CN1131672A - 氨基环戊烷衍生物 - Google Patents
氨基环戊烷衍生物 Download PDFInfo
- Publication number
- CN1131672A CN1131672A CN95121896A CN95121896A CN1131672A CN 1131672 A CN1131672 A CN 1131672A CN 95121896 A CN95121896 A CN 95121896A CN 95121896 A CN95121896 A CN 95121896A CN 1131672 A CN1131672 A CN 1131672A
- Authority
- CN
- China
- Prior art keywords
- expression
- compound
- methylol
- structural formula
- aminocyclopentane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- MGNZXYYWBUKAII-UHFFFAOYSA-N C1C=CC=CC1 Chemical compound C1C=CC=CC1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/52—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings condensed with carbocyclic rings or ring systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
本发明提供氨基环戊烷衍生物,该衍生物是具有极高的α-葡糖苷酶抑制作用并具有新型结构的糖化物类似物且可望能用于或适用于药物或农车业化学品。一种用式(1)表示的氨基环戊烷衍生物,其中R1表示H而R2表示CH2OH,或R1表示CH2OH而R2表示H和R3表示取代的或未取代的芳基或1-10个碳原子的烷基,链烯基,炔基或羟烷基,用于其合成的中间体,制备中间体的方法和制备氨基环戊烷衍生物的方法。
Description
本发明涉及新型结构的氨基环戊烷衍生物及合成该衍生物的中间体。更具体地说,涉及糖苷酶抑制剂。
最近的研究已经阐明了在细胞表面层的糖链的生物功能。结果,构成(Construct)或分解这些糖链的酶抑制剂已经引起了公众的注意并且化学家们已经对其合成进行了广泛的研究。此外,近年来,开发对于各种糖苷酶有特效的抑制剂已经成为关注的焦点。例如,人们试图开发一种不抑制麦芽糖酶但抑制蔗糖酶的化合物,尽管这些酶均是α-葡糖苷水解酶,或一种不抑制源于动物的酶但抑制源于昆虫的另一种酶的化合物。在现阶段通常使用的达到这些目的的方法包括按照在自然条件下被目标酶实际水解的糖链之后仿制抑制剂的结构。这样将所谓的糖化物类似物用于具有简单结构用作先导(1ead)化合物的苷水解酶抑制剂。这些糖化物类似物被分类成碳糖化物(Carbasaccharide),氮杂糖化物,硫杂糖化物,磷杂糖化物,等等。
观察到某些这样设计和合成的低聚糖链化合物似乎显示出对目标酶无抑制活性但对其他酶有抑制活性。因此,考虑到开发特定的抑制剂,至今为止的已知先导化合物有各种问题。为了解决这些问题,已经合成和筛选了很多新型的先导化合物。C-H.Wong等人现在正在将其研究范围从氮杂吡喃糖[G.C.Lock,C.H.Fotsh and C-H.Wong,Acc.Chem.Res.,26,182-190(1993)]扩展到氮杂呋喃糖[Y-F.Wang,Y.Takaoka and C-H.Wong,Angew.Chem.Int.Ed.Engl.,33,1242-1244(1994)]。最近已经有了脒骨架抑制糖苷的报导。这样已经进行了将氮杂糖化物通过该脒骨架引入低聚糖的偿试[a)G.Papandreou,M.K.Tong and B.Ganem,J.Amer.Chem.Soc.,11511682-11690(1993);b)Y.Bleriot,A.Genre-Grandpierre and C.Tellier,Tetrahedron Lett.,35,1867-1870(1994)]。
另一方面,α-葡糖苷水解酶包括α-葡糖苷酶I和II,它们对含于细胞中除了淀粉酶,麦芽糖酶,异麦芽糖酶,蔗糖酶和海藻糖酶以外的糖蛋白的生物合成过程起作用。最近,人们已经努力地试图将这些酶的抑制剂用于药物和农业化学品并且现在在实用上可从市场上购得某些抑制剂。例如,acarbose,它是α-淀粉酶和蔗糖酶的抑制剂,其作为抗糖尿试剂或抗肥胖试剂出售(商品名:Glucobay,Bayer生产),而有效霉素,它抑制海藻糖酶,其作为抵抗条纹病(Stripe)的农业化学品出售(商品名:Validacin,Takeda ChemicalIndustries,Ltd.生产)并预计作为杀虫剂是有效的。此外,在美国,脱氧野尻霉素衍生物作为药物(抗病毒试剂)进行临床试验。再有,正在进行的研究利用抑制剂作为工具弄清楚酶的未知功能。最近,还有报导说脱氧野尻霉素(DNJ)对爱滋病有效。也就是说,葡糖苷酶抑制剂已经成为人们关注的焦点,同时人们迫切需要开发适用于药物和农业化学品的新型结构的酶抑制剂。
本发明的目的是提供氨基环戊烷衍生物,它是具有新型结构有极高α-葡糖苷酶抑制作用的糖化物类似物并可望能用或适用于药物或农业化学品。本发明提供:(1)用式(1)表示的氨基环戊烷衍生物: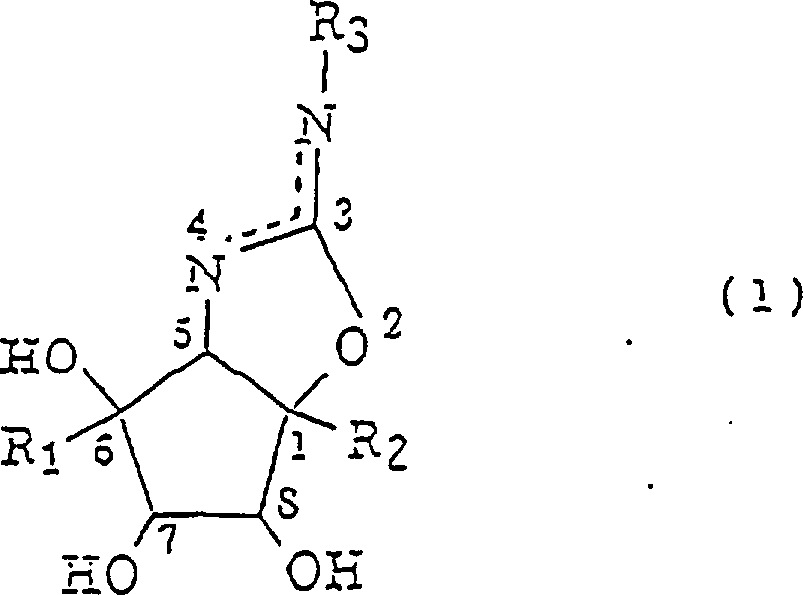 其中R1表示H而R2表示CH2OH,或R1表示CH2OH而R2表示H;和R3表示取代的或未取代的芳基基团或有1到10个碳原子的烷基、链烯基、炔基或羟烷基基团。
其中R1表示H而R2表示CH2OH,或R1表示CH2OH而R2表示H;和R3表示取代的或未取代的芳基基团或有1到10个碳原子的烷基、链烯基、炔基或羟烷基基团。
(2)如上述(1)中所述的氨基环戊烷衍生物,其中在用式(1)表示的氨基环戊烷衍生物中,R3表示取代或未取代的芳基。
(3)如上述(2)中所述的氨基环戊烷衍生物,其中用式(1)表示的氨基环戊烷衍生物选自由下面结构式(1-1L)表示的(1S,5R,6S,7S,8R)-6-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇,由下面结构式(1-1D)表示的(1R,5S,6R,7R,8S)-6-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯6,7,8-三醇,由下面结构式(1-2L)表示的(1S,5R,6S,7R,8S)-1-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇和由下面结构式(1-2D)表示的(1R,5S,6R,7S,8R)-1-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇:
(4)如上面(1)所述的氨基环戊烷衍生物,其中在由式(1)表示的氨基环戊烷衍生物中,R3表示1-10个碳原子的烷基。
(5)如上面(4)中所述的氨基环戊烷衍生物,其中由式(1)表示的氨基环戊烷衍生物选自由下面结构式(2-1L)表示的(1S,5R,6S,7S,8R)-6-羟甲基-3-丁氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇,由下面结构式(2-1D)表的(1R,5S,6R,7R,8S)-6-羟甲基-3-丁氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇,由下面结构式(2-2L)表示的(1S,5R,6S,7R,8S)-1-羟甲基-3-丁氨基-2-恶-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇和由下面结构式(2-2D)表示的(1R,5S,6R,7S,8R)-1-羟甲基-3-丁氨基-2-恶-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇:
(6)(3S,4R,5S,6R,7S)-7-乙酰胺-4,5,6-三-O-乙酰基-1-噁螺[2.4]庚烷-4,5,6-三醇,它是由下面结构式表示的化合物:
(7)由式(2)表示的氨基环戊烷衍生物: 其中R4和R5各自独立地表示H或取代的或未取代的芳基或有1-10个碳原子的烷基,链烯基,炔基或羟烷基。
其中R4和R5各自独立地表示H或取代的或未取代的芳基或有1-10个碳原子的烷基,链烯基,炔基或羟烷基。
(8)如上面(7)中所述的氨基环戊烷衍生物,其中由式(2)表示的氨基环戊烷衍生物选自由下面结构式(d)表示的1L-(1,2,4,5/3)-5-氨基-1C-羟甲基-1,2,3,4-环戊烷-四醇,由下面结构式(d-1)表示的1L-(1,2,4,5/3)-5-二丁氨基-1-羟甲基-1,2,3,4-环戊烷四醇和由下面结构式(d-2)表示的1L-(1,2,4,5/3)-5-丁氨基-1-羟甲基-1,2,3,4-环戊烷四醇:
(9)1D-(1,2,4,5/3)-5-氨基-1C-羟甲基-1,2,3,4-环戊烷四醇,它是由下面结构式表示的化合物:
(10)N-[2-羟甲基-2,3,4,5-四羟基环戊基]-N′-苯基硫脲,它是由下面结构式表示的化合物:
(11)一种含有如上面(1)或(7)所述的氨基环戊烷衍生物作为活性组份的糖苷酶抑制剂。
(12)如上面(11)所述的糖苷酶抑制剂,其中所述的糖苷酶抑制剂是α-葡糖苷酶抑制剂。
(13)如上面(12)所述的糖苷酶抑制剂,其中所述的活性组份是如上面(2),(4)或(8)所述的氨基环戊烷衍生物。
(14)如上面(13)所述的糖苷酶抑制剂,其中所述的活性组份是如上面(3)或(5)所述的氨基环戊烷衍生物。
(15)一种制备如上面(6)所述化合物的方法,包括在溶液中氧化由下面结构式表示的化合物:
(16)一种制备如上面(1)所述氨基环戊烷衍生物的方法,包括将由式(3)表示的硫脲化合物转变成环异脲:其中R1,R2和R3与上面(1)中所述的定义一样。
图1是表明对通过用HIV感染的PHA活化的PBMC产生HIV的抑制作用的图。
图2是表明对通过将HIV感染的Molt-4/C18细胞与Molt-4/IIIB细胞共培养引起的细胞融合(形成巨细胞)的抑制作用的图。
下面将详细描述本发明。
考虑到由式(1)[下文中简称为化合物(1)]表示的本发明氨基环戊烷衍生物有如下面式子所示的互变体。因此,在式(1)的结构式中的虚线代表这些互变体。此外,化合物(1)包括其同样的任意立体异构体。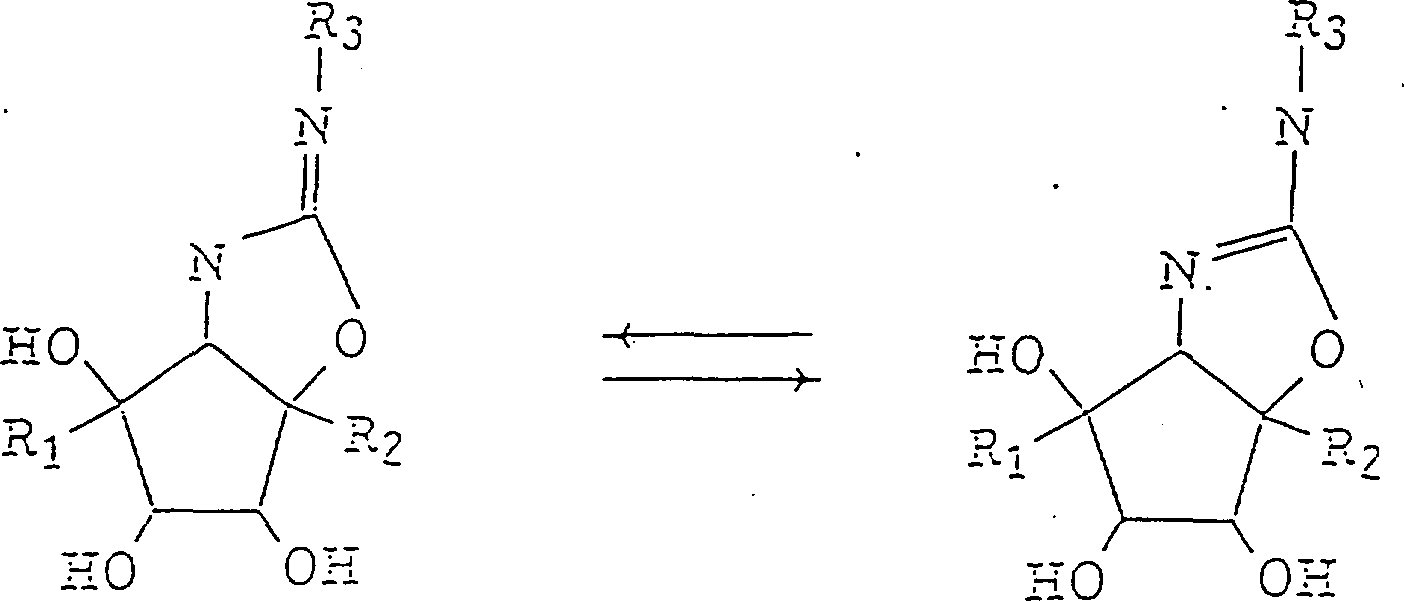
还要考虑在化合物(1)中羟基和羟甲基的指向,它有含有环异脲键的双环部分,是一个极重要的因素并且极大地影响到该化合物的酶抑制活性。假定在化合物(1)结构中有两个氮原子的异脲部分是弱碱性的并且当化合物(1)与酶形成络合物时强键合到活性中心上。这样特定地抑制酶的活性可以增强到前所未有的水平。脲在式(1)中,R3表示取代的或未取代的芳基或有1-10个碳原子,优选3-8个碳原子的烷基,链烯基,炔基或羟烷基,优选的R3是苯基,或丁基。芳基基团的取代基例子包括1-6个碳原子的烷基,链烯基和炔基,卤原子和羟基,硝基和羧基。
本发明化合物(1)的优选例子包括如下所示的化合物(1-1),(1-2),(2-1)和(2-2)。
化合物(1-1)和(1-2)彼此是结构异构的,而化合物(2-1)和(2-2)也是彼此结构异构的。如上面(3)所述,化合物(1-1)包括由结构式(1-1L)和(1-1D)表示的对映体。类似地,化合物(1-2)如在上面(3)所述包括由结构式(1-2L)和(1-2D)表示的对映体。此外,化合物(1-1)和(1-2)包括在6-,7-和8-位上由羟基产生的任意的非对映体。如上面(5)所述,化合物(2-1)包括由结构式(2-1L)和(2-1D)表示的对映体。类似地,如上面(5)所述,化合物(2-2)包括由结构式(2-2L)和(2-2D)表示的对映体。此外,化合物(2-1)和(2-2)包括在6-,7-和8-位上由羟基产生的任意的非对映体。
化合物(1-1)和(1-2)有抑制葡糖苷酶的活性。特别地,它们显示出极高的抑制α-葡糖苷酶的活性。它们还有抗HIV活性。类似地,化合物(2-1)和(2-2)有抑制葡糖苷酶的活性。特别地,它们显示出极高的抑制α-葡糖苷酶的活性。它们也有抗HIV活性。
本发明的化合物(1-1L)和(1-2L)可以根据下面反应图解1合成。
路线1
将链烯化合物(即化合物(a))(用在C.Uchida,T.Yamagishiand S.Ogawa,J.Chem.Soc.Perkin Trans.1,1994,589-602中所述的方法合成)溶解在有机溶剂(优选1,2-二氯乙烷)而不是酮中,同时在存在含水缓冲溶液中维持pH在中性,在0℃至约反应混合物沸点温度(在回流条件下)加入相当于链烯化合物1-10当量的氧化试剂例如过酸(例如间-氯代过苯甲酸,过苯甲酸,单过氧邻苯二甲酸,三氟过乙酸,过甲酸)或过氧化物(例如二环氧乙烷)。另一方面,在0℃至约反应混合物沸点温度(在回流条件下)向其加入能够生成氢过氧化物的金属催化剂[例如,五氧化二钒(V2O5)/叔丁基氢过氧化物(tBuOOH),钨酸钠(Na2WO4)/过氧化氢(H2O2),五氧化钼(MoO5)·六甲基磷酰胺(HMPA)/tBuOOH)。然后得到的混合物在黑暗中搅拌0.5-120小时从而氧化起始化合物(a)。反应体系用非醇有机溶剂(氯仿,乙酸乙酯等)稀释并用含水溶剂(硫代硫酸钠的饱和水溶液,碳酸氢钠的饱和水溶液,等等)洗涤从而得到化合物(b)[(3S,4R,5S,6R,7S)-7-乙酰胺-4,5,6-三-O-乙酰基-1-噁螺[2.4]庚烷-4,5,6-三醇]。得到的产物可以进一步用色谱等纯化。Ac表示乙酰基。
将化合物(b)溶于有机溶剂的水溶液,例如N,N-二甲基甲酰胺(DMF)水溶液,二甲基亚砜(DMSO)水溶液或2-甲氧基乙醇水溶液中,接着向其加入乙酸钠,苯甲酸钠等。在室温至150℃温度下搅拌1-48小时之后,反应混合物在减压下浓缩。然后,得到的残余物用碱,例如吡啶,4-二甲基氨基吡啶,二甲基吡啶或三乙基胺,和乙酸酐,乙酰氯等乙酰化。用色谱等纯化后,得到浆状化合物(c)[1L-(1,2,4,5/3)-5-乙酰氨基-1-乙酸基甲基-2,3,4-三-O-乙酰基-1,2,3,4-环戊烷四醇]。
化合物(c)通过溶解在0.5-6M盐酸或硫酸脱乙酰基并在室温至100℃温度下搅拌0.5-5小时。在减压条件下浓缩后,得到的残余物用阳离子交换树脂[例如,Dowex 50W-X2,X4,X8(商品名),Amberlite IR-120B,CG50(商品名)]等纯化,从而得到浆状化合物(d)[表海藻糖胺=1L-(1,2,4,5/3)-5-氨基-1C-羟甲基-1,2,3,4-环戊烷-四醇]。
另一方面,在化合物(d)的合成路线中,化合物(a)也可以用氧化锇(VIII)(OsO4)在丙酮水溶液中,其用量为相当于化合物(a)的1-5当量,或0.05-1当量的OsO4和1-5当量的氧化剂例如N-甲基吗啉-N-氧化物,羟基化1-72小时,然后乙酰化接着用硅胶色谱分离化合物(c)的非对映体从而不形成化合物(b)而得到作为中间体的化合物(c)(J.Chem.Soc.Perkin Trans.1,1994,589-602)。
将化合物(d)溶解在醇等的水溶液中。然后向其加入相当于化合物(d)1-5当量的异硫氰酸苯酯并将得到的混合物在0-80℃搅拌1-72小时。在减压条件下浓缩反应混合物后,所得的残余物通常用分配柱色谱纯化从而得到苯基硫脲化合物,即白色固体的化合物(e)[N-[(1R),(1,2,4,5/4)-2-羟甲基-2,3,4,5-四羟基环戊基]-N′-苯基硫脲]。
将化合物(e)溶解在酮或醇溶剂中。然后向其加入相当于化合物(e)1-10当量的催化剂[汞氧化物(黄色或红色),氧化铅,烷基碘,三乙基锇四氟硼酸盐,三氟酸甲酯(methyl triflate),次氯酸钠与氢氧化钠的混合物,三苯基膦与偶氮二羧酸二乙酯混合物,等等],并将该混合物在0℃至该反应混合物的沸点(在回流条件下)温度下搅拌1-72小时,从而得到环异脲化合物。反应体系通过硅藻土等过滤并用甲醇等洗涤。将滤液与洗涤液合并在一起并在减压条件下浓缩。将由此得到的残余物用薄层色谱等处理,从而将异构体彼此粗分开来。首先得到上述粗6-羟甲基异脲,即粗化合物(1-1L)。该粗产物用如上所述的阳离子交换树脂纯化从而得到白色固体的化合物(1-1L)。
接下来,从上述薄层色谱得到粗1-羟甲基异脲,即粗化合物(1-2L)并用上述阳离子交换树脂纯化从而得到白色固体的化合物(1-2L)。
为了合成化合物(1-1D)和(1-2D),重复上述过程但用下面立体异构体(ad)(用J.Chem,Soc.Perkin Trans.1,1992,1939-1942所述的方法合成)替代化合物(a)。这样用路线1和2的方法能够分别得到化合物(1-1D)和(1-2D)。
路线2
在路线1和2中,具有苯基作为式(1)中R3的化合物(e)或(ed)是通过用异硫氰酸苯酯处理化合物(d)或(dd)合成的。用同样的方法,具有不同R3的硫脲化合物可以通过使用异硫氰酸苯酯的苯基基团已经被所希望的取代基(R3)取代了的异氰酸酯衍生物得到。然后用与上述同样的方法将该硫脲化合物转变成环异脲并纯化。这样可以得到本发明所需要的氨基环戊烷衍生物。换言之,通过重复上述方法但用异硫氰酸丁酯替代异硫氰酸苯酯可以得到化合物(2-1L),(2-2L),(2-1D)和(2-2D)。
在用式(2)[下文中简称为化合物(2)]表示的本发明的氨基环戊烷衍生物中,R4和R5各自独立地表示H或取代的或未取代的芳基或有1-10个碳原子的烷基,链烯基,炔基,羟烷基。芳基的取代基的例子包括具有1-6个碳原子的烷基,链烯基和炔基,卤原子和羟基,硝基和羧基。
本发明化合物(2)的优选例子包括化合物(d),(d-1)和(d-2)。
本发明的化合物(d-1)和(d-2)可以合成如下。将化合物(d)溶解在醇的水溶液中,如果需要,也可以加入分子筛等,接着在4-40℃搅拌混合物0.5-5小时,然后加入相当于化合物(d)的1-10当量,优选1当量的丁醛。在0℃至约反应混合物沸点(在回流条件下)温度搅拌0.5-5小时之后,加入相当于化合物(d)的1-10当量的还原试剂,例如氰基硼氢化钠,该混合物在0℃至约该反应混合物的沸点(回流条件下)温度搅拌1-72小时。该反应体系通过硅藻土等过滤并用甲醇等洗涤。将滤液与洗涤液体合并在减压条件下浓缩。将如此得到的残余物用薄层色谱等处理。这样首先得到粗化合物(d-1)。这一粗产物用如上所述的阳离子交换树脂纯化从而得到化合物(d-1)。
接下来,从上述薄层色谱得到粗化合物(d-2)并用如上所述的阳离子交换树脂纯化从而得到化合物(d-2)。
通过将化合物(d)与丁醛反应,制备式(2)中R4和/或R5是丁基的化合物。类似地,通过使用丁醛的丁基基团被所需要的取代基取代的醛衍生物可以得到本发明所需要的氨基环戊烷衍生物。
预计本发明的氨基环戊烷衍生物,尤其是化合物(1-1),(1-2),(2-1)和(2-2),它们是具有类似于DJN的内氮和类似于mannostatin的外氮的新型化合物,可用于新型药物和农业化学品。还预计从本发明的新型化合物能够合成各种类似物从而得到改进了特性的衍生物。
因此,本发明的化合物能够有助于对看上去影响涉及葡糖苷酶抑制活性的各种生化相互反应体系的研究。因此这些化合物适用于开发新型药物,例如,抗病毒试剂,如抗HIV试剂,对涉及糖化物和脂新陈代谢的疾病(例如,肥胖和糖尿病)的治疗,调节免疫系统的药物例如免疫辅药,癌转移抑制剂,抗条纹病(由稻纹枯病,棉立枯病等引起)的农业化学品,抗菌剂和杀虫剂。此外,由于具有抗HIV活性,本发明的化合物作为抗HIV试剂是有效的。
为了更详细地而不是以限制的方式进一步说明本发明,将给出下面实施例。
用于下面实施例中的步骤,测定方法和样品如下。
合成
1)薄层色谱(TLC)
将用于色谱的硅胶(Kieselgel60GF 254,Merck& Co.Inc.生产)涂敷在玻璃板上并在70℃活化30分钟。通过将浓硫酸喷在该板上接着加热诱发着色。在喷浓硫酸之前用紫外灯(254nm,由Hirai Rika Kenkyusho制造)进行紫外吸收测定。
2)旋光率([α]D)
使用数字旋光仪(DIP-370,由Nippon Bunko-sha制造)。使用石英池(10×10mm)用钠的D射线进行测量。
3)核磁共振谱(1H-NMR,13C-NMR)
在1H-NMR测定中,使用NMR波谱仪(JNM-270FT,270MHz,由JEOLLtd制造)。重氯仿或重水用作溶剂,而四甲基硅烷(δ0.00)(在重氯仿情况下)或丙酮(δ2.08)(在重水情况下)用作内标。
在13C-NMR测定中,使用NMR波谱仪(JNM-400FT,由JEOL Ltd制造)。重甲醇用作溶剂而四甲基硅烷(δ0.00)用作内标。
4)红外吸收光谱(IR)
在粘合方法(纯的)的情况下,将样品粘接到KBr晶片上并使用红外光谱仪(IR-810型,由Nippon Bunko-sha制造)测定。在压片方法(KBr片)的情况下,使用Hitachi 225衍射光栅BIO-RAD DIGITALFTS-65富里叶变换红外光谱仪进行测定。
5)质谱分析
在HR-FAB-MS中,使用JEOL JMS HX-110(由JEOL,Ltd.制造)。离子检测模式是FAB正并用甘油(离子群[93+(92)n]+)作为基质。使用碘化铯(CsI)作为质量校正样品。
6)制备性薄怪色谱(PTLC)
使用一平板作为制备性薄层色谱(Merck Art5744,由Merck&Co.,Inc.制造)。使用甲醇进行洗脱。
7)硅胶柱色谱
使用Wakogel C-300(200-300目,由Wako Pure ChemicalIndestries,Ltd.生产)。
8)减压浓缩
减压条件下的浓缩是在约40℃温度下使用旋转式汽化器用一吸气器在减压条件下进行的。
9)反应溶剂
在反应中使用的每一溶剂都是未蒸馏的。
10)反应试剂
纯度70%的mCPBA(间-氯代过苯甲酸)是从Tokyo Kasei购买的。
乙酸钠是从Wako Pure Chemical Industries,Ltd.购买的。
异硫氰酸苯酯是从Tokyo Kasei购买的。
汞氧化物是从氢氧化钠和氯化汞(II)制备的。
就这样使用这些市售的试剂。
酶抑制测定
1)吸光度
使用96孔(Well)微板,用微板读数器(MPR-4Ai,由TosohCorporation制造)测定释放的对-或邻-硝基苯酚的吸光度(在405nm)。
2)培育
在37℃用自然培育箱(Compact NIB-10,由Iwaki Glass Co.Ltd.制造)进行培育。
3)酶
本文中所用的酶[α-葡糖苷酶(发面酵母),β-葡糖苷酶(杏仁),α-半乳糖苷酶(E.coli),β-半乳糖苷酶(E.Coli)和β-半乳糖苷酶(牛肝)]均从Sigma购买。
4)酶作用物
这些酶的酶作用物(对-或间-硝基苯基苷)均购自Sigma。
实施例1
(3S,4R,5S,6R,7S)-7-乙酰胺-4,5,6-三-O-乙酰基-1-噁螺[2.4]庚烷-4,5,6-三醇(化合物(b))的合成:
将化合物(a)(21.7mg,0.0725mol)溶解于1,2-二氯乙烷(2ml)中。在存在磷酸盐缓冲溶液的条件下,在室温下加入70%mCPBA(53.4mg,0.218mmol,3当量)。在黑暗中搅拌26小进后,反应体系用氯仿(30ml)稀释并接着用硫代硫酸钠饱和水溶液(5ml)和碳酸氢钠饱和水溶液(5ml)洗涤。有机层在芒硝上干燥并过滤。在减压条件下浓缩滤液并将如此得到的残余物用硅胶柱色谱(Wakogel C-300,1g,丙酮/甲苯=1/2)纯化。这样得到浆状的化合物(b)(19.8mg,收率86.5%)。
Rf 0.41(乙醇/甲苯=1/5,双展开)
[α]27 D-63.7°(C 0.96,氯仿)。
IR(纯的)3360(OH和NH),1740(OAc)和1670(NAc)cm-1。
1H-NMR(270MHz,CDCl3)
δ5.59(1H,d,J7,NH9.2Hz,NH),5.26(1H,dd,J4,5~1,J5,62.9Hz,5-H),5.21-5.17(2H,m,4和6-H),5.00(1H,dd,J6,75.1,J7,NH9.2Hz,7-H),2.98和2.68(每个1H,ABq,Jgem4.8Hz,2×2-H),2.14,2.12,2.09和2.00(每个3H,4s,4Ac)。
作为C14H19NO8的元素分析:
计算值(%):C51.06,H5.82,N4.25
实验值(%):C51.61,H5.82,N4.20
实施例2
1L-(1,2,4,5/3)-5-氨基-1-羟甲基-1,2,3,4-环戊烷四醇(化合物(d)的合成:
将化合物(b)(19.8mg,0.0628mmol)溶于80%的DMF水溶液(1ml)中并加入乙酸钠(30.9mg,0.377mmol,6当量)。在120℃搅拌20小时后,将反应混合物在减压条件下浓缩。这样得到的残余物用吡啶(1ml)和乙酸酐(0.5ml)乙酰化。得到的产物用硅胶柱色谱(Wakogel C-300 1g,丙酮/甲苯=1/2)纯化从而得到浆状五乙酸酯(化合物(c))(16.0mg收率:65.3%)。
化合物(c)的数据与文献(C.Uchida,T.Yamagishi和S.Ogawa,J.Chem.Soc.,Perkin Trans,1,1994,589-602)中所述的一致。
将化合物(c)(42.5mg,0.109mmol)溶于2M盐酸中。然后该溶液在80℃搅拌2小时并在减压条件下浓缩。这样得到的残余物用Dowex50W-X2(H+,1ml,1M氨水)纯化从而得到浆状表海藻糖胺(化合物(d))(19.7mg,收率:最高达100%)。
Rf0.38(水/乙腈=1/4)。
[α]23 D-3.8°(C0.98,水)。
IR(纯的)3350(OH和NH2)cm-1。
1H-NMR(270MHz,D2O,基准,丙酮)。
δ3.96(1H,dd,J2,38.1,J3,44.8Hz,3-H),3.87(1H,dd,J3,44.8,J4,57.7Hz,4-H),3.69(1H,d,J2,38.1Hz,2-H),3.51(2H,s,2×6-H),3.27(1H,d,J4,57.7Hz,5-H)
实施例3
N-[(1R)-(1,2,3,5/4)-2-羟甲基-2,3,4,5-四羟基环戊基]-N′-苯基硫脲(化合物(e))的合成:
将化合物(d)(15.3mg,0.0854mmol)溶于60%乙醇水溶液(1ml)中并加入异硫氰酸苯酯(23μl,0.171mmol,2.0当量)。搅拌3小时后,反应混合物在减压下浓缩并且得到的残余物用硅胶柱色谱(Wakogel C-300,2g,甲苯-乙醇/甲苯=1/5)纯化从而得到白色固体苯基硫脲化合物,即化合物(e)(23.6mg,收率:88.1%)
Rf0.41(乙醇/甲苯=1/2)。
[α]22 D+43.9°(C 1.18,丙酮)。
IR(KBr片)3280(OH和NH)和1540(NH)cm-1。
1H-NMR(270MHz,D2O,基准,丙酮)。
δ7.38-7.17(5H,m,Ph),4.66(1H,m,1-H),3.97(1H,dd,J1,58.1,J4,54.6Hz,5-H),3.87(1H,dd,J3,48.4,J4,54.6Hz,4-H),3.67(1H,d,J3,48.4Hz,3-H),3.38和3.33(每个1H,ABq,Jgem12.1Hz,2×6-H)。
作为C13H18N2O5S的元素分析:
计算值(%):C 49.67,H5.77,N8.91
实验值(%):C 49.93,H6.17,N8.59
实施例4
(1S,5R,6S,7S,8R)-6-羟甲基-3-苯基氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇[化合物(1-1L)]和(1S,5R,6S,7R,8S)-1-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇[化合物(1-2L)]的合成:
将化合物(e)(23.6mg,0.075mmol)溶于丙酮/乙醇(1.5ml,V/V)中并加入汞氧化物(黄色)(55.2mg,0.256mmol,3当量)。在室温搅拌2小时后,反应体系通过硅藻土过滤并用甲醇充分洗涤。将滤液与洗涤液体合并在减压条件下浓缩。这样得到的残余物用PTLC(乙酸/乙醇/甲苯=1/2/4,4次展开)分离。这样首先得到粗6-羟甲基异脲[化合物(1-1L)]。然后用Dowex50W-X2(H+,1ml,14M氨水∶甲醇=1/28)将其纯化从而得到白色固体的化合物(1-1L)(9.2mg,收率:43.8%)。
Rf0.22(乙醇/甲苯=1/2)。
[α]28 D+50.1°(C0.15,甲醇)。
IR(KBr片)3430(OH和NH),1660(C=N)和1580(NH)cm-1。
1H-NMR(270MHz,D2O,基准,丙酮)。
δ7.26-6.97(5H,m,Ph),4.56(1H,dd,J1,59.5,J1,84.2Hz,1-H),4.25(1H,d,J1,59.5Hz,5-H),4.13(1H,dd,J1,84.2,J7,89.5Hz,8-H),3.67(1H,d,J7,89.5Hz,7-H),3.46(2H,s,2×9-H)。
作为C13H16N2O5的元素分析:
计算值(%):C 55.71,H5.75,N9.99
实验值(%):C 55.30,H5.81,N9.84
其次,得到粗1-羟甲基异脲[化合物(1-2L)]并用Dowex50W-X2(H+,1ml,14M氨水:50%甲醇水溶液=1/28)纯化从而得到白色固体的化合物(1-2L)(10.6mg,收率:50.5%)。
Rf0.17(乙醇/甲苯=1/2)。
[α]28 D-27.5°(C0.26,甲醇)。
IR(KBr片)3420(OH和NH),1670(C=N)和1560(NH)cm-1。
1H-NMR(270MHz,D2O,基准,丙酮)。
δ7.29-6.97(5H,m,Ph),4.06(1H,d,J5,65.9Hz,5-H),3.76和3.56(每个1H,ABq,Jgem12.5Hz,2X9-H),3.72-3.61(3H,m,6.7和8-H)。
作为C13H16N2O5的元素分析:
计算值(%):C 55.71,H5.75,N9.99
实验值(%):C 55.25,H5.75,N10.01
实施例5
重复上面实施例1-4的方法步骤,根据路线2得到化合物(1-1D)和(1-2D)。
实施例6
N-[(1R)-(1,2,3,5/4)-2-羟甲基-2,3,4,5-四羟基环戊基]-N′-丁基硫脲的合成:
根据Furukawa等人[Nippon Kagaku Kaishi(1989),P.822-825]的方法采用下列方式制备异硫氰酸丁酯。将三苯基膦(31.47g,0.12mol)溶解于苯(20ml)和三乙胺(41.8ml,0.30mol)中并加入二硫化碳(7.3ml,0.12mol)。加入乙腈(150ml)后,将反应体系冷至-15℃并加入丁胺(9.9ml,0.10mol)和四氯化碳(9.7ml,0.10mol),接着搅拌。30分钟后,将反应体系加热至0℃并搅拌1小时。然后将反应混合物进一步加热至室温并搅拌5小时。将反应混合物过滤弃去不溶物质。然后将滤液在减压条件下浓缩至约50ml并用己烷(100ml×5)萃取。合并有机层并在芒硝上干燥。重复地过滤和减压浓缩后,得到的液体残余物通过减压蒸馏纯化。这样得到液体的异硫氰酸丁酯(6.34g,收率55.0%)。
沸点:75-76℃(31mmHg)[上面引用文献报导的沸点为76-77℃(22mmHg)]。
将化合物(d)(表海藻糖胺)(21.4mg,0.119mmol)溶解于60%乙醇水溶液(1.5ml)中。然后加入从丁胺制备的上述异硫氰酸丁酯(44μl,0.358mmol,3当量)并将所得混合物在室温搅拌。3,5和19小时之后,加入同样量(即总共4次,176ml,1.43mmol,12当量)的异硫氰酸丁酯并搅拌混合物22小时。减压浓缩反应混合物并用硅胶柱色谱(Katayama60(商品名,由Katayama Chemical出售)8g,乙醇/甲苯=1/3)纯化所得的残余物从而得到白色固体的丁基硫脲衍生物(35.2mg,收率:最高达100%)。
Rf0.36(乙醇/甲苯=1/2)。
[α]21 D+38.1°(C2.06,甲醇)。
IR(KBr片)3370(OH和NH)和1560(NH)cm-1。
1H-NMR(270MHz,D2O,基准,丙酮)。
δ4.70-4.60(1H,m,1-H),3.95-3.87(2H,m,4和5-H),3.67(1H,d,J3,4 8.4Hz,3-H)3.40-3.25(2H,m,NCH2CH2CH2CH3),3.37和3.30(每个1H,ABq,Jgem11.9Hz,2×6-H),1.49-1.37(2H,m,NCH2CH2CH2CH3),1.29-1.14(2H,m,NCH2CH2CH2CH3),0.76(3H,t,J3′,4′7.3Hz,NCH2CH2CH2CH3)。
作为C11H22N2O5S的元素分析:
计算值(%):C 44.88,H7.53,N9.52
实验值(%):C 44.26,H7.94,N9.38
实施例7
(1S,5R,6S,7S,8R)-6-羟甲基-3-丁氨基-2-噁-4-氮杂双环(3.3.0]辛-3-烯-6,7,8-三醇[化合物(2-1L)]和(1S,5R,6S,7R,8S)-1-羟甲基-3-丁氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇[化合物(2-2L)]的合成。
将在实施例6中得到的丁基硫脲衍生物(35.2mg,0.119mmol)溶解于丙酮/乙醇(2ml,1/1,V/V)中并加入汞氧化物(77.0mg,0.357mmol,3当量)。在室温下搅拌所得混合物。6小时后,再加入同样量(77mg)的汞氧化物继续搅拌3小时。然后反应体系通过硅藻土过滤并用甲醇彻底洗涤。将滤液与洗涤液体合并减压浓缩。用硅胶柱色谱(Katayama60(商品名,由Katayama Chemical出售),8g,乙酸/乙醇/甲苯=1/2/4)分离所得的残余物。这样首先得到粗6-羟甲基异脲[化合物(2-1L)]并用Dowex50W-X2(H+,1ml,14M氨水∶甲醇=1/28)纯化从而得到白色固体的化合物(2-1L)(8.4mg,收率:23.1%)。
RF0.14(乙酸/乙醇/甲苯=1/2/4)。
[α]23 D+35.2°(c0.41,甲醇)。
IR(KBr片)3400(OH和NH),1655(C=N)和1555(NH)cm-1。
1H-NMR(270MHz,D2O,基准,丙酮)
δ4.45(1H,dd,J1,59.2,J1,84.4Hz,1-H),4.18(1H,d,J1,59.2Hz,5-H),4.02(1H,dd,J1,84.4J7,89.7Hz,8-H),3.63(1H,d,J7,89.7Hz,7-H),3.44和3.39(每个1H,ABq,Jgem11.7Hz,2×9-H),3.01(2H,t,J1′,2′7.0Hz,NCH2CH2CH2CH3),1.41-1.31(2H,m,NCH2CH2CH2CH3),1.25-1.11(2H,m,NCH2CH2CH2CH3),0.74(3H,t,J3′,4′7.3Hz,NCH2CH2CH2CH3)。
13C-NMR(100MHz,CD3OD,基准TMS)
δ164.89,87.34,82.24,78.57,77.19,67.78,64.23,43.47.,32.83,21.01,14.16。
作为C11H20N2O5的元素分析:
计算值(%):C50.76,H7.74,N10.76
实验值(%):C50.35,H8.09,N10.46
其次,得到1-羟甲基异脲[化合物(2-2L)]并用Dowex50W-X2(H+,1ml,14M氨水∶50%甲醇水溶液=1/28)纯化从而得到白色固体的化合物(2-2L)(11.7mg,收率:32.1%)。
Rf0.12(乙酸/乙醇/甲苯=1/2/4)。
[α]23 D+16.7°(c0.58,甲醇)。
IR(KBr片)3400(OH和NH),1660(C=N)和1560(NH)cm-1。
1H-NMR(270MHz,D2O,基准,丙酮)
δ3.97(1H,d,J1,56.2Hz,5-H),3.68和3.49(每个1H,ABq,Jgem12.5Hz,2×9-H),3.64-3.54(3H,m,6,7和8-H),3.02(2H,t,J1′,2′6.8Hz,NCH2CH2CH2CH3),1.42-1.32(2H,m,NCH 2CH2CH2CH3 ),1.26-1.02(2H,m,NCH2CH2CH2CH3),0.74(3H,t,J3′,4′7.1Hz,NCH2CH2CH2CH3)。
13C-NMR(100MHz,CD3OD,基准,TMS)
δ163.59,91.02,80.12,75.90,75.16,68.78,63.31,43.49,32.86,21.05,14.19。
作为C11H20N2O5的元素分析:
计算值(%):C50.76,H7.74,N10.76
实验值(%):C50.41,H8.16,N10.61
实施例8
1L-(1,2,4,5/3)-5-二丁氨基-1-羟甲基-1,2,3,4-环戊烷四醇[化合物(d-1)]和1L-(1,2,4,5/3)-5-丁氨基-1-羟甲基-1,2,3,4-环戊烷四醇[化合物(d-2)]的合成:
将化合物(c)(表海藻糖胺五乙酸酯;51.8mg,0.1331mmol)溶解于2M盐酸(2ml)中并在80℃搅拌2小时。冷至室温后,反应体系在减压条件下浓缩从而得到残余物表海藻糖胺氢氯化物。然后将该残余物溶于甲醇(1.5ml)中并加入分子筛4A(50.0mg)。在室温搅拌2小时后,加入丁醛(13.2μl,0.1464mmol,1.1当量)并在室温下再搅拌所得混合物1小时。接下来,加入氰基硼氢化钠(26.4mg,0.3992mmol,3当量)并在同样温度下搅拌混合物3小时。通过硅藻土过滤反应体系并用甲醇充分洗涤。然后减压浓缩滤液,并用硅胶柱色谱(WakogelC-300,2g,乙酸/甲醇/氯仿=1/4/12)分离这样得到的残余物。这样首先得到粗二丁基表海藻糖胺[化合物(d-1)]。该粗产物进一步用Dowex 50W-X2(H+,1ml)吸附,用水充分洗涤并用14M氨水/甲醇(1∶13)洗脱从而得到浆状化合物(d-1)(4.2mg,收率:10.9%)。
Rf0.47(12M盐酸/乙腈=1/8)。
[α]23 D-18.5°(c0.21,甲醇)。
IR(KBr片)3400(OH和NH),1620(NH)cm-1。
1H-NMR(270MHz,D2O,基准,丙酮)
δ3.95(1H,dd,J3,44.4,J4,56.4Hz,4-H),3.89(1H,dd,J2,36.0,J3,44.4Hz,3-H),3.53(1H,d,J2,36.0Hz,2-H),3.49和3.43(每个1H,ABq,Jgem11.7Hz,2X6-H),3.06(1H,d,J4,56.4Hz,5-H),2.83-2.65(4H,m,NCH2CH2CH2CH3),1.42-1.28(4H,m,NCH2CH2CH2CH3),1.22-1.08(4H,m,NCH2CH2CH2CH3),0.76(6H,t,J3′,4′,7.1Hz,NCH2CH2CH2CH3)。
13C-NMR(100MHz,CD3OD,基准,TMS)
δ84.77,80.28,78.84,78.20,67.12,64.05,53.33,(2C),31.70(2C),21.58(2C),14.50(2C)。
HR-FAB-MS[M+H]+
实验值:292.2124
计算值:292.2124
其次得到粗单丁基表海藻糖胺[化合物(d-2)]并用Dowex50W-X2(H+,1ml)以与二丁基化合物情况下使用的同样方法纯化从而得到浆状的化合物(d-2)(4.6mg,收率14.7%)。
Rf0.43(12M盐酸/乙腈=1/8)。
[α]23 D+10.9(c0.23,甲醇)。
IR(KBr片)3350(OH和N)cm-1
1H-NMR(270MHz,D2O,基准,丙酮)
δ3.84(1H,dd,J3,44.8,J4,57.3Hz,4-H),3.78(1H,dd,J2,37.7,J3,4,4.8Hz,3-H),3.54(1H,d,J2,37,7Hz,2-H),3.42和3.38(每个1H,ABq,Jgem12.3Hz,2×6-H),2.94(1H,d,J4,57.3Hz,5-H),2.59-2.41(2H,m,NCH2CH2CH2CH3),1.39-1.26(2H,m,NCH2CH2CH2CH3),1.23-1.12(2H,m,NCH2CH2CH2CH3),0.75(3H,t,J3′,4′7.1Hz,NCH2CH2CH2CH3)。
13C-NMR(100MHz,CD3OD,基准,TMS)
δ83.25,77.93,76.85,74.27,55.76,60.75,53.29,33.32,21.40,14.32。
HR-FAB-MS[M+H]+
实验值:236.1492
计算值:236.1498
实验实施例1
用上面实施例得到的化合物(d),(d-1),(d-2),(1-1L),(1-1D),(1-2L),(1-2D)和(2-2L),脱氧野尻霉素(DNJ),N-丁基脱氧野尻霉素(称之为N-B-DNJ)和海藻糖胺作为试样测定对下列酶的抑制活性。1)α-葡糖苷酶(发面酵母,EC3.2.1.20)[在H.Halvorson andL.Ellias,Biochim.Biophys.Acta.30,28-40,(1958)中有叙述]
在每个孔(Well)中引入试样的水溶液(1μl),0.66mM对-硝基苯基α-D-吡喃葡糖苷在缓冲剂(98μl)中的溶液和(0.4mg/ml)在缓冲剂中的酶溶液(1μl)从而得到总体积100μl。在37℃培育30分钟后,通过加入1M碳酸钠水溶液(100μl)使反应停止。然后测定由该酶反应释放的对-硝基苯酚的吸光度(A405)。为了得到空白吸光度(A空白),试样(1μl)和酶作用物(99μl)以同样的方式培育并加入碳酸钠水溶液(100μl),接着测定吸光度。另一方面,为了得到对比吸光度(A对比),水(1μl),酶作用物(98μl)和酶溶液(1μl)以同样的方式培育并加入碳酸钠水溶液接着测定吸光度。同时在两个各自的孔中处理每个浓度的试样、空白和对比(试验)。使用0.1M磷酸盐缓冲溶液(pH6.8,从KH2PO4和K2HPO4制备)作为缓冲剂。
根据在连续稀释的试样浓度[即100,50,25,12.5,6.25,…(μg/μl)]下的吸光度确定酶活性的50%抑制浓度[IC50(M)]。2)β-葡糖苷酶(杏仁,EC3.2.1.21)[在A.Kobayashi,Agr.Biol.Chem.,26,203-207(1962)中有描述]
将试样水溶液(1μl),0.33mM对-硝基苯基β-D-吡喃葡糖苷在缓冲剂中的溶液(98μl)和(0.4mg/ml)酶在缓冲剂中的溶液(1μl)引入每个孔中得到总体积100μl。在37℃培育45分钟后,加入1M碳酸钠水溶液(100μl)中止反应。然后测定由该酶反应释放的对-硝基苯酚的吸光度(A405)。为了得到空白吸光度(A空白),该试样(1μl)和该酶作用物(99μl)以同样方式培育并加入碳酸钠水溶液(100μl)接着测定吸光度。另一方面,为了得到对比吸光度(A对比),水(1μl),该酶作用物(98μl)和该酶溶液(1μl)以同样方式培育并加入碳酸钠水溶液(100μl)接着测定吸光度。使用0.1M乙酸缓冲溶液(pH5.0,由NaOAc和AcOH制备)作为缓冲剂。以与α-葡糖苷酶的情况下用的同样方法确定IC50。3)α-半乳糖苷酶(E.Coli.,EC.3.2.1.22)[在H.Suzuki,S-C.Liand Y-T.Li,J.Biol.Chem.,245,781-786(1970)中有描述]
在每一个孔中引入试样水溶液(1μl),缓冲剂(74μl),9.9mM对-硝基苯基α-D-吡喃半乳糖苷酶在缓冲剂中的溶液(20μl)和(50μg/ml)酶在缓冲剂中的溶液(5μl)得到总体积100μl。在室温(20-30℃)培育所得混合物15分钟后,加入0.2M硼酸盐缓冲剂(pH9.8,由NaOH和H3BO3制备,100μl)中止反应。然后测定由该酶反应释放的对-硝基苯酚的吸光度(A405)。该试样(1μl),该酶作用物(20μl)和该缓冲溶液(79μl)以同样的方式被培育并加0.2M硼酸盐缓冲溶液(100μl)接着测定吸光度得到空白吸光度(A空白)。另一方面,以同样的方式培育水(1μl),该酶作用物(20μl),该缓冲溶液(74μl)和该酶溶液(5μl)并加入该硼酸盐缓冲溶液(100μl)接着测定吸光度得到对比吸光度(A对比)。使用0.1M磷酸盐缓冲溶液(pH6.5,由KH2PO4和K2HPO4制备)作为酶反应缓冲剂。以与在α-葡糖苷酶情况下所用的同样方式确定IC50。4)β-半乳糖苷酶(E.Coli,EC.3.2.1.23][在G.R.Graven,E.Steers,Jr.and C.B.Anfinsen,J.Biol.Chem.,240,2468-2477(1965)中有描述]
在每个孔中引入试样水溶液(2μl),缓冲剂(153μl),20mM邻-硝基苯基β-D-吡喃半乳糖苷在缓冲剂中的溶液(25μl),100mM 2-巯基乙醇(10μl)和(10μg/ml)酶在缓冲剂中的溶液(10μl)得到总体积200μl。在室温(20-30℃)下培育30分钟后,测定该酶反应释放的邻-硝基苯酚的吸光度(A405)。以同样的方式培育该试样(2μl),该酶作用物(25μl),该缓冲溶液(163μl)和2-巯基乙醇(10μl),接着测定吸光度得到空白吸光度(A空白)。另一方面,以同样的方式培育水(2μl),该酶作用物(25μl),该缓冲溶液(153μl),2-巯基乙醇(10μl)和该酶溶液(10μl)接着测定吸光度得到对比吸光度(A对比)。使用50mM磷酸盐缓冲溶液(pH7.3,由KH2PO4和K2HPO4制备,含1.3mM MgCl2)作为酶反应缓冲剂。以与在α-葡糖苷酶情况下所用的同样方式确定IC50。5)β-半乳糖苷酶(牛肝,EC3.2.1.23)[在T.Aoyagi,T.Hazato,M.Kumagai,M.Hamada,T.Takeuchi and H.Umezawa,J.Antibiot.,28,1006-1008(1975)中有描述]
在每个孔中引入试样水溶液(2μl),缓冲剂(158μl),20mM邻-硝基苯基β-D-吡喃半乳糖苷在缓冲剂中的溶液(25μl),100mM 2-巯基乙醇(10μl)和(5mg/ml)酶在缓冲剂中的溶液(5μl)得到总体积200μl。在室温(20-30℃)培育30分钟后,测定该酶反应释放的邻-硝基苯酚的吸光度(A405)。以同样的方式培育该试样(2μl),该酶作用物(25μl),该缓冲溶液(163μl)和2-巯基乙醇(10μl),接着测定吸光度得到空白吸光度(A空白)。另一方面,以同样方式培育水(2μl),该酶作用物(25μl),该缓冲溶液(158μl),2-巯基乙醇(10μl)和该酶溶液(5μl)接着测定吸光度得到对比吸光度(A对比)。50mM磷酸盐缓冲溶液(pH7.3,由KH2PO4和K2HPO4制备,含有1.3mM MgCl2)用作该酶反应的缓冲剂。以与α-萄糖苷酶所用的同样方式确定IC50。
在表1中给出了结果。
表1:IC50(M)化合物 α-葡糖苷酶 α-葡糖苷酶 α-半乳糖苷酶 β-半乳糖 β-半乳糖苷酶
(发面酵母) (杏仁) (E.coli) 苷酶 (牛肝)
(E.coli)d 4.02×10-7 2.93×10-5 - - 3.63×10-41-1D 2.32×10-6 - - - 3.00×10-61-1L 2.93×10-8 - - - 1.53×10-41-2D 9.99×10-7 - - - 1.89 ×10-61-2L 5.0×10-12 - - - 5.17×10-52-2L 7.0×10-14 - … … …d-1 5.0×10-7 1.3×10-4 … … …d-2 1.7×10-8 3.0×10-6 … … …DNJ 9.19×10-5 1.47×10-4 … … …N-B-DNJ 8.0×10-5 5.0×10-4 … … …海藻糖胺5.0×10-4 9.9×10-6 … … …
注)-:超过3.0×10-4M
…:未测定
如上表所示,包括化合物(d)(表海藻糖胺),(d-1)和(d-2)和5个异脲化合物[即化合物(1-1L),(1-1D),(1-2L),(1-2D)和(2-2L)]的本发明化合物的α-葡糖苷酶抑制活性高达脱氧野尻霉素的102-2×109倍。还表明这些化合物对于α-葡糖苷酶是高度有特效的。此外,上述5个异脲化合物均比表海藻糖胺有更高的抑制活性和更高的α,β-选择性,这表明它们是非常有用的化合物。仲羟基参与(participates in)异脲键的化合物(1-1L)与叔羟基参与异脲键的化合物(1-2L)之间抑制活性方面的比较表明后一化合物活性略微高一些。这一事实假定相应于葡萄糖2-位的羟基也许提供作为质子给体的氢键键合到本研究中所用的酶上。再有,通过在其中引入苯基可以显著地提高化合物(d)的α,β-选择性。根据这一事实,假定疏水部分会极大地影响选择性。
实验实施例2
用实施例中得到的化合物(d),(1-2L)和(2-2L)及比较化合物海藻糖胺和N-丁基脱氧野尻霉素测定对α-葡糖苷酶I的抑制活性。
根据I.Neverova等人在Anal.Biochem.,222,190-196(1994)中的方法制备本文中使用的α-葡糖苷酶I(发面酵母)和其酶作用物(α-D-Glc1→2α-D-Glc1→3α-D-Glc-O(CH2)6COOCH3)。
用该文献所述的方法进行下面的测定。
也就是说,将25μl含有α-葡糖苷酶I的缓冲溶液加入到含有上述试样和酶作用物(α-D-Glcl→2α-D-Glc1→3α-D-Glc-O(CH2)6COOCH3)的微管中。在37℃培育1小时后,向其加入1.25M三盐酸化物缓冲溶液(pH7.6,25μl)中止反应。然后将反应混合物转移到微板上,接着加入250μl反应缓冲溶液[展开溶液;1M含有葡萄糖氧化酶(5单位/毫升,Sigma生产),辣根过氧化物酶(1红棓酚单位,Sigma生产)和邻-联(二)茴香胺二盐酸化物(40μg/ml)的三盐酸化物缓冲溶液(40μg/ml)(pH7.2)]。然后,在37℃培育该混合物30分钟或直至吸光度升高达到一平稳段。然后测定由该酶反应形成和氧化的邻-联(二)茴香胺的吸光度(A450-650)。重复上面的反应但既不加上述试样也不加上述酶作用物并测定光度作为空白。并且将上述反应缓冲溶液加入到已知浓度的D-葡萄糖中并培育所得混合物接着测定吸光度从而测定D-葡萄糖浓度和吸光度之间的关系。同时在两个孔中完成每一测定。
上面的测定是在酶作用物浓度范围(0.25-4.0mM)内和试样浓度范围在每个试样给出的IC50附近进行的。根据上述D-葡萄糖浓度与吸光度之间的关系,确定存在试样时由于α-葡糖苷酶的酶反应从酶作用物释放出的葡萄糖量。从这样得到的结果确定Km,然后从试样的量的Km值确定Ki。
表2给出了这些结果。如表2所示,本发明的化合物抑制α-葡糖苷酶I的酶活性。它们之中,化合物(2-2L)在活性方面可与已经研究并开发成为抗HIV试剂的N-丁基脱氧野尻霉素相比。因此预计该化合物(2-2L)也能用作抗HIV试剂。
表2
化合物 Ki (μM)
d 300
1-2L 51
2-2L 4.2
N-丁基脱氧野尻霉素 3.6
海藻糖胺 1500
实验实施例3
氨基环戊烷衍生物对HIV产生的抑制作用(HIV中和试验):
通过用人正常外部血液单核细胞(PBMC)病毒中和试验测定本发明化合物的HIV中和活性。
将在实施例中得到的化合物(d),(1-1L),(1-2L)和(2-2L)及比较化合物N-甲基脱氧野尻霉素(称之为N-mDNJ),N-丁基脱氧野尻霉素(称之为N-B-DNJ),海藻糖胺和叠氮胸苷(AZT)作为试样,每个的量为0.1μm和1μM,与TCID50100倍量(中等组织培养感染剂量)的HIVMN混合,然后在37℃培育60分钟。将混合物移至含有1×106已经用5μg植物血球凝集素(PHA)活化了24小时的PBMC的Eppendorf管中,在37℃水浴中摇动60分钟。用PHA的活化是通过在刺激介质中培育PBMC24小时进行的。该刺激介质是通过将谷氨酰胺(2mM),热失活的10%胎腓肠血清,0.01%PHA(Difco生产),抗INFα抗体(50单位/毫升,从Cosmobio购得),青霉素(50单位/毫升)和链霉素(50μg/ml)加入到RPMI1640介质中制备的。用PBS洗涤三次之后,将该细胞悬浮在1ml的生长介质中,然后将悬浮液移到培育管(A-S Nunc,Roskilde,Denmark)并培育7天。该生长介质是通过将谷氨酰胺(2μM),热失活的10%胎腓肠血清,T细胞生长因子(IL-2)(40单位/毫升,由Shionogi&Co.,Ltd,生产),抗INFα-抗体(50单位/毫升,从Cosmobio购得),青霉素(50单位/毫升)和链霉素(50μg/ml)加入到RPMI1640中制备的。然后用HIV-1p24抗原ELISA(Dinabot生产)测定在上层清液中产生的HIV[J.Immunol.,142,4248-4255(1989);J.Immunol.,148,2175-2180(1992)]并以抑制率(%)表示试样对HIV产生的抑制作用。
用于中和试验的HIVMN是通过将已经用5μg/ml PHA活化了7天的PBMC与其量为TCID50100倍的HIVMN(H9/HTLV-IIIMN,AIDSResearch and Reference Reagent Program,NIH,Rockvill,MD)培育7天并从培养物上层清液弃去细胞制备的,接着在使用前在-130℃保存。用PHA活化的PBMC滴定该HIVMN从而确定每毫升TCID50。
图1给出了这些结果。纵坐标表示每个试样的抑制程度,它是以通过将1μM AZT的抑制作用作为70%计算出的抑制率(%)表示的。如图1所示,本发明的化合物有抗HIV活性。
实验实施例4
氨基环戊烷衍生物对由HIV诱发的细胞融合(形成巨大细胞)的抑制作用:
向含有106Molt4细胞或106Molt4/HIV-IIIB细胞的介质中分别加入在实施例中得到的化合物(1-2L),(2-2L),(d),(d-2)和(d-1)及比较化合物N-mDNJ,N-B-DNJ和AZT用作试样,其量为0.1μm和1μm,接着培育3天。本文中使用的每一介质是通过将谷氨酰胺(2mM),热失活的10%胎腓肠血清,青霉素(50单位/毫升)和链霉素(50mg/ml)加入到RPMI1640介质中制备的。完成培育后,Molt4细胞与同样数量的Molt4/HIV-III B细胞混合并在37℃培育24小时。然后用多重筛分机(Coulter制造)测定细胞尺寸大小和细胞数。直径超过20μm的细胞称为由细胞融合形成的巨细胞。这样测定每个试样对细胞融合(形成巨细胞)的抑制率(%)。
图2给出了这些结果。纵坐标表示抑制率(%)。如图2所示,本发明化合物有抗HIV活性。
实验实施例5
如表1和2所示,它证明了本发明的化合物(d),(1-1L),(1-2L),(2-2L),(d-1)和(d-2)与已知具有抑制活性的脱氧野尻霉素相比,具有非常强的对α-葡糖苷酶的抑制活性。特别地,化合物(1-1L),(1-2L),和(2-2L)对β-葡糖苷酶有弱抑制活性,这表明这些化合物是对α-葡糖苷酶非常有特效的抑制剂。此外,在细胞毒性方面也检测了这些化合物。
制备了20mM化合物(d)水溶液,10mM化合物(1-1L)在50%DMSO中的溶液和10mM化合物(1-2L)在50%DMSO中的溶液,并将其用作实验试样。
使用12-孔微板(3.8平方厘米/孔,由Corning生产),在37℃、5%CO2、密度为2×105细胞/毫升/孔条件下,在含有10%胎腓肠血清(FCS)的Dulbecco改性的Eagle介质(由Gibco实验室生产)中培育B16黑素瘤细胞。24小时后,该介质用含有每个试样的Dulbecco改性的Eagle介质替换。在同样条件下继续培育24小时。然后通过加入EDTA收获该细胞,用PBS洗涤并用0.3%锥虫蓝溶液染色从而确定该细胞的存活率。
如表3所示,在测定的任何浓度范围内没有观察到细胞毒性。化合物(1-2L)甚至在高达50%α-葡糖苷酶抑制浓度(IC50)的10000倍浓度下也没有显示出细胞毒性,这意谓着它可以用作高度安全的药物。
表3
化合物 化合物浓度(μM)
100 50 10 5
d - - - -
1-1L - - - -
1-2L - - - -
通过用0.3%锥虫蓝溶液染色确定的存活率(对比:100%)。+:25%,±:50%,-:>95%。
尽管已经详细地并参考其具体的实施方案描述了本发明,但是显然本领域普通技术人员可以在不脱离其精神和范围的条件下作各种改变和修改。
Claims (16)
1.一种由式(1)表示的氨基环戊烷衍生物:其中R1表示H而R2表示CH2OH,或R1表示CH2OH而R2表示H;和R3表示取代的或未取代的芳基或具有1-10个碳原子的烷基,链烯基,炔基或羟烷基。
2.如权利要求1所述的氨基环戊烷衍生物,其中在用上面式(1)表示的氨基环戊烷衍生物中,R3表示取代的或未取代的芳基。
3.如权利要求2所述的氨基环戊烷衍生物,其中用上面式(1)表示的氨基环戊烷衍生物选自用下面结构式(1-1L)表示的(1S,5R,6S,7S,8R)-6-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇,用下面结构式(1-1D)表示的(1R,5S,6R,7R,8S)-6-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇,用下面结构式(1-2L)表示的(1S,5R,6S,7R,8S)-1-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇和用下面结构式(1-2D)表示的(1R,5S,6R,7S,8R)-1-羟甲基-3-苯氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇:
4.如权利要求1所述的氨基环戊烷衍生物,其中,在用上面式(1)表示的氨基环戊烷衍生物中,R3表示有1-10个碳原子的烷基。
5.如权利要求4所述的氨基环戊烷衍生物,其中用上面式(1)表示的氨基环戊烷衍生物选自用下面结构式(2-1L)表示的(1S,5R,6S,7S,8R)-6-羟甲基-3-丁氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇,用下面结构式(2-1D)表示的(1R,5S,6R,7R,8S)-6-羟甲基-3-丁氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇,用下面结构式(2-2L)表示的(1S,5R,6S,7R,8S)-1-羟甲基-3-丁氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇和用下面结构式(2-2D)表示的(1R,5S,6R,7S,8R)-1-羟甲基-3-丁氨基-2-噁-4-氮杂双环[3.3.0]辛-3-烯-6,7,8-三醇:
6.(3S,4R,5S,6R,7S)-7-乙酰胺-4,5,6-三-O-乙酰基-噁螺[2.4]庚烷-4,5,6-三醇,它是用下面结构式表示的化合物:
7.一种由式(2)表示的氨基环戊烷衍生物:其中R4和R5各自独立地表示H或取代的或未取代的芳基或具有1-10个碳原子的烷基,链烯基,炔基或羟烷基。
8.如权利要求7所述的氨基环戊烷衍生物,其中由上面式(2)表示的氨基环戊烷衍生物选自用下面结构式(d)表示的1L-(1,2,4,5/3)-5-氨基-1C-羟甲基-1,2,3,4-环戊烷四醇,用下面结构式(d-1)表示的1L-(1,2,4,5/3)-5-二丁氨基-1-羟甲基-1,2,3,4-环戊烷四醇和用下面结构式(d-2)表示的1L-(1,2,4,5/3)-5-丁氨基-1-羟甲基-1,2,3,4-环戊烷四醇:
9. 1D-(1,2,4,5/3)-5-氨基-1C-羟甲基-1,2,3,4-环戊烷四醇,它是由下面结构式表示的化合物:
10.N-[2-羟甲基-2,3,4,5-四羟基环戊基]-N′-苯基硫脲,它是由下面结构式表示的化合物:
11.一种含有如权利要求1或7所述的氨基环戊烷衍生物作为活性组份的糖苷酶抑制剂。
12.如权利要求11所述的糖苷酶抑制剂。其中所述的糖苷酶抑制剂是α-葡糖苷酶抑制剂。
13.如权利要求12所述的糖苷酶抑制剂,其中所述的活性组份是如权利要求2,4或8中所述的氨基环戊烷衍生物。
14.如权利要求13所述的糖苷酶抑制剂,其中所述的活性组份是如权利要求3或5中所述的氨基环戊烷衍生物。
15.一种制备如权利要求6所述化合物的方法,包括在溶液状态下氧化由下面结构式表示的化合物:
16.一种制备如权利要求1所述的氨基环戊烷衍生物的方法,包括将式(3)表示的硫脲化合物转变成环异脲: 其中R1,R2和R3的定义与上面(1)所述的相同。
其中R1,R2和R3的定义与上面(1)所述的相同。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP31126494 | 1994-11-22 | ||
| JP311264/1994 | 1994-11-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1131672A true CN1131672A (zh) | 1996-09-25 |
Family
ID=18015053
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN95121896A Pending CN1131672A (zh) | 1994-11-22 | 1995-11-21 | 氨基环戊烷衍生物 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US5684026A (zh) |
| EP (1) | EP0713873B1 (zh) |
| KR (1) | KR960017649A (zh) |
| CN (1) | CN1131672A (zh) |
| AU (1) | AU3900995A (zh) |
| CA (1) | CA2163503A1 (zh) |
| DE (1) | DE69529799T2 (zh) |
| HU (1) | HUT77355A (zh) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0020331D0 (en) * | 2000-08-17 | 2000-10-04 | Imperial College | Enzyme |
| US7480854B2 (en) * | 2001-10-02 | 2009-01-20 | International Business Machines Corporation | Data conversion system and method |
| AU2009276223B2 (en) * | 2008-08-01 | 2015-04-02 | Simon Fraser University | Selective glycosidase inhibitors and uses thereof |
| KR101074954B1 (ko) * | 2010-01-27 | 2011-10-18 | 재단법인 경기과학기술진흥원 | (1s,2s,3s,4s)―5―아미노사이클로펜탄―1,2,3,4―테트라올을 포함하는 항비만 조성물 |
| ES2589164T3 (es) | 2010-12-23 | 2016-11-10 | Alectos Therapeutics Inc. | Inhibidores selectivos de glucosidasas y usos de los mismos |
| EP2691407B1 (en) | 2011-03-31 | 2017-02-22 | Alectos Therapeutics Inc. | Selective glycosidase inhibitors and uses thereof |
| KR101303963B1 (ko) * | 2011-04-08 | 2013-09-05 | 재단법인 경기과학기술진흥원 | (1r,2r,3r)-3-아미노사이클로펜탄-1,2-디올을 포함하는 항알러지용 조성물 |
| US9701693B2 (en) | 2011-06-27 | 2017-07-11 | Alectos Therapeutics Inc. | Selective glycosidase inhibitors and uses thereof |
| EP2890676B1 (en) | 2012-08-31 | 2018-12-05 | Alectos Therapeutics Inc. | Glycosidase inhibitors and uses thereof |
| US9809537B2 (en) | 2012-08-31 | 2017-11-07 | Alectos Therapeutics Inc. | Glycosidase inhibitors and uses thereof |
| EP2914604A4 (en) | 2012-10-31 | 2016-07-20 | Alectos Therapeutics Inc | GLYCOSIDASE INHIBITORS AND ASSOCIATED APPLICATIONS |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CZ51795A3 (en) * | 1991-02-15 | 1996-09-11 | Sankyo Co | 2-AMINO-4-(HYDROXYMETHYL)-3A,5,6,6A-TETRAHYDRO-4H-CYCLOPENT/d/OXAZOLE- -4,5,6-TRIOL, PROCESS OF ITS PREPARATION AND PRODUCTION MICRO-ORGANISMS |
| JPH06100549A (ja) * | 1992-07-17 | 1994-04-12 | Sankyo Co Ltd | 2−アミノオキサゾリン誘導体 |
-
1995
- 1995-11-21 CN CN95121896A patent/CN1131672A/zh active Pending
- 1995-11-22 HU HU9503339A patent/HUT77355A/hu unknown
- 1995-11-22 DE DE69529799T patent/DE69529799T2/de not_active Expired - Lifetime
- 1995-11-22 CA CA002163503A patent/CA2163503A1/en not_active Abandoned
- 1995-11-22 KR KR1019950042699A patent/KR960017649A/ko not_active Application Discontinuation
- 1995-11-22 US US08/561,947 patent/US5684026A/en not_active Expired - Lifetime
- 1995-11-22 AU AU39009/95A patent/AU3900995A/en not_active Abandoned
- 1995-11-22 EP EP95118369A patent/EP0713873B1/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| DE69529799D1 (de) | 2003-04-10 |
| CA2163503A1 (en) | 1996-05-23 |
| US5684026A (en) | 1997-11-04 |
| EP0713873A3 (en) | 1999-04-07 |
| HUT77355A (hu) | 1998-03-30 |
| EP0713873A2 (en) | 1996-05-29 |
| KR960017649A (ko) | 1996-06-17 |
| HU9503339D0 (en) | 1996-01-29 |
| DE69529799T2 (de) | 2003-11-13 |
| AU3900995A (en) | 1996-05-30 |
| EP0713873B1 (en) | 2003-03-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6040157B2 (ja) | セレンテラジン誘導体およびその使用方法 | |
| CN1131672A (zh) | 氨基环戊烷衍生物 | |
| CN1582277A (zh) | 用作糖原合酶激酶3β抑制剂的酰胺衍生物 | |
| CN1185212C (zh) | 水杨酸酰胺衍生物 | |
| CN1151209A (zh) | 用双-二阳离子芳基呋喃荧光检测核酸及细胞骨架成分的方法 | |
| CN88102182A (zh) | 4-脱甲氧基-4-氨基蒽环类的制备方法 | |
| CN88102932A (zh) | 新的嘌呤取代的氧杂环丁烷类化合物 | |
| CN1024197C (zh) | 表鬼臼毒吡喃葡糖甙的酰化衍生物的制备方法 | |
| CN1525962A (zh) | 广谱的2-(取代-氨基)-苯并噻唑磺酰胺hiv蛋白酶抑制剂 | |
| CN1108658A (zh) | 雪花胺衍生物及其制备方法和其作为药剂的用途 | |
| CN1027371C (zh) | 二萜内酯化合物的合成方法 | |
| CN1058016C (zh) | 氨基糖硫酸酯及其药学上可用的盐、其制备方法及其用途 | |
| CN1020610C (zh) | 取代的苯并嗪并利福霉素的制备方法 | |
| CN1278825A (zh) | 可用于稳定微管的珊瑚素和五加苷素 | |
| CN1155601C (zh) | 二氧杂环戊二烯-6(5aH)-酮化合物、其制备方法及其药物组合物 | |
| CN1636006A (zh) | 广谱2-(取代的-氨基)-苯并噁唑磺酰胺hiv蛋白酶抑制剂 | |
| CN1990482A (zh) | 一类新型的二氢黄酮醇类化合物及其制备方法和用途 | |
| CN1668605A (zh) | 取代的苯并异孔噁唑磺酰胺广谱hiv蛋白酶抑制剂 | |
| CN1030079A (zh) | 制备2-甲氧基甲基-青霉烯衍生物的方法 | |
| CN1721416A (zh) | 取代亚甲基吡喃酮类衍生物及其制备方法和用途 | |
| CN1028998C (zh) | 唾液酸糖基胆甾醇制备方法 | |
| CN1028531C (zh) | 1-氧杂-2-氧代-8-氮杂螺[4,5]癸烷衍生物及其盐的制备方法 | |
| CN1663955A (zh) | 吡啶类鬼臼毒素化合物及其制备方法和在制备杀虫剂中的应用 | |
| CN1192999C (zh) | 用作葡萄糖-6-磷酸移位酶抑制剂的芳族二酮基衍生物 | |
| CN1018646B (zh) | 苷的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C01 | Deemed withdrawal of patent application (patent law 1993) | ||
| WD01 | Invention patent application deemed withdrawn after publication |