CN108256286B - Research and analysis method for reaction mechanism of aerobic oxidation of benzyl alcohol into benzaldehyde by using amphoteric water-soluble catalyst - Google Patents
Research and analysis method for reaction mechanism of aerobic oxidation of benzyl alcohol into benzaldehyde by using amphoteric water-soluble catalyst Download PDFInfo
- Publication number
- CN108256286B CN108256286B CN201810022826.6A CN201810022826A CN108256286B CN 108256286 B CN108256286 B CN 108256286B CN 201810022826 A CN201810022826 A CN 201810022826A CN 108256286 B CN108256286 B CN 108256286B
- Authority
- CN
- China
- Prior art keywords
- bond
- complex
- reaction
- path
- energy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images








Classifications
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16C—COMPUTATIONAL CHEMISTRY; CHEMOINFORMATICS; COMPUTATIONAL MATERIALS SCIENCE
- G16C20/00—Chemoinformatics, i.e. ICT specially adapted for the handling of physicochemical or structural data of chemical particles, elements, compounds or mixtures
- G16C20/10—Analysis or design of chemical reactions, syntheses or processes
Landscapes
- Chemical & Material Sciences (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Crystallography & Structural Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Bioinformatics & Computational Biology (AREA)
- Computing Systems (AREA)
- Theoretical Computer Science (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
The invention discloses a reaction mechanism research and analysis method for oxidizing benzyl alcohol into benzaldehyde by using an amphoteric water-soluble catalyst through oxygen, which adopts a Density Functional Theory (DFT) to react amphoteric water-soluble CuIIThe reaction mechanism of the TEMPO catalytic system for catalyzing and oxidizing alcohol into aldehyde in an alkaline aqueous solution is researched. The calculation result shows that: the zwitterionic nature of the catalyst has no effect on the alcohol oxidation reaction; the catalytic reaction mechanism comprises the steps of catalyst activation, substrate oxidation and catalyst regeneration; conversion efficiency (TOF 3.89 h) is calculated based on an energy span model‑1) And experimental measurement results (TOF 5.40 h)‑1) And (5) the consistency is achieved. The rate control step is a substrate oxidation step, i.e. a process in which hydrogen atoms are transferred from the alkoxide to the oxygen atoms of TEMPO. By exploring the reaction mechanism of the catalytic system, not only can the reaction process be observed, but also a theoretical basis can be provided for designing a novel high-efficiency catalyst.
Description
Technical Field
The present invention relates to the computational chemistry of the aerobic oxidation of alcohols to aldehydes. More particularly, relates to a research and analysis method for the reaction mechanism of oxidizing benzyl alcohol into benzaldehyde by oxygen in an amphoteric water-soluble catalyst.
Background
The selective oxidation of primary alcohols to the corresponding aldehydes is one of the most important transformations in industrial production and fine chemistry. For example, benzaldehyde is widely used in perfumery and fineIn chemical synthesis. Currently, methods for effecting alcohol oxidation require the use of stoichiometric amounts of oxidizing agents such as ruthenium (VIII) oxide, sodium hypochlorite, potassium dichromate, and chromium (VI) oxide, among others. In recent years, a goose biomimetic catalytic system for catalytically oxidizing primary alcohols into aldehydes by using oxygen as an oxidant has been remarkably developed. Currently, reports are concentrated on homogeneous biomimetic catalytic systems using transition metal Cu and TEMPO (TEMPO ═ 2,2,6, 6-tetramethylpiperidine-1-oxide, a stable nitroxide radical reaction reagent). But due to the weak oxidizing power of Cu (Cu)2+/Cu Eox+0.34V) and therefore require a suitable ligand to achieve efficient oxidation of the alcohol. Most ligands are currently limited to phenanthroline, 2' -bipyridine and derivatives thereof. Due to the hydrophobic nature of these ligands, most existing catalytic systems require the use of organic solvents or mixed solvents of organic solvents and water. For example, a series of CuBr's reported by Sheldon's group to be very active towards allyl alcohol and benzyl alcohol2TEMPO catalytic systems. Such systems use a mixed solvent of acetonitrile-water. In addition, Stahl group and Koskinen group report a series of Cu-TEMPO catalytic systems with good activity on primary alcohols, including allyl alcohol, benzyl alcohol, primary aliphatic alcohols and corresponding derivatives. Although such systems extend the substrate range, acetonitrile is still employed as the solvent. It is well known that oxygen and organic solvents can form explosive mixtures in the gas phase, presenting a combustion and explosion safety hazard. Therefore, catalytic systems using organic solvents may be limited in large-scale industrial applications. In this respect, the use of water as solvent, which is inexpensive, green, recyclable and free from explosion potential, is a better choice. However, relatively few homogeneous catalytic systems have been reported for transition metal complexes using oxygen as the oxidant and water as the sole solvent. The main reasons are the hydrophobicity of most catalyst ligands, the water sensitivity of the catalytically active species and the limitation of the solubility of the substrate alcohol in water (most alcohols are only partially soluble in water). Thus, to expand the substrate range for the aerobic oxidation of primary alcohols and to enable the future industrial application of alcohol aerobic oxidation processes, the synthesis is designed to enable the aerobic oxidation of primary alcohols in aqueous solutionNovel biomimetic catalysts of grade alcohols are very critical.
In the further development of novel biomimetic catalysts capable of oxidating primary alcohols to aldehydes by oxygen in aqueous solution, we must understand the structure-activity relationship between the structure of the active center of the catalyst and the catalytic activity. However, so far, the experimental study on the existing Cu-TEMPO biomimetic catalytic system for primary alcohol aerobic oxidation by using water as a solvent is mostly limited to the consideration of catalytic reaction conditions (reaction temperature, pressure, catalyst dosage), substrate range, product selectivity, yield and other factors. The research on the relationship between the catalytic activity and the factors such as the geometric and electronic structure of the active center of the catalyst, the space and electronic effect of the ligand, the synergistic effect between the ligands, the coordination mode of the central metal Cu ion and the ligand and the bonding property thereof, and the like, and the reaction mechanism on the molecular level is still few, so that the design and synthesis of the new catalyst are limited.
2011 Pombeiro et al report [ (L) CuII(Hen)(H2O)]TEMPO catalyst system (en: ethylenediamine) [ Eur.J.Inorg.chem.2011,27,4175-4181 ] is an amphoteric, water-soluble catalyst and is capable of catalyzing the oxidation of alcohols to aldehydes in aqueous solution; but whether the zwitterionic nature affects the efficiency of the reaction system and CαThe research on the reaction mechanism of the system such as the-H activation mechanism and the like is not carried out, because a theoretical basis cannot be provided for designing a novel high-efficiency catalyst.
Disclosure of Invention
An object of the present invention is to provide [ (L) Cu ] which is water-soluble to amphoteric propertyII(Hen)(H2O)]The TEMPO catalyst system is a reaction mechanism research and analysis method for catalyzing the aerobic oxidation of the benzyl alcohol to the benzaldehyde in the water solution, and further provides a theoretical basis for designing a novel efficient catalyst.
In order to achieve the purpose, the invention adopts the following technical scheme:
the research and analysis method of the reaction mechanism of the benzene methanol oxygenated by the amphoteric water-soluble catalyst to the benzaldehyde comprises the following steps:
(1) constructing a calculation model of the catalyst activity center structure: based on the formula [ (L) CuII(Hen)(H2O)]TEMPO is reaction condition information provided by an experiment for oxidizing benzyl alcohol into benzaldehyde by oxygen in the presence of a catalyst, and a calculation model of a catalyst active center is constructed; performing geometric optimization by adopting a density functional method, and comprehensively considering the configuration stability, the energy level, the calculation result and the calculation cost according to the calculation result to select an optimal model;
searching 809721.cif files of CCDC-809721 from a Cambridge crystal database in England, and reading X-ray crystallography data provided by the 809721.cif files through Materials Studio software to initially construct a catalytic precursor model; the bond length and bond angle of the constructed preliminary catalytic precursor model were adjusted: the bond length of all Cu-N bonds is increased Increased bond length of Cu-O bond
Increased bond length of Cu-O bond Modulation of N-N bond
Modulation of N-N bond S-O bond increase
S-O bond increase Modulation of C-N bond
Modulation of C-N bond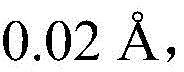 Modulation of C-C bond
Modulation of C-C bond The bond angle of O-Cu-N and O-Cu-O is increased by 0.5-1.5 degrees; optimizing the bond length, bond angle and dihedral angle of the complex by combining M06L, B3LYP, OPBE and M06 functional groups with 6-31+ G (d) group based on the adjusted structure;
The bond angle of O-Cu-N and O-Cu-O is increased by 0.5-1.5 degrees; optimizing the bond length, bond angle and dihedral angle of the complex by combining M06L, B3LYP, OPBE and M06 functional groups with 6-31+ G (d) group based on the adjusted structure;
the maximum difference between the bond lengths of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3 calculated at the level of M06L functional and the corresponding bond lengths in the experiment is Maximum difference between bond angles of O1-Cu-N2 and O2-Cu-N2 and corresponding bond angles of experimentThe value is 1 °;
Maximum difference between bond angles of O1-Cu-N2 and O2-Cu-N2 and corresponding bond angles of experimentThe value is 1 °;
the maximum difference between the bond length of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3 calculated at the level of B3LYP functional and the corresponding bond length of the experiment is The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1.8 degrees;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1.8 degrees;
the bond length calculated at the level of OPBE functional is within the experimental error The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 2.6 degrees;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 2.6 degrees;
the maximum difference between the bond lengths of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3 calculated at the level of M06 functional and the corresponding bond lengths in the experiment is The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 0.9 degrees;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 0.9 degrees;
determining the result at the level of M06L/6-31+ G (d) according to the calculation result and in consideration of the precision and calculation cost of the M06L functional;
(2) designing a reaction path;
performing possible reaction path design according to the optimal model selected in the step (1);
reaction intermediates and transition state modeling were performed:
(a) construction of Cα-intermediates and transition states of H bond cleavage:
modeling an intermediate: the three-coordinate main ligand and substrate alkoxide exactly form a four-coordinate structure of a Cu center, no vacancy is available at the moment for a TEMPO free radical, the TEMPO free radical is coordinated to a second coordination layer of the Cu active center in a hydrogen bond form during modeling, and the length range of the hydrogen bond is set to be within the range And setting Cu-O and Cu-N bond length ranges respectively
And setting Cu-O and Cu-N bond length ranges respectively And
And after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
modeling a transition state: set by flexible scanning methods, i.e. Cα-H bond length from To be provided with
To be provided with Increase the amplification to
Increase the amplification to Calculating the energy of each step, finding out the corresponding structure of the highest point of the energy, and then adjusting and establishing C in the modelαDistances of-H and O-H, respectively, are set at
Calculating the energy of each step, finding out the corresponding structure of the highest point of the energy, and then adjusting and establishing C in the modelαDistances of-H and O-H, respectively, are set at And
And the following inputs are used for the geometric optimization of the transition state model: # frequm uM06L/6-31+ g (d) get ═ connectivity opt ═ (calfc, ts, noneigen, gdis) int ═ acc2e ═ 11int ═ ultrafine; obtaining an optimized structure after geometric optimization, and carrying out frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Cα-H-O, proving that the transition state is correctly sought;
the following inputs are used for the geometric optimization of the transition state model: # frequm uM06L/6-31+ g (d) get ═ connectivity opt ═ (calfc, ts, noneigen, gdis) int ═ acc2e ═ 11int ═ ultrafine; obtaining an optimized structure after geometric optimization, and carrying out frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Cα-H-O, proving that the transition state is correctly sought;
(b) construction of intermediates and transition states for O-H bond cleavage:
modeling an intermediate: principal ligand and OH-A four-coordinate structure constituting a Cu center, the Cu-OH bond length being adjusted to Cu-OLThe bond length is
Cu-OLThe bond length is Cu-ONThe bond length is
Cu-ONThe bond length is Adding substrate alcohol to the center of four-coordinate Cu, wherein the distance between H on substrate alcohol OH and O of OH is set
Adding substrate alcohol to the center of four-coordinate Cu, wherein the distance between H on substrate alcohol OH and O of OH is set And after the setting is finished, carrying out geometric optimization, and carrying out geometric optimization on the intermediate model by adopting the following inputs: # frequm uM06L/6-31+ g (d) get ═ connectivity opt ═ gdis int ═ acc2e ═ 11int ═ ultrafine; obtaining a corresponding intermediate structure;
And after the setting is finished, carrying out geometric optimization, and carrying out geometric optimization on the intermediate model by adopting the following inputs: # frequm uM06L/6-31+ g (d) get ═ connectivity opt ═ gdis int ═ acc2e ═ 11int ═ ultrafine; obtaining a corresponding intermediate structure;
modeling a transition state: adjusting the key bond length to make the Cu-OH bond length from Is adjusted to
Is adjusted to Mixing O withsub-H bond length from
Mixing O withsub-H bond length from Is adjusted to
Is adjusted to The bond length of H-OH is increased from
The bond length of H-OH is increased from Is adjusted to
Is adjusted to Performing transition state geometric optimization on the adjusted transition state model to obtain an optimized structure, and performing frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Osub-H-OH, proving that the transition state is correctly sought;
Performing transition state geometric optimization on the adjusted transition state model to obtain an optimized structure, and performing frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Osub-H-OH, proving that the transition state is correctly sought;
(c) constructing intermediates and transition states generated by OO-H bonds:
modeling an intermediate: principal ligand and O2A four-coordinate structure constituting the Cu center, and adjusting Cu-OO-The bond length is Cu-OLThe bond length is
Cu-OLThe bond length is TEMPOH-OO·-A distance of
TEMPOH-OO·-A distance of After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure; (ii) a
After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure; (ii) a
Modeling a transition state: adjusting the key bond length, adjusting Cu-OO-Key length is from Is adjusted to
Is adjusted to Cu-OLKey length is from
Cu-OLKey length is from Is adjusted to
Is adjusted to TEMPOH-OO·-Distance from
TEMPOH-OO·-Distance from Is adjusted to
Is adjusted to H-OO·-Key length is from
H-OO·-Key length is from Is adjusted to
Is adjusted to Performing transition state geometric optimization and frequency calculation on the adjusted transition state model to obtain a transition state structure with one virtual frequency; further analyzing that the vibration direction of the virtual frequency is consistent with OO--H-TEMPO, proving that the transition state is correctly sought;
Performing transition state geometric optimization and frequency calculation on the adjusted transition state model to obtain a transition state structure with one virtual frequency; further analyzing that the vibration direction of the virtual frequency is consistent with OO--H-TEMPO, proving that the transition state is correctly sought;
(d) construction of H2O2Intermediate and transition states generated:
modeling an intermediate: the main ligand and OOH form a four-coordination structure of a Cu center, and substrate alcohol is coordinated through a hydrogen bond; adjusting the Cu-OOH bond length to Cu-OLThe bond length is
Cu-OLThe bond length is PhCH2OH-OOH distance of
PhCH2OH-OOH distance of After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure; (ii) a
After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure; (ii) a
Modeling a transition state: adjusting key length, adjusting PhCH2OH-OOH bond length of Is adjusted to
Is adjusted to Cu-OLKey length is from
Cu-OLKey length is from Is adjusted to
Is adjusted to PhCH2O-H distance from
PhCH2O-H distance from Is adjusted to
Is adjusted to Carrying out geometric optimization and frequency calculation on the transition state model to obtain a transition state structure with one virtual frequency; further analyzing that the vibration direction of the virtual frequency accords with PhCH2O-H-OOH, which proves that the transition state is correctly searched;
Carrying out geometric optimization and frequency calculation on the transition state model to obtain a transition state structure with one virtual frequency; further analyzing that the vibration direction of the virtual frequency accords with PhCH2O-H-OOH, which proves that the transition state is correctly searched;
adopting a density functional method to carry out geometric structure adjustment on reactants, intermediates, transition states and products on each designed reaction path based on the generation and the fracture form of chemical bonds and the coordination condition of ligands, then carrying out geometric optimization at the calculation level of M06L/6-31+ G (d), obtaining the activation energy and reaction enthalpy change of each elementary reaction, and drawing a corresponding potential energy surface diagram;
during calculation, a wave function stability test is carried out, the geometrical structures of an intermediate body of an open shell layer single state and a transition state are calculated by adopting a symmetrical damage method, and energy correction is carried out; analyzing from the thermodynamic and kinetic angles to obtain a potential energy surface diagram, determining an optimal reaction path and a speed control step thereof, comparing with an experimental result, performing reliability analysis, determining a reaction mechanism and an optimal activity center structure of a catalyst, and calculating a reaction potential energy surface under the participation of water by adopting a real solvent model;
carrying out geometric optimization on an intermediate and a transition state on a potential energy surface by using a meta-GGA M06L functional and a 6-31+ G (d) group to obtain a stable structure, and then calculating the frequency by using the meta-GGA M06L functional and the 6-31+ G (d) group, wherein all minimum values have no virtual frequency, the transition state is ensured to have only one virtual frequency, and the intermediate has no virtual frequency; reading the Gibbs Thermal correction value after Thermal correction to Gibbs Free Energy; to get more accurate energy, further calculate single point energy at the M06L/6-311+ G (d, p)/SMD calculation level and water as solvent for gas phase optimized structure; the single point energy calculation input file is as follows: # uM06L/6-311+ G srf (solvent water, SMD) get (available check force) read (available int ultra), finally get output file of single-point calculation of gaussian log, and read the energy after scfdone: E (uM06L) in log file; adding the Gibbs thermal correction value to the single-point energy to obtain Gibbs free energy of each structure;
IRC methods are used to confirm the transition state; for antiferromagnetically coupled Cu complexes, i.e., open shell singlet, stable wave functions were obtained using guss ═ Mix and guss ═ Mix, Always; since some antiferromagnetically coupled open shells account for singlet states leading to a degree of spin contamination, energy correction is performed according to the following formula: deltaST=2(ES–ET)/2–<S2>SIn which ESIs the energy of the open shell singlet state, ETIs the energy of the triplet state and is,<S2>Sis spin contamination of the open shell singlet, subscript S denotes the open shell singlet, T denotes the triplet;
(3) analyzing a reaction mechanism;
analyzing the transition state and the bond length and the bond angle of the intermediate and the stereochemical change around the central metal ion in each elementary reaction, and summarizing the coordination number of the central metal ion, the spatial configuration of the ligand, the bond length and the bond angle of the ligand and the central metal ion in the reaction process and the change rule of corresponding structural parameters;
Cu-OH bond length during O-H bond cleavage Is increased to
Is increased to Osub-H bond length from
Osub-H bond length from Is increased to
Is increased to H-OH bond length of
H-OH bond length of Is increased to
Is increased to Central metal CuIIThe periphery is a three-coordinate main ligand, an OH ligand and a substrate alcohol in Cu axial coordination; after the O-H bond is broken, a three-coordinate main ligand H is formed2Tetrahedrally coordinated Cu of O and alkoxide coordinationIIAn active center structure; subsequently, CαH bond cleavage process Cu-OsubKey length is from
Central metal CuIIThe periphery is a three-coordinate main ligand, an OH ligand and a substrate alcohol in Cu axial coordination; after the O-H bond is broken, a three-coordinate main ligand H is formed2Tetrahedrally coordinated Cu of O and alkoxide coordinationIIAn active center structure; subsequently, CαH bond cleavage process Cu-OsubKey length is from Is increased to
Is increased to Csub-H bond length from
Csub-H bond length from Is increased to
Is increased to TEMPO-H bond length of
TEMPO-H bond length of Is reduced to
Is reduced to Through CαFormation of Cu coordinated with a tridentate primary ligand, the product benzaldehyde and TEMPOH after the H bond cleavage processIIAn active center structure; subsequently, Cu-OO. during the formation of OO-H bonds-Key length is from
Through CαFormation of Cu coordinated with a tridentate primary ligand, the product benzaldehyde and TEMPOH after the H bond cleavage processIIAn active center structure; subsequently, Cu-OO. during the formation of OO-H bonds-Key length is from Reduced to
Reduced to Cu-OLKey length is from
Cu-OLKey length is from Is increased to
Is increased to TEMPOH-OO·-Distance from
TEMPOH-OO·-Distance from Is increased to
Is increased to H-OO·-Key length is from
H-OO·-Key length is from Reduced to
Reduced to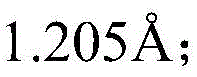 Forming a main ligand of three coordination and Cu of OOH coordination after the formation process of OO-H bondIIAn active center structure; finally, H2O2PhCH in Generation step2OH-OOH bond length of
Forming a main ligand of three coordination and Cu of OOH coordination after the formation process of OO-H bondIIAn active center structure; finally, H2O2PhCH in Generation step2OH-OOH bond length of Reduced to
Reduced to Cu-OLKey length is from
Cu-OLKey length is from Reduced to
Reduced to PhCH2O-H distance from
PhCH2O-H distance from Is increased to
Is increased to Through H2O2Formation of Cu coordinated with a tridentate primary ligand and alkoxide after the formation stepIIAn active center structure;
Through H2O2Formation of Cu coordinated with a tridentate primary ligand and alkoxide after the formation stepIIAn active center structure;
respectively calculating key elements in optimal reaction paths of different catalytic systems by adopting a natural bond orbit method and a molecular orbit theory to obtain electronic structure information of a transition state and an intermediate, and determining the bonding characteristics of a central metal ion and a ligand; the effect of solvent water on the reaction history was studied from a microscopic perspective at the molecular level: after water is added, the influence of water on the intermediate, the geometry, the electronic structure and the bonding property of the transition state and the influence of water on the reaction activation energy of the speed control step are analyzed.
(4) The conversion efficiency was calculated.
The reaction mechanism research and analysis method for oxidizing benzyl alcohol into benzaldehyde by using the amphoteric water-soluble catalyst comprises the following steps of (2): with [ (L) CuII(Hen)(H2O)]In the reaction of oxidizing benzyl alcohol into benzaldehyde by taking TEMPO as a catalyst: ([ (L) Cu)II(Hen)(H2O)]) TEMPO and an alkaline aqueous solution K are respectively used as a catalyst precursor, a cocatalyst and alkali2CO3A solution with the concentration of 0.1 mol/L; catalyst precursor ([ (L) Cu)II(Hen)(H2O)]) Has an isoelectric point of 4.96, the pH value of the solution is 11, and the catalyst precursor ([ (L) Cu)II(Hen)(H2O)]) In its anionic form [ (L) CuII(en)(H2O)](ii) present; in view of the hydrolysis of potassium carbonate, HCO3 -,CO3 2-And-three ions of OH are present in the solution, and Cu shown in the following formula (I) is present in the solution at the beginning of the reactionIIThe complex is as follows:

wherein: in the formula (I), Gibbs free energy values are shown in parentheses in kcal mol-1;
Catalyst precursor [ (L) CuII(en)(H2O)]The complex is named 0, and the complex can be prepared from OH-,CO3 2-And HCO3 -Exchange of ethylenediamine and water to form Cu, respectivelyII Complexes 1, 10 and 13, from the calculated Gibbs free energy values, gave the above-mentioned CuIIThe most stable structure in the complex is complex 1; in view of all Cu in formula (I)IIThe complex can be subjected to a catalyst activation step with a substrate alcohol, thereby giving reaction pathway A, reaction pathway B, reaction pathway C and reaction pathway D represented by the following formula (II):

for route a → C, only the catalyst activation step is different; in route A, the proton of benzyl alcohol is transferred to OH-Form H2O; in pathways B and C, protons are transferred to CO, respectively3 2-And HCO3 -Formation of HCO3 -And H2CO3(ii) a In route D, the proton of benzyl alcohol is transferred to en, while benzyl alcohol CαThe H atoms of the H bonds are transferred to the Cu centers.
The reaction mechanism research and analysis method for oxidizing benzyl alcohol into benzaldehyde by using the amphoteric water-soluble catalyst comprises the following steps of (3):
(3-1) determining possible reaction paths by calculation, converging all structures optimizing transition states into reactants, further setting the distance of H-O atoms for flexible scanning, increasing the energy by 40kcal/mol along with the reduction of the H-O distance, and showing that protons cannot be transferred to HCO3 -Upper, i.e., pathway C is a reaction pathway that is not possible;
(3-2) analyzing the path A, the path B and the path D to determine the most favorable reaction path;
in path a:
A-I, catalytic activation step
In route A, the starting complex is [ Cu ]II(L)(OH)]2-Denoted as complex 1; benzyl alcohol and complexes 21 combine to form a complex22, subsequent proton transfer of benzyl alcohol to OH-Energy barrier of 9.2kcal mol-1And the complexes formed23, i.e., [ (L) CuII(OCH2Ph)(H2O)]2-) (ii) a Then TEMPO is substituted for H2O to form active catalyst 4, in which case there are two possible isomer complexes34 and complexes34'. In the complex3In 4, TEMPO is coordinated to the second coordination sphere of the Cu center, and in the complex34' is coordinated to the first coordination sphere; it is because TEMPO coordination rotates oxygen on a ligand away from a Cu active center to cause structural distortion so that the complex34' energy ratio complex34 high 10.2kcal mol-1(ii) a Thus based on complexes34 is a favorable path;
A-II, alcohol Oxidation
After the formation of the active catalyst 4, H on the alcohol is transferred to the nitrogen atom or oxygen atom of TEMPO; the calculation results show that the path for the H atom to transfer to the oxygen atom in TEMPO hasA lower energy barrier; to verify this conclusion, active catalyst 4 and transition state TS were synthesized using the M06 functional, the OPBE functional, and the TPSSh functional4a-5a_OAnd TS4a-5a_NThe calculation is carried out again; the calculations show that for the H-transfer step, consistent results are observed even with different DFT methods, i.e. the energy barrier for the transfer of H atoms to O atoms is lower than to N atoms; thus, the H atom in the catalyst system is transferred to the O atom of TEMPO; the reaction involves a transition from the S-1 ground state to the S-0 ground state1μTS4-5_OSpin crossing of (3), i.e., a two-state reaction; the reaction can be subjected to a complex14 and complexes34, and the probability of spin flipping due to spin-crossings from S-1 to S-0; due to the complex14 and complexes3The band gap between 4 is 1.3kcal mol-1A spin balance can be established so that a spin crossover occurs; from complexes34 to1uTS4-5_OHThe energy barrier of the transfer step is 16.1kcal mol-1Subsequent production of benzaldehyde and TEMPOH;
A-III, catalyst regeneration
To complete the catalytic cycling reaction, TEMPOH and complexes 21, regeneration is needed; assuming that the catalyst regeneration step has O2Participation; triplet O2The substituted benzaldehyde will produce a complex1,36; in the complex36 → complexes3The triplet state in the 7 process is the ground state; for the complex36,Cu、O2And the spin densities of the ligands were +0.55, +1.17 and +0.27, respectively, which means that the complexes 36 has CuII-OO·-Structure; then, OO·-Can abstract H atom from TEMPOH to regenerate TEMPO, and can be used in complex37 generation of CuII-an OOH structure; this step is required to overcome 3.6kcal mol-1Energy barrier of (a), indicating that such H transfer is likely to occur; in the complex36 → complexes 37 in the process, oxidation state of Cu is changed from CuIIReduction to CuI(ii) a After regeneration of TEMPO, another benzyl alcohol is substituted for TEMPO, followed by proton transfer from the benzyl alcohol to OOH-And form H2O2I.e. the compound 37 → complexes 29; OH-substituted H2O2And PhCH2O-Regeneration of Complex 1, with H2O2Can be decomposed into H2O+1/2O2(ii) a Calculating the energy distribution of the path A;
in path B: in route B, the starting structure is [ Cu ]II(L)(CO3)]3-Denoted as complex 10; from complex 10, protons are transferred from benzyl alcohol to CO3 2-The radicals forming complexes212 ([(L)CuII(OCH2Ph)(HCO3)]3-) (ii) a TEMPO substituted HCO3 -Thereafter, an active catalyst complex 4 is produced; then after the alcohol oxidation step, i.e. Complex 4 → Complex 5 and the catalyst regeneration step, i.e. Complex 6 → Complex 9, CO3 2-Substitution of substance for H2O2And PhCH2O-Regenerating the complex 10; the calculated energy distribution of the path B;
in path D: in route D, the starting structure is [ (L) CuII(H2O)(en)]-Denoted as complex 0; starting from complex 0, benzyl alcohol replaces H2O forming a complex216, i.e., [ (L) Cu)II(HOCH2Ph)(en)]-(ii) a Followed by compounding in step (b)216 → complexes2In 17, PhCH2Protons of OH are transferred to en, with PhCH2C of OHαThe H atom of the-H bond is transferred to the Cu center, this step gives the product PhCHO, a step complex216 → complexes217 has an energy barrier of 23.9kcal mol-1(ii) a Formation of complexes2After 17, the product PhCHO is substituted by TEMPO to form a complex3,118, followed by transfer of the H atom from the Cu-H bond to TEMPO; for the triplet state, this H atom transfer step needs to overcome 44.8kcal mol-1Energy barrier of (d); however, for the open shell singlet state, the transition state1uTS18-19Direct formation of complexes119; therefore, the H atom transfer step is likely to occur on the open shell singlet potential surface; then, O2Formation of complex 6 instead of Hen; then theComplex 6 → complex 9; same procedure in Path A and Path B, en and H2O instead of H2O2And PhCH2O-Regenerating the complex 0; the calculated energy distribution of the path D;
the reaction mechanism research and analysis method for oxidizing benzyl alcohol into benzaldehyde by using the amphoteric water-soluble catalyst comprises the following steps of (4): calculating the energy span delta E of catalytic reaction of the path A, the path B and the path C based on an energy span model according to the calculated reaction potential energy surface of the dominant path of the path A, the path B and the path C, and calculating the conversion frequency TOF value of the catalyst by the following formula:

wherein: kbBoltzmann constant, h planck constant, R ideal gas constant, T reaction temperature, δ E energy span of reaction path;
delta E of route A is 27.2kcal mol-1Delta E of route B is 36.1kcal mol-1δ E of pathway C is 52.1kcal mhol-1Path a is the dominant path; TOF values of the path Ah, the path B and the path C calculated according to the formula are respectively 3.89h-1、1.2×10-4h-1And 1.4X 10-22h-1Wherein the TOF value of the path A is 5.40h from the experimental TOF value-1The fit was very good.
The reaction mechanism research and analysis method for oxidizing benzyl alcohol into benzaldehyde by using the amphoteric water-soluble catalyst comprises the following steps of (3): the TDTS and TDI on the path A potential surface are respectively an intermediate with the highest energy transition state and the lowest energy on the potential surface; analyzing the geometric structure and the electronic structure of the TDTS of the path A in the determined rate transition state in view of the fact that the TDTS is a key factor influencing TOF; firstly, reading an output file of a geometric structure through Gaussview software to obtain the bond length, the bond angle and the stereochemical change condition around a central metal ion; secondly, calculating to obtain a Weber bond WBI based on a natural bond orbit method NBO, and further analyzing the bonding and breaking conditions of chemical bonds; finally, reading the chk file of the single-point result of the TDTS structure by adopting Gaussview software, and analyzing by a visual module in a current surface analysis module to obtain a molecular orbit diagram to obtain two single-electron single occupied orbits (SOMOs); analyzing by combining a spin density analysis result to obtain the bonding characteristics of the central metal ions and the ligands in the TDTS step;
in Path A, the rate transition state TDTS is determined to be the transition state1uTS4-5_ODue to a transition state1uTS4-5_OIs the transition state of the H transfer step, i.e. complex 4 → transition state TS4-5_OTherefore, the complex was analyzed14 → transition state1uTS4-5_O→ complex1Electron transfer process during 5; for the complex14, Cu, ligand, TEMPO and substrate-OCH2The spin densities of Ph are +0.57, +0.32, -1.00, and +0.11, respectively, which means that the complex14 contains [ (L) (TEMPO) CuII(OCH2Ph)]Structure; in the transition state1uTS4-5_OIn the above formula, the spin density of Cu is +0.32, the spin density of TEMPOH is-0.32,-OCH2the spin density of Ph is-0.17, and the spin density of the ligand is + 0.17; binding complexes14 → transition state1uTS4-5_OMolecular orbital analysis of the process, resulting in the formation of TEMPO-H bond moieties and-OCH2c of Phα-H bond occurrence partial homolytic cleavage; a part of the beta spintronics on the substrate increases, namely from 0.0% → 33.1%; the part of the beta-spin electrons is derived from-OCH2C of Phα-homolytic cleavage of H bonds; also, from-OCH2C of PhαThe corresponding alpha spin electrons generated by homolytic cleavage of the H bond are transferred to TEMPO, resulting in an increase in the alpha spin density of TEMPO, i.e. from 0.0% → 15.3%; in the complex14→1uTS4-5_OIn the process, Cα-H bond weakening, TEMPO-H bond strengthening; in the complex14 → transition state1uTS4-5_OIn the process, Cu-OsubBonds are not homolytic but in a transition state1uTS4-5_O→ complex15 in process, Cu-OsubThe bond is homolytic and finally benzaldehyde and TEMPOH are obtained.
The invention has the following beneficial effects:
the invention adopts the Density Functional Theory (DFT) to couple with [ (L) CuII(Hen)(H2O)]The catalytic mechanism of the oxidative oxidation of benzyl alcohol to benzaldehyde using TEMPO as catalyst is studied, and four routes are included (route A → D), route A being the most favorable route (Δ E ═ 27.2kcal mol ═ D)-1;TOF=3.89h-1). The calculation result shows that: the zwitter-ionic property of the catalyst does not influence the alcohol oxidation process of the system. ② the catalytic cycle comprises catalyst activation, substrate oxidation and catalyst regeneration. ③ the reaction path A is a dominant path, and the conversion frequency (TOF) is 3.89h-1Consistent with the experimental results (TOF 5.40 h)-1). The rate control step is a substrate oxidation step, i.e. a process in which hydrogen atoms are transferred from the alkoxide to oxygen atoms of TEMPO. The invention is to [ (L) CuII(Hen)(H2O)]The TEMPO catalyst system carries out quantum chemical calculation, and researches the reaction mechanism of the catalytic system for oxidizing primary alcohol into aldehyde by oxygen and the structure-activity relationship of the catalyst on an atomic level; and effectively calculating the conversion efficiency TOF of the catalyst through an energy span model, and evaluating the activity of the catalyst. By exploring the reaction mechanism of the catalytic system, not only can the reaction process be observed, but also a theoretical basis can be provided for designing a novel high-efficiency catalyst.
Due to the limitation of the existing experimental technology and the self condition of the instrument, a complete reaction mechanism and a catalyst structure-activity relationship cannot be obtained by only depending on an experimental method. The invention can obtain the relationship between the reaction mechanism and the catalyst structure activity from the atomic level by means of quantum chemistry technology, finally establishes a method for calculating the conversion efficiency of the water-soluble Cu-TEMPO catalytic system based on an energy span model to further evaluate the catalyst activity, provides basic data and theoretical guidance for designing a novel green and environment-friendly bionic catalyst, and helps to shorten the research and development period of the catalyst and reduce the investment. The method can be completed only by a computer, so that expensive large-scale equipment or high test analysis cost is not required to be purchased, the design cost of the catalyst is reduced, and the research and development speed of the catalyst is accelerated. And the calculation result is accurate and reliable in accordance with the experimental result.
A reaction mechanism of catalyzing and oxidizing alcohol into aldehyde by a water-soluble Cu-TEMPO catalysis system is researched by utilizing a quantum chemistry method (such as a density functional theory), and a method for calculating conversion efficiency of the water-soluble Cu-TEMPO catalysis system based on an energy span model is established by researching the relation between factors such as geometric and electronic structure of a catalyst active center, space and electronic effect of ligands, synergistic effect among the ligands, coordination mode of central metal Cu ions and the ligands, bonding property of the central metal Cu ions and the ligands and the catalytic activity.
Drawings
The following describes embodiments of the present invention in further detail with reference to the accompanying drawings.
FIG. 1 complexes34, optimizing the structure;
FIG. 2 Complex34' is optimized;
FIG. 3 calculated Gibbs free energy profile for Path A: the solid line represents the triplet state; the dotted line represents the singlet state, the superscripts 1u and 1r represent the open-shell singlet state and the constrained singlet state, respectively, and the energy value is given in kcal mol-1Is a unit;
FIG. 4 Complex14 (value ± 0.04);
FIG. 5 transition state1uTS4-5_OMolecular orbital diagram (numerical value ═ 0.04);
FIG. 6 energy distribution for path B;
FIG. 7 energy distribution for path D;
FIG. 8 is a diagram of the corresponding complex structure in Table S2;
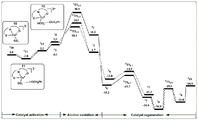
FIG. 9 is a technical flow chart of a reaction mechanism research and analysis method for oxidizing benzyl alcohol into benzaldehyde by oxygen in an amphoteric water-soluble catalyst.
Detailed Description
In order to more clearly illustrate the invention, the invention is further described below with reference to preferred embodiments and the accompanying drawings. Similar parts in the figures are denoted by the same reference numerals. It is to be understood by persons skilled in the art that the following detailed description is illustrative and not restrictive, and is not to be taken as limiting the scope of the invention.
Research and analysis method for reaction mechanism of oxidizing benzyl alcohol into benzaldehyde by oxygen in amphoteric water-soluble catalyst, [ (L) CuII(Hen)(H2O)]As shown in the following formula [ compound 4 in eur.j.inorg.chem.2011,27,4175-4181 ]:

the method specifically comprises the following steps:
(1) constructing a calculation model of the catalyst activity center structure: based on reaction condition information provided by experiments, a calculation model of the catalyst active center is constructed, a density functional method is adopted for geometric optimization, and the optimal model is selected by comprehensively considering configuration stability, energy level, calculation result and calculation cost according to the calculation result.
(1-1) modeling of a catalytic precursor model:
searching 809721.cif files of CCDC-809721 from a Cambridge crystal database in England, and reading X-ray crystallography data provided by the 809721.cif files through Materials Studio software to initially construct a catalytic precursor model; the bond length and bond angle of the constructed preliminary catalytic precursor model were adjusted due to the crystal packing effect: the bond length of all Cu-N bonds is increased Increased bond length of Cu-O bond
Increased bond length of Cu-O bond
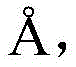 Modulation of N-N bond
Modulation of N-N bond S-O bond increase
S-O bond increase Modulation of C-N bond
Modulation of C-N bond Modulation of C-C bond
Modulation of C-C bond The bond angle of O-Cu-N and O-Cu-O is increased by 0.5-1.5 degrees;
The bond angle of O-Cu-N and O-Cu-O is increased by 0.5-1.5 degrees;
(1-2) optimizing the bond length, bond angle and dihedral angle of the complex (bond length of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3, bond angle of O1-Cu-N2 and O2-Cu-N2, dihedral angle of O3-Cu-N2-N3) based on the adjusted structure using M06L, B3LYP, OPBE and M06 functional in combination with 6-31+ G (d) group;
the maximum difference between the bond lengths of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3 calculated at the level of M06L functional and the corresponding bond lengths in the experiment is The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1 degree;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1 degree;
the maximum difference between the bond length of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3 calculated at the level of B3LYP functional and the corresponding bond length of the experiment is The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1.8 degrees;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1.8 degrees;
the bond length calculated at the level of OPBE functional is within the experimental error The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 2.6 degrees;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 2.6 degrees;
the maximum difference between the bond lengths of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3 calculated at the level of M06 functional and the corresponding bond lengths in the experiment is Bonds of O1-Cu-N2 and O2-Cu-N2The maximum difference between the angle and the corresponding key angle of the experiment is 0.9 degrees;
Bonds of O1-Cu-N2 and O2-Cu-N2The maximum difference between the angle and the corresponding key angle of the experiment is 0.9 degrees;
according to the calculation result: it can be seen that the M06L and M06 functionals gave more reliable results. Furthermore, considering that the M06L functional represents a good balance between accuracy and computational cost, the results at the level of M06L/6-31+ G (d) were ultimately determined.
(2) Designing a reaction path;
performing possible reaction path design according to the optimal model selected in the step (1);
reaction intermediates and transition state modeling were performed:
(a) construction of Cα-intermediates and transition states of H bond cleavage:
modeling an intermediate: the three-coordinate main ligand and substrate alkoxide exactly form a four-coordinate structure of a Cu center, no vacancy is available at the moment for a TEMPO free radical, the TEMPO free radical is coordinated to a second coordination layer of the Cu active center in a hydrogen bond form during modeling, and the length range of the hydrogen bond is set to be within the range And setting Cu-O and Cu-N bond length ranges respectively
And setting Cu-O and Cu-N bond length ranges respectively And
And and after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure.
and after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure.
Modeling a transition state: set by flexible scanning methods, i.e. Cα-H bond length from To be provided with
To be provided with Increase the amplification to
Increase the amplification to Calculating the energy of each step, and finding out the corresponding junction with the highest energy pointConstruct, then adjust C in the building modelαDistances of-H and O-H, respectively, are set at
Calculating the energy of each step, and finding out the corresponding junction with the highest energy pointConstruct, then adjust C in the building modelαDistances of-H and O-H, respectively, are set at And
And the following inputs are used for the geometric optimization of the transition state model: # frequm uM06L/6-31+ g (d) get ═ connectivity opt ═ (calfc, ts, noneigen, gdis) int ═ acc2e ═ 11int ═ ultrafine. Obtaining an optimized structure after geometric optimization, and carrying out frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Cα-H-O, proving that the transition state is correctly sought.
the following inputs are used for the geometric optimization of the transition state model: # frequm uM06L/6-31+ g (d) get ═ connectivity opt ═ (calfc, ts, noneigen, gdis) int ═ acc2e ═ 11int ═ ultrafine. Obtaining an optimized structure after geometric optimization, and carrying out frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Cα-H-O, proving that the transition state is correctly sought.
(b) Construction of intermediates and transition states for O-H bond cleavage:
modeling an intermediate: principal ligand and OH-A four-coordinate structure constituting a Cu center, the Cu-OH bond length being adjusted to Cu-OLThe bond length is
Cu-OLThe bond length is Cu-ONThe bond length is
Cu-ONThe bond length is Adding substrate alcohol to the center of four-coordinate Cu, wherein the distance between H on substrate alcohol OH and O of OH is set
Adding substrate alcohol to the center of four-coordinate Cu, wherein the distance between H on substrate alcohol OH and O of OH is set And after the setting is finished, performing geometric optimization. Intermediate model geometric optimization was performed with the following inputs: # frequm uM06L/6-31+ g (d) get ═ connectivity opt ═ gdis int ═ acc2e ═ 11int ═ ultrafine, resulting in the corresponding intermediate structure.
And after the setting is finished, performing geometric optimization. Intermediate model geometric optimization was performed with the following inputs: # frequm uM06L/6-31+ g (d) get ═ connectivity opt ═ gdis int ═ acc2e ═ 11int ═ ultrafine, resulting in the corresponding intermediate structure.
Modeling a transition state: adjusting the key bond length to make the Cu-OH bond length from Is adjusted to
Is adjusted to Mixing O withsub-H bond length from
Mixing O withsub-H bond length from Is adjusted to
Is adjusted to The bond length of H-OH is increased from
The bond length of H-OH is increased from Is adjusted to
Is adjusted to And performing transition state geometric optimization on the adjusted transition state model to obtain an optimized structure, and performing frequency calculation to obtain a virtual frequency. And analyzing the vibration direction of the virtual frequency to conform to Osub-H-OH, proving that the transition state is correctly sought.
And performing transition state geometric optimization on the adjusted transition state model to obtain an optimized structure, and performing frequency calculation to obtain a virtual frequency. And analyzing the vibration direction of the virtual frequency to conform to Osub-H-OH, proving that the transition state is correctly sought.
(c) Constructing intermediates and transition states generated by OO-H bonds:
modeling an intermediate: principal ligand and O2A four-coordinate structure constituting the Cu center, and adjusting Cu-OO-The bond length is Cu-OLThe bond length is
Cu-OLThe bond length is TEMPOH-OO·-A distance of
TEMPOH-OO·-A distance of And after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure.
And after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure.
Modeling a transition state: adjusting the key bond length, adjusting Cu-OO-Key length is from Is adjusted to
Is adjusted to Cu-OLKey length is from
Cu-OLKey length is from Is adjusted to
Is adjusted to TEMPOH-OO·-Distance from
TEMPOH-OO·-Distance from Is adjusted to
Is adjusted to H-OO·-Key length is from
H-OO·-Key length is from Is adjusted to
Is adjusted to And performing transition state geometric optimization and frequency calculation on the adjusted transition state model to obtain a transition state structure with one virtual frequency. Further analyzing that the vibration direction of the virtual frequency is consistent with OO--H-TEMPO, which proves that the transition is looking for the right.
And performing transition state geometric optimization and frequency calculation on the adjusted transition state model to obtain a transition state structure with one virtual frequency. Further analyzing that the vibration direction of the virtual frequency is consistent with OO--H-TEMPO, which proves that the transition is looking for the right.
(d) Construction of H2O2Intermediate and transition states generated:
modeling an intermediate: the main ligand and OOH form a four-coordination structure of a Cu center, and substrate alcohol coordinates through a hydrogen bond. Adjusting the Cu-OOH bond length to Cu-OLThe bond length is
Cu-OLThe bond length is PhCH2OH-OOH distance of
PhCH2OH-OOH distance of And after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure.
And after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure.
Modeling a transition state: adjusting key length, adjusting PhCH2OH-OOH bond length of Is adjusted to
Is adjusted to Cu-OLKey length is from
Cu-OLKey length is from Is adjusted to
Is adjusted to PhCH2O-H distance from
PhCH2O-H distance from Is adjusted to
Is adjusted to And carrying out geometric optimization and frequency calculation on the transition state model to obtain a transition state structure with one virtual frequency. Further analyzing that the vibration direction of the virtual frequency accords with PhCH2O-H-OOH, proving that the transition state is correctly found.
And carrying out geometric optimization and frequency calculation on the transition state model to obtain a transition state structure with one virtual frequency. Further analyzing that the vibration direction of the virtual frequency accords with PhCH2O-H-OOH, proving that the transition state is correctly found.
Adopting a density functional method to carry out geometric structure adjustment on reactants, intermediates, transition states and products on each designed reaction path based on the generation and the fracture form of chemical bonds and the coordination condition of key ligands, then carrying out geometric optimization at the calculation level of M06L/6-31+ G (d), obtaining the activation energy and reaction enthalpy change of each elementary reaction, and drawing a corresponding potential energy surface diagram;
during calculation, a wave function stability test is carried out, the geometrical structures of an intermediate body of an open shell layer single state and a transition state are calculated by adopting a symmetrical damage method, and energy correction is carried out; analyzing from the thermodynamic and kinetic angles to obtain a potential energy surface diagram, determining an optimal reaction path and a speed control step thereof, comparing with an experimental result, performing reliability analysis, determining a reaction mechanism and an optimal activity center structure of a catalyst, and calculating a reaction potential energy surface under the participation of water by adopting a real solvent model;
carrying out geometric optimization on an intermediate and a transition state on a potential energy surface by using a meta-GGA M06L functional and a 6-31+ G (d) group to obtain a stable structure, and then calculating the frequency by using the meta-GGA M06L functional and the 6-31+ G (d) group, wherein all minimum values have no virtual frequency, the transition state is ensured to have only one virtual frequency, and the intermediate has no virtual frequency; reading the Gibbs Thermal correction value after Thermal correction to Gibbs Free Energy; to get more accurate energy, further calculate single point energy at the M06L/6-311+ G (d, p)/SMD calculation level and water as solvent for gas phase optimized structure; the single point energy calculation input file is as follows: # uM06L/6-311+ G srf (solvent water, SMD) get (available check force) read (available int ultra), finally get output file of single-point calculation of gaussian log, and read the energy after scfdone: E (uM06L) in log file; adding the Gibbs thermal correction value to the single-point energy to obtain Gibbs free energy of each structure;
IRC methods are used to confirm the transition state; for antiferromagnetically coupled Cu complexes, i.e., open shell singlet, stable wave functions were obtained using guss ═ Mix and guss ═ Mix, Always; since some antiferromagnetically coupled open shells account for singlet states leading to a degree of spin contamination, energy correction is performed according to the following formula: deltaST=2(ES–ET)/2–<S2>SIn which ESIs the energy of the open shell singlet state, ETIs the energy of the triplet state and is,<S2>Sis spin contamination of the open shell singlet, subscript S denotes the open shell singlet, T denotes the triplet;
with [ (L) CuII(Hen)(H2O)]In the reaction of oxidizing benzyl alcohol into benzaldehyde by taking TEMPO as a catalyst: ([ (L) Cu)II(Hen)(H2O)]) TEMPO and an aqueous alkaline solution (K)2CO3Concentration of 0.1mol/L) were used as catalyst precursor, cocatalyst and base, respectively. In addition, catalyst precursor ([ (L) Cu)II(Hen)(H2O)]) Has an isoelectric point of 4.96. Since the pH of the solution was 11, the catalyst precursor ([ (L) Cu)II(Hen)(H2O)]) In its anionic form [ (L) CuII(en)(H2O)]Are present. That is, the zwitterionic nature may not affect the efficiency of alcohol oxidation. Even so, stillIt is necessary to find out whether the ethylenediamine molecule is directly involved in the catalytic cycle. In addition, HCO is formed due to hydrolysis of potassium carbonate3 -,CO3 2-And-OH plasma is present in the solution, so some Cu is present in the solution at the beginning of the reactionIIThe complex [ as shown in formula (I) ]:

in formula (I): gibbs free energy values in kcal mol-1。
Catalyst precursor [ (L) CuII(en)(H2O)]The complex was named complex 0. The complexes may be substituted with OH-,CO3 2-And HCO3 -Exchange of ethylenediamine and water to form Cu, respectivelyIIComplex 1, complex 10 and complex 13. Notably, these Cu's are calculated based on the gibbs free energy valuesIIThe most stable structure in the complex is complex 1. Due to all Cu in the formula (I)IIThe complex can be subjected to a catalyst activation step with a substrate alcohol, thus calculating all of these possible catalyst activation processes, such as reaction pathway A, reaction pathway B, reaction pathway C and reaction pathway D represented by the following formula (II):

for route a → C, only the catalyst activation step is different; in route A, the proton of benzyl alcohol is transferred to OH-Form H2O; in pathways B and C, protons are transferred to CO, respectively3 2-And HCO3 -Formation of HCO3 -And H2CO3(ii) a In route D, the proton of benzyl alcohol is transferred to en, while benzyl alcohol CαThe H atoms of the H bonds are transferred to the Cu centers.
In the calculation process of the step (1) and the step (2), the accuracy and the calculation of the result are consideredCost, geometric optimization and frequency calculation using meta-GGA M06L functional and 6-31+ G (d) basis set; all the minimum values have no virtual frequency, and the transition state has only one virtual frequency; IRC methods are used to confirm the transition state; for antiferromagnetically coupled Cu complexes, i.e., open shell singlet, stable wave functions were obtained using guss ═ Mix and guss ═ Mix, Always; since some antiferromagnetically coupled open shells account for singlet states leading to a degree of spin contamination, energy correction is performed according to the following formula: deltaST= 2(ES–ET)/2–<S2>SIn which ESIs the energy of the open shell singlet state, ETIs the energy of the triplet state and is,<S2>Sis spin contamination of the open-shell singlet state, the subscript S denoting the open-shell singlet state and T denoting the triplet state.
(3) Analyzing a reaction mechanism;
analyzing geometrical structures such as bond length and bond angle of transition states and intermediates in each elementary reaction and stereochemical change around the central metal ion, and summarizing coordination number of the central metal ion in the reaction process, spatial configuration of ligands, coordination condition information such as bond length and bond angle of the ligands and the central metal ion, and change rules of corresponding structural parameters; Cu-OH bond length during O-H bond cleavage Is increased to
Is increased to Osub-H bond length from
Osub-H bond length from Is increased to
Is increased to H-OH bond length of
H-OH bond length of Is increased to
Is increased to Central metal CuIIThe periphery is a three-coordinate main ligand, an OH ligand and a substrate alcohol which is axially coordinated in Cu. After the O-H bond is broken, a three-coordinate main ligand H is formed2Tetrahedrally coordinated Cu of O and alkoxide coordinationIIAn active center structure. Subsequently, CαH bond cleavage process Cu-OsubKey length is from
Central metal CuIIThe periphery is a three-coordinate main ligand, an OH ligand and a substrate alcohol which is axially coordinated in Cu. After the O-H bond is broken, a three-coordinate main ligand H is formed2Tetrahedrally coordinated Cu of O and alkoxide coordinationIIAn active center structure. Subsequently, CαH bond cleavage process Cu-OsubKey length is from Is increased to
Is increased to Csub-H bond length from
Csub-H bond length from Is increased to
Is increased to TEMPO-H bond length of
TEMPO-H bond length of Is reduced to
Is reduced to Through CαFormation of Cu coordinated with a tridentate primary ligand, the product benzaldehyde and TEMPOH after the H bond cleavage processIIAn active center structure. Subsequently, Cu-OO. during the formation of OO-H bonds-Key length is from
Through CαFormation of Cu coordinated with a tridentate primary ligand, the product benzaldehyde and TEMPOH after the H bond cleavage processIIAn active center structure. Subsequently, Cu-OO. during the formation of OO-H bonds-Key length is from Reduced to
Reduced to Cu-OLKey length is from
Cu-OLKey length is from Is increased to
Is increased to TEMPOH-OO·-Distance from
TEMPOH-OO·-Distance from Is increased to
Is increased to H-OO·-Key length is from
H-OO·-Key length is from Reduced to
Reduced to Forming a main ligand of three coordination and Cu of OOH coordination after the formation process of OO-H bondIIAn active center structure. Finally, H2O2PhCH in Generation step2OH-OOH bond length of
Forming a main ligand of three coordination and Cu of OOH coordination after the formation process of OO-H bondIIAn active center structure. Finally, H2O2PhCH in Generation step2OH-OOH bond length of Reduced to
Reduced to Cu-OLKey length is from
Cu-OLKey length is from Reduced to
Reduced to PhCH2O-H distance from
PhCH2O-H distance from Is increased to
Is increased to Through H2O2Formation of Cu coordinated with a tridentate primary ligand and alkoxide after the formation stepIIAn active center structure.
Through H2O2Formation of Cu coordinated with a tridentate primary ligand and alkoxide after the formation stepIIAn active center structure.
Respectively calculating key elements in optimal reaction paths of different catalytic systems by adopting a natural bond orbit method and a molecular orbit theory to obtain electronic structure information of a transition state and an intermediate, and determining the bonding characteristics of a central metal ion and a ligand; the effect of solvent water on the reaction history was studied from a microscopic perspective at the molecular level: mainly analyzes the influence of water on the intermediate, the geometry, the electronic structure and the bonding property of the transition state and the influence on the reaction activation energy of the speed control step after the water is added.
(3-1) by computationally determining the reaction paths that may exist, all structures that optimize the transition state converge to the reactants, for which we further set the distance of the H-O atoms for flexible scanning, and found that the energy increases with decreasing H-O distance, about 40kcal/mol, thus indicating that protons cannot be transferred to HCO3 -Upper, i.e., pathway C is a reaction pathway that is not possible;
(3-2) analyzing the path A, the path B and the path D to determine the most favorable reaction path;
in path a:
A-I, catalytic activation step
In route A, the starting complex is [ Cu ]II(L)(OH)]2-Denoted as complex 1; benzyl alcohol and complexes 21 combine to form a complex22, subsequent proton transfer of benzyl alcohol to OH-Energy barrier of 9.2kcal mol-1[ see FIG. 3 ] and the complexes formed23, i.e., [ (L) CuII(OCH2Ph)(H2O)]2-) (ii) a Then TEMPO is substituted for H2O to form active catalyst 4, in which case there are two possible isomer complexes34 and complexes34' [ see fig. 1 and 2 ]. In the complex3In 4, TEMPO is coordinated to the second coordination sphere of the Cu center, and in the complex34' is coordinated to the first coordination sphere; it is because TEMPO coordination rotates oxygen on a ligand away from a Cu active center to cause structural distortion so that the complex34' energy ratio complex34 high 10.2kcal mol-1(the triplet state is the ground state); thus based on complexes34 is a favorable path;
A-II, alcohol Oxidation
After formation of the active catalyst 4, H on the alcohol may be transferred to the nitrogen atom or oxygen atom of TEMPO; the calculation results show that the path for transferring the H atom to the oxygen atom in TEMPO has lower energyBase (see fig. 3); to verify this conclusion, active catalyst 4 and transition state TS were synthesized using the M06 functional, the OPBE functional, and the TPSSh functional4a-5a_OAnd TS4a-5a_NThe calculation is carried out again; the calculations show that consistent results are observed for the H-transfer step even with different DFT methods (see table S3A), i.e. the energy barrier for the transfer of H atoms to O atoms is lower than to N atoms.
TABLE S3A
| Species | OPBE | | M06 | M06L | |
| 21 | 0.0 | 0.0 | 0.0 | 0.0 | |
| 3TS4-5_O | 24.7 | 24.0 | 25.2 | 29.0 | |
| 1uTS4-5_O | --- | 20.2 | 21.3 | 16.1 | |
| 1rTS4-5_O | 19.9 | 28.0 | 29.6 | 22.5 | |
| 3TS4-5_N | 40.4 | 31.4 | 27.7 | 25.8 | |
| 1uTS4-5_N | --- | --- | 28.2 | 24.4 | |
| 1rTS4-5_N | 32.8 | 35.0 | 41.8 | 26.8 | |
| Energy difference | 12.9 | 11.2 | 6.9 | 8.3 |
Thus, the catalyst system is obtained, wherein H atoms are transferred to O atoms of TEMPO; as shown in fig. 3, the reaction involves a transition state from the S-1 ground state to the S-0 transition state1μTS4-5_OSpin crossing of (a), i.e., Two State Reaction (TSR); the reaction can be subjected to a complex14 and complexes34, and the spin flip probability (SIP) due to the spin-cross from S-1 to S-0; due to the complex14 and complexes3The band gap between 4 is 1.3kcal mol-1A spin balance can be established so that a spin crossover occurs; from complexes34 to1uTS4-5_OHThe energy barrier of the transfer step is 16.1kcal mol-1Subsequent production of benzaldehyde and TEMPOH;
A-III, catalyst regeneration
To complete the catalytic cycling reaction, TEMPOH and complexes 21, regeneration is needed; assuming that the catalyst regeneration step has O2Participation; triplet O2The substituted benzaldehyde will produce a complex1,36; as shown in FIG. 3, in the complex36 → complexes3The triplet state in the 7 process is the ground state; for the complex36,Cu、O2And the spin densities of the ligands were +0.55, +1.17 and +0.27, respectively, which means that the complexes 36 has CuII-OO·-Structure; then, OO·-Can abstract H atom from TEMPOH to regenerate TEMPO, and can be used in complex37 generation of CuII-an OOH structure; this step is required to overcome 3.6kcal mol-1Energy barrier of (a), indicating that such H transfer is likely to occur; in the complex36 → complexes 37 in the process, oxidation state of Cu is changed from CuIIReduction to CuI(ii) a After regeneration of TEMPO, another benzyl alcohol is substituted for TEMPO, followed by the removal of the TEMPO from the benzyl alcoholProton transfer to OOH-And form H2O2I.e. the compound 37 → complexes 29; OH-substituted H2O2And PhCH2O-Regeneration of Complex 1, with H2O2Can be decomposed into H2O+1/2O2(ii) a Calculating the energy distribution of the path A;
A-IV, catalytic cycle: energy span approximation
The TDTS and TDI on the path A potential surface are respectively an intermediate with the highest energy transition state and the lowest energy on the potential surface; analyzing the geometric structure and the electronic structure of the TDTS of the path A in the determined rate transition state in view of the fact that the TDTS is a key factor influencing TOF; firstly, reading an output file of a geometric structure through Gaussview software to obtain the geometric structures such as bond length and bond angle and the stereochemical change condition around a central metal ion; secondly, calculating to obtain a Weber bond WBI based on a natural bond orbit method NBO, and further analyzing the bonding and breaking conditions of chemical bonds; finally, reading the chk file of the single-point result of the TDTS structure by adopting Gaussview software, and analyzing by a visual module in a current surface analysis module to obtain a molecular orbit diagram to obtain two single-electron single occupied orbits (SOMOs); analyzing by combining a spin density analysis result to obtain the bonding characteristics of the central metal ions and the ligands in the TDTS step;
the energy span (δ E) of the catalytic reaction was calculated using the energy span model of Kozuch and Shaik, and the conversion efficiency (TOF) of the Cu/TEMPO catalytic system was calculated. For route A, δ E is 27.2kcal mol-1TOF of 3.89h-1And TOF (time of flight) of experimental result is 5.40h-1And (5) the consistency is achieved.
In Path A, the determination of the rate transition state TDTS is1uTS4-5_ODue to the fact that1uTS4-5_OIs a transition state of the H transfer step, i.e. complex 4 → TS4-5_OTherefore, the complex was analyzed14→1uTS4-5_O→ complex1Electron transfer process during 5; for the complex14, Cu, ligand, TEMPO and substrate-OCH2The spin densities of Ph are +0.57, +0.32, -1.00, and +0.11, respectively, which means that the complex14 contains [ (L) (TEMPO) CuII(OCH2Ph)]Structure; in that1uTS4-5_OIn the above formula, the spin density of Cu is +0.32, the spin density of TEMPOH is-0.32,-OCH2the spin density of Ph is-0.17, and the spin density of the ligand is + 0.17; binding complexes14→1uTS4-5_OMolecular orbital analysis of the procedure (see FIGS. 4 and 5) to yield TEMPO-H bond moiety formation and-OCH2c of Phα-H bond occurrence partial homolytic cleavage; as shown in fig. 4 and 5, a portion of the β spinelectrons on the substrate increases, i.e., from 0.0% → 33.1%; the part of the beta-spin electrons is derived from-OCH2C of Phα-homolytic cleavage of H bonds; also consist of-OCH2C of PhαThe corresponding alpha spin electrons generated by homolytic cleavage of the H bond are transferred to TEMPO, resulting in an increase in the alpha spin density of TEMPO, i.e. from 0.0% → 15.3%; this is also confirmed by WBI and bond length changes (see table 1).
TABLE 114→1TS4-5 O→15. Cu-O of processsub、Cα-H and OTEMPOBond length of-H and WBI

In the complex14→1uTS4-5_OIn the process, Cα-H bond weakening (WBI: C)α-H: 0.85 → 0.42; bond length: ) TEMPO-H bond enhancement (WBI: TEMPO-H: 0.00 → 0.57; bond length:
) TEMPO-H bond enhancement (WBI: TEMPO-H: 0.00 → 0.57; bond length: ) (ii) a In the complex14→1uTS4-5_OIn the process, Cu-OsubThe bond is not homolytic but is in1uTS4-5_O→ complex15 in process, Cu-OsubThe bond is homolytic and finally benzaldehyde and TEMPOH are obtained. This result indicates that1uTS4-5_O→ complex15, the valence change of Cu is critical for the production of the product PhCHO. Since the reaction was carried out in the aqueous phase, the present invention also calculates the effect of water molecules on the possible presence of the reaction. The result shows that the energy barrier of the water ginseng is 30.5kcal mol-1Is less than the amount of water (27.2kcal mol)-1) 3.3kcal mol higher-1Thus, the participation of water molecules reduces the activity of the reaction.
) (ii) a In the complex14→1uTS4-5_OIn the process, Cu-OsubThe bond is not homolytic but is in1uTS4-5_O→ complex15 in process, Cu-OsubThe bond is homolytic and finally benzaldehyde and TEMPOH are obtained. This result indicates that1uTS4-5_O→ complex15, the valence change of Cu is critical for the production of the product PhCHO. Since the reaction was carried out in the aqueous phase, the present invention also calculates the effect of water molecules on the possible presence of the reaction. The result shows that the energy barrier of the water ginseng is 30.5kcal mol-1Is less than the amount of water (27.2kcal mol)-1) 3.3kcal mol higher-1Thus, the participation of water molecules reduces the activity of the reaction.
In path B: in route B, the starting structure is [ Cu ]II(L)(CO3)]3-Denoted as complex 10; from complex 10, protons are transferred from benzyl alcohol to CO3 2-The radicals forming complexes212 ([(L)CuII(OCH2Ph)(HCO3)]3-) (ii) a TEMPO substituted HCO3 -Thereafter, an active catalyst complex 4 is produced; then after the alcohol oxidation step, i.e. Complex 4 → Complex 5 and the catalyst regeneration step, i.e. Complex 6 → Complex 9, CO3 2-Substitution of substance for H2O2And PhCH2O-Regenerating the complex 10; the calculated energy distribution of path B is detailed in fig. 6.
In this case, δ E is rather high (δ E ═ 36.1kcal mol)-1) TOF value in Path B is 1.2 × 10-4h-1. Thus, path B is not the dominant path.
In path D: in route D, the starting structure is [ (L) CuII(H2O)(en)]-Denoted as complex 0; starting from complex 0, benzyl alcohol replaces H2O forming a complex216, i.e., [ (L) Cu)II(HOCH2Ph)(en)]-(ii) a Followed by compounding in step (b)216 → complexes2In 17, PhCH2Protons of OH are transferred to en, with PhCH2C of OHαThe H atom of the-H bond is transferred to the Cu center, this step gives the product PhCHO, a step complex216 → complexes217 has an energy barrier of 23.9kcal mol-1(ii) a Formation of complexes2After 17, the product PhCHO is substituted by TEMPO to form a complex3,118, followed by transfer of the H atom from the Cu-H bond to TEMPO; for the triplet state, this H atom transfer step needs to overcome 44.8kcal mol-1Energy barrier of (d); however, for the open shell singlet state, the transition state1uTS18-19Direct formation of complexes119; therefore, the H atom transfer step is likely to occur on the open shell singlet potential surface; then, O2Formation of complex 6 instead of Hen; then, in the catalyst regeneration step, complex 6 → complex 9; same procedure in Path A and Path B, en and H2O instead of H2O2And PhCH2O-Regenerating the complex 0; the energy distribution of the calculated path D is detailed in fig. 7.
Similarly, the calculated energy span δ E and TOF value of the path D are 52.1kcal mol respectively-1And 1.4X 10- 22h-1. The results clearly demonstrate the infeasibility of pathway D and suggest that the en molecule does not participate in the catalytic cycle.
(4) The conversion efficiency was calculated.
Calculating the energy span delta E of catalytic reaction of the path A, the path B and the path C based on an energy span model according to the calculated reaction potential energy surface of the dominant path of the path A, the path B and the path C, and calculating the conversion frequency TOF value of the catalyst by the following formula:

wherein: kbBoltzmann constant, h planck constant, R ideal gas constant, T reaction temperature, δ E energy span of reaction path;
delta E of route A is 27.2kcal mol-1Delta E of route B is 36.1kcal mol-1δ E of pathway C is 52.1kcal mhol-1Path a is the dominant path; TOF values of the path Ah, the path B and the path C calculated according to the formula are respectively 3.89h-1、1.2×10-4h-1And 1.4X 10-22h-1Wherein the TOF value of the path A is 5.40h from the experimental TOF value-1The fit was very good.
In aqueous alkaline solutions, the Density Functional Theory (DFT) studies the catalytic mechanism of alcohol oxidation to the corresponding benzaldehyde, which includes four pathways (pathway a → D). Calculations indicate that route a is the most favorable route (δ E ═ 27.2kcal mol)-1;TOF=3.89h-1). The calculation result shows that: the zwitter-ionic property of the catalyst does not influence the alcohol oxidation process of the system. ② the catalytic cycle comprises catalyst activation, substrate oxidation and catalyst regeneration. ③ the reaction path A is a dominant path, and the conversion frequency (TOF) is 3.89h-1Consistent with the experimental results (TOF 5.40 h)-1). The rate control step is a substrate oxidation step, i.e. a process in which hydrogen atoms are transferred from the alkoxide to oxygen atoms of TEMPO.
All calculations in this invention are performed with the Gaussian 09 series of known complexes [ (L) Cu [ ]II(Hen)(H2O)]Thus, the bond length and bond angle of the complex are optimized by using M06L, B3LYP, OPBE and M06 (see Table S2), and the M06L functional is finally selected in consideration of the accuracy of the result and the calculation cost.
TABLE S2

In the gas phase, the meta-GGA M06L functional and the 6-31+ G (d) basis set were used for geometric optimization and frequency calculation. All minimum values have no virtual frequency, and the transition state has only one virtual frequency. To further determine the energy, single point energy was calculated using M06L/6-311+ G (d, p) with SMD solvent model and water as the solvent for the gas phase optimized structure, re-optimized by SMD model using solvent (water) for comparison of some key intermediates and transition states, and single point energy calculation was performed at the same level, structure and energy barrier and M06L/6-311+ G (d, p)/SMD (H)2O)// M06L/6-31+ G (d) consensus (see Table S1).
TABLE S1

Moreover, to ensure that the conclusions are not influenced by the calculation method used, key intermediates and transition states (see in particular II alcohol oxidation) were also recalculated using different DFT methods. Furthermore, TOF in path A is M06/6-311+ G (d, p)/SMD (H)2O)// M06L/6-31+ G (d) for details in Table S3B).
TABLE S3B

The IRC method is used to identify the transition state, connect two related minima, and derive the spin placement from Mulliken spin analysis in single point energy calculations. For antiferromagnetically coupled Cu complexes (open-shell singlet), in order to avoid unstable wave functions, use is made of stress Mix and stress (Mix, Always). Some antiferromagnetically coupled open shell layers account for the singlet state leading to a degree of spin contamination. Therefore, the energy correction is derived from Heisenberg spin-Hamiltonian formalisms and is estimated as follows: deltaST=2(ES–ET)/2–<S2>SWherein ESIs the energy of the open shell singlet state, ETIs the energy of the triplet state and is,<S2>Sis spin contamination of the open shell singlet state. The subscript S represents the open-shell singlet state and T the triplet state.
It should be understood that the above-mentioned embodiments of the present invention are only examples for clearly illustrating the present invention, and are not intended to limit the embodiments of the present invention, and it will be obvious to those skilled in the art that other variations or modifications may be made on the basis of the above description, and all embodiments may not be exhaustive, and all obvious variations or modifications may be included within the scope of the present invention.
Claims (5)
1. The research and analysis method for the reaction mechanism of oxidizing benzyl alcohol into benzaldehyde by using the amphoteric water-soluble catalyst is characterized by comprising the following steps of:
(1) constructing a calculation model of the catalyst activity center structure: based on the formula [ (L) CuII(Hen)(H2O)]TEMPO is reaction condition information provided by an experiment for oxidizing benzyl alcohol into benzaldehyde by oxygen in the presence of a catalyst, and a calculation model of a catalyst active center is constructed; performing geometric optimization by adopting a density functional method, and comprehensively considering the configuration stability, the energy level, the calculation result and the calculation cost according to the calculation result to select an optimal model;
searching 809721.cif files of CCDC-809721 from a Cambridge crystal database in England, and reading X-ray crystallography data provided by the 809721.cif files through Materials Studio software to initially construct a catalytic precursor model; the bond length and bond angle of the constructed preliminary catalytic precursor model were adjusted: the bond length of all Cu-N bonds is increased Increased bond length of Cu-O bond
Increased bond length of Cu-O bond Modulation of N-N bond
Modulation of N-N bond S-O bond increase
S-O bond increase Modulation of C-N bond
Modulation of C-N bond Modulation of C-C bond
Modulation of C-C bond The bond angle of O-Cu-N and O-Cu-O is increased by 0.5-1.5 degrees; optimizing the bond length, bond angle and dihedral angle of the complex by combining M06L, B3LYP, OPBE and M06 functional groups with 6-31+ G (d) group based on the adjusted structure;
The bond angle of O-Cu-N and O-Cu-O is increased by 0.5-1.5 degrees; optimizing the bond length, bond angle and dihedral angle of the complex by combining M06L, B3LYP, OPBE and M06 functional groups with 6-31+ G (d) group based on the adjusted structure;
calculated Cu-N1 at the level of M06L functional,The maximum difference between the bond lengths of Cu-N2, Cu-O1, Cu-O2 and Cu-O3 and the corresponding bond lengths in the experiment is The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1 degree;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1 degree;
the maximum difference between the bond length of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3 calculated at the level of B3LYP functional and the corresponding bond length of the experiment is The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1.8 degrees;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 1.8 degrees;
the bond length calculated at the level of OPBE functional is within the experimental error The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 2.6 degrees;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 2.6 degrees;
the maximum difference between the bond lengths of Cu-N1, Cu-N2, Cu-O1, Cu-O2 and Cu-O3 calculated at the level of M06 functional and the corresponding bond lengths in the experiment is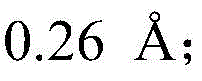 The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 0.9 degrees;
The maximum difference between the bond angles of O1-Cu-N2 and O2-Cu-N2 and the corresponding bond angles in the experiment is 0.9 degrees;
determining the result at the level of M06L/6-31+ G (d) according to the calculation result and in consideration of the precision and calculation cost of the M06L functional;
(2) designing a reaction path;
performing possible reaction path design according to the optimal model selected in the step (1); then carrying out reaction intermediate and transition state modeling:
(a) construction of Cα-intermediates and transition states of H bond cleavage:
modeling an intermediate: the three-coordinate main ligand and the substrate alkoxide exactly form a four-coordinate structure of a Cu center, no vacancy is left for a TEMPO free radical, and the TEMPO free radical is coordinated to the Cu center in a hydrogen bond mode during modelingA second coordination layer of Cu active center, the length range of hydrogen bond is set in And setting Cu-O and Cu-N bond length ranges respectively
And setting Cu-O and Cu-N bond length ranges respectively And
And after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
after the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
modeling a transition state: set by flexible scanning methods, i.e. Cα-H bond length from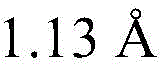 To be provided with
To be provided with Increase the amplification to
Increase the amplification to Calculating the energy of each step, finding out the corresponding structure of the highest point of the energy, and then adjusting and establishing C in the modelαDistances of-H and O-H, respectively, are set at
Calculating the energy of each step, finding out the corresponding structure of the highest point of the energy, and then adjusting and establishing C in the modelαDistances of-H and O-H, respectively, are set at And
And obtaining an optimized structure after geometric optimization, and carrying out frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Cα-H-O, proving that the transition state is correctly sought;
obtaining an optimized structure after geometric optimization, and carrying out frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Cα-H-O, proving that the transition state is correctly sought;
(b) construction of intermediates and transition states for O-H bond cleavage:
modeling an intermediate: principal ligand and OH-A four-coordinate structure constituting a Cu center, the Cu-OH bond length being adjusted to Cu-OLThe bond length is
Cu-OLThe bond length is Cu-ONThe bond length is
Cu-ONThe bond length is Adding substrate alcohol to the center of four-coordinate Cu, wherein the distance between H on substrate alcohol OH and O of OH is set
Adding substrate alcohol to the center of four-coordinate Cu, wherein the distance between H on substrate alcohol OH and O of OH is set After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
modeling a transition state: adjusting the key bond length to make the Cu-OH bond length from Is adjusted to
Is adjusted to Mixing O withsub-H bond length from
Mixing O withsub-H bond length from Is adjusted to
Is adjusted to The bond length of H-OH is increased from
The bond length of H-OH is increased from Is adjusted to
Is adjusted to Performing transition state geometric optimization on the adjusted transition state model to obtain an optimized structure, and performing frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Osub-H-OH, proving that the transition state is correctly sought;
Performing transition state geometric optimization on the adjusted transition state model to obtain an optimized structure, and performing frequency calculation to obtain a virtual frequency; and analyzing the vibration direction of the virtual frequency to conform to Osub-H-OH, proving that the transition state is correctly sought;
(c) constructing intermediates and transition states generated by OO-H bonds:
modeling an intermediate: principal ligand and O2A four-coordinate structure constituting the Cu center, and adjusting Cu-OO-The bond length is Cu-OLThe bond length is
Cu-OLThe bond length is TEMPOH-OO·-A distance of
TEMPOH-OO·-A distance of After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
modeling a transition state: adjusting the key bond length, adjusting Cu-OO-Key length is from Is adjusted to
Is adjusted to Cu-OLKey length is from
Cu-OLKey length is from Is adjusted to
Is adjusted to TEMPOH-OO·-Distance from
TEMPOH-OO·-Distance from Is adjusted to
Is adjusted to H-OO·-Key length is from
H-OO·-Key length is from Is adjusted to
Is adjusted to Performing transition state geometric optimization and frequency calculation on the adjusted transition state model to obtain a transition state structure with one virtual frequency; further analyzing that the vibration direction of the virtual frequency is consistent with OO--H-TEMPO, proving that the transition state is correctly sought;
Performing transition state geometric optimization and frequency calculation on the adjusted transition state model to obtain a transition state structure with one virtual frequency; further analyzing that the vibration direction of the virtual frequency is consistent with OO--H-TEMPO, proving that the transition state is correctly sought;
(d) construction of H2O2Intermediate and transition states generated:
modeling an intermediate: the main ligand and OOH form a four-coordination structure of a Cu center, and substrate alcohol is coordinated through a hydrogen bond; adjusting the Cu-OOH bond length to Cu-OLThe bond length is
Cu-OLThe bond length is PhCH2OH-OOH distance of
PhCH2OH-OOH distance of After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
After the setting is finished, carrying out geometric optimization to obtain a corresponding intermediate structure;
modeling a transition state: adjusting key length, adjusting PhCH2OH-OOH bond length of Is adjusted to
Is adjusted to Cu-OLKey length is from
Cu-OLKey length is from Is adjusted to
Is adjusted to PhCH2O-H distance from
PhCH2O-H distance from Is adjusted to
Is adjusted to Carrying out geometric optimization and frequency calculation on the transition state model to obtain a transition state structure with one virtual frequency; further analyzing that the vibration direction of the virtual frequency accords with PhCH2O-H-OOH, which proves that the transition state is correctly searched;
Carrying out geometric optimization and frequency calculation on the transition state model to obtain a transition state structure with one virtual frequency; further analyzing that the vibration direction of the virtual frequency accords with PhCH2O-H-OOH, which proves that the transition state is correctly searched;
based on the generation and the breakage form of chemical bonds and the coordination condition of ligands, adopting a density functional method to carry out geometric structure adjustment on reactants, intermediates, transition states and products on each designed reaction path, then carrying out geometric optimization at the level of M06L/6-31+ G (d) calculation, obtaining the activation energy and reaction enthalpy change of each elementary reaction, and drawing a corresponding potential energy surface diagram;
during calculation, a wave function stability test is carried out, the geometrical structures of an intermediate body of an open shell layer single state and a transition state are calculated by adopting a symmetrical damage method, and energy correction is carried out; analyzing from the thermodynamic and kinetic angles to obtain a potential energy surface diagram, determining an optimal reaction path and a speed control step thereof, comparing with an experimental result, performing reliability analysis, determining a reaction mechanism and an optimal activity center structure of a catalyst, and calculating a reaction potential energy surface under the participation of water by adopting a real solvent model;
carrying out geometric optimization on an intermediate and a transition state on a potential energy surface by using a meta-GGA M06L functional and a 6-31+ G (d) group to obtain a stable structure, and then calculating the frequency by using the meta-GGA M06L functional and the 6-31+ G (d) group, wherein all minimum values have no virtual frequency, the transition state is ensured to have only one virtual frequency, and the intermediate has no virtual frequency; reading the Gibbs Thermal correction value after Thermal correction to Gibbs Free Energy; to get more accurate energy, further calculate single point energy at the M06L/6-311+ G (d, p)/SMD calculation level and water as solvent for gas phase optimized structure; the single point energy calculation input file is as follows: # uM06L/6-311+ G srf (solvent water, SMD) get (available check force) read (available int ultra), finally get output file of single-point calculation of gaussian log, and read the energy after scfdone: E (uM06L) in log file; adding the Gibbs thermal correction value to the single-point energy to obtain Gibbs free energy of each structure;
IRC methods are used to confirm the transition state; for antiferromagnetically coupled Cu complexes, i.e., open shell singlet, stable wave functions were obtained using guss ═ Mix and guss ═ Mix, Always; since some antiferromagnetically coupled open shells account for singlet states leading to a degree of spin contamination, energy correction is performed according to the following formula: deltaST=2(ES–ET)/2–<S2>SIn which ESIs the energy of the open shell singlet state, ETIs the energy of the triplet state and is,<S2>Sis spin contamination of the open shell singlet, subscript S denotes the open shell singlet, T denotes the triplet;
(3) analyzing a reaction mechanism;
analyzing the transition state and the bond length and the bond angle of the intermediate and the stereochemical change around the central metal ion in each elementary reaction, and summarizing the coordination number of the central metal ion, the spatial configuration of the ligand, the bond length and the bond angle of the ligand and the central metal ion in the reaction process and the change rule of corresponding structural parameters;
Cu-OH bond length during O-H bond cleavage Is increased to
Is increased to Osub-H bond length from
Osub-H bond length from Is increased to
Is increased to H-OH bond length of
H-OH bond length of Is increased to
Is increased to Center metalCuIIThe periphery is a three-coordinate main ligand, an OH ligand and a substrate alcohol in Cu axial coordination; after the O-H bond is broken, a three-coordinate main ligand H is formed2Tetrahedrally coordinated Cu of O and alkoxide coordinationIIAn active center structure; subsequently, CαH bond cleavage process Cu-OsubKey length is from
Center metalCuIIThe periphery is a three-coordinate main ligand, an OH ligand and a substrate alcohol in Cu axial coordination; after the O-H bond is broken, a three-coordinate main ligand H is formed2Tetrahedrally coordinated Cu of O and alkoxide coordinationIIAn active center structure; subsequently, CαH bond cleavage process Cu-OsubKey length is from Is increased to
Is increased to Csub-H bond length from
Csub-H bond length from Is increased to
Is increased to TEMPO-H bond length of
TEMPO-H bond length of Is reduced to
Is reduced to Through CαFormation of Cu coordinated with a tridentate primary ligand, the product benzaldehyde and TEMPOH after the H bond cleavage processIIAn active center structure; subsequently, Cu-OO. during the formation of OO-H bonds-Key length is from
Through CαFormation of Cu coordinated with a tridentate primary ligand, the product benzaldehyde and TEMPOH after the H bond cleavage processIIAn active center structure; subsequently, Cu-OO. during the formation of OO-H bonds-Key length is from Reduced to
Reduced to Cu-OLKey length is from
Cu-OLKey length is from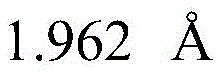 Is increased to
Is increased to TEMPOH-OO·-Distance from
TEMPOH-OO·-Distance from Is increased to
Is increased to H-OO·-Key length is from
H-OO·-Key length is from Reduced to
Reduced to Forming a main ligand of three coordination and Cu of OOH coordination after the formation process of OO-H bondIIAn active center structure; finally, H2O2PhCH in Generation step2OH-OOH bond length of
Forming a main ligand of three coordination and Cu of OOH coordination after the formation process of OO-H bondIIAn active center structure; finally, H2O2PhCH in Generation step2OH-OOH bond length of Reduced to
Reduced to Cu-OLKey length is from
Cu-OLKey length is from Reduced to
Reduced to PhCH2O-H distance from
PhCH2O-H distance from Is increased to
Is increased to Through H2O2Formation of Cu coordinated with a tridentate primary ligand and alkoxide after the formation stepIIAn active center structure;
Through H2O2Formation of Cu coordinated with a tridentate primary ligand and alkoxide after the formation stepIIAn active center structure;
respectively calculating key elements in optimal reaction paths of different catalytic systems by adopting a natural bond orbit method and a molecular orbit theory to obtain electronic structure information of a transition state and an intermediate, and determining the bonding characteristics of a central metal ion and a ligand; the effect of solvent water on the reaction history was studied from a microscopic perspective at the molecular level: analyzing the influence of water on the intermediate, the geometry, the electronic structure and the bonding characteristic of a transition state and the influence of water on the reaction activation energy of the speed control step after the water is added;
(4) the conversion efficiency was calculated.
2. The method for analyzing the reaction mechanism of oxidizing benzyl alcohol into benzaldehyde with oxygen by using the amphoteric water-soluble catalyst according to claim 1, wherein in the step (2): with [ (L) CuII(Hen)(H2O)]In the reaction of oxidizing benzyl alcohol into benzaldehyde by taking TEMPO as a catalyst: ([ (L) Cu)II(Hen)(H2O)]) TEMPO and an alkaline aqueous solution K are respectively used as a catalyst precursor, a cocatalyst and alkali2CO3A solution with the concentration of 0.1 mol/L; catalyst precursor ([ (L) Cu)II(Hen)(H2O)]) Has an isoelectric point of 4.96, the pH value of the solution is 11, and the catalyst precursor ([ (L) Cu)II(Hen)(H2O)]) In its anionic form [ (L) CuII(en)(H2O)](ii) present; in view of the hydrolysis of potassium carbonate, HCO3 -,CO3 2-And-three ions of OH are present in the solution, and Cu shown in the following formula (I) is present in the solution at the beginning of the reactionIIThe complex is as follows:

wherein: in the formula (I), Gibbs free energy values are shown in parentheses in kcal mol-1;
Catalyst precursor [ (L) CuII(en)(H2O)]The complex is named 0, and OH is used as the complex-,CO3 2-And HCO3 -Exchange of ethylenediamine and water to form Cu, respectivelyIIComplexes 1, 10 and 13, from the calculated Gibbs free energy values, gave the above-mentioned CuIIThe most stable structure in the complex is complex 1; in view of all Cu in formula (I)IIThe complex can be subjected to a catalyst activation step with a substrate alcohol, thereby giving reaction pathway A, reaction pathway B, reaction pathway C and reaction pathway D represented by the following formula (II):

for route a → C, only the catalyst activation step is different; in route A, the proton of benzyl alcohol is transferred to OH-Form H2O; in pathways B and C, protons are transferred to CO, respectively3 2-And HCO3 -Formation of HCO3 -And H2CO3(ii) a In route D, the proton of benzyl alcohol is transferred to en, while benzyl alcohol CαThe H atoms of the H bonds are transferred to the Cu centers.
3. The method for analyzing the reaction mechanism of oxidizing benzyl alcohol into benzaldehyde with oxygen in the presence of the amphoteric water-soluble catalyst according to claim 2, wherein in the step (3):
(3-1) determining possible reaction paths by calculation, converging all structures optimizing transition states into reactants, further setting the distance of H-O atoms for flexible scanning, increasing the energy to 40kcal/mol along with the reduction of the H-O distance, and showing that protons cannot be transferred to HCO3 -Upper, i.e., pathway C is a reaction pathway that is not possible;
(3-2) analyzing the path A, the path B and the path D to determine the most favorable reaction path;
in path a:
A-I, catalytic activation step
In route A, the starting complex is [ Cu ]II(L)(OH)]2-Denoted as complex 1; benzyl alcohol and complexes21 combine to form a complex22, then protons of benzyl alcoholTransfer to OH-Energy barrier of 9.2kcal mol-1And the complexes formed23, i.e., [ (L) CuII(OCH2Ph)(H2O)]2-) (ii) a Then TEMPO is substituted for H2O to form active catalyst 4, in which case there are two possible isomer complexes34 and complexes34'; in the complex3In 4, TEMPO is coordinated to the second coordination sphere of the Cu center, and in the complex34' is coordinated to the first coordination sphere; it is because TEMPO coordination rotates oxygen on a ligand away from a Cu active center to cause structural distortion so that the complex34' energy ratio complex34 high 10.2kcal mol-1(ii) a Thus based on complexes34 is a favorable path;
A-II, alcohol Oxidation
After the formation of the active catalyst 4, H on the alcohol is transferred to the nitrogen atom or oxygen atom of TEMPO; the calculation result shows that the path of the H atom transferred to the oxygen atom in TEMPO has a lower energy barrier; to verify this conclusion, active catalyst 4 and transition state TS were synthesized using the M06 functional, the OPBE functional, and the TPSSh functional4a-5a_OAnd TS4a-5a_NThe calculation is carried out again; the calculations show that for the H-transfer step, consistent results are observed even with different DFT methods, i.e. the energy barrier for the transfer of H atoms to O atoms is lower than to N atoms; thus, the catalyst system is obtained, wherein H atoms are transferred to O atoms of TEMPO; the reaction involves a transition from the S-1 ground state to the S-0 ground state1μTS4-5_OSpin crossing of (3), i.e., a two-state reaction; the reaction can be subjected to a complex14 and complexes34, and the probability of spin flipping due to spin-crossings from S-1 to S-0; due to the complex14 and complexes3The band gap between 4 is 1.3kcal mol-1A spin balance can be established so that a spin crossover occurs; from complexes34 to transition state1uTS4-5_OHThe energy barrier of the transfer step is 16.1kcal mol-1Subsequent production of benzaldehyde and TEMPOH;
A-III, catalyst regeneration
To complete the catalytic cycling reaction, TEMPOH and complexes21, regeneration is needed; assuming that the catalyst regeneration step has O2Participation; triplet O2The substituted benzaldehyde will produce a complex1,36; in the complex36 → complexes3The triplet state in the 7 process is the ground state; for the complex36,Cu、O2And the spin densities of the ligands were +0.55, +1.17 and +0.27, respectively, which means that the complexes36 has CuII-OO·-Structure; then, OO·-Can abstract H atom from TEMPOH to regenerate TEMPO, and can be used in complex37 generation of CuII-an OOH structure; this step is required to overcome 3.6kcal mol-1Energy barrier of (a), indicating that such H transfer is likely to occur; in the complex36 → complexes37 in the process, oxidation state of Cu is changed from CuIIReduction to CuI(ii) a After regeneration of TEMPO, another benzyl alcohol is substituted for TEMPO, followed by proton transfer from the benzyl alcohol to OOH-And form H2O2I.e. the compound37 → complexes29; OH-substituted H2O2And PhCH2O-Regeneration of Complex 1, with H2O2Can be decomposed into H2O+1/2O2(ii) a Calculating the energy distribution of the path A;
in path B: in route B, the starting structure is [ Cu ]II(L)(CO3)]3-Denoted as complex 10; from complex 10, protons are transferred from benzyl alcohol to CO3 2-The radicals forming complexes212([(L)CuII(OCH2Ph)(HCO3)]3-) (ii) a TEMPO substituted HCO3 -Thereafter, an active catalyst complex 4 is produced; then after the alcohol oxidation step, i.e. Complex 4 → Complex 5 and the catalyst regeneration step, i.e. Complex 6 → Complex 9, CO3 2-Substitution of substance for H2O2And PhCH2O-Regenerating the complex 10; the calculated energy distribution of the path B;
in path D: in route D, the starting structure is [ (L) CuII(H2O)(en)]-Is denoted as a complex0; starting from complex 0, benzyl alcohol replaces H2O forming a complex216, i.e., [ (L) Cu)II(HOCH2Ph)(en)]-(ii) a Followed by compounding in step (b)216 → complexes2In 17, PhCH2Protons of OH are transferred to en, with PhCH2C of OHαThe H atom of the-H bond is transferred to the Cu center, this step gives the product PhCHO, a step complex216 → complexes217 has an energy barrier of 23.9kcal mol-1(ii) a Formation of complexes2After 17, the product PhCHO is substituted by TEMPO to form a complex3,118, followed by transfer of the H atom from the Cu-H bond to TEMPO; for the triplet state, this H atom transfer step needs to overcome 44.8kcal mol-1Energy barrier of (d); however, for the open shell singlet state, the transition state1uTS18-19Direct formation of complexes119; therefore, the H atom transfer step is likely to occur on the open shell singlet potential surface; then, O2Formation of complex 6 instead of Hen; then, in the catalyst regeneration step, complex 6 → complex 9; same procedure in Path A and Path B, en and H2O instead of H2O2And PhCH2O-Regenerating the complex 0; the calculated energy distribution of path D.
4. The method for analyzing the reaction mechanism of oxidizing benzyl alcohol into benzaldehyde with oxygen by using the amphoteric water-soluble catalyst according to claim 3, wherein in the step (4): calculating the energy span delta E of catalytic reaction of the path A, the path B and the path C based on an energy span model according to the calculated reaction potential energy surface of the dominant path of the path A, the path B and the path C, and calculating the conversion frequency TOF value of the catalyst by the following formula:

wherein: kbBoltzmann constant, h planck constant, R ideal gas constant, T reaction temperature, δ E energy span of reaction path;
delta E of route A is 27.2kcal mol-1Delta E of route B is 36.1kcal mol-1δ E of route C is 52.1kcal mol-1Path a is the dominant path; TOF values of the path A, the path B and the path C calculated according to the formula are respectively 3.89h-1、1.2×10-4h-1And 1.4X 10-22h-1Wherein the TOF value of the path A is 5.40h from the experimental TOF value-1The fit was very good.
5. The method for analyzing the reaction mechanism of oxidizing benzyl alcohol into benzaldehyde with oxygen by using the amphoteric water-soluble catalyst according to claim 4, wherein in the step (3): the TDTS and TDI on the path A potential surface are respectively an intermediate with the highest energy transition state and the lowest energy on the potential surface; analyzing the geometric structure and the electronic structure of the TDTS of the path A in the determined rate transition state in view of the fact that the TDTS is a key factor influencing TOF; firstly, reading an output file of a geometric structure through Gaussview software to obtain the bond length, the bond angle and the stereochemical change condition around a central metal ion; secondly, calculating to obtain a Weber bond WBI based on a natural bond orbit method NBO, and further analyzing the bonding and breaking conditions of chemical bonds; finally, reading the chk file of the single-point result of the TDTS structure by adopting Gaussview software, and analyzing by a visual module in a current surface analysis module to obtain a molecular orbit diagram to obtain two single-electron single-occupied orbit SOMOs; analyzing by combining a spin density analysis result to obtain the bonding characteristics of the central metal ions and the ligands in the TDTS step;
in Path A, the rate transition state TDTS is determined to be the transition state1uTS4-5_ODue to a transition state1uTS4-5_OIs a transition state of the H transfer step, i.e. complex 4 → TS4-5_OTherefore, the complex was analyzed14 → transition state1uTS4-5_O→ complex1Electron transfer process during 5; for the complex14, Cu, ligand, TEMPO and substrate-OCH2The spin densities of Ph are +0.57, +0.32, -1.00, and+0.11, which means that the complex14 contains [ (L) (TEMPO) CuII(OCH2Ph)]Structure; in the transition state1uTS4-5_OIn the above formula, the spin density of Cu is +0.32, the spin density of TEMPOH is-0.32,-OCH2the spin density of Ph is-0.17, and the spin density of the ligand is + 0.17; binding complexes14 → transition state1uTS4-5_OMolecular orbital analysis of the process, resulting in the formation of TEMPO-H bond moieties and-OCH2c of Phα-H bond occurrence partial homolytic cleavage; a part of the beta spintronics on the substrate increases, namely from 0.0% → 33.1%; the part of the beta-spin electrons is derived from-OCH2C of Phα-homolytic cleavage of H bonds; also, from-OCH2C of PhαThe corresponding alpha spin electrons generated by homolytic cleavage of the H bond are transferred to TEMPO, resulting in an increase in the alpha spin density of TEMPO, i.e. from 0.0% → 15.3%; in the complex14 → transition state1uTS4-5_OIn the process, Cα-H bond weakening, TEMPO-H bond strengthening; in the complex14 → transition state1uTS4-5_OIn the process, Cu-OsubBonds are not homolytic but in a transition state1uTS4-5_O→ complex15 in process, Cu-OsubThe bond is homolytic and finally benzaldehyde and TEMPOH are obtained.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810022826.6A CN108256286B (en) | 2018-01-10 | 2018-01-10 | Research and analysis method for reaction mechanism of aerobic oxidation of benzyl alcohol into benzaldehyde by using amphoteric water-soluble catalyst |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810022826.6A CN108256286B (en) | 2018-01-10 | 2018-01-10 | Research and analysis method for reaction mechanism of aerobic oxidation of benzyl alcohol into benzaldehyde by using amphoteric water-soluble catalyst |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN108256286A CN108256286A (en) | 2018-07-06 |
| CN108256286B true CN108256286B (en) | 2021-07-02 |
Family
ID=62726284
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201810022826.6A Active CN108256286B (en) | 2018-01-10 | 2018-01-10 | Research and analysis method for reaction mechanism of aerobic oxidation of benzyl alcohol into benzaldehyde by using amphoteric water-soluble catalyst |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN108256286B (en) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109406737A (en) * | 2018-12-21 | 2019-03-01 | 齐鲁师范学院 | A kind of BN investigation of materials method |
| CN111696619B (en) * | 2019-03-13 | 2023-06-20 | 赣南师范大学 | Method for predicting influence degree of reaction environment on reaction activation energy |
| CN110364230B (en) * | 2019-08-21 | 2022-09-16 | 安阳工学院 | Method for rapidly screening organic base in reaction of preparing formic acid from carbon dioxide and hydrogen under catalysis of copper |
| CN110646562B (en) * | 2019-09-27 | 2021-06-08 | 华南理工大学 | TiO for revealing humidity influence2Method for photocatalytic degradation of polluted gas mechanism |
| CN110975936B (en) * | 2019-11-11 | 2023-01-03 | 桂林理工大学 | Copper-based catalytic system for efficiently catalyzing and oxidizing alcohol at room temperature without solvent and method thereof |
| CN113674811B (en) * | 2021-08-24 | 2024-08-06 | 西安热工研究院有限公司 | Method for calculating standard electrode potential in wet desulfurization slurry oxidation-reduction process |
| CN114067923A (en) * | 2021-11-17 | 2022-02-18 | 郑州轻工业大学 | Screening method for Lewis acid catalyzed diazo compound activation mode |
| CN114582431B (en) * | 2022-03-16 | 2023-03-17 | 内蒙古工业大学 | (bipy) Cu Ⅱ Reaction mechanism research and analysis method for catalytic oxidation of-TEMPO/organic base catalytic system |
| CN118005492B (en) * | 2024-04-08 | 2024-06-11 | 天津农学院 | Method for preparing benzaldehyde by friction catalytic oxidation of benzyl alcohol based on barium strontium titanyl oxalate |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1805695A (en) * | 2003-06-13 | 2006-07-19 | 菲利普莫里斯生产公司 | Catalyst to reduce carbon monoxide and nitric oxide from the mainstream smoke of a cigarette |
| CN103469284A (en) * | 2013-08-09 | 2013-12-25 | 内蒙古工业大学 | Preparation method of carbon nanotube/titania nanotube bio-composite coat material |
| CN103870644A (en) * | 2014-03-11 | 2014-06-18 | 中国石油大学(华东) | Method for identifying MoP catalyst denitrification active sites |
| CN107042108A (en) * | 2017-06-08 | 2017-08-15 | 杭州电子科技大学 | The method that Pt/BiOCl metal oxides prepare catalytic oxidation of benzyl alcohol as catalyst benzaldehyde |
| CN107330254A (en) * | 2017-06-16 | 2017-11-07 | 大连理工大学 | Predict organic matter and the quantitative structure activity relationship model of sulphuric acid free radical aqueous phase reactions speed constant in aqueous phase |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6743952B2 (en) * | 2002-03-28 | 2004-06-01 | Council Of Scientific And Industrial Research | Selective liquid phase air oxidation of toluene catalysed by composite catalytic system |
| US9901909B2 (en) * | 2015-02-24 | 2018-02-27 | California Institute Of Technology | Processes for preparing zincoaluminosilicates with AEI, CHA, and GME topologies and compositions derived therefrom |
-
2018
- 2018-01-10 CN CN201810022826.6A patent/CN108256286B/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1805695A (en) * | 2003-06-13 | 2006-07-19 | 菲利普莫里斯生产公司 | Catalyst to reduce carbon monoxide and nitric oxide from the mainstream smoke of a cigarette |
| CN103469284A (en) * | 2013-08-09 | 2013-12-25 | 内蒙古工业大学 | Preparation method of carbon nanotube/titania nanotube bio-composite coat material |
| CN103870644A (en) * | 2014-03-11 | 2014-06-18 | 中国石油大学(华东) | Method for identifying MoP catalyst denitrification active sites |
| CN107042108A (en) * | 2017-06-08 | 2017-08-15 | 杭州电子科技大学 | The method that Pt/BiOCl metal oxides prepare catalytic oxidation of benzyl alcohol as catalyst benzaldehyde |
| CN107330254A (en) * | 2017-06-16 | 2017-11-07 | 大连理工大学 | Predict organic matter and the quantitative structure activity relationship model of sulphuric acid free radical aqueous phase reactions speed constant in aqueous phase |
Non-Patent Citations (5)
| Title |
|---|
| "Cu-BTC及其衍生物在苯甲醇选择氧化反应中的催化活性";李和健等;《分子催化》;20170831;第31卷(第4期);第341-347页 * |
| "DFT studies on the mechanism of alcohol oxidation by (bpy)CuI-TEMPO/NMI catalytic system ";Lin Cheng等;《Dalton Transactions》;20150311(第16期);第1-12页 * |
| "Green Catalytic Oxidations in Water";Roger A.Sheldon;《Metal-Catalyzed Reactions in Water》;20131231;第139-172页 * |
| "Highly Practical Copper(I)/TEMPO Catalyst System for Chemoselective Aerobic Oxidation of Primary Alcohols";HOOVER,J.M等;《ChemInform》;20120301;第43卷(第13期);第1-2页 * |
| "Mechanistic Insight into the 2 Alcohol Oxidation Mediated by an Efficient CuI/L-Proline-TEMPO Catalyst—A Density Functional Theory Study";Siyu Li等;《Catalysts》;20170905;第7卷(第9期);第1-15页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN108256286A (en) | 2018-07-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108256286B (en) | Research and analysis method for reaction mechanism of aerobic oxidation of benzyl alcohol into benzaldehyde by using amphoteric water-soluble catalyst | |
| Roy et al. | Reduction of CO 2 by a masked two-coordinate cobalt (i) complex and characterization of a proposed oxodicobalt (ii) intermediate | |
| Niu et al. | Heterogeneous carbon nitride photocatalyst for C–C bond oxidative cleavage of vicinal diols in aerobic micellar medium | |
| Liu et al. | Unraveling molecular transformations on surfaces: a critical comparison of oxidation reactions on coinage metals | |
| Wu et al. | Intermetallic PdIn catalyst for CO 2 hydrogenation to methanol: mechanistic studies with a combined DFT and microkinetic modeling method | |
| Tan et al. | Catalytic oxidation of water and alcohols by a robust iron (III) complex bearing a cross-bridged cyclam ligand | |
| Jiang et al. | Reactivity of Neutral Tantalum Sulfide Clusters Ta3S n (n= 0–4) with N2 | |
| Wang et al. | Exploring the reaction mechanism of H2S decomposition with MS3 (M= Mo, W) clusters | |
| Jackson et al. | Weak-field ligands enable inert early transition metal oxides to convert methane to methanol: the case of ZrO | |
| Aniagyei et al. | A density functional theory study of the mechanisms of oxidation of ethylene by technetium oxo complexes | |
| Topolski et al. | Hydrogen evolution from water reactions with molybdenum sulfide cluster anions | |
| Gallego et al. | Sub-nanometer Copper Clusters as Alternative Catalysts for the Selective Oxidation of Methane to Methanol with Molecular O2 | |
| CN110364230A (en) | A kind of quick screening copper catalysis carbon dioxide reacted with hydrogen formic acid in organic base method | |
| Pembere et al. | Guerbet coupling of methanol catalysed by titanium clusters | |
| CN114582431B (en) | (bipy) Cu Ⅱ Reaction mechanism research and analysis method for catalytic oxidation of-TEMPO/organic base catalytic system | |
| Qin et al. | Effect of the bipodal Ti species on activity and selectivity of propylene epoxidation with H2O2 over TS-1: A theoretical study | |
| CN113707230A (en) | Method for calculating Gibbs free energy change in liquid acid dissociation process | |
| Aniagyei et al. | A density functional theory study of the mechanisms of oxidation of ethylene by rhenium oxide complexes | |
| CN113707226B (en) | Method for establishing micro-dynamics model during interconversion of xylose-xylulose | |
| Dalessandro et al. | Hydrogenation of CO2 to CH3OH catalyzed by a ruthenium-triphos complex: Theoretical free energy profile and microkinetic modeling | |
| Xu et al. | Chemical Activation of Water Molecule by Collision with Spin–Orbit-State-Selected Vanadium Cation: Quantum-Electronic-State Control of Chemical Reactivity | |
| Zhang et al. | A neural network potential energy surface for the F+ H 2 O↔ HF+ OH reaction and quantum dynamics study of the isotopic effect | |
| Kraemer et al. | SCF LCAO MO studies on the hydration of ions: The system F−· 2H 2 O | |
| Wang et al. | Theoretical study of partial oxidation of ethylene by vanadium trioxide cluster cation | |
| Schilling et al. | How ab initio molecular dynamics can change the understanding on transition metal catalysed water oxidation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |