CN108291152B - 生物质向液态烃物质的转化 - Google Patents
生物质向液态烃物质的转化 Download PDFInfo
- Publication number
- CN108291152B CN108291152B CN201680068105.8A CN201680068105A CN108291152B CN 108291152 B CN108291152 B CN 108291152B CN 201680068105 A CN201680068105 A CN 201680068105A CN 108291152 B CN108291152 B CN 108291152B
- Authority
- CN
- China
- Prior art keywords
- product
- biomass
- catalyst
- reactor
- hydrocarbon
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/42—Catalytic treatment
- C10G3/44—Catalytic treatment characterised by the catalyst used
- C10G3/45—Catalytic treatment characterised by the catalyst used containing iron group metals or compounds thereof
- C10G3/46—Catalytic treatment characterised by the catalyst used containing iron group metals or compounds thereof in combination with chromium, molybdenum, tungsten metals or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G1/00—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal
- C10G1/06—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal by destructive hydrogenation
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G1/00—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal
- C10G1/08—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal with moving catalysts
- C10G1/086—Characterised by the catalyst used
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/42—Catalytic treatment
- C10G3/44—Catalytic treatment characterised by the catalyst used
- C10G3/45—Catalytic treatment characterised by the catalyst used containing iron group metals or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/50—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids in the presence of hydrogen, hydrogen donors or hydrogen generating compounds
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G45/00—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds
- C10G45/02—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing
- C10G45/04—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used
- C10G45/06—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used containing nickel or cobalt metal, or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G45/00—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds
- C10G45/02—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing
- C10G45/04—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used
- C10G45/10—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds to eliminate hetero atoms without changing the skeleton of the hydrocarbon involved and without cracking into lower boiling hydrocarbons; Hydrofinishing characterised by the catalyst used containing platinum group metals or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G45/00—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds
- C10G45/44—Hydrogenation of the aromatic hydrocarbons
- C10G45/46—Hydrogenation of the aromatic hydrocarbons characterised by the catalyst used
- C10G45/48—Hydrogenation of the aromatic hydrocarbons characterised by the catalyst used containing nickel or cobalt metal, or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G45/00—Refining of hydrocarbon oils using hydrogen or hydrogen-generating compounds
- C10G45/44—Hydrogenation of the aromatic hydrocarbons
- C10G45/46—Hydrogenation of the aromatic hydrocarbons characterised by the catalyst used
- C10G45/52—Hydrogenation of the aromatic hydrocarbons characterised by the catalyst used containing platinum group metals or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G7/00—Distillation of hydrocarbon oils
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1011—Biomass
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/20—Characteristics of the feedstock or the products
- C10G2300/201—Impurities
- C10G2300/207—Acid gases, e.g. H2S, COS, SO2, HCN
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P30/00—Technologies relating to oil refining and petrochemical industry
- Y02P30/20—Technologies relating to oil refining and petrochemical industry using bio-feedstock
Landscapes
- Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
Abstract
本发明提供用于从含生物质原料和生物质衍生的原料中的至少一种产生液体烃产物的方法,所述方法包含以下步骤:a)使所述含生物质原料和/或生物质衍生的原料与加氢热解催化剂组合物和分子氢在加氢热解反应器容器中在350到600℃范围内的温度,在0.50到7.50MPa范围内的压力以及在大于2.0千克(生物质)/小时/千克(催化剂)范围内的WHSV下接触,以产生包含部分脱氧烃产物、H2O、H2、CO2、CO、C1‑C3气体、炭以及催化剂细粒的产物物料流;b)从所述产物物料流去除全部或一部分所述炭和催化剂细粒;c)将剩余产物物料流冷却到在150到400℃范围内的温度;以及d)在一种或多种适用于在步骤a)中所产生的H2O、CO2、CO、H2和C1‑C3气体存在下氢化脱氧和芳香族饱和所述部分脱氧烃产物的催化剂组合物存在下,在加氢转化反应器中加氢转化所有或一部分所述部分脱氧烃产物,以产生包含C4+烃产物、H2O、CO、CO2和C1‑C3气体的气相产物。
Description
技术领域
本发明涉及用于将含生物质或生物质衍生的原料转化成液态烃物质的方法,所述液态烃物质适用作燃料或燃料中的掺合组分。
背景技术
随着对液体运输燃料、降低‘简单油’(易于获得和回收的粗石油)的储备和增加对这类燃料的碳足迹的限制的需求增加,研发以有效方式从替代性来源产生液体运输燃料的途径变得越来越重要。
生物质提供可再生碳的来源并且是指来源于活的或已故的生物体的生物材料,并且包括木质纤维素材料(例如木材)、水生材料(例如海藻、水生植物和海菜)以及动物副产物和废料(例如下水、脂肪和水肥)。由生物质产生的液体运输燃料有时称为生物燃料。因此,当使用这类生物燃料时,与石油衍生的燃料相比,有可能实现持续时间更长的CO2排放。
然而,在生物质的常规热解中,通常在惰性气氛中进行快速热解,获得稠密、酸性、反应性液体生物油产物。这种产物含有在所述过程期间形成的水、油和炭。因此,使用经由常规热解产生的生物油可能具有若干缺陷。这些缺陷包括产物的提高的化学反应性、水可混合性、高氧含量和低热值。通常,这些产物难以升级成可替代的液态烃燃料。
在WO2010117437中以美国天然气工艺研究院(Gas Technology Institute)的名义描述了将生物质加工成高质量液体燃料的有效方法。
固体原料,如含有废弃塑料的原料和含有木质纤维素的原料(例如本质生物质、农业残余物、林业残余物、来自木制品以及纸浆和纸张工业的残余物,以及含有木质纤维素材料、废弃塑料和/或食物废料的城市固体废料),由于其可以大规模获得而成为用于生物质转化成燃料过程的重要原料。木质纤维素包含任何比例的木质素、纤维素和半纤维素的混合物并且通常还含有灰渣和水分。
WO2010117437中描述的用于将生物质转化成液态烃燃料的方法使用加氢热解和加氢转化催化剂。尽管不受任何特定催化剂限制,但用于这类方法的示例性催化剂包括含有镍、钼、钴或其混合物作为活性金属的硫化催化剂。用于用以将生物质转化为液体烃燃料的加氢热解和加氢转化步骤的其它催化剂描述于共同未决申请IN 4737/CHE/15、PCT/EP2015/064749、PCT/EP2015/064691和PCT/EP2015/064732中。
用于从生物质制备液态烃的方法描述于WO2015114008中,所述方法利用加氢热解/加氢转化方法和含有还原金属催化剂的另一下游加氢处理反应器。
可以进一步分离来自这些方法中的任一个的产物以产生柴油、汽油或汽油和柴油的掺合组分。
不同地方可能需要汽油和柴油的不同规格。不满足这些规格的物质可以用作燃料中的掺合组分或可能需要升级用作掺合组分或自身作为燃料。
藉由基于加氢热解的方法由生物质产生的烃液体产物可能不满足多个地方柴油和汽油范围产品所需的规格。举例来说,这类物质可能具有各种类别的烃分子(芳香族、链烷烃和环烷烃)的不期望的分布,从而导致例如汽油的不良辛烷值和柴油产物的不良十六烷值。
因此,加氢热解的所属领域将从提供具有所需产品品质属性的改良的产物的方法显著受益。最终目标为以简单方法从生物质产生完全可替代的汽油和/或柴油范围产物的能力。使用可以高百分比与现有汽油和柴油燃料(即来源于原油的那些)掺和而不影响品质或性能的产物的方法也是高度期望的。
发明内容
因此,本发明提供用于从含生物质原料和生物质衍生的原料中的至少一种产生液体烃产物的方法,所述方法包含以下步骤:
a)使所述含生物质原料和/或生物质衍生的原料与加氢热解催化剂组合物和分子氢在加氢热解反应器容器中在350至600℃范围内的温度,在0.50至7.50MPa范围内的压力以及在大于2.0千克(生物质)/小时/千克(催化剂)范围内的WHSV下接触,以产生包含部分脱氧烃产物、H2O、H2、CO2、CO、C1-C3气体、炭以及催化剂细粒的产物物料流;
b)从所述产物物料流去除全部或一部分所述炭和催化剂细粒;
c)将剩余产物物料流冷却到在150到400℃范围内的温度;以及
d)在一种或多种适用于在步骤a)中所产生的H2O、CO2、CO、H2和C1-C3气体存在下所述部分脱氧烃产物的氢化脱氧和芳香族饱和的催化剂组合物存在下,在加氢转化反应器中加氢转化所有或一部分所述部分脱氧烃产物,以产生包含C4+烃产物、H2O、CO、CO2和C1-C3气体的气相产物。
附图说明
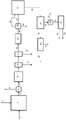
图1和2展示现有技术方法的实施例的图示。
图3和4展示本发明方法的实施例的非限制性图示。
图5到10展示本文所描述的实例的结果。
具体实施方式
本发明人已发现,可以通过在第一步骤中在加氢热解催化剂存在下使生物质原料进行加氢热解来进行用于生产液体烃产物的有效和高产率方法。这一步骤在大于2.0h-1的足够高的重量每小时空间速度(weight hourly space velocity;WHSV)下进行,使得产物烃仅部分脱氧。在去除固体副产物之后,将产物物料流冷却到在150到400℃范围内的温度并随后使其与加氢转化反应器中的一种或多种催化剂组合物接触。一种或多种催化剂组合物允许任何存在的芳香族的进一步脱氧和饱和。所得液体烃产物具有其中含有的低含量的芳香族物质。
生物质原料
本发明的方法中使用的原料含有含生物质和/或生物质衍生的原料的任何组合。
术语“生物质”是指来自生活在地球表面或地球海洋、河流和/或湖泊内的有机体的物质。代表性生物质可以包括任何植物材料,或植物材料的混合物,如硬木(例如白木树)、软木、硬木或软木树皮、木质素、藻类和/或浮萍(海草)。能源作物或者农业残余物(例如采伐残余物)或其它类型的植物废料或植物源废料也可用作植物材料。除‘目的’能源作物,如柳枝稷、芒草和海藻以外,具体示例性植物材料包括玉米纤维、玉米秆、蓖麻籽杆、甘蔗渣、甘蔗梢/夹杂物和高粱。短轮伐期林业产物,如能源作物,包括桤木、白蜡木、南方山毛榉、桦木、桉树、白杨、柳树、构树、澳洲黑檀、美国梧桐和各种各样的兰考泡桐。适合的生物质的其它实例包括有机含氧化合物,如碳水化合物(例如糖)、醇和酮,以及有机废料,如废纸、建筑废料、建筑垃圾和生物污泥。
尤其受关注的有机含氧化合物包括含三酸甘油酯组分中所含的有机含氧化合物,例如天然存在的植物(例如蔬菜)油和动物脂肪,或这类油和脂肪的混合物(例如潲水油或油脂)。含三酸甘油酯组分代表特定类型的生物质,其通常包含游离脂肪酸和三酸甘油酯,并且还可能存在单酸甘油酯酯和二酸甘油酯。含三酸甘油酯组分还可以包括包含如脂肪酸烷基酯(fatty acid alkyl esters;FAAE)的衍生物等级的化合物的组分,所述脂肪酸烷基酯涵盖脂肪酸甲酯(fatty acid methyl esters;FAME)和脂肪酸乙酯(fatty acid ethylesters;FAEE)。
植物油的实例包括油菜籽(包括芥花)油、玉米油、菜籽油、海甘蓝油、葵花油、大豆油、木麻籽油、橄榄油、亚麻籽油、芥子油、棕榈油、花生油、蓖麻油、椰子油、麻风果油、亚麻荠油、棉籽油、海蓬子油、菥蓂油、藻类油和其它坚果油,以及其混合物。动物脂肪的实例包括猪油、下水、牛脂、鲸油、乳脂、鱼油、水肥和/或食品工业的回收脂肪,包括各种废料流,如黄色和褐色油脂。一种或多种这些动物脂肪和一种或多种这些植物油的混合物也是特定类型生物质的代表。典型植物油、动物脂肪或其混合物的三酸甘油酯和游离脂肪酸可以在其结构中包括脂族烃链,其中大部分这些链具有约8到约24个碳原子。用作含三酸甘油酯组分的代表性植物油和/或动物脂肪可以包括显著比例(例如至少约30%,或至少约50%)的具有16和18个碳原子的脂族(例如,链烷烃或烯烃)烃链。含三酸甘油酯的组分在室温下可以是液体或固体。代表性含三酸甘油酯组分包括植物油和动物脂肪,呈其粗物质形式或经预处理的形式,通常具有约10-12重量%的总氧含量。在代表性实施例中,任选地干燥到低水分含量的固体粒化藻类可以是适合类型的生物质并且尤其为含有三酸甘油酯的组分。
低质量和/或粗的含三酸甘油酯的组分,如棕色油脂,是生物质的代表。有利的是,根据具体实施例,可以将这类含三酸甘油酯组分直接引入加氢热解反应器而无需预处理,使得反应器本身有效地进行所需转型,所述转型使这类低质量和/或粗的含三酸甘油酯组分的加氢热解产物在加氢转化反应器中以有效方式被进一步处理。代表性含三酸甘油酯组分,例如包括具有大于约10ppm(例如约10ppm至约500ppm)或大于约25ppm(例如约25ppm至约250ppm)的总氯化物或金属含量,在一些情况下总碱金属和碱土金属含量的组分。这类含量的污染性氯化物或金属并且尤其碱金属和碱土金属在许多类型的常规加氢处理操作中对催化剂活性不利。
含生物质原料可以包含所有或基本上所有的生物质,但还可以含有非生物材料(例如来源于石油的材料,如塑料;或来源于从土壤萃取的矿物质的材料,如金属和金属氧化物,包括玻璃)。可能包括一种或多种非生物材料的“含生物质”原料的实例是城市固体废料(municipal solid waste;MSW)。
这类城市固体废料可以包含木质纤维素材料(庭院废弃物、经加压处理的木材,如栅栏柱、胶合板)、废弃的纸张和卡纸板、食物废料、纺织物废料以及耐火材料(如玻璃、金属)的任何组合。在用于本发明的方法之前,在去除至少一部分任何耐火材料(如玻璃或金属)之后,可以任选地将城市固体废料转化成球粒或团块形式。也设想了MSW与木质纤维素废料的共同处理。某些食物废料可以与锯末或其它物料结合,并且任选地在用于本发明的方法之前粒化。
如本文中所描述,含生物质原料的另一具体实例包含除一种或多种在其重复单体取代基的官能团中含有氧的含氧聚合物(例如塑料)以外的生物质。通过产生H2O、CO和/或CO2,在本文中所描述的方法的加氢热解和/或加氢转化反应器中进行的脱氧反应中至少部分地去除氧。剩余的聚合物结构可以用于在基本完全脱氧的烃产物或液态烃燃料中产生脂族或芳族烃。代表性含氧塑料的氧含量是至少10重量%(例如在约10到约45重量%范围内),其中含氧塑料共同进料的具体实例是聚碳酸酯(例如(C15H16O2)n,约14重量%O)、聚(甲基丙烯酸甲酯)(PMMA,(C5H8O2)n,约32重量%O)、聚对苯二甲酸乙二醇酯(PET,(C10H8O4)n,约33重量%O)和多元胺(例如(CONH2)n,约36重量%的O)。由于某些含氧聚合物(例如PET和聚碳酸酯)中存在烃环结构,在本文中所描述的方法中,与脂肪族烃相比,这些含氧聚合物可以产生相对更高的芳香族烃产量,而与芳香族烃相比,其它含氧聚合物可以产生相对更高的脂肪族烃产量。
术语‘生物质来源的’,例如当用于短语生物质来源的原料时,指对上文所定义的生物质或含生物质的原料进行热和/或化学转化得到或获得的产物。代表性生物质衍生原料因此包括但不限于热解(例如生物油)、焙烧(例如经焙烧并且任选地致密化的木材)、水热碳化(例如经预处理并且通过在热压缩水中酸水解而致密化的生物质)和聚合(例如来源于植物单体的有机聚合物)的产物。生物质衍生的产物(例如,用作原料)的其它具体实例包括黑液、纯木质素以及木质素磺酸盐。
生物质的热和/或化学转化可以在将所得生物质衍生的原料用于本文中所描述的方法之前或上游的预处理步骤中进行,包括在加氢热解或加氢转化步骤中。代表性预处理步骤可在加氢热解反应器上游使用预处理反应器(预反应器),并且涉及含生物质原料的脱除挥发性成分和/或至少一定程度的加氢热解。这类脱除挥发性成分和任选的加氢热解可以伴有其它有利的转化,例如降低腐蚀性物种含量、降低加氢热解催化剂毒物含量(例如降低钠)和/或降低加氢转化催化剂毒物含量。预反应器中的预处理可以在适合的固体床材料(例如预处理催化剂、吸附剂、传热介质和其混合物)存在下进行,以帮助实现这类补充转化并且由此改良生物质衍生原料的质量。合适的固体床材料包括具有双重或多重功能的材料。关于预处理催化剂,本文所述的具有生物质加氢处理活性的那些是有代表性的。某些预处理原料,例如由脱除挥发性成分和/或至少一定程度的加氢热解产生或获得,也是生物质衍生的原料,而其它预处理原料,例如在不存在热或化学转型作用情况下由分类产生或获得,是含生物质原料,但不是生物质衍生的原料。
生物质衍生的原料还包括生物质到液体(BTL)路径的产物,其可以是费舍尔-托普希(Fischer-Tropsch;F-T)合成的产物且更具体地说,是气化接着进行F-T合成的产物。这些产物与其对应物(用于燃料掺合的富含链烷烃的石油衍生的产物)相比通常具有显著更低的质量。这种质量缺陷由存在生物质衍生的脂肪醇和其它生物质衍生的有机含氧副产物化合物以及可能的反应性烯烃引起,其中这些非链烷烃杂质的量取决于所使用的F-T催化剂系统和处理条件。作为生物质衍生的原料,F-T合成产物的代表性总氧含量通常在约0.25重量%到约10重量%,并且通常约0.5重量%到约5重量%范围内。另外,包括F-T蜡的F-T合成产物具有宽的碳数(且因此,分子量)分布和极差的低温流动属性。这些特征均可以通过以下来改进:使用本文所描述的方法(例如加氢转化步骤)中的合适转化将F-T蜡转化为富含链烷烃的组分,所述富含链烷烃的组分具有较低的平均分子量(和较窄的分子量分布)和/或具有较高的支化度(或异烷烃含量),以便满足基本上完全脱氧的烃产物或液体烃的馏出物燃料馏分(如柴油沸腾范围部分和/或航空(例如喷射机)燃料沸腾范围部分)的规格。
多种含碳原料(包括如上文所定义的生物质)的气化(例如非催化部分氧化)可以提供用于F-T合成的合成气。F-T合成是指根据以下反应将合成气,即CO和H2的混合物,转化为改进分子量的烃的方法:
n(CO+2H2)→(-CH2-)n+nH2O+热量。
F-T合成反应产生具有从甲烷分子量到重链烷烃蜡分子量的广泛范围的分子量的反应产物。一般非环状链烷烃和烯族烃的特定混合物以及这些反应产物的比例基本上由所用的催化剂系统调节。一般,甲烷的产生降到最低并且所产生的烃的相当大部分具有最少5个碳原子的碳链长。因此,C5 +烃呈一般至少约60重量%(例如约60重量%到约99重量%)和通常至少约70重量%(例如约70重量%到约95重量%)的量存在于F-T合成产物中。F-T合成产物可以进行预处理以去除轻质烃(例如C1-C4烃)和水。然而,由于这些组分在本文中所描述的方法中良好耐受,并且甚至在一些情况下是有利的(例如,经由重整用于产生所需的氢),因此F-T合成的原始产物(即,无预处理)也适用作生物质衍生的原料。这类原始产物可具有大于约占1体积%并且甚至大于5体积%的组合C1到C4烃和含氧烃含量。
如在某些F-T合成产物的情况下,可以预处理其它类型的粗或低质量生物质或生物质衍生的原料,例如特定的含三酸甘油酯组分,如棕色油脂。棕色脂膏包括固体颗粒,如腐败食物颗粒。粗的含三酸甘油酯组分可另外包括磷脂(胶)和金属污染物,包括碱金属和碱土金属。由于高固体含量、高加氢转化催化剂毒物含量和/或引起加氢转化催化剂堵塞的倾向,可以通过预处理来适当地升级低质量和/或粗的含三酸甘油酯组分,以降低固体或其它这些不合需要的材料的含量。经预处理的含三酸甘油酯组分代表特定类型的生物质衍生的原料。
生物质衍生的原料还扩展到在使用其作为本文中所描述的方法的原料之前或上游,由热和/或化学转化作用产生或获得的预处理原料。特定的生物质衍生的原料是常规热解油,即常规热解工艺的产物,包括如US5961786、CA1283880以及Bridgwater,A.V.,《生物质快速热解(Biomass Fast Pyrolysis)》综述论文BIBLID:0354-9836,8(2004),2,21-49)中所述的快速热解工艺。其中原始木质纤维素组分已经转化的代表性生物质衍生的原料可以包含显著量,例如,一般约5重量%到约85重量%,并且通常约10重量%到约75重量%的环状化合物,包括环状有机含氧物。术语“环状有机含氧物”意在包括其中氧并入到环状结构(例如,吡喃环)中的化合物,以及具有环结构并且氧并入到环状结构外部的化合物(例如,酚)在任一情况下,环结构可具有3到8个环成员,可稠合到其它环结构,并且可完全饱和(例如环烷)、完全不饱和(例如芳族)或部分不饱和。在经历本文中所描述的方法中的加氢转化之后,这些环状化合物,包括环状有机含氧物,可以有助于基本上完全脱氧的烃产物或液态烃燃料的总芳香族烃含量。如上文所描述,优选地从如木质纤维素生物质的天然来源获得这些环状化合物,所述天然来源已热解以解聚并且碎片化纤维素、半纤维素和木质素的环状构筑嵌段。
代表性的生物质衍生的原料因此是常规的热解油(生物油),其包含显著量的环状化合物(例如,通常为约10重量%到约90重量%,并且通常为约20重量%到约80重量%),如上所述,在本文所述的方法中,所述环状化合物作为芳香烃的前体。热解油常常含有约30重量%到约40重量%的总氧量,例如呈以下两种形式:(i)有机含氧物,如羟基醛、羟基酮、糖、羧酸和酚寡聚物;和(ii)溶解水。出于这一原因,尽管是可倾倒和可运输的液体燃料,热解油(且具体地说,未经预处理的原始热解油)仅具有粗油类燃料油的约55%到60%的能量含量。能量含量的代表性值在约19.0毫焦/升(69,800BTU/gal)到约25.0毫焦/升(91,800BTU/gal)范围内。此外,归因于存在如烯烃(包括二烯烃)和烯基芳烃的高度不饱和化合物,此原始产物通常具有腐蚀性并展现化学不稳定性。在如本文中所描述的方法中,在从加氢转化步骤回收的基本上完全脱氧的烃液体或液态烃燃料中,热解油可进一步脱氧和经历其它转化以产生烃。根据一些实施例,在分馏基本上完全脱氧烃液体之后,可以在液体产物中浓缩来源于常规热解油的芳香族烃,由此产物适于在燃料(如汽油)中掺合,或以其它方式在不掺合的情况下适用作这类燃料(例如符合一种或多种并且可能所有可适用的汽油规格的汽油沸腾范围部分)。
生物质衍生的原料的其它具体实例包括用于将木材转化成纸浆的牛皮纸(Kraft)或硫酸盐处理的副产物。这些副产物包括黑液、妥尔油、纯木质素和木质素磺酸盐。妥尔油是指树脂状黄黑色油性液体,即松木处理的酸化副产物。妥尔油在精炼之前通常是松香酸、脂肪酸、固醇、高分子量醇和其它烷基链材料的混合物。粗妥尔油的蒸馏可用于回收松香酸中富含的松油部分(脱沥青松油),以用作与脂肪族烃相比产生相对更高产量的芳香族烃的生物质衍生的原料。
如本文中所描述的富含环状化合物并且因此适用作生物质衍生的原料的天然衍生(例如非化石衍生)油(包括热解油)和牛皮纸或硫酸盐处理副产物(例如黑液、粗妥尔油和脱沥青妥尔油)在不脱氧情况下,具有对其用作生物燃料的价值不利的高氧含量。在妥尔油情况下,举例来说,存在相当大浓度的松香酸(皆是多环有机酸)。这些含氧环状化合物在加氢热解和/或加氢转化条件下的脱氧有利地产生芳香族烃。与除氧组合,多环化合物的至少一个环(但并非所有环)的环饱和和/或开环分别引起形成环烷烃和/或烷基化环状烃。重要的是,在所使用的特定加氢热解和/或加氢转化条件下的环烷烃/芳香族烃平衡可用于管理这些物种的相对比例,并且由此符合具体应用的所需规格,例如基本上完全脱氧的烃产物或液态烃的汽油沸腾范围部分或航空燃料沸腾范围部分中芳香族烃的产量或含量,如需要符合所需规格(例如在汽油规格情况下,辛烷值,或在航空(非涡轮机或喷气式)燃料规格情况下,芳香族烃含量)。
生物质衍生的原料的又一实例包括由芳香的叶子获得的油,如桉树脑,以及任选地干燥到低水分含量的固体粒化木质素。这些实例还可以最终引起形成基本上完全脱氧烃产物或液态烃燃料中的芳香族烃。
在引入如本文中所描述的过程之前,可以预处理代表性生物质衍生的原料以改善质量。举例来说,妥尔油可以其粗制形式使用,或可另外通过蒸馏(例如真空蒸馏)预处理以去除沥青(即,提供脱沥青妥尔油)和/或浓缩松香酸,所述松香酸主要是枞酸和脱氢枞酸但包括其它环状羧酸。生物质衍生的原料通常可以通过涉及分离以例如从粗妥尔或粗热解油(生物油)去除不合需要的材料的预处理来获得。在粗生物油的情况下,举例来说,在将经预处理的生物油引入如本文中所描述的方法之前,通过过滤和/或离子交换进行的预处理可以用于去除固体和/或可溶性金属。根据其它实施例,也可以将呈粗或低质量形式的生物质衍生的原料(如粗生物油或黑液)有利地直接引入如本文中所描述的方法中而无这类预处理步骤,使得一个或多个过程步骤(例如加氢热解和/或加氢转化)本身可进行必需的预处理和/或所需进一步转化,以最后产生液态烃。在进行预处理步骤的加氢热解反应器的情况下,可以有效方式在加氢转化步骤中进一步处理脱氧烃产物,包括粗或低质量生物质衍生的原料的加氢热解的产物。
可以将本文中所描述的任何类型的含生物质和生物质衍生的原料组合并且引入如本文中所描述的方法中,或以其它方式单独地引入,例如在不同的轴位置进入加氢热解和/或加氢转化反应器。可以将不同类型的含生物质和/或生物质衍生的原料引入加氢热解反应器或加氢转化反应器中,但是,根据上述所描述的特定实施例,引入这些反应器中的一个(例如,将粗或低质量生物质衍生的原料引入到加氢热解反应容器的情况)可能是优选的。
加氢热解步骤
本发明的加氢热解催化剂组合物优选地包含一种或多种选自以下的活性金属:钴、钼、镍、钨、钌、铂、钯、铱和铁。优选地,一种或多种活性金属选自钴、钼、镍和钨。
存在于本发明的方法中所用的加氢热解催化剂组合物中的金属受支撑,优选地位于金属氧化物载体上。适用作加氢热解催化剂组合物的载体的金属氧化物包括氧化铝、二氧化硅、二氧化钛、二氧化铈、氧化锆以及二元氧化物,如二氧化硅-氧化铝、二氧化硅-二氧化钛和二氧化铈-氧化锆。优选的载体包括氧化铝、二氧化硅和二氧化钛。最优选的载体是氧化铝。载体可以任选地含有废加氢处理催化剂的再循环、再生和再恢复细粒(例如氧化载体上的CoMo、氧化载体上的NiMo的细粒和氧化载剂和沸石的混合物上的含有NiW的加氢裂解催化剂的细粒)。
加氢热解催化剂组合物上的总金属负载量优选地对于贵金属(例如钌、铂、钯和铱)在0.05wt%到3wt%范围内并且对于贱金属(例如钴、钼、镍、钨和铁)在1wt%到75wt%范围内(重量百分比表示为呈其还原(金属)形式的煅烧催化剂上的所有活性金属的总重量百分比)。
可以将其它元素,如磷、硼和镍中的一种或多种并入催化剂以改善活性金属的分散性。
本发明的方法中所用的加氢热解催化剂组合物可以通过本领域中已知的任何适合方法制备。适合的方法包括但不限于:活性金属和载体从溶液的共沉淀;活性金属在载体上的均匀沉积沉淀;使用活性金属溶液的载体的孔体积浸渍;通过活性金属溶液对载体进行连续和多孔体积浸渍,其中在连续的孔体积浸渍之间进行干燥或煅烧步骤;用含有活性金属的溶液或细粉共同研磨载体。此外,也可以使用这些方法中的两种或更多种的组合。
可以向反应器提供呈氧化态、硫化(sulfided或sulfurised)态或呈预还原态的加氢热解催化剂组合物。优选地,在本发明的方法中,向反应器提供呈氧化或预还原状态,更优选呈氧化态的加氢热解催化剂组合物。
因此,在本发明的一个实施例中,在通过上述或另一种方法制备之后,由此形成的组合物在空气或氧气的存在下适当煅烧以获得氧化态。通过本文所用的术语“氧化态”意味着95%或更多的存在的活性金属原子以氧化态大于零的形式作为氧化物存在。举例来说,负载的氧化钴催化剂具有多于95%的钴以+2或+3的氧化态作为氧化物存在,负载的氧化镍催化剂具有多于95%的镍以+2的氧化态作为氧化物存在。
在本发明的另一实施例中,在通过上述或另一种方法制备之后,将由此形成的组合物适当地进行还原,以便将至少一部分活性金属转化为其完全还原态。这可以通过在高温和高压下使催化剂经历还原气体(例如含有氢气的气体)进行。还原步骤的温度可以在120℃到450℃范围内变化,并且压力可以在0.1兆帕到10兆帕范围内变化。
在本发明的另一实施例中,在通过上述或另一种方法制备之后,将由此形成的组合物适当地进行硫化步骤,以便将至少一部分活性金属转化为其硫化形式。这可以通过在高温和高压下使催化剂经历含硫硫体进行。典型的含硫流体包括含有硫掺杂剂的液态烃或天然存在于烃中的硫化合物,以及含有硫化氢的气态流。在本发明的此实施例中,一种或多种活性金属优选地选自钴、钼、镍、铁和钨。硫化步骤的典型压力在0.5MPa到10MPa范围内,同时典型的温度在150℃到450℃范围内。或者,催化剂可以经硫化,使得硫物种存在于催化剂上,所述硫物种将在反应器容器中在条件下与活性金属反应以便形成硫化催化剂。
显而易见的是,虽然加氢热解反应器中提供的加氢热解催化剂组合物最初将包含呈其氧化态、硫化态或还原态的(一种或多种)金属,但催化剂组合物的化学形式将在方法的操作环境下发生变化,使得催化剂上活性金属和载体的化学形式也发生变化。这种变化将涉及催化剂与反应气体(氢气、一氧化碳、二氧化碳)、产物(烃)以及副产物(水、一氧化碳、二氧化碳、氨、硫化氢等)在所述方法的温度和压力条件相互作用所产生的现象。
不希望受理论束缚,假设初始化学组合物将在本发明方法的条件下转化成组合物,在所述组合物中,一部分活性金属可以呈还原形式(氧化数为零),另一部分活性金属可以处于硫化形式的较高氧化态(与生物质原料中存在的硫原子形成化学键),还有一部分活性金属可以处于氧化形式的较高氧化态(氧原子可以从生物质原料获得或从催化剂本身获得)。
随着方法的进展,可以将更多催化剂加入到所述方法中,以替代消耗的催化剂。这类催化剂也将首先提供给反应器,其中取决于原始催化剂组合物的状态,活性金属以其氧化态、硫化态或预还原态存在。
加氢热解催化剂组合物优选地呈球形催化剂颗粒形式存在。对于用于加氢热解步骤中的市售反应器,催化剂粒度优选在0.3mm到4.0mm范围内,更优选在0.6mm到3.0mm范围内,并且最优选在1mm到2.4mm范围内。
尽管可以使用任何类型的适用于加氢热解的反应器,但优选反应器类型是鼓泡流化床反应器。选择流化速度、催化剂尺寸和体密度以及原料尺寸和体密度,使得催化剂留存在鼓泡流体化床中,同时所产生的炭被带出反应器。
加氢热解适当地在加氢热解反应器容器中在350℃到600℃范围内的温度和0.50MPa到7.50MPa范围内的压力下进行。生物质的加热速率优选大于约100W/m2。以克(生物质)/克(催化剂)/小时为单位的这一步骤的重量每小时空间速度(WHSV)大于2.0h-1。优选地,加氢热解反应器容器中的WHSV不超过6h-1,更优选地不超过5h-1。
这类高WHSV限制可通过加氢热解催化剂来实现的氢化脱氧的量。应容易理解,应限制WHSV以防止来自加氢热解反应器的产物物料流经历过少脱除挥发性成分或过少氢化脱氧,因此反应性过高。
炭去除、冷却和其它方法步骤
从加氢热解步骤的产物物料流去除炭和催化剂细粒。通常还将在这一阶段去除任何存在的灰渣。从蒸气物料流去除炭和催化剂细粒的最优选方法是通过旋风分离来进行。还可以在加氢处理反应器内部(密相床上方)使用固体分离设备(例如旋风分离器)以防止夹带超过某一粒度的固体颗粒。
还可以根据本发明的方法,通过从蒸气物料流过滤,或借助于从洗涤步骤-沸腾床过滤来去除炭。反脉冲可以用于从过滤器去除炭,只要在本发明的方法中使用的氢足以降低热解蒸气的反应性并且使所产生的炭可以自由流动即可。在液体产物的进一步加氢精制、冷却和冷凝之前,还可使用静电沉淀、惯性分离、磁性分离或这些技术的组合从热蒸气物料流去除炭和催化剂细粒。
根据本发明的一个实施例,相继进行的用于去除未在旋风分离器中去除的细粒的旋风分离和热气过滤可用于去除炭。在这种情况下,因为氢使自由基稳定并且使烯烃饱和,所以与在常规快速热解中产生的气雾剂的热过滤中去除的炭相比,过滤器上捕获的粉尘饼更易于清洗。根据本发明的另一实施例,通过使第一阶段产物气体鼓泡通过再循环液体来去除炭和催化剂细粒。所使用的再循环液体是来自这一过程的成品油的高沸点部分,并且因此是通常具有高于370℃的沸点的完全饱和(氢化)、稳定的油。在这一液体中捕捉来自第一反应阶段的炭或催化剂细粒。可以过滤一部分液体以去除细粒,并且一部分液体可以再循环回到第一阶段加氢热解反应器中。使用再循环液体的一个优势在于其提供一种使来自第一反应阶段的炭满载过程蒸气的温度降低到第二反应阶段加氢转化步骤所需的温度,同时去除炭和催化剂细粒的方法。使用液体过滤的另一优势在于可以完全避免使用热气过滤以及其附带的、充分证明的过滤器清洗问题。
根据本发明的一个实施例,使用旋风分离,接着在高孔隙度固体吸附剂床中捕获炭和催化剂细粒以从蒸气物料流去除炭和催化剂细粒。适用于捕获炭和催化剂细粒的高孔隙度固体吸附剂的实例包括可从Crystaphase购得的CatTrap(R)材料。
还可以使用惰性递变床材料从蒸气物料流去除炭和催化剂细粒。
根据本发明的另一实施例,使用部署在沸腾床中的大尺寸NiMo或CoMo催化剂进行炭去除,以在去除细粒的同时提供进一步脱氧。这种催化剂的颗粒应较大,优选尺寸在15到30mm范围内,由此使得其易于与来自第一反应阶段的通常小于200筛目(小于70微米)的负载的细粒炭分离。
也可以在炭去除步骤中去除任何存在的灰渣和催化剂细粒。
随后将剩余产物物料流冷却到温度150到400℃。优选地,将物料流冷却到在250到350℃范围内的温度。
取决于剩余产物物料流的组成,这类冷却可引起水溶液和/或有机材料的冷凝。任选地,在此阶段,因此可以例如通过使用三相气/液分离器去除在此冷却步骤中产生的任何非有机液体。可以向具有蒸气相脱氧烃产物的其余部分的加氢转化反应器提供任何冷凝有机材料以进一步加工。
任选地在冷却之前或之后,如果剩余产物物料流中存在任何硫,那么所述产物物料流可以进行脱硫。在其中向加氢热解反应器容器提供呈硫化(sulfided或sulfurised)形式的加氢热解催化剂组合物的实施例中,将需要这类脱硫步骤。在本发明的其它实施例中,脱硫步骤的需求将取决于存在于含生物质或生物质衍生的原料中的硫量。
可以在含有加氢脱硫(HDS)催化剂的独立反应器中去除有机硫。本领域中已知适合的HDS反应器和催化剂并且适合的催化剂包括氧化载体上的硫化NiMo或硫化CoMo。氧化载体的实例包括氧化铝、二氧化硅、二氧化钛以及二氧化硅-氧化铝。进料的加氢脱硫的优选催化剂是负载于氧化铝载体上的硫化CoMo。可以在低于加氢热解反应器中的温度的温度下操作HDS反应器,以避免在此反应器中形成芳香族。这类脱硫也可以导致部分氧气去除。
另外,可以使用气体净化系统脱硫(呈H2S形式)。这可以例如通过使物料流与硫保护床接触来实现。用于这类硫保护床的适合材料包括氧化载体或金属氧化物上的高度分散的金属或金属氧化物。适用作保护床的金属氧化物的实例包括氧化锌和氧化铁。氧化载体的实例包括二氧化硅、氧化铝和混合二氧化硅-氧化铝。分散于氧化载体上的适合金属包括镍、铁和铜。分散于氧化载体上的适合金属氧化物包括氧化铁、氧化锌和氧化铜。载体上的活性金属或金属氧化物的适合负载量以保护床物质的煅烧氧化形式计在2wt%到70wt%范围内。
加氢转化反应器
在去除炭并冷却之后,部分脱氧烃产物与来自加氢热解步骤的H2、CO、CO2、H2O和C1-C3气体一起与一种或多种催化剂组合物在加氢转化反应器中接触。所述一种或多种催化剂组合物包含适用于氢化脱氧和芳香族饱和的催化剂。这一步骤适当地在150℃到400℃范围内的温度和在0.50MPa到7.50MPa范围内的压力下进行。这一步骤的重量每小时空间速度(WHSV)在约0.1h-1到约2h-1范围内。
加氢转化反应器在反应器容器内可包含一个或多个反应器容器和/或一个或多个反应区。每个反应器容器和/或反应区可以在不同反应条件,例如温度和压力下操作。优选地,加氢转化反应器是单一反应器容器。
保护这一步骤中所用的催化剂组合物免于接触可另外使催化剂中毒的生物质中存在的Na、K、Ca、P和其它金属,因为这些金属主要用第一加氢热解阶段的炭和灰渣产物去除,在使部分脱氧加氢热解产物进行加氢转化之前,使这些金属与所述部分脱氧加氢热解产物分离。另外,由于在加氢热解步骤中使用非硫化催化剂或由于脱硫步骤使用或两个步骤而保护此步骤中所用的催化剂组合物免于接触硫。
适用于氢化脱氧和芳香族饱和的一种或多种催化剂组合物优选地包含具有针对氢化脱氧和芳香族饱和的活性的单一催化剂组合物。然而,设想在本发明的范围内使用在加氢转化反应器内的混合床或堆叠床中的两种分离催化剂组合物。
在本发明的一个实施例中,加氢转化反应器中存在的催化剂组合物优选地包含选自由钴、钼、镍和钨组成的碱金属的一种或多种活性金属。
在本发明的这一实施例中,加氢转化反应器中存在的催化剂组合物中存在的金属负载于优选地金属氧化物载体上。适用作加氢热解催化剂组合物的载体的金属氧化物包括氧化铝、二氧化硅、二氧化钛、二氧化铈、氧化锆以及二元氧化物,如二氧化硅-氧化铝、二氧化硅-二氧化钛和二氧化铈-氧化锆。优选的载体包括氧化铝、二氧化硅和二氧化钛。最优选的载体是氧化铝。载体可以任选地含有废加氢处理催化剂的再循环、再生和再恢复细粒(例如氧化载体上的CoMo、氧化载体上的NiMo的细粒和氧化载剂和沸石的混合物上的含有NiW的加氢裂解催化剂的细粒)。适用作载体的另一类别材料包括碳基材料,所述碳基材料包括活性碳、有序介孔碳和无序或蠕虫洞状介孔碳。
另外,在本发明的这一实施例中,可以向加氢转化反应器提供呈氧化态或呈预还原态的加氢热解催化剂组合物。
因此,在通过以上或另一方法中的一种制备之后,由此形成的组合物适当地在空气或氧气存在下煅烧以便获得氧化态。通过本文所用的术语“氧化态”意味着95%或更多的存在的活性金属原子以氧化态大于零的形式作为氧化物存在。举例来说,负载的氧化钴催化剂具有多于95%的钴以+2或+3的氧化态作为氧化物存在,负载的氧化镍催化剂具有多于95%的镍以+2的氧化态作为氧化物存在。
或者,在通过上文或另一方法中的一种制备之后,使由此形成的组合物适当地进行还原步骤以便将至少一部分活性金属转化为其完全还原态。这可以通过在高温和高压下使催化剂经历还原气体(例如含有氢气的气体)进行。还原步骤的温度可以在120℃到450℃范围内变化,并且压力可以在0.1兆帕到10兆帕范围内变化。
显而易见的是,虽然加氢热解反应器中提供的加氢热解催化剂组合物最初将包含呈其氧化态或还原态的(一种或多种)金属,但催化剂组合物的化学形式将在方法的操作环境下发生变化,使得催化剂上活性金属和载体的化学形式也发生变化。这种变化将涉及催化剂与反应气体(氢气、一氧化碳、二氧化碳)、产物(烃)以及副产物(水、一氧化碳、二氧化碳、氨、痕量硫化氢等)在所述方法的温度和压力条件相互作用所产生的现象。
不希望受理论束缚,假设初始化学组合物将在本发明方法的条件下转化成组合物,在所述组合物中,一部分活性金属可以呈还原形式(氧化数为零),并且另一部分活性金属可以处于氧化形式的较高氧化态(氧原子可以从生物质原料获得或从催化剂本身获得)。由于痕量硫存在于加氢转化反应器中,少量活性金属还可以呈硫化形式存在。
在本发明的替代实施例中,加氢转化反应器中存在的催化剂组合物优选地包含一种或多种选自铂族金属(钌、铑、钯、锇、铱和铂)的活性金属并且负载于氧化物载体上。适用于此实施例的催化剂组合物的氧化物载体的实例包括氧化铝、二氧化硅、二氧化钛、二氧化铈、氧化锆以及二元氧化物,如二氧化硅-氧化铝、二氧化硅-二氧化钛和二氧化铈-氧化锆。适用作催化剂载体的另一类别材料包括碳基材料,所述碳基材料包括活性碳、有序介孔碳和无序或蠕虫洞状介孔碳。优选的载体包括氧化铝、二氧化硅和二氧化硅-氧化铝,并且最优选的载体是二氧化硅-氧化铝。载体上的金属负载量表示为在催化剂上呈其还原形式的金属的重量%,在0.05wt%到3wt%范围内。
对于本发明的任一实施例,关于加氢转化反应器中存在的催化剂组合物,所述催化剂组合物上的总金属负载量优选地对于贵金属(例如钌、铂、钯和铱)在0.05wt%到3wt%范围内并且对于贱金属(例如钴、钼、镍、钨和铁)在1wt%到75wt%范围内(重量百分比表示为呈其还原(金属)形式的煅烧催化剂上的所有活性金属的总重量百分比)。
此外,在任一实施例中,可以将如磷、硼和镍中的一种或多种的其它元素并入加氢转化反应器中存在的催化剂组合物以便改善活性金属的分散性。
用于在本发明的方法中的加氢转化反应器的催化剂组合物可以通过本领域中已知的任何适合方法制备。适合的方法包括但不限于:活性金属和载体从溶液的共沉淀;活性金属在载体上的均匀沉积沉淀;使用活性金属溶液的载体的孔体积浸渍;通过活性金属溶液对载体进行连续和多孔体积浸渍,其中在连续的孔体积浸渍之间进行干燥或煅烧步骤;用含有活性金属的溶液或细粉共同研磨载体。此外,也可以使用这些方法中的两种或更多种的组合。
加氢转化反应器中存在的催化剂组合物优选地以挤出物催化剂颗粒存在。可以使用任何适合的挤出物形状,例如圆形、三叶性、四叶形。
用于加氢转化步骤中商业反应器的催化剂颗粒尺寸优选地具有在0.3mm到4.0mm范围内,更优选在0.6mm到3.0毫米范围内,并且最优选在1mm到2.4mm范围内的标称直径。挤出物催化剂颗粒的适合长度在3到6mm范围内。
加氢转化反应器优选地是以下流式或上流式,优选地下流式操作模式操作的固定床反应器。取决于向此反应器进料的物理状态,其可在滴流或气流方案下操作。
在加氢转化步骤之后,可以使步骤d)的气相产物冷凝以提供液相产物,所述液相产物包含基本上完全脱氧的C4+烃液体和水性材料。剩余气相适当地主要包含H2、CO、CO2和轻质烃气体(通常C1到C3,但此物料流也可以含有某些C4+烃)并且可以分离。
可以将这一剩余的气相任选地传送到气体净化系统以去除H2S、氨和痕量的有机含硫化合物(如果作为过程的副产物存在)。接着可以将含有CO、CO2、H2和轻质烃的物料流传送到过程中的分离、重整和水-气体偏移区段,其中由轻质气体产生氢并且可以重新用于过程中。优选地,这一过程提供足以供整个本发明的方法使用的氢。可再生CO2作为过程的副产物排放。
接着分离液相产物以去除水性材料,适当地通过相分离进行,并且提供呈脱氧烃液体形式的烃产物。
本文中的液体脱氧烃产物优选地包含不超过5wt%、更优选不超过1wt%存在于原始含生物质和/或生物质衍生的原料中的氧气。液体脱氧烃产物含有小于2wt%,优选小于1wt%,并且最优选小于0.1wt%氧。
适当地,液体脱氧烃产物接着经历进一步分离和纯化步骤以提供合乎需要的产物。
在本发明的一个实施例中,使液体脱氧烃产物进行蒸馏以便根据其中含有的液体产物的沸点范围将液体脱氧烃产物分离为C4+部分。
液体脱氧烃产物包含石脑油范围烃、中间馏出物范围烃和真空粗柴油(VGO)范围烃,其可以通过蒸馏来分离。出于清楚的目的,中间馏出物在本文中定义为通过根据标准ASTM蒸馏方法测量的大气压等效初始沸点(initial boiling point;IBP)与最终沸点(final boiling point;FBP)之间的蒸馏来回收的烃或氧化烃。中间馏出物的ASTM D86初始沸点可以在150℃到220℃范围内。根据ASTM D86蒸馏,中间馏出物的最终沸点可以在350℃到380℃范围内。石脑油定义为具有四个或更多个碳原子并且具有高过90℃但低于200℃的大气压等效最终沸点的烃或氧化烃。在过程中产生的少量烃(通常小于总C4+烃的10重量%,优选小于总C4+烃的3重量%,并且最优选小于总C4+烃的1重量%)在高于如上文所定义的中间馏出物的沸腾温度的温度下沸腾,即,其是具有与通过石油蒸馏产生的真空瓦斯油类似的沸腾范围的烃。
汽油是在火花点火内燃发动机中使用的主要包含石脑油范围烃的汽车燃料。在美国,ASTM D4814标准确定对用于具有火花点火内燃发动机的地面车辆的汽油的要求。
柴油是在压缩点火内燃发动机中使用的主要由中间馏出物范围烃组成的汽车燃料。在美国,ASTM D975标准涵盖对适用于不同类型的柴油发动机的若干等级的柴油的要求。
本发明的优势在于在适合操作条件下,由含生物质和/或生物质衍生的原料产生的液体脱氧烃产物可以基本上完全不含氧、硫和氮并且具有低含量芳香族化合物。优选地,这一产物的氧含量小于1.50重量%并且更优选小于0.50重量%,并且最优选小于0.10重量%。硫含量优选小于100ppmw,更优选小于10ppmw,并且最优选小于5ppmw。氮含量优选小于1000ppmw,更优选小于100ppmw,并且最优选小于10ppmw。芳香族含量优选低于10wt%、更优选低于7wt%、甚至更优选地低于5wt%。
附图详细说明
图1展示现有技术加氢热解/加氢转化方法的实施例。
使含有木质纤维素物质的原料1与含氢气流2在加氢热解反应器容器3中接触。这一反应器容器的产物4是混合固体和气相产物,所述气相产物含有氢、轻质气体(C1-C3烃、CO、CO2、H2S、氨、水蒸气)、C4+烃和含氧烃的蒸气。气相产物夹带有炭、灰渣和催化剂细粒。固体分离器5分离炭、灰渣和催化剂细粒6与气相产物物料流7。接着,气相产物物料流7进入催化加氢转化反应器8。这一反应器8是固定床反应器。这一反应器的产物9含有轻质气态烃(甲烷、乙烷、乙烯、丙烷和丙烯)、石脑油范围烃、中间馏出物范围烃、沸点高于370℃(根据ASTM D86)的烃、氢和升级反应的副产物,如H2O、H2S、NH3、CO和CO2。蒸气在在一个或多个冷凝器中冷凝,接着进入催化加氢转化反应器8下游的气体-液体分离器10,并且回收液体产物21。将不可冷凝气体11传送到气体净化系统,所述气体净化系统包含气相加氢脱硫反应器12、去除H2S物料流14的H2S去除单元13和去除作为过程的副产物的氨物料流16的氨去除单元15。将含有轻质烃的剩余物料流17传送到过程的分离、重整和水-气体偏移区段18,其中由轻质气体产生的氢19和可再生CO2作为过程20的副产物排放。也可以从这一区段以副产物形式回收燃料气体物料流。
将从冷凝和气体-液体分离系统10回收的可冷凝物21传送到液体/液体分离器23,其中分离水性产物22与烃液体产物24。接着传送烃液体产物24以用于蒸馏25,以回收汽油产物26和中间馏出物产物27。如果需要,可以从蒸馏塔以单独的物料流形式回收煤油/喷射燃料和柴油。
图2示例具有额外升级步骤的现有技术加氢热解/加氢转化方法,如WO 2015/114008中所描述。图2中所展示的实施例包括与图1中所展示的现有技术的实施例相同的步骤。然而,额外的固定床氢化反应器35用于升级通过蒸馏加氢转化反应器8的烃产物24回收的中间馏出物馏分28。泵30用于将物料流31提供到反应器35,所述物料流31包含中间馏出物物料流和来自蒸馏25的富含苯的C6物料流29。将氢物料流32压缩于压缩机33以便提供处于类似于或高于加氢热解和加氢转化反应器(3,8)操作所处于的压力的压力下的氢气流34。将物料流31泵送至反应器35并历经氢化(或氢化&开环)催化剂系统加工以产生升级的产物物料流36。
使升级的产物物料流36进行进一步蒸馏37以提供升级的中间馏出物产物38和具有还原苯的C6物料流39。可以将C6物料流提供到混合器40并在其中与来自蒸馏25的汽油产物26混合以便提供升级的汽油物料流41。
图3示例本文所描述的本发明的一个非限制性实施例。在这一实施例中,使来自固体分离器5的气相产物7经历净化系统,所述净化系统包含气相加氢脱硫反应器12,去除H2S物料流14的H2S去除单元13和去除作为过程的副产物的氨物料流16的氨去除单元15。随后在冷却器43中冷却所得气流42并向固定床反应器45提供气态冷却物料流44。固定床反应器45含有一种或多种适用于部分脱氧烃产物的氢化脱氧和芳香族饱和的催化剂组合物。
使来自反应器45的蒸气产物物料流46在反应器45下游的冷凝器和气-液分离器47中冷凝并且回收液体产物48。
将液体产物48传送到液体/液体分离器49,其中使水性产物51与烃液体产物50分离。接着传送烃液体产物50以用于蒸馏51,以回收汽油产物54和中间馏出物产物53。如果需要,可以从蒸馏塔以单独的物料流形式回收煤油/喷射燃料和柴油。
将不可冷凝气体55传送到过程的分离、重整和水-气体偏移区段56,其中由轻质气体产生的氢58和可再生CO2作为过程的副产物57排放。
在图4中展示本发明的另一非限制性实施例。在这一实施例中,冷却器43中的物料流42的冷却产生含有冷凝物质的冷却物料流44。随后在三相分离器59中分离冷却物料流44以分离冷凝含水流62、冷凝有机物物料流61和气流60。向固定床反应器45提供冷凝有机物物料流61和气流60两者,所述固定床反应器45在此实施例中作为滴流反应器运行。
图5到10展示本文所描述的实例的结果。
实例
现将借助于以下实例说明本发明,所述实例并不意图限制本发明。
实例1(比较)
将S-4211催化剂(可购自CRI催化剂公司(CRI Catalyst Co)的钴/钼催化剂)压碎并筛分到300μm到500μm的粒度范围。使催化剂经历非原位硫化程序从而将钴和钼金属转化为其硫化物形式。将210g此催化剂用作第一鼓泡流体化床加氢热解反应器中的催化剂。
使S-4212催化剂(可购自CRI催化剂公司的镍/钼催化剂)经历原位硫化步骤从而将镍和钼金属转化为其硫化形式。在第二固定床反应器中,装载705g呈标称1.3mm直径和约3mm到6mm长度的挤出物形式的硫化S-4212催化剂。
所用的固体原料是研磨并筛分到低于500μm粒度的樟子松(Pinus sylvestris)的锯末。使用预加热到约435℃温度的氢气流将第一鼓泡流体化反应器中的催化剂流体化。在第一阶段催化剂流体化之后,将生物质引入反应器并且以连续方式处理。在实验期间,基于不含水分和灰渣,生物质的处理速率是约4.42g/min。这一进料速率对应于每小时每千克催化剂约1.26千克生物质进料的重量每小时空间速度(在无水分和灰渣的基础上)。历经生物质处理的持续时间,催化剂的流体化床的加权平均温度是443.7℃。在第一加氢热解阶段中将生物质原料转化成炭、灰渣和蒸气的混合物。以使得将固体产物(炭,灰渣)和气相产物带出反应器,同时将催化剂保持在反应器中的方式调节流体化速度。将某种催化剂磨至细粒,并且也将细粒带出床。
使固体产物与气相产物在热过滤装备中分离并且将蒸气传送到第二阶段固定床反应器。在实验期间将第二阶段催化剂的平均温度维持在410.5℃。第二阶段的平均重量每小时空间速度是每小时每千克催化剂0.38千克生物质进料(在无水分和灰渣的基础上)。第一和第二阶段的操作压力都是2.25MPa(表压)。
将第二阶段的气相产物分段冷却到-41.8℃并且回收在水层上浮动的含有烃层的双层液体产物。分离烃液体与水液,并且进行分析。将过程产生的废气传送到在线气相色谱仪,并在整个运行过程中分析气体的组成。过程的质量平衡和碳平衡由液体产物的质量和分析以及气体产物的组成信息计算,据此计算产率曲线。
发现烃液体产物具有极低的氧含量(基本上低于0.01wt%的仪器的检测限),并且所产生的水性产物仅含有0.03wt%碳。因此,实现生物质的完全氢化脱氧,其产生无氧烃产物和无碳水相。发现烃产物的总酸值极低,低于0.1mg KOH/g。
使烃和水相进行进一步分析。烃产物的详细烃分析(detailed hydrocarbonanalysis;DHA)(图5)展示这种产物主要由环状物种组成。在环状物种之间,发现环烷烃在低碳数范围(7并且更低的碳数)中占主导,而芳香族在较高碳数范围(8并且高于8的碳数)下占主导。链烷烃和异烷烃主要存在于低碳数分子(7并且更低的碳数)中。6碳分子是液体产物中最丰富的分子。
烃产物的SIMDIS(图6)展示产物主要在汽油和柴油范围内沸腾,其中基本上不产生重质烃(沸点高于370℃)。在此实施例中发现C4+烃(具有4个或更多个碳原子的产物中的烃)的产率在无水分和灰渣的基础上为原料重量的26.6wt%。表1到8展示此实例的细节。
还使用IP-391分析方法测量总液体产物(total liquid product;TLP)的芳香族含量。这种方法展示产物含有约53.6wt%芳香族,其中单芳香族的比重为总液体的41.4wt%,二芳香族的比重为总液体的7.4wt%,并且三芳香族的比重为总液体的4.8wt%。
实例2(本发明)
将S-4261催化剂(可购自CRI催化剂公司的钴/钼催化剂)压碎并筛分到300μm到500μm的粒度范围。将100g此催化剂用作第一鼓泡流体化床加氢热解反应器中的催化剂。这一发明实例中所用的催化剂重量低于比较实例中的催化剂重量。这样做是为了在第一阶段中获得较高的生物质原料的重量每小时空间速度。因为预期反应器中存在的催化剂或无机固体量影响反应器中的生物质/炭的驻留时间,所以通过向实例2的加氢热解反应器添加约84.3g不含任何活性金属的氧化铝粉末来维持实例1和实例2的第一阶段中的近似相似的无机固体载荷。所使用的氧化铝的筛分粒度级是355μm到600μm。预期这种氧化铝粉末有助于避免从反应器快速夹带生物炭,这种情况将在反应器中装载仅100g催化剂时发生。
在第二固定床反应器中,使用两个不同催化剂系统的堆叠床,其包含处于顶部的S-4252催化剂(可购自CRI催化剂公司的钴/钼催化剂)和处于底部的S-4213催化剂(可购自CRI催化剂公司的Pt/Pd类氢化催化剂)。两种催化剂的重量比是1:3,并且所装载催化剂的总质量是520g。在第二固定床中装载呈标称1.3mm直径和约3mm到6毫米长度的挤出物形式的催化剂。在将生物质引入单元之前,在约2.25MPa的压力和约400℃的温度下在流动氢下还原第二固定床反应器中的堆叠床催化剂系统。
所用的固体原料是研磨并筛分到250μm到500μm粒度的樟子松的锯末。使用预加热到约435℃温度的氢气流将第一鼓泡流体化反应器中的催化剂流体化。在第一阶段催化剂流体化之后,将生物质引入反应器并且以连续方式处理。在实验期间,在无水分和灰渣的基础上,生物质的处理速率是约5.33g/min。这一进料速率对应于每小时每千克催化剂约3.20千克生物质进料的重量每小时空间速度(在无水分和灰渣的基础上)。历经生物质处理的持续时间,催化剂的流体化床的加权平均温度是433.7℃。在第一加氢热解阶段中将生物质原料转化成炭、灰渣和蒸气的混合物。以使得将固体产物(炭,灰渣)和气相产物带出反应器,同时将催化剂保持在反应器中的方式调节流体化速度。将某种催化剂磨至细粒,并且也将细粒带出床。
使固体产物与气相产物在热过滤装备中分离并且将蒸气传送到第二阶段固定床反应器。在炭过滤装备与第二阶段之间,使蒸气冷却,并且将实验期间第二阶段催化剂的平均温度维持在265.4℃下。第二阶段的平均重量每小时空间速度是每小时每千克催化剂0.62千克生物质进料(在无水分和灰渣的基础上)。第一和第二阶段的操作压力都是2.70MPa(表压)。
将第二阶段的气相产物分段冷却到-39.3℃并且回收在水层上浮动的含有烃层的双层液体产物。分离烃液体与水液,并且进行分析。将过程产生的废气传送到在线气相色谱仪,并在整个运行过程中分析气体的组成。过程的质量平衡和碳平衡由液体产物的质量和分析以及气体产物的组成信息计算,据此计算产率曲线。
发现烃液体产物具有极低氧含量(约0.01wt%),并且所产生的水性产物含有约0.28wt%碳。因此,实现生物质的完全氢化脱氧,其产生无氧烃产物和接近无碳水相。发现烃产物的总酸值极低,为0.018mg KOH/g。
使烃和水相进行进一步分析。烃产物的详细烃分析(DHA)(图7)展示这种产物主要由环烷烃,接着链烷烃(正和异)组成。6碳分子是液体产物中最丰富的分子。烃产物的SIMDIS(图8)展示产物主要在汽油和柴油范围内沸腾,其中基本上不产生重质烃(沸点高于370℃)。在此实施例中发现C4+烃(具有4个或更多个碳原子的产物中的烃)的产率在无水分和灰渣的基础上为原料重量的24.6wt%。表1到8展示此实例的细节。
使用IP-391分析方法测量总液体产物(TLP)的芳香族含量。这种方法展示产物具有极低芳香族含量。单芳香族、二芳香族和三芳香族含量各自是方法的检测限约0.1wt%。本发明方法流程中的总液体产物中的总芳香族含量(<0.3wt%)因此显著低于实例1的比较实例方法流程中的总芳香族含量(53.6wt%)。
实例3(本发明)
将S-4261催化剂(可购自CRI催化剂公司的钴/钼催化剂)压碎并筛分到300μm到500μm的粒度范围。将135g此催化剂用作第一鼓泡流体化床加氢热解反应器中的催化剂。这一发明实例中所用的催化剂重量低于比较实例中的催化剂重量。这样做是为了在第一阶段中获得较高的生物质原料的重量每小时空间速度。因为预期反应器中存在的催化剂或无机固体量影响反应器中的生物质/炭的驻留时间,所以通过向实例3的加氢热解反应器添加约65g不含任何活性金属的氧化铝粉末来维持实例1和实例3的第一阶段中的近似相似的无机固体载荷。所使用的氧化铝的筛分粒度级是355μm到600μm。预期这种氧化铝粉末有助于避免从反应器快速夹带生物炭,这种情况将在反应器中装载仅135g催化剂时发生。
在第二固定床反应器中,使用两个不同催化剂系统的堆叠床,其包含处于顶部的S-4252催化剂(可购自CRI催化剂公司的钴/钼催化剂)和处于底部的S-4213催化剂(可购自CRI催化剂公司的Pt/Pd类氢化催化剂)。两种催化剂的重量比是1:3,并且所装载催化剂的总质量是520g。在第二固定床中装载呈标称1.3mm直径和约3mm到6毫米长度的挤出物形式的催化剂。在将生物质引入单元之前,在约2.25MPa的压力和约400℃的温度下在流动氢下还原第二固定床反应器中的堆叠床催化剂系统。
所用的固体原料是研磨并筛分到250μm到500μm粒度的樟子松的锯末。使用预加热到约435℃温度的氢气流将第一鼓泡流体化反应器中的催化剂流体化。在第一阶段催化剂流体化之后,将生物质引入反应器并且以连续方式处理。在实验期间,在无水分和灰渣的基础上,生物质的处理速率是约4.97g/min。这一进料速率对应于每小时每千克催化剂约2.21千克生物质进料的重量每小时空间速度(在无水分和灰渣的基础上)。历经生物质处理的持续时间,催化剂的流体化床的加权平均温度是440.6℃。在第一加氢热解阶段中将生物质原料转化成炭、灰渣和蒸气的混合物。以使得将固体产物(炭,灰渣)和气相产物带出反应器,同时将催化剂保持在反应器中的方式调节流体化速度。将某种催化剂磨至细粒,并且也将细粒带出床。
使固体产物与气相产物在热过滤装备中分离并且将蒸气传送到第二阶段固定床反应器。在炭过滤装备与第二阶段之间,使蒸气冷却,并且将实验期间第二阶段催化剂的平均温度维持在307.2℃下。第二阶段的平均重量每小时空间速度是每小时每千克催化剂0.57千克生物质进料(在无水分和灰渣的基础上)。第一和第二阶段的操作压力都是2.70MPa(表压)。
将第二阶段的气相产物分段冷却到-42.3℃并且回收在水层上浮动的含有烃层的双层液体产物。分离烃液体与水液,并且进行分析。将过程产生的废气传送到在线气相色谱仪,并在整个运行过程中分析气体的组成。过程的质量平衡和碳平衡由液体产物的质量和分析以及气体产物的组成信息计算,据此计算产率曲线。
发现烃液体产物具有极低氧含量(接近在约0.01wt%下的仪器的检测下限),并且所产生的水性产物仅含有约0.15wt%碳。因此,实现生物质的完全氢化脱氧,其产生无氧烃产物和接近无碳水相。发现烃产物的总酸值极低,为0.01mg KOH/g。
使烃和水相进行进一步分析。烃产物的详细烃分析(DHA)(图9)展示这种产物主要由环烷烃,接着链烷烃(正和异)组成。6碳分子是液体产物中最丰富的分子。烃产物的SIMDIS(图10)展示产物主要在汽油和柴油范围内沸腾,其中基本上不产生重质烃(沸点高于370℃)。在此实施例中发现C4+烃(具有4个或更多个碳原子的产物中的烃)的产率在无水分和灰渣的基础上为原料重量的25.7wt%。表1到8展示此实例的细节。
使用IP-391分析方法测量总液体产物(TLP)的芳香族含量。这种方法展示产物具有极低芳香族含量。单芳香族含量是约1.4wt%,而二芳香族和三芳香族含量各自低于方法的检测限0.1wt%。本发明方法流程中的总液体产物中的总芳香族含量(约1.6wt%)因此显著低于实例1的比较实例方法流程中的总芳香族含量(53.6wt%)。
表1

表2
| 原料分析 | 实例1 | 实例2 | 实例3 |
| <sup>1</sup>水分,wt% | 6.51 | 4.96 | 3.46 |
| 灰分,wt%(干基) | 0.34 | 0.34 | 0.12 |
1水分含量根据在103±2℃干燥后样品的重量损失估计
表3-原料元素分析(在无水分和灰渣(Moisture and ash-free;MAF)的基础上)
| 实例1 | 实例2 | 实例3 | |
| 碳,wt% | 47.2 | 47.2 | 47.2 |
| 氢,wt% | 6.5 | 6.5 | 6.5 |
| 氧,wt% | 46.2 | 46.2 | 46.2 |
| 硫,wt% | 0.030 | 0.030 | 0.030 |
| 氮,wt% | 0.027 | 0.027 | 0.027 |
| 原料H:C原子比 | 1.64 | 1.64 | 1.64 |
表4-操作条件
| 实例1 | 实例2 | 实例3 | |
| 第1阶段的平均温度(℃) | 443.7 | 433.7 | 440.6 |
| 第2阶段的平均温度(℃) | 410.5 | 265.4 | 307.2 |
| 平均压力(MPa(表压)) | 2.25 | 2.70 | 2.70 |
表5-产率细节


表6-冷凝烃液体分析
| 实例1 | 实例2 | 实例3 | |
| 氧含量(wt%) | <sup>4</sup>BDL | <sup>4</sup>BDL | <sup>4</sup>BDL |
| 碳含量(wt%) | 88.76 | 86.01 | 85.92 |
| 氢含量(wt%) | 11.43 | 14.19 | 14.13 |
| 密度(g/mL,15℃) | 0.8365 | 0.7955 | 0.7923 |
| C4+烃中的汽油<sup>2</sup>(%) | 69 | 72 | 74 |
| C4+烃中的柴油<sup>3</sup>(%) | 31 | 28 | 26 |
| 总酸值(Total Acid Number;TAN) | <0.01 | 0.018 | <0.01 |
| H/C原子比 | 1.53 | 1.97 | 1.96 |
2汽油在本文中定义为含有具有在4与10个之间的碳原子的烃。
3柴油在本文中定义为含有具有11或更多个碳原子的烃。
4BDL=低于检测限(氧气测量为0.01wt%)
表7-C1-C3气体组成*
| 实例1 | 实例2 | 实例3 | |
| 甲烷wt% | 25.5 | 32.7 | 38.2 |
| 乙烷wt% | 44.1 | 39.0 | 36.4 |
| 丙烷wt% | 30.4 | 28.3 | 25.4 |
*归一化为100%
表8-水分析
| 实例1 | 实例2 | 实例3 | |
| pH | 9.2 | 8.8 | 9.4 |
| 密度(g/mL,15℃) | 1.0006 | 0.9971 | 0.9990 |
| 硫含量(ppmw) | 264 | 3.8 | 3.9 |
| 氮含量(ppmw) | 1132 | 171.8 | 654.1 |
| 碳含量(wt%) | 0.03 | 0.28 | 0.15 |
Claims (8)
1.一种用于从含生物质原料和/或生物质衍生的原料中的至少一种制备液体烃产物的方法,所述方法包含以下步骤:
a)使所述含生物质原料和/或生物质衍生的原料与加氢热解催化剂组合物和分子氢在加氢热解反应器容器中在350到600℃范围内的温度,在0.50到7.50MPa范围内的压力以及在2.0至6千克(生物质)/小时/千克(催化剂)范围内的WHSV下接触,以产生包含部分脱氧烃产物、H2O、H2、CO2、CO、C1-C3气体、炭以及催化剂细粒的产物物料流;
b)从所述产物物料流去除全部或一部分所述炭和催化剂细粒;
c)将剩余产物物料流冷却到在150到400℃范围内的温度;以及
d)在一种或多种适用于在步骤a)中所产生的所述H2O、CO2、CO、H2和C1-C3气体存在下氢化脱氧和芳香族饱和所述部分脱氧烃产物的催化剂组合物存在下,在加氢转化反应器中加氢转化所有或一部分所述部分脱氧烃产物,以产生包含C4+烃产物、H2O、CO、CO2和C1-C3气体的气相产物;其中
所述加氢转化反应器是固定床反应器;和
所述加氢转化反应器含有包含一种或多种负载于金属氧化物载体上的选自钴、钼、镍和钨的活性金属的催化剂组合物和包含一种或多种负载于氧化物载体上的选自以下的活性金属:钌、铑、钯、锇、铱和铂的催化剂组合物。
2.根据权利要求1所述的方法,其中所述加氢热解催化剂组合物包含一种或多种负载于金属氧化物载体上的选自以下的活性金属:钴、钼、镍、钨、钌、铂、钯、铱和铁。
3.根据权利要求1或权利要求2所述的方法,其中所述加氢热解反应器容器中的所述WHSV不超过5h-1。
4.根据权利要求1或2所述的方法,其中在步骤c)中,将所述剩余产物物料流冷却到在250到350℃范围内的温度。
5.根据权利要求1或2所述的方法,其中在冷却之前或之后使所述产物物料流进行脱硫。
6.根据权利要求1或2所述的方法,其中在步骤d)之后,使所述气相产物冷凝以提供包含随后分离的基本上完全脱氧的C4+烃液体和水性物质的液相产物。
7.根据权利要求6所述的方法,其中所述基本上完全脱氧的C4+烃液体包含不超过1wt%的存在于原始含生物质原料和/或生物质衍生的原料中的氧气。
8.根据权利要求6所述的方法,其中所述基本上完全脱氧的C4+烃液体随后进行蒸馏。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN6292CH2015 | 2015-11-23 | ||
| IN6292/CHE/2015 | 2015-11-23 | ||
| PCT/EP2016/078411 WO2017089339A1 (en) | 2015-11-23 | 2016-11-22 | Conversion of biomass into a liquid hydrocarbon material |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN108291152A CN108291152A (zh) | 2018-07-17 |
| CN108291152B true CN108291152B (zh) | 2021-01-08 |
Family
ID=57354387
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680068105.8A Active CN108291152B (zh) | 2015-11-23 | 2016-11-22 | 生物质向液态烃物质的转化 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10822546B2 (zh) |
| EP (1) | EP3380585B1 (zh) |
| CN (1) | CN108291152B (zh) |
| AU (1) | AU2016359795B2 (zh) |
| BR (1) | BR112018010521B1 (zh) |
| CA (1) | CA3005405C (zh) |
| MY (1) | MY196728A (zh) |
| PL (1) | PL3380585T3 (zh) |
| WO (1) | WO2017089339A1 (zh) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110548879A (zh) * | 2019-06-04 | 2019-12-10 | 北京化工大学 | 一种绿色合成新型铁炭材料的制备方法及其应用 |
| EP3786356A1 (en) * | 2019-09-02 | 2021-03-03 | Huntsman International LLC | A process for manufacturing an upgraded bio-oil from black liquor |
| CA3197958A1 (en) * | 2020-11-12 | 2022-05-19 | Hyung R. Kim | Fcc co-processing of biomass oil |
| AU2021401076B2 (en) | 2020-12-18 | 2024-09-05 | Shell Internationale Research Maatschappij Bv | Fluidized bed reactor and associated hydropyrolysis processes |
| DE102021104733A1 (de) | 2021-02-26 | 2022-09-01 | Ipi.Ag | Beladene kohle und verfahren zu deren herstellung aus biomasse |
| US20230017405A1 (en) | 2021-07-15 | 2023-01-19 | Shell Usa, Inc. | Method of subjecting a biomass feedstock to hydropyrolysis |
| WO2024151690A2 (en) | 2023-01-13 | 2024-07-18 | Shell Usa, Inc. | Catalyst preparation |
| CN116286067B (zh) * | 2023-02-01 | 2024-03-15 | 四川大学 | 生物质生产汽柴油的长周期稳定运行方法与装置 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015114008A1 (en) * | 2014-01-28 | 2015-08-06 | Shell Internationale Research Maatschappij B.V. | Conversion of biomass or residual waste material to biofuels |
| CN104845654A (zh) * | 2009-04-07 | 2015-08-19 | 瓦斯技术研究所 | 用于生产高质量液体燃料的生物质的加氢热解 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3767562A (en) | 1971-09-02 | 1973-10-23 | Lummus Co | Production of jet fuel |
| CA1283880C (en) | 1987-05-07 | 1991-05-07 | Dave A. Berg | Method and apparatus for rapid thermal processing |
| US5961786A (en) | 1990-01-31 | 1999-10-05 | Ensyn Technologies Inc. | Apparatus for a circulating bed transport fast pyrolysis reactor system |
| US7964761B2 (en) * | 2005-05-02 | 2011-06-21 | University Of Utah Research Foundation | Processes for catalytic conversion of lignin to liquid bio-fuels and novel bio-fuels |
| US8198492B2 (en) * | 2008-03-17 | 2012-06-12 | Uop Llc | Production of transportation fuel from renewable feedstocks |
| US8492600B2 (en) * | 2009-04-07 | 2013-07-23 | Gas Technology Institute | Hydropyrolysis of biomass for producing high quality fuels |
| BR112013029901B1 (pt) * | 2011-05-23 | 2019-10-15 | Virent, Inc. | Método para converter biomassa em produtos químicos e combustíveis derivados de biomassa, composição química, composição de gasolina, composição de querosene, e composição de diesel |
| US8859831B2 (en) * | 2011-08-02 | 2014-10-14 | Gas Technology Institute | Removal of hydrogen sulfide as ammonium sulfate from hydropyrolysis product vapors |
| US8816144B2 (en) * | 2012-10-04 | 2014-08-26 | Gas Technology Institute | Direct production of fractionated and upgraded hydrocarbon fuels from biomass |
| CN104755593A (zh) * | 2012-10-08 | 2015-07-01 | 国际壳牌研究有限公司 | 木质纤维素类生物质转化 |
| CA2953855C (en) | 2014-07-01 | 2022-07-12 | Shell Internationale Research Maatschappij B.V. | Conversion of solid biomass into a liquid hydrocarbon material |
| BR112016030879B1 (pt) | 2014-07-01 | 2021-01-12 | Shell Internationale Research Maatschappij B.V. | Processo para produzir produtos de hidrocarboneto líquido a partir de uma matériaprima de biomassa sólida |
| CN115418253A (zh) | 2014-07-01 | 2022-12-02 | 国际壳牌研究有限公司 | 固体生物质到液体烃材料的转化 |
| AU2016320326B2 (en) | 2015-09-07 | 2019-05-02 | Shell Internationale Research Maatschappij B.V. | Conversion of biomass into a liquid hydrocarbon material |
-
2016
- 2016-11-22 CN CN201680068105.8A patent/CN108291152B/zh active Active
- 2016-11-22 WO PCT/EP2016/078411 patent/WO2017089339A1/en active Application Filing
- 2016-11-22 PL PL16798527T patent/PL3380585T3/pl unknown
- 2016-11-22 BR BR112018010521-0A patent/BR112018010521B1/pt active IP Right Grant
- 2016-11-22 EP EP16798527.4A patent/EP3380585B1/en active Active
- 2016-11-22 MY MYPI2018701916A patent/MY196728A/en unknown
- 2016-11-22 US US15/777,867 patent/US10822546B2/en active Active
- 2016-11-22 CA CA3005405A patent/CA3005405C/en active Active
- 2016-11-22 AU AU2016359795A patent/AU2016359795B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104845654A (zh) * | 2009-04-07 | 2015-08-19 | 瓦斯技术研究所 | 用于生产高质量液体燃料的生物质的加氢热解 |
| WO2015114008A1 (en) * | 2014-01-28 | 2015-08-06 | Shell Internationale Research Maatschappij B.V. | Conversion of biomass or residual waste material to biofuels |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3380585B1 (en) | 2020-05-27 |
| EP3380585A1 (en) | 2018-10-03 |
| BR112018010521B1 (pt) | 2021-10-26 |
| CA3005405A1 (en) | 2017-06-01 |
| CN108291152A (zh) | 2018-07-17 |
| CA3005405C (en) | 2023-10-10 |
| US10822546B2 (en) | 2020-11-03 |
| PL3380585T3 (pl) | 2021-01-11 |
| BR112018010521A2 (pt) | 2018-11-13 |
| AU2016359795B2 (en) | 2019-08-15 |
| US20180346823A1 (en) | 2018-12-06 |
| AU2016359795A1 (en) | 2018-05-24 |
| WO2017089339A1 (en) | 2017-06-01 |
| MY196728A (en) | 2023-05-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108291152B (zh) | 生物质向液态烃物质的转化 | |
| CN108026450B (zh) | 将生物质转化成液态烃物质 | |
| CA2983021C (en) | Co-processing for control of hydropyrolysis processes and products thereof | |
| CN109153920B (zh) | 生物质向液体烃材料的转化 | |
| CA2998055A1 (en) | Conversion of biomass into methane | |
| CN108350365B (zh) | 生物质向液态烃材料的转化 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |