CN112168972A - 使用pde7抑制剂治疗成瘾和冲动控制障碍 - Google Patents
使用pde7抑制剂治疗成瘾和冲动控制障碍 Download PDFInfo
- Publication number
- CN112168972A CN112168972A CN202011054538.2A CN202011054538A CN112168972A CN 112168972 A CN112168972 A CN 112168972A CN 202011054538 A CN202011054538 A CN 202011054538A CN 112168972 A CN112168972 A CN 112168972A
- Authority
- CN
- China
- Prior art keywords
- formula
- alkyl
- substituted
- pde7
- addiction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 206010012335 Dependence Diseases 0.000 title claims abstract description 128
- 238000011282 treatment Methods 0.000 title claims abstract description 42
- 208000030990 Impulse-control disease Diseases 0.000 title claims abstract description 31
- 208000026331 Disruptive, Impulse Control, and Conduct disease Diseases 0.000 title claims abstract description 23
- 239000002606 phosphodiesterase VII inhibitor Substances 0.000 title claims description 255
- 229940123304 Phosphodiesterase 7 inhibitor Drugs 0.000 title claims description 254
- 239000003112 inhibitor Substances 0.000 claims abstract description 34
- 108010037622 Type 7 Cyclic Nucleotide Phosphodiesterases Proteins 0.000 claims abstract description 12
- 102000010984 Type 7 Cyclic Nucleotide Phosphodiesterases Human genes 0.000 claims abstract description 12
- 239000003795 chemical substances by application Substances 0.000 claims description 134
- 238000000034 method Methods 0.000 claims description 105
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 claims description 102
- 230000000694 effects Effects 0.000 claims description 100
- 150000001875 compounds Chemical class 0.000 claims description 77
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 71
- 101001117266 Homo sapiens cAMP-specific 3',5'-cyclic phosphodiesterase 7B Proteins 0.000 claims description 69
- 102100024232 cAMP-specific 3',5'-cyclic phosphodiesterase 7B Human genes 0.000 claims description 66
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 claims description 59
- 229960002715 nicotine Drugs 0.000 claims description 59
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 claims description 59
- 230000002401 inhibitory effect Effects 0.000 claims description 58
- 230000006399 behavior Effects 0.000 claims description 51
- 229960003638 dopamine Drugs 0.000 claims description 51
- 208000032841 Bulimia Diseases 0.000 claims description 41
- 208000014679 binge eating disease Diseases 0.000 claims description 41
- 239000003136 dopamine receptor stimulating agent Substances 0.000 claims description 38
- 231100000867 compulsive behavior Toxicity 0.000 claims description 31
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 31
- 230000001575 pathological effect Effects 0.000 claims description 31
- 206010004716 Binge eating Diseases 0.000 claims description 30
- 210000004556 brain Anatomy 0.000 claims description 28
- 235000013305 food Nutrition 0.000 claims description 26
- 239000003368 psychostimulant agent Substances 0.000 claims description 26
- 229940005501 dopaminergic agent Drugs 0.000 claims description 23
- 239000000126 substance Substances 0.000 claims description 23
- 239000008194 pharmaceutical composition Substances 0.000 claims description 20
- 102000004190 Enzymes Human genes 0.000 claims description 19
- 108090000790 Enzymes Proteins 0.000 claims description 19
- 208000035475 disorder Diseases 0.000 claims description 18
- 208000010235 Food Addiction Diseases 0.000 claims description 17
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 claims description 15
- 229940127450 Opioid Agonists Drugs 0.000 claims description 15
- 206010006550 Bulimia nervosa Diseases 0.000 claims description 13
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 claims description 13
- 230000002255 enzymatic effect Effects 0.000 claims description 13
- 230000002829 reductive effect Effects 0.000 claims description 12
- 229940125717 barbiturate Drugs 0.000 claims description 10
- 208000001613 Gambling Diseases 0.000 claims description 9
- 206010034158 Pathological gambling Diseases 0.000 claims description 9
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical group OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 claims description 8
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 claims description 8
- 239000002243 precursor Substances 0.000 claims description 8
- 238000004891 communication Methods 0.000 claims description 7
- 206010023461 kleptomania Diseases 0.000 claims description 7
- 230000001568 sexual effect Effects 0.000 claims description 7
- 208000002271 trichotillomania Diseases 0.000 claims description 7
- 208000022531 anorexia Diseases 0.000 claims description 6
- 235000019788 craving Nutrition 0.000 claims description 6
- 206010061428 decreased appetite Diseases 0.000 claims description 6
- FASDKYOPVNHBLU-ZETCQYMHSA-N pramipexole Chemical compound C1[C@@H](NCCC)CCC2=C1SC(N)=N2 FASDKYOPVNHBLU-ZETCQYMHSA-N 0.000 claims description 6
- 229960003089 pramipexole Drugs 0.000 claims description 6
- VMWNQDUVQKEIOC-CYBMUJFWSA-N apomorphine Chemical compound C([C@H]1N(C)CC2)C3=CC=C(O)C(O)=C3C3=C1C2=CC=C3 VMWNQDUVQKEIOC-CYBMUJFWSA-N 0.000 claims description 5
- 229960004046 apomorphine Drugs 0.000 claims description 5
- 229940049706 benzodiazepine Drugs 0.000 claims description 5
- 229960002802 bromocriptine Drugs 0.000 claims description 5
- OZVBMTJYIDMWIL-AYFBDAFISA-N bromocriptine Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 OZVBMTJYIDMWIL-AYFBDAFISA-N 0.000 claims description 5
- 229960004502 levodopa Drugs 0.000 claims description 5
- 230000009467 reduction Effects 0.000 claims description 5
- GDFGTRDCCWFXTG-SCTDSRPQSA-N (3r,4ar,10as)-3-(diethylsulfamoylamino)-6-hydroxy-1-propyl-3,4,4a,5,10,10a-hexahydro-2h-benzo[g]quinoline Chemical compound C1=CC=C2C[C@@H]3N(CCC)C[C@H](NS(=O)(=O)N(CC)CC)C[C@H]3CC2=C1O GDFGTRDCCWFXTG-SCTDSRPQSA-N 0.000 claims description 4
- OVSKIKFHRZPJSS-UHFFFAOYSA-N 2,4-D Chemical compound OC(=O)COC1=CC=C(Cl)C=C1Cl OVSKIKFHRZPJSS-UHFFFAOYSA-N 0.000 claims description 4
- KORNTPPJEAJQIU-KJXAQDMKSA-N Cabaser Chemical compound C1=CC([C@H]2C[C@H](CN(CC=C)[C@@H]2C2)C(=O)N(CCCN(C)C)C(=O)NCC)=C3C2=CNC3=C1 KORNTPPJEAJQIU-KJXAQDMKSA-N 0.000 claims description 4
- 102100020802 D(1A) dopamine receptor Human genes 0.000 claims description 4
- 101000931925 Homo sapiens D(1A) dopamine receptor Proteins 0.000 claims description 4
- BKRGVLQUQGGVSM-KBXCAEBGSA-N Revanil Chemical compound C1=CC(C=2[C@H](N(C)C[C@H](C=2)NC(=O)N(CC)CC)C2)=C3C2=CNC3=C1 BKRGVLQUQGGVSM-KBXCAEBGSA-N 0.000 claims description 4
- 229960004596 cabergoline Drugs 0.000 claims description 4
- 229960004205 carbidopa Drugs 0.000 claims description 4
- 229940125904 compound 1 Drugs 0.000 claims description 4
- 229940125782 compound 2 Drugs 0.000 claims description 4
- 229940126214 compound 3 Drugs 0.000 claims description 4
- 239000000221 dopamine uptake inhibitor Substances 0.000 claims description 4
- 229960002724 fenoldopam Drugs 0.000 claims description 4
- TVURRHSHRRELCG-UHFFFAOYSA-N fenoldopam Chemical compound C1=CC(O)=CC=C1C1C2=CC(O)=C(O)C(Cl)=C2CCNC1 TVURRHSHRRELCG-UHFFFAOYSA-N 0.000 claims description 4
- 229960003587 lisuride Drugs 0.000 claims description 4
- 229960004851 pergolide Drugs 0.000 claims description 4
- 229960000924 quinagolide Drugs 0.000 claims description 4
- 229960001879 ropinirole Drugs 0.000 claims description 4
- UHSKFQJFRQCDBE-UHFFFAOYSA-N ropinirole Chemical compound CCCN(CCC)CCC1=CC=CC2=C1CC(=O)N2 UHSKFQJFRQCDBE-UHFFFAOYSA-N 0.000 claims description 4
- 229960003179 rotigotine Drugs 0.000 claims description 4
- KFQYTPMOWPVWEJ-INIZCTEOSA-N rotigotine Chemical compound CCCN([C@@H]1CC2=CC=CC(O)=C2CC1)CCC1=CC=CS1 KFQYTPMOWPVWEJ-INIZCTEOSA-N 0.000 claims description 4
- 206010037211 Psychomotor hyperactivity Diseases 0.000 claims description 3
- 230000004888 barrier function Effects 0.000 claims description 3
- 210000004369 blood Anatomy 0.000 claims description 3
- 239000008280 blood Substances 0.000 claims description 3
- 230000000149 penetrating effect Effects 0.000 claims description 2
- 102100024233 High affinity cAMP-specific 3',5'-cyclic phosphodiesterase 7A Human genes 0.000 claims 3
- 101001117267 Homo sapiens High affinity cAMP-specific 3',5'-cyclic phosphodiesterase 7A Proteins 0.000 claims 3
- TZFNLOMSOLWIDK-JTQLQIEISA-N carbidopa (anhydrous) Chemical compound NN[C@@](C(O)=O)(C)CC1=CC=C(O)C(O)=C1 TZFNLOMSOLWIDK-JTQLQIEISA-N 0.000 claims 2
- YEHCICAEULNIGD-MZMPZRCHSA-N pergolide Chemical compound C1=CC([C@H]2C[C@@H](CSC)CN([C@@H]2C2)CCC)=C3C2=CNC3=C1 YEHCICAEULNIGD-MZMPZRCHSA-N 0.000 claims 2
- 240000004308 marijuana Species 0.000 claims 1
- 239000003814 drug Substances 0.000 abstract description 157
- 229940124597 therapeutic agent Drugs 0.000 abstract description 84
- 101100407335 Dictyostelium discoideum pde7 gene Proteins 0.000 abstract 1
- 125000004432 carbon atom Chemical group C* 0.000 description 251
- 125000000217 alkyl group Chemical group 0.000 description 186
- -1 PDE10 Proteins 0.000 description 137
- 229910052717 sulfur Inorganic materials 0.000 description 106
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 92
- 229910052760 oxygen Inorganic materials 0.000 description 91
- 125000005842 heteroatom Chemical group 0.000 description 90
- 125000003118 aryl group Chemical group 0.000 description 88
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 87
- 125000001072 heteroaryl group Chemical group 0.000 description 84
- 229910052757 nitrogen Inorganic materials 0.000 description 81
- 229940079593 drug Drugs 0.000 description 72
- 229910052736 halogen Inorganic materials 0.000 description 67
- 150000002367 halogens Chemical class 0.000 description 64
- 125000000623 heterocyclic group Chemical group 0.000 description 59
- 125000000753 cycloalkyl group Chemical group 0.000 description 58
- 125000003342 alkenyl group Chemical group 0.000 description 53
- 229910052739 hydrogen Inorganic materials 0.000 description 53
- 239000001257 hydrogen Substances 0.000 description 52
- 229960003920 cocaine Drugs 0.000 description 46
- 125000000304 alkynyl group Chemical group 0.000 description 44
- 125000005843 halogen group Chemical group 0.000 description 43
- 150000003254 radicals Chemical class 0.000 description 43
- 230000035882 stress Effects 0.000 description 43
- 101710135349 Venom phosphodiesterase Proteins 0.000 description 41
- 125000003545 alkoxy group Chemical group 0.000 description 37
- 239000000935 antidepressant agent Substances 0.000 description 37
- 229940005513 antidepressants Drugs 0.000 description 36
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 36
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 34
- 239000003401 opiate antagonist Substances 0.000 description 32
- 239000005557 antagonist Substances 0.000 description 31
- 125000001188 haloalkyl group Chemical group 0.000 description 31
- 125000001424 substituent group Chemical group 0.000 description 31
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 30
- 208000011117 substance-related disease Diseases 0.000 description 29
- 125000004093 cyano group Chemical group *C#N 0.000 description 28
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 28
- 241000700159 Rattus Species 0.000 description 27
- 239000001961 anticonvulsive agent Substances 0.000 description 27
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 27
- 239000002464 receptor antagonist Substances 0.000 description 23
- 229940044551 receptor antagonist Drugs 0.000 description 23
- 125000004433 nitrogen atom Chemical group N* 0.000 description 22
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 description 21
- 230000001430 anti-depressive effect Effects 0.000 description 20
- 235000005911 diet Nutrition 0.000 description 20
- 230000005764 inhibitory process Effects 0.000 description 20
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 description 20
- 229960003086 naltrexone Drugs 0.000 description 20
- 239000004031 partial agonist Substances 0.000 description 20
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 20
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 19
- 229910052799 carbon Inorganic materials 0.000 description 19
- 230000037213 diet Effects 0.000 description 19
- 241000218236 Cannabis Species 0.000 description 18
- 101100296720 Dictyostelium discoideum Pde4 gene Proteins 0.000 description 18
- 101100082610 Plasmodium falciparum (isolate 3D7) PDEdelta gene Proteins 0.000 description 18
- 230000027455 binding Effects 0.000 description 18
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 18
- 108090000623 proteins and genes Proteins 0.000 description 18
- 125000004438 haloalkoxy group Chemical group 0.000 description 17
- 229940127240 opiate Drugs 0.000 description 17
- GVGLGOZIDCSQPN-PVHGPHFFSA-N Heroin Chemical compound O([C@H]1[C@H](C=C[C@H]23)OC(C)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4OC(C)=O GVGLGOZIDCSQPN-PVHGPHFFSA-N 0.000 description 16
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 description 16
- 229960001736 buprenorphine Drugs 0.000 description 16
- 239000000203 mixture Substances 0.000 description 16
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 16
- 125000002252 acyl group Chemical group 0.000 description 15
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 15
- 125000000392 cycloalkenyl group Chemical group 0.000 description 15
- 229960002069 diamorphine Drugs 0.000 description 15
- 230000007613 environmental effect Effects 0.000 description 15
- 230000001965 increasing effect Effects 0.000 description 15
- 229960005181 morphine Drugs 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- 229920006395 saturated elastomer Polymers 0.000 description 15
- 230000003474 anti-emetic effect Effects 0.000 description 14
- 239000002111 antiemetic agent Substances 0.000 description 14
- 229960003965 antiepileptics Drugs 0.000 description 14
- 229960001058 bupropion Drugs 0.000 description 14
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 description 14
- 239000002552 dosage form Substances 0.000 description 14
- 206010013663 drug dependence Diseases 0.000 description 14
- 235000005686 eating Nutrition 0.000 description 14
- 230000007937 eating Effects 0.000 description 14
- 239000003887 narcotic antagonist Substances 0.000 description 14
- 230000001225 therapeutic effect Effects 0.000 description 14
- 201000010099 disease Diseases 0.000 description 13
- 230000020595 eating behavior Effects 0.000 description 13
- 229960004756 ethanol Drugs 0.000 description 13
- 125000004043 oxo group Chemical group O=* 0.000 description 13
- 125000003367 polycyclic group Chemical group 0.000 description 13
- 102000004169 proteins and genes Human genes 0.000 description 13
- 201000009032 substance abuse Diseases 0.000 description 13
- USSIQXCVUWKGNF-UHFFFAOYSA-N 6-(dimethylamino)-4,4-diphenylheptan-3-one Chemical compound C=1C=CC=CC=1C(CC(C)N(C)C)(C(=O)CC)C1=CC=CC=C1 USSIQXCVUWKGNF-UHFFFAOYSA-N 0.000 description 12
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 12
- 125000003710 aryl alkyl group Chemical group 0.000 description 12
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 12
- 229960001797 methadone Drugs 0.000 description 12
- 239000003402 opiate agonist Substances 0.000 description 12
- 239000001301 oxygen Substances 0.000 description 12
- 235000018102 proteins Nutrition 0.000 description 12
- 150000003839 salts Chemical class 0.000 description 12
- 210000004515 ventral tegmental area Anatomy 0.000 description 12
- 101100135868 Dictyostelium discoideum pde3 gene Proteins 0.000 description 11
- 206010013654 Drug abuse Diseases 0.000 description 11
- 206010020710 Hyperphagia Diseases 0.000 description 11
- 229940025084 amphetamine Drugs 0.000 description 11
- 125000004122 cyclic group Chemical group 0.000 description 11
- 230000019637 foraging behavior Effects 0.000 description 11
- 230000002431 foraging effect Effects 0.000 description 11
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 11
- 229940005483 opioid analgesics Drugs 0.000 description 11
- 238000011084 recovery Methods 0.000 description 11
- 230000000306 recurrent effect Effects 0.000 description 11
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 11
- 208000007848 Alcoholism Diseases 0.000 description 10
- 101100407340 Drosophila melanogaster Pde8 gene Proteins 0.000 description 10
- 101100407337 Mus musculus Pde8a gene Proteins 0.000 description 10
- KJADKKWYZYXHBB-XBWDGYHZSA-N Topiramic acid Chemical compound C1O[C@@]2(COS(N)(=O)=O)OC(C)(C)O[C@H]2[C@@H]2OC(C)(C)O[C@@H]21 KJADKKWYZYXHBB-XBWDGYHZSA-N 0.000 description 10
- 125000003158 alcohol group Chemical group 0.000 description 10
- 238000010171 animal model Methods 0.000 description 10
- 230000003556 anti-epileptic effect Effects 0.000 description 10
- 229960002428 fentanyl Drugs 0.000 description 10
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 10
- 125000002950 monocyclic group Chemical group 0.000 description 10
- 230000002265 prevention Effects 0.000 description 10
- 230000004044 response Effects 0.000 description 10
- 229960004394 topiramate Drugs 0.000 description 10
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 9
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 9
- 102000012289 Corticotropin-Releasing Hormone Human genes 0.000 description 9
- 108010022152 Corticotropin-Releasing Hormone Proteins 0.000 description 9
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 description 9
- JAQUASYNZVUNQP-USXIJHARSA-N Levorphanol Chemical compound C1C2=CC=C(O)C=C2[C@]23CCN(C)[C@H]1[C@@H]2CCCC3 JAQUASYNZVUNQP-USXIJHARSA-N 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 229940125683 antiemetic agent Drugs 0.000 description 9
- 150000001721 carbon Chemical group 0.000 description 9
- 230000001419 dependent effect Effects 0.000 description 9
- 229910052731 fluorine Inorganic materials 0.000 description 9
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 9
- 230000014509 gene expression Effects 0.000 description 9
- 230000036541 health Effects 0.000 description 9
- 229960003406 levorphanol Drugs 0.000 description 9
- 230000007774 longterm Effects 0.000 description 9
- 210000002569 neuron Anatomy 0.000 description 9
- 208000037920 primary disease Diseases 0.000 description 9
- 102000005962 receptors Human genes 0.000 description 9
- 108020003175 receptors Proteins 0.000 description 9
- GGCSSNBKKAUURC-UHFFFAOYSA-N sufentanil Chemical compound C1CN(CCC=2SC=CC=2)CCC1(COC)N(C(=O)CC)C1=CC=CC=C1 GGCSSNBKKAUURC-UHFFFAOYSA-N 0.000 description 9
- 229960004739 sufentanil Drugs 0.000 description 9
- 208000024891 symptom Diseases 0.000 description 9
- 102000009132 CB1 Cannabinoid Receptor Human genes 0.000 description 8
- 108010073366 CB1 Cannabinoid Receptor Proteins 0.000 description 8
- 101000957724 Catostomus commersonii Corticoliberin-1 Proteins 0.000 description 8
- UGJMXCAKCUNAIE-UHFFFAOYSA-N Gabapentin Chemical compound OC(=O)CC1(CN)CCCCC1 UGJMXCAKCUNAIE-UHFFFAOYSA-N 0.000 description 8
- 229940124639 Selective inhibitor Drugs 0.000 description 8
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 8
- 230000001684 chronic effect Effects 0.000 description 8
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 8
- AUZONCFQVSMFAP-UHFFFAOYSA-N disulfiram Chemical compound CCN(CC)C(=S)SSC(=S)N(CC)CC AUZONCFQVSMFAP-UHFFFAOYSA-N 0.000 description 8
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 8
- 229960001252 methamphetamine Drugs 0.000 description 8
- 229960000482 pethidine Drugs 0.000 description 8
- JZCPYUJPEARBJL-UHFFFAOYSA-N rimonabant Chemical compound CC=1C(C(=O)NN2CCCCC2)=NN(C=2C(=CC(Cl)=CC=2)Cl)C=1C1=CC=C(Cl)C=C1 JZCPYUJPEARBJL-UHFFFAOYSA-N 0.000 description 8
- 125000004434 sulfur atom Chemical group 0.000 description 8
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical group [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 7
- XADCESSVHJOZHK-UHFFFAOYSA-N Meperidine Chemical compound C=1C=CC=CC=1C1(C(=O)OCC)CCN(C)CC1 XADCESSVHJOZHK-UHFFFAOYSA-N 0.000 description 7
- BLGXFZZNTVWLAY-CCZXDCJGSA-N Yohimbine Natural products C1=CC=C2C(CCN3C[C@@H]4CC[C@@H](O)[C@H]([C@H]4C[C@H]33)C(=O)OC)=C3NC2=C1 BLGXFZZNTVWLAY-CCZXDCJGSA-N 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- 239000000556 agonist Substances 0.000 description 7
- 125000002947 alkylene group Chemical group 0.000 description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 7
- BLGXFZZNTVWLAY-UHFFFAOYSA-N beta-Yohimbin Natural products C1=CC=C2C(CCN3CC4CCC(O)C(C4CC33)C(=O)OC)=C3NC2=C1 BLGXFZZNTVWLAY-UHFFFAOYSA-N 0.000 description 7
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 239000000510 dopamine 1 receptor stimulating agent Substances 0.000 description 7
- 229960004242 dronabinol Drugs 0.000 description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 7
- 239000011737 fluorine Substances 0.000 description 7
- 229960004002 levetiracetam Drugs 0.000 description 7
- HPHUVLMMVZITSG-ZCFIWIBFSA-N levetiracetam Chemical compound CC[C@H](C(N)=O)N1CCCC1=O HPHUVLMMVZITSG-ZCFIWIBFSA-N 0.000 description 7
- 229960003015 rimonabant Drugs 0.000 description 7
- 239000011593 sulfur Substances 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- GQDDNDAYOVNZPG-SCYLSFHTSA-N yohimbine Chemical compound C1=CC=C[C]2C(CCN3C[C@@H]4CC[C@H](O)[C@@H]([C@H]4C[C@H]33)C(=O)OC)=C3N=C21 GQDDNDAYOVNZPG-SCYLSFHTSA-N 0.000 description 7
- 229960000317 yohimbine Drugs 0.000 description 7
- AADVZSXPNRLYLV-UHFFFAOYSA-N yohimbine carboxylic acid Natural products C1=CC=C2C(CCN3CC4CCC(C(C4CC33)C(O)=O)O)=C3NC2=C1 AADVZSXPNRLYLV-UHFFFAOYSA-N 0.000 description 7
- FELGMEQIXOGIFQ-CYBMUJFWSA-N (3r)-9-methyl-3-[(2-methylimidazol-1-yl)methyl]-2,3-dihydro-1h-carbazol-4-one Chemical compound CC1=NC=CN1C[C@@H]1C(=O)C(C=2C(=CC=CC=2)N2C)=C2CC1 FELGMEQIXOGIFQ-CYBMUJFWSA-N 0.000 description 6
- WRRSFOZOETZUPG-FFHNEAJVSA-N (4r,4ar,7s,7ar,12bs)-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7-ol;hydrate Chemical compound O.C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC WRRSFOZOETZUPG-FFHNEAJVSA-N 0.000 description 6
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 6
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 6
- HJWHHQIVUHOBQN-UHFFFAOYSA-N 9-chloro-5-phenyl-3-prop-2-enyl-1,2,4,5-tetrahydro-3-benzazepine-7,8-diol Chemical compound C1N(CC=C)CCC=2C(Cl)=C(O)C(O)=CC=2C1C1=CC=CC=C1 HJWHHQIVUHOBQN-UHFFFAOYSA-N 0.000 description 6
- 208000019901 Anxiety disease Diseases 0.000 description 6
- 102100024318 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1B Human genes 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- 101100135859 Dictyostelium discoideum regA gene Proteins 0.000 description 6
- 101001117089 Drosophila melanogaster Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1 Proteins 0.000 description 6
- 101001072031 Drosophila melanogaster Dual 3',5'-cyclic-AMP and -GMP phosphodiesterase 11 Proteins 0.000 description 6
- 208000001836 Firesetting Behavior Diseases 0.000 description 6
- 241000282412 Homo Species 0.000 description 6
- 101001117099 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1B Proteins 0.000 description 6
- 101100082606 Plasmodium falciparum (isolate 3D7) PDEbeta gene Proteins 0.000 description 6
- 101100135860 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) PDE2 gene Proteins 0.000 description 6
- 230000001154 acute effect Effects 0.000 description 6
- 239000000654 additive Substances 0.000 description 6
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 6
- 230000036506 anxiety Effects 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- 229910052801 chlorine Inorganic materials 0.000 description 6
- 229960004126 codeine Drugs 0.000 description 6
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Natural products C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 6
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 6
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 6
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 description 6
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 6
- 230000002996 emotional effect Effects 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 229960002464 fluoxetine Drugs 0.000 description 6
- 208000015046 intermittent explosive disease Diseases 0.000 description 6
- 230000007246 mechanism Effects 0.000 description 6
- 229960001785 mirtazapine Drugs 0.000 description 6
- RONZAEMNMFQXRA-UHFFFAOYSA-N mirtazapine Chemical compound C1C2=CC=CN=C2N2CCN(C)CC2C2=CC=CC=C21 RONZAEMNMFQXRA-UHFFFAOYSA-N 0.000 description 6
- 210000001009 nucleus accumben Anatomy 0.000 description 6
- 229960005343 ondansetron Drugs 0.000 description 6
- 201000004645 pyromania Diseases 0.000 description 6
- 125000003003 spiro group Chemical group 0.000 description 6
- 125000003107 substituted aryl group Chemical group 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 108091005471 CRHR1 Proteins 0.000 description 5
- 206010010904 Convulsion Diseases 0.000 description 5
- 102000003840 Opioid Receptors Human genes 0.000 description 5
- 108090000137 Opioid Receptors Proteins 0.000 description 5
- BRUQQQPBMZOVGD-XFKAJCMBSA-N Oxycodone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C BRUQQQPBMZOVGD-XFKAJCMBSA-N 0.000 description 5
- 208000002193 Pain Diseases 0.000 description 5
- 229910006074 SO2NH2 Inorganic materials 0.000 description 5
- 230000000996 additive effect Effects 0.000 description 5
- 125000003282 alkyl amino group Chemical group 0.000 description 5
- 150000001413 amino acids Chemical group 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 125000002619 bicyclic group Chemical group 0.000 description 5
- 239000003557 cannabinoid Substances 0.000 description 5
- 229930003827 cannabinoid Natural products 0.000 description 5
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 5
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 5
- 230000034994 death Effects 0.000 description 5
- 231100000517 death Toxicity 0.000 description 5
- LLPOLZWFYMWNKH-CMKMFDCUSA-N hydrocodone Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)CC(=O)[C@@H]1OC1=C2C3=CC=C1OC LLPOLZWFYMWNKH-CMKMFDCUSA-N 0.000 description 5
- 229960000240 hydrocodone Drugs 0.000 description 5
- WVLOADHCBXTIJK-YNHQPCIGSA-N hydromorphone Chemical compound O([C@H]1C(CC[C@H]23)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O WVLOADHCBXTIJK-YNHQPCIGSA-N 0.000 description 5
- 229960001410 hydromorphone Drugs 0.000 description 5
- 125000001041 indolyl group Chemical group 0.000 description 5
- 125000002757 morpholinyl group Chemical group 0.000 description 5
- UZHSEJADLWPNLE-GRGSLBFTSA-N naloxone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(O)C2=C5[C@@]13CCN4CC=C UZHSEJADLWPNLE-GRGSLBFTSA-N 0.000 description 5
- 229960004127 naloxone Drugs 0.000 description 5
- 125000001624 naphthyl group Chemical group 0.000 description 5
- 230000001537 neural effect Effects 0.000 description 5
- 239000002767 noradrenalin uptake inhibitor Substances 0.000 description 5
- 229960004013 normethadone Drugs 0.000 description 5
- WCJFBSYALHQBSK-UHFFFAOYSA-N normethadone Chemical compound C=1C=CC=CC=1C(CCN(C)C)(C(=O)CC)C1=CC=CC=C1 WCJFBSYALHQBSK-UHFFFAOYSA-N 0.000 description 5
- 235000020830 overeating Nutrition 0.000 description 5
- 229960002085 oxycodone Drugs 0.000 description 5
- JTJMJGYZQZDUJJ-UHFFFAOYSA-N phencyclidine Chemical compound C1CCCCN1C1(C=2C=CC=CC=2)CCCCC1 JTJMJGYZQZDUJJ-UHFFFAOYSA-N 0.000 description 5
- 125000004193 piperazinyl group Chemical group 0.000 description 5
- 125000003386 piperidinyl group Chemical group 0.000 description 5
- 230000003518 presynaptic effect Effects 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 108090000765 processed proteins & peptides Proteins 0.000 description 5
- 125000003226 pyrazolyl group Chemical group 0.000 description 5
- 125000004076 pyridyl group Chemical group 0.000 description 5
- 230000001105 regulatory effect Effects 0.000 description 5
- 125000003831 tetrazolyl group Chemical group 0.000 description 5
- LLPOLZWFYMWNKH-UHFFFAOYSA-N trans-dihydrocodeinone Natural products C1C(N(CCC234)C)C2CCC(=O)C3OC2=C4C1=CC=C2OC LLPOLZWFYMWNKH-UHFFFAOYSA-N 0.000 description 5
- TVYLLZQTGLZFBW-ZBFHGGJFSA-N (R,R)-tramadol Chemical compound COC1=CC=CC([C@]2(O)[C@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-ZBFHGGJFSA-N 0.000 description 4
- SJZRECIVHVDYJC-UHFFFAOYSA-N 4-hydroxybutyric acid Chemical compound OCCCC(O)=O SJZRECIVHVDYJC-UHFFFAOYSA-N 0.000 description 4
- 102100036321 5-hydroxytryptamine receptor 2A Human genes 0.000 description 4
- 101710138091 5-hydroxytryptamine receptor 2A Proteins 0.000 description 4
- 125000005330 8 membered heterocyclic group Chemical group 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- 229940122820 Cannabinoid receptor antagonist Drugs 0.000 description 4
- 244000025254 Cannabis sativa Species 0.000 description 4
- 235000012766 Cannabis sativa ssp. sativa var. sativa Nutrition 0.000 description 4
- 235000012765 Cannabis sativa ssp. sativa var. spontanea Nutrition 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- MKXZASYAUGDDCJ-SZMVWBNQSA-N LSM-2525 Chemical compound C1CCC[C@H]2[C@@]3([H])N(C)CC[C@]21C1=CC(OC)=CC=C1C3 MKXZASYAUGDDCJ-SZMVWBNQSA-N 0.000 description 4
- UEQUQVLFIPOEMF-UHFFFAOYSA-N Mianserin Chemical compound C1C2=CC=CC=C2N2CCN(C)CC2C2=CC=CC=C21 UEQUQVLFIPOEMF-UHFFFAOYSA-N 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- IDBPHNDTYPBSNI-UHFFFAOYSA-N N-(1-(2-(4-Ethyl-5-oxo-2-tetrazolin-1-yl)ethyl)-4-(methoxymethyl)-4-piperidyl)propionanilide Chemical compound C1CN(CCN2C(N(CC)N=N2)=O)CCC1(COC)N(C(=O)CC)C1=CC=CC=C1 IDBPHNDTYPBSNI-UHFFFAOYSA-N 0.000 description 4
- WJBLNOPPDWQMCH-MBPVOVBZSA-N Nalmefene Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=C)O)CC1)O)CC1CC1 WJBLNOPPDWQMCH-MBPVOVBZSA-N 0.000 description 4
- 206010057852 Nicotine dependence Diseases 0.000 description 4
- ONBWJWYUHXVEJS-ZTYRTETDSA-N Normorphine Chemical compound C([C@@H](NCC1)[C@@H]2C=C[C@@H]3O)C4=CC=C(O)C5=C4[C@@]21[C@H]3O5 ONBWJWYUHXVEJS-ZTYRTETDSA-N 0.000 description 4
- 208000008589 Obesity Diseases 0.000 description 4
- UQCNKQCJZOAFTQ-ISWURRPUSA-N Oxymorphone Chemical compound O([C@H]1C(CC[C@]23O)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O UQCNKQCJZOAFTQ-ISWURRPUSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- 229910006069 SO3H Inorganic materials 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 208000025569 Tobacco Use disease Diseases 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 201000007930 alcohol dependence Diseases 0.000 description 4
- 229960001391 alfentanil Drugs 0.000 description 4
- 125000003368 amide group Chemical group 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- 230000000202 analgesic effect Effects 0.000 description 4
- 229940035676 analgesics Drugs 0.000 description 4
- 239000000730 antalgic agent Substances 0.000 description 4
- 125000002393 azetidinyl group Chemical group 0.000 description 4
- 230000003542 behavioural effect Effects 0.000 description 4
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 4
- 230000033228 biological regulation Effects 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 239000003536 cannabinoid receptor antagonist Substances 0.000 description 4
- 229940065144 cannabinoids Drugs 0.000 description 4
- 125000002837 carbocyclic group Chemical group 0.000 description 4
- 238000005520 cutting process Methods 0.000 description 4
- CYQFCXCEBYINGO-IAGOWNOFSA-N delta1-THC Chemical compound C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@@H]21 CYQFCXCEBYINGO-IAGOWNOFSA-N 0.000 description 4
- 229960001985 dextromethorphan Drugs 0.000 description 4
- XLMALTXPSGQGBX-GCJKJVERSA-N dextropropoxyphene Chemical compound C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 XLMALTXPSGQGBX-GCJKJVERSA-N 0.000 description 4
- 229960004193 dextropropoxyphene Drugs 0.000 description 4
- 229960002563 disulfiram Drugs 0.000 description 4
- 230000003291 dopaminomimetic effect Effects 0.000 description 4
- 230000037406 food intake Effects 0.000 description 4
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 229960002870 gabapentin Drugs 0.000 description 4
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 4
- 125000002883 imidazolyl group Chemical group 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- 230000033001 locomotion Effects 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 229960003955 mianserin Drugs 0.000 description 4
- 230000036651 mood Effects 0.000 description 4
- 229960005297 nalmefene Drugs 0.000 description 4
- 210000001577 neostriatum Anatomy 0.000 description 4
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 4
- 229940127221 norepinephrine reuptake inhibitor Drugs 0.000 description 4
- 229950006134 normorphine Drugs 0.000 description 4
- 235000020824 obesity Nutrition 0.000 description 4
- 229960005118 oxymorphone Drugs 0.000 description 4
- XQYZDYMELSJDRZ-UHFFFAOYSA-N papaverine Chemical compound C1=C(OC)C(OC)=CC=C1CC1=NC=CC2=CC(OC)=C(OC)C=C12 XQYZDYMELSJDRZ-UHFFFAOYSA-N 0.000 description 4
- 229950010883 phencyclidine Drugs 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 230000001737 promoting effect Effects 0.000 description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- 229960004425 sibutramine Drugs 0.000 description 4
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 4
- 239000012453 solvate Substances 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 4
- 229940124530 sulfonamide Drugs 0.000 description 4
- 150000003456 sulfonamides Chemical class 0.000 description 4
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 4
- 230000005062 synaptic transmission Effects 0.000 description 4
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 125000000335 thiazolyl group Chemical group 0.000 description 4
- 125000001544 thienyl group Chemical group 0.000 description 4
- 125000003396 thiol group Chemical class [H]S* 0.000 description 4
- 229960004380 tramadol Drugs 0.000 description 4
- TVYLLZQTGLZFBW-GOEBONIOSA-N tramadol Natural products COC1=CC=CC([C@@]2(O)[C@@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-GOEBONIOSA-N 0.000 description 4
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 4
- YQYVFVRQLZMJKJ-JBBXEZCESA-N (+)-cyclazocine Chemical compound C([C@@]1(C)C2=CC(O)=CC=C2C[C@@H]2[C@@H]1C)CN2CC1CC1 YQYVFVRQLZMJKJ-JBBXEZCESA-N 0.000 description 3
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 3
- GJJFMKBJSRMPLA-HIFRSBDPSA-N (1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenyl-1-cyclopropanecarboxamide Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)N(CC)CC)C[C@@H]1CN GJJFMKBJSRMPLA-HIFRSBDPSA-N 0.000 description 3
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical compound C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- 206010001488 Aggression Diseases 0.000 description 3
- 235000005781 Avena Nutrition 0.000 description 3
- 244000075850 Avena orientalis Species 0.000 description 3
- 108090000994 Catalytic RNA Proteins 0.000 description 3
- 102000053642 Catalytic RNA Human genes 0.000 description 3
- 206010010219 Compulsions Diseases 0.000 description 3
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 description 3
- IJVCSMSMFSCRME-KBQPJGBKSA-N Dihydromorphine Chemical compound O([C@H]1[C@H](CC[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O IJVCSMSMFSCRME-KBQPJGBKSA-N 0.000 description 3
- 208000012661 Dyskinesia Diseases 0.000 description 3
- OGDVEMNWJVYAJL-LEPYJNQMSA-N Ethyl morphine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OCC OGDVEMNWJVYAJL-LEPYJNQMSA-N 0.000 description 3
- OGDVEMNWJVYAJL-UHFFFAOYSA-N Ethylmorphine Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OCC OGDVEMNWJVYAJL-UHFFFAOYSA-N 0.000 description 3
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 3
- 108010044467 Isoenzymes Proteins 0.000 description 3
- 229940123685 Monoamine oxidase inhibitor Drugs 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 229910003827 NRaRb Inorganic materials 0.000 description 3
- UIQMVEYFGZJHCZ-SSTWWWIQSA-N Nalorphine Chemical compound C([C@@H](N(CC1)CC=C)[C@@H]2C=C[C@@H]3O)C4=CC=C(O)C5=C4[C@@]21[C@H]3O5 UIQMVEYFGZJHCZ-SSTWWWIQSA-N 0.000 description 3
- 241000208125 Nicotiana Species 0.000 description 3
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 3
- 102000019315 Nicotinic acetylcholine receptors Human genes 0.000 description 3
- 108050006807 Nicotinic acetylcholine receptors Proteins 0.000 description 3
- PHVGLTMQBUFIQQ-UHFFFAOYSA-N Nortryptiline Chemical compound C1CC2=CC=CC=C2C(=CCCNC)C2=CC=CC=C21 PHVGLTMQBUFIQQ-UHFFFAOYSA-N 0.000 description 3
- 239000008896 Opium Substances 0.000 description 3
- 241000283984 Rodentia Species 0.000 description 3
- 229940121991 Serotonin and norepinephrine reuptake inhibitor Drugs 0.000 description 3
- CYQFCXCEBYINGO-UHFFFAOYSA-N THC Natural products C1=C(C)CCC2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3C21 CYQFCXCEBYINGO-UHFFFAOYSA-N 0.000 description 3
- AFCGFAGUEYAMAO-UHFFFAOYSA-N acamprosate Chemical compound CC(=O)NCCCS(O)(=O)=O AFCGFAGUEYAMAO-UHFFFAOYSA-N 0.000 description 3
- 229960004047 acamprosate Drugs 0.000 description 3
- 125000004442 acylamino group Chemical group 0.000 description 3
- 229960002430 atomoxetine Drugs 0.000 description 3
- VHGCDTVCOLNTBX-QGZVFWFLSA-N atomoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=CC=C1C VHGCDTVCOLNTBX-QGZVFWFLSA-N 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000005540 biological transmission Effects 0.000 description 3
- 239000004305 biphenyl Substances 0.000 description 3
- 235000010290 biphenyl Nutrition 0.000 description 3
- 230000008499 blood brain barrier function Effects 0.000 description 3
- 210000001218 blood-brain barrier Anatomy 0.000 description 3
- QWCRAEMEVRGPNT-UHFFFAOYSA-N buspirone Chemical compound C1C(=O)N(CCCCN2CCN(CC2)C=2N=CC=CN=2)C(=O)CC21CCCC2 QWCRAEMEVRGPNT-UHFFFAOYSA-N 0.000 description 3
- 229960002495 buspirone Drugs 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 229960001948 caffeine Drugs 0.000 description 3
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 210000003169 central nervous system Anatomy 0.000 description 3
- 238000007906 compression Methods 0.000 description 3
- 230000006835 compression Effects 0.000 description 3
- 230000001143 conditioned effect Effects 0.000 description 3
- 229950002213 cyclazocine Drugs 0.000 description 3
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 3
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 229960003914 desipramine Drugs 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- WDEFBBTXULIOBB-WBVHZDCISA-N dextilidine Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)OCC)CCC=C[C@H]1N(C)C WDEFBBTXULIOBB-WBVHZDCISA-N 0.000 description 3
- 229960003701 dextromoramide Drugs 0.000 description 3
- INUNXTSAACVKJS-OAQYLSRUSA-N dextromoramide Chemical compound C([C@@H](C)C(C(=O)N1CCCC1)(C=1C=CC=CC=1)C=1C=CC=CC=1)N1CCOCC1 INUNXTSAACVKJS-OAQYLSRUSA-N 0.000 description 3
- 235000001916 dieting Nutrition 0.000 description 3
- 230000037228 dieting effect Effects 0.000 description 3
- 239000003210 dopamine receptor blocking agent Substances 0.000 description 3
- 230000009977 dual effect Effects 0.000 description 3
- JYRBQCWXZNDERM-XIRDDKMYSA-N etazocine Chemical compound C1C2=CC=C(O)C=C2[C@]2(CC)[C@@H](CC)[C@H]1N(C)CC2 JYRBQCWXZNDERM-XIRDDKMYSA-N 0.000 description 3
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 3
- 229960004578 ethylmorphine Drugs 0.000 description 3
- 230000005284 excitation Effects 0.000 description 3
- 238000010304 firing Methods 0.000 description 3
- 235000012631 food intake Nutrition 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 125000002541 furyl group Chemical group 0.000 description 3
- 230000000848 glutamatergic effect Effects 0.000 description 3
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 3
- 239000010931 gold Substances 0.000 description 3
- 229910052737 gold Inorganic materials 0.000 description 3
- 230000003284 homeostatic effect Effects 0.000 description 3
- 230000000642 iatrogenic effect Effects 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 125000005956 isoquinolyl group Chemical group 0.000 description 3
- IMYHGORQCPYVBZ-NLFFAJNJSA-N lofentanil Chemical compound CCC(=O)N([C@@]1([C@@H](CN(CCC=2C=CC=CC=2)CC1)C)C(=O)OC)C1=CC=CC=C1 IMYHGORQCPYVBZ-NLFFAJNJSA-N 0.000 description 3
- 229950010274 lofentanil Drugs 0.000 description 3
- 208000020816 lung neoplasm Diseases 0.000 description 3
- 229950009131 metazocine Drugs 0.000 description 3
- YGSVZRIZCHZUHB-COLVAYQJSA-N metazocine Chemical compound C1C2=CC=C(O)C=C2[C@]2(C)CCN(C)[C@@]1([H])[C@@H]2C YGSVZRIZCHZUHB-COLVAYQJSA-N 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 229960000600 milnacipran Drugs 0.000 description 3
- 239000002899 monoamine oxidase inhibitor Substances 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- NETZHAKZCGBWSS-CEDHKZHLSA-N nalbuphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]1(O)CC[C@@H]3O)CN2CC1CCC1 NETZHAKZCGBWSS-CEDHKZHLSA-N 0.000 description 3
- 229960000805 nalbuphine Drugs 0.000 description 3
- 229960000938 nalorphine Drugs 0.000 description 3
- 239000002858 neurotransmitter agent Substances 0.000 description 3
- 229960002748 norepinephrine Drugs 0.000 description 3
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 3
- 229960001158 nortriptyline Drugs 0.000 description 3
- 102000039446 nucleic acids Human genes 0.000 description 3
- 108020004707 nucleic acids Proteins 0.000 description 3
- 150000007523 nucleic acids Chemical class 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 239000000014 opioid analgesic Substances 0.000 description 3
- 229960001027 opium Drugs 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 230000036407 pain Effects 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- VOKSWYLNZZRQPF-GDIGMMSISA-N pentazocine Chemical compound C1C2=CC=C(O)C=C2[C@@]2(C)[C@@H](C)[C@@H]1N(CC=C(C)C)CC2 VOKSWYLNZZRQPF-GDIGMMSISA-N 0.000 description 3
- 229960005301 pentazocine Drugs 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- ZQHYKVKNPWDQSL-KNXBSLHKSA-N phenazocine Chemical compound C([C@@]1(C)C2=CC(O)=CC=C2C[C@@H]2[C@@H]1C)CN2CCC1=CC=CC=C1 ZQHYKVKNPWDQSL-KNXBSLHKSA-N 0.000 description 3
- 229960000897 phenazocine Drugs 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 125000005493 quinolyl group Chemical group 0.000 description 3
- 229940044601 receptor agonist Drugs 0.000 description 3
- 239000000018 receptor agonist Substances 0.000 description 3
- 230000003578 releasing effect Effects 0.000 description 3
- 108091092562 ribozyme Proteins 0.000 description 3
- 238000002821 scintillation proximity assay Methods 0.000 description 3
- 230000000391 smoking effect Effects 0.000 description 3
- 231100000736 substance abuse Toxicity 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 125000001174 sulfone group Chemical group 0.000 description 3
- 210000000225 synapse Anatomy 0.000 description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 3
- 229960001402 tilidine Drugs 0.000 description 3
- JQSHBVHOMNKWFT-DTORHVGOSA-N varenicline Chemical compound C12=CC3=NC=CN=C3C=C2[C@H]2C[C@@H]1CNC2 JQSHBVHOMNKWFT-DTORHVGOSA-N 0.000 description 3
- 229960004751 varenicline Drugs 0.000 description 3
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 2
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 2
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 2
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 description 2
- QEDVGROSOZBGOZ-WXXKFALUSA-N (e)-but-2-enedioic acid;n-[2-[[2-hydroxy-3-(4-hydroxyphenoxy)propyl]amino]ethyl]morpholine-4-carboxamide Chemical compound OC(=O)\C=C\C(O)=O.C=1C=C(O)C=CC=1OCC(O)CNCCNC(=O)N1CCOCC1.C=1C=C(O)C=CC=1OCC(O)CNCCNC(=O)N1CCOCC1 QEDVGROSOZBGOZ-WXXKFALUSA-N 0.000 description 2
- LLSKXGRDUPMXLC-UHFFFAOYSA-N 1-phenylpiperidine Chemical class C1CCCCN1C1=CC=CC=C1 LLSKXGRDUPMXLC-UHFFFAOYSA-N 0.000 description 2
- SLRMQYXOBQWXCR-UHFFFAOYSA-N 2154-56-5 Chemical compound [CH2]C1=CC=CC=C1 SLRMQYXOBQWXCR-UHFFFAOYSA-N 0.000 description 2
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 2
- SHXWCVYOXRDMCX-UHFFFAOYSA-N 3,4-methylenedioxymethamphetamine Chemical compound CNC(C)CC1=CC=C2OCOC2=C1 SHXWCVYOXRDMCX-UHFFFAOYSA-N 0.000 description 2
- IYNWSQDZXMGGGI-NUEKZKHPSA-N 3-hydroxymorphinan Chemical compound C1CCC[C@H]2[C@H]3CC4=CC=C(O)C=C4[C@]21CCN3 IYNWSQDZXMGGGI-NUEKZKHPSA-N 0.000 description 2
- 239000003477 4 aminobutyric acid receptor stimulating agent Substances 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- WXGBMVAPOXRLDB-UHFFFAOYSA-N 6-(2-phenylethenyl)cyclohexa-2,4-dien-1-imine Chemical class N=C1C=CC=CC1C=CC1=CC=CC=C1 WXGBMVAPOXRLDB-UHFFFAOYSA-N 0.000 description 2
- 229930008281 A03AD01 - Papaverine Natural products 0.000 description 2
- 102000007527 Autoreceptors Human genes 0.000 description 2
- 108010071131 Autoreceptors Proteins 0.000 description 2
- 125000005865 C2-C10alkynyl group Chemical group 0.000 description 2
- 229940124802 CB1 antagonist Drugs 0.000 description 2
- 102000009135 CB2 Cannabinoid Receptor Human genes 0.000 description 2
- 108010073376 CB2 Cannabinoid Receptor Proteins 0.000 description 2
- 229940123158 Cannabinoid CB1 receptor antagonist Drugs 0.000 description 2
- 102000018208 Cannabinoid Receptor Human genes 0.000 description 2
- 108050007331 Cannabinoid receptor Proteins 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- GDLIGKIOYRNHDA-UHFFFAOYSA-N Clomipramine Chemical compound C1CC2=CC=C(Cl)C=C2N(CCCN(C)C)C2=CC=CC=C21 GDLIGKIOYRNHDA-UHFFFAOYSA-N 0.000 description 2
- 208000022497 Cocaine-Related disease Diseases 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- 206010010144 Completed suicide Diseases 0.000 description 2
- 208000020401 Depressive disease Diseases 0.000 description 2
- 229940121891 Dopamine receptor antagonist Drugs 0.000 description 2
- CYQFCXCEBYINGO-DLBZAZTESA-N Dronabinol Natural products C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@H]21 CYQFCXCEBYINGO-DLBZAZTESA-N 0.000 description 2
- 208000030814 Eating disease Diseases 0.000 description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 2
- FNELVJVBIYMIMC-UHFFFAOYSA-N Ethiprole Chemical compound N1=C(C#N)C(S(=O)CC)=C(N)N1C1=C(Cl)C=C(C(F)(F)F)C=C1Cl FNELVJVBIYMIMC-UHFFFAOYSA-N 0.000 description 2
- 208000019454 Feeding and Eating disease Diseases 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 208000004547 Hallucinations Diseases 0.000 description 2
- 229940122236 Histamine receptor antagonist Drugs 0.000 description 2
- 206010022998 Irritability Diseases 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 208000002720 Malnutrition Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 101000909851 Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) cAMP/cGMP dual specificity phosphodiesterase Rv0805 Proteins 0.000 description 2
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 2
- 150000001204 N-oxides Chemical class 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 108010040718 Neurokinin-1 Receptors Proteins 0.000 description 2
- 229910004727 OSO3H Inorganic materials 0.000 description 2
- 208000026251 Opioid-Related disease Diseases 0.000 description 2
- 229940099471 Phosphodiesterase inhibitor Drugs 0.000 description 2
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 2
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- QPCVHQBVMYCJOM-UHFFFAOYSA-N Propiverine Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(OCCC)C(=O)OC1CCN(C)CC1 QPCVHQBVMYCJOM-UHFFFAOYSA-N 0.000 description 2
- 241000700157 Rattus norvegicus Species 0.000 description 2
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 2
- 102100037346 Substance-P receptor Human genes 0.000 description 2
- 208000001871 Tachycardia Diseases 0.000 description 2
- 206010043903 Tobacco abuse Diseases 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 230000016571 aggressive behavior Effects 0.000 description 2
- 206010001584 alcohol abuse Diseases 0.000 description 2
- 208000025746 alcohol use disease Diseases 0.000 description 2
- 208000029650 alcohol withdrawal Diseases 0.000 description 2
- 229930013930 alkaloid Natural products 0.000 description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- 229960004538 alprazolam Drugs 0.000 description 2
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 2
- VIROVYVQCGLCII-UHFFFAOYSA-N amobarbital Chemical compound CC(C)CCC1(CC)C(=O)NC(=O)NC1=O VIROVYVQCGLCII-UHFFFAOYSA-N 0.000 description 2
- 229960002519 amoxapine Drugs 0.000 description 2
- QWGDMFLQWFTERH-UHFFFAOYSA-N amoxapine Chemical compound C12=CC(Cl)=CC=C2OC2=CC=CC=C2N=C1N1CCNCC1 QWGDMFLQWFTERH-UHFFFAOYSA-N 0.000 description 2
- 210000004727 amygdala Anatomy 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- YUENFNPLGJCNRB-UHFFFAOYSA-N anthracen-1-amine Chemical compound C1=CC=C2C=C3C(N)=CC=CC3=CC2=C1 YUENFNPLGJCNRB-UHFFFAOYSA-N 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 229940125681 anticonvulsant agent Drugs 0.000 description 2
- 230000000338 anxiogenic effect Effects 0.000 description 2
- 230000036528 appetite Effects 0.000 description 2
- 235000019789 appetite Nutrition 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 125000000732 arylene group Chemical group 0.000 description 2
- 210000004227 basal ganglia Anatomy 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 238000013542 behavioral therapy Methods 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 150000001557 benzodiazepines Chemical class 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- RDJGWRFTDZZXSM-RNWLQCGYSA-N benzylmorphine Chemical compound O([C@@H]1[C@]23CCN([C@H](C4)[C@@H]3C=C[C@@H]1O)C)C1=C2C4=CC=C1OCC1=CC=CC=C1 RDJGWRFTDZZXSM-RNWLQCGYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- IFKLAQQSCNILHL-QHAWAJNXSA-N butorphanol Chemical compound N1([C@@H]2CC3=CC=C(C=C3[C@@]3([C@]2(CCCC3)O)CC1)O)CC1CCC1 IFKLAQQSCNILHL-QHAWAJNXSA-N 0.000 description 2
- 229960001113 butorphanol Drugs 0.000 description 2
- 239000003555 cannabinoid 1 receptor antagonist Substances 0.000 description 2
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 2
- QTAOMKOIBXZKND-PPHPATTJSA-N carbidopa Chemical compound O.NN[C@@](C(O)=O)(C)CC1=CC=C(O)C(O)=C1 QTAOMKOIBXZKND-PPHPATTJSA-N 0.000 description 2
- 125000004181 carboxyalkyl group Chemical group 0.000 description 2
- 150000007942 carboxylates Chemical class 0.000 description 2
- YDSDEBIZUNNPOB-UHFFFAOYSA-N carfentanil Chemical compound C1CN(CCC=2C=CC=CC=2)CCC1(C(=O)OC)N(C(=O)CC)C1=CC=CC=C1 YDSDEBIZUNNPOB-UHFFFAOYSA-N 0.000 description 2
- 229950004689 carfentanil Drugs 0.000 description 2
- 238000005266 casting Methods 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 2
- 229960001076 chlorpromazine Drugs 0.000 description 2
- 229960004606 clomipramine Drugs 0.000 description 2
- 230000001447 compensatory effect Effects 0.000 description 2
- 239000003246 corticosteroid Substances 0.000 description 2
- 125000002993 cycloalkylene group Chemical group 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000002939 deleterious effect Effects 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 229960000632 dexamfetamine Drugs 0.000 description 2
- 229960003461 dezocine Drugs 0.000 description 2
- VTMVHDZWSFQSQP-VBNZEHGJSA-N dezocine Chemical compound C1CCCC[C@H]2CC3=CC=C(O)C=C3[C@]1(C)[C@H]2N VTMVHDZWSFQSQP-VBNZEHGJSA-N 0.000 description 2
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 235000014632 disordered eating Nutrition 0.000 description 2
- 229960002866 duloxetine Drugs 0.000 description 2
- 208000024732 dysthymic disease Diseases 0.000 description 2
- 230000006353 environmental stress Effects 0.000 description 2
- CAHCBJPUTCKATP-FAWZKKEFSA-N etorphine Chemical compound O([C@H]1[C@@]2(OC)C=C[C@@]34C[C@@H]2[C@](C)(O)CCC)C2=C5[C@]41CCN(C)[C@@H]3CC5=CC=C2O CAHCBJPUTCKATP-FAWZKKEFSA-N 0.000 description 2
- 229950004155 etorphine Drugs 0.000 description 2
- 230000002964 excitative effect Effects 0.000 description 2
- 235000019138 food restriction Nutrition 0.000 description 2
- 229930195712 glutamate Natural products 0.000 description 2
- 229960003878 haloperidol Drugs 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 210000002216 heart Anatomy 0.000 description 2
- 208000019622 heart disease Diseases 0.000 description 2
- 235000003642 hunger Nutrition 0.000 description 2
- 208000013403 hyperactivity Diseases 0.000 description 2
- 229960004801 imipramine Drugs 0.000 description 2
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 206010022437 insomnia Diseases 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 229940125425 inverse agonist Drugs 0.000 description 2
- IFKPLJWIEQBPGG-UHFFFAOYSA-N isomethadone Chemical compound C=1C=CC=CC=1C(C(C)CN(C)C)(C(=O)CC)C1=CC=CC=C1 IFKPLJWIEQBPGG-UHFFFAOYSA-N 0.000 description 2
- 229950009272 isomethadone Drugs 0.000 description 2
- 102000048260 kappa Opioid Receptors Human genes 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 229960001848 lamotrigine Drugs 0.000 description 2
- PYZRQGJRPPTADH-UHFFFAOYSA-N lamotrigine Chemical compound NC1=NC(N)=NN=C1C1=CC=CC(Cl)=C1Cl PYZRQGJRPPTADH-UHFFFAOYSA-N 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 229960002813 lofepramine Drugs 0.000 description 2
- SAPNXPWPAUFAJU-UHFFFAOYSA-N lofepramine Chemical compound C12=CC=CC=C2CCC2=CC=CC=C2N1CCCN(C)CC(=O)C1=CC=C(Cl)C=C1 SAPNXPWPAUFAJU-UHFFFAOYSA-N 0.000 description 2
- 229960004391 lorazepam Drugs 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 230000001071 malnutrition Effects 0.000 description 2
- 235000000824 malnutrition Nutrition 0.000 description 2
- 229960004090 maprotiline Drugs 0.000 description 2
- QSLMDECMDJKHMQ-GSXCWMCISA-N maprotiline Chemical compound C12=CC=CC=C2[C@@]2(CCCNC)C3=CC=CC=C3[C@@H]1CC2 QSLMDECMDJKHMQ-GSXCWMCISA-N 0.000 description 2
- FQXXSQDCDRQNQE-UHFFFAOYSA-N markiertes Thebain Natural products COC1=CC=C2C(N(CC3)C)CC4=CC=C(OC)C5=C4C23C1O5 FQXXSQDCDRQNQE-UHFFFAOYSA-N 0.000 description 2
- 230000010534 mechanism of action Effects 0.000 description 2
- 229960000365 meptazinol Drugs 0.000 description 2
- JLICHNCFTLFZJN-HNNXBMFYSA-N meptazinol Chemical compound C=1C=CC(O)=CC=1[C@@]1(CC)CCCCN(C)C1 JLICHNCFTLFZJN-HNNXBMFYSA-N 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 229960003793 midazolam Drugs 0.000 description 2
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 description 2
- 230000009456 molecular mechanism Effects 0.000 description 2
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 2
- 102000051367 mu Opioid Receptors Human genes 0.000 description 2
- 210000003061 neural cell Anatomy 0.000 description 2
- 230000000955 neuroendocrine Effects 0.000 description 2
- 229960004300 nicomorphine Drugs 0.000 description 2
- HNDXBGYRMHRUFN-CIVUWBIHSA-N nicomorphine Chemical compound O([C@H]1C=C[C@H]2[C@H]3CC=4C5=C(C(=CC=4)OC(=O)C=4C=NC=CC=4)O[C@@H]1[C@]52CCN3C)C(=O)C1=CC=CN=C1 HNDXBGYRMHRUFN-CIVUWBIHSA-N 0.000 description 2
- 229950011519 norlevorphanol Drugs 0.000 description 2
- 208000015380 nutritional deficiency disease Diseases 0.000 description 2
- 201000005040 opiate dependence Diseases 0.000 description 2
- 229960001789 papaverine Drugs 0.000 description 2
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 2
- 239000000816 peptidomimetic Substances 0.000 description 2
- YYPWGCZOLGTTER-MZMPZRCHSA-N pergolide Chemical compound C1=CC=C2[C@H]3C[C@@H](CSC)CN(CCC)[C@@H]3CC3=CN=C1[C]32 YYPWGCZOLGTTER-MZMPZRCHSA-N 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 150000002987 phenanthrenes Chemical class 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 230000036470 plasma concentration Effects 0.000 description 2
- 229950009416 polatuzumab vedotin Drugs 0.000 description 2
- 210000002442 prefrontal cortex Anatomy 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229960003510 propiverine Drugs 0.000 description 2
- 208000020016 psychiatric disease Diseases 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- 229960003770 reboxetine Drugs 0.000 description 2
- CBQGYUDMJHNJBX-RTBURBONSA-N reboxetine Chemical compound CCOC1=CC=CC=C1O[C@H](C=1C=CC=CC=1)[C@@H]1OCCNC1 CBQGYUDMJHNJBX-RTBURBONSA-N 0.000 description 2
- 239000002469 receptor inverse agonist Substances 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- HJORMJIFDVBMOB-UHFFFAOYSA-N rolipram Chemical compound COC1=CC=C(C2CC(=O)NC2)C=C1OC1CCCC1 HJORMJIFDVBMOB-UHFFFAOYSA-N 0.000 description 2
- 229950005741 rolipram Drugs 0.000 description 2
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 2
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 2
- 239000003772 serotonin uptake inhibitor Substances 0.000 description 2
- 210000002027 skeletal muscle Anatomy 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 239000003195 sodium channel blocking agent Substances 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 229940084026 sodium valproate Drugs 0.000 description 2
- AEQFSUDEHCCHBT-UHFFFAOYSA-M sodium valproate Chemical class [Na+].CCCC(C([O-])=O)CCC AEQFSUDEHCCHBT-UHFFFAOYSA-M 0.000 description 2
- 210000003523 substantia nigra Anatomy 0.000 description 2
- 125000000547 substituted alkyl group Chemical group 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 230000006794 tachycardia Effects 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- 150000003536 tetrazoles Chemical class 0.000 description 2
- FQXXSQDCDRQNQE-VMDGZTHMSA-N thebaine Chemical compound C([C@@H](N(CC1)C)C2=CC=C3OC)C4=CC=C(OC)C5=C4[C@@]21[C@H]3O5 FQXXSQDCDRQNQE-VMDGZTHMSA-N 0.000 description 2
- 229930003945 thebaine Natural products 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- 229960003991 trazodone Drugs 0.000 description 2
- PHLBKPHSAVXXEF-UHFFFAOYSA-N trazodone Chemical compound ClC1=CC=CC(N2CCN(CCCN3C(N4C=CC=CC4=N3)=O)CC2)=C1 PHLBKPHSAVXXEF-UHFFFAOYSA-N 0.000 description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 2
- 230000001960 triggered effect Effects 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 2
- 229960001930 valpromide Drugs 0.000 description 2
- OMOMUFTZPTXCHP-UHFFFAOYSA-N valpromide Chemical compound CCCC(C(N)=O)CCC OMOMUFTZPTXCHP-UHFFFAOYSA-N 0.000 description 2
- 229960004688 venlafaxine Drugs 0.000 description 2
- PNVNVHUZROJLTJ-UHFFFAOYSA-N venlafaxine Chemical compound C1=CC(OC)=CC=C1C(CN(C)C)C1(O)CCCCC1 PNVNVHUZROJLTJ-UHFFFAOYSA-N 0.000 description 2
- 108020001588 κ-opioid receptors Proteins 0.000 description 2
- 108020001612 μ-opioid receptors Proteins 0.000 description 2
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 description 1
- DBGIVFWFUFKIQN-UHFFFAOYSA-N (+-)-Fenfluramine Chemical compound CCNC(C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-UHFFFAOYSA-N 0.000 description 1
- AKNNEGZIBPJZJG-MSOLQXFVSA-N (-)-noscapine Chemical compound CN1CCC2=CC=3OCOC=3C(OC)=C2[C@@H]1[C@@H]1C2=CC=C(OC)C(OC)=C2C(=O)O1 AKNNEGZIBPJZJG-MSOLQXFVSA-N 0.000 description 1
- IGLYMJRIWWIQQE-QUOODJBBSA-N (1S,2R)-2-phenylcyclopropan-1-amine (1R,2S)-2-phenylcyclopropan-1-amine Chemical compound N[C@H]1C[C@@H]1C1=CC=CC=C1.N[C@@H]1C[C@H]1C1=CC=CC=C1 IGLYMJRIWWIQQE-QUOODJBBSA-N 0.000 description 1
- QDZOEBFLNHCSSF-PFFBOGFISA-N (2S)-2-[[(2R)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-1-[(2R)-2-amino-5-carbamimidamidopentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-N-[(2R)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]pentanediamide Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CCCNC(N)=N)C1=CC=CC=C1 QDZOEBFLNHCSSF-PFFBOGFISA-N 0.000 description 1
- YWPHCCPCQOJSGZ-LLVKDONJSA-N (2r)-2-[(2-ethoxyphenoxy)methyl]morpholine Chemical compound CCOC1=CC=CC=C1OC[C@@H]1OCCNC1 YWPHCCPCQOJSGZ-LLVKDONJSA-N 0.000 description 1
- JVLBPIPGETUEET-WIXLDOGYSA-O (3r,4r,4as,7ar,12bs)-3-(cyclopropylmethyl)-4a,9-dihydroxy-3-methyl-2,4,5,6,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-3-ium-7-one Chemical compound C([N@+]1(C)[C@@H]2CC=3C4=C(C(=CC=3)O)O[C@@H]3[C@]4([C@@]2(O)CCC3=O)CC1)C1CC1 JVLBPIPGETUEET-WIXLDOGYSA-O 0.000 description 1
- IMEOKOKCPWJLBY-RLTLFZQJSA-N (4r,4ar,7s,7ar,12bs)-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7,11-diol Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=C(O)C=C1OC IMEOKOKCPWJLBY-RLTLFZQJSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- 125000006645 (C3-C4) cycloalkyl group Chemical group 0.000 description 1
- WSEQXVZVJXJVFP-HXUWFJFHSA-N (R)-citalopram Chemical compound C1([C@@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-HXUWFJFHSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- CUJMCPPBTUATEJ-UHFFFAOYSA-N 1-(3-methyl-4-oxido-1-oxoquinoxalin-1-ium-2-yl)ethanone Chemical compound C1=CC=C2[N+](=O)C(C(=O)C)=C(C)N([O-])C2=C1 CUJMCPPBTUATEJ-UHFFFAOYSA-N 0.000 description 1
- HLCOZRNCPOJQOI-UHFFFAOYSA-N 1-chloro-1-propan-2-ylhydrazine Chemical compound CC(C)N(N)Cl HLCOZRNCPOJQOI-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- OPWDXUHOIPSREO-UHFFFAOYSA-N 1-propan-2-yl-1-(pyridin-3-ylmethyl)hydrazine Chemical compound C(C)(C)N(N)CC1=CN=CC=C1 OPWDXUHOIPSREO-UHFFFAOYSA-N 0.000 description 1
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Substances C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 description 1
- PDQRQJVPEFGVRK-UHFFFAOYSA-N 2,1,3-benzothiadiazole Chemical compound C1=CC=CC2=NSN=C21 PDQRQJVPEFGVRK-UHFFFAOYSA-N 0.000 description 1
- TXQPXJKRNHJWAX-UHFFFAOYSA-N 2-(3-aminopropylamino)ethylsulfanylphosphonic acid;trihydrate Chemical compound O.O.O.NCCCNCCSP(O)(O)=O TXQPXJKRNHJWAX-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- USPVLEIQIUNQGE-DBFLIVQGSA-N 2-[[(2s)-2-benzyl-3-[(3r,4r)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]propanoyl]amino]acetic acid;dihydrate Chemical compound O.O.C([C@@H](CN1C[C@@H]([C@](CC1)(C)C=1C=C(O)C=CC=1)C)C(=O)NCC(O)=O)C1=CC=CC=C1 USPVLEIQIUNQGE-DBFLIVQGSA-N 0.000 description 1
- SPCKHVPPRJWQRZ-UHFFFAOYSA-N 2-benzhydryloxy-n,n-dimethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 SPCKHVPPRJWQRZ-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- DVWTWOHVDUVPJV-JTQLQIEISA-N 2-n-[(1s)-1-(5-fluoropyrimidin-2-yl)ethyl]-7-methyl-4-n-(1-methylimidazol-4-yl)thieno[3,2-d]pyrimidine-2,4-diamine Chemical compound N([C@@H](C)C=1N=CC(F)=CN=1)C(N=C1C(C)=CSC1=1)=NC=1NC1=CN(C)C=N1 DVWTWOHVDUVPJV-JTQLQIEISA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- NOIIUHRQUVNIDD-UHFFFAOYSA-N 3-[[oxo(pyridin-4-yl)methyl]hydrazo]-N-(phenylmethyl)propanamide Chemical compound C=1C=CC=CC=1CNC(=O)CCNNC(=O)C1=CC=NC=C1 NOIIUHRQUVNIDD-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- QZHXKQKKEBXYRG-UHFFFAOYSA-N 4-n-(4-aminophenyl)benzene-1,4-diamine Chemical compound C1=CC(N)=CC=C1NC1=CC=C(N)C=C1 QZHXKQKKEBXYRG-UHFFFAOYSA-N 0.000 description 1
- MXUNKHLAEDCYJL-UHFFFAOYSA-N 5-(hydroxymethyl)-3-(3-methylphenyl)-1,3-oxazolidin-2-one Chemical compound CC1=CC=CC(N2C(OC(CO)C2)=O)=C1 MXUNKHLAEDCYJL-UHFFFAOYSA-N 0.000 description 1
- 102000035037 5-HT3 receptors Human genes 0.000 description 1
- 108091005477 5-HT3 receptors Proteins 0.000 description 1
- QPGGEKPRGVJKQB-UHFFFAOYSA-N 5-[2-(dimethylamino)ethyl]-11-methyl-6-benzo[b][1,4]benzodiazepinone Chemical compound O=C1N(CCN(C)C)C2=CC=CC=C2N(C)C2=CC=CC=C21 QPGGEKPRGVJKQB-UHFFFAOYSA-N 0.000 description 1
- LDCYZAJDBXYCGN-VIFPVBQESA-N 5-hydroxy-L-tryptophan Chemical compound C1=C(O)C=C2C(C[C@H](N)C(O)=O)=CNC2=C1 LDCYZAJDBXYCGN-VIFPVBQESA-N 0.000 description 1
- XKFPYPQQHFEXRZ-UHFFFAOYSA-N 5-methyl-N'-(phenylmethyl)-3-isoxazolecarbohydrazide Chemical compound O1C(C)=CC(C(=O)NNCC=2C=CC=CC=2)=N1 XKFPYPQQHFEXRZ-UHFFFAOYSA-N 0.000 description 1
- 125000003341 7 membered heterocyclic group Chemical group 0.000 description 1
- JICJBGPOMZQUBB-UHFFFAOYSA-N 7-[(3-chloro-6-methyl-5,5-dioxido-6,11-dihydrodibenzo[c,f][1,2]thiazepin-11-yl)amino]heptanoic acid Chemical compound O=S1(=O)N(C)C2=CC=CC=C2C(NCCCCCCC(O)=O)C2=CC=C(Cl)C=C21 JICJBGPOMZQUBB-UHFFFAOYSA-N 0.000 description 1
- 229930000680 A04AD01 - Scopolamine Natural products 0.000 description 1
- AEOBEOJCBAYXBA-UHFFFAOYSA-N A2P5P Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1OP(O)(O)=O AEOBEOJCBAYXBA-UHFFFAOYSA-N 0.000 description 1
- 102000003678 AMPA Receptors Human genes 0.000 description 1
- 108090000078 AMPA Receptors Proteins 0.000 description 1
- 206010000234 Abortion spontaneous Diseases 0.000 description 1
- 229940121683 Acetylcholine receptor antagonist Drugs 0.000 description 1
- 229940104915 Alpha 2 adrenoreceptor antagonist Drugs 0.000 description 1
- 208000000044 Amnesia Diseases 0.000 description 1
- 208000029197 Amphetamine-Related disease Diseases 0.000 description 1
- 206010002329 Aneurysm Diseases 0.000 description 1
- 208000000103 Anorexia Nervosa Diseases 0.000 description 1
- 206010063659 Aversion Diseases 0.000 description 1
- 208000020925 Bipolar disease Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006458 Bronchitis chronic Diseases 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- 101100296719 Caenorhabditis elegans pde-4 gene Proteins 0.000 description 1
- 108090000312 Calcium Channels Proteins 0.000 description 1
- 102000003922 Calcium Channels Human genes 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 102100033868 Cannabinoid receptor 1 Human genes 0.000 description 1
- 101710187010 Cannabinoid receptor 1 Proteins 0.000 description 1
- 102100036214 Cannabinoid receptor 2 Human genes 0.000 description 1
- 101710187022 Cannabinoid receptor 2 Proteins 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- QDHHCQZDFGDHMP-UHFFFAOYSA-N Chloramine Chemical compound ClN QDHHCQZDFGDHMP-UHFFFAOYSA-N 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- 208000014085 Chronic respiratory disease Diseases 0.000 description 1
- UDMBCSSLTHHNCD-UHFFFAOYSA-N Coenzym Q(11) Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1O UDMBCSSLTHHNCD-UHFFFAOYSA-N 0.000 description 1
- 208000028698 Cognitive impairment Diseases 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 206010012218 Delirium Diseases 0.000 description 1
- 206010012239 Delusion Diseases 0.000 description 1
- 101100189582 Dictyostelium discoideum pdeD gene Proteins 0.000 description 1
- 101100351286 Dictyostelium discoideum pdeE gene Proteins 0.000 description 1
- 102000004076 Dopamine D1 Receptors Human genes 0.000 description 1
- 108090000511 Dopamine D1 Receptors Proteins 0.000 description 1
- 229940098305 Dopamine D1 receptor agonist Drugs 0.000 description 1
- 108091007267 Dopamine D1-Like Receptors Proteins 0.000 description 1
- 108090001111 Dopamine D2 Receptors Proteins 0.000 description 1
- 102000004980 Dopamine D2 Receptors Human genes 0.000 description 1
- 108091007265 Dopamine D2-Like Receptors Proteins 0.000 description 1
- 102000015554 Dopamine receptor Human genes 0.000 description 1
- 108050004812 Dopamine receptor Proteins 0.000 description 1
- 229940098778 Dopamine receptor agonist Drugs 0.000 description 1
- 101100407341 Drosophila melanogaster Pde9 gene Proteins 0.000 description 1
- 206010052804 Drug tolerance Diseases 0.000 description 1
- 206010013954 Dysphoria Diseases 0.000 description 1
- 206010014561 Emphysema Diseases 0.000 description 1
- 108010049140 Endorphins Proteins 0.000 description 1
- 102000009025 Endorphins Human genes 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- WVVSZNPYNCNODU-CJBNDPTMSA-N Ergometrine Natural products C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@@H](CO)C)C2)=C3C2=CNC3=C1 WVVSZNPYNCNODU-CJBNDPTMSA-N 0.000 description 1
- 206010015535 Euphoric mood Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 206010056465 Food craving Diseases 0.000 description 1
- XWLUWCNOOVRFPX-UHFFFAOYSA-N Fosphenytoin Chemical compound O=C1N(COP(O)(=O)O)C(=O)NC1(C=1C=CC=CC=1)C1=CC=CC=C1 XWLUWCNOOVRFPX-UHFFFAOYSA-N 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 102000004300 GABA-A Receptors Human genes 0.000 description 1
- 108090000839 GABA-A Receptors Proteins 0.000 description 1
- 208000034826 Genetic Predisposition to Disease Diseases 0.000 description 1
- WYCLKVQLVUQKNZ-UHFFFAOYSA-N Halazepam Chemical compound N=1CC(=O)N(CC(F)(F)F)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 WYCLKVQLVUQKNZ-UHFFFAOYSA-N 0.000 description 1
- 206010019079 Hallucinations, mixed Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical class NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 1
- 206010020400 Hostility Diseases 0.000 description 1
- 101100450577 Human herpesvirus 6B (strain Z29) U74 gene Proteins 0.000 description 1
- AKOAEVOSDHIVFX-UHFFFAOYSA-N Hydroxybupropion Chemical compound OCC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 AKOAEVOSDHIVFX-UHFFFAOYSA-N 0.000 description 1
- STECJAGHUSJQJN-GAUPFVANSA-N Hyoscine Natural products C1([C@H](CO)C(=O)OC2C[C@@H]3N([C@H](C2)[C@@H]2[C@H]3O2)C)=CC=CC=C1 STECJAGHUSJQJN-GAUPFVANSA-N 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- IWVRVEIKCBFZNF-UHFFFAOYSA-N LSM-1636 Chemical compound C1CNC2CCCC3=C2N1C1=CC=C(C)C=C13 IWVRVEIKCBFZNF-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 208000032376 Lung infection Diseases 0.000 description 1
- WVVSZNPYNCNODU-XTQGRXLLSA-N Lysergic acid propanolamide Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@H](CO)C)C2)=C3C2=CNC3=C1 WVVSZNPYNCNODU-XTQGRXLLSA-N 0.000 description 1
- OCJYIGYOJCODJL-UHFFFAOYSA-N Meclizine Chemical compound CC1=CC=CC(CN2CCN(CC2)C(C=2C=CC=CC=2)C=2C=CC(Cl)=CC=2)=C1 OCJYIGYOJCODJL-UHFFFAOYSA-N 0.000 description 1
- 208000026139 Memory disease Diseases 0.000 description 1
- 241001511582 Metapone Species 0.000 description 1
- AJXPJJZHWIXJCJ-UHFFFAOYSA-N Methsuximide Chemical compound O=C1N(C)C(=O)CC1(C)C1=CC=CC=C1 AJXPJJZHWIXJCJ-UHFFFAOYSA-N 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- 208000019022 Mood disease Diseases 0.000 description 1
- 208000003445 Mouth Neoplasms Diseases 0.000 description 1
- 208000001089 Multiple system atrophy Diseases 0.000 description 1
- 229940121948 Muscarinic receptor antagonist Drugs 0.000 description 1
- 208000006550 Mydriasis Diseases 0.000 description 1
- LBHLFPGPEGDCJG-UHFFFAOYSA-N N(4)-{2,6-dimethoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinolin-8-yl}pentane-1,4-diamine Chemical compound COC=1C=C(NC(C)CCCN)C2=NC(OC)=CC(C)=C2C=1OC1=CC=CC(C(F)(F)F)=C1 LBHLFPGPEGDCJG-UHFFFAOYSA-N 0.000 description 1
- WUKZPHOXUVCQOR-UHFFFAOYSA-N N-(1-azabicyclo[2.2.2]octan-3-yl)-6-chloro-4-methyl-3-oxo-1,4-benzoxazine-8-carboxamide Chemical compound C1N(CC2)CCC2C1NC(=O)C1=CC(Cl)=CC2=C1OCC(=O)N2C WUKZPHOXUVCQOR-UHFFFAOYSA-N 0.000 description 1
- 102000004868 N-Methyl-D-Aspartate Receptors Human genes 0.000 description 1
- 108090001041 N-Methyl-D-Aspartate Receptors Proteins 0.000 description 1
- STECJAGHUSJQJN-UHFFFAOYSA-N N-Methyl-scopolamin Natural products C1C(C2C3O2)N(C)C3CC1OC(=O)C(CO)C1=CC=CC=C1 STECJAGHUSJQJN-UHFFFAOYSA-N 0.000 description 1
- WXNXCEHXYPACJF-ZETCQYMHSA-N N-acetyl-L-leucine Chemical compound CC(C)C[C@@H](C(O)=O)NC(C)=O WXNXCEHXYPACJF-ZETCQYMHSA-N 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 206010029216 Nervousness Diseases 0.000 description 1
- KYYIDSXMWOZKMP-UHFFFAOYSA-N O-desmethylvenlafaxine Chemical compound C1CCCCC1(O)C(CN(C)C)C1=CC=C(O)C=C1 KYYIDSXMWOZKMP-UHFFFAOYSA-N 0.000 description 1
- FRPRNNRJTCONEC-UHFFFAOYSA-N Ohmefentanyl Chemical group C=1C=CC=CC=1N(C(=O)CC)C(C(C1)C)CCN1CC(O)C1=CC=CC=C1 FRPRNNRJTCONEC-UHFFFAOYSA-N 0.000 description 1
- 208000012488 Opiate Overdose Diseases 0.000 description 1
- 229940121954 Opioid receptor agonist Drugs 0.000 description 1
- 229940123257 Opioid receptor antagonist Drugs 0.000 description 1
- 206010031127 Orthostatic hypotension Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 101150106395 PDE1B gene Proteins 0.000 description 1
- 101150098694 PDE5A gene Proteins 0.000 description 1
- 102000039035 PDE7 family Human genes 0.000 description 1
- 108091065699 PDE7 family Proteins 0.000 description 1
- 206010033864 Paranoia Diseases 0.000 description 1
- 208000027099 Paranoid disease Diseases 0.000 description 1
- DPWPWRLQFGFJFI-UHFFFAOYSA-N Pargyline Chemical compound C#CCN(C)CC1=CC=CC=C1 DPWPWRLQFGFJFI-UHFFFAOYSA-N 0.000 description 1
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 208000009565 Pharyngeal Neoplasms Diseases 0.000 description 1
- RMUCZJUITONUFY-UHFFFAOYSA-N Phenelzine Chemical compound NNCCC1=CC=CC=C1 RMUCZJUITONUFY-UHFFFAOYSA-N 0.000 description 1
- WLWFNJKHKGIJNW-UHFFFAOYSA-N Phensuximide Chemical compound O=C1N(C)C(=O)CC1C1=CC=CC=C1 WLWFNJKHKGIJNW-UHFFFAOYSA-N 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 208000005107 Premature Birth Diseases 0.000 description 1
- 206010036590 Premature baby Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- 101100244562 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) oprD gene Proteins 0.000 description 1
- 208000028017 Psychotic disease Diseases 0.000 description 1
- ZTVQQQVZCWLTDF-UHFFFAOYSA-N Remifentanil Chemical compound C1CN(CCC(=O)OC)CCC1(C(=O)OC)N(C(=O)CC)C1=CC=CC=C1 ZTVQQQVZCWLTDF-UHFFFAOYSA-N 0.000 description 1
- 206010039203 Road traffic accident Diseases 0.000 description 1
- 206010039897 Sedation Diseases 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 108010052164 Sodium Channels Proteins 0.000 description 1
- 102000018674 Sodium Channels Human genes 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 102400000096 Substance P Human genes 0.000 description 1
- 101800003906 Substance P Proteins 0.000 description 1
- 206010042458 Suicidal ideation Diseases 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 102000003141 Tachykinin Human genes 0.000 description 1
- JACAAXNEHGBPOQ-LLVKDONJSA-N Talampanel Chemical compound C([C@H](N(N=1)C(C)=O)C)C2=CC=3OCOC=3C=C2C=1C1=CC=C(N)C=C1 JACAAXNEHGBPOQ-LLVKDONJSA-N 0.000 description 1
- 206010044565 Tremor Diseases 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 208000023915 Ureteral Neoplasms Diseases 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- BZKPWHYZMXOIDC-UHFFFAOYSA-N acetazolamide Chemical compound CC(=O)NC1=NN=C(S(N)(=O)=O)S1 BZKPWHYZMXOIDC-UHFFFAOYSA-N 0.000 description 1
- 229960000571 acetazolamide Drugs 0.000 description 1
- 229960000669 acetylleucine Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 208000005298 acute pain Diseases 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 230000003044 adaptive effect Effects 0.000 description 1
- 229940047812 adderall Drugs 0.000 description 1
- UDMBCSSLTHHNCD-KQYNXXCUSA-N adenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O UDMBCSSLTHHNCD-KQYNXXCUSA-N 0.000 description 1
- 229950006790 adenosine phosphate Drugs 0.000 description 1
- 238000011360 adjunctive therapy Methods 0.000 description 1
- 208000012761 aggressive behavior Diseases 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 230000036626 alertness Effects 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 125000004419 alkynylene group Chemical group 0.000 description 1
- 229960003550 alosetron Drugs 0.000 description 1
- FLZQKRKHLSUHOR-UHFFFAOYSA-N alosetron Chemical compound CC1=NC=N[C]1CN1C(=O)C(C=2C(=CC=CC=2)N2C)=C2CC1 FLZQKRKHLSUHOR-UHFFFAOYSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AKNNEGZIBPJZJG-UHFFFAOYSA-N alpha-noscapine Natural products CN1CCC2=CC=3OCOC=3C(OC)=C2C1C1C2=CC=C(OC)C(OC)=C2C(=O)O1 AKNNEGZIBPJZJG-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229960004516 alvimopan Drugs 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960001097 amifostine Drugs 0.000 description 1
- HAMNKKUPIHEESI-UHFFFAOYSA-N aminoguanidine Chemical compound NNC(N)=N HAMNKKUPIHEESI-UHFFFAOYSA-N 0.000 description 1
- 229960000836 amitriptyline Drugs 0.000 description 1
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 description 1
- 229960001301 amobarbital Drugs 0.000 description 1
- 230000036592 analgesia Effects 0.000 description 1
- LKYQLAWMNBFNJT-UHFFFAOYSA-N anileridine Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCC1=CC=C(N)C=C1 LKYQLAWMNBFNJT-UHFFFAOYSA-N 0.000 description 1
- 229960002512 anileridine Drugs 0.000 description 1
- IXPROWGEHNVJOY-UHFFFAOYSA-N antalarmin Chemical compound CC1=C(C)C=2C(N(CC)CCCC)=NC(C)=NC=2N1C1=C(C)C=C(C)C=C1C IXPROWGEHNVJOY-UHFFFAOYSA-N 0.000 description 1
- 229940125680 anti-addiction agent Drugs 0.000 description 1
- 230000001078 anti-cholinergic effect Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000000043 antiallergic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000002249 anxiolytic agent Substances 0.000 description 1
- 230000000949 anxiolytic effect Effects 0.000 description 1
- ATALOFNDEOCMKK-OITMNORJSA-N aprepitant Chemical compound O([C@@H]([C@@H]1C=2C=CC(F)=CC=2)O[C@H](C)C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)CCN1CC1=NNC(=O)N1 ATALOFNDEOCMKK-OITMNORJSA-N 0.000 description 1
- 229960001372 aprepitant Drugs 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 125000005418 aryl aryl group Chemical group 0.000 description 1
- 150000005840 aryl radicals Chemical class 0.000 description 1
- 230000035045 associative learning Effects 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 229950005951 azasetron Drugs 0.000 description 1
- 238000003287 bathing Methods 0.000 description 1
- MNJNPLVXBISNSX-WDNDVIMCSA-N bemesetron Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C1=CC(Cl)=CC(Cl)=C1 MNJNPLVXBISNSX-WDNDVIMCSA-N 0.000 description 1
- 229950007840 bemesetron Drugs 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- OFYVIGTWSQPCLF-NWDGAFQWSA-N bicifadine Chemical compound C1=CC(C)=CC=C1[C@@]1(CNC2)[C@H]2C1 OFYVIGTWSQPCLF-NWDGAFQWSA-N 0.000 description 1
- 229950010365 bicifadine Drugs 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000006931 brain damage Effects 0.000 description 1
- 231100000874 brain damage Toxicity 0.000 description 1
- 230000003925 brain function Effects 0.000 description 1
- 208000029028 brain injury Diseases 0.000 description 1
- 230000008309 brain mechanism Effects 0.000 description 1
- 230000010336 brain pathway Effects 0.000 description 1
- 229960002161 brivaracetam Drugs 0.000 description 1
- MSYKRHVOOPPJKU-BDAKNGLRSA-N brivaracetam Chemical compound CCC[C@H]1CN([C@@H](CC)C(N)=O)C(=O)C1 MSYKRHVOOPPJKU-BDAKNGLRSA-N 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- SNCZNSNPXMPCGN-UHFFFAOYSA-N butanediamide Chemical class NC(=O)CCC(N)=O SNCZNSNPXMPCGN-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 102100029175 cGMP-specific 3',5'-cyclic phosphodiesterase Human genes 0.000 description 1
- 235000019577 caloric intake Nutrition 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- QHMBSVQNZZTUGM-ZWKOTPCHSA-N cannabidiol Chemical compound OC1=CC(CCCCC)=CC(O)=C1[C@H]1[C@H](C(C)=C)CCC(C)=C1 QHMBSVQNZZTUGM-ZWKOTPCHSA-N 0.000 description 1
- 229950011318 cannabidiol Drugs 0.000 description 1
- 229940121376 cannabinoid receptor agonist Drugs 0.000 description 1
- 239000003537 cannabinoid receptor agonist Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229960000623 carbamazepine Drugs 0.000 description 1
- FFGPTBGBLSHEPO-UHFFFAOYSA-N carbamazepine Chemical compound C1=CC2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 FFGPTBGBLSHEPO-UHFFFAOYSA-N 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 230000003778 catagen phase Effects 0.000 description 1
- 108020001778 catalytic domains Proteins 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000007248 cellular mechanism Effects 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- SPKBYQZELVEOLL-QDMKHBRRSA-N chembl2111147 Chemical compound N([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C1=CC(Cl)=CC2=C1OC(C)(C)C2 SPKBYQZELVEOLL-QDMKHBRRSA-N 0.000 description 1
- UKTAZPQNNNJVKR-KJGYPYNMSA-N chembl2368925 Chemical compound C1=CC=C2C(C(O[C@@H]3C[C@@H]4C[C@H]5C[C@@H](N4CC5=O)C3)=O)=CNC2=C1 UKTAZPQNNNJVKR-KJGYPYNMSA-N 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229960004782 chlordiazepoxide Drugs 0.000 description 1
- ANTSCNMPPGJYLG-UHFFFAOYSA-N chlordiazepoxide Chemical compound O=N=1CC(NC)=NC2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 ANTSCNMPPGJYLG-UHFFFAOYSA-N 0.000 description 1
- 208000007451 chronic bronchitis Diseases 0.000 description 1
- 229960002099 cilansetron Drugs 0.000 description 1
- NCNFDKWULDWJDS-OAHLLOKOSA-N cilansetron Chemical compound CC1=NC=CN1C[C@@H]1C(=O)C(C=2C=3N4CCCC=3C=CC=2)=C4CC1 NCNFDKWULDWJDS-OAHLLOKOSA-N 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 229960001653 citalopram Drugs 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 229960001403 clobazam Drugs 0.000 description 1
- CXOXHMZGEKVPMT-UHFFFAOYSA-N clobazam Chemical compound O=C1CC(=O)N(C)C2=CC=C(Cl)C=C2N1C1=CC=CC=C1 CXOXHMZGEKVPMT-UHFFFAOYSA-N 0.000 description 1
- 229960003120 clonazepam Drugs 0.000 description 1
- DGBIGWXXNGSACT-UHFFFAOYSA-N clonazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1Cl DGBIGWXXNGSACT-UHFFFAOYSA-N 0.000 description 1
- 229960004170 clozapine Drugs 0.000 description 1
- QZUDBNBUXVUHMW-UHFFFAOYSA-N clozapine Chemical compound C1CN(C)CCN1C1=NC2=CC(Cl)=CC=C2NC2=CC=CC=C12 QZUDBNBUXVUHMW-UHFFFAOYSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 201000001272 cocaine abuse Diseases 0.000 description 1
- 201000006145 cocaine dependence Diseases 0.000 description 1
- 208000010877 cognitive disease Diseases 0.000 description 1
- 230000001427 coherent effect Effects 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 230000008094 contradictory effect Effects 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 230000036461 convulsion Effects 0.000 description 1
- 230000010485 coping Effects 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 239000002769 corticotropin releasing factor antagonist Substances 0.000 description 1
- 238000009223 counseling Methods 0.000 description 1
- FSIRGTNPQFDCCD-QGMBQPNBSA-N cyanodothiepin Chemical compound C1SC2=CC=C(C#N)C=C2C(=C/CCN(C)C)/C2=CC=CC=C21 FSIRGTNPQFDCCD-QGMBQPNBSA-N 0.000 description 1
- 101150113005 cyc2 gene Proteins 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 229960003564 cyclizine Drugs 0.000 description 1
- UVKZSORBKUEBAZ-UHFFFAOYSA-N cyclizine Chemical compound C1CN(C)CCN1C(C=1C=CC=CC=1)C1=CC=CC=C1 UVKZSORBKUEBAZ-UHFFFAOYSA-N 0.000 description 1
- 150000001925 cycloalkenes Chemical class 0.000 description 1
- ZXIJMRYMVAMXQP-UHFFFAOYSA-N cycloheptene Chemical compound C1CCC=CCC1 ZXIJMRYMVAMXQP-UHFFFAOYSA-N 0.000 description 1
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical group C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- USRHYDPUVLEVMC-FQEVSTJZSA-N dapoxetine Chemical compound C1([C@H](CCOC=2C3=CC=CC=C3C=CC=2)N(C)C)=CC=CC=C1 USRHYDPUVLEVMC-FQEVSTJZSA-N 0.000 description 1
- 229960005217 dapoxetine Drugs 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 108700023159 delta Opioid Receptors Proteins 0.000 description 1
- 102000048124 delta Opioid Receptors Human genes 0.000 description 1
- 231100000868 delusion Toxicity 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- LNNWVNGFPYWNQE-GMIGKAJZSA-N desomorphine Chemical compound C1C2=CC=C(O)C3=C2[C@]24CCN(C)[C@H]1[C@@H]2CCC[C@@H]4O3 LNNWVNGFPYWNQE-GMIGKAJZSA-N 0.000 description 1
- 229950003851 desomorphine Drugs 0.000 description 1
- 229960001623 desvenlafaxine Drugs 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 229960003075 dibenzepin Drugs 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- LJXTYJXBORAIHX-UHFFFAOYSA-N diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OCC)C1 LJXTYJXBORAIHX-UHFFFAOYSA-N 0.000 description 1
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 1
- 125000004212 difluorophenyl group Chemical group 0.000 description 1
- 229960000920 dihydrocodeine Drugs 0.000 description 1
- RBOXVHNMENFORY-DNJOTXNNSA-N dihydrocodeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC RBOXVHNMENFORY-DNJOTXNNSA-N 0.000 description 1
- HESHRHUZIWVEAJ-JGRZULCMSA-N dihydroergotamine Chemical compound C([C@H]1C(=O)N2CCC[C@H]2[C@]2(O)O[C@@](C(N21)=O)(C)NC(=O)[C@H]1CN([C@H]2[C@@H](C3=CC=CC4=NC=C([C]34)C2)C1)C)C1=CC=CC=C1 HESHRHUZIWVEAJ-JGRZULCMSA-N 0.000 description 1
- 229960004704 dihydroergotamine Drugs 0.000 description 1
- BRTSNYPDACNMIP-FAWZKKEFSA-N dihydroetorphine Chemical compound O([C@H]1[C@@]2(OC)CC[C@@]34C[C@@H]2[C@](C)(O)CCC)C2=C5[C@]41CCN(C)[C@@H]3CC5=CC=C2O BRTSNYPDACNMIP-FAWZKKEFSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- MZDOIJOUFRQXHC-UHFFFAOYSA-N dimenhydrinate Chemical compound O=C1N(C)C(=O)N(C)C2=NC(Cl)=N[C]21.C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 MZDOIJOUFRQXHC-UHFFFAOYSA-N 0.000 description 1
- 229960004993 dimenhydrinate Drugs 0.000 description 1
- 229960000520 diphenhydramine Drugs 0.000 description 1
- 229960003520 diphenidol Drugs 0.000 description 1
- OGAKLTJNUQRZJU-UHFFFAOYSA-N diphenidol Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(O)CCCN1CCCCC1 OGAKLTJNUQRZJU-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229940028937 divalproex sodium Drugs 0.000 description 1
- 229960003413 dolasetron Drugs 0.000 description 1
- FGXWKSZFVQUSTL-UHFFFAOYSA-N domperidone Chemical compound C12=CC=CC=C2NC(=O)N1CCCN(CC1)CCC1N1C2=CC=C(Cl)C=C2NC1=O FGXWKSZFVQUSTL-UHFFFAOYSA-N 0.000 description 1
- 229960001253 domperidone Drugs 0.000 description 1
- 229940052760 dopamine agonists Drugs 0.000 description 1
- 230000002825 dopamine reuptake Effects 0.000 description 1
- 210000004002 dopaminergic cell Anatomy 0.000 description 1
- 229960001393 dosulepin Drugs 0.000 description 1
- 229960005426 doxepin Drugs 0.000 description 1
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 1
- 230000035622 drinking Effects 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000008482 dysregulation Effects 0.000 description 1
- 235000006694 eating habits Nutrition 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000008451 emotion Effects 0.000 description 1
- 230000006397 emotional response Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 229960000740 enrofloxacin Drugs 0.000 description 1
- 238000011841 epidemiological investigation Methods 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- 230000001667 episodic effect Effects 0.000 description 1
- 229960001405 ergometrine Drugs 0.000 description 1
- WSEQXVZVJXJVFP-FQEVSTJZSA-N escitalopram Chemical compound C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-FQEVSTJZSA-N 0.000 description 1
- 229960004341 escitalopram Drugs 0.000 description 1
- SUBDBMMJDZJVOS-DEOSSOPVSA-N esomeprazole Chemical compound C([S@](=O)C1=NC2=CC=C(C=C2N1)OC)C1=NC=C(C)C(OC)=C1C SUBDBMMJDZJVOS-DEOSSOPVSA-N 0.000 description 1
- 229960004770 esomeprazole Drugs 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 229960002767 ethosuximide Drugs 0.000 description 1
- HAPOVYFOVVWLRS-UHFFFAOYSA-N ethosuximide Chemical compound CCC1(C)CC(=O)NC1=O HAPOVYFOVVWLRS-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- ZXUMUPVQYAFTLF-UHFFFAOYSA-N etryptamine Chemical compound C1=CC=C2C(CC(N)CC)=CNC2=C1 ZXUMUPVQYAFTLF-UHFFFAOYSA-N 0.000 description 1
- 229950005957 etryptamine Drugs 0.000 description 1
- 230000002743 euphoric effect Effects 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000010228 ex vivo assay Methods 0.000 description 1
- 230000003090 exacerbative effect Effects 0.000 description 1
- 230000003492 excitotoxic effect Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- PCOBBVZJEWWZFR-UHFFFAOYSA-N ezogabine Chemical compound C1=C(N)C(NC(=O)OCC)=CC=C1NCC1=CC=C(F)C=C1 PCOBBVZJEWWZFR-UHFFFAOYSA-N 0.000 description 1
- 229960003472 felbamate Drugs 0.000 description 1
- WKGXYQFOCVYPAC-UHFFFAOYSA-N felbamate Chemical compound NC(=O)OCC(COC(N)=O)C1=CC=CC=C1 WKGXYQFOCVYPAC-UHFFFAOYSA-N 0.000 description 1
- 229960001582 fenfluramine Drugs 0.000 description 1
- 238000009093 first-line therapy Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 125000001207 fluorophenyl group Chemical group 0.000 description 1
- 229960004038 fluvoxamine Drugs 0.000 description 1
- CJOFXWAVKWHTFT-XSFVSMFZSA-N fluvoxamine Chemical compound COCCCC\C(=N/OCCN)C1=CC=C(C(F)(F)F)C=C1 CJOFXWAVKWHTFT-XSFVSMFZSA-N 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 229960000693 fosphenytoin Drugs 0.000 description 1
- 238000002599 functional magnetic resonance imaging Methods 0.000 description 1
- 230000003371 gabaergic effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000003633 gene expression assay Methods 0.000 description 1
- 230000009368 gene silencing by RNA Effects 0.000 description 1
- 239000003193 general anesthetic agent Substances 0.000 description 1
- 229940005494 general anesthetics Drugs 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229960000647 gepirone Drugs 0.000 description 1
- QOIGKGMMAGJZNZ-UHFFFAOYSA-N gepirone Chemical compound O=C1CC(C)(C)CC(=O)N1CCCCN1CCN(C=2N=CC=CN=2)CC1 QOIGKGMMAGJZNZ-UHFFFAOYSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-L glutamate group Chemical group N[C@@H](CCC(=O)[O-])C(=O)[O-] WHUUTDBJXJRKMK-VKHMYHEASA-L 0.000 description 1
- MFWNKCLOYSRHCJ-BTTYYORXSA-N granisetron Chemical compound C1=CC=C2C(C(=O)N[C@H]3C[C@H]4CCC[C@@H](C3)N4C)=NN(C)C2=C1 MFWNKCLOYSRHCJ-BTTYYORXSA-N 0.000 description 1
- 229960003727 granisetron Drugs 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 230000002650 habitual effect Effects 0.000 description 1
- 229960002158 halazepam Drugs 0.000 description 1
- 239000000380 hallucinogen Substances 0.000 description 1
- 235000015220 hamburgers Nutrition 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 230000005802 health problem Effects 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000009097 homeostatic mechanism Effects 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 102000050549 human PDE7B Human genes 0.000 description 1
- 150000001469 hydantoins Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- JUMYIBMBTDDLNG-OJERSXHUSA-N hydron;methyl (2r)-2-phenyl-2-[(2r)-piperidin-2-yl]acetate;chloride Chemical compound Cl.C([C@@H]1[C@H](C(=O)OC)C=2C=CC=CC=2)CCCN1 JUMYIBMBTDDLNG-OJERSXHUSA-N 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 1
- 239000003326 hypnotic agent Substances 0.000 description 1
- 230000000147 hypnotic effect Effects 0.000 description 1
- 230000002267 hypothalamic effect Effects 0.000 description 1
- 230000004179 hypothalamic–pituitary–adrenal axis Effects 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- MHNNVDILNTUWNS-XYYAHUGASA-N indisetron Chemical compound C1=CC=C2C(C(=O)N[C@H]3C[C@H]4CN(C[C@@H](C3)N4C)C)=NNC2=C1 MHNNVDILNTUWNS-XYYAHUGASA-N 0.000 description 1
- 229950007467 indisetron Drugs 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 238000009413 insulation Methods 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 229960002844 iprindole Drugs 0.000 description 1
- PLIGPBGDXASWPX-UHFFFAOYSA-N iprindole Chemical compound C1CCCCCC2=C1N(CCCN(C)C)C1=CC=CC=C12 PLIGPBGDXASWPX-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 229960002672 isocarboxazid Drugs 0.000 description 1
- SANOUVWGPVYVAV-UHFFFAOYSA-N isovaleramide Chemical compound CC(C)CC(N)=O SANOUVWGPVYVAV-UHFFFAOYSA-N 0.000 description 1
- 125000004284 isoxazol-3-yl group Chemical group [H]C1=C([H])C(*)=NO1 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229950007654 itasetron Drugs 0.000 description 1
- RWXRJSRJIITQAK-ZSBIGDGJSA-N itasetron Chemical compound C12=CC=CC=C2NC(=O)N1C(=O)N[C@H](C1)C[C@H]2CC[C@@H]1N2C RWXRJSRJIITQAK-ZSBIGDGJSA-N 0.000 description 1
- FABUFPQFXZVHFB-CFWQTKTJSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@H](C)C(=O)C(C)(C)[C@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-CFWQTKTJSA-N 0.000 description 1
- 229960002014 ixabepilone Drugs 0.000 description 1
- 229960005417 ketanserin Drugs 0.000 description 1
- FPCCSQOGAWCVBH-UHFFFAOYSA-N ketanserin Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCN2C(C3=CC=CC=C3NC2=O)=O)CC1 FPCCSQOGAWCVBH-UHFFFAOYSA-N 0.000 description 1
- VPPJLAIAVCUEMN-GFCCVEGCSA-N lacosamide Chemical compound COC[C@@H](NC(C)=O)C(=O)NCC1=CC=CC=C1 VPPJLAIAVCUEMN-GFCCVEGCSA-N 0.000 description 1
- 229960002623 lacosamide Drugs 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 206010023841 laryngeal neoplasm Diseases 0.000 description 1
- 230000013016 learning Effects 0.000 description 1
- 201000003723 learning disability Diseases 0.000 description 1
- FDXQKWSTUZCCTM-ZUIJCZDSSA-N levoprotiline Chemical compound C12=CC=CC=C2C2(C[C@@H](O)CNC)C3=CC=CC=C3C1CC2 FDXQKWSTUZCCTM-ZUIJCZDSSA-N 0.000 description 1
- 229950003041 levoprotiline Drugs 0.000 description 1
- 230000002197 limbic effect Effects 0.000 description 1
- 210000003715 limbic system Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 208000026037 malignant tumor of neck Diseases 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 229960001474 meclozine Drugs 0.000 description 1
- 229960004794 melitracen Drugs 0.000 description 1
- GWWLWDURRGNSRS-UHFFFAOYSA-N melitracen Chemical compound C1=CC=C2C(=CCCN(C)C)C3=CC=CC=C3C(C)(C)C2=C1 GWWLWDURRGNSRS-UHFFFAOYSA-N 0.000 description 1
- 230000006984 memory degeneration Effects 0.000 description 1
- 208000023060 memory loss Diseases 0.000 description 1
- 201000003102 mental depression Diseases 0.000 description 1
- 230000004630 mental health Effects 0.000 description 1
- 230000006996 mental state Effects 0.000 description 1
- GMHKMTDVRCWUDX-UHFFFAOYSA-N mephenytoin Chemical compound C=1C=CC=CC=1C1(CC)NC(=O)N(C)C1=O GMHKMTDVRCWUDX-UHFFFAOYSA-N 0.000 description 1
- 229960000906 mephenytoin Drugs 0.000 description 1
- 229960003729 mesuximide Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- YXVZOBVWVRFPTE-UHFFFAOYSA-N metapramine Chemical compound CNC1CC2=CC=CC=C2N(C)C2=CC=CC=C12 YXVZOBVWVRFPTE-UHFFFAOYSA-N 0.000 description 1
- 229950006180 metapramine Drugs 0.000 description 1
- 125000005394 methallyl group Chemical group 0.000 description 1
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 1
- PMRYVIKBURPHAH-UHFFFAOYSA-N methimazole Chemical compound CN1C=CNC1=S PMRYVIKBURPHAH-UHFFFAOYSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 229960002921 methylnaltrexone Drugs 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- TTWJBBZEZQICBI-UHFFFAOYSA-N metoclopramide Chemical compound CCN(CC)CCNC(=O)C1=CC(Cl)=C(N)C=C1OC TTWJBBZEZQICBI-UHFFFAOYSA-N 0.000 description 1
- 229960004503 metoclopramide Drugs 0.000 description 1
- 229960000767 metopimazine Drugs 0.000 description 1
- BQDBKDMTIJBJLA-UHFFFAOYSA-N metopimazine Chemical compound C12=CC(S(=O)(=O)C)=CC=C2SC2=CC=CC=C2N1CCCN1CCC(C(N)=O)CC1 BQDBKDMTIJBJLA-UHFFFAOYSA-N 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- 238000001690 micro-dialysis Methods 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- YHXISWVBGDMDLQ-UHFFFAOYSA-N moclobemide Chemical compound C1=CC(Cl)=CC=C1C(=O)NCCN1CCOCC1 YHXISWVBGDMDLQ-UHFFFAOYSA-N 0.000 description 1
- 229960004644 moclobemide Drugs 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 201000003152 motion sickness Diseases 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- LKPFBGKZCCBZDK-UHFFFAOYSA-N n-hydroxypiperidine Chemical compound ON1CCCCC1 LKPFBGKZCCBZDK-UHFFFAOYSA-N 0.000 description 1
- BFYWWTIGNJJAHF-LTQSXOHQSA-N nalorphine dinicotinate Chemical compound O([C@H]1C=C[C@H]2[C@H]3CC=4C5=C(C(=CC=4)OC(=O)C=4C=NC=CC=4)O[C@@H]1[C@]52CCN3CC=C)C(=O)C1=CC=CN=C1 BFYWWTIGNJJAHF-LTQSXOHQSA-N 0.000 description 1
- PLPRGLOFPNJOTN-UHFFFAOYSA-N narcotine Natural products COc1ccc2C(OC(=O)c2c1OC)C3Cc4c(CN3C)cc5OCOc5c4OC PLPRGLOFPNJOTN-UHFFFAOYSA-N 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 229960001800 nefazodone Drugs 0.000 description 1
- VRBKIVRKKCLPHA-UHFFFAOYSA-N nefazodone Chemical compound O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 VRBKIVRKKCLPHA-UHFFFAOYSA-N 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 230000001722 neurochemical effect Effects 0.000 description 1
- 229960003057 nialamide Drugs 0.000 description 1
- 229950004211 nisoxetine Drugs 0.000 description 1
- ITJNARMNRKSWTA-UHFFFAOYSA-N nisoxetine Chemical compound C=1C=CC=CC=1C(CCNC)OC1=CC=CC=C1OC ITJNARMNRKSWTA-UHFFFAOYSA-N 0.000 description 1
- 229960004708 noscapine Drugs 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 229960005017 olanzapine Drugs 0.000 description 1
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- XSXHWVKGUXMUQE-UHFFFAOYSA-N osmium dioxide Inorganic materials O=[Os]=O XSXHWVKGUXMUQE-UHFFFAOYSA-N 0.000 description 1
- 229940078646 other antiemetics in atc Drugs 0.000 description 1
- 229940053180 other antiepileptics in atc Drugs 0.000 description 1
- 229940052740 other dopaminergic agent in atc Drugs 0.000 description 1
- ADIMAYPTOBDMTL-UHFFFAOYSA-N oxazepam Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(O)N=C1C1=CC=CC=C1 ADIMAYPTOBDMTL-UHFFFAOYSA-N 0.000 description 1
- 229960004535 oxazepam Drugs 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 229960001816 oxcarbazepine Drugs 0.000 description 1
- CTRLABGOLIVAIY-UHFFFAOYSA-N oxcarbazepine Chemical compound C1C(=O)C2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 CTRLABGOLIVAIY-UHFFFAOYSA-N 0.000 description 1
- 229950005217 oxypendyl Drugs 0.000 description 1
- RUPOLIZWSDDWNJ-UHFFFAOYSA-N oxypendyl Chemical group C1CN(CCO)CCN1CCCN1C2=NC=CC=C2SC2=CC=CC=C21 RUPOLIZWSDDWNJ-UHFFFAOYSA-N 0.000 description 1
- 229960002131 palonosetron Drugs 0.000 description 1
- CPZBLNMUGSZIPR-NVXWUHKLSA-N palonosetron Chemical compound C1N(CC2)CCC2[C@@H]1N1C(=O)C(C=CC=C2CCC3)=C2[C@H]3C1 CPZBLNMUGSZIPR-NVXWUHKLSA-N 0.000 description 1
- 229960001779 pargyline Drugs 0.000 description 1
- 229960002296 paroxetine Drugs 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 101150037969 pde-6 gene Proteins 0.000 description 1
- 229960001412 pentobarbital Drugs 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 238000011422 pharmacological therapy Methods 0.000 description 1
- 230000000079 pharmacotherapeutic effect Effects 0.000 description 1
- 238000001050 pharmacotherapy Methods 0.000 description 1
- 229960000964 phenelzine Drugs 0.000 description 1
- DDBREPKUVSBGFI-UHFFFAOYSA-N phenobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O DDBREPKUVSBGFI-UHFFFAOYSA-N 0.000 description 1
- 229960002695 phenobarbital Drugs 0.000 description 1
- 150000002990 phenothiazines Chemical class 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 229960004227 phensuximide Drugs 0.000 description 1
- 125000003356 phenylsulfanyl group Chemical group [*]SC1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- ACVYVLVWPXVTIT-UHFFFAOYSA-N phosphinic acid Chemical compound O[PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000036314 physical performance Effects 0.000 description 1
- PXXKIYPSXYFATG-UHFFFAOYSA-N piminodine Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCCNC1=CC=CC=C1 PXXKIYPSXYFATG-UHFFFAOYSA-N 0.000 description 1
- 229950006445 piminodine Drugs 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229950008580 pipamazine Drugs 0.000 description 1
- OSJJYEUEJRVVOD-UHFFFAOYSA-N pipamazine Chemical compound C1CC(C(=O)N)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 OSJJYEUEJRVVOD-UHFFFAOYSA-N 0.000 description 1
- 229950002220 pirlindole Drugs 0.000 description 1
- 125000005592 polycycloalkyl group Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 229960001233 pregabalin Drugs 0.000 description 1
- AYXYPKUFHZROOJ-ZETCQYMHSA-N pregabalin Chemical compound CC(C)C[C@H](CN)CC(O)=O AYXYPKUFHZROOJ-ZETCQYMHSA-N 0.000 description 1
- 210000005215 presynaptic neuron Anatomy 0.000 description 1
- 210000000063 presynaptic terminal Anatomy 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- WIKYUJGCLQQFNW-UHFFFAOYSA-N prochlorperazine Chemical compound C1CN(C)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 WIKYUJGCLQQFNW-UHFFFAOYSA-N 0.000 description 1
- 229960003111 prochlorperazine Drugs 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Chemical group 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- KJAQRHMKLVGSCG-UHFFFAOYSA-N propan-2-ylhydrazine Chemical compound CC(C)NN KJAQRHMKLVGSCG-UHFFFAOYSA-N 0.000 description 1
- ZBAFFZBKCMWUHM-UHFFFAOYSA-N propiram Chemical compound C=1C=CC=NC=1N(C(=O)CC)C(C)CN1CCCCC1 ZBAFFZBKCMWUHM-UHFFFAOYSA-N 0.000 description 1
- 229950003779 propiram Drugs 0.000 description 1
- 229960004134 propofol Drugs 0.000 description 1
- OLBCVFGFOZPWHH-UHFFFAOYSA-N propofol Chemical compound CC(C)C1=CC=CC(C(C)C)=C1O OLBCVFGFOZPWHH-UHFFFAOYSA-N 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 238000002731 protein assay Methods 0.000 description 1
- 229960002601 protriptyline Drugs 0.000 description 1
- BWPIARFWQZKAIA-UHFFFAOYSA-N protriptyline Chemical compound C1=CC2=CC=CC=C2C(CCCNC)C2=CC=CC=C21 BWPIARFWQZKAIA-UHFFFAOYSA-N 0.000 description 1
- 230000004800 psychological effect Effects 0.000 description 1
- 238000001671 psychotherapy Methods 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 229960000279 quinupramine Drugs 0.000 description 1
- JCBQCKFFSPGEDY-UHFFFAOYSA-N quinupramine Chemical compound C12=CC=CC=C2CCC2=CC=CC=C2N1C(C1)C2CCN1CC2 JCBQCKFFSPGEDY-UHFFFAOYSA-N 0.000 description 1
- NTHPAPBPFQJABD-LLVKDONJSA-N ramosetron Chemical compound C12=CC=CC=C2N(C)C=C1C(=O)[C@H]1CC(NC=N2)=C2CC1 NTHPAPBPFQJABD-LLVKDONJSA-N 0.000 description 1
- 229950001588 ramosetron Drugs 0.000 description 1
- 210000001609 raphe nuclei Anatomy 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 230000002787 reinforcement Effects 0.000 description 1
- 229960003394 remifentanil Drugs 0.000 description 1
- GZSKEXSLDPEFPT-IINYFYTJSA-N renzapride Chemical compound COC1=CC(N)=C(Cl)C=C1C(=O)N[C@H]1[C@H](C2)CCC[N@]2CC1 GZSKEXSLDPEFPT-IINYFYTJSA-N 0.000 description 1
- 229950003039 renzapride Drugs 0.000 description 1
- 238000009256 replacement therapy Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 229960003312 retigabine Drugs 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229940099204 ritalin Drugs 0.000 description 1
- 229960003014 rufinamide Drugs 0.000 description 1
- POGQSBRIGCQNEG-UHFFFAOYSA-N rufinamide Chemical compound N1=NC(C(=O)N)=CN1CC1=C(F)C=CC=C1F POGQSBRIGCQNEG-UHFFFAOYSA-N 0.000 description 1
- NEMGRZFTLSKBAP-LBPRGKRZSA-N safinamide Chemical compound C1=CC(CN[C@@H](C)C(N)=O)=CC=C1OCC1=CC=CC(F)=C1 NEMGRZFTLSKBAP-LBPRGKRZSA-N 0.000 description 1
- 229950002652 safinamide Drugs 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 230000036186 satiety Effects 0.000 description 1
- 235000019627 satiety Nutrition 0.000 description 1
- STECJAGHUSJQJN-FWXGHANASA-N scopolamine Chemical compound C1([C@@H](CO)C(=O)O[C@H]2C[C@@H]3N([C@H](C2)[C@@H]2[C@H]3O2)C)=CC=CC=C1 STECJAGHUSJQJN-FWXGHANASA-N 0.000 description 1
- 229960002646 scopolamine Drugs 0.000 description 1
- 230000036280 sedation Effects 0.000 description 1
- 230000004799 sedative–hypnotic effect Effects 0.000 description 1
- MEZLKOACVSPNER-GFCCVEGCSA-N selegiline Chemical compound C#CCN(C)[C@H](C)CC1=CC=CC=C1 MEZLKOACVSPNER-GFCCVEGCSA-N 0.000 description 1
- 229960003946 selegiline Drugs 0.000 description 1
- ANWPENAPCIFDSZ-BQBZGAKWSA-N seletracetam Chemical compound CC[C@@H](C(N)=O)N1C[C@@H](C=C(F)F)CC1=O ANWPENAPCIFDSZ-BQBZGAKWSA-N 0.000 description 1
- 229950000852 seletracetam Drugs 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 235000019615 sensations Nutrition 0.000 description 1
- 239000000952 serotonin receptor agonist Substances 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000004088 simulation Methods 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 230000005586 smoking cessation Effects 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229940083542 sodium Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- FJPYVLNWWICYDW-UHFFFAOYSA-M sodium;5,5-diphenylimidazolidin-1-ide-2,4-dione Chemical compound [Na+].O=C1[N-]C(=O)NC1(C=1C=CC=CC=1)C1=CC=CC=C1 FJPYVLNWWICYDW-UHFFFAOYSA-M 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 208000000995 spontaneous abortion Diseases 0.000 description 1
- 230000008925 spontaneous activity Effects 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000000021 stimulant Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 229960001897 stiripentol Drugs 0.000 description 1
- IBLNKMRFIPWSOY-FNORWQNLSA-N stiripentol Chemical compound CC(C)(C)C(O)\C=C\C1=CC=C2OCOC2=C1 IBLNKMRFIPWSOY-FNORWQNLSA-N 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical class O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- KQKPFRSPSRPDEB-UHFFFAOYSA-N sumatriptan Chemical compound CNS(=O)(=O)CC1=CC=C2NC=C(CCN(C)C)C2=C1 KQKPFRSPSRPDEB-UHFFFAOYSA-N 0.000 description 1
- 229960003708 sumatriptan Drugs 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 108060008037 tachykinin Proteins 0.000 description 1
- 229950004608 talampanel Drugs 0.000 description 1
- 229950000505 tandospirone Drugs 0.000 description 1
- CEIJFEGBUDEYSX-FZDBZEDMSA-N tandospirone Chemical compound O=C([C@@H]1[C@H]2CC[C@H](C2)[C@@H]1C1=O)N1CCCCN(CC1)CCN1C1=NC=CC=N1 CEIJFEGBUDEYSX-FZDBZEDMSA-N 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- VCVWXKKWDOJNIT-ZOMKSWQUSA-N tesofensine Chemical compound C1([C@H]2C[C@@H]3CC[C@@H](N3C)[C@@H]2COCC)=CC=C(Cl)C(Cl)=C1 VCVWXKKWDOJNIT-ZOMKSWQUSA-N 0.000 description 1
- 229950009970 tesofensine Drugs 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000004299 tetrazol-5-yl group Chemical group [H]N1N=NC(*)=N1 0.000 description 1
- VZYCZNZBPPHOFY-UHFFFAOYSA-N thioproperazine Chemical compound C12=CC(S(=O)(=O)N(C)C)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(C)CC1 VZYCZNZBPPHOFY-UHFFFAOYSA-N 0.000 description 1
- 229960003397 thioproperazine Drugs 0.000 description 1
- 230000035922 thirst Effects 0.000 description 1
- PBJUNZJWGZTSKL-MRXNPFEDSA-N tiagabine Chemical compound C1=CSC(C(=CCCN2C[C@@H](CCC2)C(O)=O)C2=C(C=CS2)C)=C1C PBJUNZJWGZTSKL-MRXNPFEDSA-N 0.000 description 1
- 229960001918 tiagabine Drugs 0.000 description 1
- 229960005138 tianeptine Drugs 0.000 description 1
- 229960002309 toloxatone Drugs 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- PHTUQLWOUWZIMZ-GZTJUZNOSA-N trans-dothiepin Chemical compound C1SC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 PHTUQLWOUWZIMZ-GZTJUZNOSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 229960003741 tranylcypromine Drugs 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 125000006168 tricyclic group Chemical group 0.000 description 1
- FEZBIKUBAYAZIU-UHFFFAOYSA-N trimethobenzamide Chemical compound COC1=C(OC)C(OC)=CC(C(=O)NCC=2C=CC(OCCN(C)C)=CC=2)=C1 FEZBIKUBAYAZIU-UHFFFAOYSA-N 0.000 description 1
- 229960004161 trimethobenzamide Drugs 0.000 description 1
- 229960002431 trimipramine Drugs 0.000 description 1
- ZSCDBOWYZJWBIY-UHFFFAOYSA-N trimipramine Chemical compound C1CC2=CC=CC=C2N(CC(CN(C)C)C)C2=CC=CC=C21 ZSCDBOWYZJWBIY-UHFFFAOYSA-N 0.000 description 1
- UIVFDCIXTSJXBB-ITGUQSILSA-N tropisetron Chemical compound C1=CC=C[C]2C(C(=O)O[C@H]3C[C@H]4CC[C@@H](C3)N4C)=CN=C21 UIVFDCIXTSJXBB-ITGUQSILSA-N 0.000 description 1
- 229960003688 tropisetron Drugs 0.000 description 1
- 229960004799 tryptophan Drugs 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N urea group Chemical group NC(=O)N XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 229960000604 valproic acid Drugs 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 229960005318 vigabatrin Drugs 0.000 description 1
- PJDFLNIOAUIZSL-UHFFFAOYSA-N vigabatrin Chemical compound C=CC(N)CCC(O)=O PJDFLNIOAUIZSL-UHFFFAOYSA-N 0.000 description 1
- 229960001255 viloxazine Drugs 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 125000005023 xylyl group Chemical group 0.000 description 1
- 229950001074 zatosetron Drugs 0.000 description 1
- 229960002791 zimeldine Drugs 0.000 description 1
- OYPPVKRFBIWMSX-SXGWCWSVSA-N zimeldine Chemical compound C=1C=CN=CC=1C(=C/CN(C)C)\C1=CC=C(Br)C=C1 OYPPVKRFBIWMSX-SXGWCWSVSA-N 0.000 description 1
- 229960002911 zonisamide Drugs 0.000 description 1
- UBQNRHZMVUUOMG-UHFFFAOYSA-N zonisamide Chemical compound C1=CC=C2C(CS(=O)(=O)N)=NOC2=C1 UBQNRHZMVUUOMG-UHFFFAOYSA-N 0.000 description 1
Images









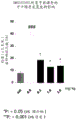


































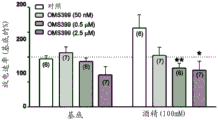


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/34—Tobacco-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Addiction (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Psychiatry (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
本公开涉及单独或与其它治疗剂联合使用磷酸二酯酶7(PDE7)抑制剂治疗成瘾和原发性冲动控制障碍。
Description
本申请是申请日为2013年5月7日,申请号为201380036048.1,发明名称为“使用PDE7抑制剂治疗成瘾和冲动控制障碍”的发明专利申请的分案申请。
关于序列表的声明
与本申请相关的序列表以文本格式代替纸质副本来提供并特此通过引用并入本说明书。含有所述序列表的文本文件的名称是NE_1_0172_SequenceListingFinal_ST25,经由EFS-Web在提交本说明书时递交。
技术领域
本发明涉及单独或联合其它治疗剂或成瘾剂,使用磷酸二酯酶7(PDE7)抑制剂预防和治疗物质和行为成瘾。
背景技术
世界卫生组织(WHO)将物质成瘾定义为反复地使用某种物质,尽管知道并经历其有害作用。物质成瘾是慢性、复发性疾病,特征为失控的药物使用,强迫觅药和渴求物质,不管不良后果坚持使用,以及对物质的身体和/或心理依赖。物质成瘾一般遵循忍耐、戒断、强迫性用药行为、觅药行为、成瘾行为实施和复发的过程。由于在暴力犯罪和传染性疾病传播方面起重要作用,物质滥用和成瘾是对成瘾者和社会都具有重大社会和经济影响的公共卫生事件。成瘾物质包括酒精、咖啡因、尼古丁、大麻(cannabis或marijuana)和大麻衍生物、阿片剂和其它吗啡样阿片类激动剂如海洛因、苯环利定和苯环利定样化合物、镇静催眠药如苯并二氮杂 类和巴比妥酸盐类以及精神兴奋药如可卡因、安非他明和安非他明相关药物如右旋安非他明和甲基安非他明。
类和巴比妥酸盐类以及精神兴奋药如可卡因、安非他明和安非他明相关药物如右旋安非他明和甲基安非他明。
酒精是全球最常见的滥用物质之一。此外,酒精中毒导致严重的肝和心血管疾病并产生依赖,造成重度精神障碍、社会问题和不良后果,包括家庭解体、悲惨事故和工作效能降低。根据WHO,酒精饮用导致了世界范围20-30%的食道癌和肝癌、肝硬化、杀人、癫痫和机动车事故。酒精滥用在全球每年导致约180万人死亡。对酒精饮用的强迫性行为是这一障碍的核心症状。近年来,已经研究了几种手段来帮助酗酒患者不仅控制饮酒,还控制酒精渴求和复发(Monti等人,J Stud Alcohol 54:235-45(1993);Volpicelli等人,Arch GemPsychiatry 49:876-880(1992);和0′Brien,Science 278:66-70(1997))。
测试了药物如纳曲酮、阿坎酸、昂丹司琼、戒酒硫、Y-羟基丁酸(GHB)、和托吡酯对酒精滥用的潜在治疗效果,这些药物属于不同类别(Volpicelli等人,1992;0′Brien等人,1997)。这些药物治疗剂中的几种如纳曲酮、阿坎酸和戒酒硫被证实有一定功用并被批准用于治疗酒精中毒。这些药物中,非选择性的阿片类拮抗剂纳曲酮目前被认为是药理学最佳的选择。然而,尽管有一些有希望的结果,但是这些药物(包括纳曲酮)中没有一种在酒精中毒方面足够有效并且预后仍较差。
尼古丁是最广泛使用的成瘾药物之一,并且尼古丁滥用是物质滥用的最常见形式。WHO估计,全球有12.5亿吸烟者,代表了年龄超过15岁的全球人口的1/3。WHO还估计,作为烟草使用的直接结果,全世界每年有5百万人死亡,使得尼古丁滥用成为全球最大的单一可预防死因。在工业化国家,70-90%的肺癌、56-80%慢性呼吸系统疾病和22%的心血管疾病病例都归因于尼古丁成瘾。吸烟仅在美国每年就与430,000人死亡相关,并且估计在卫生保健费用方面每年花费国家800亿美元。烟草使用对全部癌症的1/3负有责任,包括肺癌、口腔癌、咽癌、喉癌、食道癌、宫颈癌、肾癌、输尿管癌和膀胱癌。吸烟者中癌症的总体死亡率是不吸烟者的两倍高。吸烟还引起肺部疾病,如慢性支气管炎和肺气肿;恶化哮喘症状;并增加心脏疾病风险,包括中风、心脏病发作、血管病和动脉瘤。估计20%心脏病死亡可归因于吸烟。吸烟的怀孕妇女与不吸烟者相比有更大的早产、自然流产和婴儿出生重量减小的风险。
尼古丁使用导致神经递质多巴胺水平升高,它激活奖赏途径(reward pathways)来调节愉悦感觉并介导消耗尼古丁的愿望。与尼古丁戒断相关的症状包括渴求、易怒、发怒、敌意、攻击性、疲劳、抑郁和认知损伤,这导致滥用者寻求更多的尼古丁。环境条件因素和暴露于心理应激代表了诱发吸烟者使用尼古丁的其它因素。反复的尼古丁使用导致出现耐受,要求更高剂量的尼古丁来产生相同的初始刺激。
针对尼古丁成瘾开发的大多数疗法在预防复发方面仅显示出一般的成功,导致戒烟尝试的高失败率。治疗包括使用尼古丁替代产品、抗抑郁剂、抗过敏剂和行为疗法。
国立药物滥用研究所估计7200万美国人(约人口的1/3)尝试过大麻。使用大麻的急性效果包括记忆和学习障碍、扭曲直觉、问题解决困难、丧失协调性和心率增加。长期滥用会引起与在吸烟者中观察到的相同的呼吸问题,例如每天咳嗽、生痰、增加的肺感染风险和出现头、颈和肺癌的风险增高。抑郁、焦虑和与工作相关的问题与大麻使用有关。长期使用大麻会导致成瘾,其强迫性使用干扰日常活动。渴求和戒断症状如易怒、增加的攻击性、失眠和焦虑使得成瘾者难以停止使用大麻。没有可用于治疗大麻成瘾和复发的药物疗法。
根据WHO,估计全球有1300万人滥用阿片类物质,包括900万海洛因成瘾者。高于25%的阿片类滥用者在成瘾后10-20年内死于自杀、他杀或传染病,如HIV和肝炎。耐受和身体依赖可在二至三天内出现。对阿片剂的滥用和成瘾是已知现象,近年来的新现象是该问题日益恶化(Compton和Volkow,Drug Alcohol Depend 83suppl1:S4-7(2006A);和Compton和Volkow,Drug Alcohol Depend 81(2):103-7(2006B))。2003年在美国青年中的流行病学调查显示,在中学生(高三学生)中阿片类镇痛剂在最频繁滥用的非法药物中仅次于大麻(Delva等人,Am J Public Health95(4):696-702(2005))。此外,过去几年阿片类药物在美国的使用已显著增加,而与这种使用相关的问题甚至增加得更快。阿片类药物的使用和问题的增长特别受到关注,因为其似乎表示阿片剂成瘾途径的扩张(Siegal等人,Am FamPhysician 67:942-945(2003))。
根据近来的流行病学数据,2002年12岁以上的美国家庭居民中4.7%(即1100万人)滥用阿片类药物,并且这些人群中13.7%(即150万人)符合DSM-IV阿片剂使用障碍的标准(American Psychiatric Associaion,Diagnostic and Statistical Manual ofMental Disorders,第四版(1994);Substance Abuse and Mental Health ServicesAdministration,Mortality Data from the Drug Abuse Warning Network,2002,(2004))。如Compton和Volkow最近评估的,阿片类镇痛剂滥用的年度发生率从1990年的628,000名增加到2001年的240万名新成员(Substance Abuse and Mental HealthServices Administration,Overview of Findings from the 2002Naional Survey onDrug Use and Health,(2003);Substance Abuse and Mental Health ServicesAdministration,Emergency Department Trends From the Drug Abuse WarningNetwork,Final Estimates 1995-2002,(2003))。促进阿片剂成瘾扩张的原因之一是辅助处方药物的镇痛剂的增加使用。阿片类药物的短期使用几乎与成瘾无关。相反,长期使用这些药物治疗则与高达18%的患者成瘾的发生有关。
如同其它类型的物质成瘾一样,治疗阿片剂成瘾的目标是终止使用阿片剂,同时将痛苦的戒断症状减至最小并预防复发。当前的治疗涉及以阿片类受体激动剂或混合型激动剂/拮抗剂的替代品来代替成瘾性药物。其它的手段包括使用阿片类受体拮抗剂来阻断激动剂的作用。拮抗剂不缓解疼痛或其它戒断症状;相反,其可促成戒断,并且它们的治疗使用与意外阿片类激动剂用药过量增多和致死率增高有关。使用与受体亲和力较低的激动剂导致的重度戒断症状最少,但是会引起对替代品阿片剂的依赖。还有,很多替代疗法耗时3-6个月,为成瘾者中途停止治疗提供了时间。
精神兴奋药如可卡因和安非他明能暂时使人欣快、增加机敏性并提高人的体能。这些物质首先增加多巴胺传递,但是长期的药物使用导致多巴胺活性的降低,引起脑奖赏系统的失调和心境恶劣。WHO估计全世界有3300万人滥用安非他明。
长期可卡因滥用会导致过度刺激、心动过速、高血压、瞳孔扩大、肌颤搐、失眠、极度神经紧张、幻觉、偏执狂、攻击性行为和抑郁。可卡因用药过量会引起震颤、惊厥、谵妄以及由于心律失常和心血管衰竭导致的死亡。地昔帕明、金刚烷胺和溴隐亭已显示会减少可卡因戒断症状。
安非他明戒断症状包括EEG改变、疲劳和精神抑郁。随时间会产生耐受性,并可以与心动过速、幻听和幻视、妄想、焦虑反应、偏执型精神病、虚脱、精神错乱、记忆丧失和具有自杀倾向的长期抑郁相关。目前的安非他明成瘾的治疗包括用于幻觉的吩噻嗪类、氟哌啶醇和氯丙嗪,但是这些药物的潜在副作用包括体位性低血压和重度锥体外系运动障碍。对精神兴奋药成瘾者有时会通过心理戒断和生理戒断,使得潜在复发更有可能。
过去,物质成瘾的治疗集中在行为疗法,但是对很多这些高度成瘾物质的依赖却难以摆脱。特别是,对酒精、可卡因和海洛因的成瘾被认为是慢性、复发性障碍。另外,同时滥用多种物质如尼古丁、海洛因、可卡因和酒精也是常见的。
很多成瘾的长期、慢性性质和高再犯率对药物和酒精成瘾的治疗带来重大挑战,以致了解复发的神经生物学基础成为成瘾研究的中心课题。情绪和环境因素(条件刺激物)位列复发的主因之中。例如,已知特定的应激条件如失业和经济困难,或先前与酒精使用相关的预示其存在的刺激物如偏爱的酒的酒瓶和类似酒吧的环境,可以强烈促进戒酒的早先酗酒者复饮。
美国、欧洲和其他西方社会中肥胖的逐渐增长的发病率也表明食瘾类行为的流行。与正常进食行为的区别在于,食瘾是适应不良性行为,使得因成瘾而非饥饿感而进食的人们感觉越来越糟而不是越来越好。食瘾者的过度进食还是持久和习惯性的,经常进食过量的食物,并且通常进食过量的不健康食物。食瘾不仅导致肥胖,还可能反常地导致营养不良。食瘾和药物成瘾之间存在某些相似性,包括对情绪的影响、引起成瘾行为的外部诱因、期待、克制、矛盾情绪和归因。
存在两种主要的理论见解来解释成瘾行为的坚持和与药物和酒精成瘾相关的复发的易患性:稳态假说和条件作用假说。
稳态假说将复发风险与神经适应性变化和神经内分泌稳态的破坏联系起来,认为它们是伴随急性戒断产生的焦虑、情绪失调和身体症状的原因,这些症状可在称为“稽延性戒断(protracted withdrawal)”阶段期间持续相当长时期。因此,这种观点暗示改善不适和负面情感是复发的动机基础。
条件作用假说基于这样的观察结果:复发通常与暴露于药物相关环境刺激物有关。这种观点主张,通过经典的条件作用而与药物的奖赏作用变得相关的特定的环境刺激物可引起触发恢复药物使用的主观状态。稳态假说和条件作用假说并非相互排斥的。事实上,稳态和条件作用因素很可能发挥累加效应,因为暴露于药物相关环境刺激物可以增强稳态扰乱导致的复发易患性。
显然,本领域需要治疗和预防成瘾和复发使用成瘾剂的新方法。本发明通过提供可用于治疗和预防成瘾和再犯的方法和药物组合物满足了这些需求。
发明内容
本发明提供一种治疗或预防成瘾的方法,该方法确定受试者患有成瘾或有发生成瘾的危险,然后向该受试者施用对治疗或预防成瘾有效的磷酸二酯酶7(PDE7)抑制剂。
在本发明的一个方面,受试者对成瘾剂成瘾。成瘾剂的实例包括酒精、尼古丁、大麻、大麻衍生物、阿片类激动剂、苯并二氮杂 巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精。在另一实施方案中,成瘾剂是尼古丁。在又一实施方案中,成瘾剂是阿片类物质,例如吗啡、美沙酮、芬太尼、舒芬太尼、可待因、羟可待因和海洛因。在另一实施方案中,成瘾剂是精神兴奋药,例如,可卡因、安非他明或安非他明衍生物。在另一实施方案中,成瘾剂是可卡因。
巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精。在另一实施方案中,成瘾剂是尼古丁。在又一实施方案中,成瘾剂是阿片类物质,例如吗啡、美沙酮、芬太尼、舒芬太尼、可待因、羟可待因和海洛因。在另一实施方案中,成瘾剂是精神兴奋药,例如,可卡因、安非他明或安非他明衍生物。在另一实施方案中,成瘾剂是可卡因。
在本发明的一个方面,受试者对成瘾或强迫行为成瘾,或患有冲动控制障碍。在本发明的另一方面,受试者患有原发性冲动控制障碍,即,是其中该障碍是原发性障碍,而不是医源性的(继发于医学治疗)或继发于其他原发性疾病或障碍的冲动控制障碍。属于原发性冲动控制障碍的成瘾或强迫行为包括以下行为:暴食、病理性赌博、病理性使用电子设备、病理性使用电子视频游戏、病理性使用电子通讯设备、病理性使用移动电话、对色情作品成瘾、性成瘾、强迫性消费、厌食症、贪食症、间歇性暴发性障碍、盗窃癖、纵火癖、拔毛发癖、强迫过度运动和强迫过度工作。在另一实施方案中,成瘾或强迫行为是食瘾。在另一实施方案中,成瘾或强迫行为是暴食。在本发明的另一方面,成瘾或强迫障碍是强迫症。
在本发明的一个方面,用于治疗成瘾的PDE7抑制剂选自本文公开的以下化合物:式1A、式1B、式29、式30、式31、式32、式33、式34、式35、式36、式37、式38、式39、式40、式41、式42、式43A、式43B、式44、式45、式46、式47、式48、式49、式50、式51、式52、式53、式54、式6A、式6B、式6C、式6D、式6E、式6F、式6G、式6H、式16A、化合物1、化合物2、化合物3和化合物4。
在本发明的一个方面,该PDE7抑制剂抑制PDE7A和/或PDE7B活性的IC50小于约1μM。在一项实施方案中,PDE7抑制剂抑制PDE7A和/或PDE7B活性的IC50小于约100nM。在另一实施方案中,PDE7抑制剂抑制PDE1B活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的5倍更大。在另一实施方案中,PDE7抑制剂抑制PDE10活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的5倍更大。在又一实施方案中,该PDE7抑制剂抑制PDE3活性的IC50比抑制PDE7A活性的IC50和/或抑制PDE7B活性的IC50中的较小者的10倍更大。在另一实施方案中,该PDE7抑制剂抑制PDE3和PDE4活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大。在又一实施方案中,该PDE7抑制剂抑制PDE4和PDE8活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大。在另一实施方案中,该PDE7剂抑制PDE1、PDE2、PDE3、PDE4、PDE8、PDE10和PDE11活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大。在又一实施方案中,该PDE7抑制剂是选择性PDE7抑制剂,其抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者小于该试剂抑制PDE1-6和PDE8-11酶家族中任何其它PDE酶活性的IC50的十分之一。在另一实施方案中,该PDE7抑制剂是高选择性PDE7抑制剂,其抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者小于该试剂抑制PDE1-6和PDE8-11酶家族中任何其它PDE酶活性的IC50的五十分之一。在另一实施方案中,该PDE7抑制剂的分子量小于约450g/摩尔。在另一实施方案中。该PDE7抑制剂能穿透血/脑屏障。
本发明提供一种治疗或预防成瘾的方法,该方法确定受试者患有成瘾或有发生成瘾的危险,然后施用抑制PDE7活性的化合物。该化合物具有以下特点:(i)抑制PDE7A和/或PDE7B活性的IC50小于约1μM;和(ii)抑制PDE3的IC50比抑制PDE7A活性的IC50和/或抑制PDE7B活性的IC50中的较小者的10倍更大。
在一项实施方案中,该化合物抑制PDE7A和/或PDE7B活性的IC50小于约100nM。在另一实施方案中,PDE7抑制剂抑制PDE1B活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的5倍更大。在另一实施方案中,PDE7抑制剂抑制PDE10活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的5倍更大。在又一实施方案中,该PDE7抑制剂抑制PDE4活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大。在又一实施方案中,该PDE7抑制剂抑制PDE8活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大。在另一实施方案中,该PDE7剂抑制PDE1、PDE2、PDE3、PDE4、PDE8、PDE10和PDE11活性的IC50比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大。在又一实施方案中,该PDE7抑制剂是选择性PDE7抑制剂,其抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者小于该试剂抑制PDE1-6和PDE8-11酶家族中任何其它PDE酶活性的IC50的十分之一。在另一实施方案中,该PDE7抑制剂是高选择性PDE7抑制剂,其抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者小于该试剂抑制PDE1-6和PDE8-11酶家族中任何其它PDE酶活性的IC50的五十分之一。在另一实施方案中,该PDE7抑制剂的分子量小于约450g/摩尔。在另一实施方案中,该PDE7抑制剂能穿透血/脑屏障。
在本发明的一个方面,受试者对成瘾剂成瘾。成瘾剂的实例包括酒精、尼古丁、大麻、大麻衍生物、阿片类激动剂、苯并二氮杂 巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精。在另一实施方案中,成瘾剂是尼古丁。在又一实施方案中,成瘾剂是阿片类物质,例如吗啡、美沙酮、芬太尼、舒芬太尼、和海洛因。在另一实施方案中,成瘾剂是精神兴奋药,例如,可卡因、安非他明或安非他明衍生物。在另一实施方案中,成瘾剂是可卡因。
巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精。在另一实施方案中,成瘾剂是尼古丁。在又一实施方案中,成瘾剂是阿片类物质,例如吗啡、美沙酮、芬太尼、舒芬太尼、和海洛因。在另一实施方案中,成瘾剂是精神兴奋药,例如,可卡因、安非他明或安非他明衍生物。在另一实施方案中,成瘾剂是可卡因。
在本发明的一个方面,用PDE7抑制剂治疗的受试者对成瘾或强迫行为成瘾,或患有冲动控制障碍。在本发明的另一方面,受试者患有原发性冲动控制障碍,即,是其中该障碍是原发性障碍,而不是医源性的(继发于医学治疗)或继发于其他原发性疾病或障碍的冲动控制障碍。属于原发性冲动控制障碍的成瘾或强迫行为包括以下行为:暴食、病理性赌博、病理性使用电子设备、病理性使用电子视频游戏、病理性使用电子通讯设备、病理性使用移动电话、对色情作品成瘾、性成瘾、强迫性消费、厌食症、贪食症、间歇性暴发性障碍、盗窃癖、纵火癖、拔毛发癖、强迫过度运动和强迫过度工作。在另一实施方案中,受试者患有食瘾。在另一实施方案中,成瘾或强迫行为是暴食。在本发明的另一方面,待根据本发明治疗的受试者患有强迫症。
本发明还提供一种治疗或预防成瘾的方法,包括向患有成瘾的受试者提供磷酸二酯酶7(PDE7)的抑制剂和一种其他治疗剂,其中所述PDE7抑制剂和其他治疗剂各自有助于有效治疗或预防成瘾。所述的其他治疗剂包括,例如,阿片类拮抗剂、混合型阿片类部分激动剂/拮抗剂、抗抑郁药、抗癫痫药、止吐药、促肾上腺皮质素释放因子-1(CRF-1)受体拮抗剂、选择性5-羟色胺-3(5-HT3)拮抗剂、5-HT2A/2C拮抗剂、大麻素-1(CB-1)受体拮抗剂和多巴胺受体激动剂或其他多巴胺能剂。
阿片类拮抗剂的实例包括纳曲酮或纳美芬。抗抑郁药的实例包括氟西汀、米氮平和安非他酮。抗癫痫药的实例包括托吡酯、左乙拉西坦和加巴喷丁。安他拉明(Antalarmin)是CRF-1受体拮抗剂的一个实例。昂丹司琼是选择性5-羟色胺-3(5-HT-3)拮抗剂的实例。大麻素-1(CB1)受体拮抗剂的实例是利莫那班和泰伦那班(tanarabant)。丁丙诺啡是混合型阿片类激动剂/拮抗剂的实例。示例性的阿片类激动剂包括吗啡、美沙酮、芬太尼、舒芬太尼和海洛因。
示例性的多巴胺能剂包括,例如,左旋多巴(也称为“L-多巴”)、卡比多巴,和多巴胺受体激动剂和前体如溴隐亭、培高利特、普拉克索、罗匹尼罗、卡麦角林、阿扑吗啡、麦角乙脲、罗替戈汀和喹高利特,以及非诺多泮,其对多巴胺受体D1具有选择性。
在一个方面,受试者对成瘾剂成瘾,例如酒精、尼古丁、大麻、大麻衍生物、阿片类激动剂、苯并二氮杂 巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或是混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。在另一实施方案中,受试者对精神兴奋药成瘾,例如可卡因、安非他明、安非他明衍生物或去氧麻黄碱,而其他治疗剂是抗抑郁药,例如安非他酮。在又一实施方案中,受试者对尼古丁成瘾,其他治疗剂是抗抑郁药,例如安非他酮。
巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或是混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。在另一实施方案中,受试者对精神兴奋药成瘾,例如可卡因、安非他明、安非他明衍生物或去氧麻黄碱,而其他治疗剂是抗抑郁药,例如安非他酮。在又一实施方案中,受试者对尼古丁成瘾,其他治疗剂是抗抑郁药,例如安非他酮。
在另一方面,受试者对成瘾或强迫性行为成瘾,例如原发性冲动控制障碍,包括例如病理性赌博、暴食、病理性使用电子设备、病理性使用电子视频游戏、病理性使用电子通讯设备、病理性使用移动电话、对色情作品成瘾、性成瘾、强迫性消费、厌食症、贪食症、间歇性暴发性障碍、盗窃癖、纵火癖、拔毛发癖、强迫过度运动和强迫过度工作。在一项实施方案中,成瘾或强迫行为是暴食,其他治疗剂是托吡酯。在本发明的另一方面,待根据本发明治疗的受试者患有强迫症。
本发明提供一种预防成瘾剂的使用或成瘾或强迫行为的实施复发的方法,其包括治疗已经历一段时间的成瘾剂或者成瘾或强迫行为戒断,或者其使用受限或减少的受试者,其通过向所述受试者施用PDE7抑制剂进行。本发明还提供一种预防与原发性冲动控制障碍相关的成瘾或强迫行为复发的方法,其包括治疗已经历一段时间的与原发性冲动控制障碍相关的成瘾或强迫行为的免除,或者所述行为的实施受限或减少的受试者,其通过向所述受试者施用PDE7抑制剂进行。本发明还提供一种预防与强迫症相关的成瘾或强迫行为复发的方法,其包括治疗已经历一段时间的与强迫症相关的成瘾或强迫行为的免除,或者所述行为的实施受限或减少的受试者,其通过向所述受试者施用PDE7抑制剂进行。有助于有效预防复发的其他治疗剂可以与PDE7抑制剂一起施用。这一治疗方法可施用于曾经用(现已不用)不同的抗成瘾疗法治疗过的受试者。
在一个方面,通过施用PDE7抑制剂,预防了成瘾剂如酒精、尼古丁、大麻、大麻衍生物、阿片类激动剂、苯并二氮杂 巴比妥酸盐和精神兴奋药的复发使用。在优选的实施方案中,预防了可卡因、安非他明或去氧麻黄碱的复发使用。
巴比妥酸盐和精神兴奋药的复发使用。在优选的实施方案中,预防了可卡因、安非他明或去氧麻黄碱的复发使用。
在另一方面,通过施用PDE7抑制剂,预防了成瘾或强迫行为,特别是与原发性冲动控制障碍相关的成瘾或强迫行为的复发。在优选的实施方案中,预防了以下行为的复发:暴食、病理性赌博、病理性使用电子设备、病理性使用电子视频游戏、病理性使用电子通讯设备、病理性使用移动电话、对色情作品成瘾、性成瘾、强迫性消费、厌食症、贪食症、间歇性暴发性障碍、盗窃癖、纵火癖、拔毛发癖、强迫过度运动和强迫过度工作。在一项实施方案中,成瘾或强迫行为是由应激诱发的暴食。在另一实施方案中,对受试者进行治疗以预防与强迫症相关的成瘾或强迫行为的复发。
本发明提供一种药物组合物,其包含PDE7抑制剂和其他治疗剂,其中PDE7抑制剂和其他治疗剂均有助于有效治疗或预防成瘾。还提供了单位剂量的该药物组合物。
在本发明的一个方面,受试者对成瘾剂成瘾。成瘾剂的实例包括酒精、尼古丁、大麻、大麻衍生物、阿片类激动剂、苯并二氮杂 巴比妥酸盐、可卡因和其它精神兴奋药。在一项实施方案中,成瘾剂是酒精。在另一实施方案中,成瘾剂是尼古丁。在又一实施方案中,成瘾剂是阿片类物质,例如吗啡、美沙酮、芬太尼、舒芬太尼和海洛因。在另一实施方案中,成瘾剂是精神兴奋药,例如,可卡因、安非他明或安非他明衍生物。在另一实施方案中,成瘾剂是可卡因。
巴比妥酸盐、可卡因和其它精神兴奋药。在一项实施方案中,成瘾剂是酒精。在另一实施方案中,成瘾剂是尼古丁。在又一实施方案中,成瘾剂是阿片类物质,例如吗啡、美沙酮、芬太尼、舒芬太尼和海洛因。在另一实施方案中,成瘾剂是精神兴奋药,例如,可卡因、安非他明或安非他明衍生物。在另一实施方案中,成瘾剂是可卡因。
在本发明的一个方面,受试者对成瘾或强迫行为成瘾,或患有冲动控制障碍。在本发明的另一方面,受试者患有原发性冲动控制障碍,即,是其中该障碍是原发性障碍,而不是医源性的(继发于医学治疗)或继发于其他原发性疾病或障碍的冲动控制障碍。属于原发性冲动控制障碍的成瘾或强迫行为包括以下行为:暴食、病理性赌博、病理性使用电子设备、病理性使用电子视频游戏、病理性使用电子通讯设备、病理性使用移动电话、对色情作品成瘾、性成瘾、强迫性消费、厌食症、贪食症、间歇性暴发性障碍、盗窃癖、纵火癖、拔毛发癖、强迫过度运动和强迫过度工作。在优选实施方案中,成瘾或强迫行为是暴食。在本发明的另一方面,待根据本发明治疗的受试者患有强迫症。
在一项实施方案中,药物组合物中的其他治疗剂是阿片类拮抗剂、混合型阿片类部分激动剂/拮抗剂、抗抑郁药、抗癫痫药、止吐药、促肾上腺皮质素释放因子-1(CRF-1)受体拮抗剂、选择性5-羟色胺-3(5-HT3)拮抗剂、5-HT2A/2C拮抗剂或大麻素-1(CB-1)受体拮抗剂。
阿片类拮抗剂的实例包括纳曲酮或纳美芬。抗抑郁药的实例包括氟西汀、米氮平或安非他酮。抗癫痫药的实例包括托吡酯、左乙拉西坦和加巴喷丁。安他拉明是CRF-1受体拮抗剂的一个实例。昂丹司琼是选择性5-羟色胺-3(5-HT-3)拮抗剂的实例。大麻素-1(CB1)受体拮抗剂的实例是利莫那班和泰伦那班。丁丙诺啡是混合型阿片类激动剂/拮抗剂的实例。
在一个方面,受试者对成瘾剂成瘾,例如酒精、尼古丁、大麻、大麻衍生物、阿片类激动剂、苯并二氮杂 巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。在另一实施方案中,成瘾剂是尼古丁,其他治疗剂是伐尼克兰。在另一实施方案中,受试者对精神兴奋药成瘾,例如可卡因、安非他明、安非他明衍生物或去氧麻黄碱,其他治疗剂是抗抑郁药,例如安非他酮。在又一实施方案中,受试者对尼古丁成瘾,其他治疗剂是抗抑郁药,例如安非他酮。在另一实施方案中,受试者对多于一种的成瘾剂成瘾,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。
巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。在另一实施方案中,成瘾剂是尼古丁,其他治疗剂是伐尼克兰。在另一实施方案中,受试者对精神兴奋药成瘾,例如可卡因、安非他明、安非他明衍生物或去氧麻黄碱,其他治疗剂是抗抑郁药,例如安非他酮。在又一实施方案中,受试者对尼古丁成瘾,其他治疗剂是抗抑郁药,例如安非他酮。在另一实施方案中,受试者对多于一种的成瘾剂成瘾,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。
本发明提供一种用于治疗或预防成瘾的药盒。该药盒包括装有PDE7抑制剂的第一容器和装有其他治疗剂的第二容器。PDE7抑制剂和其他治疗剂均有助于有效治疗或预防成瘾。
在一项实施方案中,药物组合物中的其他治疗剂是阿片类拮抗剂、混合型阿片类部分激动剂/拮抗剂、抗抑郁药、抗癫痫药、止吐药、促肾上腺皮质素释放因子-1(CRF-1)受体拮抗剂、选择性5-羟色胺-3(5-HT3)拮抗剂、5-HT2A/2C拮抗剂或大麻素-1(CB-1)受体拮抗剂。
阿片类拮抗剂的实例包括纳曲酮和纳美芬。抗抑郁药的实例包括氟西汀、米氮平和安非他酮。抗癫痫药的实例包括托吡酯、左乙拉西坦和加巴喷丁。安他拉明是CRF-1受体拮抗剂的一个实例。昂丹司琼是选择性5-羟色胺-3(5-HT-3)拮抗剂的实例。大麻素-1(CB1)受体拮抗剂的实例是利莫那班和泰伦那班。丁丙诺啡是混合型阿片类激动剂/拮抗剂的实例。
在一个方面,受试者对成瘾剂成瘾,例如酒精、尼古丁、大麻、大麻衍生物、阿片类激动剂、苯并二氮杂 巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或是混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。在另一实施方案中,受试者对精神兴奋药成瘾,例如可卡因、安非他明、安非他明衍生物或去氧麻黄碱,而其他治疗剂是抗抑郁药,例如安非他酮。在又一实施方案中,受试者对尼古丁成瘾,其他治疗剂是抗抑郁药,例如安非他酮。在另一实施方案中,受试者对多于一种的成瘾剂成瘾,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。
巴比妥酸盐和精神兴奋药。在一项实施方案中,成瘾剂是酒精,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或是混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。在另一实施方案中,受试者对精神兴奋药成瘾,例如可卡因、安非他明、安非他明衍生物或去氧麻黄碱,而其他治疗剂是抗抑郁药,例如安非他酮。在又一实施方案中,受试者对尼古丁成瘾,其他治疗剂是抗抑郁药,例如安非他酮。在另一实施方案中,受试者对多于一种的成瘾剂成瘾,其他治疗剂是阿片类拮抗剂,例如纳曲酮,或混合型阿片类拮抗剂/部分激动剂,例如丁丙诺啡。
在本发明的另一方面,向对成瘾物质有成瘾危险的受试者施用与PDE7抑制剂相组合的该成瘾物质。例如,对于要施用阿片类激动剂以缓解急性或慢性疼痛的受试者,与PDE7抑制剂一起联合施用阿片类激动剂,以便提供不成瘾或少成瘾的止痛作用。可以与PDE7抑制剂联合(固定剂量组合或以药盒的形式)施用的成瘾剂的实例包括:苯并二氮杂 巴比妥酸盐及止痛药,包括阿芬太尼、烯丙罗定、阿法罗定、阿尼利定、苄吗啡、贝齐米特、丁丙诺啡、布托啡诺、氯尼他秦、可待因、环佐辛、地素吗啡、右吗拉胺、地佐辛、地恩丙胺、双氢可待因、双氢吗啡、地美沙朵、地美庚醇、二甲噻丁、吗苯丁酯、地匹哌酮、依他佐辛、依索庚嗪、乙甲噻丁、乙基吗啡、依托尼秦、芬太尼、海洛因、氢可酮、氢吗啡酮、羟哌替啶、异美沙酮、凯托米酮、左洛啡烷、左啡诺、左芬啡烷、洛芬太尼、哌替啶、美普他酚、美他佐辛、美沙酮、美托酮、吗啡、麦罗啡、纳布啡、那碎因、尼可吗啡、去甲左啡诺、去甲美沙酮、烯丙吗啡、去甲吗啡、诺匹哌酮、鸦片、羟考酮、奥斯康定
巴比妥酸盐及止痛药,包括阿芬太尼、烯丙罗定、阿法罗定、阿尼利定、苄吗啡、贝齐米特、丁丙诺啡、布托啡诺、氯尼他秦、可待因、环佐辛、地素吗啡、右吗拉胺、地佐辛、地恩丙胺、双氢可待因、双氢吗啡、地美沙朵、地美庚醇、二甲噻丁、吗苯丁酯、地匹哌酮、依他佐辛、依索庚嗪、乙甲噻丁、乙基吗啡、依托尼秦、芬太尼、海洛因、氢可酮、氢吗啡酮、羟哌替啶、异美沙酮、凯托米酮、左洛啡烷、左啡诺、左芬啡烷、洛芬太尼、哌替啶、美普他酚、美他佐辛、美沙酮、美托酮、吗啡、麦罗啡、纳布啡、那碎因、尼可吗啡、去甲左啡诺、去甲美沙酮、烯丙吗啡、去甲吗啡、诺匹哌酮、鸦片、羟考酮、奥斯康定 羟吗啡酮、阿片全碱、喷他佐辛、苯吗庚酮、非诺啡烷、非那佐辛、苯哌利定、匹米诺定、哌腈米特、普罗庚嗪、二甲哌替啶、丙哌利定、丙吡兰、右丙氧芬、舒芬太尼、曲马多、替利定,它们的盐,前述任何药物的混合物,以及混合型μ-激动剂/拮抗剂。
羟吗啡酮、阿片全碱、喷他佐辛、苯吗庚酮、非诺啡烷、非那佐辛、苯哌利定、匹米诺定、哌腈米特、普罗庚嗪、二甲哌替啶、丙哌利定、丙吡兰、右丙氧芬、舒芬太尼、曲马多、替利定,它们的盐,前述任何药物的混合物,以及混合型μ-激动剂/拮抗剂。
在一些实施方案中,对于本文描述的任何方法和组合物,均使用以下PDE7抑制剂:式1A、式1B、式29、式30、式31、式32、式33、式34、式35、式36、式37、式38、式39、式40、式41、式42、式43A、式43B、式44、式45、式46、式47、式48、式49、式50、式51、式52、式53、式54、式6A、式6B、式6C、式6D、式6E、式6F、式6G、式6H、式16A、化合物1、化合物2、化合物3和化合物4。
在本发明的另一实施方案中,向对成瘾物质成瘾或处于其成瘾风险中的受试者提供PDE7抑制剂以降低该受试者寻求或获得成瘾物质的积极性。
附图说明
本发明现在将例如参照附图予以更详细的说明,在附图中:
图1显示了PDE7抑制剂OMS182056对于大鼠可卡因自身给药的影响。
图2显示了PDE7抑制剂OMS181869对于大鼠可卡因自身给药的影响。
图3显示了PDE7抑制剂OMS182401对于大鼠可卡因自身给药的影响。
图4显示了多巴胺D1激动剂SKF82958对于大鼠可卡因自身给药的影响。
图5显示了PDE7抑制剂OMS182056对于大鼠的非强化压杆响应的影响。
图6显示了多巴胺D1激动剂SKF82958对于大鼠的非强化压杆响应的影响。
图7显示了PDE7抑制剂OMS182401对于大鼠可卡因自身给药的影响。
图8显示了PDE7抑制剂OMS182056在可卡因成瘾后消退期的第一天对大鼠的压杆响应的影响。
图9显示了PDE7抑制剂OMS182056对大鼠由育亨宾诱导的可卡因寻觅复发的影响。
图10显示了PDE7抑制剂OMS182401对大鼠由育亨宾诱导的可卡因寻觅复发的影响。
图11显示了PDE7抑制剂OMS182056对大鼠由信号(cue)诱导的可卡因寻觅复发的影响。
图12显示了PDE7抑制剂OMS182401对大鼠由信号诱导的可卡因寻觅复发的影响。
图13显示了PDE7抑制剂OMS182056对大鼠由可卡因点燃诱发的复发的影响。
图14显示了多巴胺D1激动剂SKF82958对大鼠由可卡因点燃诱发的复发的影响。
图15显示了采用短时程给药模型,PDE7抑制剂OMS182401对大鼠尼古丁自身给药的影响。
图16显示了采用长时程给药模型,PDE7抑制剂OMS182401对大鼠尼古丁自身给药的影响。
图17显示了采用短时程给药模型,PDE7抑制剂OMS182399对大鼠尼古丁自身给药的影响。
图18A和18B分别显示了PDE7抑制剂OMS182399和OMS182401对食物自身给予没有影响。
图19A-19C显示了在自身给药渐进比率研究中PDE7抑制剂OMS182401对尼古丁成瘾性小鼠获取尼古丁的积极性的影响。
图20显示了PDE7抑制剂OMS182401对尼古丁自身给药消退期的第一天的影响。
图21显示了PDE7抑制剂OMS182401对信号诱导的寻觅尼古丁行为的恢复的影响。
图22显示了PDE7抑制剂OMS182401对育亨宾诱导的寻觅尼古丁行为的恢复的影响。
图23A-23D显示了PDE7抑制剂OMS182401对大鼠由应激诱导的暴食行为的影响。图23A显示未受应激或遭受饮食限制的对照动物的结果。图23B显示未受应激但遭受饮食限制的实验动物的结果。图23C显示受应激但未遭受饮食限制的实验动物的结果。图23D显示受应激且遭受饮食限制的实验动物的结果。
图24A-24D显示了PDE7抑制剂OMS182056对大鼠由应激诱导的暴食行为的影响。图24A显示未受应激或遭受饮食限制的对照动物的结果。图24B显示未受应激但遭受饮食限制的实验动物的结果。图24C显示受应激但未遭受饮食限制的实验动物的结果。图24D显示受应激且遭受饮食限制的实验动物的结果。
图25A-25D显示作为对比物的托吡酯对大鼠由应激诱导的暴食行为的影响。图25A显示未受应激或遭受饮食限制的对照动物的结果。图25B显示未受应激但遭受饮食限制的实验动物的结果。图25C显示受应激但未遭受饮食限制的实验动物的结果。图25D显示受应激且遭受饮食限制的实验动物的结果。
图26显示在食瘾模型中PDE7抑制剂OMS182399对育亨宾诱导的觅食复发的影响。
图27和28分别显示体内微量透析研究中PDE7抑制剂OMS182399对Wistar大鼠伏隔核中的基底多巴胺释放和尼古丁诱导的多巴胺释放的影响。
图29A-29B显示PDE7抑制剂OMS182401抑制多巴胺能腹侧被盖区(VTA)神经元的自发性活动并增强多巴胺D1激动剂SKF82958的抑制作用。
图30显示PDE7抑制剂OMS182401对VTA GABA释放的影响。
图31显示PDE7抑制剂OMS182401抑制尼古丁对多巴胺能VTA神经元的激活并与多巴胺D1激动剂SKF82958协同作用。
图32显示将PDE7抑制剂OMS182401直接给药于大鼠VTA的影响。
图33显示PDE7抑制剂OMS182399剂量依赖性地阻断吗啡诱导的VTA多巴胺细胞的兴奋。
图34显示PDE7抑制剂OMS182399的急性施用剂量依赖性地阻断酒精诱导的VTA多巴胺细胞的兴奋。
图35显示PDE7抑制剂OMS182399的急性施用剂量依赖性地逆转可卡因诱导的VTA多巴胺细胞的兴奋。
图36显示PDE7抑制剂OMS182401在旷场活动模型中增强多巴胺D1激动剂SKF82958的活性。
具体实施方案
本发明是基于本发明人的令人惊奇的以下发现:7型环状核苷酸磷酸二酯酶(PDE7)的选择性抑制剂引起成瘾复发的显著减少。使用大鼠模型,在对成瘾剂成瘾的受试者和显示强迫行为的受试者中证实了这种减少。
A.用PDE7抑制剂治疗和预防成瘾的方法
因此,本发明包括治疗或预防成瘾的方法,该方法包括向成瘾的或有发生成瘾危险的受试者施用一种或多种PDE7抑制剂。在各种实施方案中,受试者是对成瘾剂或行为成瘾,包括但不限于,本文中描述的任何成瘾剂和行为。受试者可以是身体上或生理上对物质或行为依赖;受试者可以是心理上依赖;或者受试者可以是身体上或心理上都依赖。受试者可以对一种或多于一种的成瘾剂或行为成瘾。
用在本文时,除非上下文另外清楚地表明,“治疗(treat)”和类似词语如“治疗(treatment)”、“治疗(treating)”等,是用来获得有益或者期望的结果(包括并优选是临床结果)的方法。治疗可涉及任选地减弱或改善疾病或疾病状态(例如成瘾或者使用或行为复发),或者延缓疾病或者疾病状态(例如成瘾,或者使用或行为复发)的进展。
用在本文时,除非上下文另外清楚地表明,“预防(prevent)”和类似词语如“预防(prevention)”、“预防(preventing)”等是为防止疾病或疾病状态(例如成瘾或者使用或行为复发)的发作或再发,或者防止疾病或疾病状态的症状的出现或再发的方法,或者任选地为延缓疾病或疾病状态的发作或再发或者延缓疾病或疾病状态的症状的出现或再发的方法。
用在本文时,术语“PDE7”通常是指被以下两个基因:PDE7A和/或PDE7B之一或二者的转录物编码的所有的翻译产物。
用在本文时,术语“PDE7抑制剂”或“PDE7的抑制剂”是指直接或间接抑制或阻断PDE7A,PDE7B或者PDE7A和PDE7B的磷酸二酯酶活性的试剂,例如化合物、肽或核酸分子。在一些情形中,该试剂可以直接与PDE7蛋白结合或相互作用。与PDE7结合的试剂可通过任何合适的方式起作用以抑制或阻断PDE7活化,例如通过抑制cAMP或底物配体与PDE7的结合。在其它情形中,PDE7抑制剂可以间接地抑制PDE7活性,例如通过减少PDE7蛋白的表达。在一些情形中,PDE7抑制剂可以通过改变PDE7的细胞分布,例如通过干扰PDE7与细胞内锚定蛋白之间的结合,抑制PDE7活性。
用在本文时,术语“多巴胺能剂”是指功能为增强或复制中枢神经系统中由多巴胺介导的作用的试剂,包括多巴胺(如果应开发临床有效的递送方法的话)、多巴胺前体,如左旋多巴(L-多巴)、多巴胺辅因子,代谢多巴胺的酶的抑制剂,其他多巴胺受体激动剂和通过代谢转化为多巴胺受体激动剂的前体化合物,以及多巴胺再摄取抑制剂和多巴胺释放促进剂。
用在本文时,术语“多巴胺受体激动剂”是指引起多巴胺受体蛋白质家族的一种或多种亚型的活化的任何分子。
用在本文时,术语“哺乳动物受试者”包括所有的哺乳动物,包括但不限于:人、非人灵长动物、犬、猫、马、绵羊、山羊、牛、兔、猪和啮齿动物。
通常,向受试者提供有效量的PDE7抑制剂。用在本文时,物质(例如PDE7抑制剂)的“有效量”或“治疗有效量”,是足以实现预期的生物或心理作用(例如有利结果,包括临床结果)的量。例如,就使用本发明的方法治疗成瘾而言,PDE7抑制剂的有效量是足以使受试者减少或终止使用成瘾剂的量。在成瘾行为的情形下,PDE7抑制剂的有效量是足以使受试者减少或终止成瘾行为的量。
在一项实施方案中,治疗有效剂量是足以抑制神经元细胞中的PDE7酶活性的PDE7抑制剂的量。在本发明方法的另一实施方案中,治疗有效剂量是足以抑制纹状体神经元或伏隔核中的PDE7酶活性的PDE7抑制剂的量。足以穿过细胞膜并抑制细胞内的PDE7酶活性的PDE7抑制剂的有效剂量的确定可以用PDE7抑制的细胞试验法确定,如Smith S.J.等人,Molecular Pharmacology 66(6):1679-1689(2004)所述,该文在此并入作为参考。足以抑制纹状体中PDE7酶活性的PDE7抑制剂的有效剂量的确定可以用测量PDE抑制剂对纹状体内cAMP水平的影响的试验方法确定,如Siuciak JA.等人,Neuropharmacology 51:386-396(2006)所述,该文在此并入作为参考。
根据本发明的某些实施方案,向受试者只提供PDE7抑制剂,而在其它实施方案中,向受试者提供与其他治疗剂联合的PDE7抑制剂。应当清楚,当PDE7抑制剂和其他治疗剂中的任何一个单独提供时,与联合提供时相比,PDE7抑制剂和其他治疗剂中的一个或二者的有效量可以不同。例如,当PDE7抑制剂和其他治疗剂协同作用时,为达到PDE7抑制剂或其他治疗剂单独提供的相同疗效,可能需要较少量的PDE7抑制剂,较少量的其他治疗剂,或者PDE7抑制剂和其他治疗剂二者量均较少。在其它实施方案中,使用相同量的PDE7抑制剂和其他治疗剂以提供比单独的PDE7抑制剂或其他治疗剂提供的疗效更高的疗效。
根据本发明的某些实施方案,向受试者提供与成瘾性治疗剂联合的PDE7抑制剂,其中该成瘾性治疗剂的剂量经测定能实现预期的疗效,而PDE7抑制剂的剂量经测定能消除或减少对成瘾性治疗剂成瘾的可能性。
受试者可以是任何动物,包括哺乳动物,特别是人。
在本发明的一个方面,先利用诊断试验、医护人员的观察或分析,确定或诊断受试者患有成瘾,或有发生成瘾的危险。然后向受试者提供有效量的PDE7抑制剂,或有效量的PDE7抑制剂和一种其他治疗剂,用以治疗或预防成瘾。在本发明的另一方面,先利用诊断试验、医护人员的观察或分析,确定或诊断受试者患有成瘾,或有发生成瘾的危险,但该受试者尚未被诊断或确定患有糖尿病或其它胰岛素障碍。然后向受试者提供有效量的PDE7抑制剂,或有效量的PDE7抑制剂和一种其他治疗剂,用以治疗或预防成瘾。PDE7抑制剂或PDE7抑制剂和一种其他治疗剂的剂量可由医师具体确定,用于治疗或预防成瘾,而不是用于任何其它障碍或疾病。
在特定的实施方案中,受试者对任何身体成瘾剂或者成瘾或强迫行为(包括,例如任意下面描述的那些)成瘾,或有成瘾的危险。在特定的实施方案中,受试者对以下物质成瘾:酒精、可卡因、尼古丁、大麻、鸦片剂或其它阿片类激动剂、或去氧麻黄碱或其它精神兴奋药、或苯环利定和苯环利定衍生物。在另一实施方案中,受试者患有原发性冲动控制障碍。在又一实施方案中,受试者患有强迫症。在另一实施方案中,受试者有反复节食的历史,处在暴食的危险之中。
在特定的实施方案中,当受试者先前曾对相同或不同的成瘾剂或者成瘾或强迫行为成瘾时,该受试者被认为处在对成瘾剂的使用或成瘾行为的实施成瘾或者复发的危险之中。在某些实施方案中,当受试者心理上对成瘾剂或者成瘾或强迫行为成瘾时,即使该受试者身体上不再成瘾,该受试者仍被认为处在对成瘾剂的使用或成瘾行为的实施成瘾或者复发的危险之中。在一项实施方案中,该受试者患有食瘾。在另一实施方案中,成瘾行为是暴食。有暴食危险的受试者通常其历史中有以下的至少一项:经常性节食或悠悠球式节食(yo-yo dieting),响应环境应激而进食,偏爱极美味和高热量的食物,饱后进食,以及进食到不舒服为止。在另一实施方案中,受试者患有原发性冲动控制障碍。
在某些实施方案中,受试者对提供给患者用于治疗疾病或障碍的治疗剂(例如止痛药物)成瘾或有变得成瘾的危险。在相关的实施方案中,受试者可能有滥用成瘾性治疗剂(例如止痛药物)的危险。在某些实施方案中,滥用成瘾性治疗剂应理解成为了不同于或附加于其处方用途的其它原因而使用该药剂。在这种情形下,可以单独地或与其他治疗剂相联合,向受试者同时提供成瘾性治疗剂和PDE7抑制剂。例如,可以向患有疼痛或有疼痛危险的受试者提供阿片类激动剂和PDE7抑制剂,以便同时提供止痛作用和预防或治疗对阿片类激动剂的成瘾。
在各种实施方案中,向受试者提供PDE7抑制剂可以是在其使用成瘾剂的同时,在其终止使用成瘾剂之后,或在其开始使用成瘾剂之前。
成瘾剂和冲动控制障碍
术语“成瘾”被用来描述个体为从事某种特定活动而反复出现的强迫性冲动,不顾对该个体的健康、精神状态或社会生活的有害后果。该术语常常专用于药物成瘾,但是有时也用于其它强迫性冲动,例如问题赌博和暴食。已提出的作为成瘾原因的因素包括遗传、生物学/药理学和社会因素。
医学界现在对身体或生理依赖(以戒断症状为特征)和心理依赖(有时简单地称为成瘾)作了仔细的理论区分。现在狭义上将成瘾定义为“失控的强迫使用”。如果没有损害或者伤及患者或另一方,则在临床上可以将其认为是强迫的,但是就某种定义来说其并不归类为“成瘾”。实际上,这两类成瘾(生理依赖和心理依赖)不总是容易区分。成瘾常常同时具有身体和心理成分。
“身体依赖”(或“药物依赖”)指习惯性使用药物而导致的状态,其中突然中止会导致负面的身体戒断症状。使用者可能发生身体依赖的成瘾剂的实例包括尼古丁、阿片类物质、巴比妥酸盐类、苯并二氮杂 类、酒精即乙醇、GHB和甲喹酮。
类、酒精即乙醇、GHB和甲喹酮。
通常滥用的兴奋药如可卡因或安非他明类药物不被认为会引起明显的身体依赖。但是,它们的极端心理成瘾的可能性会驱使使用者消耗损害身体的数量,但是尚未观察到危及生命的戒断效应。
用在本文时,“成瘾剂”包括个体可对其成瘾的任何和所有试剂,无论是身体上或是心理上成瘾,或者二者。如上文所提到的,成瘾包括对化学实体的成瘾,例如药物,以及对行为的成瘾,如在冲动控制障碍中。
成瘾剂包括消遣性成瘾药物,以及成瘾性药物。成瘾剂的实例包括但不限于:酒精如乙醇、γ-羟基丁酸(GHB)、咖啡因、尼古丁、大麻(marijuana)和大麻衍生物、鸦片剂和其它吗啡样阿片类激动剂如海洛因、苯环利定和苯环利定样化合物,镇静催眠药如苯并二氮杂 类、甲喹酮、甲氯喹酮、依他喹酮和巴比妥酸盐类,及精神兴奋药如可卡因、安非他明和安非他明相关药物如右旋安非他明和甲基苯丙胺。其它的实例包括LSD、赛洛西宾、摇头丸和其它致幻药。成瘾性药物的实例包括,例如,苯并二氮杂
类、甲喹酮、甲氯喹酮、依他喹酮和巴比妥酸盐类,及精神兴奋药如可卡因、安非他明和安非他明相关药物如右旋安非他明和甲基苯丙胺。其它的实例包括LSD、赛洛西宾、摇头丸和其它致幻药。成瘾性药物的实例包括,例如,苯并二氮杂 类、巴比妥酸盐类和止痛药物,包括阿芬太尼、烯丙罗定、阿法罗定、阿尼利定、苄吗啡、贝齐米特、丁丙诺啡、布托啡诺、氯尼他秦、可待因、环佐辛、地素吗啡、右吗拉胺、地佐辛、地恩丙胺、双氢可待因、双氢吗啡、地美沙朵、地美庚醇、二甲噻丁、吗苯丁酯、地匹哌酮、依他佐辛、依索庚嗪、乙甲噻丁、乙基吗啡、依托尼秦、芬太尼、海洛因、氢可酮、氢吗啡酮、羟哌替啶、异美沙酮、凯托米酮、左洛啡烷、左啡诺、左芬啡烷、洛芬太尼、哌替啶、美普他酚、美他佐辛、美沙酮、美托酮、吗啡、麦罗啡、纳布啡、那碎因、尼可吗啡、去甲左啡诺、去甲美沙酮、烯丙吗啡、去甲吗啡、诺匹哌酮、鸦片、羟考酮、奥斯康定
类、巴比妥酸盐类和止痛药物,包括阿芬太尼、烯丙罗定、阿法罗定、阿尼利定、苄吗啡、贝齐米特、丁丙诺啡、布托啡诺、氯尼他秦、可待因、环佐辛、地素吗啡、右吗拉胺、地佐辛、地恩丙胺、双氢可待因、双氢吗啡、地美沙朵、地美庚醇、二甲噻丁、吗苯丁酯、地匹哌酮、依他佐辛、依索庚嗪、乙甲噻丁、乙基吗啡、依托尼秦、芬太尼、海洛因、氢可酮、氢吗啡酮、羟哌替啶、异美沙酮、凯托米酮、左洛啡烷、左啡诺、左芬啡烷、洛芬太尼、哌替啶、美普他酚、美他佐辛、美沙酮、美托酮、吗啡、麦罗啡、纳布啡、那碎因、尼可吗啡、去甲左啡诺、去甲美沙酮、烯丙吗啡、去甲吗啡、诺匹哌酮、鸦片、羟考酮、奥斯康定 羟吗啡酮、阿片全碱、喷他佐辛、苯吗庚酮、非诺啡烷、非那佐辛、苯哌利定、匹米诺定、哌腈米特、普罗庚嗪、二甲哌替啶、丙哌利定、丙吡兰、右丙氧芬、舒芬太尼、曲马多、替利定,它们的盐,前述任何药物的混合物,混合型μ-激动剂/拮抗剂等。
羟吗啡酮、阿片全碱、喷他佐辛、苯吗庚酮、非诺啡烷、非那佐辛、苯哌利定、匹米诺定、哌腈米特、普罗庚嗪、二甲哌替啶、丙哌利定、丙吡兰、右丙氧芬、舒芬太尼、曲马多、替利定,它们的盐,前述任何药物的混合物,混合型μ-激动剂/拮抗剂等。
在某些实施方案中,受试者可能对阿片类激动剂成瘾。术语“阿片类激动剂”、“阿片类物质”和“鸦片剂”在本文中可互换使用,并用于代表一组药物,它们在性质上不同程度地是鸦片样或吗啡样药物。它们的主要用途是缓解疼痛。这些试剂通过与阿片类受体结合而起作用,阿片类受体主要存在于中枢神经系统和胃肠道中。鸦片剂也是成瘾剂。鸦片剂包括阿芬太尼、烯丙罗定、阿法罗定、阿尼利定、阿朴吗啡、苄吗啡、β-羟基3-甲基芬太尼、贝齐米特、卡芬太尼、氯尼他秦、可待因、地索吗啡、右吗拉胺、二乙酰吗啡(海洛因)、地恩丙胺、双氢可待因、二氢埃托啡、双氢吗啡、地美沙朵、地美庚醇、二甲噻丁、吗苯丁酯、地匹哌酮、依他佐辛、依索庚嗪、乙甲噻丁、乙基吗啡、依托尼秦、埃托啡、芬太尼、氢可酮、氢吗啡酮、羟哌替啶、异美沙酮、凯托米酮、LMM、左啡诺、左芬啡烷、洛芬太尼、哌替啶、metapon、美他佐辛、美沙酮、醋美沙朵、美托酮、吗啡、麦罗啡、那碎因、尼可吗啡、去甲左诺啡、去甲美沙酮、去甲吗啡、诺匹呱酮、鸦片、羟考酮、羟吗啡酮、婴粟碱、苯吗庚酮、非诺啡烷、苯哌利定、匹米诺定、呱腈米特、普罗庚嗪、二甲哌替啶、丙哌利定、右丙氧芬、瑞芬太尼、舒芬太尼、蒂巴因、替利定(tildine)及曲马多。
天然存在的鸦片剂包括可待因、吗啡、诺斯卡品、婴粟碱、以及蒂巴因。半合成的鸦片类包括二乙酰吗啡、氢可酮、氢吗啡酮、左啡诺、metapon、烯丙吗啡、纳洛酮、纳曲酮、羟考酮、羟吗啡酮以及曲马多。合成的鸦片类包括依索庚嗪、芬太尼、左啡诺、哌替啶、美沙酮、非那佐辛、右丙氧芬和舒芬太尼。
鸦片剂的三个大类为菲类、苯基庚胺类和苯基哌啶类。菲类的实例包括可待因、埃托啡、氢可酮、氢吗啡酮、吗啡、羟考酮、以及羟吗啡酮。苯基庚胺类的实例包括地美庚醇、地美沙朵、地匹哌酮、异美沙酮、美沙酮、醋美沙朵、以及右丙氧芬。苯基哌啶类的实例包括阿芬太尼、阿法罗定、β-二甲哌替啶、卡芬太尼、芬太尼、洛芬太尼、哌替啶、丙哌利定及舒芬太尼。
作为实例,具体的精神兴奋药包括安非他明、可卡因、右旋安非他明、去氧麻黄碱、匹莫林、利他林、Adderall以及亚甲二氧基去氧麻黄碱。
如上所述,成瘾包括行为成瘾,例如食瘾、暴食症、病理性赌博、病理性使用电子设备(例如 病理性使用电子视频游戏、病理性使用电子通讯设备、病理性使用移动电话、对色情作品成瘾、性成瘾、强迫症、强迫性消费、间歇性暴发性障碍、盗窃癖、纵火癖、拔毛发癖、强迫过度运动、和强迫过度工作。
病理性使用电子视频游戏、病理性使用电子通讯设备、病理性使用移动电话、对色情作品成瘾、性成瘾、强迫症、强迫性消费、间歇性暴发性障碍、盗窃癖、纵火癖、拔毛发癖、强迫过度运动、和强迫过度工作。
在本发明的一方面,用PDE7抑制剂治疗的受试者患有食瘾或暴食症,通常存在继发性健康问题如由于过度进食导致的肥胖和/或由于过度进食高脂肪和/或糖分而维生素和矿物质含量低的食物而导致的营养不良。
本文所用的“暴食症”或“暴食”包括以下阵发式症状中的至少一种:进食大量的食物,即使饱了仍进食,迅速进食、感觉进食行为不受控制、不饿时进食大量的食物、频繁节食可能体重不减、单独进食、对饮食习惯感到沮丧或厌恶、因应激而进食。暴食症区别于其他类型的进食障碍,包括食瘾、贪食症和易饥症(binge-purge syndromes)。与贪食症和易饥症不同,患有暴食症和食瘾类行为的个体是不进行代偿性行为以减弱过量卡路里消耗的患有暴食症和食瘾类行为的个体。
考虑到暴食和长期过量进食与成瘾行为共有许多特征(例如控制减弱,持续使用而不管负面影响),正在形成大量科学文献来支持将某些类型的有问题的进食行为表征为成瘾行为。尽管具有相似性,暴食症和食瘾可能代表独特但有所重叠的状况。Gearhardt,A.N.等人,Binge Eating Disorder and Food Addiction,Curr.Drug Abuse Rev,4:201-207(2011)。
本文所用的术语“食瘾”是指经常性的、持久的且习惯性的过度进食模式,其特征在于渴望和寻求高卡路里食物、由于饥饿之外的刺激而过度进食、对食物摄入的控制减弱、持续进食而不管负面影响以及减少和放弃进食过量食物的能力减弱。食瘾是一种慢性复发性障碍,其通常遵循过度进食、忍耐、戒断、高卡路里食物寻求行为和复发(在一段时间的禁食后又开始过量进食)的过程。
由于与富含脂肪和碳水化合物的极美味食物相关的高卡路里支持的进化原因,产生了对这类食物的天生偏好。尽管毫无疑问进食行为受体内平衡机制调节,但进食和过度进食也受情绪、情感和学习过程控制。Polivy等人,“Food restriction and bingeeating:a study of a former prisoner of war,”J Abnorm Psychol 103:409-411(1994)。在这方面,过量进食和药物滥用之间存在一些共性(奖赏、强化、对情绪的影响、外部信号控制的食欲、应激诱导的动机)。越来越多的证据表明在特定条件下过量摄入某些食物会在大脑中产生类似于成瘾样状态的行为和变化。Gold等人,“Overeating,bingeeating,and eating disorders as addictions,”Psychiatr Ann 33:112-116(2003);Kenny等人,“Common cellular and molecular mechanisms in obesity and drugaddiction,”Nat Rev Neurosci 12:638-651(2011);Pelchat等人,“Images of desire:food-craving activation during fMRI,”Neuroimage 23:1486-1493(2004);Avena等人,“Evidence for sugar addiction:behavioral and neurochemical effects ofintermittent,excessive sugar intake,”Neurosci Biobehav Rev 32:20-39(2008);Ifland等人,“Refined food addiction:a classic substance use disorder,”MedHypotheses72:518-526(2009);Gearhardt等人,“Food addiction:an examination ofthe diagnostic criteria for dependence,”J Addict Med 3:1-7(2011)。
药物成瘾的特征在于药物使用的渐进性增加、产生耐受性、突然停止其使用后的节制以及反复复发,对于高蔗糖高脂肪食物已经描述了类似的现象。例如,喂食极美味蔗糖溶液的大鼠在移走蔗糖后经历了鸦片样戒断症状。也已经描述了蔗糖和药物滥用之间的交叉敏感。Avena等人,“Evidence for sugar addiction:behavioral and neurochemicaleffects of intermittent,excessive sugar intake,”Neurosci Biobehav Rev 32:20-39(2008);Avena等人,“Dysregulation of brain reward systems in eatingdisorders:Neurochemical information from animal models of binge eating,bulimia nervosa,and anorexia nervosa,”Neuropharmacology(印刷前的Epub)(Nov.27,2011)。而且,已经有大量的文献记载心理应激和心境烦躁不安的状态在促进过度进食以及过量药物使用方面起到主要作用,并促成了这两种障碍特有的高再犯率。Corwin等人,“Feeling and reward:perspective from three rat models of binge eating,”Physiol Behav 104:87-97(2011);Ghitza等人,“The anxiogenic drug yohimbinereinstates palatable food seeking in a rat relapse model:a role of CRF1receptors,”Neuropsychopharmacology 31(10):2188-2196(2006);Mizes等人,“Bulimia:A review of its symptomatology and treatment,”Adv.Behav.Res.Ther.7:91-142(1985);Crowther等人.,“The role of daily hassles in binge eating,”Int J EatDisord 29:449-454(2001);Herman等人,“Anxiety,restraint,and eating behaviour,”JAbnorm Psychol 84:66-72(1975)。
还已经显示暴露于环境条件刺激在诱发成瘾个体的药物渴求以及符合食瘾标准的肥胖患者的食物渴求方面起到至关重要的作用。Gearhardt等人,“Food addiction:anexamination of the diagnostic criteria for dependence,”J Addict Med 3:1-7(2011)。与人类的研究结果相一致,在实验动物中已经显示暴露于环境调节因素和暴露于应激(即育亨宾)可同样有效地诱发药物滥用和极美味食物寻求的复发行为。Cifani等人,“Preclinical model of binge-eating elicited by yo-yo dieting and stressfulexposure to food:effect of sibutramine,fluoxetine,topiramate and midazolam,”Psychopharmacology 204:113-25(2009);Cifani等人,“Pre-exposure to environmentalcues predictive of food availability elicits hypothalamic-pituitary-adrenalaxis activation and increases operant responding for food in female rats,”Addict Biol.14(4):397-407(Sept.2009);Pickens等人,“Effect of fenfluramine onreinstatement of food seeking in female and male rats:implications for thepredictive validity of the reinstatement model,”Psychopharmacology(Berl)221(2):341-353(May 2012)。还已经提出激发和加强药物滥用的神经系统是与强迫性食物寻觅和过量食物摄入相关的行为的基础。Johnson等人,“Dopamine D2 receptors inaddiction-like reward dysfunction and compulsive eating in obese rats,”NatNeurosci 13:635-641(2010);Hoebel等人,“Brain neurotransmitters in food anddrug reward,”Am J Clin Nutr 42(5 Suppl):1133-1150(1985);Vo1kow等人,“How candrug addiction help us understand obesity?”Nat Neurosci 8:555-560(2005);Corwin等人.,“Feeling and reward:perspective from three rat models of bingeeating,”Physiol Behav 104:87-97(2011);Gearhardt等人,“Neural correlates offood addiction,”Arch Gen Psychiatry 68:808-816(2011);Wang等人,“Enhancedstriatal dopamine release during food stimulation during binge eatingdisorder,”Obesity 19(8):1601-1608(Aug.2011)。例如,研究表明与药物成瘾者类似,在神经性贪食症患者中可能存在纹状体多巴胺(DA)的调节改变。
对食瘾概念的兴趣近来受到更多的关注,在很大程度上是由于成瘾与暴食症(BED)的行为表征之间的相似性。人类暴食行为发作的特征在于在短时期内强迫性、非自我平衡的食用不寻常的大量的极美味食物。尽管他们并不饥饿,个体比正常更快地进食直到感觉不舒服的饱腹感。如DMS-IV-TR(American Psychiatric Association,“Diagnosticand statistic manual of mental disorders,”Washington,DC.(2000))所述,这类发作伴随有对进食失去控制的主观感觉,并且与悲痛、厌恶、沮丧、对过度进食感到内疚以及因窘而独自进食的感觉相关。
由Stunkard,“Eating patterns and obesity,”Psychiatry Q 33:284-295(1959)首次描述的BED可能是最普遍的饮食障碍。Hudson等人,“The prevalence andcorrelates of eating disorders in the National Comorbidity SurveyReplication,”Biol Psychiatry 61:348-58(2007)。其特征在于暴食行为的反复发作而没有代偿行为来避免体重增加。在DSM-IV-TR中对于BED的诊断标准包括应当每周至少2天发生暴食行为的发作且持续6个月。BED与明显的医学和精神病共病相关。Javaras等人,“Co-occurrence of binge eating disorder with psychiatric and medical disorders,”JClin Psychiatry 269:266-273(2008);Grucza等人,“Prevalence and correlates ofbinge eating disorder in a community sample,”Compr Psychiatry 48:124-131(2007)。预计大约5%的美国成年人群在他们一生中的某段时间遭受暴食行为(Foulds等人,“The biology of binge eating,”Appetite 52:545-553(2009))并且这有助于加剧肥胖及相关的病症。Hudson等人,“The prevalence and correlates of eating disordersin the National Comorbidity Survey Replication,”Biol Psychiatry 61:348-58(2007);Yanovski,“Binge eating disorder and obesity in 2003:could treating aneating disorder have a positive effect on the obesity epidemic?”Int J EatDisord 34 Suppl:S117-S1120(2003)。
大量证据表明节食、应激和负面情感状态是患有BED或神经性贪食症的患者的暴食行为的重要触发因素。Wardle等人,“Stress,dietary restraint and food intake,”JPsychosom Res 48:195-202(2000);Freeman等人,“Daily stress,coping,and dietaryrestraint in binge eating,”Int J Eat Disord 36:204-212(2004)。事实上,节食期在暴食者的历史上是常见的,尽管在没有应激和负面情感状态的情况下饥饿本身似乎不足以诱发过量进食。Polivy等人,“Food restriction and binge eating:a study of formerprisoner of war,”J Abnorm Psychol 103:409-411(1994);Waters等人,“Internal andexternal antecedents of binge eating episodes in a group of women withbulimia nervosa,”Int J Eat Disord 29:17-22(2001)。相当多的证据表明暴食行为可由节食与应激之间的独特相互作用而引起;因此,环境应激和周期性的节食史可能是其出现和维持的原因。Stice等人,“Subtyping binge eating-disordered women along dietingand negative affect dimensions,”Int J Eat Disord 30:11-27(2001);Crowther等人,“The role of daily hassles in binge eating,”Int J Eat Disord 29:449-454(2001)。因此,经常性的节食始终是因应激而过度进食的最强有力的预测因素。Wardle等人,“Stress,dietary restraint and food intake,”J Psychosom Res 48:195-202(2000)。
尽管受试者可能对单一的成瘾剂或成瘾行为成瘾,但经常是对两种或更多种成瘾剂或成瘾行为成瘾。对两种或更多种成瘾剂或成瘾行为成瘾被称作多重成瘾(polyaddiction)。
B.使用与其它治疗剂联合的PDE7抑制剂治疗和预防成瘾和冲动控制障碍的方法PDE7抑制剂可以有效地与一种或多种其他治疗剂联合使用来治疗或预防成瘾,包括对一种或多种下文描述的成瘾剂以及强迫或成瘾行为成瘾。相应地,本发明包括治疗或预防成瘾的方法,包括向对成瘾剂成瘾的受试者施用一种或多种PDE7抑制剂和一种或多种其他治疗剂,其中所述的PDE7抑制剂和其他治疗剂各自有助于有效治疗或预防成瘾。在一项实施方案中,向受试者提供或施用一种PDE7抑制剂和一种其他治疗剂。在另一实施方案中,受试者对两种或更多种成瘾剂成瘾。
PDE7抑制剂和其他治疗剂可以一起给药(即同时地),或者一种在另一种之前给药(即顺序地)。一般地,PDE7抑制剂和其他治疗剂在一段时间内同时存在于受试者内,其水平足以向受试者提供治疗好处,即,治疗或预防对成瘾剂或者强迫或成瘾行为成瘾,或者预防成瘾剂或强迫或成瘾行为的使用复发(或恢复)。PDE7抑制剂和其他治疗剂可以通过相同或不同给药途径给药。典型地,按照市售的或其它药物组合物的标准给药途径,将PDE7抑制剂和其他治疗药各自提供给受试者。在一项实施方案中,PDE7抑制剂和其他治疗剂用包含该两种药剂的组合物共给药。
与PDE7抑制剂联合提供的其他治疗剂可以是有助于有效治疗或预防成瘾方面的任何治疗剂。例如,所述的其他治疗剂可以是用于治疗成瘾的药物或是用于减轻与成瘾剂的生理戒断相关的副作用的药物。另外,该其他治疗剂可以是影响脑5-羟色胺神经传递的任何药物,例如下文描述的选择性5-羟色胺再摄取抑制剂(SSRI)、三环和四环5-羟色胺和去甲肾上腺素再摄取抑制剂(SNRI),以及5-羟色胺激动剂如舒马曲坦、麦角新碱、双氢麦角胺和丁螺环酮。在某些实施方案中,所述的其他治疗剂为阿片类拮抗剂,包括混合型阿片类部分激动剂/拮抗剂,抗抑郁药,抗癫痫药,止吐药,多巴胺能剂如多巴胺D1受体激动剂,促肾上腺皮质激素释放因子-1(CRF-1)受体拮抗剂,选择性5-羟色胺3(5-HT3)拮抗剂,5-HT2A/2C拮抗剂如米安色林、米氮平和酮舍林,或大麻素-1(CB1)受体拮抗剂,包括但不限于在本文中具体描述的那些治疗剂。
在一项实施方案中,成瘾剂为酒精,其他治疗剂为阿片类拮抗剂或混合型阿片类拮抗剂/部分激动剂。在特定的实施方案中,该阿片类拮抗剂为纳曲酮。在另一实施方案中,该混合型阿片类部分激动剂/拮抗剂为丁丙诺啡。
在一项实施方案中,成瘾剂为酒精,其他治疗剂为托吡酯或左乙拉西坦。
在一项实施方案中,成瘾剂为尼古丁,其他治疗剂为抗抑郁药。在特定的实施方案,抗抑郁药是安非他酮。
在一项实施方案中,所述成瘾剂为可卡因,其他治疗剂为丁丙诺啡。
在一项实施方案中,成瘾剂为精神兴奋药,其他治疗剂为抗抑郁药。在特定的实施方案中,该抗抑郁药为安非他酮。
在一项实施方案中,成瘾行为是暴食,其他治疗剂是抗抑郁药或抗癫痫药。在一项特定的实施方案中,该抗抑郁药是西布曲明。在另一特定的实施方案中,该抗抑郁药是氟西汀。在一项特定的实施方案中,抗癫痫药是托吡酯。
在一项实施方案中,所述成瘾剂为尼古丁,其他治疗药为抗癫痫药。在特定的实施方案中,该抗癫痫药为左乙拉西坦。在另一特定的实施方案中,抗癫痫药为纳曲酮。
在一项实施方案中,受试者对两种或更多种成瘾剂成瘾,其他治疗剂为阿片类拮抗剂或混合型阿片类部分激动剂/拮抗剂。在特定的实施方案中,该混合型阿片类部分激动剂/拮抗剂为丁丙诺啡。
在一项实施方案中,受试者对酒精和尼古丁二者成瘾,其他治疗剂为抗癫痫药。在特定的实施方案中,抗癫痫药为纳曲酮。
为治疗酒精成瘾,根据本发明施用的联合药物包括PDE7抑制剂和阿片类激动剂或混合型阿片类拮抗剂/部分拮抗剂,PDE7抑制剂和抗抑郁药,PDE7抑制剂和CB1受体拮抗剂/逆激动剂,PDE7抑制剂和伐尼克兰,PDE7抑制剂和阿坎酸,以及PDE7抑制剂和戒酒硫。
为治疗精神兴奋药成瘾,根据本发明施用的联合药物包括,例如,PDE7抑制剂和抗抑郁药,或者PDE7抑制剂和部分阿片类激动剂/拮抗剂,例如丁丙诺啡。
为治疗尼古丁成瘾,根据本发明施用的联合药物包括,例如,PDE7抑制剂和抗抑郁药,PDE7抑制剂和尼古丁(作为替代,以口服、经皮或其它常规制剂的形式),PDE7抑制剂和阿片类拮抗剂,PDE7抑制剂和CB1受体拮抗剂/逆激动剂,及PDE7抑制剂和伐尼克兰。在一项实施方案中,一种成瘾剂例如尼古丁与PDE7抑制剂利用透皮贴剂递送体系一起施用。在本发明的另一方面,提供一种装有多个透皮贴剂的药盒,其含有递减水平的尼古丁剂量和恒定或递减水平的PDE7抑制剂剂量,供尼古丁成瘾的患者顺序使用以使其戒断尼古丁成瘾。
为治疗多重物质成瘾,根据本发明施用的联合药物包括,例如,PDE7抑制剂和阿片类激动剂或混合型阿片类拮抗剂/部分拮抗剂。
为治疗赌博成瘾,根据本发明施用的联合药物包括,例如,PDE7抑制剂和抗抑郁药,或PDE7抑制剂和影响多巴胺神经传递的试剂,例如,直接或间接的多巴胺拮抗剂。
当PDE7抑制剂和其他治疗剂联合施用时,与每一种单独施用相比,其中之一或二者的有效量可以减少。例如,当PDE7抑制剂和其他治疗剂累加地或协同地起作用时,为达到PDE7抑制剂或其他治疗剂单独提供的同样疗效,可能需要较少量的PDE7抑制剂,较少量的其他治疗剂,或者PDE7抑制剂和其他治疗剂的量都较少。
1.阿片类拮抗剂
阿片类拮抗剂作用于一种或多种阿片类受体。已经报道了至少三种类型的阿片类受体:μ、κ和δ阿片类受体,阿片类拮抗剂通常根据它们对阿片类受体的作用分类。阿片类拮抗剂可以拮抗中枢受体、外周受体或二者。纳洛酮和纳曲酮是常用的阿片类拮抗剂药物,它们以比激动剂更高的亲和性使其竞争性结合阿片类受体,但是不激活这些受体。这有效地阻断了受体,阻止了身体对鸦片剂和内啡肽作出响应。
很多阿片类拮抗剂都不是纯的拮抗剂,而是还产生一些弱的阿片类部分激动剂作用,当以高剂量向无阿片类经历的个体施用时,可产生止痛效果。这样的化合物的实例包括烯丙吗啡和左洛啡烷。但是,这些药物的止痛效果有限,并有伴随心境恶劣的倾向,很可能是由于对κ阿片类受体的作用。因为它们在正使用或曾使用过阿片类完全激动剂的人群中诱发阿片类戒断效应,所以这些药物被认为是拮抗剂。
纳洛酮是不具有部分激动剂作用的阿片类拮抗剂的一个实例。相反,它是μ阿片类受体的弱的逆激动剂,并被用于治疗阿片类用药过量。
根据本发明可以使用的阿片类拮抗剂的具体实例包括阿维莫泮、binaltorphimine、丁丙诺啡、环佐辛、赛科罗酚(cyclorphan)、cypridime、dinicotinate、β-富纳曲胺、左洛啡烷、甲基纳曲酮、纳布啡、nalide、纳美芬、纳美酮、烯丙吗啡、二烟酸烯丙吗啡、纳洛酮、纳洛肼、naltrendol、纳曲酮、纳曲吲哚、奥昔啡烷和喷他佐辛。
2.抗抑郁药
抗抑郁药是用于治疗抑郁的药物。被认为与抑郁有关的三种神经递质是5-羟色胺、多巴胺和去甲肾上腺素。某些类型的抗抑郁药通过阻断它们的再吸收而增加这些神经递质中的一种或多种在脑内的水平。
已经鉴定了数种不同种类的抗抑郁药,包括选择性5-羟色胺再摄取抑制剂(SSRI)、三环和四环5-羟色胺和去甲肾上腺素再摄取抑制剂(SNRI)、去甲肾上腺素再摄取抑制剂(NRI)、去甲肾上腺素和多巴胺再摄取抑制剂(NDRI)、azaspirones、单胺氧化酶抑制剂(MAOI)以及非典型抗抑郁药。
SSRI包括例如西文氯胺、西酞普兰、氯米帕明、cyanodothiepin、达泊西汀、度洛西汀、依他普仑、非莫西汀、氟西汀、氟伏沙明、伊福西汀、丙咪嗪、吲达品、茚洛秦、利托西汀、洛非帕明、米安色林、米那普仑、米氮平、萘发扎酮、去甲替林、帕罗西汀、舍曲林、西布曲明、托莫西汀、曲唑酮、文拉法辛和齐美定。
阿密曲替林、阿莫沙平、布替林、氯米帕明、地美替林、地昔帕明、二苯西平、二甲他林、度硫平、多塞平、丙咪嗪、伊普吲哚、洛非帕明、马普替林、美利曲辛、美他帕明、米安色林、米氮平、去甲替林、丙吡西平、普罗替林、奎纽帕明、司普替林、噻奈普汀和曲米帕明均为三环和四环抗抑郁药。
SNRI包括,例如,阿莫沙平、阿托西汀、比西发定、地昔帕明、去甲文拉法辛、度洛西汀、马普替林、米那普仑、奈法唑酮、瑞波西汀、西布曲明和文拉法辛。
尼索西汀、去甲替林、瑞波西汀、他舒普仑和托莫西汀均为NRI的实例。
NDRI包括,例如,安非他酮、羟基安非他酮和特索芬辛。
azaspirones包括例如丁螺环酮、吉哌隆、伊沙匹隆、坦度螺酮和噻斯匹隆。丁螺环酮是抗焦虑药(5-HT1自身受体的部分激动剂),其可以与抗抑郁药如SSRI一同提供。
具体的MAOI包括,例如,阿米夫胺、溴法罗明、氯吉灵、α-乙基色胺、异丙氯肼、异丙烟肼、异卡波肼、美巴那肼、吗氯贝胺、尼亚拉胺、帕吉林、苯乙肼、苯异丙肼、吡吲哚、呱异丙肼、司立吉林、托洛沙酮和反苯环丙胺。
非典型抗抑郁剂包括,例如,安麦角、阿米庚酸、苯乃静、安非他酮、氯氮平、非唑拉明、左丙替林、锂、美地沙明、米安色林、米那普林、奥氮平、奥沙氟生、羟色氨酸、咯利普兰、替尼沙秦、托芬那辛、曲唑酮、色氨酸和维洛沙秦。
3.抗癫痫药
抗惊厥药,也称为抗癫痫药(AED),是用于防止癫痫发作和双相型障碍发生的不同类别的药物。AED遏制开始发作的神经元的快速和过度放电和/或阻止发作在脑内的传播,并针对可以导致脑损伤的可能的兴奋性毒性效应提供保护。很多抗惊厥药阻断钠通道、钙通道、AMPA受体或NMDA受体。
抗癫痫药包括但不限于,苯并二氮杂 类、巴比妥酸盐类、丙戊酸类、GABA药物、亚氨基茋类、乙内酞脲类、NMDA拮抗剂、钠通道阻断剂和琥珀酰胺类。
类、巴比妥酸盐类、丙戊酸类、GABA药物、亚氨基茋类、乙内酞脲类、NMDA拮抗剂、钠通道阻断剂和琥珀酰胺类。
苯并二氮杂 类,包括,例如,阿普唑仑、利眠宁、氯氮
类,包括,例如,阿普唑仑、利眠宁、氯氮 (cholrazepate)、氯巴占、氯硝西泮、地西泮、哈拉西泮、劳拉西泮、奥沙西泮和普拉西泮。
(cholrazepate)、氯巴占、氯硝西泮、地西泮、哈拉西泮、劳拉西泮、奥沙西泮和普拉西泮。
用作抗癫痫药的巴比妥酸盐类包括,例如,异戊巴比妥、甲苯巴比妥(mepobarbital)、甲苯比妥、戊巴比妥、苯巴比妥和普里米酮。
用作抗癫痫药的丙戊酸盐类包括,例如,丙戊酸钠、丙戊酸、丙戊酸半钠、丙戊酰胺。
抗癫痫GABA药物包括例如加巴喷丁、氯西加酮、普瑞巴林、瑞替加滨、卢非酰胺和氨己烯酸。
卡马西平和奥卡西平为亚氨基茋类的实例。
乙内酰脲类包括,例如,磷苯妥英钠、美芬妥英和苯妥英钠。
NMDA拮抗剂如harkoseramide被用作抗癫痫药。
钠通道阻断剂如拉莫三嗪也是抗癫痫药。
琥珀酰亚胺类包括,例如,乙琥胺、甲琥胺和苯琥胺。
其它抗癫痫药包括乙酰唑胺、布立西坦、CBD大麻衍生物、乙二磺酸氯美噻唑、双丙戊酸钠、非尔氨酯、异戊酰胺、拉科酰胺、拉莫三嗪、左乙拉西坦、甲磺酰胺、他仑帕奈、噻加宾、托吡酯、沙芬酰胺、塞曲西坦、索瑞托胺、司替戊醇、舒噻美、丙戊塞胺和唑尼沙胺。
4.止吐药
止吐药是对呕吐和恶心有效的药物。止吐药通常用于治疗晕动病和阿片类止痛药、全身麻醉药和化学疗法的副作用。
止吐药的分类包括,例如,5-羟色胺3(5-HT3)受体拮抗剂、组胺受体拮抗剂、多巴胺受体拮抗剂、毒蕈碱受体拮抗剂、乙酰胆碱受体拮抗剂、大麻素受体拮抗剂、边缘系统抑制剂、NK-1受体拮抗剂、皮质类固醇、速激肽拮抗剂、GABA激动剂、大麻素、苯并二氮杂 类、抗胆碱能药和物质P抑制剂。
类、抗胆碱能药和物质P抑制剂。
5-HT3受体拮抗剂包括,例如,阿洛司琼、阿扎司琼、贝美司琼、西兰司琼、多拉司琼、格拉司琼、吲地司琼、伊他司琼、昂丹司琼、帕洛诺司琼、propisetron、雷莫司琼、伦扎必利、托烷司琼和扎托司琼。
皮质类固醇止吐药包括地塞米松和甲基强的松龙。
边缘系统抑制剂包括阿普唑仑、劳拉西泮和咪达唑仑。
多巴胺受体拮抗剂包括苯海拉明、屈大麻酚、氟哌啶醇、甲氧氯普胺和丙氯拉嗪。
用作止吐药的NK-1受体拮抗剂包括阿瑞匹坦和吗啉,GABA激动剂的实例为异丙酚。
硫乙拉嗪是一类组胺受体拮抗剂。
用作止吐药的大麻素受体拮抗剂或激动剂包括屈大麻酚、大麻隆、利莫那班、tanarabout和四氢大麻酚。
其它止吐药的实例包括乙酰亮氨酸、单乙醇胺、阿立必利、苯喹胺、氨醇醋茶碱、嗅必利、氯苯丁嗪、氯丙嗪、氯波必利、赛克利嗪、茶苯海明、地芬尼多、多潘立酮、dranisetron、美其敏、methalltal、美托哌丙嗪、奥昔喷地、匹哌马嗪、哌海茶碱、东莨菪碱、硫丙拉嗪和曲美苄胺。
5.大麻素受体拮抗剂
大麻素受体为一类G蛋白偶联受体超家族。它们的配体称为大麻素。目前有两种已知亚型:CB1,其主要在脑内但也在肺、肝和肾中表达,以及CB2,其主要在免疫系统和造血细胞内表达。还认为存在有新的大麻素受体,即,非CB1和非CB2,它们在内皮细胞中和CNS中表达。大麻素受体拮抗剂可以是对CB1或CB2受体选择性的。本发明考虑使用CB1和CB2受体拮抗剂之一或二者。
成瘾剂(例如酒精、鸦片剂、δ(9)-四氢大麻酚(δ(9)-THC)和精神兴奋药,包括尼古丁)通过与脑内的内源神经通道相互作用,引起多种慢性复发性病症。特别是,它们都有激活中脑缘多巴胺脑奖赏系统的共同性质,并且实际上所有的滥用药物都升高伏隔核内多巴胺水平。大麻素-1(CB1)受体在该脑奖赏回路中表达并调节δ(9)-THC和尼古丁的多巴胺释放效应。
CB1受体拮抗剂利莫那班(SR141716)阻断动物中的多巴胺释放和δ(9)-THC的识别和奖赏效应。尽管CB1受体阻断剂通常在减少啮齿动物和灵长类动物的可卡因自身给药方面无效,但其减少可卡因相关条件刺激物和可卡因点燃注射产生的已消退的可卡因觅药行为的恢复。类似地,CB1受体阻断剂在减少由再暴露于尼古丁相关刺激物诱导的尼古丁觅药行为方面有效。在人临床试验中,利莫那班显示出阻断δ(9)-THC在人体内的自觉效果并预防戒烟者复吸。
大麻素受体CB1拮抗剂的其它实例包括SR141716A(利莫那班)、rosanabant、泰伦那班和CP-945598。
6.多巴胺能剂
药物成瘾是一种慢性复发性疾病,其特征在于对药物使用失去控制、强迫性觅药和渴望某种物质、持续使用而不管负面影响、以及对物质产生生理和/心理依赖。成瘾发病机理中的基本作用已归因于多巴胺。实际上,多巴胺已经在各个层次上渗透到药物成瘾的自然史中,从其参与形成个体对易损因素(即遗传学、环境和应激)的响应到其在药物滥用的作用机理中的作用。
中脑皮层边缘多巴胺系统(mesocorticolimbic dopamine system)起源于腹测被盖区(VTA),其主要投射到伏核(NAc)和前额皮质(PFC)。所有成瘾性药物的一个显著共性是至少最初它们刺激中脑边缘系统的末端区域中且特别是核NAc壳中的多巴胺传递。Nestler,E.J.,“Is there a common molecular pathway for addiction?”Nat Neurosci8:1445-1449(2005);Pierce,R.C.等人,“The mesolimbic dopamine system:The finalcommon pathway for the reinforcing effect of drugs of abuse?”NeurosciBiobehav Rev 30:215-238(2006)。脑成像研究已经将这些观察结果扩展至人类。Drevets,W.C.等人,“Amphetamine-induced dopamine release in human ventral striatumcorrelates with euphoria,”Biol Psychiatry 49:81-96(2001);Brody A.L.等人,“Smoking-induced ventral striatum dopamine release,”Am J Psychiatry 161:1211-1218(2004)。认为多巴胺从这些投射的释放在介导药物奖赏、强化和在诱导强迫性成瘾行为中起重要作用。尽管多巴胺涉及药物成瘾的倾向和药物成瘾形成的最初阶段,该病症一旦建立,就涉及与长期暴露于药物本身相关的长期变化。
这些变化共同指示为“神经适应性的”并被认为是行为敏感(对药物的行为刺激性质的敏感度长期增加)和愉悦状态(愉悦非稳态)基底变化的基础。Berridge K.C.等人,“What is the role of dopamine in reward:Hedonic impact,reward learning,orincentive salience?”Brain Res Rev 28:309-369(1998);Koob,G.F.,等人,“Neurobiological mechanisms for opponent motivational processes inaddiction,”Philos Trans R Soc Lond B Biol Sci.363:3113-3123(2008)。神经适应性调节也在多巴胺系统的层面下发生,其中文献记载了腹测纹状体区中多巴胺传递活性的基底水平的相对减少和多巴胺D2-受体水平的减少。Volkow,ND.等人,“Dopamine in drugabuse and addiction:results from imaging studies and treatment implications,”Mol Psychiatry 9:557-569(2004);Volkow,N.D.等人,“Dopamine in drug abuse andaddiction:results from imaging studies and treatment implications,”ArchNeurol 4:1575-1579(2007);Fehr,C.等人,“Association of low striatal dopamine d2receptor availability with nicotine dependence similar to that seen withother drugs of abuse,”Am J Psychiatry 65:507-514(2008)。
药物滥用导致多巴胺神经传递的最常见的机理是通过调节VTA突触前γ-氨基丁酸(GABA)的活性。Lüscher,C.等人,“The mechanistic classification of addictivedrugs,”PLoS Med 3:e437(2006),在VTA中,GABA神经元充当局部抑制性中间神经元对中脑皮质边缘多巴胺细胞进行补充控制。GABA神经传递的减少导致多巴胺神经元的纯去抑制以及末端区域如NAc和PFC中的多巴胺释放增加。
该机理的一个原型实例由鸦片剂提供,其在激活位于突触前GABA细胞上的μ阿片样受体后导致GABA能神经传递的显著抑制,从而引起VTA多巴胺能细胞的去抑制。Johnson,S.W等人,“Opioids excite dopamine neurons by hyperpolarization of localinterneurons,”J Neurosci 2:483-488(1992)。大麻类衍生物似乎以类似机理但通过选择性激活位于突触前GABA神经元上的大麻素受体1(CB1R)来增加VTA多巴胺放电速率。尼古丁同样增加多巴胺神经传递,但通过烟碱受体对GABA和对多巴胺神经元的谷氨酸能输入的作用的复杂相互影响。Lüscher C等人,The mechanistic classification of addictivedrugs.,PLoS Med.3(11):e437(2006)。事实上,尼古丁经由含β2的nAChRs减少突触前GABA释放从而导致多巴胺的长期去抑制,同时作用于含同数α7的nAChRs(主要表达于VTA中多巴胺神经元上的兴奋性谷氨酸能传入神经的突触末端上),促进谷氨酸释放。这种作用也可能有助于尼古丁诱发的多巴胺释放。并且,近来的证据表明尼古丁直接调节NAc中的多巴胺释放。乙醇和苯并二氮杂 类似乎也通过经由调节特定的GABA-A受体亚单位抑制突触前GABA活性来刺激VTA多巴胺神经传递。
类似乎也通过经由调节特定的GABA-A受体亚单位抑制突触前GABA活性来刺激VTA多巴胺神经传递。
精神兴奋药如安非他明衍生物和可卡因,包含唯一一类的通过抑制多巴胺再摄取机制或通过促进多巴胺从突触释放而直接作用于多巴胺能末端的成瘾性药物。通过这些机制,它们增加了大脑多巴胺系统的末端区域(即NAc和MPF)中的细胞外多巴胺水平。然而,更特别地,多巴胺末端还存在于VTA,其中D1-和D2-类受体表达于多巴胺能细胞体(自受体)以及谷氨酸能和GABA能突触前神经元上。在VTA中直接施用可卡因导致多巴胺放电速率的减少或增加(取决于剂量),这种作用可被分别位于突触前GABA和谷氨酸细胞上的D1受体潜在介导。因此VTA DA活性的突触前调节也可能在调节精神兴奋药的成瘾性方面起作用。Brodie,M.S.等人,“Cocaine effects in the ventral tegmental area:Evidence foran indirect dopaminergic mechanism of action,”Naunyn Schmiedebergs ArchPharmacol 342:660-665(1990);Bonci,A.等人,“Increased probability of GABArelease during withdrawal from morphine,”J Neurosci 17:796-803(1997)。
因此,在本发明的一项实施方案中,与PDE7抑制剂联合、同时或顺序给药的其他治疗剂是治疗患有成瘾或强迫症的患者的多巴胺能剂。在本发明的另一实施方案中,所述PDE7抑制剂与多巴胺受体激动剂联合、同时或顺序给药以治疗患有成瘾或冲动控制障碍的患者。在本发明的另一实施方案中,所述PDE7抑制剂与多巴胺D1受体激动剂(即多巴胺亚型D1受体激动剂)联合、同时或顺序给药以治疗患有成瘾或冲动控制障碍的患者。
适用于与PDE7抑制剂联合给药的示例性多巴胺能剂包括,例如,左旋多巴(也称为″L-多巴″)、卡比多巴、和多巴胺受体激动剂及前体如溴隐亭、培高利特、普拉克索、罗匹尼罗、卡麦角林、阿扑吗啡、麦角乙脲、罗替戈汀和喹高利特,以及对多巴胺受体D1具有选择性的非诺多泮。
根据本发明与PDE7抑制剂联合给药的D1受体激动剂的合适剂量可由执业医师决定,但可以是例如每天或两周一次0.1mg至1,000mg,或每天或两周一次0.25mg至100mg。
C.治疗和预防复发的方法
复发使用(或者恢复)是指,在戒断、或限制或减少使用成瘾剂或实施成瘾行为一段时间后,再回到使用酒精或其它成瘾剂或实施成瘾行为的过程。在某些情况下,复发使用成瘾剂是指经历了成瘾剂身体戒断的受试者重新使用该成瘾剂。通常,该受试者在不使用或者限制或减少使用成瘾剂的一段时间内会经历身体戒断成瘾剂。在一项实施方案中,复发使用发生在这样的受试者中,其之前经历过以有效量抗成瘾剂来减少或消除成瘾剂使用的治疗方案,但是现在不再使用有效量的抗成瘾剂。抗成瘾剂包括用于治疗或预防成瘾或戒断症状的任何和所有试剂。
如同很多其它成瘾一样,酗酒是以高再犯率为特征的慢性复发性病症。触发复发行为的两个主要因素是应激和环境条件经历(O′Brien等人1997;Monti等人1993;Shaham等人1995),它们很可能通过不同的脑机制促成寻觅酒精的复发。例如,通过阿片类依赖机制(或者通过杏仁核的基底外侧核内多巴胺传递的直接改变)激活中脑缘多巴胺系统似乎介导了药物相关信号的作用(Liu and Wiess 2002;Ciccocioppo等人2001),而终纹床核和中缝正中核内的下丘脑外侧CRF很可能介导了应激诱导的觅药行为恢复(Erb等人1998;Shaham等人1995;Lê等人2000)。
几个方面的证据提示,成瘾复发背后的分子机制对不同类别的药物滥用是共同的。与复发相关的药物渴求和无节制药物摄取行为受到应激和环境条件刺激物(影响药物使用重新开始的两个主要因素)的直接影响。
慢性药物滥用不仅在涉及乙醇的急性增强效应的系统内,而且还在其它动机系统、特别是脑应激调节机制内产生神经适应性变化。应激在药物滥用的起始和维持中具有公认的作用,并且是戒断个体中复发的主要决定因素(Brown等人,J Studies Alcohol 56:538(1995);Marlatt,Relapse prevention:introduction and overview of the model,in Relapse Prevention:Maintenance Strategies in the Treatment of AddictiveBehaviours,Guilford,London,(1985);McKay等人,Drug Alcohol Dep.,38,35,(1995);和Wallace,J Subst Abuse Treat,6:95(1989))。应激在觅药行为中的重要性还在动物文献中有广泛记载。身体、社会和情绪应激可促使啮齿动物和非人灵长类动物对可卡因、海洛因及乙醇的获取或增加其自身给药(Goeders and Guerin,Psychopharmacology,114,63,(1994);Haney等人,Brain Res.,698,46,(1995);Ramsey and Van Ree,Brain Res.,608,216,(1993);Ahmed and Koob,Psychopharmacology,132,289,(1997);Shaham andStewart,Psychopharmacology 119:334(1995);Nash and Maickel,ProgNeuropsychopharmacol Biol Psychiatry,12,653,(1988);Mollenauer等人,Pharmacol.Biochem.Behav.,46,35,(1993);Blanchard等人,Pharmacol.Biochem.Behav.28,437,(1987);和Higley等人,Proc.Natl.Acad.Sci.USA,88,7261,(1991))。还证实应激刺激物会引起消退后无药动物的可卡因、海洛因和乙醇寻觅行为的恢复,这些发现对应激在复发中的作用提供了实验支持(Ahmed and Koob(1997);Shaham,Psychopharmacology,111,477,(1993);和Shaham and Stewart(1995))。
传统上,认为应激相关的觅药行为是通过对下丘脑-垂体-肾上腺(HPA)轴的激活而介导的。但是,越来越多的证据表明,杏仁核的中央核(CeA)内非神经内分泌促肾上腺皮质激素释放因子(CRF)系统可能在与应激相关的成瘾行为的调节中起独立的重要作用。CeA富含CRF免疫反应性细胞体、末端和受体,并且这种神经元CRF系统还涉及对应激刺激物的行为和情绪应答的介导(Dunn and Berridge,Brain Res Brain Res Rev 15,71,(1990);和Koob等人,Semin Neurosci 6:221(1994))。例如,制动应激(immobilization stress)升高了CeA内胞外CRF水平,而CeA内注射CRF受体拮抗剂α-螺旋CRF9-41减少由于社会和环境应激物导致的焦虑的行为迹象(Merali等人,J.Neurosci.,18,4758,(1998);Merlo Pich等人,J.Neurosci.,15,5439,(1995);Heinrichs等人,Brain Res.,581,190(1992);Swiergiel等人,Brain Res,623,229(1993))。焦虑和应激样症状是药物和酒精戒断症状的核心。考虑到CeA内CRF神经元在调节应激的情绪和致焦虑效应方面的作用的证据,戒断药物滥用的致焦虑和应激样后果很可能也通过CeA内的CRF系统介导。
CeA内CRF系统的活性调节的变化可能代表了负责发展依赖性和强迫觅药行为的关键的神经适应性机制。
以上讨论的数据确定了脑通路内神经适应性变化和应激系统内的扰动是强迫觅药行为和依赖性中的重要因素。药物滥用的长期成瘾可能性的另一重要因素是它们的奖赏行为与特定的环境刺激物的条件形成。重复地与包括酒精在内的药物滥用的自觉效应相关联的环境信号可诱发药物渴求或引起自动行为响应(Miller and Gold 1994;Tiffany andCarter 1998),这最终会导致复发(Childress等人,Conditioned craving and arousalin cocaine addiction:A preliminary report,in NIDA Research Monograph 81,(1988);Ehrman等人,Psychopharmacology,107,523(1992);Monti等人,J Stud Alcohol54:235-45(1993);Pomerleau等人,Addict.Behav.,8,1,(1983);Stormark等人,Addict.Behav.,20,571,(1995);Miller and Gold Ann.Clin.Psychiatry,6,99,(1994);和Tiffany and Carter,J Psychopharmacol.12,23,(1998))。因此,习得的对药物相关刺激物的响应可能关键性地促成了与可卡因和其它药物成瘾相关的高复发率。
从操作性响应-恢复模型(被开发用来研究大鼠中与暴露于药物相关环境信号有关的觅药行为)得到的数据表明,预兆可卡因、乙醇或海洛因可得性的区辨刺激物(discriminative stimuli)可靠地引起在没有进一步药物可得性情况下已消退的觅药行为的强力恢复。(Weiss等人,Proc.Natl.Acad.Sci.USA,97,4321,(2000);Katner等人,Neuropsychopharmacology,20,471,(1999);Katner and Weiss,Alcohol Clin ExpRes.23:1751(1999);和Gracy等人,Pharmacol.Biochem.Behav.,65,489,(2000))。这些刺激物的响应-恢复效应在反复暴露下显示出引人注目的对消退的抗性,对于可卡因,在强制戒断几个月后仍可观察到该效应。另外,对于乙醇,与酒精非偏爱(NP)和非选择的Wistar大鼠相比,发现由乙醇预示性区辨刺激物诱导的觅药行为在遗传上酒精偏爱(P)大鼠中增强(Weiss and Ciccocioppo,Soc.Neurosci.Abstr.,25,1081,(1999))。这一观察结果证实,增强乙醇摄取的遗传倾向也通过对乙醇信号的激发效应的更大易感性(即,在行为不是直接由乙醇本身强化的条件下的觅药增强)而反映出来。合起来,这些发现强烈支持这样的假设,即,对药物相关刺激物的习得反应是长期易复发性的重要因素。
对于人,复发风险涉及多个很可能相互作用的决定因素。例如,暴露于药物信号可以增强依赖性个体中神经适应性变化导致的稽延性戒断症状引起的复发易感性。加剧复发风险的相互作用效应还可以存在于应激和药物相关信号的激发效应之间。针对这些问题的最近工作确认,乙醇相关信号的响应-恢复效应和应激之间的累加相互作用确实能被证实,并且这些效应在有乙醇依赖史的大鼠中得到增强(Liu and Weiss,Soc.Neurosci.Abstr.26,786(2000))。
在实验室中,通过施用α-2肾上腺素受体拮抗剂育亨宾而实现觅药恢复,育亨宾增强了脑去甲肾上腺素细胞放电和释放,起着药理学应激物的作用。足部电击应激和育亨宾诱导的觅药行为恢复都代表了研究应激诱导的酒精复发的有效的实验模型(Lee等人,Neuropsychopharmacology 29:686-93(2004)和Le等人,Psychopharmacology 150:317-24(2000))。
如所附实施例所显示的,PDE7抑制剂明显减少应激诱导的成瘾剂的复发使用(实施例1)。因此,这些数据指示,PDE7抑制剂具有抗复发性能。
有趣的是,多种报告已显示,非选择性鸦片剂受体拮抗剂纳曲酮减少人酗酒者由于酒精信号的展示而引起的饮酒冲动(Monti等人1993,上文),并在大鼠中降低酒精信号在恢复已消退的先前药物配对杆的响应方面的效力(Katner等人1999,上文)。但是,纳曲酮不减少应激引起的复发行为(Le A.D.Psychopharmacology 1998)。
在一项相关的实施方案中,本发明包括治疗或预防复发使用成瘾剂或实施成瘾或强迫行为的方法,包括向之前响应暴露于有效量的其它抗成瘾治疗而减少或排除了使用成瘾剂或实施成瘾性或强迫性行为的受试者施用有效量的PDE7抑制剂,其中该受试者不再暴露于有效量的该抗成瘾治疗。该抗成瘾治疗可以是抗成瘾药物或者可以是非药理学疗法例如咨询、心理疗法或催眠疗法。所述的复发使用可以通过应激触发。
在某些实施方案中,所述受试者不再暴露于有效量的抗成瘾剂,因为该受试者已变得耐受该试剂,以致于之前在治疗该成瘾方面有效的抗成瘾剂血浆浓度不再有效。在其它实施方案中,受试者不再暴露于有效量的抗成瘾剂,因为该受试者现在暴露于较低血浆浓度的抗成瘾剂,而该较低的血浆浓度不是有效的。
在本发明方法的某些实施方案中,受试者经历了一段时间的戒断、或限制或减少使用成瘾剂或实施成瘾或强迫行为。该戒断、或限制或减少使用的时间段可以例如为至少24小时、至少48小时、至少3天、至少5天、至少1周、至少2周、至少1月、至少2月、至少4月、至少6月、至少9月、至少1年、至少2年、或至少5年。
在另一实施方案中,本发明包括治疗或预防复发使用成瘾剂的方法,包括向经历了生理戒断成瘾剂的受试者提供PDE7抑制剂和阿片类拮抗剂。
在又一实施方案中,本发明包括治疗或预防复发使用成瘾剂的方法,包括向经历了生理戒断成瘾剂的受试者施用PDE7抑制剂和CB1拮抗剂,如戒酒硫、托吡酯、左乙拉西坦、SSRI或昂丹司琼。
在特定的实施方案中,该复发使用通过应激、环境条件因素或二者触发。
尽管本发明的方法可以在对于单一成瘾剂成瘾的受试者中实施,但是它们也可以用于对两种或更多种成瘾剂成瘾的受试者。类似地,虽然这些方法可以用来预防经历了戒断的受试者复发使用成瘾剂,但是它们也可适用于预防复发使用或开始使用与该受试者经历生理戒断的成瘾剂不同的成瘾剂。
D.药物组合物,给药途径,单位剂型,试剂盒
本发明确定了采用PDE7抑制剂联合一种或多种其他治疗剂的联合药物的功效,所述的其他治疗剂是例如阿片类拮抗剂、抗抑郁药、抗癫痫药、止吐药和CB1受体拮抗剂。因此,本发明还包括组合物,其包含一种或多种PDE7抑制剂和一种或多种其他治疗剂,例如阿片类拮抗剂、混合型阿片类拮抗剂/部分激动剂、抗抑郁药、抗癫痫药、止吐药、CRF1受体拮抗剂和CB1受体拮抗剂。
在特定的实施方案中,该组合物包含一种PDE7抑制剂和一种其他治疗剂。在某些实施方案中,所述的其他治疗剂为阿片类拮抗剂或混合型阿片类拮抗剂/部分激动剂。在一项实施方案中,所述的阿片类拮抗剂是纳曲酮。在另一实施方案中,所述混合型阿片类部分激动剂/拮抗剂是丁丙诺啡。在某些实施方案中,所述其他治疗剂为抗抑郁药。在特定的实施方案中,所述抗抑郁药为安非他酮。在某些实施方案中,所述其他治疗剂为抗癫痫药、止吐药、或是阿片类拮抗剂或混合型阿片类部分激动剂/拮抗剂。
本发明的组合物可以作为药物组合物或者制剂向受试者给药。在特定的实施方案中,本发明的药物组合物可以是能使该组合物向受试者给药的任何形式。例如,该组合物可以为固体、液体或者气体(气溶胶)形式。典型的给药途径包括但不限于:口服、局部、肠胃外、舌下、直肠、阴道和鼻内给药。用在本文时术语肠胃外包括皮下注射、静脉内、肌内、硬膜外、胸骨内注射或输注技术。
根据本发明使用的药物组合物包含PDE7抑制剂,其它治疗剂和药学上可接受的稀释剂、赋形剂或载体。治疗用途的“药学上可接受的载体”在制药领域是公知的,并在例如Remingtons Pharmaceutical Sciences,Mack Publishing Co.(A.R.Gennaro编.1985)中描述。例如,可以使用生理pH的无菌盐水和磷酸盐缓冲盐水。防腐剂、稳定剂、染料和甚至调味剂都可以在该药物组合物中提供。例如,苯甲酸钠、山梨酸和对羟基苯甲酸的酯可以作为防腐剂添加(上述文献第1449页)。另外,可以使用抗氧化剂和悬浮剂(上述文献)。
本发明的药物组合物通常配制成使得将该组合物对受试者给药时其中含有的活性成分是生物可利用的。待向受试者给药的组合物可以采取一种或多种剂量单位的形式,例如,片剂、胶囊剂、或扁囊剂可以为单个剂量单位,而包含气雾剂形式的本发明联合药物的容器可以容纳多个剂量单位。
在特定实施方案中,包含PDE7抑制剂和其它治疗剂的组合物以一个或多个剂量的片剂给药,通常用于口服给药。该片剂可以是例如即释剂、控释剂或延释剂。在一项实施方案中,片剂含有有效量的含PDE7抑制剂和其它治疗剂的组合物。在特定实施方案中,片剂包含约1、5、10、20、30、50、100、150、200、250或300mg的PDE7抑制剂,和约1、5、10、20、30、50、100、150、200、250或300mg的其它治疗剂。
本发明还包括药物组合物的单位剂型,所述药物组合物包含PDE7抑制剂和另一治疗剂。每一单位剂型当以推荐量使用时包含治疗有效量的本发明药物组合物。例如,单位剂型可以在单个片剂中包含治疗有效量,或者单位剂型可以在两个或多个片剂中包含治疗有效量,使得处方量包含治疗有效量。
在特定的实施方案中,向受试者提供数量为每天0.1-1000mg,每天1-1000mg,每天10-100mg或每天25-50mg的PDE7抑制剂。在一项实施方案中,向患者每天提供约30mg的吡格列酮。
PDE7抑制剂和其它治疗剂的某些组合可能不容易适应共同配制。例如,药剂之一可能更易于静脉内给药,而另一种可能更易于口服给药。或者,两种药剂的血清半衰期可以是使得其中之一必须比另一种更频繁地给药。因此,本发明考虑了包含一种或多种单位剂型的PDE7抑制剂和一种或多种单位剂型的另一种治疗剂的药盒,以便可以以治疗有效的方式向受试者提供这两种单位剂型。
在一项实施方案中,本发明包括药盒,所述药盒包含PDE7抑制剂的单位剂型和尼古丁的单位剂型。在一项实施方案中,尼古丁的单位剂型包含尼古丁的多个不同的单位剂型,其中该不同剂型中的尼古丁的含量递减,它们可以在一段时间内相继使用,以便克服成瘾和实现尼古丁戒断。尼古丁的单位剂型可采取例如透皮贴剂、口胶剂或锭剂的形式。
E.PDE7蛋白和抑制剂
环核苷酸磷酸二酯酶7型(PDE7)根据其主要的氨基酸序列和与众不同的酶活性被确定为独特的家族。鉴定为PDE7(PDE7A和PDE7B)的PDE基因为cAMP特异性PDE编码。PDE7的生物化学和药理学鉴定显示出高亲和性cAMP特异性PDE(Km=0.2μM),它既不受cGMP也不受其它PDE选择性抑制剂的影响。PDE7酶选择性地分解cAMP,其特征是不受咯利普兰(PDE4的选择性抑制剂)抑制的酶,是明显不同的cAMP特异性PDE家族。在PDE7家族内已经确定了两种亚型:PDE7A(Michael,T.等人,J Biol.Chem.268(17):12925-12932,1993;Han,P.等人,JBiol.Chem.272(26):16152-16157,1997)和PDE7B(美国专利No.6,146,876;Gardner,C.等人,Biochem.Biophys.Res.Commun.272(1):186-192,2000;和Saski,T.等人,Biochem.Biophys.Res.Commun.271(3):575-583,2000)。这两种基因产物在其C端催化域显示70%同一性(Hetman J.M.等人,PNAS 97(1):472-476(2000))。
PDE7A有三个剪接变体(PDE7A1,PDE7A2和PDE7A3);这些变体通过在N-端和C-端的交替剪接产生(Bloom,T.J.和J.A.Beavo,Proc.Natl.Acad.Sci.USA.93:14188-14192,1996)。PDE7A转录变体1的核苷酸序列可利用存取号NM_002603从公共数据库得到。人PDE7A1蛋白(SEQ ID NO:2,被SEQ ID NO:1编码)有456个氨基酸,并在还原性SDS-PAGE上以53-55KDa的表观分子量迁移。
PDE7A转录变体2的核苷酸序列可利用存取号NM_002604从公共数据库得到。人PDE7A2蛋白(SEQ ID NO:4,被SEQ ID NO:3编码)有424个氨基酸。
PDE7A蛋白在羧基端有一个约270个氨基酸的区域,它显示出与其它cAMP-水解PDE的类似区域有显著的相似性(约23%同源性)。这一区域起催化域的作用。此蛋白的氨基端区域与其它PDE的不同,估计可能介导了此酶家族独有的明显不同的调控性能。
人PDE7B的蛋白序列可利用存取号NM_018945(作为SEQ ID NO:6提供,由SEQ IDNO:5编码)从公共数据库得到。已报道了PDE7B的三种剪接变体:PDE7B1,PDE7B2和PDE7B3。PDE7B公布在WO 01/62904,美国专利No.6,146,876中。
PDE7B2和PDE7B3都具有独特的N-端序列。人PDE7B基因产物在还原性SDS-PAGE上的表观分子量为53-55kDa(Sasaki,T.,Kotera,J.,Omori,K.,Biochemical J.361:211-220,2002)。与在PDE7A中一样,PDE7B在羧基端有一个对所有PDE都共同的约270个氨基酸的显著保守区,它作为催化域起作用。与PDE7A蛋白相似,PDE7B蛋白的氨基端区域是不同的,并可能是各个PDE家族独有的与众不同的调控性能的原因。PDE7B蛋白在催化域内显示与其它cAMP依赖性PDE的同源性(23%)。根据WO 2004/044196,PDE7B多肽与PDE7A有61%同源。
与其它PDE家族相比,PDE7还独特地位于哺乳动物受试者中。已在大多数被分析的组织(包括脑、心脏、肾、骨骼肌、脾和子宫)中检测到PDE7A表达(Bloom等人,PNAS93:14188,1996)。在脑中,PDE7A广泛地分布在神经细胞和非神经细胞群中(Miro等人,Synapse 40:201,2001)。PDE7A在脑内(包括基底神经节和黑质)的广泛表达为PDE7A在脑功能中的作用提供了理论基础。
在本发明方法的实施中,抑制PDE7的磷酸二酯酶活性的代表性PDE7抑制剂包括:与PDE7结合并抑制PDE7的酶活性的分子(例如与PDE7结合并降低酶活性的小分子抑制剂或阻抑肽),及在转录和/或翻译水平上减少PDE7的表达,从而阻止PDE7切割cAMP的分子(例如PDE7反义核酸分子,PDE7特异性RNAi分子和PDE7核酶)。如上所述,PDE7抑制剂可以作为主要疗法单独使用,或是与作为辅助治疗的其它治疗疗法(例如多巴胺受体激动剂)联合使用,以提高疗效。
PDE7的抑制的特征是,按照本发明方法施用PDE7抑制剂会发生至少一种以下变化:PDE7依赖性酶切割cAMP中的3’-磷酸二酯键形成5’-一磷酸腺苷(5’-AMP)被抑制,用例如基因表达分析(如RT-PCR分析)或蛋白分析(如Western印迹法)测得的PDE7的基因或蛋白表达水平降低。
在一些实施方案中,PDE7抑制剂是抑制PDE7A、PDE7B、或PDE7A和PDE7B二者的表达的分子或组合物,例如反义的或小的抑制性核苷酸(如siRNA),它与细胞mRNA和/或基因组DNA(与靶物PDE7的基因相应)特异地杂交,从而抑制它们的转录和/或翻译,或特异地切割靶物PDE7的mRNA的核酶。
PDE7抑制剂的效力
在一项实施方案中,可用于本发明方法的PDE7抑制剂是足以有效抑制PDE7(PDE7A、PDE7B、或PDE7A和PDE7B)的酶活性的化合物,其IC50≤1μM,优选小于或约0.1μM。在一项实施方案中,PDE7抑制剂足以有效抑制PDE7(PDE7A、PDE7B、或PDE7A和PDE7B)的酶活性,其IC50为约0.1至约500nM。在一项实施方案中,PDE7抑制剂有效抑制PDE7(PDE7A、PDE7B、或PDE7A和PDE7B)的酶活性,其IC50为约1至约100nM。
测定PDE7(PDE7A或PDE7B)抑制剂的IC50的典型方法是本领域所熟知的,例如在Bardelle等人,Anal Biochem 15:275(2):148-55(1999)中公开的闪烁亲近测定法(SPA)。
PDE7A或PDE7B选择性抑制剂
在一项实施方案中,适用于本发明方法的PDE7抑制剂是PDE7A抑制剂。在一项实施方案中,该PDE7A抑制剂有效抑制PDE7A的酶活性,IC50为约0.1至约500nM。在一项实施方案中,该PDE7A抑制剂具有约1至约100nM的IC50。一种适合测定PDE7A抑制剂的IC50的试验方法使用在杆状病毒体系中表达的重组人PDE7A2酶。该试验方法是以上Bardelle等报道的SPA试验法的改良。
在一些实施方案中,PDE7抑制剂显示出对于PDE7A的同工酶选择性活性。PDE7A选择性抑制剂降低PDE7A活性是降低PDE7B活性的至少2倍,更优选是至少10倍,至少20倍,至少50倍,或至少100倍。在一些实施方案中,PDE7A抑制剂是这样一种抑制剂,它抑制PDE7A活性的选择性是抑制任何其它PDE(PDE1-6,7B和8-11)的酶活性的至少10倍(例如至少20倍,或至少50倍,或至少100倍)。
在一项实施方案中,该PDE7B抑制剂的IC50为约0.1-约500nM。在一项实施方案中,该PDE7B抑制剂有足够的效力在约0.1-约500nM的IC50下抑制PDE7B的酶活性。在一项实施方案中,该PDE7B抑制剂具有约1-约100nM的IC50。测定PDE7B抑制剂的IC50的试验方法是本领域熟知的,例如以上Bardelle等报道的试验方法。
在一些实施方案中,PDE7抑制剂显示出对于PDE7B的同工酶选择性活性。PDE7B选择性抑制剂降低PDE7B活性是降低PDE7A活性的至少2倍,更优选是至少10倍,至少20倍,至少50倍,或至少100倍。在一些实施方案中,PDE7B抑制剂是这样一种抑制剂,它抑制PDE7B活性的选择性是抑制任何其它PDE(PDE1-6,7A和8-11)的酶活性的至少10倍(例如至少20倍,或至少50倍,或至少100倍)。
与其它PDE相比的PDE7选择性
在一些实施方案中,PDE7抑制剂对于抑制PDE1B活性的IC50要比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的5倍更大(例如至少10倍,至少20倍,或至少50倍,或至少100倍)。换言之,该PDE7抑制剂在抑制PDE7A或PDE7B的活性方面(不管该PDE7抑制剂对PDE7A或PDE7B同工酶中的哪一个更有效)比其抑制PDE1B活性更有效力(5倍,10倍,20倍,50倍或100倍)。就本说明书而言,作为实例,这一性质仍然可以更简单地说成是该PDE7抑制剂在抑制PDE7活性方面比其抑制PDE1B的活性方面更有效力(5倍,10倍,20倍,50倍或100倍)。
对PDE7和PDE1B的双重抑制可以为运动障碍的治疗带来另外的好处,其根据是有报道指出,小鼠中PDE1B基因的删除刺激了多巴胺的代谢并使该动物对多巴胺能激动剂的作用敏感(Siuciak等人,Neuropharmacology 53(1):113-23(2007))。
在一些实施方案中,PDE7抑制剂抑制PDE10活性的IC50要比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的5倍更大(例如至少10倍,或至少20倍,或至少50倍,或至少100倍)。对PDE7和PDE10的双重抑制可以为运动障碍的治疗带来另外的好处,其根据是有报道指出,PDE10的选择性抑制剂引起纹状体中cAMP水平的增高(Siuciak J.A.等人,Neuropharmacology 51(2):386-96(2006))。
在一些实施方案中,PDE7抑制剂抑制PDE3活性的IC50要比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大(例如至少20倍,至少50倍,或至少100倍)。这是因为已显示对心力衰竭患者施用PDE3的选择性抑制剂会增加其死亡率(Packer M.等人,N Engl J Med.325(21):1468-75(1991))。
在一些实施方案中,PDE7抑制剂抑制PDE4活性的IC50要比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大(例如至少20倍,至少50倍,或至少100倍)。这是因为删除小鼠中的PDE4基因之一已显示会引起心肌病(Lehnart S.E.等人,Cell 123(1):25-35(2005))。
在一些实施方案中,PDE7抑制剂在PDE4抑制(例如,在内毒素治疗后镇静或抑制TNF-α水平)的体内试验中的半最大有效剂量(“ED50”)要比PDE7A和PDE7B抑制(例如,预防可卡因或其它精神兴奋药成瘾复发)的体内试验中的ED50的较小者的10倍更大(例如至少20倍,至少50倍,或至少100倍)。根据这类实施方案,也已确定,与在体外试验中确定的化合物的PDE4/PDE7选择性相比,一些具有PDE4/PDE7双重抑制活性的化合物在体内对PDE7比PDE4有更高的选择性。
在一些实施方案中,PDE7抑制剂抑制PDE3活性和PDE4活性的IC50要比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大(例如至少20倍,至少50倍,或至少100倍)。
在一些实施方案中,PDE7抑制剂抑制PDE8活性的IC50要比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大(例如至少20倍,至少50倍,或至少100倍)。
在一些实施方案中,PDE7抑制剂抑制PDE4活性和PDE8活性的IC50要比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大(例如至少20倍,至少50倍,或至少100倍)。根据这一实施方案,获知特异/优选地水解cAMP的PDE家族包括PDE4、PDE7和PDE8。
在一些实施方案中,PDE7抑制剂抑制PDE1、PDE2、PDE3、PDE4和PDE8、PDE10、及PDE11活性的IC50要比抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者的10倍更大。根据这一实施方案,获知特异/优选地水解cAMP的PDE家族包括PDE4、PDE7和PDE8,并且PDE1、PDE2、PDE3、PDE10、和PDE11家族对cAMP和cGMP均表现出显著的活性。
在一些实施方案中,PDE抑制剂是选择性PDE7抑制剂,其抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者小于该试剂抑制PDE1-6和PDE8-11酶家族中的任何其它PDE酶的IC50的十分之一(例如1/20,1/50或1/100)。
选择性PDE7抑制剂可以例如通过将该试剂抑制PDE7(PDE7A、PDE7B或PDE7A和PDE7B)酶活性的能力与其抑制其它PDE家族的PDE酶的能力相比较来确定。例如,可以测定试剂抑制PDE7活性以及抑制PDE1、PDE2、PDE3、PDE4、PDE5、PDE6、PDE8、PDE9、PDE10和PDE11的能力。抑制各个PDE(1-6和8-11)同工酶的IC50与抑制PDE7(即对PDE7A或PDE7B更敏感)的IC50之比可以利用标准的体外、体内或离体的试验方法(例如本文描述的方法)确定。
在一些实施方案中,PDE7抑制剂对于PDE7具有选择性,但对其它PDE(例如PDE1、PDE2、PDE3、PDE4和PDE8、PDE10及PDE11),由于该PDE7抑制剂对一个或多个靶组织(例如脑和/或骨骼肌)的寻靶作用而基本上无活性。如本文所述,和其它PDE家族相比,PDE7独特地位于哺乳动物受试者中。在脑中,PDE7A广泛地分布在神经细胞和非神经细胞群中,包括基底神经节和黑质(Miro等人,Synapse 40:201,2001)。PDE7B在脑内表达在纹状体中(Reyes-Irisarri等人,Neuroscience 132:1173,2005)。
PDE7抑制剂的类型
PDE7抑制剂可以是任何类型的试剂,包括但不限于:化合物,蛋白质或多肽,肽模拟物,核酸分子或核酶。在一些实施方案中,PDE7抑制剂是小分子抑制剂,包括低分子量(即,小于约450g/摩尔)的天然和合成物质,例如,肽、肽模拟物和非肽抑制剂,如化合物。
化合物:
可用于本发明方法的PDE7抑制剂包括用常规途径(例如,口服、肌内、皮下、透皮、经颊、静脉内等)施用到血流中并最终经由脉管系统穿过血脑屏障输送以抑制脑内PDE7的试剂。因此,对于这些给药方法,PDE7抑制剂有穿透血脑屏障的能力。下面描述的那些能够穿过血脑屏障的PDE7抑制剂(例如分子量小于约450g/摩尔的和足够亲脂的抑制剂),当该抑制剂以最终将其输送到脑血流中的途径给药时,可用于本发明的方法。
以下是对可用于本发明方法的PDE7抑制剂实例的说明。
在一项实施方案中,可用于本发明方法的PDE7抑制剂选自在EP 1 454 897、WO2003/053975和US 20050148604中一般地或具体地公开的那些化合物,这些文献均特地以参考引用的方式全文并入本申请。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式

以上化合物的取代基定义如下:
A代表N或CR4,
B代表氢原子或卤素原子,
R1代表任选取代的C3-7环烷基或叔丁基,
R2代表氢、甲基或乙基,
R3代表氢、硝基、氰基或卤素原子、NR5R6、C(=X)R7、SO2NR5R6、OR8、NR8CONR5R6、NR8SO2R9、NR8CO2R9、杂芳基、任选取代的C1-3烷基、任选取代的C1-6烯基或任选取代的饱和或不饱和的杂环烷基,
R4代表氢或如需要被一个或多个氟原子取代的C1-3烷氧基,
R5和R6相同或不同,代表氢原子、任选取代的C1-6烷基、任选取代的杂环烷基,或任选取代的酰基,或者与它们所结合的氮原子一起形成氮杂环丁基、吡咯烷基、哌啶基、吗啉代、硫代吗啉代、哌嗪基或高哌嗪基,这些基团各自可任选地被任选取代的C1-4烷基、OH、C1-3烷氧基、CO2H、NR5R6、氧代基团、NR9COR7或C(=O)R7取代,
R7代表任选取代的C1-6烷基、OH、OR8、或NR5R6,
R8代表氢、任选取代的C1-6烷基或任选取代的杂环烷基,
R9代表任选取代的C1-6烷基,和
X代表O、S或NH。
就以上化合物而言,“任选取代的”是指任选取代的直链、支链或环状烷基基团,例如甲基、乙基、丙基或环己基;羟基;氰基;烷氧基,例如甲氧基或乙氧基;任选取代的氨基基团,例如氨基、甲氨基或二甲氨基;任选取代的酰基基团,例如乙酰基或丙酰基;羧基;任选取代的芳基,例如苯基或萘基;任选取代的杂芳基,例如吡啶基、噻唑基、咪唑基或吡唑基;任选取代的饱和或不饱和的杂环烷基,例如哌嗪基或吗啉基(morphonyl);任选取代的氨甲酰基;任选取代的酰氨基;卤素原子,例如氯、氟或溴;硝基;任选取代的砜基;任选取代的磺酰胺基(sulfonylamido);氧代基团,脲基;以及任选取代的直链、支链或环状烯基,例如乙烯基、丙烯基或环己烯基。
作为R3的杂芳基的实例包括含有2-8个碳原子并含1-4个由氧原子、氮原子或硫原子组成的杂原子的5-7元单环杂芳基,和包含2个或更多个稠合在一起的这类相同或不同的单环化合物的多环杂芳基,单环和多环杂芳基的实例是吡咯基、呋喃基、噻吩基、咪唑基、噻唑基、吡啶基、吡唑基、吲哚基、喹啉基、异喹啉基和四唑基。
在一项实施方案中,可用于本发明的PDE7抑制剂具有下式:

在其它实施方案中,可用于本发明方法的PDE7抑制剂具有下式。
在另一实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

在其它实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

在EP 1 454 897、WO2003/053975和US 20050148604中描述了以上化合物的制备。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 2002/0198198,WO2002/076953,WO 2002/074754,WO 2006/092691,Bioorganic&Medicinal ChemistryLetters 14(2004)4627-4631中一般地或具体地公开的那些化合物,上述文献均以参考引用的方式全文并入本申请。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:


以上化合物的取代基定义如下:
(a)X1、X2、X3和X4相同或不同,选自:
N,条件是,基团X1、X2、X3和X4中同时代表氮原子的不超过2个,或
C-R1,其中R1选自:
Q1,或
低级烷基、低级烯基或低级炔基,这些基团是未取代的或被一个或几个Q2基团取代;
基团X5-R5,其中X5是选自:
单键,
低级亚烷基、低级亚烯基或低级亚炔基,可任选地插入选自O、S、S(=O)、SO2或N的1或2个杂原子,这些基团的碳原子是未取代的,或是被选自SR6、OR6、NR6R7、=O、=S或=NR6的一个或几个相同或不同的基团取代,其中R6和R7相同或不同,选自氢或低级烷基,和
R5选自芳基、杂芳基、任选插入C(=O)或插入选自O、S、S(=O)、SO2或N的1、2或3个杂原子的环烷基、任选插入C(=O)或插入选自O、S、S(=O)、SO2或N的1、2或3个杂原子的环烯基,或双环基团,这些基团是未取代的,或是被选自Q3、杂芳基或任选被Q3取代的低级烷基的一个或几个基团取代;
其中Q1、Q2和Q3相同或不同,选自:
氢、卤素、CN、NO2、SO3H、P(=O)(OH)2、OR2、OC(=O)R2、C(=O)OR2、SR2、S(=O)R2、NR3R4、Q-R2、Q-NR3R4、NR2-Q-NR3R4或NR3-Q-R2,其中Q选自C(=NR)、C(=O)、C(=S)或SO2,R选自氢或低级烷基,R2、R3和R4相同或不同,选自:
氢,任选插入C(=O)的低级烷基、(CH2)n-芳基、(CH2)n-杂芳基、任选插入C(=O)或插入选自O、S、S(=O)、SO2或N的1或2个杂原子的(CH2)n-环烷基,其中n是选自0、1、2、3、或4的整数;
这些基团是未取代的,或是被选自以下基团的一个或几个基团取代:低级烷基、卤素、CN、CH3、SO3H、SO2CH3、CF3、C(=O)NHSO2CH3、OR6、COOR6、C(=O)R6、NR6R7、C(=O)NR6R7或SO2NR6R7,其中R6和R7相同或不同,选自氢或任选被1或2个选自OR、COOR或NRR8的基团取代的低级烷基,其中R和R8是氢或低级烷基,和
R6和R7,和/或R3和R4,与它们所连接的氮原子一起能够形成一个4-8元的杂环,它可含有选自O、S、S(=O)、SO2或N的1或2个杂原子,并可被以下基团取代:
4-8元杂环,它可含有选自O、S或N的1或2个杂原子,并可被低级烷基取代,或任选被OR’、NR’R”、C(=O)NR’R”或COOR’取代的低级烷基,其中R’和R”相同或不同,选自氢、任选被OR或COOR取代的低级烷基,其中R是氢或低级烷基,R’和R”与它们所连接的氮原子一起能形成一个4-8元杂环,该杂环可含1或2个选自O、S或N的杂原子;或
(b)X是O、S或NR9,其中R9选自氢、CN、OH、NH2、低级烷基、低级烯基或低级炔基,这些基团是未取代的,或是被任选插入选自O、S、S(=O)、SO2或N的1或2个杂原子的环烷基、任选插入选自O、S、S(=O)、SO2或N的1或2个杂原子的环烯基、芳基、杂芳基、OR10、或NR10R11取代,其中R10和R11相同或不同,选自氢或低级烷基;
(c)Y选自O、S或N-R12,其中R12选自氢、CN、OH、NH2、低级烷基、低级烯基或低级炔基,这些基团是未取代的,或是被任选插入选自O、S、S(=O)、SO2或N的1或2个杂原子的环烷基、任选插入选自O、S、S(=O)、SO2或N的1或2个杂原子的环烯基、芳基、杂芳基、OR10或NR10R11取代,其中R10和R11相同或不同,选自氢或低级烷基;
(d)Z选自CH-NO2、O、S或NR13,其中R13选自氢、CN、OH、NH2、芳基、杂芳基,任选插入选自O、S、S(=O)、SO2或N的1个或几个杂原子的环烷基、任选插入选自O、S、S(=O)、SO2或N的1个或几个杂原子的环烯基、C(=O)R14、C(=O)NR14R15、OR14或者未取代的或被一个或几个选自OR14或NR14R15的相同或不同的基团取代的低级烷基;R14和R15独立地选自氢或低级烷基,或者,R14和R15与它们所连接的氮原子一起能够形成一个4-8元的杂环,该杂环可含有选自O、S或N的1或2个杂原子,并可被低级烷基取代;
(e)Z1选自H、CH3或NR16R17,其中R16和R17相同或不同,选自氢、CN、芳基、杂芳基、任选插入选自O、S、S(=O)、SO2或N的一个或几个杂原子的环烷基、任选插入选自O、S、S(=O)、SO2或N的一个或几个杂原子的环烯基、C(=O)R14、C(=O)NR14R15、OR14或者未取代的或被一个或几个选自OR14或NR14R15的基团取代的低级烷基;
R14和R15选自氢或低级烷基,并且,R14和R15,和/或,R16和R17,与它们所连接的氮原子一起能够形成一个4-8元的杂环,该杂环可含有选自O、S或N的1或2个杂原子,并可被低级烷基取代;
(f)A是一个环,选自:

其中,
A1、A2、A3、A4、A5和A6相同或不同,选自O、S、C、C(=O)、SO、SO2或NR18,其中R18选自氢,芳基,杂芳基,任选插入选自O、S、S(=O)、SO2或N的一个或几个杂原子的环烷基,任选插入选自O、S、S(=O)、SO2或N的一个或几个杂原子的环烯基,未取代的或被以下基团取代的低级烷基:芳基、杂芳基、任选插入选自O、S、S(=O)、SO2或N的一个或几个杂原子的环烷基、任选插入选自O、S、S(=O)、SO2或N的一个或几个杂原子的环烯基、CN、NR19R20、C(=O)NR19R20、OR19、C(=O)R19或C(=O)OR19,其中R19和R20相同或不同,并且选自氢或低级烷基;
*代表环A和含X和/或Y的骨架环之间共有的碳原子;
环A的各碳原子均为未取代的,或被选自以下1或2个相同或不同的基团取代:任选被OR21、NR21R22、COOR21或CONR21R22取代的低级烷基,低级卤代烷基,CN,F,=O,SO2NR19R20,OR19,SR19,C(=O)OR19,C(=O)NR19R20,或NR19R20,其中R19和R20相同或不同,选自氢或任选被OR21、NR21R22、COOR21或CONR21R22取代的低级烷基,其中R21和R22相同或不同,选自氢或低级烷基,并且R19和R20,和/或R21和R22,与它们所连接的氮原子一起能够形成一个4-8元的杂环;
环A的两个不相邻原子可以通过2、3或4碳原子链连接,该碳原子链中可以插入一个选自O、S或N的杂原子;条件是,基团A1、A2、A3、A4、A5和A6中同时代表杂原子的基团不超过两个;和
其互变异构形式、其外消旋形式、其异构体及其药学上可接受的衍生物。
就以上化合物而言,卤素包括氟、氯、溴和碘。优选的卤素是F和Cl。低级烷基包括含有1-6个碳原子的直链和支链碳链。这样的烷基的实例包括甲基、乙基、异丙基和叔丁基。低级烯基包括含有2-6个碳原子和至少一个双键的直链和支链烃基。这样的烯基的实例是乙烯基、3-丁烯-1-基、2-乙烯基丁基和3-己烯-1-基。低级炔基包括含有2-6个碳原子和至少一个三键的直链和支链烃基。这样的炔基的实例是乙炔基、3-丁炔-1-基、丙炔基、2-丁炔-1-基和3-戊炔-1-基。低级卤代烷基包括被一个或几个卤素取代的如上定义的低级烷基。卤代烷基的一个实例是三氟甲基。芳基应理解为是指含6-10个碳原子的芳族碳环。芳基的一个实例是苯基。杂芳基包括含有5-10个环原子的芳族环,其中1-4个独立地选自O、S和N。代表性的杂芳基在5或6元芳族环中有1、2、3或4个杂原子。这类基团的实例是四唑、吡啶基和噻吩基。代表性的环烷基含3-8个碳原子。这类基团的实例是环丙基、环丁基、环戊基、环己基、环庚基和环辛基。术语“插入”是指在骨架链中的碳原子被本文定义的杂原子或基团代替。例如,在“任选插入C(=O)或插入一个选自O、S、S(=O)、SO2或N的杂原子的环烷基或环烯基”中,术语“插入”是指C(=O)或杂原子可以替代该环的碳原子。这类基团的实例是吗啉或哌嗪。环烯基包括其中含至少一个双键的3-10元环烷基。杂环包括如上定义的杂芳基和插入1、2或3个选自O、S、S(=O)、SO2或N的杂原子的如上定义的环烷基或环烯基。双环取代基指选自芳基、杂环、环烷基或环烯基的两个相同或不同的环稠合在一起,形成所述的双环取代基。双环取代基的一个实例是吲哚基。
在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

在其它实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的制备在US 2002/0198198,WO2002/076953,WO2002/074754,WO2006/092691,Bioorganic&Medicinal Chemistry Letters 14(2004)4623-4626,和Bioorganic&Medicinal Chemistry Letters 14(2004)4627-4631中有说明。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在EP 1 193 261,WO2002/28847,US 20030045557,美国专利No.7,122,565,Bioorganic&Medicinal ChemistryLetters 14(2004)4607-4613,和Bioorganic&Medicinal Chemistry Letters 14(2004)4615-4621中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
Y是S或O;
R1是C1-C10烷基、C2-C10烯基、C2-C10炔基、环烷基、环烯基、杂环、芳基或多环基团;它们各自任选地被一个或几个相同或不同的基团X1-R4取代,其中X1是单键、低级亚烷基、C2-C6亚烯基、亚环烷基、亚芳基或二价的杂环,R4是:
(1)H,=O,NO2,CN,卤素,低级卤代烷基,低级烷基,羧酸生物电子等排体;
(2)COOR5,C(=O)R5,C(=S)R5,SO2R5,SOR5,SO3R5,SR5,OR5;
(3)C(=O)NR7R8,C(=S)NR7R8,C(=CH-NO2)NR7R8,C(=N-CN)NR7R8,C(=N-SO2NH2)NR7R8,C(=NR7)NHR8,C(=NR7)R8,C(=NR9)NHR8,C(=NR9)R8,SO2NR7R8或NR7R8,其中R7和R8相同或不同,选自OH,R5,R6,C(=O)NR5R6,C(=O)R5,SO2R5,C(=NR9)NHR10,C(=NR9)R10,C(=CH-NO2)NR9R10,C(=N-SO2NH2)NR9R10,C(=N-CN)NR9R10或C(=S)NR9R10;
R2是低级烷基,C2-C10烯基,C2-C10炔基,环烷基,环烯基,杂环,芳基;它们各自任选地被一个或几个选自以下的相同或不同的基团取代:
(1)H,羧酸生物电子等排体,低级卤代烷基,卤素,
(2)COOR5,OR5,SO2R5,
(3)SO2NR11R12,C(=O)NR11R12,NR11R12,其中R11和R12相同或不同,选自OH,R5,R6,C(=O)NR5R6,C(=O)R5,SO2R5,C(=S)NR9R10,C(=CH-NO2)NR9R10,C(=N-CN)NR9R10,C(=N-SO2NH2)NR9R10,C(=NR9)NHR10或C(=NR9)R10;
R3是X2-R3’,其中X2是单键或选自C1-C4亚烷基、C2-C6亚烯基、C2-C6亚炔基的基团,它们各自任选地被一个或几个选自以下的相同或不同的基团取代:
(1)H,C1-C3烷基,C3-C4环烷基,芳基,杂环,=O,CN;
(2)OR5,=NR5;或
(3)NR13R14,其中R13和R14相同或不同,选自R5,R6,C(=O)NR5R6,C(=O)R5,SO2R5,C(=S)NR9R10,C(=CH-NO2)NR9R10,C(=NR9)NHR10或C(=NR9)R10;
R3’是环烷基,环烯基,芳基,杂环或多环基团;它们各自任选地被一个或几个基团X3-R17取代,其中X3是单键、低级亚烷基、C2-C6亚烯基、C2-C6亚炔基、亚环烷基、亚芳基、二价的杂环或二价的多环基团,而R17是:
(1)H,=O,NO2,CN,低级卤代烷基,卤素,羧酸生物电子等排体,环烷基;
(2)COOR5,C(=O)R5,C(=S)R5,SO2R5,SOR5,SO3R5,SR5,OR5;
(3)C(=O)NR15R16,C(=S)NR15R16,C(=N-CN)NR15R16,C(=N-SO2NH2)NR15R16,C(=CH-NO2)NR15R16,SO2NR15R16,C(=NR15)NHR16,C(=NR15)R16,C(=NR9)NHR16,C(=NR9)R16或NR15R16,其中R15和R16相同或不同,选自OH,R5,R6,C(=O)NR5R6,C(=O)R5,SO2R5,C(=S)NR9R10,C(=CH-NO2)NR9R10,C(=N-CN)NR9R10,C(=N-SO2NH2)NR9R10,C(=NR9)NHR10或C(=NR9)R10;
(4)任选被一个或几个基团R5取代的杂环;
其中R5和R6相同或不同,选自氢、低级烷基,C2-C6烯基,C2-C6炔基,X4-环烷基,X4-环烯基,X4-芳基,X4-杂环,或X4-多环基,其中X4是单键、低级亚烷基或C2-C6亚烯基,它们各自任选地被选自以下的一个或几个相同或不同的基团取代:卤素,=O,COOR20,CN,OR20,任选被OR20取代的O-低级烷基,C(=O)-低级烷基,低级卤代烷基,

其中X5是单键或低级亚烷基,和R18、R19和R20相同或不同,选自H或低级烷基;X6-杂环,X6-芳基,X6-环烷基,X6-环烯基,或X6-多环基,其中X6是单键或低级亚烷基,这些基团任选地被选自卤素、COOR21、OR21或(CH2)nNR21R22的一个或几个相同或不同的基团取代,其中n是0、1或2,R21和R22相同或不同,选自H或低级烷基;
R9选自H、CN、OH、低级烷基、O-低级烷基、芳基、杂环、SO2NH2或

其中X5是单键或低级亚烷基,R18和R19相同或不同,选自H或低级烷基;
R10选自氢、低级烷基、环丙基或杂环;
或它们的药学上可接受的衍生物。
就以上化合物而言,芳基是指环状结构中只含数目为5-10个碳原子的不饱和的碳环,包括苯基、萘基或四氢萘基。杂环指不饱和或饱和的单环,其环状结构中含1-7个碳原子和环状结构中含至少一个杂原子,例如氮、氧或硫,优选含1-4个选自氮、硫和氧原子的相同或不同的杂原子。合适的杂环包括吗啉基、哌嗪基、吡咯烷基、哌啶基、嘧啶基、2-和3-呋喃基、2-和3-噻吩基、2-吡啶基、2-和3-吡喃基、羟基吡啶基、吡唑基、异噁唑基、四唑、咪唑、三唑等。多环基团包含稠合在一起的选自芳基、杂环、环烷基、环烯基的至少两个相同或不同的环,形成该多环基团,例如,2-和3-苯并噻吩基、2-和3-苯并呋喃基、2-吲哚基、2-和3-喹啉基、吖啶基、喹唑啉基、吲哚基、苯并[1,3]间二氧杂环戊烯基和9-thioxantanyl。双环基团是指选自芳基、杂环、环烷基或环烯基的两个相同或不同的环稠合在一起,形成所述双环基团。卤素指氟、氯、溴或碘。低级烷基指含1-6个碳原子的直链或支链烷基。低级烷基的实例包括甲基、乙基、丙基、丁基、异丙基、叔丁基、异丁基、正丁基、戊基、己基等。烯基指含一个或几个双键、优选1或2个双键的直链或支链的不饱和碳原子链。炔基指含一个或几个三键、优选1或2个三键的直链或支链的不饱和碳原子链。低级卤代烷基指被一个或几个卤素取代的低级烷基,优选的低级卤代烷基包括全卤代烷基例如CF3。环烷基指含有3-10个碳原子的饱和单环碳环,包括环丙基、环丁基、环戊基、环己基和环庚基。环烯基指含有3-10个碳原子的不饱和单环碳环。合适的环烯基的实例是3-环己烯和3-环庚烯。羧酸生物电子等排体具有通常的含义,常见的羧酸生物电子等排体是四唑-5-基、C(=O)N(H)OH、异噁唑-3-基、羟基噻二唑基、磺酰胺基、磺酰基甲酰胺基、膦酸、磷酰胺基、次膦酸、磺酸、酰基磺酰胺基、巯基作(mercaptoazole)、酰基氰胺。
在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

在其它实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

在EP 1 193 261,WO 02/28847,US 20030045557,美国专利No.7,122,565,Bioorganic&Medicinal Chemistry Letters 14(2004)4607-4613,和Bioorganic&Medicinal Chemistry Letters 14(2004)4615-4621中描述了以上化合物的制备。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO 2004/111054,US20060128728和US 20070270419中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

和

以上化合物的取代基定义如下:
R1是取代或未取代的C3-8环烷基或叔丁基;
R2是氢原子或C1-3烷基;
R3是基团NR5R6、C(=O)R7或S(O)0-2R8;
R4是氢原子或是未取代的或被一个或多个氟原子取代的C1-3烷氧基;
R5和R6彼此相同或不同,是氢原子、取代或未取代的C1-6烷基、取代或未取代的酰基、取代或未取代的杂环烷基、及与连接R5和R6的氮原子形成的取代或未取代的杂环烷基环;
R7是基团OR9或NR5R6;
R8是氢原子、卤素原子和以下基团:NR5R6,取代或未取代的C1-6烷基,或取代或未取代的芳基;
R9是氢原子或取代或未取代的C1-6烷基;
或其药学上可接受的盐或溶剂化物。
就以上化合物而言,术语“C1-C3烷基”包括含有1-3个碳原子的直链或支链烷基。术语“C3-C8环烷基”包括含有3-8个碳原子的环烷基,例如环丙基、环丁基、环戊基、环己基和环辛基。术语“杂环烷基”是含有1-4个相同或不同的杂原子(例如氧、氮或硫原子)的3-7元杂环基团,实例可以包括吡咯烷基、哌啶基、哌嗪基、高哌嗪基、四氢呋喃基、四氢吡喃基、吗啉基和氮杂环丁基。术语“C1-C3烷氧基”是指含有1-3个碳原子的烷氧基。术语“酰基”是指含有1-8个碳原子的酰基。术语“芳基”是含有6-12个碳原子的苯基、萘基、联苯基,术语“杂芳基”是含2-8个碳原子和1-4个相同或不同的杂原子(例如氧、氮、硫原子)的5-7元单环或多环基团。其实例包括吡咯、呋喃基、噻吩基、咪唑基、噻唑基、吡嗪基、吲哚基、喹啉基、异喹啉基、四唑基、吡啶基、吡唑基、哒嗪基和嘧啶基。“取代或未取代的C1-C6烷基”的合适取代基的实例包括羟基和卤素原子,而“取代或未取代的酰基”的合适取代基的实例包括卤素原子和硝基。另外,“取代或未取代的芳基”的合适取代基的实例包括C1-C3烷基、卤素原子、氨基、酰基、酰胺基、羟基、酰基氨基、羧基和磺酰基。“取代或未取代的C3-C8环烷基”的合适取代基的实例是C1-C3烷基、羟基和氧代基团,而“取代或未取代的杂环烷基”的合适取代基的实例包括羧基、酰基、烷氧基、氨基、烷基氨基、酰基氨基、羟基、氧代基团、亚乙二氧基、甲基、乙基和羟乙基。
在其它实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的制备在WO 2004/111054,US 20060128728和US 20070270419中有说明。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,903,109,US 20040082578,WO 2003/088963和US 20060154949中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
(a)R1选自以下基团:
(i)COR5,其中R5选自氢,任选取代的C1-8直链或支链烷基,任选取代的芳基和任选取代的芳烷基;其中该烷基、芳基和芳烷基上的取代基选自C1-8烷氧基、苯基乙酰氧基、羟基、卤素、对甲苯磺酰氧基、甲磺酰氧基、氨基、氰基、烷氧羰基或NR20R21,其中R20和R21独立地选自氢、C1-8直链或支链烷基、C3-7环烷基、苄基或芳基;
(ii)COOR6,其中R6选自氢、任选取代的C1-8直链或支链烷基、任选取代的芳基和任选取代的芳烷基;其中该烷基、芳基和芳烷基上的取代基选自C1-8烷氧基、苯基乙酰氧基、羟基、卤素、对甲苯磺酰氧基、甲磺酰氧基、氨基、氰基、烷氧羰基或NR20R21,其中R20和R21独立地选自氢、C1-8直链或支链烷基、C3-7环烷基、苄基或芳基;
(iii)氰基;
(iv)与R4形成的内酯或内酰胺;
(v)CONR7R8,其中R7和R8独立地选自氢、C1-8直链或支链烷基、C3-7环烷基、三氟甲基、羟基、烷氧基、酰基、烷基羰基、羧基、芳烷基、芳基、杂芳基和杂环基;其中的烷基、环烷基、烷氧基、酰基、烷基羰基、羧基、芳烷基、芳基、杂芳基和杂环基可以被羧基、烷基、芳基、取代的芳基、杂环基、取代的杂环基、杂芳基、取代的杂芳基、异羟肟酸、磺酰胺、磺酰基、羟基、硫醇、烷氧基或芳烷基取代;
或者R7和R8与它们所连接的氮合起来形成一个杂环基或杂芳基;
(vi)羧酸酯或羧酸生物电子等排体,包括任选取代的杂芳基;
(b)R2选自任选取代的烷基,任选取代的芳基,任选取代的杂芳基,任选取代的C3-7环烷基,任选取代的杂环基,其中该杂环基是1,3-二氧杂环戊烷或呋喃,或者R2是

(c)R3是独立选自以下基团的1-4个基团:
(i)氢、卤素、C1-8直链或支链烷基、芳烷基、C3-7环烷基、C1-8烷氧基、氰基、C1-4烷氧羰基、三氟甲基、C1-8烷磺酰基、卤素、硝基、羟基、三氟甲氧基、C1-8羧酸酯、芳基、杂芳基和杂环基;
(ii)NR10R11,其中R10和R11独立地选自氢、C1-8直链或支链烷基、芳烷基、C3-7环烷基、羧基烷基、芳基、杂芳基或杂环基,或者R10和R11与它们所连接的氮一起形成一个杂环基或杂芳基;
(iii)NR12COR13,其中R12选自氢或烷基,R13选自氢、烷基、取代的烷基、C1-3烷氧基、羧基烷基、R30R31N(CH2)p、R30R31NCO(CH2)p、芳基、芳烷基、杂芳基或杂环基,或者R12和R13与所述羰基合起来形成一个含羰基的杂环基团,其中R30和R31独立地选自H、OH、烷基和烷氧基,p是1-6的整数,其中该烷基可以被羧基、烷基、芳基、取代的芳基、杂环基、取代的杂环基、杂芳基、取代的杂芳基、异羟肟酸、磺酰胺、磺酰基、羟基、硫醇、烷氧基或芳烷基取代;
(d)R4选自以下基团:(i)氢,(ii)C1-3直链或支链烷基,(iii)苄基,和(iv)NR13R14,其中R13和R14独立地选自氢和C1-6烷基;其中该C1-3烷基和苄基任选地被一个或多个选自以下的基团取代:C3-7环烷基、C1-8烷氧基、氰基、C1-4烷氧羰基、三氟甲基、C1-8烷磺酰基、卤素、硝基、羟基、三氟甲氧基、C1-8羧酸酯、氨基、NR13R14、芳基和杂芳基;和
(e)X选自S和O;
以及它们的药学上可接受的盐、酯和前药形式。
在另一实施方案中,R1、R3和R4如上,R2是NR15R16,其中R15和R16独立地选自氢、C1-8直链或支链烷基、芳烷基、C3-7环烷基、芳基、杂芳基和杂环基,或者R15和R16与它们所连接的氮合起来形成一个杂环基或杂芳基。
就以上化合物而言,“烷基”指直链、环状和支链烷基。该烷基可以任选地被一个或多个以下基团取代:例如,卤素、OH、CN、巯基、硝基、氨基、C1-C8烷基、C1-C8烷氧基、C1-C8烷硫基、C1-C8烷基氨基、二(C1-C8烷基)氨基、(一、二、三和全)卤代烷基、甲酰基、羧基、烷氧羰基、C1-C8烷基-CO-O-、C1-C8烷基-CONH-、甲酰胺、异羟肟酸、磺酰胺、磺酰基、硫醇、芳基、芳基(C1-C8)烷基、杂环基和杂芳基。术语“生物电子等排体”定义为“具有产生大体类似的生物性质的化学和物理性质的基团或分子”(Burger′s Medicinal Chemistry and DrugDiscovery,M.E.Wolff编,第5版,vol.1,1995,pg.785)。术语“酰基”在本文中使用时,无论是单独使用或是作为某个取代基的一部分,均指由有机酸除去羟基衍生形成的有2-6个碳原子的(支链或直链)有机基团。“芳基”或“Ar”,无论是单独使用或是作为某个取代基的一部分,均指碳环芳族基团,包括但不限于,苯基、1-或2-萘基等。该碳环芳族基团上的1-5个氢原子可以独立地被以下基团替代:卤素、OH、CN、巯基、硝基、氨基、C1-C8烷基、C1-C8烷氧基、C1-C8烷硫基、C1-C8烷基氨基、二(C1-C8烷基)氨基、(一、二、三和全)卤代烷基、甲酰基、羧基、烷氧羰基、C1-C8烷基-CO-O-、C1-C8烷基-CONH-或甲酰胺。芳基的实例包括,例如,苯基、萘基、联苯、氟苯基、二氟苯基、苄基、苯甲酰氧基苯基、乙氧羰基苯基、乙酰苯基、乙氧基苯基、苯氧基苯基、羟苯基、羧基苯基、三氟甲基苯基、甲氧基乙基苯基、乙酰胺基苯基、甲苯基、二甲苯基、二甲基氨甲酰苯基等。术语“杂芳基”指有5-10个环原子的环状完全不饱和基团,其中一个环原子选自S、O和N,0-2个环原子是独立选自S、O和N的另外杂原子,其余的环原子是碳。该基团可以通过任何环原子与分子的其余部分连接。术语“杂环”、“杂环的”和“杂环”是指任选取代的完全饱和或部分饱和的环状基团,其是例如4-7元单环、7-11元双环或10-15元三环系统,在含至少一个碳原子的环内有至少一个杂原子。含杂原子的杂环基团的各个环可以有选自氮原子、氧原子和硫原子的1、2或3个杂原子,其中氮和硫杂原子还可以任选地被氧化。氮原子可以任选地被季铵化。该杂环基团可以在任何杂原子或碳原子处连接。
在其它实施方案中,可用于本发明方法的PDE7抑制剂具有下式:


以上化合物的制备在美国专利No.6,903,109,US 20040082578,WO 2003/088963和US 20060154949中有说明。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,958,328,WO 2002/085894和US 20030212089中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。这些PDE7抑制剂具有和上述(例如美国专利No.6,903,109)相同的化学式,只是R1不是羧酸酯或羧酸生物电子等排体。这些化合物的制备在美国专利No.6,958,328,US 20030212089和WO 2002/085894中有说明。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自WO 2006/004040和EP1775 298中一般地或具体地公开的那些化合物,这些文献均特次全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1是取代或未取代的C3-8烷基,取代或未取代的环烷基,或是取代或未取代的杂环烷基(例如,环己基、环庚基或四氢吡喃基);
R2是氢原子或取代或未取代的C1-3烷基(例如甲基);
R3是氢原子,取代或未取代的C1-3烷基,或卤素原子;和
R4是取代或未取代的芳基,取代或未取代的杂芳基,或是基团CONR5R6或CO2R7,
其中R5和R6彼此相同或不同,代表氢原子;可以被卤素原子、取代或未取代的芳基、取代或未取代的杂芳基、取代或未取代的杂环烷基、取代或未取代的环烷基、基团NR7COR8、COR8、NR9R10取代的C1-6烷基;取代或未取代的环烷基;取代或未取代的杂环烷基;取代或未取代的芳基;取代或未取代的杂芳基;或取代或未取代的杂环烷基,其中该环是与结合R5及R6的氮原子一起形成的;
其中R7是氢原子或是取代或未取代的C1-3烷基;
其中R8是取代或未取代的杂环烷基,或是基团OH、OR7或NR9R10;
其中R9和R10彼此相同或不同,代表氢原子、取代或未取代的C1-3烷基、取代或未取代的杂环烷基、取代或未取代的酰基、基团SO2R7、或取代或未取代的杂环烷基,其中该环是与结合R5及R6的氮原子一起形成的;
或其药学上可接受的盐或溶剂化物。
就以上化合物而言,术语“环烷基”指有3-8个碳原子的环烷基。术语“杂环烷基”可以是含有相同或不同的1-4个杂原子(例如氧、氮或硫原子)的3-7元的单环或多环杂环基团,实例可以包括哌啶基、吡咯烷基、哌嗪基、四氢呋喃基、四氢吡喃基、吗啉基、氮杂环丁基、咪唑烷基、噁唑烷基、六氢吡咯烷基、octahydroindolidinyl、octahydroquinolidinyl、八氢吲哚基和它们的氧代衍生物。术语“芳基”可以是由单个苯环或者结合的或稠合的苯环构成的芳族烃基,例如苯基、萘基、联苯等;以及由苯环与环烷基或杂环稠合形成的二环或三环基团,例如,1,2,3,4-四氢萘、2,3-二氢化茚,二氢吲哚、苯并呋喃等。术语“杂芳基”可以是有2-8个碳原子和1-4个杂原子(例如氧、氮、硫原子)的5-7元单环杂芳基或多环杂芳基,其中该多环杂芳基是由相同或不同的单环杂芳基或苯环彼此稠合形成的稠环系统,或是由杂芳基与环烷基或杂环烷基环稠合形成的多环基团。本发明的合适取代基的实例可以包括:直链、支链或环状的C1-C8烷基,它可以被一个或多个甲基、乙基、丙基、异丙基、正丁基、叔丁基、环己基、环庚基、甲氧基甲基、羟甲基、三氟甲基、C1-C3烷氧基、卤素原子和羟基取代;羟基;氰基;取代或未取代的烷氧基,例如甲氧基、乙氧基;可以被C1-C6烷基或酰基取代的氨基,例如氨基、甲基氨基、乙基氨基、二甲基氨基、酰基氨基等;羧基;取代或未取代的酯基;磷酸酯基;磺酸基;取代或未取代的芳基;取代或未取代的杂芳基;可以被取代的饱和或不饱和的杂环烷基;取代或未取代的氨甲酰基;取代或未取代的酰胺基;取代或未取代的硫代酰胺基;卤素原子;硝基;取代或未取代的砜基;取代或未取代的磺酰胺基;氧代基团;取代或未取代的脲基;直链、支链或环状的烯基,例如乙烯基、丙烯基、环己烯基等。
在其它实施方案中,可用于本发明方法的PDE7抑制剂具有下式:


在EP 1 775 298和WO2006/004040中描述了以上化合物的制备。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO 2004/111053和US20060128707中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
A是N或CR4;
B是N或CH;
R1是取代或未取代的C3-8环烷基或叔丁基;
R2是氢原子或C1-6烷基;
R3是氢原子;硝基;氰基;卤素原子;杂芳基;取代或未取代的C1-6烷基;取代或未取代的C2-6烯基;取代或未取代的饱和或不饱和的杂环烷基;以下基团:NR5R6,C(O)R7,SO2R7,OR8,NR8COR7,NR8SO2R7;
R4是氢原子或是未取代的或被一个或多个氟原子取代的C1-3烷氧基;
R5和R6彼此相同或不同,代表氢原子,取代或未取代的C1-6烷基,取代或未取代的酰基,或是取代或未取代的杂环烷基;
R7是氢原子,取代或未取代的C1-6烷基,取代或未取代的杂环烷基,OH,OR8,或NR5R6;
R8是氢原子,取代或未取代的C1-6烷基,或取代或未取代的杂环烷基;
或其药学上可接受的盐或溶剂化物。
就以上化合物而言,术语“C1-C6烷基”指有1-6个碳原子的直链或支链烷基,术语“C2-C6烯基”指有2-6个碳原子的直链或支链烯基。术语“环烷基”指有3-8个碳原子的环烷基,例如环丙基、环丁基、环戊基、环己基、环庚基和环辛基。术语“杂环烷基”是含有相同或不同的1-4个杂原子(例如氧、氮或硫原子)的3-7元杂环基团,实例可以包括哌啶基、吡咯烷基、哌嗪基、四氢呋喃基、四氢吡喃基、吗啉基、氮杂环丁基和高哌嗪基。术语“杂芳基”是含2-8个碳原子和1-4个相同或不同的杂原子(例如氧、氮、或硫原子)的5-7元单环或多环基团。其实例包括吡咯、呋喃基、噻吩基、咪唑基、噻唑基、吡嗪基、吲哚基、喹啉基、异喹啉基、四唑基、吡啶基、吡唑基、哒嗪基和嘧啶基。“卤素原子”包括氟、氯、溴和碘。“取代或未取代的C1-C6烷基”、“取代或未取代的的C3-C8环烷基”、“取代或未取代的烯基”、“取代或未取代的杂环烷基”和“取代或未取代的酰基”的合适取代基的实例包括直链或支链、或取代或未取代的烷基,例如甲基、乙基、丙基、异丙基、正丁基、叔丁基;取代或未取代的环烷基,例如环丙基、环丁基、环戊基、环己基和环庚基;羟基;氰基;烷氧基,例如甲氧基和乙氧基;取代或未取代的氨基,例如氨基、甲基氨基、乙基氨基和二甲基氨基;取代或未取代的酰基,例如乙酰基和丙酰基;取代或未取代的芳基;取代或未取代的杂芳基;取代或未取代、饱和或不饱和的杂环烷基;取代或未取代的氨甲酰基;取代或未取代的酰胺基;卤素原子;硝基;取代或未取代的砜基;氧代基团;脲基;取代或未取代的直链或支链或环状烯基,例如乙烯基、丙烯基和环己烯基。
在其它实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的制备在US 20060128707和WO 2004/111053中有说明。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,617,357,US 20020156064和Molecular Pharmacology,66:1679-1689,2004中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1是NRaRb,其中Ra和Rb独立地是H或C1-6烷基,或代表一个含有碳或者碳与一个或多个选自O、N或S的其它杂原子的5-7元环;
R2是H、C1-8烷基、C1-3烷基-Ar、C1-3烷基-C3-6环烷基、C2-8烯基、C2-4烯基-Ar或C2-4烯基-C3-6环烷基,其中Ar是取代或未取代的苯基;
R3是NO2、卤素、CN、C(O)OR7、COR1或NRaRb,其中Ra和Rb独立地是H或C1-6烷基;
R4是H、OC1-6烷基、卤素、C(O)NRaRb、C(O)OR7、C1-8烷基、OCHF2、CH2OR8、OC1-3烷基-Ar或CH2NHC(O)CH3;
R5是H、卤素或烷基;
R6是C1-8烷基、OC1-4烷基或卤素;
R7是氢或是成酯或成酰胺基团;
R8是氢或C1-6烷基;
或其药学上可接受的盐或溶剂化物。
在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的制备在美国专利No.6,617,357,US 20020156064和MolecularPharmacology,66:1679-1689,2004中有说明。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,852,720,EP 1 348 433和WO2003/082277中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1是选自环烷基、杂环烷基、芳基和杂芳基的一个基团,这些基团任选地被彼此独立地选自以下基团的一个或多个、相同或不同的基团取代:卤素,三氟甲基,硝基,氰基,氧代,NR4R5,CO2R4,CONR4R5,OR4,S(O)nR4,S(O)nNR4R5,四唑基和(C1-C6)烷基(任选地被1-3个彼此独立地选自OR4、NR4R5和CO2R4的相同或不同的基团取代);其中n是0到2(包括0和2在内)的整数,R4和R5相同或不同,彼此独立地是氢原子或式X1-Ra基团,其中X1是单键或(C1-C6)亚烷基,Ra是选自(C1-C6)烷基、环烷基、杂环烷基、芳基和杂芳基的基团,
R2是选自(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、芳基和环烷基的基团,
R3是选自环烷基、杂环烷基、芳基和杂芳基的基团,这些基团任选地被一个或多个彼此独立地选自以下基团的相同或不同的基团取代:卤素,硝基,氰基,三氟甲基,氧代,(C1-C6)烷基,OR6,NR6R7,COR6,CO2R6,CONHOH,CONR6R7,S(O)mR6,S(O)mNR6R7,NR6COR7,NR6SO2R7,N(SO2R7)2,NR6CONR7R8,C(=NCN)NR6R7,NR8C(=NCN)NR6R7和任选被(C1-C4)烷基取代的四唑基,其中m是0-2(包括0和2在内)的整数,R6和R7相同或不同,彼此独立地是氢原子或式X2Rb的基团,其中X2是单键或(C1-C6)亚烷基,Rb是选自(C1-C6)烷基、环烷基、杂环烷基、芳基和杂芳基的一个基团,这些基团任选地被1-3个彼此独立地选自以下基团的相同或不相同的基团取代:羟基,(C1-C6)烷氧基,(C1-C6)烷基,氨基,一(C1-C6)烷基氨基,二(C1-C6)烷基氨基(各烷基氨基相同或不同,彼此独立),羧基,(C1-C6)烷氧羰基和苄基,R8则代表氢原子或(C1-C6)烷基;
其外消旋形式、其异构体、其N-氧化物或其药学上可接受的酸或碱盐。
在美国专利No.6,852,720,EP 1 348 433和WO 2003/082277中描述了以上化合物的制备。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,753,340,US 20030191167,EP 1 348 701和WO2003/082839中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1a是选自氢、(C1-C6)烷基和芳基(C1-C6)烷基的基团,
R1b是选自环烷基、杂环烷基、芳基和杂芳基的基团,这些基团任选地被彼此独立地选自以下基团的一个或多个相同或不同的基团取代:卤素,三氟甲基,硝基,氰基,氧代,NR4R5,CO2R4,CONR4R5,OR4,S(O)nR4,S(O)nNR4R5,四唑基和(C1-C6)烷基(任选地被1-3个彼此独立地选自OR4、NR4R5和CO2R4的相同或不同的基团取代);其中n是0到2(包括0和2在内)的整数,R4和R5相同或不同,彼此独立地是氢原子或式X1-Ra基团,其中X1是单键或(C1-C6)亚烷基,Ra是选自(C1-C6)烷基、环烷基、杂环烷基、芳基和杂芳基的基团,
R2是选自(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、芳基和环烷基的基团,
R3是选自环烷基、杂环烷基、芳基和杂芳基的基团,这些基团任选地被一个或多个彼此独立地选自以下基团的相同或不同的基团取代:卤素,硝基,氰基,三氟甲基,氧代,(C1-C6)烷基,OR6,NR6R7,COR6,CO2R6,CONHOH,CONR6R7,S(O)mR6,S(O)mNR6R7,NR6COR7,NR6SO2R7,N(SO2R7)2,NR6CONR7R8,C(=N-CN)NR6R7,NR8C(=N-CN)NR6R7和任选被(C1-C4)烷基取代的四唑基,其中m是0-2(包括0和2在内)的整数,R6和R7相同或不同,彼此独立地是氢原子或式X2-Rb的基团,其中X2是单键或(C1-C6)亚烷基,Rb是选自(C1-C6)烷基、环烷基、杂环烷基、芳基和杂芳基的一个基团,这些基团任选地被1-3个彼此独立地选自以下基团的相同或不同的基团取代:羟基,(C1-C6)烷氧基,(C1-C6)烷基,氨基,一(C1-C6)烷基氨基,二(C1-C6)烷基氨基(各烷基氨基相同或不同,彼此独立),羧基,(C1-C6)烷氧羰基和苄基,R8则代表氢原子或(C1-C6)烷基,或
其外消旋形式、其异构体、其N-氧化物或其药学上可接受的酸或碱盐。
这些化合物的制备在美国专利No.6,753,340,US 20030191167,EP1 348 701和WO2003/082839中说明。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,849,638,US 20030119829和WO2002/088138中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1和R2独立地选自以下基团:氢,1-8个碳原子的烷基,2-8个碳原子的烯基,2-8个碳原子的炔基,3-7个碳原子的环烷基,含2-6个碳原子和1-2个选自NH、S和O的杂原子的全饱和杂环;6-12个碳原子的芳基,它可以被以下基团取代:1-6个碳原子的烷基,2-6个碳原子的烯基,2-6个碳原子的炔基,1-6个碳原子的烷氧基,卤素,含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基,含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基,6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1或2个杂原子的杂芳基;含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基,它可以被以下基团取代:1-6个碳原子的烷基,2-6个碳原子的烯基,2-6个碳原子的炔基,1-6个碳原子的烷氧基,卤素,含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基,含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基,6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基;和R4-R5;或者R1和R2与它们所连接的氮原子结合在一起形成一个可以含1-2个选自NH、NR8、S和O的另外的杂原子的5-7元饱和环,或与它们所连接的氮原子结合在一起形成一个可以含1-2个选自N、S和O的另外的杂原子的5-7元不饱和环;
其中所述的饱和或不饱和环可以被选自以下基团的1-2个取代基取代:OH,1-6个碳原子的烷基,2-6个碳原子的烯基,2-6个碳原子的炔基,3-7个碳原子的环烷基,含2-6个碳原子和1-2个选自NH、S和O的杂原子的全饱和杂环,卤素,含1-2个碳原子和卤素原子数最高达到全卤水平的卤代烷基,1-6个碳原子的烷氧基,含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基,以及R9-R10;或
R1和R2与它们所连接的氮原子结合在一起形成一个8-10元双环饱和环;
R3选自NH、S、S(=O)2和O;
R4选自1-8个碳原子的烷基,2-8个碳原子的烯基,2-8个碳原子的炔基,C(=C),S(=O)2和C(=O)O;
R5选自氢,OH,1-8个碳原子的烷基,2-8个碳原子的烯基,2-8个碳原子的炔基,1-8个碳原子的烷氧基;6-12个碳原子的芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、6-12个碳原子的芳基和含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基和含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;3-7个碳原子的环烷基;含2-6个碳原子和1-2个选自NH、S和O的杂原子的全饱和杂环;及NR6R7;
R6和R7独立地选自氢,1-8个碳原子的烷基,2-8个碳原子的烯基和2-8个碳原子的炔基,或者R6和R7与它们所连接的氮原子结合在一起形成一个可以含1-2个选自N、S和O的另外的杂原子的5-7元不饱和环,或形成可以含1-2个选自NH、S和O的另外的杂原子的5-7元饱和环;
R8选自1-8个碳原子的烷基,2-8个碳原子的烯基,2-8个碳原子的炔基,R11-R12,3-7个碳原子的环烷基,含2-6个碳原子和1-2个选自NH、S和O的杂原子的全饱和杂环;6-12个碳原子的芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;
R9选自1-8个碳原子的烷基,2-8个碳原子的烯基和2-8个碳原子的炔基;
R10选自OH;6-12个碳原子的芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;以及含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;
R11选自1-8个碳原子的烷基,2-8个碳原子的烯基和2-8个碳原子的炔基;和
R12选自3-7个碳原子的环烷基,含2-6个碳原子和1-2个选自NH、S和O的杂原子的全饱和杂环;6-12个碳原子的芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;以及含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;
及其药学上可接受的盐。
在美国专利No.6,849,638,US 20030119829和WO 2002/088138中描述了这些化合物的制备。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 2005222138和WO2003/064389中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1和R2各自独立地是(1)氢原子,或(2)C1-8烷基,或
R1和R2可以与它们所连接的碳原子一起形成Cyc1,其中R1和R2不同时代表氢原子;
Z是(1)CR3R4,(2)O,(3)S,或(4)键;
R3和R4彼此独立地是(1)氢原子,(2)C1-8烷基,(3)C1-8烷氧基,或(4)羟基,或
R3和R4可以与它们所连接的碳原子一起形成Cyc1或C(O);
R5和R6各自独立地是(1)氢原子,或(2)C1-8烷基,或
R5和R6可以与它们所连接的碳原子一起形成Cyc1;
Cyc1(由R1和R2、R3和R4、R5和R6代表)各自独立地是:(1)C3-10环烷基,或(2)含1-2个选自氧、氮和硫的杂原子的3-10元单环杂环,并且Cyc1可以被R10取代;
R10是(1)C1-8烷基,(2)C1-8烷氧基,(3)羟基,(4)COOR11,(5)氧代,(6)SO2R12或(7)COR13;
R11是氢原子或C1-8烷基;
R12和R13是(1)C1-8烷基或(2)可以被C1-8烷基取代的苯基;
R7和R8各自独立地是:(1)氢原子,(2)C1-8烷基,(3)C1-8烷氧基,(4)羟基,(5)氰基,(6)卤素原子,(7)COOR14,(8)CONR15R16,(9)Cyc2,(10)C2-8烯基,(11)C2-8炔基,(12)NR51R52,(13)硝基,(14)甲酰基,(15)C2-8酰基,(16)被羟基、C1-8烷氧基、Cyc2、NR51R52或NR53-Cyc2取代的C1-8烷基,(17)NR54COR55,(18)NR56SO2R57,(19)SO2NR58R59,(20)被COOR14取代的C2-8烯基,(21)CH=N-OH,(22)C1-8亚烷基-NR60-(C1-8亚烷基)-R61,(23)C1-8烷硫基,(24)被1-3个卤素原子取代的C1-8烷基,(25)被1-3个卤素原子取代的C1-8烷氧基,(26)被Cyc2取代的C1-8烷氧基,(27)O-Cyc2,(28)OSO2R65,或(29)CH=N-OR137;
R14是氢原子或C1-8烷基;
R15和R16各自独立地是氢原子或C1-8烷基;
R51和R52,R58和R59,各自独立地是氢原子或C1-8烷基;
R53、R54、R56和R60各自独立地是氢原子或C1-8烷基;
R55是氢原子、C1-8烷基或C1-8烷氧基;R57是C1-8烷基;
R61是NR62R63或羟基;
R62和R63各自独立地是氢原子或C1-8烷基;
R65是C1-8烷基;
R137是C1-8烷基;

R7、R8和环代表的Cyc2各自独立地是:(1)C3-15单、双或三环(稠合的或螺)碳环,或(2)含1-4个选自氧、氮和硫的杂原子的3-15元的单、双或三环(稠合的或螺)杂环;
Cyc2可以被1-5个R17或R17,取代;
R17是(1)C1-8烷基,(2)C2-8烯基,(3)C2-8炔基,(4)C1-8烷氧基,(5)C1-8烷硫基,(6)羟基,(7)卤素原子,(8)硝基,(9)氧代,(10)羧基,(11)甲酰基,(12)氰基,(13)NR18R19,(14)可以被1-5个R20取代的苯基、苯氧基或苯硫基,(15)可以被1-5个R21取代的C1-8烷基、C2-8烯基、C1-8烷氧基或C1-8烷硫基,(16)OCOR22,(17)CONR23R24,(18)SO2NR25R26,(19)COOR27,(20)COCOOR28,(21)COR29,(22)COCOR30,(23)NR31COR32,(24)SO2R33,(25)NR34SO2R35或(26)SOR64;
R18和R19,R31和R34,各自独立地是氢原子或C1-8烷基;
R20和R21是C1-8烷基、C1-8烷氧基、羟基、卤素原子、硝基或COOR36;
R22和R64各自独立地是C1-8烷基;
R23、R24、R25和R26各自独立地是氢原子、C1-8烷基或苯基;
R27、R28、R29、R30、R32、R33和R35是(1)C1-8烷基,(2)C2-8烯基,(3)被1-5个R37取代的C1-8烷基,(4)二苯甲基,(5)三苯甲基,(6)Cyc3,(7)被Cyc3取代的C1-8烷基或C2-8烯基,(8)被O-Cyc3、S-Cyc3或SO2-Cyc3取代的C1-8烷基;
R36是氢原子或C1-8烷基;
R37是C1-8烷氧基、C1-8烷硫基,苄氧基、卤素原子、硝基或COOR38;
R38是氢原子、C1-8烷基或C2-8烯基;
Cyc3是(1)C3-15单、双或三环(稠合的或螺)碳环,或(2)含1-4个选自氧、氮和硫的杂原子的3-15元的单、双或三环(稠合的或螺)杂环;
Cyc3可以被1-5个R39取代;
R39是(1)C1-8烷基,(2)C2-8烯基,(3)C2-8炔基,(4)C1-8烷氧基,(5)C1-8烷硫基,(6)羟基,(7)卤素原子,(8)硝基,(9)氧代,(10)氰基,(11)苄基,(12)苄氧基,(13)被1-5个R40取代的C1-8烷基、C1-8烷氧基或C1-8烷硫基,(14)可以被1-5个R41取代的苯基、苯氧基、苯硫基、苯磺酰基或苯甲酰基,(15)OCOR42,(16)SO2R43,(17)NR44COR45,(18)SO2NR46R47,(19)COOR48或(20)NR49R50;
R40是卤素原子;
R41是C1-8烷基、C1-8烷氧基、卤素原子或硝基;
R42、R43和R45是C1-8烷基;
R44和R48是氢原子或C1-8烷基;
R46和R47,R49和R50,各自独立地是氢原子或C1-8烷基;
R17’是:(1)SH,(2)NR66CHO,(3)Cyc5,(4)被Cyc5取代的C1-8烷基、C2-8烯基或C2-8炔基,(5)CO-(NH-氨基酸残基-CO)n-OH,(6)NR67CONR68R69,(7)CONR70NR71R72,(8)CONR73OR74,(9)CONR75COR76,(10)C(S)NR77R78,(11)CONR79C(S)COOR80,(12)NR81COCOOR82,(13)NR83COOR84,,(14)CONR85C(S)R86,(15)OCOR87,(16)SOR88,(17)CONR89R90,(18)SO2NR91R92,(19)COOR93,(20)COCOOR94,(21)COR95,(22)COCOR96,(23)NR97COR98,(24)SO2R99,(25)NR100SO2R101或(26)NR102R103;
n是1或2的整数;
R66、R73、R75、R77、R79、R81、R83、R85、R97、R100和R102是氢原子或C1-8烷基;
R67和R68,R70和R71,各自独立地是氢原子或C1-8烷基;
R89和R91是(1)氢原子,(2)C1-8烷基,(3)苯基,或(4)被氰基或C1-8烷氧基取代的C1-8烷基;
R103是Cyc6;
R69、R72、R74、R76、R78、R80、R82、R84、R86、R87、R88、R90和R92是:(1)氢原子,(2)C1-8烷基,(3)C2-8烯基,(4)C2-8炔基,(5)被1-5个R104取代的C1-8烷基,(6)二苯甲基,(7)三苯甲基,(8)Cyc6,(9)被Cyc6取代的C1-8烷基或C2-8烯基,或(10)被O-Cyc6、S-Cyc6或SO2-Cyc6取代的C1-8烷基;
R104是(1)C1-8烷氧基,(2)C1-8烷硫基,(3)苄氧基,(4)卤素原子,(5)硝基,(6)COOR105,(7)氰基,(8)NR106R107,(9)NR108COR109,(10)羟基,(11)SH,(12)SO3H,(13)S(O)OH,(14)OSO3H,(15)C2-8烯氧基,(16)C2-8炔氧基,(17)COR110,(18)SO2R111或(19)被羟基取代的C1-8烷氧基或C1-8烷硫基;
R105是氢原子、C1-8烷基或C2-8烯基;
R106和R107各自独立地是氢原子或C1-8烷基;
R108是氢原子或C1-8烷基;
R109和R111是C1-8烷基;
R110是C1-8烷基或卤素原子;
R93、R94、R95、R96、R98、R99和R101是:(1)C2-8炔基,(2)被R128取代的C1-8烷基,其可以被1-4个R29取代,(3)Cyc8,(4)被Cyc8取代的C1-8烷基或C2-8烯基,或(5)被O-Cyc8、S-Cyc8或SO2-Cyc8取代的C1-8烷基;R128是:(1)氰基,(2)NR106 R107,(3)NR108COR109,(4)羟基,(5)SH,(6)SO3H,(7)S(O)OH,(8)OSO3H,(9)C2-8烯氧基,(10)C2-8炔氧基,(11)COR110,(12)SO2R111或(13)被羟基取代的C1-8烷氧基或C1-8烷硫基;
R129的含义与R104相同;
Cyc5和Cyc6可以被1-5个R112取代;
R112是:(1)C1-8烷基,(2)C2-8烯基,(3)C2-8炔基,(4)C1-8烷氧基,(5)C1-8烷硫基,(6)羟基,(7)卤素原子,(8)硝基,(9)氧代,(10)氰基,(11)苄基,(12)苄氧基,(13)被1-5个R113取代的C1-8烷基、C1-8烷氧基或C1-8烷硫基,(14)可以被1-5个R114取代的苯基、苯氧基、苯硫基或苯甲酰基,(15)COR115,(16)SO2R116,(17)NR117COR118,(18)SO2NR119R120,(19)COOR121,(20)NR122R123,(21)COR124,(22)CONR125R126,(23)SH,(24)被羟基或NR127-苯甲酰基取代的C1-8烷基,或(25)Cyc7;
R113是卤素原子;
R114是C1-8烷基、C1-8烷氧基、卤素原子或硝基;
R115、R116和R118是C1-8烷基;
R117、R121、R124和R127是氢原子或C1-8烷基;
R119和R120,R122和R123以及R125和R126,各自独立地是氢原子或C1-8烷基;
Cyc7可以被选自(1)C1-8烷基,(2)C1-8烷氧基,(3)卤素原子或(4)硝基的1-5个基团取代;
Cyc8可以被R130取代,并且其可进一步被1-4个R131取代;
R130是:(1)COR124,(2)CONR125R126,(3)SH,(4)被羟基或NR127-苯甲酰基取代的C1-8烷基,或(5)Cyc7;
R131的含义与R112相同;
Cyc5、Cyc6、Cyc7和Cyc8是:(1)C3-15单、双或三环(稠合的或螺)碳环,或(2)含1-4个选自氧、氮或硫的杂原子的3-15元单、双或三环(稠合的或螺)杂环;
其中当R17’是Cyc5时,Cyc5不是可以被选自C1-8烷基、C1-8烷氧基、羟基、卤素原子、硝基、COOH或COO(C1-8烷基)的1-5个基团取代的苯基;
其中Cyc7不是苯基;
Cyc4是:(1)C5-7单环碳环,或(2)含1-2个选自氧、氮和硫的杂原子的5-7元单环杂环;(后文简称为虚线a)和(后文简称为虚线b)是(1)键,或(2)双键;
R9是(1)不存在或(2)氢原子;
其中
(1)当虚线a是键时,虚线b是双键,R9不存在,
(2)当虚线a是双键时,虚线b是键,R9是氢原子,R6不存在;和
(3)2-(3,3-二甲基-3,4-二氢-(2H)-异喹啉-1-亚基)-1-苯基乙-1-酮被排除在外;
或其药学上可接受的盐。
在US 2005222138和WO 2003/064389中描述了这些化合物的制备。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2003/057149中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
(1)X选自卤素和NR1R2,
(2)Y选自NR3,S和O,条件是,当X是Cl时,Y不是S,
(3)R1和R2独立地选自:氢,1-8个碳原子的烷基,2-8个碳原子的烯基,2-8个碳原子的炔基,3-7个碳原子的环烷基,5-9个碳原子的多环烷基,含2-6个碳原子和1-2个选自NH、S和O的杂原子的杂环烷基;6-12个碳原子的芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基、1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和1-2个选自N、S和O的杂原子的杂芳基取代;含4-11个碳原子和1-2个选自N、S和O的杂原子的杂芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基,1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和1-2个选自N、S和O的杂原子的杂芳基取代;及R4R5;或者R1和R2与它们所连接的氮原子结合在一起形成一个任选地含1-2个选自NH、NR6、S和O的另外的杂原子的5-7元的单环饱和环,或者与它们所连接的氮原子结合在一起形成一个任选地含1-2个选自NH、NR6、S和O的另外的杂原子的6-10元稠合的多环饱和环,或者与它们所连接的氮原子结合在一起形成一个任选地含1-2个选自N、S和O的另外的杂原子的5-7元不饱和环,其中所述单环饱和环、多环饱和环或不饱和环可以被选自以下基团的1-2个取代基取代:OH,1-6个碳原子的烷基,2-6个碳原子的烯基,2-6个碳原子的炔基,3-7个碳原子的环烷基,含2-6个碳原子和1-2个选自NH、S和O的杂原子的杂环烷基,卤素,含1-2个碳原子和卤素原子数最高达到全卤水平的卤代烷基,1-6个碳原子的烷氧基,含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基,和R7R8,
(4)R3选自氢,1-8个碳原子的烷基,2-8个碳原子的烯基,2-8个碳原子的炔基,3-7个碳原子的环烷基,和含4-11个碳原子和1-2个选自N、S和O的杂原子的杂芳基,该杂芳基可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基,1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和1-2个选自N、S和O的杂原子的杂芳基取代,
(5)R4选自1-8个碳原子的烷基,2-8个碳原子的烯基,2-8个碳原子的炔基,C(=O),S(=O)2和C(=O)O,
(6)R5选自氢,OH,1-8个碳原子的烷基,2-8个碳原子的烯基,2-8个碳原子的炔基,1-8个碳原子的烷氧基,1-8个碳原子的硫代氧基(thioxy);6-12个碳原子的芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基,1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和1-2个选自N、S和O的杂原子的杂芳基取代;含4-11个碳原子和1-2个选自N、S和O的杂原子的杂芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基,1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和1-2个选自N、S和O的杂原子的杂芳基取代;3-7个碳原子的环烷基;含2-6个碳原子和1-2个选自NH、S和O的杂原子的杂环烷基及NR9R10,
(7)R6和R7独立地选自1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基,
(8)R8选自OH;6-12个碳原子的芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基,1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷氧基、6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;和含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基,它可以被1-6个碳原子的烷基、2-6个碳原子的烯基、2-6个碳原子的炔基,1-6个碳原子的烷氧基、卤素、含1-6个碳原子和卤素原子数最高达到全卤水平的卤代烷基、6-12个碳原子的芳基或含4-11个碳原子和选自N、S和O的1-2个杂原子的杂芳基取代;
(9)R9和R10独立地选自氢、1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基,或者R9和R10与它们所连接的氮原子结合在一起形成一个可以含1-2个选自N、S和O的另外的杂原子的5-7元不饱和环,或者形成一个可以含1-2个选自NH、NR11、S和O的另外的杂原子的5-7元饱和环,
(10)R1选自1-8个碳原子的烷基、2-8个碳原子的烯基和2-8个碳原子的炔基,
以及它们的药学上可接受的盐。
这些化合物的制备描述在WO 2003/057149中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 20030092721,美国专利No.7,022,849,WO2002/102315和US2006116516中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1是氢或烷基;
R2是(a)杂芳基或杂环基,它们各自任选地被1-3个基团T1、T2、T3取代;或(b)与杂芳基或杂环基环稠合的芳基,其中该组合环系可以任选地被1-3个基团T1、T2、T3取代;
L是(a)OR4,C(O)R4,C(O)OR4,SR4,NR3R4,C(O)NR3R4,NR3SO2R4b,卤素,硝基或卤代烷基;或(b)烷基、芳基、杂芳基、杂环基或环烷基,它们任一个可任选地被1-3个基团T1a、T2a和/或T3a取代;
Y1、Y2和Y3独立地是(a)氢、卤素或-OR4a;或(b)烷基、烯基或炔基,它们任一个可任选地被1-3个基团T1b、T2b和/或T3b取代;
R3和R4独立地是H、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂环基)烷基、环烷基、(环烷基)烷基、杂环基、或(杂环基)烷基,它们任一个可任选地被1-3个基团T1a、T2a和/或T3a取代;或
R3和R4与它们所连接的氮原子结合在一起形成一个可以任选被1-3个基团T1a、T2a和/或T3a取代的4-8元杂环;
R4a是氢、烷基、烯基、芳基、杂芳基、(芳基)烷基、(杂芳基)烷基、杂环基、(杂环基)烷基、环烷基或(环烷基)烷基,它们任一个可任选地被1-3个基团T1b、T2b和/或T3b取代;
R4b是烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,它们任一个可任选地被1-3个基团T1a、T2a和/或T3a取代;
Z是N或CH;
T1-1b、T2-2b和T3-3b各自独立地是:
(1)氢或T6,其中T6是(i)烷基、(羟)烷基、(烷氧基)烷基、烯基、炔基、环烷基、(环烷基)烷基、环烯基、(环烯基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基或(杂芳基)烷基;(ii)本身被一个或多个相同或不同的基团(i)取代的基团(i);或(iii)被T1-1b、T2-2b和T3-3b定义中的以下一个或多个基团(2)-(13)独立取代的基团(i)或(ii);
(2)-OH或-OT6;
(3)-SH或-ST6;
(4)-C(O)tH,-C(O)tT6,或-O-C(O)T6,其中t是1或2;
(5)-SO3H,-S(O)tT6或-S(O)tN(T9)T6;
(6)卤素;
(7)氰基;
(8)硝基;
(9)-T4-NT7T8;
(10)-T4-N(T9)-T5-NT7T8;
(11)-T4-N(T10)-T5-T6;
(12)-T4-N(T10)-T5-H;和
(13)氧代;
T4和T5各自独立地是单键,T11S(O)tT12-,T11C(O)T12-,T11C(S)T12,T11OT12,T11ST12,T11OC(O)T12,T11C(O)OT12,T11C(=NT9a)T12或T11C(O)C(O)T12;
T7,T8,T9,T9a和T10是:
(1)各自独立地是氢或T6定义中提供的基团,或
(2)T7和T8可以合起来是亚烷基或亚烯基,与它们所连接的原子一起构成一个3-8元的饱和或不饱和环,该环是未取代的或被T1-1b、T2-2b和T3-3b的描述中列出的一个或多个基团取代,或
(3)T7或T8,与T9一起,可以是亚烷基或亚烯基,和它们所连接的氮原子合起来构成一个3-8元的饱和或不饱和环,该环是未取代的或被T1-1b、T2-2b和T3-3b的描述中列出的一个或多个基团取代,或
(4)T7和T8,或T9和T10,可以与它们所连接的氮原子合起来构成一个基团N=CT13T14,其中T13和T14各自独立地是H或T6的定义中提供的基团;T11和T12则各自独立地是单键、亚烷基、亚烯基或亚炔基。
这些化合物的制备描述于US 20030092721,美国专利No.7,022,849,WO 2002/102315和US 2006116516中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,838,559,US 20030100571和WO 2002/102314中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

和

以上化合物的取代基定义如下:
R1是H或烷基;
R2是(a)杂芳基或杂环基,它们各自可任选地被1-3个基团T1、T2、T3取代;(b)被1-3个基团T1、T2、T3取代的芳基,条件是,T1、T2、T3中至少一个不是H;或(c)与杂芳基或杂环基环稠合的芳基,其中该组合环系可以任选地被1-3个基团T1、T2、T3取代;
Y是烷基、烯基、炔基、环烷基、芳基、杂环基、杂芳基、(芳基)烷基或(杂芳基)烷基,它们任一个可任选地被1-3个基团T1a、T2a、T3a取代;
J是(a)氢、卤素或OR4,或(b)烷基、烯基、炔基、芳基、杂芳基、杂环基或环烷基,它们任一个可任选地被1-3个基团T1b、T2b、T3b取代;
Z是(a)OR4,SR4,NR3R4,NR3SO2R4a,卤素,硝基,卤代烷基;或(b)烷基,芳基、杂芳基,杂环基或环烷基,它们任一个可任选地被1-3个基团T1c、T2c、T3c取代;
R3是H、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,它们任一个在价数许可时可任选地被1-3个基团T1c、T2c、T3c独立取代;
R4是烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,它们任一个在价数许可时可任选地被1-3个基团T1d、T2d或T3d独立取代;或
R3与R4和它们所连接的氮原子合起来形成一个任选被1-3个基团T1c、T2c、或T3c取代的4-8元杂环;
R4a是氢、烷基、烯基、芳基、杂芳基、(芳基)烷基、(杂芳基)烷基、杂环基、(杂环基)烷基、环烷基或(环烷基)烷基,它们任一个可任选地被1-3个基团T1d、T2d或T3d取代;
T1,T1a,T1b,T1c,T1d,T2,T2a,T2b,T2c,T2d,T3,T3a,T3b,T3c和T3d(以后简称为T1-1d,T2-2d和T3-3d)独立地是:
(1)氢或T6,其中T6是
(a)烷基、(羟)烷基、(烷氧基)烷基、烯基、炔基、环烷基、(环烷基)烷基、环烯基、(环烯基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基或(杂芳基)烷基;
(b)基团(a),它本身又被一个或多个相同或不同的基团(a)取代;或
(c)基团(a)或(b),它独立地被T1-1d、T2-2d或T3-3d定义中的一个或多个(优选1-3个)以下基团(2)至(13)取代,
(2)OH或OT6,
(3)SH或ST6,
(4)C(O)tH,C(O)tT6或OC(O)T6,其中t是1或2;
(5)SO3H,-S(O)tT6或S(O)tN(T9)T6;
(6)卤素;
(7)氰基;
(8)硝基;
(9)T4NT7T8;
(10)T4N(T9)-T5NT7T8;
(11)T4N(T10)-T5-T6;
(12)T4-N(T10)-T5H;和
(13)氧代;
T4和T5各自独立地是单键,T11-S(O)t-T12,T11-C(O)-T12,T11-C(S)-T12,T11-O-T12,-T11S-T12,-T11OC(O)-T12,-T11-C(O)O-T12,-T11C(=NT9a)-T12或T11-C(O)-C(O)-T12;
T7,T8,T9,T9a和T10是:
(1)各自独立地是氢或T6定义中提供的基团,或
(2)T7和T8可以合起来是亚烷基或亚烯基,与它们所连接的原子一起构成一个3-8元的饱和或不饱和环,该环是未取代的或被T1-1d、T2-2d和T3-3d的描述中列出的一个或多个基团取代,或
(3)T7或T8,与T9一起,可以是亚烷基或亚烯基,与它们所连接的氮原子合起来构成一个3-8元的饱和或不饱和环,该环是未取代的或被T1-1d、T2-2d和T3-3d的描述中列出的一个或多个基团取代,或
(4)T7和T8,或T9和T10,可以与它们所连接的氮原子合起来构成一个基团N=CT13T14,
其中
T13和T14各自独立地是H或T6的定义中提供的基团;和
T11和T12则各自独立地是单键、亚烷基、亚烯基或亚炔基。
这些化合物的制备描述于美国专利No.6,838,559,US 20030100571和WO 2002/102314中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.7,087,614,US 20030162802和WO2002/102313中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1a是氢或烷基;R2a是 W是S;X1是烷氧基;X2是烷基;
W是S;X1是烷氧基;X2是烷基;
Z*是卤素、卤代烷基、噁唑基、NR3aR4a、C(O)-N(H)-亚烷基-COOH或未取代的或被杂芳基、COtH或COtT6取代的苯基;
R3a是氢或烷基;
R4a是烷基、烷氧基、未取代的或取代的(杂芳基)烷基、未取代的或取代的杂环基、未取代的或取代的(杂环基)烷基、或(芳基)烷基,其中该芳基被一或二个基团T1和/或T2取代,和/或进一步被基团T3取代;或者R3a和R4a与它们所连接的氮原子结合在一起形成一个未取代的或取代的杂环基环;
R5a是未取代的或取代的(杂芳基)烷基或(芳基)烷基,其中该芳基被一或二个基团T1和/或T2取代,和/或进一步被基团T3取代;或者R5a和R6a与它们所连接的氮原子结合在一起形成一个未取代的或取代的杂环基环,R6a是氢或烷基;J*是氢或烷基;T1和T2独立地是烷氧基、烷氧羰基、杂芳基、SO3H或SO2R8a,其中R8a是烷基、氨基、烷基氨基或二烷基氨基;或者T1和T2与它们所连接的芳基环结合在一起形成一个双环;T3是H、烷基、卤素、卤代烷基或氰基;t是1或2;T6是烷基、卤代烷基、环烷基、烷氧基或杂芳基。
这些化合物的制备描述于美国专利No.7,087,614,US 20030162802和WO 2002/102313中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 20030104974,WO2002/088080和WO2002/088079中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

和

以上化合物的取代基定义如下:
R1是H或烷基;R2是任选取代的杂芳基或4-取代的芳基;R3是氢或烷基;R4是烷基,任选取代的(芳基)烷基,任选取代的(杂芳基)烷基,任选取代的杂环基,或任选取代的(杂环基)烷基;或者R3和R4与它们所连接的氮原子可以结合在一起,形成一个任选取代的杂环基环;R5是烷基,任选取代的(芳基)烷基,或任选取代的(杂芳基)烷基;和R6是氢或烷基。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中R1a是氢或烷基;R2a是任选取代的杂芳基;Z是卤素,烷基,取代的烷基,卤代烷基或NR3aR4aR3a是氢或烷基;R4a是烷基,任选取代的(杂芳基)烷基,任选取代的杂环基,任选取代的(杂环基)烷基,或(芳基)烷基;其中该芳基被一或二个基团T1和T2取代,并且任选地进一步被基团T3取代;或者R3a和R4a与它们所连接的氮原子可以结合在一起,形成一个任选取代的杂环基环;R5a是(芳基)烷基;其中该芳基被一或二个基团T1和T2取代,并任选地进一步被基团T3取代;R6a是氢或烷基;R7a是氢或烷基;T1和T2独立地是烷氧基、烷氧羰基、杂芳基或SO2R8a,其中R8a是烷基、氨基、烷基氨基或二烷基氨基;或者T1和T2与它们所连接的原子可以结合在一起,形成一个环(例如苯并间二氧杂环戊烯);T3是H、烷基、卤素、卤代烷基或氰基。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中R1b是氢或烷基;R2b是任选取代的杂芳基;R3b是氢或烷基;R4b是任选取代的(芳基)烷基;R5b是H,烷基或C(O)(CH2)vOYR6b,其中Y是键或C(O),R6b是氢或烷基,v是0-2的整数;J1和J2独立地是任选取代的C1-13亚烷基,条件是,J1和J2不能都大于C2亚烷基;X4和X5是任选的与J1和J2之一或二者中的任何可利用的碳原子键合的取代基,独立选自以下基团:氢,OR7,NR8R9,烷基,取代的烷基,烯基,取代的烯基,炔基,取代的炔基,环烷基,取代的环烷基,芳基,取代的芳基,杂环烷基或杂芳基;R7是氢,烷基,取代的烷基,烯基,炔基,环烷基,取代的环烷基,C(O)烷基,C(O)取代的烷基,C(O)环烷基,C(O)取代的环烷基,C(O)芳基,C(O)取代的芳基,C(O)O-烷基,C(O)O-取代的烷基,C(O)杂环烷基,C(O)杂芳基,芳基,取代的芳基,杂环烷基和杂芳基;R8和R9独立地选自以下基团:氢,烷基,取代的烷基,环烷基,取代的环烷基,烯基,炔基,C(O)烷基,C(O)取代的烷基,C(O)环烷基,C(O)取代的环烷基,C(O)芳基,C(O)取代的芳基,C(O)O-烷基,C(O)O-取代的烷基,C(O)杂环烷基,C(O)杂芳基,S(O)2烷基,S(O)2取代的烷基,S(O)2环烷基,S(O)2取代的环烷基,S(O)2芳基,S(O)2取代的芳基,S(O)2杂环烷基,S(O)2杂芳基,芳基,取代的芳基,杂环烷基和杂芳基;或者R8和R9与它们所连接的氮原子一起形成一个任选取代的杂环烷基或杂芳基环。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中R1c是H或烷基;R2c是任选取代的杂芳基;R3c是H或烷基;R4c是任选取代的(芳基)烷基;X4和X5是任选的与J1和J2之一或二者中的任何可利用的碳原子键合的取代基,独立选自以下基团:氢,OR7,NR8R9,烷基,取代的烷基,烯基,取代的烯基,炔基,取代的炔基,环烷基,取代的环烷基,芳基,取代的芳基,杂环烷基或杂芳基。
这些化合物的制备描述于US 20030104974、WO 2002/088080和WO 2002/088079中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 20030092908和WO2002/087513中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1是H或烷基;
R2是(a)杂芳基或杂环基,它们各自可任选地被1-3个基团T1、T2、T3取代;(b)被1-3个基团T1、T2、T3取代的芳基,条件是,T1、T2、T3中至少一个不是H;或(c)与杂芳基或杂环稠合的芳基,其中该组合环系可以任选地被1-3个基团T1、T2、T3取代;
Z是NR3R4,NR3SO2R4a,OR4,SR4,卤代烷基或卤素;
R3和R4独立地是H、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,它们任一个在价数允许时可任选地被1-3个基团T1a、T2a、或T3a独立取代;或
R3与R4可以和它们所连接的氮原子一起形成一个杂环基或杂芳基环,该环在价数允许时可任选地被1-3个基团T1a、T2a、或T3a独立取代;
R4a是烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基,它们任一个在价数允许时可任选地被1-3个基团T1a、T2a、或T3a独立取代;
R3b和R4b独立地是H、烷基、烯基、芳基、(芳基)烷基、杂芳基、(杂芳基)烷基、环烷基、(环烷基)烷基、杂环基或(杂环基)烷基;
R5是
(1)氢或氰基;
(2)烷基、烯基、炔基、环烷基、(环烷基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基、或(杂芳基)烷基,它们任一个在价数允许时可任选地被1-3个基团T1b、T2b、或T3b独立取代;或
(3)C(O)R6,C(O)OR6,C(O)-C(O)OR或SO2R6a;
R6是H、烷基、烯基、NR3bR4b、杂环基、(杂环基)烷基、(羟基)烷基、(烷氧基)烷基、(芳氧基)烷基、(NR3bR4b)烷基、杂芳基、芳基或(芳基)烷基,它们任一个在价数允许时可任选地被1-3个基团T1b、T2b、或T3b独立取代;
R6a是烷基、烯基、NR3bR4b、杂环基、(杂环基)烷基、(羟基)烷基、(烷氧基)烷基、(芳氧基)烷基、(NR3bR4b)烷基、杂芳基、芳基或(芳基)烷基,它们任一个在价数允许时可任选地被1-3个基团T1b、T2b、或T3b独立取代;
J1和J2独立地是任选取代的C1-3亚烷基,条件是J1和J2不都大于C2亚烷基;和
T1-1b、T2-2b和T3-3b各自独立地是:
(1)氢或T6,其中T6是(i)烷基、(羟)烷基、(烷氧基)烷基、烯基、炔基、环烷基、(环烷基)烷基、环烯基、(环烯基)烷基、芳基、(芳基)烷基、杂环基、(杂环基)烷基、杂芳基或(杂芳基)烷基;(ii)本身被一个或多个相同或不同的基团(i)取代的基团(i);或(iii)被T1-1b、T2-2b和T3-3b定义中的以下一个或多个(优选1-3个)基团(2)-(13)独立取代的基团(i)或(ii);
(2)OH或OT6;
(3)SH或ST6;
(4)C(O)tH,C(O)tT6或O-C(O)T6,其中t是1或2;
(5)SO3H,S(O)tT6或-S(O)tN(T9)T6;
(6)卤素;
(7)氰基;
(8)硝基;
(9)T4-NT7T8;
(10)T4-N(T9)-T5-NT7T8;
(11)T4-N(T10)-T5-T6;
(12)T4-N(T10)-T5-H;
(13)氧代;
T4和T5各自独立地是(1)单键,(2)T11-S(O)t-T12,(3)T11-C(O)-T12,(4)T11-C(S)-T12,(5)-T11-O-T12,(6)T11-S-T12,(7)T11-O-C(O)-T12,(8)T11-C(O)-O-T12,(9)T11-C(=NT9a)-T12或(10)T11-C(O)-C(O)-T12;
T7,T8,T9,T9a和T10是:
(1)各自独立地是氢或T6定义中提供的基团,或
(2)T7和T8可以合起来是亚烷基或亚烯基,与它们所连接的原子一起构成一个3-8元的饱和或不饱和环,该环是未取代的或被T1-1b、T2-2b和T3-3b的描述中列出的一个或多个基团取代,或
(3)T7或T8,与T9一起,可以是亚烷基或亚烯基,与它们所连接的氮原子合起来构成一个3-8元的饱和或不饱和环,该环是未取代的或被T1-1b、T2-2b和T3-3b的描述中列出的一个或多个基团取代,或
(4)T7和T8,或T9和T10,可以与它们所连接的氮原子合起来构成一个基团N=CT13T14,其中T13和T14各自独立地是H或T6的定义中提供的基团;和
T11和T12各自独立地是单键、亚烷基、亚烯基或亚炔基。
这些化合物的制备描述于US 20030092908和WO 2002/087513中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 20040127707和WO2002/085906中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1是1-2C-烷氧基或是完全或基本上被氟取代的1-2C-烷氧基,
R2是氟、溴或氯,
R3和R4都是氢或者合起来形成一个附加键,
R5是R6、CmH2m-R7、CnH2n-C(O)R8、CH(R9)2、CpH2p-Y-Aryl1、R12或R26,
R6是1-8C-烷基,3-10C-环烷基,3-7C-环烷基甲基,3-7C-烯基,3-7C-炔基,苯基-3-4C-烯基,7-10C-多环烷基,萘基,吡啶基,吡嗪基,哒嗪基,嘧啶基,喹唑啉基,喹喔啉基,噌啉基,异喹啉基,喹啉基,茚满基,吲唑基,苯并噁唑基,苯并噻唑基,噁唑基,噻唑基,N-甲基哌啶基,四氢吡喃基,6-甲基-3-三氟甲基-吡啶-2-基,1,3,4-三甲基-1H-吡唑并[3,4-b]吡啶-6-基,3-噻吩-2-基[1,2,4]噻二唑-5-基,1,1-二氧化四氢噻吩-3-基,1-氧代-1,3-二氢-异苯并呋喃-5-基,4-(4-基-丁-1-氧基)苯甲酸,或未取代的或被R61和/或R62取代的苯基,其中R61是羟基,1-4C-烷基,1-4C-烷氧基,硝基,氰基,卤素,羧基,羟基羰基-1-4C-烷基,1-4C-烷氧羰基,羟基-1-4C-烷基,氨基,一或二-1-4C-烷基氨基,1-4C-烷基羰基氨基,氨基羰基,一或二-1-4C-烷基氨基羰基,氨磺酰基,一或二-1-4C-烷基氨基磺酰基,4-甲基苯磺酰胺基,咪唑基,四唑-5-基,2-(1-4C-烷基)四唑-5-基或2-苄基四唑-5-基,和
R62是1-4C-烷基,1-4C-烷氧基,硝基或卤素,
R7是羟基,卤素,氰基,硝基,硝酰氧基(O-NO2),羧基,羧基苯氧基,苯氧基,1-4C-烷氧基,3-7C-环烷氧基,3-7C-环烷基甲氧基,1-4C-烷基羰基,1-4C-烷基羰氧基,1-4C-烷基羰基氨基,1-4C-烷氧羰基,氨基羰基,一或二-1-4C-烷基氨基羰基,氨基,一或二-1-4C-烷基氨基,或未取代的或被R71和/或R72取代的哌啶基、哌嗪基、吡咯烷基或吗啉基,其中
R71是羟基,1-4C-烷基,羟基-1-4C-烷基或1-4C-烷氧羰基,和
R72是1-4C-烷基,羧基,氨基羰基或1-4C-烷氧羰基,
R8是未取代的或被R81和/或R82取代的苯基、萘基、菲基或蒽基,其中
R81是羟基,卤素,氰基,1-4C-烷基,1-4C-烷氧基,羧基,氨基羰基,一或二-1-4C-烷基氨基羰基,1-4C-烷基羰氧基,1-4C-烷氧羰基,氨基,一或二-1-4C-烷基氨基,1-4C-烷基羰基氨基,或完全或基本上被氟取代的1-4C-烷氧基,和
R82是羟基,卤素,1-4C-烷基,1-4C-烷氧基,或完全或基本上被氟取代的1-4C-烷氧基,
R9是CqH2q-苯基,
Y是键或O(氧),
Aryl1是未取代的苯基,萘基,吡啶基,吡嗪基,哒嗪基,嘧啶基,喹唑啉基,喹喔啉基,噌啉基,异喹啉基,喹啉基,香豆素基,苯并咪唑基,苯并噁唑基,苯并噻唑基,苯并三唑基,N-苯并丁二酰亚胺基,咪唑基,吡唑基,噁唑基,噻唑基,呋喃基,噻吩基,吡咯基,2-(1-4C-烷基)-噻唑-4-基,或被R10和/或R11取代的苯基,其中
R10是羟基,卤素,硝基,氰基,1-4C-烷基,三氟甲基,1-4C-烷氧基,羧基,羟基羰基-1-4C-烷基,1-4C-烷基羰氧基,1-4C-烷氧羰基,氨基,一或二-1-4C-烷基氨基,1-4C-烷基羰基氨基,氨基羰基,一或二-1-4C-烷基氨基羰基,咪唑基或四唑基,
R11是羟基,卤素,硝基,1-4C-烷基或1-4C-烷氧基,
m是1-8的整数,n是1-4的整数,p是1-6的整数,q是0-2的整数,
R12是式(a)基团
其中R13是S(O)2-R14,S(O)2-(CH2)r-R15,(CH2)s-S(O)2R16,C(O)R17,C(O)-(CH2)r-R18,(CH2)s-C(O)-R19,Hetaryl1,Aryl2或Aryl3-1-4C-烷基;R14是1-4C-烷基,5-二甲基氨基萘-1-基,N(R20)R21,苯基或被R22和/或R23取代的苯基,R15是N(R20)R21,R16是N(R20)R21,
R17是1-4C-烷基、羟基羰基-1-4C-烷基、苯基、吡啶基、4-乙基哌嗪-2,3-二酮-1-基、2-氧代-咪唑烷-1-基或N(R20)R21,R18是N(R20)R21,R19是N(R20)R21、苯基、被R22和/或R23和/或R24取代的苯基,R20和R21彼此独立地是氢、1-7C-烷基、3-7C-环烷基、3-7C-环烷基甲基或苯基,或者R20和R21与它们所键合的氮原子一起形成一个4-吗啉基环、1-吡咯烷基环、1-哌啶基环、1-六氢氮杂环庚三烯基环或式(b) 的1-哌嗪基环,
的1-哌嗪基环,
其中R25是吡啶-4-基、吡啶-4-基甲基、1-4C-烷基二甲基氨基、二甲基氨基羰基甲基、N-甲基哌啶-4-基、4-吗啉代乙基或四氢呋喃-2-基甲基,R22是卤素、硝基、氰基、羧基、1-4C-烷基、三氟甲基、1-4C-烷氧基、1-4C-烷基羰基、氨基、一或二-1-4C-烷基氨基、氨基羰基、1-4C-烷基羰基氨基或一或二-1-4C-烷基氨基羰基,R23是卤素、氨基、硝基、1-4C-烷基或1-4C-烷氧基,R24是卤素,
Hetaryl1是嘧啶-2-基、噻吩并[2,3-d]嘧啶-4-基、1-甲基-1H-吡唑并[3,4-d]嘧啶-4-基、噻唑基、咪唑基或呋喃基,Aryl2是吡啶基、苯基或被R22和/或R23取代的苯基,Aryl3是吡啶基、苯基、被R22和/或R23取代的苯基、2-氧代-2H-色烯-7-基或4-(1,2,3-噻二唑-4-基)苯基,r是1-4的整数,s是1-4的整数,
R26是式(c)基团
其中R27是C(O)R28、(CH2)t-C(O)R29、(CH2)uR30、Aryl4、Hetaryl2、苯基丙-1-烯-3-基或1-甲基哌啶-4-基,R28是氢、1-4C-烷基、OR31、呋喃基、吲哚基、苯基、吡啶基、被R34和/或R35取代的苯基或被R36和/或R37取代的吡啶基,R29是N(R32)R33,R30是N(R32)R33、四氢呋喃基或吡啶基,R31是1-4C-烷基,R32是氢、1-4C-烷基、3-7C-环烷基或3-7C-环烷基甲基,R33是氢、1-4C-烷基、3-7C-环烷基或3-7C-环烷基甲基,或
R32和R33与它们所键合的氮原子合起来形成一个4-吗啉基环、1-吡咯烷基环、1-哌啶基环或1-六氢氮杂环庚三烯基环,
Aryl4是苯基、吡啶基、嘧啶基、被R34和/或R35取代的苯基、被R36和/或R37取代的吡啶基,R34是卤素、硝基、1-4C-烷基、三氟甲基或1-4C-烷氧基,R35是卤素或1-4C-烷基,R36是卤素、硝基、1-4C-烷基、三氟甲基或1-4C-烷氧基,R37是卤素或1-4C-烷基,Hetaryl2是吲哚-4-基、2-甲基喹啉-4-基、5-氯-6-氧代-1-苯基-1,6-二氢哒嗪-4-基-1,3-苯基-1,2,4-噻二唑-5-基或3-邻甲苯基-1,2,4-噻二唑-5-基,
t是1-4的整数,u是1-4的整数,v是1-2的整数,X是-C(O)-或-S(O)2-,
以及这些化合物的盐。
这些化合物的制备描述于US 20040127707和WO 2002/085906中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,818,651、US 20040044212和WO2002/040450中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1代表氢,R2代表氟、氯、溴、氰基、三氟甲基或苯氧基,或者R1代表氢、氟、氯、溴、三氟甲基或氰基,R2代表氢;R’和R”都代表氢或者合起来代表一个键;Ar代表式IIa、IIb或IIc的苯基基团

其中R3代表氢、羟基、硝基、氨基、羧基、氨基羰基、1-4C-烷氧基、三氟甲氧基、1-4C-烷氧羰基或一或二-1-4C-烷基氨基羰基,
R4代表1-4C-烷基、萘基、5-二甲基氨基萘-1-基、苯基乙烯-2-基、3,5-二甲基异噁唑-4-基、5-氯-3-甲基苯并[b]噻吩-2-基、6-氯-咪唑并[2,1-b]-噻唑-5-基,或代表未取代的或被选自以下的一个或多个相同或不同的基团取代的苯基或噻吩基:卤素,氰基,1-4C-烷基,三氟甲基,完全或基本上被氟取代的1-4C-烷氧基,1-4C-烷氧基,1-4C-烷基羰基氨基,1-4C-烷氧羰基,苯磺酰基或异噁唑基,
或其水合物、溶剂化物、盐、盐的水合物或盐的溶剂化物。
这些化合物的制备描述于美国专利No.6,818,651,US 20040044212和WO 2002/040450中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2002/040449中一般地或具体地公开的那些化合物,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1代表氢,R2代表氟、氯、溴、氰基、三氟甲基或苯氧基,或者
R1代表氢、氟、氯、溴、三氟甲基或氰基,R2代表氢,
R’和R”都代表氢或者合起来代表一个键,
R3代表氢、羟基、硝基、氨基、羧基、氨基羰基、1-4C-烷氧基、三氟甲氧基、1-4C-烷氧羰基或者一或二-1-4C-烷基氨基羰基,
R4代表C(O)-X-R5,N(H)-C(O)-R6或N(H)-C(O)-N(H)-R2,其中
X代表O或N(H),
R5代表氢,1-4C-烷基,3-7C-环烷基甲基,6,6-二甲基双环[3,3,1]庚-2-基,3-7C-炔基,1-4C-烷基羰基-1-4C-烷基,氨基羰基-1-4C-烷基,呋喃-2-基甲基,2-吡啶-2-基乙-1-基,2-吡啶-3-基甲基,N-甲基哌啶-3-基,1-苄基哌啶-4-基,吗啉-4-基-乙-2-基,吗啉-4-基-乙-1-基,2-苯并[1,3]间二氧杂环戊烯-4-基-乙-1-基,色满-4-基,1-甲氧基羰基-2-吲哚-3-基-乙-1-基,1,3-双甲氧羰基丙-1-基,1-甲氧羰基-3-甲基硫烷基-乙-1-基,1-甲氧羰基-2-噻唑-2-基-乙-1-基或4-甲基噻唑-5-基-乙-2-基,或者代表未取代的或被选自卤素、三氟甲基和苯基的一个或多个基团取代的苄基-、苯基-乙-1-基或1-甲氧羰基-2-苯基-乙-2-基,
R6代表2,4-二氯苯氧甲基,2-叔丁氧羰基氨基-乙-1-基,1-乙酰哌啶-4-基,Ar1或Ar2-CH=CH-,
其中Ar1代表3-氯苯基,4-三氟甲氧基苯基,3-苯氧基苯基,吲哚-5-基,2-甲基吡啶-5-基,喹啉-6-基或2-苯并噻唑-6-基,
Ar2代表呋喃-2-基,呋喃-3-基,噻吩-2-基,吲哚-3-基,3-三氟甲基苯基,3-甲氧基苯基或吡啶-3-基,
R7代表1-4C-烷基,3-7C-烯基,3-7C-环烷基,1-乙氧羰基-2-苯基-乙-1-基,噻吩-2-基乙-1-基或是未取代的或被选自以下基团的一个或多个基团取代的苯基:卤素、氰基、1-4C-烷基、三氟甲基、1-4C-烷硫基、1-4C-烷氧基、完全或基本上被氟取代的1-4C-烷氧基、1-4C-烷基羰基和苯氧基,
或其盐。
这些化合物的制备描述于WO 2002/040449中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2001/098274中一般地或具体地公开的那些化合物,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
W、X、Y和Z可以相同或不同,各代表氮原子或基团C(R5)[其中R5是氢或卤素原子,或是烷基、卤代烷基、烷氧基、卤代烷氧基、羟基、-NO2或-CN基团],条件是,W、X、Y和Z中的两个或更多个是C(R5)基团;
R1、R2和R3可以相同或不同,各是一个原子或基团-L1(Alk1)rL2(R6)s,其中L1和L2可以相同或不同,各为一个共价键或连接原子或基团,r是零或整数1,Alk1是一个脂族或杂脂族链,s是整数1、2或3,R6是氢或卤素原子,或是选自以下基团的一个基团:烷基,-OR7[其中R7是氢原子或任选取代的烷基],-SR7,NR7R8[其中R8的定义与R7一样,二者可以相同或不同],-NO2,CN,CO2R7,SO3H,S(O)R7,SO2R7,OCO2R7,CONR7R8,OCONR7R8,CSNR7R8,OCR7,OCOR7,N(R7)COR8,N(R7)CSR8,S(O)NR7R8,SO2NR7R8,N(R7)SO2R8,N(R7)CON(R8)(R9)[其中R9是氢原子或任选取代的烷基],N(R7)CSN(R8)(R9),N(R7)SO2N(R8)(R9),C(R7)=NO(R8),脂环基,杂脂环基,芳基或杂芳基;条件是,R1、R2或R3中的一个或多个是非氢原子取代基;
R4代表任选取代的苯基,1-或2-萘基,吡啶基,嘧啶基,哒嗪基或吡嗪基;和
其盐、溶剂化物、水合物和N-氧化物。
这些化合物的制备描述于WO 2001/098274中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2001/074786中一般地或具体地公开的那些化合物,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1代表芳基或杂芳基;
A,B,P和E可以相同或不同,各代表氮原子或基团C(R2)[其中R2是氢或卤素原子,或是烷基、卤代烷基、烷氧基、卤代烷氧基、羟基、-NO2或-CN基团],条件是,A、B、D和E中的两个或更多个是C(R2)基团;X代表氧或硫原子或N(R3)基团,其中R3是氢原子或烷基;
Q,R,S和T可以相同或不同,各代表氮原子或基团C(R4)[其中R4是一个原子或基团-L1(Alk1)rL2(R5)s,其中L1和L2可以相同或不同,各代表一个共价键或连接原子或基团,r是零或整数1,Alk1是一个脂族或杂脂族链,s是整数1、2或3,R5是氢或卤素原子,或选自以下基团的一个基团:烷基,OR6[其中R6是氢原子或任选取代的烷基],SR6,NR6R7[其中R7的定义与R6一样,二者可以相同或不同],NO2,CN,CO2R6,SO3H,S(O)R6,SO2R6,OCO2R6,CONR6R7,OCONR6R7,CSNR7R7,OCR6,OCOR6,N(R6)COR7,N(R6)CSR7,S(O)NR6R7,SO2NR6R7,N(R6)SO2R7;N(R6)CON(R7)(R8)[其中R8是氢原子或任选取代的烷基],N(R6)CSN(R7)(R8),N(R6)SO2N(R7)(R8),C(R6)=NO(R7),脂环基,杂脂环基,芳基或杂芳基,条件是,Q、R、S和T中的两个或更多个是C(R4)基团;
以及它们的盐、溶剂化物、水合物和N-氧化物。
这些化合物的制备描述于WO 2001/074786中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2000/068230中一般地或具体地公开的那些化合物,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
X-Y-Z代表NR4-C=N或N=C-NR4;
R1代表H,烷基,环烷基,环烷基烷基,芳烷基,杂芳基烷基或杂环烷基;
R2代表OR8,NR8R9,SR13,烷基或CF3;
R3代表卤素,烷基,CF3或OR8;
R4可以与X或Z连接,代表选自以下的残基:

其中连接是通过饱和环上的任何位置完成的,只要该连接不是在与V相邻的位置,并且该饱和环可以在任何位置被一个或多个R6取代;
A、B、D和E相同或不同,各代表ClnR5、N或N-O;
V代表O、S、NR7或C(L1 mR14)(L2 nR14),
Q和W相同或不同,各代表CLnR5或N;
T代表O、S或NR7;
L1和L2相同或不同,各代表C(R15)2;
m和n相同或不同,各代表0、1、2、3、4或5;
各R5相同或不同,各代表H,卤素,烷基,环烷基,OR8,NR8R9,CO2R10,CONR11R12,CONHOH,SO2NR11R12,SON11R12,COR13,SO2R13,SOR13,SR13,CF3,NO2或CN;
R6代表H,烷基,环烷基,OR8,NR8R9,CO2R10,CONR11R12,SO2NR11R12,SON11R12,COR13,SO2R13,SOR13,SR13,CF3,CN或=O;
R7代表H或烷基;
R8代表H,烷基,环烷基,环烷基烷基,芳基,芳烷基,杂芳基,杂芳基烷基,杂环基或杂环烷基;
R9代表R8或烷基羰基,烷氧羰基,烷基磺酰基,环烷基羰基,环烷氧基羰基,环烷基磺酰基,环烷基烷基羰基,环烷基烷氧羰基,环烷基烷磺酰基,芳基羰基,芳磺酰基,杂芳基羰基,杂芳基磺酰基,杂环基羰基,杂环基磺酰基,芳烷基羰基,芳基烷氧羰基,芳基烷磺酰基,杂芳基烷基羰基,杂芳基烷氧羰基,杂芳基磺酰基,杂环烷基羰基,杂环基烷氧羰基或杂环基烷磺酰基;或
NR8R9代表一个杂环,例如吗啉;
R10代表H,烷基,环烷基,环烷基烷基,芳烷基,杂芳基烷基,或杂环烷基;
R11和R12相同或不同,各为R8,或者NR11R12代表一个杂环,例如吗啉;
R13代表烷基,环烷基,环烷基烷基,芳基,芳烷基,杂芳基,杂芳基烷基,杂环基或杂环基烷基;
各R14相同或不同,各自选自H,烷基,环烷基,OR8,NR8R9,CO2R10,CONR11R12,CONHOH,SO2NR11R12,SON11R12,COR13,SO2R13,SOR13,SR13,CF3,NO2和CN,条件是,当m和n都代表0时,如果一个R14是OR8,NR8R9或SR13,则其他的不是OR8,NR8R9或SR13;和
R15代表H,烷基或F;或
其药学上可接受的盐。
这些化合物的制备描述于WO 2000/068230中,该文献整体并入本申请作为参考。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 20040106631,EP1 400 244和WO2004/026818中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
m是1,2或3;R1是甲基,氯,溴或氟;R2是-Q1-Q2-Q3-Q4或(C1-C6)烷基,该(C1-C6)烷基被1-3个OR4,COOR4,NR4R5,NRC(=O)R4,C(=O)NR4R5,或SO2NR4R5取代;
R4是被1-3个F,,CN,S(=O)R6,SO3H,SO2R6,SR7,C(=O)-NH-SO2-CH3,C(=O)R7,NR′C(=O)R7,NR′SO2R6,C(=O)NR7R8,O-C(=O)NR7R8或SO2NR7R8取代的(C1-C6)烷基;
R5是H或任选被1-3个F,CN,S(=O)R6,SO3H,SO2R6,SR7,C(=O)-NH-SO2-CH3,C(=O)R7,NR′C(=O)R7,NR′SO2R6,C(=O)NR7R8,O-C(=O)NR7R8或SO2NR7R8取代的(C1-C6)烷基;或所述的(C1-C6)烷基是:
(1)被1-3个OC(=O)R4a,SR4a,S(=O)R3,C(=NR9)R4a,C(=NR9)-NR4aR5a,NR-C(=NR9)-NR4aR5a,NRCOOR4a,NR-C(=O)NR4aR5a,NR-SO2-NR4aR5a,NR-C(=NR9)-R4a或NR-SO2-R3取代;和
(2)任选被1或2个OR4a,COOR4a,C(=O)-R4a,NR4aR5a,NRC(=O)R4a,C(=O)NR4R5a或SO2NR4aR5a取代;
R9是H,CN,OH,OCH3,SO2CH3,SO2NH2或(C1-C6)烷基;R3是任选被1-3个F,CN,S(=O)R6,SO3H,SO2R6,C(=O)-NH-SO2-CH3,OR7,SR7,COOR7,C(=O)R7,O-C(=O)NR7R8,NR7R8,NR′C(=O)R7,NR′SO2R6,C(=O)NR7R8,或SO2NR7R8取代的(C1-C6)烷基;
R4a和R5a相同或不同,是氢或任选被1-3个F,CN,S(=O)R6,SO3H,SO2R6,C(=O)-NH-SO2-CH3,OR7,SR7,COOR7,C(=O)R7,O-C(=O)NR7R8,NR7R8,NR′C(=O)R7,NR′SO2R6,C(=O)NR7R8,或SO2NR7R8取代的(C1-C6)烷基;
Q1是单键或(C1-C6)亚烷基;Q2是含1或2个O或N的饱和的4-6元杂环基;Q3是(C1-C6)亚烷基;Q4是含1-4个O,S,S(=O),SO2或N的4-8元的芳族或非芳族杂环基,该杂环基任选地被1-3个OR、NRR’、-CN或(C1-C6)烷基取代;
R是H或(C1-C6)烷基;
R6是任选被1或2个OR’取代的(C1-C6)烷基;
R7和R8相同或不同,是H或任选被1或2个OR’取代的(C1-C6)烷基;
R9是H,CN,OH,OCH3,SO2CH3,SO2NH2或(C1-C6)烷基;
R’是H或(C1-C6)烷基;R”是H或(C1-C6)烷基;
条件是,(1)与Q1键合的Q2的原子是碳原子;和(2)与Q3键合的Q4的原子是碳原子;
或其外消旋形式,异构体,药学上可接受的衍生物。
这些化合物的制备描述于US 20040106631,EP 1 400 244和WO 2004/026818中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,936,609和US 20040249148中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1代表(C6-C10)-芳基,它可任选地被选自卤素、甲酰基、氨甲酰基、氰基、羟基、三氟甲基、三氟甲氧基、硝基、(C1-C6)-烷基或(C1-C6)-烷氧基的相同或不同的基团取代,并可任选地被式SO2NR5R6的基团取代,其中R5和R6彼此独立地代表氢或(C1-C6)-烷基,或者NR5R6代表经由氮原子键合的4-8元杂环基,其可任选地被选自氧代、卤素、(C1-C6)-烷基和(C1-C6)-酰基的相同或不同的基团取代;
R2代表饱和或部分不饱和的1-10个碳原子的烃基;
R3代表甲基或乙基;
A代表O、S或NR7,其中R7代表氢或任选被(C1-C3)-烷氧基取代的(C1-C6)-烷基;
E代表键或(C1-C3)-链烷二基;
R4代表(C6-C10)-芳基或5-10元杂芳基,该芳基和杂芳基任选地被选自以下的相同或不同的基团取代:卤素,甲酰基,羧基,氨甲酰基,-SO3H,氨磺酰基,氰基,羟基,三氟甲基,三氟甲氧基,硝基,(C1-C6)-烷基,(C1-C6)-烷氧基,1,3-二氧杂丙烷-1,3-二基,(C1-C6)-烷硫基,(C1-C6)-烷基亚磺酰基和(C1-C6)-烷基磺酰基,-NR8R9以及任选被甲基取代的5-6元杂芳基或苯基;
其中R8和R9彼此独立地代表氢,(C1-C6)-烷基或(C1-C6)-酰基;
或其盐。
这些化合物的制备描述于美国专利No.6,936,609和US 20040249148中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2006/092692中一般地或具体地公开的那些化合物,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:


其中n是1-4的整数,并且在存在立构中心的情形下,各中心可以独立地是R或S。
这些化合物的制备描述于WO 2006/092692中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 2006229306和WO2004/065391中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1和R2
(1)独立地代表:
(a)氢原子;
(b)选自烷基、烯基和炔基的基团,其中各烷基、烯基和炔基独立地任选被一个或多个以下取代基取代:卤素原子、羟基、烷氧基、芳氧基、烷硫基、羟基羰基、烷氧羰基、一和二烷基氨基酰基、氧代、氨基及一和二烷基氨基;或
(c)式(CH2)n-R6基团,其中n是0-4的整数,R6代表环烷基或环烯基;
(2)R1和R2与它们所连接的氮原子一起形成一个含1-4个选自氮、氧和硫的杂原子的3-8元环,该环是饱和的或不饱和的,可任选地被选自以下基团的一个或多个取代基取代:卤素原子,烷基,羟基,烷氧基,酰基,羟基羰基,烷氧羰基,亚烷二氧基,氨基,一和二烷基氨基,一和二烷基氨基酰基,硝基,氰基和三氟甲基;
R3是式(CH2)n-G基团,其中n是0-4的整数,G代表含0-4个杂原子的单环或双环芳基或杂芳基,该基团任选地被一个或多个选自以下基团的取代基取代:
(1)卤素原子;
(2)烷基和亚烷基,其中各个烷基和亚烷基独立地任选被一个或多个选自卤素原子的取代基取代;和
(3)苯基,羟基,羟烷基,烷氧基,亚烷二氧基,芳氧基,烷硫基,氨基,一和二烷基氨基,酰基氨基,硝基,酰基,羟基羰基,烷氧羰基,氰基,二氟甲氧基和三氟甲氧基;
R4代表氢原子、烷基或芳基。
这些化合物的制备描述于US 2006229306和WO 2004/065391中。
可用于本发明方法的其它化合物包括咪唑并吡啶衍生物(WO 2001/34601),二氢嘌呤衍生物(WO 2000/68203),吡咯衍生物(WO 2001/32618),苯并噻喃并咪唑啉酮衍生物(DE19950647),杂环化合物(WO 2002/87519),鸟嘌呤衍生物(Bioorg.Med.Chem.Lett.11:1081-1083,2001),和苯并噻吩并噻二嗪衍生物(Eur.J.Med.Chem.36:333,2001)。以上列出的各个公布的专利申请和期刊文章的内容都特此全文并入本申请作为参考。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2008/130619中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
X是SO或SO2,
R1是H或烷基,
R2是烷基或卤素。
在特定的实施方案中,R1是Me。在其它的特定实施方案中,R1是F。在某些实施方案中,R2是叔丁基。在特定的实施方案中,R1是甲基。在更特定的实施方案中,该化合物选自:

在相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中,
R1是烷基,
R2是芳基或杂芳基,
R3是烷基,芳基、环烷基或烷基芳基。
在特定的实施方案中,R1是甲基。在某些实施方案中,R2是呋喃基或噻吩基。在其它特定的实施方案中,R2是取代的苯基或苄基。在优选的实施方案中,R3是异丁基。在更特定的实施方案中,该化合物选自:

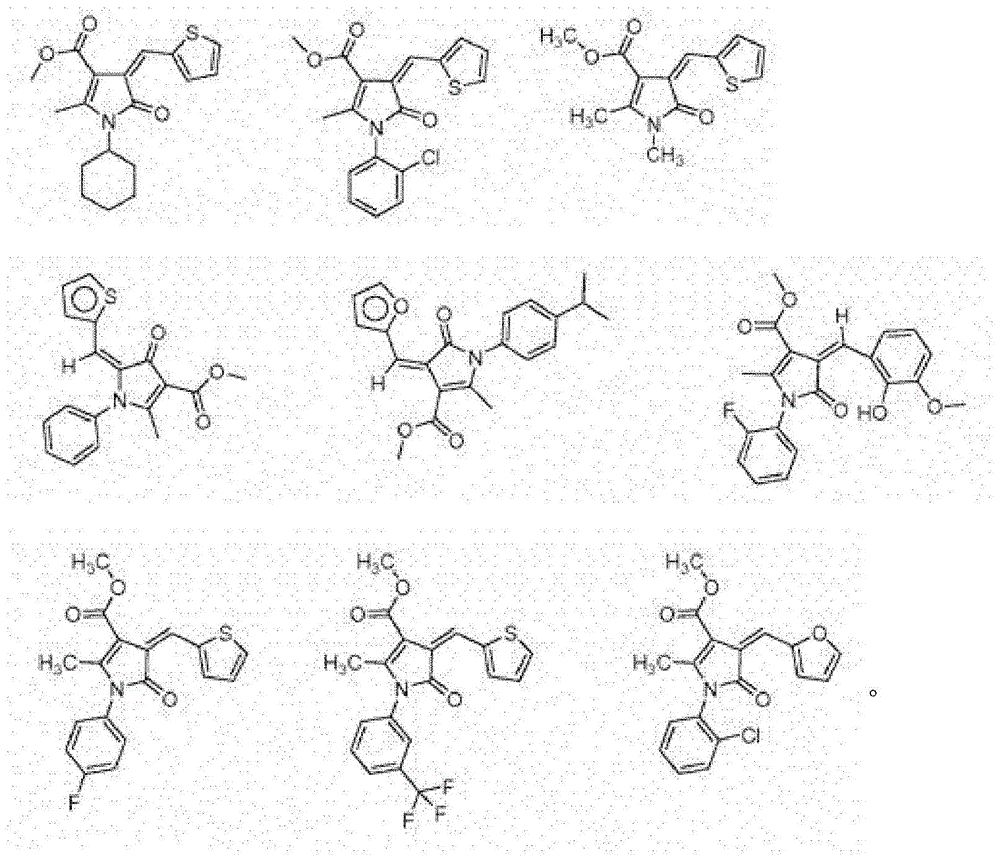
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中
R1是腈,或烷基羧酸酯,
R2是烷基、芳基或杂芳基。
在特定的实施方案中,R1是腈或甲基羧酸酯。在某些实施方案中,R2是5元杂芳基。在更特定的实施方案中,R2是呋喃基或噻吩基。在其它实施方案中,R2是6元芳基。
在更特定的实施方案中,R2是取代的苯基。

在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中
R1是烷基、烯基或烷基羧酸,
R2是卤素。
在某些实施方案中,R1是丁基。在其它实施方案中,R1是末端烯基。在更特定的实施方案中,R1是烯丙基或乙烯基。在其它实施方案中,R1是C1-C4烷基。在特定的实施方案中,R1是甲基羧酸。在某些实施方案中,R2是Cl或Br。在更特定的实施方案中,该化合物选自:


在其它相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中
R1是CO或烷基醇,R2是烷基,R3是烷氧基,而C4和C9立构中心独立地是(R)或(S)。
在某些实施方案中,R1是羰基或2-甲基丙-1-醇。在特定的实施方案中,R2是甲基。在某些实施方案中,R3是甲氧基。在更特定的实施方案中,该化合物选自:

在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中,
R1是氢、羟基、羰基或烷基醇,
R2和R3独立地选自氢、烷基、烷基羧酸酯或羧酸,
R4是氢或烷基,
R5是氢、烷基、羟基或乙酸酯,
R6是氢或烷氧基,且C4和C9立构中心独立地是(R)或(S)。
在某些实施方案中,R1是2-甲基丙-1-醇。在特定的实施方案中,R2是甲基。在某些实施方案中,R2是甲基羧酸酯。在特定的实施方案中,R2和R3都是甲基。在其它实施方案中,R2是甲基,R3是甲基羧酸酯。在特定的实施方案中,R4是异丙基。在特定的实施方案中,R5是甲基。在某些实施方案中,R6是甲氧基。在更特定的实施方案中,该化合物选自:

就以上化合物而言,术语“烷基”、“烯基”和前缀“烷-”包括直链、支链和环状(即,环烷基和环烯基)基团。除非另外指明,这些基团含1-20个碳原子,其中烯基含2-20个碳原子。优选的基团有总计最高达10个碳原子。环状基团可以是单环或多环基团,优选有3-10个环碳原子。环状基团的实例包括环丙基、环戊基、环己基、环丙基甲基、金刚烷基、降冰片烷和降冰片烯。包含前缀“烷基-”的基团也是这样,例如烷基羧酸、烷基醇、烷基羧酸酯、烷基芳基等。合适的烷基羧酸基团的实例是甲基羧酸、乙基羧酸等。合适的烷基醇的实例是甲醇、乙醇、异丙醇、2-甲基丙-1-醇等。合适的烷基羧酸酯的实例是甲基羧酸酯、乙基羧酸酯等。合适的烷基芳基的实例是苄基、苯丙基等。
术语“芳基”在本文中使用时包括碳环芳族环或环系。芳基的实例包括苯基、萘基、联苯、芴基和茚基。术语“杂芳基”包括含至少一个环杂原子(例如O,S,N)的芳族环或环系。合适的杂芳基的实例包括呋喃基、噻吩基、吡啶基、喹啉基、异喹啉基、吲哚基、异吲哚基、噻唑基、吡咯基、四唑基、咪唑基、吡唑基、噁唑基、噻唑基、苯并呋喃基、苯并噻吩基、咔唑基、苯并噁唑基、嘧啶基、苯并咪唑基、喹喔啉基、苯并噻唑基、萘啶基、异噁唑基、异噻唑基、嘌呤基、喹唑啉基等。
芳基和杂芳基可以是未取代的或被一个或多个独立选自以下基团的取代基取代:烷基,烷氧基,亚甲二氧基,亚乙二氧基,烷硫基,卤代烷基,卤代烷氧基,卤代烷硫基,卤素,硝基,羟基、巯基,氰基,羧基,甲酰基,芳基,芳氧基,芳硫基,芳基烷氧基,芳基烷硫基,杂芳基,杂芳氧基,杂芳基烷氧基,杂芳基烷硫基,氨基,烷基氨基,二烷基氨基,杂环基,杂环烷基,烷基羰基,烯基羰基,烷氧羰基,卤代烷基羰基,卤代烷氧基羰基,烷硫基羰基,芳基羰基,杂芳基羰基,芳氧基羰基,杂芳氧基羰基,芳硫基羰基,杂芳硫基羰基,烷酰氧基,烷酰硫基,烷酰氨基,芳基羰氧基,芳基羰硫基,烷基氨磺酰基,烷基磺酰基,芳基磺酰基,杂芳基磺酰基,芳基二嗪基,烷基磺酰胺基,芳基磺酰胺基,芳烷基磺酰胺基,烷基羰基氨基,烯基羰基氨基,芳基羰基氨基,芳烷基羰基氨基,芳基羰基氨基烷基,杂芳基羰基氨基,杂芳基烷基羰基氨基,烷基磺酰胺基,烯基磺酰胺基,芳基磺酰胺基,芳烷基磺酰胺基,杂芳基磺酰胺基,杂芳基烷基磺酰胺基,烷基氨基羰基氨基,烯基氨基羰基氨基,芳基氨基羰基氨基,芳烷基氨基羰基氨基,杂芳基氨基羰基氨基,杂芳基烷基氨基羰基氨基,和在杂环基的情形下,氧代。如果其它基团被说成是“取代的”或“任选取代的”,则这些基团也可以被一个或多个以上列举的取代基取代。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2008/142550中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
m是0、1或2,n是0、1、2或3,
X是O,S或N-CN,
R1是卤素或CN,
A是单键,CH2,O或S,
B是单键,CH2,或OCH2,各R2独立地是卤素,(C1-6)烷基(任选被1-3个氟原子取代),OH,(C1-6)烷硫基或CN,
R3选自以下基团(i)至(x):

R是H或(C1-6)烷基(任选被1-3个氟原子取代),R’是(C1-6)烷基(任选被1-3个氟原子取代),
或其药学上可接受的盐、溶剂化物、多晶形物或前药。
就以上化合物而言,术语“烷基”代表含1-6个碳原子的直链或支链的一价饱和烃链。烷基的实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、2-甲基丁基、3-甲基丁基、新戊基、正己基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2-乙基丁基和2,2-二甲基丁基。优选的烷基特别是甲基和乙基,尤其是甲基。
所提到的烷基可以被1-3个氟原子取代。这种取代可以在烷基链的任何位置。优选的是,这种氟代的烷基基团有1-4个碳原子,更优选有1或2个碳原子。特别优选的是一、二和三氟甲基(尤其是三氟甲基)及一、二和三氟乙基(尤其是2,2,2-三氟乙基)。
术语“烷氧基”代表“烷基-O-”,其中“烷基”的定义,无论是在其最宽的方面或是优选的方面,均与以上定义的相同。优选的烷氧基是基团,特别是甲氧基和乙氧基。术语“烷硫基”代表“烷基-S-”,其中“烷基”的定义,无论是在其最宽的方面或是优选的方面,均与以上定义的相同。优选的烷硫基是(C1-4)烷硫基,特别是甲硫基和乙硫基。术语“卤素”指氟、氯、溴或碘。优选的卤素基团是氟和氯。
优选m是0或1,更优选1。
优选n是0或1,更优选0。
优选X是O或N-CN,更优选O。
优选R1是F或Cl,更优选Cl。
优选A是单键或O,更优选是O。
当基团B是OCH2时,该氧原子与苯环键合,亚甲基与基团R3键合。
优选B是单键。
优选R2是F或Cl,更优选F。
优选R3是基团(i)、(ii)、(iii)、(iv)、(v)或(vi),更优选是基团(i)或(ii),尤其是基团(ii)。
在一项实施方案中,基团-B-R3位于苯环的2位(基团A的位置是1位)。在其它实施方案中,基团-B-R3位于3位。在又一实施方案中,基团-B-R3位于4位。
可用于本发明方法的PDE7抑制剂包括其中上式中的各个变量都选自各变量的合适的和/或优选的基团的那些化合物。可用于本发明方法的更优选的PDE7抑制剂包括其中上式中的各个变量都选自各变量的更优选或最优选的基团的那些化合物。
在一项相关的实施方案中,以下PDE7抑制剂可用于本发明的方法:
5-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)]-2-氟苯甲酸,
3-(8′-氯-2-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)苯甲酸,
5-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-4′-基)]-2-氟苯甲酸,
8-氯-5′-[4-氟-3-(2H-四唑-5-基)苯基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
[3-(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)苯氧基]乙酸,
2-{(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基}-3-氟苯甲酸,
2-{(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环戊烷-1,4′-喹唑啉]-5′-基)氧基}-3-氟苯甲酸,
3-氯-2-{(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基}苯甲酸,
3-氯-2-{(8′-氟-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基}苯甲酸,
8′-氯-5′-[2-氟-6-(2H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[4-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[6-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[4-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[6-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[6-氯-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[2-(1H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[2-氟-6-(5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[2-氟-6-(5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
2-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]-3-氟-N-(甲磺酰基)苯甲酰胺,
N-{2-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]-3-氟苯基}-1,1,1-三氟甲烷磺酰胺,
{2-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]-3-氟苯基}乙酸,
{2-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]苯氧基}乙酸,
{4-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]苯氧基}乙酸,
2-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]-3-氟苯甲酸甲酯,
及其药学上可接受的盐、溶剂化物和前药。
在另一相关的实施方案中,以下PDE7抑制剂可用于本发明的方法:
8′-氯-5′-[2-氟-6-(2H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[4-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[6-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[4-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[6-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[6-氯-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[2-(1H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
及其药学上可接受的盐、溶剂化物和前药。
以下化合物是最优选的化合物:
8′-氯-5′-[2-氟-6-(2H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[4-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[6-氟-2-(1H-四唑-5-基)苯氧基]-1′H-螺[环戊烷-1,4′-喹唑啉]-2′(3′H)-酮,
8′-氯-5′-[2-(1H-四唑-5-基)苯氧基]-1′H-螺[环己烷-1,4′-喹唑啉]-2′(3′H)-酮,
及其药学上可接受的盐、溶剂化物和前药。
这些化合物的制备描述在WO 2008/142550中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 7498334,US2005/0059686和WO2003/055882中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
X是苯基或Het,它们各自是未取代的或被R1和/或R2单取代或多取代;R1和R2各自彼此独立地是:A,OH,OA,SA,SOA,SO2A,SO2NH2,S02NHA,SO2AA’,CN,NO2,NH2,NHA,NAA′,NHCOA,NHCOOA,COOH,COOA,CONH2,CONHA,CONAA′或Hal,或者R1和R2合起来是-OCH2O-或-OCH2CH2O-;R3是A,OH,OA,SA,SOA,SO2A,SO2NH2,SO2NHA,SO2AA’,CN,NO2,NH2,NHA,NHB,NAA′,NHCOA,NHCOOA,NHCOB,NHCOOB,COOH,COOA,COOB,CONH2,CONHA,CONHB,CONAA′或Hal;R4是有最多10个碳原子的直链或支链烷基或烯基,它可以被1-5个F和/或Cl原子取代,和/或其中一个或多个CH2基团可以被O,S,SO,SO2,NH,NA,NHCO,NACO,NHCOO或NACOO代替,或是有3-7个碳原子的环烷基或环烯基,其中一个或二个CH2基团可以被O,S,SO,SO2,SO2NH,SO2NA,NH,NHA,NHCONH,NACONH,NACONA,NHCO,NACO,NHCOO或NACOO代替;R5是OH,OA,SA,SOA,SO2A,SO2NH2,SO2NHA,SO2AA’,CN,NO2,NH2,NHA,NAA’,NHCOA,NHCOOA,COOH,COOA,CONH2,CONHA,CONAA′或Hal;R6是H,OH,OA,SA,SOA,SO2A,SO2NH2,SO2NHA,SO2AA′,CN,NO2,NH2,NHA,NAA’,NHCOA,NHCOOA,COOH,COOA,CONH2,CONHA,CONAA′或Hal;A和A’各自彼此独立地是有最多10个碳原子的直链或支链烷基或烯基,它可以被1-5个F和/或Cl原子取代,和/或其中一个或多个CH2基团可以被O,S,SO,SO2,NH,NR7,NHCO,NR7CO,NHCOO或NR7COO代替。或者A和A’合起来是有3-7个碳原子的亚烷基,其中1或2个CH2基团可以被CHR7,CHR7R8,O,S,SO,SO2,NH,NR7,NHCO,NR7CO,NHCOO或NR7COO代替;B是苯基或Het,它们均为未取代的或被R1和/或R2单取代或多取代,Het是有1-3个N、O和/或S原子的芳族5或6元杂环,其为未取代的或被A”,Hal或CF3单取代、二取代或三取代;R7和R8各自彼此独立地是有最多5个碳原子的支链或直链烷基或烯基,它可被1-5个F和/或Cl原子取代,和/或其中一个或多个CH2基团可以被O、S、SO、SO2或NH代替;A”是有1-6个碳原子的烷基,Hal是F、Cl、Br或I;及其可药用的衍生物、溶剂化物和立体异构体,包括其所有比例的混合物。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂包括其中R5是OH的上式化合物,它也可以是下式的其互变异构形式:

就以上化合物而言,可用于本发明方法的PDE7抑制剂包括这些化合物的光学活性形式(立体异构体)、对映异构体、外消旋物、非对映异构体和水合物及溶剂化物。术语化合物的溶剂化物是指惰性溶剂分子和化合物由于它们的相互吸引力而形成的加合物。溶剂化物是例如一水合物、二水合物或醇化物。
就以上化合物而言,术语可药用的衍生物是指,例如,以上化合物的盐和所谓的前药化合物。术语前药衍生物是指,例如,已被改性,例如,用烷基或酰基、糖或低聚肽改性的以上化合物,它在生物体内迅速裂解,从而释放出活性化合物。这也包括以上化合物的可生物降解的聚合物衍生物,如在Int.J.Pharm.115,61-67(1995)中所述。
就以上化合物而言,所有出现一次以上的基团的含义在每次出现时都是彼此独立的。
A和A’优选是烷基,更优选是被1-5个氟和/或氯原子取代的烷基,还优选是烯基。
在以上化学式中,烷基优选是直链,有1、2、3、4、5、6、7、8、9、或10个碳原子,优选有1、2、3、4、5、或6个碳原子,优选的是甲基、乙基、三氟甲基、五氟乙基或丙基,还优选是异丙基、丁基、异丁基、仲丁基或叔丁基,而且也优选是正戊基、新戊基、异戊基或正己基。特别优选的是甲基、乙基、三氟甲基、丙基、异丙基、丁基、正戊基、正己基或正癸基。
A”优选是有1、2、3、4、5、或6个碳原子的烷基,例如甲基、乙基或丙基,还优选是异丙基、丁基、异丁基、仲丁基或叔丁基,而且还优选是正戊基、新戊基、异戊基或正己基。特别优选的是甲基、乙基、丙基、异丙基或丁基。
环烷基优选有3-7个碳原子,优选的是环丙基或环丁基,还优选是环戊基或环己基,也优选是环庚基;特别优选的是环戊基。
烯基优选是乙烯基、烯丙基、2-或3-丁烯基、异丁烯基或仲丁烯基,还优选是4-戊烯基、异戊烯基或5-己烯基。
亚烷基优选是直链的,优选是亚甲基或亚乙基,还优选是亚丙基或亚丁基。
Hal优选是F、Cl、或Br,也优选是I。
基团R1和R2可以相同或不同,优选是在苯基环的2-或4-位。它们彼此独立地是例如A或Hal,或者合起来是亚甲二氧基。
但是,它们优选各为甲基、乙基、丙基、甲氧基、乙氧基、丙氧基、异丙氧基、苄氧基,而且还包括氟-、二氟-或三氟-甲氧基,或1-氟-、2-氟-、1,2-二氟-、2,2-二氟-、1,2,2-三氟-或2,2,2-三氟乙氧基,以及氟或氯。
R1特别优选是氟、氯、甲基、乙基或丙基。
R2特别优选是氟、氯、甲基、乙基或丙基。
X优选是被R1单取代的苯基,或是未取代的Het。
X特别优选是2-氯苯基、2-氟苯基、4-甲基苯基、3-氯苯基或4-氯苯基。
Het优选是,例如,未取代的2-或3-呋喃基,2-或3-噻吩基,1-、2-或3-吡咯基,1-、2-、4-、或5-咪唑基,2-、3-或4-吡啶基,2-、4-、5-或6-嘧啶基,还优选是1,2,3-三唑-1-、-4-或-5-基,1,2,4-三唑-1-、-3-或-5-基,1,2,3-噁二唑-4-或-5-基,1,2,4-噁二唑-3-或-5-基,1,3,4-噻二唑-2-或-5-基,1,2,4-噻二唑-3-或-5-基,或1,2,3-噻二唑-4-或-5-基。
R3优选是,例如,COOA”或COOH。
R4优选是,例如,有1、2、3、4、5或6个碳原子的直链或支链烷基,它可以被1-5个F或Cl原子取代,优选是甲基、乙基、三氟甲基、五氟乙基或丙基,还优选是异丙基、丁基、异丁基、仲丁基或叔丁基,而且也优选是正戊基、新戊基、异戊基或正己基。特别优选的是甲基、乙基、三氟甲基、丙基、异丙基、丁基、正戊基、正己基或正癸基。
R5优选是Cl或OH。
R6优选是H。
就以上化合物而言,所述基团中至少有一个具有以上指出的优选含义之一。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物,其中X是被R1单取代的苯基或是未取代的Het;R1是A或Hal;R3是COOA”或COOH;R4是有1、2、3、4、5或6个碳原子的直链或支链烷基,它可以被1-5个F或Cl原子取代;R5是Cl或OH;R6是H。
在其它相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物,其中X是被R1单取代的苯基或是未取代的Het;R1是A或Hal;R3是COOA”或COOH;R4是有1、2、3、4、5或6个碳原子的直链或支链烷基,它可以被1-5个F或Cl原子取代;R5是Cl或OH;R6是H;Het是呋喃基、噻吩基、吡咯基、咪唑基、吡啶基或嘧啶基;A和A”各自彼此独立地是有1、2、3、4、5或6个碳原子的直链或支链烷基,它可以被1-5个F或Cl原子取代;Hal是F、Cl、或Br;及其可药用的衍生物、溶剂化物和立体异构体,包括其任何比例的混合物。
以上化合物的制备及用于其制备的起始物在文献中有报道(例如在经典著作中,如Houben-Weyl,Methoden der Organischen Chemie[有机化学方法],Georg-Thieme-Verlag,Stuttgart),确切地说,在已知的适合所述反应的反应条件下。这里也可以使用本身已知,但文中未详细说明的各种方法的变型。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括:
5-异丙基-4-氧代-7-对甲苯基-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸乙酯,
5-甲基-4-氧代-7-(3-氯苯基)-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸乙酯,
5-甲基-4-氧代-7-(2-氯苯基)-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸乙酯,
5-甲基-4-氧代-7-(2-氟苯基)-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸乙酯,
5-丙基-4-氧代-7-(2-氯苯基)-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸乙酯,
5-甲基-4-氧代-7-(4-氯苯基)-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸乙酯,
5-甲基-4-氧代-7-对甲苯基-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸乙酯,
5-甲基-4-氧代-7-(2-氯苯基)-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸甲酯,
5-甲基-4-氧代-7-苯基-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸甲酯,
5-甲基-4-氧代-7-(2-噻吩基)-4,7-二氢-3H-吡咯并[2,3-d]-嘧啶-6-甲酸甲酯,及其可药用的衍生物、溶剂化物和立体异构体,包括其所有比例的混合物。
以上化合物的制备描述于US 7498334和WO 2003/055882中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6884800和WO01/36425中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1和R2彼此独立地各自代表A1、OA1、SA1或Hal,A1代表H、A、烯基、环烷基或亚烷基环烷基,A代表有1-10个碳原子的烷基,Hal代表F、Cl、Br或I,X代表O、S、SO或SO2,以及它们的药学上可接受的盐和/或溶剂化物。
就以上化合物而言,A代表有1-10个碳原子的烷基,有1、2、3、4、5、6、7、8、9、或10个碳原子,优选代表甲基、乙基或丙基,还优选代表异丙基、丁基、异丁基、仲丁基或叔丁基,而且也优选是正戊基、新戊基、异戊基或己基。在这些基团中,1-7个H原子也可以被F和/或Cl置换。因此A也代表例如三氟甲基或五氟乙基。环烷基有3-9个碳原子,优选代表例如环戊基或环己基。烯基有2-10个碳原子,是支链或直链,优选代表乙烯基、丙烯基或丁烯基。亚烷基环烷基有4-10个碳原子,代表例如亚甲基环戊基、亚乙基环戊基、亚甲基环己基或亚乙基环己基。R1和R2在每种情形下都彼此独立地优选代表H、氟、氯、甲基、乙基、丙基、甲氧基、乙氧基、丙氧基、甲硫基、环戊基或环己基。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物,其中X是S;
X是S,R1是H;
X是S,R1是F或Cl;
X是S,R2是H;
X是S,R2是F或Cl;
X是S,R1是H,R2是F或Cl;
X是S,R1是F或Cl,R2是H;
X是S,A1是H或A,A是有1、2、3或4个碳原子的烷基;
X是S,R1和R2彼此独立地各自代表A1或Hal,A1是H或A,A是有1、2、3或4个碳原子的烷基,Hal是F或Cl;
以及它们的生理上可接受的盐和溶剂化物。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:10-氯-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-kl]吩噻嗪,4-氯-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-kl]吩噻嗪,10-甲氧基-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-kl]吩噻嗪,10-丙氧基-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-kl]吩噻嗪,10-甲硫基-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-kl]吩噻嗪,10-氟-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-kl]吩噻嗪,4,10-二氯-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-k1]吩噻嗪,10-三氟甲基-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-k1]吩噻嗪,4-环戊氧基-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-k1]吩噻嗪,10-氯-3-咪唑-1-基-2,3-二氢-1H-7-氧杂-11b-氮杂苯并[de]-蒽,和10-氯-3-咪唑-1-基-2,3-二氢-1H-吡啶并[3,2,1-k1]吩噻嗪7,7-二氧化物。
这些化合物的制备描述于美国专利No.6,884,800和WO 01/36425中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,531,498和WO01/32175中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1、R2、R3和R4彼此独立地各自是Hal,OA1,SA1,A,H,COOA1,CN或CONA1A2;
R5是COOA1,CN或CONA1A2;
A1、A2彼此独立地各自是H,A,烯基,环戊基或亚烷基环烷基;
A是有1-10个碳原子的烷基;
Hal是F,Cl,Br或I;
及其生理上可接受的盐和/或溶剂化物。
就以上化合物而言,A代表有1-10个碳原子的烷基,有1、2、3、4、5、6、7、8、9、或10个碳原子,优选是甲基、乙基或丙基,还优选是异丙基、丁基、异丁基、仲丁基或叔丁基,而且也优选是正戊基、新戊基、异戊基或己基。这些基团中的1-7个H原子也可以被F和/或Cl置换。因此A也是例如三氟甲基或五氟乙基。
环烷基有3-9个碳原子,优选是例如环戊基或环己基。烯基有2-10个碳原子,是支链或直链,优选是乙烯基、丙烯基或丁烯基。
亚烷基环烷基有4-10个碳原子,是例如亚甲基环戊基、亚乙基环戊基、亚甲基环己基或亚乙基环己基。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物,其中
R1是H;
R1和R2是H;
R1是H,R2是F或Cl;
R1、R2各自彼此独立地是H或Hal;
R1、R2各自彼此独立地是H或Hal,A1、A2各自彼此独立地是H或A;
A1、A2各自彼此独立地是H或A;
R1、R2各自彼此独立地是H或Hal,A1、A2各自彼此独立地是H或A,A是有1、2、3或4个碳原子的烷基,Hal是F或Cl。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:
5-[2-(2-氟-4-羟基苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(2,4-二氟苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(3-甲硫基苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(2,4-二甲氧基苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-(2-氨基-2-苯基乙烯基)-4-甲基氨基羰基-3-苯基异噁唑,
5-(2-苯基氨基乙烯基)-4-甲氧基羰基-3-苯基异噁唑,
5-[2-(4-羧基苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(4-羧基苯基氨基)乙烯基]-4-甲氧基羰基-3-苯基异噁唑,
5-[2-(5-氯-2-羟基苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(3,4-二甲基苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-[2-(4-氯苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-(2-苯基氨基乙烯基)-4-氰基-3-(2-氯苯基)异噁唑,
5-[2-(4-甲氧基苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-[2-(4-羧基苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-[2-(2-氟-4-羟基苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-[2-(4-氟苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-[2-(3,5-二氯苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-[2-(3-氯苯基氨基)乙烯基]-4-氰基-3-(2-氯苯基)异噁唑,
5-[2-苯基氨基乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(4-氯苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-苯基氨基乙烯基]-4-甲氧羰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(4-氯苯基氨基)乙烯基]-4-甲氧羰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(4-羧基苯基氨基)乙烯基]-4-甲氧羰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(2,4-二氟苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(2,4-二氯苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(4-羧基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(3,5-二氯苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(4-甲氧基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(2,4-二甲氧基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(2-苯基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-[2-(4-甲基苯基氨基)乙烯基]-4-氰基-3-(2,6-二氯苯基)异噁唑,
5-(2-苯基氨基乙烯基)-4-氰基-3-(2-氯-6-氟苯基)异噁唑,
5-[2-(4-羧基苯基氨基)乙烯基]-4-氰基-3-(2-氯-6-氟苯基)异噁唑,
5-[2-(4-氯苯基氨基)乙烯基]-4-氰基-3-(2-氯-6-氟苯基)异噁唑,
5-[2-(3-甲氧基苯基氨基)乙烯基]-4-氰基-3-(2-氯-6-氟苯基)异噁唑,
5-[2-(4-氯苯基氨基)乙烯基]-4-甲氧羰基-3-(2-氯-6-氟苯基)异噁唑,
5-(2-苯基氨基乙烯基)-4-甲氧羰基-3-(2-氯-6-氟苯基)异噁唑,
5-[2-(2,4-二氯苯基氨基)乙烯基]-4-甲氧羰基-3-(2-氯-6-氟苯基)异噁唑,
5-(2-苯基氨基乙烯基)-4-氰基-3-苯基异噁唑,
5-[2-(3-三氟甲氧基苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(4-甲氧基苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(4-甲氧基苯基氨基)乙烯基]-4-甲氧羰基-3-(2-氯-6-氟苯基)异噁唑,
5-[2-(3-甲硫基苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(2,4-二氟苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑,
5-[2-(2-氟-4-羟基苯基氨基)乙烯基]-4-氰基-3-苯基异噁唑。
这些化合物的制备描述于美国专利No.6531498和WO 01/32175中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US 7491742和WO2001/29049中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1是H、A、苄基、茚满-5-基、1,2,3,4-四氢萘-5-基、二苯并噻吩-2-基,或是未取代的或被Hal、A、A-CO-NH、苄氧基、烷氧基、COOH或COOA单取代、二取代或三取代的苯基,R2是H或A,X是O或S,Hal是F、Cl、Br或I,A是有1-6个碳原子的烷基,
及其生理上可接受的盐和/或溶剂化物。
就以上化合物而言,A是有1-6个碳原子的烷基,有1、2、3、4、5或6个碳原子,优选是甲基、乙基或丙基,还优选是异丙基、丁基、异丁基、仲丁基或叔丁基,但也优选是正戊基、新戊基、异戊基或己基。A还是环烷基,例如环己基。烷氧基优选是甲氧基、乙氧基、丙氧基或丁氧基。Hal优选是F或Cl。A-CO-NH优选是乙酰胺基。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂选自以下化合物:1-苯基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-苄基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-环己基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-环戊基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-丁基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-异丙基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-丙基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-乙基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-甲基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,2-甲基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-苯基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-苄基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-环己基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-环戊基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-丁基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-异丙基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-丙基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-乙基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-甲基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,2-甲基-[1]苯并噻喃并[3,4-d]咪唑-4-(1H)-酮,1-(2-氯苯基-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(4-甲基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(4-氟苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2,4-二甲基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(3-氯苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2,4-二氯苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2,5-二氯苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(4-乙酰胺基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2-氟苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(3-氟苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2-苄氧基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2,6-二甲基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(茚满-5-基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2-甲氧基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2,3-二甲基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2,3-二氯苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(3-氯-4-甲基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(2,5-二甲基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(4-氯苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(1,2,3,4-四氢萘-5-基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(二苯并噻吩-2-基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(3-甲氧基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,1-(4-羧基-2-甲基苯基)-[1]苯并吡喃并[3,4-d]咪唑-4-(1H)-酮,及其生理上可接受的盐和/或溶剂化物。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,737,436和WO 01/32618中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1和R2彼此独立地各自代表H、A、OA、SA或Hal,
R3代表H或A,
R4代表A或NH2,
R5代表H、NH2、NHA或NA2,
A代表有1-10个碳原子的烷基,烯基、环烷基或亚烷基环烷基,
Hal代表F、Cl、Br或I,
及它们的生理上可接受的盐和/或溶剂化物。
就以上化合物而言,A代表有1-10个碳原子的烷基,有1、2、3、4、5、6、7、8、9、或10个碳原子,优选代表甲基、乙基或丙基,还优选代表异丙基、丁基、异丁基、仲丁基或叔丁基,而且还优选是正戊基、新戊基、异戊基或己基。在这些基团中,1-7个H原子也可以被F和/或Cl置换。因此A也代表例如三氟甲基或五氟乙基。
A还代表有3-8个碳原子的环烷基,优选代表例如环戊基或环己基。
A还代表烯基。烯基有2-10个碳原子,是直链或支链,代表例如乙烯基、丙烯基或丁烯基。A还代表亚烷基环烷基。亚烷基环烷基有4-10个碳原子,优选代表例如亚甲基环戊基、亚乙基环戊基、亚甲基环己基或亚乙基环己基。
R1和R2各自彼此独立地优选代表H、甲基、乙基、丙基、丁基、异丙基、叔丁基、甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、S-甲基、S-乙基、F或Cl。
R3优选代表H、甲基或乙基。
R4优选代表甲基、乙基、丙基、丁基或NH2。
R5优选代表H、氨基、甲基氨基、乙基氨基、二甲基氨基或二乙基氨基。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂包括上式化合物,其中R1和R2不都是氢,并且其中当R1或R2中的一个是H时,另一个不能是CH3、OCH3或Cl。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物,其中:
R1、R2、R3和R5是H,R4是甲基;
R1是4-Cl,R2是H,R3是乙基,R4是氨基,和R5是H;
R1和R2是H,R3是乙基,R4是甲基,和R5是氨基;
R1和R2是H,R3是乙基,R4是氨基,和R5是H;
R1和R2是H,R3是乙基,R4是H,和R5是氨基;
R1是3-Cl,R2是4-O-甲基,R3是乙基,R4是氨基,和R5是H;
R1是3-Cl,R2是4-O-甲基,R3是乙基,R4是甲基,和R5是氨基;
R1是4-OCF3,R2是H,R3是乙基,R4是氨基,和R5是H;
R1是3-Cl,R2是4-O-甲基,R3是乙基,R4是氨基,和R5是H;
R1是3-Cl,R2是4-O-甲基,R3是乙基,R4是甲基,和R5是氨基;
R1是4-OCF3,R2是H,R3是乙基,并且R4是氨基,和R5是H。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.6,613,778和WO01/34601中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1代表CONR4R5,
R2代表H或A,
R4和R5彼此独立地各自代表H或A1,
R3代表Hal,
Hal代表F、Cl、Br或I,
A代表有1-4个碳原子的烷基,
A1代表有1-10个碳原子的烷基,
X代表有1-4个碳原子的亚烷基,其中一个亚乙基也可以被双键或三键代替,
及其生理上可接受的盐和/或溶剂化物。
就以上化合物而言,A代表有1-4个碳原子的烷基,有1、2、3或4个碳原子,优选代表甲基、乙基或丙基,还优选代表异丙基、丁基、异丁基、仲丁基或叔丁基。这些基团中的1-7个H原子也可以被F和/或Cl置换。因此A也代表例如三氟甲基或五氟乙基。
A1代表有1-10个碳原子的烷基,有1、2、3、4、5、6、7、8、9、或10个碳原子,优选代表甲基、乙基或丙基,还优选代表异丙基、丁基、异丁基、仲丁基或叔丁基,但也优选是正戊基、新戊基、异戊基或己基。在这些基团中,1-7个H原子也可以被F和/或Cl置换。因此A1也代表例如三氟甲基或五氟乙基。
X代表有1-4个碳原子的亚烷基,优选亚甲基、亚乙基、亚丙基或亚丁基,其中一个亚乙基也可以被一个双键或三键代替。因此X也代表例如-CH2-CH=CH-CH2-或-C≡C-。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:
2-(3-丁基-7-氯-3H-咪唑并[4,5-c]吡啶-4-基硫烷基)-N,N-二甲基乙酰胺

2-(3-丁基-7-氯-3H-咪唑并[4,5-c]吡啶-4-基硫烷基)乙酰胺,
2-(3-丁基-7-氯-3H-咪唑并[4,5-c]吡啶-4-基硫烷基)丙酰胺,
2-(3-丁基-7-氯-3H-咪唑并[4,5-c]吡啶-4-基硫烷基)丁酰胺,
2-(3-丁基-7-氯-3H-咪唑并[4,5-c]吡啶-4-基硫烷基)-N-己基乙酰胺,
2-(3-丁基-7-氯-3H-咪唑并[4,5-c]吡啶-4-基硫烷基)-N-辛基乙酰胺,
2-(3-丁基-7-氯-3H-咪唑并[4,5-c]吡啶-4-基硫烷基)-丁-2-烯酸二甲基酰胺。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物,其中
R3是Cl;
R3是Cl,和X是有1-4个碳原子的亚烷基;
R3是Cl,X是有1、2、3、或4个碳原子的亚烷基,和A1是有1、2、3、或4个碳原子的烷基。
这些化合物的制备描述于美国专利No.6,613,778和WO 01/34601中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在WO2008/113881和ESP 200700762中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
A是5、6或7元的稠合碳环或杂环,可以是饱和的或不饱和的;各虚线独立地表示单键或双键;X和Y独立地选自以下基团:烷基,氢,=O,=S,-N(烷基),-N(芳基),芳基,O-烷基,O-芳基,烷基-S和-S-芳基;R1和R2独立地选自以下基团:氢,卤素,烷基,卤代烷基,芳基,环烷基,(Z)n-芳基,杂芳基,-OR3,-C(O)OR3,-(Z)n-C(O)OR3和-S(O);或其药学上可接受的盐、衍生物、前药、溶剂化物或立体异构体。
例外:当A是未取代的苯时,X=O,Y=S;当A是未取代的苯时,X=O,Y=O;当A是未取代的苯时,X=O,Y=S-Me;当A是未取代的噻吩时,X=O,Y=S;和当A是未取代的苯并噻吩时,X=O,Y=S。
在一项相关的实施方案中,以上化合物构成一种用于向患者IV给药的有用药物组合物,其包含治疗有效量的以上化合物或其混合物,其盐、衍生物、前药、溶剂化物或药学上可接受的立体异构体,及载体、辅助剂或药学上可接受的媒介物。
在其它相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉及其衍生物,选自以下:

6-溴-2,3,4-四氢喹唑啉,6-溴-(2,6-二氟苯基)-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉,
6-溴-(2,3,4-三氟苯基)-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉,6-溴-(2-溴苯基)-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉,3-(2,6-二氟苯基)-8-甲基4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉,
3-(2,3,4-三氟苯基)-8-甲基-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉和3-(2-溴苯基)-8-甲基-4-氧代-2-二氧代-1,2,3,4-四氢喹唑啉。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:2-甲硫基-4-氧代-3,4-二氢喹唑啉及其衍生物,选自以下:

6-溴-(2,6-二氟苯基)-2-甲硫基-4-氧代-3,4-二氢喹唑啉,6-溴-(2,3,4-三氟苯基)-2-甲硫基-4-氧代-3,4-二氢喹唑啉,6-溴-(2-溴苯基)-2-甲硫基-4-氧代-3,4-二氢喹唑啉,3-苯基-8-甲基-2-甲硫基-4-氧代-3,4-二氢喹唑啉,3-(2,6-二氟苯基)-8-甲基-2-甲硫基-4-氧代-3,4-二氢喹唑啉,3-(2,3,4-三氟苯基)-8-甲基-2-甲硫基-4-氧代-3,4-二氢喹唑啉和3-(2-溴苯基)-8-甲基-2-甲硫基-4-氧代-3,4-二氢喹唑啉。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:2,4-二硫代-1,2,3,4-四氢喹唑啉及其衍生物,选自以下:

3-苯基-2,4-二硫代-1,2,3,4-四氢喹唑啉,3-(2,6-二氟苯基)-2,4-二硫代-1,2,3,4-四氢喹唑啉,和3-(2,3,4-三氟苯基)-2,4-二硫代-1,2,3,4-四氢喹唑啉。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:(2-甲硫基-4-硫代-3,4-二氢喹唑啉)及其衍生物,选自以下:

3-苯基-2-甲硫基-4-硫代-3,4-二氢喹唑啉,3-(2,6-二氟苯基)-2-甲硫基-4-硫代-3,4-二氢喹唑啉,3-(2,3,4-三氟苯基)-2-甲硫基-4-硫代-3,4-二氢喹唑啉和3-(2-溴苯基)-2-甲硫基-4-硫代-3,4-二氢喹唑啉。
以上化合物的制备描述在WO 2008/113881中。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂描述在ES P200700762中,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在美国专利No.7,214,676和US 2007/0049558中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:
螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
螺[环庚烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
7′-甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-苯基螺[环庚烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
7′-氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
5′-氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-甲基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-溴螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氟螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-甲基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
5′,8′-二氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′,7′-二氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
5′,6′-二氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-苯基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-碘螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-溴螺[环丁烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-溴螺[环庚烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-溴-4-甲基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-溴螺[双环[3,2,1]辛烷-2-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′,8′-二氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-碘螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-苯基螺[环庚烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-苯基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-甲基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-(3-吡啶基)螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-(4-吡啶基)螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-(4-羧基苯基)-8′-氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-(3-羧基苯基)-8′-氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-(1H-吲哚-5-基)螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-(2-吡啶基)螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-(3-二甲基氨基丙-1-炔基)螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-(3-甲基氨基丙-1-炔基)螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-(4-甲基哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-(3-N-二甲基氨基丙基甲酰胺)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-(2-N-二甲基氨基乙基甲酰胺)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[3-(3-N-二甲基氨基丙基甲酰胺)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[3-(4-甲基哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[3-(2-N-二甲基氨基乙基甲酰胺)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-硫酮,
8′-氯-2′-氰基亚氨基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉,
8′-氯-6′-[4-(4-嘧啶-2-基哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-(4-(2-吗啉-4-基乙基)哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-(4-(2-吗啉-4-基-2-氧代-乙基)哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-(4-(2-羟基乙氧基)乙基)哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
螺[环己烷-1,9′-(8′,9′-二氢)吡唑并[4′,3′-f]喹唑啉]-7′(6′H)-酮,
8′-氯-5′-甲氧基螺[环己烷-1,4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
5′,8′-二氟螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-甲基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-(吗啉-4-基)甲基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-羟基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-羟基-6′-碘螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-碘-5′-甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-氰基-5′-甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[2-(4-吗啉代)乙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[2-二甲基氨基乙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[2-氨基乙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[2-(甲基氨基)乙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[2-(2-氨基乙氧基)乙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[3-二甲基氨基丙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-乙氧羰基甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
5′-羧基甲氧基-8′-氯-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
5′-羧基丙氧基-8′-氯-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(3-磺基丙氧基)螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[2-(四氢吡喃-2-基氧基)乙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(2-羟基乙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(5-乙氧羰基呋喃-2-基甲氧基)螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(5-羧基呋喃-2-基甲氧基)-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-氰基甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(1H-四唑-5-基甲氧基)-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(5-羟基-[1,2,4]噁二唑-3-基甲氧基)-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-碘-5′-[2-二甲基氨基乙氧基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-(4-羧基苯基)-8′-氯-5′-甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
6′-(3-羧基苯基)-8′-氯-5′-甲氧基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[2-(4-甲基哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[2-甲基-4-(4-甲基哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-(哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-氨甲酰基苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-[4-((1-甲基哌啶-4-基)哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-甲氧基-6-[4-(4-甲基哌嗪-1-羰基)苯基]螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-三氟甲基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-6′-氰基甲基螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(3-二甲基氨基-2-羟基丙氧基)-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(3-甲基氨基-2-羟基丙氧基)-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[2-(乙氧羰基甲基氨基)乙氧基]-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-[2-(羧甲基氨基)乙氧基]-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮盐酸盐,
8′-氯-5′-(2-甲磺酰胺基-2-氧代-乙氧基)-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮,
8′-氯-5′-(2-[(5-甲基异噁唑-3-基甲基)氨基]乙氧基)-螺[环己烷-1-4′-(3′,4′-二氢)喹唑啉]-2′(1′H)-酮。
这些化合物的制备描述于美国专利No.7,087,614,US 2007/0049558和WO 2002/074754中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂和双重PDE4/7抑制剂选自在US 7,087,614,US 2003/0162802和WO 2002/102313中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

可用于本发明方法的PDE7抑制剂包括上式化合物的对映异构体、非对映异构体、互变异构体和药学上可接受的盐、前药和溶剂化物。
以上化合物的取代基定义如下:
R1是H或烷基;
R2是(a)杂芳基,或杂环基,它们各自可任选地被1-3个基团T1、T2、T3取代;(b)被1-3个基团T1、T2、T3取代的芳基,条件是T1、T2、T3中至少一个不是H;或(c)与杂芳基或杂环基环稠合的芳基,其中该组合环系可以任选地被1-3个基团T1、T2、T3取代;
Z是(a)-OR4,-C(O)R4,-C(O)OR4,-SR4,-NR3R4,-C(O)NR3R4,-NR3SO2R4c,卤素,硝基,卤代烷基;或(b)烷基,芳基,杂芳基,杂环基或环烷基,它们任一个可任选地被1-3个基团T1a、T2a、T3a取代;
J是(a)氢,卤素,-OR4a,或(b)烷基,烯基或炔基,它们任一个可任选地被1-3个基团T1b、T2b、或T3b取代;
L是(a)氢,-OR4b,-C(O)R4b,-C(O)OR4b,-SR4b,-NR5R6,-C(O)NR5R6,-NR5SO2R4d,卤素,卤代烷基,硝基,或(b)烷基,芳基,杂芳基,杂环基或环烷基,它们任一个可任选地被1-3个基团T1c、T2c、或T3c取代;
R3和R4独立地是H,烷基,烯基,芳基,(芳基)烷基,杂芳基,(杂芳基)烷基,环烷基,(环烷基)烷基,杂环基或(杂环基)烷基,它们任一个可任选地被1-3个基团T1a、T2a、或T3a取代;
或者R3和R4与它们所连接的氮原子合起来可以结合形成一个任选被1-3个基团T1a、T2a、或T3a取代的4-8元杂环基环;
R4a是H,烷基,烯基,芳基,杂芳基,(芳基)烷基,(杂芳基)烷基,杂环基,(杂环基)烷基,环烷基或(环烷基)烷基,它们任一个可任选地被1-3个基团T1b、T2b、或T3b取代;
R4b是H,烷基,烯基,芳基,杂芳基,(芳基)烷基,(杂芳基)烷基,杂环基,(杂环基)烷基,环烷基或(环烷基)烷基,它们任一个可任选地被1-3个基团T1c、T2c、或T3c取代;
R4c和R4d独立地是烷基,烯基,芳基,(芳基)烷基,杂芳基,(杂芳基)烷基,环烷基,(环烷基)烷基,杂环基或(杂环基)烷基,它们任一个可任选地被1-3个基团T1a、T2a、或T3a取代;
R5和R6独立地是H,烷基,烯基,芳基,(芳基)烷基,杂芳基,(杂芳基)烷基,环烷基,(环烷基)烷基,杂环基或(杂环基)烷基,价态允许时它们任一个可任选地被1-3个基团T1c、T2c、或T3c独立取代;
或者R5和R6与它们所连接的氮原子合起来可以结合形成一个任选被1-3个基团T1c、T2c、或T3c取代的4-8元杂环基环;
T1-1c、T2-2c和T3-3c各自独立地是(1)氢或T6,其中T6是(i)烷基,(羟基)烷基,(烷氧基)烷基,烯基,炔基,环烷基,(环烷基)烷基,环烯基,(环烯基)烷基,芳基,(芳基)烷基,杂环基,(杂环基)烷基,杂芳基或(杂芳基)烷基;(ii)基团(i),其本身被一个或多个相同或不同的基团(i)取代;或(iii)基团(i)或(ii),它独立地被T1-1c、T2-2c和T3-3c定义中的一个或多个(优选1-3个)以下基团(2)至(13)取代:(2)-OH或-OT6,(3)-SH或-ST6,(4)-C(O)tH,-C(O)tT6,或-O-C(O)T6,其中t是1或2;(5)-SO3H,-S(O)T6,或S(O)tN(T9)T6,(6)卤素,(7)氰基,(8)硝基,(9)-T4-NT7T8,(10)-T4-N(T9)-T5-NT7T8,(11)-T4-N(T10)-T5-T6,(12)-T4-N(T10)-T5-H,(13)氧代;
T4和T5各自独立地是(1)单键,(2)-T11-S(O)t-T12-,(3)-T11-C(O)-T12-,(4)-T11-C(S)-T12-,(5)-T11-O-T12-,(6)-T11-S-T12-,(7)-T11-O-C(O)-T12-,(8)-T11-C(O)-O-T12-,(9)-T11-C(=NT9a)-T12-,或(10)-T11-C(O)-C(O)-T12,
T7、T8、T9、T9a和T10:(1)各自独立地是氢或在T6的定义中提供的基团,或(2)T7和T8可以合起来是亚烷基或亚烯基,与它们所连接的原子一起构成一个3-8元的饱和或不饱和环,该环是未取代的或被在T1-1c、T2-2c和T3-3c的描述中列出的一个或多个基团取代,或(3)T7或T8,与T9一起,可以是亚烷基或亚烯基,与它们所连接的氮原子一起构成一个3-8元的饱和或不饱和的环,该环是未取代的或被在T1-1c、T2-2c和T3-3c的描述中列出的一个或多个基团取代,或(4)T7和T8,或T9和T10,与它们所连接的氮原子一起可以合起来形成基团-N=CT13T14,其中T13和T14各自独立地是H或T6的定义中提供的基团;
T11和T12各自独立地是(1)单键,(2)亚烷基,(3)亚烯基,或(4)亚炔基。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以上化合物,其中:
Z是(a)卤素,烷氧基,卤代烷基,-NR3R4,-C(O)OR4,-C(O)NR3R4;(b)芳基或杂芳基,它们各自任选地被一或多个T1a、T2a或T3a(尤其是氰基、任选取代的烷基、(羟)烷基、-OH、-OT6、-ST6、-SOtT6、-COtH、-COtT6、-T4NT7T8、或-T4-N(T10)-T5-T6)取代;(c)任选取代的烷基(尤其是被一个或多个-OH、-COtH、-COtT6、-T4-NT7T8、-T4-N(T10)-T5-H或-T4-N(T10)-T5-T6取代);
J是(a)H,或(b)烷基或烯基,它们各自任选地被取代(尤其是被一个或多个-OH、-OT6、-COtH或-COtT6取代);
L是(a)H;(b)卤素、烷氧基、卤代烷基、-NR5R6,-C(O)OR4b、-C(O)NR5R6;(c)芳基或杂芳基,它们各自任选地被一个或多个T1c、T2c、T3c(尤其是氰基、任选取代的烷基、(羟)烷基、-OH、-OT6、-ST6、-SOtT6、-COtH、-COtT6、-T4NT7T8或-T4-N(T10)-T5-T6)取代;或(d)任选取代的烷基(尤其是被一个或多个-OH、-COtH、-COtT6、-T4-NT7T8、-T4-N(T10)-T5-H或-T4-N(T10)-T5-T6取代);
R1是H或烷基;
R2是(a)杂芳基(更优选噻唑基或噁唑基),任选地被一个或多个基团T1、T2、T3(优选包括H、烷基、卤代烷基、卤素、杂芳基、氰基、-C(O)tT6、-OT6、-T4NT7T8)取代;(b)被1-3个基团T1、T2、T3(优选包括杂芳基(优选咪唑基、噁唑基或噻唑基,各自任选地进一步被取代)、氰基、-C(O)tT6、-S(O)tN(T9)T6、卤素、烷基和卤代烷基)取代的芳基;或(c)与杂环基环稠合的芳基(例如,通过芳基环结合的2,3-二氢-1H-吲哚,通过芳基环结合的喹啉基(尤其是喹啉-6-基),通过芳基环结合的喹唑啉基(尤其是喹唑啉-7-基),通过芳基环结合的噌啉基(尤其是噌啉-6-基),通过芳基环结合的异喹啉基(尤其是异喹啉-6-基),和通过芳基环结合的酞嗪基(尤其是酞嗪-6-基),其中该组合环系可以任选地被1-3个基团T1、T2、T3(尤其是卤素、OH、OT6、烷基、-COtH、-COtT6或-C(O)NT7T8)取代;
R3是H或任选取代的烷基(尤其是被一个或多个-OH或-OT6取代);
R4是(a)氢;(b)(芳基)烷基,其中该芳基任选地独立被一个或多个基团T1a、T2a、T3a(尤其是任选取代的烷基、卤素、氰基、硝基、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4NT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代;(c)(杂芳基)烷基,其中该杂芳基任选地独立被一个或多个基团T1a、T2a、T3a(尤其是任选取代的烷基、卤素、氰基、硝基、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4NT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代;(d)(杂环基)烷基,其中该杂环基任选地独立被一个或多个基团T1a、T2a、T3a(尤其是任选取代的烷基、卤素、氰基、硝基、氧代、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4NT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代;(e)任选地独立被一个或多个基团T1a、T2a、T3a(尤其是-OH、-OT6、-COtH、-COtT6、-T4NTT8或-T4-N(T10)-T5-T6)取代的烷基;(f)任选地独立被一个或多个基团T1a、T2a、T3a(尤其是任选取代的烷基、任选取代的芳基、任选取代的杂芳基、任选取代的芳烷基、任选取代的杂环基、氰基、-OH、-OT6、-COtH、-COtT6、氧代、羟(烷基)、(烷氧基)烷基、-T4-N(T10)-T5-T6或-T4-NT2T8)取代的杂环基;
或者R3和R4与它们所连接的氮原子一起,结合形成一个4-8元的杂环基环(尤其是吡咯烷基、哌啶基、哌嗪基、吗啉基、二氮杂环庚基(diazapanyl)或1,4-二氧杂-8-氮杂螺[4,5]癸烷-8-基),它可任选地被1-3个基团T1a、T2a、T3a(尤其是任选取代的烷基、任选取代的芳基、任选取代的杂芳基、任选取代的芳烷基、任选取代的杂环基、氰基、-OH、-OT6、-COtH、-COtT6、氧代、羟(烷基)、(烷氧基)烷基、-T4-N(T10)-T5-T6或-T4-NT7T8)取代;
R5是氢或烷基;
R6是(a)氢;(b)(芳基)烷基,其中该芳基任选地独立被一个或多个基团T1c、T2c、T3c(尤其是任选取代的烷基、卤素、氰基、硝基、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代;(c)(杂芳基)烷基,其中该杂芳基任选地独立被一个或多个基团T1c、T2c、T3c(尤其是任选取代的烷基、卤素、氰基、硝基、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代;(d)(杂环基)烷基,其中该杂环基任选地独立被一个或多个基团T1c、T2c、T3c(尤其是任选取代的烷基、卤素、氰基、硝基、氧代、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代;(e)任选地独立被一个或多个基团T1c、T2c、T3c(尤其是-OH、-OT6、-COtH、-COtT6、-T4NT7T8或-T4-N(T10)-T5-T6)取代的烷基;(f)任选地独立被一个或多个基团T1c、T2c、T3c(尤其是任选取代的烷基、任选取代的芳基、任选取代的杂芳基、任选取代的芳烷基、任选取代的杂环基、氰基、-OH、-OT6、-COtH、-COtT6、氧代、羟(烷基)、(烷氧基)烷基、-T4-N(T10)-T5-T6或-T4-NT7T8)取代的杂环基;
或者R5和R6与它们所连接的氮原子合起来形成一个4-8元的杂环基环(尤其是吡咯烷基、哌啶基、哌嗪基、吗啉基、二氮杂环庚烷基(diazapanyl)或1,4-二氧杂-8-氮杂螺[4,5]癸烷-8-基),它可任选地被1-3个基团T1c、T2c、T3c(尤其是任选取代的烷基、任选取代的芳基、任选取代的杂芳基、任选取代的芳烷基、任选取代的杂环基、氰基、-OH、-OT6、-COtH、-COtT6、氧代、羟基(烷基)、(烷氧基)烷基、-T4-N(T10)-T5-T6或-T4-NT7T8)取代。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以上化合物,其中:
Z是(a)卤素,烷氧基,卤代烷基,-NR3R4,-C(O)OR4,-C(O)NR3R4;(b)芳基或杂芳基,它们各自任选地被一或多个T1a、T2a或T3a取代,该T1a、T2a或T3a选自氰基、任选取代的烷基、(羟)烷基、-OH、-OT6、-ST6、-SOtT6、-COtH、-COtT6、-T4NT7T8或-T4-N(T10)-T5-T6,其中T4是键或-C(O)-,T5是-C(O)-或-C(O)O-,T6是烷基或卤代烷基;T7和T8独立地是H,任选被环烷基、杂芳基、羟基或-NT7T8环烷基取代的烷基,或任选被卤素取代的芳基;或者T7和T8与它们所连接的氮原子合起来形成一个任选被(羟基)烷基、-COtH或-COtT6取代的杂环基环;T10是氢;(c)任选被一个或多个-OH、-COtH、-COtT6、-T4-NT7T8、-T4-N(T10)-T5-H或-T4-N(T10)-T5-T6取代的烷基,其中;T4是-C(O)-,T5是-亚烷基-O-,T6是烷基,T7和T8独立地是H、烷基、环烷基、芳基、(芳基)烷基(任选地如R4的定义中所述地被取代)或杂环基(任选地如结合形成杂环基环的R3和R4的定义中所述地被取代);T10是H;
J是(a)H,或(b)烷基或烯基,它们各自任选地被一个或多个-OH、-OT6、-COtH、或-COtT6取代,其中T6是烷基;
L是(a)H,或(b)卤素、烷氧基、卤代烷基、-NR5R6,-C(O)OR4b、-C(O)NR5R6;(c)芳基或杂芳基,它们各自任选地被一个或多个T1c、T2c、T3c取代,该T1c、T2c、T3c选自氰基、任选取代的烷基(尤其是被-COtH或-COtT6取代)、(羟)烷基、-OH、-OT6、-ST6、-SOtT6、-COtH、-COtT6、-T4NT7T8或-T4-N(T10)-T5-T6,其中T4是键或-C(O)-,T5是-C(O)-或-C(O)O-,T6是烷基或卤代烷基;T7和T8独立地是H,任选被环烷基、杂芳基、羟基或-NT7T8取代的烷基;环烷基;或任选被卤素取代的芳基;或者T7和T8与它们所连接的氮原子合起来形成一个任选被(羟基)烷基、-COtH或-COtT6取代的杂环基环;T10是氢;(d)任选被一个或多个-OH、-COtH、-COtT6、-T4-NT7T8、-T4-N(T10)-T5-H或-T4-N(T10)-T5-T6取代的烷基,其中T4是-C(O)-,T5是-亚烷基-O-,T6是烷基,T7和T8独立地是H、烷基、环烷基、芳基、(芳基)烷基(任选地如R4的定义中所述被取代)或杂环基(任选地如结合形成杂环基环的R3和R4的定义中所述被取代),T10是H;
R1是H或烷基;
R2是(a)杂芳基(更优选噻唑基或噁唑基),任选地被1-3个基团T1、T2、T3(优选包括H、烷基、卤代烷基、卤素、杂芳基、氰基、-C(O)tT6、-OT6、-T4NT7T8)取代;(b)被1-3个基团T1、T2、T3(优选包括杂芳基(优选咪唑基、噁唑基或噻唑基,各自任选地进一步被取代)、氰基、-C(O)tT6、-SOtN(T9)T6、卤素、烷基和卤代烷基)取代的芳基;或(c)与杂环基环稠合的芳基(例如,通过芳基环结合的2,3-二氢-1H-吲哚),其中该组合环系可以任选地被1-3个基团T1、T2、T3(尤其是卤素、-OH、-OT6、烷基、-COtH、-COtT6或-C(O)NT7T8)取代;
R3是H或任选取代的烷基(尤其是被一个或多个-OH或-OT6取代);
R4是(a)氢;(b)(芳基)烷基,其中该芳基任选地独立被一个或多个基团T1a、T2a、T3a(选自任选取代的烷基、卤素、氰基、硝基、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4NT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代,其中T4是键、-SO2-或-C(O)-,T5是-SO2-或-亚烷基-O-,T6是烷基或环烷基,T7和T8独立地是H或烷基,T9和T10是氢;(c)(杂芳基)烷基,其中该杂芳基任选地独立被一个或多个基团T1a、T2a、T3a(选自任选取代的烷基、卤素、氰基、硝基、氧代、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4NT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代,其中T4是键、-SO2-或-C(O)-,T5是-SO2-或-亚烷基-O-,T6是烷基或环烷基,T7和T8独立地是H或烷基,T9和T10是氢;(d)(杂环基)烷基,其中该杂环基任选地独立被一个或多个基团T1a、T2a、T3a(选自任选取代的烷基、卤素、氰基、硝基、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-T4NT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基)取代,其中T4是键、-SO2-或-C(O)-,T5是-SO2-或-亚烷基-O-,T6是烷基或环烷基,T7和T8独立地是H或烷基,T9和T10是氢;(e)任选地独立被一个或多个基团T1a、T2a、T3a(选自-OH、-OT6、-COtH、-COtT6、-T4NT7T8或-T4-N(T10)-T5-T6)取代的烷基,其中T4是键,T5是-C(O)-,T6是烷基,T7和T8独立地是H或烷基,T10是氢;(f)任选地独立被一个或多个基团T1a、T2a、T3a取代的杂环基,基团T1a、T2a、T3a选自任选取代的烷基(尤其是被-T4NT7T8取代)、任选取代的芳基(尤其是被卤素或卤代烷基取代)、氰基、-OH、-OT6、-COtH、-COtT6、氧代、羟(烷基)、(烷氧基)烷基、-T4-N(T10)-T5-T6或-T4-NT7T8,其中T4是键或-C(O)-,T5是-C(O)-、-SO2-或-亚烷基-C(O)O-,T6是烷基、烷氧基或杂芳基,T7和T8独立地是H、烷基或环烷基,或者T7和T8与它们所连接的氮原子合起来形成一个任选取代的杂环基环;
或者R3和R4与它们所连接的氮原子合起来形成一个杂环基环,选自吡咯烷基、哌啶基、哌嗪基、吗啉基、二氮杂环庚烷基(diazapanyl)或1,4-二氧杂-8-氮杂螺[4,5]癸烷-8-基,它们任一个可任选地独立被1-3个基团T1a、T2a、T3a取代,基团T1a、T2a、T3a选自任选取代的烷基(尤其是被-T4NT7T8取代)、任选取代的芳基(尤其是被卤素或卤代烷基取代)、氰基、-OH、-OT6、-COtH、-COtT6、氧代、羟(烷基)、(烷氧基)烷基、-T4-N(T10)-T5-T6或-T4-NT7T8,其中T4是键或-C(O)-,T5是-C(O)-、-SO2-或-亚烷基-C(O)O-,T6是烷基、烷氧基或杂芳基,T7和T8独立地是H、烷基或环烷基,或者T7和T8与它们所连接的氮原子合起来形成一个任选取代的杂环基环;
R5是氢或烷基;
R6是(a)氢;(b)(芳基)烷基,其中该芳基任选地独立被一个或多个基团T1c、T2c、T3c取代,基团T1c、T2c、T3c选自任选取代的烷基、卤素、氰基、硝基、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-TNT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基,其中T4是键、-SO2-或-C(O)-,T5是-SO2-或-亚烷基-O-,T6是烷基或环烷基,T7和T8独立地是H或烷基,T9和T10是氢;(c)(杂芳基)烷基,其中该杂芳基任选地独立被一个或多个基团T1c、T2c、T3c取代,基团T1c、T2c、T3c选自任选取代的烷基、卤素、氰基、硝基、氧代、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-SOtN(T9)(T6)、-T4NT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基,其中T4是键、-SO2-或-C(O)-,T5是-SO2-或-亚烷基-O-,T6是烷基或环烷基,T7和T8独立地是H或烷基,T9和T10是氢;(d)(杂环基)烷基,其中该杂环基任选地独立被一个或多个基团T1c、T2c、T3c取代,基团T1c、T2c、T3c选自任选取代的烷基、卤素、氰基、硝基、(羟)烷基、-OH、-OT6、-ST6、-COtH、-COtT6、-SO3H、-SOtT6、-T4NT7T8、-T4-N(T10)-T5-T6、杂环基或杂芳基,其中T4是键、-SO2-或-C(O)-,T5是-SO2-或-亚烷基-O-,T6是烷基或环烷基,T7和T8独立地是H或烷基,T9和T10是氢;(e)任选地独立被一个或多个基团T1c、T2c、T3c取代的烷基,基团T1c、T2c、T3c选自-OH、-OT6、-OCtH、-COtT6、-T4NT7T8或-T4-N(T10)-T5-T6,其中T4是键,T5是-C(O)-,T6是烷基,T7和T8独立地是H或烷基,T10是氢;(f)任选地独立被一个或多个基团T1c、T2c、T3c取代的杂环基,基团T1c、T2c、T3c选自任选取代的烷基(尤其是被-T4NT7T8取代)、任选取代的芳基(尤其是被卤素或卤代烷基取代)、氰基、-OH、-OT6、-COtH、-COtT6、氧代、羟(烷基)、(烷氧基)烷基、-T4-N(T 10)-T5-T6或-T4-NT7T8,其中T4是键或-C(O)-,T5是-C(O)-、-SO2-或-亚烷基-C(O)O-,T6是烷基、烷氧基或杂芳基,T7和T8独立地是H、烷基或环烷基,或者T7和T8与它们所连接的氮原子合起来形成一个任选取代的杂环基环;
或者R5和R6与它们所连接的氮原子合起来形成一个杂环基环,选自吡咯烷基、哌啶基、哌嗪基、吗啉基、二氮杂环庚烷基(diazapanyl)或1,4-二氧杂-8-氮杂螺[4,5]癸烷-8-基,它们任一个可任选地独立被1-3个基团T1a、T2a、T3a取代,基团T1a、T2a、T3a选自任选取代的烷基(尤其是被-T4NT7T8取代)、任选取代的芳基(尤其是被卤素或卤代烷基取代)、氰基、-OH、-OT6、-COtH、-COtT6、氧代、羟(烷基)、(烷氧基)烷基、-T4-N(T10)-T5-T6或-T4-NT7T8,其中T4是键或-C(O)-,T5是-C(O)-、-SO2-或-亚烷基-C(O)O-,T6是烷基、烷氧基或杂芳基,T7和T8独立地是H、烷基或环烷基,或者T7和T8与它们所连接的氮原子合起来形成一个任选取代的杂环基环;
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:
2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(3,4-二甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐;
2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[[[4-(甲基磺酰基)苯基]甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[[(4-甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(3-甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(2-甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-(1-哌嗪基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[[(2-乙氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(2,5-二甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(3,5-二甲氧基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(2,6-二甲基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[[4-(甲氧羰基)苯基]甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(3-溴苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[(1,3-苯并间二氧杂环戊烯-5-基甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[甲基(3-吡啶基甲基)氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
4-甲基-2-[[4-(1-哌嗪基)-6-[[[4-(1,2,3-噻二唑-4-基)苯基]甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[[[3-(环戊氧基)-4-甲氧基苯基]甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[(苯基甲基)氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
4-甲基-2-[[4-(4-甲基-1-哌嗪基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-(4-羟基-1-哌啶基)-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[[2-(1-甲基乙氧基)乙基]氨基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[[3-(氨基羰基)-1-哌啶基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(2-(1H-咪唑-4-基)乙基]氨基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[[[4-(甲磺酰基)苯基]甲基]氨基]-6-[[3-(4-吗啉基)丙基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[(2-甲氧基-1-甲基乙基)氨基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[[[4-(甲磺酰基)苯基]甲基]氨基]-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[4-(2-羟乙基)-1-哌嗪基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-(氨基羰基)-1-吡咯烷基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[甲基(3-吡啶基甲基)氨基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[4-(羟甲基)-1-哌啶基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-(二乙基氨基)乙基]甲基氨基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[[[4-(甲磺酰基)苯基]甲基]氨基]-6-[[3-(2-氧代-1-吡咯烷基)丙基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[3-(羟甲基)-1-哌啶基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-(4-甲基-1-哌嗪基)-6-[[[(4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[[2-[(乙酰基氨基)乙基]氨基]-6-[[[(4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(4-乙基-1-哌嗪基)-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(4-乙酰基-1-哌嗪基)-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[2-(二甲基氨基)乙基]氨基]-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(3-羟基-1-吡咯烷基)-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[(4-羟基丁基)氨基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[(2,3-二羟基丙基)氨基-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[(4-氨基-1-哌啶基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-3-(羟甲基)-1-哌啶基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(4-二甲基氨基-1-哌啶基)-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-(甲基氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4,6-双-(4-甲基哌嗪-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(3-羟甲基哌啶-1-基)-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[4-(4-甲基哌嗪-1-基)-6-吗啉-4-基-嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
2-[4-(4-氨基哌啶-1-基)-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4,6-双-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-氧代哌啶-1-基)-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-甲基-4-羟基哌啶-1-基)-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[(4-羟基哌啶-1-基)-6-(4-二甲基甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟甲基哌啶-1-基)-6-(4-二甲基甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(3-羟甲基哌啶-1-基)-6-(4-二甲基甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟甲基哌啶-1-基)-6-(4-羟基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[4-(4-羟基哌嗪-1-基)-6-吗啉-4-基-嘧啶-2-基氨基]-噻唑-5-甲酸乙酯;
2-[[(4-[[[4-(甲磺酰基)苯基]甲基]氨基]-6-氯-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-氯-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(二甲基氨基)-1-哌啶基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[1-哌嗪基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(4-氨基-1-哌啶基)-6-[[[4-(甲磺酰基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(羟甲基)-1-哌啶基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基]-6-(3-氧代哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[1-吗啉基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[[(1,1-二氧化-3-氧代-1,2-苯并异噻唑-2-(3H)-基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(乙磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(羟基磺酰基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-甲基-3-氧代哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-(二甲基氨基)-哌嗪-1-基)-6-(4-((1-吡咯烷基)羰基甲基)哌嗪-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-氨基-1-哌啶基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(羟甲基)-1-哌啶基)-6-[4-[四唑-5-基]-4-羟基哌啶-1-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基-1-哌嗪基]-6-[N-甲基-N-[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(羟基磺酰基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(4-氰基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐(1∶1);
2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-吗啉基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基)-6-[(1-氧杂-3,8-二氮杂螺[4,5]癸-2,4-二酮-8-基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(2-(二甲基氨基)乙基)-哌嗪-1-基]-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基)-6-[甲基(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-3-羟甲基哌啶-1-基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(3,4-二氢-6,7-二羟基-2(1H)-异喹啉基)-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐(1∶1);
2-[[4-[4-[(甲氧乙酰基)氨基]-1-哌啶基]-6-[[4-(甲磺酰基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(3,4-二甲氧基苯基)甲基]氨基]-6-[4-(二甲基氨基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基噻唑甲酸乙酯;
2-[[4-[4-(羟乙基)哌啶-1-基]-6-[4-(二甲基氨基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(二甲基氨基)-1-哌啶基]-6-[甲基(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(羟基)哌啶-1-基]-6-[4-(甲氧羰基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(羟基)哌啶-1-基]-6-[4-(甲基)-4-(羟基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(3-氧代哌嗪-1-基]-6-(4-甲基哌嗪-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;2-[[4-[[(4-氰基苯基)甲基]氨基]-6-[4-(二甲基氨基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[[(3-硝基苯基)甲基]氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑甲酸乙酯三氟乙酸盐(1∶1);
2-[[4-(4-羟基-1-哌啶基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(二甲基氨基)-哌嗪-1-基]-6-(4-甲基哌嗪-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(二甲基氨基)-哌啶-1-基]-6-(3-(氨基羰基)-1-哌嗪基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(2-羟乙基)-哌嗪-1-基]-6-(4-甲基-1-哌嗪基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[4-(氨基羰基)-1-哌啶基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(羟甲基)-1-哌啶基]-6-[N-甲基-N-(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基哌嗪-1-基]-6-[[(3,4-二甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[哌嗪-1-基]-6-[[(4-羧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-羟甲基哌啶-1-基]-6-[[N-[(3,4,5-三甲氧基苯基)甲基]]-N-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-(羟基哌啶-1-基)-6-(4-羧基哌啶-1-基)嘧啶-2-基]氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[哌嗪-1-基]-6-[[(3,4-二甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(4-甲酰基-1-哌嗪基]-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-(羟基)-4-(4-氯苯基)哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[[4-[4-二甲基氨基-1-哌啶基]-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[哌嗪-1-基]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-吗啉基]-6-[4-[四唑-5-基]-4-羟基哌啶-1-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(1,1-二氧化-3-氧代-1,2-苯并异噻唑-2-(3H)-基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-(1-甲基-1-羟乙基)哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌啶基]-6-[[N-甲基-N-(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟甲基-1-哌啶基]-6-[[(4-(乙磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[4-[四唑-5-基]-4-羟基哌啶-1-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-叔丁氧羰基氨基-1-哌啶基]-6-[[N-[(3,4,5-三甲氧基苯基)甲基]]-N-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(4-氰基苯基)甲基]氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐(1∶1);
2-[[4-[4-[[(2-乙氧基-2-氧代乙基)氨基]羰基]-1-哌嗪基]-6-[甲基(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐(1∶1);
2-[4-(4-羟基哌啶-1-基)-6-(3-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-羟基-4-苯基-1-哌啶基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[[4-[4-吗啉基]-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[(四氢-2-呋喃基)甲基]氨基]-6-[[N-[(3,4,5-三甲氧基苯基)甲基]]-N-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-吗啉基]-6-[[(4-(羟基磺酰基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[双-4,6-(4-氰基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[4-(环戊基氨基羰基)-1-哌嗪基]-6-[N-甲基-N-(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(2-甲氧基乙基)-哌嗪-1-基)-6-(4-甲基-1-哌嗪基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(3-羧基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[4-甲基哌嗪-1-基]-6-[3-(乙酰胺基)-1-吡咯烷基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[N-甲基-N-(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-甲基-3-氧代-1-哌嗪基]-6-[4-甲基-1-哌嗪基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌啶基]-6-(4-二甲基氨基-1-哌啶基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[1-哌嗪基]-6-[[N-甲基-N-(2-呋喃甲基)]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(4-甲氧基羰基苯基)甲基]氨基]-6-(4-二甲基-1-哌啶基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐(1∶1);
2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(甲磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(丙磺酰基氨基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌啶基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[双-4,6-(4-羟基-4-甲基-1-哌啶基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[[4-[4-二甲基氨基-1-哌啶基]-6-[[(2-氧代-1-吡咯烷基)丙基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(异丙基磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(3-羟甲基-1-哌啶基)嘧啶-2-基]氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[[4-[4-羟基-1-哌啶基]-6-[[(2-(4-吗啉基)乙基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[[[4-(乙基氨基磺酰基)苯基]甲基]氨基]-6-甲氧基-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸甲酯三氟乙酸盐(1∶1);
2-[[4-[4-吗啉基]-6-[(1-氧杂-3,8-二氮杂螺[4,5]癸-2,4-二酮-8-基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(乙磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[叔丁氧基羰基-1-哌嗪基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌啶基]-6-[[(3,4-二甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-乙氧羰基-1-哌嗪基]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[[(4-(环丙基磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟甲基-1-哌啶基]-6-[[(4-(甲磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-二甲基氨基-1-哌嗪基)-6-(4-叔丁氧羰基氨基-1-哌啶基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-甲氧基甲基-1-哌啶基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-羟乙基-1-哌啶基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-(羟基)-4-(3-三氟甲基苯基)哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[4-吗啉基]-6-[4-[1-甲基-1-羟乙基]-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[[3-吡啶基]氧基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基-1-哌嗪基]-6-[(1,4-二氧杂螺[4,5]癸-8-基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-吗啉基]-6-[[(4-(甲磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[(1-氧杂-3,8-二氮杂螺[4,5]癸-2,4-二酮-8-基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(羧基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-(羟基)-4-(4-溴苯基)哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[4-吗啉基]-6-[[(4-乙磺酰胺基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基]-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[(3,4-二甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲酰基-1-哌嗪基]-6-[[(3-(5-(1H)四唑基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(羟甲基)-1-哌啶基]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基-1-哌嗪基]-6-[[(2,5-二甲基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(2-氧代-1-吡咯烷基)丙基]氨基]-6-[N-甲基-N-(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[1-吗啉基]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基-1-哌嗪基]-6-[4-[甲磺酰胺基]-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(2,5-二甲基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-(4-吗啉基)-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-(4-羟基哌啶-1-基)-6-(3-羟基-1-哌啶基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[[4-(4-甲基-1-哌嗪基)-6-[甲基(3-吡啶基甲基)氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[3-氧代-1-哌嗪基]-6-[[(2-(5-(1H)四唑基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[(2-呋喃甲基)氨基]-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐(1∶1);
2-[[4-[[(3,4-二甲氧基苯基)甲基]氨基]-6-(4-吗啉基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[甲基(3-吡啶基甲基)氨基]-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[(4-羟基-1-哌啶基)]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-[(4-(羟基)-4-(苯基甲基)哌啶-1-基)]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-二甲基氨基-1-哌嗪基)-6-[[2-(1-吗啉基)乙基]氨基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(3-吡啶基甲基)]氧基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌啶基]-6-[[(2,6-二甲基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(甲基磺酰胺基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(4-(丙基磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌啶基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(3,4-二氢-6,7-二甲氧基-2(1H)-异喹啉基)-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲酰基-1-哌嗪基]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(4-羧基苯基)甲基]氨基]-6-[4-(羟甲基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(4-羧基苯基)甲基]氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯,一盐酸盐;
4-甲基-2-[[4-(4-甲基-1-哌嗪基)-6-[[(四氢-2-呋喃基)甲基]氨基]-2-嘧啶基]氨基]-5-噻唑甲酸乙酯,;
2-[[4-[[(4-羧基苯基)甲基]氨基]-6-[3-(羟甲基)-1-哌啶基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[[4-[[(2-甲氧基乙基)氨基]羰基]苯基]甲基]氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯,三氟乙酸盐(1∶1);
2-[4,6-双-(1-吗啉基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[甲基(3-吡啶基甲基)氨基]-6-[4-吗啉基]-2吡啶基甲基]氨基]-5-噻唑甲酸乙酯;
2-[[4-[3-(氨基羰基)-1-哌嗪基]-6-[[[4-(甲氧羰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-氯-6-[(1-氧杂-3,8-二氮杂螺[4,5]癸-2,4-二酮-8-基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-(羟甲基)-1-哌啶基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(羟甲基)-1-哌啶基]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(羟甲基)-1-吡咯烷基]-6-[[N-甲基-N-(5-四唑基甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[[4-[甲基(苯甲基)氨基]-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-5-噻唑甲酸乙酯;
2-[[4-(二甲基氨基)-6-[[[4-(甲磺酰基)苯基]甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-1-哌啶基]-6-[[(3-(5-(1H)四唑基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟甲基-1-哌啶基]-6-[[(4-(丙磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟甲基-1-哌啶基]-6-[[(4-(环丙基磺酰胺基)苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[3-(羟甲基)-1-哌啶基]-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-四氢吡喃基]氧基-6-[[N-[(3,4,5-三甲氧基苯基)甲基]]-N-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基-1-哌嗪基]-6-[(4-甲氧基苯基)氧基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
4-甲基-2-[4-(4-甲基哌嗪-1-基)-6-[[[4-(氨基磺酰基)苯基]甲基]氨基]嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
2-[4-异丙基-6-(4-氨磺酰-苄氨基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[4-(4-氨磺酰-苄氨基)-6-甲基嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
4-甲基-2-[4-(4-氨磺酰-苄氨基)-6-羟甲基嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
4-甲基-2-[4-(4-甲基哌嗪-1-基)-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[4-[(四氢呋喃-2-基甲基)氨基]-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
4-甲基-2-[4-吗啉-4-基-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
2-[4-(3-氨甲酰哌啶-1-基)-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟甲基哌啶-1-基)-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(2-羟甲基-1-吡咯烷基)-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(3-N,N-二乙基氨甲酰-1-哌啶基)-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(3-羟基-1-吡咯烷基)-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
4-甲基-2-[[[2-[4-吗啉-4-基]乙基]氨基]-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
4-甲基-2-[[[4-羟基]丁基]氨基]-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]噻唑-5-甲酸乙酯;
2-[4-(4-甲酰基-1-哌嗪基)-6-[4-(1H-四唑-5-基)苄氨基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[[(4-氯苯基)甲基]氨基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(4-氨基磺酰基苯基)甲基]氨基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-吗啉代-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(3,4-二甲氧基苯基)甲基]氨基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(1,4-二氧杂-8-氮杂螺[4,5]癸-8-基)-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基-4-苯基哌啶基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(4-甲基磺酰基苯基)甲基]氨基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-羟基哌啶基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-乙氧基羰基哌啶基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-哌啶基-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[N-甲基哌嗪基-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[N-(2-呋喃基羰基)哌嗪基-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[N-乙酰基-[1,4-二氮杂环庚三烯基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[N-甲基-N-(N-甲基-4-哌啶基)氨基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[N-甲基-[1,4]-二氮杂环庚三烯基]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-N,N-二甲氧基乙基氨基-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[(1’,4)-联哌啶]-6-(5-噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(3,4,5-三甲氧基苯基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[(4-(4-羟基哌啶-1-基)-6-[4-(1H-四唑-5-基)苯基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-吡啶-3-基嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-甲磺酰基苄氨基)-6-吡啶-3-基嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-嘧啶-4-基嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-氰基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-乙酰苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟甲基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羟基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-甲磺酰基苄氨基)-6-(3,4,5-三甲氧基苯基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯
2-[4-(4-甲亚磺酰基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-(氨基)苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧甲基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-(三氟甲基羰基氨基)苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-(乙氧基羰基甲基)苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(1,2,3,6-四氢吡啶-4-基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(3-(氰基)苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-(甲氧基羰基)苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(2-(甲氧基)-5-吡啶基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-叔丁氧基羰基-1,2,3,6-四氢吡啶-4-基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(1,4-二氧杂螺[4,5]癸-7-烯-8-基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-甲基-1-哌嗪基)-6-(3,4,5-三甲氧基苯基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-吗啉基)-6-(3,4,5-三甲氧基苯基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-吗啉基)-6-(3-吡啶基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(哌啶-4-基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[4-羟基哌啶基]-6-(3,5-二甲基-4-异噁唑基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-叔丁氧羰基氨基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-氰基苯基)-6-(4-甲磺酰基-苄氨基)嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-甲磺酰基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-甲硫烷基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-(4-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-(3-氧代-哌嗪-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-(4-甲基哌嗪-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-吗啉-4-基-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-(4-甲基-[1,4]二氮杂环庚烷-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-(3-R-羟基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-(3-羟甲基哌啶-1-基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-乙酰基-[1,4]二氮杂环庚烷-1-基)-6-(4-羧基苯基)嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-[N-甲基-N-(1-N-甲基哌啶-4-基)-氨基]-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-哌嗪-1-基-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4-(4-羧基苯基)-6-(4-氨磺酰-苄氨基)-嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[5-烯丙基[4-(氨基磺酰基)苯基]甲基]氨基]-6-氯-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-5-甲基-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐(1∶3);
2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-5-甲基-6-(4-吗啉基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[5-烯丙基[4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-甲基哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[5-[2-[2-甲基丙-3-烯]]-4-[4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-甲基哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[[(3,4,5-(三甲氧基)苯基)甲基]氨基]-5-甲基-6-(1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐;
2-[[4-[[5-[2,3-丙二醇][4-(氨基磺酰基)苯基]甲基]氨基]-6-(4-甲基哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[[3,4,5-(三甲氧基)苯基]甲基]氨基]-5-甲基-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯三氟乙酸盐;
2-[[4-[[5-[2-[2-甲基丙-3-烯]]-4-[4-(氨基磺酰基)苯基]甲基]氨基]-6-氯-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[[4-(氨基磺酰基)苯基]甲基]氨基]-5-甲基-6-(4-叔丁氧羰基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[N-[[3,4,5-(三甲氧基)苯基]甲基]-N-甲基氨基]-5-甲基-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4,6-双-(4-羟基-哌啶-1-基)-5-甲基嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4,6-双-(3-氧代-哌嗪-1-基)-5-[乙氧羰基甲基]嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[4,6-双-(4-羟基-哌啶-1-基)-5-甲氧基嘧啶-2-基氨基]-4-甲基噻唑-5-甲酸乙酯;
2-[[4-[N-[[3,4,5-(三甲氧基)苯基]甲基]-N-甲基氨基]-5-甲氧基-6-(4-甲基-1-哌嗪基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[3-吡啶基]甲氧基]-5-(2-丙烯基-6-(4-吗啉基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[(4-乙氧羰基甲基-6-吗啉-4-基-嘧啶-2-基)氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[(4-乙氧羰基甲基-6-[3-氧代-1-哌嗪基]-嘧啶-2-基)氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[(4-羧甲基-6-吗啉-4-基-嘧啶-2-基)氨基]-4-甲基-5-噻唑甲酸;
2-[4-吗啉-4-基-6-[(3,4,5-三甲氧基苯基氨甲酰基)甲基]-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-氧代-2-(3-氧代-哌嗪-1-基)乙基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(4-氨磺酰基苄氨基)-6-[(4-氨磺酰基苄基氨甲酰基)甲基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[2-(1,4-二氧杂-8-氮杂螺[4,5]癸-8-基)-2-氧代乙基]-6-(4-氨磺酰基-苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[(4-氯苯基)甲基-氨甲酰]甲基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-(4-羟基哌啶-1-基)-2-氧代-乙基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-(4-乙氧羰基-哌啶-1-基)-2-氧代-乙基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(2-氧代-2-哌啶-1-基乙基)-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-[4-(呋喃-2-羰基)-哌嗪-1-基]-2-氧代乙基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[(环己基甲基-氨甲酰)甲基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-(4-乙酰基-[1,4]二氮杂环庚-1-基)-2-氧代乙基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[甲基-(1-甲基哌啶-4-基)氨甲酰]甲基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-(4-甲基-[1,4]二氮杂环庚-1-基)-2-氧代乙基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[[双-(2-甲氧基乙基)氨甲酰]甲基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(2-[1,4′]联哌啶-1’-基-2-氧代乙基)-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[2-(4-羟基-4-苯基哌啶-1-基)-2-氧代乙基]-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-乙氧羰基-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-羧基-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(羧甲基-氨甲酰)-6-(4-氨磺酰基苄氨基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-甲硫烷基-苄基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-(4-羟基哌啶-1-基)-6-(4-甲亚磺酰基-苄基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-(4-羟基哌啶-1-基)-6-(4-甲基磺酰基-苄基)-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基-1-哌嗪基]-6-[N-甲基-N-[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-三氟甲基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基哌嗪-1-基]-6-(N-甲基-N-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-氰基噻唑;
2-[[4-[4-甲基哌嗪-1-基]-6-甲基-6-[[(3,4,5-三甲氧基苯基)甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸2-甲氧基乙酯;
2-[[4-[4-羟基哌啶-1-基]-6-[N-甲基-[[N-[(3,4,5-三甲氧基苯基)甲基][N-甲基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸丁酯;
2-[[4-[1-吗啉基]-6-[[2-[1-吗啉基]乙基]氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸丁酯;
2-[[4-[4-甲基-1-哌嗪基]-6-[[N-[(3,4,5-三甲氧基苯基)甲基]]-N-(甲基)氨基]-2-嘧啶基]氨基]-4-异丙基-5-噻唑甲酸乙酯;
2-[[4-[4-甲基-1-哌嗪基]-6-[[N-[(3,4,5-三甲氧基苯基)甲基]]-N-(甲基)氨基]-2-嘧啶基]氨基]-4-甲基-5-噻唑甲酸,甲酰胺;
2-[4-[4-(2-二异丙基氨基-乙基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(3-二甲基氨基-丙基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(环己基甲基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(吡啶-4-基甲基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(异丁基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(N-环己基-N-甲基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(N-环丙基甲基-N-丙基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(4-乙氧羰基哌啶-1-氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(3-羟甲基-哌啶-1-羰基)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(N-2-羟乙基-N-乙基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(硫代吗啉-1-羰基)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
2-[4-[4-(吗啉-1-羰基)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;和
2-[4-[4-(4-氯苯基氨甲酰)苯基]-6-(4-羟基哌啶-1-基)-嘧啶-2-基氨基]-4-甲基-5-噻唑甲酸乙酯;
或其立体异构体,药学上可接受的盐,或水合物。
在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:
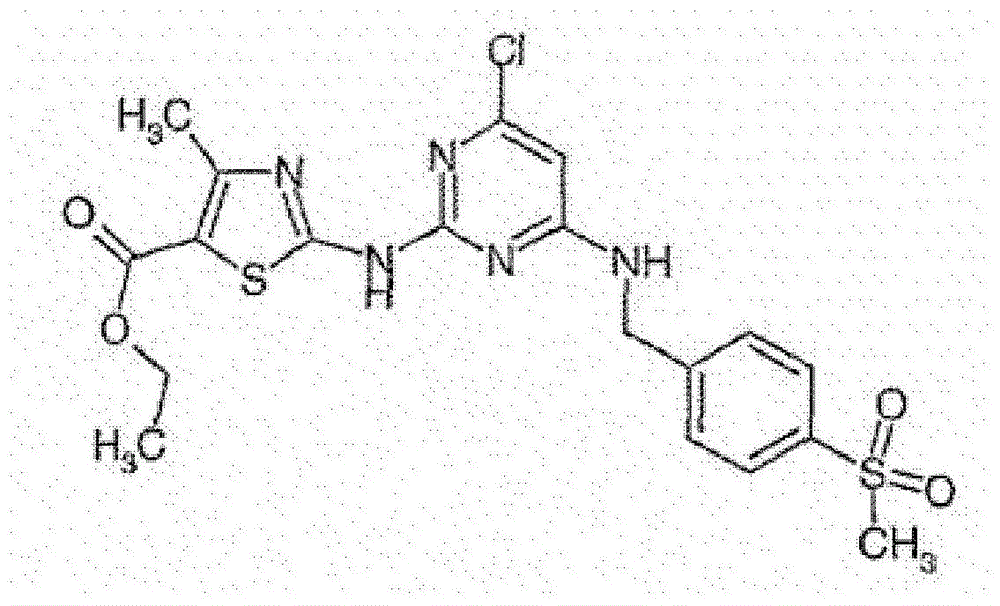












和

或其立体异构体,药学上可接受的盐或水合物。
这些化合物的制备描述在美国专利No.7,087,614,US 20030162802和WO 2002/102313中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在US2007/0129388和WO2007/063391中一般地或具体地公开的那些化合物,这些文献均特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
m是0,1或2;X是O,S或N-CN;R是F,Cl或CN;A是任选被C1-4烷基取代的C3-6亚环烷基;B是单键或C1-2亚烷基;或其药学上可接受的盐、溶剂化物、多晶形物或前药。
就以上化合物而言,术语“亚烷基”代表一个有1或2个碳原子的二价饱和烃链。亚烷基的实例包括亚甲基、亚乙基和甲基亚甲基,其中优选的是亚甲基。
术语“亚环烷基”代表有3-6个碳原子的二价饱和碳环。亚环烷基的实例包括亚环丙基(例如1,1-亚环丙基和顺-及反-1,2-亚环丙基)、亚环丁基(例如1,1-亚环丁基,顺-及反-1,2-亚环丁基,和顺-及反-1,3-亚环丁基)、亚环戊基(例如1,1-亚环戊基,顺-及反-1,2-亚环戊基,和顺-及反-1,3-亚环戊基)和亚环己基(例如1,1-亚环己基,顺-及反-1,2-亚环己基,顺-及反-1,3-亚环己基,和顺-及反-1,4-亚环己基)。优选的实例包括亚环丁基和亚环己基,更优选的是亚环丁基,还要优选的是1,3-亚环丁基,最优选的是反-1,3-亚环丁基。
术语“烷基”代表含有1-4个碳原子的直链或支链的一价饱和烃链。烷基的实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基和叔丁基。优选的实例包括甲基和乙基,尤其是甲基。
亚环烷基可任选地被C1-4烷基取代。该烷基取代基(如果存在)优选是甲基或乙基,更优选是甲基。该烷基取代基(如果存在)可以位于环上的任何位置,但优选位于1-位(即,与羧酸基团同一位置)。
优选的是,m是1或2,更优选是1。
优选的是,X是O或N-CN,更优选是O。
优选的是,R是F或Cl,更优选是Cl。
优选的是,A是任选被甲基取代的亚环丁基或亚环己基。更优选的是,A是亚环丁基。还要优选的是,A是1,3-亚环丁基,尤其是反-1,3-亚环丁基。
优选的是,B是单键或亚甲基。更优选的是,B是单键。
在另一实施方案中,可用于本发明方法的PDE 7抑制剂选自以下化合物:
顺-3-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]环丁烷甲酸;
反-3-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]环丁烷甲酸;
3-[(8′-氟-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基甲基]环丁烷甲酸;
反-3-[(8′-氰基-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基]环丁烷甲酸;
1-[(8′-氟-2′-氧代-2′,3′-二氢-1′H-螺[环己烷-1,4′-喹唑啉]-5′-基)氧基甲基]环丁烷甲酸;
反-3-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环庚基-1,4′-喹唑啉]-5′-基)氧基]环丁烷甲酸;和
反-3-[(8′-氯-2′-氧代-2′,3′-二氢-1′H-螺[环戊基-1,4′-喹唑啉]-5′-基)氧基]环丁烷甲酸;或其药学上可接受的盐、溶剂化物、多晶形物或前药。
以上化合物的制备描述在US 2007/0129388和WO 2007/063391中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂包括在Kadoshima-Yamaoka,K.等人,″ASB16165,a novel inhibitor for phosphodiesterase 7A(PDE7A),suppressesIL-12-induced IFN-g production by mouse activated T lymphocytes,″ImmunologyLetters 122:193-197,2009中描述的化合物ASB16165(1-环己基-N-[6-(4-羟基-1-哌啶基)-3-吡啶基]-3-甲基-1H-噻吩并[2,3-c]吡唑-5-甲酰胺一水合物),该文献特此并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

制备以上化合物的方法描述于WO2006/004040中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂包括在Yamamoto,S.等人,″The effects of a novel phosphodiesterase 7A and -4 dual inhibitor,YM-393059,on T-cell-related cytokine production in vitro and in vivo.″European Journalof Pharmacology 541:106-114,2006中描述的化合物YM-393059((±)-N-(4,6-二甲基嘧啶-2-基)-4-[2-(4-甲氧基-3-甲基苯基)-5-(4-甲基哌嗪-1-基)-4,5,6,7-四氢-1H-吲哚-1-基]苯磺酰胺二富马酸盐),该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在Martinez等人″Benzyl derivatives of 2,1,3-benzo-and benzothieno 3,2-aathiadiazine 2,2-dioxides:first phosphodiesterase 7 inhibitors,″J.Med.Chem.43:683-689,2000中一般地或具体地公开的那些化合物,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:
1-[(4-甲氧基苯基)羰基甲基]苯并噻吩并[3,2-a]-1,2,6-噻二嗪-4(3H)-酮2,2-二氧化物;和1-[(3,4-二氯苯基)甲基]-2,1,3-苯并噻二嗪-4(3H)-酮2,2-二氧化物。
以上化合物的制备描述在J.Med.Chem.43:683-689,2000中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在Castro,A等人,″CODES,a novel procedure for ligand-based virtual screening:PDE7 inhibitors asan application example,″J.Med.Chem.43:1349-1359,2008中一般地或具体地公开的那些化合物,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:


在另一实施方案中,可用于本发明方法的PDE7抑制剂具有下式:


以上化合物的取代基定义如下:
X=O或S,
R=H,苯基,4-甲氧基苯基,2,6-二氟苯基,2,3,4-三氟苯基,2-溴苯基,苄基,萘基或甲基。
在另一实施方案中,可用于本发明方法的PDE7抑制剂包括以下化合物:
5.2.4.3-(2,3,4-三氟苯基)-2-硫代-(1H)-喹唑啉-4-酮;
5.3.2.3-苯基-2-硫代-(1H)-噻吩并[3,2-d]嘧啶-4-酮;
5.3.3.3-(2,6-二氟苯基)-2-硫代-(1H)-噻吩并[3,2-d]嘧啶-4-酮;和
5.4.2.3-(2,6-二氟苯基)-2-硫代-(1H)-苯并[4,5]-噻吩并[3,2-d]嘧啶-4-酮。
以上化合物的制备描述在J.Med.Chem.43:1349-1359,2008中。
在另一实施方案中,可用于本发明方法的PDE7抑制剂包括Yang,G.等人,″Phosphodiesterase 7A-deficient mice have functional T cells,″J.Immunol171:6414-6420,2003中描述的BMS-586353,该文献特此全文并入本申请作为参考。
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在Pitts,W.J.等人,″Identification of purine inhibitors of phosphodiesterase7(PDE7)″,Bioorg.Med.Chem.Lett.14:2955-2958,2004,和Kempson,J.等人,″Fused pyrimidinebased inhibitors of phosphodiesterase 7(PDE7):synthesis and initialstructure-activity relationships,″Bioorg.Med.Chem.Lett.15:1829-1833,2005中一般地或具体地公开的那些化合物,上述文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

以上化合物的取代基定义如下:
R1是

或

R2是

或

其中乙基可以连接到7或9位。
在一项相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

和

在另一相关的实施方案中,可用于本发明方法的PDE7抑制剂具有下式:

其中,X=CH2,CH2CH2或OCH2;
Ar是

或

NRR’是
在另一实施方案中,可用于本发明方法的PDE7抑制剂选自在Kang,N.S.等人,″Docking and 3-D QSAR studies of dual PDE4-PDE7 inhibitors″,MolecularSimulation 33:1109-1117,2007中一般地或具体地公开的那些化合物,该文献特此全文并入本申请作为参考。在一项实施方案中,可用于本发明方法的PDE7抑制剂具有下式:


和

制备以上化合物的方法描述在Molecular Simulation 33:1109-1117,2007中。
多肽或肽抑制剂
在一些实施方案中,PDE7抑制剂包括分离的PDE7多肽或肽抑制剂,包括抑制PDE7活性的分离的天然肽抑制剂和合成的肽抑制剂。在本文中使用时,术语“分离的PDE7多肽或肽抑制剂”是指通过与PDE7结合,与PDE7竞争与底物的结合,和/或直接与PDE7相互作用以抑制依赖于PDE7的cAMP裂解的抑制依赖于PDE7的cAMP裂解的多肽或肽,它们是大体上纯的,基本上不含在天然状态可能存在的其它物质,从而对其预期用途是实用和适当的。
肽抑制剂已经成功地用于在体内干扰蛋白-蛋白相互作用和催化位点。例如,近来已批准结构和LFA-1相关的粘附分子的肽抑制剂在凝血病中临床应用(Ohman,E.M.等人,European Heart J.16:50-55,1995)。短直链肽(<30个氨基酸)被指出能防止或干扰整联蛋白依赖性粘附(Murayama,O.等人,J.Biochem.120:445-51,1996)。长度范围从25到200个氨基酸残基的较长的肽,也已成功地用于阻断整联蛋白依赖性粘附(Zhang,L.等人,J.Biol.Chem.271(47):29953-57,1996)。通常,较长的肽抑制剂比短肽有较高的亲和性和/或较慢的解离速率,因此可以是更有效的抑制剂。环肽抑制剂在体内也已显示出是用于治疗人免疫性疾病的有效的整联蛋白抑制剂(Jackson,D.Y.等人,J.Med.Chem.40:3359-68,1997)。制备环肽的一种方法包括合成其中肽的末端氨基酸是半胱氨酸的肽,从而使该肽能够通过在末端氨基酸之间形成二硫键而以环状形式存在,这已表明会在用于造血组织肿瘤的治疗中提高在活体内的亲和性和半衰期(例如Larson的美国专利No.6,649,592)。
合成的PDE7肽抑制剂
可用于本发明方法的PDE7抑制肽用模拟对于PDE7酶活性重要的靶区(例如PDE7的催化域)的氨基酸序列举例说明。PDE7A和PDE7B在催化域中有70%的同一性。(Hetman,J.M.等人,PNAS 97(1):472-476,2000)。PDE7A1的催化域是从SEQ ID NO:2的氨基酸残基185至456。PDE7A2的催化域是从SEQ ID NO:4的氨基酸残基211至424。PDEB的催化域是从SEQ IDNO:6的氨基酸残基172至420。可用于实施本发明方法的抑制肽的大小是从约5个氨基酸至约250个氨基酸。也可以使用分子模拟和合理分子设计来产生和筛选那些模拟PDE7催化区域的分子结构和抑制PDE7的酶活性的肽。用来模拟的分子结构包括抗-PDE7单克隆抗体的CDR区域。鉴别与特定靶结合的肽的方法是本领域熟知的。例如,可以使用分子印迹法从头构建大分子结构,例如与特定分子结合的肽。参见,例如,Shea,K.J.,“MolecularImprinting of Synthetic Network Polymers:The De Novo Synthesis ofMacromolecular Binding and Catalytic Sties”,TRIP 2(5),1994。
作为示例,一种制备PDE7结合肽的模拟物的方法如下。使显示PDE7抑制作用的抗PDE7抗体的结合域(模板)的功能性单体聚合。然后去除模板,接着在该模板留下的空隙聚合第二类单体,以提供具有一种或多种与该模板相似的预期性质的新分子。除了以这种方式制备肽以外,也可以制备作为PDE7抑制剂的其它PDE7结合分子,例如多糖、核苷、药物、核蛋白、脂蛋白、碳水化合物、糖蛋白、类固醇、脂质和其它生物活性物质。此方法可用于设计多种比其天然对应物更稳定的生物模拟物,因为它们通常通过功能性单体的自由基聚合制备,结果形成具有不能生物降解的骨架的化合物。
PDE7抑制肽可以用本领域熟知的方法制备,例如最早由Merrifield在J.Amer.Chem.Soc.85:2149-2154,1963中描述的固相合成技术。使用例如AppliedBiosystems 431A肽合成仪(Foster City,Calif.),按照制造商提供的说明,可以实现自动化合成。其它的技术可以在例如Bodanszky,M.等人,Peptide Synthesis,第二版,JohnWiley&Sons,1976以及本领域技术人员已知的其它参考书中查到。这些肽也可以用本领域技术人员已知的标准的基因工程技术制备。
候选的PDE7抑制肽可以用几种试验方法中的一种,包括例如PDE7磷酸二酯酶试验法,测试其作为PDE7抑制剂起作用的能力。
PDE7的表达抑制剂
在本发明方法的一些实施方案中,PDE7抑制剂是能够抑制依赖于PDE7的cAMP裂解的PDE7表达抑制剂(PDE7A,PDE7B,或二者)。在本发明这一实施方案的实施中,代表性的PDE7表达抑制剂包括PDE7反义核酸分子(例如反义mRNA,反义DNA,或反义寡核苷酸)、PDE7核酶和PDE7 RNAi分子。
反义RNA和DNA分子通过与PDE7 mRNA杂交和阻止PDE7蛋白的翻译,起着直接阻抑PDE7 mRNA的翻译的作用。反义核酸分子可以以多种不同的方式构筑,只要其能够干扰PDE7的表达。例如,反义核酸分子可以通过将PDE7A1 cDNA(SEQ ID NO:1)、PDE7A2 cDNA(SEQ IDNO:3)或PDE7B cDNA(SEQ ID NO:5)的编码区(或其一部分)相对于其正常的转录定向反转,以使其补体得以转录来完成构筑。设计和操控反义寡核苷酸的方法是本领域熟知的,描述在例如Mautino等人,Hum Gene Ther 13:1027-37,2002;和Pachori等人,Hypertension39:969-75,2002中,它们均并入本申请作为参考。
反义核酸分子通常与靶基因的至少一部分基本相同。但是,核酸不需要完全相同才能抑制表达。一般,可以利用较高的同源性来补偿较短的反义核酸分子的使用。最低同一性百分数通常大于约65%,但较高的同一性百分数可以对内源序列的表达发挥更有效的阻抑作用。通常优选高于约80%的显著较高的同一性百分数,但通常最优选约95%的同一性至绝对同一。
反义核酸分子不需要与靶基因有相同的内含子或外显子模式,而且靶基因的非编码区段在实现靶基因表达的反义抑制方面可以与编码区段同样有效。可以使用至少约8个左右的核苷酸的DNA序列作为反义核酸分子,但优选的是更长的序列。在本发明中,适用的PDE7抑制剂的代表性实例是反义PDE7核酸分子,它与由SEQ ID NO:1中列出的核酸序列组成的PDE7A1 cDNA的一部分的补体有至少90%的同一性。适用的PDE7抑制剂的另一个代表性实例是反义PDE7核酸分子,它与由SEQ ID NO:3中列出的核酸序列组成的PDE7A2cDNA的一部分的补体有至少90%的同一性。再一个适用的PDE7抑制剂的代表性实例是反义PDE7核酸分子,它与由SEQ ID NO:5中列出的核酸序列组成的PDE7B cDNA的一部分的补体有至少90%的同一性。
反义寡核苷酸寻靶结合PDE7 mRNA是可以用来降低PDE7蛋白合成的水平的另一机制。例如,聚半乳糖醛酸酶和毒蕈碱2型乙酰胆碱受体的合成受到指向其各自mRNA序列的反义寡核苷酸的抑制(Cheng的美国专利No.5,739,119和Shewmaker的美国专利No.5,759,829)。另外,反义抑制的实例已由核蛋白周期蛋白、多药抗药性基因(MDG1)、ICAM-1、E-选择蛋白、STK-1、纹状体GABAA受体和人EGF证实(例如参见,Baracchini的美国专利No.5,801,154;Baker的美国专利No.5,789,573;Considine的美国专利No.5,718,709;和Reubenstein的美国专利No.5,610,288)。
已描述的一个系统能使本领域普通技术人员确定何种寡核苷酸可用于本发明,它包括使用核糖核酸酶H裂解作为转录物内序列可及性的指示来探测靶物mRNA内的合适位点。Scherr,M.等人,Nucleic Acids Res.26:5079-5085,1998;Lloyd等人,Nucleic AcidsRes.29:3665-3673,2001。向表达PDE7的细胞提取物中加入与该PDE7转录物的一定区域互补的反义寡核苷酸的混合物并且杂交,以便产生RNAse H易攻击位点。这一方法可以与计算机辅助的序列筛选联合,后者能根据其形成二聚体、发夹结构或其它会降低或抑制与宿主细胞内的靶mRNA特异结合的二级结构的相对能力,预测反义组合物的最佳序列选择。这些二级结构分析和靶位选择的考虑可以用OLIGO引物分析软件(Rychlik,I.,1997)和BLASTN2.0.5算法软件(Altschul,S.F.等人,Nucl.Acids Res.25:3389-3402,1997)完成。以靶序列为目标的反义化合物优选包含约8-约50个核苷酸长度。特别优选的是含约9-约35个左右核苷酸的反义核苷酸。本发明人考虑范围在9-35个核苷酸(即,长度为9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34或35个左右的碱基的核苷酸)的所有寡核苷酸组合物对于实施本发明的基于反义寡核苷酸的方法都是高度优选的。高度优选的PDE7 mRNA的靶区是位于或靠近AUG翻译起始密码子的那些,和基本上与mRNA的5‘区互补,例如位于PDE7基因核苷酸序列的0和+10区之间的那些序列(SEQ IDNO:1、SEQ ID NO:3、SEQ ID NO:5)。
术语“寡核苷酸”在本文中使用时是指核糖核酸(RNA)或脱氧核糖核酸(DNA)或其模拟物的低聚物或聚合物。这一术语还包括由天然存在的核苷酸、糖和核苷间(骨架)共价连接以及具有非天然存在的改性的寡核苷酸组成的那些寡核碱基。上述的改性使得可以引入天然存在的寡核苷酸中不具备的某些所要的性质,例如降低的毒性、提高的对抗核酸酶降解的稳定性和增强的细胞摄取。在示例性的实施方案中,本发明的反义化合物由于磷酸二酯骨架的改性而与天然DNA不同,这延长了反义寡核苷酸(其中磷酸取代基被硫代磷酸代替)的寿命。类似地,寡核苷酸的一端或二端可以被一个或多个吖啶衍生物取代,它嵌入到核酸链内的相邻的碱基对之间。
反义的另一替代物是利用“RNA干扰”(RNAi)。双链RNAs(dsRNAs)能在体内在哺乳动物内诱发基因沉默。RNAi和共抑制的天然功能似乎保护基因组对抗可动遗传元件的侵袭,可动遗传元件的实例是反转录转座子和病毒,当它们变得活化时,会在宿主细胞内产生异常的RNA或dsRNA(参见,例如,Jensen,J.等人,Nat.Genet.21:209-12,1999)。双链RNA分子可以通过合成两个各自长约19-25(例如19-23个核苷酸)的能形成双链RNA分子的RNA链来制备。例如,一种可用于本发明方法的dsRNA分子可以含有与SEQ ID NO:1、SEQ ID NO:3、SEQ ID NO:5中至少一个的一部分及其补体对应的RNA。优选的是,RNA的至少一个链有1-5个核苷酸的3’突出端。所合成的RNA链在形成双链分子的条件下相结合。该RNA序列可以含有SEQ ID NO:1、SEQ ID NO:3或SEQ ID NO:5的至少一个8核苷酸部分,总长度为25个核苷酸或更短。对于指定的靶物设计siRNA序列属于本领域技术人员的普通技能。可获得设计siRNA序列的商业服务并保证抑制至少70%表达(Qiagen,Valencia,CA)。示例性的PDE7shRNAs和siRNAs可以从Sigma-Aldrich Company商购得到(产品#SHDNA_-NM_002603;SASI_Hs01_00183420至SASI_Hs01_00010490)。
dsRNA可以以药物组合物的形式施用,并用已知的方法实施,其中核酸被引入到预定的靶细胞中。通常使用的基因转移法包括磷酸钙法、DEAE-葡聚糖法、电穿孔法、显微注射法和病毒法。这些方法在Ausubel等人,Current Protocols in Molecular Biology,JohnWiley&Sons,Inc.,1993中有教导。治疗性的核酸分子可以被改性以穿透血脑屏障。例如,已证实,i.c.v.施用指向淀粉样蛋白前体(APP)的Abeta中央区的硫代磷酸反义寡核苷酸可以逆转阿尔兹海默小鼠模型的学习和记忆缺损。Banks W.A.等人,Journal of Pharm.andExp.Therapeutics,297(3):1113-1121,2001。
核酶:
在一些实施方案中,PDE7抑制剂是特异地裂解靶物PDE7(例如PDE7A,PDE7B或二者)的mRNA的核酶。以PDE7为靶的核酶可以作为PDE7抑制剂使用以便减少PDE7的量和/或生物活性。核酶是催化性RNA分子,它能裂解序列与该核酶的序列完全或部分同源的核酸分子。有可能设计出编码RNA核酶的核酶转基因,该RNA核酶特异地与靶RNA配对并在特定位置裂解磷酸二酯骨架,从而使靶RNA功能性失活。在进行这一裂解时,核酶本身不改变,于是能再循环和裂解其它分子。在反义RNA中包含核酶序列使其具有RNA裂解活性,由此提高了反义构建物的活性。
可用于实施本发明的核酶通常包含一个在核苷酸序列方面与靶PDE7 mRNA的至少一部分互补的至少约9个核苷酸的杂交区,和一个适合裂解靶PDE7 mRNA的催化区(一般可参看,欧洲专利No.0 321 201;WO 88/04300;Haseloff,J.等人,Nature 334:585-591,1988;Fedor,M.J.等人,Proc.Natl.Acad.Sci.USA 87:1668-1672,1990;Cech,T.R.等人,Ann.Rev.Biochem.55:599-629,1986)。
核酶可以以并入核酶序列的RNA寡核苷酸的形式直接寻靶细胞,或是作为编码预定的核酶RNA的表达载体被引入细胞。核酶可以按照与对反义多核苷酸所述非常相似的方式使用和施加。
可用于本发明方法的反义RNA和DNA、核酶和RNAi分子可以用本领域已知的用于合成DNA和RNA分子的任何方法制备。这些方法包括本领域熟知的化学合成寡脱氧核糖核苷酸和寡核糖核苷酸的技术,例如固相亚磷酰胺化学合成。或者是,RNA分子可以通过编码反义RNA分子的DNA序列的体外和体内转录来产生。所述的DNA序列可以插入到多种结合了合适的RNA聚合酶启动子(例如T7或SP6聚合酶启动子)的载体中。或者是,可以将组成性或诱导性(取决于所用的启动子)合成反义RNA的反义cDNA构建物稳定地引入到细胞系中。
可以对DNA分子进行各式各样众所周知的改性作为增加稳定性和半衰期的手段。适用的改性包括,但不限于,在分子的5’和/或3’端加入核糖核苷酸或脱氧核糖核苷酸的侧翼序列,或在寡脱氧核糖核苷酸骨架中使用硫代磷酸或2’O-甲基代替磷酸二酯酶键。
iv.可用于治疗成瘾的PDE7抑制剂的筛选方法
另一方面,提供了鉴别可用于治疗需要其的哺乳动物受试者中成瘾的抑制PDE7活性的试剂的方法。本发明这一方面的方法包括:(a)对大量试剂的每一种都测定抑制PDE7活性的IC50;(b)从大量试剂中选择抑制PDE7活性的IC50小于约1μM的试剂;(c)对于抑制PDE7活性的IC50小于约1μM的试剂,测定其抑制PDE4活性的IC50;(d)选择抑制PDE4活性的IC50比抑制PDE7活性的IC50的10倍更大的化合物,鉴定出可用于治疗成瘾的试剂;和(e)在成瘾模型体系中评价所鉴定的化合物的活性,其中抑制PDE7的IC50小于约1μM,抑制PDE4活性的IC50比抑制PDE7的IC50的10倍更大,并且已确定在动物模型中对治疗成瘾有效的试剂,表现出是可用于治疗哺乳动物受试者成瘾的PDE7抑制剂。
可以用于实施本发明这方面的方法的代表性试剂包括与PDE7结合并抑制PDE7的酶活性的分子(例如与PDE7结合并降低酶活性的小分子抑制剂或阻抑肽),和在转录和/或翻译水平上减少PDE7的表达的分子(例如PDE7反义核酸分子,PDE7特异性RNAi分子和PDE7核酶),从而阻止PDE7裂解cAMP。
V组合物的一般描述和定义
在一个方面,本发明提供了一种治疗成瘾的方法,包括向需要其的患者施用对抑制PDE7酶活性有效的一定量的PDE7抑制剂,其中这种对PDE7酶活性的抑制是PDE7抑制剂在治疗成瘾方面的主要治疗作用模式。成瘾包括对成瘾剂或对成瘾行为成瘾。成瘾剂包括,但不限于,精神兴奋药、酒精、阿片类物质和尼古丁。对于用PDE7抑制剂治疗成瘾,优选的精神兴奋药是可卡因或去氧麻黄碱。对于用PDE7抑制剂治疗,优选的成瘾行为是暴食。
对于可用于本发明方法的每种PDE7抑制化合物,其所有可能的立体异构体和几何异构体都包括在内。所述化合物不仅包括外消旋化合物,而且还包括光学活性异构体。当希望PDE7抑制剂是单个对映异构体时,可以通过拆分最终产物或者通过从异构纯的起始物出发或是使用手性辅助试剂来立体特异性合成得到,例如参看Ma,Z.等人,Tetrahedron:Asymmetry 8(6):883-888,1997。终产物、中间体或起始物的拆分可以用本领域已知的任何合适的方法完成。另外,在化合物可能有互变异构体的情形下,本发明打算涵盖该化合物的所有的互变异构形式。
含酸性部分的PDE7抑制剂可以与合适的阳离子形成药学上可接受的盐。合适的药学上可接受的阳离子包括碱金属(例如钠或钾)和碱土金属(例如钙或镁)阳离子。含碱性中心的PDE7抑制剂的药学上可接受的盐是与药学上可接受的酸形成的酸加成盐。其实例包括盐酸盐、氢溴酸盐、硫酸盐或硫酸氢盐、磷酸盐或磷酸氢盐、乙酸盐、苯甲酸盐、琥珀酸盐、富马酸盐、马来酸盐、乳酸盐、柠檬酸盐、酒石酸盐、葡糖酸盐、methanefulgonate、苯磺酸盐和对甲苯磺酸盐。根据上述,本文中出现的可用于本发明方法的任何提及的化合物均打算包括PDE7抑制剂及其药学上可接受的盐和溶剂化物。
本发明化合物能以纯净化合物的形式在治疗上施用,但优选以药物组合物或制剂的形式施用PDE7抑制剂。因此,本发明还提供了药物组合物或制剂,其含有PDE7抑制剂或其药学上可接受的盐,和一种或多种药学上可接受的载体,以及任选其它治疗和/或预防成分。合适的载体与制剂中的其它成分相容,并且对其接受者无害。本发明化合物还可以承载在递送系统中,以提供化合物的持续释放或增强的摄入或活性,例如用于注射的脂质体或水凝胶体系,用于口服或肠胃外递送的微粒、纳米颗粒或胶束体系,或用于口服递送的分段胶囊体系(staged capsule system)。
血-脑屏障:
在一些实施方案中,施用PDE7抑制剂以便穿过或绕过血-脑屏障。优选的是,在治疗方法中施用的抑制剂、化合物或组合物能够以足够的量和足够的速率穿透血-脑屏障,从而使运动障碍得到治疗。允许试剂穿过血-脑屏障的方法是本领域已知的,包括将试剂的尺寸减至最小,提供促进穿透的疏水因素,和与具有显著的血-脑屏障穿透性的载体分子缀合。
在一些实施方案中,有效量的PDE7抑制剂是在脑组织中的浓度能达到或超过指定的PDE7抑制剂活性的IC50的量。在一些实施方案中,以一定的方式和剂量施用PDE7抑制剂,使得峰值浓度约为抑制PDE7A或PDE7B的IC50浓度中较大者的1、1.5、2、2.5、5、10、20或更高倍。
实施例
实施例1:
PDE7抑制减轻可卡因成瘾
a.可卡因自身给药的减少
PDE7抑制通过减少可卡因成瘾复发和减少长期可卡因自身给药来减少可卡因使用的能力在可卡因成瘾大鼠模型中得到证实。将可卡因盐酸盐(得自National Institute onDrug Abuse,Bethesda,MD)以0.25mg/0.1ml的浓度溶在无菌的生理盐水中。在4秒内输注体积为0.1ml的药物或媒介物溶液。测试符合本文上述式1A(OMS182056)和1B(OMS181869)的两种PDE7抑制剂和符合本文上述式6(OMS182401)的一种PDE7抑制剂对可卡因自身给药的作用。在开始可卡因自身给药之前12小时和1小时,通过腹腔内(i.p.)手术给予OMS182056和OMS181869。腹腔内给予OMS182401。
使用到达实验室时重180-200g的雄性Wistar大鼠。将大鼠按3只一组安置在控湿(45-55%)和控温(22℃)的饲养箱中,实行12小时:12小时的逆向明/暗循环(开,17:00;关,05:00),可随意接近食物和水。在明/暗循环的黑暗期间进行实验。到达后一周,对大鼠施以手术,在右颈静脉内植入硅橡胶导管。实验台由装在隔音、通风的环境隔间中的操作性调节室(Med Associate Inc.)构成。在试验阶段开始前通过与导管相连的塑料管递送可卡因。通过有效杆的响应来激活输液泵,同时无效杆的响应被记录但不产生任何程序性后果。泵的激活引起0.1mL液体递送。IBM兼容性计算机控制液体的递送并记录行为数据。
对于评价PDE7抑制剂OMS182056和OMS181869的研究,按照固定比率5强化程序,每天在2小时的一段时间内训练大鼠自身给药可卡因,其中每次响应会导致递送0.25mg/0.1ml的液体可卡因溶液。继续可卡因自身给药训练,直到达到稳定的响应基线(每单只大鼠连续3天计算值变化小于10%)。此时开始药物试验。
大鼠用在自身给药阶段开始之前12小时和1小时i.p.给予的PDE7抑制剂(0.0、0.3、1.0和3.0mg/kg)治疗。记录对有效和无效杆的响应次数。药物试验之间允许有3天间隔。在这些间隔期中,继续可卡因的自身给药以重新建立基线杆响应。
结果示于图1和2。用OMS182056或OMS181869治疗都没有明显减少可卡因的自身给药,尽管在较高剂量时有减少的趋势。
对于评价OMS182401的研究,每天在2小时(短时程给药)或6小时(长时程给药)的训练期内训练大鼠自身给药可卡因,其中有效杆的每次按压会触发递送0.25mg可卡因,而无效杆的按压不递送可卡因;一周后,提高比率使得需要5次杆按压才能得到相同量的可卡因。继续可卡因自身给药训练,直到达到稳定的响应基线(每单只大鼠连续3天计算值变化小于10%)。此时开始药物试验。在试验期前15分钟给动物注射媒介物或药物。记录对有效和无效杆的响应次数。
结果示于图3。用OMS182401治疗在长时程给药模型中明显减少可卡因的自身给药,而在短时程给药模型中没有减少。
PDE7抑制剂通过减少cAMP因磷酸二酯酶-7活性的降解,提高了cAMP的细胞水平。细胞cAMP水平还通过Gs选择性G蛋白偶联受体(例如多巴胺D1受体(DRD1))的激活而提高。为了测试通过DRD1的激活而提高cAMP水平对可卡因自身给药的影响,将DRD1激动剂SKF82958 i.p.给予已训练为自身给药可卡因的大鼠。(±)-SKF-82958氢溴酸盐[(±)-6-氯-7,8-二羟基-3-烯丙基-1-苯基-2,3,4,5-四氢-1H-3-苯并氮杂 氢溴酸盐6-氯-N-烯丙基-SKF-38393氢溴酸盐]或“SKF82958”获自Sigma Aldrich。SKF82958按1.0mg/kg给药。结果示于图4。在短时程给药模型中,用SKF82958治疗明显减少可卡因的自身给药。在该例中未评价长时程给药模型。在治疗后第1小时,在两个剂量都观察到自身给药的显著减少。治疗后2小时,自身给药减少的趋势不再显著(数据未显示)。但是,动物还表现出明显反常的行为并且极具攻击性,表明由于和可卡因一起施用SKF82958抵消了它们的压杆能力。表现出的行为与过量服用可卡因后所显示的相似(数据未显示)。
氢溴酸盐6-氯-N-烯丙基-SKF-38393氢溴酸盐]或“SKF82958”获自Sigma Aldrich。SKF82958按1.0mg/kg给药。结果示于图4。在短时程给药模型中,用SKF82958治疗明显减少可卡因的自身给药。在该例中未评价长时程给药模型。在治疗后第1小时,在两个剂量都观察到自身给药的显著减少。治疗后2小时,自身给药减少的趋势不再显著(数据未显示)。但是,动物还表现出明显反常的行为并且极具攻击性,表明由于和可卡因一起施用SKF82958抵消了它们的压杆能力。表现出的行为与过量服用可卡因后所显示的相似(数据未显示)。
在一段消退期内,施用PDE7抑制剂OMS182056和多巴胺D1激动剂SKF82958,但不施用可卡因,以便确定这些试剂是否有成瘾性质。结果示于图5和6中。没有一种试剂当其单独施用时会引起大鼠的压杆增加。OMS182056的确显示出轻微的但非统计显著性的压杆减少的倾向。因此,这些试剂都没有成瘾性质。
尽管PDE7抑制剂和多巴胺D1激动剂都能增加cAMP的细胞水平,但当与可卡因一起施用时,这两类试剂产生非常不同的作用。
还在更长一段时间内测试了OMS182401对可卡因自身给药的影响。大鼠被手术植入颈静脉导管并令其恢复一周。然后动物进行每天6小时(长时程给药)的训练阶段,其中每次压动有效杆都触发递送0.25mg可卡因。一周后,将该比率提高,使得需要5次压杆才能得到同样量的可卡因。动物训练继续约6周。在达到连续6天稳定速率的有效压杆之后,对动物每天2次i.p.注射媒介物或药物(4.5mg/kg)。在2小时的一段时间内评估强化的响应。
结果示于图7。长期的OMS182401治疗在长时程给药的大鼠模型中减少可卡因自身给药。OMS182401的作用在至少6个治疗日内保持稳定。当停止用OMS182401治疗时,可卡因自身给药恢复到基线水平。OMS182401不改变无效杆的压动(数据未显示)。此实验证实了在可卡因成瘾治疗的急性模型中观察到的PDE7抑制剂的效力,并指示OMS182401适宜长期用药。
b.消退后可卡因寻觅的减少
测试OMS182056对消退后不久可卡因寻觅行为的影响。如上所述将大鼠训练为自身给药可卡因,然后暴露于消退条件。在第一天,用盐水溶液代替可卡因,向大鼠i.p.给药OMS182056(1.0和3.0mg/kg)并计数压杆数。结果示于图8。3mg/kg的OMS182056明显减少消退期开始时可卡因寻觅行为的量,表明OMS182056促进可卡因消退。
c.复发后可卡因寻觅的减少
还测试了OMS182056和OMS182401在应激诱导的成瘾复发后对可卡因寻觅行为的影响。如上所述将大鼠训练为自身给药可卡因,然后暴露于消退条件。对于恢复期(最后的消退阶段后的那一天),给大鼠注射育亨宾(1.25mg/kg)并在30分钟后置于操作室,监测压杆行为30分钟。已知给药α-2肾上腺素受体拮抗剂育亨宾(增加大脑去甲肾上腺素细胞放电和释放)可起到药理应激原的作用并促进酒精寻觅复发。(Lê等人,Psychopharmacology 179:366-73(2005))。结果示于图9和10。在剂量为1.0和3.0mg/kg(图9)时,OMS182056表现出在预防应激诱导的可卡因成瘾复发方面的显著作用。同样地,在剂量0.3、1.0和3.0mg/kg(图10)时,OMS182401表现出在预防应激诱导的可卡因成瘾复发方面的显著作用。
测试OMS182056对信号诱导的成瘾复发后可卡因寻觅行为的影响。将大鼠训练为自身给药可卡因,外加视觉或嗅觉信号,然后暴露于没有可卡因的消退条件。对于恢复期(最后的消退阶段后的那一天),将大鼠再暴露于先前获知的信号。监测压杆行为,结果示于图11。尽管结果不是统计显著的,给药增加量的OMS182056(1.0或3.0mg/kg)导致动物的信号诱导的复发的剂量相关性减少的趋势。还测试了OMS182401对信号诱导的成瘾复发后可卡因寻觅行为的影响。结果示于图12。测试了三个浓度的OMS182401:0.3mg/kg、1.0mg/kg和3.0mg/kg,并且在三个浓度下结果都是统计显著的。给药增加量的OMS182401导致动物的信号诱导的复发以剂量相关的方式统计显著性减少。
接着测试PDE7抑制剂和DRD1激动剂对可卡因点燃诱导的复发的影响。如上所述将大鼠训练为自身给药可卡因,然后暴露于消退条件。用盐水溶液代替可卡因。在压杆显著减少后,开始恢复程序。用媒介物或试剂处理大鼠并在10分钟后给药可卡因。在可卡因给药后计数压杆行为1个小时。PDE7抑制剂OMS182056的结果示于图13。给药OMS182056对可卡因诱导的点燃响应没有明显作用。DRD1激动剂SKF82958的结果示于图14。用SKF82958处理显著减少了由可卡因点燃诱导的压杆行为。然而,如上所述,用SKF82958处理的动物表现出明显异常的行为。给药PDE7抑制剂和多巴胺D1激动剂又一次在可卡因成瘾动物中具有非常不同的结果。
实施例2
PDE7抑制减轻尼古丁成瘾
a.尼古丁自身给药的减少
PDE7抑制减少尼古丁使用的能力在尼古丁成瘾的大鼠模型中得到证实。使用到达实验室时重180-200g的雄性Wistar大鼠。将大鼠按3只一组安置在控湿和控温(22℃)的饲养箱中,实行12小时:12小时的逆向明/暗循环(开,17:00;关,05:00),可随意接近食物和水。
大鼠被手术植入颈静脉导管并令其恢复一周。然后动物进行每天2小时(短时程给药)或6小时(长时程给药)的训练阶段,其中有效杆每压动3次触发递送0.03mg尼古丁。在达到稳定的有效杆压动速率之后,在试验阶段之前15分钟对动物i.p.注射媒介物或药物(OMS182401或OMS182399,符合以上式6的另一种PDE7抑制剂)。测定的读数是在2小时内强化响应的数目。OMS182401在大鼠内的半衰期是1.7-4.9小时。结果示于图15(OMS182401,短时程给药),16(OMS182401,长时程给药)和17(OMS182399,短时程给药)。OMS182401对PDE7的抑制在短时程给药和长时程给药大鼠模型中都以剂量依赖方式减少尼古丁自身给药。在3.0mg/kg和9.0mg/kg的剂量下,OMS182399对PDE7的抑制在短时程给药大鼠模型中减少尼古丁的自身给药。这些化合物在每种情形下都不改变无效杆的压动。
b.PDE7抑制对食物自身给予的影响
评估PDE7抑制剂OMS182399和OMS1823401对食物自身给予的影响以评估PDE7抑制的作用是否对药物滥用是特异性的。每种化合物测试八只雄性Wistar大鼠。每种药物以配制在0.03M酒石酸中的0.0、3.0和9.0mg/kg的浓度测试。腹腔内(i.p.)给药各溶液。
自身给药台由装在光亮的、隔音、通风的环境隔间中的操作调节室(MedAssociate Inc.)构成。该室的前门和背墙由透明塑料制成,其它墙由不透明金属制成。各室配备有位于该室的前面板的两个可伸缩的杆。杆上的最小向下压力(约25g)足以产生程序性响应。其中食物丸粒(各45mg;Bio-ServInc.,Frenchtown,NJ)可由丸粒分配器分配的嵌入式食物容器位于两个杆之间。IBM兼容性计算机控制液体的递送并记录行为数据。
对于数据评估,使用方差分析(ANOVA),之后适当的话进行事后(Newman-Keuls)检验。对数据进行处理结果的分析。统计显著性设为p<0.05。
图18A和18B分别显示OMS182399和OMS182401获得的结果。两个化合物都没有表现出与媒介物相比的对食物自身给予的统计显著性作用,证实PDE7抑制的作用对药物滥用是特异性的。该研究提供证据证明先前观察到的这些PDE7抑制剂对尼古丁和可卡因相关的操作响应的抑制是选择性的并且不依赖于自发活动或积极性的一般抑制。
c.累进比率研究中尼古丁自身给药的减少
首先在强化的特征释放1(FR1)程序下如上述实施例2中所述将雄性Wistar大鼠训练为自身给药尼古丁。在建立尼古丁的稳定响应基线后,在累进比率条件下测试动物,其中逐步增加响应要求(即,得到一剂0.03mg尼古丁所需的杆响应数)。
每次尼古丁加强的响应引起20秒的室内灯的照明。最后一次加强响应过去后多于60分钟,终止该阶段。收集的数据有断点(最后一个阶段中的压杆数),整个研究过程中的总压杆数(有效杆)和总奖赏数。利用拉丁方设计测试15只动物,即每只动物接受各自的剂量。将PDE7抑制剂OMS182401在不同剂量下的作用与单独的媒介物比较。
总奖赏数、断点和总有效杆按压的结果分别示于图19A-19C。这些数据说明PDE7抑制剂以剂量依赖性方式减少成瘾动物获得尼古丁的积极性,即对尼古丁的积极性。
实施例3
尼古丁消退的自身给药的减少
PDE7抑制对于加速尼古丁消退的能力在尼古丁成瘾大鼠模型中得到证实。将大鼠训练到稳定水平的尼古丁自身给药。有效杆不与任何强化的奖赏相联系。在第一消退期之前,对动物注射媒介物或OMS182401。计数在第一消退期的第一小时内有效杆的总响应数。结果示于图20。OMS182401以统计显著性的方式促进了尼古丁消退。
另一项研究评估OMS182401对已训练适应食物丸粒的大鼠在消退第一天的非强化响应的影响以证实OMS182401对药物滥用的作用特异性。将16只雄性Wistar大鼠进行为期8天的自身给予食物丸粒的训练,然后分为两组并在第一消退阶段前15分钟注射OMS182401(9.0mg/kg于0.03M酒石酸中)或媒介物。结果证实OMS 182401不减少经食物丸粒训练的大鼠的非强化响应(数据未显示),进一步提供了证据支持OMS182401对药物滥用的作用特异性。
实施例4
恢复后尼古丁使用的减少
PDE7抑制对于减少信号诱导恢复后的尼古丁使用的能力在尼古丁成瘾的大鼠模型中得到证实。将大鼠训练到稳定速率的尼古丁自身给药和能区分尼古丁与盐水可得到性。在尼古丁阶段,始终存在一个音调(7kHz,70dB),并在响应后亮起信号灯(在有效杆上方)。在盐水阶段,试验箱总是用饲养箱灯光照明,并在每次响应后响起白噪音。区分期之后是消退期,它继续到压杆少于稳定速率的20%。为测试化合物,注射媒介物或OMS182401,并将动物暴露于尼古丁刺激条件下。在尼古丁刺激条件的第一小时内计数有效杆总响应数。结果示于图21。OMS182401对PDE7的抑制在受试动物中统计显著性减少了信号诱导的尼古丁复发。无效杆的响应不受OMS182401给药的影响。
PDE7抑制以减少应激诱导恢复后尼古丁使用的能力在使用育亨宾(一种α2-肾上腺素能拮抗剂)作为应激物的大鼠模型中得到证实。育亨宾起着药理性应激物的作用,促进尼古丁寻觅的复发。测试OMS182401对寻觅尼古丁行为的影响。将大鼠训练到稳定速率的尼古丁自身给药。有效杆不与任何强化的奖赏相联系(消退),而且在几个阶段内“有效”杆压动的速率下降。当压杆速率下降到小于稳定速率的20%时,与媒介物或测试化合物一起向动物腹腔内(i.p.)注射1.25mg/kg的育亨宾。在施用育亨宾之后的第一小时计数有效杆的总响应数。结果示于图22。OMS182401对PDE7的抑制统计显著性减少了受试动物由应激诱导的尼古丁寻觅复发。无效杆响应不受施用育亨宾或OMS182401的影响。
实施例5
PDE7抑制减少应激响应的暴食行为
为了在实验动物中模拟暴食症,建立了暴食模型,其中通过“悠悠球”式节食和应激暴露于极美味食物(HPF)来引发暴食行为。在该模型中,雌性大鼠暴露于特征为将动物暴露于HPF但没有得到它的可能的限制与应激性程序的反复循环。该程序引起非常短的时期内过度食用的极美味食物摄入的选择性增加。如下所述,利用具有重要的预测性和表面有效性的该模型,研究了PDE7抑制剂对暴食行为(食瘾最重要的形式之一)的影响。
通过反复的食物限制和应激的组合在实验动物中获得暴食行为的恢复。[Cifani等人,Psychopharmacology 204:113-125(2009)]。对于本发明,如Cifani所述测试了应激诱导的暴食行为。评估了两种不同的PDE7抑制剂:OMS182401和OMS182056的作用并与媒介物和与托吡酯(已在临床试验中显示抑制暴食行为发作的抗癫痫药物)比较。
将大鼠饲养在独立笼中并在实验前两周可随意获得食物和水。在实验期间,向大鼠给予两种食物源之一:标准大鼠食物丸粒(食物)或极美味食物(HPF)(由52%Nutella TM巧克力酱,33%大鼠食物丸粒和15%水的混合物组成(5.33kcal/g;碳水化合物、脂肪和蛋白质分别为56%、31%和7%))。
将大鼠分为四组。使各组经历以下的8-天循环,连续三次。在第25天,向大鼠给药PDE7抑制剂或媒介物。
(1)对照组-非限制,非应激(NR+NS)。大鼠随意进食4天。第5天和第6天,它们自由获取食物和两小时的HPF。第7天和第8天,大鼠随意进食。第25天,动物不暴露于应激。
(2)限制、非应激(R+NS)。连续4天大鼠获得限制到其正常摄入量的66%的食物。第5天和第6天,它们自由获取食物和两小时的HPF。第7天和第8天,大鼠摄入正常食物摄入的66%。第25天,动物不暴露于应激。
(3)非限制、应激(NR+S)。大鼠随意进食4天。第5天和第6天,它们自由获取食物和两小时的HPF。第7天和第8天,大鼠随意进食。第25天,动物暴露于应激。
(4)限制和应激(R+S)。连续四天将大鼠的食物限制到正常摄入的66%。第5天和第6天,它们自由获取食物和两小时的HPF。第7天和第8天,大鼠完成正常食物摄入的66%。第25天,动物暴露于应激。
通过使大鼠在进食HPF之前将HPF置于动物可看见和闻到但不能达到的容器中15分钟来诱导应激。
在第25天,将适当的大鼠应激后,向动物给药PDE7抑制剂OMS182401(1.0或3.0mg/kg i.p.)、PDE7抑制剂OMS182056(1.0或3.0mg/kg i.p.)、托吡酯(60mg/kg i.p.)或对照媒介物。一小时后,向动物给予HPF且随意进食。两小时后测量HPF的摄入。
结果示于图23A-23D(OMS182401)、图24A-24D(OMS182056)和图25A-25D(托吡酯)。
在所有组中,动物在两个小时内进食增加量的HPF。在NR+NS、NR+S和R+NS组中,给药OMS182401没有明显影响动物消耗的HPF的量。在R+S组中,HPF的最初消耗速率和两小时内进食的总量都比其它三组更高。因此R+S条件模拟了人类暴食行为。此外,在R+S组中,给药3.0mg/kg OMS182401的动物相比其它动物消耗更少的HPF。到两小时为止,对照动物与给予3.0mg/kg OMS182401的动物之间的HPF消耗的差异是明显的。因此,PDE7抑制剂OMS182401的给药在该条件的大鼠模型中减少了应激诱导的暴食行为。
在四组大鼠中只有一组(R+S)观察到统计显著的OMS182056作用:R+S组[F(2,22)=10.79,p<0.001],NR+NS组[F(2,24)=0.31,p>0.05];R+NS组[F(2,24)=0.84,p>0.05];NR+S组[F(2,24)=1.41,p>0.05]。事后比较显示R+S组中响应于两个剂量的OMS182056的HPF摄入方面的统计显著性差异。在所有观察时间观察到更高剂量(3mg/kgi.p.)的作用,而1mg/kg i.p.的剂量仅在15和120min有效(*p<0.05)。
托吡酯(60mg/kg i.p.)也仅在R+S组中减少HPF摄入:[F(1,15)=9.03,p<0.01],但在其它组中未减少:NR+NS组[F(1,16)=0.84,p>0.05];R+NS组[F(1,16)=1.59,p>0.05];NR+S组[F(1,16)=0.49,p>0.05]。事后比较显示在所有观察时间作用都是明显的(*p<0.05)。
统计分析显示有食物限制史的大鼠的应激暴露引起显著的暴食行为。这些动物(R+S组)与未经受食物限制经历和应激的对照大鼠(NR+NS)相比进食显著更高量的HPF。OMS182401和OMS182056在R+S组而非其他组(其中动物未表现出暴食行为)中减少HPF的效力进一步证实模型的有效性。这些药物似乎减少暴食行为而不影响一般的食物消耗。有趣的是,在最高剂量,OMS182056与对照药物托吡酯相比,似乎更有效力地减少强迫性进食。
实施例6
PDE7抑制减少应激诱导的觅食复发
为了评估PDE7抑制剂预防食物成瘾者的过量食物摄入的复发的能力,在育亨宾诱导的觅食复发模型中评估PDE7抑制剂OMS182399。首先使16只雄性Long Evans大鼠适应,然后在两个月的训练期内每天喂食正常食物摄入的60%的饮食。在训练期内,将大鼠训练为稳定速率的极美味食物自身给予。训练大鼠按压试验箱内的有效杆以递送45mg含有高含量脂肪(35%w/w)和中等含量碳水化合物(45%w/w)的极美味食物(HPF)丸粒(获自Bioserve,Frenchtown NJ)。每隔一天,每天三小时根据固定比率1的强化程序训练大鼠,每次压杆得到HPF丸粒和按压间20秒暂停的强化。按压无效杆不引起食物强化。
该自我给予期后接着是消退期,其中有效杆的按压不引起任何HPF或其他奖赏。每天持续该消退期达十四个阶段。在最后四个阶段期间,大鼠习惯于注射。
消退期后,开始测试期,其中给大鼠注射育亨宾,其充当药理应激原并促进过量食物寻觅复发。给大鼠i.p.注射OMS182399(0.3,1.0或3.0mg/kg)或盐水,10分钟后注射育亨宾或蒸馏水。育亨宾或蒸馏水注射后30分钟开始测试阶段,其中再次将大鼠置于具有递送HPF的有效杆和无效杆的测试箱。在14-,30-,45-,60-和180-分钟时间点测量强化行为(有效杆的按压)。当将第一个小时内的总有效杆响应与育亨宾处理的大鼠相比时,OMS182399显示以统计显著和剂量依赖性方式减少应激诱导的觅食复发,如图26所示。无效杆的响应不受育亨宾或OMS182399的影响(数据未显示)。与育亨宾处理大鼠中OMS182399的作用相反,在另一实验中,OMS182399注射在媒介物处理的大鼠中不减少压杆数(数据未显示)。
实施例7
对伏隔核中基底和尼古丁诱导的多巴胺释放的影响
为了阐明PDE7抑制剂对成瘾行为的作用机制,我们检测了它们对伏隔核(NAc)中的多巴胺(DA)水平的影响。已知药物滥用增加NAc壳中的DA水平,这种效应直接涉及它们的成瘾性(Laviolette,S.R.,Van der Kooy,D.,2004.The neurobiology of nicotineaddiction:bridging the gap from molecules to behavior.Nat.Rev.Neurosci.5,55-65)。进行一项研究以通过体内微量透析研究评估OMS182399对Wistar大鼠的NAc中基底和尼古丁诱导的DA释放的影响。将重275-300g的雄性Wistar大鼠(Harlan,Italy)以每笼六只在恒温(23℃)、恒湿(60%)和12小时明/暗循环(从8a.m.至8p.m.照明)在中心动物室中分组饲养至少5天,可随意获取标准食物(Morini,Italy)和水。
根据De Luca等人(2007)的方法,用AN69(丙烯腈甲基丙烯磺酸钠共聚物)透析纤维(310μm o.d.220μm i.d.Hospal,Dasco,Italy)制备具有1.5mm透析部分的同轴透析探针。大鼠用水合氯醛(400mg/kg i.p.)麻醉并置于立体定位仪中。切开头皮并将lambda与前囟之间的头盖骨弄平。根据Paxinos和Watson(1998)的图集钻两个小孔以暴露硬膜并将两个探针在NAc壳和核的水平垂直插入(坐标 壳:A:2.2,L:距离前囟1.0,V:距离硬膜-7.8;坐标核:A,1.4;L,距离前囟1.6;V距离硬膜-7.6)。将探针用glasionomeric接合剂(CX-Plus,Shofu Inc.,Japan)固定于头盖骨。探针植入后24小时,在自由活动的大鼠上进行实验。通过透析探针以1μl/min的恒定速率泵入林格氏溶液(147mM NaCl,4mM KCl,2.2mM CaCl2)。每10或20分钟(取决于实验)从各NAc壳和核采集样品并分析。
将透析液样品(10或20μl)注入配备有反相柱(C83.5um,Waters,Mildford,MA,USA)和库仑检测器(ESA,Coulochem II,Bedford,MA)的HPLC以定量DA。第一电极设定为+125mV且第二电极设定为-175mV。以下溶液用作流动相:50mM NaH2PO4,0.1mM Na2-EDTA,0.5mM正辛基硫酸钠,15%(v/v)甲醇,pH 5.5。用Jasco泵以0.6ml/min的流速泵入流动相。DA的分析灵敏度为每份样品5fmol。实验结束时,通过穿心灌注100ml盐水(0.9%NaCl)和100ml甲醛(10%)处死动物。移去探针,在振动切片机上将大脑切为连续冠状切片以定位微量透析探针的位置。对由每次处理后DA的连续分析获得的原始数据适用重复测量后的方差分析(ANOVA)。对显示明显的整体变化的处理结果施以事后Tukey检验,p<0.05视为统计显著的。相差不超过10%的三个连续样品的均值视为基础值。
为了确定OMS182399对NAc壳和核中基底和尼古丁诱导的DA释放的影响,对大鼠植入两个微量透析探针并且,24小时后,给药媒介物或OMS182399(9mg/kg,i.p.)。检测NAc壳和核透析液DA至少三个小时。如果没有观察到对细胞外DA水平的影响,或者,如果有任何影响,恢复至基底DA水平后至少两个小时,在注射尼古丁(0.4mg/kg皮下(sc))之前10分钟,向相同的动物注射媒介物或OMS182399。在尼古丁处理后监测透析液DA至少三个小时。
图27显示OMS 182399(9mg/kg i.p.)对NAc壳/核细胞外DA水平产生的影响。结果表示为以基底值的百分数表示的DA细胞外水平变化的均值±SEM。OMS182399对NAc的任一区域的基底DA水平没有影响。
图28显示尼古丁(0.4mg/kg sc)对OMS 182399(9mg/kg i.p.;尼古丁之前10分钟)预处理大鼠的NAc壳细胞外DA水平产生的影响。结果表示为以基底值的百分数表示的DA细胞外水平变化的均值±SEM。给药尼古丁增加NAc壳中DA释放,并且这种效应被OMS182399显著减小。
实施例8
PDE7抑制对多巴胺能腹侧被盖区神经元的自发活动的影响和对抗尼古丁的作用下述电生理学研究证实选择性PDE7抑制剂在尼古丁成瘾模型中是有效的并通过增强经由DRD1的多巴胺信号传导而起作用。据信尼古丁的强化效应由其对腹侧被盖区(VTA)中多巴胺能神经元的活化而引起(Pidoplichko VI等人,Nature 390:401-404(1997);Liu L等人,Nicotine persistently activates ventral tegmental area dopaminergic neuronsvia nicotinic acetylcholine receptors containing α 4and α6 subunits.MolPharmacol,81(4):541-8(2012))。发明人试图通过进行离体大鼠VTA水平切片的全细胞电流钳记录以研究PDE7抑制剂改变基底和滥用药物诱导的VTA多巴胺活性的变化的能力来评估PDE7抑制剂的抗成瘾潜能。出于此目的,尼古丁用作滥用药物的一个例子。并且,考虑到PDE7同工型在所有主要的大脑区域中广泛表达并通常与DRD1在相同神经元内共表达[Reyes-Irisarri,E等人,“Neuronal expression of cAMP-specific phosphodiesterase7B mRNA in the rat brain,”Neuroscience 132:1173-85(2005);Mir6,X.等人,“Differential distribution of cAMP-specific phosphodiesterase 7A mRNA in ratbrain and peripheral organs,”Synapse 40:201-214(2001)],以及在旷场活动试验中DRD1激动活性被PDE7抑制加强(以下实施例10),还在电生理学研究中比较这些试剂的作用。
通过大超级化电流的存在识别多巴胺能神经元。Johnson,S.W.等人,“Opioidsexcite dopamine neurons by hyperpolarization of local interneurons,”JNeurosci 12:483-488(1992)。以0.3和1.0μM施用PDE7抑制剂OMS182401明显降低了多巴胺能VTA神经元的自发放电速率(图29A)。特别地,还如图29A所示,以3.0和10.0uM的浓度施用DRD1激动剂SKF82958观察到类似的效应。
并且,当一起施用非有效浓度(即过低的浓度)的SKF82958和OMS182399时,该组合也产生活动的明显减少(图29B),表明PDE7抑制加强了DRD1活化作用并且PDE7抑制剂和DRD1激动剂在这方面协同作用。
由于DRD1刺激增加GABAB-介导的抑制性突触后电位(IPSPs)的幅度并且cAMP-PKA增强GABAA-介导的抑制性突触后电流(IPSCs)[Bonci,A.等人,“Increased probabilityof GABA release during withdrawal from morphine,”J Neurosci 17:796-803(1997)],发明人假定该观察结果(即,PDE7抑制或DRD1活化后VTA DA神经元的自发活动的减少)可归因于GABA自突触前末梢释放的可能性增加。该假说部分得到微型抑制性突触后电位(mIPSPs)分析的证实(图30),其证实PDE7抑制后mIPSPs的频率而非幅度的增加。此外,当在GABA-A受体拮抗剂印防己毒素(100microM,数据未显示)存在下施用PDE7抑制剂时,该抑制任然存在,表明GABA-B受体参与PDE7抑制剂的效应。实际上,在GABA-B拮抗剂CGP35348(100uM)存在的情况下,不再观察到OMS182401对VTA DA神经元自发放电的抑制作用(数据未显示)。
在另外的研究中,观察到OMS182401逆转尼古丁对VTA多巴胺能神经元的活化(图31)。另外,虽然单独的30nM OMS182401或1uM SKF82958是无效的,30nM OMS182401与1uMSKF82958的组合逆转尼古丁诱导的活化(p<0.05)。这证实PDE7抑制加强DRD1信号传导并且PDE7抑制剂与DRD1激动剂的组合在这方面的作用大于累加效应。
为了体内证实VTA是PDE7抑制效应的作用位点,将OMS182401直接递送至大鼠VTA并评估其对尼古丁自身给药的影响。对大鼠手术植入颈静脉导管并使其恢复一周。然后对大鼠施以颅内手术以在VTA中双侧植入引导插管。恢复后,大鼠经历每天两小时(短时程给药)的训练阶段,其中每次有效杆按压触发递送0.03mg尼古丁。在达到稳定速率的有效杆按压后,在测试阶段前不久将媒介物或药物注射到VTA中。在拉丁方设计(即,每只动物接受各自的剂量)实验中记录强化响应。在实验结束时组织学确认颈静脉导管的位置。如图32所示,直接给药PDE7抑制剂至大鼠VTA减少了尼古丁自身给药,证实VTA是动物中PDE7抑制效应的作用位点。
实施例9
PDE7抑制对腹侧被盖区多巴胺神经元活动的影响和对抗可卡因、吗啡和酒精的作用已完成另外的电生理学研究来评估PDE7抑制剂OMS182399在可卡因、吗啡或酒精的情况下对VTADA神经元活动的影响。如上述实施例8中所述制备VTA切片。
通过平均给药(5分钟)后的效应并相对于给药前基线归一化来计算药物诱导的放电速率的变化。所有数值数据以均值±s.e.m给出。通过利用重复测量的双因素ANOVA(处理×时间)或(如果合适)重复测量的Student’s t检验来比较和分析数据。借助于GraphPadPrism程序进行统计分析。显著性水平确立为P<0.05。
研究OMS182399对含有中脑的大鼠水平脑切片制品中的VTA多巴胺神经元的急性效应。自VTA多巴胺神经元进行全细胞电流钳记录。如图33所示,急性浴施用OMS182399(0.05,0.5和2.5μM,5min)以剂量依赖性方式显著减少VTA多巴胺神经元的自发活动。重复测量的单因素ANOVA揭示均值显著不同:F3,42=5.67,P=0.002(n=15)。数据表示为均值±SEM.**P<0.005。
还如图33所示,急性施用PDE7抑制剂OMS182399(在吗啡施用前5分钟)能够以剂量依赖性方式阻断吗啡诱导的VTA DA细胞兴奋(吗啡1μM,3min:基底放电速率的224.4±19.5%,n=7),统计显著性(非成对t-检验)为剂量50nM(P<0.0001,n=7);0.5μM(P=0.0001,n=8);2.5μM(P=0.0002,n=7);其本身不影响DA神经元的基底自发活动。图33中的虚线表示VTA DA细胞的归一化的基底自发活动。数据表示为均值±SEM,***P<0.0001。特别地,统计分析(配对t-检验)未揭示存在或不存在吗啡时OMS182399对VTADA细胞的放电活动的影响方面的任何差异,即添加吗啡不能克服OMS182399的作用(在所有检验浓度下P>0.05)。
如图34所示,急性施用PDE7抑制剂OMS182399(在浴施用酒精前5分钟)以剂量依赖性方式阻断酒精诱导的VTA DA细胞兴奋(酒精100mM,5min:基底放电速率的158.3±26.7%,n=6),统计显著性(非成对t-检验)为剂量500nM(P=0.007,n=8);0.05μM(P>0.05,n=7);2.5μM(P=0.02,n=7);其本身减少DA神经元的基底自发活动。图34中的虚线表示VTA DA细胞的归一化的基底自发活动。数据表示为均值±SEM,**P<0.001,*P<0.05。特别地,统计分析(配对t-检验)揭示OMS182399剂量为500nM时在存在和不存在酒精时OMS182399对VTA DA细胞的放电活动的影响方面的差异(P=0.01,n=8),而对于其它两个测试的剂量未揭示统计学差异(在两个测试浓度下P=0.6)。
如图35所示,急性施用PDE7抑制剂OMS182399(在浴施用可卡因前5分钟)在剂量50nM(P=0.036,n=7)和0.5μM(P=0.049,n=7)下能够阻断可卡因诱导的VTA DA细胞抑制(可卡因1μM,5min:基底放电速率的36.4±9.1%,n=7)。图35中的虚线表示VTA DA细胞的归一化的基底自发活动。数据表示为均值±SEM,*P<0.05。特别地,在本身产生与可卡因性质类似效应(即,降低多巴胺神经元的放电速率)的最高测试剂量(2.5μM)下,OMS182399未能阻止可卡因诱导的多巴胺神经元的自发活动抑制(非配对t-检验vs可卡因P=0.5;配对t-检验vs OMS182399 P=0.8)。显著地,在存在测试的最低剂量的情况下(即50nM),可卡因对多巴胺神经元活动的效应与对照相反(即,与抑制相反产生兴奋)。
如上述电生理学研究所示,PDE7抑制阻断常用的滥用药物的效应而不管该滥用药物的化学结构和作用机理如何。特别地,如上所示,OMS182401能阻断尼古丁诱导的多巴胺细胞放电速率的激发,这可归因于位于多巴胺体细胞上的α4β2nAChRs的活化[PicciottoMR等人,Nature,391:173-177(1998)]。类似地,结构相关化合物OMS182399能阻断吗啡和酒精诱导的多巴胺神经元的兴奋,两者都是由于对多巴胺细胞的去抑制机理[Theile JW等人,Neuroscience,172:94-103;Jalabert M等人,Proc Natl Acad Sci U S A,108:16446-16450(2011)]。有趣的是,OMS182399还可恢复可卡因效应。对可卡因的效应更复杂,因为可卡因诱导纹状体区中的快速多巴胺增加和中脑多巴胺细胞放电的减少(通过激活位于体细胞内的D2受体)。
实施例10
PDE7抑制对多巴胺D1亚型受体-介导的旷场活动刺激的影响
为了评估PDE7抑制剂增强体内DRD1信号传导的作用,利用野生型C57BL/6小鼠在旷场活动模型中评估了单独和组合的PDE7抑制剂OMS182401和DRD1激动剂SKF82958的作用。如图36所示,DRD1激动剂以1mg/kg刺激旷场活动。OMS182401作为单独试剂没有作用(甚至在高于1mg/kg的剂量下,数据未显示),但显著增强DRD1激动剂SKF82958的刺激作用。
尽管已经举例说明和描述了示例性实施方案,但是应该理解,可以在不偏离本发明的精神和范围的情况下作出各式各样的变动。
序列表
<110> Omeros Corporation
<120> 使用PDE7抑制剂治疗成瘾和冲动控制障碍
<130> NE.1.0172.PCT
<140>
<141>
<150> 61/643,611
<151> 2012-05-07
<150> 13/835,607
<151> 2013-03-15
<160> 6
<170> PatentIn version 3.5
<210> 1
<211> 1739
<212> DNA
<213> 人类
<220>
<221> CDS
<222> (157)..(1527)
<400> 1
ggggatcact gttggaaggc agctgcttga ggtccaaggc agtcagtgtc ccctctcttt 60
tgcctcggga cagctggtat ttatcagact cctaagaagt tttccttgct ccctagtaga 120
agagagagat tatgcagcgg gcttttgatt gatcca atg gga att aca ttg atc 174
Met Gly Ile Thr Leu Ile
1 5
tgg tgt ctg gcc ttg gtt ctt atc aag tgg atc acc tct aag agg cgt 222
Trp Cys Leu Ala Leu Val Leu Ile Lys Trp Ile Thr Ser Lys Arg Arg
10 15 20
gga gct att tcc tat gac agt tct gat cag act gca tta tac att cgt 270
Gly Ala Ile Ser Tyr Asp Ser Ser Asp Gln Thr Ala Leu Tyr Ile Arg
25 30 35
atg cta gga gat gta cgt gta agg agc cga gca gga ttt gaa tca gaa 318
Met Leu Gly Asp Val Arg Val Arg Ser Arg Ala Gly Phe Glu Ser Glu
40 45 50
aga aga ggt tct cac cca tat att gat ttt cgt att ttc cac tct caa 366
Arg Arg Gly Ser His Pro Tyr Ile Asp Phe Arg Ile Phe His Ser Gln
55 60 65 70
tct gaa att gaa gtg tct gtc tct gca agg aat atc aga agg cta cta 414
Ser Glu Ile Glu Val Ser Val Ser Ala Arg Asn Ile Arg Arg Leu Leu
75 80 85
agt ttc cag cga tat ctt aga tct tca cgc ttt ttt cgt ggt act gcg 462
Ser Phe Gln Arg Tyr Leu Arg Ser Ser Arg Phe Phe Arg Gly Thr Ala
90 95 100
gtt tca aat tcc cta aac att tta gat gat gat tat aat gga caa gcc 510
Val Ser Asn Ser Leu Asn Ile Leu Asp Asp Asp Tyr Asn Gly Gln Ala
105 110 115
aag tgt atg ctg gaa aaa gtt gga aat tgg aat ttt gat atc ttt cta 558
Lys Cys Met Leu Glu Lys Val Gly Asn Trp Asn Phe Asp Ile Phe Leu
120 125 130
ttt gat aga cta aca aat gga aat agt cta gta agc tta acc ttt cat 606
Phe Asp Arg Leu Thr Asn Gly Asn Ser Leu Val Ser Leu Thr Phe His
135 140 145 150
tta ttt agt ctt cat gga tta att gag tac ttc cat tta gat atg atg 654
Leu Phe Ser Leu His Gly Leu Ile Glu Tyr Phe His Leu Asp Met Met
155 160 165
aaa ctt cgt aga ttt tta gtt atg att caa gaa gat tac cac agt caa 702
Lys Leu Arg Arg Phe Leu Val Met Ile Gln Glu Asp Tyr His Ser Gln
170 175 180
aat cct tac cat aac gca gtc cac gct gcg gat gtt act cag gcc atg 750
Asn Pro Tyr His Asn Ala Val His Ala Ala Asp Val Thr Gln Ala Met
185 190 195
cac tgt tac tta aag gaa cct aag ctt gcc aat tct gta act cct tgg 798
His Cys Tyr Leu Lys Glu Pro Lys Leu Ala Asn Ser Val Thr Pro Trp
200 205 210
gat atc ttg ctg agc tta att gca gct gcc act cat gat ctg gat cat 846
Asp Ile Leu Leu Ser Leu Ile Ala Ala Ala Thr His Asp Leu Asp His
215 220 225 230
cca ggt gtt aat caa cct ttc ctt att aaa act aac cat tac ttg gca 894
Pro Gly Val Asn Gln Pro Phe Leu Ile Lys Thr Asn His Tyr Leu Ala
235 240 245
act tta tac aag aat acc tca gta ctg gaa aat cac cac tgg aga tct 942
Thr Leu Tyr Lys Asn Thr Ser Val Leu Glu Asn His His Trp Arg Ser
250 255 260
gca gtg ggc tta ttg aga gaa tca ggc tta ttc tca cat ctg cca tta 990
Ala Val Gly Leu Leu Arg Glu Ser Gly Leu Phe Ser His Leu Pro Leu
265 270 275
gaa agc agg caa caa atg gag aca cag ata ggt gct ctg ata cta gcc 1038
Glu Ser Arg Gln Gln Met Glu Thr Gln Ile Gly Ala Leu Ile Leu Ala
280 285 290
aca gac atc agt cgc cag aat gag tat ctg tct ttg ttt agg tcc cat 1086
Thr Asp Ile Ser Arg Gln Asn Glu Tyr Leu Ser Leu Phe Arg Ser His
295 300 305 310
ttg gat aga ggt gat tta tgc cta gaa gac acc aga cac aga cat ttg 1134
Leu Asp Arg Gly Asp Leu Cys Leu Glu Asp Thr Arg His Arg His Leu
315 320 325
gtt tta cag atg gct ttg aaa tgt gct gat att tgt aac cca tgt cgg 1182
Val Leu Gln Met Ala Leu Lys Cys Ala Asp Ile Cys Asn Pro Cys Arg
330 335 340
acg tgg gaa tta agc aag cag tgg agt gaa aaa gta acg gag gaa ttc 1230
Thr Trp Glu Leu Ser Lys Gln Trp Ser Glu Lys Val Thr Glu Glu Phe
345 350 355
ttc cat caa gga gat ata gaa aaa aaa tat cat ttg ggt gtg agt cca 1278
Phe His Gln Gly Asp Ile Glu Lys Lys Tyr His Leu Gly Val Ser Pro
360 365 370
ctt tgc gat cgt cac act gaa tct att gcc aac atc cag att ggt ttt 1326
Leu Cys Asp Arg His Thr Glu Ser Ile Ala Asn Ile Gln Ile Gly Phe
375 380 385 390
atg act tac cta gtg gag cct tta ttt aca gaa tgg gcc agg ttt tcc 1374
Met Thr Tyr Leu Val Glu Pro Leu Phe Thr Glu Trp Ala Arg Phe Ser
395 400 405
aat aca agg cta tcc cag aca atg ctt gga cac gtg ggg ctg aat aaa 1422
Asn Thr Arg Leu Ser Gln Thr Met Leu Gly His Val Gly Leu Asn Lys
410 415 420
gcc agc tgg aag gga ctg cag aga gaa cag tcg agc agt gag gac act 1470
Ala Ser Trp Lys Gly Leu Gln Arg Glu Gln Ser Ser Ser Glu Asp Thr
425 430 435
gat gct gca ttt gag ttg aac tca cag tta tta cct cag gaa aat cgg 1518
Asp Ala Ala Phe Glu Leu Asn Ser Gln Leu Leu Pro Gln Glu Asn Arg
440 445 450
tta tca taa cccccagaac cagtgggaca aactgcctcc tggaggtttt 1567
Leu Ser
455
tagaaatgtg aaatggggtc ttgaggtgag agaacttaac tcttgactgc caaggtttcc 1627
aagtgagtga tgccagccag cattatttat ttccaagatt tcctctgttg gatcatttga 1687
acccacttgt taattgcaag acccgaacat acagcaatat gaatttggct tt 1739
<210> 2
<211> 456
<212> PRT
<213> 人类
<400> 2
Met Gly Ile Thr Leu Ile Trp Cys Leu Ala Leu Val Leu Ile Lys Trp
1 5 10 15
Ile Thr Ser Lys Arg Arg Gly Ala Ile Ser Tyr Asp Ser Ser Asp Gln
20 25 30
Thr Ala Leu Tyr Ile Arg Met Leu Gly Asp Val Arg Val Arg Ser Arg
35 40 45
Ala Gly Phe Glu Ser Glu Arg Arg Gly Ser His Pro Tyr Ile Asp Phe
50 55 60
Arg Ile Phe His Ser Gln Ser Glu Ile Glu Val Ser Val Ser Ala Arg
65 70 75 80
Asn Ile Arg Arg Leu Leu Ser Phe Gln Arg Tyr Leu Arg Ser Ser Arg
85 90 95
Phe Phe Arg Gly Thr Ala Val Ser Asn Ser Leu Asn Ile Leu Asp Asp
100 105 110
Asp Tyr Asn Gly Gln Ala Lys Cys Met Leu Glu Lys Val Gly Asn Trp
115 120 125
Asn Phe Asp Ile Phe Leu Phe Asp Arg Leu Thr Asn Gly Asn Ser Leu
130 135 140
Val Ser Leu Thr Phe His Leu Phe Ser Leu His Gly Leu Ile Glu Tyr
145 150 155 160
Phe His Leu Asp Met Met Lys Leu Arg Arg Phe Leu Val Met Ile Gln
165 170 175
Glu Asp Tyr His Ser Gln Asn Pro Tyr His Asn Ala Val His Ala Ala
180 185 190
Asp Val Thr Gln Ala Met His Cys Tyr Leu Lys Glu Pro Lys Leu Ala
195 200 205
Asn Ser Val Thr Pro Trp Asp Ile Leu Leu Ser Leu Ile Ala Ala Ala
210 215 220
Thr His Asp Leu Asp His Pro Gly Val Asn Gln Pro Phe Leu Ile Lys
225 230 235 240
Thr Asn His Tyr Leu Ala Thr Leu Tyr Lys Asn Thr Ser Val Leu Glu
245 250 255
Asn His His Trp Arg Ser Ala Val Gly Leu Leu Arg Glu Ser Gly Leu
260 265 270
Phe Ser His Leu Pro Leu Glu Ser Arg Gln Gln Met Glu Thr Gln Ile
275 280 285
Gly Ala Leu Ile Leu Ala Thr Asp Ile Ser Arg Gln Asn Glu Tyr Leu
290 295 300
Ser Leu Phe Arg Ser His Leu Asp Arg Gly Asp Leu Cys Leu Glu Asp
305 310 315 320
Thr Arg His Arg His Leu Val Leu Gln Met Ala Leu Lys Cys Ala Asp
325 330 335
Ile Cys Asn Pro Cys Arg Thr Trp Glu Leu Ser Lys Gln Trp Ser Glu
340 345 350
Lys Val Thr Glu Glu Phe Phe His Gln Gly Asp Ile Glu Lys Lys Tyr
355 360 365
His Leu Gly Val Ser Pro Leu Cys Asp Arg His Thr Glu Ser Ile Ala
370 375 380
Asn Ile Gln Ile Gly Phe Met Thr Tyr Leu Val Glu Pro Leu Phe Thr
385 390 395 400
Glu Trp Ala Arg Phe Ser Asn Thr Arg Leu Ser Gln Thr Met Leu Gly
405 410 415
His Val Gly Leu Asn Lys Ala Ser Trp Lys Gly Leu Gln Arg Glu Gln
420 425 430
Ser Ser Ser Glu Asp Thr Asp Ala Ala Phe Glu Leu Asn Ser Gln Leu
435 440 445
Leu Pro Gln Glu Asn Arg Leu Ser
450 455
<210> 3
<211> 2990
<212> DNA
<213> 人类
<220>
<221> CDS
<222> (1)..(1275)
<400> 3
atg gaa gtg tgt tac cag ctg ccg gta ctg ccc ctg gac agg ccg gtc 48
Met Glu Val Cys Tyr Gln Leu Pro Val Leu Pro Leu Asp Arg Pro Val
1 5 10 15
ccc cag cac gtc ctc agc cgc cga gga gcc atc agc ttc agc tcc agc 96
Pro Gln His Val Leu Ser Arg Arg Gly Ala Ile Ser Phe Ser Ser Ser
20 25 30
tcc gct ctc ttc ggc tgc ccc aat ccc cgg cag ctc tct cag agg cgt 144
Ser Ala Leu Phe Gly Cys Pro Asn Pro Arg Gln Leu Ser Gln Arg Arg
35 40 45
gga gct att tcc tat gac agt tct gat cag act gca tta tac att cgt 192
Gly Ala Ile Ser Tyr Asp Ser Ser Asp Gln Thr Ala Leu Tyr Ile Arg
50 55 60
atg cta gga gat gta cgt gta agg agc cga gca gga ttt gaa tca gaa 240
Met Leu Gly Asp Val Arg Val Arg Ser Arg Ala Gly Phe Glu Ser Glu
65 70 75 80
aga aga ggt tct cac cca tat att gat ttt cgt att ttc cac tct caa 288
Arg Arg Gly Ser His Pro Tyr Ile Asp Phe Arg Ile Phe His Ser Gln
85 90 95
tct gaa att gaa gtg tct gtc tct gca agg aat atc aga agg cta cta 336
Ser Glu Ile Glu Val Ser Val Ser Ala Arg Asn Ile Arg Arg Leu Leu
100 105 110
agt ttc cag cga tat ctt aga tct tca cgc ttt ttt cgt ggt act gcg 384
Ser Phe Gln Arg Tyr Leu Arg Ser Ser Arg Phe Phe Arg Gly Thr Ala
115 120 125
gtt tca aat tcc cta aac att tta gat gat gat tat aat gga caa gcc 432
Val Ser Asn Ser Leu Asn Ile Leu Asp Asp Asp Tyr Asn Gly Gln Ala
130 135 140
aag tgt atg ctg gaa aaa gtt gga aat tgg aat ttt gat atc ttt cta 480
Lys Cys Met Leu Glu Lys Val Gly Asn Trp Asn Phe Asp Ile Phe Leu
145 150 155 160
ttt gat aga cta aca aat gga aat agt cta gta agc tta acc ttt cat 528
Phe Asp Arg Leu Thr Asn Gly Asn Ser Leu Val Ser Leu Thr Phe His
165 170 175
tta ttt agt ctt cat gga tta att gag tac ttc cat tta gat atg atg 576
Leu Phe Ser Leu His Gly Leu Ile Glu Tyr Phe His Leu Asp Met Met
180 185 190
aaa ctt cgt aga ttt tta gtt atg att caa gaa gat tac cac agt caa 624
Lys Leu Arg Arg Phe Leu Val Met Ile Gln Glu Asp Tyr His Ser Gln
195 200 205
aat cct tac cat aac gca gtc cac gct gcg gat gtt act cag gcc atg 672
Asn Pro Tyr His Asn Ala Val His Ala Ala Asp Val Thr Gln Ala Met
210 215 220
cac tgt tac tta aag gaa cct aag ctt gcc aat tct gta act cct tgg 720
His Cys Tyr Leu Lys Glu Pro Lys Leu Ala Asn Ser Val Thr Pro Trp
225 230 235 240
gat atc ttg ctg agc tta att gca gct gcc act cat gat ctg gat cat 768
Asp Ile Leu Leu Ser Leu Ile Ala Ala Ala Thr His Asp Leu Asp His
245 250 255
cca ggt gtt aat caa cct ttc ctt att aaa act aac cat tac ttg gca 816
Pro Gly Val Asn Gln Pro Phe Leu Ile Lys Thr Asn His Tyr Leu Ala
260 265 270
act tta tac aag aat acc tca gta ctg gaa aat cac cac tgg aga tct 864
Thr Leu Tyr Lys Asn Thr Ser Val Leu Glu Asn His His Trp Arg Ser
275 280 285
gca gtg ggc tta ttg aga gaa tca ggc tta ttc tca cat ctg cca tta 912
Ala Val Gly Leu Leu Arg Glu Ser Gly Leu Phe Ser His Leu Pro Leu
290 295 300
gaa agc agg caa caa atg gag aca cag ata ggt gct ctg ata cta gcc 960
Glu Ser Arg Gln Gln Met Glu Thr Gln Ile Gly Ala Leu Ile Leu Ala
305 310 315 320
aca gac atc agt cgc cag aat gag tat ctg tct ttg ttt agg tcc cat 1008
Thr Asp Ile Ser Arg Gln Asn Glu Tyr Leu Ser Leu Phe Arg Ser His
325 330 335
ttg gat aga ggt gat tta tgc cta gaa gac acc aga cac aga cat ttg 1056
Leu Asp Arg Gly Asp Leu Cys Leu Glu Asp Thr Arg His Arg His Leu
340 345 350
gtt tta cag atg gct ttg aaa tgt gct gat att tgt aac cca tgt cgg 1104
Val Leu Gln Met Ala Leu Lys Cys Ala Asp Ile Cys Asn Pro Cys Arg
355 360 365
acg tgg gaa tta agc aag cag tgg agt gaa aaa gta acg gag gaa ttc 1152
Thr Trp Glu Leu Ser Lys Gln Trp Ser Glu Lys Val Thr Glu Glu Phe
370 375 380
ttc cat caa gga gat ata gaa aaa aaa tat cat ttg ggt gtg agt cca 1200
Phe His Gln Gly Asp Ile Glu Lys Lys Tyr His Leu Gly Val Ser Pro
385 390 395 400
ctt tgc gat cgt cac act gaa tct att gcc aac atc cag att ggt aac 1248
Leu Cys Asp Arg His Thr Glu Ser Ile Ala Asn Ile Gln Ile Gly Asn
405 410 415
tat aca tat tta gat ata gct ggt tag aaaaatgcca ctgtttttat 1295
Tyr Thr Tyr Leu Asp Ile Ala Gly
420
caagaaggga aatatatttg aaatataaaa tattaaaatt atgctcattt ctatttttaa 1355
aaataattta agaaatttta cccttgtttt cccttgttat ggctcttcta attctcattt 1415
aattttagga tgtaaaaagt atatttttgc agaacaggca gcagcaataa cttgtttctg 1475
ttcttatgta aataagaatc cattattcgc tcatgtggaa gcttcttttg catcatttgg 1535
gactgccatt taaaaaagga taggtaaaca aagaaatgac aaaaataaaa taaataaaat 1595
aaaaatggat aggtggtgac ccactgagcc tgatcataat acgaagacca gcttctgcca 1655
ctgcctttcc agactcttac cactgcctgt tgattaaatc taactcttca acatcctaga 1715
caggccctta taatcttgct tcaaatgctg tgcagccatc ttgcctcaac ttccctctca 1775
tttgcctaca gcatctcggg acgcttctgt gtttcccaag tatacgctgt tctttcgctc 1835
tttgtgcttc gccagtgctt tccatgtgcc tcgtagagtt atttttcttg aagaggcagc 1895
tcaaatgtca ccttctccag aagctgctct ccacttgctt taggcagagt cagtcacttt 1955
tcttctagat tccaaagtgc ctgatccact tggttgtgga ttcctggagc ctagcaccac 2015
accagaagca cgaggccctt gagaactgtg tgttgagtga actaataact gtattataga 2075
aagcataatg aaaatgtcct gtgactgaag tatgtgtagc ttgttgcagg agtcacagga 2135
aagttgacta ggattgagtg tgttgggctt tgggtataaa ggagggggat tctacggggg 2195
cagtagctca acaaggaata gagggaggag tgtaattttg gtagctggtg ttgaataggg 2255
cctttgagaa tcagactgaa cacagtgaaa tatgtgccca aagttcagaa agatgaagtt 2315
tccagaaact aagaaggtag cacaatatgt ggcatcatac tcagaaagga agaccatgcc 2375
atggggccag aaattcagaa acgtaattct tacattgtga ttgcaatgga tactcatgaa 2435
agaaagtggg tagtggccga tttgccttca gagtgacagg tagagaaggg aagagcgtgt 2495
agaactgtgg ccatacttta ggagtgtgag ggatgctgaa tctcccagag agctcacact 2555
ggccaggaat gctgagagta gcagatgctt ttcttttggg aggatagtaa aacaatttag 2615
aaccagatat gctttgtctt gattctcaag tagaataatc ttcaaatgca aaagaataca 2675
ttagaaatgg acaaaagtgg ccaggagcgg tagctcatac ttgtaaccca gcactttggg 2735
aagccgaggc gggctgatcg cttgaggtca ggagttcgag accagcctgg ccaaaatagt 2795
gaaactcacg tttctactaa aaatacaaaa attagctggg tgtgatggcc acttgggagg 2855
ctgagatagg agaatcgctt gaacctggga ggcagaggtt gcagtgagcc aatatcgtgc 2915
cactgcattc cagcctgggt gacagaatga aactccatca ctccatctca aaaaaaaaaa 2975
aaaaaaaaaa aaaaa 2990
<210> 4
<211> 424
<212> PRT
<213> 人类
<400> 4
Met Glu Val Cys Tyr Gln Leu Pro Val Leu Pro Leu Asp Arg Pro Val
1 5 10 15
Pro Gln His Val Leu Ser Arg Arg Gly Ala Ile Ser Phe Ser Ser Ser
20 25 30
Ser Ala Leu Phe Gly Cys Pro Asn Pro Arg Gln Leu Ser Gln Arg Arg
35 40 45
Gly Ala Ile Ser Tyr Asp Ser Ser Asp Gln Thr Ala Leu Tyr Ile Arg
50 55 60
Met Leu Gly Asp Val Arg Val Arg Ser Arg Ala Gly Phe Glu Ser Glu
65 70 75 80
Arg Arg Gly Ser His Pro Tyr Ile Asp Phe Arg Ile Phe His Ser Gln
85 90 95
Ser Glu Ile Glu Val Ser Val Ser Ala Arg Asn Ile Arg Arg Leu Leu
100 105 110
Ser Phe Gln Arg Tyr Leu Arg Ser Ser Arg Phe Phe Arg Gly Thr Ala
115 120 125
Val Ser Asn Ser Leu Asn Ile Leu Asp Asp Asp Tyr Asn Gly Gln Ala
130 135 140
Lys Cys Met Leu Glu Lys Val Gly Asn Trp Asn Phe Asp Ile Phe Leu
145 150 155 160
Phe Asp Arg Leu Thr Asn Gly Asn Ser Leu Val Ser Leu Thr Phe His
165 170 175
Leu Phe Ser Leu His Gly Leu Ile Glu Tyr Phe His Leu Asp Met Met
180 185 190
Lys Leu Arg Arg Phe Leu Val Met Ile Gln Glu Asp Tyr His Ser Gln
195 200 205
Asn Pro Tyr His Asn Ala Val His Ala Ala Asp Val Thr Gln Ala Met
210 215 220
His Cys Tyr Leu Lys Glu Pro Lys Leu Ala Asn Ser Val Thr Pro Trp
225 230 235 240
Asp Ile Leu Leu Ser Leu Ile Ala Ala Ala Thr His Asp Leu Asp His
245 250 255
Pro Gly Val Asn Gln Pro Phe Leu Ile Lys Thr Asn His Tyr Leu Ala
260 265 270
Thr Leu Tyr Lys Asn Thr Ser Val Leu Glu Asn His His Trp Arg Ser
275 280 285
Ala Val Gly Leu Leu Arg Glu Ser Gly Leu Phe Ser His Leu Pro Leu
290 295 300
Glu Ser Arg Gln Gln Met Glu Thr Gln Ile Gly Ala Leu Ile Leu Ala
305 310 315 320
Thr Asp Ile Ser Arg Gln Asn Glu Tyr Leu Ser Leu Phe Arg Ser His
325 330 335
Leu Asp Arg Gly Asp Leu Cys Leu Glu Asp Thr Arg His Arg His Leu
340 345 350
Val Leu Gln Met Ala Leu Lys Cys Ala Asp Ile Cys Asn Pro Cys Arg
355 360 365
Thr Trp Glu Leu Ser Lys Gln Trp Ser Glu Lys Val Thr Glu Glu Phe
370 375 380
Phe His Gln Gly Asp Ile Glu Lys Lys Tyr His Leu Gly Val Ser Pro
385 390 395 400
Leu Cys Asp Arg His Thr Glu Ser Ile Ala Asn Ile Gln Ile Gly Asn
405 410 415
Tyr Thr Tyr Leu Asp Ile Ala Gly
420
<210> 5
<211> 2100
<212> DNA
<213> 人类
<220>
<221> CDS
<222> (304)..(1656)
<400> 5
cagtcagttg gtctgggcac tgcagcaggc tcggctctgt cccagcactt gtctgggaga 60
aaagtggtgt tactcaccca gggagagtct ctctttctac cttccttctt tctcgatctc 120
cttgtgtgct tttgtgtttc tttatttctt ttcctttttt ttcttttttt ttttttgtta 180
cttaattata ttcctaatcc tggatgaagt tgctggattc tgcagcacaa gtcttcatga 240
acaagcagca ccgctcagag atttcacggc attcaaaggt cacagaactg ccactatggt 300
taa atg tct tgt tta atg gtt gag agg tgt ggc gaa atc ttg ttt gag 348
Met Ser Cys Leu Met Val Glu Arg Cys Gly Glu Ile Leu Phe Glu
1 5 10 15
aac ccc gat cag aat gcc aaa tgt gtt tgc atg ctg gga gat ata cga 396
Asn Pro Asp Gln Asn Ala Lys Cys Val Cys Met Leu Gly Asp Ile Arg
20 25 30
cta agg ggt cag acg ggg gtt cgt gct gaa cgc cgt ggc tcc tac cca 444
Leu Arg Gly Gln Thr Gly Val Arg Ala Glu Arg Arg Gly Ser Tyr Pro
35 40 45
ttc att gac ttc cgc cta ctt aac agt aca aca tac tca ggg gag att 492
Phe Ile Asp Phe Arg Leu Leu Asn Ser Thr Thr Tyr Ser Gly Glu Ile
50 55 60
ggc acc aag aaa aag gtg aaa aga cta tta agc ttt caa aga tac ttc 540
Gly Thr Lys Lys Lys Val Lys Arg Leu Leu Ser Phe Gln Arg Tyr Phe
65 70 75
cat gca tca agg ctg ctt cgt gga att ata cca caa gcc cct ctg cac 588
His Ala Ser Arg Leu Leu Arg Gly Ile Ile Pro Gln Ala Pro Leu His
80 85 90 95
ctg ctg gat gaa gac tac ctt gga caa gca agg cat atg ctc tcc aaa 636
Leu Leu Asp Glu Asp Tyr Leu Gly Gln Ala Arg His Met Leu Ser Lys
100 105 110
gtg gga atg tgg gat ttt gac att ttc ttg ttt gat cgc ttg aca aat 684
Val Gly Met Trp Asp Phe Asp Ile Phe Leu Phe Asp Arg Leu Thr Asn
115 120 125
gga aac agc ctg gta aca ctg ttg tgc cac ctc ttc aat acc cat gga 732
Gly Asn Ser Leu Val Thr Leu Leu Cys His Leu Phe Asn Thr His Gly
130 135 140
ctc att cac cat ttc aag tta gat atg gtg acc tta cac cga ttt tta 780
Leu Ile His His Phe Lys Leu Asp Met Val Thr Leu His Arg Phe Leu
145 150 155
gtc atg gtt caa gaa gat tac cac agc caa aac ccg tat cac aat gct 828
Val Met Val Gln Glu Asp Tyr His Ser Gln Asn Pro Tyr His Asn Ala
160 165 170 175
gtt cac gca gcc gac gtc acc cag gcc atg cac tgc tac ctg aaa gag 876
Val His Ala Ala Asp Val Thr Gln Ala Met His Cys Tyr Leu Lys Glu
180 185 190
cca aag ctt gcc agc ttc ctc acg cct ctg gac atc atg ctt gga ctg 924
Pro Lys Leu Ala Ser Phe Leu Thr Pro Leu Asp Ile Met Leu Gly Leu
195 200 205
ctg gct gca gca gca cac gat gtg gac cac cca ggg gtg aac cag cca 972
Leu Ala Ala Ala Ala His Asp Val Asp His Pro Gly Val Asn Gln Pro
210 215 220
ttt ttg ata aaa act aac cac cat ctt gca aac cta tat cag aat atg 1020
Phe Leu Ile Lys Thr Asn His His Leu Ala Asn Leu Tyr Gln Asn Met
225 230 235
tct gtg ctg gag aat cat cac tgg cga tct aca att ggc atg ctt cga 1068
Ser Val Leu Glu Asn His His Trp Arg Ser Thr Ile Gly Met Leu Arg
240 245 250 255
gaa tca agg ctt ctt gct cat ttg cca aag gaa atg aca cag gat att 1116
Glu Ser Arg Leu Leu Ala His Leu Pro Lys Glu Met Thr Gln Asp Ile
260 265 270
gaa cag cag ctg ggc tcc ttg atc ttg gca aca gac atc aac agg cag 1164
Glu Gln Gln Leu Gly Ser Leu Ile Leu Ala Thr Asp Ile Asn Arg Gln
275 280 285
aat gaa ttt ttg acc aga ttg aaa gct cac ctc cac aat aaa gac tta 1212
Asn Glu Phe Leu Thr Arg Leu Lys Ala His Leu His Asn Lys Asp Leu
290 295 300
aga ctg gag gat gca cag gac agg cac ttt atg ctt cag atc gcc ttg 1260
Arg Leu Glu Asp Ala Gln Asp Arg His Phe Met Leu Gln Ile Ala Leu
305 310 315
aag tgt gct gac att tgc aat cct tgt aga atc tgg gag atg agc aag 1308
Lys Cys Ala Asp Ile Cys Asn Pro Cys Arg Ile Trp Glu Met Ser Lys
320 325 330 335
cag tgg agt gaa agg gtc tgt gaa gaa ttc tac agg caa ggt gaa ctt 1356
Gln Trp Ser Glu Arg Val Cys Glu Glu Phe Tyr Arg Gln Gly Glu Leu
340 345 350
gaa cag aaa ttt gaa ctg gaa atc agt cct ctt tgt aat caa cag aaa 1404
Glu Gln Lys Phe Glu Leu Glu Ile Ser Pro Leu Cys Asn Gln Gln Lys
355 360 365
gat tcc atc cct agt ata caa att ggt ttc atg agc tac atc gtg gag 1452
Asp Ser Ile Pro Ser Ile Gln Ile Gly Phe Met Ser Tyr Ile Val Glu
370 375 380
ccg ctc ttc cgg gaa tgg gcc cat ttc acg ggt aac agc acc ctg tcg 1500
Pro Leu Phe Arg Glu Trp Ala His Phe Thr Gly Asn Ser Thr Leu Ser
385 390 395
gag aac atg ctg ggc cac ctc gca cac aac aag gcc cag tgg aag agc 1548
Glu Asn Met Leu Gly His Leu Ala His Asn Lys Ala Gln Trp Lys Ser
400 405 410 415
ctg ttg ccc agg cag cac aga agc agg ggc agc agt ggc agc ggg cct 1596
Leu Leu Pro Arg Gln His Arg Ser Arg Gly Ser Ser Gly Ser Gly Pro
420 425 430
gac cac gac cac gca ggc caa ggg act gag agc gag gag cag gaa ggc 1644
Asp His Asp His Ala Gly Gln Gly Thr Glu Ser Glu Glu Gln Glu Gly
435 440 445
gac agc ccc tag gggccggccc aacttagacg cggctctcct ccggcagggc 1696
Asp Ser Pro
450
ccccagaggg cagaagcagc gtggaggggc cctcacgcag cagcccagcc actttctgag 1756
tgttgtcctg gggctctttg gaacgccatc ttcctcccac ttacctgcct cccctccttt 1816
tcgcaaatgt acagaagcca tttgtcacct cagcattcgc tgccgaaatg agcaactcca 1876
ttcagtaacg tgggagctga tcccacgggc aggctctccc tgctccagga gaagactagg 1936
aggaagaatg aggtgctcct gccgtgtccg ccttgttccg ggtcgcactg gaacaggcag 1996
caattcctaa gtccggagcg tttgagcgtt tgctatctga ctgctgatct gcgtgacaga 2056
aacaccagca tatttgcaac gccaaggata ttggtcttaa gtgc 2100
<210> 6
<211> 450
<212> PRT
<213> 人类
<400> 6
Met Ser Cys Leu Met Val Glu Arg Cys Gly Glu Ile Leu Phe Glu Asn
1 5 10 15
Pro Asp Gln Asn Ala Lys Cys Val Cys Met Leu Gly Asp Ile Arg Leu
20 25 30
Arg Gly Gln Thr Gly Val Arg Ala Glu Arg Arg Gly Ser Tyr Pro Phe
35 40 45
Ile Asp Phe Arg Leu Leu Asn Ser Thr Thr Tyr Ser Gly Glu Ile Gly
50 55 60
Thr Lys Lys Lys Val Lys Arg Leu Leu Ser Phe Gln Arg Tyr Phe His
65 70 75 80
Ala Ser Arg Leu Leu Arg Gly Ile Ile Pro Gln Ala Pro Leu His Leu
85 90 95
Leu Asp Glu Asp Tyr Leu Gly Gln Ala Arg His Met Leu Ser Lys Val
100 105 110
Gly Met Trp Asp Phe Asp Ile Phe Leu Phe Asp Arg Leu Thr Asn Gly
115 120 125
Asn Ser Leu Val Thr Leu Leu Cys His Leu Phe Asn Thr His Gly Leu
130 135 140
Ile His His Phe Lys Leu Asp Met Val Thr Leu His Arg Phe Leu Val
145 150 155 160
Met Val Gln Glu Asp Tyr His Ser Gln Asn Pro Tyr His Asn Ala Val
165 170 175
His Ala Ala Asp Val Thr Gln Ala Met His Cys Tyr Leu Lys Glu Pro
180 185 190
Lys Leu Ala Ser Phe Leu Thr Pro Leu Asp Ile Met Leu Gly Leu Leu
195 200 205
Ala Ala Ala Ala His Asp Val Asp His Pro Gly Val Asn Gln Pro Phe
210 215 220
Leu Ile Lys Thr Asn His His Leu Ala Asn Leu Tyr Gln Asn Met Ser
225 230 235 240
Val Leu Glu Asn His His Trp Arg Ser Thr Ile Gly Met Leu Arg Glu
245 250 255
Ser Arg Leu Leu Ala His Leu Pro Lys Glu Met Thr Gln Asp Ile Glu
260 265 270
Gln Gln Leu Gly Ser Leu Ile Leu Ala Thr Asp Ile Asn Arg Gln Asn
275 280 285
Glu Phe Leu Thr Arg Leu Lys Ala His Leu His Asn Lys Asp Leu Arg
290 295 300
Leu Glu Asp Ala Gln Asp Arg His Phe Met Leu Gln Ile Ala Leu Lys
305 310 315 320
Cys Ala Asp Ile Cys Asn Pro Cys Arg Ile Trp Glu Met Ser Lys Gln
325 330 335
Trp Ser Glu Arg Val Cys Glu Glu Phe Tyr Arg Gln Gly Glu Leu Glu
340 345 350
Gln Lys Phe Glu Leu Glu Ile Ser Pro Leu Cys Asn Gln Gln Lys Asp
355 360 365
Ser Ile Pro Ser Ile Gln Ile Gly Phe Met Ser Tyr Ile Val Glu Pro
370 375 380
Leu Phe Arg Glu Trp Ala His Phe Thr Gly Asn Ser Thr Leu Ser Glu
385 390 395 400
Asn Met Leu Gly His Leu Ala His Asn Lys Ala Gln Trp Lys Ser Leu
405 410 415
Leu Pro Arg Gln His Arg Ser Arg Gly Ser Ser Gly Ser Gly Pro Asp
420 425 430
His Asp His Ala Gly Gln Gly Thr Glu Ser Glu Glu Gln Glu Gly Asp
435 440 445
Ser Pro
450
Claims (24)
1.成瘾的治疗方法,所述成瘾是对成瘾物质成瘾或是与原发性冲动控制障碍或强迫症有关的强迫行为的实施,所述方法包括:
确定受试者患有成瘾或有发生成瘾的危险;和
向所述受试者施用一定量的磷酸二酯酶7(PDE7)抑制剂和多巴胺能剂,其中所述PDE7抑制剂和所述多巴胺能剂可有效治疗或减少成瘾剂的使用或强迫行为的实施。
2.权利要求1的方法,其中所述受试者对成瘾剂成瘾。
3.权利要求2的方法,其中所述受试者对选自酒精、尼古丁、大麻、大麻衍生物、阿片类激动剂、苯并二氮杂䓬、巴比妥酸盐和精神兴奋药的成瘾剂成瘾。
4.权利要求1的方法,其中所述成瘾是与原发性冲动控制障碍或强迫症有关的强迫行为。
5.权利要求4的方法,其中所述原发性冲动控制障碍选自暴食、食瘾、病理性赌博、病理性使用电子设备、病理性使用电子视频游戏、病理性使用电子通讯设备、病理性使用移动电话、对色情作品成瘾、性成瘾、强迫性消费、厌食症、贪食症、间歇性暴发性障碍、盗窃癖、纵火癖、拔毛发癖、强迫过度运动和强迫过度工作。
6.权利要求5的方法,其中所述原发性冲动控制障碍是暴食或食瘾。
7.权利要求1的方法,其中所述多巴胺能剂选自多巴胺前体、多巴胺辅因子、代谢多巴胺的酶的抑制剂、多巴胺受体激动剂、通过代谢转化为多巴胺受体激动剂的前体化合物、多巴胺再摄取抑制剂和多巴胺释放促进剂。
8.权利要求1的方法,其中所述多巴胺能剂选自左旋多巴、卡比多巴、溴隐亭、培高利特、普拉克索、罗匹尼罗、卡麦角林、阿扑吗啡、麦角乙脲、罗替戈汀、喹高利特和非诺多泮。
9.权利要求1的方法,其中所述多巴胺能剂对多巴胺受体D1具有选择性。
10.权利要求9的方法,其中所述多巴胺能剂是多巴胺激动剂。
11.权利要求1的方法,其中所述PDE7抑制剂抑制PDE7A和/或PDE7B活性的IC50小于约1μM。
12.权利要求1的方法,其中所述PDE7抑制剂抑制PDE7A和/或PDE7B活性的IC50小于约100 nM。
13.权利要求1的方法,其中所述PDE7抑制剂是选择性PDE7抑制剂,其抑制PDE7A活性的IC50和抑制PDE7B活性的IC50中的较小者小于该试剂抑制PDE1-6和PDE8-11酶家族中任何其它PDE酶活性的IC50的十分之一。
14.权利要求1的方法,其中所述PDE7抑制剂是选自以下的成员:式1A、式1B、式29、式30、式31、式32、式33、式34、式35、式36、式37、式38、式39、式40、式41、式42、式43A、式43B、式44、式45、式46、式47、式48、式49、式50、式51、式52、式53、式54、式6A、式6B、式6C、式6D、式6E、式6F、式6G、式6H、式16A、化合物1、化合物2、化合物3和化合物4。
15.权利要求1的方法,其中所述PDE7抑制剂能够穿透血/脑屏障。
16.权利要求1的方法,其中所述受试者已经经历了一段时间的成瘾剂或者成瘾或强迫行为实施的戒断、或其限制或减少使用,且所述PDE7抑制剂和所述多巴胺能剂可有效减少该成瘾实施的复发使用。
17.药物组合物,其包含磷酸二酯酶7 (PDE7)抑制剂和多巴胺能剂,其中所述PDE7抑制剂和所述多巴胺能剂各自有助于有效治疗或减少成瘾剂的使用或强迫行为的实施。
18.权利要求17的药物组合物,其中所述多巴胺能剂选自多巴胺前体、多巴胺辅因子、代谢多巴胺的酶的抑制剂、多巴胺受体激动剂、通过代谢转化为多巴胺受体激动剂的前体化合物、多巴胺再摄取抑制剂和多巴胺释放促进剂。
19.权利要求17的药物组合物,其中所述多巴胺能剂选自左旋多巴、卡比多巴、溴隐亭、培高利特、普拉克索、罗匹尼罗、卡麦角林、阿扑吗啡、麦角乙脲、罗替戈汀、喹高利特和非诺多泮。
20.权利要求17的药物组合物,其中所述多巴胺能剂对多巴胺受体D1具有选择性。
21.权利要求20的方法,其中所述多巴胺能剂是多巴胺激动剂。
22.权利要求17的药物组合物,其中所述PDE7抑制剂是选自以下的成员:式1A、式1B、式29、式30、式31、式32、式33、式34、式35、式36、式37、式38、式39、式40、式41、式42、式43A、式43B、式44、式45、式46、式47、式48、式49、式50、式51、式52、式53、式54、式6A、式6B、式6C、式6D、式6E、式6F、式6G、式6H、式16A、化合物1、化合物2、化合物3和化合物4。
23.用于治疗成瘾的药盒,其包括包含磷酸二酯酶7 (PDE7)抑制剂的第一容器和包含多巴胺能剂的第二容器,其中所述PDE7抑制剂和所述多巴胺能剂各自有助于有效治疗或减少成瘾剂的使用或强迫行为的实施。
24.食瘾的治疗方法,其包括:
确定受试者患有食瘾或有产生食瘾的危险;和
向所述受试者施用一定量的磷酸二酯酶7(PDE7)抑制剂和多巴胺能剂,其中所述PDE7抑制剂和所述多巴胺能剂可有效治疗或减少食瘾行为的实施。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261643611P | 2012-05-07 | 2012-05-07 | |
| US61/643611 | 2012-05-07 | ||
| US13/835607 | 2013-03-15 | ||
| US13/835,607 US9220715B2 (en) | 2010-11-08 | 2013-03-15 | Treatment of addiction and impulse-control disorders using PDE7 inhibitors |
| CN201380036048.1A CN104768556A (zh) | 2012-05-07 | 2013-05-07 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201380036048.1A Division CN104768556A (zh) | 2012-05-07 | 2013-05-07 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112168972A true CN112168972A (zh) | 2021-01-05 |
Family
ID=49624481
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202011054538.2A Pending CN112168972A (zh) | 2012-05-07 | 2013-05-07 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
| CN201380036048.1A Pending CN104768556A (zh) | 2012-05-07 | 2013-05-07 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
| CN202210569437.1A Pending CN114984229A (zh) | 2012-05-07 | 2013-05-07 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201380036048.1A Pending CN104768556A (zh) | 2012-05-07 | 2013-05-07 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
| CN202210569437.1A Pending CN114984229A (zh) | 2012-05-07 | 2013-05-07 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
Country Status (10)
| Country | Link |
|---|---|
| EP (1) | EP2846805B1 (zh) |
| JP (1) | JP2015521177A (zh) |
| CN (3) | CN112168972A (zh) |
| AU (1) | AU2013266793B2 (zh) |
| CA (1) | CA2871151C (zh) |
| IN (1) | IN2014DN10404A (zh) |
| PL (1) | PL2846805T3 (zh) |
| RS (1) | RS58381B1 (zh) |
| SI (1) | SI2846805T1 (zh) |
| WO (1) | WO2013176877A2 (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9220715B2 (en) | 2010-11-08 | 2015-12-29 | Omeros Corporation | Treatment of addiction and impulse-control disorders using PDE7 inhibitors |
| CN107375296A (zh) | 2010-11-08 | 2017-11-24 | 奥默罗斯公司 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
| KR20190040054A (ko) * | 2016-08-26 | 2019-04-16 | 미쓰비시 타나베 파마 코퍼레이션 | 이환식 함질소 헤테로환 화합물 |
| WO2023243659A1 (ja) * | 2022-06-15 | 2023-12-21 | 真一 松本 | 線条体ストリオソームのドパミンd1シグナルを標的とした強迫性障害の薬物療法 |
| WO2024038089A1 (en) | 2022-08-18 | 2024-02-22 | Mitodicure Gmbh | Use of a therapeutic agent with phosphodiesterase-7 inhibitory activity for the treatment and prevention of diseases associated with chronic fatigue, exhaustion and/or exertional intolerance |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007038551A2 (en) * | 2005-09-26 | 2007-04-05 | Avigen, Inc. | Use of ibudilast for treating drug and behavioral addictions |
| CN1976938A (zh) * | 2004-07-01 | 2007-06-06 | 第一阿斯比奥制药株式会社 | 具有pde7抑制活性的噻吩并吡唑衍生物 |
| CN101087778A (zh) * | 2004-12-31 | 2007-12-12 | 辉瑞产品有限公司 | 芳香杂环化合物吡咯烷基衍生物磷酸二酯酶抑制剂 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8637528B2 (en) * | 2007-03-27 | 2014-01-28 | Omeros Corporation | Use of PDE7 inhibitors for the treatment of movement disorders |
| CN101917992A (zh) * | 2007-03-27 | 2010-12-15 | 奥默罗斯公司 | 用于治疗运动障碍的pde7抑制剂的用途 |
| KR101622403B1 (ko) * | 2007-04-11 | 2016-05-31 | 오메로스 코포레이션 | 중독의 예방 및 치료용 조성물 및 방법 |
| FR2943673B1 (fr) * | 2009-03-27 | 2013-03-29 | Sanofi Aventis | Applications therapeutiques de derives de quinazolinedione |
| FR2944282B1 (fr) * | 2009-04-09 | 2013-05-03 | Sanofi Aventis | Derives de quinazolinedione, leur preparation et leurs diverses applications therapeutiques |
| CN107375296A (zh) * | 2010-11-08 | 2017-11-24 | 奥默罗斯公司 | 使用pde7抑制剂治疗成瘾和冲动控制障碍 |
-
2013
- 2013-05-07 WO PCT/US2013/039866 patent/WO2013176877A2/en active Application Filing
- 2013-05-07 CN CN202011054538.2A patent/CN112168972A/zh active Pending
- 2013-05-07 SI SI201331354T patent/SI2846805T1/sl unknown
- 2013-05-07 AU AU2013266793A patent/AU2013266793B2/en active Active
- 2013-05-07 CN CN201380036048.1A patent/CN104768556A/zh active Pending
- 2013-05-07 JP JP2015511605A patent/JP2015521177A/ja active Pending
- 2013-05-07 EP EP13793319.8A patent/EP2846805B1/en active Active
- 2013-05-07 CN CN202210569437.1A patent/CN114984229A/zh active Pending
- 2013-05-07 PL PL13793319T patent/PL2846805T3/pl unknown
- 2013-05-07 IN IN10404DEN2014 patent/IN2014DN10404A/en unknown
- 2013-05-07 RS RS20190228A patent/RS58381B1/sr unknown
- 2013-05-07 CA CA2871151A patent/CA2871151C/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1976938A (zh) * | 2004-07-01 | 2007-06-06 | 第一阿斯比奥制药株式会社 | 具有pde7抑制活性的噻吩并吡唑衍生物 |
| CN101087778A (zh) * | 2004-12-31 | 2007-12-12 | 辉瑞产品有限公司 | 芳香杂环化合物吡咯烷基衍生物磷酸二酯酶抑制剂 |
| WO2007038551A2 (en) * | 2005-09-26 | 2007-04-05 | Avigen, Inc. | Use of ibudilast for treating drug and behavioral addictions |
Also Published As
| Publication number | Publication date |
|---|---|
| RS58381B1 (sr) | 2019-04-30 |
| CN104768556A (zh) | 2015-07-08 |
| WO2013176877A2 (en) | 2013-11-28 |
| EP2846805B1 (en) | 2018-11-21 |
| CN114984229A (zh) | 2022-09-02 |
| IN2014DN10404A (zh) | 2015-08-14 |
| PL2846805T3 (pl) | 2019-05-31 |
| CA2871151A1 (en) | 2013-11-28 |
| EP2846805A4 (en) | 2016-08-10 |
| WO2013176877A3 (en) | 2015-06-18 |
| EP2846805A2 (en) | 2015-03-18 |
| AU2013266793B2 (en) | 2016-08-04 |
| JP2015521177A (ja) | 2015-07-27 |
| SI2846805T1 (sl) | 2019-04-30 |
| AU2013266793A1 (en) | 2014-11-27 |
| CA2871151C (en) | 2019-03-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220062194A1 (en) | Treatment of Addiction and Impulse-Control Disorders Using PDE7 Inhibitors | |
| EP2846805B1 (en) | Treatment of addiction and impulse-control disorders using pde7 inhibitors | |
| US11464785B2 (en) | Treatment of addiction and impulse-control disorders using PDE7 inhibitors | |
| JP2021138766A (ja) | Pde7インヒビターを用いる嗜癖および衝動制御障害の処置 | |
| US20230110784A1 (en) | Treatment of addiction and impulse-control disorders using pde7 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| WD01 | Invention patent application deemed withdrawn after publication | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20210105 |