MI-S-C-A: coupling C-A obtained in the step 1 and MI-S obtained in the step 2 through amino and carboxyl to obtain an intermediate (MI-S-C-A);
and 4, step 4: and (3) covalently combining the carboxyl or hydroxyl activated product at the A end of the compound MI-S-C-A obtained in the step (3) with the amino of an optional drug to form the targeted delivery and activation immunostimulatory adriamycin conjugate complex.
When A is PABC-OH, the synthetic route comprises: coupling the amino acid residue suitable for the invention and PABC by using the known chemical and biological recombinant coupling technology, and then purifying and separating to obtain C-PABC containing proper amino acid protecting groups; the reaction may be carried out in the presence of a condensing agent, a base, a polar aprotic solvent. Then removing the protecting group to obtain the C-PABC. Then reacting the C-PABC with acid or ester or acyl chloride containing MI-S group in the presence of condensing agent, alkali and polar aprotic solvent to form MI-S-C-A shown in the formula (I) of the invention, and then reacting with the interested drug compound or salt thereof in the presence of condensing agent, alkali and polar aprotic solvent to form the drug compound shown in the formula (II) of the invention
Examples of the base used in the preparation method include, for example, organic bases such as triethylamine, pyridine, N-diisopropylethylamine, 4-dimethylaminopyridine, 1,2,2,6, 6-pentamethylpiperidine and the like, or inorganic bases such as sodium carbonate, potassium carbonate, sodium hydrogencarbonate, potassium hydrogencarbonate and the like. Examples of the condensing agent used in the production method include, for example, HBTU, DMC, HATU, HOBT, DIC, DCC, EDCI, deptt and the like, and the solvent used in the production method may be any solvent as long as the solvent itself is inert in the reaction and does not inhibit the reaction. Such solvents include halogenated hydrocarbon solvents such as dichloromethane and chloroform, etc., aromatic hydrocarbon solvents such as benzene and toluene, etc., aprotic solvents such as acetonitrile, N-dimethylformamide, dimethyl sulfoxide, etc., ester solvents such as methyl acetate and ethyl acetate, etc., ether solvents such as tetrahydrofuran, or mixtures of these solvents. The reaction in the preparation method may be carried out at a temperature ranging from 150 ℃ under ice-cooling.
Medicine composition
The invention includes a pharmaceutical composition comprising a compound of formula (II) according to the invention or a pharmaceutically acceptable salt thereof, or a platinum derivative according to the invention or a pharmaceutically acceptable salt thereof, or a compound of formula (II) covalently linked to albumin or a pharmaceutically acceptable salt thereof, or comprising EMC-AANL-DOX covalently coupled to albumin or a pharmaceutically acceptable salt thereof.
The pharmaceutical composition may further comprise a pharmaceutically acceptable carrier or excipient. The carrier or excipient may be any of a variety of pharmaceutically acceptable carriers or excipients known in the art and will vary with the pharmaceutical dosage form or mode of administration.
In one embodiment, the pharmaceutical composition comprises one or more of a vehicle, a solubilizer/cosolvent, a pH adjuster, a lyophilization excipient, and an osmotic pressure adjuster.
Freeze-drying excipients suitable for use in the present invention include one or more of sugars (e.g. lactose, maltose, dextran, glucose, fructose), amino acids (e.g. arginine, lysine, histidine), mannitol, tartaric acid, maleic acid, citric acid, sodium chloride and cyclodextrins (e.g. hydroxypropyl beta cyclodextrin, sulfobutyl beta cyclodextrin).
The pH regulator suitable for the invention comprises one or more of hydrochloric acid, phosphoric acid, sulfuric acid, carbonic acid, nitric acid, acetic acid, citric acid, DL-tartaric acid, D-tartaric acid, L-tartaric acid, sodium hydroxide, potassium hydroxide, meglumine, maleic acid, ethylenediamine, triethylamine, arginine, lysine, histidine, sodium dihydrogen phosphate and disodium hydrogen phosphate.
The solvent suitable for the invention is preferably an organic solvent, and comprises one or more of ethanol, propylene glycol, polyethylene glycol 300, polyethylene glycol 400, tert-butyl alcohol, glycerol, tween, soybean oil, hydroxypropyl beta cyclodextrin solution and sulfobutyl beta cyclodextrin solution.
Osmo-regulators suitable for use in the present invention include one or more of glucose, sodium chloride, mannitol and sodium lactate.
Solubilizers/co-solvents suitable for use in the present invention include one or more of tween 80, tween 60, poloxamers, hydroxypropyl beta cyclodextrin, polyethylene glycol (PEG), lithium lauryl stearate, sulfobutyl beta cyclodextrin, PVP, glycerol and polyoxyethylated castor oil.
In general, a compound of the present invention or a pharmaceutically acceptable salt thereof is administered orally to a mammal daily, usually in an amount of about 0.0025 to 50mg/kg body weight, preferably about 0.01 to 10 mg/kg body weight. If a known anti-cancer drug is administered simultaneously or other treatments are administered, the dosage should be effective to achieve its intended purpose. Optimal dosages of these known anti-cancer drugs are well known to those skilled in the art.
A unit oral dosage may include from about 0.01 to 50mg, preferably from about 0.1 to 10mg, of a compound of the present invention or a pharmaceutically acceptable salt thereof. A unit dose may be administered one or more times daily in one or more doses, each dose containing from about 0.1 to 50mg, conveniently from about 0.25 to 10mg, of a compound of the invention or a pharmaceutically acceptable salt thereof.
The pharmaceutical composition of the present invention may be prepared in any suitable dosage form including, but not limited to, tablets, capsules, injections, and the like. The pharmaceutical compositions of the present invention can be administered by routes well known in the art, such as oral, intravenous, intramuscular, and the like.
Use of compound and pharmaceutical composition
The cytokines secreted by the tumor induce the transformation of the monocytes into tumor-associated macrophages (TAM), which can stimulate the generation of strong immunosuppression and can directly help the infiltration and metastasis of tumor cells. A confirmation that tumor-associated macrophages (type M2) differ from monocytes and inflammatory macrophages (type M1) is the expression of asparaginyl endopeptidase. The compounds of the invention can be activated to release in the presence of aspartic endopeptidase. Because different parts of the couplet specifically activated by the asparagines endopeptidase have great influence on the functions of the final medicament such as targeting, activation, stability, toxicity, medicament effect and the like, the couplet specifically activated by the asparagines endopeptidase can effectively reduce the toxicity of the connected medicament, so that the final medicament has new targeting, activation and metabolic characteristics, the effect of treating the tumor is improved, new tumor indications and the effect of resisting tumor metastasis are generated, and brand new structures and functions are generated.
The invention also discovers that the compound shown as the formula (II) has the effects of killing tumor-related macrophages, weakening immunosuppressive cytokines in microenvironment and promoting immunopotentiation of toxic CD8 cells. More importantly, the compounds released by the tumor microenvironment are only activated locally in the tumor, which is different from the damage of the whole immune system caused by the traditional chemotherapy drugs. In the experiment, the tumor microenvironment releasable compound and the PD-1 (programmed death molecule 1) inhibitory antibody (anti-PD-L1 antibody, which is commercially available and is a candidate drug with the existing immunotherapy effect) have strong synergistic therapy effect, and can overcome the defect that immunotherapy is difficult to combine with chemotherapy drugs.
Accordingly, the compound, its pharmaceutically acceptable salt, or the pharmaceutical composition of the present invention can be used for treating or preventing various diseases known in the art to be treatable by Racemide, prednisone, T3, doxorubicin, daunorubicin, epirubicin, methotrexate, fludarabine, gemcitabine, cytarabine, melphalan, nimustine, mitoxantrone, camptothecin, 10-hydroxycamptothecin, topotecan, fluorouracil, doxifluridine, etoposide, fludarabine, capecitabine, vincristine, epothilone B, paclitaxel, docetaxel, dabrafenil, doxertinib, motricib, compound a, compound B, and platinum compounds (e.g., carboplatin, cisplatin, oxaliplatin), including cancer, ophthalmic diseases, liver diseases, and the like.
For example, it is known in the art that camptothecin can be used to treat or prevent malignant tumors, psoriasis, warts, acute/chronic leukemia, and hepatosplenomegaly caused by schistosomiasis; the 10-hydroxycamptothecin can be used for treating gastric cancer, hepatocarcinoma, head and neck cancer, leukemia, etc.; paclitaxel is mainly used for treating ovarian cancer and breast cancer, and also has therapeutic effects on lung cancer, carcinoma of large intestine, melanoma, head and neck cancer, lymphoma, cerebroma, etc.; mitomycin can be used for treating chronic lymphoma, chronic myelogenous leukemia, esophageal cancer, gastric cancer, colon cancer, rectal cancer, lung cancer, pancreatic cancer, liver cancer, cervical cancer, uterine corpus cancer, ovarian cancer, breast cancer, head and neck tumor, bladder tumor, malignant intracavity hydrops, etc.
Thus, for example, diseases that can be treated or prevented with a compound of the present invention, a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof include, but are not limited to, cancers of the bladder, brain, breast/mammary gland, cervix, colon-rectum, esophagus, kidney, liver, lung, nasopharynx, pancreas, prostate, skin, stomach, uterus, ovary, testis, and blood. In particular, these cancers are selected from: liver cancer, kidney cancer, thyroid cancer, colorectal cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, rectal cancer, esophageal cancer, lung cancer (e.g., bronchogenic carcinoma, including undifferentiated small cell and non-small cell), nasopharyngeal cancer, pancreatic cancer, prostate cancer, skin cancer, stomach cancer, uterine cancer, ovarian cancer, testicular cancer, blood cancer (e.g., chronic or acute leukemia, including lymphocytic and myelocytic leukemias), malignant lymphoma, fibrosarcoma, soft tissue sarcoma, osteogenic sarcoma, rhabdomyosarcoma, ewing's sarcoma, wilms' tumor, neuroblastoma, thyroid cancer, and head and neck squamous carcinoma.
In one embodiment, the pharmaceutical compound of the present invention, wherein D is mitomycin, or a pharmaceutically acceptable salt thereof, of formula (II) is also useful for treating or preventing ophthalmic diseases, including treating or preventing scar or choroidal neovascularization, or inhibiting macrophages. In other embodiments, the pharmaceutical compound represented by formula (II) wherein D is mitomycin or a pharmaceutically acceptable salt thereof can also be used for the treatment or prevention of corneal transplantation, glaucoma, sequela of pterygium surgery, and the like.
The compounds or pharmaceutical compositions of the invention may also be used to prevent tumor metastasis, especially tumor lung metastasis. In one embodiment, the compounds or pharmaceutical compositions of the present invention can be used to prevent breast cancer lung metastasis.
The liver diseases include, but are not limited to fatty liver (including alcoholic and non-alcoholic fatty liver), steatohepatitis, fatty liver disease, liver fibrosis, liver inflammation, and fatty degeneration phenomenon of hepatocyte injury.
Accordingly, the present invention includes a method for treating or preventing a disease, preferably cancer, an ophthalmic disease and a liver disease according to any of the embodiments of the present invention, comprising administering to a subject in need thereof a therapeutically or prophylactically effective amount of a compound of formula (II) of the present invention or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of formula (II) of the present invention or a pharmaceutically acceptable salt thereof. In some embodiments, a platinum derivative or a pharmaceutically acceptable salt thereof, or a compound of formula (II) or a pharmaceutically acceptable salt thereof covalently linked to albumin, or EMC-AANL-DOX or a pharmaceutically acceptable salt thereof covalently coupled to albumin, or a pharmaceutical composition of each thereof, is administered or described herein.
The invention also includes a method of preventing tumor metastasis comprising administering to a subject in need thereof an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of the invention or a pharmaceutically acceptable salt thereof. Preventing tumor metastasis includes, but is not limited to, preventing tumor lung metastasis and/or bone metastasis.
Tumor-associated macrophages (TAMs), a key inflammatory cell, play an extremely important role in tumor-associated inflammation. In the tumor microenvironment, TAMs promote tumor development by affecting various biological properties of the tumor. It secretes some molecules (such as EGF) to directly promote the growth of tumor cells and promote angiogenesis, thus creating conditions for infiltration and metastasis of cancer cells, and simultaneously inhibiting the function of acquired immunity. Accordingly, the present invention also includes a method of inhibiting tumor-associated macrophages comprising administering to a subject in need thereof an effective amount of a compound of the present invention or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition containing a compound of the present invention or a pharmaceutically acceptable salt thereof. By inhibiting tumor-related macrophages, tumor growth and angiogenesis can be inhibited, infiltration and metastasis of cancer cells can be inhibited, and anti-tumor immunity can be promoted, so that prevention and/or treatment of cancer can be realized. In one embodiment, the tumor-associated macrophage expresses aspartate endopeptidase, type M2.
The above methods of the invention may be used in combination with radiotherapy or immunotherapy as known in the art.
Accordingly, the present invention also includes a compound of the present invention, a pharmaceutically acceptable salt thereof, or a pharmaceutical composition of the present invention for use in the various methods or uses described above.
The invention also includes the use of a compound of the invention or a pharmaceutically acceptable salt thereof or a pharmaceutical composition of the invention in the manufacture of a medicament for the treatment or prevention of the above-mentioned diseases (e.g. cancer and cancer metastasis). The invention also comprises the application of the compound or the pharmaceutically acceptable salt thereof or the pharmaceutical composition in preparing medicines for inhibiting tumor-related macrophages, inhibiting tumor growth, inhibiting angiogenesis, inhibiting infiltration and metastasis of cancer cells and/or promoting antitumor immunity.
The present invention also provides a method of reducing the toxic side effects of an anticancer compound, particularly an anticancer compound described herein, comprising linking the anticancer compound to a linker arm compound of the present invention represented by formula (I).
The therapeutic or prophylactic methods of the present invention comprise administering a compound or pharmaceutical composition of the present invention to a subject in need thereof. Methods of administration include, but are not limited to, oral, intravenous, intramuscular, and the like. The subject includes a mammal, particularly a human.
In some embodiments, the invention also provides an EMC-AANL-DOX compound having a structure represented by the following formula or an albumin-conjugated drug thereof for use in the preparation of a medicament for treating liver cancer:

it is to be understood that the terms "comprising", "including" and "including" also include "consisting of … … and" consisting of … … ". The sum of all weight percentages or volume percentages should equal 100%. The various reagents and products used in the examples are, unless otherwise indicated, commercially available products; for the methods involved, they are carried out according to conventional techniques, unless otherwise indicated. The following examples are not intended to limit the scope of the present invention.
Example 1: QHL-095-DOX Synthesis
QHL-095-DOX was synthesized as follows:

1. synthesis of intermediate 1
Taking a dry clean 2L reaction bottle, adding 500ml of THF, weighing 80g of Fmoc-Asn (Trt) -OH, adding into the reaction bottle, stirring for dissolving, adding 46.6g of DEPBT, stirring for 15 minutes at room temperature, adding 16g of PABC, reacting for 30 minutes at room temperature, adding 45ml of DIPEA, reacting for 3 hours at room temperature under the protection of nitrogen gas exchange, and monitoring by TLC that the reaction is finished (the reaction of Fmoc-Asn (Trt) -OH is complete).
The reaction solution was evaporated under reduced pressure, dissolved in a small amount of DMF (180ml), added dropwise to 3L of water under stirring to precipitate a pale yellow solid, washed with water for 2-3 times, filtered, collected, and vacuum dried to obtain an off-white solid (yield over 90%).
2. Synthesis of intermediate 2
And (3) sequentially adding 500ml of THF (tetrahydrofuran) and the white-like solid obtained in the previous step into a 2L single-mouth reaction bottle, stirring and dissolving, cooling to 0-5 ℃ by using an ice salt bath, dropwise adding 100ml of piperidine, gradually returning to room temperature for reaction for 1h after the dropwise addition is finished, and monitoring the reaction completion by TLC. Evaporating the solvent under reduced pressure, adding a small amount of DMF for dissolving, dropwise adding into 2L of water obtained by stirring, mechanically stirring for 30min, performing suction filtration, repeatedly washing for 2-3 times, performing suction filtration, adding 800ml of methyl tert-butyl ether into a filter cake, stirring for 30min, performing suction filtration, adding PE into the filter cake: EA is 10: washing for 2 times, filtering, collecting filter cake, and vacuum drying to obtain white solid 80g with purity of 70%.
3. Synthesis of intermediate 3
50ml THF, 5.04g Boc-Ala-Ala-OH, 3.89g DEPBT were added to a dry, clean 250ml single-neck reaction flask in sequence, the mixture was reacted at room temperature for 10min, 2.6g NH was added2H2H2-Asn (Trt) -PABC, performing nitrogen gas exchange protection, reacting for 15min at room temperature, dropwise adding 3.5ml of DIPEA, performing nitrogen gas exchange protection, reacting for 3h at room temperature, evaporating the solvent under reduced pressure, adding water, pulping for 2-3 times, performing suction filtration to obtain light yellow solid, 3.7g of the light yellow solid, and purifying by a column to obtain 2.0g of a product with the purity of 94.8% and the yield of 26.6%.
4. Synthesis of intermediate 4
Adding 1.8g of the intermediate 3 into a 250ml single-mouth reaction bottle, adding 28.5ml of TFA, dropwise adding 1.5ml of water, reacting at room temperature for 30min, monitoring by TLC, evaporating under reduced pressure to remove a solvent, adding methyl tert-butyl ether, pulping, performing suction filtration to obtain a solid, adding dioxane: water 1: dissolving the solution 1, adding 1N sodium hydroxide to adjust the pH value to 13, stirring at room temperature for 40min, evaporating the solvent under reduced pressure, mixing the sample with silica gel, and passing through a column to obtain 450mg of a product with the yield of 47.5%.
Synthesis of MI-S intermediates
MI-S1(338mg, 2mmol) and DEPBT (717.6mg, 2.4mmol) are added into a 100ml single-neck bottle, DMF (15ml) is added for dissolution, nitrogen gas exchange protection is carried out, reaction is carried out for 15min at room temperature, R3-b (819mg, 2mmol) is added, stirring is carried out for dissolution, reaction is carried out for 15min at room temperature, DIPEA 137 mu l is added dropwise, nitrogen gas exchange protection is carried out, reaction is carried out for 3h at room temperature, TLC is used for monitoring the completion of the reaction of R3-a, the solvent is removed by reduced pressure distillation, the crude product is dissolved by methanol, and an intermediate of R3-1 is obtained by a reversed-phase high-pressure column (720mg, yield: 64.3%).
Synthesis of MI-S
Adding the intermediate (720mg, 1.28mmol) obtained in the previous step into a 100ml single-mouth reaction bottle, adding 15ml of dichloromethane for dissolving, dropwise adding TFA5ml, dropwise adding 0.25ml of water, reacting at room temperature for 30min, monitoring by TLC after the reaction is finished, evaporating under reduced pressure to remove the solvent, adding methyl tert-butyl ether for pulping, performing suction filtration to obtain a solid, mixing the silica gel with a sample, and passing through a reverse phase column to obtain 242mg of a product. The yield thereof was found to be 37.5%.
7. Synthesis of intermediate 5
Adding the intermediate 4(150mg, 0.395mmol) and EMC-6Peg-COOH (239mg, 0.474mmol) into a 100ml single-neck bottle, adding DMF (15ml) for dissolving, carrying out nitrogen exchange protection, reacting at room temperature for 15min, dropwise adding DIPEA 137 ul, carrying out nitrogen exchange protection, reacting at room temperature for 3h, monitoring by TLC that the reaction of the intermediate 4 is finished, distilling under reduced pressure to remove the solvent, dissolving the crude product with methanol, and passing through a reverse-phase high-pressure column to obtain an intermediate 5(95mg, yield: 21%).
8. Synthesis of intermediate 6
Adding 25ml of DMF, an intermediate 5(300mg, 0.346mmol), a Bis-PNP (316mg, 1.04mmol), nitrogen gas exchange protection, reacting at room temperature for 15min, dropwise adding 258 mu l of DIPEA, nitrogen gas exchange protection, reacting at room temperature for 3h, monitoring 7% of the raw material remained by HPLC, stopping the reaction, evaporating the solvent under reduced pressure, and purifying by a column to obtain a product 150mg with a yield of 42%.
9. Synthesis of end product QHL-095-DOX
A100 mL reaction flask was charged with 84mg of doxorubicin hydrochloride (1.0eq, 0.145mmol) and 150mg of intermediate 6(1.0eq, 0.145mmol) and reacted at room temperature for 15min under nitrogen protection, 75. mu.L of DIPEA was added dropwise and reacted at room temperature for 4 hours, the solvent was distilled off under reduced pressure, the crude product was dissolved in methanol, and QHL-095-DOX (49mg of a red solid, yield: 23.8%) was obtained by passing through a reverse phase high pressure column.
EXAMPLE 2 Synthesis of QHL-116-DOX

1. Synthesis of intermediate 1
Taking a dry clean 2L reaction bottle, adding 500ml of THF, weighing 80g of Fmoc-Asn (Trt) -OH, adding into the reaction bottle, stirring for dissolving, adding 46.6g of DEPBT, stirring for 15 minutes at room temperature, adding 16g of PABC, reacting for 30 minutes at room temperature, adding 45ml of DIPEA, reacting for 3 hours at room temperature under the protection of nitrogen gas exchange, and monitoring by TLC that the reaction is finished (the reaction of Fmoc-Asn (Trt) -OH is complete).
The reaction solution was evaporated under reduced pressure, dissolved in a small amount of DMF (180ml), added dropwise to 3L of water under stirring to precipitate a pale yellow solid, washed with water for 2-3 times, filtered, collected, and vacuum dried to obtain an off-white solid (yield over 90%).
2. Synthesis of intermediate 2
And (3) sequentially adding 500ml of THF (tetrahydrofuran) and the white-like solid obtained in the previous step into a 2L single-mouth reaction bottle, stirring and dissolving, cooling to 0-5 ℃ by using an ice salt bath, dropwise adding 100ml of piperidine, gradually returning to room temperature for reaction for 1h after the dropwise addition is finished, and monitoring the reaction completion by TLC. Evaporating the solvent under reduced pressure, adding a small amount of DMF for dissolving, dropwise adding into 2L of water in stirring, mechanically stirring for 30min, performing suction filtration, repeatedly washing for 2-3 times, performing suction filtration, adding 800ml of methyl tert-butyl ether into a filter cake, stirring for 30min, performing suction filtration, adding PE into the filter cake: EA is 10: washing for 2 times, filtering, collecting filter cake, and vacuum drying to obtain white solid 80g with purity of 70%.
3. Synthesis of intermediate 3
50ml THF, 5.04g Boc-Ala-Ala-OH, 3.89g DEPBT were added to a dry, clean 250ml single-neck reaction flask in sequence, the mixture was reacted at room temperature for 10min, 2.6g NH was added2H2H2-Asn (Trt) -PABC, performing nitrogen gas exchange protection, reacting for 15min at room temperature, dropwise adding DIPEA3.5ml, performing nitrogen gas exchange protection, reacting for 3h at room temperature, evaporating the solvent under reduced pressure, adding water, pulping for 2-3 times, performing suction filtration to obtain light yellow solid, 3.7g, and purifying by a column to obtain 2.0g of a product with the purity of 94.8% and the yield of 26.6%.
4. Synthesis of intermediate 4
Adding 1.8g of the intermediate 3 into a 250ml single-mouth reaction bottle, adding 28.5ml of TFA, dropwise adding 1.5ml of water, reacting at room temperature for 30min, monitoring by TLC, evaporating under reduced pressure to remove a solvent, adding methyl tert-butyl ether, pulping, performing suction filtration to obtain a solid, adding dioxane: water 1: dissolving the solution 1, adding 1N sodium hydroxide to adjust the pH value to 13, stirring at room temperature for 40min, evaporating the solvent under reduced pressure, mixing the sample with silica gel, and passing through a column to obtain 450mg of a product with the yield of 47.5%.
5. Synthesis of intermediate 5
Fmoc-Glu (OAll) -COOH (1.554g, 3.79mmol) was weighed, dissolved in 10ml of a mixed solution of DCM and THF, and 2.72ml of HOtBu was added dropwise with stirring, after completion of the addition, N was added2And (3) ventilation protection, reacting for 16 hours at room temperature, monitoring by TLC after the reaction is finished, evaporating the solvent under reduced pressure, mixing the silica gel with a sample, and passing the mixture through a column to obtain 1.4g of a product, wherein the yield is as follows: 79.5 percent.
6. Synthesis of intermediate 6
And sequentially adding 10ml of THF (tetrahydrofuran) into a dry and clean 250ml single-mouth reaction bottle, stirring and dissolving the intermediate 5(1.4g and 3mmol) obtained in the previous step, cooling to 0-5 ℃ by an ice salt bath, dropwise adding 3ml of piperidine, gradually heating to room temperature after the dropwise adding is finished, reacting for 2 hours, monitoring the reaction by TLC (thin layer chromatography), evaporating under reduced pressure to remove the solvent, mixing and purifying silica gel, collecting eluent containing the product, and drying under reduced pressure in vacuum to constant weight to obtain 583mg of the product with the yield of 80%.
7. Synthesis of intermediate 7
Adding 15ml of THF, 583mg of intermediate 6 and 932.8mg of DEPBT into a dry and clean 250ml single-mouth reaction bottle in sequence, reacting for 10min at room temperature, adding 506.4mg of maleimide caproic acid, performing nitrogen exchange protection, reacting for 15min at room temperature, dropwise adding 1.3ml of DIPEA, performing nitrogen exchange protection, reacting for 3 hours at room temperature, evaporating the solvent under reduced pressure, adding water, pulping for 2-3 times, performing suction filtration to obtain pale yellow solid, 800mg, purifying by passing through a column to obtain 628mg of a product with the purity of 94.8 percent and the yield of 59.9 percent.
8. Synthesis of intermediate 8
Adding 10ml of dichloromethane and 872mg of intermediate 7 into a clean 100ml single-mouth reaction bottle in sequence, stirring uniformly, dropwise adding 3ml of TFA, reacting at room temperature for 2 hours, monitoring the completion of the reaction of raw materials by TLC, evaporating the solvent under reduced pressure in vacuum, adding methyl tert-butyl ether for pulping, performing suction filtration to obtain a solid, stirring and purifying the solid with silica gel, collecting eluent containing the product, and drying under reduced pressure in vacuum to constant weight to obtain 459mg of the product with the yield of 60.3%.
9. Synthesis of intermediate 9
Adding 15ml of THF, 459mg of intermediate 8 and 434mg of DEPBT into a dry and clean 250ml single-mouth reaction bottle in sequence, reacting for 10min at room temperature, adding 457.8mg of intermediate 4, carrying out nitrogen gas exchange protection, reacting for 15min at room temperature, dropwise adding 627 microliter of DIPEA, adding the rest, carrying out nitrogen gas exchange protection, reacting for 3 hours at room temperature, evaporating the solvent under reduced pressure, adding water, pulping for 2-3 times, carrying out suction filtration to obtain pale yellow solid, 750mg, and purifying by passing through a column to obtain 655mg of the product, wherein the yield is 63.2%.
10. Synthesis of intermediate 10
Adding 25ml of DMF, an intermediate 9(655mg, 0.88mmol), a Bis-PNP (804mg, 2.64mmol), nitrogen gas exchange protection, reacting at room temperature for 15min, dropwise adding 258 mu l of DIPEA, nitrogen gas exchange protection, reacting at room temperature for 3h, monitoring 7% of the raw material remained by HPLC, stopping the reaction, evaporating the solvent under reduced pressure, and purifying by a column to obtain 335mg of a product with the yield of 42%.
11. Synthesis of intermediate 11
214.3mg of doxorubicin hydrochloride (1.0eq, 0.369mmol) and 335mg of intermediate 10(1.0eq, 0.369mmol) were charged into a 100mL reaction flask, and the mixture was reacted at room temperature for 15min under nitrogen protection, followed by dropwise addition of 190. mu.L of DIPEA, followed by reaction at room temperature for 4 hours, evaporation of the solvent under reduced pressure to dissolve the crude product in methanol, and then the crude product was passed through a reverse-phase autoclave column to obtain intermediate 11(115mg of a red solid, yield: 23.8%).
12. Synthesis of the end product
To a 100mL reaction flask were added, in order, 15mL of THF, intermediate 11(115mg, 0.0877mmol), and tri-n-butylstannyl hydride (76mg, 0.2631mmol), and the reaction solution was saturated with nitrogen. Tetrakis (triphenylphosphine) palladium (0) (14.2mg, 0.012mmol) was then added, and the mixture was stirred at room temperature overnight. Monitored by TLC until conversion was complete. The flask contents were then filtered through celite and the residue was washed with THF. The filtrate was concentrated under reduced pressure. The resulting crude product was purified by column chromatography to obtain 100mg (yield: 90%) of the objective compound.
Example 3: synthesis of N-CBP

1. Synthesis of intermediate 1
Adding 300mg of raw materials into a 100ml three-necked bottle, adding 15ml of THF/ETOH (4: 1) for dissolution, cooling to-5-0 ℃ through an ice salt bath, stirring and controlling the temperature, dropwise adding 210mg of LiOH (5ml) aqueous solution into the bottle in batches, naturally heating for reaction for 1h after dropwise adding, sending the reaction solution to HPLC, controlling the temperature to-5-0 ℃ when the raw materials completely react, adjusting the pH of the reaction solution to 3-4 by using 1mol/L of HCl, controlling the temperature to 25-30 ℃ and removing the solvent in a rotary manner to obtain a crude product of the intermediate 1, and directly using the crude product in the next step.
2. Synthesis of intermediate 2
Adding 15ml of 1mol/L dioxane hydrochloride solution into the intermediate 1, stirring at room temperature for reaction for 1h, sending the reaction liquid to HPLC, controlling the temperature to 25-30 ℃ to remove the solvent to obtain a crude product of the intermediate 2, and directly carrying out the next step.
3. Synthesis of intermediate 3
Adding the crude product of the intermediate 2 into a 100ml three-necked bottle, adding 20ml dioxane for dissolution, cooling to-5-0 ℃ in an ice salt bath, dropwise adding 159mg of sodium carbonate aqueous solution (pH is about 8) into the bottle at the controlled temperature, carrying out nitrogen protection, dropwise adding 311mg of Fmoc-Cl dioxane solution at the controlled temperature of-5-0 ℃, naturally heating for reaction for 1h after dropwise adding, sending HPLC (high performance liquid chromatography) for detection, completely reacting the intermediate 2, removing the solvent by rotation, mixing the crude product with silica gel, and passing through a reverse phase medium pressure column to obtain 107mg of the intermediate 3.
4. Synthesis of intermediate 4
Adding 107mg of the intermediate 3 into a 50ml single-mouth bottle, adding 15ml of methanol for dissolving, cooling to-20 ℃ by using liquid nitrogen, dropwise adding 302ul of tetrabutylammonium hydroxide (25% methanol solution) into the bottle, and naturally heating for reaction for 1h after dropwise adding, wherein the reaction solution is a standby solution 1.
Adding 140.8mg of diiododiammineplatinum into a 50ml single-mouth bottle, adding 10ml of ultrapure water for dissolving, heating to 50 ℃, keeping out of the sun, dropwise adding 49.5mg of silver nitrate aqueous solution into the bottle under the protection of nitrogen, reacting for 15min, continuously dropwise adding 49.5mg of silver nitrate aqueous solution into the bottle, reacting for 15min after dropwise adding, filtering the reaction solution with a filter membrane, transferring the filtrate into a 100ml single-mouth bottle, dropwise adding the standby solution 1 into the bottle at room temperature, replacing with nitrogen for three times, transferring the reaction solution into an oil bath, heating to 50 ℃, reacting overnight (generally 16h) in the sun, stopping the reaction, centrifuging the reaction solution, directly passing the supernatant through a high-pressure reverse phase column, and freeze-drying the preparation solution to obtain 79mg of an intermediate 4 with the yield of 45.7%.
Synthesis of N-CBP
Adding 5mg of the intermediate 4 into a 10ml single-neck bottle, adding 2ml of MeOH/ACN (1: 1), stirring and dissolving, dripping 2ul of DBU into the reaction liquid at room temperature, reacting for half an hour under the protection of nitrogen, detecting by HPLC, completely reacting the intermediate 4, dripping the reaction liquid into 6ml of methyl tert-butyl ether, separating out a white-like solid, centrifuging, removing a supernatant, dissolving the solid with water/tert-butyl alcohol, and passing through a column to obtain 1.8mg of the product N-CBP.
Example 4: QHL-140-N-CBP Synthesis

1. Synthesis of intermediate 1
Adding 500mg of raw material into a 100ml three-necked bottle, adding 10ml of DCM for dissolving, cooling to-5 ℃ -0 ℃, dropwise adding 5ml of TFA under stirring, after reacting for 1h, monitoring the completion of the reaction of the raw material by HPLC, removing the solvent in the reaction solution by screwing, and obtaining the residual oily substance as an intermediate 1.
2. Synthesis of intermediate 2
Adding the intermediate 1 and 1.15g of raw material Fmoc-AAN-PABC-PNP into a 100ml single-mouth bottle, dissolving the mixture with 20ml of DMF, activating the mixture for 10min under the protection of nitrogen and stirring, dropwise adding 0.87ml of DIPEA into a reaction bottle, reacting for 0.5h, sending the mixture to HPLC (high performance liquid chromatography) for detection, completely reacting the raw material Fmoc-AAN-PABC-PNP, removing the DMF in a reaction solution by spinning, dissolving a crude product with water/DMF, and passing through a high-pressure reverse phase column to obtain 975mg of an intermediate 2, wherein the yield is as follows: 78.6 percent.
3. Synthesis of intermediate 3
Adding 400mg of the intermediate 2 into a 250ml three-necked bottle, adding THF/ETOH (4: 1)35ml for dissolution, cooling to-5-0 ℃ by using an ice salt bath, controlling the temperature to-5-0 ℃, dropwise adding 202mg of LiOH aqueous solution in batches, controlling the temperature for reaction for 3h after completing dropwise addition, sending HPLC (high performance liquid chromatography) for detection, controlling the temperature to-5-0 ℃ when the intermediate 2 is completely reacted, adjusting the pH of a reaction solution to 6-7 by using 1mol/L of HCL, removing a solvent by rotation at 25-30 ℃, pulping a crude product twice by using methyl tert-butyl ether, dissolving a solid by using methanol/water, passing through a high-pressure reverse phase column to obtain 230mg of the intermediate 3, wherein the yield is 86.7%.
4. Synthesis of intermediate 4
Adding 235mg of intermediate 3 and 222mg of EMC-OSU into a 100ml single-neck bottle, adding 30ml of DMF, stirring and dissolving, heating to 50 ℃, reacting overnight (generally 16h) under nitrogen protection, sending HPLC (high performance liquid chromatography) for detection, completely reacting the intermediate 3, removing DMF by spinning, dissolving the crude product with methanol/water, and passing through a high-pressure reverse phase column to obtain 200mg of intermediate 4 with the yield of 53.6%.
5. Synthesis of the end product QHL-140-N-CBP
Adding 200mg of the intermediate 4 into a 100ml single-mouth bottle, adding 20ml of methanol for dissolving, cooling to-20 ℃ by using liquid nitrogen, dropwise adding 279ul tetrabutyl ammonium hydroxide (25% methanol solution) into the bottle, and naturally heating for reaction for 1h after dropwise adding, wherein the reaction liquid is a standby liquid 1.
Adding 130mg of diiododiammineplatinum into a 100ml single-mouth bottle, adding 30ml of ultrapure water for dissolving, heating to 50 ℃, keeping out of the sun, dropwise adding 46mg of silver nitrate aqueous solution into the bottle under the protection of nitrogen, reacting for 15min, continuously dropwise adding 46mg of silver nitrate aqueous solution into the bottle, reacting for 15min after dropwise adding, filtering the reaction solution with a filter membrane, transferring the filtrate into a 250ml single-mouth bottle, dropwise adding the standby solution 1 into the bottle at room temperature, carrying out dropwise adding, carrying out nitrogen replacement for three times, transferring the reaction solution into an oil bath, heating to 50 ℃, reacting overnight (generally 16h) in the absence of the sun, centrifuging the reaction solution, directly passing the supernatant through a high-pressure reverse phase column, and freeze-drying the preparation solution to obtain 90mg of products QHL-140-N-CBP with the yield of 34.5%.
Example 5: QHL-086-N-CBP Synthesis

1. Synthesis of intermediate 1
Adding 500mg of raw material into a 100ml three-necked bottle, adding 10ml of DCM for dissolving, cooling to-5 ℃ -0 ℃, dropwise adding 5ml of TFA under stirring, after reacting for 1h, monitoring the completion of the reaction of the raw material by HPLC, removing the solvent in the reaction solution by screwing, and obtaining the residual oily substance as an intermediate 1.
2. Synthesis of intermediate 2
Adding the intermediate 1 and 1.15g of raw material Fmoc-AAN-PABC-PNP into a 100ml single-neck bottle, dissolving the mixture with 20ml of DMF, activating the mixture for 10min under the protection of nitrogen, dropwise adding 0.87ml of DIPEA into the reaction bottle, reacting for 0.5h, sending the mixture to HPLC (high performance liquid chromatography) for detection, completely reacting the raw material Fmoc-AAN-PABC-PNP, removing the DMF in the reaction liquid by spinning, dissolving the crude product with water/DMF, and passing through a high-pressure reverse phase column to obtain 975mg of intermediate 2 with the yield of 78.6%.
3. Synthesis of intermediate 3
Adding 400mg of the intermediate 2 into a 250ml three-necked bottle, adding THF/ETOH (4: 1)35ml for dissolution, cooling to-5-0 ℃ by using an ice salt bath, controlling the temperature to-5-0 ℃, dropwise adding 202mg of LiOH aqueous solution in batches, controlling the temperature for reaction for 3h after completing dropwise addition, sending HPLC (high performance liquid chromatography) for detection, controlling the temperature to-5-0 ℃ when the intermediate 2 is completely reacted, adjusting the pH of a reaction solution to 6-7 by using 1mol/L of HCL, removing a solvent by rotation at 25-30 ℃, pulping a crude product twice by using methyl tert-butyl ether, dissolving a solid by using methanol/water, passing through a high-pressure reverse phase column to obtain 235mg of the intermediate 3, and obtaining the yield of 88.6%.
4. Synthesis of intermediate 4
Adding 89mg of EMC-2Peg-OH into a 100ml single-neck bottle, dissolving DMF, adding 97mg of DEPBT into the bottle, stirring and activating at room temperature for 1h, dropwise adding 95ul of DEPBT into the bottle, continuously stirring for 1h, dropwise adding 150mg of DMF solution of the intermediate 3 into the bottle in batches, stirring and reacting at room temperature after dropwise adding, and detecting by HPLC. After the reaction is finished, DMF is removed by rotation, and the crude product is dissolved by water/methanol and passes through a reversed phase high pressure column to obtain 88mg of a product with the yield of 37.6 percent.
5. Synthesis of end product QHL-086-N-CBP
Adding 88mg of the intermediate 4 into a 50ml single-mouth bottle, adding 10ml of methanol for dissolving, cooling to-20 ℃ by using liquid nitrogen, dropwise adding 106ul of tetrabutylammonium hydroxide (25% methanol solution) into the bottle, and naturally heating for reaction for 1h after dropwise adding, wherein the reaction solution is a standby solution 1.
Adding 49mg of diiododiammineplatinum into a 50ml single-mouth bottle, adding 10ml of ultrapure water for dissolving, heating to 50 ℃, keeping out of the sun, dropwise adding 17mg of silver nitrate aqueous solution into the bottle under the protection of nitrogen, reacting for 15min, continuously dropwise adding 17mg of silver nitrate aqueous solution into the bottle, reacting for 15min after dropwise adding, filtering the reaction solution with a filter membrane, transferring the filtrate into a 100ml single-mouth bottle, dropwise adding the standby solution 1 into the bottle at room temperature, carrying out nitrogen replacement for three times, transferring the reaction solution into an oil bath, heating to 50 ℃, reacting overnight (generally 16h) in the sun, sending HPLC (high performance liquid chromatography) for detection, stopping the reaction, centrifuging the reaction solution, directly passing the supernatant through a high-pressure reverse phase column, and freeze-drying the preparation solution to obtain 54mg of a product QHL-086-N-CBP with the yield of 48.6%.
Example 6: QHL-095-N-CBP Synthesis

1. Synthesis of intermediate 1
Adding 500mg of raw material into a 100ml three-necked bottle, adding 10ml of DCM for dissolving, cooling to-5 ℃ -0 ℃, dropwise adding 5ml of TFA under stirring, after reacting for 1h, monitoring the completion of the reaction of the raw material by HPLC, removing the solvent in the reaction solution by screwing, and obtaining the residual oily substance as an intermediate 1.
2. Synthesis of intermediate 2
Adding the intermediate 1 and 1.15g of raw material Fmoc-AAN-PABC-PNP into a 100ml single-mouth bottle, dissolving the mixture with 20ml of DMF, activating the mixture for 10min under the protection of nitrogen and stirring, dropwise adding 0.87ml of DIPEA into a reaction bottle, reacting for 0.5h, sending the mixture to HPLC (high performance liquid chromatography) for detection, completely reacting the raw material Fmoc-AAN-PABC-PNP, removing the DMF in a reaction solution by spinning, dissolving a crude product with water/DMF, and passing through a high-pressure reverse phase column to obtain 975mg of an intermediate 2, wherein the yield is as follows: 78.6 percent.
3. Synthesis of intermediate 3
Adding 400mg of the intermediate 2 into a 250ml three-necked bottle, adding THF/ETOH (4: 1)35ml for dissolution, cooling to-5-0 ℃ by using an ice salt bath, controlling the temperature to-5-0 ℃, dropwise adding 202mg of LiOH aqueous solution in batches, controlling the temperature for reaction for 3h after completing dropwise addition, sending HPLC (high performance liquid chromatography) for detection, controlling the temperature to-5-0 ℃ when the intermediate 2 is completely reacted, adjusting the pH of a reaction solution to 6-7 by using 1mol/L of HCL, removing a solvent by rotation at 25-30 ℃, pulping a crude product twice by using methyl tert-butyl ether, dissolving a solid by using methanol/water, passing through a high-pressure reverse phase column to obtain 235mg of the intermediate 3, and obtaining the yield of 88.6%.
4. Synthesis of intermediate 4
Adding 180mg of intermediate 3 and 240mg of EMC-6Peg-OSU into a 100ml single-mouth bottle, adding 20ml of DMF, stirring for dissolving, heating to 50 ℃, reacting overnight (generally 16h) under the protection of nitrogen, sending HPLC (high performance liquid chromatography) for detection, completely reacting the intermediate 3, removing DMF by spinning, dissolving the crude product with methanol/water, and passing through a high-pressure reverse phase column to obtain 234mg of intermediate 4 with the yield of 69.2%.
5. Synthesis of end product QHL-095-N-CBP
Adding 234mg of the intermediate 4 into a 100ml single-mouth bottle, adding 15ml of methanol for dissolving, cooling to-20 ℃ by using liquid nitrogen, dropwise adding 234ul of tetrabutylammonium hydroxide (25% methanol solution) into the bottle, and naturally heating for reaction for 1h after dropwise adding, wherein the reaction liquid is a standby liquid 1.
Adding 109mg of diiododiammineplatinum into a 100ml single-mouth bottle, adding 20ml of ultrapure water for dissolving, heating to 50 ℃, keeping out of the sun, dropwise adding 38mg of silver nitrate aqueous solution into the bottle under the protection of nitrogen, reacting for 15min, continuously dropwise adding 38mg of silver nitrate aqueous solution into the bottle, reacting for 15min after dropwise adding, filtering the reaction solution with a filter membrane, transferring the filtrate into a 250ml single-mouth bottle, dropwise adding the standby solution 1 into the bottle at room temperature, after dropwise adding, performing nitrogen replacement for three times, transferring the reaction solution into an oil bath, heating to 50 ℃, reacting overnight (generally 16h) in the absence of the sun, centrifuging the reaction solution, directly passing the supernatant through a high-pressure reverse phase column, and freeze-drying the preparation solution to obtain 138mg of a final product with the yield of 48%.
Example 7: QHL-006-DOX Synthesis
QHL-006, the synthetic route is shown below:

synthesis of MI-S intermediate-1 in QHL-006-DOX
Weighing maleic anhydride (245mg, 2.5mmol) in a dry and clean 100ml single-mouth reaction bottle, adding 10ml dichloromethane, stirring for dissolving, weighing NH2H2H2-3Peg-COOtBu (624mg, 2.25mmol) at room temperature for 6 hours, monitoring by LC-MS that the maleic anhydride reaction is completed, spin-drying the reaction solution, stirring with silica gel and passing through a column to obtain MI-S intermediate-1 (456mg, yield 48.6%).
Synthesis of MI-S intermediate-2 in QHL-006-DOX
Adding 456mg of MI-S intermediate-1 obtained in the step into a 100ml single-mouth reaction bottle, adding 10ml of acetic anhydride, stirring and dissolving, weighing NaOAC (98.7mg, 1.216mmol), adding in batches slowly, heating to 110 ℃ in an oil bath, reacting for 3h, monitoring by LC-MS that the reaction of the MI-S intermediate-1 is finished, cooling to room temperature, drying the reaction liquid in a spinning mode, and purifying through a column to obtain MI-S intermediate-2 (312, yield 70%).
Synthesis of MI-S in QHL-006-DOX
Adding the MI-S intermediate-2 (312mg, 0.87mmol) obtained in the previous step into a 100ml single-mouth reaction bottle, adding 10ml of dichloromethane for dissolving, dropwise adding TFA2ml, dropwise adding 0.15ml of water, reacting at room temperature for 30min, monitoring by TLC after the reaction is finished, evaporating under reduced pressure to remove the solvent, adding methyl tert-butyl ether for pulping, performing suction filtration to obtain a solid, mixing the silica gel with a sample, and passing through a reverse phase column to obtain 196mg of a product. The yield thereof was found to be 75%.
The final product was prepared by a method analogous to the synthesis of QHL-095-DOX, using different MI-S for ligation (preparation of MI-S is referred to the synthesis of MI-S in QHL-006-DOX).
Example 8: QHL-096-DOX Synthesis

1) Synthesis of intermediate 1
Dissolving N-benzyloxycarbonyl-L-alanine (100g, 0.45mol) in dry N, N-dimethylformamide (3L), adding 1-hydroxybenzotriazole (72.6g, 0.54mol) and 1-ethyl- (3-dimethylaminopropyl) carbodiimide hydrochloride (103.3g, 0.54mol) under stirring, stirring for reaction for 1 hour, dropwise adding L-alanine methyl ester (46.2g, 0.45mol) and N, N-diisopropylethylamine (173.8g, 1.34mol) in N, N-dimethylformamide (1L) at 0 ℃, stirring for 10 hours at room temperature after dropwise adding, evaporating the solvent under reduced pressure, dissolving the crude product in dichloromethane (2L), washing with saturated ammonium chloride solution, water and saturated sodium chloride solution in turn, drying the organic phase with anhydrous sodium sulfate, evaporating the solvent under reduced pressure, recrystallizing the crude product with ethyl acetate/petroleum ether to obtain a pure product as an intermediate 1(101g of a white solid, yield: 73.1%).
2) Synthesis of intermediate 2
Intermediate 1(100g, 0.34mol) was dissolved in a mixed solution of tetrahydrofuran (2L) and water (1L), cooled to 0 ℃ and 1mol/L lithium hydroxide solution (400mL) was added dropwise, the reaction was stirred for 10 hours, concentrated hydrochloric acid was added dropwise to neutralize the solution to PH <6, tetrahydrofuran was evaporated under reduced pressure, the remaining aqueous phase was extracted with dichloromethane (1L × 3), the organic phase was dried over anhydrous sodium sulfate, and evaporated under reduced pressure to dryness to give intermediate 2(88g of white solid, yield: 92.2%).
3) Synthesis of intermediate 3
In a three-necked flask, tert-butyl L-leucine (22.4g, 0.1mol), N-Fmoc-N' -trityl asparagine (59.6g, 0.1mol) and N, N-dimethylformamide (1000mL) were dissolved, 1-hydroxybenzotriazole (14.85g, 0.11mol) and 1-ethyl- (3-dimethylaminopropyl) carbonyldiimine hydrochloride (23g, 0..12mol) were added with stirring, N-diisopropylethylamine (25.8g, 0.2mol) was added after cooling in ice to 0 ℃, the solvent was distilled off under reduced pressure after stirring for 10 hours, the crude product was dissolved in chloroform (1000mL), washed with a saturated ammonium chloride solution, a saturated sodium chloride solution and water in this order, the organic phase was dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure after filtration to obtain intermediate 3(42.4g of a white solid) after purification (dichloromethane: ethyl acetate 1: 1 by volume ratio), yield: 55.4%).
4) Synthesis of intermediate 4
Intermediate 3(7.65g, 0.01mol) was dissolved in a mixed solution of dichloromethane (100mL) and N, N-dimethylformamide (100mL), piperidine (40mL) was added, and after stirring at room temperature for 5 hours, the solvent was distilled off under reduced pressure, and then placed in a vacuum oven to be dried under high vacuum to remove a small amount of piperidine, to give intermediate 4 as a pale yellow solid which was used in the next step without purification.
5) Synthesis of intermediate 5
The crude intermediate 4 obtained in the above step was dissolved in N, N-dimethylformamide (200mL), and intermediate 2(2.94g, 0.012mol), benzotriazole-N, N, N ', N' -tetramethyluronium Hexafluorophosphate (HBTU) (6.07g, 0.016mol) were added thereto, and after ice-cooling to 0 ℃ N, N-diisopropylethylamine (2.6g, 0.02mol) was added thereto, and the mixture was stirred at room temperature overnight, the solvent was evaporated under reduced pressure, and the residue was dissolved in chloroform (100mL), washed with a saturated ammonium chloride solution and a saturated sodium chloride solution in this order, dried over anhydrous sodium sulfate, filtered, and the solvent was evaporated, and the crude product obtained was subjected to silica gel column chromatography to give intermediate 5(3.1g of a white solid, two-step total yield: 37.8%).
6) Synthesis of intermediate 6
Cbz-AAN (trt) -L-Otbu (3.00g, 3.65mmol) was dissolved in methanol (100mL), 10% palladium on carbon (0.3g) was added, hydrogen was introduced, the reaction was stirred at normal temperature and pressure for 4 hours, the palladium on carbon was removed by filtration, washing with methanol was conducted, the filtrate and the washing liquid were combined, and the solvent was distilled off under reduced pressure to obtain intermediate 6(2.38g of a white solid, yield: 95.2%).
7) Synthesis of intermediate 7
Intermediate 6(2.38g,3.4mmol) and EMC-6Peg-OSu (2.4g,4.08mmol) were added to a 250ml single neck flask, dissolved in DMF (30ml) and heated to 50 ℃ for 6 h. The solvent was removed by distillation under the reduced pressure, and the crude product was dissolved in methanol and passed through a reverse-phase high-pressure column to give intermediate 7(2.5g, yield: 63.2%).
8) Synthesis of intermediate 8
Intermediate 7(1.00g,0.852mmol) was dissolved in DCM (20mL), trifluoroacetic acid (10mL) was added dropwise at room temperature, the reaction was stirred for 2h, the reaction solution was monitored by HPLC, intermediate 1 reacted completely, the solvent was removed by distillation under reduced pressure, the crude product was washed twice with methyl tert-butyl ether, the solid was dissolved with methanol, and intermediate 8(721mg of white solid, yield: 96.8%) was obtained by reverse phase high pressure column.
9) Synthesis of end product QHL-096-DOX
A100 mL reaction flask was charged with 63mg of doxorubicin hydrochloride (1.0eq), 95mg of intermediate 8(1eq), 39mg of DEPBT (1.2eq) and 10mL of DMF, 60ul of DIPEA (3eq) was added to the reaction mixture under nitrogen protection, and after 4 hours at room temperature, the solvent was distilled off under reduced pressure, the crude product was dissolved in methanol, and passed through a reverse phase high pressure column to obtain QHL-096-DOX (52mg of red solid, yield: 34.2%). 7) Synthesis of intermediate 7
Intermediate 6(1.00g, 1.46mmol) was dissolved in DCM (20mL), trifluoroacetic acid (10mL) was added dropwise at room temperature, the reaction was stirred for 2h, the reaction solution was monitored by HPLC, intermediate 1 reacted completely, the solvent was removed by distillation under reduced pressure, the crude product was washed twice with methyl tert-butyl ether, the solid was dissolved in methanol, and passed through a reverse phase high pressure column to give intermediate 7(546mg of a white solid, yield: 96.8%).
Example 9: QHL-117-DOX Synthesis
QHL-117 is synthesized as follows:

1) synthesis of intermediate 1
Dissolving N-benzyloxycarbonyl-L-alanine (100g, 0.45mol) in dry N, N-dimethylformamide (3L), adding 1-hydroxybenzotriazole (72.6g, 0.54mol) and 1-ethyl- (3-dimethylaminopropyl) carbodiimide hydrochloride (103.3g, 0.54mol) under stirring, stirring for reaction for 1 hour, dropwise adding L-alanine methyl ester (46.2g, 0.45mol) and N, N-diisopropylethylamine (173.8g, 1.34mol) in N, N-dimethylformamide (1L) at 0 ℃, stirring for 10 hours at room temperature after dropwise adding, evaporating the solvent under reduced pressure, dissolving the crude product in dichloromethane (2L), washing with saturated ammonium chloride solution, water and saturated sodium chloride solution in turn, drying the organic phase with anhydrous sodium sulfate, evaporating the solvent under reduced pressure, recrystallizing the crude product with ethyl acetate/petroleum ether to obtain a pure product as an intermediate 1(101g of a white solid, yield: 73.1%).
2) Synthesis of intermediate 2
Intermediate 1(100g, 0.34mol) was dissolved in a mixed solution of tetrahydrofuran (2L) and water (1L), cooled to 0 ℃ and 1mol/L lithium hydroxide solution (400mL) was added dropwise, the reaction was stirred for 10 hours, concentrated hydrochloric acid was added dropwise to neutralize the solution to PH <6, tetrahydrofuran was evaporated under reduced pressure, the remaining aqueous phase was extracted with dichloromethane (1L × 3), the organic phase was dried over anhydrous sodium sulfate, and evaporated under reduced pressure to dryness to give intermediate 2(88g of white solid, yield: 92.2%).
3) Synthesis of intermediate 3
In a three-necked flask, tert-butyl L-leucine (22.4g, 0.1mol), N-Fmoc-N' -trityl asparagine (59.6g, 0.1mol) and N, N-dimethylformamide (1000mL) were dissolved, 1-hydroxybenzotriazole (14.85g, 0.11mol) and 1-ethyl- (3-dimethylaminopropyl) carbonyldiimine hydrochloride (23g, 0..12mol) were added with stirring, N-diisopropylethylamine (25.8g, 0.2mol) was added after cooling in ice to 0 ℃, the solvent was distilled off under reduced pressure after stirring for 10 hours, the crude product was dissolved in chloroform (1000mL), washed with a saturated ammonium chloride solution, a saturated sodium chloride solution and water in this order, the organic phase was dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure after filtration to obtain intermediate 3(42.4g of a white solid) after purification (dichloromethane: ethyl acetate 1: 1 by volume ratio), yield: 55.4%).
4) Synthesis of intermediate 4
Intermediate 3(7.65g, 0.01mol) was dissolved in a mixed solution of dichloromethane (100mL) and N, N-dimethylformamide (100mL), piperidine (40mL) was added, and after stirring at room temperature for 5 hours, the solvent was distilled off under reduced pressure, and then placed in a vacuum oven to be dried under high vacuum to remove a small amount of piperidine, to give intermediate 4 as a pale yellow solid which was used in the next step without purification.
5) Synthesis of intermediate 5
The crude intermediate 4 obtained in the above step was dissolved in N, N-dimethylformamide (200mL), and intermediate 2(2.94g, 0.012mol), benzotriazole-N, N, N ', N' -tetramethyluronium Hexafluorophosphate (HBTU) (6.07g, 0.016mol) were added thereto, and after ice-cooling to 0 ℃ N, N-diisopropylethylamine (2.6g, 0.02mol) was added thereto, and the mixture was stirred at room temperature overnight, the solvent was evaporated under reduced pressure, and the residue was dissolved in chloroform (100mL), washed with a saturated ammonium chloride solution and a saturated sodium chloride solution in this order, dried over anhydrous sodium sulfate, filtered, and the solvent was evaporated, and the crude product obtained was subjected to silica gel column chromatography to give intermediate 5(3.1g of a white solid, two-step total yield: 37.8%).
6) Synthesis of intermediate 6
Cbz-AAN (trt) -L-Otbu (3.00g, 3.65mmol) was dissolved in methanol (100mL), 10% palladium on carbon (0.3g) was added, hydrogen was introduced, the reaction was stirred at normal temperature and pressure for 4 hours, the palladium on carbon was removed by filtration, washing with methanol was conducted, the filtrate and the washing liquid were combined, and the solvent was distilled off under reduced pressure to obtain intermediate 6(2.38g of a white solid, yield: 95.2%).
7) Synthesis of intermediate 7
15ml of THF, (2.387g,3.4mmol) intermediate 6 and 1.35g of DEPBT are sequentially added into a dry and clean 250ml single-mouth reaction bottle, the mixture is reacted for 10min at room temperature, EMC-Glu (OAll) -COOH (1.3g,3.4mmol) is added, nitrogen gas exchange protection is carried out, the mixture is reacted for 15min at room temperature, 1.8ml of DIPEA is added dropwise, after the addition is finished, nitrogen gas exchange protection is carried out, the mixture is reacted for 3h at room temperature, the solvent is evaporated under reduced pressure, water is added for pulping for 2-3 times, suction filtration is carried out, light yellow solid is obtained, 700mg is obtained, column chromatography purification is carried out, 2.2g of the product is obtained, and the yield is 63.2%.
8) Synthesis of intermediate 8
Intermediate 7(1.53g,1.46mmol) was dissolved in DCM (20mL), trifluoroacetic acid (10mL) was added dropwise at room temperature, the reaction was stirred for 2h, the reaction solution was monitored by HPLC, intermediate 1 reacted completely, the solvent was removed by distillation under reduced pressure, the crude product was washed twice with methyl tert-butyl ether, the solid was dissolved with methanol, and the intermediate 8 was obtained by reverse phase high pressure column (928mg white solid, yield: 84.8%).
9) Synthesis of intermediate 9
A100 mL reaction flask was charged with 510.4mg of doxorubicin hydrochloride (1.0eq, 0.88mmol) and 659mg of intermediate 8(1.0eq, 0.88mmol), reacted at room temperature for 15min under nitrogen protection, 78. mu.L of DIPEA was added dropwise, reacted at room temperature for 4 hours, the solvent was distilled off under reduced pressure, the crude product was dissolved in methanol, and the resulting solution was passed through a reverse-phase high-pressure column to obtain intermediate 9(258mg of a red solid, yield: 23.8%). 10) Synthesis of the end product
To a 100mL reaction flask were added in this order 15mL of THF, intermediate 9(258mg, 0.202mmol), and n-butylstannyl hydride (175.7mg, 0.606mmol), and the reaction solution was saturated with nitrogen. Tetrakis (triphenylphosphine) palladium (0) (32.7mg, 0.028mmol) was then added, and the mixture was stirred at room temperature overnight. Monitored by TLC until conversion was complete. The flask contents were then filtered through celite and the residue was washed with THF. The filtrate was concentrated under reduced pressure. The resulting crude product was purified by column chromatography to obtain 224mg (yield: 90%) of the objective compound.
The other compounds in Table 1 below were prepared using similar methods to examples 1-2, 4-9, by using different MI, S, C, A and D moieties.
The compounds were verified by Mass Spectrometry (MS) and their molecular weights are shown in table 1, which are consistent with the calculated molecular weights based on their structures.
TABLE 1









The present invention also provides the following comparative compounds, of the formula:
compound C1: doxorubicin

Compound C2: AANL-DOX

Compound C3: EMC-AANL-DOX
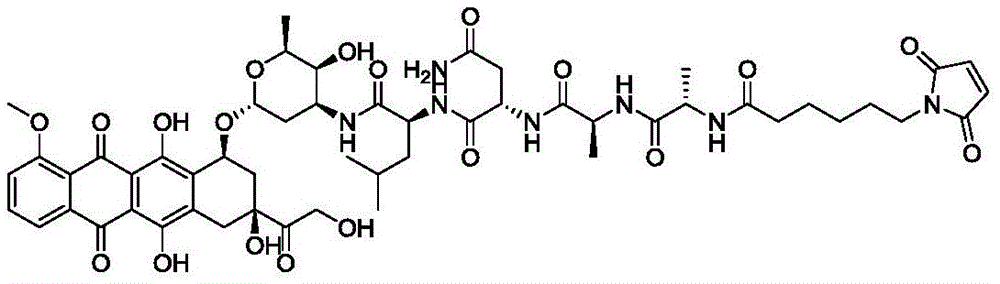
Compound C4: Peg-AANL-DOX

Example 10: preparation of human serum Albumin-conjugated HSA-EMC-AANL-DOX, HSA-QHL-087-DOX and HSA-QHL-087-N-CBP drugs
EMC-AANL-DOX, QHL-087-DOX and QHL-087-N-CBP were formulated, wherein EMC-AANL-DOX was dissolved using DMSO, QHL-087-DOX and QHL-087-N-CBP were dissolved with sterile water. HSA was dissolved in sterile water. Compound with HSA at 3: 1(4.8umol/mL, 1.6umol/mL), reacting in water bath at 37 ℃ for 3h, taking out the reaction solution, filtering unbound compounds by using a pressurized ultrafiltration membrane, adding physiological saline for dilution, and filtering for 3 times to obtain a semi-finished product. Human serum albumin-conjugated doxorubicin antitumor agents are isolated using, for example, chromatographic methods such as DEAE ion exchange, gel filtration and hydroxyapatite chromatography. And (5) packaging the semi-finished product in time, and performing rotary freezing and freeze-drying. The freeze-drying process of the product can be formulated according to the performance characteristics of a machine, but the preparation quality and the preservation quality of the product are ensured to meet the requirements. The experiment compares the binding of the LAGUBIXIN and HSA at different ratios and different times, and the result shows that EMC-AANL-DOX, QHL-087-DOX and QHL-087-N-CBP are mixed with HSA in a ratio of 3: 1, and the binding is carried out in a water bath at 37 ℃ for 3h, and the binding rates of HSA are respectively 62%, 99.6% and 99.7%.
Example 11: selection of chemically modified linker arms for optimized activation efficiency
S-C-A is a chemically modified linker and shows high activation efficiency compared to the native peptide sequence linker cleaved by Legumain. When AAN is selected for C, the activation of the different S-C-A linkers and the control linker is evaluated in an activation assay. S-C-A conjugates were used to solubilize and dilute them ten-fold to a concentration of 0.1 mM/ml. Sample compound was added at a concentration of 1mg/ml to 100 μ g of acidified human breast cancer (MDA-MB435) tumor homogenate (pH6.0) at 37 ℃. Enzymes in the tumor homogenate were released and detected by HPLC, comparing the efficiency of tumor tissue activation of the linker. The results are shown in tables 2-1, 2-2, 2-3 and 2-4.
TABLE 2-1


Tables 2 to 2

Tables 2 to 3


Detection of the Effect of the D-type tripeptide on activation efficiency compared to the native peptide sequence linker cleaved by Legumain, reptazing, S1 being-CH in MI-S2CH2-CONH-, S2: 2 peg. The results are shown in tables 2-4 below.
Tables 2 to 4
| C | A | Activation efficiency (%) |
| Ala-Ala-Asn | Leu | 96.8 |
| Thr-Ala-Asn | Leu | 90.2 |
| Val-Ala-Asn | Leu | 78.9 |
| D-Thr-L-Val-L-Asn | Leu | 73.5 |
| D-Thr-L-Ala-L-Asn | Leu | 89.6 |
| D-Ala-L-Val-L-Asn | Leu | 93.5 |
| L-Thr-D-Val-L-Asn | Leu | 90.6 |
| L-Thr-D-Ala-L-Asn | Leu | 72.4 |
| L-Ala-D-Val-L-Asn | Leu | 83.4 |
| D-Thr-D-Val-L-Asn | Leu | 66.1 |
| D-Thr-D-Ala-L-Asn | Leu | 78.4 |
| D-Ala-D-Val-L-Asn | Leu | 61.5 |
| L-Ala-L-Val-D-Asn | Leu | 22.4 |
As can be seen from the tabulated data, under the condition that S and A are designed to be highly activated, when the variable is different tripeptides, different amino acid selections and configurations have influence on activation efficiency, especially D-Asn causes the loss of activation capability, and the other two positions of amino acid are adjusted to be D-type and still have activation activity.
Example 12: comparison of cleavage kinetics rates for preferred Compounds
Precisely weighing 10mg of each of C3, QHL-087-DOX, QHL-090-DOX, QHL-093-DOX, QHL-094-DOX, QHL-093-DOX and QHL-096-DOX, adding a proper amount of water to prepare a sample stock solution of 4umol/mL, and adding water to gradually dilute the sample stock solution into sample solutions of various concentrations in the following table 3; respectively measuring 20ul of sample solution with different concentrations, adding 80ul of Legumain, and carrying out water bath at 37 ℃; taking out after water bath for 2h, injecting 10ul of sample, and detecting by HPLC; reading the area of each corresponding product, calculating the product concentration according to the linear equation of the product, and substituting the product concentration into a formula to obtain the corresponding V:
V(umoL/mL/min)=C(umoL/mL)/120min
by plotting V against [ C ], the intersection of the intercept Km/Vmax and the line with the x-axis can be obtained as-Km and [ C ] as the concentration of each substrate, i.e., the concentration of the sample solution, in umoL/mL.
TABLE 3
| [C1] | [C2] | [C3] | [C4] | [C5] | [C6] | [C7] |
| 2 | 1.6 | 0.8 | 0.7 | 0.35 | 0.175 | 0.0875 |
The results of the experiment are shown in FIG. 3Under the condition of consistent other structures: QHL-087, the 2PEG group significantly increased the activation efficiency, but instead the efficiency decreased as the amount of PEG increased. Compare under the same conditions for 6PEG group attachment: h2PABC-NH2H2Replacement leu significantly improves activation efficiency.
Example 13:
separation and culture of mouse spleen and CD8+ T cells, separation of mouse bone marrow mononuclear cells and induced differentiation of M2 macrophages
1. Mouse spleen cell isolation
1) Spleen from C57BL/6 mouse was placed on a petri dish (ice bath) with a 40uM mesh screen, and approximately 10mL of physiological saline was added and gently triturated with a sterile syringe core.
2) Transferring the ground cell suspension into a 50mL centrifugal tube, adding about 5mL of physiological saline to wash the culture dish, transferring into the 50mL centrifugal tube, and combining the suspensions.
3) Centrifuging the cell suspension at 1000r/min for 10min, discarding the supernatant, resuspending with a proper volume of normal saline, adding 3 times volume of ammonium chloride erythrocyte lysate, and blowing to obtain the final product. After cracking on ice for about 10min, adding 10mL of physiological saline to stop cracking, and centrifuging at 1000r/min for 5 min.
4) After centrifugation, the supernatant is discarded, 10mL of physiological saline is added, the mixture is evenly blown and beaten, the mixture is centrifuged at 1000r/min for 5min, the operation is repeated once, and then the cells are resuspended in 10% RMPI1640 culture medium for subsequent culture or 0.5% BSA for subsequent T cell sorting.
CD8+ T cell sorting
Resuspending the mouse splenocytes obtained by the separation to 1E8/mL, adding 100ul of Miltenyi biotec CD8a (Ly-2) microBeads into each 1E8 cell, uniformly mixing, incubating for 15 minutes at 4 ℃ in the dark, adding PBS (5-10 times volume), fully mixing and washing, centrifuging for 5 minutes at 300g, removing supernatant, repeatedly washing once, resuspending the cells to 2E8/mL to be separated from the column, placing the cell suspension on an LS column on a magnet plate (the LS column is balanced by a washing buffer (pH7.2 PBS + 0.5% BSA +2mM EDTA) in advance), after the cell suspension slowly flows through the LS column and CD8+ T cells are combined with magnetic particles in the LS column, washing the LS column by using the washing buffer of 3 times of the volume cells, taking out the LS column from the magnet plate after the washing is finished, placing the LS column in a 15mL centrifuge tube, adding 5mL of the washing buffer into the LS column, and then rapidly extruding and eluting the combined cells in the LS column core to elute in the centrifuge tube, all cells passed through the column were collected, the resulting cells were centrifuged to remove the supernatant, washed repeatedly once with washing buffer, the cells were resuspended in the appropriate volume of 10% RMPI1640 medium, and the cells were counted for use.
Activation and expansion of CD8+ T positive cells
A. Washing of CD3/CD28 magnetic beads: a. the immunomagnetic beads in the small tube are shaken up and suspended (vortex for more than 30s, or inclined rotation for 5 min). b. The required amount of immunomagnetic beads was taken out into a 1.5ml tube, and 1ml of 1640 containing serum was added and suspended, vortexed for more than 30s, or kept rolling for at least 5 min.
B. Activation of T cells: a. an appropriate number of cells is plated on a plate (e.g., 6 well plate 1E6/ml, 2ml of medium, maintaining a T cell density above 2E6/ml, but not exceeding 2E 6/ml). b. Adding washed magnetic beads to ensure that the magnetic beads: cells (number ratio) ═ 1: 1. c. placing into an incubator for 3 days.
C. Amplification: a. the culture solution does not need to be replaced within 3 days, 30U/ml IL-2 is added (can be increased properly according to actual conditions) on the 3 rd day, and the culture medium is replaced (the number of cells can be increased by about 1 time after 48 hours, and the size and the shape of the cells are monitored in real time). b. After 4-5 days of stimulation, the beads were removed (to avoid over-activation) and the cells were moved up and down 5-10 times to avoid air bubbles and turbulence to separate the cells from the beads. The cells were collected in a 1.5ml tube, placed on a magnet for 1min to attach the beads to the tube wall, moved to another tube to remove the beads as completely as possible, and the cells with the beads removed were cultured in a medium containing 30U/ml IL-2 (which may be increased as appropriate) and monitored for status, proliferation and viability.
4. Mouse bone marrow mononuclear cell separation and induced differentiation of M2 macrophages
Taking out bilateral femurs and tibiae of 2C 57BL/6 mice under aseptic condition, cutting metaphysis in a super clean bench, extracting serum-free MEM culture solution by using a5mL aseptic injector, repeatedly and softly flushing a marrow cavity for 4 times, and collecting all cell suspensions; centrifuging at 1000r/min for 10min, removing supernatant to obtain cell precipitate, resuspending in serum-free MEM culture solution with appropriate volume, repeatedly blowing off and homogenizing, filtering with 40uM filter screen, adding 3 times volume of erythrocyte lysate, and lysing on ice for 10 min; centrifuging at 1000r/min for 5min, removing supernatant to obtain cell precipitate, cleaning with serum-free MEM culture medium for 2 times, and collecting cell precipitate; resuspending the cells in the appropriate volume of MEM containing 10% FBS, 1% PS by volume in complete medium and counting the cells for subsequent differentiation; 100uL of the cells are inoculated into a 96-well cell culture plate at 20000 cells/well, 100ng of M-CSF is added into a culture solution for differentiation of M2 macrophages, the mixture is statically cultured in an incubator with the volume fraction of 5% CO2 at 37 ℃ for 7 days to induce differentiation, and the cell morphology is observed. Induced cell morphology referring to fig. 4, M2 macrophages were differentiated from monocytes, DC and GM-macrophages and identified as M2 macrophages.
Example 14: MTT method for determining inhibition effect of drug on cell growth
The cells of example 13 were counted and then plated at 100. mu.l cell suspension per well in 96-well plates using medium to adjust cell concentration, CD8+ T cells were plated at 100000 cells/well and M2 macrophages at 20000 cells/well. The 96-well plate was incubated overnight for 24 hours at 37 ℃ in a carbon dioxide (5%) incubator. After 24 hours, the 96-well plate was filled with 100ul of cell culture medium containing different concentrations of the drug, and control wells (0.1% DMSO) containing no drug and corresponding drug solvent were set, and zero-adjusted wells (Blank) containing no cells were set in culture medium. Each group was set up with 3 parallel wells and the plates were then incubated for 48 hours at 37 ℃ in a carbon dioxide (5%) incubator. After 48 hours, 20. mu.l MTT (5 mg/ml) was added to each well and incubation was continued for 4 h. Then, the culture medium was gently aspirated, 150. mu.l of DMSO as a solvent was added to each well to dissolve the culture medium, and the absorbance at 490nm was measured using a microplate reader after the dissolution.
And calculating the survival rate of the cells and the half inhibition concentration of the drug on the cells. Cell viability = (OD assay-OD blank)/(OD assay control-OD blank) × 100%. The survival (%) was calculated using Excel software, a dose-response curve of drug versus cells was plotted using Prism 5 software, each index was expressed as a mean value, and the Coefficient of Variation (CV) was used to evaluate the consistency of the data.
According to the experimental schematic diagram and the cell dosing concentration setting in the experimental method, the maximum initial concentration of the drug to be tested is set to be 14uM, and the ratio of 1: the 3-ratio gradient dilution was performed to 9 dose groups (3 replicates in each group), the concentration of the drug solvent (DMSO) was controlled to 0.1% in all the wells, the drug solvent (0.1% DMSO) alone was added as the test Control group (Control), and the Blank group (Blank) to which the cells were added only with the culture medium was not added, and then the tumor cell survival (%) of each dose group relative to the Control group (Control) was calculated according to the following method.
Cell survival (%) for each dose group (OD dose-OD blank)/(OD 0.1% DMSO-OD blank) 100%
The results of the experiment are shown in FIGS. 5 and 6. Under otherwise structurally identical conditions, QHL-087-DOX has significantly increased cytotoxicity against M2 macrophages relative to C3 (i.e., EMC-AANL-DOX) and is less cytotoxic to CD8+ T, thus resulting in selectivity for immunosuppressive cells.
Example 15: cytotoxicity screening of multiple drugs on M2 macrophages
A partial compound cytotoxicity screening assay was performed on M2 macrophage inhibition as in example 14. Each drug is detected in 3 holes, 10uM of the following drugs are added into each hole, the inhibition rate of the relative drug-free medicines is detected, and the experimental result is shown in table 4.
Example 16: the water solubility of the water-soluble high-efficiency targeting activated adriamycin derivative and the like prepared by the embodiment of the invention is compared with that of a control compound
The compounds prepared according to the present invention examples, the compounds prepared above and the reference compounds C1, C2, C3 and C4 were lyophilized (-70 ℃). Compounds were dissolved in different concentrations of water and water solubility was checked by observation and HPLC testing (> 95%). The results are shown in Table 4.
Table 4: screening of drug Water solubility test data and M2 macrophage inhibition Rate








The test results show that under otherwise structurally identical conditions, the 2PEG group significantly increased water solubility, changing from water insoluble to water soluble, and increased solubility with increasing amounts of PEG. Under the same conditions of PEG ligation, the addition of Glu and Asp increases water solubility. Through the change of the groups, the water solubility of the coupling drug is changed, so that the permeability of the blood outlet membrane of the drug and the membrane of the tumor cell can be greatly influenced, and the curative effect is influenced. The compound has enhanced water solubility, and provides necessary conditions for the production of drug property of the drug and the production of coupled drugs.
Example 17: study on drug effect of C3, QHL-085-DOX, QHL-087-DOX, QHL-091-DOX and QHL-094-DOX injection in nude mouse HT1080 model
The purpose of the test is as follows: c3, QHL-085-DOX, QHL-087-DOX, QHL-091-DOX and QHL-94-DOX were investigated for anti-tumor efficacy in a mouse model during tumor treatment.
Test drugs: c3, QHL-085-DOX, QHL-087-DOX, QHL-091-DOX and QHL-094-DOX were used as injections, and were diluted with physiological saline to the corresponding concentrations at the time of the test.
The method and the result are as follows:
1. animals: 6-8 weeks old nude mice are all female.
2. Preparation of tumor model
1) HT1080 cells were purchased from ATCC and identified according to the instructions provided by ATCC. Cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ with 5% CO2Culturing in medium. Passages were performed every three days and cells within 15 passages were used.
2) Tumor generation: 5X 106HT1080 cells were injected subcutaneously into the back of nude mice. When the tumor size reaches 100mm3And then, random grouping is carried out. Treatment was then started and the day on which treatment was started was recorded as day 1.
3) Course of treatment
Drug (IV) was injected intravenously according to the clinical application of C3, QHL-085-DOX, QHL-087-DOX, QHL-091-DOX and QHL-094-DOX. C3, QHL-085-DOX, QHL-087-DOX, QHL-091-DOX and QHL-094-DOX were administered at low and the same dose of 18umol/kg, respectively. The control group was given physiological saline. Once weekly dosing for 3 weeks.
4) Results and discussion: grouping and test results as shown in fig. 7, the tumor suppressive effects were sequentially enhanced in the 4peg and 2peg groups compared to the equimolar dose low dose treatment group.
Example 18: pharmacodynamic study of C1, C2, C3, QHL-086-DOX, QHL-092-DOX, QHL-095-DOX, QHL-087-DOX, QHL-010-DOX and QHL-117-DOX injection in nude mouse HT1080 model
The purpose of the test is as follows: the antitumor efficacy of the above compounds in a mouse model during tumor therapy was investigated.
Test drugs: c1, C2, C3, and the corresponding compound injections were diluted with physiological saline to the corresponding concentrations at the time of the test.
The method and the result are as follows:
1. animals: 6-8 weeks old nude mice are all female.
2. Preparation of tumor model
1) HT1080 cells were purchased from ATCC and identified according to the instructions provided by ATCC. Cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ with 5% CO2Culturing in medium. Passages were performed every three days and cells within 15 passages were used.
2) Tumor generation: 5X 106HT1080 cells were injected subcutaneously into the back of nude mice. When the tumor size reaches 100mm3And then, random grouping is carried out. Treatment was then started and the day on which treatment was started was recorded as day 1.
3) Course of treatment
The drug (IV) was injected intravenously according to the clinical use of the corresponding compound. The compounds shown in the tables were administered at low doses and at the same dose of 36 umol/kg. The control group was given physiological saline. Once weekly dosing for 3 weeks.
5) The grouping and test results are shown in table 5.
Table 5: c1, C2, C3 and treatment of nude mouse tumor by partial compound and mitomycin

5) Results and discussion: as shown in Table 5, compared with the treatment group of equimolar AANL-DOX, QHL-086-DOX, QHL-092-DOX, QHL-095-DOX, QHL-087-DOX, QHL-010-DOX and QHL-117-DOX high dose treatment greatly improves the growth inhibition and disappearance of tumors and achieves the effect of healing.
Example 19: QHL-087-DOX and EMC-AANL-DOX in liver transplantation in situ CT26 tumor tissue distribution studies.
The purpose of the test is as follows: study of the tissue distribution of active drugs in liver tumors.
Test animals: BALB/c mice, 6-8 weeks old, were all female.
Preparing a tumor model:
1) CT26 cells were purchased from ATCC and the cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ with 5% CO2Culturing in medium. Passages were performed every three days and cells within 15 passages were used.
2) Tumor generation: 5X 106The CT26 cells of (a) were injected subcutaneously into the back of nude mice. When the size of the tumor reaches 800-3And then, random grouping is carried out. Then extracting tumor tissue and cutting into 100mm3And transplanted in situ into the liver of BALB/c mice.
3) The administration process comprises the following steps: after 14 days, when the orthotopic transplanted tumor grew, a group of 36 orthotopic transplanted tumor mice was treated with the drug. Different tissues were then harvested at 1, 6, 12, 24, 36, 72hr to examine the concentration of doxorubicin released in the different tissues. AUClast h nmol/g was calculated, and the mean and SEM are shown in FIGS. 8 and 9.
4) Results and discussion: as shown in FIGS. 8 and 9, active doxorubicin of QHL-087-DOX and EMC-AANL-DOX was distributed predominantly in liver and liver tumors in situ. The previously published use of EMC-AANL-DOX was for breast cancer treatment, and it was found through further studies that QHL-087-DOX has the property of delivering more drug to the liver, while tumor activation in situ in the liver resulted in higher doxorubicin exposure. In situ liver cancer delivery and activation doxorubicin exposure enhancement of QHL-087-DOX versus EMC-AANL-DOX.
Example 20: pharmacodynamic study of QHL-087-DOX in orthotopic liver transplantation CT26 tumor.
The purpose of the test is as follows: QHL-087-DOX, PD-1 and combinations thereof were investigated for their efficacy in orthotopic transplantation of CT26 tumors.
Test drugs: QHL-087-DOX 18. mu. mol/kg, mouse PD-15 mg/kg.
Animals: BALB/c mice 6-8 weeks old were all female.
1) Generation of tumor model: CT26 tumor cells were from ATCC. Cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ and 5% CO2And (5) culturing. Cells were passaged every three days and cells within passage 15 were used. Will be 5X 105CT26 cancer cell subcutaneously injected into nude miceA back part. The tumor reaches 800-3Thereafter, mice were randomly grouped. Then extracting tumor tissue and cutting into 100mm3And transplanted in situ into the liver of BALB/c mice. After 1 week, when the orthotopic transplanted tumor grew, mice with orthotopic transplanted tumors were randomly grouped.
2) The treatment process comprises the following steps: 6 mice in one group were treated with drug. The day of treatment was day 1. According to the clinical application of QHL-087-DOX, the (IV) drug was injected intravenously once a week for 3 weeks. Mouse PD-1 antibody was injected Intravenously (IV) twice weekly for 3 weeks. The grouping and test results are shown in FIG. 10.
3) Results and discussion: QHL-087-DOX is combined with PD-1 for the first time to treat the hepatoma, so that the immunopotentiation effect is achieved on the hepatoma, and QHL-087-DOX is combined with PD-1, so that the single drug of QHL-087-DOX has better curative effect and the immunotherapy property.
Example 21: effect of EMC-AANL-DOX (Legubicin), Rivatinib and PD-1 in combination with each other on treatment of in situ liver cancer
The purpose of the test is as follows: EMC-AANL-DOX, Lunvatinib and PD-1 in combination with each other for the treatment of in situ liver cancer.
Test drugs: EMC-AANL-DOX 18 micromoles/kg, mouse PD-15 mg/kg.
Animals: BALB/c mice 6-8 weeks old were all female.
Generation of tumor model: CT26 tumor cells were from ATCC. Cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ and 5% CO2And (5) culturing. Cells were passaged every three days and cells within passage 15 were used. Will be 5X 105CT26 cancer cells were injected subcutaneously into the back of nude mice. The tumor reaches 800-3Thereafter, mice were randomly grouped. Then extracting tumor tissue and cutting into 100mm3And transplanted in situ into the liver of BALB/c mice. After 1 week, when the orthotopic transplanted tumor grew, mice with orthotopic transplanted tumors were randomly grouped.
The treatment process comprises the following steps: 6 mice in one group were treated with drug. The day of treatment was day 1. According to the clinical application of EMC-AANL-DOX, the (IV) drug was injected intravenously once a week for 3 weeks. Mouse PD-1 antibody was injected Intravenously (IV) twice weekly for 3 weeks. The grouping and test results are shown in FIG. 11.
Results and discussion: the combination of EMC-AANL-DOX and PD-1 and the treatment of liver orthotopic tumor is found to have better curative effect than the combination treatment of the Rankine and the combination PD-1 for the first time, and the combination treatment of EMC-AANL-DOX and the Rankine is found to have better curative effect than the single-drug treatment of the EMC-AANL-DOX and the Rankine.
Example 22: the research on the curative effect of the partial compound injection on the liver cancer HepG2 cells of nude mice
The purpose of the test is as follows: the antitumor efficacy of some of the compounds of the invention in a mouse tumor therapy model was investigated.
Test drugs: the table corresponds to compound injection and control injection, and the test is diluted with normal saline to corresponding concentration.
The method and the result are as follows:
1. test animals: 6-8 weeks old nude mice are all female.
2. Preparation of tumor model
1) Human liver cancer HepG2 cells were purchased from ATCC and identified according to the instructions provided by ATCC. Cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ with 5% CO2Culturing in medium. Passages were performed every three days and cells within 15 passages were used.
2) Tumor generation: 5X 106The HepG2 cells were injected subcutaneously into the back of nude mice. When the tumor size reached 100mm3, random groupings were made. Treatment was then started and the day on which treatment was started was recorded as day 1.
3) Course of treatment
The drug (IV) was injected intravenously according to the clinical use of the corresponding compound. The compound and control drug were administered at a dose of 54umol/kg, and DOX was only used at a dose of 18umol/kg due to toxicity limitations. The control group was given physiological saline. Once weekly dosing for 4 weeks.
6) The grouping and test results are shown in table 6.
Table 6: effect of partial Compounds of the invention and control groups on nude mouse tumor treatment


5) Results and discussion: as shown in Table 6, EMC-AANL-DOX has a better effect on liver cancer, and the compounds of the present invention have an increased therapeutic effect on tumor growth compared to the same molar dose of EMC-AANL-DOX.
Example 23: QHL-096-DOX, QHL-087-DOX, QHL-090-DOX, QHL-093-DOX, QHL-117-DOX in CT26 tumor immune model treatment
The purpose of the test is as follows: the antitumor effect of the compound in the immunotherapy of a CT26 tumor model is researched.
Test drugs: QHL-096-DOX, QHL-087-DOX, QHL-090-DOX, QHL-093-DOX, QHL-117-DOX and a control group, at a dose of 36umol/k, murine PD-1 antibody, 5 mg/kg.
Test animals: BALB/c mice of 6-8 weeks old are all female mice.
Preparing a tumor model:
1) CT26 cells were purchased from ATCC and the cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ with 5% CO2Culturing in medium. Passages were performed every three days and cells within 15 passages were used.
2) The generation of tumors. Will be 5X 106Individual corresponding cells were injected subcutaneously into the back of nude mice. The tumor reaches at least 100mm3Thereafter, mice were randomly grouped. Treatment was then started, the day of treatment initiation being the first day.
3) And (5) a treatment process. The drug was administered at a dose of equimolar 36 umol/kg. The control group was given physiological saline. Once weekly for three weeks.
4) Tumor CD8+T cell analysis. Tumor tissue was homogenized, individual cells in the tumor were filtered, separated and washed twice with buffer, and then labeled with leukocyte common antigens CD45-PE and CD8-FITC at room temperatureThe antibody was incubated for 1 hour. The cells were washed twice with phosphate buffer containing 1% fetal bovine serum, and then the proportion of T lymphocyte antigen (CD8) positive cells among leukocyte common antigen (CD45) positive cells was analyzed by flow cytometry. An increase in the ratio indicates an increase in T lymphocytes and thus an improvement in the immunity of the animal to the tumor.
4) The grouping and test results are shown in table 7.
Table 7: effect of corresponding Compounds on tumor suppression and immune activation with control group


5) Results and discussion: the treatment effect of the combination of C3, QHL-096-DOX, QHL-087-DOX, QHL-090-DOX, QHL-093-DOX and QHL-117-DOX and PD-1 is improved relatively to that of a single medicine, and tumors can be cured. QHL-090-dabrafenib, QHL-090-dabrafenib also has certain synergistic effect when combined with PD-1.
Example 24: QHL-087-DOX injection for treating various tumor models
The purpose of the test is as follows: the anti-tumor spectrum of QHL-087 was studied by various tumor models in mice.
Test drugs: QHL-087-DOX was injected and diluted with physiological saline to the corresponding concentration at the time of the experiment.
The method and the result are as follows:
1. animals: 6-8 weeks old nude mice were all female.
2. Generation of tumor models
1) Corresponding tumor cells were purchased from the American Type Culture Collection (ATCC) and identified according to the specifications provided by the ATCC. Cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ and 5% CO2And (5) culturing. Cells were passaged every three days and cells within passage 15 were used.
2) The generation of tumors. Will 5 generate106Individual corresponding cells were injected subcutaneously into the back of nude mice. The tumor reaches at least 100mm3Thereafter, mice were randomly grouped. Treatment was then started, the day of treatment initiation being the first day.
3) And (5) a treatment process. According to the clinical application of QHL-087-DOX, QHL-087-DOX was administered at a dose of 36 umol/kg. The control group was given physiological saline. Once weekly for three weeks.
4) The grouping and test results are shown in table 8.
Table 8: QHL-087-DOX therapeutic Effect in various tumor models
| Group of | Tumor cells | Tumor inhibition rate (day 26) | |
| Human breast cancer | MDA-MB435 | 91.5% | |
| Human ovarian cancer | SK-OV-3 | 78.7% | |
| Human colon cancer | HT-29 | 85.3% | |
| Chronic leukemia of human | K562 | 79.4% | |
| Human body knotIntestinal cancer | HT1080 | 90.5% | |
| Human pancreatic cancer | Panc-1 | 75.7% | |
| Human non-small cell lung cancer | A549 | 75.8% | |
| Human | Hepg2 | 100% | |
| Human kidney cancer | OS-RC-2 | 87.4% |
5) Results and discussion: QHL-087-DOX showed excellent therapeutic effects in various tumor models, indicating that the drug has a broad anti-tumor spectrum.
Example 25: comparison of solubility of HSA-EMC-AANL-DOX, HSA-QHL-087-DOX, and HSA-QHL-087-N-CBP to control Compounds
The freeze-dried products of EMC-AANL-DOX, HSA-EMC-AANL-DOX, HSA-QHL-087-DOX and HSA-QHL-087-N-CBP prepared in the embodiment of the invention are subpackaged in a sterile room and are re-dissolved by water for injection. HSA-EMC-AANL-DOX, HSA-QHL-087-DOX and HSA-QHL-087-N-CBP were all able to dissolve completely as shown in Table 9.
TABLE 9
| Compound (I) | Water for injection |
| EMC-AANL-DOX | Insoluble matter |
| HSA-EMC-AANL-DOX | Dissolution, 5mg/ml |
| HSA-QHL-087-DOX | Dissolution, 35mg/ml |
| HSA-QHL-087-N-CBP | Dissolution, 35mg/ml |
As can be seen from Table 9, when human serum albumin was coupled to the compound to further improve the solubility, HSA-EMC-AANL-DOX, HSA-QHL-087-DOX and HSA-QHL-087-N-CBP as macromolecular proteinoid drugs could be dissolved directly to high concentration with water for injection or physiological saline without using an irritant organic solvent required for dissolution of EMC-AANL-DOX. Different from the EMC-AANL-DOX small molecular compound drug which is insoluble in water, the change of the solubility property has great influence on the distribution metabolism and the drug action mode of the drug.
Example 26: solution stability comparison of water-soluble high-efficiency target-activated adriamycin derivative prepared in embodiment of the invention and a control compound
Compounds EMC-AANL-DOX, HSA-EMC-AANL-DOX, QHL-087-DOX, HSA-QHL-087-DOX, QHL-087-N-CBP and HSA-QHL-087-N-CBP were accurately weighed, and 5.0mg of each sample was dispensed in a sterile room, and 0.5ml of sterile water for injection was added to prepare a 10mg/ml mother solution, and 50% ethanol was required for dissolution of EMC-AANL-DOX. 30ul of the mother liquor was taken and 570ul of buffer solutions with different pH values of 5.5 were added to prepare 0.5mg/ml sample solutions. After the sample is clarified, the sample is placed in a water bath at 25 ℃/37 ℃, 8H sampling is carried out, HPLC and electrophoresis are used for detecting the content of the sample for 0 hour, and then the solution stability data of different compounds can be obtained. The results are shown in Table 10.
Table 10: solution stability data
| Compound (I) | 8hr |
| EMC-AANL-DOX | 94.3% |
| HSA-EMC-AANL-DOX | 97.8% |
| QHL-087-DOX | 98.2% |
| HSA-QHL-087-DOX | 100.2% |
| QHL-087-N-CBP | 85.3% |
| HSA-QHL-087-N-CBP | 99.8% |
As can be seen from the data in the above table, the stability of the albumin-coupled compound increased at 25 ℃ and pH 5.5, which is more significant for QHL-087-N-CBP and HSA-QHL-087-N-CBP.
Example 27: HSA-EMC-AANL-DOX, HSA-QHL-087-DOX and HSA-QHL-087-N-CBP activation efficiency assays
EMC-AANL-DOX was dissolved using a solvent (50% water for injection + 50% ethanol), HSA-EMC-AANL-DOX, HSA-QHL-087-DOX and HSA-QHL-087-N-CBP were dissolved collectively using water for injection, and diluted 10-fold to 1mg/ml with water. In the present experiment, 1mg/ml of the sample compound was added to 100. mu.g of acidified tumor homogenate (pH6.0) at 37 ℃ and the enzyme in the tumor homogenate was able to cause release of doxorubicin, and decrease in the compound and increase in doxorubicin were able to be detected by HPLC to compare the activation efficiency of the drug in the tumor tissue, and the compounds EMC-AANL-DOX, HSA-EMC-AANL-DOX, HSA-QHL-087-DOX, and HSA-QHL-087-N-CBP of the present invention were found to have the highest activation efficiency among the compounds screened by screening. The results are shown in Table 11.
Table 11: compound activation ratios (%), of EMC-AANL-DOX, HSA-EMC-AANL-DOX, HSA-QHL-087-DOX, and HSA-QHL-087-N-CBP in different tumor homogenates

Example 28: toxicity assays for EMC-AANL-DOX, HSA-EMC-AANL-DOX, QHL-087-DOX, HSA-QHL-087-DOX, QHL-087-N-CBP, and HSA-QHL-087-N-CBP
The purpose of the test is as follows: the acute toxicity of the drug organism of the invention is known by measuring the MTD experiment of intravenous medication of mice.
Test drugs: EMC-AANL-DOX was dissolved with a solvent (50% water for injection, 50% alcohol), HSA-EMC-AANL-DOX, QHL-087-DOX, HSA-QHL-087-DOX, QHL-087-N-CBP and HSA-QHL-087-N-CBP were dissolved with water for injection, and diluted with physiological saline at the time of the test to the corresponding doses.
Animals: first-class Barex race (BALB/C) mice (purchased from Shanghai Spiker laboratory animals, Inc.) weighed 19-21g and were all female.
The method and the result are as follows: the test BALB/C mice were 36, 19-21g in weight, all female, and randomly divided into 7 groups of 6 mice each based on body weight. EMC-AANL-DOX, HSA-EMC-AANL-DOX, QHL-087-DOX, HSA-QHL-087-DOX, QHL-087-N-CBP and HSA-QHL-087-N-CBP were each injected intravenously in one dose as shown in Table 12. A control experiment was performed with a saline group and paclitaxel injection (commercially available, Beijing Yuekang), and a volume of 0.2ml was administered to each mouse. Continuously observing for 17 days, observing whether animals have standing hair, disorder and no luster, lethargy, stoop and humpback, overexcitation reaction and the like every day, and recording the weight and death condition. On days 3, 5, and 14, blood samples were collected for complete blood count, and on day 14, animals were dissected and examined for HE staining of heart, liver, kidney, lung, spleen, and pancreas.
Table 12: the test mice respectively receive different dosages of compound injection and the mortality result of physiological saline and paclitaxel injection
| Group of | Dosage (micromotion/kilogram) | Animals (only) | Death number (only) | |
| 1 | |
0 | 10 | 0 |
| 2 | EMC-AANL-DOX | 38.4 | 10 | 8 |
| 3 | HSA-EMC-AANL-DOX | 38.4 | 10 | 0 |
| 4 | QHL-087-DOX | 38.4 | 10 | 4 |
| 5 | HSA-QHL-087-DOX | 38.4 | 10 | 0 |
| 6 | QHL-087-N-CBP | 19.2 | 10 | 7 |
| 7 | HSA-QHL-087-N-CBP | 19.2 | 10 | 0 |
Results and discussion: when HSA-EMC-AANL-DOX, HSA-QHL-087-DOX and HSA-QHL-087-N-CBP are injected into animals, the animals do not have the conditions of pilaster, messy and lusterless, lethargy, stoop and humpback, over-stimulation reaction and death, and the toxicity of the albumin coupling medicament is remarkably reduced compared with that of unconjugated medicaments.
Example 29: combination therapeutic efficacy of HSA-EMC-AANL-DOX, HSA-QHL-087-DOX with anti-PD-1 antibodies
The purpose of the test is as follows: the effect of EMC-AANL-DOX, HSA-QHL-087-DOX in combination with anti-PD-1 antibodies was investigated comparatively.
Test drugs: HSA-EMC-AANL-DOX and HSA-QHL-087-DOX at a dose of 18. mu. mol/kg, respectively; murine PD-1 antibody, 5 mg/kg.
Test animals: BALB/c mice of 6-8 weeks old are all female mice.
Preparing a tumor model: CT26 cells were purchased from ATCC and the cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ with 5% CO2Culturing in medium. Passages were performed every three days and cells within 15 passages were used. Will be 5X 106CT26 cancer cells were injected subcutaneously into mice. Mice were injected 3 times weekly with anti-PD-1 antibody twice a week for 8 times.
Results and discussion: the test results are shown in figure 12, and the therapeutic effect of the combination of HSA-QHL-087-DOX and the anti-PD-1 antibody is improved compared with that of HSA-EMC-AANL-DOX, and the cure rate is higher.
Example 30: MTT method for determining inhibition effect of N-CBP and HSA-QHL-095-N-CBP on growth of tumor cells
After the cell concentration was adjusted by using a medium after the cell isolation and the cell culture were counted, the cells were seeded in a 96-well plate at 100. mu.l cell suspension per well, 100000 cells/well for CD8+ T cells, and 20000 cells/well for CT26 tumor cells. The 96-well plate was incubated overnight for 24 hours at 37 ℃ in a carbon dioxide (5%) incubator. After 24 hours, the 96-well plate was filled with 100ul of cell culture medium containing drugs at different concentrations, and control wells (0.1% DMSO) containing no drug and corresponding drug solvent were set, while zero-adjusted wells (Blank) containing no cells were set in medium. Each group was set up with 3 parallel wells and the plates were then incubated for 48 hours at 37 ℃ in a carbon dioxide (5%) incubator. After 48 hours, 20. mu.l MTT (5 mg/ml) was added to each well and incubation was continued for 4 h. Then, the culture medium was gently aspirated, 150. mu.l of DMSO as a solvent was added to each well to dissolve the culture medium, and the absorbance at 490nm was measured using a microplate reader after the dissolution.
Results and discussion: the test results are shown in FIGS. 13 and 14. N-CBP was more cytotoxic than carboplatin and oxaliplatin in vitro and less cytotoxic than cisplatin in vitro (figure 13); HSA-QHL-095-N-CBP had reduced cytotoxicity in vitro relative to N-CBP and carboplatin (FIG. 14).
Example 31: single agent of HSA-QHL-095-N-CBP and combined therapeutic effect with anti-PD-1 antibody
The purpose of the test is as follows: the effect of carboplatin, HSA-QHL-095-N-CBP, and combination therapy with anti-PD-1 antibodies was compared.
Test drugs: carboplatin, HSA-QHL-095-N-CBP, dose 18. mu. mol/kg, murine PD-1 antibody, 5 mg/kg.
Test animals: BALB/c mice of 6-8 weeks old are all female mice.
Preparing a tumor model: CT26 cells were purchased from ATCC and the cells were cultured in DMEM medium containing 10% fetal bovine serum at 37 ℃ with 5% CO2Culturing in medium. Passages were performed every three days and cells within 15 passages were used. Will be 5X 106CT26 cancer cells were injected subcutaneously into mice.
The treatment process comprises the following steps: injecting the corresponding medicine once a week for 3 weeks; anti-PD-1 antibody was administered once a week for 4 weeks.
Results and discussion: the test result is shown in figure 15, the equimolar dosage of HSA-QHL-095-N-CBP is obviously superior to that of carboplatin, and the treatment effect of HSA-QHL-095-N-CBP combined with anti-PD-1 antibody is improved relatively to that of single medicine, so that the cure effect is obtained.
Example 32: non Alcoholic Fatty Liver Disease (NAFLD) model mouse inflammatory response.
The experimental method comprises the following steps: the C57 mice were randomly divided into a normal group (standard diet), a model group (high-fat diet), a simvastatin group (positive control, 3mg/kg) and a drug group dose group (50mg/kg), with 6 mice per group. Normal groups of mice were fed with standard diet, and the remaining groups of mice were fed with high fat diet to induce NAFLD model. At the same time of molding, the mice in each group IV were dosed with a corresponding dose of 48umol/kg of drug 2 times per week for a total of 8 weeks. 12h after the last administration, the full-automatic biochemical analyzer measures the serum biochemical indexes: high density lipoprotein cholesterol (HDL-C), low density lipoprotein (LDL-C). The results are shown in Table 13.


As a result: compared with the normal group, the HDL-C content in the serum of the model group is remarkably reduced to 66.2 percent, and the LDL-C content is remarkably increased to 135.6 percent (P is less than 0.05); QHL-158-T3 and QHL-159-T3 are better for restoring normal level of HDL-C and LDL-C in the treatment group.
Claims (20)
1. A compound having a structure represented by the following formula (I):
MI-S-C-A (I)
in the formula (I), the compound is shown in the specification,
MI is a maleimide group;
s is a group for improving the enzyme digestion efficiency or the selectivity;
c is an amino acid connecting arm which can be broken by proteolytic enzyme; and
a is an auxiliary connecting arm.
2. The compound of claim 1, wherein MI is a maleimide group of the formula:

wherein the wavy line indicates the position of connection to S.
3. The compound of claim 1, wherein S is represented by S1-S2-S3, wherein S1 is selected from the group consisting of:

wherein Rx is absent or selected from: c1-6Alkylene radical, C1-6Alkylene amino group, C1-6Alkylene carboxyl and C1-6An alkylenecarbonylamino group, the wavy line indicating the position of attachment to the adjacent moiety; s2 is absent or is- [ (CH)2)pO]q-, wherein p is an integer of 1 to 4, preferably 2, q is an integer of 0 to 15, preferably 1 to 15, more preferably 2 to 6; s3 is absent or selected from polar amino acid residues such as: glu, Asp, Gly, Ala, Val, Leu, Ile, Met, Phe, Trp, Ser, Thr, Cys, Tyr, Asn, Gln, Lys, Arg, and His, preferably Glu and Asp;
wherein S is attached to C via a group selected from:

wherein, the wavy line indicates the connecting portion with the adjacent portion;
and wherein at least one of S1, S2, and S3 is present.
4. The compound of claim 1, wherein S is-R1-[(CH2)pO]q-R2-R3-, in which R1Is linked to MI, is absent or selected from C1-6Alkylene or C1-6An alkylenecarbonylamino group; r2Is selected from C1-6An alkylene group; r3Selected from-C (O) O-, -NH-, -O-or-C (O) -R4Wherein R is4Is an amino acid residue selected from Glu, Asp, Gly, Ala, Val, Leu, Ile, Met, Phe, Trp, Ser, Thr, Cys, Tyr, Asn, Gln, Lys, Arg and His, and preferably Glu and Asp, and R4Forming an amide bond with the-c (o) -through an amino group thereof; p is an integer of 1 to 4; q is an integer of 0 to 15, preferably 1 to 15, more preferably 2 to 6.
5. The compound of any one of claims 1-4, wherein MI, S1, S2, S3, C, and a are linked to each other by any one of:

wherein the wavy line indicates the connecting portion with the adjacent portion.
6. The compound of claim 1, wherein MI-S is selected from the group consisting of:



7. the compound of claim 1, wherein C is selected from the group consisting of a group that cleaves asparagine endopeptidase when expressed in a tumor microenvironment, and wherein the group comprises an Asn residue.
8. The compound of claim 7, wherein C is X1X2X3Wherein X is1Selected from L or D form Ala, Thr, Val and Asn; x2Selected from L or D form Ala, Thr, Val and Ile; x3Asn, preferably not D-Asn;
preferably, C is selected from: Ala-Ala-Asn, Thr-Ala-Asn, Val-Ala-Asn, Asn-Ala-Asn, Thr-Thr-Asn, Val-Thr-Asn, Asn-Thr-Asn, Ala-Val-Asn, Thr-Val-Asn, Asn-Val-Asn, Ala-Ile-Asn, Thr-Ile-Asn, Val-Ile-Asn, Asn-Ile-Asn, Ala-Thr-Asn, D-Thr-L-Val-L-Asn, D-Thr-L-Ala-L-Asn, D-Ala-L-Val-L-Asn, L-Thr-D-Val-L-Asn, L-Thr-D-Ala-L-Asn, L-Ala-D-Val-L-Asn, D-Thr-D-Val-L-Asn, D-Thr-D-Ala-L-Asn, D-Ala-D-Val-L-Asn.
9. The compound of claim 1, wherein a is selected from the group consisting of:

wherein the wavy line indicates the position of the connection to C.
10. The compound of claim 1, wherein S and a are selected from any one of the following groups QHL-001 to QHL-162:






preferably, C is AAN.
11. A conjugate represented by the following formula (II):
MI-S-C-A-D (II)
wherein MI, S, C and A are as described in any one of claims 1-10; d is a drug, preferably an anticancer compound;
more preferably, D is selected from the group consisting of doxorubicin, daunorubicin, epirubicin, methotrexate, fludarabine, gemcitabine, cytarabine, melphalan, nimustine, mitoxantrone, mitomycin, camptothecin, 10-hydroxycamptothecin, topotecan, fluorouracil, doxifluridine, etoposide, fludarabine, capecitabine, vincristine, epothilone B, paclitaxel, docetaxel, darafenib, doxertinib, motoney, prednisone, triiodothyronine, ranisimetride, platinum derivatives of the formula:

and the following compounds a and b:

preferably, A and D are connected by any one of the following means:

wherein the wavy line indicates the connecting portion with the adjacent portion.
12. The conjugate of claim 11, or a pharmaceutically acceptable salt thereof, wherein the compound is selected from the group consisting of:











13. a platinum derivative having the structure:

14. a conjugate of claim 11 or 12, or a pharmaceutically acceptable salt thereof, formed by covalent conjugation of the conjugate to albumin.
15. A pharmaceutical composition comprising the conjugate of claim 11 or 12 or a pharmaceutically acceptable salt thereof, the platinum derivative of claim 13 or a pharmaceutically acceptable salt thereof, or the conjugate of claim 14 or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
16. Use of the conjugate of claim 11 or 12 or a pharmaceutically acceptable salt thereof, the platinum derivative of claim 13 or a pharmaceutically acceptable salt thereof, or the conjugate of claim 14 or a pharmaceutically acceptable salt thereof, for the manufacture of a medicament for the treatment or prevention of the steatosis phenomenon of cancer, fatty liver (including alcoholic and non-alcoholic fatty liver), steatohepatitis, fatty liver disease, liver fibrosis, liver inflammation, hepatocyte injury; preferably, the cancer is a solid cancer or a hematological tumor, preferably of the bladder, brain, breast/breast, cervix, colon, rectum, esophagus, kidney, liver, lung, nasopharynx, pancreas, prostate, skin, stomach, uterus, ovary, testis, and hematological location.
17. Use of a compound of any one of claims 1-10 to enhance the drug's aqueous solubility, reduce drug toxicity, improve drug efficacy, and/or improve drug selectivity for immune cells of a compound, or to prepare a drug molecule for delivering a drug to the liver with improved aqueous solubility, reduced drug toxicity, improved drug efficacy, and/or improved drug selectivity for immune cells.
18. An EMC-AANL-DOX compound with a structure shown in the following formula or an albumin-coupled drug thereof is applied to the preparation of a liver cancer treatment drug, or is applied to the preparation of a drug for combined treatment of tumors together with an anti-PD-1 antibody:

19. use of the conjugate of claim 11 or 12 or a pharmaceutically acceptable salt thereof, the platinum derivative of claim 13 or a pharmaceutically acceptable salt thereof, or the conjugate of claim 14 or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for inhibiting immunosuppressive cells, inhibiting tumor-associated macrophages, inhibiting MDSC cells, inhibiting angiogenesis, promoting anti-tumor immunity, and/or promoting T-lymphocyte proliferation.
20. Use of a conjugate according to claim 11 or 12 or a pharmaceutically acceptable salt thereof, a platinum derivative according to claim 13 or a pharmaceutically acceptable salt thereof, or a conjugate according to claim 14 or a pharmaceutically acceptable salt thereof, and an anti-PD-1 antibody for the manufacture of a medicament for the combined treatment of a tumour.
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010106067.9A CN113274507A (en) | 2020-02-20 | 2020-02-20 | Preparation and use of immunostimulatory conjugate complexes for targeted delivery and activation |
| US17/800,691 US20230414771A1 (en) | 2020-02-20 | 2021-02-20 | Preparation and use of immunostimulatory conjugated complexes for targeted delivery and activation |
| KR1020227032497A KR20220143908A (en) | 2020-02-20 | 2021-02-20 | Preparation and Use of Immunostimulatory Conjugation Complexes of Targeted Delivery and Activation |
| JP2022549600A JP2023515034A (en) | 2020-02-20 | 2021-02-20 | Preparation and Use of Targeted Delivery and Activated Immunostimulatory Complexes |
| CA3167564A CA3167564A1 (en) | 2020-02-20 | 2021-02-20 | Preparation and use of immunostimulatory conjugated complexes for targeted delivery and activation |
| PCT/CN2021/077056 WO2021164765A1 (en) | 2020-02-20 | 2021-02-20 | Preparation and use of immunostimulatory coupling complex which is delivered and activated in targeted manner |
| EP21756482.2A EP4108675A4 (en) | 2020-02-20 | 2021-02-20 | Preparation and use of immunostimulatory coupling complex which is delivered and activated in targeted manner |
| AU2021222203A AU2021222203A1 (en) | 2020-02-20 | 2021-02-20 | Preparation and use of immunostimulatory coupling complex which is delivered and activated in targeted manner |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010106067.9A CN113274507A (en) | 2020-02-20 | 2020-02-20 | Preparation and use of immunostimulatory conjugate complexes for targeted delivery and activation |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN113274507A true CN113274507A (en) | 2021-08-20 |
Family
ID=77275295
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010106067.9A Pending CN113274507A (en) | 2020-02-20 | 2020-02-20 | Preparation and use of immunostimulatory conjugate complexes for targeted delivery and activation |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20230414771A1 (en) |
| EP (1) | EP4108675A4 (en) |
| JP (1) | JP2023515034A (en) |
| KR (1) | KR20220143908A (en) |
| CN (1) | CN113274507A (en) |
| AU (1) | AU2021222203A1 (en) |
| CA (1) | CA3167564A1 (en) |
| WO (1) | WO2021164765A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115353476A (en) * | 2022-08-19 | 2022-11-18 | 安阳工学院 | Synthesis method of maleimide-amide-oligo (ethylene glycol) -propionic acid |
| CN116041421A (en) * | 2023-04-03 | 2023-05-02 | 上海亲合力生物医药科技股份有限公司 | Tumor targeted activated platinum compound, preparation method and application thereof |
| WO2023241632A1 (en) * | 2022-06-16 | 2023-12-21 | 亚飞(上海)生物医药科技有限公司 | Conjugate of anti-ctla-4 antibody activated by tumor microenvironment and use thereof |
| WO2024188314A1 (en) * | 2023-03-15 | 2024-09-19 | 上海亲合力生物医药科技股份有限公司 | Linker and drug conjugate using same, and antibody-drug conjugate and use thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024182569A2 (en) | 2023-02-28 | 2024-09-06 | Eisai R&D Management Co., Ltd. | Anti-psma antibodies, conjugates, and methods of use |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060281897A1 (en) * | 2003-08-22 | 2006-12-14 | Andre Trouet | Potentialization of the activation of high molecular weight prodrugs |
| CN103044521A (en) * | 2012-12-26 | 2013-04-17 | 亚飞(上海)生物医药科技有限公司 | Aspartase-targeted activated adriamycin derivative as well as preparation method and application thereof |
| WO2014203691A1 (en) * | 2013-06-18 | 2014-12-24 | 株式会社ヤクルト本社 | Novel medicine containing platinum complex |
| CN104262455A (en) * | 2014-08-22 | 2015-01-07 | 亚飞(上海)生物医药科技有限公司 | Tumor microenvironment targeted activation docetaxel derivatives, preparation thereof and uses of the derivatives |
| CN106715457A (en) * | 2014-08-22 | 2017-05-24 | 亚飞(上海)生物医药科技有限公司 | Specifically activated micromolecular target coupling body in tumor microenvironment and use thereof |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100381459C (en) * | 2004-12-03 | 2008-04-16 | 成都南山药业有限公司 | Adriamycin derivative and its preparing method and use |
| US9982011B2 (en) * | 2012-12-26 | 2018-05-29 | Yafei (Shanghai) Biopharmaceutical Co., Ltd. | Legumain activated doxorubicin derivative as well as preparation method and application thereof |
| CN104147612B (en) * | 2014-08-22 | 2017-05-24 | 亚飞(上海)生物医药科技有限公司 | Tumor microenvironment specific activated micromolecular targeted conjugate and application thereof |
| CA2895779A1 (en) * | 2015-06-26 | 2016-12-26 | Yafei (Shanghai) Biopharmaceutical Co., Ltd. | Legumain activated doxorubicin derivative as well as preparation method and application thereof |
| PE20231050A1 (en) * | 2016-03-02 | 2023-07-11 | Eisai Randd Man Co Ltd | ERIBULIN-BASED ANTIBODY-DRUG CONJUGATES AND METHODS FOR THEIR USE |
| US11319341B2 (en) * | 2016-12-21 | 2022-05-03 | Yafei (Shanghai) Biopharmaceutical Co., Ltd. | Immune-stimulating soluble doxorubicin-conjugated complex |
| MX2020010458A (en) * | 2018-04-06 | 2021-01-29 | Seattle Genetics Inc | Camptothecin peptide conjugates. |
| WO2019204364A1 (en) * | 2018-04-16 | 2019-10-24 | Avelas Biosciences, Inc. | Compositions and methods for the selective delivery of therapeutic and imaging agents |
| WO2020014306A1 (en) * | 2018-07-10 | 2020-01-16 | Immunogen, Inc. | Met antibodies and immunoconjugates and uses thereof |
| CA3107732A1 (en) * | 2018-07-27 | 2020-01-30 | Daiichi Sankyo Company, Limited | Protein recognizing drug moiety of antibody-drug conjugate |
-
2020
- 2020-02-20 CN CN202010106067.9A patent/CN113274507A/en active Pending
-
2021
- 2021-02-20 CA CA3167564A patent/CA3167564A1/en active Pending
- 2021-02-20 AU AU2021222203A patent/AU2021222203A1/en active Pending
- 2021-02-20 EP EP21756482.2A patent/EP4108675A4/en active Pending
- 2021-02-20 US US17/800,691 patent/US20230414771A1/en active Pending
- 2021-02-20 JP JP2022549600A patent/JP2023515034A/en active Pending
- 2021-02-20 WO PCT/CN2021/077056 patent/WO2021164765A1/en unknown
- 2021-02-20 KR KR1020227032497A patent/KR20220143908A/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060281897A1 (en) * | 2003-08-22 | 2006-12-14 | Andre Trouet | Potentialization of the activation of high molecular weight prodrugs |
| CN103044521A (en) * | 2012-12-26 | 2013-04-17 | 亚飞(上海)生物医药科技有限公司 | Aspartase-targeted activated adriamycin derivative as well as preparation method and application thereof |
| WO2014203691A1 (en) * | 2013-06-18 | 2014-12-24 | 株式会社ヤクルト本社 | Novel medicine containing platinum complex |
| CN104262455A (en) * | 2014-08-22 | 2015-01-07 | 亚飞(上海)生物医药科技有限公司 | Tumor microenvironment targeted activation docetaxel derivatives, preparation thereof and uses of the derivatives |
| CN106715457A (en) * | 2014-08-22 | 2017-05-24 | 亚飞(上海)生物医药科技有限公司 | Specifically activated micromolecular target coupling body in tumor microenvironment and use thereof |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023241632A1 (en) * | 2022-06-16 | 2023-12-21 | 亚飞(上海)生物医药科技有限公司 | Conjugate of anti-ctla-4 antibody activated by tumor microenvironment and use thereof |
| CN115353476A (en) * | 2022-08-19 | 2022-11-18 | 安阳工学院 | Synthesis method of maleimide-amide-oligo (ethylene glycol) -propionic acid |
| CN115353476B (en) * | 2022-08-19 | 2024-03-26 | 安阳工学院 | Synthesis method of maleimide-amide-oligoethylene glycol-propionic acid |
| WO2024188314A1 (en) * | 2023-03-15 | 2024-09-19 | 上海亲合力生物医药科技股份有限公司 | Linker and drug conjugate using same, and antibody-drug conjugate and use thereof |
| CN116041421A (en) * | 2023-04-03 | 2023-05-02 | 上海亲合力生物医药科技股份有限公司 | Tumor targeted activated platinum compound, preparation method and application thereof |
| CN116041421B (en) * | 2023-04-03 | 2023-07-28 | 上海亲合力生物医药科技股份有限公司 | Tumor targeted activated platinum compound, preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2021222203A1 (en) | 2022-09-08 |
| WO2021164765A1 (en) | 2021-08-26 |
| US20230414771A1 (en) | 2023-12-28 |
| JP2023515034A (en) | 2023-04-12 |
| KR20220143908A (en) | 2022-10-25 |
| CA3167564A1 (en) | 2021-08-26 |
| EP4108675A1 (en) | 2022-12-28 |
| EP4108675A4 (en) | 2024-05-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN113274507A (en) | Preparation and use of immunostimulatory conjugate complexes for targeted delivery and activation | |
| US6825166B2 (en) | Molecular conjugates for use in treatment of cancer | |
| JP6854759B2 (en) | Small molecule target conjugates specifically activated by the tumor microenvironment and their use | |
| CN111689980A (en) | Camptothecin drug and antibody conjugate thereof | |
| US6156754A (en) | Glycoconjugates of modified camptothecin derivatives (A-or B-ring linkage) | |
| AU2002254400A1 (en) | Molecular conjugates for use in treatment of cancer | |
| EA000006B1 (en) | Increased bioavailability of biologically active compounds by linking to polypyrrolecarboxamidonaphthalene derivatives | |
| CN116726192A (en) | Antibody conjugated drugs of N-alkoxyalkyl substituted camptothecin derivatives | |
| WO2024140449A1 (en) | Antibody-drug conjugate from n-oxycycloalkyl substituted camptothecin derivative | |
| CN111001012A (en) | Hydrophilic carbonate type antibody coupling drug | |
| CN116712562A (en) | Antibody conjugated drugs of N-azidoalkyl substituted camptothecin derivatives | |
| TW202216724A (en) | A camptothecin drug and its antibody conjugate thereof | |
| JPH07157428A (en) | Spongostachin 5,7,8 and 9 | |
| US11319341B2 (en) | Immune-stimulating soluble doxorubicin-conjugated complex | |
| JP3058188B2 (en) | Anti-cancer, anti-viral and therapeutic cacinoid preparations with plant growth inhibitory action | |
| US20020173468A1 (en) | Modified cytostatic agents | |
| WO2024012569A9 (en) | Linkers, conjugates and applications thereof | |
| CN106344930B (en) | Preparation and application of molecular site-specific targeting and activating short peptide adriamycin | |
| EP3572420B1 (en) | Preparation and use of molecular site targeted and activated kinase inhibitor | |
| KR20120016050A (en) | Prodrugs | |
| JP2019515025A (en) | Topoisomerase poison | |
| CN117916253A (en) | Alpha fetoprotein bioconjugates for the treatment of diseases | |
| CN110627876A (en) | A7R glycopeptide and its application in preparing medicine for treating tumor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |