通过下面的实施例和参考实施例更详细地阐述本发明,但不应当认为本发明仅限于它。
实施例1
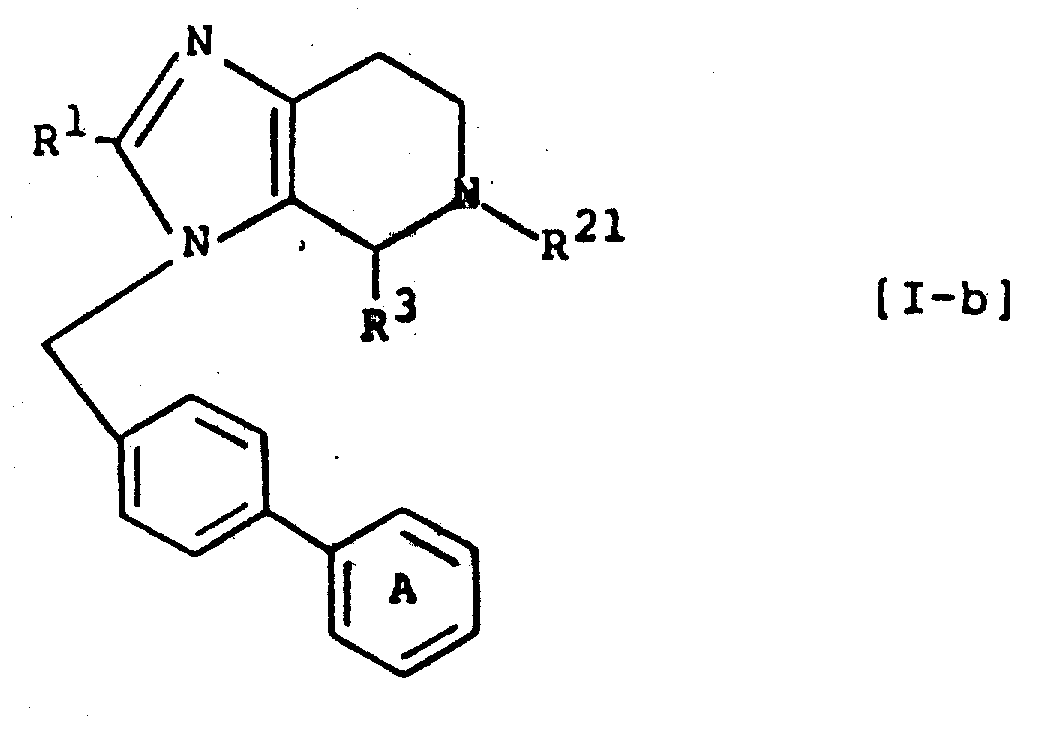
将5-二苯乙酰基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(1.60g)溶于二甲基甲酰胺(20ml)中,在冰冷却下向其中加入氢化钠(60%油分散物,176mg)。混合物在0℃搅拌20分钟,向其中加入[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基溴(2.40g),混合物在冰冷却下搅拌1小时,进一步在室温下搅拌1小时。减压浓缩反应溶液,向残留物中加入氯仿和水。干燥有机层,蒸去溶剂。所得残留物用硅胶柱色谱(溶剂;氯仿/甲醇=100∶1)纯化得5-二苯乙酰基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(430mg),为无色泡沫状物。
FAB-MS(m/z):852(M+H),792,610,567,244
NMR(CDCl3)δ:3.56(3H,s),5.12(2H,ABq)
结果,得到5-二苯基乙酰基-1-[2′-(1-三苯甲基-1H-四唑-5-基]联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(1.17g),为无色泡沫状物。
FAB-MS(m/z):852(M+H),792,610,567,244
NMR(CDCl3)δ:3.79(3H,s),4.81(2H,s)
实施例2
向5-二苯乙酰基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(400mg)和氯仿(1ml)的混合物中加入18%盐酸-甲醇(5ml),混合物在室温下搅拌30分钟。反应完成后,蒸去溶剂,所得残留物溶于甲醇(5ml),用1N氢氧化钠水溶液(5ml)调节至pH10-12,再在室温下搅拌3小时。用乙醚萃取除去三苯甲烷,减压蒸馏水层。所得残留物溶于少量水中,用非离子吸附树脂(商品名;HP-20日本Mitsubishi Kasei Corporation制造)柱色谱法纯化并冷冻干燥得以下化合物。
5-二苯乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸二钠盐(170mg)。
IR(Nujol;cm-1):1630
实施例3
盐酸2-正丁基-4-(2-氨基乙基)-1-[2′-(1H-四唑-5-基)联苯-4-基]甲基咪唑(8.09g),水合二羟乙酸(1.73g),1N NaOH水溶液(53ml)和二噁烷(50ml)的混合物在50℃搅拌两天。用盐酸酸化反应溶液,并减压蒸发。残留物溶于甲醇(100ml),并将混合物冷至-30℃,向其中滴加亚硫酰氯(12.4g)。加完后,在60℃搅拌混合物2天。减压蒸发混合物除去溶剂。用NaHCO3水溶液中和混合物,用氯仿萃取。干燥萃取液,减压蒸发,所得油状产物用硅胶柱色谱法(溶剂;氯仿/甲醇=15∶1)纯化,得2-正丁基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(3.74g),为粉末。
FAB-MS(m/z):472(M+H)(碱)
NMR(DMSO-d6)δ:0.90(3H,t),3.72(3H,s),5.20(2H,s)
实施例4
向实施例3得到的化合物(296mg),三乙胺(317mg)和氯仿(10ml)的混合物中在冰冷却下滴加在氯仿(1ml)中的乙酰氯(148mg)。混合物在室温下搅拌2小时,洗涤反应溶液,干燥并蒸去溶剂。所得残留物用硅胶柱色谱法(溶剂;氯仿/甲醇=30∶1)纯化得5-乙酰基-2-正丁基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(158mg)。
FAB-MS(m/z):514(M+H),119(碱)
NMR(CDCl3)δ:0.94(3H,t),2.16(3H,s),3.78(3H,s)
实施例5
实施例3得到的化合物(361mg),二氯甲烷(10ml),三乙胺(85mg),苯甲酸(103mg),盐酸N-羟基苯并三唑(114mg)和1-乙基-3-(3-二甲氨基丙基)碳二亚胺盐酸盐(162mg)的混合物在室温下搅拌过液。洗涤反应溶液,干燥并蒸去溶剂。所得油状残留物用硅胶柱色谱法(溶剂;氯仿/甲醇=20∶1)纯化得2-丁基-5-苯甲酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(297mg)。
FAB-MS(m/z):576(M+H),105(碱)
NMR(CDCl3)δ:0.97(3H,t),3.79(3H,s)
实施例6
实施例3得到的化合物(483mg)和噻吩-2-羧酸(158mg)用与实施例5相同的方法处理得2-正丁基-5-(2-噻吩基)羰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(300mg)。
FAB-MS(m/z):582(M+H),111(碱)
NMR(CDCl3)δ:0.95(3H,t),3.73(3H,s)
实施例7
将实施例3得到的化合物(2.98g),1N NaOH水溶液(14ml)和甲醇(30ml)的混合物在室温下搅拌过夜,蒸去溶剂。
用甲醇水溶液重结晶残留物并收集得2-正丁基-3-[2′-(1H-四唑-5-基)联苯-4基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸(2.11g)。
m.p.216-217℃(分解)
NMR(DMSO-d6)δ:0.97(3H,t),4.37(1H,s),5.17(1H,d),6.01(1H,d)
实施例8
实施例4得到的化合物(142mg),1N NaOH水溶液(0.60ml)和甲醇(5ml)的混合物在室温下搅拌过夜,减压蒸馏除去溶剂。所得残留物用非离子型吸附树脂(商品名;HP-20,日本Mitsubishi Kasei Corporation制造)柱色谱法纯化,冷冻干燥得2-正丁基-5-乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸二钠盐(100mg)。
m.p.>265℃
IR(Nujol;cm-1:1620
实施例9-10
用本与实施例8相同的方法处理实施例5-6得到的化合物得以下列于表1的化合物。
(Tet:1H-四唑-5-基基团)
实施例11
(1)向2-正丙基-4-(2-氨乙基)-1-[2′-(1-三苯甲基基-1H-四唑-5-基)联苯-4-基]甲基咪唑(3.66g)和甲醇(30ml)的混合物中加入9%盐酸-甲醇液(50ml),混合物在室温下搅拌40分钟。减压蒸发混合物除去溶剂,向残留物中加水。用乙酸乙酯洗涤混合物,减压蒸馏水层,进一步用甲苯共沸蒸馏处理,得粗的盐酸2-正丙基-4-(2-氨乙基)-1-[2′-四唑-5-基)联苯-4-基]甲基咪唑(2.68g)。
(2)用与实施例3相同的方法处理上述化合物(2.15g),得2-正丙基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(0.76g),为泡沫状物。
FAB-MS(m/z):458(M+H),207(碱)
NMR(DMSO-d6)δ:0.98(3H,t),3.84(3H,s),5.06(2H,ABq)
实施例12
盐酸2-正丁基-4-(2-氨乙基)-1-(2′-甲氧基羰基联苯-4-基)甲基咪唑(1.80g)用与实施例3相同的办法处理,得粗的2-正丁基-3-(2′-甲氧基羰基联苯-4-基)甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(1.70g)。
实施例13
向实施例12得到的化合物(1.70g)和吡啶(20ml)的混合物中加入乙酸酐(5ml),并将混合物搅拌过夜。蒸去溶剂,向所得残留物加氯仿,洗涤混合物,干燥,蒸去溶剂。残留物用硅胶柱色谱法(溶剂;氯仿/甲醇=10∶1)纯化,得2-正丁基-5-乙酰基-3-(2′-甲氧基羰基联苯-4-基)甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(0.47g),为油状物。
NMR(CDCl3)δ:0.88(3H,t),2.21(3H,s),3.46(3H,s),3.64(3H,s),5.33(2H,ABq)
实施例14
盐酸2-正丁基-4-(2-氨乙基)-1-[2′-(1H-四唑-5-基)联苯-4-基]甲基咪唑(0.278g),二羟基乙酸乙酯(0.179g)和乙醇(5ml)的混合物回流三天。向混合物中加入氯仿,洗涤混合物,干燥并蒸去溶剂。残留物用硅胶柱色谱法(溶剂;氯仿/甲醇=10∶1)纯化,得2-正丁基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.136g),为泡沫状物。
NMR(CDCl3)δ:0.92(3H,t),1.35(3H,t),5.08(2H,ABq)
实施例15
向2-正丁基-4-(2-叔丁氧基羰基氨基乙基)-1-(2′-甲氧基羰基联苯-4-基)甲基咪唑(2.02g)和氯仿(50ml)的混合物中加入三氟乙酸(25ml),混合物在室温搅拌30分钟。减压蒸发混合物除去溶剂,向残留物加入四氢呋喃(30ml),水(1ml)和NaHCO3(1.13g),搅拌混合物。向混合物加入氯仿,干燥混合物,减压蒸去溶剂。向残留物加入二羟乙酸乙酯水合物(0.570g)和乙酸(40ml),混合物回流15分钟。减压蒸去溶剂,所得残留物用硅胶柱色谱法(溶剂;氯仿/甲醇=20∶1)纯化得2-正丁基-3-(2′-甲氧基羰基联苯-4-基)甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.551g),为泡沫状物。
NMR(CDCl3)δ:0.89(3H,t),1.23(3H,t),3.65(3H,s)5.24(2H,ABq)
实施例16-31
实施例3,11和15得到的化合物用与实施例4或5相同的方法处理,得以下列于表2和3的化合物。
表2
Tet:1H-四唑-5-基基团
表3
Tet:1H-四唑-5-基基团
实施例32
向实施例3得到的化合物(0.50g)和甲醇(10ml)的混合物中加入双烯酮(0.20g),混合物在室温搅拌2小时。混合物减压蒸去溶剂,向残留物加入氯仿。洗涤混合物,干燥并减压蒸发。所得残留物通过硅胶柱色谱法(溶剂;氯仿/乙酸乙酯/甲醇=10∶10∶1)纯化,得2-正丁基-5-乙酰乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(0.29g),为泡沫。
FAB-MS(m/z):556(M+1),207(碱)
NMR(CDCl3)δ:0.96(3H,t),2.22(3H,s),3.74(3H,s)
实施例33
向实施例16得到的化合物(0.131g)和甲醇(2ml)的混合物中加入0.5M NaHCO3水溶液(0.53ml)。过5分钟后,混合物减压蒸去溶剂,残留物用非离子吸附树脂(商品名;HP-20,日本Mitsubishi Kasei Corporation制造)柱色谱法纯化并冷冻干燥,得2-正丙基-5-乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯钠盐(0.089g),为粉末。
m.p.>196℃
NMR(DMSO-d6)δ:0.83-0.92(3H,m),1.92和2.12(3H,s),3.27和3.32(3H,s)
实施例34-37
用与实施例33相同的方法处理实施例4、20、21和24得到的化合物,得以下列于表4的化合物。
表4
(Tet:1H-四唑-5-基基团)
实施例38-46
用与实施例8相同的方法处理实施例13、16、20-25和27得到的化合物,得到以下列于表5的化合物。
表5
*:实施例42的化合物是三钠盐
Tet:1H-四唑-5-基基团
实施例47
(1)向2-正丁基-5-乙氧基羰基乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(2.0g),三乙胺(0.69g)和氯仿(20ml)的混合物中加入三苯甲基氯(1.43g),混合物在室温搅拌30分钟。洗涤反应溶液,干燥,并减压蒸发。所得残留物用硅胶柱色谱法(溶剂;乙酸乙酯/正己烷)纯化,得2-正丁基-5-乙氧基羰基乙酰基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(1.29g),为针状。
m.p.124-126℃(分解)
(2)用HPLC(商品名;Chiralcel OD,日本Daical Chemical Zndustries,Ltd.制造)柱色谱法(溶剂∶正己烷/乙醇=7∶3)分离光学活性异构体以光学拆分上述产物,得分离的(+)-异构体和(-)-异构体。
(+)-异构体:
[α]D:-25.2°(C=0.5,氯仿,25℃)
(-)-异构体:
[α]D:-22.8°(C=0.5,氯仿,25℃)
实施例48
向(+)-2-正丁基-5-乙氧基羰基乙酰基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(395mg)的四氢呋喃(4ml)的混合物中在冰冷却下加入90%甲酸(8ml),混合物在室温下搅拌30分钟,减压蒸去溶剂。所得残留物用硅胶柱色谱法(溶剂;氯仿/甲醇)纯化,得(+)-2-正丁基-5-乙氧基羰基乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯(270mg),为泡沫状物。
[α]D:+70.4°(C=0.5,氯仿,20℃)
实施例49
(-)将-2-正丁基-5-乙氧基羰基乙酰基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯用与实施例48相同的方法处理得到(-)-2-正丁基-5-乙氧基羰基乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯,为泡沫状物。
[α]D:-70.8°(C=0.5,氯仿,25℃)
实施例50
2-正丙基-5-(2-氨乙基)-3-[2′-(1-三苯甲-1H-四唑-5-基)联苯-4-基]甲基咪唑(1.01g)溶于四氢呋喃(10ml)中,向其中加入水合二羟乙酸乙酯(195mg),混合物在室温搅拌过夜。再在50℃加热30分钟,冷却后,向混合物加入7%盐酸-甲醇溶液(3ml)和氯仿(10ml),回流20分钟。混合物减压蒸发除去溶剂,所得残留物溶于氯仿(30ml)。向混合物中加入乙酸酐(290mg)和在水(20ml)中的NaCHO3(1.2g),混合物在室温搅拌过夜。混合物用柠檬酸酸化,并用氯仿萃取。水洗萃取液,干燥,蒸发。向残留物加入富马酸(100mg)和乙醇(35ml),混合物回流5小时。然后,减压蒸馏反应混合物,用甲醇/乙醚重结晶混合物,得2-正丙基-5-乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酸1/2富马酸盐(728mg)。
m.p.184-185℃
产率:80%
实施例51
向2-正丙基-4-(2-氨乙基)-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑(21.95g)和四氢呋喃(200ml)的混合物中在5℃加入水合二羟乙酸乙酯(4.25g)在四氢呋喃(20ml)中的溶液。混合物在室温搅拌过夜,回流30分钟。在室温向混合物加入8%HCl乙醇溶液(100ml)。反应混合物搅拌30分钟,然后蒸发。将残留物溶于氯仿并用饱和NaHCO3溶液和盐水充分洗涤。干燥有机层并蒸发。将该草酸盐在乙醇中重结晶得2-正丙基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯草酸盐(10.84g)。
m.p.140-142℃
NMR(DMSO-d6)δ:0.84(3H,t),4.95(1H,s)5.29(2H,brs)
游离羧酸
NMR(CDCl3):δ:1.00(3H,t),3.98(1H,s)5.09(2H,q)
实施例52-59
用与实施例4或5相同的方法处理实施例11得到的化合物和相应的起始化合物,得以下表6和表7列出的化合物。
表6
(Tet:1H-四唑-5-基基团)
表7
(Tet:1H-四唑-5-基基团)
实施例60
用与实施例32相同的方法用乙醇代替甲醇处理2-正丙基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.00g),得2-正丙基-5-乙酰乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.00g),为白色泡沫状物。
NMR(CDCl3)δ:1.03(3H,t),2.22(3H,s),3.61(2H,ABq),5.30(2H,ABq),5.36(1H,s)
实施例61
在冰冷却下向2-正丙基-5-乙酰乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(200mg),MgCl2(34mg),吡啶(58μl)和乙腈(2ml)的混合物中加入苯甲酰氯(42μl)。反应混合物在室温搅拌过夜,然后用氯仿(50ml)稀释。用10%HCl和盐水洗涤溶液,干燥,蒸馏得粗产物(266mg),为油状物。将上述产物(260mg)与在乙醇(5.0ml)中的10%HCl(1.0ml)回流1小时。反应混合物用氯仿(30ml)稀释,用盐水洗涤,干燥,然后蒸发。用硅胶柱色谱法(溶剂;氯仿/乙醇)纯化,得2-正丙基-5-苯甲酰基乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(144mg),为白色泡沫。
FAB-MS(m/z):618(MH+),207(碱)
NMR(CDCl3)δ:1.04(3H,t),4.00-4.28(5H,m),5.30(2H,s),5.40(1H,s)
实施例62
在室温下向2-正丙基-4-(2-氨乙基)-1-[2′-(叔丁氧基羰基)联苯-4-基]甲基咪唑(10.0g)和四氢呋喃(100ml)的混合物中加入水合二羟乙酸乙酯(2.90g)在四氢呋喃中的溶液。将反应混合物搅拌过夜,再回流30分钟,蒸发。残留物溶于氯仿中,用2%HCl溶液,饱和NaHCO3溶液和盐水洗涤溶液。干燥有机层并蒸馏。粗产物与草酸在乙醇-乙醚中重结晶,得2-正丙基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯草酸盐(8.45g)。
m.p.166-168℃
NMR(DMSO-d6)δ:0.87(3H,t),1.25(9H,s),5.10(1H,s),5.40(2H,s)
实施例63
将实施例62得到的化合物(1.00g)悬浮于氯仿中。用饱和NaHCO3和盐水洗涤悬浮液,用MgSO4干燥并蒸发。向粗产物(0.75g),乙氧基羰基乙酸(0.33g)和二氯甲烷(10ml)中在室温加入盐酸1-乙基-3-(3-二甲氨基丙基)碳二亚胺(0.48g)。将反应混合物搅拌1小时,并用10%柠檬酸溶液,饱和NaHCO3溶液和盐水洗涤。干燥有机层并蒸发。粗产物用硅胶柱色谱法(溶剂;氯仿/乙酸乙酯)纯化,得2-正丙基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-5-乙氧基羰基乙酰基-4-羧酸乙酯(0.70g)。
FAB-MS(m/z):618(MH+),211(碱)
NMR(CDCl3)δ:1.12(3H,t),1.28(9H,s),3.57(2H,s),5.37(2H,ABq)
实施例64
在室温下向实施例62得到的化合物(1.00g),NaHCO3(1.41g),氯仿(20ml)和水(10ml)的混合物中加入乙酸酐(516mg)。混合物搅拌过夜。分离有机层,用盐水洗涤,干燥,蒸馏。用硅胶柱色谱法(溶剂∶氯仿/甲醇=20∶1)得到2-正丙基-5-乙酰基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.90g)为白色泡沫状物。
FAB-MS(m/z):546(MH+),211(碱)
NMR(CDCl3)δ:0.94(3H,t),1.28(9H,s),5.37(2H,ABq),6.02(1H,s)
实施例65
用与实施例64相同的方法,只是用丙酰氯(0.23g)代替乙酸酐处理实施例62得到的化合物(1.0g),得2-正丙基-5-丙酰基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.82g),为白色泡沫。
FAB-MS(m/z):560(MH+),211(碱)
NMR(CDCl3)δ:0.94(3H,t),1.12(3H,t),1.18(3H,t),1.29(9H,s)
实施例66
用与实施例64相同的方法,只是用氯甲酸乙酯(0.27g)代替乙酸酐处理实施例62所得的化合物(1.00g),得到2-正丙基-5-乙氯基羰基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.82g),为白色泡沫状物。
FAB-MS(m/z):576(MH+),211(碱)
NMR(CDCl3)δ:0.95,0.96(3H,t),1.12-1.31(15H,m),5.22-5.60(3H,m)
实施例67
将2-正丙基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-5-乙氧基羰基乙酰基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(657mg),三氟乙酸(3ml)和二氯甲烷(10ml)的混合物在室温搅拌过夜。用饱和NaHCO3溶液和盐水洗涤反应混合物,干燥,蒸发。粗产物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得到2-正丙基-3-(2′-羧酸联苯-4-基)甲基-5-乙氧基羰基乙酰基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(516mg)。
FAB-MS(m/z):562(MH+),211(碱)
NMR(CDCl3)δ:0.76(3H,t),3.35(2H,s),5.34(2H,ABq)
实施例68-70
用与实施例67相同的方法处理实施例64到66得到的化合物得以下列于表8的化合物。
表8
实施例71
将2-乙基-4-(2-氨乙基-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑(7.48g)溶于四氢呋喃(60ml)中,向其中加入水合二羟乙酸乙酯(1.56g)。反应混合物在室温搅拌过夜,然后回流1小时。蒸去溶剂,残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化得2-乙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(5.54g),为泡沫状物表现成它的草酸盐的特性。
m.p.142-146℃
NMR(DMSO-d6)δ:4.93(1H,s),5.23(2H,s)
实施例72
2-乙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.94g),丙二酸单乙酯(0.74g),盐酸1-(3-二甲氨基丙基)-3-乙基碳二亚胺(0.80g),三乙胺(1.40g)在二氯甲烷(20ml)中的溶液在室温搅拌过夜。水洗反应混合物,用Na2SO4干燥,然后蒸发。残留物溶于乙醇(30ml),加入富马酸(2.00g),并将溶液回流3小时。蒸去溶剂,用饱和NaHCO3溶液处理残留物,然后用氯仿萃取。干燥有机层并蒸馏。用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得2-乙基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-5-乙氧基羰基乙酰基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.07g),为泡沫状物。
NMR(CDCl3)δ:5.29(2H,s),5.48(1H,s),6.92(2H,d),7.10(2H,d)
实施例73
2-乙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.51g),乙酸酐(0.44g),NaHCO3(1.09g),氯仿(18ml)和水(18ml)的混合物在室温搅拌过夜。用氯仿萃取水层,并将合并的有机层用柠檬酸水溶液和盐水洗涤,干燥,蒸馏。向残留物(1.46g)加入富马酸(1.2g)和乙醇(20ml),混合物回流4小时,然后蒸发。残留物用饱和NaHCO3水溶液洗涤,然后用氯仿萃取。干燥有机层并蒸发。用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得2-乙基-5-乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.76g),为白色泡沫状物。
FAB-MS(m/z):500(M+1碱),207
NMR(CDCl3)δ:1.30(3H,t),2.16(3H,s),5.30(2H,s),5.45(1H,s)
实施例74
向2-乙基-4-(2-氨乙基)-1-[2′-(叔丁氧基羰基)联苯-4-基]甲基咪唑(5.30g)在四氢呋喃(40ml)中的溶液中加入水合二羟乙酸乙酯(1.67g)。反应混合物在室温搅拌过夜,然后回流两小时。蒸去溶剂,残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得2-乙基-3-[2′-(叔丁氧羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(5.90g),为黄色油状物。
FAB-MS(m/z):490(M+1),211(碱),
NMR(CDCl3)δ:1.22(3H,t),1.28(9H,s),1.31(3H,t),4.36(1H,s)
实施例75
2-乙基-3-[2′-叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(2.26g),乙酸酐(0.94g),NaHCO3(2.23g),氯仿(18ml)和水(18ml)的混合物在室温搅拌过夜。分离有机层,盐水洗涤,干燥,蒸馏。残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得2-乙基-5-乙酰基-3-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(2.10g),为白色泡沫状物。
FAB-MS(m/z):532(M+1),211(碱)
NMR(CDCl3)δ:1.13(3H,t),1.28(9H,s),2.22(3H,t),6.05(1H,s)
实施例76
将2-乙基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.72g),丙二酸单乙酯(0.94g),盐酸1-(3-二甲氨基丙基)-3-乙基碳二亚胺(1.02g),三乙胺(1.78g)在二氯甲烷(20ml)中的溶液在室温下搅拌过夜。水洗反应混合物,用Na2SO4干燥然后蒸发。残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得2-乙基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-5-乙氧基羰基乙酰基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.57g),为油状物。
NMR(CDCl3)δ:1.12(3H,t),1.28(9H,s),5.35(2H,q),6.00(1H,s)
实施例77
向上述产物(2.05g)在二氯甲烷(20ml)中的溶液中加入三氟乙酸(6ml)。反应混合物在室温下搅拌过夜,然后蒸发。残留物溶于氯仿并用饱和NaHCO3溶液洗涤。用Na2SO4干燥有机层,然后浓缩得到粗产物,用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得到2-乙基-3-(2′-羧基联苯-4-基)甲基-5-乙氧基羰基乙酰基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.17g),为白色泡沫状物。
NMR(CDCl3)δ:0.98(3H,t),1.16(3H,s),1.27(3H,t),5.33(2H,q),6.00(1H,s)
实施例78
用与实施例67相同的方法处理实施例75得到的化合物(2.05g),得2-乙基-5-乙酰基-3-(2′-羧基联苯-4-基)甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.53g),为白色泡沫状物。
FAB-MS(m/z):476(MH+1),211(碱)
NMR(CDCl3)δ:1.00(3H,t),2.20(3H,s),6.00(1H,s),6.95(2H,d)
实施例79
将2-正丙基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.20g),K2CO3(0.082g),烯丙基溴(0.057g)和四氢呋喃(2ml)的混合物在室温搅拌过夜。向混合物加入氯仿,洗涤混合物,干燥并蒸发。残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得2-正丙基-5-烯丙基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.128g)。
FAB-MS(m/z):544(M+1),211(碱)
NMR(CDCl3)δ:0.96(3H,t),1.19(3H,t),1.30(9H,s),4.25(1H,s)
实施例80
向2-正丙基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.65g),K2CO3(0.533g)和二甲基甲酰胺(6ml)的混合物加入苄基溴(0.33g)。混合物在室温搅拌1小时,然后用乙酸乙酯稀释。水洗溶液,干燥,并蒸发。残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得2-正丙基-5-苄基-3-[2′-(叔丁氧基羰基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.49g)。
FAB-MS(m/z):594(M+1),91(碱)
NMR(CDCl3)δ:0.96(3H,t),1.16(3H,t),1.28(9H,s),4.22(1H,s)
实施例81
用与实施例67相同的方法处理实施例79得到的化合物,得2-正丙基-5-烯丙基-3-(2′-羧基联苯-4-基)甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯。
FAB-MS(m/z):488(M+1),43(碱)
NMR(CDCl3)δ:0.74(3H,t),1.18(3H,t),4.34(1H,s)
实施例82
用与实施例67相同的方法处理实施例80得到的化合物,得2-正丙基-5-苄基-3-(2′-羧基联苯-4-基)甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯。
FAB-MS(m/z):538(M+1),91(碱)
NMR(CDCl3)δ:0.77(3H,t),1.09(3H,t),3.63(1H,d),3.76(1H,d),4.18(1H,s)
实施例83
2-正丙基-4-(2-氨乙基)-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑(6.92g),水合二羟乙酸乙酯(1.30g)和四氢呋喃(70ml)的混合物在室温搅拌过夜。蒸发混合物,残留物用硅胶柱色谱法(溶剂:氯仿/乙醇)纯化,)得2-正丙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(5.32g),为泡沫状物。
FAB-MS(m/z):714(M+1),243(碱)
NMR(CDCl3)δ:0.90(3H,t),1.21(3H,t),1.92(brs),4.17(1H,s),5.10(2H,ABq)
实施例84
2-正丙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(2.00g),K2CO3(1.16g),乙基溴(0.61g)和二甲基甲酰胺(10ml)的混合物在室温搅拌过夜。混合物用乙酸乙酯稀释,水洗溶液,干燥并蒸发。残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得5-乙基-2-正丙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.12g),为白色泡沫状物。
FAB-MS(m/z):742(M+1),243(碱)
NMR(CDCl3)δ:0.88(3H,t),0.97(3H,t),1.15(3H,t),4.15(1H,s)
实施例85
2-正丙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(2.00g),K2CO3(1.16g),苄基溴(0.72g)和二甲基甲酰胺(10ml)的混合物在冰冷却下搅拌2小时。混合物用乙酸乙酯稀释,水洗溶液,干燥并蒸发。残留物用硅胶柱色谱法(溶剂:正己烷/乙酸乙酯)纯化,得-5-苄基-2-正丙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.50g),为白色泡沫状物。
FAB-MS(m/z):804(M+1),243(碱)
NMR(CDCl3)δ:0.87(3H,t),1.13(3H,t),3.64(1H,d),3.77(1H,d),4.13(1H,s)
实施例86
5-乙基-2-正丙基-3-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(1.10g),富马酸(1.2g)和乙醇(20ml)的混合物回流1小时。蒸发混合物,残留物溶于氯仿,用饱和NaHCO3和盐水洗涤混合物,干燥并蒸馏。残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得5-乙基-2-正丙基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(0.65g),为白色泡沫状物。
FAB-MS(m/z):500(M+1),43(碱)
NMR(CDCl3)δ:0.88(3H,t),0.99(3H,t),1.05(3H,t),4.05(1H,s)
实施例87
用与实施例86相同的方法处理实施例85所得的化合物,得到5-苄基-2-正丙基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯。
FAB-MS(m/z):562(M+1),91(碱)
NMR(CDCl3)δ:0.89(3H,t),1.03(3H,t),3.54(1H,d),3.68(1H,d),4.02(1H,s)
实施例88-102
用与实施例8相同的方法处理实施例52到56、59、68到70、73、78、81、82、86和87所得的化合物,得到以下列于表9到12中的化合物。
表9
(Tet:1H-四唑-5-基基团)
Tet:1H-四唑-5-基基团
Tet:1H-四唑-5-基基团
实施例103
向2-正丙基-5-乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸乙酯(50.0g)在甲醇(500ml)中的溶液在冰冷却下加入4N NaOH水溶液(50ml)。混合物在室温搅拌过夜,然后蒸发。残留物从乙醇重结晶,得2-正丙基-5-乙酰基-3-[2′-(1H-四唑-5-基)联苯-4-基]甲基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸二钠盐(44.3g)。
m.p.>300℃
FAB-MS(m/z):552(M+Na),530(M+1),177(碱)
NMR(D2O)δ:0.81-0.93(3H,m),1.67,2.02(3H,每个s),4.53,5.44(1H,每个s)
参考实施例1
用与实施例5相同的方法处理4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯,得5-二苯基乙酰基-4,5,6,7-四氢咪唑并[4,5-c]吡啶-4-羧酸甲酯,为淡黄色泡沫状物。
NMR(CDCl3)δ:3.68(3H,s),6.07(1H,s)
参考实施例2
(1)1-叔丁氧基羰基-4-[2-(叔丁氧基羰基氨基)乙基]咪唑(78.1g)溶于乙腈(500ml)中,并向其中加入甲氧基氯甲烷(22.2g)。混合物在室温搅拌过夜,倒入10%Na2CO3水溶液中,用乙酸乙酯萃取。洗涤萃取液,干燥并蒸馏,得5-[2-(叔丁氧基羰基氨基)乙基]-1-甲氧基甲基咪唑(54.4g),为油状物。
NMR(CDCl3)δ:1.43(9H,s),3.27(3H,s),5.20(2H,s)
(2)将上述化合物55g)溶于四氢呋喃(1.5升),混合物冷却至-40℃。向混合物滴加1.6M正丁基锂/正己烷溶液(150ml),混合物搅拌30分钟。向混合物加入六甲基磷酰胺(150ml),再加入正丁基锂(137ml),在-30℃向混合物滴加正丁基碘(37.5g)。将混合物搅拌10分钟,用氯化铵水溶液终止反应。向反应混合物加入乙酸乙酯,收集有机层,洗涤,干燥并蒸去溶剂。所得油状残留物用硅胶柱色谱法(溶剂:氯仿/乙酸乙酯/甲醇=32∶8∶1)纯化,得5-[2-(叔丁氧基羰基氨基)乙基]-2-正丁基-1-甲氧基甲基咪唑(44.8g),为油状。
NMR(CDCl3)δ:0.94(3H,t),1.44(9H,s),3.27(3H,s),5.09(2H,s)
(3)将上述化合物(80.7g),氯甲酸乙酯(84.5g)和氯仿(1.3升)的混合物回流2.5小时,并蒸去溶剂。向残留物加入乙醇(300ml)和10%NaOH水溶液(200ml),混合物在冰冷却下搅拌20分钟。蒸去溶剂,向残留物加入氯仿和水。将氯仿层干燥并蒸发。所得残留物从异丙醚重结晶,得4-[2-(叔丁氧基羰基氨基)乙基]-2-正丁基咪唑(50.3g)。
m.p.118-120℃
(4)用与实施例1相同的处理方法上述化合物,得到4-[2-(叔丁氧基羰基氨基)乙基]-2-正丁基-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑。
NMR(CDCl3)δ:0.89(3H,t),1.43(9H,s),4.85(2H,s)
(5)上述化合物(15.2g),10%HCl(40ml)和甲醇(60ml)的混合物回流1小时。反应完成后,混合物蒸去甲醇,洗涤水层并减压浓缩至干。所得残留物经与干甲苯共沸蒸馏,得粗的盐酸2-正丁基-4-(2-氨乙基)-1-[2′-(1H-四唑-5-基)联苯-4-基]甲基咪唑(9.7g),为酱色状物。
FAB-MS(m/z):402(M+H)(碱)
NMR(DMSO-d3)δ:0.84(3H,t),1.43(9H,s),5.40(2H,s)
参考实施例3
(1)2-丙基-4-羟甲基咪唑(2.61g)加入到亚硫酰氯(4.5ml)中,混合物在50℃加热2小时。蒸去溶剂,将残留物溶于二甲基甲酰胺(20ml)中,并将混合物滴加到氰化钠(5.47g)在二甲基甲酰胺(120ml)中的溶液中。混合物在室温下搅拌过夜,蒸去溶剂。向所得残留物加乙酸乙酯,洗涤混合物,干燥并蒸发。残留物用硅胶柱色谱法(溶剂:乙酸乙酯)纯化,得到2-正丙基-4-氰基甲基咪唑(3.08g),为油状物。
NMR(CDCl3)δ:0.95(3H,t),3.67(2H,d)
(2)将上述产物(3.08g)溶于乙酸(30ml)中,向其中加入10%HCl(10ml)。混合物用氧化铂作催化剂催化加氢。反应完成后,过滤除去氧化铂,减压蒸发滤液,得盐酸2-正丙基组胺(4.83g)。
(3)上述化合物(4.83g),邻苯二甲酸酐(3.04g),乙酸钠(6.10g)和乙酸(50ml)的混合物回流19小时。减压蒸发该混合物,向所得残留物加水。用NaHCO3中和混合物,并用氯仿萃取。干燥萃取液,蒸发,所得残留物用硅胶柱色谱法(溶剂∶氯仿/甲醇=20∶1)纯化,得2-正丙基-4-(2-邻苯二甲酰亚胺基乙基)咪唑。
m.p.137-139℃
NMR(CDCl3)δ:0.90(3H,t),3.95(2H,t),7.6,7,86(4H,m)
参考实施例4
以实施例如同样的方法处理参考实施例子所得的化合物,得2-正丙基-4-(2-邻苯二甲酰亚胺基乙基)-1-〔2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基〕甲基咪唑为泡沫状物
NMR(CDCl3)δ:0.86(3H,t)4.82(2H,s).
参考实施例5
向参考实施例4所得化合物(4.11g)和乙醇(100ml)的混合物中加10%水合肼(2ml),混合物在室温下搅拌5小时。反应完毕后,向反应混合物加入氯仿,洗涤混合物,干燥并蒸发,得粗2-正丙基-4-(2-氨乙基)-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑(3.68g),为油状。
参考实施例6
用与实施例1相同的方法处理4-[2-(叔丁氧基羰基氨基)乙基]-2-正丁基咪唑(3.0g)和4′-溴甲基二苯基-2-羧酸甲酯(3.75g),得到2-正丁基-4-(2-叔丁氧基羰基氨基乙基)-1-[2′-甲氧基羰基联苯-4-基]甲基咪唑(2.65g),为油状物。
EI-MS(m/z):491(M+),225(碱)
NMR(CDCl3)δ:0.91(3H,t),3.66(3H,s),5.02(2H,s)
参考实施例7
将参考实施例6所得的化合物,10%HCl(40ml)和甲醇(60ml)的混合物回流1小时。反应完毕后,混合物蒸去甲醇,洗涤水层并减压浓缩至干。所得残余物经与干甲苯共沸蒸馏得粗盐酸2-正丁基-4-(2-氨乙基)-1-(2′-甲氧基羰基联苯-4-基)甲基咪唑。
参考实施例8
丁脒(5.0g)和K
2CO
3(11.4g)悬浮在乙腈(100
℃-90℃加热。在搅拌下向混合物中滴加1-
酮(10.0g)在乙腈(200ml)中的溶液,
,滤去不溶物,浓缩滤液。所得残留
4干燥,蒸馏。向残留物加入富马酸,
到2-正丙基-4-(2-邻苯二甲酰亚胺基
9.9g)。
酸酯(13.7g)悬浮于乙酸乙酯/水中,向其中
g),得到2-正丙基-4-(2-邻苯二甲酰亚胺基乙基)咪唑(9.24g)。向该产物(9,24g)和4-(2′-氰基苯基)苄基溴(9.3g)在四氢呋喃(150ml)和二甲基甲酰胺(10ml)中的溶液中在50℃滴加叔丁基氧化钾(3.84g)在四氢呋喃中的溶液。将混合物冷却至20℃,搅拌2小时。用NH4Cl水溶液终止反应,用乙酸乙酯萃取混合物,水洗乙酸乙酯层,用Na2SO4干燥,蒸发得到无色油状产物(15.3g)。将该产物溶于乙醇中,向其中加入草酸,用乙醇重结晶产物,得2-正丙基-4-(2-邻苯二甲酰亚胺基乙基)-1-(2′-氰基联苯-4-基)甲基咪唑草酸盐(13.44g)。
m.p.162-166℃
NMR(DMSO-d6)δ:0.82(3H,t),2.60-3.10(4H,m),5.34(2H,s)
(3)2-正丙基-4-(2-邻苯二甲酰亚胺基乙基)-1-(2′-氰基联苯-4-基)甲基咪唑(0.5g)和三丁基叠氮化锡(0.70g)的混合物在110℃加热过夜。向反应混合物加8%HCl乙醇溶液(5ml),在室温下搅拌溶液30分钟,然后蒸发。将残留物溶于水(30ml),用乙醚洗涤溶液。用NaHCO3中和水层,并用氯仿萃取。干燥氯仿溶液,蒸馏。向残留物(575mg)加入三苯基氯甲烷(0.38g),三乙胺(0.20ml)和氯仿(5.0ml)。溶液在室温搅拌2小时,然后用饱和碳酸氢钠溶液和盐水洗涤,用Na2SO4干燥,蒸发。向残留物加入草酸(0.10g),混合物用乙醇重结晶,得到2-正丙基-4-(2-邻苯二甲酰亚胺基乙基)-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑草酸盐(0.70g)。
m.p.112℃(烧结)
参考实施例9
向1-(二乙氧基)甲基咪唑(260g)在四氢呋喃(5升)中的溶液中在-45℃下滴加入1.6M的正丁基锂在己烷(1升)中的溶液。30分钟后,在相同温度下向混合物滴加正丁基碘(294g)。反应混合物在室温搅拌过夜然后蒸发。向残留物加乙醚(2升),用10%HCl萃取该溶液。水溶液用10%NaOH溶液碱化,然后用氯仿萃取。洗涤有机层,干燥并蒸发。向粗产品(204g),三乙胺(170g)和氯仿(2升)在冰冷却下滴加二甲基氨磺酰氯(200g)在氯仿(200ml)中的溶液。混合物在室温搅拌过夜,以盐水洗涤,干燥,蒸发。并蒸馏纯化粗产品,得2-正丁基-1-二甲基氨磺酰基咪唑(249.4g)。
b.p.124℃(1mmHg)
参考实施例10
用与参考实施例9相同的方法处理1-(二乙氧基)甲基咪唑和正丙基锂,得到2-正丙基-1-二甲基氨磺酰基咪唑。
b.p.141-143℃(3mmHg)
参考实施例11
在0℃下向搅拌的2-乙基咪唑(100g)和三乙胺(115g)在氯仿(800ml)中的溶液加入二甲基氨磺酰氯(153g)在氯仿(200ml)中的溶液。混合物在室温搅拌过夜。向反应混合物加水(1.5升),然后分离有机层并浓缩。将残留物溶于乙酸乙酯(1升)并水洗,然后用Na2SO4干燥并浓缩。蒸馏得到1-二甲基氨磺酰基-2-乙基咪唑(182g),为无色液体。
b.p.139-142℃(5mmHg)
NMR(CDCl3)δ:1.37(3H,t),2.89(6H,s),6.94(1H,d),7.23(1H,d)
参考实施例12
在-78℃向上述产品(53g)在四氢呋喃(1升)中的溶液中加入1.6M的正丁基锂在己烷(185ml)中的溶液。在-78℃搅拌溶液1小时,向其中加入1-叔丁氧基羰基氮丙啶(52g)在四氢呋喃(300ml)中的溶液,再向其中加入乙醚合三氟化硼(147g)。在-78℃再搅拌反应混合物2小时,然后倒入冰冷却的饱和K2CO3水溶液(2升)中。蒸去剩余的四氢呋喃后,用乙酸乙酯萃取水层,水洗并用Na2SO4干燥,然后浓缩。残留物用硅胶柱色谱法(溶剂:氯仿/甲醇)纯化,得到1-二甲基氨磺酰基-2-乙基-5-[2-(叔丁氧基羰基氨基)]乙基咪唑(67g),为黄色油状物。
NMR(CDCl3)δ:1.35(3H,t),1.43(9H,s),2.87(6H,s),6.72(1H,s)
参考实施例13
上述产物(67g)在10%HCl(600ml)中的溶液回流2小时。减压蒸去溶剂,将所得油状黑色残留物溶于乙酸(300ml)中。加入NaOAc(62g)和邻苯二甲酸酐(34g)后,反应混合物回流过夜。减压浓缩反应混合物,将残留物在丙酮(300ml)中研制,得到粗的2-乙基-4-(2-邻苯二甲酰亚胺基乙基)咪唑(26g),为白色粉末。该产物用于下面的反应,无需进一步纯化。
参考实施例14
在-60℃下向上述产物(6.56g)和2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基甲基溴(16.3g)在四氢呋喃(100ml)二甲基甲酰胺(50ml)中的溶液中加入正丁基氯化钾(3.01g)。在5小时内让反应混合物缓慢暖至室温,然后倒入盐水(2升)中,用乙酸乙酯萃取,用盐水洗涤并用Na2SO4干燥。浓缩后,残留物用硅胶柱色谱法(溶剂;己烷/乙酸乙酯)纯化,得2-乙基-4-(2-邻苯二甲酰亚胺基乙基)-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑(10.69g),为白色泡沫状物,系以其富马酸盐表示特性。
m.p.173-174℃
NMR(DMSO-d6)δ:0.92(3H,t),2.35(2H,q),2.73(2H,t),3.78(2H,t),4.99(2H,s)
参考实施例15
在0℃向上述产物(10.69g)在乙醇(150ml)和四氢呋喃(90ml)中的溶液中加入水合肼(6.25g)。反应混合物在室温搅拌过夜,用硅藻土垫过滤并蒸发。用0.5N NaOH溶液(300ml)处理残留物并用氯仿萃取。用No2SO4干燥有机层并浓缩得到粗的2-乙基-4-(2-氨乙基)-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑(7.48g),为泡沫状物。使用该产物无需进一步纯化。
参考实施例16
在-60℃下向2-乙基-4-(2-邻苯二甲酰亚胺基乙基)咪唑(9.00g)和2′-(叔丁氧基羰基)联苯-4-基甲基溴(13.93g)在四氢呋喃(150ml)和二甲基甲酰胺(100ml)中的溶液中加入正丁基氯化钾(4.12g)。在4小时内让反应混合物缓慢暖至室温,然后倒入水(100ml)中。减压除去四氢呋喃,然后用乙酸乙酯萃取水层。用盐水洗涤有机层,用Na2SO4干燥,然后蒸发得油状残留物。用硅胶柱色谱法(溶剂:己烷/乙酸乙酯)纯化,得到2-乙基-4-(2-邻苯二甲酰亚胺基乙基)-1-[2′-(叔丁氧基羰基)联苯-4-基]甲基咪唑(9.39g),为油状物。
NMR(CDCl3)δ:1.20(3H,t),1.25(9H,s),5.02(2H,s),6.64(1H,s)
参考实施例17
用与参考实施例15相同的方法处理参考实施例16所得化合物,得到2-乙基-4-(2-氨乙基)-1-[2′-(叔丁氧羰基)联苯-4-基]甲基咪唑。
FAB-MS(m/s):406(M+1),211(碱)
NMR(CDCl3)δ:1.26(9H,s),1.29(3H,t),5.06(2H,s),6.60(1H,s)
参考实施例18
用与参考实施例12相同的方法处理2-正丁基-1-二甲氨磺酰基咪唑(46.0g),得2-正丁基-1-二甲氨磺酰基-5-[2′-(叔丁氧基羰基)氨乙基]咪唑(69g),为油状物。
NMR(CDCl3)δ:0.95(3H,t),1.43(9H,s),2.86(6H,s)
参考实施例19
用与参考实施例15相同的方法处理2-正丁基-1-二甲氨磺酰基-5-[2′-(叔丁氧基羰基)氨乙基]咪唑(15.3g),得2-正丁基-4-(2-邻苯二甲酰亚胺乙基)咪唑(64g)。
m.p.114-117℃
NMR(CDCl3)δ:0.88(3H,t),2.66(2H,t),2.97(2H,t),3.95(2H,t),6.67(1H,s)
参考实施例20
用与参考实施例14相同的方法处理参考实施例19所得的化合物,得2-正丁基-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基-4-(2-邻苯二甲酰亚胺乙基)咪唑。
NMR(CDCl3)δ:0.84(3H,t),4.81(2H,s)
参考实施例21
将2-正丁基-4-(2-邻苯二甲酰亚胺乙基)-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基唑唑(96.0g),水合肼(44.7g)和乙醇(1.3升)的混合物在室温搅拌过夜,然后用硅藻土垫过滤并蒸发。用5%NaOH溶液(200ml)处理残留物并用氯仿萃取。干燥有机层并浓缩得到粗的2-正丁基-4-(2-氨乙基)-1-[2′-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲基咪唑(84.0g)。向上述产物(84.0g)在甲醇中的溶液中加入24%的HCl甲醇溶液(200ml),并蒸发混合物。向残留物加水(2升),并用乙醚洗涤混合物。浓缩水层得到盐酸2-正丁基-4-(2-氨乙基)-1-[2′-(1-四唑-5-基)联苯-4-基]甲基咪唑(52.8g),为泡沫状物。
NMR(DMSO-d6)δ:0.83(3H,t),2.92(2H,t)5.41(2H,s)
参考实施例22
用与参考实施例12相同的方法处理2-正丙基-1-二甲氨磺酰基咪唑(20.0g)得2-正丙基-1-二甲氨磺酰-5-(2-叔丁氧基羰基氨基乙基)咪唑(24.4g),为油状物。
NMR(CDCl3)δ:1.01(3H,t),1.43(9H,s),2.86(6H,s),6.71(1H,s)
参考实施例23
用与参考实施例15相同的方法处理参考实施例22所得的产物,得到2-正丙基-4-(2-邻苯二甲酰亚胺基乙基)咪唑。
m.p.137-139℃
NMR(CDCl3)δ:0.90(3H,t),3.95(2H,t),6.68(1H,s)
参考实施例24
在-60℃向2-正丙基-4-(2-邻苯二甲酰亚胺乙基)咪唑(10.0g),2′-(叔丁氧基羰基)联苯-4-基甲基溴(13.5g),四氢呋喃(150ml)和二甲基甲酰胺(15ml)的混合物中加入正丁基氯化钾(4.16g)在四氢呋喃(40ml)中的溶液。使混合物缓慢暖至室温,然后用水终止反应。用乙酸乙酯萃取混合物,洗涤有机层,干燥并蒸馏。用草酸从乙醇/乙醚重结晶粗产物,得到2-正丙基-4-(2-邻苯二甲酰亚胺乙基)-1-[2′-(叔丁氧基羰基)联苯-4-基]甲基咪唑草酸盐(15.6g)。
m.p.128-131℃
NMR(DMSO-d6)δ:0.84(3H,t),1.19(9H,s),5.30(2H,s)
参考实施例25
在室温下向2-正丙基-4-(2-邻苯二甲酰亚胺乙基)-1-[2′-(叔丁氧基羰基)联苯-4-基]甲基咪唑(15.5g)和乙酸(150ml)的混合物中加入水合肼(8.45g)。混合物搅拌过夜,滤去沉淀。蒸发滤液,将残留物溶于氯仿并用3%NaOH溶液和盐水洗涤。干燥有机层并蒸馏,得到粗的2-正丙基-4-氨乙基-1-[2′-(叔丁氧基羰基)联苯-4-基]甲基咪唑(10.0g)。该化合物用于下一步,不必进一步纯化。