CN108004259B - Chimeric antigen receptor targeting B cell maturation antigen and uses thereof - Google Patents
Chimeric antigen receptor targeting B cell maturation antigen and uses thereof Download PDFInfo
- Publication number
- CN108004259B CN108004259B CN201610943985.0A CN201610943985A CN108004259B CN 108004259 B CN108004259 B CN 108004259B CN 201610943985 A CN201610943985 A CN 201610943985A CN 108004259 B CN108004259 B CN 108004259B
- Authority
- CN
- China
- Prior art keywords
- sequence
- human
- amino acid
- seq
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/70517—CD8
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/17—Lymphocytes; B-cells; T-cells; Natural killer cells; Interferon-activated or cytokine-activated lymphocytes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/7051—T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70596—Molecules with a "CD"-designation not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/02—Fusion polypeptide containing a localisation/targetting motif containing a signal sequence
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/03—Fusion polypeptide containing a localisation/targetting motif containing a transmembrane segment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/33—Fusion polypeptide fusions for targeting to specific cell types, e.g. tissue specific targeting, targeting of a bacterial subspecies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/70—Fusion polypeptide containing domain for protein-protein interaction
- C07K2319/74—Fusion polypeptide containing domain for protein-protein interaction containing a fusion for binding to a cell surface receptor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/10041—Use of virus, viral particle or viral elements as a vector
- C12N2740/10043—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Cell Biology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Toxicology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biomedical Technology (AREA)
- Gastroenterology & Hepatology (AREA)
- General Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Virology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Physics & Mathematics (AREA)
- Epidemiology (AREA)
- Plant Pathology (AREA)
- Pharmacology & Pharmacy (AREA)
- Microbiology (AREA)
- Developmental Biology & Embryology (AREA)
- Peptides Or Proteins (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
The invention relates to a chimeric antigen receptor targeting BCMA (J22.9) and application thereof, in particular to a polynucleotide sequence selected from (1) a coding sequence containing an anti-BCMA single-chain antibody, a coding sequence of a human CD8 α hinge region, a coding sequence of a human CD8 transmembrane region, a coding sequence of a human 41BB intracellular region and (2) a complementary sequence of the polynucleotide sequence, wherein the anti-BCMA single-chain antibody, the coding sequence of the human CD8 α hinge region, the coding sequence of the human CD8 transmembrane region, and the coding sequence of the human 41BB intracellular region are connected in sequence, and the invention also provides a related fusion protein, a vector containing the coding sequence, and application of the.
Description
Technical Field
The invention belongs to the field of chimeric antigen receptors, and particularly relates to a BCMA (brain cell activating antigen) targeted chimeric antigen receptor and application thereof.
Background
Multiple myeloma is a malignant plasma cell disease, which is characterized by malignant clonal proliferation of bone marrow plasma cells, secretion of monoclonal immunoglobulin or a fragment thereof (M protein), and damage to relevant target organs or tissues such as bones and kidneys, and is commonly and clinically manifested by bone pain, anemia, renal insufficiency, infection and the like [ Multiple myelotoma.N Engl J Med,2011.364(11): p.1046-60 ]. Multiple myeloma is the second most serious malignancy in the blood system, accounting for 10% of the malignancy in the blood system, and is frequently developed in men, the incidence rate of which increases year by year with the increase of age, and the multiple myeloma is more likely to become younger in recent years [ Siegel, R., et al, Cancer statistics,2014.CA Cancer J Clin,2014.64(1): p.9-29 ].
The B Cell Maturation Antigen (BCMA), also called CD269, consists of 184 amino acid residues, the intracellular region of which contains 80 amino acid residues and has a very short sequence, and only one carbohydrate recognition domain is a B cell surface molecule. BCMA belongs to the type I transmembrane signal protein lacking a signal peptide, and is a member of the Tumor necrosis factor receptor family (TNFR), which binds to the B cell activator factor BAFF or proliferation-induced ligand (APRIL), respectively [ Tumor necrosis factor receptor ligand-receptor binding. current Opin Struct Biol,2004.14(2): p.154-60 ]. In normal tissues, BCMA is expressed on the surface of mature B cells and plasma cells, BCMA knockout mice have normal immune system, normal spleen structure, normal development of B lymphocytes but obviously reduced plasma cell number, and the BCMA plays an important role in maintaining the survival of plasma cells, and the mechanism mainly comprises the combination of BCMA and BAFF protein, and up-regulation of anti-apoptosis genes Bcl-2, Mcl-1, Bclw and the like, and the maintenance of cell growth [ BCMA is the important for the survival of the low-live bone marrow cells plasma. J Exp Med,2004.199(1): p.91-8 ]. Similarly, this mechanism also functions in myeloma cells and plays an important role in promoting malignant proliferation of myeloma cells [ BAFF and APRIL protected myelomas cells from apoptosis induced by interferon 6 depletion and demethasone. blood,2004.103(8): p.3148-57 ]. It has been shown that BCMA is ubiquitously expressed in multiple myeloma cell lines and that detection in multiple myeloma patients also gives consistent results [ Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. blood,2004.103(2): p.689-94 ]. Based on the reports, Kochenderfer et al further applied Q-PCR, Flow Cytometry and immunohistochemistry to study the expression characteristics of BCMA, and confirmed that BCMA is not expressed in normal human tissues except mature B cells and plasma cells, and also not expressed in CD34+ hematopoietic cells [ B-cell growth inhibitor is a developmental target for additive T-cell therapy of multiple myocyte. Clin Cancer Res,2013.19(8): p.2048-60 ]. Combined with the high similarity of BCMA expression profiles to CD19, and the successful progress of anti-CD 19CAR T cell therapy, suggests that our BCMA can be used as one of the CAR-T cell targets for cellular immunotherapy of multiple myeloma.
Chimeric Antigen Receptor-T cell (CAR-T) T cell refers to a T cell that is genetically modified to recognize a specific Antigen of interest in an MHC non-limiting manner and to continuously activate expanded T cells. The international cell therapy association (interna) in 2012 indicates that biological immune cell therapy has become a fourth means for treating tumors besides surgery, radiotherapy and chemotherapy, and will become a necessary means for treating tumors in the future. CAR-T cell back-infusion therapy is the most clearly effective form of immunotherapy in current tumor therapy. A large number of studies show that the CAR-T cells can effectively recognize tumor antigens, cause specific anti-tumor immune response and remarkably improve the survival condition of patients.
Chimeric Antigen Receptors (CARs) are a core component of CAR-T, conferring on T cells the ability to recognize tumor antigens in an HLA-independent manner, which enables CAR-engineered T cells to recognize a broader range of targets than native T cell surface receptor TCRs. The basic design of a CAR includes a tumor-associated antigen (TAA) binding region (usually the scFV fragment from the antigen binding region of a monoclonal antibody), an extracellular hinge region, a transmembrane region, and an intracellular signaling region. The choice of antigen of interest is a key determinant for the specificity, efficacy of the CAR and safety of the genetically engineered T cells themselves.
With the continuous development of Chimeric Antigen Receptor T cell (CAR-T) technology, CAR-T can be divided into four generations.
The first generation CAR-T cells consist of an extracellular binding domain-single chain antibody (scFV), a transmembrane domain (TM), and an intracellular signaling domain-Immunoreceptor Tyrosine Activation Motif (ITAM), wherein the chimeric antigen receptor portions are linked as follows: scFv-TM-CD3 ζ. Although some specific cytotoxicity could be seen in the first generation CARs, it was found to be less effective when summarized in 2006 in clinical trials. The reason for this is because the first generation of CAR-T cells are rapidly depleted in the patient and have so poor persistence that CAR-T cells already apoptotic when they have not yet come into contact with a large number of tumor cells can elicit an anti-tumor cytotoxic effect, but rather less cytokine secretion, but their short survival time in vivo fails to elicit a persistent anti-tumor effect [ simple g2D-modified T cells inhibition system T-cell lymphoma growth in a mannenrinating multiple cytokines and cytotoxic pathways, Cancer 2007, 67 (22): 11029 vs 11036).
Optimization of T cell activation signaling regions in CAR design of second generation CAR-T cells remains a hotspot of research. Complete activation of T cells relies on dual signaling and cytokine action. Wherein the first signal is a specific signal initiated by the recognition of an antigen peptide-MHC complex on the surface of an antigen presenting cell by the TCR; the second signal is a co-stimulatory signal. Second generation CAR appeared as early as 1998 (J Immunol.1998; 161 (6): 2791-7). The 2 nd generation CAR adds a costimulatory molecule in the intracellular signal peptide region, namely the costimulatory signal is assembled into the CAR, and can better provide an activation signal for CAR-T cells, so that the CAR can simultaneously activate the costimulatory molecule and the intracellular signal after identifying tumor cells, double activation is realized, and the proliferation and secretion capacity of the T cells and the anti-tumor effect can be obviously improved. The first well-studied T cell costimulatory signal receptor was CD28, which was capable of binding to a B7 family member on the surface of target cells. Co-stimulation of CD28 promotes T cell proliferation, IL-2 synthesis and expression, and enhances T cell resistance to apoptosis. Costimulatory molecules such as CD134(OX40) and 41BB (4-1BB) are subsequently presented to increase cytotoxicity and proliferative activity of T cells, maintain T cell responses, prolong T cell survival, and the like. Such second generation CARs produced unexpected results in subsequent clinical trials, with shaking frequently triggered since 2010 based on clinical reports of second generation CARs, with complete remission rates of up to 90% and above, especially for relapsed, refractory ALL patients.
The third generation CAR signal peptide region integrates more than 2 costimulatory molecules, which can lead the T cells to continuously activate and proliferate, lead the cytokines to be continuously secreted, and lead the capability of the T cells to kill tumor cells to be more remarkable, namely, the new generation CAR can obtain stronger anti-tumor response (Mol ther., 2005, 12 (5): 933-941). Most typically, U Pen Carl June is added with a 41BB stimulating factor under the action of CD28 stimulating factor.
The fourth generation CAR-T cells are supplemented with cytokines or co-stimulatory ligands, for example the fourth generation CAR can produce IL-12, which can modulate the immune microenvironment-increase the activation of T cells, and simultaneously activate innate immune cells to act to eliminate target antigen negative cancer cells, thus achieving a bi-directional regulatory effect [ TRUCKs: the four generation of cars, Expert Opin Biol ther, 2015; 15(8): 1145-54 ].
A successful treatment of 1 relapsing refractory Multiple Myeloma (MM) patient with CD19 molecular targeted CAR-T cell therapy was published by Carl June research group in New England journal, top-level journal of the medical community at 9.2015 [ Chimeric Antigen Receptor T Cells against CD19for Multiple Myeloma.N Engl JMed,2015.373(11): p.1040-7 ]. While multiple myeloma does not typically express CD19 as a B cell line tumor, CD19 is not a target for multiple myeloma immunotherapy. A trace of multiple myeloma clones with drug resistance and disease recurrence properties have been reported to have a B cell phenotype (i.e., CD19 positive). After it was clear that BCMA could be a target for CAR T cells, the american national cancer institute Kochenderfer et al successfully constructed anti-BCMA CAR T cells, and preclinical studies showed that the CAR T cells specifically recognized BCMA, and after activation by BCMA, amplified in large amounts, secreted cytokines and exerted killing function, and also had anti-tumor effect in mouse tumorigenesis models [ B-cell growth inhibitors i s apoptosis target for adaptive T-cell therapy of multiple myelomas. Phase I clinical studies of anti-BCMA CAR T cell therapy for multiple myeloma were performed by the national cancer institute in 2014, verifying the clinical safety and efficacy of anti-BCMA CAR T cells against multiple myeloma patients that are unresponsive to current standard treatment protocols (Cl inorganic trials. gov Identifier: NCT 02215967). At 57 th American blood year date, beginning at 12 months of 2015, the professor team of Syed Abbas Al I, national cancer institute medical oncology, has reported phase I clinical trial results for CAR-T cell therapy in multiple myeloma patients. The study included 12 patients with refractory relapsed multiple myeloma who failed over-line 3 chemotherapy, with some patients having more than 50% myeloma cells in their bone marrow. After infusion of BCMA CAR-T cells into these patients, 1 patient was completely relieved, 3 patients were partially relieved, and the rest were stable, thus demonstrating for the first time that anti-BCMA CAR-T cell therapy was effective in multiple myeloma without major side effects and was evaluated as one of the most influential clinical studies in the year of ASH (Late-Breaking extracts, Abstract number of university: LAB-1). Currently, the Abelmoscon cancer center, university of Abelmoschus, has also registered a phase I clinical trial of anti-BCMA CART cells for the treatment of multiple myeloma and was in the development of a Ting Rong Drum study (clinical trials. gov Identifier: NCT 02546167).
The BCMA-J22.9 antibody is a human-murine chimeric antibody that serves as a target for targeted Multiple Myeloma (MM) immunotherapy, and this high affinity antibody blocks BCMA binding to the natural ligand B cell activator (BAFF) or proliferation-induced ligand (APRIL). The amino acid residues of BCMA recognized by this antibody are positions 6-41. The experimental result shows that the antibody has good specificity in experiments such as Elisa and flow detection. The results of in vivo experiments showed that intraperitoneal injection of BCMA-J22.9 antibody reduced tumor burden and significantly prolonged survival in mice. BCMA-J22.9 chimeric antibody therapy is not limited to the treatment of Multiple Myeloma (MM), as BCMA is also an immune target for the treatment of autoimmune diseases such as systemic lupus erythematosus or rheumatoid arthritis [ buttera latent-tumor by targeting B cell growth antigen (BCMA) in a molar of multiple myeloma.1tmol oncol.2015aug; 9(7):1348-58].
Our patent uses the heavy chain and light chain of scFv of BCMA-J22.9 antibody as the structure of CAR, and the CART cell in vitro experiment of the invention shows strong killing effect on target cells. The preclinical experimental result lays a good foundation for clinical experiments and clinical treatment.
Disclosure of Invention
In a first aspect, the present invention provides a polynucleotide sequence selected from the group consisting of:
(1) a polynucleotide sequence comprising the coding sequence of an anti-BCMA single-chain antibody, the coding sequence of a human CD8 α hinge region, the coding sequence of a human CD8 transmembrane region, the coding sequence of a human 41BB intracellular region, the coding sequence of a human CD3 zeta intracellular region, and optionally the coding sequence of a fragment of EGFR comprising extracellular domain III and extracellular domain IV, which are linked in sequence, and
(2) (1) the complement of the polynucleotide sequence.
In one or more embodiments, the polynucleotide sequence further comprises a coding sequence for a signal peptide prior to the coding sequence for the anti-BCMA single chain antibody in one or more embodiments, the amino acid sequence of the signal peptide is represented by amino acids 1-21 of SEQ ID NO. 2 in one or more embodiments, the amino acid sequence of the light chain variable region of the anti-BCMA single chain antibody is represented by amino acids 22-128 of SEQ ID NO. 2 in one or more embodiments, the amino acid sequence of the heavy chain variable region of the anti-BCMA single chain antibody is represented by amino acids 144-263 of SEQ ID NO. 2 in one or more embodiments, the amino acid sequence of the hinge region of human CD8 α is represented by amino acids 264-310 of SEQ ID NO. 2 in one or more embodiments, the amino acid sequence of the transmembrane region of human CD8 is represented by amino acids 311 of SEQ ID NO. 2 in one or more embodiments, the amino acid sequence of the intracellular domain of SEQ ID NO. 332 of SEQ ID NO. 41 is represented by amino acids 333-380 of SEQ ID NO. 2 in one or more embodiments, the amino acid sequence of amino acids 381 of SEQ ID NO. 380-380.
In one or more embodiments, the coding sequence for the signal peptide preceding the coding sequence for the anti-BCMA single-chain antibody is represented by the nucleotide sequence at positions 1-63 of SEQ ID NO.1 in one or more embodiments, the coding sequence for the light chain variable region of the anti-BCMA single-chain antibody is represented by the nucleotide sequence at positions 64-384 of SEQ ID NO.1 in one or more embodiments, the coding sequence for the heavy chain variable region of the anti-BCMA single-chain antibody is represented by the nucleotide sequence at positions 430-789 of SEQ ID NO.1 in one or more embodiments, the coding sequence for the hinge region of human CD8 α is represented by the nucleotide sequence at positions 790-930 of SEQ ID NO.1 in one or more embodiments, the coding sequence for the transmembrane region of human CD8 is represented by the nucleotide sequence at positions 996 of SEQ ID NO.1 in one or more embodiments, the coding sequence for the intracellular region of human 41BB is represented by the nucleotide sequence at positions 997 of SEQ ID NO.1 in one or more embodiments, the coding sequence at positions 1140 of the intracellular region of SEQ ID NO.1, the fusion protein of SEQ ID NO. 1143, the first fusion protein of SEQ ID NO.1, wherein the coding sequence of SEQ ID NO.1, the fusion protein:
(1) a fusion protein comprising an anti-BCMA single-chain antibody, a human CD8 α hinge region, a human CD8 transmembrane region, a human 41BB intracellular region and a human CD3 zeta intracellular region linked in this order, and
(2) a fusion protein derived from (1) by substituting, deleting or adding one or more amino acids in the amino acid sequence defined in (1) and retaining the activity of activated T cells;
preferably, the anti-BCMA single chain antibody is anti-BCMA monoclonal antibody J22.9.
In one or more embodiments, the fusion protein further comprises a signal peptide at the N-terminus of the anti-BCMA single chain antibody, in one or more embodiments, the amino acid sequence of the signal peptide is as set forth in amino acids 1-21 of SEQ ID NO 2 in one or more embodiments, the amino acid sequence of the light chain variable region of the anti-BCMA single chain antibody is as set forth in amino acids 22-128 of SEQ ID NO 1 in one or more embodiments, the amino acid sequence of the heavy chain variable region of the anti-BCMA single chain antibody is as set forth in amino acids 144 of SEQ ID NO 1 in one or more embodiments, the amino acid sequence of the human CD8 α hinge region is as set forth in amino acids 264-310 of SEQ ID NO 1 in one or more embodiments, the amino acid sequence of the human CD8 transmembrane region is as set forth in amino acids 311 of SEQ ID NO 1-332 of SEQ ID NO 1 in one or more embodiments, the amino acid sequence of the human CD8 transmembrane region is as set forth in amino acids 311 of SEQ ID NO 1-333 of SEQ ID NO 1 in one or more embodiments, the polynucleotide constructions provided in the polynucleotide sequences of SEQ ID NO 1, 380, the polynucleotide constructions of the invention.
In one or more embodiments, the nucleic acid construct is a vector. In one or more embodiments, the nucleic acid construct is a retroviral vector comprising a replication initiation site, a 3 'LTR, a 5' LTR, a polynucleotide sequence described herein, and optionally a selectable marker.
In a fourth aspect, the invention provides a retrovirus containing a nucleic acid construct as described herein, preferably containing the vector, more preferably containing the retroviral vector.
In a fifth aspect, the invention provides a genetically modified T cell comprising a polynucleotide sequence as described herein, or comprising a nucleic acid construct as described herein, or infected with a retrovirus as described herein, or stably expressing a fusion protein as described herein and optionally a fragment of EGFR comprising extracellular domain III, extracellular domain IV and optionally a transmembrane region.
In a sixth aspect, the invention provides a pharmaceutical composition comprising a genetically modified T cell as described herein.
In a seventh aspect, the invention provides the use of a polynucleotide sequence, fusion protein, nucleic acid construct or retrovirus as described herein in the preparation of an activated T cell.
In an eighth aspect, the invention provides the use of a polynucleotide sequence, fusion protein, nucleic acid construct, retrovirus, or genetically modified T cell as described herein, or a pharmaceutical composition thereof, in the manufacture of a medicament for the treatment of a BCMA mediated disease.
In one or more embodiments, the BCMA-mediated disease is multiple myeloma.
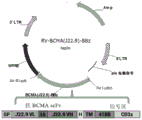
Drawings
FIG. 1 is a schematic representation of a BCMA-CAR retroviral expression vector (BCMA-41 BBz). SP: a signal peptide; VL: a light chain variable region; and Lk: joint (G)4S)3VH is the heavy chain variable region, H is the CD8 α hinge region, TM is the CD8 transmembrane region.
FIG. 2 is a peak plot of the partial sequencing results of the BCMA-CAR retroviral expression vector (BCMA-41 BBz).
FIG. 3 shows the BCMA (J22.9) CART and BCMA (C11d5.3) CART expression efficiencies of retroviral-infected T cells for 72 hours by flow cytometry.
Figure 4 shows target cell BCMA expression by flow cytometry.
FIG. 5 is a graph of CD107a expression prepared by co-culturing 5-day BCMA (J22.9) CART and BCMA (C111D5.3) CART cells with target cells for 4 hours.
FIG. 6 shows INF γ secretion by 5-day preparation of BCMA (J22.9) CART and BCMA (C111D5.3) CART cells co-cultured with target cells for 4 hours.
FIG. 7 is a graph of the killing effect on tumor cells after 5 days of preparation of BCMA (J22.9) CART and BCMA (C111D5.3) CART cells co-cultured with target cells for 5 hours.
Detailed Description
The invention provides a Chimeric Antigen Receptor (CAR) targeting BCMA, wherein the CAR comprises fragments of an anti-BCMA single-chain antibody, a human CD8 α hinge region, a human CD8 transmembrane region, a human 41BB intracellular region and a human CD3 zeta intracellular region which are connected in sequence.
anti-BCMA single chain antibodies suitable for use in the present invention can be derived from a variety of anti-BCMA monoclonal antibodies known in the art.
Optionally, the light chain variable region and the heavy chain variable region may be linked together by a linker sequence. Such single chain antibodies that may be exemplified include, but are not limited to, C11D5.3, J22.9. In certain embodiments, the monoclonal antibody is the monoclonal antibody clone No. J22.9. In certain embodiments, the amino acid sequence of the variable region of the light chain of the anti-BCMA single chain antibody is represented by amino acid residues 22 to 128 of SEQ ID No. 2. In other embodiments, the amino acid sequence of the heavy chain variable region of the anti-BCMA single chain antibody is as shown in amino acid residues 144-263 of SEQ ID NO: 2.
The amino acid sequence of the human CD8 α hinge region suitable for use in the present invention can be shown as amino acids 264 and 310 of SEQ ID NO 2.
The human CD8 transmembrane region suitable for use in the present invention can be the various human CD8 transmembrane region sequences commonly used in the art for CARs. In certain embodiments, the amino acid sequence of the transmembrane region of human CD8 is depicted as amino acids 311-332 of SEQ ID NO 2.
The 41BB suitable for use in the present invention can be any of the various 41 BBs known in the art for use in CARs. As an illustrative example, the present invention uses the 41BB shown in the amino acid sequence 333-380 of SEQ ID NO: 2.
The intracellular domain of human CD3 ζ suitable for use in the present invention may be various intracellular domains of human CD3 ζ conventionally used in CARs in the art. In certain embodiments, the amino acid sequence of the intracellular domain of human CD3 ζ is as set forth in amino acids 381-491 of SEQ ID NO 2.
The above-mentioned portions forming the fusion protein of the present invention, such as the light chain variable region and the heavy chain variable region of an anti-BCMA single chain antibody, the human CD8 α hinge region, the human CD8 transmembrane region, 41BB, and the human CD3 zeta intracellular region, etc., may be directly linked to each other, or may be linked by a linker sequence, which may be a linker sequence known in the art, such as a linker sequence comprising G and S, typically, a linker comprises one or more motifs repeating back and forth, e.g., GGGS, GGGGS, SSSGS, GSGSA, and GGSGG, preferably, the motifs are adjacent in the linker sequence without intervening amino acid residues between the repeats, the linker sequence may comprise 1, 2,3, 4, or 5 repeating motifs, the length of the linker may be 3 to 25 amino acid residues, e.g., 3 to 15, 5 to 15, 10 to 20 amino acid residues, in some embodiments, the linker sequence is a polyglycine linker sequence, the number of glycine residues in the linker sequence is not particularly limited, typically 2, 2 to 2 amino acid residues, e.g., 15 to 20 amino acid residues in the linker sequence, e.g., 2 to 15 amino acid residues in the linker sequence, e.g., 15 amino acid residues in the amino acid sequence (amino acid sequence), and the amino acid sequence of the heavy chain variable region of the linker sequence of the present invention, e.g., preferably, L, the amino acid sequence of the present invention, and the heavy chain variable region of the present invention may comprise amino acid sequence of the present invention (e.g., preferably, EnAnd (b) connecting, wherein n is an integer of 1-5.
It will be appreciated that in gene cloning procedures it is often necessary to design appropriate cleavage sites which will introduce one or more irrelevant residues at the end of the expressed amino acid sequence without affecting the activity of the sequence of interest. In order to construct a fusion protein, facilitate expression of a recombinant protein, obtain a recombinant protein that is automatically secreted outside of a host cell, or facilitate purification of a recombinant protein, it is often necessary to add some amino acids to the N-terminus, C-terminus, or other suitable regions within the recombinant protein, for example, including, but not limited to, suitable linker peptides, signal peptides, leader peptides, terminal extensions, and the like. Thus, the amino-terminus or the carboxy-terminus of the fusion protein of the invention (i.e., the CAR) may also contain one or more polypeptide fragments as protein tags. Any suitable label may be used herein. For example, the tag may be FLAG, HA, HA1, c-Myc, Poly-His, Poly-Arg, Strep-TagII, AU1, EE, T7, 4A6, ε, B, gE, and Ty 1. These tags can be used to purify proteins.
The invention also includes the CAR shown in the amino acid sequence of SEQ ID NO. 2 at positions 22-491 or a mutant of the CAR shown in SEQ ID NO. 2 at positions 1-491. These mutants include: an amino acid sequence that has at least 80%, preferably at least 85%, preferably at least 90%, preferably at least 95%, preferably at least 97% sequence identity to the CAR and retains the biological activity (e.g., activating T cells) of the CAR. Sequence identity between two aligned sequences can be calculated using, for example, BLASTp from NCBI.
Mutants also include amino acid sequences having one or several mutations (insertions, deletions or substitutions) in the amino acid sequence shown in positions 22-491 of SEQ ID No. 2, the amino acid sequence shown in positions 1-491 of SEQ ID No. 2 or the amino acid sequence shown in SEQ ID No. 2, while still retaining the biological activity of the CAR, said several mutations typically referring to within 1-10, such as 1-8, 1-5 or 1-3 substitutions are preferably conservative substitutions, e.g. in the art conservative substitutions with similarly performing or similar amino acids do not typically alter the function of the protein or polypeptide.
The present invention includes polynucleotide sequences encoding the fusion proteins of the present invention. The polynucleotide sequences of the invention may be in the form of DNA or RNA. The form of DNA includes cDNA, genomic DNA or artificially synthesized DNA. The DNA may be single-stranded or double-stranded. The DNA may be the coding strand or the non-coding strand. The invention also includes degenerate variants of the polynucleotide sequences encoding the fusion proteins, i.e., nucleotide sequences which encode the same amino acid sequence but differ in nucleotide sequence.
The polynucleotide sequences described herein can generally be obtained by PCR amplification. Specifically, primers can be designed based on the nucleotide sequences disclosed herein, particularly open reading frame sequences, and the relevant sequences can be amplified using commercially available cDNA libraries or cDNA libraries prepared by conventional methods known to those skilled in the art as templates. When the sequence is long, two or more PCR amplifications are often required, and then the amplified fragments are spliced together in the correct order. For example, in certain embodiments, the polynucleotide sequence encoding the fusion proteins described herein is as set forth in nucleotides 64 to 1473 of SEQ ID NO.1, or as set forth in nucleotides 1 to 1473 of SEQ ID NO. 1.
In certain embodiments, the polynucleotide sequences of the invention further comprise nucleotide sequences encoding fragments of EGFR.
Thus, in certain embodiments, the polynucleotide sequence of the invention comprises a coding sequence for a CAR of the invention, a coding sequence for a P2A polypeptide, a coding sequence for a signal peptide from the chain of GM-CSF receptor α, and a coding sequence for tEGFR.
The control sequence may be an appropriate promoter sequence. The promoter sequence is typically operably linked to the coding sequence of the protein to be expressed. The promoter may be any nucleotide sequence which shows transcriptional activity in the host cell of choice including mutant, truncated, and hybrid promoters, and may be obtained from genes encoding extracellular or intracellular polypeptides either homologous or heterologous to the host cell. The control sequence may also be a suitable transcription terminator sequence, a sequence recognized by a host cell to terminate transcription. The terminator sequence is operably linked to the 3' terminus of the nucleotide sequence encoding the polypeptide. Any terminator which is functional in the host cell of choice may be used in the present invention. The control sequence may also be a suitable leader sequence, a nontranslated region of an mRNA which is important for translation by the host cell. The leader sequence is operably linked to the 5' terminus of the nucleotide sequence encoding the polypeptide. Any terminator which is functional in the host cell of choice may be used in the present invention.
In certain embodiments, the nucleic acid construct is a vector. Expression of a polynucleotide sequence of the invention is typically achieved by operably linking the polynucleotide sequence to a promoter and incorporating the construct into an expression vector. The vector may be suitable for replication and integration into eukaryotic cells. Typical cloning vectors contain transcriptional and translational terminators, initiation sequences, and promoters that may be used to regulate the expression of the desired nucleic acid sequence.
The polynucleotide sequences of the present invention can be cloned into many types of vectors. For example, it can be cloned into plasmids, phagemids, phage derivatives, animal viruses and cosmids. Further, the vector is an expression vector. The expression vector may be provided to the cell in the form of a viral vector. Viral vector technology is well known in the art and is described, for example, in Sambrook et al (2001, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, New York) and other virology and Molecular biology manuals. Viruses that can be used as vectors include, but are not limited to, retroviruses, adenoviruses, adeno-associated viruses, herpes viruses, and lentiviruses.
Generally, suitable vectors comprise an origin of replication, a promoter sequence, a convenient restriction enzyme site, and one or more selectable markers that function in at least one organism (e.g., WO 01/96584; WO 01/29058; and U.S. Pat. No. 6,326,193).
For example, in certain embodiments, the invention uses a retroviral vector that contains a replication initiation site, a 3 'LTR, a 5' LTR, polynucleotide sequences described herein, and optionally a selectable marker.
Another example of a suitable promoter is the extended growth factor-1 α (EF-1 α). however, other constitutive promoter sequences can also be used, including but not limited to the simian virus 40(SV40) early promoter, mouse breast cancer virus (MMTV), Human Immunodeficiency Virus (HIV) Long Terminal Repeat (LTR) promoter, the MoLV promoter, the avian leukemia virus promoter, the EB virus immediate early promoter, the rous sarcoma virus promoter, and human gene promoters such as but not limited to the actin promoter, myosin promoter, heme promoter, and creatine kinase promoter.
To assess the expression of the CAR polypeptide or portion thereof, the expression vector introduced into the cells can also comprise either or both of a selectable marker gene or a reporter gene to facilitate identification and selection of expressing cells from a population of cells sought to be transfected or infected by the viral vector. In other aspects, the selectable marker may be carried on a separate piece of DNA and used in a co-transfection procedure. Both the selectable marker and the reporter gene may be flanked by appropriate regulatory sequences to enable expression in a host cell. Useful selectable markers include, for example, antibiotic resistance genes, such as neo and the like.
Suitable reporter genes may include genes encoding luciferase, β -galactosidase, chloramphenicol acetyltransferase, secreted alkaline phosphatase, or green fluorescent protein.
Methods for introducing and expressing genes into cells are known in the art. The vector may be readily introduced into a host cell by any method known in the art, for example, mammalian, bacterial, yeast or insect cells. For example, the expression vector may be transferred into a host cell by physical, chemical or biological means.
Physical methods for introducing polynucleotides into host cells include calcium phosphate precipitation, lipofection, particle bombardment, microinjection, electroporation, and the like. Biological methods for introducing a polynucleotide of interest into a host cell include the use of DNA and RNA vectors. Chemical means of introducing polynucleotides into host cells include colloidal dispersion systems such as macromolecular complexes, nanocapsules, microspheres, beads; and lipid-based systems including oil-in-water emulsions, micelles, mixed micelles, and liposomes.
Biological methods for introducing polynucleotides into host cells include the use of viral vectors, particularly retroviral vectors, which have become the most widely used method for inserting genes into mammalian, e.g., human, cells. Other viral vectors may be derived from lentiviruses, poxviruses, herpes simplex virus I, adenoviruses, adeno-associated viruses, and the like. Many virus-based systems have been developed for gene transfer into mammalian cells. For example, retroviruses provide a convenient platform for gene delivery systems. The selected gene can be inserted into a vector and packaged into a retroviral particle using techniques known in the art. The recombinant virus can then be isolated and delivered to the subject cells in vivo or ex vivo. Many retroviral systems are known in the art. In some embodiments, an adenoviral vector is used. Many adenoviral vectors are known in the art. In one embodiment, a lentiviral vector is used.
Thus, in certain embodiments, the invention also provides a retrovirus for activating T cells, the virus comprising a retroviral vector as described herein and corresponding packaging genes, such as gag, pol and vsvg.
T cells suitable for use in the present invention may be of various types from various sources. For example, T cells may be derived from PBMCs of B cell malignancy patients.
In certain embodiments, after T cells are obtained, activation may be stimulated with an appropriate amount (e.g., 30-80 ng/ml, such as 50ng/ml) of CD3 antibody prior to culturing in an appropriate amount (e.g., 30-80 IU/ml, such as 50IU/ml) of IL2 medium for use.
The CAR-T cells of the invention can undergo robust in vivo T cell expansion and sustained at high levels in the blood and bone marrow for extended amounts of time, and form specific memory T cells.
The anti-tumor immune response elicited by the CAR-T cells can be an active or passive immune response. Additionally, the CAR-mediated immune response can be part of an adoptive immunotherapy step, in which the CAR-T cells induce an immune response specific for the antigen-binding portion in the CAR.
Thus, the diseases that can be treated with the CARs, their coding sequences, nucleic acid constructs, expression vectors, viruses, and CAR-T cells of the invention are preferably BCMA-mediated diseases.
The CAR-modified T cells of the invention can be administered alone or as a pharmaceutical composition in combination with diluents and/or with other components such as relevant cytokines or cell populations. Briefly, a pharmaceutical composition of the invention may comprise CAR-T cells as described herein, in combination with one or more pharmaceutically or physiologically acceptable carriers, diluents, or excipients. Such compositions may include buffers such as neutral buffered saline, sulfate buffered saline, and the like; carbohydrates such as glucose, mannose, sucrose or dextran, mannitol; a protein; polypeptides or amino acids such as glycine; an antioxidant; chelating agents such as EDTA or glutathione; adjuvants (e.g., aluminum hydroxide); and a preservative.
The pharmaceutical compositions of the present invention may be administered in a manner suitable for the disease to be treated (or prevented). The amount and frequency of administration will be determined by such factors as the condition of the patient, and the type and severity of the patient's disease.
When referring to an "immunologically effective amount", "an anti-tumor effective amount", "a tumor-inhibiting effective amount", or a "therapeutic amount", the precise amount of the composition of the invention to be administered can be determined by a physician, taking into account the age, weight, tumor size, extent of infection or metastasis, and individual differences in the condition of the patient (subject). It can be generally pointed out that: pharmaceutical compositions comprising T cells described herein can be in the range of 104To 109Dosage of individual cells/kg body weight, preferably 105To 106Dosage of individual cells/kg body weight. The T cell composition may also be administered multiple times at these doses. Cells can be administered by using infusion techniques well known in immunotherapy (see, e.g., Rosenberg et al, New Eng.J.of Med.319:1676, 1988). Optimal dosages and treatment regimens for a particular patient can be readily determined by those skilled in the medical arts by monitoring the patient for signs of disease and adjusting the treatment accordingly.
Administration of the subject composition may be carried out in any convenient manner, including by spraying, injection, swallowing, infusion, implantation or transplantation. The compositions described herein can be administered to a patient subcutaneously, intradermally, intratumorally, intranodal, intraspinally, intramuscularly, by intravenous injection, or intraperitoneally. In one embodiment, the T cell composition of the invention is administered to a patient by intradermal or subcutaneous injection. In another embodiment, the T cell composition of the invention is preferably administered by intravenous injection. The composition of T cells can be injected directly into the tumor, lymph node or site of infection.
In some embodiments of the invention, the CAR-T cells of the invention or compositions thereof can be combined with other therapies known in the art. Such therapies include, but are not limited to, chemotherapy, radiation therapy, and immunosuppressive agents. For example, treatment may be in conjunction with radiation or chemotherapeutic agents known in the art for the treatment of BCMA mediated diseases.
Herein, "anti-tumor effect" refers to a biological effect that can be represented by a reduction in tumor volume, a reduction in tumor cell number, a reduction in the number of metastases, an increase in life expectancy, or an improvement in various physiological symptoms associated with cancer.
"patient," "subject," "individual," and the like are used interchangeably herein and refer to a living organism, such as a mammal, that can elicit an immune response. Examples include, but are not limited to, humans, dogs, cats, mice, rats, and transgenic species thereof.
The present invention adopts the gene sequence of anti-BCMA antibody (specifically, scFv derived from clone number J22.9), and searches the gene sequence information of human CD8 α hinge region, human CD8 transmembrane region, human 41BB intracellular region and human CD3 zeta intracellular region from NCBI GenBank database, synthesizes chimeric antigen receptor anti-BCMA scFv-CD8 hinge region-CD 8TM-41BB-CD3 zeta gene fragment by whole gene synthesis, inserts into retroviral vector, recombinant plasmid packages virus in 293T cell, infects T cell, makes T cell express the chimeric antigen receptor.
The present invention is described in further detail by referring to the following experimental examples. These examples are provided for illustrative purposes only and are not intended to be limiting unless otherwise specified. Accordingly, the present invention should in no way be construed as limited to the following examples, but rather should be construed to include any and all variations which become apparent in light of the teachings provided herein. The methods and reagents used in the examples are, unless otherwise indicated, conventional in the art.
Example 1 determination of the sequence of the mBCMA (J22.9) scFv-CD8 α -41BB-CD3 zeta Gene
1.1 search of sequence information of human CD8 α hinge region, human CD8 α transmembrane region, human 41BB intracellular region and human CD3 zeta intracellular region from NCBI website database, anti-BCMA (J22.9) single chain antibody clone number J22.9https://sg.idtdna.com/siteThe codon optimization is carried out, so that the expression of the human cells is more suitable under the condition of unchanging the coded amino acid sequence.
The information of each amino acid and gene sequence is shown in SEQENCE LISTING (SEQINCE ID NO. 1).
The sequences are connected according to the sequence of anti-BCMA (J22.9) scFv, human CD8 α hinge region gene, human CD8 α transmembrane region gene, human 41BB intracellular region gene and human CD3 zeta intracellular region gene in sequence, and enzyme cutting sites are introduced at the sequence connection positions to form complete mBCMA (J22.9) -CAR gene sequence information.
1.2 sequencing of recombinant plasmids
The recombinant plasmid is sent to Shanghai Biotechnology Limited for sequencing, and the sequencing result is compared with the sequence of the synthesized mBCMA (J22.9) -CAR to verify whether the sequence is correct. The sequencing primer is as follows:
sense of justice AGCATCGTTCTGTGTTGTCTC
Antisense TGTTTGTCTTGTGGCAATACAC
The plasmid map constructed in this example is shown in FIG. 1. FIG. 2 shows a partial sequencing peak plot of the retroviral expression plasmid.
Example 2: construction of viral vectors comprising the nucleic acid sequence of the CAR molecule
The nucleotide sequence of the CAR molecule prepared in example 1 was double-digested with NotI (NEB) and EcoRI (NEB), ligated with T4 ligase (NEB) into the NotI-EcoRI site of the retroviral prero vector, transformed into competent e.coli (DH5 α), and after correct sequencing, plasmids were extracted and purified using a plasmid purification kit from Qiagen, and 293T cells were transfected with the plasmid calcium phosphate method for plasmid purification for retroviral packaging experiments.
Example 3: retroviral packaging
1. Day 1 293T cells should be less than 20 passages, but overgrown. Plating the cells in 0.6 x 10^6cells/ml, adding 10ml of DMEM medium into a 10cm dish, fully mixing the cells, and culturing at 37 ℃ overnight;
2. on day 2, 293T cells are transfected to a confluence of about 90% (usually, plating for about 14-18 h); plasmid complexes were prepared with 12.5ug of retrointerbone, 10ug of Gag-pol, 6.25ug of VSVg, CaCl for each plasmid2250ul,H2O is 1ml, and the total volume is 1.25 ml; in another tube, an equal volume of HBS to plasmid complex was added, and the plasmid complex was vortexed for 20 seconds. Adding the mixture into a 293T dish gently along the edge, culturing at 37 ℃ for 4h, removing the culture medium, washing with PBS once, and adding the preheated fresh culture medium again;
3. day 4: after transfection for 48h, the supernatant was collected, filtered through a 0.45um filter, split-charged and stored at-80 ℃, and preheated fresh DMEM medium was added continuously.
Example 4: retroviral infection of human T cells
1. Separating with Ficcol separation solution (tertiary sea of Tianjin) to obtain relatively pure CD3+ T cells, and adjusting cell density to 1 × 10 with medium containing 5% AB serum X-VIVO (LONZA)6and/mL. The cells were inoculated at 1 ml/well with anti-human 50ng/ml CD3 antibody (Beijing Hokkimei) and 50ng/ml CD28 antibody (Beijing Hokkimei), and 100IU/ml interleukin 2 (Beijing double Lut) was added to stimulate the culture for 48 hours to cause viral infection.
Every other day after T cell activation culture, the non-ti ssue treated plates were coated with 250. mu.l/well of retronectin (Takara) diluted in PBS to a final concentration of 15. mu.g/ml. Protected from light and kept at 4 ℃ overnight for use.
And 3, after the T cells are activated and cultured for two days, taking out 2 coated 24-well plates, sucking and removing the coating solution, adding HBSS containing 2% BSA, and sealing at room temperature for 30 min. The volume of blocking solution was 500. mu.l per well, and the blocking solution was aspirated and the plate washed twice with HBSS containing 2.5% HEPES.
4. Adding the virus solution into each well, adding 2ml of virus solution into each well, centrifuging at 32 ℃ for 2000g, and centrifuging for 2 h.
5. The supernatant was discarded and activated T cells 1 × 10 were added to each well of a 24-well plate6The volume is 1ml, and the culture medium is T cell culture medium added with IL-2200 IU/ml. Centrifuge at 30 ℃ for 10min at 1000 g.
6. After centrifugation, the plates were incubated at 37 ℃ in a 5% CO2 incubator.
7. 24h after infection, the cell suspension was aspirated and centrifuged at 1200rpm, 4 ℃ for 7 min.
8. After the cells are infected, the density of the cells is observed every day, and a T cell culture solution containing IL-2100 IU/ml is supplemented at a proper time to maintain the density of the T cells at 5 × 105Cells were expanded at around/ml.
Thus, CART cells each infected with the retrovirus shown in example 3, named BCMA CART cells (expressing the BCMA CAR of example 1), were obtained
Example 5: flow cytometry for detecting proportion of infected T lymphocytes and expression of surface CAR protein
And respectively centrifuging to collect CAR-T cells and NT cells (control group) 72 hours after infection, washing with PBS 1 time, discarding supernatant, adding corresponding antibody, washing with PBS 30min in the dark, resuspending, and finally detecting the CAR positive rate by a flow cytometer. The antibody is anti-mouse IgG F (ab') antibody (Jackson Immunoresearch).
FIG. 3 shows that, 72 hours after T cells were infected with the retrovirus prepared in example 3, the expression efficiency of BCMA (J22.9) CART was 51.3% and that of BCMA (C11d5.3) CART was 85.3%
Fig. 4 shows that the percentage of BCMA in the target cells, which was detected by BCMA antibody, was 95.5% in U266 cells, demonstrating that the target cells are highly expressing BCMA.
Example 6: detection of CD107a expression following coculture of CAR-T cells with target cells
1. Adding CART/NT cells 2 x 10 to each V-bottom 96-well plate5Sum target cells (U266)/control cells (K562)2 x 105Resuspending to 100ul of IL-2 free X-VIVO complete medium, adding BD GolgiStop (containing monesin, 1. mu.l BD GolgiStop per 1ml medium), and adding 2ul CD107 per wellantibody a (1:50), incubated at 37 ℃ for 4 hours, and cells were collected.
2. The samples were centrifuged to remove the medium, washed once with PBS, 400g, and centrifuged at 4 ℃ for 5 minutes. The supernatant was discarded, and appropriate amounts of specific surface antibodies CD3, CD4, and CD8 were added to each tube, and the volume of the suspension was 100ul, followed by incubation for 30 minutes on ice in the absence of light.
3. Cells were washed 1 time with 3mL PBS per tube and centrifuged at 400g for 5 min. The supernatant was carefully aspirated.
4. The appropriate amount of PBS was resuspended and CD107a was detected by flow cytometry.
Shown in fig. 5. Fig. 5 shows that the percentage of CD107a secretion by BCMA (J22.9) CART cells in CD8 positive U266 cells was 45.1%, respectively, and the percentage of CD107a secretion by BCMA (J22.9) CART cells in CD4 positive U266 cells was 39.4%, respectively.
Example 7: INF-gamma secretion assay after co-culture of CAR-T cells with target cells
1. Taking prepared CAR-T cells, resuspending the CAR-T cells in Lonza culture medium, and adjusting the cell concentration to be 1 × 106/mL。
2. Each well of the experimental group contained either target cells (U266) or negative control cells (K562)2 × 105CAR-T cell 2 × 105200. mu.l of Lonza medium without IL-2. Mix well and add to 96-well plate. BD GolgiPlug (containing BFA, 1. mu.l BD GolgiPlug per 1ml cell culture medium) was added at the same time, mixed well and incubated at 37 ℃ for 5-6 hours. Cells were collected as experimental groups.
3. Cells were washed 1 time with 1mL of PBS per tube and centrifuged at 300g for 5 minutes. The supernatant was carefully aspirated or decanted.
After washing the cells with PBS, 250. mu.l/EP tube Fixation/Permeabilization solution was added and incubated at 4 ℃ for 20 minutes to fix the cells and rupture the membranes using 1 × BD Perm/WashTMbuffer washes cells 2 times, 1 mL/time.
5. Staining with intracellular factor, taking appropriate amount of IFN-gamma cytokine fluorescent antibody or negative control, and performing BD Perm/WashTMDiluting to 50 μ l with buffer, resuspending the fixed and ruptured cells with the antibody diluent, incubating at 4 deg.C in the dark for 30min, 1 × BD Perm/WashTMbuffer 1 mL/cell wash 2 times, then use PBS heavy suspension。
6. And (4) detecting by using a flow cytometer.
Shown in fig. 6. FIG. 6 shows that the percentage of INF- γ secretion by BCMA (J22.9) CART cells in U266 cells positive for CD8 was 8.11%, and the percentage of INF- γ secretion by BCMA (J22.9) CART cells in U266 cells positive for CD4 was 10.7%.
Example 8: detection of tumor-specific cell killing after Co-culture of CAR-T cells with target cells
K562 cells (negative control cells as target cells without BCMA target protein) were resuspended in serum-free medium (1640) adjusted to a cell concentration of 1 × 106Perml, the fluorescent dye BMQC (2,3,6,7-tetrahydro-9-bromomethyl-1H,5Hquinolizino (9,1-gh) coumarins) was added to a final concentration of 5. mu.M.
2. Mixing, and incubating at 37 deg.C for 30 min.
3. Centrifugation was carried out at 1500rpm for 5min at room temperature, the supernatant was discarded and the cells resuspended in cytotoxic medium (phenol red-free 1640+ 5% AB serum) and incubated for 60min at 37 ℃.
4. Fresh cytotoxic Medium cells were washed twice and resuspended in fresh cytotoxic Medium at a density of 1 × 106/ml。
U266 cells (containing BCMA target protein, as target cells) were suspended in PBS containing 0.1% BSA and adjusted to a concentration of 1 × 106/ml。
6. The fluorescent dye CFSE (fluorescent dye) (CFSE) was added to a final concentration of 1. mu.M.
7. Mixing, and incubating at 37 deg.C for 10 min.
8. After the incubation was completed, FBS in an equal volume to the cell suspension was added and incubated at room temperature for 2min to terminate the labeling reaction.
9. Cells were washed and resuspended in fresh cytotoxic medium at a density of 1 × 106/ml。
10. Effector T cells were washed and suspended in cytotoxic medium at a concentration of 5 × 106/ml。
11. In all experiments, the cytotoxicity of CAR-T cells was compared to that of uninfected negative control effector T cells (ntcells), and these effector T cells were from the same patient.
CAR-T and NT, according to effector cell: target cells were cultured in 5ml sterile test tubes (BDBiosciences) at a ratio of 10:1, 2:1, with two duplicate wells per group. In each co-culture group, 50,000 (50. mu.l) U266 cells were targeted, and 50,000K 562 cells (50. mu.l) were negative control cells. A panel was set up containing only U266 target cells and K562 negative control cells.
13. The co-cultured cells were incubated at 37 ℃ for 5 h.
14. After incubation was complete, cells were washed with PBS and immediately followed by rapid addition of 7-AAD (7-aminoactomycin D) at the concentrations recommended by the instructions and incubation on ice for 30 min.
15. The Flow-type detection is directly carried out without cleaning, and the data is analyzed by Flow Jo.
16. Assay the proportion of viable U266 target cells and viable K562 negative control cells after co-culture of T cells and target cells was determined using 7AAD negative viable cell gating.
a) For each group of co-cultured T cells and target cells
The% cytotoxic killer cells is 100-the% calibrated target cell survival, i.e. (ratio of Raji viable cell number when no effector cells were present-Raji viable cell number when effector cells were present)/K562 viable cell number.
The results are shown in fig. 7. Figure 7 shows that BCMA (J22.9) CART cells kill 40% of U266 cells at an effective target ratio of 10: 1.
Sequence listing
<110> Shanghai Hengrunheng Dasheng Biotech Co., Ltd
<120> chimeric antigen receptor targeting B cell maturation antigen and use thereof
<170>PatentIn version 3.3
<210>1
<211>1473
<212>DNA
<213> Artificial sequence
<400>1
atggctctgc ctgtgaccgc cctgctgctg cctctggctc tgctgctgca cgccgctcgg 60
cctgatatag tgatgactcaaagtcaaaga tttatgacca catctgtcgg agatcgggtc 120
tctgtgacct gtaaggcatc ccagagtgtt gactccaacg tagcctggta ccagcagaaa 180
ccgcgacagt ctcccaaggc attgatattt tcagctagtc tgaggttttc aggtgtacct 240
gctcggttca ccgggtctgg tagcggaaca gacttcactt tgacaattag taatcttcaa 300
agtgaagacc ttgcggaata cttctgtcag cagtacaaca attatcccct tacctttggg 360
gcaggaacaa agcttgaatt gaagggcggc gggggttctg gtggcggcgg cagcggcggt 420
ggaggatcac aggtacagct tcagcagagc gggggaggtt tggtacaacc tggcggatct 480
ttgaaacttt cctgtgcagc ttcaggaata gacttttcac ggtactggat gagctgggtc 540
cgccgagcac ctgggaaagg tcttgaatgg attggggaga taaatccaga ttcttccaca 600
attaactatg ctcccagttt gaaggacaag ttcatcatta gccgcgataa cgctaaaaac 660
actttgtact tgcagatgag taaagtacgg agtgaggata cagcgttgta ctactgcgcg 720
agcttgtatt atgattacgg agatgccatg gattactggg gccaaggcac gtctgtgact 780
gtatcttcta ctacaactcc agcacccaga ccccctacac ctgctccaac tatcgcaagt 840
cagcccctgt cactgcgccc tgaagcctgt cgccctgctg ccgggggagc tgtgcatact 900
cggggactgg actttgcctg tgatatctac atctgggcgc ccttggccgg gacttgtggg 960
gtccttctcc tgtcactggt tatcaccctt tactgcaggt tcagtgtcgt gaagagaggc 1020
cggaagaagc tgctgtacat cttcaagcag cctttcatga ggcccgtgca gactacccag 1080
gaggaagatg gatgcagctg tagattccct gaagaggagg aaggaggctg tgagctgaga 1140
gtgaagttct cccgaagcgc agatgcccca gcctatcagc agggacagaa tcagctgtac 1200
aacgagctga acctgggaag acgggaggaa tacgatgtgc tggacaaaag gcggggcaga 1260
gatcctgaga tgggcggcaa accaagacgg aagaaccccc aggaaggtct gtataatgag 1320
ctgcagaaag acaagatggc tgaggcctac tcagaaatcg ggatgaaggg cgaaagaagg 1380
agaggaaaag gccacgacgg actgtaccag gggctgagta cagcaacaaa agacacctat 1440
gacgctctgc acatgcaggc tctgccacca aga 1473
<210>2
<211>491
<212>PRT
<213> Artificial sequence
<400>2
Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu
1 5 10 15
His Ala Ala Arg Pro Asp Ile Val Met Thr Gln Ser Gln Arg Phe Met
20 25 30
Thr Thr Ser Val Gly Asp Arg Val Ser Val Thr Cys Lys Ala Ser Gln
35 40 45
Ser Val Asp Ser Asn Val Ala Trp Tyr Gln Gln Lys Pro Arg Gln Ser
50 55 60
Pro Lys Ala Leu Ile Phe Ser Ala Ser Leu Arg Phe Ser Gly Val Pro
65 70 75 80
Ala Arg Phe Thr Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile
85 90 95
Ser Asn Leu Gln Ser Glu Asp Leu Ala Glu Tyr Phe Cys Gln Gln Tyr
100 105 110
Asn Asn Tyr Pro Leu Thr Phe Gly Ala Gly Thr Lys Leu Glu Leu Lys
115 120 125
Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gln
130 135 140
Val Gln Leu Gln Gln Ser Gly Gly Gly Leu Val Gln Pro Gly Gly Ser
145 150 155 160
Leu Lys Leu Ser Cys Ala Ala Ser Gly Ile Asp Phe Ser Arg Tyr Trp
165 170 175
Met Ser Trp Val Arg Arg Ala Pro Gly Lys Gly Leu Glu Trp Ile Gly
180 185 190
Glu Ile Asn Pro Asp Ser Ser Thr Ile Asn Tyr Ala Pro Ser Leu Lys
195 200 205
Asp Lys Phe Ile Ile Ser Arg Asp Asn Ala Lys Asn Thr Leu Tyr Leu
210 215 220
Gln Met Ser Lys Val Arg Ser Glu Asp Thr Ala Leu Tyr Tyr Cys Ala
225 230 235 240
Ser Leu Tyr Tyr Asp Tyr Gly Asp Ala Met Asp Tyr Trp Gly Gln Gly
245 250 255
Thr Ser Val Thr Val Ser Ser Thr Thr Thr Pro Ala Pro Arg Pro Pro
260 265 270
Thr Pro Ala Pro Thr Ile Ala Ser Gln Pro Leu Ser Leu Arg Pro Glu
275 280 285
Ala Cys Arg Pro Ala Ala Gly Gly Ala Val His Thr Arg Gly Leu Asp
290 295 300
Phe Ala Cys Asp Ile Tyr Ile Trp Ala Pro Leu Ala Gly Thr Cys Gly
305 310 315 320
Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr Cys Arg Phe Ser Val
325 330 335
Val Lys Arg Gly Arg Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe
340 345 350
Met Arg Pro Val Gln Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg
355 360 365
Phe Pro Glu Glu Glu Glu Gly Gly Cys Glu Leu Arg Val Lys Phe Ser
370 375 380
Arg Ser Ala Asp Ala Pro Ala Tyr Gln Gln Gly Gln Asn Gln Leu Tyr
385 390 395 400
Asn Glu Leu Asn Leu Gly Arg Arg Glu Glu Tyr Asp Val Leu Asp Lys
405410 415
Arg Arg Gly Arg Asp Pro Glu Met Gly Gly Lys Pro Arg Arg Lys Asn
420 425 430
Pro Gln Glu Gly Leu Tyr Asn Glu Leu Gln Lys Asp Lys Met Ala Glu
435 440 445
Ala Tyr Ser Glu Ile Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly
450 455 460
His Asp Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr
465 470 475 480
Asp Ala Leu His Met Gln Ala Leu Pro Pro Arg
485 490
<210>3
<211>21
<212>DNA
<213> Artificial sequence
<220>
<223> primer
<400>3
agcatcgttc tgtgttgtct c 21
<210>4
<211>22
<212>DNA
<213> Artificial sequence
<220>
<223> primer
<400>4
tgtttgtctt gtggcaatac ac 22
Claims (16)
1. An isolated polynucleotide, the sequence of which is selected from the group consisting of:
(1) a polynucleotide sequence comprising the coding sequence of an anti-BCMA single-chain antibody, the coding sequence of a human CD8 α hinge region, the coding sequence of a human CD8 transmembrane region, the coding sequence of a human 41BB intracellular region and the coding sequence of a human CD3 zeta intracellular region which are connected in sequence, wherein the amino acid sequence of the light chain variable region of the anti-BCMA single-chain antibody is shown as the 22 nd to 128 th amino acids in SEQ ID NO:2, the amino acid sequence of the heavy chain variable region of the anti-BCMA single-chain antibody is shown as the 144 nd and 263 nd amino acids in SEQ ID NO:2, and
(2) (1) the complement of the polynucleotide sequence,
wherein the amino acid sequence of the hinge region of the human CD8 α is shown as the amino acid at the 264 th-310 nd position of SEQ ID NO. 2, the amino acid sequence of the transmembrane region of the human CD8 is shown as the amino acid at the 311 st-332 nd position of SEQ ID NO. 2, the amino acid sequence of the intracellular region of the human CD 41BB is shown as the amino acid at the 333 rd-380 rd position of SEQ ID NO. 2, and the amino acid sequence of the intracellular region of the human CD3 zeta is shown as the amino acid at the 381 rd-491 nd position of SEQ ID NO. 2.
2. The polynucleotide of claim 1, wherein the sequence of said polynucleotide further comprises a coding sequence for a signal peptide prior to the coding sequence for said anti-BCMA single chain antibody.
3. The polynucleotide of claim 2, wherein the amino acid sequence of said signal peptide is as set forth in amino acids 1-21 of SEQ ID No. 2.
4. The polynucleotide of claim 2, wherein the coding sequence for said signal peptide preceding the coding sequence for said anti-BCMA single chain antibody is as set forth in the nucleotide sequence of SEQ ID No.1 to 63.
5. The polynucleotide of claim 1,
the coding sequence of the light chain variable region of the anti-BCMA single-chain antibody is shown as the nucleotide sequence of 64 th-384 th site of SEQ ID NO.1, the coding sequence of the heavy chain variable region of the anti-BCMA single-chain antibody is shown as the nucleotide sequence of 430 th-789 th site of SEQ ID NO.1, the coding sequence of the hinge region of the human CD8 α is shown as the nucleotide sequence of 790 th-930 th site of SEQ ID NO.1, the coding sequence of the transmembrane region of the human CD8 is shown as the nucleotide sequence of 931 rd-996 th site of SEQ ID NO.1, the coding sequence of the intracellular region of the human CD 41 is shown as the nucleotide sequence of 997 th-1140 th site of SEQ ID NO.1, and the coding sequence of the intracellular region of the human CD3 is shown as the nucleotide sequence of 1141 st-1473 site of SEQ ID NO.
6. A fusion protein comprises an anti-BCMA single-chain antibody, a human CD8 α hinge region, a human CD8 transmembrane region, a human 41BB intracellular region and a human CD3 zeta intracellular region which are connected in sequence, wherein the amino acid sequence of a light chain variable region of the anti-BCMA single-chain antibody is shown as amino acids 22-128 of SEQ ID NO. 2, the amino acid sequence of a heavy chain variable region of the anti-BCMA single-chain antibody is shown as amino acid 144-263 of SEQ ID NO. 2, the amino acid sequence of the human CD8 α hinge region is shown as amino acid 264-310 of SEQ ID NO. 2, the amino acid sequence of the human CD8 transmembrane region is shown as amino acid 311-332 of SEQ ID NO. 2, the amino acid sequence of the human 41BB intracellular region is shown as amino acid 333-380 of SEQ ID NO. 2, and the amino acid sequence of the intracellular region of the human CD3 is shown as amino acid 491-381 of SEQ ID NO. 2.
7. The fusion protein of claim 6, further comprising a signal peptide at the N-terminus of the anti-BCMA single chain antibody.
8. The fusion protein of claim 7, wherein the signal peptide has the amino acid sequence shown as amino acids 1-21 of SEQ ID NO 2.
9. A nucleic acid construct comprising the polynucleotide of any one of claims 1-5.
10. The nucleic acid construct of claim 9, wherein said nucleic acid construct is a vector.
11. The nucleic acid construct of claim 9, wherein the nucleic acid construct is a retroviral vector comprising a replication initiation site, a 3 'LTR, a 5' LTR, and the polynucleotide of any one of claims 1-5.
12. A retrovirus comprising the nucleic acid construct of any one of claims 9 to 11.
13. A genetically modified T-cell or a pharmaceutical composition comprising a genetically modified T-cell, wherein said cell comprises the polynucleotide of any one of claims 1 to 5, or comprises the nucleic acid construct of any one of claims 9 to 11, or is infected with the retrovirus of claim 12.
14. Use of the polynucleotide of any one of claims 1-5, the fusion protein of any one of claims 6-8, the nucleic acid construct of any one of claims 9-11, or the retrovirus of claim 12 in the preparation of an agent for activating T cells.
15. Use of the polynucleotide of any one of claims 1-5, the fusion protein of any one of claims 6-8, the nucleic acid construct of any one of claims 9-11, the retrovirus of claim 12, or the genetically modified T-cell of claim 13, or a pharmaceutical composition thereof, in the preparation of a medicament for treating a BCMA-mediated disease.
16. The use of claim 15, wherein the BCMA-mediated disease is multiple myeloma.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610943985.0A CN108004259B (en) | 2016-11-02 | 2016-11-02 | Chimeric antigen receptor targeting B cell maturation antigen and uses thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610943985.0A CN108004259B (en) | 2016-11-02 | 2016-11-02 | Chimeric antigen receptor targeting B cell maturation antigen and uses thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN108004259A CN108004259A (en) | 2018-05-08 |
| CN108004259B true CN108004259B (en) | 2020-06-30 |
Family
ID=62048247
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201610943985.0A Active CN108004259B (en) | 2016-11-02 | 2016-11-02 | Chimeric antigen receptor targeting B cell maturation antigen and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN108004259B (en) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108728460A (en) * | 2018-05-25 | 2018-11-02 | 上海恒润达生生物科技有限公司 | Target the Chimeric antigen receptor and application thereof of GD2 |
| CN108707619B (en) * | 2018-05-28 | 2023-07-18 | 上海恒润达生生物科技股份有限公司 | Chimeric antigen receptor targeting ROR1 and application thereof |
| CN110551740A (en) * | 2018-06-01 | 2019-12-10 | 上海恒润达生生物科技有限公司 | chimeric antigen receptor targeting CD30 and uses thereof |
| CN110551743B (en) * | 2018-06-04 | 2023-10-03 | 上海恒润达生生物科技股份有限公司 | Chimeric antigen receptor targeting CD30 and application thereof |
| CN110714018B (en) * | 2018-07-11 | 2023-10-03 | 上海恒润达生生物科技股份有限公司 | Chimeric antigen receptor targeting EGFRVIII and application thereof |
| CN110850067B (en) * | 2018-08-21 | 2023-08-15 | 上海恒润达生生物科技股份有限公司 | Chimeric antigen receptor affinity detection method |
| CN110923255B (en) * | 2018-09-19 | 2023-08-29 | 上海恒润达生生物科技股份有限公司 | Chimeric antigen receptor targeting BCMA and CD19 and uses thereof |
| CN111218465A (en) * | 2018-11-26 | 2020-06-02 | 上海恒润达生生物科技有限公司 | Chimeric antigen receptor method targeting CD22 and application |
| WO2020224605A1 (en) * | 2019-05-07 | 2020-11-12 | 亘喜生物科技(上海)有限公司 | Bcma-targeting engineered immune cell and use thereof |
| EP4007777B1 (en) * | 2019-08-02 | 2023-10-11 | Fundació de Recerca Clínic Barcelona-Institut d'Investigacions Biomèdiques August Pi i Sunyer (FRCB-IDIBAPS) | Car t-cells against bcma for the treatment of multiple myeloma |
| CN110540997B (en) * | 2019-08-29 | 2024-02-13 | 浙江煦顼技术有限公司 | BCMA chimeric antigen receptor, nucleic acid sequence, vector and application |
| CN118206658A (en) * | 2021-08-10 | 2024-06-18 | 上海恒润达生生物科技股份有限公司 | Chimeric antigen receptor of targeted BCMA based on fully human and murine single chain antibodies and application thereof |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX357823B (en) * | 2012-04-11 | 2018-07-25 | Us Health | Chimeric antigen receptors targeting b-cell maturation antigen. |
| CN105837693A (en) * | 2016-05-30 | 2016-08-10 | 李斯文 | BCMA-based (B cell maturation antigen-based) chimeric antigen receptor and preparation method and application thereof |
| CN105949324B (en) * | 2016-06-30 | 2019-08-27 | 上海恒润达生生物科技有限公司 | Target the Chimeric antigen receptor and application thereof of GPC3 |
| CN108018299B (en) * | 2016-11-01 | 2020-03-17 | 上海恒润达生生物科技有限公司 | Chimeric antigen receptor targeting BCMA and uses thereof |
-
2016
- 2016-11-02 CN CN201610943985.0A patent/CN108004259B/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN108004259A (en) | 2018-05-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108018299B (en) | Chimeric antigen receptor targeting BCMA and uses thereof | |
| CN108004259B (en) | Chimeric antigen receptor targeting B cell maturation antigen and uses thereof | |
| CN108504668B (en) | Chimeric antigen receptor targeting CD19 and CD22 and uses thereof | |
| CN108070607B (en) | Chimeric antigen receptor targeting CD19-41BB-tEGFR and application thereof | |
| CN107964549B (en) | Chimeric antigen receptor targeting CD22 and uses thereof | |
| CN108728459B (en) | Method and use of chimeric antigen receptor targeting CD19 and co-expressing IL-15 | |
| CN109320615B (en) | Chimeric antigen receptor targeting novel BCMA and uses thereof | |
| CN108330133B (en) | Methods of targeting and double-modifying CD19 chimeric antigen receptors and uses thereof | |
| CN109503721B (en) | Chimeric antigen receptor targeting CD19 and uses thereof | |
| WO2017219936A1 (en) | Car-t cell capable of efficiently and stably expressing activated antibody, and uses thereof | |
| CN107841506B (en) | Mesothelin-targeted chimeric antigen receptors and uses thereof | |
| CN108441505B (en) | Chimeric antigen receptor targeting ROR1 and application thereof | |
| CN108070608B (en) | Chimeric antigen receptor targeting CD19-CD28-tEGFR and application thereof | |
| CN108864286B (en) | Chimeric antigen receptor targeting CD19, method for combined expression of anti-PD 1 antibody variable region and application thereof | |
| CN110923255B (en) | Chimeric antigen receptor targeting BCMA and CD19 and uses thereof | |
| CN108866088B (en) | Targeting CLL-1 chimeric antigen receptor and uses thereof | |
| CN108707619B (en) | Chimeric antigen receptor targeting ROR1 and application thereof | |
| CN110857319A (en) | Isolated T cell receptor, modified cell thereof, encoding nucleic acid and application thereof | |
| WO2020061796A1 (en) | Bcma-and-cd19-targeting chimeric antigen receptor and uses thereof | |
| CN108239623B (en) | Preparation method and application of mixed CART cells | |
| WO2020034081A1 (en) | Bcma-targeting chimeric antigen receptor and uses thereof | |
| CN108728458B (en) | Methods and uses of chimeric antigen receptors targeting mesothelin in combination with IL-15 expression | |
| CN110845621A (en) | Chimeric antigen receptor method targeting EGFR and CD19 double targets | |
| CN108624607B (en) | Methods and uses of chimeric antigen receptors targeting mesothelin and dual modifications thereof | |
| CN108624608B (en) | Preparation method and application of fourth generation chimeric antigen receptor targeting mesothelin |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| CP03 | Change of name, title or address |
Address after: 201210 block D, 1st floor, building 1, Lane 1238, Zhangjiang Road, China (Shanghai) free trade zone, Pudong New Area, Shanghai Patentee after: Shanghai Hengrun Dasheng Biotechnology Co.,Ltd. Address before: 201210 floor 8, building 1, Hengyue international building, Lane 1238, Zhangjiang Road, Pudong New Area, Shanghai Patentee before: SHANGHAI HRAIN BIOTECHNOLOGY Co.,Ltd. |
|
| CP03 | Change of name, title or address |