A tool for domain-based annotation with databases from the Conserved Domains Database.
- Features
- Installing reCOGnizer
- Annotation with reCOGnizer
- Output
- Other parameters
- Referencing reCOGnizer
reCOGnizer performs domain-based annotation with RPS-BLAST and databases from CDD as reference.
- Reference databases currently implemented: CDD, NCBIfam, Pfam, TIGRFAM, Protein Clusters, SMART, COG and KOG.
- reCOGnizer performs multithread annotation with RPS-BLAST, significantly increasing the speed of annotation.
- After domain assignment to proteins, reCOGnizer converts CDD IDs to the IDs of the respective DBs, and obtains domain descriptions available at CDD.
- Further information is retrieved depending on the database in question:
- NCBIfam, Pfam, TIGRFAM and Protein Clusters annotations are complemented with taxonomic classifications and EC numbers.
- SMART annotations are complemented with SMART descriptions.
- COG and KOG annotations are complemented with COG/KOG categories, EC numbers and KEGG Orthologs.
A detailed representation of reCOGnizer's workflow is presented in Fig. 1.
To install reCOGnizer, simply run: conda install -c conda-forge -c bioconda recognizer
The simplest way to run reCOGnizer is to just specify the fasta filename and an output directory - though even the output directory is not mandatory.
recognizer -f input_file.faa -o output_directory
reCOGnizer takes a FASTA file (of aminoacids, commonly either .fasta or .faa) as input and produces two main outputs into the output directory:
reCOGnizer_results.tsvandreCOGnizer_results.xlsx, tables with the annotations from every database for each proteincog_quantification.tsvand respective Krona representation (Fig. 2), which describes the functional landscape of the proteins in the input file
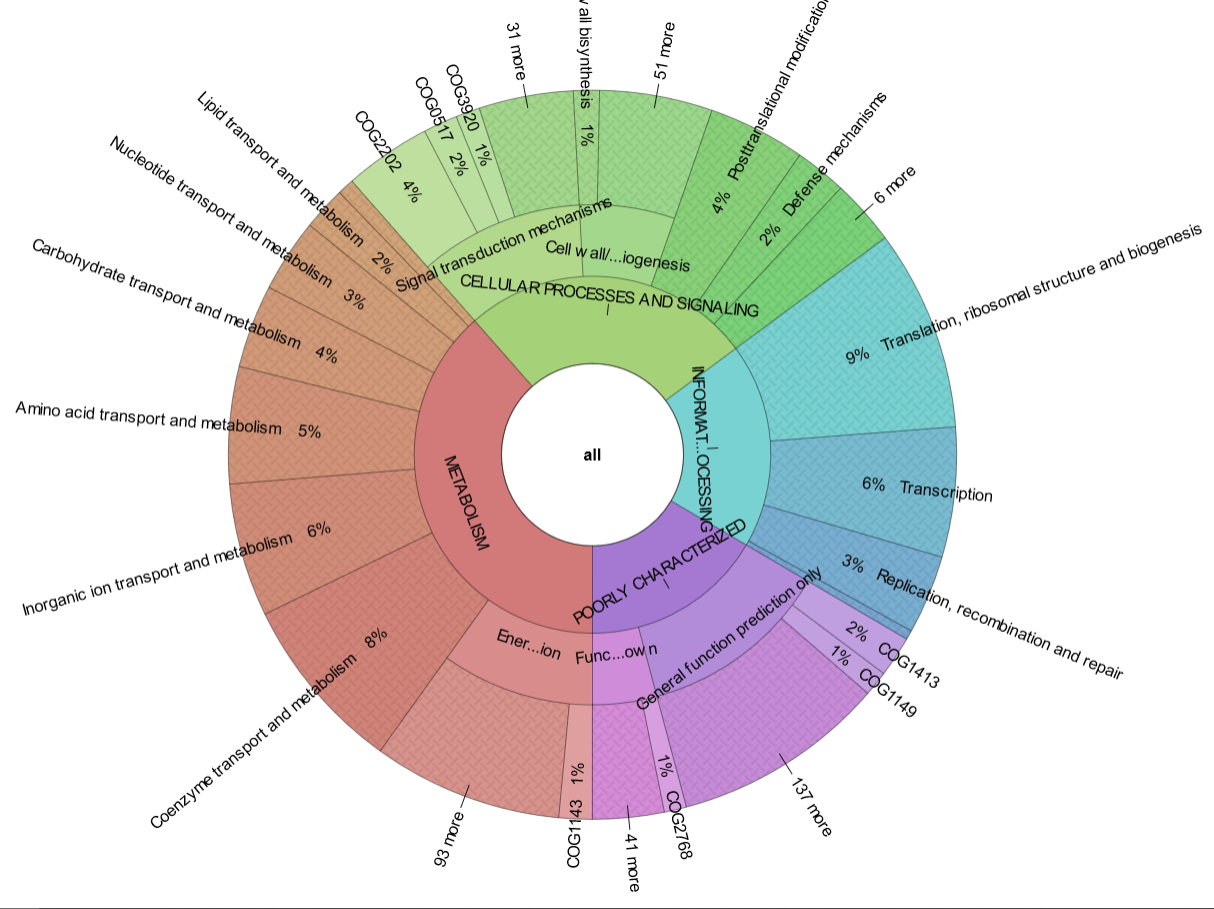
Fig. 2. Krona plot with the quantification of COGs identified in the simulated dataset used to test MOSCA and reCOGnizer. Click in the plot to see the interactive version that is outputed by reCOGnizer.
reCOGnizer can make use of taxonomic information by filtering Markov Models for the specific taxa of interest. This can be done by providing a file with the taxonomic information of the proteins. To simulate this, run the following commands, after installing reCOGnizer:
git clone https://github.com/iquasere/reCOGnizer.git
cd reCOGnizer/ci
recognizer -f proteomes.fasta --f UPIMAPI_results.tsv --tax-col 'Taxonomic lineage IDs (SPECIES)' --protein-id-col qseqid --species-taxids
Running reCOGnizer this way will usually obtain better results, but will likely take much longer to finish.
options:
-h, --help show this help message and exit
-f FILE, --file FILE Fasta file with protein sequences for annotation
-t THREADS, --threads THREADS
Number of threads for reCOGnizer to use [max available]
--evalue EVALUE Maximum e-value to report annotations for [1e-3]
-o OUTPUT, --output OUTPUT
Output directory [reCOGnizer_results]
-dr DOWNLOAD_RESOURCES, --download-resources DOWNLOAD_RESOURCES
This parameter is deprecated. Please do not use it [None]
-rd RESOURCES_DIRECTORY, --resources-directory RESOURCES_DIRECTORY
Output directory for storing databases and other resources [~/recognizer_resources]
-dbs DATABASES, --databases DATABASES
Databases to include in functional annotation (comma-separated) [all available]
--custom-databases If databases inputted were NOT produced by reCOGnizer [False]. Default databases of reCOGnizer (e.g., COG, TIGRFAM, ...) can't be used simultaneously with custom
databases. Use together with the '--databases' parameter.
-mts MAX_TARGET_SEQS, --max-target-seqs MAX_TARGET_SEQS
Number of maximum identifications for each protein [1]
--keep-spaces BLAST ignores sequences IDs after the first space. This option changes all spaces to underscores to keep the full IDs.
--no-output-sequences
Protein sequences from the FASTA input will be stored in their own column.
--no-blast-info Information from the alignment will be stored in their own columns.
--output-rpsbproc-cols
Output columns obtained with RPSBPROC - 'Superfamilies', 'Sites' and 'Motifs'.
-sd SKIP_DOWNLOADED, --skip-downloaded SKIP_DOWNLOADED
This parameter is deprecated. Please do not use it [None]
--keep-intermediates Keep intermediate annotation files generated in reCOGnizer's workflow, i.e., ASN, RPSBPROC and BLAST reports and split FASTA inputs.
--quiet Don't output download information, used mainly for CI.
--debug Print all commands running in the background, including those of rpsblast and rpsbproc.
--test-run This parameter is only appropriate for reCOGnizer's tests on GitHub. Should not be used.
-v, --version show program's version number and exit
Taxonomy Arguments:
--tax-file TAX_FILE File with taxonomic identification of proteins inputted (TSV). Must have one line per query, query name on first column, taxid on second.
--protein-id-col PROTEIN_ID_COL
Name of column with protein headers as in supplied FASTA file [qseqid]
--tax-col TAX_COL Name of column with tax IDs of proteins [Taxonomic identifier (SPECIES)]
--species-taxids If tax col contains Tax IDs of species (required for running COG taxonomic)
If you use reCOGnizer, please cite its publication.