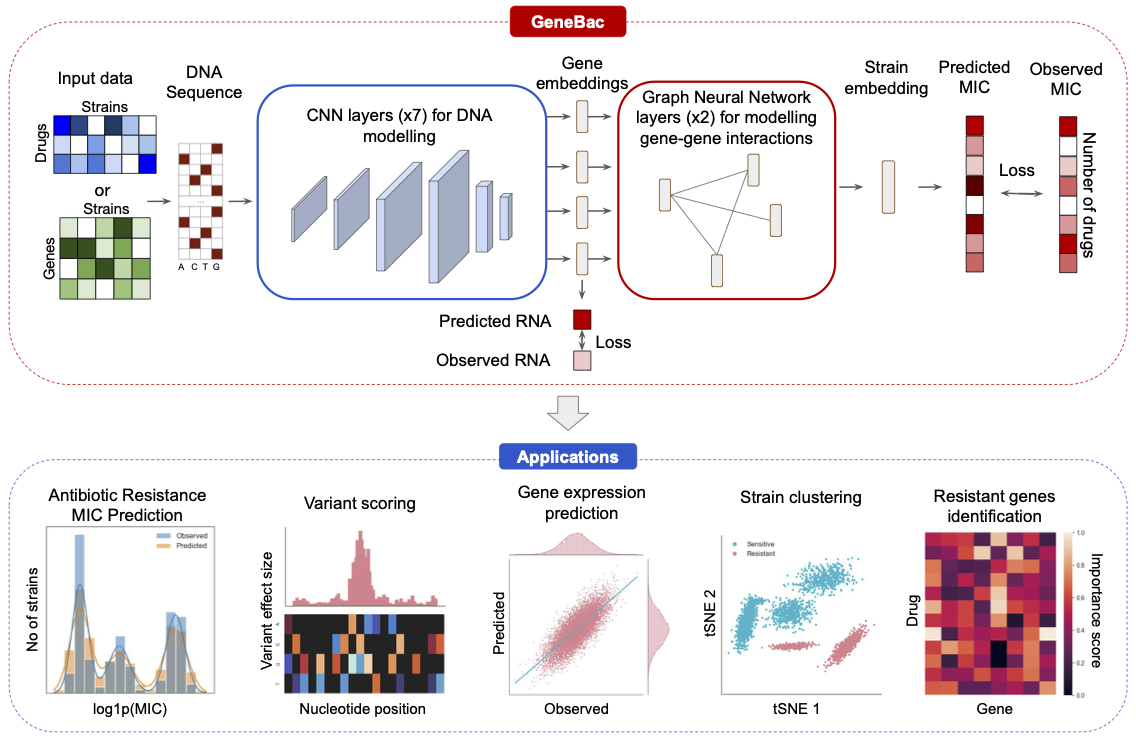
GeneBac is a modular framework for predicting antibiotic resistance in bacteria from DNA sequence. It takes as input the DNA sequence of selected genes with variants integrated into the sequence and outputs the predicted MIC to a set of drugs. GeneBac can also be used for other tasks, including variant effect scoring, gene expression prediction, resistant genes identification, and strain clustering.
This repository contains the training and code for the GeneBac model described in "Sequence-based modelling of bacterial genomes enables accurate antibiotic resistance prediction".
Table of contents
GeneBac is implemented in Python 3.9. To setup with the repo with all dependencies, clone the repository locally and create
an environment, for example with virtualenwrapper or conda. To install the dependencies, navigate to the directory
of the repository and run:
pip install .To train the GeneBac model for antibiotic resistance prediction, run:
python deep_bac/train_gene_pheno.py \
--regression \
--input-dir <input_dir> \
--output-dir <output_dir>For other arguments like batch size etc., see deep_bac/argparser.py or run python deep_bac/train_gene_pheno.py -h.
We provide a processed CRyPTIC Mycobacterium Tuberculosis dataset of 12,460 strains for training and evaluation on
Google drive (6.7 GB). The folder contains processed DNA sequences with variants integrated into the sequence as well as
training, test and cross-validation splits.
All the training were done with CUDA 10.2 on a single V100 or A100 GPU and takes less then 12 hours to train.
To evaluate the GeneBac model for antibiotic resistance prediction, run:
python deep_bac/train_gene_pheno.py \
--test \
--regression \
--input-dir <input_dir> \
--output-dir <output_dir> \
--ckpt-path <path_to_the_trained_model_checkpoint>To compute variant effect scores for a single variant, run:
import pandas as pd
from deep_bac.experiments.variant_scoring.variant_scoring import compute_variant_effect_size
from deep_bac.utils import load_trained_pheno_model
ckpt_path = "files/checkpoints/abr_mtb_regression.ckpt"
gene_interactions_file_dir = "files/gene_interactions/mtb/"
ref_gene_data_df = pd.read_parquet("files/reference_gene_data_mtb.parquet").set_index("gene")
model = load_trained_pheno_model(
ckpt_path=ckpt_path,
gene_interactions_file_dir=gene_interactions_file_dir,
)
# compute the variant effect size
variant_effect_size = compute_variant_effect_size(
model=model,
reference_gene_data_df=ref_gene_data_df,
gene="rpoB",
variant="g",
start_idx=1332,
end_idx=1333,
drug="RIF", # if None, will return all drug scores
)It is also possible to compute variant effect scores for a batch of variants.
To do it you firstly need to create a file with variants. An example file is provided in
files/example_variants.parquet.
Then, run:
python deep_bac/experiments/variant_scoring/run_batch_variant_scoring.py \
--output-dir <output_dir> \
--ckpt-path <path_to_the_trained_model_checkpoint> \
--variants-file-path <path_to_the_variants_file_in_parquet_format>One can use the trained MTB model checkpoint provided in files/checkpoints/ and the example variants file provided in files/example_variants.parquet
to compute the variant effect scores for the variants in the example file.
We also provide a set of variants with variant effect size computed using trained GeneBac model on CRyPTIC Mycobacterium Tuberculosis dataset for:
- variants present in WHO Mycobacterium Tuberculosis Catalogue
- SNP variants exceeding a threshold of
0.1variant effect size using In Silico Mutagenesis (ISM) method infiles/variant_effect_scores/.
To train the GeneBac model for gene expression prediction, run:
python deep_bac/train_gene_expr.py \
--input-dir <input_dir> \
--output-dir <output_dir> \
--lr 0.001 \
--max-epochs 100 \
--batch-size 256 \
--monitor-metric val_r2We provide a processed gene expression dataset of 386 Pseudomonas aeruginosa strains
on Google drive (220 MB), where each gene in a strain is a separate example.
The folder contains processed DNA sequences with variants integrated into the sequence as well as
training, validation and test splits.
To evaluate the model run:
python deep_bac/train_gene_expr.py \
--input-dir <input_dir> \
--output-dir <output_dir> \
--batch-size 256 \
--monitor-metric val_r2 \
--ckpt-path <path_to_the_trained_model_checkpoint> \
--testGeneBac can also be used to identify genes that are associated with antibiotic resistance to particular drugs. We compute the association using the DeepLift algorithm. To compute the gene-drug scores on a test set using a trained model, run:
python deep_bac/experiments/loci_importance/run_loci_importance.py \
--input-dir <input_dir> \
--output-dir <output_dir> \
--ckpt-path <path_to_the_trained_model_checkpoint>Here, the <input-dir> can be the same as the one used for antibiotic resistance prediction.
To cluster strains, we compute the strain embeddings using a trained GeneBac model. The embeddings can be then used for visualisation in two dimensions using a tSNE algorithm. To get the embeddings, run:
python deep_bac/experiments/latent_representations/latent_strain_representations.py
--input-dir <input_dir> \
--output-dir <output_dir> \
--ckpt-path <path_to_the_trained_model_checkpoint>We provide trained model checkpoints for antibiotic resistance prediction on the CRyPTIC Mycobacterium tuberculosis dataset
and gene expression prediction on the Pseudomonas aeruginosa dataset. The checkpoints can be found in files/checkpoints/.
If you find GeneBac useful in your work, please cite our paper:
@article{wiatrak2024genebac,
author = {Wiatrak, Maciej and Weimann, Aaron and Dinan, Adam M and Brbić, Maria and Floto, R. Andres},
title={Sequence-based modelling of bacterial genomes enables accurate antibiotic resistance prediction},
year={2024},
doi={10.1101/2024.01.03.574022},
url={https://www.biorxiv.org/content/10.1101/2024.01.03.574022v1},
journal={bioRxiv}
}GeneBac is licensed under the MIT License.