CN110023290B - 作为ripk2抑制剂的杂芳基甲酰胺化合物 - Google Patents
作为ripk2抑制剂的杂芳基甲酰胺化合物 Download PDFInfo
- Publication number
- CN110023290B CN110023290B CN201780056054.1A CN201780056054A CN110023290B CN 110023290 B CN110023290 B CN 110023290B CN 201780056054 A CN201780056054 A CN 201780056054A CN 110023290 B CN110023290 B CN 110023290B
- Authority
- CN
- China
- Prior art keywords
- methyl
- imidazo
- alkyl
- mixture
- pyridin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5355—Non-condensed oxazines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Transplantation (AREA)
- Physical Education & Sports Medicine (AREA)
- Emergency Medicine (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Otolaryngology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Steroid Compounds (AREA)
Abstract
本发明涉及式(I)化合物: 或其药学上可接受的盐,其中R1、R2、X、Y及HET如本文所定义。本发明也涉及包含这些化合物的药物组合物、使用这些化合物治疗各种疾病及病症的方法、制备这些化合物的方法及用于这些方法的中间体。
或其药学上可接受的盐,其中R1、R2、X、Y及HET如本文所定义。本发明也涉及包含这些化合物的药物组合物、使用这些化合物治疗各种疾病及病症的方法、制备这些化合物的方法及用于这些方法的中间体。
Description
发明背景
1.技术领域
本发明涉及一系列新颖杂芳基甲酰胺化合物、这些化合物的合成、其治疗发炎性疾病的用途及包含这些化合物的药物组合物。
2.背景信息
RIPK2(也称为RICK、CARDIAK、CARD3或RIP2)为双特异性丝胺酸/苏胺酸及酪胺酸激酶,其为通过NOD1及NOD2信号传导途径的促炎信号的关键组分(Inohara等人1998;McCarthy等人1998;Thome等人1998;Tigno-Aranjuez等人2010)。NOD受体为用于监测细胞内细菌病原体的机理之一。细菌细胞壁组分通过使NOD1细菌配体D-谷氨酰基-内消旋-二氨基庚二酸及NOD2配体胞壁酰二肽结合到适当的细胞内NOD受体来引发信号通过NOD1及NOD2途径(Girardin等人2003a;Girardin等人2003b;Girardin等人2003c;Chamaillard等人2003;Inohara等人2003)。此结合通过同型CARD/CARD域相互作用诱导NOD蛋白质的寡聚(Inohara等人2000;Ogura等人2001)。此NOD受体的活化通过泛素E3连接酶(诸如XIAP、cIAP1、cIAP2、TRAF2、TRAF5及TRAF6)的活化导致RIPK2的Lys63联结多遍在蛋白化(Krieg等人2009;Bertrand等人2009;Yang等人2007;Hasegawa等人2008)且募集线性泛素系统(LUBAC)(Damgaard等人2012;Ver Heul等人2013)。另外,作为其活化及至NOD信号传导复合物的组装的部分,RIPK2经历酪胺酸474的自磷酸化(Tigno-Aranjuez等人2010)。依赖于信号传导复合物的组装的其他RIPK2导致IKKα/β/γ及TAK1的活化,引起NF-κB及MAPK途径的活化,从而导致产生促炎细胞因子(Yang等人2007)。
NOD2中的突变与多种疾病有关。活化突变与影响皮肤、关节及眼睛的早发型结节病(Kanazawa等人,2005)及Blau综合征(Miceli-Richard等人,2001)有关。这些活化突变导致基础NF-κB活性增加(Kanazawa等人,2005)。NOD2LRR中的功能丧失型突变与克罗恩氏病(Ogura等人2001;Hugot等人2001;Hampe等人2001;Hampe 2002;Lesange 2002)有关。另外,NOD1中的多态现象与特应性(Weidinger等人2005)及哮喘(Hysi等人2005)有关。细胞及活体内小鼠模型中的另外研究已表明NOD1及NOD2信号传导在各种疾病(诸如移植物抗宿主疾病、关节炎、多发性硬化症及糖尿病肾病变)中的作用(Peaneck等人2009;Saha等人2009;Vieira等人2012;Rosenzweig等人2010;Joosten等人2008;Shaw等人2011;Du等人2013)。RIP2激酶(RIPK2)的小分子抑制剂公开于US2013/0023532A1中但似乎效能有限。
通过强力的及选择性的小分子抑制剂的RIPK2的药理学抑制将减弱通过NOD1及NOD2刺激引发的通过细菌感应途径的促炎信号。炎症信号中的这种减少将在各种自身炎症性疾病中提供治疗效益。因此,出于药物目的,需要RIPK2的有效抑制剂。
发明内容
本发明提供新颖杂芳基甲酰胺系列化合物,其抑制受体相互作用丝胺酸/苏胺酸蛋白激酶2(RIPK2),且因此可用于治疗由RIPK2的活性介导或维持的各种疾病及病症,包括炎性疾病、心血管代谢疾病及心血管疾病以及癌症。本发明也涉及包含这些化合物的药物组合物、将这些化合物用于治疗各种疾病及病症的方法、制备这些化合物的方法及可用于这些方法的中间体。
在本发明的一方面,本发明的化合物具有良好结合效能。
在本发明的另一方面,本发明的化合物展现良好细胞效能。
在又一方面,本发明的化合物展现良好稳定性。
在另一方面,本发明的化合物展现良好细胞渗透性。
具体实施方式
在本发明的最广泛实施方案中,本发明涉及式I化合物:

或其药学上可接受的盐,其中:
X为N且Y为CH;或
X为CH且Y为N;
HET为含有选自氮及硫的一至三个杂原子的5元杂芳环,其中各杂芳环任选地经独立地选自R3及R4的一至两个取代基取代;或
HET为含有选自氮及硫的一至三个杂原子的5元杂芳环,其中各杂芳环经选自Ra及Rb的两个取代基取代,其中Ra及Rb与其所连接的原子一起形成5元至6元杂环或杂芳环,所述5元至6元杂环或杂芳环可任选地经选自R3及R4的一至两个取代基取代;
R1为氢或F;
R2为C1-3烷基或Cl;
R3及R4各自独立地选自:
(a)-H,
(b)-OR5,
(c)-O-C1-6烷基-O-C1-3烷基,
(d)-O-C3-6环烷基,
(e)-C(O)R5,
(f)任选地经以下取代的C1-6烷基:一至三个-OH、氟、任选地经氧代取代的杂环基、C3-6环烷基、-CO2R5、-O-C1-6烷基、芳基、-N(R5)(R6)或-C(O)N(R5)(R6),
(g)任选地经以下取代的C3-6环烷基:一至三个-OH、一至三个氟、C1-6烷基、-OC1-6烷基、C1-6烷基-OC1-6烷基、C1-6烷基-OH、CF3、-OC3-6环烷基、-CO2H、-CO2R5、C3-6环烷基、5元至6元杂芳基、C3-6杂环基、N(R5)(R6)或-C(O)N(R5)(R6),
(h)-CO2R5,
(i)-C(O)N(R5)(R6),
(j)-S(O)2N(R5)(R6),
(k)-S(O)n-R5,
(l)任选地经选自以下的一至三个基团取代的5元至6元杂芳基:C1-6烷基、C3-6环烷基、卤素、-CF3、-OH、-(CH2)nCO2R5、-C(O)N(R5)(R6)、-N(R5)(R6)、-NH-SO2C1-6烷基、C1-6烷氧基、C1-6烷基-O-C1-3烷基、C1-6烷基羟基、C1-3烷基-CN、氧代、任选地经卤素取代的苯基及-S(O)nC1-6烷基,
(m)含有N、S或O的4元至10元单环、双环或螺环杂环基,其中各杂环任选地经选自以下的1至3个取代基取代:3元至6元杂环、卤素、-C1-3烷基、-C1-3烷基-O-C1-3烷基及-C1-3烷基-C(O)N(R5)(R6),
(n)芳基,
(o)-N(R5)(R6);
R5及R6各自独立地选自-H、4元至6元杂环基、-C(O)-C1-3烷基-C(O)-C1-3环烷基及-(C1-C6)烷基,其中各R5及R6任选独立地经以下取代:-OH、C3-6环烷基、-C1-3烷基、-O-C1-3烷基、-NH-C1-3烷基或-N-(C1-3-烷基)2;或
R5及R6与其所连接的氮原子一起形成任选地经甲基取代的4元至6元杂环;且
n为0、1或2。
在第二实施方案中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
X为N且Y为CH;或
X为CH且Y为N;
Het为选自吡唑基、咪唑基、噻唑基及噻二唑基的5元杂芳环,其中各杂芳环任选地经独立地选自R3及R4的一至两个取代基取代;或
Het为选自吡唑基及咪唑基的5元杂芳环,其中各杂芳环经选自Ra及Rb的两个取代基取代,其中Ra及Rb与其所连接的原子一起形成5元至6元杂环或杂芳环,所述5元至6元杂环或杂芳环可任选地经选自R3及R4的一至两个取代基取代;
R1为氢或F;
R2为C1-3烷基或Cl;
R3及R4各自独立地选自:
(a)-H,
(b)-OR5,
(c)-O-C1-6烷基-O-C1-3烷基,
(d)-O-C3-6环烷基,
(e)-C(O)R5,
(f)任选地经以下取代的C1-6烷基:一至三个-OH、氟、任选地经氧代取代的杂环基、C3-6环烷基、-CO2R5、-O-C1-6烷基、芳基、-N(R5)(R6)或-C(O)N(R5)(R6),
(g)任选地经以下取代的C3-6环烷基:一至三个-OH、一至三个氟、C1-6烷基、-OC1-6烷基、C1-6烷基-OC1-6烷基、C1-6烷基-OH、CF3、-OC3-6环烷基、-CO2H、-CO2R5、C3-6环烷基、5元至6元杂芳基、C3-6杂环基、N(R5)(R6)或-C(O)N(R5)(R6),
(h)-CO2R5,
(i)-C(O)N(R5)(R6),
(j)-S(O)2N(R5)(R6),
(k)-S(O)n-R5,
(l)任选地经选自以下的一至三个基团取代的5元至6元杂芳基:C1-6烷基、C3-6环烷基、卤素、-CF3、-OH、-(CH2)nCO2R5、-C(O)N(R5)(R6)、-N(R5)(R6)、-NH-SO2C1-6烷基、C1-6烷氧基、C1-6烷基-O-C1-3烷基、C1-6烷基羟基、C1-3烷基-CN、氧代、任选地经卤素取代的苯基及-S(O)nC1-6烷基,
(m)含有N、S或O的4元至10元单环、双环或螺环杂环基,其中各杂环任选地经选自以下的1至3个取代基取代:3元至6元杂环、卤素、C1-3烷基及C1-3烷基-C(O)N(R5)(R6),
(n)芳基,
(o)-N(R5)(R6);
R5及R6各自独立地选自-H、4元至6元杂环基、-C(O)-C1-3烷基-C(O)-C1-3环烷基及-(C1-C6)烷基,任选地经以下取代:-OH、C3-6环烷基、-NH-C1-3烷基或-N-(C1-3-烷基)2;或
R5及R6与其所连接的氮原子一起形成任选地经甲基取代的5元至6元杂环;且
n为0或2。
在第三实施方案中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
X为N且Y为CH。
在第四实施方案中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
X为CH且Y为N。
在第五实施方案中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET为选自吡唑基、咪唑基、噻唑基及噻二唑基的5元杂芳环,其中各杂芳环任选地经独立地选自R3及R4的一至两个取代基取代;
R3及R4各自独立地选自:
(a)-H,
(b)-OR5,
(c)-O-C1-6烷基-O-C1-3烷基,
(d)-O-C3-6环烷基,
(e)-C(O)R5,
(f)任选地经以下取代的C1-6烷基:一至三个-OH、氟、任选地经氧代取代的杂环基、C3-6环烷基、-CO2R5、-O-C1-6烷基、芳基、-N(R5)(R6)或-C(O)N(R5)(R6),
(g)任选地经以下取代的C3-6环烷基:一至三个-OH、一至三个氟、C1-6烷基、-OC1-6烷基、C1-6烷基-OC1-6烷基、C1-6烷基-OH、CF3、-OC3-6环烷基、-CO2H、-CO2R5、C3-6环烷基、5元至6元杂芳基、C3-6杂环基、N(R5)(R6)或-C(O)N(R5)(R6),
(h)-CO2R5,
(i)-C(O)N(R5)(R6),
(j)-S(O)2N(R5)(R6),
(k)-S(O)n-R5,
(l)任选地经选自以下的一至三个基团取代的5元至6元杂芳基:C1-6烷基、C3-6环烷基、卤素、-CF3、-OH、-(CH2)nCO2R5、-C(O)N(R5)(R6)、-N(R5)(R6)、-NH-SO2C1-6烷基、C1-6烷氧基、C1-6烷基-O-C1-3烷基、C1-6烷基羟基、C1-3烷基-CN、氧代、任选地经卤素取代的苯基及-S(O)nC1-6烷基,
(m)含有N、S或O的4元至10元单环、双环或螺环杂环基,其中各杂环任选地经选自以下的1至3个取代基取代:3元至6元杂环、卤素、C1-3烷基及C1-3烷基-C(O)N(R5)(R6),
(n)芳基,
(o)-N(R5)(R6);
R5及R6各自独立地选自-H、4元至6元杂环基、-C(O)-C1-3烷基-C(O)-C1-3环烷基及-(C1-C6)烷基,任选地经以下取代:-OH、C3-6环烷基、-NH-C1-3烷基或-N-(C1-3-烷基)2;或
R5及R6与其所连接的氮原子一起形成任选地经甲基取代的5元至6元杂环;且
n为0或2。
在第六实施方案中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET为选自吡唑基及咪唑基的5元杂芳环,其中各杂芳环经选自Ra及Rb的两个取代基取代;其中
Ra及Rb与其所连接的原子一起形成5元至6元杂环或杂芳环,所述5元至6元杂环或杂芳环可任选地经选自R3及R4的一至两个取代基取代;
R3及R4各自独立地选自:
(a)-H,
(b)-OR5,
(c)-O-C1-6烷基-O-C1-3烷基,
(d)-O-C3-6环烷基,
(e)-C(O)R5,
(f)任选地经以下取代的C1-6烷基:一至三个-OH、氟、任选地经氧代取代的杂环基、C3-6环烷基、-CO2R5、-O-C1-6烷基、芳基、-N(R5)(R6)或-C(O)N(R5)(R6),
(g)任选地经以下取代的C3-6环烷基:一至三个-OH、一至三个氟、C1-6烷基、-OC1-6烷基、C1-6烷基-OC1-6烷基、C1-6烷基-OH、CF3、-OC3-6环烷基、-CO2H、-CO2R5、C3-6环烷基、5元至6元杂芳基、C3-6杂环基、N(R5)(R6)或-C(O)N(R5)(R6),
(h)-CO2R5,
(i)-C(O)N(R5)(R6),
(j)-S(O)2N(R5)(R6),
(k)-S(O)n-R5,
(l)任选地经选自以下的一至三个基团取代的5元至6元杂芳基:C1-6烷基、C3-6环烷基、卤素、-CF3、-OH、-(CH2)nCO2R5、-C(O)N(R5)(R6)、-N(R5)(R6)、-NH-SO2C1-6烷基、C1-6烷氧基、C1-6烷基-O-C1-3烷基、C1-6烷基羟基、C1-3烷基-CN、氧代、任选地经卤素取代的苯基及-S(O)nC1-6烷基,
(m)含有N、S或O的4元至10元单环、双环或螺环杂环基,其中各杂环任选地经选自以下的1至3个取代基取代:3元至6元杂环、卤素、C1-3烷基及C1-3烷基-C(O)N(R5)(R6),
(n)芳基,
(o)-N(R5)(R6);
R5及R6各自独立地选自-H、4元至6元杂环基、-C(O)-C1-3烷基-C(O)-C1-3环烷基及-(C1-C6)烷基,任选地经以下取代:-OH、C3-6环烷基、-NH-C1-3烷基或-N-(C1-3-烷基)2;或
R5及R6与其所连接的氮原子一起形成任选地经甲基取代的6元杂环;且
n为0或2。
在第七实施方案中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET为任选地经选自R3及R4的一至两个取代基取代的吡唑基。
在实施方案八中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET为选自吡唑基及咪唑基的5元杂芳环,其中各杂芳环经选自Ra及Rb的两个取代基取代;其中
Ra及Rb与其所连接的原子一起形成5元至6元杂芳环,以使得HET为选自咪唑并吡啶及吡唑并吡啶的双环杂芳环,该双环杂芳环可任选地经选自R3及R4的一至两个取代基取代。
在实施方案九中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
X为N;
Y为CH;
R1为F;
R2选自甲基及Cl;
HET选自可任选地经选自R3及R4的一至两个取代基取代的咪唑并吡啶及吡唑并吡啶;
R3及R4各自独立地选自:
(a)-H,
(b)-OR5,
(c)-O-C1-6烷基-O-C1-3烷基,
(d)-O-C3-6环烷基,
(e)-C(O)R5,
(f)任选地经以下取代的C1-6烷基:一至三个-OH、氟、任选地经氧代取代的杂环基、C3-6环烷基、-CO2R5、-O-C1-6烷基、芳基、-N(R5)(R6)或-C(O)N(R5)(R6),
(g)任选地经以下取代的C3-6环烷基:一至三个-OH、一至三个氟、C1-6烷基、-OC1-6烷基、C1-6烷基-OC1-6烷基、C1-6烷基-OH、CF3、-OC3-6环烷基、-CO2H、-CO2R5、C3-6环烷基、5元至6元杂芳基、C3-6杂环基、N(R5)(R6)或-C(O)N(R5)(R6),
(h)-CO2R5,
(i)-C(O)N(R5)(R6),
(j)-S(O)2N(R5)(R6),
(k)-S(O)2-R5,
(l)任选地经选自以下的一至三个基团取代的5元至6元杂芳基:C1-6烷基、C3-6环烷基、卤素、-CF3、-OH、-(CH2)CO2R5、-C(O)N(R5)(R6)、-N(R5)(R6)、-NH-SO2C1-6烷基、C1-6烷氧基、C1-6烷基-O-C1-3烷基、C1-6烷基羟基、C1-3烷基-CN、氧代、任选地经卤素取代的苯基及-S(O)2C1-6烷基,
(m)含有N、S或O的4元至10元单环、双环或螺环杂环基,其中各杂环任选地经选自以下的1至3个取代基取代:3元至6元杂环、卤素、C1-3烷基及C1-3烷基-C(O)N(R5)(R6),
(n)芳基,
(o)-N(R5)(R6);
R5及R6各自独立地选自-H、4元至6元杂环基、-C(O)-C1-3烷基-C(O)-C1-3环烷基及-(C1-C6)烷基,任选地经以下取代:-OH、C3-6环烷基、-NH-C1-3烷基或-N-(C1-3-烷基)2;或
R5及R6与其所连接的氮原子一起形成任选地经甲基取代的6元杂环。
在实施方案十中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
X为N;
Y为CH;
R1为F;
R2选自甲基及Cl;
HET为可任选地经选自R3及R4的一至两个取代基取代的咪唑并吡啶;
R3及R4各自独立地选自:
(a)-H,
(b)-OR5,
(c)-O-C1-6烷基-O-C1-3烷基,
(d)-O-C3-6环烷基,
(e)-C(O)R5,
(f)任选地经以下取代的C1-6烷基:一至三个-OH、氟、任选地经氧代取代的杂环基、C3-6环烷基、-CO2R5、-O-C1-6烷基、芳基、-N(R5)(R6)或-C(O)N(R5)(R6),
(g)任选地经以下取代的C3-6环烷基:一至三个-OH、一至三个氟、C1-6烷基、-OC1-6烷基、C1-6烷基-OC1-6烷基、C1-6烷基-OH、CF3、-OC3-6环烷基、-CO2H、-CO2R5、C3-6环烷基、5元至6元杂芳基、C3-6杂环基、N(R5)(R6)或-C(O)N(R5)(R6),
(h)-CO2R5,
(i)-C(O)N(R5)(R6),
(j)-S(O)2N(R5)(R6),
(k)-S(O)2-R5,
(l)任选地经选自以下的一至三个基团取代的5元至6元杂芳基:C1-6烷基、C3-6环烷基、卤素、-CF3、-OH、-(CH2)CO2R5、-C(O)N(R5)(R6)、-N(R5)(R6)、-NH-SO2C1-6烷基、C1-6烷氧基、C1-6烷基-O-C1-3烷基、C1-6烷基羟基、C1-3烷基-CN、氧代、任选地经卤素取代的苯基及-S(O)2C1-6烷基,
(m)含有N的6元单环杂环基,其中该杂环任选地经选自以下的1至3个取代基取代:3元至6元杂环、卤素、C1-3烷基及C1-3烷基-C(O)N(R5)(R6),
(n)芳基,
(o)-N(R5)(R6);
R5及R6各自独立地选自-H、4元至6元杂环基、-C(O)-C1-3烷基-C(O)-C1-3环烷基及-(C1-C6)烷基,任选地经以下取代:-OH、C3-6环烷基、-NH-C1-3烷基或-N-(C1-3-烷基)2;或
R5及R6与其所连接的氮原子一起形成任选地经甲基取代的6元杂环。
在实施方案十一中,本发明涉及如上文第九实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET为:

任选地经选自R3及R4的一至两个取代基取代;
R3及R4各自独立地选自:
(a)-H,
(b)-OR5,
(c)-O-C1-6烷基-O-C1-3烷基,
(d)-O-C3-6环烷基,
(e)-C(O)R5,
(f)任选地经以下取代的C1-6烷基:一至三个-OH、氟、任选地经氧代取代的杂环基、C3-6环烷基、-CO2R5、-O-C1-6烷基、芳基、-N(R5)(R6)或-C(O)N(R5)(R6),
(g)任选地经以下取代的C3-6环烷基:一至三个-OH、一至三个氟、C1-6烷基、-OC1-6烷基、C1-6烷基-OC1-6烷基、C1-6烷基-OH、CF3、-OC3-6环烷基、-CO2H、-CO2R5、C3-6环烷基、5元至6元杂芳基、C3-6杂环基、N(R5)(R6)或-C(O)N(R5)(R6),
(h)-CO2R5,
(i)-C(O)N(R5)(R6),
(j)-S(O)2N(R5)(R6),
(k)-S(O)2-R5,
(l)任选地经选自以下的一至三个基团取代的5元至6元杂芳基:C1-6烷基、C3-6环烷基、卤素、-CF3、-OH、-(CH2)CO2R5、-C(O)N(R5)(R6)、-N(R5)(R6)、-NH-SO2C1-6烷基、C1-6烷氧基、C1-6烷基-O-C1-3烷基、C1-6烷基羟基、C1-3烷基-CN、氧代、任选地经卤素取代的苯基及-S(O)2C1-6烷基,
(m)含有N、S或O的4元至10元单环、双环或螺环杂环基,其中各杂环任选地经选自以下的1至3个取代基取代:3元至6元杂环、卤素、C1-3烷基及C1-3烷基-C(O)N(R5)(R6),
(n)芳基,
(o)-N(R5)(R6);
R5及R6各自独立地选自-H、4元至6元杂环基、-C(O)-C1-3烷基-C(O)-C1-3环烷基及-(C1-C6)烷基,任选地经以下取代:-OH、C3-6环烷基、-NH-C1-3烷基或-N-(C1-3-烷基)2;或
R5及R6与其所连接的氮原子一起形成任选地经甲基取代的6元杂环。
在实施方案十二中,本发明涉及如上文第九实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET为:

任选地经选自R3及R4的一至两个取代基取代。
在实施方案十三中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET为:

任选地经选自R3及R4的一至两个取代基取代。
在实施方案十四中,本发明涉及如上文最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET为:

任选地经选自R3及R4的一至两个取代基取代。
在实施方案十五中,本发明涉及如最广泛实施方案中所描述的化合物,或其药学上可接受的盐,其中:
HET选自:

任选地经选自R3及R4的一至两个取代基取代。
在实施方案十六中,本发明涉及式I化合物或其药学上可接受的盐,其中:
X为N;
Y为CH;
R1为F;
R2为甲基;
HET为:

R3为甲氧基;且
R4为
表1:以下为本发明的代表性化合物,其可通过本领域的一般合成流程、实例及已知的方法制得。


























或其药学上可接受的盐。
在一个实施方案中,本发明涉及上文表1中所描绘的化合物中的任一种及其药学上可接受的盐。
对于本申请中于上文所公开的所有化合物,在命名法与结构冲突的情况下,将理解为化合物由结构定义。对于具有立体中心的化合物,结构展示绝对立体化学。
本发明也涉及药物制剂,其含有一或多种本发明的化合物作为活性物质,或其药学上可接受的衍生物,所述化合物及衍生物任选地与常规赋形剂及/或载体组合。
本发明的化合物也包括其经同位素标记的形式。本发明组合的活性剂的经同位素标记的形式与该活性剂相同,但事实为该活性剂的一或多个原子已经原子质量或质量数不同于该原子的原子质量或质量数的一或多个原子(通常发现于自然界中)置换。易于商购可得且可根据完善程序并入本发明组合的活性剂中的同位素的实例包括氢、碳、氮、氧、磷、氟及氯的同位素,分别例如2H、3H、13C、14C、15N、18O、17O、31P、32P、35S、18F及36Cl。含有上文所提及的同位素及/或其他原子的其他同位素中的一或多者的本发明组合的活性剂、其前药或任一者的药学上可接受的盐预想落入本发明的范围内。
本发明包括含有一或多个不对称碳原子的上文所描述的任何化合物的用途,该化合物可以外消旋体及外消旋混合物、单一对映异构体、非对映异构混合物及单独非对映异构体存在。异构体应定义为对映异构体及非对映异构体。这些化合物的所有这些异构形式均明确包括于本发明内。各立体碳可呈R或S构型或构型的组合。
本发明的化合物中的一些可以多于一种互变异构形式存在。本发明包括使用所有这些互变异构体的方法。
除非另外说明,否则如在本说明书中所用的所有术语应理解为如本领域已知的该术语的一般含义。例如,“C1-6烷氧基”为具有末端氧的C1-6烷基,诸如甲氧基、乙氧基、丙氧基、丁氧基。除非另外说明,否则所有烷基、烯基及炔基皆应理解为在结构上可能为支化的或未支化的。其他更具体定义如下:
术语“烷基”是指支化及未支化烷基。应理解,使用“烷”或“烷基”前缀的任何组合术语是指根据“烷基”的以上定义的类似物。例如,诸如“烷氧基”、“烷硫基”的术语是指经由氧或硫原子与第二基团连接的烷基。“烷酰基”是指与羰基(C=O)连接的烷基。
应理解,如果N未经取代,则其为NH。如本文所用,“氮”及“硫”包括氮及硫的任何氧化形式以及任何碱性氮的季铵化形式。例如,对于-S-C1-6烷基,除非另外说明,否则应理解为包括-S(O)-C1-6烷基及-S(O)2-C1-6烷基。
术语“C3-10碳环”或“C3-10环烷基”是指非芳族3元至10元(但优选为3元至6元)单环碳环/环烷基或非芳族6元至10元稠合双环、桥连双环或螺环碳环基。C3-10碳环/环烷基环可为饱和或部分不饱和的,且碳环/环烷基环可通过该环的任何原子连接,从而产生稳定结构。3元至10元单环碳环/环烷基环的非限制性实例包括环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环庚基、环庚烯基及环己酮。6元至10元稠合双环碳环/环烷基的非限制性实例包括双环[3.3.0]辛烷、双环[4.3.0]壬烷及双环[4.4.0]癸基(十氢萘基)。6至10元桥连双环碳环基的非限制性实例包括双环[2.2.2]庚基、双环[2.2.2]辛基及双环[3.2.1]辛基。6至10元螺环碳环基的非限制性实例包括(但不限于)螺[3,3]庚基、螺[3,4]辛基及螺[4,4]庚基。
术语“芳基”是指含有六至十个碳环原子的芳族烃环。术语芳基包括环中至少之一为芳族的单环及双环。C6-10芳基的非限制性实例包括苯基、茚满基、茚基、苯并环丁烷基、二氢萘基、四氢萘基、萘基、苯并环庚烷基及苯并环庚烯基。
术语“杂环”是指稳定非芳族4元至8元单环杂环基或稳定非芳族6元至11元稠合双环、桥连双环或螺环杂环基。5至11元杂环由碳原子及选自氮、氧及硫的一或多个(优选为一至四个)杂原子构成。杂环可为饱和或部分不饱和的。非芳族4元至8元单环杂环基的非限制性实例包括四氢呋喃基、氮杂环丁基、吡咯烷基、吡喃基、四氢吡喃基、二噁烷基、硫代吗啉基(thiomorpholinyl)、1,1-二氧代-1λ6-硫代吗啉基、吗啉基、哌啶基、哌嗪基及氮杂卓基(azepinyl)。非芳族6元至11元稠合双环基的非限制性实例包括八氢吲哚基、八氢苯并呋喃基及八氢苯并噻吩基。非芳族6元至11元桥连双环基的非限制性实例包括2-氮杂双环[2.2.1]庚基、3-氮杂双环[3.1.0]己基及3-氮杂双环[3.2.1]辛基。非芳族6元至11元螺环杂环基的非限制性实例包括7-氮杂-螺[3,3]庚基、7-螺[3,4]辛基及7-氮杂-螺[3,4]辛基。
术语“杂芳基”应理解为意指环中至少之一为芳族的芳族5元至6元单环杂芳基或芳族7元至11元杂芳基双环,其中杂芳基环含有诸如N、O及S的1至4个杂原子。5元至6元单环杂芳基环的非限制性实例包括呋喃基、噁唑基、异噁唑基、噁二唑基、噻唑基、吡唑基、吡咯基、咪唑基、四唑基、三唑基、噻吩基、噻二唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、三嗪基及嘌呤基。7元至11元杂芳基双环杂芳环的非限制性实例包括苯并咪唑基、喹啉基、二氢-2H-喹啉基、异喹啉基、喹唑啉基、吲唑基、噻吩并[2,3-d]嘧啶基、吲哚基、异吲哚基、苯并呋喃基、苯并吡喃基、苯并间二氧杂环戊烯基(benzodioxolyl)、苯并噁唑基、苯并噻唑基、二氢吲哚基、氮杂吲哚基、苯并噻唑基、苯并吡咯基、苯并吡唑基、吡啶并吡唑基、二氢苯并呋喃基、苯并噻吩基、苯并二噁烷基、二氢苯并[1,4]二噁烷基及苯并[1,3]间二氧杂环戊烯基。
如本文所用的术语“杂原子”应理解为意指除碳以外的原子,诸如O、N及S。
如本说明书所用的术语“卤素”应理解为意指溴、氯、氟或碘。定义“卤化”、“部分或完全卤化”、部分或完全氟化、“经一或多个卤原子取代”包括例如一或多个碳原子上的单、二或三卤代衍生物。对于烷基,非限制性实例将会为-CH2CHF2、-CF3等。
本文所描述的各烷基、芳基、环烷基/碳环、杂环或杂芳基或其类似物应理解为任选地部分或完全卤化。
本发明的化合物仅为如本领域技术人员所将了解而预期为“在化学上稳定的”化合物。例如,将具有“悬空化合价(dangling valency)”或“碳阴离子”的化合物不为通过本文所公开的本发明方法预期的化合物。
本发明包括式(I)化合物的药学上可接受的衍生物。“药学上可接受的衍生物”是指任何药学上可接受的盐或酯、或其任何其他化合物,在向患者给药时,其能够(直接或间接地)提供适用于本发明的化合物或其药理学上活性的代谢物或药理学上活性的残余物。药理学上活性的代谢物应理解为意指能够以酶方式或以化学方式代谢的任何本发明化合物。这包括例如本发明的羟基化或氧化衍生化合物。
药学上可接受的盐包括衍生自药学上可接受的无机酸及无机碱以及有机酸及有机碱的盐。适合的酸的实例包括盐酸、氢溴酸、硫酸、硝酸、高氯酸、反丁烯二酸、顺丁烯二酸、磷酸、乙醇酸、乳酸、水杨酸、丁二酸、甲苯-对硫酸、酒石酸、乙酸、柠檬酸、甲磺酸、甲酸、苯甲酸、丙二酸、萘-2-硫酸及苯磺酸。诸如乙二酸的其他酸尽管自身非药学上可接受的酸,但可用于在获得化合物及其药学上可接受的酸加成盐中制备用作中间体的盐。衍生自适当碱的盐包括碱金属(例如钠)、碱土金属(例如镁)、铵及N-(C1-C4烷基)4+盐。
另外,在本发明的范围内使用本发明的化合物的前药。前药包括在简单化学转化时经改性以产生本发明的化合物的那些化合物。简单化学转化包括水解、氧化及还原。具体来说,当向患者给药前药时,前药可转化为上文所公开的化合物,由此赋予所需药理学作用。
可使用下文所描述的通用合成方法来制备式I化合物,该方法也构成本发明的一部分。
合成实例
缩写列表



中间体的通用合成方法及合成
本发明的化合物可通过下文呈现的方法及实例以及本领域技术人员已知的方法来制备。在下文实例中的每一者中,除非指出,否则针对通式I,基团R1至R7如上文所定义。最佳反应条件及反应时间可取决于所用特定反应物而改变。除非另外说明,否则本领域技术人员可易于选择溶剂、温度、压力及其他反应条件。具体程序提供于下文中。用于下文合成中的中间体为商购可得的或易于通过本领域技术人员已知的方法来制备。反应进程可通过常规方法来监测,该方法诸如薄层色谱法(TLC)或高压液相色谱-质谱分析法(HPLC-MS)。中间体及产物可通过本领域已知的方法来纯化,该方法包括柱色谱法、HPLC、制备型TLC或制备型HPLC。
中间体
5-叔丁基硫烷基-吡啶-2-基胺(I-1)的合成

向5-溴-吡啶-2-基胺(300mg,1.73mmol)于DMSO(3mL)中的混合物中添加2-甲基-2-丙硫醇钠(388mg,3.47mmol)及NaOH(35mg,0.87mmol)。将混合物用Ar脱气20分钟。向反应混合物添加L-脯胺酸(100mg,0.870mmol)及CuI(330mg,1.73mmol)且在密封管中将反应物加热至120℃持续12h。随后使反应物冷却至室温,倒入冰水中且用EtOAc(2×)萃取。在减压下移除溶剂以提供200mg无需进一步纯化即使用的粗5-叔丁基硫烷基-吡啶-2-基胺(I-1)。
5-叔丁基硫烷基-吡唑并[1,5-a]吡啶(I-2)的合成

在0℃下向5-溴-吡唑并[1,5-a]吡啶(2.0g,10.1mmol)于DME(30mL)中的搅拌溶液中添加作为于THF(24mL,25mmol)中的1M溶液的双(三甲基甲硅烷基)酰胺锂。在另一烧瓶中,将乙酸钯(II)(227mg,1.01mmol)及dppf(2.0g,3.6mmol)合并至DME(30mL)中且在室温下搅拌10分钟。随后在室温下合并两种溶液,且用2-甲基-2-丙硫醇(2.0g,22mmol)处理。将反应物加热至90℃且搅拌2h。随后使反应物冷却至室温,用水稀释且用EtOAc(2×)萃取。将有机层用盐水洗涤,经Na2SO4干燥,并且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以提供1g 5-叔丁基硫烷基-吡唑并[1,5-a]吡啶(I-2)。
6-(氧杂环丁-3-基硫烷基)-咪唑并[1,2-a]吡啶(I-3)的合成

用N2冲洗6-碘-咪唑并[1,2-a]吡啶(800mg,3.30mmol)、碘化铜(62mg,0.33mmol)、碳酸铯(2.1g,6.6mmol)、NMP(8mL)及氧杂环丁烷-3-硫醇(325mg,3.6mmol)的混合物且将混合物密封在微波反应管中。在130℃下将混合物在微波反应器中加热30分钟。使反应物冷却至室温,经由硅藻土过滤且用EtOAc冲洗。用水稀释且用EtOAc(3×)萃取滤液。经合并的有机层用盐水洗涤,经Na2SO4干燥,过滤且在减压下浓缩。通过快速硅胶柱色谱法来纯化所得残余物以提供283mg 6-(氧杂环丁-3-基硫烷基)-咪唑并[1,2-a]吡啶(I-3)。
使用商购可得的起始物质以类似于如上文所描述的方式合成以下中间体:
3-(咪唑并[1,2-a]吡啶-6-基硫烷基)-氮杂环丁烷-1-甲酸叔丁基(I-4)
2-环丙基-1-甲基-1H-咪唑(I-5)的合成

在0℃下向于二噁烷(7.3mL,29mmol)中的4N HCl缓慢地添加作为于无水甲醇中的溶液(1.2mL)的环丙烷甲腈(1mL,13.5mmol)。将混合物在0至5℃之间搅拌3h,在此期间形成沉淀物。使混合物升温至室温且在减压下浓缩溶液。添加MTBE(10mL),且将混合物搅拌数分钟,随后过滤以提供783mg环丙烷甲酰亚胺酸甲酯HCl(I-5-1)。
向于异丙醇(2.0mL)中的I-5-1(0.50g,3.9mmol)的搅拌悬浮液中添加(2,2-二甲氧基-乙基)-甲基-胺(0.50mL,4.0mmol)。混合物变得几乎均相;随后形成凝胶状沉淀物。使混合物升温至80℃持续4h,随后使其冷却至室温且持续搅拌额外12h。随后添加浓缩HCl(1.1mL,13mmol)且将混合物加热至80℃持续45分钟。使混合物冷却至室温且倒入至NaHCO3的饱和水溶液(20mL)中。将混合物用EtOAc(3×)萃取且有机层用盐水洗涤,经Na2SO4干燥,过滤且浓缩以提供212mg 2-环丙基-1-甲基-1H-咪唑(I-5)。
3-溴-6-(氧杂环丁-3-基硫烷基)-咪唑并[1,2-a]吡啶(I-6)的合成
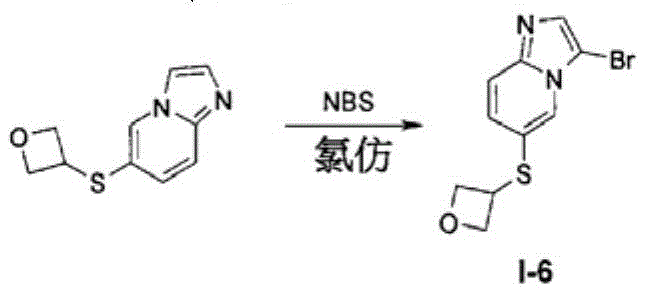
向I-3(242mg,1.20mmol)于氯仿(4.2mL)中的搅拌溶液中添加NBS(208mg,1.20mmol)。将反应物在室温下搅拌30分钟并且随后在减压下浓缩。通过快速硅胶柱色谱法来纯化所得残余物以提供294mg 3-溴-6-(氧杂环丁-3-基硫烷基)-咪唑并[1,2-a]吡啶(I-6)。
使用I-4以类似于如上文所描述的方式合成以下中间体:3-(3-溴-咪唑并[1,2-a]吡啶-6-磺酰基)-氮杂环丁烷-1-甲酸叔丁酯(I-7)
使用I-17以类似于如上文所描述的方式合成以下中间体:
3-溴-6-(吗啉-4-磺酰基)-咪唑并[1,2-a]吡啶(I-8)
使用I-18以类似于如上文所描述的方式合成以下中间体:
3-溴-6-甲磺酰基-咪唑并[1,2-a]吡啶(I-9)
使用I-20以类似于如上文所描述的方式合成以下中间体:
3-溴-6-叔丁基硫烷基-咪唑并[1,2-a]吡啶(I-10)
使用I-23以类似于如上文所描述的方式合成以下中间体:
3-溴-5-(2-甲基-丙烷-2-磺酰基)-吡唑并[1,5-a]吡啶(I-11)
使用I-16以类似于如上文所描述的方式合成以下中间体:
3-溴-5-乙磺酰基-吡唑并[1,5-a]吡啶(I-12)
使用I-19以类似于如上文所描述的方式合成以下中间体:
3-溴-6-(4-甲基-哌嗪-1-磺酰基)-咪唑并[1,2-a]吡啶(I-13)
使用I-5以类似于如上文所描述的方式合成以下中间体:
5-溴-2-环丙基-1-甲基-1H-咪唑(I-14)
使用I-32以类似于如上文所描述的方式合成以下中间体:
3-溴-6-[1-(叔丁基-二甲基-硅烷基氧基)-1-甲基-乙基]-咪唑并[1,2-a]吡啶(I-15)
5-乙磺酰基-吡唑并[1,5-a]吡啶(I-16)的合成

在0℃下向乙基硫烷基-吡啶(5.0g,36mmol)于乙腈(50mL)中的搅拌溶液中分批添加2,4-二-硝苯基羟基胺(DNPH)(7.0g,35mmol)。使反应物缓慢地升温至室温并且随后加热至40℃持续15h。在减压下浓缩反应物以提供5.0g 4-乙基硫烷基-2H-吡啶-1-基胺(I-16-1)。
向粗I-16-1(5.0g,32mmol)于DMF(50mL)中的搅拌溶液(冷却至0℃)中添加K2CO3(4.0g,29mmol)。向反应混合物中逐滴添加丙酸乙酯(3.0g,29mmol)且使反应物缓慢地升温至室温。2h后,用水稀释且用EtOAc(3×)萃取反应物。在减压下浓缩经合并的有机层以得到2.0g粗5-乙基硫烷基-吡唑并[1,5-a]吡啶-3-甲酸乙酯(I-16-2)。
向I-16-2(1.0g,4.0mmol)于乙腈(5.0mL)及水(10.0mL)的混合物中的搅拌溶液(冷却至0℃)中添加氯化钌(III)水合物(826mg,4.0mmol)及偏高碘酸钠(2.0mg,9.0mmol)。30分钟后,用水稀释且用EtOAc萃取反应物。在减压下浓缩有机层以提供500mg粗5-乙磺酰基-吡唑并[1,5-a]吡啶-3-甲酸乙酯(I-16-3)。
在0℃下向硫酸(2.0g)于水(4.0mL)中的搅拌溶液缓慢地添加I-16-3(1.0g,3.6mmol)。将反应物在90℃下搅拌2h,随后冷却至室温。通过添加2NNaOH溶液来将混合物的pH调节为中性并且随后用DCM(2×)进行萃取。将经合并的有机层用盐水洗涤,经Na2SO4干燥,并且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以提供298mg 5-乙磺酰基-吡唑并[1,5-a]吡啶(I-16)。
6-(吗啉-4-磺酰基)-咪唑并[1,2-a]吡啶(I-17)的合成

在室温下将溴乙醛缩二乙醇(383μL,2.50mmol)及2M HCl水溶液(1.4mL,2.7mmol)的混合物搅拌2h。随后将反应物加热至80℃持续1h。使反应物冷却至5℃且通过添加固体碳酸氢钠来将混合物的pH调节为pH 8。向反应混合物中添加5-(吗啉-4-磺酰基)-吡啶-2-基胺(300mg,1.2mmol)且使所得溶液升温至室温并且搅拌过夜。混合物在减压下浓缩且用EtOAc(10mL)稀释。超声处理并过滤混合物。在减压下浓缩滤液且通过快速硅胶柱色谱法来纯化所得残余物以提供165mg 6-(吗啉-4-磺酰基)-咪唑并[1,2-a]吡啶(I-17)。
使用商购可得的物质以类似于如上文所描述的方式合成以下中间体:
6-甲磺酰基-咪唑并[1,2-a]吡啶(I-18)
6-(4-甲基-哌嗪-1-磺酰基)-咪唑并[1,2-a]吡啶(I-19)
使用I-1以类似于如上文所描述的方式合成以下中间体:
6-叔丁基硫烷基-咪唑并[1,2-a]吡啶(I-20)
3-(咪唑并[1,2-a]吡啶-6-磺酰基)-氮杂环丁烷-1-甲酸叔丁酯(I-21)的合成

在室温下向I-4(480mg,1.6mmol)于乙腈(20mL)及水(10mL)的混合物中的搅拌溶液中添加氯化钌(III)水合物(19mg;0.1mmol)及偏高碘酸钠(2.0g,9.4mmol)。1h后,将反应物用EtOAc(3×)萃取,用盐水洗涤,经Ns2SO4干燥且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以获得100mg3-(咪唑并[1,2-a]吡啶-6-磺酰基)-氮杂环丁烷-1-甲酸叔丁酯(I-21)。
使用I-10根据上文所描述的中间体合成以下中间体:
3-溴-6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶(I-22)
使用I-2根据上文所描述的中间体合成以下中间体:
5-(2-甲基-丙烷-2-磺酰基)-吡唑并[1,5-a]吡啶(I-23)
3-溴-7-甲氧基-6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]-吡啶(I-24)的合成

在室温下向4-甲氧基吡啶-2-基胺(15g,121mmol)于乙酸(490mL)中的搅拌溶液中缓慢地添加作为于乙酸(120mL,120mmol)中的1M溶液的溴。1.5h后,过滤反应混合物且将所收集的固体溶解于EtOAc中。用饱和NaHCO3然后以水并且随后用盐水洗涤混合物。有机层经无水Na2SO4干燥,过滤,且在减压下浓缩滤液以提供15.1g 5-溴-4-甲氧基吡啶-2-基胺(I-24-1)。
向I-24-1(15g,74mmol)于乙醇比水为4:1的混合物(150mL)中的搅拌溶液中添加氯乙醛水溶液(55%水溶液,15mL,88.7mmol),然后添加固体NaHCO3(7.4g,89mmol)。使所得溶液回流持续4h,随后冷却至室温,用水稀释且用EtOAc(2×)萃取。经合并的有机层经无水Na2SO4干燥,过滤且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以提供10.6g 6-溴-7-甲氧基-咪唑并[1,2-a]吡啶(I-24-2)。
向1,4-二噁烷(360mL)中的I-24-2(13g,56mmol)的搅拌悬浮液中添加NaOtBu(6.6g,69mmol)及2-甲基-2-丙硫醇(7.74g,85.9mmol)。所得混合物用Ar脱气5分钟,随后用Pd(OAc)2(250mg,1.14mmol)及dppf(760mg,1.37mmol)处理。将反应物在90℃下加热12h,随后冷却至室温并过滤。用水稀释且用EtOAc(3×)萃取滤液。将经合并的有机层用水洗涤然后用盐水洗涤,随后经无水Na2SO4干燥,过滤且在减压下浓缩。通过快速柱色谱法来纯化残余物以提供10.4g 6-叔丁基硫烷基-7-甲氧基-咪唑并[1,2-a]吡啶(I-24-3)。
在0℃下向I-24-3(6.0g,25mmol)于MeOH:水为1:1的混合物(60mL)中的搅拌溶液中添加 (47g,76mmol)。使所得混合物搅拌且在1h内升温至室温。过滤混合物且用EtOAc洗涤滤垫。通过添加NaHCO3饱和溶液来将经合并的滤液的pH调节为中性,随后用EtOAc萃取。将经合并的有机层用水洗涤,经无水Na2SO4干燥,过滤且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以提供6.0g 7-甲氧基-6-(2-甲基-丙烷-2磺酰基)咪唑[1,2-a]吡啶(I-24-4)。
(47g,76mmol)。使所得混合物搅拌且在1h内升温至室温。过滤混合物且用EtOAc洗涤滤垫。通过添加NaHCO3饱和溶液来将经合并的滤液的pH调节为中性,随后用EtOAc萃取。将经合并的有机层用水洗涤,经无水Na2SO4干燥,过滤且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以提供6.0g 7-甲氧基-6-(2-甲基-丙烷-2磺酰基)咪唑[1,2-a]吡啶(I-24-4)。
向I-24-4(6.0g,18.6mmol)于DMF(30mL)中的搅拌溶液中添加NBS(3.3g,18.6mmol)。将所得混合物在室温下搅拌30分钟,随后用水稀释且用EtOAc萃取。有机层用水(4×)洗涤然后用盐水洗涤,随后经无水Na2SO4干燥,过滤且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以提供5.5g 3-溴-7-甲氧基-6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶(I-24)。
3-溴-咪唑并[1,2-a]吡啶-6-甲酸甲基酰胺(I-25)的合成

在室温下向3-溴-咪唑并[1,2-a]吡啶-6-甲酸(130mg,0.540mmol)于DMF(2.1mL)中的搅拌溶液中添加Et3N(376μL,2.7mmol),然后以甲胺于EtOH(33wt%,101mg,1.1mmol)及HATU(308mg,0.81mmol)中的溶液。将反应物在室温下搅拌18h,随后用水稀释且用EtOAc(3×)萃取。有机层用水然后用盐水洗涤,随后经Na2SO4干燥,过滤且在减压下浓缩。通过快速硅胶色谱法来纯化所得残余物以提供83mg 3-溴-咪唑并[1,2-a]吡啶-6-甲酸甲基酰胺。
使用商购可得的胺以类似于如上文所描述的方式合成以下中间体:
3-溴-咪唑并[1,2-a]吡啶-6-甲酸二甲酰胺(I-26)
5-溴-噻唑-2-甲酸酰胺(I-27)的合成

向5-溴-噻唑-2-甲酸乙酯(100mg,0.42mmol)的搅拌溶液中添加氨于MeOH(3.0ml,21mmol)中的7M溶液。将反应物在密封管中在80℃下搅拌12h,随后冷却至室温并且在减压下浓缩以提供88mg 5-溴-噻唑-2-甲酸酰胺(I-28)。
使用商购可得的胺以类似于如上文所描述的方式合成以下中间体:
5-溴-噻唑-2-甲酸甲基酰胺(I-29)
3-溴-6-[1-(叔丁基-二甲基-硅烷基氧基)-乙基]-咪唑并[1,2-a]吡啶(I-30)的合成

在-20℃下向6-碘-咪唑并[1,2-a]吡啶(3.8g,16mmol)于THF(204mL)中的搅拌溶液中添加作为于THF(14.5mL,18.8mmol)中的1.3M溶液的iPrMgCl*LiCl。搅拌20分钟后,添加于THF(4.1mL,20mmol)中的5M乙醛的溶液。将反应在-20℃下搅拌5分钟并且随后移除冷浴且使混合物升温至室温。1.5h后,将反应用饱和碳酸氢钠水溶液(2mL)稀释,随后在减压下浓缩。通过快速硅胶柱色谱法来纯化所得残余物以提供2.1g 1-咪唑并[1,2-a]吡啶-6-基-乙醇(I-30-1)。
将I-30-1(3.0g,18.5mmol)、TBDMSCl(4.0g,27mmol)及咪唑(2.0g,30mmol)的混合物溶解于DMF:DCM为9:1的混合物(60mL)中。将反应在室温下搅拌3h并且随后在减压下浓缩。通过快速硅胶柱色谱法来纯化所得残余物以提供4.26g 6-[1-(叔丁基-二甲基-硅烷基氧基)-乙基]-咪唑并[1,2-a]吡啶(I-30-2)。
在室温下向I-30-2(4.3g,15mmol)于氯仿(55mL)中的搅拌溶液中添加NBS(2.7g,15mmol)。25分钟后,反应用饱和NaHCO3溶液稀释且用EtOAc(3×)萃取。将经合并的有机层用盐水洗涤,经无水Na2SO4干燥且在减压下浓缩。通过快速硅胶色谱法析来纯化残余物以提供5.3g 3-溴-6-[1-(叔丁基-二甲基-硅烷基氧基)-乙基]-咪唑并[1,2-a]吡啶(I-30)。
使用商购可得的醛以类似于如上文所描述的方式合成以下中间体:
3-溴-6-[(叔丁基-二甲基-硅烷基氧基)-(四氢-吡喃-4-基)-甲基]-咪唑并[1,2-a]吡啶(I-31)
2-咪唑并[1,2-a]吡啶-6-基-丙-2-醇(I-31)的合成

在-78℃下向1-咪唑并[1,2-a]吡啶-6-基-乙酮(710mg,4.4mmol)于THF(35mL)中的搅拌溶液中添加作为于THF(3M,1.6mL,4.8mmol)中的溶液的MeMgBr。使混合物升温至室温过夜。混合物随后用饱和NH4Cl溶液淬灭,用EtOAc(3×)萃取并且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以提供576mg 2-咪唑并[1,2-a]吡啶-6-基-丙-2-醇(I-31)。
6-[1-(叔丁基-二甲基-硅烷基氧基)-1-甲基-乙基]-咪唑并[1,2-a]吡啶(I-32)的合成

在室温下向I-31(467mg,2.70mmol)于THF(6.0mL)中的搅拌溶液逐滴添加作为于甲苯(0.5M,5.3mL,2.6mmol)中的溶液的KHMDS。向此混合物中添加TBDMSCl(400mg,2.65mmol)于THF(4mL)中的溶液。1.5h后,将反应用水稀释且用EtOAc萃取(3×)。将有机层用盐水洗涤,经Na2SO4干燥,过滤且浓缩。通过快速硅胶柱色谱法来纯化残余物以得到400mg6-[1-(叔丁基-二甲基-硅烷基氧基)-1-甲基-乙基]-咪唑并[1,2-a]吡啶(I-32)。
3-溴-5-(2-甲基-丙烷-2-亚磺酰基)-4,5,6,7-四氢-吡唑并[1,5-a]吡嗪(I-33)的合成

在0℃下向3-溴-4,5,6,7-四氢-吡唑并[1,5-a]吡嗪盐酸盐(0.90g,3.8mmol)与三乙胺(2.3g,22.6mmol)于无水二氯甲烷(20mL)中的混合物中添加2-甲基-丙烷-2-亚磺酰氯(1.3g,9.4mmol)。在持续搅拌16小时之后,将反应用水(5mL)稀释且用DCM(3×)萃取。经合并的有机萃取物经(Na2SO4)干燥,过滤且在减压下浓缩。通过快速硅胶色谱法来纯化残余物以获得0.9g 3-溴-5-(2-甲基-丙烷-2-亚磺酰基)-4,5,6,7-四氢-吡唑并[1,5-a]吡嗪(I-33)。
3-溴-5-(2-甲基-丙烷-2-磺酰基)-4,5,6,7-四氢-吡唑并[1,5-a]吡嗪(I-34)的合成

向I-33(0.9g,2.9mmol)于无水DCM(20mL)中的溶液中添加mCPBA(0.76g,4.4mmol)。在持续搅拌16h之后,添加亚硫酸钠饱和水溶液(2mL)且将反应搅拌30分钟。随后用DCM(3×20mL)萃取反应且经合并的有机萃取物经(Na2SO4)干燥,过滤且在减压下浓缩。通过快速硅胶色谱法来纯化残余物以获得0.7g 3-溴-5-(2-甲基-丙烷-2-磺酰基)-4,5,6,7-四氢-吡唑并[1,5-a]吡嗪(I-34)。
3-溴-7-(2-甲氧基乙氧基)咪唑并[1,2-a]吡啶(I-35)的合成

在0℃下向2-氯-4-硝基-吡啶(8.0g,50mmol)于2-甲氧基乙醇(9.9mL,130mmol)中的搅拌溶液中分批添加KOt-Bu(6.2g,55mmol)。在添加之后,将反应混合物在室温下搅拌3h。使反应混合物分配于DCM与水之间,并且随后分离各层。用EtOAc(2×)萃取含水层且经合并的有机层用盐水洗涤,经无水MgSO4干燥并且在减压下浓缩以获得5.2g 2-[(2-氯-4-吡啶基)氧基]乙醇(I-35-1),其无需进一步纯化即用于后续步骤中。
将I-35-1(2.9g,15mmol)、Pd2(dba)3(0.28g,0.31mmol)与X-Phos(0.29g,0.62mmol)于干燥THF(30mL)中的混合物用氩脱气10至15分钟。向此混合物中逐滴添加LiHMDS于THF(32.5mL,32.6mmol,1M)中的溶液。随后将所得混合物加热至60℃。18h后,使反应混合物冷却至室温且添加1M HCl(20mL,20mmol)。将所得溶液用MTBE(50mL)洗涤且分离有机层。通过添加6M NaOH水溶液来使得含水层为碱性pH~11,且随后用EtOAc(3×)萃取。将经合并的有机层用水洗涤,经无水MgSO4干燥且在减压下浓缩以获得2.1g 2-[(2-氨基-4-吡啶基)氧基]乙醇(I-35-2),其无需进一步纯化即用于后续步骤中。
向I-35-2(4.0g,24mmol)于THF(40mL)中的搅拌溶液中添加2-氯乙醛的水溶液(6.8g,48mmol,55%水溶液)。将混合物在密封管中加热至75℃持续18h。随后使混合物冷却至室温且分配于EtOAc(3×50mL)与NaHCO3饱和水溶液(100mL)之间。将经合并的有机层用盐水(100mL)洗涤,经无水MgSO4干燥且在减压下浓缩以获得1.8g 7-(2-甲氧基乙氧基)咪唑并[1,2-a]吡啶(I-35-3),其无需进一步纯化用于后续步骤中。
向I-35-3(1.3g,6.8mmol)于DMF(15mL)中的搅拌溶液一次性添加NBS(1.2g,6.8mmol)。将所得混合物在室温下搅拌5分钟,随后用硫代硫酸钠饱和水溶液(150mL)稀释并且随后用EtOAc(3×50mL)萃取。将经合并的有机层用盐水洗涤,经无水MgSO4干燥,在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以获得1.1g 3-溴-7-(2-甲氧基乙氧基)咪唑并[1,2-a]吡啶(I-35)。
N-环丙基-4-甲基-3-(1H-吡唑-4-基)苯甲酰胺(I-36)的合成
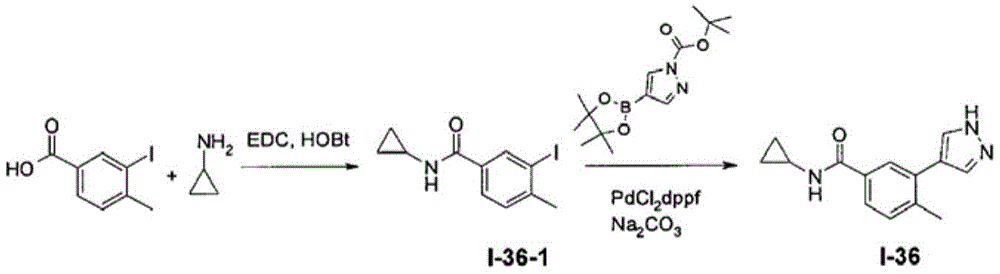
在室温下向3-碘-4-甲基-苯甲酸(42g,160mmol)于DMF(400mL)中的搅拌溶液中添加EDC HCl(92g,481mmol)然后添加HOBt(32g,240mmol)。将反应混合物搅拌30分钟,随后添加环丙胺(13.3mL,192mmol)及DIPEA(140mL,802mmol)。16h后,将反应用水淬灭且用EtOAc萃取。将经合并的有机层用盐水洗涤,经无水MgSO4干燥且在减压下蒸发。将粗制物质用20%EtOAc的己烷溶液(200mL)洗涤以获得45g N-环丙基-3-碘-4-甲基-苯甲酰胺(I-36-1)。
在室温下向I-36-1(20g,66.4mmol)于1,4-二噁烷(500mL)中的溶液中添加4-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)吡唑-1-甲酸叔丁酯(23.4g,79.7mmol)然后添加Na2CO3(21.1g,199mmol)及水(150mL)。将反应混合物脱气且用氮气再填充两次。添加PdCl2(dppf)(5.4g,6.6mmol)且在110℃下加热反应混合物4h。冷却且在减压下蒸发反应混合物。通过快速硅胶柱色谱法(3%MeOH于EtOAc中的洗脱液)来纯化粗残余物,以得到15.2g N-环丙基-4-甲基-3-(1H-吡唑-4-基)苯甲酰胺(I-36)。
使用商购可得的苯甲酸根据上文所描述的通用程序合成以下中间体:
N-环丙基-2-氟-4-甲基-5-(1H-吡唑-4-基)苯甲酰胺(I-37)
4-氯-N-环丙基-2-氟-5-(1H-吡唑-4-基)苯甲酰胺(I-38)
以下方式制备用于合成中间体I-37的起始苯甲酸:
2-氟-5-碘-4-甲基-苯甲酸(I-1A)的合成

在0℃下经45分钟向2-氟-4-甲基苯甲酸(26g,168mmol)于浓缩H2SO4(260mL)中的搅拌溶液中逐滴添加新制备的硝化混合物[浓缩H2SO4(10.7mL)+70%HNO3(11.9mL)]。在0℃下搅拌所得溶液3h。用冰水淬灭反应混合物。用乙酸乙酯萃取所得非均相溶液。将经合并的有机层用水、盐水洗涤,经无水MgSO4干燥,过滤且在减压下浓缩以获得30g粗2-氟-4-甲基-5-硝基-苯甲酸(I-1-1)。
在10℃下向I-1-1(30g,150mmol)于甲醇(300mL)中的搅拌溶液中逐滴添加亚硫酰氯(22.5mL,301mmol)。使所得溶液升温以回流。12h后,在减压下浓缩溶剂且使粗残余物分配于乙酸乙酯与水之间。有机层经分离且用饱和NaHCO3溶液、水、盐水洗涤,经无水MgSO4干燥,过滤且在减压下浓缩以获得30g 2-氟-4-甲基-5-硝基-苯甲酸甲酯(I-1-2)。
将甲基I-1-2(30g,141mmol)于甲醇(600mL)中的溶液装入2升帕尔(Parr)压力容器。随后在氮气气氛下添加10%钯/碳(3g,28mmol)。将帕尔容器置于氢气气氛(45psi)下。12h后,将反应物质经由硅藻土过滤,且在减压下浓缩滤液以获得26g 5-氨基-2-氟-4-甲基-苯甲酸甲酯(I-1-3)。
在-5℃下向I-1-3(26g,142mmol)于乙腈(540mL)中的搅拌溶液中逐滴添加亚硝酸异戊酯(21.7g,184mmol)。5分钟后,将碘化铜(I)(56g,369mmol)逐滴添加至反应混合物中,且将所得混合物缓慢加热至65℃持续2小时。溶液经由硅藻土过滤,且在减压下浓缩滤液。快速柱色谱法(硅胶,5%乙酸乙酯于己烷中的洗脱液)得到20g 2-氟-5-碘-4-甲基-苯甲酸甲酯(I-1-4)。
在室温下向I-1-4(20g,68mmol)于THF:MeOH:H2O(1:1:1,300mL)中的搅拌溶液中添加固体NaOH(4g,102mmol)。在室温下搅拌所得溶液3h。在减压下浓缩溶剂,且将残余物用水(500mL)稀释且用乙酸乙酯(2×150mL)洗涤。通过添加10%HCl水溶液将含水层的pH调节至pH为2且随后用DCM(3×150mL)萃取。将经合并的有机层用水(2×100mL)、盐水(200mL)洗涤,经无水MgSO4干燥,过滤且在减压下浓缩以获得2-氟-5-碘-4-甲基-苯甲酸(I-1A)。
N-环丙基-3-(1H-咪唑-4-基)-4-甲基-苯甲酰胺(I-39)的合成

将N-环丙基-4-甲基-3-(4,4,5,5-四甲基-[1,3,2]二噁硼烷-2-基)-苯甲酰胺(1.0g;3.2mmol)、4-溴-1-三苯甲基-1H-咪唑(1.4g;3.54mmol)、2M K2CO3水溶液(4.0ml;8.05mmol)与四(三苯基膦)钯(0)(428.0mg;0.37mmol)于二噁烷(13.4ml)中的混合物在微波瓶中脱气且用氮再填充。在微波反应器中在130℃下加热反应持续30分钟。将混合物用水稀释且用EtOAc萃取。将有机相用盐水洗涤,经Na2SO4干燥,过滤且在减压下浓缩。通过快速硅胶色谱法来纯化所得残余物以提供N-环丙基-4-甲基-3-(1-三苯甲基-1H-咪唑-4-基)-苯甲酰胺I-39-1(1.0g)。
向I-39-1(1g;2.1mmol)于二氯甲烷(11.7ml)中的溶液中添加TFA(2.0ml)。将混合物在室温下搅拌2h。添加额外0.5mL TFA且再将反应搅拌1h。通过添加NaHCO3饱和溶液来将混合物的pH调节为pH=8。将混合物用盐水洗涤,经由Na2SO4干燥,过滤且在减压下浓缩。通过快速硅胶柱色谱法来纯化所得残余物以提供标题化合物(I-39)(500mg)。
使用商购可得的苯甲酸根据上文所描述的通用程序合成以下中间体:
N-环丙基-2-氟-4-甲基-5-(1H-咪唑-4-基)苯甲酰胺(I-39-2)
3-溴-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸二甲酰胺(I-40)的合成

在0.5h内向4-甲氧基-吡啶-2-基胺(45g;0.362mol;1.0当量)于HOAc(1000mL)中的溶液中逐滴添加Br2(57.9g;0.362mol;1.0当量)于HOAc(260mL)中的溶液。产生大量白色固体。将所得混合物在18℃下搅拌1.5h。在过滤之后,将滤饼用EtOAc(1500mL)溶解且用饱和NaHCO3(500mL×2)、水(300mL)及盐水(200mL)洗涤,经Na2SO4干燥,过滤并浓缩以获得呈白色固体状的5-溴-4-甲氧基-吡啶-2-基胺(53.0;0.261mol),其无需进一步纯化用于下一步骤中。
向5-溴-4-甲氧基-吡啶-2-基胺(53g,0.261mol,1.0当量)于EtOH:H2O=4:1(500mL)中的溶液中添加氯乙醛(24.589g,0.313mol,1.2当量),随后添加NaHCO3(26.3g,0.313mol,1.2当量)。将所得混合物加热至90℃持续4h。在冷却至室温之后,使有机溶剂蒸发。用DCM(200mL×3)萃取残余物。有机层经合并,经Na2SO4干燥,过滤且浓缩。通过硅胶色谱法(DCM:MeOH=50:1)来纯化粗产物以获得为褐色固体的化合物6-溴-7-甲氧基-咪唑并[1,2-a]吡啶(39g,66%)。
向6-溴-7-甲氧基-咪唑并[1,2-a]吡啶(34.9g,0.154mol,1.0当量)于MeOH(350ml)及甲苯(350ml)中的溶液中添加TEA(23g,0.231mol,1.5当量),随后在N2气氛下添加Pd(dppf)Cl2(11.2g,0.015mol,0.1当量)。在CO气氛(3MPa)下将所得混合物加热至80℃持续16h。在真空下移除溶剂。通过柱色谱法(DCM)来纯化残余物且用PE:EA=1:1(20mL)来洗涤该残余物以获得为褐色固体的7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸甲酯(20g,63%)。
在-10℃下在N2气氛下向7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸甲酯(20g,97mmol,1.0当量)于CHCl3(400ml)中的溶液中添加NBS(17g,97mmol,1.0当量)。使所得溶液升温至0℃且搅拌20分钟。在用DCM(400mL)稀释之后,用水(200mL×2)及盐水(300mL)洗涤所得溶液。有机层经分离,经Na2SO4干燥,过滤且浓缩。用混合物溶剂PE:EA=1:1(500mL)及DCM(100mL)洗涤残余物以获得为浅色固体的化合物3-溴-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸甲酯(15.5g,54mmol)。
3-溴-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸甲酯(2g,7mmol)溶解于THF(40mL)中,随后添加3.5mL 6N HCl水溶液。在60℃下将混合物加热2天。添加额外3.5mL 6N HCl。将反应物再加热一天。在冷却至室温之后,蒸发溶剂以获得为HCl盐(2.2g,60%)的粗产物3-溴-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸。
用饱和NaHCO3中和上文的3-溴-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸的HCl盐(2.2g)直到pH=7为止。过滤固体,且用水冲洗。在高度真空下干燥固体。随后将固体溶解于DMF(20mL)中,添加Et3N(3mL,21mmol)及二甲胺HCl(700mg,8.6mmol),然后添加HATU(2.4g,6.4mmol)。在室温下持续搅拌过夜。向反应中添加水,用EtOAc萃取,用水、盐水洗涤,经Na2SO4干燥,过滤且浓缩。通过硅胶色谱法(0%至10%MeOH/DCM)来纯化残余物以获得化合物3-溴-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸二甲酰胺(I-40)(1.24g,76%)。
N-环丙基-2-氟-4-甲基-5-[1-(6-哌啶-4-基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-苯甲酰胺二-盐酸盐(I-41)的合成

在室温下于N2下向6-溴-咪唑并[1,2-a]吡啶(20g,0.102mol,1.0当量)、硼酸酯(37.66g,0.122mol,1.2当量)与Cs2CO3(65.98g,0.203mol,2.0当量)于二噁烷(300mL)与H2O(30mL)中的混合物中添加Pd(PPh3)2Cl2(7.13g,0.01mol,0.1当量)。在N2下将混合物加热至100℃且搅拌15h。TLC及LCMS显示反应完成。反应经由硅藻土垫过滤且用DCM(3×500mL)洗涤。在真空中浓缩滤液以得到粗产物,其通过硅胶柱色谱法(DCM:MeOH=100:1至30:1)来纯化以获得为褐色油的4-咪唑并[1,2-a]吡啶-6-基-3,6-二氢-2H-吡啶-1-甲酸叔丁酯(26g,0.087mol)。
在Ar2下向4-咪唑并[1,2-a]吡啶-6-基-3,6-二氢-2H-吡啶-1-甲酸叔丁酯(52g,173.7mmol,1.0当量)于MeOH(3000mL)中的溶液中添加10%Pd/C(20g)。在20℃下在H2(50psi)下将反应混合物搅拌10h。混合物经由硅藻土垫过滤且用MeOH(1500mL)洗涤,在真空中浓缩滤液以得到粗产物,其通过硅胶柱色谱法(DCM:MeOH=300:1至20:1)来纯化为褐色油的4-咪唑并[1,2-a]吡啶-6-基-哌啶-1-甲酸叔丁酯(50g,165.9mmol)。
向4-咪唑并[1,2-a]吡啶-6-基-哌啶-1-甲酸叔丁酯(1.5g,5mmol)于氯仿(15mL)中的搅拌溶液中添加NBS(890mg,5mmol)。在室温下搅拌溶液2h并且随后浓缩。残余物用EtOAc稀释,用NaHCO3、H2O、盐水洗涤且浓缩以得到残余物,其通过快速色谱法(25g,0%至80%EtOAc/庚烷)然后以反相色谱法(100g,10%至100%H2O/ACN,两者均含有0.5%甲酸)来纯化以获得化合物4-(3-溴-咪唑并[1,2-a]吡啶-6-基)-哌啶-1-甲酸叔丁基(1.6g,85%)。
将4-(3-溴-咪唑并[1,2-a]吡啶-6-基)-哌啶-1-甲酸叔丁基(1.55,4.1mmol)、I-37(1.3g,4.9mmol)、CuI(388mg,2.0mmol)、K3PO4(1.7g,8.2mmol)及反式-二甲基氨基环己烷(463mg,3.3mmol)悬浮于DMF(15mL)中且用Ar冲洗混合物。随后将混合物加热至65℃且将其搅拌过夜。将混合物冷却至室温,用EtOAc稀释,用H2O、盐水洗涤且浓缩以得到残余物,其通过快速色谱法(50g,0%至100%EtOAc/庚烷)来纯化以获得化合物4-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-基}-哌啶-1-甲酸叔丁酯(1.4g,62%)。
向4-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-基}-哌啶-1-甲酸叔丁酯(1.4g,2.5mmol)于DCM(15mL)与MeOH(5mL)中的搅拌且冷却(0℃)溶液中添加含4N HCl的二噁烷(10mL)。使混合物升温至室温过夜。浓缩溶液以得到残余物,其在真空中干燥以获得化合物I-41(1.32g,99%)。
最终化合物
实施例1:3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸二甲酰胺

将I-40(217mg,0.7mmol)、I-37(170mg,0.7mmol)、CuI(50mg,0.3mmol)、K3PO4(278mg,1.3mmol)及反式-二甲基氨基环己烷(74mg,0.5mmol)悬浮于DMF(10mL)中且用Ar冲洗混合物。随后将混合物加热至60℃持续6h,随后在室温下过夜。将反应过滤,用EtOAc冲洗且浓缩。向残余物中添加水,用EtOAc萃取,用水洗涤且浓缩。通过反相HPLC(5%至50%ACN于含甲酸的水中)来纯化残余物。合并且浓缩纯级分。使残余物溶解于MeOH中且穿过碳酸氢盐滤筒以获得为白色固体的1(75mg,24%)。
实施例2:3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸甲酯

将3-溴-咪唑并[1,2-a]吡啶-6-甲酸甲酯(492mg,1.93mmol)、I-37(500mg,1.93mmol)、磷酸钾(819mg,3.86mmol)、反式-N,N'-二甲基-环己烷-1,2-二胺(0.2ml,1.54mmol)于无水DMF(8.0ml)中的混合物用氮气脱气。向此混合物中添加CuI(147mg,0.77mmol)。将反应放置在氮气下且在60℃下将反应加热持续4h。随后使混合物冷却至室温且搅拌18h。反应随后用水稀释且用EtOAc(3×)萃取。有机层经合并,用水、随后用盐水洗涤且经硫酸钠干燥。随后将溶液过滤且在减压下浓缩。通过快速硅胶色谱法来纯化所得残余物以提供374mg标题化合物(2)。
使用商购可得的杂芳基溴化物及/或本文所描述的中间体以类似于实施例2的方式合成以下化合物:
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-咪唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸甲基酰胺(3)
4-氯-N-环丙基-2-氟-5-(1-咪唑并[1,2-a]吡啶-3-基-1H-吡唑-4-基)-苯甲酰胺(5)
4-氯-N-环丙基-2-氟-5-(1-咪唑并[1,2-a]吡嗪-3-基-1H-吡唑-4-基)-苯甲酰胺(6)
5-[1-(2-乙酰氨基-噻唑-5-基)-1H-吡唑-4-基]-4-氯-N-环丙基-2-氟-苯甲酰胺(7)
5-[1-(8-氨基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-4-氯-N-环丙基-2-氟-苯甲酰胺(8)
4-氯-N-环丙基-2-氟-5-[1-(6-甲氧基-咪唑并[1,2-a]吡嗪-3-基)-1H-吡唑-4-基]-苯甲酰胺(9)
4-氯-N-环丙基-2-氟-5-{1-[6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(10)
4-氯-N-环丙基-2-氟-5-(1-吡唑并[1,5-a]吡啶-3-基-1H-吡唑-4-基)-苯甲酰胺(11)
4-氯-N-环丙基-2-氟-5-(1-咪唑并[1,2-a]吡嗪-6-基-1H-吡唑-4-基)-苯甲酰胺(12)。注意:作为以6-溴-3-碘-咪唑并[1,2-a]吡嗪为起始物,且反应在100℃下进行18h的副产物而获得。
N-环丙基-3-(1-咪唑并[1,2-a]吡嗪-3-基-1H-吡唑-4-基)-4-甲基-苯甲酰胺(13)
3-[1-(2-乙酰氨基-噻唑-5-基)-1H-吡唑-4-基]-N-环丙基-4-甲基-苯甲酰胺(14)
N-环丙基-3-(1-咪唑并[1,2-a]吡啶-3-基-1H-吡唑-4-基)-4-甲基-苯甲酰胺(15)
3-[1-(2-乙酰氨基-噻唑-5-基)-1H-咪唑-4-基]-N-环丙基-4-甲基-苯甲酰胺(17)
N-环丙基-4-甲基-3-{1-[6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(19)
N-环丙基-4-甲基-3-{1-[6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶-2-基]-1H-吡唑-4-基}-苯甲酰胺(20)。注意:作为在反应在100℃下进行18h时来自19的合成的副产物而获得。
4-氯-N-环丙基-2-氟-5-[1-(6-甲磺酰基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-苯甲酰胺(21)
N-环丙基-4-甲基-3-{1-[6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-咪唑-4-基}-苯甲酰胺(22)
N-环丙基-4-甲基-3-{1-[6-(氧杂环丁-3-基硫烷基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(23)
5-[4-(2-氯-5-环丙基胺甲酰基-4-氟-苯基)-吡唑-1-基]-噻唑-2-甲酸酰胺(25)
5-[4-(2-氯-5-环丙基胺甲酰基-4-氟-苯基)-吡唑-1-基]-噻唑-2-甲酸甲基酰胺(26)
N-环丙基-3-[1-(6-乙磺酰基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(27)
N-环丙基-3-{1-[7-甲氧基-6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶-2-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺(28)。注意:作为在35的合成期间在反应在100℃下进行18h时的副产物而获得。
N-环丙基-4-甲基-3-{1-[5-(2-甲基-丙烷-2-磺酰基)-吡唑并[1,5-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(29)
N-环丙基-3-[1-(6-乙磺酰基-7-甲氧基-咪唑并[1,2-a]吡啶-2-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(30)。注意:作为在反应在100℃下进行18h时的副产物而获得。
N-环丙基-3-[1-(7-乙氧基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(31)
N-环丙基-2-氟-5-(1-咪唑并[1,2-a]吡啶-3-基-1H-吡唑-4-基)-4-甲基-苯甲酰胺(32)
N-环丙基-2-氟-4-甲基-5-{1-[6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(33)
N-环丙基-2-氟-5-[1-(6-甲氧基-咪唑并[1,2-a]吡嗪-3-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(34)
N-环丙基-3-{1-[7-甲氧基-6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺(35)
N-环丙基-2-氟-4-甲基-5-{1-[6-(吗啉-4-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(36)
N-环丙基-4-甲基-3-{1-[6-(吗啉-4-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(37)
N-环丙基-4-甲基-3-{1-[6-(4-甲基-哌嗪-1-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(38)
N-环丙基-2-氟-4-甲基-5-{1-[6-(4-甲基-哌嗪-1-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(39)
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-咪唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸二甲酰胺(41)
3-(1'-叔丁基-1'H-[1,4']二吡唑基-4-基)-N-环丙基-4-甲基-苯甲酰胺(42)
N-环丙基-4-甲基-3-(1-噻唑-5-基-1H-吡唑-4-基)-苯甲酰胺(43)
3-[1-(2-环丁氧基-噻唑-5-基)-1H-吡唑-4-基]-N-环丙基-4-甲基-苯甲酰胺(44)
3-{1-[2-(环丙羰基-氨基)-噻唑-5-基]-1H-吡唑-4-基}-N-环丙基-4-甲基-苯甲酰胺(45)
N-环丙基-4-甲基-3-[1-(2-吗啉-4-基-噻唑-5-基)-1H-吡唑-4-基]-苯甲酰胺(46)
N-环丙基-4-甲基-3-[1-(2-苯基-噻唑-5-基)-1H-吡唑-4-基]-苯甲酰胺(47)
N-环丙基-4-甲基-3-{1-[2-(2-氧代-2H-吡啶-1-基)-噻唑-5-基]-1H-吡唑-4-基}-苯甲酰胺(48)
N-环丙基-4-甲基-3-[1-(2-吡咯烷-1-基-噻唑-5-基)-1H-吡唑-4-基]-苯甲酰胺(49)
N-环丙基-4-甲基-3-[1-(2-哌啶-1-基-噻唑-5-基)-1H-吡唑-4-基]-苯甲酰胺(50)
N-环丙基-3-[1-(2-羟基甲基-3-甲基-3H-咪唑-4-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(53)
N-环丙基-4-甲基-3-[1-(5-甲基-[1,3,4]噻二唑-2-基)-1H-吡唑-4-基]-苯甲酰胺(54)
N-环丙基-3-[1-(2,3-二甲基-3H-咪唑-4-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(55)
实施例16:N-环丙基-3-(1-咪唑并[1,2-a]吡啶-3-基-1H-咪唑-4-基)-4-甲基-苯甲酰胺

将3-溴-咪唑并[1,2-a]吡啶-6-甲酸甲酯(43mg,0.22mmol)、I-39(0.63mg,0.26mmol)、磷酸钾(0.93mg,0.44mmol)、反式-N,N'-二甲基-环己烷-1,2-二胺(0.03mL,0.18mmol)于无水DMF(1.0ml)中的混合物用氮气脱气。向此混合物中添加CuI(0.17mg,0.09mmol)。将反应置于氮气下且在100℃下将反应加热24h。随后将混合物冷却至室温,用水稀释且用EtOAc(3×50mL)萃取。有机层经合并,用水、随后用盐水洗涤且经硫酸钠干燥。随后将溶液过滤且在减压下浓缩。通过快速硅胶色谱法来纯化所得残余物以提供0.17mg标题化合物(16)。
使用商购可得的杂芳基溴化物及/或本文所描述的中间体以类似于实施例16的方式合成以下化合物:
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-咪唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸甲酯(40)
实施例56:3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸

向2(354mg,0.82mmol)于甲醇(15ml)及水(5ml)中的悬浮液中添加氢氧化锂一水合物(274mg,6.53mmol)。2h后,反应逐渐变成透明且将其搅拌18h。随后在减压下浓缩反应。向残余物中添加2M HCl水溶液(3mL)且浓缩混合物以提供610mg标题化合物(56)。残余物无需进一步纯化即用于后续步骤中。
使用本文所描述的中间体以类似于实施例56中所描述的步骤的方式合成以下化合物:
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸(57)
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-咪唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸(58)
实施例59:3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸(2-羟基-乙基)-酰胺:

向56(178mg,56%纯度,0.24mmol)于DMF(3.7ml)中的搅拌溶液中添加Et3N(0.10mL,0.72mmol)、氨基乙醇(29mg,0.48mmol)及HATU(136mg,0.36mmol)。18h后,通过反相HPLC然后以快速硅胶柱色谱法来纯化反应以提供82mg标题化合物(59)。
使用商购可得的胺以类似于实施例59的方式合成以下化合物:
3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸(2-甲胺基-乙基)-酰胺(60)
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-7-甲酸(2-羟基-乙基)-酰胺(61)
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-7-甲酸酰胺(62)
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-7-甲酸(2-甲胺基-乙基)-酰胺(63)
N-环丙基-4-甲基-3-{1-[6-(吗啉-4-羰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(64)
N-环丙基-4-甲基-3-{1-[6-(4-甲基-哌嗪-1-羰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(65)
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸二甲酰胺(66)
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸甲基酰胺(67)
3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-7-甲酸甲基酰胺(68)
3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-甲酸二甲酰胺(69)
N-环丙基-2-氟-4-甲基-5-{1-[6-(4-甲基-哌嗪-1-羰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(70)
实施例71:N-环丙基-2-氟-5-{1-[6-(1-羟基-乙基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺

使用中间体I-30根据实施例2合成5-(1-{6-[1-(叔丁基-二甲基-硅烷基氧基)-乙基]-咪唑并[1,2-a]吡啶-3-基}-1H-吡唑-4-基)-N-环丙基-2-氟-4-甲基-苯甲酰胺(71-1)。
向71-1(100mg,0.16mmol)于THF(4mL)中的搅拌溶液中添加作为于THF(1M,160μL,0.16mmol)中的溶液的四丁基氟化铵。将所得溶液在室温下搅拌2.5h,并且随后在减压下浓缩。通过快速硅胶色谱法来纯化所得残余物。通过制备型反相色谱法来进一步纯化经分离物质以提供66mg标题化合物(71)。
使用本文所描述的中间体以类似于实施例71中所描述的步骤的方式合成以下化合物。
N-环丙基-3-{1-[6-(1-羟基-乙基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺(72)
N-环丙基-3-(1-{6-[羟基-(四氢-吡喃-4-基)-甲基]-咪唑并[1,2-a]吡啶-3-基}-1H-吡唑-4-基)-4-甲基-苯甲酰胺(73)
N-环丙基-3-{1-[6-(1-羟基-乙基)-咪唑并[1,2-a]吡啶-3-基]-1H-咪唑-4-基}-4-甲基-苯甲酰胺(74)
N-环丙基-2-氟-5-{1-[6-(1-羟基-1-甲基-乙基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺(75)
实施例76:3-[1-(6-乙酰基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-N-环丙基-4-甲基-苯甲酰胺

向72(660mg,1.6mmol)于无水DCM(30mL)中的溶液中添加戴斯-马丁试剂(Dess-Martin reagent)(725mg,1.70mmol)。30分钟后,将反应用NaHCO3饱和水溶液(5mL)稀释,且搅拌20分钟。混合物经由硅藻土过滤,且分离这些相。将有机层用盐水(50mL)洗涤,经MgSO4干燥,过滤且在减压下浓缩。通过快速硅胶柱色谱法来纯化残余物以提供618mg标题化合物(76)。
实施例77:N-环丙基-4-甲基-3-{1-[6-(氧杂环丁烷-3-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺

向28(70mg,0.16mmol)于乙腈(2.0mL)与水(1.0mL)的混合物中的搅拌溶液中添加氯化钌(III)水合物(2mg,0.01mmol)及偏高碘酸钠(202mg,0.94mmol)。1h后,用EtOAc(10mL)及水(5mL)稀释反应。分离各层且有机层用EtOAc(3×)萃取,用盐水(5mL)洗涤,经Na2SO4干燥,过滤且在减压下浓缩。通过反相HPLC(29%至49%ACN于含NH4HCO3的水中)来纯化所得残余物以提供11mg标题化合物(77)。
实施例78:N-环丙基-3-{1-[7-羟基-6-(2-甲基-丙烷-2-磺酰基)-咪唑并[1,2-a]吡啶-2-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺

向35(200mg,0.39mmol)于DMF(5.0mL)中的搅拌溶液中添加NaSi-Pr(387mg,3.94mmol)。将反应在150℃下搅拌1h。将反应冷却至室温并且在减压下浓缩。通过反相色谱法来纯化所得残余物以提供35mg标题化合物(78)。
实施例79:3-{1-[6-(氮杂环丁烷-3-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-N-环丙基-4-甲基-苯甲酰胺

使用本文所描述的中间体根据实施例2合成3-{3-[4-(5-环丙基胺甲酰基-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-磺酰基}-氮杂环丁烷-1-甲酸叔丁酯(79-1)。
向79-1(15mg,0.03mmol)于MeOH(1.0mL)中的搅拌溶液中添加4MHCl于二噁烷(130μL,0.52mmol)中的溶液。将反应在室温下搅拌18h并且随后在减压下浓缩。通过制备型反相HPLC来纯化所得残余物以提供10mg标题化合物(79)。
使用本文所描述的中间体以类似于如实施例79中所描述的方式合成以下化合物。
N-环丙基-4-甲基-3-{1-[6-(哌啶-4-磺酰基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(80)
实施例81:N-环丙基-2-氟-5-{1-[6-(1-羟基-乙基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺

通过手性hplc(ChiralCel OJ-H 20×250mm,22%EtOH:庚烷,10毫升/分钟,38℃)分离50(250mg,0.6mmol)的样品。将洗脱液的第一峰指定为81-1(52mg)且将第二峰指定为81-2(56mg)。
实施例82:N-环丙基-3-{1-[6-(1-羟基-乙基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺

通过手性hplc(ChiralCel OJ-H 20×250mm,20%EtOH(.1%DEA):庚烷,以10毫升/分钟,35℃)分离N-环丙基-3-{1-[6-(1-羟基-乙基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺(100mg,0.25mmol)的样品。将洗脱液的第一峰指定为82-1(39mg)且将第二峰指定为82-2(41mg)。
(82-1及82-2)
实施例84:N-环丙基-4-甲基-3-(1'-甲基-1'H-[1,4']二吡唑基-4-基)-苯甲酰胺
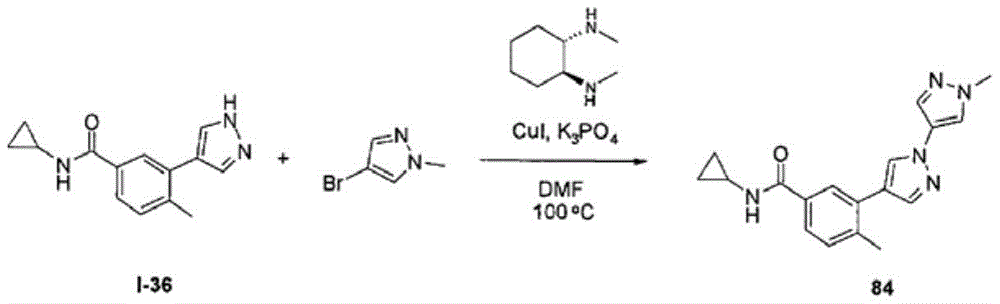
将I-36(75mg,0.31mmol)、4-溴-1-甲基-1H-吡唑(48mg,0.47mmol)、CuI(24mg,0.12mmol)及磷酸钾(132mg,0.62mmol)的样品合并至经脱气DMF(1.5mL)中。向此混合物中添加反式-1,2-双(甲胺基)环己烷(0.04mL,0.25mmol)且在100℃下加热悬浮液。18h后,添加水(0.15mL),然后添加3mL 10%水于DMF中的混合物。过滤反应且通过反相HPLC来纯化洗脱液以获得38mg(84)。
使用商业来源的或如本文中所描述的芳基溴化物以类似于如实施例84中所描述的方式合成以下化合物:
N-环丙基-3-{1-[7-(2-甲氧基-乙氧基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺(85)
N-环丙基-3-[1-(7-甲氧基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(86)
N-环丙基-4-甲基-3-[1-(5,6,7,8-四氢-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]苯甲酰胺(87)
N-环丙基-4-甲基-3-[1-(2-吡唑-1-基-噻唑-5-基)-1H-吡唑-4-基]-苯甲酰胺(88)
N-环丙基-3-[1-(2-羟基甲基-噻唑-4-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(89)
实施例90:N-环丙基-4-甲基-3-{1-[5-(2-甲基-丙烷-2-磺酰基)-4,5,6,7-四氢-吡唑并[1,5-a]吡嗪-3-基]-1H-吡唑-4-基}-苯甲酰胺

将I-36、I-34(134mg,0.41mmol)、CuI(2mg,0.012mmol)及磷酸钾(176mg,0.83mmol)的样品合并至经脱气二噁烷(1.4mL)及DMSO(0.5mL)中。添加乙二胺(0.83L,0.012mmol)且在60℃下加热悬浮液。16h后,再添加I-34(134mg,0.41mmol)、碘化铜(20mg,0.10mmol)及反式-1,2-双(甲胺基)环己烷(0.05mL,0.33mmol)且在120℃下加热反应。在加热额外16小时之后,用EtOAc(4mL)稀释反应且经由短硅胶柱(12mm宽×15mm高)过滤悬浮液且用EtOAc(2×2mL)洗脱硅胶柱。在减压下浓缩经合并的洗脱液且将残余物溶解于10%水与DMSO(2mL)中的混合物中且通过反相HPLC来纯化以产生20mg标题化合物(90)。
实施例91:N-环丙基-3-[1-(6-甲氧基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺

将I-36、3-溴-6-甲氧基-咪唑并[1,2-a]吡啶(134mg,0.41mmol)、碘化铜(24mg,0.12mmol)及磷酸钾(132mg,0.62mmol)的样品合并至经脱气DMF(1.5mL)中。向此混合物中添加反式-1,2-双(甲胺基)环己烷(0.04mL,0.25mmol)且将悬浮液加热至100℃。18h后,再添加CuI(24mg,0.12mmol)及磷酸钾(66mg,0.31mmol)且持续加热。在加热额外16小时之后,用EtOAc(1mL)稀释反应且经由短硅胶柱过滤悬浮液且用EtOAc(2×2mL)洗脱硅胶柱。在减压下浓缩经合并的洗脱液且将残余物溶解于10%水于DMSO(2mL)中的混合物中且通过反相HPLC来纯化以产生26mg(91)。
实施例92:N-环丙基-2-氟-4-甲基-5-{1-[6-(1-氧杂环丁-3-基-哌啶-4-基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺

将胺盐I-41(400mg,0.8mmol)溶解于MeOH(10mL)中,随后添加HOAc(97mg,1.6mmol)及酮(0.29mL,4mmol)。将溶液搅拌30分钟。随后添加NaBH3CN(0.5g,8.1mmol)且将反应在50℃下搅拌过夜。在冷却至室温之后,通过添加NaHCO3淬灭反应,用EtOAc萃取。浓缩经合并的萃取物且通过快速色谱法(25g,0至5%MeOH/DCM)来纯化残余物以获得化合物92(220mg,53%)。
实施例93:N-环丙基-5-{1-[6-(1-二甲基胺甲酰基甲基-哌啶-4-基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-2-氟-4-甲基-苯甲酰胺

将I-41(1.3g,2.6mmol)溶解于DMF(10mL)中。添加iPr2NEt(4.8mL,260mmol)及烷基溴化物(520mg,3.2mmol)且将溶液在50℃下搅拌2h。移除溶剂且将残余物用EtOAc稀释,用NaHCO3、盐水洗涤且浓缩以得到残余物,其通过快速色谱法(25g,0%至10%MeOH/DCM)纯化得到混杂有碱的粗产物。随后通过反相柱(60g,0%至60%ACN/H2O)来纯化此粗制品,得到产物FO盐。将此盐再溶解于DCM中,用Na2CO3洗涤,浓缩且通过快速色谱法(25g,0%至10%MeOH/DCM)来纯化以获得为游离碱的93(810mg,57%)。
实施例94:N-环丙基-2-氟-5-{1-[6-(4-氟-1-甲基哌啶-4-基)-7-甲氧基咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺

在-15℃下经30分钟向I-24-2(50.0g,220mmol)于THF(1L)中的溶液逐滴添加i-PrMgCl-LiCl(1.3M THF,338mL,440mmol)。将混合物搅拌额外30分钟且经20分钟逐滴添加于THF(180mL)中的酮(92.1g,462mmol)。在室温下搅拌所得混合物24h。用饱和NH4Cl水溶液(400mL)淬灭混合物且浓缩。添加水(400mL)且用EtOAc(2×600mL)萃取。用盐水(400mL)洗涤且浓缩经合并的萃取物。将粗残余物溶解于DCM中且通过硅胶色谱法(1%至8%MeOH/DCM)来纯化以获得为固体的羟基哌啶产物(30.7g,68.1mmol)。
在0℃下在氩气下经10分钟向于DCM(470mL)中的上述羟基哌啶产物(24.6g,70.9mmol)逐滴添加NBS(11.4g,63.8mmol)。将所得暗绿色溶液在0℃下搅拌45分钟。用饱和NaHCO3水溶液(380mL)淬灭0℃混合物。随后添加水(190mL)且用DCM(2×250mL)萃取混合物。浓缩经合并的有机层且通过硅胶色谱法(0%至5%MeOH/DCM)来纯化粗产物以得到为白色固体的4-{3-溴-7-甲氧基咪唑并[1,2-a]吡啶-6-基}-4-羟基哌啶-1-甲酸叔丁酯(22.5g,52.8mmol)。
在具有200mL经脱气DMF(200mL)的耐压烧瓶中添加4-{3-溴-7-甲氧基咪唑并[1,2-a]吡啶-6-基}-4-羟基哌啶-1-甲酸叔丁酯(19.9g,46.7mmol)、碘化铜(4.4g,23.3mmol)、(1S,2S)-N1,N2-二甲基环己烷-1,2-二胺(7.3mL,46.7mmol)、I-37(14.5g,56.0mmol)及磷酸钾(19.8g,93.4mmol)。在90℃下加热反应混合物24h。在水浴中将混合物冷却至室温且用EtOAc(420mL)稀释。经由硅藻土过滤且蒸发混合物。在DCM与水之间分配所得残余物。有机层经MgSO4干燥且通过硅藻土过滤且蒸发。将所得黏稠残余物溶解于MeOH(400mL)中且逐滴添加水(1.4L)。过滤所得固体沉淀物并且随后通过硅胶色谱法(100%EtOAc持续10分钟,随后0%至10%MeOH/EtOAc经过35分钟)来纯化以得到4-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-7-甲氧基-哌啶-1-甲酸叔丁酯(15g,20.8mmol,84%纯度)。
在-78℃下向4-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-7-甲氧基-哌啶-1-甲酸叔丁酯(15g,20.8mmol)于DCM(250mL)中的溶液中逐滴添加[双(2-甲氧乙基)氨基]三氟化硫(50%于THF中,21.3mL,49.6mmol)。将混合物在-78℃下搅拌10分钟。将混合物经1h升温至-30℃并且随后在室温下搅拌1.5h。将混合物冷却至0℃且缓慢添加NaHCO3饱和水溶液(150mL)来淬灭。搅拌混合物5分钟,发生鼓泡。分离有机层且用DCM(2×50mL)萃取含水层。将经合并的有机物用水(100mL)洗涤且经硫酸钠干燥,过滤且浓缩。通过硅胶色谱法(0%至10%MeOH/DCM)来纯化所得粗物质以得到为固体的4-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-7-甲氧基-咪唑并[1,2-a]吡啶-6-基}-4-氟-哌啶-1-甲酸叔丁酯(10.4g,17.2mmol)。
向4-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-7-甲氧基-咪唑并[1,2-a]吡啶-6-基}-4-氟-哌啶-1-甲酸叔丁酯(17.8g,29.4mmol)于MeOH(120mL)中的溶液中添加含4M HCl的二噁烷(73.5mL,293.9mmol)且搅拌混合物1h。浓缩混合物且在50℃下在真空中干燥残余物2h以得到粗产物N-环丙基-2-氟-5-{1-[6-(4-氟-哌啶-4-基)-7-甲氧基-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-甲基-苯甲酰胺盐酸盐(17.6g,32.5mmol),其无需进一步纯化即使用。
向N-环丙基-2-氟-5-{1-[6-(4-氟-哌啶-4-基)-7-甲氧基-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-甲基-苯甲酰胺盐酸盐(17.6g,32.5mmol)于DCM(335mL)中的溶液中添加甲醛(9.7mL,130.2mmol)及三乙酰氧基硼氢化钠(27.6g,130.2mmol)且搅拌45分钟。用NaHCO3饱和水溶液(350mL)经30分钟淬灭混合物。分离各层且用DCM(2×200mL)萃取含水层。将经合并的有机物经水(2×200mL)洗涤,经Na2SO4干燥,过滤且浓缩。通过制备型HPLC来纯化粗残余物以得到为白色固体的N-环丙基-2-氟-5-{1-[6-(4-氟-1-甲基哌啶-4-基)-7-甲氧基咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-4-甲基-苯甲酰胺(15.0g,28.8mmol)。
实施例95:N-环丙基-5-[1-(6-二甲胺基甲基-7-甲氧基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-2-氟-4-甲基-苯甲酰胺

在0.5h内向4-甲氧基-吡啶-2-基胺(45g;0.362mol;1.0当量)于HOAc(1000mL)中的溶液中逐滴添加Br2(57.9g;0.362mol;1.0当量)于HOAc(260mL)中的溶液。产生大量白色固体。将所得混合物在18℃下搅拌1.5h。在过滤之后,用EtOAc(1500mL)溶解滤饼且用饱和NaHCO3(500mL×2)、水(300mL)及盐水(200mL)洗涤,经Na2SO4干燥,过滤且浓缩以获得为白色固体的5-溴-4-甲氧基-吡啶-2-基胺(53.0g;0.26mol),其无需纯化即用于下一步骤中。
向5-溴-4-甲氧基-吡啶-2-基胺(53g,0.261mol,1.0当量)于EtOH:H2O=4:1(500mL)中的溶液中添加氯乙醛(24.6g,0.31mol),随后添加NaHCO3(26.3g,0.313mol)。将所得混合物加热至90℃持续4h。在冷却至室温之后,蒸发有机溶剂。用DCM(200mL×3)萃取残余物。有机层经合并,经Na2SO4干燥,过滤且浓缩。通过硅胶色谱法(DCM:MeOH=50:1)来纯化粗产物以获得为褐色固体的化合物6-溴-7-甲氧基-咪唑并[1,2-a]吡啶(39g,66%)。
向6-溴-7-甲氧基-咪唑并[1,2-a]吡啶(34.9g,0.154mol,1.0当量)于MeOH(350ml)及甲苯(350ml)中的溶液中添加TEA(23g,0.231mol,1.5当量),随后在N2气氛下添加Pd(dppf)Cl2(11.2g,0.015mol,0.1当量)。在80℃在CO气氛(3MPa)之下将所得混合物加热持续16h。在真空下移除溶剂。通过柱色谱法(DCM)来纯化残余物且用PE:EA=1:1(20mL)来洗涤该残余物以获得为褐色固体的7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸甲酯(20g,63%)。
在-10℃下在N2气氛下向7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸甲酯(20g,97mmol,1.0当量)于CHCl3(400ml)中的溶液中添加NBS(17g,97mmol,1.0当量)。使所得溶液升温至0℃且搅拌20分钟。在用DCM(400mL)稀释之后,用水(200mL×2)及盐水(300mL)洗涤所得溶液。有机层经分离,经Na2SO4干燥,过滤且浓缩。用混合物溶剂PE:EA=1:1(500mL)及DCM(100mL)洗涤残余物以获得为浅色固体的化合物3-溴-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸甲酯(15.5g,54mmol)。
将3-溴-7-甲氧基-咪唑并[1,2-a]吡啶-6-甲酸甲酯(10g,35mmol)悬浮于干燥THF(200mL)中且在室温下经由加料漏斗向此中逐滴添加LAH(1M,105mL,105mmol)。使反应在室温下搅拌2h。向混合物中添加水(2mL),然后添加15%NaOH水溶液(2mL)且再次添加水(2mL)。将混合物搅拌1h且通过过滤移除固体。用热甲醇/DCM洗涤固体且在减压下浓缩滤液以得到粗物质,通过硅胶色谱法(2%至10%MeOH/DCM)将该粗物质纯化以得到(7-甲氧基咪唑并[1,2-a]吡啶-6-基)-甲醇(1.7g,9.5mmol)。
使用加热及超声处理将(7-甲氧基咪唑并[1,2-a]吡啶-6-基)-甲醇(2.7g,13.8mmol)溶解于MeCN(150mL)及MeOH(10mL)中。随后添加NIS(4.3g,19.3mmol)且将其搅拌30分钟。添加Na2CO3饱和水溶液且用EtOAc(3×100mL)萃取混合物。将经合并的有机萃取物用饱和Na2CO3、盐水洗涤且经MgSO4干燥。过滤且浓缩混合物,得到(3-碘-7-甲氧基咪唑并[1,2-a]吡啶-6-基)甲醇,其无需进一步纯化即使用。
将3-碘-7-甲氧基咪唑并[1,2-a]吡啶-6-基)甲醇(2.6g,8.5mmol)溶解于DMF(25mL)中且向其中添加I-37(2.8g,11.1mmol)、反式-1,2-双(甲胺基)环己烷(1.1mL,6.8mmol)、CuI(0.84g,4.3mmol)、磷酸三钾(4.5g,21.4mmol)且在75℃下加热混合物过夜。使反应冷却至室温且用EtOAc稀释且通过硅藻土过滤、用EtOAc及水冲洗。使有机层分离、浓缩且通过制备型HPLC来纯化粗残余物以得到N-环丙基-2-氟-5[1-(6-羟基甲基-7-甲氧基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(1.3g,3.0mmol)。
将(6-羟基甲基-7-甲氧基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-4-甲基-苯甲酰胺(0.69,1.6mmol)溶解于DCM(30mL)中且添加DIPEA(0.414mL,2.4mmol)且在冰浴中使反应物冷却至0℃。添加亚硫酰氯(0.14mL,1.9mmol)于DCM(1mL)中的溶液且将反应搅拌过夜。添加额外当量的含亚硫酰氯的DCM(1mL)且搅拌混合物4h。添加后续额外当量的含亚硫酰氯的DCM(1mL)且搅拌混合物4h。随后向混合物中添加饱和Na2CO3且用DCM萃取。分离且浓缩有机层以得到粗产物,其通过硅胶色谱法(1%至5%MeOH/DCM)来纯化以得到为白色固体的5-[1-(6-氯甲基-7-甲氧基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-N-环丙基-2-氟-4-甲基-苯甲酰胺(0.15g,0.33mmol)。
将5-[1-(6-氯甲基-7-甲氧基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-N-环丙基-2-氟-4-甲基-苯甲酰胺(0.11g,0.24mmol)溶解于DCM中且向其中添加二甲胺(2.0M于DCM中,1.7mL,3.3mmol)且使反应在室温下搅拌过夜。浓缩混合物且直接地通过柱色谱法(25%至100%EtOAc/庚烷)纯化以得到为白色固体的95(0.11g,0.24mmol)。
实施例96:N-环丙基-5-{1-[7-乙氧基-6-(4-氟-1-甲基-哌啶-4-基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-2-氟-4-甲基-苯甲酰胺

将4-乙氧基-吡啶-2-基胺(15g,109mmol)溶解于HOAc(100mL)中且冷却至0℃。在剧烈搅拌下逐滴添加溴。将混合物在室温下搅拌30分钟,此时形成沉淀物。将混合物搅拌30分钟且通过过滤收集固体,用EtOAc洗涤,在真空烘箱中干燥以得到5-溴-4-乙氧基-吡啶-2-基胺氢溴酸盐(23.3g,78.2mmol)。
将5-溴-4-乙氧基-吡啶-2-基胺氢溴酸盐(23.4g,78.5mmol)溶解于EtOH(500mL)中且向其中添加碳酸氢钠(26.4g,314mmol)。随后逐滴添加2-氯乙醛(14.9mL,118mmol)。在添加之后,在115℃下加热混合物1h并且随后冷却至室温且将其搅拌过夜。过滤混合物且在减压下浓缩滤液。使残余物分配于EtOAc与水之间。有机层经分离,经MgSO4干燥,过滤且浓缩,得到6-溴-7-乙氧基-咪唑并[1,2-a]吡啶(6.8g,28.2mmol)。
将6-溴-7-乙氧基-咪唑并[1,2-a]吡啶(0.92g,3.8mmol)溶解于THF(40mL)中。使溶液冷却至-20℃并且随后逐滴添加iPrMgCl LiCL络合物(1.3M于THF中,5.9mL,7.6mmol)。30分钟后,添加1-Boc-4-哌啶酮(1.6g,8.0mmol)且使反应物升温至室温。混合物用饱和NH4Cl淬灭且用EtOAc萃取。有机物经分离且经MgSO4干燥,过滤且浓缩,得到粗产物,该粗产物通过硅胶色谱法(0%至10%MeOH/DCM)纯化以得到4-(7-乙氧基咪唑并[1,2-a]吡啶-6-基)-4-羟基哌啶-1-甲酸叔丁酯(0.38g,1.0mmol)。
将4-(7-乙氧基咪唑并[1,2-a]吡啶-6-基)-4-羟基哌啶-1-甲酸叔丁酯(0.38g,1.0mmol)溶解于DCM(8mL)中且冷却至0℃。向其中添加NBS(0.19g,1.1mmol)且将混合物在室温下搅拌过夜。用DCM稀释且用饱和NaHCO3洗涤混合物。有机层经分离,经MgSO4干燥并且在减压下浓缩以得到粗产物,该粗产物通过硅胶色谱法(0%至10%MeOH/DCM)纯化以得到4-(3-溴-7-乙氧基咪唑并[1,2-a]吡啶-6-基)-4-羟基哌啶-1-甲酸叔丁酯(0.39g,0.87mmol)。
将4-(3-溴-7-乙氧基咪唑并[1,2-a]吡啶-6-基)-4-羟基哌啶-1-甲酸叔丁酯(0.39g,0.87mmol)溶解于DMF(20mL)中且向其中添加CuI(0.066g,0.35mmol)、I-37(0.25g,0.95mmol)、磷酸三钾(0.37g,1.7mmol)及(1S,2S)-N1,N2-二甲基环己烷-1,2-二胺(0.11mL,0.69mmol)且用氩气脱气。密封反应容器,在85℃下加热混合物过夜。使混合物冷却至室温且用EtOAc(150mL)稀释。经由硅藻土过滤混合物且蒸发滤液。使残余物分配于EtOAc与水之间。有机层经分离,经MgSO4干燥且浓缩以得到粗产物,该粗产物通过硅胶色谱法(0%至5%MeOH/DCM)纯化以得到4-(3-{4-[5-(环丙基胺甲酰基)-4-氟-2-甲基苯基]-1H-吡唑-1-基}-7-乙氧基咪唑并[1,2-a]吡啶-6-基)-4-羟基哌啶-1-甲酸叔丁酯(0.36g,0.58mmol)。
将4-(3-{4-[5-(环丙基胺甲酰基)-4-氟-2-甲基苯基]-1H-吡唑-1-基}-7-乙氧基咪唑并[1,2-a]吡啶-6-基)-4-羟基哌啶-1-甲酸叔丁酯(0.36g,0.58mmol)溶解于DCM(5mL)中且在干冰/丙酮浴中冷却。向其中逐滴添加双(2-甲氧基乙基)氨基]三氟化硫(50%于THF中,0.32mL,0.76mmol)。将混合物在该浴中搅拌30分钟并且随后转移至水浴持续1h。在0℃下用NaHCO3淬灭混合物。混合物用DCM稀释且经萃取。有机相经分离且在减压下浓缩以得到粗产物,该粗产物通过制备型HPLC纯化以得到4-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基苯基)-吡唑-1-基]-7-乙氧基-咪唑并[1,2-a]吡啶-6-基}-4-氟-哌啶-1-甲酸叔丁酯(0.15g,0.25mmol)。
将4-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基苯基)-吡唑-1-基]-7-乙氧基-咪唑并[1,2-a]吡啶-6-基}-4-氟-哌啶-1-甲酸叔丁酯(0.15g,0.25mmol)溶解于DCM(5mL)及MeOH(1mL)中且向其中添加含HCl的二噁烷(4M,4mL)。将混合物在室温下搅拌2h。在减压下浓缩混合物以得到N-环丙基-5-{1-[7-乙氧基-6-(4-氟-哌啶-4-基)-咪唑并[1,2-a]吡啶-3-基]-1H吡唑-4-基}-2-氟-4-甲基-苯甲酰胺盐酸盐(0.14g,0.24mmol)。
将N-环丙基-5-{1-[7-乙氧基-6-(4-氟-哌啶-4-基)-咪唑并[1,2-a]吡啶-3-基]-1H吡唑-4-基}-2-氟-4-甲基-苯甲酰胺盐酸盐(0.14g,0.24mmol)溶解于DCM(5mL)及MeOH(1mL)中。向其中添加甲醛(0.071mL,0.98mmol)及双(乙酰氧基)甲硼烷乙酸钠(sodium bis(acetyloxy)boranuidyl acetate)(0.21g,0.98mmol)。将混合物在室温下搅拌15分钟并且随后用NaHCO3淬灭且用DCM萃取。经合并的有机萃取物经MgSO4干燥,过滤且在减压下浓缩。用MeOH研磨残余物且过滤以得到为白色固体的96(0.081g,0.15mmol)。
实施例97:N-环丙基-2-氟-4-甲基-5-{1-[6-(1-甲基-氮杂环丁-3-基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺

将6-溴-咪唑并[1,2-a]吡啶(1.0g,0.005mol)装入微波瓶中且向其中添加4,4'-二叔丁基-2,2'-二吡啶(0.14g,0.001mol)、3-溴-氮杂环丁烷-1-甲酸叔丁酯(1.2g,0.005mol)、锌粉(0.66g,0.01mol)、NiI2(0.16g,0.001mol)及MgCl2(0.48,0.005mol)。向其中添加吡啶(0.4g,0.005mol)及DMA(15mL)。密闭反应且将其在65℃下加热16h。将混合物冷却至室温,用EtOAc稀释且用饱和NaHCO3及盐水洗涤。浓缩有机层且通过硅胶色谱法(0%至100%EtOAc/庚烷,然后以5%MeOH/DCM)来纯化残余物以得到粗产物,该粗产物接着通过制备型HPLC纯化以得到3-咪唑并[1,2-a]吡啶-6-基-氮杂环丁烷-1-甲酸叔丁酯(0.85g,3.1mmol)。
将3-咪唑并[1,2-a]吡啶-6-基-氮杂环丁烷-1-甲酸叔丁酯(0.85g,3.0mmol)溶解于CHCl3(15mL)中且向其中添加NBS且将混合物在室温下搅拌2h。在减压下浓缩反应且将残余物用EtOAc稀释且用饱和NaHCO3、盐水洗涤,且浓缩有机层。通过硅胶色谱法(0%至100%EtOAc/庚烷)来纯化所得粗产物以得到3-(3-溴-咪唑并[1,2-a]吡啶-6-基)-氮杂环丁烷-1-甲酸叔丁酯(0.84g,2.4mmol)。
将3-(3-溴-咪唑并[1,2-a]吡啶-6-基)-氮杂环丁烷-1-甲酸叔丁酯(0.84g,2.4mmol)溶解于DMF(8.0mL)中且向其中添加CuI(0.23g,1.2mmol)、(1R,2R)-二甲基氨基环己烷(0.27g,1.9mmol)、I-37(0.74g,2.9mmol)及磷酸三钾(1.0g,4.8mmol)且在65℃下加热混合物过夜。将混合物冷却至室温且用EtOAc稀释。用水、盐水洗涤混合物且浓缩有机层以得到粗产物,该粗产物通过硅胶色谱法(0%至100%EtOAc/庚烷)然后以反相色谱法(含0.5%甲酸的10%至100%水/MeCN)纯化以得到3-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-基}-氮杂环丁烷-1-甲酸叔丁酯(0.62g,1.2mmol)。
将3-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-基}-氮杂环丁烷-1-甲酸叔丁酯(0.62g,1.2mmol)溶解于DCM(5.0mL)及MeOH(2.0mL)中且向其中添加含4M HCl的二噁烷(1.8mL,7.0mmol)。在冰浴中冷却混合物且将其搅拌过夜。混合物经浓缩,用DCM/MeOH稀释且用饱和NaHCO3洗涤。有机层经浓缩且通过硅胶色谱法(含1%NH4OH的0%至10%MeOH/DCM)来纯化残余物以得到5-[1-(6-氮杂环丁-3-基-咪唑并[1,2-a]吡啶-4-基)-N-环丙基-2-氟-4-甲基-苯甲酰胺(0.39g,0.91mmol)。
将5-[1-(6-氮杂环丁-3-基-咪唑并[1,2-a]吡啶-4-基)-N-环丙基-2-氟-4-甲基-苯甲酰胺(0.07g)溶解于MeOH(2mL)中且向其中添加甲醛(0.13mL,1.6mmol)。将混合物搅拌30分钟,随后添加NaBH3CN(0.10g,1.6mmol)及乙酸(0.019g,0.33mmol)且在50℃下加热反应过夜。将混合物冷却至室温,经饱和NaHCO3中和且分离有机层。浓缩有机物且通过硅胶色谱法(含1%NH4OH的0%至5%MeOH/DCM)来纯化粗产物以得到97(0.032g,0.072mmol)。
实施例98:N-环丙基-2-氟-4-甲基-5-{1-[6-((R)-1-甲基-吡咯烷-3-基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺(99)

将6-溴咪唑并[1,2-a]吡啶(3.9g,20mmol)、3-溴-吡咯烷-1-甲酸叔丁酯(5.0g,20mmol)、NiI2(0.62g,2.0mmol)、4,4'-二叔丁基-2,2'-二吡啶(0.54g,2.0mmol)及MgCl2(0.19g,20mmol)在耐压烧瓶中溶解于吡啶及DMA中。在65℃下加热混合物3天。将混合物冷却至室温且用EtOAc稀释。将混合物用饱和NaHCO3、水及盐水洗涤。浓缩有机层且通过硅胶色谱法(0%至10%MeOH/DCM)来纯化粗产物以得到3-咪唑[1,2a]吡啶-6-基-吡咯烷-1-甲酸叔丁酯(2.7g,6.1mmol)。
将3-咪唑[1,2a]吡啶-6-基-吡咯烷-1-甲酸叔丁酯(2.7g,6.1mmol)溶解于CHCl3(30mL)中且冷却至0℃。向其中添加NBS(1.1g,6.1mmol)且将其搅拌30分钟。浓缩且通过硅胶色谱法来纯化混合物。随后通过手性HPLC分离对映异构体以得到(R)-3-(3-溴-咪唑并[1,2-a]吡啶-6-基)-吡咯烷-1-甲酸叔丁酯(0.65mmol,1.6mmol)及(S)-3-(3-溴-咪唑并[1,2-a]吡啶-6-基)-吡咯烷-1-甲酸叔丁酯(0.59g,1.6mmol)。任意地指定立体化学。
将(R)-3-(3-溴-咪唑并[1,2-a]吡啶-6-基)-吡咯烷-1-甲酸叔丁酯(0.65mmol,1.6mmol)溶解于DMF(5.0mL)中且向其中添加CuI(0.15g,0.81mmol)、1R,2R-二甲基氨基环己烷(0.20mL,1.3mmol)、I-37(0.5g,1.9mmol)及磷酸三钾(0.69g,3.3mmol)且在65℃下加热混合物过夜。将混合物冷却至室温,用EtOAc稀释且用水(2×50mL)及盐水洗涤。浓缩有机层且通过硅胶色谱法首先用0%至10%MeOH/DCM、随后用于含0.1%甲酸的10%至95%MeCN/水来纯化以得到(R)-3-{3-[4-(5-环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-基}-吡咯烷-1-甲酸叔丁酯(0.62g,1.1mmol)。
将环丙基胺甲酰基-4-氟-2-甲基-苯基)-吡唑-1-基]-咪唑并[1,2-a]吡啶-6-基}-吡咯烷-1-甲酸叔丁酯(0.23g,0.42mmol)溶解于MeOH(1mL)中且向其中添加含4N HCl的二噁烷(0.53mL,2.1mmol)且将混合物在室温下搅拌过夜。浓缩反应以得到N-环丙基-2-氟-4-甲基-5-[1-((R)-6-吡咯烷-3基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-苯甲酰胺盐酸盐(0.26g,0.53mmol)。
将N-环丙基-2-氟-4-甲基-5-[1-((R)-6-吡咯烷-3基-咪唑并[1,2-a]吡啶-3-基)-1H-吡唑-4-基]-苯甲酰胺盐酸盐(0.20g,0.42mmol)溶解于DCM(4mL)中且向其中添加甲醛(0.31mL,4.2mmol)及NaBH(OAc)3(1.3g,6.4mmol)且将其搅拌过夜。混合物用饱和NaHCO3淬灭且用DCM(3×50mL)萃取。将经合并的有机层用水、盐水洗涤且在减压下浓缩。通过硅胶色谱法(10%MeOH/DCM)来纯化粗物质以得到98(0.096g,0.21mmol)。
实施例99:N-环丙基-2-氟-4-甲基-5-{1-[6-((S)-1-甲基-吡咯烷-3-基)-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-苯甲酰胺
以类似于98的方式但利用(S)-3-(3-溴-咪唑并[1,2-a]吡啶-6-基)-吡咯烷-1-甲酸叔丁酯来制备99。
实施例100:N-环丙基-2-氟-5-{1-[6-(4-氟-1-甲基-哌啶-4-基)-7-甲氧基-咪唑并[1,2-a]吡啶-3-基]-1H-咪唑-4-基}-4-甲基-苯甲酰胺
使用I-39-2以类似于实例94的方式制备标题化合物。
实施例101:N-环丙基-5-{1-[6-(1-乙基-4-氟-哌啶-4-基)-7-甲氧基-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-2-氟-4-甲基-苯甲酰胺

将N-环丙基-2-氟-5-{1-[6-(4-氟-哌啶-4-基)-7-甲氧基-咪唑并[1,2-a]吡啶-3-基]-1H-吡唑-4-基}-甲基-苯甲酰胺盐酸盐(0.060g,0.12mmol)溶解于DCM(2.0mL)中且向其中添加乙醛(0.019mL,0.36mmol)及NaBH(OAc)3(0.10g,0.45mmol)且将其在室温下搅拌过夜。将混合物用饱和NaHCO3淬灭且用DCM萃取。将经合并的有机萃取物用盐水洗涤且浓缩以得到粗产物,该粗产物通过制备型HPLC纯化以得到101(0.008g,0.016mmol)。
用于表1中的化合物的HPLC及MS数据显示于表3中,其使用阐述于下表2中的方法来测量。
表2:HPLC方法

生物特性的评估
用于表1中的化合物的RIPK2抑制显示于表3中,且使用以下方法来测量:
物质:白色,384孔optiplates(型号6007290)购自PerkinElmer。V9103XADP-Glo激酶检定定制(Kinase Assay Custom)(包括超纯ATP)购自Promega。自制8His-RIPK2FL。所有其他物质为商购可得的最高级别。
方法:在384孔盘中,将在分析缓冲液(1%DMSO最终)中稀释的测试化合物与8His-RIPK2FL酶(最终浓度为8nM)混合。在室温下预培育15分钟后,添加溶解于分析缓冲液的ATP(最终浓度为5μM)中。将混合物在37℃下于含湿气培育箱中培育60分钟。随后,添加ADP Glo试剂,然后在室温下培育40分钟。最后,添加激酶检测试剂(Kinase Detection Reagent),且将全部混合物在室温下培育40分钟。用Envision读取器测量发光信号以确定产生的ADP的量。分析缓冲液:25mM HEPES(4-(2-羟乙基)-1-哌嗪乙磺酸)、0.1%BSA(牛血清白蛋白)、10mM MgCl2、5mM MnCl2、50mM KCl、0.01%CHAPS(3-[(3-胆酰氨丙基)二甲基铵基]-1-丙磺酸盐)(3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate)、10μMNa3VO4、1mM DTT(二硫苏糖醇),pH 7.5。所有板含有:有代替化合物(1%DMSO)的媒介物对照的孔,作为高信号的参考(100%CTL(100%对照),高信号);及不含酶的孔,作为低信号的参考(0%CTL,低信号)。产生的发光信号与产生的ADP成正比,且与酶活性相关。对数据的分析通过计算在测试化合物及RIPK2存在下的ADP产量相比于在RIPK2加50μM Gefitinib存在下的ADP产量的百分比来进行。(RLU(相对发光单位)(样品)-RLU(低对照))*100/(RLU高值)-RLU(低对照))[RLU=相对发光单位]。
表3




进行额外分析,诸如人全血TNF抑制、人肝细胞稳定性及CACO-2渗透性,以分别获得细胞效能、稳定性及细胞渗透性。
使用方法
本发明的化合物为RIPK2的有效抑制剂。因此,在本发明的一个实施方案中,提供使用本发明的化合物治疗RIPK2介导的病症的方法。在另一实施方案中,提供使用本发明的化合物治疗心血管、炎症、过敏性、肺及纤维化疾病、肾病及癌症的方法。
在不希望受理论所束缚的情况下,RIPK2的药理学抑制将减弱通过NOD1及NOD2刺激引发的通过细菌感应途径的促炎信号。炎症信号中的此减少将在各种自身炎症性疾病中提供治疗效益。
这些自身炎症性疾病包括:
心血管疾病,包括动脉粥样硬化、心肌梗塞、中风、主动脉瘤、镰状细胞危象、缺血-再灌注损伤、肺动脉高血压及败血症;
过敏性疾病,包括哮喘、过敏性鼻炎、鼻窦炎、特应性皮肤炎及荨麻疹;
纤维化疾病,包括哮喘气道重塑、特发性肺纤维化、硬皮病、石棉沉滞症;
肺综合征,包括成人呼吸窘迫综合征、病毒性细支气管炎、阻塞性睡眠呼吸暂停、慢性阻塞性肺病、囊肿性纤维化及肺支气管发育不良;
炎症性疾病,包括类风湿性关节炎、骨关节炎、痛风、肾小球肾炎、间质性膀胱炎、牛皮癣、炎症性肠病(溃疡性结肠炎及克罗恩氏病)、Blau综合征、全身性红斑性狼疮症、移植排斥反应、多发性硬化症、炎性疼痛、炎症及过敏性眼病;
自体免疫性疾病或过敏性病症,选自类风湿性关节炎(rheumatoid arthritis)、牛皮癣(psoriasis)、全身性红斑狼疮(systemic lupus erythromatosis)、狼疮性肾炎(lupus nephritis)、硬皮病(scleroderma)、哮喘(asthma)、慢性阻塞性肺病(ChronicObstructive Pulmonary Disease)(COPD)、过敏性鼻炎(allergic rhinitis)、过敏性湿疹(allergic eczema)、多发性硬化症(multiple sclerosis)、青少年类风湿性关节炎(juvenile rheumatoid arthritis)、青少年特发性关节炎(juvenile idiopathicarthritis)、I型糖尿病(type I diabetes)、炎症性肠病(inflammatory bowel disease)、移植物抗宿主疾病(graft versus host disease)、牛皮癣性关节炎(psoriaticarthritis)、反应性关节炎(reactive arthritis)、僵直性脊椎炎(ankylosingspondylitis)、克罗恩氏病(Crohn’s disease)、溃疡性结肠炎(ulcerative colitis)、葡萄膜炎(uveitis)及非放射性脊椎关节病(non-radiographic spondyloarthropathy)。
癌症,包括实体肿瘤、白血病及淋巴瘤;及
肾病,诸如肾小球肾炎或糖尿病肾病变或糖尿病性肾病。
肝病,诸如肝的非酒精性脂肪肝病或非酒精性脂肪性肝炎(NASH)或肝硬化。
为治疗上文所描述的疾病及病况,治疗有效的剂量将通常在每公斤体重每剂量本发明化合物约0.01mg至约100mg的范围内;优选为每公斤体重每剂量约0.1mg至约20mg。例如,对70kg的个人给药时,剂量范围将为每剂量本发明化合物约0.7mg至约7000mg,优选为每剂量约7.0mg至约1400mg。可能需要进行一定程度的常规剂量优化以确定最佳剂量水平及模式。活性成分可一日给药1至6次。
通用给药及药物组合物
在用作药剂时,本发明的化合物通常以药物组合物形式给药。该组合物可使用药物领域中熟知的步骤制备,且包含本发明的至少一种化合物。本发明的化合物也可单独或与佐剂组合给药,该佐剂提高本发明的化合物的稳定性,有助于在某些实施方案中给药含有佐剂的药物组合物,提供提高的溶解度或分散度、提高的拮抗剂活性,提供辅助疗法等等。根据本发明的化合物可独立或与根据本发明的其他活性物质组合、任选地也与其他药理学活性物质组合使用。一般而言,本发明的化合物以治疗有效量或药物有效量给药,但出于诊断性或其他目的可以较少量给药。
以纯形式或以适当的药物组合物形式给药本发明的化合物可使用药物组合物给药的任何可接受模式进行。因此,给药可为例如以固体、半固体、冻干粉剂或液体剂型的形式(诸如片剂、栓剂、丸剂、软弹性及硬明胶胶囊、粉剂、溶液、悬浮液或气溶胶或类似形式),优选地以适合于简单给药精确剂量的单位剂型经口、经颊(例如舌下)、经鼻、胃肠外、局部、经皮、经阴道或经直肠给药。药物组合物通常将包括常规医药载体或赋形剂及作为活性剂的本发明的化合物,且另外可包括其他药剂、医药剂、载体、佐剂、稀释剂、媒介物或其组合。用于各种模式或给药的该药学上可接受的赋形剂、载体或添加剂以及制备药物组合物的方法为本领域技术人员所熟知。技术状况通过以下证明,例如:Remington:The Science andPractice of Pharmacy,第20版,A.Gennaro(编),Lippincott Williams及Wilkins,2000;Handbook of Pharmaceutical Additives,Michael及Irene Ash(编),Gower,1995;Handbook of Pharmaceutical Excipients,A.H.Kibbe(编),美国医药协会(AmericanPharmaceutical Ass'n),2000;H.C.Ansel及N.G.Popovish,Pharmaceutical DosageForms and Drug Delivery Systems,第5版,Lea及Febiger,1990;其各者以其全文引用的方式并入本文中以更好地描述技术状况。
如本领域技术人员所期望,将选择特定药物制剂中采用的本发明的化合物的形式(例如盐),其具有有效制剂所需的合适物理特性(例如水溶性)。
Claims (15)
1.一种化合物,其选自由以下组成的群:












2.根据权利要求1所述的化合物,其具有以下结构:

3.根据权利要求1所述的化合物,其具有以下结构:

4.根据权利要求1所述的化合物,其具有以下结构:

5.根据权利要求1所述的化合物,其具有以下结构:

6.根据权利要求1所述的化合物,其具有以下结构:

7.根据权利要求1所述的化合物,其具有以下结构:

8.根据权利要求1所述的化合物,其具有以下结构:

9.根据权利要求1所述的化合物,其具有以下结构:

10.根据权利要求1所述的化合物,其具有以下结构:

11.根据权利要求1所述的化合物,其具有以下结构:

12.根据权利要求1至11中任一项的化合物的药学上可接受的盐。
13.一种药物组合物,其包含根据权利要求1所述的化合物或其药学上可接受的盐及至少一种药学上可接受的载体。
14.根据权利要求1至11中任一项的化合物或其药学上可接受的盐在制备用于治疗自体免疫疾病或过敏性病症的药物中的用途。
15.根据权利要求14所述的在制备用于治疗自体免疫疾病或过敏性病症的药物中的用途,该自体免疫疾病或过敏性病症选自:类风湿性关节炎、牛皮癣、全身性红斑狼疮、狼疮性肾炎、硬皮病、哮喘、慢性阻塞性肺病(COPD)、过敏性鼻炎、过敏性湿疹、多发性硬化症、青少年类风湿性关节炎、青少年特发性关节炎、I型糖尿病、炎症性肠病、移植物抗宿主疾病、牛皮癣性关节炎、反应性关节炎、僵直性脊椎炎、克罗恩氏病(Crohn's disease)、溃疡性结肠炎、葡萄膜炎及非放射性脊椎关节病。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662394779P | 2016-09-15 | 2016-09-15 | |
| US62/394,779 | 2016-09-15 | ||
| PCT/US2017/050197 WO2018052772A1 (en) | 2016-09-15 | 2017-09-06 | Heteroaryl carboxamide compounds as inhibitors of ripk2 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN110023290A CN110023290A (zh) | 2019-07-16 |
| CN110023290B true CN110023290B (zh) | 2022-08-12 |
Family
ID=59901598
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201780056054.1A Active CN110023290B (zh) | 2016-09-15 | 2017-09-06 | 作为ripk2抑制剂的杂芳基甲酰胺化合物 |
Country Status (32)
| Country | Link |
|---|---|
| US (2) | US10138241B2 (zh) |
| EP (1) | EP3512833B1 (zh) |
| JP (1) | JP6733050B2 (zh) |
| KR (1) | KR102472736B1 (zh) |
| CN (1) | CN110023290B (zh) |
| AR (1) | AR109650A1 (zh) |
| AU (1) | AU2017327539B2 (zh) |
| BR (1) | BR112019003320B1 (zh) |
| CA (1) | CA3037141A1 (zh) |
| CL (1) | CL2019000476A1 (zh) |
| CO (1) | CO2019001181A2 (zh) |
| CY (1) | CY1123494T1 (zh) |
| DK (1) | DK3512833T3 (zh) |
| EA (1) | EA038128B1 (zh) |
| ES (1) | ES2816003T3 (zh) |
| HR (1) | HRP20201494T1 (zh) |
| HU (1) | HUE051551T2 (zh) |
| IL (1) | IL265062B (zh) |
| LT (1) | LT3512833T (zh) |
| MA (1) | MA46229B1 (zh) |
| MX (1) | MX2019003026A (zh) |
| NZ (1) | NZ750416A (zh) |
| PE (1) | PE20190979A1 (zh) |
| PH (1) | PH12019500497A1 (zh) |
| PL (1) | PL3512833T3 (zh) |
| PT (1) | PT3512833T (zh) |
| RS (1) | RS60729B1 (zh) |
| SA (1) | SA519401322B1 (zh) |
| SI (1) | SI3512833T1 (zh) |
| TW (1) | TWI749062B (zh) |
| UA (1) | UA123287C2 (zh) |
| WO (1) | WO2018052772A1 (zh) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3464336B1 (en) | 2016-06-01 | 2022-03-16 | Athira Pharma, Inc. | Compounds |
| JP6932771B2 (ja) * | 2016-09-15 | 2021-09-08 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Ripk2の阻害剤としてのピリジンおよびピラジン化合物 |
| CN110023290B (zh) | 2016-09-15 | 2022-08-12 | 勃林格殷格翰国际有限公司 | 作为ripk2抑制剂的杂芳基甲酰胺化合物 |
| CA3121202A1 (en) | 2018-11-30 | 2020-06-04 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| CN113874015B (zh) * | 2018-12-21 | 2024-05-24 | 细胞基因公司 | Ripk2的噻吩并吡啶抑制剂 |
| AR118471A1 (es) * | 2019-03-22 | 2021-10-06 | Takeda Pharmaceuticals Co | Derivados de pirrol e imidazol fusionados con piridina como inhibidores de ripk2 |
| WO2023239941A1 (en) | 2022-06-10 | 2023-12-14 | Interline Therapeutics Inc. | Imidazo(1,2-a)pyridine derivatives as ripk2 inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011123609A1 (en) * | 2010-03-31 | 2011-10-06 | Glaxo Group Limited | Imidazolyl-imidazoles as kinase inhibitors |
| CN105073735A (zh) * | 2013-02-06 | 2015-11-18 | 拜耳作物科学股份公司 | 作为害虫防治剂的卤素取代的吡唑衍生物 |
| WO2016065461A1 (en) * | 2014-10-27 | 2016-05-06 | University Health Network | Ripk2 inhibitors and method of treating cancer with same |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK1618092T3 (da) | 2003-05-01 | 2011-01-31 | Bristol Myers Squibb Co | Aryl-substituerede pyrazol-amidforbindelser, der er anvendelige som kinasehæmmere |
| PE20080695A1 (es) | 2006-04-27 | 2008-06-28 | Banyu Pharma Co Ltd | Derivados de dihidropirazolopirimidinona como inhibidores de quinasa weel |
| EP2041116A1 (de) | 2006-07-07 | 2009-04-01 | Boehringer Ingelheim International GmbH | Phenyl substituierte heteroaryl-derivate und deren verwendung als antitumormittel |
| JP5084839B2 (ja) | 2006-11-09 | 2012-11-28 | エフ.ホフマン−ラ ロシュ アーゲー | チアゾール及びオキサゾール置換アリールアミド |
| KR20100117137A (ko) | 2008-02-27 | 2010-11-02 | 메르크 파텐트 게엠베하 | 당뇨병 치료를 위한 카르복사마이드-헤테로아릴유도체 |
| CN101671336B (zh) | 2009-09-23 | 2013-11-13 | 辽宁利锋科技开发有限公司 | 芳杂环并嘧啶衍生物和类似物及其制备方法和用途 |
| US8377970B2 (en) | 2009-10-08 | 2013-02-19 | Rhizen Pharmaceuticals Sa | Modulators of calcium release-activated calcium channel |
| JP2013523657A (ja) | 2010-03-26 | 2013-06-17 | グラクソ グループ リミテッド | キナーゼ阻害剤としてのインダゾリル‐ピリミジン |
| UY34863A (es) | 2012-06-19 | 2013-12-31 | Bristol Myers Squibb Company Una Corporacion Del Estado De Delaware | Antagonistas de iap |
| TWI592417B (zh) | 2012-09-13 | 2017-07-21 | 葛蘭素史克智慧財產發展有限公司 | 胺基喹唑啉激酶抑制劑之前藥 |
| TW201427668A (zh) | 2012-11-20 | 2014-07-16 | Celgene Avilomics Res Inc | 治療和布魯頓(bruton’s)酪胺酸激酶相關之疾病或失調的方法 |
| WO2014145022A1 (en) | 2013-03-15 | 2014-09-18 | President And Fellows Of Harvard College | Hybrid necroptosis inhibitors |
| CA2927920A1 (en) | 2013-10-18 | 2015-04-23 | Dana-Farber Cancer Institute, Inc. | Polycyclic inhibitors of cyclin-dependent kinase 7 (cdk7) |
| AU2014375265B2 (en) | 2014-01-03 | 2018-11-01 | Elanco Animal Health Gmbh | Novel pyrazolyl-heteroarylamides as pesticides |
| MX2019002265A (es) | 2016-08-24 | 2019-10-30 | Prilenia Therapeutics Dev Ltd | Uso de pridopidina para el tratamiento de la disminucion funcional. |
| CN110023290B (zh) | 2016-09-15 | 2022-08-12 | 勃林格殷格翰国际有限公司 | 作为ripk2抑制剂的杂芳基甲酰胺化合物 |
| JP6932771B2 (ja) | 2016-09-15 | 2021-09-08 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Ripk2の阻害剤としてのピリジンおよびピラジン化合物 |
-
2017
- 2017-09-06 CN CN201780056054.1A patent/CN110023290B/zh active Active
- 2017-09-06 AU AU2017327539A patent/AU2017327539B2/en active Active
- 2017-09-06 WO PCT/US2017/050197 patent/WO2018052772A1/en active Application Filing
- 2017-09-06 KR KR1020197010671A patent/KR102472736B1/ko active IP Right Grant
- 2017-09-06 DK DK17768914.8T patent/DK3512833T3/da active
- 2017-09-06 PT PT177689148T patent/PT3512833T/pt unknown
- 2017-09-06 ES ES17768914T patent/ES2816003T3/es active Active
- 2017-09-06 MX MX2019003026A patent/MX2019003026A/es unknown
- 2017-09-06 MA MA46229A patent/MA46229B1/fr unknown
- 2017-09-06 EP EP17768914.8A patent/EP3512833B1/en active Active
- 2017-09-06 CA CA3037141A patent/CA3037141A1/en active Pending
- 2017-09-06 NZ NZ750416A patent/NZ750416A/en unknown
- 2017-09-06 PE PE2019000500A patent/PE20190979A1/es unknown
- 2017-09-06 LT LTEP17768914.8T patent/LT3512833T/lt unknown
- 2017-09-06 RS RS20201022A patent/RS60729B1/sr unknown
- 2017-09-06 PL PL17768914T patent/PL3512833T3/pl unknown
- 2017-09-06 HU HUE17768914A patent/HUE051551T2/hu unknown
- 2017-09-06 US US15/696,540 patent/US10138241B2/en active Active
- 2017-09-06 JP JP2019514206A patent/JP6733050B2/ja active Active
- 2017-09-06 SI SI201730450T patent/SI3512833T1/sl unknown
- 2017-09-06 EA EA201990678A patent/EA038128B1/ru not_active IP Right Cessation
- 2017-09-06 UA UAA201903410A patent/UA123287C2/uk unknown
- 2017-09-06 BR BR112019003320-3A patent/BR112019003320B1/pt active IP Right Grant
- 2017-09-14 TW TW106131512A patent/TWI749062B/zh active
- 2017-09-14 AR ARP170102539A patent/AR109650A1/es unknown
-
2018
- 2018-10-12 US US16/158,407 patent/US11130754B2/en active Active
-
2019
- 2019-02-08 CO CONC2019/0001181A patent/CO2019001181A2/es unknown
- 2019-02-22 CL CL2019000476A patent/CL2019000476A1/es unknown
- 2019-02-26 IL IL265062A patent/IL265062B/en active IP Right Grant
- 2019-03-07 PH PH12019500497A patent/PH12019500497A1/en unknown
- 2019-03-14 SA SA519401322A patent/SA519401322B1/ar unknown
-
2020
- 2020-09-18 HR HRP20201494TT patent/HRP20201494T1/hr unknown
- 2020-10-22 CY CY20201101004T patent/CY1123494T1/el unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011123609A1 (en) * | 2010-03-31 | 2011-10-06 | Glaxo Group Limited | Imidazolyl-imidazoles as kinase inhibitors |
| CN105073735A (zh) * | 2013-02-06 | 2015-11-18 | 拜耳作物科学股份公司 | 作为害虫防治剂的卤素取代的吡唑衍生物 |
| WO2016065461A1 (en) * | 2014-10-27 | 2016-05-06 | University Health Network | Ripk2 inhibitors and method of treating cancer with same |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN110023290B (zh) | 作为ripk2抑制剂的杂芳基甲酰胺化合物 | |
| TWI766281B (zh) | 作為jak1抑制劑之哌啶-4-基三亞甲亞胺衍生物 | |
| JP6437452B2 (ja) | Pimキナーゼ阻害剤として有用な二環式芳香族カルボキサミド化合物 | |
| WO2020239103A1 (zh) | 一种btk抑制剂环衍生物及其制备方法和药学上的应用 | |
| KR20160058188A (ko) | Btk의 치환된 니코틴이미드 저해제 및 그의 제조 방법 및 암, 염증 및 자가면역 질환에의 용도 | |
| CA2897333A1 (en) | Thiazolecarboxamides and pyridinecarboxamide compounds useful as pim kinase inhibitors | |
| CN112105357A (zh) | 药用6,5杂双环衍生物 | |
| KR101921486B1 (ko) | PARP억제제인 4H-피라졸로[1,5-a]벤즈이미다졸 화합물의 아날로그 | |
| TW202035409A (zh) | 單醯基甘油脂肪酶調節劑 | |
| KR20160086930A (ko) | 피롤로피롤론 유도체 및 bet 억제제로서의 그의 용도 | |
| WO2019013562A1 (ko) | 신규한 1h-피라졸로피리딘 유도체 및 이를 포함하는 약학 조성물 | |
| TW202317106A (zh) | 作為egfr抑制劑之取代胺基吡啶化合物 | |
| US12139484B2 (en) | Substituted benzamides as RIPK2 inhibitors for treatment of inflammatory bowel disease | |
| WO2022137082A1 (en) | Pyrrolo[3,2-b]pyridine derivatives useful in treating conditions associated with cgas | |
| AU2017201801B2 (en) | Piperidin-4-yl azetidine derivatives as jak1 inhibitors | |
| CN117957225A (zh) | 用于靶向irak4蛋白的降解的化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |