

![]()

A Gaussian Process Library for Molecules, Proteins and Reactions.
| BNN Regression on Molecules | |
| Bayesian Optimisation Over Molecules |
We recommend using a conda virtual environment:.
conda env create -f conda_env.yml
pip install --no-deps rxnfp
pip install --no-deps drfp
pip install transformers
Optional for running tests.
pip install gpflow grakel
| Tutorial (BNN Regression on Molecules) | Docs |
from gauche.dataloader import DataLoaderMP
from gauche.dataloader.data_utils import transform_data
from sklearn.model_selection import train_test_split
loader = DataLoaderMP()
loader.load_benchmark(dataset, dataset_paths[dataset])
loader.featurize(feature)
X = loader.features
y = loader.labels
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=test_set_size, random_state=i)
# We standardise the outputs but leave the inputs unchanged
_, y_train, _, y_test, y_scaler = transform_data(X_train, y_train, X_test, y_test)| Tutorial (Bayesian Optimisation Over Molecules) | Docs |
from botorch.models.gp_regression import SingleTaskGP
from gprotorch.kernels.fingerprint_kernels.tanimoto_kernel import TanimotoKernel
# We define our custom GP surrogate model using the Tanimoto kernel
class TanimotoGP(SingleTaskGP):
def __init__(self, train_X, train_Y):
super().__init__(train_X, train_Y, GaussianLikelihood())
self.mean_module = ConstantMean()
self.covar_module = ScaleKernel(base_kernel=TanimotoKernel())
self.to(train_X) # make sure we're on the right device/dtype
def forward(self, x):
mean_x = self.mean_module(x)
covar_x = self.covar_module(x)
return MultivariateNormal(mean_x, covar_x)The representations considered are summarised graphically in the figure with the tabulated references included below. For molecular graph representations, all featurisations currently included in PyTorch Geometric [2] are supported.
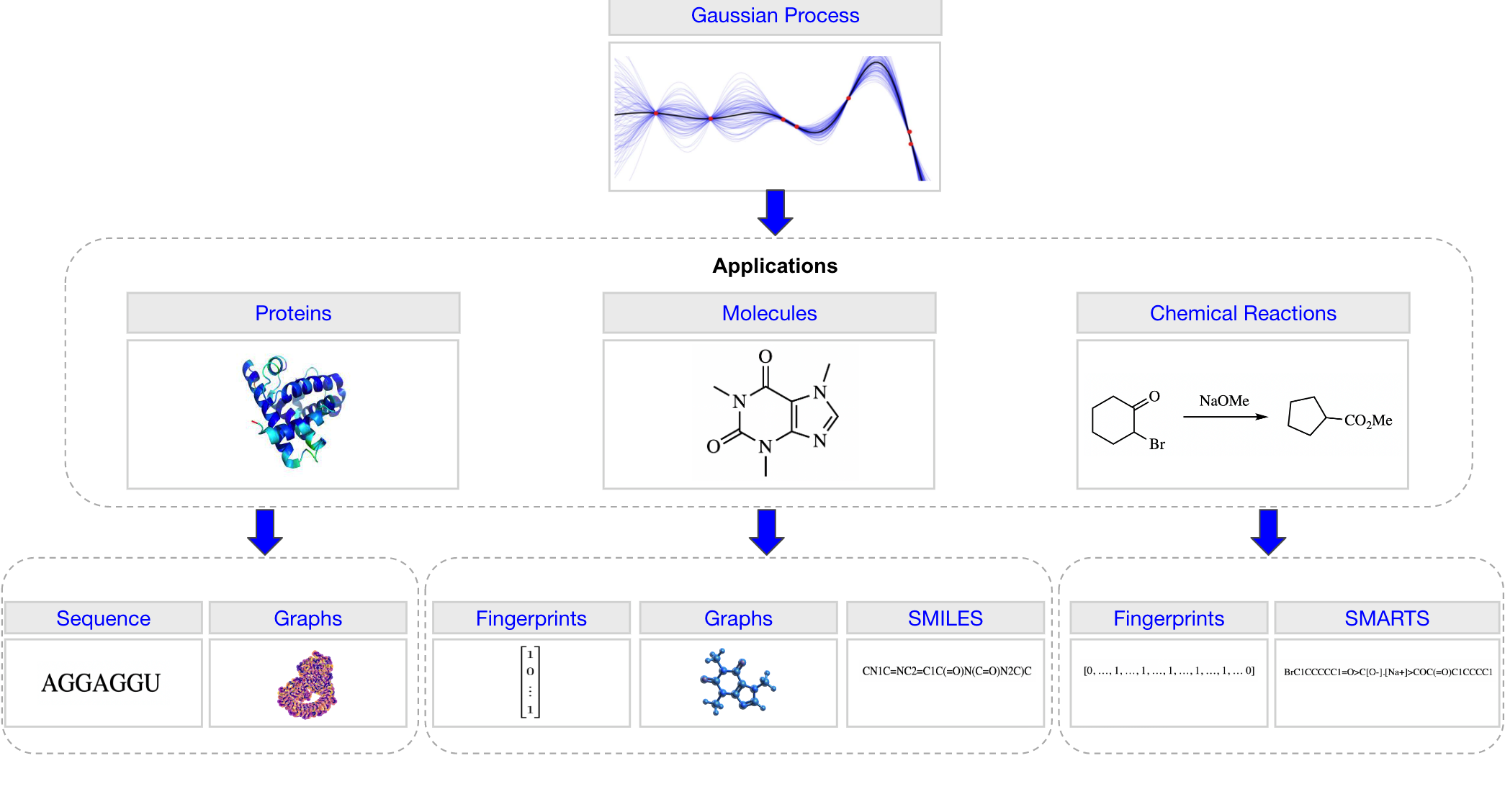
| Application | Representation |
|---|---|
| Molecules | ECFP Fingerprints [1] |
| Graphs [2] | |
| SMILES [3, 4] | |
| SELFIES [5] | |
| Chemical Reactions | One-Hot Encoding |
| Data-Driven Reaction Fingerprints [6] | |
| Differential Reaction Fingerprints [7] | |
| Reaction SMARTS | |
| Proteins | Sequences |
| Graphs [8] |
[1] Rogers, D. and Hahn, M., 2010. Extended-connectivity fingerprints. Journal of Chemical Information and Modeling, 50(5), pp.742-754.
[2] Fey, M., & Lenssen, J. E. (2019). Fast graph representation learning with PyTorch Geometric. arXiv preprint arXiv:1903.02428.
[3] Weininger, D., 1988. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of Chemical Information and Computer Sciences, 28(1), pp.31-36.
[4] Weininger, D., Weininger, A. and Weininger, J.L., 1989. SMILES. 2. Algorithm for generation of unique SMILES notation. Journal of Chemical Information and Computer Sciences, 29(2), pp.97-101.
[5] Krenn, M., Häse, F., Nigam, A., Friederich, P. and Aspuru-Guzik, A., 2020. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Machine Learning: Science and Technology, 1(4), p.045024.
[6] Probst, D., Schwaller, P. and Reymond, J.L., 2022. Reaction classification and yield prediction using the differential reaction fingerprint DRFP. Digital Discovery, 1(2), pp.91-97.
[7] Schwaller, P., Probst, D., Vaucher, A.C., Nair, V.H., Kreutter, D., Laino, T. and Reymond, J.L., 2021. Mapping the space of chemical reactions using attention-based neural networks. Nature Machine Intelligence, 3(2), pp.144-152.
[8] Jamasb, A., Viñas Torné, R., Ma, E., Du, Y., Harris, C., Huang, K., Hall, D., Lió, P. and Blundell, T., 2022. Graphein-a Python library for geometric deep learning and network analysis on biomolecular structures and interaction networks. Advances in Neural Information Processing Systems, 35, pp.27153-27167.
If GAUCHE is useful for your work please consider citing the following paper:
@misc{griffiths2022gauche,
title={GAUCHE: A Library for Gaussian Processes in Chemistry},
author={Ryan-Rhys Griffiths and Leo Klarner and Henry B. Moss and Aditya Ravuri and Sang Truong and Bojana Rankovic and Yuanqi Du and Arian Jamasb and Julius Schwartz and Austin Tripp and Gregory Kell and Anthony Bourached and Alex Chan and Jacob Moss and Chengzhi Guo and Alpha A. Lee and Philippe Schwaller and Jian Tang},
year={2022},
eprint={2212.04450},
archivePrefix={arXiv},
primaryClass={physics.chem-ph}
}