CN1049500A - 苯并吡喃化合物,其生产方法及药物组合物 - Google Patents
苯并吡喃化合物,其生产方法及药物组合物 Download PDFInfo
- Publication number
- CN1049500A CN1049500A CN90107121A CN90107121A CN1049500A CN 1049500 A CN1049500 A CN 1049500A CN 90107121 A CN90107121 A CN 90107121A CN 90107121 A CN90107121 A CN 90107121A CN 1049500 A CN1049500 A CN 1049500A
- Authority
- CN
- China
- Prior art keywords
- formula
- alkyl
- compound
- cyano group
- dihydro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/58—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4
- C07D311/70—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4 with two hydrocarbon radicals attached in position 2 and elements other than carbon and hydrogen in position 6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/58—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4
- C07D311/68—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4 with nitrogen atoms directly attached in position 4
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Steroid Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
式(I)的化合物或其药学上合格的盐。
其中:
R1为氢原子,取代或未取代的C1-6烷基,C2-6链
烯基,C2-6炔基,或R7-CO-基团(其中R7是C1-6烷
基,苯基,可被苯基取代的C2-6链烯基,或C1-6烷氧
基);
R2为氢原子,取代的或未取代的C1-8烷基,或苯
基;
R3为氢原子,及R4为羟基,或R3和R4一起形
成键;
R5和R6各为C1-4烷基;及
Y为氰基,卤原子,硝基,C1-6烷基,C2-6链烯基,
C1-6烷羰基C1-6烷氧基,二-C1-6烷基氨基羰基,芳
基,C1-6烷氧羰基,羧基或吗啉代羰基。
Description
本发明涉及新颖苯并吡喃化合物,其生产方法,及含这些苯并吡喃化合物作为活性成分的药物组合物。
近年来,随着高龄人口的增长,人们开始注意高血压,它对健康可为很危险的因子。作为治疗高血压的制剂,临床上已使用许多具各种活性机理的药物。从现在起,需要发展一种新的降血压药,它具有钾通道激活作用(具高级活性机理)。
本发明的目的是提供具优秀钾通道激活作用的新颖化合物,因此可用来治疗各种疾病。
本申请的发明人已合成一种新颖的苯并吡喃衍生物,并研究了其钾通道激活作用,发现下式(Ⅰ)的苯并吡喃化合物具优秀的钾通道激活作用,并由于这种活性机理,表现出强的血压降低能力及支气管扩张能力。从而达到了上述的目的。
本发明提供式(Ⅰ)的苯并吡喃化合物:

或其药学上合格的盐,其中:
R1为氢原子,取代或未取代的C1-6烷基,C2-6链烯基,C2-6炔基,或R7-CO-基团(其中R7是C1-6烷基,苯基,可被苯基取代的C2-6链烯基,或C1-6烷氧基);
R2为氢原子,取代的或未取代的C1-8烷基,或苯基;
R3为氢原子,及R4为羟基,或R3和R4一起形成键;
R5和R6各为C1-4烷基;及
Y为氰基,卤原子,硝基,C1-6烷基,C2-6链烯基,C1-6烷羰基,C1-6烷氧基,二-C1-6烷基氨基羰基,芳基,C1-6烷氧羰基,羧基或吗啉代羰基。
本发明还提供一种制备式(Ⅰ)化合物或其药学上合格的盐的方法,它包括:
(1)将式(Ⅱ)的化合物:

其中R1、R5、R6和Y如上所定义,以除去R1为R7-CO-基团,及Y为羧基的情况为条件,与式(Ⅲ)化合物反应:
其中R2如上所述,R8为C1-6烷基,以获得式(Ⅰ)的一种化合物,其中R3为氢原子,R4为羟基;
(2)将式(Ⅰa)化合物:

其中R1、R2、R5、R6和Y均如上所定义,以除去R1为R7-CO-基团,及Y为羧基的情况为条件,与式(Ⅳ)化合物反应:
其中R9为烷基或取代或未取代的芳基,X为卤原子,以形成式(Ⅴ)化合物:

其中R1、R2、R5、R6、R9和Y如上所定义,然后式(Ⅴ)化合物用式(Ⅵ)化合物处理:
其中R10为烷基,M为碱金属,以得到一种式(Ⅰ)的化合物,其中R3和R4一起形成键;
(3)将式(Ⅴ)化合物与1,8-二氮杂二环[5,4,0]-7-十一碳烯反应,以得到一种式(Ⅰ)的化合物,其中R3和R4一起形成键;
(4)使式(Ⅰa)化合物用氢化钠、盐酸或对甲苯磺酸处理以脱水,获得一种式(Ⅰ)的化合物,其中R3和R4一起形成键;
(5)将式(Ⅶ)化合物:

其中R2、R5、R6和Y如上所定义,与式(Ⅷ)化合物反应:
其中R11为取代或未取代的C1-6烷基,C2-6链烯基,C2-6炔基,及X为卤原子,以获得一种式(Ⅰ)化合物,其中R1为取代或未取代C1-6烷基,C2-6链烯基,C2-6炔基,及R3和R4一起形成键;
(6)使式(Ⅶ)化合物与式(Ⅸ)化合物反应:
其中R7和X如前所定义,以获得一种式(Ⅰ)化合物,其中R1为R7-CO-基团,(其中R7如前所定义),R3和R4一起形成键;
(7)水解式(Ⅹ)化合物:

其中R1、R2、R3、R4、R5和R6如前所定义,R12为C1-16烷基,以得到一种式(Ⅰ)化合物,其中Y为羧基;或
(8)将式(Ⅰ)化合物,其中Y为羧基,与吗啉或二烷基胺反应,以获得一种式(Ⅰ)化合物,其中Y为吗啉代羰基,或二-C1-6烷基氨基羰基。
另外,本发明提供具有钾通道激活作用的药物组合物,它含有式(Ⅰ)化合物或其药学上合格的盐作为活性成分。
本发明进一步提供一种降血压或抗气喘药,它含式(Ⅰ)化合物或其药学上合格的盐作为活性成分。
更进一步地,本发明提供C2-6炔基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并(b)吡喃-3-醇作为中间产物,并作为一个新颖化合物。
现在,将参考实施例详细描述本发明。
在代表本发明的苯并吡喃化合物的式(Ⅰ)中,R1、R7和Y的C1-6烷基包括,如甲基、乙基、丙基、异丙基、正丁基、仲丁基、叔丁基、戊基和己基。相似地,R5或R6的C1-4烷基包括,如甲基、乙基、丙基、异丙基、正丁基、仲丁基、叔丁基。R2的C1-8烷基包括,如甲基、乙基、丙基、异丙基、正丁基、仲丁基、叔丁基、戊基、己基、庚基及辛基。R1或R7的C2-6链烯基包括,如烯丙基、甲代烯丙基、丁烯基、1-甲代烯丙基、异戊二烯基、3-甲基-3-丁烯基,及3-戊烯基。R1或Y的C2-6炔基包括,如乙炔基、炔丙基、1-甲代炔丙基、2-丁炔基、1-甲基-2-丁炔基、丁炔基、2-戊炔基及3-戊炔基。R7或Y的C1-6烷氧基包括:如甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、戊氧基及己氧基。
R1的取代C1-6烷基的取代基可为C1-6烷氧基、芳基、羟基、C1-6烷氧羰基或二-C1-6烷基氨基。C1-6烷氧基作为这种取代基包括,如甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、戊氧基及己氧基。芳基包括,如苯基、萘基及二甲苯基。C1-6烷氧羰基包括,如甲氧羰基、乙氧羰基、丙氧羰基、异丙氧羰基、正丁氧羰基、仲丁氧羰基、叔丁氧羰基、戊氧羰基及己氧羰基。在二-C1-6烷基氨基中的C1-6烷基包括,如甲基、乙基、丙基、异丙基、正丁基、仲丁基、叔丁基、戊基和己基。
R2的取代C1-8烷基的取代基可为C1-6烷氧基或芳基。作为此取代基的C1-6烷氧基包括,如甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、戊氧基及己氧基。芳基包括,如苯基、萘基及二甲苯基。
Y的卤原子包括,如氯原子、氟原子、及碘原子。C1-6烷氧羰基包括,如甲氧羰基、乙氧羰基、丙氧羰基、异丙氧羰基、正丁氧羰基、仲丁氧羰基、叔丁氧羰基、戊氧羰基及己氧羰基。C1-6烷基羰基包括,如甲基羰基、乙基羰基、丙基羰基、丁基羰基、戊基羰基及己基羰基。二-C1-6烷基氨基羰基包括,如二甲基氨基羰基及二乙基氨基羰基。芳基包括,如苯基、萘基和二甲苯基。
除在以后的实施例中给出的那些化合物以外,还有以下化合物可被称作式(Ⅰ)的较佳化合物。
(1)6-氰基-2,2-二甲基-4-[(N-氰基-丁酰亚氨基)氨基]-二氢-苯并[b]吡喃
(2)6-氰基-2,2-二甲基-4-[(N-氰基-异丁酰亚氨基)氨基]-二氢-苯并[b]吡喃
(3)6-氰基-2,2-二甲基-4-[(N-氰基-异戊酰亚氨基)氨基]-二氢-苯并[b]吡喃
(4)6-氰基-2,2-二甲基-4-[(N-氰基-甲基乙基乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(5)6-氰基-2,2-二甲基-4-[(N-氰基-新戊酰亚氨基)氨基]-二氢-苯并[b]吡喃
(6)6-氰基-2,2-二甲基-4-[(N-氰基-己酰亚氨基)氨基]-二氢-苯并[b]吡喃
(7)6-氰基-2,2-二甲基-4-[(N-氰基-庚酰亚氨基)氨基]-二氢-苯并[b]吡喃
(8)6-氰基-2,2-二甲基-4-[(N-氰基-2-苯乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(9)6-氰基-2,2-二甲基-4-[(N-氰基-3-苯丙酰亚氨基)氨基]二氢-苯并[b]吡喃
(10)6-氰基-2,2-二甲基-4-[(N-甲基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
(11)6-氰基-2,2-二甲基-4-[(N-甲基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(12)6-氰基-2,2-二甲基-4-[(N-甲基-(N-氰基-丙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(13)6-氰基-2,2-二甲基-4-[(N-甲基-(N-氰基-丁酰亚氨基)氨基]-二氢-苯并[b]吡喃
(14)6-氰基-2,2-二甲基-4-[(N-甲基-(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃
(15)6-氰基-2,2-二甲基-4-[(N-乙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
(16)6-氰基-2,2-二甲基-4-[(N-乙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(17)6-氰基-2,2-二甲基-4-[(N-乙基-(N-氰基-丙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(18)6-氰基-2,2-二甲基-4-[(N-乙基-(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃
(19)6-氰基-2,2-二甲基-4-[(N-丙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
(20)6-氰基-2,2-二甲基-4-[(N-丙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(21)6-氰基-2,2-二甲基-4-[(N-丙基-(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃
(22)6-氰基-2,2-二甲基-4-[(N-氰基-2-甲氧基乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(23)6-氰基-2,2-二甲基-4-[(N-氰基-3-甲氧基丙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(24)6-氰基-2,2-二甲基-4-[(N-丁基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
(25)6-氰基-2,2-二甲基-4-[(N-丁基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(26)6-氰基-2,2-二甲基-4-[(N-苄基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
(27)6-氰基-2,2-二甲基-4-[(N-苄基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(28)6-氰基-2,2-二甲基-4-[(N-烯丙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
(29)6-氰基-2,2-二甲基-4-[(N-烯丙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(30)6-氰基-2,2-二甲基-4-[(N-甲氧基乙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
(31)6-氰基-2,2-二甲基-4-[(N-2-甲氧基乙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(32)6-氰基-2,2-二甲基-4-[(N-炔丙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
(33)6-氰基-2,2-二甲基-4-[(N-炔丙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(34)6-硝基-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(35)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-异丁酰亚氨基]-二氢-苯并[b]吡喃
(36)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-异戊酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(37)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-甲基乙基乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(38)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-新戊酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(39)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-己酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(40)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-庚酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(41)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-2-甲氧基乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(42)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-3-甲氧基丙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(43)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-2-苯基乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(44)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-3-苯基丙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-酮
(45)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-甲基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
(46)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-甲基-(N-氰基-丙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(47)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-甲基-(N-氰基-丁酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(48)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-甲基-(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃-3-醇
(49)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-乙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
(50)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-乙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(51)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-乙基-(N-氰基-丙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(52)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-乙基-(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃-3-醇
(53)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-丙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
(54)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-丙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(55)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-丙基-(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃-3-醇
(56)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-丁基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
(57)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-苄基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
(58)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-烯丙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
(59)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-2-甲氧乙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
(60)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-2-甲氧基乙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(61)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-炔丙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
(62)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-2-羟基乙基-(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇(63)6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-2-羟基乙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
根据情况需要,式(Ⅰ)的化合物可以转变成药学上合格的盐。这些盐包括,如无机基的盐如碱金属盐(如钠盐、钾盐及铯盐),碱土金属盐(如钙盐和镁盐)和铵盐;有有机基的盐如有机胺盐(如三乙胺盐、吡啶盐、甲基吡啶盐、乙醇胺盐、三乙醇胺盐、二环己胺盐,及N,N-二苄乙二胺盐);无机酸加成盐(如,盐酸盐、溴酸盐、硫酸盐和磷酸盐);有机羧酸加成盐或有机磺酸加成盐(如甲酸盐、乙酸盐、三氟乙酸盐、马来酸盐、酒石酸盐、甲磺酸盐、苯磺酸盐及对甲苯磺酸盐);及带碱性氨基酸或酸性氨基酸的盐(如精氨酸(盐)、天冬氨酸(盐)和谷氨酸(盐))。
另外,在许多情况下,式(Ⅰ)化合物在分子中具不对称碳原子。本发明的苯并吡喃化合物包括这些旋光异构体及其混合物。
本发明式(Ⅰ)的苯并吡喃化合物可以通过以下方法制备。
方法1(生产式(Ⅰ)化合物,其中R3是氢原子,R4是羟基的方法)
可通过将式(Ⅱ)的化合物:
其中R1、R5、R6和Y如上所定义,以排除R1为R7-CO-,及Y是羧基的情况为条件,与式(Ⅲ)化合物反应。
其中R2和R8如上所定义,得到上述的化合物。这里,式(Ⅱ)和式(Ⅲ)中R1、R2和Y的较佳例子是如同对式(Ⅰ)化合物所描述的。R8的较佳例子为,具有1到6个碳原子的低级烷基,如甲基和乙基。这反应为缩合反应,涉及一低级醇(R8OH)的释放,如以下反应式所示:
在这反应中,反应温度较佳为0-200℃,尤其是50-150℃,反应时间较佳为约30分钟到12小时,尤其是1-6小时。反应可在溶剂存在下或不存在下进行。作为溶剂,例如可用甲醇、乙醇、丙醇、苯、甲苯、二甲基甲酰胺(DMF)、二甲亚砜(DMSO)、或乙酸乙酯。
方法2(生产式(Ⅰ)化合物,其中R3和R4一起形成键,的方法)
可通过将式(Ⅰa)的化合物:

其中R1、R2、R5、R6和Y如上所定义,以排除R1是R7-CO-基团,及Y是羧基的情况为条件,经受脱水作用得到上述的化合物:

即,第一步反应,其中式(Ⅳ)化合物R9SO2X与式(Ⅰa)化合物起反应,是在溶剂如吡啶、喹啉或三乙胺中进行的,反应温度较佳为-10-50℃,更佳的是在-5-10℃,反应时间较佳为30分钟到12小时,更佳的是1-6小时。
在式(Ⅳ)中,R9为烷基或取代的或未取代的芳基,X为卤原子。式(Ⅳ)代表的特定化合物包括,如甲磺酰氯、甲磺酰溴、对甲苯磺酰氯及对甲苯磺酰溴。
然后,第二步反应,其中所得式(Ⅴ)化合物用式(Ⅵ)化合物R10OM处理,以获得所需式(Ⅰb)化合物,在溶剂如二甲基甲酰胺,二甲亚砜、乙腈、苯或甲苯中进行,反应温度较佳为0-100℃,更佳的在20-40℃,反应时间较佳为30分钟到24小时,更佳为3-12小时。
在式(Ⅵ)中,R10为烷基。作为这样的烷基,较佳的例如有甲基、乙基、丙基或叔丁基。M为碱金属。
式(Ⅵ)代表的特定化合物包括,如叔丁酸钾、乙醇钾、甲醇钾、乙醇钠和甲醇钠。
方法3(生产式(Ⅰ)化合物,其中R3和R4一起形成键,的方法)
可通过将从上述方法2所得的式(Ⅴ)化合物,与1,8-二氮杂二环[5,4,0]-7-十一碳烯在有机溶剂如苯中反应,得到上述式(Ⅰb)化合物。
方法4(生产式(Ⅰ)化合物,其中R3和R4一起形成键,的方法)
可通过将式(Ⅰa)化合物1)与氢化钠(NaH)在溶剂如四氢呋喃中反应,2)与盐酸在溶剂如乙醇中反应,或3)与对甲苯磺酸在溶剂如苯中反应,以进行脱水处理,得到式(Ⅰb)化合物。
方法5(生产式(Ⅰ)化合物,其中R1是取代或未取代的C1-6烷基、C2-6链烯基、或C2-6炔基,R3和R4一起形成键,的方法)
式(Ⅶ)化合物:

其中R2、R5、R6和Y如上所定义,与式(Ⅷ)化合物反应:
其中R11和X如上所定义,在有机溶剂如乙腈、甲醇、苯或DMF中,在碱性化合物如碳酸钾、碳酸钠、三乙胺或甲醇钠中反应,可得到式(Ⅰb)化合物,其中R1为取代或未取代C1-6烷基,C2-6链烯基或C2-6炔基。
方法6(生产式(Ⅰ)化合物,其中R1为R7-CO-基团,R3和R4一起形成键,的方法)
式(Ⅶ)化合物与式(Ⅸ)化合物反应:
其中R7和X如上所定义,在有机溶剂如吡啶或三乙胺中进行,以获得式(Ⅰb)化合物,其中R1是R7-CO-基团。
方法7(生产式(Ⅰ)化合物,其中Y是羧基,的方法)
式(Ⅰ)化合物,其中Y为羧基,可通过在碱性条件下水解式(Ⅹ)化合物而得到:

其中R1、R2、R3、R4、R5、R6和R12如上所述。
方法8(生产式(Ⅰ)化合物,其中Y为吗啉代羰基或二-C1-6烷基氨基羰基,的方法)
上述方法7获得的化合物(式(Ⅰ)化合物,其中Y是羧基),与吗啉或二烷基胺在如二环己基碳二亚胺存在下反应,以获得式(Ⅰ)化合物,其中Y为吗啉代羰基或二-C1-6烷基氨基羰基。
本发明还提供式(Ⅺ)化合物:

其中Y1为C2-6炔基,R5和R6如上所定义,作为生产式(Ⅰ)化合物的有用的起始材料。
式(Ⅺ)化合物是新颖化合物,可通过例如下式表示的反应得到:
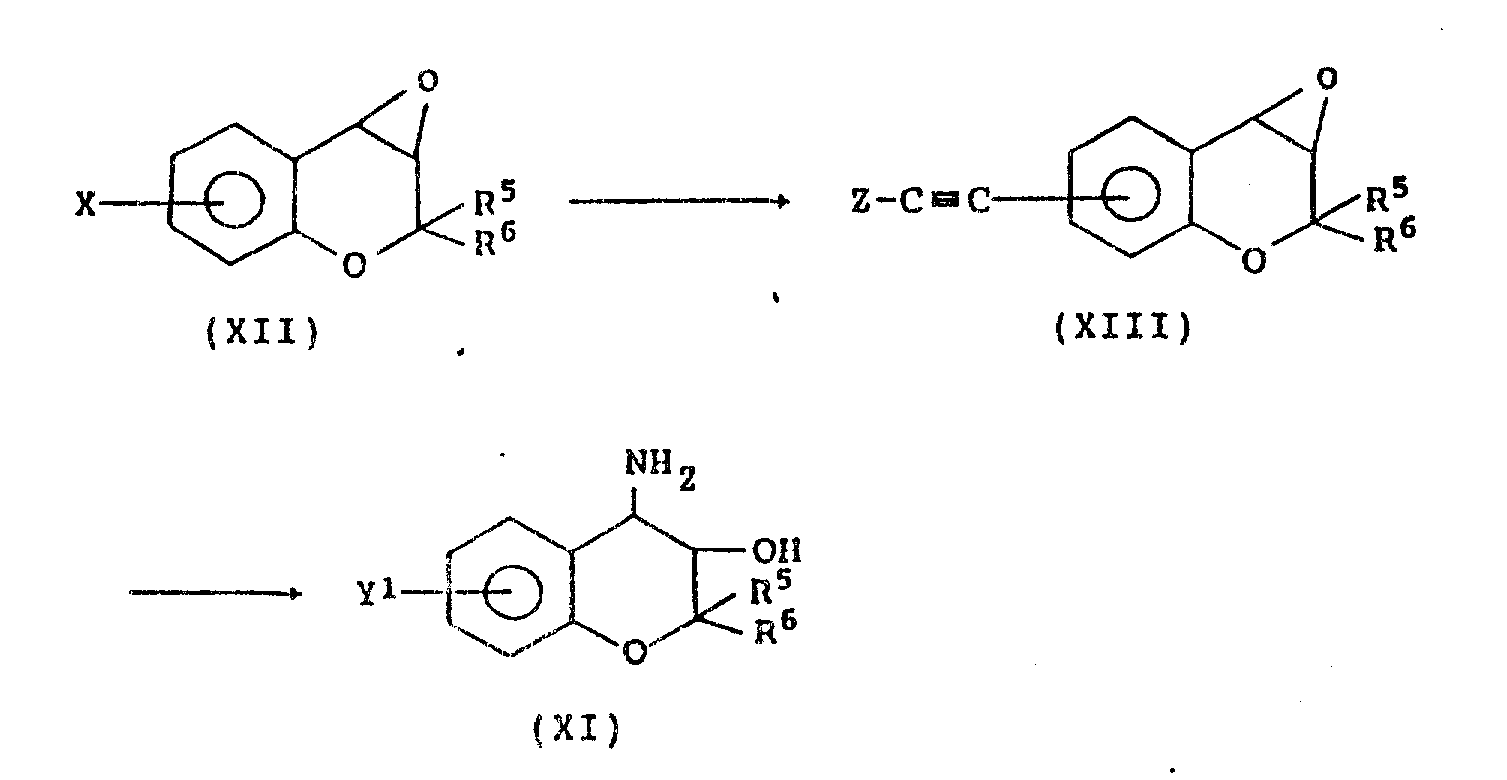
在上式中,X为卤原子,Z为C1-4烷基或三烷基甲硅烷基。
特定地,对式(Ⅻ)化合物,加入叔胺如三乙胺、三苯基膦、乙酸钯、或氯化钯,及C3-C6炔化合物或三甲基硅炔,在氮气流中于50-150℃之间温度下反应6-24小时以获得式(ⅩⅢ)化合物。然后,式(ⅩⅢ)化合物溶解于含氨的醇中,在5-50℃温度下反应进行6-48小时,以获得式(Ⅺ)的C2-6炔基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇。
本发明的苯并吡喃化合物具卓越的钾通道激活作用,能使各种平滑肌松驰,并改进钾通过平滑肌细胞膜的渗透性。通过这类活动来对付疾病包括如高血压、瞬间脑局部缺血发作、脑梗死、脑动脉硬化、心绞痛、慢性心机能不全、心肌梗塞及心律不齐的预防和治疗。另外,它们能有效地松驰气管、胃肠道、膀胱和子宫的平滑肌。因此,他们能有效地用来治疗气喘、呼吸系统障碍疾病、胃肠道疾病和子宫疾病。另外,它还可用来治疗间歇性跛行。
此外,由于钾通道激活作用,它们还具有血管舒张活性,这样它们有潜在的作为毛发促进剂的用途。
本发明的药物组合物可以单纯地由式(Ⅰ)苯并吡喃化合物组成。但是,较佳的常常是使它与合适的载体或赋体剂一起制成适于口服的合适制剂。然而,它还可以通过非口服的其它途径给药,例如,对有心脏病的病人。口服的药物制剂包括片剂、胶囊剂、颗粒剂、溶液剂和悬浮液剂。这些药物制剂可通过普通方法制备。
用于这类药物制剂的药学上合格的赋形剂包括,如明胶、乳糖、葡萄糖、氯化钠、淀粉、硬脂酸镁、滑石、植物油,及其它药物赋形剂。
本发明的苯并吡喃化合物的剂量根据所用的给药途径、制剂类型、病人情况等而变化,但常常是在0.002-2毫克/千克体重范围内,优先在0.01-0.2毫克/千克体重范围内。
现在,根据以下实施例和试验实施例详细地描述本发明。当然,本发明决不受以下特定实施例的限制。
实施例1
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃-3-醇
把2.29克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇加入到1.18克N-氰基亚胺甲酸乙酯中,混合物在搅拌下于温度100-120℃之间反应2小时。冷却反应混合物,然后使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤二次,然后用无水硫酸钠干燥。随后,在减压下蒸馏去除乙酸乙酯,残余物固体在二氯甲烷/异丙醇/石油醚中重结晶,得到结晶状的上述化合物1.75克。
熔点:155-158℃
NMR(CDCl3)δ:
1.27(s,3H),1.48(s,3H),3.66(q,1H),5.10(t,1H),
5.13(d,1H),6.80(d,1H),7.28-7.55(m,2H),8.33(d,1H),
8.57-8.97(br,1H)
实施例2
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
在1.23克N-氰基乙酰亚氨酸乙酯中,加入2.18克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇,混合物在100-120℃温度下搅拌2小时。冷却反应混合物,并使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤二次,用无水硫酸钠干燥。然后,减压下蒸馏去除乙酸乙酯。残余油状物在乙酸乙酯/正己烷中重结晶,得到1.75克结晶状的上述化合物。
熔点:240-243℃
NMR(CDCl3)δ:
1.26(s,3H),1.50(s,3H),2.45(s,3H),3.70(q,1H),
4.86(d,1H),5.12(t,1H),6.86(d,1H),7.32-7.60(m,2H),
8.53(d,1H)
实施例3
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-丙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
在1.39克N-氰基丙酰亚氨酸乙酯中,加入2.17克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇,混合物在搅拌下于温度100-120℃反应2小时。冷却反应混合物,并使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤二次,用无水硫酸钠干燥。然后,减压蒸馏去除乙酸乙酯。残余油状物用硅胶柱层析法纯化,得到1.48克结晶状的上述化合物。
熔点:113-115℃
NMR(CDCl3)δ:
1.28(s,3H),1.52(s,3H),2.70(q,2H),3.75(d,1H),
4.00(br,1H),5.12(t,1H),6.80(d,1H),7.29-7.61(m,2H),
7.5-7.6(br,1H)
实施例4
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-戊酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
在3.35克N-氰基戊酰亚氨酸乙酯中加入4.37克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇,混合物在搅拌下于温度100-120℃反应2小时。冷却反应混合物,并使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤二次,用无水硫酸钠干燥。然后,减压下蒸馏去除乙酸乙酯。残余油状物用硅胶柱层析法纯化,得到1.25克上述油状化合物。
NMR(CDCl3)δ:
0.95(t,3H),1.27(s,3H),1.50(s,3H),1.13-1.95(br,4H),
2.35(m,2H),3.62(d,1H),5.07(t,1H),6.51(d,1H),
6.86(d,1H),7.34(m,1H),7.50(s,1H)
实施例5
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-丁酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
在3.09克N-氰基丁酰亚氨酸乙酯中,加入4.38克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇,混合物在搅拌下于温度100-120℃反应2小时。冷却反应混合物,并使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤二次,然后用无水硫酸钠干燥。减压下蒸馏去除乙酸乙酯。通过硅胶柱层析法纯化残余油状物,得到3.54克结晶状上述化合物。
熔点:138-140℃
NMR(CDCl3)δ:
1.04(t,3H),1.27(s,3H),1.50(s,3H),1.75(m,2H),
2.66(t,1H),3.75(d,1H),5.08(t,1H),6.80(d,1H),
7.32(m,2H),7.82(d,1H)
实施例6
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃-3-醇
在1.89克N-氰基苄基亚氨酸乙酯中,加入2.15克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇,混合物在搅拌下于温度100-120℃反应2小时。冷却反应混合物,并使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤四次,然后用无水硫酸钠干燥。减压下蒸馏去除乙酸乙酯。通过硅胶柱层析法纯化残余油状物,得到1.23克结晶状上述化合物。
熔点:184-185℃
NMR(CDCl3)δ:
1.32(s,3H),1.51(s,3H),3.65-3.95(m,1H),4.45-
4.75(br,1H),5.27(t,1H),6.80(d,1H),7.67-7.80(m,7H),
8.10(d,1H)
实施例7
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-甲基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
在2.80克N-氰基乙酰亚氨酸乙酯中,加入4.65克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-甲氨基-二氢-苯并[b]吡喃-3-醇,混合物在搅拌下于温度100-120℃反应2小时。冷却反应混合物,并使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤二次,然后用无水硫酸钠干燥。减压下蒸馏去除乙酸乙酯。残余固体在乙醇中重结晶,得到2.34克结晶状上述化合物。
熔点:250-251℃
NMR(CDCl3)δ:
1.29(s,3H),1.53(s,3H),2.60-2.91(m,6H),3.58(q,1H),
4.88(d,1H),5.50(d,1H),6.80(d,1H),6.86(d,1H),
7.20-7.63(m,2H)
实施例8
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-丁基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
在1.13克N-氰基乙酰亚氨酸乙酯中,加入2.3克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-二丁氨基-二氢-苯并[b]吡喃-3-醇,混合物在搅拌下于温度100-120℃反应2小时。冷却反应混合物,并使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤二次,然后用无水硫酸钠干燥。减压下蒸馏去除乙酸乙酯。通过硅胶柱层析法纯化残余固体,得到1.43克结晶状上述化合物。
熔点:163-165℃
NMR(CDCl3)δ:
0.91(t,3H),1.15-2.00(m,4H),1.28(s,3H),1.58(s,3H),
6.76-7.64(m,3H)
实施例9
制备6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-苄基(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
在1.54克N-氰基乙酰亚氨酸乙酯中,加入3.86克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-苄氨基-二氢-苯并[b]吡喃-3-醇,混合物在搅拌下于温度100-120℃反应2小时。冷却反应混合物,并使其溶解于100毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤二次,然后用无水硫酸钠干燥。减压下蒸馏去除乙酸乙酯。通过硅胶柱层析法纯化残余固体,得到1.65克结晶状上述化合物。
熔点:201-203℃
NMR(CDCl3)δ:
1.28(s,3H),1.47(s,3H),2.60(d,3H),3.70(br,1H),
4.45(s,2H),5.00(d,1H),6.85(d,1H),7.05-7.55(m,7H)
实施例10
制备6-乙炔基-3,4-二氢-2,2-甲基-反(式)-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
反应如同实施例1中所述的方法进行,除了用6-乙炔基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇作为起始材料,以得到上述化合物。
熔点:265℃(分解)
NMR(DMSO-d6)δ:
1.25(s,3H),1.50(s,3H),2.40(s,3H),3.26(s,1H),
3.63(d,1H),5.13(d,1H),6.70(d,1H),7.60-7.83(m,2H)
实施例11到19
反应如同实施例1中所述进行,除了换用不同的起始材料,以得到以下本发明的苯并吡喃化合物。在实施例11-19中获得的化合物如下所示。
(实施例11)
6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-烯丙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(实施例12)
6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-炔丙基-(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(实施例13)
6-氟代-3,4-二氢-2,2-二甲基-反(式)-4-[N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(实施例14)
6-氯代-3,4-二氢-2,2-二甲基-反(式)-4-[N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(实施例15)
6-甲氧基-3,4-二氢-2,2-二甲基-反(式)-4-[N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(实施例16)
6-乙酰基-3,4-二氢-2,2-二甲基-反(式)-4-[N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(实施例17)
6-硝基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(实施例18)
6-苯基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
(实施例19)
6-乙氧羰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇
在实施例1-19中获得的苯并吡喃化合物的熔点及NMR分析结果示于表1中。
实施例20到33
用以上所述的同样方法,制备各种式(Ⅰ)苯并吡喃化合物,其中R3是氢原子,R4为羟基,每个R5和R6为甲基。实施例20-33中获得的化合物的熔点和NMR分析结果示于表2中。
(实施例34)
制备6-氰基-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
(1)把17.25克6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇溶解于130毫升吡啶中,在搅拌下及用冰冷却下,加入9.0克甲磺酰氯。让混合物反应4小时。反应混合物倾倒入冰水中,这样,有固体沉淀下来。过滤收集此固体,在乙醇中重结晶,得28.5克结晶状的6-氰基-3,4-二氰-3-甲磺酰氧基-2,2-二甲基-反(式)-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃。
熔点:202-204℃
NMR(CDCl3)δ:
1.38(s,3H),1.56(s,3H),2.41(s,3H),3.15(t,3H),
4.82(d,1H),5.46(t,1H),6.90(d,1H),7.27-7.61(m,3H)
(2)然后,将9.3克叔丁酸钾溶于60毫升二甲基甲酰胺中。然后加入0.3克步骤(1)中得到的6-氰基-3,4-二氢-3-甲磺酰氧基-2,2-二甲基-反(式)-4-[(N-氰基-乙酰亚氧基)氨基]-二氢-苯并[b]吡喃,混合物在室温下搅拌4小时。反应混合物倾倒入150毫升冰水中,用稀盐酸将pH调到2-3。然后,用200毫升乙酸乙酯萃取混合物,萃取液用饱和氯化钠水溶液洗涤二次,用无水硫酸钠干燥。然后,减压蒸馏去乙酸乙酯。残余固体在50毫升苯/乙酸乙酯(1/1)中重结晶,得1.9克结晶状上述化合物
熔点:214-215℃(分解)
NMR(DMSO-d6)δ:
1.50(s,3H),2.52(s,3H),6.23(s,1H),6.87(d,1H),
7.48(dd,1H),7.60(d,1H),9.30(b,1H)
实施例35到40
以下化合物用如实施例34中所述的方法制备,除了换用不同的起始材料
实施例35
6-氰基-2,2-二甲基-4-[(N-氰基-亚胺甲基)氨基]-二氢-苯并[b]吡喃
实施例36
6-氰基-2,2-二甲基-4-[(N-氰基-丙酰亚氨基)氨基]-二氢-苯并[b]吡喃
实施例37
6-氰基-2,2-二甲基-4-[(N-氰基-戊酰亚氨基)氨基]-二氢-苯并[b]吡喃
实施例38
6-氰基-2,2-二甲基-4-[(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃
实施例39
6-氯代-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
实施例40
6-氟代-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
实施例35-40中获得的化合物的熔点和NMR(DMSOd6)分析结果示于表3中。
实施例41到49
以同样的方法,制备各种式(Ⅰ)苯并吡喃化合物,其中R3和R4一起形成键,各个R5和R6为甲基。实施例41-49中获得的化合物的熔点和NMR分析结果示于表4中。
实施例50
制备6-氰基-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃
在15克实施例34的步骤1中获得的6-氰基-3,4-二氢-3-甲磺酰氧基-2,2-二甲基-反(式)-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃中,先后加入300毫升苯,及18克1,8-二氮杂二环[5,4,0]-7-十-碳烯,混合物在回流下反应3小时。冷却反应混合物至室温,加入200毫升乙酸乙酯稀释,然后用水洗涤三次。苯/乙酸乙酯萃取液用无水硫酸钠干燥。减压蒸馏去除苯和乙酸乙酯。残余固体在乙醇中重结晶,得9.3克结晶状的上述化合物。
熔点:214-215℃。
实施例51
制备6-氰基-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)-乙氧羧基甲氨基]-二氢-苯并[b]吡喃
将6.0克实施例34中获得的6-氰基-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃悬浮于200毫升乙腈中,加入6.12克碳酸钾,0.75克碘化钾和7.53克溴乙酸乙酯。混合物回流下搅拌2小时。然后,冷却反应混合物,过滤以去除不溶解的碳酸钾和溴化钾。滤液在减压下蒸馏去除乙腈。残余物溶解于250毫升乙酸乙酯中,用饱和氯化钠水溶液洗涤,接着用无水硫酸钠干燥。然后减压蒸馏去除乙酸乙酯。残余固体在乙酸乙酯/正己烷中重结晶,得到4.8克结晶状的上述化合物。
熔点:180.0-182.0℃
NMR(DMSO-d6)δ:
1.31(t,3H),1.54(s,6H),2.36(s,3H),3.79(d,1H),
4.24(q,2H),4.70(d,1H),6.08(s,1H),6.94(d,1H),
7.31(d,1H),7.53(dd,1H)
实施例52到73
以同样的方法,制备各种式(Ⅰ)苯并吡喃化合物,其中R3和R4一起形成键,各个R5和R6为甲基。实施例52-73中获得的化合物的熔点和NMR分析结果示于表5中。
实施例74
制备6-氰基-2,2-二甲基-4-{[(N-氰基-乙酰亚氨基)-N'-乙酰基]氨基}-二氢-苯并[b]吡喃
将4.0克实施例34中获得的6-氰基-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃溶解于60毫升吡啶中。向这溶液中,在用冰冷却的情况下,滴加入1.77克乙酰氯。反应混合物在用冰冷却条件下搅拌2.5小时。然后,加入250毫升冰水,这样有固体沉淀下来。过滤收集固体,用水洗涤并干燥,此固体在乙酸乙酯中重结晶,得到3.4克结晶状的上述化合物。
熔点:214.5-216℃
NMR(DMSO-d6)δ:
1.56(s,6H),2.29(s,3H),2.91(s,3H),5.71(s,1H),
6.90(s,1H),7.03(d,1H),7.51(dd,1H)
实施例75到81
以同样的方法,制备各种式(Ⅰ)苯并吡喃化合物,其中R3和R4一起形成键,各个R5和R6为甲基。实施例75-81中获得的化合物的熔点和NMR分析结果示于表6中。
实施例82
制备6-羧基-4-[(N-氰基乙酰亚氨基)氨基]-2,2-二甲基-二氢-1-苯并[b]吡喃
将5克(16.7毫摩尔)6-甲氧基羰基-4-[(N-氰基乙酰亚氨基)氨基]-2,2-二甲基-二氢-1-苯并[b]吡喃溶解于200毫升甲醇中,将溶液冷却到0℃。在此溶液中,加入60毫升饱和氢氧化锂,混合物在室温下搅拌17小时。反应混合物倾倒入400毫升25%的磷酸二氢钠水溶液中,并中和,然后用1升乙酸乙酯萃取。萃取液依次用水及饱和氯化钠水溶液洗涤,随后用无水硫酸钠干燥。蒸馏去除溶剂,所得白色粉末用乙酸乙酯/己烷(5/1)洗涤,并干燥,得4.7克(得率:98%)6-羧基-4-[(N-氰基乙酰亚氨基)氨基]-2,2-二甲基-二氢-1-苯并[b]吡喃。
熔点:194-196℃。
NMR(DMSO-d6)δ:
1.40(6H,s),2.40(s,3H),6.08(s,1H),6.98(d,1H),
7.82(s,1H),7.92(dd,1H)
实施例83
制备6-(4-吗啉代羰基-4-[(N-氰基乙酰亚氨基)氨基]-2,2-二甲基-二氢-1-苯并[b]吡喃
将5.6克(19.6毫摩尔)6-羧基-4-[(N-氰基乙酰亚氨基)氨基]-2,2-二甲基-二氢-1-苯并[b]吡喃溶解于由120毫升四氢呋喃和120毫升乙腈组成的溶剂混合物中,并使该溶液冷却至0℃。在此溶液中,加入8.0克(39.3毫摩尔)二环己基碳化二亚胺,搅拌混合物15分钟。然后,加入5.1克(58.9毫摩尔)吗啉,混合物在室温下继续搅拌4小时。用500毫升乙酸乙酯稀释反应混合物,然后依次用水、30%磷酸二氢钠水溶液及饱和氯化钠水溶液洗涤,用无水硫酸钠干燥。蒸馏去除溶剂。然后,用硅胶柱层析法(展开溶剂:乙酸乙酯,硅胶:150克)纯化,得到3.6克白色粉末状(得率:52.0%)6-(4-吗啉代)羰基-4-[(N-氰基乙酰亚氨基)氨基]-2,2-二甲基-二氢-1-苯并[b]吡喃。
熔点:126-128℃
NMR(DMSO-d6)δ:
1.44(s,6H),2.42(s,3H),3.49(br,s,4H),3.60(br,s,4H),
5.96(s,1H),6.86(d,1H),7.18(d,1H),7.25(dd,1H),
10.06(br,s,1H)
实施例84
制备6-(二乙氨基)羰基-4-[(N-氰基乙酰亚氨基)氨基]-2,2-二甲基-二氢-1-苯并[b]吡喃
以实施例83中所述的方法,制备上述化合物,其熔点和NMR分析结果如下所示。
熔点:184-186℃
NMR(DMSO-d6)δ:
1.15(t,6H),1.43(s,6H),2.54(s,3H),3.34(m,4H),
6.00(s,1H),6.64(d,1H),6.84(d,1H),6.97(dd,1H),
9.79(s,1H)
实施例85
制备6-甲氧羧基-4-[(N-氰基乙酰亚氨基)氨基]-2,2-二甲基-二氢-1-苯并[b]吡喃
以实施例19和34中的相同方法制备上述化合物,其熔点和NMR分析如下所示。
熔点:193-195℃
NMR(DMSO-d6)δ:
1.49(s,6H),2.54(s,3H),3.84(s,3H),6.32(s,1H),
6.85(d,1H),7.82(m,2H)
实施例86
制备6-乙炔基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇(实施例10中的起始材料)
1)在35毫升三乙胺中,加入100毫克乙酸钯和200毫克三苯膦。在此反应溶液中,加入10.0克6-溴代-3,4-环氧-2,2-二甲基-二氢-苯并[b]吡喃,然后加入6.5克三甲基硅乙炔,然后混合物在氮气流下在温度70-85℃下反应12小时。使反应混合物减压下浓缩,然后溶于300毫升乙酸乙酯中。溶液用水洗涤,用无水硫酸钠干燥。然后,减压蒸馏去除乙酸乙酯。用硅胶层析法纯化残余物,得到10.0克油状物的6-(2'-三甲基硅)乙炔基-3,4-环氧-2,2-二甲基-二氢-苯并[b]吡喃。
NMR(CDCl3)δ:
0.23(s,9H),1.20(s,3H),1.53(s,3H),3.40(d,1H),
3.80(d,1H),6.63(d,1H),7.20(d,1H),7.43(d,1H)
10.0g 6-(2-trimethylsilyl)ethynyl-3,4-epoxy-2,2-
把10.0克6-(2-三甲基硅)乙炔基-3,4-环氧-2,2-二甲基-二氢-苯并[b]吡喃溶解于500毫升饱和氨的甲醇溶液中。以上溶液留置于室温7天,然后,减压浓缩。残余物通过硅胶层析法纯化,得到5.5克上述化合物。
NMR(CDCl3)δ:
1.20(s,3H),1.50(s,3H),2.23(br,3H),2.97(s,1H),
3.31(d,1H),3.61(d,1H),7.16(dd,1H),7.53(d,1H)
现在,参照试验实施例来描述本发明化合物的药学性质。
试验实施例1
钾通道激活作用
从体重约为240克的雄性Wister鼠上切取主动脉,并使之形成盘旋形标本,它在0.5克负载下悬吊以95%氧气+5%二氧化碳混合气体饱和的Krebs Henselate溶液中(37℃)。然后,加入20mM或60mM的氯化钾,通过等长换能器(由Nippon Koden K.K.生产)测量其收缩反应。当由氯化钾引起的收缩反应达最大值时,累积地加入本发明的一个化合物,同时测量舒张反应。由药物引起的舒张反应被测定为与氯化钾引起的最大收缩有关的舒张,结果示于表7中。
试验实施例2
降低血压活性
一只20周龄的雄性自发高血压鼠(SHR)放入笼中,在加热箱中于45℃加热5分钟。加热5分钟的SHR固定于测量板上,尾巴上套一套箍以加压并测量血压。当加压开始,测量心搏率。当鼠安静后,测量最高血压。
另外,本发明的一个化合物溶解于或悬浮在0.5%甲基纤维素溶液中,并口服给药,然后在给药后满1小时、3小时和6小时测量血压。与给药前的值相对的变动
(%)= (给药前血压-给药后血压)/(给药前血压) ×100
结果示于表8中:
气管平滑肌的松驰活性
通过常规方法,切取Hartley雄性豚鼠的气管平滑肌,在负载0.3克下垂直悬吊在2毫升Magnus浴中,该浴中盛满37℃台罗德氏溶液,并以95%氧气和5%二氧化碳混合气体不停地吹泡。当自发紧张平稳后,累积地从10-8M(起始浓度)开始加入本发明的化合物,在每一浓度观察反应约10分钟。最后,加入10-6克/毫升(最终浓度)异丙肾上腺素,以取得最大松驰。然后,由以下公式决定松驰率:
松驰率(%)= (由测试化合物引起的松驰长度(cm))/(由10-6克/毫克异丙肾上腺素引起的最大松驰长度(cm)) ×100
结果示于表9中。
试验实施例3
毒性试验
为进行急性毒性试验,把体重为22到25克的十只雄鼠作为一组,以本发明的一个化合物按相应于体重的剂量口服给药。从72小时的死亡率中,通过区域方法计算LD50。
实施例2和34中获得的本发明的化合物的LD50至少为2,000毫克/千克。
现在,将描述制剂实施例。
制备片剂
用以下组分,以常规方法制备片剂。
活性成分(实施例2中
获得的本发明的化合物) 2毫克
乳糖 150毫克
结晶纤维素 100毫克
硬脂酸镁 3毫克
如以上所详尽描述,本发明提供苯并吡喃化合物(具有卓越的钾通道激活作用,能用以治疗多种疾病),它们的制备方法,及含这些苯并吡喃化合物的药物组合物。
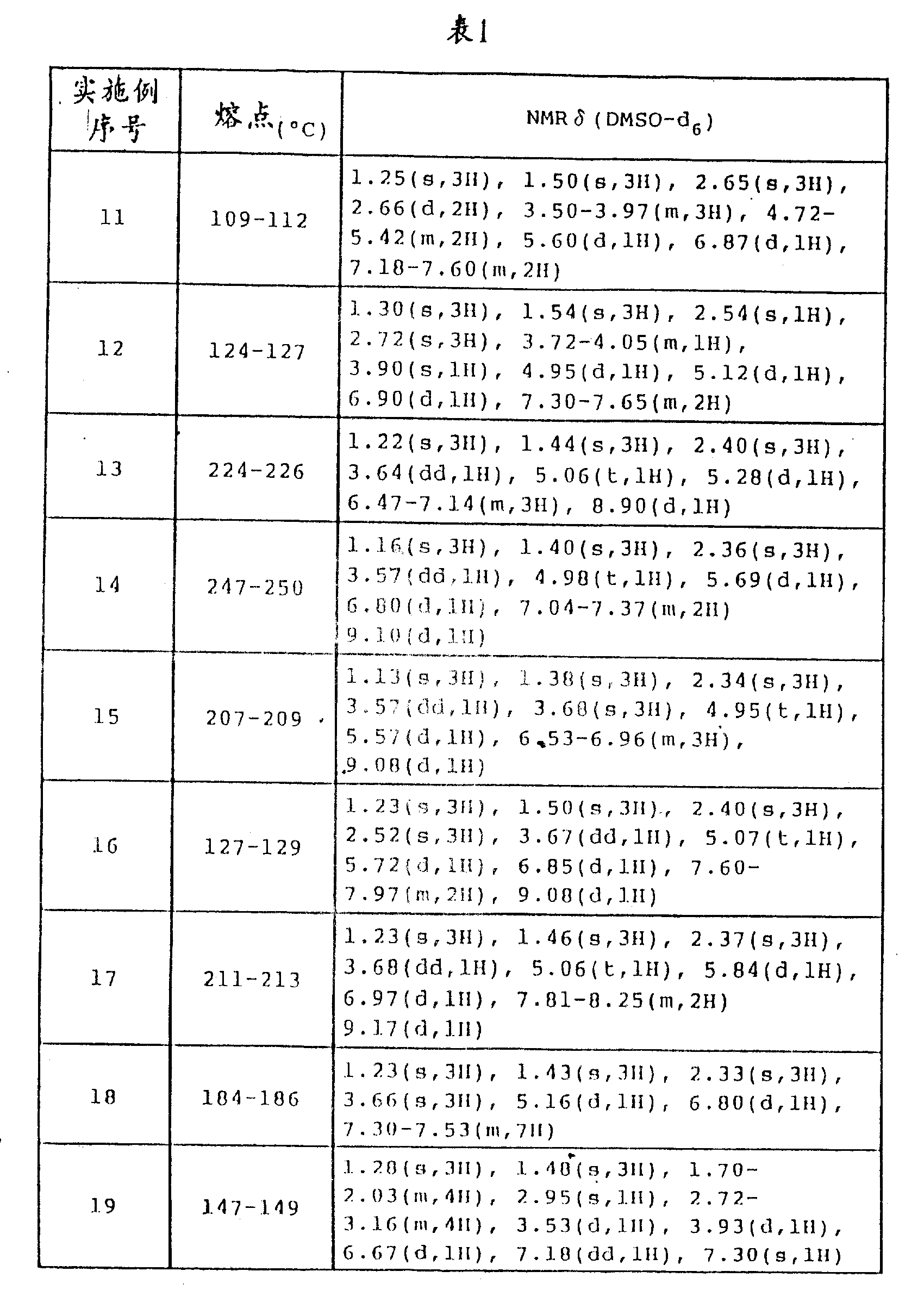





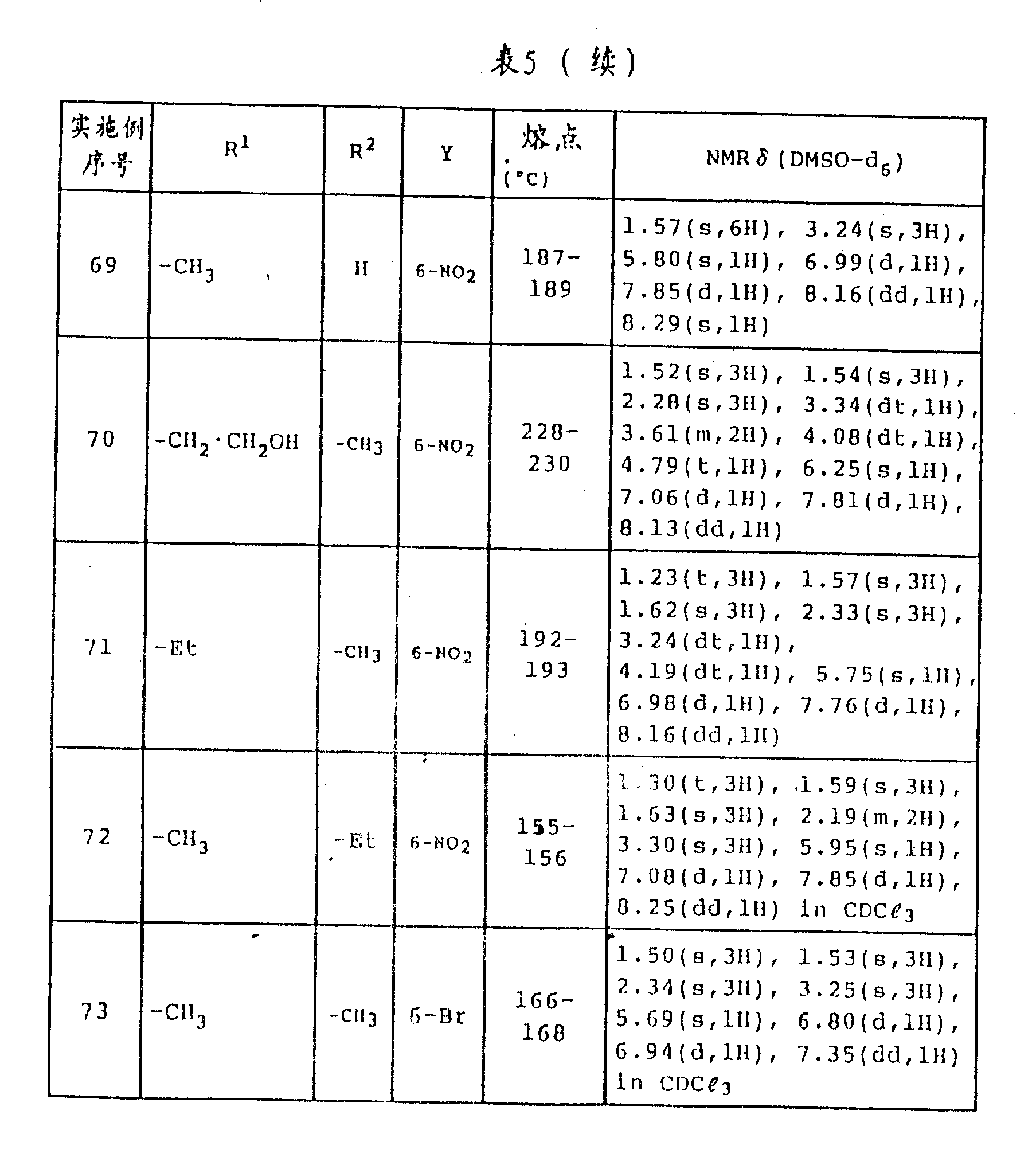

表7钾通道激活作用
| 化合物序号 | 浓度(mol) | 在20mMKCl中的舒张(%) | 在60mMKCl中的舒张率(%) |
| 实施例2 | 10-810-710-610-5 | 6.830.695.6100 | 001.05.6 |
| 实施例34 | 10-810-710-610-5 | 28.794.3100100 | 04.513.221.8 |
表8降低血压活性
| 化合物序号 | 剂量(mg/kg) | 与给药前的值相对的变动(%) | ||
| 1小时后 | 3小时后 | 6小时后 | ||
| 实施例2 | 10310.30.1 | 6460422610 | 615645175 | 615826120 |
| 实施例34 | 10310.30.1 | 6465624112 | 646360455 | 656357230 |
| 比较药硝吡胺甲酯 | 103 | 4020 | 98 | 70 |
表9气管平滑肌的松驰活性
| 化合物序号 | 剂量 | 松驰率(%) |
| 实施例2 | 10-73×10-710-63×10-610-53×10-5 | 0020.741.667.568.2 |
| 实施例34 | 10-73×10-710-63×10-610-53×10-5 | 010.552.374.880.282.1 |
Claims (10)
1、式(Ⅰ)的化合物或其药学上合格的盐,
其特征在于:
R1为氢原子,取代或未取代的C1-6烷基,C2-6链烯基,C2-6炔基,或R7-CO-基团(其中R7是C1-6烷基,苯基,可被苯基取代的C2-6链烯基,或C1-6烷氧基);
R2为氢原子,取代的或未取代的C1-8烷基,或苯基;
R3为氢原子,及R4为羟基,或R3和R4一起形成键;
R5和R6各为C1-4烷基;及
Y为氰基,卤原子,硝基,C1-6烷基,C2-6链烯基,C1-6烷羰基,C1-6烷氧基,二-C1-6烷基氨基羰基,芳基,C1-6烷氧羰基,羧基或吗啉代羰基。
2、根据权利要求1所述的化合物或其药学上合格的盐,其特征在于R1的取代的C1-6烷基的取代基为C1-6烷氧基,芳基,羟基,C1-6烷氧羰基或二-C1-6烷基氨基。
3、根据权利要求1所述的化合物或其药学上合格的盐,其特征在于R2的取代的C1-8烷基的取代基为C1-6烷氧基或芳基。
4、根据权利要求1所述的化合物或其药学上合格的盐,其特征在于式(Ⅰ)中,R1为氢原子;R2为C1-8烷基或苯基;R3为氢原子及R4为羟基,或R3和R4一起形成键;每个R5和R6为C1-4烷基;Y是氰基或硝基。
5、根据权利要求1所述的化合物或其药学上合格的盐,为6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇或其药学上合格的盐,6-氰基-3,4-二氢-2,2-二甲基-反(式)-4-[(N-氰基-亚氨苄基)氨基]-二氢-苯并[b]吡喃-3-醇或其药学上合格的盐,6-硝基-3,4-二氢-2,2-二甲基-反(式)-4-[(-N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃-3-醇或其药学上合格的盐,6-氰基-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃或其药学上合格的盐,6-氰基-2,2-二甲基-4-[(N-氰基-丙酰亚氨基)氨基]-二氢-苯并[b]吡喃或其药学上合格的盐,或6-硝基-2,2-二甲基-4-[(N-氰基-乙酰亚氨基)氨基]-二氢-苯并[b]吡喃或其药学上合格的盐。
6、式(Ⅰ)化合物,或其

药学上合格的盐:
R1为氢原子,取代或未取代的C1-6烷基,C2-6链烯基,C2-6炔基,或R7-CO-基团(其中R7是C1-6烷基,苯基,可被苯基取代的C2-6链烯基,或C1-6烷氧基);
R2为氢原子,取代的或未取代的C1-8烷基,或苯基;
R3为氢原子,及R4为羟基,或R3和R4一起形成键;
R5和R6各为C1-4烷基;及
Y为氰基,卤原子,硝基,C1-6烷基,C2-6链烯基,C1-6烷羰基,C1-6烷氧基,二-C1-6烷基氨基羰基,芳基,C1-6烷氧羰基,羧基或吗啉代羰基,的生产方法,其特征在于:
(1)将式(Ⅱ)的化合物:

其中R1、R5、R6和Y如上所定义,以除去R1为R7-CO-基团,及Y为羧基的情况为条件,与式(Ⅲ)化合物反应:
其中R2如上所述,R8为C1-6烷基,以获得式(Ⅰ)的一种化合物,其中R3为氢原子,R4为羟基;
(2)将式(Ⅰa)化合物:

其中R1、R2、R5、R6和Y均如上所述,以除去R1为R7-CO基团,及Y为羧基的情况为条件,与式(Ⅳ)化合物反应:
其中R9为烷基或取代或未取代的芳基,X为卤原子,以形成式(Ⅴ)化合物:

其中R1、R2、R5、R6、R9和Y如上所定义,然后式(Ⅴ)化合物用式(Ⅵ)化合物处理:
其中R10为烷基,M为碱金属,以得到一种式(Ⅰ)的化合物,其中R3和R4一起形成键;
(3)将式(Ⅴ)化合物与1,8-二氮杂二环[5,4,0]-7-十-碳烯反应,以得到一种式(Ⅰ)的化合物,其中R3和R4一起形成键;
(4)使式(Ⅰa)化合物用氢化物、盐酸或对甲苯磺酸处理以脱水,获得一种式(Ⅰ)的化合物,其中R3和R4一起形成键;
(5)将式(Ⅶ)化合物:
其中R2、R5、R6和Y如上所定义,与式(Ⅶ)合化物反应:
其中R11为取代或未取代的C1-6烷基,C2-6烯烯基,C2-6炔基,及X为卤原子,以获得一种式(Ⅰ)化合物,其中R1为取代或未取代C1-6烷基,C2-6链烯基,C2-6炔基,及R3为R4一起形成键;
(6)使式(Ⅶ)化合物与式(Ⅸ)化合物反应:
其中R7和X如前所定义,以获得一种式(Ⅰ)化合物,其中R1为R7-CO-基团(其中R7如前所述定义),R3和R4一起形成键;
(7)水解式(Ⅹ)化合物:

其中R1、R2、R3、R4、R5和R6如前所定义,R12为C1-6烷基,以得到一种式(Ⅰ)化合物,其中Y为羧基;或
(8)将式(Ⅰ)化合物,其中Y为羧基,与吗啉或二烷基胺反应,以获得一种式(Ⅰ)化合物,其中Y为吗啉代羰基,或二-C1-6烷基氨基羰基。
7、一种具有钾通道激活作用的药物组合物,它含有式(Ⅰ)的化合物,或其药学上合格的盐,作为活性成份,

其特征在于:
R1为氢原子,取代或未取代的C1-6烷基,C2-6链烯基,C2-6炔基,或R7-CO-基团(其中R7是C1-6烷基,苯基,可被苯基取代的C2-6链烯基,或C1-6烷氧基);
R2为氢原子,取代的或未取代的C1-8烷基,或苯基;
R3为氢原子,及R4为羟基,或R3为和R4一起形成键;
R5和R6各为C1-4烷基;及
Y为氰基,卤原子,硝基,C1-6烷基,C2-6链烯基,C1-6烷羰基,C1-6烷氧基,二-C1-6烷基氨基羰基,芳基,C1-6烷氧羰基,羧基或吗啉代羰基。
8、一种降血压的或抗气喘的药,它的活性成分为式(Ⅰ)化合物或其药学上合格的盐,
其特征在于:
R1为氢原子,取代或未取代的C1-6烷基,C2-6链烯基,C2-6炔基,或R7-CO-基团(其中R7是C1-6烷基,苯基,可被苯基取代的C2-6链烯基,或C1-6烷氧基);
R2为氢原子,取代的或未取代的C1-8烷基,或苯基;
R3为氢原子,及R4为羟基,或R3和R4一起形成键;
R5为R6各为C1-4烷基;及
Y为氰基,卤原子,硝基,C1-6烷基,C2-6链烯基,C1-6烷羰基,C1-6烷氧基,二-C1-6烷基氨基羰基,芳基,C1-6烷氧羰基,羧基或吗啉代羰基。
9、C2-6炔基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇。
10、根据权利要求9所述的化合物,为6-乙炔基-3,4-二氢-2,2-二甲基-反(式)-4-氨基-二氢-苯并[b]吡喃-3-醇。
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP208547/89 | 1989-08-11 | ||
| JP20854789 | 1989-08-11 | ||
| JP341528/89 | 1989-12-29 | ||
| JP34152889 | 1989-12-29 | ||
| JP73653/90 | 1990-03-23 | ||
| JP7365490 | 1990-03-23 | ||
| JP73654/90 | 1990-03-23 | ||
| JP7365390 | 1990-03-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1049500A true CN1049500A (zh) | 1991-02-27 |
Family
ID=27465604
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN90107121A Pending CN1049500A (zh) | 1989-08-11 | 1990-08-11 | 苯并吡喃化合物,其生产方法及药物组合物 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US5095016A (zh) |
| EP (1) | EP0412531B1 (zh) |
| KR (1) | KR910004592A (zh) |
| CN (1) | CN1049500A (zh) |
| AT (1) | ATE105556T1 (zh) |
| AU (1) | AU624526B2 (zh) |
| CA (1) | CA2022882A1 (zh) |
| CZ (1) | CZ393390A3 (zh) |
| DE (1) | DE69008791T2 (zh) |
| FI (1) | FI903939A0 (zh) |
| HU (1) | HUT54673A (zh) |
| NO (1) | NO174050C (zh) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5140031A (en) * | 1989-05-31 | 1992-08-18 | E. R. Squibb & Sons, Inc. | Pyranyl cyanoguanidine derivatives |
| US5061813A (en) * | 1990-04-02 | 1991-10-29 | E. R. Squibb & Sons, Inc. | Substituted cyanoimino benzopyranes |
| US5276168A (en) * | 1990-06-18 | 1994-01-04 | E. R. Squibb & Sons, Inc. | Benzopyran derivatives and heterocyclic analogs thereof as antiischemic agents |
| JPH05186458A (ja) * | 1991-04-26 | 1993-07-27 | Japan Tobacco Inc | 新規なベンゾピラン誘導体 |
| US5374643A (en) * | 1992-09-11 | 1994-12-20 | E. R. Squibb & Sons, Inc. | Aryl urea (thiourea) and cyanoguanidine derivatives |
| US5453421A (en) * | 1992-09-11 | 1995-09-26 | E. R. Squibb & Sons, Inc. | Aryl and heterocyclic substituted propenamide derivatives |
| US5514690A (en) * | 1992-11-17 | 1996-05-07 | E. R. Squibb & Sons, Inc. | Aminocarbonyl (thiocarbonyl) and cyanoguanidine derivatives of quinoline and indoline |
| IL109229A0 (en) * | 1993-05-11 | 1994-07-31 | Bristol Myers Squibb Co | Heterocyclic compounds and processes for the preparation of pyranyl cyanoguanidine derivatives using the same |
| US5393771A (en) * | 1993-05-12 | 1995-02-28 | Brisol-Myers Squibb Company | 4-substituted benzopyran and related compounds |
| US5837702A (en) * | 1993-10-07 | 1998-11-17 | Bristol-Myers Squibb Co. | 4-arylamino-benzopyran and related compounds |
| US5547966A (en) * | 1993-10-07 | 1996-08-20 | Bristol-Myers Squibb Company | Aryl urea and related compounds |
| US5401758A (en) * | 1993-10-07 | 1995-03-28 | Bristol-Myers Squibb Company | Pyridinyl cyanoguanidine compounds |
| US5612323A (en) * | 1995-06-07 | 1997-03-18 | Bristol-Myers Squibb Company | Phosphinic ester substituted benzopyran derivatives |
| US5869478A (en) * | 1995-06-07 | 1999-02-09 | Bristol-Myers Squibb Company | Sulfonamido substituted benzopyran derivatives |
| US5629429A (en) * | 1995-06-07 | 1997-05-13 | Bristol-Myers Squibb Company | Process for preparing 4-arylamino-benzopyran and related compounds |
| US5612370A (en) * | 1995-06-07 | 1997-03-18 | Bristol-Myers Squibb Company | Phenylglycine and phenylalaninen amido benzopyran derivatives |
| DE19921886A1 (de) * | 1999-05-12 | 2000-11-16 | Bayer Ag | Substituierte N-Cyano-amidine |
| WO2003000675A1 (en) * | 2001-06-25 | 2003-01-03 | Nissan Chemical Industries, Ltd. | Substituted benzopyran derivatives against arrhythmia |
| WO2003014113A1 (en) * | 2001-08-06 | 2003-02-20 | Glenmark Pharmaceuticals Limited | Novel benzopyran compounds and process for their preparation and use |
| KR100439972B1 (ko) * | 2002-02-19 | 2004-07-14 | 주식회사 심바이오테인 | 공생과 먹이사슬관계를 이용한 수산생물의 육상생산시설 |
| KR100458861B1 (ko) * | 2002-11-07 | 2004-12-03 | 주식회사 심바이오테인 | 미세조류-어류 공생 생산공장 |
| US7241776B2 (en) | 2004-08-02 | 2007-07-10 | Abbott Laboratories | Cyanoamidine P2X7 antagonists for the treatment of pain |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8419515D0 (en) * | 1984-07-31 | 1984-09-05 | Beecham Group Plc | Treatment |
| GB8521857D0 (en) * | 1985-09-03 | 1985-10-09 | Beecham Group Plc | Active compounds |
| IL84809A (en) * | 1987-11-06 | 1992-12-01 | Ciba Geigy | Process for the synthesis of 1-substituted imidazole- 5-carboxylic acids and derivatives thereof |
| JPH03500542A (ja) * | 1988-05-09 | 1991-02-07 | ビーチャム グループ ピーエルシー | 新規な化合物及び治療法 |
| US4988723A (en) * | 1988-06-02 | 1991-01-29 | Fujisawa Pharmaceutical Co., Ltd. | Benzopyran derivatives and their use as anti-hypertensives |
| GB8902118D0 (en) * | 1989-02-01 | 1989-03-22 | Beecham Group Plc | Chemical process |
-
1990
- 1990-08-03 US US07/562,577 patent/US5095016A/en not_active Expired - Fee Related
- 1990-08-08 EP EP90115242A patent/EP0412531B1/en not_active Expired - Lifetime
- 1990-08-08 CA CA002022882A patent/CA2022882A1/en not_active Abandoned
- 1990-08-08 DE DE69008791T patent/DE69008791T2/de not_active Expired - Fee Related
- 1990-08-08 AT AT9090115242T patent/ATE105556T1/de not_active IP Right Cessation
- 1990-08-09 AU AU60844/90A patent/AU624526B2/en not_active Ceased
- 1990-08-09 FI FI903939A patent/FI903939A0/fi not_active IP Right Cessation
- 1990-08-09 CZ CS903933A patent/CZ393390A3/cs unknown
- 1990-08-10 NO NO903524A patent/NO174050C/no unknown
- 1990-08-10 HU HU904956A patent/HUT54673A/hu unknown
- 1990-08-11 CN CN90107121A patent/CN1049500A/zh active Pending
- 1990-08-11 KR KR1019900012382A patent/KR910004592A/ko not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| US5095016A (en) | 1992-03-10 |
| NO903524D0 (no) | 1990-08-10 |
| NO903524L (no) | 1991-02-12 |
| DE69008791T2 (de) | 1995-01-12 |
| EP0412531B1 (en) | 1994-05-11 |
| EP0412531A1 (en) | 1991-02-13 |
| CA2022882A1 (en) | 1991-02-12 |
| ATE105556T1 (de) | 1994-05-15 |
| AU6084490A (en) | 1991-02-14 |
| DE69008791D1 (de) | 1994-06-16 |
| HU904956D0 (en) | 1991-01-28 |
| CZ393390A3 (en) | 1997-02-12 |
| AU624526B2 (en) | 1992-06-11 |
| NO174050C (no) | 1994-03-09 |
| HUT54673A (en) | 1991-03-28 |
| FI903939A0 (fi) | 1990-08-09 |
| KR910004592A (ko) | 1991-03-29 |
| NO174050B (no) | 1993-11-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1049500A (zh) | 苯并吡喃化合物,其生产方法及药物组合物 | |
| CN1027068C (zh) | α-取代的4-(喹啉-2-基-甲氧基)苯乙酸和酯的制备方法 | |
| CN1159311C (zh) | 二氢嘧啶类化合物 | |
| CN1146567C (zh) | 奥氮平二水合物d | |
| CN1068879C (zh) | 二氢苯并呋喃 | |
| CN1031992C (zh) | 苯醌衍生物及其药用 | |
| CN1109052A (zh) | 嘧啶和吡啶衍生物,它们的生产和应用 | |
| CN1106806A (zh) | 粘结受体拮抗剂iii | |
| CN1122036C (zh) | 用于制备奥氮平的中间体和方法 | |
| CN1016507B (zh) | 新的福斯克林衍生物的制备方法 | |
| CN1071427A (zh) | 6-苯并嗪基和6-苯并噻嗪基-2,3,4,5-四氢哒嗪-3-酮 | |
| CN87100658A (zh) | 1,4-苯并嗪类衍生物,它们的制备和应用 | |
| CN87104319A (zh) | 苯并吡喃衍生物 | |
| CN1169797C (zh) | 抗病毒的嘧啶二酮衍生物及其制备方法 | |
| CN1516695A (zh) | 用作抗病毒剂的芳基磺酰胺类化合物 | |
| CN1086817A (zh) | 苯并吡喃和苯并噁嗪衍生物 | |
| CN1097591C (zh) | 制备嘧啶衍生物的方法 | |
| CN1413205A (zh) | 具有抗肿瘤活性的2-(1h-吲哚-3-基)-2-氧代-乙酰胺 | |
| CN1138583A (zh) | 9-取代的2-(2-正烷氧基苯基)-嘌呤-6-酮类化合物 | |
| CN1079745A (zh) | 新的9-氟-7-氧代-7H-吡啶并[1,2,3-d,e][1,4]苯并嗪-6-羧酸及其酯 | |
| CN1653081A (zh) | 甾族化合物的c-17螺甾内酯化和6,7氧化 | |
| CN1083812A (zh) | 苯并咪唑类,含有这些化合物的药物组合物和它们的制备 | |
| CN1038583C (zh) | 酰基苯基甘氨酸衍生物及以其为有效成分的胶原酶活性亢进所致疾病的防治药物 | |
| CN1040192A (zh) | 新的氟甲氧苯基二氢吡啶、其制备方法和在药物方面的应用 | |
| CN101033224A (zh) | 一类新型嘧啶类小分子化合物、其制备方法及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C01 | Deemed withdrawal of patent application (patent law 1993) | ||
| WD01 | Invention patent application deemed withdrawn after publication |