QUANTITATIVE HUMAN
PHYSIOLOGY
QUANTITATIVE
HUMAN PHYSIOLOGY
AN INTRODUCTION
Second Edition
Joseph Feher
Department of Physiology and Biophysics,
Virginia Commonwealth University School of Medicine
AMSTERDAM • BOSTON • HEIDELBERG • LONDON • NEW YORK • OXFORD • PARIS
SAN DIEGO • SAN FRANCISCO • SINGAPORE • SYDNEY • TOKYO
Academic Press is an imprint of Elsevier
Academic Press is an imprint of Elsevier
125 London Wall, London EC2Y 5AS, United Kingdom
525 B Street, Suite 1800, San Diego, CA 92101-4495, United States
50 Hampshire Street, 5th Floor, Cambridge, MA 02139, United States
The Boulevard, Langford Lane, Kidlington, Oxford OX5 1GB, United Kingdom
Copyright r 2012, 2017 Elsevier Inc. All rights reserved.
No part of this publication may be reproduced or transmitted in any form or by any means, electronic
or mechanical, including photocopying, recording, or any information storage and retrieval system, without
permission in writing from the publisher. Details on how to seek permission, further information about the
Publisher’s permissions policies and our arrangements with organizations such as the Copyright Clearance Center
and the Copyright Licensing Agency, can be found at our website: www.elsevier.com/permissions.
This book and the individual contributions contained in it are protected under copyright by the Publisher
(other than as may be noted herein).
Notices
Knowledge and best practice in this field are constantly changing. As new research and experience broaden our
understanding, changes in research methods, professional practices, or medical treatment may become necessary.
Practitioners and researchers must always rely on their own experience and knowledge in evaluating and using any
information, methods, compounds, or experiments described herein. In using such information or methods they
should be mindful of their own safety and the safety of others, including parties for whom they have a
professional responsibility.
To the fullest extent of the law, neither the Publisher nor the authors, contributors, or editors, assume any liability
for any injury and/or damage to persons or property as a matter of products liability, negligence or otherwise, or
from any use or operation of any methods, products, instructions, or ideas contained in the material herein.
British Library Cataloguing-in-Publication Data
A catalogue record for this book is available from the British Library
Library of Congress Cataloging-in-Publication Data
A catalog record for this book is available from the Library of Congress
ISBN: 978-0-12-800883-6
For Information on all Academic Press publications
visit our website at https://www.elsevier.com
Publisher: Katey Birtcher
Acquisition Editor: Steve Merken
Editorial Project Manager: Nate McFadden
Production Project Manager: Stalin Viswanathan
Designer: Maria Ines Cruz
Typeset by MPS Limited, Chennai, India
Preface
Welcome to the second edition of Quantitative Human
Physiology! This new edition has been updated with
numerous enhancements, many of which were suggested or inspired by instructors and students who used
the first edition. These important changes include
(but are not limited to):
G
G
G
G
G
G
Substantial updating of the text throughout to reflect
the latest research results, with many sections expanded
to include relevant and important information
Enhanced, updated, and improved figures for better
understanding and clarification of challenging topics
Addition of several new appendices covering statistics, nomenclature of transport carriers, and structural biology of important items such as the
neuromuscular junction and calcium release unit
Addition of new problems within the problem sets
and example calculations in the text
Addition of some Clinical Applications such as dual
energy X-ray bone densitometry
Addition of commentary to power point presentations.
The goal of this new edition was to make important
improvements while retaining the features that make
this text uniquely suited to the needs of undergraduate
bioengineering students. While it is sometimes very
tempting to make drastic and sweeping changes in an
attempt satisfy everyone, this new edition focuses on
refinements and updates that, we hope, most instructors
and students will find helpful, informative, and instructionally sound.
THIS TEXT IS A PHYSIOLOGY TEXT
FIRST, AND QUANTITATIVE SECOND
The second edition of this text remains faithful to its primary goals: it remains, first and foremost, a physiology
text. It is explicitly designed for a certain class of students, those majoring in Biomedical Engineering at
Virginia Commonwealth University, and their suggestions, limitations, and desires have shaped the text from
the outset. Specifically, the text is designed for students
who have never been exposed to Physiology, students
who know neither the language nor the concepts of the
subject. The text contains all elements of physiology in
nine units: physical and chemical foundations; cell physiology; excitable tissue physiology; neurophysiology;
cardiovascular physiology; respiratory physiology; renal
physiology; gastrointestinal physiology; and endocrinology. The course is best taught in the order of the text but
it is possible to present the material in other sequences.
Secondly, the text affirms its aim to be quantitative.
Being quantitative has two aspects. The first is about
knowing the numerical value for the ranges of crucial
aspects of physiology, such as the flows or forces within
the body. The second is about discovering the relations
between physiological parameters. For many aspects of
physiology, there currently is not enough information
to make a detailed quantitative analysis, or the analysis
at that depth is beyond the scope of this text. In these
cases, this text is not very different from more traditional texts. Where possible, the text takes an analytical
and quantitative approach.
THE TEXT USES MATHEMATICS
EXTENSIVELY
Carl Frederick Gauss famously said, “Mathematics is the
queen of sciences.” Mathematics is not just about the
numerical value of something, such as the magnitude of
the arterial blood pressure or the rate of salivary secretion, although that is what many people think of when
they think of quantitation. Rather, mathematics is about
the relationship between things that can vary with time
or position. These relationships cannot be fully understood with words alone. They require the language of
mathematics. Students should be able to articulate these
relationships with words, but this text demands more.
Mathematical statements—equations—are simply logical sentences. You can read an equation in words. But
the mathematical statement uses an economy of words.
Mathematics also has specific rules for the manipulation
of the logical parts of the sentences, so that rearrangement or combination of equations leads to new insight.
NOT ALL THINGS WORTH KNOWING
ARE WORTH KNOWING WELL
This text uses lots of mathematics at the level of the
calculus and elementary differential equations. These
mathematical tools are used to encode the physics or
chemistry of processes into mathematical symbolism, and
mathematical manipulation leads to useful equations.
ix
x
PREFACE
The point of the derivations is the useful equation, not
the derivations themselves. The derivations are included,
sometimes as an appendix, to satisfy the students that the
final equations are not magical, but come from the application of mathematics to physical and chemical principles
that apply to the body. The point is to be able to apply
the final equations. Physics, chemistry, and math at the
level of calculus are used to get the equations, but generally algebra is all that is required to apply them. In my
view, the derivations are important to teach students the
process of encoding the physics and chemistry and deriving the relationship between variables that constitute the
useful equations, but rote memorization of derivations is
boring and useless.
PERFECT IS THE ENEMY OF GOOD:
EQUATIONS AREN’T PERFECT,
BUT THEY’RE OFTEN GOOD ENOUGH
The text does not say what I tell my students about the
applicability of equations in general. I tell my students in
lecture that all of these equations are wrong. They are
wrong in something of the same sense that Newtonian
mechanics is wrong. Relativity and quantum mechanics
supplant Newtonian mechanics (even in the macroscopic
world) but Newtonian mechanics will still get the rocket
ship to the moon. So I tell them that these equations are
theoretically wrong but they give satisfactorily correct
answers, in much the same way that Newtonian mechanics still does, even though theoretically incomplete. Fick’s
Law of Diffusion depends on continuum mathematics
for an inherently discrete process. But the discreteness is
so finely divided that the distinction is unimportant.
EXAMPLES AND PROBLEM
SETS ALLOW APPLICATION
OF THE USEFUL EQUATIONS
There are several aids in the text to foster a quantitative
and analytical understanding. First, there are some
worked calculations in the text that are set apart in text
boxes as Examples. These aim to show the students how
to apply some of the ideas presented in the text. Second,
there are a total of 17 problem sets scattered throughout
the text. All units have at least one, most have two, and
the cardiovascular section has three. These are meant to
cover about three chapters each, so that a problem set
can be assigned approximately once a week, for three lectures per week. Students have repeatedly told me that
they want a solution set to the problems to see how they
can do them, but this makes them useless as a graded
assignment. There is no better teacher than wrestling
with a problem. The second edition has expanded on
these problem sets with new problems.
The problems themselves are generally meant to cover
some new idea or concept that could not be, or was not,
effectively covered in the text itself, and to expand on the
student’s understanding of the material. They are not
merely busy work or “plug and chug” exercises. The alert
student should not merely do the problem, but think
about what the result means. In many cases, the problems are written as a sequence of questions, each of
which sets up the student for further questions or
insights. These illustrate the process necessary for answering a larger question, breaking it up into parts of the
answer that must come first. This method aids the students in thinking about larger problems: break it down
into its simpler components. Some subject matter unfortunately does not lend itself easily to such problems, but
I have attempted to find problems that students can do.
The students in my classes find the problems challenging.
I allow them to work on them collaboratively, because
they are meant to be part of the instruction and less of
the evaluation, but the problem sets are graded and contribute significantly to the final grades. Of course, such
policies are set at the discretion of the instructor.
LEARNING OBJECTIVES, SUMMARIES,
AND REVIEW QUESTIONS GUIDE
STUDENT LEARNING
The Learning Objectives are meant as a guide to the
construction of examination questions, either directly or
indirectly. These learning objectives are fairly broad and
together they cover the breadth of physiology. This is
my advice to students: read the Learning Objectives first,
the chapter Summary second, and then read the text.
Next, attempt to answer the Review Questions and
return to the Learning Objectives. If you are mystified
by any of the Review Questions or Learning Objectives,
read the pertinent part of the text again.
CLINICAL APPLICATIONS
PIQUE INTEREST
Pathological situations often illuminate normal physiology. Clinical Applications are scattered throughout the text.
Because it takes a lot of background material to understand
these clinical applications, Clinical Applications are less
prevalent in the early parts of the text, which are foundational, than in the latter parts of the text. There are more
than 50 such Clinical Applications, with several new additions in the second edition. The students like them because
these Clinical Applications tell them that there is a reason
for learning what might otherwise appear arcane.
HOW INSTRUCTORS CAN USE THIS TEXT
This text is meant for anyone with a fairly modest level of
mathematical skill, at the level of the calculus with elementary differential equations. Students without this level of
training will find this book too difficult. I have developed
this book specifically with undergraduate Biomedical
PR E FACE
Engineering students in mind, and have taught this material for 16 years. Each Chapter is intended to be a single
lecture, and the length of the chapter is meant to be readable in a single sitting. There are 77 chapters, so it is most
useful in a two-semester sequence. I cover all chapters in
that time, and all problem sets. The text would also be useful for instructors with less time by adjusting the breadth
and depth of coverage. Units 1, 8, and 9, (Physical and
Chemical Foundations, Gastrointestinal Physiology
and Endocrinology, respectively) for example, could be
eliminated or covered more superficially. Unit 1 could be
eliminated because it is a review, Units 8 and 9 because
they are relatively peripheral to Biomedical Engineering.
Unit 2, Cell Physiology, could be covered more superficially or eliminated if students previously have had Cell
Biology. Alternatively, it is possible to cover the breadth of
physiology if the depth is reduced. This can be done by
focusing on the chapter summaries, which give a broach
picture of what is happening with less detail, and combining lectures or omitting some topics. In this regard, the text
is useful similarly to a cafeteria: the instructor is free to
choose those sections of most interest and to downplay
those of lesser interest. Some instructors may feel that
knowing how to think about problems is the most important thing, and that the details of the physiology are secondary. Such an instructor may want to focus more on the
appendices than on the chapter matter themselves, and
more on the problems and how to solve them.
ANCILLARY MATERIALS
FOR INSTRUCTORS
For instructors adopting this text for use in a course, the
following ancillaries are available: Power Point lecture
slides, electronic images from the text, solutions manual
for the problem sets, laboratory manuals with example
data, and examination questions with answers. The
Power Point slides have been updated to include the
new figures and more commentary as appropriate for
the lectures. Please visit https://textbooks.elsevier.com/
9780128008836.
HOW STUDENTS CAN USE THIS TEXT
A student’s goal ought to be to learn as much
physiology as possible within the constraints of available time. This text is written with considerable detail.
The Learning Objectives and Review Questions set the
stage for the kinds of things you should be able to do,
and the kinds of questions you should be able to
answer. The Chapter Summaries encapsulate each
chapter in an economy of words and detail. Start
with the Learning Objectives, read the Summary, and
then look at the Review Questions. Then read the chapter and repeat Learning Objectives, Summary, and
Review Questions. What you cannot answer at that
point requires you to re-read the pertinent sections a
second time.
The approach to the problem sets is different. The first
job is: find a bright fellow student to work with.
Second, read the question for understanding of what it
is asking. Then ask yourself, what is needed to answer
this question? How can you find out what is needed? If
it is a physical constant, where can you find it? Do you
know a relationship or equation that relates what is
being asked to what is given? Write it down and rearrange it to give the desired answer. Plug and chug what
is given and what else you have looked up to get a
numerical answer. It is very simple in principle but
sometimes very difficult in execution.
ANCILLARY MATERIALS
FOR STUDENTS
Student resources available with this text include a set of
online flashcards, a selection of animations based on the
figures in the text, and online quizzes for self-study.
Please visit https://booksite.elsevier.com/9780128008836.
STUDENT FEEDBACK
Students who have completed the course regularly tell me
that it is both one of the most challenging and one of the
most rewarding courses of their undergraduate career.
This is generally the case: what you get out of an educational enterprise is proportionate to what you put in.
Physiology is an integrative science. There is great satisfaction in understanding how a system works from the cellular and subcellular level all the way up to the organism
level. Human beings do not come with an owner’s
manual. The idea that this text is a first approximation
to an owner’s manual resonates with the students.
Joseph Feher
xi
Acknowledgments
I would like to thank all the reviewers of the proposal
and drafts of this project. Their feedback was very
helpful in improving the final version:
Brett BuSha
Jingjiao Guan
Nuran M. Kumbaraci
Marie Luby
Ken Yoshida
Ryan Zurakowski
The College of New Jersey
Florida State University
Stevens Institute of Technology
University of Connecticut
Indiana University—Purdue University
Indianapolis
University of Delaware
I would also like to acknowledge the help and support
of my colleagues at the VCU School of Medicine, especially Clive Baumgarten and Ray Witorsch, for their criticism of early drafts, and Margaret Biber, who expressed
the confidence in publishing this text that kept me
going. I would like to thank George Ford, VCU School
of Medicine, who helped develop the early outlines of
the cardiovascular section; Scott Walsh, who provided
the basis for some of the endocrine chapters; and Andy
Anderson, who helped critique multiple teaching efforts
of mine. I especially acknowledge the feedback from
many years of sophomore and junior BME students
who pointed out errors and difficulties in the material
and suggested improvements to help their learning
experience, which is really what this text is all about.
Special thanks go to students Woon Chow, Yasha
Mohajer, Matthew Caldwell, Matthew Painter, Mary
Beth Bird, Richard Boe, Linda Scheider, William
Eggleston, Alex Sherwood, Ross Pollansch, Kate Proffitt,
and Roshan George. Teaching these students and many
more like them, and getting to know them, has been
tremendously rewarding.
I would like to thank the publishing team at Elsevier
including Katey Birtcher, Publisher; Steve Merken,
Senior Acquisitions Editor; Nate McFadden, Senior
Developmental Editor; Maria Inês Cruz, Cover Designer;
and Stalin Viswanathan, Production Project Manager.
Much more indirectly, I wish to thank a long line of
scientific mentors who instilled in me the academic
integrity and the desire to be thorough and correct.
Included in this list are Lemuel Wright, and Donald B.
McCormick, of Cornell University, who advised me on
my master’s thesis; Robert Hall of Upstate Medical
Center, with whom I worked for a short but meaningful time and who provided the epiphany for my pursuit
of an academic career; Robert Wasserman, of Cornell
University, who first inspired me to apply mathematics
to transport processes; Norm Briggs, of VCU School of
Medicine, who first thought I would be a good bet
for a tenure track position; Don Mikulecky, who introduced me to many ideas of theoretical biology;
and Margaret Biber, who first presented me with the
daunting task of developing a year-long physiology
sequence for BME students, with laboratories, that
formed the text.
Although I desire to be thorough and correct, often I
am mistaken. I acknowledge the help of all those listed
above, but reserve to myself the blame for any errors in
the text. If the reader finds any errors of fact or analysis,
I would appreciate them letting me know so that I can
evaluate and correct it.
Lastly, I express my deep appreciation for my wife, Lee,
who put up with innumerable late nights and weekends
with a husband glued to the chair in front of the computer screen. Her support and encouragement made the
work possible. The first edition of this text was dedicated
to the memory of Karen Esterline Feher, who died tragically during the writing of the first edition. The second
edition was written shortly after the death of Mildred
Hastings Feher, who was a most gracious human being.
Most of it was also written while my daughter, Teresa,
gave birth to three boys. These four ladies in my life
have made me understand more fully the circle of life
and death, and have taught me to value all parts of it.
They are the bringers of life and I can only hope to
preserve and protect it, and to finally let it go. I dedicate
this effort to Mildred, Lee, Karen and Teresa.
xiii
The Core Principles of Physiology
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
Define the discipline of physiology
Describe in general terms how each organ system
contributes to homeostasis
Define reductionism and compare it to holism
Describe what is meant by emergent properties
Define homeostasis
List the four Aristotelian causes and define teleology
Define mechanism
Describe how evolution is a cause of human form and
function
Write equations for the conservation of mass and energy
for the body
Give an example of signaling at the organ or cellular level
of organization
List the core principles of physiology
Contrast feed-forward or anticipatory control to negative
feedback control
HUMAN PHYSIOLOGY IS THE
INTEGRATED STUDY OF THE NORMAL
FUNCTION OF THE HUMAN BODY
ORGAN SYSTEMS WORK TOGETHER TO
PRODUCE OVERALL BODY BEHAVIOR
Almost any explanation of something begins with a
description of that something’s parts. The human body
consists of many parts. We consider an assortment of
parts that usually relate to each other in defined ways to
be a system. In physiology, a system is usually considered to be a group of organs that serve some welldefined function of the body. The parts of these systems
can be described separately, but they work together to
produce the overall system behavior. That is, the individual behavior of the parts is integrated to produce
overall behavior. The various organ systems and their
functions in the body are summarized in Table 1.1.1.
These systems are further integrated to produce overall
bodily function. Physiology is the study of the integrated normal function of the human body.
Each of these organ systems is essential to the survival
of the organism, the living human being. It is possible
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00001-X
1.1
in an artificial environment to survive with a single
compromised system—such as persons with failed
kidneys or failed immunological systems—but these
persons could not survive in natural ecosystems.
REDUCTIONISM EXPLAINS SOMETHING
ON THE BASIS OF ITS PARTS
The process of explaining something on the basis of
its parts is called reductionism. Thus the behavior of
the body can be explained by the coordinated behavior
of its component organ systems. In turn, each organ
system can be explained in terms of the behavior of
the component organs. In this reduction recursion, the
behavior of the component organs can be explained by
their components, the individual cells that make up the
organ. These cells, in turn, can be explained by the
behavior of their component subcellular organelles;
the subcellular organelles can be explained by the
macrochemicals and biochemicals that make up these
organelles; the biochemicals can be explained by their
component atoms; the atoms can be explained by their
component subatomic particles; the subatomic particles
can be explained by fundamental particles. According
to this reductionist recursion, we might anticipate
that the final explanation of our own bodies lies in the
physics of the fundamental particles. Beyond being
impractical, there is a growing realization that it is theoretically impossible to describe complex and complicated living beings solely on this basis of fundamental
physics, because at each step in the process some information is lost.
This situation should not trouble us too much. In
physics, we speak of the force of friction even though
friction is not a fundamental force. It results from
tremendous numbers of electromagnetic interactions
between particles on two surfaces so that friction really
is just a trace of all of those microscopic electromagnetic
interactions onto the macroscopic bodies. The details
of the surfaces produce those forces and we can experimentally reduce the force simply by polishing the surfaces that interact, making the tiny bumps and valleys
on the surfaces smaller. We don’t know the details of
the surface and so we ignore the reality and lump all of
those interactions together and call it the “force” of friction. We have lost some information and abandon
the idea of recursing reductionism down to the level of
fundamental physics.
3
4
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
parts, the molecules that make it up, and how the
organelle’s function is regulated by and contributes to
the function of the cell.
TABLE 1.1.1 Organ Systems of the Body
Organ System
Function
Nervous system/
endocrine system
Sensory input and integration; command
and control
Musculoskeletal
system
Support and movement
Cardiovascular
system
Transportation between tissues and
environmental interfaces
Gastrointestinal
system
Digestion of food and absorption of
nutrients
Respiratory system
Regulation of blood gases and exchange of
gas with the air
Renal system
Regulation of volume and composition of
body fluids
Integumentary
system (skin)
Protection from microbial invasion, water
vapor barrier, and temperature control
Reproductive
system
Pass life on to the next generation
Immune system
Removal of microbes and other foreign
materials
Level
Discipline
Universe
Cosmology
Society
Sociology
Body
Explanation
Physiological systems
Function
Physiology
Organs
Cells and cell products
Cell physiology
Subcellular organelles
Biochemistry
Molecules
Chemistry
Atoms
Subatomic particles
High-energy physics
FIGURE 1.1.1 Hierarchical description of physical reality as applied to
physiological systems. We attempt to “explain” something in terms of its
component parts and describe a function for a part in terms of its role
in the “higher” organizational entity.
PHYSIOLOGICAL SYSTEMS ARE PART OF A
HIERARCHY OF LEVELS OF ORGANIZATION
The recursion of explanation described above for
reductionism involves various levels of complexity in a
hierarchical description of living beings, as shown in
Figure 1.1.1. Understanding any particular level entails
relating that level to the one immediately above it and
the one immediately below it. For example, scientists
studying a particular subcellular organelle can be said to
have mastered it when they can explain how the function of the organelle derives from the activities of its
REDUCTIONISM IS AN EXPERIMENTAL
PROCEDURE; RECONSTITUTION IS A
THEORETICAL PROCEDURE
The processes used in going “down” or “up” in this
hierarchy are not the same. We use reductionism to
explain the function of the whole in terms of its parts,
by going “down” in the levels of organization. We
describe the function of the parts at one level by showing how they contribute to the behavior of the larger
level of organization, going “up” in Figure 1.1.1. These
processes are fundamentally different. Reductionism
involves actually breaking the system into its parts and
studying the parts’ behavior in isolation under controlled conditions. For example, we can take a sample
of tissue and disrupt its cells so that the cell membranes
are ruptured. We can then isolate various subcellular
organelles and study their behavior. This procedure
characterizes the behavior of the subcellular organelle.
Knowing the behavior of the individual parts and paying close attention to how these parts are connected,
it is possible to predict system behavior from the parts’
behavior using simulation or other techniques. Because
it is impossible, except in rare and limited cases, to
reassemble broken systems (we cannot unscramble the
egg!), we must test our ideas of subcellular function
theoretically.
HOLISM PROPOSES THAT THE BEHAVIOR
OF THE PARTS IS ALTERED BY THEIR CONTEXT
IN THE WHOLE
Holism conveys the idea that the parts of an organism
are interconnected and that each part affects others. The
parts cannot be studied in isolation because important
aspects of their behavior depend solely on their interaction with other parts. Reductionism seems to imply that
the whole is the sum of the parts, whereas holism suggests that the whole is greater than the sum of the parts.
The system takes on new properties, emergent properties, that arise from complex interactions among the
parts. Examples of emergent properties include selfreplication. The ability of cells to form daughter cells is
a system property that does not belong to any one part
but belongs to the entire system. Consciousness is also
an emergent property that arises from neuronal function
but at a much higher level of organization.
PHYSIOLOGICAL SYSTEMS OPERATE AT
MULTIPLE LEVELS OF ORGANIZATION
SIMULTANEOUSLY
It should be clear from Figure 1.1.1 that all the levels of
organization simultaneously operate in the living
human being. Processes occur at the molecular, subcellular, cellular, organ, and system levels simultaneously
and dynamically.
The C ore P rinciples of Physiology
THE BODY CONSISTS OF CAUSAL
MECHANISMS THAT OBEY THE LAWS
OF PHYSICS AND CHEMISTRY
When we say that we explain something, usually we
mean that we can trace the output of the system—its
behavior—to some cause. That is, we can seamlessly
trace cause and effect from some starting point to
some ending point. Aristotle (384322 BC) posited
four different kinds of causality:
1. Material Cause
A house is a house because of the boards, nails,
shingles, and so on that make it up. We are what
we are because of the cells and the cell products
that make us up.
2. Efficient Cause
A house is a house because of the laborers who
assembled the materials to make the house. We
are what we are because of the developmental
processes that produced us and because of all of
the experiences we have had that alter us.
3. Formal Cause
A house is a house because of the blueprint that
directed the laborers to assemble the materials in
a particular way. We are what we are because of
the DNA that directs our cells to make some proteins and not others, and because of epigenetic
effects—those effects resulting from the environment interacting with the genome.
4. Final Cause
A house is a house because someone needed
shelter. We are what we are because. . .
The final cause for humans has a variety of possible answers. This is the only cause that addresses
the idea of a purpose. We make a house for a
purpose: to provide someone with shelter. What
is the purpose of human beings? This cause asks
the question of why rather than how.
TELEOLOGY IS AN EXPLANATION IN
REFERENCE TO A FINAL CAUSE
A description or explanation of a system based on the reference to the final cause is called a teleological explanation. Teleology has long been ridiculed by scientists
because it appears to reverse the scientific notion of cause
and effect. In normal usage, cause-and-effect linkages
describe only the efficient cause. When a force acts on
something, that something reacts in a predictable way. Its
predictability is encoded in physical law. Teleology
describes the behavior in terms of its final purpose,
and not its driving force, which reverses the cause-andeffect link.
HOW? QUESTIONS ADDRESS CAUSAL
LINKAGES. WHY? QUESTIONS ADDRESS
FUNCTION
To clarify this process, let us ask some questions about
blood pressure. How does your body regulate the average aterial blood pressure? This answer takes some time
to develop fully (see Unit 5) but we can simplify the
answer by saying: by increasing or decreasing the caliber
of the arteries, by increasing or decreasing the output of
the heart, and by increasing or decreasing the volume of
fluid in the arteries. All of these actions—and more—
interact to determine the arterial blood pressure. All of
these parts to the answer involve actions, and actions
are efficient causes. For example, increasing or decreasing the caliber of the arteries depends on a complicated
network of signaling pathways and biochemical reactions, but each starts with a cause and ends with an
effect. Now we ask: why does your body regulate average aterial pressure at the level it does? The answer is
again not so simple but we can simplify it as: to assure
perfusion of the tissues. This addresses a purpose. In
this sense, it is teleological, but it also makes sense to
us. It is not that the cardiovascular system knows what it
is doing, but its operation has been selected to produce
the desired output. Within the framework of the body,
organs do serve a purpose and that purpose is their
function. All of the functions listed in Table 1.1.1 are
final causes for the organ systems.
HUMAN BEINGS ARE NOT MACHINES BUT
STILL OBEY PHYSICAL LAW
Aristotle had a different idea about the mind. He posited that the mind was not a material entity, much like
an idea is not a material thing. He posited that the
mind was what perceived, imagined, thought, emoted,
desired, felt pain, reasoned, remembered, and controlled the body. This philosophy in which the mind
controls behavior is called mentalism. These ideas
went largely unchallenged until the Renaissance. René
Descartes (15961650) wrote a book, Treatise on Man,
in which he tried to explain how the nonmaterial mind
might interact with the material body. In this process,
he constructed mechanical analogues to explain sensation and command of movement. He thought that the
operation of all things, however complex, could be
explained by some mechanism. Each mechanism consists of a sequence of events that link an initial causal
input to an effect that becomes the cause of the next
step in the mechanism. In this view, human beings are
very complex machines that obey natural law. In the
late 1800s, W.O. Atwater (18441907) built a calorimeter to study heat production, gas exchange, and fuel
consumption in humans. He found that the energy output of humans matched the chemical energy of the
food consumed, within narrow experimental error. This
result confirmed that the law of conservation of energy
held for the transformation of energy by the human
body as well as for inanimate transformations.
IS THERE A GHOST IN THE MACHINE?
The core principle of physiology that states the human
body is a mechanism strikes at the heart of the concept
of vitalism. Vitalism states that living things cannot
be described in mechanistic terms alone, and that
some organizing force or vital principle forever distinguishes living things from nonliving things. For human
beings, we could call this the soul and be in reasonable
agreement with the vitalists. So far, science has found
5
6
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
no reliable, scientific verification that the human body
violates any physical law. The existence of emergent
properties gives credence to the idea of vitalism.
Emergent properties are new properties that arise from
complex systems because of their complexity and topology—the way that subsystems are connected. These
emergent properties do not appear to be predictable.
That is, we cannot see how we would predict their
emergence given the fundamental laws of physics.
Examples of these emergent properties are life itself and
consciousness. These properties appear to arise from
interactions of parts that appear to obey physical law
alone, but how these properties arise remains mysterious. These emergent properties are system properties,
not mechanism properties. Because of overwhelming
evidence that specific deficits in brain function produce
specific deficits in mental function, we have come to
believe that the brain somehow produces the mystical
thing that is conscious and self-aware. This thing is
not material in the ordinary sense of the word, just like
an idea is not a material thing. The new mindbody
problem is the inverse of Descarte’s mindbody problem: how can a material thing (the brain) produce
the nonmaterial thing that we identify as self. Is
this relationship a one-way street, or does the mind
have a reciprocal effect on the brain? Although these are
extremely important questions, most physiologists take
a narrower aim of explaining only those phenomena for
which we have satisfactory mechanistic models and
attempt to extend the range to include all physiologic
phenomena, including consciousness.
UNDERSTANDING A SYSTEM IS EQUIVALENT
TO MAKING A MODEL OF IT
What we have been discussing so far is what it means to
say that we understand something. The something we
are trying to understand we will label “the real system.”
To understand this system, we make a model of it in a
process that we will call encoding (B in Figure 1.1.2).
The model does not have to remain solely in our minds.
It could be written down as a set of equations or as a
computer algorithm, for example. In the real system,
perturbations cause changes in the real system. This
causal link (A in Figure 1.1.2) between perturbation
and effect is what we desire to predict or explain using
the model. In the model, the cause is also encoded, and
its predicted effect we will call an inference (C in
Figure 1.1.2). That is, some perturbation of the model is
predicted to cause some effect in the model. When we
decode this effect (D in Figure 1.1.2), the inference of
the model is translated as our prediction of the behavior
of the real system. A correct model is one that correctly
predicts the behavior of the real system. That is
½1:1:1
A5B1C1D
THE CORE PRINCIPLES OF
PHYSIOLOGY
The rest of this chapter will discuss several Core
Principles of Physiology. These are:
G
G
G
G
G
G
Cells are the organizational unit of Life.
Homeostasis is a central theme of physiology.
We have evolved from prior life forms and our pedigree is revealed in our genome.
Physiological systems transform matter and energy
while obeying the conservation laws.
Coordinated command and control requires signaling at all levels of organization.
Control systems using negative feedback, positive
feedback, anticipatory and threshold mechanisms.
CELLS ARE THE ORGANIZATIONAL
UNIT OF LIFE
THE CELL THEORY IS A UNIFYING PRINCIPLE
OF BIOLOGY
The cell theory states that all biological organisms are
composed of cells; cells are the unit of life and all life
come from preexisting life. The cell theory is so established today that it forms one of the unifying principles
of biology.
The word cell was first used by Robert Hooke
(16351703) when he looked at cork with a simple
microscope and found what appeared to be blocks of
material making up the cork. The term today describes a
microscopic unit of life that separates itself from its surroundings by a thin partition, the cell membrane.
D: Decoding step
FIGURE 1.1.2 The modeling relation. The real
system is characterized by causal links, A, that
correspond to the behavior of the real system to
external or internal perturbation. The real system is
converted by an encoding step to a model (B). In
the model, the response of the model to a
perturbation is determined as an inference (C). The
behavior of the model is then decoded (D) in the
last step in testing the validity of the model. When
B 1 C 1 D 5 A, the model has successfully predicted
the behavior of the real system and we say that it is
a valid model. Adapted from R. Rosen, Theoretical
Biology and Complexity, 1985, Academic Press, NY.
Perturbation
A: Causal link
Effect
Real system
Effect
Model
C: Inference
Perturbation
B: Encoding step
The C ore P rinciples of Physiology
Erythrocyte Leukocyte
Eosinophil
Inner hair cell
Hepatocyte
Enterocyte
Parietal cell
Motor neuron
Cardiomyocyte
FIGURE 1.1.3 Examples of the different cells that populate the human body. Motor neurons such as the one illustrated are found in the ventral horn
of the spinal cord. The inner hair cells are found in the cochlea and form part of our response to sound. Erythrocytes, leukocytes, and eosinophils are
all found in the blood. Cells typically are not colored but may be seen in color by their adsorption of histological stains. Hepatocytes in the liver help
package nutrients, form bile, and detoxify foreign chemicals. The enterocytes line the small intestine and absorb nutrients from the food into the
blood. The parietal cells secrete HCl in the stomach. The cardiomyocyte shown is a ventricular cell whose contraction contributes to the pumping
action of the heart. This is a small sampling of the diversity of cell forms in the human body.
Most biologists believe that life arose spontaneously from
inanimate matter, but the details of how this could have
happened remain unknown, and the time scale was long.
Rudolf Virchow, a German pathologist (18211902),
famously wrote “omnis cellula e cellula”—all cells come
from other cells—meaning that spontaneous generation
of living things from inanimate matter does not occur
over periods as short as our lifetimes.
CELLS WITHIN THE BODY SHOW A MULTITUDE
OF FORMS
Large multicellular organisms such as ourselves consist
of a vast number of different cells that share some features but vary in size, structure, biochemical makeup,
and functions. A sampling of the spectrum of cells that
make up the body is illustrated in Figure 1.1.3. Almost
all cells in the body have a cell membrane, also called
the plasma membrane, and most contain a nucleus.
The simplest cell in the human is probably the erythrocyte, which is the only cell in the body that lacks a
nucleus.
THE DIVERSITY OF CELLS IN THE BODY
DERIVES FROM DIFFERENTIAL EXPRESSION
OF THE GENOME
The outward appearance and behavior of an organism
define its phenotype, which is related to but not identical to the organism’s genetic material, its genotype. The
genotype consists of the set of alternate forms of genes,
called alleles, that the organism has, and these alternate
forms of genes are further defined by the sequence of
nucleotides in their DNA. DNA is the genetic material
that is passed on through the generations. It determines
the kind of proteins that cells can produce, and these
materials make up the phenotype. The genome is the
entirety of the hereditary information, including all of
the genes and regions of the DNA that are not involved
in producing proteins. Nearly all cells in the body contain the entire genome. The exceptions include the erythrocytes and the reproductive cells. Those cells that
are not reproductive cells are called somatic cells (from
the Greek soma, meaning body). Thus the great majority
of body cells are somatic cells, and they all contain
the same amount and kind of DNA. The astounding
diversity of the types of human cells derives from their
expression of different parts of the genome. Here
expression means using DNA to produce proteins.
THE CONCEPT OF HOMEOSTASIS IS A
CENTRAL THEME OF PHYSIOLOGY
EXTRACELLULAR FLUID SURROUNDS ALL
SOMATIC CELLS
As described above, each cell in the body is surrounded
by a cell membrane that defines the limits of the cell
and separates the interior of the cell from its exterior.
The interior consists of a number of subcellular organelles suspended in a fluid, the intracellular fluid. The
exterior consists of an extracellular matrix that holds
things in place and an extracellular fluid. The extracellular fluid has two components: the plasma and the
interstitial fluid. The plasma is that part of the extracellular fluid that is contained in the blood vessels. The
interstitial fluid is that part of the extracellular fluid
between the cells and the walls of the vasculature.
Nearly all cells of the body come in intimate contact
7
8
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Red blood cells
Body cells
Plasma
Intracellular fluid
Nutrients
EVOLUTION RESULTS FROM CAUSE AND
EFFECT SUMMED OVER LONGTIME PERIODS
Wastes
Capillary walls
would shift to those better suited to the environment.
New variations in the genotype arise by mutation.
Over geological time, such natural selection gradually
changes the population. Darwin believed that such slow
changes in the genetic makeup of populations could
eventually produce new species, and he termed this slow
formation of new species evolution.
Interstitial fluid
FIGURE 1.1.4 Relationship between cells and the extracellular fluid. All
cells of the body are surrounded by a thin layer of extracellular fluid
from which they immediately derive nutrients such as amino acids,
sugars, and oxygen, and to which they discharge wastes such as carbon
dioxide and other end products of metabolism. Nutrients are delivered
to the cells and waste products are removed through the circulation,
which does not make direct contact with the interstitial fluid, but is
separated from it by the walls of the vascular system.
with the extracellular fluid. The last step in the delivery
of nutrients and the first step in removal of wastes is
achieved through the extracellular fluid (see
Figure 1.1.4). The extracellular fluid was called the
milieu interieur, or the internal environment, by the
great
French
physiologist,
Claude
Bernard
(18131878). Survival of the cells depends on the
maintenance of a constant internal environment. The
maintenance of a constant internal environment is
called homeostasis, which is literally translated as same
standing. Contributing to the maintenance of a constant
internal environment appears to the “goal” (or final
cause or purpose) of many physiological systems, and
this homeostasis is the central theme of physiology.
EVOLUTION IS AN EFFICIENT CAUSE
OF THE HUMAN BODY WORKING OVER
LONGTIME SCALES
EVOLUTION WAS POSTULATED TO EXPLAIN
THE DIVERSITY OF LIFE FORMS
Charles Darwin (18091892) wrote On The Origin of
Species in 1858 as his attempt to explain the origin
of the tremendous variety of animals and plants in
today’s ecosystems. He noted that any one species consists of a population of individuals that are capable of
breeding among themselves but not with members
of other species. The similarity among members of a
species defines the species; the differences between them
define the individual. These outward appearances constitute the phenotype, as described earlier, which arises
from the response of the genotype to the environment.
Some of the individual members of a species are better
suited to their environment than others and produce
more offspring as a consequence. With sufficient time,
the frequency of genotypes represented in the population
Evolution is like a higher level on the hierarchy of
cause-and-effect relationships. As an example, consider
a mutation that alters the structure of a critical protein
located in a selected group of cells in the body that
enhances the function of these cells. The mutation
causes an altered protein, which in turn causes
enhanced behavior of the organism. This altered behavior of the organism causes greater success in reproduction. Over time, greater success in reproduction replaces
the less-fit genotype with the mutated, superior genotype. Thus evolution results from thousands of independent cause-and-effect linkages played out over a
population of individuals, over longtime periods.
EVOLUTION WORKS ON PREEXISTING FORMS:
COMPARATIVE GENOMICS REVEALS PEDIGREE
At some time in the distant past, there was no life on
earth. The origin of life is unknown and, in some sense,
how it arose is not a scientific question because we cannot test any hypothesis of events in the past. We can,
however, search for the trace of past events in the world
today, much like a detective searches for clues to determine what happened earlier. This search has some of the
character of an experiment. In this way, the fossil record
illuminates the march of evolution to the present day.
Similarly, we carry traces of our evolution in our own
genome in the form of “fossil genes.” Because evolution
works on preexisting forms, and because the multicellular organism plan entails the same challenges to homeostasis, and the same problems of cell maintenance,
the genomes for many diverse animals and plants share
profound similarities. For this reason, similarities in
the genome can be used to trace the evolution of the
proteins and shed light on the pedigree of species.
EVOLUTION TAILORS THE PHENOTYPE
TO THE ECOSYSTEM
Humans live and reproduce within the context of
an ecosystem. Our evolution has occurred because of
our fit, or lack of it, with a specific environment. This
explains some of the diversity of human forms within
our species. Skin color and overall body shape, for
example, are adaptations that arose to better fit the different levels of sunlight and air temperatures at different
latitudes. Evolution has prepared us to meet the challenges of our environment but has not prepared us
for unusual challenges. For example, we are adapted to
survive short periods without water or longer periods
without food, but we cannot do without air even for
short periods.
The C ore P rinciples of Physiology
ROBUSTNESS MEASURES THE ABILITY OF THE
BODY TO RESPOND TO ENVIRONMENTAL
CHALLENGES
A robust system is one that continues to function even
when faced with difficult challenges. A fragile system
fails easily. Engineered systems aim for a degree of
robustness and usually achieve that end by adding
redundant or back-up control systems and by building
in safety factors. These systems are robust for some
kinds of failure while remaining vulnerable to others.
For example, autopilot systems in aircraft use multiple
computers with different programs so that failure
of one does not cause failure of the entire system.
But these systems remain vulnerable to general power
failure. We also have redundant or reserve function for
several physiological systems. We have two kidneys,
yet generally we can survive with only one. Liver, intestine, brain, and heart have more functional capacity
than generally used so that we can survive if part of
these organs fails. Strokes and heart attacks damage
parts of the brain or the heart, respectively. Persons
who suffer these cardiovascular accidents often recover
much of their function, depending on the degree of
damage and its location. If the damage is severe or
involves a critical area, the victim may be permanently
impaired or they may die. History is replete with astonishing stories of the tenacity of humans for life under
amazingly harsh conditions. On the other hand, history also tells of the crumbling of civilizations when
exposed to unfamiliar pathogens. Thus the human
body may surprise us either because of its robustness
or its fragility.
REGULATION OF THE GENOME MAY EXPLAIN
THE FAST PACE OF EVOLUTION
There is a growing realization among evolutionary biologists that mutations in the genes that encode for
somatic proteins—the ones that make us up—are only a
small part of the story and cannot account for the rapid
pace of evolution. Instead, much of evolution is
accounted for in the genes that regulate the expression
of other genes. Many modern birds, for example, do not
have teeth. Yet it is possible experimentally to induce
birds to make teeth, because they retain the genes for
making teeth but also have genes that suppress the
expression of the genes for teeth. Many diverse groups
of animals share most of the genes involved in body
building but differ in how and when these genes are
used. The result is the differing body forms that are
found in the animal kingdom.
EVOLUTION HELPS LITTLE IN EXPLAINING THE
NORMAL FUNCTION OF THE BODY
Although evolution is one of the few unifying principles
of biology and is one cause of the structure and function of the human body, it answers the question of the
efficient cause of the body on a different time scale than
the normal operating time scale of the body. Evolution
does not aid us very much when trying to explain how
our bodies work on a minute-to-minute basis. Instead,
we look to control theory to explain the normal functioning of the human body.
LIVING BEINGS TRANSFORM ENERGY
AND MATTER
Repair, maintenance, growth, activity, and reproduction
all require input of energy and matter in the form of
food. The gastrointestinal system breaks down the food,
which is absorbed into the blood and distributed
among the tissues according to need. The available
building blocks must be transformed into cellular or
extracellular components, and all of this metabolism
requires energy. The energy comes from the oxidation
of food and subsequent production of wastes. In addition to this conversion of chemical energy of one compound to another, we also transform chemical energy
into other forms of energy, including electrical and
mechanical energy. These processes obey physical and
chemical laws that govern the transformation of energy
and matter. In particular, we can write two equations
that describe overall mass and energy balance in the
body:
Min 5 Mout 1 ΔMbody
Mfood 1 Mdrink 1 Minspiredair 5 Mfeces 1 Murine 1 Mexpiredair
1 Mexfoliation 1 ΔMbody
½1:1:2
where M indicates mass and the subscripts indicate the
origin of the mass (in 5 input; out 5 output; most of
these are self-explanatory) and ΔMbody indicates change
in body mass. These equations describe mass balance
and simply indicate that all of the mass that enters the
body must either stay there (ΔMbody) or exit the body
through one of several routes. A similar equation can be
written for energy balance:
Ein 5 Eout 1 ΔEbody
Efood 1 Edrink 1 Einspiredair 5 Efeces 1 Eurine 1 Eexpiredair
1 Eexfoliation 1 ΔEbody 1 Eheat
1 Ework
½1:1:3
The overall mass and energy balance is shown schematically in Figure 1.1.5.
FUNCTION FOLLOWS FORM
Almost all processes carried out by the body, at all
levels of organization, depend on the three-dimensional
structure of some component. The structure both
9
10
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Air
system level at which the structure and arrangement of
nerves and tissues is vital for the proper coordination of
activity such as the heart beat or gastrointestinal motility. As another example, both the lungs and the gastrointestinal tract involve transfer of gas or nutrients from
the environment to the blood. Both lungs and intestine
have enormous surface areas and thin barriers—consequences of their structure—to maximize the rate of
transport.
Food and drink
Lungs
Skin
COORDINATED COMMAND AND CONTROL
REQUIRES SIGNALING AT ALL LEVELS OF
ORGANIZATION
Gastrointestinal
system
Heart
Nutrients and water
Internal
work
Kidney
Heat
Work
Fat
Tissues
Energy released
in metabolism
Feces
Urine
FIGURE 1.1.5 Overall mass and energy balance in the body. The lighter
arrows indicate energy transfer. The black arrows denote transfer of
mass. The chemical energy of the ingested food is released by oxidation
in metabolism, as indicated by the starburst in the tissues. This released
chemical energy is used for internal work, which usually eventually
degrades to heat, for external work and for storage in the chemical
energy of body components such as glycogen and fat. Growth also
entails a form of storage of ingested mass and chemical energy. The
laws of conservation of mass and energy (in ordinary chemical reactions)
require that the matter and energy that enter the body must be equal
to the matter and energy that leave the body plus any change in the
matter and energy content of the body.
enables function and constrains it, by determining what
can be done and how fast it can be accomplished.
These structural considerations apply at the molecular
level, at which the three-dimensional shape of protein
surfaces determines what binds to the protein, how it is
chemically altered, and how it interacts with other surfaces. These structural considerations apply at the subcellular level, at which the organelles themselves can
compartmentalize chemicals and so determine or limit
rates of reactions by regulating transfer between the
compartments. Structural considerations are also important at the tissue level, at which the topology or spatial
distribution of cellular processes allows countercurrent
flows, for example, that are crucial in clearing metabolites from the blood or concentrating the urine.
Structural considerations are important at the organ
Success of an organism requires adaptive responses
to change in the environment. This, in turn, requires
sensory apparatus that senses both the external environment (exteroreceptors) and the interior environment (interoreceptors). These originate signals that
pass either to nearby cells or to the central nervous system either for specific, reflex responses or for global
responses. These signals are important at all levels
of organization. At the subcellular level, these signals
regulate the activities of subcellular components such
as the expression of specific genes or the regulation of
the rates of energy transformation. At the tissue level,
local signals can regulate smooth muscle contraction
to regulate blood flow within the organ or secretion
into ducts; at the organ system level, signals traveling
through the blood (hormones) or over nerves can
coordinate activity of the system. At the whole
organism level, signals at all levels must be used to
adapt to whole-body responses such as running to
avoid predators. Coordinating command and control
for muscle contraction using sensory information
from the environment (exteroreceptors) and from the
muscle (interoreceptors) is illustrated in Figure 1.1.6.
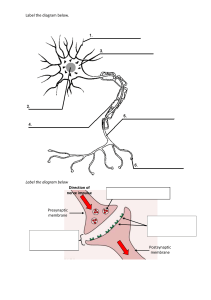
Signaling at the cellular level is illustrated schematically
in Figure 1.1.7.
MANY CONTROL SYSTEMS OF THE BODY USE
NEGATIVE FEEDBACK LOOPS
One of the main themes of physiological control systems comprises a negative feedback loop. This consists
of controlled parameters such as plasma calcium concentration, body temperature, plasma glucose concentration, and plasma pH, a sensor for that parameter, a
comparator, and an effector. For many physiologically
controlled parameters, there is a set point or reference
(see Figure 1.1.8). This is the desired value for the controlled parameter. Its value can change under some circumstances. When the controlled parameter varies from
its set point, the variation is detected by the sensor and
comparator. The comparator then engages some effector
or actuator mechanism to correct the departure of the
parameter from its normal, set-point value. An example
of this is core body temperature, whose normal setpoint value is about 37 C. Whenever heat loss exceeds
heat production, body temperature falls below the
set point and the person shivers. In this case, the sensor
The C ore P rinciples of Physiology
POSITIVE FEEDBACK CONTROL
SYSTEMS HAVE DIFFERENT SIGNS FOR
THE ADJUSTMENT TO PERTURBATIONS
Central nervous
system
Command signal
Exteroreceptor
sensory signal
Skin
Positive feedback systems exist for the mechanism of
blood clotting, parts of the menstrual cycle, aspects of
the action potential, and for parturition (child birth). In
these cases, the disturbance is followed by an adjustment in the same direction of the disturbance, so that
there is a rapid increase in some component. These positive feedback systems generally are self-limiting and,
after the rapid increase in some component, there is a
gradual return to baseline levels.
ANTICIPATORY OR FEED-FORWARD CONTROL
AVOIDS WIDE SWINGS IN CONTROLLED
PARAMETERS
Muscle
Interoreceptor
sensory signal
Motor signal
Spinal cord
FIGURE 1.1.6 Neural signaling in the control of muscle. Neurons consist
of cell bodies (dark circles) that have long processes (black lines) that
bring signals into the central nervous system (dark lines with arrows) or
take signals out toward the periphery (light lines with arrows). Branches at
the ends of the long processes signify the junction of one neuron with
another or with muscle. Neural signal transmission across these junctions
is discussed in Chapter 4.2. Muscles are controlled by motor neurons
whose cell bodies lie in the spinal cord. These can be activated in reflexes
initiated by exteroreceptors that sense perturbations on the skin and
send signals to the spinal cord and eventually activate the motor neurons
by a simple reflex involving just a few interneurons in the spinal cord.
Muscles can also be activated by another reflex involving a stretch
receptor internal to the muscle (interoreceptor). In a third pathway,
motor neurons can be activated by command signals originating in the
brain. Nervous control of muscle is considered in Chapters 4.4 and 4.5.
detects the temperature, the comparator determines that
it has fallen below the set point, and it engages the skeletal muscles as an effector to produce heat by shivering
to help raise the temperature back to the set point. In a
fever, the set point is elevated and the individual feels
chilled even when the temperature is elevated to, say,
40 C. When the fever “breaks,” the set point is reset
back to 37 C and the person perspires because now the
body temperature is elevated above the set point. When
the controlled parameter varies from its set point, the
variation is detected by the comparator. The comparator
then engages some actuator mechanism to correct the
departure of the parameter from its normal, set-point
value. The controlled parameter can vary from its set
point by the action of some physiological disturbance.
These control systems are called negative feedback loops
because the causality forms a loop (y.x.u.a.y)
and the adjustment is typically the opposite sign, or the
negative, of the disturbance. If d adds to y, the adjustment a subtracts from y; if d subtracts from y, a adds to
it. There are many systems that operate through negative
feedback loops.
Sometimes rapid changes in a controlled parameter can
outstrip the physiological mechanisms for reacting
to these changes, resulting in potentially catastrophic
changes in the internal environment. To avoid this,
some physiological systems anticipate changes in controlled parameters and begin to do something about it
even before the parameter changes. Most wide swings in
controlled parameters have to do with behavior. Eating,
for example, is followed by an influx of nutrients into
the blood. The nervous system prepares the gastrointestinal tract for a meal by using sensory cues—the sight,
aroma, and taste of food—to induce the secretion of
gastrointestinal fluids even before food is swallowed.
In another example, controlled parameters including
blood pH, PCO2 (the partial pressure of CO2 in the
blood—a measure of CO2 concentration) and blood
PO2 help regulate the depth and frequency of breathing.
Negative feedback mechanisms keep these controlled
parameters within narrow ranges during normal activity.
During strenuous activity, there appears to be little or
no error in these controlled parameters, though the
depth and frequency of breathing is markedly increased.
This occurs through an anticipatory response of the central nervous system in which the depth and frequency
of breathing is activated simultaneously with activity.
DEVELOPMENTAL AND THRESHOLD CONTROL
MECHANISMS REGULATE NONCYCLICAL AND
CYCLICAL PHYSIOLOGICAL SYSTEMS
Although negative feedback control is a major theme in
physiology, it does not account for a variety of important physiological events. Developmental events include
the onset of puberty and menopause. Pregnancy, parturition (birth), and cyclical events such as the menstrual
cycle and the sleep/wake cycle are episodic events that
do not obey negative feedback mechanisms and may
involve positive feedback mechanisms.
WE ARE NOT ALONE: THE MICROBIOTA
In natural ecosystems, human beings are literally covered with tiny contaminating organisms. We typically
have 10 times more bacteria than body cells, but each
11
12
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Small molecular weight,
lipophilic signal
Polypeptide
Polypeptide
hormone
4
hormone
3
Nuclear
receptors
Trimeric G-proteins
Amplifying enzyme
Catalytic receptors
AC
PLC
5
Ligand-gated channels
P
JAK
2
IP3
Ca2+
Ca2+
ATP
cAMP
ATP
Ca2+
PKA
Ca2+
1
P
Voltage-gated
channels
+
mRNA
Secretory protein
Ribosome
Proteins
RNA
polymerase II
+
-
Bind to response
element
Nuclear receptors
Nucleus
FIGURE 1.1.7 Synopsis of signaling mechanisms on the cellular level. Cells receive electrical signals that can be converted into chemical signals
through voltage-gated channels (1). The voltage-gated calcium channel is shown. Chemical signals released from nearby cells can also open ion
channels, producing electrical signals in the cell (2). Cells receive chemical signals in the form of polypeptide hormones that cannot penetrate the cell
membrane. These can affect the cell by coupling to heterotrimeric G-proteins (3) or to catalytic receptors on the surface of the cell (5). These are
coupled to amplifying enzymes or to kinases that phosphorylate intracellular proteins. Small molecular weight, permeant chemical messengers (4) can
enter the cell and bind to receptors in the nucleus, which then alter the kind or amount of specific proteins made by the cell. These signaling
mechanisms are discussed in detail in Chapter 2.8.
4
5
Reference
value or set point
The actuator makes adjustment, a,
based on input u from the comparator
Disturbance
r
Comparator
+
–
d
u
x
Actuator
a
+
FIGURE 1.1.8 Component parts of a
negative feedback loop. A controlled
parameter, y, is sensed by a sensor
that releases a signal that related
to the value of y [x 5 g(y)]. This signal
is fed into a comparator, which
compares the signal x to some
reference or set-point value, causing
the comparator to produce a signal u
that is a function of the error of x
from its reference [u 5 h(r 2 x), where
u is the signal from the comparator,
r 2 x is the error and h(r 2 x) gives
the functional dependence of u on
the error]. The signal u turns on an
actuator that makes an adjustment,
a, to the value of y. In negative
feedback, the value of a reduces the
error (r 2 x) so that the value of y
returns towards its normal, set-point
level. Disturbances, d, can alter the
value of y and the negative feedback
loop is engaged to minimize the
departure of y from its set-point level.
The reference value, r, is like a set point; its input
into the comparator results in a signal u = h(r–x)
1
y is the controlled parameter
y
Controlled
parameter
Sensor
3
The comparator compares the
sensor's signal, x, to a reference
signal, r, to produce an output signal
u, to the actuator
2
y is sensed by a sensor which produces
an output signal, x, related to y
(x = g(y))
The C ore P rinciples of Physiology
of these is small, and so the total mass of these bacteria
amounts to 0.51.5 kg. These organisms are present as
surface contaminants, but they are the usual surface
contaminants and so they can be considered to be part
of us. They live on the surface of the body, and invasion
within the body constitutes infection, and must be
fought off by our immune systems. They live on the
skin, in the oral cavity, in the airways, and in the gastrointestinal system and on the terminal parts of the reproductive system where it melds with the skin. Most of
these are within the gastrointestinal system, and fully
half of the feces is estimated to consist of bacteria. In
addition to the bacteria, many natural ecosystems provide us with a load of other organisms: tapeworms,
flukes, helminths (hookworms, pinworms, and roundworms), leeches, fleas, various fungal organisms, and a
host of viruses. Although in natural ecosystems, infections with some of the multicellular parasites may be
unusual, the load of bacterial contaminants is unavoidable. The aggregate of these hangers-on is called the
microbiota. The microbiota engage signaling systems of
the body and thereby alter our physiological states. This
is true of the rhinoviruses that make us sneeze, thereby
spreading them around to other individual hosts, and
intestinal bugs that induce diarrhea, using the host signaling systems, to likewise spread them to other hosts.
Evidence is accruing that even those “benevolent”
strains that do not make us frankly sick still manipulate
our physiology to their advantage. Thus the microbiota
become part of our physiology.
PHYSIOLOGY IS A QUANTITATIVE
SCIENCE
As described earlier, homeostasis refers to the maintenance of a constant internal environment, where the
internal environment refers to the extracellular fluid
that surrounds the cells. This internal environment is
characterized by the concentration of a host of materials, and each of these concentrations has a unit and a
numerical value. Many of these materials are metabolized by the tissues so that maintaining constant values
requires matching supply to consumption. The rates of
supply and consumption also have units and numerical
values. As Figure 1.1.5 shows, the circulatory system
unites all organs of the body by virtue of their perfusion
with a common fluid, the blood. Maintenance of this
flow requires pressure differences that also have units
and magnitudes. Understanding the flows and forces
that keep the blood moving and keep its composition
relatively constant requires a quantitative approach.
SUMMARY
Physiology is the integrated study of the normal function of the human body. Like many complicated things,
the body can be viewed as a set of subcomponents that
interact by linking the output of one component to the
input of another. These subcomponents are the organ
systems. These include the cardiovascular system, the
respiratory system, the renal system, the gastrointestinal
system, the neuroendocrine system, the musculoskeletal
system, the integument, and the reproductive system.
Understanding how the body works as a whole requires
us to make a model, either implicit or explicit, that
explains the integration of the structures that make
up organ systems, and the integration of the organ
systems that produces the overall system behavior.
Explanation requires that cause and effect in the model
faithfully predicts cause and effect in the real system.
Understanding can occur on different hierarchical levels
of integration: the systems level, the organ level, the cell
level, and the subcellular level. Each level seeks to
explain behavior at that level on the basis of the components that make up that level. This is reductionism, the
explanation of the behavior of a complicated object on
the basis of its parts. We say that we understand something when we can explain function in terms of the
parts one level below and we can show how behavior at
that level contributes to behavior one level above.
Aristotle identified four classes of causality: the material
cause, the efficient cause, the formal cause, and the final
cause. Explanation of something on the basis of the
final cause is called teleology. Although science presumes that each component of living things obeys physical law alone, systems produce emergent properties
that we seem to be unable to predict. These emergent
properties belong to the system as a whole rather than
to individual parts within it.
Evolution is one cause of the human form and function, but it aids us only a little in understanding how
physiological systems work.
In the hierarchy of levels of organization, cells are the
fundamental unit of life. The various cells of the body
show a remarkable diversity of form and function, but
they all carry the complete genome, with the exception
of erythrocytes and reproductive cells. The diversity of
form arises from the use of only parts of the genome for
each type of cell.
The overriding principle of human physiology is
homeostasis, meaning the maintenance of a constant
internal environment. Our internal environment is the
extracellular fluid that bathes all cells in the body.
A combination of internal control systems and external
behavior maintains homeostasis. In the final analysis,
our life depends on inputs from the environment and
so our behavior (feeding, drinking, and temperature
control) is crucial to our survival. Internal regulation
of the internal environment relies primarily on negative
feedback control, in which controlled parameters feed
into a sensor that compares the value of the controlled
parameter to the desired set point or reference levels.
Variation from the set point results in the activation
of effector mechanisms that increase or decrease the
controlled parameter so that it more closely approximates the set-point level. Anticipatory control also contributes to homeostasis without allowing wide swings
in the values of controlled parameters. Some important
physiological events, such as puberty, menstruation,
pregnancy, parturition, menopause, and the sleep/wake
cycle, are not homeostatic. Instead, they incorporate
switches between one physiological state and another.
13
14
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
These control systems contribute to the robust control
of bodily functions that enable homeostasis under
harsh environmental conditions.
REVIEW QUESTIONS
1. What argument does holism make against
reductionism?
2. Give some examples of emergent properties
3. What is a cell? Why is it considered to be a fundamental unit of organization of life?
4. Contrast genotype with phenotype
5. What is homeostasis? Why is it central to
physiology?
6. What is the internal environment in large multicellular animals such as ourselves?
7. What would constitute proof for the theory of
evolution? Do you think science has provided
it? Why or why not?
8. From Einstein’s equation E 5 mc2, you have
learned in physics that mass and energy are
interconvertible. Why can we say that mass and
energy are conserved in physiological systems?
What does it mean to say that living things
“transform” matter and energy?
9. Describe the hierarchical organization of the
body.
10. Give examples of signaling at the organism level
and cellular level.
Physical Foundations of
Physiology I: Pressure-Driven Flow
Learning Objectives
G
G
G
G
G
G
G
G
G
G
Define intensive and extensive variables
Define flow and flux
Describe the driving principle for heat flow, electrical
current, diffusive flow, and volume flow
Explain what is meant by fluxes moving downhill
Write a continuity equation and describe its meaning
Explain why steady-state flux requires a linear gradient of
T, Ψ, C, or P
List four capacitances commonly encountered in
physiology
Define pressure and be able to convert pressure between
atm, mmHg, and Pa
Write Poiseuille’s law, state its assumptions, and be able to
calculate flow using it
Write the Law of Laplace for cylindrical tubes and for
spheres
FORCES PRODUCE FLOWS
EXTERNAL AND INTERNAL MOVEMENT IS A
HALLMARK OF HUMAN LIFE
For humans in their natural environment, movement
is essential for survival. This movement refers to translation of the body from one location to another,
and movement of the limbs relative to one another. In
addition to this movement of body parts with respect
to the external world, movement of materials within
the body is also essential. Most important among
these internal movements are the movement of the
blood, movement of the air in and out of the lungs,
movement of food and fecal material along the gastrointestinal tract, and movement of the urine from its formation to elimination. In addition, the body transports
materials across barriers such as the gastrointestinal
tract lining, lungs, and kidney tubule. Transport also
occurs within cells. All of these movements require the
continued application of force to overcome inertia and
friction.
1.2
TRANSPORT OF MATERIAL IS DESCRIBED
AS A FLOW OR A FLUX
The transport of material is quantitatively expressed
as a flow, which we will symbolize by the variable Q.
The flow can be expressed as
G
G
G
G
volume of material or fluid transported per unit time;
mass of material transported per unit time;
number of particles or moles transported per unit
time;
number of ions or unit charges transported per unit
time (electrical current).
FLOW DEPENDS ON THE AREA;
FLUX IS FLOW PER UNIT AREA
The total flow of volume or solute is an extensive variable: the flow depends on the extent or the amount of
the system that gives rise to the flow. In the case of two
compartments separated by a membrane, doubling the
area or extent of the membrane would produce twice as
much flow between the two compartments. Dividing
the flows by the area normalizes the flows. The normalized flow is the flux, and the flux is an intensive
variable whose value is independent of the extent of the
system. Flux is defined as
QV
A
QS
JS 5
A
JV 5
½1:2:1
where QV is the volume flow, JV is the volume flux, A is
the cross-sectional area through which flow occurs, oriented at right angles to the direction of flow, QS is the
amount of material (solute) transported per unit time,
JS is the solute flux, and A is the area. The units of flux
are amount or volume or mass or charge per unit time
per unit area.
Strictly speaking, fluxes and flows are vectors, consisting
of the magnitude of the flux or flow and its direction.
Unless otherwise noted, we will consider flux or flow
only in one direction and therefore we will suppress the
vector nature of flux and flow.
15
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00002-1
16
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
FLUX DEPENDS LINEARLY ON ITS
CONJUGATE FORCE
For a variety of forces and fluxes, the flux that results
from a net force varies linearly with the force:
½1:2:2
Jx 5 LFx
½1:2:4
where Jx is the flux of something, L is a phenomenological coefficient, and Fx is the net force that drives the flux.
This generic equation holds for a variety of kinds of
fluxes. The flux of heat energy, electrical flux (the current
density), diffusion of solute and pressure-driven flow all
obey this general phenomenological law. In each of these
cases, the net force is proportional to the gradient of an
intensive variable. Strictly speaking, the gradient is a
vector quantity, but we use it here to denote the slope of
these intensive variables along one dimension:
½1:2:3
diffusion gradient will also modify the flux of solute. In
general, flows produced by multiple flow processes are
not independent. If there are two forces driving flows,
we write
Fourier0 s law of heat conduction: JH 5 2λ
dT
dx
Ohm0 s current law:
Je 5 2σ
dψ
dx
Fick0 s first law of diffusion:
JS 5 2D
Pressure-driven flow:
JV 5 2LP
dC
dx
dP
dx
where JH is the flux of heat energy, dT/dx is the temperature gradient, λ is the coefficient of thermal conductivity,
Je is the electrical current flux, dψ/dx is the voltage gradient, σ is the electrical conductivity, JS is the solute flux,
dC/dx is the concentration gradient, D is the diffusion
coefficient, JV is the volume flux, dP/dx is the pressure gradient, and LP is the hydraulic conductivity. All of these
phenomenological equations find application in physiological systems. They are all analogues of Ohm’s law.
These equations are true only if the only driving force is
the one specified. For example, diffusion of electrolytes,
charged solutes, is influenced by electric fields. If a voltage gradient is also present along with a concentration
gradient, Fick’s first law of diffusion would need to be
modified to reflect that influence. A pressure difference
that produces a volume flow in the presence of a
J1 5 L11 F1 1 L12 F2
J2 5 L21 F1 1 L22 F2
where L11 is the coefficient relating flux 1 to its primary
driving force 1 and L22 relates flux 2 to its primary driving force 2, and L12 and L21 are the coupling coefficients
that describe how secondary forces affect the flows. An
example of this is a bimetallic junction. When two
unlike metals are joined together, passing a current
through the junction causes it to either heat up or cool,
and this is called the Peltier effect. The coupling coefficient implies that if you heat up or cool the junction, a
current will flow. This is the basis of the thermocouple. In
this case, the two fluxes are heat and current and the
two forces are temperature gradient and voltage gradient. Because of a principle called microscopic reversibility, it turns out that the cross-coupling coefficients are
equal: L12 5 L21. This is called Onsager reciprocity, in
honor of Lars Onsager (19031976) who earned the
Nobel Prize in Chemistry in 1968 for this discovery.
FLUX MOVES DOWNHILL
The relations in Eqn [1.2.3] describe fluxes in one
dimension. Both fluxes and gradients are actually vectors, but we consider a single direction here for simplicity. Consider Fick’s law of diffusion for solutes. If the
gradient of concentration is constant, we may write
½1:2:5
JS 5 2D
ðC1 2 C2 Þ
ðx1 2 x2 Þ
for two points (C1, x1) and (C2, x2). If C1 . C2 and
x1 , x2, the slope is negative and the flux is positive. If
C1 , C2 and x1 , x2, then the slope is positive and the
flux is negative. Thus, the flux always goes from regions
of high concentration to regions of low concentration
(see Figure 1.2.1). This is true for all the intensive variables for the fluxes in Eqn [1.2.3]. These fluxes always
move downhill, unless acted upon by additional forces.
x-axis
dC < 0
dx
C
J>0
dC > 0
dx
J<0
C
X (distance, m)
X (distance, m)
FIGURE 1.2.1 Flux moves downhill. Consider one-dimensional flux, with positive flux defined as in the direction of the x-axis. In this case, we consider
diffusion that is driven only by a concentration gradient. If the gradient is negative (higher concentrations at lower values of x), then by Eqn [1.2.3], the
flux is positive and directed to the right (middle panel). If the gradient is positive, then the flux is negative and directed to the left. In each case, the
flux of solute is from the region of high concentration toward the region of lower concentration.
Phys ical Foundat ions of P hysiology I: P ressure-Dr iven Flow
CONSERVATION OF MATTER OR
ENERGY LEADS TO THE CONTINUITY
EQUATION
Here we consider that a concentration gradient exists
and produces a solute flux as a consequence. We consider a cylindrical tube as shown in Figure 1.2.2, having
a cross-sectional area A, which is intersected at right
angles by planes at x 5 x and x 5 x 1 Δx. so that the
tube is cut into three compartments, the left, middle,
and right compartments.
The concentration may vary with time and distance.
We define the concentration in any volume element as
Nðx; tÞ
½1:2:6
Cðx; tÞ 5
V
where N(x, t) is the number of solute particles in the
volume element and V is the volume element. We
define J(x) as the net number of solute particles crossing
the plane at x 5 x per unit time per unit area, with positive being directed along the x-axis, to the right. The
number of particles entering the middle compartment
from the left in time Δt is AJ(x)Δt and the number leaving the middle compartment by crossing the plane at
x 5 x 1 Δx is AJ(x 1 Δx)Δt. Here the parenthesis means
“function of” and not multiplication. If there is no
chemical transformation of the solute particles, their
number is conserved and we can write
ΔN 5 AJðxÞΔt 2 AJðx 1 ΔxÞΔt
½1:2:7
ΔN
5 AJðxÞt 2 AJðx 1 ΔxÞ
Δt
Dividing by the volume element V 5 AΔx and rearranging, we have
1 ΔN
AJðxÞ 2 AJðx 1 ΔxÞ
5
AΔx Δt
AΔx
½1:2:8
N
V
Δ
Δt
52
Similar continuity equations can be written for the flux
of heat energy, charge, and volume. Their form is given
in Eqn [1.2.10]:
Heat: ρCP
C @ψ
@Je
52
V @t
@x
Charge:
½1:2:10
Solute:
@T
@JH
52
@t
@x
@CS
@JS
52
@t
@x
Volume:
C @P
@JV
52
V @t
@x
where ρ is the density of matter through which heat
flows and Cp is its specific heat capacity. In the next
equation, C is the electrical capacitance, V is the volume. Next, Cs is the concentration. In the last line C is
the compliance, and V is the volume. Here we have the
unfortunate situation in which single variables denote
different quantities: C stands for electrical capacitance
(in farads), concentration (usually in molar) and compliance (5ΔV/ΔP). Generally the meaning of the variable is clear from its context.
STEADY-STATE FLOWS REQUIRE
LINEAR GRADIENTS
AJðx 1 ΔxÞ 2 AJðxÞ
AΔx
In the limit of Δt-0 and Δx-0, this becomes
½1:2:9
This equation is called the continuity equation. What
this equation says is that if the flux of solute is not the
same everywhere, then the amount of solute must be
building up or becoming depleted somewhere, and
this buildup or depletion changes the concentration of
solute. It is a straightforward consequence of the conservation of material. This equation is not true if the
diffusing chemical undergoes chemical transformation.
In this case, it is not conserved.
@Cðx; tÞ
@JðxÞ
52
@t
@x
Cross-sectional
area, A
In homeostasis, there is a steady supply of nutrients and
removal of wastes and a steady withdrawal of nutrients
and a steady production of wastes by the tissues. This
steady state in which all flows are constant is more easily amenable to mathematical analysis. What “steady
state” means is that each of the variables on the lefthand side of Eqn [1.2.10] is zero because at the steady
state there are no changes in temperature, charge, concentration, or pressure with time:
Heat: ρCP
J(x)
J(x +Δx)
C @ψ
@Je
52
50
V @t
@x
Charge:
½1:2:11
x
x +Δx
x axis
FIGURE 1.2.2 Fluxes as a function of distance in the presence of a
concentration gradient.
Solute:
@T
@JH
52
50
@t
@x
@CS
@JS
52
50
@t
@x
Volume:
C @P
@JV
52
50
V @t
@x
17
18
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Substituting in from Eqn [1.2.3] for JH, Je, JS, and JV,
we have
@2 T
50
@x2
½1:2:12
@2 ψ
50
@x2
@2 C
50
@x2
capacitance is an important concept for physiologists
as well, because membrane potential derives from a separation of electrical charges across the membrane, and
the membrane itself acts like a tiny capacitor with two conducting plates, separated by a dielectric. We will discuss
this further in the sections on membrane potential, action
potential, and the cable properties of nerves (Chapters
3.13.3). The other relationships completely analogous to
the relation between charge, capacitance, and voltage, are
H 5 CP MT: Heat energy 5 heat capacity 3 mass
3 temperature
@ P
50
@x2
2
This condition is met only if the gradient of T, ψ, C, or
P is constant; thus, the slope of T, ψ, C, and P at steady
state is constant, and each of these intensive variables
varies linearly with distance.
HEAT, CHARGE, SOLUTE, AND VOLUME
CAN BE STORED: ANALOGUES OF
CAPACITANCE
The steady state is often approximated in the body but
rarely achieved. At rest heat production balances heat
exhausted to the environment. When we begin exercising,
heat production rises rapidly and the temperature of the
body rises accordingly until, once again, heat production
matches heat transfer to the environment, achieved by
using other forces besides the conduction described in
Fourier’s law. This new steady state of temperature during
exercise is achieved at different operating conditions than
at rest. In another example, transport of blood through the
cardiovascular system is pulsatile, because the pressure that
drives transport comes from the heart, and the heart produces force rhythmically. Each of the main four variables
we have been discussing, heat, charge, amount of chemicals, and volume, can be temporarily stored or depleted.
Electrical charge can be stored in capacitors. The constitutive relation between charge, voltage, and capacitance
is given as
½1:2:13
Q 5 CV
where Q here stands for charge, C is the capacitance, and V
is the voltage. Here we are victims of the use of the same
variables to denote entirely different quantities. We will
use Q most often to signify a flow, but here it signifies
charge, in coulombs. In physiology, we often use C to
denote concentration, but here it means capacitance, in
farads (5CV21); in physiology, V usually signifies volume,
but here it means electrical potential, in volts. Electrical
M 5 VC: Amount 5 volume 3 concentration
V 5 CP: Volume 5 compliance 3 pressure
½1:2:14
where the capacitance-like elements include electrical
capacitance (C in Eqn [1.2.13]), thermal mass (CpM,
the specific heat capacity times the mass), volume (V),
and compliance (C). Note again the multiplicity of uses
of a single notation. C variously stands for capacitance,
heat capacity, concentration, or compliance.
The capacitances are all expressed as the ratio of an
extensive variable and an intensive variable and are all
themselves extensive variables. Table 1.2.1 summarizes
the four kinds of capacitances.
PRESSURE DRIVES FLUID FLOW
In the case of fluid or air flow, pressure differences drive
the flow. The SI unit of pressure is the pascal, Pa, equal to
1 N m22. However, physiologists still use other units, notably the atmosphere and mmHg. The atmospheric pressure
is the weight of a column of air equal to the height of the
atmosphere in the earth’s gravitational field per unit area
of the earth’s surface. The actual pressure in the atmosphere
decreases as you ascend, but the unit of 1 atm is defined
for a standard condition of the air and standard altitude at
sea level. The conversion between atmospheres and mmHg
is an observed phenomenon. Atmospheric pressure can be
measured in units of mmHg as described in Figure 1.2.3.
Figure 1.2.3 illustrates that atmospheric pressure supports a column of 760 mmHg high. The pressure of this
column of Hg is equal to atmospheric pressure, and the
pressure of the column is simply its weight divided by
its area. The weight is the force of gravity acting on the
column, and is given as
½1:2:15
W 5 F 5 mg
5 ρVg
5 ρAhg
TABLE 1.2.1 Four Kinds of Capacitances
Capacitance
Expression Units
Application
Electrical capacitance, C
Q/V
Nerve conduction, membrane potential
Farads 5 coulombs/volt
21
Thermal capacitance, CpM
H/T
JK
Chemical capacitance, V, volume
M/C
Volume 5 moles/molarity, L
Mechanical capacitance, C, compliance V/P
5 joules/temperature
Heat production/loss temperature regulation
Metabolism, filtration
Compliance 5 volume/pressure, m3 Pa21 Blood pressure, breathing
Phys ical Foundat ions of P hysiology I: P ressure-Dr iven Flow
High vacuum
High vacuum pump
h = 760 mm
Atmospheric pressure
supports a column of
Hg
Weight of column of Hg
= mg
Pressure force up
= PA
Hg
At equilibrium, the weight of the column of
Hg equals the force of atmospheric pressure:
mg = P A
FIGURE 1.2.3 Measurement of atmospheric pressure. A closed vertical tube is connected to a high vacuum pump and evacuated air. When inverted
into a dish of mercury, the atmospheric pressure forces mercury up the tube until mechanical equilibrium is achieved when the weight of the column
of mercury exerts a pressure equal to the atmospheric pressure. At sea level in dry air, 1 atm will support a column 760 mmHg high.
TABLE 1.2.2 Conversion Between Pressure Units
atm
mmHg
Pa
1
760
1.013 3 105
1
133.29
0.00750
1
0.00132
26
9.87 3 10
The pressure is just the force per unit area. Dividing
Eqn [1.2.15] by the area, we get
F
5 ρgh
A
Thus, the height of the column of mercury in equilibrium with the atmospheric pressure is independent
of its area. We need to specify only the height of the column of mercury. Thus, at sea level, the atmospheric
pressure supports a column of 760 mmHg high and we
say that 760 mmHg 5 1 atm.
½1:2:16
P5
The value of atmospheric pressure in pascals 5 N m22 can
be calculated from 760 mmHg by using the density of Hg
(13.59 g cm23) and the acceleration due to gravity
(9.81 m s2). Inserting these values into Eqn [1.2.16], we get
P 5 13:59 g cm23 3 9:81 m s22 3 0:76 m
5 13:59 g cm23 3 ð100 cm m21 Þ3 3 1023 kg g21
3 9:81 m s22 3 0:76 m
5 13:59 3 103 kg m23 3 9:81 m s22 3 0:76 m
1 atm 5 101:3 3 103 kg m s22 m22 5 1:013
3 105 N m22 5 1:013 3 105 Pa
We can therefore complete a conversion table for
pressure units (Table 1.2.2).
POISEUILLE’S LAW GOVERNS
STEADY-STATE LAMINAR FLOW
IN NARROW TUBES
In 1835, Jean Leonard Marie Poiseuille experimentally
established the relationship between flow through
narrow pipes and the pressure that drives the flow. The
relationship is
½1:2:17
πa4 ΔP
Qv 5
8η Δx
where QV is the flow, in units of volume per unit
time, π is the geometric ratio, a is the radius of the
pipe, η is the viscosity, ΔP is pressure difference
between the beginning and end of the pipe, and Δx is
the length of the pipe. This equation describes the relationship between flow and pressure difference only for
laminar flow. Laminar flow is steady, streamlined flow,
and it is distinguished from turbulent or chaotic flow.
This equation is often applied to problems in physiology even though the conditions for its valid application
are missing. Its application requires us to understand
viscosity.
Consider two parallel plates separated by a fluid, as
shown in Figure 1.2.4. The top plate can be moved at a
constant velocity relative to the stationary bottom plate
only if the plate is subjected to a force that continuously
overcomes the frictional resistance on the plate caused
by its contact with the adjacent fluid.
The viscosity is the resistance of a fluid to shear forces.
It is defined by the equation
½1:2:18
F
dv
5η
A
dy
19
20
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Moving plate
Force applied to the moving plate
at constant v is opposed by an
equal drag force of the fluid on
the plate
F
FD
Thin fluid layer
The layer of fluid adjacent to the
plate has the same velocity as
the plate
The layer of fluid adjacent to
the stationary plate has
zero velocity
Stationary plate
FIGURE 1.2.4 Definition of viscosity. Two plates are separated by a fluid. The top plate moves with constant velocity, v, with respect to the stationary
bottom plate. The fluid adheres to the plates and a thin layer of fluid immediately adjacent to the plates has the same velocity as the plates.
This results in a velocity profile in the fluid. The steepness of this velocity profile, dv/dy, is the velocity gradient.
where F is the shear force, A is the area, v is the velocity,
and y is the dimension perpendicular to the plate.
The ratio F/A is called the shear stress and the quantity
dv/dy is called the velocity gradient. F/A in this equation has the units of pressure, Pa 5 Nm22 and dv/dy has
the units of m s21 m21 5 s21, so the units of η in SI are
Pa s. Thus, the viscosity is the ratio of the shear stress,
F/A, dvided by the velocity gradient, dv/dy. In older
texts viscosity is sometimes given in units of poise 5 1
dyne cm22 s. These can be converted to Pa s by using
the definition of Pa 5 1 N m22, 1 N 5 1 kg m s22 and 1
dyne 5 1 g cm s22: Thus, 1 N 5 105 dyne.
1 Pas 5 1 N m22 s 5 1 kg m s22 m22 s 5 1 kg
3 103 g kg21 m 3 100 cm m21 3 s22 3 m22
3 ð0:01 cm m21 Þ2 s 5 103 g 3 100 cm 3 s22
3 1024 cm22 s 5 10 g cm s22 2 cm22 s 5
10 dyne cm22 s 5 10 poise
So 1 Pa s 5 10 poise
EXAMPLE 1.2.1 Ultrafiltration in the Kidney
The kidney has a structure called the glomerulus that consists
of combined layers of cells and extracellular matrix—bundles of
fibers in the extracellular space—that together form an ultrafilter (see Chapter 6.2 for further description). It is called an ultrafilter because the combined layers retain proteins while letting
most small solutes pass into the ultrafiltrate. We model the
membrane here as a flat membrane that is pierced by many
identical right cylindrical holes, or pores. Assume that the radius
of the pores is 3.5 nm and the pore length is 50 nm. The viscosity of the fluid is taken to be the same as plasma, 0.02 poise.
The aggregate area of the pores makes up 5% of the total surface area of the membrane. The total pressure on the input
side, the side of the blood, averages 60 mmHg and on the ultrafiltrate side the total pressure averages 45 mmHg (see
Chapter 6.2 for a discussion of the origin of these pressures).
The total available area of the membrane is 1.5 m2. What is the
filtration rate in cm3 min21?
The situation is depicted schematically in Figure 1.2.5. We use
Poiseuille’s equation here. The total flow, QV, is the sum of the
flow through all of the pores:
Qv 5 Nqv 5 Nπa4 =8η ðΔP=ΔxÞ
Pore
Pultrafiltrate
Pblood
qV
n = N pores /unit area
Δx
FIGURE 1.2.5 Model of the kidney ultrafilter.
where N is the number of pores and qV denotes the flow through
a single pore. Here a is the radius, a 5 3.5 3 1029 m, η is the viscosity (η 5 0.02 poise 3 1 Pa s/10 poise 5 0.002 Pa s), ΔP is the
pressure difference 5 15 mmHg 3 133.3 Pa mmHg21 5 1999.5 Pa,
Phys ical Foundat ions of P hysiology I: P ressure-Dr iven Flow
and Δx 5 60 3 1029 m. Now that all the units are compatible, we
plug them into the equation and get:
29
qV 5 π3ð3:53 10
236
5 471:44310
23
mÞ =83 23 10
4
23
m =16310
4
29
Pa s3 ð1999:5 Pa=60310
mÞ
21
Pa s 333:33310 Pa3m
9
5 982:13 10224 m3 3s21 3ð100 cm3m21 Þ3 3 60 s3 min21
N 3 πa2 5 0:05 3 1:5 m2
Knowing that a 5 3.5 3 1029 m, we solve for N:
N 5 0:075 m2 =π 3 ð3:5 3 1029 mÞ2 5 1:95 3 1015
which is a lot of pores! Multiplying N 3 qV, we get
Qv 5 N 3 qv 5 1:9 3 1015 pores 3 5:89 3 10214 cm3 min21 pore21
5 5:893 10214 cm3 3 min21
5 111:9 cm3 min21
This is the flow through a single pore. We need to know how
many of them are there. If their aggregate area is 5% of the
total, then the number of pores can be calculated from
THE LAW OF LAPLACE RELATES
PRESSURE TO TENSION IN HOLLOW
ORGANS
Rearrangement of this equation gives the Law of Laplace
for a cylinder:
½1:2:20
The blood vessels maintain a pressure difference across
their walls. These vessels approximate hollow cylinders.
The gallbladder, urinary bladder, heart, and lung alveoli
also maintain a pressure difference, and approximate
hollow spheres. The Law of Laplace can be derived for
these ideal geometries by considering mechanical equilibrium. Consider first a circular cylinder of radius r
subjected to an external pressure Po and internal pressure Pi, as shown in Figure 1.2.6.
In Figure 1.2.6, the internal pressure acting on the upper
half of the cylinder by the lower half cylinder produces a
force on the upper half that is given as Pi 3 2r 3 L. The
external pressure acting over the surface of the upper half
of the cylinder produces a force that is equal to
Po 3 2r 3 L, as can be deduced from the situation where
Po 5 Pi in a mechanically stable fluid with no walls. The
balance of the internal and external pressures produces a
net force of ΔP 3 2r 3 L where Δ P 5 Pi 2 Po. This is balanced by the force of the walls of the lower half acting
on the upper half. These forces are the wall tension, in
units of N 3 m21, acting along the length of the wall.
These forces are directed downward, as shown. For
mechanical equilibrium to occur
½1:2:19
Fnet 5 0 5 T2L 5 ΔP2rL
r
T
T
T
L
Pi
This is a reasonable approximation to the filtration rate in an
adult human.
ΔP
Po
FIGURE 1.2.6 Right circular cylinder of radius r and length L is subjected
to a transmural pressure difference. For mechanical equilibrium, the
net force on any part of the cylinder must be zero. We split the cylinder
in half down its axis. The sum of forces on the upper half must sum
to zero.
ΔP 5
T
r
An exactly analogous argument can be made for a sphere.
In this case, the pressure is distributed over the crosssectional area of the hemisphere, and the wall tension
is distributed around the circumference. The result gives
½1:2:21
Fnet 5 0 5 T2πr 5 ΔPπr 2
which is easily rearranged to
½1:2:22
ΔP 5
2T
r
SUMMARY
Flow consists of the movement of something from one
place to another in the body. We identify four classes of
flow of major importance in physiology: flow of heat
energy, electric current, solute, and volume (gas or body
fluids). Flow is an extensive variable, depending on the
extent of the system. Flux is the flow divided by the area
through which the flow occurs. Fluxes are driven by
forces. Heat flux is driven by a temperature gradient,
electrical current by a potential gradient, solute flux by a
concentration gradient, and volume flux by a pressure
gradient. Gradients are vector quantities. Here we consider one-dimensional flow and flux and suppress the
vector nature of these gradients. In each of these cases,
flux occurs “downhill,” meaning from regions of high
temperature, potential, concentration or pressure, to
regions of low temperature, potential, concentration, or
pressure. Each flux has the form
Jx 5 2K
dψx
dx
The continuity equation for each of these fluxes states
that a gradient of flux produces a time dependence of
the driving force. If the flux is not the same everywhere,
then temperature, voltage, concentration, or pressure
varies with time. The continuity equation is a consequence of conservation of energy, electric charge, chemical species, or volume.
21
22
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Steady-state flows require linear gradients of temperature, potential, concentration, or pressure.
Heat energy, electric charge, concentration, and volume
can all be stored in the body. The ability to store these
things is quantified by capacitances. These include thermal capacitance, electrical capacitance, chemical capacitance, and mechanical capacitance. The thermal
capacitance is MCp, where Cp is the specific heat capacity; the units of MCp are in J K21; electrical capacitance
is Q/V, in F; chemical capacitance is V, in L; mechanical
capacitance is compliance, in L Pa21.
Pressure drives flow. Pressure is measured in pascals
5 1 N m22. This unit is equal to 9.87 3 1026 atm or
0.0075 mmHg. Steady-state pressure-driven flow
through narrow pipes is described by Poiseuille’s law:
πa4 ΔP
QV 5
8η Δx
where a is the radius of the pipe, η is the viscosity,
in Pa s, π is the geometric ratio of circumference to
diameter of a circle, ΔP is the pressure difference in
the pipe, and Δx is the length of the pipe. Viscosity is
the resistance of a fluid to shear forces and is defined
mathematically as the ratio of the shear stress to the
velocity gradient.
A transmural pressure within hollow organs or tubes
requires tension in the walls of these organs. So far as
these organs approximate thin-walled cylinders or
spheres, the wall tension obeys the Law of Laplace:
T
r
2T
ΔP 5
r
Cylinder: ΔP 5
Sphere:
where ΔP is the transmural pressure difference, T is the
wall tension, in N m21, and r is the radius of the cylinder or sphere.
REVIEW QUESTIONS
1. Is density an intensive or extensive variable? Is
temperature intensive or extensive?
2. What drives current flow? What drives solute
flow? If solute is charged, would its movement
make a current?
3. What does it mean to say that fluxes “move
downhill”?
4. What does capacitance mean for charge? Solute?
Heat? Volume?
5. How is Ohm’s law like Fick’s law of diffusion?
6. What are the units of viscosity?
7. What are the assumptions in the derivation of
Poiseuille’s law? Are these reasonable?
8. What is the relationship between velocity and
flux in fluid flow?
9. What is the relationship between capacitance,
charge, and voltage?
10. What is the relationship between volume,
amount, and concentration?
11. What is the relationship between compliance,
volume, and pressure?
12. What is the relationship between heat capacity,
heat energy, and temperature?
13. What is the relationship between wall tension
and pressure in a sphere? In a cylinder?
APPENDIX 1.2.A1 DERIVATION OF
POISEUILLE’S LAW
POISEUILLE’S LAW DESCRIBES PRESSUREDRIVEN FLOW THROUGH A CYLINDRICAL PIPE
Consider two fluid compartments which are joined by a
right cylindrical pipe, as shown below, of area A. Since
pressure is defined as a force per unit area, the total
force acting on the fluid in the pipe on the left-hand
surface is just PLA, and the total force acting on the fluid
in the pipe on the right-hand side is PRA. If PL and PR
are not equal, then the fluid within the pipe will be subject to a net force and therefore this volume of fluid will
be accelerated. The result will be a movement of fluid
in the direction of the net force. What we wish to establish is the quantitative relationship between the resulting flow and the pressure difference which drives this
flow.
SHEAR STRESS IS THE VISCOSITY TIMES THE
VELOCITY GRADIENT
The movement of fluid through the pipe shown in
Figure 1.2.A1.1 encounters resistance along the cylindrical surfaces of the pipe. This resistance is key to deriving
an expression for the flow as a function of pressure. It is
due to the viscosity of the fluid, as shown in
Figure 1.2.4. We consider that the flow of fluid occurs
in layers, or laminae, and that each layer exerts a viscous
drag on the layer immediately above and below. The
fluid remains in contact with the walls on either side of
the tube, and so has a velocity v 5 0 at the walls.
In order to achieve a constant velocity, we need to apply
a continual force, F, in order to overcome the viscous
drag of the fluid. The shear stress is the force exerted by
one lamina on an adjacent one, and is given by
½1:2:A1:1
F
dv
5η
A
dy
PL
Area
PR
FIGURE 1.2.A1.1 Flow is driven by pressure differences.
Phys ical Foundat ions of P hysiology I: P ressure-Dr iven Flow
where F is the force, A is the area, η is the coefficient of
viscosity of the fluid, and dv/dy is the gradient of velocity. This equation makes good common sense: the force
necessary to keep the lamina moving depends on how
much area is exposed to the resistance, it depends on
how sticky the fluid is, and it depends on how fast it is
going.
Insertion of this result into Eqn [1.10.A1.2] gives
½1:2:A1:4
0 5 ðPðxÞ 2 Pðx 1 ΔxÞÞ2πr dr
dv
1 2πrΔxη
dr r
1 2πðr 1 drÞΔxη
FLOW THROUGH A PIPE
We consider here the flow through a tube or pipe of
radius a and length Δx. The pressure at the left end
of the pipe is P(x) and the pressure at the right end is
P(x 1 Δx). We consider here a constant flow which
therefore has a constant velocity. This means that the
fluid is not subjected to any net force. Consider the
forces experienced by a hollow shell of inner radius r
and outer radius r 1 dr, as shown in Figure 1.2.A1.2.
The fluid in contact with the walls of the pipe does not
move, so that v 5 0 at r 5 a. The velocity of the fluid
increases as one approaches the center of the tube. The
actual velocity profile will be solved on the way to deriving an expression for the total flow through the pipe.
Because the fluid flows through the pipe at a constant
velocity along the pipe (but which depends on the distance from the walls of the pipe), the sum of the forces
on the hollow shell shown in Figure 1.2.A1.2 must be
zero. The forces to the right include the force of the
pressure on the left and the drag force of the inner layer
of fluid, and the forces to the left include the pressure
on the right and the drag force of the outer fluid. Thus,
we have
½1:2:A1:2
F1
P(x)
which can be re-written as
0 5 2ðΔPÞ2πr dr
dv
1 2πrΔxη
dr r
dv
dv
2 2πrΔxη
2 2π drΔxη
dr r1dr
dr r1dr
½1:2:A1:5
where ΔP 5 P(x 1 Δx) 2 P(x). We next approximate the
gradient of velocity at (r 1 dr) by the first two terms of a
Taylor’s series:
½1:2:A1:6
r
F2
P(x + dx)
r + dr
x
x + dx
FIGURE 1.2.A1.2 Balance of forces on a hollow cylinder of inner radius r
and outer radius r 1 dr. F1 is the drag force of the lamina with inner
radius r 1 dr on the hollow cylinder; F2 is the drag force on the hollow
cylinder by the lamina immediately inside, with outer radius r.
2 dv
dv
d v
C
1 dr
dr r1dr
dr r
dr 2 r
Substitution of Eqn [1.2.A1.6] into Eqn [1.2.A1.5] gives
ΔP
2πr drΔx
052
Δx
dv
1 2πrΔxη
dr r
0 5 PðxÞA 1 F2 2 Pðx 1 ΔxÞA 2 F1
The area of the hollow sphere on which the pressures
act is 2πrdr. The drag forces F1 and F2 are given by
Eqn [1.2.A1.1] as
dv
F1 5 2πðr 1 drÞΔxη
dr r1dr
½1:2:A1:3
dv
F2 5 2πrΔxη
dr r
dv
dr r1dr
dv
dv
2 2πrΔxη
2 2π dr Δxη
dr r
dr r
2 2πr drΔxη
2 2 d v
d v
2
2
2πr
dr
Δxη
dr 2 r
dr 2 r
½1:2:A1:7
Canceling out the like terms, and factoring out the term
2πrdr Δx, we obtain
2 ΔP
η dv
d v
η d2 v
2η
2 dr
052
2
Δx
r dr r
dr 2 r
r dr 2 r
½1:2:A1:8
In the limit as dr-0, the last term in Eqn [1.2.A1.8]
vanishes. Multiplying through by 21, we are left with
the differential equation
2 ΔP
η dv
d v
2
05
1η
½1:2:A1:9
Δx
r dr
dr 2
This second-order differential equation can be solved by
converting it to a first order differential equation with
the substitution y 5 dv/dr, and then multiplying through
23
24
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
by an integrating factor, ρ. Re-arranging Eqn [1.2.A1.9],
we have
dy
ρ
ρ ΔP
½1:2:A1:10
1 y1
50
ρ
dr
r
η Δx
We choose ρ so that the first two terms are an exact
differential. This is true if ρ 5 r. Thus we have
dy
dðryÞ
r ΔP
52
r
½1:2:A1:11
1y5
dr
dr
η Δx
Integrating Eqn [1.2.A1.11], we obtain
ð
ð
r ΔP
dðryÞ 5 2
dr
η Δx
½1:2:A1:12
1 ΔP 2
ry 5 2
r
2η Δx
Canceling the r factor on both sides of Eqn [1.2.A1.12]
and recalling that y 5 dv/dr, we integrate Eqn [1.2.A1.12]
again:
dv
1 ΔP
52
r
dr
2η Δx
ð
ð
1 ΔP
dv
5
2
r dr
½1:2:A1:13
2η Δx
1 ΔP 2
v52
r 1C
4η Δx
where C is a constant of integration which can be evaluated from the boundary conditions. The boundary conditions are that v 5 0 when r 5 a. That is, the velocity of
the fluid immediately adjacent to the walls of the pipe
is zero. Insertion of v 5 0 and r 5 a into Eqn [1.2.A1.13]
gives
a2 ΔP
C5
4η Δx
½1:2:A1:14
1 ΔP
v5
ða2 2 r 2 Þ
4η Δx
This equation gives the velocity of fluid flow as a function of the radial distance from the center (r 5 0) to the
edge (r 5 a) of the pipe. This equation says that the
velocity profile is parabolic.
What we wanted to do at the outset was to calculate the
total flow through the pipe, QV. This is the volume of
fluid which crosses the total cross-section of the pipe
per unit time. The volume flux, JV, is the volume moving through a small increment of unit area per unit
time. In fact JV is the velocity of fluid movement. To see
this, consider a block of fluid moving at constant velocity, v. The block has a cross-sectional area, A. In time t
the block moves a distance vΔt. The volume of
fluid moving in this time is AvΔt. The volume flux, JV,
is the volume of fluid moved per unit area per unit
time. This is
½1:2:A1:15
JV 5
AvΔt
5v
AΔt
Thus, the volume flux is the velocity of fluid movement.
To find the total fluid flow, we integrate the flow as a
function of distance from the center of the pipe. Thus
ða
ða
ða
QV 5 dQV 5 JV dA 5 JV 2πr dr
0
0
ð0a
5 v2πr dr
0
ða ½1:2:A1:16
1 ΔP
5
ða2 2 r 2 Þ2πr dr
4η
Δx
0
ða
2π ΔP
5
ða2 2 r 2 Þr dr
4η Δx
0
The last integral can be evaluated by making the substitution u 5 (a2 2 r2), so du 5 22rdr, to obtain
ð r5a ð r5a
du
2
2
ða 2 r Þr dr 5
u 2
2
r50
r50
ð u50
1
52
u du
½1:2:A1:17
2 u5a2
5
a4
4
Inserting this result of the integration into Eqn [1.2.A1.16],
we obtain
πa4 ΔP
QV 5
½1:2:A1:18
8η Δx
This last equation gives the total flow through the pipe,
QV. This equation is called Poiseuille’s Law, in honor of
the French physician Jean Leonard Marie Poiseuille,
who experimentally established the law in 1835. The
relation shows that the total flow is linearly dependent
on the driving force for the flow, the pressure difference
between the left and right ends of the pipe. Further, it is
inversely related to the length of the pipe, Δx, and to
the viscosity, η. Most importantly, the flow is proportional to the fourth power of the radius of the pipe.
One of the points of the derivation given above was to
show the mathematical origin of this very steep dependence on the size of the pipe.
APPENDIX 1.2.A2 INTRODUCTORY
STATISTICS AND LINEAR REGRESSSION
INTRODUCTION
Statistics have two functions: (1) to describe the variation in some data and (2) perform tests on sets of data
to determine the cause of the variation in that data. As
an example, we know that people of the same age are
not the same height or weight. The set of heights for a
population can be described by a statistic that describes
the center (mean, median or mode) and by a statistic
that describes the variation around that center. Typically
the mean is used to describe the center and the standard
deviation is used to describe the variation, or spread
around that center. These are examples of descriptive
statistics. Suppose we wanted to know the causes of
Phys ical Foundat ions of P hysiology I: P ressure-Dr iven Flow
variation in human height. Possible causes might
include diet, genetics, sleep patterns. It is extremely difficult to determine the cause of variation in humans
because these possible causes are not controlled.
However, there are populations that differ in a variety
of ways. We may ask the question, do two different
populations have different average heights? These kinds
of questions involve statistical tests.
n
P
½1:2:A2:5
s2 5
i51
Xi2 2 2X
Suppose we have a set of variables {X1, X2, . . ., Xi, . . ., Xn}.
The mean is defined as
n
P
n
X
½1:2:A2:6
X 5 μX 5
n
P
i51
Xi 5 n X
n
P
i51
2
σ 5
2
i51
n
ðXi 2XÞ2
i51
The sample variance is a measure of the spread of a population when some sub-set of the population is used so
that the mean is estimated rather than known. Its formula is similar to that of the population variance, except
that one “degree of freedom” is used for the estimation
of the mean. The sample variance is calculated as
s2 5
i51
n21
2
here s is used as a symbol for the sample variance,
whereas σ2 is used as a symbol for the population variance. Note that the formula for s2 is very similar to the
formula for the variance except that n 2 1 is used as the
divisor instead of n, the number of measurements used
to estimate the mean and variance of the sample. The
term (n 2 1) is the degrees of freedom that refers to the
number of independent pieces of information that goes
into the estimation of the statistic. Because the mean is
used in the calculations, each measurement contributes
to the mean and, if you know the mean, the last value
can be calculated from all of the other values. Thus the
degrees of freedom when the data is used to estimate
the mean is n 2 1. Expanding the square term in Eqn
[1.2.A2.3], we have
s2 5
i51
i51
Xi2 2 n X
2
n
½1:2:A2:8
s2 5
n
P
i51
Xi2 2 n2 X
2
n ðn 2 1Þ
Again making use of Eqn [1.2.A2.6] we arrive at the
computational formula for the sample variance:
n 2
n
P
P
2
n Xi 2
Xi
i51
i51
½1:2:A2:9
s2 5
n ðn 2 1Þ
2
ðXi2 2 2X Xi 1 X Þ
n21
The sample standard deviation, denoted by the statistic s,
is defined to be the positive square root of the sample
variance:
vffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
n 2ffi
u n
P
u P 2
un Xi 2
Xi
t i51
i51
½1:2:A2:10
s5
n ðn 2 1Þ
THE STANDARD ERROR OF THE MEAN
ðXi 2XÞ2
n
P
n
P
THE SAMPLE STANDARD DEVIATION
n
n
P
2
Xi2 2 2 n X 1 n X Þ
5
n21
n21
Multiplying both numerator and denominator by n, we
reach
THE SAMPLE VARIANCE
½1:2:A2:4
2
Inserting this into Eqn [1.2.A2.5] gives
½1:2:A2:7 s2 5
Xi
The variance is a measure of the spread of a population.
It is the average of the squared deviations from the
mean. The square is taken so that all deviations from
the mean contribute to the variance; otherwise, negative
deviations would cancel positive deviations: the average
deviation from the mean is zero. For a population the
variance is given by
½1:2:A2:3
Xi 1 n X
n21
Recognizing from Eqn [1.2.A2.1] that
THE POPULATION VARIANCE
½1:2:A2:2
n
P
i51
THE MEAN
½1:2:A2:1
This is rewritten as
The standard deviation of a sampling distribution of a
statistic is called the standard error of that statistic. A
sampling distribution of a statistic is its probability distribution. What this means is that if we estimate the
mean of a population several times using different random samples, we will not get identical answers. There
will be some distribution of the mean. In the same way,
there will be some variation in our sample variance calculated from the different samples. The standard deviation of the distribution of means is the standard error of
the mean. Note that the sample standard deviation does
not change with the sample size, because each additional
member of the sample contributes both to n and to the
squared deviation from the mean. On the other hand,
we ought to expect that our estimate of the mean
improves with the number of observations in the sample. Thus, the standard error of the mean gets smaller as
the number of observations in the sample increases. The
formula for the standard error of the mean is
½1:2:A2:11
s
SEM 5 pffiffiffi
n
25
26
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
PROBABILITY
Probability theory began with the analysis of games of
chance. The probability of an event happening, such as
drawing a particular card, or a card that belongs to a
particular suit, or the appearance of a particular face on
a die after a roll, is defined as the ways in which the
event can happen compared to the total possible number of outcomes. For example, there are six faces on a
die and the likelihood that any particular face ends
upright is one out six, or 1/6 5 0.1625. We can write
this as
P ð4Þ 5 0:1625; Pð5Þ 5 0:1625; P ð6Þ 5 0:1625
where P(i) refers to the probability of the face with
i dots (pips) on it. If we have two dice, the probability
of having a total number of dots depends on how many
outcomes produce that number of dots. For example,
we can find 12 dots only by having 6 on one die and 6
on the other. There are a total of 36 possible outcomes
as shown in Table 1.2.A2.1 below.
Since there are 36 possible outcomes, the probability of
getting a 12 is 1/36 5 0.0278. The probability of getting
a total of 7, however, is higher. We can get a 7 from the
following combinations: {(4,3), (3,4), (5,2), (2,5),
(6,1), and (1,6)}. The probability of getting a 7 is thus
6/36 5 0.1625. Note the probability of getting some
outcome is 1.0, and this is the sum of each of the probabilities for the individual outcomes:
i5n
X
The validity of these conclusions depends on fair dice
that are independent. A die is fair if and only if the
probabilities of all six outcomes are the same, and they
are independent if and only if the outcome of one die
does not influence the outcome of the other.
HYPOTHESIS TESTING
Pð1Þ 5 0:1625; Pð2Þ 5 0:1625; P ð3Þ 5 0:1625;
½1:2:A2:12
there are two ways to get (4,3), without regard to order.
In this case P(4,3) 5 P(3,4) and the probability of getting a 7 with a 4 and a 3 is P{4,3} 5 P(4,3) 1 P(3,4) 5
2/36.
PðiÞ 5 1:0
i51
where there are a total of n distinguishable outcomes.
The probability of observing a particular outcome for a
roll of the dice is the product of the individual outcomes for each die. Thus, the probability of a {4,3} outcome is given as
Pð4; 3Þ 5 Pð4Þ 3 Pð3Þ 5 1=6 3 1=6 5 1=36
Here we are making the distinction of which die shows
4 and which shows 3. For example, if the dice were colored this would correspond to a red die with a 4 and a
white die with a 3. If they are not distinguishable, then
Often in scientific investigations or engineering it is necessary to obtain data to test whether or not some treatment had an effect, or whether or not the data fit some
equation whose derivation is based on some theory. The
derivation of the equation may have required some
assumptions that limit the conditions under which the
equation is valid. An excellent fit of the data to the predicted values based on the theory gives us confidence
that the theory is valid and the assumptions used
were met. There is no guarantee that this is so. The philosophy of science tells us that all we can do is disprove
hypotheses. If the data do not agree with the theory, one
of two things must be true: either there is something
“wrong” with the data or there is something wrong
with the theory. The data must be taken at face value but
perhaps the data is not what you think it is. For example,
suppose you want to investigate the relationship between
pressure and flow at steady-state and you make measurements of the flow and pressure in a system. You make
some measurements, but do not realize that the system
is not at steady-state. Thus the data cannot be analyzed
using an equation that assumes steady-state. The data
are not “wrong,” but you are wrong in thinking that the
data represent the steady-state condition. Failure of the
equation to match your data is not a problem with the
data or the validity of the data, it is a problem of trying
to fit data to conditions that do not pertain to how the
data was actually obtained. Other possible problems
exist. For example, perhaps you measured the pressure
with an incorrectly calibrated meter, so the pressures
that you report are not the true pressures. There is generally far more ways of making mistakes than there are
ways of making the measurements correctly.
TABLE 1.2.A2.1 Possible Outcomes from a Roll of Two Dice
Outcome of First Die
Outcome of Second Die
1
2
3
4
5
6
1
(1,1)
(2,1)
(3,1)
(4,1)
(5,1)
(6,1)
2
(1,2)
(2,2)
(3.2)
(4,2)
(5,2)
(6,2)
3
(1,3)
(2,3)
(3,3)
(4,3)
(5,3)
(6,3)
4
(1,4)
(2,4)
(3,4)
(4,4)
(5,4)
(6,4)
5
(1,5)
(2,5)
(3,5)
(4,5)
(5,5)
(6,5)
6
(1,6)
(2,6)
(3,6)
(4,6)
(5,6)
(6,6)
Phys ical Foundat ions of P hysiology I: P ressure-Dr iven Flow
TABLE 1.2.A2.2 Type I and Type II Errors
Inference
Null Hypothesis
True
False
Reject H0
Type I error
Correct inference
Accept H0
Correct Inference
Type II error
So one kind of hypothesis is this: the derived equation
is a good fit for the data. We can call this the “null
hypothesis,” H0: there is no difference between the data
and what is predicted from the equation. We suppose
that the data is not a perfect fit. There are differences
between the data and the predicted value for the data. If
the differences are small, we may think that the fit is
good and we are likely to accept the null hypothesis as
being true. If the differences are large, we begin to think
something is “wrong” with either the data or the equation, and we reject the null hypothesis as being false. It
is possible to make two distinct types of errors in this
testing of hypotheses. The Type I error is the incorrect
rejection of a true null hypothesis. The Type II error is
the failure to reject a false null hypothesis. These types
of errors are clarified in Table 1.2.A2.2.
Another way of describing these is that the Type I error
is a false positive: you conclude there is an effect when
there is none present. A Type II error is a false negative.
You fail to detect an effect when it is actually present.
The probability of making a Type I error is designated
α. The probability of making a Type II error is designated as β. α is generally accepted as the level of significance of a statistical test of H0. This is to say, we have to
agree on how different the data can be from what is predicted from the null hypothesis before we can reject the
null hypothesis. There is some probability that the differences that you obtained arose by random chance, and
this is the probability of making a Type I error.
In other types of experiments we may want to know if
some treatment or variable has some effect in a population of individuals. Suppose we do an experiment in
which we measure some variable X for a set of individuals who are not treated, the control group, and we
measure the same variable for a set of individuals
who receive some treatment. Did the treatment affect
the value of X? What we generally have is a set of
measurements of variable X from the control group
G0 5 {X01, . . ., X0n} and a second set of measurements
from the experimental group Ge 5 {XE1, . . ., XEn}. The
mean for G0 is designated μ0 and the mean for the
experimental group is μE. The null hypothesis is H0:
μ0 5 μE. Generally the means will not be exactly equal.
There is some probability that a given difference μ0 2 μE
will arise by chance. The t-statistic for comparison of
two means is given as
μ0 2 μE
ffi
½1:2:A2:13
t 5 qffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
SEM20 1 SEM2E
Clearly, the larger the value of t the more likely that the
difference in the means does not arise by chance. If the
difference arises by chance, then we would be wrong to
reject the null hypothesis. The probability that the difference arises by chance, and we reject the null hypothesis, is the probability of the Type I error, and this is
called the level of significance of the test. Typically it is
accepted that α 5 0.05, which means there is a 5%
chance that we will reject the null hypothesis even
though it is true. Values of t for given values of α are
published, and these depend on the degrees of freedom
for the t-statistic. The degrees of freedom are the number of independent pieces of information that contribute to the estimation of the statistic, which is the t-value
in this case. In a two-sample comparison such as the
one described here, the degrees of freedom (symbolized
as df or ν) is given as n0 1 nE 2 2. We subtract 2 because
the means are derived from the data and so the means
and the values themselves are not independent, and we
have used 2 means in the calculation of the t-statistic.
We could apply these or other types of statistical test to
groups of people to compare them and to make statistical
statements about them, such as “men are more prone
to heart attacks than women”, or “black american
males have a higher incidence of hypertension than white
american males,” or “obese white females with multiple
children are most prone to gallstones.” These statistical
statements mean nothing when you are faced with a single incidence of the population: population averages and
trends tells you nothing about any specific member of the
group. You don’t know, without making some measurements, how far away from average this specific person
will be. However, we can make general statements about
the populations that may guide us in understanding or
treating a condition or setting public health policy.
THE NORMAL PROBABILITY DENSITY
FUNCTION
A probability density function is one in which the
probability of an outcome being in some interval is
the product of the density function and the interval.
Mathematically, it is given as
ðb
½1:2:A2:14
Pða # x # bÞ 5 pðxÞdx
a
where P(a # x # b) is the probability of x being between
a and b and p(x) is the probability density function.
There are several kinds of probability distribution functions. We will derive the normal or gaussian probability
density distribution by considering a random throw of
a dart aimed at the origin of a Cartesian plane. We
make some basic assumptions of the probability of the
dart landing in a particular area. These are:
G
the distribution is symmetrical: the probability of
being high by some distance is the same as being
low by the same distance, and this is equal to the
probability of being right by the same distance, or
by being left by the same distance. In fact, the probability of being off-center by some distance, r, is independent of the angle θ from the origin.
27
28
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
This differential equation is true for all x and y, and x and
y are independent. This can happen only if the ratio
defined on each side of the equation is constant. We let
y
dpðxÞ
dx 5 C
pðxÞx
Δy
r
θ
Δx
dpðxÞ
5 C x dx
pðxÞ
ð
ð
dpðxÞ
5 C x dx
px
x
½1:2:A2:20
FIGURE 1.2.A2.1 Cartesian coordinate and polar coordinate systems
for the analysis of the normal probability distribution function. The
probability of a dart hitting the gray area, dxdy, is the probability of
it landing in the vertical stripe of width Δx times the probability of it
landing in the horizontal stripe with width Δy.
G
G
G
errors in particular directions are independent. That
is, the probability of being off-center by x in the horizontal direction does not alter the probability of
being off-center by y in the vertical direction.
large errors are less likely than small errors.
our aim is unbiased. That is, on average, the distance
from the origin, taking x and y as positive and negative, is zero.
Consider the Cartesian coordinate system shown in
Fig. 1.2.A2.1. The probability of the dart landing in the
vertical stripe between x and x 1 Δx is given by p(x)Δx,
where p(x) is the probability density function. Similarly,
the probability of the dart landing in the horizontal stripe
between y and y 1 Δy is p(y)Δy. What we want to do is
determine the mathematical form of the function p.
From the independence assumption, the probability of the
dart landing in the shaded region is the product of the probabilties of landing in the horizontal or vertical stripes: p
(x)Δxp(y)Δy. Because of symmetry, the probability of the
dart landing in the area the size of ΔxΔy located r distance
from the origin does not depend on θ and we can write
½1:2:A2:15
gðrÞΔxΔy 5 pðxÞΔx pðyÞΔy
which gives
gðrÞ 5 pðxÞ pðyÞ
Differentiating with respect to θ, we obtain
dgðrÞ
dpðyÞ
dpðxÞ
5 0 5 pðxÞ
1 pðyÞ
dθ
dθ
dθ
dg(r)/dθ 5 0 because g(r) is independent of θ. Inserting
x 5 r cosθ and y 5 r sinθ, we can re-write Eqn [1.2.A2.17] as
0 5 pðxÞ
½1:2:A2:18
½1:2:A2:19
dpðyÞ dr sinθ
dpðxÞ dr cosθ
1 pðyÞ
dy
dθ
dx
dθ
dpðyÞ
dpx
r cosθ 2 pðyÞ
r sinθ
0 5 pðxÞ
dy
dx
dpðxÞ dpðyÞ
dx 5 dy
pðyÞy
pðxÞx
x2
1c
2
Taking the exponent of e of both sides of the last equation gives
k 2
pðxÞ 5 Ae2 x
½1:2:A2:21
Because large x is less likely than small x, we
know that C must be negative and we can re-write this
equation as
k 2
pðxÞ 5 Ae2 2 x
½1:2:A2:22
We can evaluate A by the requirement that the total
probability for all outcomes is 1.0. This is expressed
mathematically as
ðN
½1:2:A2:23
k 2
2N
Ae2 2 x dx 5 1:0
Since A is a constant, it can be removed from the integrand. Due to the symmetry of the problem, this integral
is twice the integral from zero to infinity. We write this as
ðN
½1:2:A2:24
k 2
e2 2 x dx 5
0
½1:2:A2:16
½1:2:A2:17
ln pðxÞ 5 C
1
2A
We can combine this with the distribution about y, as
these are symmetrical:
½1:2:A2:25
ðN
k 2
e2 2 x dxU
0
ðN
k 2
e2 2 y dy 5
0
1
4A2
Since x and y are independent, we can re-write this
product of integrals as the integral of their products:
½1:2:A2:26
ðN ðN
0
0
k 2
2
1
e2 2 ðx 1y Þ dx dy 5
4A2
This can be converted to polar coordinates, recognizing
that (x2 1 y2) 5 r2 and dxdy 5 rdrdθ. We obtain
½1:2:A2:27
ðπ ðN
2
0 0
k 2
e2 2 r r dr dθ 5
1
4A2
Phys ical Foundat ions of P hysiology I: P ressure-Dr iven Flow
Evaluation of the interior integral is 21. Eqn [1.2.
A2.28] thus becomes
ðπ
2 1
π
1
dθ 5
5
½1:2:A2:29
2k 4A2
0k
The value of A is thus given as
rffiffiffiffiffiffi
k
A5
½1:2:A2:30
2π
The probability distribution in Eqn [1.2.A2.22] becomes
sffiffiffiffiffiffi 2 k x2
2
k
½1:2:A2:31
pðxÞ 5
e
2π
We can evaluate k from the variance of the probability
distribution. The average value for x is given as
ðN
μ5
½1:2:A2:32
x pðxÞ dx
2N
The variance, similar to our earlier definition in Eqn
[1.2.A2.2] is given as
ðN
½1:2:A2:33
ðx2μÞ2 pðxÞ dx
σ2 5
2N
Because of the symmetry of the coordinates, and the fact
that p(x) 5 p(2x), we know that the mean, μ, is zero.
With Eqn [1.2.A2.31], and μ 5 0, the variance is given as
k 2
sffiffiffiffiffiffi
k ÐN 2 22x
2
½1:2:A2:34
σ 52
x e
dx
2π 0
We use symmetry as we did before to integrate from 0
to N:
sffiffiffiffiffiffi
k 2
k ÐN 2 22x
2
½1:2:A2:35
x e
dx
σ 52
2π 0
We can integrate this by parts by identifying
½1:2:A2:36
k 2
22x
u 5 x dv 5 x e
k 2
1 22x
dx v 5 2 e
k
Substituting these into Eqn [1.2.A2.35] we obtain
sffiffiffiffiffiffi ð N
k 2
22x
k
2
x2 e
dx
σ 52
2π 0
sffiffiffiffiffiffi ð N
k
2
σ 52
udv
2π 0
sffiffiffiffiffiffi N ð N
k
2
uv 2
vdu
σ 52
2π 0
0
3
sffiffiffiffiffiffi2
k 2
ð N 2 k x2
2 2 x N
2
k
x
1
1
42 e
σ2 5 2
e
dx5
2π
k
0 k
0
½1:2:A2:37
0.05
μ=0
0.04
σ = 10
μ = 50
0.03
P(x)
We can evaluate the integrals by making the
substitution u 5 2kr2/2. It follows that du 5 2krdr, and
thus 2du/k 5 rdr. Making these substitutions in the
interior integral, we derive
ð 2N
ðπ
2 21
1
½1:2:A2:28
eu du dθ 5
4A2
0 k
0
σ = 15
0.02
σ = 25
0.01
σ = 50
0.00
–150
–100
–50
0
50
100
150
X
FIGURE 1.2.A2.2 Gaussian probability distribution functions for varying
values of σ and μ. Different values of μ move the distribution to the
right or to the left; different values of σ influence the shape of the
curve, with smaller values resulting in sharper curves and larger values
creating more spread out curves.
The first term in brackets is evaluated from x 5 0 to
x 5 M in the limit as M-N, and it is zero. The second
term in the brackets has already been done in this
derivation (Eqn [1.2.A2.24] and Eqn [1.2.A2.30]). This
last equation in Eqn [1.2.A2.37] becomes
rffiffiffiffiffiffi
pffiffiffiffiffiffi
k
1 2π
1
2
pffiffiffi 5
½1:2:A2:38
σ 52
01
2π
k2 k
k
2
This last equation gives k 5 1/σ . Inserting this into the
probability density function (Eqn. [1.2.A2.31]) gives
1
2
pðxÞ 5 2 pffiffiffiffiffiffi e22σ
½1:2:A2:39
σ 2π
This is the normal probability distribution centered at
μ 5 0. The general equation with mean μ is achieved by
a horizontal shift in x:
½1:2:A2:40
2
1
pðxÞ 5 2 pffiffiffiffiffiffi e
σ 2π
ðx2μÞ2
2 σ2
Calculated Gaussian or normal probability distribution
functions are shown in Figure 1.2.A2.2. Note that the
value of σ determines the spread of the distribution.
Smaller values of σ result in a sharper distribution.
LINEAR REGRESSION
For linear regression, the data we’re interested in is
generally numerical and comes in sets of ordered pairs:
{(x1,y1), . . ., (xn,yn)}. What we desire to know is what is
the best linear fit to the ordered pairs? There are many different ways of doing this. What we are going to go over is
the least squares linear regression. The first thing we are
going to do is to make a few assumptions. These are:
1. there is no error in the values of Xi. All of the
error is in the values of Yi
2. the values of Yi are distributed normally. That is,
they obey the normal probability distribution. This
is the same as the Gaussian probability distribution
and is commonly referred to as the “bell curve”.
This curve has been described in Eqn [1.2.A2.40]
29
30
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
We introduce a model equation
Y^ 5 m X 5 b
½1:2:A2:41
where Ŷ is the predicted value of Y by this model equation. Since we have assumed that the values of X are
determined perfectly, with no error, all of the error is in
the set of Yi and how it differs from the predicted values,
Ŷi. What we want to do is minimize the squared error
between the set of Yi and Ŷi. The total squared error is
X
Error 5
½1:2:A2:42
ðYi 2 Y^ i Þ2
i
Substituting in from Eqn [1.2.A2.41] we have
X
Error 5
½1:2:A2:43
ðYi 2mXi 2bÞ2
i
The error estimated as the square of the deviations from
the observed values, the set of Yi, from the predicted
values, mXi 1 b, varies with m and b used in the calculations. We require this sum of square errors to be minimized in order to have the best fit line. We achieve this
by looking for minima in the square errors as we vary m
and b. That is, we look for the m and b that has the least
squared error. This occurs when the partial derivatives
of the error with respect to m and b are minima:
P
@ i ðYi 2mXi 2bÞ2
50
@m
½1:2:A2:44
P
@ i ðYi 2mXi 2bÞ2
50
@b
These two form a pair of simultaneous equations in m
and b; all of the pairs of (xi, yi) are known. Expanding
the square term, we get
P
@ i ðYi2 2 2mYi Xi 2 2Yi b 1 m2 Xi2 1 2mbXi 1 b2 Þ
50
@m
P
@ i ðYi2 2 2mYi Xi 2 2Yi b 1 m2 Xi2 1 2mbXi 1 b2 Þ
50
@b
canceling out the common factors of 2, this is recognized as a system of two simultaneous equations in two
unknowns:
X
X
X
Xi2 m 1
Xi b 5
Yi Xi
½1:2:A2:47
i
i
X
Xi m 1 n b 5
i
i
X
Yi
i
This system of simultaneous equations in two
unknowns (m and b) can be solved by application of
Cramer’s Rule:
P P
i Xi Y i
i Xi P
n i Yi
P
P
½1:2:A2:48
5m
i Xi2
i Xi P
X
n i
i
and the expression for b is
P 2 P
i Xi
i Xi Yi P
P
i Xi
i Yi
P 2 P 5b
½1:2:A2:49
i Xi
i Xi P
n i Xi
Evaluating the determinants in the numerator and
denominator for m in Eqn [1.2.A2.48], we get the
computational formula for the slope of the least-squares
best fit line:
P
P P
n
Xi Y i 2
Xi
Yi
½1:2:A2:50
m 5 Pi 2 P i P i
n i Xi 2 i Xi i Xi
½1:2:A2:45
Evaluating the determinants from Eqn [1.2.A2.49]
we get
P P
P 2P
Xi
Yi 2
Xi
Xi Y i
b5 i P i2 P i P i
½1:2:A2:51
n i X i 2 i Xi i Xi
Performing the partial differentiation gives
X
X
X
2m
Xi2 1 2b
Xi 2 2
Y i Xi 5 0
By algebraic manipulation, it can be shown that this
last equation for the intercept of the least-squares line is
given also as
½1:2:A2:46
i
2m
X
i
i
Xi 1 2nb 2 2
X
i
i
Yi 5 0
½1:2:A2:52
b 5 Y 2 mX
Physical Foundations of Physiology
II: Electrical Force, Potential,
Capacitance, and Current
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
Write Coulomb’s law for electrostatic forces
Define electrical potential at x as the work done moving a
unit positive charge from infinity to x
Write three equivalent but different descriptions of a
conservative force
Define electric field as the electric force per unit charge
Describe the electric field as the negative gradient of the
potential
Recognize Gauss’s law
Write the formula for capacitance in terms of charge and
voltage
Write the formula for capacitance in terms of area, dielectric
constant, and plate separation
Describe how capacitance varies with area, dielectric, and
plate separation
Be able to calculate the capacitance of biological
membranes given k, δ, and physical dimensions
Be able to calculate electric field intensity and force on a
charged particle given V(x, y, z)
Write Kirchhoff’s Current Law and Kirchhoff’s Voltage Law
Be able to calculate the time constant for a simple RC circuit.
COULOMB’S LAW DESCRIBES
ELECTRICAL FORCES
Electric charge is a fundamental property of some subatomic particles. Electrons have negative charge and
protons have positive charge. These designations of positive and negative are arbitrary but rigidly accepted by
convention. Separated electrical charges in a vacuum
(see Figure 1.3.1) experience a force that is described by
Coulomb’s law:
F5
½1:3:1
F1 on 2 5
q1 q2
4πε0 r 2
q1 q2 r12
5 2F2 on 1
4πε0 r 3
The bold face symbols signify vector quantities. F is the
force; q1 and q2 are electrical point charges, in coulombs, that are separated by the distance r, in meter; ε0
is a constant, the electrical permittivity of space, which
has the value of 8.85 3 10212 C2 N21 m22. The lower
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00003-3
1.3
equation signifies that the direction of the force is along
the line between the two charges. The magnitude of the
force is proportional to the product of the charges and
inversely proportional to the square of their separation.
Its sign depends on the signs of the point charges. Two
positive charges, or two negative charges, result in a positive force which is repulsive and directed away from
their center as indicated in Figure 1.3.1. Two charges of
opposite sign experience a negative force which is attractive along a line connecting them.
If the intervening space is not vacuum, but some
medium, the equation is altered slightly by the inclusion of a dielectric constant, κ, whose value depends
on the medium:
½1:3:2
F1 on 2 5
q1 q2 r12
κ4πε0 r 3
The dielectric constant for the vacuum is 1.0. For all
other materials, κ . 1.0. The reduced force in the presence of a dielectric material is due to charges present
in the material that reorient themselves in the presence
of the external point charges and thereby screen the
charges from each other. Materials with asymmetric
charge distributions within their materials typically have
large dielectric constants.
THE ELECTRIC POTENTIAL IS THE WORK
PER UNIT CHARGE
Suppose there is a positive charge of magnitude qfixed fixed
in space at some location. We have another charge, a unit
positive charge, located infinitely far away so that the force
between the charges initially is effectively zero. If we bring
the unit positive charge qtest toward the fixed charge at a
constant velocity, then its kinetic energy does not change.
As we approach the fixed charge, the repulsive force
becomes larger and larger and we must apply an external
force to keep the qtest at constant velocity. Because our
applied external force, Fext, has moved through a distance,
we have performed work on the body, given as
ðf
½1:3:3
Worki.f 5 Fext Uds
i
This work is the amount of energy we have expended in
moving the positive qtest toward the positive qfixed.
Where did that energy go? If we release qtest, we find
that it moves away from qfixed and gains kinetic energy
which is exactly equal to the energy we used to move 31
32
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
qtest toward qfixed. We say that the energy we used to
move qtest was stored as potential energy, U. We define
the potential at any place A in space as being the work
done to bring a positive unit charge from infinite separation to point A:
ðA
WorkN.A
Fext Uds
½1:3:4
5
UA 5
qtest
N qtest
The unit of potential here is joules coulomb21 5 volts. The
usefulness of the potential is that the work can be determined easily by multiplying the potential times the charge.
Fext in this equation is the external force required to move
the positive test charge with no change in velocity. It is
exactly equal to and opposite in sign to Fint, the interacting
electrostatic force. This is, in turn, given by Coulomb’s law
(see Eqn [1.3.1]). Since it is directed along ds, we can write
ðA
ðA
Fint Uds
qqtest
52
dr
UA 5 2
2
q
4πε
test
0 r qtest
N
N
½1:3:5
1 q
UA 5
4πε0 rA
F2 on 1
F1 on 2
+
q1
r12
F2 on 1
+
q1
THE IDEA OF POTENTIAL IS LIMITED
TO CONSERVATIVE FORCES
THE CONSERVATION OF ENERGY THEOREM
STATES THAT ENERGY MAY BE CONVERTED
BUT NOT DESTROYED
The First Law of Thermodynamics is the conservation
of energy theorem. It states that in ordinary mechanical
events, the total energy is constant. It is written in differential form as
+
q2
F1 on 2
r12
From this definition of the potential, it should be clear
that the potential surrounding a positive charge is positive: it takes work to bring a positive charge toward it.
The potential surrounding a negative charge is negative,
as we can get energy out of bringing a positive charge
toward it. These lead us to an important conclusion: a
separation of charge produces an electric potential.
The potential defined in this way is a scalar quantity,
having magnitude but not a direction, whereas the electrical force is a vector. This comes about from integrating the dot product of Fint with ds where ds is the
distance increment that points along the pathway taken
from infinite separation to point A. The dot product
means that we add only those components of the
force that are directed on the line connecting the centers
of the charges. These conclusions are illustrated in
Figure 1.3.2.
–
q2
½1:3:6
FIGURE 1.3.1 Electrical forces between separated point charges. q
indicates charge. Like charges repel, so that the force of q1 on q2 is
directed away from q1 on a line connecting their centers. The force of q1
on q2 is exactly opposite to the force of q2 on q1. Unlike charges attract
with forces opposite but in line with the vector connecting their centers.
dE 5 dq 2 dw
where E is the total energy, q in this case is the heat
energy, and w is the work. This is another unfortunate
case where variables are used to denote completely different quantities. In thermodynamics, q symbolizes
The potential at A is the work required to bring a
unit positive charge from infinity to A; it is independent
of path
For a positive fixed charge,
the energy required to bring a
positive charge to A is positive:
the potential around a positive
charge is positive
A
+ +
+ + +
+ +
qfixed
FIGURE 1.3.2 Definition of the electrical potential.
The potential at a point A is defined as the work
required to bring a unit positive charge (qtest) from
infinite separation to point A. If there is a fixed
positive charge near A, it takes work to bring qtest
to A (we must apply a force to overcome the
repulsive force and we move that force through a
distance) and the potential is positive. If there is a
fixed negative charge near A, then qtest is attracted
to it and it takes energy (work) to slow qtest—the
work is negative because the applied force is
opposite to the direction of movement.
- - - - -
qfixed
Fapplied Felectrical
+
For a negative fixed charge,
the energy required to bring a
positive charge to A is negative:
the potential around a negative
charge is negative
A
qtest
Felectrical Fapplied
+
qtest
Physical Foundations of Physiology II: Electrical Force, Potential, Capacitance, and Current
heat, and in electrostatics it symbolizes charge. The
appearance of heat in this equation is extremely important, because it turns out that there are theoretical limits
in the conversion of heat energy to useful work, and
this gives rise to the concepts of entropy and free
energy.
This equation assumes that heat and work are alternate
forms of energy. We take the equivalence of mechanical,
thermal, chemical, and electrical energies for granted but
historically this idea took some time to develop. In writing that any energy change in the system is the balance
between work output and heat input, it is assumed that
work is equivalent to heat. The equality of mechanical
work and heat was established in 1845 by Joule.
The signs of dq and dw in this equation are important,
and are consequences of the definitions of heat and
work. The quantity dq is defined as the heat absorbed by
the system from its surroundings, and dw is defined as
the work done by the system on its surroundings (see
Figure 1.3.3). The “system” here is anything we have
drawn a conceptual line around, usually in agreement
with some physical boundary, that sets part of the universe off from the rest of it. In the case of electrostatics,
the system is the set of charges distributed in space.
THE WORK DONE BY A CONSERVATIVE
FORCE IS PATH INDEPENDENT
The work done by moving qtest toward qfixed is the work
done by the surroundings (us) on the system of interacting charged particles. It is equal but opposite in sign
to the work done by the interacting force. There may be
heat generated by the necessity to apply more force
than the interacting force in order to overcome friction,
if the charges move through some medium, but this is
separate from the interacting force (the coulombic or
electrostatic force) itself. The coulombic force itself generates no heat at all. It belongs to a class of forces called
conservative forces that do not dissipate energy as heat.
Conservative forces are characterized by three equivalent
statements:
1. The work done by a conservative force depends
only on the initial and final positions, and not
on the path (see Figure 1.3.4).
2. The potential difference between two points
depends only on the end points and not the
path.
3. The total work done by a conservative force
acting around a closed loop is zero.
The positive increment in heat, dq,
is defined as the heat taken up by
the system from its surroundings
Heat energy
Work energy
dq +
dw
+
System
dE = dq – dw
Surroundings The positive increment in work, dw,
is defined as the work done by
the system on its surroundings
FIGURE 1.3.3 Theorem of the Conservation of Energy.
The system is any part of the universe that we have
enclosed by some boundary, real or imagined. Positive
heat flow is defined as heat energy that is absorbed by
the system from its surroundings. Similarly, positive
work is defined as work that is done by the system on
its surroundings. By these definitions, conservation of
energy means that dE 5 dq 2 dw. In order to write this
equation, it is assumed that heat and work have the
same units, that of energy. Here work can be electrical,
mechanical, or chemical.
Components of path normal to r
require no force and entail no work
+
q2
+
q1
Components of the path along r
contribute F dr to the work
A
... so the work required to get to point
A depends only on the radial separation,
and not the path
FIGURE 1.3.4 Potential is independent of the path.
Any path from start to finish can be successively
approximated by a series of paths oriented either
parallel to the vector connecting the charges
or perpendicular to it. Those components of the
path perpendicular to the vector require no force
and therefore contribute nothing to the potential at
point A, which is defined as the work necessary to
bring a unit positive charge (here shown as q2) from
infinite separation to point A. Components of the
path oriented parallel to the vector connecting
the point charges contribute F dr to the force.
Therefore, the total work (potential) moving the
charge depends only on the radial separation and
not the path taken.
33
34
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
POTENTIAL DIFFERENCE DEPENDS
ONLY ON THE INITIAL AND FINAL
STATES
If the potential depends only on the position, then
it is a state function, one that is independent of
path and dependent only on the state of the system.
Thus we can associate a potential with any position,
A and B:
ðA
Fint Uds
UA 5 2
N qtest
ðB
Fint Uds
UB 5 2
½1:3:7
N qtest
ðB
Fint Uds
UA.B 5 2
5 UB 2 U A
A qtest
If A 5 B, where A is the initial state and B is the final
state, we can write
I final
Uinitial.final 5 0 5 2
½1:3:8
Fint Uds
initial
This is the mathematical statement that the work performed by the system around any closed loop is zero.
This turns out to be equivalent to the statement that the
potential is a function of position (state) only and not
of the path used to get to that position.
THE ELECTRIC FIELD IS THE NEGATIVE
GRADIENT OF THE POTENTIAL
The electric field intensity is defined as the electric force
per unit charge:
E5
½1:3:9
Fint
qtest
Insertion of this into Eqn [1.3.7] and differentiating, we
get
½1:3:10
E52
dU
dS
This equation is not correct as written yet, because we
have a vector (the electric field intensity) on one side
and a scalar on the other! We need to take a particular
kind of derivative, the gradient, to convert the scalar
potential into a vector force. Equation [1.3.10] is correct
as written as long as the axis of ds corresponds with
the direction of F. The full three-dimensional vector
equation is
@U
@U
@U
2j
2k
@x
@y
@z
E 5 2 rU
where i, j, and k are unit vectors in the x, y, and z directions. The expression on the right-hand side of
Eqn [1.3.11] is called the gradient of the function U.
It is a vector whose components on each axis are
½1:3:11
E52i
the slope of the potential projected onto that axis.
Generally, the gradient is a vector that does not align
with any axis. Instead, it points in the direction of the
steepest slope of the potential surface in three dimensions. The force points down this slope. It is the negative of the gradient of the potential. The last equation
shows the gradient written in operator notation. The
operator r is called del and is defined as
½1:3:12
r5i
@
@
@
1j 1k
@x
@y
@z
FORCE AND ENERGY ARE SIMPLE
CONSEQUENCES OF POTENTIAL
The usefulness of potential is that it simplifies the
idea of force and energy. The force on a charge q is
given easily by multiplying it times the electric field,
which is 2 grad U. The energy cost in moving a charge
from one potential to another is just qΔU:
½1:3:13
F 5 2 qrU
Δ Energy 5 qΔU
where r is the del operator and Δ signifies the difference between final and initial states. The electric potential, U, here is in units of volts. If charge is in coulombs,
the force is in units of coulomb-volt per meter; a voltcoulomb is a joule 5 1 N m 5 1 kg m2 s22; therefore, the
force is in units of 1 N m/m 5 N. The energy is in units
of joules. From now on we will abandon use of U as a
symbol for the potential; physiologists typically use V,
E, or ψ as symbols of potential.
GAUSS’S LAW IS A CONSEQUENCE OF
COULOMB’S LAW
Gauss’s law is written as
I
q
EUds 5
½1:3:14
ε0
where the integral is taken over any closed surface of
the dot product of the electric field and the area vector,
equal to the area increment ds and oriented perpendicular to the surface. What this says is that this dot product, summed over any closed surface, is equal to
the charge enclosed by the surface divided by ε0, the
electrical permittivity of space. If there is no enclosed
charge, the surface integral is zero. This equation is
a variant of Coulomb’s law (see Eqn [1.3.1]). To see
how this equation works, we consider a spherically
symmetrical distribution of positive charges as shown
in Figure 1.3.5.
The evaluation of the surface integral is simplified
by choosing an appropriate surface. In this case, we
choose a sphere centered on the symmetrical charge.
By symmetry, the electric field is directed radially
outward, pointing along the vector ds. Similarly, the
electric field is everywhere constant in magnitude at a
Physical Foundations of Physiology II: Electrical Force, Potential, Capacitance, and Current
Area = A
Electric field lines
are radially directed
and symmetrical
E
E = kq
r2
ds
Gaussian surface
+ + +
+ + ++
+ + +
δ
r
E
FIGURE 1.3.6 A parallel plate capacitor. Two plates, each of area A,
are separated by a distance δ. They are charged by connecting them
to a battery that produces a capacitance current until the potential
difference between the two plates is equal to that across the two
poles of the battery, so that the net potential difference across the
entire circuit loop is zero. At this point, there is no more current flow.
The separation of charges produces a uniform electric field between
the two plates.
FIGURE 1.3.5 Electric field surrounding a spherically symmetrical
distribution of positive charge. ds is a vector having a magnitude of the
area increment and directed normal to the closed surface. In this case,
we take the Gaussian surface, indicated here by a dashed line, to be a
sphere centered on the symmetrically distributed charge. The electric
field vector and the surface normal vector are pointing in the same
direction, so that the angle between them, θ, is zero and the dot
product of E and ds is E ds, because cos θ 5 1.
In the plate, E = 0
Area = A
δ
prescribed distance, r, from the center of the charged
body. Thus we can write
I
E
q
EUds 5 E4πr 5
ε0
q
E5
4πr 2 ε0
2
½1:3:15
which is the magnitude of the electric field (E 5 F/q,
the electric force per charge) from Coulomb’s law (see
Eqn [1.3.1]).
THE CAPACITANCE OF A PARALLEL
PLATE CAPACITOR DEPENDS ON ITS
AREA AND PLATE SEPARATION
As described in Chapter 1.2, the ability to store electric
charge is characterized by the capacitance, defined as
½1:3:16
C5
Q
V
where C is the capacitance, Q is the charge, and V is the
potential, in volts. We now consider a particular type of
device to store charge, a parallel plate capacitor, as
described in Figure 1.3.6.
The two charged plates will be attracted to each other
and so must be held apart by some dielectric material
that insulates the plates and keeps the charges separated. There will be some fringing of the electric field
FIGURE 1.3.7 Parallel plate capacitor with a Gaussian surface. The
Gaussian surface is the box indicated by the dashed lines. The electric
field is constant within the capacitor and oriented as shown. The
integral of E ds in the plate is zero because E is zero there. The integral
of E ds in the dielectric between the plates is EA. The integral of E ds
on the sides of the enclosed surface is zero because E and ds are
orthogonal in this region.
around the edges of the plate, which we shall ignore.
The resulting electric field within the capacitor is uniform, which can be proved by integrating the
Coulomb force over the uniformly distributed charge
on a plane, which we will not do here. We draw a
rectangular closed surface, one side of which is in the
middle of the dielectric and the other in the middle of
the plate. Since the plate is a good conductor, the electric field within the plate is zero—the voltage difference in the plate is zero. The closed surface integral is
just the constant electric field times the area of the
surface in the dielectric. The situation is illustrated in
Figure 1.3.7.
Application of Gauss’s law according to the description
in the legend of Figure 1.3.5 gives
I
q
EUds 5 EA 5
ε0
½1:3:17
q
E5
ε0 A
35
36
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Since the electric field is uniform, its relation to V from
Eqn [1.3.10] is given as
½1:3:18
E52
dV
V1 2 V2
5
dx
δ
The capacitance is calculated as
½1:3:19
C5
q
V1 2 V2
Substituting in for q from Eqn [1.3.17] and for V1 2 V2
from Eqn [1.3.18], we have
½1:3:20
C5
ε0 A
δ
The presence of a dielectric between the plates reduces
E for a given charge, and therefore increases the
capacitance. The formula for parallel plates with a
dielectric is
½1:3:21
κε0 A
C5
δ
where κ is the dielectric constant, a dimensionless
ratio.
According to Eqn [1.3.21], the capacitance increases linearly with the area and inversely with the separation
between the plates, and is increased by materials with
high dielectric constants.
BIOLOGICAL MEMBRANES ARE
ELECTRICAL CAPACITORS
Biological membranes share some of the features of
parallel plate capacitors and act as electrical capacitors.
Their structure is detailed in Chapter 2.4. Briefly, biological membranes consist of an asymmetric bilayer of
lipid molecules that assemble to form an interior insulating core. This effectively separates two plates—the
surfaces of each bilayer—from each other. The separation distance is typically quite small, on the order of
7 nm. This bilayer structure is shown in Figure 1.3.8.
From it, you can see the resemblance of the bilayer to a
parallel plate capacitor.
The dielectric constant of some materials is shown
in Table 1.3.1. This constant varies with temperature and
the chemical make-up of the dielectric. Materials that
are polar and mobile, such as water, can orient their
partial charges with the electric field, and reduce the
field within the dielectric. In this way, more charge can
be added to the surfaces of the plates and therefore
these dielectrics have a high dielectric constant.
TABLE 1.3.1 Dielectric Constant of Some Materials
Material
Dielectric Constant, κ
Air
1.00059
Water
80
Glycerol
43
Acetic acid
6.2
Benzene
2.3
CCl4
2.2
Oleic acid
2.46
Hexanol
13.3
Hexane
1.89
Stearic acid
2.29
Monopalmitin
5.34
Aqueous layer
Insulating hydrocarbon layer
FIGURE 1.3.8 Lipid bilayer membrane consisting of
various lipid molecules arranged with their
hydrocarbon tails toward the interior of the bilayer
and their water-soluble parts facing the water phase.
Aqueous layer
Physical Foundations of Physiology II: Electrical Force, Potential, Capacitance, and Current
EXAMPLE 1.3.1 Capacitance of Planar Lipid Bilayers
Dr. Alexandre Fabiato at VCU drilled a narrow and clean hole
with a diameter of 250 μm 5 0.25 mm into a Lexan partition that
separated two electrolyte solutions. He “painted” some phospholipids over the hole using a Teflon stick cut at an angle and
dipped into a solution of lipids dissolved in hexane. After “thinning” (passive removal of the hexane through the aqueous
phase), the membranes form a planar lipid bilayer (see
Figure 1.3.9). Dr. Fabiato measured the capacitance of the membrane using an AC signal connected to electrodes immersed in
the solutions. He derived a capacitance of 350 pF
(350 3 10212 F). Calculate the specific capacitance of the membrane Dr. Fabiato made, in F cm22.
The specific capacitance is just the capacitance per unit
area of membrane: Cm 5 C/A. The measured capacitance is
350 3 10212 F and the area A is πr2, where r 5 0.0125 cm;
therefore,
Cm 5 340 3 10212 F=4:9 3 1024 cm2 5 0:71 µF cm22
Barrier
What Is the Approximate Thickness of the Bilayer?
Equation [1.3.19] allows us to calculate the thickness as
δ 5 κε0/Cm, where Cm is the specific capacitance. We do not
know κ, but the dielectric constant for lipid-like substances has
been determined, as examples shown in Table 1.3.1. Here we use
ε0 5 8.85 3 10212 C2 J21 m21 and Cm 5 0.71 3 1026 C V21 cm22
that we calculated earlier. Using the dielectric constant for n-hexane as an example, we calculate
δ 5 1:89 3 8:85 3 10212 C2 J21 m21 =0:71
26
3 10
CV21 cm22 3 ð100 cm m21 Þ2 5 2:36 3 1029 m
The calculated values of δ (Table 1.3.2) are of the same order as
expected from electron micrographs of membranes.
If the potential across a membrane is 80 mV, and its thickness is
7 nm, What is the electric field intensity?
The field is uniform inside a capacitor, and so is given by
E 5 2ΔV/Δx, where ΔV is the potential difference and Δx is the
separation of the plates. Thus the electric field is
E 5 2 80 3 1023 V=7 3 1029 m 5 2 11:4 3 106 V m21
Water
Water
TABLE 1.3.2 Calculated δ for Various κ
Narrow
aperture
κ
δ (nm)
1.89
2.36
2.29
2.85
2.46
3.07
5.34
6.67
FIGURE 1.3.9 Planar lipid bilayer formed in a narrow hole between
two aqueous compartments.
ELECTRIC CHARGES MOVE IN
RESPONSE TO ELECTRIC FORCES
As mentioned in the section “Force and Energy Are
Simple Consequences of Potential,” the usefulness of
the concept of potential lies in the ease of calculating
the force on a charged particle or the energy needed to
move from one region to another. The electrical force
on a charged particle is given as
½1:3:22
F 52qrU 5 qE 5 zeE
where U is the potential, often written as V, q is the
charge, E is the electric field, z is the valence (1/ 2 integral number of charges per particle), and e is the unit
charge of the electron. Thus, a charged particle, of either
sign, in an electric field is subjected to an accelerating
force. Ions in solution are subjected to these forces and
accelerate on account of them. These ions accelerate
until they reach a terminal velocity, v, at which point
the electrical force is matched by a drag force on the
particle by the surrounding solution. Figure 1.3.10 illustrates this situation.
MOVEMENT OF IONS IN RESPONSE TO
ELECTRICAL FORCES MAKES A
CURRENT AND A SOLUTE FLUX
The drag force on a particle moving through a solution
is proportional to its velocity and directed opposite to
it. Further, the electrical force given in Eqn [1.3.22] at
37
38
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
E
Fe
Fd
+
V
½1:2:3
q
FIGURE 1.3.10 Forces on a charged particle in solution subjected to a
constant electric field. The electrical force, Fe, is the product of the
charge, q, on the particle and the electric field, E. This electrical force
accelerates the charged particle and it moves through the solution. This
movement produces a drag force, Fd, which is proportional to the
velocity, v. The particle reaches a terminal velocity when the net force
on the particle is zero: Fe 1 Fd 5 0.
the terminal velocity is equal but opposite to Fd.
Therefore, we can write
Fd 5 2βv
Fe 5 2Fd
Fe 5 βv
½1:3:23
where β is a drag coefficient or frictional coefficient.
Thus ions subjected to a constant electrical field will
move at a constant terminal velocity, v, and that velocity
will be proportional to the electric field. This movement
of charged particles constitutes a movement of charge
from place to place, and so it is an electrical current.
Further, because solute particles carry the charge, the
movement also forms a solute flow. The solute flux is
related to the velocity by
½1:3:24
Js 5 vC
where J and v are written as vectors and C is the concentration of the solute (see Figure 1.3.11). Because each solute
particle carries the charge ze, the current density is
i 5 zeJs
½1:3:25
This expression can be converted into Ohm’s law by
using Eqns [1.3.22][1.3.24]:
½1:3:26
i 5 zeJs 5 zevC
Fe
i 5 zeC
β
i5
The last equation is an analogue of Eqn [1.2.3] for the
one-dimensional form of Ohm’s law:
z2 e2 C
ð2rVÞ
β
Je 5 2σ
EXAMPLE 1.3.2 Forces on Charged Particles
Consider the planar lipid bilayer in Example 1.3.1, which has a
potential difference of 80 mV across it. What would the electric
force be on a Na1 ion in the middle of the bilayer?
The electric force on a charged particle is given as Fe 5 qE.
We calculated the electric field intensity, E, in this case to
be 211.4 3 106 V m21. The charge on any ion is ze, where z is
the valence or integral number of charges on the particle and e
is the charge on the electron. In this case, z 5 11 and the
charge on the electron is given in various units.
The most useful unit here is the coulomb: 1e 5 1.6 3 10219 C.
The force is thus given as
Fe 5 1:6 3 10219 C 3 2 11:4 3 106 V m21
5 2 1:82 3 10212 V C m21
5 2 1:82 3 10212 J m21
5 2 1:82 3 10212 N m m21
5 2 1:82 3 10212 N
THE RELATIONSHIP BETWEEN J AND C
DEFINES AN AVERAGE VELOCITY
Consider the right cylindrical tube shown in
Figure 1.3.11 that contains solute particles moving to
the right at average velocity v. In time Δt, the particles
travel a horizontal distance Δx 5 vΔt. All of the solute
particles in the volume AΔx will have crossed a crosssectional plane in the cylinder. Thus the flux will be
the total number of solute particles in that volume,
per unit area per unit time. The number of solute
In time Δt the solute particles move
Δx = vΔt; the flux is therefore C A Δx / AΔt
= CΔx/Δt = Cv
Cross-sectional
area, A
J(x)
FIGURE 1.3.11 Relationship among J, C, and v.
If solutes have an average velocity v, they
sweep out a distance vΔt in time Δt, and this
corresponds to an entire volume of solute,
equal to AvΔt, moving to the right. The
number of solute particles in this volume is
CAvΔt. The flux is this number per unit area,
per unit time: J 5 CAvΔt/AΔt 5 Cv.
dψ
dx
v
Δx = vΔt
x axis
Physical Foundations of Physiology II: Electrical Force, Potential, Capacitance, and Current
particles in the volume AΔx is CAΔx. Thus the flux is
given as
½1:3:27
CAΔx
AΔt
J 5 Cv
J5
From this equation, it is clear that the ratio of J/C
defines an average velocity for solute particles.
OHM’S LAW RELATES CURRENT
TO POTENTIAL
According to the discussion earlier, a difference in
potential produces a force on charged particles, and
the force is proportional to the negative gradient of
the potential. The movement of charges in response
to a potential makes a current, a flow of charges. It is
given as
½1:3:28
I5
Δq
Δt
The current I is given in amperes 5 coulomb s21.
Current is defined as the movement of positive
charge, so that the movement of cations (positive ions)
constitutes a current in the direction of the flow; the
movement of anions (negative ions) makes a current in
the opposite direction to the flow. The current due to
an ion is related to its flow by
½1:3:29
Ix 5 z ℑ Qx
where Ix is the current of ion x, in coulombs s21,
Qx is the flow of ion x in mol s21, z is the integral
charge per ion (1/ 2 1, 2, ...) and ℑ is the Faraday
(9.649 3 104 coulombs mol21 5 6.02 3 1023 electrons mol21 3 1.6 3 10219 coulombs electron21); zℑ converts
mol to coulombs. Note that current is an extensive variable, while current density is an intensive variable.
The movement of charge through matter-filled space
encounters resistance from the matter. Those materials
that offer little resistance are called conductors. Other
materials, such as membrane lipids and the myelin
sheath that surrounds nerve axons, offer high resistance
and are called insulators. The current is greater if the
potential driving it is greater and is less according to
the resistance of the material through which the current
flows. This is Ohm’s Law:
½1:3:30
I5
Δψ
E
5
R
R
where I is the current, Δψ is the potential difference,
often symbolized as E or V, and R is the resistance.
Resistance has the units of ohms 5 volts/amps, symbolized as Ω. Ohm’s law can also be written as
½1:3:31
I5g Δ ψ
where g 5 1/R is the conductance. The SI unit for conductance is the siemen 5 amp/volt.
A battery is a device for using chemical reactions to
create a voltage difference. In effect, chemicals
trap electrons, with their negative charges, at fixed distances from their positively charged nuclei. Different
chemicals will then have different potentials for their
electrons. In chemistry these are called oxidation
potentials. These refer to the energy required to
remove the electron from the chemical. The movement
of electrons from one chemical to another can then
release energy equal to the difference in the oxidation
potentials times the number of charges that move. If
we hook up a battery in series with a resistance, we
can produce a current which is given by Ohm’s Law
(see Eqn [1.3.30]). This situation is shown schematically in Figure 1.3.12.
KIRCHHOFF’S CURRENT LAW AND
KIRCHHOFF’S VOLTAGE LAW
The circuit shown in Figure 1.3.12 is a simple circuit
in which a resistor is placed over the terminals of
a battery. When the circuit is completed, current
flows and this can be measured with an ammeter.
Kirchhoff’s Voltage Law states that the total voltage
differences around any loop must be zero. This is a
restatement of the conservative nature of the electric
force: the work done in any loop is zero. Since
the only resistance is between nodes 2 and 4, the
voltage differences around the loop are ΔV23 and
ΔV71, where ΔV71 is the voltage across the battery
from the negative electrode to the positive electrode.
Thus, ΔV71 5 2ΔV17 5 2E, the voltage provided by
the battery. The total voltage drop around the loop is
given as 0 5 ΔV23 1 ΔV71 5 ΔV23 2 E. Solving this for
ΔV23, we find ΔV23 5 E and therefore the current
through the resistor is I 5 E/R.
Kirchhoff’s Current Law states that the sum of current
into any node must be zero. Thus if there is a current
I23 5 E/R that enters node 3, there must be a current
I34 5 I23 that leaves node 3. The total current into node
3 is I23 2 I34 5 0. I34 is negative in this last equation
because it leaves node 3—it is the opposite direction
(with respect to node 3) of the current I23, but it is
equal in magnitude.
THE TIME CONSTANT CHARACTERIZES
THE CHARGING OF A CAPACITOR IN A
SIMPLE RC CIRCUIT
Suppose now that we include a capacitor in the circuit
shown in Figure 1.3.12. This expanded circuit is shown
in Figure 1.3.13. Initially, with the switch open, there
will be no potential across the capacitor, and no current
in the circuit. If we flip the switch, current will begin to
flow because there will be potential differences in the
circuit. But the capacitor is filled with a dielectric that
disallows current flow! How can current flow across the
39
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Kirchoff's Voltage Law:
ΣV
R
3
Ammeter
–
I=0
+
1
–
I = E/R
4
E
+
5
–
I
E
–
5
2
+
+
+
+
R
3
Ammeter
Ammeter
4
0
The sum of the voltage drops around
any loop is zero
R
1
=
loop
Closing the circuit produces
current I = E/R
When the circuit is open, there is
no current
–
40
E
–
I = E/R
I
I56
7
6
6
7
I67
Kirchoff's Current Law:
ΣI
node
=
0
The sum of the currents into a node is zero
FIGURE 1.3.12 Ohm’s Law, Kirchhoff’s Current Law, and Kirchhoff’s Voltage Law. When the circuit is completed by closing the switch between nodes 1
and 2, current flows as described by Ohm’s Law: I 5 ΔV23/R, where ΔV23 is the voltage difference across the resistor (between nodes 2 and 3) and R is
the resistance. In this circuit, the only resistance is between nodes 2 and 3 and all other resistances are negligible. Kirchhoff’s Current Law (KCL) states
that the sum of currents into a node is zero. At node 6, for example, the sum of the currents is I56I67, the negative sign indicating that the current
is out of the node. Thus, I56I67 5 0 or I56 5 I67. Kirchhoff’s Voltage Law (KVL) states that the sum of voltage drops around any closed loop is zero.
For the circuit shown, the voltage drops are from nodes 2 to 3, where ΔV23 5 I R 5 E; and from nodes 7 to 1 where ΔV71 5 2ΔV17 5 2E. Thus the
voltage drop around the loop is ΔV23 1 ΔV71 5 E 2 E 5 0. All of the other voltage drops between nodes (ΔV45, ΔV56, ΔV67, and ΔV12) are zero
because the resistances in this part of the circuit are negligible.
capacitor? The current in this case is a capacitive current, not a resistive current, like the kind shown in
Figure 1.3.12. The flow of positive ions onto the top
plate of the capacitor produces an electric field (a force
per unit charge) that repels positive charges from the
bottom plate. This movement of charges away from
the bottom plate is the capacitive current. As the charges
move away, there is a separation of charges on the
capacitor and it now has a potential difference given by
V 5 q/C. Charges continue to move until the potential
across the capacitor is equal but opposite to the potential across the battery, E.
To analyze the time course of current and voltage in the
circuit, we make use of Kirchhoff’s Voltage Law that says
the sum of the voltage drops in the circuit must be zero.
We write
ΔV61 1 ΔV23 1 ΔV45
½1:3:32
2E1
dq
q
R1
dt
C
5
0
5
0
where q is the charge on the capacitor and dq/dt is the current through the resistor, which is also the current across
the capacitor. We can separate variables in Eqn [1.3.32]
and integrate to solve this equation for q as a function of
t. The rate of charging of the capacitor is given as
t
q 5 EC 1 2 e2RC
½1:3:33
where t is the time and the combined terms RC is called
the time constant because it describes the time taken to
charge the capacitor. The current can be obtained from
differentiation of q to give
½1:3:34
I5
E 2t
e RC
R
The time course of charge and current is shown in
Figure 1.3.14. In Eqn [1.3.33], if t 5 RC, then q 5 EC
(1 2 1/e), so the time constant is the time required for
the charge to be 1/e 5 0.37 of its final value.
SUMMARY
Some particles in nature either repel or attract other
particles, and the force developed between them varies
inversely with the square of their separation. These
particles are said to be “charged,” and there is no more
basic description or explanation of their interaction
than Coulomb’s law that quantifies it. Charges have two
types: positive and negative. Like charges repel, unlike
charges attract.
This electrostatic force is a conservative force, meaning
that the work performed in moving a charge around
any closed loop is zero. Conservative forces also
mean that the work done in moving a particle around
depends only on the initial and final states, and
not the path. Equivalently, the potential energy associated with a distributed set of charges depends only
on the position and not on the path it takes to get
there.
Physical Foundations of Physiology II: Electrical Force, Potential, Capacitance, and Current
V=0
voltmeter
Area = A
-
R
3
+
2
1
4
+
E
*
The capacitance is defined as: C 5 Q/V, where Q is the
charge and V is the potential difference across the capacitor. The capacitance of a parallel plate capacitor
depends on several physical characteristics of the capacitor and is given as
E
5
V = V (t)
voltmeter
Area = A
-
+
+
6
R
3
4
2
1
C5
I
+
+
+
+
E
+
*
-
E
-
-
V=E
voltmeter
Area = A
-
+
6
R
3
2
1
4
+
+ + + + + + + ++
+ + + + + + + +
*
- - -
E
- - -
-
E
-
5
kε0 A
δ
where κ is the dielectric constant that depends on material between the plates, ε0 is a physical constant, A is
the area of the plates, and δ is the spatial separation of
the plates. Biological membranes form capacitors with
very small δ.
Ions move in response to electrical forces. This movement forms both a solute flux and an electrical
current. Movement in response to electrical forces
accelerates ions in solution until a terminal velocity
is reached. At this point, the net force on the ion is
zero and is the balance between the electrical
force and the drag or frictional force on the solute
particle by the solution. The average terminal velocity
is the flux divided by the concentration. Ohm’s Law
is given as I 5 E/R.
I
5
is measured in volts. The charge is measured in coulombs. Energy is measured in joules or volt-coulombs.
Because the potential is defined as the integral of the
work to move a unit positive charge from infinity to A,
the electric force per unit positive charge is the negative
derivative of the potential. The potential is a scalar
whereas the force is a vector. The derivative here is the
gradient, which converts the scalar potential into a
force vector. The electric force per unit positive charge is
the electric field.
6
FIGURE 1.3.13 Charging of a capacitor. The capacitor consists of two
parallel conducting plates separated by a dielectric, or insulating,
material. At the start, top, the circuit is broken by a switch and there is
no potential difference across the capacitor. When the switch is closed,
middle panel, charge begins to move, making a current. The positive
charges on the top plate repel positive charges on the bottom plate,
which move back to the battery, completing the circuit for the current.
This is a capacitive current, because there is no flow across the dielectric
but there is a flow in the circuit. The separation of charges across the
capacitor creates a potential difference related to the capacitance of the
capacitor: V 5 qC. This builds up as current continues to flow until V is
exactly opposite to E. At this point, current flow stops and the capacitor
is fully charged.
The electrical potential at a point, A, is defined as the
work necessary to bring a unit positive charge from infinite separation to that point. Therefore, positive fixed
charges are associated with positive potential and negative fixed charges are associated with negative potential.
Separation of charge produces a potential. The potential
Conservation of charge and the conservative nature of
the electric force give rise to Kirchhoff’s Current Law
and Kirchhoff’s Voltage Law. Kirchhoff’s Current Law
states that the sum of all currents into any node of a circuit must be zero. Kirchhoff’s Voltage Law states that
the potential differences around any closed loop must
sum to zero. When a capacitor is connected in series
with a resistor and a voltage source (a battery), the
capacitor gradually becomes charged until the potential
across it exactly opposes the potential of the battery.
The time course of charging depends on RC, the product of the resistance and the capacitance. When the
capacitor is fully charged, current in the circuit goes to
zero. The value of RC is the time constant for the circuit,
which is the time that the charge takes to get to within
1/e of its final value.
REVIEW QUESTIONS
1. What do we mean by “electric potential”?
2. What makes an electrical potential difference
between two points?
3. How much energy is gained by charge q over a
potential difference V?
4. What is meant by “electric field intensity”?
5. What is the relationship between charge and
voltage for a parallel plate capacitor?
41
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
FIGURE 1.3.14 Charging of a capacitor in
series with a resistor. Here the specific
capacitance was taken as 1 μf cm22 and the
specific resistance was taken as 1000 Ω cm2
and the time constant RC 5 1 ms. The charge
rises exponentially to reach a maximum
and the current simultaneously decreases
exponentially from an initial value to zero.
The time constant is the time required for q to
rise to with 1/e of its final value, which is the
same as the time required for I to decrease to
within 1/e of its final value or for I to decrease
to 1/e I0.
1.20e-4
1.2e-7
1.00e-4
1.0e-7
8.00e-5
8.0e-8
6.00e-5
4.00e-5
q (coulombs cm-2)
I (amps cm–2)
42
Charge rises exponential to a maximum when
the switch is closed; voltage on the capacitor
follows the charge
q ( e–1) = 0.37 q
At t = RC, q rises to within 1/e
of its final value
6.0e-8
q ( 1– e–1) = 0.63 q
4.0e-8
2.00e-5
2.0e-8
0.00
0.0
6. Why do biological membranes act as tiny
capacitors?
7. How does capacitance depend on membrane
thickness and surface area? What is the dielectric
constant?
8. What is the relationship between average solute
velocity and solute flux?
9. What is the frictional coefficient and how does
it relate to velocity and flux?
0.000
0.002
0.004
0.006
0.008
0.010
Time (s)
Current is initially maximum and falls exponentially
to zero as the capacitor charges
10. What is Kirchhoff’s Current Law? What is
Kirchhoff’s Voltage Law?
11. What is meant by “resistive current”?
“Capacitive current”?
12. Why does current stop in an RC circuit?
13. What is meant by the term “time constant”?
What is the time constant in a simple RC
circuit?
Problem Set
Physical Foundations: Pressure and Electrical
Forces and Flows
1. Identify whether the following variables are intensive or extensive and explain your reasoning.
A. Temperature
B. Heat content
C. Volume
D. Density
E. Mass
F. Concentration
G. Moles
H. Pressure
I. Area
J. Flow
K. Flux
L. Viscosity
2. Normal systolic blood pressure is about
120 mmHg
A. Convert this to atmospheres.
B. Convert this to pascals
3. Normal diastolic blood pressure is about
80 mmHg
A. Convert this to atmospheres
B. Convert this to pascals
4. The density of whole blood is typically
1.055 g cm23. The density of Hg is 13.6 g cm23.
A. Derive an equation to express the hydrostatic
pressure of a column of blood as the height
of Hg that would produce the same hydrostatic pressure as that column of blood
B. Use the equation to determine the pressure
of a 20-cm column of blood, expressed in
mmHg
C. If systolic blood pressure is 120 mmHg,
what is the height of blood that this could
support (if it was constant)?
5. For dialysis membranes, the Lp was determined
to be 6.34 3 1027 cm min21 mmHg21. A cylindrical hole 1 cm in diameter was cut into two
Lexan pieces that were then bolted together with
the membrane between. Fluid entered one side
through a pump and a pressure transducer was
affixed. The flow was adjusted until the pressure
(above atmospheric) was 20,000 pascals. The
pressure on the opposite side was atmospheric
(zero). Assume steady-state flow. What was the
flow through the membrane?
1.1
6. The viscosity of water at 25 C is about
0.00089 Pa s. The inner diameter of a PE160
polyethylene tubing is 1.14 mm. Assume steadystate laminar flow.
A. What pressure is necessary to get a flow of
5 mL min21 through a 20-cm length of this
tubing?
B. What is the velocity of the flow?
C. What pressure is necessary to get a flow
of 5 mL min21 through the same PE160
tubing if plasma is used, with viscosity of
0.002 Pa s?
D. What pressure would be needed for the
same flow of water through a 20-cm length
of PE60 tubing with i.d. 5 0.76 mm?
7. After the end of a normal inspiration, the volume of air in the lungs is about 2.8 L. Normally
quiet inspiration is driven by a pressure difference of about 2 mmHg. The air in the lungs is at
37 C and after normal expiration it is at atmospheric pressure. Quiet inspiration is driven by
the expansion of the chest cavity by contraction
of the diaphragm, which expands the air in the
lungs. How much is the air expanded to produce
an decrease of 2 mmHg in pressure? Use the
ideal gas equation, PV 5 nRT, where P is the pressure, V is the volume, n is the number of moles,
R 5 0.082 L atm mol21 K21 is the gas constant,
and T is the absolute temperature.
8. A burette is a vertical, right circular cylinder that
is open at the top and has a stopcock valve at
the bottom to let fluid out. Assume that you
have a burette that has an inner diameter of
1 cm and a height of 100 cm.
A. Assume that you fill the burette with water
to some height, h. What is the relation
between h and the pressure at the base of
the burette?
B. What is the relation between volume of
water in the burette and the pressure?
C. How does this relation map onto the
relation between charge and voltage on a
capacitor?
D. What is the hydraulic analogue of voltage?
E. What is the hydraulic analogue of charge?
43
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00004-5
44
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
9.
10.
11.
12.
13.
F. Suppose you open the stopcock fully. The
fluid will drain out. Assume a constant
diameter of the opening of the stopcock that
provides a constant resistance, R. Derive an
equation that describes the time course of
draining the burette.
G. Identify the time constant for the burette
emptying.
You are given a long vertical tube filled with
fluid of viscosity η at sea level where the acceleration due to gravity is g 5 9.81 m s22. You have
a steel ball or radius r and density ρsteel that you
carefully drop into the fluid. Assume that the
drag force on the ball obeys Stokes’ Law: the
drag coefficient is 6 πrη, where Fdrag 5 26 πrη v.
Remember that the steel ball is subjected to a
buoyant force equal to the volume of the ball
times the density of the fluid, ρfluid, times the
acceleration of gravity.
A. Derive an expression for the time of
approach of the steel ball to terminal
velocity.
B. Derive an expression for the viscosity of the
fluid as a function of the terminal velocity.
(This is how you can determine the viscosity
of a fluid.)
The Poiseuille equation that relates flow in narrow tubes to the pressure difference is analogous
to Ohm’s Law for current flow.
A. What is the resistance to flow in terms of the
parameters of the tube?
B. What happens to resistance if the radius of
the tube is halved?
C. What happens to resistance if the radius of
the tube is doubled?
Suppose that you begin to exercise and as a consequence you begin to produce more heat.
A. What do you suppose will happen to the
body temperature if, when environmental
temperature does not change, the rate of
heat production increases.
B. How is this change in body temperature
described by the continuity equation applied
to heat?
C. How do you suppose the body will shed this
excess heat production?
You are out camping and it is very cold
outside. You are losing heat faster than you can
produce it.
A. What happens to body temperature?
B. How is this change in body temperature
described by the continuity equation applied
to heat?
C. How do you suppose you can prevent
hypothermia?
Consider a capacitor with a capacitance of
10 μF. You connect it to a variable DC voltage
source with a switch in the circuit and a resistor
of 1000Ω in series with the capacitor.
A. What is the relation between the steady-state
voltage across the capacitor and the charge?
14.
15.
16.
17.
18.
B. Before you close the switch, there is no
charge on the capacitor. When you close the
switch, current begins to flow from the voltage source at E volts. What is the relation
between current and the voltage drop across
the resistor in terms of the current and the
resistance?
C. What is the voltage drop across the capacitor
in terms of charge and capacitance?
D. Kirchoff’s voltage law says that the voltage
drop around any loop must be zero. Write
the equation for the voltage drop across the
resistor, capacitor, and voltage source.
E. Solve the equation in part D to derive the
time course of charging of the capacitor.
F. Solve the equation in part C to derive the
time course of the current.
G. Identify the time constant for the charging
of the capacitor.
An unmyelinated axon can be considered to be
a long right circular cylinder. Consider that an
axon is 10 cm long with a diameter of 1.0 μ
(1 μ 5 1026 m).
A. If the specific capacitance of the membrane
is 1 μF cm22, what is the capacitance of the
axon membrane?
B. How much charge is separated by this membrane to give a potential of 70 mV?
A muscle cell approximates a right circular cylinder 10 cm long and 70 μ in diameter. The specific capacitance of the membrane (the
capacitance per unit area) is 1 μF cm22.
A. What is the capacitance of the muscle
membrane?
B. How much charge is separated by this membrane to give a potential of 285 mV?
A bubble is held at a radius of 250 μm. The
transmural pressure difference is 2 mm Hg.
What is the tension in the wall?
The thickness of a single membrane is about
7 nm with a specific capacitance Cm 5 1 μF cm22.
A myelin sheath consists of multiple membranes
produced by coils of Schwann cell or oligodendroglia cells. Suppose a myelin sheath results
from 100 membranes stacked on top of each
other.
A. What is the specific capacitance of the
myelin?
B. If the myelin sheath is a right circular cylinder 1 mm long with a radius of 3 m, what is
its total capacitance?
C. How much charge does it take to produce a
voltage of 270 mV across this capacitor?
D. If the myelin were just 1 membrane—i.e.,
not myelin—what would be its capacitance?
How much charge would it take to
produce a voltage of 270 mV across the single membrane?
The transmural pressure difference across a
small vein is 20 mmHg. The radius is 1 mm.
What is the wall tension?
Problem Set
19. The heat capacity of the human body is about
3500 joules kg21 C21. Suppose that a person
weighs 75 kg. How much energy does it take to
raise the body temperature from 98 C to 102 C?
20. The charge on the electron is 1.602 3 10219 coulombs. How much energy, in joules, is gained
when an electron is accelerated across a potential difference of 1 v in a vacuum? This amount
of energy is called the electron-volt.
21. Suppose that the average diameter of the aorta
is 1.1 cm. The flow through the aorta is nearly
the entirety of the cardiac output.
A. If the cardiac output is 5 L/min, what is the
average flow through the aorta?
B. Suppose further that the cardiovascular
system is nearly closed to fluid transfer.
That is, that on a short-term basis the
volume of the blood does not change. This
means that ALL of the blood that leaves
the heart goes through the arteries, and
then capillaries, and then returns to the
heart through the veins. Using the continuity equation, what is the flow through the
aggregate capillaries?
C. If the average diameter of the capillary is
4 3 1024 cm, and flow through the capillary
is 0.1 cm s21, how many capillaries are
there?
45
1.4
Chemical Foundations of
Physiology I: Chemical Energy and
Intermolecular Forces
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
List the chemical elements that make up the organic part
of the body
List the major chemical elements found as electrolytes in
the body
List the chemical elements found in trace quantities and
used as cofactors for enzymes
Explain why single CC bonds rotate easily whereas double
CC bonds do not
Define and give examples of structural isomerism, geometric isomerism, and optical isomerism
Write correct estimates of bond length and energy (within
a factor of 2) for covalent bonds
Define electronegativity
Describe what is meant by a polar bond
Distinguish between covalent and ionic bonds
Define dipole moment and be able to calculate the energy
of dipoledipole interactions
Describe the hydrogen bond and recognize its typical
energy
Describe what is meant by London dispersion forces
Draw the Lennard-Jones Potential and label the axes
ATOMS CONTAIN DISTRIBUTED
ELECTRICAL CHARGES
In ordinary chemical reactions, atoms are the fundamental particles. The word atom derives from the Greek
atomos, which means indivisible. The atoms themselves
are composed of simpler subatomic particles, the neutrons, protons, and electrons. These particles are characterized by their rest mass and electrical charge,
as shown in Table 1.4.1.
Ernest Rutherford showed that all of the positive
charges in an atom are concentrated within a very small
volume, called the nucleus, and was awarded the 1908
Nobel Prize in Chemistry for the work. The nucleus has
dimensions on the order of 10215 m! This requires
some new thinking: if positive charges repel each other
according to Coulomb’s law, how can they be concentrated in the nucleus? The answer is that there are
other fundamental forces at work here, the strong
46 nuclear force and weak nuclear force, that have effects
only over very short distances (,10214 m) and account
for the stability of atomic nuclei.
Each atom has a definite number of neutrons, protons,
and electrons. The number of protons in the nucleus is
called the atomic number, Z, and this number defines
the chemical element that describes the atom. In a
neutral atom, the number of electrons is equal to Z, and
these orbit the nucleus. The behavior of the electrons
defines the chemical reactivities of the elements, and the
concentrated positive charge of the nucleus, in turn,
determines the behavior of the surrounding electrons.
ELECTRON ORBITALS HAVE SPECIFIC,
QUANTIZED ENERGIES
Although we refer to electrons as particles, in fact they
have wave-like characteristics such as constructive and
destructive interference. The structure of the atom
cannot be explained using classical physics. Instead, it
requires quantum mechanics. Quantum mechanics
posits that the “orbit” of electrons around the nucleus is
described by a wave function, which has been interpreted as being related to the probability of finding
the electron in some volume. The wave function has
quantum numbers in that it allow electron orbitals to
have only specific energy levels, and transitions between
them can occur only when the exchange of energy is
exactly equal to the difference in the two energy levels.
A set of quantum numbers uniquely describes the
energy state of each individual electron in an atom.
One quantum number describes the electron “shell,”
a second describes the “orbital” within that shell, and a
third describes the spin of the electron. The Pauli exclusion principle states that no two electrons can share the
same set of quantum numbers within an atom. These
orbitals are generally described as “clouds,” indicating
the distributed nature of the orbital electrons.
TABLE 1.4.1 Mass and Charge of Subatomic Particles
Particle
Rest Mass (g)
224
Electrical Charge (C)
Neutron
1.6747 3 10
0
Proton
1.6726 3 10224
11.602176 3 10219
Electron
9.132 3 10228
21.602176 3 10219
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00005-7
Che mical Foundations of Physiology I: Chemical E nergy a nd Inter mol ecular Forces
Group
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
Period
1
1
H
2
3
Li
3
4
11
Na
19
K
p Orbital
s Orbital
d Orbital
4
Be
12
Mg
17
Cl
33
As
34
Se
35
Br
35
Kr
47
Ag
48
Cd
49
In
50
Sn
51
Sb
52
Te
53
I
54
Xe
76
Os
77
Ir
78
Pt
79
Au
80
Hg
81
Tl
82
Pb
83
Bi
84
Po
85
At
86
Rn
65
Tb
66
Dy
67
Ho
68
Er
69
Tm
70
Yb
71
Lu
40
Zr
41
Nb
42
Mo
43
Tc
72
Hf
73
Ta
74
W
75
Re
88
Ra
16
S
46
Pd
39
Y
87
Fr
15
P
44
Ru
26
Fe
7
13
Al
10
Ne
18
Ar
31
Ga
25
Mn
56
Ba
9
F
30
Zn
24
Cr
55
Cs
8
O
29
Cu
23
V
6
7
N
28
Ni
22
Ti
38
Sr
6
C
27
Co
45
Rh
21
Sc
36
Rb
5
B
14
Si
32
Ge
Transition elements
20
Ca
5
2
He
57
La
58
Ce
59
Pr
60
Nd
61
Pm
62
Sm
89
Ac
90
Th
91
Pa
92
U
93
Np
94
Pu
63
Eu
64
Gd
FIGURE 1.4.1 Periodic table of the elements. Each element is distinguished by the atomic number, the number of protons in the nucleus of each
atom. Each chemical element is symbolized by a one or two letter abbreviation, as indicated in the figure. Some elements have special biological
significance. C, H, O, and N (carbon, hydrogen, oxygen, and nitrogen, respectively) form the basic organic structures of the body. Phosphorus and
sulfur (P and S, respectively) are less common parts of the organic substance. Sodium (Na), potassium (K), magnesium (Mg), calcium (Ca), and chlorine
(Cl) highlighted in gray make up the electrolytes of the body fluids and are necessary in large amounts. Other elements, particularly transition
elements, bind to organic structures and enable their activities. These are required in trace amounts and include iron (Fe), manganese (Mn), cobalt
(Co), nickel (Ni), copper, (Cu), zinc (Zn), selenium (Se), molybdenum (Mo), and iodine (I).
HUMAN LIFE REQUIRES RELATIVELY
FEW OF THE CHEMICAL ELEMENTS
As noted above, each chemical element consists of
atoms whose nuclei contain a definite number of protons and some number of neutrons, which typically is
about the same as the number of protons, and an equal
number of electrons distributed among the atomic
orbitals. There are 94 naturally occurring elements, but
relatively few of these are essential to human life, as
illustrated in Figure 1.4.1.
ATOMIC ORBITALS EXPLAIN THE
PERIODICITY OF CHEMICAL
REACTIVITIES
There are eight main “shells,” referring to the principal
quantum number, n 5 (1,2,3,4,5,6,7,8) that describes
atomic orbitals. There are four major subshells: s, p, d,
and f, whose names derive from spectroscopic descriptions of sharp, principal, diffuse, and fundamental. These
orbitals are described by the azimuthal quantum number, l 5 (0,1,2,3) for (s,p,d,f), respectively. Each subshell
has a structure and a capacity for electrons that is
described by the magnetic quantum number, m, and
the spin quantum number, s. The s subshell is spherically symmetrical and holds only 2 electrons; each set
of p orbitals holds 6 electrons, the d orbitals hold 10,
and the f orbitals hold 14. The sequential filling of these
Principal
quantum
number
n
Azimuthal quantum number, l
l=0
l=1
l=2
l=3
1
1s
2
2s
2p
3
3s
3p
3d
4
4s
4p
4d
4f
5
5s
5p
5d
5f
6
6s
6p
6d
7
7s
7p
FIGURE 1.4.2 Order of filling of atomic orbitals. Electronic orbits are
characterized by a principal quantum number that determines the main
shell, an azimuthal quantum number that determines the subshell, a
magnetic quantum number that determines the orbital, and the spin
quantum number that determines the spin of the electron. There are
four subshells: s, p, d, and f. These have 1, 3, 5, and 7 orbitals that each
can hold up to two electrons of opposite spin. The order of filling with
increasing number of electrons follows the blue diagonal arrows in the
diagram: 1s fills first, followed by 2s and 2p; next is 3s followed by 3p
and 4s, followed by 3d, 4p, and 5s; next is 4d, 5p, and 6s; then 4f, 5d,
6p, and 7s.
orbitals accounts for the periodic chemical behavior
of the elements with their atomic number. This order of
filling is shown in Figure 1.4.2. Each subshell (s, p, d, f)
is typically filled with the requisite number of electrons
47
48
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
before filling the remaining subshells. Each electron
has a spin quantum number, s, that is represented as
“up” or “down.” The orbitals in the subshells are
typically filled singly with electrons of parallel spin
before double occupancy begins. This is the so-called
“bus seat rule,” analogous to the filling of a bus where
double seats tend to fill with single individuals before
double occupancy occurs.
is entirely different from the two elements themselves.
A classic example is water. Two volumes of hydrogen gas
will combine with one volume of oxygen gas to produce
water, which at the same temperature is a liquid and
behaves altogether differently from either the hydrogen
or oxygen gas. Such combinations of elements are
called compounds, and the fundamental unit of them
is the molecule. Molecules consist of atoms that are
bonded together through the sharing of electrons
in their outer atomic orbitals. The electrostatic shielding and energy involved in the orbital electrons
overcome the repulsive forces between the positively
charged nuclei. The resulting molecule is generally more
stable than the starting materials. In order to break apart
the molecule, energy must be supplied. This energy is
called the bond energy, and its magnitude depends
on the compound.
Full orbitals are inherently stable, because they have
low energy, and atoms having full orbitals are chemically unreactive. These correspond to the noble gases,
helium (He), neon (Ne), argon (Ar), krypton (Kr),
xenon (Xe), and radon (Rn). The electronic structure
of some of these stable atoms is shown in
Figure 1.4.3. All of the other elements can react with
other atoms, in order to become more stable by
attempting to fill their orbitals. They do this by sharing electrons, a process that constitutes chemical
bonding. This sharing can be equal or very unequal,
corresponding to the extremes of covalent bonding
and ionic bonding.
COMPOUNDS HAVE CHARACTERISTIC
GEOMETRIES AND SURFACES
Carbon has an atomic number of 6. Its electronic
structure is 1s2 2s2 2p2: there are two electrons in the 1s
orbital, two in the 2s orbital, and two in the 2p orbitals,
as shown in Figure 1.4.4. Carbon can achieve the
stable neon configuration of 1s2 2s2 2p6 by sharing
electrons with four hydrogen atoms. The resulting
ATOMS BIND TOGETHER IN DEFINITE
PROPORTIONS TO FORM MOLECULES
Two or more elements can combine to form a compound, and the resulting character of the compound
Helium (2)
1s2
s
Neon (10)
1s2 2s2 2p6
Argon (18)
1s2 2s2 2p6 3s23p6
Krypton (36)
1s2 2s2 2p63s23p64s23d10 4p6
s
s
s
p
p
d
p
Xenon (54)
1s2 2s2 2p63s23p64s23d10 4p65s24d105p6
s
d
1
1
1
1
1
2
2
2
2
2
3
3
3
3
3
4
4
4
4
4
p
d
5
6
FIGURE 1.4.3 Electronic structure of the inert gases. These inert gases are chemically unreactive because their orbitals are already filled. Helium, with
n 5 2 protons in its nucleus, fills the 1s orbital with 2 electrons of opposite spin. Spin is indicated in the drawing by an arrow pointed upward or
downward. Neon (n 5 10) fills the 2s and 2p orbitals with a total of 8 electrons. Each orbital in the subshells carries at most two electrons. The order of
filling of the orbitals corresponds to that shown in Figure 1.4.2.
Carbon (6)
1s2 2s2 2p2
s
p
1
2
3
4
FIGURE 1.4.4 Electronic structure of carbon. Carbon has 6 protons in its nucleus and 6 electrons that occupy the orbitals, 2 in the 1s orbital, 2 in the
2s orbital, and 2 more in the p orbitals. Only two of the three p orbitals are occupied by electrons. Carbon requires four more electrons to reach the
stable configuration of Neon.
Che mical Foundations of Physiology I: Chemical E nergy a nd Inter mol ecular Forces
compound is methane, written as CH4 to convey the definite and fixed proportion of 1C for 4H atoms. All molecules in methane have this compositional stoichiometry.
The close approach of the H nuclei and C nucleus
alters the electronic structure of both the carbon and
hydrogen. In molecular orbital theory, both the 2s and
2p orbitals of carbon participate in bonding by forming
four hybrid molecular orbitals, termed sp3, meaning
the hybrid of the 2s orbital with 3p orbitals (see
Figure 1.4.5). The angle between the neighboring CH
bonds is 109 280 . The shape-filling model of methane
shows the edges of the carbon and hydrogen atoms as if
they were hard spheres, but really the orbitals do not
have such definite boundaries. The electron orbitals
define these soft edges. All compounds are defined
by the relative locations of their atomic nuclei and
the three-dimensional distribution of their electronic
charges. These make up a three-dimensional surface
that can interact with other three-dimensional surfaces.
The bedrock of all of chemistry and physiology is the
interaction of these surfaces.
SINGLE CC BONDS CAN FREELY
ROTATE
Carbon can also form bonds with other carbon atoms.
Ethane has the compositional stoichiometry of C2H6
(see Figure 1.4.6). It is two methane molecules in which
two CH bonds are replaced by a single CC bond. In the
single CC bond, the sp3 hybrid orbitals overlap along
their axis and form a circularly symmetric sigma bond.
There is relatively free rotation around the axis of symmetry of this single bond, with three dips of about
12 kJ mol21 for each rotation when the H atom from
one methyl group aligns with the space between the
H atoms of the opposite methyl group. These ideas are
shown schematically in Figure 1.4.6.
1s orbital from H
sp3 from C
H
H
0.11 nm
H
C
H
C
109.5o
H
H
H
H
0.1 nm
Chemical structure
Ball-and-stick
model
Orbital picture
Space-filling model
FIGURE 1.4.5 Structure of methane. The compositional stoichiometry of methane is CH4—one carbon atom bonded to four hydrogen atoms. It arises
from the sharing of the 1s electron of H with the 2s and 2p electrons of carbon. The bonding arises from overlap of the 1s H electron with electrons
with hybrid C orbitals called sp3—formed from one s orbital and three p orbitals.
Sigma bond forms by overlap of
carbon sp3 orbitals
Rotation about the C—C bond
is relatively free, except for slight
energy variations due to positions
of H atoms in the two methyl groups
H
H
H
H
C
H
C
H
C
H
H
C
H
109.3°
H
H
H
Chemical structure
Orbital picture
Ball-and-stick
model
Space-filling model
FIGURE 1.4.6 Structure of ethane. Here 1C atom binds to another C and 3H atoms. The CC bond forms by overlap of the sp3 orbital along its axis to
form a sigma bond that has circular symmetry. This bond can rotate about its axis, with some resulting configurations having just a little more stability
than others. The most stable arrangement is shown, with the H atom of one methyl group aligned with the space between the H atoms in the
opposing methyl group.
49
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
DOUBLE CC BONDS PROHIBIT FREE
ROTATION
of bonds that connect atoms together to make molecules, and each of these bonds has definite average
bond lengths, bond angles, and bond energies.
Table 1.4.2 summarizes approximate values for some of
these bonds. These are approximate because atoms
bound to other parts of the molecule can influence the
molecular orbitals some distance away, thereby altering
the angle, or length, or energy of any particular bond.
Carbon can form double bonds by altering its molecular orbitals. Ethylene is ethane in which another pair
of CH bonds converts to a second CC bond (see
Figure 1.4.7). Instead of combining 2s2 with 2px1,
2py0, and 2pz1 to form the 4sp3 orbitals, it can arrange
the electrons in a planar trigonal geometry by hybridizing the 2s2 with the 2px1 and 2py0 orbitals to form
three sp2 orbitals with one electron each, and another
pz1 orbital that can form a second bond, a pi bond,
out of the plane of the sp2 orbitals. This bond resists
twisting and a 90 twist breaks the overlap of the p
orbitals, and hence breaks the second bond. Thus double bonds such as that in ethylene, shown in
Figure 1.4.7, produce a somewhat rigid plane in any
molecule in which they are found.
BOND ENERGY IS EXPRESSED
AS ENTHALPY CHANGES
Earlier we wrote the conservation of energy theorem as
½1:3:6
where dE was the incremental change in the energy of a
system, dq is defined as the heat absorbed by the system
from its surroundings, and dw is defined as the work
performed by the system on its surroundings. The total
energy content of the system, E, does not depend on the
path taken to get to its configuration. It is a state variable.
If we conduct a change of state at constant pressure,
then Eqn [1.3.6] can be written as
CHEMICAL BONDS HAVE BOND
ENERGIES, BOND LENGTHS, AND
BOND ANGLES
½1:4:1
So far we have discussed CH bonds, CC single bonds,
and CC double bonds. There are a host of other kinds
Pi bond forms by overlap of
carbon py orbitals
H
dE 5 dq 2 dW
dE 5 dqp 2 P dV
Rotation about the C—C double bond
is restricted.
H
H
C
H
C
C
C
H
H
H
H
H
H
H
pi bond
C
C
H
H
C
H
H
sigma bond
d
H
C
C
C
C
Space-filling model
H
C
H
Ball-and-stick
model
Orbital picture
Chemical structure
H
50
H
Carbon 1s orbitals Carbon sp2 orbitals
H
H
Carbon pz orbitals
H
H
Hydrogen 1s orbitals
FIGURE 1.4.7 Structure of ethylene. The double CC bond is stronger than the single bond (it requires more energy to break) and locks the C atoms
and all of the bonded groups into a single plane. This plane resists twisting because twist along the CC axis rotates the p orbitals away from overlap,
breaking the pi bond. The space-filling model is not oriented the same way as the ball-and-stick model; the plane of the space-filling model is parallel
to the plane of the paper. The lower part of the diagram shows the component orbitals. The 1s orbitals from carbon do not participate in molecular
bonding. The sp2 orbitals bond to the H 1s orbitals through sigma bonds and form the CC sigma bond. The pz orbitals perpendicular to the plane of
the sp2 orbitals form a second CC bond, a pi bond.
Che mical Foundations of Physiology I: Chemical E nergy a nd Inter mol ecular Forces
TABLE 1.4.2 Typical Bond Length and Bond Energy for Some Bonds Important in Physiology
Bond Length (pm) Energy (kJ mol21) Bond Length (pm) Energy (kJ mol21) Bond Length (pm) Energy (kJ mol21)
CC
154
346
CN
147
308
CO
143
360
CS
182
272
HC
109
411
HN
101
386
HO
96
459
HvS
134
363
PO
163
335
SS
205
226
CC
134
602
CO
120
799
PO
150
544
CC
120
835
Bond lengths are given in pm (510212 m) and bond energy is reported in kilojoule per mole (kJ mol21). Many tables of bond energies report them in kcal mol21.
The calorie is defined as the amount of heat energy required to raise the temperature of 1 g of water from 14.5 C to 15.5 C. The calorie is readily converted to the
joule using the conversion factor 1 cal 5 4.184 J. Calorie derives from the Latin calor, meaning to heat.
if the only work is pressurevolume work. Note here that
½1:4:2
F dx 5
F
A dx 5 P dV
A
clarifies that a pressure moving a volume has the
same work units as a force moving through a distance.
Integrating Eqn [1.4.1] between two states, we obtain
Ðf
Ðf
ÐV
5 i dqp 2 Vif P dV
i dE
½1:4:3
Ef 2 Ei
5 qp 2 PðVf 2 Vi Þ
ðEf 1 PVf Þ 2 ðEi 1 PVi Þ
5 qp
Hf 2 Hi
5 qp
ΔH
5 qp
Here we make the definition
½1:4:4
H 5 E 1 PV
where H is the enthalpy. Since E, P, and V are all state
variables, depending only on the state and not the
path taken to that state, enthalpy is also a state variable.
The bond energies are differences in the enthalpy
of formation of the products and reactants in any
chemical reaction.
THE MULTIPLICITY OF CX BONDS
PRODUCES ISOMERISM
Some chemical compounds with identical compositional stoichiometry behave differently. Their different
behaviors can be obvious and large, or quite subtle,
depending on how the compounds differ. These compounds with identical composition differ in the way the
atoms are arranged in the molecule. They are called
isomers, meaning same weight. There are three major
classes of isomers, structural, geometric, and optical,
which are described below and shown schematically in
Figures 1.4.81.4.10:
G
G
G
G
Structural isomers differ in the connectivity of the
atoms in the molecule.
Stereoisomers have identical connectivity but the
atoms are arranged differently in space. These consist
of two classes:
G Geometric isomers, involving a double CC bond
that does not allow free rotation
G Optical isomers, existing in two types.
Enantiomers are mirror images of each other. They
have identical physical characteristics such as melting point and density but are not superimposable.
This requires an asymmetric carbon atom in which
four nonidentical groups are bonded. They differ in
their ability to rotate the plane of polarized light.
Diasteriomers are optical isomers that are not enantiomers. They typically differ in the spatial distribution about one or more asymmetric carbons, while
not being mirror images.
UNEQUAL SHARING MAKES POLAR
COVALENT BONDS
The electrons that are shared in covalent bonds distribute
themselves according to the charges on the nuclei within
the molecule and the other electrons in the unshared
orbitals that shield that charge from the electron. In
almost all cases, the electrons are not shared equally but
tend to spend more time near one or the other of the
nuclei involved in the bond. The ability of an atom in a
molecule to attract shared electrons is called its electronegativity. It generally increases going up the periodic
table and going to the right, so that F has the highest
electronegativity and Fr has the lowest. The electronegativity of some common elements is shown in Table 1.4.3
in arbitrary Pauling units scaled to F at 4.0.
51
Structural isomerism: same formula, different connectivity
An aldehyde is a carbonyl group
—O) bonded to an H and a C
(C—
Hydroxyl group is an —OH
group bonded to another atom
H
H
H
C
H
A ketone is a carbonyl group
—O) bonded to two C atoms
(C—
C
O
O
C
O
P
H
C
OH
H
O
_
H
_
O
O
C
OH
O
C
O
P
H
O
O
_
_
Glyceraldehyde 3-phosphate
Dihydroxyacetone phosphate
Phosphate groups bind H2PO4 to C
with high energy store
FIGURE 1.4.8 An example of structural isomerism. Both glyceraldehyde 3-phosphate and dihydroxyacetone phosphate contain the same number of
each type of atom. However, the connectivity of the atoms differs. Glyceraldehyde 3-phosphate contains an aldehyde group, which is defined as a C
atom with a double bond to O and a single bond to H. Dihydroxyacetone phosphate contains a ketone group, which is a C atom with a double bond
to O and the remaining bonds to C atoms. Conversion of the two chemicals requires breaking and reforming chemical bonds.
Geometric isomerism: same connectivity but different spatial distribution
Carboxyl group gives acid character to
organic compounds
HO
HO
O
O
C
H
C
C
FIGURE 1.4.9 An example of geometric
isomerism. Both elaidic acid and oleic acid
belong to a class of organic compounds
called fatty acids, characterized by a
carboxyl group at one end of an
unbranched hydrocarbon chain. In these
molecules, the chains are 18 carbons long.
Both have one double bond beginning at
the ninth carbon from the carboxyl end or
the ninth carbon from the terminal methyl
end. The spatial arrangement of the carbon
chain and hydrogens at the double bond
can be achieved in two ways: either cis or
trans. In the cis arrangement, found in oleic
acid, the two hydrogens are on the same
side of the double bond, meaning that the
hydrocarbon chain toward the carboxyl
end and the hydrocarbon chain toward the
methyl end are also on the same side,
opposite to the hydrogens. This produces a
kink in the hydrocarbon chain. In the trans
arrangement, in the case of elaidic acid,
the two H atoms on the doubly bonded
C atoms are on opposite sides of the
double bond. This has the effect of keeping
the hydrocarbon chain straighter. Both
compounds have the same composition
but different physical properties due to this
geometric isomerism.
H
C
C
C
C
H
C
H
H
H
H
H
C
H
C
H
H
C
H
H
H
C
H
C
H
H
H
H
Oleic acid
C
H
H
C
H
C
H
C
H
C
C
H
H
H
C
H
H
C
H
H
H
H
H
H
H
C
H
H
C
C
H
H
H
H
Elaidic acid
H
H
H
C
H
C
C
H
Trans double bond has H groups
on opposite sides of the double
bond
H
H
C
H
C
H
H
H
C
C
H
H
H
Cis double bond has
H
H groups on the same
side of the double bond;
connectivity is identical to
the trans configuration, but
spatial difference is great
C
C
H
H
H
H
C
C
H
H
H
Che mical Foundations of Physiology I: Chemical E nergy a nd Inter mol ecular Forces
Optical isomerism: same connectivity, not superimposable
L-alanine
D-alanine
H
O
H
H
C
O
H
C
H
H
C
C
H
C
C
N
H
H
O
H
H
N
H
H
The amino group gives a basic
character to the amino acid
H
The α carbon is asymmetric; it has four
different groups and no plane of symmetry;
it can form mirror image arrangements that
cannot be superimposed
WATER PROVIDES AN EXAMPLE
OF A POLAR BOND
TABLE 1.4.3 Electronegativity of Common Atoms
Atoms
H
O
C
N
O
F
Na Mg P
Electronegativity 2.2 2.5 3.0 3.5 4.0 0.9 1.2
S
FIGURE 1.4.10 An example of optical isomerism.
Amino acids consist of a backbone of a carboxyl group
bonded to a central C atom, called the α carbon, and
an amino group bonded to the other side of the
α carbon. Typically the α carbon has two more bonds,
one to an H atom and the other to a variable group,
called R for residue, whose composition determines
the kind of amino acid. There are 20 different R groups
and 20 different amino acids. The R group for alanine,
shown here, is a methyl group. Because the α C atom
is asymmetric—there is no plane of symmetry—the
four groups can be arranged in two nonequivalent
ways. Our bodies use only the L-amino acids. In the
L-amino acids, starting from the carboxyl group and
moving toward the amino group, the R group is to the
left. This is very important, as D-amino acids in proteins
would have their R groups pointing the wrong way.
Cl
2.1 2.5 3.0
Source: L. Pauling, The Nature of the Chemical Bond. Cornell University
Press, 1960.
Atoms with similar electronegativities will share bonding
electrons equally and will produce nonpolar bonds.
Examples include the CC bond, CH bond, and HS bond.
Bonds such as OH will be polar bonds.
IONIC BONDS RESULT FROM ATOMS
WITH HIGHLY UNEQUAL
ELECTRONEGATIVITIES
If the difference in electronegativity is too great, an
ionic bond will form in which the strongly electronegative atom strips an electron from the weakly electronegative atom. Examples include NaCl, KCl, CaCl2,
MgCl2, and a host of other physiologically relevant
compounds. These are noted for their dissociation in
water to form ions, Na1, Cl2, K1, Ca21, and Mg21, for
example. These isolated ions are stabilized by their
interaction with the polar water molecule, which presents a negative side towards the positive ions
(cations) and a positive side towards the negative ions
(anions).
The bond angle defined by HOH is 104.5 , which is
close to the tetrahedral angle. In this case, O forms 3
orbitals by hybridization of the 2s2 orbitals with the
2px and 2py orbitals. This leaves 2 O electrons in an sp2
orbital that are unshared and 2 electrons in a pz2 orbital
that are also unshared. These form the lone electron
pairs of water that participate in yet another kind of
bonding, the hydrogen bond, discussed later. The electronegative O atom attracts the electrons away from the
H nuclei, forming a partial separation of charge in the
molecule itself. Thus the bond is said to be polar (see
Figure 1.4.11).
The estimated charge separation is about 20.67 on the
O atom and about 10.33 on each of the H atoms. The
dipole moment is defined as
½1:4:5
p 5 qd
where p is a vector, the dipole moment, q, is the charge
divided into equal q2 and q1, and d is the vector pointing from q2 to q1. The dipole moment of a single
water molecule is 1.855 debye (1 debye 5 3.33564 3
10230 C m), but the dipole moment of water varies with
the size of the water cluster, because nearby water molecules rearrange themselves in the presence of an electric
field. Dipoles themselves produce an electric field and
therefore interact with electric charges in its vicinity.
These electrostatic interactions are part of the forces that
govern the interaction of surfaces. In an electric field,
the dipole experiences a torque given by
53
54
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
δ+
H
0.096 nm
H
O
O
δ–
104.5o
H
δ+
H
Chemical structure
Ball-and-stick
model
Orbital picture
Space-filling model
FIGURE 1.4.11 Structure of water. The HOH angle is 104.5 and the bond length is about 96 pm. The O atom is far more electronegative than the H
atoms, thereby causing an asymmetric redistribution of the electrical charges away from the H atoms and surrounding the O atom. These partial
separations of charges produce a polar compound and a net dipole moment.
δ–
δ+
δ–
δ+
O
H
δ+
δ+
H
δ–
H
O
H
δ+
FIGURE 1.4.12 The hydrogen bond in water. The polar OH bond involves a separation of charge. The partial positive charge on the H atom in water is
attracted to the lone electron pairs on the opposite side of the molecule on adjacent molecules. This forms a weak bond that is easy to form and easy
to break. In many situations, the number of hydrogen bonds significantly stabilizes large structures. In water, there are many hydrogen bonds because
each water molecule has the potential of participating in four of them: two because of the H atoms and two more because of the two lone electron
pairs.
½1:4:6
τ5p3E
where τ is the torque, p is the dipole moment, and E is
the electric field strength.
INTERMOLECULAR FORCES ARISE FROM
ELECTROSTATIC INTERACTIONS
Chemical bonds join atoms together to form molecules.
Molecules can also be attracted to each other through a
variety of intermolecular forces that include:
G
G
G
Hydrogen bonding
Dipoledipole interactions
London dispersion forces.
HYDROGEN BONDING OCCURS
BETWEEN TWO ELECTRONEGATIVE
CENTERS
The hydrogen bond involves the sharing of the positive
charge of hydrogen between two electronegative centers
such as oxygen and nitrogen. It requires proximity and
proper orientation of the two electronegative centers.
In the case of water, the bond is strongest when
the OH bond on one water molecule aligns with the
lone electron pair orbital of the adjacent water molecule, as shown in Figure 1.4.12. The hydrogen bond
requires only 840 kJ mol21 to break, compared to
much higher values for covalent bonds such as CC
(346 kJ mol21) or CH (411 kJ mol21). This low bond
energy makes it useful, because it means that the bond
can form and break under the influence of normal thermal agitation. At the same time, a large number of
hydrogen bonds can stabilize structures such as proteins
and DNA.
DIPOLEDIPOLE INTERACTIONS ARE
EFFECTIVE ONLY OVER SHORT
DISTANCES
By virtue of their spatial separation of charges, dipoles
produce an electric field surrounding them whose
magnitude is given by
½1:4:7
Uðr; θÞ 5
p cos θ
4πε0 κr 2
An elementary proof of this result is given in Appendix
1.4.A1. The angle θ is defined as the angle between the
point at which potential is given and the midpoint
between the two separated charges within the dipole.
Note that this is a potential, not the force. In Coulomb’s
law, the potential varied with 1/r (see Eqn [1.3.5]) but
here it varies with 1/r2. Replacing one ion with a dipole
causes the interaction to be shorter range. At short range,
Che mical Foundations of Physiology I: Chemical E nergy a nd Inter mol ecular Forces
a nearby charge “sees” both charges; at longer range the
charge “sees” two opposing dipole charges that tend to
neutralize each other; the interaction becomes weaker at
longer distances.
Because they both produce an electric field, pairs of
dipoles interact with each other. The energy of interaction
is given by
2
pA pB 2 1
½1:4:8
UðrÞ 5 2
3kT 4πε0 κ r 6
where k is a new physical constant, Boltzmann’s constant,
which is the gas constant per molecule. Its value is
1.38 3 10223 J K21; T is the temperature in K, pA and
pB are the dipole moments of the two dipoles, ε0 is
the electrical permittivity of space, given earlier as
8.85 3 10212 C2 N21 m22, and r is the separation of the
centers of the dipoles.
LONDON DISPERSION FORCES INVOLVE
INDUCED DIPOLES
Water has a permanent dipole moment. Symmetrical
compounds such as methane and H2 have no permanent dipole moment, but these can be induced to form
a dipole by the presence of an externally applied electric
field. A polarizable atom redistributes its internal
charge in response to an electric field to form a dipole
moment aligned with the applied field. For small electric field strength, the induced dipole is approximately
proportional to the applied field:
½1:4:9
pind 5 αE
where pind is the induced dipole moment, E is the
electric field, and α is the polarizability. The SI unit for
α is C m2 V21, but it is often converted to units of
volume, cm3, by multiplying by 1/4πε0 3 1026, where
ε0 is the electrical permittivity of space. This effect,
shown in Figure 1.4.13, results in the attraction of a
neutral molecule to a charged molecule.
The totality of forces between atoms or molecules
due to dipoledipole, dipole-induced dipole, and
instantaneously induced dipoles (London dispersion
force) is called the van der Waals force. It excludes
the interaction due to covalent bonds or electrostatic
interactions.
CLOSE APPROACH OF MOLECULES
RESULTS IN A REPULSIVE FORCE THAT
COMBINES WITH THE VAN DER WAALS
FORCES IN THE LENNARDJONES
POTENTIAL
Imagine two atoms or molecules separated by a large
distance. Because of the large distance, their interaction is
very small—there is little force between them. Even if
they have very little dipole moment, as they approach
one another they will experience attractive forces due to
London dispersion forces, and these are attractive. As the
distance between their atomic nuclei shortens, they begin
to experience repulsive forces due to the interpenetration
of their atomic or molecular orbitals. This repulsive
force varies quite steeply with separation. The overall
potential energy for the interaction of nonbonding particles has been mathematically approximated by the
LennardJones potential. It is given as
r 6 σ 12 σ6
rm 12
m
2
22
5ε
UðrÞ 5 4ε
r
r
r
r
½1:4:10
where U(r) is the potential energy of the interaction, ε
is depth of the potential well (a measure of the strength
of the attractive forces), and σ is the distance at which
the intermolecular potential is zero; rm is the distance of
separation when the potential is 2 ε (see Figure 1.4.14).
E
+
Even molecules that do not have a permanent dipole
moment can transiently produce dipole moments that
result in their attraction. We imagine that electrons orbit
their nucleus in an “electron cloud” but this picture is
an average distribution. At any instant the electrons are
separated from their nucleus, producing a transient
dipole. When nearby atoms synchronize the distribution of electrons in their clouds, they can produce
attractive forces first described by F. London in 1937
and called London dispersion forces.
r
–
–
–
–
–
+
+
+
+
+
q–
q+
Δr
FIGURE 1.4.13 Electrical polarizability. The presence of a charge
establishes a local electrical field. Electrons within nearby molecules can
respond to this field by redistribution of charge, causing an induced
dipole. Because of the separation of charge, the induced dipole
experiences unequal forces from the fixed charge. The result is a net
movement of the induced dipole.
ATOMS WITHIN MOLECULES WIGGLE
AND JIGGLE, AND BONDS STRETCH
AND BEND
The bond lengths, angles, and energies listed in
Table 1.4.2 are averages. Two atoms involved in a
bond actually oscillate back and forth around the
average bond length. In addition, the angles defined
by, say, HCH are not fixed, but the three atoms oscillate around the average bond angle. The molecules
also translate through the solution or gas, and rotate.
Some of these motions affect others, as rapid rotation
about an axis perpendicular to a bond tends to
55
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
stretch the bond. All of these modes of action within
molecules store kinetic and potential energy on a submolecular scale. When a moving body stops because of
friction, the kinetic energy does not disappear; it is
converted from the macromolecular to the molecular
scale—the kinetic energy appears as heat and the
temperature of the surfaces increases. This conversion
is a one-way street: we can never fully recover that
molecular energy back into macromolecular action.
The temperature is, in one sense, a measure of this
dynamic motion. These various modes of thermal
motion are shown in Figure 1.4.15.
Intermolecular potential
U(r)
56
rm
0
σ
r
ε
F >0 F=0
Repulsive
force
F<0
Attractive force
FIGURE 1.4.14 The LennardJones potential. At far separation, there is
little force and no potential. Because of the inverse 6th power
dependence on the separation, this force becomes larger as particles
approach one another, reaching a minimum at U(r) 5 ε and r 5 rm. The
separation at which U(r) 5 0 is r 5 σ. The equilibrium for the particles
occurs at the minimum potential.
SUMMARY
Atoms are the fundamental units of the elements and
are defined by the number of protons in the atomic
nucleus. Only relatively few of the chemical elements
have active roles in the body. The electrons orbit the
nucleus in electron orbitals that have definite energy
levels. These electron orbitals describe the distributed
nature of the electrons within their atoms. Electrons
populate the orbitals in a defined sequence that gives
rise to the periodic behavior of the chemical elements
within the periodic table.
The chemical elements can combine to form compounds in fixed ratios to each other, often by sharing
the outer orbital electrons. These bonds form by combination of their atomic orbitals or by the formation of
molecular orbitals in which the electrons are distributed
among the nuclei that make up the molecule. Most
organic compounds form covalent bonds in which the
electrons are shared approximately equally among
the nuclei. These bonds have energies in the range
200500 kJ mol21 and have bond lengths on the order
of 0.10.2 nm. The bond angles are determined by the
kind of bonds that form. Many of the biochemicals
within the human body are organic compounds that
have CC, CC, CN, COH, CO, and CH bonds. The single
bonds typically freely rotate about the axis connecting
the two nuclei. Double bonds do not permit free
rotation because the rotation would break the second
bond and this requires energy.
The rigid form of the CC bond gives rise to geometric
isomerism. The different arrangement of the same
nuclei around the CC bond can cause large differences
in the overall shape of molecules. Isomerism also arises
from the different spatial arrangement of chemical
groups around an asymmetric C atom. Such isomers
can be mirror images of each other (enantiomers) or
not. All of the amino acids (except glycine) exist in
enantiomeric forms, but only the L-type of amino acids
are used to make proteins.
Rotation about the y axis
Bending of the bond
Stretching of the bond
Rotation about the x axis
Rotation about the z axis
Three translational degrees
of freedom
FIGURE 1.4.15 Degrees of freedom of motion in water. The HOH bond can bend, changing its angle; the OH bond can oscillate, stretch, and compress
its bond length; the entire molecule can rotate around several independent axes; the molecule can translate in three independent directions. Each of
these modes of movement is independent and each carries some kinetic energy. The energy distributed amongst the various modes of motion
increases with increase in temperature.
Che mical Foundations of Physiology I: Chemical E nergy a nd Inter mol ecular Forces
Polar bonds involve unequal sharing of bonding
electrons between the two nuclei involved in the
bond. A good example is the OH bond in which the
oxygen atom is more electronegative. Electronegativity
refers to the ability of a nucleus to attract shared
electrons. Because O is more electronegative than H,
the resulting OH bond is polar, with more negative
charge around the O atom than the H atom. This partial
separation of charge produces a dipole which is
described by its dipole moment, equal to the charge
times its separation directed from the 2 to the 1 charge.
The dipole moment produces an electric field that can
interact with other nearby atoms. The OH group is very
important because it forms hydrogen bonds with other
electronegative atoms. This low energy bond is easy
to form and easy to break and can stabilize biological
structures.
Ionic bonds occur when two atoms or chemical groups
differ greatly in their electronegativities. The more electronegative atom or group effectively “steals” an electron
from the less electronegative atom. When dissolved in
water, these types of compounds typically dissociate into
a cation and anion.
Molecules can interact with each other through ionion
interactions, described by Coulomb’s law, hydrogen
bonding, dipoledipole interactions, and London dispersion forces. The London dispersion forces arise
from transient dipoles in atoms that induce transient
dipoles in nearby atoms, producing an attractive force.
The LennardJones potential describes the overall
interaction between nonbonded particles.
Atoms and molecules form surfaces that interact
with other surfaces. Almost all of physiology is about
how these surfaces interact to produce catalysis or tight
binding or loose binding or specificity of binding.
REVIEW QUESTIONS
1. What is the fundamental unit of an element?
What is the fundamental unit of a chemical
compound?
2. Why do atoms form compounds in definite and
fixed proportions?
3. What forms the surfaces of atoms and molecules?
4. Why are the CH bonds in methane arranged to
point toward the vertices of a tetrahedron?
5. What is the tetrahedral angle?
6. What are typical energies for CH and CC bonds?
7. What are typical bond distances for CH and CC
bonds?
8. What is meant by structural isomerism?
9. What is meant by geometric isomerism?
10. What is optical isomerism?
11. Why does water form polar covalent bonds?
12. What is hydrogen bonding? How much energy
is in a hydrogen bond?
13. What is an electric dipole?
14. What are dipoledipole interactions?
15. What are London dispersion forces?
APPENDIX 1.4.A1 DIPOLE MOMENT
A DIPOLE CONSISTS OF TWO EQUAL
CHARGES SEPARATED BY A DISTANCE, D,
AND IS DESCRIBED BY ITS ELECTRIC
DIPOLE MOMENT
An electric dipole consists of two equal charges, q1 and
q2, separated by a distance d, as shown in Figure 1.4.
A1.1. These are typically molecules whose separation
distance is small compared to the distance at which
electrical effects are noted. As we shall see, cardiomyocytes can also act as electric dipoles. The electric dipole
moment is defined as
½1:4:A1:1
p 5 q1 d
where d is a vector pointing from q2 to q1, as shown in
Figure 1.4.A1.1. The net force on a unit positive test
charge at any point surrounding the dipole can be
determined by the vectorial sum of the component
forces from q1 and q2, as shown in Figure 1.4.A1.1. We
can determine a set of points surrounding the dipole
that has the same magnitude of force, but in varying
directions. This set forms a curve. The family of lines for
a set of force magnitudes, shown in Figure 1.4.A1.2,
represents the electric field surrounding the dipole.
Moving a positive unit charge from a large distance
away (N) to any point within the electric field entails
p = q+ d
d
q+
q–
θ
r+
r––r+
r–
F–
F–
Ftot
qtest
F+
Ftot
qtest
F+
FIGURE 1.4.A1.1 Origin of the electrical forces near an electric dipole
determined at two different locations. The electric dipole consists of two
equal charges (q1 and q2) separated by a distance, d. Charges placed
nearby experience a force as a result of the electric dipole. The net force
is the vector sum of the forces exerted by the two charges, as shown for
two different positions. Because the force declines as 1/r2, the direction
of the force changes with distance and angle, θ, made between the line
joining the center of the dipole and the test charge, and the electric
dipole moment.
57
58
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
V=0
in Figure 1.4.A1.2. These lines intersect the lines of
electric force at right angles.
The electric potential surrounding a dipole is just the
sum of the potentials associated with each charge. Thus
we write:
-
+
+
½1:4:A1:2
FIGURE 1.4.A1.2 Electric field and electrical potential surrounding a
dipole. The electric field lines are shown in dashed lines with arrows.
They represent the lines of equal force that would be felt by a positive
unit test charge as it approaches the dipole. The electric field is a vector
which, when magnified by the size of a charge, gives the magnitude
and direction of the force. The electric potential contour lines are shown
in solid. The negative gradient of these contour lines are the electric
field lines. Thus the electric field is the steepest slope down the
potential surface. The lines of equal potential intersect the electric field
lines at right angles, much like the steepest descent off a hill intersects
its altitude contour lines at right angles.
expending energy that is stored as potential energy,
which is identified as the electrical potential. The potential at any point is the scalar sum of the potential energy
associated with each charge. That is, the potential at any
point is just the sum of the potential energy of q1 and
that of q2. Contour lines of equal potential are shown
Vtotal 5 V1 1 V2
q1
q1
5
1
4πε0 r1
4πε0 r2
q1
1
1
5
2
r2
4πε0 r1
q1 r2 2 r1
5
4πε0 r1 r2
q1 d cos θ p cos θ
5
4πε0 r 2
4πε0 r 2
where the quantity q1d appears. We have identified this
as the electric dipole moment, p 5 q1d. Here d cos θ
enters the equation as an approximation of r2r1,
as suggested by the geometry of Figure 1.4.A1.1.
This assumption is generally made when rcd. Thus
the voltage at any point is inversely proportional to the
square of the distance and varies with the relative position of the point with respect to p, the electric dipole
moment. The point of these calculations is to show
that the electric dipole creates an electric field and a
potential field that can be measured some distance away.
The value of the potential depends on the distance and
the relative position compared to the dipole.
Chemical Foundations of
Physiology II: Concentration and
Kinetics
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
Be able to calculate the molar concentration given the
mass of solute, formula weight, and volume of solution
Be able to calculate the number of moles of solute in an
aliquot of solution given the volume and concentration
Be able to determine the gram molecular weight of simple
compounds
Know the prefix notations for scientific notation
Be able to calculate dilutions of stock solutions to form
working solutions
Be able to calculate the volume of distribution using Fick’s
dilution principle
Define rate constant
Be able to derive a first-order rate equation and calculate
its half-life
Define equilibrium constant
Describe how an enzyme can change the rate of a reaction
without changing its equilibrium
Write the MichaelisMenten equation and draw a saturation plot. Identify Km and Vmax
Draw a double-reciprocal plot and identify Km and Vmax on
this plot
mixtures of isotopes that differ slightly in their atomic
mass. These are atoms that possess the characteristic
number of protons (the Z number) but differ in their
numbers of neutrons.
Each molecule has a definite mass that depends on the
atoms that make it up. If we add up the atomic masses
of the constituent atoms, we get the molecular weight
in daltons. If we express this molecular weight in
grams, we will have the gram molecular weight (just
scratch out “daltons” and replace it with “grams”).
A pile of molecules whose mass in grams is the gram
molecular weight is called a mole. Such a pile will
consist of not one molecule but very many of them,
and the number will be the number of daltons in a
gram.
To see this, suppose we take a substance with a molecular weight of Z daltons. How many of these molecules
will we have to pile up in order to make up a pile with
a mass of Z grams? Let N be the number of molecules
in the pile of Z grams. The mass of one molecule is Z
daltons. The mass of N of them is just N 3 Z daltons.
This mass will be the gram molecular weight. Thus,
we have
Z daltons 3 N 5 Z grams
N5
AVOGADRO’S NUMBER COUNTS
THE PARTICLES IN A MOLE
As described in Chapter 1.4, each chemical species is
composed of a definite number of atoms of each element. This is Dalton’s law of fixed proportions, which
states that a molecule contains integral numbers of each
kind of atom. Under ordinary chemical reactions, these
atoms cannot be converted into each other. Each of
these atoms contributes a tiny but definite mass to the
molecule. The atomic mass unit is defined as 1/12 of
the mass of a carbon-12 atom, the one containing 6
protons and 6 neutrons in its nucleus. This unit of mass
is also called the dalton, Da. The atomic weight of
a carbon-12 atom is defined as being 12 daltons. The
atomic weight of most elements, including carbon,
is not exactly integral because most elements consist of
1.5
Z grams
1g
5
Z daltons
1 Da
The actual mass of 1 Da is 1.66 3 10224 g. Thus, the
number of molecules in a gram molecular weight is
N5
1g
5 6:02 3 1023
1:66 3 10224 g
This is, of course, Avogadro’s number. It is the number
of molecules per mole. A mole is a pile of molecules
of a single substance whose mass in grams is equal to
its gram molecular weight and which contains
Avogadro’s number of molecules. We can turn this definition around and define the mole as Avogadro’s number of particles. This definition is completely general.
We did not specify how big the molecule is in the
above calculation. No matter the size of the compound, one mole contains Avogadro’s number of
molecules.
59
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00006-9
60
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
CONCENTRATION IS THE AMOUNT
PER UNIT VOLUME
In Chapter 1.2, we defined extensive and intensive variables as those that depend on the extent of the system
and those whose value is independent of the extent of
the system, respectively. The amount of a solute, and
the volume in which it is distributed, are both extensive
variables. If you have twice the volume of solution,
you have twice the amount of solute dissolved in it. The
concentration, on the other hand, is an intensive variable which is given as the ratio of two extensive
variables:
amount
Concentration 5
volume
½1:5:1
m
C5
V
where C is the concentration, m is the amount of solute,
and V is the volume in which that amount of solute
is dissolved.
SCIENTIFIC PREFIXES INDICATE ORDER
OF MAGNITUDE
Often concentrations of physiologically relevant materials in physiological fluids are quite small and must be
expressed in scientific notation, as in the example of
hemoglobin in Example 1.5.3. It is useful to know the
established prefixes for units of volume, mass, or length.
The standard units are the liter, L, the gram, g, and the
meter, m, respectively. The standard prefixes are shown
in Table 1.5.1.
EXAMPLE 1.5.1 Calculating Molar Concentration
Suppose we dissolve 18 g of NaCl in water in a 2-L volumetric
flask and then add water to the mark so that the solution
volume is 2 L. What is its concentration?
We can report this concentration in mass per liter as 9 g L21. For
many solutes, this is an acceptable way of expressing the concentration. For example, the concentration of hemoglobin in whole
blood is 15 g dL21. Here dL means “deciliter” and is one-tenth of
a liter or 0.1 L. Since 1 L 5 1000 mL, 1 dL 5 1000 mL/10 5 100 mL.
Thus, a hemoglobin concentration of 15 g dL21 means that there
is 15 g of hemoglobin in every 100 mL of whole blood.
The concentration of many solutes is not reported in these units,
however, but in units of moles per liter, or molar. We can
convert from mass to moles by dividing by the gram molecular
weight. In the case of NaCl given above, the atomic weight of Na is
22.99 daltons and the atomic weight of chlorine is 34.45 daltons,
giving a molecular weight for NaCl of 22.99 1 35.45 5 58.44
daltons; its gram molecular weight is 58.44 g. Thus 58.44 g of
NaCl constitutes 1 mole of NaCl. The number of moles of NaCl
in 18 g is calculated as
X moles 5
18 g NaCl
5 0:308 mol
58:44 g NaCl mol21
Since this amount was dissolved in 2 L of solution, its concentration 5 amount/volume.
Concentration 5
0:308 mol
5 0:154 M
2L
where M designates molarity or moles per liter.
EXAMPLE 1.5.2 How to Make Up a Solution
You need to make up 500 mL of a solution containing 0.3 M
urea. How much urea do you need?
Here we rearrange the equation C 5 m/V to read m 5 C 3 V.
In this case, C 5 0.3 M and V 5 0.5 L. The number of moles of
urea is thus
m 5 0:3 M 3 0:5 L 5 0:15 mol
We need to convert this into grams so that we can accurately
weigh out the required amount of urea on a good balance. To
do this, we need the gram molecular weight of urea. We can
find this out several ways. We can look it up in a CRC handbook
or similar source. Another way is to write out the formula and
add up all of the atomic weights of all the atoms in the molecule
times their compositional stoichiometry. This could be tedious
for a large molecule. We could look on the bottle, because most
chemical companies publish the formula weight on the bottle.
This formula weight may be different from the gram molecular
weight because the chemical might have waters of hydration
with it or ions to counterbalance charges on the chemical.
ATP, for example, is usually sold as Na2ATP 3H2O and its
formula weight is not the gram molecular weight of ATP alone.
Looking on the bottle seems like a winner, as we must go find
the bottle to weigh out the urea. We find that the formula
weight for urea is 60.08 g mol21. The mass of urea is found by
multiplying the moles by the gram molecular weight:
m 5 0:15 mol 3 60:08 g mol21 5 9:012 g
This is the amount you need to weigh on an accurate balance.
Chemical Foundations of Physiology II: Concentration and Kinetics
EXAMPLE 1.5.3 Blood Concentration of Hemoglobin
The blood content of hemoglobin is 15 g dL21. The molecular
weight of hemoglobin is 66,500 daltons. What is its average
molar concentration in the blood?
If the molecular weight is 66,500 daltons, then 1 mole has a
mass of 66,500 g. The question asks for the average concentration because hemoglobin is not uniformly distributed within
the blood, but is contained within red blood cells only.
Therefore, its concentration in the red blood cells exceeds its
average concentration in the blood. The concentration in
blood is given by
½Hb 5
15 g dL21 3 1 dL=0:1 L
5 0:00266 M 5 2:66 3 1023 M
66; 500 g mol21
These examples should enable you to calculate:
G
G
G
the number of moles in a given volume of solution of given
concentration;
the mass of material in a given volume of solution of given
concentration;
the concentration of solution containing a known mass of
material in a given volume.
EXAMPLE 1.5.4 Make a Dilution of a Stock Solution
Suppose we have a stock solution of 0.1 M MgCl2 and we want
to make up 50 mL of solution with a final concentration of 5 mM
MgCl2. How much of the stock solution should we add to a 50mL volumetric flask?
We use Eqn [1.5.3] directly here:
0:1 M 3 X mL 5 0:005 M 3 50 mL
Solving for X, we find X 5 2.5 mL
TABLE 1.5.1 Prefixes Used with Powers of Ten, in 103 Ratios
Atto
10218
Femto
10215
Pico
10212
Nano
1029
Micro
1026
Milli
1023
100
Kilo
103
6
Mega
10
Giga
10
9
Thus we can write:
Tera
12
10
DILUTION OF SOLUTIONS IS
CALCULATED USING CONSERVATION
OF SOLUTE
1 picoliter
(1 pL) 5 10212 L
1 picogram
(1 pg) 5 10212 g
1 picometer
(1 pm) 5 10212 m
1 nanoliter
(1 nL) 5 1029 L
1 nanogram
(1 ng) 5 1029 g
1 nanometer
(1 nm) 5 1029 m
1 microliter
(1 μL) 5 1026 L
1 microgram
(1 μg) 5 1026 g
1 micrometer
(1 μm) 5 1026 m
1 milliliter
(1 mL) 5 1023 L
1 milligram
(1 mg) 5 1023 g
1 millimeter
(1 mm) 5 1023 m
Often it is necessary or easier to prepare solutions
from more concentrated stock solutions by removing an
aliquot (a fraction of the solution) of the more concentrated solution and placing it in a volumetric flask.
We then add solvent to bring the volume up to the final
solution volume, as shown in Figure 1.5.1.
1 liter (1 L) 5 100 L
1 gram
(1 g) 5 100 g
1 meter
(1 m) 5 100 m
1 kilogram
(1 kg) 5 103 g
1 kilometer
(1 km) 5 103 m
Let V1 be the volume of the aliquot of the more highly
concentrated solution, with concentration C1. The amount
of solute in this aliquot is given by Eqn [1.5.2]:
½1:5:2
The prefixes mega-, giga-, and tera- are not typically
used for units of volume, mass, or distance, but often
find use with other units such as hertz or watts. There
are additional prefixes in the SI. These include:
centi
1022
deci
1021
deca
101
hecto
102
m 5 C 1 3 V1
This amount is also the amount in the final solution
with volume V2 and final concentration C2. Since the
amount in the aliquot is still in the final solution, we
can write
½1:5:3
C1 V1 5 C2 V2
61
62
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
1 Remove aliquot of known
2 Transfer aliquot to empty
3 Add water to volume;
volumetric flask; m1 = V1 C1
volume, V1; m1 = V1 C1
m1 = V2 C2 = V1 C1
Stock solution
Flask 1
Flask 2
Flask 1
Flask 2
Flask 1
Flask 2
Flask 1
Flask 2
FIGURE 1.5.1 Dilution of a stock solution. Flask 1 contains a stock solution of concentration C1. We remove a known volume, V1, with a calibrated
pipette and place it in an empty volumetric flask. We then add water, which adds solvent but no solute, to bring the volume up to the final volume.
The amount of solute in the second flask is the same amount that we added in the aliquot, V1, but now it is distributed in the volume V2.
CALCULATION OF FLUID VOLUMES
BY THE FICK DILUTION PRINCIPLE
The equation describing dilution of stock solutions can
be used to determine the volume of distribution of materials in the body. Suppose we inject a known amount
of a substance, Y, into a person and this substance Y
remains in the plasma because it cannot get out of the
vascular system and it cannot get into the cells suspended
in the blood. We wait a few minutes for Y to become
evenly distributed, and then take a sample of plasma and
measure Y’s concentration. Then we can calculate the
volume of the plasma as
amount
Volume 5
concentration
½1:5:4
m
V5
C
This is just a variant of Eqn [1.5.2] in which we solve
for V instead of C or m. In our particular case in this
example, we write
V 5 mY =½Y
where mY is the amount of substance Y and [Y] is its
concentration.
CHEMICAL REACTIONS HAVE
FORWARD AND REVERSE RATE
CONSTANTS
Suppose that we observe a simple chemical reaction
that can be described as
½1:5:5
A 1 B"C
where A, B, and C denote different chemicals and the
arrows indicate that the reaction proceeds in the direction of the arrow. The reaction actually consists of two
separate reactions:
A 1 B,C
A 1 B&C
½1:5:6
The top reaction is the forward reaction and the bottom
reaction is the reverse reaction. Both are characterized
by a rate constant. The rate constant gives the rate of
the reaction when the rate constant is multiplied by the
concentration of reactants:
Jf 5 kf ½A½B
Jr 5 kr ½C
½1:5:7
EXAMPLE 1.5.5 Estimation of Plasma Volume
Suppose that we inject a 50-kg person with 2 mL of a solution of
Evans’ Blue Dye, at 5 mg mL21. Evans’ Blue Dye is restricted to the
plasma because it tightly binds to a plasma protein, albumin, that
ordinarily does not escape from the circulation and which only
slowly is removed or added to the plasma. We wait 10 min and
then obtain a sample of blood and measure the concentration
of Evans’ Blue Dye in the plasma and find that it is 0.4 mg dL21.
What is the person’s plasma volume?
The amount of Evans’ Blue Dye that was injected is calculated as
minj 5 Cinj Vinj 5 5 mg mL21 3 2 mL 5 10 mg
The concentration of Evans’ Blue Dye after mixing was
0.4 mg dL21. We calculate its volume of distribution as
V5
10 mg
5 25 dL 5 2:5 L
0:4 mg dL21
Chemical Foundations of Physiology II: Concentration and Kinetics
where Jf is the forward reaction rate, Jr is the reverse
reaction rate, kf is the forward reaction rate constant,
and kr is the reverse rate constant, and [A], [B], and [C]
are the concentrations of the indicated reactants.
Although this form of the reaction rate is true for
elementary reactions, more complicated reactions could
have a form that appears to have little to do with the
overall reaction as written—its stoichiometric relation
or accounting of the number of each kind of molecule
that participates in the reaction. This is because complicated reactions occur with intermediary steps that may
involve chemicals that do not appear in the overall
balanced reaction. Nearly all enzymatic reactions, for
example, do not obey Eqn [1.5.7] because the reaction
rate is determined largely by the concentration of enzyme.
For the moment, we will consider only elementary
reactions that obey Eqn [1.5.7].
The rate of a reaction is the number of completed
reactions that take place per unit time. You should
recognize this as an extensive property. If we had twice
the volume of a solution, with the same concentrations
of reactants and products, we would have twice the
number of reactions taking place per unit time. So we
often convert reaction rates into intensive variables
by dividing by the volume. The forward rate is thus the
number of completed reactions per unit time per unit
volume. We express these numbers of reactions in terms
of moles, which are related by Avogadro’s number to a
real number of completed reactions. Thus the forward
rate constant has units of M21 s21. By similar reasoning,
the reverse rate constant in Eqn [1.5.7] has the units
of s21. The rate of change of reactants A and C can be
given as
½1:5:8
d½C
5 2 kf ½ A½B 2 kr ½C
dt
d½A
5 0 5 2kf ½A½B 1 kr ½C
dt
FIRST-ORDER RATE EQUATIONS SHOW
EXPONENTIAL DECAY
Some kinds of chemical reactions, such as decomposition
reactions, obey the equation
½1:5:11
The rate constants, kf and kr, are characteristic of the
reaction path involved and the experimental conditions
such as ionic strength and temperature of the reactants.
They typically do not vary with the concentrations of
A, B, and C. Thus, at equilibrium, the middle expression
in Eqn [1.5.9] is true for any set of [A], [B], and [C].
We rearrange this to get:
A-B
Often this reaction is strongly directed to the right,
meaning that kf .. kr. The reaction rate is approximated as
½1:5:12
J 5 2 kf ½A
Here we imagine that initially [B] 5 0 and there is no
reverse reaction. Some reactions occur with such completeness that the reverse reaction is negligible. We can
rewrite Eqn [1.5.12] as
d½A
5 2 kf ½ A
dt
This is called a first-order rate equation because the rate
of reaction is proportional to the first order or the concentration of reactant—it is proportional to the first
power of its concentration. We can separate variables
and integrate this equation, as follows:
½1:5:13
ðt
½1:5:14
0
ðt
d½A
5 2k dt
½A
0
In½A 2 In½A0 5 2 kt
½A 5 ½A0 e2kt
The last equation describes the concentration of A with
time—it decays exponentially. This equation is described
as a first-order exponential decay. Many reactions and
processes are described by this type of analysis, such as
radioactive decay and the disappearance of many different hormones from the circulation. This type of reaction
is characterized by its half-life, the time required for [A]
to fall from its initial value, [A]0, to one-half of its initial
value, [A]0/2. In this case, Eqn [1.5.13] gives
½A0
5 ½A0 e2kt1=2
2
kf ½A½B 5 kr ½C
Jf 5 Jr
kf
½C
5 Keq
5
½A½B
kr
where Keq is the equilibrium constant. From the units
of kf as M21 s21 and kr as s21, we see that Keq has the
units of M21.
d½A
5 2 kf ½ A½B 1 kr ½C
dt
The negative sign before Jf(5kf[A][B]) in the above
equation indicates that this reaction reduces the concentration of reactant A; the positive sign of Jr indicates that
this reaction flux adds to the concentration of reactant
A. Similar reasoning gives us the rate of change of [C].
At equilibrium, the concentrations of A, B, and C are
no longer changing. The values of [A], [B], and [C]
are altered from their original concentrations so that the
forward and reverse rates are equal:
½1:5:9
½1:5:10
½1:5:15
1
ln 5 ln e2kt1=2
2
2ln2 5 2 kt1=2
ln 2
t1=2 5
k
Thus the half-life of a first-order reaction is inversely
proportional to the first-order rate constant.
63
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
RATES OF CHEMICAL REACTIONS
DEPEND ON THE ACTIVATION ENERGY
The values of kf and kr depend on the reaction path
taken for the reaction, but Keq is not affected by the
reaction path. We shall not prove this result here but
will give some rationale for it. A more complete description is given in Appendix 1.5.A1.
The reactants (A and B) and the product, C, can be
viewed as possessing some degree of energy. This energy
is a potential energy that consists of the potential energy
of all of the interactions of their orbital electrons with
the positive nuclei. The set of nuclei has some spatial
arrangement which changes during the course of the
reaction. This is the essence of a chemical reaction, in
which the relative positions of the nuclei are altered.
The orbital electrons, of course, follow the nuclei so
that the energy of the ensemble changes during the
course of the reaction. We call this energy the potential
energy, and it includes the potential energy of the electrons and the nuclei and the kinetic energy of the
electrons, but does not include the kinetic energy of
the nuclei. We can plot this potential energy against the
“reaction coordinate,” which is the actual distance along
the minimum energy path from reactants to products.
An example of such a graph is shown in Figure 1.5.2.
The rate constants that govern the rates of reaction
depend on the energy required to reach the activated
complex intermediate between reactants and products.
Large activation constants are associated with small
rate constants. The relationship is expressed by the
Arrhenius equation:
–300
½1:5:16
ln k 5 ln A 2
Ea
RT
where k is the rate constant for the reaction, A is a preexponential factor that has to do with the orientation of the
reaction and not its temperature dependence, Ea is the
activation energy in J mol21, R is the gas constant
(58.314 J mol21 K21), and T is the absolute temperature (K). The equilibrium constant, however, depends
only on the initial and final energy levels. Note that the
Arrhenius equation means that a larger Ea would cause a
lower rate constant, and a smaller Ea would increase the
rate constant.
ENZYMES SPEED UP REACTIONS
BY LOWERING EA
It is possible for the reaction to take a different path.
For example, A and B could be absorbed onto the
surface of an enzyme. The forces that aid in this
binding have been discussed in Chapter 1.4: electrostatic interaction between ions in the substrate and
enzyme; hydrogen bonding; dipoledipole interactions; and London dispersion forces. This binding
to the enzyme changes the configuration of nuclei
and alters the energy of the activated complex. In this
way, the enzyme offers an alternative pathway for the
reaction that involves far less activation energy. This
increases the reaction rates without altering the final
energetics of reactants and products. Thus the rates
increase without changing the equilibrium constant.
Figure 1.5.3 illustrates this idea.
Activated complex
A–B
Activated complex
–350
Uncatalyzed Ea
Ea
–400
–450
Initial state
A+B
Final state
C
Potential energy
Potential energy (kJ mol–1)
64
Ea
Initial state
Intermediate
steps
ΔG
–500
0
200
400
600
800
1000
Distance along the reaction coordinate (pm)
Enzyme reduces
Ea
ΔG is unaltered
by the enzyme
Final state
Distance along the reaction coordinate
FIGURE 1.5.2 Potential energy along the reaction coordinate for
the reaction A 1 B-C. Reactants A and B are at low potential energy.
The activated complex is a form intermediate between reactants and
products, and can be attained by converting kinetic energy into
potential energy by a collision between A and B. The difference between
the energy of the activated complex and the reactants is the activation
energy that must be supplied for the reaction to proceed. The “reaction
coordinate” is the distance along the minimum free-energy path from
reactants to products.
FIGURE 1.5.3 The effect of an enzyme on the overall activation energy
for a reaction. The uncatalyzed reaction requires a large activation
energy, Ea, and so the reaction occurs slowly. The enzyme offers an
alternative path, often by breaking the reaction into a number of small
steps, so that the reaction occurs more quickly. Enzymes do not change
the overall energetics of the products with respect to the reactants (ΔG
in the figure is the change in free energy in the transition between
reactants and products) and so the equilibrium constant is unaffected.
Chemical Foundations of Physiology II: Concentration and Kinetics
THE MICHAELISMENTEN
FORMULATION OF ENZYME KINETICS
and dividing numerator and denominator by k1, we
obtain
Catalysis is the speeding up of a chemical reaction by a
chemical species that does not enter into the stoichiometry of the reaction. Enzymes are catalysts because they
speed up reactions without themselves being altered.
Enzymes typically bind the reactants and alter their
shape (the three-dimensional arrangement of all of the
atomic nuclei in the reactants) by virtue of their being
attracted to or bound to the surface of the enzyme. The
enzyme changes the reaction from a homogeneous
reaction in the solution to a heterogeneous reaction
occurring on the surface of the enzyme. In this process,
the enzyme itself is unchanged. It participates in the
reaction but does not show up in an accounting of
the reactants and products because it is unchanged.
½1:5:24
Figure 1.5.3 shows that enzymes speed up reactions by
providing an alternate path for the reaction to take, and
that this path requires less activation energy. Michaelis
and Menten provided a simple analysis of this reaction
path by imagining it to take place in two steps:
k1
E 1 S " E 2 Sk3 E 1 P
-
k2
where E is the enzyme, S is the substrate, ES is
the substrateenzyme complex, and P is the product.
This is a very simple reaction mechanism. The rate of
the enzyme-catalyzed reaction is defined by the rate
of product release:
½1:5:18
J 5 k3 ½E 2 S
If we know [ES] and k3, we can calculate the reaction
rate. Here k3 is in units of s21. If we keep [S] and [P]
constant, we can define a steady-state rate in which S
is converted to P at a constant rate. Under these conditions, we can solve for [ES]. The rate of change of
[ES] is given by
½1:5:19
½1:5:20
k1 ½E½S 5 ðk2 1 k3 Þ½E 2 S
Since the enzyme can exist in only two states, E and
ES, we have a conservation relation
½1:5:21
½Etotal 5 ½E 1 ½E 2 S
where [E]total is the total concentration of enzyme.
Inserting this relation for [E] in Eqn [1.5.20], we get
½1:5:22
k1 ð½Etotal 2 ½E 2 S½SÞ 5 ðk2 1 k3 Þ½E 2 S
Solving for [ES], we find
½1:5:23
½1:5:25
Jmax 5 k3 ½Etotal
Inserting this definition into Eqn [1.5.24], we finally
obtain
½1:5:26
J5
½E 2 S 5
k1 ½Etotal ½S
k2 1 k3 1 k1 ½S
From Eqn [1.5.18], the rate of the reaction is just
k3[ES]. Multiplying both sides of Eqn [1.5.23] by k3,
Jmax ½S
Km 1 ½S
where Km is a newly defined constant called the
MichaelisMenten constant. It is given as
½1:5:27
Km 5
k2 1 k3
k1
The units of k2 and k3 are both in s21, whereas the unit
of k1 is M21 s21; thus, the unit of Km is M. If k3{k2, Km
approximates the value of k2/k1, which is the dissociation constant for binding of S to the enzyme. It can be
obtained experimentally as the value of the substrate
concentration at which the enzyme exhibits one-half
maximal velocity. This can be seen from Eqn [1.5.26]
by inserting J 5 1/2Jmax and finding that [S] 5 Km when
the rate of the reaction is one-half maximal.
The saturation curve for a MichaelisMenten type reaction is shown in Figure 1.5.4. In this case, Jmax was
8 μmol min21 mL21. At one-half of this maximal velocity,
the substrate concentration was 1.5 mM.
The curve shown in Figure 1.5.4 often lacks sufficient
points to accurately extrapolate the observed velocity
Vmax
10
d½E 2 S
5 k1 ½E½S 2 k2 ½E 2 S 2 k3 ½E 2 S
dt
At steady state, d[E 2 S]/dt 5 0, so that
k3 ½Etotal ½S
ðk2 1 k3 Þ=k1 1 ½S
The maximum velocity occurs when all of the enzyme is
present as [ES]. The maximum velocity or rate of the
reaction is thus given as
Velocity (μmol mL–1 min–1)
½1:5:17
J 5 k3 ½E 2 S 5
8
1/2V max
6
Km is the concentration of
S when V is 1/2 maximum
4
2
0
0
5
10
15
[S] (mM)
20
25
FIGURE 1.5.4 Saturation curve for an enzyme that obeys Michaelis
Menten kinetics. The velocity at low [S] is nearly linear with [S] but quickly
levels off. This behavior is called saturation kinetics. The concentration of
substrate at one-half maximal velocity (5one-half saturation) is equal to
the Km, the MichaelisMenten constant.
65
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
to the maximal velocity. To aid in this graphical determination of enzyme kinetics, the MichaelisMenten
equation is transformed. If we take the inverse of
Eqn [1.5.26], we obtain
J5
Jmax ½S
Km 1 ½S
1 Km 1 ½S
5
J
Jmax ½S
½1:5:28
1
1
Km 1
5
1
J
Jmax
Jmax ½S
This equation suggests that a plot of 1/J versus 1/[S]
should be linear with an intercept on the 1/J axis of
1/Jmax and a slope of Km/Jmax. The intercept on the
1/[S]-axis is 21/Km. Such a plot is called a doublereciprocal plot, also known as a LineweaverBurk plot.
An example for the results shown in Figure 1.5.4 is
shown in Figure 1.5.5.
Throughout this discussion of enzyme rates, we have
used J as the symbol of the rate of product release, or
the rate of the reaction. J has the units of moles per unit
volume, per unit time. Often this is taken to be the
same as the reaction velocity, and the usual symbol
in the MichaelisMenten equation is V, and not J.
Sometimes, the reaction velocity in these units is
normalized by dividing by the enzyme concentration,
so that the reaction velocity is given as a specific activity
of the enzyme, in units of moles of reaction per unit
enzyme per unit time. These various representations of
the velocity all obey the same general form of equations
and can be subjected to the same analysis.
1/V intercept = 1/Jmax
0.4
Slope = Km/Jmax
0.3
0.2
X-intercept = –1/Km
0.1
–1.0 –0.5
0.0
0.5 1.0
1/[S]
PHYSIOLOGY IS ALL ABOUT SURFACES
As noted above, enzyme catalysis derives from the
reaction occurring on the surface of the enzyme rather
than in homogeneous solution. Physiology is all about
surfaces and their interactions. Proteins have complicated three-dimensional shapes that closely appose
the surfaces of some materials and not others—they are
specific. Sometimes the fit is very close and therefore
the binding strength is very high—the two surfaces form
a tight association. Which proteins stick to which other
proteins, or which other substrates, or ligands, determines the activity of the proteins, which determines the
activities of the cells, and then the organs, and, finally,
the organism. Ultimately, physiology is all about what
happens on the surfaces of molecules.
SUMMARY
The concentration of a solute in solution is the amount
of that solute per unit volume of solution. It can be
expressed as the mass of the solute per unit volume or
the number of moles of solute per unit volume. A mole
of any substance is Avogadro’s number of particles.
Avogadro’s number originates in the ratio of the mass
of a carbon-12 atom to 12 g. The dalton is defined as
1/12 of the mass of a carbon-12 atom. The number of
daltons in 1 g is Avogadro’s number, 6.02 3 1023.
The concentration in molar is the number of moles per
liter of solution. Small concentrations use established
prefixes to describe them, in increments of 1000. A millimolar solution is 1023 M, micromolar is 1026 M,
nanomolar is 1029 M, and picomolar is 10212 M. These
same prefixes are used for units of g, L, and m.
The relationship among concentration, amount of solute,
and volume of solution can be used to determine the
volume of physiological fluids. Evans’ Blue Dye is an
example of a solute that can be used to estimate plasma
volume, because the dye enters the plasma but cannot
leave it easily.
0.5
1/Velocity
66
1.5
2.0
2.5
FIGURE 1.5.5 LineweaverBurk plot. Values of 1/[S] are plotted along
the abscissa while values of 1/J are plotted on the ordinate. Here J is
the velocity, or rate, of the enzyme reaction and [S] is the substrate
concentration. The slope of the line is Km/Jmax, where J is the enzyme
flux enzyme velocity and Jmax is the theoretical maximal rate. Km is
the MichaelisMenten constant. It is also determined as the
X-intercept 5 21/Km. The Y-intercept is 1/Jmax. Deviations from linearity
on the LineweaverBurk plot suggest that the enzyme does not obey
MichaelisMenten kinetics.
Elementary chemical reactions have forward and reverse
rate constants that govern the rate of conversion in
either the forward or reverse reaction. The rates of
reaction have the units of moles per unit time per unit
volume of solution. The ratio of the forward and reverse
rate constants is the equilibrium constant. Conversion
of reactants to products requires an activation energy,
and because kinetic energy increases with temperature,
the rate of the reaction also varies with the temperature.
The relationship between temperature and rate is
described by the Arrhenius equation, which incorporates
the activation energy, Ea.
Enzymes speed chemical reactions by altering the path of
the reaction by allowing it to proceed on the surface
of the enzyme. Thus enzymes convert homogeneous reactions in the fluid phase to heterogeneous reactions on the
surface of the enzyme. The alternate path reduces the activation energy for the reaction, thereby allowing it to proceed quicker.
Chemical Foundations of Physiology II: Concentration and Kinetics
The MichaelisMenten formulation of enzyme kinetics
describes the steady-state rate of enzyme reactions and
is derived for a particular kind of enzyme mechanism.
Nevertheless, the resulting equation often fits many
enzyme-catalyzed reactions. It is given as
J5
Jmax ½S
Km 1 ½S
where J is the reaction flux or velocity, in units of moles
of completed reaction per unit time per unit volume,
Jmax is the asymptotic maximal rate achievable extrapolated at infinite concentration, Km is the Michaelis
Menten constant, equal to the concentration of substrate
at half maximal velocity, and [S] is the substrate concentration. This equation describes saturation kinetics.
It can be analyzed more easily using the inverse of the
equation, rearranged, to give
1
1
Km 1
5
1
J
Jmax
Jmax ½S
Plots of 1/J against 1/[S] yield 1/Jmax as the intercept on
the abscissa and 21/Km as the extrapolated intercept on
the ordinate.
REVIEW QUESTIONS
1. What is a dalton? What is meant by molecular
weight?
2. What is a mole? What is meant by the gram
molecular weight? How would you determine
the gram molecular weight of small compounds?
3. Write the relationship among concentration,
volume, and amount.
4. What is meant by “first-order reaction”?
5. How do you calculate the half-life of a reaction?
6. In the plot of potential energy against reaction
coordinate, what is meant by “reaction coordinate”? What units does it have?
7. What is the activation energy?
8. Why do reaction rates generally depend on the
temperature?
9. How could you determine the activation energy
for a reaction?
10. In general, how do enzymes speed up biochemical reactions?
11. How would you determine Km and Vmax for an
enzyme?
APPENDIX 1.5.A1 TRANSITION STATE
THEORY EXPLAINS REACTION RATES
IN TERMS OF AN ACTIVATION ENERGY
TRANSITION STATE THEORY CALCULATES THE
POTENTIAL ENERGY OF REACTANTS AS A
FUNCTION OF SEPARATION
Transition state theory gives an expression for the rate
constant of a chemical reaction by applying statistical
mechanics to quantum mechanical calculations of various configurations of reactant and product. In this
procedure, a collection of nuclei with their attendant
electrons is treated as a “supermolecule.” Quantum
mechanical calculations are performed in which the
potential energy of the supermolecule is calculated as a
function of the relative positions of the nuclei. An exact
solution requires solving for the total energy including
the kinetic energy terms of all of the nuclei and electrons
and the potential energy terms for all electronelectron,
electronnucleus, and nucleusnucleus pairs. Since this
solution is extremely difficult, some simplifying assumptions are made. One is that the electronic motion is
extremely rapid compared to translation of the nuclei
and that the electrons adjust instantly to any change in
the positions of the nuclei. Thus the energy of the electrons and potential energy of the nuclei can be calculated
as if the nuclei were at rest. The energy calculated in this
way includes everything but the kinetic energy of the
nuclei. It is called the potential energy even though it
includes the kinetic energy of the electrons.
POTENTIAL ENERGY CAN BE GRAPHED
AGAINST SEPARATION IN A SINGLE MOLECULE
The potential energy of a configuration of nuclei with
their electrons may be represented as a point on an
f-dimensional surface in an f 1 1-dimensional space,
where f is the number of independent variables required
to specify the relative positions of all nuclei. For a
diatomic molecule (like H2 or HF), f 5 6 because we
need six variables to specify the Cartesian coordinates
of the two nuclei. However, three of these can be
considered to locate the center of mass of the molecule
and another two specify the orientation of the molecular axis. As these do not really concern us, we have
only one remaining variable, the internuclear distance,
to specify the relative positions of the two nuclei.
Since f 5 1, we have a potential surface which is the
one-dimensional potential energy curve in the twodimensional graph for a diatomic molecule (H2) as
shown in Figure 1.5.A1.1.
POTENTIAL ENERGY CAN BE GRAPHED
AGAINST NUCLEI SEPARATION IN A CHEMICAL
REACTION
In a configuration of three nuclei, the potential energy
surface can be shown as a fixed-angle surface, where the
angle is defined as the angle between one bond and
the approaching reactant, as shown in Figure 1.5.A1.2.
This allows us to plot the potential surface on paper;
otherwise we need another physical dimension. The entire
potential surface consists of an infinite number of these
fixed-angle surfaces, one for each angle of approach.
Such a fixed-angle surface is shown in Figure 1.5.A1.3
for the reaction
F 1 H 2 H0 -F 2 H 1 H0
The surface itself is three-dimensional. What Figure 1.5.
A1.3 shows is the two-dimensional projection of the
surface onto the plane of the paper. Here every point
on a given line has the same potential energy. Thus
these lines represent potential energy contours in much
67
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
broken. The horizontal cross-section at “Z” gives the
potential energy dependence of FH on its bond
distance.
+300
+200
Potential energy (kJ mol-1)
68
THE ACTIVATED COMPLEX IS A
METASTABLE COMPLEX OF REACTANTS
+100
0
–100
–200
–300
–400
–500
0
100
200
300
400
rHH' Internuclear distance (pm)
500
FIGURE 1.5.A1.1 Potential energy diagram for a diatomic molecule, H2.
Potential energy is plotted as a function of the separation of the two H
nuclei. The average bond length corresponds to the minimum energy
level, which in this case occurs at about 80 pm. The horizontal lines
indicate quantum vibrational states. Rotation of the molecule alters the
potential energy profile due to a centrifugal effect (rotation stretches the
bond length). Adapted from J.W. Moore and R.G. Pearson, Kinetics and
Mechanism, John Wiley & Sons, New York, NY, 1981.
C
135°
A
B
FIGURE 1.5.A1.2 Fixed angle of approach of reactant C to the diatomic
molecule AB. The potential energy surface varies with the angle of
approach.
the same way as contour lines on a topographic map
represent lines of equal altitude.
In the contour map for the reaction F 1 H 2 H0 F 2 H 1 H0 , several regions are labeled. Region “W”
corresponds to separated F, H, and H0 nuclei and this is
a plateau region where potential energy varies little with
the distance between the nuclei. At region “X,” F is far
removed from HH0 and the potential energy, U, is
affected little by changes in rFH, the distance between
the F nucleus and the H nucleus. The effect of rHH on
U is that shown earlier for the diatomic molecule, so
that a vertical cross-section at “X” would show a deep
valley with steep sides. At region “Y,” all three nuclei are
close together and the potential energy has increased
due to van der Waals repulsion. A collision of F with
HH0 thus corresponds to the movement of the configuration of nuclei from region “X” to region “Y” where
potential energy is increased. If the reaction is completed, then the configuration moves on to region “Z.”
In this region, rFH is short and rHH’ is long, indicating
that a bond has formed between FH and the HH is
The movement of the configuration of nuclei from X to
Y to Z in Figure 1.5.A1.3 traces a completed reaction
from the initial reactants. At the point Y, there is a
saddle point represented by higher potential energy, in
this case, than for either reactants or products. The configuration of nuclei at this point is called the activated
complex which is said to be in the transition state.
Here all of the nuclei are close together, forming a
complex. The path of steepest ascent from reactants to
activated complex and on to products is called the
reaction coordinate. This path is perpendicular to each
contour it crosses. It is the minimum energy path from
reactants to products.
Recall that the potential energy surface is just that—a
potential energy. Collisions between reactants can overcome the potential energy barrier between the reactant
and the activated complex if there is sufficient kinetic
energy of the molecules that collide. The potential
energy can be plotted against the distance along the
reaction coordinate. This is the familiar diagram
encountered in general chemistry, without any kind of
explanation of what the reaction coordinate actually is.
For our example reaction, F 1 HH0 -FH 1 H0 , the
potential energy versus reaction coordinate plot is
shown in Figure 1.5.A1.4. This plot is the conceptual
origin of Figure 1.5.2 in the text.
REACTIONS GENERALLY DO NOT FOLLOW
A MINIMUM ENERGY PATH
Reactions in general do not follow the reaction coordinate. Two alternative trajectories are shown in Figure 1.5.
A1.5. Because the HH0 bond vibrates, the configuration
of all three nuclei while F approaches HH0 oscillates.
In Figure 1.5.A1.5A, the reaction ascends near to the
activated complex but does not go to completion.
This trajectory represents a nonreactive, inelastic collision
between F and HH0 . In this case, there is a transfer
of energy from translational kinetic energy to vibrational
energy. In Figure 1.5.A1.5B, a completed reaction is
shown. Here the oscillations correspond to vibrations
of reactant HH0 and product FH.
THE TRANSITION STATE THEORY SAYS THAT
THE RATE CONSTANT VARIES WITH THE
EXPONENT OF THE ACTIVATION ENERGY
The derivation of a rate equation from the transition
state theory is a bit complicated and we will not
attempt it in any detail. However, an elementary
understanding of it can be provided by thinking of the
activated complex as a separate, transient species.
Then the rate of completed reaction will be proportional to the amount of activated complex, and the
rate per unit volume will be proportional to its
concentration. If we imagine that the reactants and
+200
l
mo
(kJ
y
g
ner
–1)
+300
W
+100
0
ia l e
ent
Pot
-100
W
-200
-300
This point is H + FH
-400
500
Potential energy
isoclines (kJ mol–1)
Z
400
-510
-470
W
-440
This point is the
activated complex
+100
300
200
-290
-200
-100
0
100
0
100
200
300
400
rFH Internuclear distance (pm)
-500
0
-400
X
-300
Y
-100
X
y
rg
J
+200
-320
+300
-410
-380
-350
-200
rHH' Internuclear distance (pm)
This point is H + F + H
Z
-500
-1)
ol
m
(k
ne
e
al
ti
en
t
Po
This point is the reactants,
F + H-H
FIGURE 1.5.A1.3 Potential energy contour diagram for a fixed angle (180 ) for the reaction F 1 HH0 -FH 1 H0 . The lines represent a set of points of
equal potential much like contour lines on a topographic map represent a set of points of equal altitude. The value of the potential energy (in
kJ mol21) for each line is indicated. Point W lies on a plateau of high potential energy corresponding to dissociated F, H, and H0 nuclei. X corresponds
to the configuration in which F is far from HH0 ; it is at a potential energy minimum. Y is the saddle point at which all three nuclei are close; it
corresponds to the activated complex. Z corresponds to the configuration in which H0 is far from FH. It represents the products of the completed
reaction. The graph at the right is a vertical cross-section through the three-dimensional surface at point X. The graph at the top is a vertical crosssection (orthogonal to the one at the right) through the three-dimensional surface at point Z. Adapted from J.W. Moore and R.G. Pearson, Kinetics and
Mechanism, John Wiley & Sons, New York, NY, 1981.
–300
the activated complex are in equilibrium, then the rate
constant for the overall reaction will be proportional
to the equilibrium constant.
Activated complex
Y
Potential energy (kJ mol–1)
–350
½1:5:A1:1
Ea
–400
–450
Initial state
X
Final state
Z
–500
0
200
400
600
800
1000
Distance along the reaction coordinate (pm)
FIGURE 1.5.A1.4 Potential energy along the reaction coordinate for the
reaction F 1 HH0 -FH 1 H0 . Reactants F 1 HH0 are at a low potential
energy. The activated complex is at higher potential energy, which can be
attained by converting kinetic energy into potential energy through a
collision of F and HH0 . The reactants are at lower potential energy. The
difference between the energy of the activated complex and the reactants
is the activation energy that must be supplied for the reaction to proceed.
Kr 5 κ
kT K
h
There are a lot of k’s in this equation. Kr is the rate constant for the reaction; κ is a transmission coefficient
that tells us what fraction of activated complexes goes
on to complete the reaction; k in kT is Boltzmann’s constant, which is equal to the gas constant per molecule,
or R/No, the gas constant divided by Avogadro’s number; h is Planck’s constant; and K* is a constant that has
the form of an equilibrium constant for the formation
of the activated complex, but strictly speaking it is not
an equilibrium constant. The equilibrium constant is
related to the change in free energy for the reaction (see
Chapter 1.7) according to
½1:5:A1:2
2ΔG
RT
K 5 e
where ΔG* is the free-energy change per mole for the
formation of the activated complex from the reactants.
This energy change is identified with the activation
energy, Ea, as described in Figure 1.5.A1.4.
500
400
rHH'' Internuclear distance (pm)
300
Y
X
0
X
100
400
300
200
Y
(B)
Z
200
(A)
Z
100
rHH'' Internuclear distance (pm)
500
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
0
70
0
100
200
300
400
rFH Internuclear distance (pm)
500
0
100
200
300
400
rFH Internuclear distance (pm)
500
FIGURE 1.5.A1.5 Hypothetical trajectories of reactions. (A) An inelastic collision that does not result in a completed reaction. (B) A completed reaction.
The oscillations are due to vibration of the molecule in which bond length oscillates around an average value.
In 1884, van’t Hoff proposed that the temperature dependence of equilibrium constants could be described by
½1:5:A1:3
d ln Keq
ΔG0
5
dT
RT 2
It was discovered shortly thereafter that the rates of
chemical reactions approximately double for every 10 C
increase in temperature. By analogy to the van’t Hoff
law, Arrhenius proposed in 1889 that the temperature
dependence of the rate constant could be given as
½1:5:A1:4
d ln Kr
ΔE0
5
dT
RT 2
The integrated form of this equation is similar to what
we see from transition state theory:
½1:5:A1:5
Kr 5 Ae2Ea =RT
where A is the preexponential factor that incorporates
the transmission coefficient. This is close to what one
would expect by inserting the definition of K* into
Eqn [1.5.A1.1]. The natural log of both sides gives
½1:5:A1:6
Ea
ln Kr 5 ln A 2
RT
Thus the plots of the logarithm of the rate constant
against 1/T should be linear with a slope 5 2Ea/R. Such a
plot is an Arrhenius plot. Although transition state theory
does not predict a perfectly linear relation between ln Kr
and 1/T, most reactions obey Eqn [1.5.A1.6] well.
more complicated because of the large number of
atomic nuclei involved in typical biochemical reactions.
However, the idea of the reaction coordinate and activation energy remains. By absorbing reactants onto their
surfaces, enzymes completely alter the potential energy
surfaces of a reaction. The net effect is to lower Ea.
According to Eqn [1.5.A1.6], lower activation energies
mean larger rate constants. Thus enzymes and other catalysts increase the speed of reactions by providing an
alternate mechanism such that Ea is reduced. This idea
is depicted graphically in Figure 1.5.3 in the text.
APPENDIX 1.5.A2 UNIDIRECTIONAL
FLUXES OVER A SERIES OF
INTERMEDIATES DEPEND ON ALL OF
THE INDIVIDUAL UNIDIRECTIONAL
FLUXES
UNIDIRECTIONAL FLUXES DIFFER FROM NET
FLUXES
Consider the diagram shown in Figure 1.5.A2.1. Each
node, indicated by a dot, represents a state, compartment, or individual chemical species in a series.
Movement between nodes indicates either a flux of
material between compartments or the transition of one
chemical species into another, or the transition between
intermediate states of an enzyme, for example.
We will treat all of the transitions between states as
being an elementary reaction so that the flux obeys the
relation
THE ACTIVATION ENERGY DEPENDS
ON THE PATH
½1:5:A2:1
The potential energy surfaces described in Figure 1.5.
A1.3 pertain only to the collisions between the molecules HH0 and F, which is a simple system. Similar
potential energy surfaces could be described for more
complicated reactions, but the surface becomes much
where Jij is the flux from node i to node j, Ni is the population of node i, and αij is a pseudo-first order rate
constant. It is called a pseudo-first order rate constant
because it may incorporate the concentration of a ligand
if the transition between node i and node j requires it.
Jij 5 αij Ni
Chemical Foundations of Physiology II: Concentration and Kinetics
J12
Jnet
N1
J21
Jnet
J23
N2
J32
N3
FIGURE 1.5.A2.1 Chemical species can be transformed into various
intermediates here represented as three nodes, N1, N2, and N3. The
fluxes between the nodes are indexed with the source node first and
sink node second. Alternatively, the nodes can represent compartments
into which the same chemical is transported, or different states of the
same object, such as an enzyme.
At steady state, there could be a throughput of material
such that sum flux, Jnet, of material enters node 1 and
the same flux exits the end, at node 3 in this case. At
steady state, this requires that the net flux between any
two nodes is the same, equal to Jnet. However, there can
be a higher unidirectional flux of material from node 1
to node 2 than this net flux. All that steady state
requires is that
½1:5:A2:2
Jij 2 Jji 5 Jnet
for each ij pair. The unidirectional flux, Jij, can be determined by somehow labeling the population of state i
and seeing how fast they are converted to state j. One
way this can be done is by adding a pulse of radioactive
material of state i, and watching its conversion to state j.
We will note the population of radioactive tracer in each
state or compartment with an asterisk, which is added
into state i alone. We can write the following sets of
equations that govern the time change in the population
of radioactivity in each state:
½1:5:A2:3
Measuring the unidirectional flux from state 1 to state 3
requires a similar condition as the unidirectional flux
from state 1 to 2; that is, there must be negligible back
flux during the measurement. In this case, this means
that N3*/N3 is negligible. We also require a steady state.
In this case, steady state means dN2*/dt 5 0. What we
are searching for is an overall unidirectional flux that
obeys the relation:
dN3
N
5 J13 1
dt
N1
½1:5:A2:5
Because N3*/N3 0 as a criterion for measuring unidirectional flux, the bottom part of Eqn [1.5.A2.3]
becomes
dN3
N
5 J23 2
dt
N2
½1:5:A2:6
If dN2*/dt 5 0 as a condition of steady state, then by
the middle part of Eqn [1.5.A2.3] we can write
N1
ðJ21 1 J23 Þ N2
5
J12
N1
N2
½1:5:A2:7
Substituting in for N2*/N2 from Eqn [1.5.A2.3] into
Eqn [1.5.A2.7], we obtain
dN3
J12 J23 N1
5
dt
ðJ21 1 J23 Þ N1
½1:5:A2:8
dN1
N
N
5 2 J12 1 1 J21 2
dt
N1
N2
Comparison of Eqn [1.5.A2.5] with Eqn [1.5.A2.8]
allows us to identify J13 as
dN2
N
N
N
5 J12 1 2 ð J21 1 J23 Þ 2 1 J32 3
dt
N1
N2
N3
½1:5:A2:9
dN3
N
N
5 J23 2 2 J32 3
dt
N2
N3
This result has an intuitive interpretation. It says that
the unidirectional flux from node 1 to node 3 is the
flux from node 1 to node 2 times the proportion of
the flux that goes on to node 3. This proportion is the
flux from node 2 to node 3 divided by the total flux
away from node 2: the sum of J21 and J23.
The idea about unidirectional flux is that there is
no back flux. Thus, for the transition between state 1
and state 2, we can measure unidirectional flux J12 only
when N2* is negligible. For the top equation in
Eqn [1.5.A2.3], we have
dN1
N
N
5 2 J12 1 1 J21 2
dt
N1
N2
N2
½1:5:A2:4
is called the specific activity of the added radioactivity.
The units of J12 are in units of N per unit time.
N2
dN1
dt
0
5 2 J12
N1
N1
dN1 N1
J12 5 2
dt
N1
The last equation in Eqn [1.5.A2.4] defines what is
meant by a unidirectional flux obtained by tracer
means. In this case, dN1*/dt is negative—the amount of
tracer in state 1 decreases with time—and thus the flux
J12 is positive. The factor N1*/N1 in the denominator
J13 5
J12 J23
J23
5 J12
ð J21 1 J23 Þ
ðJ21 1 J23 Þ
It can be seen readily that we could just as easily added
radioactivity at node 3 and watch its appearance in
node 1. From the symmetry, we can write J31 as
½1:5:A2:10
J31 5
J32 J21
J21
5 J32
ðJ21 1 J23 Þ
ðJ21 1 J23 Þ
These results are completely general. Suppose that the
sequence of nodes, states, or compartments was longer
than just three, as shown in Figure 1.5.A2.2. We could use
the unidirectional flux to reduce the diagram to one fewer
states, and then do so again using the same principle that
we discovered here. The results for J14 and J41 are given as
J13 J34
5
J14 5
ðJ31 1J34 Þ
½1:5:A2:11
J23
J34
J12 J23 J34
ðJ21 1 J23 Þ
5
J21
J32 J21 1J21 J34 1J23 J34
J32
1J34
J21 1 J23
J12
71
72
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
J12
Jnet
N1
Jnet
J21
J23
N2
J32
J34
N3
J31
Jnet
N4
J34
J13
N1
J43
N3
J43
J41
J54
N4
J54
J54
A
P
B
P
Jnet
FIGURE 1.5.A3.1 Two-pool sequential model. Pool A of size A is
converted to pool B of size B with a steady-state rate of P.
Jnet
turnover or the rate of renewal of the pool. The fractional
turnover rate is the fraction of the total pool which is
replaced per unit time. If A is the size of pool (in g or
moles) and P g or moles of A are renewed each minute,
then the fractional turnover rate is given as
N5
J45
N4
P
N5
J45
J14
N1
Jnet
J45
N5
FIGURE 1.5.A2.2 Unidirectional fluxes over a series of intermediate
states. The flux between states 1 and 3 can be replaced by two
unidirectional fluxes. The process can be repeated over any number of
intermediates.
J21
J32
J43 J31
J43 J32 J21
ð J211J23 Þ
5
J41 5
5
J21
ð J31 1J34 Þ
J32 J211 J21 J34 1J23 J34
J32
1J34
J211J23
½1:5:A2:12
J43
In this way, the unidirectional flux over any number
of intermediate states can be calculated from the set of
individual unidirectional fluxes.
APPENDIX 1.5.A3 SIMPLE
COMPARTMENTAL ANALYSIS
THE AGGREGATE OF A CHEMICAL SPECIES
CONSTITUTES ITS BODILY “POOL”
Compounds in the body are normally in a dynamic state
in which materials are converted from precursors to
products. For example, there is a steady state between the
phospholipids in the cytoplasm and in the mitochondria,
with continuing interconversion between both. The aggregate of the cytoplasmic phospholipids in the its pool, as
is the aggregate of the mitochondrial phospholipids.
A “pool” or “compartment” is defined as the set of molecules of a specific compound or group of compounds
found in a specific part of the organism. For example, we
may speak of “liver glycogen” or “muscle glycogen” or
“liver phospholipid” or “plasma phospholipid.” In investigating these pools, we are most interested in the identity
of the pools, their size, and the rate at which they
exchange material with other pools. Radioactive labeling
is an effective way of determining these parameters.
Generally these pools change only slowly with time.
Their maintenance at a fairly steady level is a consequence
of homeostasis, the maintenance of a constant internal
environment, which is the hallmark of physiological
systems. Though blood glucose or blood [Ca21] is maintained fairly constant, there is always material entering
and leaving the plasma pool.
THE TURNOVER DESCRIBES THE RATE
OF EXCHANGE OF A POOL
The rate of exchange of material is the rate at which
material enters or leaves the pool, and can be given in
units of amount per unit time. This is also called the
½1:5:A3:1
k1 5
P
A
where the units of k are reciprocal time. Here we
consider pool A of size A which is a precursor of pool B
of size B, as shown in Figure 1.5.A3.1. We will assume
that the pools are in a steady state (their sizes do not
change with time); there is rapid and uniform mixing
within the pools and that the rate of transfer does not
change with time. Suppose we inject an amount of
radioactive compound A, which we denote as A*, into
pool A at time t 5 0. If the radioactivity mixes well and
uniformly, we may describe the change in radioactivity
in pool A as
dA
A
52P
dt
A
This equation derives from the idea that the radioactive
portion of pool A will be turned over in proportion to
its concentration in pool A. Thus when P unit of pool A
turns over per unit time, PA*/A unit is radioactive and P
(A 2 A*)/A unit is not radioactive. The quantity A*/A is
called the specific activity of pool A, denoted as SA.
Equation [1.5.A3.2] can be rewritten as
½1:5:A3:2
½1:5:A3:3
1 dA
P
5 2 SA
A dt
A
Since A is constant under steady-state conditions, we
may rewrite Eqn [1.5.A3.3] as
½1:5:A3:4
dðA =AÞ
P
5 2 SA
dt
A
dSA
5 2 k1 SA
dt
Here we have used the definition of the fractional turnover rate given in Eqn [1.5.A3.1]. The last equation is of
the form of first-order decay and is easily integrated to
give
½1:5:A3:5
SA 5 SA0 e2k1 t
where SA0 is the specific activity of pool A at time t 5 0.
Plots of ln SA against time will allow the calculation of
the fractional turnover rate as the negative of the slope.
Extrapolation back to time zero gives the pool size: if
A*/A is known, and the amount of radioactivity
injected, A*, is known, then A can be calculated. It is
important to note that the determination of the specific
Chemical Foundations of Physiology II: Concentration and Kinetics
activity, SA 5 A*/A, does not require knowing A; it does
require knowing A* and A in an aliquot of the pool.
Thus the specific activity can be determined even if the
pool size A cannot be directly measured. The validity
of estimating pool sizes in this way depends on
the assumptions of uniform and rapid mixing of the
radioactive label with the endogenous material.
Integration from t 5 t to t 5 t gives
dðρ SB Þ
dB
5 P SA 2 P SB
dt
1 dB
P
5 ðSA 2 SB Þ
B dt
B
dB =B
5 k2 ðSA 2 SB Þ
dt
dSB
5 k2 ðSA 2 SB Þ
dt
We can rewrite the last part of this equation as
½1:5:A3:8
dSB
1 k2 SB 5 k2 SA
dt
This equation cannot be solved by integration directly,
because SA is a function of time and we don’t know SB
as a function of time. We can multiply both sides by an
integrating factor, ρ, such that
½1:5:A3:9
dðρSB Þ 5 ρ dSB 1 ρ k2 SB dt
We choose ρ such that ρSB is an exact differential, so
that
½1:5:A3:10
dðρSB Þ 5 ρ dSB 1 SB dρ
Comparison of Eqns [1.5.A3.9] and [1.5.A3.10] indicates that
½1:5:A3:11
½1:5:A3:16
SB ek2 t
5
k2 SA0 ðk2 2k1 Þt
ðe
21Þ
k2 2 k1
k2
SA ðe2k1 t 2 e2k2 t Þ
k2 2 k1 0
We have now derived equations for SA and SB for the
conditions shown in Figure 1.5.A3.1. The graph of ln SA
versus time will give ln SA0 as the intercept and 2 k1 as
the slope. Eqn [1.5.A3.6] shows that the maximum of
the graph of ln SB will occur with SA 5 SB, because at
this point dB*/dt 5 0, which means dSB/dt 5 0 and
therefore d ln SB/dt 5 0. P may be obtained from A and
k1. Figure 1.5.A3.2 shows the results of calculations for
Eqns [1.5.A3.5] and [1.5.A3.16] for assumed values of
A, A*, k1 and k2. The plot of ln SA gives an intercept
of 9.2103. Thus SA0 5 10,000 cpm μmol21. Since the
amount of injected radioactivity was 1 3 108 cpm, we
can calculate the pool size, A, as
A 5 A =SA0 5 1 3 108 cpm=104 cpm μmol21
5 104 μmol 5 0:02 mol; thus
A 5 0:02 mol
The fractional turnover rate is given as the negative of
the slope: k1 5 0.02 min21. The turnover of the pool at
steady state can be calculated as
P 5 k1 A 5 4 3 1024 mol min21 ; P 5 4 3 104 mol min1
ln SA0 = 9.2103
9.0
ρ5e
k2 t
dðρ SB Þ 5 ρ k2 SA dt
2k1 t
dðρ SB Þ 5 k2 e SA0 e
k2 t
ln SA vs Time
ln SB vs Time
8.5
ln SA or ln SB
ρ dSB 1 ρ k2 SB dt 5 ρ k2 SA dt
8.0
7.5
7.0
–k1 = –0.0200
6.5
6.0
Integration of this equation is now possible because
the left-hand side is a function of ρSB alone and the
right-hand side is a function of t alone. Substitution of
ρ from Eqn [1.5.A3.12] and for SA from Eqn [1.5.A3.5]
gives
½1:5:A3:15
k2 SA0 ðk2 2k1 Þt t
e
j
k2 2 k1
0
SB 5
9.5
We have chosen ρ so that Eqn [1.5.A3.9] is valid,
so we can substitute d(ρSB) for the left-hand side of
Eqn [1.5.A3.13]:
½1:5:A3:14
5
dρ 5 ρk2 dt
Multiplying both sides of Eqn [1.5.A3.8] by this multiplication factor gives
½1:5:A3:13
ρ SB j
0
Solution of this equation gives
½1:5:A3:12
0
t
Dividing both sides by the exponent on the left finally
leaves us with
We can divide both sides of the equation by B to obtain
½1:5:A3:7
5 k2 ek2 t SA0 e2k1 t dt
0
½1:5:A3:16
The specific activity of pool B is affected by influx from
pool A and efflux from pool B. Here we write
½1:5:A3:6
ðt
ðt
dt
5.5
0
20
40
60 80 100 120 140 160
Time (min)
FIGURE 1.5.A3.2 Plot of ln SA and ln SB against time. The plot of ln SA is
linear with a Y-intercept of 9.2103 and a slope of 20.02. The plot of ln SB
intersects that of ln SA at the point where SA 5 SB.
73
74
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Note that the plot of ln SB peaks at the intersection
of the ln SA and ln SB curve. This occurs at 40.55 minutes and SA 5 SB 5 4444.2 cpm μmol21. Eqn [1.5.A3.16]
can be reorganized to attempt to solve for k2. It can be
rewritten as
½1:5:A3:17
ðk2 2 k1 Þ
SB 5 SA0 e2k1 t 2 SA0 e2k2 t
k2
k1
12
SB 5 SA 2 SA0 e2k2 t
k2
k1
12
SB 5 SB 2 SB ek1 t e2k2 t
k2
2k1 t
k1 e
2k2 t
5 k2 e
This result predicts that the point of intersection of
the two curves depends only on the two fractional
turnover rates and not on the amount of radioactivity
injected. This makes intuitive sense. We know k1 and
the time of intersection t 5 40.55 min. Thus k1 e2k1t 5
0.008888 min21 and we search for k2 such that k2 e2k2 t 5
0.008888 min21. The solution is
k2 5 0:03 min1
which can be verified by substitution. This solution can
be obtained graphically by the intersection of the line
y 5 ln k2 with the line y 5 40.55k2 2 4.723.
The pool size B can be calculated from k2 5 P/B; we have
k2 5 0.03 min21 and P 5 4 3 1024 mol min21, giving B as
B 5 4 3 1024 mol min21 =0:03 min21 5 0:0133 mol;
B 5 0:0133 mol
Diffusion
Learning Objectives
G
G
G
G
G
G
G
G
G
G
Define flow and flux
Describe the meaning of the continuity equation
Write Fick’s First Law of Diffusion
Recognize Fick’s Second Law of Diffusion
Identify the units of the diffusion coefficient
Identify the three major assumptions of the onedimensional random walk
Describe the diffusion coefficient in terms of the parameters
of the one-dimensional random walk
Describe what is meant by the time of diffusion and its
dependence on distance
Describe the diffusion coefficient in the cytoplasm compared
to that in water
Write the StokesEinstein equation and identify the
parameters in it
FICK’S FIRST LAW OF DIFFUSION WAS
PROPOSED IN ANALOGY TO FOURIER’S
LAW OF HEAT TRANSFER
Adolph Fick (18291901) was a German physiologist
who enunciated what we now call Fick’s First Law
of Diffusion in 1855. Fick argued from analogy to two
well-known laws of physics involving flows and their
driving forces. The first was Fourier’s Law of Heat
Transfer:
½1:6:1
@T
JH 5 2λ
@x
where JH is the rate of heat energy transfer per unit area
per unit time and @T/@x is the temperature gradient.
The second law was Ohm’s law:
½1:6:2
Je 5 2 σ
@ψ
@x
where Je is the electrical flux and @ψ/@x is the voltage or
potential gradient. By analogy, Fick wrote:
½1:6:3
Js 5 2D
@C
@x
1.6
This is Fick’s First Law of Diffusion in one dimension.
This law says that the positive J is in the direction of the
negative spatial slope of the concentration. In analogy
to the other laws, this law says that solutes move from
regions of high concentrations to low concentrations,
and that the driving force for such a movement is the
concentration gradient. The term gradient has a
specific meaning that is defined by vector calculus, as
described in Chapter 1.3. Fluxes are usually expressed in
units of moles per square centimeter per second. Since
concentration is in units of moles cm23, and x is in
units of cm, the gradient is in units of moles cm24.
Dividing the units of J by the units of the gradient,
we have the units of the diffusion coefficient as
cm2 s21. In free water solutions, most low molecular
weight materials have diffusion coefficients on the
order of 1 3 1025 cm2 s21. Larger materials diffuse more
slowly, as we will see.
FICK’S SECOND LAW OF DIFFUSION
FOLLOWS FROM THE CONTINUITY
EQUATION AND FICK’S FIRST LAW
In Chapter 1.2, we derived the continuity equation
from the conservation of material flowing along one
dimension. It was given as
½1:6:4
@Cðx; tÞ
@JðxÞ
52
@t
@x
If we differentiate Fick’s First Law (see Eqn [1.6.3]) with
respect to x, we obtain
½1:6:5
@JS
@2 C
5 2D 2
@x
@x
By substitution into the continuity equation, we obtain
½1:6:6
@C
@2 C
5D 2
@t
@x
This is Fick’s Second Law of Diffusion. It follows from
the First Law of Diffusion by application of the continuity equation. It relates changes in concentration with
time with the spatial distribution of solute particles.
Given initial conditions of C(x,0) and boundary conditions, solutions of this equation, or its three-dimensional
analogue, allow one to determine concentration as a
function of time and position (C(x,t)).
75
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00007-0
76
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
FICK’S SECOND LAW CAN BE DERIVED
FROM THE ONE-DIMENSIONAL
RANDOM WALK
Fick’s Second Law of Diffusion can be derived from
molecular kinetic theory elucidated by Maxwell and
Boltzmann in the latter half of the 19th century. This can
be accomplished by analyzing the statistics of the
random walk. Application of the continuity equation to
the Second Law can then result in the derivation of Fick’s
First Law. It should become clear, then, that both these
laws describing diffusion are a consequence of the
random motion of particles. The process of diffusion thus
appears to be a statistical result only, there being no literal
“driving force” for the diffusive flux. Despite this, we will
find that there is energy associated with concentration
or dilution of solutes, and that opposition of diffusion,
e.g., during active transport, requires real forces.
The random walk we will discuss is a one-dimensional
model of real events. We choose to simplify the analysis
because otherwise it is intractable and because it turns
out to be a good model due to the enormously large
number of collisions which occur during diffusion
of real solute particles. The main assumptions of the
model are:
G
G
G
Each particle moves in a straight line between
collisions.
The motion consists of steps of length λ taken either
to the left or to the right.
The probability of making a step to the right is equal
to the probability of taking a step to the left.
This surprisingly simple model will allow us to derive a
one-dimensional form of Fick’s Second Law.
If the probability of taking a step to the right is p and
the probability of taking a step to the left is q, the third
assumption gives p 5 q 5 1/2. The question we ask first
is this: if a particle starts out at x 5 0, what is the probability that, after some elapsed time, t, it will be found
some distance x away, where x is an integral multiple of
λ? We can take λ to be a measure of the mean free
path. If tc is the time between collisions, and we wait
for the interval t, then there will be t/tc collisions in this
time. These simple assumptions allow us to derive an
expression for the probability density that a particle
starting at x 5 0 will be found in a distance interval centered at x at time t.
First we will convert the elapsed time to the number of
steps taken in that interval. The size of the steps to the
right and to the left is λ, the mean free path between
collisions. If the average speed is v, then the average
speed times the time between collisions, tc, will be equal
to λ. Thus
½1:6:7
tc 5
λ
hvi
The number of collisions in the time t is given as the
time t divided by the time between collisions:
½1:6:8
N5
t
tc
Note that we are using N now to count the number of
steps a particle of average speed v and mean free path λ
will make in the interval t. In this derivation, it does
not signify the number of solute molecules. Now the
original problem can be reformulated: what is the probability that, after N steps, the particle will be found at a
distance x 5 mλ, where m is an integer, away from its
starting place?
Let R be the number of steps to the right and L be the
number of steps to the left. Then
½1:6:9
N5R1L
R2L5m
where the total number of steps is N. To travel a distance
mλ away from the starting point in steps of size λ,
R 2 L 5 m. The desired probability of making R steps to
the right and L steps to the left out of a total of N steps
is given by the binomial probability distribution:
N
1
N!
½1:6:10
PN ðR; LÞ 5
2
R!L!
This is simply the probability that with N trials, R trials
will be to the right and L trials will be to the left. From
Eqn [1.6.9], we have
N1m
2
N2m
L5
2
R5
½1:6:11
which can be substituted back into Eqn [1.6.10] to give
N
1
N!
PN ðR; LÞ 5 PN ðmÞ 5
2
ððN 1 mÞ=2Þ!ððN 2 mÞ=2Þ!
½1:6:12
In this form the probability distribution is a discrete
variable, with only integral values of m. This random
walk model of diffusion is shown in Figure 1.6.1 where
the distribution models diffusion from an initially sharp
distribution at m 5 0 and P 5 1.0. After 10 steps, the
material is distributed between 210 # m # 10; after 20
steps, the material is distributed between 220 # m # 20,
but the probability at the extreme ends of the distribution is small. The probability profile loses density in the
center and gradually spreads out with the number of
steps. Remember here that the number of steps is
directly proportional to the time according to Eqn
[1.6.8]. This behavior corresponds subjectively to the
idea of diffusion: the material gradually diminishes at
its source and spreads out with time.
If you look carefully at the binomial probability distribution in Figure 1.6.1, you will notice that its profile
appears to be a fairly well-behaved function. As written,
it is a discrete probability function, being defined for
only integral values of N and m. What we would like is a
continuous distribution that describes the envelope of
the binomial probability distribution. Now normally the
value of N, the number of collisions in the time interval
t is extraordinarily large. In a gas at room temperature,
there are typically 5 3 109 collisions per second, while
Di ffusion
0.25
Binomial distribution, N = 10 steps
0.10
Gaussian distribution
(N = 50)
0.20
0.08
PN (m)
0.15
0.10
0.06
0.04
0.05
Binomial
distribution
0.02
0.00
Binomial distribution, N = 20 steps
0.20
0.00
–40
–20
0
m
0.15
40
FIGURE 1.6.2 Binomial probability distribution for N 5 50, with the
envelope of the Gaussian distribution with N 5 50. The binomial
probability distribution is discrete, with only integral values of m
allowed. The Gaussian distribution is continuous.
0.10
0.05
PN (m)
20
0.005
0.00
Binomial distribution, N = 50 steps
0.20
0.004
PN (m)
0.15
0.10
Gaussian distribution
(N = 50)
0.003
Δx
0.002
X
0.05
0.001
0.00
Binomial distribution, N = 100 steps
Binomial distribution
0.000
0.20
18
0.15
20
22
24
m
26
28
30
FIGURE 1.6.3 Enlargement from Figure 1.6.2.
0.10
Stirling’s formula for n factorial is
pffiffiffiffiffiffiffiffiffi
½1:6:13
n! 5 2πnen lnðn2nÞ
0.05
0.00
–100–80 –60 –40 –20 0
20 40 60 80 100
m
FIGURE 1.6.1 Binomial probability density for the probability of finding
a particle that initially was placed at m 5 0, as a function of the number
of steps, N 5 10, 20, 50, and 100. Note that material initially present in a
narrow band centered at the origin spreads out with increasing number
of steps, corresponding to increasing time.
the mean free path is typically 1025 cm or so. We cannot count the number of steps accurately nor can
we measure the distance accurately, so we resort to a
simpler question: What is the probability that a particle starting at x 5 0 and t 5 0 will end up in the
displacement interval Δx centered at x at time t?
We can get this probability from PN(m) first by letting
N get very large to justify the use of Stirling’s approximation, and then by counting the number of m’s
which land the particle in the displacement interval Δx
centered at x.
This truly amazing formula is the key to converting the
discrete probability distribution into a continuous one.
Using Stirling’s formula in Eqn [1.6.13], plus the
approximation that ln(1 1 α) α for small values of α,
it is possible to convert Eqn [1.6.12], by a lot of algebraic manipulation, to
rffiffiffiffiffiffiffi
2 2ðm2 =2NÞ
e
½1:6:14
PN ðmÞ 5
πN
This is the Gaussian approximation to the probability
distribution function PN(m). This function provides a
continuous envelope to the discrete probability function given in Eqn [1.6.12], as shown schematically in
Figure 1.6.2. The Gaussian probability distribution is
discussed in Appendix 1.2.A2.
As mentioned earlier, what we desire is to find the probability that a particle starting at x 5 0 and t 5 0 will end up
in the interval of Δx centered at x at some time t later.
How we can accomplish this can be made clearer if we
enlarge part of Figure 1.6.2, as shown in Figure 1.6.3.
77
78
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Since each step is of length λ, a net displacement of
R 2 L 5 m steps leads to the displacement x 5 mλ. Thus,
m 5 x/λ, and we may replace m in Eqn [1.6.14] with x/λ
to obtain
rffiffiffiffiffiffiffi
x
2 2ðx2 =2Nλ2 Þ
e
PN ðmÞ 5 PN
½1:6:15
5
λ
πN
What we desired was the probability that a particle will
be in the interval Δx centered at x at some time t after
beginning the random walk. Specifying the time t is
equivalent to specifying the value of N, as these are
related according to Eqn [1.6.8] as N 5 t/tc, where tc is
the time between collisions. We may substitute this
value of N into Eqn [1.6.15] to obtain
rffiffiffiffiffiffi
x
x
2tc 2ðx2 tc =2λ2 tÞ
;t 5
PN ðmÞ 5 PN
e
5 Pðm; tÞ 5 P
λ
λ
πt
½1:6:16
The probability the particle will be in the interval Δx
centered at x is the sum of the probabilities that the
value of mλ will fall in this interval. For a given N
(which is the same as for a given time, t) values of
m 5 R 2 L are either all odd or all even. Thus the possible values of displacement are separated by 2λ. The
number of values of m consistent with landing in the
interval Δx is thus Δx/2λ. If we take P(x/λ,t) given in
Eqn [1.6.16] as the average probability in the interval
Δx, then the probability of finding the particle in the
interval is
X
Δx
x
P
;t
Pðm; tÞ 5
2λ
λ
mAΔx
1
x
5
P
;
t
Δx
½1:6:17
2λ
λ
sffiffiffiffiffiffiffiffiffiffiffiffi
tc
2
2
5
e2tc x =2λ t Δx
2πλ2 t
From this result, we define a probability density function:
rffiffiffiffiffiffiffiffiffiffiffiffi
tc
2ð2tc x2 =2λ2 tÞ
½1:6:18
Pðx; tÞ 5
2 e
2πλ t
When multiplied by the length of the interval, Δx, this
function gives the probability of finding the particle in
the interval Δx at time t. The diffusion coefficient is
defined as
½1:6:19
D5
λ2
2tc
Note that this definition is consistent with the units of
the diffusion coefficient of cm2 s21 that we obtained
from Fick’s First Law of Diffusion. Using this definition,
Eqn [1.6.18] becomes
rffiffiffiffiffiffiffiffiffiffiffi
1
2
e2ð2x =4DtÞ
Pðx; tÞ 5
½1:6:20
4πDt
This is still the probability density function for finding a
particle in an interval at some time after beginning the
random walk, but all of the parameters of the random
walk, the mean free path, and the time between collisions
are submerged into a single constant, D. Thus the diffusion coefficient derives its values from microscopic characteristics of the diffusing substance. Since we have defined
the average velocity as hvi 5 λ/tc (see Eqn [1.6.7]), we see
that the diffusion coefficient is related to the square of the
average velocity. Thus thermal agitation should increase
the average velocity and thereby increase diffusion.
We can use Eqn [1.6.20] to derive an expression for the
concentration of solute particles at position x at time t,
which we denote as C(x,t). Ordinarily the concentration
is the number of particles per unit volume. In our onedimensional analogue, it is the number of particles per
unit length. At any time, t, the number of particles
per unit length, Δx, at position x is the sum of all particles which have random walked into the displacement
interval from all other areas. This is the probability
density function times the initial concentration summed
over all intervals. This can be written as
ðN
½1:6:21
C0 ðx0 ÞPðx 2 x0 ; tÞdx0
Cðx; tÞ 5
2N
0
where C0(x ) is the initial concentration at point x0 . Here
rffiffiffiffiffiffiffiffiffiffiffi
1
0 2
e2ðx2x Þ =4Dt
Pðx 2 x0 ; tÞ 5
½1:6:22
4πDt
Eqn [1.6.21] expresses the concentration at position x
and time t in terms of the concentration everywhere
at an initial time t 5 0. The initial time t 5 0 is chosen
arbitrarily. We can write
ðN
½1:6:23
Cðx0 ; tÞPðx 2 x0 ; ΔtÞdx0
Cðx; t 1 ΔtÞ 5
2N
0
Let us substitute in s 5 x 2 x, so that C(x0 ,t) 5 C(x 1 s,t)
and P(x 2 x0 ,Δt) 5 P(2s,Δt) 5 P(s,Δt) (because of symmetry in x in the probability distribution function) and
ds 5 dx0 . Then Eqn [1.6.23] becomes
ðN
Cðx; t 1 ΔtÞ 5
½1:6:24
Cðx 1 s; tÞPðs; ΔtÞds
2N
We let Δt be small, so that the concentration is altered
only by local events. We approximate C(x 1 s,t) as
Cðx 1 s; tÞ 5 Cðx; tÞ 1 s
½1:6:25
@Cðx; tÞ 1 2 @2 Cðx; tÞ
1 s
1?
@x
2
@x2
which is a Taylor’s series expansion of C(x 1 s,t).
Inserting Eqn [1.6.25] back into Eqn [1.6.24], we obtain
ðN
Cðx 1 t; ΔtÞ 5 Cðx; tÞ
Pðs; ΔtÞds
2N
ðN
½1:6:26
@Cðx; tÞ
1
@x
2N
1 @2 Cðx; tÞ
1
2 @x2
sPðs; ΔtÞds
ðN
2N
s2 Pðs; ΔtÞds
Di ffusion
The function P(s,Δt) is given from Eqn [1.6.22] and our
definition of s 5 x0 2 x as
rffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
1
2
e2s =4DΔt
Pðs; ΔtÞ 5
½1:6:27
4πDΔt
Evaluation of the integrals gives
ÐN
2N Pðs; ΔtÞds 5 1
ÐN
½1:6:28
2N sPðs; ΔtÞds 5 0
ÐN 2
2N s Pðs; ΔtÞds 5 2DΔt
We shall not prove these integration results. You should
note their meaning, however. The first integration result
is the normalization of the probability distribution,
which means that the particle must be somewhere with
the probability equal to 1. The second result is the average displacement about the initial zero displacement s.
The zero value of the average displacement means that
the distribution is symmetrical: displacement to the
right and to the left are equally likely in the random
walk. Recall by the definition of s 5 x0 2 x that s may be
both negative and positive, so that the average is zero.
The last integration gives the average square displacement. Both positive and negative values of s
contribute to s2. This integration gives the variance, or
the average squared displacement, for the distribution.
For the case of this one-dimensional model of diffusion,
the variance is 2DΔt. Inserting these integration results
into Eqn [1.6.26] gives
½1:6:29
Cðx; t 1 ΔtÞ 5 Cðx; tÞ 1
1 @2 Cðx; tÞ
2DΔt
2 @x2
This may be rewritten as
½1:6:30
Cðx; t 1 ΔtÞ 2 Cðx; tÞ
@2 Cðx; tÞ
5D
Δt
@x2
If we take the limit as Δt-0, we recognize the lefthand side of Eqn [1.6.30] as the partial derivative of the
concentration with respect to time. We then have
½1:6:31
@Cðx; tÞ
@2 Cðx; tÞ
5D
@t
@x2
This is Fick’s Second Law of Diffusion. From the continuity equation, Fick’s First Law can be derived. Although
Fick’s First Law was originally derived on phenomenological grounds and the Second Law followed it from
the continuity equation, this derivation shows that it can
be done the other way using a quite simple model which
nevertheless embodies the main ideas giving rise to
diffusion: there are an enormous number of collisions
which give rise to a random motion of particles from
one region of space to another.
THE TIME FOR ONE-DIMENSIONAL
DIFFUSION INCREASES WITH THE
SQUARE OF DISTANCE
How long does it take for a given material to diffuse
some distance, x? This question is deceptively simple.
It is not asking how long it takes the first molecule to
get to x, or how long it takes for the concentration at x
to reach a particular value. It is asking about the population of molecules that are moving from high concentration to low concentration. From Figure 1.6.1, you can
see that an initially sharp distribution gradually broadens with time because of diffusion. What we want is a
quantitative measure of the shape of the concentration
profile. The time of diffusion is usually calculated from
the variance of the Gaussian distribution:
ð 1N rffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
1
2
e2x =4DΔt dx 5 2DΔt
½1:6:32
x2 5
x2
4πDΔt
2N
where the elapsed time of diffusion is Δt. For twodimensional diffusion, the variance is 4DΔt; for threedimensional diffusion it is 6DΔt. The elapsed time for
diffusion, Δt, is the time taken for the inflection point
of the distribution to move from x 5 0 to x 5 x, given an
initially sharp distribution at x 5 0. The time taken to
diffuse a given distance, x, is the mean square displacement divided by 2D. Alternatively, we can calculate the
distance of diffusion in time Δt as
pffiffiffiffiffi pffiffiffiffiffiffiffiffiffiffiffiffi
½1:6:33
x 5 x2 5 2DΔt
For distances smaller than the cell (010 μm), diffusion
takes less than a ms up to a few ms. For distances on
the order of the diameter of muscle cells (40100 μm),
diffusion takes several seconds.
DIFFUSION COEFFICIENTS IN CELLS
ARE LESS THAN THE FREE DIFFUSION
COEFFICIENT IN WATER
If the initial concentration of a substance is very narrow,
then the subsequent distribution some time later due to
diffusion will be given by Eqn [1.6.20] as
rffiffiffiffiffiffiffiffiffiffiffi
1 2x2 =4Dt
e
Cðx; tÞ 5 C0
½1:6:34
4πDt
This equation can be shown to obey Fick’s Second Law
of Diffusion, Eqn [1.6.31], which is left as an exercise
for the student (see Problem 15 in Problem Set 1.2).
Dividing Eqn [1.6.34] by C0 and taking the logarithm
of both sides, we obtain:
sffiffiffiffiffiffiffiffiffiffiffi
Cðx; tÞ
1
2
5 ln
1 lne2x =4Dt
ln
C0
4πDt
½1:6:35
pffiffiffiffiffiffiffiffi
x2
2 ln ð2 πDt Þ
52
4Dt
Kushmerick and Podolsky (Ionic mobility in cells, Science
166:12971298, 1969) microinjected a 36 mm segment of muscle fiber from the semitendinosus muscle of
the frog with small amounts of tracer materials, and then
allowed the materials to diffuse along the fibers for
various periods of time. They immersed the fibers in oil
to avoid diffusion through the water phase outside the
muscle. After the prescribed period, the muscles were
dehydrated in acetone, stained, embedded in paraffin,
79
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Example 1.6.1 Time of One-Dimensional Diffusion
The
diffusion
coefficient
for
Ca21
in
water
is
25
2 21
DCa 5 0.8 3 10 cm s ; for calbindin, a 9500-Da protein that binds
Ca21 with high affinity, DP 5 0.12 3 1025 cm2 s21. How long does it
take for Ca21 or calbindin to diffuse x 5 {0.1, 1, 10, 20, 50, 100 μm}?
Here we use Eqn [1.6.32] to calculate Δt 5 x2/2D. As a representative calculation, we find Δt for Ca21 for 0.1 μm:
Δt 5
which is amazingly quick. The other times can be calculated by
the same method, and we can fill out Table 1.6.1.
Variation of this calculation was performed by A.V. Hill in 1949
to show that a diffusable substance could not be the activator of
muscle cells because the delay between nervous excitation and
contraction of the muscle was too fast, only a few ms.
ð0:1 3 1026 m 3 102 cm m21 Þ2
5 6:25 3 1026 s
2 3 0:8 3 1025 cm2 s21
TABLE 1.6.1 The Calculated Time for One-Dimensional Diffusion of Free Ca21 and Calbindin in Water for Various
Distances
Distance (µm)
Calcium DCa 5 0.8 3 1025 cm2 s21
0.1
6.3 3 1026
1.0
0.6 3 1023
10.0
23
62.5 3 10
Calbindin DP 5 0.12 3 1025 cm2 s21
42 3 1026
4.2 3 1023
420 3 1023
20.0
0.25
1.67
50.0
1.56
10.4
100.0
6.25
41.7
and cut into 25 μm sections, which were then counted.
The plots of ln (counts at distance x/total counts) were
linear with the square of the distance, as predicted by
Eqn [1.6.35], and the diffusion coefficient in the muscle
was estimated from the slope. An example of their results
for 42K is shown in Figure 1.6.4. Kushmerick and
Podolsky found that the diffusion coefficient for most
substances they injected was about one-half of the free
water diffusion coefficient. These materials included K1,
Na1, SO422, sorbitol, sucrose, and ATP. For Ca21, however, the apparent diffusion coefficient was about 50 times
less than the free water diffusion. From this they concluded that Ca21 was retarded by interaction with fixed
components within the muscle cell. It is important to
recognize that this effect is a reduction in the apparent diffusion coefficient because the total Ca21 is being partitioned between a freely diffusable form and a bound or
fixed form. Thus the apparent diffusion coefficient is
reduced because they calculated the diffusion from the
total concentration and not just the free concentration.
EXTERNAL FORCES CAN MOVE
PARTICLES AND ALTER THE DIFFUSIVE
FLUX
The mathematical relations describing the diffusion
of nonelectrolytes have been presented to you in the
form of Fick’s First and Second Laws of Diffusion.
These expressions were derived from the one-
2
Ln (42K per 0.012 cm length/total 42K)
80
4
6
8
0.03
0.06
0.09
Length2 (cm2)
FIGURE 1.6.4 Typical results for the Kushmerick and Podolsky
experiment. The longitudinal distribution of 42K1 at 0.02 cm intervals
after diffusion for 320 s at 20 C was plotted as a logarithmic transform
of the diffusion equation for an infinite slab from an infinitely thin
distribution: ln (counts at distance x/ total cts) 5 2x2/4Dt 2 ln 2(πDt)1/2.
dimensional random walk, which considered that the
probability of making a step to the right and to the left
was the same. Under some circumstances, this is
not true. For example, diffusive flux is altered when a
Di ffusion
bulk flux of fluid occurs simultaneously with the
diffusive flux. In this case, we write
½1:6:36
Js ðx; tÞ 5 2D
@Cðx; tÞ
1 Jv ðtÞCðx; tÞ
@x
The first term on the right describes the diffusive flux
and the second term describes the flux of solute due to
solvent drag. Here Jv is the volume flux, equal to the
volume of fluid moving across an area per unit area per
unit time. This volume times the concentration of solute
will give the amount of solute moving across that area
per unit area per unit time. Jv is equal to the velocity
of fluid flow: the volume flux is V/(AΔt) 5 (AΔx)/
(AΔt) 5 Δx/Δt, the velocity of fluid flow. Eqn [1.6.36]
is the convectiondiffusion equation, because the
bulk flow is described as convection.
There are other circumstances which alter the flux from
that described by Fick’s First Law. These circumstances
occur when there are external forces applied to the solute
particles. Examples of these forces include electrical forces
and gravitational forces. Recall in Chapter 1.3 that we
considered that electrical forces accelerate ions in solution
until they reach a terminal velocity in which the electrical
force is balanced by the drag force. The drag force, Fd,
is proportional to the terminal velocity. We wrote:
Fe 5 zeE
½1:6:37
Fd 52β v
Fe 1 Fd 5 0
Fe 5 β v
where β is the frictional coefficient or drag coefficient.
It is given as
½1:6:38
β5
kT
D
as originally proposed by Einstein in 1905 (see Appendix
1.6.A1). Here k is Boltzmann’s constant, the ideal gas
constant divided by Avogadro’s number: k 5 R/N0, and D
is the diffusion coefficient. This result makes sense: a
large diffusion coefficient is usually associated with
small particles, and these would have a small frictional
coefficient, encountering less resistance to movement.
Incorporating this definition of β into the last equation
in Eqn [1.6.37], we have
½1:6:39
Fe 5
kT
v
D
In Chapter 1.3, we also established that the ratio of J to
C defines an average velocity of movement of particles:
½1:6:40
J5v C
Substituting for v from Eqn [1.6.40] into Eqn [1.6.39]
and rearranging, we obtain
D
Fe C
kT
What this equation means is that, in the absence of a
concentration gradient, an external force will produce
a flux that is linearly related to the magnitude of the
½1:6:41
J5
force per particle and the concentration, with a coefficient related to the diffusion coefficient. We have
derived this for an electric force, but the result is
completely general for any external force.
In the presence of a concentration gradient, we expect
the diffusive flux to add to the flux caused by the
application of an external force. From Fick’s First Law
of Diffusion and Eqn [1.6.41], we obtain, for one
dimension, Fick’s First Law of Diffusion for solutes
subjected to an external force:
½1:6:42
J 5 2D
@C
D
1
fC
@x
kT
This can be written in vector notation as
½1:6:43
J 5 2DrC 1
D
CF
kT
THE STOKESEINSTEIN EQUATION
RELATES THE DIFFUSION COEFFICIENT
TO MOLECULAR SIZE
Stokes showed that for a spherical particle, the drag
force was related to its size and to the viscosity of the
medium:
½1:6:44
Fd 5 2β v 5 2 6πηas v
where η is the viscosity of the fluid and as is the radius
of the sphere. Clearly, Stokes derived an expression
for the frictional coefficient. From Eqns [1.6.38] and
[1.6.44], we can write
6πηas 5
½1:6:45
D5
kT
D
kT
6πηas
The last expression is the StokesEinstein equation.
It indicates that the diffusion coefficient for a spherical
particle should be a function of its radius, the absolute
temperature, and the viscosity of the fluid in which it is
diffusing.
Recall earlier that Kushmerick and Podolsky found that
the apparent diffusion coefficient of readily diffusable
substances like K1, Na1, and sucrose inside cells was
about one-half of their diffusion coefficients in water.
From the StokesEinstein equation, it would seem that
the variable most likely responsible for these decreases
in diffusion coefficients is the viscosity of the medium.
Thus the cytoplasm appears to be a watery environment, where most small molecular weight materials are
free to diffuse but with reduced diffusion coefficients
owing to the greater viscosity of the cytoplasm. The tortuosity of the path is not included in this analysis. This
refers to the blockade of direct diffusion by large structures in the cytoplasm, including organelles and cytoskeleton. Because materials cannot diffuse in a straight
line, the diffusion apparently takes longer because the
actual path length in the microscopic domain is larger
than the apparent path length. In fact, tortuosity and
81
82
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
increased viscosity are both partial explanations for the
reduced diffusion coefficient inside cells.
expression for the drag force in terms of the diffusion
constant and gave us the StokesEinstein equation:
D5
SUMMARY
Solutes move by diffusion from regions of high concentration to regions of low concentration. Fick’s First Law
of Diffusion states that the flux is proportional to the
negative of the gradient of C:
D @C
Js 5 2
@x
where Js is the solute flux, D is the diffusion coefficient,
and @C/@x is the one-dimensional gradient. The continuity equation states that changes in concentration with
time must be due to changes in flux with distance:
@C
@Js
52
@t
@x
Fick’s Second Law of Diffusion derives from his First
Law and the Continuity Equation:
@C D @2 C
5
@t
@x2
Fick’s Second Law can be derived from a random walk
model of diffusion in which molecules take large numbers
of small steps. Using Stirling’s approximation, the discrete
binomial probability distribution can be converted to a
continuous one, resulting in a Gaussian probability distribution. For a narrow starting distribution at time t 5 0,
the distribution of solute at time t is given as
pffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
2
Cðx; tÞ 5 C0 1=4πDt e2x =4Dt
From the random walk, D is identified as λ2/2tc, where
λ is the distance between collisions and tc is the time
between collisions. The time of diffusion is typically
estimated as the variance of the Gaussian distribution,
which gives
x2
2D
Although diffusion is a statistical result, it is equivalent
to a force in that it produces a flow of material. Other
forces can also make solutes move. These forces include
electrical forces on charged solutes, solvent drag
(convection), and gravitational forces. An external force
applied to solute particles causes a flux given by
t5
J5
D
fC
kT
where f is the force per molecule, k is Boltzmann’s constant (k 5 R/N0), T is the absolute temperature, and C is
the concentration. In the presence of a concentration
gradient, the total diffusive flux in the presence of an
external force is
J52
D@C DfC
1
@x
kT
Stokes derived an equation for the drag force on
a spherical object. Einstein combined this with his
kT
6πηas
where η is the viscosity of the medium in which diffusion occurs and as is the radius of a spherical solute.
REVIEW QUESTIONS
1. Why is diffusive flux proportional to the negative
of the gradient and not the gradient?
2. What are the assumptions of the random walk
derivation?
3. What are the units of the diffusion coefficient?
4. How does the time of diffusion vary with
distance?
5. Why are diffusion coefficients slower in the cytoplasm of cells than in water?
6. What is solvent drag?
7. What is meant by convective flow?
8. How does the diffusion coefficient vary with
molecular size? With viscosity of the medium?
With temperature? What is Boltzmann’s constant?
9. How is Fick’s First Law of Diffusion altered in
the presence of additional forces acting on the
diffusing particles?
APPENDIX 1.6.A1 DERIVATION OF
EINSTEIN’S FRICTIONAL COEFFICIENT
FROM MOMENTUM TRANSFER IN
SOLUTION
Here we consider a right cylindrical volume V of crosssectional area A and thickness Δx, so that V 5 AΔx. We
imagine that solute particles may move with velocity 1 v
in the x-direction (to the right) and velocity 2 v (velocity
v to the left), as shown in Figure 1.6.A1.1. Let the number of particles in V with velocity v be N1(t) and the
number with velocity 2 v be N2(t). The concentration of
particles with velocity 1 v or 2 v in V at any time will be
given by
½1:6:A1:1
N1 ðtÞ
5 C1 ðtÞ
V
N2 ðtÞ
5 C2ðtÞ
V
The number of particles with a given velocity may change
with time. This may happen in four different ways: (1)
particles with velocity 1 v may enter the volume element
from the left; (2) particles with velocity 1 v may leave
the volume element at the right; (3) particles with
velocity 1 v within the volume V may convert to particles
with velocity 2 v by colliding with solvent particles;
(4) particles with velocity 2 v could convert to velocity
1 v by collisions with solvent particles. The entry of
particles from the left is given by
½1:6:A1:2
Qi ðxÞ 5 v A C1 ðxÞ
Di ffusion
Cross-sectional
area, A
J(x)
+v
+v
J(x +Δx)
–v
–v
x
x +Δx
x axis
FIGURE 1.6.A1.1 Solutes within a hypothetical volume. Solutes have a velocity 1 v in the positive x-direction or 2 v in the opposite direction. All
particles have velocity 1 v or 2 v, although only a few are shown.
where we recognize that vC1 is the flux of particles with
velocity 1 v. The exit of particles at the right is
½1:6:A1:3
Qo ðxÞ 5 v A C1 ðx 1 ΔxÞ
The rate of conversion of N1 to N2 is proportional to
N1 within the volume V, with the proportionality
constant having dimensions of reciprocal time. This proportionality constant is 1/tc, where tc is the time between
collisions. This conversion reduces N1 within V, so we
may write:
@N1
N1
V C1
½1:6:A1:4
52
5
@t -collision
tc
tc
The rate of conversion of N2 to N1 adds to the value of
N1 and is given by a similar expression:
@N1
N2
V C2
½1:6:A1:5
5
5
@t 1collision
tc
tc
The total net change of N1 is given by the sum of Eqns
[1.6.A1.2, 1.6.A1.3, 1.6.A1.4, 1.6.A1.5]:
@N1
V
5 vA C1 ðxÞ 2 vA C1 ðx 1 ΔxÞ 1 ðC2 2 C1 Þ
tc
@t
½1:6:A1:6
where v means velocity and V means volume. We can
approximate C1(x 1 Δx) by the first two terms of a
Taylor’s series expansion:
@C1
C1 ðx 1 ΔxÞ 5 C1 ðxÞ 1 Δx
½1:6:A1:7
1?
@C
Insertion of Eqn [1.6.A1.7] into Eqn [1.6.A1.6] gives
"
#
@N1
@CðxÞ
V
1 ðC2 2C1Þ
5vA C1ðxÞ2C1ðxÞ2Δx
@x
tc
@t
!
@N1
@C1ðxÞ
V
2 ðC1 2C2Þ
52vAΔx
@x
tc
@t
½1:6:A1:8
Since AΔx 5 V, we write
½1:6:A1:9
1 @N1
@C1
1
5 2v
2 ðC1 2 C2 Þ
V @t
tc
@x
@C1
@C1
1
5 2v
2 ðC1 2 C2 Þ
tc
@t
@x
By completely analogous reasoning, we can determine
the rate of change in C2 in V as
½1:6:A1:10
@C2
@C2
1
5v
1 ðC1 2 C2 Þ
tc
@t
@x
Since the total concentration, C(x) is the sum of C1 and
C2, then
½1:6:A1:11
@CðxÞ
@t
5
@C1 ðxÞ @C2 ðxÞ
1
@t
@t
and substituting into Eqn [1.6.A1.11] from Eqns
[1.6.A1.9] and [1.6.A1.10], we obtain
@CðxÞ
@C1
1
@C1
1
5 2v
2 ðC1 2 C2 Þ 1 v
1 ðC1 2 C2 Þ
@t
tc
tc
@x
@x
!
@C1
@C2
5 2v
2
@x
@x
52
@ðvðC1 2 C2 ÞÞ
@x
½1:6:A1:12
Now the net flux across any area element within the
volume V can be described as the difference between
two unidirectional fluxes:
½1:6:A1:13
J 5 J1 2 J2
5 v C1 2 v C2
5 v ðC1 2 C2 Þ
Substitution of this relation into Eqn [1.6.A1.12] gives
½1:6:A1:14
@CðxÞ
@JðxÞ
52
@t
@x
83
84
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
which is the continuity equation we derived earlier
(see Eqn [1.2.9]). The purpose of deriving the continuity equation in this way was to familiarize you with this
method of accounting for all of the particles and to
impress upon you that the collisions with the solvent
involving changes in the velocity canceled. These collisions did not affect the total number of particles since
a particle contributing to N1 before collision still
contributes to N2 after collision.
Now let us consider what happens to the total momentum, P, of the particles in volume V. Let m be the mass
of each solute particle. The total momentum of the
particles is
½1:6:A1:15
P 5 ðmvÞN1 1 ð2mvÞN2
Because N1 and N2 change with time, so does P. We
write
@P
@N1
@N2
5 mv
½1:6:A1:16
2 mv
@t
@t
@t
Since N1 5 VC1 and N2 5 VC2, we have
½1:6:A1:17
@P
@C1
@C2
5 mvV
2 mvV
@t
@t
@t
Inserting our earlier results from Eqns [1.6.A1.9] and
[1.6.A1.10], we obtain
1 @P
@C1
@C2
2mv
5 2mv2
1
ðC1 2 C2 Þ
2
V @t
tc
@x
@x
½1:6:A1:18
Recalling that C 5 C1 1 C2, and using Eqns [1.6.A1.11]
and [1.6.A1.13], we obtain
½1:6:A1:19
1 @P
@C 2m
5 2 mv2
2
J
V @t
@x
tc
The two terms on the right-hand side of Eqn [1.6.A1.19]
have specific interpretations. The first term, 2 mv2@C/@x
V, represents the net flow of momentum carried by particles with velocity 1 v or 2 v. The second term, 22m/tc JV,
is the average force exerted by the solvent particles on the
solute particles. To see these interpretations, we write the
flow of momentum across area A at any point x within
the volume V as
mv ðvC1ðxÞÞA 1 ð2 mvÞð2 vC2ðxÞÞA 5 mv2 ðC1ðxÞ 1 C2ðxÞÞA
½1:6:A1:20
The net flow of momentum into volume V across the
boundaries between x and x 1 Δx is
mv2 ðC1 ðxÞ 1 C2 ðxÞÞA 2 mv2 ðC1 ðx 1 ΔxÞ 1 C2 ðx 1 ΔxÞÞA
½1:6:A1:21
Since C(x) 5 C1(x) 1 C2(x), the expression in Eqn [1.6.
A1.21] becomes
½1:6:A1:22
mv2 A½CðxÞ 2 Cðx 1 ΔxÞ
Expanding C(x 1 Δx) as C(x) 1 Δx @C/@x, the expression
in Eqn [1.6.A1.22] becomes
½1:6:A1:23
2mv2
@C
@C
ΔxA 5 2 mv2
V
@x
@x
Thus, in Eqn [1.6.A1.19], 2 mv2 @C/@x is that part of
1/V @P/@t which is due to the net momentum change of
the particles in V due to what they carried into or out
of the volume.
For the second term in Eqn [1.6.A1.19], note that every
time a particle moving with velocity 1 v collides with a
solvent obstacle, its velocity becomes 2 v, and thus
it experiences a net momentum change of 22mv.
The total number of such changes per unit time in the
volume V is N1/tc. So the time-averaged change for
the momentum change per unit time due to this
collision is 22mv N1/tc. In a similar way, particles
traveling with velocity 2 v experience a momentum
change of 12mv upon collision with the solvent and
the time-averaged total change in momentum per unit
time for this type of collision in the volume V is 12mv
N2/V. The time-averaged rate of net change in momentum for these types of collisions is just their sum:
@Pc
N1
N2
5 Fc 5 2 2mv
1 2mv
@t
tc
tc
½1:6:A1:24
5 Fc 5 2
2mv
VðC1 2 C2 Þ
tc
1 @Pc Fc
2m
5
52
J
V @t
tc
V
where the subscript c denotes that the momentum
change and force are due to collisions of the solute
molecules with the solvent. Thus the second term in
Eqn [1.6.A1.19] is identified as the change in momentum produced by collisions with the solvent. It is equivalent to a force per unit volume exerted by the solvent
particles on the solute particles. Equation [1.6.A1.24]
can be rewritten as a differential equation in J(x,t).
Recall that the total momentum, P, of the solute
particles in the volume V is given by Eqn [1.6.A1.15];
substituting in for the definition of the concentration
(Eqn [1.6.A1.1] and (J) Eqn [1.6.A1.13]), we have:
P 5 ðmvÞN1 1 ð2 mvÞN2
½1:6:A1:25
5 mV vðC1 2 C2 Þ
P 5 mV Jðx; tÞ
Insertion of this result into Eqn [1.6.A1.19] gives
½1:6:A1:26
1
@Jðx; tÞ
@Cðx; tÞ 2m
mV
52mv2
2
Jðx; tÞ
V
@t
@x
tc
This can be rearranged to
½1:6:A1:27
@Jðx; tÞ
2
tc v2 @Cðx; tÞ
5
2 Jðx; tÞ
2
@t
tc
@x
2
Eqn [1.6.A1.27] describes the buildup of J(x,t)
in time. We will pay particular attention to the situation
Di ffusion
where the concentration gradient, @C(x,t)/@x, is constant
and steady-state flux is achieved. Steady-state flux means
that @J(x,t)/@t 5 0. That is, the flux no longer changes
with time. Under these circumstances, by Eqn [1.6.
A1.27], we obtain the steady-state flux as
tc v2 @C
½1:6:A1:28
J52
2 @x
Comparing this to Fick’s First Law of Diffusion,
½1:6:A1:29
@C
J 5 2D
@x
we can identify
tc v2
2
In the one-dimensional random walk model of diffusion, we defined the diffusion coefficient to be
½1:6:A1:30
D5
D5
½1:6:A1:31
5
5
λ2
2tc
ðvtc Þ2
2tc
2
tc v
2
Thus the derivation performed here is completely consistent with the random walk model of diffusion.
Eqn [1.6.A1.24] describes the momentum change of the
solute particles that result from the collisions with solvent. It is given per unit volume as
½1:6:A1:32
1 @Pc
Fc
2m
5
52
J
V @t
tc
V
In this equation, Fc is the force per unit volume on the
solute particles. This is the same as the drag force on
the solute particles when they move through the solution. If we imagine that the particles are subjected to a
uniform external force, then the particles will accelerate
until they reach a terminal velocity, v. The force FC will
be the sum of all of the drag forces on the particles
within the volume, which is the drag force per particle
times the number of particles in the volume. The flux, J,
will be given as vC. Eqn [1.6.A1.32] can then be rewritten as
½1:6:A1:33
fc N
2m
52
vC
V
tc
where fC is the force on a single particle, N is the number of particles in the volume, V, v is the terminal velocity, and C is the concentration. Since N/V 5 C, the
equation is further simplified to
½1:6:A1:34
fc 5 2
2m
v
tc
Here the force fC is equal to the drag force on the particle traveling at velocity v, and the coefficient is the drag
or frictional coefficient:
½1:6:A1:35
FD 5 2β v
which allows us to identify the drag coefficient as
½1:6:A1:36
2m
tc
β5
The equipartition theorem of thermodynamics gives
1 2 1
mv 5 kT
2
2
½1:6:A1:37
v2 5
kT
m
where k is Boltzmann’s constant (51.38 310223 J K21,
which is equal to the gas constant, R, divided by
Avogadro’s number). Insertion of this result into the
equation for the diffusion coefficient, Eqn [1.6.A1.31]
gives
D5
tc kT
2m
tc 5
2mD
kT
½1:6:A1:38
Insertion of this into Eqn [1.6.A1.36] gives
½1:6:A1:39
β5
2m
2m
kT
5
5
2mD
tc
D
kT
This is Einstein’s frictional coefficient, given as
½1:6:A1:39
β5
kT
D
Recall Eqn [1.6.A1.19] reproduced here:
½1:6:A1:19
1 @P
@C 2m
5 2 mv2
2
J
V @t
@x
tc
substituting in v2 5 kT/m from Eqn [1.6.A1.37] and
tc 5 2mD/kT from Eqn [1.6.A1.38], we obtain
½1:6:A1:40
1 @P
@C kT
5 2 kT
2
J
V @t
@x
D
Recall that this equation describes the rate of momentum change of the solute per unit volume V. It has two
components: the first is the net momentum carried into
the volume by the diffusing solutes, and the second
is the momentum change produced upon collisions
with the solvent. Note that if C(x) is decreasing with
increasing x then the gradient, @C/@x, will be negative
and the first term on the right-hand side of Eqn [1.6.
A1.40] will be positive. In this case of diffusion of
solute towards increasing values of x, J will be positive
and the second term, denoting the change in
momentum by collisions with solvent, will be negative.
This second term denotes the drag of solvent on solute
movement. Steady-state diffusion occurs when @P/@t
is zero and @C/@x is constant. Under these conditions,
Eqn [1.6.A1.40] becomes Fick’s First Law of Diffusion.
Suppose now that we add an additional force to the
solute particles in the volume V. Let the force acting
on each particle be f. In the volume element, there are
85
86
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
N(x) 5 C(x) V particles. The total force acting on these
particles is f C(x) V. This force clearly contributes to
the rate of change of the momentum of the solute
particles in the volume. Eqn [1.6.A1.40] becomes
½1:6:A1:41
1 @P
@C kT
52 kT
2
J 1 fC
V @t
@x
D
To recapitulate, the first term on the right-hand side of
Eqn [1.6.A1.41] represents the rate of momentum
change by particles entering or leaving the volume V;
the second term is due to collisions with the solvent
particles; the third term is due to the action of some
external force. At steady state, @J/@t 5 0, and this implies
by Eqn [1.6.A1.25] that @P/@t also is zero. Under this
constraint, Eqn [1.6.A1.41] gives
½1:6:A1:42
J52D
@C
D
1
fC
@x
kT
This is Fick’s First Law of Diffusion for solutes subjected
to an external force, which is Eqn [1.6.42]. Thus this
equation, which forms the basis of the derivation of
the electrochemical potential, can be derived using
momentum transfer of solutes in a solution and the
equipartition theorem of thermodynamics.
Electrochemical Potential and
Free Energy
derivative, we retain the three-dimensional vector that
describes diffusive flux in three dimensions.
Learning Objectives
G
G
G
G
G
G
G
G
G
G
Write Fick’s First Law of Diffusion and explain how a concentration gradient makes a flux
Describe how an external force such as electrostatic force
can make a flux of charged solute
Write the formula for the electrochemical potential
Explain the formula for electrochemical potential in terms
of the component driving forces
Explain how the driving forces that produce a flux equal
the negative gradient of the electrochemical potential
Define the term free energy
Describe the relationship between the free energy and the
direction of any process
Be able to calculate the free energy of ATP hydrolysis under
specified conditions of temperature and concentrations of
reactants ATP, ADP, and Pi
Write the relationship between the standard free energy
change and the equilibrium constant
Know the approximate value for the free energy of ATP
hydrolysis under cellular conditions
Third, in the absence of a concentration gradient, any
external force acting on solute particles causes them
to be accelerated until they reach a terminal velocity.
At this terminal velocity, the external force is balanced
by the drag force on the particle by the solvent. This
drag force is proportional to the velocity. Because of
this, there is a relationship between the flux and the
external force, given as
½1:7:3
J5
DIFFUSIVE AND ELECTRICAL FORCES
CAN BE UNIFIED IN THE
ELECTROCHEMICAL POTENTIAL
Let us recapitulate what we learned in Chapters 1.3
and 1.6 about the movement of charged particles in
solution. First, the relationship between the flux and the
concentration defines an average velocity, given as
½1:7:4
J 5 2D
@C
@x
where D is the diffusion coefficient. Here we drop the
vector notation because the equation used here is one
dimensional and the direction is assumed to be along
the x-axis. If we use the gradient of C, instead of the
½1:7:2
J 5 2D
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00008-2
@C
D
1
Cf
@x
kT
If the force is an electrical force, its magnitude is given by
F 5 FE 5 zeE
where z is the valence of the particle ( 6 integer), e is
the charge on the electron, and E is the electric field.
Assuming one dimension, we drop the vector notation
for force and insert Eqn [1.7.5] into Eqn [1.7.4] to give
½1:7:6
J 5 2D
@C
D
1
CzeE
@x
kT
Thus we have these two equations:
J 5 vC
where J is the one-dimensional flux, v is the velocity,
and C is the concentration. Second, the presence of a
concentration gradient, in the absence of any other
forces, produces a flux given by Fick’s First Law of
Diffusion:
D
Cf
kT
where f is the force acting on the solute particles, per
particle. The concentration gradient has some of the
appearances of a force in that it causes a flux. So does an
externally applied force. What we seek to do is to combine diffusion and other forces into a single equivalent
force. When both are operating, the flux is given as
½1:7:5
½1:7:1
1.7
D
Cf
kT
@C
D
1
CzeE
J 5 2D
@x
kT
J5
½1:7:7
The top equation says that there is a flux produced by
some unknown force. The bottom equation says that
now we know what these forces are and we have parceled them out. Part of the flux is caused by diffusion
and part of the flux is caused by an electrical force.
What we desire is an expression for the total force in
the top equation that will produce the flux in the
bottom equation. To unite the diffusive force and 87
88
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
the electrical force, we solve these two equations for the
unknown force, f, and find that
½1:7:8
f 52
kT @C
1 zeE
C @x
The electric field, E, is the electric force per unit charge.
This electric force is the negative gradient of its potential
(see Chapter 1.3 for a definition of potential, gradients,
and conservative forces). In a three-dimensional model,
the electric field is a vector and the potential is a scalar.
We will ignore these realities in this one-dimensional
model because in one dimension the force and gradient
have a single direction. Using Ψ as the symbol for electrical potential energy, we can rewrite Eqn [1.7.8] as
½1:7:9
f 52
kT @C
@Ψ
2 ze
C @x
@x
N0 kT @C
@Ψ
2 z N0 e
C @x
@x
RT @C
@Ψ
F5 2
2 z`
C @x
@x
F 5 N0 f 5 2
21
where R is the gas constant (58.314 J mol K )
and ` is the Faraday (9.649 3 104 C mol21 5 6.02 3
1023 electrons mol2131.6310219 C electron21). We define
an electrochemical potential so that the overall force
(produced by both diffusion and electrical forces, in this
case) is the negative derivative of the electrochemical
potential:
F52
@µ
@x
Combining Eqns [1.7.10] and [1.7.11], we get
½1:7:12
2
@µ
RT @C
@Ψ
52
2 z`
@x
C @x
@x
Integrating, we obtain
½1:7:13
µi 5 µ0i 1 RT ln Ci 1 zi `Ψ
THE OVERALL FORCE THAT DRIVES
FLUX IS THE NEGATIVE GRADIENT OF
THE ELECTROCHEMICAL POTENTIAL
The flux produced by a concentration difference and
electric potential difference is given by
21
½1:7:11
In a typical physiological solution, several kinds of
particles are dissolved. Since their concentrations
are nearly independent of one another, they each have
an identifiable electrochemical potential. Each solute
within a local region, on the other hand, experiences
the same electrical potential. Therefore, we can write
an electrochemical potential for each material in the
solution:
½1:7:14
The units of Ψ are volts. The force in this equation is
the force per particle. It is customary to convert this to
the force per mole of particles by multiplying by
Avogadro’s number:
½1:7:10
by Eqn [1.7.13]. The second term, RT ln C, refers to the
work necessary to concentrate the solute, per mole;
the third term, z`Ψ, is an electrical work term. It is the
work necessary to bring one mole of particles from zero
potential to Ψ. If other kinds of work are involved,
we would need to expand Eqn [1.7.13] to include the
other work terms.
µ 5 µ0 1 RT ln C 1 z`Ψ
This is the electrochemical potential. We will use it to
make calculations about membrane potential and the
energetics involved in physiological processes. It is a
potential in the same sense as the electrical potential.
It has the units of energy per mole. The electrical
potential, in volts, is equivalent to joules per coulomb,
which can be converted to units of energy per mole.
Similarly, the electrochemical potential can be converted
into volts.
As can be seen from Eqn [1.7.13], the electrochemical
potential has three components. The first component,
µ0, has the sense of a constant of integration. It sets the
zero of the electrochemical potential and its reference is
a standard state. For solutions, µ0 refers to the chemical
potential of a hypothetical solution of unit molarity
and no potential. That is, when C 5 1 and Ψ 5 0, µ 5 µ0
½1:7:3
J5
D
Cf
kT
where J and f are both vectors. f is the force per molecule. We convert to molar dimensions by multiplying
and dividing by Avogadro’s number:
D
CN0 f
N0 kT
D
CF
J5
RT
J5
½1:7:15
This emphasizes that J is a vector in the same direction
as F, the force on the solute particles per mole. The
force per mole is, in turn, given by the negative gradient
of the electrochemical potential:
@µ
@µ
@µ
1j
1k
½1:7:16
F 5 2rµ 5 i
@x
@y
@z
Inserting in µ from Eqn [1.7.13] and F into Eqn
[1.7.15], we get
D
RT @C
@C
@C
C 2
i
1j
1k
J5
RT
C
@x
@y
@z
@Ψ
@Ψ
@Ψ
1j
1k
½1:7:17
2 z` i
@x
@y
@z
which is simplified to
½1:7:18
z`
CrΨ
J 5 2D rC 1
RT
The one-dimensional version of this equation is
½1:7:19
J 5 2D
@C
D
@Ψ
2
z`C
@x
RT
@x
Electr ochemical P otentia l and Fre e E nergy
THE ELECTROCHEMICAL POTENTIAL IS
THE GIBBS FREE ENERGY PER MOLE
Throughout the derivation of Fick’s Laws of Diffusion,
we made an unstated assumption that the pressure and
temperature of the diffusing particles were constant.
Under these conditions, we could derive the electrochemical potential. The constraints of constant temperature
and pressure are useful when applied to problems in
mammalian physiology, where these conditions are
usually met. Under these conditions, it is useful to define
a thermodynamic variable called the Gibbs free energy:
½1:7:20
G 5 E 1 PV 2 TS
where E is the internal energy, P is the pressure, V is the
volume, T is the temperature, and S is the entropy.
The internal energy consists of all of the myriads of
movements of the particles, including their internal
motions. This is the part of the free energy where
chemical bonding energy is stored. When compounds
undergo chemical transformations, energy is either
released or stored, depending on whether the reaction is
exothermic or endothermic, respectively. The entropy has
a specific thermodynamic definition which has been
shown to be related to the number of ways that the particles can be arranged in the system which are consistent
with the state of the system (its temperature, pressure,
volume, and number of particles). This is related to the
probability of finding the state for randomly arranged
particles. Thus states with high probabilities have high
entropy. Highly organized states, which can be accomplished in only a few ways, have a low probability and a
low entropy. For example, a concentrated solution has
only a few ways to crowd all the solute particles together
compared to a dilute solution, so a concentrated solution
has less entropy than a dilute solution.
As mentioned above, the Gibbs free energy is used to
describe transformations that occur at constant temperature and under constant pressure. It is a state variable,
meaning that it depends only on the state of the system
and not on the path required to get there. Thus there is
a defined difference in the free energy between an initial
state and a final state:
½1:7:21
ΔG 5 Gfinal 2 Ginitial
The energy involved in a chemical transformation can
be harnessed to do useful work. For example, a reaction
that gives off heat can be used to expand a gas to move
a piston, producing work. The relationship between the
free energy and the work that can be accomplished by a
chemical transformation is
½1:7:22
2ΔG $ W
where W is the work energy. That is the decrease in free
energy in any transformation at constant T and P is
equal to the maximum amount of work that can be performed by that transformation. It is for this reason that
the function G is called the “free energy”; it is the energy
which is available to perform work. The work that can
be performed could be mechanical or electrical or
chemical or concentration work.
Another variable determining the state of a system is
its composition. Suppose that the system is composed
of a number of substances, i, j, k, . . . of mole amounts
ni, nj, nk, . . . If a small amount of substance i is added,
then the system will experience a change in its free
energy, G. The chemical potential is
@G
½1:7:23
µi 5
@ni T;P;nj1i
Thus the chemical potential is the free energy per mole.
The Gibbs free energy is an extensive variable that
increases with the volume of the system or the number
of moles of materials in the system. The chemical potential is an intensive variable, meaning that it depends
on the state of the system and not its extent. However,
usually ΔG values are calculated per mole so that G
and µ are often used interchangeably. Equation [1.7.23]
can be integrated to give
X
½1:7:24
G5
µi ni
i
THE SIGN OF ΔG DETERMINES THE
DIRECTION OF A REACTION
One of the conclusions of thermodynamics is that natural processes always occur in such a way that the free
energy of the universe decreases. That is, in all natural
processes, ΔG in Eqn [1.7.21] is negative. A corollary of
this conclusion is the condition of equilibrium.
Equilibrium occurs when no further change can occur.
In this case, the free energy has reached a minimum,
and the free energy change is zero with respect to this
process. On the other hand, if ΔG is positive, then the
reverse reaction will occur spontaneously:
ΔG , 0.spontaneous reaction
ΔG 5 0.reaction is at equilibrium
ΔG . 0.opposite reaction occurs spontaneously
½1:7:25
PROCESSES WITH ΔG . 0 CAN
PROCEED ONLY BY LINKING THEM
WITH ANOTHER PROCESS WITH ΔG , 0
As described earlier, a natural process can proceed only
if ΔG , 0 for that process. Many processes that occur in
biological systems require energy, meaning that ΔG . 0.
These processes cannot proceed on their own. They can
be made to proceed, however, by linking the process for
which ΔG . 0 with another process for which ΔG , 0.
The combined processes will proceed spontaneously
only if the global ΔG for both of them is less than zero.
We will consider these kinds of processes in detail when
we consider active transport in Chapter 2.6. “Proceed
spontaneously” here means that the linked processes
will occur with no further input of energy outside the
combined processes. It does not mean that the processes
will occur rapidly or slowly. Thermodynamics tells us
whether or not a process will occur, and with what
energy changes, but it does not speak of the kinetics of
the processes. In this sense, the term “thermodynamics”
89
90
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Wcounter
currency. Energy derived from the oxidation of foodstuffs is stored in this terminal phosphate bond, and it is
used in myriad reactions involving synthesis of materials, transport of materials, and the performance of
mechanical work. The hydrolysis reaction of ATP to ADP
and Pi is shown in Figure 1.7.3.
The free energy change for the ATP hydrolysis reaction is
given by a combination of Eqns [1.7.21] and [1.7.24]:
ΔG 5 Gfinal 2 Ginitial
nfinal ATP µATP 1 nfinal ADP µADP 1 nfinal Pi µPi
5 2ninitial ATP µATP 2ninitial ADP µADP 2ninitial Pi µPi
Welevator
5 ΔnATP µATP 1 ΔnADP µADP 1 ΔnPi µPi
5 Δnrxn µADP 1 Δnrxn µPi 2 Δnrxn µATP
½1:7:26
FIGURE 1.7.1 Coupling of a spontaneous process to a nonspontaneous
process. An elevator sits on the ground floor. It will not rise spontaneously
because the free energy for that process is positive: ΔGelevator . 0. A large
counterweight at the top floor will fall spontaneously; the free
energy change for this process is negative: ΔGcounter , 0. The elevator can
be raised if it is coupled to the larger negative free energy of the
counterweight’s fall: ΔGelevator 1 ΔGcounter , 0. In this case, the elevator
can be raised only once. Repetition of the process would require coupling
to yet another process that provides the free energy to raise the
counterweight back up again. In a real process, some of the free energy
for the counterweight’s fall would be dissipated. One can never recover
100% of the energy as useful work.
is something of a misnomer, as really it is about the
statics of energy transformation.
An example of coupling is lifting a weight against gravity
by using another weight, as shown in Figure 1.7.1.
The coupling of biological process can be visualized
using the energy diagrams for chemical reactions such
as those shown in Chapter 1.5. Reactions that involve
a decrease in free energy (Gfinal 2 Ginitial , 0 and thus
Gfinal , Ginitial) are called exergonic reactions, and
they proceed spontaneously. A schematic example of
an exergonic reaction is shown in Figure 1.7.2A.
Reactions that involve an increase in free energy
(Gfinal 2 Ginitial . 0 and thus Gfinal . Ginitial) are called
endergonic reactions, and they do not proceed spontaneously (see Figure 1.7.2B). If an exergonic reaction
can be coupled to an endergonic reaction, and the sum
of the ΔG for the two reactions is less than zero, then
the combined processes will both proceed spontaneously (see Figure 1.7.2C).
THE LARGE NEGATIVE FREE ENERGY
OF ATP HYDROLYSIS POWERS MANY
BIOLOGICAL PROCESSES
ATP is adenosine triphosphate and its structure is
shown in Figure 1.7.3. It occupies a special position in
the cellular flow of energy because of the energy stored
in its terminal phosphate bond. It takes a lot of energy
to add a phosphate to ADP to form ATP, and that
chemical energy becomes available to do work when
the bond is split. The cell uses ATP as its energy
where the subscripts denote the chemical species, µ is the
chemical potential, and Δn is the number of moles of
each material that participates in the reaction. The last
line in Eqn [1.7.26] relates the change in the number
of moles of participating reactants to the number of
completed reactions. This is just the stoichiometry of the
reaction. We may write it as follows:
½1:7:27
1 ATP 5 1 ADP 1 1 Pi
2 1 ATP 1 1 ADP 1 1 Pi 5 0
The coefficients here are the stoichiometry. Here they
indicate that, for every completed reaction, the number of molecules of ATP decreases by one and the
number of molecules of ADP and Pi increases by one.
Thus the last line in Eqn [1.7.26] relates the change
in free energy upon completion of a Δn number of
reactions.
This number can be specified in moles because, as
described in Chapter 1.5, using moles is just another
way of counting a large number of things.
We divide the last line of Eqn [1.7.26] by the number
of completed reactions to obtain
½1:7:28
ΔG
5 µADP 1 µPi 2 µATP
Δnrxn
Substituting in for the chemical potentials, we get
ΔG
½1:7:29a Δn
½1:7:29b
5 µ0ADP 1 RT ln½ADP 1 µ0Pi 1 RT ln½Pi
2 µ0ATP 2 RT ln½ATP
ΔG
½ADP½Pi
5 Δµ0 1 RT ln
Δn
½ATP
If we will let Δn 5 1.0 to indicate that we are speaking
of the free energy change for Avogadro’s number of
completed reactions, then we have
½ADP½Pi
0
½1:7:30
ΔG 5 ΔG 1 RT ln
½ATP
where ΔG0 is the standard free energy change. It refers
to the free energy change per mole under standard
Gibbs free energy (kJ mol–1)
(A)
(B)
Exergonic process ΔGA < 0
process is spontaneous
Endergonic process ΔGB > 0
process is not spontaneous
Activated complex, A*
Final state, D
Initial state, B
Initial state, A
Final state, C
ΔGB > 0
ΔGA < 0
Progress of the reaction
Sum of A and B
(C)
Coupling of an exergonic process with an
endergonic process allows both to occur.
The free energy change of the exergonic
process drives the endergonic process.
A+B
Gibbs free energy (kJ mol–1)
Progress of the reaction
C+D
ΔGA+B < 0
Progress of the reaction
FIGURE 1.7.2 Coupling of an endergonic reaction with an exergonic reaction. Exergonic reactions are those for which ΔGA 5 Gfinal 2 Ginitial , 0, and so
these reactions proceed spontaneously (panel A). Endergonic reactions are those for which ΔGB 5 Gfinal 2 Ginitial . 0, and these reactions do not
proceed spontaneously (panel B). However, an endergonic reaction can be made to proceed if it can obtain a decrease in free energy by linking it to
the exergonic reaction. In essence, a coupled reaction involves a completion of both the exergonic and endergonic reactions. If the combined
ΔGA1B 5 ΔGA 1 ΔGB , 0, then the combined reaction will occur spontaneously (panel C).
NH2
O–
NH2
O–
O–
–O P
O P
O
O
O P O
O
N
N
O–
–O P
O
N
N
O–
CH2
O
O P O
O
N
N
H2O
OH OH
CH2
O
N
N
Adenosine diphosphate, ADP
OH OH
Adenosine triphosphate, ATP
O–
+
–O P
OH
O
Phosphate
FIGURE 1.7.3 ATP and its hydrolysis to ADP and Pi. Chemical energy is stored in each of the phosphate bonds of ATP. The last one, the γ-phosphate,
is typically used to power mechanical, electrical, and chemical energy needs of the cell.
92
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
EXAMPLE 1.7.1 Free Energy of ATP Hydrolysis Under “Typical” Cell Conditions
“Typical” values for the concentrations of ATP, ADP, and Pi are
about 5 mM for ATP, 5 mM for Pi, and 40 μM for ADP. Calculate
the free energy of ATP hydrolysis under these conditions.
0
Here we use Eqn [1.7.33]. The value of ΔG0 is given in the text
as 27.4 kcal mol21 or 230.9 kJ mol21. The value of R we use is
8.314 J mol21 K21 and T 5 37 C 5 310 K. In the calculation, all
concentrations must be expressed in M. We insert these
values and calculate:
ΔG 5 231:0 kJ mol21 1 8:314 J mol21 K21 3 310 K
5 231:0 kJ mol21 1 2:58 kJ mol21 3 ð210:12Þ
5 257:1 kJ mol21 5 213:6 kcal mol21
This free energy of ATP hydrolysis under cell conditions is the
energy available for the various kinds of work undertaken by
the cell, including chemical work (synthesis), mechanical work
(movement and transport), and electrical work. In the final
analysis, nearly all of the work produced by the cells is
eventually degraded and appears as heat.
3 ln½40 3 1026 M 3 5 3 1023 M=5 3 1023 M
conditions of 25 C, 1 atmosphere pressure and unit
concentration. Note that we have said all along that ΔG
is an extensive variable, and now it seems that we have
transformed it into an intensive variable. Tabulated
values of ΔG0 necessarily report it in units of energy per
mole, so these values are actually values of Δµ0.
You can see that if all species were at unit concentration,
the second term on the right-hand side of Eqn [1.7.25]
would be zero. Under these conditions, ΔG5ΔG0.
Experimental determination of ΔG0 takes advantage of
the fact that at equilibrium ΔG 5 0 (see Eqn [1.7.25])
and the fact that the second term in Eqn [1.7.30] incorporates the equilibrium constant for the reaction:
½ADP½Pi
Keq 5
½1:7:31
½ATP
So that at equilibrium (ΔG 5 0), Eqn [1.7.30] becomes
½1:7:32
ΔG0 5 2 RT ln Keq
MEASUREMENT OF THE EQUILIBRIUM
CONCENTRATIONS OF ADP, ATP, AND
PI ALLOWS US TO CALCULATE ΔG0
It turns out that the free energy of ATP hydrolysis is
a bit more complicated than we have let on here.
The chemical species produced have different ionizations at different pH values, and ATP and ADP both
bind Mg21 ions. The energy of binding of H1 and
Mg21 ions should be incorporated into the reaction.
In addition, we are interested in the hydrolysis of ATP
under physiological conditions of 37 C. The free energy
of ATP hydrolysis has been determined for these different
conditions. Since the details of these conditions differ
from cell to cell, no one value can be used. However,
a
0
“typical” value is given a special symbol, ΔG0 , and signifies the free energy of ATP hydrolysis under the “typical”
cell conditions. Its value is about 27.4 kcal mol21
or 231.0 kJ mol21.
The units of ΔG are those of RT. Values of R usually
used here are 1.987 cal mol21 K21 5 8.314 J mol21 K21.
The conversion
1 J 50.239 cal.
between
joule
and
calorie
is
Under cellular conditions, [ATP], [ADP], and [Pi] are
not at their equilibrium values, nor are they unit
concentrations. The free energy under these conditions
is given by
½ADP½Pi
0
½1:7:33
ΔG 5 ΔG0 1 RT ln
½ATP
It is important to realize that the argument of the logarithm ([ADP][Pi]/[ATP]) is generally not equal to the
equilibrium constant, but only when the reaction is at
equilibrium.
SUMMARY
A difference of concentration produces a flow of
material in solution. Application of an electric force
to charged particles also produces a flow of material
in solution. These two forces can be united in the
definition of a single force that is proportional to
the negative gradient of a potential. This potential is the
electrochemical potential written as
µ 5 µ0 1 RT ln C 1 z`Ψ
This electrochemical potential is the Gibbs free energy
per mole. The Gibbs free energy, G, is an extensive
variable, whereas µ is an intensive variable. G is also a
state variable, depending only on the state of a system
and not on the path it took to reach that state. The free
energy is the maximum energy that can be extracted
to do useful work. For all spontaneous reactions, the
change in free energy, ΔG 5 Gfinal 2 Ginitial, is negative.
Processes that require work occur spontaneously only if
they are linked to other processes that lose free energy,
so that for the overall process ΔG , 0. For any process
at equilibrium, ΔG 5 0.
Many cellular processes require energy for the synthesis
of materials, transport, or mechanical or electrical work.
These occur because energy is supplied by the hydrolysis
of ATP. ATP hydrolysis to ADP and Pi has a large
Electr ochemical P otentia l and Fre e E nergy
negative ΔG under cellular conditions. The free energy
of ATP hydrolysis per mole is given as
½ADP½Pi
0
ΔGATP hydrolysis 5 ΔG0 1 RT ln
½ATP
Under “typical” cellular conditions, ΔGATP hydrolysis 5213.6
kcal mol21 5 257.1 kJ mol21.
REVIEW QUESTIONS
1. In the formula for the electrochemical potential,
what is R? What is T? What are the units of
RT ln C? What units must C be in? What is z?
What is the Faraday? What is Ψ?
2. What is the Gibbs free energy?
3. What is the relationship between the electrochemical
potential and the Gibbs free energy?
4. How does the sign of ΔG determine the direction
of a process?
5. Describe thermodynamic coupling.
6. What is the relationship between the free energy
change and the equilibrium constant?
7. How would you determine the free energy change
for ATP hydrolysis under cellular conditions?
8. What is the approximate free energy change of
ATP hydrolysis in cells?
93
1.2
Problem Set
Kinetics and Diffusion
1. A. The empirical formula of glucose is
C6H12O6. What is its molecular weight?
B. Isotonic glucose is 5% (w/v) glucose. How
much glucose would we need to make 100 mL
of isotonic glucose?
2. A. You need to make 250 mL of a stock solution
of 0.1 M Na2 ATP. Its formula weight is
605.2 g mol21. How much Na2 ATP should you
weigh out?
B. Your advisor is skeptical of your abilities.
He wants you to check out the 0.1 M ATP
solution and tells you to do it spectrophotometrically. Spectrophotometry relies on the
different abilities of chemicals to absorb light
of specific wavelengths. A diagram of a spectrophotometer is shown in Figure 1.PS2.1.
At particular wavelengths, chemicals absorb
light according to their chemical structure and
their concentration. The law governing the
absorption of light is the BeerLambert Law:
A 5 εCd
where A is the absorbance; ε is a constant that
depends on the chemical and typically varies
with the wavelength of light—it is the molar
extinction coefficient and is in units of M21;
C is the concentration of the chemical (in M);
and d is the path length. The molar extinction
coefficient is defined for a path length of 1 cm.
The absorbance is defined as
A 5 logðI0 =IÞ
where I0 is the incident light intensity and I is
the transmitted light intensity. Your advisor tells
you that ε259 5 15.4 3 103 M21; this is the molar
extinction coefficient of ATP at a wavelength of
incident light of 259 nm. He tells you to make
a dilution of the stock by taking 25 μL of
the stock solution and diluting it to 100 mL.
What absorbance do you expect of the final
diluted solution, if you made it up correctly, at
λ 5 259 nm?
3. A. The molecular weight of ryanodine is
493.54 g mol21. You want to make 10 mL of a
10-mM stock solution. How much ryanodine
should you weigh out?
B. You make a dilution of the 10-mM ryanodine
stock by pipetting 10 μL of the stock solution
into a 10-mL volumetric flask and adding
Light source
Mirror
Motor to set
wavelength
Mirror
Sample cuvette
PMT
I0
I
Diffraction grating
FIGURE 1.PS2.1 Light path in a single beam spectrophotometer. The
view is from above. Light from a source is collimated (making a narrow
beam) and passed through a monochromator that selects a narrow
band of wavelength of light to be passed through the sample. A
photomultiplier tube (PMT) detects the light and measures its intensity.
Comparison of this intensity, I, to the intensity when the sample is
missing, I0, allows the calculation of the absorbance. Absorbance is
recorded with time or as a function of wavelength.
water to the mark. You measure the absorbance as a function of wavelength (against
a water blank, using a standard 1 cm path
length optical cell) and find a peak at 271 nm
with an absorbance of 0.179. What is ε271 for
ryanodine? (See Problem #2 for a discussion
of the BeerLambert Law and a definition of
the molar extinction coefficient.)
4. A. Magnesium chloride has a formula of
MgCl2 6H2O. What is its formula weight?
B. You desire to make 1 L of 0.1 MgCl2 solution. How much MgCl2 6H2O should you
weigh?
C. You need to make 25 mL of a 25-mM solution of MgCl2. How much of the 0.1 M stock
solution do you add to the 25 mL volumetric
flask?
5. The extracellular fluid volume varies with the
size of the person. Suppose in an individual
we determine that the ECF is 14 L. The average
[Na1] in the ECF is about 143 mM.
A. What is the total amount of Na1 in the ECF,
in moles? in grams?
B. Suppose this person works out and sweats
1.5 L with an average [Na1] of 50 mM.
During this time the urine output is 30 mL
94
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00009-4
Problem Set
with an average [Na1] of 600 mM. How
much Na1 is lost during the workout?
C. If the person does not drink fluids at all
during the workout, what will be the [Na1]
in the plasma at the end of the workout?
Assume that all of the fluid in the sweat and
urine originated from the ECF.
6. The body normally produces about 2 g of
creatinine per day. The amount varies with
individuals and is approximately proportional
to the muscle mass. It is excreted through
the kidneys according to urinary excretion
of creatinine 5 GFR 3 plasma concentration of
creatinine, where GFR is an abbreviation for
“glomerular filtration rate.” If the GFR is
120 mL min21, what is the plasma concentration of creatinine at steady state? Hint: Assume
the body is at steady state with respect to
creatinine.
7. Just before noon, your plasma glucose concentration was 100 mg dL21. This plasma glucose is
approximately evenly distributed among 3.5 L
of plasma and 10.5 L of interstitial fluid that
comprises your 14 L of ECF. Glucose is readily
distributed in both compartments. You drink a
can of soda that contains 35 g of glucose.
A. How much would your blood glucose rise if
all the glucose in the soda was absorbed and
none of it was metabolized?
B. Given that postprandial (after eating)
increases in blood glucose amount to maybe
40 mg dL21, depending on the meal, over a
period of an hour, give a crude estimate of
the rate of glucose uptake by the peripheral
tissues. Assume that the meal contains 100 g
of carbohydrates and all of it is absorbed in
1 hour.
8. The association reaction for Ca21 and EGTA
(a chemical that binds Ca21) is written as
Ca21 1 EGTA$CaUEGTA
Under defined and particular conditions of
temperature and ionic mixture, the association
constant was determined to be KA 5 2.52 3
106 M21. In a chemical mixture, 400 μM total
EGTA was included and the free [Ca21] determined by a Ca21-selective electrode was found
to be 4 3 1027 M. Assuming that there are no
other binding agents for Ca21, what is the total
[Ca21] in the mixture?
9. 2,4-Dinitrophenyl acetate decomposes in alkaline
solution with a pseudo-first-order rate constant
of 11.7 s21 at 25 C. It is a “pseudo”-first-order
rate constant because it depends on the pH.
A. If the initial concentration of DNPA is
1 mM, what is its concentration after
15 seconds?
B. At what time is the concentration reduced
to 0.5 mM (i.e., what is the half-life of the
reaction)?
C. After 5 minutes of reaction, what is the concentration of DNPA?
10. The following data were obtained for the rate of
the Mg, Ca-ATPase activity of vesicles of cardiac
sarcoplasmic reticulum as a function of temperature. What can you tell about the activation
energy?
Temperature ( C)
ATPase Rate (µmol min21 mg21)
6.9
11.5
15.8
19.8
20.2
25.6
26.1
31.0
34.8
39.2
0.068
0.138
0.300
0.568
0.585
1.236
1.154
2.238
3.030
4.220
11. Superoxide reduces cytochrome C in the reaction
21
Cyt CUFe31 1 O2
1 O2
2 .Cyt CUFe
where Cyt C Fe31 is the oxidized form and Cyt C Fe21
is the reduced form of cytochrome C. The reaction
can be followed spectrophotometrically at 550 nm.
The extinction coefficient for the reduced form of cytochrome C is εRED 5 2.99 3 104 M21 and the extinction
coefficient for the oxidized form εOX 5 0.89 3 104 M21
(V. Massey, The microestimation of succinate and the
extinction coefficient of cytochrome C. Biochimica et
Biophysica Acta, 34:255256, 1959). See Problem #2 for
a discussion of extinction coefficients and spectrophotometry. When xanthine oxidase converts xanthine to
uric acid, it produces superoxide that can be measured
using cytochrome C reduction. The following data were
obtained for A550:
Time (min)
A550
0
1
2
3
4
5
6
0.1326
0.1478
0.1637
0.1791
0.1941
0.2073
0.2202
A. Calculate the rate of cytochrome C reduction.
B. The xanthine oxidase was added in 75 μL of
6.5 mg XO per mL into a 3-mL reaction mixture. Calculate the specific activity of cytochrome C reduction (moles of cytochrome C
reduced per min per mg of XO protein).
12. You suspect you are anemic and your physician
orders some tests. He finds that your hemoglobin
is 13 g%. The molecular weight of hemoglobin is
66,500 g mol21.
A. What is the concentration of hemoglobin in
molar in your blood?
B. Each hemoglobin binds four oxygen molecules. If the hemoglobin is saturated with
oxygen, what is the concentration of O2
bound to Hb, in molar?
95
96
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
ATPase
ATP
CH3
CH2
C
O
Pyruvate
kinase
HO
Pyruvate
NADH + H+
O
P
OH
O–
C
O
Protein
O
C
C
HO
TABLE 1.PS2.1 Diffusion Coefficients and Mr for a
Variety of Proteins
ADP + Pi
Lactate dehydrogenase
(LDH)
CH3
H
C
D 3 107
(cm2 s21)
Milk lipase
6600
14.5
Metallothionein
9700
12.4
Cytochrome C
12,000
12.9
Ribonuclease
12,600
13.1
Myoglobin
16,890
11.3
Chymotrypsinogen
23,200
9.5
Carbonic anhydrase
30,600
10.0
O
Phosphoenol pyruvate
NAD+
Molecular
Weight
OH
C
HO
O
Lactic acid
FIGURE 1.PS2.2 ATP hydrolysis by pyruvate kinase converts
phosphoenolpyruvate to pyruvic acid. This is coupled by lactate
dehydrogenase to the conversion of pyruvic acid to lactic acid and
conversion of NADH to NAD1. The progress of the reaction can be
followed spectrophotometrically by the change in absorbance of NADH.
C. Convert the answer in B to volume using the
ideal gas equation, PV 5 nRT, where T is
the absolute temperature, R 5 0.082 L atm
mol21 K21, V is the volume that we seek,
and P 5 1 atm. The conditions for volume of
gas are usually STPD—standard temperature
and pressure, dry. The standard temperature is
0 C and pressure is 1 atm.
13. The rate of ATP hydrolysis by ATPases can be
followed by the coupled enzyme assay shown in
Figure 1.PS2.2. The progress of the reaction can
be followed by A340. The extinction coefficient of
NAD1 at 340 nm is negligible. The extinction
coefficient of NADH at 340 nm is 6.2 3 103 M21.
See Problem #2 for a discussion of extinction
coefficients and spectrophotometry. In one reaction, the concentration of Ca-ATPase was
0.22 mg mL21 and A340 was 0.65 at t 5 0 min
and 0.455 at t 5 2.0 min. What is the activity of
the Ca-ATPase in units of μmol min21 mg21?
14. Show by representative calculations that Stirling’s
formula
pffiffiffiffiffiffiffiffiffi
0
n! 5 2πn en ln ðn 2nÞ
is a good approximation for n! Use n 5 1, 2, 3,
4, 5.
15. Show that the equation
rffiffiffiffiffiffiffiffiffiffiffi
1 2X2
e 4Dt
Cðx; tÞ 5 C0
4πDt
obeys Fick’s Second Law of Diffusion.
16. The intestinal enterocytes form a covering over
the intestinal lining which, to the first approximation, can be considered to be a plane.
Assuming no binding or sequestration within
the cell, what is the estimated time of diffusion
Peroxidase II
44,050
6.8
Albumin
68,500
6.1
Lactoperoxidase
92,620
6.0
149,100
4.6
Aldolase
17.
18.
19.
20.
of Ca21 across the intestinal enterocyte? The
length of the enterocyte is 20 μm and assume
that the effective diffusion coefficient of Ca21 is
about 0.4 3 1025 cm2 s21.
Table 1.PS2.1 lists the diffusion coefficients and
the molecular weight of a variety of proteins.
What relationship can you deduce between the
size and the diffusion coefficients of these soluble proteins? (Hint: regress ln D against ln Mr).
Is the relationship you found consistent with
the StokesEinstein equation?
The free diffusion coefficient of oxygen in
aqueous solutions is about 1.5 3 1025 cm2 s21.
If the diffusion distance between air and blood
is 0.5 μm, about how long is the diffusion time?
Suppose a soluble protein has a molecular weight
of 45 kDa and a density of 1.06 g cm23. Suppose
further that the viscosity of the cytoplasm has
a viscosity of 0.005 Pa s (about five times that
of water—there is debate about the viscosity of
cytoplasm with numbers varying from 0.001 to
over 0.1 Pa s).
A. Estimate the diffusion coefficient for the
protein in the cytoplasm at 37 C.
B. If the proteins were synthesized in the cell
body, or soma, of a neuron in the spinal
cord, about how long would it take to
diffuse to the axon terminal 75 cm away?
Diffusion coefficients in cytoplasm have been estimated by a technique of photobleaching recovery.
In this technique, an area of the cytoplasm is irradiated with light to photobleach a fluorescent
probe. Recovery of fluorescence in the region is
achieved by diffusion of unbleached probes from
adjacent areas of the cytoplasm. The translational
diffusion coefficient can be estimated from the
half-time of fluorescent recovery. (D. Axelrod
et al., Mobility measurements by analysis of
fluorescence photobleaching recovery kinetics.
Problem Set
Biophysical Journal 16:10551069, 1976.) This
technique was applied to estimate the relative
viscosity of cytoplasm and nucleoplasm by microinjecting fluorescein isothiocyanate-labeled dextrans of varying molecular sizes and measuring the
fluorescence photobleaching recovery (I. Lang
et al., Molecular mobility and nucleoplasmic
flux in hepatoma cells. Journal of Cell Biology
102:11831190, 1986). These authors obtained
the following data:
Probe Molecular
Weight
(kD)
Equivalent
Radius
(nm)
D in
Dilute
Solution
D in
Cytoplasm
D in
Nucleoplasm
D is in units of 1026 cm2 s21
FD20
FD40
FD70
FD150
17.5
41.0
62.0
156.9
3.30
4.64
5.51
9.07
0.651
0.463
0.390
0.237
0.080
0.044
0.029
0.015
0.069
0.056
0.036
A. Plot D against 1/a, where a is the molecular
radius, for each of the solutions. From the
StokesEinstein relation, you would expect
the resulting curves to pass through the origin of zero diffusion coefficient with infinite
radius. Do the curves extrapolate back in this
way? Why or why not?
B. Regardless of the intercept, the slope of the
plot from part A ought to be related to the
viscosity of the medium. Use the slopes to
estimate the relative viscosity of the dilute
solution, cytoplasm, and nucleoplasm.
97
Cell Structure
Learning Objectives
organisms and cells in multicellular organisms must
solve a number of problems in order to survive. These
include:
G
G
G
G
G
G
G
G
G
G
G
G
G
List the main categories of cellular function
Describe the general structure, location, and function of the
plasma membrane
Describe the composition, location, and function of the
cytosol
Compare the general structure and function of microtubules, microfilaments, and intermediate filaments
Describe the general structure, location, and function of the
nucleus and its envelope
Describe the general structure, location, and function of the
mitochondria
Describe the general structure, location, and function of the
endoplasmic reticulum
Describe the general structure, location, and function of the
Golgi apparatus
Describe the general structure, location, and function of
lysosomes and peroxisomes
Describe the general structure, location, and function of
proteasomes
Explain the general purpose of ubiquitinylation of proteins
List the different types of cell-to-cell junctions
G
G
G
G
G
G
FOR CELLS, FORM FOLLOWS
FUNCTION
In Chapter 1.1, we discussed the following points:
G
G
G
G
2.1
Cells are the organization unit of life.
Cells come in a multitude of forms, specialized for
their function.
The vast majority of cells are somatic cells, and all of
these contain the same set of genetic information,
present in DNA and organized into genes.
The multitude of forms comes from using only specific sets of the genes to make proteins.
Multicellular organisms such as ourselves evolved because
multicellular structures can provide an internal environment that is more stable than the natural environment,
and thereby enhance the survival of the component
cells and of the organism. Free-floating, single-celled
organisms live at the mercy of environmental conditions,
whereas protected, multicellular organisms can better
withstand changes in the environment. Single-celled
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00010-0
Catalysis: Cells must be able to change one metabolite into another in order to synthesize cellular
constituents, degrade them, or provide energy.
Transport: Cells must be able to move things from
outside the cell to inside or from one compartment
to another within the cell. This includes bulk secretion or uptake of materials from the extracellular
fluid.
Signal transduction: Cells must have mechanisms
for responding to signals from other cells or from
within the cell. These may be chemical signals or
electrical signals.
Recognition: Cells attach to other cells and to extracellular structures. They must be able to recognize
where they should form attachments.
Movement: At some stage of their development, all
cells must be able to move so as to position themselves
properly within the cellular matrix that makes us up.
Control: All of the activities of the cell must be coordinated. Control here also means that cells must
select the parts of the genome that they will use.
“Control” thus implies differentiation—the formation of specialized cells uniquely suited to their task.
Proliferation: At appropriate times of development,
cells must make new cells. This involves cell division
and its control.
ORGANELLES MAKE UP THE CELL LIKE
THE ORGANS MAKE UP THE BODY
The cells of the body typically are composed of a
relatively small number of organelles that carry out
specific functions of the cell, much like our organs do
for us. These are called organelles because their relation
to the cell is like the organs’ relation to the body.
Table 2.1.1 lists these organelles along with their major
function. The disposition of these organelles in a “typical”
cell is shown in Figure 2.1.1.
THE CELL MEMBRANE MARKS THE
LIMITS OF THE CELL
The cell membrane, also known as the plasma
membrane, defines the inside and outside of the cell.
Like all biological membranes, it consists of two lipid 101
102
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Microvilli
TABLE 2.1.1 Major Organelles of the Cell and Their
Function
Organelles Function
Plasma
membrane
Customs officer of the cell: determines what gets
into or out of the cell, also signal transduction
and cell recognition
Cytosol
Cell sap: the fluid medium in which soluble
biochemicals diffuse and in which the other
organelles are suspended
Cytoskeleton Support, movement, and cell attachment
Cytoskeleton
Endocytotic
vesicle
Golgi
Transport vesicles
Secondary
lysosome
Polysome
CIS
face
Rough ER
Nucleus
Command center: contains the hereditary
material and organizes and controls differentiation
Free
ribosomes
Factory of the cell for soluble proteins
Rough ER
Factory of the cell for membrane proteins and
secreted proteins
Nucleolus
Smooth ER
Synthesis of lipids and steroids
Nucleus
Golgi
apparatus
Shipping department: finishing and targeting of
proteins to specific locations
Plasma membrane
Free ribosomes
Smooth ER
Peroxisome
Centrioles
Mitochondria Powerhouse of the cell: site of oxidation and
energy transfer
Lysosomes
Exocytotic
vesicle
Primary lysosome
Trans face
Nuclear membrane
Outer membrane
Inner membrane
Garbage disposal: destruction of worn-out
organelles
Mitochondria
Proteasomes Destruction of tagged proteins
Peroxisomes
Oxidation of fatty acids and detoxification of
xenobiotics
layers, or leaflets, in which are embedded a variety of
proteins that serve specific functions (see Chapter 2.4
for a discussion of the structure of lipids in biological
membranes). Both the inside and outside layers may be
coated with carbohydrate units that partly define the
function of the lipids. An important function of this
membrane is to determine what goes into and what
comes out of the cell. The “customs officer” of the cell
has several mechanisms that can transport materials
into or out of the cell. These include:
G
G
G
G
Passive transport
Active transport
Exocytosis
Endocytosis.
We will discuss all of these in more detail in Chapters
2.5 and 2.6. Passive transport requires no metabolic
energy and may involve diffusion through the lipid
layer of the membrane or through water-like channels
that are established by proteins that span the membrane. Active transport requires the input of cellular
metabolic energy and may be primary or secondary.
Primary active transport directly couples transport to
metabolic energy. Secondary active transport indirectly
links transport of a material to metabolic energy.
Exocytosis and endocytosis refer to the movement of
materials that are enclosed in vesicles. These vesicles are
tiny hollow spheres of membrane. Secretory vesicles
1×10–6 m
FIGURE 2.1.1 A “typical” human cell showing various subcellular
organelles including the plasma or cell membrane, nucleus, nuclear
membrane, rough ER, smooth ER, transport vesicles, Golgi apparatus,
mitochondria, cytoskeletal elements, lysosomes, peroxisomes, and
endocytotic and exocytotic vesicles. Although there is no “typical” cell,
most cells contain this set of organelles. Exceptions to this rule include
the adult erythrocytes.
are filled with some material by the cell. These vesicles
can fuse with the plasma membrane, and, in so doing,
they dump their contents into the extracellular space.
This process is called exocytosis. Endocytosis is similar
but occurs in the opposite direction. In this case, parts
of the plasma membrane invaginate and pinch off
to form endocytotic vesicles inside the cell. Endocytosis
of fluid is called pinocytosis or “cell drinking”; endocytosis of particulate stuff is called phagocytosis or
“cell eating.”
In addition to these functions, the cell membrane forms
the hub of signal transduction and surface recognition. It must transfer extracellular signals originating
from other cells to an intracellular signal inside the cell.
This is what is meant by “signal transduction.” The cell
membrane receives the first messenger in the form of a
chemical or electrical signal, and receipt of the first messenger causes the formation of a second messenger
inside the cell. Surface recognition occurs through surface proteins that are members of the major histocompatibility complex or MHC. These proteins are
responsible for beginning transplant rejections by recognizing the transplants as foreign matter.
Cell St ructur e
THE CYTOSOL PROVIDES A MEDIUM
FOR DISSOLUTION AND TRANSPORT
OF MATERIALS
The cell membrane surrounds all the constituents of the
cell, which themselves exist in a fluid medium that
allows transfer of materials among them. This fluid
medium is the cytoplasm, which literally means “cell
fluid.” It includes the cytosol and all of the organelles
suspended in it. The cytosol itself is the fluid that
contains dissolved ions and organic compounds of a
bewildering variety. These include amino acids for
building proteins, glucose for energy, a tremendous variety of metabolic intermediates, and cytoplasmic enzymes
for glycolysis, the first stage in burning carbohydrates for
energy. The ionic composition of the cytoplasm varies
with different cell types, but Table 2.1.2 gives some
reasonable approximate numbers for the “typical” cell.
The cytoplasm also serves as a medium for the transmission of control signals from the outer surface of the cell
to the interior, and from the nucleus to the rest of the
TABLE 2.1.2 Selected Components of the Cytosol
Component
1
Concentration
Na
14 3 1023 M
K1
120 3 1023 M
Cl2
10 3 1023 M
HCO32
10 3 1023 M
ATP
5 3 1023 M
ADP
50 3 1026 M
Mg
0.5 3 1023 M
Ca21
0.1 3 1026 M
21
pH
7.17.2
Osmolarity
295 mOsm L21
α-Tubulin
cell. Although we describe it here as a fluid, the cytosol
is not like water: ions diffuse through the cytosol slower
than they do through water (see Chapters 1.6 and 1.PS2,
Problem #20). Cutting a muscle fiber, for example, does
not cause its fluid to leak out like water. The cytoplasm
is more akin to a gel. Most of this behavior is due to
the small volume of fluid and the abundance of cell
surfaces. Although bulk water flows, a thin film adheres
to any wettable surface. This thin film generally exceeds
the thickness of a cell. Thus, at the cell level, surface
forces govern much of bulk fluid flow.
THE CYTOSKELETON SUPPORTS THE
CELL AND FORMS A NETWORK FOR
VESICULAR TRANSPORT
Arrays of protein filaments form a network within
the cytoplasm. These filaments determine the shape of
the cell and provide for the movement of the cell as a
whole or for the movement of organelles from one
part of the cell to another. There are four major types of
filaments comprising the cytoskeleton:
1.
2.
3.
4.
Microtubules
Intermediate filaments
Actin filaments or microfilaments
Myosin filaments.
MICROTUBULES ARE THE LARGEST
CYTOSKELETAL FILAMENTS
The microtubules are about 25 nm in diameter and
are constructed of heterodimers of tubulin, a globular
polypeptide of 50,000 Da. These dimers are assembled
into protofilaments of tubulin dimers with the
β-tubulin of one joined to the α-tubulin of the next.
The microtubules are assembled from 13 such protofilaments arranged in a cylinder with a hollow core
(see Figure 2.1.2).
A number of microtubule-associated proteins (MAPs)
bind to microtubules. Some of these MAPs are “motor
β-Tubulin
GTP GDP
8 nm
Singlet
Doublet
Protofilament
– End 25 nm
+ End
GTP
FIGURE 2.1.2 Schematic diagram of the structure of a microtubule. Microtubules consist of protofilaments composed of tubulin dimers, one α-tubulin
and one β-tubulin. Both bind GTP, guanosine triphosphate. GTP, like ATP, stores chemical energy in its phosphate bonds. Hydrolysis of GTP to GDP can
be used to stabilize protein shapes. The α-tubulin does not hydrolyze its GTP, whereas the β-tubulin hydrolyzes its GTP to GDP; 13 of the
protofilaments assemble to form a singlet microtubule. Because of the asymmetrical arrangement of the tubulin monomers, the microtubule has an
asymmetry, with a plus (1) end and a minus (2) end. The plus (1) end is the end pointing away from the origin of the microtubule and is the end to
which monomers add to the growing microtubule. These ends differ in the rates of tubulin association and dissociation. Because these rates differ, the
microtubule can treadmill—dissolve at one end while lengthening at the other. Tubulin can also form doublet and triplet structures. Cross-sections of
a singlet microtubule and a doublet microtubule are shown.
103
104
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
proteins” that can “walk” along the microtubule. Two
of these motor proteins are named kinesin and dynein.
Kinesin forms a family of motor proteins with about
40 members. Most of these are “ 1 directed” motors,
moving along the microtubules toward the 1 end.
Dyneins comprise a family of 2 directed motors. These
motor proteins can attach vesicles and then move along
the microtubules, carrying their vesicles along. In this
way, the microtubules can provide a track along which
intracellular transport occurs. This is especially important in neurons in which transport must occur down a
long narrow process of the cell. Figure 2.1.3 shows a
highly schematic cartoon illustrating kinesin and dynein
movement along a microtubule.
the cilia to bend. The waving cilia move the extracellular
fluid past the fixed cell. In this way, the movement of
the cilia causes the movement of extracellular fluid.
In the lungs, the cilia move mucus, trapped dust, and
foreign material toward the pharynx where they can be
expelled. In the Fallopian tubes, the cilia help move the
ovum toward the uterus. Figure 2.1.4 shows a schematic
diagram of the structure of a cilium. Activation of the
dynein arms linking two isolated microtubules will
ordinarily cause the two microtubules to slide past each
other. In the cilia, linking proteins turn this motion
into bending of the cilia.
Microtubules also form the mitotic spindle during cell
division. Nearly all cells possess two centrioles
oriented at right angles. These form a microtubule
organizing center (MTOC) that provides a scaffold
for the assembly of microtubules. Another kind of
tubulin, γ-tubulin, binds to accessory proteins to form
a γ-tubulin ring complex (γ-TuRC) that acts as a nucleation site for microtubules. The centrosome consists
Microtubules can also form the interior of larger structures called cilia that extend out from the cell into the
extracellular fluid. These cilia have a special arrangement of nine doublets arrayed circumferentially around
two central microtubules. In this case, the microtubules
are cross-linked such that the action of dynein causes
Cargo
Ankyrin
Dynactin
complex
Spectrin
Light chains
Dynein
Kinesin
– End
+ End
FIGURE 2.1.3 Schematic model of microtubule motor proteins. Kinesins typically have two globular heads and an elongated coiled tail. The tail regions
of most kinesins bind cargo, either membrane-enclosed vesicles or microtubules. Dyneins may contain two or three globular heads and a large
number of accessory proteins that bind vesicle cargo. Dynein itself is a complex assembly that requires a second complex assembly, dynactin, to
transport cargo. A possible arrangement of some of these structures is shown.
+
+
–
–
Plasma membrane
Radial spoke
Doublet microtubule
Outer dynein arm
Inner dynein arm
Linking protein
Cell membrane
FIGURE 2.1.4 Structure of a cilium. A cilium contains a “9 1 2” arrangement of microtubules. Nine doublet microtubules containing A and B
microtubules with 13 and 11 protomers, respectively, surround a central pair of microtubules. Multiple structures link these microtubules. The dynein
arms move toward the minus end of the microtubule. In isolated tubules, this would cause sliding of one microtubule past another. The linking
proteins turn this motion into a bending of the cilium.
Cell St ructur e
of the two centrioles surrounded
matrix containing many copies of
microtubules that grow out of this
plex provide tracks for chromosome
cell division.
by a centrosome
the γ-TuRC. The
centrosome commovement during
ACTIN FILAMENTS ARISE FROM
NUCLEATION SITES USUALLY IN THE
CELL CORTEX
Actin filaments are present in most cells but are
especially abundant in muscle cells. The monomer is a
globular protein called G-actin, with a molecular
weight of 41,800 Da. G-actin polymerizes noncovalently into actin filaments, called F-actin. Actin filaments consist of two strands of globular molecules
twisted into a helix with a repeat distance of about
36 nm. The filament is asymmetric having distinguishable ends that are detectable by the way in which it
interacts with myosin, another protein that is present
in many cell types but is especially abundant in
muscle. Thus the actin filament also has a plus end
(the growing end) and a minus end (the nucleation
or beginning end). Each individual actin filament is
about 3.5 nm across, so that F-actin has a diameter of
about 7 nm. Assembly and stabilization of filamentous,
or F-actin, is described in Figure 2.1.5.
Actin filaments determine the shape and movement
of the cell’s surface, including structures such as
microvilli, which are fingerlike extensions of epithelial
cells that line internal structures like the intestinal villi
and kidney tubules. The membrane of these cells
anchors the actin filaments and extends them into
a web of cytoskeletal elements in the main body of the
cell. Other proteins can cross-link actin microfilaments
together to form bundles of filaments or gel-like networks. These cross-linking proteins include α-actinin,
fimbrin, and villin, which bundle actin filaments
together. Spectrin and filamin both have two actinbinding sites. They join two actin filaments together to
form a web of supporting filaments.
INTERMEDIATE FILAMENTS
ARE DIVERSE
Intermediate filaments were originally named because
with diameters between 8 and 10 nm, they are intermediate in size between the microtubules (at 25 nm) and
the microfilaments at 7 nm. These intermediate filaments are composed of a number of different proteins.
They play some structural or tension-bearing role. They
differ from microtubules and microfilaments in that:
G
Both microtubules and microfilaments are made
by the polymerization of globular monomeric
Thymosin binding inhibits actin
association
Thymosin
ARP-2, ARP-3 complex
nucleates actin polymerization
G-actin is activated by
binding ATP
G-actin
ATP
Profilin binding activates
actin binding to the plus end
ARP complex
After polymerization, actin
hydrolyzes its ATP but keeps
ADP tightly bound
Profilin
Capping protein (Cap Z )
+
–
–
+
Tropomodulin caps some
F-actin at the minus end
Cap Z caps some F-actin
at the plus end
FIGURE 2.1.5 Assembly and stabilization of microfilaments (actin filaments). Actin binds ATP and begins assembly by binding to actin-related proteins
(ARPs) that serve as a nucleation site, usually just under the cell membrane in the cortex of the cell. The ARP complex can also bind F-actin on the side
of the filament, so it can build a tree-like web from individual actin filaments. After assembly, actin hydrolyzes its bound ATP, but the ADP remains
tightly bound. Formation and stabilization of F-actin is regulated by proteins that bind the free monomer. Thymosin binds to the free monomer and
inhibits its association with either the minus or plus end of the F-actin. Profilin binds to the free monomer and inhibits its association with the minus
end but markedly enhances its association with the plus end. Cap Z binds to the plus end of the F-actin and stabilizes it. The minus end can be
stabilized by remaining bound to the ARP complex. In muscle cells, tropomodulin binds to the minus end and stabilizes it.
105
106
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Head
Rod
Tail
45 nm dimer
N terminal
C terminal
60 nm tetramer
2 dimers
60 nm unit-length filament
8 tetramers
Full length
filament
> 240 nm
FIGURE 2.1.6 Highly schematic representation of the structure of intermediate filaments. The elementary subunit of the intermediate filaments
consists of an elongated rod with an N-terminal “head” and a C-terminal “tail.” A variety of these elementary subunits are made by the body. These
associated laterally to form a homo- or heterodimer, approximately 45 nm long. These dimers further associate laterally, with an offset, to form a 60nm-long tetramer. Eight of these tetramers further associate laterally to form a unit length filament. These unit length filaments can associate end to
end to form longer filaments. The width of the mature filaments is not eight times the width of the tetramer, as these associate in three dimensions to
form a mature filament about 11 nm in diameter. After the initial association into full length filaments, the filaments are radially compacted to form
the mature filaments.
G
G
G
proteins, but the intermediate filaments are made of
elongated (45 nm) and thin (23 nm) rod-like
dimers. The intermediate filament units align with
their long axis parallel to the filament axis, and
filament width is determined by lateral association
of the dimers (see Figure 2.1.6)
Both microtubules and microfilaments are polar,
which allows the active movement of motor proteins
with their associated cargo along the filaments.
Assembled intermediate filaments have no polarity
because individual monomers are oriented in both
directions along the axis of the filament.
Intermediate filaments differ from both microtubules
and microfilaments in that reversible association
and dissociation of intermediate filament dimers can
occur all along the length of the filament, whereas
association and dissociation of microtubules and
microfilaments occur only at their ends. This process
is called dynamic subunit exchange. However, the
exchange occurs much slower than the exchange of
subunits in microtubules and microfilaments.
Unlike tubulin and actin, the subunits of the
intermediate filaments do not bind a nucleotide.
The intermediate filaments are diverse; some 73 separate proteins in humans have been identified encoded
by over 70 genes. They all consist of three parts: a
“head,” a long rod-like central part, and a “tail.” The
members of the IF family have been subdivided into
five distinct groups based on their structure, mode of
assembly, and developmental expression in different tissues. These groups and their types are summarized in
Table 2.1.3. There is considerable variation within types.
For example, there are over 50 different varieties of
keratin.
CYTOSKELETAL UNITS FORM FREEFLOATING STRUCTURES BASED ON
TENSEGRITY
Buckminster Fuller in the 1960s invented the word
“tensegrity” as a blend of tension and integrity. He used
it to describe architectural structures of remarkable
rigidity that were composed of compressive rods and
elastic cables. These two elements can be combined to
form stable structures. Cells cannot rely on their membranes for structural stability because the membranes
themselves are weak. But if you drape the membranes
over cytoskeletal elements, structural strength can be
achieved. In this view, the microtubules are the rigid
rods and intermediate filaments are the tension-bearing
elements. The actin and myosin elements allow for the
movement of the cytoskeleton and the consequent
movement of the attached membrane. In this way, the
cell can extend processes or move from one place to
another.
Cell St ructur e
TABLE 2.1.3 Classification of the Intermediate Filaments
Types of IFs Protein
Tissue Distribution
Proposed Function
Associated Diseases
Type I
Type II
Acidic keratins
Basic keratins
Epithelial tissues
Epithelial tissues
Tissues strength and
integrity
Epidermolytic hyperkeratosis
Type III
Desmin
GFAP
Peripherin
Vimentin
Syncoilin
Muscle
Glial cells
Sarcomere organization
Dilated cardiomyopathy
Alexander disease
Type IV
Neurofilaments
NF-L, NF-M, and
NF-H
Nestin
Synemin α, β
Neurons
Axon organization
Amyotrophic lateral sclerosis; Parkinson disease
Type V
Nuclear lamins type
A, B1, B2, C1,C2
Nucleus
Nuclear organization and
signaling
HutchinsonGilford progeria; limbgirdle muscle
dystrophy; EmeryDreifuss muscular dystrophy
Type VI
Filensin
Phakinin
Lens
Cataracts
Cytoplasmic filaments
Outer nuclear membrane
Transporter
subunit
Ring subunit
Column subunit
Lumenal subunit
Inner nuclear membrane
50 nm
Nuclear lamina proteins
Basket
FIGURE 2.1.7 Cartoon of the structure of the nuclear pore. The nuclear pores consist of a complex of more than 50 different proteins that form a
complicated structure with octagonal symmetry. Small molecular weight materials (, 20 kD) can pass through these pores in both directions, but the
movement of larger materials, such as RNA and ribosomes, is regulated. The outer membrane faces the cytosolic compartment and the inner
membrane faces the nuclear compartment. Two rings made of eight subunits each are connected by columnar scaffold subunits. Lumenal subunits
anchor the scaffold to the membrane. The ring subunits connect to fibrils that form a basket structure on the nuclear side. Transport subunits in the
interior of the pore actively transport materials into or out of the nucleus.
MYOSIN INTERACTS WITH ACTIN TO
PRODUCE FORCE OR SHORTENING
THE NUCLEUS IS THE COMMAND
CENTER OF THE CELL
Myosin also exists in multiple isoforms. A major form is
a protein of about 200 kDa that forms a homodimer
with two long tails forming a coil, a hinge region, and a
head region that binds actin filaments, hydrolyzes ATP,
and “walks” along the actin filament. The interaction of
the actin filament relative to the myosin filament causes
either shortening of the acto-myosin thread or production of force. This mechanism is responsible for muscle
force and also produces movement in nonmuscle cells.
Most cells have linear dimensions on the order of
2050 μm. The nucleus is the largest organelle, with a
diameter of about 35 μm. The nucleus is bounded by
a double membrane, the nuclear envelope, that has
pores for materials to move between nucleus and cytoplasm, as shown in Figure 2.1.7. The nucleus contains
nearly all of the DNA of the cell. As described in
Chapter 2.2, this DNA carries the information that
allows the synthesis of specific proteins. The nucleus
107
108
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
also contains a specialized region called the nucleolus.
This is a diffuse region that is not delimited by a
membrane. The nucleolus is involved in the synthesis of
ribosomes.
RIBOSOMES ARE THE SITE OF PROTEIN
SYNTHESIS
Ribosomes exist either free in the cytoplasm or bound
to membranes of the rough endoplasmic reticulum
(ER). Both types consist of two main subunits, designated 60S and 40S, where the S refers to Svedbergs and
describes how fast the particles sediment during centrifugation (see Appendix 2.1.A1). The larger subunit consists of three different strands of ribonucleic acid (RNA)
and about 49 different proteins. The smaller subunit
has a single RNA strand and about 33 other proteins.
Both are assembled in the nucleolus, a specialized
region of the nucleus that is not membrane bound.
There, large loops of DNA containing ribosomal RNA
genes are transcribed by the enzyme RNA polymerase I
to ribosomal RNA or rRNA. Ribosomal proteins made
in the cytoplasm are imported back into the nucleus
and assembled, along with the rRNA, into the two subunits. The two subunits join to form a functional ribosome in the cytoplasm that makes a platform on which
proteins are synthesized, as described more fully in
Chapter 2.2.
THE ER IS THE SITE OF PROTEIN AND
LIPID SYNTHESIS AND PROCESSING
The ER is a membranous network within the cell
fluid, or cytoplasm, that is continuous with both
the outer nuclear membrane and the plasma membrane. The membrane forms flattened disks with an
enclosed space called cisternae. Electron micrographs
and density gradient centrifugation reveal two functionally distinct regions of the ER; these are rough
ER and smooth ER. The “rough” ER was given that
name because the many ribosomes attached to the
membrane give it a granular appearance in electron
micrographs. The smooth ER lacks these attached
ribosomes.
Special proteins, called translocons, span the ER
membrane and bind ribosomes to the cytoplasmic
face of the rough ER. Protein synthesis occurs on
the ribosomes. Some of these are free within the
cytoplasm, and the proteins made there are generally
soluble proteins that remain in the cytoplasm. The
ribosomes on the ER synthesize proteins that pass
through the ER membrane as they are being synthesized. These proteins may become embedded in
membranes or they may be destined for secretion from
the cell. After synthesis, many proteins are further
processed within the ER cisternae, preparing them for
secretion or targeting them for some location within
the cell.
THE GOLGI APPARATUS PACKAGES
SECRETORY MATERIALS
The Golgi apparatus consists of sets of membranedelimited smooth-surfaced cisternae. Each set of flattened, disk-shaped cisternae resembles a stack of pancakes. This structure is called a Golgi stack or
dictyosome. It is about 1 μm in diameter and is usually
located near the nucleus and near the centrioles that
define the cell center. The number of cisternae in a stack
varies from 6 to 30, and the number of Golgi stacks in
the cell varies enormously with the biochemical activity
of the cell.
Golgi stacks are polarized with two distinct faces. The cis
or forming face is nearest a smooth transitional portion
of the rough ER. The trans or maturing face typically
faces the plasma membrane. Swarms of small vesicles
(about 50 nm in diameter) cluster on the cis face of the
Golgi stack. A large number of vesicles associate with the
sides of the stack near the dilated rims of each cisternae.
In electron micrographs, these vesicles sometimes appear
to bud off the Golgi cisternae. In secretory cells, larger
vesicles containing high concentrations of secreted
proteins appear to originate from the trans face of a
Golgi stack. The Golgi stacks are a processing station for
proteins manufactured in the rough ER. Proteins made
in the rough ER travel to the Golgi through the small
transport vesicles. In the Golgi, the proteins are
processed and packaged for delivery to various locations
throughout the cell, including packaging for eventual
fusion with the plasma membrane and secretion into
the extracellular space (see Figure 2.1.8).
THE MITOCHONDRION IS THE
POWERHOUSE OF THE CELL
Mitochondria produce much of the cell’s ATP by
coupling the chemical energy of oxidation of metabolites to the synthesis of ATP. Their main structural
features are shown schematically in Figure 2.1.9. The
matrix contains many different enzymes required for
the oxidation of pyruvic acid and fatty acids, including
those involved in the tricarboxylic acid cycle. The mitochondrial matrix also includes mitochondrial DNA,
special mitochondrial ribosomes, tRNAs, and enzymes
that are required for the expression of mitochondrial
genes. The inner mitochondrial membrane contains a
number of important proteins that collectively comprise
the electron transport chain and another enzyme
complex called the ATP synthetase that makes ATP
from ADP and Pi, the reverse of the ATP hydrolysis
reaction discussed in Chapter 1.7. These complexes
are discussed further in Chapter 2.10. Briefly, these
complexes couple the chemical energy derived from the
oxidation of fuels obtained from food to the synthesis
of ATP. This is the site of oxygen consumption by
aerobic cells.
Lynn Margulis originally postulated that the mitochondria
in aerobic cells that contain a nucleus (eukaryotic cells)
Cell St ructur e
Apical cell membrane
Zymogen granules
Condensing vacuole
trans face
originated from the engulfment of aerobic bacteria
by anaerobic single-celled organisms. This hypothesis,
called the endosymbiotic hypothesis, derives from
the similarity of mitochondria to bacteria. Both have
circular DNA; both are approximately the same size;
both reproduce by dividing into two, asexually; mitochondrial ribosomes resemble bacterial ribosomes
rather than eukaryotic ribosomes, and both bacteria
and mitochondria share a slightly different genetic
code from that in the nucleus.
LYSOSOMES AND PEROXISOMES ARE
BAGS OF ENZYMES
Retrograde
transport
vesicles
Golgi stack
cis face
Rough ER
Ribosomes + mRNA
FIGURE 2.1.8 Packaging of secreted proteins in secretory cells of the
pancreas. Proteins are synthesized on the membrane of the rough ER
and translocated into the lumen of the ER as they are being made. The
ER forms transport vesicles that fuse to form the cis face of the Golgi
stack. These membranes progress through the stack as new layers are
added on the cis face and taken away on the trans face. On the trans,
or maturing, face of the Golgi stack, the enclosed proteins are
collected in secretory vesicles. These then fuse into condensing
vacuoles that concentrate the proteins to form zymogen granules.
These granules lie in the apical aspect of the secretory cells, adjacent
to the plasma membrane. Upon stimulation, these granules fuse with
the apical membrane and release their contents of secretory enzymes
into the lumen of a duct, or channel, that takes the enzymes into the
intestine.
Lysosomes are membranous bags of hydrolytic enzymes
including proteases, nucleases, glycosidases, lipases,
phospholipases, and phosphatases. These hydrolytic
enzymes are acid hydrolases, being optimally active
in an acid environment. Lysosomes are typically
0.20.3 μm in diameter. They originate from the trans
face of the Golgi stack and are formed first as primary
lysosomes. The primary lysosome fuses repeatedly with
a variety of membrane-bound substrates including
endocytotic vesicles, phagocytotic vesicles, and worn-out
intracellular organelles. After fusion, the combined
vesicle forms a secondary lysosome. Because of its
diverse substrate contents, the secondary lysosomes have
a diverse morphology. Lysosomes degrade phagocytosed
material and worn-out parts of the cell.
The peroxisome is another membrane-bounded vesicle,
with a diameter of about 0.5 μm. It contains oxidative
enzymes such as catalase, d-amino acid oxidase, and
urate oxidase. Like the mitochondria, the peroxisomes
are a major site of O2 utilization. The peroxisome
detoxifies foreign chemicals and metabolizes fatty
acids. Beta-oxidation, a process in which fatty acids are
shortened by two carbons to form acetyl-coenzyme A,
occurs in both mitochondria and peroxisomes (see
Chapter 2.11).
Outer mitochondrial membrane
Matrix
Cristae
Intermembrane space
Inner mitochondrial membrane
FIGURE 2.1.9 Typical features of a mitochondrion. The mitochondria, shown cut longitudinally, are membrane-delimited structures about 0.51.0 μm
wide and 14 μm long. The outer mitochondrial membrane is permeable to many materials with molecular weights below 10 kDa. The inner
mitochondrial membrane is impermeable to most materials but can transport specific materials. It is folded to form the shelf-like cristae and
encloses the matrix. The intermembrane space lies between the inner and outer membranes.
109
110
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
PROTEASOMES DEGRADE MARKED
PROTEINS
Proteasomes are large, multisubunit protein complexes
that are scattered throughout the cytoplasm and that
degrade cell proteins. Proteins are tagged for degradation by the attachment of a ubiquitin molecule to the
proteins. Ubiquitin is a protein consisting of 76 amino
acids. Proteins can be marked by more than one ubiquitin chain through a complex series of reactions involving several different enzymes. This process is called
ubiquitinylation, in which a series of enzymes recognizes the proteins to be degraded and adds activated
ubiquitin onto them. Different sets of enzymes in this
pathway recognize different degradation signals on proteins. In this way, each set of enzymes targets distinct
subsets of proteins that bear particular degradation signals. The process of ubiquitinylation is illustrated in
Figure 2.1.10.
1. a barrier function that disallows free movement of
materials between cells in an epithelial sheet. This
is the so-called “tight” part of tight junctions, but
in reality the junctions vary considerably in their
permeabilities.
2. a fence function that prevents the free migration
of membrane components from the apical surface
Ub
ATP
AMP + PPi
Ub
E1
E1
E2
E3
Ub
E2
The proteasome consists of a central hollow cylinder
capped at both ends. The central cylinder consists of a
stack of four 7-membered rings. The caps recognize
the ubiquitin coats of proteins that have been tagged for
degradation. Each cap contains ATPase activity, releasing
chemical energy in the process. These caps use the energy
to unfold the ubiquitinylated proteins and move them
into the central cylinder where proteases cleave the
protein into its constituent amino acids. The structure
of the proteasome is shown in Figure 2.1.11.
E3
Ub
Ub
Ub
Substrate
E2
E3
Proteasome
CELLS ATTACH TO EACH OTHER
THROUGH A VARIETY OF JUNCTIONS
Cells form a variety of attachments to each other or to
the extracellular matrix. These include:
G
Zonula occludens: This “tight” junction joins epithelial cells in an occluding zone at one pole of the
epithelium. It has three functions:
FIGURE 2.1.10 The ubiquitinylation reaction. Ubiquitin, noted as Ub in
the figure, is a 76 amino acid protein that is used to “tag” proteins as
being ready for degradation. First, Ub is attached to E1 through an ATPrequiring reaction. It is then passed from E1 to E2, and from there to the
protein substrate being tagged for demolition. The ubiquitinylated
protein is then degraded by a specialized cell structure called the
proteasome.
19S regulatory particle
Lid
Base
α ring
β ring
β ring
α ring
20S core particle
Chambered protease
Complete proteasome
FIGURE 2.1.11 Structure of the proteasome. The overall structure consists of a 20S core particle of 28 subunits capped on one or both ends by a 19S
regulatory particle containing at least 19 subunits. The core particle consists of four 7-membered rings that is symmetrical about a plane perpendicular
to the long axis of the particle. Proteolytic activity resides in the two middle layers of the core particle.
Cell St ructur e
G
G
G
of the cell to the lateral surface. This effectively
partitions the cell membrane into components.
3. a signal function that help regulate cell proliferation, differentiation, and polarity.
Zonula adherens: This is a belt of attachment that
typically surrounds epithelial cells just below the
zonula occludens—meaning toward the basolateral
pole of the cell.
Desmosomes: These are “spot welds” between cells
and are constructed of different proteins than those
that make up zonula adherens.
Gap junctions: These junctions serve to electrically
connect cells because they allow small ions to pass
from one cell to another, and these ions carry electrical
current.
Each of these junctions is made up of complexes
of many proteins and a variety of proteins can associate
with these. Figure 2.1.12 illustrates the structure of the
desmosome, zonula adherens, zonula occludens, and
gap junction, and Figure 2.1.13 shows their use in an
epithelial sheet in the small intestine, consisting of a
lining of cells that separate the ingested food and gastrointestinal secretions from the blood. Many of the
proteins that make up these structures are present in the
body in various isoforms—variants of the protein that
serve the same basic function but in different tissues.
For example, there are some 20 different types of connexins that associate as hexamers to form the connexons in gap junctions.
Adherens junction
Gap junction
Microtubule
Plekha7
Nezha
β-catenin
p-120 α-catenin
Vinculin
Actin filament
Cadherins
α-actinin
Connexon
Plakoglobin
Connexin
Nectin
Afadin/AF6
Desmosome
Desmocolin
Desmoglein
Tight junction (zonula occludens)
Cell membranes
Plakophilin
Plakoglobin Intermediate
filaments
Jam
ZO1
Claudin
ZO2
Inner dense
Outer dense
plaque
plaque
Desmoplakin
Dense midline
Occludin
Jam
FIGURE 2.1.12 Schematic diagram of the proposed structures of gap junctions, adherens junctions, and desmosomes. Gap junctions form by linking
of connexons on opposing membranes. Each connexon consists of a hexamer of connexin units that form a central pore. When two connexons link,
the pore of one lines up with the pore of another, forming a watery path between the cells. This allows diffusion of small, soluble materials from
one cell to another without crossing the cells’ membranes. Adherens junctions form by interaction of extracellular parts of cadherin molecules.
These proteins are embedded in the cell membrane and have a short cytoplasmic tail. This cytoplasmic tail binds to a variety of proteins including
p-120, plakoglobin, and β-catenin. These in turn bind other proteins that eventually form initiation sites for actin polymerization. Cadherin also
directly binds microtubules, although other proteins may stabilize this interaction. Desmosomes adhere two cells together because of the
interaction of extracellular domains of desmoglein and desmocolin. These penetrate the membrane and their cytoplasmic domains bind other
proteins, plakophilin, plakoglobin, and desmoplakin, that eventually connect to intermediate filaments. Tight junctions consist of binding
of claudins and occludins, along with junctional adhesion molecule (JAM). There are multiple isoforms of each of these that produce junctions of
varying permeability. These are stabilized by multiple cytoplasmic accessory proteins, including zonula occludens proteins (ZO-1, ZO-2, and ZO-3).
These can connect to the cytoskeleton through other proteins such as afadin. In the junction between three cells, a special protein called tricellulin
is required to seal the gap between the cells.
111
112
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Tight junction: seals adjacent cells in
an epithelial sheet
Adherens junction: joins actin bundles
in adjacent cells
Actin
Desmosome: joins intermediate filaments
in adjacent cells
Junctional
complex
Intermediate filaments
Gap junction: joins cells with a watery
channel
Connexons
Hemidesmosome: anchors the cell to
extracellular matrix through intermediate
filaments
Basal lamina
FIGURE 2.1.13 Cell attachments found in an epithelial sheet. The intestinal cells form a layer, referred to as an epithelium, that lines the intestine. The
intestinal cells are bound at their apical pole, the side facing the gastrointestinal lumen, by a junctional complex consisting of tight junction, adherens
junction, and desmosomes. The tight junction and adherens junction form a belt completely surrounding the cell. The desmosomes are located in
spots. The cells are also joined by gap junctions that allow free passage of small molecular weight materials between the cells. The cells attach to the
extracellular basal lamina through hemidesmosomes that connect the fibers in the extracellular matrix with the cytoskeletal intermediate filaments.
SUMMARY
The cell is the fundamental organizational unit of the
body. Although cells come in many different forms,
they share many features. Each cell consists of a number
of organelles, so named because they contribute to
overall cell function in much the same way as our
organs contribute to bodily function.
The cell membrane determines the inside and outside of a
cell. This is the “customs officer” of the cell, determining
what enters or exits the cell. Materials move through the
cytoplasm, the watery cell fluid that transports materials
from one place to another, largely by diffusion. A cytoskeleton maintains cell shape and provides movement.
Cytoskeletal elements include microtubules, which are
hollow rods formed from 13 protomer filaments of a heterodimer of tubulin, actin filaments, composed of a double helix of actin monomers strung together, intermediate
filaments of various descriptions, and myosin filaments.
These cytoskeletal elements are dynamic and allow the
cell to change shape and to transport materials along cytoskeletal tracks. The nucleus is the single largest organelle,
and it is the “command post” of the cell. It is enclosed by
a double membrane that is pierced by numerous nuclear
pores. In response to signals, the nucleus “expresses”
select regions of the genome. This means that the nucleus
specifically converts some DNA into mRNA, but not all.
The mRNA then makes proteins in the “factory” of the
cell located on ribosomes either free in the cytoplasm
or bound to the surface of the rough ER, another membranous network in the cytoplasm. The smooth ER makes
lipids; the rough ER makes membrane-bound proteins
and proteins destined for export from the cell. The rough
ER transfers its protein content to the cis side of the
Golgi apparatus, which is the “shipping and packaging”
department of the cell. The materials move from one
part of the Golgi apparatus to another in tiny membrane
spheres called vesicles. At each stage, the proteins are
processed further. The final vesicles leaving the trans
face of the Golgi stack are ready for export from the cell
by exocytosis.
All of these activities of the cell require chemical energy
supplied as ATP. The mitochondria make ATP by coupling the chemical energy liberated by oxidation of
foodstuffs to the synthesis of ATP. Thus the mitochondrion is the “powerhouse” of the cell. It consists of a
double membrane structure. The electron transport
chain is on the highly folded inner membrane. This
membrane synthesizes ATP from chemical precursors
and is the main site of oxygen consumption by the cell.
The cell also contains a variety of other organelles
including the lysosome, peroxisome, and proteasome.
These structures degrade material engulfed by the cell
Cell St ructur e
by endocytosis and also degrade worn-out organelles
and cell proteins.
Cells form a variety of attachments to other cells
and to the extracellular matrix. These include zona
occludens and adherens junctions, gap junctions, and
desmosomes. These form by the complex association of
a variety of different proteins: connexons on opposite
membranes link up to form the gap junction; cadherins
link up to form adherens junctions; desmoglein and
desmocolin form the desmosomes; claudin and occludin link up in the tight junction. These junctions
form by the association of extracellular parts of these
transmembrane proteins. The intracellular parts join
up with still other proteins that eventually connect the
junctions with the cell’s cytoskeleton.
REVIEW QUESTIONS
1. What is the plasma membrane? What compartments does it separate? What is endocytosis?
Exocytosis? Pinocytosis? Phagocytosis?
2. What is the major cation (positively charged ion
that migrates toward the cathode, the negative
electrode) of the cytosol? Is the cytosol comparable
to water?
3. What makes up the cytoskeleton? What is a
microtubule? What is a microfilament? What
is an intermediate filament? How do intermediate filaments differ from microtubules and
microfilaments?
4. What does the plus (1) end of a microtubule or
microfilament mean? What does the minus (2)
end mean? What are “motor proteins”? What
does it mean to be “ 1 directed”? Name the
family of 1 directed motor proteins. Name
the family of 2 directed motor proteins. How do
motor proteins carry cargo?
5. Describe the structure of a nuclear membrane.
Why does the nuclear membrane have an elaborate pore structure? How big is the nucleus?
What is the nucleolus?
6. What do ribosomes do? What are they made of?
Where are they made? What do “40S” and
“60S” descriptions of the major ribosomal
subunits mean?
7. What distinguishes “rough ER” from “smooth
ER”? What does each make?
8. What is the Golgi apparatus? What is the cis
face? Trans face? What are all those vesicles
doing hanging around the Golgi rims and cis
and trans faces? What goes on inside the Golgi
cisternae?
9. How big is a mitochondrion? Why do you
suppose it has two membranes? Where is the
electron transport chain? What do the mitochondria do?
10. What do lysosomes contain? What is the difference between a primary and a secondary lysosome? What goes on inside peroxisomes?
11. What do proteasomes do? How do they know
which proteins to degrade?
12. Name four junctions between cells. Which ones
involve connexons? Which ones join actin
filaments from one cell to another? Which ones
join intermediate filaments from one cell to
another?
APPENDIX 2.1.A1 SOME METHODS
FOR STUDYING CELL STRUCTURE AND
FUNCTION
THE MICROSCOPE HAS REVOLUTIONIZED OUR
UNDERSTANDING OF BIOLOGY
The invention of the microscope literally opened up a
new world view in biology. For the first time, we could
look upon the microscopic world and we discovered
that it was full of animated objects. Robert Hooke
(16351703) first used the microscope to study biological material in 1665 and coined the term “cell” from
the likeness of the empty cells in cork to the monks’
cells within a monastery. Antoine van Leeuwenhoek
(16321723) was the first to observe live cells with
hand-made microscopes that could magnify up to 500
times. He observed an astonishing array of cells from
single-celled protists to red blood cells, bacteria, sperm,
and muscle fibers. These observations, along with many
others, prompted Theodor Schwann to enunciate the
Cell Theory in 1839, based in part on conversations
with Matthias Schleiden, whom Schwann did not credit.
It contained three important elements:
1. The cell is the unit of structure, physiology, and
organization in living things.
2. The cell is a distinct entity but is the building
block of complicated organisms.
3. Cells form from cell-free material, as in
crystallization.
We now know that this last point is incorrect. Its first
correction was famously uttered by the German
pathologist, Rudolf Virchow, when he said “Omnis
cellula e cellula”—all cells come from cells. The entire
development of the cell theory was supported by a single
advance—the light microscope.
MICROSCOPIC RESOLUTION IS THE
ABILITY TO DISTINGUISH BETWEEN
TWO SEPARATED OBJECTS
What do we mean when we say that red blood cells,
erythrocytes, are invisible to the naked eye? Surely we
can see blood. What we mean here is that we cannot
make out the individual cells. The ability to distinguish
two objects separated by a distance is called the
resolution.
Light produces a diffraction pattern around all objects.
This pattern consists of areas of maximum and minimum intensity of light. The resolution of optical devices
is determined by considering the diffraction pattern produced by a circular aperture of radius a. This circular
aperture is called an Airy disk. The convention is that
two apertures can be distinguished if the first maximum
113
114
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
structure of cells only after artificial preparation.
Without functional studies, the activity going on in the
structures remains elusive.
Objective
α
Specimen
Condensor
Light
FIGURE 2.1.A1.1 Angle of the cone of light passing through the
specimen and incident to the objective.
of the diffraction pattern from one pattern falls on the
first minimum of the second. This is called the Rayleigh
criterion. The diffraction pattern can be described
analytically using Bessel functions, which we will not
do here. The result is given by
½2:1:A1:1
Resolution 5
0:61 λ
η sin α
where λ is the wavelength of light, η is the refractive
index of the medium between the specimen and the
lens, and α is the angle of the cone of light that passes
between the specimen and the lens (see Figure 2.1.
A1.1). The expression η sin α is called the numerical
aperture of the lens being used. This description of resolution is the inverse of what is ordinarily meant.
Eqn [2.1.A1.1] defines resolution in terms of a distance.
However, when we can resolve two objects that are
close together, we ordinarily speak of a high resolution.
According to Eqn [2.1.A1.1], the resolved distance is
smallest (and the resolution is highest) when α is 90
and the refractive index is increased by placing oil
between the specimen and lens. The refractive index of
air is close to 1.0, whereas that of oil is 1.41.5.
THE ELECTRON MICROSCOPE HAS
ADVANCED OUR UNDERSTANDING OF
CELL STRUCTURE
Until the 1950s, the workhorse of cellular structure was
the optical microscope. Our ideas of the structure
and composition of cells were further enhanced by the
electron microscope. This device allows tremendous
magnification of cell components but suffers from the
disadvantage that the cells must be fixed and stained
before viewing their structures because the electron
microscope views specimens in a vacuum. Otherwise,
the incident electron beam would be scattered by air
molecules. The electron microscope illuminates the
The electron microscope can achieve much higher
resolution (it can resolve objects that are separated by a
smaller distance) than the optical microscope because the
wavelength of electrons is so much shorter than the wavelength of visible light. The fact that electrons have a wavelength at all was an astounding discovery. The first clue to
the wavelength of electrons was the Compton effect,
reported by Arthur H. Compton in 1922. Compton found
that incident X-rays were scattered by a carbon target, subsequently shifting the X-rays to a lower wavelength and
dislodging an electron from the crystal. The dislodged
electron has a momentum. To describe this collision of
light with electrons, it was necessary to postulate that the
incident light possessed momentum, in order to preserve
the conservation of momentum theorem of physics.
Compton produced a quantum-mechanical analysis of
the scattering that differed from classical explanations,
but agreed with the experimental observations.
The photoelectric effect and Compton effects showed
that light had distinctive particle-like aspects. It was
already known, from interference and diffraction experiments, that light also had distinctive wave-like aspects.
Louis de Broglie in 1924 postulated that the wave
particle duality of photons might also apply to particles;
if photons have momentum, particles such as the electrons ought to have a wavelength. From the Compton
experiments, the wavelength would be given by
½2:1:A1:2
λ5
h
mv
where h is Planck’s constant 5 6.625 3 10234 J s. The
wave nature of electrons was confirmed in 1927 by
Davisson and Germer, who exposed a single crystal
of nickel to electrons having 54 electron volts of kinetic
energy. They observed an electron diffraction pattern
that confirmed de Broglie’s relation.
The short wavelength of electrons opened the possibility
of a microscope with astounding resolution. The electron
microscope, invented in the 1930s, was first applied
to living tissues by Albert Claude, Keith Porter, and
George Palade in the late 1940s and 1950s.
The resolution of the electron microscope is determined
not only by the wavelength of the incident radiation,
which in this case is a beam of electrons, but also
by the numerical aperture. Although theoretically the
resolution of the electron microscope should be close to
0.002 nm, in principle it is much larger than this
because the inherent properties of magnetic lenses limit
the aperture angle to about 0.5 .
Because biological specimens lack inherent contrast, the
practical resolution is further reduced to about 12 nm.
Nevertheless, this is a marked improvement over
the optical microscope. The optical microscope was useful to a magnification of about 1000 3 ; the electron
microscope could attain more than a 100-fold better
magnification.
Cell St ructur e
SUBCELLULAR FRACTIONATION
ALLOWS STUDIES OF ISOLATED
ORGANELLE BUT REQUIRES
DISRUPTION OF CELL FUNCTION AND
STRUCTURE
Although the optical microscope and electron microscope provided keen insights into the structure of living
things, the function of the structures could not be
directly investigated. In the late 1940s and 1950s,
Albert Claude and Christian de Duve developed
methods for subcellular fractionation for separating
cells into their component parts and elucidating the
function of the cellular constituents. This method disrupted the cells and separated the parts by differential
centrifugation.
DIFFERENTIAL CENTRIFUGATION
PRODUCES ENRICHED FRACTIONS OF
SUBCELLULAR ORGANELLES
The first step in subcellular fractionation is the disruption of the cell into its component subcellular organelles. This process usually uses homogenization, and
its aim is to break the plasma membrane that delimits
the cell, thereby releasing the cellular contents, without
damaging those contents. Cells can be homogenized by
sonication (exposure to high-frequency sound waves),
shearing the cells between two surfaces such as a Teflon
mortar and glass pestle, or placing the cells in a
high-speed blender. These treatments break the cell
membrane and leave the remaining parts of the cells
relatively intact. These relatively harsh treatments
obviously disrupt some of the normal relationships
between parts of the cell, and may scramble normal
constituents of cells.
The resulting mixture, the homogenate, can be separated into its component parts on the basis of their size
and density (see Figures 2.1.A1.2 and 2.1.A1.3).
Following lysis of the cells, the homogenate is placed in
a tube in a centrifuge and spun at relatively low speed
(1000 3 gravity) for a short time (1020 minutes). The
centrifugation causes materials to move away from the
axis of centrifugation. When the particles reach the bottom of the tube, they form a pellet. This process is
called sedimentation. Particles that sediment quickly
reach a terminal velocity in the centrifuge tube. At this
terminal velocity, the frictional drag on the particle provides the acceleration necessary to keep the particle in
approximate uniform circular motion (see below for an
explanation of the forces during sedimentation). The
frictional drag depends on the density of the particle,
viscosity of the medium, and speed of centrifugation.
The first centrifugation, at low speed and short times,
sediments unbroken cells, and the nuclei because these
Separate supernatant
and pellet
Homogenization
1
4
2
Supernatant
5
Pellet
Centrifugation
at higher speed
Centrifugation
at low speed
6
3
Rotor
FIGURE 2.1.A1.2 Separation of subcellular organelles by differential centrifugation. Whole tissue is first homogenized, which disrupts cell membranes
and releases subcellular organelles. The homogenate is then centrifuged to separate out particles on the basis of their sedimentation. In general, large
particles sediment with small centrifugal forces and smaller particles require larger forces. Successive centrifugation at progressively faster revolutions
per minute (RPM) separates organelles on the basis of their sedimentation characteristics.
115
116
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
2 Separate supernatant
and pellet
1 Low-speed
centrifugation
3 Medium-speed
centrifugation
4 Separate supernatant
and pellet
5
6 Separate supernatant
and pellet
High-speed
centrifugation
Homogenate
Pellet contains
whole cells, nuclei
Pellet contains
mitochondria,
lysosomes
Pellet contains
microsomes
FIGURE 2.1.A1.3 Differential centrifugation. The first slow speed spin causes large and heavy particles to sediment. These are separated from the
particles that do not sediment. Successively faster centrifugations cause progressively smaller and lighter particles to sediment.
are the largest and heaviest of subcellular structures.
Therefore, a fraction of the homogenate is produced
that is enriched in nuclei. The supernatant fraction, the
suspension that lies above the first pellet, is cleared of
nuclei but still contains other subcellular particles.
Successive centrifugations at higher speeds and longer
times produce fractions enriched in the mitochondria,
lysosomes, and peroxisomes. Further centrifugation at
still higher speeds sediments the broken fragments of
the plasma membrane and endoplasmic reticulum. De
Duve identified these subcellular fractions through the
use of marker enzymes. The basic idea here is that each
of the subcellular structures has a unique biochemical
composition that enables their unique biochemical
function within the cell. Part of their biochemical composition is their component enzymes. By measuring the
distribution of an enzyme, the scientist can track the
distribution of the particles that contain it. Markers for
the mitochondria, for example, include cytochrome C
oxidase and succinate dehydrogenase, among others.
By measuring cytochrome C oxidase in the various fractions, one can estimate the amount of mitochondria in
those fractions. De Duve deduced the existence of the
lysosome on the basis of an enzyme that distributed
itself differently from all other known markers.
DENSITY GRADIENT CENTRIFUGATION
ENHANCES PURITY OF THE FRACTIONS
The centrifuge is a crude instrument for the separation
of subcellular fractions because of the way in which it
separates the different subcellular organelles. Differential
centrifugation can produce fractions enriched in one
particle or another, but the fractions are not pure. This is
due to the fact that sedimentation occurs over the
considerable length of the centrifuge tube. When the
tube is spun, particles throughout the tube are subject to
centrifugal forces that cause them to sediment. Although
heavier particles sediment more quickly, the heavier particles at the top of the tube have much further to travel
than those near the bottom. By the time all of the heavy
particles sediment, some of the lighter particles near the
bottom of the tube, or at the middle, also sediment.
Therefore, pure fractions of subcellular particles cannot
be achieved easily by simple differential centrifugation.
Further purification can be achieved by using density
gradient centrifugation. In this method, subcellular
organelles are separated by centrifugation through a
gradient of a dense substance, such as sucrose. In
velocity centrifugation, the material to be separated is
layered on top of a sucrose gradient, and then centrifuged. Particles of different sizes and density sediment
through the gradient at different rates moving as discrete
bands. At the end of the centrifugation, the different
layers consist of purified organelles, and they can be collected for further experiments. In equilibrium centrifugation, the density gradient is used to separate particles
based on their buoyant density. Instead of being
separated by their sedimentation velocity, particles will
sediment until they reach a layer with the same density
as the particles. At this point, sedimentation stops and
the purified organelles can be collected at the equilibrium position. These methods of separating subcellular
organelles are illustrated in Figure 2.1.A1.4.
ANALYSIS OF CENTRIFUGATION
SEPARATION
CIRCULAR MOTION REQUIRES AN INWARD
CENTRIPETAL FORCE
Centrifugation typically involves spinning tubes of material at a constant angular velocity, except for the angular
acceleration to that velocity, and the deceleration when
the spin stops. The position of any particle in the tube at
any time may be represented by the vector r, as shown
in Figure 2.1.A1.5.
Cell St ructur e
Velocity sedimentation
Centrifugation
Equilibrium sedimentation
Slow-sedimenting
component
Fast-sedimenting
component
Shallow sucrose
gradient (5–20%)
Centrifugation
Low buoyant
density component
High buoyant
density component
Steep sucrose
gradient (20–60%)
FIGURE 2.1.A1.4 Comparison of velocity sedimentation and equilibrium sedimentation for the separation of subcellular particles. In velocity
sedimentation, separation relies on different velocity of sedimentation through a shallow sucrose (or other material) gradient. The sucrose is added to
prevent mixing by convection. In equilibrium sedimentation, subcellular particles sediment until they reach a zone of solution that matches their
density. At equilibrium, the organelles distribute themselves over a narrow region of the tube corresponding to the density of the organelle. The
organelles can then be harvested from a narrow band.
v = dr
dt
r = i cos ωt + j sin ωt
θ = ωt
FIGURE 2.1.A1.5 A particle in uniform circular motion around a central
pivot point. The angular displacement is a linear function of time.
This vector makes the angle θ from an arbitrary zero
reference, the x-axis, and this angle increases with time.
The angular velocity is defined as
dθ
dt
By integrating this we see easily that:
½2:1:A1:3
ω5
½2:1:A1:4
The position vector indicating the location of the point
relative to the center of rotation is thus given as
-
-
-
r 5 i cos ωt 1 j sin ωt
where r is the position vector and i and j are unit vectors
along the x-axis and y-axis, respectively. The velocity vector at any time is the derivative of this position vector:
~
r
5 2 i ω sin ωt 1 j ω cos ωt
dt
This velocity vector is orthogonal to the position vector
as seen by the dot product: it is given as
½2:1:A1:6
-
-
½2:1:A1:8
-
V 5d
r U V 5 2 ω sin ωt cos ωt 1 ω sin ωt cos ωt 5 0
½2:1:A1:7
~
a 5d
V
5 2 i ω2 cos ωt 2 j ω2 sin ωt
dt
~
r
a 5 2 ω2~
We see here that the magnitude of the acceleration is
ω2r and its direction is directly opposite that of the position vector (the negative sign in Eqn [2.1.A1.8]).
(Compare Eqn [2.1.A1.8] to [2.1.A1.5] for the identity
of r in a.) This is the centripetal acceleration. In order
for the particle to remain in uniform circular motion,
the velocity must be continuously bent toward the center. The acceleration vector is orthogonal to the velocity
vector, so that all of the acceleration is used to change
the direction of the velocity, and not its magnitude.
CENTRIPETAL FORCE IN A SPINNING
TUBE IS PROVIDED BY THE SOLVENT
As described above, particles that are spinning in a rotor
at a constant angular velocity, ω, are subjected to a
centripetal acceleration given by
½2:1:A1:9
θ 5 ωt
½2:1:A1:5
This means that the velocity vector is oriented at 90 to
the position vector, as shown in Figure 2.1.A1.5. The
acceleration at any time is the derivative of the
velocity:
a 5 ω2 r
where a is the acceleration, r is the radius, and ω is
the angular velocity, in radians per second (52π 3
revolutions per second). This centripetal acceleration is
the acceleration necessary to keep a particle rotating
about the axis, at a distance r. If the actual force is less
than this, the particle will move away from the axis of
rotation. In the centrifuge, the centripetal acceleration
is provided by collisions with solvent particles. The net
force of these collisions under ordinary conditions
(i.e., not in the centrifuge) adds to the force of gravity
on the particle. In the centrifuge, the centripetal force
necessary to keep the particle rotating at angular velocity
ω at distance r from the axis is
½2:1:A1:10
Fc 5 ðmparticle 2 msolution Þω2 r
Here the mass of solution is the mass of the volume
of solution which is displaced by the particle and is
117
118
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
the origin of the buoyant force. Eqn [2.1.A1.10] can be
rewritten as
Fc 5 ðm 2 Vp ρÞω2 r
½2:1:A1:11
5 ðm 2 mvρÞω2 r
The relative centrifugal field (RCF) is given as
ω2 r
½2:1:A1:15
RCF 5
g
22
where g 5 981 cm s or 9.81 m s22.
5 mð1 2 vρÞω2 r
where m is the mass of the particle, V is the partial
specific volume, equal to 1/ρ for the particle, and ρ is
the density of the solution.
As pointed out above, this centripetal force is the force
required to maintain an orbit at angular velocity ω at a
distance r from the axis of revolution. The source of this
force is the collisions with solvent molecules, which
have a net direction toward the axis of revolution
only when there is a net velocity of the particle in the
opposite direction. That is, a particle more dense than
the solution will move toward the bottom of the tube
(away from the axis of revolution) with some velocity
relative to the tube. Because it is moving through the
solution, it experiences a drag force that is proportional
to the velocity. This drag force on a particle moving
outward from the axis of revolution is given by
½2:1:A1:12
FD 5 2βV
where β is a frictional coefficient. This equation says
that the drag force is proportional to the velocity but
opposite in direction. A terminal velocity, v, is reached
when there is a balance between Fc and FD. From Eqns
[2.1.A1.11] and [2.1.A1.12], this is
½2:1:A1:13
mð1 2 vρÞω r 5 βv
2
THE MAGNITUDE OF THE CENTRIPETAL
FORCE CAN BE EXPRESSED AS
RELATIVE CENTRIFUGAL FORCE
The frame of reference for the analysis presented so far
is the nonrotating frame of the laboratory. One can
also view the situation from the accelerated, rotating
frame of reference of the solution within the rotor.
In this case, there is an apparent force on every particle,
the centrifugal force, which is equal but opposite to the
centripetal force, which appears to drive particles
heavier than the solution to the bottom of the tube.
This motion within the tube is opposed by the frictional
force, given above, which is opposite to the direction of
motion. The terminal velocity is reached when the
centrifugal force is equal to the frictional force, as given
by Eqn [2.1.A1.13]. Note that the centrifugal force is a
fictional force which must be invented in order to apply
Newton’s laws in a uniformly accelerated frame of
reference. The centrifugal force is just that force given
by Eqns [2.1.A1.10] and [2.1.A1.11]. Usually the centrifugal force is given in multiples of g, the acceleration
due to gravity:
½2:1:A1:14
a5
ω2 r
g
g
THE VELOCITY OF SEDIMENTATION IS
MEASURED IN SVEDBERGS OR S UNITS
The terminal velocity within the tube can be written as
dr
½2:1:A1:16
V5
dt
where r is the distance from a sedimenting particle and
the center of rotation. Insertion of this definition into
Eqn [2.1.A1.13] and rearranging, we obtain:
2 3
dr
6 7
mð1 2 vρÞ 5 β 4 dt
½2:1:A1:17
5
ω2 r
The term in the brackets on the right-hand side of the
equation defines the sedimentation coefficient, s. The
sedimentation coefficient is the rate of sedimentation
per unit of centrifugal force. Sedimentation coefficients
are usually of the order of 10213 s. They are reported in
Svedberg units, where 1 Svedberg 5 10213 s. This unit
is named after T. Svedberg, an early pioneer in the
design of centrifuges and their use in investigations of
biological material.
The sedimentation coefficient is obtained experimentally by plotting the logarithm of the radius of the maximum of the concentration profile against the time. To
see this, consider again the definition of the sedimentation coefficient:
dt
½2:1:A1:18
s 5 dr
ω2 r
multiplying through by ω2.
dr
ω s 5 dt
r
2
½2:1:A1:19
5
d ln r
dt
Thus the slope of a plot of ln r against t will give ω2s.
The sedimentation coefficient varies with the concentration of solute, usually decreasing as the total protein
concentration increases. The sedimentation coefficient
at infinite dilution, s0, is usually obtained by extrapolation of plots of 1/s against protein concentration to zero
protein concentration.
Sedimentation information can sometimes inform us
about molecular dimensions. Eqn [2.1.A1.17] can be
rearranged to give
½2:1:A1:20
mð1 2 vρÞ
5β
s
Cell St ructur e
where β is the frictional coefficient described in Eqn
[1.2.A1.12]. The Stokes equation gives the frictional
coefficient as
fluorescence microscopy and confocal microscopy. The
main advantage in these techniques is that intact and
living cells can be investigated.
½2:1:A1:21
In fluorescence microscopy, specific components of the
cells can be labeled by attaching a fluorophore to
them. The fluorophore is a fluorescent molecule.
It absorbs light at one wavelength and emits it at a second, lower wavelength. The location of the fluorescent
molecule is achieved by illuminating the specimen at
the excitation wavelength and using filters to collect the
emitted light. For example, incubating cells with a
fluorophore-tagged antibody directed against a specific
protein allows the study of the distribution of the
protein. This technique can be used with either living
or fixed cells. In other experiments, native proteins
expressed by cells can be "tagged" with a fluorescent
protein, green fluorescent protein (GFP by incorporating the gene for GFP into the gene for the protein. Thus
the location of the tagged protein can be followed by
fluorescent microscopy.
β 5 6πηas
where η is the viscosity of the solution and as is the
radius of the molecule, assuming spherical geometry.
Thus the measurement of s and the knowledge of the
molecular mass, m, and its partial molar volume allow
estimate of its size.
DENSITY GRADIENT CENTRIFUGATION
Macromolecules or subcellular organelles can be sedimented through gradients of increasing density on a
preparative scale to purify them. There are a variety of
materials which can be used to prepare such density
gradients, including CsCl, D2O, Ficoll, glycerol, sorbitol,
sucrose, and percoll. The gradients usually are formed
by mixing two limiting solutions in varying proportions
in order to produce the desired gradient. Gradients
can be discontinuous, in which solutions of varying
densities are layered on top of each other manually, or
continuous. Continuous gradients can have a variety
of shapes, although linear gradients are most common.
Materials will sediment until they reach a solution with
the same density as the particles, and then they stop
sedimenting.
OTHER OPTICAL METHODS
A number of other optical approaches have proven useful
for the modern investigation of cell function. These
include phase-contrast microscopy and differential
interference microscopy. Both of these optical techniques
use variation in the refractive index of cell structures,
rather than variation in light absorption, to produce
contrast between the structures. These images can be
clarified by using video cameras and computerized image
analysis and processing. Other techniques include
Confocal microscopy allows for optical sectioning of
live cells. Confocal microscopy refers to the idea that
both incident and emitted light are in focus. In a bright
field, light illuminates the specimen and the objective
lens is moved to focus the light. However, out-of-focus
light still reaches the detector. In confocal microscopy,
an aperture is placed in front of the detector so that
out-of-focus light is eliminated. The result is that the
image is formed from a narrow plane of in-focus light.
By moving the plane up and down, one can obtain a
series of optical sections. By computer techniques,
three-dimensional reconstructions of the object can be
obtained. Because the image is obtained and stored
digitally, the resulting image can be viewed from any
perspective. Confocal microscopy can be combined
with fluorescent probes to achieve outstanding detail of
localization.
119
2.2
DNA and Protein Synthesis
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
G
G
Compare genotype and phenotype
Describe the components of a nucleotide
Name the different nucleotides that comprise DNA and
RNA
Describe how the nucleotides are connected to form
single-stranded DNA or RNA
Describe how hydrogen bonding is useful in combining
antiparallel nucleotide strands to form the double helix
Explain why antiparallel strands require special arrangement
for DNA replication
List the ways RNA differs from DNA
List the various kinds of RNA and their function in the cell
Know the origin of the RNA polymerases responsible for
formation of the various RNA classes
Describe what is meant by “the genetic code”
Define and contrast “transcription” and “translation”
Distinguish among “response elements,” “intron,” and
“exon”
Describe histones and their postulated function in the
structure of chromosomes
Describe what is meant by the “histone code”
Describe how DNA is methylated and how this methylation
can be passed from parent to daughter cells
DNA MAKES UP THE GENOME
As described in Chapter 1.1, almost all cells in the
body have the same amount and kind of DNA.
The diversity of human cell forms derives from their
expression of different parts of the DNA. The total
DNA with its division into units, called genes, constitutes the genome. Expression of a gene means that
the DNA that makes up the gene is used to direct the
synthesis of a specific protein. As we will see in this
chapter, genes associate with a host of proteins that
regulate the expression of the genes. The human
genome refers to the set of genes that are normally
present in humans.
120
The DNA in human cells is organized into compact
units called chromosomes, meaning “colored body,”
which refers to their appearance in fixed and stained
preparations. Each chromosome carries a defined set of
genes that carries the instructions for making a set of
proteins. Because each chromosome is paired, nearly
every gene comes in pairs, but the two pairs are usually
not identical. Paired genes carry the instructions for the
synthesis of analogous materials, but they differ in the
details. These alternate forms of the genes in a single
person are called alleles. The set of alleles of a particular
person is called the genotype. The set of proteins
and other materials that the person actually makes, and
which determine their outward appearance and behavior, is called the phenotype. Humans have 23 pairs of
chromosomes.
Two chromosomes determine the sex of the individual.
These are the X and Y chromosomes. Persons with
two X chromosomes are genotypic females; having
one X and one Y makes a genotypic male. Because the
Y chromosome is smaller than the X, some of the genes
carried on the X chromosome are not paired. This is the
one exception to the rule that all genes are paired.
DNA CONSISTS OF TWO INTERTWINED
SEQUENCES OF NUCLEOTIDES
DNA IS BUILT FROM NUCLEOTIDES
DNA stands for deoxyribonucleic acid. It is located
primarily in the nucleus of cells but important parts are
also present in the mitochondria. It is composed of
a sequence of building blocks called nucleotides.
These nucleotides come in two different types and four
varieties. The types are the purines and pyrimidines.
The purines in DNA are adenine and guanine, and the
pyrimidines are thymine and cytosine. Each of these
nucleotides consists of the base (adenine, guanine, thymine, and cytosine) linked to a sugar, deoxyribose, and
phosphate. The chemical structures of the nucleotides
are shown in Figure 2.2.1.
NUCLEOTIDES ARE LINKED TOGETHER
TO FORM A CHAIN
These four bases are linked together to form a long
sequence of nucleotides. The DNA is elongated by
reacting a nucleotide triphosphate (with two more
phosphates linked to the phosphate shown in
Figure 2.2.1) on the 30 end of an existing chain. This
reaction is catalyzed by an enzyme called DNA polymerase. This enzyme is involved in the replication of
DNA, where two complete DNA strands are made from
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00011-2
DNA and P rot ein Sy nt hes is
NH2
O
H
N
5′ End
–O
P
O
O
Phosphate
CH2
5
N
O
N
N
OH
OH
N
–O
NH2
3
2
Deoxyribose
P
5
O
1
4
5′ End
O CH2
3
Base
2
3′ End
OH
Deoxy guanosine monophosphate
Deoxy cytidine monophosphate
O
NH2
H3C
N
OH
–O
P
O
5′ End
O CH2
5
N
O
3
NH
N
OH
–O
N
P
5′ End
O CH2
5
O
1
4
O
1
4
3′ End
OH
N
O
2
O
1
4
3
3′ End
OH
Deoxy adenosine monophosphate
N
O
2
3′ End
OH
Deoxy thymidine monophosphate
FIGURE 2.2.1 Structures of the nucleoside monophosphates.
one, using the original DNA as a template. The structure
of single-stranded DNA is shown in Figure 2.2.2.
The single-stranded DNA described in Figure 2.2.2
is just half of the story. In humans, DNA is normally
present as double strands that are held together by
hydrogen bonds, as shown in Figure 2.2.3. In doublestranded DNA, adenine on one strand pairs with
thymine on the opposite strand and guanine on one
strand pairs with cytosine on the opposite strand.
The two strands have opposite polarity: the 50 end of
one strand is opposite to the 30 end of the other.
O
–O
P
THE DNA TEMPLATE SETS THE SEQUENCE
OF NUCLEOTIDES
0
DNA polymerase adds nucleotides to the 3 end, using
a nucleotide triphosphate as a substrate. The base
on the opposite strand determines which nucleotide is
incorporated. Thus DNA polymerase replicates DNA on
the basis of the DNA already present. The DNA strand
unwinds to form two single strands. The DNA polymerase adds nucleotides on both strands to form two
complete DNA double strands. The hydrogen bonding
between nucleotides is crucial to the ability of DNA to
N
Guanine
5′ End
O
CH2
5
O
N
O
N
NH2
1
4
3
NH2
2
3′ End
N
N
O
Adenine
5′ End
–O
P
O
CH2
5
O
N
O
N
1
4
3
HYDROGEN BONDING ALLOWS FOR DNA
STABILITY WITH RAPID DISSOCIATION
As discussed in Chapter 1.4, hydrogen bonds involve
sharing of the positive H atom between two electronegative centers. It requires the right spatial separation and
orientation of these centers, and has low dissociation
energy. This allows the H-bond to form or break rapidly. However, many hydrogen bonds can stabilize large
structures like DNA and proteins.
H
N
OH
2
O
H2C
3′ End
Thymine
NH
O
5′ End
P
–O
O
CH2
5
O
N
O
O
1
4
3
2
OH 3′ End
NH2
DNA polymerase
OH
–O
P
O
N
OH
O
P
O
Cytosine
OH
O
P
O
5′ End
O
CH2
5
N
O
4
O
1
3
2
3′ End
OH
FIGURE 2.2.2 Arrangement of bases in single-stranded DNA. The
phosphatedeoxyribose part of the nucleotide triphosphates forms a
backbone of alternating phosphate and deoxyribose molecules.
Attached to this backbone are the four bases: guanine, adenine,
thymine, and cytosine. The 30 and 50 ends of the strand derive from the
numbering of the ribose carbons.
121
122
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
H
3′ End OH
Hydrogen bonds
N
H
C
O
–O
OH
5′ End
P
O
CH2
N
N
O
O
H
N
H2C
O
N
N
H
N
H3C
N
N
H
O
O
N
O
N
O
CH2
T
O
O
N
O–
O
H
N
H
P
P
G
O
O
–O
O
H
A
O
H2C
O
N
5′ End
P
O–
HO
N
Hydrogen bonds
O
OH
3′ End
FIGURE 2.2.3 Pairing of bases in double-stranded DNA.
serve as a template for its own replication and for the
synthesis of RNA.
THE DOUBLE HELIX IS SUPERCOILED
IN CHROMOSOMES
The two DNA strands intertwine around each other to
form a double helix. This helix can be further wrapped
around proteins called histones. The complex of DNA
and its associated proteins is called chromatin. The
complex resembles “beads on a string” that are visible
in electron micrographs (see Figure 2.2.9). The entire
structure can be further coiled and coiled again to form
the chromosomes. All of the DNA becomes condensed
like this when the cell divides. Between divisions, the
chromosomes partially “unravel” to form a less dense
form of chromatin that is the working state of DNA.
THE DOUBLE HELIX POSES SPECIAL
CHALLENGES FOR DNA REPLICATION
The replication of DNA by adding nucleotides only to
the 30 end of the growing DNA strand poses a problem
for the duplication of double-stranded DNA because
the strands are of opposite polarity. This leads to the
replication of DNA in spurts, as shown in Figure 2.2.4
and described in the legend.
RNA IS CLOSELY RELATED TO DNA
RNA is ribonucleic acid. Structurally, it is very similar
to DNA but it differs in several ways. First, the sugar in
the backbone in DNA is deoxyribose, whereas in RNA
it is ribose. Second, the nucleotide base thymine in
DNA is replaced by uracil in RNA. Uracil hydrogen
bonds with adenine, taking the place of thymine. Third,
RNA in eukaryotic cells is single stranded. Fourth, RNA
is not replicated. All of the RNA is produced from DNA
using DNA as a template.
There are different kinds of RNA:
G
G
G
G
G
mRNA: “messenger” RNA
tRNA: “transfer” RNA
rRNA: “ribosomal” RNA
snRNA, scRNA: “small nuclear” and “small cytoplasmic” RNA
Mitochondrial RNA.
MESSENGER RNA CARRIES THE INSTRUCTIONS
FOR MAKING PROTEINS
mRNA is “messenger” RNA. mRNA is synthesized in the
nucleus using the nucleotide sequence of DNA as a template. This process requires nucleotide triphosphates as
substrates and is catalyzed by the enzyme RNA polymerase II. The process of making mRNA from DNA is called
transcription, and it occurs in the nucleus. The mRNA
directs the synthesis of proteins, which occurs in the
cytoplasm. mRNA formed in the nucleus is transported
out of the nucleus and into the cytoplasm where it
attaches to the ribosomes. Proteins are assembled on the
ribosomes using the mRNA nucleotide sequence as a
guide. Thus mRNA carries a “message” from the nucleus
to the cytoplasm. The message is encoded in the nucleotide sequence of the mRNA, which is complementary
to the nucleotide sequence of the DNA that served as a
template for synthesizing the mRNA. Making proteins
from mRNA is called translation.
RIBOSOMAL RNA IS ASSEMBLED IN THE
NUCLEOLUS FROM A DNA TEMPLATE
As discussed in Chapter 2.1, ribosomes are complex
structures comprised of ribosomal RNA (rRNA) and a
number of proteins. RNA polymerase I makes rRNA
form a large loop of DNA called the nucleolar organizer
region. The rRNAs then combine with proteins that
migrate into the nucleolus from the cytoplasm to form
the small and large ribosomal subunits. These ribosomal
DNA and P rot ein Sy nt hes is
DNA polymerase
adds nucleotides
to the 3′ end of
the leading strand
The leading strand
is 3′ to 5′
3′
DNA helicase
unwinds the
two strands
5′
DNA polymerase
cannot fill in the entire
complement—makes
Okazaki fragments
Complementary DNA
double helix; the two
strands have opposite
polarity
5′
3′
5′
3′
3′
5′
3′
Okazaki
fragments
DNA primase adds short
RNA strand to prime DNA
polymerase
5′
The lagging strand
is 5′ to 3′
DNA ligase joins
Okazaki fragments
FIGURE 2.2.4 Replication fork of DNA. The double-stranded DNA consists of two strands of opposite polarity. DNA helicase unwinds the strands,
forming two single-stranded DNAs. The leading strand is 30 to 50 so its complementary strand being newly synthesized is 50 to 30 and DNA
polymerase adds nucleotides to the 30 end as they become available from the helicase. The lagging strand is 50 to 30 and its complementary strand is
30 to 50 . DNA primase adds a short RNA strand that primes the DNA polymerase. DNA polymerase then makes the complement by progressing away
from the replication fork. While it is making DNA, the helicase unwinds more DNA so another DNA polymerase starts replication nearer the
replication fork. In this way, the lagging strand is filled in with Okazaki fragments that bind to the lagging strand but are not connected. DNA ligase
connects the Okazaki fragments to complete replication of the lagging strand.
subunits are then transferred to the cytoplasm where
they are fully assembled to form an 80S functional
ribosome and become protein factories.
TRANSFER RNA COVALENTLY BINDS AMINO
ACIDS AND RECOGNIZES SPECIFIC REGIONS
OF MRNA
How does mRNA specify the sequence of amino acids
in a protein? Which amino acid is to be incorporated
into the protein is specified by a sequence of three
nucleotides called a codon. The mRNA triplets do not
directly recognize and specify the amino acids; they do
so through the use of another kind of RNA called transfer RNA or tRNA. These remarkable molecules are adapters that can link with an amino acid and recognize the
triplets of nucleotides on the mRNA, the codons. They
do this by containing a sequence complementary to the
codon: the anticodon. The function that maps triplets
of nucleotides on the mRNA to specific amino acids is
called the genetic code. Figure 2.2.5 shows the genetic
code in look-up table format.
The tRNA consists of a single strand of RNA from 70 to
90 nucleotides long that is held together by hydrogen
bonding within nucleotides on the same chain. One
end of the tRNA allows for covalent attachment of an
amino acid. Another section of the tRNA contains a
sequence of three nucleotides that forms the anticodon.
Precursors to the tRNA are transcribed from DNA by
RNA polymerase III.
Another key in the formation of proteins is the attachment of amino acids to the specific tRNA. Specific
enzymes called aminoacyl-tRNA synthetases couple the
amino acid to the appropriate tRNA. There is a different
synthetase for each amino acid. One attaches glycine
to tRNAGly, another attaches alanine to tRNAAla, and so
on. These synthetases must recognize both the amino
acid and the tRNA that contain the right anticodon.
The overall processing of RNA and protein synthesis
is shown in Figure 2.2.6. Translation is shown in
Figure 2.2.7.
THE GENETIC CODE IS A SYSTEM
PROPERTY
The genetic code shown in Figure 2.2.5 lists an amino
acid or other signal (such as STOP) for every triplet
nucleotide in mRNA. The code is the function that
maps the nucleotide sequence onto instructions for
protein synthesis. That is,
½2:2:1
f fNi : fA; U; C; Ggg
5 fAj : fcontrol steps; amino acidsgg
This describes the function that turns a set of Ni nucleotides on the mRNA (selected from adenosine, uracil,
cytosine, and guanosine nucleotides) into a set of Aj
amino acids. Where does this code reside? The code
should not be confused with the message or the means
of writing the message. Therefore, the code itself is not a
123
124
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Second position
First position
5′ end
Amino acid
Third position
3′ end
U
C
A
G
U
Phe
Phe
Leu
Leu
Ser
Ser
Ser
Ser
Tyr
Tyr
STOP
STOP
Cys
Cys
STOP
Trp
U
C
A
G
C
Leu
Leu
Leu
Leu
Pro
Pro
Pro
Pro
His
His
Gln
Gln
Arg
Arg
Arg
Arg
U
C
A
G
A
Ile
Ile
Ile
Met
Thr
Thr
Thr
Thr
Asn
Asn
Lys
Lys
Ser
Ser
Arg
Arg
U
C
A
G
G
Val
Val
Val
Val
Ala
Ala
Ala
Ala
Asp
Asp
Glu
Glu
Gly
Gly
Gly
Gly
U
C
A
G
ThreeOneletter code letter code
Alanine
Arginine
Asparagine
Aspartic acid
Cysteine
Glutamic acid
Glutamine
Glycine
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Proline
Serine
Threonine
Tryptophan
Tyrosine
Valine
Ala
Arg
Asn
Asp
Cys
Glu
Gln
Gly
His
Ile
Leu
Lys
Met
Phe
Pro
Ser
Thr
Trp
Tyr
Val
A
R
N
D
C
E
Q
G
H
I
L
K
M
F
P
S
T
W
Y
V
Codons
GCA GCC GCG GCU
AGA AGG CGA CGC CGG CGU
AAC AAU
GAC GAG
UGC UGU
GAA GAG
CAA CAG
GGA GGC GGG GGU
CAC CAU
AUA AUC AUU
UUA UUG CUA CUC CUG CUU
AAA AAG
AUG
UUC UUU
CCA CCC CCG CCU
AGU AGC UCU UCC UCA UCG
ACA ACC ACG ACU
UGG
UAC UAU
GUA GUC GUG GUU
FIGURE 2.2.5 The genetic code. Each amino acid that is incorporated into a protein is specified by a triplet sequence of nucleotides on the mRNA.
These triplets are called codons. Which codons specify which amino acids is shown here in two formats. The left format shows which amino acids
correspond to which codons given as a first, second, and third position. Here we use the Biochemists’ shorthand for RNA bases and amino acids,
where U 5 uracil, C 5 cytosine, A 5 adenine, and G 5 guanosine, and each amino acid is given by its three-letter shorthand designation, where
Phe 5 phenylalanine, Ser 5 serine, Tyr 5 tyrosine, Cys 5 cysteine, Leu 5 leucine, Pro 5 proline, His 5 histidine, Arg 5 arginine, Gln 5 glutamine,
Ile 5 isoleucine, Thr 5 threonine, Asn 5 asparagine, Lys 5 lysine, Met 5 methionine, Val 5 valine, Ala 5 alanine, Asp 5 aspartic acid, Gly 5 glycine, and
Glu 5 glutamic acid. The right format lists the amino acids together with their three-letter designation and single-letter designation, with a list of the
codons that specify them.
Replication: DNA polymerase
DNA
RNA polymerase III
in the nucleus
RNA polymerase I
in the nucleolus
Pre-tRNA
Transcription: RNA polymerase II
in the nucleus
Pre-mRNA (nucleus)
45S pre-rRNA
snRNA
5S rRNA
Processing and splicing
rRNA 28S
Ribosomal
18S
proteins
5.8S
mRNA: sequence of codons
Cytoplasm
Ribosomes
40S with 18S rRNA
60S with 5S, 5.8S, and
28S rRNA
tRNA: anticodon
Aminoacyl-tRNA
synthetases
Amino acids
Translation: free in
the cytoplasm or on
the ER
Proteins
FIGURE 2.2.6 Processing of RNA and DNA. Replication of DNA is accomplished by DNA polymerase using the original DNA as a template. Messenger
RNA is synthesized in the nucleus as a precursor that is processed to form the final mRNA. The synthesis of mRNA is called transcription and is
accomplished by RNA polymerase II. The final mRNA travels to the cytoplasm where it binds to ribosomes. The ribosomal subunits are formed in the
nucleolus from proteins and ribosomal RNA that is made as a precursor and cut into the final rRNA strands. rRNA is made from DNA by RNA
polymerase I. The mRNA directs the sequential addition of amino acids to form proteins in a process called translation. This requires tRNA, made from
DNA by RNA polymerase III.
DNA and P rot ein Sy nt hes is
Growing peptide chain
is attached at the P site
Next charged tRNA
binds to the A site
Peptide bond forms and
peptide chain moves to
new carboxy terminus AA
Trp COOH
Aminoacyl tRNA
NH2 Ala Trp
Phe
Met
Met
Phe
Phe
P site
His
Met
NH2 Ala Trp
His
NH2 Ala Trp
His
Trp COOH
Ribosome moves down
one codon
Trp
ACC
E site
A site
UAC AAG
GCACAUGUUCUGGCAGAGGCACAUGU
5′ End
mRNA
3′ End
UAC AAG ACC
GCACAUGUUCUGGCAGA GGCACAUGU
5′ End
mRNA
A AG ACC
GCACAUGUUCUGGCAGAGGCACAUGU
5′ End
3′ End
mRNA
3′ End
tRNA on E site
leaves
UAC
FIGURE 2.2.7 Elongation of a growing polypeptide chain. The mRNA binds to a ribosome consisting of some 82 proteins and 4 separate rRNA strands.
There are 3 tRNA binding sites: an aminoacyl or A site, a peptidyl or P site, and an exit or E site. The aminoacyl tRNA is escorted to the ribosome by an
elongation factor that hydrolyzes GTP. The scheme begins with a short polypeptide bound to a tRNA at the peptidyl site along with the tRNAMet that
remains at the exit site. The next aminoacyl tRNA binds to the aminoacyl site; in this case it is tRNATrp that is charged with its amino acid. The peptide bond
is formed between the peptide and its next amino acid on the carboxy terminus. The ribosome shifts over one codon; the tRNA at the exit site leaves and
the former occupant of the peptidyl site now occupies the exit site. The peptide now occupies the peptidyl site one codon further along the mRNA.
property of the mRNA. The DNA itself also does not
contain the code, as its message has no meaning without the tRNA. The tRNA is synthesized from other parts
of the DNA that are not directly transcribed for protein
synthesis. Is the code in the tRNA that links the triplets
of mRNA to a specific amino acid? Or is the code in the
aminoacyl-tRNA synthetases, the enzymes that couple
the amino acid to the specific tRNA? If the code is in
the aminoacyl-tRNA synthetases, then the code is in
some sense also in the DNA because the DNA directs
the synthesis of the aminoacyl-tRNA synthetases! But
the DNA does not “make” proteins; mRNA does not
“make” proteins. The proper proteins are synthesized
with DNA as the store of information, mRNA carrying
the message, tRNA converting, in a single step, the
nucleotide information into the protein information,
and preexisting proteins catalyzing the entire series of
events. The code itself is an emergent property. The
genetic code does not exist in any single component of
the mechanism for making proteins. It is not “in” the
DNA, or the mRNA or the tRNA or the tRNA synthetases. Rather, it is a system property that emerges from
the interactions of all of these parts.
REGULATION OF DNA TRANSCRIPTION
DEFINES THE CELL TYPE
The differentiation of cell types produces the wide spectrum of cell types in the body, but our understanding of
the process is still rudimentary: we do not know how to
change one cell type into another. We do know,
however, that the hallmark of differentiation is selective
expression of the cell’s DNA. Selective expression of DNA
as proteins requires selective transcription. Initiation of
transcription by RNA polymerase II requires a number of
specific proteins called transcription factors. These come
in two flavors: some are required for activity at all genes
and are therefore called general transcription factors.
Other transcription factors bind to DNA sequences that
control the expression of specific genes. The overall process is shown in Figure 2.2.8.
Most genes contain both transcribed and untranscribed
regions. On the 30 end of the DNA gene is a specialized
sequence called the promoter that helps regulate gene
expression. This region contains a TATAA sequence
(called a TATA box) some 2530 nucleotides upstream
from the initiation site. Initiation begins when a transcription factor TFIID (transcription factor polymerase II)
binds to the DNA TATAA sequence. Some promoters do
not contain a TATA box, but instead contain an initiator
sequence. Nevertheless, TFIID is involved in initiation of
transcription even on promoters that lack the TATA box.
In addition to these general transcription factors, there
are a number of transcription factors that may act as
enhancers or repressors of gene expression. These factors interact with specific sites on the DNA that are generally further away from the gene than the promoter
region and act as regulatory elements for gene transcription. The signals that turn on or turn off the production
of transcription factors ultimately determine the phenotypic fate of cells. How these transcription factors work
is still being investigated. An example of this is the
125
126
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Regulatory sites
Promoter
Introns
DNA strand
5′ End
3′ End
Exons
Primary RNA transcript
5′
AAAAAA3′
Cap
Poly A tail
Splicing
5′
AAAAAA
mRNA
Translation
N
C
Protein product
Posttranslational
modification
S S
Processed protein
FIGURE 2.2.8 Overall processes of transcription and translation. The gene has a promoter that often contains a TATAA sequence for initiation of
transcription by RNA polymerase II, several exons (expressed or coding regions of the DNA sequence) and introns (intervening or noncoding regions),
and regions that bind transcription factors to either enhance or repress expression. The primary gene transcript contains the sequence of bases
complementary to the introns and exons. This primary RNA transcript is then further processed on large complexes called spliceosomes, composed of
protein and small nuclear RNAs or snRNA. The spliceosomes remove the introns by clipping them out and then splice together the remaining exons
to form a sequential mRNA. This mRNA is then translated in the cytosol to a primary protein product which may then require further processing.
steroid hormone receptors. Steroid hormones and similar materials like vitamin D bind to receptor proteins
that in turn bind to specific sequences on the DNA
called response elements. Binding of the receptor proteins then activates gene expression. A number of accessory proteins are required for this process.
THE HISTONE CODE PROVIDES
ANOTHER LEVEL OF REGULATION
OF GENE TRANSCRIPTION
As described earlier, the double helix of DNA winds
around specific proteins called histones. These histones
form the basic unit of chromatin called the nucleosome.
A schematic of this structure is shown in Figure 2.2.9.
When wrapped up in this way, DNA is inaccessible to
RNA polymerase II and so cannot be transcribed to form
mRNA. Most cells sequester away large portions of their
DNA in this way. In order to express DNA, cells must
unwrap it from the chromatin. Determining which sections of DNA should be unwrapped is the first step in
regulating gene transcription. This is accomplished
through covalent modifications of the histones. Covalent
modification of histones after synthesis is called posttranslational modification. Histones undergo
G
G
acetylation of lysine and arginine amino acids in the
histones;
methylation of lysine and arginine amino acids;
G
G
phosphorylation of serine and threonine amino
acids in the histones;
ubiquitinylation of lysines.
All of these modifications are accomplished by enzymes
that must themselves be regulated. These enzymes possess histone acetyltransferase (HAT) activity, histone
deacetylation (HDAC) activity, histone methyltransferase (HMT) activity, and histone kinase activity. The
function of these enzymes is evident from their names:
HAT adds acetyl groups to the histones; HDAC removes
them; HMT adds methyl groups; and histone kinase
phosphorylates the histones. Figure 2.2.10 summarizes
the various known modifications of the core histones.
These posttranslational modifications of the histones have
functional consequences. For example, the combination
of acetylation at lysine at position 8 on H4 and lysine
at position 14 on H3 with phosphorylation of serine at
position 10 on H3 is associated with transcription.
Trimethylation of lysine at position 9 on H3 with the lack
of acetylation of H3 and H4 correlates with transcriptional
repression. These observations have led to the hypothesis
that gene expression is regulated in part by a “histone
code.” This hypothesis requires two components:
1. Specific enzymes write the code by adding or removing modifications at specific sites in the histones.
2. Other proteins recognize the histone markers
and interact with histones and other factors to
mediate functional effects.
DNA and P rot ein Sy nt hes is
Sequence-specific
DNA-binding protein
H2A
H4
H3
H1
Nucleosome
DNA strand
H2B
30 nm DNA fiber
H3
FIGURE 2.2.9 Schematic diagram of nucleosome structure and 30-nm DNA fiber. DNA is wrapped around a complex of eight histone proteins: a
tetramer of H3 and H4 and two dimers of H2A and H2B, to form the nucleosome. Strands of DNA bound to nucleosomes resemble beads on a string.
The interaction of DNA with the nucleosomes is altered by H1, a linker histone, that enables the nucleosomes to condense to form a 30-nm DNA
strand. H1 interacts with both the histones in the core and DNA. The nucleosome contains 147 base pairs of DNA wrapped nearly twice around the
core histones. A short “linker” of 1060 base pairs separates each nucleosome from its neighbor. The linear sequence of nucleosomes forms “beads on
a string” in electron micrographs. This becomes more highly condensed to form 30-nm-thick fibers that are stabilized by the H1 histones.
Ac
P
H2A
Ub
Ac-S G R G K Q G G K A R A ... A V L L P K K T E S H H K A K G K-COOH
1
5
119
Ac P Ac
Ac
H2B
5
12
MeP Ac
AcP P
14 15
20
Me Ac
Ac
120
Ac
MeAc
P
Me
Me
NH2 -A R T K Q T A R K S T G G K A P R K Q L A S K A A R K S A ... G V K K ... E F K T D...
2 3 4
P
H4
Ub
NH2 - P E P V K S A P V P K K G S K K A I N K ... V K Y T S S K-COOH
1
H3
Ac
Me
9 10 11
Ac
Ac
14
17 18
Ac
Ac
23
26 27 28
36
79
Me
Ac -S G R G K G G K G L G K G G A K R H R K V L R D N I Q G I T...
1
3
Ac
P
5
8
12
16
Acetyl group added
Phosphate group added
20
Me
Ub
Methyl group added
Ubiquitin protein attached
FIGURE 2.2.10 Posttranslational modifications of the core histones. The one-letter amino acid abbreviation follows that in Figure 1.3.5. The numbers
below the amino acids refer to the number of amino acids in the sequence, starting from the amino terminus. Source: From C.L. Peterson and M.-A. Lanie,
Histones and histone modifications, Current Biology 14:R546R551, 2004.
The histone code may be a misnomer if it is viewed as
being like the genetic code. In the genetic code, given
triplets of nucleotides on mRNA always produce the
same result, independent of which cell or tissue is
being analyzed. The term “histone code” implies that a
particular combination of histone modifications will
always produce the same biological result. Evidence suggests that the same pattern of histone modifications can
be interpreted differently by different cells depending
on the gene and its cellular context.
127
128
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
DNA METHYLATION REPRESSES
TRANSCRIPTION
A methyl group can be added to the 5 position of cytosine
to form 5 methyl cytosine by the action of DNMT, DNA
methyltransferase, as shown in Figure 2.2.11. These
enzymes come in two classes. DNMT1 is responsible for
maintenance methylation, which methylates the new
strand of recently replicated DNA, so that the methylation
pattern is passed down from stem cells to daughter cells
(see Figure 2.2.12). This explains the unidirectionality of
most developmental processes. Stem cells become
differentiated cells and the differentiated cells maintain
their differentiation, partly through a pattern of DNA
methylation. The second class of DNA methylation
transferases, represented by DNMT3a and DNMT3b, is
responsible for de novo methylation. Both the maintenance methylation enzymes and de novo methylation
enzymes methylate cytosine in a CpG sequence in the
DNA (see Figure 2.2.11).
The consequence of DNA methylation is typically
repression of transcription for the genes that are methylated. The methyl group does two things: it interferes
with the binding of transcription factors that eventually
recruit RNA polymerase II, and it also allows the binding of a set of proteins that specifically recognize the
methylated DNA, and these proteins recruit histone deacetylases that modify the histones associated with the
DNA. These actions result in a repressed transcription
of the methylated parts of the DNA.
Cytosine NH2
DNA methyltransferase
(DNMT)
Cytosine NH2
CH3
6
O
-O
P
5
1
4 3 2
5′ end
O
CH2
5
O
O
-O
N
O
3
O
2
P
O
CH2
5
CH2
5
O
1
4
3
O
CpG
-O
N
O
1
3
O
2
H
N
N
O
N
N
O
N
O
4
3′ end
H
N
6
N
5
1
4 3 2
5′ end
O
O
O
-O
P
O
1
4
3′ end
N
NH2
P
O
CH2
5
O
Guanosine
3
3′ end
N
1
4
2
N
O
NH2
Guanosine
2
3′ end
O
Methylated CpG
FIGURE 2.2.11 DNA methylation. DNA is methylated at sites with the sequence CpG by DNA methyltransferase. It can also be demethylated, but the
process is not simply a reversal of the methylation reaction. Demethylases form hydroxymethyl cytosine, which is then cut out and replaced with
cytosine by DNA repair mechanisms.
CH3
Parent strand
T A C G T T G T A G A C G T A C
CH3
Parent strand
T A C G T T G T A G A C G T A C
3′
5′
Nonmethylated CpG
Methylation
3′
Methylated CpG
3′
A T G C A A G A T C T G C A T G
Daughter strand
CH3
T A C G T T G T A G A C G T A C
3′
5′
3′
3′
5′
DNA replication
Not recognized by
maintenance DNA
methyl transferase
A T G C A A G A T C T G C A T G
Daughter strand
CH3
Recognized by DNA
methyl transferase
Parent pattern of methylation
is preserved in both daughters
5′
A T G C A A G A T C T G C A T G
CH3
CH3
Parent strand
T A C G T T G T A G A C G T A C
Daughter strand
T A C G T T G T A G A C G T A C
3′
5′
3′
5′
Methylation
3′
3′
A T G C A A G A T C T G C A T G
Parent strand
CH3
A T G C A A G A T C T G C A T G
Daughter strand
CH3
FIGURE 2.2.12 Maintenance methylation. The parent DNA has some CpG sequences that are methylated, and some that are not. These are
determined by de novo methylation reactions catalyzed by DNA methyl transferase 3A and 3B (DNMT3a and DNMT3b). When the DNA is replicated,
the daughter strands are not methylated. The maintenance DNA methyl transferase (DNMT1) recognizes the CpG sequence on the daughter strand
that corresponds to the methylated CpG sequence on the parent strand, and then methylates the daughter strand cytosine. DNMT1 does not bind to
the unmethylated CpG sequences and therefore does not methylated these. The result is that the methylated pattern of the parent strand is replicated
in the methylated pattern of the two daughter strands. In this way, the pattern of gene repression in a differentiated cell line is passed onto its
descendants.
DNA and P rot ein Sy nt hes is
SUMMARY
Genetic information is stored in DNA in the nucleus
and mitochondria of cells. DNA consists of two strands
of nucleotides on a phosphodeoxyribose backbone.
The two strands form a double helix that is stabilized
by the formation of hydrogen bonds between nucleotide
bases on the two strands. Replication of DNA is based
on making strands complementary to the two strands
produced when the double-stranded DNA is unwound.
The four bases include two purines (adenine and guanine) and two pyrimidines (cytosine and thymine).
They pair up as A:T and G:C, with two hydrogen bonds
between A and T and three between G and C.
DNA serves as a template for making a variety of RNA
types: RNA polymerase I makes ribosomal RNA (rRNA)
from nuclear DNA; RNA polymerase II makes messenger RNA (mRNA); RNA polymerase III makes transfer
RNA (tRNA). All of these contribute to the synthesis of
proteins and control of gene expression.
Protein synthesis occurs on the ribosomes, using mRNA
as a template for tRNA. The ribosomes themselves are
large and complex structures consisting of rRNA and a
number of proteins. The ribosomes consist of 60S
and 40S subunits. The 60S subunit has three rRNAs
(28S, 5S, and 5.8S) and 49 other proteins; the 40S
subunit has an 18S rRNA and another 33 proteins.
The ribosome binds to mRNA and provides a reaction
site for the peptide bound to tRNA (P site), a second
reaction site for the next amino acid covalently attached
to its tRNA (A site), and a third site for the tRNA about
to leave the ribosome (E site). The ribosome brings the
peptide carboxyl terminal close to the amino terminal
of the next amino acid, and then forms the peptide
bond, simultaneously shifting the peptide from the P
to the A site. The mRNA provides a sequence of nucleotides. Groups of three nucleotides form codons that are
recognized by complementary nucleotide sequences
on the tRNA—the anticodons. The specific binding of
anticodon to codon allows the mRNA to determine the
sequence of amino acids in the proteins being made.
The attachment of specific amino acids to specific
tRNAs, however, is assured by the tRNA synthetases that
hook the amino acids onto tRNA.
mRNA directs the synthesis of specific proteins by virtue
of its sequence of codons. The set of proteins that are
made determines the type of cell, because cell structure
and activity derives from the kinds and amounts of proteins expressed by the cell. Cells have developed elaborate methods for determining what parts of the DNA
are transcribed into mRNA. These methods involve transcription factors, enhancers and repressors, and the
histone code. The DNA in cells is wrapped around a
complex of histone proteins, forming a nucleosome.
Modification of the histones allows specific sections of
the DNA to be either used to make proteins or silenced.
A second method for repression of gene transcription
is methylation of cytosines in the DNA in sequences of
CpG. These are originally methylated by de novo methylation, but the pattern of methylation can survive DNA
replication through a maintenance methyltransferase.
The result is a stable pattern of gene repression that
survives proliferation. DNA methylation interacts with
histone modification to determine which DNA sequences
will be silenced.
REVIEW QUESTIONS
1. What is the genotype? What is the phenotype?
What is an allele? What is the usefulness in
having two copies of each gene?
2. What are the parts of a nucleotide? Name the
purine bases. Name the pyrimidine bases.
3. How are nucleotides linked together to form
single-stranded DNA or RNA? What distinguishes
the 50 end from the 30 end? What enzyme makes
DNA from nucleotides?
4. What holds double-stranded DNA together?
Why are hydrogen bonds useful? How many
hydrogen bonds link guanine to cytosine? How
many such bonds link adenine to thymine?
5. During DNA replication, what determines the
sequence of DNA in the new strands?
6. Does DNA polymerase add nucleotides at
the 50 end or the 30 end of the strand? What
problem does this make for DNA replication?
What is an Okazaki fragment?
7. How does RNA differ from DNA? What is
mRNA? What is tRNA? What is rRNA? What
RNA polymerase makes mRNA? Which makes
tRNA? rRNA?
8. What is a codon? Is it on mRNA, DNA, or
tRNA? What is an anticodon?
9. What couples tRNA with amino acids? Are these
enzymes specific for the anticodon?
10. What is a ribosome? What is the A site? What is
the P site? What is the E site?
11. What is the genetic code? Where is it located in
the cell?
12. What is meant by “transcription”? What is
meant by “translation”?
13. How are inactive portions of DNA locked up by
the cell? How do they get unlocked? What is a
“response element”? What is an “intron”? What
is an “exon”?
14. What are histones? What promotes DNA binding?
What promotes DNA unraveling from the histones? What is meant by “the histone code”?
15. What is DNA methylation? Where does it occur?
What is the consequence of DNA methylation?
How does DNA methylation pattern get passed
on to daughter cells?
129
2.3
Protein Structure
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
Draw the basic structure of an amino acid, specifying the
amino group, carboxyl group, alpha carbon, and R group
For each amino acid, tell whether it is polar, nonpolar,
acidic, or basic
Draw the reaction describing the formation of a peptide
bond
Describe what is meant by the primary structure of a protein
Describe what is meant by the secondary, tertiary, and
quaternary structure of a protein
Describe the four kinds of noncovalent interactions that
stabilize protein structure
Define posttranslational modification
List four major classes of posttranslational modification
List the major kinds of chemical modification of proteins
List the amino acids involved in glycosylation and its overall
function in proteins
Describe the consequence of gamma carboxylation of
proteins and name the vitamin involved
Describe two distinct ways of varying the activity of
proteins in cells
Describe three distinct ways membrane proteins can be
anchored in the membrane by covalent modification
AMINO ACIDS MAKE UP PROTEINS
In Chapter 2.2, we described how proteins are made on
ribosomes by linking amino acids together in long
chains. Which amino acids make up a specific protein,
and in which order, is determined by the sequence of
triplet nucleotide codons residing on the mRNA, which
in turn is produced on a DNA template in the nucleus.
Because these constituent amino acids determine the
detailed shape of the protein surface, we should find
the key to protein activity in the three-dimensional
arrangement of these amino acids.
The general chemical structure of the amino acids is
shown in Figure 2.3.1. The amino acids are named for
the two functional groups each of them possesses: an
amino group (NH) and a carboxylic acid group
(COOH). These two groups have widely different reactivities. The amino group is basic and will be positively
charged at neutral pH, forming an ammonium ion
(NH31). The carboxyl group is acidic and will form a
negatively charged carboxyl ion (COO2) at neutral
pH. Thus at neutral pH, many amino acids will have a
positive charge on the amino end and a negative charge
on the carboxyl end. Compounds possessing both
positive and negative charges simultaneously are called
zwitterions, as shown in Figure 2.3.2.
Although the amino and carboxyl groups are important
for the structure of proteins, the tremendous variety of
structure and function of proteins is produced by the R
groups (R stands for “residue” and refers to the part of the
amino acid other than the amino or carboxyl group covalently bonded to the α carbon). There are 20 different
amino acids differing only in their R groups. The R groups
confer properties on the amino acids that classify them as
nonpolar, polar, basic, or acidic. Each of these groups is
shown in Figures 2.3.32.3.5. Biochemists often use a
three-letter abbreviation for the amino acids or a oneletter code for a further shorthand. These abbreviations are
shown under each structural formula (see Figure 2.3.6).
As their name implies, the nonpolar amino acids contain
side groups that are nonpolar and therefore not attracted
to water. Because water is so attracted to itself, these
groups naturally are repulsed by the water and are said
to be hydrophobic, or water hating. For glycine and
alanine, the nonpolar side groups are so small that the
amino acid has little preference for either a watery
(hydrophilic) or nonwatery (hydrophobic) environment.
Phenylalanine and tryptophan are highly hydrophobic
and seek environments away from water.
HYDROPHOBIC INTERACTIONS CAN BE
ASSESSED FROM THE PARTITION
COEFFICIENT
We can estimate the strength of hydrophobic interactions by measuring the partitioning of a material
between the water phase and an immiscible solvent
such as N-octanol or ethyl acetate, whose properties
may be regarded as being similar to the interior of
protein away from the water phase. The partition
coefficient is
½2:3:1
ks 5
½xorganic phase
½xwater phase
130
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00012-4
Prot ein St ructur e
L-amino
D-amino
acid
where the concentrations are the equilibrium concentrations, as shown in Figure 2.3.7. This equilibrium condition obeys Eqn [1.7.32]:
acid
Amino group
NH2
NH2
½2:3:2
R
H
C
H
R
C
Alpha carbon
C
O
C
OH
O
HO
Carboxyl group
FIGURE 2.3.1 Chemical structure of the amino acids. Amino acids
consist of a central carbon, the alpha carbon, to which are bonded a
carboxyl group, an amino group, a hydrogen atom, and a variable
group, called R. Because the alpha carbon has four different groups
bonded to it, it is an asymmetric carbon that is capable of
stereoisomerism. All of the amino acids can be made in L- and D-form
(L and D originally described the ability of compounds to rotate the plane
of polarized light; L for levo, meaning “left,” and D for dextro meaning
“right.” The symbols L and D for the amino acids do not actually refer to
the direction of rotation of the plane of polarized light). All naturally
occurring amino acids in higher organisms are the L-form. The L and D
forms are mirror images of each other and are not superimposable.
+
NH3
R
H
C
C
O
–
O
FIGURE 2.3.2 Zwitterion form of an amino acid. At neutral pH (pH 5 7),
the carboxyl group is dissociated to form the anionic COO2 group,
and the amino group binds a H1 ion to form NH31. Thus both ends of
the amino acid are charged but with opposite polarity.
O
OH
C
O
OH
CH2
C
CH2
H2N
C
H
CH2
O
H2N
C
OH
Aspartic acid (Asp) D
C
H
O
C
OH
Glutamic acid (Glu) E
FIGURE 2.3.3 The acidic amino acids aspartic acid and glutamic acid.
The R group in both cases contains another carboxylic acid group that
dissociates to form an H1 ion and a negatively charged carboxyl group
(COO2). The presence of this group classifies these amino acids as
acidic amino acids. The negatively charged carboxyl group imparts a
negative charge to the region of any protein that contains these groups.
ΔG0 5 2RT ln Keq
½xorganic phase
ΔG0T 5 2RT ln
½xwater phase
where ΔG0T is the standard Gibbs free energy change for
transfer of substance X from water to the organic phase.
Although this can represent the strength of hydrophobic
interaction of a small molecule, it cannot reliably predict the behavior of a polymer of the material or of the
material when it participates in a heteropolymer such as
a protein. This is true for amino acids in particular
because some of their functional groups that determine
the octanol/water partition are altered when the amino
acids are linked up to make proteins. Nevertheless, parts
of molecules can be described as being hydrophobic or
hydrophilic, depending on whether or not they would
partition themselves into the organic phase.
Hydrophobicity has two components: the “squeezing
out” of water-insoluble components due to the attraction of water for itself and the self-association of nonpolar materials due to dipoledipole interactions,
dipoleinduced-dipole, and induced-dipoleinduceddipole interactions (London dispersion forces) discussed in Chapter 1.4. Water repels nonpolar materials
because the surface of the nonpolar material cannot
form hydrogen bonds with the water, and its shape
therefore reduces the number of hydrogen bonds that
the water can form at the surface—the water molecules
at the surface are in a higher energetic state, having
some of their hydrogen bonds broken. Thus it takes
energy to insert a nonpolar material into the water
phase.
The amino acids can be classified solely on the basis of
their hydrophobicity or hydrophilicity. In this case, we
have three categories: the hydrophilic amino acids
include aspartic acid, glutamic acid, lysine, and arginine;
the hydrophobic amino acids include valine, leucine,
isoleucine, methionine, phenylalanine, and tryptophan;
the “neutral” amino acids, those that are neither
strongly hydrophilic nor strongly hydrophobic, include
glycine, alanine, serine, histidine, proline, threonine,
cysteine, glutamine, and asparagine.
PEPTIDE BONDS LINK AMINO ACIDS
TOGETHER IN PROTEINS
Proteins are formed as a linear, unbranched chain of
amino acids. The amino acids are covalently linked by a
peptide bond formed between the amino group of one
amino acid and the carboxyl group of the next. The formation of the peptide bond is a dehydration reaction,
as shown in Figure 2.3.8.
Cells make proteins by the sequential addition of amino
acids to the carboxyl terminus of the growing chain.
This is accomplished on the ribosome when specific
tRNAs (transfer RNAs) with their specific bound amino
131
132
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
NH2
NH2
C
CH2
NH
CH2
CH2
CH2
CH2
+
NH2
HN
CH2
H2N
CH2
O
C
C
H2N
C
OH
H
NH+
CH2
O
C
H2N
C
OH
H
OH
H
Arginine (Arg) R
Lysine (Lys) K
C
O
Histidine (His) H
FIGURE 2.3.4 The chemical structures of the basic amino acids, lysine, arginine, and histidine. These contain ionizable NH groups so that at neutral pH,
these residues would contribute positive charge to the protein.
OH
CH3
OH
HC
CH2
H2N
C
OH
O
H2N
C
C
CH2
O
C
H2N
C
C
OH
OH
H
H
Serine (Ser) S
H
Threonine (Thr) T
OH
Tyrosine (Tyr) Y
O
NH2
C
O
C
NH2
CH2
H2N
O
C
CH2
CH2
O
C
H2N
C
O
C
OH
H
Asparagine (Asn) N
H
OH
Glutamine (Gln) Q
FIGURE 2.3.5 The chemical structure of the polar amino acids. The polar amino acids contain groups that can form hydrogen bonds with water. These
groups are soluble in water. As a result, those portions of proteins which contain large numbers of polar residues will usually be exposed to water.
They help solubilize proteins in solution, so we might expect soluble proteins to be coated with polar, acid, or basic R groups.
Prot ein St ructur e
H3C
H3C
CH3
H
O
H2N
C
H2N
C
OH
H
Glycine (Gly) G
C
C
H2N
OH
H
C
Alanine (Ala) A
CH2
O
H 2N
C
OH
H
H2C
CH
CH3
CH
O
CH3
CH3
C
C
H 2N
OH
H
Leucine (Leu) L
Valine (Val) V
H3C
O
CH
C
O
C
OH
H
Isoleucine (Ile) I
CH3
NH
S
CH2
SH
CH2
H2C
CH2
HN
C
CH2
O
H2N
C
H
Proline (Pro) P
OH
C
CH2
O
C
H
H 2N
OH
Cysteine (Cys) C
C
H
CH2
O
C
H 2N
OH
Methionine (Met) M
C
H
CH2
O
C
H2N
OH
Phenylalanine (Phe) F
C
O
C
H
OH
Tryptophan (Trp) W
FIGURE 2.3.6 Chemical structures of the nonpolar amino acids.
PROTEIN FUNCTION CENTERS ON
THEIR ABILITY TO FORM REACTIVE
SURFACES
n-Octanol
Hydrophobic molecules
Proteins perform a variety of functions in cells, and
these can be broadly classified as structural, catalytic,
transport, or regulatory. All of these functions require
the surface of the protein to interact with other surfaces.
FIGURE 2.3.7 Assessment of hydrophobicity by the partition coefficient
between octanol and water. A hydrophobic molecule is dissolved in
n-octanol and then an aliquot of the solution is placed in contact with
water and shaken. The material will distribute itself between the two
phases. The ratio of the concentrations at equilibrium in the organic
phase to the water phase defines the partition coefficient, which is an
equilibrium constant that can be used to calculate the free energy of
transfer from the organic phase to the water phase. Hydrophobic
materials will have a higher concentration in the n-octanol phase;
hydrophilic materials will have a higher concentration in the water phase.
As discussed in Chapter 1.5, enzymes provide a surface
on which biochemical reactions can occur. These reactions can occur in the water phase, but only slowly
because of the large activation energy required. Binding
of the substrates to the enzyme surface alters the path of
the reaction, enabling it to proceed more quickly. This
lower energy pathway is produced by the interaction of
the protein surface with the substrates of the biochemical reaction. The details of the protein surface enable
catalysis, the speeding up of a reaction without appearing in the overall stoichiometry of the reaction. The surface of the protein must closely match the surface of the
substrates, which means that it cannot match other substrates well, because the protein surface cannot match
two different surfaces. Thus the protein surface simultaneously enables catalysis while it confers specificity—
the enzyme works only with specific substrates.
acid bind to the appropriate triplet codon of the mRNA.
The amino acid is attached to the tRNA via its carboxyl
group. When the peptide bond is formed, the entire
growing chain is transferred to the free amino terminal
of the next amino acid. The ribosome then moves one
frame (nucleotide triplet) and the process is repeated
(see Chapter 2.2).
In the same way that protein enzymes interact with biochemical substrates on the surfaces of the proteins,
structural proteins interact with other components by
virtue of their surfaces. The proteins that make up connective tissue are sticky. They bind to themselves and to
a variety of other proteins. Their surfaces make them
sticky. Closely matching surfaces allow proteins to interact with other proteins, thereby allowing one protein to
Water
133
134
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
H3C
CH3
CH
H2N
H3C
CH2
O
C
C
C
H2N
OH
H
O
C
H2N
CH3
CH
O
C
C
CH2
NH
O
C
C
OH
H
OH
H
H
+
Valine
Phenylalanine
Peptide bond
H2O
FIGURE 2.3.8 Formation of a peptide bond between two amino acids. The overall reaction is shown. The actual reaction involves many intermediary
steps, catalyzed by enzymes.
COOH
Ser Leu
Ile
Val Glu Gln
Cys
NH2 Gly
Thr
Tyr
Cys
S S
Ile
His
S
Leu Cys
Lys
Gln
Pro
Thr
Leu
Cys
Ser
S
Thr
Tyr
Glu
Phe
Asn
COOH
Gly
Gln
Ser
Asn
Tyr
Cys
His
Val Phe
Asn
Arg
Glu
S
NH2
S
Leu
Phe
Gly
Gly
Cys
Leu Val
Val
Tyr
Glu
Ala
Leu
FIGURE 2.3.9 Primary structure of insulin. Insulin is a protein hormone secreted by the pancreas in response to high blood glucose. It causes
peripheral tissues to take up the glucose, thereby lowering the plasma glucose concentration back toward normal. Insulin is synthesized as a larger,
single polypeptide chain that is modified by excision of two peptides to form the A chain, with 21 amino acids, and the B chain with 30. This is one
example of posttranslational modification that occurs with many proteins.
regulate another through the binding together of matching surfaces.
THERE ARE FOUR LEVELS OF
DESCRIPTION FOR PROTEIN
STRUCTURE
Proteins take on their remarkably diverse functions
because they can fold to form specific shapes and
because some of the amino acids that make up the proteins have inherent chemical reactivity. These shapes
provide a surface for the binding of materials, and this
binding originates all of the functions of proteins. The
shape of proteins has four levels of description:
1.
2.
3.
4.
Primary
Secondary
Tertiary
Quaternary.
THE PRIMARY STRUCTURE OF A PROTEIN
IS ITS AMINO ACID SEQUENCE
The primary structure of a protein refers to its amino
acid sequence. It defines the chemical connectivity of
the constituent amino acids. In 1953, Sanger determined the amino acid sequence of the A and B chain of
insulin. This was the first protein whose entire amino
acid sequence was determined and the work was a
major milestone in biochemistry. The chemical structure
of insulin is shown in Figure 2.3.9.
THE SECONDARY STRUCTURE OF PROTEIN
REFERS TO THE FOLDING OF AMINO ACIDS
IN ADJACENT SEQUENCES
Proteins have three regular secondary structures: the
α-helix, the β-sheet, and the β-turn. These are complicated, three-dimensional structures. The α-helix was first
postulated by Linus Pauling, Robert Corey, and Herman
Prot ein St ructur e
Cα
H
Amino N of
next amino acid
Carboxyl C of
one amino acid
Distributed pi electrons in double bonds
require trigonal geometry of both C–O
bond and C–N bond.This makes O, C,
N, H and both Cα nuclei coplanar
Cα
H
Cα
Cα
H
Cα
Cα
Electrons establish a rapid equilibrium
between two alternate forms of bonding.
This is called resonance. This is equivalent
to partial double bonds for both the C–O and
C–N bonds
FIGURE 2.3.10 Planar peptide bond. The peptide bond involves a combination of an carboxyl group of one amino acid with the amino group of the
next amino acid. The CN bond and C 5 O bond resonate: the electrons alternate between a single CN and double C 5 N bond, and simultaneously
between a double C 5 O bond and a CO2 bond. This is equivalent to a distributed electron density over both bonds. This restricts free rotation
about the CN bond, locking all six nuclei into a single plane.
Branson (The Structure of Proteins: Two HydrogenBonded Helical Configurations of the Polypeptide
Chain, Proc. Natl. Acad. Sci. USA 37:205211, 1951) on
the basis of a planar peptide bond and linear hydrogen
bond length of 0.272 nm, and no requirement for an
integral number of amino acids per turn of the helix.
The planar nature of the peptide bond was key. Pauling
realized that resonance between the carbonyl oxygen
and the CN bond would produce a partial double
bond character to the CN bond that would prohibit
free rotation about its axis. The H bonding would be trigonal and therefore planar. The carboxyl C would be
similarly planar, and because both the N and the carboxyl C were connected, all six nuclei connected to the
peptide bond would be coplanar. This idea is shown in
Figure 2.3.10.
Although the CN bond cannot freely rotate, due to its
partial double bond nature, the NCα bond is not so
restricted. This Cα nucleus is connected to the next C
that participates in a peptide bond, and this CαC
bond can also rotate. The result is that the next peptide
bond, which also forms a plane, is rotated relative to the
first. Two dihedral angles are defined to describe this
rotation: ϕ is the dihedral angle about the NCα bond
and ψ is the dihedral angle about the CαC bond.
These are shown diagrammatically in Figure 2.3.11.
Pauling and co-workers used the idea of the stiff plane
of the peptide bond and the known length of the
hydrogen bond to deduce the structure of the alpha
helix, shown in Figure 2.3.12. In this structure, the polypeptide backbone traces a right-handed helix (going
from the amino terminus to the carboxy terminus).
Note that the C 5 O bonds point towards the carboxy
terminus, where they hydrogen bond with the amino
hydrogen. The protein forms a rod with the side groups
of the amino acid sticking out more or less radially.
Rotation around the N–Cα
bond is called phi, φ
Cα
H
N
φ ψ
C
The orientation of successive
planes of the peptide bonds
are determined by φ and ψ
Cα
Rotation about the Cα–C
bond is called ψ
FIGURE 2.3.11 Rotation of successive peptide bonds. Each peptide
bond defines a plane. Between each bond there is allowable rotation
around the NCα bond (defined as ϕ) and rotation about the CαC
bond (defined as ψ). The result is a change in direction of the
polypeptide chain.
The second major secondary structure in proteins is the
beta sheet, shown schematically in Figure 2.3.13. These
result from sideways hydrogen bonding of a linear
chain of amino acids whose peptide plane is bent at the
Cα carbon. There are two ways for this to be accomplished: parallel beta sheets join segments of the polypeptide chain whose amino to carboxyl direction is
going in the same direction. Antiparallel beta sheets
join polypeptide chains of opposite N to C polarity.
Structures not fitting into these categories are often
called “random coils,” although they may not be random at all. There are four main principles involved in
the formation of these secondary structures.
Hydrogen Bonding Stabilizes Structure
The polar centers of the carbonyl oxygen and the amide
nitrogen are hydrogen bonded to other structures in the
135
136
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
protein, either another amide bond or a polar side
group of an amino acid. These hydrogen bonds stabilize
protein structure and are instrumental in forming the
α-helix.
Carboxy terminus
Hydrophilic Groups Face Water; Hydrophobic
Groups Face Away from Water
The protein will fold so that highly charged groups and
polar groups are on the outside of the protein facing
water. Here the interaction between these hydrophilic
groups and the hydroxyl groups of water stabilizes the
structure.
Carbon
Side Groups Cannot Occupy the Same Space
Hydrogen
This is a simple way of saying there is steric hindrance
in the structure. Atoms in the amino acids exhibit repulsive forces when placed too close together. These forces
ultimately arise from the interpenetration of atomic or
molecular electron orbitals.
Nitrogen
Oxygen
R groups
Single bond
Hydrogen bonds
C–C and C–N bonds in
the alpha helix backbone
0.54 nm = 3.7 amino acid residues
Electrostatic Interactions Stabilize Structures
Groups with opposite electric charge attract each other
and this attractive force can stabilize the threedimensional arrangement of amino acids. Similarly,
groups with the same sign electric charge repel each
other and this repulsive force can also stabilize the
structure by preventing closer movement of the electrically charged groups.
TERTIARY STRUCTURE DEALS WITH THE
THREE-DIMENSIONAL ARRANGEMENT OF ALL
OF THE AMINO ACIDS
Amino terminus
FIGURE 2.3.12 The alpha helix. The dark bonds indicate those in the
polypeptide backbone that are directly involved in the peptide bonds.
The dashed lines indicate hydrogen bonds that bond successive turns of
the helix with each other. These hydrogen bonds connect the carbonyl
of one amino acid with the amino hydrogen of the fourth amino acid
down the chain.
The tertiary structure of proteins deals with how the
regional structures are put together in space. For example, the α-helices may be oriented parallel to each other
or at right angles. So the tertiary structure refers to the
folding of the different segments of helices, sheets,
turns, and the remainder of the protein into its native
three-dimensional structure. Commonly, membrane
proteins are anchored in the membrane by hydrophobic
Carboxy terminus
Carbon
Amino
terminus
N
C
Hydrogen
Nitrogen
Parallel
Oxygen
Carboxy terminus
R groups
Amino
terminus
N
C
Antiparallel
Carboxy
terminus
Single bond
Hydrogen bonds
C–C and C–N bonds in
the beta sheet backbone
Amino terminus
C
N
FIGURE 2.3.13 Structure of the beta sheet. Strings of amino acids are bonded laterally through hydrogen bonds. The arrangement can be between
strings that have the same amino to carboxy orientation, called parallel, or between strings with opposite orientation, called antiparallel. The planar
peptide bonds line up to form a kind of pleated sheet.
Prot ein St ructur e
alpha helices that may be far removed in the primary
sequence but closely apposed in the tertiary structure of
the protein.
the interconversion of disulfide bonds until the right
ones are formed.
CHAPERONES AND CHAPERONINS HELP
PROTEINS FOLD
QUATERNARY STRUCTURE REFERS TO THE
INTERACTION OF A PROTEIN WITH OTHER
PROTEINS
As they are made on the ribosome, proteins begin to
fold up. Sometimes the primary structure of the protein
alone can determine the proper final shape, and denaturing the protein (adding materials or heat that disrupts its shape) is reversible. In other cases, proteins
need help in determining their shape, and they are synthesized on a kind of scaffold that ensures that they
fold properly. These scaffolds are generally other proteins called chaperones. There are two kinds of chaperones. Molecular chaperones bind to unfolded or
partially unfolded proteins and stabilize their structure,
preventing them from being degraded. Chaperonins
directly facilitate the folding of proteins. Molecular chaperones are members of the Hsp70 family of proteins;
hsp means “heat shock protein” because these increase
after heat stress to an animal that would denature
proteins. Complexes of eight Hsp60 molecules form a
barrel-shaped chaperonin that aids protein folding.
The quaternary structure refers to the interaction of a
protein with other proteins or other components of the
cell. This association of the protein with other elements
of the cell can alter its three-dimensional shape and its
activities because the close proximity of another protein’s surface can alter the shape of the protein. Many
proteins exist in the cell in complex macromolecular
assemblies in which quaternary structure enables or
regulates the function of the component proteins.
POSTTRANSLATIONAL MODIFICATION
REGULATES AND REFINES PROTEIN
STRUCTURE AND FUNCTION
Proteins that come off the ribosome are not yet
finished. The cell processes the newly made proteins in
several ways, including:
G
G
G
G
PROTEOLYTIC CLEAVAGE
Cells make many proteins in precursor form with
longer primary sequences than the finished product.
Proteolytic cleavage forms the final protein by chopping
off unwanted parts of the protein.
formation of disulfide bonds
folding into the functional form
cleaving the proteins at specific sites
chemical modification.
PROTEINS ARE CHEMICALLY MODIFIED IN A
VARIETY OF WAYS
PROTEIN DISULFIDE ISOMERASE CATALYZES
DISULFIDE EXCHANGE
Cells chemically modify proteins after translation in a variety of ways, some of which are shown in Figure 2.3.14.
These include:
Proteins often are stabilized by disulfide bonds between
cysteine residues in the protein. These cysteines are
not necessarily close to each other on the primary
sequence but must be close in the tertiary structure of
the protein. The protein disulfide isomerase catalyzes
G
acetylation
methylation
G
NH2
CH3
CH2
S
CH
CH2
CH2
H3C
N
O
H3C C
CH2 O
NH
C
H
N-acetylation
C
CH2 O
NH
C
NH+
H2
C
H2C
C
H
N
CH O
C
H
5-Hydroxy lysine
CH2 O
H
N
C
H2C
C
H
3-Methyl histidine
3-Hydroxy proline
N
CH
CH2 O
C
H
OH
C
C
CH
OH
C
OH O
O
OH
OH
C
CH2
HN
C
O
C
H
OH
Gamma carboxy
4-Hydroxy proline glutamic acid
FIGURE 2.3.14 Examples of some chemical posttranslational modifications of proteins. The N-terminal amino acid of many proteins (about 80% of
them) is acetylated. Lysine groups can also be acetylated (not shown). Acetylation may regulate the life span of proteins as nonacetylated proteins
rapidly degrade. Hydroxylation of lysine in the 5 position occurs in collagen. In collagen, proline is converted to 3 hydroxyproline or 4 hydroxyproline.
Histidine, particularly in actin, is methylated. Arginine can also be methylated (not shown) and this modification is part of the histone code (see
Chapter 2.2). Glutamic acid residues in prothrombin (a protein involved in clotting of blood) and in bone proteins are carboxylated at the γ-side chain
carbon to form gamma carboxy glutamic acid. The close proximity of the two carboxyl groups in gamma carboxy glutamic acid creates a binding site
for calcium ions, and this confers Ca21 sensitivity to the coagulation process. The gamma carboxylation reaction requires vitamin K, which is necessary
for normal blood coagulation.
137
138
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Rest of the
protein
GPI-linked protein
Mannose
O
–
O P O
Mannose
H2
C
H2
C
O
HC
NH2
O
Mannose
O
O P O
Mannose
N-acetyl galactosamine
O–
N-acetyl glucosamine
Inositol
O
–
O P O
H
H
C
C
H
O
H
C
Extracellular face
H
O
O C
C O
CH2
CH2
H2C
HC
O
2
CH2
H2C
CH2
H2C
CH2
H2C
CH2
H2C
CH2
H2C
CH2
Membrane bilayer lipid core
H2C
CH2
H2C
CH2
H2C
CH2
H2C
CH2
H2C
CH3
CH3
H3C
H3C
FIGURE 2.3.15 Anchors of membrane proteins.
Some membrane proteins are anchored in the
membrane by attachment of hydrophobic parts
such as myristic acid or palmitic acid. Myristic
acid is a 14-carbon hydrocarbon chain with a
carboxyl group on one end that can covalently
attach to the N-terminal glycine of membrane
proteins. Palmitic acid is a 16-carbon saturated
monocarboxylic fatty acid and attaches to
proteins similar to myristic acid. The process
of attaching myristic acid or palmitic acid is called
myristoylation or palmitoylation. Farnesyl is a
polymer of three 5-carbon units called isoprene.
Farnesyl attaches covalently via a thioester
bond to cysteine residues somewhere in the
middle of the protein and helps anchor some
proteins in the membranes. Other proteins
link to GPI or glycosylphosphatidylinositol, a
glycosylated membrane lipid that helps keep
proteins in the membrane.
G
G
G
G
G
CH2
CH3
C
H2C
HC
CH2
CH2
H2C
H2C
C
CH2
CH3
H2C
HC
Farnesyl
H 2C
C
CH2
CH3
H 2C
HC
CH2
CH2
H2C
H2C
C
CH2
Cysteine residue of
membrane protein
O
C
N
H
O
hydroxylation
gamma carboxylation
glycosylation
myristoylation or palmitoylation
phosphorylation.
Proteins in the endoplasmic reticulum are often glycosylated, meaning that sugars or sugar derivatives are covalently attached to the proteins. N-linked glycosylation
CH
N-terminal glycine of
membrane protein
NH
S
CH2
HC
Myristic acid
CH2
CH2
H2C
2HC
C
C
NH
NH
Intracellular face
O
O
occurs when carbohydrate branches are added to the side
chain NH2 of asparagine through N-acetylglucosamine.
O-glycosylation occurs on the side chain OH of serine,
threonine, or hydroxylysine and the connecting carbohydrate is N-acetylgalactosamine.
Proteins stick in membranes because they are anchored
there by a variety of posttranslational modifications, as
shown in Figure 2.3.15.
Prot ein St ructur e
PROTEIN ACTIVITY IS REGULATED BY
THE NUMBER OF MOLECULES OR BY
REVERSIBLE ACTIVATION/
INACTIVATION
The cell can alter the activity of its component proteins
by altering the number of copies of the protein, or by
activating or inactivating the proteins that are already
present.
Regulating the number of protein molecules requires
synthesis of new protein molecules or degradation of
existing ones. This takes time and degrading existing
proteins wastes energy. Reversible activation or inactivation of proteins can achieve rapid regulation that also
conserves energy. Cells use phosphorylation/dephosphorylation of proteins to regulate their activities.
Attachment of a phosphate group changes the charge
on a local region of the protein, which alters its threedimensional shape and changes its activity. Serine, threonine, and tyrosine residues are the targets for these
phosphorylation reactions (see Figure 2.3.16). An
enzyme that phosphorylates proteins is called a protein
kinase; one that dephosphorylates proteins is called a
protein phosphatase. Cells contain a variety of protein
kinases and protein phosphatases (see Figure 2.3.17).
OH
–O
P
O
Protein state A
O
OH
–O
P
OH
O
CH2
C
P
H3C
O
O
Protein kinase
Protein phosphatase
O
HN
–O
ATP
Pi
CH
O
C
H
N
C
CH2
O
C
NH
C
O
C
ADP
Protein state B
PO4
H
H
H
Serine (Ser) S
Threonine (Thr) T
Tyrosine (Tyr) Y
FIGURE 2.3.16 Phosphorylation sites of proteins. Serine, threonine, and
tyrosine all have hydroxyl groups that can be esterified with phosphate.
This alters the shape and charge of the protein surface in that area,
leading to changes in protein activity.
FIGURE 2.3.17 Phosphorylation cycle for protein regulation. Some
proteins have phosphorylation sites that can be covalently linked to
phosphate from ATP through the action of a variety of protein kinases.
This alters the local charge of the protein, which in turn changes its
shape and its activity. The protein returns to its dephosphorylated state
by the action of protein phosphatases.
Clinical Applications: Protein Folding Diseases
With the advent of the microscope in the mid-1800s, Pasteur,
Koch, and many others formulated the germ theory of infectious
diseases. We now know that microscopic viral, bacterial, protozoan, or parasitic agents cause a long list of diseases: smallpox,
polio, rabies, HIV, yellow fever, anthrax, bubonic plague, syphilis,
tuberculosis, cholera, gonorrhea, malaria, sleeping sickness,
schistosomiasis, to name but a few. Each of these infectious
agents contains DNA or RNA that codes for the organism’s own
proteins and enables replication of its nucleic acid. Viruses do
this by using the host’s machinery. Bacteria, protozoans, and
parasites are self-contained organisms that use the host’s
environment for their own reproduction. Investigation of a group
of diseases called transmissible spongiform encephalopathies
(TSE) required revolutionarily new thinking about infection.
These diseases include CreutzfeldtJakob disease, kuru,
GerstmannStraussler syndrome (GSS), and fatal familial insomnia (FFI) in humans, and scrapies and bovine spongiform
encephalopathy (BSE) in animals.
Kuru is a neurological disease among the Fore, a linguistic group
of people in Papua New Guinea. In ritual cannabalism, these
people ate the bodies of dead relatives. After long incubations,
they developed partial paralysis and loss of motor control and
eventually died. Because the disease seemed to run in families, a
genetic cause was first proposed, but later rejected. Carleton
Gajdusek, a pediatrician and virologist, was unable to transmit
the disease to any animal, including primates. Igor Klatzo, a neuropathologist, examined tissues sent by Gajdusek and found that
kuru was a unique disease without precedent, the closest condition being CreutzfeldtJakob disease. In a bizarre twist, William
Hadlow, a veterinarian neuropathologist, saw some of Klatzo’s
photomicrographs in an exhibit at the Wellcome Medical
Museum in London, and noted a startling resemblance between
neurohistological changes in kuru and those in scrapie, a neurological disease in sheep first described in 1732. It was known to
be infectious, but the infectious agent was not yet identified.
Hadlow wrote to Gajdusek and published a letter in The Lancet
(Continued)
139
140
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Clinical Applications: Protein Folding Diseases (Continued)
in 1959. The similarity of kuru to infectious scrapie prompted
Gajdusek to further attempt inoculating animals with kuru. In
1966, he and his co-workers transmitted kuru to chimpanzees
and then CreutzfeldtJakob disease to chimpanzees in 1968.
Gajdusek earned the 1976 Nobel Prize in Medicine.
Although it was initially assumed that the infectious agent must
be some virus, investigators failed to identify any virus or any
immunological response to one. The agent was not destroyed
by UV irradiation or nucleases that typically inactivate nucleic
acids. Tikvah Alper and her co-workers in 1966 found that the
infectious agent of scrapie was too small to provide a nucleic
acid code. In 1967, J.S. Griffith proposed that a protein alone
could be the infectious agent of TSE if it was a pathogenic form
of a host protein that could convert normal host protein to the
pathogenic form. Finally, in 1982, Stanley Prusiner identified the
infectious agent as a protein devoid of nucleic acids. He coined
the term “prion” which stands for proteinaceous infectious particle. Prusiner earned the 1997 Nobel Prize in Medicine.
SUMMARY
The core of amino acids is their asymmetric alpha
carbon. Attached to one side is an amino group. On the
other side is a carboxyl group. The third group is hydrogen and the fourth group is a variable group, usually
referred to as a side chain or R group. These variable
groups define the class of the amino acids. Glutamic
acid and aspartic acid have a carboxyl group on the side
chain. At neutral pH, this group ionizes and therefore
has a negative charge. Lysine, arginine, and histidine are
basic amino acids because their side chains have a basic
chemical character. At neutral pH, these are positively
charged. Serine, threonine, and tyrosine have hydroxyl
groups that confer polar character. Asparagine and
glutamine have an amide group that is also polar.
Nonpolar amino acids include glycine, alanine, valine,
leucine, isoleucine, proline, cysteine, methionine, tryptophan, and phenylalanine. Some of these are highly
hydrophobic, such as tryptophan, isoleucine, leucine,
and phenylalanine.
Proteins are made by the formation of peptide bonds
between the amino group of one amino acid and the
carboxyl group of another. Because of this bond, the
basic character of the amino group is neutralized, and
the acidic character of the carboxyl group is neutralized,
and the character of the chain of amino acids is determined by the sequence of the side chains.
We describe protein structure on four levels: the primary
sequence describes the linear sequence of amino acids
along the peptide chain backbone, proceeding from the
amino terminus to the carboxy terminus. The protein
folds into secondary local structures such as α-helices,
β-sheets, and β-turns. The arrangement of these secondary structures in three-dimensional space produces the
tertiary structure. Combination of proteins with other
structures produces macromolecular complexes with
quaternary structure. Some proteins spontaneously fold
Prions are infectious proteins. They “reproduce” by causing normal cellular prion protein (PrPC) to fold up differently, converting
it into the pathogenic, or scrapie, isoform (PrPSc). The native PrPC
appears to have three α-helices and two short β-strands; PrPSc
has two α-helices and much more β-sheet. This transition from
α-helix to β-sheet is the fundamental event underlying prion diseases. Proteolysis of PrPSc produces a smaller, protease-resistant
molecule of about 142 amino acids (Prp 2730) which
polymerizes into amyloid that presents itself in the disease state.
Ritualistic cannabalism transmitted kuru among the Fore people
of New Guinea; industrial cannabalism spread BSE (“mad cow
disease”) in Europe. There is more than one bad way to fold a
protein. Increasing numbers of patients have contracted a new
variant of CreutzfeldtJakob disease (vCJD) from prion-tainted
beef. Because of its long incubation time, we do not yet know
the price of mad cow disease. Current thinking that the disease
can be eradicated completely by control of infection is wrong.
The disease can spontaneously appear without infection.
into their “native” shape, whereas others are assembled
on a kind of scaffold that helps them fold up properly
into their active form. Denaturation of proteins occurs
when the protein loses its normal shape. In some cases
this is reversible, but usually loss of the proper folding
causes irreversible loss of function. Hydrogen bonding,
electrostatic interactions, hydrophobic interactions, and
steric hindrance all help stabilize proteins in their
secondary structures. Disulfide bonds between cysteine
side chains help stabilize higher order structure.
Proteins undergo posttranslational modifications after
they are synthesized. These include N-glycosylation
or O-glycosylation, proteolytic cleavage, hydroxylation,
methylation, acetylation, γ-carboxylation, covalent
attachment of hydrophobic molecules that anchor proteins in membranes, and phosphorylation of specific
hydroxyl groups on side chains of serine, threonine,
and tyrosine.
Protein function depends on the way their surfaces
interact with the surfaces of other materials—substrates,
structural elements, or other materials to which the proteins bind. Catalytic activity or structural or regulatory
roles of proteins depend on the close match of their
surface with the surface of the things they bind to. This
also determines the specificity of the protein’s action.
In general, activity of proteins in the cell can be regulated by altering the amount of protein or by altering its
intrinsic activity. Reversible regulation can be achieved
by phosphorylation/dephosphorylation of proteins.
REVIEW QUESTIONS
1. Name the two acidic amino acids. What makes
them acidic? Name three basic amino acids.
What makes them basic?
2. Name five polar amino acids. What makes them
polar? Name the nonpolar amino acids.
Prot ein St ructur e
3. What does “hydrophobic” mean? What does
“hydrophilic” mean? What is the partition coefficient? How does it measure hydrophobicity?
4. What is a peptide bond? Where do you find it?
5. What is the primary structure of a protein?
6. What is an α-helix? β-sheet? What interactions
among protein side chains stabilize these
structures?
7. What is “posttranslational modification”? Name
four different kinds of posttranslational
modification.
8. What residues are most often acetylated?
What residues are hydroxylated? What is “γ-carboxylation”? What amino acid is γ-carboxylated?
What is glycosylation? What is myristoylation or
palmitoylation?
9. How can proteins be anchored in hydrophobic
membranes?
10. Describe the phosphorylation/dephosphorylation
cycle for regulating activity of proteins in the
cell.
141
2.4
Biological Membranes
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
Describe fatty acids and what is meant by saturated and
unsaturated fatty acids
Distinguish between cis and trans arrangement around a
double bond in an unsaturated fatty acid
Describe the constituents of phosphatidic acid, phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine,
and phosphatidylinositol
Identify hydrophilic and hydrophobic groups in membrane
lipids
Recognize the steroid ring structure of cholesterol
Recognize the chemical structures of cardiolipin, sphingosine, sphingomyelin, and ceramide
Define surface tension
Describe how amphipathic lipids reduce surface tension
Describe motion in the plane of a lipid bilayer
Describe the fluid mosaic model of biological membranes
Distinguish between integral and peripheral proteins
Describe caveolae and clathrin-coated pits
Describe how secreted proteins are synthesized on the ER
membrane
BIOLOGICAL MEMBRANES
SURROUND MOST INTRACELLULAR
ORGANELLES
As discussed in Chapter 2.1, cells contain a variety
of subcellular organelles and the hallmark of most of
them is that they are surrounded by a membrane.
These membranes divide the cell into several compartments in which enzymes and substrates are sequestered away from the rest of the cell. This separation
into compartments is required for the functioning
of these organelles. Maintenance of this compartmentalization requires selective transport of materials
across the membranes. Table 2.4.1 lists the various
subcellular organelles with their approximate contributions to the cell volume and membrane area.
The proportions of cell volume and area represented
by the subcellular organelles vary markedly with cell
type and activity.
BIOLOGICAL MEMBRANES CONSIST
OF A LIPID BILAYER CORE
WITH EMBEDDED PROTEINS
AND CARBOHYDRATE COATS
The composition of biological membranes varies
enormously among the different subcellular organelles,
but all biological membranes share a basic structure.
The core of the membrane is a lipid bilayer. Embedded
in this core are a variety of proteins that carry out many
of the activities of the membrane, including selective
membrane transport, and some of both the lipids and
proteins have carbohydrate coats. This basic structure is
shown in Figure 2.4.1. The rest of this chapter expands
on this general description.
ORGANIC SOLVENTS CAN EXTRACT
LIPIDS FROM MEMBRANES
Gorter and Grendel, in 1925, provided early evidence
for the lipid bilayer structure of membranes when they
extracted the lipids from erythrocytes with acetone and
spread them over the surface of water. They noted that
the area occupied by the lipids was about twice the calculated area of the surface of the erythrocytes. Because
the only membrane in the erythrocytes is the plasma
membrane, they concluded that the membrane was a
lipid bilayer. This confirmed earlier observations by
Ernst Overton in the 1890s that there is an excellent
correlation between the ability of a number of solutes
to enter cells and their solubility in olive oil. Overton
concluded that the surface of the cell was made up of
lipids similar to olive oil.
BIOLOGICAL MEMBRANES CONTAIN
MOSTLY PHOSPHOLIPIDS
Organelles can be isolated by cellular disruption and differential centrifugation, as described in Appendix 2.1.A1.
The lipids in the membranes can be extracted into
an organic phase, typically a chloroform/methanol mixture, because these lipids are hydrophobic (see
Figure 2.3.7), and then the lipids can be separated into
their component classes by chromatography, and the
amounts can be measured. Approximate lipid composition of some membranes is given in Table 2.4.2.
142
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00013-6
Biological Membra nes
However, it should be kept in mind that the lipid composition varies not only with the kind of organelle but
also with the kind of cell.
TABLE 2.4.1 The Relative Area and Enclosed Volumes of
the Major Subcellular Membranes in a Typical Liver Cell
Membrane Percent of
Total Cell
Volume
Enclosed
Plasma
membrane
Approximate Percent of
Number per Total Cell
Cell
Membrane
100
1
2
22
1700
39
Rough ER
9
1
35
Smooth ER
2
1
16
Golgi
apparatus
4
1
7
Nuclear
inner
membrane
6
1
0.2
Lysosomes
1
300
0.4
Peroxisomes
1
400
0.4
Mitochondria
PHOSPHOLIPIDS CONTAIN FATTY ACYL
CHAINS, GLYCEROL, PHOSPHATE,
AND A HYDROPHILIC GROUP
Table 2.4.2 shows that phospholipids comprise the
most abundant class of lipids in membranes. Each consists of four parts: a glycerol backbone, a phosphate
esterified to one end of the glycerol molecule, a polar
head group attached to the phosphate, and two fatty
acids esterified to the other two hydroxyl groups of the
glycerol. The glycerol phospholipids form the major subclass of the phospholipids, and they contain phosphate
esterified to the C-3 hydroxyl group of glycerol and two
fatty acids esterified to the C-2 and C-1 hydroxyl groups.
Fatty acids consist of two distinct regions (see
Figure 2.4.2): a long hydrocarbon chain and a carboxylic
acid group (COOH). The most frequent length of the
carbon chain is 16 or 18 carbon atoms. As discussed in
Chapter 1.4, carbon can form four single bonds with
bond angles that approximate those between the center
of a tetrahedron and its vertices. In a hydrocarbon chain,
two of these bonds link a carbon with adjacent carbons
and two remain for other bonds. When hydrogen is
covalently bound to all of the two remaining bonding
orbitals, the hydrocarbon is saturated. Unsaturated fatty
acids contain one or more double bonds between carbon atoms. Those containing more than one double
bond are polyunsaturated. The variety of phospholipids
is produced from the variety of fatty acids and from the
different polar head groups. The most common lipid
constituent of membranes is phosphatidylcholine, with
other phospholipids making major contributions. The
components of the simplest phospholipid, phosphatidic
acid, are shown in Figure 2.4.2.
Values were estimated by quantitative electron microscopy.
Carbohydrate coats
Proteins
Lipid bilayer
core
FIGURE 2.4.1 Basic structure of biological membranes. Membranes
consist of a lipid bilayer core to which proteins are attached in a variety
of ways. In addition, some lipids and proteins have carbohydrate groups
attached to them.
A variety of polar groups can be attached to the phosphate of phospholipids. These groups include serine,
inositol, and ethanolamine, which can be methylated
to form choline. The resulting phospholipids are shown
in Figure 2.4.3.
TABLE 2.4.2 Approximate Lipid Composition of Different Cell Membranes
Lipids
Percentage of Total Lipids by Weight
Plasma
Membrane
Phosphatidylcholine
Mitochondrial
Membrane
Endoplasmic Reticulum
Membrane
24
39
40
Phosphatidylethanolamine
7
35
17
Phosphatidylserine
4
2
5
Cholesterol
17
3
6
Sphingomyelin
19
0
5
7
0
0
22
21
27
Glycolipids
Other
143
144
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Glycerol
H
H
1
C
OH
Palmitic acid
(C16:0)
OH
H
2C
Phosphate
OH
-O
C
3
OH
H
Stearic acid
(C18:0)
OH
CH2
CH2
HC
CH2
CH2
H2C
H2C
CH2
CH3
H2C
CH2
CH2
H2C
H2C
Nonpolar tail
HC
H2C
CH2
CH2
CH2
CH2
CH2
H2C
CH2
H2C
H2C
CH2
HC
CH2
H2C
H3C
CH3
C O
CH2
H2C
H2C
CH2
H2C
H2C
H
CH2
CH2
CH2
CH2
3
CH2
H2C
H2C
H2C
C
H2C
H2C
CH2
Polar head
H
CH2
HC
CH2
O
O
H2C
H2C
H2C
H2C
2C
CH2
CH2
CH2
C
O C
H2C
H2C
H2C
1
CH2
CH2
CH2
H
O
H2C
H2C
H
H
CH2
CH2
CH2
H2C
OH
O C
H2C
O-O P O-
Oleic acid
(C18:1, n9)
CH2
CH2
H2C
Phosphatidic
acid
OH
O C
O C
O
P OH
H
H2C
CH3
H2C
CH2
H2C
CH2
H3C
FIGURE 2.4.2 Components of a simple phospholipid, phosphatidic acid. Glycerol forms the backbone. Each of the three carbons in glycerol covalently
bonds to a hydroxyl group (OH). Rotation about the CC single bonds can position the C1 OH or C3 OH in any orientation. The central carbon, C2, is
an asymmetric carbon when C1 and C3 are differently substituted, and will show stereoisomerism when different groups attach to the two ends. To
distinguish the 1 and 3 positions, the numbering nomenclature shown in the figure is used. The structure of inorganic phosphate is also shown. Fatty
acids form the hydrophobic core of phospholipids. The chemical structure of three common fatty acids: palmitic acid, stearic acid, and oleic acid is
shown. The single carboncarbon bonds allow free rotation about the axis connecting the two carbon nuclei. Each carbon atom forms bonds that
nearly line up the centers of two tetrahedrons with their respective apices. Palmitic acid is a 16-carbon saturated fatty acid, meaning that every
carbon’s bonds other than the carboxyl carbon are fully occupied with single bonds to carbon or hydrogen. In the nomenclature of fatty acids, it is
designated 16:0, indicating a 16-carbon fatty acid with no double CC bonds. Stearic acid is a 18-carbon saturated fatty acid, designated as 18:0. Oleic
acid is a mono-unsaturated fatty acid, meaning that one pair of carbon atoms are joined by a double bond. In this case, the double bond is between
carbons 9 and 10, numbering down from the carboxyl carbon. Its designation is 18:1 Δ9, where the 1 indicates one double bond and the Δ9 indicates
the double bond begins at Carbon 9. There is an alternate number system starting from the terminal methyl group of the fatty acid, instead of starting
at the carboxyl group. This nomenclature uses the prefix omega (ω); thus oleic acid is 18:1 ω 2 9. Here the 18 stands for the length of the hydrocarbon
chain, 1 indicates the number of double bonds, and ω 2 9 indicates the position of the double bond numbering from the methyl end. Substitution of
the omega with the letter n is becoming popularized, with the same meaning. As discussed in Chapter 1.4, double bonds produce kinks in the
hydrocarbon chain due to restricted rotation about C 5 C double bonds. Oleic acid has a cis orientation of the H atoms around the double bond,
meaning the two hydrogens are on the same side of the double bond. The structure of a phosphatidic acid is shown on the right. This particular
phosphatidic acid has an oleic acid molecule esterified to C-2 of glycerol and a palmitic acid molecule esterified to C-1. The fatty acids differ from
molecule to molecule, but this is a typical arrangement in which saturated fatty acids occupy the C-1 position and unsaturated or polyunsaturated
fatty acids occupy the C-2 position. Phosphatidic acid illustrates a common property of this class of lipids in that it consists of spatially separated
water-soluble polar or hydrophilic groups and water-insoluble nonpolar or hydrophobic groups.
Phosphatidic
acid
Phosphatidyl
ethanolamine
Phosphatidyl choline
Polar group
H
1
C
C
3
CH2
C O
CH2
H2C
H2C
CH2
H2C
H2C
H2C
CH2
H2C
CH2
H2C
CH2
H2C
CH3
CH2
CH2
H2C
CH2
H2C
CH2
CH2
H2C
CH2
H3C
CH2
H 2C
CH2
H 3C
Fatty acid: palmitic acid C16:0
H 2C
CH3
H 2C
CH2
CH2
H 2C
CH2
H 3C
H 2C
CH3
CH2
H 2C
CH2
H 2C
CH2
H 2C
CH2
CH2
H 2C
CH2
H 2C
CH2
HC
CH2
HC
Nonpolar tail
HC
H 2C
H 2C
CH2
CH2
C O
CH2
CH2
H 2C
3
H
H 2C
HC
CH2
H 2C
CH2
CH2
H 2C
H2C
CH3
CH2
H 2C
CH2
H 2C
H
C
O
H 2C
H 2C
CH2
HC
C
2
CH2
CH2
CH2
CH2
H 2C
H2C
CH2
H 2C
CH2
H 2C
H 2C
Polar head
O
CH2
H 2C
H 2C
HC
CH2
H 2C
CH2
CH2
CH2
C
O C
H 2C
H 2C
H
O
C O
CH2
CH2
1
H
H 2C
H
H
3
CH2
CH2
H 2C
H
C
O
H 2C
H 2C
CH2
HC
O
CH2
H 2C
H 2C
2C
CH2
CH2
CH2
HC
CH2
HC
H2C
CH2
H 2C
1C
O C
H 2C
H 2C
H
O
C O
CH2
CH2
CH2
CH2
HC
CH2
H 2C
H2C
H
H 2C
H
H
3
CH2
CH2
CH2
CH2
H
C
O
H 2C
O
P O-
O
O
CH2
CH2
CH2
H2C
2C
O C
H 2C
H2C
1C
O
C O
H2C
CH2
CH2
H
CH2
H2C
CH2
3
O
O C
H
H
C
2C
O
H
H
H
CH CH OH
HC OH
CH
C
CH
O H
OH
O P O-
CH2 OH
Phosphate
O
P O-
O
O
H
H
1C
O
O C
H2C
H
H
2C
O
H2C
O
H
H
Glycerol
O
P O-
O
O
C
HC
H 2C
CH2
OH OH
NH2
+
H 2C
CH2
Phosphatidyl inositol
Polar group
CH3
H3C N CH3
NH2
O-O P O-
Phosphatidyl serine
CH3
H3C
H 2C
CH2
H 2C
CH2
H 3C
Fatty acid: oleic acid C18:1 n9
FIGURE 2.4.3 Chemical structures of some common glycerophospholipids. The phosphate group in phosphatidic acid is esterified to the hydroxyl group of several other hydrophilic molecules
including ethanolamine, choline, serine, and inositol. These form the lipids shown. Each of these are named for the hydrophilic group and the fatty acids, as in 1-palmitoyl, 2-oleolyl
phosphatidylcholine.
146
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
PLASMANYL PHOSPHOLIPIDS
AND PLASMENYL PHOSPHOLIPIDS
USE FATTY ALCOHOLS INSTEAD
OF FATTY ACIDS
A major subclass of the phospholipids use fatty alcohols
instead of fatty acids, forming an ether linkage with
glycerol instead of an ester. These are called plasmanyl
glycerol phospholipids. Most of these are modified to
contain a vinyl ether linkage, in which the alcohol
group is doubly bonded to the rest of the fatty alcohol
chain. These are termed plasmenyl glyercol phospholipids or plasmalogens. Their structures are shown in
Figure 2.4.4. They make up 1520% of the total phospholipids of cell membranes.
SPHINGOLIPIDS USE SPHINGOSINE AS
A BACKBONE AND ARE PARTICULARLY
RICH IN BRAIN AND NERVE TISSUES
Sphingolipids are present in many membranes but they
are particularly rich in brain and nerve tissues. There
are three classes of sphingolipids: sphingomyelin, cerebrosides, and gangliosides. Sphingomyelin is the only
one of these that is a sphingophosphatide. It is analogous to the glycerophosphatides except that it contains
sphingosine instead of glycerol as the core structure
that links the hydrophilic phosphate and choline to the
hydrophobic hydrocarbon chains. Sphingosine is a
derivative of the amino acid, serine. The chemical structures of sphingosine and sphingomyelin are shown in
Figure 2.4.5.
The fatty acid amide of sphingosine alone is called a
ceramide (see Figure 2.4.5). Ceramides can be linked
through the hydroxyl group to sugar groups to form
Diacyl glyercol
phospholipid
H
H
1
C
O
O C
R1
H
2C
CARDIOLIPIN IS TWO GLYCEROLIPIDS LINKED
BACK TO BACK
The structure of cardiolipin is shown in Figure 2.4.6. It
consists of two phosphatidic acid molecules linked
through another glycerol. Mitochondrial membranes
are particularly rich in cardiolipin.
CHOLESTEROL CONDENSES MEMBRANES
Cholesterol, shown in Figure 2.4.7, is the most abundant steroid in animal tissues. All steroid hormones are
derived from cholesterol. It possesses a rigid ring structure that attracts normally flexible phospholipid chains
to itself, causing membranes to become more rigid in
the vicinity of this molecule.
PHOSPHOLIPIDS IN WATER SELF-ORGANIZE
INTO LAYERED STRUCTURES
All of the glycerophospholipids and sphingolipids we
have discussed are characterized by the spatial separation of a polar head group, consisting of ionized
groups, hydroxyls and carbonyl oxygens, and a long tail
consisting mainly of hydrocarbons. The polar head
group is capable of interacting with water and each of
the materials there individually is water soluble. This
(Plasmalogen)
Plasmenyl
glycerol phospholipid
X
O
P O–
–O
O
H
C
H
H
1
3
O
OTHER LIPID COMPONENTS OF
MEMBRANES INCLUDE CARDIOLIPIN,
SPHINGOLIPIDS, AND CHOLESTEROL
Plasmanyl
glycerol phospholipid
X
–O
another class of sphingolipids, the cerebrosides. The
sugar part contains a number of hydroxyl groups and is
hydrophilic. In some cases the sugar part is quite large
and branched, forming a ganglioside. Both cerebrosides
and gangliosides form another class of lipids called glycolipids because they incorporate sugar derivatives.
H
C
O
H
2C
X
O
P O–
–O
O
H
C
O
H
H
C
2C
H
1
3
H
O
O
P O–
O
H
C
3
O
C O
CH2
C O
CH
C O
R2
CH2
R2
CH
R2
R1
Fatty acids
X = polar head group:
ethanolamine, choline
or inositol
H
R1
Vinyl ether bond
Fatty alcohol
FIGURE 2.4.4 Plasmanyl glycerol phospholipids and plasmenyl glycerol phospholipids. Some long-chain hydrocarbons have an alcohol and not a
carboxyl group at their end. These can be joined to glycerol through an ether linkage, forming a plasmanyl glycerol phospholipid which is typically
enriched in the sn-2 position with polyunsaturated fatty acids. In most cases, the carbon adjacent to the ether is joined in a double bond, forming a
vinyl ether bond that characterizes the plasmenyl glycerol phospholipids, also known as the plasmalogens.
Biological Membra nes
Sphingomyelin
CH3
H3C N CH3
Choline
+
H 2C
CH2
Sphingosine
O
P O-
Ceramide
O
Serine
H
HO
C
H
HO
H
C
H
C
C
H
HC
H
HO
NH2
HC
H2C
CH2
H2C
Different from
glycerol
CH3
CH3
C O
CH2
H 2C
H 2C
H 2C
CH2
H 2C
H 2C
CH2
HC
H 2C
CH2
CH2
H 2C
CH3
CH2
H 2C
CH2
H 2C
H 2C
Nonpolar tail
HC
CH2
HC
Amide linkage
CH2
CH2
CH2
H 2C
H2C
H
CH2
CH2
CH2
CH2
H
C
CH2
CH2
H 2C
H2C
O
HN
H 2C
HC
CH2
CH2
Amide linkage
H 2C
H 2C
C
H 2C
CH2
CH2
C
CH2
CH2
CH2
H
HC
H 2C
H 2C
H2C
HC
H 2C
H 2C
CH2
H
C O
CH2
CH2
CH2
H2C
C
HN
H 2C
H
O
H
CH2
CH2
H
C
HC
HC
HO
Polar head
H 2C
CH2
H2C
H2C
CH2
CH2
H3C
H3C
FIGURE 2.4.5 Chemical structures of sphingosine, ceramide, and sphingomyelin. Note that sphingosine does not have glycerol as a core structure and
joins a hydrocarbon through an amide linkage instead of an ester to form sphingomyelin.
region of the molecule is hydrophilic, meaning water
loving. The hydrocarbon tail is not soluble in water; it is
hydrophobic or water hating. Hydrophobic parts are
also described as lipophilic or fat loving. Molecules
having both of these separate domains are said to be
amphipathic, from the Latin and Greek amphi meaning
having two sides. These two sides are illustrated by the
space-filling model in Figure 2.4.8. These two separate
domains of phospholipids are crucial to their behavior
in cells. When placed in water, the polar heads of these
molecules remain associated with water and form
hydrogen bonds with it; the long hydrocarbon, nonpolar tails repel the water and associate with each other,
forming a self-organized structure, the lipid bilayer.
Much of this behavior of the lipids resides in the nature
of water. To see this, we need to learn more about the
behavior of water at hydrophobic interfaces.
SURFACE TENSION OF WATER RESULTS
FROM ASYMMETRIC FORCES AT THE
INTERFACE
At the airwater interface, water molecules are subjected
to asymmetric forces, as shown in Figure 2.4.9. These
molecules have lost some of their bonds connecting
them to the bulk phase and are, therefore, in a higher
energy state than water in the bulk phase. These molecules are partially evaporated. Thus it takes energy to
promote water molecules to the interface and the energy
of the surface increases with its area. At constant temperature and pressure, the change in surface energy is
the change in the Gibbs free energy, G. The change is
given by:
½2:4:1
dG 5 γ dA
where dA is the increment in area, dG is the increment
in Gibbs free energy, and γ is the surface tension. Since
the energy has units of force 3 distance, the tension has
units of force per unit length. Typical units of γ are
dynes cm21. In SI units, γ is expressed in N m21 or
J m22. The surface tension is a measure of how much
more a water molecule at the surface is attracted to the
bulk water phase because of the increase in intermolecular forces compared to the surface.
WATER “SQUEEZES OUT”
AMPHIPATHIC MOLECULES
Recall that amphipathic molecules consist of spatially
separated water-loving or hydrophilic head group and a
147
148
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Cardiolipin
Phosphatidylcholine
Water
CH
H2C
H
H
C
2C
H
1
CH2
O
–O P O
O
P O–
O
O
O
H H
C
3
H2C
CH2
H2C
CH2
CH
HC
H2C
CH
CH2
H2C
CH2
H2C
H2C
CH2
CH2
H2C
Nonpolar tail
H2C
H2C
H2C
CH2
CH2
H3C
H2C
H2C
CH2
FIGURE 2.4.8 Space-filling model of phosphatidylcholine. Nonpolar
surfaces are shown in gray or white. Polar surfaces are dark blue
(oxygen), light blue (phosphorus), or intermediate blue (nitrogen).
Charged surfaces attract water by dipoledipole interactions. The
hydrophilic or water-loving parts of the molecule are all concentrated at
one end. The hydrophobic (water-hating) or lipophilic (fat-loving) groups
are located at the opposite end.
CH2
CH2
CH2
CH3
CH2
H2C
H2C
CH2 H2C
CH2
CH2
CH2
H2C
H2C
H2C
H2C
HC
Fatty acids
CH2
CH2
CH2
Hydrophilic groups
–
Hydrophobic groups
(= Lipophilic groups)
H2C
H2C
CH2
+
Glycerol
O
C O
H2C
CH2
CH2
C1
CH2
CH2
H2C
C2
H2C
H2C
H2C
H
O C
H2C
CH2
H2C
H
O
C O
CH2
CH2
Phosphate
Polar head
H
O
O C
Choline
H
C
3
H
O
OH
Air
CH3
H3C
FIGURE 2.4.6 Chemical structure of cardiolipin.
Steroid nucleus
Cholesterol
18
12
11
13
C
19
14
1
2
9
10
A
B
5
3
4
Water
17
D 16
15
H3C
CH3
8
CH3
7
Molecules at the surface have
fewer attractive forces than molecules
in the bulk phase; they are
partially evaporated and have
a higher energy
6
HO
FIGURE 2.4.7 Chemical structure of the steroid nucleus and cholesterol.
The steroids are all derived from the steroid nucleus, a
perhydrocyclopentanophenanthrene nucleus of four rings: three fused
rings in the phenanthrene arrangement and a five-carbon ring attached.
Therefore, the nucleus has the name “cyclopentano” to refer to the fivecarbon ring; “phenanthrene” refers to the three six-membered rings and
“perhydro” indicates that the double bonds of phenanthrene are
saturated with hydrogen. The steroid nucleus is numbered as shown.
FIGURE 2.4.9 Asymmetric forces of water molecules at the airwater
interface. In the bulk phase of liquid water, the intermolecular forces
acting on any water molecule are on average equal in all directions. In
the air, intermolecular interactions are markedly reduced. In order to
evaporate, energy must be added to the water molecules to break the
attractive intermolecular forces. At the airwater interface, water
molecules are subject to more intermolecular forces than those water
molecules in the air phase, and fewer forces than water molecules in the
bulk liquid phase. Therefore, water molecules at the surface have more
energy than those in the bulk phase. In a sense, they are partially
evaporated, having lost some but not all of their intermolecular bonds. If
we were to increase the surface area, we would have to put in energy
proportional to the area of increase.
Biological Membra nes
Hydrophobic fatty acyl tails
Water has no attractive forces to
hydrocarbon chains; being
near them is the same as being
partially evaporated—they
have a higher energy;
it takes energy to insert lipid into water
Hydrophilic phosphate
Air
Hydrophilic choline
Water
Because it takes energy for the lipid to be in
the middle of the water phase compared to
at the surface, most lipids are found at the
hydrophobic/hydrophilic surface where
hydrophilic groups interact with hydrophilic
water and hydrophobic groups interact with
other hydrophobic groups. In essence, the
water squeezes out the lipid to the air/water
interface
Water binds to hydrophilic head groups of
phospholipids—they have the same number
of attractive forces and have the same
energy as water in the bulk phase
hydrophobic or water-fearing hydrocarbon tail.
Dissolving an amphipathic molecule in the bulk water
phase disrupts the self-association of water, creating an
interface within the bulk phase that requires energy.
Because of this, the amphipathic molecule is “squeezed
out” of the bulk water phase to the surface of the solution adjacent to the air. In this situation, the water
molecules at the surface interact with hydrophilic
groups in the amphipathic molecule, which lowers the
energy of the surface, and the hydrophobic groups
in the amphipathic molecule can interact with
other hydrophobic groups through London dispersion
forces. These ideas are shown diagrammatically in
Figures 2.4.10 and 2.4.11.
The free energy change for the transfer of phospholipid
from the bulk phase to the surface is given as
½2:4:2
ΔGbulk.surface 5 Gsurface 2 Gbulk
Since Gsurface is less than Gbulk, the free energy change
for the transfer is negative, and therefore the transfer
occurs spontaneously.
AMPHIPATHIC MOLECULES SPREAD
OVER A WATER SURFACE, REDUCE
SURFACE TENSION, AND PRODUCE
AN APPARENT SURFACE PRESSURE
When amphipathic molecules such as phosphatidylcholine or oleic acid are dissolved in a volatile organic solvent (e.g., hexane, decane) and then layered over water,
FIGURE 2.4.10 Water “squeezes out” phospholipids.
The hydrocarbon parts of a phospholipid molecule
within the bulk aqueous phase cannot form
hydrogen bonds with the adjacent water
molecules, so these water molecules cannot form
as many hydrogen bonds as the other water
molecules in the bulk phase. The set of water
molecules surrounding the hydrocarbon are
essentially partially evaporated and have a higher
energy than the water molecules in the bulk phase.
Therefore, to insert the hydrocarbon in the water
takes energy. Phospholipids on the surface of the
water, on the other hand, are in a lower energy
state. The self-association of water thus “squeezes
out” the phospholipid to the surface.
the organic solvent evaporates and leaves a thin film
of the lipid. As shown in Figures 2.4.10 and 2.4.11, these
amphipathic molecules are squeezed out to the surface
of the water, forming a layer a single molecule thick. The
lipids form a monolayer. These lower the surface tension
according to Eqn [2.4.1] because they lower the energy
required to move water molecules to the surface (see
Figure 2.4.11). This lowering of the surface tension can
be measured using a Langmuir trough, as shown in
Figure 2.4.12. This device has two barriers on the surface
of the water. One barrier is fixed, the other is movable.
Since the monolayer decreases the surface tension, the
movable barrier feels a net force in the direction of
the clean surfaces. By lowering the surface tension, the
lipids appear to exert a surface pressure defined as
½2:4:3
π 5 γ0 2 γ
where π is the surface pressure, γ 0 is the surface tension
of the clean surface, and γ is the surface tension in the
presence of the monolayer. Surface pressure has the
same units as the surface tension.
PHOSPHOLIPIDS FORM BILAYER
MEMBRANES BETWEEN TWO
AQUEOUS COMPARTMENTS
Two monolayers can orient themselves back to back to
form a bilayer between two aqueous compartments, as
shown in Figure 2.4.13. This is the low energy state for
this situation for the reasons we have already described.
149
150
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
If the phospholipids were dispersed throughout the
solution, the surface area between the water phase and
the hydrophobic, hydrocarbon phase would be large.
Since it takes energy to produce this surface, the dispersed hydrocarbon is a high-energy state compared to
the condensed one. A secondary cause of this spontaneous organization of the lipids is the attractive interactions among the hydrocarbon chains.
LIPID BILAYERS CAN ALSO FORM
LIPOSOMES
The macroscopic lipid bilayer shown in Figure 2.4.14
is unstable to mechanical forces. When disrupted, the
membrane breaks apart to form spherical bilayers
separating an internal and external watery compartment. These hollow spheres are called liposomes (see
Figure 2.4.15). These structures can also be generated
directly from phospholipids by soaking them in water
and adding sonic energy.
Phospholipid at the air–water interface lowers the energy
of the surface by providing attractive forces to the surface
molecules; the hydrocarbon chains also attract each other
by London dispersion forces
Phosphatidyl choline
Air
Macroscopic planar bilayer membranes can be formed
across a narrow aperture separating two solutions, as
shown in Figure 2.4.14. These membranes are useful
because they allow the study of single membrane channels incorporated into the membrane.
ALTHOUGH LIPIDS FORM THE CORE,
MEMBRANE PROTEINS CARRY OUT
MANY OF THE FUNCTIONS
OF MEMBRANES
Phosphatidyl
inositol
So far we have been describing membrane lipids that
form the core of biological membranes. These contribute to the barrier function of membranes, but many
other functions of biological membranes are performed
by protein constituents of the membranes. These functions include:
Water
Molecules at the surface have
fewer attractive forces than
molecules in the bulk phase; they
are partially evaporated and have
a higher energy
G
G
FIGURE 2.4.11 Lowering of the surface energy by amphipathic
molecules. Water at its ordinary surface is in a higher energy state—it
takes energy to break the hydrogen bonds that binds the water
molecules in the bulk aqueous phase. When phospholipids are present
at the surface, water binds to the hydrophilic groups of the
phospholipids, thereby reducing the energy needed to form additional
surface. Because the energy of the surface is related to its area by the
surface tension, reducing the energy of the surface per unit area is the
same as reducing the surface tension.
G
G
Transport: Cells must be able to move things into
and out of the cell.
Signal transduction: Cell must have mechanisms for
responding to signals from other cells or from within
the cell. These may be chemical, electrical, or
mechanical signals.
Recognition: Cells attach to other cells and to extracellular structures. They must be able to recognize
where they should form attachments.
Attachment: Cells must anchor themselves to the
extracellular matrix or to each other. Often these
attachments also provide a signaling pathway.
Hydrophobic tails
Fixed barrier
FIGURE 2.4.12 The Langmuir trough.
A clean water surface has a fixed barrier
and a movable barrier. When only clean
water forms the surface, the surface
tension is γ0. Adding a lipid film reduces
the surface tension to γ. The movable
barrier experiences a net force toward the
clean surface without lipid. Thus the lipid
appears to exert a surface pressure.
Air phase
Movable barrier
γ
Water phase
Polar head groups
γ0
Biological Membra nes
Phosphatidylcholine
Cholesterol
Phosphatidylethanolamine
Aqueous layer
Insulating hydrocarbon layer
Aqueous layer
Phosphatidic acid
FIGURE 2.4.13 Structure of the lipid bilayer. Only some lipids are shown
and in expanded format for clarity. In reality, the lipid bilayer forms a
closed surface that approximates a plane. The interior of the lipid bilayer
is fluid, consisting of hydrocarbon chains that are saturated (the straight
chains in the figure) or unsaturated (bent chains in the figure). The
phospholipids form the bulk of the bilayer. Lipids such as cholesterol
have a rigid backbone that partially stiffens and solidifies the
membrane. Cholesterol may accumulate in heterogeneous patches of
membrane called lipid rafts.
Barrier
Water
Water
Narrow
aperture
FIGURE 2.4.14 Planar lipid bilayer between two aqueous compartments.
Phospholipids were dissolved in hexane and “painted” over a small
aperture drilled in a Lexan partition that separated two electrolyte
solutions. After thinning, with the passive removal of hexane through the
aqueous phase, a lipid bilayer forms between the two compartments.
G
Movement or force production: Cells often must
move or transmit a force from inside the cell to the
extracellular matrix. This requires connection of the cell’s
cytoskeleton through the membrane to the extracellular
matrix.
FIGURE 2.4.15 A schematic drawing of a cross-section of a liposome.
These are small structures about 50150 nm across. Because each
bilayer is about 7.510 nm across, the thickness of the bilayer occupies
a considerable portion of the entire liposome volume and the enclosed
volume, the lumen, is small. Because of the high curvature and small
size, the area on the outside of the liposome is nearly twice the area on
the inside, and therefore significantly more lipid faces the outside
compared to the inside surface of the liposome. Liposomes may find use
someday to deliver drugs to specific locations within the body by
incorporating recognition signals into the lipid bilayer.
MEMBRANE PROTEINS BIND
TO MEMBRANES WITH VARYING
AFFINITY
The proteins that perform the various functions listed
above can be loosely classified according to how tightly
they are bound to the membrane. Loosely bound proteins are called peripheral proteins, also sometimes
called extrinsic proteins. They can be released from the
membrane by relatively gentle procedures such as washing with a salt solution. Other proteins are called integral proteins, also sometimes called intrinsic proteins.
These are tightly bound by the membrane and can be
released only by resorting to drastic measures such as
dissolving the membrane with a detergent. By coating
the hydrophobic parts of the membrane proteins with
hydrophilic material, detergents solubilize the membrane proteins. Examples of useful detergents in membrane research include the ionic sodium dodecyl
sulfate (SDS) and the nonionic family of Triton detergents. Figure 2.4.16 illustrates integral and peripheral
proteins.
Proteins are held in membranes by the same kinds of
interactions that hold lipids in the bilayer: hydrophobic
and hydrophilic interactions. Many proteins have
sequences of amino acids that penetrate all the way
across the membrane. These are transmembrane proteins. In the parts of the protein exposed to lipid,
hydrophobic amino acid side chains appose the lipid
core: valine, leucine, isoleucine, phenylalanine, tryptophan, and methionine. Those parts of the protein
exposed to water have a preponderance of hydrophilic
amino acids: aspartic acid, glutamic acid, lysine, and
arginine. The neutral amino acids can be present in
151
152
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Peripheral protein
Glycolipids
GPI anchors protein
in the membrane
Myristyl anchor
Lipid bilayer
core
FIGURE
2.4.16
Schematic
arrangement of membrane proteins
in the lipid bilayer. Darker areas
of proteins are predominantly
hydrophilic; lighter areas denote
hydrophobic areas. Hexagons signify
hydrophilic carbohydrates. Proteins
are usually anchored by hydrophobic
sequences of amino acids; less often
they are anchored by covalent
attachment to hydrophobic materials
such as GPI, farnesyl, palmitic acid, or
myristic acid.
Farnesyl anchor
Integral, transmembrane
protein
either domain. Some proteins have multiple sequences
that cross the membrane, with domains facing each of
the watery solutions on the two sides of the bilayer. The
sequences that cross the membrane are typically
arranged as alpha helices, with the hydrophobic amino
acids facing the hydrophobic lipid core. Other proteins
may bind to the membrane by covalently attached
hydrophobic groups. These include myristic acid (14:0
fatty acid), palmitic acid, (16:0) farnesyl, and glycosylphosphatidylinositol (GPI) (see Figure 2.4.16).
Lateral diffusion
Stretch
Flip-flop
Flexion
Rotation
LIPIDS MAINTAIN DYNAMIC MOTION
WITHIN THE BILAYER
Researchers have labeled phospholipids with probes
that are sensitive indicators of molecular motion and
have tracked the mobility of lipids and proteins within
the plane of the bilayer. The results of these experiments
show a variety of molecular motions, shown in
Figure 2.4.17. This has given rise to the fluid mosaic
model of biological membranes. The term fluid mosaic
model describes a dynamic system in which the lipids
form a plane that gradually curves around to form a
closed surface—there are no exposed lipid edges. Lipid
motion in the plane of the membrane includes rotation,
flexion, stretch, and lateral diffusion in the plane. All of
these movements are rapid. The proteins in the membrane can also move, unless they are anchored by binding to other components of the membrane. There is a
gradient of fluidity of motion from the polar head
group to the center of the hydrophobic interior of the
membrane, being most fluid in the center and most
anchored at the head group. Lipid movement from one
side of the membrane to the other—the “flip-flop”
FIGURE 2.4.17 Possible motions of lipids within a bilayer. Lipid
molecules can move laterally within the plane of the membrane, rotate
about their long axis, flex within the fluid interior of the membrane, or
move from one side of the bilayer to the other. Most of these motions
are fast, but the “flip-flop” reaction is very slow, occurring less than
once in 2 weeks for any individual lipid molecule. Exchange of
neighbors occurs very fast, on the order of 107 times per second. This
rapid exchange gives rise to a rapid lateral movement within each half
of the bilayer.
reaction—is slow because it requires lipids to bring their
hydrophilic head group through the lipid phase, which
is energetically costly. Because of the very slow movement of lipids from one half of the bilayer to another,
membranes can maintain an asymmetric composition,
but it must actively make and sustain it. In most biological membranes, the two half bilayers differ in their
composition. Cells add lipids only to the cytoplasmic
side of membranes. An enzyme called a “scramblase”
flips lipids from the cytoplasmic half to the extracellular
half, but this enzyme is not very specific. The different
composition of membranes arises from a second
enzyme, a “flippase”, that flips only some phospholipids from the cytoplasmic half to the extracellular half.
Biological Membra nes
The fluidity of membrane lipids depends on their composition. Saturated fatty acids are known to be very stiff
compared to unsaturated fatty acids. Saturated fatty acyl
chains can be packed closely together, whereas the kinks
produced by cis double bonds make it difficult to pack
these chains close together. Because of this, unsaturated
fatty acids promote fluid movement within the bilayer.
Cholesterol is also a very rigid molecule. Cholesterol
molecules orient themselves in the membrane with their
steroid nucleus adjacent to the hydrophobic tails and
the hydroxyl group adjacent to the polar head groups.
The steroid nucleus is a flat, plate-like structure that partially immobilizes the nearby fatty acyl chain, thereby
reducing the fluidity of the bilayer and increasing the
mechanical stability of the bilayer.
The motions of the lipids in the bilayer makes it appear
as a two-dimensional fluid. It is fluid within the plane
of the membrane, but relatively rigid perpendicular to
this plane. The membrane proteins more or less “float”
in this lipid see like so many icebergs in the North
Atlantic. This combination of fluid lipids and iceberg
proteins was the origin of the descriptive term, fluid
mosaic model.
LIPID RAFTS ARE SPECIAL AREAS OF
LIPID AND PROTEIN COMPOSITION
Lipid rafts are microdomains in biological membranes
that contain different proportions of lipids and proteins
from the rest of the membrane. They were discovered
when portions of membranes were found to be less easily solubilized by detergents. Detergents are chemicals
that dissolve membranes by providing their constituents
with flotation devices: they bind the hydrophobic
domains and coat them with water-soluble material.
These detergent-resistant areas of membrane accumulate
cholesterol and sphingolipids. Sphingolipids generally
contain longer and straighter fatty acyl chains. These
attract each other more forcefully than do unsaturated
fatty acids, because the straight chains can pack more
closely without the kinks in their chains. These aggregate into the raft microdomain. Because these chains are
straighter, the membranes are also thicker at the rafts.
The plasma membrane is thought to have many such
rafts about 70 nm in diameter.
CAVEOLAE AND CLATHRIN-COATED
PITS ARE STABILIZED BY INTEGRAL
PROTEINS
The surface of cells forms a variety of specializations that
curve inwardly, forming an indentation of the membrane. Caveolae are one of these. Caveolae are a subset
of lipid rafts, but not all lipid rafts are caveolae.
Caveolae are 6080 nm pits in the membrane that contain some 140150 oligomeric caveolin molecules.
There are three mammalian caveolins: CAV1, CAV2, and
CAV3, all have parts that bind to membranes. Their oligomeric structure is stabilized by a family of cytoplasmic
proteins called cavins. Caveolar membranes are enriched
in both cholesterol and phosphatidylserine (another
phospholipid in which the head group is serine instead
of ethanolamine, choline, or inositol). Depletion of the
cholesterol or mechanical flattening of caveolae causes
dissociation of cavin from caveolin (see Figure 2.4.18).
Flattening of the caveolae occurs upon stretch of skeletal
muscle, cardiac myocytes, endothelial cells, and fibroblasts. It may be that caveolae are involved in the sensing or response to mechanical stretch.
Membranes can also form clathrin-coated pits that are
involved in receptor-mediated endocytosis, in which
parts of the membrane invaginate and pinch off, forming
an interior vesicle with enclosed extracellular material.
Clathrin consists of three heavy chain subunits (CHC17
or CHC22) and three light chains (CLC) that trimerize to
form a triskelion, the unit of clathrin. These units then
associate on membranes to form a clathrate (lattice)
structure. The lattice structure consists of a number of
pentagons and hexagons. The clathrin protein itself has a
curvature to it, and this imparts a curvature to the clathrate and stabilizes the budding part of the membrane.
The membrane is then pinched off by another protein
complex called dynamin (see Figure 2.4.19).
CAV1
Shear stress
Cavin complex
Plasma membrane
FIGURE 2.4.18 Caveolae response to stretch. Caveolae are indentations
or pits in the plasma membrane that are stabilized by a network of
integral proteins that include oligomers of caveolin (CAV1). Cytoplasmic
proteins called cavins stabilize the caveolin structure. When the
membrane is stretched, the caveolae flatten and cavin dissociates from
the caveolin. This may be part of how cells sense or respond to stretch.
H+
ATP
Clathrin heavy chains
Light chains
Clathrin coat
Triskelion structure
Dynamin
FIGURE 2.4.19 Clathrin-coated pits and endocytosis. Clathrin consists of a
trimer of heavy chains each of which binds a light chain. These selfassemble to form a clathrin coat that stabilizes budding membranes. The
budding membranes are pinched off through the actions of dynamin.
153
154
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
mRNA
1. Protein synthesis begins on
free ribosomes
40S
3. Complex of ribosome, peptide, and SRP
binds to SRP receptor on the ER
SRP
Ribosome
60S
1
Signal
sequence
4. Complex transfers to a
translocon and elongation
resumes
NH2
2
3
ER membrane
α
3'
β
6 and 7. Chaperones bind
to the elongating chain
and help fold it properly
SRP receptor
2. SRP recognizes the signal
sequence, binds to it and
the large ribosomal subunit,
and stops elongation
4
Translocon
5
6
7
Folded protein
Signal peptidase
Cleaved
signal sequence
5. Signal peptidases cleave
off the signal sequence
Chaperone protein
FIGURE 2.4.20 Mechanism of synthesis of secretory proteins into the ER lumen in the pancreas. Synthesis begins on free ribosomes in the cytoplasm
(1). The initial N-terminus of the protein contains a signal sequence that is recognized and bound to by cytosolic SRP, signal recognition particle, a
nucleoprotein consisting of RNA and a set of six separate proteins (2). Binding of SRP stops elongation. The complex of nascent polypeptide, ribosome,
and SRP is bound to the ER by the SRP receptor consisting of a peripheral α-subunit and an integral β-subunit (3). The SRP receptor transfers
the ribosome to a translocon, a complex of proteins that spans the ER membrane and provides an aqueous channel for the protein across the
membrane (4). The SRP dissociates from the complex in the process. The polypeptide chain grows and translocates across the ER membrane at
the same time. The signal sequence is cleaved off (5) by signal peptidases in the ER lumen. The growing polypeptide chain is eventually completed
and folds up into its active conformation (7). The folding is assisted by chaperone proteins and enzymes such as Hsp-70, protein disulfide isomerase,
and peptidyl prolyl isomerase (6). Source: Modified from Lodish et al., Molecular Cell Biology, 4th Ed, W.H. Freeman, New York, NY, 2000.
SECRETED PROTEINS HAVE SPECIAL
MECHANISMS FOR GETTING INSIDE
THE ENDOPLASMIC RETICULUM
The synthesis of membrane proteins or proteins destined for secretion poses a problem for cells because the
ribosomes on which the proteins are made are in the
cytosolic compartment but their products are either in
the membranes or in the extracellular compartment.
This problem is solved by an elaborate mechanism,
shown in Figure 2.4.20.
SUMMARY
Lipids readily dissolve in organic solvents such as chloroform while they are sparingly soluble in water. Several
classes make up biological lipids including fatty acids,
phospholipids, and steroids. They are typically made up
of long chains of hydrocarbons or carbon rings. The
major constituent of biological membranes are the
phospholipids. These consist of a polar head group connected covalently to a nonpolar tail. The polar head
group in turn consists of a hydrophilic group like inositol, serine, choline, or ethanolamine linked to a phosphate group, which in turn is esterified to glycerol. All
of these are highly water soluble. The other hydroxyls of
the glycerol are esterified to two fatty acids, which are
highly water insoluble. Thus these phospholipids
spatially separate hydrophilic and hydrophobic parts.
When placed in water, the hydrophilic parts associate
with the water while the hydrophobic parts associate
with other hydrophobic molecules. When placed on top
of water, these amphipathic molecules form a lipid
monolayer.
The surface tension of the water results from asymmetric
forces on the surface from the bulk water phase and the
air phase. Because the hydrophilic parts of the lipids
attract water molecules on the surface, they reduce the
asymmetry in forces. Accordingly, lipids reduce the surface tension. Experimentally, this appears as a surface
pressure. Folding such a monolayer back on itself produces a bilayer membrane. This consists of a double
layer of molecules in which the hydrophilic domain
faces the water phase and the hydrophobic domain
faces the interior of the membrane, occupied by other
hydrophobic parts of lipid molecules. Other lipid aggregates include the liposome. The liposome is a bilayer
that forms a hollow sphere.
As in all molecules, chemical bonds in lipids can
stretch, rotate, and flex. These motions along long
chains produce motion within the hydrophobic interior
of membranes. The hydrophobic chains are relatively
well anchored at the polar head group, so there is a gradient of fluidity in the membrane, it being most fluid in
the center and less at the periphery. Double bonds in
Biological Membra nes
the hydrocarbon chains make a kink in the chain that
disallows close packing of the chains. Thus double
bonds promote fluidity within the bilayer. Saturated
fats, those that contain no double bonds, make membranes more rigid. Cholesterol is a rigid molecule composed of a plate-like steroid nucleus and a hydrophobic
tail. It generally makes membranes more rigid.
Lipids freely move in the plane of the membrane while
motion across the membrane, the “flip-flop” reaction, is
slow. Cells take advantage of this slow flip-flop to maintain asymmetric distributions of lipids in the two halves
of the bilayers. Proteins embed in the membrane and
move around, something like icebergs floating in a lipid
sea. Thus the membrane is described as a “fluid mosaic.”
However, thicker microdomains of the membrane contain concentrations of sphingolipids and cholesterol.
These microdomains are called lipid rafts.
Proteins can be loosely associated with membranes or
tightly bound. The loosely bound proteins are called
peripheral or extrinsic proteins and the tightly bound proteins are called integral or intrinsic proteins. Proteins are
held in the membrane either by hydrophilichydrophobic interactions between their amino acids and the lipid
and water phases or by attachment of hydrophobic
groups such as myristic acid, palmitic acid, farnesyl, or
GPI. Many proteins and lipids have carbohydrate coats.
The synthesis of secreted proteins requires the synthesis
of an endoplasmic reticulum (ER) signal sequence that
is recognized by a signal recognition particle (SRP),
which then binds to a receptor on the ER membrane.
This enables transfer of the cytosolic ribosome to the ER
membrane and subsequent simultaneous translation
and translocation of protein across the ER membrane.
Once inside the ER, the synthesized protein undergoes
posttranslational modification.
REVIEW QUESTIONS
1. Name the major membranes in the cell. Which
membrane accounts for most of the membranes
in the cell?
2. What is a saturated fatty acid? What is an unsaturated fatty acid? What effect does unsaturation
have on the structure of the fatty acid? What do
fatty acids attach to in phospholipids?
3. What is a phospholipid? What are the major
types of phospholipids? Which chemical groups
on the phospholipid are hydrophilic? Which
groups are hydrophobic? What is the significance of spatial separation of hydrophilic and
hydrophobic character in lipids?
4. What is the general structure of cardiolipin?
What is sphingosine? What is sphingomyelin?
What is a ceramide? How do these differ from
phosphatidylcholine?
5. What is surface tension? What are its units?
What do amphipathic lipids do to the surface
tension? Why?
6. Name the various degrees of freedom of lipid
motion in a bilayer. Which is the slowest?
Which is the fastest?
7. What is meant by the term “fluid mosaic
model”?
8. What is an integral protein? What is a peripheral
protein? In what ways can proteins be anchored
to membranes?
9. What is a liposome? What is a planar lipid
bilayer?
10. What are lipid rafts?
11. What is meant by “caveolae”? What is a
clathrin-coated pit?
12. How do secreted proteins get inside secretory
vesicles? What is meant by “signal sequence”?
155
Problem Set
2.1
Surface Tension, Membrane Surface Tension,
Membrane Structure, Microscopic Resolution,
and Cell Fractionation
1. Consider a soap film stretched over a wire
frame, one end of which is movable.
Experimentally, one observes that there is a
force exerted on the movable member as indicated in Figure 2.PS1.1. Clearly, this force
depends on the dimensions of the wire frame.
Therefore, we express the force per unit length
as γ. Write an expression for the work performed in expanding the film a distance dx.
Rewrite this in terms of the area increment, dA,
by which the film is expanded.
2. Consider a soap film again in the form of a bubble as shown in Figure 2.PS1.2. The surface tension can be thought of as either the force per unit
length or the energy per unit area. The minimal
energy form for a soap film is the minimum area
for a given volume. This is the sphere. So, in the
absence of other effects, including gravity, the
soap bubble should be a sphere.
A. What is the total surface energy of the
sphere? Remember that the variable we have
been using for surface tension is γ.
B. If the radius were to decrease by dr, what
would be the change in the surface energy?
C. Since shrinking decreases the surface energy,
at equilibrium the tendency to shrink must
be balanced by a pressure difference across
the film, ΔP. At equilibrium, the work
against this pressure for an increment in
radius dr is exactly equal to the decrease in
surface energy. That is, at equilibrium the
free energy change is zero. Otherwise, the
bubble would not be stable and it would
shrink. What is the work that must be done
against this pressure difference? Hint:
Pressure is force per unit area, so the total
force must be the area times the pressure.
Work is force times distance.
D. Equate the pressurevolume work in part C
to the surface energy decrease in part B.
From this equation, derive an expression for
ΔP in terms of γ and r. This result is a
famous equation, the Law of Laplace, which
finds application in respiratory physiology
and cardiovascular physiology.
3. When heart cells are exposed to a hypotonic
medium, they swell and measurements show
that their volume has increased. Measurements
of their membrane capacitance, however, do not
change. How can this happen?
4. Liposomes form structures 100 nm across their
outside diameter. The average density of the
lipids used to form the liposomes is
0.89 g cm23. Assume that the thickness of the
bilayer is 8 nm.
A. What is the volume of the lipid shell? What
is its mass?
B. What is the ratio of the outer surface area to
the inner surface area of the liposomes?
C. What is the enclosed volume of the
liposome?
Po
Pi
dr
Movable barrier
Surface tension
l
Direction of expansion
r
Soap film
156
FIGURE 2.PS1.1 Soap film on a wire frame. The soap film exerts a force
per unit length on the movable barrier. This force is the surface tension.
To expand the film, we must do work.
FIGURE 2.PS1.2 A soap bubble of radius r. Because the surface tension
results in an inwardly directed force, the bubble will tend to collapse
unless there is a pressure difference across the membrane that prevents
its collapse.
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00014-8
Problem Set
D. Using the answers to A and C, what is the
overall density of the liposomes? Assume
that the liposome is filled with water with
density 5 1 g cm23.
E. How many liposomes can be derived from
100 mg of lipids?
F. A drug is soluble to 5 mM. If the liposomes
were formed in a solution of this drug, and
therefore the enclosed volume included the
drug to this concentration, how many moles
of drug would be contained in the liposomes derived from 100 mg of lipids?
5. Isolated cardiac sarcoplasmic reticulum (SR)
vesicles have an average outside diameter of
about 150 nm. The membrane itself is about
10 nm thick. The enclosed volume can be estimated by measuring the efflux of passively
loaded tracer materials such as mannitol, and
the result gives 5 μL mg21 SR protein.
A. How many vesicles are there per mg of SR
protein?
B. What is the surface area of the vesicles per
mg of SR protein?
C. If the SR Ca-ATPase Ca21 uptake activity is
4 μmol min21 mg21, what is the uptake
activity per unit surface area?
6. The method of measuring the surface tension of
a liquidair interface is the drop weight
method. In this method, drops are allowed to
form at the end of a tube of known radius, and
a number of them are collected and then
weighed so that the weight per drop can be
determined accurately. The weight per drop is
given by Tate’s Law (1864):
½2:PS1:1
W 5 2πrγ
where r is the radius of the tube. This equation
uses the idea that the surface tension is the force
per unit length and that the maximum force that
can be used to support the weight of the forming
drop is the circumference of the tube times its
surface tension. In practice, the weight of the drop
is less than that given by Tate’s Law because some
of the liquid supported by the tube remains after
the drop falls. More detailed analysis makes use of
a correction factor such that
½2:PS1:2
W 0 5 2πrγf
where W0 is the actual weight per drop and f is
the correction factor. It turns out that the correction factor f varies with rV 21/3, where V is the
volume of the drop. Approximate values of the
correction factor are given in Table 2.PS1.1:
A. Using a tip with an outside diameter of
0.40 cm and an inside diameter of 0.20 cm,
20 drops of an organic liquid weighed
0.80 g. The density of the liquid was
0.95 g cm23. This liquid wet the tip. (Hint:
This goes to determine whether you use the
inside or the outside diameter!) Use the appropriate correction factor from Table 2.PS1.1,
TABLE 2.PS1.1 Correction Factors for the Drop Weight
Method
rV21/3
f
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2
1.3
1.4
1.5
1.6
0.7256
0.6828
0.6515
0.6250
0.6093
0.6000
0.5998
0.6098
0.6280
0.6535
0.640
0.603
0.567
0.535
Source: Data from A.W. Adamson, Physical Chemistry of Surfaces,
Interscience, New York, NY, 1967.
and the drop weight, to calculate the surface
tension of the organic liquid.
B. Using a tip with and outside diameter of
0.21 cm and an inside diameter of 0.18 cm,
20 drops of a liquid had a mass of 0.766 g.
The density of the liquid was 1.00 g cm23. This
liquid wet the tip. (Hint: This goes to determine whether you use the inside or the outside
diameter!) Use the appropriate correction factor from Table 2.PS1.1, and the drop weight,
to calculate the surface tension of the organic
liquid.
7. The surface tension of pure water is
72.0 dyne cm21. 1 dyne cm21 is equivalent to
1 mN m21, which is the SI unit for surface tension. When dipalmitoyl lecithin is spread at
the
surface
pressure
is
50 Å2 mol21,
11 mN m21. What is the surface tension when
dipalmitoyl lecithin is spread on the surface?
8. The tension in a biological membrane can be
measured in a variety of ways. One way is called
the pipette aspiration technique. In this technique, a specially manufactured micropipette is
attached to a vesicle by light suction. These pipettes typically have a open diameter of 12 μm
and have a square end. Application of increasing
suction draws the vesicle into the pipette, forming a cylindrical part within the pipette and a
spherical part outside of it (see Figure 2.PS1.3).
A. Assume that the Law of Laplace holds for
both the spherical part of the vesicle outside
the pipette and the hemisphere within the
pipette. Write the two equations relating Pv,
Po to Dv and T, the tension in the vesicle, and
Pv, Pp to Dp. There is only one tension in the
membrane, which is the same everywhere.
B. Defining ΔP 5 Po 2 Pp, use the answer in part
A to solve for ΔP in terms of T, Dv, and Dp:
157
158
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Pv
TABLE 2.PS1.2 Data for the Dilatation of Sarcolemma
Vesicles Using the Micropipette Aspiration Technique
Dv
Dp
Pp
L
Po
FIGURE 2.PS1.3 Pipette aspiration technique. A vesicle obtained from a
“bleb” on a cell when the cell is exposed to hypotonic medium is
excised and attached to a micropipette by application of suction.
Increasing the suction draws the vesicle into the pipette a distance L
and reduces the diameter of the remaining vesicle. By LaPlace’s law, this
increases the tension in the membrane. Continual decreases in Pp
eventually causes the vesicle to rupture. The critical tension for rupture
can be determined in this way.
C. Solve part B to express T in terms of ΔP, Dp,
and Dv
D. A giant sarcolemmal vesicle was obtained
from a rabbit muscle and subjected to the
pipette aspiration technique (J.A. Nichol
and O.F. Hutter, Tensile strength and dilatational elasticity of giant sarcolemmal vesicles
shed from rabbit muscle, J Physiol.
493:187198 (1996)). The pipette diameter
(Dp) was 19 μm and the vesicle diameter
was 66 μm. At a pipette suction of 8 cm
H2O, what is the tension in the membrane?
E. What is the total area of the membrane in
terms of Dv and Dp and the length L?
F. When suction pressure is increased, L
increases and Dv decreases. The increased
tension stretches the membrane, causing a
dilatation. The elastic area expansion modulus is defined as
T
ΔA
K5
A
where ΔA is the expansion of the membrane
area due to dilatation and T is the tension.
Considering your answer for part E, write an
expression for the increase in area attributable to membrane dilatation, ΔA, in terms
of an initial condition with Dvi and Li and a
final condition with Dvf and Lf.
G. In practice, the increase in the length of the
projection is not long enough to cause an
easily measurable difference in Dvf compared to Dvi. Assuming that the volume of
the vesicle plus projection remains constant,
express Dvf in terms of Dvi, ΔL, and Dp.
9. Using the micropipette aspiration technique
described in Problem #8, the following data
were obtained for the tension and area dilatation for sarcolemma vesicles obtained from rabbit skeletal muscle (see Table 2.PS1.2):
The vesicle ruptured at the last point recorded.
Tension (mN m21)
ΔA/A
Tension (mN m21)
ΔA/A
1.4
2.6
3.9
5.1
6.6
7.9
0.0027
0.0058
0.0078
0.0112
0.0127
0.0160
9.1
10.5
11.9
13.1
14.5
0.0178
0.0212
0.0248
0.0261
0.0296
J.A. Nichol and O.F. Hutter, Tensile strength and dilatational elasticity of giant
sarcolemmal vesicles shed from rabbit muscle, J Physiol. 493:187198 (1996)
Calculate the elastic area expansion modulus (see
Problem 2.PS1 problem #8 for a definition of the
elastic area expansion modulus).
10. For light of wavelength 5000 Å (5500 nm),
calculate the theoretical maximum resolution of
an optical microscope.
11. Typically the energy of the electron beam in an
electron microscope is known, because the
voltage through which the electrons are accelerated is known. One electron volt is the energy
gained by an electron when it is accelerated
across a potential of 1 V. One electron has a
charge of 1.602 3 10219 C. So the electron volt is
1.6310219 V C51.6310219 J (1 J51 V C51 N m).
The rest mass of the electron is 9.109 3
10231 kg.
A. Using this information, and assuming that all
of the energy are converted to kinetic energy,
calculate the momentum of a 150-keV electron (the denominator in Eqn (1.2.A1.2)).
(Hint: Kinetic energy E 5 p2/2m.)
B. Calculate the wavelength of an electron having
a kinetic energy of 150 keV.
C. Using the result of (B), calculate the theoretical resolving power of an electron microscope using a 150-keV electron beam.
12. In a Sorvall T-865 fixed angle rotor, the distance
to the axis of rotation is 3.84 cm at the top of
the tube and 9.10 cm at the bottom of the tube.
Calculate the RCF at 20,000 rpm at the top and
bottom of the tube.
13. We are centrifuging a collection of particles
with diameter 150 nm and average density of
1.10 g cm23 through a water solution with
density 1.0 g cm23 at 20,000 rpm. The viscosity
of the water is 1 3 1023 Pa s, where Pa is the
pascal 5 1 N m22.
A. At a distance of 8.5 cm from the axis of
rotation, what is the net force on a particle?
B. From Stoke’s equation, calculate the frictional coefficient.
C. What is the particle’s terminal velocity?
D. What direction is the net force?
E. What causes this net force?
Problem Set
14. In eukaryotic cells (cells with a nucleus), ribosomes have two major subunits, a 60s and a
40s. If we assume both are spheres and have the
same average density, what are their relative
sizes?
15. Intestinal cells make a calcium-binding protein
called calbindin. Calbindin has a molecular
weight of about 9000 Da. Its synthesis requires
the active form of vitamin D, 1,25(OH)2 cholecalciferol, to turn on the gene for the protein.
When 1,25(OH)2 cholecalciferol is given to
vitamin-D-deficient people, it takes about 45
minutes for the intestinal cells to make the first
complete calbindin.
A. Identify the major steps that could account
for the 45-minute lag in appearance of
calbindin.
B. Eukaryotic cells (cells with a nucleus) attach
amino acids to new proteins at the rate of
about 2 per second. Is the synthesis of the
protein the major part of the lag? (Hint: The
average molecular weight of an amino acid
is about 100 Da. You can estimate how
many amino acids are in the protein from
this information—you could look it up
because its sequence is known, but we are
just doing a “back of the envelope” calculation here.)
16. We have a double-stranded DNA segment of
1000 base pairs. Its nucleotide composition is
randomly distributed among A, T, C, and G.
Assume that each hydrogen bond in the double
strand has an energy of 4 kcal mol21
(1 J 5 0.239 cal).
A. How many hydrogen bonds are there in the
segment? (Hint: Consult Figure 2.2.3 for the
numbers of hydrogen bonds for each base
pair.)
B. What is the total energy necessary to pry
apart the two strands, assuming that hydrogen bonding is the only force keeping them
together? (It is not.)
C. Assume that hydrogen bonds break when
they are stretched 0.2 nm. How much force
is necessary to break one?
Pump 2
Pump 1
Flow rate R1
Reservoir S1 at C1
D. If all the hydrogen bonds in our DNA segment were to be ruptured all at once, how
much force would be necessary?
17. The molecular weight of the protein myosin is
525,000 g mol21. Its sedimentation coefficient is
6.4S (this S is the Svedberg, not seconds) in
water with a density of 1.0 g cm23, and its partial specific volume is 0.73 cm3 g21. Calculate
the frictional coefficient, β. This is sometimes
called f.
A. From the molecular weight and the specific
volume, calculate the radius myosin would
have if it were spherical.
B. From the radius calculated in (B), determine
the frictional coefficient from Stoke’s equation. This is called f0. The viscosity of water
at 25 C is 1 3 1023 Pa s, where Pa is the
pascal 5 1 N m22.
C. If the protein were spherical we would
expect f 5 f0. Is myosin spherical?
18. Consider the device shown in Figure 2.PS1.4
that consists of two reservoirs, S1 and S2, that
initially contain the limiting concentrations C1
and C2, respectively, where C1 , C2. A pump
removes fluid from S1 at rate R1 and places it in
reservoir S2. Therefore, as soon as the pumping
starts the concentration in S2 begins to change.
A magnetic stir bar rapidly mixes reservoir S2,
and a second pump withdraws fluid from S2 at
rate R2 and places it in a centrifuge tube. The
total volume of S1 5 S2 and both are one-half of
the capacity of the tube, so that when all of the
solutions are pumped into the tube, the tube is
filled. Show that for R2 5 2 3 R1, the gradient is
linear in volume from C2 at the bottom of the
tube to C1 at the top.
19. The SR is a specialized endoplasmic reticulum
in skeletal, cardiac, and smooth muscle cells. It
contains a Ca-ATPase pump that actively pumps
Ca21 ions from the cytosol to an enclosed compartment within the SR, its lumen. The activity
of the SR can be estimated by the rate of
oxalate-supported Ca21 uptake. This activity is
useful because it can also be measured in homogenates of the tissue. Evidence suggests that the
Flow rate R2
Reservoir S2 at C2
Stirrer
Centrifuge tube with gradient
FIGURE 2.PS1.4 One way to make a
gradient. Two reservoirs have the limiting
concentrations C1 and C2. Pump 1
removes fluid from reservoir S1 at rate R1
and places it in reservoir S2, initially at C2
but then becomes diluted with fluid from
S1. Pump 2 removes fluid from S2 at rate
R2 and places it in a centrifuge tube.
The gradient that is formed depends on
the values of R1 and R2 and the volumes
of the reservoirs. Both S1 and S2 begin
with identical volumes equal to one-half
of the volume delivered to the centrifuge
tube.
159
160
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
oxalate-supported Ca21 uptake rate is only due
to the SR and other organelles—the surface
membrane or mitochondria—do not contribute
to it. The left ventricles from a set of dogs were
removed under general anesthesia, weighed and
homogenized in 3 volumes of buffer (3 mL of
buffer for every g wet weight of heart) and
homogenate protein, volume and oxalatesupported Ca21 uptake rate were measured. The
homogenate was subjected to differential and
then sucrose-gradient centrifugation to isolate
membrane vesicles of the SR. The following was
obtained from an average of 10 preparations:
heart homogenate volume: 4.1 mL g21 wet
weight of heart, heart homogenate protein:
46.1 mg mL21, heart homogenate oxalatesupported Ca21 uptake rate: 119 nmol min21
(mg homogenate protein)21 isolated SR oxalatesupported Ca21 uptake rate: 3.44 μmol min21
(mg SR protein)21.
A. Calculate the homogenate protein per g wet
weight of heart tissue.
B. Calculate the total homogenate Ca21 uptake
rate per g wet weight of tissue.
C. Assuming that the SR is 100% pure, how
much SR is there, in mg of SR protein, per g
of wet weight of heart tissue? (Hint: think
about the units in the calculation.)
Passive Transport and
Facilitated Diffusion
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
2.5
transport. The cell must be able to control what goes
across the membranes and how fast.
There are three main mechanisms for transport:
Describe the microporous membrane as a model of passive
membrane transport
Describe the lipid bilayer model of passive membrane
transport
Define the permeability of a membrane
Describe how the permeability depends on the microscopic
character of the membrane for a porous membrane
Describe how the permeability depends on the microscopic
character of the membrane and solute for a dissolution
model of passive transport
Be able to determine the free energy change for passive
transport
Distinguish between facilitated transport and diffusional
transport on the basis of saturability, specificity, rates, and
competition
Write an equation showing the rate of facilitated transport
as a function of its solute concentration with zero-trans
concentration. Identify the variables and describe their
meaning
Distinguish between an ionophore and a channel
Describe what is meant by channel gating
Distinguish between voltage-gated channels and ligandgated channels
MEMBRANES POSSESS A VARIETY
OF TRANSPORT MECHANISMS
As described in Chapter 2.4, membranes serve as effective barriers to the free movement of materials, thereby
dividing the cell into compartments. This compartmentalization is necessary. In muscle, for example, it allows
for the control of contraction by releasing Ca21 ions
from a store (the specialized endoplasmic reticulum of
the muscle cell) into the cytoplasmic compartment.
Relaxation is then brought about by removing Ca21
ions from the cytosol back into the storage compartment. In another example, compartmentalization allows
mitochondria to transduce the energy of oxidation of
foodstuffs into the chemical energy of ATP. However,
compartmentalization does not make sense if material
absolutely cannot travel between the compartments.
What is necessary is selective transport and regulated
A. Passive transport
1. Diffusion
2. Facilitated transport
B. Active transport
1. Primary active transport
2. Secondary active transport
C. Osmosis.
In this chapter, we will consider passive transport across
two types of hypothetical membranes: a microporous
membrane and a lipid bilayer membrane. The mechanisms of passive transport differ considerably between
these two models, but the overall form of the equations is
similar. In Chapter 2.6, we will consider active transport
and then in Chapter 2.7, we will discuss osmosis.
A MICROPOROUS MEMBRANE IS ONE
MODEL OF A PASSIVE TRANSPORT
MECHANISM
Here we introduce the porous membrane as a model for
biological membranes as shown in Figure 2.5.1. We consider here that a microporous membrane separates two
solutions of different concentrations but the same
pressure. The pores allow solute particles to pass, but the
rest of the membrane that lacks pores is impermeable to
the solute, and also to solvent water. We assume that the
solute particles are small compared to the pores.
First we write the flux, the flow per unit area within a
single pore. This is governed by Fick’s Laws of Diffusion
given as
js 5 2D
½2:5:1
@CðxÞ
@x
@CðxÞ
@2 CðxÞ
5D
@t
@x2
where js indicates the flux within the pore. We use the
lower case “j” purposefully to distinguish it from Js,
which we will use to signify the macroscopic flux across
the entire membrane. The top equation is Fick’s First
Law of Diffusion; the bottom equation is Fick’s Second
Law of Diffusion. Here we are concerned only with
flux across the membrane, in one direction, and the
161
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00015-X
162
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Membrane
Pore
CL
Constant slope
= (CL – CR)/ (0 – δ)
δ
js
Pore
CR
n = N pores/unit area
FIGURE 2.5.1 Schematic of the hypothetical microporous membrane. In
this model, the membrane is a thin sheet of thickness δ. It is pierced by
many cylindrical pores oriented perpendicular to the surface of the
membrane. The radius of each pore is a and the number of pores, N, per
unit area A is n 5 N/A. This model might not pertain to some cellular
membranes, but it may describe some extracellular membranes such as
the basement membrane, which supports many cells, especially
epithelial cells, or it may represent the filtration membrane present in
the kidney.
one-dimensional forms of Fick’s laws apply to this situation. Let CL be the concentration on the left side of the
membrane and CR be the concentration on the right.
We can arrange it so that the volumes of the two baths
are so large that CL and CR are effectively kept constant.
Under these conditions, the solute flow will come to a
steady state or stationary value. This means that neither
the fluxes nor the concentration of solute changes with
time. Fick’s Second Law of Diffusion becomes
½2:5:2
05D
@2 CðxÞ
@x2
Note that this situation cannot be literally true, as solute
is moving from one compartment to another, so there
must be some changes in C(x) with time. However, C(x)
can be so nearly constant that we can ignore the very
slight error. The solution to this equation is that @C(x)/@x
is constant. This means that the concentration within the
pores is linear with x. We solve this equation by two
successive integrations, incorporating the boundary conditions that at x 5 0, C(x) 5 CL and at x 5 δ, C(x) 5 CR
(see Figure 2.5.2). The concentration is written as
CL 2 CR
CðxÞ 5 CL 1
½2:5:3
x
02δ
and the concentration gradient is
@CðxÞ
CL 2 CR
ΔC
½2:5:4
52
52
@x
δ
δ
The flux in the pore is given from Eqns [2.5.1] and
[2.5.4] as
½2:5:5
js 5 D
ΔC
δ
The total flow of solute per pore, qs, is given by the area
of the pore times the flux within the pore:
½2:5:6
qs 5 πa2 js
δ
x=δ
x=0
FIGURE 2.5.2 Cross-section of a microporous membrane in the vicinity
of a pore. Superimposed on the cross-section is a graph of the
concentration gradient. The left compartment has a higher
concentration (CL) than the right compartment with concentration CR.
Under this situation, the flux through the pore is to the right.
The total flow across an area A of the membrane containing N pores is
½2:5:7
Qs 5 Nqs 5 Nπa2 js
The macroscopically observed flux across the membrane
is the total flow of solute (Qs) divided by the macroscopic
area of the membrane.
Js 5
½2:5:8
Qs
A
5
Nπa2 js
5 nπa2 js
A
5
nπa2 D
ΔC
δ
According to this equation, the observed macroscopic
solute flux across the membrane is linearly related to the
concentration difference by a coefficient that includes
the thickness of the membrane (δ), the density of pores
in the membrane (n), the radius of the pores (a), and
the diffusion coefficient of the solute (D). Often many
of these parameters are not known with accuracy and we
lump all of these terms together to write
½2:5:9
Js 5 pΔC
where p is the permeability of the membrane to the solute. This phenomenological coefficient has the units of
cm s21 and includes all of the microscopic parameters
of the membrane:
n πa2 D
δ
In this model, the permeability increases when the size
of the pores increases, when the number of pores per
½2:5:10
p5
Passive Tr ansport and F acilit ated Di ffusion
unit area of membrane increases, when the thickness
of the membrane decreases, and when the diffusion
coefficient of the transported solute increases. Which of
these can be regulated? Typically membranes do not
regulate their thickness, nor can the diffusion coefficient
be altered. Channels can act like pores and they can
be gated. That is, the channels can be opened or shut.
This has the effect of controlling the area through
which materials can be transported and this is a common way of regulating ion transport. Another way of
physiologically regulating passive transport is by
controlling the number of pores (or channels) in a
membrane.
The distinction between pores and channels lies in the
substrate. We think of pores as holes in a substrate that
will not collapse, as if we drilled a tiny hole in a thin
plastic sheet. Lipid bilayers, however, will not support a
watery void in their interior. Channels are proteins
embedded in the membrane that line a watery pathway
across the membrane and prevent its collapse by providing mechanical support on the sides of the pathway.
The watery path across the membrane is made by the
proteins that form the channel.
DISSOLUTION IN THE LIPID BILAYER
IS ANOTHER MODEL FOR PASSIVE
TRANSPORT
Consider now a markedly different model of the membrane. In this case, there are no pores, but we envision
that a molecule may penetrate from the left to the right
of the membrane by dissolving in the lipid bilayer
core of the membrane, diffusing across the lipid, and
then being extracted back into the aqueous phase
on the other side of the membrane. This model of
passive transport is shown schematically in Figure 2.5.3.
The dissolution of the solute in the lipid membrane
is described quantitatively by a constant called the
partition coefficient (see Chapter 2.3):
½2:5:11
ks 5
equilibrium C in the lipid phase
equilibrium C in the water phase
Partition into
membrane
Partition into
solution
Diffusion
1
2
3
CR
CL
Lipid bilayer
FIGURE 2.5.3 Cartoon of the lipid bilayer model of passive transport.
The left and right compartments are separated by a lipid bilayer
membrane. Solute, shown here as blue spheres, moves across the
membrane in three well-defined steps. In step 1, the particle partitions
itself into the lipid phase of the membrane. In step 2, the material
diffuses across the lipid bilayer. In step 3, the material partitions itself
back into the aqueous phase on the right side of the membrane. Overall
transport rates are determined by the rates of steps 1, 2, and 3.
diffusion across the lipid bilayer. This equation can be
rewritten as
½2:5:13
Js 5
ks Ds;lipid
ΔC
δ
This last equation is identical in form to that derived
earlier in the microporous membrane model:
½2:5:9
Js 5 pΔC
In the case of the dissolutiondiffusionsolution
model, we identify the permeability as
½2:5:14
p5
ks Ds;lipid
δ
If equilibrium is reached quickly at both the left and
right surface of the membrane, then diffusion through
the lipid phase would limit the rate of transport. Let the
concentration on the left side of the membrane be CL
and the concentration on the right side of the membrane be CR. The concentration immediately inside the
membrane on the left, by Eqn [2.5.11], will be ksCL,
and the concentration on the right inside the membrane
will be ksCR. The steady-state flux through the lipid
phase is
ks CL 2 ks CR
Js 5 Ds;lipid
½2:5:12
δ
Equation [2.5.14] neatly sums up many experimental
observations concerning the permeability of materials
through biological membranes. These are briefly summarized in Overton’s rules:
where Ds,lipid is the diffusion coefficient of the solute in
the lipid phase and δ is the thickness of the lipid phase.
We write Js here because the entire area of the membrane is available for dissolution of the solute and
A. The permeability is proportional to the lipid
solubility.
B. The permeability is inversely proportional to
molecular size.
Once again, the permeability is a single phenomenological parameter that relates the flux to the concentration
difference. It incorporates all of the microscopic parameters of the membranesolute pair into a single
parameter. In this case, these microscopic parameters
are the partition coefficient, the thickness of the membrane, and the diffusion coefficient of the solute in the
membrane.
163
164
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Thus we expect ethanol to permeate cell membranes
quite easily because it is small and it is lipid soluble.
On the other hand, glucose is larger and it is not readily
lipid soluble and so it requires another mechanism to
enter the cell. This mechanism is the carrier. Overton’s
rules derive from two main components of Eqn
[2.5.14]: the dependence of p on ks means that lipid solubility is a direct determinant of the permeability and
the dependence on Ds means that size is an inverse
determinant of permeability.
Lipid solubility here depends on the hydrophobicity or
lipophilicity of the solute. Different chemical groups in
any molecule confer hydrophilic or hydrophobic character to those parts of the molecule, as described in
Chapters 2.3 and 2.4. In particular, electric charge make
a solute highly hydrophilic and not lipophilic. Thus
charged or ionized solutes are generally highly impermeable by this dissolution mechanism of transport.
Solutes enriched in hydroxyl groups, carboxyl groups,
and amino groups are generally not easily permeable
through this mechanism unless they are also very small.
FACILITATED DIFFUSION USES A
MEMBRANE-BOUND CARRIER
For some valuable materials, the membrane permeability
is not large enough for the cell’s needs and facilitated
diffusion is used to carry solute across the membrane.
One possible way a carrier could operate is shown
in Figure 2.5.4. The transformations that occur to allow
this transport are not known in detail. It is likely that the
SL
EL
1
E.SL
4
2
E.SR
3
ER
SR
FIGURE 2.5.4 Schematic diagram of carrier-mediated passive transport.
The carrier is designated as “E.” In this scheme, an integral protein
molecule in the membrane binds to a solute molecule on one side of
the membrane. The carrier molecule then undergoes a transformation
that has the effect of changing the side of the membrane that is
accessible to the binding site. The solute molecule then dissociates from
the carrier on the opposite side. The carrier then returns to its original
shape.
carrier provides something like a pore for the solute, but
the pore is specifically designed to fit the solute. In this
case, the solute molecule never dissolves in the lipid
bilayer but is protected from it by a pocket of the protein
carrier.
The mechanism involved in facilitated diffusion shown
in Figure 2.5.4 involves a sequence of four reactions:
SL 1 EL -E 2 SL
E 2 SL -E 2 SR
½2:5:15
E 2 SR -ER 1 SR
ER -EL
222222222
Sum : SL -SR
The sum of these four reactions is the movement of solute from the left side of the membrane to the right side.
The carrier concentration does not enter into the overall
stoichiometry of the reaction, because its presence on
both sides of the reactions cancels itself out. It acts as a
catalyst for transport because it determines its rate without being altered by the process.
FACILITATED DIFFUSION SATURATES
WITH INCREASING SOLUTE
CONCENTRATIONS
Facilitated diffusion can be distinguished from a purely
diffusional mechanism because facilitated diffusion is
saturable and it is specific. Plots of the flux versus the
concentration are not linear, as you would expect from
the diffusion mechanisms as shown in Eqns [2.5.8] and
[2.5.13]. The rate increases with concentration but only
up to a point. This is due to the fact that there are
only so many carrier molecules in the membrane. When
they are all busy, there can be no further increase in the
rate of transport. This is analogous to the ferrying of
people across a river that is too deep for most of them to
wade. Although some can wade, most must cross only
by ferry. When there are not too many people, the ferries
can accommodate them easily and the transport rate will
increase with each increase in the number of people
waiting on shore. When the crowd on the shore gets too
great, however, the ferries become full and the rate of
transport can be increased further only by the number
of brave souls who can wade the river (diffuse across the
membrane) or by increasing the number of ferries.
Figure 2.5.5 shows the kinetics of a saturable transport
mechanism.
These curves often closely resemble the hyperbolic plots
characteristic of MichaelisMenten enzyme kinetics and
can be fit to
½2:5:16
Qtrans 5
Qmax C
Km 1 C
where Qtrans is the flow of transported material across
the membrane in moles per unit time, Qmax is the maximum flow, C is the concentration of the transported
Passive Tr ansport and F acilit ated Di ffusion
competitive inhibitors. This competitive inhibition is
closely related to that observed in enzyme kinetics. In
other cases, a compound sharing some similarity with
the natural substrate may bind to the carrier but not be
transported. If the binding is at the transport site, such
a compound might inhibit transport.
12
Qmax
10
Qtransport
8
PASSIVE TRANSPORT OCCURS
SPONTANEOUSLY WITHOUT INPUT
OF ENERGY
6
Km
4
The chemical reaction for the overall transport is written as
½2:5:17
2
SoluteL -SoluteR
and the free energy change for the reaction is
0
0
2
4
6
[S]
8
10
12
½2:5:18
ΔG 5 GR 2 GL
Substituting in with the chemical potential, we obtain
FIGURE 2.5.5 Graph of a saturable transport mechanism. The rate of
transport in moles per unit time per unit area is plotted against the
concentration of material on the feed side [S]. A maximum transport
rate, Qmax, can be identified. The substrate concentration at halfmaximal transport is used to characterize the affinity of the transport
mechanism for its substrate.
ΔG 5 nðΔμ0 1 RT ln CR Þ 2 nðΔμ0 1 RT ln CL Þ
CR
½2:5:19
5 n RT ln
CL
solute on the feed side of the membrane, keeping the concentration on the opposite side at zero, and Km is
a constant characteristic of the carrier. The term “Km”
comes from MichaelisMenten kinetics, and these carriers
almost certainly do not have the mechanism first proposed by Michaelis and Menten. Nevertheless, the term
“Km” has come to mean “the concentration of substrate at
half-maximal activity.” Sometimes this is referred to as Kt,
the concentration at half-maximal transport. Eqn [2.5.16]
is a simplified version of the exact solution of the kinetics
of the scheme shown in Figure 2.5.4.
where n is the number of moles of solute moving
from left to right. Here there is no electrical work
term because the charge on the molecule, z, is zero.
If CR . CL, ΔG calculated from Eqn [2.5.19] will be
positive. This means that the opposite process will occur.
That is, solute will move from the right to the left, opposite to the direction shown in Eqn [2.5.17]. If ΔG 5 0,
then no net movement occurs and CR 5 CL. If CR , CL,
then ΔG calculated according to Eqn [2.5.19] will be negative and the reaction will proceed as written, with solute
moving from the left to the right side of the membrane.
Thus thermodynamics tells us what process can occur and
with what change in free energy, but it does not give us
an expression for the permeability or the rate at which the
process will occur.
FACILITATED DIFFUSION SHOWS
SPECIFICITY
Another distinguishing feature of carrier-mediated facilitated diffusion is its structural specificity. The parts of
the carriers that bind transported solute are specifically
designed for that solute and not others. For example,
most cell membranes in the human contain carriers for
glucose. They will transport D-glucose but not its enantiomer (mirror image compound) L-glucose. The carrier
for glucose will not transport amino acids and vice versa.
FACILITATED DIFFUSION SHOWS
COMPETITIVE INHIBITION
The specificity of carrier-mediated facilitated diffusion
also gives rise to competitive inhibition. Compounds
that closely approximate the shape of the natural substrate may also bind to the carrier and be transported
across the membrane. Since the carrier cannot carry
both compounds at the same time, the transport of the
natural substrate is reduced by the presence of
Diffusion through aqueous pores or through the lipid
barrier of membranes or by facilitated diffusion is called
passive transport because none of these mechanisms
requires “outside” energy. These flows occur spontaneously. What this means is that the energy that drives
them is contained within the solutions themselves. This
does not mean that they occur rapidly, but only that
they occur naturally without the addition of any “outside” force. The rate at which they occur depends on the
mechanisms of transfer. The analysis of the mechanism
gives us additional information, such as what determines and regulates the rate.
The overall ΔG for facilitated diffusion is the ΔG for the
sum, which is the same in Eqns [2.5.15] and [2.5.17].
The net ΔG 5 nRT ln CL/CR. Thus the participation of
the carrier, which remains unchanged by the transport
reaction, does not alter the reaction energetics at all,
whereas it does alter the reaction kinetics. The carrier is a
catalyst. It speeds up the reaction, which in this case is
transport, without entering into the stoichiometry of the
reaction. This is an example of how thermodynamic
165
166
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Example 2.5.1 Specificity of Transport
There are a variety of transporters for glucose that are called
GLUT (for glucose transporter). The GLUT-1 transporter imports
glucose into a variety of cell types. This is an integral membrane
protein with a molecular weight of 45 kDa. Its Km for glucose is
1.5 mM. It will also transport L-glucose with a Km of 3000 mM.
Glucose is typically about 100 mg% in the extracellular fluid.
At what fraction of Qmax will glucose be transported at this
concentration?
The normal plasma [glucose] is given as 100 mg%, which is
100 mg of glucose per 100 mL of plasma. This is Pg 5 100 mg/
100 mL 3 1000 mL L21 5 1 g L21. Since the molecular weight of
glucose is 180 Da, its gram molecular weight is 180 g mol21 and
the normal plasma glucose concentration is
Pg 5 1 g L21 =180 g mol21 5 0:0056 M 5 5:6 mM
The rate of transport, assuming zero-trans glucose, is given by
Eqn [2.5.16] as
Qtrans 5 ½5:6 mM=ð1:5 mM 1 5:6 mMÞQmax 5 0:789 Qmax
At plasma [glucose], the transporters are nearly saturated and
the rate of transport could be increased mostly by affecting the
number of transporters, i.e., increasing Qmax.
What would the L-glucose rate of transport be at the same
concentration as D-glucose?
Qtrans L-glucose 5 ½5:6 mM=ð3000 mM 1 5:6 mMÞQmax
5 0:0019 Qmax
D-Glucose
is transported almost 400 times more quickly than
L-glucose.
Example 2.5.2 Effect of Substrate Concentration on Flux
GLUT-1 glucose transporter is the most ubiquitous form of
glucose transporter. It is present in high amounts in erythrocytes
and endothelial cells, the bloodbrain barrier and in the
proximal straight tubule of the nephron. If its Km for glucose is
1.5 mM, how much would transport increase if plasma glucose
were increased from 80 mg% to 120 mg%?
First, we convert the plasma glucose concentrations to mM.
80 mg% means 80 mg per 100 mL or 80 mg/0.1 L 5 800 mg/L.
The molecular weight of glucose is 180 g, so this concentration
is equivalent to 0.8 g/180 g mol21/L 5 4.44 mM. Similarly,
120 mg% is 6.67 mM.
GLUT2 is another glucose transporter that is present in beta cells
of the islets of Langerhans, in the pancreas, and also in the
kidney, intestine, and liver. Its Km for glucose is much higher,
17 mM. How much would transport increase if plasma glucose
were increased from 80 mg% to 120 mg%?
We can use the molar concentrations for glucose that we used
before for the GLUT1 calculations. The transport rates are
calculated at 80 mg% glucose as
Q 5 Qmax 3 4:4 mM=ð17 mM 1 4:44 mMÞ 5 0:207Qmax
and at 120 mg% glucose, it would be
If the Km for glucose is 1.5 mM, then the transport rate at 80 mg
% glucose would be
Q 5 Qmax 3 6:66 mM=ð17 mM 1 6:66 mMÞ 5 0:282Qmax
Q 5 Qmax 3 4:4 mM=ð1:5 mM 1 4:44 mMÞ 5 0:75Qmax
Here the transport rate increases 36% when blood glucose
increases 50%.
And at 120 mg% glucose it would be
Q 5 Qmax 3 6:66 mM=ð1:5 mM 1 6:66 mMÞ 5 0:82Qmax
The transport rate increases 9% when the blood glucose
increases by 50%. These GLUT1 transporters are insensitive to
changes in blood glucose.
analysis of the reaction is independent of the mechanism: it tells us about the energetics without telling us
anything about the reaction’s path or its rate.
The saturability of carrier-mediated facilitated diffusion
distinguishes it from the other passive transport
mechanisms that show a linear relationship between
flow and the concentration difference across the membrane. Neither mechanism can concentrate solute. Flow
of material always occurs from the side with the higher
concentration to the side with the lower concentration.
Thus GLUT1 and GLUT2 serve different functions. GLUT1 operates close to maximal rates relatively independently of blood
glucose levels. GLUT2 increases transport almost proportionately
with blood glucose, so that GLUT1 is used for basal glucose
transport into metabolizing tissues, whereas GLUT2 finds use as
part of the sensor apparatus for glucose concentrations in blood.
When the concentrations on the two sides of the membrane are equal, no further flow occurs because the two
solutions are in equilibrium.
IONS CAN BE PASSIVELY
TRANSPORTED ACROSS MEMBRANES
BY IONOPHORES OR BY CHANNELS
So far we have considered passive diffusion of nonelectrolytes. Suppose now that the diffusing species
Passive Tr ansport and F acilit ated Di ffusion
are electrically charged. Charged species are poorly
soluble in the lipid phase, and so they cannot merely
dissolve in the lipid on one side of the membrane, diffuse across, and then enter the compartment on the
opposite side of the membrane. They need either carriers or channels to get across.
IONOPHORES CARRY IONS ACROSS
MEMBRANES OR FORM CHANNELS
Fungi and bacteria make a class of poison called
ionophores. These are molecules that allow ions to cross
membranes. The fungi and bacteria make these compounds to kill off competition by disrupting the permeability barrier of their competitors’ membranes. These
ionophores are of two types: carriers and channel formers.
Figure 2.5.6 illustrates these two types of ionophores.
An example of a carrier is A23187. This material is commercially obtained from Streptomyces chartreusis and has
weak antibiotic activity against gram-positive bacteria. It
is particularly active for divalent cations with a specificity
of
Mn21 .Ca21 .Mg21 .Sr21 .Ba21 .Li1 .Na1 .K1.
It is predominantly used as a carrier for Ca21. Other
examples of natural molecules that act as carriers include
Diffusable ionophore
binds ligand and carries
it across the bilayer
1
An example of a channel former is gramicidin A. This is
an antibiotic polypeptide containing 15 amino acids
that is isolated from the bacterium Bacillus brevis. The
molecule appears to form a pore by linking two molecules of gramicidin A across the bilayer. The gramicidin
pore appears to behave like a water-filled pore. Other
examples of pore-forming antibiotics include amphotericin and nystatin. Amphotericin makes a channel by
interacting with cholesterol in cell membranes.
ION CHANNELS
A variety of integral membrane proteins form channels
for ions. These ion channels exhibit some of the characteristics of carriers in that they are highly selective. These
channels exhibit other characteristics such as gating.
Gating refers to the fact that these channels act as if they
have gates that are opened sometimes, allowing ions to
cross the membrane, and are closed at other times, preventing ions from moving. The percent of the time the
channels are opened is referred to as the open probability,
po, and can be regulated in various ways. Some channels
open when another molecule binds to the channel. These
are ligand-gated channels. Other channels sense the local
potential, probably through the presence of charged
groups on the channel, and open or close depending on
the potential. These are voltage-gated channels. A cartoon
of these types of channels is shown in Figure 2.5.7.
Voltage gates are usually charged;
they move in response to the local
electric field
4
CL
valinomycin (a K1 ionophore) and nigericin (an H1
ionophore).
3
CR
–
+
+
+
2
Lipid bilayer
+
Interstitial
fluid
Channel-forming ionophore
provides an aqueous path
across the membrane
FIGURE 2.5.6 How ionophores work. Some ionophores increase the
passive diffusion across a lipid bilayer by providing a hydrophilic pocket
that binds a solute and sequesters it away from the hydrophobic lipid
interior. These ionophores generally show specificity of transport
because the pocket binds some ions better than others. For these types
of ionophores, the ionophoreligand complex is believed to diffuse
across the lipid bilayer, carrying the ligand with the ionophore. Other
ionophores form an aqueous channel across the lipid bilayer. These
channel-forming ionophores are less specific but still show specificity
due to the size and shape of the channel.
–
+
E
–
–
+
Lipid bilayer
+
–
+
–
+
–
+
–
+
–
+
–
Other parts of the
channel confer
specificity
Cytosol
Ligand binding to
ligand-gated
channels opens or
closes a gate
FIGURE 2.5.7 Voltage- and ligand-gated channels. Voltage-gated
channels typically have a highly charged part of the protein that
responds to the local electrical field produced by the separation of
charge on the two sides of the membrane. Changes in this electric field
alter the disposition of the gate to either open or close access to a
hydrophilic pathway across the membrane. Ligand-gated channels bind
a regulatory ligand that alters the shape of the channel so as to open or
close its pathway.
167
168
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Examples of voltage-gated and ligand-gated channels
abound, and a full description of them here is premature
because we have not yet studied membrane potential or
action potentials. Voltage-gated Na1 and K1 channels
allow ions to flow across the membrane only under
specific circumstances. The flow of ions is an electric current, because charged ions are moving. The membrane
itself is a tiny capacitor, as we saw in Chapter 1.3.
The currents going through these channels can discharge
the capacitor, changing the voltage across the membrane, or they can charge it back up again. In this way,
opening of the fast Na1 channel in neurons causes
a brief, pulse-like change in the voltage across the
neuronal cell membrane. The membrane potential is
reestablished by a later opening of the K1 channels.
These actions produce the nerve impulse, the brief
change in nerve cell membrane potential that propagates
down the nerve and activates its target—either another
neuron, a muscle fiber, or some secretory cell.
Other voltage-gated channels include the T-, L-, and
N-type Ca21 channels. The designation “T” signifies
that this channel opens “transiently”; the “L” stands
for “long-lasting”; and the N indicates that this type of
channel is “neuronal.” The channels generally open
upon depolarization of the cell membrane and they
specifically transport Ca21. The consequence is that
the [Ca21] inside the cell increases in the vicinity of the
channel, and this Ca21 binds to cellular elements to
change their activity.
Ligand-gated channels may be present on the surface
membrane and also on interior membranes. The endoplasmic reticulum of many cells contains a large
tetrameric protein called the IP3 receptor. This receptor
forms a channel for Ca21 across the ER membrane
and opens in response to IP3 (inositol trisphosphate)
that is liberated from the surface membrane as part of
signal transduction. Gating by IP3 causes Ca21 release
from the ER and the increased cytoplasmic [Ca21] alters
cellular activity.
Many ligand-gated channels are present on the surface
membrane and respond to neurotransmitters or hormones. These channels are gated by the binding of a
chemical rather than by the voltage difference across the
membrane. Acetylcholine is the neurotransmitter involved
in skeletal muscle neurotransmission. When activated, the
motor neuron nerve terminal releases acetylcholine near
the skeletal muscle membrane. The acetylcholine binds to
nicotinic acetylcholine receptors, so named because of
their sensitivity to nicotine, on the muscle membrane.
Binding of acetylcholine opens a large conductance
pathway mainly for Na1. This causes a depolarization of
the muscle membrane that propagates along the muscle
surface, eventually activating muscle contraction.
WATER MOVES PASSIVELY THROUGH
AQUAPORINS
Passive transport of water across biological membranes
also occurs through water channels. These are tiny pores
formed by proteins called aquaporins. There are a
variety of aquaporins and they are present on virtually
every cell membrane. AQP1 has a molecular weight
of 29 kDa and forms a channel by the association of
four monomers. In some membranes, the number
of aquaporins is physiologically regulated so that water
movement through the cell can be regulated. This is
particularly important in the kidney, because the
kidney has the final job of retaining water when it is
scarce and excreting it when it is in excess. Although
water obeys Fick’s Laws of Diffusion, its movement
is dominated by pressure-driven flow.
SUMMARY
Materials cross biological membranes by a variety of
mechanisms including passive transport, active transport, and osmosis. Passive transport mechanisms
require no input of metabolic energy. Because of this,
passive transport always entails the movement of materials from regions of high concentration to regions of
low concentration. The free energy change per mole in
the reaction SL-SR is
Δμ 5 RT ln½CR =CL where CR and CL are the concentrations of S on the
right- and left-hand sides of the membrane, respectively. If CR , CL, then Δμ , 0 and the reaction
proceeds from the high concentration (CL) to the low
concentration (CR).
Passive diffusion across membranes is characterized
by a linear relationship between the rate of transport
and the concentration difference across the membrane:
Js 5 pΔC
where Js is the macroscopically observed flux and p
is the permeability. This equation holds true if we
envision the membrane as a microporous membrane
in which diffusion occurs through tiny pores, or if we
envision the solute as dissolving in the lipid bilayer and
diffusing across it. The dependence of p on the microscopic characteristics of the membrane differs in these
two models. For a microporous membrane
p 5 nπa2 D=δ
where n is the number of pores per unit area, a is the
radius of the pore, D is the diffusion coefficient of
the solute, and δ is the thickness of the membrane.
For a solute dissolving in the lipid bilayer
p 5 KDlipid =δ
where K is the partition coefficient of the material in
the lipid phase, Dlipid is the diffusion coefficient in the
lipid bilayer, and δ is the thickness of the bilayer.
Some membrane proteins bind solutes and provide an
alternative path across membranes. The alternative
path facilitates the diffusion of the solute across the
membrane. These proteins are carriers for the solutes.
The kinetics of transport shows specificity, saturation,
Passive Tr ansport and F acilit ated Di ffusion
and competition with similar solutes. The overall transport rate often obeys an equation of the form
Qtrans 5 Qmax C=½Km 1 C
3.
where Qmax is the maximum transport rate, typically
limited by the number of carriers in the membrane,
and Km is a measure of the dissociation constant of the
carrier for the solute.
4.
Passive transport mechanisms include lipid dissolution
and diffusion, facilitated diffusion, ligand-gated channels,
voltage-gated channels, diffusion-mediated ionophores,
and pore-forming ionophores.
5.
6.
REVIEW QUESTIONS
1. Why is the gradient for a diffusive process linear
at steady state?
2. For a microporous membrane, what effect
would increasing the number of pores have on
diffusive flux across a membrane? What effect
would increasing the size of the pores have?
What effect should result from increasing the
7.
8.
9.
10.
size of the diffusing solute? Increasing the
temperature?
For a solute dissolution model, what effect
would increasing the partition coefficient have?
Increasing the particle size of the diffusing
solute? Increasing the temperature?
Why should two models as different as the
microporous membrane and solute dissolution
model have identical relationship between J and
ΔC?
Under what conditions is the free energy for
transfer across a membrane for a solute equal to
zero?
How does the function relating rate of transport
to concentration differ between facilitated diffusion and simple diffusion?
Why does simple diffusion not show specificity
or competition?
What two general mechanisms are used to
regulate the open or closed state of channels?
What is a channel?
How would you determine Km for facilitated
diffusion? Qmax?
169
2.6
Active Transport:
Pumps and Exchangers
Learning Objectives
G
G
G
G
G
G
G
G
G
Write the equation for the electrochemical potential for an ion
Give the approximate concentration of the major ions
inside and outside of a heart cell
Be able to calculate the free energy change for ion movement across the cell membrane under given conditions of
membrane potential and ion concentrations
Be able to calculate the free energy change for ion transport when coupled to other processes
Distinguish between active and passive transport
Distinguish between primary and secondary active transport
Give an example of a primary and secondary active transport mechanism
Distinguish between P-type, V-type, F-type, and ABC-type
active transporters
Define the terms: symport, cotransporter, antiport, and
exchanger
THE ELECTROCHEMICAL POTENTIAL
DIFFERENCE MEASURES THE
ENERGETICS OF ION PERMEATION
Here we consider the transport of four ions: Na1, K1,
Cl2, and Ca21, across the surface membrane of heart cells
at rest. These ions have different concentrations inside
and outside of the cell, and are also subjected to electrical
forces because there is a potential difference across the
cell membrane and thus an electric field within the membrane. The cardiomyocytes maintain a resting membrane
potential. We will learn later how this is established, but
for now it is enough to know that there is a separation of
charge in the outside and inside compartments. At rest,
there is an accumulation of negative charges inside the
cell and positive charges outside. This separation of
charges gives rise to the membrane potential, which is
always taken as the difference between the electrical
potential inside and outside the membrane:
½2:6:1
Δψ 5 ψi 2 ψo
The membrane potential is sometimes identified with
the variables ψ, Vm, or Em. The situation is described in
Figure 2.6.1.
Let us first take the case of Na1. The concentration of
1
170 Na outside the cell is about 145 mM and inside the
cell it is 12 mM. The higher concentration outside of
the cell favors a net Na1 flow from outside to inside,
driven by diffusion. Further, the negative potential
inside the cell also favors Na1 movement into the cell.
We write the free energy change per mole for the movement of Na1 from out to in as
½2:6:2
Δμ 5 μi 2 μo
Recall here that the free energy change is the free energy
of the final state (inside the cell, in this case) minus the
free energy of the initial state (outside the cell in this
case). When we are dealing with the free energy per
mole, we write μ. This is an intensive property which is
defined by the conditions and not by the extent of the
cell or its membrane or of the amount of material being
transported. The free energy itself is an extensive property that depends on how much material is being transported. We can insert the definition of electrochemical
potential that we justified earlier (see Eqn [1.7.14]):
½2:6:3
μx 5 μ0x 1 RT ln Cx 1 zx ℑψx
where the subscript x denotes substance x to avoid confusion with the subscript i or o which denotes the inside
or outside of the cell, respectively. Substituting in for the
conditions of the inside and outside of the cell, we find
Δμ 5μi 2μo
5μ0Nai1RT ln½Na1i1ℑψi2μ0Nao2RT ln½Na1o 2ℑψo
½Na1i
5RT ln
1ℑðψi 2ψo Þ
½Na1o
½2:6:4
The standard free energy per mole (μ0) cancels out
because it is independent of condition. The μ0 is the
part of the electrochemical potential that incorporates
the chemical energy involved in the formation of
bonds. Since in this case there is no chemical transformation (the Na1 ion is not chemically transformed in
any way; it is simply transported from one side of the
membrane to the other), Δμ0 5 0. Also, the z in the formula for electrochemical potential is the integer charge,
which is 11 for the Na1 ion. Remember here that ln is
the natural logarithm, not logarithm base 10.
The calculation of Δμ for the conditions of the cell
shows that Δμ , 0 (see Example 2.6.1). This means that
the reaction as written is spontaneous: it will occur in
the direction written passively, without additional
forces. Our analysis of the energy does not tell us
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00016-1
Active Tr ans port: Pumps a nd Exchangers
anything about how fast the process will occur, because
the thermodynamic analysis is independent of the
mechanism, and the mechanism is what determines
the rate. What we can say, however, is that a channel
for Na1 that opens in this membrane will cause a rapid
inflow of Na1 from the extracellular fluid into the cell.
Net flow would not occur in the opposite direction.
What we can also say is that if the membrane has any
non-zero permeability to Na1, the flux will be from the
extracellular fluid (the outside) into the cell.
Now let us consider what happens with Ca21. In a
completely analogous way, we write the change in free
energy for Ca21 entry into the cell as
Δμ 5 μi 2 μo
5 RT ln½Ca21 i 1 2ℑψi 2 RT ln½Ca21 o 2 ℑψo
5 RT ln
½Ca21 i
1 2ℑðψi 2 ψo Þ
½Ca21 o
Membrane
160
150
[Na+]o 145 mM
140
130
Concentration (mM)
120
110
[Cl– ]o100 mM
100
90
80
70
60
50
40
30
20
10
0
[K+]o 4 mM
[Ca2+]o1.2 mM
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
Cytosol
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
Note that this process (see Example 2.6.2) has a negative
Δμ, so it also occurs spontaneously. In this case, the free
energy change is much more negative. This is due to
the fact that the concentration gradient for Ca21
contains more energy and the electrical energy gain is
twice as great because each Ca21 ion has twice the charge
of an Na1 ion. A channel for Ca21 on the cell membrane
would let Ca21 into the cell under these conditions.
Now let us calculate the free energy change for K1 entry
into the cell. The formula is:
½2:6:6
½2:6:5
Extracellular fluid
Here the 2 in the equation arises because Ca21 has two
positive charges per ion. The two charges correspond to
zx in Eqn [2.6.3]. The electrical force on a Ca21 ion is
twice the force on an Na1 ion in the same electric field
and thus movement produces twice the energy.
[K+]
i
155 mM
Voltmeter
Δμ 5 μi 2 μo
5 RT ln½K1 i 1 ℑψi 2 RT ln½K1 o 2 ℑψo
5 RT ln
½K1 i
1 ℑðψi 2 ψo Þ
½K1 o
In this case, Δμ is positive (see Example 2.6.3). This
means that K1 under these conditions does not passively enter the cell. Rather, the spontaneous process is
K1 exit from the cell. If a channel specific for K1 was to
open in the membrane, K1 would leave the cell.
In this last example, let us calculate the free energy associated with Cl2 entry into the cell. Again we write the
difference in the electrochemical potentials:
–80 mV
–
Δμ 5 μi 2 μo
5 μ0Cli 1 RT ln½Cl2 i 1 ð2 1Þℑψi 2 μ0Clo 1 RT ln½Cl2 o
2 ð2 1Þℑψo
½Cl2 5 RT ln 2 i 2 ℑðψi 2 ψo Þ
½Cl o
[Na+]i 12 mM
[Cl–]i 5 mM
[Ca2+]i 0.1 µM
FIGURE 2.6.1 Concentrations of Na1, K1, and Ca21 and the resting
membrane potential across the resting cardiac muscle cell membrane.
The subscript “o” refers to the “outside” of the cell; “i” denotes the
“inside” compartment.
½2:6:7
Note that the valence on the Cl2 ion is negative and it
is entered that way in the equation. According to the
calculation (see Example 2.6.4), at rest [Cl2] is distributed at equilibrium across the cell membrane. That is,
there is no free energy change for Cl2 transport across
the resting muscle membrane.
These calculations show how the electrochemical potential calculates the energetics of transport, and further
EXAMPLE 2.6.1 Free Energy of Na1 Transport
For the conditions shown in Figure 2.6.1, calculate the free
energy of transport of Na1 from outside to inside the
cardiomyocyte.
Inserting the values for the concentrations into Eqn [2.6.4]
and using R 5 8.314 J mol21 K21 (51.987 cal mol21 K21; 1 J 5
0.239 cal) and T 5 310 K, and remembering that the faraday (ℑ)
is 9.649 3 104 C mol21, we get
Δμ 5 8:314 J mol21 K21 3 310 K ln½ð12 3 1023 MÞ=ð145 3 1023 MÞ
1 9:649 3 104 C mol21 3 ð2 0:080 VÞ
5 2 6:42 kJ mol21 2 7:72 kJ mol21
5 2 14:14 kJ mol21 5 2 3:38 kcal mol21
171
172
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
EXAMPLE 2.6.2 Free Energy of Ca21 Transport
For the conditions shown in Figure 2.6.1, calculate the free
energy of transport of Ca21 from outside to inside the
cardiomyocyte.
Inserting the values for the concentrations and membrane
potential into Eqn [2.6.5], we get:
Δμ 5 8:314 J mol21 K21 3 310 K ln½ð0:131026 MÞ=ð1:231023 MÞ
1 2 3 9:649 3 104 C mol21 3 ð2 0:080 VÞ
5 2 24:2 kJ mol21 2 15:4 kJ mol21
5 2 39:6 kJ mol21 5 2 9:46 kcal mol21
EXAMPLE 2.6.3 Free Energy of K1 Transport
For the conditions shown in Figure 2.6.1, calculate the free
energy of transport of K1 from outside to inside the
cardiomyocyte.
We can insert the values for [K1]i 5 155 3 1023 M and
[K1]o 5 4 3 1023 M into Eqn [2.6.6] to calculate Δμ:
Δμ 5 8:314 J mol21 K21 3 310 K ln½ð155 3 1023 MÞ=ð4 3 1023 MÞ
1 9:649 3 104 C mol21 3 ð2 0:080 VÞ
5 2 9:43 kJ mol21 2 7:72 kJ mol21
5 1 1:71 kJ mol21 5 1 0:41 kcal mol21
EXAMPLE 2.6.4 Free Energy of Cl2 Transport
For the conditions shown in Figure 2.6.1, calculate the free
energy of transport of Cl2 from outside to inside the
cardiomyocyte.
2
23
We can insert the values for [Cl ]i 5 5 3 10
[Cl2]o 5 100 3 1023 M into Eqn [2.6.7] to calculate Δμ:
M
and
show that both the electrical and diffusive forces enter
into the equations that determine the direction of ion
flow. Things get a bit more complicated and interesting
when ion flow is coupled to other processes.
ACTIVE TRANSPORT MECHANISMS
LINK METABOLIC ENERGY
TO TRANSPORT OF MATERIALS
All of the ion movements we have discussed so far
involve movement “down” the electrochemical gradient: the free energy was higher for the initial condition
than for the final condition, and the free energy
change Δμ 5 μfinalμinitial is negative (Δμ , 0). Passive
diffusion through pores or the lipid bilayer, carriers,
and channels are all passive. This does not mean that
energy is not involved. What it means is that the
energy does not derive from metabolism. The energy
comes from the solutions themselves. However, cells
also concentrate some materials by moving them from
a region of low electrochemical potential to a region of
higher electrochemical potential. This movement has
Δμ . 0. It can occur spontaneously only when the
Δμ 5 8:314 J mol21 K21 3 310 K ln½ð5 3 1023 MÞ=ð100 3 1023 MÞ
2 9:649 3 104 C mol21 3 ð2 0:080 VÞ
5 2 7:72 kJ mol21 1 7:72 kJ mol21
5 0 kJ mol21 5 0 kcal mol21
positive Δμ for transport is coupled to another process
with a more negative Δμ. Typically this process is ATP
hydrolysis.
Primary active transport moves materials against an
electrochemical gradient by the direct involvement of
ATP hydrolysis. Examples of molecules that are involved
in active transport include the ion pumps: Na,
K-ATPase, Ca-ATPase, and H-ATPase.
Secondary active transport moves materials against an
electrochemical gradient by the indirect involvement of
ATP hydrolysis. ATP is used to establish an electrochemical gradient for something, usually Na1, and the energy
stored in the electrochemical gradient for Na1 is then
used to pump material “uphill.” Examples of secondary
active transport are the Naglucose cotransport in the
intestinal epithelium and renal proximal tubule and the
NaCa exchange in the heart surface membrane.
NA,K-ATPASE IS AN EXAMPLE
OF PRIMARY ACTIVE TRANSPORT
The analysis of free energy changes on ion movement
that we performed earlier in this chapter indicated that
Active Tr ans port: Pumps a nd Exchangers
EXAMPLE 2.6.5 Calculate the Free Energy for Operation of Na,K-ATPase
Δμ
For the conditions of the cell, calculate the free energy for the
Na,K-ATPase.
We also calculated
1 1:71 kJ mol21 .
We have already calculated that Δμ for ATP hydrolysis is
257.1 kJ mol21 (see Chapter 1.7).
Inserting the values for Δμ, we get
Na1
o
going into the cell: ΔμNao -Nai 5
We have calculated Δμ for
2 14:14 kJ mol21 . This is the opposite process of what the pump
does. Thus Δμ for Na1 exit is ΔμNai -Nao 51 14:14 kJ mol21 .
the distribution of Na1, K1, and Ca21 is far from equilibrium. This implies that the cell actively maintains
these concentrations away from equilibrium, or else the
equilibrium distribution would eventually occur. The
mechanism responsible for maintaining the resting concentrations of Na1 and K1 is the NaK pump. This
pump moves three Na1 ions out of the cell at the same
time that it transports two K1 ions into the cell. The
movement of these ions is coupled to the hydrolysis of
ATP. The whole process is written as
1
1
1
½2:6:8 ATP 1 3Na1
i 1 2K o .ADP 1 Pi 1 3Nao 1 2K i
The Δμ for this entire process is equal to the sum of the
Δμ for three separate processes:
½2:6:9a
ATP.ADP 1 Pi
½2:6:9b
1
3Na1
i .3Nao
½2:6:9c
1
2K1
o .2K i
The overall free energy for this coupled process is
written as
ΔμNa;K -ATPase 5 ΔμATP.ADP1Pi 1 3ΔμNai .Nao 1 2ΔμKo .Ki
½2:6:10
for
K1
entry
ΔμKo -Ki 5
as
ΔμNa;K -ATPase 5 2 57:1 kJ mol21 1 3 3 14:14 kJ mol21
12 3 1:71 kJ mol21 5 2 11:26 kJ mol21
3Nao
E2
3Nao
P
2Ko
E2
E2
P
P
2Ko
E2
2Ko
E2
2Ki
P
Plasma membrane
E1
3Nai
E1
P
ATP
ADP
3Nai
E1
E1
ATP
3Nai
ATP 2Ki
FIGURE 2.6.2 Modified post-Albers scheme for the reaction mechanism
of Na,K-ATPase. Follow the reaction scheme in the direction of the
arrows and you will see that the net reaction is the hydrolysis of ATP
and the transport of three Na1 ions from in to out and two K1 ions from
out to in. The processes are coupled in the reaction mechanism of the
pump. The pump cannot hydrolyze ATP without binding Na1 and K1 in
sequence. Neither can Na1 be transported without K1 transport and ATP
hydrolysis. The coupling is made possible in part by the phosphorylation
of the enzyme at an aspartic acid residue. This is shown by EP in the
diagram. The formation of a phosphoenzyme is common to the P-type
active transport pumps.
The negative Δμ that we have calculated for the Na,KATPase indicates that the Na,K-ATPase reaction will
occur spontaneously, but it will not tell us at what
rate. The rate is a consequence of the mechanism of
the pump.
Recall that Δμ is the free energy per mole. In this case, it
is the free energy per mole of completed reaction with the
stoichiometry given by the overall reaction in Eqn [2.6.8].
NA,K-ATPASE FORMS A
PHOSPHORYLATED INTERMEDIATE
ΔμNa,K-ATPase 5 211.26 kJ mol21 (see Example 2.6.5)
means that the net change in free energy per mole of
reaction, not per mole of Na1 or K1 or ATP, is
211.26 kJ. This is excess free energy of ATP hydrolysis
beyond that required to transport Na1 and K1. ATP
hydrolysis has a total of 57.1 kJ of energy per mole of
ATP that can be harnessed to do work, and the Na,KATPase uses 45.84 kJ of energy per mole of reaction to
do electrochemical work, under cell conditions.
According to this result, there is enough energy in ATP
hydrolysis to drive the Na,K-ATPase reaction. These calculations hold for the resting cell under the conditions
we have investigated. The free energy for the Na,KATPase reaction changes with changes in cellular [Na1],
[K1], Δψ, [ATP], [ADP], or [Pi]. Changes in [ATP],
[ADP], or [Pi] alter the energy available to the Na,KATPase to do work. Changes in [Na1], [K1], or Δψ alter
the energy necessary for transport.
The mechanism of ion pumps is generally complicated.
It is useful to think of the enzyme as being characterized
by a limited number of conformations to which we can
give identifying labels. The reaction mechanism is then
viewed as the sequential steps that occur among these
conformations to achieve ATP hydrolysis and ion transport. Transformations between these conformations
are determined by rate constants. Each step has two
rate constants, one for the forward and the other for the
reverse reaction. A simplified scheme for Na,K-ATPase is
shown in Figure 2.6.2.
THE NA,K-ATPASE IS ELECTROGENIC
The overall reaction of the Na,K-ATPase shown in Eqn
[2.6.8] indicates the stoichiometry of 3 Na1 being transported out of the cell and 2 K1 ions being transported
into the cell. Thus the numbers of charges moving in
173
174
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
PMCA
Na,K-ATPase
3Na+
2K+
H,K-ATPase
Ca2+
H + K+
Plasma membrane
ADP + Pi ATP
ATP
3Na+2K+
ATP
Ca2+ ADP + Pi
Ca2+ ADP + Pi
ATP
H+
ATP
H+ K+
ADP + Pi
ADP + Pi
ATP
Ca2+ ADP + Pi
Vacuolar H-ATPase
SERCA
SPCA
Vesicle membrane
Ca2+
H+
Ca2+
Endoplasmic
reticulum
membrane
FIGURE 2.6.3 Some types of primary active transport mechanisms. Some are located on the plasma membrane of specific cell types; others, such as
the smooth endoplasmic reticulum Ca-ATPase (SERCA), are located on subcellular organelles. All of the primary active transporters hydrolyze ATP.
PMCA is the plasma membrane Ca-ATPase; SERCA is the smooth endoplasmic reticulum Ca-ATPase; SPCA is the secretory pathway Ca-ATPase.
the two directions are not equal, and each turnover of
the pump corresponds to the movement of one net positive charge out of the cell. Thus the operation of the
pump involves a transmembrane current and a net separation of charge. For this reason, the pump is called electrogenic—it generates an electric current and produces
an electrical potential. As we will see later, its contribution to the resting membrane potential is small, but it is
not zero.
THERE ARE MANY DIFFERENT
PRIMARY ACTIVE TRANSPORT PUMPS
There are a variety of primary active transport pumps
encoded by the human genome. These can be classified
into four major groups.
P-type ATPases: These all form phosphorylated intermediates in the pump mechanism, like that shown for
the Na,K-ATPase in Figure 2.6.2. Among these are gastric H1-ATPase that is responsible for acidification of
the stomach contents; Na1,K1-ATPase that is responsible for maintaining ionic gradients in most cells; PMCA
(for plasma membrane calcium ATPase) responsible for
pumping Ca21 out of cells; the SERCA family of pumps,
where SERCA stands for smooth endoplasmic reticulum
Ca21ATPase, which is responsible for removing Ca21
from the cytosol of a variety of cell types and placing it
in storage in internal sacs within cells; and SPCA, the
secretory pathway Ca-ATPase, which pumps Ca21 and
other ions, such as Mn21 and Zn21, into the Golgi to
bind to secretory proteins. The SPCA is distinguishable
functionally from the SERCA pumps because the SERCA
pumps are inhibited by thapsigargin at low concentrations whereas SPCA is not. Figure 2.6.3 shows some of
these primary active transporters.
V-type ATPases: Membranes of lysosomes and secretory
vesicles contain a vacuolar-type H1-ATPase that pumps
H1 ions from the cytoplasm into the vesicles. This
V-type H1-ATPase differs from the gastric H1-ATPase in
that it does not require K1. The structure and mechanism of V-type ATPases differs from the P-type active
transporters.
F-type ATPases: These are more commonly referred to as
ATP-synthetases, because they usually work in the reverse
mode to make ATP rather than hydrolyze it for the purpose
of transport. The main example in the human is the
F0F1ATPase of the inner mitochondrial membrane, which
is discussed in Chapter 2.10. It uses the electrochemical gradient of H1 to make ATP, but it can also hydrolyze ATP.
ABC transporters: The ABC here stands for “ATP-binding cassette.” This is a large family of proteins that
engage in the primary active transport of a wide variety
of solutes.
THE NACA EXCHANGER AS AN
EXAMPLE OF SECONDARY ACTIVE
TRANSPORT
According to our earlier calculation, Δμ for Ca21 entry
into the heart muscle cell was 239.6 kJ mol21. If left to
Active Tr ans port: Pumps a nd Exchangers
itself Ca21 would slowly leak into the cell and disturb
the resting [Ca21]i. Heart cells have at least two
mechanisms for pumping the Ca21 back out. One of
these, mentioned above, is a Ca-ATPase that directly
couples ATP hydrolysis to the outward transport of a
single Ca21 ion. This is the PMCA Ca21 pump. A second mechanism is called the NaCa exchanger. Its
stoichiometry, or relative mole numbers, is 3Na:1Ca.
The overall reaction is
21
1
21
3Na1
o 1 Cai .3Nai 1 Cao
½2:6:11
The Δμ for this entire process is equal to the sum of the
Δμ for two separate processes:
1
3Na1
o .3Nai
½2:6:12
21
Ca21
i .Cao
So we write
½2:6:13
ΔμNa;Ca exchange 5 3ΔμNao .Nai 1 ΔμCai .Cao
According to this result (see Example 2.6.6), there is
enough energy in the Na1 electrochemical gradient to
drive Ca21 out of the cell. However, it requires coupling
the entry of three Na1 ions for each Ca21 ion that exits
the cell. If the Na1Ca21 exchange was to couple the
movement of two Na1 to each Ca21, there would
be insufficient energy to drive Ca21 efflux, and the
exchanger would actually work in reverse mode, with
the Ca21 gradient driving the efflux of Na21.
The Ca21 efflux that occurs through the NaCa
exchanger is “uphill,” meaning that it requires energy.
Therefore, it is an active transport. The energy, however,
does not come from ATP hydrolysis directly. It comes
from the energy stored in the electrochemical gradient
of Na1. This energy, in turn, comes from the operation
of the Na,K-ATPase that establishes and maintains the
Na1 and K1 gradients across the cell membrane.
Therefore, the NaCa exchange is an example of secondary active transport. It requires energy from a
source outside of the solutes themselves. The energy
is supplied by the Na1 gradient. The Na1 gradient is
established using ATP hydrolysis.
SECONDARY ACTIVE TRANSPORT
MECHANISMS ARE SYMPORTS
OR ANTIPORTS
The NaCa exchanger, NCX, described above is an
example of an antiport. It has this description because
the two materials being transported go in opposite
directions. Such a device is also called an exchanger or
a counter-transporter. There are a variety of antiport
secondary active transport mechanisms, as summarized
in Figure 2.6.4. Thus far we have largely discussed cationic transporters, those that transport the cations.
Cations are ions that migrate toward the cathode, the
negatively charged electrode, and so cations are positively charged ions. Devices also transport anions or
negatively charged ions. An important example of these
is the Cl2HCO32 exchanger. In the red blood cell
membrane this exchanger is the AE1 protein, which
comprises a large fraction of the integral proteins of the
erythrocyte membrane. This anion transporter
exchanges Cl2 for HCO32 in the ratio of 1:1. This
EXAMPLE 2.6.6 Calculate the Free Energy for Operation of the NaCa Exchanger
We have already calculated Δμ for Na1 entry as: ΔμNao -Nai 5
2 14:14 kJ mol21
We have also calculated Δμ for Ca21 entry as ΔμCao -Cai 5
2 39:6 kJ mol21 .
ΔμNCX 5 3 3 ð2 14:14 kJ mol21 Þ 1 39:6 kJ mol21 5 2 2:8 kJ mol21
Symports (=cotransporters)
Antiports (=exchangers)
NCX
AE
Ca2+ 3Na+ HCO3– Cl–
But in this case we are dealing with Ca21exit, which has
Δμ 5 139.6 kJ mol21. The overall Δμ for the Na,Ca exchanger is
thus
NHE
H+ Na+
NIS
B0AT1 Neutral
Na+ AA
2Na+ I–
SGLT1
NKCC2
2Na+ Glucose
Na+ 2Cl– K+
2Na+ Glucose
Na+ 2Cl– K+
Furosemide
Bumetanide
Plasma membrane
Ca2+ 3Na+ HCO3– Cl–
H+ Na+
Na+ Neutral 2Na+ I–
AA
FIGURE 2.6.4 Examples of secondary active transport. NCX means “Na calcium exchanger”; AE means “anion exchanger”; NHE means “Na H
exchanger”; B0 is a specific name of a type of Naamino acid transporter, of which there are several types; NIS means “Na iodine symport”; SGLT
means “sodium glucose linked transporter.” Many of these transporters exist in multiple forms or isoforms. NKCC2 means “Na, K two chloride
cotransporter”.
175
176
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
exchanger is important, as you will see later, in helping
the erythrocytes to carry waste CO2 (see Chapter 6.4).
Other secondary active transport mechanisms transport
two materials in the same direction and therefore are
categorized as symports, also called cotransporters.
Examples include the Naglucose transporter in the
intestine and kidney membranes that transports glucose
in the lumen of these organs into the absorptive cells
lining the lumen. These cells also contain a number of
Naamino acid transporters that do the same thing as
the glucose transporter: they transport amino acids from
the lumen into the cells. The amino acids and glucose
transported in this way are then transported into the
blood by another mechanism, usually by facilitated diffusion. Some examples of symports are shown in
Figure 2.6.4.
Secondary active transporters and facilitated diffusion
proteins are classified in the family of solute carriers
(SLC). There is a wide variety of these proteins, over
300 of them, and Figure 2.6.4 shows just a sampling of
them. Appendix 2.6.A2 describes the nomenclature of
these transport proteins.
SUMMARY
The movement of ions across cell membranes involves a
change in free energy that depends on the concentration
of the ion on both sides of the membrane, the charge
on the ion, and the membrane potential. The free
energy change can be calculated as
Δμ 5 μfinal 2 μinitial
where
μ 5 μ0 1 RT ln C 1 zℑψ
where R is the gas constant 5 8.314 J mol21 K21, z is the
charge on the ion, ℑ is the faraday 5 9.649 3 104 C mol21,
and T is the temperature in Kelvin.
At rest in muscle cells, the free energy per mole (Δμ)
for Na1 entry into the cell is negative, meaning that it
occurs spontaneously. Similarly, Δμ for K1 exit is negative, whereas Δμ for Cl2 is near zero, implying that Cl2
distribution is near equilibrium. Cells maintain the Na1
and K1 gradients by actively pumping out Na1 ions
and pumping in K1 ions. This movement of ions
requires free energy that is supplied by the energy in the
terminal phosphate bond of ATP. The Na,K-ATPase couples the outward movement of three Na1 ions and the
inward movement of two K1 ions to the hydrolysis of 1
ATP molecule. The enzyme mechanism is responsible
for this coupling. Under cell conditions, the overall Δμ
for the Na,K-ATPase is negative because Δμ for ATP
hydrolysis is more negative than the combined positive
Δμ for Na1 and K1 transport “uphill.”
The Na,K-ATPase is an example of primary active transport in which the transport of ions is directly linked to
the hydrolysis of ATP. Other transporters couple the
positive Δμ for solute transport with the negative Δμ
for Na1 entry into the cell. These are secondary active
transporters, because they use energy to concentrate
materials but the energy is derived directly from the Na1
gradient and indirectly from ATP hydrolysis. Examples
of secondary active transport include the surface membrane NaCa exchanger, Naglucose cotransport, and
Naamino acid cotransport.
Transporters carrying materials in the same direction are
called symports or cotransporters. Those carrying materials in opposite directions are called antiports or
exchangers. Multiple examples of both classes occur in
the body.
Clinical Applications: SGLT2 Inhibitors and Diabetes Mellitus
Diabetes mellitus is characterized by an abnormally high plasma
glucose concentration caused by insufficient production of insulin by the pancreas. Insulin is a protein hormone that increases
glucose transporters in peripheral tissues (GLUT4) that remove
glucose from the circulation. There are two major classifications
of persons with diabetes. Those with Type 1 diabetes require
insulin injections and have little or no production of insulin, generally caused by a destruction of the beta cells in the pancreas
that produce the hormone. Persons with Type 2 diabetes generally produce insulin but the body cells are resistant to the hormone and the circulating levels of insulin are inadequate to
lower plasma glucose levels. High blood glucose causes glycosylation of proteins, as measured clinically by HbA1c, glycosylated
hemoglobin. Long-term control of plasma glucose is monitored
by the HbA1c level. Diabetes mellitus is described in more detail
in Chapter 9.4.
The kidney produces urine by filtering large volumes of blood
and then reabsorbing the desired materials and discarding the
rest. Glucose is reabsorbed in the kidney in the proximal tubule
of the nephron, the functional unit of the kidney, by SGLT2
present in the apical membrane of the tubule (see Figure 2.6.5).
Final transport of glucose into the blood occurs over GLUT2, a
facilitated transport mechanism. 90% of glucose reabsorption in
the kidney occurs via the SGLT2 glucose uptake mechanism. A
relatively new class of drugs inhibits the SGLT2 so that not all of
the filtered glucose is reabsorbed, and glucose appears in the
urine. These drugs include empagliflozin, canagliflozin, and dapagliflozin. The chemical structures of these drugs are shown in
Figure 2.6.6.
The term “diabetes” derives from the Greek meaning “to siphon”
and this refers to the increased urinary flow in diabetic persons.
This is caused by the inability of the kidney to reabsorb all of the
glucose, and the excretion of this glucose in a larger volume of
water. The effect of the SGLT2 inhibitors is to (1) lower blood
glucose levels, (2) reduce HbA1c levels, (3) generally cause a
slight loss of weight, and (4) tend to cause dehydration due to
increased urine flow. These drugs are of limited value to persons
with dysfunctional kidneys (Whalen K, Miller, S, and Onge E, The
role of sodiumglucose co-transporter 2 inhibitors in the treatment of type 2 diabetes, Clin. Therap. 37:11501166, 2015).
Active Tr ans port: Pumps a nd Exchangers
Basolateral
membrane
Apical
membrane
Lumen
Na+
SGLT2
Basement
membrane
ADP
Na+
K+
ATP
Peritubular
capillary
Na+
K+
FIGURE 2.6.5 Mechanism of reabsorption of
glucose from the ultrafiltrate in the lumen of the
nephron proximal tubule. SGLT2 transports 2 Na1
ions into the cell along with glucose. The energy is
supplied by the electrochemical gradient for Na1
that is established by pumping Na1 out of the cell
at the basolateral membrane. Glucose enters the
blood through GLUT2, a facilitated diffusion carrier
on the basolateral membrane.
Glucose
Glucose
GLUT2
–4 mV
0 mV
–7 0mV
HO
Empagliflozin
O
HO
HO
Cl
O
O
OH O
HO
Canagliflozin
HO
HO
O
CH3
OH O
F
S
HO
Dapagliflozin
HO
HO
O
Cl
OH O
O
CH3
FIGURE 2.6.6 Chemical structure of the SGLT2
inhibitors.
Clinical Applications: Oral Rehydration Therapy
One of most important public health issues in the world is the availability of clean drinking water. Contaminated water supplies carry
cholera and other infectious agents that result in diarrhea and vomiting that cause dehydration that can be fatal, especially in children.
Prior to the introduction of oral rehydration therapy (ORT), diarrhea was the leading cause of infant mortality in developing nations.
ORT is estimated to have reduced world-wide infant deaths from 5 million per year to 3 million per year (2006 figures). However, diarrhea remains the second leading cause of death in children less than 5 years old (18%, after pneumonia at 19%).
The WHO (World Health Organization) and UNICEF (United Nations Children’s Fund, shortened from the original United Nationals
International Children’s Emergency Fund) jointly publish guidelines for the composition of oral rehydration solution (ORS). Its current
formulation is: 2.6 g NaCl; 2.9 g trisodium citrate dihydrate (Na3C6H5O7∙2 H2O); 1.5 g KCl; 13.5 g glucose per L of solution.
The molar ratio of sodium to glucose in this ORS is 1.0, and it is slightly hyposmotic. It should be made with clean water, but when
clean water is not available other fluids may be substituted—but not sugar-containing fluids like fruit juices.
The effectiveness of ORT relies on the SGLT1 system in the intestine that transports glucose along with 2 Na1 ions into the enterocyte
and then into the blood. The transport of nutrients creates an osmotic reabsorption of water along with the solutes. This is enough to
counteract the loss of fluid through diarrhea or vomiting. ORT was not used until the 1960s. Before that time, rehydration was accomplished by intravenous fluid administration. In the developing world, IV therapy is not widely available and ORT is a low-technology
solution that is much more readily available.
177
178
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
REVIEW QUESTIONS
1. If a Na channel was to open on the surface of a
cardiac cell, at rest, in which direction would Na
ions travel? If the channels were K-specific, in
which direction would K ions travel? If it were
Ca21-specific, which way would Ca21 go? If it
was a Cl2 channel, which way would Cl2 ions
go?
2. Which part of Eqn [2.6.4] gives the energy due to
diffusion? Which part gives the energy due to
electrical forces? For Na1, which is bigger, the
part due to concentration differences or the part
due to electrical forces? Which part of the total
energy, concentration or electrical, is bigger for
Ca21?
3. Where does the energy come from for the movement of materials by passive transport?
4. Where does the energy come from for the movement of materials by primary active transport?
5. Where does the energy come from for the movement of materials by secondary active transport?
6. Do you think that the NCX can go in reverse?
7. Why cannot thermodynamics predict reaction
rates?
8. What is a symport? What is an antiport? What is
a cotransporter?
APPENDIX 2.6.A1 DERIVATION OF
THE USSING FLUX RATIO EQUATION
The passive flux of an ion in a solution is given by
Fick’s First Law of Diffusion with an external force,
which was introduced in Eqn [1.7.19]. The onedimensional form of this equation is reproduced here:
½2:6:A1:1
Js 5 2 D
@C
D
@ψ
2
Czℑ
@x
RT
@x
where J is the flux, in mol cm22 s21, D is the diffusion
coefficient in cm2 s21, C is the concentration (converted
to units of mol cm23), R is the gas constant
(58.314 J mol21 oK21 5 1.987 cal mol21 oK21), T is the
absolute temperature (oK), z is the integer of the charge
on the ion ( 6 1 or 2, generally), ℑ is the Faraday
(596,489 coulomb mol21) and ψ is the electrical
potential, in joules (5volt-coulomb). The quantity @C/
@x is the magnitude of the gradient of C, and @ψ/@x is
the magnitude of the electric field. The flux can be
obtained by integrating this equation. To begin, we
multiply both sides of the equation by an integrating
factor, ρ, and we choose ρ so that the right-hand side of
the equation becomes an exact differential:
@C Czℑρ @ψ
1
½2:6:A1:2
Js ρ 5 2 D ρ
@x
RT @x
We choose ρ so that the terms in brackets are an exact
differential. Thus we want
½2:6:A1:3
ρ
@C Czℑρ @ψ dðρ CÞ
@C
@ρ
1
5
5ρ
1C
@x
RT @x
dx
@x
@x
From comparing the left of Eqn [2.6.A1.3] to the right,
we see that multiplication by ρ transforms the equation
into an exact differential if
@ρ zℑρ @ψ
5
@x
RT @x
½2:6:A1:4
We rearrange this to get
@ρ
zℑ
5
@ψ
ρ
RT
½2:6:A1:5
A solution to Eqn [2.6.A1.5] is
ρ 5 eRT ψ
zℑ
½2:6:A1:6
We may insert this result back into Eqn [2.6.A1.2], with
Eqn [2.6.A1.3], to obtain
zℑ
ψ
RT
d Ce
zℑ
½2:6:A1:7
Js eRT ψ 5 2 D
dx
which may be rewritten as
zℑ
Js eRT ψ dx 5
½2:6:A1:8
zℑ
ψ
2 D d CeRT
If we are considering passive transport across a membrane, we can determine the passive flux by integrating
this equation from x 5 0 (one side of the membrane) to
x 5 δ, the other side of the membrane for a membrane
with thickness δ:
ðδ
ð@ zℑ
zℑ
ψ
ψ
RT
RT
Js e
½2:6:A1:9
dx 5 2 D δ Ce
0
0
Here we limit ourselves to the steady-state condition. In
this case, Js does not vary with distance across the membrane—it is constant. Therefore, Js may be removed
from the integral and we get
zℑ
Ð@
2 D 0 δ CeRT ψ
Js 5
½2:6:A1:10
Ð δ zℑ ψ
RT dx
0 e
The numerator in this equation is the integral of an
exact differential and can be immediately evaluated
between the boundaries. This gives
zℑ
zℑ
ψðδÞ
ψð0Þ
2 D CðδÞeRT
2 Cð0ÞeRT
Js 5
½2:6:A1:11
Ð δ zℑ ψ
RT dx
0 e
The denominator in this equation can be evaluated
only if ψ(x) is known. However, generally ψ(x) is
unknown. Ussing made the observation that the presence of an active transport mechanism would not obey
Eqn [2.6.A1.11], because the flux would not be passive,
and this equation describes the passive flux. He further
made the observation that we don’t need to know how
ψ varies with x if we take the ratio of the unidirectional
fluxes. The unidirectional flux is the flux that you would
observe if the concentration on the other side was zero.
Active Tr ans port: Pumps a nd Exchangers
We define here the unidirectional flux Ji-o to be the
flux from inside to outside, with x 5 0 on the inside
and x 5 δ on the outside. This flux is given from
Eqn [2.6.A1.11] by setting C(δ) 5 0, and we obtain
½2:6:A1:12
zℑ
eRT ψð0Þ
D Cð0Þ
Ji-o 5 Ð zℑ
δ
RT ψ dx
0 e
We further define the unidirectional flux Jo-i to be the
unidirectional flux from outside with x 5 δ to the inside,
with x 5 0. This flux is also given from Eqn [2.6.A1.11]
by setting C(0) 5 0; we obtain
½2:6:A1:13
Jo-i 5
zℑ
2 D CðδÞ eRT ψðδÞ
Ð δ zℑ ψ
eRT dx
0
Here the minus sign conveys a convention that the outward flux is taken as positive. Thus a negative flux simply means that the flux is directed inward, and a
positive flux is directed outward. The magnitude of the
fluxes is given by the absolute values of the fluxes. If we
take the ratio of the unidirectional fluxes, the denominators in each cancel each other out, and we don’t need
to do the integration that requires knowledge of ψ(x).
Taking the ratio of the two unidirectional fluxes, we
arrive at
½2:6:A1:14
zℑ
Ji-o
D Cð0Þ eRT ψð0Þ
5
zℑ
Jo-i
2 D CðδÞ eRT ψðδÞ
Using the notation that C(0) 5 C(i), the inside concentration, and C(δ) 5 C(o), the outside concentration, this
equation can be simplified to
½2:6:A1:15
Ji-o
CðiÞ zℑ ψð0Þ2ψðδÞ
eRT
52
CðoÞ
Jo-i
Using the definition that the difference in potential
across the membrane, ψ(0) 2 ψ(δ) 5 Em, the membrane
potential, this last Eqn [2.6.A1.15] becomes
½2:6:A1:16
Ji-o
CðiÞ zℑ Em
eRT
52
CðoÞ
Jo-i
This last equation is the Ussing Flux Ratio Equation.
It describes the expected ratio of the unidirectional
fluxes if the ions are transported passively across the
membrane. Deviations from the flux ratio equation are
taken to indicate that the fluxes are not transported
passively. That is, deviations from the expected flux
ratio can be taken to indicate that an active transport
mechanism is present. Ussing considered other possible deviations from the expected flux ratio such as
single-file transport.
Hans Ussing (19112000) derived his flux ratio equation in the late 1940s, soon after radioactive isotopes
became available to measure unidirectional ion fluxes.
He was the first to prove the existence of active transport mechanisms, using the frog skin as a model. The
Na,K-ATPase was later discovered, in 1957, by Jens
Skou, who earned a Nobel Prize for the discovery.
APPENDIX 2.6.A2 NOMENCLATURE
OF TRANSPORT PROTEINS
HUGO NOMENCLATURE
Proteins have “trivial” or common names that were generally first provided by their discoverers. These names
have often been changed when a new function for the
protein was discovered or its relationship to other proteins was discovered. Since these proteins derive from
genes, it is now increasingly useful to use the name for
the gene to also describe the resulting protein. The
HUGO Gene Nomenclature Committee (HGNC) provides a unique identifier for each gene. This committee
is a part of the Human Gene Organization (HUGO).
Full and updated databases are available at www.genenames.org.
CARRIER CLASSIFICATIONS
The transport proteins as described in Chapters 2.5 and
2.6 can be generally classified as belonging to a small
number of types. These are:
Passive Transporters:
Facilitated transporters
Ion channels
Water channels
Active Transporters:
Secondary active transporters: exchangers and
cotransporters
ATPase pumps (P, V, and F-types)
ABC (ATP-binding cassette) transporters.
These functional classifications do not map precisely
onto the HUGO nomenclature. The HUGO classification lumps facilitated transporters and secondary active
transporters into one group, the solute carriers, SLC.
Carriers are often grouped into families based on
sequence homology rather than functionality. An example of this is the sodiumiodine exchanger located in
the thyroid gland, which is part of the sodiumglucose
cotransport family.
SOLUTE CARRIERS
The solute carriers include the facilitated diffusional carriers such as GLUT1 and GLUT2 and the secondary
active transport carriers such as NCX, NHE, SGLT, NIS,
and AE. All members of the SLC superfamily are named
according to
SLC n X m
where SLC indicates the superfamily, n is an integer that
denotes the family, X is a letter that denotes the subfamily, and m is a second integer that denotes the isoform.
As an example, the facilitated glucose carriers are all
members of the SLC2A subfamily of which there are 14
members: SLC2A{1. . .14}. These correspond to their
common names, as shown in Table 2.6.A2.1.
There are an enormous number of these SLC genes.
There are at least 52 families (n) with a total of 396
genes (Σmi) that encode for transporter proteins. It is
179
180
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
TABLE 2.6.A2.1 Nomenclature of the Facilitated Glucose Transporters (Augustin, R. “The protein family of glucose
transport facilitators; It’s not about glucose after all.” Life 62:315333, 2010)
HUGO
Name
Common
Name
Location and Function
SLC2A1
GLUT1
Km for glucose 5 1.5 mM; ubiquitous but important for erythrocytes and uptake of glucose into the cerebrospinal
fluid (CSF)
SLC2A2
GLUT2
Km for glucose 5 17 mM; liver, kidney, intestine, β cells of pancreas; transports glucose out of intestine and
kidney cells
SLC2A3
GLUT3
Km for 2-deoxyglucose 5 1.4 mM; present in neurons, spermatozoa, placenta
SLC2A4
GLUT4
Km for glucose 5 5 mM; skeletal and cardiac muscle, adipose tissue; rate-limiting step in insulin-stimulated
glucose uptake into tissues
SLC2A5
GLUT5
Km for fructose 5 6 mM; mainly jejunum of small intestine but also found in kidney, brain, muscle and fat;
primarily a fructose transporter
SLC2A6
GLUT6
brain, spleen, and leukocytes
SLC2A7
GLUT7
Km for glucose 5 0.3 mM; apical membrane of intestine and colon; also transports fructose
SLC2A8
GLUT8
Km for glucose 5 2 mM; testis, cerebellum, liver, spleen, lung, fat; intracellular without insulin-stimulated
translocation to the surface
SLC2A9
GLUT9
Km for glucose 5 0.6 mM but also transports urate; kidney and liver and β cells of pancreas; exchanges urate for
glucose or fructose
SLC2A10
GLUT10
Present in heart, lung, brain, liver, muscle, pancreas, placenta, and kidney; may be intracellular
SLC2A11
GLUT11
Three isoforms (GLUT11{A,B,C}); in humans GLUT11 is exclusively expressed in slow-twitch muscle fibers
SLC2A12
GLUT12
SLC2A13
GLUT13; HMIT
SLC2A14
GLUT14
Does not transport sugar; H1-coupled myoinositol symporter; Km 5 0.1 mM for myoinositol; located
intracellularly; highest in brain
entirely unreasonable to attempt memorization of all of
them. The list of these and other gene families is available in tabular format on the HUGO website.
other solutes that need carrying, there are a large number
of these kinds of carriers. They belong to the SLC1, SLC3,
SLC6, SLC7, SLC36, SLC38, and SLC43 subfamilies.
Note that some designations within a family can be
confusing in that the function of the carrier is not
related to its stated family! For example, GLUT 9
(SLC2A9) is primarily regarded as a urate transporter
rather than a glucose transporter, but it is a member of
the facilitated diffusion carriers for glucose.
The anion exchanger shown in Figure 2.6.4, AE, also
has three subtypes (AE13) that correspond to
SLC4A13. These are all electroneutral Cl2 HCO32
exchangers. The SLC4 family contains 10 members
(SLC4A{1. . .5; 7. . .11} that include Na1 and HCO32 or
CO322 cotransporters.
A second example of this is found in the sodiumglucose transport family. SGLT1 and SGLT2 are part of the
SLC5A subfamily that includes 12 members (SLC5A
{1. . .12}. SLC5A1 corresponds to SGLT1 and SLC5A2
corresponds to SGLT2, but SLC5A5 corresponds to the
sodiumiodine exchanger (NIS), shown in Figure 2.6.4,
that does not transport glucose! The NIS is grouped in
this way because of structural similarities in the transport proteins that leads researchers to suppose that they
belong to the same subfamily of transporters. SGLT1 is
the main transporter in the intestine whereas SGLT2
is the main transporter in the kidney, although SGLT1 is
also found in the kidney.
The amino acid transporter shown in Figure 2.6.4 has
three members (B0AT13) that correspond to genes
SLC6A19, SLC6A15, and SLC6A18, respectively. These all
carry neutral amino acids. Because there are so many
amino acids and also a variety of neurotransmitters and
The Na1Ca21 exchanger (NCX) of Figure 2.6.4 also
has three members (NCX13) encoded by genes
SLC8A13, respectively. There is also a mitochondrial
Na1Ca21 exchanger, NCLX, as a gene product of
SLC8B1. There are additional transporters for Ca21
including NCKX (Na1Ca21K1 exchanger, SLC24
family) and CCX (Ca21-cation exchanger).
The Na1H1 exchanger (NHE) of Figure 2.6.4 has several members. NHE15 are all present on the surface
membrane, and NHE3 and NHE5 recycle between the
surface and intracellular membranes. They correspond
to SLC9A15, respectively. NHE6, NHE7, and NHE9
appear to be located in intracellular membranes along
the secretory pathway. They correspond to SLC9A6,
SLC9A7, and CLC9A9, respectively.
The NaK2Cl cotransporter shown in Figure 2.6.4
comes in two forms, NKCC1 and NKCC2, corresponding to genes SLC12A1 and SLC12A2.
Active Tr ans port: Pumps a nd Exchangers
There are a large number of other transporters of the
SLC gene superfamily that transport neurotransmitters
or ions, or vitamins, or metabolites across plasma membranes or internal membranes. A complete listing is
available on the HUGO website.
ATP-DRIVEN ION PUMPS
Names for the genes for the ATP-driven ion pumps have
the form
ATP n X m
complex. The mitochondrial F-type ATPase consists of
an F1 and an FO subunit that themselves are complexes
of additional subunits. These are encoded by the ATP5
family of genes, with subfamilies denoted by the letters
A, B, C, D, E, F, G, H, I, J, L, and O.
Somewhat differently, the V-type ATPase that acidifies
lysosome contents by pumping in H1 ions is designated ATP6V0 and ATP6V1 for the V0 and V1 subunits, and letters corresponding to the subunits within
V0 and V1.
where ATP indicates the superfamily, n is an integer that
denotes the family, X is a letter that denotes the subfamily, and m is an integer that denotes the member. Many
of the ATP-driven pumps consist of multiple subunits,
and these are generally organized by having the same
family integer, n, and a different subfamily name. For
example, the Na,K-ATPase has an α and a β subunit,
and these are indicated as ATP1A and ATP1B. There are
four varieties of each, so that the α subunit of the Na,KATPase corresponds to ATP1A{1. . .4} and the β subunit
has genes ATP1B{1. . .4}.
ABC TRANSPORTERS
The Ca21 pumps are members of the ATP2 family.
ATP2A{1,2,3} correspond to SERCA1, SERCA2, and
SERCA3, respectively, that are located on the internal
membranes of the cell, the endoplasmic reticulum, or
the sarcoplasmic reticulum. The plasma membrane CaATPases, PMCA{1. . .4} are encoded by the ATP2B
{1. . .4} genes, respectively. SPCA1 and SPCA2 are
encoded by ATP2C1 and ATP2C2. The gastric H1K1ATPase is encoded by ATP4A (the α subunit) and
ATP4B (the β subunit}. All of the above-mentioned
pumps constitute the P-class of ATP-driven ion pumps.
AQUAPORINS
The F-type and V-type ATPase ion pumps are much
more complex, consisting of multiple subunits and multiple copies of some of these subunits. Therefore, there
is no one gene that encodes the entire operating
ABC transporters hydrolyze ATP to transport a wide
variety of substrates. There are 48 transporters classified
in 7 families denoted in this case by letters alone:
ABC X m
where ABC denotes “ATP-binding cassette”, X is a letter
indicating the family, and m is an integer indicating the
member.
Aquaporins are proteins that increase water movement
across biological membranes. They have a molecular
weight of around 30 kDa and associate as tetramers,
although each monomer has a water channel. Several of
the aquaporins will transport other small molecular
weight, electrically neutral substrates such as glycerol or
urea. There are a variety of aquaporins named AQP n
where n is the member of the family. There are 14
members of the family, named AQP{1. . .12}. AQP0 has
been renamed MIP for “major intrinsic protein” of the
lens, and AQP12 has A and B subtypes. In the HUGO
classification, aquaporins are considered to be a subtype
of ion channels.
181
2.7
Osmosis and Osmotic Pressure
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
G
G
G
Define osmosis and osmotic pressure
Write van’t Hoff’s limiting law for osmotic pressure dependent on concentration
List the colligative properties of solutions and explain why
vapor pressure depression perfectly predicts osmotic pressure
Describe the osmotic coefficient for correction of nonideality in solutions
Be able to calculate the predicted osmotic pressure for a
solution
Define hydraulic conductivity or hydraulic permeability
Define the reflection coefficient
Explain how the hydraulic conductivity depends on the
microscopic parameters of the membrane
Write an equation for volume flow across a microporous
membrane in the presence of hydraulic and osmotic
pressures
Be able to calculate the hydraulic conductivity and reflection coefficient given appropriate data
Describe the origin of the osmotic pressure for microporous
membranes
Describe how erythrocyte cell volume changes when
placed in contact with solutions of varying [NaCl] or
[glucose]
Contrast the concepts of tonicity and osmolarity
Describe the behavior of a perfect osmometer
Explain why cells are not perfect osmometers
Define RVD and RVI
OSMOSIS IS THE FLOW OF WATER
DRIVEN BY SOLUTE CONCENTRATION
DIFFERENCES
Probably no concept is more confusing to beginning
students than osmosis and osmotic pressure, partly
because it is defined backwards, as you will see.
We begin with the experiments of Pfeffer in 1877.
Pfeffer made a precipitation membrane in the walls
of unglazed porcelain cups by reacting copper salts
with potassium ferricyanide. He used the precipitation
membrane that resulted to separate a sucrose solution
on the inside of the cup from water on the outside.
182 He observed that water flowed from the outside to the
inside. This is the primary observation of osmosis.
Osmosis refers to the movement of fluid across a
membrane in response to differing concentrations of
solute on the two sides of the membrane. The word
“osmosis” originates from the Greek, meaning “thrust”
or “impulse.”
Pfeffer also observed that the flow was proportional to
the sucrose concentration inside the cup. When water
was inside the cup, he observed that a pressure applied
to the inside compartment would force water out of the
cup, and this flow was proportional to the pressure.
When sucrose was inside a closed cup, a pressure
would develop inside the cup and this pressure was
proportional to the sucrose concentration. He recognized
this as an equilibrium state in which the outward filtration of water balanced the inward movement caused
by osmosis. He defined the osmotic pressure as the pressure necessary to stop osmotic flow across a barrier
that is impermeable to the solute. The osmotic pressure
historically is given the symbol π (see Figure 2.7.1).
This definition is the key: it defines the osmotic pressure
as the pressure needed to stop osmotic flow rather
than the pressure that drives osmotic flow. It is also
defined only for a semipermeable membrane, one that
is impermeable to solute but permeable to water.
THE VAN’T HOFF EQUATION
RELATES OSMOTIC PRESSURE
TO CONCENTRATION
Pfeffer’s data (see Figure 2.7.2) showed that the osmotic
pressure of solutions was linearly related to their concentration. In 1887, van’t Hoff argued from Pfeffer’s
results, and from thought experiments considering gases
in equilibrium with solutions, that the osmotic pressure
should be given by
½2:7:1
π 5 RTCs
where R is the gas constant (0.082 L atm mol21 K21),
T is the absolute temperature, and C is the molar
concentration of solute in the inner compartment.
Equation [2.7.1] is known as van’t Hoff’s Law. It gives
the osmotic pressure due to the solute, s. In a mixture,
the osmotic pressures due to each solute particle add
up, much like Dalton’s Law of Partial Pressures in gases.
The result is that
X
½2:7:2
π 5 RT
Cs
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00017-3
Pressure-driven flow: ΔP > 0
Osmotic flow: ΔP = 0
Qv = A LpΔP
Qv = -A LpΔπ
Osmotic pressure: Qv = 0
ΔP = Δπ
Qv = A Lp(ΔP - Δπ)
Applied pressure, PL
Applied pressure, PL
Piston
Piston
Piston
Pure water
Pure water
Pure water
Pure water
Solution
Solution
Semipermeable membrane
Semipermeable membrane
Semipermeable membrane
FIGURE 2.7.1 Equivalence of hydrostatic and osmotic pressures in driving fluid flow across a membrane. Left panel: An ideal, semipermeable
membrane is freely permeable to water but is impermeable to solute. When the membrane separates pure water on the right from pure water on
the left, application of a pressure, PL, to the left compartment forces water across the semipermeable membrane. The flow is linearly related to the
pressure difference by the area of the membrane (A) and a proportionality constant, LP, that is characteristic of the membrane. This constant is
variously called the hydraulic conductivity, hydraulic permeability, or filtration coefficient. Positive QV is taken as flow to the right. Middle panel:
The ideal, semipermeable membrane separates a solution on the left from pure water on the right, and water moves to the solution side by osmosis.
The flow, QV, is linearly related to the difference in osmotic pressure, Δπ, by the area of the membrane and the hydraulic conductivity, LP. The flow
causes expansion of the left compartment and movement of the piston, which is assumed here to be weightless. Right panel: Application of a
pressure, PL, to a solution so that ΔP 5 Δπ results in no net flow across the membrane. The osmotic pressure of a solution is defined as the pressure
necessary to stop fluid flow when an ideal semipermeable membrane separates pure water from the solution.
3500
Pressure (mmHg)
3000
2500
2000
1500
1000
500
0
0
1
2
3
4
5
6
7
Sucrose concentration (%w/w)
FIGURE 2.7.2 Plot of the data of Pfeffer (1877) for the osmotic pressure of sucrose solutions. A copper ferrocyanide precipitation membrane was
formed in the walls of an unglazed porcelain cup. The membrane separated a sucrose solution in the inner chamber from water outside the cup. The
inner chamber was then attached to a manometer and sealed. The linear relationship between the pressure measured with this device and the
sucrose concentration was the experimental impetus for deriving van’t Hoff’s Law. [Data from Pfeffer, W. Osmotische Untersuchungen Studien zur
Zellmechanik, Leipzig [translated by G.R. Kepner and E.J. Tadelmann: Osmotic investigations: Studies on cell membranes, 1985, Van Nostrand Reinhold, New York].
184
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
The concentration in van’t Hoff’s law, ΣCs, refers to the
concentration of osmotically active solute particles in
the solution. Organic compounds such as glucose typically dissolve to form one particle for each molecule of
solute, and for these compounds Cs is the same as the
molar concentration. Strong salts, on the other hand,
dissociate to form more than one particle for each
mole of salt. NaCl, for example, dissociates in solution
to form one Na1 and one Cl2 ion. The concentration of
osmotically active particles is twice the concentration
of NaCl. Similarly, CaCl2 dissociates nearly completely to
form one Ca21 ion and two Cl2 ions, and the total
concentration of particles is 3 times the concentration of
CaCl2. The osmolarity of a solution equals ΣCs and is
expressed in osmoles per liter to indicate clearly that we
are referring to the number of osmotically active solute
particles, called osmolytes, rather than the concentration
of the solute.
of work such as pressurevolume work. The general
equation for the chemical potential is
THERMODYNAMIC DERIVATION
OF VAN’T HOFF’S LAW
½2:7:5
We have learned that all spontaneous processes (those
that occur naturally without any additional forces)
are accompanied by a decrease in free energy. We have
also learned that the free energy per mole is the chemical potential (see Chapter 1.7). In the case of a solution
separated from pure water by a semipermeable membrane, water movement will occur when there is a
difference in the chemical potential of water on the two
sides of the membrane, such that water movement
results in a decrease in the free energy. At equilibrium,
when the pressure applied to the solution is equal to
the osmotic pressure, the chemical potential of water
is equal on both sides of the membrane, and no net
movement of water occurs.
In the derivation that follows, we consider that a
semipermeable membrane separates two compartments. On the right is pure water and on the left is
some solution with concentration Cs. We know that
there will be flow of water from the pure water to the
solution side and that application of pressure, π, to
the solution side will stop the flow. What is the
relation between π and Cs? We can discover this by
looking at the equilibrium condition when osmotic
pressure balances hydrostatic pressure. At this point,
the free energy change for water across the membrane
is zero.
THE CHEMICAL POTENTIAL INCLUDES
PRESSUREVOLUME WORK
The equation we have used for chemical potential of a
solute, the free energy per mole, is
½2:7:3
μs 5 μ0s
1 RT ln Cs 1 z`ψ
In this equation, z`ψ represents the work done, per
mole, in moving the material from the standard state to
the condition in which the material is placed. In this
case, it is the electrical work. There are other kinds
½2:7:4
μs 5 μ0s 1 RT ln Cs 1 work terms
where the work terms include all work (except concentration work, which is included explicitly in RT ln Cs) necessary to bring the material from the standard state to its
present state.
THE ACTIVITY CORRECTS THE CHEMICAL
POTENTIAL FOR INTERACTIONS BETWEEN
SOLUTE PARTICLES
It turns out that Eqn [2.7.4] is an approximation. In our
derivation of the general Fick’s Law, we did
not consider some other kinds of interactions, such as
solute molecules bumping into each other. The accurate
equation is
μs 5 μ0s 1 RT ln as 1 work terms
where as is the activity of the solute. In the case of
osmosis, the work term is the pressurevolume work
and there is no electrical work term. At equilibrium,
where the pressure across the semipermeable membrane
is the osmotic pressure, the chemical potential of water
on the two sides of the semipermeable membrane must
be equal (because the free energy change at equilibrium
is zero). Therefore, we write the equality of chemical
potential for water on the left and right sides as
μ0w 1 RT ln aw;L 1 V w PL 5 μ0w 1 RT ln aw;R 1 V w PR
½2:7:6
where the subscripts L and R refer to the left and
right sides of the semipermeable membrane, μw0 is the
chemical potential of liquid water in its standard state
(pure water at 1 atm pressure), Vw is the volume of
water per mole (the partial molar volume), P is the
pressure, and aw is the activity of water. For an ideal
solution, the activity of water is its mole fraction:
½2:7:7
aw 5 Xw 5
nw
nw 1 ns
where Xw is the usual variable denoting the mole
fraction of water, and nw and ns are the moles of water
and solute, respectively, in any aliquot of the solution.
Substituting in for aw and canceling the μw0 on both
sides of Eqn [2.7.6], we come to
½2:7:8
V w PL 2 V w PR 5 RT ln Xw;R 2 RT ln Xw;L
Since the right-hand solution is pure water, Xw,R 5 1.0
and ln Xw,R 5 0. Thus we have
½2:7:9
V w ðPL 2 PR Þ 5 2 RT ln Xw;L
Now the mole fractions of solute and water in a
solution must sum to 1.0. This is expressed as
½2:7:10
Xw;L 1 Xs;L 5 1:0
ln Xw;L 5 lnð1 2 Xs;L Þ
Os mos is an d Os motic P res sur e
In dilute solutions, Xs,L ,, 1.0, so we may approximate
ln(1 2 Xs,L) 2Xs,L. Substitution of this result into
Eqn (2.7.9) gives
½2:7:11
ðPL 2 PR Þ 5
RT
Xs;L
Vw
The left-hand side of Eqn [2.7.11] is just the osmotic
pressure, π, which is equal to the extra pressure that
must be applied to the left-hand side in order to establish equality of the chemical potential of water on the
two sides of the membrane. From the definition of
mole fraction, Eqn [2.7.11] becomes
½2:7:12
ðPL 2 PR Þ 5 π 5
π5
Water
Solution
Water
RT ns
V w nw
Again for a dilute solution, nwVw V, the volume of the
solution. Thus Eqn [2.7.13] gives
½2:7:14
Solution
RT ns
V w ns 1 nw
For a dilute solution, ns ,, nw, so we approximate this
result as
½2:7:13
Water vapor
ns
π 5 RT
V
Air
This last equation is the van’t Hoff equation for the
osmotic pressure. This thermodynamic derivation
entails two assumptions: the solution is dilute enough
to approach ideality and that the solution is incompressible so that the pressurevolume work is VwΔP.
It is important to recognize that the van’t Hoff equation
is not exact for physiological solutions. Rather, it is
an approximation that is strictly true only for dilute
ideal solutions.
FIGURE 2.7.3 Equivalence of vapor pressure and osmotic pressure.
Two beakers containing a solution or pure water are both placed in
a single, sealed compartment. The air above the fluids contains air
molecules plus water vapor. The partial pressure of water is its
contribution to the total pressure, and it is proportional to the water
concentration in the gas phase. The vapor pressure is defined as the
partial pressure of water in equilibrium with the liquid phase. Water
molecules will leave the liquid to moisten a dry gas. At equilibrium,
molecules will evaporate from the liquid phase and condense from the
gas phase at equal rates, so that a dynamic equilibrium is established.
The vapor pressure of pure water is higher than the vapor pressure of
the solution. This vapor pressure depression is one of the colligative
properties of solutions. Because there is only one vapor pressure, it
cannot simultaneously be in equilibrium with the water and with the
solution. Thus water from the pure water beaker will continue to
evaporate, because its vapor pressure is higher than that in the air, and
water will continue to condense into the solution, because the partial
pressure of water is higher than the vapor pressure of the solution.
OSMOTIC PRESSURE IS A PROPERTY
OF SOLUTIONS RELATED TO OTHER
COLLIGATIVE PROPERTIES
Thus osmotic pressure and vapor pressure depression
are perfect predictors of each other because essentially
they are the same phenomenon.
or
½2:7:15
π 5 RTCs
Osmotic pressure is closely related to some other
properties of solutions, the colligative properties.
These include the freezing point depression, the
boiling point elevation, and the vapor pressure
depression, all caused by dissolving solutes in a
solution. The osmolarity is often determined from
vapor pressure depression or freezing point depression,
rather than from direct osmotic pressure measurements. The osmolarity is the concentration necessary
to observe these phenomenon.
To see the connection between osmotic pressure
and vapor pressure depression, consider Figure 2.7.3.
A solution placed in a sealed container with a source of
pure water will gain water because its vapor pressure is
lower than that of the water. This situation is formally
equivalent to osmosis, where the semipermeable membrane is the intervening air between the two surfaces.
THE OSMOTIC COEFFICIENT ϕ
CORRECTS FOR THE ASSUMPTION
OF DILUTE SOLUTION AND FOR
NONIDEAL BEHAVIOR
As noted above, the van’t Hoff equation makes two
assumptions: the solution is dilute and it is ideal. The
assumption of ideality enters when we equate the activity
of water with its mole fraction. The assumption of dilute
solutions allows us to identify ln(1 2 Xs) with 2 Xs.
We can correct for both assumptions by identifying
πobserved
ϕ5
½2:7:16
RTCs
Here ϕ is the osmotic coefficient. The osmotic
coefficient can be less than or greater than 1.0.
185
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
THE RATIONAL OSMOTIC COEFFICIENT
CORRECTS FOR THE ASSUMPTION
OF IDEALITY
3.5
Equation [2.7.9] gives the osmotic pressure in terms of
the mole fraction of water:
2.5
½2:7:17
π52
RT
ln Xw
Vw
But the true equation for osmotic pressure is given by
the manipulation of Eqn [2.7.6] as
½2:7:18
π52
3.0
π observed / π calculated
186
π52
RT
RT
p
ln aw 5 2
ln 0
Vw
Vw p
Figure 2.7.4 shows the ratio of the observed osmotic
pressure to that calculated by Eqn [2.7.15], Eqn
[2.7.17], or Eqn [2.7.21]. These give an estimate of ϕ
and g. The approximate values of ϕ at physiological
concentrations for a variety of common solutes are
given in Table 2.7.1. Units used in the calculation of
osmotic pressure and appropriate values for R are given
in Table 2.7.2.
EQUIVALENCE OF OSMOTIC
AND HYDROSTATIC PRESSURES
As mentioned earlier, Pfeffer observed a linear relationship between the flow across the membrane and the
pressure when water was on both sides of the membrane. This relationship can be described as
½2:7:22
g
1.5
1.0
0.5
0.0
0.0
0.5
g ln Xw 5 ln aw
The value of g can be calculated from Eqns [2.7.17] and
[2.7.19] as
πobserved
g5
½2:7:20
RT
2
ln Xw
Vw
Thus the rational osmotic coefficient corrects for the discrepancy between the osmotic pressure and the osmotic
pressure calculated from the mole fraction of water.
This assumes ideality, in which the activity is equal to
the mole fraction, but not dilution. Thus the calculations of osmotic pressure based on Eqn [2.7.17] are better than those calculated using van’t Hoff’s Law because
calculations based on Eqn [2.7.17] are valid even for
solutions that are not dilute. However, Eqn [2.7.17] still
requires the assumption of ideal solution behavior, or
that the activity of water is equal to its mole fraction.
Eqn [2.7.18] gives the osmotic pressure without assuming either a dilute solution or ideality. It can be calculated from vapor pressure measurements as
½2:7:21
2.0
πobserved / (-RT/VW ln p/p0)
RT
ln aw
Vw
We introduce the rational osmotic coefficient, g, to
make these two equations give the same result:
½2:7:19
φ
QV 5 A LP ðPL 2 PR Þ 5 ALP ΔP
JV 5 LP ðPL 2 PR Þ 5 LP ΔP
1.0
1.5
2.0
Sucrose concentration (M)
2.5
3.0
FIGURE 2.7.4 Osmotic coefficients as a function of sucrose
concentration. The osmotic coefficient, ϕ, was calculated according to
Eqn [2.7.16] by dividing the observed osmotic pressure by RTC (circles).
The rational osmotic coefficient was obtained according to Eqn [2.7.20]
by dividing the observed osmotic pressure by 2 RT/VW ln XW (squares).
The correlation of vapor pressure to osmotic pressure was tested by
dividing the observed osmotic pressure by 2 RT/VW ln p/p0 (triangles).
This figure shows that the van’t Hoff equation is good for dilute
solutions but fails at high solute concentrations, due to the failure of the
assumptions that solutions are dilute and ideal. The osmotic coefficient
corrects for these failures. The rational osmotic coefficient deviates
significantly at higher sucrose solutions where solution behavior is
further from ideal. The ratio of the observed osmotic pressure to that
calculated from vapor pressure measurements is close to 1.0 over the
entire concentration range. Data from Glasstone, S. Textbook of Physical
Chemistry. Princeton: Van Nostrand, 1946.
TABLE 2.7.1 Approximate Values of the Osmotic
Coefficient for Common Solutes Under Physiological
Conditions
Solute
Number of
Particles Formed
upon Solution
Molecular
Weight
(g mol21)
NaCl
2
58.4
0.93
KCl
2
74.6
0.92
Osmotic
Coefficient
(ϕ)
CaCl2
3
111.0
0.85
Na2SO4
3
142.0
0.74
MgCl2
3
95.2
0.89
MgSO4
2
120.4
0.58
NaHCO3
2
84.0
0.96
Alanine
1
89.1
1.00
Mannitol 1
182.2
1.00
Glucose
1
180.2
1.01
Sucrose
1
342.3
1.02
where QV is the flow in units of volume per unit time
and JV is the flux, or flow per unit area, in units of
velocity. Here the positive flow is taken from left to
right and pressure drives the flow. Thus if PL . PR, then
Os mos is an d Os motic P res sur e
TABLE 2.7.2 Units for the Calculation of Osmotic Pressure
Pressure Units
1 atm Equivalent
Gas Constant (R)
Solute Osmolyte Concentration (ΣCs)a
atm
1
0.082 L atm mol21 K21
mol L21
mm Hg
760
62.36 L mm Hg mol21 K21
mol L21
Pa 5 N m22
22
dyne cm
1.013 3 105
8.314 N m mol21 K21
1.013 3 10
8.314 3 10 dyne cm mol
6
7
mol m23 5 mol (1000 L)21
21
21
K
mol cm23
a
Osmolarity (osmol L21) is defined as the concentration of osmotically active particles, osmolytes, in mol L21. Therefore, the units osmoles and moles cancel in the
calculation of osmotic pressure.
Example 2.7.1 Calculate the Osmotic Pressure of 0.9% NaCl
0.9% NaCl means 0.9 g per 100 mL of solution. This is 9 g L21.
We can convert this to molarity by dividing by the molecular
weight, 58.4 g mol21: [NaCl] 5 9 g L21/58.4 g mol21 5 0.154 M.
The osmotic pressure is calculated as π 5 RT ϕC 5
0.082 L atm mol21 K21 3 310 K 3 0.2866 mol L21 5 7.29 atm.
This is an enormous pressure on the physiological scale.
The effective osmolarity of this solution is 2 osmol mol21 3
0.154 M 3 0.93 5 0.2866 osmol L21.
QV is positive and flow is to the right. If PL , PR, then
(PL 2 PR) is negative, QV is negative and flow is to the
left. Lp is a coefficient characteristic of the membrane,
variously called the hydraulic conductivity, hydraulic
permeability, or filtration coefficient.
When Pfeffer added impermeant solutes to the inner
chamber (left chamber), flow was observed into the
solution in the absence of any macroscopic hydrostatic
pressure differences. That is, when ΔP 5 0 there was a
negative flow, and that flow was proportional to the
osmotic pressure. Thus it appears that the solute caused
a reduction in the pressure on the solution side because
the flow is inward and additional pressure on the solution side, the osmotic pressure, is necessary to stop the
osmotic flow. The osmotic pressure, which is characteristic of the solution, is equivalent to a reduction in the
hydrostatic pressure. If solution is present on both sides
of the membrane, we write
½2:7:23
QV 5 A LP ½ðPL 2 πL Þ 2 ðPR 2 πR Þ
QV 5 A LP ½ðPL 2 PR Þ 2 ðπL 2 πR Þ
QV 5 A LP ðΔP 2 ΔπÞ
THE REFLECTION COEFFICIENT
CORRECTS VAN’T HOFF’S EQUATION
FOR PERMEABLE SOLUTES
When membranes are partially permeable to the solute
(leaky membranes), the measured osmotic pressure is
less than that predicted by van’t Hoff Law. This has led
to the definition of another membrane parameter, σ,
the reflection coefficient, which is defined as
πeff
½2:7:24
σ5
ϕRTC
where πeff is the effective or observed osmotic pressure
and ϕRTC is the theoretical osmotic pressure that would
be observed if the membrane was perfectly semipermeable. The reflection coefficient is different for each pair
of solute and membrane. Its range is 0 # σ # 1.0.
Rewriting Eqn [2.7.24], we have
½2:7:25
πeff 5 σϕRTC 5 σπ
It is important to note that the osmotic pressure is a
characteristic of the solution, and it is defined as the
pressure necessary to stop osmotic flow when a solution
is separated by an ideal, perfectly semipermeable membrane from pure water. Recall that a semipermeable
membrane is defined as one that has zero permeability
to solute. Every aqueous solution has an osmotic
pressure—it is a concentration measure like molarity
is—and it can be measured by any of the colligative
properties. When a solution is placed in contact with
a membrane that is not semipermeable, its osmotic
pressure will be reduced by the membrane’s reflection
coefficient, which is characteristic not only of the membrane but also of the solute. As a consequence of this,
flow across a real membrane that is not semipermeable
will be altered. It is governed by the equation
½2:7:26
QV 5 A LP ½ðPL 2 ΣL σi πi Þ 2 ðPR 2 ΣR σi πi Þ
QV 5 A LP ðΔP 2 ΣΔσi πi Þ
where the summation sign means that all osmotically
active solutes in the solution contribute to the total
osmotic pressure.
We now have three phenomenological coefficients that
describe volume and solute flux across a membrane.
These are summarized in Table 2.7.3. The question
before us now is: what is the physical meaning of LP
and σ?
187
188
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Example 2.7.2 Calculate the Net Driving Force and Flow Across a Dialysis Membrane.
A semipermeable dialysis membrane has an area of 90.5 cm2
and Lp 5 6.46 3 1026 cm min21 mmHg21. Inside the dialysis
membrane was a solution of 5% (w/v) sucrose at a pressure of
10 mmHg. Outside was a solution of 2% sucrose at a pressure of
50 mmHg. The entire apparatus was equilibrated to room temperature, 20 C. What is the net pressure across the membrane?
What is the net flow across the membrane? Assume ϕ for
sucrose is 1.0. The molecular weight of sucrose is 342 g mol21.
First, we calculate the osmotic pressures of the solutions. 5%
sucrose means 5 g of sucrose per 100 mL of solution or 50 g L21.
We can convert this to molarity by dividing by the molecular
weight, 342 g mol21:
½sucrosein 5 50 g L21 =342g mol21 5 0:146 M:
The molar concentration of 2% sucrose is similarly calculated as
½sucroseout 5 20 g L21 =342g mol21 5 0:058 M
The osmotic pressure of the two solutions is calculated as
πout 5 ϕ RT 3 0:058 M
5 62:36 L mmHg mol21 K21 3 293 K 3 0:058 mol L21
5 1060 mmHg
The net driving force is calculated as
ðPin 2 Pout Þ 2 ðπin 2 πout Þ 5 ð10 mmHg 2 50 mmHgÞ
2 ð2668 mmHg 2 1060 mmHgÞ
5 2 1648 mmHg
Positive pressure would force fluid outward; negative net pressure means flow is inward. Note that the systematic insertion of
the pressure values is essential to obtaining the correct result.
The inward flow is given by
QV 5 A LpðΔP 2 ΔπÞ
5 90:5 cm2 3 6:46 3 1026 cm min21 mmHg21 3 2 1648 mmHg
5 2 0:963 cm3 min21 :
πin 5 ϕ RT 3 0:146 M
5 62:36 L mmHg mol21 K21 3 293 K 3 0:146 mol L21
5 2668 mmHg
Pore
TABLE 2.7.3 Phenomenological Membrane
Coefficients
Coefficient Parameter
Calculated as
p
Permeability
( Js/ΔC)Jv50
Lp
Hydraulic
conductivity
( Jv/ΔP)Δπ50 ;
2 ( Jv/σΔπ)ΔP50
σ
Reflection coefficient 2( Jv/LpΔπ)ΔP50
δ
n = N pores/unit area
LP FOR A MICROPOROUS MEMBRANE
DEPENDS ON THE MICROSCOPIC
CHARACTERISTICS OF THE MEMBRANE
Here we consider fluid flow across a microporous
membrane such as that shown in Figure 2.7.5. We
consider that the membrane itself is impermeant to
water and solute, but both may go through tiny pores
in the membrane. We consider three cases:
1. There is a pressure difference across the membrane but either there is water on both sides of
the membrane or the solute particles are very
small compared to the size of the pore. Pressuredriven flow will occur.
2. There is no pressure difference across the membrane that separates water from solution, but
the solutes on one side of the membrane are
FIGURE 2.7.5 Schematic drawing of the hypothetical microporous
membrane. We imagine that this membrane is a thin, flat sheet that
is pierced by right cylindrical pores of radius a and depth δ, which is
equivalent to the thickness of the membrane. There are n 5 N/A
pores per unit area of the membrane. The membrane separates two
aqueous solutions with a solute concentration CL on the left and CR on
the right. We further suppose that the left chamber is subject to the
pressure PL and the right side to the pressure PR.
too large to fit through the pores. This will
produce an osmotic pressure difference and an
osmotic flow.
3. There is no pressure difference across the membrane that separates water from solution, but the
solutes are small enough to fit through the pore,
but not as easily as water. This will produce a
smaller osmotic pressure and a smaller osmotic
flow than in case 2.
Os mos is an d Os motic P res sur e
CASE 1: THE SOLUTE IS VERY SMALL
COMPARED TO THE PORE
The volume flow through the membrane is the number
of pores times the volume flow per pore:
½2:7:27
Qv 5 N qv
where QV signifies the overall observed macroscopic
flow, N is the number of pores, and qv is the flow
through a single pore. If we assume laminar flow (see
Chapter 1.2), then the flow through each pore will be
given by Poiseuille’s Law, which gives the flow through
a right cylindrical pipe of radius a and length δ as
½2:7:28
qv 5
πa4
ΔP
8ηδ
where η is the viscosity of the fluid. The use of
Poiseuille’s law in this situation requires the assumptions of laminar flow, Newtonian fluids, and a pore
length that is long compared to the entrance length of
the pore. The entrance length is the distance it takes
for the fluid to establish a parabolic velocity profile
within the pore. The observed macroscopic flux, Jv, is
the flow per unit area of membrane per unit time.
Substituting Poiseuille’s law into Eqn [2.7.27] gives the
volume flux as
½2:7:29
Jv 5
Nqv
nπa4
5 nqv 5
ΔP
A
8ηδ
where n 5 N/A is the density of pores in the membrane 2
the number of pores per unit area. Comparing this to
Eqn [2.7.22], JV 5 LP ΔP, we can identify
½2:7:30
Lp 5
nπa4
8ηδ
According to this equation, the observed macroscopic
flux is linearly related to the pressure difference by
a coefficient which includes the thickness of the
membrane, δ, the density of pores in the membrane, n,
the radius of the pores, a, and the viscosity of the fluid,
η. Lp has units of volume per unit time per unit area per
unit pressure.
CASE 2: THE SOLUTE IS LARGER THAN
THE PORE: THE MECHANISM
OF OSMOSIS FOR MICROPOROUS
MEMBRANES
If the solute particles are larger than the pore, then they
cannot get through the membrane and the membrane is
perfectly semipermeable. Pfeffer has already experimentally determined what happens in this case, and his
experimental results permit several conclusions. First,
the solute in the water causes the flow because there is
no flow when pure water is on both sides of the membrane. Second, the pressure at equilibrium or the flow is
directly proportional to the concentration of the solute,
if the solutions are sufficiently dilute. Third, the solute
causes a reduction in the pressure of the solution
because the flow is inward and additional pressure on
the solution side is necessary to stop the osmotic flow.
Fourth, because the observed osmotic pressure obeys
van’t Hoff’s Law only when the membrane is impermeable to solute, the osmotic pressure and the osmotic
flow result from the interaction of the solute with the
membrane. If the membrane is highly permeable to
solute also, no osmosis and no osmotic pressure are
observed. We should look to the interaction of the
membrane with the solute to explain the mechanism of
osmosis. The derivation for one mechanism of osmotic
pressure flow and pressure generation is given in
Appendix 2.7.A1. Briefly, this derivation recognizes that
the total pressure in the bulk solution results from contributions of both solute and solvent molecules.
However, solute molecules cannot enter the pores and
so they cannot contribute to the pressure within the
pore. Thus there is a pressure deficit on the solution
side immediately upon entering the pore from that side.
Since pressure results from collisions of molecules,
resulting in momentum change in the molecules, there
is a momentum deficit within the pores due to restricted
entry of solute particles into the pores.
In Case 2, we have the situation where PL 5 PR, CL 5 CL,
and CR 5 0 for a membrane which is impermeant to
solute. From Eqn [2.7.A1.9], the pressure immediately
within the pore, PL’, is given by
½2:7:31
P 0L 5 PR 2 RTCL
assuming the validity of van’t Hoff’s law. A plug of
water extending the length of the pore is subjected to a
pressure difference, which is given by
½2:7:32
P0L 2 PR 5 2 RTCL
because, in this case, PL 5 PR. The negative sign indicates
the pressure is higher on the right than on the left. Thus
fluid flow should occur from right to left according to
Poiseuille’s law:
½2:7:33
qv 5
πa4
ð2πÞ
8ηδ
where qv is the flow per pore. In this equation, we see
the absurd situation in which one symbol is used to
signify two entirely different quantities. The π in the
fraction is the geometric ratio, while the π in parenthesis signifies the osmotic pressure. The negative sign
indicates flow to the left. The overall volume flux for
the membrane is then given as
½2:7:34
Jv 5 N
qv
nπa4
5
ð2πÞ
A
8ηδ
It is clear, then, that Jv in the presence of osmotic flow
is given by
½2:7:35
Jv 5 2 L p π
where Lp is identical to that described earlier for
pressure-driven flow (see Eqn [2.7.30]).
189
190
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
PL > PR + RTCL
PL = PR + RTCL
Membrane
CL
CL
PL
Membrane
CL
RTCL
PR
RTCL
RTCL
Membrane
PR
PR
Solute particles
qv < 0
Js = 0
CR = 0
x=0 x=δ
qv = 0
qv > 0
CR = 0
CR = 0
x=0 x=δ
x=0 x=δ
FIGURE 2.7.6 The direction of flow in the presence of hydrostatic and osmotic pressure differences across the membrane. Far left: The hydrostatic
pressure is the same on both sides of the membrane but the left side contains a solution of concentration CL. In the pore, there is a pressure gradient
and flow is toward the solution. Middle: The pressure on the left was increased by RTCL 5 π, the osmotic pressure. This is the equilibrium situation and
no flow occurs. Right: Pressure on the left is more than RTCL greater than pressure on the right, and flow is to the right.
It is instructive to consider the pressure profiles within
the pore which are established due to the osmotic pressure. Poiseuille’s Law is given as
πa4 ΔP
½2:7:36
Qv 5
8η ΔX
Since @Qv/@x 5 0 at steady state, it follows that ΔP/Δx
must be a constant in x and in t: the pressure gradient is
linear in x. This allows us to draw the schematic diagrams of pressure and concentration gradients during
osmotic flow as shown in Figure 2.7.6.
CASE 3: THE REFLECTION COEFFICIENT
RESULTS FROM PARTIALLY
RESTRICTED ENTRY OF SOLUTES
INTO THE PORES
When the membrane is freely permeable to the
solute (Case 1), there is no osmosis and no osmotic
pressure and hydrostatic pressure drives fluid flow.
When the membrane is completely impermeable to the
solute (Case 2), and there is no hydrostatic pressure,
the osmotic pressure is ideally given by van’t Hoff’s Law
and drives fluid flow. Here we consider Case 3, a membrane which distinguishes between solute and solvent
but which is not completely impermeable to solute.
Here we consider pores that are large compared to the
solvent particles but they partially exclude solute particles. The partial exclusion is due to the fact that the
effective area of the pore is reduced compared to the
area available to the solvent. When solute particles enter
the pore, it reduces the effective osmotic pressure in
direct proportion to the fraction of solute particles that
can enter the pore. Consider that the pores have a
radius a and that the solute particles are spherical with
a radius as. If as , a, then at least some of the solute particles can get through the pore. Suppose that if a solute
Solute particle
Area available
to solvent
Membrane
surface
a
a – as
as
Area available to
solute
FIGURE 2.7.7 Relative areas of the pore available to solvent water and
solute particles. The figure depicts a single pore, looking down along its
axis normal or perpendicular to the surface of the membrane. The outer
circle is the dimension of the pore that is available to the solvent water.
The inner circle represents the dimensions of the pore available to the
solute, which is less than that available to solvent.
particle hits the rim of the pore prior to entry, then it is
reflected back into the bulk solution. The effective pore
area will be reduced due to this reflection. The situation
is depicted schematically in Figure 2.7.7, looking perpendicular to the membrane along the axis of the pore. The
area of the pore which is accessible to solute is given by
as 2
As 5 πða2as Þ2 5 πa2 12
½2:7:37
a
The relative area available to solute compared to that
available to solvent is
as 2
2
πa
12
As
a 5 12 as 2
5
½2:7:38
A
πa2
a
Os mos is an d Os motic P res sur e
The fraction of solute particles which are reflected by
the pore is approximated by the ratio of the area in light
blue in Figure 2.7.7 to the total area. This is identified
with σ, the reflection coefficient:
A 2 As
As
512
A
A
2
as
σ 5 1 2 12
a
σ5
½2:7:39
SOLUTIONS MAY BE HYPERTONIC
OR HYPOTONIC
If ξ 5 as /a, then
σ 5 2ξ 2 ξ2
½2:7:40
We may expect the ratio of the concentration in the
pore to the concentration in the bulk solution to be
the same as the ratio of the pore area available to
solute to the area available to solvent, As/A. This is given
by Eqn [2.7.39] to be
½2:7:41
C0L
As
512σ
5
CL
A
In the absence of solvent drag (when Jv 5 0), the
concentration profile may be drawn as shown in
Figure 2.7.8.
In this model, some of the solute molecules can enter
the pore and therefore they contribute to the pressure in
the pore. We expect the pressure deficit within the pore
to be due only to those molecules which are reflected.
Thus the osmotic pressure should be σRTΔC 5 σΔπ.
The expression for laminar flow in the pore is
½2:7:42
Jv 5
physical meaning only in the range 0 # σ # 1.0.
If As 5 A, then the pore is large enough so that the
membrane does not discriminate between solute and
solvent, and σ 5 0 according to Eqn [2.7.39]. If as $ a,
then As 5 0 and σ 5 1.0.
When cells are placed in contact with a solution, they
may either swell or shrink as shown in Figures 2.7.9
and 2.7.10. These observations introduce the idea of
tonicity, which is operationally defined. If we place a
cell in a solution and the cell swells, the solution is
called hypotonic. If we place a cell in a solution and the
cell shrinks, we call that solution hypertonic. If the cell
neither swells nor shrinks, the solution is isotonic.
OSMOSIS, OSMOTIC PRESSURE,
AND TONICITY ARE RELATED BUT
DISTINCT CONCEPTS
Osmotic pressure is a theoretical concept. It is the pressure necessary to stop osmotic flow if a solution is separated from pure water by a semipermeable membrane.
A semipermeable membrane is defined as a membrane
that allows the passage of some molecules but not
others. It is freely permeable to water but impermeable
1.4
nπa4
½ΔP 2 σΔπ
8ηδ
Solutions that cause cells to swell
are hypotonic
1.3
Jv 5 Lp ½ΔP 2 σΔπ
Membrane
V/V0 (relative volume)
1.2
In the above model, σ is viewed as being due to a
hindered entry into the pore. There may be additional
hindrance to solute flow through the pore due to interactions with the pore wall, but these are ignored here.
According to this view, the reflection coefficient has
Solutions that cause cells to shrink
are hypertonic
1.1
1.0
0.9
CL
Solutions that cause cells
to neither shrink nor swell
are isotonic
σCL
0.8
(1 – σ) CL
CR
Hypotonic
Isotonic
0.7
0.2
(1 – σ) CR
x=0
x=δ
FIGURE 2.7.8 Concentration profile within a pore that partially excludes
solute particles. This profile pertains only when solvent drag is zero.
Solvent drag washes the concentration gradient in the direction of
volume flow and alters Js.
Hypertonic
0.4
0.6
0.8
1.0 1.2 1.4
[NaCl] (%)
1.6
1.8
2.0
2.2
FIGURE 2.7.9 Swelling or shrinking of red blood cells in contact with
different concentrations of NaCl. Whole blood was centrifuged to
separate the red blood cells from the plasma. Then an aliquot of the
plasma was removed and replaced with an equal volume of the
indicated concentration of NaCl. When the plasma was replaced with
NaCl solution less than about 0.9%, the cells’ volume increased relative
to that in normal plasma. These solutions are described as being
hypotonic. When the [NaCl] that replaced plasma was greater than
0.9%, the red blood cells shrank. These solutions are called hypertonic.
Replacement with 0.9% NaCl caused the cells to neither swell nor shrink:
this solution is isotonic.
191
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
to solute. All solutions have an osmotic pressure. In the
approximation of the ideal, dilute solution, the magnitude of the osmotic pressure is given by van’t Hoff’s
Equation (see Eqn 2.7.7), where C is the molar concentration of osmotically active solutes. This C is the
osmolarity.
Both osmolarity and osmotic pressure are properties of
a solution, but tonicity is not. Tonicity refers to the
direction of osmotic flow when a particular solution is
placed in contact with a particular cell. Tonicity involves
real membranes rather than an ideal, semipermeable
membrane. The real membrane has a set of reflection
coefficients, one for each solute on each side of the
membrane. Whether shrinking or swelling occurs
depends on the osmotic concentrations and compositions of the two solutions on the two sides of the cell
membrane, and also on how these solutes interact with
the real biological membrane.
1.4
Solutions that cause cells to swell
are hypotonic
1.3
CELLS BEHAVE LIKE OSMOMETERS
1.2
V/V0 (relative volume)
192
1.1
1.0
Solutions that cause cells to shrink
are hypertonic
0.9
Solutions that cause cells
to neither shrink nor swell
0.8are isotonic
0.7
Hypotonic
Hypertonic
Isotonic
0.6
0
2
4
6
8
10
12
14
16
[Glucose] (%)
FIGURE 2.7.10 Swelling or shrinking of red blood cells in contact with
different concentrations of glucose. Whole blood was centrifuged to
separate the red blood cells from the plasma. Then an aliquot of the
plasma was removed and replaced with an equal volume of the
indicated concentration of glucose. When the plasma was replaced with
solutions of glucose less than about 5%, the cells’ volume increased
relative to that in normal plasma. These solutions are described as being
hypotonic. When the [glucose] that replaced plasma was greater than
5%, the red blood cells shrank. These solutions are called hypertonic.
Replacement with 5% glucose caused the cells to neither swell nor
shrink: this solution is isotonic.
Consider a cell with a volume V0 and a total concentration of osmotically active solutes, ΣCi, resting in an
isotonic medium. Now suppose that we rapidly replace
the medium with another with a different osmotic pressure which we will symbolize as π. If the new medium
is hypotonic, then by definition the cell will swell to
a new volume, at which point the cell’s cytoplasmic
tonicity will match the medium’s. Similarly, if the new
medium is hypertonic, then the cell will shrink to a
new volume that will match the tonicity of the medium.
We assume here that the volume of the medium is so
large that water movement into the cell or out of the
cell does not appreciably affect the osmolarity of
the medium. We further assume that water movement
is fast compared to the movement of solutes or ions
across the membrane, and that we can measure the
cell’s volume after water movement has occurred but
before solute movement. What is the relationship
between the medium osmotic pressure and the final
cell volume, Vc?
In this situation, the total amount of osmotically active
solutes in the cell is constant—the intracellular solutes
do not move. The total intracellular osmolytes are equal
to V0ΣCi, where V0 is the volume under isotonic
conditions and Ci is the concentration of solute i in the
cytoplasm in the isotonic condition. Now the total
amount of osmotically active solutes at any time is
Example 2.7.3 Isosmotic Solutions May Not Be Isotonic
A. Calculate the osmolarity and osmotic pressure of isotonic
saline (NaCl):
Isotonic saline is 0.9% NaCl. This is 0.9 g NaCl per dL or
100 mL of solution 5 0.9/0.1 L or 9.0 g L21.
The formula weight for NaCl is 58.44 g mol21. The molarity
of NaCl is 9 g L21/58.44 g mol21 5 0.154 M.
Since NaCl dissociates into two particles per mole, the
osmolarity is 2 3 0.154 M 5 0.308 OsM.
The osmotic coefficient of NaCl is ϕNaCl 5 0.93, so that
the measured osmolarity of this solution would be
ϕ 3 C 5 0.93 3 0.308 OsM 5 0.286 OsM. The osmotic
pressure at 37 C is 7.27 atm.
B. Calculate the osmolarity and osmotic pressure of 1.8% urea
in water:
The formula weight for urea is 60.0 g mol21 and
ϕurea 5 0.95.
Its osmolarity is ϕC 5 0.95 3 18 g L21/60 g mol21 5
0.285 OsM. Its osmotic pressure is 7.24 atm.
C. What happens when red blood cells are placed in 0.9%
NaCl? 1.8% urea?
Red blood cells placed in contact with 0.9% NaCl neither
shrink nor swell. The solution is isotonic. When placed in
1.8% urea, the cells swell so much that the cells lyse
or break open. Although 0.9% NaCl and 1.8% urea are
isoosmotic with red blood cell contents, 0.9% NaCl is
isotonic and 1.8% urea is not isotonic.
Os mos is an d Os motic P res sur e
distributed in the volume of the cell, Vc. So the osmolarity at any time is
P
V0
Ci
½2:7:43
Vc
When the osmotic pressure of the medium equilibrates
with that of the cell, with the assumption of no solute
movement (which is equivalent to assuming σi 5 1.0),
we get
P
V0
Ci
½2:7:44
π 5 RT
Vc
This can be rewritten as
π5
½2:7:45
V0 Σ RTCi
V0
5
πisotonic
Vc
Vc
which we can rearrange to obtain
πisotonic
Vc 5 V0
½2:7:46
π
According to Eqn [2.7.46], a cell acting as a perfect
osmometer would show a cell volume that was inversely
proportional to the osmolarity of the external medium,
with an intercept of zero. Typically the cell’s volume
under isotonic conditions, V0, is the control for their
volume under nonisotonic conditions. Thus we rewrite
Eqn [2.7.46] as
Vc
πisotonic
5
V0
π
½2:7:47
Figure 2.7.11 shows the plot of the volume of cardiac
cells exposed to various osmolar solutions, with volume
normalized to the volume under isotonic conditions.
According to Eqn [2.7.47], we expect the data to be
linear with an intercept of zero. Actual data from
these cardiomyocytes show that the response of relative
volume (Vc /V0) is linear within a considerable range,
1.6
but the curve does not extrapolate to zero volume.
These real cells are not perfect osmometers.
The response of real cells is described by the empirical
equation
Vc
Vb πisotonic
Vb
1
½2:7:48
5 12
V0
V0
π
V0
The intercept of this line on the volume axis is Vb/V0,
which in the case of cardiomyocytes shown in
Figure 2.7.11 is 0.34. This is interpreted to mean that
there is a fraction of the cell’s volume that is osmotically inactive. This is partly to be expected. Not all of
the volume of the cell is water, and it is the volume
of water that dissolves osmotically active solutes and
is responsive to changes in the medium osmolarity.
Thus the sum of all of the volumes of large particles
such as DNA, RNA ribosomes, and membranes contributes to an osmotically inactive volume. Although this
is certainly part of the explanation for the osmotically
inactive volume, the issue is by no means settled. Other
components of the inactive volume might include the
volume of a variety of macromolecular assemblies such
as the cytoskeleton.
CELLS ACTIVELY REGULATE THEIR
VOLUME THROUGH RVDs AND RVIs
Although the presence of a hypotonic or hypertonic
solution initiates swelling or shrinking, respectively,
often the volume change is not maintained. A cell that
initially swells when placed in a hypotonic medium
may eventually lose some of its acquired volume: it
undergoes a regulatory volume decrease or RVD. The
swelling of the cell activates compensatory mechanisms that cause transport of osmotically active solutes
(osmolytes) out of the cell. Similarly, cell shrinking
can activate an influx of osmolytes in some cells
leading to a compensatory swelling that is called a
regulatory volume increase or RVI. These RVDs and
RVIs are accomplished by altering the cell’s contents of
osmolytes.
Relative volume (V/V0)
1.4
1.2
SUMMARY
1.0
Osmosis refers to the movement of fluid across a
membrane in response to different concentrations of
solutes on the two sides of the membrane. The movement of fluid is toward the more concentrated solution. Osmotic pressure is defined as the pressure that
must be applied to the solution side to stop fluid
movement when a semipermeable membrane separates
a solution from pure water. Here, the semipermeable
membrane is permeable to water but not to solute.
The osmotic pressure for dilute ideal solutions obeys
van’t Hoff’s Law:
0.8
0.6
0.4
0.2
0.0
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0
πisotonic/π
FIGURE 2.7.11 Relative volume of isolated cardiac cells exposed to test
solutions of different osmolarities. Source: Data from Drewnowska and
Baumgarten, American Journal of Physiology 260:C122131, 1991.
π 5 RTΣ Cs
which can be derived on thermodynamic grounds.
Because the solutions are not ideal, the equation is
193
194
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
refined by including an osmotic coefficient, ϕs,
characteristic of each solute:
πobserved 5 RTΣ ϕs Cs
Defined in this way, the osmotic pressure is a pressure
deficit caused by dissolving solutes. However, membranes that are somewhat permeable to the solute
develop a transient osmotic pressure that is less than
that predicted by van’t Hoff’s Law. Thus the actual pressure developed across a membrane that separates a
solution from pure water depends on the interaction of
the solute with the membrane. The correction for
partially permeable membranes requires the reflection
coefficient, σ:
πobserved 5 RTΣ σs ϕs Cs
The magnitude of the pressure tells us nothing of the
flow. Osmotic pressure and hydrostatic pressure add to
drive fluid flow across the membrane, with a proportionality constant, Lp. The phenomenological equation
describing fluid flow is
Qv 5 A Lp ðΔP 2 σΔπÞ
Osmosis and osmotic pressure is a thermodynamic
concept which exists independently of mechanism. In
microporous membranes, osmosis is caused by a
momentum deficit within the pores due to the reflection of solute molecules by the membrane. This reduces
the pressure on the solution side of the pore by π for a
semipermeable membrane.
Thus there are three characteristic parameters for describing passive material transfer across membranes: the permeability, p, the hydraulic conductivity, Lp, and the
reflection coefficient, σ.
Osmolarity is a kind of concentration measure, distinct
from molarity. It is related to other colligative properties
of solutions including freezing point depression, vapor
pressure depression, and boiling point elevation.
Tonicity is a related concept but involves a real, biological membrane that may not be semipermeable. Tonicity
makes reference to a particular cell and its membrane.
Solutions may be isoosmotic but not isotonic.
3. What is the reflection coefficient? How does it
relate to permeability?
4. The equations in this chapter were derived for a
microporous membrane. Would they still hold
for a lipid dissolution model of a membrane?
5. How does tonicity differ from osmolarity? Define
hypotonic, hypertonic, and isotonic.
6. What is the relationship between volume and
osmotic pressure in a perfect osmometer?
7. What is meant by “osmotically inactive” volume?
8. How do cells regulate their volume?
APPENDIX 2.7.A1 MECHANISM
OF OSMOSIS: FILTRATION VERSUS
DIFFUSION DOWN A CONCENTRATION
GRADIENT
PHYSICAL ORIGIN OF THE OSMOTIC PRESSURE
ACROSS A MICROPOROUS MEMBRANE
Consider what happens in the vicinity of a single pore,
as shown in Figure 2.7.A1.1. Since the pore excludes
solute particles, the average concentration must decrease
from CL in the bulk solution to zero in moving along
the axis of the pore. Thus there is a concentration gradient near the opening of the pore. By Fick’s First Law of
Diffusion, we should expect a net diffusion of solute
particles into the pore. However, the particles hit the
rim of the pore and are reflected back into the bulk
solution. On the average, these particles experience a
force in the 2x direction. The magnitude of this average
force can be obtained from the generalized Fick’s
equation (see Eqn [1.6.42]):
½2:7:A1:1
js 5 2 D
CL
@CðxÞ
D
1
F CðxÞ
@x
RT
Membrane
Cells respond to swelling or shrinking according to the
empirical relation:
Pore
V=V0 5 ð1 2 Vb =V0 Þ πisotonic =π 1 Vb =V0
where V0 is the volume of the cell under isotonic conditions. Vb is interpreted as the osmotically inactive
volume. If Vb 5 0, the cell would behave like an ideal
osmometer.
Solute particles
REVIEW QUESTIONS
1. In which direction does osmotic flow occur?
Why?
2. What equation gives the magnitude of the
osmotic pressure? What are the limitations of this
equation? How would you correct for its errors?
CR = 0
x=0
x=δ
FIGURE 2.7.A1.1 Concentration gradient for solute particles near a pore.
In this case, the solute particles are large compared to the pore and
cannot penetrate into the membrane.
Os mos is an d Os motic P res sur e
We write R in place of k and F in place of f in
Eqn [2.7.A1.1] because we are speaking of the force
per mole rather than the force per molecule, as was
done in Eqn [1.6.42].
Since js 5 0 for an impermeant membrane, Eqn [2.7.A1.1]
becomes
½2:7:A1:2
FCðxÞ 5 RT
Here we consider the forces acting on an element of fluid
with an area A from a point x well within the
bulk solution to a point x 1 Δx within the pore near its
surface. We consider that the volume element is in
mechanical equilibrium. The forces acting on this volume
are the forces acting on the solute particles and the forces
acting on the surfaces in contact with adjacent fluid. The
sum of these forces must be zero to meet the condition of
mechanical equilibrium. We identify these forces with the
“body” forces, FB, and “contact” forces, FC. Thus we write:
0 5 FB 1 FC
for the condition of mechanical equilibrium. The net
contact force is the balance of the pressure acting on the
surface area to the right and left of the volume element:
½2:7:A1:4
FC 5 A PðxÞ 2 A Pðx 1 ΔxÞ
Membrane
Solute particle
½2:7:A1:5
FB 5
dV = Adx
F CðxÞdV
x
FB 5 ART
½2:7:A1:6
P(x + Δx)
ð x1Δx
x
@CðxÞ
dx
@x
5 ART½Cðx 1 ΔxÞ 2 CðxÞ
Since C(x 1 Δx) 5 0 because solute particles are not in
the pore, Eqn [2.7.A1.6] becomes
½2:7:A1:7
FB 5 2 ART CðxÞ
The negative sign in Eqn [2.7.A1.7] indicates that FB
is directed to the left. Inserting Eqns [2.7.A1.4] and
[2.7.A1.7] into Eqn [2.7.A1.3], we obtain
½2:7:A1:8
A PðxÞ 2 A Pðx 1 ΔxÞ 5 ART CðxÞ
or
½2:7:A1:9
PðxÞ 2 Pðx 1 ΔxÞ 5 RTCL
This equation says that the net pressure experienced
by the volume of fluid immediately adjacent to the
pore decreases as one moves from the left compartment
into the pore, and that the drop in pressure is RTCL.
This analysis suggests that the osmotic pressure develops
as a consequence of the interactions between the solute
particles and the membrane. The solute particles contribute momentum in the solution. When the particles
collide with the membrane, that momentum is transferred to the membrane and not to the fluid within the
pore. As a consequence, there is a momentum deficit
within the pore (compared to the bulk solution). Since
the pressure is the average momentum change per unit
area experienced by particles colliding within the fluid,
this momentum deficit shows up as a pressure deficit.
The resulting pressure difference produces a flow that is
given as
Pore
P(x)
ð x1Δx
Inserting dV 5 Adx and FC(x) 5 RT @C(x)/@x from
Eqn [2.7.A1.2], we obtain
@CðxÞ
@x
The force F is an external force that acts on the solute
particles. The total force acting on the solute particles is
just Fn(x), where n is the number of moles of particles.
Dividing by the volume, V, we have Fn(x)/V 5 FC(x),
which is the average force per unit volume. This is the
force that the membrane is exerting on the solute bodies, per unit volume, located in the volume of fluid
directly in front of the pore. The consequence of this is
that there will be a pressure drop at the pore entrance.
To see this, we will analyze the forces acting on an element of fluid located directly in front of a pore, as
shown in Figure 2.7.A1.2.
½2:7:A1:3
The total body force is the body force per unit volume
integrated over the volume.
½2:7:A1:9
Jv 5
nπa4
Δπ
8ηδ
where we identify
x
x + Δx
½2:7:A1:10
x=0
x=δ
FIGURE 2.7.A1.2 Forces acting on the plug of fluid immediately in front
of a pore in a porous membrane.
Lp 5
nπa4
8ηδ
DIFFUSIONAL PERMEABILITY
OF MICROPOROUS MEMBRANES
In the absence of a hydrostatic or osmotic pressure
gradient, water will diffuse across a microporous membrane. If we assume the membrane is impermeable
195
196
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
except at the pores, the diffusion through the pores will
obey Fick’s Laws of diffusion:
jw 5 2 D
½2:7:A1:11
@C
@x
@C
@C2
5D 2
@t
@x
where jW is the flux through a single pore. At steady
state, the flux does not change with time, or with
distance, and so the gradient is linear in x and we can
write
½2:7:A1:12
πa2 Dw
ΔCw
jw 5
δ
where jw is the flux of water through the pore, a is the
radius of the pore, DW is the diffusion coefficient of
water in the pore, δ is the length of the pore (equal
to the thickness of the membrane), and ΔCW is the concentration difference of water. The overall macroscopic
flux for a microporous membrane is given as
½2:7:A1:13
Jw 5
n πa2 Dw
ΔCw
δ
where n is the density of the pores, n 5 N/A, the
number of pores per unit area of the membrane. This is
analogous to the passive solute flux discussed in
Chapter 2.5. We can similarly define a diffusional
permeability for water from the relation:
½2:7:A1:14
Jw 5 Pd ΔCw
and we can identify a diffusional permeability coefficient for water from Eqns [2.7.A1.13] and [2.7.A1.14] as
½2:7:A1:15
Pd 5
n πa2 Dw
δ
From the thermodynamic derivation of van’t Hoff’s
Law, we write for the chemical potential of water
½2:7:A1:19
μw 5 μ0w 1 RT ln aw 1 V w P
insertion of this into Eqn [2.7.A1.18] and at steady-state
flow, we obtain for the macroscopic membrane
½2:7:A1:20
Jw 5
nπ2 Dw
Cw V w ΔP
RTδ
Vw is the partial molar volume (in volume per mole,
L mol21), and CW is the water concentration, in
moles L21, and these cancel: VWCW 5 1.0. The flux given
here is in mole per unit area per unit time. To convert
to JV we multiply by VW:
½2:7:A1:21
JV 5
V w nπa2 Dw
ΔP
RTδ
This describes pressure-driven flow across the membrane. We recognize that the same terms for Pd are present in this equation. We recognize Eqn [2.7.A1.21] as
JV 5 LP ΔP and identify LP as
½2:7:A1:22
LP 5
V w nπa2 Dw
V w Pf
5
RTδ
RT
where Pf is the filtration permeability for water. It has
the same expression for Pd given in Eqn [2.7.A1.15] but
we obtain it experimentally from LP as
½2:7:A1:23
Pf 5 LP
RT
n πa2 Dw
5
δ
Vw
Thus comparing Eqn [2.7.A1.15] to Eqn [2.7.A1.23], Pd
and Pf should be the same if the mechanism of osmosis
is by diffusion.
FILTRATION PERMEABILITY IN THE PRESENCE
OF A PRESSURE DIFFERENCE IN A
MICROPOROUS MEMBRANE
PRESSURE- AND OSMOSIS-DRIVEN FLOW
ACROSS A LIPID BILAYER
BY DISSOLUTIONDIFFUSION
In Chapter 1.6, we found that diffusional flux can
be altered by the application of an additional force.
In particular, a flux of solute obeyed the relation:
Here we consider a membrane that presents all of its
area to the solution phase, and water crosses by dissolving in the lipid of the membrane on one side of the
membrane, diffusing across the membrane essentially
like a vapor, and then coming out of lipid solution
back into the aqueous phase on the other side. We
imagine that dissolution is rapid (the solution phase
and membrane phase are in equilibrium) and that diffusion is comparatively slow. Equilibrium of water in
the solution phase with water in the membrane phase is
described by equating the chemical potential of water in
the two phases:
½2:7:A1:16
Js 5 2
D
CF
RT
where JS is the flux of solute, D is its diffusion coefficient,
R is the gas constant, T is the temperature (K), C is the
concentration, and F is the total force on the particle per
mole. We went on to define the electrochemical potential
so that
½2:7:A1:17
F52
dμ
dx
These equations have general validity and are also true
for water. We can write for the flux in the pore that
½2:7:A1:18
Dw dμw
jw 5
C
RT dx
μ0wðsolutionÞ 1 RT ln XwðsolutionÞ 1 V wðsolutionÞ P
5 μ0wðmembranceÞ 1 RT ln XwðmembraneÞ 1 V wðmembraneÞ P
½2:7:A1:24
where Xw(solution) and Xw(membrane) are the mole fractions of water in the solution in equilibrium with the
Os mos is an d Os motic P res sur e
membrane and in the membrane phase, respectively.
The partition coefficient is defined as
½2:7:A1:25
Kw 5
XwðmembraneÞ
XwðsolutionÞ
½2:7:A1:23
Consider the case where only osmotic pressure drives
water flow and the hydrostatic pressure difference across
the membrane is zero. Since water generally partitions
poorly into hydrocarbon solvents, we may assume that
the mole fraction of water in the membrane phase is
low. Thus the water concentration is dilute, and we may
replace the mole fraction of water with its
concentration:
½2:7:A1:26
CWðmembraneÞ XwðmembraneÞ
V lipid
If equilibrium is rapid, we can combine Eqn [2.7.A1.25]
and Eqn [2.7.A1.26] to get
½2:7:A1:27
CwðmembraneÞ Kw
XwðsolutionÞ
V lipid
For dilute solutions, XW(solution) can be replaced by
1 2 V w CS where CS is the solute concentration:
½2:7:A1:28
CwðmembraneÞ Kw
1 2 V w CS
V lipid
The concentration of water immediately inside the lefthand side of the membrane is given by Eqn [2.7.A1.28]
where CS is the concentration of solute in the left
compartment. A similar expression pertains to the water
concentration immediately inside on the right side. The
net diffusive flux of water across the membrane is given as
m
½2:7:A1:29
Jw 5 2 D w
Cw;L 2 Cw;R
02δ
where Dwm is the diffusion coefficient of water in the
membrane phase. Substituting in for the concentrations
from Eqn [2.7.A1.28], we get
Jw 5 2
Dm
K V ðCS;L 2 CS;R Þ
w w w
V lipid δ
52
In Eqn [2.7.A1.23], we described Pf, the filtration water
permeability, as
Dm
K V ΔCS
w w w
V lipid δ
RT
Vw
P f 5 LP
Applying this result to Eqn [2.7.A1.32], we obtain
½2:7:A1:33
Dm
K V
w w w
Pf 5
V lipid δ
This equation was derived for the situation in which
there was an osmotic gradient (ΔCS . 0) in the absence
of a hydrostatic pressure gradient (ΔP 5 0). The expression for the situation where ΔCS 5 0 and ΔP . 0 can be
obtained by returning to Eqn [2.7.24] and setting
the mole fractions of water to 1.0 while the pressures
differ. The result is that exactly the same LP is derived
for pressure-driven flow as for osmotic flow when the
mechanism is by rapid dissolution of water followed
by slow diffusion of water through the membrane
phase.
DIFFUSIONAL PERMEABILITY
BY THE DISSOLUTIONDIFFUSION
MECHANISM: PD
The permeability of lipid membranes to a diffusional
water flux is expressed by the equation:
½2:7:A1:34
Jw 5 Pd ΔCw
where Pd is the diffusional permeability and ΔCw is the
difference in water concentration across the membrane.
The overall permeation of water takes three steps: dissolution into the membrane phase at the left interface,
diffusion across the membrane phase, and reversal of
the dissolution at the right interface. If we assume, as
we did in the derivation of Pf, that the rate-limiting step
is diffusion through the membrane phase, then we may
rewrite Eqn [2.7.A1.34] as
½2:7:A1:35
ΔCwðmembraneÞ
δ
m
Jw 5 Dw
½2:7:A1:30
Substituting in for ΔCw(membrane) from Eqn [2.7.A1.27],
this is
The flux given here is in mole per unit area per unit
time. To convert to JV, we multiply by Vw:
½2:7:A1:36
2
JV 5 V w Jw 5 2
Dm
K V ΔCS
w w w
V lipid δ
2
52
Dm
K V
w w w
V lipid RT δ
RTΔCS
The last term on the far right is the osmotic pressure difference, Δπ. Equation [2.7.A1.31] relates the volume
flux to the osmotic pressure when the mechanism of
water flow is dissolution and diffusion. We recognize it
as a form of the phenomenological equation,
JV 5 2LPΔπ and therefore we identify LP as
2
½2:7:A1:32
LP 5
Dm
K V
w w w
V lipid RT δ
Kw ΔXwðsolutionÞ
V lipid δ
since ΔXw 5 Vw ΔCw, this becomes
½2:7:A1:37
½2:7:A1:31
m
Jw 5 Dw
m
Jw 5 Dw
Kw V w
ΔCw
V lipid δ
and we can identify Pd by comparing Eqns [2.7.A1.37]
and [2.7.A1.34] as
½2:7:A1:38
Pd 5
Dm
K V
w w w
V lipid δ
Comparing the results for Pf (see Eqn [2.7.A1.33]) and
for Pd (see Eqn [2.7.A1.38]), the dissolutiondiffusion
mechanism of water transport indicates that Pf/Pd 5 1.0.
197
198
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
If we measure the diffusional permeability of a lipid
membrane and LP, and calculate Pf from the LP according to Eqn [2.7.A1.23], we should expect them to be
equal provided that the mechanism is dissolution and
diffusion. Our earlier results with microporous membranes also concluded that Pf/Pd 5 1.0 if the mechanism
of transport was by diffusion.
EXPERIMENTS CONFIRM PF/PD 5 1.0 FOR LIPID
BILAYERS BUT PF/PD . . 1.0 FOR MOST
BIOLOGICAL MEMBRANES
The measurement of Pf (from LP) and Pd in planar lipid
bilayers confirm that they are the same, indicating that
water transfer across pure lipid bilayer membranes is by
diffusion. However, in most biological membranes,
Pf/Pd is much greater than 1.0, suggesting that the
mechanism is not diffusional, but hydrodynamic.
The erythrocyte membrane was used in many of these
studies, and it was shown that compounds that interact
with protein sulfhydryl groups markedly reduced Pf
while leaving Pd relatively unchanged, and causing the
ratio of Pf/Pd to become 1.0. These early studies pointed
to the existence of proteinaceous pores in the erythrocyte membrane, and subsequently these were identified
as AQP1, the first in the family of aquaporins. Peter
Agre earned the Nobel Prize in 2003 for his discovery of
the aquaporins.
AQUAPORINS ACCOUNT FOR MOST WATER
TRANSPORT IN CELLS
Aquaporins are a family of proteins, all about 28 kDa,
that are found in a variety of tissues that have high
water transport rates such as the intestines and kidneys,
salivary glands, pancreas, and more. There are 13
human varieties, labeled AQP0, AQP112. The proteins
all have six transmembrane domains and associate
to form a functional tetramer, although each part has its
own water channel. Although water can permeate
through lipid membranes, this pathway is much slower
than the AQP-mediated pathway. Thus real biological
membranes are a mosaic of lipid pathways for diffusional transport of water and pores for pressure-driven
transport of water.
Problem Set
Membrane Transport
1. The GLUT-1 transporter has a Km for glucose of
1.5 mM. The normal, resting plasma glucose
concentration is about 90 mg%. This is a clinical
unit that is not part of the ISI but you have to get
familiar with it anyway. X mg% means that the
solute has that many mg in a deciliter of plasma.
One deciliter is 0.1 L 5 100 mL. The molecular
weight of glucose is 180 g mol21. From this information, calculate the rate of glucose transport by
GLUT-1, as a percent of its maximum, when
exposed to normal plasma. Assume saturation
kinetics.
2. When CL was 2.3 3 1026 M and CR was zero, the
flux across a microporous membrane was found
to be 0.234 pmol cm22 s21. The free diffusion
coefficient of the material being measured was
0.8 3 1025 cm2 s21.
A. What is the permeability of the membrane
to the material?
B. If the thickness of the membrane is
10 3 1026 m, what is the equivalent relative
area available for diffusion of the material?
3. Two membranes, A and B, have permeabilities
PA and PB for a given solute. These two membranes are joined together to form a single
composite, two-layered membrane, as shown in
Figure 2.PS2.1. Examples are the successive filtration/diffusion barriers in the kidney glomerulus,
successive permeability barriers in the lung, and
successive permeability barriers of unstirred layers
adjacent to intestinal epithelial membranes.
A
B
CL
CR
FIGURE 2.PS2.1 Composite membrane formed from the sandwich of
two membranes, A and B, with different characteristics.
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00018-5
2.2
A. How would you define the overall permeability P of the two-layered membrane?
Hint: Think about how you would define
permeability for any membrane. Think
about what you need to know to calculate P.
Use CL for the left concentration and CR for
the concentration on the right.
B. Why is the steady-state solute flux through
the composite membrane the same through
the membrane A layer and the membrane B
layer? Hint: Think of the continuity equation
and what it means.
C. What is the concentration profile through
the composite membrane? Hint: Calculate
the concentration at the interface of membranes A and B; call it Cm, in terms of CL
and CR. Hint: Equate the flux through the
two membranes.
D. Find an expression for P in terms of PA and
PB.
E. Do the permeabilities act like inverse resistances in a series arrangement?
4. When CL was 2.3 3 1026 M and CR was zero, the
flux across a microporous membrane was found
to be 0.234 pmol cm22 s21. This flux was determined with vigorous stirring, which virtually
eliminated any unstirred layers adjacent to the
membrane. When the stirrer was turned off,
the flux decreased to 0.157 pmol cm22 s21. The
free diffusion coefficient of the material being
measured was 0.8 3 1025 cm2 s21.
A. What is the permeability of the membrane
plus unstirred layer?
B. What is the permeability of the membrane
alone?
C. Using the information in Problem #3, what
is the thickness of the unstirred layer?
(Assume that the diffusion coefficient in the
unstirred layer is equal to the free diffusion
coefficient.)
5. The surface area of the lungs is about 75 m2,
and the thickness of the alveolar diffusion
layer is about 0.5 μm. The PO2 in alveolar air
is 100 mmHg, while the PO2 of venous blood is
40 mmHg. The diffusion coefficient of O2 in
water is about 1.5 3 1025 cm2 s21. The solubility
of O2 is given by Henry’s Law as [O2] 5 0.024
PO2 ; here [O2] is expressed in mL O2 at STPD
(standard temperature and pressure, dry: 0 C and
1 atm pressure) per mL of water and PO2 is in 199
200
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
atmospheres. Only the dissolved O2 diffuses.
What is the initial rate of O2 diffusion from the
aggregate alveoli to the blood? (Initial rate means
to pretend that the venous PO2 is clamped at
40 mmHg.) Give the answer in mL O2 per minute
and in mol O2 per minute using the Ideal Gas
Law. Assume 37 C, R 5 0.082 L atm mol21 K21.
Remember that 1 atm 5 760 mmHg.
6. Heart cells contain a NaCa exchanger with a
stoichiometry of 3Na:1Ca. The following questions pertain to this transporter.
A. The free energy of transport of Ca21 across
the sarcolemma of the heart cell can be calculated from the following conditions during the rest phase of the heart beat:
10.
½Ca21 o 5 1:2 3 1023 M
½Ca21 i 5 0:1 3 1028 M
Em 5 20:085 V
Calculate the free energy for the reaction
Caout-Cain. Recall that R58.314 J mol21 K21,
T5310 K, `59.6493104 C mol21. Remember
that Ca21 has two electrical charges per atom.
B. Calculate the free energy for the reaction
Nain-Naout for the following conditions
during the rest phase of the heart:
11.
½Na1 o 5 145 3 1023 M
½Na1 i 5 14 3 1023 M
Em 5 20:085 V
C. Which way does the NaCa exchange proceed at rest?
7. Ischemia refers to the condition of no blood
flow. When the artery perfusing an area of tissue
is blocked, oxygen can no longer be delivered to
support energy metabolism. Under these conditions, the ATP concentration falls and ADP and
Pi concentrations rise. What will this do to the
free energy of ATP hydrolysis (also called the
affinity of ATP hydrolysis)? What do you think
will be the consequence of ischemia on the
effectiveness of the ion pumps? (Kammermeir,
Schmidt, and Jungling, Free energy change of
ATP hydrolysis: a causal factor of early hypoxic
failure of the myocardium? J. Mol. Cell. Cardiol.
14:267277, 1982).
8. The membrane of the sarcoplasmic reticulum
(an internal membrane in muscle cells) has
multiple ion channels, so the membrane potential across this membrane is believed to be zero.
This membrane has a Ca-ATPase pump that
links two Ca21 atoms to the hydrolysis of ATP.
Using a free energy of ATP hydrolysis of
257.1 kJ mol21, what is the thermodynamic
limit of Ca21 accumulation if the free [Ca21] on
the cytosolic face is 1 3 1027 M? (Hint: The thermodynamic limit is when the free energy change
for the transport reaction is zero.)
9. Each of the reactions shown in Eqn [2.5.15] has
a forward and reverse rate constant. Show that
12.
13.
for a passive transport process the product of all
the forward rate constants is equal to the product of all the reverse rate constants. (Hint: Use a
principle called detailed balance, which states
that at equilibrium all steps in a reaction
sequence must also be at equilibrium.)
Consider that a membrane separates two compartments, each containing a solution of 10 mL.
The left compartment has an initial concentration CL. The membrane has permeability p to the
solute and area A.
A. Derive an expression for CL and CR as a
function of time.
B. Suppose that you obtained experimental
values for CL(t) or CR(t). What plot of the
data could you make to determine p?
C. What would happen if we doubled both the
volume and the surface area to the time
course of equilibration of CL and CR.
The surface area of the lungs is about 75 m2,
and the thickness of the alveolar diffusion layer
is about 0.5 μm. The PO2 in alveolar air is
100 mmHg, while the PO2 of venous blood
is 40 mmHg. The diffusion coefficient of O2 in
water is about 1.5 3 1025 cm2 s21. The solubility
of O2 is given by Henry’s Law as [O2] 5 0.024
PO2 ; here [O2] is expressed in mL O2 per mL
of water and PO2 is in atmospheres. Only the
dissolved O2 diffuses. The volume of blood in
the lung is 70 mL, and the volume of air at the
end of normal inspiration volume is about 2.8 L
(at body temperature of 37 C).
A. Derive an equation for the time course of
oxygen equilibration between blood and air
in the lungs.
B. Using the values given here, estimate the
half-time of equilibration.
C. Most of the oxygen in the blood is not free
but is bound to hemoglobin within the red
blood cells. Do you think this would accelerate or decelerate the rate of equilibration of
blood and air oxygen pressures?
Vesicles of the sarcoplasmic reticulum have
embedded in their membrane an active CaATPase pump. Several different isoforms of this
primary active pump are expressed in different
tissues. When exposed to ATP, Mg, and Ca,
Ca21 ions are accumulated and eventually reach
a steady-state uptake. If the pump is quickly
quenched by adding extravesicular EGTA, which
complexes activator Ca21 and thereby stops the
pump, the accumulated Ca21 will leak back out.
Monitoring the intravesicular Ca21 with time
allows one to estimate the permeability of the
vesicles. Derive an expression for the amount
of Ca21 remaining in the vesicle as a function of
time and suggest a plot to determine the permeability. What other information might you need
to know to determine the permeability?
The osmotic coefficient for CaCl2 under physiological conditions is 0.85. Calculate the osmotic
pressure of a solution of 10 mM CaCl2 at 37 C.
Problem Set
Recall here that R 5 0.082 L atm mol21 K21.
Give the answer in both atm and mmHg
(1 atm 5 760 mmHg).
14. The kidney filters plasma to produce an
ultrafiltrate, which is the first step in the
formation of urine. This filtrate is called an
ultrafiltrate because the kidney can retain even
small particles like plasma proteins. The force
behind ultrafiltration is the blood pressure. The
kidney ultrafiltration occurs at a structure called
the glomerulus, which is a group of small
blood vessels (capillaries) that are closely joined
to another structure, Bowman’s capsule, that
forms a double-walled cup for the collection
of the ultrafiltrate. The filtration barrier is a
combination of the capillary walls and structure
in Bowman’s capsule.
A. Calculate the filtration coefficient (Lp) for
the basement membrane of kidney glomeruli
using the following approximations:
1. Pore radius 5 35 Å
2. Pore length 5 600 A
3. Fractional pore area 5 5%
4. Blood plasma viscosity 5 0.02 poise
(dyne s cm22)
The “fractional pore” is the total area of
the pores divided by the total area of the
membrane. A dyne is a g cm s22.
B. Calculate the glomerular filtration rate (GFR)
assuming a total area for both kidneys of
1.5 m2 and a driving force ΔP 5 20 mmHg.
The GFR is the total volume of ultrafiltrate
produced per minute. Its units should be in
cm3 min21. Make it so.
15. The observed osmotic pressure of solutions of
plasma proteins increases more rapidly than
concentration. Empirical fits to the concentration
dependence of osmotic pressure are given by
Landis and Pappenheimer (Handbook of Physiology,
vol 2, section 2, pp. 9611034, 1963):
πalbumin 5 2:8C 1 0:18C2 1 0:012C3
where π is in units of mmHg and C is in units
of g% (i.e., g of protein per deciliter of plasma).
A. According to this equation, as C becomes
more dilute the relation approaches van’t
Hoff’s Equation. Keeping in mind the units
of the variables, what is the molecular
weight of albumin? Assume that the temperature is 37 C. (By the way, osmotic measurements were the first measurements of protein
molecular weights.)
B. What is the contribution of albumin to the
osmotic pressure of plasma when it contains
4.0 g% of albumin?
C. The osmotic pressure of plasma proteins
and associated ions is called the oncotic
pressure. If the plasma oncotic pressure is
25 mmHg, how much of the oncotic pressure is contributed by globulins, fibrinogen,
and other components?
16. Assume that serum albumin is a sphere of
diameter 31 Å. Assume that the glomerular
membrane is pierced by pores of equivalent
diameter of 35 Å.
A. Give an estimate of σ for albumin for the
glomerular membrane.
B. Calculate the concentration of albumin in
the ultrafiltrate.
C. If the GFR is 120 mL min21, calculate the
daily filtered load of albumin (how much is
filtered every day). How does this compare
with the recommended dietary intake of
0.8 g protein per kg body weight per day?
17. The value of Lp for the red blood cell is about
1.8 3 10211 cm3 dyne21 s21. Its surface area is
about 1.35 3 1026 cm2 (Solomon, Methods in
Enzymology, pp. 192222, 1989).
A. What is the initial osmotic flow if the osmolarity inside is initially 300 mOs M and the
osmolarity outside is 275 mOs M? (Assume
σ 5 1.0 for all solutes.)
B. If the volume of the cell is 100 3 10212 cm3,
how long would it take to double its volume
provided that the osmotic pressure and area
of the membrane and Lp did not change?
C. In the case described, how much water would
be required to enter the cell to equilibrate the
osmotic pressure between inside and outside?
Assume that the outside bath is essentially
infinite so that its osmotic pressure is kept
constant.
18. Osmotic pressure is one of a class of properties
of solutions that are called colligative properties. The others in this class include vapor
pressure depression, boiling point elevation,
and freezing point depression. These properties
are different expressions of the same phenomenon: the lowering of the activity of water by
dissolution of solutes. Various osmometers have
been made using one or another or these properties. Table 2.PS2.1 shows several solutions of
sucrose and glucose, their water concentrations,
TABLE 2.PS2.1 Solute Concentration, Water
Concentration, and Freezing Point Depression in
Sucrose and Glucose Solutions
Sucrose Solutions
Glucose Solutions
[Sucrose]
(M)
[Water]
(M)
Δ
( C)
[Glucose]
(M)
[Water]
(M)
Δ
( C)
0
55.45
0
0
55.46
0
0.029
55.12
0.06
0.028
55.28
0.05
0.059
54.77
0.11
0.056
55.11
0.10
0.081
54.42
0.16
0.084
54.94
0.16
0.118
54.07
0.23
0.112
54.76
0.21
0.179
53.72
0.35
0.140
54.59
0.27
Source: Data from Handbook of Chemistry and Physics, CRC Co, Cleveland,
OH, 1965.
201
202
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
and their freezing point depression. Because dissolving solutes invariably dilutes the solvent,
water, you can see from the table that the water
concentration also decreases with increasing
solute concentration. Sucrose, however, is twice
as large as glucose, so we might expect that
dissolution of sucrose would dilute the water
further. Therefore, the colligative properties of
solutions cannot be proportional to both solute
and solvent concentration. Plot separately the
freezing point depression against the water
concentration and against the solute concentration. Which relationship shows the same dependence? From this result, do you expect osmosis
to be dependent on solvent water concentration
or on solute concentration?
19. From the data in Table 2.PS2.1, calculate the
coefficient relating freezing point depression to
molarity in the equation:
½2:PS2:1
PL 2 PR 5 π 5
RT apure water
ln
asolution
Vw
Here the subscripts L and R refer to the left and
right sides of the semipermeable membrane,
respectively, where pure water is on the right
side. If the solution was ideal, we can use the
mole fraction for the activity and this equation
becomes
½2:PS2:3
π52
RT
ln Xw
Vw
In the further approximation of a dilute solution,
this equation was transformed to
½2:PS2:4
[Sucrose] Observed RT Cs
(M)
π (atm)
(atm)
0.098
2.47
2.40
2RT/Vw ln RT/Vw ln
xw (atm)
(P0/Ps)
(atm)
2.48
2.50
0.824
27.2
20.4
24.6
26.6
1.399
58.4
35.1
48.8
57.3
1.823
95.2
45.5
72.6
93.2
2.146
139.0
55.7
96.0
135.6
2.55
187.3
64.5
118.9
186.5
Source: Data from Glasstone, Textbook of Physical Chemistry, van Nostrand,
1946.
Tf 2 T 5 Δ 5 kf C
where Tf is the freezing point of the pure liquid,
T is the freezing point of the solution, kf is the
coefficient, and C is the concentration.
20. Chapter 2.7 gives us several expressions for the
osmotic pressure. The most complete is derived
from Eqn [2.7.6] and is
½2:PS2:2
TABLE 2.PS2.2 Values for the Observed Osmotic
Pressure and the Osmotic Pressure Calculated from the
van’t Hoff Equation, from the Mole Fraction of Water,
and from the Measured Vapor Pressure of Solutions
π 5 RT Cs
The activity of water in Eqn [2.PS2.2] is measured
by the vapor pressure. Table 2.PS2.2 tabulates
the calculations of the osmotic pressure from the
vapor pressure measurements, the mole fraction
of water and from the concentration of solute.
Plot the ratio of each of the observed osmotic
pressures to the calculated osmotic pressure,
in columns 3, 4, and 5, against the sucrose
concentration. The values you calculate for the
ratio of the observed π to the calculated π in
column 3 is ϕ(5π/RT ln Cs), the osmotic coefficient. The ratio of the observed π to column 4
(i.e., (π/[ 2 RT/V w ln xw]) defines the rational
osmotic coefficient, g.
A. Why is the equation using the mole fraction
of water a better predictor of the osmotic
pressure than the van’t Hoff Equation?
TABLE 2.PS2.3 Molecular Weight and Concentration
of Plasma Proteins.
Protein
Average Molecular Concentration in
Weight
Plasma (g dL21)
Albumin
69,000
4.2
Fibrinogen
330,000
0.3
Immunoglobin G 150,000
0.8
Immunoglobin M 750,000
0.3
α2 Globulins
100,000
0.7
α1 Globulins
50,000
0.5
β Globulins
100,000
0.8
B. Why is the equation using the vapor pressure
a better predictor of the osmotic pressure
than the equation using the mole fraction of
water?
C. Are these methods good predictors of the
osmotic pressure in dilute solutions?
21. KrebsHenseleit buffer has the following composition: 119 mM NaCl, 25 mM NaHCO3,
3.2 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4,
11 mM glucose, and 1.4 mM CaCl2. Calculate
its osmolarity and osmotic pressure, at 37 C,
assuming ϕ 5 1 for all solutes. Would you
expect this solution to be hypertonic, hypotonic,
or isotonic to mammalian cells?
22. Plasma contains a variety of proteins that exert
osmotic pressure. A list of these, with their
molecular weights and approximate protein concentrations, is given in Table 2.PS2.3.
From these data, calculate the approximate
osmotic pressure in plasma that is due just to
plasma proteins.
23. A. The device shown in Figure 2.PS2.2 was used
to determine the steady-state flow and pressure across a dialysis membrane. The data
Problem Set
Syringe drives fluid
into the compartment
Syringe pump
O ring retainers
Motor
Dialysis membrane
1 L beaker
Pressure transducer
measures pressure
Water
Pressure increases and forces
fluid out to match inflow
Recorder shows pressure
increase until steady - state
is reached
FIGURE 2.PS2.2 Device used to determine LP. Water was injected into the inner chamber at a known rate using a syringe pump. This increases the
pressure within the chamber and forces fluid out through the membrane. The pressure increases as more water is injected and eventually a steady
state is reached in which the rate of injection matches the rate of filtration through the membrane. The steady-state pressure is measured
continuously by a pressure transducer and recorded.
TABLE 2.PS2.4 Pressure at Steady-State Flow Across a
Dialysis Membrane
Flow Rate (cm3 min21)
0
Syringe for addition or removal
of solutions
Pressure (mmHg)
0
0.0097
180
0.0194
360
0.0388
680
that were obtained are given in Table 2.PS2.4.
The area of the membrane that was exposed
to flow was 90.5 cm2. Determine LP for the
membrane.
B. The membrane in part A was used to separate pure water on the outside from a 0.75 M
sucrose solution on the inside. The flow
across the membrane was measured using
the device shown in Figure 2.PS2.3.
24. When vesicles of the cardiac sarcoplasmic
reticulum (CSR) are incubated with ATP, Mg21
and Ca21, they take up Ca21 and reach a pseudo
steady state. This is a steady state that changes,
but slowly. The uptake of Ca21 is mediated by
the SERCA2a Ca-ATPase. The uptake reaction can
be quenched by adding EGTA to the external
solution, which binds the Ca21 outside of the
vesicles, or by adding glucose plus hexokinase,
that converts the ATP to ADP and glucose-6
phosphate. When the uptake reaction is stopped,
Ca21 that was already taken up by the vesicles
leaks out passively.
A. The amount of Ca21 taken up by the vesicles
is generally normalized to the amount
Graduated tube
O ring retainers
1 L beaker
Dialysis membrane
Water
Magnetic stir bar
Magnetic stirrer
FIGURE 2.PS2.3 Device for measuring osmotic flow at constant ΔP 5 0.
The inner compartment could be drained and then filled with various
experimental solutions. The outer compartment contained pure water.
As fluid enters the compartment across the dialysis membrane as a result
of the osmotic pressure difference, it forces fluid down the horizontal
tube without any increase in the hydrostatic pressure. The rate of fluid
flow can be estimated from the rate of fluid movement down the tube.
of CSR protein in mg rather than being
expressed as a concentration. A typical
steady-state Ca21 uptake is 40 nmol mg21.
In separate experiments, the enclosed
203
204
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
volume of the CSR vesicles was determined
to be 5 μL mg21. What is the approximate
concentration of Ca21 inside the vesicles at
steady state?
B. The average vesicle size determined by electron microscopy is about 150 nm. What is
the volume and surface area of a vesicle this
size, assuming it is a sphere?
C. Given that the enclosed volume of the aggregate vesicles is 5 μL mg21, how many vesicles
are there per mg of CSR protein? How much
surface area is there per mg of CSR protein?
D. The initial passive efflux at a load of
40 nmol mg21 when the pump is stopped is
16 nmol min21 mg21. Convert this to a flux
in units of nmol cm22 s21 by dividing by
the surface area per mg of CSR protein and
converting min to s.
E. From the information in part D, What is the
passive permeability to Ca21 in units of
cm s21?
25. Oral Rehydration Solution used for Oral
Rehydration Therapy, as recommended by
WHO/UNICEF, has the following composition:
2.6 g NaCl
2.9 g Na3C6H5O7∙2 H2O (trisodium citrate dihydrate)
1.5 g KCl
13.5 g glucose
per L of solution.
Calculate the osmolarity of this solution. Trisodium citrate will dissociate fully at neutral pH to 3 Na1 ions
and one citrate23 ion. Assume complete dissociation of
NaCl and KCl.
Cell Signaling
Learning Objectives
G
G
G
G
G
G
G
G
Distinguish among autocrine, paracrine, endocrine, neural,
and neuroendocrine signaling
Explain why Ca21 is a special ion with respect to signaling
Describe in general terms what ligand-gated ion
channels do
List the major steps in turning on and off of heterotrimeric
G-protein-coupled receptors
Distinguish among Gs, Gi, Gq, and G12/13 mechanisms
Describe the steps in a Gs mechanism’s activation of PKA
and how they differ among tissues
Describe the steps in a Gq mechanism’s activation of CAM
kinase
List the four types of catalytic receptors
Describe the steps in a JAKSTAT pathway
Describe in general terms lipophilic signaling molecules'
effects on gene transcription
2.8
CELL-TO-CELL COMMUNICATION
CAN ALSO USE DIRECT MECHANICAL,
ELECTRICAL, OR CHEMICAL SIGNALS
Mechanical signals can originate in the external environment, as in the case of sensory transduction, or they can
be the signals from another cell. Mechanical signaling
requires close contact of cells and generally occurs
through cell junctions as discussed in Chapter 2.2.
Mechanical force originates on filaments within cells that
eventually connect to the extracellular matrix through
cytoskeletal elements. Transmission of these forces occurs
through the extracellular matrix, but it is also sensed by
neighboring cells. All of the pressure sensors in the body
are really stretch receptors, in which mechanical stretch is
transduced into electrical signals or chemical signals.
SIGNALING TRANSDUCES ONE EVENT
INTO ANOTHER
Electrical signals are usually used within the cell, as
part of a signaling pathway to communicate intracellularly, and most often to move the signal rapidly from
one place in the cell to another. Less frequently, direct
electrical coupling occurs between cells. Such electrical
coupling uses gap junctions, whose structure was discussed in Chapter 2.1. This mechanism is vitally important in coordinating some smooth muscle contraction
and cardiac contraction.
In its broadest context, cell signaling involves the
transduction of some event into another event.
In sensory transduction, a sensory cell is exposed to
some external signal that is transduced to produce a
nervous signal, the action potential. As we will see
later in Chapter 3.2, this action potential can move
along cell membranes to rapidly convey the signal,
the action potential, to remote parts of the sensory
neuron. The action potential is then transduced to
release neurotransmitter at the synapse—the gap
between one neuron and another. The neurotransmitter is then transduced to form the response of the
postsynaptic cell, the one on the other side of the
synapse. In the case of cutaneous (skin) senses, the
original sensory signal is mechanical—a push or a
pull on the nerves in the skin. The mechanical signal
is transduced to an electrical signal, and the electrical
signal is then transduced to a chemical signal.
This simple series of events illustrates the use of
mechanical, electrical, and chemical signals in the
body (see Figure 2.8.1).
Chemical signals do not require close contact and
can be classified according to the distances involved,
the mechanism of transmission, and the target of the
chemical signals. These various classes of inter- and intracellular communication are shown in Figure 2.8.2.
In some cases the signaling molecule remains bound to
the cell and so transmission of this signal requires contact
between the signaling cell and its target. This contactdependent signaling is important in development and
in immune responses. In other cases, the signaling cell
releases a chemical that either acts locally (a paracrine or
autocrine signal) or travels through the blood to act on
remote targets (an endocrine signal). Autocrine signals
have receptors on the signaling cell itself or others like it.
Paracrine signals affect other types of cells located in the
neighborhood of the signaling cell. Neurons also release
signaling molecules, usually at the end of a long extension of the cell, the axon. When the neural chemical
signal enters the blood and acts on distant targets, it is
called a neuroendocrine signal. When it acts in the
vicinity of its release site, it is a neurotransmitter.
G
G
205
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00019-7
206
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
The sensory cell transduces
a mechanical stimulus to an
electrical signal, the action
potential
At a terminal, the action
potential is transduced to
a chemical signal,
a neurotransmitter
Sensory neuron cell body
Interneuron
Mechanical stimulus
Action potential
The final response can be
a mechanical, chemical, or
electrical response
The action potential moves
along neuron cell processes
to convey the signal to distant
parts of the cell
The postsynaptic cell
transduces the chemical
signal to another electrical
signal
FIGURE 2.8.1 Transduction of signals. Some kinds of sensory cells can transduce mechanical stimuli to electrical signals which can be conveyed along
their surface for rapid spatial relay of the signal. At the end of the cell, the electrical signal is transduced to a chemical signal to convey the signal
across the gap between the cells. The postsynaptic cell transduces this chemical signal back to an electrical signal.
SIGNALS ELICIT A VARIETY OF
CLASSES OF CELLULAR RESPONSES
Intra- and intercellular signals begin a cascade of events
that eventually changes cell behavior. The response of
cells to signaling events includes altered:
G
G
G
G
G
G
ion transport;
metabolism;
gene expression or differentiation;
shape, movement, or force production;
cell growth or cell division;
apoptosis or programmed cell death.
ELECTRICAL SIGNALS AND
NEUROTRANSMITTERS ARE FASTEST;
ENDOCRINE SIGNALS ARE SLOWEST
The speed of response to an initial stimulus depends on
the mode of delivery of the signal and the mechanism
of the response in the target cells. Electrical signals
are the fastest way to transmit a signal from one place
in the body to another, in milliseconds, but the overall
response depends on what happens in the target cell.
If the response involves changes in activity of proteins
already present in the target cell, the response can be
rapid. If the response involves altered gene expression
that requires synthesis of new protein, response can
take hours. If it involves altered cell growth, it can take
days to complete. Neurotransmitter signaling is the
fastest response, followed by changes in cell shape or
the development of force. Endocrine signals are slowest
but last longer.
VOLTAGE-GATED ION CHANNELS
CONVEY ELECTRICAL SIGNALS
Ion channels allow ions to cross biological membranes
that they otherwise cannot penetrate. Because they
are electrically charged, movement of ions makes a
current, and the current either charges or discharges the
membrane—the current alters the membrane potential.
Thus the voltage-gated ion channels are largely responsible for alterations in the membrane potential that is
rapidly conveyed through the cell.
VOLTAGE-GATED Ca21 CHANNELS
TRANSDUCE AN ELECTRICAL SIGNAL
TO AN INTRACELLULAR Ca21 SIGNAL
Cells maintain a very low intracellular [Ca21] (,100 nM)
to avoid Ca21 precipitation with phosphate and organic
phosphates (ATP, etc.) present in high concentrations
in the cytoplasm. The low cytoplasmic [Ca21] allows
increases in cytoplasmic [Ca21] to be used as a signal.
Multiple types of voltage-gated Ca21 channels (voltagedependent calcium channel, VDCCs) reside on the surface
membrane of many cells. Depolarization of the cell
membrane opens these channels, causing Ca21 to move
from the ECF, with 1.2 mM [Ca21], to the intracellular
compartment. The cytoplasm contains a number of
proteins that bind Ca21 with high affinity and that change
shape or activity upon Ca21 binding. The effects of
increasing cytoplasmic [Ca21] include the following:
1. Stimulussecretion coupling: The increased
[Ca21] binds to Ca21 sensors on vesicles, causing
the fusion of the secretory vesicles with the plasma
membrane and release of secreted products into
the ECF.
2. Excitationcontraction coupling: The increased
[Ca21] binds to Ca-sensitive elements on contractile filaments or cytoskeletal elements, causing
either force development or shortening by muscle
cells.
3. Calmodulin-dependent activation of enzymes:
Calmodulin is a small cytosolic protein that
binds four Ca21 molecules and then activates
many enzymes such as myosin light chain kinase
in smooth muscle.
C ell Signal ing
Mechanical signals from the cytoskeleton are
transferred to neighboring cells through the
extracellular matrix that links to the cytoskeleton
Mechanical signals
Electrical signals
Electrical signals can be directly transferred
to a neighboring cell through gap junctions
Chemical signals
Receptor
Signaling cell
Contact dependent
Target cells
Signaling cell
Response
Signaling cell
Target cell
ECF
ECF
Response
Response
Chemical signal
(local hormone)
Paracrine
Autocrine
Signaling cell
Target cell
Endocrine
ECF
Bloodstream
ECF
Neuron
Neuroendocrine
Rapid electrical signal
Response
ECF
Bloodstream
Target cell
ECF
Response
Axon
Neurotransmitter
Neuron
Target cell
Neuron
Response
Diffuse
Response
Discrete
FIGURE 2.8.2 Main classes of signaling. Mechanical signals can pass from cell to cell through filaments in the extracellular matrix attached to membranebound proteins in the surfaces of cells, particularly at cell junctions such as desmosomes. Electrical signals can also pass directly from one cell to another
through gap junctions. Some signaling molecules remain bound to the surface and so the signal affects the target cell only by direct contact of the
signaling cell with the target cell. Cells can release chemical signals that act locally. When they affect the signaling cell, or others like it, they are autocrine
signals. When they affect other nearby cells, they are paracrine signals. Signals that are released into the bloodstream to affect distant target cells are
endocrine signals. If they are released from long processes by neurons, they are neuroendocrine signals. Nerve cells release a variety of chemical signals
at terminals near target cells. These are neurotransmitters. If they are released very close to clustered receptors on target cells, they are discrete
neurotransmitters. If they are released into a general area to affect multiple cells, they are diffuse neurotransmitters. Electrical signals are most often used
intracellularly to rapidly convey the action potential from one part of the cell to another. This is the fastest movement of a signal in the body.
4. Direct activation of enzymes: Ca21 can directly
bind to some enzymes, such as PKC, and activate
them. Figure 2.8.3 illustrates these aspects of
Ca21 signaling in cells.
LIGAND-GATED ION CHANNELS
OPEN UPON BINDING WITH
CHEMICAL SIGNALS
Fast release of chemical signals by electrical signals
in nerve terminals, followed by an electrical signal in the
target cell, is the fastest mechanism of signaling used in
the body. This is the classic neurotransmitter mechanism:
the electrical signal (the action potential) on the
presynaptic cell propagates down the axon to the
nerve terminal where it causes a local intracellular Ca21
signal that releases chemical neurotransmitter into the
ECF immediately adjacent to the postsynaptic cell.
The chemical signal then binds to a receptor which is also
an ion channel, causing a flux of ions across the postsynaptic cell membrane and a resulting electrical signal in
the postsynaptic cell. Three major classes of surface membrane, ligand-gated ion channels (LGICs) have been
identified, as shown in Figure 2.8.4, and many of these
have multiple subtypes. The major types are distinguished by their structures. Receptors for acetylcholine,
serotonin (55 hydroxy tryptamine), gamma amino
butyric acid (GABA), and glycine all consist of five subunits. Each family, such as nicotinic ACh receptor, has
207
208
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
1 An action potential or depolarization of the
surface membrane opens voltagedependent Ca channels on the surface
2
Ca rushes in from the high ECF
[Ca2+] to the low cytoplasmic [Ca2+]
3
Increased cytoplasmic [Ca2+] binds to
Ca sites on target proteins
Ca2+
VDCC
Synaptotagmin
4D
... directly activating some enzymes
Ca2+
4A
... causing stimulus-secretion
coupling in neurons or most
endocrine cells
RyR1
PKC
Ca2+
CSQ
Ca2+
Ca2+
Calmodulin
Ca2+
Ca2+
TnC
MLCK
Ca2+
4B
... activating contraction in muscle
cells
Ca2+
Ca2+
2+
Ca
Inactive
Active
4C... binding to calmodulin, activating
a variety of enzymes
FIGURE 2.8.3 Electrically coupled calcium signaling. Depolarization of the cell membrane opens a calcium channel that lets Ca21 into the cell. At rest,
the cytoplasmic [Ca21] is very low. Upon stimulation, influx of Ca21 raises the [Ca21] enough for Ca21 to bind to Ca21-binding sites on specific
proteins. Ca21 binding to synaptotagmin causes fusion of secretory vesicles with the plasma membrane. Binding to troponin C (TnC) activates force in
skeletal or cardiac muscle. Ca21 binding to calmodulin activates a number of enzymes such as MLCK (myosin light chain kinase) involved in smooth
muscle contraction. In other cases, Ca21 directly activates some enzymes; PKC is shown. RyR1 is the ryanodine receptor on the endoplasmic reticulum
membrane; CSQ is calsequestrin, a calcium-binding protein in the lumen of some ER membranes.
Tetrameric channels
Pentameric channels
Acetylcholine
Serotonin
Na+,K+
Nicotinic ACh
GABA
Glycine
Na+
Cl–
5-HT3
GABAA
Trimeric channels
Glutamate
Cl–
Gly
Ca2+
Na+,K+
NMDA
AMPA
GluN1–3
GluA1–4
ATP
Na+,K+
Kainate
GluK1–5
Na+,K+, Ca2+
P2x
P2x1–7
FIGURE 2.8.4 Ligand-gated ion channels. These channels reside in the plasma membrane and respond to specific ligands by allowing specific ions to
cross the membrane. The channels are classified according to their structure and agonist or chemical signal that opens the channel. The names of the
channels are at the bottom of the figure, and alternate naming conventions have been proposed. Each family of channels has multiple isoforms that
depend on the subunit make-up of the channels.
C ell Signal ing
4
1
Chemical signal binds
to its receptor
Chemical signal
2
α Subunit hydrolyzes GTP and
dissociates from effector
3
α Subunit dissociates from βγ
GTP exchanges for subunit and both may bind to and
GDP on the α subunit change effector behavior
Activated state
Basal state
5 α Subunit reassociates with βγ
subunit and with the receptor
to return to the basal state
Effector 1 Effector 2
Receptor
GDP
Gα Subunit βγ Subunits
Pi
GTP
GDP
FIGURE 2.8.5 General scheme for heterotrimeric GPCRs. The receptors are membrane-bound proteins that bind chemical signals. The heterotrimeric Gprotein consists of an α subunit that binds and hydrolyzes GTP and a βγ subunit that does not dissociate. Binding of the ligand to its receptor triggers the
exchange of GTP for GDP and subsequent dissociation of the α subunit and βγ subunit and both are then able to alter the behavior of effector targets in the
cell. The α subunit spontaneously hydrolyzes its GTP, and the α subunit reassociates with the βγ subunit to return to the basal, unstimulated state.
multiple subtypes consisting of different subunits, but
each member of the subtype responds to one chemical
signal, acetylcholine in this case. Receptors for glutamate
each have four subunits, and this family of LGIC has further subtypes distinguished by artificial agonists (stimulators of the receptor), NMDA (N-methyl D-aspartic acid),
AMPA (α-amino-3-hydroxy-5-methyl-4 isoxazole propionic acid), and kainate.
HETEROTRIMERIC G-PROTEINCOUPLED RECEPTORS (GPCRS) ARE
VERSATILE
G-protein-coupled signaling pathways are versatile
because of their modular structure: they consist of receptors, heterotrimeric G-proteins, and effectors. Receptors
are membrane-bound proteins that bind signaling molecules on the external surface of cells. Binding alters the
conformation of the receptor, and this change is transferred to a heterotrimeric G-protein, consisting of α, β,
and γ subunits. In the unstimulated state, the Gα subunit
binds GDP. Upon ligand binding to its receptor, the
receptor causes the Gα GDP to exchange GTP for the
bound GDP, and the Gα GTP dissociates from the βγ
subunits. The dissociated subunits can then bind to effector molecules, exerting some change in their behavior.
The Gα GTP has inherent GTPase activity, so it reverts
back to Gα GDP and reassociates with the βγ subunit.
The overall plan of the G-protein signaling pathway is
shown in Figure 2.8.5.
THERE ARE FOUR CLASSES OF
G-PROTEINS: GαS, GαI/GαO, GαQ,
AND Gα12/Gα13
β ADRENERGIC STIMULATION IS AN EXAMPLE
OF A GαS MECHANISM
A number of signaling molecules can bind to a receptor that is coupled to a Gαs-protein. All Gαs-protein’s
α subunit binds to a membrane-bound enzyme,
adenylyl cyclase, which converts ATP into 30 ,50 cyclic
AMP, or cAMP. Increasing the concentration of cAMP
activates protein kinase A (PKA), which phosphorylates (adds a phosphate to) specific target proteins.
Phosphorylation of these target proteins alters cell
behavior, the final consequence of exposure to the
chemical signal. The basic pathway for this signaling
mechanism is shown in Figure 2.8.6. This signal is
turned on by activation of adenylyl cyclase and then
PK and then phosphorylation of proteins. It is turned
off by the simultaneous inactivation of all three of
these. The α subunit of the G-protein spontaneously
hydrolyzes its GTP and reassociates with the βγ
subunit. This inactivates adenylyl cyclase and stops its
synthesis of cAMP. The cAMP is also broken down
by phosphodiesterase (PDE) so that the increased
[cAMP] is removed. The cAMP dissociates from the
regulatory subunits of the PKA, causing it to inactivate
so that target proteins are no longer phosphorylated.
However, this does not reverse phosphorylation
of proteins. The phosphorylated proteins are dephosphorylated by specific enzymes called protein
phosphatases. There are four classes of serine/threonine phosphoprotein phosphatases: PP1, PP2a,
PP2b, and PP2c, which dephosphorylate proteins
phosphorylated at serine and threonine residues. Fine
control of this system is provided by the regulation
of both adenylyl cyclase, as described, and by
the regulation of both PDE and the protein
phosphatases.
α2 ADRENERGIC STIMULATION IS AN EXAMPLE
OF A GαI/GαO MECHANISM
Other molecules bind to their G-protein-coupled receptor (GPCR) and release a Gα subunit that inhibits adenylyl cyclase. These are referred to as Gi mechanisms.
An example is epinephrine binding to α2 receptors on
neurons and is illustrated in Figure 2.8.7. Other members of this class achieve the same end—reduction in
cAMP levels—by activating PDE. Retinal photoreceptor
209
210
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
1
Epinephrine binds to
2
β1 or β2 receptor
Epinephrine
3
GTP exchanges for
GDP on the α subunit
Activated state
α Subunit dissociates from βγ
subunit, binds to adenylyl cyclase
and activates it
4 Adenylyl cyclase converts
ATP to cAMP
Basal state
Adenylyl cyclase
β receptor
GDP
Gα Subunit βγ Subunits
Phosphodiesterase
hydrolyzes cAMP
Pi
GTP
GDP
5 cAMP binds to regulatory ATP
subunit of PKA, releasing
the active catalytic subunit
cAMP Phosphodiesterase
AMP
Regulatory subunit
Inactive PKA
Catalytic subunit
PKA catalytic subunit phosphorylates
target proteins, changing their behavior
Active PKA
ATP
ADP
P
P
6
Protein phosphatase
FIGURE 2.8.6 Mechanism of Gs-coupled receptors. Many different kinds of ligands bind to Gs-coupled receptors. Epinephrine is shown, which binds to
β1 and β2 receptors that are coupled to Gs proteins. Activation follows the general scheme shown in Figure 2.8.5. Here the Gαs subunit binds to
adenylyl cyclase, activating it and increasing the cytoplasmic concentration of cAMP. This activates PKA that phosphorylates a variety of target
proteins. The signal is turned off by (1) hydrolysis of GTP by the Gα subunit and dissociation and removal of activation of adenylyl cyclase; (2)
hydrolysis of cAMP by PDE and removal of activation of PKA; (3) dephosphorylation of target proteins by protein phosphatases.
1
Epinephrine binds to
2
α2 receptor
GTP exchanges for
Epinephrine
GDP on the α subunit
3
α Subunit dissociates from βγ
subunit, binds to adenylyl cyclase
and inhibits it
Activated state
Adenylyl cyclase
Basal state
α2 Receptor
Gα Subunit GDP
–
αγ Subunits
GTP
GDP
ATP
cAMP
FIGURE 2.8.7 Mechanism of Gi-coupled receptors. Here epinephrine binds to α2 receptors, which is followed by the inhibition of adenylyl cyclase and
a reduction in cytoplasmic [cAMP].
cells, for example, activate cGMP PDE. This mechanism
is illustrated in Figure 2.8.8. Still other ligands, such as
acetylcholine, bind to M2 receptors that cause inhibition of adenylyl cyclase, and the βγ subunit activates a
K1 channel (see Figure 2.8.9). Thus this class of GPCR
exerts a variety of effects including direct inhibition of
adenylyl cyclase, activation of PDE, and direct activation
of K1 channels.
C ell Signal ing
Light
α Subunit dissociates from βγ subunit
binds to cGMP phosphodiesterase and
activates it
Metarhodopsin II
cGMP PDE
βγ Subunits Activated state
Basal state
+
Transducin
GDP
GMP
Gα subunit
GDP
GTP
Na+
Guanylyl cyclase
+
cGMP
FIGURE 2.8.8 GPCR involved in retinal signal transduction.
Through a series of steps, light converts rhodopsin
to metarhodopsin II, which binds to a heterotrimeric
G-protein called transducin. The Gα subunit exchanges
GTP for GDP, dissociates from the βγ subunit, and activates
cGMP PDE. Guanylyl cyclase in these cells makes cGMP
continuously, and cGMP opens a Na1 channel. Degrading
the cGMP reduces [cGMP] and therefore regulates the
open state of the channel, producing an electrical signal.
α Subunit binds to adenylyl cyclase
and inhibits it
Acetylcholine
βγ Subunits
K+
Activated state
Adenylyl cyclase
Basal state
M2 receptor
GDP
Gα Subunit
GTP
βγ Subunit binds to K
channel and activates it
GDP
ATP
cAMP
FIGURE 2.8.9 Gi mechanism involved in M2 GPCR. Acetylcholine binds to a variety of receptors. The M2 receptor, present in heart, couples binding of
acetylcholine to a Gαi subunit that inhibits adenylyl cyclase. The βγ subunit released by acetylcholine binding directly activates a K1 channel.
GαQ/Gα11 GPCR ACTIVATES PHOSPHOLIPASE
C AND RELEASES CA FROM INTRACELLULAR
STORES
modulatory proteins, called GAPs, for GTPase Activating
Proteins, facilitate the inactivation of these small monomeric GTPases. The overall plan is shown in Figure 2.8.11.
A third class of GPCR activates phospholipase C
on the surface membrane, which cleaves phosphatidyl
inositol bisphosphate to produce diacylglycerol (DAG)
and inositol triphosphate (IP3). The IP3 releases Ca21
from the endoplasmic reticulum, while the DAG activates protein kinase C (PKC) (see Figure 2.8.10).
The superfamily of small monomeric GTPases is divided
into several major subfamilies, including:
Gα12/Gα13-COUPLED RECEPTORS ACTIVATE
SMALL MONOMERIC GTPASES
The Gα12 is the last of the four major families of
heterotrimeric G-proteins (Gs, Gi, Gq, and G12) that we
will discuss. Gα12 and Gα13 are linked to GTP exchange
factors (GEFs) that activate small monomeric G-proteins
by exchanging their bound GDP with GTP. A second set of
Ras, Rho, Rab, Ran, and Arf
These are all small (2040 kDa) proteins that are
membrane bound due to covalent attachment of lipids
(such as N-myristoylation on Arf proteins). The Rho
GTPases regulate the cytoskeleton and play a role in
the regulation of smooth muscle contraction. One of
its effectors is Rho Kinase, a serinethreonine protein
kinase that phosphorylates myosin light chain phosphatase and inactivates it (see Chapter 3.8). Rho is also
involved in cell cycle progression and gene expression.
The Rab family of monomeric GTPases regulates
vesicular traffic as well as modulation of actin
211
212
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
α Subunit dissociates from βγ
subunit, activates PLC
Epinephrine
Activated state
DAG activates PKC
Diacylglycerol
Phospholipase C
PIP2
Basal state
α1 Receptor
PKC
Gα subunit GDP
βγ Subunits
P
GTP
GDP
IP3
ATP
Ca2+
Ca2+
Ca2+
IP3 binds to IP3 receptor on ER
and releases stored Ca2+ to the
cytoplasm
ADP + Pi
FIGURE 2.8.10 Mechanism of Gαq signaling. Binding of ligand to its GPCR results in release of the Gαq and βγ subunits. The Gαq subunit activates
phospholipase C, which cleaves phosphatidyl inositol bisphosphate in the surface membrane, liberating DAG and IP3. IP3 releases Ca21 stored in the
ER and DAG activates PKC that phosphorylates sets of target proteins.
Pi
GTPase activating proteins (GAPs)
Active
Inactive
GTP
GDP
GTP
GDP
GTP exchange factors
(GEFs)
FIGURE 2.8.11 Heterotrimeric G-proteins with Gα12-activated GEFs that
promote the exchange of GTP for GDP on small monomeric G-proteins.
This converts the small monomeric G-proteins into an active form.
Reversion to the inactive form is catalyzed by GAPs, GTPase activating
proteins. The GEF binds to the Gα12 subunit.
dynamics. Members of the Ran family regulate transport
of materials between the cytoplasm and nucleus. Arf
stands for “ADP-ribosylation factor.” All six mammalian
Arfs are located in the Golgi and regulate vesicular
transport there.
THE RESPONSE OF A CELL TO A
CHEMICAL SIGNAL DEPENDS ON THE
RECEPTOR AND ITS EFFECTORS
According to Figures 2.8.6, 2.8.7, and 2.8.10, epinephrine can bind to β GPCRs, with the final activation of
adenylyl cyclase, increased [cAMP], and activation
of PKA; or it can bind to α2 receptors with the
inhibition of adenylyl cyclase, decreased [cAMP], and
no activation of PKA; or it can bind to α1 receptors
with subsequent activation of PLC, release of Ca21
from intracellular stores, and activation of PKC. These
different effects typically occur in separate cells. Thus
the response of the cell depends on the receptor for
the chemical signal and not on the chemical signal
alone. Further, some cells respond differently than
others because they express entirely different sets of
target proteins. In the liver, for example, the primary
target for PKA from the stimulation of β adrenergic
receptors is phosphorylase kinase, which then phosphorylates phosphorylase, activating it, and glycogen
synthetase inactivating it. In the heart, the primary targets of β adrenergic stimulation are the voltagedependent Ca21 channels in the surface membrane,
phospholamban on the sarcoplasmic reticulum, ryanodine receptors (RyR2) on the sarcoplasmic reticulum,
and troponin I (TnI). These are illustrated in
Figure 2.8.12. Thus the effects of epinephrine using
the same type of receptor are glycogenolysis in the
liver and increased contractile strength in the heart.
These differences indicate that the final effect is a function of (1) the chemical signal, (2) its receptor, (3)
the effector, and (4) the targets within the cell.
CHEMICAL SIGNALS CAN BIND TO AND
DIRECTLY ACTIVATE MEMBRANEBOUND ENZYMES
A variety of extracellular chemical signals can bind to
receptors on the surface membrane; these are amplifying enzymes or directly activate an amplifying enzyme.
They are of four main types: receptor guanylyl cyclase,
receptor serine/threonine kinase, receptor tyrosine
kinase, and receptor-associated tyrosine kinase. These
are illustrated in Figure 2.8.13.
C ell Signal ing
Ca2+
Epinephrine
βγ Subunits
Activated state
Adenylyl cyclase
Basal state
Inactive PKA
β receptor
Ca2+
GDP
Gβ subunit
cAMP
TnI
ATP
GTP
GDP
Regulatory subunit
Catalytic subunit
Myosin filament
Actin filament
Active PKA
ATP
Phosphorylase kinase
(inactive)
Ca2+
ADP
P
ATP
Phosphorylase kinaase
(active)
ADP
Phosphorylase
(active)
Phosphorylase
(inactive)
ADP + Pi
ATP
SERCA2a
Phospholamban
Glucose -1-Pi
Ryanodine receptor
Glycogen
Glycogen synthase P
(inactive)
Glycogen synthase
(active)
ADP
ATP
Liver
Heart
FIGURE 2.8.12 Different effects of beta adrenergic stimulation of liver cells versus heart cells. In the liver, the primary response of beta adrenergic
stimulation is activation of glycogenolysis through activation of glycogen phosphorylase through a cascade of protein phosphorylation reactions. In
the heart, the primary response of beta adrenergic stimulation is faster activation and relaxation of the muscle through control of cytoplasmic [Ca21]
by phosphorylation of voltage-dependent Ca21 channels on the surface of the cell, phosphorylation of the ryanodine receptor on the surface of the
sarcoplasmic reticulum, phosphorylation of TnI on the contractile filaments, and phosphorylation of phospholamban to relieve inhibition of the
SERCA2a Ca21 pump on the surface of the SR.
MANY SIGNALS ALTER GENE
EXPRESSION
So far we have discussed signaling molecules that cannot
penetrate the cell membrane. These bind to receptors
on the surface of the cell, and the binding is transduced
into a second messenger such as Ca21, cAMP, cGMP, IP3,
and DAG, one of a number of small monomeric
GTPases, or causes phosphorylation of intracellular proteins. A number of lipophilic signaling molecules penetrate the cell membrane and bind to receptors either
in the cytoplasm or in the nucleus and alter the expression of specific genes in the cell. Hormones that alter
gene expression this way include the sex hormones
testosterone, estrogen, and progesterone; corticosteroids, including glucocorticoids, produced by the adrenal
cortex; mineralocorticoids, produced by the adrenal
gland that regulate electrolyte and water balance; vitamin
D, which regulates calcium and phosphate balance,
among other effects; and thyroid hormone and retinoic
acid, which have effects in almost all cells.
NUCLEAR RECEPTORS ALTER GENE
TRANSCRIPTION
The nuclear receptors constitute a superfamily of
proteins that are structurally related and perform
similar functions, but they exhibit specificity for
binding ligands and specificity of action. These nuclear
receptors include the following: estrogen receptors α
and β (ERα and ERβ); androgen receptor (AR); progesterone receptor (PR); glucocorticoid receptor (GR);
mineralocorticoid receptor (MR); vitamin D receptor
(VDR); thyroid receptors α and β (TRα and TRβ);
retinoic acid receptor types α, β, and γ (RARα, RARβ,
and RARγ); and 9-cis-retinoic acid receptors (RXRα,
RXRβ, and RXRγ). These nuclear receptors are
restricted to the nucleus with the exception of the
mineralocorticoid receptor (MR) and the GR which
reside in the cytoplasm. Upon binding with its ligand,
MR and GR move into the nucleus where they bind
to the regulatory region of a modulated gene. Binding
of the lipophilic ligand with its receptor changes
the conformation of the receptorligand complex,
allowing the receptor to bind to specific nucleotide
sequences on the DNA, called response elements.
Binding of these receptors begins a cascade of events
that eventually activates the transcription of specific
genes. The newly transcribed mRNA is transferred
to the cytoplasm where it is translated into a protein.
The set of proteins regulated by the hormones
confers specific capabilities on the cells in which
they are expressed. Thus cell function is regulated
by controlling cell concentration of specific active
proteins.
213
214
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Receptor guanylyl Receptor serine/threonine
cyclase
kinase
Receptor tyrosine kinase
TGFβ
Insulin
Receptor-associated
ANP
tyrosine kinase
Insulin
Type II receptor
Growth hormone
receptor
Type I receptor
GHR
+
P
P
PI-3K
ATP
GTP
P
cGMP
PKB
smad2/3
P
P
P
IRS-2
IRS-1
P
P JAK2
Grb-2
+
P
ATP
Ras
P
STAT5
smad4
P
P
STAT or GR
P
DNA
RNA polymerase II
Nucleus
FIGURE 2.8.13 Catalytic receptors. Chemical signals binding to the extracellular surface of the cell are coupled to enzymes that alter intracellular
components. Receptor guanylate cyclases respond to atrial natriuretic peptide or brain natriuretic peptide, and binding increases the concentration
of cGMP in the cell, which increases activity of cGMP-dependent protein kinase. Receptor serine/threonine kinases: dimers of a variety of growth
factors including transforming growth factor beta (TGF-β), myostatin, and bone morphogenic protein (BMP) bind to an activin type II receptor that
recruits an activin type I receptor and phosphorylates it. This active serine/threonine kinase then phosphorylates one of a family of proteins called
smads (smad2 or smad3 is shown), which binds to smad4. The complex enters the nucleus and regulates gene expression. Receptor tyrosine kinases:
Insulin or insulin-like growth factor (e.g., IGF-1 and IGF-2) binds to the insulin receptor. The activated receptor phosphorylates several intracellular
substrates. Insulin receptor substrate-1 (IRS-1) is shown. The phosphorylated proteins can activate two main pathways: the PI-3K (phosphatidyl inositol
3 kinase) pathway activates PKB and PKC downstream. The Ras pathway is activated by Grb-2 that binds to IRS and then activates a Ras GTP exchange
protein leading eventually to transcription factors. Receptor-associated tyrosine kinases are used by growth hormone (shown), prolactin and
erythropoietin, and most interleukins. Their binding induces close proximity of two receptors that bind members of the Janus family of tyrosine
kinases (JAKs: JAK1-3 and TYK2). These transphosphorylate themselves and their receptor and phosphorylate proteins called STAT (for signal
transduction and activation of transcription). These form dimers with other transcription factors and regulate gene transcription.
NUCLEAR RECEPTORS RECRUIT
HISTONE ACETYLASE TO UNWRAP
THE DNA FROM THE HISTONES
Heterochromatin is highly condensed DNA that cannot
be transcribed. Euchromatin is more easily accessible
for the assembly of transcriptional subunits, and DNA
in this configuration has a higher rate of transcription.
The configuration of chromatin is regulated by the
acetylation of histones. Histones are a family of proteins, described in Chapter 2.2, that form a complex
with DNA called a nucleosome that is stabilized by the
attraction of the negatively charged DNA to the positively charged histones. In this form, the DNA cannot
be transcribed. Acetylation reduces the association of
the DNA with the histones by reducing the positive
charge on the histones. Deacetylation promotes condensation of the DNA into heterochromatin. The enzyme
histone acetyl transferase (HAT) sticks acetyl groups
on the histones, and histone deacetylase removes
them. Both of these enzymes associate with coactivators
and repressors of transcription. A variety of proteins
associated with nuclear receptors possess HAT activity.
NUCLEAR RECEPTORS RECRUIT
TRANSCRIPTION FACTORS
Once acetylation of the histones has allowed the reorganization of chromatin, several other complexes of
proteins bind to the DNA to initiate transcription.
The transcription factor TFIID (for transcription factor
polymerase II) is a complex of proteins that binds to a
TATAA sequence on the DNA some 2530 nucleotides
upstream of the initiation site (see Chapter 1.3). TFIID
contains a TATAA-binding protein (TBP), which
binds directly to the TATAA sequence, and a series of
other factors called TBP-associated factors (TAFs). TBP
then binds a second basal transcription factor, TFIIB.
This allows the binding of RNA polymerase II to the
complex, which is then fully activated by the binding of
assorted other transcription factors, TFIIF, TFIIE, and
TFIIH. The nuclear receptor influences transcription
through specific proteins that interact both with the
nuclear receptor on its recognition site and with
the RNA polymerase complex on its initiation site. This
interaction is possible because the DNA can form
loops that closely appose the nuclear receptors and the
C ell Signal ing
Nucleosome
H2A
H2B
H3
H4
RNA polymerase II
6
TFIIE
4
TFIIF
TFIIH
TATAA
H1
TFIIB
TBP
TAF
HAT
3
SRC-1
TFIID
5
1,25 (OH)2D3
1
DRIP
Ligand binding site
2
VDR
RXR
ACTTGG
ACTGGG
3′
3nt
VDRE
RXR
VDR
5′
AF-1
Dimerization site
DNA binding site
FIGURE 2.8.14 Simplified model of activation of transcription of DNA by vitamin D3. Not all steps in this process are established, and the figure is
meant to convey some of the players and their postulated roles. It is not to be taken too seriously. The active form of vitamin D3, 1,25(OH)2D3, binds
to its receptor, VDR (vitamin D receptor), which is nonspecifically bound to DNA. Binding of the ligand (1) results in dimerization of the VDR with RXR
(9-cis-retinoic acid receptor) and binding of the dimer to specific vitamin D-responsive elements on the DNA (VDRE) (2). These are similar repeat motifs
on the DNA as indicated by the sequences ACTTGG and ACTGGG. Transactivation begins with the recruitment of coactivators with HAT activity (3).
One of these is SRC-1 for the steroid receptor coactivator. Acetylation of histones causes chromatin remodeling that facilitates transcription. Actual
initiation of transcription requires the binding of TBP to the TATAA box, along with several TAFs (4). This complex is TFIID, for transcription factor for
RNA polymerase II. TFIID then binds TFIIB, which forms a bridge to RNA polymerase II. Several proteins, called vitamin D-receptor interacting proteins,
or DRIPs, form a bridge to RNA polymerase II, stabilizing the preinitiation complex (5). Transcription is then initiated (6).
preinitiation complex. This process is illustrated for
VDR in Figure 2.8.14. In other cases, nuclear receptors
can also regulate gene expression by suppressing transcription. For example, glucocorticoids suppress the
effects of transcription factor nuclear factor κB (NF-κB),
which stimulates genes as part of the inflammatory
response. Glucocorticoids reduce inflammation by this
effect (see Chapter 9.5).
cAMP
Phosphodiesterase
AMP
Cytoplasm
Regulatory subunit
Inactive PKA
Catalytic subunit
OTHER SIGNALING PATHWAYS ALSO
REGULATE GENE EXPRESSION
The binding of signaling molecules to nuclear receptors is only one of many routes for the regulation
of gene expression. As noted earlier, receptor serine/
threonine kinases can alter gene expression through
phosphorylation of smads; receptor tyrosine kinases
alter gene expression through Ras, and receptorassociated tyrosine kinase alters gene expression
through phosphorylation of STATs. Signals that affect
30 ,50 cyclic AMP levels also regulate gene expression.
Specific genes possess a regulatory sequence called
the cAMP response element or CRE. PKA phosphorylates CREB (CRE-binding protein), a transcription
factor that binds to the CRE. CREB can also be phosphorylated by CaM kinase II and CaM kinase IV.
Transcriptional activation by CREB requires coactivators including CREB binding protein (CBP) with a
molecular weight of 300 kDa. CREB forms homodimers to activate transcription, but it can also form
heterodimers with CREM (CRE modifier) that either
activate or inhibit transcription. A schematic of CREB’s
involvement in the regulation of transcription is
shown in Figure 2.8.15.
Active PKA
ATP
ADP
CREB
CREB
Pi
CBP
Nucleus
CREB
Pi
CREB
CREB
Pi
Pi
DNA
CRE
FIGURE 2.8.15 Modulation of gene transcription by cyclic AMP-dependent
PKA. Activation of adenylyl cyclase occurs on the surface membrane of the
cell through a Gs-coupled receptor for a hormone. The increased cAMP
activates PKA by dissociating the regulatory subunit from the catalytic
subunit. The catalytic subunit translocates to the nucleus where it
phosphorylates CREB, a protein that binds to cyclic AMP responsive
elements (CRE) on the DNA strand. CREB binds to CBP and may form
homodimers or heterodimers and may activate or inhibit transcription. A
variety of cofactors are recruited to complete the mechanism.
215
216
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Small molecular weight,
llipophilic signal
Polypeptide
hormone
3
2
1
4
Trimeric G-proteins
Amplifying enzyme
DAG
PLC
PKC
AC
+
NMDA
Ca2+
Ca2+
VDCC
Secretory protein
Ca2+
IP3
P ATP
cAMP
JAK
PP
CREB
PP
P
STAT5
CREBP
mRNA
+
Growth hormone
ATP
PKA
Ca2+
5A
P
STAT or GR
+
Proteins
RNA
Polymerase II
-
Bind to response
element
Nuclear receptors
Nucleus
PKB
P
Insulin
+
Ribosome
PI-3K 5B
IRS-1
+
P
Ras
FIGURE 2.8.16 Summary of major signaling pathways. (1) Voltage-dependent channels open to convey electrical responses. (2) LGICs convert chemical
signals into electrical signals. (3) Four different classes of heterotrimeric GPCRs convert extracellular chemical signals into intracellular chemical signals.
The GPCR coupled to phospholipase C (Gq) and to adenylyl cyclase (Gs) is shown, but other responses also occur. (4) Small lipophilic signaling
molecules enter the cell and affect gene transcription and other processes. (5) Extracellular chemical signals, larger proteins, activate enzymes that
produce intracellular signals. Growth hormone and insulin receptor types are shown.
SUMMARY OF SIGNALING
MECHANISMS
A synopsis of the signaling mechanisms discussed
in this chapter is shown in Figure 2.8.16. The main classes of signaling include the following: (1) voltage-gated
ion channels, including the fast Na1 channel and K1
channels involved in action potential origination
and propagation. These channels maintain electrical signaling that is rapidly conveyed over the surface of the
cell. (2) LGICs, the mainstay of synaptic transmission
between neurons and between nerve and muscle.
(3) Heterotrimeric GPCR, including four main subtypes:
those that excite adenylyl cyclase (Gs mechanisms);
those that inhibit adenylyl cyclase (GI mechanisms);
those that activate phospholipase C, releasing IP3 and
DAG (Gq mechanisms); and those that stimulate GEFs
to activate one of a family of small GTPase proteins.
(4) Extracellular signals, which directly activate enzymes
such as guanylyl cyclase, receptor serine/threonine
kinase, receptor tyrosine kinase, and receptor-associated
tyrosine kinase. (5) Signaling molecules that bind
cytoplasmic or nuclear receptors. Other signaling
mechanisms, such as those involving sphingosine
phosphate, are not shown here.
SUMMARY
Mechanical, electrical, or chemical signals are used by
cells to communicate. Mechanical signaling and some
chemical signaling require close contact. Electrical
signaling is the fastest way to move a signal from one
part of a cell to another and to adjacent cells. Chemical
signals can be used by a cell to regulate itself (autocrine), its near neighbors (paracrine) or distant cells
through the medium of the blood (endocrine). Nerve
cells use electrical signals over long cell processes to
cause release of chemical signals near their target cells.
Neuroendocrine signals are chemical signals released
by neurons into the blood.
Voltage-gated ion channels make electrical signals possible. The influx of Ca21 ions carries an electrical and a
chemical signal because Ca21 binds to specific receptors
inside the cell to initiate secretion, to activate enzymes
indirectly through CAM kinase or directly through
activation of other enzymes, or to activate contraction.
LGICs convert chemical into electrical signals. This is
used in neurotransmission: an action potential on one
cell is converted to an electrical signal on another.
Many chemical signals have multiple types of receptors,
so that the effect in the postsynaptic cell depends on
the chemical released by the presynaptic cell and the
receptor expressed by the postsynaptic cell.
Many chemical signals bind to receptors on the surface
membrane that are linked to heterotrimeric G-proteins
(GPCRs). These dissociate upon ligand binding and
generally the α subunit activates an amplifying enzyme
such as adenylyl cyclase (for Gs mechanisms) or phospholipase C (for Gq mechanisms), which increase the
C ell Signal ing
formation of 30 ,50 cyclic AMP or IP3, respectively. Other
GPCRs inhibit adenylyl cyclase (Gi) or recruit a number
of small monomeric G-proteins such as Ras and Rho
(G11/12). The βγ subunit can also affect intracellular
targets. Increased cytosolic cAMP activates PKA that
phosphorylates specific target proteins. IP3 released by
Gq-coupled receptors causes Ca21 release from ER stores
and activation of CaM kinase.
Other chemical signals bind to surface receptors that
are catalytic. The four classes are receptor guanylyl
cyclase, receptor serine/threonine kinase, receptor tyrosine kinase, and receptor-associated tyrosine kinase.
Examples of these signals include insulin and growth
hormone. Insulin binding activates an intrinsic tyrosine
kinase that phosphorylates insulin receptor substrates
that bind phosphatidyl inositol 3 kinase, PI-3K. This
forms PIP3, which activates a phosphoinositidedependent protein kinase (PDK). Growth hormone activates a receptor that in turn activates a member of the
Janus Kinase family of proteins, which then phosphorylates STAT5 (signal transduction and activation of
transcription). The phosphorylated STAT molecule turns
on specific genes.
Small lipophilic chemical signals such as thyroxine,
vitamin D, and the steroid hormones penetrate the cell
membrane and bind to receptors either in the cytosol or
in the nucleus. These receptors bind to specific regions
of DNA called response elements. The receptors recruit
a large number of accessory proteins that unravel the
DNA and direct the synthesis of mRNA that codes for
specific proteins.
The phosphorylation state of a set of regulated
proteins depends not only on the activity of the protein
kinase but also on the activity of the protein phosphatases. Both the kinases and the phosphatases may be
regulated to alter the phosphorylation state of cellular
proteins.
REVIEW QUESTIONS
1. What is an autocrine hormone? Paracrine hormone? Endocrine hormone?
2. What is the fastest way to convey a signal from
one part of the body to another?
3. How does an electrical signal on the surface of
a cell become a Ca21 signal in its interior? List
four distinct ways that Ca21 can affect cell
function.
4. What is a ligand-gated ion channel? What is the
source of the extracellular ligand? Is the ligand a
chemical, electrical, or mechanical signal?
5. What is meant by “G-protein-coupled receptor”?
A Gαs mechanism couples to what amplifying
enzyme? What is the product of this enzyme?
What does this product do? What is a Gαi
mechanism?
6. What amplifying enzyme is activated by a Gαq
mechanism? What are its products? What do
these products do?
7. Name the four classes of catalytic receptors.
8. Why do peptide hormones have receptors facing
the extracellular space?
9. How do small lipophilic signals affect cell
function? What is meant by “nuclear receptor”?
What do these receptors do? What is meant by
the term “response element”?
10. How does cAMP alter gene expression?
217
2.9
ATP Production I: Glycolysis
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
Be able to draw a diagram showing the relationship among
glycolysis, tricarboxylic acid cycle, and electron transport
chain
Explain what is meant in describing ATP as the “energy currency” of the cell
Write the empirical formula for glucose
List three sources of glucose in the body
Define glycogenolysis
Define gluconeogenesis
Describe how glycogenolysis is regulated in the liver by
epinephrine
Explain why muscle tissue does not contribute to plasma
glucose directly
Describe how glucose gets into cells
Describe what is meant by substrate-level phosphorylation
Explain the function of lactate dehydrogenase during rapid
glycolysis
Describe how the rate of gluconeogenesis can be
increased
TAKE A GLOBAL VIEW
OF METABOLISM
Intermediary metabolism comprises all of the transformations of biological chemicals that allow the cell to
produce energy and synthesize materials that make
it up. It is a bewildering array of chemicals and
their interconnected pathways. Within this, there are
processes that are the composite of many of the individual processes. Glycolysis, for example, occupies a
special place in the metabolic scheme. We ought to
have some appreciation of its place without having
to recall all of the transformations that occur within it.
The same is true of the citric acid cycle, also known
as the Kreb’s cycle or the tricarboxylic acid cycle.
This set of metabolic transformations is central to
energy production in cells. We ought to understand
the role of the metabolic pathways without necessarily
knowing all of the individual transformations that
occur within them.
ENERGY PRODUCTION OCCURS
IN THREE STAGES: BREAKDOWN
INTO UNITS, FORMATION OF
ACETYL COA, AND COMPLETE
OXIDATION OF ACETYL COA
Figure 2.9.1 shows the overall plan of energy-producing
reactions in cells. These occur in three stages. In Stage 1,
foodstuffs consisting of proteins, lipids, and carbohydrates are broken down into their constituent subunits.
These are the amino acids, simple sugars like glucose,
and fatty acids and glycerol.
In the second stage, these simple subunits are broken
down to form acetyl coenzyme A. Coenzyme A is a
chemical that acts as a carrier for the two-carbon acetyl
group, but it is not a carrier in the sense of being transported across a membrane. It is being carried forward in
a biochemical reaction. The formation of acetyl CoA is
accompanied by the incorporation of some of the
energy of the food into ATP, and some limited formation of another compound, NADH. NADH is nicotinamide adenine dinucleotide. It acts as a carrier for
reducing equivalents. We will learn more about NADH
later on in this chapter. These reducing equivalents are
later used to produce ATP in the mitochondria.
The third stage of energy production takes place in the
mitochondria and involves the complete oxidation of
acetyl CoA to water and CO2 and produces the major
proportion of reducing equivalents. The energy stored
in NADH produced in this stage is converted to energy
stored in ATP via the electron transport chain which is
coupled indirectly to the ATP synthetase in the inner
mitochondrial membrane.
ATP IS THE ENERGY CURRENCY
OF THE CELL
Electricity is a very versatile form of energy that has
come to dominate modern society. We use it to operate heavy machinery, melt metal for casting or extrusion, power drills, pumps, and saws; run television,
toasters, ovens, and computers—we use it for almost
everything. We generate this electric power by burning coal, natural gas, and even public refuse, but we
can also “burn” nuclear material. These methods
218
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00020-3
ATP P roduction I: G lycol ysis
Food
Proteins
Amino acids
Fats
Polysaccharides
Stage 1: Breakdown of foodstuffs
to simple units
Fatty acids,
glycerol
Simple sugars
ADP + Pi
Glycolysis
ATP
NADH
Stage 2: Breakdown of simple units to
acetyl CoA with limited production
of ATP and NADH
Pyruvate
NADH
CO2
Acetyl CoA
Citric acid
cycle
CO2
Stage 3: Complete oxidation of acetyl CoA
with the production of lots of NADH
and ATP
Reducing power:
NADH
ADP + Pi
Electron
transport
chain (ETC)
ATP synthetase
ATP
O2
H2O
NH3, urea
CO2
Waste products
FIGURE 2.9.1 Overall scheme of intermediary metabolism. In the first stage, macronutrients found in food are broken down into their constituent
subunits. In the second stage, these are converted to acetyl CoA in a process that produces only a little ATP and NADH. In the third stage, the acetyl
CoA is completely oxidized, accompanied by the production of lots of ATP and NADH.
generate power by boiling water to turn a turbine
connected to a dynamo. We can also generate electric
power by turning a dynamo by moving water or
wind. We can also use solar radiation to generate
useful electrical power.
Our cells have an analogue of the power plant: the
mitochondrion. It does not make electric power, but it
does generate chemical energy in the form of ATP. Just
like electric power, ATP can be generated from multiple kinds of fuel. Carbohydrates, fats, or amino acids
can all be “burned” to produce energy that is stored in
the terminal phosphate bond of ATP. Just like societal
production of electrical energy, ATP formation has a
final common pathway in the mitochondria, the
“power house of the cell.” Analogous to electricity,
ATP can also be produced outside of the mitochondria.
Like electricity, this form of chemical energy is very
versatile. ATP fuels chemical work such as the synthesis
of materials. It fuels mechanical work such as muscle
contraction and movement of the cytoskeleton. It fuels
electrical work in moving ions across membranes. The
extra energy not directly captured by these processes is
used to heat the body. All of these activities require
ATP to be split into ADP and inorganic phosphate, Pi.
The human body continuously splits ATP, and the
steady state requires that this continuous splitting is
matched to a continuous reformation of ATP from
ADP and Pi. This idea is shown in Figure 2.9.2.
FUEL RESERVES ARE STORED
IN THE BODY PRIMARILY IN FAT
DEPOTS AND GLYCOGEN
Energy that the body uses for movement, biochemical
synthesis, and transport all ultimately derives from
chemical energy stored in food. However, the body
stores some of this energy in its own materials. These
include the fat deposits in adipose tissue and glycogen
granules stored in the muscles and liver. Energy is not
stored as protein deposits, but body proteins can be
219
220
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Food
Proteins
Polysaccharides
Amino acids
Simple sugars
Glycolysis
Fats
Fatty acids,
glycerol
ADP + Pi
ADP + Pi
ATP
NADH
ATP
Chemical work
Electrical work
Mechanical work
Heat
Pyruvate
NADH
CO2
Acetyl CoA
Citric acid
cycle
CO2
Reducing power:
NADH
ADP + Pi
Electron
transport
chain (ETC)
ATP synthetase
ATP
O2
Chemical work
Electrical work
Mechanical work
Heat
H2O
NH3, urea
CO2
Waste products
FIGURE 2.9.2 ATP as the energy currency of the cell. ATP is continuously being used for a variety of purposes that include chemical synthesis,
production of mechanical force and transport of materials, and movement of ions that constitutes an electrical current. ATP hydrolysis also generates
heat. This continuous use of ATP varies with the state of activation. ATP hydrolysis is coupled to resynthesis of ATP in order to maintain a constant
supply of energy so that activation that is coupled to increased rates of ATP hydrolysis is simultaneously linked to increased rates of ATP synthesis.
and are continuously used as energy sources. We begin
our discussion of energy metabolism with glucose.
H
HOH2C6
HO
1
5
GLUCOSE IS A READILY AVAILABLE
SOURCE OF ENERGY
Many cells of the body, particularly those in the central
nervous system, depend crucially on glucose as an
energy source. Glucose is a six-carbon compound with
the empirical formula of C6H12O6. It is called a carbohydrate because its chemical formula is close to
Cn(H2O)n, indicating a 1:1 ratio between carbon and
water. Thus its empirical formula is equivalent to a
hydrated carbon atom. The chemical structure of glucose is shown in Figure 2.9.3. The blood plasma
O
H 4
3
H
OH
2
H
OH
OH
α-D glucopyranose
FIGURE 2.9.3 Chemical structure of α-D-glucopyranose. Glucose can
exist in several configurations, one of which is shown here. The glucose
atoms within the molecule are numbered 1 through 6 as shown in the
figure. The pyranose ring forms a six-membered structure that
approximates a plane. The hydroxyl side groups project from the plane
either up or down. At C-1, C-2, and C-4 it is down and at C-3 it is up.
When the hydroxyl group is down it is designated as α; when it is up it
is designated as β.
ATP P roduction I: G lycol ysis
typically contains glucose at levels between 80 and
120 mg%. Recall that mg% is mg of glucose per 100 mL
of plasma (51 dL). Glucose enters the circulation from
several sources. The first source is directly from foodstuffs. Plant starches in the food we eat are broken
down to glucose which is absorbed from the intestine
into the portal blood (blood that flows from intestine
to liver) and then into the general circulation.
Another source of glucose is from glycogen stored in
the liver and in muscle. Glycogen is a polymer of glucose in which the glucose subunits are stuck together
end to end. There are two ways of doing this, called an
α-1,4 glucosidic bond and an α-1,6 glucosidic bond.
This nomenclature merely names the numbers of the
carbon atoms that are attached to one another and the
α signifies the stereochemistry of how the bond is
formed. The chemical structure of glycogen is shown in
Figure 2.9.4. Glucose is stored as glycogen in many
cells, but in large quantities in the muscles and liver.
The glucose in glycogen cannot release its chemical
energy while it is bound in the glycogen. It must first
be broken down to the constituent subunits, the glucose molecules, by a process called glycogenolysis. The
root word “lysis” means “break down,” so glycogenolysis means “glycogen break down.” Liver glycogen can
contribute to blood glucose, whereas muscle glycogen
is converted to glucose in the muscle fiber and used
only for muscle activities.
A third source of glucose is from amino acids. Some
amino acids can be used to produce glucose through a
process called gluconeogenesis. Literally, this means
“new glucose formation.”
HO
HO
HO
4
6
5
1
HO
O
HO
4
HO
HO
HO
α–1,4-bond
O
3 2
O
HO
HO
O
HO
O
1
OH O
HO
HO
O
OH O
HO
4
5
α–1,6-bond
O
3 2
1
OH O
6
O
OH O
HO
HO
O
OH O
FIGURE 2.9.4 Structure of glycogen. Note that glycogen is a branched
polymer of glucose. The α-1,4 glucosidic bond connects linear chains of
glucose molecules. The α-1,6 glucosidic bond causes the chain to branch.
GLUCOSE RELEASE BY THE LIVER IS
CONTROLLED BY HORMONES
THROUGH A SECOND MESSENGER
SYSTEM
Glycogenolysis in the liver is controlled partly by
hormones. A hormone is a material which is released
from secretory cells in the body that travels through
the body via the blood, and has an effect on target
cells located some distance away (see Chapter 2.8).
One of the important hormones regulating glycogenolysis in the liver is epinephrine. Epinephrine does
not enter the liver cell. It binds to a receptor on the
hepatocyte (liver cell) surface and a “second messenger” is produced within the cell. The receptor for
epinephrine is a G-protein-coupled receptor (GPCR),
as discussed in Chapter 2.8. The receptor is coupled to
a heterotrimeric G-protein, a class of protein that
binds GTP, guanosine triphosphate. In the case of the
epinephrine receptor, the G protein is a Gαs, meaning
that the α subunit of the heterotrimeric G-protein
stimulates adenylyl cyclase to increase the cytosolic
concentration of cyclic AMP (30 ,50 cyclic adenosine
monophosphate). The cAMP is the “second messenger” within the hepatocyte.
The cAMP then activates an enzyme, protein kinase A
(PKA), in the liver cell. PKA begins a cascade of phosphorylation reactions that shuts down glycogen synthesis and activates glycogen breakdown according to the
scheme shown in Figure 2.9.5.
After activation by cAMP, the system returns to its
inactivated state in two ways. First, the cAMP produced by adenylyl cyclase is degraded to AMP (adenosine monophosphate) by another enzyme, cAMP
phosphodiesterase. This turns off the second messenger signal. Second, protein phosphatases dephosphorylate the proteins that were phosphorylated
during activation of the cascade. There are four classes
of serine/threonine phosphoprotein phosphatases:
PP1, PP2a, PP2b, and PP2c. PP1 dephosphorylates
many of the proteins phosphorylated by PKA. The balance between phosphorylated and dephosphorylated
proteins is set by the competing activity of the kinases
and phosphatases.
THE LIVER EXPORTS GLUCOSE
INTO THE BLOOD BECAUSE IT CAN
DEPHOSPHORYLATE GLUCOSE-6-P
In the liver, glycogenolysis ends at glucose-1-P. This
is converted to glucose-6-P by another enzyme, phosphoglucomutase. The glucose-6-P is then converted to
glucose by glucose-6-phosphatase. This enzyme is
extremely important because only the liver, kidney,
and intestine have it, allowing them to release glucose
into the blood from glucose-6-P; neither glucose-1-P
nor glucose-6-P can exit the cell. Muscle cells have
glycogen stores that can be broken down to provide
energy, but only for the muscle cell because they lack
221
222
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
cAMP
+
Protein kinase A
ATP
ADP
Phosphorylase kinase
(inactive)
Phosphorylase kinase-P
(active)
Phosphorylase -P
(active)
Phosphorylase
(inactive)
FIGURE 2.9.5 Cascade of activation events to shut down
glycogen synthesis and activate glycogenolysis upon
stimulation of the liver with epinephrine. Epinephrine binds
to a G-Protein-Coupled Receptor on the surface of the
hepatocytes which stimulates adenylyl cyclase to increase
formation of 30 ,50 cyclic adenosine monophosphate (cAMP).
The increased cAMP stimulates protein kinase A, which then
phosphorylates the enzyme phosphorylase kinase, so-named
because it phosphorylates another enzyme, phosphorylase.
Phosphorylase has its name because it phosphorylates
glycogen during glycogenolysis to produce glucose-1phosphate. PKA also phosphorylates glycogen synthase,
converting it from its active form to an inactive form.
glucose-6-phosphatase. Muscles cannot contribute glucose to the blood.
A SPECIFIC GLUCOSE CARRIER TAKES
GLUCOSE UP INTO CELLS
In muscle cells, glucose can be taken up from the
blood by a glucose transporter, GLUT, of which there
are multiple isoforms. The one in muscle and fat is
GLUT4. The number of these receptors is regulated
hormonally, and they exist in a latent form in vesicles
stored within the cell. The GLUT4 transporters are
particularly sensitive to the hormone insulin. Brain,
liver, and red blood cells have GLUT transporters that
are not regulated by insulin, and therefore these tissues are insensitive to insulin. Muscle cells can also
derive glucose from glycogenolysis within the cell. The
fate of glucose, whether derived from blood or glycogen, is conversion to pyruvate through the process of
glycolysis.
GLYCOLYSIS IS A SERIES
OF BIOCHEMICAL TRANSFORMATIONS
LEADING FROM GLUCOSE
TO PYRUVATE
Figures 2.9.6 and 2.9.7 show the reactions of glycolysis
that produce pyruvate from glucose. These reactions
occur in the cytoplasm. The pyruvate then enters the
mitochondria where it is completely oxidized and produces a number of ATP molecules per molecule of
pyruvate.
Glycogen
Glycogen synthase-P
(inactive)
Glucose-1-P
Glycogen synthase
(active)
ATP
ADP
Protein kinase A
+
cAMP
GLYCOLYSIS GENERATES ATP QUICKLY
IN THE ABSENCE OF OXYGEN
Glycolysis can generate ATP in the absence of oxygen.
This is described as anaerobic metabolism. It results
from substrate-level phosphorylation. This is distinct from oxidative phosphorylation that occurs in
the mitochondria. Substrate-level phosphorylation
refers to the formation of ATP from ADP and a phosphorylated intermediate, rather than from ADP and
inorganic phosphate, Pi, as is done in oxidative
phosphorylation.
The amount of ATP that is generated by glycolysis is relatively low. Two ATP molecules are required to start glycolysis (from glucose), and four are generated by
substrate-level phosphorylation. An additional two
NADH molecules are generated, which can be used to
generate another three to five ATP molecules through
the electron transport chain in the mitochondria. So a
net gain of 57 moles of ATP can be generated from
the conversion of 1 mole of glucose to 2 moles of pyruvate. The total energy in the oxidation of glucose is
2867 kJ mol21. The energy in 7 moles of ATP is about
7 3 57.1 kJ mol21 5 399.7 kJ mol21. This represents capture of only some 14% of the total energy available
from glucose oxidation.
GLYCOLYSIS REQUIRES NAD 1
Glycolysis occurs in the cytoplasm and it generates
some NADH from NAD1. The NAD1 is an obligatory substrate for the reaction of glyceraldehyde-3phosphate to 1,3-diphosphoglycerate. If NAD1 is not
ATP P roduction I: G lycol ysis
CH2OH
OH
O
Glucose
OH
OH
OH
CH2OH
OH
O
ATP
Glucokinase (liver)
Hexokinase (muscle)
ADP
CH2OPO3
Phosphoglucomutase
O
OH
Glucose-6-phosphate
Glycogen
OH
OH
OPO3
OH
OH
OH
Glucose-1-phosphate
Phosphoglucose isomerase
CH2OPO3
OH
O OH
Fructose-6-phosphate
OH
Pi
Fructose-1,6-diphosphatase
H2O
CH2OH
ATP
Phosphofructokinase
ADP
CH2OPO3
OH
O OH
OH
Fructose-1,6-diphosphate
CH2OPO3
Glyceraldehyde-3-phosphate
Aldolase
Dihydroxyacetone phosphate
CH2OPO3
HC
O
C
HC
OH
O
CH2OH
Triose phosphate isomerase
CH2OPO3
FIGURE 2.9.6 First part of glycolysis, leading from glucose to two three-carbon intermediates that are readily interconvertible. Chemical structures and
names of the intermediates are shown in black. The enzymes that participate in the interconversions are shown in blue. Glycolysis begins by
phosphorylation of glucose in two successive steps, forming glucose-6-phosphate and then forming fructose-1,6-diphosphate. These steps in glycolysis
require ATP to “prime” the process.
regenerated, glycolysis will halt. In the presence of
oxygen, NADH is oxidized in the mitochondria to
regenerate NAD1, but NADH itself cannot cross the
mitochondrial membrane. Two shuttles transfer the
“reducing equivalents” across the mitochondrial
membrane. These are the glycerolphosphate shuttle
and the malate/aspartate shuttle (see Chapter 2.10).
Figure 2.9.8 illustrates the requirement of glycolysis
for NAD1.
If the glycolytic generation of NADH exceeds the
mitochondrial oxidation of cytoplasmic NADH, then
cytoplasmic NAD1 will become depleted and its
absence will limit the metabolic flux through glycolysis. Under these conditions, the cell must regenerate
NAD1 from NADH in order to allow glycolysis to continue. This is achieved by making lactic acid from
pyruvate through the enzyme lactate dehydrogenase,
LDH. Lactic acid production occurs all the time, but
increases when glycolysis is going faster than the
mitochondria can accommodate the metabolic flux of
cytoplasmic NADH, regardless of the state of oxygenation of the tissue.
A good example of this occurring physiologically is in
muscle during brief strenuous exercise such as a 200-m
sprint. In this case, nearly all of the energy will be supplied by glycolysis. In order for glycolysis to continue,
the muscle will produce lactic acid, which will leave the
muscle and travel to the liver. The oxygen necessary to
oxidize the accumulated lactic acid constitutes part of
the “oxygen debt” that must be repaid when oxygen is
available.
Provided the liver is adequately oxygenated, the liver
will reoxidize the lactic acid to pyruvate, which can then
223
224
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
HC
O
HC
OH
Glyceraldehyde-3-phosphate
is converted to muscle glucose is called the Cori cycle
(see Chapter 3.7).
CH2OPO3
NAD+
Pi
Glyceraldehyde-3-phosphate
dehydrogenase
NADH + H+
OPO3
C
O
1,3-Diphosphoglycerate
OH
HC
Substrate-level
phosphorylation
CH2OPO3
ADP
Phosphoglycerate kinase
ATP
–
C
O
O
HC
3-Phosphoglycerate
OH
CH2OPO3
Phosphoglycerate mutase
–
O
C
O
OPO3
HC
2-Phosphoglycerate
CH2OH
Enolase
O
Substrate-level
phosphorylation
–
C
O
C
OPO3
Phosphoenolpyruvate
CH2
ADP
Pyruvate kinase
ATP
O
–
C
O
C
O
Pyruvate
CH3
FIGURE 2.9.7 Second part of glycolysis leading from glyceraldehyde-3-P
to pyruvate. Chemical structures and names of the intermediates are
shown in black. The enzymes that participate in the interconversions
are shown in blue. ATP is formed twice in this sequence, once in the
conversion of 1,3-diphosphoglycerate to 3-phosphoglycerate and for a
second time in the conversion of phosphoenolpyruvate to pyruvate. The
formation of ATP directly from phosphorylated intermediary metabolites
is called substrate-level phosphorylation. Two molecules of
glyceraldehyde-3-phosphate are formed from every molecule of glucose.
Thus glycolysis produces 4 ATP per molecule of glucose.
be converted to glucose by gluconeogenesis. The glucose
so formed can be released by the liver into the blood
for use again by the muscle. The overall process by
which muscle glucose becomes blood lactic acid which
GLUCONEOGENESIS REQUIRES
REVERSAL OF GLYCOLYSIS
Energy transduction in cells involves glycolysis, as we
have reviewed it, plus the complete oxidation of pyruvate in the mitochondria, plus the oxidation of other
fuels such as fats and proteins. Some tissues (liver, intestine, kidney) export glucose into the blood for the muscles to use during exercise. As mentioned earlier, the
liver can mobilize glycogen stores for this purpose, but
it can also make new glucose from the amino acids
derived from proteins. The process of making new glucose from proteins is called gluconeogenesis. It
involves chemically transforming the hydrocarbon parts
of amino acids into intermediates of carbohydrate
metabolism, and then running glycolysis backwards to
form glucose. How this is accomplished is illustrated in
Figure 2.9.9 for the effect of glucagon on liver cells.
Briefly, glucagon activates glycogenolysis through means
similar to what we have described earlier for epinephrine. This produces glucose-1-phosphate. In the liver,
phosphoglucomutase converts glucose-1-phosphate to
glucose-6-phosphate. Glucose-6-phosphatase removes
the phosphate from glucose-6-phosphate to produce
glucose, which is then released into the blood stream.
Activated PKA also phosphorylates CREB, the cyclic
AMP responsive element binding protein. This activates its binding to the CRE, cAMP responsive element.
Activation of CRE increases the transcription of another
transcriptional activator that then turns on the synthesis
of PEPCK, phosphoenolpyruvate carboxy kinase. This
enzyme converts oxaloacetate to phosphoenolpyruvate. The oxaloacetate is a common carbohydrate
intermediate formed from the glucogenic amino acids.
These are amino acids that form glucose (see
Chapter 2.11). PKA also indirectly regulates a key controlling enzyme in glycolysis: phosphofructokinase
1/fructose biphosphatase 1(PFK1/FBPase1). PFK converts fructose-6-phosphate to fructose-1,6-biphosphate;
FBPase converts fructose-1,6-biphosphate to fructose-6phosphate. The FBPase1 activity and PFK1 activities are
regulated by cytosolic levels of fructose-2,6-biphosphate (FBP). Fructose-2,6-biphosphate stimulates PFK
activity and it inhibits FBPase activity. Fructose-2,6biphosphate levels are determined by the activity of
phosphofructose kinase 2 and fructose-2,6-biphosphatase (FBPase2) which convert fructose-6phosphate to fructose-2,6-biphosphate. The activities of
PFK2/FBPase 2 reside on a single polypeptide chain.
PKA phosphorylates PFK2/FBPase2, stimulating the
FBPase2 activity and inhibiting the PFK2 activity. This
reduces the level of fructose-2,6-biphosphate, which
subsequently removes activation of PFK1 and removes
inhibition of FBPase1. The net result is an inhibition of
PFK1, which thereby slows glycolysis, and activation of
FBPase1, which increases gluconeogenesis.
ATP P roduction I: G lycol ysis
Glucose
ATP
Glucose-6-P
ATP
Fructose-6-P
Fructose-1,6-diP
2 NAD+
2 NADH
2 Glyceraldehyde-3-P
2 1,3-Diphosphoglycerate
2 3-Phosphoglycerate
2 ATP
2 2-Phosphoglycerate
2 NAD+
2 NADH
2 Lactate
2 Phosphoenolpyruvate
2 Pyruvate
2 ATP
Lactate dehydrogenase
1
FIGURE 2.9.8 Necessity for regenerating NAD during rapid glycolysis. When NADH oxidation by the mitochondria cannot keep pace with glycolysis,
[NAD1] falls and [NADH] rises. The oxidation of NADH by lactate dehydrogenase, converting pyruvate to lactate, occurs to regenerate NAD1 so that
glycolysis can continue to generate some ATP.
SUMMARY
Cells use chemical energy to power their synthetic,
mechanical, and transport work. The chemical energy
stored in the terminal phosphate bond of ATP is used
as a common energy source for all of these processes.
Cells produce ATP by linking the energy of oxidation of
foodstuffs to the chemical synthesis of ATP. Oxidation
of carbohydrates, fats, and proteins all give rise to ATP
as a common energy currency for the cell.
The overall process of energy production occurs in three
stages: (1) breakdown of foodstuffs into component
units (amino acids for the proteins; fatty acids and glycerol for fats; glucose and fructose for carbohydrates);
(2) formation of acetyl CoA with limited formation of
ATP and NADH; (3) complete oxidation of acetyl CoA
with the production of lots of NADH and ATP through
the electron transport chain in the mitochondria.
Carbohydrates provide the most rapid source of ATP.
Glucose in the blood can be taken up by tissues
through specific glucose transporters in their cell membranes (GLUT1, GLUT4) to provide a ready source of
energy. Liver and muscle cells store carbohydrates in a
readily usable form called glycogen. Liver can convert
glycogen stores to blood glucose but muscle uses its
glycogen stores inside the muscle by converting it to
glucose-1-phosphate. Glycogen utilization begins with
its breakdown into component glucose molecules, a
process called glycogenolysis. In liver cells this is regulated by hormones. One important hormone, epinephrine, helps raise blood glucose by mobilizing liver
glycogen. It achieves this task by binding to a receptor
on the outside surface of hepatocytes. This receptor is
coupled to a G-protein, so-named because it binds
and then hydrolyzes guanosine triphosphate, GTP.
This G-protein consists of three subunits: α, β, and γ.
Upon binding of hormone, the α subunit dissociates
and activates adenylyl cyclase, which converts intracellular ATP to 30 ,50 -cyclic AMP (cAMP). The cAMP then
activates protein kinase A (PKA), which phosphorylates a number of target proteins involved in glycogen
metabolism. PKA phosphorylates phosphorylase
kinase, which then phosphorylates phosphorylase, the
enzyme that breaks down glycogen. It also phosphorylates glycogen synthase, inactivating it. In this way,
increasing cAMP turns on glycogenolysis and inhibits
glycogen synthesis.
The end product of glycogenolysis is glucose-1phosphate. This is converted to glucose-6-phosphate
by phosphoglucomutase. Glucose-6-phosphate can then
enter glycolysis, the conversion of glucose to pyruvic
acid that occurs in the cytoplasm. Liver cells can convert
225
226
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Active PKA
Active PKA
+
+
Glycolysis
Glucose-6-phosphatase
Glucose-1-Pi
Glucose
Glucose-6-Pi
Plasma
glucose
Glycogen
Hexokinase
+
Active PKA
FBPase2
Fructose-6-P
ATP
PFK2
Fructose-2,6 P2
PFK2
CREB
mRNA
−
−
Fructose-2,6 P2
–Pi
RNA
Polymerase II
+
FBPase2
+
PFK1
FBPase1
−
Fructose-1,6-diP
+
-Pi -Pi
Active PKA
Active PKA
Bind to CRE
+
Nucleus
PEPCK
Gluconeogenesis
2 Phosphoenolpyruvate
Oxaloacetate
Ribosome
2 Pyruvate
Malate
Amino acids
FIGURE 2.9.9 Mechanism of action of glucagon on liver cells to put glucose into the blood. Glucagon increases key processes, indicated by the circled
1 signs, through increasing cAMP in the hepatocytes. Gluconeogenesis is the synthesis of new glucose from amino acids, indicated by the pathway
highlighted in blue. Gluconeogenesis requires the conversion of fructose-1,6 diphosphate to fructose-6-phosphate, the reverse of the reaction that
occurs during glycolysis. This is accomplished by inhibiting PFK1 and activating FBPase. Inhibition of PFK1 and activation of FBPase are brought about
by decreasing fructose-2,6 diphosphate levels by stimulating FBPase2 and inhibiting PFK2 through phosphorylation mediated by PKA.
glucose-6-phosphate to glucose, which it then exports
into the blood. Muscle cells lack glucose-6-phosphatase
and so cannot export glucose into the blood.
is produced to regenerate NAD1 so that glycolysis can
continue.
The first stage in glucose oxidation is glycolysis, in
which one molecule of glucose is converted to two
molecules of pyruvic acid. Glycolysis generates some
ATP by substrate-level phosphorylation (occurring at
the level of the phosphorylated intermediates of glycolysis as opposed to synthesis from ADP and Pi). It also
requires another compound, nicotinamide adenine
dinucleotide (NAD1). This compound is converted to
NADH during glycolysis, and under aerobic conditions
is regenerated from NADH by the mitochondria.
When glycolysis outstrips the ability of mitochondria
to regenerate NAD1, NADH can be converted to NAD1
by linking this conversion to the production of
lactic acid from pyruvate through the enzyme lactic
dehydrogenase. Thus in strenuous activity lactic acid
REVIEW QUESTIONS
1. What is glucose? Name three processes that supply body cells with glucose.
2. What is glycogen? What is glycolysis? Why can
liver convert glycogen to blood glucose but muscle
cannot?
3. Why is ATP first consumed in glycolysis instead
of being produced? What is substrate-level
phosphorylation?
4. Why do muscle cells produce lactic acid during
bursts of activity?
5. What is lactate dehydrogenase?
6. What is the Cori cycle?
7. What is gluconeogenesis?
ATP Production II:
The TCA Cycle and
Oxidative Phosphorylation
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
G
G
G
G
Describe the reaction catalyzed by pyruvate dehydrogenase
Describe in general terms the function of the water-soluble
vitamins
Describe what it means to say that NADH is a carrier for
reducing equivalents
Define reduction potential and describe how it can be
measured
Be able to calculate the energy released in a reduction
oxidation reaction
List the number of NADH molecules generated per turn of
the TCA cycle
List the number of FADH2 molecules generated per turn of
the TCA cycle
List the number of ATP molecules (or equivalent) produced
by substrate-level phosphorylation per turn of the TCA
cycle
Indicate where CO2 is released during glucose oxidation
Describe what is meant by the “electron transport chain”
Tell the approximate magnitude and sign of the membrane
potential across the inner mitochondrial membrane
Give the stoichiometry of ATP formation from NADH; from
FADH2
Be able to calculate the electrochemical potential difference
for H1 ions across the inner mitochondrial membrane
Describe in words how the ATP synthase makes ATP
Describe the chemiosmotic hypothesis for oxidative
phosphorylation
Describe in general terms how cytoplasmic NADH enters
the mitochondria
Describe how ADP and Pi get into the mitochondrion and
how ATP leaves it
OXIDATION OF PYRUVATE OCCURS
IN THE MITOCHONDRIA VIA THE
TCA CYCLE
Pyruvate is the end product of glycolysis. Its metabolism
continues in the mitochondria via the “TCA cycle,” the
tricarboxylic acid cycle, so named because many of
the intermediates have three carboxyl groups. It is also
2.10
referred to as the Krebs cycle in honor of Sir Hans
Krebs, who did much of the pioneering work in describing it, and it is also referred to as the citric acid cycle
because citric acid is formed in it. This series of metabolic transformations occurs in the inner mitochondria
of cells. Its fuel source is pyruvic acid derived from
glycolysis in the cytosol. The TCA cycle can also be
initiated within the mitochondria by the oxidation of
fatty acids to form acetyl CoA (see Chapter 2.11).
PYRUVATE ENTERS THE
MITOCHONDRIA AND IS CONVERTED
TO ACETYL CoA
The mitochondria have two membranes, an outer membrane and an inner membrane. The outer membrane
is relatively permeable, whereas the inner membrane is
highly impermeable to most materials. Pyruvate produced in the cytosol by glycolysis crosses the inner
mitochondrial membrane by facilitated diffusion on its
own pyruvate carrier. Inside the matrix of the mitochondria, pyruvate is converted to acetyl coenzyme A.
This conversion of pyruvate to acetyl CoA requires three
different enzymes and five different coenzymes, which
are organized into a multienzyme complex called pyruvate dehydrogenase. Three of the coenzymes required
here are vitamins: thiamine, riboflavin, and niacin.
The water-soluble vitamins all find their use in
mammals as part of enzymatic reactions, and most of
the B vitamins are involved in carbohydrate metabolism. The overall reaction is shown in Figure 2.10.1
along with the structure of coenzyme A.
PYRUVATE DEHYDROGENASE
RELEASES CO2 AND MAKES NADH
The production of acetyl CoA from pyruvate is noteworthy because here is the first production of CO2 from
glucose. This gas forms a major waste product that must
be eliminated, largely through the lungs. Second, the
reaction produces NADH from NAD1. NADH is a carrier for reducing equivalents in the cell. The structures
of NAD1 and NADH are shown in Figure 2.10.2.
The conversion of NAD1 to NADH is a reduction reaction. Oxidation and reduction are two halves of the
same process, dealing with the exchange of electrons
227
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00021-5
CH3
Pyruvate
dehydrogenase
NAD+
C
NADH + H
COO–
S
C
CH2
O
Acetyl group
O
CH2
CH3
Pyruvate CoASH
CO2
Thiamine PP
Lipoic acid
Riboflavin
NH
C
O
HC
H3C
OH
C
CH3
CH2
O
–O
P
O
Coenzyme A
O
NH2
–O
P
O
N
N
O
CH2
O
–O
N
O
OH
P
O
N
OH
FIGURE 2.10.1 Conversion of pyruvate to acetyl CoA by pyruvate dehydrogenase, indicated in blue. Note that the reaction releases CO2. Coenzyme A is
a complex of ATP with pantothenic acid. It carries the acetyl group on through biochemical reactions in the cell. The reaction requires five different
coenzymes (coenzyme A, NAD1, thiamine pyrophosphate, lipoic acid, and riboflavin). Three of these coenzymes are vitamins (niacin, thiamine, and
riboflavin).
Reducing equivalents
Niacin
H
O
C
O
O
NH2
O
P O–
+
OH OH
O–
N
P
O O
CH2
N
O
NH2
N
N
O
NH2
C
CH2
N+
O
O
H
H+ + 2e–
O
CH2 N
O
P O–
O
OH OH
O–
N
P
O O
CH2
N
O
OH OH
OH OH
NAD+
Nicotinamide adenine dinucleotide
NADH
Oxidized
Reduced
FIGURE 2.10.2 Formation of NADH from NAD1. NAD1 is the oxidized form; NADH is the reduced form.
NH2
N
N
AT P P roduc ti on II : T h e T CA Cy c le a nd O xidat ive P hos phor yla tion
between chemicals. A mnemonic device for oxidation/
reduction reactions is
LEO GER
which stands for “Loss of Electrons, Oxidation; Gain of
Electrons, Reduction.” When NAD1 gains two electrons
in the form of H2, it is reduced. At the same time, the
chemical from which the H2 is extracted is oxidized.
In oxidation/reduction reactions, one chemical is
reduced while the other is oxidized. Now the electrons
in the reduction reaction had to come from someplace,
and that someplace is another chemical. So in a reductionoxidation reaction there are always two redox
pairs, one being reduced and the other being oxidized
in the process.
THE AFFINITY OF A CHEMICAL FOR
ELECTRONS IS MEASURED BY ITS
STANDARD REDUCTION POTENTIAL
If compound A binds electrons more tightly than
compound B, we expect that A will take electrons from
B in the reaction A 1 B-A2 1 B1. In this reaction, A is
reduced and B is oxidized. This can be written as the
sum of two half-reactions:
A 1 e2 -A2 and B-B1 1 e2
The relative tendency for a compound to be reduced is
called its reduction potential. The standard redox
potential is measured against a standard half-reaction,
arbitrarily assigned the reduction potential of zero.
This is the reduction/oxidation of hydrogen:
2H1 ðaqÞ 1 2e2 -H2 ðgÞ E0 5 0:00
Here E0 is referred to as the standard reduction potential
of hydrogen. Because oxidation is the reverse reaction
of reduction, the standard oxidation potential is the
negative of the standard reduction potential.
Standard reduction potentials are measured as shown in
Figure 2.10.3 against a Standard Hydrogen Electrode.
It is measured under standard conditions of 1 M concentration of all reactants and 1 atm pressure of H2 gas. The
standard reduction potential is given the symbol E0.
Recall from Chapter 1.3 that the potential is the work
done in bringing a positive unit charge from infinite
separation to the point at which the potential is to be
defined. The work done in moving a charge through a
potential is just the charge times the potential. This was
expressed in Eqn [1.3.13]:
½2:10:1
ΔEnergy 5 qΔU
where U is the potential and q is the total charge.
In electrochemistry we generally use E for the potential,
as we have done above, in units of volts, and q is in
coulombs. This gives the energy change in volt-coulombs
or joules. This energy change is the change in free energy,
and so Eqn [2.10.1] can be rewritten as
½2:10:2
ΔG 5 z` ΔE00
Voltmeter
E0 > 0
E0 = 0
–
H2 gas (1 atm) in
Salt bridge
Pt black electrode
1M
Reactant
Test half cell
1M H+
Standard hydrogen electrode
FIGURE 2.10.3 Measurement of the standard electrode potential.
One half-cell containing standard concentrations of reactant (1 M) is
connected to the standard hydrogen electrode (SHE) with a finely
divided Pt black electrode bubbled with 1 atm of H2 gas and 1 M H1 in
solution. The voltage between the two half-cells is the reduction
potential. If electrons flow to the reactant, then the reactant is being
reduced and has a positive standard reduction potential—it has a higher
affinity for electrons than hydrogen. Note that current is defined as
positive charge flow, which is opposite to electron flow. Voltages
are typically measured with a potentiometer that finds the voltage
necessary to stop current flow, so that the measurement occurs at
equilibrium when no current flows.
where ΔE00 is the difference in reduction potential
between the two half-cells, z is the valence of the carrier,
and ` is the Faraday 5 98,500 coulombs mol21. The
Faraday converts the charges to coulombs and normalizes the free energy change to the free energy change
per mole. Since the charge carrier is the electron,
z 5 21, this equation becomes
½2:10:3
ΔG 5 2 `ΔE00
This is the free energy change per mole of electrons. If
there are n electrons involved per reaction, the free
energy change per mole of reaction is
½2:10:4
ΔG 5 2 n`ΔE00
THE REDUCTION POTENTIAL DEPENDS
ON THE CONCENTRATION OF
OXIDIZED AND REDUCED FORMS, AND
THE TEMPERATURE
The standard reduction potential is defined at unit concentrations of all reactants. When the concentrations are
not 1 M, the measured reduction potential changes.
Consider the reduction of A in contact with a Standard
Hydrogen Electrode, as described earlier. We can write
the two half-cell reactions as
2
A 1 n H1
test 1 n e -AHn
n=2 H2 -n HSHE 1 n e2
1
A 1 n=2 H2 1 n H1
test -AHn 1 n HSHE
229
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
where SHE denotes Standard Hydrogen Electrode.
The free energy per mole for the overall reaction is
calculated as
0.4
Δμ 5 μ0AHn 1 RT ln½AHn 1 n μ0HSHE 1 n RT ln½H1
SHE n
n
2μ0A 2 RT ln½A 2 μ0H2 2 RT ln fH2
2
2
2n
μ0Htest
2 n RT
ln½H1
test ½2:10:5
where fH2 is the fugacity of hydrogen, analogous to the
activity in aqueous solutions. Since the pressure of H2
gas at the Standard Hydrogen Electrode is 1 atm, fH2 is
1.0. Collecting terms in Eqn [2.10.5], we get
½2:10:6
The top line of Eqn [2.10.6] is the free energy per mole
for the standard reduction potential for A. This is given
by Eqn [2.10.4]. Remembering that [H1] for the
Standard Hydrogen Electrode 5 1 M, its term drops out,
because ln [H1] 5 ln 1 5 0, and we write
½AHn 2 n RT ln H1 test
½A
Δμ 5 2 n`ΔE0 1 RT ln
½2:10:7
The observed potential is related to the free energy
change of the reaction through Eqn [2.10.4] and so
we have
½AHn 2n`ΔE 5 2n`ΔE0 1 n RT ln H1 test 1 RT ln
½A
½2:10:8
1
In most situations, the [H ] in the test half-cell can
be kept nearly constant by the use of chemical buffers.
In this case, its contribution to the free energy will
also be constant, and we can define a practical reduction potential (ΔE0’) that incorporates the standard
reduction potential and the pH term. Doing this, plus
dividing both sides by 2 n`, we come to
½2:10:9
ΔE 5 ΔE00 1
RT
½A
ln
n` ½AHn The argument of the logarithm is inverted from
Eqn [2.10.8] because of multiplying through by 21 to
convert the minus sign in 2 n` to positive values.
What this equation means is that the actual reduction
potential depends on the relative concentrations of
the oxidized form ([A]) and reduced form ([AHn]). The
reduction potential as a function of the oxidation
state of a redox reaction is shown in Figure 2.10.4
for NADH, Ubiquinone, and Cytochrome C (more on
these later). Note that when [A] 5 [AH], which occurs
Cyt Cox + e–
CoQred
CoQox + 2H+ + 2e–
0.0
–0.2
–0.4
n
Δμ 5 μ0AHn 1 n μ0HSHE 2 μ0A2 μ0H2 2 n μ0Htest 1 RT ln½AHn 2
1
2 RT ln½A 1 n RT ln½H1
SHE 2 n RT ln½H test Cyt Cred
0.2
Reduction potential
230
–0.6
0.0
NADH
0.2
NAD+ + H+ + 2e–
0.4
0.6
Fractionof oxidation
0.8
1.0
FIGURE 2.10.4 Reduction potential for various redox reactions found
in the cell as a function of their oxidation state. The reduced form is AHn
in Eqn [2.10.9] and the oxidized form is A. As AHn is oxidized by addition
of a strong oxidant, it is converted to A and the reduction potential
changes according to Eqn [2.10.9]. When the reaction is 50% complete,
[A] 5 [AHn] and the argument of the logarithm becomes 1.0, and
ln 1.0 5 0. At this point, the measured reduction potential is the practical
standard reduction potential: ΔE 5 ΔE0'. For NADH this is 20.32 volts;
for Coenzyme Q (CoQ) this is 10.030 volts; for cytochrome C this is
10.23 volts. The higher reduction potential means that oxidized CoQ
will oxidize NADH by taking electrons from it, and oxidized Cyt C will
oxidize CoQ by taking electrons from it.
when the reactant is 50% oxidized, the measured ΔE
is equal to ΔE00 .
THE TCA CYCLE IS A CATALYTIC CYCLE
The biochemical transformations that constitute the
TCA cycle are shown in Figure 2.10.5. This is a catalytic
cycle in that the intermediates themselves are not
altered by the cycle. It starts with oxaloacetate, a
4-carbon dicarboxylic acid, condensing with acetyl CoA
to produce citrate. As the cycle continues, NADH is
generated in each of three separate reactions (at isocitrate dehydrogenase, α-ketoglutarate dehydrogenase,
and malate dehydrogenase) and FADH2 is generated
at succinate dehydrogenase. FADH2 is the oxidized
form of FAD, flavin adenine dinucleotide. It is another
chemical carrier of reducing equivalents whose structure
is shown in Figure 2.10.6. GTP is generated at succinyl
CoA synthetase, and CO2 is generated twice, at isocitrate
dehydrogenase and α-ketoglutarate dehydrogenase.
The GTP generated in the cycle by substrate-level
phosphorylation is formally equivalent to ATP as the
two high-energy compounds are readily interconverted.
The overall TCA cycle is
Acetyl CoA 1 2H2 O 1 GDP 1 Pi 1 FAD 1 3NAD1 CoASH 1 2 CO2 1 GTP 1 3 NADH 1 3 H1 1 FADH2
AT P P roduc ti on II : T h e T CA Cy c le a nd O xidat ive P hos phor yla tion
Acetyl CoA
O
H3C
Malate
HC
HO
O
C
COO–
H2C
OH
H2C
COO–
C
COO–
H2C
COO–
Aconitase
Citrate synthase
CoASH
NADH + H
COO–
Citrate
SCoA
COO–
Oxaloacetate
Malate dehydrogenase
C
H2C
COO–
HC
COO–
H
C
COO–
NAD+
HO
H2C
–
COO
Isocitrate
NAD+
Isocitrate dehydrogenase
Fumarase
TCA Cycle
HC
COO–
HC
COO–
CO2
NADH + H
Fumarate
H2C
FADH2
H2C
NAD+
O
FAD
C
COO–
Alpha ketoglutarate
COO–
NADH + H
Succinate dehydrogenase
CoASH
H2C
COO–
–
H2C
COO
H2C
COO–
CH2
Succinate
O
GTP
C
CoASH
Alpha ketoglutarate
dehydrogenase
SCoA
CO2
GDP Pi Succinyl CoA
Succinyl CoA synthetase
Pyruvate
dehydrogenase
FIGURE 2.10.5 Metabolic interconversions in the TCA cycle. Note that the two-carbon fragment, acetic acid, is carried by CoA to combine with
oxaloacetate to form citrate. The oxaloacetate is regenerated by the cycle. Thus the cycle’s intermediates are catalysts in that they are not consumed.
The two carbons in acetate are converted to CO2 by the cycle.
The result is that the two carbons in acetate are converted to CO2 and a bunch of reducing equivalents
(8 per 2-carbon acetate). Note that O2 is not explicitly
required for this process, but it is required for the continued operation of this cycle. If we add in the formation of acetyl CoA from pyruvate, the overall reaction is
Pyruvate 1 3 H2 O 1 GDP 1 Pi 1 FAD 1 4 NAD1 3 CO2 1 GTP 1 4 NADH 1 4 H1 1 FADH2
The NADH and FADH2 produced during this overall
series of reactions must be returned to NAD1 and FAD,
or the process will stop. The reduced NADH and
FADH2 are oxidized by a special system called the electron transport chain (ETC). In the process of being
oxidized, the energy stored in these compounds enables
the synthesis of ATP through a process called oxidative
phosphorylation.
THE ETC LINKS CHEMICAL ENERGY
TO H 1 PUMPING OUT OF THE
MITOCHONDRIA
The ETC consists of an array of proteins inserted in the
inner mitochondrial membrane. The overall plan is
this: NADH delivers two electrons to a series of chemicals that differ in their chemical affinity for these
electrons (see Figure 2.10.7). This is expressed in their
reduction potential (see above) which is related to
231
232
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
O
O
O
Ribose
O–
CH2
HO
O
P
O
O–
P
O
N
O
Adenine
CH2
N
CH
CH2
Flavin
NH2
HO
CH
HO
CH
N
H3C
N
N
CH
HO
CH
HO
CH
P
O
–
N
CH2
H
H3C
N
N
H3C
N
NH
NH2
N
CH2
N
+ 2H+ + 2e–
O
O–
P
O
O
OH OH
CH2
H3C
O
HO
O
O
N
N
N
OH OH
O
NH
O
Reducing equivalents
H
O
Flavin adenine dinucleotide
Oxidized
Reduced
FIGURE 2.10.6 Structure of oxidized and reduced FAD. FAD stands for “flavin adenine dinucleotide.” It consists of a flavin part, a ribose part, and an
adenine part. The ribose 1 flavin is better known as riboflavin or vitamin B2. The binding of reducing equivalents is associated with gain of energy,
which can be released on oxidation.
Electron transport through
complexes I, III, and IV is linked
to H+ pumping from the matrix
to the intermembrane space
Outer mitochondrial membrane
H+
H+
H+
Complex I
2e– Cyt C
CoQ
2e–
Inner mitochondrial membrane
CoQ
2e–
NADH
Complex IV
Complex III
Complex II
NAD+ H+
Intermembrane space
2e–
FADH2
FAD
H+
H+
Mitochondrial matrix
NADH feeds into the
beginning of the ETC—
three complexes pump H+
1/2 O2 + 2H+
FADH2 feeds into the ETC
after complex I; only two
complexes pump H+
H2O
The final electron acceptor is
oxygen; without it, the whole
ETC backs up
FIGURE 2.10.7 The electron transport chain (ETC). NADH feeds in reducing equivalents at the beginning of the ETC, which hands them on to proteins
with progressively higher affinity until at the end of the chain the electrons are combined with oxygen. Complexes I, III, and IV use the chemical
energy of oxidation to pump H1 ions from the mitochondrial matrix to the intermembrane space. This makes an electrical current that separates
charge and produces a potential difference across the mitochondrial membrane.
their free energy. The energy is released gradually, in
steps, and the ETC complexes use the decrease in free
energy to pump hydrogen ions from the matrix space to
the intermembrane space between the inner and outer
mitochondrial membranes. This pumping of hydrogen
ions produces an electrochemical gradient for hydrogen
ions and the energy in this gradient is used to generate
ATP from ADP and Pi.
AT P P roduc ti on II : T h e T CA Cy c le a nd O xidat ive P hos phor yla tion
OXYGEN ACCEPTS ELECTRONS
AT THE END OF THE ETC
Molecular oxygen oxidizes the last step in the ETC. This
is the point at which oxygen is consumed by the mitochondria, producing water. Without oxygen to finally
oxidize the ETC, the chain itself will remain reduced
and no further reducing agents can be fed through it.
That is, NADH cannot be converted to NAD1 (at the far
left of the chain) in the absence of oxygen. FADH2 also
cannot be converted to FAD in the absence of oxygen.
The ΔE0’ for the redox reaction 1/2O2 1 2H1 1 2e2H2O is 10.82 volts. The cascade of electrons down
the ETC is shown diagrammatically in Figure 2.10.8.
Note that the electrons travel down the cascade towards
ever more positive reduction potentials, as these signify
increasing affinity for electrons.
PROTON PUMPING AND ELECTRON
TRANSPORT ARE TIGHTLY COUPLED
If the proton gradient is at equilibrium with the free
energy of electron transport, then the electrons cannot
be transported through the ETC. The energy stored in
the electrochemical gradient of protons across the inner
mitochondrial membrane must also be drained in some
way for the ETC to continue operating. The energy in
the proton electrochemical gradient is used to make
ATP. The coupling of the electrochemical gradient of
H1 across the inner mitochondrial membrane with ATP
synthesis is called chemiosmotic coupling (because
–0.5
NADH
–0.4
–0.3
THE ATP SYNTHASE COUPLES INWARD
H 1 FLUX TO ATP SYNTHESIS
The inner mitochondrial membrane contains many
copies of a protein called the F0F1ATPase. This is also
called ATP synthase. It consists of two parts: the F0
component spans the membrane and provides a
channel for protons to move into the matrix from the
intermembrane space. The F1 component is a complex
of five proteins with the composition α3β3γδε, with a
molecular weight of about 360,000. The F0 Part of the
complex consists of an integral membrane, a subunit,
a b dimer, and 815 small c-subunits. The structure
of the ATP synthase is shown in Figure 2.10.9. This
remarkable complex couples the movement of H1 to
the synthesis of ATP through mechanical intermediates.
Hydrogen ions from the inner matrix access the c
subunit via a channel in the a subunit, causing a rotation of the c subunit turbine. This rotates the γ subunit,
which has a cam-like protrusion that deforms the α and
β subunits. Each time the cam passes one of the three
αβ complexes, ATP is formed from ADP and Pi bound
to the αβ subunits. Because there are three of these αβ
subunits, each turn of the c-protein turbine produces
3 ATP molecules.
NAD+
4H+
–0.320 volts
NADH dehydrogenase
complex
–0.2
Reduction potential (E0', volts)
there is a concentration difference across the membrane
and an electric potential). It was first proposed by Peter
Mitchell in 1961, who was awarded the Nobel Prize for
the work in 1978.
2H+
–0.1
0.0
0.030 volts
CoQ
Cytochrome C reductase
complex
0.1
0.2
0.230 volts
Cyt C
4H+
0.3
0.4
0.5
Cytochrome C oxidase
complex
0.6
0.7
0.8
0.820 volts
0.9
2H+ + 1/2 O2
H2O
FIGURE 2.10.8 Cascade of electrons in the electron transport chain. It begins with the production of NADH by reactions in glycolysis or TCA cycle. The
reduction potential of NADH/NAD1 is 20.320 volts. Electrons are passed to the NADH dehydrogenase complex that pumps 4H1 per 2 e2 out of the
mitochondrial matrix to the intermembrane space. Electrons are then passed to Coenzyme Q, with a reduction potential of 0.030 volts. Coenzyme Q
passes the electrons to the cytochrome C reductase complex that pumps 2H1 per 2 e2. Electrons then are transferred to cytochrome C with a
reduction potential of 0.230 volts. The electrons that are taken by the Cytochrome C oxidase complex that pumps 4H1 per 2 e2. Cytochrome C
oxidase is finally oxidized by molecular oxygen, whose reduction potential is 0.820 volts.
233
234
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Binding of H+ to the c unit causes
rotation of the c complex, which is
connected to the γ subunit
H+
c
c
c
a
c
c
+ H+
H+ H
ε
H+
H+
Inner mitochondrial membrane
H+
The shaft has a cam-like projection
that interacts with the αβ subunits
that form ATP from ADP and Pi
γ
b dimer
ATP
ADP + Pi
β
α
δ
ATP
β
So if there are 10 C units then 10 H+
cause a complete rotation of the C rotor
and one rotation of the γ shaft that makes
3 ATP because there are three αβ
subunits on each ATP synthase
α
α
ADP + Pi
β
ADP + Pi
ATP
FIGURE 2.10.9 The F0F1ATPase or the ATP synthase of mitochondria. It consists of a complex of proteins that make up a tiny H1-driven turbine. H1
ions enter from the intermembrane space through protein a. They bind to a c unit in the rotator, which cause a rotation of the c-complex. After a
nearly complete rotation, the H1 ion is removed to the mitochondrial matrix. The c-complex binds the γ subunit and rotates it. The αβ subunits of the
head are kept steady by the stator components, δ and the b dimer. The γ subunit has a projection that interacts with each of the αβ subunits, and this
mechanical interaction is used to synthesize ATP from ADP and Pi. Complete rotation of the c-complex requires as many H1 as c-subunits. For each
complete rotation, the ATP synthase makes 3 ATP molecules.
THE PROTON ELECTROCHEMICAL
GRADIENT PROVIDES THE ENERGY
FOR ATP SYNTHESIS
The ETC pumps H1 ions out of the matrix into the
intermembrane space. The stoichiometry is about 10H1
ions per 2e2 when they originate from NADH, and
about 6H1 when the 2e2 originate from FADH2 (see
Figure 2.10.7). Because the H1 ions move without
counter ions, this movement is an outward current that
separates charge, and therefore there is a potential
developed across the inner mitochondrial membrane.
This potential varies depending on the state of mitochondrial activity, but a typical value is about 160 mV,
negative inside. In addition to the potential, there is a
concentration difference in H1 established across the
membrane. The pH of the intermembrane space is
about 7.0, whereas the pH of the matrix is about 8.0.
Recall that pH 5 2log [H1], so that pH 5 7.0 implies
that [H1] 5 1027 M and pH 5 8 means [H1] 5 1028 M.
Thus there is a 10-fold difference in the [H1] established by the ETC. When H1 ions travel from the intermembrane space to the matrix, they release the free
energy stored in the electrochemical gradient for H1,
enabling the F1 subunit to synthesize ATP from ADP
and Pi. This proton electrochemical gradient is sometimes called the proton motive force. The free energy
for H1 transfer from the intermembrane space to the
mitochondrial matrix is calculated as
Δμout-in 5 μin 2 μout
5 μ0 1 RT ln½H1 in 1 `ψin 2 μ0
½2:10:10
2 RT ln½H1 out 2 `ψout
5 RT ln
½H1 in
1 `ðψin 2 ψout Þ
½H1 out
The free energy change for ATP synthesis under the
conditions of the cell varies from cell to cell and from
place to place within the cell because the local concentrations of ADP, Pi, ATP, and ions that bind to them
(H1, Ca21, and Mg21) also vary from place to place.
Nevertheless, we have already calculated an approximate free energy change for ATP hydrolysis under
conditions of the cell to be 257.1 kJ mol21. The free
energy of ATP synthesis should be the opposite of
this, 157.1 kJ mol21.
According to the result in Example 2.10.1, there is not
enough energy in one H1 transport to synthesize ATP.
If we assume integral stoichiometry, we need at least
three of them. The free energy for the reaction
½2:10:11
1
ADP 1 Pi 1 3H1
out -ATP 1 3Hin
AT P P roduc ti on II : T h e T CA Cy c le a nd O xidat ive P hos phor yla tion
EXAMPLE 2.10.1 Calculate the Free Energy in the Mitochondrial H1 Electrochemical Gradient
The free energy per mole of H1 is given by Eqn [2.10.1] as
1 ½H in
ΔμHout -Hin 5 RT ln
1 `ðψin ψout Þ
½H1 out
21
21
T 5 310 K,
Inserting
values
of
R 5 8.314 J mol K ,
[H1]in 5 1028 M, [H1]out 51027 M, we calculate the chemical part as
!
!
½H1 in
1028 M
21 21
K
3
310
K
3
ln
RT ln
5
8:314
J
mol
½H1 out
1027 M
Using ` 5 9.649 3 104 C mol21 and ψin 5 20.16 V (with ψout 5 0),
the electrical part of the free energy change is
`ðψin ψout Þ 5 9:649 3 104 C mol21 3 ð2 0:16 V 0Þ
5 2 15:44 kJ mol21
Thus the total free energy change for H1 transfer from the
intermembrane space to the matrix, for this condition given
here, is 2 21.37 kJ mol21.
5 2 5:93 kJ mol21
is the sum of the free energy of two processes:
½2:10:12
ADP 1 Pi-ATP
1
3H1
out -3Hin
We add the two to get
Δμ 5 ΔμADP1Pi-ATP 1 3ΔμH1out -H1in
½2:10:13
5 57:1 kJ mol21 1 3ð2 21:37 kJ mol21 Þ
5 27:01 kJ mol21
The negative free energy change for this coupled
reaction indicates that this process will proceed
spontaneously. That is, there is enough energy in the
electrochemical gradient of H1 across the inner mitochondrial membrane to synthesize 1 ATP for every 3H1
ions transported. As it turns out, the stoichiometry is not
integral.
NADH FORMS ABOUT 2.5 ATP
MOLECULES; FADH2 FORMS ABOUT
1.5 ATP MOLECULES
The amount of ATP formed from oxidative phosphorylation has been controversial but a consensus seems to
have been reached. Measurements show that electron
transport beginning with NADH results in 10H1 ions
being transported from matrix to intermembrane space,
and with FADH2 the number is 6, because the first
complex is bypassed. What happens to these H1 ions?
Most are used to drive the ATP synthase as described in
Figure 2.10.9, but some are used to bring the phosphate
into the mitochondria from the cytosolic compartment
(see Figure 2.10.10) and some are used for other
transport processes. Mitochondria from the heart of cows
has 8 c-subunits in their ATP synthase, suggesting that
8H1 ions are needed for one complete rotation of the
rotor and synthesis of 3 ATP molecules. This gives
a nonintegral stoichiometry: each ATP requires
8/3 5 2.67H1 ions! Our calculation above indicates that
the minimum number is 57.1 kJ mol21/21.87 kJ mol21 5
2.61. Because 1H1 is required to import Pi, the number
of ATP produced by NADH becomes 10/(2.67 1 1) 5 2.7
ATP/NADH. If the proton motive force is used to drive
other processes, the ATP yield will be lower. Recent studies suggest a relatively constant H1/ATP ratio of 4.0,
including transport processes. In this case ATP production
from NADH would be 10H1 per NADH/4H1 per
ATP 5 2.5 ATP per NADH. For FADH2, the ratio would
be 6H1 per FADH2/4H1 per ATP 5 1.5 ATP per FADH2.
These numbers are approximate and tentative. The
ATPase from different sources has different c-ring sizes
that may cause differences in the H1/ATP ratio and
therefore the ATP/NADH ratio.
ATP CAN BE PRODUCED FROM
CYTOSOLIC NADH
The NADH produced in the cytosol by glycolysis cannot
enter the mitochondrial matrix, yet it must be oxidized
back to NAD1 to allow glycolysis to proceed. This can
be accomplished by lactate dehydrogenase, as described
in Chapter 2.9, but this does not extract the energy of
combustion remaining in the lactic acid. Two types of
shuttle mechanisms have the effect of bringing cytosolic
reducing equivalents into the matrix, without NADH
itself actually entering the matrix. These shuttles are the
glycerol phosphate shuttle and the malate/aspartate
shuttle.
In the glycerol phosphate shuttle, NADH is oxidized
to NAD1 by the cytosolic glycerol-3-phosphate
dehydrogenase, while dihydroxyacetone phosphate is
simultaneously reduced to glycerol-3-phosphate.
Glycerol-3-phosphate then penetrates the mitochondrial
outer membrane and reduces FAD to FADH2 by the
mitochondrial glycerol-3-phosphate dehydrogenase to
form dihydroxyacetone phosphate. In this way, we start
with cytoplasmic NADH and dihydroxyacetone
phosphate and we end up with mitochondrial FADH2
and cytoplasmic NAD1 and dihydroxyacetone phosphate. So the reducing power of NADH is transferred to
FADH2, which then enters the ETC to generate 1.5 ATP
molecules. Because complex I is bypassed, only 1.5 ATP
are made per molecule of NADH passed on to the
mitochondria by the glycerol P shuttle. Figure 2.10.11
illustrates the glycerol phosphate shuttle.
235
236
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
3
Some additional H+ is used to import Pi and
other substrates, such as pyruvate, for the
mitochondria. The overall requirement for
ATP synthesis, including transport, is about
4H+ per ATP
1
The ETC uses the energy in electron binding to NADH
and FADH2 to create a concentration difference in [H+]
and a membrane potential across the inner mitochondrial
membrane. Three complexes of the ETC pump a total of
10 H+ per NADH or 6H+ per FADH2
4H+ 2H+ 4H+
Outer mitochondrial membrane
–
10H+/NADH
ADP3ATP4-
1H+/ATP
Pyruvate
–3
10
–7
+
Intermembrane space
0.3H+/ATP
ETC
–160 mV
OH–
M = pH 7.0
OH–
2.7H+/ATP
PO4
++++++++
F0
Inner mitochondrial
membrane
------------
NADH
10–8 M = pH 8.0
F1
NAD+
1.3 OH–/ATP
ATP
Pi + ADP
H2O
1.3 H+/ATP
Mitochondrial matrix
2.7H+/ATP
2
The ATP synthase uses the energy
in the H+ gradient and membrane
potential to synthesize ATP from ADP
and Pi. About 2.7H+ ions are required
for each ATP synthesized
FIGURE 2.10.10 Overall coupling of the ETC to the ATP synthase. The ETC pumps electrons from the mitochondrial matrix to the intermembrane
space, creating a [H1] concentration gradient and an electrical potential difference. The ATP synthase uses the energy in this gradient to link ATP
synthesis to H1 ions going down their electrochemical gradient. Because of its mechanism (see Figure 2.10.9) in heart mitochondria 8H1 ions make 3
ATP molecules, for a stoichiometry of 2.7H1/ATP. However, the H1 gradient is used for mitochondrial transport as well. For each Pi that enters the
mitochondria, 1.0H1 ion is used—the exit of OH2 is equivalent to the entry of H1. Small amounts of H1 flow is also used for other transport processes.
The total H1 required for ATP synthesis, including transport, is about 4.0. Because each NADH causes ETC to pump 10H1 ions, this means that about
2.5 ATP molecules are formed per NADH. ATP exit and ADP entry do not require energy, as shown.
In the malate/aspartate shuttle, cytoplasmic NADH is
used to convert cytoplasmic oxaloacetate to malate,
which can be carried across the mitochondrial inner
membrane by a dicarboxylate carrier by facilitated diffusion with no metabolic energy expenditure. Inside the
matrix, the malate is converted back to oxaloacetate,
generating the NADH back, which then transfers
electrons to the ETC. To complete the cycle, oxaloacetate must get back outside. This is accomplished by converting oxaloacetate to aspartate (using glutamate as a
substrate). The aspartate is transported out of the
mitochondria where it is converted back to oxaloacetate
and glutamate. The cycle is completed when glutamate
goes back into the mitochondria. In this shuttle, 2.5
ATP molecules are produced for each cytosolic NADH
because it is effectively transferred into the matrix
as NADH. Figure 2.10.12 illustrates the malate/aspartate
shuttle.
MOST OF THE ATP PRODUCED DURING
COMPLETE GLUCOSE OXIDATION
COMES FROM OXIDATIVE
PHOSPHORYLATION
Figure 2.10.13 shows the production of ATP throughout
glycolysis and the TCA cycle. Glycolysis utilizes 2 moles
of ATP per mole of glucose and then produces 4 moles
ATP per mole by substrate-level phosphorylation. The 2
moles of NADH produced by glyceraldehyde-3-P
dehydrogenase can be converted to either 5 moles of
NAD+
NADH + H+
Glycerol-3-P dehydrogenase
Cytosolic compartment
Dihydroxyacetone P
Glycerol-3-P
Outer mitochondrial membrane
Dihydroxyacetone P
Glycerol-3-P
H+
Complex I
H+
E-FADH2
E-FAD
2e–
Inner mitochondrial membrane
Complex IV
Complex III
NAD+
Complex II
H+
Intermembrane space
2e–Cyt C
CoQ
2e–
CoQ
NADH
H+
2e–
FADH2
FAD
H+
H+
Mitochondrial matrix
1/2 O2 + 2H+
H2O
FIGURE 2.10.11 The glycerol phosphate shuttle for transferring cytosolic reducing equivalents to the mitochondria. Cytosolic NADH 1 H1 is converted to
NAD1 by glycerol P dehydrogenase. Glycerol-3-P crosses the outer mitochondrial membrane and is converted back to dihydroxyacetone P by a
mitochondrial glycerol-3-P dehydrogenase in the inner mitochondrial membrane. The dihydroxyacetone P goes back into the cytosol. The reduced
mitochondrial glycerol-3-P dehydrogenase reduces ubiquinone in the inner membrane, which passes the reducing equivalents on to complex III of the ETC.
NAD+
NADH + H+ Cytosolic compartment
Cytosolic malate
dehydrogenase
Malate
Oxaloacetate
Aspartate
Aspartate
aminotransferase
α-ketoglutarate
Glutamate
Outer mitochondrial membrane
Malate α-ketoglutarate
Glutamate Aspartate
Intermembrane space
Inner mitochondrial membrane
α-ketoglutarate
Malate
Glutamate
Oxaloacetate Aspartate Aspartate
aminotransferase
Mitochondrial malate
dehydrogenase
NAD+
NADH + H+
Mitochondrial matrix
FIGURE 2.10.12 The malate shuttle. Two cycles running in opposite directions have the net effect of transferring NADH from cytosol to mitochondrial
matrix. In one cycle, oxaloacetate is converted to malate while NADH is converted to NAD1. Malate crosses the inner mitochondrial membrane where it is
converted back to oxaloacetate and NADH. The oxaloacetate is then converted to aspartate, which leaves the mitochondria and passes back into the
cytosol where the aspartate is converted back to oxaloacetate. A second cycle runs in the opposite direction. Malate entry into the mitochondrial matrix is
accompanied by α-ketoglutarate exit. In the cytosol, the latter is converted to glutamate by aspartate amino transferase, which links the reaction
α-ketoglutarate-glutamate to the reaction aspartate-oxaloacetate. The glutamate exchanges with aspartate across the inner mitochondrial membrane.
238
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Glucose
ATP
Glucose 6-P
Fructose 6-P
ATP
Fructose 1,6 diP
ATP synthase
2 NADH
ETC
3-5 ATP
2 Glyceraldehyde 3-P
2 NAD+
2 1,3 Diphosphoglycerate
2 ATP
2 3-Phosphoglycerate
2 2-Phosphoglycerate
2 Phosphoenol pyruvate
ATP synthase
5 ATP
ETC
2 ATP
2 Pyruvate
2 NAD+
2 NADH
2 Acetate
5 ATP
2 NADH
Oxaloacetate
ATP synthase 2 NAD+
ETC
Malate
Citrate
2 NAD+
Isocitrate
Fumarate
2 NADH
Alpha ketoglutarate
ATP synthase
2
3 ATP
ETC
2 FADH2
Succinate
NAD+
5 ATP
ETC
ATP synthase
Succinyl CoA
2 NADH
2 FAD
2 GTP
ATP synthase
ETC
5 ATP
2 GDP + 2 Pi
FIGURE 2.10.13 Overall ATP production from glycolysis, TCA cycle, and ETC. Glycolysis produces 4 ATP, but consumes 2 ATP molecules, per mole of
glucose. It also produces 2 moles NADH per mole of glucose. These reducing equivalents are cytosolic and can result in 35 moles of ATP depending
on how the NADH enters the mitochondria. In the mitochondria, a total of 8 moles of NADH are produced per mole of glucose, including 2 in the
pyruvate dehydrogenase step and 6 in the TCA cycle. These each produce 2.5 moles of ATP per mole of NADH, so the NADH produces 8 moles
NADH/mol glucose 3 2.5 moles ATP/mol NADH 5 20 moles ATP/mol glucose. The 2 FADH2 produced at succinate dehydrogenase produce 1.5 moles of
ATP per mole of FADH2 for a total of 3 moles ATP/mol glucose. Succinyl CoA synthetase produces 2 moles GTP/mol glucose, which is energetically
equivalent to 2 moles ATP/mol glucose. Total ATP production is thus 57 moles in glycolysis 125 moles in the mitochondria, for a total of
3032 moles of ATP per mole of glucose.
ATP through the malate/aspartate shuttle or 3 moles of
ATP through the glycerol phosphate shuttle.
MITOCHONDRIA HAVE SPECIFIC
TRANSPORT MECHANISMS
In order for oxidative phosphorylation to work, the
inner mitochondrial membrane must be impermeable
to H1 ions. H1 ions in solution are usually bound to
water as H3O1, the hydronium ion, which is very small.
Therefore, the inner mitochondrial membrane must be
relatively impermeable to most ions in order for the
membrane to establish the potential difference and
concentration difference in [H1]. At the same time,
things have to be able to get in and out. We have
already discussed two such carriers—those that
operate the shuttles that allow cytosolic reducing
equivalents to enter the matrix. Several other carriers are
shown schematically in Figure 2.10.14.
ATP produced in the matrix must leave the matrix to
power cellular activities. This occurs through facilitated
diffusion by the ATPADP translocase. Since ATP has
AT P P roduc ti on II : T h e T CA Cy c le a nd O xidat ive P hos phor yla tion
–160 mV
–
Outer mitochondrial
membrane
+
ATP/ADP Pi transporter Pyruvate
translocase
carrier
Intermembrane space
H+
ADP3–
10–7 M = pH 7.0
PO4–3
Pyruvate
+++++++++
ETC
Inner mitochondrial
membrane
-----------
NADH
NAD+
10–8 M = pH 8.0
ATP4–
H+
OH–
OH–
Mitochondrial matrix
–160 mV
–
Outer mitochondrial
membrane
+
Intermembrane space
H+
ETC
Na/H
Na/Ca
Electrophoretic
Exchanger Exchanger uniport
10–7 M = pH 7.0
Na+
Na+
Ca2+
+ + + + + + + + +
Inner mitochondrial
membrane
– – – – – – – – –
NADH
Na+
NAD+
H+
10–8 M = pH 8.0
Ca2+
Ca2+
Mitochondrial matrix
FIGURE 2.10.14 Selected transport mechanisms in mitochondria. See text for details.
more negative charges, and its concentration is higher
in the mitochondria where it is produced, the transport
is “downhill” in both directions. This translocase is
poisoned by atractyloside.
Phosphate must also enter the matrix in order to be
incorporated into ATP. Since phosphate is highly
charged, movement across the membrane against a
strong electric force is energetically unfavorable.
Phosphate is carried across in exchange for OH2 ions.
The outward flow of OH2 is equivalent to an inward
flow of H1. Therefore, some of the energy of the electrochemical H1 gradient is used to transport phosphate
into the mitochondria.
Mitochondria also take up Ca21 because of the large
negative potential inside. This uptake occurs through a
channel called the electrophoretic uniport. This name
signifies that the electrical gradient is the driving force
for Ca21 movement and that only one ion moves
(uniport). Ca21 taken up this way must be able to exit.
This is accomplished by a Na/Ca exchanger that
couples Ca21 exit with Na1 entry. This is another
example of secondary active transport, in which “uphill”
transport of Ca21 is linked to “downhill” movement of
Na1. Of course, the Na1 taken up by this process must
also have an exit. Na1 efflux from the mitochondria is
through a Na/H exchanger. The Na/H exchanger is also
an example of secondary active transport, in which
uphill movement of Na1 is coupled to downhill movement of H1. In the mitochondria neither Na1 efflux
nor H1 entry is linked to ATP hydrolysis. They are powered by the ETC pumping H1 out of the mitochondrial
matrix into the intermembrane space, thereby creating
a large potential and a concentration gradient for
H1 ions.
SUMMARY
Glycolysis converts glucose into pyruvate, which enters
the mitochondria by facilitated diffusion on its own
carrier. The mitochondria couples the oxidation of
pyruvate to the formation of the high-energy chemical
bond in ATP, a process called oxidative phosphorylation.
Inside the mitochondrial matrix, pyruvate enters into
a series of reactions, beginning with pyruvate dehydrogenase. This series of reactions converts the three-carbon
pyruvate molecule into three molecules of CO2. In the
first step, pyruvate dehydrogenase converts pyruvate into
acetyl coenzyme A, producing a molecule of CO2 and a
molecule of NADH, nicotinamide adenine dinucleotide.
Some of the chemical energy of oxidation of pyruvate is
captured in the energy of electrons binding to NADH.
239
240
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
This energy is converted to ATP later on by the ATP
synthase that relies on the electrochemical gradient of
H1 that is established by the ETC.
The second reaction combines acetyl CoA with oxaloacetate to form citric acid. This is the first step in the citric
acid cycle, or the tricarboxylic acid cycle (TCA cycle),
also called the Krebs cycle for Sir Hans Krebs. This cycle
is a catalytic cycle in that it regenerates oxaloacetate and
all parts of the acetate part of acetyl CoA are converted
to CO2 or H2O. The cycle captures energy of oxidation
by forming NADH or FADH2, flavin adenine dinucleotide, or by forming GTP directly in the conversion of
succinyl CoA to succinate by succinyl CoA synthetase.
Each turn of the cycle produces 3 NADH molecules,
1 FADH2 molecule, 1 molecule of GTP, and 2 molecules
of CO2.
The mitochondria make additional ATP molecules
from NADH and FADH2. These transfer their electrons
to the ETC, which uses the energy of binding of the
electrons, expressed as the reduction potential, to
pump hydrogen ions from the mitochondrial matrix
to the intermembrane space. This active pumping
produces a concentration difference of H1 ions and a
large electrical potential. The ATP synthase in the inner
mitochondrial membrane uses the energy in the electrochemical potential difference for H1 to make ATP
by a process that converts electrochemical energy to
mechanical energy and back again to ATP. NADH
enters the ETC early and its energy causes H1 pumping
at complexes I, III, and IV of the ETC. A total of 10H1
are pumped out per NADH molecule. The ATP
synthase requires 4H1 ions to make ATP (about 3 for
the direct use of the ATP synthase and 1 for transport
of reactants into the mitochondria). As a consequence,
each NADH produces about 2.5 ATP molecules.
FADH2 enters the ETC after complex I, and so it makes
only 1.5 ATP molecules.
Thus complete oxidation of pyruvate to CO2 and H2O
forms 4 NADH, 1 FADH2, and 1 GTP for a total equivalent of 4 3 2.5 1 1 3 1.5 1 1 5 12.5 ATP molecules.
Since there are two pyruvate molecules formed from
glucose, the TCA cycle accounts for 25 ATP molecules
per glucose molecule.
NADH is also generated by glycolysis in the cytoplasm.
This NADH is oxidized in the mitochondria, but
indirectly because NADH itself cannot cross the inner
mitochondrial membrane. Instead, two shuttle systems
have the effect of transferring cytosolic NADH to mitochondrial matrix NADH. The glycerol phosphate shuttle
converts cytosolic NADH to mitochondrial FADH2,
whereas the malate shuttle converts it to mitochondrial
NADH. Glycolysis produces a net gain of 2 ATP and
2 NADH per glucose molecule.
REVIEW QUESTIONS
1. How does pyruvate get into the mitochondria? How
does cytosolic NADH get into the mitochondria?
How do ADP and Pi get into the mitochondria?
How do ATP get out of the mitochondria?
2. Is NADH reduced or oxidized? Is FAD reduced or
oxidized? Why does NADH make more ATP than
FADH2?
3. What is the TCA cycle? How many CO2 molecules are released per pyruvate? Per acetate? In
general, where is CO2 released?
4. How many ATP molecules are produced during
glycolysis per mole of glucose? Does this ATP
production require oxygen?
5. What determines the direction of electron flow in
the ETC? How do you calculate the energy of a
reductionoxidation reaction?
6. How many ATP molecules are produced in
mitochondria during oxidative phosphorylation?
Does this ATP production require oxygen?
7. Why is there a membrane potential across the
inner mitochondrial membrane? If the ATP
synthetase lets in H1, why does not this current
depolarize the mitochondria?
8. What is the chemiosmotic hypothesis? Some
materials are proton ionophores. What effect
would these have on oxidative phosphorylation?
ATP Production III: Fatty Acid
Oxidation and Amino Acid
Oxidation
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
Describe the chemical structure of a triglyceride
Describe how adipocyte lipolysis is regulated by
catecholamines and insulin
Describe the main route of glycerol oxidation
Describe the role of carnitine in the import of fatty acids
into mitochondria
List the number of NADH, FADH2, and acetyl CoA produced
for each turn of the beta oxidation cycle
Account for the number of beta oxidation cycles for
palmitic acid
List the number of NADH, FADH2, and GTP produced for
oxidation of acetyl CoA
Account for the total numbers of ATP molecules produced
by oxidation of palmitic acid
List three chemicals that comprise the ketone bodies
Distinguish between the terms glucogenic and ketogenic
for amino acids
List the amino acids that are exclusively ketogenic
Recognize the amino acids that are exclusively glucogenic
Name the amino acid that is required for feed into the urea
cycle
2.11
DEPOT FAT IS STORED AS
TRIGLYCERIDES AND BROKEN
DOWN TO GLYCEROL AND FATTY
ACIDS FOR ENERGY
Most fatty acids in the body are stored as triglycerides
(triacylglycerol, TAG; see Figure 2.11.1), the acyl esters
of three fatty acids with a glycerol molecule. Blood carries triglycerides in special structures called lipoproteins, in which the water-insoluble lipids are coated
with special proteins. These lipoproteins are made
solely in the intestines and the liver. All tissues can also
store triglycerides in lipid droplets in the cytoplasm,
Tripalmitin
H
H
H
H
1C
2C
O
Glycerol
C
3
O
O
C O
C O
CH2
CH2
CH2
H2C
H2C
H2C
CH2
CH2
CH
O C
2
H2C
FATS AND PROTEINS CONTRIBUTE
50% OF THE ENERGY CONTENT
OF MANY DIETS
In the previous chapters, we saw how carbohydrates are
metabolized through glycolysis to form pyruvate, producing some energy in the process. Pyruvate is then
converted to acetyl CoA, which enters the TCA cycle to
produce reducing equivalents. These reduced compounds, NADH and FADH2, are then oxidized by the
respiratory chain of the mitochondria to produce a proton electrochemical gradient that is then used to
produce ATP. The typical American diet contains only
about 49% of its calories as carbohydrates, with another
35% coming from fat and 16% from protein. Thus the
fats and proteins must also be used to generate cellular
energy. How are fats and proteins used to make ATP?
H2C
H2C
CH2
CH2
H2C
H2C
H2C
CH2
CH2
CH2
CH2
H2C
H2C
H2C
H2C
H2C
CH2
CH2
CH2
CH2
CH2
H2C
H2C
CH2
H2C
H2C
Palmitic acid
CH2
CH2
H2C
H2C
H2C
CH3
CH3
CH3
CH2
FIGURE 2.11.1 Structure of tripalmitin as an example of a triglyceride.
These are stored in adipose tissue and released into the circulation.
Lipase breaks down the triglyceride by hydrolyzing the ester bonds
between the fatty acids carboxyl group and the glycerol hydroxyl group.
241
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00022-7
242
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Epinephrine
-
+
TAG
DAG
ATGL
Glycerol
Natriuretic peptide
Insulin
+
+
MAG
HSL
MGL
Glycerol
Fatty acids
Triglyceride
Diacylglycerol
Free fatty acid
Monoacylglycerol
Free fatty acid
Free fatty acid
FIGURE 2.11.2 Adipocyte lipolysis. Adipose triglyceride lipase (ATGL) begins lipolysis by converting triglycerides (TAG) in the lipid droplet to diacylglycerol
(DAG) and a free fatty acid. Hormone-sensitive lipase (HSL) continues the process by hydrolyzing a second fatty acid, producing monoacylglycerol (MAG).
Monoacylglycerol lipase (MGL) then hydrolyzes the last fatty acid ester bond to produce glycerol and a third free fatty acid. Hormones and nerves control
lipolysis by activating or inhibiting ATGL and HSL. Epinephrine can activate ATGL and HSL through β receptors and this action is blocked by insulin. Epinephrine
can inhibit ATGL and HSL through α2 receptors. Natriuretic peptides also can activate lipolysis through independent mechanisms.
but adipocytes contain very large lipid droplets that
may occupy most of the cell. The adipocytes are specialized for storage and release of lipids for energy. The
circulating triglycerides can be hydrolyzed to form glycerol and three fatty acids through the action of lipoprotein lipase that both hydrolyzes the lipid and acts as a
bridge in lipoprotein uptake. Lipid droplets can also be
hydrolyzed for energy within the cell, but the adipocyte
alone can export the glycerol and free fatty acids
derived from the lipid store.
Lipid droplets are surrounded by a phospholipid
monolayer to which is absorbed the protein perilipin,
which has five members (PLIN1-5) that are distributed
in different tissues. In adipose tissue, the major form is
PLIN1. PLIN1 stabilizes the lipid droplet against lipolysis. Lipolysis begins by ATGL, adipose triglyceride
lipase, that converts tricacylglycerides (TAG) into
diacylglycerol (DAG) and a free fatty acid (FA).
Hormone-sensitive lipase (HSL) then converts the
DAG into monoacylglycerol (MAG) and a free fatty
acid. The MAG is further converted to glycerol and an
FA by the enzyme monoglycerol lipase. Control of
this process is hormonal and neural, as shown in
Figures 2.11.2 and 2.11.3. The catecholamines, derived
either from circulating epinephrine or from sympathetic
nervous stimulation, can either activate or inhibit lipolysis. Insulin inhibits lipolysis, and natriuretic peptide
stimulates it.
In Chapter 2.9, we learned that epinephrine mobilizes
liver glycogen stores by activating glycogenolysis
through a Gs mechanism. Epinephrine can also mobilize lipid stores by activating lipolysis through a Gs
mechanism, as shown in Figure 2.11.3. Binding of
catecholamines to β1 or β2 receptors on the adipocyte is
followed by the activation of adenylyl cyclase and an
increase in 3’,5’-cyclic AMP in the adipocyte cytosol.
This activates protein kinase A (PKA) that phosphorylates a number of targets including perilipin, PLIN1,
and HSL (hormone-sensitive lipase). PLIN1 binds
ABHD5 (α/β hydrolase domain containing 5) that activates ATGL. Phosphorylation of PLIN1 releases the
ABHD5 to activate ATGL. Simultaneously, HSL that
was previously located in the cytosol binds to the lipid
droplet where it participates in lipolysis.
Adipocytes also have α2 receptors that are coupled to a
Gi mechanism that inhibits adenylyl cyclase activity.
Occupancy of these receptors has the opposite effect, an
inhibition of lipolysis, as occupancy of the β receptors.
Insulin inhibits lipolysis by activating cAMP phosphodiesterase, type 3B, by phosphorylation by PKB. This activated PDE-3B decreases the cAMP concentration,
leading to inactivation of lipolysis.
Natriuretic peptide increases lipolysis by activation of
HSL through phosphorylation by PKG, a cGMPactivated protein kinase.
GLYCEROL IS CONVERTED
TO AN INTERMEDIATE OF
GLYCOLYSIS
Plasma glycerol is taken up by tissues and converted
to α-glycerophosphate by an enzyme called glycerol
kinase, by phosphorylating the glycerol with the
terminal phosphate group of ATP. The glycerophosphate is converted to dihydroxyacetone phosphate
ATP P roduction III: F att y Acid O xidat ion and A mino Aci d Oxi da tion
Epinephrine
βγ subunits
Adenylyl cyclase
Epinephrine
α2 receptor
βγ subunits
-
β1 or β2 receptor
+
Gα subunit
PDE-3B
P
cAMP
GDP ATP
+
PKB
Gα subunit Inactive PKA
IRS-1
+
P
+
AMP
Active PKA
HSL
Insulin
PI-3K
+
MGL
ATGL
ABHD5
P
P PLIN1
P
ABHD5
Insulin receptor
P IRS-2
TAG
PLIN1
ATGL
DAG
DAG
MAG
MGL
TAG
Basal state
P
HSL
P
P
Glycerol
+ 3 FA
Stimulated state
Lipid droplet
FIGURE 2.11.3 Control of lipolysis by hormones. Binding of epinephrine to β1 or β2 receptors activates adenylyl cyclase by the Gαs subunit. This increases
formation of cAMP and therefore increases the concentration of cAMP in the cell. Increased cAMP activates protein kinase A that phosphorylates PLIN1 and
HSL. Phosphorylation of PLIN1 releases ABHD5 that then binds to ATGL and activates it. PKA phosphorylation of HSL results in its translocation from the
cytosol to the lipid droplet surface. In this position, the enzymes completely hydrolyze TAG to glycerol and 3 fatty acids, which are exported out of the
adipocyte. Lipolysis is inhibited by binding of epinephrine to α2 receptors that inhibit adenylyl cyclase. Insulin binding to its receptor results in a cascade
that activates PKB that phosphorylates cAMP phosphodiesterase. This lowers the cAMP concentration and inhibits lipolysis.
by another enzyme, α-glycerophosphate dehydrogenase, requiring NAD1, and thus the glycerol can enter
into glycolysis and the TCA cycle to be fully oxidized
to CO2 and H2O to provide energy as ATP to the cell
(see Figure 2.11.4).
FATTY ACIDS ARE METABOLIZED
IN THE MITOCHONDRIA
AND PEROXISOMES
Free fatty acids are formed in the cytoplasm by the
action of lipase on stored triglycerides, but the fatty
acids themselves are degraded and oxidized only in the
mitochondria and peroxisomes. The fatty acids have
surface activity (they lower the surface tension) and can
impair membrane integrity. Therefore, the fatty acids
are carried in solution by fatty acid binding proteins.
These are low-molecular-weight proteins (about
14,000 Da) that probably have a dual function of
decreasing the concentration of the free fatty acids and
of enhancing the diffusion through the cytoplasm by
carrying the fatty acids.
The first step in the metabolism of free fatty acids is
their import into the inner mitochondrial matrix by
combining with a carrier substance, carnitine (see
Figure 2.11.5). First, the fatty acid is combined with
coenzyme A by the enzyme thiokinase, which hydrolyzes ATP to AMP and PPi. This fatty acyl CoA is then
transferred to carnitine by the enzyme carnitine fatty
acyl transferase. Once the fatty acyl carnitine is transferred to the mitochondrial matrix, it is once again combined with CoA to form fatty acyl CoA. In this form
and in this place, the fatty acid can be oxidized in a systematic way to produce energy.
BETA OXIDATION CLEAVES TWO
CARBON PIECES OFF FATTY ACIDS
Beta oxidation is the process by which fatty acids are
processed progressively to release two-carbon segments
in the form of acetyl CoA. This series of reactions is
summarized in Figure 2.11.6. These reactions produce
acetyl CoA and 1 FADH2 and 1 NADH per turn of the
beta oxidation cycle.
243
CH2OH
OH
O
Glucose
OH
OH
OH
ATP
Glucokinase (liver)
Hexokinase (muscle)
ADP
CH2OH
OH
CH2OPO3
Phosphoglucomutase
O
OH
O
Glucose-6-phosphate
Glycogen
OH
OH
OPO3
OH
OH
OH
Glucose-1-phosphate
Phosphoglucose isomerase
CH2OPO3
OH
O
OH
Pi
H2C
Glycerol
OH
HC
OH
H2C
OH
OH
Phosphofructokinase
ADP
CH2OPO3
OH
O
HO
ATP
Glycerol kinase
ADP
CH2OPO3
Glycerophosphate
HC
OH
H2C
OH
ATP
Fructose-1,6-diphosphatase
H2O
NAD+
OH
NADH
+ H+
Fructose-6-phosphate
CH2OH
Fructose-1,6-diphosphate
CH2OPO3
Aldolase
CH2OPO3
HC
O
C
HC
OH
O
Glycerophosphate
dehydrogenase CH OH Triose phosphate isomerase
2
Dihydroxyacetone phosphate
CH2OPO3
Glyceraldehyde-3-phosphate
Pyruvate
FIGURE 2.11.4 Metabolism of glycerol. Glycerol is phosphorylated and then converted into dihydroxyacetone phosphate, an intermediate in glycolysis.
EXAMPLE 2.11.1 ATP Yield from Glycerol
How much ATP is made from glycerol, under optimal conditions?
Glycerol requires 1 ATP per molecule for the conversion into
glycerol phosphate. The glycerol phosphate is then converted to
dihydroxyacetone phosphate with the production of 1 molecule
of NADH. The dihydroxyacetone phosphate can then be oxidized
fully to CO2 through glycolysis and the TCA cycle as outlined in
Chapters 2.9 and 2.10.
In the conversion to pyruvic acid, dihydroxyacetone phosphate
generates 1 NADH and 2 ATP molecules per molecule of glycerol.
The net effect of converting glycerol to pyruvic acid is 21 ATP 1
2 ATP 1 2 NADH 5 1 ATP 1 2 NADH 5 6 ATP, assuming that
the NADH are both oxidized with the generation of 2.5 ATP per
NADH. The complete oxidation of pyruvate produces 4 NADH,
1 FADH2, and 1 GTP. When the reducing equivalents are oxidized
through the electron transport chain, this produces
4 NADH 3 2:5 ATP=NADH 1 1 FADH2 3 1:5ATP=FADH2 1 1 GTP
3 1 ATP=GTP 5 12:5 ATP
Each glycerol molecule liberated from a triglyceride thus produces at most 18.5 molecules ATP per molecule glycerol.
Glycerol has a gram molecular weight of 92 g mol21. It produces
18.5 moles ATP/92 g mol21 5 0.20 mol g21.
This is a little more than the energy derived from glucose:
32 moles ATP/180 g mol21 5 0.18 mol g21.
ATP P roduction III: F att y Acid O xidat ion and A mino Aci d Oxi da tion
O
O
H2
R C C C OH
H2
Free fatty acid
R
C
H2
ATP
AMP + PPi
O
H2
C C S CoA
AMP + PPi
O
CH3
H
C C
H2
CH3 O
CH3 N
Carnitine
CH3
O
H
C C C C OH
H2
H2
CH3 OH
C
N
H2
C
R
H2
C
H2
C
C
S
CoA
Fatty acyl CoA
O
FAD
Acyl CoA dehydrogenase
C C OH
H2
FADH2
O
C
O
C
R
R
H2
C
C
H
H
C
C
Cytoplasm
Carnitine acyltransferase I
S
CoA
Trans enoyl CoA
H2O
Outer mitochondrial
membrane
Enoyl CoA hydratase
Intermembrane
space
Acylcarnitine translocase
OH
Inner mitochondrial
membrane
R
Carnitine acyltransferase II
CH3
CH3
O
H
CH3 N C C C C OH
H2
H2
CH3 OH
H2
C
C
H
O
H2
C
H
C C
H2
CH3 O
N
C
CoA
3-Hydroxy acyl CoA
NAD+
3-Hydroxy acyl CoA dehydrogenase
O
NADH + H+
C C OH
H2
O
S
C
Mitochondrial
matrix
CoA-SH
Carnitine
CH3
Free fatty acid
OH
Thiokinase
Fatty acyl carnitine
CH3
C
CoA
Thiokinase
R
H2
C
ATP
CoA
Fatty acyl CoA
H2
C
C
H2
O
R
H2
C
C
O
H2
C
C
C
S
CoA
3-Keto acyl CoA
CoA
C
R
C
H2
O
H2
C C S CoA
Thiolase
R
Fatty acyl CoA
O
R
H2
C
C
O
S
CoA
+
H3C
Fatty acyl CoA
FIGURE 2.11.5 Involvement of carnitine in the entry of free fatty acids
into the inner mitochondrial space. Fatty acids are combined with
coenzyme A in a reaction that effectively costs 2 ATP molecules per
reaction. The fatty acyl CoA is then converted to fatty acyl carnitine,
which can penetrate the inner mitochondrial membrane. In the
mitochondrial matrix, the carnitine is removed by a second, different
carnitine acyl transferase.
Once coupled with coenzyme A, the fatty acyl chain is
oxidized, producing FADH2. The fatty acyl chain is further oxidized by adding water and then removing two
more hydrogens by the 3-hydroxy acyl CoA dehydrogenase, this time producing NADH. The final step in
each turn of the beta oxidation cycle is the production
of acetyl CoA and the regeneration of fatty acyl CoA,
shortened by two carbon atoms. This shorter fatty acyl
CoA reenters the beta oxidation cycle until, at the end,
all of the chain is converted to acetyl CoA. In this
way, palmitic acid, for example, will produce 8 acetyl
CoA molecules and 7 FADH2 molecules and 7 NADH
molecules (there are only 7 because the last turn of
the cycle produces two acetyl CoA molecules and so
the last two carbons do not enter the cycle again to
produce FADH2 and NADH).
C
S
CoA
Acetyl CoA
FIGURE 2.11.6 Beta oxidation of free fatty acid.
THE LIVER PACKAGES SUBSTRATES
FOR ENERGY PRODUCTION BY OTHER
TISSUES
During exercise, when fatty oxidation occurs rapidly,
the liver packages acetyl CoA in a form that can be readily used by other tissues for the generation of energy. In
the liver, two molecules of acetyl CoA combine to form
acetoacetate. The acetoacetate that forms can then be
converted to β-hydroxybutyric acid and, to a lesser
extent, to acetone. All three of these compounds, acetoacetate, β-hydroxybutyric acid, and acetone, are
referred to as ketone bodies (see Figure 2.11.7). They
leave the liver cell and travel in the blood to the peripheral tissues, which take up the compounds and metabolize them for energy.
The combined concentration of the ketone bodies is
typically less than about 3 mg%. Despite these low
245
246
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
EXAMPLE 2.11.2 ATP Yield from Fatty Acids
Each turn of the beta oxidation cycle produces NADH and FADH2.
For palmitic acid, C16H32O2, a total of eight acetyl CoA molecules
are produced from seven turns of the beta oxidation cycle.
The NADH and FADH2 feed into the electron transport chain
to produce ATP. The total ATP produced from beta oxidation is
thus
7 NADH=palmitate 3 2:5 ATP=NADH 1 7 FADH2 =palmitate
3 1:5 ATP=FADH2 5 28 ATP=palmitate
Each acetyl CoA produced by beta oxidation enters the TCA
cycle where it is further oxidized to CO2 and produces reducing
equivalents that are used by the ETC to make ATP. Each acetyl
CoA molecule produces 3 NADH, 1 FADH2, and 1 GTP molecule
during a single turn of the TCA cycle. These are eventually used
by the ETC to produce
3 NADH=acetyl CoA32:5 ATP=NADH11 FADH2 =acetylCoA
31:5 ATP=FADH2 11 GTP=acetyl31 ATP=GTP
510 ATP=acetyl CoA
Since there are eight acetyl CoA molecules per palmitic acid produced from beta oxidation, the total ATP produced from acetyl
CoA from palmitic acid is
10 ATP=acetyl CoA 3 8 acetyl CoA=palmitate 5 80 ATP=palmitate
We add this to the ATP produced from beta oxidation and subtract the two ATP needed to start beta oxidation from the initial
thiokinase reaction to get
28 ATP from beta oxidation 1 80 ATP from TCA cycle
2 ATP from thiokinase reaction 5 106 ATP=palmitic acid
EXAMPLE 2.11.3 Compare ATP Yield from Glucose to that of Tripalmitin
In Chapter 2.10, we found that the maximum yield of ATP from
the complete oxidation of glucose was 32 ATP per glucose. This
corresponds to 7 ATP per glucose from the oxidation of glucose
to pyruvate, and 25 ATP from the complete oxidation of pyruvate to CO2 and H2O. The gram molecular weight of glucose is
180 g mol21, so the ATP production is
glycerol. We have calculated that glycerol gives rise to at most
18.5 ATP per glycerol, and each palmitic acid produces at most
106 ATP per palmitic. The total for tripalmitin is thus
32 moles ATP=mol glucose 5 32 moles ATP=180 g
5 0:18 mol ATP per g of glucose
Thus fat has about 2.3 times as much energy stored per unit
weight.
336:5 moles ATP=mole tripalmitin 5 336:5 moles=807:3 g
5 0:42 ATP per g of tripalmitin
The gram molecular weight of tripalmitin is 807.3 g mol21, tripalmitin consisting of three palmitic acid molecules and one
concentrations, the flux of energy to the metabolizing
tissues can be great because the ketone bodies are
taken up so quickly. The concentrations of the ketone
bodies can occasionally rise to very high levels, a
condition known as ketosis. This condition occurs
whenever metabolism of fats is emphasized such as
in starvation and in diabetes mellitus. In this case,
the urine can contain ketones and the presence of
acetone is sometimes detectable by its odor in the
exhaled air.
In the peripheral tissues, acetoacetic acid is taken up
and converted in the mitochondria to acetoacetyl CoA
by the transfer of a CoA moiety from succinyl CoA, an
intermediate in the TCA cycle. Since succinyl CoA is
usually converted to succinate with the formation of 1
molecule of GTP, conversion of succinyl CoA to succinate in this reaction removes the potential synthesis of
1 molecule of GTP. So, although no energy in the form
of ATP is directly involved in this transfer, it has a net
cost of 1 molecule of ATP. The acetoacetyl CoA can
then be converted to two molecules of acetyl CoA by
thiolase, the same enzyme involved in the production
of acetyl CoA from fatty acyl CoA during the beta oxidation pathway of fatty acids.
AMINO ACIDS CAN BE USED
TO GENERATE ATP
Amino acids can be used to build body proteins and
they can be broken down to yield energy. In the steady state of the adult, the body store of proteins
remains constant and there is a constant throughput
of amino acids, equal to the dietary intake, that is
converted to metabolic energy. In the typical
American diet, about 16% of the calories are provided
by dietary protein.
Because the hepatic portal blood leaves the intestines
and travels to the liver, the liver has the first opportunity to metabolize all the nutrients, including the amino
acids absorbed from digested proteins in the intestinal
lumen. The liver does several things: it catabolizes a
ATP P roduction III: F att y Acid O xidat ion and A mino Aci d Oxi da tion
CoA-SH
O
H3C
C
Acetyl CoA
S
2
H3C
C
CoA
+
CoA
Thiolase
+
O
HS
Acetoacetate
Acetyl CoA
S
O
H3C
CoA
C
O
H2
C
C
OH
NAD + H+
Beta-hydroxy butyric acid
dehydrogenase
CO2
NADH
OH
H3C
C
H
O
H2
C
O
OH
C
Beta-hydroxy butyric acid
H3C
C
CH3
Acetone
FIGURE 2.11.7 Formation of ketone bodies by the liver. During rapid lipid metabolism, when production of acetyl CoA outstrips the liver’s ability to
oxidize it, acetyl CoA coalesces to form acetoacetate, beta-hydroxybutyric acid, and acetone.
large fraction of the amino acids (57%), releases some
unchanged into the general circulation (23%), and utilizes some 20% for synthesis of proteins that either
remain in the liver or are released into the blood.
Catabolism of amino acids can be broadly categorized
into two processes: the breakdown of amino acids to
carbohydrate precursors and potentially leading to the
formation of glucose; and transformations leading to
acetyl CoA that result in the potential formation of
ketone bodies. Amino acids that break down into carbohydrate precursors are called glucogenic; those leading
to acetyl CoA are called ketogenic.
G
G
G
Leucine and lysine are the only exclusively ketogenic
amino acids.
Isoleucine, threonine, phenylalanine, tyrosine, and
tryptophan are both glucogenic and ketogenic.
Aspartatic acid, asparagine, glutamic acid, glutamine, alanine, arginine, histidine, glycine, serine, proline, valine, methionine, and cysteine are
glucogenic.
Because each amino acid has a different side chain, each
amino acid is catabolized differently to produce energy
and waste products. We will not go through all of these
reactions for each of the amino acids. The overall fate of
the amino acids is shown in Figure 2.11.8.
AMINO ACIDS ARE DEAMINATED
TO ENABLE OXIDATION
Many amino acids share a common mechanism for the
removal of the amino group to form intermediates
in the TCA cycle or glycolytic cascade. This is a transamination reaction followed by a dehydrogenation reaction, as shown in Figure 2.11.9. The α-ketoglutarate
formed in the transamination reaction in the mitochondria can then enter the TCA cycle. The deamination
results in the liberation of ammonia. The reaction
sequence shown is one of many such involved in the
deamination of a variety of amino acids including phenylalanine, tyrosine, aspartate, cysteine, lysine, arginine,
alanine, isoleucine, leucine, and valine. The reactions
differ only in the α-keto acid formed following the
transamination.
UREA IS PRODUCED DURING
DEAMINATION AND IS ELIMINATED
AS A WASTE PRODUCT
The ammonia released during deamination is removed
from the blood almost entirely by conversion into urea
in the liver. This occurs through another metabolic process called the urea cycle (see Figure 2.11.10). In this
process, the ammonia is combined with bicarbonate
ion to form carbamoyl phosphate. The complete
operation of the cycle requires continual input of
aspartate. This can be derived from transamination of
oxaloacetic acid by glutamic acid, the reverse of the
process shown in Figure 2.11.9. Since glutamate is
the product of transamination with several amino
acids, it can be replenished. Thus one of the amino
groups of urea is derived from ammonia and the other
is derived from amino groups on various amino acids,
transaminated to glutamate.
247
248
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Aspartic acid
Asparagine
GDP
GTP
CO2
Phosphoenolpyruvate
Oxaloacetate
Phenylalanine
tyrosine
Phosphoenolpyruvate
carboxykinase
ADP
NADH + H
Pyruvate kinase
Tryptophan
Threonine
Cysteine
Serine
Glycine
Alanine
Malate dehydrogenase
NAD
ATP
+
Pyruvate
Malate
Fumarate
Cytoplasm
Outer mitochondrial
membrane
Dicarboxylic acid carrier
Intermembrane
space
Pyruvate carrier
Inner mitochondrial
membrane
Mitochondrial
matrix
CoA-SH
–
OH
NAD
+
Phenylalanine
Tyrosine
Tryptophan
Threonine
Isoleucine
Lysine
Leucine
Pyruvate dehydrogenase
CO2
NADH + H
Acetyl CoA
Citrate
Oxaloacetate
Aconitase
Citrate synthase
Malate dehydrogenase
CoA-SH
NADH + H
Isocitrate
Malate
NAD
+
NAD
+
Isocitrate dehydrogenase
Fumarase
TCA cycle
CO2
NADH + H
Fumarate
FADH2
Alpha-ketoglutarate
+
NAD
FAD
Succinate dehydrogenase
CoA-SH
NADH + H
Succinyl CoA
Glutamic acid
Glutamine
Proline
Histidine
Arginine
CoA-SH
Alpha-ketoglutarate
dehydrogenase
CO2
Succinate
GTP
GDP Pi
Succinyl CoA synthetase
Pyruvate
dehydrogenase
Isoleucine
Valine
Methionine
FIGURE 2.11.8 Metabolic entry points for the catabolism of the amino acids. Those amino acids that produce acetyl CoA are called ketogenic. These
include leucine, lysine, phenylalanine, tyrosine, tryptophan, and isoleucine. Those amino acids that produce carbohydrate precursors that can be
converted to glucose are called glucogenic. These include aspartic acid, asparagine, phenylalanine, tyrosine, tryptophan, alanine, cysteine, serine,
threonine, glycine, glutamic acid, glutamine, proline, histidine, arginine, isoleucine, valine, and methionine. Only lysine and leucine are exclusively
ketogenic. Exclusively ketogenic amino acids are in light blue italic; exclusively glucogenic are in dark blue; both ketogenic and glucogenic are in
black.
ATP P roduction III: F att y Acid O xidat ion and A mino Aci d Oxi da tion
COO–
Aspartate
+H N
3
COO–
CH
O
C
CH2
CH2
COO–
COO–
Aspartate
aminotransferase
COO–
H2C
Glutamate
CH2
CH2
COO–
C
COO–
H2C
Alpha-ketoglutarate
O
Oxaloacetate
+H
3N
COO–
C
H
Cytoplasm
Outer mitochondrial
membrane
Intermembrane
space
Dicarboxylic acid carrier
Inner mitochondrial
membrane
Mitochondrial
matrix
COO–
H2C
Glutamate
dehydrogenase
CH2
Alpha-ketoglutarate
O
COO–
C
COO–
H2C
CH2
+H
3N
C
H
Glutamate
COO–
H2O
+
NH4
NADH + H+
NAD+
FIGURE 2.11.9 Deamination of amino acids by aminotransferase and glutamate dehydrogenase action.
249
250
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
COO–
+
H3N
Aspartate
CH
CH2
COO–
AMP + PPi
ATP
COO–
COO–
+
+
H3N
H3N
CH
(CH2)3
Citrulline
CH
Arginosuccinate
(CH2)3
Arginosuccinate synthetase
COO–
NH
NH
CH
HCO3–
NH2
NH2
Pi
Bicarbonate
NH
CH
O
C
CH2
COO–
2ADP + Pi
Urea cycle
Lyase
2 ATP
Fumarate
COO–
–
PO4
C
Ornithine transcarbamylase
CH
O
COO–
Carbamoyl phosphate
NH4+
synthetase
+
NH4+
Carbamoyl phosphate
+
Ammonia
H3N
Ornithine
H3N
COO–
CH
CH
COO–
(CH2)3
CH
NH
(CH2)3
C
NH2
H2O
Arginine
NH2
NH
O
NH2
C
NH2
Urea
FIGURE 2.11.10 Formation of urea from the urea cycle.
Clinical Applications: Nonshivering Thermogenesis
Immediately after birth, the baby is thrust into a cool and dry
environment. It loses heat rapidly by evaporation. Even when its
skin dries, the baby continues to lose heat because it has a large
surface area relative to its small body mass, it cannot put on
warmer clothes, it has poor thermal insulation in the form of fat,
and it has limited ability to generate heat by muscular contractions. However, the baby is able to generate heat by nonshivering thermogenesis, also called metabolic thermogenesis.
Babies have specialized fat tissue called brown fat, located
mainly in the neck and in the midline of the back.
Mitochondria contain cytochromes that contain iron, giving
the mitochondria a reddish-brown color. Brown fat contains
lots of mitochondria so it takes on a brownish color from
mitochondrial pigments. The mitochondria in this fat make
ATP primarily from the oxidation of fatty acids that are
produced by hydrolysis of stored triglycerides. Under certain
circumstances, these mitochondria can become uncoupled.
Uncoupling of oxidative phosphorylation in brown fat mitochondria generates heat.
Oxidative phosphorylation couples an exothermic reaction, one
that releases heat, to an endothermic reaction, one that requires
heat. Pumping of H1 ions out of the mitochondrial matrix couples the exothermic oxidation reactions to the energy of the H1
electrochemical gradient. This in turn is coupled to the endothermic synthesis of ATP. The oxidation of foodstuffs (carbohydrates,
fats, and amino acids) reduces NAD1 to NADH, whose oxidation
powers the H1 pumping by the electron transport chain. The
ATP synthase couples the energy stored in the electrochemical
potential of H1 to ATP synthesis. This coupling can never be
100% efficient, so that cellular respiration releases some energy
ATP P roduction III: F att y Acid O xidat ion and A mino Aci d Oxi da tion
as heat. Under certain circumstances a special protein, called the
uncoupling protein, short-circuits the synthesis of ATP by allowing H1 ions to cross the inner mitochondrial membrane without
making ATP. Then more of the energy stored in the H1 gradient
is dissipated as heat and less is captured by ATP. The exothermic
reactions are uncoupled from ATP synthesis and the mitochondria generate heat proportionate to the number of mitochondria
and the rate of oxygen consumption.
content of the diet increases the tissue content of UCP2. Thus
these proteins are believed to participate in basal or regulatory
thermogenesis, but their exact functions are not yet worked out.
The mechanism of brown fat thermogenesis in response to
cold is shown in Figure 2.11.11. Adipose tissue is supplied by
sympathetic nerves that release noradrenaline onto the fat
cells. This stimulates the breakdown of stored triglycerides to
fatty acids, which in turn activate UCP1. The effect depends on
the tissue content of UCP1, which is also increased by cold
exposure. How UCP1 increases the H1 flux across the membrane is also not yet known. UCP1 may form a H1-specific pore
or it may transport H1 ions by cycling an H1 carrier, most likely
fatty acids. Current work favors the H1 carrier mechanism
because some carboxylic acid groups can activate transport
without being transported.
UCP1, a 32-kDa protein isolated from brown fat mitochondria,
was the first uncoupling protein to be described. UCP1 is located
mainly in brown fat, whereas UCP2 is expressed in many tissues
and UCP3 is found mainly in skeletal muscle. BMCP1 is specific
to the brain. UCP1 is activated by fatty acids and inhibited by
purine nucleotides (ATP, ADP, GTP, and GDP). Exposure to cold
increases UCP1 content in brown fat, and increased caloric
Cold
Diet
Sympathetic nerves
Norepinephrine
Adenylyl cyclase
Cell membrane
β-Receptor
GDP
Gα subunit βγ subunits
cAMP
ATP
+
Triglyceride
droplet
+
ATP
Lipase, inactive
ADP
CREB Pi
CREB
+
Active PKA
Pi CREB CREB Pi
DNA
CRE
Lipase, active
mRNA
FA
Nucleus
Outer mitochondrial membrane
–160 mV
H+
H+
++++++
–
+
+
ETC
F0
NADH
NAD
---------
Inner mitochondrial membrane
UCP1
F1
+
ATP
ADP + Pi
H+
H+
Heat
FIGURE 2.11.11 Postulated mechanisms for nonshivering thermogenesis in brown adipose tissue. Noradrenaline released from sympathetic nerve
terminals in adipose tissue binds to β receptors on the surface of the adipocytes, leading to increased cytosolic cAMP by activating adenylyl cyclase.
cAMP activates protein kinase A (PKA), which phosphorylates several targets, thereby changing their activity. PKA activates hormone-sensitive lipase
that increases triglyceride breakdown and increases cytoplasmic fatty acid concentration. The increased fatty acids activate UCP1, short-circuiting
the H1 electrochemical gradient and reducing the synthesis of ATP from ADP and Pi. This uncouples oxidative phosphorylation and the energy of
oxidation of fats appears as heat. PKA also phosphorylates a transcription factor, CREB (cyclic AMP response element binding protein), that binds to
specific regions of the DNA (CRE—cAMP response element) and activates their transcription into mRNA. This leads to increased numbers of UCP1
protein in the mitochondria, which adapts the body to exposure to cold or some other stress. Diet-induced thermogenesis probably involves
increased expression of UCP2.
251
252
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
SUMMARY
The oxidation of fatty acids and amino acids produces
ATP through some of the same reactions used to produce ATP by the oxidation of carbohydrates. Fatty
acids are released from triglycerides by hormonesensitive lipase. The fatty acids are bound in the cytoplasm by fatty acid binding protein and carried into
the mitochondria by being complexed with carnitine.
Inside the mitochondria, fatty acids undergo beta oxidation in which two carbons at a time are cleaved off
the carboxyl end and converted into acetyl Coenzyme
A. Each turn of the beta oxidation cycle produces one
NADH and one FADH2. These feed reducing equivalents into the electron transport chain, which pumps
H1 ions out of the mitochondrial matrix and produces the electrochemical gradient of H1 that drives
ATP synthesis through the F0F1ATPase. Beta oxidation
also produces acetyl CoA that enters the TCA cycle
by combining with oxaloacetate to form citric acid.
Each turn of the TCA cycle converts the acetyl CoA
into 2 molecules of CO2, 3 NADH, 1 FADH2, and 1
GTP. Thus palmitic acid (16:0) produces 7 NADH and
7 FADH2 from beta oxidation, and 24 NADH, 8
FADH2, and 8 GTP from the complete oxidation of
acetyl CoA. Since NADH produces 2.5 ATP molecules
and FADH2 produces 1.5, the ATP count from palmitic acid is 28 from beta oxidation and 80 from the
TCA cycle. Two ATP molecules are used to prime the
palmitic acid in the thiokinase reaction that converts
palmitic acid to palmitoyl CoA.
The liver packages lipid metabolites into ketone bodies,
which collectively consist of acetoacetic acid, betahydroxybutyric acid, and acetone. Build-up of these
ketone bodies during starvation or other metabolic conditions such as diabetes is called ketosis.
Each amino acid has its own metabolic pathway because
they all differ chemically. However, they feed into the
main metabolic pathways in a limited number of places.
Those amino acids that can be used to make glucose are
called glucogenic. Those that can be used to make ketone
bodies by producing acetyl CoA are called ketogenic.
Many amino acids are both glucogenic and ketogenic.
Only leucine and lysine are exclusively ketogenic.
Many amino acids are deaminated by a combination of
transamination and dehydrogenation. In this reaction,
the amino group is transferred from the amino acid to
alpha-ketoglutaric acid, forming glutamic acid. The glutamic acid is then converted back to alpha-ketoglutaric
acid by glutamate dehydrogenase, resulting in release of
ammonia and formation of NADH. The ammonium
formed in this way is converted to urea through the
urea cycle. The urea cycle begins with the formation
of carbamoyl phosphate by condensing ammonium
with HCO32. Carbamoyl phosphate then combines
with ornithine to form citrulline. Citrulline combines
with aspartic acid to form arginosuccinate and arginine
in sequence. Arginine gives rise to urea and ornithine to
begin the cycle again.
REVIEW QUESTIONS
1. What components make up a triglyceride?
Where are triglycerides found in the body?
Can they be metabolized as is? What enzyme
breaks triglycerides down into components?
What activates this enzyme? What inhibits the
enzyme?
2. How is glycerol oxidized? How much ATP is
produced from glycerol, mole per mole?
3. How are fatty acids carried in the cytosol?
Where does oxidation of fatty acids take place?
How do the fatty acids get into the
mitochondria?
4. What is meant by beta oxidation? What are the
main products? How many beta oxidation cycles
are there for palmitic acid? Stearic acid? Oleic
acid?
5. How much ATP is produced from the complete
oxidation of palmitic acid, mole per mole?
6. What are the ketone bodies? Where are they
produced?
7. What is a glucogenic amino acid? Which amino
acids are glucogenic?
8. What is a ketogenic amino acid? Which amino
acids are exclusively ketogenic?
9. What is transamination? What are the major
receptors for transamination?
10. What is urea? Where is it produced?
The Origin of the Resting
Membrane Potential
Learning Objectives
G
G
G
G
G
G
G
G
Write the Nernst equation for any given ion
Define equilibrium potential
Be able to calculate the equilibrium potential for any ion
Recognize the proper form of the GoldmanHodgkinKatz
equation
Explain why in the GoldmanHodgkinKatz equation the
anion concentration in one compartment appears on the
opposite side of the argument from the cation concentration in the same compartment
Define and distinguish between slope conductance and
chord conductance
Write the chord conductance equation
Explain what happens to membrane potential when the
conductance to a particular ion changes
INTRODUCTION
In Chapter 2.6, we analyzed the energetics of membrane
transport across resting cardiac muscle cells that have a
membrane potential of about 280 mV, negative inside.
At rest, the concentration gradient favored Na1 and
Ca21 entry into the cell and K1 exit. These gradients
produced slow leaks of ions that were continually
balanced by active transport mechanisms such as the
Na,K-ATPase and NaCa exchanger so that the ionic
composition of the cytosol stayed constant.
The resting membrane potential is extremely important
because, first, all cells have a membrane potential (but
not the same value!), and modulation of the membrane
potential is associated with modulation of cellular
activity. Certain cells of the body, called excitable cells,
can use rapid changes in their membrane potential as a
signal. These cells include nerve cells and muscle cells.
Now we ask the question, where did the resting
membrane potential come from? To answer this question, we will consider hypothetical membranes. These
membranes are not like any biological membrane, but
we consider them because they will clarify how the resting membrane potential comes to be what it is. Here we
will use concepts of potential and capacitance already
covered in Chapter 1.3, and the concept of the electrochemical potential discussed in Chapter 1.7.
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00023-9
3.1
THE EQUILIBRIUM POTENTIAL ARISES
FROM THE BALANCE BETWEEN
ELECTRICAL FORCE AND DIFFUSION
First we consider a hypothetical membrane that is
permeable only to Na1 ions. Suppose that [Na1]o,
the outside or extracellular sodium concentration, is
145 3 1023 M, and [Na1]i 5 12 3 1023 M. And suppose
that initially there is no membrane potential. What happens? As shown in Figure 3.1.1, the diffusion gradient
for Na1 favors Na1 entry into the cell, and the initial
Na1 influx carries a charge that builds up on the inside
of the cell. This produces a potential (recall that separation of charge produces a potential) across the membrane that impedes further Na1 ion movement because
the positive charges repel the positively charged Na1
ion. The potential that develops is related to the electric
force that now works against further Na1 movement.
Eventually the electric force will get so large that the
diffusive flow will be exactly counterbalanced, and net
flow will stop. This will occur at the Na equilibrium
potential. We can calculate what that potential should
be in two ways: first by looking at Fick’s law and second
by analyzing the energetics. We get the same answer
either way. The situation for the development of the
Na1 equilibrium potential is shown in Figure 3.1.1.
Fick’s law with an electrical force is given as
½3:1:1
Js 5 2D
@C
D
@ψ
2
Cz`
@x RT
@x
This is Eqn [1.7.19]. Here Js is the solute flux, in this case
the flux of Na1, D is the diffusion coefficient, C is the concentration of Na1, R is the gas constant, T is the absolute
temperature, z is the charge on the ion (11 for Na1),
and ` is the faraday, the number of coulombs per mole.
At equilibrium, Js 5 0, and we equate the diffusive force
and the electrical force:
½3:1:2
RT @C
@ψ
5 2z`
C @x
@x
Integrating both sides from outside to inside of the
membrane, we get
ði
ði
RT @C
@ψ
dx 5 2z` dx
C
@x
@x
o
o
½3:1:3
Ci
RT ln 5 z`ðψo 2 ψi Þ
Co
255
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
160
160
[Na+]o 145 mM
Membrane
Na+ flux is down its
concentration gradient
130
Concentration (mM)
120
J = –D dC/dx
100
Na+
90
80
70
60
50
Extracellular fluid
Cytosol
120
110
60
50
[Na+]i 12 mM
20
[Na+]i 12 mM
10
0
10
0
160
160
[Na+ ]o 145 mM
140
150
3
120
J = –D dC/dx
100
Na+
–
+
70
J = – D/RT CzF dV/dx
Concentration (mM)
The current builds up charge
on the two sides of the membrane
that oppose further Na+ flux
130
60
i
70
30
90
80
J = –D dC/dx
90
80
30
110
Na+ carries a charge;
its flux makes a current
100
40
20
2
130
40
150
[Na+]o 145 mM
140
1
140
110
150
Concentration (mM)
150
Concentration (mM)
256
4
130
At equilibrium, the electrical
force exactly balances diffusion
120
110
100
90
80
70
–
+
–
+
–
+
–
+
60
50
40
40
30
30
[Na+]i 12 mM
Membrane
140
50
20
[Na+]o 145 mM
Extracellular fluid
Cytosol
[Na+]i 12 mM
20
10
0
10
0
FIGURE 3.1.1 Generation of the Na equilibrium potential across a hypothetical membrane that is permeable only to Na. The [Na]o in this case is high,
about 145 mM, whereas [Na]i is 12 mM. Thus the diffusion gradient favors Na1 flux from the outside to the inside of the cell. Since Na1 is electrically
charged, flux of only Na1 makes a current across the membrane that separates charges and thus produces an electrical potential. The electric field
exerts forces on Na1 that retards its movement. When the diffusive force exactly balances the electrical force, flux is zero and the potential is ENa, the
sodium equilibrium potential.
This gives the equilibrium membrane potential as
½3:1:4
RT Ci
ln
5 ðψo 2 ψi Þ
z` Co
It is usual for physiologists to take the outside solution
as ground (ψo 5 0) because it is potential differences
that we are concerned with. Using this standard ψo 5 0,
we rewrite Eqn [3.1.4] as
½3:1:5
RT Co
ln
5 ðψi 2 ψo Þ
z` Ci
If you always put the outside ion in the numerator of
the argument of the logarithm, then the sign of the
membrane potential will be correct. This is a famous
equation, the Nernst equation, named for Walther
Nernst (18641941), a physical chemist from Berlin,
Germany. It calculates the potential at which net flux
is zero, which occurs at equilibrium for the ion. The
membrane potential at which the diffusive force is
exactly balanced by the electrical force is called the
equilibrium potential for that ion. For sodium, it is
usually symbolized as ENa.
The Nernst Equation can also be derived easily from
considering the electrochemical potentials. At equilibrium we have
Δμ 5 μNai 2 μNao
½3:1:6
0 5 μoNai 1 RT ln½Na1 i 1 z`ψi 2 μoNao
2 RT ln½Na1 o 2 z`ψo
T he O rig in of the Res tin g Membra ne P oten tial
EXAMPLE 3.1.1 Calculate the Equilibrium Potential for Na1
Inserting values for [Na1]i 5 12 3 1023 M and [Na1]o 5 145 3
1023 M, into Eqn [3.1.5], we calculate
145 3 1023 M
ψi ψo 5 8:314 J mol21 K21 3 310 K 3 ln
5 0:0666 V
123 1023 M
Note that the potential inside is positive, as it should be to
impede further influx of a positive ion.
1 3 9:649 3 104 C mol21
Canceling out the standard free energies, which are
equal, we can arrange Eqn [3.1.6] to give
½3:1:7
RT ½Na1 o
ln
5 ðψi 2 ψo Þ
z` ½Na1 i
The Nernst equation is often written with log10 instead
of the natural logarithm. At 37 C 5 310 K, Eqn [3.1.7]
can be written as
½3:1:8
0:0615 log
½Na1 o
5 ðψi 2 ψo Þ
½Na1 i
where the log is now log10. The term 0.0615 is the evaluation of the expression RT/z` and conversion of the
natural log, ln, to log10. What this means is that every
10-fold gradient in concentration of a singly charged
ion gives an equilibrium potential of 61.5 mV at 37 C.
THE EQUILIBRIUM POTENTIAL
FOR K 1 IS NEGATIVE
Now we suppose that the membrane is impermeable
to Na1 and Cl2 ions but it is permeable only to K1.
The [K1] concentrations on the two sides of the
membrane are those in a muscle cell, namely,
[K1]o 5 4 3 1023 M and [K1]i 5 155 3 1023 M. Because
of its concentration gradient, K1 will diffuse out of the
cell, causing an outward current and accumulation of
positive charges on the outside of the cell. The Nernst
equation is
½3:1:9
RT ½K1 o
ln
5 ðψi 2 ψo Þ
z` ½K1 i
The result of the calculation using [K1]o 5 4 3 1023 M and
[K1]i 5 155 3 1023 M is EK 5 20.0977 V or 297.7 mV.
Here the potential is strongly negative inside.
In this case, z 5 21 because the Cl2 ion has a negative
charge. This is equivalent to inverting the argument
of the logarithm. Here we get ψi 2 ψo 5 20.0615 log
(100/5) 5 20.080 V. Thus ECl 5 20.080 V 5 280 mV.
Figure 3.1.2 shows the concentration differences for
Na1, K1, and Cl2 and the equilibrium potential
for each of these ions. It is important to remember what
the equilibrium potential is. It is the potential at which
the electrical force exactly balances diffusion so that the
net flux of the ion across the membrane is zero. Since
there is no flux, there is no change in the concentrations
on the two sides of the membrane and the ion is at
equilibrium.
Such hypothetical membranes as the ones we have been
considering that are permeable only to Na1, or only to
K1 or only to Cl2 do not exist. Real membranes have
some nonzero permeability to all of these ions. So what
is the membrane potential across a membrane that is
permeable to all three? The short answer is: it depends
on how permeable the membrane is to each of the
ions. What we need is some expression that tells us
what the magnitude of the membrane potential will
be, given the equilibrium potentials and the relative
permeabilities of the ions.
INTEGRATION OF THE
NERNSTPLANCK ELECTRODIFFUSION
EQUATION GIVES THE
GOLDMANHODGKINKATZ EQUATION
When there is a flux of solute, and the solute is charged,
there is a current. The relationship between current
density and flux is
½3:1:11
Ii 5 z`Ji
We can repeat this calculation for any ion to obtain its
equilibrium potential. We must remember what goes
into the equations, however. As an example, consider
that the membrane is not permeable to either K1 or
Na1, but is freely permeable only to Cl2. Suppose
further that Cl2 is 100 3 1023 M outside the cell
and 5 3 1023 M inside the cell. The Nernst equation for
Cl is
where the subscript i denotes the particular ion that is
carrying the current. The total current is the sum of all
the ionic currents. Now we substitute in for the flux
using Eqn [3.1.1] to get
@Ci
Di
@ψ
1
Ci zi `
½3:1:12
Ii 5 zi `Ji 5 2zi ` Di
@x
@x
RT
RT ½Cl2 o
ln
5 ðψi 2 ψo Þ
z` ½Cl2 i
This is the NernstPlanck electrodiffusion equation.
We can obtain an expression for Ii by integrating this
equation over the thickness of the membrane (from
½3:1:10
257
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Membrane
160
150
[K+]i 155 mM
[Na+]o 145 mM
130
[Cl–]o100 mM
0
100
90
80
70
–20
Extracellular fluid
Cytosol
60
50
–40
–60
40
ECl = –80 mV
30
20
10
ENa = +66.7 mV
+20
120
110
+60
+40
140
Concentration (mM)
258
[K+]o 4 mM
0
[Na+]i 12 mM
[Cl–]i 5 mM
–80
–100
EK = –97.7 mV
FIGURE 3.1.2 Equilibrium potentials for Na, K, and Cl in a muscle cell. A positive equilibrium potential is needed to prevent Na1 ions from entering the
cell from the extracellular fluid; a negative equilibrium potential is needed to prevent Cl2 ions from entering; a negative equilibrium potential is
needed to prevent K1 ions from exiting the cell. The solid lines represent ion concentrations.
x 5 0 to δ, where δ is the thickness of the membrane),
but this integration can be accomplished in closed
form only if we assume that the potential gradient is
linear. This is called the constant field assumption.
The integration gives
ðzi `Em =RTÞ
z2 `2 Di
Ci e
2 Co
Em
Ii 5 i
½3:1:13
RTδ
eðzi `Em =RTÞ 2 1
as detailed in Appendix 3.1.A1. This is often written in
an alternate form by multiplying both numerator and
denominator by eð2Z`=RTÞEm :
½3:1:14
Iion 5
ðD=δÞ½ðz2 `2 =RTÞEm ½Ci 2 Co eð2z`=RTÞEm ½1 2 eð2z`=RTÞEm This is the GoldmanHodgkinKatz (GHK) current
equation. It relates the current carried by each ion to
its concentration on both sides of the membrane
(Ci and Co) and to the membrane potential, Em. Now at
steady-state resting membrane potential, the total
current across the membrane must be zero, or otherwise
Em would be changing:
½3:1:15
Itotal 5 INa 1 IK 1 ICl 5 0
By substituting in for the expression for INa, IK, and ICl,
we can derive an expression for Em:
RT
PK ½K1 o 1 PNa ½Na1 o 1 PCl ½Cl2 i
ln
½3:1:16 Em 5
`
PK ½K1 i 1 PNa ½Na1 i 1 PCl ½Cl2 o
This is the GHK equation. It describes the resting
membrane potential when only Na1, K1, and Cl2 are
permeant, but it can be expanded to include other ions.
The inside concentration of Cl2 appears in the numerator with the outside concentrations of Na1 and K1
because z for Cl2 is 21.0 and z for Na1 and K1 is 11.0.
Additional ions can be added to this equation if
they contribute significantly to the currents across
the membrane. This equation shows that the resting
membrane potential results from the concentrationweighted permeabilities across the membrane because
each permeability is multiplied by the concentration of
the ion. Appendix 3.1.A1 presents a full derivation
of this equation.
SLOPE CONDUCTANCE AND CHORD
CONDUCTANCE RELATE ION FLOWS
TO THE NET DRIVING FORCE
The GHK current equation (Eqn [3.1.14]) describes
the current carried by any given ion in terms of its
concentration on both sides of the membrane and the
membrane potential. If we assume that the permeability, D/δ, is constant, we can calculate the current carried
by each ion as a function of the membrane potential.
The currents carried by K1 and Na1 for a muscle cell
containing 155 mM [K1]i and 12 mM [Na1]i and 4 mM
[K1]o and 145 mM [Na1]o as predicted from the GHK
current equation are shown in Figure 3.1.3. At the
equilibrium potential for each ion, there is no current.
This equilibrium potential is also called the reversal
potential, because at this point the current changes
from negative (positive charges enter the cell—by
convention this is an inward current) to positive
(positive charges exits the cellthis is an outward
current).
T he O rig in of the Res tin g Membra ne P oten tial
At high Em the IK approaches
ohmic because [K+]i dominates
the outward current and K+
o
carries little inward current
The slope of the chord connecting
the curve to its reversal potential is
the chord conductance
30
30
IK
INa
Slope of the curve at any Em
20
(dI/dEm) gives the slope
conductance
The reversal potential20
is the Em
at which current falls to zero.
For K this is –98 mV
10
10
Em
0
0
–10
–10
–20
–20
–30
–0.2
The dashed line
is Ohm's law which
occurs only
–0.1
0.0 if
0.1
[K+]i = [K+]o = 155 mM
0.2
Em
–30
–0.2
Em (V)
Reversal potential for
Na is +67 mV
–0.1
0.0
0.1
0.2
Em (V)
FIGURE 3.1.3 Currents carried by K1 (left) and Na1 (right) as predicted by the GHK current equation if the membrane was permeable only to K1 (left)
or to Na1 (right).
The relationship between current and voltage can be
described by a conductance. Ohm’s law states
½3:1:17
Ii 5
E
5 gi E
Ri
where E is the potential difference that drives current
flow (NOT the electric field intensity!), Ri is the resistance to the ion i, and gi is the conductance.
Resistances have units of ohms. Conductances have
units of ohm21, which is a siemen, equal to 1 AV21.
According to Ohm’s law, the conductance is defined for
a line that passes through the origin. The origin is the
point at which there is no net driving force for current
flow. For ions that are not uniformly distributed across
the membrane, however, the point of no net current
flow occurs at the reversal potential. Thus we define a
chord conductance:
½3:1:18
Ii 5 gi ðEm 2 Ei Þ
where gi is the chord conductance at Em, Em is the membrane potential at any point in the currentvoltage
curve, and Ei is the equilibrium potential 5 reversal
potential for a single ion, i. This conductance is the
slope of the chord joining the curve to its reversal
potential, as shown in Figure 3.1.3. Thus the chord conductance is not constant but varies with membrane
potential, Em.
We can also define a slope conductance that relates I to
E. This is obtained by differentiating Eqn [3.1.17]:
½3:1:19
g5
dIi
dEm
Neither the slope conductance nor the chord conductance is a constant. Even for a straight cylindrical pore
that is specific for some ion, the conductance varies
with voltage because the current is carried only by one
ion and its concentration is not the same on the two
sides of the membrane.
THE CHORD CONDUCTANCE
EQUATION RELATES MEMBRANE
POTENTIAL TO ALL ION FLOWS
There are two ways to answer the problem of finding
the resting membrane potential when the membrane
is permeable to several ions. One way focuses on the
permeabilities as we have already defined them, and a
second focuses on the conductances, which are related,
but not identical, to the permeabilities. We will use conductances here because it makes it easier to understand
other electrical phenomena in cells. We begin with the
fact that the total current at the resting membrane
potential is zero. This is Eqn [3.1.15]:
½3:1:15
Itotal 5 INa 1 IK 1 ICl 5 0
Substituting in from Eqn [3.1.18] for the individual
currents, we get
½3:1:20 I 5 gNa ðEm 2 ENa Þ 1 gK ðEm 2 EK Þ 1 gCl ðEm 2 ECl Þ
At rest, I 5 0, and so we collect terms in Em on the lefthand side to find
½3:1:21 ðgNa 1 gK 1 gCl ÞEm 5 gNa ENa 1 gK EK 1 gCl ECl
Solving for Em, we obtain
259
260
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
gNa
gK
gCl
ENa 1
EK 1
ECl
gNa 1gK 1gCl
gNa 1gK 1gCl
gNa 1gK 1gCl
½3:1:22
Em 5
This is the chord conductance equation. What it
says is that the resting membrane potential is the
conductance-weighted average of the equilibrium
potentials for the ions that have any conductance
across the membrane. We can clearly enlarge Eqn
[3.1.22] to include other ions such as Ca21.
Both the chord conductance equation and the GHK
equation indicate that all permeant ions contribute to
the resting membrane potential. Those ions with the
largest conductances, or the largest permeabilities, are
correspondingly greater determinants of the resting
membrane potential.
INa 5 gNa ð285 mV 2 66:7 mVÞ 5 2gNa 3 151:7 mV
Which way does the Na1 ion go? Its concentration
is higher outside the cell than inside. A potential
of 166.6 mV inside would be necessary to stop the
diffusive flow down its concentration gradient, but the
resting membrane potential is 285 mV. Thus the negative membrane potential further drives Na1 influx. Na1
goes into the cell, but the current is negative, as indicated
by the sign and the fact that gNa is always positive.
The current is negative because that is our convention
for determining the direction of current. An inward
current (flux of a positive ion) is a negative current.
From Eqn [3.1.11], we see that the current is in the
same direction as the flux:
½3:1:11
THE CURRENT CONVENTION IS THAT
AN OUTWARD CURRENT IS POSITIVE
As mentioned earlier, the reversal potential is the
potential at which the current reverses direction: it
changes from positive to negative. It is important here
to remember the sign convention for these currents.
First, current is taken as the direction of positive
charge flow. Second, a positive current is taken as an
outward current—it goes from the inside of the cell to
the outside. Third, a negative current is taken as an
inward current. This convention becomes apparent
when we consider the individual ionic currents.
According to Eqn [3.1.18], the individual ionic currents
are
INa 5 gNa ðEm 2 ENa Þ
ICl 5 gCl ðEm 2 ECl Þ
Recall that g stands for a conductance. Conductances
are always positive.
We consider the example given above for the concentrations and equilibrium potentials. These values are
recapitulated in Table 3.1.1.
Suppose that the resting membrane potential is 285 mV.
The Na current could then be calculated as
TABLE 3.1.1 Concentrations of Ions and Their
Equilibrium Potentials
n
[Ion]out
1
[Ion]in
23
Na
145 3 10
1
23
M
K
4 3 10
Cl2
100 3 1023 M
M
Ei
23
12 3 10
M
23
155 3 10
5 3 1023 M
M
Thus inward Na flux is also a negative flux. The convention for positive flux is also from inside the cell to
outside.
In the case of K1, the current is given as
IK 5 gK ð285 mV 1 97:7 mVÞ 5 gNa 3 8:7 mV
which is positive. It takes 297.7 mV to stop K1 exit from
the cell. The resting membrane potential of 285 mV
is insufficient to stop K1 exit, so at rest there is some
outward IK. Because it is an outward current, it is
positive. The JK is similarly positive.
In the case of Cl2, the current is
ICl 5 gCl ð285 mV 1 80 mVÞ 5 2gCl 3 5 mV
IK 5 gK ðEm 2 EK Þ
½3:1:23
Ii 5 zi `Ji
166.6 mV
297.7 mV
280.0 mV
So the current is negative. Here 280 mV is enough to
stop Cl entry into the cell. This is the ECl and you can
see that if the membrane potential was 280 mV, ICl
would be zero. But the membrane potential is 285 mV,
which is more negative than ECl; thus the negative
inside potential forces Cl2 out of the cell. There is an
outward flux of Cl2, which is a positive flux. But the
charge on Cl2, zCl 5 21 and so the current carried by
Cl2 is opposite to its flux! (see Eqn [3.1.11]). The negative current is an inward current carried by the outward
flux of Cl2.
This current convention and the convention that
membrane potential is defined as Em 5 ψi 2 ψo are true
conventions in that the opposite conventions do
not violate any physical law. These conventions are
equivalent to the orientation of the x-axis perpendicular
to the surface of the cell membrane. The convention is
that x 5 0 is on the inside of the cell and x 5 δ, where δ
is the thickness of the membrane, is on the outside
surface. Figure 3.1.4 illustrates this convention and
what it means for the gradients in C and ψ.
T he O rig in of the Res tin g Membra ne P oten tial
The concentration gradient is positive,
so the diffusive force is negative or inward
160
+
–
150
+
–
+
–
+
–
+
–
140
130
[Na+]o145 mM
Concentration (mM)
120
110
100
90
80
ψo = 0 mV
+
dC
=-
+
dx
-
+
–
70
+
–
60
+
–
Extracellular fluid
50
+
The voltage
gradient is positive,
so the40
electrical force is negative
+
or inward
30
20
10
0
dψ
+40
Diffusive force
+20
[Na]o – [Na]i
0
δ–0
Cytosol
–20
Electrical force
–40
–60
–
– ψ
i = –85 mV
ψo – ψ
Em = –85 mV
–
+ i
δ-0
dx
ENa = +66.7 mV
–
+
=
+60
[Na+]i 12 mM
–100
x=0
x =δ
X-axis
The concentration gradient is negative
so the diffusive force is positive or outward
+
160
150
+
140
+
+60
– [K+]i 155 mM
+40
–
+
–
+
+
dC =
dx -
+
–
+
–
70
+
–
60
+
–
50
+
–
+
–
130
Concentration (mM)
120
110
100
90
80
ψo = 0 mV
Extracellular fluid
40
30
dψ
20
dx
10
0
=
ψ
+o – ψi
+δ –
[K]o – [K]i
+20
δ–0
Cytosol
0
–20
[K+]o 4 mM
–60
–100
x =δ
Electrical force
–40
– ψ
i = –85 mV
–
0
Diffusive force
Em = –85 mV
EK = –97.7 mV
x=0
X-axis
FIGURE 3.1.4 Cartoon of the concentration and voltage gradients and their resulting parts of the ion flux. Top, situation with Na1 ions. The [Na1]o is
about 145 mM, whereas the [Na1]i is about 12 mM. Thus the concentration gradient is positive (it slopes up with x; the direction of the x-axis is from
inside to outside of the cell as indicated at the bottom of the figure). Fick’s law gives the diffusive flux as being proportional to the negative of the
gradient. Thus the diffusive force favors a negative flux, which is directed inward. The outside potential, ψo, is taken as zero. At rest, ψi is about
285 mV. Thus the voltage gradient is also positive. The flux produced by the electrical force is also proportional to the negative of the electrical
gradient. Thus the positive voltage gradient produces a negative flux, which is inward. Thus for Na1, both the diffusive force and electrical force
produce an inward flux and an inward current. The bottom panel illustrates the situation with K1 ions. [K1]o is just 4 mM and [K1]i 5 155 mM, so
the concentration gradient is negative. Thus the diffusive force is positive or directed outward. The negative electrical potential inside makes a positive
electrical potential gradient, which favors a negative K1 movement, or an inward current and flux. Thus here the diffusive force and the electrical
force oppose each other, but their magnitudes are not equal. Another way of looking at the equilibrium potential is that it is the magnitude of the
diffusive force expressed in electrical terms. The net effect on movement is obtained by subtracting the two forces.
261
262
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
SUMMARY
Separation of charges produces an electric field defined
as the force experienced by a test positive unit charge.
The field at any point is also characterized by a potential, defined as the work necessary to move a positive
unit test charge from infinite separation to that point.
The potential surrounding positive charge is positive,
and that surrounding negative charge is negative. Thus
the separation of charges across a membrane produces
a potential difference between the two sides of the
membrane.
Ions move in response to concentration differences
and in response to electrical forces. If a membrane is permeable only to a single ion, and the concentration of the
ion is different on the two sides of the membrane, then
the ion will diffuse from its high concentration toward
its low concentration. Since it is charged, the ion movement makes a current and there is a separation of charge.
This separation of charge produces an electrical force that
opposes further diffusion. The electric field is the force
per unit test charge, and this is the negative spatial derivative of the potential. The membrane potential at which
diffusion is exactly balanced by the electrical forces is
called the equilibrium potential. It can be calculated by
using the Nernst equation:
RT Co
ln
5 ðψi 2 ψo Þ
z` Ci
The equilibrium potentials for K1, Na1, and Cl2
depend on their concentrations on both sides of the
membrane and they are typically different from each
other. In the Nernst equation, put the outside concentration in the numerator to agree with the sign
convention.
Real membranes are permeable to a variety of ions, but
with different permeabilities. The permeability can be
expressed either as a permeability relating flux to driving
force in concentration units or as conductance relating
current to driving force in voltage units. The resting membrane potential occurs at a net zero membrane current.
This occurs at a membrane potential that is the
conductance-weighted average of the equilibrium potentials, as described by the chord conductance equation:
Em 5
gNa
gK
ENa 1
EK
ð gNa 1 gK 1 gCl Þ
ð gNa 1 gK 1 gCl Þ
1
gCl
ECl
ð gNa 1 gK 1 gCl Þ
Thus the resting membrane potential is closest to the
equilibrium potential of the ion with the highest conductance. The resting membrane potential also can be
expressed in terms of the permeabilities: it depends on
the concentration-weighted permeabilities as expressed
in the GHK equation.
Membrane potential is defined as Em 5 ψi 2 ψo. An outward flow of positive ions is taken as a positive current.
The current for any ion is given as
Ii 5 gi ðEm 2 Ei Þ
where gi is the conductance to the particular ion, in
siemens. This is a chord conductance which is distinguished from a slope conductance. Here Ei is the equilibrium potential for the ion in question. Conductances
are always positive. A negative current means that the
current is directed inward.
REVIEW QUESTIONS
1. What is the current of an ion at its equilibrium
potential? What would the current look like as
a function of membrane potential if the ion
had the same concentration on both sides of the
membrane?
2. If the conductance of the membrane to K1 was
increased, what would happen to the equilibrium
potential for K1 (EK)? What would happen to
Em?
3. Why is Clo in the denominator of the GHK equation when Nao and Ko are in the numerator?
4. What potential is calculated by the Nernst
equation?
5. What potential is calculated by the GHK
equation?
6. What is a siemen?
7. What is meant by an inward current? Outward
current? What sign does an inward current have?
If Cl2 exits the cell, does it make a positive or
negative current?
APPENDIX 3.1.A1 DERIVATION
OF THE GHK EQUATION
The flux of an ion through a water-filled channel
should be given by the generalized Fick’s law given by
Eqn [3.1.1]:
½3:1:A1:1
Js 5 2D
@C
D
@ψ
2
Cz`
@x
RT
@x
The flux can be obtained by integrating this equation.
To begin, we multiply both sides of the equation by
an integrating factor, ρ, and we choose ρ so that the
right-hand side of the equation becomes an exact
differential:
@C Cz`ρ @ψ
2
Js ρ 5 2D ρ
½3:1:A1:2
@x
RT @x
We choose ρ so that the terms in brackets are an exact
differential. Thus we want
½3:1:A1:3
ρ
@C Cz`ρ @ψ dðρCÞ
@C
@ρ
1
5
5ρ
1C
@x
RT @x
dx
@x
@x
From comparing the extreme left of Eqn [3.1.A1.3] to
the extreme right, we see that the condition that multiplication by ρ transforms the equation into an exact differential is met if
½3:1:A1:4
@ρ z`ρ @ψ
5
@x
RT @x
T he O rig in of the Res tin g Membra ne P oten tial
We rearrange this to get
+60
@ρ
z`
5
@ψ
ρ
RT
½3:1:A1:5
160
150
140
A solution to Eqn [3.1.A1.5] is
130
ρ 5 eðz`=RTÞψ
120
We may insert this result back into Eqn [3.1.A1.2] to
obtain
Js eðz`=RTÞψ 5 2D
½3:1:A1:7
dðCeðz`=RTÞψ Þ
dx
which may be rewritten as
Js eðz`=RTÞψ dx 5 2DdðCeðz`=RTÞψ Þ
½3:1:A1:8
The flux can be obtained by integrating this equation
from x 5 0 (one side of the membrane) to x 5 δ,
the other side of the membrane for a membrane with
thickness δ:
ðδ
ðδ
½3:1:A1:9
Js eðz`=RTÞψ dx 5 2D dðCeðz`=RTÞψ Þ
0
The numerator in this equation is the integral of an
exact differential and can be immediately evaluated
between the boundaries. This gives
Js 5
2D½CðδÞeðz`=RTÞψðδÞ 2 Cð0Þeðz`=RTÞψð0Þ Ðδ
ðz`=RTÞψ dx
0 e
The denominator in this equation can be evaluated only
if ψ(x) is known. However, generally ψ(x) is unknown.
We can integrate the denominator if we assume that ψ is
a linear function of x. This is true if the electric field
between x 5 0 and δ is constant. Recall that the electric
field intensity is the negative derivative of the potential.
For this reason, the assumption of a linear potential
gradient is called the constant field assumption. If the
potential is linear, then we can write
½3:1:A1:12
ψ 5 ψð0Þ 1
ψð0Þ 2 ψðδÞ
x
02δ
The situation of C(0), C(δ), ψ(0), and ψ(δ) is illustrated
in Figure 3.1.A1.1. The difference in potential across the
membrane, ψ(0) 2 ψ(δ), is the the membrane potential
Em. Here ψ(0) is the inside potential and ψ(δ) is the
outside potential. Thus Eqn [3.1.A1.12] becomes
½3:1:A1:13
100
–
+
–
+
–
+
–
+
–
dC
ψo = ψ (δ ) = 0 mV +
=–
dx
+
90
–
80 Extracellular fluid
70
+
–
+
–
60
+
–
50
+
–
40
+
–
+
–
30
20
dψ
10
=
–
ψ 5 ψð0Þ 1
Em
x
δ
+40
+20
Co – Ci
0
δ – 0
Cytosol
–20
–40
–60
ψi = ψ (0)
Ci
δ – 0
dx
0
ψ – +ψ
o
i
–100
0
In this case, we limit ourselves to the steady-state
condition. In this case, Js does not vary with distance
across the membrane; it is constant. Therefore Js may be
removed from the integral and we get
Ðδ
2D 0 dðCeðz`=RTÞψ Þ
½3:1:A1:10
Js 5
Ðδ
ðz`=RTÞψ dx
0 e
½3:1:A1:11
110
Concentration (mM)
½3:1:A1:6
Co
+
x=0
x =δ
X–axis
FIGURE 3.1.A1.1 Concentration and potential profile across the
membrane along with the convention for the location of the X-axis.
This result can be substituted into the denominator of
Eqn [3.1.A1.11] to give
ðδ
ðδ
ðz`=RTÞψ
e
dx 5 eðz`=RTÞðψð0Þ2ðEm =δÞxÞ dx
0
0
5e
ðz`=RTÞψð0Þ
ðδ
e2ðz`=RTÞðEm =δÞx dx
0
½3:1:A1:14
Evaluation of the integral gives
eðz`=RTÞψð0Þ
ðe2ðz`=RTÞEm 21Þ
2ðz`=RTÞðEm =δÞ
½3:1:A1:15
Inserting the result of Eqn [3.1.A1.15] back into
Eqn [3.1.A1.11]
Js 5
2D½CðδÞeðz`=RTÞψðδÞ 2 Cð0Þeðz`=RTÞψð0Þ ðz`=RTÞEm 21
m =δÞÞ½e
ðeðz`=RTÞψð0Þ Þ=ð2ðz`=RTÞðE
½3:1:A1:16
This can be simplified to
Js 5
ðD=δÞ½ðz`=RTÞEm ½CðδÞeðz`=RTÞψðδÞ 2 Cð0Þeðz`=RTÞψð0Þ e2ðz`=RTÞψð0Þ
½e2ðz`=RTÞEm 2 1
½3:1:A1:17
Multiplying through by the exponent in the numerator,
and recalling that Em 5 ψ(0) 2 ψ(δ), we convert Eqn
[3.1.A1.17] into
½3:1:A1:18 Js 5
ðD=δÞ½ðz`=RTÞEm ½CðδÞe2ðz`=RTÞEm 2 C0 ½e2ðz`=RTÞEm 2 1
263
264
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
This equation is typically rewritten by multiplying
numerator and denominator by 21, with no change in
meaning:
ðD=δÞ½ðz`=RTÞEm ½Cð0Þ 2 CðδÞe2ðz`=RTÞEm ½1 2 e2ðz`=RTÞEm ½3:1:A1:19
Js 5
This flux equation holds true for any ion including
Na1, K1, and Cl2. The current carried by each ion is
given by Eqn [3.1.19] by multiplying the flux by z`. We
identify C(0) with the inside concentration of the ion,
and C(δ) with the outside concentration, as described in
Figure 3.1.A1.1 which of course are different for each
of the ions. For each ion, we can write an equation for
the current that has the form
Iion 5
ðD=δÞ½ðz2 `2 =RTÞEm ½Ci 2 Co e2ðz`=RTÞEm ½1 2 e2ðz`=RTÞEm ½3:1:A1:20
We now insert the expressions for each of the individual currents. Here, however, we make an additional
substitution that the diffusion coefficient for each ion
divided by the thickness of the membrane, D/δ, is
equal to the permeability of the membrane to each
ion. The result is
PNa ð`2 =RTÞEm ½Nain 2 ½Naout e2ð`=RTÞEm
Itotal 5 0 5
1 2 e2ð`=RTÞEm
PK ð`2 =RTÞEm ½Kin 2 ½Kout e2ð`=RTÞEm
1
1 2 e2ð`=RTÞEm
PCl ð`2 =RTÞEm ½Clin 2 ½Clout eð`=RTÞEm
1
1 2 e ð`=RTÞEm
½3:1:A1:25
Solving this equation, we find
0 5 PNa ½½Nai 2 ½Nao e2ð`=RTÞEm Thus we can write
ðD=δÞ½ð`2 =RTÞEm ½½Nai 2 ½Nao e2ð`=RTÞEm INa 5
½1 2 e2ð`=RTÞEm ½3:1:A1:21
½3:1:A1:22 IK 5
ðD=δÞ½ð`2 =RTÞEm ½½Ki 2 ½Ko e2ð`=RTÞEm ½1 2 e2ð`=RTÞEm ðD=δÞ½ð`2 =RTÞEm ½½Clin 2 ½Clout eð`=RTÞEm ½1 2 eð`=RTÞEm ½3:1:A1:23
1 PK ½½Ki 2 ½Ko e2ð`=RTÞEm ½3:1:A1:26
1 PCl ½½Clo 2 ½Cli e2ð`=RTÞEm where we have multiplied the numerator and denominator of the Chloride contribution to the current by exp
(2`/RT Em) and rearranged the terms. Solving for Em,
we finally obtain
ICl 5
Note that the exponent in the numerator and denominator of ICl is positive instead of negative! This is
because the z term in the exponent in Eqn [3.1.A1.20] is
21 for Cl. The total current across the membrane is the
sum of the currents carried by each ion. At steady state
(the resting membrane potential), the current is zero.
Thus we can write
½3:1:A1:24
Itotal 5 INa 1 IK 1 ICl 5 0
½3:1:A1:27
Em 5
RT ½PNa ½Nao 1 PK ½Ko 1 PCl ½Cli ln
`
½PNa ½Nai 1 PK ½Ki 1 PCl ½Clo This is the GHK equation. It tells us that the resting
membrane potential (at which point the net current
across the membrane is zero) is determined by the
concentrations of ions on both sides of the membrane
and by their respective permeabilities. The ion with
the largest permeability dominates the membrane potential by moving the potential closer to its equilibrium
potential.
The Action Potential
Learning Objectives
G
G
G
G
G
G
G
G
G
G
G
G
G
Draw a picture of a motor neuron and identify soma, dendrites, axon, myelin sheath, and terminals
Describe what is meant by depolarization and hyperpolarization and what currents achieve it
Explain what is meant by the “all or none” law
Define latency, absolute refractory period, relative refractory
period, and overshoot in an action potential on a nerve
Define rheobase and chronaxie and how they can be determined from a strengthduration curve
Recognize the Weiss relation between strength and
duration
Draw a graph showing the conductance changes with time
during an action potential on a nerve
Explain what is meant by the “activation gate” and “inactivation gate” of the Na channel
Describe the state of the activation gate and inactivation
gate during an action potential
Describe how the inactivation gate is reset
Distinguish between unitary current and ensemble current
Using the whole-cell iv curve, explain the effect of small
depolarizations below threshold and of depolarizations
above threshold
Explain why stronger stimuli need shorter durations to
achieve an action potential
CELLS USE ACTION POTENTIALS
AS FAST SIGNALS
Certain cells in the body are capable of initiating and
propagating an action potential over their surface.
These cells are called excitable cells and they include
muscle and nerve cells. The action potential is a brief,
pulse-like change in the membrane potential. Because
it can be propagated rapidly over the surface of the
cell, it conveys a fast signal from one place to another.
We will use a motor neuron as an example of an
excitable cell.
THE MOTOR NEURON HAS DENDRITES,
A CELL BODY, AND AN AXON
Motor neurons are large cells in the ventral horn of the
spinal cord as shown in Figure 3.2.1. They have a
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00024-0
3.2
number of processes called dendrites that bring signals
to the motor neuron. The motor neuron also has one
large process, the axon, that connects the motor neuron
on one end with a muscle fiber on the other. Action
potentials move along the axon so that activity in the
motor neuron alters activity in the muscle.
Axons from neurons can be myelinated or unmyelinated. Myelin refers to a sheath that covers the axon, but
not entirely. In the peripheral nervous system, Schwann
cells make the myelin by wrapping themselves around
the axon, forming a multilayered structure of multiple
cell membranes of the Schwann cell. In the central nervous system, oligodendroglial cells make the myelin.
The sheath is not continuous in either the peripheral or
central nervous system. At the end of each Schwann
cell, there is a gap in the myelin. This gap is called the
Node of Ranvier (see Figure 3.2.2).
Like all cells, the motor neuron has a nucleus located in
its cell body or soma. The soma is also sometimes referred
to as the perikaryon from the Greek root “peri” meaning
“around” or “surrounding” and “karyon” meaning “nut”
or “kernel,” and referring to the nucleus. There is only one
nucleus in the motor neuron and it is the site of mRNA
transcription.
PASSING A CURRENT ACROSS THE
MEMBRANE CHANGES THE MEMBRANE
POTENTIAL
Figure 3.2.3 shows a highly schematic diagram of how
the membrane potential can be measured by inserting a
microelectrode through the membrane of the axon. The
resting membrane potential in these axons is about
260 mV. Recall that the membrane potential is defined
as ψi 2 ψo.
It is possible to pass current across the membrane
through the arrangements shown in Figure 3.2.3. In one
case, the battery is hooked up so that current will pass
into the cell. Recall here that current is defined as the
direction of positive charge flow and an outward flow is
positive. An inward current, shown in the middle of
Figure 3.2.3, will depolarize the cell. The cell is already
polarized; an inward current would make the cell less
polarized and thus it would depolarize it. If the battery
was connected with the opposite polarity, the resulting
current would be outward and this would make the
membrane more polarized. This is a hyperpolarizing
265
current.
266
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
AN OUTWARD CURRENT
HYPERPOLARIZES THE MEMBRANE
POTENTIAL
When an outward current is passed across the membrane, the recorded membrane potential is a distorted
version of the stimulus. As the magnitude of the current
is increased (stimulus intensity is increased), the recorded
Dorsal
Dorsal root
White matter
hyperpolarization grows larger (see Figure 3.2.4). It takes
some time for the current to reach a new steady-state
membrane potential. In addition, the magnitude of the
hyperpolarization depends on the distance from the
stimulating electrode to the recording electrode. These
phenomena are consequences of the electrical characteristics of the axon, its cable properties. In brief, the cable
properties define a time constant and a length constant
which describe how voltage builds up (or falls off) with
time and with distance. The membrane parameters which
influence these cable characteristics include the membrane resistance, the membrane capacitance, and the
electrical resistance of the axoplasm.
Gray matter
THE RESULT OF DEPOLARIZING
STIMULUS OF ADEQUATE SIZE IS
A NEW PHENOMENON—THE ACTION
POTENTIAL
Motor neuron
Ventral root
Ventral
FIGURE 3.2.1 Location of the motor neuron in the spinal cord. The spinal
cord is shown in cross-section. The dorsal aspect is toward the back; ventral
is toward the front. The dorsal and ventral roots are paired, with one on
each side of the cord, but only one side is shown here. The motor neuron is
shown in dark blue. The motor neuron’s cell body is located in gray matter
in the ventral horn, and its long axon leaves the cord via the ventral root
and continues on to a muscle where it makes a neuromuscular junction.
These cells produce an action potential that propagates along the axon,
excites the nerve at the neuromuscular junction, and conveys that
excitation to the muscle in order to activate the muscle.
Depolarization to a small degree produces a change in
Em that mirrors hyperpolarization—the change is a distorted version of the stimulus. With larger depolarization, however, a new phenomenon arises. When Em
exceeds a threshold value, there is an abrupt rise in Em,
reaching positive values near 130 mV. Just as quickly,
Em returns to near normal values. This abrupt change in
the membrane potential brought about by depolarization is the action potential (see Figure 3.2.5).
Dendrites
Axon
terminals
Node of Ranvier
Soma
Axon
Myelin sheath
FIGURE 3.2.2 Parts of the motor neuron. Dendrites are multiple processes of the neuron that bring signals to the cell body, or soma. A single long
axon exits the cell on one pole and reaches all the way to its target cell, the muscle fiber. The long axon is covered by a myelin sheath made by
Schwann cells. The sheath is interrupted at regular intervals at the nodes of Ranvier.
Negative (inward) current
depolarizes the membrane
Positive (outward) current
hyperpolarizes the membrane
Battery
+
−
Battery
−
+
Voltmeter
FIGURE 3.2.3 Arrangement of
electrodes to record membrane
potential
(left);
to
inject
a
depolarizing current (middle) or to
pass a hyperpolarizing current (right).
IM refers to “membrane current,” or
current across the membrane.
+
−
IM
IM
++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
Axoplasm
The Action P otential
THE ACTION POTENTIAL IS ALL
OR NONE
When the strength of depolarization is increased further, the resulting wave form in Em remains remarkably
constant. That is, if the depolarization is enough to trigger an action potential, the resulting action potential is
independent of the stimulus. The action potential is “all
or none.” The action potential is not graded, meaning
that it does not vary from a minimum to a maximum;
rather, a given stimulus will either produce an action
potential or it will not, and the resulting shape of the
plot of Em against time is approximately the same.
THE LATENCY DECREASES WITH
INCREASING STIMULUS STRENGTH
+60
+40
Once initiated, the action potential moves away from its
point of origin, and this is called propagation. At some
point away from its origin, the action potentials are
indistinguishable. However, near the stimulus there is a
slight difference, having to do with the time it takes to
reach threshold, the latency. The membrane potential is
not a completely faithful representation of the stimulus:
the rise in Em lags behind the stimulus rise and the fall
in Em lags behind the stimulus fall. The membrane distorts the stimulus. When the stimulus strength is
increased above threshold, the time to reach threshold,
the latency, decreases. The relationship between latency
and stimulus strength is shown in Figure 3.2.6.
+20
0
Em
(mV) −20
−40
−60
−80
Stimulus
intensity
0
1
2
3
4 5 6 7
Time (ms)
8
9 10
FIGURE 3.2.4 Results of hyperpolarizing stimuli of varying intensity on the
axon membrane potential, Em. The effect of hyperpolarizing stimuli on
the membrane potential is a distorted version of the stimulus wave form.
The rise time and fall time of Em are part of the cable properties of the axon.
THRESHOLD IS THE MEMBRANE
POTENTIAL AT WHICH AN ACTION
POTENTIAL IS ELICITED 50%
OF THE TIME
The threshold for a nerve is the membrane potential
which must be reached in order to “fire” an action
Myelinated squid axon
Rabbit motor neuron
+60
Absolute refractory
period
+40
+60
+20
+40
0
Em
(mV) −20
Em
Relative refractory
period
Overshoot
+20
0
(mV)
−40
–20
−60
–40
−80
–60
Hyperpolarizing
after potential
–80
Stimulus
intensity
Stimulus
intensity
Stimulus–response
latency
0
1
2
3
4 5 6 7
Time (ms)
8
9 10
FIGURE 3.2.5 Results of depolarizing stimuli of varying intensity on the
axon membrane potential. At low stimulus strength, Em is a distorted
version of the stimulus, as in the case of hyperpolarization. When the
depolarization exceeds a threshold value, there is an abrupt rise in Em,
reaching positive values. This abrupt change in the membrane potential
brought about by depolarization is the action potential.
0 1 2
3 4
5 6 7 8
Time (ms)
9 10
FIGURE 3.2.6 Effect of stimulus strength above threshold on resulting
wave forms in a myelinated squid motor neuron axon. Note that the
wave form here differs somewhat from that in mammalian motor
neurons. It is shorter in the squid and shows a hyperpolarization after
potential that is largely lacking in mammalian motor neurons.
267
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
potential. Operationally, it is defined as the membrane
potential at which the nerve will fire an action potential
50% of the time. However, it is possible to initiate an
action potential even when the membrane potential is
below threshold, but the probability of this happening
is much reduced. Similarly, it is possible to fail to initiate an action potential even when the membrane
potential is above threshold, but with a much reduced
probability. The probability of initiating an action
potential is a steep function of the membrane potential.
THE NERVE CANNOT PRODUCE
A SECOND EXCITATION DURING
THE ABSOLUTE REFRACTORY PERIOD
While the first action potential is occurring, it is impossible to begin a second action potential, no matter how
powerful the second stimulus. The time during which
the nerve is refractory to a second stimulus is called the
absolute refractory period. It typically lasts for 12 ms.
Following the absolute refractory period is a second,
relative refractory period. This typically lasts some
4 ms or so, and during this time it is possible to stimulate the nerve cell to make another action potential, but
it is more difficult to do so than in the resting neuron.
That is, the threshold is elevated during the relative
refractory period.
THE ACTION POTENTIAL REVERSES
TO POSITIVE VALUES, CALLED
THE OVERSHOOT
The first recordings of action potentials with intracellular
electrodes were accomplished in 1939 and 1940 by A.L.
Hodgkin and Andrew Huxley and by K.S. Cole and H.J.
Curtis, respectively, using the squid giant axon. They
found results similar to those shown in Figure 3.2.6. In
particular, they found that the membrane potential not
only went to zero, but it took on positive values. This is
called the overshoot. Given the fact that the equilibrium
potentials for all known ions were negative except for
Na1 and Ca21, these pioneers suggested that the membrane could have a positive potential by becoming selectively permeable to Na1.
THE STRENGTHDURATION
RELATIONSHIP IS HYPERBOLIC
The relationship between the current necessary to reach
threshold and the duration of the current for a compound action potential is shown in Figure 3.2.7. This is
related to the strengthlatency relationship, as the
latency is the time required to elicit an action potential
for a given strength of stimulus. The strengthduration
relationship shown in Figure 3.2.7 asks the question,
for a given strength of stimulus, how long must it
continue to produce an action potential? For some
strengths of stimulus, no duration is sufficient—you
never get an action potential. For a critical strength of
stimulus, you get an action potential only if the stimulus lasts infinitely long. This strength of stimulus is
50
40
Threshold current (mA)
268
30
20
10
0
0
0.2
0.4
0.6
Stimulus duration (ms)
0.8
1.0
FIGURE. 3.2.7 Strengthduration relationship in human peripheral nerve.
The median nerve was stimulated using surface electrodes 1 cm in
diameter taped to the skin over the median nerve at the wrist, 4 cm apart
oriented along the course of the nerve. Stimulus was a square wave with
rise and fall times of 10 μs. The antidromic compound sensory action
potential was recorded at the index finger using ring electrodes with
23 mm diameter set 23 cm apart on the finger. Stimulus intensity was
reduced 25% until the amplitude of the compound action potential (the
aggregate of a bundle of axons) was reduced to 30% of maximum (From
Mogyoros, I, Kiernan, MC, and Burke, D. Strengthduration properties of
human peripheral nerve. Brain 119:439447, 1996).
called the rheobase (see Figure 3.2.8). Weiss described
this relationship by the equation:
½3:2:1
Q 5 Irh ðt 1 τSD Þ
where Q is the charge, I is the rheobase current, t is the
time the current is on, and τ SD is a strengthduration
time constant, often called the chronaxie. Because the
total charge is the current times the time, this equation
becomes
½3:2:2
I 5 Irh
ðt 1 τSD Þ
τSD
5 Irh 1 Irh
t
t
This last equation describes a rectangular hyperbole offset
from the x-axis by Irh. Also, when t 5 τ SD, the current
I 5 2 Irh; the current is twice the rheobase. These relationships are shown diagrammatically in Figure 3.2.8. We
will seek to further understand this relationship after we
establish how depolarization induces an action potential.
VOLTAGE-DEPENDENT CHANGES
IN ION CONDUCTANCE CAUSE
THE ACTION POTENTIAL
INCREASE IN ION CONDUCTANCE BEGINS
AFTER THE MEMBRANE BEGINS
TO DEPOLARIZE
K.S. Cole and H.J. Curtis in 1939 studied the impedance
properties of the squid giant axon and discovered a
marked decrease in the impedance (equivalently,
an increase in the conductance) of the squid axon
The Action P otential
140
Current calculated as I = Irh (t + τSD) / t
with Irh = 10 mA and τSD = 0.2 s
Threshold current (mA)
120
The chronaxie is the time at which
threshold current is twice the
rheobase
The rheobase, Irh, is the current
that requires infinite time to elicit
an action potential
100
80
60
τSD
40
20
Irh
0
0.0
0.2
0.4
0.6
Stimulus duration (ms)
0.8
membrane during the upstroke of the action potential.
This increase begins only after the membrane potential
rises many millivolts above the resting membrane
potential. They argued that the foot of the action potential resulted from the discharging of the membrane
from local currents from elsewhere in the cell. At the
inflection point on the rising phase of the action potential, the cell generated its own net inward current. Such
a current must be carried by an ion, and the most likely
candidate is Na1, because of its high extracellular
concentration.
THE ACTION POTENTIAL IS
ACCOMPANIED BY NA 1 INFLUX
To test the idea that increases in Na1 conductance
might cause the action potential, Alan Hodgkin and
Bernard Katz replaced some of the NaCl in seawater
with choline chloride. Replacing the Na1 reduced the
upstroke of the action potential and markedly reduced
the size of the action potential. Later experiments using
radioactive tracers showed that Na1 influx accompanies
the action potential.
THE CHORD CONDUCTANCE
EQUATION PREDICTS THAT CHANGES
IN CONDUCTANCE WILL CHANGE
THE MEMBRANE POTENTIAL
In Chapter 3.1, we developed the chord conductance
equation that shows that the resting membrane potential is the conductance-weighted average of the equilibrium potentials for all ions (see Eqn [3.2.3]). At rest,
the membrane potential is closer to the K1 equilibrium
potential because conductance K1 is higher than the
conductance of the other ions.
Em 5
½3:2:3
gNa
gK
ENa 1
EK
ðgNa 1 gK 1 gCl Þ
ðgNa 1 gK 1 gCl Þ
gCl
ECl
1
ð gNa 1 gK 1 gCl Þ
1.0
FIGURE 3.2.8 Definition of rheobase and chronaxie. The rheobase
current is the current which takes an infinite time to elicit an
action potential—it is obtained by extrapolation or curve fitting.
The chronaxie is the time at which threshold current is twice the
rheobase. These two parameters are parameters of Eqn [3.2.2]
that can be obtained by fitting the equation to experimental
data. In the figure, the current was calculated according to Eqn
[3.2.2] using a rheobase of 10 mA and a chronaxie of 200 ms.
Note that the calculated curve resembles the form of the
experimental curve shown in Figure 3.2.7.
During the rising phase of the action potential, the conductance to Na1 increases, changing Em from its resting,
polarized value toward more positive values. If the
conductance to Na1 becomes large enough, relative to
the conductances for K1 and Cl2, then Em will be
driven toward the equilibrium potential for Na1, ENa.
Since ENa is positive, Em will be driven to positive values
and will exhibit the overshoot.
GNA INCREASES TRANSIENTLY DURING
THE ACTION POTENTIAL; GK
INCREASES LATER AND STAYS
ELEVATED LONGER
The results described earlier show that the rapid depolarization and overshoot in the action potential is due
to a transient increase in membrane conductance, and
this is accompanied by an Na1 influx. The rapid repolarization of the membrane afterward is due to shutting
off the increased Na1 conductance and increasing the
K1 conductance. These results are consistent with the
chord conductance equation. Hodgkin and Huxley
succeeded in calculating gNa and gK during different
parts of the action potential in the squid axon, and their
results are summarized schematically in Figure 3.2.9.
Although these results were determined in the squid,
the principles remain the same for action potentials in
mammalian excitable cells.
CONDUCTANCE AND EQUILIBRIUM
POTENTIALS FOR NA 1 AND K 1
ACCOUNT FOR ALL OF THE FEATURES
OF THE ACTION POTENTIAL
The origin of the action potential can be explained on the
basis of the equilibrium potential for Na1 and K1 and
the time course of their conductances. In the squid axon,
the resting potential is on the order of 260 mV, with EK
about 275 mV. When the membrane is depolarized by
some means, a conductance pathway for Na1 begins to
open. At this time, Na1 is far away from its equilibrium
269
270
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Em
+60
ENa
30
+40
+20
Ionic
gNa
0
Em
(mV) –20
20 conductance
gK
10
–40
(ms cm–2)
–60
–80
FIGURE 3.2.9 Changes in gNa and gK
during the propagated action potential
as calculated by Hodgkin and Huxley.
EK
0
1
2
potential, so the driving force for Na1 is large and the
opening of the conductance pathway causes a Na1 influx
and a further depolarization of the membrane. This additional depolarization further opens additional conductance pathways. This positive feedback explains the
explosive increase in Na1 conductance and the rapid
increase in the membrane potential toward ENa.
3
4 5 6
Time (ms)
G
The pathway for Na1 conductance next undergoes a
time-dependent inactivation. This by itself reduces the
inward Na1 current, except that the inactivation is
accompanied by a reduction in Em (i.e., a repolarization
of the membrane) and a subsequent increase in the
driving force. The Na1 current is complicated, being the
product of the conductance and the net driving force
(see Eqn [3.1.18]).
Repolarization of the membrane would occur with a
return of Na1 conductance to normal, but it is hastened
by a delayed increase in gK followed by its gradual
return to resting levels. The increased gK increases an
outward current carried by K1, and this current more
quickly repolarizes the cell. Because gK remains elevated
even when gNa has returned to normal, the cell is hyperpolarized for several ms following the action potential
until gK returns to normal.
G
GNA IS A FUNCTION OF A NA 1 SELECTIVE CHANNEL
G
The identification of two major components, INa and IK,
in the ionic currents by Hodgkin and Huxley, has
allowed electrophysiologists to characterize the gating
properties of the channels that pass these currents. The
present view is that the Na1 channel has at least four
distinct components:
G
The Selectivity Filter Enables the Na1 Channel to
Pass Na1 Preferentially
The relative permeabilities of ions in ion channels
can be calculated by measuring the reversal potential
when the ion composition is changed. The results
show that the Na1 channel actually conducts H1
ions much more readily than it does Na1 ions. The
current through the Na1 channel, however, is dominated by Na1 because of its much higher concentration. The [H1] is around 1027 M, whereas [Na1] is
around 0.1 M, or 106-fold higher than [H1]. It is
7
8
0
9 10
believed that the selectivity relies on a combination
of hydrated ion size and the free energy of hydration. Most other ions have a lower permeability
through the Na1 channel. Thus the Na1 channel is
selective for Na1.
The Na1 Channel Possesses an Activation Gate
At rest the membrane has a low gNa because the Na1
channel is blocked by its activation gate. This is a
part of the Na1 channel that can be moved to allow
Na1 to conduct through the channel. The opening
of this gate is voltage dependent: the activation gate
begins to open only when the membrane depolarizes. This voltage-dependent opening of the activation gates causes the explosive increase in Na1
conductance in the rising phase of the action
potential.
The Na1 Channel Possesses an Inactivation Gate
Na1 channels also inactivate. This is the function of
a separate gate that closes according to time and
voltage. When it closes, the Na1 channel is nonconducting and, most importantly, it is inactivatable.
Conductance through the Na1 channel requires that
both the inactivation gate and the activation gate are
open. When the inactivation gate is closed, opening
of the activation gate alone does not allow the channel to conduct ions.
Specific Toxins Bind to the Na1 Channel
The puffer fish produces a potent toxin, tetrodotoxin, or TTX, that binds to Na1 channels and
blocks them. As a result, action potential conduction
in nerve and muscle is blocked, with lethal consequences. Another natural toxin, saxitoxin, has similar properties. It derives its name from the Alaskan
butter clam, Saxidomus. Eating a single contaminated
shellfish can be fatal. The Na1 channel contains
regions that bind these toxins.
THE INACTIVATION GATES MUST BE
RESET BEFORE ANOTHER ACTION
POTENTIAL CAN BE FIRED
An action potential can occur in a nerve only when the
Na1 channels can open. If they are blocked, for example, by tetrodotoxin or saxitoxin, then no action potentials are possible. Similarly, if the channels are in the
inactivatable state because the inactivation gate is
The Action P otential
Na+ channel
+
Selectivity filter
+
+
TTX binding site
+
–
–
Activation gate
–
Inactivation gate
–
TTX
Extracellular fluid
+
–
+
–
Cytosol
TEA
–
–
+ filter
Selectivity
+
K+
channel
Activation gate
–
+
–
+
–
FIGURE 3.2.10 Hypothetical and conceptual model of the voltage-gated
Na1 channel and K1 channel. The selectivity filter allows Na1 to pass but
not K1 (top). When TTX (black circle) binds to its site, the channel is
blocked. Closure of either the activation gate or inactivation gate will
block Na1 conductance. The K1 channel also possesses an activation
gate and is blocked by tetraethylammonium (TEA).
closed, they also cannot be activated by opening the
activation gates. Following a normal action potential,
the Na1 channels are reset when the activation gates
close and the inactivation gates open. This takes some
time. Part of the refractoriness of the nerve cell immediately following the action potential is due to the lower
conductance of the membrane to Na1 because the inactivation gates are closed.
Figure 3.2.10 shows a conceptual model of the Na1 and
K1 channels with their activation and inactivation gates
and their binding sites for specific blockers. Figure 3.2.11
shows how these channels change during the action
potential. Figure 3.2.12 shows the timing of opening of
the activation and inactivation gates and that Na1 entry
is possible only when both are open.
CONDUCTANCE DEPENDS ON THE
NUMBER AND STATE OF THE
CHANNELS
Figure 3.2.11 shows a cartoon of the states of the Na1 and
K1 channels in axon membranes that give rise to the
action potential. Out of necessity, only representative
channels are shown in the figure. They are meant to represent what a population of channels is doing. The overall
current carried through a patch of membrane is given by
½3:2:4
Ii 5 NPo i
where I is the current, N is the number of channels, Po
is the probability of a channel being open, and i is the
unitary current, the current carried by the open channel
under physiological conditions. Cells possess thousands
of channels, each of whose behavior is stochastic,
meaning that their opening and closing are not deterministic but probabilistic. The average behavior of
many channels or the behavior of a large number of
them is predictable, but the behavior of a single channel
appears to be erratic and unpredictable. The sum of the
currents of a population of channels is called the
ensemble current.
PATCH CLAMP EXPERIMENTS
MEASURE UNITARY CONDUCTANCES
It is possible to study the behavior of single channels
using a patch clamp technique, shown in Figure 3.2.13.
In this method, one clamps the voltage across the patch
and measures current across it at that voltage. One can
also step the potential from some holding value to a new
value and measure the currents. Figure 3.2.14 shows the
unitary Na1 and K1 currents in successive sweeps from
patch clamp recordings in neuroblastoma cells (left) and
squid giant axon (right). Distinction can be made
between INa and IK by judicious choice of ionic conditions
and use of specific inhibitors. The Na1 channels open
briefly upon depolarization and then do not open later
on. Note that the individual channels open and close
rather erratically. It is not possible to predict exactly when
a particular channel will either open or close. The presence of a large number of channels, however, will smooth
out these discontinuities. The ensemble average is
obtained by averaging many sweeps. This describes the
average behavior of a single channel over many experiments, which is equivalent to the average behavior of a
population of channels in a single sweep. The average
behavior shows that Na1 channels open upon depolarization and then close, or inactivate. K1 channels, on the
other hand, open after a slight delay and stay open during
depolarization. Repolarization closes the K1 channels.
THE CURRENTVOLTAGE
RELATIONSHIP FOR THE WHOLE CELL
DETERMINES THE THRESHOLD
FOR EXCITATION
Now that we know the ionic basis of both the resting
membrane potential and the action potential, we are in
a better position to understand why there is a critical
level of depolarization. The whole-cell currentvoltage
relationship in a cell is approximated by the curve
shown in Figure 3.2.15. This curve results mainly from
the iv relationship shown for the K1 channels and for
the Na1 channels shown in Figure 3.1.3. These currents
were for the open channels, and the whole-cell iv relationship is not just a sum of these, but also reflects the
probability of the channel opening at the particular
membrane potential. The resulting curve has negative
currents at hyperpolarized membrane potentials, positive current at slight depolarizations, and negative currents at further depolarizations. Recall our convention
for current sign: a positive current is an outward current,
271
272
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
3
2
1
–
+
+
–
+
+
– Cytosol
–
+
–
ECF
+
–
–
+
–
Cytosol
–
+
–
+
Cytosol
ECF
–
+
Na+
+
Na+ channel
Na+ channel
Na+ channel
ECF
+40 mV
–55 mV
–70 mV
Na+
Na+
–
–
+
K+
+
K+
–
FIGURE 3.2.11 Proposed conceptual
changes in the Na1 and K1 channels
that give rise to the action potential. At
rest (1), fluxes through both Na1 and K1
channels are small and the membrane is
polarized. The Na1 activation gate is
closed and the inactivation gate is open.
The K1 activation gate is closed. During
threshold depolarization (2), the Na1
activation gate opens and inward Na1
current further depolarizes the cell,
leading to the upstroke of the action
potential and the overshoot. Inward Na1
slows as Na1 approaches its equilibrium
potential. Meanwhile, the K1 activation
gate opens, leading to an outward K1
current (3). This outward K1 current
repolarizes the cell and the Na1
inactivation gate closes (4). The
continued outward K1 current may lead
to a hyperpolarization. The Na1
activation gate closes (5). Upon
repolarization, the K1 activation gate
closes and the Na1 inactivation gate
opens, resetting to the resting condition.
The diagram here shows a single
channel, but membrane conductance is
governed by an ensemble of channels
whose states are not necessarily
identical.
+
–
+
–
K+ channel
+
–
+
–
+
–
channel –
–
–
+
–
+
–
Cytosol
ECF
+
6
Na+ channel
–
channel
–70 mV
5
Na+ channel
+
K+
–80 mV
4
ECF
K+
+
–50 mV
+
–
+
Na+ channel
+
–
+
–
+
+
+
– Cytosol
–
+
– Cytosol
–
+
–
+
–
ECF
Na+
Na+
+
+
+
–
+
–
–
–
+
–
+
–
K+
K+
–
+
–
+
K+ channel –
+
–
and current is in the direction of positive ion flow. Thus a
positive current removes positive ions from the cell, which
hyperpolarizes the cell. Thus at membrane potentials
lower than the resting membrane potential there is a negative total current carried by Na1 ions into the cell, the
result being a depolarization back toward the resting
membrane potential. The total current is zero at the resting membrane potential. At the resting membrane potential, the total iv curve crosses the x-axis. At slight
depolarizations, the current is positive, meaning that positive ions exit the cell and cause a return towards the resting membrane potential. At the uniform threshold, the
iv curve again crosses the x-axis. At this point, the current
shifts from positive to negative. A slightly more positive
+
–
+ K+ channel –
–
+
–
+
–
+
–
+ K+ channel –
–
+
–
+
membrane potential shifts the current from positive,
which returns the membrane to the resting membrane
potential, to negative, caused by influx of Na1, and the
membrane depolarizes. This depolarization results in progressively more negative currents. This is the influx of Na1
ions that constitutes the rising phase of the action potential. The point where the iv curve crosses the x-axis on
the high potential side is the “uniform threshold,” meaning that if the membrane was uniformly depolarized to
this point, an action potential would ensue 50% of the
time. It turns out that the membrane is seldom uniformly
depolarized: the potential varies with distance from the
point of excitation. As a consequence, the actual threshold
is generally higher than the uniform threshold.
The Action P otential
Upstroke of the
action potential
Resetting of the resting
condition
Repolarization of the
membrane
Rest
A
I
Time
1
FIGURE 3.2.12 Timing of the opening of the Na channel activation gate (A) and inactivation gate (I). The activation gate is closed at rest while the
inactivation gate is open. The activation gate opens briefly during excitation, and Na1 can cross the membrane because both gates are open.
The inactivation gate closes, partly contributing to repolarization of the membrane. This first closes the activation gate, followed by a later resetting of
the inactivation gate to the open state. Blue indicates closed state; white indicates open state.
Pull
Suction
Cell
1. Polished glass patch clamp
makes contact with the cell
membrane, forming a high
resistance seal
2. Gentle suction seals the
membrane to the patch clamp
pipette
3. Pulling the patch clamp pipette
away breaks the membrane and
forms the excised patch. Current
measured from the patch pipette inside
to the outside must go through the patch
FIGURE 3.2.13 Method for making an excised cell patch. A polished glass microelectrode is brought near to a cell while positive pressure is applied to
keep the microelectrode contents uncontaminated by the cell bathing solution. As the microelectrode approaches the cell, the resistance increases,
indicating close approach. Positive pressure is turned off and the microelectrode advances to make contact with the membrane. Clean membranes will
form a high-resistance seal. Suction is then applied and the microelectrode is reversed to pull off a small patch of membrane. In this configuration, any
current that passes across the microelectrode tip must pass through the patch. In this way, single channel currents can be measured. The patch may
have none, one, two, or several channels. Experiments can also be performed using cell-attached patches.
THRESHOLD DEPOLARIZATION
REQUIRES A THRESHOLD CHARGE
MOVEMENT, WHICH EXPLAINS THE
STRENGTHDURATION RELATIONSHIP
Depolarization to threshold requires enough charge to
change the voltage across the membrane to the threshold
voltage. When stimulation is at a point, the threshold
voltage is actually higher because the negative current in
the patch of membrane above threshold is partially offset
by positive currents in nearby membrane that is below
threshold. The condition for threshold is that the total
current of the cable is inward. The area of membrane
that supplies inward current must be large enough so
that outward currents supplied by the rest of the membrane are counterbalanced. The minimum length of fiber
that must be depolarized to threshold is called the liminal length. This idea is shown diagrammatically in
Figure 3.2.16.
273
274
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Voltage clamp record shows the holding potential
of –130 mV was stepped to –40 mV at time zero in
neuroblastoma cells
Holding potential of –100 mV was stepped
to +50 mV in squid giant axons
+50 mV
Sweeps show long openings and
outward currents carried by K+
–40 mV
–100 mV
Individual sweeps show
discrete channel openings
–130 mV
Downward deflection is an
inward current. Here it is
carried by Na+
2 pA
5 ms
1 pA
0.5 pA
0
5
10
Time (ms)
15
20
Ensemble average shows
opening early after depolarization
and later closing of the Na+ channel
0
5
10
Time (ms)
15
20
Ensemble average shows delayed
opening and continued conductance
until membrane is repolarized
FIGURE 3.2.14 Patch clamp currents of Na1 channels in neuroblastoma cells (left) and K1 channels of squid giant axon (right). (Left) Adapted from
data of C.M. Baumgarten, S.C. Dudley, R.B. Rogart and H.A. Fozzard, Unitary conductance of Na1 channel isoforms in cardiac and NB2a neuroblastoma
cells. Am. J. Physiol. 269: C1356C1363, 1995. (Right) Adapted from data of F. Bezanilla and C.K. Augustine cited in B. Hille, Ionic Channels in
Excitable Membranes, Sinauer, 1992.
THE AMOUNT OF CHARGE NECESSARY
TO REACH THRESHOLD EXPLAINS THE
STRENGTHDURATION RELATIONSHIP
Figure 3.2.16 indicates that depolarization to threshold
requires movement of sufficient charge according to the
capacitance of the membrane: ΔV 5 Δq/C. Thus reaching threshold (a ΔV from the resting potential) requires
a defined amount of charge movement. This explains
the inverse relationship between stimulus strength (its
current) and the duration, according to the Weiss
Equation (see Eqn [3.2.1]).
SUMMARY
Resting nerve cells are polarized with a negative resting
membrane potential caused by greater K1 conductance
in the resting cell. Application of an outward current further polarizes the membrane, and the recording membrane potential is a distorted version of the stimulus.
Application of an inward current depolarizes the membrane. If depolarization reaches threshold, nerve cells fire
an action potential. The action potential is a brief, pulselike change in the membrane potential which can move
from one area of the cell membrane to another and so it
can be used to signal distant parts of the neuron.
The Action P otential
Depolarization above resting membrane but
below threshold causes a positive current that
depolarizes the membrane back towards the
resting potential
+1
Depolarization above threshold
results in a negative current—an inward
flow of + ions—that further depolarizes
the membrane, leading to the action
potential
0
Resting membrane
potential
Current
-1
Uniform voltage threshold
–2
Hyperpolarization below
resting potential causes
negative currents—an inward
flow of + ions that depolarize
the membrane back to the
resting potential
–3
–4
–5
–100
–80
–60
–40
–20
0
20
40
60
80
Membrane potential (volts)
FIGURE 3.2.15 Whole-cell currentvoltage relationship. Positive currents above the equilibrium potential for K1 are mainly due to K1 exit, mainly due
to the increased driving force for K1. Negative currents are mainly due to Na1 entry, due to reduction in the driving force for K1. Hyperpolarization
below the resting membrane potential causes a negative current (inward flow of positive ions) that depolarizes the cell back towards the resting
membrane potential. Slight depolarizations produce a positive current (outward flow of positive ions) that repolarizes the cell back towards the resting
membrane potential. Thus slight hyperpolarization or depolarization returns the cell to rest. Larger depolarizations cause a negative current due to
inward flow of Na1 ions that further depolarize the cell in the action potential.
+1
0
Current
–1
–3
+1
–4
0
Outward current in nearby
slightly depolarized patch
tends to limit depolarization
–1
Current
–2
–2
–5
–100
–80
–60
–40
–20
0
20
40
60
80
Membrane potential (volts)
–3
Depolarizing, inward current
–4
–5
–100
-80
+ + + + + + + + + + + + + + + + + +
+ + + + + + + + + +
+ + + + +
Membrane potential (volts)
+
+ + + + + + + + +
+
+
+
+
+
area
- - Less +depolarized
+
+ + + + + + + + + + + + + + +
+
+
Slightly depolarized area
+ +
+
+ -- ++ ++ ++ + ++ + ++ + + ++ ++ ++ + + ++ +
+ - +
+ + + + + + + +
+
+ + + +
+ +
- + + + + + + + + + + + + + + +
+ + + + + + + + +
- + + +movement
+
+ + within
+ -- + +Charge
+ + + + + + + + + +
+ + depolarization
+ reduces
fiber
+
- - - + +the
+
+
+
+ of current injection+ + + + + + + +
+ point
at the
+
+ + + + + + +++ ++ +
+ + + + + ++
+
+ + +
+ + + + + + + +
-60
-40
-20
0
20
40
60
80
+
FIGURE 3.2.16 Consequence of spatial differences in membrane potential. Excitation occurs at a location in the membrane that causes a
depolarization. This depolarization is sufficiently large to produce a negative or inward current that would further depolarize the cell. However, nearby
patches of membrane are depolarized less and they are located on the part of the iv curve that carries a positive current. This positive current lowers
the depolarization at the point of excitation.
275
276
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Action potentials are all or none: you either get one or
you don’t. Increasing stimulus strength shortens the
delay before the start of the action potential, but it does
not alter its peak or its duration. The threshold is the
potential at which an action potential is triggered 50%
of the time, but this depends on the rate of depolarization. For some period of time after the start of an action
potential, nerve cells will not begin another action
potential. This period is the absolute refractory period.
The relative refractory period follows the absolute refractory period. During the relative refractory period, the
threshold for initiating a second action potential is
higher.
During the action potential, the membrane potential
overshoots zero and becomes positive. Since only Na1
and Ca21 have positive equilibrium potentials, the
membrane potential can become positive only if the
permeability to one of these increases. The ionic currents giving rise to the action potential for squid giant
axons were described by Alan Hodgkin and Andrew
Huxley, who received the Nobel Prize for the work in
1963. They developed a kinetic model that explained
the action potential in terms of a single voltage-gated
K1 channel and a Na1 channel governed by voltagedependent and time-dependent activation and inactivation gates. During rest, the Na1 inactivation gate is
open and the activation gate is closed. Upon depolarization, the activation gate opens and Na1 conductance
increases markedly. This causes an inward current (carried by Na1) that further depolarizes the membrane
and causes more Na1 channels to open. The explosive
increase in gNa causes the rising phase of the action
potential. With time, the Na1 inactivation gate closes,
the K1 activation gate opens, and the Na1 activation
gate closes. Opening of the K1 activation gate and closing of the Na1 inactivation gate cause an outward current that repolarizes the membrane. With further time,
the Na1 inactivation gates reopen and this reestablishes
the resting condition.
The squid axon is presented here as an example of how
to think about excitable cells. Each excitable cell, and in
fact each part of an excitable cell, can have different sets
of channels with distinct activation and inactivation
properties. The height of the action potential and its
duration depend on these characteristics, as well as the
electrical characteristics of the cell and the ionic conditions inside and outside the cell.
Channels have discrete states with discrete conductances.
Each channel undergoes transitions between conducting
and nonconducting or subconducting states. Such discontinuous conductance changes cannot be described by
continuum mathematics but rely on probabilistic
descriptions. Continuum mathematics can describe the
average or ensemble behavior of a population of channels, provided the population is sufficiently large.
Depolarization to threshold requires the movement of
sufficient charge to cause the depolarization. This charge
can be delivered over a short time at high current or
long time at low current. This is the basic nature of the
strengthduration relationship. At lower currents there
is more time for redistribution of charges within the
nerve fiber so that the relationship is not purely reciprocal. The strengthduration relationship is adequately
described by the Weiss equation: I 5 Irh(t 1 τ SD)/t, where
Irh is the rheobase and τ SD is the chronaxie.
REVIEW QUESTIONS
1. If a Na-selective channel was to open on the
membrane of a motor neuron at rest, which
way would current flow? Would this depolarize
or hyperpolarize the cell?
2. If a K-selective channel was to open on the
membrane of a motor neuron at rest, which
way would current flow? Would this depolarize
or hyperpolarize the cell?
3. What is an action potential? Why does the
membrane potential become positive during the
action potential?
4. What do we mean when we say that action
potentials are “all or none”? What is the absolute refractory period? Relative refractory period?
5. What is the “activation gate” of the Na1 channel? When is it open? When does it close? What
is the “inactivation gate” of the Na1 channel?
When is it open? When does it close?
6. What is tetrodotoxin? Why does it block action
potentials?
7. What is a patch clamp? Why is it useful? What is
an ensemble current?
8. How do gNa and gK change during the action
potential? What causes these changes?
9. What is the total current across the membrane at
the resting membrane potential? Why does a
slight depolarization, below threshold, come
back to the resting membrane potential? Why
does a slight hyperpolarization return to the resting membrane potential?
10. Why is there an inverse relationship between
current and time to reach threshold?
APPENDIX 3.2.
A1 THE HODGKINHUXLEY MODEL
OF THE ACTION POTENTIAL
ALAN HODGKIN AND ANDREW HUXLEY’S
GOAL WAS TO ACCOUNT FOR THE ACTION
POTENTIAL BY MOLECULAR MECHANISMS
Hodgkin and Huxley’s goal was to explain the ionic
fluxes and conductance changes during the action potential in terms of molecular mechanisms. After trying some
different mechanisms, they concluded that not enough
was known to determine a unique mechanism. Instead,
they tried to develop an empirical kinetic description
which would allow them to calculate electrical responses
and which would correctly predict the shape of the
action potential and its conduction velocity. Today this
model is referred to as the HodgkinHuxley or HH
model.
The Action P otential
THE HH MODEL DIVIDES THE TOTAL
CURRENT INTO SEPARATE NA 1 , K 1 ,
AND LEAK CURRENTS
An alternative way of expressing Eqn [3.2.A1.3] is as
follows:
½3:2:A1:5
dn ðnN 2 nÞ
5
dt
Tn
Hodgkin and Huxley wrote the Na1 and K1 currents in
terms of their maximum conductances which are multiplied by coefficients that vary continually between 0
and 1. The overall conductance, then, varies between 0
and the maximum conductance. All of the kinetic properties of the conductances are embedded in the characteristics of the coefficients. The conductances in the
model vary with voltage and time but not with concentration of either Na1 or K1.
where nN is the steady-state value of n at any particular
voltage and Tn is a time constant. The values of nN and
Tn are given by
αn
nN 5
αn 1 β n
½3:2:A1:6
1
Tn 5
αn 1 β n
THE HH MODEL OF THE K 1
CONDUCTANCE INCORPORATES FOUR
INDEPENDENT “PARTICLES”
THE HH MODEL OF NA 1
CONDUCTANCES USES ACTIVATING
AND INACTIVATING PARTICLES
On depolarization, the increase in gK follows an
S-shaped curve, whereas gK decreases exponentially
upon repolarization. Hodgkin and Huxley proposed
that the gating of the K1 channel could be modeled by
four identical membrane “particles.” The probability
that each channel is in the position to allow K1 conductance is n. All four particles must be correctly situated to allow conductance. The probability that all
four are positioned for conductance is n4. The K1 current is given as
Similar to the case with the K1 conductance model, HH
empirically modeled the Na1 conductance with four
hypothetical gating particles that make first-order transitions between conductive and nonconductive states.
Because the Na1 conductance has two opposing actions,
activation and inactivation, Hodgkin and Huxley used
two kinds of gating particles, called m and h. Here the
probability of an open configuration is m and h, respectively. The probability that all four gates are open is
m3h. In this case, the Na1 current is given as
½3:2:A1:1
½3:2:A1:7
IK 5 n4 gKmax ðEm 2 EK Þ
Here the probability n depends on time and voltage. In
the HH model, values of n are determined by a firstorder reaction, written as
½3:2:A1:2
αn
12n"n
½3:2:A1:3
dn
5 αn ð1 2 nÞ 2 β n n
dt
Hodgkin and Huxley determined empirical relationships
between the rate constants, αn and β n, and the membrane potential. For the squid axon at 6 C, these were:
½3:2:A1:4
The same formalism for transitions between open and
closed states of the n gates for the K1 channels also governs transitions between the open and closed states of
the m and h gates of the Na1 channel:
βn
where αn and β n depend on voltage. The reaction in
Eqn [3.2.A1.2] can be written in differential form as
αn 5
0:01ð10 2 ðEm 2 Er ÞÞ
eð102ðEm 2Er ÞÞ=10 2 1
β n 5 0:125eð2ðEm 2Er Þ=80Þ
where the membrane potential is in millivolts and the
rate constants have units of M s21. These are not the
exact forms originally used by Hodgkin and Huxley,
because their sign convention for membrane potential
is the reverse of that used today. In addition, Hodgkin
and Huxley derived their empirical equations based on
voltage clamp experiments in which the degree of variation from resting membrane potential, here symbolized
as Er, was clamped. Equation [3.2.A1.4] is transformed
to agree with today’s conventions.
INa 5 m3 hgNamax ðEm 2 ENa Þ
αm
½3:2:A1:8
12m"m
βm
αh
12h"h
βh
The rate constants all depend on voltage, according to
the following empirical relations:
αm 5
½3:2:A1:9
0:1ð25 2 ðEm 2 Er ÞÞ
eð252ðEm 2Er Þ=10Þ 2 1
β m 5 4eð2ðEm 2Er Þ=18Þ
αh 5 0:07eð2ðEm 2Er Þ=20Þ
1
β h 5 ðð302ðE 2E ÞÞ=10Þ
m
r
11
e
Relationships among mN, hN, Tm, and Th are given in
analogy to Eqn [3.2.A1.6].
CALCULATION OF GNA(T) AND GK(T)
FOR A VOLTAGE CLAMP EXPERIMENT
To calculate the time dependence of Na1 and K1 conductances, we need to know the set of rate constants
277
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
which describe the transitions between states of the conductances in the HH model: (αn, β n, αm, β m, αh, β h).
Further, we need to know this set at the two voltages. In
the voltage clamp experiment, the nerve is held at some
voltage and then is rapidly switched to another voltage.
As a result, the conductances go through a transition
from one state to another. Because the gates have different kinetics, the overall behavior can be complex. Each
of the gates relaxes between the steady-state value before
the voltage jump, and the second steady-state value at
the new voltage. The equation describing the time
course from an initial value of n, m, and h (n0, m0, and
h0) and a final value of n, m, and h (nN, mN, and hN)
can be derived from integrating Eqn [3.2.A1.3]:
1.0
9
n∞
8
0.8
7
6
n∞
0.6
5
4
0.4
τn
3
2
0.2
1
dn
dt
5 αn ð1 2 nÞ 2 β n n
0
5@
½3:2:A1:10
10
τn
278
0
–100
1
3n
α
t
n
ln4
2n5 5 2
Tn
αn 1β n
½3:2:A1:11
2
2
0
50
Em (mV)
αn
2 nAðαn 1 β n Þ
αn 1 β n
dn
5 ðαn 1 β n Þdt
ðαn =αn 1 β n Þ 2 n
ðn
ðt
dn
5 ðαn 1 β n Þdt
n0 ðαn =αn 1 β n Þ 2 n
0
Integrating, we obtain
0.0
–50
FIGURE 3.2.A1.1 The dependence of τ n and the steady-state value of
n (nN) as a function of the membrane potential, Em. These values of
n control the conductance through the K1 channel in the HH formalism.
nN, and Tn, we can calculate the n(t). All of these values
depend on the α and β for the given gates. The procedure is entirely analogous for m and h.
RESULTS OF THE CALCULATIONS
Figure 3.2.A1.1 shows the voltage dependence of nN
and Tn.
n0
3
n
2
n
N
55 2 t
ln4
nN 2 n0
Tn
n 5 nN 2 ðnN 2 n0 Þe2ðt=Tn Þ
Here we have identified Tn and nN according to Eqn
[3.2.A1.6]. It is easy to justify the assignment of nN:
simply let dn/dt in Eqn [3.2.A1.3] go to zero at infinite
time (at which point the steady-state value of n should
have been reached). From that constraint, we find
nN 5 αn/(αn 1 β n).
The last equation tells us that at t 5 0, n 5 n0 and at
t 5 N, n 5 nN. In between, n relaxes between n0 and
nN with an exponential time course. If we know n0,
The voltage clamp experiments performed by Hodgkin
and Huxley involved changing the membrane potential
essentially instantaneously from the resting potential
(265 mV) to some set potential and clamping it there.
What happens is that the resting values of n, m, and h
(called n0, m0, and h0) relax to their new values at the
clamped voltage, which we will call nN, mN, and hN.
The resulting time course of n, m, and h generates a
time course for the conductances, calculated according
to Eqns [3.2.A1.1] and [3.2.A1.7]. For a 188-mV clamp
from 265 to 123 mV, the relevant values are given in
Table 3.2.A1.1 (see Figure 3.2.A1.2).
These values are substituted into equations of the form
of Eqn [3.2.A1.11] to derive n(t), m(t), and h(t), from
which the instantaneous conductances can be calculated
TABLE 3.2.A1.1 Values of n, m, and h for Voltage Clamp from 265 to 123 mV
K Channel
Na Channel
n0 5 0.3177
m0 5 0.0529
h0 5 0.5961
nN 5 0.9494
mN 5 0.9953
hN 5 0.0009
Tn 5 1.2028 ms
Tm 5 0.1577 ms
Th 5 1.0022 ms
gKmax 5 36 ms cm22
gNamax 5 120 ms cm22
Derived from Figures 3.2.A1.1 and 3.2.A1.2.
The Action P otential
1.0
m∞
0.8
1.0
10
1.0
0.8
8
0.8
τm
0.4
0.4
4
0.4
0.2
0.2
2
h∞
0.6
τm (ms)
6
0.6
0.6
m∞
τm (ms)
τh
0.2
h∞
0.0
–100
0
–100
0.0
–50
0
50
0.0
–50
Em (mV)
0
50
Em (mV)
FIGURE 3.2.A1.2 The dependence of τ m and mN on the membrane potential. These values control the opening of the activation gate of the
Na1 channel in the HH fomalism.
50
Time after voltage clamp from –65 mV to +23 mV
45
gK or gNa (ms cm–2)
40
35
gK
30
25
20
15
10
gNa
5
0
0
1
2
3
4
5
Time (ms)
6
7
8
FIGURE 3.2.A1.3 Calculated changes in gNa and gK during a voltage clamp from 265 mV to 123 mV using the HH formalism.
according to Eqns [3.2.A1.1] and [3.2.A1.7]. The results
of these calculations for the given voltage clamp are
shown in Figure 3.2.A1.3.
Of course, Hodgkin and Huxley had it much more difficult than this, because they had to find the original
equations and parameters to fit their voltage clamp
results, whereas here we are simply confirming that the
equations and parameters they found do, indeed, look
like their voltage clamp records. It is important to
remember that the HodgkinHuxley formalism is an
empirical model, designed to fit the data. Although
there are deficiencies in the model, it succeeds admirably well in predicting the wave form of the action
potential. It is now generally accepted that the basis of
the action potential is a rapid switching on of the Na1
conductance, followed by its inactivation and more
slowly turning on of the K1 conductance.
279
3.3
Propagation of the Action
Potential
Learning Objectives
G
G
G
G
G
G
G
G
G
Define propagation of the action potential
Define conduction velocity
Describe how conduction velocity varies with axon diameter and with myelination
Using the formula for a parallel plate capacitor, explain how
myelin decreases membrane capacitance
Using the formula for resistances in parallel, explain how
internal resistance of the axoplasm varies with axon
diameter
Define the space constant and time constant
Describe how the space constant and time constant vary
with axon diameter and myelination
Qualitatively account for how membrane capacitance and
axoplasmic resistance explain the dependence of conduction velocity on myelination and axon diameter
Describe saltatory conduction and explain what “jumps”
from node to node
THE ACTION POTENTIAL MOVES
ALONG THE AXON
Consider Figure 3.3.1, which shows an axon of a motor
neuron that has been impaled at intervals by recording
electrodes. If an action potential is begun at the far left
by depolarization to threshold, each succeeding recording electrode records an action potential. Note that the
successive action potentials have similar waveforms but
they are observed at each electrode at successively later
times. The action potential is propagated along the
surface of the nerve. The action potential moves over
the surface of the cell, appearing some distance away
after some elapsed time.
THE VELOCITY OF NERVE
CONDUCTION VARIES DIRECTLY WITH
THE AXON DIAMETER
The action potentials shown in Figure 3.3.1 do not
have identical waveforms due to the stimulation artifact
that dies out with distance along the axon. After this initial stimulation artifact decays away, all subsequent
action potentials are essentially identical. The identical
280 waveform of the action potential as it travels over the
axon is a variant of the “all-or-none” description of the
action potential. As the action potential appears later at
longer distances from the point of initiation, we can
define a conduction velocity of action potential propagation equal to the distance between the recording
electrodes divided by the delay in time between action
potentials recorded at the two sites. The velocity of
action potential conduction has been determined for
myelinated and unmyelinated fibers of different sizes
(see Table 3.3.1).
Within each category of nerve fiber, myelinated or
unmyelinated, the conduction velocity varies with the
diameter of the nerve. For myelinated fibers, the conduction velocity varies approximately in proportion to
the diameter. In unmyelinated fibers, the conduction
velocity varies approximately with the square root of the
diameter.
THE ACTION POTENTIAL IS
PROPAGATED BY CURRENT MOVING
AXIALLY DOWN THE AXON
How is the action potential conducted down the length
of the axon? Recall that the action potential is triggered
by a depolarization of the membrane to threshold.
In order for an action potential to move from one place
to another along the axon, the depolarization that triggers the action potential must precede it. Depolarization
of the membrane proceeds electrotonically or passively.
As shown in Figure 3.3.2, the local depolarization of the
neuron’s axon membrane spreads out from the origin
of the depolarization.
THE TIME COURSE AND DISTANCE OF
ELECTROTONIC SPREAD DEPEND ON
THE CABLE PROPERTIES OF THE NERVE
Recall from Figures 3.2.4 and 3.2.5 that a hyperpolarizing or depolarizing stimulus was not faithfully reproduced in the axon: the signal was distorted. This
distortion is a consequence of the cable properties of
the nerve. The cable properties of the nerve refer to the
passive or electrotonic properties and not to the active
properties that give rise to the action potential. Each
length of axon is characterized by an Ohmic resistance
to current across the membrane (Rm), a capacitance
(Cm), a resistance through the external solution that
bathes the membrane (Ro), and a resistance through the
© 2017 Elsevier Inc. All rights reserved.
DOI: https://dx.doi.org/10.1016/B978-0-12-800883-6.00025-2
Propa gat ion of t he Ac ti on Potential
Voltmeter
–
+
+
–
+
+ + + + + + + + + +
–
+
+ + + + + + + + + +
–
+
+ + + + + + + + + +
+ + + + + + + + + +
– – – – – – – – – – – – – – – – – – – – – – – – – – – – – – – – – – –
Axoplasm
Direction of action potential
propagation
Em
+60
+60
+60
+60
+40
+40
+40
+40
+20
+20
+20
Em
0
(mV)
Em
(mV)
–20
Em
0
0
(mV)
–20
+20
0
(mV)
–20
–20
–40
–40
–40
–40
–60
–60
–60
–60
–80
–80
–80
0
2 4 6 8 10
Time (ms)
0
2 4 6 8 10
Time (ms)
–80
0
2 4 6 8 10
Time (ms)
0
2 4 6 8 10
Time (ms)
FIGURE 3.3.1 Appearance of action potentials at later times down the axon from the point of stimulation. The output of each voltmeter is shown
below it.
Depolarized region
TABLE 3.3.1 Velocity of Nerve Impulse Conduction
as a Function of Axon Size
Nerve
Fiber
Type
Diameter Conduction
(µm)
Velocity (m s21)
Physiological
Function
Aα
1222
70120
Somatic motor
Aδ
15
1230
Pain, sharp
C
0.51.2
0.22
Pain, ache
Axoplasm
axoplasm that fills the axon (Ri). A schematic diagram
of this electrical model is shown in Figure 3.3.3.
If we pass a constant current across the membrane
between nodes A and B, for example, so that some new
membrane potential E is established, we should expect
that the membrane potential Ex at some point x away
from the current source will depend on the distance
from the current source and, because of the capacitances, it will also depend on the time since the current
was turned on. The cable properties determine this
dependence on position x and time t.
CAPACITANCE DEPENDS ON THE AREA,
THICKNESS, AND DIELECTRIC
CONSTANT
The membrane acts much like a parallel plate capacitor.
The expression for the capacitance of a parallel plate
capacitor is given as
½3:3:1
C5
κεo A
δ
Passive spread of current
depolarizes adjacent patches
of membrane
Axoplasm
In the next patch of
membrane, passive
spread continues
Newly depolarized regions
Axoplasm
FIGURE 3.3.2 Passive spread of a depolarization to adjacent areas of
membrane. Depolarization of a patch of membrane spreads to adjacent
areas. If the depolarization reaches threshold in the nearby patch, an
action potential will be initiated. If there is no action potential, the
spread of depolarization will decay away with time and distance from
the original depolarized area.
where C is the capacitance (in F 5 C V21), κ is the
dielectric constant characteristic of the material between
the plates (a dimensionless ratio), ε0 is the electrical
permittivity of the vacuum 5 8.85 3 10212 C2 J21 m21,
281
282
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
External solution
A
Rm
Ro
E
C
Cm
Membrane
B
Ri
D
F
Axoplasm
FIGURE 3.3.3 Schematic diagram of the electrical model of an axon.
A is the area (m2), and δ is the distance between the
plates (m). Since 1 J 5 1 V C, the units of C come out
in C V21. From this equation, it should be clear that the
capacitance depends directly on the area of the membrane. It is common to normalize the capacitance by
dividing by the area. The specific capacitance Cm 5 C/A
is this normalized capacitance.
and GM be the specific conductance per unit area.
Similarly, let R be the total resistance, which is the
inverse of the conductance, and RM be the specific resistance. Then the resistance of a patch of membrane is
given as
The real membrane is not a parallel plate capacitor,
but a concentric, coaxial capacitor. This does not materially affect the analysis presented here, as detailed in
Appendix 3.3.A1.
1
1
1 1
5
5
G Gm A Gm A
CHARGE BUILDUP OR DEPLETION
FROM A CAPACITOR CONSTITUTES
A CAPACITATIVE CURRENT
The relationship among charge, volts, and capacitance is
½3:3:2
C5
Q
V
where C is the capacitance, Q is the charge (in coulombs)
on the capacitor, and V is the voltage difference across
the capacitor. When the capacitor is charging or discharging, there is a capacitative current given as
½3:3:3
i5
dQ
dV
5C
dt
dt
The capacitive current is given this special name because
current does not pass through the dielectric, but charge
builds up on one side and is taken away from the other,
so effectively there is a charge movement across the
capacitor without a charge flow through the dielectric.
G 5 Gm A
½3:3:4
R5
Here the specific resistance, Rm, is given in units of
Ω cm2, and R is in Ω. These units may seem peculiar,
that the resistance per unit area is given as Ω cm2, but
this is a consequence of Ohm’s Law that gives the current
as being inversely proportional to the resistance.
THE AXOPLASMIC RESISTANCE
DEPENDS ON THE DISTANCE, AREA,
AND SPECIFIC RESISTANCE
The resistance of an electrolyte solution such as that in
the axon is typically given as its specific resistance, ρ.
This is the resistance between two faces of a cube 1 cm
on a side. Since resistances in series add, the resistance
of a length of solution is just the length times the specific resistance. The resistance of an area of electrolyte
solution is the specific resistance divided by the area,
as in Eqn [3.3.4]. So the equivalent resistance of the
axoplasm is given as
½3:3:5
THE TRANSMEMBRANE RESISTANCE
DEPENDS ON THE AREA OF THE
MEMBRANE
Adding membrane area is like adding resistances in
parallel—the overall resistance actually decreases. The
conductances add, whereas the inverse of the resistances
add. The total conductance of a patch of membrane is
the conductance per unit area times the area. Let G
be the total conductance of a membrane of area A,
Rm
A
Ri 5
ρi d
A
where d is the distance and A is the cross-sectional area.
Since Ri has the units Ω, ρi has the units Ω cm.
THE EXTRACELLULAR RESISTANCE
ALSO DEPENDS ON THE DISTANCE,
AREA, AND SPECIFIC RESISTANCE
The resistance provided by the extracellular electrolyte
solution is entirely analogous to the axoplasmic
Propa gat ion of t he Ac ti on Potential
resistance. However, the area involved here is not
precisely known and it is large. Because the area
appears in the denominator of Eqn [3.3.5], typically
the outside resistance is small compared to the axoplasmic resistance. We will treat Ro as being zero,
so that the voltage everywhere along the axon on the
outside is zero.
CABLE PROPERTIES DEFINE A SPACE
CONSTANT AND A TIME CONSTANT
Consider part of the schematic diagram of Figure 3.3.3
shown in Figure 3.3.4. Analysis of the currents as a
function of distance will allow us to characterize the
axon in terms of its cable properties. These include a
space constant and a time constant.
One of Kirchoff’s circuit laws states that the sum of all
currents out of any node must be zero. This is just
another way of saying that there is a conservation of
total charge. Applying this principle to node (x) in
Figure 3.3.4, we have
iðxÞ 5 iðx 1 dxÞ 1 im
½3:3:6
where i(x) is the current passing down the axon into the
node at (x), im is the current that passes through the
membrane at this node, and i(x 1 dx) is the current that
passes down to the next node at (x 1 dx). The current
across the membrane, im, has two parts: one part
that passes through the resistance and a second
part that either charges or discharges the capacitor.
External solution
Ro
The current im can be written as
½3:3:7
im 5
where V(x) is the membrane potential at position x and Vr
is the membrane potential at rest at which the net
membrane current is zero. The first part of the right-hand
side of this equation is just Ohm’s law for the current
through the resistance across the membrane. The second
part is from Eqn [3.3.3] and describes that part of the
current that either charges or discharges the capacitor.
Substituting Eqn [3.3.7] into Eqn [3.3.6], we have
½3:3:8
iðxÞ 5 iðx 1 dxÞ 1
VðxÞ 2 Vr
dV
A 1 Cm A
dt
Rm
This equation can be rearranged, using A 5 2πa dx as the
surface area of the membrane element, where a is the
radius of the axon. Insertion of the area in Eqn [3.3.8]
and rearranging, we obtain
½3:3:9
iðx 1 dxÞ 2 iðxÞ
VðxÞ 2 Vr
dV
522πa
A 22πaCm A
dx
dt
Rm
In the limit as dx-0, the left-hand side of this equation
is the definition of the derivative. Taking this limit, we
derive
di
VðxÞ 2 Vr
dV
522πa
22πaCm
dx
dt
Rm
½3:3:10
There is another relationship between i and V(x) that
we can use here, and that is Ohm’s law through Ri.
There would be no longitudinal current unless there is a
voltage gradient in x. Ohm’s law gives the longitudinal
current, i, in Eqn [3.3.10] as
i5
R=
VðxÞ 2 Vr
dV
A 1 Cm A
dt
Rm
VðxÞ 2 Vðx 1 dxÞ
Ri
Vðx 1 dxÞ 2 VðxÞ
ðρi dx=πa2 Þ
πa2
Vðx 1 dxÞ 2VðxÞ
i52
limdx-0
dx
ρi
i52
Rm
½3:3:11
A
C = CmA
i52
im
V(x)
Ri =
Substituting this result for i into Eqn [3.3.10], we derive
ρd
A
V(x + dx)
i(x + dx)
i(x)
πa2 dV
ρi dx
Axoplasm
FIGURE 3.3.4 Currents at a patch of membrane area of the axon. C is
the capacitance of the membrane; Cm is the specific capacitance; R is
the resistance across the axon membrane; and Rm is the resistance per
unit area; im is the current across the membrane; i(x) is the current down
the axon at node x. Ri is the internal resistance of the axoplasm and ρi is
its specific resistance.
½3:3:12
2
πa2 d2 V
V 2 Vr
dV
5 22πa
2 2πaCm
2
dt
ρi dx
Rm
which can be rearranged to
½3:3:13
V 2 Vr 5
Rm πa2 d2 V
Rm
dV
2πaCm
2
dt
2πaρi dx2
2πa
If we let V0 5 V 2 Vr, this equation has the form
½3:3:14
V 0 5 λ2
d2 V 0
dV 0
2
τ
dx2
dt
283
284
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Here λ is identified as a space constant and τ is a time
constant, so named because they govern the spatial
and time derivatives of the voltage when a constant
current is injected across the membrane. By comparison
with Eqn [3.3.13], their values are given as
½3:3:15
sffiffiffiffiffiffiffiffiffiffiffiffiffiffi sffiffiffiffiffiffiffi
Rm πa2
Rm a
5
λ5
ρi 2πa
ρi 2
τ 5 Rm Cm
Note that at steady state, when dV/dt 5 0, the space
constant defines the way in which the voltage varies
with distance. This would be the case when a constant
current has been passed across the membrane for a
sufficiently long time to charge all the capacitors to their
steady-state level. In this case, Eqn [3.3.14] becomes
½3:3:16
V 0 5 λ2
d2 V 0
dx2
The relevant solution to this differential equation is
½3:3:17
V 5 ðV0 2 Vr Þeð2x=λÞ 1 Vr
This equation means that the voltage falls off exponentially from the point of current application to the
nerve to its value some large distance away. Here the
resting variable V0 refers to the voltage at x 5 0 and
Vr is the resting voltage some large distance away.
The equation looks this way because of the boundary
conditions that V 5 V0 when x 5 0, and V 5 Vr when
x 5 N. These boundary conditions arise because of
the way that we set up the situation: the current at a
specified point on the axon is constant and it is
applied for sufficiently long times that the capacitors
are all charged and the capacitative currents go to
zero. Under these conditions, at steady state, the
voltage at the point of current application will be V0
and the voltage far away (actually, infinitely far away)
will be the resting membrane potential. This condition
is called electrotonus.
Note that the space constant described in Eqn [3.3.15]
consists of two resistances: the resistance to current flow
across the membrane and the resistance to current
flow down the interior of the axon. If the resistance
across the membrane becomes larger, as occurs in
myelinated fibers, then the space constant will be larger.
If the resistance of the axoplasm is smaller, as occurs
when the axon is larger (and therefore has a larger
cross-sectional area, which participates in the determination of Ri), then the space constant will be larger.
Figure 3.3.5 shows the voltage as a function of distance
from the point of application of an inward current
sufficient to depolarize the cell to 240 mV without the
engagement of the active behavior of the axon, say
in a TTX-poisoned nerve. In myelinated fibers, the
voltage decreases slowly away from the point of current injection because λ, the space constant, is large.
In contrast, the steady-state voltage in unmyelinated
fibers decays rapidly away from the point of injection
of the current.
From Eqn [3.3.17], the space constant is the distance
for the voltage difference, (V 2 Vr), to fall from V0 2 Vr
to within 1/e of V0 2 Vr, which is 0.367 3 [V0 2 Vr].
This can be readily seen by letting x 5 λ, at which
point V 2 Vr 5 [V0 2 Vr] 3 1/e. The larger space constant
in myelinated fibers means that a depolarization maintains a higher voltage at longer distances, because more of
the current travel down the axon to change the membrane
potential as opposed to going through the membrane.
Equation [3.3.15] predicts that the space constant will
vary with the square root of the axon diameter. This
is the case with unmyelinated fibers. In myelinated
fibers, the value of Rm increases proportionate to the
radius. So the overall length constant would be proportional to the square root of the square of the radius or
approximately proportionate to the radius.
Substituting in for Rm and ρi from Eqns [3.3.4] and
[3.3.5] into Eqn [3.3.15], and using the area terms 2πad
for the surface area (relevant to Rm) or πa2 for the crosssectional area (relevant to Ri), we obtain
sffiffiffiffiffiffiffiffiffiffi
Rm a
λ5
ρi 2
sffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
2πadR a
5
ðπa2 =dÞRi 2
½3:3:18
sffiffiffiffiffiffiffiffiffiffiffiffiffi
dR
5
ðRi =dÞ
5
sffiffiffiffiffiffiffiffiffiffi
R 2
d
Ri
It is important to note that the units of these resistances are different because of the way in which they
have been normalized. Rm is the resistance per unit
area, and so its units are Ω cm2. The specific resistivity,
ρi, is in units of resistance per unit area per unit length,
so its units are Ω cm. Insertion of these units in
Eqn [3.3.18] gives the proper units of distance in the
calculation of λ. R and Ri are resistances, in Ω.
Since these resistances depend on the distance down
the axon, the distance used is included in the calculation of R and Ri. Sometimes it is convenient to express
the resistances as resistances per unit length. These
variables are commonly noted as rm and ri, with units
of Ω cm and Ω cm21, respectively. This is equivalent to
identifying
½3:3:19
rm 5 dR
Ri
ri 5
d
in Eqn [3.3.18]. The utility of this nomenclature is that
the space constant is then given simply as
rffiffiffiffiffi
rm
½3:3:20
λ5
ri
The temporal properties of cables can be made readily
apparent by using a space clamp. This is accomplished
Propa gat ion of t he Ac ti on Potential
–5
–4
Ro
Ro
–3
Cm
Cm
Ro
4–
Cm
Ro
Cm
Extracellular space
–1 Ro
Ro
Ro
1
Cm
Cm
2
Ro
Cm
3
Ro
Cm
4
Ro
Cm
5
Ro
Cm
Iin
Rm
Rm
Ri
Rm
Ri
Rm
Ri
Rm
Ri
Rm
Ri
Ri
Rm
Ri
Rm
Ri
Rm
R3
Rm
Ri
Ri
Axoplasm
–30
V0
–40
Myelinated fiber
Em (mV)
–50
Unmyelinated fiber
–60
–70
1/e(V0 – V)
V
–80
λ
–90
–6
–4
–2
0
2
4
6
Displacement from X = 0 (cm)
FIGURE 3.3.5 Steady-state voltage as a function of distance from the point of injection of current in myelinated and unmyelinated nerve fibers. In
unmyelinated nerves, the voltage drops off quickly with distance, implying a small space constant because Rm is small and Ri is large. In myelinated
fibers, the voltage drops off much less with distance, indicating a large space constant.
experimentally by passing a thin wire down the axon,
effectively making Ri 5 0. This removes the spatial
dependence of V(x,t) caused by passing a current. Here
dV/dx 5 0 and Eqn [3.3.14] becomes
½3:3:21
V0 5 2 τ
dV 0
dt
This is a familiar equation for exponential decay.
The applicable solution is
½3:3:22
V 5 ðV0 2 Vr Þe2ðt=τÞ 1 Vr
where the time constant, τ 5 RmCm does not vary very
much from myelinated to unmyelinated fibers because
myelination increases Rm according to the thickness
of the myelin layer, but it decreases Cm inversely with
the thickness of the myelin layer. The time constant
describes the characteristic time for the capacitor to
charge or discharge from the initial value to the final
value. Here V0 2 Vr refers to the difference between the
membrane potential initially and the membrane potential at steady state after injection of current. Note that,
as with the space constant, the time constant is defined
as the time necessary for the voltage difference to decay
to 1/e 5 0.367 of the difference to its new value. Each
patch of membrane is identical in this way, but the
distributed nature of the capacitors and resistances
makes the response of each membrane dependent on
its distance from the current source. In this way,
the patches of membrane nearer the current source
charge faster than exponentially, and those further away
charge slower than exponentially. This phenomenon is
easily modeled using electronic networks.
THE CABLE PROPERTIES EXPLAIN
THE VELOCITY OF ACTION POTENTIAL
CONDUCTION
As we discussed in the section “The Action Potential Is
Propagated by Current Moving Axially Down the Axon,”
the action potential occurs at a specified location x
because the membrane is depolarized to threshold at
that location. If the depolarization occurs earlier at location x, then the action potential also occurs earlier at that
location and the velocity of nerve impulse conduction is
faster. Myelinated nerves have a longer space constant.
Thus the depolarization occurring some distance x away
285
286
QUANTITATIVE HUMAN PHYSIOLOGY: AN INTRODUCTION
Action potential occurs only at
nodes of Ranvier because this is
the location of the regenerative
Na+ and K+ channels
Jumping of the current from
node to node is "saltatory
conduction"
Waveform propagates
passively with decrement
between nodes
Axon
Myelin sheath
Node of Ranvier
Passive spread of current is fast
because of large Rm, small Cm, and
small Ri
FIGURE 3.3.6 Saltatory conduction. The inward current accompanying an action potential travels down the axon to depolarize adjacent parts of the
cell. In myelinated fibers, little of the current is lost across the membrane because the transmembrane resistance is high (myelin insulates the axon)
and little is lost to discharging the capacitor of the axon because the capacitance is low. Enough current remains to depolarize the axon at the next
node of Ranvier where voltage-gated Na1 channels reside. In this way, myelin increases conduction velocity. In between nodes, the “action potential”
does not involve active currents across the membrane: it is a passive spread of the waveform of the action potential. Its decrement shown here is
exaggerated for illustrative purposes.
from the point of stimulation is greater in the myelinated
nerve. This is due to the fact that:
G
G
G
more of the current can travel down the axon,
because its resistance is less due to its larger size;
less of the current crosses the membrane, because its
resistance is larger in the myelinated fibers;
less current is required to depolarize the membrane,
because the membrane capacitance is smaller due to
the greater thickness of the myelin (see Eqn 3.3.