CN107922407B - 呼吸道合胞病毒抑制剂 - Google Patents
呼吸道合胞病毒抑制剂 Download PDFInfo
- Publication number
- CN107922407B CN107922407B CN201680044547.9A CN201680044547A CN107922407B CN 107922407 B CN107922407 B CN 107922407B CN 201680044547 A CN201680044547 A CN 201680044547A CN 107922407 B CN107922407 B CN 107922407B
- Authority
- CN
- China
- Prior art keywords
- compound
- alkyl
- stirred
- added
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本发明公开了式(I)的化合物,其中Z1为NR1A、CHR1A或CR1BR1B;Z2和Z3中的一个为CH或CR1A’,另一个为N、CH或CR1A’;n为0、1或2;q为0、1或2;R1A、R1A’、R1B、R2和R3如本文所定义,它们作为RSV的抑制剂的用途以及相关方面。
Description
技术领域
本发明涉及异喹啉类似物和它们作为呼吸道合胞病毒(RSV)的复制的抑制剂的用途、包含此类类似物的药物组合物,以及使用这些类似物治疗和预防RSV感染的方法。
背景技术
在全球范围内,由RSV造成的年死亡率估计超过160,000例并且RSV感染的临床负担与流感的临床负担相当(Bourgeois等人,2009;Boyce等人,2000;Hall等人,2009;Stockman等人,2012)。RSV的流行季节从晚秋直到早春。有不良结果风险的主要人群是5岁以下的儿童、免疫受损患者和老年人,尤其是被送入社会事业机构或患有慢性潜在疾病的那些(Hall等人,2009;Falsey等人,2005)。除了支持性护理(supportive care)之外,通常没有可用于RSV感染的疗法。吸入性利巴韦林经批准用于实验室诊断的RSV感染的治疗,但是因为其有限的效果、施用的复杂性以及对于患者和工作人员潜在的致突变性而仅施用于一些骨髓移植和免疫受损患者。由于不存在针对RSV感染的有效疗法,并且由于RSV发病率的显著性和/或处于风险中的人群的伦理学,有效的RSV药剂的引入将被认为是这些患者的护理中的一大突破。
发明内容
本发明提供了一系列新型化合物,所述化合物对于RSV的复制表现出抑制活性。
由以下说明和实例,本领域技术人员将会明白本发明的另外的目标。
本发明的一个方面提供了一种通过式(I)表示的化合物,或其外消旋体、对映体、非对映异构体或互变异构体。
在一个实施方案中,本发明涉及一种具有式(I)的化合物,或其外消旋体、对映体、非对映异构体或互变异构体:

其中
Z1为NR1A、CHR1A、CR1BR1B;
Z2和Z3中的一个为CH或CR1A’,另一个为N、CH或CR1A’;
R1A为C1-C6烷基、C3-C7环烷基、-S(=O)2R1C、芳基、杂芳基或杂环基,其中每个所述烷基、环烷基、芳基、杂芳基和杂环基任选地被各自独立地选自由以下各项组成的组的取代基单、二或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、氨基、-NHR1C、-NR1DR1D’、-C(=O)OH、
-C(=O)R1C、-C(=O)C1-C6亚烷基NH2、-C(=O)OR1C、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、-S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、-OC(=O)NHR1C、-NHC(=O)R1C、-NHC(=O)NHR1C、-NHC(=O)OR1C或-NHS(=O)2R1C;
两个R1B与它们所附接的碳原子组合在一起并形成C3-C6环烷基或杂环基,其中环烷基和杂环基任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、氨基、-NHR1C、-NR1DR1D’、-C(=O)OH、-C(=O)R1C、-C(=O)OR1C、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、-S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、-OC(=O)NHR1C、-NHC(=O)R1C、-NHC(=O)NHR1C、-NHC(=O)OR1C或-NHS(=O)2R1C;
每个R1A’独立地选自卤代基、羟基、氰基、C1-C3卤代烷基、C1-C3烷氧基。
R1C为C1-C6烷基或C3-C7环烷基,其中的任一个任选地被选自以下的一个或两个取代基取代:卤代基、羟基、氰基、氨基、三氟甲基、C1-C3烷基、C1-C3烷氧基、C1-C3卤代烷基、C1-C3烷基氨基以及C1-C3二烷基氨基;
R1D和R1D’各自独立地为H或C1-C6烷基,或
R1D和R1D’与它们所附接的氮原子一起形成4至6元环,所述环任选地被独立地选自以下的一个或两个取代基取代:卤代基、羟基、氰基和氨基;
R2为C1-C6烷基,其被各自独立地选自以下的一个、两个或三个取代基取代:卤代基、羟基、氰基、三氟甲基、氨基、-NHR2A、-NR2BR2B’、C1-C3烷氧基、S(=O)2R2A、C3-C4环烷氧基、杂环氧基,其中每个所述烷氧基、环烷氧基和杂环氧基任选地被各自独立地选自以下的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、C1-C3烷基氨基以及S(=O)2R2A,或
R2为C2-C6烷基、C3-C7环烷基C0-C5烷基、杂环基C0-C5烷基、芳基C0-C5烷基或杂芳基C0-C5烷基,其中杂环基为4至8元饱和的单环、双环或螺环,并且其中每个所述环烷基、杂环基、芳基和杂芳基任选地被各自独立地选自以下的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3羟基烷基、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、C1-C3烷基氨基以及S(=O)2R2A;
R2A为C1-C3烷基、C3-C4环烷基、芳基、杂芳基或杂环基;
R2B和R2B’各自独立地为C1-C3烷基,或
R2B和R2B’与它们所附接的氮原子组合在一起并形成4至6元杂环基,所述杂环基任选地被独立地选自以下的一个或两个取代基取代:氨基、卤代基、C1-C3烷基和三氟甲基;
R3各自独立地选自由以下各项组成的组:卤代基、羟基、氰基、氨基、C1-C6烷基、C1-C6烷氧基、C3-C7环烷基C0-C2烷基或杂环基C0-C2烷基,其中烷基、烷氧基、环烷基和杂环基任选地被独立地选自以下的1、2或3个取代基取代:-NR3AR3B、卤代基、羟基和三氟甲基;
R3A和R3B各自独立地为H或C1-C6烷基,其中烷基任选地被一个或两个卤代基取代;
n为0、1或2;
q为0、1或2;
杂环基为含有1、2或3个杂原子的饱和的4至7元单环、双环或螺环,所述杂原子各自独立地选自O、S和N,除非另外指明;
或其盐。
在另一个实施方案中,本发明涉及一种具有式(I)的化合物,或其外消旋体、对映体、非对映异构体或互变异构体:

其中
Z1为NR1A、CHR1A、CR1BR1B;
Z2和Z3中的一个为CH或CR1A’,另一个为N、CH或CR1A’;
R1A为C1-C6烷基、C3-C7环烷基、S(=O)2R1C、芳基、杂芳基或杂环基,其中每个所述烷基、环烷基、芳基、杂芳基和杂环基任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、氨基、-NHR1C、-NR1DR1D’、-C(=O)OH、
-C(=O)R1C、-C(=O)C1-C6亚烷基NH2、-C(=O)OR1C、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、-S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、-OC(=O)NHR1C、NHC(=O)R1C、NHC(=O)NHR1C、-NHC(=O)OR1C或-NHS(=O)2R1C;
两个R1B与它们所附接的碳原子组合在一起并形成C3-C6环烷基或杂环基,其中环烷基和杂环基任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、氨基、-NHR1C、-NR1DR1D’、-C(=O)OH、-C(=O)R1C、-C(=O)OR1C、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、-OC(=O)NHR1C、-NHC(=O)R1C、-NHC(=O)NHR1C、NHC(=O)OR1C或NHS(=O)2R1C;
每个R1A’独立地选自卤代基、羟基、氰基、C1-C3卤代烷基、C1-C3烷氧基。
R1C为C1-C6烷基、C3-C7环烷基或杂环基,其中的任一个任选地被独立地选自以下的一个或两个取代基取代:卤代基、羟基、氰基、氨基、三氟甲基、C1-C3烷基、C1-C3烷氧基、C1-C3卤代烷基、C1-C3烷基氨基以及C1-C3二烷基氨基;
R1D和R1D’各自独立地为H或C1-C6烷基,或
R1D和R1D’与它们所附接的氮原子一起形成4至6元环,所述环任选地被独立地选自以下的一个或两个取代基取代:卤代基、羟基、氰基和氨基;
R2为C1-C6烷基,其被各自独立地选自以下的一个、两个或三个取代基取代:卤代基、羟基、氰基、三氟甲基、氨基、-NHR2A、-NR2BR2B’、C1-C3烷氧基、S(=O)2R2A、S(=O)2NH2、C3-C4环烷氧基、杂环氧基,其中每个所述烷氧基、环烷氧基和杂环氧基任选地被各自独立地选自以下的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、C1-C3烷基氨基和S(=O)2R2A以及S(=O)2NH2,或
R2为C2-C6烷基、C3-C7环烷基C0-C5烷基、杂环基C0-C5烷基、芳基C0-C5烷基或杂芳基C0-C5烷基,其中杂环基为4至8元饱和的单环、双环或螺环,并且其中每个所述环烷基、杂环基、芳基和杂芳基任选地被各自独立地选自以下的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3羟基烷基、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、C1-C3烷基氨基、C3-C4环烷基、氧杂环丁烷基(oxetanyl)、S(=O)2R2A、S(=O)2NH2、NHS(=O)2R2A以及C(=O)NH2,并且环烷基和氧杂环丁烷基任选地被氨基或甲基取代;
R2A为C1-C3烷基、C3-C4环烷基、芳基、杂芳基或杂环基;
R2B和R2B’各自独立地为C1-C3烷基,或
R2B和R2B’与它们所附接的氮原子组合在一起并形成4至6元杂环基,所述杂环基任选地被独立地选自以下的一个或两个取代基取代:氨基、卤代基、C1-C3烷基和三氟甲基;
R3各自独立地选自由以下各项组成的组:卤代基、羟基、氰基、氨基、C1-C6烷基、C1-C6烷氧基、C3-C7环烷基C0-C2烷基或杂环基C0-C2烷基,其中烷基、烷氧基、环烷基和杂环基任选地被独立地选自以下的1、2或3个取代基取代:-NR3AR3B、卤代羟基和三氟甲基;
R3A和R3B各自独立地为H或C1-C6烷基,其中烷基任选地被一个或两个卤代基取代;
n为0、1或2;
q为0、1或2;
杂环基除非另外指明否则为含有1、2或3个杂原子的饱和的4至7元单环、双环或螺环,所述杂原子各自独立地选自O、S和N;
或其盐。
在另一个实施方案中,本发明涉及一种具有式(I)的化合物,或其外消旋体、对映体、非对映异构体或互变异构体:

其中
Z1为NR1A、CHR1A、CR1BR1B;
Z2和Z3中的一个为CH或CR1A’,另一个为N、CH或CR1A’;
R1A为C1-C6烷基、C3-C7环烷基、S(=O)2R1C、芳基、杂芳基、杂环基或7或8元螺杂环基,其中每个所述烷基、环烷基、芳基、杂芳基、杂环基和螺杂环基任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、氨基、-NHR1C、-NR1DR1D’、
-C(=O)OH、-C(=O)R1C、-C(=O)C1-C6亚烷基NH2、-C(=O)OR1C、-C(=O)NHR1C、
-C(=O)NR1DR1D’、-S(=O)2R1C、S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、
-OC(=O)NHR1C、-NHC(=O)R1C、-NHC(=O)NHR1C、-NHC(=O)OR1C或-NHS(=O)2R1C;
两个R1B与它们所附接的碳原子组合在一起并形成C3-C6环烷基或杂环基,其中环烷基和杂环基任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、氨基、-NHR1C、-NR1DR1D’、-C(=O)OH、-C(=O)R1C、-C(=O)OR1C、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、-OC(=O)NHR1C、-NHC(=O)R1C、
-NHC(=O)NHR1C、-NHC(=O)OR1C或-NHS(=O)2R1C;
每个R1A’独立地选自卤代基、羟基、氰基、C1-C3卤代烷基、C1-C3烷氧基;
R1C为C1-C6烷基、C3-C7环烷基或杂环基,其中的任一个任选地被独立地选自以下的一个或两个取代基取代:卤代基、羟基、氰基、氨基、三氟甲基、C1-C3烷基、C1-C3烷氧基、C1-C3卤代烷基、C1-C3烷基氨基以及C1-C3二烷基氨基;
R1D和R1D’各自独立地为H或C1-C6烷基,或
R1D和R1D’与它们所附接的氮原子一起形成4至6元环,所述环任选地被独立地选自以下的一个或两个取代基取代:卤代基、羟基、氰基和氨基;
R2为C1-C6烷基,其被各自独立地选自以下的一个、两个或三个取代基取代:卤代基、羟基、氰基、三氟甲基、氨基、-NHR2A、-NR2BR2B’、C1-C3烷氧基、S(=O)2R2A、C3-C4环烷氧基以及杂环氧基,其中每个所述烷氧基、环烷氧基和杂环氧基任选地被各自独立地选自以下的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、C1-C3烷基氨基以及-S(=O)2R2A,或
R2为C2-C6烷基、C3-C7环烷基C0-C5烷基、杂环基C0-C5烷基、芳基C0-C5烷基或杂芳基C0-C5烷基,其中杂环基为4至8元饱和的单环、双环或螺环,并且其中每个所述环烷基、杂环基、芳基和杂芳基任选地被各自独立地选自以下的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3羟基烷基、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、C1-C3烷基氨基、C3-C4环烷基、氧杂环丁烷基、-S(=O)2R2A、-S(=O)2NH2、-NHS(=O)2R2A以及-C(=O)NH2,并且环烷基和氧杂环丁烷基任选地被氨基或甲基取代;
R2A为C1-C3烷基、C3-C4环烷基、氨基、芳基、杂芳基或杂环基;
R2B和R2B’各自独立地为C1-C3烷基,或
R2B和R2B’与它们所附接的氮原子组合在一起并形成4至6元杂环基,所述杂环基任选地被独立地选自以下的一个或两个取代基取代:氨基、卤代基、C1-C3烷基和三氟甲基;
R3各自独立地选自由以下各项组成的组:卤代基、羟基、氰基、氨基、C1-C6烷基、C1-C6烷氧基、C3-C7环烷基C0-C2烷基或杂环基C0-C2烷基,其中烷基、烷氧基、环烷基和杂环基任选地被独立地选自以下的1、2或3个取代基取代:-NR3AR3B、卤代基、羟基和三氟甲基;
R3A和R3B各自独立地为H或C1-C6烷基,其中烷基任选地被一个或两个卤代基取代;
n为0、1或2;
q为0、1或2;
杂环基为含有1、2或3个杂原子的饱和的4至7元单环或双环,所述杂原子各自独立地选自O、S和N,除非另外指明;
或其盐。
本发明的另一个方面提供了一种用作药物的式(I)的化合物,或其药学上可接受的盐。
式(I)的化合物,或其药学上可接受的盐用于制造在人类中治疗或预防RSV感染的药物的用途也在本发明的范围内。
一种包含式(I)的化合物、或其药学上可接受的盐,以及药学上可接受的载体的药物组合物包括在本发明的范围之内。
根据该实施方案的另一方面,根据本发明的药物组合物还包含治疗有效量的至少一种其他抗病毒剂。
本发明还提供了如上文所述的药物组合物用于在患有感染或处于患感染风险的人类中治疗RSV感染的用途。
本发明的另一方面涉及一种在人类中治疗或预防RSV感染的方法,其通过单独或与至少一种其他抗病毒剂组合地(一起或分开施用)向人类施用抗RSV病毒有效量的本发明的化合物、其药学上可接受的盐、或如上所述的组合物实现。
本发明的另一方面涉及一种制品,其包括有效治疗RSV感染的组合物;以及包括指示所述组合物可用于治疗由RSV造成的感染的标签的包装材料;其中所述组合物包含根据本发明的式(I)的化合物或其药学上可接受的盐。
本发明的另一方面涉及一种抑制RSV的复制的方法,其包括在RSV的复制被抑制的条件下将病毒暴露于有效量的式(I)的化合物或其盐。
还包括在本发明的范围中的是式(I)的化合物或其盐用以抑制RSV的复制的用途。
具体实施方式
定义
本文未具体定义的术语应具有由本领域技术人员根据本公开和上下文所给出的含义。然而,如本说明书中所用,除非有相反规定,否则以下术语具有所指明的含义并且遵守以下惯例。在下文定义的基团、自由基或部分中,通常在基团的前面指明碳原子的数目,例如,C1-C6烷基意指具有1至6个碳原子的烷基基团或自由基。一般来讲,对于包括两个或更多个子基团的基团而言,最后命名的子基团为自由基附接点。例如,取代基“芳基C1-C3-烷基”意指结合至C1-C3烷基基团的芳基基团,其中C1-C3烷基基团结合至核心。
自由基的定义前面的符号“-”指示自由基与核心附接的点。例如,标记“-C(=O)NHC1-C6烷基”表示通过羰基碳连接至核心的伯酰胺,“-C(=O)OC1-C6烷基”指示通过羰基碳连接至核心的酯,-NHC(=O)C1-C6烷基表示通过氮原子连接的伯酰胺并且“-OC(=O)C1-C6烷基”指示通过氧原子连接至核心的酯。
在以化学名称的形式描述本发明的化合物和将本发明的化合物描述为式的情况下,当存在任何不一致时,以式为准。命名----可以用在部分式中以指示与定义的核心分子连接的键。
除非具体指出,否则在本说明书通篇和随附的权利要求中,给定的化学式或名称应涵盖其互变异构体和所有的立体、光学和几何异构体(例如对映体、非对映体、E/Z异构体、阻转异构体)和外消旋体,以及不同比例的单独对映体的混合物、非对映体的混合物,或在存在此类异构体和对映体的情况下任何前述形式的混合物,以及盐,包括其药学上可接受的盐及其溶剂化物,诸如例如水合物,包括游离化合物的溶剂化物或化合物的盐的溶剂化物。
本领域的技术人员将知道如何分离、富集或选择性地制备本发明的化合物的对映体。纯立体异构体(例如对映体和非对映体)或具有期望的对映体过量(ee)或对映体纯度的混合物的制备通过本领域的技术人员已知的许多(a)对映体的分离或拆分、或(b)对映选择性合成的方法中的一种或多种,或它们的组合实现。这些拆分方法通常依赖于手性识别并且包括但不限于使用手性固定相的色谱法、对映选择性主-客络合、使用手性助剂的拆分或合成、对映选择性合成、酶促和非酶促动力学拆分,或自发的对映选择性结晶。此类方法通常公开于Chiral Separation Techniques:A Practical Approach(第2版),G.Subramanian(编辑),Wiley-VCH,2000;T.E.Beesley和R.P.W.Scott,ChiralChromatography,John Wiley&Sons,1999;以及Satinder Ahuja,Chiral Separations byChromatography,Am.Chem.Soc.,2000。此外,存在同样为人们所熟知的用于对映体过量或纯度的定量的方法,包括但不限于GC、HPLC、CE或NMR,以及绝对构型和构象的排布(assignment),包括但不限于CD、ORD、X-射线结晶法或NMR。
术语“卤代基”通常表示氟、氯、溴和碘。
术语“C1-Cn烷基”(其中n为2至n的整数,单独或与另一个自由基组合)意指具有1至n个C原子的非环状、饱和、支链或直链的一价烃自由基。例如,术语C1-3烷基涵盖自由基H3C-、H3C-CH2-、H3C-CH2-CH2-和H3C-CH(CH3)-。
术语C1-Cn亚烷基(其中n为2至n的整数)意指具有1至n个C原子的非环状、饱和、支链或直链的二价烃自由基。例如,术语C1-C3亚烷基涵盖自由基-CH2-、-CH2CH2-、-CH(CH3)-、-CH2CH2CH2-、-CH2CH(CH3)-和-CH(CH3)CH2-。术语C1-Cn卤代烷基是指C1-Cn烷基,其中至少一个C原子被卤素,优选氯或氟取代。示例性C1-Cn卤代烷基为三氟甲基。
术语C1-Cn烷氧基或C1-Cn烷氧基意指通过氧原子连接的自由基-O-C1-Cn烷基,其中C1-Cn烷基如上文所定义,并且包括例如甲氧基、乙氧基、正丙氧基、异丙氧基、叔丁氧基、正丁氧基和异丁氧基。
术语“氨基”意指NH2。
术语“氨基C1-Cn烷基”意指被NH2取代的C1-Cn烷基,其中C1-Cn烷基如上文所定义。
术语“C1-Cn烷基氨基”意指被C1-Cn烷基取代的氨基基团,其中C1-Cn烷基如上文所定义。
术语“卤代基”或“卤素”包括氟、氯、溴和碘。
如本文所用,术语“碳环基”或“碳环”单独或与另一个自由基组合地意指由3至14个碳原子组成的单环、双环或三环环结构。术语“碳环基”或“碳环”是指完全饱和的芳族环系和部分饱和的环系。术语“碳环基”或“碳环”涵盖稠合的、桥连的以及螺环的体系。
术语“C3-Cm环烷基”(其中m为整数3至m,单独或与另一个自由基组合)意指具有3至m个C原子的环状的、饱和的、非支化的烃自由基。例如,术语C3-7环烷基包括环丙基、环丁基、环戊基、环己基和环庚基。
术语“C3-Cm环烷氧基”意指通过氧原子连接的自由基-O-C3-Cm环烷基,其中C3-Cm环烷基如上文所定义。
术语“氧代基”或(=O)用于指示以双键键合至碳或硫原子,从而提供羰基C(=O)、亚砜S(=O)或磺酰基S(=O)2部分的氧原子。
如本文所用,术语“芳基”单独或与另一个自由基组合地意指含有6个碳原子的碳环芳族单环基团,其可进一步与可为芳族、饱和或不饱和的一个或多个5或6元碳环基团稠合。芳基包括但不限于苯基、茚满基、茚基、萘基、蒽基、菲基、四氢萘基以及二氢萘基。
术语“杂环基”或“杂环”意指饱和或不饱和的单环、双环或三环环系,其包括由3至14个环原子组成的芳族环系并且含有各自独立地选自N、O和S的一个、两个、三个或四个杂原子。术语“杂环基”或“杂环”旨在包括所有可能的异构形式和所有稠合、桥连和螺形式。“杂环基”可任选地被一个或多个取代基取代。
术语“杂环氧基”意指通过氧原子连接的自由基-O-杂环基,其中杂环基如上文所定义。
术语“杂芳基”意指含有各自独立地选自N、O和S的一个、两个、三个或四个杂原子的单环、双环或三环环系,其由5至14个环原子组成,其中杂原子中的至少一个是芳环的一部分。术语“杂芳基”旨在包括所有可能的异构形式和所有稠合、桥连和螺形式。典型的杂芳基为吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、噁唑基、异噁唑基、吡咯基、吡唑基。“杂芳基”可任选地被一个或多个取代基取代。
如本文所用,表述“C3-Cm环烷基C0-Cn烷基”(其中m为3至m的整数,并且n为1至n的整数)意在包括直接键合(C0)或通过如上文所定义的中间C1-Cn亚烷基接头键合的如上文所定义的C3-Cm环烷基部分。
如本文所用,表述“碳环基C0-Cn烷基”(其中n为1至n的整数)意在包括直接键合(C0)或通过如上文所定义的中间C1-Cn亚烷基接头键合的碳环基部分。
如本文所用,表述“杂环基C0-Cn烷基”(其中n为1至n的整数)意在包括直接键合(C0)或通过如上文所定义的中间C1-Cn亚烷基接头键合的杂环基部分。
如本文所用,表述“杂芳基C0-Cn烷基”(其中n为1至n的整数)意在包括直接键合(C0)或通过如上文所定义的中间C1-Cn亚烷基接头键合的杂芳基部分。
如本文所用,表述“芳基C0-Cn烷基”(其中n为1至n的整数)意在包括直接键合(C0)或通过如上文所定义的中间C1-Cn亚烷基接头键合的芳基部分。
上文给出的许多术语可重复用于式或基团的定义并且在每种情况下彼此独立地具有上文给出的含义中的一个。
如本文所用,短语“药学上可接受的”是指在合理的医学判断范围内,适用于与人类和动物的组织接触而不产生过量毒性、刺激、过敏反应或其他问题或并发症,且与合理的收益/风险比相称的化合物、材料、组合物和/或剂型。
如本文所用,短语“药学上可接受的盐”是指所公开的化合物的衍生物,其中母体化合物通过制备其酸或碱盐来改性。药学上可接受的盐的实例包括但不限于碱性残基诸如胺的矿物酸或有机酸盐;酸性残基诸如羧酸的碱或有机盐;等等。例如,此类盐包括乙酸盐、抗坏血酸盐、苯磺酸盐(benzenesulfonate)、苯甲酸盐、苯磺酸盐(besylate)、碳酸氢盐、酒石酸氢盐、溴化物/氢溴酸盐、Ca-乙二胺四乙酸盐/乙二胺四乙酸盐、樟脑磺酸盐、碳酸盐、氯化物/盐酸盐、柠檬酸盐、乙二磺酸盐(edisylate)、乙二磺酸盐(ethane disulfonate)、丙酸酯月桂硫酸盐(estolate)、乙磺酸盐、富马酸盐、葡庚糖酸盐、葡糖酸盐、谷氨酸盐、羟乙酸盐、乙内酰胺苯胂酸盐(glycollylarsnilate)、己基间苯二酚盐、海巴明(hydrabamine)、羟基马来酸盐、羟基萘甲酸盐、碘化物、异硫代硫酸盐、乳酸盐、乳糖醛酸盐(lactobionate)、苹果酸盐、马来酸盐、扁桃酸盐、甲磺酸盐(methanesulfonate)、甲磺酸盐(mesylate)、甲基溴化物、甲基硝酸盐、甲基硫酸盐、粘酸盐、萘磺酸盐、硝酸盐、草酸盐、双羟萘酸盐、泛酸盐、苯基乙酸盐、磷酸盐/二磷酸盐、聚半乳糖醛酸盐、丙酸盐、水杨酸盐、硬脂酸盐、次乙酸盐(subacetate)、琥珀酸盐、磺酰胺、硫酸盐、丹宁酸盐、酒石酸盐、茶氯酸盐(teoclate)、甲苯磺酸盐、三乙基碘化物(triethiodide)、铵、苄星(benzathine)、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、葡甲胺以及普鲁卡因。另外的药学上可接受的盐可使用来自金属的阳离子形成,所述金属如铝、钙、锂、镁、钾、钠、锌等等。(另见Pharmaceutical salts,Birge,S.M.等人,J.Pharm.Sci.,(1977),66,1-19)。
本发明的药学上可接受的盐可通过常规化学方法由含有碱性或酸性部分的母体化合物合成。一般而言,这样的盐可由这些化合物的游离酸或碱形式与足量的适当碱或酸在水或有机稀释剂(如醚、乙酸乙酯、乙醇、异丙醇或乙腈或其混合物)中反应来制备。
比上文提到的那些例如更适用于纯化或分离本发明的化合物的其他酸的盐也构成本发明的一部分。
如本文所用,术语“治疗”意指施用根据本发明的化合物或组合物,以减轻或消除患者的RSV疾病的症状和/或减少病毒载量。
如本文所用,术语“预防”意指在个体暴露于病毒之后但是在出现疾病症状之前和/或在检测到病毒之前施用根据本发明的化合物或组合物,以预防疾病症状的出现。
术语“治疗有效量”意指当向有需要的患者施用时,足以有效治疗根据本发明的化合物对其具有效用的疾病状态、病状或病症的所述化合物的量。这样的量将足以引起研究人员或医生所寻求的组织系统或患者的生物学或医学反应。构成治疗有效量的根据本发明的化合物的量将根据诸如以下各项的因素而变化:化合物及其生物活性、用于施用的组合物、施用时间、施用途径、化合物的排泄速度、治疗的持续时间、正在治疗的疾病状态或病症的类型及其严重程度、与本发明的化合物组合或同时使用的药物,以及患者的年龄、体重、一般健康状况、性别和饮食。这一治疗有效量可以由本领域普通技术人员根据他们自己的知识、现有技术和本公开按常规进行确定。
在以下实施方案中,详细描述根据本发明的式(I)的化合物的基团和取代基。以下定义中的任一个和每一个可彼此组合。
在式(I)的化合物的一个实施方案中,Z1为NR1A,从而提供式(Ia)的化合物:

在一个构型中,R1A为未取代的C3-C6环烷基,诸如环丙基。在式(Ia)的化合物的另一个构型中,R1A为被甲基或氟取代的C3-C6环烷基。
在式(Ia)的化合物的另一个构型中,R1A为任选被以下各项取代的4至6元杂环基:-C(=O)R1C、-C(=O)OR1C、-C(=O)NH2、-C(=O)NHR1C、
-C(=O)NR1DR1D’和-S(=O)2R1C;
R1C为C1-C4烷基、C3-C6环烷基,其中的任一个任选被甲基或氟取代;
R1D和R1D’为C1-C4烷基,或R1D和R1D’与它们所附接的氮原子一起形成4至6元杂环基;
在式(Ia)的化合物的一个典型构型中,R1A为在N原子上被取代的氮杂环丁烷基或哌啶基。在此构型中的典型取代基包括-C(=O)OR1C和-S(=O)2R1C,其中R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被甲基或氟取代;
在式(Ia)的化合物的另选构型中,R1A为C1-C6烷基或C1-C6卤代烷基。
在式(I)的化合物的另选的实施方案中,Z1为CR1BR1B。在此实施方案中,两个R1B与它们所附接的碳原子组合在一起并且形成C3-C6环烷基或4至7元杂环基,其中的任一个任选被取代,从而提供式(Ib)的化合物:

环W的代表性取代基选自C1-C4烷基、C1-C4卤代烷基、卤代基、羟基、C1-C4烷氧基、-C(=O)R1C、-C(=O)OR1C、-C(=O)NH2、-C(=O)NHR1C、-C(=O)NR1DR1D’和-S(=O)2R1C,其中
R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被独立地选自甲基、氟、氨基和羟基的一个或两个取代基取代;
R1D和R1D’各自独立地为C1-C4烷基,或R1D和R1D’与它们所附接的氮原子组合在一起并且形成4至6元任选取代的环;
在式(Ib)的化合物的一个实施方案中,环W为任选取代的4至6元杂环基。
在式(Ib)的化合物的一个实施方案中,环W为未取代的杂环基。
在式(Ib)的化合物的一个实施方案中,环W为任选取代的C3-C6环烷基。根据该实施方案的代表性取代基包括羟基、NHR1C、-C(=O)OR1C、-OC(=O)NHR1C和-NHC(=O)OR1C;
R1C为C1-C3烷基,其任选被氟取代,或环丙基,其任选被甲基或氟取代。通常,在该实施方案中,R1C为甲基;
通常,式(Ib)的化合物中的杂环基为含氮环,诸如氮杂环丁烷或哌啶,其通常如上文所定义的在氮原子上被取代,从而提供分别具有结构(Ib’)和(Ib”)的化合物:

根据该实施方案的R1CC的代表值包括:
-C(=O)R1C、-C(=O)OR1C、-C(=O)NH2、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、S(=O)2NHR1C、-S(=O)(=NH)R1C,尤其是-C(=O)R1C或-S(=O)2R1C,其中
R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被甲基氨基或氟取代。
R1D和R1D’为C1-C4烷基,诸如甲基。
根据该实施方案的R1CC的另一组代表值包括:
-C(=O)R1C、-C(=O)OR1C、-C(=O)NH2、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C,尤其是-C(=O)R1C、C(=O)OR1C或-S(=O)2R1C,其中
R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被甲基氨基或三氟甲基取代。
R1D和R1D’为C1-C4烷基,诸如甲基。
根据该实施方案的R1CC的另一组代表值包括:
-C(=O)R1C、-C(=O)OR1C、-S(=O)2R1C,其中
R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被甲基氨基或三氟甲基取代。
在式(Ib’)和(Ib”)的化合物的一个实施方案中,R2为杂芳基,诸如吡啶基、嘧啶基、哒嗪基、吡嗪基或噻唑基。通常,在该实施方案中,Z2为CH并且Z3为N或CH。
在式(Ib’)和(Ib”)的化合物的一个实施方案中,
Z2为CH;
Z3为N或CH;
R1CC为-C(=O)R1C、-C(=O)OR1C、-C(=O)NH2、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C,其中
R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被甲基、氨基或三氟甲基取代。
R2为杂芳基,其任选被氰基、一个或两个氟、-C(=O)NH2、
-NHS(=O)2Me或-S(=O)2NH2取代;
n为0或1;
q为0;
在式(Ib’)和(Ib”)的化合物的一个实施方案中,
Z2为CH;
Z3为N或CH;
R1CC为-C(=O)R1C、-C(=O)OR1C、-C(=O)NH2、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C,其中
R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被甲基、氨基或三氟甲基取代。
R2为杂芳基;
n为0或1;
q为0;
通常,在该实施方案中,R1CC为-C(=O)R1C、-C(=O)OR1C或-S(=O)2R1C,其中
R1C为甲基或环丙基,其中环丙基任选被氨基或三氟甲基取代;
R2为吡啶基、嘧啶基、哒嗪基、吡嗪基或噻唑基。
在式I的化合物的另一个另选的实施方案中,Z1为CHR1A;
在本发明的一个实施方案中,q为0。
在本发明的一个实施方案中,Z2为CR1A’并且Z3为N。通常,Z2为CH并且Z3为N。
在本发明的一个实施方案中,Z2为CH并且Z3为N并且q为0。
在一个另选的实施方案中,Z2和Z3均为CH。
在本发明的一个实施方案中,Z2和Z3均为CH并且q为0。
在式(I)的化合物或式(I)的任何子基团的典型实施方案中,q为0。
在另选的实施方案中,q为1或2。
在本发明的一个实施方案中,R2为C1-C6烷基,其被各自独立地选自以下的一个、两个或三个取代基取代:卤代基、羟基、三氟甲基、氨基、-NHR2A、
-NR2BR2B’、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、S(=O)2R2A;
R2A为C1-C3烷基、C3-C4环烷基;
R2B和R2B’各自独立地为C1-C3烷基。
在本发明的一个实施方案中,R2为C1-C6烷基,其被羟基或三氟甲基取代。
根据该实施方案的R2的代表性构型为被羟基取代的C1-C6烷基,诸如羟丙基、羟丁基或羟戊基,通常为羟丁基。
根据该实施方案的R2的另一个代表性构型为被一个、两个或三个氟取代的C1-C6烷基。
根据该实施方案的R2的另一个代表性构型为被氰基取代的C1-C6烷基。
在本发明的一个另选的实施方案中,R2选自C2-C6烷基、C3-C7环烷基C0-C5烷基、杂环基C0-C5烷基、芳基C0-C5烷基或杂芳基C0-C5烷基,其中每个所述环烷基、芳基、杂芳基和杂环基任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3羟基烷基、C1-C3烷氧基、C1-C3卤代烷氧基、C1-C3烷基氨基以及-S(=O)2R2A,其中R2A如上文所定义。通常,在该实施方案中,R2A为C1-C3烷基,诸如甲基。
在本发明的一个实施方案中,R2为任选取代的C3-C7环烷基、杂环基、苯基或杂芳基,其直接连接至异喹啉部分。
根据该实施方案的R2的一个代表性构型为被氰基、C1-C3烷基诸如甲基或被-S(=O)2R2A取代的苯基,其中R2A通常为C1-C3烷基诸如甲基。
根据该实施方案的R2的另一个代表性构型为C3-C6环烷基或4至6元杂环基,其中的每一个被氧代基、一个或两个卤代基、羟基、C1-C3烷基或-S(=O)2R2A取代,其中R2A如上文所定义。通常,在该实施方案中,R2A为C1-C3烷基,诸如甲基。
在本发明的一个实施方案中,R2为杂芳基,其任选被一个或两个取代基取代。根据该实施方案的典型的杂芳基包括吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、噁唑基、异噁唑基、吡咯基、吡唑基。根据该实施方案的代表性取代基包括C3-C4环烷基,诸如环丙基;被氨基或卤代基取代的C3-C4环烷基,诸如被氨基或氟取代的环丙基。另外的代表性取代基包括卤代基诸如氟、氰基、三氟甲基、氨基、-NHR2A、-NR2BR2B’、-S(=O)2R2A、-NHS(=O)2R2A、-S(=O)2NH2、
-S(=O)2NHR2A、-C(=O)NHR2A、-C(=O)NR2BR2B’
其中R2A为C1-C3烷基、C3-C4环烷基,诸如甲基或环丙基;
R2B和R2B’各自独立地为C1-C3烷基。
在本发明的一个实施方案中,R2为杂芳基。
在本发明的一个典型的实施方案中,R2为选自吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、噁唑基、异噁唑基、吡咯基、吡唑基的杂芳基。通常,根据该实施方案,R2为嘧啶基、吡啶基或哒嗪基。
在本发明的一个实施方案中,R2为噻唑基或任选取代的吡啶基。
在本发明的一个实施方案中,R2为任选取代的吡啶基。
在本发明的一个实施方案中,R2为吡啶基。
在本发明的一个代表性实施方案中,R2为吡啶-3-基或吡啶-4-基,其中的任一个为任选取代的。
在根据该实施方案的一个代表性构型中,R2为吡啶-3-基。
在根据该实施方案的另一个代表性构型中,R2为吡啶-4-基。
在本发明的一个另选实施方案中,R2为噻唑基。通常,根据该实施方案,R2为噻唑-5-基。
在其中n为1或2的本发明的一个实施方案中,每个R3独立地选自由以下各项组成的组:卤代基、氰基、羟基、C1-C6烷基、C1-C6烷氧基、C3-C7环烷基和C1-C6卤代烷基。通常,根据该实施方案,R3为氟、氯或氰基。
在本发明的一个实施方案中,n为1,R3为C1-C3烷基,其被-NR3AR3B并且任选被1、2或3个氟或被C1-C3烷氧基取代。R3A和R3B独立地为H或C1-C3烷基。通常,根据该实施方案,R3为被NH2并且任选被一个氟取代的甲基。
在其中n为1的本发明的一个另选实施方案中,R3为C1-C3烷基或卤代基。通常,根据该实施方案,R3为甲基、氯或氟。
在本发明的一个实施方案中,n为1,R3为C1-C3烷基、卤代基或三氟甲基。通常,根据该实施方案,R3为甲基、氯、氟或三氟甲基。
在本发明的一个实施方案中,n为1并且R3位于异喹啉部分的7位,从而提供以下通式的化合物:

根据该实施方案的R3的一个典型值为氨基甲基。
根据该实施方案的R3的另一个典型值为卤代基,诸如氯或氟,优选氯。
根据该实施方案的R3的另一个典型值为C1-C3烷基诸如甲基。
在本发明的一个实施方案中,n为0。在本发明的一个另选的实施方案中,n为1或2。通常,n为0。
在本发明的一个实施方案中,式I的化合物为式IIb’或IIb”的化合物:

其中
Z3为N或CH;
R1CC为-C(=O)R1C、-C(=O)OR1C、-S(=O)2R1C,其中
R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被甲基、氨基或三氟甲基取代;
R2为噻唑基、吡啶基或被氰基、-NHS(=O)2Me或氟取代的吡啶基;
R3为C1-C3烷基、卤代基、氰基或C1-C3卤代烷基;
n为0或1。
通常,根据该实施方案,R1C为甲基、环丙基或被甲基、氨基或三氟甲基取代的环丙基。
根据该实施方案的R1CC的代表值包括-C(=O)Me、-S(=O)2Me。
在根据该实施方案的一个构型中,n为1并且R3为甲基。
在根据该实施方案的一个另选构型中,n为1并且R3为氟。
在根据该实施方案的另一个另选构型中,n为1并且R3为氯。
在根据该实施方案的一个构型中,R2为吡啶-3-基。
在根据该实施方案的一个另选构型中,R2为吡啶-4-基。
在根据该实施方案的另一个另选构型中,R2为噻唑基,通常为噻唑-5-基。
在式IIb’或IIb”的化合物的一个实施方案中,
R1C为甲基或环丙基,其中环丙基任选被甲基、氨基或三氟甲基取代;并且
R2为噻唑-5-基、吡啶-3-基或吡啶-4-基;
R3为甲基、氯、氟或三氟甲基。
在式IIb’或IIb”的化合物的一个代表性构型中,Z3为N。
通用合成方法
本发明的化合物可通过多种方法制备,例如,如在下文示出和描述的例示性合成方案中所描绘的。所用的起始物质和试剂可得自商业供应商或可根据参考文献中阐述的文献过程使用本领域技术人员熟知的方法制备。
为简便起见,在以下通用合成方案中,将使用以下名称:

本发明的化合物通常通过异喹啉部分(异喹啉-bb)的氯甲基或溴甲基与R1构件(R1-bb)的偶联获得。偶联步骤通常在碱性条件下使用碱如碳酸铯、氢化钠或叔丁醇钾或类似物在有机溶剂如DMF或乙腈等中进行。策略在方案1中大体描绘。

方案1
将对核心结构的取代基在偶联之前引入异喹啉和R1构件上,或者它们可在偶联步骤之后引入。另选地,最终取代基的前体可存在于构件上并且在最终化合物的合成的后一阶段转化成期望的取代基。
可用于制备本发明的化合物的合适的异喹啉构件为4-溴-3-(溴甲基)异喹啉。构件可商购获得或者可如方案2中所概述的那样进行制备。

方案2
使用例如如J.Org.Chem.,1991,56(8),2805-2809中所述的过程,通过用氢溴酸和溴进行处理实现的可商购获得的3-甲基异喹啉的溴化提供溴衍生物(2A),随后通过用N-溴代琥珀酰亚胺和四氯化碳进行处理实现的苄型溴化提供期望的构件(2B)。
卤代基取代的异喹啉构件的另选方法例示于方案3中。

方案3
任选取代的苯甲醛与可为羟基保护的丙-2-炔-1-醇在Sonogashira条件下,即使用Pd催化剂诸如双(三苯基膦)氯化钯(II)或等效物以及卤化铜诸如碘化铜或类似物,在诸如三乙胺或类似物的碱的存在下,在交叉偶联反应中的反应提供乙炔衍生物(3A)。在硫酸镁或类似物的存在下与叔丁胺的反应,随后与碘的反应,以及任选的羟基去保护,提供异喹啉衍生物(3B)。苄型羟基基团接着可被转化成合适的离去基团诸如氯或溴。通常,氯衍生物(3C)通过用光气处理醇实现,而溴衍生物(3D)通常通过在三苯基膦的存在下用四氯化碳处理醇实现。另选地,碘衍生物(3B)可进一步反应,以引入期望的R2取代基或其合适的前体。
R2为任选取代的芳基或杂芳基的本发明的化合物使用携带与R1构件偶联的期望的R2基团的异喹啉部分便利地制备。此类异喹啉可例如通过钯催化的交叉偶联反应制备,如方案4中所例示。

方案4
使用催化剂如双(三苯基膦)氯化钯(II)或等效物在诸如碳酸钾或类似物的碱的存在下进行的卤代基取代的异喹啉(4A)和期望的芳基或杂芳基的硼酸(4B)的反应提供联芳基衍生物(4C)。使用如上所述的条件进行的苄型羟基基团到氯化物或溴化物的转化提供期望的异喹啉衍生物(4D),以备用于与R1构件偶联。
其中R2为烷基或取代烷基的异喹啉例如通过异喹啉卤化物与期望的烯烃在Heck偶联条件下的反应、以及随后双键的还原获得,如方案5中所示。

方案5
另选地,异喹啉构件(5C)可使用9-BBN或类似物通过烯烃(5B)的硼氢化以及随后的所得硼酸酯,即利用钯催化剂在三苯基膦或类似物和诸如三乙胺或碳酸钾或类似物的碱的存在下的Suzuki偶联获得。
可用于使用任选取代的苯甲醛作为起始物质制备其中R2为烷基或取代烷基的本发明的化合物的异喹啉的另一个另选制备路线例示于方案6中。

方案6
通过与叔丁胺的反应由任选取代的苯甲醛制备的亚胺(6A)与适当地羟基保护的乙炔衍生物在使用如二溴双(三苯基膦)镍l(II)或等效物的催化剂的交叉偶联反应中的反应提供喹啉衍生物(6B)。使用对于所用的保护基适当的条件去除羟基保护基,例如在THP基团的情况下通过用酸进行处理,接着如上文所述进行卤化,提供异喹啉(6D),以备用于与期望的R1构件偶联。在化合物6D中的R为羟基的情况下,在与化合物6A的偶联中使用的乙炔二醇的羟基基团被适当地保护。优选地,在这种情况下,利用正交保护基以便实现选择性去保护,例如可使用甲硅烷基基团,例如TBDMS以及缩醛基团诸如四氢吡喃基基团。
以类似的方式,可使用取代的乙炔衍生物制备携带直接连接的任选取代的环烷基或杂环烷基的异喹啉,如方案7中所示。

方案7
R1构件可根据文献过程或如以下本文所公开的那样进行制备。例如,WO2003/053344和WO2013/186335公开了R1构件的制备,其中Z1为N,Z2为CH并且Z3为CH或N。在J.OrgChem.60(1995)1565-1582中,公开了用于制备R1’构件的方法,其中q为0,Z1为N。螺环R1构件(即其中Z1为CR1BR1B并且两个R1B与它们所附接的碳原子一起形成C3-C6环烷基或4-6元杂环基)公开于例如WO2014/060411和WO2015/022301中。其中Z2为CH且Z3为CH或N的R1构件可根据例如WO2014/009302或Tetrahedron 70(2014)8413-8418中所述的过程制备。
例如,其中Z1为CR1BR1B并且两个R1B与它们所附接的碳原子一起形成4至6元杂环基的R1构件可如方案8中所示进行制备。

方案8
期望的氨基衍生物(8A)和酸(8B)在肽偶联条件下诸如在偶联剂如HATU、EDC或类似物的存在下在胺如DIEA或类似物的存在下的偶联提供酰胺(7c)。使用例如对甲氧基苄基基团(其可通过与对甲氧基苄基氯在碱如碳酸钾或类似物的存在下的反应引入)保护酰胺氮,接着使用例如乙酸钯和三环己基膦或类似物以及碱诸如叔丁醇钠或类似物进行钯催化的环闭合来提供双环(8E)。根据所用的保护基使用适当的条件去除两个N-保护基,诸如在Boc和对甲氧基苄基的情况下进行酸处理,提供未保护的螺环(8F)。然后可通过与酸R1CC(=O)OH在肽偶联条件下反应,或与酰氯R1CC(=O)Cl反应将所得的胺转化成酰胺(8G),或通过与氯甲酸酯R1COC(=O)Cl或酸酐R1COC(=O)OC(=O)R1C或类似物反应将所得的胺转化成氨基甲酸酯(8H)。胺与磺酰氯R1CS(=O)2Cl的反应提供磺酰胺(3i),而与羰基二咪唑或光气或类似物,接着与胺H2NR1CC或HNR1DR1D’C的反应分别提供脲(8J)或(8K)。
可用于制备其中Z1为NR1A并且R1A为4至6元环胺的式I的化合物的R1构件的制备路线描绘于方案9中。

方案9
使用如在碱诸如二异丙基乙胺或类似物的存在下的条件在如DMF的溶剂中并且通常在升高的温度下硝基取代的芳基卤化物(9A)在取代反应中与适当保护的杂环基胺(9B)的反应提供取代的苯胺衍生物(9C)。使用如碳载钯的催化剂在如MeOH或EtOH等的溶剂中或类似的条件下,通过例如催化氢化实现的硝基基团的还原提供苯胺(9D)。接着通过在碱如三乙胺或类似物的存在下与羰基二咪唑或光气或三光气的反应进行环形成,从而提供双环化合物(9E)。
类似地,其中Z1为NR1A并且R1A为任选取代的C3-C7环烷基的本发明的化合物可使用与芳基卤化物9A偶联的期望的取代的环烷基胺以类似方式制备。此构件的制备路线例示于方案10中。

方案10
其中Z1为NR1A并且R1A为在氮原子上被取代的环胺的本发明的化合物的制备路线大体描绘于方案11中。

方案11
通过用如上所述的Cs2CO3处理实现的喹啉衍生物(11A)与适当地N-保护的R1构件的偶联,以及随后N-保护基的去除提供化合物(11B),以备用于使用期望基团取代环胺。例如,可使胺与酸R1CC(=O)OH在肽偶联条件下反应或与酰氯R1CC(=O)Cl反应,从而提供酰胺(11C)。与磺酰氯R1CS(=O)2Cl的反应提供磺酰胺(11D),而与氯甲酸酯R1CC(=O)Cl或酸酐R1CC(=O)OC(=O)R1C或类似物的反应提供氨基甲酸酯(11E)。与羰基二咪唑或光气或类似物的反应以及随后与胺H2NR1C或HNR1DR1D’C的反应分别提供脲(11F)或(11G)。
其中n≥1并且R3为氨基取代的C3-C6环烷基或4至6元杂环基的式I的化合物的制备路线大体例示于方案12中。

方案12
使用正丁基锂或叔丁基锂将任选取代的卤代基取代的异喹啉(12A)锂化,接着与期望的环烷基或杂环基的亚磺酰胺衍生物反应提供酰胺(12B)。接着如上文所述引入期望的R1构件,即去除羟基保护基,如上文所述分别使用例如CBr4或光气将因此释放的羟基基团转化成溴或氯提供异喹啉衍生物(12C)以及期望的R1构件的最终偶联。
药物组合物
适用于施用本发明的化合物的制剂将对本领域普通技术人员显而易见并且包括例如片剂、丸剂、胶囊、栓剂、锭剂、糖锭剂、溶液、糖浆、酏剂、小药囊、注射剂、吸入剂以及粉剂等。一种或多种药物活性化合物的含量作为整体应在组合物的0.05至90重量%,优选地0.1至50重量%的范围内。
合适的片剂可例如通过将一种或多种本发明的化合物与已知的赋形剂,例如惰性稀释剂、载体、粘结剂、崩解剂、佐剂、表面活性剂和/或润滑剂混合来获得。片剂也可由数层组成。
合适的吸入剂可例如通过施用呈溶液、干粉或混悬液形式的一种或多种本发明的化合物来获得。本发明的化合物可以雾化或气溶胶化剂量的形式通过溶液的吸入施用。
每天适用的本发明的化合物的剂量范围通常为0.01至100mg/kg体重,优选地0.1至50mg/kg体重。每个剂量单位可便利地包含5%至95%的活性化合物(w/w)。优选地,此类制剂包含20%至80%的活性化合物。
实际的药物有效量或治疗剂量当然将取决于本领域技术人员已知的因素,诸如患者的年龄和体重、施用途径以及疾病的严重程度。在任何情况下,将基于患者的独特状况以允许递送药物有效量的剂量和方式施用组合。
组合疗法
当本发明的组合物包含本发明的化合物和一种或多种另外的治疗或预防药剂的组合时,化合物和另外的药剂两者应以介于通常在单一治疗方案中施用的剂量的约10至100%之间并且更优选地介于约10与80%之间的剂量水平存在。因此,根据一个实施方案,本发明的药物组合物另外包含一种或多种抗病毒剂。
设想用于这样的组合疗法的抗病毒剂包括有效抑制病毒在人类中的产生和/或复制的药剂(化合物或生物制品),包括但不限于干扰病毒在人类中的产生和/或复制所需的宿主或病毒机制的药剂。此类药剂可选自:RSV融合物抑制剂,诸如MDT-637(MicroDose)、BTA-9881(Biota);RSV聚合酶抑制剂,诸如ALS-8112(Alios)、ALS-8176(Alios)和Virazole(利巴韦林(ribavirin));其他药剂,诸如GS-5806(Gilead Sciences)和RSV-604(Novartis);抗体,诸如 (palimizumab)、
(palimizumab)、 (RSV-IG)、MEDI-557(MedImmune/AstraZeneca)、ALX-0171(Ablynx)、莫维珠单抗(motavizumab)(MedImmune/AstraZeneca);其他生物制品,诸如ALN-RSV-01(Alnylam)和疫苗,诸如MEDI-559(MedImmune/AstraZeneca)、RSV F(Novavax)、MEDI-534(MedImmune/AstraZeneca)。
(RSV-IG)、MEDI-557(MedImmune/AstraZeneca)、ALX-0171(Ablynx)、莫维珠单抗(motavizumab)(MedImmune/AstraZeneca);其他生物制品,诸如ALN-RSV-01(Alnylam)和疫苗,诸如MEDI-559(MedImmune/AstraZeneca)、RSV F(Novavax)、MEDI-534(MedImmune/AstraZeneca)。
实施例
本发明的其他特征将由阐述本发明原理的以下非限制性实施例变得显而易见。如本领域技术人员熟知的,在需要保护反应组分不受空气或湿气影响的情况下,在惰性气氛(包括但不限于氮气或氩气)中进行反应。温度以摄氏度(℃)给出。溶液百分比和比率表示体积与体积的关系,除非另行指出。在以下实施例中使用的反应物可由商业来源获得或者它们可由如本文所述的可商购获得的起始物质或通过本领域已知的方法制备。
本发明的所有化合物均根据本文所述的实施例合成。对技术人员将显而易见的是,可在适当的修改下使用类似的合成路线制备如本文所述的本发明的化合物。本文所述的反应的进展在适当时通过例如LC、GC或TLC来跟踪,并且如技术人员将容易地认识到的,可相应地调整反应时间和温度。
除了以上定义之外,在以上合成方案和以下实施例中还使用了以下缩写。如果本文所用的缩写未定义,那么它具有其公认的含义
ABC 碳酸氢铵
Ac 乙酰基
ACN 乙腈
AcOH 乙酸
Bn 苄基
Boc 叔丁氧羰基
BOP-Cl 双(2-氧代-3-噁唑烷基)次膦酰氯
CDI 1,1’-羰基二咪唑
DCC 二环己基碳二亚胺
DCM 二氯甲烷
DIEA 二异丙基乙胺
DIPEA 二异丙基乙胺
DMAP 4-二甲氨基吡啶
DME 1,2-二甲氧基乙烷
DMEM 杜贝科氏改良的伊戈尔氏培养基
DMF N,N-二甲基甲酰胺
DMSO 二甲亚砜
EC50 50%有效浓度
EDAC 1-乙基-3-(3-二甲氨基丙基)碳二亚胺
Et 乙基
EtOAc 乙酸乙酯
Et3N 三乙胺
EtOH 乙醇
Et2O 乙醚
LC 液相色谱
HATU [O-(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐]
HDMS 六甲基二硅氮烷
HOAc 乙酸
HOBt 羟基苯并三唑
HPLC 高效液相色谱
Me 甲基
MeCN 乙腈
MeO 甲氧基
MeOH 甲醇
MS 质谱
PCC 氯铬酸吡啶鎓
Pg 保护基
Ph 苯基
PCC 氯铬酸吡啶鎓
rt 室温(18至22℃)
TBAF 氟化四丁基铵
TEA 三乙胺
TEST 双(三乙氧基甲硅烷基)丙基-四硫化物
TFA 三氟乙酸
TFAA 三氟乙酸酐
THF 四氢呋喃
在表1中例示的化合物在文献中有所描述并在本发明的化合物的合成中用作中间体且按照所指示的那样来提及
表1


中间体的制备
中间体1

步骤a)2-硝基-N-(2,2,2-三氟乙基)苯胺(I-1a)
向2,2,2-三氟乙胺的HCl盐(1.0g,7.38mmol,1当量)在DMF(8.4mL)中的溶液中添加DIPEA(1.6mL,9.2mmol,1.25当量)和1-氟-2-硝基-苯(0.65mL,6.15mmol,0.83当量)。将混合物在80℃下搅拌16h,然后倾注到柠檬酸水溶液(10%)中并且搅拌5分钟。用EtOAc(3x30mL)萃取混合物并且用盐水(2x10mL)洗涤。将有机层经无水Na2SO4干燥、过滤并且减压浓缩。使用20%EtOAc/己烷,在硅胶(100-200目)上通过柱色谱纯化所得的粗料,得到为固体的标题化合物(0.84g,51.7%)。
步骤b)N1-(2,2,2-三氟乙基)苯-1,2-二胺(I-1b)
向化合物I-1a(5.0g,22.7mmol)的乙醇溶液(50mL)中添加Pd/C(50%水分,500mg)并且将混合物在室温下在40psi的氢气压力下在帕尔振荡器(Parr shaker)中搅拌4h。通过硅藻土垫过滤反应混合物并且将滤液真空浓缩。使用在己烷中的30%EtOAc作为洗脱液通过柱色谱(100-200目硅胶)纯化粗化合物,得到标题化合物(2.8g,65%)。
步骤c)1-(2,2,2-三氟乙基)-1H-苯并[d]咪唑-2(3H)-酮(I-1c)
在环境温度、氮气气氛下向化合物I-1b(4.5g,23.5mmol,1当量)在DCM(50mL)中的经搅拌溶液中添加DIPEA(12.3mL,70mmol,3当量),接着添加CDI(6.1g,37.7mmol,1.6当量)。将反应混合物搅拌3h,接着用水稀释并且用DCM萃取。将有机层用盐水洗涤,经无水Na2SO4干燥,并且减压浓缩。使用在己烷中的40%EtOAc,通过硅胶(100-200目)柱色谱纯化所得的粗化合物,得到为固体的标题化合物(1.2g,23%)。
1H NMR 400MHz,DMSO-d6δ4.68-4.75(q,J=9.4Hz,2H),7.01-7.10(m,3H),7.21-7.22(m,1H),11.11(s,1H)。
中间体2

步骤A)环丙基(4-硝基苯基)碳酸酯(I-2A)
向0℃的环丙基硼酸(5g,58mmol)在10%NaOH水溶液中的经搅拌溶液中缓慢添加50%H2O2并且在0℃下搅拌2h。向反应混合物中添加饱和的硫代硫酸钠水溶液,用乙醚萃取产物并且用饱和的硫代硫酸钠水溶液、水和盐水洗涤醚层。将有机层干燥(Na2SO4)并且在低温下真空浓缩以获得无色液体,其直接使用而无需进一步纯化。
将所得的液体(4.17g,包含乙醚)溶解于乙腈(40mL)中并且冷却至0℃。添加吡啶(9.3g,119mmol)并且将混合物在0℃下搅拌15min并且在N2气氛下添加4-硝基苯基氯甲酸酯(8g,39.8mmol)的乙腈(40mL)溶液。接着将反应混合物在环境温度下搅拌12h。将反应混合物用水稀释并且将有机组分用EtOAc萃取、用盐水洗涤并干燥(Na2SO4)并且减压浓缩以获得粗化合物。使用4%EtOAc/己烷作为洗脱液,通过硅胶(100-200目)柱色谱纯化粗化合物,获得为固体的标题化合物(1.5g)。

步骤a)4-((3-硝基吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(I-2a)
向4-氯-3-硝基吡啶(15g,94.9mmol,1当量)在DMF(100mL)中的经搅拌溶液中添加DIPEA(19.9mL,114mmol,1.2当量),接着添加4-氨基哌啶-1-羧酸叔丁酯(18.9g,94.9mmol,1当量)并且将所得混合物在80℃下搅拌16h。将混合物冷却至环境温度并且用冷却水稀释并用EtOAc萃取,将有机相用水、盐水洗涤并干燥(Na2SO4)。将有机层减压浓缩并且使用20%EtOAc/己烷,通过硅胶(100-200目)柱色谱纯化残余物,获得为固体的标题化合物(10g,33%)。MS 323.3[M+H]+。
步骤b)4-((3-氨基吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(I-2b)
向化合物I-2a(12g,41.1mmol)在甲醇(100mL)中的经搅拌溶液中添加披钯炭(2.5g,50%水分)并且将混合物在氢气(气球压力)下搅拌2h。通过硅藻土过滤反应混合物并且将滤液真空浓缩,获得为固体的粗标题化合物(8g),其无需进一步纯化即用于下一步骤。MS 292.9[M+H]+。
步骤c)4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸叔丁酯
(I-2c)
在氮气气氛下向化合物I-2b(5g,17.06mmol,1当量)在DCM(60mL)中的经搅拌溶液中添加DIPEA(4mL,42.7mmol,2.5当量),接着添加CDI(4.3g,26.6mmol,1.6当量)并且将所得反应混合物在环境温度下搅拌12h。用水稀释反应混合物并且将有机组分萃取到DCM中。将有机层用盐水洗涤、干燥(Na2SO4)并且减压浓缩。将粗化合物在30%EtOAc/己烷中搅拌30min,然后过滤,得到为固体的标题化合物(3.5g,64%)。MS 319.2[M+H]+。
步骤d)1-(哌啶-4-基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(I-2d)
在0℃下将HCl(4M,在二噁烷中,50mL)添加到化合物I-2c(8.5g,26.7mmol,1当量)在二噁烷(20mL)中的经搅拌溶液中。将溶液在环境温度下搅拌16h,接着真空浓缩并且将所得固体用乙醚研磨,获得为固体的标题化合物(4.8g,82%)。MS 219.3[M+H]+。
步骤e)4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸环丙基
酯(I-2e)
在0℃下将TEA(4.17mL,41.2mmol,3当量)添加到化合物I-2d(3.0g,13.76mmol,1当量)在DCM(50mL)中的经搅拌溶液中,接着添加化合物I-2A(3.07g,13.7mmol,1当量)。将混合物在环境温度下搅拌12h,接着用水稀释并且用EtOAc萃取。将有机层用盐水洗涤、干燥(Na2SO4)并且减压浓缩。使用5%MeOH/DCM作为洗脱液,通过硅胶(100-200目)柱色谱纯化所得的粗化合物,获得为固体的标题化合物(1.8g,43%)。MS(ES+)303.2[M+H]+。
中间体3

步骤a)1-叔丁基3-乙基2-(3-硝基吡啶-4-基)丙二酸酯(I-3a)
在0℃、N2气氛下向4-氯-3-硝基吡啶(11g,69.6mmol)在THF(100mL)中的经搅拌溶液中添加NaH(60%,在矿物油中,8.3g,208.8mmol)。将混合物在15℃下搅拌1h,接着冷却至0℃,并且逐滴添加乙基丙二酸叔丁酯(14.5g,76.5mmol)的THF(50mL)溶液。将所得反应混合物在15℃下搅拌1h,接着用饱和的NH4Cl水溶液淬灭。将反应混合物用水稀释并且用EtOAc萃取。将有机层用水、盐水洗涤、干燥(Na2SO4)并且减压浓缩,得到为油状物的粗标题化合物(8g),其无需进一步纯化即用于下一步骤。
步骤b)2-(3-硝基吡啶-4-基)乙酸乙酯(I-3b)
在0℃、N2气氛下向化合物I-3a(25g,80.64mmol)在DCM(150mL)中的经搅拌溶液中逐滴添加TFA(25mL),并且将所得混合物在60℃下搅拌16h。通过完全蒸馏出挥发物来浓缩反应混合物并且通过冰冻的水缓慢稀释。添加NaHCO3水溶液并且用EtOAc萃取混合物。接着将有机层用水和盐水洗涤、干燥(Na2SO4)并且真空浓缩。使用25%EtOAc/己烷作为洗脱液,在硅胶(100-200目)上通过柱色谱纯化所得的粗化合物,获得为液体的标题化合物(12.5g,74%)。MS(ES+)211.0[M+H]+。
步骤c)2-(3-氨基吡啶-4-基)乙酸乙酯(I-3c)
在氮气下将10%Pd/C(50%水分)添加到化合物I-3b(1g,4.7mmol)的甲醇(10mL)溶液中。将混合物在氢气气氛(50psi)、环境温度下在帕尔振荡器中搅拌4h,接着通过硅藻土过滤,并且将滤液真空浓缩,得到为油状物的标题化合物(550mg,64%),其无需进一步纯化即用于下一步骤。MS(ES+)181.2[M+H]+。
步骤d)1H-吡咯并[2,3-c]吡啶-2(3H)-酮盐酸盐(I-3d)
向化合物I-3c(30g,167mmol)在1.4N HCl水溶液中的经搅拌溶液中添加二异丙醚并且将所得混合物在环境温度下搅拌16h。从反应混合物中分离二异丙醚并且用DCM洗涤含水部分。收集水层并真空浓缩。将所得的粗化合物用乙酸乙酯研磨并过滤,得到为固体的标题化合物(15g,67%)。
步骤e)1-苄基螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(I-3e)
在-78℃下经15min的时间段向化合物I-3d(11g,82mmol)在THF(200mL)中的经搅拌溶液中添加1M LiHMDS/THF溶液(234mL)。使所得反应混合物缓慢升温至0℃并且在氮气气氛、0℃下添加N-苄基-2-氯-N-(2-氯乙基)乙胺。接着将所得反应混合物在回流下搅拌12h。将反应混合物用10%NH4Cl水溶液淬灭,用水稀释并且用EtOAc萃取有机组分。将有机部分用水和盐水洗涤,干燥(Na2SO4)并且减压浓缩,得到为浅褐色固体的粗化合物。使用5%MeOH/DCM,通过硅胶(100-200目)柱色谱纯化粗化合物,获得为米白色固体的化合物8(3g,12%)。MS(ES+)293.7[M+H]+。
步骤f)螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(I-3f)
在氮气下将10%Pd/C(50%水分)添加到化合物I-3e(5g,17mmol)的甲醇(80mL)溶液中。将所得反应混合物在氢气气氛(50psi,50℃)下在帕尔振荡器中搅拌15h,接着通过硅藻土过滤并且将滤液真空浓缩,获得为油状物的粗标题化合物(3g,86%)。粗化合物无需进一步纯化即用于下一步骤。MS(ES+)204.3[M+H]+。
步骤g)2'-氧代-1',2'-二氢螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯
(I-3g)
在0℃下向化合物I-3f(3.5g,17.2mmol)在甲醇(40mL)中的经搅拌溶液中添加二碳酸二叔丁酯(3.9g,17.2mmol)并且将混合物在环境温度下搅拌16h。将反应混合物真空浓缩并且使用5%MeOH/DCM,通过硅胶(100-200目)柱色谱纯化粗料,得到为固体的标题化合物(3.9g,75%)。MS(ES+)304.2[M+H]+。
中间体4

步骤a)2-(3-((四氢-2H-吡喃-2-基)氧基)丙-1-炔-1-基)苯甲醛(I-4a)
将2-溴苯甲醛(20.0g,108.11mmol,1当量)和2-(丙-2-炔-1-基氧基)四氢-2H-吡喃(17.90g,129.73mmol,1.2当量)在Et3N(300mL)中的溶液用氩气脱气,接着添加PdCl2(PPh3)2(1.52g,2.16mmol,0.02当量)和CuI(0.205g,1.08mmol,0.01当量)并且将混合物再次用氩气脱气10min,接着在Ar气氛下在80℃下加热4h。真空浓缩混合物并且将残余物用水(30mL)溶解并且用乙酸乙酯(2x 250mL)萃取。将合并的有机层用水和盐水洗涤,经无水Na2SO4干燥,并且减压浓缩。使用EtOAc,通过硅胶(100-200目)柱色谱纯化所得的残余物,得到标题化合物(21g,80%)。
步骤b)3-(羟甲基)-4-碘代异喹啉2-氧化物(I-4b)
向化合物I-4a(0.5g,2.05mmol,1当量)在EtOH(10mL)中的经搅拌悬浮液中添加NH2OH(0.213g,3.07mmol,1.5当量)和吡啶(0.24mL,4.10mmol,2.0当量)并且将反应混合物在环境温度下搅拌40min。添加I2(2.6g,10.2mmol,5当量)并且继续搅拌15min。如通过TLC所确认的反应完全之后,将产物萃取到DCM中。使用在DCM中的2%MeOH,通过硅胶(100-200目)柱色谱纯化粗产物,获得为固体的标题化合物(240mg,30%)。
步骤c)3-(羟甲基)-4-碘代异喹啉2-氧化物(I-4c)
向化合物I-4b(8.0g,26.6mmol,1当量)和TBDPSCl(9.82g,31.9mmol,1.2当量)在TEA(50.0mL)和DMF(10.0mL)中的经脱气溶液中添加PdCl2(PPh3)2(0.187g,0.266mmol,0.01当量)和CuI(0.101g,0.532mmol,0.02当量)并且再次用氩气脱气10min。将所得混合物在Ar气氛下加热至80℃,持续4h。在反应完全(通过TLC监测)后,将反应混合物用DCM稀释并用水和盐水洗涤。将合并的有机层经无水Na2SO4干燥,过滤并且减压浓缩。使用在己烷中的70%乙酸乙酯,通过硅胶(100-200目)柱色谱纯化所得粗料,得到为固体的标题化合物(8.0g,62%)。
步骤d)3-(羟甲基)-4-碘代异喹啉2-氧化物(I-4d)
向化合物I-4c(3.0g,6.2mmol,1当量)在MeOH(70mL)中的经搅拌的用氩气脱气的溶液中添加炭中钯(10%,4.2g,50%水分)。通过气球压力将混合物置于氢气气体下并且在环境温度下搅拌。14h之后,通过硅藻土过滤反应混合物。将滤液减压浓缩,与甲苯共沸蒸馏并浓缩,得到为固体的标题化合物(2.8g,96%)。
步骤e)4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-3-(氯甲基)异喹啉(I-4e)
在0℃下将光气(2.16mL,29.81mmol,2当量)逐滴添加到化合物I-4d(7.0g,14.9mmol,1当量)在DCM(200mL)中的经搅拌溶液中。然后将反应混合物在环境温度下搅拌17h,接着减压浓缩,用DCM稀释并且用水和盐水洗涤。将合并的有机层经无水Na2SO4干燥,并且减压浓缩。使用在DCM中的1%MeOH,通过硅胶(100-200目)柱色谱纯化所得的残余物,得到为固体的标题化合物(5.5g,76%)。MS(ES+)488.0[M+H]+。
中间体5

步骤a)4-((3-氯-5-硝基吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(I-5a)
向4-氨基哌啶-1-羧酸叔丁酯(4.47g,22.39mmol)在DMF(60mL)中的经搅拌溶液中添加DIPEA(4.68mL,26.87mmol)和3,4-二氯-5-硝基吡啶(4.3g,22.39mmol)并且将所得反应混合物在80℃下搅拌16h。将反应混合物冷却至室温,用冷却水稀释并且将有机组分萃取到EtOAc中。将有机层用水、盐水洗涤,并且经无水Na2SO4干燥。接着将有机层真空浓缩,获得为深褐色油状物的粗产物。使用20%EtOAc/己烷作为洗脱液,通过硅胶(100-200目)柱色谱纯化粗料,得到为固体的标题化合物(2g,25%)。
步骤b)4-((3-氨基-5-氯吡啶-4-基)氨基)哌啶-1-羧酸叔丁酯(I-5b)
向化合物I-5a(5g,14.1mmol,1当量)在THF(80mL)和水(20mL)的混合物中的经搅拌溶液中缓慢添加NH4Cl(7.5g,141mmol,10当量)和Zn粉(7.3g,113mmol,8当量)并且将所得反应物料在100℃下搅拌2h。通过硅藻土过滤反应混合物并且将滤液真空浓缩,接着用水和EtOAc稀释。将有机组分用EtOAc(3x 150mL)萃取,接着用水和盐水洗涤。将有机层经无水硫酸钠干燥并且减压浓缩,获得褐色油状物作为粗标题化合物(3.5g,76%),其无需进一步纯化即用于下一步骤。
步骤c)4-(7-氯-2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸叔
丁酯(I-5c)
在环境温度、氮气气氛下向化合物I-5b(5g,15.34mmol,1当量)在DCM(150mL)中的经搅拌溶液中添加DIPEA(21mL,123mmol,8当量)和CDI(9.9g,61.3mmol,4当量)并且继续搅拌12h。用水稀释反应混合物并且将有机组分萃取到DCM中。将有机层用盐水洗涤,并经无水Na2SO4干燥,并且减压浓缩。将所得的粗料在30%EtOAc/己烷中搅拌30min并过滤,获得为固体的标题化合物(3g,55%)。
中间体6

步骤a)6-((2-氨基苯基)氨基)-2-氮杂螺[3.3]庚烷-2-羧酸叔丁酯(I-6a)
在氮气气氛下向在甲醇(50mL)中的1,2-苯二胺(1,54g,14,2mmol)中顺序添加6-氧代-2-氮杂螺[3.3]庚烷-2-羧酸叔丁酯(1.00g,4,73mmol)、乙酸(0,38mL,6,64mmol)和氰基硼氢化钠(447mg,7,11mmol)。将所得混合物在室温下搅拌1h 10min,接着减压浓缩。使残余物在饱和碳酸氢钠水溶液与乙酸乙酯之间分配。将层分离并且用新鲜的乙酸乙酯萃取水相。使有机萃取物通过相分离器并且减压浓缩。使用在庚烷中的0-100%乙酸乙酯的梯度通过快速色谱纯化所得残余物。将期望级分汇集并减压浓缩,通过快速色谱进一步纯化残余物,使用二氯甲烷(2CV)第一等度洗脱,接着利用在二氯甲烷(6CV)中的5%甲醇等度洗脱。将期望级分汇集并减压浓缩,得到标题化合物和副产物(957mg,57%),其无需进一步纯化原样用于下一步骤。
MS(ES+)m/z 304[M+H]+,(ES-)m/z 362[M+OAc]-。
步骤b)6-(2-氧代-2,3-二氢-1H-苯并[d]咪唑-1-基)-2-氮杂螺[3.3]庚烷-2-羧
酸叔丁酯(I-6b)
将I-6a(957mg,3,15mmol)和1,1'-羰基二咪唑(613mg,3,78mmol)在DCM(40mL)中的混合物在室温下搅拌5天。添加水并且分离层。用乙酸乙酯萃取水相两次。使有机萃取物通过相分离器并且减压浓缩。使用在庚烷中的0-70%乙酸乙酯的梯度通过快速色谱纯化。将期望的级分汇集并且减压浓缩,得到标题化合物(340mg,31%)。MS(ES-)m/z 328[M-1]-。
中间体7

步骤a)(E)-N-(2-溴-5-氯亚苄基)-2-甲基丙-2-胺
将2-溴-5-氯苯甲醛(7g,31.9mmol)在叔丁胺(25mL)中的溶液在密封管中在80℃下加热17h,接着将反应混合物冷却并减压浓缩,得到标题化合物(8.5g,97%)。MS(ES+)273.8&275.8[M+H]+。

步骤b)叔丁基二苯基((7-((四氢-2H-吡喃-2-基)氧基)庚-5-炔-1-基)氧基)硅烷
(I-7b)
将正丁基锂(2.2M,在己烷中,6.8mL,1.05当量)逐滴添加到-78℃的叔丁基(4-碘代丁氧基)二苯基硅烷(6.25g,14.3mmol,1当量)在THF(80mL)中的经搅拌溶液中,接着添加DMPU(15mL)。30min后,经2h的时间段使反应混合物升温至-10℃并且在-10℃保持20min。接着使反应混合物再次冷却至-78℃并且添加2-(丙-2-炔-1-基氧基)四氢-2H-吡喃(2g,14.3mmol,1当量)的THF(10mL)溶液。使反应混合物缓慢达到室温并且搅拌17h。将饱和的氯化铵水溶液添加到反应混合物中并且将有机组分萃取到乙醚中。将有机层用水和盐水洗涤并干燥(Na2SO4)并且减压浓缩。使用在己烷中的10%EtOAc,通过硅胶(100-200目)柱色谱纯化所得的残余物,得到标题化合物(5.5g,85%)。
步骤c)4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-7-氯-3-(((四氢-2H-吡喃-
2-基)氧基)甲基)异喹啉(I-7c)
将包含Zn(43.6mg,0.67mmol,3当量)和NiBr2(PPh3)2(8.25mg,0.011mmol,0.05当量)的圆底烧瓶抽真空并且用氩气重新填充。这个过程重复三次,然后添加化合物I-7b(0.1g,450mmol,0.22当量)和化合物I-7a(0.06g,0.22mmol,1当量)在乙腈(5mL)中的经脱气溶液。将反应混合物在80℃、氩气下加热3h,接着通过硅藻土过滤。将滤液减压浓缩并且使用在己烷中的0-15%EtOAc,通过硅胶(100-200目)柱色谱纯化,得到标题化合物(60mg,46%)以及另一种区域异构体(regioisomer)。两种异构体均进行下一步骤并且在那个阶段通过1D NOE确认构型。
步骤d)(4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-7-氯异喹啉-3-基)甲醇(I-
7d)
将pTSA一水合物(77.6mg,0.4mmol,0.3当量)添加到化合物I-7c(800mg,1.36mmol,1当量)在MeOH(10mL)中的经搅拌溶液中。将混合物在回流下搅拌6h,接着添加固体NaHCO3并且将混合物减压浓缩。将残余物用乙酸乙酯稀释并且用饱和NaHCO3、水和盐水洗涤。将合并的有机层干燥(Na2SO4)并真空浓缩。使用在己烷中的30%EtOAc,通过硅胶(100-200目)柱色谱纯化粗料,获得为固体的标题化合物(370mg,54%)。MS(ES+)504.0[M+H]+。
步骤e)4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-7-氯-3-(氯甲基)异喹啉(I-
7e)
将光气(0.76mL,10.5mmol,2当量)逐滴添加到化合物I-7d(2.65g,5.25mmol,1当量)在DCM(15mL)中的冰冷溶液中。将反应混合物在环境温度下搅拌5h,接着减压浓缩,用DCM稀释并且用饱和碳酸氢钠水溶液、水和盐水洗涤。将合并的有机层干燥(Na2SO4)并减压浓缩,得到为固体的标题化合物(2.7g,98%)。MS(ES+)522.1[M+H]+。
中间体8

步骤a)4-((4,5-二氟-2-硝基苯基)氨基)哌啶-1-羧酸叔丁酯(I-8a)
将K2CO3(4.9g,35.6mmol,1.26当量)分批添加到1,2,4-三氟-5-硝基苯(5g,28.2mmol,1当量)和4-氨基哌啶-1-羧酸叔丁酯(7.91g,39.5mmol,1.4当量)在1,4-二噁烷(50mL)中的经搅拌溶液中。将所得混合物在环境温度下搅拌12h,接着用水稀释并且将有机组分萃取到EtOAc中。用水和盐水洗涤有机层。将合并的有机层经无水Na2SO4干燥,并且减压浓缩。使用10%的己烷中的EtOAc,通过硅胶(100-200目)柱色谱纯化所得的粗化合物,得到呈固体形式的标题化合物(4.2g,42%)。MS(ES+)358.2[M+H]+。
步骤b)4-((2-氨基-4,5-二氟苯基)氨基)哌啶-1-羧酸叔丁酯(I-8b)
将Pd/C(10%,350.0mg,50%水分)添加到化合物I-8a(3.5g,9.80mmol,1.0当量)在MeOH(60.0mL)中的经搅拌且用氩气脱气的溶液中。将混合物在氢气气氛(50psi)下在帕尔振荡器中于环境温度下振荡4h,接着通过硅藻土垫过滤并且用MeOH彻底洗涤。将滤液减压浓缩,得到为固体的标题化合物(2.21g,69%)。MS(ES+)328.3[M+H]+。
步骤c)4-(5,6-二氟-2-氧代-2,3-二氢-1H-苯并[d]咪唑-1-基)哌啶-1-羧酸叔丁
酯(I-8c)
在环境温度、氮气气氛下向化合物I-8b(6.0g,18.39mmol,1当量)在DCM(50.0mL)中的经搅拌溶液中添加DIPEA(8.0mL,45.87mmol,2.5当量),之后添加CDI(5.94g,36.7mmol,2.0当量)。将混合物在环境温度下搅拌12h,接着用水稀释并且将有机组分萃取到DCM中。用水和盐水洗涤有机层。将合并的有机层经无水Na2SO4干燥,并且减压浓缩。将所得粗产物溶解于在己烷中的30%EtOAc中,搅拌30min,然后过滤,得到为固体的标题化合物(3.2g,49%)。MS(ES+)352.3[M-H]-。
1H NMR DMSO-d6δ1.426(s,9H),1.644-1.670(d,2H),2.146-2.240(m,2H),2.828(bs,2H),4.074-4.092(m,2H),4.249-4.310(m,1H),7.014-7.058(t,1H),7.425-7.471(t,1H),11.0393(s,1H)。
中间体9

步骤a)4-((2-氯-6-硝基苯基)氨基)哌啶-1-羧酸叔丁酯(I-9a)
向1-氯-2-氟-3-硝基苯(3.64g,20.72mmol,1当量)在DMF(42.0mL)中的经搅拌溶液中添加DIPEA(8.7mL,49.93mmol,2.4当量),之后添加4-氨基哌啶-1-羧酸叔丁酯(5.0g,24.96mmol,1.2当量)。将所得反应混合物在80℃下搅拌16h,接着倾注到10%柠檬酸水溶液中并且将有机组分萃取到EtOAc中。用水和盐水洗涤有机层。将合并的有机层经无水Na2SO4干燥并减压浓缩,得到为固体的期望化合物3(4.2g,47%)。MS(ES+)356.2[M+H]+。
步骤b)4-((2-氨基-6-氯苯基)氨基)哌啶-1-羧酸叔丁酯(I-9b)
向化合物I-9a(10.0g,28.16mmol,1当量)在THF:水(4:1)(90.0mL)中的经搅拌溶液中添加NH4Cl(15.1g,281.7mmol,10当量),之后添加锌粉(14.7g,225mmol,8当量)。将混合物在80℃下搅拌1h,接着通过硅藻土过滤并且用EtOAc洗涤硅藻土床。将有机层减压浓缩并且使粗化合物在水与乙酸乙酯之间分配。将合并的有机层用水和盐水洗涤,且经无水Na2SO4干燥并减压浓缩,得到为液体的标题化合物(8.0g,87%)。
步骤c)4-(7-氯-2-氧代-2,3-二氢-1H-苯并[d]咪唑-1-基)哌啶-1-羧酸叔丁酯
(I-9c)
在环境温度、氮气气氛下向化合物I-9b(8.0mg,24.62mmol,1当量)在DCM(80.0mL)中的经搅拌溶液中添加DIPEA(15.9mL,123mmol,5.0当量),之后添加CDI(11.97g,73.8mmol,3.0当量)。将混合物搅拌3h,接着用水稀释并且将有机组分萃取到DCM中。将有机层用水和盐水洗涤,经无水Na2SO4干燥,并且减压浓缩。使用在己烷中的30%乙酸乙酯,通过硅胶(100-200目)柱色谱纯化所得粗料,获得纯产物,将所述纯产物用乙醚和戊烷研磨,得到为固体的标题化合物(5.6g,65%)。MS(ES+)352.1[M+H]+。1H NMR DMSO-d6δ1.421(s,9H),1.719-1.749(m,2H),2.413-2.438(m,2H),2.796(m,2H),4.071-4.091(m,2H),5.0274(bs,1H),6.931-7.023(m,3H),11.1873(s,1H)。
中间体10

4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-3-(氯甲基)异喹啉-7-甲腈(I-10)
根据针对中间体7所述的过程制备标题化合物,但是使用4-溴-3-甲酰基苄腈代替2-溴-5-氯苯甲醛。MS(ES+)513.1[M+H]+。1H NMR DMSO-d6δ0.95(s,9H),1.73(m,4H),3.17(m,2H),3.71(m,2H),5.02(s,2H),7.40-7.45(m,6H),7.57-7.59(m,4H),8.05-8.07(d,1H),8.31-8.33(d,1H),8.82(s,1H),9.31(s,1H)。
中间体11

步骤a)3-((2-溴苯基)氨基甲酰基)氮杂环丁烷-1-羧酸叔丁酯(I-11a)
在23℃下向2-溴苯胺(5g,29.42mmol,1当量)和DMAP(4.6g,37.5mmol,1.3当量)在DCM(83mL)中的经搅拌溶液中添加1-(叔丁氧羰基)氮杂环丁烷-3-羧酸(5.85g,29.10mmol,1当量),之后添加EDCIxHCl(7.24g,37.93mmol,1.3当量)并且将所得反应混合物在环境温度下搅拌12h。接着用10%柠檬酸水溶液、水、饱和Na2CO3水溶液、盐水洗涤反应混合物,并且将有机组分萃取到DCM中。将有机层经无水Na2SO4干燥,并且减压蒸发,得到粗化合物。使用在己烷中的15%EtOAc作为洗脱液,通过硅胶(100-200目)柱色谱纯化粗化合物,得到为米白色固体的标题化合物(6g,57.6%)。
步骤b)3-((2-溴苯基)(4-甲氧基苄基)氨基甲酰基)氮杂环丁烷-1-羧酸叔丁酯
(I-11b)
向化合物I-11a(6g,16.9mmol,1当量)和1-(氯甲基)-4-甲氧基苯(4.02g,25.7mmol)在乙腈(80mL)中的经搅拌溶液中添加K2CO3(7.24g,50.72mmol,3当量)并且将所得反应混合物在回流下搅拌12h。将反应混合物过滤并且用乙腈洗涤固体残余物。将滤液真空浓缩并且用己烷/乙酸乙酯(30:1)研磨粗产物,得到为米白色固体的标题化合物(6.5g,81.25%)。
步骤c)1'-(4-甲氧基苄基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-羧酸叔
丁酯(I-11c)
向化合物I-11b(0.5g,1.27mmol,1当量)和tBuONa(0.182g,1.9mmol,1.5当量)在二噁烷(4mL)中的经搅拌的用氩气脱气的溶液中添加Pd(OAc)2(0.0071g,0.03mmol,0.025当量)和PCy3(0.0088g,0.03mmol,0.025当量),并且将所得混合物用氩气进一步脱气5min。接着将反应混合物在微波下在120℃下搅拌1h。接着通过硅藻土过滤反应混合物并且将滤液减压浓缩,获得粗物料,将其用水和盐水洗涤。将有机组分萃取到EtOAc中并且经无水硫酸钠干燥并真空浓缩至干燥。接着使用7%EtOAc-己烷作为洗脱液,在硅胶(230-400目)重力柱上纯化粗料,得到标题化合物。
步骤d)螺[氮杂环丁烷-3,3'-二氢吲哚]-2'-酮(I-11d)
向化合物I-11c(0.3g,0.76mmol,1当量)在TFA(1.2mL)中的溶液中添加CF3SO3H(0.20mL,2.28mmol,3当量),并且将所得混合物在23℃下搅拌12h。接着将反应混合物减压浓缩并且将残余物溶解于水中并用DCM洗涤。将水从水层中完全蒸馏出去,获得标题化合物的对应的盐,其直接用于下一步骤。
步骤e)2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-羧酸甲酯(I-11e)
在0℃下向化合物I-11d(0.167g,0.96mmol,1当量)在DMF中的经搅拌溶液中添加TEA(0.404mL,2.89mmol,3当量)和氯甲酸甲酯(0.075mL,0.95mmol,1当量)并且将所得反应混合物在室温下搅拌4h,接着用水稀释反应混合物并用乙酸乙酯萃取有机组分。将有机层用盐水洗涤并且经无水硫酸钠干燥并真空浓缩,以获得粗标题化合物。
中间体12

4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-3-(氯甲基)-5-氟异喹啉(I-12)
根据针对中间体7所述的过程制备标题化合物,但是使用2-溴-3-氟苯甲醛代替2-溴-5-氯苯甲醛。MS(ES+)506.2[M+H]+。1H NMR DMSO-d6δ0.954(s,9H),1.73(m,4H),3.18(m,2H),3.72-3.73(m,2H),5.00-5.02(s,2H),7.38-7.46(m,6H),7.58-7.74(m,6H),8.00-8.02(d,1H),9.24(d,1H)。
中间体13

步骤a)N-(3-硝基吡啶-4-基)环丙烷磺酰胺(I-13a)
向化合物3-硝基吡啶-4-胺(3.0g,24.8mmol,1当量)在DMSO(45.0mL)中的经搅拌溶液中添加NaH(60%,1.2g,29.8mmol,1.2当量)并且将反应混合物在环境温度下搅拌15min。向反应混合物中添加化合物环丙烷磺酰氯(3.92g,24.8mmol,1当量)。将混合物在80℃下搅拌16h,接着冷却至环境温度,添加冰冷的水并且将有机组分萃取到EtOAc中。将有机层用水和盐水洗涤、干燥(Na2SO4)、过滤并且减压浓缩。使用在己烷中的50%EtOAc,通过硅胶(100-200目)柱色谱纯化所得的粗料,得到为固体的标题化合物(2g,33%)。MS(ES+)243.8[M+H]+。
步骤b)N-(3-氨基吡啶-4-基)环丙烷磺酰胺(I-13b)
向化合物I-13a(2.5g,10.28mmol,1当量)在MeOH(50mL)中的经搅拌的用氩气脱气的溶液中添加Pd/C(10%,300mg,50%水分)并且将混合物在环境温度下在帕尔振荡器中在氢气气氛(40psi)下振荡12h。接着通过硅藻土过滤反应混合物并且用MeOH彻底洗涤。将滤液减压浓缩,得到为固体的粗标题化合物4(2.1g,96%),其无需任何纯化直接用于下一步骤。MS(ES+)214.1[M+H]+。
步骤c)1-(环丙基磺酰基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(I-13c)
在氮气气氛下向化合物I-13b(1.3g,6.10mmol,1当量)在DCM(30mL)中的经搅拌溶液中添加DIPEA(6.3mL,65.4mmol,8当量),之后添加CDI(1.98g,12.2mmol,2.0当量)并且将所得混合物在环境温度下搅拌12h。接着用水稀释反应混合物并且将有机组分萃取到DCM中。将有机层用水和盐水洗涤,经无水Na2SO4干燥,并且减压浓缩。将所得的粗料与乙腈一起搅拌30min,接着过滤,得到为固体的标题化合物(800mg,55%)。MS(ES+)240.0[M+H]+。1HNMR DMSO-d6δ1.20-1.21(m,2H),1.27-1.31(m,2H),7.49-7.50(d,1H),8.28-8.33(m,2H),11.8836(s,1H)。
中间体14

4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-3-(氯甲基)-8-氟异喹啉(I-14)
根据针对中间体4所述的过程制备标题化合物,但是代替地以2-溴苯甲醛起始。总收率:8.5%。MS(ES+)506.2[M+H]+。1H NMR DMSO-d6δ0.87-0.96(s,9H),1.75(m,4H),3.16(m,2H),3.73(m,2H),5.02(m,2H),7.40-7.45(m,6H),7.50-7.60(m,5H),7.79-7.83(m,1H),7.99(m,1H),8.00(m,1H),9.35(s,1H)。
中间体15

步骤a)3-((3-硝基吡啶-4-基)氨基)氮杂环丁烷-1-羧酸叔丁酯(I-15a)
向4-氯-3-硝基吡啶(2.5g,15.8mmol,1当量)在乙醇中的经搅拌溶液中添加三乙胺(4.4mL,31.5mmol,2当量)并且使反应混合物冷却至0℃。接着在0℃下向反应混合物中逐滴添加3-氨基氮杂环丁烷-1-羧酸叔丁酯(2.98g,17.3mmol,1.1当量)并且使反应混合物缓慢升温至80℃,在80℃下搅拌12h。将反应混合物减压浓缩并用水稀释并且将有机组分萃取到EtOAc中。将有机层用盐水洗涤,经无水Na2SO4干燥并真空浓缩。使用50%乙酸乙酯/己烷,通过硅胶(100-200目)柱色谱纯化粗料,获得为固体的标题化合物(3.5g,75%)。MS(ES+)294.8[M+H]+。
步骤b)3-((3-氨基吡啶-4-基)氨基)氮杂环丁烷-1-羧酸叔丁酯(I-15b)
向化合物I-15a(13.5g,45.9mmol)在EtOAc(200mL)中的经搅拌溶液中添加10%披钯炭(2.5g,50%水分)并且在环境温度下在帕尔振荡器中在氢气气氛(50psi)下搅拌2h。通过硅藻土过滤反应混合物并且将滤液真空浓缩。将获得的粗料用MTBE研磨以获得为固体的标题化合物(12.1g,99%)。MS(ES+)264.9[M+H]+。
步骤c)3-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)氮杂环丁烷-1-羧酸
叔丁酯(I-15c)
在0℃下将CDI(2.38g,14.7mmol,1.05当量)添加到化合物I-15b(3.7g,14mmol,1当量)在乙腈(40mL)中的经搅拌溶液中。将反应混合物在环境温度下搅拌1h,接着过滤并且将固体残余物用乙腈研磨并减压干燥以得到为固体的标题化合物(2.7g,66%)。MS(ES+)291.2[M+H]+。
步骤d)1-(氮杂环丁烷-3-基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(I-15d)
在0℃下将HCl(4M,在二噁烷中,20mL)添加到化合物I-15c(4.2g,14.5mmol,1当量)在二噁烷(5mL)中的经搅拌溶液中。将反应混合物在环境温度下搅拌1h,接着过滤并且将固体残余物用MTBE洗涤以得到标题化合物(3.6g)。
步骤e)1-(1-(环丙基磺酰基)氮杂环丁烷-3-基)-1H-咪唑并[4,5-c]吡啶-2(3H)-
酮(I-15e)
在环境温度下将DIPEA(2.5当量)添加到化合物I-15d(0.4g,0.53mmol)在DCM中的经搅拌溶液中。使混合物冷却至0℃,接着缓慢添加环丙烷磺酰氯并且将混合物在0℃下搅拌1h,接着在室温下搅拌12h。将反应混合物用水稀释并且用EtOAc萃取。将有机层经无水Na2SO4干燥并减压浓缩,得到标题化合物(73mg)。
中间体16

步骤a)N-(1-甲基环丙基)-3-硝基吡啶-4-胺(I-16a)
在N2气氛下,在冰冷的条件下将TEA(1.8mL,12.66mmol)添加到4-氯-3-硝基吡啶(1.0g,6.32mmol)在EtOH(20mL)中的经搅拌溶液中,接着添加1-甲基环丙胺的HCl盐(0.74g,6.96mmol)。将所得混合物在80℃下搅拌16h,接着减压浓缩。将残余物用水稀释并且用EtOAc萃取。将有机层用水和盐水洗涤、干燥(Na2SO4)并且减压浓缩。使用在己烷中的30%EtOAc,通过硅胶柱色谱纯化所得的粗产物,得到为固体的标题化合物(410mg,60%)。MS(ES+)193.6[M+H]+。
步骤b)N4-(1-甲基环丙基)吡啶-3,4-二胺(I-16b)
将Pd/C(10%,50%水分,50mg)添加到化合物I-16a(0.38g,1.96mmol)在EtOH(10mL)中的经搅拌的用氩气脱气的溶液中。将所得反应混合物在氢气气氛(40psi)、环境温度下在帕尔振荡器中振荡16h,接着通过硅藻土垫过滤并且将滤液减压浓缩,得到为固体的标题化合物(290mg,90%)。MS(ES+)164.0[M+H]+。
步骤c)1-(1-甲基环丙基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(I-16c)
在氮气气氛下将DIPEA(0.9mL,5.34mmol)添加到化合物I-16b(0.29g,1.8mmol)在DCM(10mL)中的经搅拌溶液中,接着添加CDI(0.346g,2.14mmol)。将反应混合物在环境温度下搅拌16h,接着用水稀释并且用DCM萃取混合物。将有机层用水和盐水洗涤,干燥(Na2SO4)、过滤并且减压浓缩,得到为深褐色固体的粗化合物。使用在DCM中的4%MeOH,通过硅胶(100-200目)柱色谱纯化粗化合物,获得为米白色固体的化合物RSV_BB132(150mg,45%)。MS(ES+)189.09[M+H]+。
中间体17

步骤a)3-((2-氟乙基)氨基)螺[环丁烷-1,3'-二氢吲哚]-2'-酮(I-17a)
将2-氟乙胺盐酸盐(691mg,6.94mmol)和TEA(1.94mL,13.9mmol)在氮气、室温下添加到螺[环丁烷-1,3'-二氢吲哚]-2',3-二酮(1.3g,6.94mmol)在MeOH(20mL)中的经搅拌溶液中。将反应混合物在室温下搅拌1h,接着添加氰基硼氢化钠(873mg,13.9mmol),将所得反应混合物在室温下搅拌12h。将反应混合物用水(50mL)稀释、用EtOAc(2x 50mL)萃取并且用水(20mL)和盐水(10mL)洗涤合并的有机层。将有机层经硫酸钠干燥、过滤并且减压浓缩。在硅胶上通过快速色谱纯化粗料,使用在DCM中的4%MeOH洗脱,得到为固体的标题化合物(1.3g,60%)。MS(ES+)235.13[M+H]+。
步骤b)(2-氟乙基)(2'-氧代螺[环丁烷-1,3'-二氢吲哚]-3-基)氨基甲酸叔丁酯
(I-17b)
在氮气、0℃下将Boc酸酐(1.29mL,5.64mmol)添加到I-17a(1.1g,4.70mmol)和TEA(1.31mL)在DCM(30mL)中的经搅拌溶液中。将所得的混合物在室温下搅拌3h,接着用水(50mL)稀释并且用DCM(2x 50mL)萃取。将合并的有机层用水(50mL)和盐水(50mL)洗涤,经硫酸钠干燥、过滤并减压浓缩。使用在DCM中的3%MeOH,通过快速色谱纯化粗料,得到标题化合物(650mg,41%)。
MS(ES+)335.26[M+H]+。
中间体18

1-((1-甲基环丙基)磺酰基)螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(I-
18)
在氮气气氛、0℃下向化合物I-3f(0.50g,2.46mmol)在吡啶(6mL)中的经搅拌溶液中添加在吡啶(2mL)中的1-甲基环丙烷-1-磺酰氯(0.61g,3.94mmol)。将反应混合物在室温下6h,接着用冷水(30mL)稀释并且用EtOAc(2x50mL)萃取。将合并的有机层用盐水和冷水(2x30mL)洗涤、经无水硫酸钠干燥并减压浓缩。在中性氧化铝上通过柱色谱纯化所得的粗料,使用2%的MeOH:DCM洗脱,得到标题化合物(110mg,10%)。MS(ES+)322.98[M+H]+。
中间体19

2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-羧基酰胺(I-19)
在室温下向I-11d的TFA盐(1.1g)在DCM(20mL)中的经搅拌溶液中添加三乙胺(1.9mL)和(三甲基甲硅烷基)异氰酸酯(0.67mL)。将所得混合物在室温下搅拌1h,然后将形成的固体过滤并干燥,得到标题化合物(400mg,46%)。MS(ES+)218.26[M+H]+。
中间体20

N-(叔丁基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-羧基酰胺(I-20)
在0℃下向I-11d的TFA盐(300mg,1.72mmol)在DCM(10mL)中的经搅拌悬浮液中添加三乙胺(0.266mL)。将悬浮液搅拌5min,接着添加叔丁基异氰酸酯(256mg,2.58mmol)并且在室温下继续搅拌2h。将反应混合物真空浓缩,得到粗标题化合物(300mg,73%)。MS(ES+)274.24[M+H]+。
中间体21

1-(甲基磺酰基)螺[氮杂环丁烷-3,3'-二氢吲哚]-2'-酮(I-21)
在0℃下向I-11c(1.8g)在DMF(15mL)中的经搅拌溶液中添加三乙胺(2.6mL)和甲磺酰氯(0.48mL)。将所得混合物在0℃下搅拌1h,接着用冰水稀释并搅拌10分钟,形成固体并过滤和干燥,得到为固体的标题化合物(480mg,30%)。MS(ES+)253.14[M+H]+。
中间体22

步骤a)4-(6-氟吡啶-3-基)-3-甲基异喹啉(I-22a)
在氩气下将(6-氟吡啶-3-基)硼酸(1.05g,7.43mmol)和碳酸钠(1.97g,18.6mmol)添加到4-碘-3-甲基异喹啉(2.00g,7.43mmol)在1,2-二甲氧基乙烷(25mL)和水(10mL)的溶液中。将反应混合物用氩气脱气15分钟,接着在氩气、室温下添加与二氯甲烷(1.09g,1.49mmol)络合的1,1′-双(二苯基膦基)二茂铁]-二氯化钯(II)。将反应混合物在密封管中在100℃下搅拌。当如通过TLC所判断的确认反应完全时(~6h),将混合物冷却至室温,添加水(100mL)并且用EtOAc(2X100mL)萃取混合物。将合并的有机相干燥(Na2SO4)、过滤并浓缩。在硅胶上通过柱色谱纯化所得的粗化合物,使用在石油醚(p.ether)中的20-30%EtOAc洗脱,得到为固体的标题化合物(1.4g,72%)。MS(ES+)239.43[M+H]+。
步骤b)3-(溴甲基)-4-(6-氟吡啶-3-基)异喹啉(I-22b)
在0℃下将偶氮二异丁腈(103mg)和N-溴琥珀酰亚胺(822mg)添加到化合物I-22a(1.00g)在四氯化碳(50mL)的经搅拌溶液中。在70℃下搅拌反应混合物并且通过TLC监测进展。6h后,用饱和碳酸氢钠溶液(100mL)稀释混合物并用DCM(2X50mL)萃取。将合并的有机层干燥(Na2SO4),过滤并且减压浓缩。在硅胶上通过柱色谱纯化所得的粗料,使用在石油醚中的24%EtOAc洗脱,得到为固体的标题化合物(700mg,43%)。MS(ES+)318.85[M+H]+。
使用适当的硼酸和通过TLC判断的反应时间,用针对中间体22所述的方法制备以下异喹啉衍生物。


1在步骤a中使用四(三苯基膦)-钯(0)作为催化剂。
2在步骤a中使用1,4-二噁烷/H2O作为溶剂。
3在步骤a中使用(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)衍生物代替对应的硼酸衍生物。
中间体31

螺[环丁烷-1,3'-二氢吲哚]-2',3-二酮(I-31)
在室温、氮气下向3-氨基螺[环丁烷-1,3'-二氢吲哚]-2'-酮(1,0g,5.3mmol,如WO2014/060411中所述进行制备)在THF(30mL)中的经搅拌溶液中添加TEA(1.48mL,10.6mol)。将反应混合物冷却至0℃,接着添加氯甲酸甲酯(493μl,6.38mmol)。将所得反应混合物在室温下搅拌1h,用水(100mL)稀释,用EtOAc(2x 200mL)萃取。将合并的有机层用水(100mL)和饱和的碳酸氢盐溶液(50mL)洗涤,经硫酸钠干燥、过滤并减压浓缩。通过硅胶柱色谱(洗脱液在DCM中的1%MeOH)纯化粗料,得到为固体的标题化合物(700mg,49%)。MS(ES+)247.20[M+H]+。
中间体32

(1-(2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-基羰基)环丙基)氨基甲酸叔丁
酯(I-32)
在室温下向1-((叔丁氧羰基)氨基)环丙烷羧酸(1.36g,6.77mmol)在DMF(20mL)中的经搅拌溶液中添加EDAC(1.20g,6.24mmol)、HOBt(0.960g,6.24mmol)、Et3N(2.1mL,20.8mmol)和I-11d(1.50g,5.20mmol)。将所得反应混合物在室温下搅拌16h,接着用水(30mL)稀释并且用EtOAc(2x30mL)萃取。将有机层用水(3x50mL)洗涤,干燥(Na2SO4)、过滤并且减压浓缩,得到为固体的标题化合物(800mg,41%)。MS(ES+)358.26[M+H]+。
中间体33

甲基(1-(2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-基羰基)环丙基)氨基甲酸
叔丁酯(I-33)
根据针对中间体32所述的方法使化合物I-11d的TFA盐(536mg,2.0mmol)与1-((叔丁氧羰基)(甲基)氨基)环丙烷羧酸反应,得到标题化合物(350mg,41%)。MS(ES+)372.27[M+H]+。
中间体34

1-(吗啉-4-羰基)螺[氮杂环丁烷-3,3'-二氢吲哚]-2'-酮(I-34)
在5min的过程中将吗啉-4-羰基氯(519mg)在DCM(10mL)中的溶液添加到化合物I-11d(500mg)在DCM(10mL)和10%氢氧化钠(10mL)的混合物中的经搅拌溶液中。将反应混合物在室温下搅拌3h,接着将相分离并且用DCM(50mL)洗涤氢氧化钠层。将合并的DCM层用盐水(20mL)洗涤,经硫酸钠干燥、过滤、浓缩并真空干燥,用MeCN(4mL)洗涤所得的固体,得到为固体的标题化合物(400mg,69%收率)。MS(ES+)288.42[M+H]+。
中间体35

步骤a)4-碘-3-甲基异喹啉(I-35a)
向7-氯-3-甲基异喹啉(4.80g,20.8mmol)在乙酸(50mL)中的经搅拌溶液中添加N-碘琥珀酰亚胺(5.7g,25.3mmol)。将混合物在80℃下加热3天,接着冷却至室温,用饱和氢氧化钠溶液(100mL)淬灭反应并且用乙酸乙酯(2x300mL)萃取。将合并的有机相用饱和硫代硫酸钠溶液(100mL)洗涤,经硫酸钠干燥、过滤并减压浓缩。在硅胶上通过柱色谱纯化所得的粗化合物,使用在石油醚中的10%EtOAc洗脱。收集纯级分并且减压浓缩,得到标题化合物(4g,62%)。通过1H NMR确认结构。
步骤b)4-溴-3-(溴甲基)-7-氯异喹啉(I-35b)
在室温、氮气下向化合物I-35a(4g)在CCl4(400mL)中的经搅拌溶液中添加偶氮二异丁腈(425mg)和N-溴琥珀酰亚胺(9.3g)。将所得反应混合物回流18h,接着冷却至室温。添加Na2S2O3饱和溶液(100mL)并且用DCM(2x250mL)萃取混合物。将合并的有机相经硫酸钠干燥、过滤并且减压浓缩。通过硅胶柱色谱纯化粗产物,使用在石油醚中的15-20%EtOAc洗脱,得到标题化合物以及不可分离的副产物(2.1g)。材料在下面的步骤中原样使用,无需进一步纯化。
中间体36

步骤a)N-(3-氟代苄基)-1,1-二甲氧基丙-2-胺(I-36a)
在室温下向(3-氟苯基)甲胺(20.0g)在1,2-二氯乙烷(800mL)中的经搅拌溶液中添加丙酮醛二甲基缩醛(28.3g),之后添加三乙酰氧基硼氢化钠(67.7g),并且将混合物搅拌16h,接着添加2N NaOH(100mL)水溶液并且搅拌混合物直到有机层几乎透明为止。将层分离,用DCM(2x50mL)萃取水层。将合并的有机物经Na2SO4干燥、过滤并减压浓缩,以获得粗标题化合物(32g)。MS(ES+)228.17[M+H]+。
步骤b)7-氟-3-甲基异喹啉(I-36b)
在<5℃下将化合物36a(32g,141mmol)逐滴添加到氯磺酸(96.5mL,1.41mmol)中。将混合物加热至100℃,持续10min,接着冷却并倾注到冰(700g)中。用MTBE(700mL)洗涤混合物,将水层冷却至5℃并且用50%NaOH水溶液(400mL)碱化至pH 14。用DCM(2x 100mL)萃取水层。将合并的有机层经Na2SO4干燥、过滤并减压浓缩,以获得粗标题化合物(11g,34%)。MS(ES+)162.09[M+H]+。
步骤c)3-(溴甲基)-7-氟-4-碘异喹啉(I-36c)
将N-碘琥珀酰亚胺(8.38g)添加到I-36b(5g)在乙酸(50mL)中的经搅拌溶液中。将反应混合物在80℃下加热3天,接着冷却至室温。添加氢氧化钠溶液(150mL)并且用乙酸乙酯(2x 200mL)萃取混合物。将合并的有机相用饱和硫代硫酸钠溶液(100mL)洗涤,经硫酸钠干燥、过滤并减压浓缩。在硅胶上通过柱色谱纯化获得的粗化合物,使用在石油醚中的10%EtOAc洗脱。汇集纯级分并且减压浓缩,得到标题化合物(2.3g,33%)。MS(ES+)288.01[M+H]+。
步骤d)4-溴-3-(溴甲基)-7-氟异喹啉(I-36d)
在室温、氮气下将偶氮二异丁腈(343mg)和N-溴琥珀酰亚胺(7.44g)添加到化合物I-36c(3.0g,10.4mmol)在CCl4(300mL)中的经搅拌溶液中。将所得混合物在回流下搅拌18h,接着冷却至室温。添加Na2S2O7(100mL)的溶液并且用DCM(2x 250mL)萃取混合物。将合并的有机层经硫酸钠干燥、过滤并且减压浓缩。将获得的粗产物与先前制备的批次合并并且在二氧化硅上通过柱色谱纯化,使用在石油醚中的15-20%EtOAc洗脱。汇集纯级分并且减压浓缩,得到标题化合物。
中间体37

步骤a)3-((4-溴吡啶-3-基)氨基甲酰基)氮杂环丁烷-1-羧酸叔丁酯(I-37a)
向3-氨基-4-溴吡啶(5.05g,29.2mmol)和N-boc-氮杂环丁烷-3-羧酸(6.17g,30.6mmol)在无水DCM(100mL)中的溶液中添加DMAP(4.64g,38.0mmol)和EDC-盐酸盐(7.27g,38.0mmol)。将混合物在室温下搅拌三天,接着用乙酸乙酯稀释并且用水和盐水洗涤两次。用乙酸乙酯萃取水相一次,将合并的有机相经硫酸钠干燥并减压浓缩。通过硅胶色谱分离产物,用DCM和0至3%MeOH洗脱,得到标题化合物(9.6g,92%)。MS(ES+)356.2&358.2[M+H]+。
步骤b)3-((4-溴吡啶-3-基)(2,4-二甲氧基苄基)氨基甲酰基)氮杂环丁烷-1-羧
酸叔丁酯(I-37b)
向I-37a(7.12g,20.0mmol)在干燥DMF(25mL)中的溶液中添加碳酸铯(1.63g,50.0mmol)并且将混合物在室温下搅拌30分钟。添加1-(氯甲基)-2,4-二甲氧基苯(8.77g,47.0mmol)在苯(~10mL)中的溶液并且将混合物在室温下搅拌两小时。添加水并且用乙酸乙酯萃取混合物三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。通过硅胶色谱纯化产物,用DCM和10至50%EtOAc洗脱,得到标题化合物(10.1g,79%)。
步骤c)1'-(2,4-二甲氧基苄基)-2'-氧代-1',2'-二氢螺[氮杂环丁烷-3,3'-吡咯
并[2,3-c]吡啶]-1-羧酸叔丁酯(I-37c)
在氩气下将叔丁醇钠(2.28g,23.7mmol)、乙酸钯(II)(355mg,1.58mmol)和三环己基膦(443mg,1.58mmol)添加到I-37b在无水二噁烷(85mL)中的溶液中。将混合物在氩气、95℃下搅拌两小时,接着冷却至室温并且添加到饱和氯化铵溶液中。用DCM萃取混合物四次,将有机相经硫酸钠干燥并减压浓缩。通过硅胶色谱分离产物,用DCM和20至60%EtOAc洗脱,得到标题化合物(6.72g,80%)。MS(ES+)426.4[M+H]+。
步骤d)2'-氧代-1',2'-二氢螺[氮杂环丁烷-3,3'-吡咯并[2,3-c]吡啶]-1-羧酸
叔丁酯(I-37d)
将I-37c(3.22g,7.58mmol)在乙腈(80mL)中的溶液添加到硝酸铈铵(3.29g)的冰冷水(40mL)溶液中。将反应混合物在室温下搅拌两小时,接着添加另外的硝酸铈铵(1.64g)并且将混合物在室温下再搅拌两小时。添加5%碳酸钾溶液(400mL)并且用乙酸乙酯萃取混合物四次。将有机相经硫酸钠干燥并且减压浓缩。通过硅胶色谱分离产物,用DCM和2至8%MeOH洗脱,得到标题化合物(1.05g,50%)。MS(ES+)276.3[M+H]+。
中间体38

步骤a)N-((5-溴吡啶-2-基)亚甲基)-2-甲基丙烷-2-亚磺酰胺(I-38a)
将碳酸铯(2.5g,7.68mmol)添加到5-溴-2-吡啶羧醛(1.19g,6.4mmol)和2-甲基-2-丙烷亚磺酰胺(0.78g,6.4mmol)在DCM(6mL)中的混合物中。将混合物在室温下搅拌20h,接着用DCM稀释,用H2O洗涤,经Na2SO4干燥并浓缩。在硅胶上通过柱色谱纯化,利用在正庚烷中的0至51%EtOAc梯度洗脱,得到标题化合物(1.76g,95%)。MS(ES+)289.1;291.1[M+H]+。
步骤b)N-(1-(5-溴吡啶-2-基)-2,2,2-三氟乙基)-2-甲基丙烷-2-亚磺酰胺(I-
38b)
将N-((5-溴吡啶-2-基)亚甲基)-2-甲基丙烷-2-亚磺酰胺(1.91g,6.6mmol)和四甲基氟化铵(738mg,7.93mmol)在THF(20mL)中的溶液在室温下用Ar净化15min。在-55℃/-60℃下向其中添加三氟甲基三甲基硅烷TMSCF3(2.44mL,16.5mmol)。将反应混合物在相同的温度下搅拌1h,接着通过在0℃下添加饱和NH4Cl水溶液(15mL)淬灭反应。分离有机层,用EtOAc(2x 20mL)萃取水相并且将合并的有机层经Na2SO4干燥并减压浓缩。在硅胶上通过柱色谱纯化,利用在正庚烷中的EtOAc梯度洗脱,得到标题化合物(1.80g,76%)。MS(ES+)359.1;361.2[M+H]+。
步骤c)1-(5-溴吡啶-2-基)-2,2,2-三氟乙胺盐酸盐(I-38c)
将在1,4-二噁烷(0.52ml)中的4M HCl添加到N-(1-(5-溴吡啶-2-基)-2,2,2-三氟乙基)-2-甲基丙烷-2-亚磺酰胺(148mg,0.41mmol)在MeOH(0.5mL)中的溶液中并且将反应物搅拌2h。将混合物减压浓缩,与甲苯共蒸发并且减压干燥18h,得到标题化合物(120mg,99%)。
MS(ES+)255.1;257.1[M+H]+。
步骤d)(1-(5-溴吡啶-2-基)-2,2,2-三氟乙基)氨基甲酸叔丁酯(I-38d)
将二碳酸二叔丁酯(180mg,0.82mmol)添加到1-(5-溴吡啶-2-基)-2,2,2-三氟乙胺盐酸盐(120mg,0.41mmol)、吡啶(0.07ml,0.82mmol)和4-4-二甲基氨基吡啶(10.1mg,0.08mmol)在1,4-dioxane(3mL)中的溶液中。将获得的反应混合物搅拌24h。将固体过滤出来并且将滤液减压浓缩。在硅胶上通过柱色谱纯化,利用在正庚烷中的EtOAc梯度洗脱,得到标题化合物(146mg,99%)。MS(ES+)299.1;301.1[M+H]+。
步骤e)(2,2,2-三氟-1-(5-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)吡啶-
2-基)乙基)氨基甲酸叔丁酯(I-38e)
通过使N2气体通过3-5min对(1-(5-溴吡啶-2-基)-2,2,2-三氟乙基)氨基甲酸叔丁酯(146mg,0.41mmol)、双(频哪醇)二硼(113mg,0.44mmol)、Pd(dppf)Cl2(15.04mg,0.02mmol)和乙酸钾(80.69mg,0.82mmol)在1,4-二噁烷(3mL)中的悬浮液脱气。接着在130℃下通过微波辐射加热反应物1h。然后使混合物在EtOAc与5%NaHCO3之间分配。将有机层经Na2SO4干燥并且减压浓缩。使用硅胶色谱纯化,利用在正庚烷中的EtOAc梯度洗脱,得到标题化合物(140mg,85%)。MS(ES+)403.4[M+H]+。
中间体39

步骤a)1'-((4-溴-7-氯异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-4,3'-
吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(I-39a)
将碳酸铯(2.1g,6.45mmol)添加到I-3g(633mg,2.09mmol)在DMF(20ml)中的溶液中。将浆液在室温下搅拌1h,接着添加I-35b(700mg,2,09mmol)并且将混合物搅拌过夜。在EtOAc与H2O之间萃取反应混合物。用EtOAc(x 2)进一步萃取水相。将汇集的有机相经MgSO4干燥、浓缩并真空干燥。在硅胶上通过色谱纯化粗料,使用在DCM中的0-5%MeOH洗脱,得到标题化合物(843mg,72%)。MS(ES+)559.33[M+H]+。
步骤b)1'-((4-溴-7-氯异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-4,3'-
吡咯并[2,3-c]吡啶]-1-羧酸甲酯(I-39b)
将化合物I-39a(501mg,0.898mmol)与在二噁烷中的4M HCl(6.75mL)、MeOH(3mL)和DCM(11mL)在室温下搅拌1h 15min。将混合物真空浓缩,接着从甲苯中共蒸发2x,得到HCl盐的淡黄色固体。添加DCM(4mL),之后添加DIEA(0.8mL,4.6mmol)和氯甲酸甲酯。将混合物搅拌1h 45min,接着在真空下,与更多DCM共蒸发数次。通过二氧化硅色谱(梯度在DCM中的1%至7%MeOH)纯化所得油状物,得到为固体的标题化合物(410mg,88%)。MS(ES+)517.3[M+H]。
中间体40

步骤a)1'-((4-溴-7-氯异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[氮杂环丁
烷-3,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(I-40a)
在氮气、室温下向I-37d(2.0g,7.26mmol)在乙腈(30mL)中的经搅拌溶液中添加碳酸铯(7.2g,22.1mmol)。将反应混合物搅拌15min,接着添加I-35b(2.5g,7.45mmol)。将所得反应混合物搅拌3h,接着减压浓缩。将残余物用水(80mL)稀释,用在DCM中的5%MeOH(3x100mL)萃取。将合并的有机层用水(80mL)洗涤、经硫酸钠干燥、过滤并减压浓缩。将获得的固体用乙醚(30mL)研磨、搅拌15min并过滤,得到为固体的标题化合物(2.9g,68%)。化合物无需进一步纯化即用于下一步骤。MS(ES+)531.07[M+H]+。
步骤b)1'-((4-溴-7-氯异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[氮杂环丁
烷-3,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(I-40b)
在室温下将TFA(5mL,64.9mmol)添加到I-40a(1.0g,1.88mmol)在DCM(5mL)中的经搅拌溶液中。将混合物搅拌2h,接着减压浓缩,用DCM(80mL)稀释并用饱和碳酸氢钠溶液(2x30mL)洗涤。将有机层用水(15mL)洗涤、经硫酸钠干燥、过滤并减压浓缩。将残余物溶解于DCM(20mL)中并且在0℃、氮气下添加ET3N(2.6mL,18.7mmol)和氯甲酸甲酯(220mg,2.33mmol)。将所得混合物在室温下搅拌2h,接着用DCM(50mL)稀释并且用水(30mL)洗涤。将有机层经硫酸钠干燥、过滤并且减压浓缩。在硅胶上通过快速色谱纯化粗料,使用在DCM中的5%MeOH洗脱,得到为固体的标题化合物(620mg,57%)。MS(ES+)489.25[M+H]+。
实施例1

步骤a)3-((4-溴异喹啉-3-基)甲基)-1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-
酮(1a)
将碳酸铯(2.44g,7.50mmol)添加到4-溴-3-(溴甲基)异喹啉(755mg,2.51mmol)和1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(442mg,2.52mmol)在DMF(24mL)中的溶液中并且将混合物在室温下搅拌3h。添加冰水(150mL)并且将混合物搅拌30min,接着过滤并且用水洗涤形成的沉淀。将沉淀溶解于具有一些MeOH的CH2Cl2中,真空蒸发溶剂并且在二氧化硅柱上纯化所得的粗产物,得到为固体的标题化合物(0.542g,55%)。MS(ES+)396.95[M+H]+。
步骤b)1-环丙基-3-((4-(4-羟基丁-1-炔-1-基)异喹啉-3-基)甲基)-1H-咪唑并
[4,5-c]吡啶-2(3H)-酮(1b)
向溴代喹啉1a(100mg,0.254mmol)在DMF(3mL)中的悬浮液中顺序添加PPh3(14.2mg,0.054mmol)、CuI(7.40mg,0.039mmol)、Et2NH(276mg,3.77mmol)、丁-3-炔-1-醇(23.3mg,0.333mmol),最后添加Pd(PPh3)2Cl2(18.7mg,0.027mmol)。将悬浮液在120℃下在微波反应器中加热30min,接着使其冷却至室温。以双倍大小重复以上过程一次,将两种溶液合并,用水稀释并且用EtOAc(3x30mL)萃取。将有机相合并,用水(2x 20mL)洗涤、干燥(Na2SO4)并真空浓缩。在35克二氧化硅上通过快速色谱纯化所得的粗料,使用在DCM中的2-6%MeOH的梯度洗脱,得到标题化合物(217.6mg,74.5%)。
LCMS(ES+)385.06[M+H]+。CDCl 3中的1H NMR与结构一致。
步骤c)1-环丙基-3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1H-咪唑并[4,5-c]吡
啶-2(3H)-酮(1c)
使用气球中的H2(g)和10%碳载钯(105mg),在室温下将炔烃1b(108mg,0.282mmol)在95%EtOH(19mL)+MeOH(1mL)中的溶液氢化。18h后,添加另外的Pd/C(21mg)、MeOH(1mL)和95%EtOH(2mL),并且将混合物再搅拌24h。通过槽纹滤纸过滤混合物,并且用95%EtOH(20mL)和MeOH(30mL)洗涤沉淀数次。将溶剂真空移除,得到为固体的标题化合物(76.2mg,70%)。MS(ES+)389.1[M+H]+。通过1H和13C NMR确认结构。
实施例2

步骤a)4-(3-((4-溴异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡
啶-1-基)哌啶-1-羧酸叔丁酯(2a)
将碳酸铯(650mg,1.99mmol)添加到4-溴-3-(溴甲基)异喹啉(200mg,0.665mmol)和I-2c(212mg,0.665mmol)在DMF(7.0mL)中的溶液中。将获得的浆液在室温下搅拌2h,接着添加水(10mL)和EtOAc(20mL)。用EtOAc(2x10mL)萃取水相两次,并且将有机相用盐水洗涤两次,接着干燥(Na2SO4)并浓缩。在二氧化硅上通过色谱纯化获得的粗料,使用DCM:MeOH97:3洗脱,得到标题化合物(210mg,59%)。MS(ES+)540.15[M+H]+。通过1H NMR确认结构。
步骤b)4-(3-((4-(4-羟基丁-1-炔-1-基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-
1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸叔丁酯(2b)
将PdCl2(PPh3)2(13.7mg,0.020mmol)、碘化铜(14.9mg,0.078mmol)和化合物2a(210mg,0.390mmol)溶解于微量小瓶(microvial)中的DMF(3.0mL)中,并且搅拌直到溶液澄清。使用氮气气体将小瓶抽真空,然后添加3-丁炔-1-醇(54.7mg,0.78mmol)和TEA(98.7mg,0.975mmol)。将小瓶再次抽真空,接着使用微波辐射加热,110℃持续75min,接着常规加热,80℃持续16小时。再添加1当量3-丁炔-1-醇、0.05当量PdCl2(PPh3)2和0.2当量碘化铜,并且通过微波辐射在110℃下辐射混合物120min。将混合物
浓缩至干燥,之后在二氧化硅上通过色谱纯化残余物,使用DCM:MeOH95:5洗脱,得到标题化合物(151mg,73%)。MS(ES+)528.21[M+H]+。
步骤c)4-(3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-咪唑
并[4,5-c]吡啶-1-基)哌啶-1-羧酸叔丁酯(2c)
在10%Pd/C的存在下将化合物2b(40.0mg,0.076mmol)在MeOH(10mL)中的溶液在H-cube(1mL/min,30℃,30巴)中氢化两个循环。混合物接着直接用于下一步骤。
步骤d)4-(3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-咪唑
并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(2d)
将化合物2c(28mg,0.053mmol)溶解于MeOH(2.0mL)中,添加在二噁烷中的4M HCl(3mL),将澄清溶液在室温下搅拌30min,接着浓缩。用DMF稀释粗HCl盐并添加DIPEA(68.9mg,0.533mmol)。将混合物在室温下搅拌15分钟,接着在冰浴中冷却并且添加氯甲酸甲酯(10.1mg,0.107mmol)。将混合物在0℃下搅拌90分钟,接着用2mL 1M NaOH(水溶液)淬灭反应,用EtOAc稀释并且用NaHCO3和盐水洗涤,经Na2SO4干燥、过滤并浓缩。在二氧化硅上通过柱色谱纯化残余物并且将适当的级分冷冻干燥,得到标题化合物(3mg,12%)。MS(ES+)490.2[M+H]+。通过1H和13C NMR确认结构。
实施例3

步骤a)3-((4-溴异喹啉-3-基)甲基)-1-(2,2,2-三氟乙基)-1H-咪唑并[4,5-c]吡
啶-2(3H)-酮(3a)
使4-溴-3-(溴甲基)异喹啉(300mg,0.997mmol)和1-(2,2,2-三氟乙基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(217mg,0.997mmol)根据实施例1步骤a的过程反应,得到标题化合物(305.7mg,70%)。MS(ES+)436.94&438.92[M+H]+。
步骤b)3-((4-(4-羟基丁-1-炔-1-基)异喹啉-3-基)甲基)-1-(2,2,2-三氟乙基)-
1H-咪唑并[4,5-c]吡啶-2(3H)-酮(3b)
使化合物3a(299mg,0.684mmol)和3-丁炔-1-醇(62.3mg,0.889mmol)根据实施例1步骤b的过程反应,得到标题化合物(191mg,66%)。MS(ES+)427.09[M+H]+。
步骤c)3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1-(2,2,2-三氟乙基)-1H-咪唑
并[4,5-c]吡啶-2(3H)-酮(3c)
根据实施例1步骤c的过程氢化炔烃3b,得到标题化合物(78mg,41%)。MS(ES+)431.1[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.35(d,J=0.7Hz,1H),8.26(d,J=5.3Hz,1H),8.19–8.13(m,1H),8.12–8.04(m,1H),7.83(ddd,J=8.4,6.9,1.3Hz,1H),7.67(ddd,J=7.9,6.9,0.9Hz,1H),7.40(d,J=5.3Hz,1H),5.45(s,2H),4.90(q,J=9.3Hz,2H),4.43(t,J=5.1Hz,1H),3.47(q,J=6.1Hz,2H),3.26–3.19(m,2H),1.70–1.52(m,4H)。
实施例4

步骤a)3-((4-溴异喹啉-3-基)甲基)-1-(4-氟苯基)-1H-咪唑并[4,5-c]吡啶b2(3H)-酮(4a)
将1-(4-氟苯基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(171mg,0.757mmol)和Cs2CO3(728mg,2.24mmol)悬浮于DMF(7.0mL)中并且在室温下搅拌30min。接着添加为固体的4-溴-3-(溴甲基)异喹啉(228mg,0.757mmol)并且继续搅拌3h。将冰水(50mL)倾注到反应物中并且将混合物搅拌30min,接着过滤。用共计50mL水洗涤沉淀数次,接着将其溶解于DCM中。通过旋转蒸发仪除去溶剂并且真空干燥固体。将粗料溶解于DCM(5mL)中并且在二氧化硅上通过柱色谱进行纯化,使用在DCM中的0.4%至8%MeOH梯度洗脱,得到标题化合物(256mg,75%)。MS(ES+)450.86[M+H]+。
步骤b)1-(4-氟苯基)-3-((4-(4-羟基丁-1-炔-1-基)异喹啉-3-基)甲基)-1H-咪
唑并[4,5-c]吡啶-2(3H)-酮(4b)
使化合物4a(239mg,0.532mmol)和3-丁炔-1-醇(48.5mg,0.692mmol)根据实施例1步骤b的过程反应,得到标题化合物(128mg,55%)。MS(ES+)439.03[M+H]+。通过NMR确认结构。
步骤c)1-(4-氟苯基)-3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1H-咪唑并[4,5-
c]吡啶-2(3H)-酮(4c)
根据实施例1步骤c的过程使化合物4b进行氢化,得到标题化合物(51mg,40%)。MS(ES+)443.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.14(s,1H),8.46–8.41(m,1H),8.24–8.14(m,2H),8.09(dd,J=8.2,1.3Hz,1H),7.84(ddd,J=8.4,6.8,1.3Hz,1H),7.73–7.59(m,3H),7.49–7.40(m,2H),7.09(dd,J=5.3,0.8Hz,1H),5.49(s,2H),4.43(t,J=5.1Hz,1H),3.47(q,J=5.8Hz,2H),3.25(dd,J=9.3,6.4Hz,2H),1.69–1.54(m,3H)。
实施例5

步骤a)1-((4-溴异喹啉-3-基)甲基)-3-(2,2,2-三氟乙基)-1H-苯并[d]咪唑-2
(3H)-酮(5a)
使4-溴-3-(溴甲基)异喹啉(300mg,0.997mmol)和I-1c(216mg,1.00mmol)根据实施例1步骤a的过程反应,得到标题化合物(388mg,89%)。MS(ES+)436.01&437.93[M+H]+。
步骤b)1-((4-(4-羟基丁-1-炔-1-基)异喹啉-3-基)甲基)-3-(2,2,2-三氟乙基)-
1H-苯并[d]咪唑-2(3H)-酮(5b)
使化合物5a和3-丁炔-1-醇根据实施例1步骤b的过程反应,得到标题化合物(69%)。MS(ES+)426.05[M+H]+。
步骤c)1-((4-(4-羟基丁基)异喹啉-3-基)甲基)-3-(2,2,2-三氟乙基)-1H-苯并
[d]咪唑-2(3H)-酮(5c)
根据实施例1步骤c的过程氢化炔烃5b(247mg,0.58mmol),得到标题化合物(141mg,57%)。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.18–8.11(m,1H),8.09–8.03(m,1H),7.82(ddd,J=8.4,6.8,1.3Hz,1H),7.66(ddd,J=7.9,6.8,0.9Hz,1H),7.30(d,J=7.7Hz,1H),7.12(dd,J=7.6,1.2Hz,1H),7.03(dtd,J=27.2,7.7,1.2Hz,2H),5.40(s,2H),4.84(q,J=9.3Hz,2H),4.42(t,J=5.1Hz,1H),3.46(td,J=6.3,5.0Hz,2H),3.25–3.18(m,2H),1.64(p,J=6.7Hz,2H),1.53(ddt,J=9.4,6.1,3.2Hz,2H)。
实施例6
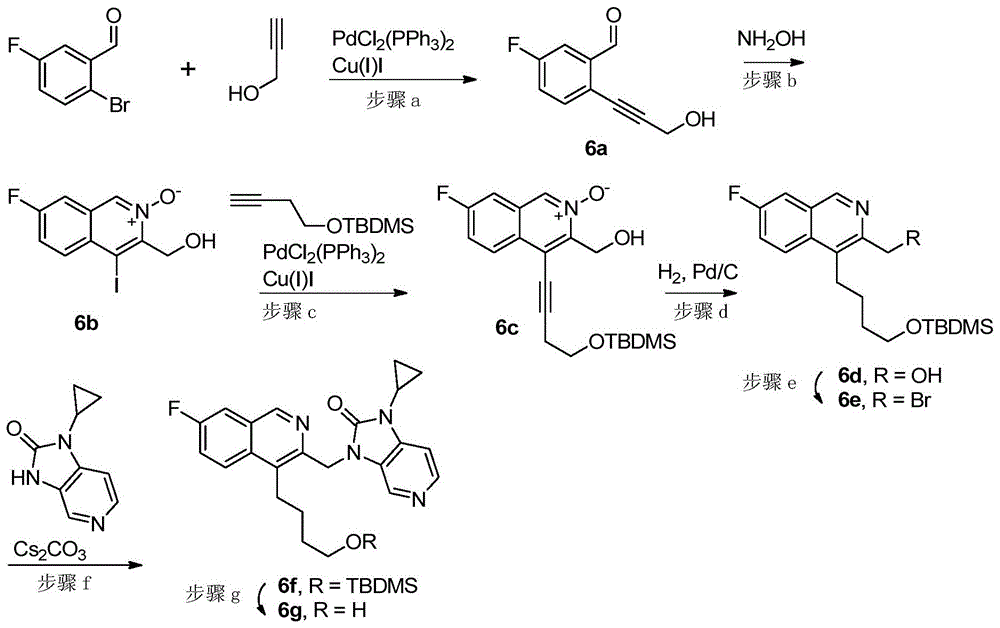
步骤a)5-氟-2-(3-羟基丙-1-炔-1-基)苯甲醛(6a)
在氮气下将3-丁炔-1-醇(331mg,5.91mmol)添加到PdCl2(PPh3)2(86.4mg,0.123mmol)、碘化铜(37.5mg,0.197mmol)和2-溴-5-氟苯甲醛(1.00g,4.93mmol)在TEA(5mL)和DMF(5mL)中的溶液中。通过微波辐射在40℃下将溶液加热整个周末,接着通过硅藻土垫过滤。用EtOAc将滤液稀释至约20mL,接着用NaHCO3和盐水洗涤。将有机层经Na2SO4干燥并浓缩,并且将所得的粗化合物在二氧化硅上纯化,利用庚烷:EtOAC(4:1)洗脱,得到为固体的标题化合物(570mg,65%)。
步骤b)(4-碘异喹啉-3-基)甲醇N-氧化物(6b)
将6a(550mg,3.09mmol)在乙醇(10mL)中的溶液在室温下逐滴添加到羟胺(322mg,4.63mmol)在乙醇(10mL)和吡啶(0.5mL)中的经搅拌溶液中。将溶液在室温下搅拌30min,接着添加碘(3.13g,12.4mmol)并且继续搅拌15分钟。添加硫代硫酸钠(饱和水溶液,10mL)并且搅拌混合物直到其变成澄清溶液。用DCM(3x20mL)萃取溶液,并且将合并的有机相用盐水洗涤、干燥(Na2SO4)并浓缩。
添加少量DCM,接着添加Et2O(20mL)。标题化合物立即析出,将其过滤出来(467mg,47%)。
LC-MS(ES+)319.82[M+H]+。
步骤c)(4-(4-((叔丁基二甲基甲硅烷基)氧基)丁-1-炔-1-基)-7-氟异喹啉-3-
基)甲醇N-氧化物(6c)
在氮气下将4-TBDMS-丁炔(260mg,1.41mmol)添加到PdCl2(PPh3)2(19.8mg,0.028mmol)、碘化铜(8.95mg,0.047mmol)和6b(300mg,0.94mmol)在DMF/TEA(3+3mL)中的混合物中。将混合物在50℃、氮气下加热24h。将混合物浓缩、用EtOAc(10mL)稀释、用NaHCO3和盐水洗涤、干燥(Na2SO4)并浓缩。在二氧化硅上通过柱色谱纯化所得的粗化合物,使用EtOAc:庚烷1:1洗脱,得到为固体的标题化合物(252mg,71%)。
LC-MS(ES+)377.01[M+H]+。
步骤d)(4-(4-((叔丁基二甲基甲硅烷基)氧基)丁基)-7-氟异喹啉-3-基)甲醇
(6d)
将化合物6c(252mg,0.671mmol)溶解于EtOH(15mL)中,添加10%碳载钯(250mg,0.211mmol)并且使混合物在室温下氢化3h,接着添加另外的Pd/C(50mg)并且再继续氢化2h。通过硅藻土过滤混合物,将滤液浓缩并且在二氧化硅上通过柱色谱纯化残余物,使用EtOAC:庚烷1:1洗脱,得到标题化合物(113mg,46%)。
步骤e)3-(溴甲基)-4-(4-((叔丁基二甲基甲硅烷基)氧基)丁基)-7-氟异喹啉
(6e)
在0℃下将三苯基膦(111mg,0.424mmol)添加到化合物6d(110mg,0.303mmol)在无水DCM(16mL)中的溶液中。5min后,添加CBr4(151mg,0.454mmol)并且将溶液在0℃下搅拌10min,接着在室温下搅拌1.5h。将反应混合物浓缩,并且在二氧化硅上通过柱色谱纯化所得的粗料,使用EtOAc:己烷1:9洗脱,得到标题化合物(98mg,76%)。
步骤f)3-((4-(4-((叔丁基二甲基甲硅烷基)氧基)丁基)-7-氟异喹啉-3-基)甲
基)-1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(6f)
将碳酸铯(103mg,0.317mmol)添加到1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(18.5mg,0.106mmol)在DMF中的溶液中。将浆液在室温下搅拌15min,接着冷却至0℃并且缓慢添加溶解于DMF中的化合物6e(45mg,0.106mmol)。使混合物达到室温并且搅拌16h。添加水(5mL)和EtOAc(10mL)。用EtOAc(5x 2mL)萃取水相两次。将合并的有机相用盐水洗涤两次,接着干燥(Na2SO4)并浓缩。在二氧化硅上通过柱色谱纯化所得的粗料,使用EtOAC:MeOH98:2洗脱,得到标题化合物(43mg,78%)。
步骤g)1-环丙基-3-((7-氟-4-(4-羟基丁基)异喹啉-3-基)甲基)-1H-咪唑并[4,
5-c]吡啶-2(3H)-酮(6g)
将在二噁烷中的4M HCl(2mL)添加到化合物6e(43mg,0.083mmol)在MeOH(2mL)中的溶液中。将溶液搅拌30min,接着通过添加几滴1M NaOH将pH调节至~7。用EtOAc稀释溶液并且将有机相用NaHCO3和盐水洗涤、干燥(Na2SO4)并浓缩。在二氧化硅上通过柱色谱纯化所得的粗料,用DCM:MeOH96:4洗脱。将适当的级分合并,并且冷冻干燥,得到为白色粉末的标题化合物(26mg,78%)。MS(ES+)407.1[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.09(s,1H),8.25(s,1H),8.28–8.21(m,1H),8.20(d,J=5.2Hz,1H),7.88(dd,J=9.1,2.7Hz,1H),7.74(td,J=9.0,2.8Hz,1H),7.25(dd,J=5.2,0.8Hz,1H),5.35(s,2H),4.43(t,J=5.0Hz,1H),3.49–3.40(m,2H),3.25–3.17(m,2H),2.98(tt,J=7.1,3.7Hz,1H),1.62(p,J=6.7Hz,2H),1.48(tt,J=9.7,6.1Hz,2H),1.10–1.00(m,2H),0.94–0.87(m,2H)。
实施例7

步骤a)4-(3-((4-(4-((叔丁基二甲基甲硅烷基)氧基)丁基)-7-氟异喹啉-3-基)
甲基)-2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸叔丁酯(7a)
使化合物6e(45mg,0.106mmol)与I-2c(34mg,0.106mmol)根据实施例6步骤f中所述的方法反应,得到标题化合物(49mg,70%)。MS(ES+)664.37[M+H]+。
步骤b)4-(3-((7-氟-4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-
咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(7b)
将在二噁烷中的4M HCl添加到化合物7a(35mg,0.053mmol)在MeOH(2mL)中的溶液中并且将澄清溶液在室温下搅拌90min,接着浓缩。将残余物溶解于DMF(3mL)中,添加DIPEA(57.5mg,0.445mmol)并且将混合物在室温下搅拌15分钟,接着在冰浴中冷却。添加氯甲酸甲酯(8.41mmol,0.089mmol)并且在0℃下继续搅拌90min。添加1M NaOH(水溶液,2mL)并且将混合物用EtOAc稀释,用NaHCO3和盐水洗涤、干燥(Na2SO4)、过滤并浓缩。在硅胶上通过柱色谱纯化所得的粗产物,用DCM:MeOH 96:4洗脱。将适当的级分合并,并且冷冻干燥,得到为固体的标题化合物(14mg,62%)。MS(ES+)508.1[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.09(s,1H),8.31(s,1H),8.25(dd,J=9.4,5.1Hz,1H),8.17(d,J=4.9Hz,1H),7.88(dd,J=9.1,2.7Hz,1H),7.74(td,J=9.0,2.8Hz,1H),7.41(d,J=5.3Hz,1H),5.38(s,2H),4.52–4.40(m,2H),4.14(s,2H),3.63(s,3H),3.49–3.41(m,2H),3.30(s,1H),3.26–3.18(m,2H),2.21(qd,J=12.5,4.5Hz,2H),1.79–1.72(m,2H),1.62(p,J=6.7Hz,2H),1.51(ddt,J=15.7,9.8,5.7Hz,2H)。
实施例8

步骤a)2-((7,7,7-三氟庚-2-炔-1-基)氧基)四氢-2H-吡喃(8a)
在-78℃下,将DMPU(1.10g,8.56mmol)以及随后的正丁基锂(548mg,8.56mmol)逐滴添加到2-(丙-2-炔-1-基氧基)四氢-2H-吡喃(1.00g,7.13mmol)在THF(20mL)中的经搅拌溶液中。将溶液在-78℃下搅拌30分钟,接着逐滴添加4-溴-1,1,1-三氟丁烷(1.43g,7.49mmol)。移除冰浴并且使反应混合物达到室温。18后,添加NaHCO3并且用EtOAc(30mL)稀释混合物。分离有机相并且用水和盐水洗涤、干燥(Na2SO4)、过滤并浓缩。通过二氧化硅塞过滤所得的粗料,用4:1庚烷EtOAc洗脱,得到标题化合物(1.29g,72%)。
步骤b)3-(((四氢-2H-吡喃-2-基)氧基)甲基)-4-(4,4,4-三氟丁基)异喹啉(8b)
在氩气下向(E)-N-(2-溴亚苄基)-2-甲基丙-2-胺和化合物8a(1.25g,5.00mmol)在乙腈(100mL)中的溶液中添加锌粉(817mg,12.5mmol),之后添加二溴双(三苯基膦)镍(II)(309mg,0.416mmol)。用冷凝器在80℃、氩气下加热反应混合物。3h后,通过小硅藻土垫将催化剂过滤出来。浓缩滤液,并且利用二氧化硅纯化残余物,使用庚烷:EtOAc 9:1洗脱,得到标题化合物(505mg,34%)。
步骤c)(4-(4,4,4-三氟丁基)异喹啉-3-基)甲醇(8c)
将化合物8b(550mg,1.56mmol)在乙酸/水(8:2,10mL)中的混合物加热至60℃,持续16h,接着浓缩并用EtOAc稀释。用1M NaOH和盐水洗涤3次。用EtOAc萃取水相,直到检测不到UV活性组分。将合并的有机层使用(Na2SO4)干燥、过滤并浓缩。在硅胶上通过柱色谱纯化所得的粗化合物,使用EtOAC:庚烷2:1洗脱,得到标题化合物(200mg,48%)。MS(ES+)270.42[M+H]+。
步骤d)3-(溴甲基)-4-(4,4,4-三氟丁基)异喹啉(8d)
根据实施例6步骤e中所述的方法将化合物8c(200mg,0.743mmol)转化成对应的溴代衍生物。收率204mg,83%。
步骤e)1-环丙基-3-((4-(4,4,4-三氟丁基)异喹啉-3-基)甲基)-1H-咪唑并[4,5-
c]吡啶-2(3H)-酮(8e)
使化合物8d(30mg,0.090mmol)与1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(15.8mg,0.090mmol)根据实施例6步骤f中所述的方法反应,得到标题化合物(21mg,55%)。MS(ES+)427.09[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.14(s,1H),8.32(s,1H),8.23–8.17(m,2H),8.09(dd,J=8.2,1.2Hz,1H),7.86(ddd,J=8.4,6.8,1.3Hz,1H),7.69(ddd,J=8.0,6.9,0.9Hz,1H),7.25(dd,J=5.2,0.8Hz,1H),5.38(s,2H),3.32(s,2H),2.96(tt,J=7.1,3.7Hz,1H),2.54(dd,J=8.2,3.5Hz,1H),1.74–1.63(m,2H),1.12–1.00(m,2H),0.93–0.86(m,2H)。
实施例9

步骤a)4-(2-氧代-3-((4-(4,4,4-三氟丁基)异喹啉-3-基)甲基)-2,3-二氢-1H-
咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸叔丁酯(9a)
使化合物8d(35.0mg,0.105mmol)与I-2c(40.3mg,0.126mmol)根据实施例6步骤f中所述的方法反应,得到标题化合物(42mg,70%)。MS(ES+)570.18[M+H]+。
步骤b)4-(2-氧代-3-((4-(4,4,4-三氟丁基)异喹啉-3-基)甲基)-2,3-二氢-1H-
咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(9b)
使化合物9a(42mg,0.074mmol)根据实施例7步骤b中所述的过程反应,得到标题化合物(18mg,46%)。MS(ES+)528.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.12(s,1H),8.40(s,1H),8.24–8.15(m,2H),8.09(dd,J=8.2,1.2Hz,1H),7.86(ddd,J=8.4,6.8,1.3Hz,1H),7.69(ddd,J=8.0,6.9,0.9Hz,1H),7.45–7.39(m,1H),5.41(s,2H),4.46(tt,J=12.3,4.0Hz,1H),3.63(s,3H),3.31(d,J=11.8Hz,6H),2.96(s,2H),2.60–2.51(m,1H),2.21(qd,J=12.5,4.5Hz,2H),1.74(dq,J=11.7,4.0Hz,4H)。
实施例10

1-环丙基-3-((4-(3-(羟基甲基)苯基)异喹啉-3-基)甲基)-1H-咪唑并[4,5-c]吡
啶-2(3H)-酮(10)
将化合物1a(110mg、0.278mmol)、(3-(羟基甲基)苯基)硼酸(63.4mg,0.417mmol)、碳酸钾(154mg,1.11mmol)和PdCl2(PPh3)2(9.77mg,0.014mmol)在DMF:水4:1(2.5mL)中的混合物加热至80℃,持续18h,接着过滤出固体并且添加EtOAc 10mL和水。将相分离并且用EtOAc(5mL)萃取水相一次。将合并的有机相用饱和NaHCO3和盐水洗涤、干燥(Na2SO4)、过滤并浓缩。在硅胶上通过柱色谱纯化所得的粗料,用DCM:MeOH(98:2)洗脱。将适当的级分合并,并且冷冻干燥,得到标题化合物(25mg,21%)。MS(ES+)423.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.25(s,1H),8.21–8.12(m,2H),7.96(s,1H),7.70(dddd,J=16.5,8.0,6.9,1.4Hz,2H),7.51(t,J=7.5Hz,1H),7.44(dt,J=7.7,1.5Hz,1H),7.36(dd,J=8.3,1.4Hz,1H),7.31–7.23(m,2H),7.23–7.17(m,1H),5.27(t,J=5.7Hz,1H),5.09(d,J=2.5Hz,2H),4.58(d,J=5.6Hz,2H),2.88(tt,J=7.0,3.6Hz,1H),1.01(dt,J=7.1,3.4Hz,2H),0.87–0.80(m,2H)。
实施例11

步骤a)1'-((4-溴异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-4,3'-吡咯
并[2,3-c]吡啶]-1-羧酸叔丁酯(11a)
将碳酸铯(1.62g,4.98mmol)在真空下加热5min,接着使其冷却至室温并且用氮气吹扫烧瓶。添加MeCN(10mL)和I-3g(504mg,1.66mmol)并且将溶液在室温下搅拌30分钟,接着逐滴添加4-溴-3-(溴甲基)异喹啉(500mg,1.66mmol)。将反应混合物搅拌18h,接着添加水(5mL)并且通过蒸发去除MeCN,用EtOAc稀释所得浆液。分离相并且将有机相用盐水洗涤并且(Na2SO4)、过滤并浓缩。在硅胶上通过柱色谱纯化所得的粗料,使用EtOAC洗脱,得到标题化合物(759mg,87%)。MS(ES+)523.10&525.09[M+H]+。
步骤b)1'-((4-(3-(甲基磺酰基)丙基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢
螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(11b)
将3-(甲基磺酰基)丙-1-烯(33.5mg,0.279mmol)和0.5M 9-硼杂二环[3.3.1]壬烷(0.56mL,0.279mmol)在四氢呋喃(2mL)中的溶液在室温、氮气下搅拌60分钟。添加水(100μl)并且继续搅拌10分钟,接着添加2M碳酸钾水溶液。将溶液再搅拌30分钟,之后添加化合物11a(112mg,0.214mmol)和Pd(PPh3)4(24.8mg,0.021mmol)。将氮气鼓泡通过溶液15分钟,接着给小瓶盖上盖子并且在120℃下在微波反应器中加热30分钟。
添加水(5mL)和EtOAc(10mL),分离相并且将有机相用盐水洗涤、干燥并浓缩。在C18,2cm,Gemini柱上通过制备型HPLC纯化所得的粗料,在pH 7下用NH4OAc洗脱。将纯级分汇集并浓缩、溶解于水:MeCN 1:1中并且冷冻干燥,得到为固体的标题化合物(29mg,24%)。MS(ES+)566.23[M+H]+。
步骤c)1'-((4-(3-(甲基磺酰基)丙基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢
螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(11c)
将在二噁烷中的4M HCl添加到化合物11b在MeOH中的溶液中。将溶液在室温下搅拌90min,接着浓缩。将所得残余物溶解于DMF中,添加DIPEA(55.6mg,0.43mmol)并且将反应混合物在室温下搅拌15分钟,接着在冰浴中冷却,添加氯甲酸甲酯(4.07mg,0.043mmol)并且在0℃下继续搅拌90分钟。用1M NaOH(水溶液,2mL)淬灭反应,添加EtOAc并且将混合物用NaHCO3和盐水洗涤、干燥(Na2SO4)、过滤并浓缩。在硅胶上通过柱色谱纯化所得的粗料,用DCM:MeOH 96:4洗脱。将纯级分汇集,并且冷冻干燥,得到标题化合物(8mg,36%)。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.38–8.34(m,1H),8.30(s,1H),8.24(d,J=8.6Hz,1H),8.07(dd,J=8.3,1.3Hz,1H),7.86(ddd,J=8.4,6.8,1.3Hz,1H),7.73–7.64(m,2H),5.29(s,2H),3.74(tt,J=13.5,6.2Hz,4H),3.65(s,3H),3.38(dt,J=19.1,7.9Hz,4H),3.02(s,3H),2.10–1.99(m,2H),1.80(tp,J=13.5,4.9Hz,4H)。
实施例12

步骤a)1'-((4-(4-氰基苯基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(12a)
将化合物11a(100mg,0.191mmol)、(4-氰基苯基)硼酸(42.1mg,0.287mmol)、碳酸钾(106mg,0.764mmol)和PdCl2(PPh3)2(13.4mg,0.019mmol)溶解于MeCN(2mL)中,将反应容器密封并且将混合物加热至80℃。当如通过TLC所判断确认反应完全时(约~18h),将固体过滤出来,添加EtOAc(10mL)并且用水洗涤溶液。用EtOAc(5mL)萃取水相一次并且将合并的有机层用饱和NaHCO3和盐水洗涤、干燥(Na2SO4)、过滤并浓缩。在硅胶上通过柱色谱纯化所得的粗料,用EtOAc:庚烷1:1且接着用EtOAc洗脱。将纯级分汇集,并且冷冻干燥,得到标题化合物(78mg,74%)。MS(ES+)546.25[M+H]+。
步骤b)1'-((4-(4-氰基苯基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(12b)
将在二噁烷中的4M HCl(3mL)添加到化合物12a(78.0mg,0.143mmol)在MeOH(2mL)中的溶液中。将溶液在室温下搅拌90min,接着浓缩。将所得残余物溶解于DMF(5mL)中,添加DIPEA(186mg,1.46mmol)并且将反应混合物在室温下搅拌15分钟,接着在冰浴中冷却,添加氯甲酸甲酯(13.8mg,0.146mmol)并且在0℃下继续搅拌90分钟。用1M NaOH(水溶液,2mL)淬灭反应,添加EtOAc并且将混合物用NaHCO3和盐水洗涤、干燥(Na2SO4)、过滤并浓缩。通过制备型HPLC C18,2cm,Gemini胶纯化所得的粗料,在pH 7下使用NH4OAc洗脱。将纯级分汇集,并且冷冻干燥,得到标题化合物(32mg,44%)。MS(ES+)504.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.31(d,J=0.9Hz,1H),8.27(s,1H),8.21–8.15(m,1H),8.11–8.06(m,2H),7.79–7.68(m,5H),7.61(d,J=4.3Hz,1H),7.31(dd,J=8.3,1.4Hz,1H),4.95(s,2H),3.76–3.65(m,1H),3.70(s,1H),3.64(s,4H),1.76(ddd,J=13.2,8.1,4.9Hz,2H),1.65(dt,J=13.4,5.0Hz,2H)。
实施例13

1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(13b)
如实施例12中所述的那样制备标题化合物,但是在步骤a中使用(6-氰基吡啶-3-基)硼酸。MS(ES+)505[M+H]+。通过1H和13C NMR确认结构。
实施例14

步骤a)4-(3-((4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)异喹啉-3-基)甲基)-
5,6-二氟-2-氧代-2,3-二氢-1H-苯并[d]咪唑-1-基)哌啶-1-羧酸叔丁酯(14a)
在氮气下将氢化钠(60%,分散在矿物油中,9mg,0.23mmol)添加到化合物I-8c(66mg,0,19mmol)在DMF(1mL)中的经搅拌溶液中。将所得混合物在室温下搅拌30min,接着将温度降低至0℃并且添加化合物I-4e(100mg,0,2mmol)。将反应混合物在室温下搅拌22h,接着使其在水与DCM之间分配。将层分离,将水相用新鲜的二氯甲烷萃取两次并用乙酸乙酯萃取三次。将合并的有机萃取物经Na2SO4干燥并减压浓缩,得到粗标题化合物136mg(90.4%),其无需进一步纯化即用于下一步骤。MS(ES+)805.37&806.30[M+H]+。
步骤b)4-(5,6-二氟-3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二
氢-1H-苯并[d]咪唑-1-基)哌啶-1-羧酸叔丁酯(14b)
向0℃的在THF(1mL)中的化合物14a(136mg,0,17mmol)中添加在THF(253μl)中的1M四丁基氟化铵。将所得混合物在室温下搅拌过夜,接着通过添加饱和NH4Cl水溶液淬灭反应。将有机组分萃取到乙酸乙酯中并用盐水洗涤。将有机萃取物经Na2SO4干燥并且减压浓缩。使用在庚烷中的0-100%乙酸乙酯的梯度通过快速色谱纯化残余物。将期望的级分汇集并且减压浓缩,得到标题化合物(65mg,62%)。MS(ES+)567.21[M+H]+。
步骤c)4-(5,6-二氟-3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二
氢-1H-苯并[d]咪唑-1-基)哌啶-1-羧酸甲酯(14c)
将甲醇(0.5mL)添加到化合物14b(119mg,0,19mmol)在处于二噁烷中的4M盐酸(1.8mL)中的溶液中。将所得混合物在室温下搅拌1h,接着添加甲苯并且将混合物减压浓缩。将甲醇和甲苯添加到残余物中并且将所得混合物减压浓缩。重复两次。将残余物溶解于DCM(1mL)中并且在氮气气氛下顺序添加N,N-二异丙基乙胺(187μl,1.07mmol)和氯甲酸甲酯(9μl,0.12mmol)。将所得混合物在室温下搅拌过夜,接着减压浓缩。通过快速色谱纯化所得的残余物,使用在二氯甲烷中的甲醇洗脱。将期望级分汇集并减压浓缩,并且在C18Phenomenex Kinetex 5μXB-100A,150x21.20mm柱上通过制备型HPLC进一步纯化,使用水/MeCN洗脱。将期望级分汇集并减压浓缩。将残余物溶解于乙腈中并且转移到4mL小瓶中。添加水并且将所得混合物冷冻干燥,得到标题化合物(22mg,37,6%)。MS(ES+)m/z 525[M+H]+;MS(ES-)m/z 583[M+OAc]-。
1H NMR(500MHz,DMSO-d6)δ9.10(s,1H),8.17–8.11(m,1H),8.10–8.04(m,1H),7.82(ddd,J=8.4,6.8,1.3Hz,1H),7.67(ddd,J=8.0,6.9,0.9Hz,1H),7.61(dd,J=10.8,7.3Hz,1H),7.31(dd,J=10.5,7.4Hz,1H),5.32(s,2H),4.46–4.35(m,2H),3.63(s,3H),3.46(q,J=6.2Hz,2H),3.33(s,3H),3.20(d,J=16.3Hz,1H),3.20(s,1H),2.93(s,2H),2.25(qd,J=12.5,4.5Hz,2H),1.71(d,J=11.5Hz,2H),1.62(p,J=6.7Hz,2H),1.52(ddt,J=15.4,9.6,5.7Hz,2H)。
实施例15

4-(7-氯-3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-咪唑并
[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(15)
如实施例14中所述的那样制备标题化合物,但是使用I-5c代替I-8c。总收率40%。MS(ES+)m/z 524[M+H]+;MS(ES-)m/z 582[M+OAc]-。
1H NMR(500MHz,DMSO-d6)δ9.07(s,1H),8.33(s,1H),8.21–8.13(m,2H),8.07(dt,J=8.2,0.9Hz,1H),7.83(ddd,J=8.4,6.8,1.3Hz,1H),7.67(ddd,J=7.9,6.8,0.9Hz,1H),5.40(s,2H),5.08(s,1H),4.44(t,J=5.0Hz,1H),4.11(d,J=12.8Hz,2H),3.62(s,3H),3.48(q,J=5.8Hz,2H),3.24–3.14(m,1H),2.92(s,3H),2.41(qd,J=12.6,4.7Hz,2H),1.85(d,J=11.4Hz,2H),1.69–1.51(m,4H)。
实施例16

4-(7-氯-3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-苯并
[d]咪唑-1-基)哌啶-1-羧酸甲酯(16)
如实施例14中所述的那样制备标题化合物,但是使用I-9c代替I-8c。总收率42%。MS(ES+)m/z 523[M+H]+,MS(ES-)m/z 581[M+OAc]-。
1H NMR(500MHz,DMSO-d6)δ9.07(s,1H),8.17–8.11(m,1H),8.09–8.03(m,1H),7.82(ddd,J=8.4,6.8,1.3Hz,1H),7.66(ddd,J=7.9,6.8,0.9Hz,1H),7.14–7.08(m,1H),7.04(dd,J=8.2,1.1Hz,1H),6.97(t,J=8.0Hz,1H),5.34(s,2H),5.17(s,1H),4.43(t,J=5.1Hz,1H),3.62(s,3H),3.46(td,J=6.3,5.0Hz,2H),3.33(s,6H),3.23–3.16(m,2H),2.91(s,2H),2.45(td,J=12.4,4.4Hz,1H),1.82(d,J=11.9Hz,2H),1.63(p,J=6.7Hz,2H),1.57–1.46(m,1H),1.52(s,1H)。
实施例17

6-(3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-苯并[d]咪
唑-1-基)-2-氮杂螺[3.3]庚烷-2-羧酸甲酯(17)
基本上如实施例14中所述的那样制备标题化合物,但是使用I-6b代替I-8c。总收率1.5%。MS(ES+)m/z 501[M+H]+;MS(ES-)m/z 559[M+OAc]-。
1H NMR(500MHz,DMSO-d6)δ9.10(s,1H),8.15–8.09(m,1H),8.07(dd,J=8.3,1.3Hz,1H),7.81(ddd,J=8.5,6.8,1.3Hz,1H),7.66(ddd,J=7.9,6.8,0.9Hz,1H),7.26(d,J=7.8Hz,1H),7.07–6.96(m,2H),6.93(td,J=7.7,1.1Hz,1H),5.33(s,2H),4.78(p,J=8.8Hz,1H),4.41(t,J=4.9Hz,1H),4.08(s,2H),3.56(s,3H),3.42(q,J=6.3Hz,2H),3.23–3.14(m,2H),3.02(td,J=9.4,2.9Hz,2H),2.61(ddd,J=9.8,8.3,3.1Hz,2H),1.61(p,J=6.8Hz,2H),1.47–1.33(m,2H)。
实施例18

步骤a)4-(3-((4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)异喹啉-3-基)甲基)-
2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸叔丁酯(18a)
使4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸叔丁酯(161mg,0.506mmol)和I-4e(184mg,0.377mmol)如实施例14步骤a中所述的那样反应,得到标题化合物(162mg,56%)。MS(ES+)770.44[M+H]+。
步骤b)3-((4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)异喹啉-3-基)甲基)-1-
(1-(甲基磺酰基)哌啶-4-基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(18b)
将TFA逐滴添加到化合物18a(158mg,0.205mmol)在DCM(2mL)中的溶液中。将溶液在室温下搅拌75min,接着真空浓缩,用DCM(5mL)稀释并且用饱和NaHCO3(5mL)碱化。用2x5mL DCM萃取水相。将有机相合并,并且真空浓缩。将所得的残余物溶解于DCM(5mL)中并且添加MsCl(35.1mg,0.307mmol)和DIEA(134mg,1.04mmol)。将反应混合物在室温下搅拌18h,接着用DCM(10mL)稀释并且用2x 15mL饱和NaCl洗涤。将有机相真空浓缩。将粗料悬浮于DCM(6mL)+MeOH(0.8mL)中并且在硅胶上通过柱色谱进行纯化,使用在DCM中的MeOH的梯度洗脱,得到不完全纯的标题化合物(79mg,52%)。MS(ES+)748.37[M+H]+。
步骤c)3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1-(1-(甲基磺酰基)哌啶-4-
基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(18c)
将TBAF(1M,在THF中)添加到化合物18b(79mg,0.106mmol)在THF(2mL)中的溶液中,在冰浴中冷却。将混合物在室温下搅拌过夜,接着浓缩,用饱和NH4Cl(5mL)淬灭并且用EtOAc(3x 5mL)萃取。将有机相合并,用饱和NaCl(5mL)洗涤、干燥(Na2SO4)并真空浓缩。将所得的粗料溶解于MeCN(2.2mL)+水(0.4mL)中,并且在Phenomenex Gemini-NX 5μC18,110A,AX,100x 30mm柱上通过制备型HPLC进行纯化,
使用溶剂A:在95/5H2O-MeCN中的10mM NH4OAc和溶剂B:在90/10MeCN-H2O中的10mMNH4OAc的梯度洗脱。将适当的级分合并,并且冷冻干燥,得到标题化合物(17.5mg,32%)。MS(ES+)510.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.11(s,1H),8.32(s,1H),8.20–8.12(m,2H),8.08(dd,J=8.3,1.2Hz,1H),7.83(ddd,J=8.4,6.8,1.3Hz,1H),7.67(ddd,J=8.0,6.8,0.9Hz,1H),7.39(d,J=5.2Hz,1H),5.40(s,2H),4.42(tt,J=12.3,4.2Hz,2H),3.78–3.69(m,2H),3.46(dt,J=10.3,5.0Hz,2H),3.26–3.18(m,2H),2.95(s,3H),2.95(t,J=11.2Hz,1H),2.38(qd,J=12.5,4.3Hz,2H),1.87(dt,J=12.9,2.8Hz,2H),1.63(p,J=6.7Hz,2H),1.53(ddt,J=15.4,9.7,5.7Hz,2H)。
实施例19

步骤a)3-((4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-7-氯异喹啉-3-基)甲
基)-1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(19a)
向1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(65mg,0.37mmol,1当量)在DMF(7mL)中的经搅拌溶液中添加Cs2CO3(362mg,1.11mmol,3当量),之后添加化合物I-7e(194mg,0.37mmol,1当量)。将反应混合物在环境温度下搅拌5h,接着用乙酸乙酯稀释并且用水和盐水洗涤。将合并的有机层干燥(Na2SO4)并减压浓缩,得到粗标题化合物(240mg),其无需进一步纯化即用于下一步骤。MS(ES+)661.0[M+H]+。
步骤b)3-((7-氯-4-(4-羟基丁基)异喹啉-3-基)甲基)-1-环丙基-1H-咪唑并[4,
5-c]吡啶-2(3H)-酮(19b)
在0℃下向化合物19a(450mg,0.68mmol,1当量)在THF(3.0mL)中的经搅拌溶液中逐滴添加TBAF(1M,在THF中,1.02mL,1.02mmol,1.5当量)并且将反应混合物在环境温度下搅拌5h,接着用饱和NH4Cl水溶液淬灭反应,将有机组分萃取到乙酸乙酯中并且用水和盐水彻底洗涤。将合并的有机层干燥(Na2SO4)并浓缩。通过制备型HPLC纯化所得的粗料,得到标题化合物(50mg,17%)。MS(ES+)423.1[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.09(s,1H),8.25(s,1H),8.23–8.16(m,3H),7.82(dd,J=9.1,2.3Hz,1H),7.25(d,J=5.2Hz,1H),5.36(s,2H),4.44(t,J=5.1Hz,1H),3.45(q,J=6.0Hz,2H),3.19(s,0H),2.98(tt,J=7.1,3.6Hz,1H),1.62(p,J=6.8Hz,2H),1.55–1.44(m,2H),1.24(d,J=4.3Hz,0H),1.06(dt,J=7.1,3.4Hz,2H),0.91(p,J=5.3,4.8Hz,2H)。
实施例20

4-(3-((7-氯-4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-咪唑并
[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(20)
使4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯与I-7e根据实施例19步骤a中所述的方法反应,然后根据实施例19步骤b中所述的方法去除SiTBDP基团。收率51%。MS(ES+)524.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.31(s,1H),8.23–8.14(m,3H),7.83(dd,J=9.1,2.3Hz,1H),7.42(d,J=5.4Hz,1H),5.39(s,2H),4.52–4.40(m,2H),4.14(s,2H),3.63(s,3H),3.45(td,J=6.3,5.0Hz,2H),3.21(s,1H),3.25–3.12(m,1H),2.96(s,2H),2.21(qd,J=12.5,4.5Hz,2H),1.76(dd,J=12.9,3.6Hz,2H),1.62(p,J=6.6Hz,2H),1.52(ddt,J=15.4,9.8,5.7Hz,2H),1.23(s,0H)。
实施例21

步骤a)2'-氧代-1',2'-二氢螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯
(21a)
向化合物I-2d(0.5g,2.46mmol,1当量)在THF(3mL)中的经搅拌溶液中添加TEA(1mL,7.38mmol,3当量),之后添加氯甲酸甲酯(0.2mL,2.46mmol,1当量)。将混合物在环境温度下搅拌16h,接着用水稀释并且将有机组分萃取到乙酸乙酯中并用水和盐水洗涤。将有机层经无水Na2SO4干燥,并且减压浓缩。使用在DCM中的5%MeOH,通过硅胶(100-200目)柱色谱纯化所得的粗产物,得到为固体的标题化合物(250mg,39%)。MS(ES+)262.1[M+H]+。
步骤b)1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(21b)
在0℃下将NaH(60%,分散在矿物油中,13.8mg,0.345mmol,1.5当量)添加到化合物21a(60mg,0.23mmol,1当量)在DMF(4mL)中的经搅拌溶液中。将混合物在0℃下搅拌15min,然后添加化合物I-4e(123mg,0.25mmol,1.1当量)。使反应混合物达到环境温度并且搅拌17h,接着冷却至0℃并且添加冷却水。将有机组分萃取到EtOAc中。将有机部分用水和盐水洗涤,经无水Na2SO4干燥,并且减压浓缩。通过反相制备型HPLC纯化所得的粗化合物,得到为固体的标题化合物(0.06g,55%)。MS(ES+)475.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.06(s,1H),8.32–8.24(m,2H),8.16(d,J=8.6Hz,1H),8.05(d,J=8.1Hz,1H),7.83(ddd,J=8.4,6.7,1.3Hz,1H),7.66(dd,J=11.2,6.0Hz,2H),5.26(s,2H),4.46(t,J=5.1Hz,1H),3.75(qd,J=16.6,13.5,8.7Hz,4H),3.66(s,3H),3.50(q,J=5.6Hz,2H),3.20(d,J=15.2Hz,1H),3.20(s,1H),1.88–1.74(m,4H),1.65(dq,J=10.2,6.1Hz,4H)。
实施例22

步骤a)3-((4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)异喹啉-3-基)甲基)-1-
(环丙基磺酰基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(22a)
向化合物1-(环丙基磺酰基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮I-13c(0.2g,0.84mmol,1当量)在DMF(8mL)中的经搅拌溶液中添加K2CO3(0.173g,1.2mmol,1.4当量)和KI(1.6mg,0.01mmol,0.01当量),之后添加化合物I-4e(0.41g,0.84mmol,1当量)。将混合物在环境温度下搅拌12h,接着用水稀释并且用乙酸乙酯萃取。将有机层用水和盐水洗涤,经无水Na2SO4干燥,并且减压浓缩。使用在DCM中的2%MeOH,通过硅胶(100-200目)柱色谱纯化粗产物,得到为固体的标题化合物(150mg,40%)。MS(ES+)691.3[M+H]+。
步骤b)1-(环丙基磺酰基)-3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1H-咪唑并
[4,5-c]吡啶-2(3H)-酮(22b)
在0℃下向化合物22a(200mg,0.3mmol)在MeOH(2mL)中的经搅拌溶液中添加在水(2mL)中的HCl(0.5mL)。将溶液在环境温度下搅拌12h,接着真空浓缩。用水稀释残余物,通过添加NaHCO3水溶液将pH调节至8并且用EtOAc萃取溶液。将有机层经无水Na2SO4干燥,并且减压浓缩。通过反相制备型HPLC纯化粗化合物,得到为固体的标题化合物(29mg,21%)。MS(ES+)453.1[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.07(s,1H),8.47(s,1H),8.33(d,J=5.4Hz,1H),8.18(d,J=8.6Hz,1H),8.06(d,J=8.0Hz,1H),7.84(ddd,J=8.4,6.8,1.3Hz,1H),7.68(t,J=7.5Hz,1H),7.58(d,J=5.4Hz,1H),5.46(s,2H),4.45(t,J=5.1Hz,1H),3.50(q,J=5.7Hz,2H),3.40(tt,J=7.9,4.7Hz,1H),3.22(d,J=15.2Hz,1H),3.22(s,1H),1.65(dp,J=14.1,7.3,6.8Hz,4H),1.36(dt,J=6.9,3.5Hz,2H),1.24(qd,J=6.1,5.7,1.3Hz,2H)
实施例23

步骤a)4-(3-((4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-7-氰基异喹啉-3-
基)甲基)-2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(23a)
向4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(175mg,0.63mmol,1当量)在DMF(5.0mL)中的经搅拌溶液中添加Cs2CO3(619mg,1.9mmol,3.0当量),之后添加化合物I-10(325mg,0.63mmol,1当量)。将反应混合物在80℃下搅拌90min,接着用乙酸乙酯稀释并且用水和盐水洗涤。将合并的有机层经无水硫酸钠干燥并减压浓缩,得到粗标题化合物(475mg,99%)。MS(ES+)753.5[M+H]+。
步骤b)4-(3-((7-氰基-4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-
1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(23b)
在冰冷条件下向化合物23a(475mg,0.63mmol,1当量)在MeOH(10.0mL)中的经搅拌溶液中添加1:1HCl水溶液(4.0mL)。将反应混合物在环境温度下搅拌5h,接着减压浓缩并且与甲苯共沸蒸馏。将获得的粘性固体溶解于水中并且用乙酸乙酯萃取。将有机层用水和盐水洗涤,经无水Na2SO4干燥并浓缩,得到为固体的标题化合物(320mg,98%)。MS(ES+)515.2[M+H]+。1H NMR(500MHz,DMSO-d6)δ9.19(s,1H),8.74(d,J=1.7Hz,1H),8.36–8.29(m,2H),8.18(d,J=5.4Hz,1H),8.09(dd,J=8.9,1.8Hz,1H),7.42(d,J=5.4Hz,1H),5.43(s,2H),4.46(dt,J=10.1,4.5Hz,2H),4.14(s,2H),3.63(s,3H),3.46(q,J=5.9Hz,2H),3.33(s,5H),3.24(d,J=16.3Hz,1H),2.21(qd,J=12.5,4.6Hz,2H),1.76(dd,J=12.6,3.8Hz,2H),1.63(p,J=6.6Hz,2H),1.55(tq,J=9.8,6.5,5.4Hz,2H)。
实施例24

4-(3-((7-(氨基甲基)-4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-
1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(24)
向化合物23b(700mg,1.36mmol,1当量)在乙醇(2.0mL)中的经搅拌溶液中添加雷尼镍(2.0g),之后添加乙醇NH3(20mL)并且将混合物在环境温度、氢气气氛(气球压力)下搅拌17h,接着通过硅藻土过滤并且用在DCM中的10%MeOH彻底洗涤并真空浓缩。通过反相制备型HPLC纯化所得的粗产物,将纯级分汇集并浓缩。将所得的化合物溶解于最小体积的MeOH中,添加在无水醚中的HCl并且将溶液冻干,得到标题化合物的HCl盐(40mg)。MS(ES+)519.34[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.86(s,1H),8.63(d,J=5.8Hz,2H),8.27(d,J=8.9Hz,1H),8.17(d,J=1.8Hz,1H),8.08(d,J=6.5Hz,1H),8.02(dd,J=8.9,1.8Hz,1H),5.55(s,2H),4.67(tt,J=12.2,4.0Hz,1H),4.23(q,J=5.8Hz,2H),4.15(s,2H),3.62(s,3H),3.49(s,2H),3.49(d,J=11.7Hz,0H),3.24(dt,J=8.1,4.2Hz,2H),2.97(s,3H),2.22(qd,J=12.4,4.5Hz,2H),1.83(dd,J=12.6,3.6Hz,2H),1.64(dq,J=6.6,3.3Hz,4H),1.23(s,1H)。
实施例25

3-((1-环丙基-2-氧代-1H-咪唑并[4,5-c]吡啶-3(2H)-基)甲基)-4-(4-羟基丁
基)异喹啉-7-甲腈(25)
根据实施例23中所述的方法制备标题化合物,但是使用1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮代替4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯。收率98%。MS(ES+)414.1[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.20(s,1H),8.74(d,J=1.7Hz,1H),8.33(d,J=8.9Hz,1H),8.25(s,1H),8.21(d,J=5.2Hz,1H),8.09(dd,J=8.9,1.7Hz,1H),7.26(d,J=5.2Hz,1H),5.41(s,2H),4.44(t,J=5.1Hz,1H),3.46(q,J=6.0Hz,2H),3.25(s,0H),3.24(s,1H),3.22(s,0H),2.98(tt,J=7.1,3.7Hz,1H),1.63(p,J=6.7Hz,2H),1.57–1.47(m,2H),1.06(dt,J=7.1,3.5Hz,2H),0.91(p,J=5.3,4.9Hz,2H)。
实施例26

3-((7-(氨基甲基)-4-(4-羟基丁基)异喹啉-3-基)甲基)-1-环丙基-1H-咪唑并
[4,5-c]吡啶-2(3H)-酮(26)
根据实施例24中所述的过程还原化合物25(170mg,0.41mmol),得到为固体的标题化合物(40mg)。MS(ES+)418.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.12(s,1H),8.82(s,1H),8.66(d,J=5.9Hz,2H),8.58(d,J=6.4Hz,1H),8.29(d,J=8.9Hz,1H),8.20(d,J=1.8Hz,1H),8.04(dd,J=8.9,1.8Hz,1H),7.81(d,J=6.3Hz,1H),5.54(s,2H),4.23(q,J=5.8Hz,2H),3.49(d,J=5.8Hz,2H),3.25(s,2H),3.15(tt,J=7.1,3.6Hz,1H),1.70–1.58(m,4H),1.25(d,J=11.0Hz,1H),1.13(dt,J=7.2,3.5Hz,2H),1.03–0.96(m,2H)。
实施例27

3-((7-(氨基甲基)-4-(4-羟基丁基)异喹啉-3-基)甲基)-1-环丙基-1H-咪唑并
[4,5-c]吡啶-2(3H)-酮(27)
根据实施例23和24中所述的方法制备标题化合物(51mg),但是使用1-环丙基-1H-苯并[d]咪唑-2(3H)-酮代替4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯。MS(ES+)417.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.25(s,1H),8.61(s,1H),8.25(d,J=16.3Hz,2H),8.03(dd,J=8.7,1.8Hz,1H),7.25–7.19(m,1H),7.04(ddd,J=7.2,3.8,2.7Hz,2H),6.99–6.92(m,1H),5.39(s,2H),4.25(q,J=5.8Hz,2H),3.40(t,J=6.4Hz,2H),3.24–3.17(m,2H),2.92(tt,J=7.0,3.6Hz,1H),1.57(p,J=6.7Hz,2H),1.45–1.33(m,2H),1.23(d,J=4.6Hz,1H),1.05(td,J=7.2,5.1Hz,2H),0.90(p,J=5.3,4.8Hz,2H)。
实施例28

步骤a)1'-((4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)异喹啉-3-基)甲基)-
2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-羧酸甲酯(28a)
在0℃下向化合物I-11e(0.222g,0.96mmol,1当量)在DMF中的经搅拌溶液中添加Cs2CO3(0.936g,2.88mmol,3当量)和化合物I-4e(0.467g,0.96mmol,1当量)。将混合物在室温下搅拌12h,接着用水稀释并且用乙酸乙酯萃取。将有机层用盐水洗涤、干燥(Na2SO4)并且减压浓缩。粗化合物无需进一步纯化即用于下一步骤。
步骤b)1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-
二氢吲哚]-1-羧酸甲酯(28b)
在0℃下向化合物28a(0.3g,0.44mmol,1当量)在MeOH(2mL)中的经搅拌溶液中添加HCl水溶液(2mL,3N)。将所得反应混合物在室温下搅拌12h,接着真空浓缩并且将残余物溶解于水中。通过添加NaHCO3水溶液将溶液的pH调节至8并且将有机组分萃取到乙酸乙酯中。将有机层干燥(Na2SO4)并且减压浓缩。通过反相制备型HPLC纯化获得的粗化合物,得到标题化合物(70mg)。MS(ES+)446.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.17–8.11(m,1H),8.06(dd,J=8.3,1.3Hz,1H),7.82(ddd,J=8.4,6.8,1.3Hz,1H),7.72–7.62(m,2H),7.21(td,J=7.8,1.2Hz,1H),7.07(td,J=7.5,1.0Hz,1H),7.01(d,J=7.8Hz,1H),5.21(s,2H),4.44(t,J=5.1Hz,1H),4.20(s,4H),3.65(s,3H),3.51–3.44(m,2H),3.22–3.15(m,2H),1.69–1.51(m,4H)。
实施例29

步骤a)1-环丙基-3-((8-氟-4-(4-羟基丁基)异喹啉-3-基)甲基)-1H-咪唑并[4,
5-c]吡啶-2(3H)-酮(29)
根据实施例28中所述的过程由化合物I-14和1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮制备标题化合物。收率20%。MS(ES+)407.2[M+H]+。通过1H NMR确认结构。
实施例30

4-(3-((5-氟-4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-咪唑并
[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(30)
根据实施例28中所述的过程由I-12和4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯制备标题化合物。收率21%。MS(ES+)508.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.14(d,J=2.4Hz,1H),8.31(s,1H),8.17(d,J=5.4Hz,1H),7.93(dd,J=8.0,1.3Hz,1H),7.66(td,J=7.8,4.8Hz,1H),7.62(ddd,J=13.9,7.7,1.3Hz,1H),7.45–7.39(m,1H),5.42(s,2H),4.47(tt,J=12.2,4.0Hz,1H),4.41(t,J=5.1Hz,1H),4.15(s,2H),3.63(s,3H),3.44(q,J=6.0Hz,2H),3.26(dd,J=15.6,2.4Hz,1H),3.25(s,1H),2.97(s,2H),2.22(qd,J=12.5,4.5Hz,2H),1.80–1.73(m,2H),1.66–1.50(m,4H)。
实施例31

1-环丙基-3-((5-氟-4-(4-羟基丁基)异喹啉-3-基)甲基)-1H-咪唑并[4,5-c]吡
啶-2(3H)-酮(31)
根据实施例28中所述的过程由I-12和1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮制备标题化合物。收率21%。MS(ES+)407.2[M+H]+。1H NMR(500MHz,DMSO-d6)δ9.15(d,J=2.4Hz,1H),8.24(s,1H),8.21(d,J=5.2Hz,1H),7.94(dd,J=8.0,1.3Hz,1H),7.66(td,J=7.8,4.7Hz,1H),7.61(ddd,J=13.9,7.7,1.3Hz,1H),7.26(dd,J=5.2,0.8Hz,1H),5.39(s,2H),4.41(t,J=5.1Hz,1H),3.44(td,J=6.4,5.0Hz,2H),3.25(dd,J=16.1,2.4Hz,1H),3.25(s,1H),2.99(tt,J=7.0,3.6Hz,1H),1.61(p,J=6.7Hz,2H),1.57–1.47(m,2H),1.07(td,J=7.3,5.2Hz,2H),0.91(qd,J=5.1,3.7Hz,2H)。
实施例32

步骤a)(R)-叔丁基(1-(4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)-3-((1-环丙
基-2-氧代-1H-咪唑并[4,5-c]吡啶-3(2H)-基)甲基)异喹啉-7-基)吡咯烷-3-基)氨基甲酸
酯(32a)
将微波管中的化合物19a(200mg,0.302mmol)、(R)-(+)-3-(Boc-氨基)吡咯烷(113mg,0.605mmol)、碳酸铯(148mg,0.454mmol)、乙酸钯(6.79mg,0.030mmol)和2,2’-双(二苯基膦基)-1,1’-联萘(37.7mg,0.060mmol)在1,4-二噁烷(3mL)中的溶液超声处理片刻,接着用Ar气体(x3)脱气并且在~7巴的压力下在200℃下加热50min。通过硅藻土过滤反应浆液、用DCM洗涤并且将滤液真空浓缩。将残余物溶解于DCM中并且用饱和NaHCO3水溶液萃取,将DCM相干燥(Na2SO4)并浓缩。通过硅胶柱色谱纯化所得的残余物,用梯度EtOH/DCM洗脱,得到标题化合物(17.4mg,6.6%)。MS(ES+)811.42[M+H]+。
步骤b)(R)-叔丁基(1-(3-((1-环丙基-2-氧代-1H-咪唑并[4,5-c]吡啶-3(2H)-
基)甲基)-4-(4-羟基丁基)异喹啉-7-基)吡咯烷-3-基)氨基甲酸酯(32b)
将在THF中的1M TBAF水合物(44μL)添加到化合物32a(32.5mg,0.040mmol)在THF(0.40mL)中的溶液中。将溶液在室温下搅拌4h,接着浓缩。
将残余物溶解于几毫升CHCl3中并且通过制备型TLC进行纯化,用10%MeOH/CHCl3洗脱,得到标题化合物(18mg,78%)。MS(ES+)573.26[M+H]+。
步骤c)(R)-3-((7-(3-氨基吡咯烷-1-基)-4-(4-羟基丁基)异喹啉-3-基)甲基)-
1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(32c)
将在二噁烷中的4M HCl(393μl)逐滴添加到化合物32b(18mg,0.031mmol)在MeOH(4mL)中的溶液中。将反应混合物在室温下搅拌3h,接着添加另外的在二噁烷中的4M HCl(80μL)并且将溶液搅拌18h。将反应混合物浓缩并且从MeOH(x3)中共蒸发。将所得固体溶解于水:MeCN 1:2中,通过棉花和硅藻土塞过滤出不溶物并且将滤液冷冻干燥。将残余物溶解于50%H2O/MeCN(1200μL)中并且在Gemini C18 20mm柱上通过制备型HPLC进行纯化,使用30-40%B的梯度洗脱;缓冲液A=0.1%NH4OH/水;缓冲液B:0.1%NH4OH/MeCN。将适当的级分合并并且浓缩,接着从30%H2O/MeCN中冷冻干燥,得到标题化合物(9.34mg,63%)。MS(ES+)473.25[M+H]+。
1H NMR(500MHz,DMSO-d6)δ8.84(s,1H),8.25(s,1H),8.17(d,J=5.2Hz,1H),7.93(d,J=9.3Hz,1H),7.30–7.20(m,2H),6.81(d,J=2.5Hz,1H),5.25(s,2H),4.40(s,1H),3.62(p,J=5.8Hz,1H),3.55–3.33(m,4H),3.42(s,2H),3.09(s,1H),3.09(d,J=16.5Hz,1H),3.05–2.92(m,2H),2.16–2.06(m,1H),1.76(dq,J=12.8,6.2Hz,1H),1.58(p,J=6.7Hz,2H),1.41(dq,J=9.4,6.1,5.6Hz,2H),1.24(s,1H),1.06(td,J=7.3,5.2Hz,2H),0.94–0.82(m,2H)。
实施例33

(S)-3-((7-(3-氨基吡咯烷-1-基)-4-(4-羟基丁基)异喹啉-3-基)甲基)-1-环丙
基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(33)
使用(S)-(-)-3-(Boc-氨基)吡咯烷代替(R)-(+)-3-(Boc-氨基)吡咯烷,根据实施例32中所述的过程制备标题化合物。
1H NMR(500MHz,DMSO-d6)δ8.84(s,1H),8.25(s,1H),8.17(d,J=5.2Hz,1H),7.92(d,J=9.3Hz,1H),7.30–7.20(m,2H),6.81(d,J=2.5Hz,1H),5.24(s,2H),4.40(t,J=5.0Hz,1H),3.63–3.59(m,1H),3.55–3.33(m,5H),3.13–3.05(m,2H),3.04–2.92(m,2H),2.11(dq,J=12.8,6.4Hz,1H),1.75(dq,J=12.6,6.4Hz,1H),1.58(p,J=6.7Hz,2H),1.49–1.33(m,2H),1.24(s,1H),1.06(dt,J=7.1,3.5Hz,2H),0.94–0.82(m,2H)。
实施例34

1-(1-(环丙基磺酰基)氮杂环丁烷-3-基)-3-((4-(4-羟基丁基)异喹啉-3-基)甲
基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(34)
使化合物I-4e(255mg,0.52mmol)与I-15e(0.14g,0.48mmol)根据实施例21步骤b中所述的方法反应,得到为固体的标题化合物(50g,29%)。MS(ES+)508.0[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.11(s,1H),8.35(s,1H),8.26(d,J=5.3Hz,1H),8.16(d,J=8.6Hz,1H),8.08(d,J=8.1Hz,1H),7.83(ddd,J=8.4,6.8,1.3Hz,1H),7.68(t,J=7.5Hz,1H),7.50(d,J=5.4Hz,1H),5.43–5.31(m,3H),4.58(dd,J=8.5,6.8Hz,2H),4.42(t,J=5.1Hz,1H),4.30(t,J=8.5Hz,2H),3.45(q,J=6.0Hz,2H),3.26–3.18(m,2H),2.89(tt,J=7.9,4.8Hz,1H),1.63(p,J=6.7Hz,2H),1.58–1.47(m,2H),1.10(dt,J=7.2,3.5Hz,2H),1.06–0.96(m,2H)。
实施例35

4-(3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-咪唑并[4,5-
c]吡啶-1-基)哌啶-1-羧酸环丙基酯(35)
使化合物I-4e(248mg,0.51mmol)与I-2e(140mg,0.46mmol)根据实施例21步骤b中所述的方法反应,得到为固体的标题化合物(40g,16%)。MS(ES+)515.25[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.10(s,1H),8.31(s,1H),8.19–8.10(m,2H),8.07(dd,J=8.2,1.2Hz,1H),7.83(ddd,J=8.4,6.8,1.3Hz,1H),7.71–7.64(m,1H),7.40(d,J=5.4Hz,1H),5.39(s,2H),4.51–4.40(m,2H),4.16(s,1H),4.02(ddd,J=9.3,6.3,4.2Hz,1H),3.46(q,J=6.1Hz,2H),3.21(s,1H),3.21(d,J=16.4Hz,1H),2.94(s,2H),2.26–2.14(m,2H),1.75(d,J=12.1Hz,2H),1.62(p,J=6.7Hz,2H),1.52(ddt,J=9.3,6.7,4.1Hz,2H),0.66(s,1H),0.65(s,3H)。
实施例36

步骤a)1'-((4-溴异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-
1-羧酸叔丁酯(36a)
将化合物I-11c(449mg,1.64mmol)和碳酸铯(1.6g,4.91mol)在乙腈(10mL)中的溶液搅拌15分钟,接着分批添加4-溴-3-(溴甲基)异喹啉(493mg,1.64mol)。将溶液在室温下搅拌16h,接着
浓缩,在硅胶上通过柱色谱纯化所得的粗产物,使用庚烷-EtOAC的梯度洗脱,得到标题化合物(750mg,93%)。MS(ES+)494.1&496.1[M+H]+。
步骤b)1'-((4-(4-羟基丁-1-炔-1-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁
烷-3,3'-二氢吲哚]-1-羧酸叔丁酯(36b)
将化合物36a(365mg,0.738mmol)、CuI(21.1mg,0.111mmol)和Pd(PPh3)2Cl2(51.8mg,0.074mmol)溶解于DMF(5mL)中。将烧瓶抽真空并且置于N2下,然后添加Et3N(1.03mL,7.38mmol)和3-丁炔-1-醇(0.073mL,0.960mmol)。将烧瓶抽真空并且再次用氮气吹扫。将反应物在70℃下加热过夜,接着在微波反应器中在120℃下加热1h。通过硅藻土垫过滤反应混合物,用EtOAc冲洗并浓缩。在硅胶上通过柱色谱纯化粗产物,使用在DCM中的MeOH洗脱,得到标题化合物(199mg,55%)。MS(ES+)484.3[M+H]+。
步骤c)1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-
二氢吲哚]-1-羧酸叔丁酯(36c)
使用H-cube仪和20%氢氧化钯作为催化筒在室温和环境压力下使化合物36b在MeOH(20mL)、EtOAc(5mL)和AcOH(0.5mL)中的溶液进行氢化。在40℃和10atm压力下进行通过H-cube的第二轮。将溶液浓缩并冻干,得到标题化合物(89mg,44%)。MS(ES+)488[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,0H),8.17–8.11(m,0H),8.09–8.03(m,0H),7.82(ddd,J=8.4,6.8,1.3Hz,1H),7.70–7.61(m,1H),7.20(td,J=7.8,1.2Hz,0H),7.07(td,J=7.5,1.0Hz,1H),7.01(d,J=7.9Hz,0H),5.21(s,1H),4.44(t,J=5.1Hz,1H),4.32(t,J=5.2Hz,2H),4.15(s,1H),4.10(s,1H),3.48(q,J=6.0Hz,1H),3.37(td,J=6.6,5.1Hz,4H),3.18(s,1H),1.62(dt,J=15.6,8.3Hz,1H),1.60–1.52(m,1H),1.44(s,4H),1.39(q,J=6.6Hz,4H),1.26(d,J=10.0Hz,2H),1.25(s,5H)。
实施例37

步骤a)1'-((4-(4-羟基丁-1-炔-1-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢
螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(37a)
使化合物11a(500mg,0.956mmol)与3-丁炔-1-醇(87.1mg,1.24mol)根据实施例36步骤b中所述的方法反应,得到标题化合物(490mg,84%)。MS(ES+)513.22[M+H]+。
步骤b)1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(37b)
将化合物37a(409mg,0.798mmol)溶解于EtOH(19mL)和MeOH(1mL)中。将烧瓶抽真空,之后添加氮气气体,重复两次,接着添加10%Pd/C(150mg,1.27mmol)。将烧瓶抽真空并且填充氢气气体(从气球中),将反应物在环境温度下搅拌过夜。过滤出反应催化剂并浓缩滤液。将剩余物溶解于EtOH和MeOH中,如参见上文所述再次起始反应并且使其进行72h。
通过硅藻土垫过滤反应混合物并且真空浓缩,得到定量收率的标题化合物。所得化合物无需进一步纯化即用于下一步骤。MS(ES+)517.21[M+H]+。
步骤c)1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1-(甲基磺酰基)螺[哌啶-4,
3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(37c)
将TFA(1mL,13.1mol)添加到化合物37b在DCM(3mL)中的溶液中,并且将溶液在室温下搅拌,直到完全去保护。将混合物真空浓缩,用甲苯稀释并且浓缩两次。将所得残余物和DIPEA(47μl,0.27mmol)溶解于MeCN(4mL)中,将溶液搅拌15min,接着缓慢添加甲磺酰氯(10μl,0.129mmol)在MeCN(1mL)中的溶液。当如通过LCMS判断确认反应完全时,将混合物真空浓缩。使用95%水、5%乙腈(10mM,在乙酸铵中)和10%水、90%乙腈(10mM,在乙酸铵中)的梯度,在Gemini C18100x30mm上通过制备型HPLC纯化粗产物。将适当的级分浓缩,得到标题化合物(14.5mg,38%)。MS(ES+)495.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.05(s,1H),8.33–8.26(m,2H),8.16(d,J=8.6Hz,1H),8.05(dd,J=8.2,1.2Hz,1H),7.83(ddd,J=8.4,6.8,1.3Hz,1H),7.70–7.62(m,2H),5.27(s,2H),3.61–3.41(m,5H),3.20(d,J=15.2Hz,0H),3.20(s,1H),2.99(s,3H),1.96(dddd,J=35.2,13.7,7.3,4.0Hz,4H),1.72(d,J=3.9Hz,3H),1.70–1.59(m,4H),1.29–1.21(m,1H)。
实施例38

1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-N-甲基-2'-氧代螺[氮杂环丁烷-3,
3'-二氢吲哚]-1-羧基酰胺(38)
将在二噁烷(2mL)中的4M HCl添加到化合物36b(35mg,0.072mmol)的DCM溶液中。将反应物搅拌1h,接着真空浓缩并且与甲苯共蒸发。将所得的HCl盐溶解于MeCN(4mL)中,添加碳酸氢钠(12.1mg,0.144mmol)和DIEA(43.8μl,0.251mmol)并且将混合物在0℃下搅拌15min。缓慢添加4-硝基苯基氯甲酸酯在MeCN(1mL)中的溶液。2h后,添加40%的甲胺水溶液(100μl,1.00mmol)并且将混合物在室温下搅拌,接着真空浓缩。使用从25%至35%B的梯度经12min以及40mL/min的流速,在Gemini C18 100x30mm上通过制备型HPLC纯化粗产物。溶剂A:95%水、5%乙腈(10mM,在乙酸铵中);溶剂B:10%水、90%乙腈(10mM,在乙酸铵中)。将与产物对应的级分汇集在一起并冷冻干燥。通过硅胶色谱进一步纯化所得的产物,使用DCM-MeOH洗脱,将适当的级分汇集并冻干,得到标题化合物(3.7mg,12%)。MS(ES+)445.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.17–8.11(m,1H),8.06(dd,J=8.2,1.3Hz,1H),7.82(ddd,J=8.4,6.8,1.3Hz,1H),7.66(ddd,J=7.9,6.8,0.9Hz,1H),7.58(dd,J=7.4,1.2Hz,1H),7.19(td,J=7.8,1.3Hz,1H),7.07(td,J=7.5,1.0Hz,1H),6.98(d,J=7.8Hz,1H),6.54(q,J=4.5Hz,1H),5.21(s,2H),4.44(t,J=5.1Hz,1H),4.13(d,J=7.8Hz,2H),3.98(d,J=7.7Hz,2H),3.48(td,J=6.2,5.0Hz,2H),3.18(s,1H),3.16(s,0H),2.61(d,J=4.5Hz,3H),1.68–1.50(m,4H),1.24(s,1H)。
实施例39

步骤a)(1-(1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺
[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)氨基甲酸叔丁酯(39a)
将TFA(1.0mL,13.1mmol)添加到化合物37b(120mg,0.232mmol)在DCM(3.0mL)中的溶液中。在室温下搅拌反应物直到完全去保护,接着真空浓缩,用甲苯稀释并浓缩(x2)。将一部分残余物(64mg,0.15mmol)和1-((叔丁氧羰基)氨基)环丙烷羧酸(31mg,0.15mmol)溶解于DMF(5.0mL)中,在0℃下添加DIPEA(99mg,0.77mmol)和HATU(76mg,0.20mol)并且将混合物在0℃下搅拌5分钟,接着使其达到室温并搅拌30分钟。使用28-38%B的梯度经12min以及40mL/min的流速,在Gemini C18 100x30mm上通过制备型HPLC纯化粗产物。溶剂A:95%水、5%乙腈(10mM,在乙酸铵中);溶剂B:10%水、90%乙腈(10mM,在乙酸铵中)。将适当的级分汇集并且浓缩,得到标题化合物(14mg,15%)。MS(ES+)600.4[M+H]+。
步骤b)1-(1-氨基环丙烷羰基)-1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)螺[哌
啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(39b)
将HCl在二噁烷(2.5mL)中的4M溶液添加到化合物39a(10mg)在DCM(4mL)和MeOH(2mL)中的溶液中。将反应物在室温下搅拌1h,接着真空浓缩。
将残余物溶解于MeOH中并且在Gemini-NX Prep C18 5mm OBD 21x 100mm上在pH10下通过制备型LCMS进行纯化,用水/乙腈(在两种洗脱液中均使用0.1%NH4OH)洗脱。将适当的级分汇集并冷冻干燥,得到标题化合物(5.4mg,65%)。MS(ES+)500.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.06(s,1H),8.33–8.25(m,2H),8.16(d,J=8.6Hz,1H),8.06(dd,J=8.2,1.2Hz,1H),7.83(ddd,J=8.4,6.8,1.3Hz,1H),7.70–7.61(m,2H),5.27(s,2H),4.46(t,J=5.1Hz,1H),3.50(q,J=5.4Hz,2H),3.33(s,11H),3.21(d,J=15.2Hz,0H),3.21(s,1H),1.84(s,3H),1.65(dd,J=7.7,4.4Hz,3H),0.90(q,J=4.4Hz,2H),0.68(t,J=3.3Hz,2H)。
实施例40

1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢吲哚]-2'-
酮(40)
将TFA(0.5mL)添加到化合物36c(22mg,0.045mmol)在DCM(3mL)中的溶液中。将溶液在室温下搅拌30min,接着浓缩。使用水(+0.1%NH4OH)和MeCN(+0.1%NH4OH)的梯度,通过反相制备型LCMS纯化残余物。将适当的级分汇集,并且冷冻干燥,得到标题化合物(11.4mg,65%)。MS(ES+)388.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.09(s,1H),8.12(d,J=8.6Hz,1H),8.09–8.03(m,1H),7.81(ddd,J=8.4,6.8,1.3Hz,1H),7.74(s,1H),7.66(t,J=7.4Hz,1H),7.15(t,J=7.7Hz,1H),7.06(t,J=6.6Hz,1H),6.95(d,J=7.8Hz,1H),5.19(s,2H),4.43(s,1H),4.03(s,1H),3.45(s,2H),3.45(d,J=14.9Hz,1H),3.38(d,J=7.3Hz,1H),3.16(d,J=16.4Hz,1H),3.16(s,1H),1.62(p,J=6.7Hz,2H),1.51(d,J=7.5Hz,1H),1.49(s,2H),1.39(q,J=6.7Hz,0H),1.25(q,J=7.6,6.4Hz,2H)。
实施例41

1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1-甲基螺[氮杂环丁烷-3,3'-二氢吲
哚]-2'-酮(41)
将37%甲醛的水溶液(0.041mmol)添加到化合物40(8.0mg,0.021mmol)在DCM(5mL)中的溶液中,之后添加乙酸(7.0μl,0.126mol)和三乙酰氧基硼氢化钠(13mg,0.062mol)。将反应混合物在室温下搅拌15min,接着添加MeOH并且接着将溶液真空浓缩。使用制备型Gemini-NX C185mm OBD 21x100mm柱通过制备型LCMS纯化残余物,利用水(+0.1%NH4OH)和MeCN(+0.1%NH4OH)的梯度在pH 10下洗脱。将适当的级分汇集,并且冷冻干燥,得到标题化合物(5.0mg,60%)。MS(ES+)402.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.09(s,1H),8.12(d,J=8.5Hz,1H),8.06(dd,J=8.3,1.2Hz,1H),7.81(ddd,J=8.4,6.8,1.3Hz,1H),7.66(td,J=7.3,1.0Hz,2H),7.16(td,J=7.7,1.3Hz,1H),7.05(td,J=7.5,1.0Hz,1H),6.95(d,J=7.8Hz,1H),5.19(s,2H),4.44(s,1H),3.16(dd,J=7.1,4.2Hz,2H),2.36(s,4H),1.61(p,J=6.7Hz,2H),1.50(ddd,J=13.6,10.4,5.4Hz,3H),1.26–1.21(m,1H)。
实施例42

1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1-(甲基磺酰基)螺[氮杂环丁烷-3,
3'-二氢吲哚]-2'-酮(42)
将TFA(0.5mL)添加到化合物36c在DCM(3mL)中的溶液中。将溶液在室温下搅拌30min,接着浓缩并与甲苯共蒸发。
将残余物和DIEA(28μl,0.16mol)溶解于MeCN(4mL)中,将溶液在0℃下搅拌15min,接着缓慢添加甲磺酰氯(40μl,0.054mmol)在MeCN(1mL)中的混合物。当没有起始物质剩余时,将溶液真空浓缩并且使用包含10mM乙酸铵的水和乙腈的梯度,在Gemini C18 100x30mm上通过制备型HPLC进一步纯化粗产物。将适当的级分汇集并冷冻干燥,并且通过制备型LCMS进一步纯化,使用包含0.1%NH4OH的水/乙腈在pH 10下洗脱,得到标题化合物(2.1mg,10%)。MS(ES+)466.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.15(d,J=8.6Hz,1H),8.06(d,J=8.2Hz,1H),7.83(ddd,J=8.3,6.8,1.3Hz,1H),7.70–7.62(m,1H),7.24(td,J=7.8,1.2Hz,1H),7.12(t,J=7.5Hz,1H),7.03(d,J=7.8Hz,1H),5.22(s,1H),4.45(s,0H),4.24(d,J=8.3Hz,1H),4.18(d,J=8.3Hz,1H),3.33(s,11H),3.20(s,1H),1.70–1.53(m,3H),1.24(s,1H)。
实施例43

步骤a)(E)-甲基4-溴-3-((叔丁基亚氨基)甲基)苯甲酸酯(43a)
在氩气下向4-溴-3-甲酰基苯甲酸甲酯(1.22g,5.00mmol)在无水乙醚(7.5mL)中的溶液中添加叔丁胺(1.10g,15.0mmol)并且将混合物在室温下搅拌四小时。用DCM(25mL)稀释悬浮液,添加硫酸镁(9g)并且将混合物在室温下搅拌过夜。将盐过滤出来并且用DCM洗涤。将溶液真空浓缩并且与苯共蒸发。将产物真空干燥。收率1.48g,99%。MS(ES+)299.52[M+H]+。
步骤b)4-(4-((叔丁基二甲基甲硅烷基)氧基)丁基)-3-(((四氢-2H-吡喃-2-基)
氧基)甲基)异喹啉-7-羧酸甲酯(43b)
在氩气下向化合物43a(1.38g,4.62mol)和叔丁基二甲基((7-((四氢-2H-吡喃-2-基)氧基)庚-5-炔-1-基)氧基)硅烷(1.81g,5.55mmol)在乙腈(120mL)中的溶液中添加锌粉(907mg,13.9mmol)和NiBr2(PPh3)2(343g,0.462mmol)并且使反应物回流3小时。再添加Zn(450mg)和NiBr2(PPh3)2(175mg)并且使混合物再回流一小时。将混合物冷却并且用乙腈稀释。将催化剂过滤出来并且用乙腈洗涤。将溶液浓缩并且通过硅胶色谱分离产物,用异己烷和10至30%乙酸乙酯洗脱,得到标题化合物(735mg,33%)。
步骤c)(4-(4-((叔丁基二甲基甲硅烷基)氧基)丁基)-3-(((四氢-2H-吡喃-2-基)
氧基)甲基)异喹啉-7-基)甲醇(43c)
在氩气、-70℃下向化合物43b(8700mg,1.44mmol)在无水DCM(25mL)中的溶液中逐滴添加在甲苯中的1M DIBAL溶液(3.16mmol)。将混合物在-70℃下搅拌一小时并且使其达到-50℃。用10%氯化钠溶液淬灭反应并且用乙酸乙酯萃取三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。通过硅胶柱色谱分离产物,用DCM和0至5%甲醇洗脱,得到标题化合物(470mg,71%)。
步骤d)4-(4-((叔丁基二甲基甲硅烷基)氧基)丁基)-7-(氯甲基)-3-(((四氢-2H-
吡喃-2-基)氧基)甲基)异喹啉(43d)
在氩气下将四氯化碳(108mg,0.700mmol)添加到化合物43c(230mg,0.500mol)和三苯基膦(171mg,0.650mmol)在DCM(10mL)中的冰冷溶液中。将混合物在室温下搅拌两小时。仅约10%转化率。添加四溴化碳(232mg,0.700mmol)并且将混合物在室温下搅拌30分钟。将溶液添加到装填了DCM的硅胶柱中,用2至6%的甲醇洗脱产物,得到氯代化合物和溴代化合物的混合物,其直接原样用于下一步骤。收率估计为50%。
步骤e)4-(4-((叔丁基二甲基甲硅烷基)氧基)丁基)-7-((4-甲基哌嗪-1-基)甲
基)-3-(((四氢-2H-吡喃-2-基)氧基)甲基)异喹啉(43e)
将N-甲基哌嗪(136mg,1.36mmol)添加到化合物43d(130mg,0.272mmol)的溶液中。将混合物搅拌72h,接着浓缩并且通过硅胶柱色谱分离产物,用DCM和5至20%甲醇洗脱,得到标题化合物(160mg,109%)。MS(ES+)542.35[M+H]+。
步骤f)4-(7-((4-甲基哌嗪-1-基)甲基)-3-(((四氢-2H-吡喃-2-基)氧基)甲基)
异喹啉-4-基)丁-1-醇(43f)
向化合物43e(150mg,0.277mmol)在THF(5mL)中的溶液中添加四丁基氟化铵(0.831mol)的1M溶液并且将混合物在室温下搅拌过夜。将混合物用甲醇稀释并且蒸发到二氧化硅上。通过硅胶柱色谱分离产物,用DCM和10至30%甲醇洗脱,得到被四丁基铵盐污染的标题化合物(120mg,101%)。
步骤g)4-(7-((4-甲基哌嗪-1-基)甲基)-3-(((四氢-2H-吡喃-2-基)氧基)甲基)
异喹啉-4-基)丁基乙酸酯(43g)
向化合物43f(120mg,0.281mmol)在无水DCM(5mL)中的溶液中添加TEA(199mg,1.96mmol)和乙酸酐(142mg,1.403mmol)。将溶液搅拌18h,接着浓缩并与甲苯共蒸发。通过硅胶柱色谱分离产物,用DCM和5至20%甲醇洗脱。收率125mg,95%。
MS(ES+)470.33[M+H]+。
步骤h)4-(3-(羟甲基)-7-((4-甲基哌嗪-1-基)甲基)异喹啉-4-基)丁基乙酸酯
(43h)
将化合物43g(120mg,0.256mmol)在80%乙酸(16mL)中的溶液在80℃下搅拌两小时,接着减压浓缩并且通过HPLC(Gemini-NX 100x30mm 20至40%乙腈,流速40mL/min)纯化产物。将适当的级分汇集,并且冷冻干燥,得到标题化合物(95mg,96%)。MS(ES+)386.2[M+H]+。
步骤i)4-(3-((1-环丙基-2-氧代-1H-咪唑并[4,5-c]吡啶-3(2H)-基)甲基)-7-
((4-甲基哌嗪-1-基)甲基)异喹啉-4-基)丁基乙酸酯(43i)
将化合物43h(90mg)从二噁烷甲苯中共蒸发三次,得到70mg干燥产物。在氩气下将干燥产物溶解于无水DCM中并冷却至0℃。添加亚硫酰氯(54.0mg,0.454mmol)并且将混合物在冰浴中搅拌两小时。将悬浮液浓缩并与甲苯共蒸发。将固体悬浮于约DMF(3mL)中并且添加到1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(31.8mg,0.182mmol)和碳酸铯(177mg,0.454mmol)在DMF(3mL)中的经预先搅拌(30min)的悬浮液中。将悬浮液在室温下搅拌18h,接着添加另外的碳酸铯(177mg,0.454mmol)并且继续搅拌72h。将反应用水淬灭并用DCM萃取三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。产物无需进一步纯化即用于下一步骤。
步骤j)1-环丙基-3-((4-(4-羟基丁基)-7-((4-甲基哌嗪-1-基)甲基)异喹啉-3-
基)甲基)-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(43j)
将在甲醇中的1M甲醇钠溶液添加到化合物43i(90mg,0.166mmol)在THF(1mL)和甲醇(2mL)中的溶液中。将混合物在室温下搅拌一小时,接着用甲醇稀释并且用乙酸酸化。将溶液减压浓缩并且通过HPLC分离产物
(Gemini-NX,100x20mm,20至40%乙腈的水溶液)。将适当的级分汇集,并且冷冻干燥,得到标题化合物(55mg,66%)。MS(ES+)501.23[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.07(s,1H),8.23(s,1H),8.19(d,J=5.3Hz,1H),8.10(d,J=8.8Hz,1H),7.93(d,J=1.7Hz,1H),7.76(dd,J=8.8,1.7Hz,1H),7.24(d,J=5.2Hz,1H),5.35(s,2H),3.63(s,2H),3.44(t,J=6.4Hz,2H),3.22–3.15(m,2H),2.98(tt,J=7.1,3.7Hz,1H),2.39(s,3H),2.31(s,4H),2.14(s,3H),1.86(s,2H),1.62(p,J=6.7Hz,2H),1.53–1.42(m,2H),1.06(dt,J=7.1,3.5Hz,2H),0.94–0.88(m,1H)。
实施例44

步骤a)1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-
3,3'-二氢吲哚]-1-羧酸叔丁酯(44a)
使化合物36a(200mg,0.405mmol)和(6-氰基吡啶-3-基)硼酸(89.8mg,0.607mmol)根据实施例12步骤a中所述的方法偶联在一起,得到标题化合物(130mg,62%)。
步骤c)1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-
3,3'-二氢吲哚]-1-羧酸甲酯(44b)
将TFA(1mL)添加到化合物44a在DCM(6mL)中的溶液中。将混合物在室温下搅拌一小时,接着真空浓缩,与甲苯共蒸发两次,真空干燥。将残余物溶解于DMF(20mL)中并且将10mL的所得溶液冷却至0℃,添加DIEA(151mg,1.17mmol),接着缓慢添加氯甲酸甲酯(16.5mg,0.175mmol)。将混合物在0℃下搅拌15分钟,接着添加乙醇和水。用乙酸乙酯萃取混合物三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。在硅胶上通过柱色谱纯化产物,用DCM和3%甲醇洗脱。在Gemini-NX 100x30mm柱上通过HPLC进一步纯化所得的产物,使用水和40至60%乙腈的梯度洗脱。将适当的级分汇集,浓缩并且从乙腈-水中冷冻干燥,得到标题化合物(35mg,63%)。MS(ES+)476.13[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.40(d,J=0.8Hz,1H),8.74(dd,J=2.2,0.9Hz,1H),8.26–8.18(m,2H),8.14(dd,J=7.9,2.2Hz,1H),7.79–7.70(m,2H),7.63(dd,J=7.4,1.2Hz,1H),7.29–7.23(m,1H),7.19(td,J=7.8,1.3Hz,1H),7.06(td,J=7.6,1.0Hz,1H),6.81(d,J=7.8Hz,1H),5.03–4.91(m,2H),3.64(s,3H),3.32(s,3H)。
13C NMR(126MHz,DMSO)δ41.88,44.21,51.99,57.36,109.33,117.42,122.55,123.18,123.78,126.06,126.93,127.68,127.87,128.60,128.91,129.12,131.70,132.08,134.48,135.32,139.19,142.88,145.93,151.52,152.83,156.09,175.99。
实施例45

步骤a)1'-((4-溴异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-
1-羧酸叔丁酯(45)
将10mL来自实施例44步骤b的DMF溶液置于氩气下并冷却至0℃,添加DIEA(151mg,1.17mmol),之后缓慢添加甲磺酰氯(20.5mg,0.175mmol)。将混合物在0℃下搅拌10分钟,接着添加乙醇和水。用乙酸乙酯萃取混合物三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。在硅胶上通过柱色谱纯化产物,用DCM和3%甲醇洗脱。在Gemini-NX 100x30mm柱上通过HPLC进一步纯化所得的产物,使用水和40至60%乙腈的梯度洗脱。将适当的级分汇集,浓缩并且从乙腈-水中冷冻干燥,得到标题化合物(25mg,43%)。MS(ES+)496.12[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.39(d,J=0.8Hz,1H),8.77(dd,J=2.2,0.9Hz,1H),8.28–8.19(m,2H),8.17(dd,J=7.9,2.1Hz,1H),7.79–7.70(m,2H),7.59(dd,J=7.5,1.2Hz,1H),7.32–7.24(m,1H),7.22(td,J=7.8,1.3Hz,1H),7.10(td,J=7.6,1.0Hz,1H),6.87(d,J=7.8Hz,1H),4.98(s,1H),4.94(d,J=15.9Hz,1H),4.07(s,4H),3.17(s,3H),1.91(s,0H),1.23(d,J=6.5Hz,0H)。
实施例46

步骤a)(E)-N-(5-溴-2-碘亚苄基)-2-甲基丙-2-胺(46a)
在氩气下向5-溴-1-碘苯甲醛(1.00g,3.22mmol)在无水乙醚(5mL)中的溶液中添加叔丁胺(1.01mL,9.65mmol)并且将混合物在室温下搅拌4小时。用DCM(16mL)稀释悬浮液,添加硫酸镁(4.5g)并且将混合物在室温下搅拌过夜,接着将盐过滤出来并用DCM洗涤。将溶液浓缩并且与甲苯共蒸发。将产物真空干燥,得到标题化合物(1.14g,97%)。MS(ES+)367.84[M+H]+。
步骤b)7-溴-3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-4-(4-((四氢-2H-吡喃-
2-基)氧基)丁基)异喹啉(46b)
将化合物46a(2.17g,4.81mmol)在DMF(40mL)中的溶液添加到Pd(OAc)2(29.2mg,0.130mmol)、Ph3P(63.1mg,0.240mmol)和Na2CO3(255mg,2.40mmol)中。将混合物搅拌几分钟,接着添加I-7b(880mg,2.40mmol)在DMF(8mL)中的溶液。将混合物置于氮气下,并且在100℃下搅拌18h。将混合物过滤,浓缩滤液并且使其在浓NH4Cl(~50mL)与乙醚(50-60mL)之间分配。将醚相干燥(Na2SO4)、过滤并浓缩。在硅胶上通过柱色谱纯化所得的残余物,用EtOAc:庚烷洗脱。通过二次硅胶色谱再次纯化所得的产物,用EtOAc:庚烷洗脱。获得的总量:378mg,25%。MS(ES+)634.11[M+H]+。
步骤c)N-(3-(3-(((叔丁基二苯基甲硅烷基)氧基)甲基)-4-(4-((四氢-2H-吡喃-
2-基)氧基)丁基)异喹啉-7-基)氧杂环丁烷3-基)-2-甲基丙烷-2-亚磺酰胺(46c)
接着在-78℃下将丁基锂在己烷(0.565mmol)中的1.5M溶液逐滴添加到化合物46b(376mg,0.594mmol)在无水THF(1.4mL)中的溶液中。将混合物在-78℃下搅拌~30min,接着逐滴添加3在THF(0.24mL)中的溶液,将混合物在-78℃下搅拌~20min,接着使其达到室温。在30min室温后,添加浓NH4Cl(~0.5mL)并且蒸发掉大部分THF。添加水并且用DCM(x3)萃取混合物。将有机相用盐水(x1)萃取并干燥(Na2SO4)。在硅胶上通过柱色谱纯化残余物,使用EtOH:DCM的梯度洗脱,得到标题化合物(46mg,11%)。MS(ES+)729.4[M+H]+。
步骤d)N-(3-(3-(羟甲基)-4-(4-((四氢-2H-吡喃-2-基)氧基)丁基)异喹啉-7-
基)氧杂环丁烷-3-基)-2-甲基丙烷-2-亚磺酰胺(46d)
将TBAF水合物(73μL,0.073mmol)添加到化合物46c(46.0mg,0.063mmol)在THF(0.63mL)中的溶液中。将溶液在室温下振荡8h,接着浓缩至干燥。添加分子筛(0.5mL)和无水THF并且将混合物振荡3h,接着将溶液浓缩并且通过制备型TLC纯化所得的残余物,使用在CHCl3中的10%MeOH洗脱,得到标题化合物(26mg,84%)。MS(ES+)491.2[M+H]+。
步骤e N-(3-(3-(溴甲基)-4-(4-((四氢-2H-吡喃-2-基)氧基)丁基)异喹啉-7-
基)氧杂环丁烷-3-基)-2-甲基丙烷-2-亚磺酰胺(46e)
在0℃下将三苯基膦(19.5mg,0.074mmol)添加到化合物46d(26.0mg,0.053mmol)在DCM(1.3mL)中的溶液中。将混合物在0℃下搅拌15min,接着添加四溴化碳(26.4mg,0.079mmol),使溶液达到室温并且继续搅拌60min。将溶液浓缩并且通过制备型TLC(1mm)纯化残余物,使用10%MeOH/CHCl3洗脱。刮下适当的带并且用10%MeOH/CHCl3充分洗涤凝胶,将滤液浓缩并且与DCM/庚烷共蒸发,得到标题化合物(11mg,38%)。MS(ES+)555.1[M+H]+。
步骤f N-(3-(3-((1-环丙基-2-氧代-1H-咪唑并[4,5-c]吡啶-3(2H)-基)甲基)-
4-(4-((四氢-2H-吡喃-2-基)氧基)丁基)异喹啉-7-基)氧杂环丁烷-3-基)-2-甲基丙烷-2-
亚磺酰胺(46f)
使化合物46e(11.0mg,0.020.mmol)与1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(3.66mg,0.021mmol)根据实施例4步骤a中所述的过程偶联。通过制备型TLC进行粗化合物的纯化,使用在DCM中的10%MeOH洗脱,得到标题化合物(8.5mg,66%)。MS(ES+)648.4[M+H]+。
步骤g)3-((7-(3-氨基氧杂环丁烷-3-基)-4-(4-羟基丁基)异喹啉-3-基)甲基)-
1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(46g)
将化合物46f(8.5mg,0.013mmol)在AcOH:THF:H2O(4:2:1v/v/v)(~0.7mL)中的溶液在40℃下温热16h,接着再在50℃下16h。将溶液浓缩并且与THF(x3)共蒸发。将所得的残余物溶解于MeOH(1mL)中并冷却至0℃。添加HCl(4M,在二噁烷中,250μL)并且将反应混合物搅拌大约一分钟,接着在寒冷条件下浓缩,直到检测不到起始物质。将所得粗料溶解于MeOH中并且通过制备型LCMS进行纯化,在pH 10下使用水和乙腈(两者均包含0.1%NH4OH)的梯度洗脱。收集适当的级分,并且冷冻干燥,得到标题化合物(4.5mg,75%)。LCMS:ES(+):460.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.11(s,1H),8.25–8.15(m,4H),8.08(dd,J=8.9,2.0Hz,1H),7.27–7.21(m,1H),5.36(s,2H),4.79(d,J=6.0Hz,2H),4.73(d,J=6.1Hz,2H),4.43(t,J=5.1Hz,1H),3.45(q,J=6.0Hz,2H),3.21(s,1H),2.98(tt,J=7.1,3.7Hz,1H),2.70(s,2H),1.62(p,J=6.7Hz,2H),1.58–1.44(m,2H),1.24(s,1H),1.07(td,J=7.3,5.2Hz,2H),0.95–0.82(m,2H)。
实施例47

步骤a)4-((叔丁基二苯基甲硅烷基)氧基)丁-2-炔-1-醇(47a)
在环境温度下向丁-2-炔-1,4-二醇(20g,233mmol)在CH2Cl2(250mL)中的经搅拌溶液中添加咪唑(9.5g,140mmol,0.6当量),之后缓慢添加TBDPSCl(30.2mL,116mmol)在DCM(50mL)中的溶液。将反应混合物搅拌16h,接着用CH2Cl2稀释,用水和盐水洗涤,经无水Na2SO4干燥,并且减压浓缩。使用在己烷中的10%EtOAc,通过硅胶(100-200目)柱色谱纯化所得的粗料,得到标题化合物(75.5g,16%)
步骤b)(E)-N-(2-溴亚苄基)-2-甲基丙-2-胺(47b)
将2-溴苯甲醛(5.0g,27.0mmol)和叔丁胺(25mL)放入密封管中并且在80℃下加热17h。接着将反应混合物真空浓缩,得到为液体的标题化合物(5.5g,85%)。MS(ES+)241.7[M+H]+。
步骤c)(4-(((叔丁基二苯基甲硅烷基)氧基)甲基)异喹啉-3-基)甲醇(47c)
在圆底烧瓶中放入Zn(8.17g,125mmol,3当量)和NiBr2(PPh3)2(1.55g,2.1mmol,0.05当量),抽真空并且用氩气重新填充。这个过程重复三次,然后添加化合物47a(13.5g,41.7mmol,1当量)和47b(10g,41.7mmol,1当量)在乙腈(200mL)中的经脱气溶液。将反应混合物在80℃、氩气下加热4h,接着通过硅藻土过滤,将滤液减压浓缩并且使用在己烷中的0-20%EtOAc通过硅胶(100-200目)柱色谱纯化残余物,从而提供两种分离的异构体。将所需异构体以固体的形式分离(31%)。MS(ES+)428.1[M+H]+。
步骤d)4-(((叔丁基二苯基甲硅烷基)氧基)甲基)-3-(氯甲基)异喹啉(47d):
SOCl2(0.42mL,5.85mmol,5当量)逐滴添加到化合物47c(0.5g,1.17mmol,1当量)在DCM(20mL)中的冰冷溶液中。将反应混合物在环境温度下搅拌5h,接着减压浓缩,并用DCM稀释且用饱和碳酸氢钠水溶液、水和盐水洗涤。将合并的有机层经无水Na2SO4干燥并减压浓缩,得到标题化合物(0.5g,98%)。MS(ES+)445.9[M+H]+。
步骤e)3-((4-(((叔丁基二苯基甲硅烷基)氧基)甲基)异喹啉-3-基)甲基)-1-环
丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(47e)
将Cs2CO3(2.19g,6.72mmol,3当量)添加到化合物47d(1.0g,2.24mmol,1当量)在DMF(15mL)中的经搅拌溶液中,接着添加1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(0.39g,2.24mmol,1当量)。将混合物在80℃下搅拌90min,接着用乙酸乙酯稀释并且用水和盐水洗涤。将合并的有机层经无水硫酸钠干燥并减压浓缩,得到为红色粘性固体的粗标题化合物(1.3g),其无需进一步纯化即用于下一步骤。
步骤f)1-环丙基-3-((4-(羟甲基)异喹啉-3-基)甲基)-1H-咪唑并[4,5-c]吡啶-2
(3H)-酮(47f)
将HCl水溶液(6M,2mL)添加到化合物47e(3.5g,5.98mmol,1当量)在MeOH(10mL)中的经搅拌的冰冷溶液中。将溶液在环境温度下搅拌4h,接着真空浓缩并且通过与甲苯共沸蒸馏而完全干燥。将所得固体溶解于水中并且用乙酸乙酯萃取。将NaHCO3添加到水层,接着用乙酸乙酯萃取。将合并的有机层用水和盐水洗涤,经无水Na2SO4干燥并浓缩。将所得的粗料用戊烷、乙醚和丙酮研磨,得到为固体的纯标题化合物(0.68g,33%)。MS(ES+)347.2[M+H]+。通过1H NMR确认结构。
实施例48

4-(3-((4-(羟甲基)异喹啉-3-基)甲基)-2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡
啶-1-基)哌啶-1-羧酸甲酯(48)
根据实施例47步骤e和f中所述的过程由化合物47d(2.6g,5.8mmol)和4-(2-氧代-2,3-二氢-1H-咪唑并[4,5-c]吡啶-1-基)哌啶-1-羧酸甲酯(1.6g,5.8mmol)制备标题化合物。收率:1.5g,59%。MS(ES+)448.1[M+H]+。通过1H NMR确认结构。
实施例49

步骤a)1'-((4-(4-((叔丁基二苯基甲硅烷基)氧基)丁基)异喹啉-3-基)甲基)螺
[环丙烷-1,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(49a)
将NaH(26mg,60%,分散于矿物油中,0.65mmol)在0℃、惰性气体下添加到螺[环丙烷-1,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(81.6mg,0.509mmol)在DMF(3.5mL)中的溶液中。在室温下搅拌15min之后,在冰浴中冷却的同时添加氯化物I-4e(250mg,0.512mmol)。添加DMF(1mL)以冲洗管的侧面。将溶液在室温下搅拌过夜,接着添加饱和NaCl水溶液(30mL)+水(15mL)并且用EtOAc(3x 40mL和1x 20mL)萃取混合物。将有机相合并,干燥(Na2SO4)并且真空浓缩。在硅胶上通过柱色谱纯化所得的粗产物,使用MeOH-DCM洗脱,得到标题化合物(168mg,54%)。MS(ES+)612.4[MH]+。
步骤b)1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)螺[环丙烷-1,3'-吡咯并[2,3-
c]吡啶]-2'(1'H)-酮(49b)
使化合物49a如实施例14步骤b中所述的那样反应,得到标题化合物(56.8mg,55%)。
1H NMR(500MHz,DMSO-d6)δ9.17(s,1H),9.14(s,1H),8.35(s,2H),8.26(s,1H),8.21(s,1H),8.15(d,J=8.6Hz,1H),8.08(d,J=8.0Hz,1H),7.87–7.81(m,1H),7.72–7.66(m,1H),7.19(s,1H),7.15(s,1H),5.42(s,0H),5.38(s,1H),5.35(s,1H),4.44(s,1H),3.20(s,1H),1.82(s,2H),1.70(s,2H),1.63(s,2H),1.53(s,2H)。
实施例50

步骤a)(1-(1'-((4-(6-氨基甲酰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',
2'-二氢螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)氨基甲酸叔丁酯(50a-1)
和
5-(3-((1-(1-((叔丁氧羰基)氨基)环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并
[2,3-c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶甲酸甲酯(50a-2)
将MeOH(3.0mL)添加到化合物13a(160mg,0.293mmol)在4M HCl(3mL)中的悬浮液中。将混合物在室温下搅拌90min,接着在冰浴上冷却并且添加1-boc氨基1-环丙烷羧酸、DIEA和HATU,将混合物在冰浴上搅拌两小时。将反应混合物添加到碳酸氢钠的饱和溶液中并且用EtOAc萃取三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。通过硅胶色谱纯化所得粗料,用DCM和2至6%MeOH洗脱,得到化合物50a-1(100mg,53%)。MS(ES+)648.45[MH]+和50a-2(45mg,23%)。MS(ES+)663.44[MH]+。
步骤b)5-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并[2,3-c]吡
啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶甲酸
(50b)
将TFA(864mg,7.58mmol)添加到化合物50a-1(70mg,0.108mmol)在DCM(8mL)中的溶液中。将混合物在室温下搅拌1h,接着减压浓缩并且与甲苯共蒸发。将残余物溶解于MeOH(1.5mL)和在MeOH(1mL)中的1M NaOMe中并且通过HPLC(Gemini NX100x30mm,15至35%乙腈)进行纯化,得到标题化合物(59mg,62%)。MS(ES+)548.3[MH]+。
1H NMR(500MHz,DMSO-d6)δ9.38(s,1H),8.71(d,J=2.1Hz,1H),8.30–8.12(m,7H),7.75(dddd,J=13.8,7.9,5.4,3.6Hz,3H),7.57(d,J=4.7Hz,1H),7.34(dd,J=7.9,1.6Hz,1H),5.05(d,J=15.9Hz,1H),4.98(d,J=15.9Hz,1H),3.93(s,2H),3.76(s,2H),1.88(s,2H),1.76(s,2H),1.67(s,2H),0.88(q,J=4.0Hz,2H),0.66(q,J=3.7Hz,2H)。
步骤c)5-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并[2,3-c]吡
啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶甲酸甲酯(50c)
将化合物50a-2如实施例50步骤b中所述的那样去保护,得到标题化合物(28mg,66%)。MS(ES+)563.6[MH]+。
1H NMR(500MHz,DMSO-d6)δ9.38(d,J=0.9Hz,1H),8.79(dd,J=2.2,0.9Hz,1H),8.30–8.24(m,2H),8.25–8.16(m,2H),8.18(s,1H),7.75(tt,J=6.9,5.2Hz,2H),7.57(d,J=4.7Hz,1H),7.35–7.29(m,1H),5.05(d,J=15.9Hz,1H),4.96(d,J=15.9Hz,1H),3.96(s,3H),3.92(s,2H),3.76(s,2H),1.79(d,J=8.1Hz,1H),1.70–1.65(m,2H),0.88(q,J=4.4Hz,2H),0.66(t,J=3.3Hz,2H)。
实施例51

步骤a)(4-(4-(叔丁基)苯基)异喹啉-3-基)甲醇N-氧化物(51a)
将碳酸钾(1.38g,9.96mmol)和(4-(叔丁基)苯基)硼酸(0.887g,4.98mmol)添加到化合物I-4b(1.0g,3.32mmol)在1,4-二噁烷(16mL)和水(4.0mL)中的经搅拌溶液中。将混合物用N2脱气15min,接着添加四(三苯基膦)钯(0)(0.192g,0.166mmol)并且将混合物再次用N2脱气10min。将反应混合物在N2气氛、100℃下搅拌。通过TLC监测反应的进展并且当确认完全时(~16h),将混合物冷却至室温,减压浓缩。添加水(15mL)并且用DCM(2x 25mL)萃取混合物。将有机层用盐水洗涤、经Na2SO4干燥并浓缩。将所得粗料
用10%EtOAc/石油醚沉淀,将所得浆液搅拌20min,过滤并干燥,得到为固体的标题化合物(900mg,83%)。MS(ES+)308.25[MH]+。
步骤b)(4-(4-(叔丁基)苯基)异喹啉-3-基)甲醇(51b)
将氢氧化钠(0.439g,11.0mmol)添加到化合物51a(0.900g,2.20mmol)在苄醇(16.0mL,0.154mol)中的经搅拌溶液中并且将所得混合物在120℃下搅拌16h,接着冷却。添加水(20mL)并且将混合物用DCM(2x25mL)萃取、干燥(Na2SO4)并浓缩。在硅胶(100-200目)上通过柱色谱纯化所得的粗料,使用EtOAc/石油醚洗脱,得到为固体的标题化合物(0.60g,85%收率)。MS(ES+)292.22[MH]+。
步骤c)4-(4-(叔丁基)苯基)-3-(氯甲基)异喹啉(51c)
在0℃下将亚硫酰氯(0.3mL,4.12mmol)添加到化合物51b(0.60g,2.06mmol)在DCM(10mL)中的经搅拌溶液中。将所得混合物在回流下搅拌2h,接着冷却至室温并减压浓缩。添加冷水(5mL),利用饱和NaHCO3溶液将pH调节至7-7.5,并且用DCM(2x 25mL)萃取混合物。将有机层干燥(Na2SO4)并且真空浓缩。利用正戊烷(10mL)沉淀所得的残余物,并且通过过滤收集形成的固体,得到为固体的标题化合物(580mg,91%)。MS(ES+)310.19[MH]+。
步骤d)3-((4-(4-(叔丁基)苯基)异喹啉-3-基)甲基)-1-环丙基-1H-咪唑并[4,5-
c]吡啶-2(3H)-酮(51d)
将碳酸铯(1.42g,4.36mmol)和化合物51c(0.450g,1.45mmol)在室温、氮气下添加到1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(0.280g,1.60mmol)在乙腈(15mL)中的经搅拌溶液中。将所得混合物在室温下搅拌16h,接着减压浓缩,通过冰冷的水(15mL)缓慢稀释,用DCM(2x25mL)萃取。将有机层用水(15mL)、盐水(15mL)洗涤、干燥(Na2SO4)并浓缩。用乙醚(15mL)研磨所得残余物。使用硅胶(100-200目)通过柱色谱纯化粗料,使用1-2%MeOH/DCM洗脱,得到为固体的标题化合物(0.19,29%)。MS(ES+)449.41[MH]+。
1H NMR(500MHz,DMSO-d6)δ9.25–9.21(m,1H),8.20–8.10(m,2H),8.05–8.00(m,1H),7.70(dddd,J=22.6,8.0,6.8,1.3Hz,2H),7.61–7.54(m,2H),7.35(dq,J=8.7,2.4,1.8Hz,3H),7.20(dd,J=5.2,0.8Hz,1H),5.08(s,2H),2.89(tt,J=7.0,3.6Hz,1H),1.37(s,9H),1.01(td,J=7.3,5.2Hz,2H),0.87–0.80(m,2H)。
实施例52

步骤a和b)3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1-(1-甲基环丙基)-1H-咪唑
并[4,5-c]吡啶-2(3H)-酮(52b)
使I-4e(201mg,0.413mmol)与I-16c(81.3mg,0.430mmol)如实施例14步骤a中所述的那样反应,然后如实施例14步骤b中所述的那样去除TBDSi基团,得到标题化合物(28.7mg,13%)。MS(ES+)403.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.12(s,1H),8.29(s,1H),8.21(d,J=5.2Hz,1H),8.15(d,J=8.6Hz,1H),8.12–8.06(m,1H),7.83(ddd,J=8.4,6.8,1.3Hz,1H),7.68(t,J=7.5Hz,1H),7.28(d,J=5.2Hz,1H),5.37(s,2H),4.44(s,1H),3.46(t,J=6.5Hz,2H),3.25–3.18(m,2H),1.63(p,J=6.8Hz,2H),1.50(ddt,J=15.9,10.2,5.9Hz,2H),1.42(s,3H),1.09–0.97(m,4H)。
实施例53

3-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1-(1-甲基环丙基)-1H-咪唑并[4,5-c]
吡啶-2(3H)-酮(53)
使I-4e(1.1g,2.0mmol)与A-14(469mg,2.0mmol)如实施例11步骤f中所述的那样反应,然后如实施例14步骤b中所述的那样去除TBDSi基团,得到标题化合物(8.2%)。MS(ES+)641.52[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.10(s,1H),8.15–8.09(m,1H),8.07(dd,J=8.2,1.2Hz,1H),7.81(ddd,J=8.4,6.8,1.3Hz,1H),7.66(ddd,J=7.9,6.8,0.9Hz,1H),7.46(dd,J=7.4,1.2Hz,1H),7.11(td,J=7.7,1.2Hz,1H),7.05–6.93(m,2H),5.57(d,J=6.8Hz,1H),5.21(s,2H),4.77–4.65(m,1H),4.44(t,J=5.0Hz,1H),3.46(td,J=6.4,5.0Hz,2H),3.20–3.12(m,2H),2.45–2.37(m,2H),1.62(p,J=6.7Hz,2H),1.53–1.42(m,2H)。
实施例54

1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-1-((1-甲基环丙基)磺酰基)螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(54)
使I-4e(304mg,0.622mmol)与I-18(100mg,0.311mmol)如实施例11步骤f中所述的那样反应,然后如实施例14步骤b中所述的那样去除TBDSi基团,得到标题化合物(69mg,30%)。MS(ES+)641.52[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.05(s,1H),8.32–8.25(m,2H),8.16(d,J=8.6Hz,1H),8.05(dd,J=8.3,1.2Hz,1H),7.83(ddd,J=8.4,6.8,1.3Hz,1H),7.70–7.63(m,1H),7.62(d,J=4.8Hz,1H),5.76(s,1H),5.27(s,2H),4.46(s,1H),3.76(ddd,J=12.8,9.3,3.5Hz,2H),3.61(dt,J=12.6,4.8Hz,2H),3.50(s,2H),3.50(d,J=11.5Hz,1H),3.20(d,J=15.1Hz,1H),3.20(s,1H),1.96(ddd,J=13.5,9.3,4.2Hz,2H),1.88(dt,J=13.7,4.4Hz,2H),1.65(dt,J=9.9,4.3Hz,4H),1.49(s,3H),1.20(q,J=4.5Hz,2H),0.91–0.83(m,2H)。
实施例55

1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲
哚]-1-羧基酰胺(55)
在室温下向I-19(200mg,0.921mmol)在乙腈(15mL)中的经搅拌溶液中添加三氟甲磺酸亚铁(II)(65.2mg,0.184mmol)和碳酸铯(600mg,1.84mmol)。将反应混合物在70℃下搅拌15分钟,接着添加I-43(674mg,1.38mmol),将所得反应混合物在70℃下搅拌6h,接着减压浓缩。将所得粗料用水(25mL)稀释并且搅拌10分钟,形成固体,通过过滤收集所述固体,接着在硅胶上通过柱色谱进行纯化,使用在DCM中的5%MeOH洗脱。如实施例14步骤b中所述的那样处理所得化合物,得到为固体的标题化合物(220mg,51%)。MS(ES+)431.20[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.14(d,J=8.6Hz,1H),8.06(d,J=8.1Hz,1H),7.82(t,J=7.7Hz,1H),7.66(t,J=7.5Hz,1H),7.59(d,J=7.3Hz,1H),7.19(t,J=7.7Hz,1H),7.07(t,J=7.5Hz,1H),6.98(d,J=7.8Hz,1H),6.15(s,2H),5.21(s,2H),4.45(t,J=5.1Hz,1H),4.12(dd,J=18.9,6.5Hz,2H),3.99(d,J=7.9Hz,2H),3.48(q,J=5.9Hz,2H),3.16(s,3H),1.63(t,J=7.1Hz,1H),1.63–1.55(m,0H),1.55(s,1H)。
实施例56

N-(叔丁基)-1'-((4-(4-羟基丁基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-
3,3'-二氢吲哚]-1-羧基酰胺(56)
使I-4e(400mg,0.819mmol)与I-19(246mg,0.901mmol)如实施例11步骤f中所述的那样反应,然后如实施例14步骤b中所述的那样去除TBDSi基团,得到标题化合物(55mg,20%)。MS(ES+)487.26[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.08(s,1H),8.14(d,J=8.6Hz,1H),8.06(d,J=8.1Hz,1H),7.82(t,J=7.6Hz,1H),7.66(t,J=7.4Hz,1H),7.55(d,J=7.2Hz,1H),7.19(t,J=7.6Hz,1H),7.07(t,J=7.5Hz,1H),6.99(d,J=7.8Hz,1H),5.99(s,1H),5.21(s,2H),4.45(t,J=5.2Hz,1H),4.13(d,J=7.5Hz,2H),3.96(d,J=7.9Hz,2H),3.48(q,J=6.2,5.7Hz,2H),3.41(s,1H),3.18(t,J=7.9Hz,2H),1.64(p,J=6.8,6.3Hz,2H),1.57(q,J=7.8Hz,2H),1.29(s,9H)。
实施例57

1-环丙基-3-((4-(4-氟丁基)异喹啉-3-基)甲基)-1H-咪唑并[4,5-c]吡啶-2
(3H)-酮(57)
在-78℃下向化合物1c(0.35g,0.90mmol)在DCM(10mL)中的经搅拌溶液中添加二乙氨基三氟化硫(0.73g,4.5mmol)在DCM中的溶液。将反应混合物在室温下搅拌3h,接着减压浓缩,用水(10mL)稀释并且用DCM(3x 20mL)萃取。将合并的有机层干燥(Na2SO4)并浓缩,并且通过硅胶柱色谱纯化所得粗料,使用在DCM中的3.5%MeOH洗脱,然后通过制备型HPLC进一步纯化,得到为固体的标题化合物(50mg,14%)。MS(ES+)391.26[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.12(s,1H),8.34–8.21(m,1H),8.20(d,J=5.2Hz,1H),8.23–8.08(m,1H),8.09(dd,J=8.2,1.2Hz,1H),7.84(ddd,J=8.4,6.8,1.3Hz,1H),7.68(ddd,J=7.9,6.8,0.9Hz,1H),7.25(dd,J=5.2,0.8Hz,1H),5.37(s,2H),4.51(dt,J=47.5,6.0Hz,2H),3.43–3.16(m,3H),2.97(tt,J=7.1,3.6Hz,1H),1.99–1.75(m,2H),1.65–1.45(m,2H),1.06(td,J=7.3,5.1Hz,2H),1.02–0.79(m,2H)。
实施例58和59

使用实施例57的过程分别由醇28b和42制备化合物58和59。
化合物58:收率15%,MS(ES+)448.31[M+H]+。
化合物59:收率8.7%,MS(ES+)468.59[M+H]+。
实施例60

步骤a)5-(3-((2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1'-基)甲基)异喹啉-4-
基)甲基吡啶腈(60a)
使用实施例40中所述的方法从化合物44a(210mg,0.406mmol)中去除Boc基团,得到标题化合物(170mg,100%)。
步骤b)(1-(1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环
丁烷-3,3'-二氢吲哚]-1-基羰基)环丙基)氨基甲酸叔丁酯(60b)
在氩气下将HATU(201mg,0.529mmol)和DIEA(316mg,2.44mmol)添加到化合物60a(170mg,0.406mmol)和1-((叔丁氧羰基)氨基)-环丙烷羧酸(90mg,0.448mmol)在无水DMF(8mL)中的溶液中。将混合物在冰浴上搅拌90min,接着添加到5%碳酸氢钠溶液中并且用EtOAc萃取三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。通过硅胶色谱纯化粗料,用DCM和0至4%MeOH洗脱,得到标题化合物(58mg,28%)。MS(ES+)601.37[M+H]+。
步骤c)5-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲
哚]-1'-基)甲基)异喹啉-4-基)甲基吡啶腈(60c)
使用实施例40中所述的方法从化合物44a(210mg,0.406mmol)中去除Boc基团。通过HPLC(Gemini NX,100x30mm,梯度30至50%乙腈)纯化产物,接着从乙腈水中冷冻干燥。收率20mg,24%。MS(ES+)501.5[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.39(s,1H),8.75(dd,J=2.1,0.9Hz,1H),8.26–8.18(m,2H),8.15(d,J=8.0Hz,1H),7.79–7.70(m,2H),7.58(dd,J=7.4,1.2Hz,1H),7.30–7.24(m,1H),7.19(td,J=7.8,1.3Hz,1H),7.08(td,J=7.5,1.0Hz,1H),6.79(d,J=7.8Hz,1H),5.04–4.93(m,2H),4.70(s,2H),3.97(s,2H),1.14–1.06(m,2H),0.74–0.70(m,2H)。
实施例61

步骤a)叔丁基(1-(1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺
[氮杂环丁烷-
在0℃下将Et3N(0.158mL)和1-甲基环丙烷-1-磺酰氯(0.039mL)添加到化合物60a(200mg,0.376mmol)在DMF(5mL)中的经搅拌溶液中。将所得反应混合物在0℃下搅拌1h,接着用冰水稀释并且搅拌10分钟。形成固体,通过过滤收集所述固体,接着在C18柱上通过制备型HPLC进行纯化。收集适当的级分,浓缩并冻干,得到标题化合物(58mg,28%)。(MS ES+)536.17[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.44–9.38(m,1H),8.69(dd,J=2.1,0.9Hz,1H),8.27–8.19(m,2H),8.11(dd,J=7.9,2.2Hz,1H),7.79–7.70(m,2H),7.57(dd,J=7.4,1.2Hz,1H),7.29–7.17(m,2H),7.11(td,J=7.6,1.0Hz,1H),6.80(d,J=7.8Hz,1H),5.76(s,1H),5.01(s,1H),4.97(d,J=15.8Hz,1H),4.12(dd,J=7.7,3.1Hz,2H),3.92(t,J=7.1Hz,2H),2.47(s,1H),1.55(s,3H),1.26–1.15(m,2H),0.96–0.89(m,2H)。
实施例62

5-(3-((1-(1-(二甲基氨基)环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲
哚]-1'-基)甲基)异喹啉-4-基)甲基吡啶腈(62)
将TFA添加到化合物44a(170mg,0.263mmol)在无水DCM(8mL)中的溶液中并且将溶液在室温下搅拌90min,接着浓缩并与甲苯共蒸发两次。将残余物溶解于无水DMF(10mL)中并且添加1-(二甲基氨基)环丙烷羧酸(50.9mg,0.394mmol)和DIEA(340mg,2.63mmol)。将溶液在冰浴中冷却并且添加HATU(130mg,0.342mmol)。将混合物在冰浴中搅拌一小时,接着添加水(40mL)和饱和碳酸氢钠溶液(40mL)。用EtOAc萃取混合物三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。通过硅胶色谱纯化产物,用DCM和2至4%甲醇洗脱。通过HPLC(Gemini NX 100x30,25-50%乙腈水溶液,10mmol乙酸铵缓冲液,梯度14分钟,流速40mL/min)进一步纯化所得的产物。将适当的级分汇集,浓缩并且从乙腈水中冷冻干燥,得到标题化合物(55mg,40%)。
1H NMR(500MHz,DMSO-d6)δ9.42(d,J=0.8Hz,1H),8.27–8.19(m,2H),8.12(s,1H),7.78–7.70(m,2H),7.59(dd,J=7.4,1.2Hz,1H),7.27–7.21(m,1H),7.18(td,J=7.8,1.2Hz,1H),7.07(td,J=7.5,1.0Hz,1H),6.76(d,J=7.8Hz,1H),4.40(s,2H),3.99(s,1H),2.21(s,6H),2.08(s,3H),1.24(s,1H),0.94(s,4H)。
实施例63

5-(3-((1-(1-(甲基氨基)环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲
哚]-1'-基)甲基)异喹啉-4-基)甲基吡啶腈(63)
将化合物44a(160mg,0.247mmol)N-去保护,接着使其与1-((叔丁氧羰基)(甲基)氨基)环丙烷羧酸(63.9mg,0.297mmol)如实施例62中所述的那样反应。将残余物溶解于无水DCM(8mL)中,添加TFA并且将溶液在室温下搅拌90min,接着浓缩并与甲苯共蒸发两次。将残余物溶解于DCM(5mL)中并添加TEA(0.5mL)。将溶液减压浓缩并且通过HPLC纯化残余物。(Gemini NX 100x30mm;30-50%乙腈水溶液,10mmol乙酸铵缓冲液)。将适当的级分汇集,浓缩并且从乙腈水中冷冻干燥,得到标题化合物(56mg,60%)。
1H NMR(500MHz,DMSO-d6)δ9.41(s,1H),8.75(s,1H),8.71(s,1H),8.23(dt,J=7.5,2.6Hz,2H),8.14(s,1H),7.79–7.70(m,2H),7.59(dd,J=7.4,1.2Hz,1H),7.29–7.23(m,1H),7.18(td,J=7.8,1.3Hz,1H),7.07(td,J=7.5,1.0Hz,1H),6.79(d,J=7.8Hz,1H),4.54(s,2H),4.51(s,1H),3.98(s,2H),2.28(s,3H),2.00(d,J=7.4Hz,1H),1.24(s,0H),1.04(d,J=14.6Hz,2H),0.79(s,2H)。
实施例64

2-羟乙基1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁
烷-3,3'-二氢吲哚]-1-羧酸酯(64)
将TFA添加到化合物44a(360mg,0.696mmol)在无水DCM(8mL)中的溶液中并且将溶液在室温下搅拌90min,接着浓缩并与甲苯共蒸发两次。将残余物、碳酸铯(552mg,1.69mmol)和碳酸亚乙酯(149mg,1.69mmol)在微波烘箱中在70℃下搅拌4h。用水(10mL)淬灭反应并且用乙酸乙酯(2x50mL)萃取,将有机层用盐水洗涤,经无水硫酸钠干燥、过滤并减压浓缩。使用在H2O:乙腈中的10mM碳酸氢铵作为流动相,在C18柱上通过制备型HPLC纯化所得粗料。将纯级分汇集,浓缩并冻干,得到标题化合物(60mg,21%)。(MS ES+)506.21[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.40(s,1H),8.75(d,J=2.1Hz,1H),8.27–8.18(m,2H),8.15(dd,J=7.9,2.2Hz,1H),7.79–7.70(m,3H),7.62(dd,J=7.4,1.2Hz,1H),7.30–7.23(m,1H),7.19(td,J=7.8,1.3Hz,1H),7.07(td,J=7.6,1.0Hz,1H),6.82(d,J=7.8Hz,1H),5.04–4.91(m,3H),4.84(s,1H),4.07(s,1H),3.57(t,J=5.1Hz,2H),3.39(s,1H)。
实施例65

步骤a)(1-(1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二
氢螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)氨基甲酸叔丁酯(65a)
将TFA(17.15mmol)添加到化合物13a(125mg,0.229mmol)在无水DCM(10mL)中的溶液中。将溶液在室温下搅拌90分钟,接着减压浓缩并且与甲苯共蒸发两次。
将残余物和1-(boc氨基)环丙烷羧酸(55.1mg,0.274mmol)溶解于DMF(10mL)中。将溶液置于氩气下并且在冰浴上冷却,添加DIEA(295mg,2.28mmol)和HATU(113mg,0.297mmol)。将混合物在冰浴上搅拌90min,接着添加碳酸氢钠饱和溶液并且用EtOAc萃取混合物三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。通过硅胶色谱分离产物,用DCM和0至4%MeOH洗脱。收率140mg,97%。(MS ES+)630.6[M+H]+。
步骤b)5-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并[2,3-c]吡
啶]-1'(2'H)-基)甲基)异喹啉-4-基)甲基吡啶腈(65b)
将TFA(23.5mg,0.206mmol)添加到化合物65a(130mg,0.206mmol)在无水DCM(8mL)中的溶液中。将溶液在室温下搅拌90min,接着减压浓缩并且与甲苯共蒸发两次。将残余物溶解于DCM(5mL)中,添加TEA(0.5mL)并且将溶液浓缩。通过HPLC(Gemini NX 100x30,15至45%乙腈水溶液,10mM乙酸铵)纯化产物并且从乙腈水中冷冻干燥。收率65mg,59%。MS(ES+)530.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.40(d,J=0.8Hz,1H),8.89(dd,J=1.9,1.0Hz,1H),8.34–8.24(m,3H),8.22(d,J=7.2Hz,2H),7.81–7.71(m,2H),7.60–7.54(m,1H),7.36–7.30(m,1H),5.00(d,J=11.2Hz,2H),3.94(s,2H),3.32(s,5H),1.88(s,1H),1.76(d,J=9.8Hz,2H),1.69(s,2H),0.89(q,J=4.2Hz,2H),0.67(t,J=3.2Hz,2H)。
实施例66

5-(3-((1-(甲基磺酰基)-2'-氧代螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1'(2'H)-
基)甲基)异喹啉-4-基)甲基吡啶腈(66-1)和
5-(3-((2'-氧代-1-(2,2,2-三氟乙酰基)螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1'
(2'H)-基)甲基)异喹啉-4-基)甲基吡啶腈(66-2)
将TFA(207μl,2.71mmol)在室温下逐滴添加到化合物13a(75.0mg,0.137mmol)在DCM(2mL)中的溶液中。将溶液搅拌2h,接着浓缩并与甲苯共蒸发。将残余物和DIEA(62.1mg,0.480mmol)在室温下溶解于MeCN(5mL)中,添加甲磺酰氯(18.9mg,0.165mmol)。将溶液搅拌18h,接着再添加DIEA和磺酰氯。4h后,起始物质被消耗,将反应混合物浓缩。将所得粗料溶解于MeCN/MeOH中并且在Gemini-NX Prep C18 5mm柱上通过制备型LCMS进行纯化,在pH 7下使用在水中的10nM乙酸铵/在乙腈中的10nM乙酸铵洗脱。
单独收集两种标题化合物并冷冻干燥。在硅胶上通过柱色谱进一步纯化第二峰的第二化合物,使用DCM/MeOH洗脱并且冷冻干燥。
66-1:收率16.8mg,23%,MS(ES+)525.1[M+H]+。
1H NMR(500MHz,DMSO)δ9.40(s,1H,10),8.89(m,1H,35),8.32(m,1H,32),8.27(dd,J=7.9,2.1Hz,3H,18,20,31),8.22(dd,J=6.7,2.4Hz,1H,6),7.76(m,2H,1,2),7.60(dd,J=10.8,5.8Hz,1H,21),7.34(m,1H,3),4.99(d,J=3.3Hz,2H,12),3.41(m,4H,23,25),2.97(s,3H,30),1.92(m,2H,22”,26”),1.77(m,2H,22',26')。
13C NMR(126MHz,DMSO)δ177.00,17,152.80,10,151.86,35,145.78,8,143.91,20,141.09,15,139.50,31,139.28,14,135.29,11,134.44,4,132.20,33,131.73,2,130.46,18,129.13,32,127.88,6,127.75,1,127.01,5,126.53,7,123.87,3,118.37,21,117.53,37,43.52,12,16,40.53,23,25,34.11,30,31.16,31.05,31.04,22,26。
66-2:收率31.8mg,43%,MS(ES+)543.1[M+H]+。
1H NMR(500MHz,DMSO)δ9.39(d,J=1.6Hz,1H,11),8.89(m,1H,35),8.29(m,3H,21,31,32),8.25(d,J=6.3Hz,1H,19),8.22(m,1H,7),7.76(ddt,J=10.1,6.9,3.6,3.6Hz,2H,2,3),7.64(d,J=4.7Hz,1H,22),7.33(d,J=7.9Hz,1H,4),5.00(m,2H,13),4.04(m,1H,26”),3.89(m,1H,24”),3.82(dt,J=14.7,7.2,7.2Hz,1H,24'),3.74(t,J=11.1,11.1Hz,1H,26'),1.94(ddd,J=13.6,9.7,4.0Hz,2H,23”,27”),1.76(m,2H,23',27')。
13C NMR(126MHz,DMSO)δ177.07(d,J=4.9Hz,18),154.22(d,J=34.7Hz,29),152.86,11,151.89,35,145.70,9,144.04,21,140.76,16,139.53,31,139.18,15,135.28,33,134.44,5,132.20,12,131.74,3,130.55,19,129.09,127.89,7,127.76,2,127.00,6,126.50,126.46,8,123.87,4,118.31,22,117.48,115.17,36,44.05,17,43.52(d,J=3.3Hz,13),40.66,24,38.37,26,31.45,27,30.52,23。
实施例67

5-(3-((1-(1-(甲基氨基)环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并[2,3-c]吡
啶]-1'(2'H)-基)甲基)异喹啉-4-基)甲基吡啶腈(67)
使用实施例63的过程由化合物13a(100mg,0.183mmol)和1-((叔丁氧羰基)(甲基)氨基)环丙烷羧酸(47.2mg,0.220mmol)获得标题化合物。收率45mg,46%。MS(ES+)544.5[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.40(d,J=0.9Hz,1H),8.88(dd,J=2.0,1.0Hz,1H),8.34–8.18(m,6H),7.76(tt,J=7.0,5.3Hz,2H),7.58(d,J=4.8Hz,1H),7.36–7.30(m,1H),5.00(d,J=12.2Hz,2H),2.26(s,3H),2.21(s,1H),1.77–1.66(m,5H),0.88(s,2H),0.68(s,2H)。
实施例68

2-羟乙基1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢
螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸酯(68)
使用实施例64中所述的方法由化合物13a(236mg,0.432mmol)和碳酸亚乙酯(189mg,2.14mmol)获得标题化合物,但是微波反应在130℃下进行1h。收率:25mg,13%。MS(ES+)535.18[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.39(s,1H),8.88(s,1H),8.34–8.22(m,2H),8.22(d,J=7.3Hz,1H),7.76(ddd,J=7.3,5.2,1.7Hz,1H),7.62–7.56(m,1H),7.37–7.30(m,1H),4.99(d,J=10.7Hz,1H),4.80(t,J=5.4Hz,1H),4.07(dq,J=23.7,4.6,4.0Hz,2H),3.59(q,J=5.0Hz,1H),3.36(d,J=10.1Hz,1H),3.33(s,9H),3.17(d,J=5.0Hz,1H),1.75(ddd,J=13.2,8.6,4.5Hz,1H),1.65(dd,J=12.1,6.7Hz,1H)。
实施例69

步骤a)(4-(4-(甲基磺酰基)苯基)异喹啉-3-基)甲醇N-氧化物(69a)
使在二噁烷-水(10mL-2mL)中的I-1b(500mg,1.66mmol)与4-(甲基磺酰基)苯基硼酸(498mg,20mmol)如实施例51步骤a中所述的那样反应,得到标题化合物。MS(ES+)330.22[M+H]+。
步骤b)(4-(4-(甲基磺酰基)苯基)异喹啉-3-基)甲醇(69b)
将雷尼镍(156mg,1.0当量)在室温下添加到化合物I-22a(600mg,1.0当量)在MeOH(100mL)中的溶液中。将反应混合物在室温、氢气气氛下搅拌1h,接着蒸馏出来。在硅胶上通过柱色谱纯化所得的粗产物,使用在石油醚中的30-100%EtOAc洗脱,得到为固体的标题化合物(400mg,64%)。MS(ES+)314.23[M+H]+。
步骤c)3-(氯甲基)-4-(4-(甲基磺酰基)苯基)异喹啉(69c)
使化合物I-22b(400mg,1.28mmol)与亚硫酰氯如实施例51步骤c中所述的那样反应,得到标题化合物(360mg,81%)。MS(ES+)332.20[M+H]+。
步骤d)1-环丙基-3-((4-(4-(甲基磺酰基)苯基)异喹啉-3-基)甲基)-1H-咪唑并
[4,5-c]吡啶-2(3H)-酮(69d)
使化合物69c(350mg,1.06mmol)与1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(222mg,1.27mmol)如实施例11步骤a中所述的那样反应,得到标题化合物(160mg,32%)。MS(ES+)471.24[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.33(s,1H),8.23–8.14(m,2H),8.10–8.04(m,3H),7.78–7.68(m,2H),7.70–7.63(m,2H),7.30–7.24(m,1H),7.20(d,J=5.2Hz,1H),5.10(s,2H),3.33(s,3H),2.87(tt,J=7.1,3.7Hz,1H),1.00(td,J=7.3,5.2Hz,2H),0.86–0.79(m,2H)。
实施例70

步骤a)3-甲基-4-(6-(甲基磺酰基)吡啶-3-基)异喹啉(70a)
使4-溴-3-甲基异喹啉(600mg,2.70mmol)与(6-(甲基磺酰基)吡啶-3-基)硼酸(597mg,2.97mmol)如实施例51步骤a中所述的那样反应,得到标题化合物(560mg,61%)。MS(ES+)299.21[M+H]+。
步骤b)3-(溴甲基)-4-(6-(甲基磺酰基)吡啶-3-基)异喹啉(70b)
向化合物70a(550mg,1.84mmol)在四氯化碳(10mL)中的经搅拌溶液中添加N-溴琥珀酰亚胺(361mg)和偶氮二异丁腈(30mg,0.184mmol)。将所得混合物在回流下搅拌16h,接着冷却至室温并减压浓缩。添加水(10mL)并且用DCM(2x 25mL)萃取混合物。将合并的有机层用盐水洗涤、干燥(Na2SO4)并浓缩。在硅胶上通过柱色谱纯化所得的粗料,使用在DCM中的1-2%MeOH洗脱,得到标题化合物(230mg,19%)。379.2[M+H]+。
步骤c)1-环丙基-3-((4-(6-(甲基磺酰基)吡啶-3-基)异喹啉-3-基)甲基)-1H-咪
唑并[4,5-c]吡啶-2(3H)-酮(70c)
使化合物70b(220mg,0.583mmol)与1-环丙基-1H-咪唑并[4,5-c]吡啶-2(3H)-酮(112mg,0.641mmol)如实施例11步骤a中所述的那样反应,得到标题化合物(58mg,21%)。MS(ES+)472.36[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.40(s,1H),8.80(s,1H),8.23(d,J=7.3Hz,1H),8.18(d,J=7.3Hz,3H),8.12(s,1H),7.76(p,J=6.8Hz,2H),7.28(d,J=7.8Hz,1H),7.20(d,J=5.2Hz,1H),5.13(s,2H),3.38(s,4H),2.86(tt,J=6.8,3.3Hz,1H),1.00(d,J=7.1Hz,2H),0.85–0.80(m,2H)。
实施例71

1'-((4-(4-(叔丁基)苯基)异喹啉-3-基)甲基)-1-(甲基磺酰基)螺[氮杂环丁烷-
3,3'-二氢吲哚]-2'-酮(71)
在室温下向I-21(400mg)在乙腈(30mL)中的经搅拌溶液中添加三氟甲磺酸亚铁(II)(561mg)和碳酸铯(1.03g)。将反应混合物在70℃下搅拌15min,接着添加I-24(677mg),将所得混合物在70℃下搅拌6h。将反应混合物减压浓缩并且将残余物用水(20mL)稀释并且搅拌20min。形成固体,通过过滤收集所述固体,并且在硅胶上通过柱色谱进行纯化,使用在石油醚中的40%EtOAc洗脱。将适当的级分汇集并减压浓缩,将所得的固体在乙醚和戊烷(3:1,5mL)中研磨、过滤并干燥,得到为固体的标题化合物(40mg)。MS(ES+)526.64[M+H]+。
使用所指出的中间体,使用实施例71中所述的方法制备以下化合物,反应时间通过TLC判断:



实施例79

1-(甲基磺酰基)-1'-((4-(4-(三氟甲基)苯基)异喹啉-3-基)甲基)螺[氮杂环丁
烷-3,3'-二氢吲哚]-2'-酮(79)
将碳酸铯(1.55g)在室温下添加到I-21(400mg,1.58mmol)和I-23(813mg,2.22mmol)在乙腈(30mL)中的经搅拌混合物中。将混合物在70-80℃下搅拌,直到根据TLC确认反应完全为止(~4h),接着减压浓缩。将所得粗料用水(20mL)稀释并搅拌20分钟。收集所形成的固体并且用乙腈(20mm)研磨、过滤,用乙腈(10mL)洗涤并干燥,得到为固体的标题化合物(170mg,20%)。MS(ES+)538.57[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.33(s,1H),8.19(dd,J=6.8,2.5Hz,1H),7.91(d,J=8.0Hz,2H),7.72(tt,J=7.0,5.2Hz,2H),7.60(dd,J=12.0,7.4Hz,3H),7.29–7.22(m,1H),7.19(td,J=7.8,1.2Hz,1H),7.08(t,J=7.5Hz,1H),6.72(d,J=7.8Hz,1H),4.96(s,2H),4.05(s,4H),3.15(s,3H)。
使用所指出的中间体,使用实施例79中所述的方法制备以下化合物,反应时间通过TLC判断:



实施例85

步骤a)1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,
3'-二氢吲哚]-1-羧酸叔丁酯(85a)
使化合物I-22b(2.00g,6.31mmol)和I-11c(1.73g,6.31mmol)根据实施例79中所述的方法反应,得到标题化合物(1.20g,27%)。MS(ES+)511.36[M+H]+。
步骤b)1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢
吲哚]-2'-酮(85b)
在0℃下将TFA(2.2mL,29mmol)添加到化合物85a(1.0g,2.0mmol)在DCM(20mL)中的经搅拌悬浮液中。将混合物在室温下搅拌4h,接着减压浓缩。添加NaHCO3并且用DCM(2x15mL)萃取混合物。将合并的有机层经无水Na2SO4干燥并浓缩,得到标题化合物(700mg,73%)。MS(ES+)411.25[M+H]+。
步骤c)1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)-1-(2-甲氧基乙基)螺[氮杂
环丁烷-3,3'-二氢吲哚]-2'-酮(85c)
在室温下向化合物85b(300mg,0.731mmol)在乙腈(30mL)中的经搅拌溶液中添加2-甲氧基乙基4-甲基苯磺酸酯(0.167mL,0.877mmol)和碳酸铯(714mg,2.19mmol)。将所得反应混合物在100℃下加热48h,接着减压浓缩,用水(30mL)稀释并且用DCM(2x50mL)萃取。将合并的有机层经无水Na2SO4干燥并浓缩。在硅胶上通过柱色谱纯化所得的粗料,用在DCM中的2.5%MeOH洗脱。将适当的级分汇集并浓缩,并且在X-Bridge C18,25*150,10μ柱上通过制备型HPLC进一步纯化(流动相:在H2O:乙腈中的10mM碳酸氢铵),得到为固体的标题化合物(45mg,13%)。MS(ES+)469.70[M+H]。
1H NMR(500MHz,DMSO-d6)δ9.35(s,1H),8.19(dt,J=4.4,2.2Hz,2H),7.96(td,J=8.1,2.5Hz,1H),7.79–7.68(m,2H),7.61(dd,J=7.4,1.2Hz,1H),7.35(dd,J=8.4,2.6Hz,1H),7.29(dd,J=7.9,1.6Hz,1H),7.12(td,J=7.7,1.3Hz,1H),7.04(td,J=7.5,1.0Hz,1H),6.67(d,J=7.7Hz,1H),4.97(d,J=2.4Hz,2H),3.44–3.33(m,5H),3.25(s,3H),2.65(t,J=5.7Hz,2H),2.55(d,J=16.0Hz,1H)。
实施例86

1-(2-氟乙基)-1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,
3'-二氢吲哚]-2'-酮(86)
使化合物85b(300mg,731mmol)与2-氟乙基4-甲基苯磺酸酯(191mg,877mmol)根据实施例85步骤c中所述的方法反应,得到标题化合物(30mg,8.8%)。MS(ES+)457.18[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.35(s,1H),8.19(dd,J=7.3,2.1Hz,2H),7.97(td,J=8.1,2.5Hz,1H),7.82–7.63(m,2H),7.64(dd,J=7.3,1.2Hz,1H),7.35(dd,J=8.4,2.6Hz,1H),7.29(dd,J=7.9,1.5Hz,1H),7.13(td,J=7.8,1.3Hz,1H),7.05(td,J=7.5,1.0Hz,1H),6.69(d,J=7.7Hz,1H),4.97(d,J=2.9Hz,2H),4.48(dt,J=47.7,4.8Hz,2H),3.52–3.24(m,4H),2.80(dt,J=29.3,4.8Hz,2H),2.55(s,1H)。
实施例87

步骤a)(2-氟乙基)(1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[环
丁烷-1,3'-二氢吲哚]-3-基)氨基甲酸叔丁酯(87a)
使化合物I-22b(600mg,1.89mmol)和I-17b(633mg,1.89mmol)根据实施例79中所述的方法反应,得到标题化合物(850mg,64%)。MS(ES+)571.39[M+H]+。
步骤b)3-((2-氟乙基)氨基)-1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)螺[环
丁烷-1,3'-二氢吲哚]-2'-酮(87b,异-1和异-2)
在室温、氮气下将TFA(2.21mL)添加到化合物87a(825mg,1.45mmol)在DCM(20mL)中的经搅拌溶液中。将反应混合物在室温下搅拌2h,接着减压浓缩。将所得粗料用DCM(100mL)稀释,用饱和碳酸氢钠溶液(50mL)洗涤,经硫酸钠干燥、过滤并减压浓缩。将粗料通过制备型HPLC纯化,流动相:在H2O:乙腈中的10mM碳酸氢铵,柱:X-Bridge C18,30*150,5μ。将所得固体的两种异构体通过SFC(固定相:Chiralpak IC 250x30mm,5μ;流动相:CO2,在MeOH中的30mM NH4OMe)分离,随后在硅胶(12g,在DCM中的3%MeOH)上通过快速色谱纯化。将纯级分合并、浓缩并且用戊烷研磨并冻干,得到为以下异构体的标题化合物:
异构体1:151mg,22%。
1H NMR(500MHz,DMSO-d6)δ9.40–9.26(m,1H),8.28(d,J=2.5Hz,1H),8.26–8.10(m,1H),8.07(td,J=8.1,2.5Hz,1H),7.83–7.63(m,2H),7.56(dd,J=7.4,1.3Hz,1H),7.40(dd,J=8.4,2.6Hz,1H),7.40–7.25(m,1H),7.09(td,J=7.7,1.3Hz,1H),7.00(td,J=7.5,1.0Hz,1H),6.68(d,J=7.7Hz,1H),4.92(d,J=2.6Hz,2H),4.45(dt,J=47.8,5.1Hz,2H),3.56(p,J=7.9Hz,1H),2.80(dt,J=27.5,5.1Hz,2H),2.61–2.34(m,2H),2.15(dd,J=10.9,8.2Hz,2H)。
异构体2:50mg,7.1%。MS(ES+)471.84[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.36(s,1H),8.19(h,J=5.8,5.3Hz,2H),7.96(td,J=8.1,2.5Hz,1H),7.83–7.63(m,2H),7.50(dd,J=7.4,1.2Hz,1H),7.48–7.22(m,2H),7.09(td,J=7.7,1.2Hz,1H),6.99(td,J=7.5,1.0Hz,1H),6.68(d,J=7.7Hz,1H),5.09–4.85(m,2H),4.47(dt,J=47.6,5.2Hz,2H),3.75(p,J=8.2Hz,1H),2.83(dt,J=26.5,5.2Hz,2H),2.32(qd,J=6.8,3.3Hz,2H),2.19(dt,J=11.5,8.3Hz,2H)。
实施例88

1-环丙基-3-((4-(6-羟基吡啶-3-基)异喹啉-3-基)甲基)-1H-苯并[d]咪唑-2
(3H)-酮(88)
将化合物80(140mg,0.28mmol)溶解于MeOH中,添加10%碳载钯(15mg)并且将混合物置于H2下并且在气球压力下氢化16h。通过硅藻土过滤反应混合物,用MeOH(2x10mL)洗涤滤饼。将溶液真空浓缩并且通过制备型HPLC纯化所得粗料,得到为固体的标题化合物(26mg,23%)。MS(ES+)410.14[M+H]+。
1H NMR(500MHz,DMSO-d6)δ11.91(d,J=0.7Hz,1H),9.27(s,1H),8.18(d,J=5.2Hz,1H),8.15(d,J=8.1Hz,1H),7.99(s,1H),7.78(t,J=7.5Hz,1H),7.71(t,J=7.4Hz,1H),7.57(d,J=8.4Hz,1H),7.55–7.51(m,1H),7.25(dd,J=9.3,2.2Hz,1H),7.22(d,J=5.2Hz,1H),6.42(d,J=9.3Hz,1H),5.29(d,J=15.9Hz,1H),5.15(d,J=15.9Hz,1H),2.90(tt,J=6.8,3.5Hz,1H),1.02(d,J=6.8Hz,2H),0.87(q,J=10.2,9.7Hz,2H)。
实施例89

1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[环丁烷-1,3'-二氢
吲哚]-3-基氨基甲酸甲酯(89)
在室温下添加到将化合物82(140mg,0.324mmol)、Et3N(0.226mL,1.62mmol)和双(2,5-二氧代吡咯烷-1-基)碳酸酯(415mg,1.62mmol)在DCM(20mL)中的溶液在温度下搅拌2h,接着添加甲胺在THF(1.62mmol)中的1.0M溶液并且将反应混合物搅拌16h。将反应混合物减压浓缩并且将所得的粗产物在硅胶上通过柱色谱纯化,用在石油醚中的50%EtOAc洗脱,接着使用10mM碳酸氢铵:乙腈作为流动相在XSELECT柱上通过制备型HPLC纯化,得到为固体的标题化合物(30mg,19%)。MS(ES+)490.16[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.42(s,1H),8.66(d,J=2.1Hz,1H),8.26–8.17(m,2H),8.06(dd,J=7.9,2.2Hz,1H),7.74(dt,J=6.3,3.4Hz,2H),7.48(dd,J=7.4,1.2Hz,1H),7.24(dt,J=6.2,3.6Hz,1H),7.23–7.08(m,2H),7.03(td,J=7.6,1.0Hz,1H),6.71(d,J=7.8Hz,1H),5.36(p,J=7.9Hz,1H),5.06–4.98(m,2H),2.58(d,J=4.6Hz,3H),2.49–2.36(m,3H)。
实施例90

步骤a1-((4-溴异喹啉-3-基)甲基)-3-(1-甲基环丙基)-1H-苯并[d]咪唑-2(3H)-
酮(90a)
使化合物A-1(120mg,0.399mmol)和I-16c(75.4mg,0.399mmol)根据实施例79中所述的方法反应,得到标题化合物(98mg,60%)。MS(ES+)409.3[M+H]+。
步骤b)5-(3-((3-(1-甲基环丙基)-2-氧代-2,3-二氢-1H-苯并[d]咪唑-1-基)甲
基)异喹啉-4-基)甲基吡啶腈(90b)
使用实施例12步骤a中所述的方法使化合物90a(70mg,0.17mmol)与(6-氰基吡啶-3-基)硼酸(38mg,0.26mmol)反应,得到标题化合物(43mg,58%)。MS(ES+)433.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.43(d,J=0.8Hz,1H),8.64(dd,J=2.2,0.9Hz,1H),8.27–8.20(m,1H),8.22–8.15(m,2H),8.08(dd,J=7.9,2.2Hz,1H),8.05(s,1H),7.74(dt,J=6.9,3.4Hz,2H),7.29–7.18(m,2H),5.17(s,1H),5.14(s,1H),1.34(s,3H),0.94(s,3H)。
实施例91

步骤a)1-甲基环丙基1'-((4-溴异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,
3'-二氢吲哚]-1-羧酸酯(91a)
将TFA(397μl,5.19mmol)在室温下逐滴添加到化合物36a(130mg,0.263mmol)在DCM(2mL)中的溶液中。将反应物搅拌2h,接着浓缩并与甲苯共蒸发。在室温下将残余物和DIEA(119mg,0.920mmol)溶解于DCM(3mL)中。向其中添加2,5-二氧代吡咯烷-1-基(1-甲基环丙基)碳酸酯(56.0mg,0.263mmol)并且将反应物搅拌16h,接着真空浓缩。将粗料溶解于DCM中并且通过二氧化硅柱色谱进行纯化,使用DCM:MeOH洗脱,得到标题化合物(118mg,91%)。MS(ES+)492.3[M+H]+。
步骤b)1-甲基环丙基1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺
[氮杂环丁烷-3,3'-二氢吲哚]-1-羧酸酯(91b)
使用实施例12步骤a中所述的方法使化合物91a(75mg,0.15mmol)与(6-氰基吡啶-3-基)硼酸(34mg,0.28mmol)反应,得到标题化合物(34mg,44%)。MS(ES+)516.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.40(s,1H),8.72(s,1H),8.22(dt,J=7.6,2.3Hz,2H),8.13(d,J=7.9Hz,1H),7.79–7.70(m,2H),7.60(dd,J=7.4,1.2Hz,1H),7.29–7.22(m,1H),7.18(td,J=7.8,1.3Hz,1H),7.06(td,J=7.6,1.0Hz,1H),6.80(d,J=7.8Hz,1H),4.99(t,J=14.4Hz,2H),4.04–3.94(m,4H),1.50(s,3H),0.83(s,2H),0.64(q,J=2.9Hz,2H)。
实施例92

步骤a)2'-氧代-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-
二氢吲哚]-1-羧酸叔丁酯(92a)
根据实施例51步骤a中所述的过程制备标题化合物,但是使用3-吡啶硼酸并且使用碳酸钠作为碱。收率97%。MS(ES+)493.5[M+H]+。
步骤b)2'-氧代-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-
二氢吲哚]-1-羧酸甲酯(92b)
如实施例44中所述将化合物92a N-去保护并酰化,在酰化步骤中使用DCM作为溶剂,得到标题化合物(76%)。MS(ES+)451.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(d,J=0.8Hz,1H),8.71(dd,J=4.9,1.7Hz,1H),8.53(dd,J=2.3,0.9Hz,1H),8.22–8.16(m,1H),7.84(dt,J=7.8,2.0Hz,1H),7.80–7.66(m,2H),7.66–7.55(m,2H),7.29–7.23(m,1H),7.17(td,J=7.8,1.2Hz,1H),7.05(td,J=7.6,1.0Hz,1H),6.72(d,J=7.8Hz,1H),5.03–4.90(m,2H),4.07–3.99(m,3H),3.63(s,3H)。
实施例93

1'-((4-(4-氰基-3-氟苯基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-
二氢吲哚]-1-羧酸甲酯(93)
根据实施例51步骤a中所述的过程制备标题化合物,
但是使用(4-氰基-3-氟苯基)硼酸,接着进行如实施例44中所述的N-去保护和酰化。收率27%。MS(ES+)493.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.36(d,J=0.8Hz,1H),8.24–8.16(m,1H),8.07(dd,J=7.9,6.9Hz,1H),7.78–7.67(m,3H),7.63(dd,J=7.4,1.2Hz,1H),7.37(dd,J=7.9,1.4Hz,1H),7.34–7.27(m,1H),7.18(td,J=7.8,1.2Hz,1H),7.06(td,J=7.5,1.0Hz,1H),6.76(d,J=7.8Hz,1H),5.03(d,J=16.0Hz,1H),4.94(d,J=16.0Hz,1H),3.64(s,3H),3.32(s,5H)。
实施例94

1-(1-氨基环丙烷羰基)-1'-((4-(5-氟吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环
丁烷-3,3'-二氢吲哚]-2'-酮(94)
根据实施例12步骤a中所述的过程制备标题化合物,但是使用(5-氟吡啶-3-基)硼酸,接着进行如实施例60步骤a、b和c中所述的N-去保护、酰化和N-去保护。收率85%。MS(ES+)494.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.37(d,J=0.8Hz,1H),8.72(d,J=2.8Hz,1H),8.39(s,1H),8.23–8.17(m,1H),7.94(dt,J=9.4,2.1Hz,1H),7.79–7.69(m,2H),7.59(dd,J=7.4,1.3Hz,1H),7.31(dd,J=8.1,1.5Hz,1H),7.18(td,J=7.7,1.2Hz,1H),7.07(td,J=7.5,1.0Hz,1H),6.78(d,J=7.8Hz,1H),5.05(d,J=16.1Hz,1H),4.95(d,J=16.0Hz,1H),3.96(s,1H),1.24(s,0H),1.14(s,2H),0.76(s,2H)。
实施例95

步骤a)1'-((4-溴异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-
1-羧酸甲酯(95a)
根据实施例44步骤b中所述的过程将化合物36a(450mg,0.910mmol)N-去保护并酰化,得到标题化合物(405mg,98%)。MS(ES+)452.2&454.2[M+H]+。
步骤b)1'-((4-(2-氟吡啶-4-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,
3'-二氢吲哚]-1-羧酸甲酯(95b)
使化合物95a(67mg,0.148mmol)与2-氟吡啶-4-基)硼酸(31.3mg,0.222mmol)根据实施例12步骤a中所述的过程反应,
得到标题化合物(26.7mg,38%)。MS(ES+)469.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.38(s,1H),8.36(d,J=5.0Hz,1H),8.25–8.17(m,1H),7.74(tt,J=6.9,5.2Hz,2H),7.63(dd,J=7.4,1.2Hz,1H),7.36–7.25(m,3H),7.17(td,J=7.8,1.3Hz,1H),7.05(td,J=7.5,1.0Hz,1H),6.72(d,J=7.8Hz,1H),5.06(s,1H),4.96(s,1H),4.05(s,3H),3.98(s,1H),3.64(s,3H),2.07(s,1H),1.26–1.21(m,1H)。
实施例96

1'-((4-(2,6-二氟吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,
3'-二氢吲哚]-1-羧酸甲酯(96)
使化合物95a(72mg,0.159mmol)与(2,6-二氟吡啶-3-基)硼酸根据实施例12步骤a中所述的过程反应,不同的是,在18h反应时间之后,添加另外的催化剂(0.1当量)、硼酸(1.5当量)和碳酸钾(318μL)并且将反应物在90℃下再加热4h,以实现完全转化。收率:19.8mg,26%。MS(ES+)487.1[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.42(d,J=0.8Hz,1H),8.31–8.19(m,2H),7.75(tt,J=7.0,5.3Hz,2H),7.64(dd,J=7.4,1.2Hz,1H),7.42(dd,J=8.1,2.2Hz,1H),7.39–7.32(m,1H),7.18(td,J=7.8,1.3Hz,1H),7.06(td,J=7.6,1.0Hz,1H),6.86(d,J=7.8Hz,1H),5.04(d,J=15.7Hz,1H),4.93(d,J=15.7Hz,1H),4.08(s,3H),4.03(s,1H),3.64(s,3H)。
实施例97

2'-氧代-1'-((4-(吡啶-4-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢吲
哚]-1-羧酸甲酯(97)
使化合物95a(115mg,0.254mmol)与吡啶-4-基硼酸(46.9mg,0.381mmol)根据实施例12步骤a中所述的过程反应,得到标题化合物(37.5mg,33%)。MS(ES+)451.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.35(d,J=0.8Hz,1H),8.75–8.70(m,2H),8.23–8.15(m,1H),7.78–7.68(m,2H),7.63(dd,J=7.4,1.2Hz,1H),7.41–7.36(m,2H),7.27(dt,J=7.4,1.2Hz,1H),7.16(td,J=7.8,1.3Hz,1H),7.05(td,J=7.6,1.0Hz,1H),6.64(d,J=7.8Hz,1H),4.98(s,2H),4.06(s,2H),4.00(s,2H),3.63(s,3H),1.25(d,J=11.0Hz,1H)。
实施例98

步骤a)1'-((4-溴异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'
H)-酮(98a)
将TFA(5g)在0℃、氮气下经5min的时间段逐滴添加到11a(5g)在DCM(150mL)中的经搅拌溶液中。将混合物在室温下搅拌3h,接着浓缩,用水(50mL)稀释,用饱和碳酸氢盐溶液(50mL)中和并且用在DCM中的5%MeOH(2x200mL)萃取。将合并的有机层用水(50mL)洗涤、经硫酸钠干燥、过滤并减压浓缩。将粗化合物用乙醚(50mL)研磨、搅拌15min、过滤并干燥,得到为固体的标题化合物(3.4g,94%)。MS(ES+)423.4[M+H]+。
步骤b)1'-((4-溴异喹啉-3-基)甲基)-1-(甲基磺酰基)螺[哌啶-4,3'-吡咯并[2,
3-c]吡啶]-2'(1'H)-酮(98b)
在0℃下向98a(2g)在DCM(25mL)中的溶液中添加三乙胺(2mL),接着添加甲磺酰氯(0.5mL),将所得混合物在室温下搅拌3h,接着用水(50mL)稀释。用DCM(2x 50mL)萃取水层,将合并的有机层经硫酸钠干燥、过滤并浓缩。将获得的固体用乙醚(20mL)研磨并真空干燥,得到为固体的标题化合物(1.8g,64%)。MS(ES+)503.06[M+H]+。
步骤c)1-(甲基磺酰基)-1'-((4-(噻唑-5-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-
吡咯并[2,3-c]吡啶]-2'(1'H)-酮(98c)
在室温下向98b(300mg)和噻唑(76mg)在N,N-二甲基乙酰胺(10mL)中的经搅拌溶液中添加乙酸钾(176mg)和乙酸钯(7mg)。将反应混合物在130℃下搅拌36h,接着用冰水(20mL)稀释并且用乙酸乙酯(2X30mL)萃取。将合并的有机层经Na2SO4干燥,过滤并且减压浓缩。通过制备型C18HPLC纯化所得的粗料,将包含期望产物的级分汇集,浓缩并且再次通过制备型C18HPLC纯化,得到为米白色固体的标题化合物(28mg)。MS(ES+)506.33[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.45(s,1H),9.31(s,1H),8.29(d,J=4.8Hz,1H),8.20–8.11(m,3H),7.82(ddd,J=8.3,6.9,1.3Hz,1H),7.74(ddd,J=8.0,6.9,1.1Hz,1H),7.62(d,J=4.8Hz,1H),7.56–7.50(m,1H),5.04(s,2H),3.54–3.38(m,4H),2.97(s,3H),1.96(ddd,J=13.3,8.9,4.2Hz,2H),1.86(ddd,J=13.7,6.0,3.7Hz,2H)。
实施例99

2'-氧代-1'-((4-(4-氨磺酰苯基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢
吲哚]-1-羧酸甲酯(99)
使化合物95a(300mg,0.663mmol)与(4-氨磺酰苯基)硼酸(146mg,0.730mmol)根据针对中间体22步骤a所述的方法反应,不同的是通过微波辐射在100℃下持续1h实现加热。在二氧化硅上通过柱色谱纯化粗化合物,使用在EtOAc中的2%MeOH洗脱,接着使用XSELECT柱和流动相10mM碳酸氢铵:MeCN通过制备型HPLC进行纯化,得到为固体的标题化合物(135mg,38%)。MS(ES+)529.18[M+H]+。
实施例100

2'-氧代-1'-((4-(噻唑-5-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢吲
哚]-1-羧酸甲酯(100)
在室温下向化合物95a(240mg,0.531mol)和噻唑(67.8mg,0.796mmol)在N,N-二甲基乙酰胺(8mL)中的经搅拌溶液中添加乙酸钾(156mg,1.59mmol)和乙酸钯(5.95mg,0.027mmol)。将反应混合物在130℃下搅拌16h,接着通过硅藻土过滤。将滤液减压浓缩并且在硅胶上通过柱色谱纯化获得的粗料,使用85%EtOAc:石油醚洗脱。将包含期望产物的级分汇集并浓缩,并且在X-Bridge C18柱上使用在H2O:乙腈中的10mM碳酸氢铵通过制备型HPLC进一步纯化。使用甲醇作为溶剂通过SFC进一步纯化残余物,得到为固体的标题化合物(43mg,17%)。
MS(ES+)457.59[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.40(s,1H),9.36(s,1H),8.19(d,J=8.1Hz,1H),8.02(s,1H),7.79(t,J=7.6Hz,1H),7.73(t,J=7.5Hz,1H),7.66(d,J=7.3Hz,1H),7.45(d,J=8.4Hz,1H),7.17(t,J=7.7Hz,1H),7.06(t,J=7.5Hz,1H),6.72(d,J=7.8Hz,1H),5.02(s,2H),4.10(s,4H),3.64(s,3H),3.41–3.36(m,1H),2.45(s,0H)。
实施例101

1'-((4-(5-氰基吡啶-2-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-
二氢吲哚]-1-羧酸甲酯(101)
在氩气、室温下将6-(三丁基甲锡烷基)烟腈(469mg,1.19mmol)添加到
化合物95a(450mg,0.995mmol)和碳酸铯(972mg,2.98mmol)在1,4-二噁烷中的经搅拌且经脱气的溶液中。将混合物在室温下脱气10min,接着在100℃下加热16h。将反应混合物减压浓缩,用冰冷的水(25mL)缓慢稀释并用DCM(2x25mL)萃取。将有机层用水(25mL)、盐水(25mL)洗涤、经Na2SO4干燥并浓缩。在硅胶上通过柱色谱纯化获得的粗化合物,使用30%EtOAc/石油醚洗脱。收集纯级分,浓缩并且在Kromasil C18柱上通过制备型HPLC进一步纯化。将纯级分汇集并浓缩,添加水,使用饱和NaHCO3溶液将pH调节至7.5。将混合物搅拌15min,过滤并干燥,得到为固体的标题化合物(51mg,11%)。MS(ES+)476.18[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.38(s,1H),9.20(dd,J=2.1,0.9Hz,1H),8.51(dd,J=8.1,2.2Hz,1H),8.25–8.17(m,1H),7.86(dd,J=8.1,0.9Hz,1H),7.77–7.68(m,2H),7.62(dd,J=7.4,1.2Hz,1H),7.33–7.26(m,1H),7.16(td,J=7.8,1.2Hz,1H),7.04(td,J=7.5,1.0Hz,1H),6.80(d,J=7.8Hz,1H),4.98(s,2H),4.09(s,2H),4.04(d,J=8.0Hz,2H),3.64(s,3H),3.35(s,1H)。
实施例102

2'-氧代-1'-((4-(吡啶-2-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢吲
哚]-1-羧酸甲酯(102)
使化合物95a(220mg,0.486mmol)与2-(三甲基甲锡烷基)吡啶(141mg,0.584mmol)根据实施例101中所述的过程反应,不同的是使反应混合物受到微波辐射,得到标题化合物(30mg,13%)。MS(ES+)451.70[M+H]+。
1H NMR(500MHz,DMSO)δ9.32(s,1H),8.78(m,1H),8.17(m,1H),8.00(td,J=7.7,7.7,1.7Hz,1H),7.71(pd,J=6.8,6.8,6.8,6.8,1.3Hz,2H),7.62(d,J=7.2Hz,1H),7.60(d,J=7.7Hz,1H),7.52(m,1H),7.31(d,J=8.0Hz,1H),7.14(m,1H),7.03(t,J=7.5,7.5Hz,1H),6.75(d,J=7.8Hz,1H),4.94(s,2H),4.08(s,4H),3.64(s,3H)。
13C NMR(126MHz,DMSO)δ176.13,156.10,154.39,152.21,149.85,145.26,143.09,136.90,134.31,131.19,129.90,129.15,128.68,128.56,127.72,127.31,127.06,125.61,123.94,123.13,123.01,122.35,109.10,57.48,51.99,43.50,41.91。
实施例103

2'-氧代-1'-((4-(吡嗪-2-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢吲
哚]-1-羧酸甲酯(103)
使化合物95a(200mg,0.442mmol)与2-(三丁基甲锡烷基)吡嗪(196mg,0.531mmol)根据实施例101中所述的过程反应,不同的是使反应混合物受到微波辐射,得到标题化合物(25mg,12%)。MS(ES+)452.10[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.41(s,1H),8.85(s,1H),8.78(d,J=2.4Hz,1H),8.25–8.18(m,1H),7.78–7.69(m,2H),7.62(dd,J=7.4,1.2Hz,1H),7.29(dd,J=7.9,1.9Hz,1H),7.17(td,J=7.8,1.2Hz,1H),7.04(td,J=7.6,1.0Hz,1H),6.85(d,J=7.8Hz,1H),5.00(s,2H),4.06(d,J=16.0Hz,4H),3.64(s,3H),2.54(s,0H)。
实施例104

1'-((4-(6-氨基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-
二氢吲哚]-1-羧酸甲酯(104)
将95a(200mg)、5-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)吡啶-2-胺(194mg)和碳酸钾(183mg)在乙腈(16mL)和水(4mL)中的经搅拌溶液用氩气净化5分钟,接着添加双(三苯基膦)二氯化钯(II)(62mg)。将反应混合物再次用氩气净化10分钟,接着在密封管中在100℃下搅拌7h。通过硅藻土过滤反应混合物,将滤液用水(15mL)稀释并用EtOAc(2x15mL)萃取,将合并的有机层经硫酸钠干燥、过滤并减压浓缩。在二氧化硅上通过柱色谱纯化获得的粗料,用在DCM中的5-6%MeOH洗脱。将包含期望产物的级分减压浓缩并且将所得固体用研钵和研杵碾磨30分钟,得到为固体的标题化合物(73mg,35%)。MS(ES+)466.13[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.22(s,1H),8.12(d,J=8.0Hz,1H),7.86(d,J=2.4Hz,1H),7.76–7.61(m,3H),7.48(d,J=8.4Hz,1H),7.37(dd,J=8.4,2.4Hz,1H),7.18–7.11(m,1H),7.04(t,J=7.5Hz,1H),6.60(d,J=8.1Hz,2H),6.19(s,2H),4.99(s,2H),4.11(s,4H),3.64(s,3H),3.40–3.34(m,1H)。
实施例105

1'-((4-(4-(甲基磺酰胺基)苯基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-
3,3'-二氢吲哚]-1-羧酸甲酯(105)
将化合物95a(250mg,0.553mmol)、(4-(甲基磺酰胺基)苯基)-硼酸(155mg,0.719mmol)和碳酸钠(146mg,1.38mmol)在1,4-二噁烷(10mL)和水(1mL)中的经搅拌溶液用氩气净化5分钟,接着添加四(三苯基膦)钯(63.9mg,0.055mmol)并且将混合物再次用氩气净化10分钟,在微波辐射下在110℃下搅拌1小时。通过硅藻土过滤反应混合物,将滤液用水(15mL)稀释并用EtOAc(3x15mL)萃取。将合并的有机层经硫酸钠干燥、过滤并且减压浓缩。在硅胶上通过柱色谱纯化所得的粗料,使用在DCM中的5%MeOH洗脱,得到为固体的标题化合物(192mg,63%)。MS(ES+)543.20[M+H]+。
1H NMR(500MHz,DMSO-d6)δ10.00(s,1H),9.27(s,1H),8.14(dd,J=7.8,1.6Hz,1H),7.70(dddd,J=17.6,8.0,6.9,1.4Hz,2H),7.63(dd,J=7.4,1.2Hz,1H),7.40–7.30(m,5H),7.15(td,J=7.8,1.2Hz,1H),7.04(td,J=7.5,0.9Hz,1H),6.64(d,J=7.8Hz,1H),4.95(s,2H),4.08(d,J=14.4Hz,4H),3.64(s,3H),3.39–3.32(m,1H),3.11(s,3H)。
实施例106

步骤a)(1-(1'-((4-溴异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-4,3'-
吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)氨基甲酸叔丁酯(106a)
向化合物11a在DCM(2mL)中的经搅拌溶液中添加TFA(1mL)。将溶液在室温下搅拌90min,接着减压浓缩并且与甲苯共蒸发。将残余物溶解于DMF(2mL)中,添加HATU和DIEA,接着添加Boc保护的环丙基氨基酸。在室温下搅拌16h后,将反应混合物真空浓缩,在硅胶上通过柱色谱纯化残余物,用MeOH-DCM洗脱,得到标题化合物(506mg)。MS(ES+)606.3&608.3[M+H]+。
步骤b)(1-(1'-((4-(5-氟吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢
螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)氨基甲酸叔丁酯(106b)
使化合物106a(173mg,0.285mmol)与硼酸(60mg,0.428mmol)如实施例12步骤a中所述的那样反应,得到标题化合物(158mg,89%)。MS(ES+)623.4[M+H]+。
步骤c)1-(1-氨基环丙烷羰基)-1'-((4-(5-氟吡啶-3-基)异喹啉-3-基)甲基)螺
[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(106c)
向化合物106b(158mg,0.254mmol)在DCM(2mL)中的经搅拌溶液中逐滴添加TFA(571m,5.01mmol)。将溶液在室温下搅拌2h,接着减压浓缩并且与甲苯共蒸发。在Gemini-NXPrep C18柱上通过制备型LCMS纯化残余物(流动相在水中的0.1%NH4OH-在乙腈中的0.1%NH4OH(pH 10)),将纯级分汇集并冷冻干燥,得到标题化合物(72mg,54%)。MS(ES+)523.22[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.37(d,J=0.8Hz,1H),8.77(d,J=2.8Hz,1H),8.54(d,J=3.4Hz,0H),8.54(s,1H),8.29–8.17(m,3H),8.06(ddd,J=9.5,2.8,1.7Hz,1H),7.76(dddd,J=18.2,8.0,6.9,1.4Hz,2H),7.57(dd,J=4.8,0.8Hz,1H),7.37(dd,J=8.3,1.3Hz,1H),5.02(d,J=12.0Hz,2H),2.30(s,2H),1.77(s,2H),1.70(s,2H),0.89(q,J=4.3Hz,2H),0.67(t,J=3.3Hz,2H)。
实施例107

1-(1-氨基环丙烷羰基)-1'-((4-(4-(三氟甲基)苯基)异喹啉-3-基)甲基)螺[哌
啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(107)
根据实施例106中所述的过程制备标题化合物,但是使用硼酸(6-(三氟甲基)吡啶-3-基)硼酸。收率50%,MS(ES+)573.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.42–9.37(m,1H),8.88(d,J=2.1Hz,1H),8.33–8.24(m,2H),8.26–8.19(m,1H),8.21(s,1H),8.15(dd,J=8.0,0.9Hz,1H),7.81–7.71(m,2H),7.60–7.54(m,1H),7.35–7.29(m,1H),5.04(s,1H),4.99(s,1H),3.92(s,2H),3.32(s,3H),2.28(s,2H),1.75(s,2H),1.67(s,2H),0.88(q,J=4.2Hz,2H),0.66(t,J=3.0Hz,2H)。
实施例108

步骤a)(1-(1'-((4-(6-氨基甲酰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',
2'-二氢螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)(甲基)氨基甲酸叔丁酯
(108a-1)
&
5-(3-((1-(1-((叔丁氧羰基)(甲基)氨基)环丙烷羰基)-2'-氧代螺[哌啶-4,3'-
吡咯并[2,3-c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶甲酸甲酯(108a-2)
使化合物13a与1-((叔丁氧羰基)(甲基)氨基)环丙烷羧酸根据实施例50步骤a中所述的过程反应,得到两种标题化合物
108a-1,152mg,35%,MS(ES+)662.5[M+H]+。
108a-2,160mg,36%,MS(ES+)677.6[M+H]+。
步骤b)5-(3-((1-(1-(甲基氨基)环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并[2,
3-c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶甲酰胺(108b)
如实施例50步骤b中所述的那样将化合物108a-1去保护,得到标题化合物(85mg,69.07%),MS(ES+)562.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.37(d,J=0.8Hz,1H),8.74–8.69(m,1H),8.21(dd,J=6.7,1.7Hz,1H),8.15(dd,J=7.9,2.2Hz,1H),7.80–7.70(m,4H),7.57(dd,J=4.8,0.9Hz,1H),7.37–7.31(m,1H),5.01(q,J=15.9Hz,3H),2.26(s,4H),1.73(d,J=10.3Hz,1H),1.68(s,3H),0.87(s,2H)。
实施例109

N-环丙基-5-(3-((1-(1-(甲基氨基)环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并
[2,3-c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶甲酰胺(109)
将化合物108a-2(150mg,0.222mmol)和环丙胺(253mg,4.43mmol)在THF:EtOH 1:1(5mL)中的溶液在密封管中在100℃下搅拌2天。将混合物减压浓缩并真空干燥。将残余物溶解于DCM(10mL)中,添加TFA(2.01g,17.7mmol)并且将混合物在室温下搅拌90min,接着减压浓缩并与甲苯共蒸发两次。将残余物溶解于THF:MeOH 1:1(2mL)中,添加在水(1mL)中的1MLiOH并且将混合物在室温下搅拌1h,接着在乙酸和减压条件下浓缩。通过HPLC分离产物(Gemini NX 100x30,梯度14分钟,在水中的15-45%乙腈,具有10mM乙酸铵缓冲液)。将适当的级分汇集并冷冻干燥,得到标题化合物(35mg,26%),MS(ES+)602.5[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.37(d,J=0.8Hz,1H),8.91(d,J=5.0Hz,1H),8.68(d,J=1.9Hz,1H),8.28–8.17(m,3H),8.16(s,1H),8.16(dd,J=7.9,2.2Hz,1H),7.80–7.70(m,2H),7.57(dd,J=4.8,0.8Hz,1H),7.37–7.30(m,1H),5.04(d,J=15.9Hz,1H),4.96(d,J=15.9Hz,1H),3.32(s,8H),3.02–2.93(m,1H),2.26(d,J=3.4Hz,3H),2.16(s,1H),1.70(s,1H),1.68(s,2H),1.65(s,1H),1.24(s,1H),0.87(s,2H),0.74(dt,J=3.0,1.6Hz,1H),0.72(s,2H),0.67(s,2H)。
实施例110

步骤a)1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌
啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(110a)
使化合物11a(147mg,0.281mmol)与(6-氟吡啶-3-基)硼酸(59.4mg,0.421mmol)根据实施例12步骤a中所述的过程偶联,得到标题化合物(150mg,99%),MS(ES+)540.3[M+H]+。
步骤b)1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)-1-(甲基磺酰基)螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(110b)
将在二噁烷(2mL)中的4M HCl添加到化合物110a(108mg,0.200mmol)的溶液中。将溶液在室温下搅拌30min,接着减压浓缩并且与甲苯共蒸发。将残余物溶解于MeCN(4mL)中并且在0℃下搅拌10-15min,接着缓慢添加甲磺酰氯在MeCN(1mL)中的混合物。通过LCMS监测反应的进展并且当确认完全时,将反应混合物浓缩并且使用95%水:5%乙腈(10mM,在乙酸铵中)和10%水:90%乙腈(10mM,在乙酸铵中)的梯度在Gemini C18柱上通过制备型HPLC纯化粗产物。将适当的级分汇集,冷冻干燥并且在二氧化硅上通过柱色谱进一步纯化,使用DCM MeOH洗脱,得到标题化合物(103mg,99%)。MS(ES+)518.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.33(d,J=0.8Hz,1H),8.38(d,J=2.4Hz,1H),8.29(d,J=4.7Hz,1H),8.23(s,1H),8.22–8.13(m,2H),7.75(dddd,J=20.2,8.0,6.9,1.3Hz,2H),7.60(d,J=4.7Hz,1H),7.46(dd,J=8.3,2.5Hz,1H),7.38(dd,J=8.4,1.2Hz,1H),4.99(d,J=6.4Hz,2H),3.52–3.32(m,2H),3.42(s,3H),2.97(s,3H),2.55(s,1H),1.93(ddd,J=13.2,8.8,4.3Hz,2H),1.81(dq,J=14.0,5.1Hz,2H)。
实施例111

步骤a)(1-(1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢
螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)氨基甲酸叔丁酯(111a)
使用在DCM中的TFA如实施例60步骤a中所述的那样对化合物110a(750mg,1.39mmol)进行N-去保护,接着使其与1-(叔丁氧羰基氨基)环丙烷羧酸(382mg,1.90mmol)根据针对中间体32所述的过程偶联,得到标题化合物(500mg,59%)。
步骤b)(1-(1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢
螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)(甲基)氨基甲酸叔丁酯(111b)
将NaH(96mg,2.41mmol)在0℃下添加到化合物111a(500mg,0.803mmol)在DMF(10mL)中的经搅拌溶液中,将反应混合物搅拌10min,接着在氮气下添加碘甲烷(0.333mL,3.21mmol)并且在室温下继续搅拌2h。用水(40mL)淬灭反应并且用乙酸乙酯(2x60mL)萃取混合物,将合并的有机层用盐水洗涤,经硫酸钠干燥、过滤并减压浓缩。在中性氧化铝上通过柱色谱纯化粗料,使用在DCM中的MeOH的梯度洗脱,得到标题化合物(350mg,60%)。MS(ES+)637.39[M+H]+。
步骤c)1'-((4-(6-氟吡啶-3-基)异喹啉-3-基)甲基)-1-(1-(甲基氨基)环丙烷羰
基)螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(111c)
如实施例60步骤a中所述的那样使用在DCM(10mL)中的TFA对化合物111b(330mg,0.518mmol)进行N-去保护。使用X-Bridge C18柱并且使用在MeCN中的10mM NH4HCO3作为流动相通过制备型HPLC纯化所得的粗化合物。将纯级分汇集并冻干,得到为固体的标题化合物(100mg,35%)。MS(ES+)537.24[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(d,J=0.8Hz,1H),8.37(d,J=2.4Hz,1H),8.26(d,J=4.7Hz,1H),8.26–8.13(m,2H),7.75(dddd,J=19.3,8.0,6.9,1.3Hz,1H),7.58(dd,J=4.8,0.8Hz,1H),7.45(dd,J=8.3,2.6Hz,1H),7.40–7.34(m,1H),5.00(d,J=6.2Hz,1H),3.39–3.30(m,1H),3.33(s,12H),2.26(s,2H),2.18(s,1H),1.72(s,3H),0.88(s,1H),0.68(s,1H)。
实施例112

步骤a)(E)-甲基3-(3-((1-环丙基-2-氧代-1H-咪唑并[4,5-c]吡啶-3(2H)-基)甲
基)异喹啉-4-基)丙烯酸酯(112a)
将丙烯酸甲酯(26mg,0.304mmol)和Et3N(0.142mL,1.01mmol)添加到化合物1a(100mg,0.253mmol)在DMF(5mL)中的溶液中。将溶液在密封管中用氩气脱气10分钟,接着添加乙酸钯(6mg,0.025mmol)和三(邻甲苯基)膦(39mg,0.127mmol)。将混合物脱气10min,接着在100℃下加热24h。将反应混合物冷却至室温并用EtOAc稀释。将有机层用水(10mL)、盐水(10mL)洗涤、经无水Na2SO4干燥、过滤并浓缩。通过柱色谱纯化粗料(洗脱液:10%MeOH-CHCl3),得到标题化合物(80mg,7.4%)。MS(ES+)401.29[M+H]+。
步骤b)3-(3-((1-环丙基-2-氧代-1H-咪唑并[4,5-c]吡啶-3(2H)-基)甲基)异喹
啉-4-基)-4-硝基丁酸甲酯(112b)
将化合物112a(500mg,1.25mmol)在硝基甲烷(5mL)中的溶液在-20℃下逐滴添加到1,8-二氮杂双环十一碳-7-烯(0.285g,1.87mmol)在硝基甲烷(5mL)中的溶液中。将混合物在-20℃下搅拌4h,接着使其达到室温并用DCM稀释。将有机层用水(10mL)、盐水(10mL)洗涤、经无水Na2SO4干燥、过滤并减压浓缩。通过制备型HPLC纯化所得的粗料,得到标题化合物(180mg,31%)。MS(ES+)462.29[M+H]+。
步骤c)1-环丙基-3-((4-(5-氧代吡咯烷-3-基)异喹啉-3-基)甲基)-1H-咪唑并
[4,5-c]吡啶-2(3H)-酮(112c)
将铁粉(85mg)添加到化合物112b(140mg,0.303mmol)在乙酸(5mL)中的溶液中。将混合物在80℃下加热8h,接着通过硅藻土过滤并且用DCM洗涤滤饼。将有机层减压浓缩,将所得粗料溶解于10%MeOH-CHCl3中并且用水(5mL)、盐水(5mL)洗涤,经无水Na2SO4干燥,过滤并减压浓缩。将粗料与另一批次(50mg)混合并且通过制备型HPLC C18进行纯化,使用在H2O:MeCN中的10mM ABC洗脱,得到标题化合物(40mg)。
1H NMR(500MHz,DMSO-d6)δ9.15(s,1H),8.33(s,1H),8.20(d,J=5.2Hz,1H),8.14(d,J=8.1Hz,1H),8.06(s,1H),7.94–7.80(m,2H),7.70(t,J=7.4Hz,1H),7.25(d,J=5.2Hz,1H),5.47(s,1H),4.75(qd,J=10.2,7.5Hz,1H),3.73(t,J=10.1Hz,1H),3.60(dd,J=10.0,7.5Hz,1H),2.99(tt,J=7.0,3.6Hz,1H),2.70–2.55(m,2H),1.06(dt,J=7.1,3.5Hz,2H),0.89(p,J=4.8Hz,2H)。
实施例113

步骤a)1'-((4-(6-氨基甲酰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂
环丁烷-3,3'-二氢吲哚]-1-羧酸叔丁酯(113a)
向化合物44a(400mg,0.618mmol)在EtOH中的溶液中添加1M NaOH溶液(3.1mL)。将混合物在室温下搅拌72h,接着在60℃下搅拌2.5h,冷却至室温,用乙酸酸化并浓缩到二氧化硅上。通过硅胶色谱纯化产物,用DCM和2至10%MeOH洗脱,得到标题化合物(142mg,43%)。MS(ES+)536.5[M+H]+。
步骤b)5-(3-((1-(1-(二甲基氨基)环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-
二氢吲哚]-1'-基)甲基)异喹啉-4-基)吡啶甲酰胺(113b)
使化合物113a(70mg,0.131mmol)如实施例62中所述的那样反应,得到标题化合物(71mg,42%),MS(ES+)547.5[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.40(d,J=0.8Hz,1H),8.22(dt,J=7.4,2.7Hz,2H),8.16(d,J=7.9Hz,1H),7.81–7.69(m,2H),7.57(dd,J=7.4,1.2Hz,1H),7.27–7.21(m,1H),7.16(td,J=7.7,1.3Hz,1H),7.06(td,J=7.5,1.0Hz,1H),6.68(d,J=7.8Hz,1H),4.36(s,2H),2.20(s,6H),0.92(s,4H)。
实施例114

步骤a)(1-(1'-((4-溴异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲
哚]-1-基羰基)环丙基)氨基甲酸叔丁酯(114a)
使A-1(960mg,2.91mmol)与I-32(800mg,2.24mmol)根据实施例79中所述的过程反应,得到标题化合物(1.29g,51%)。MS(ES+)579.23[M+H]+。
步骤b)(1-(1'-((4-(4-(甲基磺酰胺基)苯基)异喹啉-3-基)甲基)-2'-氧代螺[氮
杂环丁烷-3,3'-二氢吲哚]-1-基羰基)环丙基)氨基甲酸叔丁酯(114b)
使化合物114a(300mg,0.52mmol)与(4-(甲基磺酰胺基)苯基)硼酸(145mg,0.675mmol)根据实施例105中所述的过程反应,得到标题化合物(280mg,77%)。MS(ES+)668.53[M+H]+。
步骤c)N-(4-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲
哚]-1'-基)甲基)异喹啉-4-基)苯基)甲磺酰胺(114c)
将TFA(0.64mL)在0℃下添加到化合物114b(280mg)在DCM(10mL)中的经搅拌悬浮液中。将所得反应混合物在室温下搅拌8h,接着减压浓缩。将获得的残余物用饱和碳酸氢钠溶液碱化并用EtOAc(3X20mL)萃取。将有机层经Na2SO4干燥、过滤并减压浓缩,并且将所得的残余物用水:乙腈(1:1,30mL)研磨,搅拌1h,过滤并干燥,得到为固体的标题化合物(115mg,46%)。MS(ES+)568.23[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.26(s,1H),8.14(d,J=8.0Hz,1H),7.85(s,1H),7.70(dt,J=19.3,7.2Hz,2H),7.59(d,J=7.3Hz,1H),7.37(q,J=8.4Hz,5H),7.15(t,J=7.7Hz,1H),7.05(t,J=7.5Hz,1H),6.64(d,J=7.8Hz,1H),4.96(s,2H),4.75(s,2H),4.07–4.00(m,1H),3.99(s,2H),3.40(s,0H),3.12(s,3H),1.12(s,1H),1.07(s,1H),0.72(s,2H)。
实施例115

步骤a)1'-((4-(6-氨基甲酰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂
环丁烷-3,3'-二氢吲哚]-1-羧酸叔丁酯(115a)
将1M氢氧化钠(水溶液,3.1ml)添加到化合物44a(400mg,0.618mmol)的EtOH溶液中。将混合物在室温下搅拌整个周末,接着在60℃下温热2.5小时。将混合物冷却,用HOAc酸化并且浓缩到二氧化硅上。通过硅胶柱色谱纯化产物,用DCM和2至10%MeOH洗脱,得到化合物115a(142mg,43%),MS(ES+)536.5[M+H]+,以及化合物115d(66mg,20%),MS(ES+)537.4[M+H]+。
步骤b)(1-(1'-((4-(6-氨基甲酰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺
[氮杂环丁烷-3,3'-二氢吲哚]-1-基羰基)环丙基)(甲基)氨基甲酸叔丁酯(115b)
N-去保护:将TFA(1.19g,10.5mmol)添加到化合物115a(70mg,0.131mmol)在DCM(8mL)中的溶液中。将溶液在室温下搅拌90min,接着真空浓缩并且与甲苯共蒸发两次。
酰化:将残余物溶解于无水DMF(10mL)中,添加1-((叔丁氧羰基)(甲基)-氨基)环丙烷羧酸(33.8mg,0.157mmol)和DIEA(169mg,1.31mmol)并且将溶液在冰浴中冷却并添加HATU(64.6mg,0.170mmol)。将混合物在冰浴中搅拌1h,接着添加水(40mL)和饱和碳酸氢钠溶液(40ml)并且用EtOAc萃取混合物三次。将有机相用盐水洗涤、经硫酸钠干燥并且减压浓缩。通过硅胶色谱纯化产物,用DCM和2至5%MeOH洗脱,得到标题化合物(56mg,68%)。
步骤c)5-(3-((1-(1-(甲基氨基)环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二
氢吲哚]-1'-基)甲基)异喹啉-4-基)吡啶甲酰胺(115c)
将TFA(991mg,8.69mmol)添加到化合物115b(55mg,0.087mmol)在DCM(8mL)中的溶液中。将溶液在室温下搅拌90min,接着真空浓缩并且与甲苯共蒸发两次。将残余物溶解于DCM(5mL)中,添加TEA(0.5mL)并且将溶液浓缩。通过反相HPLC纯化残余物。将适当的级分汇集并且从乙腈水中冷冻干燥,得到标题化合物(28mg,60%)。MS(ES+)533.4[M+H]+。
1H NMR(500MHz,DMSO)δ9.38(s,1H),8.56(m,1H),8.21(m,3H),8.01(m,1H),7.73(m,3H),7.58(d,J=6.9Hz,1H),7.26(m,1H),7.17(m,1H),7.06(m,1H),6.71(d,J=7.8Hz,1H),4.98(s,2H),4.47(s,2H),3.96(m,2H),2.27(s,3H),1.96(s,1H),1.03(d,J=22.6Hz,2H),0.78(d,J=6.1Hz,2H)。
13C NMR(126MHz,DMSO)δ176.11,173.33,165.61,152.57,149.80,148.58,145.83,142.86,138.77,134.78,133.66,131.57,129.81,128.44,127.85,127.54,127.00,126.75,123.79,122.69,122.51,121.72,109.17,60.96,56.33,44.13,42.80,42.33,33.26,15.04,14.31。
实施例116

步骤a)1'-((4-溴异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-
1-羧基酰胺(116a)
使A-1(1.14g,3.45mmol)与I-12(500mg,2.30mmol)根据实施例79中所述的过程反应,得到标题化合物(800mg,78%)。MS(ES+)439.05[M+H]+。
步骤b)2'-氧代-1'-((4-(4-氨磺酰苯基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,
3'-二氢吲哚]-1-羧基酰胺(116b)
使用针对中间体22步骤a所述的方法使化合物116a(250mg,0.752mmol)与(4-氨磺酰苯基)硼酸(149mg,0.743mmol)反应,得到标题化合物(100mg,34%)。MS(ES+)514.20[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.30(s,1H),8.24(s,2H),8.17(d,J=7.8Hz,1H),8.01(d,J=7.7Hz,2H),7.72(p,J=7.0Hz,2H),7.64(d,J=7.8Hz,2H),7.55(d,J=6.6Hz,3H),7.27(d,J=8.2Hz,1H),7.17(t,J=7.6Hz,1H),7.09–7.02(m,1H),6.68(d,J=7.8Hz,1H),6.10(s,2H),4.92(s,2H),3.99(d,J=7.7Hz,2H),3.90(d,J=8.0Hz,2H)。
实施例117

1'-((4-(4-(甲基磺酰胺基)苯基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-
3,3'-二氢吲哚]-1-羧基酰胺(117)
使用实施例105中所述的方法使化合物116a(250mg,0.572mmol)与(4-(甲基磺酰胺基)苯基)硼酸(160mg,0.743mmol)反应,得到标题化合物(58mg,19%)。MS(ES+)528.22[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.97(s,1H),9.28–9.24(m,1H),8.17–8.11(m,1H),7.70(dddd,J=18.8,8.0,6.8,1.4Hz,2H),7.54(dd,J=7.4,1.2Hz,1H),7.42–7.32(m,5H),7.14(td,J=7.7,1.3Hz,1H),7.04(td,J=7.5,1.0Hz,1H),6.63(d,J=7.8Hz,1H),6.10(s,2H),5.76(s,0H),4.95(s,2H),4.00(d,J=7.9Hz,2H),3.91(d,J=7.9Hz,2H),3.37–3.33(m,1H),3.13(s,3H),2.54(s,0H)。
实施例118

1-(2-羟基-2-甲基丙酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环
丁烷-3,3'-二氢吲哚]-2'-酮(118)
根据实施例51步骤a中所述的过程制备标题化合物,但是使用3-吡啶硼酸,接着进行如实施例115步骤b中所述的N-去保护和使用2-羟基异丁酸进行的酰化。收率35%。MS(ES+)479.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(s,1H),8.74–8.68(m,1H),8.54(d,J=2.3Hz,1H),8.19(dd,J=7.5,1.9Hz,1H),7.86(s,1H),7.86(dt,J=14.6,1.9Hz,1H),7.78–7.68(m,2H),7.62–7.52(m,2H),7.27(dd,J=8.1,1.6Hz,1H),7.17(td,J=7.8,1.3Hz,1H),7.10–7.03(m,1H),6.71(dd,J=7.9,5.0Hz,1H),5.25(s,1H),4.97(qd,J=16.0,4.7Hz,2H),4.55(dd,J=9.7,4.9Hz,1H),4.48(dd,J=9.7,4.4Hz,1H),4.03–3.93(m,2H),1.34–1.27(m,6H)。
13C NMR(126MHz,DMSO-d6)δ200.48,176.20,175.04,152.29,149.70,149.15,145.77,143.01,137.36,137.34,134.91,131.42,131.02,129.81,128.46,127.79,127.42,127.31,127.03,123.81,123.69,122.67,122.41,109.11,74.69,73.20,61.52,56.35,43.93,42.56,27.64,27.37。
实施例119

步骤a)1'-((4-(6-(二甲基氨基甲酰基)吡啶-3-基)异喹啉-3-基)甲基)-2'-氧
代-1',2'-二氢螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(119a)
将2M碳酸钠水溶液(1.46ml,2.82mmol)和双(三苯基膦)Pd(II)Cl2(114mg,0.16mmol)添加到微波小瓶中的N,N-二甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)吡啶甲酰胺(202mg,0.73mmol)和化合物11a(390mg,0.74mmol)在CH3CN(7ml)中的混合物中。将小瓶密封,抽真空并且填充N2(x 3),接着加热至80℃,持续16h。过滤出固体并浓缩滤液。在二氧化硅上通过色谱纯化产物,使用在DCM中的1-5%MeOH洗脱,得到被例如三苯基膦氧化物污染的标题化合物(501mg,116%)。化合物无需进一步纯化即用于下一步骤。
步骤b)5-(3-((1-(1-(二甲基氨基)环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并
[2,3-c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)-N,N-二甲基吡啶甲酰胺(119b)
对来自上文的化合物119a进行N-去保护并且使用1-(二甲基氨基)环丙烷羧酸如实施例115步骤b中所述的那样将所得的胺进行酰化。两步收率46%。MS(ES+)604.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.36(d,J=0.8Hz,1H),8.69(dd,J=2.1,0.9Hz,1H),8.28–8.23(m,2H),8.23–8.17(m,1H),8.05(dd,J=7.9,2.2Hz,1H),7.81–7.70(m,3H),7.60(dd,J=4.8,0.8Hz,1H),7.37(dd,J=8.4,1.3Hz,1H),5.04(s,1H),5.01(s,1H),3.32(s,4H),3.09(d,J=11.2Hz,6H),2.21(s,6H),1.70(pd,J=14.8,12.0,5.8Hz,4H),0.88(s,2H),0.77(s,2H)。
实施例120

5-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1'
(2'H)-基)甲基)异喹啉-4-基)-N,N-二甲基吡啶甲酰胺(120)
对来自上文的化合物119a进行N-去保护并且使用1-((叔丁氧羰基)氨基)环丙烷羧酸如实施例115步骤b中所述的那样将所得的胺进行酰化。使用在DCM中的TFA对所得的Boc保护的化合物进行N-去保护,得到标题化合物(37%)。MS(ES+)576.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.36(d,J=0.8Hz,1H),8.70(dd,J=2.2,0.9Hz,1H),8.29–8.21(m,2H),8.20(dd,J=8.2,1.3Hz,1H),8.07(dd,J=7.9,2.2Hz,1H),7.81–7.70(m,3H),7.58(dd,J=4.8,0.8Hz,1H),7.37(dd,J=8.3,1.2Hz,1H),5.02(d,J=9.6Hz,2H),3.33(s,4H),3.09(d,J=8.4Hz,6H),2.30(s,2H),1.76(s,2H),1.71(s,3H),0.88(q,J=4.3Hz,2H),0.67(t,J=3.2Hz,2H)。
实施例121

步骤a)1'-((4-(6-(二甲基氨基甲酰基)吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代
螺[氮杂环丁烷-3,3'-二氢吲哚]-1-羧酸叔丁酯(121a)
将DIEA(100μl,0.57mmol)添加到化合物115d(65mg,0.12mmol)、二甲胺盐酸盐(23mg,0.28mmol)和HATU(52mg,0.14mmol)在DMF(3ml)中的混合物中。将反应混合物在环境温度下搅拌1.5h,接着添加另外的HATU(50mg,0.13mmol)和DIEA(100μl,0.57mmol)并且反应继续1h。将反应混合物用EtOAc稀释,用NaHCO3水溶液、柠檬酸水溶液和盐水洗涤。将有机相经MgSO4干燥并浓缩,并且在二氧化硅上通过柱色谱纯化产物,使用在DCM中的0-10%MeOH洗脱,得到不纯的标题化合物(78mg,114%),MS(ES+)564.4[M+H]+。
步骤b)5-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲
哚]-1'-基)甲基)异喹啉-4-基)-N,N-二甲基吡啶甲酰胺(121b)
对化合物121a(78mg,0.138mmol)进行N-去保护并且使用1-((叔丁氧羰基)氨基)环丙烷羧酸如实施例115步骤b中所述的那样将所得的胺进行酰化。使用在DCM中的TFA对所得的Boc保护的化合物进行N-去保护,得到标题化合物(26mg,34%)。MS(ES+)547.3[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.38(d,J=0.8Hz,1H),8.64(s,0H),8.54(s,1H),8.24–8.18(m,1H),7.96(s,1H),7.89(s,1H),7.81–7.68(m,3H),7.59(dd,J=7.4,1.2Hz,1H),7.35–7.28(m,1H),7.17(td,J=7.8,1.3Hz,1H),7.06(td,J=7.6,1.0Hz,1H),6.77(s,1H),5.04(d,J=8.1Hz,0H),4.68(s,2H),4.04(s,1H),3.96(s,2H),3.07(d,J=4.7Hz,6H),2.25(s,2H),1.13(s,1H),1.06(s,1H),0.72(s,2H)。
实施例122

N,N-二甲基-5-(3-((1-(1-(甲基氨基)环丙烷羰基)-2'-氧代螺[哌啶-4,3'-吡咯
并[2,3-c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶甲酰胺(122)
对化合物119a进行N-去保护并且使用1-((叔丁氧羰基)氨基)环丙烷羧酸如实施例115步骤b中所述的那样将所得的胺进行酰化。使用在DCM中的TFA对所得的Boc保护的化合物进行N-去保护,得到标题化合物(11%)。MS(ES+)590.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.38–9.34(m,1H),8.70(dd,J=2.2,0.9Hz,1H),8.26(t,J=2.3Hz,2H),8.23–8.17(m,1H),8.06(dd,J=7.9,2.2Hz,1H),7.81–7.70(m,4H),7.61–7.56(m,1H),7.37(dd,J=8.4,1.2Hz,1H),5.09–4.96(m,2H),3.33(s,4H),3.09(d,J=9.4Hz,7H),2.26(d,J=4.9Hz,3H),2.18(d,J=6.0Hz,1H),0.87(s,2H),0.68(s,2H)。
实施例123

步骤a)(1-(1'-((4-溴异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲
哚]-1-基羰基)环丙基)(甲基)氨基甲酸叔丁酯(123a)
使化合物I-33(350mg,0.94mmol)与化合物A-1(369mg,1.0mmol)根据实施例79中所述的方法反应,得到标题化合物(300mg,46mmol)。MS(ES+)593.2[M+H]+。
步骤b)5-(3-((1-(1-(甲基氨基)环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二
氢吲哚]-1'-基)甲基)异喹啉-4-基)吡啶-2-磺酰胺(123b)
使化合物123a(250mg,0.423mmol)与(5-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)吡啶-2-基)磺酰基氨基甲酸叔丁酯(216mg,0.634mmol)根据实施例51步骤a中所述的方法反应。接着通过在75℃下用在DCM中的TFA处理16h来将所得化合物N-去保护,得到标题化合物(24%)。MS(ES+)569.34[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.36(s,1H),8.78(s,1H),8.24–8.18(m,2H),8.13(dd,J=7.9,0.8Hz,1H),7.76(dddd,J=20.3,8.0,6.9,1.3Hz,2H),7.60(d,J=1.2Hz,0H),7.28–7.17(m,2H),7.08(td,J=7.5,0.9Hz,1H),6.80(d,J=7.8Hz,1H),4.98(s,1H),4.94(s,1H),4.61(s,1H),4.53(s,1H),2.55(s,1H),2.28(s,3H),1.07(s,1H),1.03(s,1H),0.79(s,2H)。
实施例124

5-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1'-
基)甲基)异喹啉-4-基)吡啶-2-磺酰胺(124)
使化合物114a(350mg,0.606mmol)如实施例123步骤b中所述的那样反应,得到标题化合物(23%)。MS(ES+)555.28[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.37(s,1H),8.79(s,1H),8.71(s,0H),8.24–8.18(m,1H),8.12(d,J=7.9Hz,1H),7.76(dddd,J=18.6,8.0,6.9,1.3Hz,2H),7.63(s,2H),7.27–7.16(m,2H),7.08(td,J=7.5,1.0Hz,1H),6.77(d,J=7.8Hz,1H),5.03(s,1H),4.97(s,1H),4.90(s,1H),4.72(s,2H),3.96(s,2H),3.38–3.33(m,1H),2.31–2.26(m,2H),1.12(s,1H),1.07(s,1H),0.71(d,J=4.7Hz,2H)。
实施例125

4-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1'-
基)甲基)异喹啉-4-基)苯磺酰胺(125)
使化合物114a(400mg,0.693mmol)与(4-氨磺酰苯基)硼酸(181mg,0.900mmol)根据针对中间体22步骤a中所述的方法反应,接着通过在室温下用在DCM中的TFA处理7h进行N-去保护,得到标题化合物(28%)。MS(ES+)554.14[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.33–9.28(m,1H),8.20–8.14(m,1H),8.01(d,J=7.9Hz,2H),7.72(dddd,J=18.2,8.0,6.9,1.4Hz,2H),7.65(s,2H),7.60(dd,J=7.4,1.2Hz,1H),7.53(s,2H),7.27(dd,J=8.4,1.2Hz,1H),7.18(td,J=7.7,1.3Hz,1H),7.07(td,J=7.5,1.0Hz,1H),6.68(d,J=7.8Hz,1H),4.97(s,1H),4.91(s,1H),4.72(s,2H),4.01(s,1H),3.96(s,1H),3.39–3.33(m,0H),2.26(s,2H),1.13(s,1H),1.07(s,1H),0.71(d,J=3.9Hz,2H)。
实施例127

1-环丙基-3-((4-((2,2,2-三氟乙氧基)甲基)异喹啉-3-基)甲基)-1H-咪唑并[4,
5-c]吡啶-2(3H)-酮(127)
将甲磺酰氯(152mg,1.0mmol)在室温、氮气下逐滴添加到化合物47f(400mg,1.16mmol)和三乙胺(351mg,3.0mmol)在DCM(20mL)中的经搅拌的悬浮液中。将所得混合物在室温下搅拌1h,接着用DCM(50mL)稀释并且用水(2x 30mL)洗涤。将有机层经硫酸钠干燥、过滤并且减压浓缩。将所得残余物在室温下添加到2,2,2-三氟乙醇(94mg,0.94mmol)、碳酸铯(614mg,2.0mmol)和碘化钠(141mg,0.94mmol)在DMF(3mL)中的经搅拌溶液中,搅拌10分钟。将混合物在室温下搅拌16h,接着倾注到水(30mL)中并用EtOAc(2X50mL)萃取。将合并的有机层用盐水(30mL)洗涤、经硫酸钠干燥、过滤并减压浓缩。通过C18制备型HPLC(在H2O:乙腈中的10mM碳酸氢铵)纯化所得的粗化合物。将适当的级分汇集并浓缩。用水(5mL)研磨所得固体,得到为固体的标题化合物(60mg,15%)。MS(ES+)429.61[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.25(s,1H),8.29(s,1H),8.28–8.22(m,1H),8.20(d,J=5.2Hz,1H),8.12(dd,J=8.0,1.4Hz,1H),7.89(ddd,J=8.4,6.8,1.3Hz,1H),7.72(ddd,J=7.9,6.8,0.9Hz,1H),7.24(dd,J=5.2,0.8Hz,1H),5.43(s,2H),5.36(s,2H),4.30(q,J=9.3Hz,2H),3.37–3.33(m,1H),2.97(tt,J=7.1,3.7Hz,1H),1.06(td,J=7.3,5.2Hz,2H),0.94–0.87(m,2H)。
实施例128

1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)-1-(2,2,2-三氟乙酰基)螺[氮杂环丁
烷-3,3'-二氢吲哚]-2'-酮(128)
在室温下将TFA逐滴添加到化合物92a(81mg,0.164mmol)在DCM(2mL)中的溶液中。将溶液在室温下搅拌30min,接着浓缩并与甲苯共蒸发。在室温下将残余物和DIEA(0.115mL,0.658mmol)溶解于DCM(3mL)中,
接着添加2,2-二氟乙磺酰氯(20μL,0.197mmol)。将混合物在室温下搅拌30min,接着真空浓缩。将粗料溶解于DCM中并在二氧化硅上通过柱色谱纯化,接着在C18柱上通过制备型LCMS进一步纯化。将适当的级分汇集并浓缩并且将残余物冻干,得到标题化合物(20.3mg,25%),MS ES(+):489.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.35(s,1H),8.75–8.70(m,1H),8.58–8.53(m,1H),8.22–8.15(m,1H),7.88(tt,J=7.9,1.8Hz,1H),7.81–7.66(m,3H),7.60(dd,J=7.9,4.8Hz,1H),7.30–7.24(m,1H),7.21(td,J=7.8,1.3Hz,1H),7.07(td,J=7.5,1.0Hz,1H),6.80–6.74(m,1H),5.05–4.88(m,2H),4.65(dd,J=9.7,3.7Hz,1H),4.48(d,J=9.5Hz,1H),4.31(dd,J=10.4,4.9Hz,1H),4.16(dd,J=10.2,3.5Hz,1H)。
实施例129

步骤a)2'-氧代-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)-1',2'-二氢螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(129a)
使化合物11a(135mg,0.258mmol)与吡啶-3-硼酸(47.6mg,0.387mmol)根据实施例12步骤a中所述的方法反应,得到标题化合物(135mg,97%),MS(ES+)522.3[M+H]+。
1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)-1-(2,2,2-三氟乙酰基)螺[哌啶-4,3'-
吡咯并[2,3-c]吡啶]-2'(1'H)-酮(129b)
使化合物129a(50mg,0.096mmol)如实施例128中所述的那样反应,但是反应温度为0℃,并且使用吡啶作为碱代替DIPEA,得到标题化合物(49.6mg,79%),MS(ES+)518.3MS[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(d,J=4.6Hz,1H),8.68(s,1H),8.29(s,1H),8.22–8.15(m,2H),7.98(ddt,J=7.5,3.6,1.9Hz,1H),7.74(dddd,J=19.5,8.0,6.9,1.3Hz,2H),7.65(q,J=4.3,3.5Hz,2H),7.36–7.30(m,1H),4.99(qd,J=16.0,9.2Hz,2H),4.03(dd,J=10.4,5.0Hz,1H),3.91(ddd,J=13.4,9.4,3.3Hz,1H),3.85–3.72(m,2H),2.05–1.89(m,2H),1.78(ddt,J=19.1,13.9,4.5Hz,2H)。
实施例130

步骤a)(1-(1'-((4-溴异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-4,3'-
吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)氨基甲酸叔丁酯(130a)
向化合物11a(800mg,1.53mmol)在1,4-二噁烷(20mL)和MeOH(6mL)中的溶液中添加浓HCl水溶液(4mL)。将混合物在环境温度下搅拌两小时,接着浓缩并与甲苯共蒸发三次,接着真空干燥。将所得的胺的二盐酸盐、1-(Boc氨基)环丙烷羧酸(320mg,1.59mmol)和HATU(680mg,1.79mmol)在DMF(12mL)中浆化并且添加DIEA(1.1mL,6.32mmol)。将所得溶液在环境温度下搅拌两小时,接着用EtOAc稀释,用NaHCO3水溶液和盐水洗涤。将有机相经MgSO4干燥并浓缩。通过硅胶柱色谱纯化产物,用DCM和1至10%MeOH洗脱,得到被一些DIEA和PF6污染的标题化合物(1.05g,112%)。
步骤b)(1-(1'-((4-(3,5-二甲基异唑-4-基)异喹啉-3-基)甲基)-2'-氧代-1',
2'-二氢螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-基羰基)环丙基)氨基甲酸叔丁酯(130b)
使化合物130a(150mg,0.247mmol)和3,5-二甲基异唑-4-硼酸(68mg,0.48mmol)根据实施例12步骤a中所述的方法偶联在一起,得到标题化合物(42mg,27%)。
步骤c)1-(1-氨基环丙烷羰基)-1'-((4-(3,5-二甲基异唑-4-基)异喹啉-3-基)甲
基)螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(130c)
将化合物130b(42mg,0.067mmol)溶解于DCM(1.5mL)中,接着根据实施例39步骤b中所述的方法用HCl在二噁烷(1.5mL)中的4M溶液进行处理,得到标题化合物(14mg,98%)。
1H NMR(500MHz,DMSO)δ0.66(d,1H),0.88(q,1H),1.73(m,2H),1.87(s,2H),2.28(d,3H),3.88(m,0H),4.95(d,1H),5.08(d,1H),7.44(d,1H),7.61(d,1H),7.73(m,1H),7.81(m,1H),8.19(m,1H),8.28(d,1H)。
实施例131

2'-氧代-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢吲
哚]-1-羧基酰胺(131)
将吡啶-3-硼酸(54.8mg,0.446mmol)、1M碳酸钠溶液(1.2mL)和四(三苯基膦)钯(0)在氩气添加到化合物116a(130mg,0.297mmol)在二噁烷(8mL)中的溶液中。将混合物在90℃下在密封管中搅拌6h,接着冷却,用二噁烷稀释并过滤。将滤液减压浓缩并且通过硅胶色谱纯化残余物,用DCM和2至8%MeOH洗脱,得到标题化合物(129mg,70%),MS(ES+)436.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.36–9.32(m,1H),8.72(dd,J=4.9,1.7Hz,1H),8.53(d,J=2.2Hz,1H),8.22–8.16(m,1H),7.83(dt,J=7.8,2.0Hz,1H),7.78–7.68(m,1H),7.61–7.50(m,1H),7.31–7.24(m,1H),7.16(td,J=7.8,1.3Hz,1H),7.05(td,J=7.5,1.0Hz,1H),6.68(d,J=7.8Hz,1H),6.11(s,1H),4.97(d,J=4.0Hz,1H),3.96(d,J=7.9Hz,1H),3.88(dd,J=7.9,2.5Hz,1H)。
实施例132

步骤a)(1-(1'-((4-(4-氨基苯基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-
3,3'-二氢吲哚]-1-基羰基)环丙基)氨基甲酸叔丁酯(132a)
使化合物114a(400mg,0.693mmol)与4-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)苯胺(305mg,1.38mmol)根据实施例12步骤a中所述的方法反应,但是使用CH3CN:水4:1作为溶剂和100℃的反应温度,得到标题化合物(300mg,72%)。MS(ES+)591.35[M+H]+。
步骤b)N-(4-(3-((1-(1-氨基环丙烷羰基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲 哚]-1'-基)甲基)异喹啉-4-基)苯基)甲磺酰胺(132b)
在0℃下将Et3N(0.35mL)和甲磺酰氯(0.09mL)添加到化合物132a(300mg)在DCM(10mL)中的经搅拌溶液中。将所得混合物在室温下搅拌2h,接着减压浓缩。将残余物溶解于1,4-二噁烷(10mL)中,将温度降低至0℃并且在搅拌的同时添加1N NaOH溶液(5mL)。将所得混合物在室温下搅拌3h,接着在0℃下用1N HCl溶液中和。用EtOAc(2x15mL)萃取混合物,将有机层用盐水洗涤,经Na2SO4干燥,过滤并减压浓缩。将所得固体悬浮于DCM(10mL)中,在0℃下添加TFA,接着将混合物在室温下搅拌6h,接着减压浓缩并与甲苯(2x5mL)共蒸发。将残余物溶解于DCM(10mL)和Et3N(1mL)中,浓缩并用乙酸乙酯(2X10mL)洗涤。将残余物用饱和碳酸氢钠溶液研磨2h、过滤、干燥并且用研钵和研杵碾磨40分钟,得到为固体的标题化合物(83mg,27%)。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.26(s,1H),8.17(d,J=8.0Hz,1H),7.73(dt,J=21.4,7.7Hz,3H),7.59(d,J=7.4Hz,1H),7.38(d,J=8.4Hz,1H),7.16(q,J=9.4,8.5Hz,2H),7.06(t,J=7.5Hz,1H),6.70(d,J=7.8Hz,1H),5.01(s,2H),4.75–4.69(m,2H),4.03(s,1H),3.97(s,1H),3.37(s,3H),1.13(s,1H),1.08(s,1H),0.72(s,2H)。
实施例133

(1R,3R)-1'-((4-(6-氰基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[环丁烷-1,
3'-二氢吲哚]-3-基氨基甲酸甲酯(133)
在室温下将Et3N(0.258mL)、双(2,5-二氧代吡咯烷-1-基)碳酸酯(474mg)添加到化合物83(160mg)在DCM(5mL)中的溶液中。将反应混合物在室温下搅拌2h,接着添加甲胺在THF(57.4mg)中的2M溶液并且继续搅拌16h,然后减压浓缩。通过制备型HPLC纯化粗产物,得到为固体的标题化合物(64mg,35%)。MS(ES+)490.61[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.37(s,1H),8.81–8.77(m,1H),8.25(dd,J=8.0,0.9Hz,1H),8.20(qd,J=4.2,1.7Hz,2H),7.74(ddt,J=9.8,7.0,3.6Hz,2H),7.46–7.40(m,1H),7.31–7.25(m,1H),7.18–7.09(m,1H),7.07–6.99(m,1H),6.75(d,J=7.8Hz,1H),5.19(p,J=7.3Hz,1H),4.93(d,J=7.7Hz,2H),2.68–2.55(m,4H),2.39(td,J=7.3,3.8Hz,2H)。
实施例134

1'-((4-(6-(甲基磺酰胺基)吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环
丁烷-3,3'-二氢吲哚]-1-羧酸甲酯(104)
在室温下将Et3N(0.33mL)添加到化合物104(220mg)在DCM(15mL)中的经搅拌溶液中。将混合物冷却至0℃,添加甲磺酰氯(0.11mL),然后将混合物在室温下搅拌3h,然后浓缩。将残余物溶解于1,4-二噁烷(10mL)中,添加1N NaOH溶液(7mL)并且将混合物在室温下搅拌2h,接着浓缩。添加水并且使用1N HCl溶液将pH调节至8-8.5。用DCM(2x 25mL)萃取混合物,将有机层用水(10mL)、盐水(10mL)洗涤,经Na2SO4干燥并浓缩,以得到粗化合物。使用1-2%MeOH/DCM作为洗脱液,在硅胶上通过柱色谱纯化所得的粗料。收集纯级分并浓缩,并且用乙醚研磨残余物,得到为固体的标题化合物(80mg,28%)。MS(ES+)544.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ10.86(s,1H),9.32(s,1H),8.25(s,1H),8.17(d,J=8.0Hz,1H),7.82(s,0H),7.73(dt,J=13.7,7.1Hz,3H),7.63(d,J=7.4Hz,1H),7.37(d,J=8.3Hz,1H),7.15(dt,J=16.0,8.0Hz,2H),7.05(t,J=7.5Hz,1H),6.72(d,J=7.8Hz,1H),5.00(s,2H),4.07(d,J=13.4Hz,4H),3.63(s,3H),3.37(s,3H)。
实施例135

步骤a)1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯并[2,3-c]吡
啶]-2'(1'H)-酮(135a)
将TFA(1.2mL)在室温下添加到化合物129a(400mg)在DCM(10mL)中的经搅拌溶液中。将反应混合物在室温下搅拌1h,接着真空浓缩。将所得粗料用DCM(50mL)稀释,用饱和碳酸氢钠溶液(40mL)洗涤。将有机层经硫酸钠干燥、过滤并且减压浓缩,得到为固体的标题化合物(325mg,90%)。MS(ES+)422.2[M+H]+。
步骤b)2'-氧代-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)-1',2'-二氢螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-1-羧基酰胺(135b)
将化合物135a(100mg,0.237mmol)溶解于DCM(10mL)中并且在室温下添加三乙胺(0.119mL)和(三甲基甲硅烷基)异氰酸酯(0.042mL)。将所得混合物在室温下搅拌5h,接着用DCM(50mL)稀释并且用水(30mL)洗涤。将有机层经硫酸钠干燥、过滤并减压浓缩。通过制备型C18HPLC纯化所得的粗化合物。将纯级分汇集、浓缩并冻干,得到为固体的标题化合物(50mg,44%)。MS(ES+)465.46[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(dd,J=4.9,1.6Hz,1H),8.67(d,J=2.1Hz,1H),8.25(d,J=4.8Hz,1H),8.21–8.14(m,2H),7.98(dt,J=7.8,2.0Hz,1H),7.74(dddd,J=19.8,7.9,6.9,1.3Hz,2H),7.65(dd,J=7.8,4.8Hz,1H),7.58(d,J=4.8Hz,1H),7.33(d,J=8.4Hz,1H),6.02(s,2H),5.01(d,J=16.0Hz,1H),4.94(d,J=16.0Hz,1H),3.64(dt,J=12.3,5.8Hz,2H),3.57(dt,J=13.6,5.5Hz,2H),1.65(t,J=5.7Hz,4H)。
实施例136

1-(甲基磺酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯并
[2,3-c]吡啶]-2'(1'H)-酮(136)
将化合物135a(80mg)溶解于DCM(3mL)中并且在0℃下添加三乙胺(0.106μL)和甲磺酰氯(0.018μL)。将反应混合物在室温下搅拌2h,接着用冷水(20mL)稀释并用DCM(2x30mL)萃取。将合并的有机层用盐水(30ml)洗涤,经无水硫酸钠干燥、过滤并浓缩。通过制备型C18HPLC纯化所得的粗化合物,得到标题化合物(45mg,47%)。MS(ES+)500.25[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.31(s,1H),8.76(d,J=4.7Hz,1H),8.68(s,1H),8.28(d,J=4.7Hz,1H),8.19(s,2H),7.98(d,J=7.6Hz,1H),7.74(dt,J=21.1,7.1Hz,2H),7.69–7.62(m,1H),7.60(d,J=4.9Hz,1H),7.34(d,J=8.3Hz,1H),4.99(s,1H),4.96(s,1H),3.45(dt,J=22.1,9.7Hz,5H),2.97(s,3H),1.97–1.88(m,2H),1.81(d,J=13.8Hz,2H)。
实施例137

1-(甲基磺酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯并
[2,3-c]吡啶]-2'(1'H)-酮(136)
将化合物135a(125mg)使用1-((叔丁氧羰基)氨基)环丙烷羧酸如实施例115步骤b中所述的那样酰化。使用在DCM中的TFA对所得的Boc保护的化合物进行N-去保护,得到标题化合物(40%)。MS(ES+)505.34[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(dd,J=4.9,1.6Hz,1H),8.68(d,J=2.1Hz,1H),8.26(d,J=4.7Hz,1H),8.18(s,1H),7.98(dt,J=7.8,2.0Hz,1H),7.74(dddd,J=19.8,8.0,6.9,1.3Hz,2H),7.65(dd,J=7.8,4.9Hz,1H),7.58(d,J=4.7Hz,1H),7.36–7.30(m,1H),5.00(s,1H),4.97(s,1H),3.92(s,2H),3.33(s,7H),2.55(s,1H),2.30(s,2H),1.73(dd,J=27.9,12.5Hz,3H),0.88(q,J=4.4Hz,1H),0.76–0.63(m,2H)。
实施例138

1-(2-羟基-2-甲基丙酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,
3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(138)
使用实施例115步骤b中所述的方法用2-羟基异丁酸(23.0mg,0.22mmol)将化合物135a酰化,得到标题化合物(51%)。MS(ES+)508.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(dd,J=4.9,1.6Hz,1H),8.68(d,J=2.2Hz,1H),8.27(d,J=4.7Hz,1H),8.21–8.15(m,2H),7.98(dt,J=7.8,2.0Hz,1H),7.74(dddd,J=19.8,8.0,6.9,1.3Hz,2H),7.65(ddd,J=7.8,4.9,0.9Hz,1H),7.58(d,J=4.8Hz,1H),7.36–7.30(m,1H),5.47(s,1H),5.00(s,1H),4.96(s,1H),4.21(s,2H),3.33(s,3H),1.72(s,4H),1.36(s,6H)。
实施例139

1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)-1-((2,2,2-三氟乙基)磺酰基)螺[哌
啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(139)
通过用在DCM/二噁烷中的HCl进行处理对化合物129a(129mg,0.247mmol)进行N-去保护。将所得的胺和吡啶(10当量)溶解于无水DCM(3mL)中,冷却至0℃并且添加2,2,2-三氟乙磺酰氯(5当量)。在0℃下10min后,使反应物达到室温,接着加热至35℃,持续72h。将溶液真空浓缩,将粗料溶解于乙腈中,使其通过过滤器并且接着在中性pH下通过制备型C18LCMS进行纯化。将纯级分汇集并且冷冻干燥,接着溶解于DCM中并且通过二氧化硅色谱进一步纯化,使用DCM:MeOH的梯度洗脱。将纯级分汇集、浓缩并冻干(水/乙腈),得到为固体的标题化合物(49.8mg,36%)。MS(ES+)568.22[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.31(s,1H),8.76(dd,J=4.9,1.6Hz,1H),8.68(d,J=2.2Hz,1H),8.29(d,J=4.7Hz,1H),8.21–8.15(m,2H),7.98(dt,J=7.8,2.0Hz,1H),7.74(dddd,J=20.5,8.0,6.9,1.3Hz,2H),7.65(ddd,J=7.8,4.9,0.8Hz,1H),7.59(d,J=4.8Hz,1H),7.37–7.31(m,1H),4.99(s,1H),4.96(s,1H),4.61(q,J=10.1Hz,2H),3.61(ddd,J=12.7,9.0,3.6Hz,2H),3.52(dt,J=12.7,5.0Hz,2H),1.91(ddd,J=13.3,8.9,4.2Hz,2H),1.81(dp,J=9.6,3.7Hz,2H)。
实施例140

1-(哌啶-4-羰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯
并[2,3-c]吡啶]-2'(1'H)-酮(140)
通过用在DCM/二噁烷中的HCl进行处理对化合物129a(129mg,0.247mmol)进行N-去保护。使用实施例115步骤b中所述的方法使126mg(0.237mmol)的所得胺与1-(叔丁氧羰基)哌啶-4-羧酸(54mg,0.237mmol)反应。通过用DCM:HCl(4M,在二噁烷中)1:1在室温下处理30min来去除Boc基团,接着真空浓缩并使用碱性pH通过制备型C18LCMS进行纯化,得到标题化合物(8.8mg,7.2%)。MS(ES+)533.32[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(dd,J=4.9,1.6Hz,1H),8.70–8.64(m,1H),8.26(d,J=4.8Hz,1H),8.21–8.15(m,1H),8.18(s,1H),8.01–7.94(m,1H),7.74(dddd,J=19.6,8.0,6.9,1.3Hz,2H),7.65(dd,J=7.8,4.9Hz,1H),7.60(d,J=4.8Hz,1H),7.33(d,J=8.3Hz,1H),6.29(s,1H),4.98(s,1H),3.86–3.80(m,2H),3.68(s,1H),2.96–2.88(m,2H),2.76–2.66(m,1H),2.60(s,5H),1.79–1.71(m,2H),1.68(s,2H),1.66(dd,J=14.7,9.2Hz,1H),1.57–1.50(m,4H),1.46(dd,J=13.0,9.2Hz,1H),1.25(d,J=13.5Hz,1H)。
实施例141

1-(3-氨基丙酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯
并[2,3-c]吡啶]-2'(1'H)-酮(141)
通过用在DCM/二噁烷中的HCl进行处理对化合物129a(0.19mmol)进行N-去保护。将所得的胺、3-((叔丁氧羰基)氨基)丙酸(40mg,0,21mmol)和HATU(85mg,0,22mmol)悬浮于DMF(3mL)中并且添加DIEA(0,17ml,0.98mmol)。将混合物在室温下搅拌2h,接着用EtOAc稀释,用NaHCO3水溶液、柠檬酸水溶液和盐水洗涤。将有机相经MgSO4干燥、过滤并浓缩。在二氧化硅上通过柱色谱纯化粗产物,用在DCM中的1-10%MeOH洗脱。将纯级分汇集并浓缩。将残余物溶解于二噁烷(5mL)中,添加浓HCl水溶液(0.8ml)并且将混合物在室温下搅拌1h,接着浓缩,与甲苯共蒸发并真空干燥。通过LC-MS纯化粗化合物,得到标题化合物(33mg,34%)。MS(ES+)493.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(dd,J=4.9,1.6Hz,1H),8.68(s,1H),8.26(d,J=4.8Hz,1H),8.18(s,2H),7.98(ddt,J=8.2,4.2,1.9Hz,1H),7.74(dddd,J=19.7,8.0,6.9,1.3Hz,2H),7.68–7.61(m,1H),7.59(d,J=4.8Hz,1H),7.36–7.30(m,1H),4.98(qd,J=16.0,9.8Hz,2H),3.91–3.83(m,1H),3.69(dd,J=14.5,8.3Hz,2H),2.82(t,J=6.5Hz,2H),2.53(d,J=6.4Hz,1H),1.83(s,5H),1.79(dd,J=8.2,3.9Hz,1H),1.79–1.60(m,2H)。
实施例142

1-(4-氨基丁酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯
并[2,3-c]吡啶]-2'(1'H)-酮(142)
如实施例141中所述的那样制备标题化合物,但是使用酸4-((叔丁氧羰基)氨基)丁酸。收率31%。MS(ES+)607.45[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(dd,J=4.9,1.6Hz,1H),8.67(s,1H),8.67(d,J=4.8Hz,0H),8.26(d,J=4.8Hz,1H),8.19(d,J=1.5Hz,1H),8.18(s,2H),7.98(ddd,J=7.6,4.8,2.2Hz,1H),7.74(dddd,J=19.7,8.0,6.9,1.3Hz,2H),7.68–7.61(m,1H),7.59(d,J=4.8Hz,1H),7.36–7.30(m,1H),4.98(qd,J=16.0,11.0Hz,2H),3.86(t,J=9.2Hz,1H),3.69(ddt,J=17.0,8.7,4.5Hz,2H),2.67–2.60(m,2H),2.42(hept,J=7.8Hz,2H),1.80(s,4H),1.84–1.72(m,0H),1.66(p,J=7.3Hz,3H)。
实施例143

1-(2-氨基乙酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯
并[2,3-c]吡啶]-2'(1'H)-酮(143)
如实施例141中所述的那样制备标题化合物,但是使用酸2-((叔丁氧羰基)氨基)乙酸。收率27%。MS(ES+)579.45[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(dd,J=4.9,1.7Hz,1H),8.68(s,1H),8.68(d,J=4.8Hz,0H),8.26(d,J=4.7Hz,1H),8.21–8.14(m,1H),8.17(s,2H),7.98(ddt,J=7.6,3.9,1.9Hz,1H),7.74(dddd,J=19.7,8.0,6.9,1.3Hz,3H),7.65(ddd,J=7.8,4.9,0.9Hz,1H),7.57(d,J=4.8Hz,1H),7.36–7.30(m,1H),5.05–4.92(m,2H),3.93–3.83(m,1H),3.87(s,1H),3.74–3.64(m,3H),3.60(d,J=14.1Hz,1H),3.40(s,1H),1.84–1.61(m,3H)。
实施例144

2'-氧代-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)-1',2'-二氢螺[哌啶-4,3'-吡
咯并[2,3-c]吡啶]-1-羧酸甲酯(144)
在0℃下将三乙胺(0.1mL)和氯甲酸甲酯(0.05mL)添加到化合物135a(80mg,0.190mmol)在DCM(10mL)中的经搅拌溶液中。将混合物在0℃下搅拌1h,接着倾注到冰水(20mL)中并且用DCM(2x 20mL)萃取。将合并的有机层用无水硫酸钠干燥、过滤并且真空浓缩。通过C18制备型HPLC纯化粗产物,得到为固体的标题化合物(50mg,55%)。MS(ES+)480.41[M+H]。
1H NMR(500MHz,DMSO-d6)δ9.32(d,J=0.8Hz,1H),8.76(dd,J=4.9,1.6Hz,1H),8.67(dd,J=2.2,0.9Hz,1H),8.26(d,J=4.8Hz,1H),8.21–8.15(m,1H),8.17(s,1H),7.97(dt,J=7.8,1.9Hz,1H),7.74(dddd,J=19.8,8.0,6.9,1.3Hz,2H),7.65(ddd,J=7.8,4.9,0.9Hz,1H),7.60(dd,J=4.8,0.8Hz,1H),7.33(dd,J=8.3,1.2Hz,1H),4.99(s,1H),4.96(s,1H),3.75–3.62(m,7H),1.75(dt,J=13.1,6.2Hz,2H),1.66(dt,J=13.4,5.0Hz,2H)。
实施例145

1-(2-(1-氨基环丙基)乙酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌
啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(145)
根据实施例39步骤a和b中所述的方法通过化合物135a的HCl盐(109mg,0.238mmol)与2-(1-((叔丁氧羰基)氨基)环丙基)乙酸(84.5mg,0.393mol)的反应以及随后的N-去保护来制备标题化合物。收率(65mg,32%)。MS(ES+)519.4[M+H]。
实施例146

步骤a)1-乙酰基-1'-((4-溴异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯并[2,3-c]吡
啶]-2'(1'H)-酮(146a)
向11a(3g)在DCM(30ml)中的溶液中添加TFA(22mL),将混合物在室温下搅拌3h,接着浓缩。将所得油状物用饱和碳酸氢钠溶液(30mL)碱化,然后用DCM(2x 50mL)萃取。将合并的有机层经硫酸钠干燥、过滤并浓缩至干燥,得到为浅褐色固体的期望产物(2.4g,93.68%)。将900mg所得的游离胺溶解于DCM(15mL)中并且在0℃下添加三乙胺(0.9mL),之后添加乙酸酐(0.3mL)。将混合物在室温下搅拌3h,接着用水(30mL)稀释,用DCM(2x 20mL)萃取水层。将有机层经硫酸钠干燥、过滤并浓缩,得到为固体的标题化合物(750mg,70%)。MS(ES+)467.02[M+H]。
步骤b)1-乙酰基-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[哌啶-4,3'-吡咯并
[2,3-c]吡啶]-2'(1'H)-酮(146b)
向化合物146a(0.4g)、吡啶-3-基硼酸(0.211g)在1,2-二甲氧基乙烷(10mL)和水(1mL)中的悬浮液中添加碳酸钠(0.273g)和[1,1'-双(二苯基膦基)二茂铁]氯化钯(II)(0.126g),将整体混合物脱气20min。将所得混合物在微波辐射、100℃下搅拌2h,然后通过硅藻土过滤。将滤液浓缩并且将获得的粗料与另一批次(0.42g)合并,并在二氧化硅上通过柱色谱进行纯化,使用在DCM中的0-5%MeOH洗脱。将纯级分汇集、浓缩并且通过制备型C18HPLC进一步纯化100mg,得到60mg的标题化合物。MS(ES+)465.04[M+H]。
1H NMR(500MHz,DMSO-d6)δ9.32(s,1H),8.76(dd,J=4.9,1.6Hz,1H),8.68(s,1H),8.26(d,J=4.7Hz,1H),8.21–8.15(m,1H),8.17(s,1H),8.02–7.94(m,1H),7.74(dddd,J=19.8,8.0,6.9,1.3Hz,2H),7.65(ddd,J=7.7,4.9,0.8Hz,1H),7.58(d,J=4.8Hz,1H),7.37–7.30(m,1H),4.98(qd,J=16.0,10.6Hz,2H),3.91–3.81(m,1H),3.77(ddd,J=12.9,8.6,3.7Hz,1H),3.72–3.59(m,1H),3.66(s,1H),2.06(s,3H),1.86–1.59(m,3H)。
实施例147

1-(1-氨基环丙烷羰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁
烷-3,3'-二氢吲哚]-2'-酮(147)
使用实施例146步骤b中所述的方法使化合物114a(400mg,0.693mmol)与吡啶-3-基硼酸(128mg,1.04mmol)反应,之后通过用在DCM中的TFA处理来进行N-去保护,得到标题化合物(63mg,19%)。MS(ES+)476.6[M+H]。
1H NMR(500MHz,DMSO-d6)δ9.34(s,1H),8.72(dd,J=4.9,1.6Hz,1H),8.56(s,1H),8.19(dd,J=7.4,1.8Hz,1H),7.86(d,J=7.8Hz,1H),7.78–7.67(m,2H),7.59(t,J=6.4Hz,2H),7.27(d,J=8.2Hz,1H),7.21–7.13(m,1H),7.07(t,J=7.5Hz,1H),6.71(d,J=7.8Hz,1H),5.03–4.91(m,2H),4.72(s,2H),4.00(s,2H),3.96(s,0H),3.35(s,1H),2.26(s,2H),1.12(s,1H),1.08(s,1H),0.72(s,2H)。
实施例148

步骤a)1'-((4-溴异喹啉-3-基)甲基)-1-(吗啉-4-羰基)螺[氮杂环丁烷-3,3'-二
氢吲哚]-2'-酮(148a)
使用实施例79中所述的方法使化合物A1(400mg,1.33mmol)与I-34(382mg,1.33mmol)反应,但是在室温下,而不是在70℃下,得到标题化合物(400mg,54%)。MS(ES+)509.1[M+H]。
步骤b)1-(吗啉-4-羰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁
烷-3,3'-二氢吲哚]-2'-酮(148b)
使化合物148a(380mg,0.749mmol)与吡啶-3-基硼酸(184mg,1.50mmol)根据实施例22步骤a中所述的方法反应,不同的是通过微波辐射实现加热并且反应时间为1h,得到标题化合物(95mg,25%)。MS(ES+)506.5[M+H]。
1H NMR(500MHz,DMSO-d6)δ9.33(s,1H),8.71(dd,J=4.8,1.6Hz,1H),8.54(d,J=2.2Hz,1H),8.22–8.16(m,1H),7.84(dt,J=7.8,2.0Hz,1H),7.78–7.68(m,2H),7.62–7.52(m,2H),7.27(d,J=8.0Hz,1H),7.17(t,J=7.7Hz,1H),7.06(t,J=7.5Hz,1H),6.70(d,J=7.8Hz,1H),5.02–4.90(m,2H),4.09(d,J=8.0Hz,2H),4.00(dd,J=8.0,4.9Hz,2H),3.56(t,J=4.7Hz,4H),3.40–3.33(m,0H),3.28(t,J=4.8Hz,4H)。
实施例149

步骤a)1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢吲
哚]-2'-酮(149a)
通过用在DCM中的TFA进行处理来对化合物92a(1.42g,2.88mmol)进行N-去保护。将TFA(11ml,144mmol)逐滴添加到化合物92a(1.42g,2.88mmol)在DCM(25ml)中的溶液中。将混合物搅拌1h,接着浓缩并与甲苯共蒸发。将粗料减压干燥2h,得到标题化合物。
步骤b)1-(哌啶-4-羰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁
烷-3,3'-二氢吲哚]-2'-酮(149b)
使用实施例115步骤b中所述的方法用1-(叔丁氧羰基)哌啶-4-羧酸(45.3mg,0.197mmol)将化合物149a(100mg)酰化。通过用DCM:HCl(4M,在二噁烷中)1:1在室温下处理30min来去除Boc基团,接着真空浓缩并使用碱性pH通过制备型C18LCMS进行纯化,得到标题化合物(75%)。MS(ES+)504.28[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(s,1H),8.72(dd,J=4.9,1.6Hz,1H),8.55(dd,J=10.9,2.2Hz,1H),8.29(s,1H),8.19(dd,J=7.4,1.9Hz,1H),7.86(ddt,J=10.1,7.7,2.0Hz,1H),7.79–7.68(m,2H),7.64–7.56(m,2H),7.34–7.24(m,1H),7.18(td,J=7.8,1.3Hz,1H),7.05(t,J=7.5Hz,1H),6.74(dd,J=7.9,1.8Hz,1H),6.29(s,0H),5.04–4.89(m,2H),4.37–4.26(m,2H),3.98(d,J=7.0Hz,1H),3.97(s,1H),2.93(d,J=12.3Hz,2H),2.66(s,1H),2.55(s,1H),2.46(d,J=13.2Hz,2H),2.40–2.30(m,1H),1.57(dd,J=27.8,12.6Hz,2H),1.42(dqt,J=19.2,14.0,6.9Hz,2H),1.25(d,J=13.4Hz,0H)。
实施例150

1-(3-氨基丙酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,
3'-二氢吲哚]-2'-酮(150)
如实施例141中所述的那样由化合物92a制备标题化合物,但是使用TFA用于Boc去除。收率38%。MS(ES+)464.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(s,1H),8.72(dd,J=4.9,1.6Hz,1H),8.54(dd,J=8.1,2.5Hz,1H),8.19(dd,J=7.5,2.0Hz,1H),7.85(ddd,J=9.4,4.7,2.0Hz,1H),7.72(dddd,J=14.2,8.9,5.2,1.8Hz,2H),7.66–7.56(m,2H),7.27(d,J=7.8Hz,1H),7.17(tt,J=7.6,1.9Hz,1H),7.05(td,J=7.5,3.6Hz,1H),6.72(t,J=7.0Hz,1H),5.04–4.90(m,2H),4.35–4.21(m,2H),3.98(d,J=4.3Hz,2H),3.17(q,J=6.6Hz,1H),2.77(t,J=6.5Hz,1H),2.29–2.14(m,2H)。
实施例151

1-(4-氨基丁酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,
3'-二氢吲哚]-2'-酮(151)
如实施例142中所述的那样由化合物92a制备标题化合物,但是使用TFA用于Boc去除。收率33%。MS(ES+)478.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(s,1H),8.72(dd,J=4.9,1.6Hz,1H),8.54(dd,J=7.6,2.4Hz,1H),8.22–8.16(m,1H),7.89–7.81(m,1H),7.78–7.66(m,2H),7.60(dq,J=8.3,4.0Hz,2H),7.27(d,J=8.1Hz,1H),7.21–7.13(m,1H),7.05(td,J=7.6,2.6Hz,1H),6.72(d,J=7.7Hz,1H),5.04–4.89(m,2H),4.33–4.21(m,2H),3.97(s,2H),2.95(q,J=6.4Hz,1H),2.56(t,J=6.8Hz,1H),2.21–2.03(m,2H),1.69–1.54(m,2H)。
实施例152

1-(2-氨基乙酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,
3'-二氢吲哚]-2'-酮(152)
如实施例143中所述的那样由化合物92a制备标题化合物,但是使用TFA用于Boc去除。收率38%。MS(ES+)450.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.36–9.32(m,1H),8.72(dd,J=4.9,1.6Hz,1H),8.54(dd,J=7.5,2.2Hz,1H),8.22–8.16(m,1H),7.86(tt,J=7.6,2.0Hz,1H),7.78–7.68(m,2H),7.60(dt,J=8.0,3.0Hz,2H),7.30–7.24(m,1H),7.18(td,J=7.8,1.3Hz,1H),7.06(td,J=7.6,1.0Hz,1H),6.75–6.67(m,1H),5.03–4.89(m,2H),4.35–4.23(m,2H),4.01(s,2H),3.19(s,2H)。
实施例153

1-乙酰基-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-二氢吲
哚]-2'-酮(153)
在0℃下将三乙胺(0.5mL)缓慢添加到化合物149a(180mg)在DCM(5mL)中的溶液中。将所得溶液在氮气下搅拌5分钟,然后添加乙酰氯(0.06mg)并且将溶液搅拌9h。将反应混合物减压浓缩,并且将所得的油状物用水(20mL)稀释并用在DCM(2x 30mL)中的5%甲醇萃取。将合并的有机层用盐水(15mL)洗涤、经Na2SO4干燥、过滤并减压浓缩。通过制备型C18HPLC纯化粗化合物。将纯级分汇集、浓缩至最小体积,然后冷冻干燥,得到为固体的标题化合物(56mg,31%)。MS(ES+)435.17[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.37–9.32(m,1H),8.72(dd,J=4.9,1.6Hz,1H),8.55(dd,J=9.9,2.2Hz,1H),8.22–8.16(m,1H),7.86(ddt,J=9.6,7.7,2.0Hz,1H),7.78–7.67(m,2H),7.64–7.55(m,2H),7.30–7.24(m,1H),7.18(td,J=7.8,1.3Hz,1H),7.06(td,J=7.6,1.0Hz,1H),6.72(d,J=7.9Hz,1H),5.04–4.89(m,2H),4.31(dd,J=8.3,3.4Hz,1H),4.25(d,J=8.1Hz,1H),3.97(s,2H),1.85(s,3H)。
实施例154

1-(甲基磺酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-
二氢吲哚]-2'-酮(154)
使用实施例134步骤b中所述的方法用甲磺酰氯将化合物149a(180mg,0.393mmol)磺酰化,得到标题化合物(42mg,22%)。MS(ES+)471.13[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(s,1H),8.82(s,1H),8.72(d,J=4.8Hz,1H),8.55(s,1H),8.19(d,J=7.7Hz,1H),7.93(s,0H),7.87(d,J=7.7Hz,1H),7.73(p,J=6.9Hz,2H),7.63–7.57(m,2H),7.27(d,J=8.1Hz,1H),7.20(t,J=7.8Hz,1H),7.09(t,J=7.4Hz,1H),6.76(d,J=7.9Hz,1H),4.98(s,1H),4.95(s,1H),4.07(s,4H),3.40(s,1H),3.17(s,3H)。
实施例155

N-(1-氨基环丙基)-2'-氧代-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环
丁烷-3,3'-二氢吲哚]-1-羧基酰胺(155)
在氮气、0℃下将三乙胺(0.8mL)和化合物149a(600mg)添加到(1-异氰酸根合环丙基)氨基甲酸叔丁酯(450mg)在甲苯(20mL)中的溶液中。将混合物在室温下搅拌16h,接着用水(100mL)稀释,用EtOAc(2x200mL)萃取。将合并的有机层经硫酸钠干燥、过滤并浓缩。将所得粗料溶解于DCM(100mL)中,在室温、氮气下添加TFA(0.8mL)并且将溶液搅拌2h,然后浓缩。添加饱和碳酸氢钠溶液(100mL)并且用DCM(2x 100mL)萃取混合物。将合并的有机层用水(100mL)洗涤、经硫酸钠干燥、过滤并减压浓缩。通过制备型C18HPLC纯化所得的残余物,得到为固体的标题化合物(96mg,60%)。MS(ES+)491.2[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(s,1H),8.71(dd,J=4.9,1.6Hz,1H),8.52(d,J=2.2Hz,1H),8.19(dd,J=7.4,2.0Hz,1H),7.83(dt,J=7.8,2.0Hz,1H),7.78(s,1H),7.78–7.67(m,2H),7.58(dd,J=7.8,4.9Hz,1H),7.51(dd,J=7.4,1.2Hz,1H),7.31–7.22(m,2H),7.16(td,J=7.7,1.2Hz,1H),7.04(t,J=7.6Hz,1H),6.68(d,J=7.8Hz,1H),5.02–4.93(m,2H),3.95(d,J=7.9Hz,2H),3.85(dd,J=7.9,2.4Hz,2H),3.38–3.32(m,1H),2.56–2.52(m,2H),0.71–0.65(m,3H)。
实施例156

1-(2-(1-氨基环丙基)乙酰基)-1'-((4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂
环丁烷-3,3'-二氢吲哚]-2'-酮(156)
使用实施例145中所述的方法使化合物149a的TFA盐(151mg,0.298mmol)与2-(1-((叔丁氧羰基)氨基)环丙基)乙酸(122mg,0.556mmol)反应,之后通过用在DCM中的TFA处理来去除N-保护基,得到标题化合物(58mg,40%)。MS(ES+)490.4[M+H]+。
1H NMR(500MHz,DMSO-d6)δ9.34(s,1H),8.72(dd,J=4.8,1.6Hz,1H),8.58–8.51(m,1H),8.22–8.16(m,1H),7.85(ddt,J=8.1,4.1,2.0Hz,1H),7.78–7.68(m,2H),7.63–7.56(m,2H),7.27(dd,J=8.1,1.5Hz,1H),7.18(td,J=7.7,1.2Hz,1H),7.05(td,J=7.5,1.0Hz,1H),6.72(d,J=7.8Hz,1H),5.03–4.90(m,2H),4.34–4.23(m,2H),4.00(s,1H),4.00(d,J=14.9Hz,1H),2.30(s,1H),2.26(s,1H),0.50–0.41(m,4H)。
实施例157

步骤a)1'-((4-(6-(1-((叔丁氧羰基)氨基)-2,2,2-三氟乙基)吡啶-3-基)异喹
啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-羧酸甲酯(157a)
使化合物95a(92mg,0.2mmol)与(2,2,2-三氟-1-(5-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)吡啶-2-基)乙基)氨基甲酸叔丁酯(98mg,0.24mmol)根据实施例12步骤a中所述的过程反应,得到标题化合物(11.6mg,9%)。MS(ES+)648.3[M+H]+。
步骤b)1'-((4-(6-(1-氨基-2,2,2-三氟乙基)吡啶-3-基)异喹啉-3-基)甲基)-
2'-氧代螺[氮杂环丁烷-3,3'-二氢吲哚]-1-羧酸甲酯(157b)
根据针对106c所述的过程,将化合物157a(11.6mg,0.018mmol)溶解于DCM(2mL)中,然后用TFA(500μL,3.5mmol)进行处理,得到为双TFA盐的标题化合物(18.3mg,132%)。MS(ES+)548.3[M+H]+。
1H NMR(500MHz,Methanol-d4)δ9.23(s,1H),9.20(s,0H),8.67(s,1H),8.13–8.06(m,1H),8.02–7.95(m,0H),7.78(t,J=8.2Hz,1H),7.64(tdd,J=7.0,5.6,5.1,2.6Hz,2H),7.50(s,0H),7.50(dd,J=14.0,1.2Hz,1H),7.18(t,J=9.0Hz,1H),7.15–7.06(m,1H),7.07–6.98(m,1H),6.85(s,1H),6.75(d,J=7.9Hz,0H),5.69(d,J=7.2Hz,1H),5.64(q,J=7.1Hz,0H),4.96(d,J=16.0Hz,1H),4.89(d,J=16.1Hz,0H),4.16(q,J=8.5,7.8Hz,2H),4.09–4.03(m,3H),3.63(d,J=4.4Hz,3H),1.26–1.21(m,1H)。
实施例158

1'-((4-溴异喹啉-3-基)甲基)-1-(甲基磺酰基)螺[哌啶-4,3'-吡咯并[2,3-c]吡
啶]-2'(1'H)-酮(158a)
在0℃下将甲磺酰氯(0.06ml,0.77mmol)逐滴添加到化合物11a(147mg,0.32mmol)和N,N-二异丙基乙胺(0.45ml,2.56mmol)在乙腈(2mL)中的悬浮液中并且将获得的反应混合物在0℃下搅拌20min。通过添加饱和NaHCO3水溶液淬灭反应,用DCM萃取产物,将有机层经Na2SO4干燥并减压浓缩。在硅胶上通过柱色谱纯化产物,用在DCM中的MeOH梯度洗脱。将具有期望化合物的级分合并,并且用活性炭处理获得的溶液。通过硅藻土垫将固体过滤出来并且将溶液减压浓缩,得到标题化合物(82mg,51%)。MS(ES+)501.2;503.2[M+H]+。
(2,2,2-三氟-1-(5-(3-((1-(甲基磺酰基)-2'-氧代螺[哌啶-4,3'-吡咯并[2,3-
c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶-2-基)乙基)氨基甲酸叔丁酯(158b)
通过使N2气体通过3-5min对化合物I-38e(98.7mg,0,25mmol)、158a(82mg,0,16mmol)、Pd(dppf)Cl2(5.98mg,0.01mmol)和2M碳酸钾水溶液(0.25mL)在1,4-二噁烷(3mL)中的混合物进行脱气。将混合物在微波烘箱中在130℃下加热1h,然后冷却并通过硅藻土垫过滤。浓缩滤液并且使用柱色谱在硅胶(使用在DCM中的0-16%MeOH梯度洗脱)上纯化产物并且通过制备型HPLC(Phenomenex Gemini-NX 5μC18,110A,AX,100x 30mm,梯度13min,流速40ml/min,在具有10mM乙酸铵缓冲液的水中的50-60%乙腈)进一步纯化,以得到标题化合物(15mg,13%)。MS(ES+)697.6[M+H]+。
1'-((4-(6-(1-氨基-2,2,2-三氟乙基)吡啶-3-基)异喹啉-3-基)甲基)-1-(甲基
磺酰基)螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(158c)
将化合物158b(15mg,0,02mmol)悬浮于1,4-二噁烷(0.5ml)中。添加4M HCl(0.54ml)。添加一些甲醇以获得溶液并且搅拌反应物1h。将混合物浓缩并且将粗料减压干燥。
通过HPLC分离产物(Gemini NX 100x30,梯度14分钟,在具有10mM乙酸铵缓冲液的水中的40-45%乙腈)。将适当的级分汇集并冷冻干燥,得到标题化合物(5mg,53%),MS(ES+)597.4[M+H]+。
1H NMR(500MHz,DMSO)δ1.82(m,2H,22',26'),1.91(dtd,J=15.6,11.4,9.9,6.5Hz,2H,22”,26”),2.71(s,2H,34),2.96(d,J=2.2Hz,3H,40),3.43(ddt,J=22.6,11.6,6.8,6.8Hz,4H,23,25),4.76(t,J=8.6,8.6Hz,1H,33),4.96(m,2H,7),6.62(s,0H),7.29(m,1H,21),7.60(d,J=4.8Hz,1H,16),7.73(t,J=7.5,7.5Hz,1H,19),7.78(m,1H,20),7.83(dd,J=8.0,3.8Hz,1H,28),8.06(dt,J=8.0,2.4,2.4Hz,1H,27),8.19(m,2H,13,18),8.29(d,J=4.8Hz,1H,15),8.73(s,1H,31),9.32(s,1H,6)。19F NMR(376MHz,d2o)δ-71.61(dd,J=29.0,8.6Hz,36,37,38)。
实施例159

步骤(a)1'-((4-(噁唑-5-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(159a)
改编自文献过程[N.Primas等人Tetrahedron 65(2009),6348-6353],将化合物11a(305mg,0.58mmol)、5-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)-2-(三异丙基甲硅烷基)噁唑(277mg,0.79mmol)、碳酸钠(158mg,1.48mmol)和Pd(PPh3)4(55mg,0.048mmol)在二噁烷-水3:1(9mL)中的混合物在密封的反应容器中在氩气下从90℃加热至100℃。通过LCMS监测反应。在3.5h后,添加更多在1.5mL溶剂中的硼酸酯(300mg,0.85mmol)。2h后,将温度增加至110至115℃。3h后,添加更多硼酸酯(150mg,0.43mmol)、Pd催化剂(45mg,0.039mmol)和碳酸钠(150mg,1.42mmol)并且继续加热4h,直到11a被消耗。此时,包含期望化合物和具有甲硅烷基基团的偶联产物的1/1混合物的反应物仍然是完整的。将反应混合物用EtOAc(20mL)稀释并过滤。将滤液真空浓缩并且使残余物在40mL EtOAc与40mL饱和NaCl(水溶液)之间分配。将有机相用2x 20mL NaCl溶液洗涤、干燥(Na2SO4)并且真空蒸发。使用在DCM中的MeOH的梯度,通过二氧化硅色谱进行纯化,得到标题化合物(212mg,70%)。MS(ES+)512.4[M+H]。
步骤(b)1-(甲基磺酰基)-1'-((4-(噁唑-5-基)异喹啉-3-基)甲基)螺[哌啶-4,
3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(159b)
为了去除Boc基团,将在二噁烷(4mL)和MeOH(2.5mL)中的4M HCl添加到化合物159a(208mg,0.41mmol)在DCM(4mL)中的溶液中并搅拌30min。将混合物与DCM共蒸发数次,得到胺HCl盐。将一半材料(0.20mmol)在冰浴中与在MeCN(4mL)中的DIEA(0.14mL,0.80mmol)一起搅拌10-15min。缓慢添加甲磺酰氯(20μL,0.26mmol)在MeCN(1mL)中的混合物。30min后,将混合物真空浓缩并且与DCM共蒸发数次。将粗料重新溶解于30mL DCM中并且用15mL NaHCO3饱和水溶液洗涤。再用10mL DCM萃取水相。将有机相合并,并且通过旋转蒸发仪去除溶剂,得到米白色固体。通过制备型C18HPLC进行纯化,得到标题化合物(13.3mg,14%收率)。ES+490.3[M+H]。
1H NMR(500MHz,DMSO-d6)δ9.36–9.31(m,1H),8.73(s,1H),8.28(d,J=4.8Hz,1H),8.19(dt,J=8.2,1.0Hz,1H),8.13(s,1H),7.86(ddd,J=8.3,6.9,1.3Hz,1H),7.81–7.71(m,3H),7.65–7.60(m,1H),5.16(s,2H),3.51(ddd,J=12.4,8.9,3.6Hz,2H),3.47–3.39(m,2H),2.98(s,3H),1.96(ddd,J=13.2,8.9,4.1Hz,2H),1.86(ddt,J=13.6,6.1,3.6Hz,2H)。
实施例160

步骤a)1'-((4-溴-7-氯异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-4,3'-
吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(160a)
使化合物I-35b(1.64g,4.0mmol)与I-3g(1.10g,4.0mmol)根据实施例3步骤a中所述的方法反应,得到标题化合物(1.96g,68%)。MS(ES+)559.1[M+H]+。
步骤b)1'-((7-氯-4-(吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺
[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(160b)
将化合物160a(500mg,0.896mmol)、吡啶-3-基硼酸(176mg,1.43mmol)和碳酸钠(285mg,2.69mmol)在1,2-二甲氧基乙烷(15mL)和水(2mL)中的经搅拌溶液用氩气净化10分钟,接着添加[1,1'-双(二苯基膦基)二茂铁]氯化钯(II)CH2Cl2(65mg,0.089mmol)并且将混合物再次用氩气净化5分钟。将所得反应混合物在密封管中在100℃下搅拌16小时,接着冷却至室温,通过硅藻土过滤,并且将滤液真空浓缩。在硅胶柱上通过快速色谱纯化所得的粗化合物,使用在DCM中的3%MeOH洗脱,得到为固体的标题化合物(300mg,54%)。MS(ES+)556.21[M+H]+。
步骤c)1'-((7-氯-4-(吡啶-3-基)异喹啉-3-基)甲基)-1-(甲基磺酰基)螺[哌啶-
4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(160c)
将在二噁烷(6mL)中的4M HCl在氮气、室温下逐滴添加到化合物160b(300mg,0.416mmol)在DCM(4mL)中的溶液中。将所得混合物在室温下搅拌1h,接着减压浓缩。将残余物悬浮于DCM(5mL)中,在0℃、氮气下添加三乙胺(0.6mL,4.32mmol)和甲磺酰氯(55mg,0.48mmol)。将所得混合物在室温下搅拌1h,接着用DCM(50mL)稀释,用水(2x 30mL)洗涤。将有机层经硫酸钠干燥、过滤并减压浓缩,并且通过C18制备型HPLC(在H2O:乙腈中的10mM碳酸氢铵)纯化所得的粗化合物,得到为固体的标题化合物(112mg,52%)。MS(ES+)534.17[M+H]+。
1H NMR(500MHz,DMSO)δ1.81(dt,J=13.8,4.5,4.5Hz,2H),1.92(ddd,J=13.4,8.9,4.3Hz,2H),2.96(s,3H),3.45(dddd,J=23.4,17.6,10.7,5.2Hz,4H),4.95(m,2H),7.36(d,J=9.1Hz,1H),7.61(m,1H),7.66(ddd,J=7.8,4.9,0.9Hz,1H),7.77(dd,J=9.1,2.2Hz,1H),7.99(dt,J=7.8,2.0,2.0Hz,1H),8.18(s,1H),8.29(d,J=4.8Hz,1H),8.32(d,J=2.2Hz,1H),8.69(d,1H),8.77(dd,J=4.9,1.6Hz,1H),9.29(s,1H)。
13C NMR(126MHz,DMSO)δ31.19,31.22,34.49,40.67,43.14,43.72,118.53,124.00,126.57,127.53,127.81,130.41,130.62,131.90,132.03,133.45,137.75,139.57,141.17,143.94,146.43,149.62,150.11,151.62,177.45。
实施例161

步骤a)1'-((7-氯-4-(吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺
[氮杂环丁烷-3,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(161a)
向化合物161a(1.1g,2.08mmol)、吡啶-3-基硼酸(330mg,2.68mmol)在DME(45mL)和水(5mL)中的混合物中添加碳酸钠(660mg,6.23mmol)。将反应混合物用氩气脱气15min,接着添加[1,1'-双(二苯基膦基)二茂铁]氯化钯(II)(160mg,0.219mmol)并且将混合物再次脱气5min。将所得混合物在100℃下搅拌16h,接着使其通过硅藻土并减压浓缩。使用硅胶通过柱色谱纯化所得的粗化合物,用在DCM中的3%MeOH洗脱。将包含化合物的级分混合并减压浓缩,得到为固体的标题化合物(800mg,49%),其无需进一步纯化即用于下一步骤。MS(ES+)528.16[M+H]+。
步骤c)1'-((7-氯-4-(吡啶-3-基)异喹啉-3-基)甲基)螺[氮杂环丁烷-3,3'-吡咯
并[2,3-c]吡啶]-2'(1'H)-酮(161b)
在0℃、氮气下将TFA(5mL,65.29mmol)添加到化合物161a(800mg,1.52mmol)在DCM(10mL)中的经搅拌溶液中。将反应混合物在室温下搅拌2h,浓缩并用饱和碳酸氢钠溶液碱化至pH 8,并且用DCM(2x 80mL)萃取。将合并的有机层经硫酸钠干燥、过滤并且减压浓缩。将获得的固体用乙醚(20mL)研磨、搅拌15min并过滤,得到为固体的标题化合物(500mg,67%)。所得材料无需任何进一步纯化即用于下一步骤。MS(ES+)428.15[M+H]+。
步骤c)1'-((7-氯-4-(吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺
[氮杂环丁烷-3,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(161c)
在0℃下将三乙胺(0.8mL,5.74mmol)添加到化合物161c(250mg,0.584mmol)在DCM(5mL)中的经搅拌溶液中。30min后,在0℃下,在氮气下添加在DCM中的氯甲酸甲酯(70mg,0.741mmol)的贮备溶液。将所得混合物在室温下搅拌2h,接着将混合物用水(20mL)稀释并且用DCM(2x50mL)萃取。将合并的有机层经硫酸钠干燥、过滤并且减压浓缩。通过制备型C18HPLC纯化获得的固体,得到为固体的标题化合物(120mg,42%)。MS(ES+)486.35[M+H]+。
1H NMR(500MHz,DMSO)δ3.63(s,3H),4.03(s,2H),4.12(s,2H),5.00(q,J=16.0,16.0,16.0Hz,2H),7.31(d,J=9.1Hz,1H),7.60(dd,J=7.8,4.8Hz,1H),7.74(d,J=4.7Hz,1H),7.76(dd,J=9.0,2.2Hz,1H),7.88(dt,J=7.8,2.0,2.0Hz,1H),8.09(s,1H),8.34(m,2H),8.57(d,J=2.2Hz,1H),8.72(dd,J=4.9,1.6Hz,1H),9.34(s,1H)。
13C NMR(126MHz,DMSO)δ41.75,44.11,52.02,118.35,123.73,126.48,127.49,127.74,130.14,130.45,131.88,131.94,133.43,137.39,137.61,140.07,144.40,146.12,149.40,149.73,151.54,156.04,175.27。
实施例162

1'-((7-氯-4-(吡啶-3-基)异喹啉-3-基)甲基)-1-(甲基磺酰基)螺[氮杂环丁烷-
3,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(162)
在0℃下向化合物161b(250mg,0.584mmol)在DCM(5mL)中的溶液中添加三乙胺(0.3g,2.96mmol),将所得混合物在0℃下搅拌20min,接着添加甲磺酰氯(80mg,0.701mmol)并且将混合物在室温下搅拌2h。用水(15mL)稀释反应混合物并且用DCM(2x 10mL)萃取水层。将合并的有机层经硫酸钠干燥、过滤并浓缩。通过制备型C18HPLC纯化获得的粗化合物。将纯级分汇集,浓缩并冻干,得到为固体的标题化合物(92mg,31%),MS(ES+)506.3[M+H]+。
1H NMR(500MHz,DMSO)δ3.18(s,3H),4.10(t,4H),5.00(m,2H),7.32(d,1H),7.61(m,1H),7.65(m,1H),7.77(dd,1H),7.90(dt,1H),8.13(s,1H),8.35(d,1H),8.38(d,1H),8.59(m,1H),8.72(dd,1H),9.33(s,1H)。
13C NMR(126MHz,DMSO)δ33.30,40.74,44.11,57.06,57.08,118.16,123.75,126.46,127.42,127.72,130.24,130.45,131.86,131.94,133.40,137.40,140.08,144.43,146.13,149.40,149.75,151.52,174.48。
实施例163

1'-((7-氯-4-(吡啶-4-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[氮杂环
丁烷-3,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(163)
将化合物I-40b(350mg,0.718mmol)、吡啶-4-基硼酸(125mg,1.02mmol)和碳酸钠(230mg,2.17mmol)在1,2-二甲氧基乙烷(10mL)和水(1.5mL)中的经搅拌溶液用氩气净化10分钟。添加[1,1'-双(二苯基膦基)二茂铁]氯化钯(II)CH2Cl2(50mg,0.068mmol)并且将混合物再次用氩气净化5分钟。将所得反应混合物在密封管中在105℃下搅拌16h,接着冷却至室温,通过硅藻土过滤并真空浓缩。通过制备型C18HPLC纯化粗化合物,得到为固体的标题化合物(112mg,25%)。MS(ES+)486.35[M+H]+。
1H NMR(500MHz,DMSO)δ3.63(s,3H),3.99(d,J=8.4Hz,2H),4.12(s,2H),5.01(s,2H),7.31(d,J=9.1Hz,1H),7.44(m,2H),7.74(dd,J=4.7,0.9Hz),7.76(dd,J=9.1,2.2Hz,1H),8.01(s,1H),8.34(d,J=4.7Hz,1H),8.35(d,J=2.2Hz,1H),8.73(m,2H),9.35(d,J=0.8Hz,1H)。
13C NMR(126MHz,DMSO)δ41.69,44.12,52.03,56.62(m),118.37,124.62,126.34,126.53,127.68,128.24,130.00,131.95,132.02,132.47,137.60,139.98,142.67,144.43,145.14,149.97,151.66,156.03,175.22。
实施例164

1'-((7-氯-4-(吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[哌啶-4,
3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(164)
化合物I-40b(350mg,0.718mmol)、吡啶-4-基硼酸(125mg,的经搅拌溶液
将在微波小瓶中的I-39b、3-吡啶硼酸、NaHCO3和与CH2Cl2络合的Pd(dppf)Cl2在经脱气溶剂(二噁烷/水/DMF 6:2:1,4.75mL)中的混合物用N2鼓泡1min,再添加0.2mL溶剂。将反应混合物在微波反应器中在110℃下加热1h,接着冷却至室温并且使其在水(15mL)与EtOAc(15mL)之间分配。用EtOAc(2x 15mL)萃取水相。将有机相合并,干燥(Na2SO4)并且真空浓缩。将所得粗料溶解于MeCN(3mL)+H2O(1.5mL)中并且通过C18制备型HPLC纯化。将产物级分冷冻干燥,得到标题化合物(66mg,45%)。MS(ES+)514.48[M+H]+。
1H NMR(500MHz,DMSO)δ1.67(dt,2H),1.75(dt,2H),3.64(s,3H),3.68(dq,4H),4.96(m,2H),7.36(d,1H),7.60(d,1H),7.65(m,1H),7.77(dd,1H),7.99(dt,1H),8.16(s,1H),8.26(d,1H),8.32(d,1H),8.69(d,1H),8.77(dd,1H),9.29(s,1H)。
13C NMR(126MHz,DMSO)δ30.95,38.54,43.03,44.34,52.26,118.41,123.84,126.43,126.46,127.47,127.69,130.26,130.49,131.78,131.89,133.34,137.63,139.34,141.22,143.79,146.31,149.47,149.99,151.48,155.04,177.41。
实施例165

步骤a)1'-((7-氯-4-(吡啶-4-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺
[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸叔丁酯(165a)
使160a(500mg,0.896mol)与吡啶-4-基硼酸(200mg,1.63mmol)根据实施例163中所述的过程反应,得到标题化合物(450mg,62%)。MS(ES+)556.44[M+H]+。
步骤b)1'-((7-氯-4-(吡啶-4-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺
[哌啶-4,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(165b)
在0℃、氮气下将在二噁烷(5mL,20mmol)中的4.0M HCl添加到化合物165a(0.45g,0.56mmol)在DCM(6mL)中的经搅拌溶液中。将所得反应物料在室温下搅拌2h。当TLC指示起始物质完全消耗时,将反应混合物减压浓缩并与1,4-二噁烷(2x 10mL)共蒸馏。将残余物溶解于DCM(5mL)中,在0℃下添加三乙胺(0.80mL,5.74mmol)并且在30min后,在氮气下添加在DCM中的氯甲酸甲酯(67.0mg,0.709mmol)的贮备溶液。将所得反应混合物在室温下搅拌1h,接着将反应混合物用水(20mL)稀释并且用DCM(2x 50mL)萃取。将合并的有机层经硫酸钠干燥、过滤并且减压浓缩。通过制备型C18HPLC纯化获得的粗料,得到为固体的标题化合物(58mg,21%)。MS(ES+)514.33[M+H]+。
1H NMR(500MHz,DMSO)δ1.67(dt,2H),1.75(dt,2H),3.64(s,3H),3.69(p,4H),4.96(s,2H),7.37(d,1H),7.58(m,2H),7.60(d,1H),7.77(dd,1H),8.14(s,1H),8.26(d,1H),8.32(d,1H),8.81(m,2H),9.29(s,1H)。
13C NMR(126MHz,DMSO)δ31.11,38.65,43.01,44.47,52.38,118.54,125.09,126.49,126.59,127.72,128.41,130.31,131.95,132.08,132.52,139.43,141.34,142.84,143.93,145.47,150.25,151.71,155.16,177.57。
实施例166

步骤a)1'-((4-溴异喹啉-3-基)甲基)-1-(1-(三氟甲基)环丙烷羰基)螺[哌啶-4,
3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(166a)
将在二噁烷(1.5mL)中的4M HCl添加到化合物11a(260mg,0.497mmol)在DCM(2mL)中的溶液中。将溶液在室温下搅拌1h,接着真空浓缩并与甲苯共蒸发。将获得的残余物溶解于DMF(1.5mL)中,添加DIEA(321mg,2.48mmol)并且将溶液搅拌15min,然后添加HATU(208mg,0.546mmol)和1-(三氟甲基)环丙烷-1-羧酸(84.2mg,0.546mmol)。将反应物在室温下搅拌1h,然后真空浓缩到二氧化硅上,接着上二氧化硅柱。使用MeOH-DCM的梯度洗脱产物,得到标题化合物(134mg,48%),MS(ES+)559.22[M+H]+。
步骤b)1'-((4-(6-氨基吡啶-3-基)异喹啉-3-基)甲基)-1-(1-(三氟甲基)环丙烷
羰基)螺[哌啶-4,3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(166b)
将5-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)吡啶-2-胺(55.1mg,0.250mmol)、双(三苯基膦)二氯化钯(II)(18mg,0.025mmol)和碳酸钾的2M溶液在氮气、室温下添加到包含化合物166a(70mg,0.125mmol)在乙腈(3mL)中的经搅拌溶液的密封管中。将反应混合物在100℃下搅拌16h。将反应混合物与另一批次的相同材料合并,过滤并减压浓缩。使用在DCM中的1-6%MeOH作为洗脱液,在硅胶上通过柱色谱纯化粗化合物,得到标题化合物(110mg,71%)。MS ES(+)573.3[M+H]+。
步骤c)N-(5-(3-((2'-氧代-1-(1-(三氟甲基)环丙烷羰基)螺[哌啶-4,3'-吡咯并
[2,3-c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶-2-基)甲磺酰胺(166c)
&
N-(甲基磺酰基)-N-(5-(3-((2'-氧代-1-(1-(三氟甲基)环丙烷羰基)螺[哌啶-4,
3'-吡咯并[2,3-c]吡啶]-1'(2'H)-基)甲基)异喹啉-4-基)吡啶-2-基)甲磺酰胺(166d)
将甲磺酰氯(20.9mg,0.183mol)在0℃下添加到化合物166b(95.2mg,0.166mmol)和TEA(37mg,0.366mmol)在DCM(3mL)中的溶液中。将反应物在0℃下搅拌1h,然后在室温下搅拌3h,并且添加更多的磺酰氯(2.2当量)和TEA(4当量),将溶液在室温下搅拌整个周末。
将反应混合物真空浓缩并且使用在DCM中的MeOH的梯度作为洗脱液,在硅胶上通过柱色谱纯化粗料,得到单磺酰化和二磺酰化的化合物。在pH 7下通过制备型LCMS进一步纯化单磺酰化的化合物,在冻干后提供纯的单磺酰化的化合物(9.3mg,7.7%),MS ES(+)651.5[M+H]+。
1H NMR(500MHz,DMSO)δ1.27(s,2H),1.32(s,2H),1.72(m,2H),1.79(d,2H),3.88(m,4H),5.03(m,2H),7.13(d,1H),7.46(m,1H),7.59(d,1H),7.72(ddd,1H),7.76(ddd,1H),7.83(dd,1H),8.12(s,1H),8.16(dd,1H),8.26(d,1H),8.32(d,1H),9.29(s,1H),11.04(s,1H)。
13C NMR(126MHz,DMSO)δ9.78,26.63(q),31.13,41.49,43.30,44.40,112.34,118.35,123.72,124.06,125.13(q),127.13,127.37,127.74,130.28,131.31,135.08,139.40,139.98,141.07,143.77,145.86,147.75,152.03,153.11,162.72,177.36。
19F NMR(376MHz,dmso)δ-66.26。
在pH 7下通过制备型LCMS进一步纯化二磺酰化的化合物,在冻干后提供纯的二磺酰化的化合物(9.5mg,7.8%)。MS ES(+)729.29[M+H]+。
1H NMR(500MHz,DMSO)δ1.30(d,4H),1.80(d,4H),3.75(s,6H),3.93(m,4H),4.95(m,2H),7.26(dd,1H),7.63(d,1H),7.75(ddd,1H),7.83(ddd,1H),8.00(dd,1H),8.19(dt,1H),8.25(s,1H),8.30(m,2H),8.79(dd,1H),9.31(m,1H)。
13C NMR(126MHz,DMSO)δ9.77,26.62(q),31.08,42.74,43.89,44.47,118.39,123.44,125.30(q),125.51,126.02,126.95,127.54,127.92,130.41,131.71,132.53,134.59,139.59,141.03,141.31,143.82,145.73,148.22,149.97,152.76,162.71,177.67。
19F NMR(376MHz,dmso)δ-66.24。
实施例167

步骤a)1'-((4-溴异喹啉-3-基)甲基)-1-(1-(三氟甲基)环丙烷羰基)螺[哌啶-4,
3'-吡咯并[2,3-c]吡啶]-2'(1'H)-酮(167a)
将碳酸铯添加到I-37d(140mg,0.356mmol)在DMF(3mL)中的溶液中。将获得的浆液搅拌1h,然后添加4-溴-3-(溴甲基)异喹啉在DMF(2mL)中的溶液。将浆液在室温下搅拌3h,然后浓缩到二氧化硅上,并且在二氧化硅上通过色谱进行纯化,使用在DCM中的1-8%MeOH洗脱,得到标题化合物(170mg,96%)。MS ES(+)495.2[M+H]+。
步骤b)1'-((4-溴异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺[氮杂环丁烷-3,
3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(167b)
在室温下将TFA逐滴添加到化合物166a(170mg,0.343mmol)在DCM(23mL)中的溶液中。18h后,将反应物浓缩并且与甲苯共蒸发两次。在室温下将残余物和DIEA(444mg,3.43mmol)溶解于DCM(3mL)中,添加氯甲酸甲酯并且将反应混合物在室温下搅拌1h,然后真空浓缩。将粗料溶解于DCM中并且使用在DCM中的MeOH的梯度作为梯度,通过硅胶柱色谱进行纯化,得到标题化合物(158mg,102%)。MS ES(+)453.18[M+H]+。
步骤c)1'-((4-(6-氨基吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',2'-二氢螺
[氮杂环丁烷-3,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(167c)
使化合物167b(158mg,0.349mmol)与5-(4,4,5,5-四甲基-1,3,2-二氧杂硼戊烷-2-基)吡啶-2-胺(153mg,0.697mmol)根据实施例166步骤b中所述的过程反应,得到标题化合物(160mg,98%)。MS ES(+)467.3[M+H]+。
步骤d)1'-((4-(6-(甲基磺酰胺基)吡啶-3-基)异喹啉-3-基)甲基)-2'-氧代-1',
2'-二氢螺[氮杂环丁烷-3,3'-吡咯并[2,3-c]吡啶]-1-羧酸甲酯(167d)
在0℃下将甲磺酰氯(43.2mg,0.377mmol)添加到化合物167c(160mg,0.343mmol)和TEA(76.4mg,0.755mmol)在DCM(3mL)中的溶液中。将反应物在0℃下搅拌30min,接着真空浓缩。
使用在DCM中的MeOH的梯度作为洗脱液,在硅胶上通过柱色谱纯化粗料。在pH 7下通过制备型LCMS进一步纯化产物,之后将纯级分冻干,得到标题化合物(20.3mg)。MS ES(+)545.5[M+H]+。
1H NMR(500MHz,DMSO)δ3.35(s,3H),3.63(s,3H),4.05(d,2H),4.14(s,2H),5.04(d,2H),7.11(d,1H),7.39(m,1H),7.74(m,4H),8.07(s,1H),8.18(dd,1H),8.26(d,1H),8.34(d,1H),9.33(s,1H),10.95(d,1H)。
13C NMR(126MHz,DMSO)δ41.61,41.77,44.25,52.00,56.60,112.01,118.34,124.02,124.23,127.13,127.23,127.45,127.76,130.14,131.37,135.15,137.58,139.90,140.12,144.34,145.70,152.10,152.41,156.04,175.29。
实施例168

步骤a)1'-((4-溴-7-氟异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-3,3'-二氢
吲哚]-1-羧酸甲酯(168a)
使用中间体41步骤a中所述的方法使化合物I-36d(1.50g,4.22mmol)与I-31(1.00g,4.22mmol)反应,得到标题化合物(1.60g,76%)。MS(ES+)471.9[M+H]+。
步骤b)1'-((7-氟-4-(噻唑-5-基)异喹啉-3-基)甲基)-2'-氧代螺[氮杂环丁烷-
3,3'-二氢吲哚]-1-羧酸甲酯(168b)
将5-(三丁基甲锡烷基)噻唑(480mg,1.27mmol)在MeCN(10mL)中的溶液和碳酸钠(2.5g,1.910mmol)在氩气下添加到化合物168a(300mg,0.637mmol)在MeCN(20mL)中的溶液中。将反应混合物用氩气净化15min,接着添加双(三叔丁基膦)钯(0)(30mg,0.057mmol),并且将反应混合物在130℃下搅拌18h。通过硅藻土过滤反应混合物并且真空浓缩。将粗料与另一批次(100mg)一起通过制备型HPLC纯化。收集适当的级分并且通过SFC进一步纯化。收集包含期望化合物的级分,真空浓缩,用水研磨并过滤出来,得到为固体的标题化合物(52mg)。MS(ES+)476.29[M+H]+。[M+H]+。
1H NMR(500MHz,DMSO)δ3.64(s,3H),4.08(d,2H),4.17(s,2H),5.06(s,2H),7.54(dd,1H),7.72(td,1H),7.76(dd,1H),8.02(dd,1H),8.05(d,1H),8.08(s,1H),8.35(d,1H),9.35(s,1H),9.41(d,1H)。
13C NMR(126MHz,DMSO)δ41.76,44.08,52.01,56.65,110.94(d),118.40,120.40,122.09(d),127.21(d),127.82(d),128.85,129.99,133.05,137.70,140.06,144.25,144.42,147.02(d),152.47(d),156.04,156.62,160.30(d),175.31。
19F NMR(376MHz,dmso)δ-111.41(m)。
实施例169

步骤a)1-((4-溴-7-氯异喹啉-3-基)甲基)-1'-(甲基磺酰基)螺[二氢吲哚-3,4'-
哌啶]-2-酮(169a)
在0℃、氮气下将在二噁烷(5mL,20mmol)中的4.0M HCl添加到化合物160a(0.3g,0.48mmol)在DCM(3mL)中的经搅拌溶液中。将所得混合物在室温下搅拌2h,接着减压浓缩并与1,4-二噁烷(2x 10mL)共蒸馏。
将残余物溶解于DCM(5mL)中,在0℃、氮气下添加三乙胺(0.4mL,2.83mmol)和甲磺酰氯(0.1mL,0.85mmol)并且将混合物在室温下搅拌3h。将反应混合物倾注到水(20mL)中并用DCM(2x 30mL)萃取。将有机层用盐水(10mL)洗涤、经硫酸钠干燥、过滤并减压浓缩。在硅胶上通过快速色谱纯化粗化合物,使用在DCM中的15%MeOH洗脱,得到为固体的标题化合物(130mg,50%)。MS(ES+)535.06[M+H]+。
步骤b)1-((7-氯-4-(噻唑-5-基)异喹啉-3-基)甲基)-1'-(甲基磺酰基)螺[二氢
吲哚-3,4'-哌啶]-2-酮(169b)
使化合物169a(110mg,0.203mmol)与5-(三丁基甲锡烷基)噻唑(134mg,0.358mmol)根据实施例168步骤b中所述的过程反应。将所得的粗化合物与使用相同方法获得的另外的粗化合物合并。将合并的粗化合物纯化,得到标题化合物(40mg)。MS(ES+)540.11[M+H]+。
1H NMR(500MHz,DMSO)δ=1.85(dt,2H),1.96(ddd,2H),2.97(s,3H),3.42(dt,2H),3.49(td,2H),5.03(s,2H),7.55(d,1H),7.63(d,1H),7.83(dd,1H),8.17(s,1H),8.18(s,1H),8.30(d,1H),8.32(d,1H),9.29(s,1H),9.46(s,1H)。
此外,表2中列出的化合物使用与上文所述的那些一致的过程,采用可商购获得的构件或根据文献过程或如本文所述的那样制备的构件制备。
表2



表2















实施例A:RSV致细胞病变效应
最初使用永生化细胞和RSV的实验室菌株(Long)在基于致细胞病变效应(CPE)的病毒复制测定中对本发明的化合物进行测试。该测定评估化合物抑制病毒复制的能力。
过程:
通过以384孔黑色、底部透明的板(Greiner Bio-One)的每个孔2,500个HEp-2细胞(ATCC)接种在20μL测定培养基(定义为补充有2%热失活胎牛血清和1%青霉素/链霉素的DMEM)中来制备测定板。将测定板在包含5%CO2的培养箱中在37℃下温育过夜。第二天,在DMSO中制备测试化合物的10点连续稀释液。随后用测定培养基稀释化合物并且将20μL经过稀释的化合物(包含1.5%DMSO)转移到测定板中以用于抗病毒活性的评估。
对于CPE测定,使用20μL稀释于测定培养基中的RSV Long(ATCC)以0.015的感染复数(MOI)感染细胞。DMSO浓度在整个测定板中是恒定的,包括阴性和阳性对照。将测定板在包含5%CO2的培养箱中在37℃下温育3天。通过添加10μL的CellTiter-Glo(ProMega)来评估细胞活力。使用EnVision读板器(Perkin Elmer)测量发光。使用来自CPE测定的原始数据计算EC50值。
在实施例A中所述的测定中测试本发明的化合物,结果如表3中所示。表3





每篇参考文献,包括在本申请中引用的所有专利、专利申请和公布均以引用方式全文并入本文,如同其中的每一篇单独被并入的那样。此外,应认识到,在本发明的以上教导中,本领域技术人员可对本发明做出某些改变或修改,并且这些等同物将仍然在由本申请的随附权利要求所限定的发明范围内。
Claims (16)
1.一种具有式(I)的化合物:

其中
Z1为NR1A、CHR1A、CR1BR1B;
Z2和Z3中的一个为CH或CR1A’,另一个为N、CH或CR1A’;
R1A为C1-C6烷基、C3-C7环烷基、S(=O)2R1C、芳基、杂芳基或杂环基,其中每个所述烷基、环烷基、芳基、杂芳基和杂环基任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、-NHR1C、-NR1DR1D’、-C(=O)OH、-C(=O)R1C、-C(=O)C1-C6亚烷基NH2、-C(=O)OR1C、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、-OC(=O)NHR1C、-NHC(=O)R1C、-NHC(=O)NHR1C、-NHC(=O)OR1C或-NHS(=O)2R1C;
两个R1B与它们所附接的碳原子组合在一起并形成C3-C6环烷基或杂环基,其中所述环烷基和所述杂环基任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、-NHR1C、-NR1DR1D’、-C(=O)OH、-C(=O)R1C、-C(=O)OR1C、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、-OC(=O)NHR1C、-NHC(=O)R1C、-NHC(=O)NHR1C、-NHC(=O)OR1C或-NHS(=O)2R1C;
每个R1A’独立地选自卤代基、羟基、氰基、C1-C3卤代烷基、C1-C3烷氧基;
R1C为C1-C6烷基、C3-C7环烷基或杂环基,其中的任一个任选地被独立地选自以下的一个或两个取代基取代:卤代基、羟基、氰基、氨基、三氟甲基、C1-C3烷基、C1-C3烷氧基、C1-C3卤代烷基、C1-C3烷基氨基以及C1-C3二烷基氨基;
R1D和R1D’各自独立地为H或C1-C6烷基,或
R1D和R1D’与它们所附接的氮原子一起形成4至6元环,所述环任选地被独立地选自以下的一个或两个取代基取代:卤代基、羟基、氰基和氨基;
R2为C1-C6烷基,其被各自独立地选自以下的一个、两个或三个取代基取代:卤代基、羟基、氰基、三氟甲基、氨基、-NHR2A、-NR2BR2B’、C1-C3烷氧基、S(=O)2R2A、C3-C4环烷氧基以及杂环氧基,其中每个所述烷氧基、环烷氧基和杂环氧基任选地被各自独立地选自以下的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、C1-C3烷基氨基以及-S(=O)2R2A,或
R2为C2-C6烷基、C3-C7环烷基C0-C5烷基、杂环基C0-C5烷基、芳基C0-C5烷基或杂芳基C0-C5烷基,其中每个所述环烷基、杂环基、芳基和杂芳基任选地被各自独立地选自以下的取代基单取代、二取代或三取代:氧代基、卤代基、羟基、氰基、氨基、C1-C3烷基、C1-C3卤代烷基、C1-C3羟基烷基、C1-C3烷氧基、C1-C3卤代烷氧基、羟基C1-C3烷氧基、C1-C3烷基氨基、C3-C4环烷基、氧杂环丁烷基、-S(=O)2R2A、-S(=O)2NH2、-NHS(=O)2R2A以及-C(=O)NH2,并且所述环烷基和氧杂环丁烷基任选地被氨基或甲基取代;
R2A为C1-C3烷基、C3-C4环烷基、氨基、芳基、杂芳基或杂环基;
R2B和R2B’各自独立地为C1-C3烷基,或
R2B和R2B’与它们所附接的氮原子组合在一起并形成含有1、2或3个杂原子的饱和的4至6元杂环基,所述杂原子各自独立地选自O、S和N,所述杂环基任选地被独立地选自以下的一个或两个取代基取代:氨基、卤代基、C1-C3烷基和三氟甲基;
R3各自独立地选自由以下各项组成的组:卤代基、羟基、氰基、C1-C6烷基、或杂环基C0-C2烷基,其中烷基或杂环基任选地被独立地选自以下的1、2或3个取代基取代:-NR3AR3B、卤代基、羟基和三氟甲基;
R3A和R3B各自独立地为H或C1-C6烷基,其中烷基任选地被一个或两个卤代基取代;
n为0、1或2;
q为0、1或2;
其中除前述R2B和R2B’之外,所述杂环基为含有1、2或3个杂原子的饱和的4至7元单环或双环,所述杂原子各自独立地选自O、S和N;
其中所述芳基为含有6个碳原子的碳环芳族单环基团,其可进一步与芳族、饱和或不饱和的一个或多个5或6元碳环基团稠合;
其中所述杂芳基为含有各自独立地选自N、O和S的一个、两个、三个或四个杂原子的单环、双环或三环环系,其由5至14个环原子组成,其中杂原子中的至少一个是芳环的一部分;
或其盐。
2.根据权利要求1所述的化合物或其盐,其中Z1为CR1BR1B。
3.根据权利要求1所述的化合物或其盐,其中q为0。
4.根据权利要求1所述的化合物或其盐,其中Z2为CH,Z3为N。
5.根据权利要求1所述的化合物或其盐,其中Z2和Z3均为CH。
6.根据权利要求1所述的化合物或其盐,其中n为1并且R3为C1-C3烷基、卤代基或三氟甲基。
7.根据权利要求6所述的化合物或其盐,其中R3为甲基、氯、氟或三氟甲基。
8.根据权利要求6或7所述的化合物或其盐,其中R3位于所述异喹啉部分的7位,从而提供以下通式的化合物:

9.根据权利要求1所述的化合物或其盐,其中R2为任选地被一个或两个取代基取代的杂芳基。
10.根据权利要求9所述的化合物或其盐,其中R2为噻唑基或任选地被一个或两个取代基取代的吡啶基。
11.根据权利要求10所述的化合物或其盐,其中R2为吡啶-3-基或吡啶-4-基,其中的任一个任选地被一个或两个取代基取代。
12.根据权利要求1所述的化合物或其盐,其中R1A为7或8元螺杂环基,其任选地被各自独立地选自由以下各项组成的组的取代基单取代、二取代或三取代:C1-C6烷基、C1-C6卤代烷基、卤代基、C1-C6烷氧基、羟基、氰基、-NHR1C、-NR1DR1D’、-C(=O)OH、-C(=O)R1C、-C(=O)C1-C6亚烷基NH2、-C(=O)OR1C、-C(=O)NHR1C、-C(=O)NR1DR1D’、-S(=O)2R1C、S(=O)2NHR1C、-S(=O)(=NH)R1C、-OC(=O)R1C、-OC(=O)NHR1C、-NHC(=O)R1C、-NHC(=O)NHR1C、-NHC(=O)OR1C或-NHS(=O)2R1C。
13.根据权利要求1所述的化合物或其盐,其具有结构IIb’或IIb”

其中
Z3为N或CH;
R1CC为-C(=O)R1C、-C(=O)OR1C、-S(=O)2R1C,其中
R1C为C1-C4烷基或C3-C6环烷基,其中的任一个任选被甲基、氨基或三氟甲基取代;
R2为噻唑基、吡啶基或被氰基、-NHS(=O)2Me、C(=O)NH2、S(=O)2NH2或氟取代的吡啶基;
R3为C1-C3烷基、卤代基、氰基或C1-C3卤代烷基;
n为0或1。
14.根据权利要求13所述的化合物或其盐,其中
R1C为甲基或环丙基,其中环丙基任选地被甲基、氨基或三氟甲基取代;
R2为噻唑-5-基、吡啶-3-基或吡啶-4-基;
R3为甲基、氯、氟或三氟甲基。
15.根据权利要求1至14中任一项所述的化合物或其药学上可接受的盐的用途,其用于制造在人类中治疗或预防RSV感染的药物。
16.一种药物组合物,其包含根据权利要求1至14中任一项所述的化合物,或其药学上可接受的盐,以及药学上可接受的载体。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SESE1551051-4 | 2015-07-30 | ||
| SE1551051 | 2015-07-30 | ||
| SESE1650843-4 | 2016-06-15 | ||
| SE1650843 | 2016-06-15 | ||
| PCT/SE2016/050733 WO2017018924A1 (en) | 2015-07-30 | 2016-07-28 | Respiratory syncytial virus inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN107922407A CN107922407A (zh) | 2018-04-17 |
| CN107922407B true CN107922407B (zh) | 2020-09-25 |
Family
ID=57884956
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680044547.9A Active CN107922407B (zh) | 2015-07-30 | 2016-07-28 | 呼吸道合胞病毒抑制剂 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20190002436A1 (zh) |
| EP (1) | EP3328859B1 (zh) |
| JP (1) | JP6739516B2 (zh) |
| CN (1) | CN107922407B (zh) |
| AU (1) | AU2016298355A1 (zh) |
| BR (1) | BR112018001726A2 (zh) |
| CA (1) | CA2993144A1 (zh) |
| HK (1) | HK1246781A1 (zh) |
| MX (1) | MX2018001317A (zh) |
| WO (1) | WO2017018924A1 (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018038667A1 (en) * | 2016-08-25 | 2018-03-01 | Medivir Ab | Respiratory syncytial virus inhibitors |
| WO2018038668A1 (en) * | 2016-08-25 | 2018-03-01 | Medivir Ab | Respiratory syncytial virus inhibitors |
| CN108358832A (zh) * | 2017-12-29 | 2018-08-03 | 上海艾康睿医药科技有限公司 | 一种合成4-[(2-氨基-4-氯)苯基]-氨基-n-哌啶甲酸乙酯的方法 |
| WO2021098732A1 (zh) * | 2019-11-18 | 2021-05-27 | 江苏恩华药业股份有限公司 | 一种1',2'-二氢-3'h-螺[环丁烷1,4'-异喹啉]-3'-酮衍生物及其应用 |
| US11351149B2 (en) | 2020-09-03 | 2022-06-07 | Pfizer Inc. | Nitrile-containing antiviral compounds |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015065338A1 (en) * | 2013-10-29 | 2015-05-07 | Medivir Ab | Quinazoline based respiratory syncytial virus inhibitors |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015065336A1 (en) * | 2013-10-29 | 2015-05-07 | Medivir Ab | Respiratory syncytial virus inhibitors |
| CN104693211A (zh) * | 2013-12-10 | 2015-06-10 | 南京明德新药研发股份有限公司 | 作为抗病毒剂的咪唑衍生物及其制药用途 |
-
2016
- 2016-07-28 WO PCT/SE2016/050733 patent/WO2017018924A1/en active Application Filing
- 2016-07-28 JP JP2018504813A patent/JP6739516B2/ja active Active
- 2016-07-28 EP EP16830925.0A patent/EP3328859B1/en active Active
- 2016-07-28 CA CA2993144A patent/CA2993144A1/en not_active Abandoned
- 2016-07-28 MX MX2018001317A patent/MX2018001317A/es unknown
- 2016-07-28 US US15/748,606 patent/US20190002436A1/en not_active Abandoned
- 2016-07-28 CN CN201680044547.9A patent/CN107922407B/zh active Active
- 2016-07-28 BR BR112018001726A patent/BR112018001726A2/pt not_active Application Discontinuation
- 2016-07-28 AU AU2016298355A patent/AU2016298355A1/en not_active Abandoned
-
2018
- 2018-05-11 HK HK18106131.7A patent/HK1246781A1/zh unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015065338A1 (en) * | 2013-10-29 | 2015-05-07 | Medivir Ab | Quinazoline based respiratory syncytial virus inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2017018924A1 (en) | 2017-02-02 |
| EP3328859A1 (en) | 2018-06-06 |
| US20190002436A1 (en) | 2019-01-03 |
| JP6739516B2 (ja) | 2020-08-12 |
| EP3328859B1 (en) | 2020-06-03 |
| EP3328859A4 (en) | 2019-03-13 |
| BR112018001726A2 (pt) | 2018-09-18 |
| CN107922407A (zh) | 2018-04-17 |
| JP2018525372A (ja) | 2018-09-06 |
| MX2018001317A (es) | 2018-04-30 |
| HK1246781A1 (zh) | 2018-09-14 |
| CA2993144A1 (en) | 2017-02-02 |
| AU2016298355A1 (en) | 2018-02-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI848954B (zh) | 作為hpk1抑制劑的吡咯並[2,3-b]吡啶或吡咯並[2,3-b]吡嗪及其用途 | |
| TWI548636B (zh) | 吡咯并[2,3-d]嘧啶基、吡咯并[2,3-b]吡基及吡咯并[2,3-d]吡啶基丙烯醯胺 | |
| TWI642657B (zh) | 作為btk抑制劑的初級甲醯胺 | |
| JP5832524B2 (ja) | ピリドン及びアザピリドン化合物、並びにそれらの使用方法 | |
| CN107922407B (zh) | 呼吸道合胞病毒抑制剂 | |
| CN111527084A (zh) | 可用作hpk1抑制剂的异呋喃酮化合物 | |
| JP2019081786A (ja) | Trkaキナーゼ阻害剤としてのn−ピロリジニル、n’−ピラゾリル尿素、チオ尿素、グアニジン、およびシアノグアニジン化合物 | |
| CN110582493A (zh) | 作为免疫调节剂的苯并噁唑衍生物 | |
| TW201819376A (zh) | 經取代之1H-咪唑並[4,5-b]吡啶-2(3H)-酮及其作為GLUN2B受體調節劑之用途 | |
| TW201803871A (zh) | 作為PI3K-γ抑制劑之雜環化合物 | |
| CN115175907A (zh) | 可用作t细胞激活剂的经取代的哌嗪衍生物 | |
| CN109121414A (zh) | 苯并吡唑化合物和其类似物 | |
| AU2016366546B2 (en) | Inhibitors of Bruton's tyrosine kinase and methods of their use | |
| TW201718536A (zh) | 可作為TNFα調節劑之雜環化合物 | |
| TW201639827A (zh) | TGF-β抑制劑 | |
| CN111247136A (zh) | 嘧啶基-吡啶氧基-萘基化合物以及治疗ire1相关疾病和病症的方法 | |
| TW202229298A (zh) | 囊腫纖維化跨膜傳導調節蛋白之調節劑 | |
| JP2021523941A (ja) | フューリンインヒビター | |
| TW202104223A (zh) | 磷脂醯肌醇3-激酶抑制劑 | |
| JP7370617B2 (ja) | アミノノルカンファン誘導体及びその製造方法と使用 | |
| TW202112774A (zh) | 經取代之吡唑并-吡啶醯胺及其作為glun2b受體調節劑之用途 | |
| CN106661032A (zh) | 治疗或预防糖尿病、肥胖症和炎性肠病的1,3‑取代的2‑氨基吲哚衍生物及类似物 | |
| CN111050765A (zh) | 螺环化合物及其制造和使用方法 | |
| WO2023045960A1 (zh) | 一种吡啶类衍生物及其用途 | |
| TW201240971A (en) | Fused heterocyclic compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1246781 Country of ref document: HK |
|
| TA01 | Transfer of patent application right | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20191115 Address after: Ingham, Germany Applicant after: Bollinger Ingham International Co., Ltd. Address before: Stockholm, Sweden Applicant before: Medivir Aktiebolag |
|
| GR01 | Patent grant | ||
| GR01 | Patent grant |