
- MassiveFold: parallelize protein structure prediction
- Installation
- Usage
- massivefold_plots: output representation
- Authors
MassiveFold is a tool that allows to massively expand the sampling of structure predictions by improving the computing of AlphaFold based predictions.
It optimizes the parallelization of the structure inference by splitting the computing on CPU for alignments, running automatically batches of structure predictions on GPU, and gathering the results in one global output directory, with a global ranking and a variety of plots.
MassiveFold uses AFmassive or ColabFold. as inference engine; AFmassive is an updated version of Björn Wallner's AFsample that offers additional diversity parameters for massive sampling.
It has been submitted for publication and a preprint is available here: https://doi.org/10.21203/rs.3.rs-4319486/v1.
MassiveFold's design (see schematic below) is optimized for GPU cluster usage. It allows fast computing for massive sampling by automatically splitting a large run of numerous predictions into several jobs. Each of these individual jobs are computed on a single GPU node and their results are then gathered as a single output with each prediction ranked on a global level instead of the level of each individual job.
This automatic splitting is also convenient for massive sampling on a single GPU server to manage jobs priorities.
MassiveFold is only available with the SLURM workload manager (Simple Linux Utility for Resource Management) as it relies heavily on its features (job array, job dependency, etc...).

A run is composed of three steps:
-
alignment: on CPU, sequence alignments is the first step (can be skipped if alignments are already computed)
-
structure prediction: on GPU, structure predictions follow the massive sampling principle. The total number of predictions is divided into smaller batches and each of them is distributed on a single GPU. These jobs wait for the alignment job to be over, if the alignments are not provided by the user.
-
post_treatment: on CPU, it finishes the job by gathering all batches outputs and produces plots with the plots module to visualize the run's performances. This job is executed only once all the structure predictions are over.
MassiveFold was developed to run massive sampling with AFmassive and ColabFold and relies on them for its installation.
- Retrieve MassiveFold
# clone MassiveFold's repository
git clone https://github.com/GBLille/MassiveFold.gitFor AFmassive runs, two additional installation steps are required to use MassiveFold for AFmassive runs:
- Download sequence databases
- Retrieve the neural network (NN) models parameters
For ColabFold runs, two additional installation steps are required to use MassiveFold for AFmassive runs:
- Download sequence databases
- Retrieve the neural network (NN) models parameters](https://github.com/GBLille/AFmassive?tab=readme-ov-file#alphafold-neural-network-model-parameters) and move them to a 'params' folder in the sequence databases folder
- Install MassiveFold
We use an installation based on conda. The install.sh script we provide installs the conda environments using the
environment.yml and mf-colabfold.yml files. One is created for MassiveFold and AFmassive, and another one is created
for ColabFold. It also creates the files architecture and set paths according to this architecture in the
AFmassive_params.json and/or ColabFold_params.json parameters file.
Help with:
cd MassiveFold
./install.sh -hInstallation with:
./install.sh [--only-envs] || --alphafold-db str --colabfold-db str [--no-env]
Options:
--alphafold-db <str>: path to AlphaFold database
--colabfold-db <str>: path to ColabFold database
--no-env: do not install the environments, only sets up the files and parameters.
At least one of --alphafold-db or colabfold-db is required with this option.
--only-envs: only install the environments (other arguments are not used)This file tree displays the files' architecture after running ./install.sh.
MassiveFold
├── install.sh
├── ...
├── examples
├── massivefold
└── massivefold_runs
├── AFmassive_params.json
├── ColabFold_params.json
├── headers/
├── example_header_alignment_jeanzay.slurm
├── example_header_jobarray_jeanzay.slurm
└── example_header_post_treatment_jeanzay.slurm
├── input/
├── log/
├── output/
└── run_massivefold.shThe directory massivefold_runs is created, which contains:
AFmassive_params.jsonto set the run parameters for AFmassive,ColabFold_params.jsonto set the run parameters for ColabFold,headers' directory, containing the headers that must be created to use MassiveFold. Examples are given for the Jean Zay national CNRS French cluster (ready to use, see the installation on Jean Zay to run MassiveFold directly on Jean Zay),inputwhich contains the FASTA sequences,logwith the logs of the MassiveFold runs (debug purposes),outputwhich contains the predictions,run_massivefold.shbeing the script to run MassiveFold
- Create header files
Refer to Jobfile's header for this installation step.
To run MassiveFold in parallel on your cluster/server, it is required to build custom jobfile headers for each step.
They are three and should be named as follows: {step}.slurm (alignment.slurm, jobarray.slurm and
post_treatment.slurm). The headers contain the parameters to give to SLURM for the jobs running (#SBATCH parameters).
They have to be added in MassiveFold/massivefold_runs/headers/ directory. Depending on your installation it can be
another path, this path has to be set in the AFmassive_params.json and/or ColabFold_params.json as
jobfile_headers_dir parameter.
Headers for Jean Zay cluster are provided as examples to follow (named example_header_<step>_jeanzay.slurm), to use
them, rename each one following the previously mentioned naming convention.
- Set custom parameters
Each cluster has its own specifications in parameterizing job files. For flexibility needs, you can add your custom
parameters in your headers, and then in the AFmassive_params.json and/or ColabFold_params.json file so that you can
dynamically change their values in the json file.
To illustrate these "special needs", here is an example of parameters that can be used on the French national Jean Zay cluster to specify GPU type, time limits or the project on which the hours are used:
Go to AFmassive_params.json and/or ColabFold_params.json location:
cd MassiveFold/massivefold_runsModify AFmassive_params.json and/or ColabFold_params.json:
{
"custom_params":
{
"jeanzay_gpu": "v100",
"jeanzay_project": "<project>",
"jeanzay_account": "<project>@v100",
"jeanzay_gpu_with_memory": "v100-32g",
"jeanzay_alignment_time": "05:00:00",
"jeanzay_jobarray_time": "15:00:00"
},
}And specify them in the jobfile headers (such as here for MassiveFold/headers/jobarray.slurm)
#SBATCH --account=$jeanzay_account
#SBATCH -C $jeanzay_gpu_with_memory
#SBATCH --time=$jeanzay_jobarray_time
The jobfiles for each step are built by combining the jobfile header that you have to create in MassiveFold/massivefold_runs/headers/ with the jobfile body in massivefold/parallelization/templates/.
Only the headers have to be adapted in function of your computing infrastructure. They contain the parameters to give to
SLURM for the job running (#SBATCH parameters).
Each of the three headers (alignment, jobarray and post treatment) must be located in the headers directory
(see File architecture section).
Their names should be identical to:
- alignment.slurm
- jobarray.slurm
- post_treatment.slurm
The templates work with the parameters provided in AFmassive_params.json and/or ColabFold_params.json file, given
as a parameter to the run_massivefold.sh script.
These parameters are substituted in the template job files thanks to the python library string.Template.
Refer to How to add a parameter for parameters substitution.
- Requirement: In the jobarray's jobfile header (massivefold_runs/headers/jobarray.slurm) should be stated that
it is a job array and the number of tasks in it has to be given. The task number argument is substituted with the
$substitute_batch_number parameter.
It should be expressed as:
#SBATCH --array=0-$substitute_batch_number
For example, if there are 45 batches, with 1 batch per task of the job array, the substituted expression will be:
#SBATCH --array=0-44
- Add these lines too in the headers, it is necessary to store MassiveFold's log:
In alignment.slurm:
#SBATCH --error=${logs_dir}/${sequence_name}/${run_name}/alignment.log
#SBATCH --output=${logs_dir}/${sequence_name}/${run_name}/alignment.log
In jobarray.slurm:
#SBATCH --error=${logs_dir}/${sequence_name}/${run_name}/jobarray_%a.log
#SBATCH --output=${logs_dir}/${sequence_name}/${run_name}/jobarray_%a.log
In post_treatment.slurm:
#SBATCH --output=${logs_dir}/${sequence_name}/${run_name}/post_treatment.log
#SBATCH --error=${logs_dir}/${sequence_name}/${run_name}/post_treatment.log
We provide headers for the Jean Zay French CNRS national GPU cluster (IDRIS,) that can also be used as examples for your own infrastructure.
- Add $new_parameter or ${new_parameter} in the template's header where you want its value to be set and
in the "custom_params" section of
AFmassive_params.jsonand/orColabFold_params.jsonwhere its value can be specified and modified conveniently for each run.
Example in the json parameters file for Jean Zay headers:
{
"custom_params":
{
"jeanzay_account": "project@v100",
"jeanzay_gpu_with_memory": "v100-32g",
"jeanzay_jobarray_time": "10:00:00"
}
}Where "project" is your 3 letter project with allocated hours on Jean Zay.
- These parameters will be substituted in the header where the parameter keys are located:
#SBATCH --account=$jeanzay_account
#SBATCH --error=${logs_dir}/${sequence_name}/${run_name}/jobarray_%a.log
#SBATCH --output=${logs_dir}/${sequence_name}/${run_name}/jobarray_%a.log
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task=8
#SBATCH --hint=nomultithread
#SBATCH --gpus-per-node=1
#SBATCH --array=0-$substitute_batch_number
#SBATCH --time=$jeanzay_jobarray_time
##SBATCH --qos=qos_gpu-t4 # Uncomment for job requiring more than 20h (max 16 GPUs)
#SBATCH -C $jeanzay_gpu_with_memory # GPU type+memory
- Never use single $ symbol for other uses than parameter/value substitution from the json file.
To use $ inside the template files (bash variables or other uses), use instead $$ as an escape following string.Template documentation.
To use it on Jean Zay, the only installation steps are:
git clone https://github.com/GBLille/MassiveFold.git
./install.shThe same file architecture is built, follow the usage section to use MassiveFold.
Edit the AFmassive_params.json or ColabFold_params.json parameters file (see file architecture).
Set first the parameters of your run in the
AFM_run section of the AFmassive_params.json, for instance:
{
"AFM_run": {
"db_preset": "full_dbs",
"use_gpu_relax": "true",
"models_to_relax": "none",
"dropout": "false",
"dropout_structure_module": "false",
"dropout_rates_filename": "",
"max_recycles": "20",
"early_stop_tolerance": "0.5",
"bfd_max_hits": "100000",
"mgnify_max_hits": "501",
"uniprot_max_hits": "50000",
"uniref_max_hits": "10000",
"model_preset": "multimer",
"templates": "true",
"stop_recycling_below": "0",
"min_score": "0",
"max_score": "1"
}
}or in the ColabFold_params.json file, for instance:
{
"CF_run": {
"model_preset": "multimer",
"use_dropout": "false",
"num_recycle": "20",
"recycle_early_stop_tolerance": "0.5",
"stop_at_score": "100",
"disable_cluster_profile": "false"
}
}Then you can set the parameters of the custom_params section if necessary and the plots section.
Activate the conda environment, then launch MassiveFold.
conda activate massivefold
./run_massivefold.sh -s <SEQUENCE_PATH> -r <RUN_NAME> -p <NUMBER_OF_PREDICTIONS_PER_MODEL> -f <JSON_PARAMETERS_FILE> N.B.: on the Jean Zay cluster, load the massivefold module instead of activating the conda environment
Example for AFmassive:
./run_massivefold.sh -s input/H1140.fasta -r afm_default_run -p 5 -f AFmassive_params.jsonExample for ColabFold:
./run_massivefold.sh -s input/H1140.fasta -r cf_default_run -p 5 -f ColabFold_params.json -t ColabFoldIf the multiple sequence alignments have already been run and are present in the output folder, they won't be computed, but you can force their recomputation with:
./run_massivefold.sh -s input/H1140.fasta -r afm_default_run -p 5 -f AFmassive_params.json -aExample to only run the alignments with AFmassive (JackHMMer and HHblits):
./run_massivefold.sh -s input/H1140.fasta -r afm_default_run -p 1 -f AFmassive_params.json -oor with ColabFold(MMseqs2):
./run_massivefold.sh -s input/H1140.fasta -r cf_default_run -p 1 -f ColabFold_params.json -t ColabFold -oFor more help and list of required and facultative parameters, run:
./run_massivefold.sh -hHere is the help message given by this command:
Usage: ./run_massivefold.sh -s str -r str -p int -f str [-t str] [ -b int | [[-C str | -c] [-w int]] ] [-m str] [-n str] [-a] [-o]
./run_massivefold.sh -h for more details
Required arguments:
-s| --sequence: path of the sequence(s) to infer, should be a 'fasta' file
-r| --run: name chosen for the run to organize in outputs.
-p| --predictions_per_model: number of predictions computed for each neural network model.
-f| --parameters: json file's path containing the parameters used for this run.
Facultative arguments:
-b| --batch_size: (default: 25) number of predictions per batch, should not be higher than -p.
-C| --calibration_from: path of a previous run to calibrate the batch size from (see --calibrate).
-w| --wall_time: (default: 20) total time available for calibration computations, unit is hours.
-m| --msas_precomputed: path to directory that contains computed msas.
-n| --top_n_models: uses the n neural network models with best ranking confidence from this run's path.
Facultative options:
-t| --tool_to_use: (default: 'AFmassive') Use either AFmassive or ColabFold in structure prediction for MassiveFold
-o| --only_msas: only compute alignments, the first step of MassiveFold
-c| --calibrate: calibrate --batch_size value. Searches from the previous runs for the same 'fasta' path given
in --sequence and uses the longest prediction time found to compute the maximal number of predictions per batch.
This maximal number depends on the total time given by --wall_time.
-a| --recompute_msas: purges previous alignment step and recomputes msas.It launches MassiveFold with the same parameters introduced above but instead of running AFmassive or ColabFold a single time, it divides it into multiple batches.
For the following examples, we assume that --model_preset=multimer as it is the majority of cases to run MassiveFold in parallel.
However, --model_preset=monomer_ptm works too and needs to be adapted accordingly, at least the models to use (if parameter not set as default).
You can decide how the run will be divided by assigning run_massivefold.sh parameters e.g.:
./run_massivefold.sh -s ./input/H1140.fasta -r 1005_preds -p 67 -b 25 -f AFmassive_params.jsonThe predictions are computed individually for each neural network (NN) model, -p or --predictions_per_model
allows to specify the number of predictions desired for each chosen model.
These --predictions_per_model are then divided into batches with a fixed -b or --batch_size to optimize the
run in parallel as each batch can be computed on a different GPU, if available.
The last batch of each NN model is generally smaller than the others to match the number of predictions fixed by
--predictions_per_model.
N.B.: an interest to use run_massivefold.sh on a single server with a single GPU is to be able to run massive
sampling for a structure in low priority, allowing other jobs with higher priority to be run in between.
For example, with -b 25 and -p 67 the predictions are divided into the following batches (separated runs), which are repeated for each NN model:
- First batch: --start_prediction=0 and --end_prediction=24
- Second batch: --start_prediction=25 and --end_prediction=49
- Third batch: --start_prediction=50 and --end_prediction=67
By default (if --models_to_use is not assigned), all NN models are used: with --model_preset=multimer,
15 models in total = 5 neural network models
The prediction number per model can be adjusted, here with 67 per model and 15 models, it amounts to 1005 predictions in total divided into 45 batches, these batches can therefore be run in parallel on a GPU cluster infrastructure.
The batch size can also be auto calibrated with the -c or -C parameters if at least one basic run has already been
performed. The -c parameter will automatically search in the output folder that corresponds to the input sequence for
the longest prediction duration. These options have to be coupled with the -w walltime parameter (it is advised to
adapt this walltime value to the one of the job). For instance:
./run_massivefold.sh -s ./input/H1140.fasta -r 1005_preds -p 67 -f AFmassive_params.json -c -w 10In addition to the parameters displayed with -h option, the json parameters file set with -f or --parameters
should be organized like the AFmassive_params.json or ColabFold_params.json file.
Each section of AFmassive_params.json or ColabFold_params.json is used for a different purpose.
The massivefold section designates the whole run parameters.
{
"massivefold":
{
"run_massivefold": "run_AFmassive.py",
"run_massivefold_plots": "../massivefold/massivefold_plots.py",
"data_dir": "/gpfsdswork/dataset/AlphaFold-2.3.1",
"jobfile_headers_dir": "./headers",
"jobfile_templates_dir": "../massivefold/parallelization/templates",
"output_dir": "./output",
"logs_dir": "./log",
"input_dir": "./input",
"models_to_use": "",
"keep_pkl": "false",
"scripts_dir": "../massivefold/parallelization"
}
}The paths in the section are filled by install.sh but can be changed here if necessary.
Headers (jobfile_headers_dir) are specified to setup the run, in order to give the parameters that are required to
run the jobs on your cluster/server.
Build your own according to the Jobfile's header building section.
models_to_use is the list of NN models to use. To select which NN models are used, separate them with a comma e.g.:
"model_3_multimer_v1,model_3_multimer_v3", by default all are used
pkl_format: how to manage pickle files
- ‘full’ to keep the pickle files generated by the inference engine,
- ‘light’ to reduce its size by selecting main components, which are: number of recycles, PAE values, max PAE,
plddt scores, ptm scores, iptm scores and ranking confidence values (stored in ./light_pkl directory)
- ‘none’ to remove them
- The custom_params section is relative to the personalized parameters that you want to add for your own cluster. For instance, for the Jean Zay GPU cluster:
{
"custom_params":
{
"jeanzay_project": "project",
"jeanzay_account": "project@v100",
"jeanzay_gpu_with_memory": "v100-32g",
"jeanzay_alignment_time": "10:00:00",
"jeanzay_jobarray_time": "10:00:00"
}
}As explained in How to add a parameter, these variables are substituted by their value when the jobfiles are created.
- For AFmassive, the AFM_run section gathers all the parameters used by MassiveFold for the run
(see AFmassive parameters
section). All parameters except --keep_pkl, --models_to_relax, --use_precomputed_msas, --alignment_only,
--start_prediction, --end_prediction, --fasta_path and --output_dir are exposed in this section.
You can adapt the parameter values in function of your needs.
The non exposed parameters mentioned before are set internally by the Massivefold's pipeline or in the massivefold
section (models_to_use and pkl_format).
{
"AFM_run":
{
"db_preset": "full_dbs",
"use_gpu_relax": "true",
"models_to_relax": "none",
"dropout": "false",
"dropout_structure_module": "false",
"dropout_rates_filename": "",
"max_recycles": "20",
"early_stop_tolerance": "0.5",
"bfd_max_hits": "100000",
"mgnify_max_hits": "501",
"uniprot_max_hits": "50000",
"uniref_max_hits": "10000",
"model_preset": "multimer",
"templates": "true",
"stop_recycling_below": "0",
"min_score": "0",
"max_score": "1"
}
}Lastly, the plots section is used for the MassiveFold plotting module.
"plots":
{
"MF_plots_top_n_predictions": "10",
"MF_plots_chosen_plots": "coverage,DM_plddt_PAE,CF_PAEs,score_distribution,recycles"
}colabfold_relax developed by the ColabFold team can be used to relax selected predictions. For help, type:
colabfold_relax -hMassiveFold plotting module can be used on a MassiveFold output to evaluate visually its predictions.
Here is an example of a basic command you can run:
conda activate massivefold
massivefold_plots.py --input_path=<path_to_MF_output> --chosen_plots=DM_plddt_PAE-
--input_path: it designates MassiveFold output dir and the directory to store the plots except if you want them in a separate directory (use
--output_pathfor this purpose) -
--chosen_plots: plots you want to get. You can give a list of plot names separated by a coma (e.g.:
--chosen_plots=coverage,DM_plddt_PAE,CF_PAEs).
Here is the list of available plots:
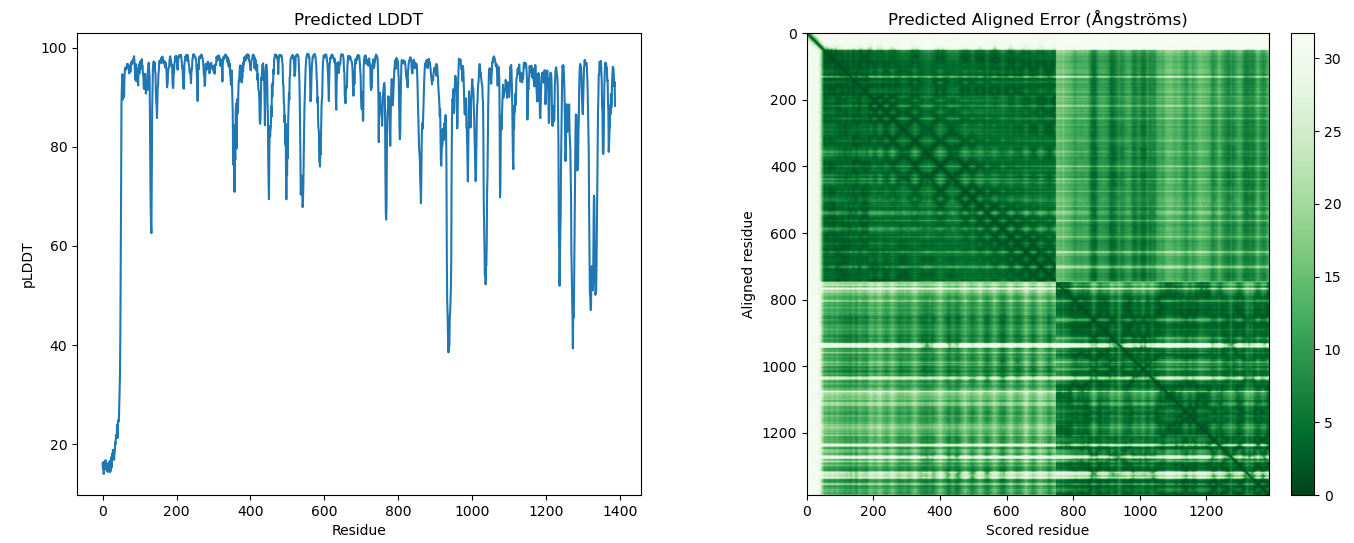
- DM_plddt_PAE: Deepmind's plots for predicted lddt per residue and predicted aligned error matrix
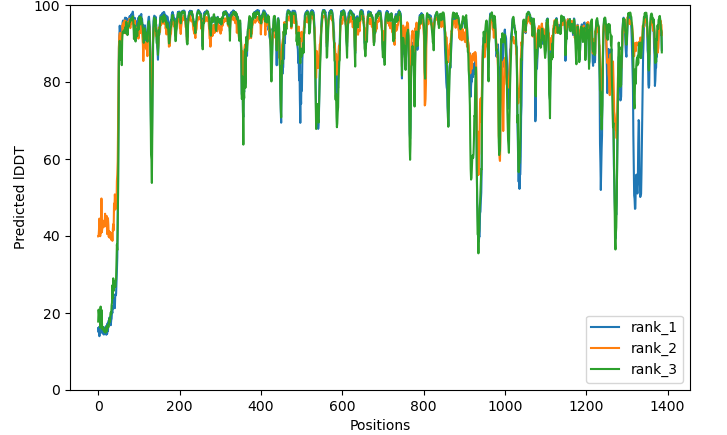
- CF_plddts: ColabFold's plot for predicted lddt per residue
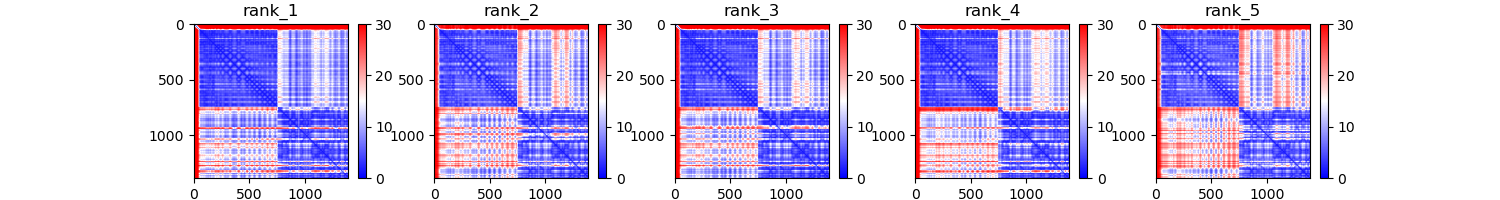
- CF_PAEs: ColabFold's plot for predicted aligned error of the n best predictions set with --top_n_predictions
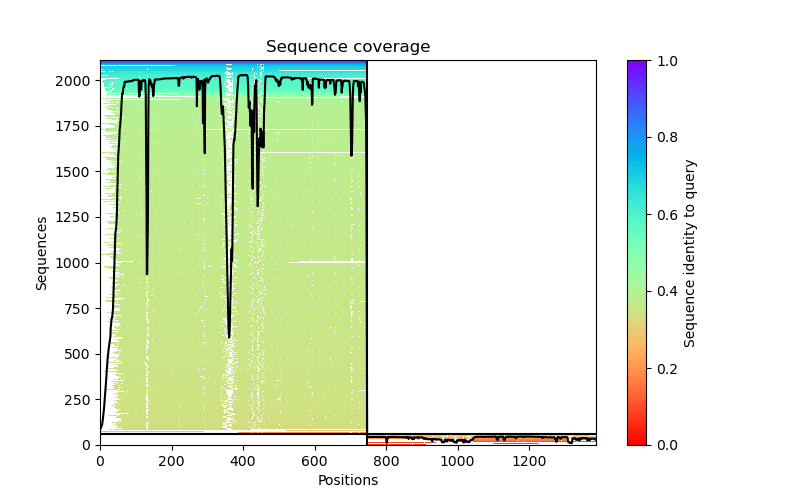
- coverage: ColabFold's plot for sequence alignment coverage
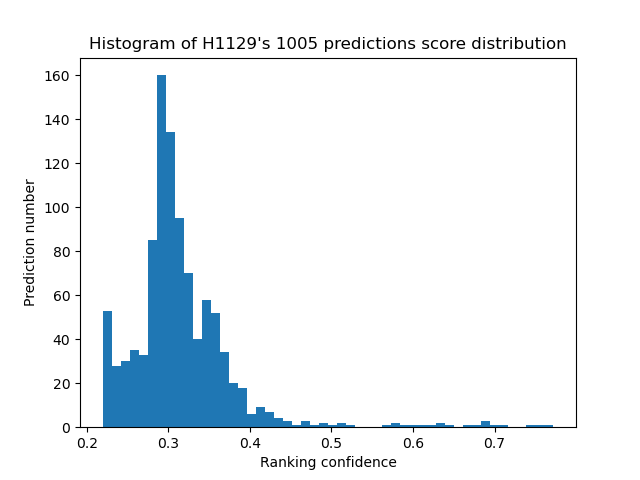
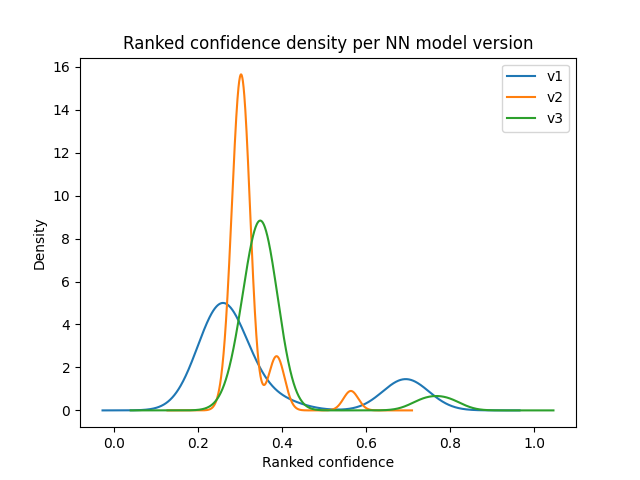
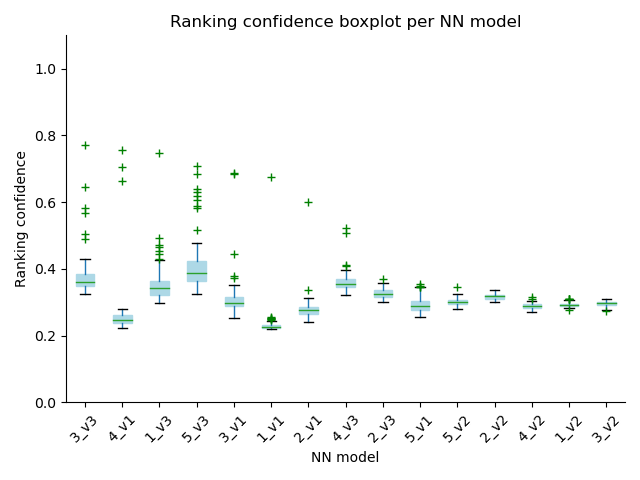
- score_distribution: performs 3 plots that summarize the score's distribution at three levels:
- an histogram of all scores indiscriminately
- a density plot for each neural network model version
- a boxplot for each neural network model
- an histogram of all scores indiscriminately
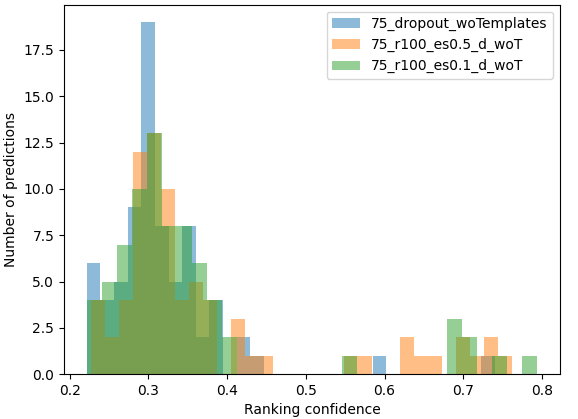
- distribution_comparison: ranking confidence distribution comparison between various MassiveFold outputs, typically
useful for runs with different sets of parameters on the same input sequence(s).
- recycles: ranking confidence during the recycle process (only for multimers and ColabFold monomers)
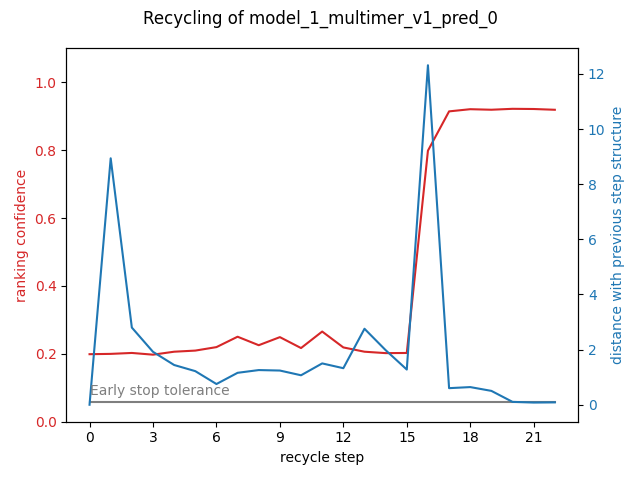









--top_n_predictions: (default 10), number of best predictions to take into account for plotting--runs_to_compare: names of the runs you want to compare on their distribution, this argument is coupled with --chosen_plots=distribution_comparison
More help with
conda activate massivefold
massivefold_plots.py --helpNessim Raouraoua (UGSF - UMR 8576, France)
Claudio Mirabello (NBIS, Sweden)
Thibaut Véry (IDRIS, France)
Christophe Blanchet (IFB, France)
Björn Wallner (Linköping University, Sweden)
Marc F Lensink (UGSF - UMR8576, France)
Guillaume Brysbaert (UGSF - UMR 8576, France)
This work was carried out as part of Work Package 4 of the MUDIS4LS project led by the French Bioinformatics Institute (IFB). It was initiated at the IDRIS Open Hackathon, part of the Open Hackathons program. The authors would like to acknowledge OpenACC-Standard.org for their support.