{kind=link}
Leveraging Machine Learning To Predict Binding Affinity Of Drugs To Their Target Based Only On Their Structural Embeddings.
The data utilized in this project was generated by the IDG-Kinase group with funding from the NIH Common Fund. It can be orginally accessed through a synapse account from the following website https://www.synapse.org/#!Synapse:syn15667962/wiki/583305
Alternatively it can also be downloaded from the link below http:https://drugtargetcommons.fimm.fi/static/Excell_files/DTC_data.csv
Data from other sources was used for building the model, they were obtained from: https://www.ebi.ac.uk/chembl/ https://www.ncbi.nlm.nih.gov/ https://www.uniprot.org/
- DeepChem: For creating molecular fingerprints
- RDKit: DeepChem requires RDKit for creating fingerprints from SMILES
- SGT: For creating protein embedding
- Other common NLP and ML packages were used such as, sklearn, wordcloud, gensim, etc.
I would like to thank my instructors from Flatiron, Dan Sanz, Dave Baker, Jon Tyler, and Wachira Ndaiga for helping me with this project.
The focus of this project is in seeking to evaluate the power of statistical and machine learning models as a systematic and cost-effecive means for catalyzing compound-target interaction mapping efforts by prioritizing most potent interactions for further experiemental evaluation. In this project the dataset used, focuses on Kinase inhibitors due to their clinical importantance. I carried out this independent project as part of Flatirons final capstone project. In order to train my models I have used the virtual machines provided by Paperspace.
Mapping the complete target space of drugs and drug-like compounds, including both intended ‘primary targets’ as well as secondary ‘off-targets’, is a critical part of drug discovery efforts. Such a map would enable one not only to explore the therapeutic potential of chemical agents but also to better predict and manage their possible adverse effects prior to clinical trials. However, the massive size of the chemical universe makes experimentally mapping the bioactivity of the full space of compound-target interactions quickly infeasible in practice, even with automated high-throughput profiling assays.

Before I begin I wanted to go over the project workflow and the order in which the different notebooks were run and steps taken.
├── Dataset <- Dataset downloaded as Zip file from drug target commons
├── README.md <- Top level overview for anyone using this repository.
├── data_filtering.ipynb
│ ├── concentration value <- Filtering compounds based on their concentration type.
│ ├── target type <- Removed mutant proteins from dataset.
│ └── droping values <- Dropping duplicates, null and columns not used for analaysis
│
├── smiles_retrieval <- Obtained SMILES data on filtered set of compounds using chemblID
│
├── protein_retrieval <- Obtained Protein sequencen FASTA files from Uniprot using TargetID
│
├── embedding.ipynb <- Embedding protein sequences and SMILES to use in ML models
│ ├── ECFP4 <- ECFP4 fingerprints turn SMILES notation into vector of dimension 1024
│ ├── PSSM <- PSSM embeddings of protein sequences obtained using NCBI blas
│ └── SGT <- SGT embeddings of protein sequences performed using SGT library
│
├── reg_models.ipynb <- Notebook containing 3 different models
│ ├── MLP
│ ├── XGBoost
│ └── Random Forest
│
├── clustering.ipynb <- using clustering methods to assing target proteins in different cluster
│
├── reg_models-on-cluster.ipynb
│ └── RF and MLP <- Final models run on the clusters for each embedding to check if there is any
│ improvement
│
└── utilities.py <- Functions used for this project
In order to build a drug-kinase prediction model its important to first represent molecules. I have used Extendent Connectivity Finger Prints to represent molecules, which is a standard way to represent structural information inside the molecule. They were implemented using deepchem package, which builds molecules from SMILES and then analyses the substructures in the molecules to create binary representation of the molecules. The resulting output is a 1024 dimension vector.
Encoding methods There are several ways to encode protein sequences in a machine readable format. One of the most basic way to represent sequences would be to using one hot encoding. It is the easiest method to implement and can be done in python in the following way.
codes = ['A', 'C', 'D', 'E', 'F', 'G', 'H', 'I', 'K', 'L',
'M', 'N', 'P', 'Q', 'R', 'S', 'T', 'V', 'W', 'Y']
def show_matrix(m):
#display a matrix
cm = sns.light_palette("seagreen", as_cmap=True)
display(m.style.background_gradient(cmap=cm))
def one_hot_encode(seq):
o = list(set(codes) - set(seq))
s = pd.DataFrame(list(seq))
x = pd.DataFrame(np.zeros((len(seq),len(o)),dtype=int),columns=o)
a = s[0].str.get_dummies(sep=',')
a = a.join(x)
a = a.sort_index(axis=1)
show_matrix(a)
e = a.values.flatten()
return eSince there are twenty amino acids we create a list containing all the letters found in peptide sequences. So if we have a sequence ''ALDFEQEMT' and run the function then find the following output.
pep='ALDFEQEMT'
e=one_hot_encode(pep)
As we see this method is not feasible for larger sequences. In the dataset we are working with the largest sequence has the length of 2500. Using one hot encoding will be computationally expensive. There are other forms of encoding available, such as NLF encoding, and Blossum Encoding. They are also computationally expesive but perform better than one hot encoding because they capture the meaning behind the amino acid sequences.
Embedding There are several embedding methods available to use to reduce the dimensions of the integer encoded sequences. Some of the common NLP embedding methods and libraries like Word2Vec, TransformerXL, Seq2Vec have been used to embed protein sequences and have proven to be useful in some cases. To embed the sequences requires the training of the embedding model on large corpuse of amino acid sequecnes. There are some common pretrained protein embedding methods available, these include:
- PSSM: Position Specific Scoring Matrix
- SGT: Sequence Graph Transform
- TAPE: Tasks Assessing Protein Embeddings
- PepNet
- SeqVec
After experiemting with all of them, I finally chose to run my models on PSSM and SGT, as they allow for creation of a 400 dimension embedding vector which is easy to work with, without having to use additional computational resources. I thought they were viable for use for this project. I have found that overall PSSM based embeddings resulted in a model with the lowest MSE score, but it would take longer to run compared to the SGT embeded model. The difference between their predictive performance were not too large.
I have ran 3 different models to create embeds for protein sequences and compared them to teh PSSM based encoding. The task was to predict the pChemBL value, which indicated the binging affinity betweeen drug and their target. We use the ECFP4 molecular fingerprint and the embeded proteins to train the model to predict . pChemBL value.
I have found that that Randomforest model after performing gridsearvhCV gave the best performance on both types of embeds. The results were almost similar, they have the same pearson's correlation coefficient of 0.86. The PSSM embedded protein sequences gave a mean squared error of 0.42.
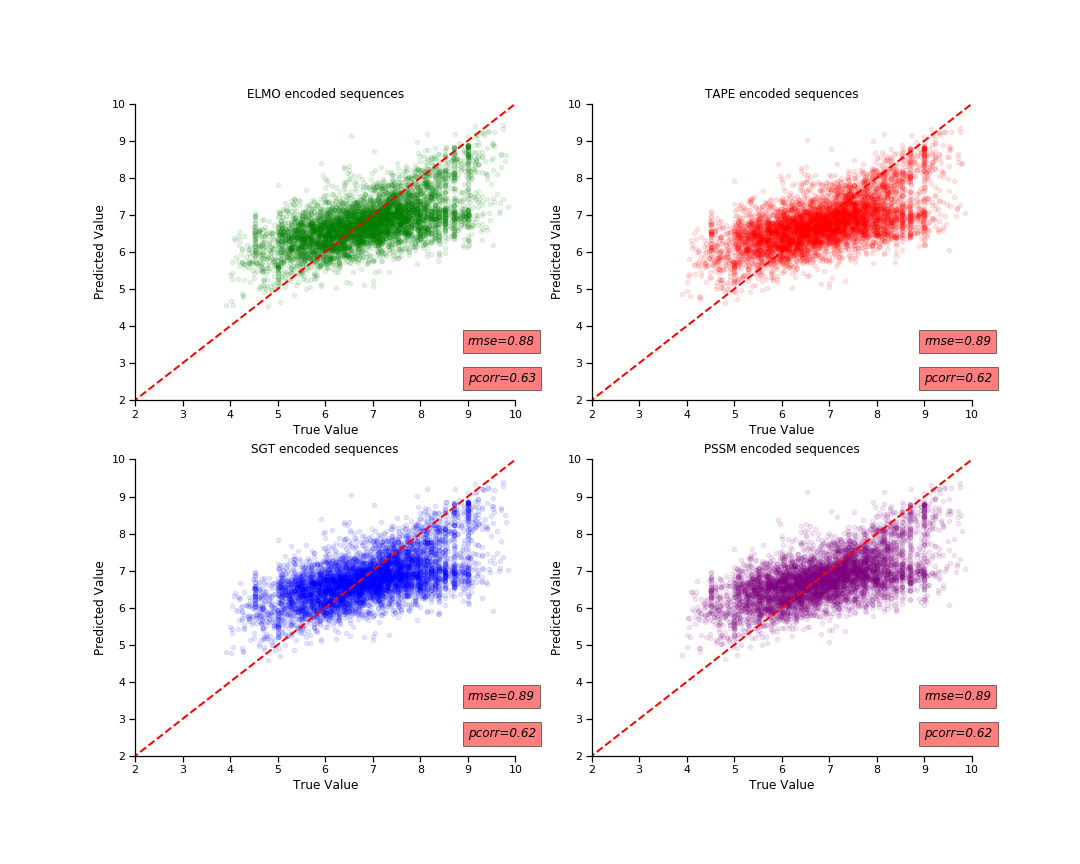
The next best model I obtained was the use of simple MLP regressor. It had a good correlation coefficient but had a lot of variance and spread resulting in poorer MSE score. I have therefore just chosen to work with random forest model
When we plot a dendrogram on our protein sequence data we see that our data can be split into 2 separate clusters.

I tried Agglomerative Clustering methods to cluster protein sequences into 3 separate groups. After having created those clusters. Clusters were created for both embedding methods, I have noticed that SGT embedded sequences were much better at clustering the proteins in different groups based on the of number of proteins present in each clusters.
After having carried out the clusters, I repeated my model training and prediction on different clusters. The results are shown below:

The figure above shows results for RandomForest model. We notice that there is no major difference in either of the metrics. Their MSE score decreases by 0.1 but their correleation coefficent remains unchanged.

The figure above shows results for MLP regressor Compared to the randomforest model, MLP regressors performance for both embedding methods have improved by a lot. There is a much better increase in metrics associated with SGT embeds, this might be because SGT embeds has been implemented initially to cluster protein sequences and so they seem to do a better job in creating clusters, which may explain why they perform better.
Summary: In short it is possible to use machine learning approaches to predict the interaction of drugs with their target. In the project we notice that the prediction is conservative at best, where the model predicts low pChemb value for when the actual is higher and for when predicts a higher pChemb value for those where the actual is lower. It can still be useful in weeding particularly low interacting drugs and help save money in the long run.