WO2023084449A1 - Diaminocyclopentylpyridine derivatives for the treatment of a disease or disorder - Google Patents
Diaminocyclopentylpyridine derivatives for the treatment of a disease or disorder Download PDFInfo
- Publication number
- WO2023084449A1 WO2023084449A1 PCT/IB2022/060845 IB2022060845W WO2023084449A1 WO 2023084449 A1 WO2023084449 A1 WO 2023084449A1 IB 2022060845 W IB2022060845 W IB 2022060845W WO 2023084449 A1 WO2023084449 A1 WO 2023084449A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- pyridin
- cyclopentyl
- oxadiazol
- pharmaceutically acceptable
- Prior art date
Links
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title claims description 177
- -1 Diaminocyclopentylpyridine derivatives Chemical class 0.000 title claims description 142
- 201000010099 disease Diseases 0.000 title claims description 89
- 208000035475 disorder Diseases 0.000 title claims description 88
- 238000011282 treatment Methods 0.000 title claims description 36
- 150000001875 compounds Chemical class 0.000 claims abstract description 394
- 150000003839 salts Chemical class 0.000 claims abstract description 183
- 238000000034 method Methods 0.000 claims abstract description 53
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 167
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 105
- 102100038955 Proprotein convertase subtilisin/kexin type 9 Human genes 0.000 claims description 105
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 98
- 239000003112 inhibitor Substances 0.000 claims description 75
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 70
- 239000003814 drug Substances 0.000 claims description 57
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 54
- 229910052736 halogen Inorganic materials 0.000 claims description 54
- 102100040214 Apolipoprotein(a) Human genes 0.000 claims description 49
- 239000008194 pharmaceutical composition Substances 0.000 claims description 49
- 101710115418 Apolipoprotein(a) Proteins 0.000 claims description 48
- AAILEWXSEQLMNI-UHFFFAOYSA-N 1h-pyridazin-6-one Chemical compound OC1=CC=CN=N1 AAILEWXSEQLMNI-UHFFFAOYSA-N 0.000 claims description 44
- 125000001424 substituent group Chemical group 0.000 claims description 43
- 230000002401 inhibitory effect Effects 0.000 claims description 38
- 239000000556 agonist Substances 0.000 claims description 37
- 125000000623 heterocyclic group Chemical group 0.000 claims description 36
- 230000008685 targeting Effects 0.000 claims description 36
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 35
- 150000002367 halogens Chemical class 0.000 claims description 35
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 33
- 201000001320 Atherosclerosis Diseases 0.000 claims description 32
- 208000029078 coronary artery disease Diseases 0.000 claims description 32
- 230000001404 mediated effect Effects 0.000 claims description 30
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 29
- 125000005842 heteroatom Chemical group 0.000 claims description 29
- 208000035150 Hypercholesterolemia Diseases 0.000 claims description 28
- 102000039446 nucleic acids Human genes 0.000 claims description 28
- 108020004707 nucleic acids Proteins 0.000 claims description 28
- 150000007523 nucleic acids Chemical class 0.000 claims description 28
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 27
- 206010048214 Xanthoma Diseases 0.000 claims description 26
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 26
- 206010063985 Phytosterolaemia Diseases 0.000 claims description 25
- 208000002227 Sitosterolemia Diseases 0.000 claims description 25
- 206010048215 Xanthomatosis Diseases 0.000 claims description 25
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 25
- 125000001072 heteroaryl group Chemical group 0.000 claims description 25
- 208000006575 hypertriglyceridemia Diseases 0.000 claims description 25
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 24
- 206010014486 Elevated triglycerides Diseases 0.000 claims description 24
- 239000003937 drug carrier Substances 0.000 claims description 24
- 208000005764 Peripheral Arterial Disease Diseases 0.000 claims description 23
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 claims description 23
- 208000018262 Peripheral vascular disease Diseases 0.000 claims description 23
- 208000035868 Vascular inflammations Diseases 0.000 claims description 23
- 108090000623 proteins and genes Proteins 0.000 claims description 23
- 125000005843 halogen group Chemical group 0.000 claims description 22
- 102000004169 proteins and genes Human genes 0.000 claims description 21
- 125000001153 fluoro group Chemical group F* 0.000 claims description 20
- 102000004217 thyroid hormone receptors Human genes 0.000 claims description 20
- 108090000721 thyroid hormone receptors Proteins 0.000 claims description 20
- 239000000411 inducer Substances 0.000 claims description 19
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 19
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 18
- 101001098868 Homo sapiens Proprotein convertase subtilisin/kexin type 9 Proteins 0.000 claims description 18
- 102100031545 Microsomal triglyceride transfer protein large subunit Human genes 0.000 claims description 18
- 108010038232 microsomal triglyceride transfer protein Proteins 0.000 claims description 18
- 230000014509 gene expression Effects 0.000 claims description 17
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 claims description 16
- 229910052757 nitrogen Inorganic materials 0.000 claims description 15
- 229940030600 antihypertensive agent Drugs 0.000 claims description 13
- 239000002220 antihypertensive agent Substances 0.000 claims description 13
- 238000004519 manufacturing process Methods 0.000 claims description 13
- 239000011664 nicotinic acid Substances 0.000 claims description 13
- 235000001968 nicotinic acid Nutrition 0.000 claims description 13
- 229910052760 oxygen Inorganic materials 0.000 claims description 13
- 101150102415 Apob gene Proteins 0.000 claims description 12
- 102000003867 Phospholipid Transfer Proteins Human genes 0.000 claims description 12
- 108090000216 Phospholipid Transfer Proteins Proteins 0.000 claims description 12
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 12
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 12
- WJRBRSLFGCUECM-UHFFFAOYSA-N hydantoin Chemical compound O=C1CNC(=O)N1 WJRBRSLFGCUECM-UHFFFAOYSA-N 0.000 claims description 12
- 229960003512 nicotinic acid Drugs 0.000 claims description 12
- 239000012190 activator Substances 0.000 claims description 11
- 229940125708 antidiabetic agent Drugs 0.000 claims description 11
- 239000003472 antidiabetic agent Substances 0.000 claims description 11
- 239000003524 antilipemic agent Substances 0.000 claims description 11
- 229920000080 bile acid sequestrant Polymers 0.000 claims description 11
- 235000021323 fish oil Nutrition 0.000 claims description 11
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 11
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical class IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 claims description 10
- 239000000883 anti-obesity agent Substances 0.000 claims description 10
- 229940125710 antiobesity agent Drugs 0.000 claims description 10
- 229940096699 bile acid sequestrants Drugs 0.000 claims description 10
- 239000003795 chemical substances by application Substances 0.000 claims description 10
- 210000002824 peroxisome Anatomy 0.000 claims description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- 239000005495 thyroid hormone Substances 0.000 claims description 10
- 229940036555 thyroid hormone Drugs 0.000 claims description 10
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 9
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims description 9
- 102000004146 ATP citrate synthases Human genes 0.000 claims description 9
- 108090000662 ATP citrate synthases Proteins 0.000 claims description 9
- 108010036824 Citrate (pro-3S)-lyase Proteins 0.000 claims description 9
- 101710088335 Diacylglycerol acyltransferase/mycolyltransferase Ag85A Proteins 0.000 claims description 9
- 101710088334 Diacylglycerol acyltransferase/mycolyltransferase Ag85B Proteins 0.000 claims description 9
- 101710088427 Diacylglycerol acyltransferase/mycolyltransferase Ag85C Proteins 0.000 claims description 9
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 claims description 9
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 claims description 9
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 claims description 9
- 102000007466 Purinergic P2 Receptors Human genes 0.000 claims description 9
- 108010085249 Purinergic P2 Receptors Proteins 0.000 claims description 9
- 239000013543 active substance Substances 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 9
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 claims description 9
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 claims description 9
- 239000003381 stabilizer Substances 0.000 claims description 9
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 8
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 6
- 102100036869 Diacylglycerol O-acyltransferase 1 Human genes 0.000 claims description 5
- 238000001802 infusion Methods 0.000 claims description 5
- 229910052727 yttrium Inorganic materials 0.000 claims description 5
- 125000002733 (C1-C6) fluoroalkyl group Chemical group 0.000 claims description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 4
- 238000002347 injection Methods 0.000 claims description 4
- 239000007924 injection Substances 0.000 claims description 4
- 239000008177 pharmaceutical agent Substances 0.000 claims description 4
- 238000007920 subcutaneous administration Methods 0.000 claims description 2
- 101000927974 Homo sapiens Diacylglycerol O-acyltransferase 1 Proteins 0.000 claims 2
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 92
- 238000003786 synthesis reaction Methods 0.000 description 115
- 230000015572 biosynthetic process Effects 0.000 description 111
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 104
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 93
- 101710180553 Proprotein convertase subtilisin/kexin type 9 Proteins 0.000 description 91
- 238000003756 stirring Methods 0.000 description 91
- 230000002829 reductive effect Effects 0.000 description 90
- 239000000243 solution Substances 0.000 description 84
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 73
- 239000007787 solid Substances 0.000 description 72
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 72
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 68
- 239000011541 reaction mixture Substances 0.000 description 66
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 64
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 60
- 239000003480 eluent Substances 0.000 description 54
- 238000003818 flash chromatography Methods 0.000 description 54
- 235000019439 ethyl acetate Nutrition 0.000 description 52
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 50
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 46
- 239000000741 silica gel Substances 0.000 description 44
- 229910002027 silica gel Inorganic materials 0.000 description 44
- 238000005481 NMR spectroscopy Methods 0.000 description 38
- 239000000047 product Substances 0.000 description 34
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 32
- 239000012074 organic phase Substances 0.000 description 32
- 229940124597 therapeutic agent Drugs 0.000 description 30
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 28
- 125000000217 alkyl group Chemical group 0.000 description 28
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 27
- 235000011114 ammonium hydroxide Nutrition 0.000 description 26
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 24
- 108010007622 LDL Lipoproteins Proteins 0.000 description 24
- 102000007330 LDL Lipoproteins Human genes 0.000 description 24
- 238000002953 preparative HPLC Methods 0.000 description 24
- 230000005764 inhibitory process Effects 0.000 description 23
- 125000003118 aryl group Chemical group 0.000 description 22
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- 229910052805 deuterium Inorganic materials 0.000 description 20
- 230000000694 effects Effects 0.000 description 19
- 235000018102 proteins Nutrition 0.000 description 19
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 18
- 239000012043 crude product Substances 0.000 description 18
- 206010040047 Sepsis Diseases 0.000 description 16
- 239000002253 acid Substances 0.000 description 16
- 235000012000 cholesterol Nutrition 0.000 description 16
- 239000003643 water by type Substances 0.000 description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 15
- 108090000028 Neprilysin Proteins 0.000 description 15
- 102000003729 Neprilysin Human genes 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 239000002585 base Substances 0.000 description 15
- 238000000746 purification Methods 0.000 description 15
- 125000004429 atom Chemical group 0.000 description 14
- 125000000753 cycloalkyl group Chemical group 0.000 description 13
- 229920006395 saturated elastomer Polymers 0.000 description 13
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 12
- 229910052786 argon Inorganic materials 0.000 description 12
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- 102000004311 liver X receptors Human genes 0.000 description 12
- 108090000865 liver X receptors Proteins 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- 229940123338 Aldosterone synthase inhibitor Drugs 0.000 description 11
- 108010028554 LDL Cholesterol Proteins 0.000 description 11
- 150000001412 amines Chemical class 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000008280 blood Substances 0.000 description 11
- 238000010348 incorporation Methods 0.000 description 11
- 210000002966 serum Anatomy 0.000 description 11
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 10
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 10
- 239000007821 HATU Substances 0.000 description 10
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- 230000002441 reversible effect Effects 0.000 description 10
- 239000012453 solvate Substances 0.000 description 10
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 9
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 9
- 108010010234 HDL Lipoproteins Proteins 0.000 description 9
- 102000015779 HDL Lipoproteins Human genes 0.000 description 9
- 102100040918 Pro-glucagon Human genes 0.000 description 9
- 125000003342 alkenyl group Chemical group 0.000 description 9
- 239000000460 chlorine Substances 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 9
- 230000000670 limiting effect Effects 0.000 description 9
- 230000002265 prevention Effects 0.000 description 9
- 125000006239 protecting group Chemical group 0.000 description 9
- 150000003626 triacylglycerols Chemical class 0.000 description 9
- 102000016622 Dipeptidyl Peptidase 4 Human genes 0.000 description 8
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 description 8
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 8
- 101001047090 Homo sapiens Potassium voltage-gated channel subfamily H member 2 Proteins 0.000 description 8
- 208000000563 Hyperlipoproteinemia Type II Diseases 0.000 description 8
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 8
- 201000004681 Psoriasis Diseases 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 description 8
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 8
- 201000001386 familial hypercholesterolemia Diseases 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- UUUHXMGGBIUAPW-UHFFFAOYSA-N 1-[1-[2-[[5-amino-2-[[1-[5-(diaminomethylideneamino)-2-[[1-[3-(1h-indol-3-yl)-2-[(5-oxopyrrolidine-2-carbonyl)amino]propanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-methylpentanoyl]pyrrolidine-2-carbon Chemical compound C1CCC(C(=O)N2C(CCC2)C(O)=O)N1C(=O)C(C(C)CC)NC(=O)C(CCC(N)=O)NC(=O)C1CCCN1C(=O)C(CCCN=C(N)N)NC(=O)C1CCCN1C(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C1CCC(=O)N1 UUUHXMGGBIUAPW-UHFFFAOYSA-N 0.000 description 7
- 101100348848 Mus musculus Notch4 gene Proteins 0.000 description 7
- 206010028980 Neoplasm Diseases 0.000 description 7
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 7
- 102000004270 Peptidyl-Dipeptidase A Human genes 0.000 description 7
- 108090000882 Peptidyl-Dipeptidase A Proteins 0.000 description 7
- 102100022807 Potassium voltage-gated channel subfamily H member 2 Human genes 0.000 description 7
- 125000000304 alkynyl group Chemical group 0.000 description 7
- 239000012267 brine Substances 0.000 description 7
- 201000011510 cancer Diseases 0.000 description 7
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 229910052739 hydrogen Inorganic materials 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- XZMCDFZZKTWFGF-UHFFFAOYSA-N Cyanamide Chemical compound NC#N XZMCDFZZKTWFGF-UHFFFAOYSA-N 0.000 description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 6
- 102000000853 LDL receptors Human genes 0.000 description 6
- 108010001831 LDL receptors Proteins 0.000 description 6
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 6
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 6
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 6
- 210000001367 artery Anatomy 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 125000002619 bicyclic group Chemical group 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 6
- 125000004122 cyclic group Chemical group 0.000 description 6
- 230000008030 elimination Effects 0.000 description 6
- 238000003379 elimination reaction Methods 0.000 description 6
- 150000002632 lipids Chemical class 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 125000004076 pyridyl group Chemical group 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- PGBVMVTUWHCOHX-YUMQZZPRSA-N tert-butyl n-[(1s,3s)-3-aminocyclopentyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@H]1CC[C@H](N)C1 PGBVMVTUWHCOHX-YUMQZZPRSA-N 0.000 description 6
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 6
- 108010074708 B7-H1 Antigen Proteins 0.000 description 5
- 102000008096 B7-H1 Antigen Human genes 0.000 description 5
- LQRNAUZEMLGYOX-LZVIIAQDSA-N CC(=O)N[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OCCCCC(=O)NCCCNC(=O)CCOCC(COCCC(=O)NCCCNC(=O)CCCCO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O)(COCCC(=O)NCCCNC(=O)CCCCO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O)NC(=O)CCCCCCCCCCC(=O)N1C[C@H](O)C[C@H]1COP(O)(O)=O Chemical compound CC(=O)N[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OCCCCC(=O)NCCCNC(=O)CCOCC(COCCC(=O)NCCCNC(=O)CCCCO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O)(COCCC(=O)NCCCNC(=O)CCCCO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O)NC(=O)CCCCCCCCCCC(=O)N1C[C@H](O)C[C@H]1COP(O)(O)=O LQRNAUZEMLGYOX-LZVIIAQDSA-N 0.000 description 5
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 5
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 5
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 125000002252 acyl group Chemical group 0.000 description 5
- 125000003545 alkoxy group Chemical group 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 5
- 230000004071 biological effect Effects 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 229910052791 calcium Inorganic materials 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 238000002648 combination therapy Methods 0.000 description 5
- 229960002027 evolocumab Drugs 0.000 description 5
- 229950005863 inclisiran Drugs 0.000 description 5
- 230000000155 isotopic effect Effects 0.000 description 5
- 125000004043 oxo group Chemical group O=* 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 229940056298 pelacarsen Drugs 0.000 description 5
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 229910052938 sodium sulfate Inorganic materials 0.000 description 5
- 235000011152 sodium sulphate Nutrition 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 4
- HBEDSQVIWPRPAY-UHFFFAOYSA-N 2,3-dihydrobenzofuran Chemical compound C1=CC=C2OCCC2=C1 HBEDSQVIWPRPAY-UHFFFAOYSA-N 0.000 description 4
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- 102100023990 60S ribosomal protein L17 Human genes 0.000 description 4
- 102000055510 ATP Binding Cassette Transporter 1 Human genes 0.000 description 4
- 108700005241 ATP Binding Cassette Transporter 1 Proteins 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 208000024172 Cardiovascular disease Diseases 0.000 description 4
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 4
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 4
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 4
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- 206010022489 Insulin Resistance Diseases 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- 102000004895 Lipoproteins Human genes 0.000 description 4
- 108090001030 Lipoproteins Proteins 0.000 description 4
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 4
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 4
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 4
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 4
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 4
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 4
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 4
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 4
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 4
- 125000005605 benzo group Chemical group 0.000 description 4
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 4
- 230000001906 cholesterol absorption Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 4
- 239000002158 endotoxin Substances 0.000 description 4
- 229940125753 fibrate Drugs 0.000 description 4
- 125000001188 haloalkyl group Chemical group 0.000 description 4
- 102000053786 human PCSK9 Human genes 0.000 description 4
- 239000003456 ion exchange resin Substances 0.000 description 4
- 229920003303 ion-exchange polymer Polymers 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- 239000011591 potassium Substances 0.000 description 4
- 229910052700 potassium Inorganic materials 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 229960003912 probucol Drugs 0.000 description 4
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical group C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 4
- 229940095574 propionic acid Drugs 0.000 description 4
- 125000000714 pyrimidinyl group Chemical group 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 235000015424 sodium Nutrition 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 125000000547 substituted alkyl group Chemical group 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 4
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 4
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 3
- HDPNBNXLBDFELL-UHFFFAOYSA-N 1,1,1-trimethoxyethane Chemical compound COC(C)(OC)OC HDPNBNXLBDFELL-UHFFFAOYSA-N 0.000 description 3
- GYZNHUNWABYAPO-UHFFFAOYSA-N 2-fluoro-5-iodopyridine Chemical compound FC1=CC=C(I)C=N1 GYZNHUNWABYAPO-UHFFFAOYSA-N 0.000 description 3
- SYHJILPZUNKNHQ-UHFFFAOYSA-N 3-phenylbenzonitrile Chemical compound N#CC1=CC=CC(C=2C=CC=CC=2)=C1 SYHJILPZUNKNHQ-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 101150092476 ABCA1 gene Proteins 0.000 description 3
- UXOWGYHJODZGMF-QORCZRPOSA-N Aliskiren Chemical compound COCCCOC1=CC(C[C@@H](C[C@H](N)[C@@H](O)C[C@@H](C(C)C)C(=O)NCC(C)(C)C(N)=O)C(C)C)=CC=C1OC UXOWGYHJODZGMF-QORCZRPOSA-N 0.000 description 3
- 229940123413 Angiotensin II antagonist Drugs 0.000 description 3
- 108010059886 Apolipoprotein A-I Proteins 0.000 description 3
- 102000005666 Apolipoprotein A-I Human genes 0.000 description 3
- PCBZRNYXXCIELG-WYFCWLEVSA-N COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 Chemical compound COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 PCBZRNYXXCIELG-WYFCWLEVSA-N 0.000 description 3
- 229940127193 DGAT1 inhibitor Drugs 0.000 description 3
- 108050004099 Diacylglycerol O-acyltransferase 1 Proteins 0.000 description 3
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 3
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 3
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 3
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 3
- HEMJJKBWTPKOJG-UHFFFAOYSA-N Gemfibrozil Chemical compound CC1=CC=C(C)C(OCCCC(C)(C)C(O)=O)=C1 HEMJJKBWTPKOJG-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 3
- 108010023302 HDL Cholesterol Proteins 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- 101100317378 Mus musculus Wnt3 gene Proteins 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 3
- 102000034527 Retinoid X Receptors Human genes 0.000 description 3
- 108010038912 Retinoid X Receptors Proteins 0.000 description 3
- 101000767160 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) Intracellular protein transport protein USO1 Proteins 0.000 description 3
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 125000004414 alkyl thio group Chemical group 0.000 description 3
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 3
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 208000037998 chronic venous disease Diseases 0.000 description 3
- 230000004087 circulation Effects 0.000 description 3
- 239000010949 copper Substances 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 125000000392 cycloalkenyl group Chemical group 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 235000005911 diet Nutrition 0.000 description 3
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 3
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 3
- 229960005156 digoxin Drugs 0.000 description 3
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 3
- 230000029142 excretion Effects 0.000 description 3
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 3
- 229960000815 ezetimibe Drugs 0.000 description 3
- 229940121360 farnesoid X receptor (fxr) agonists Drugs 0.000 description 3
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 3
- 229960002297 fenofibrate Drugs 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 229960003627 gemfibrozil Drugs 0.000 description 3
- 229940093915 gynecological organic acid Drugs 0.000 description 3
- 125000001183 hydrocarbyl group Chemical group 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 3
- 125000001041 indolyl group Chemical group 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 3
- 238000010253 intravenous injection Methods 0.000 description 3
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 3
- 238000011813 knockout mouse model Methods 0.000 description 3
- 210000000265 leukocyte Anatomy 0.000 description 3
- 229920006008 lipopolysaccharide Polymers 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 3
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 3
- 229960002510 mandelic acid Drugs 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 108020004017 nuclear receptors Proteins 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 235000020660 omega-3 fatty acid Nutrition 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 229960005141 piperazine Drugs 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 235000019260 propionic acid Nutrition 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000000171 quenching effect Effects 0.000 description 3
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 239000002464 receptor antagonist Substances 0.000 description 3
- 229940044551 receptor antagonist Drugs 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 208000013223 septicemia Diseases 0.000 description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000011975 tartaric acid Substances 0.000 description 3
- 235000002906 tartaric acid Nutrition 0.000 description 3
- 229960001367 tartaric acid Drugs 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- JWZZKOKVBUJMES-UHFFFAOYSA-N (+-)-Isoprenaline Chemical compound CC(C)NCC(O)C1=CC=C(O)C(O)=C1 JWZZKOKVBUJMES-UHFFFAOYSA-N 0.000 description 2
- LPUDGHQMOAHMMF-JBACZVJFSA-N (2s)-2-[[[(2s)-6-amino-2-(methanesulfonamido)hexanoyl]amino]methyl]-3-[1-[[(1s)-1-carboxy-2-(4-hydroxyphenyl)ethyl]carbamoyl]cyclopentyl]propanoic acid Chemical compound N([C@@H](CC=1C=CC(O)=CC=1)C(O)=O)C(=O)C1(C[C@@H](CNC(=O)[C@H](CCCCN)NS(=O)(=O)C)C(O)=O)CCCC1 LPUDGHQMOAHMMF-JBACZVJFSA-N 0.000 description 2
- FJLGEFLZQAZZCD-JUFISIKESA-N (3S,5R)-fluvastatin Chemical compound C12=CC=CC=C2N(C(C)C)C(\C=C\[C@H](O)C[C@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 FJLGEFLZQAZZCD-JUFISIKESA-N 0.000 description 2
- NGJOFQZEYQGZMB-KTKZVXAJSA-N (4S)-5-[[2-[[(2S,3R)-1-[[(2S)-1-[[(2S,3R)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[2-[[(1S)-4-carbamimidamido-1-carboxybutyl]amino]-2-oxoethyl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-2-oxoethyl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-2-oxoethyl]amino]-4-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]propanoyl]amino]-5-oxopentanoic acid Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 NGJOFQZEYQGZMB-KTKZVXAJSA-N 0.000 description 2
- OJBYZWHAPXIJID-UHFFFAOYSA-N (6-fluoropyridin-3-yl)boronic acid Chemical compound OB(O)C1=CC=C(F)N=C1 OJBYZWHAPXIJID-UHFFFAOYSA-N 0.000 description 2
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 2
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 2
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 2
- NDCPERCVXDYEFU-UHFFFAOYSA-N 1-fluorocyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(F)CC1 NDCPERCVXDYEFU-UHFFFAOYSA-N 0.000 description 2
- NKWCGTOZTHZDHB-UHFFFAOYSA-N 1h-imidazol-1-ium-4-carboxylate Chemical compound OC(=O)C1=CNC=N1 NKWCGTOZTHZDHB-UHFFFAOYSA-N 0.000 description 2
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 2
- CJFRIMRBHHUJKW-UHFFFAOYSA-N 2-chloro-5-cyclopropylpyrimidine Chemical compound C1=NC(Cl)=NC=C1C1CC1 CJFRIMRBHHUJKW-UHFFFAOYSA-N 0.000 description 2
- IDUSJBBWEKNWAK-UHFFFAOYSA-N 3,4-dihydro-2h-1,2-benzothiazine Chemical compound C1=CC=C2SNCCC2=C1 IDUSJBBWEKNWAK-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- MZQQHYDUINOMDG-UHFFFAOYSA-N 3-methylimidazolidine-2,4-dione Chemical compound CN1C(=O)CNC1=O MZQQHYDUINOMDG-UHFFFAOYSA-N 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- HVBSAKJJOYLTQU-UHFFFAOYSA-N 4-aminobenzenesulfonic acid Chemical compound NC1=CC=C(S(O)(=O)=O)C=C1 HVBSAKJJOYLTQU-UHFFFAOYSA-N 0.000 description 2
- NFFXEUUOMTXWCX-UHFFFAOYSA-N 5-[(2,4-dioxo-1,3-thiazolidin-5-yl)methyl]-2-methoxy-n-[[4-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound C1=C(C(=O)NCC=2C=CC(=CC=2)C(F)(F)F)C(OC)=CC=C1CC1SC(=O)NC1=O NFFXEUUOMTXWCX-UHFFFAOYSA-N 0.000 description 2
- GWZJXMRSPIFFAK-UHFFFAOYSA-N 5-[(2-naphthalen-2-yl-1,3-benzoxazol-5-yl)methyl]-1,3-thiazolidine-2,4-dione Chemical compound S1C(=O)NC(=O)C1CC1=CC=C(OC(=N2)C=3C=C4C=CC=CC4=CC=3)C2=C1 GWZJXMRSPIFFAK-UHFFFAOYSA-N 0.000 description 2
- YVQKIDLSVHRBGZ-UHFFFAOYSA-N 5-[[4-[2-hydroxy-2-(5-methyl-2-phenyl-1,3-oxazol-4-yl)ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione Chemical compound CC=1OC(C=2C=CC=CC=2)=NC=1C(O)COC(C=C1)=CC=C1CC1SC(=O)NC1=O YVQKIDLSVHRBGZ-UHFFFAOYSA-N 0.000 description 2
- 239000005541 ACE inhibitor Substances 0.000 description 2
- 239000012114 Alexa Fluor 647 Substances 0.000 description 2
- 108700028369 Alleles Proteins 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- 102100038495 Bile acid receptor Human genes 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- 102100033868 Cannabinoid receptor 1 Human genes 0.000 description 2
- 101710187010 Cannabinoid receptor 1 Proteins 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 108010004103 Chylomicrons Proteins 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- 102100035762 Diacylglycerol O-acyltransferase 2 Human genes 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- JRWZLRBJNMZMFE-UHFFFAOYSA-N Dobutamine Chemical compound C=1C=C(O)C(O)=CC=1CCNC(C)CCC1=CC=C(O)C=C1 JRWZLRBJNMZMFE-UHFFFAOYSA-N 0.000 description 2
- 208000032928 Dyslipidaemia Diseases 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 102000008016 Eukaryotic Initiation Factor-3 Human genes 0.000 description 2
- 108010089790 Eukaryotic Initiation Factor-3 Proteins 0.000 description 2
- 108010011459 Exenatide Proteins 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 102000001267 GSK3 Human genes 0.000 description 2
- NAXSRXHZFIBFMI-UHFFFAOYSA-N GW 3965 Chemical compound OC(=O)CC1=CC=CC(OCCCN(CC(C=2C=CC=CC=2)C=2C=CC=CC=2)CC=2C(=C(C=CC=2)C(F)(F)F)Cl)=C1 NAXSRXHZFIBFMI-UHFFFAOYSA-N 0.000 description 2
- BYTNEISLBIENSA-MDZDMXLPSA-N GW 4064 Chemical group CC(C)C=1ON=C(C=2C(=CC=CC=2Cl)Cl)C=1COC(C=C1Cl)=CC=C1\C=C\C1=CC=CC(C(O)=O)=C1 BYTNEISLBIENSA-MDZDMXLPSA-N 0.000 description 2
- 101800004295 Glucagon-like peptide 1(7-36) Proteins 0.000 description 2
- 108010014905 Glycogen Synthase Kinase 3 Proteins 0.000 description 2
- 101000603876 Homo sapiens Bile acid receptor Proteins 0.000 description 2
- 101000930020 Homo sapiens Diacylglycerol O-acyltransferase 2 Proteins 0.000 description 2
- CZGUSIXMZVURDU-JZXHSEFVSA-N Ile(5)-angiotensin II Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C([O-])=O)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=[NH2+])NC(=O)[C@@H]([NH3+])CC([O-])=O)C(C)C)C1=CC=C(O)C=C1 CZGUSIXMZVURDU-JZXHSEFVSA-N 0.000 description 2
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 208000017170 Lipid metabolism disease Diseases 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 101100446506 Mus musculus Fgf3 gene Proteins 0.000 description 2
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- 208000008589 Obesity Diseases 0.000 description 2
- 229940127355 PCSK9 Inhibitors Drugs 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 description 2
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 description 2
- 102000014190 Phosphatidylcholine-sterol O-acyltransferase Human genes 0.000 description 2
- 108010011964 Phosphatidylcholine-sterol O-acyltransferase Proteins 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 2
- 102000006437 Proprotein Convertases Human genes 0.000 description 2
- 108010044159 Proprotein Convertases Proteins 0.000 description 2
- AJLFOPYRIVGYMJ-UHFFFAOYSA-N SJ000287055 Natural products C12C(OC(=O)C(C)CC)CCC=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 AJLFOPYRIVGYMJ-UHFFFAOYSA-N 0.000 description 2
- DLSWIYLPEUIQAV-UHFFFAOYSA-N Semaglutide Chemical compound CCC(C)C(NC(=O)C(Cc1ccccc1)NC(=O)C(CCC(O)=O)NC(=O)C(CCCCNC(=O)COCCOCCNC(=O)COCCOCCNC(=O)CCC(NC(=O)CCCCCCCCCCCCCCCCC(O)=O)C(O)=O)NC(=O)C(C)NC(=O)C(C)NC(=O)C(CCC(N)=O)NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)C(CC(C)C)NC(=O)C(Cc1ccc(O)cc1)NC(=O)C(CO)NC(=O)C(CO)NC(=O)C(NC(=O)C(CC(O)=O)NC(=O)C(CO)NC(=O)C(NC(=O)C(Cc1ccccc1)NC(=O)C(NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)C(C)(C)NC(=O)C(N)Cc1cnc[nH]1)C(C)O)C(C)O)C(C)C)C(=O)NC(C)C(=O)NC(Cc1c[nH]c2ccccc12)C(=O)NC(CC(C)C)C(=O)NC(C(C)C)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CCCNC(N)=N)C(=O)NCC(O)=O DLSWIYLPEUIQAV-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 102000009822 Sterol Regulatory Element Binding Proteins Human genes 0.000 description 2
- 108010020396 Sterol Regulatory Element Binding Proteins Proteins 0.000 description 2
- 101710135785 Subtilisin-like protease Proteins 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 229940100389 Sulfonylurea Drugs 0.000 description 2
- 229910052771 Terbium Inorganic materials 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 150000001325 aldosterones Chemical class 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 229960004601 aliskiren Drugs 0.000 description 2
- JAZBEHYOTPTENJ-JLNKQSITSA-N all-cis-5,8,11,14,17-icosapentaenoic acid Chemical compound CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O JAZBEHYOTPTENJ-JLNKQSITSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- HYHMLYSLQUKXKP-UHFFFAOYSA-N bempedoic acid Chemical group OC(=O)C(C)(C)CCCCCC(O)CCCCCC(C)(C)C(O)=O HYHMLYSLQUKXKP-UHFFFAOYSA-N 0.000 description 2
- 229950002974 bempedoic acid Drugs 0.000 description 2
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- KKBIUAUSZKGNOA-HNAYVOBHSA-N benzyl (2s)-2-[[(2s)-2-(acetylsulfanylmethyl)-3-(1,3-benzodioxol-5-yl)propanoyl]amino]propanoate Chemical compound O=C([C@@H](NC(=O)[C@@H](CSC(C)=O)CC=1C=C2OCOC2=CC=1)C)OCC1=CC=CC=C1 KKBIUAUSZKGNOA-HNAYVOBHSA-N 0.000 description 2
- 230000008827 biological function Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 229960001948 caffeine Drugs 0.000 description 2
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 239000002327 cardiovascular agent Substances 0.000 description 2
- 229940125692 cardiovascular agent Drugs 0.000 description 2
- 230000007681 cardiovascular toxicity Effects 0.000 description 2
- 231100000060 cardiovascular toxicity Toxicity 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229920001429 chelating resin Polymers 0.000 description 2
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 2
- 230000031154 cholesterol homeostasis Effects 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 235000015165 citric acid Nutrition 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000003218 coronary vasodilator agent Substances 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- JBDSSBMEKXHSJF-UHFFFAOYSA-N cyclopentanecarboxylic acid Chemical compound OC(=O)C1CCCC1 JBDSSBMEKXHSJF-UHFFFAOYSA-N 0.000 description 2
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 2
- QQKNSPHAFATFNQ-UHFFFAOYSA-N darglitazone Chemical compound CC=1OC(C=2C=CC=CC=2)=NC=1CCC(=O)C(C=C1)=CC=C1CC1SC(=O)NC1=O QQKNSPHAFATFNQ-UHFFFAOYSA-N 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 150000001975 deuterium Chemical group 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- IYYZUPMFVPLQIF-UHFFFAOYSA-N dibenzothiophene Chemical compound C1=CC=C2C3=CC=CC=C3SC2=C1 IYYZUPMFVPLQIF-UHFFFAOYSA-N 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 2
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 2
- 125000005436 dihydrobenzothiophenyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000002934 diuretic Substances 0.000 description 2
- 230000001882 diuretic effect Effects 0.000 description 2
- 229960001089 dobutamine Drugs 0.000 description 2
- 229940088679 drug related substance Drugs 0.000 description 2
- 235000020673 eicosapentaenoic acid Nutrition 0.000 description 2
- 229960005135 eicosapentaenoic acid Drugs 0.000 description 2
- JAZBEHYOTPTENJ-UHFFFAOYSA-N eicosapentaenoic acid Natural products CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O JAZBEHYOTPTENJ-UHFFFAOYSA-N 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- DTMGIJFHGGCSLO-FIAQIACWSA-N ethyl (4z,7z,10z,13z,16z,19z)-docosa-4,7,10,13,16,19-hexaenoate;ethyl (5z,8z,11z,14z,17z)-icosa-5,8,11,14,17-pentaenoate Chemical compound CCOC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CC.CCOC(=O)CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CC DTMGIJFHGGCSLO-FIAQIACWSA-N 0.000 description 2
- SSQPWTVBQMWLSZ-AAQCHOMXSA-N ethyl (5Z,8Z,11Z,14Z,17Z)-icosapentaenoate Chemical compound CCOC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CC SSQPWTVBQMWLSZ-AAQCHOMXSA-N 0.000 description 2
- 229960001519 exenatide Drugs 0.000 description 2
- 229950005203 fasidotril Drugs 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 210000000497 foam cell Anatomy 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 235000011087 fumaric acid Nutrition 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 125000004613 furo[2,3-c]pyridinyl group Chemical group O1C(=CC=2C1=CN=CC2)* 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 229960004580 glibenclamide Drugs 0.000 description 2
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229960002600 icosapent ethyl Drugs 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 239000004041 inotropic agent Substances 0.000 description 2
- 230000002473 insulinotropic effect Effects 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 229940039009 isoproterenol Drugs 0.000 description 2
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 239000002171 loop diuretic Substances 0.000 description 2
- 229940115970 lovaza Drugs 0.000 description 2
- 229960003646 lysine Drugs 0.000 description 2
- 159000000003 magnesium salts Chemical class 0.000 description 2
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 239000001630 malic acid Substances 0.000 description 2
- 235000011090 malic acid Nutrition 0.000 description 2
- 210000004379 membrane Anatomy 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- AJLFOPYRIVGYMJ-INTXDZFKSA-N mevastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=CCC[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 AJLFOPYRIVGYMJ-INTXDZFKSA-N 0.000 description 2
- BOZILQFLQYBIIY-UHFFFAOYSA-N mevastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CCC=C21 BOZILQFLQYBIIY-UHFFFAOYSA-N 0.000 description 2
- PZRHRDRVRGEVNW-UHFFFAOYSA-N milrinone Chemical compound N1C(=O)C(C#N)=CC(C=2C=CN=CC=2)=C1C PZRHRDRVRGEVNW-UHFFFAOYSA-N 0.000 description 2
- 229960003574 milrinone Drugs 0.000 description 2
- 238000010172 mouse model Methods 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- SGIWFELWJPNFDH-UHFFFAOYSA-N n-(2,2,2-trifluoroethyl)-n-{4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl}benzenesulfonamide Chemical compound C1=CC(C(O)(C(F)(F)F)C(F)(F)F)=CC=C1N(CC(F)(F)F)S(=O)(=O)C1=CC=CC=C1 SGIWFELWJPNFDH-UHFFFAOYSA-N 0.000 description 2
- PKWDZWYVIHVNKS-UHFFFAOYSA-N netoglitazone Chemical compound FC1=CC=CC=C1COC1=CC=C(C=C(CC2C(NC(=O)S2)=O)C=C2)C2=C1 PKWDZWYVIHVNKS-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 238000013546 non-drug therapy Methods 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 235000020824 obesity Nutrition 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- LVRLSYPNFFBYCZ-VGWMRTNUSA-N omapatrilat Chemical compound C([C@H](S)C(=O)N[C@H]1CCS[C@H]2CCC[C@H](N2C1=O)C(=O)O)C1=CC=CC=C1 LVRLSYPNFFBYCZ-VGWMRTNUSA-N 0.000 description 2
- 229950000973 omapatrilat Drugs 0.000 description 2
- 229940012843 omega-3 fatty acid Drugs 0.000 description 2
- 239000006014 omega-3 oil Substances 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- DHHVAGZRUROJKS-UHFFFAOYSA-N phentermine Chemical compound CC(C)(N)CC1=CC=CC=C1 DHHVAGZRUROJKS-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- ACVYVLVWPXVTIT-UHFFFAOYSA-M phosphinate Chemical compound [O-][PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-M 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 2
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 2
- 229960005095 pioglitazone Drugs 0.000 description 2
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000002028 premature Effects 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 230000000135 prohibitive effect Effects 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- RASPWLYDBYZRCR-UHFFFAOYSA-N pyrrolidin-1-ium-2-one;chloride Chemical compound Cl.O=C1CCCN1 RASPWLYDBYZRCR-UHFFFAOYSA-N 0.000 description 2
- 229940107700 pyruvic acid Drugs 0.000 description 2
- 108700040249 racecadotril Proteins 0.000 description 2
- 239000002461 renin inhibitor Substances 0.000 description 2
- 229940086526 renin-inhibitors Drugs 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 229960004586 rosiglitazone Drugs 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229950001780 sampatrilat Drugs 0.000 description 2
- 150000003335 secondary amines Chemical class 0.000 description 2
- 230000003248 secreting effect Effects 0.000 description 2
- 108010060325 semaglutide Proteins 0.000 description 2
- 229950011186 semaglutide Drugs 0.000 description 2
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 2
- 238000002603 single-photon emission computed tomography Methods 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 230000003637 steroidlike Effects 0.000 description 2
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 2
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- GZCRRIHWUXGPOV-UHFFFAOYSA-N terbium atom Chemical compound [Tb] GZCRRIHWUXGPOV-UHFFFAOYSA-N 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 239000003053 toxin Substances 0.000 description 2
- 231100000765 toxin Toxicity 0.000 description 2
- 108700012359 toxins Proteins 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 235000019798 tripotassium phosphate Nutrition 0.000 description 2
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 2
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 2
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- KZJWDPNRJALLNS-VPUBHVLGSA-N (-)-beta-Sitosterol Natural products O[C@@H]1CC=2[C@@](C)([C@@H]3[C@H]([C@H]4[C@@](C)([C@H]([C@H](CC[C@@H](C(C)C)CC)C)CC4)CC3)CC=2)CC1 KZJWDPNRJALLNS-VPUBHVLGSA-N 0.000 description 1
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- SIFCHNIAAPMMKG-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) acetate Chemical compound CC(=O)ON1C(=O)CCC1=O SIFCHNIAAPMMKG-UHFFFAOYSA-N 0.000 description 1
- CSVWWLUMXNHWSU-UHFFFAOYSA-N (22E)-(24xi)-24-ethyl-5alpha-cholest-22-en-3beta-ol Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(C)C=CC(CC)C(C)C)C1(C)CC2 CSVWWLUMXNHWSU-UHFFFAOYSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- DVSZKTAMJJTWFG-SKCDLICFSA-N (2e,4e,6e,8e,10e,12e)-docosa-2,4,6,8,10,12-hexaenoic acid Chemical compound CCCCCCCCC\C=C\C=C\C=C\C=C\C=C\C=C\C(O)=O DVSZKTAMJJTWFG-SKCDLICFSA-N 0.000 description 1
- KVVODNUBDFULSC-XMMPIXPASA-N (2r)-1-[4-[[5-methyl-2-[4-(trifluoromethyl)phenyl]-1,3-oxazol-4-yl]methoxy]phenyl]sulfonyl-2,3-dihydroindole-2-carboxylic acid Chemical compound N=1C(COC=2C=CC(=CC=2)S(=O)(=O)N2C3=CC=CC=C3C[C@@H]2C(O)=O)=C(C)OC=1C1=CC=C(C(F)(F)F)C=C1 KVVODNUBDFULSC-XMMPIXPASA-N 0.000 description 1
- FONCZICQWCUXEB-RUZDIDTESA-N (2r)-2-[4-(9-bromo-2,3-dimethylbenzo[f][1]benzothiol-4-yl)-2,6-dimethylphenoxy]-3-phenylpropanoic acid Chemical compound C([C@@H](OC1=C(C)C=C(C=C1C)C=1C2=CC=CC=C2C(Br)=C2SC(=C(C2=1)C)C)C(O)=O)C1=CC=CC=C1 FONCZICQWCUXEB-RUZDIDTESA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- WOVRTBFSWOVRST-ROUUACIJSA-N (2s)-2-[[(2s)-1-(2-carboxyethylamino)-1-oxo-3-phenylpropan-2-yl]amino]-3-phenylpropanoic acid Chemical compound C([C@@H](C(=O)NCCC(=O)O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CC=CC=C1 WOVRTBFSWOVRST-ROUUACIJSA-N 0.000 description 1
- KAFZOLYKKCWUBI-HPMAGDRPSA-N (2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-3-amino-2-[[(2s)-2-[[(2s)-2-(3-cyclohexylpropanoylamino)-4-methylpentanoyl]amino]-5-methylhexanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]butanediamide Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H](CCC(C)C)C(=O)N[C@@H](CN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC(N)=O)C(N)=O)C(=O)CCC1CCCCC1 KAFZOLYKKCWUBI-HPMAGDRPSA-N 0.000 description 1
- YFDSDRDMDDGDFC-HOQQKOLYSA-N (2s)-2-benzyl-n-[(2s)-1-[[(2s,3r,4s)-1-cyclohexyl-3,4-dihydroxy-6-methylheptan-2-yl]amino]-1-oxo-3-(1,3-thiazol-4-yl)propan-2-yl]-3-(4-methylpiperazin-1-yl)sulfonylpropanamide Chemical compound C([C@@H]([C@@H](O)[C@@H](O)CC(C)C)NC(=O)[C@H](CC=1N=CSC=1)NC(=O)[C@H](CC=1C=CC=CC=1)CS(=O)(=O)N1CCN(C)CC1)C1CCCCC1 YFDSDRDMDDGDFC-HOQQKOLYSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- VUDZSIYXZUYWSC-DBRKOABJSA-N (4r)-1-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4-hydroxy-1,3-diazinan-2-one Chemical compound FC1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N[C@H](O)CC1 VUDZSIYXZUYWSC-DBRKOABJSA-N 0.000 description 1
- NDGUBXOBXSPVHJ-LXVLQKCJSA-N (4r)-4-[(3s,8s,9s,10r,13r,14s,17r)-3-hydroxy-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-17-yl]-n,n-dimethylpentanamide Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@@H](CCC(=O)N(C)C)C)[C@@]1(C)CC2 NDGUBXOBXSPVHJ-LXVLQKCJSA-N 0.000 description 1
- VDSBXXDKCUBMQC-HNGSOEQISA-N (4r,6s)-6-[(e)-2-[2-(4-fluoro-3-methylphenyl)-4,4,6,6-tetramethylcyclohexen-1-yl]ethenyl]-4-hydroxyoxan-2-one Chemical compound C1=C(F)C(C)=CC(C=2CC(C)(C)CC(C)(C)C=2\C=C\[C@H]2OC(=O)C[C@H](O)C2)=C1 VDSBXXDKCUBMQC-HNGSOEQISA-N 0.000 description 1
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 description 1
- UCTWMZQNUQWSLP-VIFPVBQESA-N (R)-adrenaline Chemical compound CNC[C@H](O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-VIFPVBQESA-N 0.000 description 1
- 229930182837 (R)-adrenaline Natural products 0.000 description 1
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 1
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- BBVIDBNAYOIXOE-UHFFFAOYSA-N 1,2,4-oxadiazole Chemical compound C=1N=CON=1 BBVIDBNAYOIXOE-UHFFFAOYSA-N 0.000 description 1
- SILNNFMWIMZVEQ-UHFFFAOYSA-N 1,3-dihydrobenzimidazol-2-one Chemical compound C1=CC=C2NC(O)=NC2=C1 SILNNFMWIMZVEQ-UHFFFAOYSA-N 0.000 description 1
- ZOBPZXTWZATXDG-UHFFFAOYSA-N 1,3-thiazolidine-2,4-dione Chemical compound O=C1CSC(=O)N1 ZOBPZXTWZATXDG-UHFFFAOYSA-N 0.000 description 1
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 1
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 1
- BOVGTQGAOIONJV-BETUJISGSA-N 1-[(3ar,6as)-3,3a,4,5,6,6a-hexahydro-1h-cyclopenta[c]pyrrol-2-yl]-3-(4-methylphenyl)sulfonylurea Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1C[C@H]2CCC[C@H]2C1 BOVGTQGAOIONJV-BETUJISGSA-N 0.000 description 1
- LLJFMFZYVVLQKT-UHFFFAOYSA-N 1-cyclohexyl-3-[4-[2-(7-methoxy-4,4-dimethyl-1,3-dioxo-2-isoquinolinyl)ethyl]phenyl]sulfonylurea Chemical compound C=1C(OC)=CC=C(C(C2=O)(C)C)C=1C(=O)N2CCC(C=C1)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 LLJFMFZYVVLQKT-UHFFFAOYSA-N 0.000 description 1
- MVASGNRQYLESCQ-UHFFFAOYSA-N 1-ethylcyclohexane-1,2-diamine Chemical compound CCC1(N)CCCCC1N MVASGNRQYLESCQ-UHFFFAOYSA-N 0.000 description 1
- JJABOWZNFOCHMN-UHFFFAOYSA-N 1-hydroxycyclopentane-1-carboxylic acid Chemical compound OC(=O)C1(O)CCCC1 JJABOWZNFOCHMN-UHFFFAOYSA-N 0.000 description 1
- GQXURJDNDYACGE-UHFFFAOYSA-N 1-hydroxycyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(O)CC1 GQXURJDNDYACGE-UHFFFAOYSA-N 0.000 description 1
- DIZKLZKLNKQFGB-UHFFFAOYSA-N 1-methylcyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(C)CC1 DIZKLZKLNKQFGB-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- LXFQSRIDYRFTJW-UHFFFAOYSA-N 2,4,6-trimethylbenzenesulfonic acid Chemical compound CC1=CC(C)=C(S(O)(=O)=O)C(C)=C1 LXFQSRIDYRFTJW-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- XMQODGUTLZXUGZ-RPBOFIJWSA-N 2-[(3s)-3-[[1-[(2r)-2-ethoxycarbonyl-4-phenylbutyl]cyclopentanecarbonyl]amino]-2-oxo-4,5-dihydro-3h-1-benzazepin-1-yl]acetic acid Chemical compound C([C@@H](C(=O)OCC)CC1(CCCC1)C(=O)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XMQODGUTLZXUGZ-RPBOFIJWSA-N 0.000 description 1
- HALJGQCLTOMLAW-UHFFFAOYSA-N 2-[1-[4-[6-[3-(trifluoromethyl)anilino]pyridazin-3-yl]phenyl]piperidin-4-yl]acetic acid Chemical compound C1CC(CC(=O)O)CCN1C1=CC=C(C=2N=NC(NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1 HALJGQCLTOMLAW-UHFFFAOYSA-N 0.000 description 1
- OXTKAAXEHMOYSB-UHFFFAOYSA-N 2-[2-(2,6-dichlorophenyl)-3h-benzimidazol-5-yl]-4,5-diphenyl-1,3-oxazole Chemical compound ClC1=CC=CC(Cl)=C1C1=NC2=CC=C(C=3OC(=C(N=3)C=3C=CC=CC=3)C=3C=CC=CC=3)C=C2N1 OXTKAAXEHMOYSB-UHFFFAOYSA-N 0.000 description 1
- XUOUFJZVPYHDRQ-UHFFFAOYSA-N 2-[4-[4-(5-anilino-3-methoxypyridin-2-yl)phenyl]cyclohexyl]acetic acid Chemical compound C=1N=C(C=2C=CC(=CC=2)C2CCC(CC(O)=O)CC2)C(OC)=CC=1NC1=CC=CC=C1 XUOUFJZVPYHDRQ-UHFFFAOYSA-N 0.000 description 1
- WTVIAORFDWMSQX-UHFFFAOYSA-N 2-[4-[4-[4-methyl-6-[[6-(trifluoromethyl)pyridin-3-yl]amino]pyridazin-3-yl]phenyl]cyclohexyl]acetic acid Chemical compound N=1N=C(C=2C=CC(=CC=2)C2CCC(CC(O)=O)CC2)C(C)=CC=1NC1=CC=C(C(F)(F)F)N=C1 WTVIAORFDWMSQX-UHFFFAOYSA-N 0.000 description 1
- AXWHXYMUDZXGAP-UHFFFAOYSA-N 2-[4-[4-[5-(1,3-benzoxazol-2-ylamino)pyridin-2-yl]phenyl]cyclohexyl]acetic acid Chemical compound C1CC(CC(=O)O)CCC1C1=CC=C(C=2N=CC(NC=3OC4=CC=CC=C4N=3)=CC=2)C=C1 AXWHXYMUDZXGAP-UHFFFAOYSA-N 0.000 description 1
- MZEZEGPBQFEKQN-UHFFFAOYSA-N 2-[4-[4-[5-[(1-methylpyrazol-3-yl)amino]pyridin-2-yl]phenyl]cyclohexyl]acetic acid Chemical compound CN1C=CC(NC=2C=NC(=CC=2)C=2C=CC(=CC=2)C2CCC(CC(O)=O)CC2)=N1 MZEZEGPBQFEKQN-UHFFFAOYSA-N 0.000 description 1
- QDIYCQQAUAKRBD-UHFFFAOYSA-N 2-[4-[4-[5-[(5-fluoro-6-methoxypyridin-3-yl)amino]pyridin-2-yl]phenyl]cyclohexyl]acetic acid Chemical compound C1=C(F)C(OC)=NC=C1NC1=CC=C(C=2C=CC(=CC=2)C2CCC(CC(O)=O)CC2)N=C1 QDIYCQQAUAKRBD-UHFFFAOYSA-N 0.000 description 1
- WRRNJBDVTFXSEI-UHFFFAOYSA-N 2-[4-[4-[5-[[6-(trifluoromethyl)pyridin-3-yl]amino]pyrazin-2-yl]phenyl]cyclohexyl]acetic acid Chemical compound C1CC(CC(=O)O)CCC1C1=CC=C(C=2N=CC(NC=3C=NC(=CC=3)C(F)(F)F)=NC=2)C=C1 WRRNJBDVTFXSEI-UHFFFAOYSA-N 0.000 description 1
- GXALXAKNHIROPE-UHFFFAOYSA-N 2-[4-[4-[5-[[6-(trifluoromethyl)pyridin-3-yl]amino]pyridin-2-yl]phenyl]cyclohexyl]acetic acid Chemical compound C1CC(CC(=O)O)CCC1C1=CC=C(C=2N=CC(NC=3C=NC(=CC=3)C(F)(F)F)=CC=2)C=C1 GXALXAKNHIROPE-UHFFFAOYSA-N 0.000 description 1
- FECDSEUAOKXYDM-IRXDYDNUSA-N 2-[6-[[(1S,3S)-3-[(5-cyclopropylpyrimidin-2-yl)amino]cyclopentyl]amino]pyridin-3-yl]pyridazin-3-one Chemical compound C1(CC1)C=1C=NC(=NC=1)N[C@@H]1C[C@H](CC1)NC1=CC=C(C=N1)N1N=CC=CC1=O FECDSEUAOKXYDM-IRXDYDNUSA-N 0.000 description 1
- ODUOJXZPIYUATO-UHFFFAOYSA-N 2-[[2-[(acetylthio)methyl]-1-oxo-3-phenylpropyl]amino]acetic acid (phenylmethyl) ester Chemical compound C=1C=CC=CC=1COC(=O)CNC(=O)C(CSC(=O)C)CC1=CC=CC=C1 ODUOJXZPIYUATO-UHFFFAOYSA-N 0.000 description 1
- WXHLLJAMBQLULT-UHFFFAOYSA-N 2-[[6-[4-(2-hydroxyethyl)piperazin-1-yl]-2-methylpyrimidin-4-yl]amino]-n-(2-methyl-6-sulfanylphenyl)-1,3-thiazole-5-carboxamide;hydrate Chemical compound O.C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1S WXHLLJAMBQLULT-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- QKRMFCXDTFLKKT-UHFFFAOYSA-N 2-hydroxyethanesulfonic acid Chemical compound OCCS(O)(=O)=O.OCCS(O)(=O)=O QKRMFCXDTFLKKT-UHFFFAOYSA-N 0.000 description 1
- ILPUOPPYSQEBNJ-UHFFFAOYSA-N 2-methyl-2-phenoxypropanoic acid Chemical class OC(=O)C(C)(C)OC1=CC=CC=C1 ILPUOPPYSQEBNJ-UHFFFAOYSA-N 0.000 description 1
- FRDZGSBXKJXGNR-UHFFFAOYSA-N 2-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CN(C)C1CCCCC1N FRDZGSBXKJXGNR-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- KLEXDBGYSOIREE-UHFFFAOYSA-N 24xi-n-propylcholesterol Natural products C1C=C2CC(O)CCC2(C)C2C1C1CCC(C(C)CCC(CCC)C(C)C)C1(C)CC2 KLEXDBGYSOIREE-UHFFFAOYSA-N 0.000 description 1
- BCHZICNRHXRCHY-UHFFFAOYSA-N 2h-oxazine Chemical compound N1OC=CC=C1 BCHZICNRHXRCHY-UHFFFAOYSA-N 0.000 description 1
- LNVAFTZCCRGKBM-UHFFFAOYSA-N 3,5-dimethyl-4-[6-[5-[4-(trifluoromethyl)anilino]-1,3,4-oxadiazol-2-yl]-1H-benzimidazol-2-yl]phenol Chemical compound CC1=CC(O)=CC(C)=C1C1=NC2=CC=C(C=3OC(NC=4C=CC(=CC=4)C(F)(F)F)=NN=3)C=C2N1 LNVAFTZCCRGKBM-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- NANIJLBTHOSADK-UHFFFAOYSA-N 3-[(6-fluoro-3-methyl-2-pyridin-3-ylindol-1-yl)methyl]benzonitrile;hydrochloride Chemical compound Cl.C=1C=CC(C#N)=CC=1CN1C2=CC(F)=CC=C2C(C)=C1C1=CC=CN=C1 NANIJLBTHOSADK-UHFFFAOYSA-N 0.000 description 1
- GHLMRBVSXAZWDQ-UHFFFAOYSA-N 3-[4-[6-[5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl]-1h-benzimidazol-2-yl]-3,5-dimethylphenyl]propylphosphonic acid Chemical compound CC1=CC(CCCP(O)(O)=O)=CC(C)=C1C1=NC2=CC=C(C=3OC(=NN=3)C=3C=CC(Cl)=CC=3)C=C2N1 GHLMRBVSXAZWDQ-UHFFFAOYSA-N 0.000 description 1
- JMHFOGLVAUTEJQ-UHFFFAOYSA-N 3-[4-[6-[5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-yl]-1h-benzimidazol-2-yl]-3,5-dimethylphenyl]-2,2-dimethylpropanoic acid Chemical compound C1=CC(OC)=CC=C1C1=NN=C(C=2C=C3N=C(NC3=CC=2)C=2C(=CC(CC(C)(C)C(O)=O)=CC=2C)C)O1 JMHFOGLVAUTEJQ-UHFFFAOYSA-N 0.000 description 1
- MINLAUDEZKJETF-UHFFFAOYSA-N 3-[4-[6-[5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-yl]-1h-benzimidazol-2-yl]-3,5-dimethylphenyl]propanoic acid Chemical compound C1=CC(OC)=CC=C1C1=NN=C(C=2C=C3NC(=NC3=CC=2)C=2C(=CC(CCC(O)=O)=CC=2C)C)O1 MINLAUDEZKJETF-UHFFFAOYSA-N 0.000 description 1
- SYVOVPVHOHPDIA-UHFFFAOYSA-N 3-[[4-[4-[6-[[6-(trifluoromethyl)pyridin-3-yl]amino]pyridazin-3-yl]phenyl]cyclohexyl]methyl]-2h-1,2,4-oxadiazol-5-one Chemical compound C1=NC(C(F)(F)F)=CC=C1NC1=CC=C(C=2C=CC(=CC=2)C2CCC(CC=3NC(=O)ON=3)CC2)N=N1 SYVOVPVHOHPDIA-UHFFFAOYSA-N 0.000 description 1
- ZSHGVMYLGGANKU-UHFFFAOYSA-N 3-hydroxycyclobutane-1-carboxylic acid Chemical compound OC1CC(C(O)=O)C1 ZSHGVMYLGGANKU-UHFFFAOYSA-N 0.000 description 1
- LIBTYOWZULVKBU-UHFFFAOYSA-N 3-methylsulfinyl-1,2,4-triazine Chemical compound CS(=O)C1=NC=CN=N1 LIBTYOWZULVKBU-UHFFFAOYSA-N 0.000 description 1
- NFRVNFNJYHDNJA-UHFFFAOYSA-N 3-methylsulfonyl-1,2,4-triazine Chemical compound CS(=O)(=O)C1=NC=CN=N1 NFRVNFNJYHDNJA-UHFFFAOYSA-N 0.000 description 1
- UIAGMCDKSXEBJQ-IBGZPJMESA-N 3-o-(2-methoxyethyl) 5-o-propan-2-yl (4s)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound COCCOC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)C)[C@H]1C1=CC=CC([N+]([O-])=O)=C1 UIAGMCDKSXEBJQ-IBGZPJMESA-N 0.000 description 1
- QSVDFJNXDKTKTJ-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1h-indene Chemical compound C1CCCC2=C1CC=C2 QSVDFJNXDKTKTJ-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- CLPFFLWZZBQMAO-UHFFFAOYSA-N 4-(5,6,7,8-tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 CLPFFLWZZBQMAO-UHFFFAOYSA-N 0.000 description 1
- SWLAMJPTOQZTAE-UHFFFAOYSA-N 4-[2-[(5-chloro-2-methoxybenzoyl)amino]ethyl]benzoic acid Chemical class COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(C(O)=O)C=C1 SWLAMJPTOQZTAE-UHFFFAOYSA-N 0.000 description 1
- KEWSCDNULKOKTG-UHFFFAOYSA-N 4-cyano-4-ethylsulfanylcarbothioylsulfanylpentanoic acid Chemical compound CCSC(=S)SC(C)(C#N)CCC(O)=O KEWSCDNULKOKTG-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- LTPVSOCPYWDIFU-UHFFFAOYSA-N 4-methoxyphenylethylamine Chemical compound COC1=CC=C(CCN)C=C1 LTPVSOCPYWDIFU-UHFFFAOYSA-N 0.000 description 1
- AWQSAIIDOMEEOD-UHFFFAOYSA-N 5,5-Dimethyl-4-(3-oxobutyl)dihydro-2(3H)-furanone Chemical compound CC(=O)CCC1CC(=O)OC1(C)C AWQSAIIDOMEEOD-UHFFFAOYSA-N 0.000 description 1
- NKOHRVBBQISBSB-UHFFFAOYSA-N 5-[(4-hydroxyphenyl)methyl]-1,3-thiazolidine-2,4-dione Chemical compound C1=CC(O)=CC=C1CC1C(=O)NC(=O)S1 NKOHRVBBQISBSB-UHFFFAOYSA-N 0.000 description 1
- MVDXXGIBARMXSA-PYUWXLGESA-N 5-[[(2r)-2-benzyl-3,4-dihydro-2h-chromen-6-yl]methyl]-1,3-thiazolidine-2,4-dione Chemical compound S1C(=O)NC(=O)C1CC1=CC=C(O[C@@H](CC=2C=CC=CC=2)CC2)C2=C1 MVDXXGIBARMXSA-PYUWXLGESA-N 0.000 description 1
- PCAZCAZVHLGDBA-UHFFFAOYSA-N 5-[[4-(2-indol-1-ylethoxy)phenyl]methyl]-1,3-thiazolidine-2,4-dione Chemical compound S1C(=O)NC(=O)C1CC(C=C1)=CC=C1OCCN1C2=CC=CC=C2C=C1 PCAZCAZVHLGDBA-UHFFFAOYSA-N 0.000 description 1
- WWGVNZWSVJDZLH-UHFFFAOYSA-N 5-[[4-[2-(2,3-dihydroindol-1-yl)ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione Chemical compound S1C(=O)NC(=O)C1CC(C=C1)=CC=C1OCCN1C2=CC=CC=C2CC1 WWGVNZWSVJDZLH-UHFFFAOYSA-N 0.000 description 1
- XPGIBDJXEVAVTO-UHFFFAOYSA-N 5-bromo-2-chloropyrimidine Chemical compound ClC1=NC=C(Br)C=N1 XPGIBDJXEVAVTO-UHFFFAOYSA-N 0.000 description 1
- ITLAZBMGSXRIEF-UHFFFAOYSA-N 5-naphthalen-2-ylsulfonyl-1,3-thiazolidine-2,4-dione Chemical compound S1C(=O)NC(=O)C1S(=O)(=O)C1=CC=C(C=CC=C2)C2=C1 ITLAZBMGSXRIEF-UHFFFAOYSA-N 0.000 description 1
- RZTAMFZIAATZDJ-HNNXBMFYSA-N 5-o-ethyl 3-o-methyl (4s)-4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OC)[C@@H]1C1=CC=CC(Cl)=C1Cl RZTAMFZIAATZDJ-HNNXBMFYSA-N 0.000 description 1
- 125000004938 5-pyridyl group Chemical group N1=CC=CC(=C1)* 0.000 description 1
- GZJLLYHBALOKEX-UHFFFAOYSA-N 6-Ketone, O18-Me-Ussuriedine Natural products CC=CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O GZJLLYHBALOKEX-UHFFFAOYSA-N 0.000 description 1
- RRELDGDKULRRDM-UHFFFAOYSA-N 6-[2-chloro-4-nitro-5-(oxan-4-yloxy)anilino]-3,4-dihydro-1H-quinolin-2-one Chemical compound [O-][N+](=O)c1cc(Cl)c(Nc2ccc3NC(=O)CCc3c2)cc1OC1CCOCC1 RRELDGDKULRRDM-UHFFFAOYSA-N 0.000 description 1
- WOKGJRGMYZPOFQ-UHFFFAOYSA-N 6-[4-[4-(2h-tetrazol-5-ylmethyl)cyclohexyl]phenyl]-n-[6-(trifluoromethyl)pyridin-3-yl]pyridazin-3-amine Chemical compound C1=NC(C(F)(F)F)=CC=C1NC1=CC=C(C=2C=CC(=CC=2)C2CCC(CC3=NNN=N3)CC2)N=N1 WOKGJRGMYZPOFQ-UHFFFAOYSA-N 0.000 description 1
- LFIBZSRMIUIDOI-UHFFFAOYSA-N 6-chloro-1-methyl-2-(5-phenylmethoxypyridin-3-yl)indole Chemical compound C=1C2=CC=C(Cl)C=C2N(C)C=1C(C=1)=CN=CC=1OCC1=CC=CC=C1 LFIBZSRMIUIDOI-UHFFFAOYSA-N 0.000 description 1
- OUPWLAZNJSBVAQ-UHFFFAOYSA-N 6-chloro-1-methyl-2-[5-[(2-pyrrolidin-1-ylethylamino)methyl]pyridin-3-yl]indole-3-carbonitrile Chemical compound N#CC=1C2=CC=C(Cl)C=C2N(C)C=1C(C=1)=CN=CC=1CNCCN1CCCC1 OUPWLAZNJSBVAQ-UHFFFAOYSA-N 0.000 description 1
- MLQBDXALRJCDJF-UHFFFAOYSA-N 6-chloro-1-methyl-2-[5-[(4-methylsulfonylpiperazin-1-yl)methyl]pyridin-3-yl]indole-3-carbonitrile Chemical compound N#CC=1C2=CC=C(Cl)C=C2N(C)C=1C(C=1)=CN=CC=1CN1CCN(S(C)(=O)=O)CC1 MLQBDXALRJCDJF-UHFFFAOYSA-N 0.000 description 1
- IRNVFWLJSRNETN-UHFFFAOYSA-N 6-chloro-1-methyl-2-[5-[[(1-methylpiperidin-4-yl)amino]methyl]pyridin-3-yl]indole-3-carbonitrile Chemical compound C1CN(C)CCC1NCC1=CN=CC(C=2N(C3=CC(Cl)=CC=C3C=2C#N)C)=C1 IRNVFWLJSRNETN-UHFFFAOYSA-N 0.000 description 1
- YTHMOBMZVVFNBE-UHFFFAOYSA-N 6-fluoropyridin-3-amine Chemical compound NC1=CC=C(F)N=C1 YTHMOBMZVVFNBE-UHFFFAOYSA-N 0.000 description 1
- 101150071783 APOA1 gene Proteins 0.000 description 1
- 108010006533 ATP-Binding Cassette Transporters Proteins 0.000 description 1
- 102000005416 ATP-Binding Cassette Transporters Human genes 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000010444 Acidosis Diseases 0.000 description 1
- 102100027840 Acyl-CoA wax alcohol acyltransferase 1 Human genes 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- FHHHOYXPRDYHEZ-COXVUDFISA-N Alacepril Chemical compound CC(=O)SC[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 FHHHOYXPRDYHEZ-COXVUDFISA-N 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 239000012099 Alexa Fluor family Substances 0.000 description 1
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 102400000344 Angiotensin-1 Human genes 0.000 description 1
- 101800000734 Angiotensin-1 Proteins 0.000 description 1
- 102400000345 Angiotensin-2 Human genes 0.000 description 1
- 101800000733 Angiotensin-2 Proteins 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 200000000007 Arterial disease Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 208000031729 Bacteremia Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 206010004053 Bacterial toxaemia Diseases 0.000 description 1
- XPCFTKFZXHTYIP-PMACEKPBSA-N Benazepril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XPCFTKFZXHTYIP-PMACEKPBSA-N 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000002080 C09CA02 - Eprosartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- 239000002081 C09CA05 - Tasosartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- 125000000172 C5-C10 aryl group Chemical group 0.000 description 1
- 101150041968 CDC13 gene Proteins 0.000 description 1
- 108090000312 Calcium Channels Proteins 0.000 description 1
- 102000003922 Calcium Channels Human genes 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N Camphoric acid Natural products CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- IFYLTXNCFVRALQ-OALUTQOASA-N Ceronapril Chemical compound O([C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)P(O)(=O)CCCCC1=CC=CC=C1 IFYLTXNCFVRALQ-OALUTQOASA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RKWGIWYCVPQPMF-UHFFFAOYSA-N Chloropropamide Chemical compound CCCNC(=O)NS(=O)(=O)C1=CC=C(Cl)C=C1 RKWGIWYCVPQPMF-UHFFFAOYSA-N 0.000 description 1
- 229940122502 Cholesterol absorption inhibitor Drugs 0.000 description 1
- 102100037637 Cholesteryl ester transfer protein Human genes 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- KPSRODZRAIWAKH-JTQLQIEISA-N Ciprofibrate Natural products C1=CC(OC(C)(C)C(O)=O)=CC=C1[C@H]1C(Cl)(Cl)C1 KPSRODZRAIWAKH-JTQLQIEISA-N 0.000 description 1
- 206010009192 Circulatory collapse Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- LPZCCMIISIBREI-MTFRKTCUSA-N Citrostadienol Natural products CC=C(CC[C@@H](C)[C@H]1CC[C@H]2C3=CC[C@H]4[C@H](C)[C@@H](O)CC[C@]4(C)[C@H]3CC[C@]12C)C(C)C LPZCCMIISIBREI-MTFRKTCUSA-N 0.000 description 1
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- VGMFHMLQOYWYHN-UHFFFAOYSA-N Compactin Natural products OCC1OC(OC2C(O)C(O)C(CO)OC2Oc3cc(O)c4C(=O)C(=COc4c3)c5ccc(O)c(O)c5)C(O)C(O)C1O VGMFHMLQOYWYHN-UHFFFAOYSA-N 0.000 description 1
- 101000785223 Crocosmia x crocosmiiflora Myricetin 3-O-glucosyl 1,2-rhamnoside 6'-O-caffeoyltransferase AT1 Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- ARVGMISWLZPBCH-UHFFFAOYSA-N Dehydro-beta-sitosterol Natural products C1C(O)CCC2(C)C(CCC3(C(C(C)CCC(CC)C(C)C)CCC33)C)C3=CC=C21 ARVGMISWLZPBCH-UHFFFAOYSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 108010067770 Endopeptidase K Proteins 0.000 description 1
- 208000037487 Endotoxemia Diseases 0.000 description 1
- 102000010911 Enzyme Precursors Human genes 0.000 description 1
- 108010062466 Enzyme Precursors Proteins 0.000 description 1
- 229920000064 Ethyl eicosapentaenoic acid Polymers 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000400611 Eucalyptus deanei Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102000009123 Fibrin Human genes 0.000 description 1
- 108010073385 Fibrin Proteins 0.000 description 1
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 1
- 206010017523 Fungaemia Diseases 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 102000007446 Glucagon-Like Peptide-1 Receptor Human genes 0.000 description 1
- 108010086246 Glucagon-Like Peptide-1 Receptor Proteins 0.000 description 1
- 229940089838 Glucagon-like peptide 1 receptor agonist Drugs 0.000 description 1
- 101800004266 Glucagon-like peptide 1(7-37) Proteins 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- UXDDRFCJKNROTO-UHFFFAOYSA-N Glycerol 1,2-diacetate Chemical compound CC(=O)OCC(CO)OC(C)=O UXDDRFCJKNROTO-UHFFFAOYSA-N 0.000 description 1
- 108010046163 Glycogen Phosphorylase Proteins 0.000 description 1
- 102000007390 Glycogen Phosphorylase Human genes 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 102100040896 Growth/differentiation factor 15 Human genes 0.000 description 1
- 101150103290 Hcar2 gene Proteins 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 101000698136 Homo sapiens Acyl-CoA wax alcohol acyltransferase 1 Proteins 0.000 description 1
- 101000880514 Homo sapiens Cholesteryl ester transfer protein Proteins 0.000 description 1
- 101000893549 Homo sapiens Growth/differentiation factor 15 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 102000004286 Hydroxymethylglutaryl CoA Reductases Human genes 0.000 description 1
- 108090000895 Hydroxymethylglutaryl CoA Reductases Proteins 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000003746 Insulin Receptor Human genes 0.000 description 1
- 108010001127 Insulin Receptor Proteins 0.000 description 1
- 229940122199 Insulin secretagogue Drugs 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 238000008214 LDL Cholesterol Methods 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 208000000501 Lipidoses Diseases 0.000 description 1
- 206010024585 Lipidosis Diseases 0.000 description 1
- 108010033266 Lipoprotein(a) Proteins 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 206010027417 Metabolic acidosis Diseases 0.000 description 1
- 102000003979 Mineralocorticoid Receptors Human genes 0.000 description 1
- 108090000375 Mineralocorticoid Receptors Proteins 0.000 description 1
- UWWDHYUMIORJTA-HSQYWUDLSA-N Moexipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC(OC)=C(OC)C=C2C1)C(O)=O)CC1=CC=CC=C1 UWWDHYUMIORJTA-HSQYWUDLSA-N 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 208000034486 Multi-organ failure Diseases 0.000 description 1
- 208000010718 Multiple Organ Failure Diseases 0.000 description 1
- IRLWJILLXJGJTD-UHFFFAOYSA-N Muraglitazar Chemical compound C1=CC(OC)=CC=C1OC(=O)N(CC(O)=O)CC(C=C1)=CC=C1OCCC1=C(C)OC(C=2C=CC=CC=2)=N1 IRLWJILLXJGJTD-UHFFFAOYSA-N 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 101100135848 Mus musculus Pcsk9 gene Proteins 0.000 description 1
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 1
- TZYWCYJVHRLUCT-VABKMULXSA-N N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal Chemical compound CC(C)C[C@@H](C=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)OCC1=CC=CC=C1 TZYWCYJVHRLUCT-VABKMULXSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- ZBBHBTPTTSWHBA-UHFFFAOYSA-N Nicardipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCCN(C)CC=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1 ZBBHBTPTTSWHBA-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 101150094724 PCSK9 gene Proteins 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 241001504519 Papio ursinus Species 0.000 description 1
- 208000009182 Parasitemia Diseases 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 101150056304 Pltp gene Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 108010085867 SCH 32615 Proteins 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 239000005478 Saprisartan Substances 0.000 description 1
- DUEWVPTZCSAMNB-UHFFFAOYSA-N Saprisartan Chemical compound NC(=O)C=1N(CC=2C=C3C(Br)=C(OC3=CC=2)C=2C(=CC=CC=2)NS(=O)(=O)C(F)(F)F)C(CC)=NC=1C1CC1 DUEWVPTZCSAMNB-UHFFFAOYSA-N 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 102100037202 Sodium/myo-inositol cotransporter 2 Human genes 0.000 description 1
- 101710090560 Sodium/myo-inositol cotransporter 2 Proteins 0.000 description 1
- 102000007451 Steroid Receptors Human genes 0.000 description 1
- 108010085012 Steroid Receptors Proteins 0.000 description 1
- 229930182558 Sterol Natural products 0.000 description 1
- 108090000787 Subtilisin Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 102000018692 Sulfonylurea Receptors Human genes 0.000 description 1
- 108010091821 Sulfonylurea Receptors Proteins 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- NGBFQHCMQULJNZ-UHFFFAOYSA-N Torsemide Chemical compound CC(C)NC(=O)NS(=O)(=O)C1=CN=CC=C1NC1=CC=CC(C)=C1 NGBFQHCMQULJNZ-UHFFFAOYSA-N 0.000 description 1
- 208000013222 Toxemia Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 206010065341 Ventricular tachyarrhythmia Diseases 0.000 description 1
- 206010058874 Viraemia Diseases 0.000 description 1
- 108090000013 Voltage-Gated Potassium Channels Proteins 0.000 description 1
- 102000003734 Voltage-Gated Potassium Channels Human genes 0.000 description 1
- VXDSGTRNDFHIJB-QQPOVDNESA-N [(1s,4ar)-8-[2-[(2r,4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1CCC[C@@H](C21)OC(=O)[C@@H](C)CC)=CC(C)C2CC[C@@H]1C[C@@H](O)CC(=O)O1 VXDSGTRNDFHIJB-QQPOVDNESA-N 0.000 description 1
- HGRTYWLJJPLSFG-UHFFFAOYSA-N [5-(6-chloro-3-cyano-1-methylindol-2-yl)pyridin-3-yl] pyrrolidine-1-sulfonate Chemical compound N#CC=1C2=CC=C(Cl)C=C2N(C)C=1C(C=1)=CN=CC=1OS(=O)(=O)N1CCCC1 HGRTYWLJJPLSFG-UHFFFAOYSA-N 0.000 description 1
- NYMQJWVULPQXBK-UHFFFAOYSA-N [[amino(methylsulfanyl)methylidene]amino]azanium;iodide Chemical compound [I-].CSC(N)=[NH+]N NYMQJWVULPQXBK-UHFFFAOYSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- GOEMGAFJFRBGGG-UHFFFAOYSA-N acebutolol Chemical compound CCCC(=O)NC1=CC=C(OCC(O)CNC(C)C)C(C(C)=O)=C1 GOEMGAFJFRBGGG-UHFFFAOYSA-N 0.000 description 1
- 229960002122 acebutolol Drugs 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 229960001466 acetohexamide Drugs 0.000 description 1
- VGZSUPCWNCWDAN-UHFFFAOYSA-N acetohexamide Chemical compound C1=CC(C(=O)C)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 VGZSUPCWNCWDAN-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 229960000250 adipic acid Drugs 0.000 description 1
- 230000001800 adrenalinergic effect Effects 0.000 description 1
- 239000000674 adrenergic antagonist Substances 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229950007884 alacepril Drugs 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 239000003888 alpha glucosidase inhibitor Substances 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 229940000806 amaryl Drugs 0.000 description 1
- 150000001408 amides Chemical group 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- ORWYRWWVDCYOMK-HBZPZAIKSA-N angiotensin I Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C1=CC=C(O)C=C1 ORWYRWWVDCYOMK-HBZPZAIKSA-N 0.000 description 1
- 229950006323 angiotensin ii Drugs 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- NETXMUIMUZJUTB-UHFFFAOYSA-N apabetalone Chemical group C=1C(OC)=CC(OC)=C(C(N2)=O)C=1N=C2C1=CC(C)=C(OCCO)C(C)=C1 NETXMUIMUZJUTB-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 208000028922 artery disease Diseases 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 229960002274 atenolol Drugs 0.000 description 1
- 230000003143 atherosclerotic effect Effects 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 229960004530 benazepril Drugs 0.000 description 1
- 229960004067 benazeprilat Drugs 0.000 description 1
- MADRIHWFJGRSBP-ROUUACIJSA-N benazeprilat Chemical compound C([C@H](N[C@H]1CCC2=CC=CC=C2N(C1=O)CC(=O)O)C(O)=O)CC1=CC=CC=C1 MADRIHWFJGRSBP-ROUUACIJSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 125000005872 benzooxazolyl group Chemical group 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- ODUOJXZPIYUATO-IBGZPJMESA-N benzyl 2-[[(2r)-2-(acetylsulfanylmethyl)-3-phenylpropanoyl]amino]acetate Chemical compound C([C@@H](CSC(=O)C)C(=O)NCC(=O)OCC=1C=CC=CC=1)C1=CC=CC=C1 ODUOJXZPIYUATO-IBGZPJMESA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 1
- UIEATEWHFDRYRU-UHFFFAOYSA-N bepridil Chemical compound C1CCCN1C(COCC(C)C)CN(C=1C=CC=CC=1)CC1=CC=CC=C1 UIEATEWHFDRYRU-UHFFFAOYSA-N 0.000 description 1
- 229960003665 bepridil Drugs 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- MJVXAPPOFPTTCA-UHFFFAOYSA-N beta-Sistosterol Natural products CCC(CCC(C)C1CCC2C3CC=C4C(C)C(O)CCC4(C)C3CCC12C)C(C)C MJVXAPPOFPTTCA-UHFFFAOYSA-N 0.000 description 1
- LGJMUZUPVCAVPU-UHFFFAOYSA-N beta-Sitostanol Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(C)CCC(CC)C(C)C)C1(C)CC2 LGJMUZUPVCAVPU-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- NJKOMDUNNDKEAI-UHFFFAOYSA-N beta-sitosterol Natural products CCC(CCC(C)C1CCC2(C)C3CC=C4CC(O)CCC4C3CCC12C)C(C)C NJKOMDUNNDKEAI-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960004324 betaxolol Drugs 0.000 description 1
- CHDPSNLJFOQTRK-UHFFFAOYSA-N betaxolol hydrochloride Chemical compound [Cl-].C1=CC(OCC(O)C[NH2+]C(C)C)=CC=C1CCOCC1CC1 CHDPSNLJFOQTRK-UHFFFAOYSA-N 0.000 description 1
- 229960000516 bezafibrate Drugs 0.000 description 1
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 1
- 150000005347 biaryls Chemical group 0.000 description 1
- JBFDZEJAJZJORO-UHFFFAOYSA-N bicyclo[4.1.0]hept-3-ene Chemical compound C1C=CCC2CC21 JBFDZEJAJZJORO-UHFFFAOYSA-N 0.000 description 1
- DCRRIOWFXXDTHV-UHFFFAOYSA-N bicyclo[4.2.0]oct-3-ene Chemical compound C1C=CCC2CCC21 DCRRIOWFXXDTHV-UHFFFAOYSA-N 0.000 description 1
- RPZUBXWEQBPUJR-UHFFFAOYSA-N bicyclo[4.2.0]octane Chemical compound C1CCCC2CCC21 RPZUBXWEQBPUJR-UHFFFAOYSA-N 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- LSPHULWDVZXLIL-QUBYGPBYSA-N camphoric acid Chemical compound CC1(C)[C@H](C(O)=O)CC[C@]1(C)C(O)=O LSPHULWDVZXLIL-QUBYGPBYSA-N 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- ZTWZVMIYIIVABD-OEMFJLHTSA-N candoxatril Chemical compound C([C@@H](COCCOC)C(=O)OC=1C=C2CCCC2=CC=1)C1(C(=O)N[C@@H]2CC[C@@H](CC2)C(O)=O)CCCC1 ZTWZVMIYIIVABD-OEMFJLHTSA-N 0.000 description 1
- 229950004548 candoxatril Drugs 0.000 description 1
- ACZWIDANLCXHBM-HRCADAONSA-N candoxatrilat Chemical compound N([C@@H]1CC[C@@H](CC1)C(O)=O)C(=O)C1(C[C@@H](COCCOC)C(O)=O)CCCC1 ACZWIDANLCXHBM-HRCADAONSA-N 0.000 description 1
- 229950001305 candoxatrilat Drugs 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 125000004452 carbocyclyl group Chemical group 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 229960003362 carbutamide Drugs 0.000 description 1
- VDTNNGKXZGSZIP-UHFFFAOYSA-N carbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(N)C=C1 VDTNNGKXZGSZIP-UHFFFAOYSA-N 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229960005110 cerivastatin Drugs 0.000 description 1
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 description 1
- GPUADMRJQVPIAS-QCVDVZFFSA-M cerivastatin sodium Chemical compound [Na+].COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 GPUADMRJQVPIAS-QCVDVZFFSA-M 0.000 description 1
- 229950005749 ceronapril Drugs 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229960001761 chlorpropamide Drugs 0.000 description 1
- 150000001840 cholesterol esters Chemical class 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000012085 chronic inflammatory response Effects 0.000 description 1
- YZFWTZACSRHJQD-UHFFFAOYSA-N ciglitazone Chemical compound C=1C=C(CC2C(NC(=O)S2)=O)C=CC=1OCC1(C)CCCCC1 YZFWTZACSRHJQD-UHFFFAOYSA-N 0.000 description 1
- 229950009226 ciglitazone Drugs 0.000 description 1
- HHHKFGXWKKUNCY-FHWLQOOXSA-N cilazapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N2[C@@H](CCCN2CCC1)C(O)=O)=O)CC1=CC=CC=C1 HHHKFGXWKKUNCY-FHWLQOOXSA-N 0.000 description 1
- 229960005025 cilazapril Drugs 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 229960002174 ciprofibrate Drugs 0.000 description 1
- KPSRODZRAIWAKH-UHFFFAOYSA-N ciprofibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1C1C(Cl)(Cl)C1 KPSRODZRAIWAKH-UHFFFAOYSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229960001214 clofibrate Drugs 0.000 description 1
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 229940126523 co-drug Drugs 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 1
- 239000005516 coenzyme A Substances 0.000 description 1
- 229940093530 coenzyme a Drugs 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 1
- 210000003618 cortical neuron Anatomy 0.000 description 1
- 150000001925 cycloalkenes Chemical group 0.000 description 1
- TXWOGHSRPAYOML-UHFFFAOYSA-N cyclobutanecarboxylic acid Chemical compound OC(=O)C1CCC1 TXWOGHSRPAYOML-UHFFFAOYSA-N 0.000 description 1
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 229950010776 daglutril Drugs 0.000 description 1
- 229950003040 dalvastatin Drugs 0.000 description 1
- 229950006689 darglitazone Drugs 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-M decanoate Chemical compound CCCCCCCCCC([O-])=O GHVNFZFCNZKVNT-UHFFFAOYSA-M 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000011903 deuterated solvents Substances 0.000 description 1
- 108700010883 dexecadotril Proteins 0.000 description 1
- 229950008996 dexecadotril Drugs 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- PBWZKZYHONABLN-UHFFFAOYSA-N difluoroacetic acid Chemical compound OC(=O)C(F)F PBWZKZYHONABLN-UHFFFAOYSA-N 0.000 description 1
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- VXDSGTRNDFHIJB-UHFFFAOYSA-N dihydrocompactin Natural products C12C(OC(=O)C(C)CC)CCCC2C=CC(C)C1CCC1CC(O)CC(=O)O1 VXDSGTRNDFHIJB-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 108010083220 ditekiren Proteins 0.000 description 1
- 229950010513 ditekiren Drugs 0.000 description 1
- 235000020669 docosahexaenoic acid Nutrition 0.000 description 1
- KAUVQQXNCKESLC-UHFFFAOYSA-N docosahexaenoic acid (DHA) Natural products COC(=O)C(C)NOCC1=CC=CC=C1 KAUVQQXNCKESLC-UHFFFAOYSA-N 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- ODUOJXZPIYUATO-LJQANCHMSA-N ecadotril Chemical compound C([C@H](CSC(=O)C)C(=O)NCC(=O)OCC=1C=CC=CC=1)C1=CC=CC=C1 ODUOJXZPIYUATO-LJQANCHMSA-N 0.000 description 1
- 229950001184 ecadotril Drugs 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 150000002066 eicosanoids Chemical class 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 229950002375 englitazone Drugs 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 210000001842 enterocyte Anatomy 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229960005139 epinephrine Drugs 0.000 description 1
- 229960004563 eprosartan Drugs 0.000 description 1
- OROAFUQRIXKEMV-LDADJPATSA-N eprosartan Chemical compound C=1C=C(C(O)=O)C=CC=1CN1C(CCCC)=NC=C1\C=C(C(O)=O)/CC1=CC=CS1 OROAFUQRIXKEMV-LDADJPATSA-N 0.000 description 1
- 229950011248 eprotirome Drugs 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- AVOLMBLBETYQHX-UHFFFAOYSA-N etacrynic acid Chemical compound CCC(=C)C(=O)C1=CC=C(OCC(O)=O)C(Cl)=C1Cl AVOLMBLBETYQHX-UHFFFAOYSA-N 0.000 description 1
- 229960003199 etacrynic acid Drugs 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethanedisulfonate group Chemical group C(CS(=O)(=O)[O-])S(=O)(=O)[O-] AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- RSOJSFYJLQADDM-UHFFFAOYSA-N ethanesulfonamide Chemical compound [CH2]CS(N)(=O)=O RSOJSFYJLQADDM-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 229940052303 ethers for general anesthesia Drugs 0.000 description 1
- ZVQXPUMRSJGLSF-MSOLQXFVSA-N ethyl (2s)-2-[[(2s)-2-(acetylsulfanylmethyl)-3-(2-methylphenyl)propanoyl]amino]-4-methylsulfanylbutanoate Chemical compound CCOC(=O)[C@H](CCSC)NC(=O)[C@@H](CSC(C)=O)CC1=CC=CC=C1C ZVQXPUMRSJGLSF-MSOLQXFVSA-N 0.000 description 1
- NIDZUMSLERGAON-UHFFFAOYSA-N ethyl 2-(methylamino)acetate;hydron;chloride Chemical compound Cl.CCOC(=O)CNC NIDZUMSLERGAON-UHFFFAOYSA-N 0.000 description 1
- OMSSNKJFNJWYFD-UHFFFAOYSA-N ethyl 5-(3-cyano-1-methylindol-2-yl)pyridine-3-carboxylate Chemical compound CCOC(=O)C1=CN=CC(C=2N(C3=CC=CC=C3C=2C#N)C)=C1 OMSSNKJFNJWYFD-UHFFFAOYSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 229950011548 fadrozole Drugs 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 150000002185 fatty acyl-CoAs Chemical class 0.000 description 1
- 229960003580 felodipine Drugs 0.000 description 1
- 229950003499 fibrin Drugs 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229940013317 fish oils Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 125000004615 furo[2,3-b]pyridinyl group Chemical group O1C(=CC=2C1=NC=CC2)* 0.000 description 1
- YRTCKZIKGWZNCU-UHFFFAOYSA-N furo[3,2-b]pyridine Chemical compound C1=CC=C2OC=CC2=N1 YRTCKZIKGWZNCU-UHFFFAOYSA-N 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- YRSVDSQRGBYVIY-GJZGRUSLSA-N gemopatrilat Chemical compound O=C1N(CC(O)=O)C(C)(C)CCC[C@@H]1NC(=O)[C@@H](S)CC1=CC=CC=C1 YRSVDSQRGBYVIY-GJZGRUSLSA-N 0.000 description 1
- 229950006480 gemopatrilat Drugs 0.000 description 1
- 238000012224 gene deletion Methods 0.000 description 1
- 229960000346 gliclazide Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical group O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 229960003468 gliquidone Drugs 0.000 description 1
- 229960003236 glisoxepide Drugs 0.000 description 1
- ZKUDBRCEOBOWLF-UHFFFAOYSA-N glisoxepide Chemical compound O1C(C)=CC(C(=O)NCCC=2C=CC(=CC=2)S(=O)(=O)NC(=O)NN2CCCCCC2)=N1 ZKUDBRCEOBOWLF-UHFFFAOYSA-N 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 230000014101 glucose homeostasis Effects 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N glutaric acid Chemical compound OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 229950011569 glybuthiazol Drugs 0.000 description 1
- DVQVBLBKEXITIK-UHFFFAOYSA-N glybuthiazol Chemical compound S1C(C(C)(C)C)=NN=C1NS(=O)(=O)C1=CC=C(N)C=C1 DVQVBLBKEXITIK-UHFFFAOYSA-N 0.000 description 1
- NFRPNQDSKJJQGV-UHFFFAOYSA-N glyhexamide Chemical compound C=1C=C2CCCC2=CC=1S(=O)(=O)NC(=O)NC1CCCCC1 NFRPNQDSKJJQGV-UHFFFAOYSA-N 0.000 description 1
- 229950008290 glyhexamide Drugs 0.000 description 1
- QFWPJPIVLCBXFJ-UHFFFAOYSA-N glymidine Chemical compound N1=CC(OCCOC)=CN=C1NS(=O)(=O)C1=CC=CC=C1 QFWPJPIVLCBXFJ-UHFFFAOYSA-N 0.000 description 1
- 229960004440 glymidine Drugs 0.000 description 1
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 1
- RHQSNARBXHRBNP-UHFFFAOYSA-N glypinamide Chemical compound C1=CC(Cl)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 RHQSNARBXHRBNP-UHFFFAOYSA-N 0.000 description 1
- 229950009188 glypinamide Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000007407 health benefit Effects 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 125000004404 heteroalkyl group Chemical group 0.000 description 1
- 125000004475 heteroaralkyl group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical group C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 235000009200 high fat diet Nutrition 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 208000020346 hyperlipoproteinemia Diseases 0.000 description 1
- 230000002218 hypoglycaemic effect Effects 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 229960001195 imidapril Drugs 0.000 description 1
- KLZWOWYOHUKJIG-BPUTZDHNSA-N imidapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1C(N(C)C[C@H]1C(O)=O)=O)CC1=CC=CC=C1 KLZWOWYOHUKJIG-BPUTZDHNSA-N 0.000 description 1
- UVNXNSUKKOLFBM-UHFFFAOYSA-N imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=CSC2=NC=CN21 UVNXNSUKKOLFBM-UHFFFAOYSA-N 0.000 description 1
- 125000000336 imidazol-5-yl group Chemical group [H]N1C([H])=NC([H])=C1[*] 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000003914 insulin secretion Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000002608 intravascular ultrasound Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-M lactobionate Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-M 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229910052747 lanthanoid Inorganic materials 0.000 description 1
- 150000002602 lanthanoids Chemical class 0.000 description 1
- WHXMKTBCFHIYNQ-SECBINFHSA-N levosimendan Chemical compound C[C@@H]1CC(=O)NN=C1C1=CC=C(NN=C(C#N)C#N)C=C1 WHXMKTBCFHIYNQ-SECBINFHSA-N 0.000 description 1
- 229960000692 levosimendan Drugs 0.000 description 1
- JFOZKMSJYSPYLN-QHCPKHFHSA-N lifitegrast Chemical compound CS(=O)(=O)C1=CC=CC(C[C@H](NC(=O)C=2C(=C3CCN(CC3=CC=2Cl)C(=O)C=2C=C3OC=CC3=CC=2)Cl)C(O)=O)=C1 JFOZKMSJYSPYLN-QHCPKHFHSA-N 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 108010053156 lipid transfer protein Proteins 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- RLAWWYSOJDYHDC-BZSNNMDCSA-N lisinopril Chemical compound C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 RLAWWYSOJDYHDC-BZSNNMDCSA-N 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 235000004213 low-fat Nutrition 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 230000006674 lysosomal degradation Effects 0.000 description 1
- FRIJBUGBVQZNTB-UHFFFAOYSA-M magnesium;ethane;bromide Chemical compound [Mg+2].[Br-].[CH2-]C FRIJBUGBVQZNTB-UHFFFAOYSA-M 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229950004994 meglitinide Drugs 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- AWIJRPNMLHPLNC-UHFFFAOYSA-N methanethioic s-acid Chemical compound SC=O AWIJRPNMLHPLNC-UHFFFAOYSA-N 0.000 description 1
- NZTCVGHPDWAALP-UHFFFAOYSA-N methyl 2,2-dimethoxyacetate Chemical compound COC(OC)C(=O)OC NZTCVGHPDWAALP-UHFFFAOYSA-N 0.000 description 1
- VKQFCGNPDRICFG-UHFFFAOYSA-N methyl 2-methylpropyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCC(C)C)C1C1=CC=CC=C1[N+]([O-])=O VKQFCGNPDRICFG-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 229950009116 mevastatin Drugs 0.000 description 1
- 238000004452 microanalysis Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 229960005170 moexipril Drugs 0.000 description 1
- STUHQDIOZQUPGP-UHFFFAOYSA-N morpholin-4-ium-4-carboxylate Chemical compound OC(=O)N1CCOCC1 STUHQDIOZQUPGP-UHFFFAOYSA-N 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 208000029744 multiple organ dysfunction syndrome Diseases 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- UQEIFYRRSNJVDO-UHFFFAOYSA-N n,n-dibenzyl-2-phenylethanamine Chemical compound C=1C=CC=CC=1CN(CC=1C=CC=CC=1)CCC1=CC=CC=C1 UQEIFYRRSNJVDO-UHFFFAOYSA-N 0.000 description 1
- MITVGNGZNBCXRB-UHFFFAOYSA-N n-[1-[5-(4-cyano-3-methoxyphenyl)pyridin-3-yl]ethyl]ethanesulfonamide Chemical compound CCS(=O)(=O)NC(C)C1=CN=CC(C=2C=C(OC)C(C#N)=CC=2)=C1 MITVGNGZNBCXRB-UHFFFAOYSA-N 0.000 description 1
- FTRMOJIRMFXZJV-UHFFFAOYSA-N n-[4-[(2,4-dioxo-1,3-thiazolidin-5-yl)methyl]phenyl]-1-phenylcyclopropane-1-carboxamide Chemical compound C1CC1(C=1C=CC=CC=1)C(=O)NC(C=C1)=CC=C1CC1SC(=O)NC1=O FTRMOJIRMFXZJV-UHFFFAOYSA-N 0.000 description 1
- OTAHELNWDDHTNJ-UHFFFAOYSA-N n-[5-(3-chloro-4-cyanophenyl)pyridin-3-yl]-1-phenylmethanesulfonamide Chemical compound C1=C(C#N)C(Cl)=CC(C=2C=C(NS(=O)(=O)CC=3C=CC=CC=3)C=NC=2)=C1 OTAHELNWDDHTNJ-UHFFFAOYSA-N 0.000 description 1
- SFHVDXYBDSETOH-UHFFFAOYSA-N n-[5-(3-chloro-4-cyanophenyl)pyridin-3-yl]-4-(trifluoromethyl)benzenesulfonamide Chemical compound C1=CC(C(F)(F)F)=CC=C1S(=O)(=O)NC1=CN=CC(C=2C=C(Cl)C(C#N)=CC=2)=C1 SFHVDXYBDSETOH-UHFFFAOYSA-N 0.000 description 1
- PWWXYPDWSQEUMH-UHFFFAOYSA-N n-[[5-(3-chloro-4-cyanophenyl)pyridin-3-yl]-cyclopropylmethyl]-n-ethylethanesulfonamide Chemical compound C=1N=CC(C=2C=C(Cl)C(C#N)=CC=2)=CC=1C(N(CC)S(=O)(=O)CC)C1CC1 PWWXYPDWSQEUMH-UHFFFAOYSA-N 0.000 description 1
- KCHHHGRFMSWSFF-UHFFFAOYSA-N n-[[5-(3-chloro-4-cyanophenyl)pyridin-3-yl]-cyclopropylmethyl]ethanesulfonamide Chemical compound C=1N=CC(C=2C=C(Cl)C(C#N)=CC=2)=CC=1C(NS(=O)(=O)CC)C1CC1 KCHHHGRFMSWSFF-UHFFFAOYSA-N 0.000 description 1
- GMQJALWDENUKHB-UHFFFAOYSA-N n-[[5-(6-chloro-1-methylindol-2-yl)pyridin-3-yl]methyl]ethanesulfonamide Chemical compound CCS(=O)(=O)NCC1=CN=CC(C=2N(C3=CC(Cl)=CC=C3C=2)C)=C1 GMQJALWDENUKHB-UHFFFAOYSA-N 0.000 description 1
- UBIOLVNQWDPYRD-UHFFFAOYSA-N n-[[5-(6-chloro-1-methylpyrrolo[2,3-b]pyridin-2-yl)pyridin-3-yl]methyl]ethanesulfonamide Chemical compound CCS(=O)(=O)NCC1=CN=CC(C=2N(C3=NC(Cl)=CC=C3C=2)C)=C1 UBIOLVNQWDPYRD-UHFFFAOYSA-N 0.000 description 1
- WAVHJLJLOKAMKK-UHFFFAOYSA-N n-[cyclopropyl-[5-(1h-indol-5-yl)pyridin-3-yl]methyl]ethanesulfonamide Chemical compound C=1N=CC(C=2C=C3C=CNC3=CC=2)=CC=1C(NS(=O)(=O)CC)C1CC1 WAVHJLJLOKAMKK-UHFFFAOYSA-N 0.000 description 1
- 125000004370 n-butenyl group Chemical group [H]\C([H])=C(/[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- YOVHPXBQZFTTFM-UHFFFAOYSA-N n-methyl-n-[[5-(1-methylindol-2-yl)pyridin-3-yl]methyl]methanesulfonamide Chemical compound CS(=O)(=O)N(C)CC1=CN=CC(C=2N(C3=CC=CC=C3C=2)C)=C1 YOVHPXBQZFTTFM-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- 238000004848 nephelometry Methods 0.000 description 1
- 229960001783 nicardipine Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- 229960000715 nimodipine Drugs 0.000 description 1
- 229960000227 nisoldipine Drugs 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-M octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC([O-])=O QIQXTHQIDYTFRH-UHFFFAOYSA-M 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000021032 oily fish Nutrition 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-M oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC([O-])=O ZQPPMHVWECSIRJ-KTKRTIGZSA-M 0.000 description 1
- 230000004768 organ dysfunction Effects 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 1
- 229960001243 orlistat Drugs 0.000 description 1
- 150000002905 orthoesters Chemical class 0.000 description 1
- PFGVNLZDWRZPJW-OPAMFIHVSA-N otamixaban Chemical compound C([C@@H](C(=O)OC)[C@@H](C)NC(=O)C=1C=CC(=CC=1)C=1C=C[N+]([O-])=CC=1)C1=CC=CC(C(N)=N)=C1 PFGVNLZDWRZPJW-OPAMFIHVSA-N 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 229920003175 pectinic acid Polymers 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 229960002582 perindopril Drugs 0.000 description 1
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000036581 peripheral resistance Effects 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001828 phenalenyl group Chemical group C1(C=CC2=CC=CC3=CC=CC1=C23)* 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- AFOGBLYPWJJVAL-UHFFFAOYSA-N phenbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=CC=C1 AFOGBLYPWJJVAL-UHFFFAOYSA-N 0.000 description 1
- 229950008557 phenbutamide Drugs 0.000 description 1
- 229960003562 phentermine Drugs 0.000 description 1
- 150000002993 phenylalanine derivatives Chemical class 0.000 description 1
- 230000012006 phospholipid homeostasis Effects 0.000 description 1
- 125000001476 phosphono group Chemical group [H]OP(*)(=O)O[H] 0.000 description 1
- XQZYPMVTSDWCCE-UHFFFAOYSA-N phthalonitrile Chemical compound N#CC1=CC=CC=C1C#N XQZYPMVTSDWCCE-UHFFFAOYSA-N 0.000 description 1
- 229920006391 phthalonitrile polymer Polymers 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical compound OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 230000000291 postprandial effect Effects 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- GCYXWQUSHADNBF-AAEALURTSA-N preproglucagon 78-108 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 GCYXWQUSHADNBF-AAEALURTSA-N 0.000 description 1
- 230000000207 pro-atherogenic effect Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229960003387 progesterone Drugs 0.000 description 1
- 239000000186 progesterone Substances 0.000 description 1
- NPUSXSOBPNHOPH-UHFFFAOYSA-N propan-2-yl 4-(2-chlorophenyl)-1-ethyl-2-methyl-5-oxo-4,7-dihydrofuro[3,4-b]pyridine-3-carboxylate Chemical compound CC(C)OC(=O)C1=C(C)N(CC)C(COC2=O)=C2C1C1=CC=CC=C1Cl NPUSXSOBPNHOPH-UHFFFAOYSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 235000004252 protein component Nutrition 0.000 description 1
- 244000000040 protozoan parasite Species 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 229960001455 quinapril Drugs 0.000 description 1
- JSDRRTOADPPCHY-HSQYWUDLSA-N quinapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC=CC=C2C1)C(O)=O)CC1=CC=CC=C1 JSDRRTOADPPCHY-HSQYWUDLSA-N 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 229960002281 racecadotril Drugs 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 230000029865 regulation of blood pressure Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- RWWYLEGWBNMMLJ-YSOARWBDSA-N remdesivir Chemical compound NC1=NC=NN2C1=CC=C2[C@]1([C@@H]([C@@H]([C@H](O1)CO[P@](=O)(OC1=CC=CC=C1)N[C@H](C(=O)OCC(CC)CC)C)O)O)C#N RWWYLEGWBNMMLJ-YSOARWBDSA-N 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 230000002336 repolarization Effects 0.000 description 1
- FDBYIYFVSAHJLY-UHFFFAOYSA-N resmetirom Chemical group N1C(=O)C(C(C)C)=CC(OC=2C(=CC(=CC=2Cl)N2C(NC(=O)C(C#N)=N2)=O)Cl)=N1 FDBYIYFVSAHJLY-UHFFFAOYSA-N 0.000 description 1
- JZCPYUJPEARBJL-UHFFFAOYSA-N rimonabant Chemical compound CC=1C(C(=O)NN2CCCCC2)=NN(C=2C(=CC(Cl)=CC=2)Cl)C=1C1=CC=C(Cl)C=C1 JZCPYUJPEARBJL-UHFFFAOYSA-N 0.000 description 1
- 229960003015 rimonabant Drugs 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- PYNXFZCZUAOOQC-UTKZUKDTSA-N sacubitril Chemical compound C1=CC(C[C@H](C[C@@H](C)C(=O)OCC)NC(=O)CCC(O)=O)=CC=C1C1=CC=CC=C1 PYNXFZCZUAOOQC-UTKZUKDTSA-N 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- MOODSJOROWROTO-UHFFFAOYSA-N salicylsulfuric acid Chemical compound OC(=O)C1=CC=CC=C1OS(O)(=O)=O MOODSJOROWROTO-UHFFFAOYSA-N 0.000 description 1
- 229950006241 saprisartan Drugs 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 229940116351 sebacate Drugs 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 1
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- KZJWDPNRJALLNS-VJSFXXLFSA-N sitosterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CC[C@@H](CC)C(C)C)[C@@]1(C)CC2 KZJWDPNRJALLNS-VJSFXXLFSA-N 0.000 description 1
- 235000015500 sitosterol Nutrition 0.000 description 1
- 229950005143 sitosterol Drugs 0.000 description 1
- NLQLSVXGSXCXFE-UHFFFAOYSA-N sitosterol Natural products CC=C(/CCC(C)C1CC2C3=CCC4C(C)C(O)CCC4(C)C3CCC2(C)C1)C(C)C NLQLSVXGSXCXFE-UHFFFAOYSA-N 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 description 1
- 229960002370 sotalol Drugs 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 229960002909 spirapril Drugs 0.000 description 1
- HRWCVUIFMSZDJS-SZMVWBNQSA-N spirapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2(C1)SCCS2)C(O)=O)CC1=CC=CC=C1 HRWCVUIFMSZDJS-SZMVWBNQSA-N 0.000 description 1
- 108700035424 spirapril Proteins 0.000 description 1
- 239000004059 squalene synthase inhibitor Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229940114926 stearate Drugs 0.000 description 1
- 229960004274 stearic acid Drugs 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 150000003432 sterols Chemical class 0.000 description 1
- 235000003702 sterols Nutrition 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000005415 substituted alkoxy group Chemical group 0.000 description 1
- 125000004426 substituted alkynyl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229950000244 sulfanilic acid Drugs 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 229940071103 sulfosalicylate Drugs 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 208000008203 tachypnea Diseases 0.000 description 1
- 206010043089 tachypnoea Diseases 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229960000651 tasosartan Drugs 0.000 description 1
- ADXGNEYLLLSOAR-UHFFFAOYSA-N tasosartan Chemical compound C12=NC(C)=NC(C)=C2CCC(=O)N1CC(C=C1)=CC=C1C1=CC=CC=C1C=1N=NNN=1 ADXGNEYLLLSOAR-UHFFFAOYSA-N 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 229960004084 temocapril Drugs 0.000 description 1
- FIQOFIRCTOWDOW-BJLQDIEVSA-N temocapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C[C@H](SC1)C=1SC=CC=1)=O)CC1=CC=CC=C1 FIQOFIRCTOWDOW-BJLQDIEVSA-N 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- UZQBKCWYZBHBOW-YIPNQBBMSA-N terlakiren Chemical compound C([C@@H](C(=O)N[C@@H](CSC)C(=O)N[C@@H](CC1CCCCC1)[C@@H](O)C(=O)OC(C)C)NC(=O)N1CCOCC1)C1=CC=CC=C1 UZQBKCWYZBHBOW-YIPNQBBMSA-N 0.000 description 1
- 108010069247 terlakiren Proteins 0.000 description 1
- 229950003204 terlakiren Drugs 0.000 description 1
- SASWSEQJAITMKS-JJNNLWIXSA-N tert-butyl (2s)-2-[[(2s)-1-[[(2s)-1-[[(4s,5s,7s)-5-hydroxy-2,8-dimethyl-7-[[(2s,3s)-3-methyl-1-oxo-1-(pyridin-2-ylmethylamino)pentan-2-yl]carbamoyl]nonan-4-yl]amino]-3-(1h-imidazol-5-yl)-1-oxopropan-2-yl]-methylamino]-1-oxo-3-phenylpropan-2-yl]carbamoyl]p Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)[C@@H](O)C[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC=1N=CC=CC=1)C(C)C)N(C)C(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H]1N(CCC1)C(=O)OC(C)(C)C)C1=CN=CN1 SASWSEQJAITMKS-JJNNLWIXSA-N 0.000 description 1
- IMCGHZIGRANKHV-AJNGGQMLSA-N tert-butyl (3s,5s)-2-oxo-5-[(2s,4s)-5-oxo-4-propan-2-yloxolan-2-yl]-3-propan-2-ylpyrrolidine-1-carboxylate Chemical compound O1C(=O)[C@H](C(C)C)C[C@H]1[C@H]1N(C(=O)OC(C)(C)C)C(=O)[C@H](C(C)C)C1 IMCGHZIGRANKHV-AJNGGQMLSA-N 0.000 description 1
- PGBVMVTUWHCOHX-UHFFFAOYSA-N tert-butyl n-(3-aminocyclopentyl)carbamate Chemical compound CC(C)(C)OC(=O)NC1CCC(N)C1 PGBVMVTUWHCOHX-UHFFFAOYSA-N 0.000 description 1
- CXGTZJYQWSUFET-IBGZPJMESA-N tesaglitazar Chemical compound C1=CC(C[C@H](OCC)C(O)=O)=CC=C1OCCC1=CC=C(OS(C)(=O)=O)C=C1 CXGTZJYQWSUFET-IBGZPJMESA-N 0.000 description 1
- 229950004704 tesaglitazar Drugs 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- CBXCPBUEXACCNR-UHFFFAOYSA-N tetraethylammonium Chemical class CC[N+](CC)(CC)CC CBXCPBUEXACCNR-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- 125000004305 thiazinyl group Chemical group S1NC(=CC=C1)* 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- VJYJJHQEVLEOFL-UHFFFAOYSA-N thieno[3,2-b]thiophene Chemical compound S1C=CC2=C1C=CS2 VJYJJHQEVLEOFL-UHFFFAOYSA-N 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229960004605 timolol Drugs 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- 229960002277 tolazamide Drugs 0.000 description 1
- OUDSBRTVNLOZBN-UHFFFAOYSA-N tolazamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 OUDSBRTVNLOZBN-UHFFFAOYSA-N 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- 229960005461 torasemide Drugs 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 229960002051 trandolapril Drugs 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- GXPHKUHSUJUWKP-UHFFFAOYSA-N troglitazone Chemical compound C1CC=2C(C)=C(O)C(C)=C(C)C=2OC1(C)COC(C=C1)=CC=C1CC1SC(=O)NC1=O GXPHKUHSUJUWKP-UHFFFAOYSA-N 0.000 description 1
- 229960001641 troglitazone Drugs 0.000 description 1
- GXPHKUHSUJUWKP-NTKDMRAZSA-N troglitazone Natural products C([C@@]1(OC=2C(C)=C(C(=C(C)C=2CC1)O)C)C)OC(C=C1)=CC=C1C[C@H]1SC(=O)NC1=O GXPHKUHSUJUWKP-NTKDMRAZSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 206010047302 ventricular tachycardia Diseases 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- SYOKIDBDQMKNDQ-XWTIBIIYSA-N vildagliptin Chemical compound C1C(O)(C2)CC(C3)CC1CC32NCC(=O)N1CCC[C@H]1C#N SYOKIDBDQMKNDQ-XWTIBIIYSA-N 0.000 description 1
- 229960001254 vildagliptin Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 229950000339 xinafoate Drugs 0.000 description 1
- 229950004219 zankiren Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- DIAMINOCYCLOPENTYLPYRIDINE DERIVATIVES FOR THE TREATMENT OF A DISEASE OR DISORDER RELATED APPLICATIONS [001] This application claims the benefit of and priority to U.S. Provisional Application Nos. 63/278,754, filed November 12, 2021, 63/325,988 filed March 31, 2022, and 63/379,562 filed October 14, 2022, the entire contents of each of which are hereby incorporated by reference in their entireties.
- TECHNICAL FIELD [002] The present disclosure is directed to modulators of proprotein convertase subtilisin/ kexin type 9 (PCSK9) useful in the treatment of diseases or disorders.
- PCSK9 Proprotein convertase subtilisin/kexin type 9
- the PCSK9 protein contains a signal sequence, a prodomain, a catalytic domain containing a conserved triad of residues (D186, H226 and S386), and a C-terminal domain and is synthesized as a soluble 74-kDa precursor that undergoes autocatalytic cleavage in the endoplasmic reticulum.
- PCSK9 has pronounced effects on plasma low density lipoprotein cholesterol (LDL-C) levels via its modulation of hepatic low density lipoprotein receptors (LDLR), the main route by which cholesterol is removed from the circulation.
- LDL-C plasma low density lipoprotein cholesterol
- LDLR hepatic low density lipoprotein receptors
- PCSK9 binds the LDLR and directs it to lysosomal degradation, thereby increasing plasma LDL-C levels and, in turn, coronary heart disease (CHD) risk.
- CHD coronary heart disease
- PCSK9 knockout mice show an approximate 50% reduction in plasma cholesterol levels and enhanced sensitivity to statins in reducing plasma cholesterol (Rashid, S., et al., Proc. Natl. Acad. Sci., 2005, 102:5374-5379). Human genetic data strongly support the role of PCSK9 in LDL homeostasis.
- PCSK9 protein-semiconductor
- plasma LDL-C levels were first established by the discovery of PCSK9 missense mutations in patients with an autosomal dominant form of familial hypercholesterolemia (Abifadel, M., et al., Nature, 2003, 34:154-6).
- Patients carrying PCSK9 gain-of-function alleles have increased plasma LDL-C levels and premature CHD, whereas those with PCSK9 loss-of-function alleles have markedly reduced plasma LDL-C and are protected from CHD.
- PCSK9 also plays a role in Lipoprotein (a) (Lp(a)) metabolism.
- Lp(a) is a proatherogenic lipoprotein comprised of an LDL particle covalently linked to apoLp(a).
- PCSK9 therapeutic antibodies have been shown to significantly reduce Lp(a) levels in patients with hypercholesterolemia. (Desai, N.R., et. al., Circulation. 2013, 128(9):962-969; Lambert, G., et. al., Clin. Sci., 2017, 131, 261-268).
- PCSK9 plays an important role in sepsis, a lifethreatening condition caused by a body’s response to infection.
- Overexpression of PSCK9 in septic mice has been shown to aggravate sepsis by increasing inflammation, while inhibition of PCSK9 has been shown to reduce mortality. (Dwivedi, D. J., et al., Shock, 2016, 46(6), 672-680).
- PCSK9 negatively regulates gram-negative lipopolysaccharide (LPS) uptake by hepatocytes through the regulation of the LDLR-mediated bacterial lipid uptake of lipoteichoic acid (LTA) and LPS through an LDL-dependent mechanism.
- LPS gram-negative lipopolysaccharide
- LTA lipoteichoic acid
- PCSK9 inhibition of PCSK9 has the potential to treat sepsis by reducing the body’s immune response to an infection.
- the disclosure relates to a compound of Formula (I):
- A is a 5 or 6 membered heterocycle or heteroaryl containing at least one N and at least one oxo at a ring carbon and is optionally substituted with (C 1 -C 6 )alkyl;
- X is N, 0, or S
- Y is CR 1 or N
- Z is CR 2 , NR 3 , 0, or S
- R 1 and R 2 are each, independently selected from H, halogen, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 - C 10 )aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from 0 and N, wherein the (C 1 - C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 -C 10 )aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C 1 -C 6 )alkyl, (C 1 - C 6 )haloalkyl, (C 1 -C 6 )alkoxy, (C 3 -C 6 )cycloalkyl, -SO 2 (
- R 3 is absent, H or (C 1 -C 6 )alkyl
- R 4 is H, (C 1 -C 6 )alkyl, (C 1 -C 6 )haloalkyl, (C 1 -C 6 )alkoxy or (C 3 -C 6 )cycloalkyl, wherein the (C 1 -C 6 )alkoxy is optionally substituted with halogen.
- the present disclosure relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable carriers.
- the present disclosure relates to a combination comprising a compound of Formula I or a pharmaceutically acceptable salt thereof and one or more pharmaceutical agents.
- the present disclosure relates to a method for treating a disease or disorder comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula I or a pharmaceutically acceptable salt thereof.
- the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the disease or disorder is selected from sepsis, psoriasis and cancer.
- the present disclosure relates to a method of modulating PCSK9 comprising administering to a patient in need thereof a compound of Formula I or a pharmaceutically acceptable salt thereof.
- the present disclosure relates to a method of inhibiting PCSK9 comprising administering to a patient in need thereof a compound of Formula I or a pharmaceutically acceptable salt thereof.
- the present disclosure relates to a compound of Formula I or a pharmaceutically acceptable salt thereof for use as a medicament.
- the present disclosure relates to a compound of Formula I or a pharmaceutically acceptable salt thereof for use in the treatment of a disease or disorder.
- the present disclosure relates to a compound of Formula I for use in the manufacture of a medicament for treating a disease or disorder.
- the present disclosure relates to use of a compound of Formula I or a pharmaceutically acceptable salt thereof in the treatment of a disease or disorder.
- the disclosure provides substituted diaminocyclopentylpyridine compounds, and pharmaceutical compositions thereof.
- substituted compounds are useful as PCSK9 inhibitors, and thus can be used to treat or prevent a disease or condition.
- the disclosure therefore provides a compound of formula (I)
- A is a 5 or 6 membered heterocycle or heteroaryl containing at least one N and at least one oxo at a ring carbon and is optionally substituted with (C 1 -C 6 )alkyl;
- X is N, 0, or S
- Y is CR 1 or N
- Z is CR 2 , NR 3 , 0, or S
- R 1 and R 2 are each, independently selected from H, halogen, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 5 - C 10 )aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from 0 and N, wherein the (C 1 - C 6 )alkyl, (C 3 -C 6 )cycloalkyl, ( C 6 - C 10 )aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C 1 -C 6 )alkyl, (C 1 - C 6 )haloalkyl, (C 1 -C 6 )alkoxy, (C 3 -C 6 )cycloalkyl, -SO 2 (
- R 3 is absent, H or (C 1 -C 6 )alkyl
- R 4 is H, (C 1 -C 6 )alkyl, (C 1 -C 6 )haloalkyl, (C 1 -C 6 )alkoxy or (C 3 -C 6 )cycloalkyl, wherein the (C 1 -C 6 )alkoxy is optionally substituted with halogen.
- the term “compounds of the present disclosure” or “compound of the present disclosure” refers to compounds of formula (I) thereof, and exemplified compounds, and salts thereof, as well as all stereoisomers (including diastereoisomers and enantiomers), rotamers, tautomers and isotopically labeled compounds (including deuterium substitutions), as well as inherently formed moieties.
- the compound is a compound of formula II or a pharmaceutically acceptable salt thereof.
- the compound is a compound of formula III or a pharmaceutically acceptable salt thereof.
- At least one of X, Y and Z is N.
- Z is CR 2 or N.
- Z is O or S, X is N.
- X is N. In some embodiments, X is N, Y is CR 1 , and Z is O or S
- B is In some embodiments, B is , particularly B is or , more particularly B is
- B is . In some embodiments, B is
- A is , or . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is , particularly A is
- R 1 is H, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, ( C 6 - C 10 )aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from 0 and N, wherein the (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 -C 10 )aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C 1 -C 6 )alkyl, (C 1 -C 6 )haloalkyl, (C 1 -C 6 )alkoxy, (C 3 - C 6 )cycloalkyl, -SO 2 (C 1 -C 6 )alkyl,
- R 1 is H, (C 1 - C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 -C 10 )aryl, a 4 to 6 membered heterocyclyl comprising 1 heteroatom selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1 or 3 heteroatoms selected from N, wherein the (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 -C 10 )aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C 1 C 6 )alkyl, (C 1 -C 6 )haloalkyl, (C 1 -C 6 )alkoxy, (C 3 -C 6 )cycloalkyl, -SO 2 (C 1 -C 6 )alkyl, -COOH, and
- R 1 is H, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, or (C 6 -C 10 )aryl, wherein the (C 1 - C 6 )alkyl, (C 3 -C 6 )cycloalkyl, or (C 6 -C 10 )aryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C 1 -C 6 )alkyl, (C 1 -C 6 )haloalkyl, and (C 1 -C 6 )alkoxy.
- R 1 is H, (C 1 -C 6 )alkyl optionally substituted with one or more halogen, -CN, (C 3 - C 6 )cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C 1 - C 6 )alkyl and (C 1 -C 6 )haloalkyl, or (C 6 -C 10 )aryl optionally substituted one or more with halogen or CN.
- R 1 is (C 1 -C 6 )alkyl optionally substituted with one or more halogen or -CN, (C 3 - C 6 )cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C 1 - C 6 )alkyl and (C 1 -C 6 )haloalkyl, or (C 6 -C 10 )aryl optionally substituted with one or more halogen or -CN.
- R 1 is H, (C 1 -C 6 )alkyl optionally substituted with one or more halogen, (C 3 - C 6 )cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C 1 - C 6 )alkyl and (C 1 -C 6 )haloalkyl, or (C 6 -C 10 )aryl optionally substituted one or more with halogen.
- R 1 is (C 1 -C 6 )alkyl optionally substituted with one or more halogen, (C 3 -C 6 )cyclo alkyl optionally substituted with one or more substituents selected from halogen, -OH, (C 1 -C 6 )alkyl and (C 1 - C 6 )haloalkyl, or (C 6 -C 10 )aryl optionally substituted with one or more halogen.
- R 1 is H, (C 1 -C 6 )alkyl optionally substituted with one or more fluoro, (C 3 -C 6 )cyclo alkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C 1 -C 6 )alkyl and (C 1 -C 6 )fluoroalkyl, or ( C 6 - C 10 )aryl optionally substituted one or more with fluorine.
- R 1 is (C 1 -C 6 )alkyl optionally substituted with fluoro, (C 3 -C 6 )cycloalkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C 1 -C 6 )alkyl and (C 1 -C 6 )haloalkyl, or (C 6 -C 10 )aryl optionally substituted with one or more fluoro.
- R 1 is (C 1 -C 6 )alkyl optionally substituted with one or more fluoro or -CN.
- R 1 is (C 1 -C 6 )alkyl optionally substituted with one or more fluoro.
- R 1 is unsubstituted (C 1 -C 6 )alkyl.
- R 1 is (C 3 -C 6 )cycloalkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C 1 -C 6 )alkyl and (C 1 - C 6 )fluoroalkyl.
- R 1 is (C 3 -C 6 )cycloalkyl.
- R 1 is phenyl optionally substituted with one or more fluoro.
- R 1 is -CH 3 , -CH 2 CH 3 , -CHF 2 , -CF 3 , -CF 2 CH 3 , -CH 2 CF 3 , -CH 2 CN, or In certain embodiments, R 1 is -CH 3 , -CH 2 CH 3 , -CHF 2 , -CF 3 , -CF 2 CH 3 , or . In certain embodiments, R 1 is -CH 3 , -CH 2 CH 3 , -CHF 2 , -CF 3 , or -CF 2 CH 3 . In certain preferred embodiments,
- R 1 is -CH 3 , -CHF 2 , or -CF 3 .
- R 1 is In some embodiments, R 1 is
- R 1 is In some embodiments, R is
- R 1 is . In some embodiments, R 1 is
- R 2 is H, halogen, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, or (C 6 -C 10 )aryl.
- R 2 is H, fluoro, chloro, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, or (C 6 -C 10 )aryl.
- R 2 is halogen, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, or phenyl.
- R 2 is fluoro, chloro, (C 1 -C 6 )alkyl, (C 3 -C 6 lcycloalkyl, or phenyl. In some embodiments, R 2 is -Cl, -CH?. or [039] In some preferred embodiments, R 3 is H.
- R 4 is (C 1 -C 6 )alkyl or (C 3 -C 6 ) cycloalkyl. In some embodiments, R 4 is (C 1 -C 6 )alkyl.
- R 4 is methyl, ethyl, or cyclopropyl. In some embodiments, R 4 is methyl or ethyl. In some embodiments, R 4 is (C 1 -C 6 )alkoxy optionally substituted with halogen. In some embodiments, R 4 is -OCF 3 .
- B is
- B is . In some embodiments, B is , or
- B is
- Embodiment 1 A compound of formula (I), or a pharmaceutically acceptable salt thereof, as described above.
- Embodiment 2 A compound according to embodiment 1 or a pharmaceutically acceptable salt thereof, wherein at least one of X, Y and Z is N.
- Embodiment 3 A compound according to embodiment 1 or embodiment 2 or a pharmaceutically acceptable salt thereof, wherein when X is 0 or S, Z is CR 2 or N.
- Embodiment 4 A compound according to any one of embodiments 1 to 3 or a pharmaceutically acceptable salt thereof, wherein when Z is 0 or S, X is N.
- Embodiment 5 A compound according to embodiment 1 or a pharmaceutically acceptable salt thereof, wherein R 4 is methyl, ethyl, or cyclopropyl. [046] Embodiment 6. A compound according to any one of embodiments 1 to 5 or a pharmaceutically acceptable salt thereof, wherein A is
- Embodiment 7 A compound according to any one of embodiments 1 to 6 or a pharmaceutically acceptable salt thereof, wherein A is
- Embodiment 8 A compound according to any one of embodiments 1 to 7 or a pharmaceutically acceptable salt thereof, wherein X is N.
- Embodiment 9 A compound according to any one of embodiments 1 to 8 or a pharmaceutically acceptable salt thereof, wherein X is N, Y is CR 1 , and Z is 0 or S.
- Embodiment 10 A compound according to any one of embodiments 1 to 9 or a pharmaceutically acceptable salt thereof, wherein X is N, Y is CR 1 , and Z is O.
- Embodiment 11 A compound according to any one of embodiments 1 to 10 or a pharmaceutically acceptable salt thereof, wherein R 1 is H, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 -C 10 )aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from O and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from O and N, wherein the (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 -C 10 )aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C 1 -C 6 )alkyl, (C 1 -C 6 )haloalkyl, (C 1 - C 6 )alkoxy, (C 3
- Embodiment 12 A compound according to any one of embodiments 1 to 11 or a pharmaceutically acceptable salt thereof, wherein R 1 is H, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 -C 10 )aryl, a 4 to 6 membered heterocyclyl comprising 1 heteroatom selected from O and N, or a 5 or 6 membered heteroaryl comprising 1 or 3 heteroatoms selected from N, wherein the (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 -C 10 )aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C 1 -C 6 )alkyl, (C 1 -C 6 )haloalkyl, (C 1 -C 6 )alkoxy, (C 3 - C 6 )cycl
- Embodiment 13 A compound according to any one of embodiments 1 to 12 or a pharmaceutically acceptable salt thereof, wherein R 1 is H, (C 1 -C 6 )alkyl optionally substituted with one or more halogen, -CN, (C 3 -C 6 )cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C 1 -C 6 )alkyl and (C 1 -C 6 )haloalkyl, or (C 6 -C 10 )aryl optionally substituted one or more with halogen or -CN.
- R 1 is H, (C 1 -C 6 )alkyl optionally substituted with one or more halogen, -CN, (C 3 -C 6 )cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C 1 -C 6 )alkyl and (C 1 -C 6 )haloalkyl, or (C 6
- Embodiment 14 A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R 1 is (C 1 -C 6 )alkyl optionally substituted with one or more halogen or -CN, (C 3 -C 6 )cyclo alkyl optionally substituted with one or more substituents selected from halogen, -OH, (C 1 -C 6 )alkyl and (C 1 -C 6 )haloalkyl, or (C 6 -C 10 )aryl optionally substituted with one or more halogen or -CN.
- R 1 is (C 1 -C 6 )alkyl optionally substituted with one or more halogen or -CN, (C 3 -C 6 )cyclo alkyl optionally substituted with one or more substituents selected from halogen, -OH, (C 1 -C 6 )alkyl and (C 1 -C 6 )haloalkyl, or (C 6 -C 10
- Embodiment 15 A compound according to embodiment 13 or a pharmaceutically acceptable salt thereof, wherein each halogen is fluoro.
- Embodiment 16 A compound according to any one of embodiments 1 to 15 or a pharmaceutically acceptable salt thereof, wherein R 1 is (C 1 -C 6 )alkyl optionally substituted with one or more fluoro or -CN.
- Embodiment 17 A compound according to any one of embodiments 1 to 16, wherein R 1 is -CH3,
- Embodiment 18 A compound according to any one of embodiments 1 to 17, wherein R 1 is -CH 3 , -CHF 2 , or -CF 3 .
- Embodiment 19 A compound according to any one of embodiments 1 to 14 or a pharmaceutically acceptable salt thereof, wherein R 1 is (C 3 -C 6 )cycloalkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C 1 -C 6 )alkyl and (C 1 -C 6 )fluoroalkyl.
- Embodiment 20 A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R 1 is
- Embodiment 21 A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R 1 is phenyl optionally substituted with one or more fluoro.
- Embodiment 22 A compound according to embodiments 21 or a pharmaceutically acceptable salt thereof, wherein R 1 is
- Embodiment 23 A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R 1 is
- Embodiment 24 A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R 1 is ,
- Embodiment 25 A compound according to any one of embodiments 1 to 3, 5 to 8, and 11 to 24 or a pharmaceutically acceptable salt thereof, wherein R 2 is H, halogen, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl, or (C 6 -C 10 )aryl.
- Embodiment 26 A compound according to any one of embodiments Ito 3, 5 to 8, and 11 to 22 or a pharmaceutically acceptable salt thereof, wherein R 2 is halogen, (C 1 -C 6 )alkyl, (C 3 -C 6 )cycloalkyl. or phenyl
- Embodiment 27 A compound according to any one of embodiments 1 to 3, 5 to 8, and 11 to 26 or a pharmaceutically acceptable salt thereof, wherein R 2 is -Cl, -CH 3 .
- Embodiment 28 A compound according to any one of embodiments 1 to 3, 5 to 8, and 11 to 27 or a pharmaceutically acceptable salt thereof, wherein R 3 is H.
- Embodiment 29 A compound according to any one of embodiments 1 to 4 or a pharmaceutically acceptable salt thereof, wherein B is
- Embodiment 30 A compound according to any one of embodiments 1 to 4 or a pharmaceutically acceptable salt thereof,, wherein B is or
- Embodiment 31 A compound according to any one of embodiments 1 to 4 or a pharmaceutically acceptable salt thereof, wherein B is
- Embodiments 32 and 33 A compound according to embodiment 1 or a pharmaceutically acceptable salt thereof selected from: [072] Additionally, Applicants have surprisingly and unexpectedly found that compounds of Formula (I) exhibit low or no inhibition of the human Ether-a-go-go-Related Gene (hERG). As discussed herein, some aspects of cardiovascular toxicity of a compound can be measured using a hERG assay.
- the hERG gene encodes the inward rectifying voltage gated potassium channel in the heart known as K V 11.1, which is involved in cardiac repolarization. Inhibition of the hERG current causes QT interval prolongation resulting in potentially fatal ventricular tachyarrhythmia. hERG inhibition an important antitarget that must be avoided.
- compounds of Formula I exhibited no measurable hERG inhibition.
- This lack of cardiovascular toxicity by the compounds of Formula I is surprising and unexpected, especially considering the inhibition of hERG by 2-(6-(((lS,3S)-3-((5- cyclopropylpyrimidin-2-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one and 3-(6- ((( IS, 3 S)-3 -((5 -(difluoromethoxy )pyrimidin-2-yl)am ino)cyclopentyl)am ino)pyridin-3-y 1)-1- methylimidazolidine-2, 4-dione.
- salt refers to an acid addition or base addition salt of a compound of the present disclosure.
- Salts include in particular “pharmaceutical acceptable salts”.
- pharmaceutically acceptable salts refers to salts that retain the biological effectiveness and properties of the compounds of this disclosure and, which typically are not biologically or otherwise undesirable.
- the compounds of the present disclosure are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- the compounds of the present disclosure may also form internal salts, e.g., zwitterionic molecules.
- compositions can be formed with inorganic acids and organic acids.
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like.
- compositions can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table.
- the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like.
- C 6 rtain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
- the present disclosure provides compounds of the present disclosure in acetate, ascorbate, adipate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfonate, caprate, chloride/hydrochloride, chlortheophyllonate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, glutamate, glutarate, glycolate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, mucate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate,
- any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms of the compounds.
- Isotopically labeled compounds have structures depicted by the formulae given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- Isotopes that can be incorporated into compounds of the disclosure include, for example, isotopes of hydrogen.
- the disclosure provides a compound of formula (la) or a pharmaceutically acceptable salt thereof, wherein each R 10a , R 10b , R 10c , R lla , R llb , R 12a , R 12b , R 12c , R 12d , R 12e and R 12 is independently selected from H or deuterium; and A and B are as defined herein.
- isotopes particularly deuterium (i.e., 2 H or D) may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements or an improvement in therapeutic index or tolerability.
- deuterium in this context is regarded as a substituent of a compound of the present disclosure.
- concentration of deuterium may be defined by the isotopic enrichment factor.
- isotopic enrichment factor as used herein means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
- a substituent in a compound of this disclosure is denoted as being deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
- isotopic enrichment factor can be applied to any isotope in the same manner as described for deuterium.
- isotopes that can be incorporated into compounds of the disclosure include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 3 H, n C, 13 C, 14 C, 15 N, 18 F 31 P, 32 P, 35 S, 36 C1, 123 I, 124 1, 125 I respectively. Accordingly it should be understood that the disclosure includes compounds that incorporate one or more of any of the aforementioned isotopes, including for example, radioactive isotopes, such as 3 H and 14 C, or those into which non-radio active isotopes, such as 2 H and 13 C are present.
- Such isotopically labelled compounds are useful in metabolic studies (with 14 C), reaction kinetic studies (with, for example 2 H or 3 H), detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in radioactive treatment of patients.
- PET positron emission tomography
- SPECT single-photon emission computed tomography
- an 18 F or labeled compound may be particularly desirable for PET or SPECT studies.
- Isotopically-labeled compounds of the present disclosure can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
- any asymmetric atom (e.g., carbon or the like) of the compound(s) of the present disclosure can be present in racemic or enantiomerically enriched, for example the (R ⁇ )-, (5)- or (R, 5)- configuration.
- each asymmetric atom has at least 50 % enantiomeric excess, at least 60 % enantiomeric excess, at least 70 % enantiomeric excess, at least 80 % enantiomeric excess, at least 90 % enantiomeric excess, at least 95 % enantiomeric excess, or at least 99 % enantiomeric excess in the (R ⁇ )- or (5)- configuration.
- Substituents at atoms with unsaturated double bonds may, if possible, be present in cis- (Z)- or trans- (E)- form.
- a compound of the present disclosure can be in the form of one of the possible stereoisomers, rotamers, atropisomers, tautomers or mixtures thereof, for example, as substantially pure geometric (cis or trans) stereoisomers, diastereomers, optical isomers (antipodes), racemates or mixtures thereof.
- Any resulting mixtures of stereoisomers can be separated on the basis of the physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization.
- any resulting racemates of compounds of the present disclosure or of intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the diastereomeric salts thereof, obtained with an optically active acid or base, and liberating the optically active acidic or basic compound.
- a basic moiety may thus be employed to resolve the compounds of the present disclosure into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl tartaric acid, diacetyl tartaric acid, di-G.G'-/)-toluoyl tartaric acid, mandelic acid, malic acid or camphor- 10-sulfonic acid.
- Racemic compounds of the present disclosure or racemic intermediates can also be resolved by chiral chromatography, e.g., high pressure liquid chromatography (HPLC) using a chiral adsorbent.
- HPLC high pressure liquid chromatography
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the present disclosure, or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable carriers.
- the composition comprises at least two pharmaceutically acceptable carriers, such as those described herein.
- pharmaceutical composition further comprises at least one additional pharmaceutically active agent.
- the additional pharmaceutically active agent is selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) 0-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA: diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl- like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9, an inhibitory nucleic acid targeting Lp(a), an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-dia
- TTR thyroid hormone receptor
- MTP
- the additional pharmaceutically active agent is selected from bempedoic acid, statins, ezetimibe, inclisiran, pelacarsen, evolocumab, PD-1, PD-L1, PD-L2 and combinations thereof [091]
- the pharmaceutical composition can be formulated for particular routes of administration such as oral administration, parenteral administration (e.g. by injection, infusion, transdermal or topical administration), and rectal administration. Topical administration may also pertain to inhalation or intranasal application.
- compositions of the present disclosure can be made up in a solid form (including, without limitation, capsules, tablets, pills, granules, powders or suppositories), or in a liquid form (including, without limitation, solutions, suspensions or emulsions). Tablets may be either film coated or enteric coated according to methods known in the art.
- the pharmaceutical compositions are tablets or gelatin capsules comprising the active ingredient together with one or more of: a) diluents, e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine; b) lubricants, e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or polyethyleneglycol; for tablets also c) binders, e.g., magnesium aluminum silicate, starch paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; if desired d) disintegrants, e.g., starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and e) absorbents, colorants, flavors and sweeteners.
- diluents e.g., lactose, dextrose
- Liquid, particularly injectable, compositions can, for example, be prepared by dissolution, dispersion, etc.
- the disclosed compound is dissolved in or mixed with a pharmaceutically acceptable solvent such as, for example, water, saline, aqueous dextrose, glycerol, ethanol, and the like, to thereby form an injectable isotonic solution or suspension.
- a pharmaceutically acceptable solvent such as, for example, water, saline, aqueous dextrose, glycerol, ethanol, and the like.
- Proteins such as albumin, chylomicron particles, or serum proteins can be used to solubilize the disclosed compounds.
- the disclosed compounds can be also formulated as a suppository that can be prepared from fatty emulsions or suspensions; using polyalkylene glycols such as propylene glycol, as the carrier.
- Parental injectable administration is generally used for subcutaneous, intramuscular or intravenous injections and infusions.
- Injectables can be prepared in conventional forms, either as liquid solutions or suspensions or solid forms suitable for dissolving in liquid prior to injection.
- compositions can be prepared according to conventional mixing, granulating or coating methods, respectively, and the present pharmaceutical compositions can contain from about 0.1% to about 99%, from about 5% to about 90%, or from about 1% to about 20% of the disclosed compound by weight or volume.
- the dosage regimen utilizing the disclosed compound is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; the renal or hepatic function of the patient; and the particular disclosed compound employed.
- a physician or veterinarian of ordinary skill in the art can readily determine and prescribe the effective amount of the drug required to prevent, counter or arrest the progress of the condition.
- the pharmaceutical composition or combination of the present disclosure may, for example, be in unit dosage of about 1-1000 mg of active ingredient(s) for a subject of about 50-70 kg.
- the compositions are in the form of a tablet that can be scored.
- the therapeutically effective dosage of a compound, the pharmaceutical composition, or the combinations thereof, is dependent on the species of the subject, the body weight, age and individual condition, the disorder or disease or the severity thereof being treated.
- Embodiment 34 A pharmaceutical composition comprising a compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable carriers.
- Embodiment 35 The pharmaceutical composition of embodiment 34, further comprising at least one additional pharmaceutically active agent.
- Embodiment 36 The pharmaceutical composition of embodiment 35, wherein the additional pharmaceutically active agent is selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression, an inhibitory nucleic acid targeting Lp(a) protein expression, an inhibitory nucleic acid targeting apoB 100, apoA-I up- regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, anti-di
- TR-FRET time resolved fluorescence resonance energy transfer
- Solutions of varying concentrations are prepared by diluting a compound of the disclosure in dimethylsulfoxide (DMSO) and the resulting solutions are pipetted into a plate.
- DMSO dimethylsulfoxide
- An intermediate plate is prepared in by transferring a known amount of each compound solution and of the control from the compound plate into a corresponding well containing assay buffer and mixing thoroughly.
- a third plate is then prepared to be used for the assay by adding Terbium labeled human PCSK9, followed by a known amount of each solution from the intermediate plate. Unlabeled human PCSK9 in assay buffer containing DMSO is used as a control for the assay.
- Alexa Fluor 647 labeled probe is added to each well of the assay plate and the resulting mixture is incubated for an additional period of time.
- the TR-FRET signal is measured and the FRET ratio (FRET/Terbium) is used to calculate the IC50 and Amax of the compounds.
- the compounds of Formula (I) may be prepared by methods known in the art of organic synthesis as set forth in part by the following synthetic schemes. In the schemes described below, it is well understood that protecting groups for sensitive or reactive groups are employed where necessary in accordance with general principles or chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection processes, as well as the reaction conditions and order of their execution, shall be consistent with the preparation of compounds of Formula (I).
- the present disclosure includes both possible stereoisomers (unless specified in the synthesis) and includes not only racemic compounds but the individual enantiomers and/or diastereomers as well.
- a compound When a compound is desired as a single enantiomer or diastereomer, it may be obtained by stereospecific synthesis or by resolution of the final product or any convenient intermediate. Resolution of the final product, an intermediate, or a starting material may be affected by any suitable method known in the art. See, for example, "Stereochemistry of Organic Compounds" by E. L. Eliel, S. H. Wilen, and L. N. Mander (Wiley-lnterscience, 1994).
- the compounds described herein may be made from commercially available starting materials or synthesized using known organic, inorganic, and/or enzymatic processes.
- the present disclosure is directed to a method of treating or preventing a disease or disorder comprising administering to a patient in need thereof an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, and a pharmaceutically acceptable carrier.
- the disclosure is directed to a method of modulating PCSK9 comprising administering to a patient in need thereof a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- the disclosure is directed to a method of inhibiting PCSK9.
- the method involves administering to a patient in need thereof an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- Another aspect of the disclosure relates to a method of treating, preventing, inhibiting, or eliminating a disease or disorder in which PCSK9 plays a role.
- the method comprises administering to a patient in need of a treatment for diseases or disorders in which PCSK9 plays a role an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- Another aspect of the present disclosure relates to a method of treating, preventing, inhibiting, or eliminating a disease or disorder in a patient associated with the inhibition of PCSK9, the method comprising administering to a patient in need thereof an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the present disclosure relates to a method of treating, preventing, inhibiting, or eliminating a PCSK9-mediated disease or disorder.
- the method comprises administering to a patient in need of a treatment for a PCSK9-mediated disease or disorder an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, and a pharmaceutically acceptable carrier.
- the present disclosure relates to a method of reducing Lp(a), reducing Lp(a) plasma levels, reducing Lp(a) serum levels, reducing serum TRL or LDL levels, reducing serum triglyceride levels, reducing LDL-C, reducing total plasma apoB concentrations, reducing LDL apoB, reducing TRL apoB, or reducing non HDL-C.
- the method comprises administering to a patient in need thereof an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof for use a medicament.
- Another aspect of the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of a PCSK9-mediated disease or disorder.
- the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of a disease or disorder in which PCSK9 plays a role.
- the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of a disease or disorder , wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, , elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, , elevated Lp(a), elevated LDL, elevated TRL, and elevated t
- the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of a disease or disorder , the disease or disorder is selected from sepsis, psoriasis, psoriasis and cancer.
- Another aspect of the present disclosure relates to the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier in the manufacture of a medicament for treating of a PCSK9-mediated disease or disorder.
- Another aspect of the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- hypercholesterolemia hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- Another aspect of the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of sepsis, psoriasis, and cancer.
- Another aspect of the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, and a pharmaceutically acceptable carrier for use in the manufacture of a medicament for treating a disease in which PCSK9 plays a role.
- the present disclosure relates to the use of a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, and a pharmaceutically acceptable carrier in the manufacture of a medicament treating a PCSK9-mediated disease or disorder.
- the present disclosure relates to the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier in the manufacture of a medicament for treating, preventing, inhibiting, or eliminating hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the present disclosure relates to the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier in the manufacture of a medicament for treating, preventing, inhibiting, or eliminating sepsis, psoriasis, and cancer.
- the disease or disorder is a PCSK9-mediated disease or disorder.
- the PCSK9-mediated disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the PCSK9-mediated disease or disorder is selected from sepsis, psoriasis, and cancer.
- the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the disease or disorder is selected from sepsis, psoriasis, and cancer.
- the disclosed compounds of the disclosure can be administered in effective amounts to treat or prevent a disorder and/or prevent the development thereof in subjects.
- Embodiment 37 The pharmaceutical composition of any one of embodiments 34 to 36 for use in the treatment of a PCSK9-mediated disease or disorder.
- Embodiment 38 The pharmaceutical composition of any one of embodiments 34 to 36 for use in the treatment of a disease or disorder, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- Embodiment 41 A method for treating or preventing a disease or disorder comprising administering to a patient in need thereof a therapeutically effective amount of a compound according to any one of the embodiments 1 to 33, or a pharmaceutically acceptable salt thereof.
- Embodiment 42 The method of embodiment 41, wherein the disease or disorder is a PCSK9- mediated disease or disorder.
- Embodiment 43 The method of embodiments 42, wherein the PCSK9-mediated disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- Embodiment 44 A method for treating a disease or disorder comprising administering to a patient in need thereof a therapeutically effective amount of a compound according to any one of the embodiments 1-33, or a pharmaceutically acceptable salt thereof, wherein the disease or disorder is selected from selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- Embodiment 45 A method of modulating PCSK9 comprising administering to a patient in need thereof a compound of any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof.
- Embodiment 46 A method of inhibiting PCSK9 comprising administering to a patient in need thereof a compound of any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof.
- Embodiment 47 The method of any one of embodiments 41 to 46, wherein administering the compound is oral, parental, subcutaneous, by injection, or by infusion.
- Embodiment 48 A compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof, for use as a medicament.
- Embodiment 49 A compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof, for use in the treatment of a PCSK9-mediated disease or disorder.
- Embodiment 50 The compound of embodiment 49, wherein the PCSK9-mediated disease or disorder selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the PCSK9-mediated disease or disorder selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- Embodiment 51 A compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof, for use in the treatment of a disease or disorder, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- Embodiment 52 A compound according to any one of the embodiments 1 to 33, or a pharmaceutically acceptable salt thereof, for use in the manufacture of a medicament for treating of a PCSK9-mediated disease or disorder.
- Embodiment 53 The compound for use in the manufacture of a medicament of embodiment 52, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- Embodiment 54 Use of a compound according to any one of embodiments 1 to 33, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating a PCSK9- mediated disease or disorder.
- Embodiment 55 The use of embodiment 54, wherein said PCSK9-mediated disease or disorder selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- said PCSK9-mediated disease or disorder selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- a compound according to any one of embodiments 1 to 33, or a pharmaceutically acceptable salt thereof, in the treatment of a disease or disorder wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- the compounds of the disclosure can be administered in therapeutically effective amounts in a combinational therapy with one or more therapeutic agents (pharmaceutical combinations) or modalities, e.g., non-drug therapies.
- therapeutic agents pharmaceutical combinations
- modalities e.g., non-drug therapies.
- synergistic effects can occur with other cardiovascular agents, antihypertensive agents, coronary vasodilators, and diuretic substances.
- dosages of the co-administered compounds will of course vary depending on the type of co-drug employed, on the specific drug employed, on the condition being treated and so forth.
- the compounds of the present disclosure may be administered either simultaneously with, or before or after, one or more other therapeutic agent.
- the compound of the present disclosure may be administered separately, by the same or different route of administration, or together in the same pharmaceutical composition as the other agents.
- a therapeutic agent is, for example, a chemical compound, peptide, antibody, antibody fragment or nucleic acid, which is therapeutically active or enhances the therapeutic activity when administered to a patient in combination with a compound of the present disclosure.
- the disclosure provides a product comprising a compound of the present disclosure and at least one other therapeutic agent as a combined preparation for simultaneous, separate or sequential use in therapy.
- the therapy is the treatment of a disease or condition mediated by PCSK9.
- Products provided as a combined preparation include a composition comprising the compound of the present disclosure and the other therapeutic agent(s) together in the same pharmaceutical composition, or the compound of the present disclosure and the other therapeutic agent(s) in separate form, e.g. in the form of a kit.
- the disclosure includes a compound of Formula (I) or a pharmaceutically acceptable salt thereof, for use in a combination therapy.
- a compound, composition, medicament and compounds for use of Formula according to any one of embodiments 1 to 33 or embodiments 41 to 44, or any embodiment of Formula (I) or a pharmaceutically acceptable salt thereof, may also be used to advantage in combination with one or more other therapeutic agents.
- Another aspect of the disclosure is directed to pharmaceutical compositions comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, a pharmaceutically acceptable carrier, and one or more therapeutic agents.
- the pharmaceutical acceptable carrier may further include an excipient, diluent, or surfactant.
- Combination therapy includes the administration of the subject compounds in further combination with other biologically active ingredients (such as, but not limited to, a second agent such as, but not limited to, a cardiovascular agent, an adrenergic blocker, an antihypertensive agent, an angiotensin system inhibitor, an angiotensin-converting enzyme (ACE) inhibitor, a coronary vasodilator, a diuretic, or an adrenergic stimulant or a second agent that targets PCSK9) and non-drug therapies (such as, but not limited to, surgery or radiation treatment).
- the compounds of the application can be used in combination with other pharmaceutically active compounds, preferably compounds that are able to enhance the effect of the compounds of the application.
- the compounds of the application can be administered simultaneously (as a single preparation or separate preparation) or sequentially to the other drug therapy or treatment modality.
- a combination therapy envisions administration of two or more drugs during a single cycle or course of therapy.
- compounds of the application can be used in combination with agents known to be beneficial for reducing cholesterol, including LDL-C, non-HDL-C, triglyceride-lowering agents, and total cholesterol and/or raising HDL-C.
- Exemplary therapeutic agents that may be used in combination with the compounds of the disclosure, include, but are not limited to, hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression (e.g., inclisiran), an inhibitory nucleic acid targeting Lp(a) protein expression (e.g., pelacarsen, an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1 stabilizer or inducer
- hypolipidemic agents examples include, but are not limited to, an HMG-CoA reductase inhibitor, squalene synthase inhibitors, LXR agonist, FXR agonist, fibrates, cholesterol absorption inhibitors, nicotinic acid bile acid binding resins, bempedoic acid, nicotinic acid and other GPR109 agonists, and aspirin.
- HMG-CoA reductase inhibitors i.e., statins
- statins are a class of drugs used to lower cholesterol levels by inhibiting the enzyme HMG-CoA reductase, which plays a central role in the production of cholesterol in the liver.
- statins include, but are not limited to, atorvastatin, cerivastatin, compactin, dalvastatin, dihydrocompactin, fluindostatin, fluvastatin, lovastatin, pitavastatin, mevastatin, pravastatin, rivastatin, simvastatin, and velostatin, or pharmaceutically acceptable salts thereof.
- Fibrates or fibric acid derivatives lower triglycerides and raise HDL cholesterol. They may have little effect on LDL cholesterol.
- Gemfibrozil or fenofibrate is prescribed for people who have very high triglycerides or who have low HDL and high triglycerides. Gemfibrozil may be used to reduce the risk of heart attack in people with coronary artery disease (CAD) who have low HDL and high triglycerides.
- fibrates include, but are not limited to, clofibrate, gemfibrozil, fenofibrate, ciprofibrate, and bezafibrate.
- Cholesterol absorption inhibitors are a class of compounds that prevents the uptake of cholesterol from the small intestine into the circulatory system, and, in turn, reduce plasma LDL-C concentrations. Increased cholesterol levels are associated with increased CVD risk; thus, cholesterol absorption inhibitors are used with the goal of reducing CVD risk.
- a non-limiting example of a cholesterol absorption inhibitor is Ezetimibe.
- bile acid sequestrants examples include, but are not limited to, cholestyramine, colestipol, and colesvelam.
- a non-limiting example of a thyroid hormone mimetic that may be used in combination with the compounds of the disclosure is compound KB2115.
- TRR thyroid hormone receptor
- MGL-3196 A non-limiting example of a thyroid hormone receptor (THR) P-selective agonist that may be used in combination with the compounds of the disclosure is MGL-3196.
- DGAT is an enzyme that catalyzes the last step in triacylglycerol biosynthesis. DGAT catalyzes the coupling of a 1,2-diacylglycerol with a fatty acyl-CoA resulting in Coenzyme A and triacylglycerol.
- DGAT1 acyl coA-diacylglycerol acyl transferase 1
- DGAT2 acyl coA- diacylglycerol acyl transferase 2 see Cases et al, J. Biol. Chem.
- DGAT1 and DGAT2 do not share significant protein sequence homology. Importantly, DGAT1 knockout mice are protected from high fat diet-induced weight gain and insulin resistance (Smith et al, Nature Genetics 25:87-90, 2000). The phenotype of the DGAT1 knockout mice suggests that a DGAT1 inhibitor has utility for the treatment of obesity and obesity-associated complications.
- DGAT1 inhibitors useful in said combination are compounds and analogs generically and specifically disclosed e.g. in WO2007/126957 and W02009/040410, in particular in the compound claims and the final products of the working examples, the subject-matter of the final products, the pharmaceutical preparations and the claims.
- Examples of DGAT1 inhibitors suitable for use in combination with compounds of the present disclosure include but are not limited to, ⁇ 4-[4-(3-Methoxy-5-phenylamino-pyridin-2-yl)-phenyl]- cyclohexyl ⁇ -acetic acid, (4- ⁇ 4-[5-(l-Methyl-lH-pyrazol-3-ylamino)-pyridin-2-yl]-phenyl ⁇ -cyclohexyl)- acetic acid, (4- ⁇ 4-[5-(5-Fluoro-6-methoxy-pyridin-3-ylamino)-pyridin-2-yl]-phenyl ⁇ -cyclohexyl)-acetic acid, (4- ⁇ 5-[5-(6-Trifluoromethyl-pyridin-3-ylamino)-pyridin-2-yl]-spirocyclohexylidenyl-l,l ’-indanyl
- a non-limiting example of a Niemann Pick Cl-like 1 (NPC1-L1) inhibitor that may be used in combination with the compounds of the disclosure is ezetimibe.
- Apolipoprotein A-I is a protein that in humans is encoded by the APOA1 gene. It has a specific role in lipid metabolism. Apolipoprotein A-I is the major protein component of high density lipoprotein (HDL) in plasma. Chylomicrons secreted from enterocytes also contain ApoA-I but it is quickly transferred to HDL in the bloodstream. The protein promotes cholesterol efflux from tissues to the liver for excretion. It is a cofactor for lecithin cholesterolacyltransferase (LCAT) which is responsible for the formation of most plasma cholesteryl esters.
- HDL high density lipoprotein
- LCAT lecithin cholesterolacyltransferase
- apoA-I Infusion of a variant of apoA-I in humans has been shown to regress atherosclerotic plaque, as assessed by intravascular ultrasound; thus, apoA-I reduces CVD risk and has the ability to both slow progression and induce regression of atherosclerosis.
- a non-limiting example of an apoA-I up-regulator/inducer is RVX208.
- ABCA1 member 1 of human transporter sub-family ABCA
- CHRP cholesterol efflux regulatory protein
- This transporter is a major regulator of cellular cholesterol and phospholipid homeostasis.
- a non-limiting example of an ABCA1 regulator is Probucol.
- Probucol lowers the level of cholesterol in the bloodstream by increasing the rate of LDL catabolism. Additionally, probucol may inhibit cholesterol synthesis and delay cholesterol absorption.
- Probucol is a powerful antioxidant, which inhibits the oxidation of cholesterol in LDLs; this slows the formation of foam cells, which contribute to atherosclerotic plaques.
- LXR liver X receptor
- LXRs Liver X receptors
- LXR agonists are effective for treatment of murine models of atherosclerosis, diabetes, anti-inflammation and Alzheimer’s disease.
- Treatment with LXR agonists include but are not limited to, hypocholamide, T0901317, GW3965, or N,N- dimethyl-3-beta-hydroxy-cholenamide (DMHCA)) lowers the cholesterol level in serum and liver and inhibits the development of atherosclerosis in murine disease models.
- LXR agonists include, but are not limited to, GW3965 (a synthetic nonsteroidal liver X receptor (LXR) agonist/activator) and T0901317 (a dual LXR, FXR agonist).
- the famesoid X receptor also known as NR1H4 (nuclear receptor subfamily 1, group H, member 4) is a nuclear hormone receptor with activity similar to that seen in other steroid receptors such as estrogen or progesterone but more similar in form to PPAR, LXR and RXR. Activation of the nuclear receptor FXR is known to improve hyperglycemia and hyperlipidemia.
- FXR agonist is GW4064 (3-(2,6-Dichlorophenyl)-4-(3'-carboxy-2-chlorostilben-4-yl)oxymethyl-5- isopropylisoxazole).
- Phospholipid transfer protein is a protein that in humans is encoded by the PLTP gene.
- the protein encoded by this gene is one of at least two lipid transfer proteins found in human plasma, with CETP being the other.
- the encoded protein transfers phospholipids from triglyceride-rich lipoproteins to HDL. In addition to regulating the size of HDL particles, this protein may be involved in cholesterol metabolism. At least two transcript variants encoding different isoforms have been found for this gene. Because PLTP influences the metabolism of both triglyceride-rich lipoproteins and HDL, modulation of this transfer protein has the potential to alter cardiovascular disease risk.
- Fish oil is derived from the tissues of oily fish.
- Fish oils contain the omega-3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), precursors of eicosanoids that are known to have many health benefits.
- Fish oil and other omega-3 sources are most highly recommended for the following conditions: hypertriglyceridemia, secondary cardiovascular disease and prevention of high blood pressure.
- Lovaza ® is used along with a low-fat and low-cholesterol diet to lower very high triglycerides (fats) in your blood.
- omega-3 fatty acids that may be used in combination with the compounds of the disclosure include, but are not limited to Lovaza ® and Vascepa ® (icosapent ethyl).
- anti-diabetic agents examples include, but are not limited to, insulin, insulin derivatives and mimetics; insulin secretagogues such as the sulfonylureas; insulinotropic sulfonylurea receptor ligands such as meglitinides, e.g., nateglinide and repaglinide; protein tyrosine phosphatase- IB (PTP-1B) inhibitors including, but not limited to, PTP-112; GSK3 (glycogen synthase kinase-3) inhibitors including, but not limited to, SB- 517955, SB-4195052, SB-216763, NN-57-05441 and NN-57-05445; RXR ligands including, but not limited to, GW-0791 and AGN- 194204; sodium -dependent glucose cotransporter inhibitors including, but not limited to, T-1095; glycogen phosphory
- sulfonylureas include, but are not limited to, tolbutamide, chlorpropamide, tolazamide, acetohexamide, 4-chloro-V-[(l-pyrolidinylamino)carbonyl]-benzenesulfonamide (glycopyramide), glibenclamide (glyburide), gliclazide, l-butyl-3-metanilylurea, carbutamide, glibonuride, glipizide, gliquidone, glisoxepid, glybuthiazole, glibuzole, glyhexamide, glymidine, glypinamide, phenbutamide, amaryl, and tolylcyclamide, or pharmaceutically acceptable salts thereof.
- DPP-IV dipeptidyl peptidase IV
- GLP-1 is a major stimulator of pancreatic insulin secretion and has direct beneficial effects on glucose disposal.
- the DPP-IV inhibitor can be peptidic or, preferably, non-peptidic.
- DPP-IV inhibitors also include, but are not limited to, generically and specifically DPP-IV inhibitors disclosed in WO 98/19998, DE 196 16 486 Al, WO 00/34241 and WO 95/15309, in each case in particular in the compound claims and the final products of the working examples, the subject-matter of the final products, the pharmaceutical preparations and the claims are hereby incorporated into the present application by reference to these publications.
- GLP-1 is an insulinotropic protein which is described, e.g., by W.E. Schmidt et al. in Diabetologia, 28, 1985, 704-707 and in US 5,705,483.
- GLP-1 agonists includes variants and analogs of GLP-1 (7-36)NHz which are disclosed in particular in U.S. 5,120,712, U.S. 5,118666, U.S. 5,512,549, WO 91/11457 and by C. Orskov, et al, in J. Biol. Chem., 264 (1989) 12826.
- GLP-l(7-37) in which compound the carboxy-terminal amide functionality of Arg 36 is displaced with Gly at the 37 th position of the GLP-1 (7-36)NHz molecule and variants and analogs thereof including GLN 9 -GLP-l(7-37), D-GLN 9 -GLP-l(7-37), acetyl LYS 9 -GLP-1(7- 37), LYS 18 -GLP-l(7-37) and, in particular, GLP-1 (7-37)OH, VAL 8 -GLP-l(7-37), GLY 8 -GLP-l(7-37), THR 8 -GLP-l(7-37), MET 8 -GLP-l(7-37) and 4-imidazopropionyl-GLP-l.
- GLP agonist analog exendin-4 described by Greig, et al., in Diabetologia, 1999, 42, 45-50.
- insulin sensitivity enhancers which restore impaired insulin receptor function to reduce insulin resistance and consequently enhance the insulin sensitivity.
- examples include hypoglycemic thiazolidinedione derivatives (e.g., glitazone, (5)- ((3 ,4-dihydro-2-(phenyl-methyl)-2W- 1 -benzopyran-6-yl)methyl-thiazolidine-2, 4-dione (englitazone), 5- ⁇ [4-(3-(5-methyl-2-phenyl-4-oxazolyl)-l-oxopropyl)-phenyl]-methyl ⁇ -thiazolidine-2, 4-dione (darglitazone), 5- ⁇ [4-(l -methyl-cyclohexyl)methoxy)-phenyl]methyl ⁇ -thiazolidine-2, 4-dione (ciglitazone), 5 - ⁇ [4 - (2 - ( 1 )
- anti-obesity agents examples include, but are not limited to, semaglutide, GDF15, orlistat, sibutramine, phentermine and Cannabinoid Receptor 1 (CB1) antagonists e.g., rimonabant.
- CB1 Cannabinoid Receptor 1
- Examples of agonists of peroxisome proliferator-activator receptors that may be used in combination with the compounds of the disclosure include, but are not limited to, fenofibrate, pioglitazone, rosiglitazone, tesaglitazar, BMS-298585, L-796449, the compounds specifically described in the patent application WO 2004/103995 i.e. compounds of examples 1 to 35 or compounds specifically listed in claim 21, or the compounds specifically described in the patent application WO 03/043985 i.e.
- anti-hypertensive agents examples include, but are not limited to, loop diuretics; angiotensin converting enzyme (ACE); inhibitors of the Na-K-ATPase membrane pump; neutralendopeptidase (NEP) inhibitors; ACE/NEP inhibitors; angiotensin II antagonists; renin inhibitors; 0-adrenergic receptor blockers; inotropic agents; calcium channel; aldosterone receptor antagonists; and aldosterone synthase inhibitors.
- ACE angiotensin converting enzyme
- NEP neutralendopeptidase
- ACE/NEP inhibitors angiotensin II antagonists
- renin inhibitors renin inhibitors
- 0-adrenergic receptor blockers inotropic agents
- calcium channel aldosterone receptor antagonists
- aldosterone synthase inhibitors aldosterone synthase inhibitors.
- loop diuretics examples include, but are not limited to, ethacrynic acid, furosemide and torsemide.
- ACE-inhibitor also called angiotensin converting enzyme inhibitors
- ACE-inhibitor includes molecules that interrupt the enzymatic degradation of angiotensin I to angiotensin II. Such compounds may be used for the regulation of blood pressure and for the treatment of congestive heart failure.
- Examples include, but are not limited to, alacepril, benazepril, benazeprilat, captopril, ceronapril, cilazapril, delapril, enalapril, enaprilat, fosinopril, imidapril, lisinopril, moexipril, moveltopril, perindopril, quinapril, ramipril, spirapril, temocapril, and trandolapril, or a pharmaceutically acceptable salt thereof.
- a non-limiting example of an inhibitor of the Na-K-ATPase membrane pump is digoxin.
- NEP inhibitor refers to a compound that inhibits neutral endopeptidase (NEP).
- Examples include, but are not limited to, Candoxatril, Candoxatrilat, Dexecadotril, Ecadotril, Racecadotril, Sampatrilat, Fasidotril, Omapatrilat, Gemopatrilat, Daglutril, SCH-42495, SCH-32615, UK- 447841, AVE-0848, PL-37, and (2R,4s)-5-Biphenyl-4-yl-4-(3-carboxy-propionylamino)-2-methyl- pentanoic acid ethyl ester, or a pharmaceutically acceptable salt thereof.
- NEP inhibitors also include Phosphono/biaryl substituted dipeptide derivatives, as disclosed in U.S. Patent 5,155,100. NEP inhibitors also include N-mercaptoacyl phenylalanine derivative as disclosed in PCT application WO 2003/104200. NEP inhibitors also include dual-acting antihypertensive agents as disclosed in PCT applications WO 2008/133896, WO 2009/035543, or WO 2009/134741. Other examples include compounds disclosed in U.S. applicationsl2/788,794; 12/788,766, and 12/947,029.
- NEP inhibitors also include compounds disclosed in WO 2010/136474, WO 2010/136493, WO 2011/061271, WO 2012/065953, WO 2012/065956, WO 2014/126979, and WO 2014/015965.
- Other examples of NEP inhibitors are compounds disclosed in WO2015116786, W02015116760, WO2014138053, WO2014025891, WO2013184934, WO2013067163, WO2012166389, WO2012166387, WO2012112742, and WO2012082853.
- ACE/NEP inhibitors refers to a compound that inhibits both angiotensin converting enzyme(ACE) and neutral endopeptidase (NEP).
- ACE/NEP inhibitors that may be used in combination with the compounds of the disclosure include, but are not limited to, omapatrilat, sampatrilat, and fasidotril.
- the class of angiotensin II antagonists or ATi receptor antagonists comprises compounds having differing structural features, essentially preferred are the non-peptidic ones.
- angiotensin II antagonists that may be used in combination with the compounds of the disclosure include, but are not limited to, valsartan, losartan, candesartan, eprosartan, irbesartan, saprisartan, tasosartan, telmisartan, the compounds with the designation E-1477 and ZD-8731 of the following formulae
- renin inhibitor includes ditekiren (chemical name: [1S-[1R,2R,4R(1R,2R)]]- l-[(l,l-dimethylethoxy)carbonyl]-L-proly l-L-phenylalanyl-N-[2-hydroxy-5-methyl-l-(2-methylpropyl)- 4-[[[2-methyl-l-[[(2 pyridinylmrthyl)amino]carbonyl]butyl]amino]carbonyl]hexyl]-N-alfa-methyl-L- histidinamide); terlakiren (chemical name: [R-(R,S)]-N-(4-morpholinylcarbonyl)-L-phenylalanyl-N-[l- (cyclohexylmethyl)-2-hydroxy-3-(l-methylethoxy)-3-oxopropyl]-S-methyl-cysteineamide
- 3-adrenergic receptor blockers examples include, but are not limited to, acebutolol, atenolol, betaxolol, bisoprolol, metoprolol, nadolol, propranolol, sotalol, and timolol.
- inotropic agents examples include, but are not limited to, digoxin, dobutamine, and milrinone; Inotropes as used herein include, for example, dobutamine, isoproterenol, milrinone, amirinone, levosimendan, epinephrine, norepinephrine, isoproterenol, and digoxin.
- Examples of calcium channel blockers that may be used in combination with the compounds of the disclosure include, but are not limited to, amlodipine, bepridil, diltiazem, felodipine, nicardipine, nimodipine, nifedipine, nisoldipine and verapamil.
- the class of aldosterone synthase inhibitors comprises both steroidal and non-steroidal aldosterone synthase inhibitors, the latter being most preferred.
- the class of aldosterone synthase inhibitors comprises compounds having differing structural features. Examples of aldosterone synthase inhibitor that can be used in combination with the compounds of the present disclosure include, but are not limited to, the (+)-enantiomer of the hydrochloride of fadrozole (U.S. patents 4,617,307 and 4,889,861) of formula or, if appropriable, a pharmaceutically acceptable salt thereof; and compounds and analogs generically and specifically disclosed e.g.
- aldosterone synthase inhibitors that can be used in combination with the compounds of the present disclosure include, but are not limited to, without limitation 4-(6,7-dihydro-5H- pyrrolo[l,2-c]imidazol-5-yl)-3-m ethylbenzonitrile; 5-(2-chloro-4-cyanophenyl)-6,7-dihydro-5H- pyrrolo[l,2-c]imidazole-5-carboxylic acid (4-methoxybenzyl)methylamide; 4 ’-fluoro -6 -(6, 7,8,9- tetrahydro-5H-imidazo[l,5-a]azepin-5-yl)biphenyl-3-carbonitrile; 5-(4-Cyan
- aldosterone synthase inhibitors also include, but are not limited to, compounds and analogs disclosed in W02008/076860, W02008/076336, W02008/076862, W02008/027284, W02004/046145, W02004/014914, and WO2001/076574.
- Aldosterone synthase inhibitors also include, but are not limited to, compounds and analogs disclosed in U.S. patent applications US2007/0225232, US2007/0208035, US2008/0318978, US2008/0076794, US2009/0012068, US20090048241 and in PCT applications W02006/005726, WO2006/128853, WO2006128851, WO2006/128852, W02007065942, W02007/116099, W02007/116908, W02008/119744 and in European patent application EP 1886695.
- Preferred aldosterone synthase inhibitors suitable for use in the present disclosure include, without limitation 8-(4-Fluorophenyl)-5,6-dihydro-8H-imidazo[5,l-cl[l ,41oxazine; 4-(5,6-Dihydro-8H- imidazo[5,l-c][l ,4]oxazin-8-yl)-2-fluorobenzonitrile; 4-(5,6-Dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8- yl)-2,6-difluorobenzonitrile; 4-(5,6-Dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8-yl)-2-methoxybenzonitrile; 3-(5,6-Dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8-yl)benzonitrile; 4-
- Aldosterone synthase inhibitors useful in said combination include, but are not limited to, compounds and analogs generically and specifically disclosed e.g. in WO 2009/156462 and WO 2010/130796, in particular in the compound claims and the final products of the working examples, the subject-matter of the final products, the pharmaceutical preparations and the claims.
- Preferred Aldosterone Synthase inhibitors suitable for combination in the present disclosure include, 3-(6-Fluoro-3- methyl-2-pyridin-3-yl-lH-indol-l-ylmethyl)-benzonitrile hydrochloride, l-(4-Methanesulfonyl-benzyl)-3- methyl -2 -pyridin-3 -yl- 1 H-indole, 2-(5 -Benzyloxy -pyridin-3 -yl)-6-chloro- 1 -methyl- 1 H-indole, 5-(3- Cyano-1 -methyl- lH-indol-2-yl)-nicotinic acid ethyl ester, N-[5-(6-chloro-3-cyano-l-methyl-lH-indol-2- yl)-pyridin-3-y Im ethyl] -ethanesulfonamide, Pyr
- Lipid-lowering agents are known in the art, and described, e.g., in Goodman and Gilman ’s The Pharmacological Basis of Therapeutics, 11th Ed., Brunton, Lazo and Parker, Eds., McGraw-Hill (2006); 2009 Physicians ’ Desk Reference (PDR), for example, in the 63rd (2008) Eds., Thomson PDR.
- Combination therapy is intended to embrace administration of these therapeutic agents in a sequential manner, wherein each therapeutic agent is administered at a different time and in any order, or in alternation and in any order, as well as administration of these therapeutic agents, or at least two of the therapeutic agents, in a substantially simultaneous manner.
- Substantially simultaneous administration can be accomplished, for example, by administering to the subject a single capsule having a fixed ratio of each therapeutic agent or in multiple, single capsules for each of the therapeutic agents.
- Sequential or substantially simultaneous administration of each therapeutic agent can be effected by any appropriate route including, but not limited to, oral routes, intravenous routes, intramuscular routes, and direct absorption through mucous membrane tissues.
- the therapeutic agents can be administered by the same route or by different routes.
- a first therapeutic agent of the combination selected may be administered by intravenous injection while the other therapeutic agents of the combination may be administered orally.
- all therapeutic agents may be administered orally or all therapeutic agents may be administered by intravenous injection.
- the sequence in which the therapeutic agents are administered is not narrowly critical.
- Embodiment 39 A combination comprising of a compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof and one or more additional pharmaceutical agents.
- Embodiment 40 The combination of embodiment 39, where the one or more agents additional pharmaceutically active agent is selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA: diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression, an inhibitory nucleic acid targeting Lp(a) protein expression, an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabe, a thyroid
- the present disclosure also provides a therapeutic combination, e.g., a kit, kit of parts, e.g., for use in any method as defined herein, comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, to be used concomitantly or in sequence with at least one pharmaceutical composition comprising at least another therapeutic agent, selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression (e.g., a kit, kit of parts, e
- kits of parts comprising: (i) a pharmaceutical composition of the disclosure; and (ii) a pharmaceutical composition comprising a compound selected from a hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA: diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl- like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression (e.g., inclisiran), an inhibitory nucleic acid targeting Lp(a) protein expression (e.g., pelacarsen, an inhibitory nucleic acid targeting apoB 100, apoA-
- the present disclosure provides a method as defined above comprising coadministration, e.g., concomitantly or in sequence, of a therapeutically effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a second drug substance, said second drug substance being hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression (e.g., inclisiran), an inhibitory nucleic acid targeting Lp(a) protein expression (e
- the include plural referents unless the context clearly dictates otherwise.
- reference to “the pharmaceutical formulation” includes reference to one or more pharmaceutical formulations; and so forth.
- acyl refers to a group represented by the general formula hydrocarbylC(O) — , preferably alkylC(O) — .
- acyloxy refers to a group represented by the general formula hydrocarbylC(O)O — , preferably alkylC(O)O — .
- alkenyl refers to an aliphatic group containing at least one double bond and is intended to include both “unsubstituted alkenyls” and “substituted alkenyls”, the latter of which refers to alkenyl moieties having substituents replacing a hydrogen on one or more carbons of the alkenyl group. Such substituents may occur on one or more carbons that are included or not included in one or more double bonds. Moreover, such substituents include all those contemplated for alkyl groups, as discussed below, except where stability is prohibitive.
- alkenyl groups substitution of alkenyl groups by one or more alkyl, carbocyclyl, aryl, heterocyclyl, or heteroaryl groups is contemplated.
- alkenyl groups include ethenyl, propenyl, n-butenyl, iso-butenyl, pentenyl, or hexenyl.
- alkoxy refers to an alkyl group, preferably a lower alkyl group, having an oxygen attached thereto, e.g., -O(alkyl).
- Representative alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, tert-butoxy and the like.
- Representative substituted alkoxy groups include, but are not limited to, — OCF3 and the like.
- alkyl group or “alkane” is a straight chained or branched non-aromatic hydrocarbon which is completely saturated. Typically, a straight chained or branched, alkyl group has from 1 to about 20 carbon atoms, preferably from 1 to about 10 unless otherwise defined. Examples of straight chained and branched alkyl groups include methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, tert-butyl, pentyl, hexyl, pentyl and octyl.
- a C 1 -C 6 straight chained or branched alkyl group is also referred to as a 'lower alkyl’’ group.
- the term '“alkyl” (or ‘‘lower alkyl”) as used throughout the specification, examples, and claims is intended to include both “unsubstituted alkyls’’ and “substituted alkyls”, the latter of which refers to alkyl moselles having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone.
- Such substituents can include, for example, a halogen., a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or .heteroaro
- the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate.
- the substituents of a substituted alkyl may include substituted and unsubstituted forms of ammo, azido, imino, amido, phosphoryl (including phosphonate and. phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), CF,, CN and the like.
- Cycloalkyls can be further substituted with alkyls, alkenyls, alkoxys. alkylthios, aminoalkyls, carbonyl-substituted alkyls, CF 3 , CN, and the like.
- alkynyl refers to an aliphatic group containing at least one triple bond and is intended to include both, “unsubstituted alkynyls” and “substituted alkynyls”, the latter of which refers to alkynyl moieties having substituents replacing a hy drogen on one or more carbons of the alkynyl group. Such substituents may occur on one or more carbons that are included or not included in one or more triple bonds. Moreover, such substituents include all those contemplated for alkyl groups, as discussed above, except where stability is prohibitive.
- alkynyl groups substitution of alkynyl groups by one or more alkyl, carbocycly 1, aryl, heterocyclyl, or heteroaryl groups is contemplated.
- alkenyl groups include ethynyl, propargyl, n-butynyl, iso-butynyl, pentynyl, or hexynyl.
- aryl include substituted or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon.
- the ring is a 5- to 7-membered ring, more preferably a 6-membered ring.
- aryl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cyclo alkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Aryl groups include, but are not limited to, phenyl, biphenyl, naphthyl, anthracenyl, phenalenyl, phenanthrenyl, indanyl, indenyl, tetrahydronaphthalenyl, tetrahydrobenzoannulenyl, and the like.
- C x . y when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups that contain from x to y carbons in the chain.
- C x . y alkyl refers to substituted or unsubstituted saturated hydrocarbon groups, including straight-chain alkyl and branched-chain alkyl groups that contain from x to y carbons in the chain, including haloalkyl groups such as trifluoromethyl and 2,2,2-trifluoroethyl, etc.
- Co alkyl indicates a hydrogen where the group is in a terminal position, a bond if internal.
- C2- y alkenyl and C2- y alkynyl refer to substituted or unsubstituted unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
- carbocycle refers to a saturated or unsaturated ring in which each atom of the ring is carbon.
- carbocycle includes both aromatic carbocycles and non-aromatic carbocycles.
- Non-aromatic carbocycles include both cycloalkane rings, in which all carbon atoms are saturated, and cycloalkene rings, which contain at least one double bond.
- Carbocycle includes 5-7 membered monocyclic and 8-12 membered bicyclic rings. Each ring of a bicyclic carbocycle may be selected from saturated, unsaturated and aromatic rings.
- Carbocycle includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings.
- the term “fused carbocycle” refers to a bicyclic carbocycle in which each of the rings shares two adjacent atoms with the other ring.
- Each ring of a fused carbocycle may be selected from saturated, unsaturated and aromatic rings.
- an aromatic ring e.g., phenyl
- an aromatic ring e.g., phenyl
- a saturated or unsaturated ring e.g., cyclohexane, cyclopentane, or cyclohexene. Any combination of saturated, unsaturated and aromatic bicyclic rings, as valence permits, is included in the definition of carbocyclic.
- Exemplary “carbocycles” include cyclopentane, cyclohexane, bicyclo[2.2.1]heptane, 1,5 -cyclooctadiene, 1, 2,3,4- tetrahydronaphthalene, bicyclo[4.2.0]oct-3-ene, naphthalene and adamantane.
- Exemplary fused carbocycles include decalin, naphthalene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]octane, 4, 5,6,7- tetrahydro-lH-indene and bicyclo[4.1.0]hept-3-ene.
- “Carbocycles” may be susbstituted at any one or more positions capable of bearing a hydrogen atom.
- a “cycloalkyl” group is a cyclic hydrocarbon which is completely saturated.
- “Cycloalkyl” includes monocyclic and bicyclic rings. Typically, a monocyclic cycloalkyl group has from 3 to about 10 carbon atoms, more typically 3 to 8 carbon atoms unless otherwise defined.
- the second ring of a bicyclic cycloalkyl may be selected from saturated, unsaturated and aromatic rings. Cycloalkyl includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings.
- the term “fused cycloalkyl” refers to a bicyclic cycloalkyl in which each of the rings shares two adjacent atoms with the other ring.
- the second ring of a fused bicyclic cycloalkyl may be selected from saturated, unsaturated and aromatic rings.
- halo and “halogen”, as used herein, means halogen and includes chloro, fluoro, bromo, and iodo.
- haloalkyl refers to an alkyl group substituted by one or more halo.
- Examples of (C 1 - 6 )haloalkyl include, but are not limited to, trifluoromethyl, difluoromethyl, fluoromethyl, trichloromethyl, 2,2,2-trifluoroethyl, l,3-dibromopropan-2-yl, 3-bromo-2-fluoropropyl and 1,4,4- trifluorobutan-2-yl.
- heteroalkyl and “hetero aralkyl”, as used herein, refers to an alkyl group substituted with a hetaryl group.
- hetero alkyl refers to a saturated or unsaturated chain of carbon atoms and at least one heteroatom, wherein no two heteroatoms are adjacent.
- heteroaryl and “hetaryl” include substituted or unsubstituted aromatic single ring structures, preferably 5- to 7 -membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms.
- heteroaryl and “hetaryl” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heteroaryl groups include, but are not limited to, furyl, thienyl, pyrrolyl, pyridyl, pyridyl N-oxide, pyrazolyl, pyrimidinyl, imidazolyl, isoxazolyl, oxazolyl, oxadiazolyl, pyrazinyl, indolyl, thiophen-2-yl, quinolyl, benzopyranyl, isothiazolyl, thiazolyl, thiadiazole, indazole, benzimidazolyl, thieno[3,2-b]thiophene, triazolyl, triazinyl, imidazo[l,2-b]pyrazolyl, furo[2,3-c]pyridinyl, imidazo[l,2-a]pyridinyl, indazolyl, pyrrolo[2,3-c]pyridinyl, pyrrolo[3,2-c]pyri
- heteroatom as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur.
- heterocyclyl refers to substituted or unsubstituted non-aromatic ring structures, preferably 3- to 10-membered rings, more preferably 3- to 7-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms.
- heterocyclyl and “heterocyclic” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heterocyclyl groups include, for example, piperidine, piperazine, pyrrolidine, morpholine, lactones, lactams, and the like. Heterocyclyl groups can also be substituted by oxo groups.
- “heterocyclyl” encompasses both pyrrolidine and pyrrolidinone.
- hydroxyalkyl refers to an alkyl group substituted with a hydroxy group.
- Haloalkyl refers to an alkyl group substituted with one or more halogens.
- haloalkyl groups include, but are not limited to, trifluoromethyl, difluoromethyl, pentafluoroethyl, trichloromethyl, etc.
- ox_ o"XxOo refers to a carbonyl group.
- an oxo substituent occurs on an otherwise saturated group, such as with an oxo-substituted cycloalkyl group (e.g., 3-oxo-cyclobutyl)
- the substituted group is still intended to be a saturated group.
- an alkyl group that is optionally substituted can be a fully saturated alkyl chain (e.g., a pure hydrocarbon).
- the same optionally substituted alkyl group can have substituents different from hydrogen. For instance, it can, at any point along the chain be bounded to a halogen atom, a hydroxyl group, or any other substituent described herein.
- the term “optionally substituted” means that a given chemical moiety has the potential to contain other functional groups, but does not necessarily have any further functional groups.
- Suitable substituents used in the optional substitution of the described groups include, without limitation, halogen, oxo, -OH, -CN, -COOH, -CH2CN, -O-(C 1 -C 6 )alkyl, (C 1 -C 6 )alkyl, (C 1 - C 6 )alkoxy, (C 1 -C 6 )haloalkyl, (C 1 -C 6 )haloalkoxy, -O-(C 2 -C 6 )alkenyl, -O-(C 2 -C 6 )alkynyl, (C 2 -C 6 )alkenyl, (C 2 -C 6 )alkynyl, -OH, -OP(O)(OH) 2 , -OC(O)(C 1 -C 6 )alkyl, -C(O)(C 1 -C 6 )alkyl, -OC(O)O(C 1 -C 6
- substituted means that the specified group or moiety bears one or more suitable substituents wherein the substituents may connect to the specified group or moiety at one or more positions.
- an aryl substituted with a cycloalkyl may indicate that the cycloalkyl connects to one atom of the aryl with a bond or by fusing with the aryl and sharing two or more common atoms.
- “Pharmaceutically-acceptable acid addition salt” means those salts which retain the biological effectiveness and properties of the free bases and which are not biologically or otherwise undesirable, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, sulfamic acid, nitric acid, phosphoric acid, and the like, and organic acids such as acetic acid, trichloroacetic acid, trifluoroacetic acid, adipic acid, alginic acid, ascorbic acid, aspartic acid, benzenesulfonic acid, benzoic acid, 2-acetoxybenzoic acid, butyric acid, camphoric acid, camphorsulfonic acid, cinnamic acid, citric acid, digluconic acid, ethanesulfonic acid, glutamic acid, glycolic acid, glycerophosphoric acid, hemisulfic acid, heptanoic acid, hexanoic acid, formic
- “Pharmaceutically-acceptable base addition salt” means those salts which retain the biological effectiveness and properties of the free acids and which are not biologically or otherwise undesirable, formed with inorganic bases such as ammonia or hydroxide, carbonate, or bicarbonate of ammonium or a metal cation such as sodium, potassium, lithium, calcium, magnesium, iron, zinc, copper, manganese, aluminum, and the like. Particularly preferred are the ammonium, potassium, sodium, calcium, and magnesium salts.
- Salts derived from pharmaceutically-acceptable organic nontoxic bases include salts of primary, secondary, and tertiary amines, quaternary amine compounds, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion-exchange resins, such as methylamine, dimethylamine, trimethylamine, ethylamine, diethylamine, triethylamine, isopropylamine, tripropylamine, tributylamine, ethanolamine, diethanolamine, 2-dimethylaminoethanol, 2- diethylaminoethanol, dicyclohexylamine, lysine, arginine, histidine, caffeine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine, purines, piperazine, piperidine, A'-cthylpipcridinc.
- tetramethylammonium compounds tetraethylammonium compounds, pyridine, N,N- dimethylaniline, A'-mcthylpipcridinc.
- A'-mcthylmorpholinc dicyclohexylamine, dibenzylamine, N,N- dibenzylphenethylamine, 1 -ephenamine, N,N ’-dibenzylethylenediamine, polyamine resins, and the like.
- Particularly preferred organic nontoxic bases are isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexylamine, choline, and caffeine.
- a “patient” or “subject” is a mammal, e.g., a human, mouse, rat, guinea pig, dog, cat, horse, cow, pig, or nonhuman primate, such as a monkey, chimpanzee, baboon or, rhesus.
- the subject is a primate.
- the subject is a human.
- pharmaceutically effective amount or “therapeutically effective amount” or “effective amount” means an amount of a compound according to the disclosure which, when administered to a patient in need thereof, is sufficient to effect treatment for disease-states, conditions, or disorders for which the compounds have utility. Such an amount would be sufficient to elicit the biological or medical response of a tissue, system, or patient that is sought by a researcher or clinician.
- the amount of a compound according to the disclosure which constitutes a therapeutically effective amount will vary depending on such factors as the compound and its biological activity, the composition used for administration, the time of administration, the route of administration, the rate of excretion of the compound, the duration of treatment, the type of disease-state or disorder being treated and its severity, drugs used in combination with or coincidentally with the compounds of the disclosure, and the age, body weight, general health, sex, and diet of the patient.
- a therapeutically effective amount can be determined routinely by one of ordinary skill in the art having regard to their own knowledge, the prior art, and this disclosure.
- composition refers to a compound of the disclosure, or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, together with at least one pharmaceutically acceptable carrier, in a form suitable for oral or parenteral administration.
- Carrier encompasses carriers, excipients, and diluents and means a material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting a pharmaceutical agent from one organ, or portion of the body, to another organ, or portion of the body of a subject.
- “Combination” refers to either a fixed combination in one dosage unit form, or a combined administration where a compound of the present disclosure and at least one combination partner (e.g.
- therapeutic agent another drug as explained below, also referred to as “therapeutic agent” or “co-agent”) may be administered independently at the same time or separately within time intervals, especially where these time intervals allow that the combination partners show a beneficial effect from the co-action of these therapeutic agents.
- the beneficial effect of the combination includes, but is not limited to, a cooperative, e.g., synergistic, effect and/or a pharmacokinetic or pharmacodynamic co-action, or any combination thereof, resulting from the combination of therapeutic agents.
- administration of these therapeutic agents in combination is carried out over a defined time period (e.g., minutes, hours, days or weeks depending upon the combination selected). “
- the single components may be packaged in a kit or separately.
- One or both of the components e.g., powders or liquids
- co -administration or “combined administration” or the like as utilized herein are meant to encompass administration of the selected combination partner to a single subject in need thereof (e.g. a patient), and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
- the term “pharmaceutical combination” as used herein means a product that results from the mixing or combining of more than one therapeutic agent and includes both fixed and non-fixed combinations of the therapeutic agents.
- the term “fixed combination” means that the therapeutic agents, e.g., a compound of the present disclosure and a combination partner, are both administered to a patient simultaneously in the form of a single entity or dosage.
- the term “non-fixed combination” means that the therapeutic agents, e.g., a compound of the present disclosure and a combination partner, are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the two compounds in the body of the patient.
- cocktail therapy e.g. the administration of three or more therapeutic agents.
- a subject is “in need of’ a treatment if such subject would benefit biologically, medically, or in quality of life from such treatment (preferably, a human).
- PCSK9 or “proprotein convertase subtilisin/kexin type 9” interchangeably refer to a naturally-occurring human proprotein convertase belonging to the proteinase K subfamily of the secretory subtilase family.
- PCSK9 is synthesized as a soluble zymogen that undergoes autocatalytic intramolecular processing in the endoplasmic reticulum, and is thought to function as a proprotein convertase.
- PCSK9 plays a role in cholesterol homeostasis and may have a role in the differentiation of cortical neurons. Mutations in the PCSK9 gene are a cause of autosomal dominant familial hypercholesterolemia. (Burnett and Hooper, Clin. Biochem. Rev. (2008) 29(1): 11 -26)
- the term “inhibit”, “inhibition”, or “inhibiting” refers to the reduction or suppression of a given condition, symptom, or disorder, or disease, or a significant decrease in the baseline activity of a biological activity or process.
- treat refers to alleviating or ameliorating the disease or disorder (i.e., slowing or arresting the development of the disease or at least one of the clinical symptoms thereof); or alleviating or ameliorating at least one physical parameter or biomarker associated with the disease or disorder, including those which may not be discernible to the patient.
- the term “prevent”, “preventing”, or “prevention” of any disease or disorder refers to the prophylactic treatment of the disease or disorder; or delaying the onset or progression of the disease or disorder.
- “Pharmaceutically acceptable” means that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
- disorder means, and is used interchangeably with, the terms disease, condition, or illness, unless otherwise indicated.
- administering means to either directly administering a disclosed compound or pharmaceutically acceptable salt of the disclosed compound or a composition to a subject, or administering a prodrug derivative or analog of the compound or pharmaceutically acceptable salt of the compound or composition to the subject, which can form an equivalent amount of active compound within the subject’s body.
- “Compounds of the present disclosure”, “Compounds of Formula (I)”, “compounds of the disclosure”, and equivalent expressions refer to compounds of Formulae (I) and (la) as herein described including the salts particularly the pharmaceutically acceptable salts thereof, where the context so permits thereof, as well as all stereoisomers (including diastereoisomers and enantiomers), rotamers, tautomers, and isotopically labelled compounds (including deuterium (“D”) substitutions).
- the term “about” or “approximately” means within 20%, preferably within 10%, and more preferably within 5% of a given value or range.
- a “modulator of PCSK9” refers to a compound or molecule that is able to modulate PCSK9 biological activity or function, and/or downstream pathway(s) mediated by PCSK9 activity.
- an “inhibitor of PCSK9” refers to a compound or molecule that is able to inhibit PCSK9 biological activity or function, and/or downstream pathway(s) mediated by PCSK9 signaling.
- An inhibitor of PCSK9 activity encompasses compounds that block, antagonize, suppress or reduce (to any degree including significantly) PCSK9 biological activity, including downstream pathways mediated by PCSK9 activity.
- disorders or diseases responsive to the inhibition of PCSK9 include, but are not limited to, hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, sepsis, cancer, psoriasis, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
- Inhibition of PCSK9 activity refers to a decrease in the PCSK9 activity, e.g., by administration of the compound of the disclosure.
- hypercholesterolemia or “dyslipidemia” includes, e.g., familial and non-familial hypercholesterolemia.
- Familial hypercholesterolemia FH is an autosomal dominant disorder characterized by elevation of serum cholesterol bound to low density lipoprotein (LDL).
- Familial hypercholesterolemia includes both heterozygous FH and homozygous FH.
- Hypercholesterolemia (or dyslipidemia) is the presence of high levels of cholesterol in the blood. It is a form of hyperlipidemia (elevated levels of lipids in the blood) and hyperlipoproteinemia (elevated levels of lipoproteins in the blood).
- Hyperlipidemia is an elevation of lipids in the bloodstream. These lipids include cholesterol, cholesterol esters, phospholipids and triglycerides. Hyperlipidemia includes for example, type I, Ila, lib, III, IV and V.
- Hypertriglyceridemia denotes high blood levels of triglycerides. Elevated levels of triglycerides are associated with atherosclerosis, even in the absence of hypercholesterolemia, and predispose to cardiovascular disease.
- “Sitosterolemia” or “phytosterolemia” is a rare autosomal recessively inherited lipid metabolic disorder characterized by hyperab sorption of sitosterol from the gastrointestinal tract and decreased biliary excretion of dietary sterols (i.e., leading to hypercholesterolemia, tendon and tuberous xanthomas, premature development of atherosclerosis) and altered cholesterol synthesis.
- “Atherosclerosis” includes hardening of arteries associated with deposition of fatty substances, cholesterol, cellular waste products, calcium and fibrin in the inner lining of an artery. The buildup that results is called plaque.
- Atherosclerosis or “arteriosclerotic vascular disease (ASVD)” is a specific form of arteriosclerosis involving thickening, hardening and loss of elasticity of the walls of arteries as a result of invasion and accumulation of white blood cells, containing both living, active white blood cells (producing inflammation) and remnants of dead cells, including cholesterol and triglycerides. Atherosclerosis is therefore a syndrome affecting arterial blood vessels due to a chronic inflammatory response of white blood cells in the walls of arteries.
- ASVD arteriosclerotic vascular disease
- Atherosclerotic artery disease also known as atherosclerotic artery disease, atherosclerotic cardiovascular disease, coronary heart disease or ischemic heart disease is the most common type of heart disease and cause of heart attacks. The disease is caused by plaque building up along the inner walls of the arteries of the heart, which narrows the lumen of arteries and reduces blood flow to the heart.
- Xanthoma is a cutaneous manifestation of lipidosis in which lipids accumulate in large foam cells within the skin. Xanthomas are associated with hyperlipidemias.
- elevated Lp(a) concentration refers to a serum Lp(a) concentration above 30 mg/dl (75 nmol/L).
- elevated serum Lp(a) means a serum Lp(a) level greater than about 14 mg/dL.
- a patient is considered to exhibit elevated serum Lp(a) if the level of serum Lp ⁇ a) measured in the patient is greater than about 15 mg/dL, about 20 mg/dL, about 25 mg/dL, about 30 mg/dL, about 35 mg/dL, about 40 mg/dL, about 45 mg/dL, about 50 mg/dL, about 60 mg/dL, about 70 mg/dL, about 80 mg/dL, about 90 mg/dL, about 100 mg/dL, about 20 mg/dL, about 140 mg/dL, about 150 mg dL, about 180 mg/dL, or about 200 mg/dL
- the serum Lp(a) level can be measured in a patient post-prandial.
- the Lp(a) level is measured after a period of time of fasting (e.g., after fasting for 8 hrs, 8 hrs, 10 hrs, 12 hrs or more).
- exemplary methods for measuring serum Lp(a) in a patient include, but are not limited to, rate immunonephelometry, ELISA, nephelometry, immunoturbidimetry, and dissociation-enhanced lanthanide fluorescent immunoassay, although any clinically acceptable diagnostic method can be used in the context of the present disclosure.
- ETL elevated triglyceride levels
- Sepsis is a systemic reaction characterized by arterial hypotension, metabolic acidosis, decreased systemic vascular resistance, tachypnea, and organ dysfunction. Sepsis can result from septicemia (i.e., organisms, their metabolic end-products or toxins in the blood stream), including bacteremia (i.e., bacteria in the blood), as well as toxemia (i.e., toxins in the blood), including endotoxemia (i.e., endotoxin in the blood).
- septicemia i.e., organisms, their metabolic end-products or toxins in the blood stream
- bacteremia i.e., bacteria in the blood
- toxemia i.e., toxins in the blood
- endotoxemia i.e., endotoxin in the blood
- septicemia also encompasses systemic reactions resulting from fungemia (i.e., fungi in the blood), viremia (i.e., viruses or virus particles in the blood), and parasitemia (i.e., helminthic or protozoan parasites in the blood).
- fungemia i.e., fungi in the blood
- viremia i.e., viruses or virus particles in the blood
- parasitemia i.e., helminthic or protozoan parasites in the blood.
- septicemia and septic shock acute circulatory failure resulting from septicemia often associated with multiple organ failure and a high mortality rate
- septicemia and septic shock acute circulatory failure resulting from septicemia often associated with multiple organ failure and a high mortality rate
- Compounds of the present disclosure may be prepared by methods known in the art of organic synthesis. In all of the methods it is understood that protecting groups for sensitive or reactive groups may be employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Green and P. G. M. Wuts (1999) Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art.
- Mass spectra were collected using a Waters System (Acquity UPLC and a Micromass ZQ mass spectrometer) or Agilent-1260 Infinity (6120 Quadrupole); all masses reported are the m/z of the protonated parent ions unless recorded otherwise.
- the chemical names were generated using ChemBioDraw Ultra vl4 from CambridgeSoft.
- Scheme 1 represents the general synthesis of a compound of formula (I). wherein R 1 , A are as defined in herein.
- Step A tert-butyl (3-aminocyclopentyl)carbamate is reacted with la in the presence of a suitable base such as diisopropylethylamine at a suitable temperature such as 120 °C to form lb.
- a suitable base such as diisopropylethylamine at a suitable temperature such as 120 °C to form lb.
- Step B Deprotection of the protecting group to form 1c in the presence of a strong acid such as hydrochloric acid or trifluoroacetic acid.
- Step C Cyanamide Id can be formed through a nucleophilic substitution reaction of 1c with cyanogen bromide and a suitable base such as sodium acetate.
- Step D Cyanamide Id is reacted with hydroxyamine to form A'-hydroxyguanidinc le.
- Step E A'-hydroxyguanidinc le can be converted to the target compound 1,2,4-oxadiazole 1g by the following two conditions.
- Condition E-2 Cyclization at a suitable temperature such as 100 °C to form 1g.
- Scheme 2 represents the alternative synthesis of a compound of formula (I) in Scheme 1. wherein R 1 , A are as defined in herein.
- Step A A'-hydroxyguanidinc le can be converted to the target compound 1,2,4-oxadiazole 1g by reacting with an orthoester 2a.
- Scheme 3 represents the alternative synthesis of a compound of formula (I) in Scheme 1. wherein R 1 , A are as defined in herein.
- Step A Amine 1c is reacted with 3-chloro-l,2,4-thiadiazole 3a to form the target compound 1g in the presence of a suitable base such as A'.
- A' diisopropylcthylaminc at a suitable temperature such as 100 °C.
- Scheme 4 represents the general synthesis of a compound of formula (I). wherein R 2 , A are as defined in herein.
- Step A Amine 1c is reacted with 5-chloro-l,2,4-thiadiazole 4a to form the target compound 4b in the presence of a suitable base such as A'.
- Scheme 5 represents the general synthesis of a compound of Formula (I). wherein R 1 , A are as defined in herein.
- Step A Amine 1c is reacted with l,3,4-oxadiazol-2)(3H )-one 5a to form the target compound 5b with a coupling reagent such as PyBroP in the presence of a suitable base such as A'.
- A' diisopropylcthylaminc.
- Scheme 6 represents the general synthesis of a compound of Formula (I). wherein R 4 , A are as defined in herein.
- Step A Amine 1c is reacted with 3 -(methylsulfinyl)- 1,2, 4-triazine 6a or 3 -(methylsulfonyl)- 1, 2, 4-triazine 6a’ to form the target compound 6b in the presence of a suitable base such as sodium carbonate.
- a suitable base such as sodium carbonate.
- Scheme 7 represents the general synthesis of a compound of formula (I). wherein A is as defined herein.
- Step A Cyanamide Id reacted with sodium azide to form the target compound 7a.
- Scheme 8 represents the general synthesis of a compound of formula (I). wherein X, Y, Z, and A are as defined in herein.
- Step A Intermediate 8a can be formed by either of the following conditions
- Step B Deprotection of the protecting group to form amine 8b in the presence of a strong acid such as hydrochloric acid or trifluoro acetic acid.
- Step C Amine 8b is reacted with la to form the target compound 8c following Scheme 1 Step A.
- Scheme 9 represents the general synthesis of a compound of formula (I). wherein R 4 , and A are as defined in in herein.
- Step A Intermediate 9a is formed following Scheme 6 Step A.
- Step B Deprotection of the protecting group to form 9 b in the presence of a strong acid such as hydrochloric acid or trifluoroacetic acid.
- Step C Amine 9b is reacted with la to form the target compound 9c following Scheme 1 Step A.
- Example 1 Synthesis of 2-(6-(((1S,3S) -3-((5-methvl-l,2,4-oxadiazol-3-vl) amino)cyclopentyl)amino) pyridin-3-vl)pvridazin-3(2H)-one
- Step-2 Synthesis of tert-butyl ((LS',3.S')-3-((5-(6-oxopyridazin-l(6H )-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-2)
- Step-3 Synthesis of 2-(6-(((1S,3S)-3-aminocyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one hydrochloride (Int-3)
- Step-4 Synthesis of /V-((LS',3.S')-3-((5-(6-oxopyridazin-l(6H )-yl)pyridin-2- yl)amino)cyclopentyl)cyanamide (Int-4)
- Step-5 Synthesis of 2-hydroxy-l-((1S,3S)-3-((5-(6-oxopyridazin-l(6H )-yl)pyridin-2-yl)amino) cyclopentyl)guanidine (Int-5)
- Step-6 Synthesis of 2-(6-(((1S,3S')-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino) pyridin-3-yl)pyridazin-3(2H)-one (Example 1)
- Step-2 Synthesis of 3-(6-fhioropyridin-3-yl)-l-methylimidazolidine-2, 4-dione (Int-7)
- Step-3 Synthesis of tert-butyl ((1S,3S)-3-((5-(3-methyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-8)
- Step-4 Synthesis of 3-(6-(((1S,3S)-3-aminocyclopentyl)amino)pyridin-3-yl)-l-methylimidazolidine- 2, 4-dione hydrochloride (Int-9)
- Step-5 Synthesis of N-((1S,3S)-3-((5-(3-methyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)cyanamide (Int-10)
- Step-6 Synthesis of 2-hydroxy-l-((1S,3S)-3-((5-(3-methyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)guanidine (Int-11)
- Step-7 Synthesis of l-methyl-3-(6-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)imidazolidine-2, 4-dione (Example 10)
- Example 13 Synthesis of 2-(6-(((1S,3S) -3-((5-(3-hvdroxvcvclobutvl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-vl)pyridazin-3(2H)-one
- Example 14 Synthesis of 2-(6-(((1S,3S) -3-((5-ethvl-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pyridin-3-vl)pyridazin-3(2H)-one
- Example 15 Synthesis of 2-(6-(((1S,3S)-3-((5-(l-(trifluoromethvl)cvclopropvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pyridin-3-yl)pyridazin-3(2H)-one
- Step-1 Synthesis of 2,5-dioxopyrrolidin-l-yl l-(trifluoromethyl)cyclopropane-l-carboxylate (Int- 12)
- Step-2 Synthesis of 2-(6-(((15,3S')-3-((5-(l-(trifhioromethyl)cyclopropyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 15)
- Example 17 Synthesis of 2-(6-(((1S,3S)-3-((3-cvclopropyl-l,2,4-thiadiazol-5- vl)amino)cvclopentvl)amino)pvridin-3-yl)pvridazin-3(2H)-one
- the crude product was purified by preparative HPLC (20-40% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 19 mm), 5.0 ⁇ m column, flow rate 15 mL/min) to provide the title compound as a yellow solid (40 mg, 31%), ESI-MS m/z: 396.25 [M+H] + .
- Step-1- Synthesis of tert-butyl ((1S,3S)-3-((5-iodopyridm-2-yl)ammo)cyclopentyl)carbamate (Int-13)
- Step-2- Synthesis of tert-butyl ((1S,3S)-3-((5-(2-oxopyrrolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-14)
- Step-4 Synthesis of V-((l.S’.3.S’)-3-((5-(2-o ⁇ opyrroli(lin-l-yl)pyriilin-2- yl)amino)cyclopentyl)cyanamide (Int-16)
- Step-5 Synthesis of 2-hydroxy-l-((15,35)-3-((5-(2-oxopyrrolidin-l-yl)pyridin-2- yl)amino)cyclopentyl) guanidine (Int-17)
- Step-6 Synthesis of l-(6-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino) cyclopentyl)amino)pyridin-3-yl)pyrrolidin-2-one (Example 20)
- Step-1 Synthesis of tert-butyl ((1S,3S)-3-((5-(3-methyl-2,4-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-18)
- Step-3 Synthesis of A-((1S,3S)-3-((5-(3-methyl-2,4-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)cyan amide
- Step-4 Synthesis of 2-hydroxy-l-((15,35)-3-((5-(3-methyl-2,4-dioxoimidazolidine-l-yl)pyridin-2- yl)amino)cyclopentyl)guanidine (Int-21) [312] To a solution of Int-20 (395 mg, 1.26 mmol) in EtOH (10 mL), was added hydroxylamine hydrochloride (96 mg, 1.38 mmol) and TEA (0.35 mL, 2.51 mmol). The mixture was heated under stirring at 50 °C for 1 h, then concentrated under reduced pressure.
- Step-5 1 Synthesis of 3-methyl-l-(6-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)imidazolidine-2, 4-dione (Example 21)
- Step-1 1 Synthesis of 2,5-dioxopyrrolidin-l-yl 1-hydroxycyclopentane-l-carboxylate
- Step-1 1 Synthesis of 6'-fhioro-27/-[l,3'-bipyridin]-2-one (Int-23)
- Step-2 Synthesis of tert-butyl ((l 1 $',3 1 S')-3-((2-oxo-27/-[l,3 , -bipyridin]-6 , -yl)amino) cyclopentyl)carbamate
- Step-3 Synthesis of 6'-(((1S,3S)-3-aminocyclopentyl)amino)-27/-[l,3'-bipyridin]-2-one hydrochloride (Int-25)
- Step-4 Synthesis of A-((1S,3S)-3-((2-oxo-277-[l,3'-bipyridin]-6'-yl)amino)cyclopentyl)cyanamide (Int-26)
- Step-5 Synthesis of 2-hydroxy-l-((1S,3S)-3-((2-oxo-27/-[l,3'-bipyridin]-6'- yl)amino)cyclopentyl)guanidine (Int-27)
- Step-6 Synthesis of bMflLS'vLS'l-d-flS-methyl-LZH-oxadiazol-d-yllamino) cyclo pentyl )am in o)-2//- [l,3'-bipyridin]-2-one (Example 23)
- Example 25 Synthesis of 2-(6-(((l 1 S , ,3 1 S)-3-((5-(4-fluorophenvl)-l,2,4-thiadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one
- Example 28 Synthesis of 3-(6-((2-(6'-(((1S,3S)-3-((5-cvcloDroDvlDvrimidin-2- vl)amino)cvcloDentvl)amino)-2-oxo-27/-[l,3'-biDvridine]-5-carboxamido)ethvl)amino)-6-oxohexvl)-
- Step-1 Synthesis of 2-chloro-5-cyclopropylpyrimidine (Int-28)
- Step-2 Synthesis of tert-butyl ((15,35)-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)carbamate (Int-29)
- Step-3 Synthesis of (15,3S')-/V 1 -(5-cyclopropylpyrimidin-2-yl)cyclopentane-l,3-diamine hydrochloride (Int-30)
- Step-4 Synthesis of tert-butyl (2-(6-oxo-l,6-dihydropyridine-3-carboxamido)ethyl)carbamate (Int- 31)
- Step-5 Synthesis ooff tert-butyl (2-(6'-fluoro-2-o ⁇ o-2//-
- Step-6 Synthesis of tert-butyl (2-(6'-(((1S,3S)-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)amino)-2-oxo-27/-[l,3'-bipyridine]-5-carboxamido)ethyl)carbamate (Int-33)
- Step-7 Synthesis of 2V-(2-aminoethyl)-6'-(((lS',3S')-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)amino)-2-oxo-27/-[l,3'-bipyridine]-5-carboxamide (Int-34)
- Step-8 Synthesis of 3-(6-((2-(6'-(((1S,3S)-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)amino)-2-oxo-27/-[l,3'-bipyridine]-5-carboxamido)ethyl)amino)-6-oxohexyl)- 2-((12j,32j)-5-((2j)-3,3-dimethyl-5-sulfo-l-(3-sulfopropyl)indolin-2-ylidene)penta-l,3-dien-l-yl)-3- methyl-5-sulfo-l-(4-sulfobutyl)-37/-indol-l-ium
- Step-3 Synthesis of 2-(6-(((1S,3S)-3-((6-methyl-l,2,4-triazin-3-yl)amino) cyclopentyl)amino)pyridin- 3-yl)pyridazin-3(2H)-one (Example 29)
- Step-2 Synthesis of tert-butyl ((ES',3,S')-3-((5-(5-methyl-6-oxopyridazin- l(6//)-yl) pyridine-2-yl) amino) cyclopentyl) carbamate (lnt-38)
- StepS Synthesis of 2-(6-(((15',35')-3-aminocyclopentyl)amino)pyridin-3-yl)-4-methylpyridazin- 3(2//)-one (Int-39) [339] To a mixture of Int-38 (1.5 g, 3.89 mmol) in MeOH (10 ml) was added 4 M HC1 in 1,4-dioxane (6 mL). After stirring for 2 h at room temperature, the mixture was concentrated under reduced pressure. The crude product was treated with Amberlyst® A21 ion exchange resin (3 g) in MeOH (20 mL) and stirred for 15 min.
- Amberlyst® A21 ion exchange resin 3 g
- Step-4 Synthesis of 4-methyl-2-(6-(((1S,3S)-3-((6-methyl-l,2,4-triazin-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 30)
- Step-2 Synthesis of tert-butyl ((l 1 S',3 1 S')-3-((l,2,4-triazin-3-yl)amino)cyclopentyl) carbamate (Int-41)
- Step-3 Synthesis of tert-butyl ((f 1 S',3 1 S')-3-((6-bromo-l,2,4-triazin-3-yl)amino)cyclopentyl)carbamate (Int-42)
- Step-4 Synthesis of tert-butyl ((LS',3.S')-3-((6-cyclopropyl-l,2,4-triazin-3-yl)amino) cyclopentyl)carbamate (Int-43)
- Step-5 Synthesis of (15',35')-A7-(6-cyclopropyl-l,2,4-triazin-3-yl)cyclopentane-l,3-diamine (Int-44)
- Step-6 Synthesis of 2-(6-(((1S,3S)-3-((6-cyclopropyl-l,2,4-triazin-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 31)
- Example 33 Synthesis of 2-(6-(((l 1 S , ,3 1 S)-3-((6-ethvl-l,2,4-triazin-3-yl)amino)cvclopentvl) amino)pvridin-3-vl)-4-methvlpyridazin-3(277)-one
- Step-2 Synthesis of /V,2,2-trimethoxy-/V-methylacetamide (Int-46)
- Step-4 Synthesis of 6-ethyl-3-(methylthio)-l,2,4-triazine (lnt-48) [351]
- a mixture of methyl hydrazinecarbimidothioate hydroiodide (5 g, 21.4 mmol) and Int-47 (2.5 g, 21.4 mmol) in EtOH (100 mL) was heated at 70 °C for 4 h.
- the reaction mixture was concentrated under reduced pressure.
- Step-5 Synthesis of 6-ethyl-3-(methylsulfonyl)-l, 2, 4-triazine and 6-ethyl-3-(methylsulfinyl)-l,2,4- triazine (Int-49 / Int-49a)
- Step-6 Synthesis of 2-(6-(((lS,3S)-3-((6-ethyl-l,2,4-triazin-3-yl)amino)cyclopentyl) amino)pyridin-3- yl)-4-methylpyridazin-3(2H)-one (Example 33)
- Batch-1 To a solution of Int-39 (330 mg, 1.16 mmol) in NMP (5 mL), was added NazCOs (243 mg, 2.32 mmol) followed by Int-49a (200 mg, 1.16 mmol). The reaction mixture was stirred at rt for 16 h.
- Batch-2 To the stirred solution of Int-39 (732 mg, 2.56 mmol) in NMP (5 mL), was added NazCOs (537 mg, 5.12 mmol) followed by Int-49 (480 mg, 2.56 mmol). The reaction mixture was stirred at rt for 16 h. The combined reaction mixtures were quenched with water and the product was extracted with EtOAc.
- Example 35 Synthesis Of 2-(6-(((15',35')-3-((5-(l-hvdroxvcvcloDentvl)-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-yl)Dvridazin-3(2H)-one
- Step-1 Synthesis of 2,5-dioxopyrrolidin-l-yl 1-hydroxycyclopentane-l-carboxylate (Int-35) lnt-60 was synthesized following the Int-12 starting from commercially available 1 -hydroxycyclopentane- 1 -carboxylic acid.
- 'H NMR 300 MHz, CDC1 3 ) 5 2.86 (s, 4H), 2.61 (s, IH), 2.38 - 2.33 (m, 2H), 2.04 - 1.84 (m, 6H).
- Step-2 Synthesis of 2-(6-(((1S,3S)-3-((5-(l-hydroxycyclopentyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 35)
- Example 35 was synthesized following the Example 15 starting from Int-5. ESI-MS m/z: 424.1 [M+H] + .
- Example 36 Synthesis of l,5,5-trimethyl-3-(6-(((l 1 S',3 1 S)-3-((5-methyl-l,2,4-oxadiazol-3- vl)amino)cvdopentvl)amino)pyridin-3-vl)imidazolidine-2, 4-dione
- Step-1 Synthesis of tert-butyl ((15',35')-3-((5-(3,4,4-trimethyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-61)
- Int-61 was synthesized following the Int-14 starting from commercially available 1,5,5- trimethylimidazolidine-2, 4-dione, ESI-MS m/z: 418.25 [M+H] + .
- Step-2 Synthesis ooff 3-(6-(((LS',3.S')-3-aminocyclopentyl) amino) pyridin-3-yl)-l ,5,5- trimethylimidazolidine-2, 4-dione hydrochloride (Int-62)
- Int-62 was synthesized following the Int-3 starting from Int-61, ESI-MS m/z: 318.15 [M+H] + .
- Step-3 Synthesis ooff N-((1S,3S)-3-((5-(3,4,4-trimethyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)cyanamide (Int-63)
- Int-63 was synthesized following the Int-4 starting from Int-62, ESI-MS m/z: 343.15 [M+H] + .
- Step-4 Synthesis of 2-hydroxy-l-((15,3 ⁇ )-3-((5-(3,4,4-trimethyl-2,5-dioxoimidazolidin-l-yl)pyridin- 2-yl)amino)cyclopentyl)guanidine (Int-64)
- Int-64 was synthesized following the Int-5 starting from Int-63, ESI-MS m/z: 375.95 [M+H] + .
- Step-5 Synthesis of l,5,5-trimethyl-3-(6-(((15,35)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)imidazolidine-2, 4-dione (Example 36)
- Example 36 was synthesized following the Example 1 starting from Int-64. ESI-MS m/z: 400.1 [M+H] + .
- Example 37 Synthesis of 4-methyl-2-(6-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-vl)Dvridazin-3(27Q-one
- Int-65 was synthesized following the Int-1 starting from commercially available 4-methylpyridazin- 3(2H)-one, ESI-MS m/z: 205.70 [M+H] + .
- Step-2 Synthesis ooff tert-butyl ((1S,3S)-3-((5-(5-methyl-6-oxopyridazin-l(67/)-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-66)
- Int-66 was synthesized following the Int-2 starting from Int-65, ESI-MS m/z: 385.75 [M+H] + .
- Step-3 Synthesis of 2-(6-(((1S,3S)-3-aminocyclopentyl) amino) pyridin-3-yl)-4-methylpyridazin- 3(2H)-one hydrochloride (lnt-67) lnt-67 was synthesized following the Int-3 starting from Int-66, ESI-MS m/z: 286.10 [M+H] + .
- Step-4 Synthesis ooff A L ((1S,3S)-3-((5-(5-methyl-6-oxopyridazin-l(67/)-yl) pyridin-2- yl)amino)cyclopentyl)cyanamide (Int-68)
- Int-68 was synthesized following the Int-4 starting from lnt-67, ESI-MS m/z: 310.95 [M+H] + .
- Step-5 Synthesis of 2-hydroxy-l-((1S,3S)-3-((5-(5-methyl-6-oxopyridazin-l(67/)-yl)pyridin-2- yl)amino)cyclopentyl)guanidine (Int-69)
- Int-69 was synthesized following the Int-5 starting from Int-68, ESI-MS m/z: 344.20 [M+H] + .
- Step-6 Synthesis ooff 4-methyl-2-(6-(((15,35)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 37)
- Example 38 Synthesis of 2-(6-(((15',35')-3-((5-(3-methvloxetan-3-vl)-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-yl)Dvridazin-3(27D-one
- Example 39 Synthesis Of 2-(6-(((15',35')-3-((5-(cvcloDroDvlmethvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(27D-one
- Step-1 Synthesis of Synthesis of 2-acetoxy-2-methylpropanoic acid (Int-70)
- Int-71 was synthesized following the Int-12 starting from Int-71.
- Step-3 Synthesis of 2-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(67/)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)propan-2-yl acetate (Int-72)
- Int-72 was synthesized following the Example 15 starting from Int-5 and Int-71.
- Step-4 Synthesis ooff 2-(6-(((15,35)-3-((5-(2-hydroxypropan-2-yl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 40)
- Int-73 was synthesized following the Int-70 starting from commercially available 1-hydroxycyclobutane- 1 -carboxylic acid.
- Int-74 was synthesized following the Int-12 starting from Int-73.
- Step-3 Synthesis of l-(3-(((l 1 S',3 1 S')-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)cyclobutyl acetate (Int-75)
- Int-75 was synthesized following the Example 15 starting from Int-5 and Int-74.
- Step-4 1 Synthesis oOff 2-(6-(((1S,3S)-3-((5-(l-hydroxycyclobutyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 41)
- Example 42 was synthesized following the Example 2 starting from Int-5 with commercially available cyclohexanecarboxylic acid. ESI-MS m/z: 422.40 [M+H] + .
- Example 43 2-(6-(((1S,3S)-3-((5-phenvl-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pyridin-3- vl)pyridazin-3(277)-one
- Example 44 2-(6-(((1S,3S)-3-((5-(pvridin-3-vl)-l,2,4-oxadiazol-3- vl)amino)cvclonentvl)amino)pvridin-3-vl)pyridazin-3(277)-one
- Example 44 was synthesized following the Example 2 starting from Int-5 with commercially available nicotinic acid. ESI-MS m/z: 416.95 [M+H] + .
- 1 H NMR 400 MHz, CD 3 OD
- Example 46 Synthesis of 2-(6-(((1S,3S)-3-((5-((methvlsulfonvl)methvl)-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-vl)Dvridazin-3(2H)-one
- Step-1 Synthesis of 3-acetoxy-2,2-dimethylpropanoic acid (Int-76)
- Int-76 was synthesized following the Int-70 starting from commercially available 3-hydroxy-2,2- dimethylpropanoic acid. 'H NMR (300 MHz, CDC1 3 ) 5 4.18 (s, 2H), 2.08 (s, 3 H), 1.35 (s, 6 H).
- Int-77 was synthesized following the Int-12 starting from Int-76. ’H NMR: (400MHz, DMSO-de) 5 4.15 (s, 2H), 3.32 (s, 4 H), 2.03 (s, 3 H), 1.33 (s, 6H)
- Step-3 Synthesis of 2-methyl-2-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)propyl acetate (Int-78)
- Int-78 was synthesized following the Example 15 starting from Int-5 and Int-77.
- Step-4 Synthesis of Synthesis of 2-(6-(((15,35)-3-((5-(l-hydroxy-2-methylpropan-2-yl)-l,2,4- oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 47)
- Example 48 Synthesis ooff 2-(6-(((1S,3S)-3-((5-(2,2,2-trifluoroethvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(27O-one
- Example _ 49 _ methyl _ l-(3-(((l 1 $',3 1 S')-3-((5-(6-oxopvridazin-l(6H)-vl)pvridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)cvclopropane-l-carboxylate
- Example 49 was synthesized following the Example 2 starting from Int-5 with commercially available 1- (methoxycarbonyl)cyclopropane-l -carboxylic acid. ESI-MS m/z: 438.15 [M+H] + .
- Example 50 Synthesis of l-methyl-6'-(((15,35)-3-((5-methyl-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)-[3,3'-bipyridin]-2(lZ7)-one Step-1; Synthesis of Synthesis of tert-butyl ((15,35)-3-((5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl) pyridin-2-yl) amino) cyclopentyl) carbamate (Int-79)
- Step-2- Synthesis of tert-butyl ((lS,35)-3-((r-methyl-2'-oxo-r,2'-dihydro-[3,3'-bipyridin]-6-yl) amino) cyclopentyl) carbamate (Int-80)
- Step-3 Synthesis of 6'-(((LS',3 > S')-3-aminocyclopcntyl) amino)-l-methyl-[3,3'-bipyridin]-2(17/)-one hydrochloride (Int-81)
- Int-81 was synthesized following the Int-3 starting from Int-80, ESI-MS m/z: 285.10 [M+H] + .
- Step-4- Synthesis of N-((15,35)-3-((l'-methyl-2'-oxo-l',2'-dihydro-[3,3'-bipyridin]-6-yl) amino) cyclopentyl) cyanamide (Int-82)
- Int-82 was synthesized following the Int-4 starting from Int-81, ESI-MS m/z: 310.05 [M+H] + .
- Step-5 Synthesis of 2-hydroxy-l-((15,35)-3-((l'-methyl-2'-oxo-l',2'-dihydro-[3,3'-bipyridin]-6- yl)amino)cyclopentyl)guanidine (Int-83)
- Int-83 was synthesized following the Int-5 starting from Int-82, ESI-MS m/z: 343.15 [M+H] + .
- Step-6 Synthesis of l-methyl-6'-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)-[3,3'-bipyridin]-2(17/)-one (Example 50)
- Example 51 Synthesis of 2-methyl-2-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(67f)-yl) pyridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-yl)propanenitrile
- Example 53 Synthesis of l-(3-(((15',35')-3-((5-(6-oxopyridazin-l(6Zf)-yl)pyridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)cvclopropane-l-carboxylic acid
- Example 49 To a solution of Example 49 (200 mg, 0.45 mmol) in MeOH (3.0 mL) and THF(7.0 mL) was added LiOH.H2O (56 mg, 1.37 mmol) in H2O (1.0 mL). After stirred for 2 h, the reaction was quenched with water and the product was extracted with EtOAc. The organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. .
- Example 54 Synthesis of 2-(6-(((1S,3S)-3-((5-(l-methvl-lH-l,2,4-triazol-3-vl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one
- Step-1 Synthesis of l-methyl-lH-l,2,4-triazole-3-carboxylic acid (Int-84)
- Step-2 Synthesis ooff 2-(6-(((1S,3S)-3-((5-(l-methyl-lH-l,2,4-triazol-3-yl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2Z/)-one (Example 54)
- Example 54 was synthesized following the Example 2 starting from Int-5 and Int-84. ESI-MS m/z: 421.10 [M+H] + .
- Example 55 Synthesis of 2-(3-(((LS',3.S')-3-((5-(6-oxopvridazin-l(6//)-vl)i)vridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)acetonitrile
- Int-85 was synthesized following the Int-12 starting from commercially available 2-cyanoacetic acid. ’H NMR (300 MHz, CD 3 OD) 5 3.68 (s, 2H), 2.67 (s, 4H).
- Step-2 Synthesis of 2-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(67/)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l ,2,4-oxadiazol-5-yl)acetonitrile (Example 55)
- Example 55 was synthesized following the Example 15 starting from Int-5 and Int-85. ESI-MS m/z: 378.90 [M+H] + .
- Example 56 Synthesis of 2 - (6 -((( l 1 S',3 1 S')-3-((5-eth yl-1 ,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-vl)-4-methvlpvridazin-3(2ZD-one
- Example 57 Synthesis of l-(3-(((1S,3S)-3-((5-(6-oxoDvridazin-l(6Zn-vl)Dvridin-2- vl)amino)cvcloDentvl)amino)-l,2,4-oxadiazol-5-vl)cvcloDropane-l-carbonitrile
- Example 57 was synthesized following the Example 2 starting from Int-5 with commercially available 1- cy anocyclopropane- 1 -carboxylic acid. ESI-MS m/z: 405.02 [M+H] + .
- 'HNMR 400 MHz, CD3OD
- Example 58 Synthesis of ethyl l-(3-(((f 1 S',3 1 S')-3-((5-(6-oxopvridazin-l(677)-vl)pvridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)cvclobutane-l-carboxylate
- Example 58 was synthesized following the Example 2 starting from Int-5 with commercially available 1- (ethoxycarbonyl)cyclobutane-l -carboxylic acid. ESI-MS m/z: 466.0 [M+H] + .
- Example 59 Synthesis of l-(3-(((1S,3S)-3-((5-(6-oxopvridazin-l(6Z7)-vl)pvridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)cvclobutane-l-carboxylic acid
- Example 60 Synthesis of l-methyl-3-(6-(((l 1 $',3 1 S')-3-((5-(l-methvlcvclonropvl)-l,2,4-oxadiazol-3- vl)amino)cvclonentvl)amino)pyridin-3-vl)imidazolidine-2,4-dione
- Example 61 Synthesis of 3-(6-(((l 1 $',3 1 S')-3-((5-cvclobutvl-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pyridin-3-vl)-l-methvlimidazolidine-2, 4-dione
- Example 62 Synthesis of l
- Example 62 was synthesized following the Example 2 starting from Int-64 with commercially available 1 -methylcyclopropane- 1 -carboxylic acid. ESI-MS m/z: 440.0 [M+H] + .
- Example 63 Synthesis of l-(3-(((15 , ,35 , )-3-((5-(6-oxopvridazin-l(6H)-vl)pyridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-yl)cvclobutane-l-carbonitrile
- Step-1 Synthesis ooff Synthesis ooff l-(3-(((LS',3.S')-3-((5-(6-oxopyridazin-l(6//)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)cyclobutane-l-carboxamide (Int-86)
- Example 59 To a solution of Example 59 (35 mg, 0.08 mmol) in DMF (1.0 mL) were added ammonium chloride (21 mg, 0.40 mmol), HATH (45 mg, 0.12 mmol) and DIPEA ( 0.06 mL, 0.40 mmol). After stirring for 48 h, the reaction was quenched with water and the product was extracted with EtOAc. The organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide crude product as a pale brown sticky solid (50 mg, Crude) . ESI-MS m/z: 436.95 [M+H] + .
- Step-2 Synthesis oOff l-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)cyclobutane-l-carbonitrile (Example 63)
- Example 64 Synthesis of 3-(6-(((lS'.3S r )-3-((5-cvclopropvl-1.2.4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)-l,5,5-trimethylimidazolidine-2,4-dione
- Example 65 Synthesis of 3-(6-(((lS , ,3S)-3-((5-cyclopropyl-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pyridin-3-vl)-l-methvlimidazolidine-2,4-dione
- Example 66 Synthesis of 2-(6-(((1S,3S)-3-((5-cvclopropvl-l,2,4-thiadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)-4-methvlpyridazin-3(2ZD-one
- Int-76 was synthesized following the Int-70 starting from commercially available 1 -hydroxy cyclopropane- 1 -carboxylic acid. *H NMR (400 MHz, DMSO-d s ) 5 12.86 (s, 1H), 2.01 (s, 3H), 1.36 - 1.33 (m, 2H), 1.17
- Step-2 Synthesis ooff l-(3-(((1S,3S)-3-((5-(3-methyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)cyclopropyl acetate (Int-77)
- Int-77 was synthesized following the Example 2 starting from Int-76 and Int-11.
- Step-3 Synthesis ooff 3-(6-(((LS',3.S')-3-((5-(l-hydroxycyclopropyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)-l-methylimidazolidine-2, 4-dione (Example 67)
- TR-FRET time resolved fluorescence resonance energy transfer
- a master compound plate was prepared in a Greiner V bottom plate by diluting compounds of the disclosure in dimethylsulfoxide to the correct concentration for the desired top concentration based on the desired final concentration: for a 30 pM final concentration the master plate concentration is 1.5 mM (68 pL DMSO + 12 pL 10 mM of a compound of the disclosure), for a 10 pM final concentration the master plate concentration is 0.5 mM (76 pL DMSO + 4 pL 10 mM of a compound of the disclosure), for a 3 pM final concentration the master plate concentration is 150 pM (69 pL DMSO + 1 pL 10 mM of a compound of the disclosure).
- An intermediate plate was generated in a Greiner V bottom plate by transferring 8 pL from each well of the master plate into a corresponding well containing 92 pL of assay buffer and mixing thoroughly.
- a Proxi plate-low volume assay plate was used for the assay. To all wells of the plate was added 10 pL of 7 nM Human PCSK9 Terbium, followed by 5 pL from the intermediate plate. For the competition displacement control wells in columns 23 and 24 of the plate, 5 pL of unlabeled human PCSK9 was added at 4 pM in assay buffer containing 8% DMSO. Following a 30 minute incubation, 5 pL of 120 pM Alexa Fluor 647 labeled probe was added and the mixture was incubated for an additional 2 hours.
- TR-FRET TR-FRET signal was measured on an EnVision instrument with a 60 ms delay, 330 nm excitation and 665 nm emission (FRET), and 330 nM excitation and 615 nm (Terbium).
- FRET 330 nm excitation and 665 nm emission
- Terbium 330 nM excitation and 615 nm
- hERG expressing cell lines were produced using CH0-K1 T-Rex inducible plasmid system (Invitrogen) as described previously (Cao et al, Assay Drug Dev. Technol. 2010, 8, 766-780).
- C 6 ll lines were maintained in Ham’s F12 nutrient mixture containing 10% FBS, blasticidin (10 mg/mL; InvivoGen), hygromycin B (200 mg/mL;InvivoGen), Zeocin (200 mg/mL, Invitrogen), and neomycin (200 mg/mL, Invitrogen) using SelecT automated cell culture system (TAP Biosystems, Cambridge, U.K.).
- hERG and hCavl.2 channels expression was induced with tetracycline (0.25-1 ⁇ g/mL, Invitrogen) at least 24 h prior to the experiment.
- hERG currents were recorded using the Qpatch automated patch clamp systems (Sophion Bioscience Inc., North Brunswick, NJ) in the whole (single) cell configuration.
- hERG expressing CHO-K1 cells were harvested with Detachin (Genlantis) and stored in the modified serum-free SFM-2 media (Life Technologies) at room temperature.
- the extracellular solution contained (in mM) NaCl (145), KC1 (4), MgC12 (1), CaC12 (2), and HEPES (10), pH 7.4, with NaOH.
- the intracellular solution contained KC1 (135), MgC12 (1.75), CaC12 (5.4), EGTA (10), K2-ATP (4), and HEPES (10), pH 7.2, with KOH.
- Reference Compound 1 is 2-(6-(((lS,3S)-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one. The compound was prepared following the procedure reported in W02020150473.
- Reference Compound 2 is 3-(6-(((lS,3S)-3-((5- (difluoromethoxy)pyrimidin-2-yl)amino)cyclopentyl)amino)pyridin-3-yl)-l-methylimidazolidine-2,4- dione The compound was prepared following the procedure reported in W02020150473.
- Examples 1, 2, 5, 6, 10, 24, 29 and 31 showed unexpectedly reduced inhibition of the hERG channel, compared to the reference compounds (Reference compounds 1 and 2). Reduced inhibition of the hERG channel may translate into an improved safety profile for the compounds as compared to reference compounds.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The disclosure relates to a compound of Formula (I) or a pharmaceutically acceptable salt thereof wherein A and B are as described herein, as well as compositions and methods of using such compounds.
Description
DIAMINOCYCLOPENTYLPYRIDINE DERIVATIVES FOR THE TREATMENT OF A DISEASE OR DISORDER RELATED APPLICATIONS [001] This application claims the benefit of and priority to U.S. Provisional Application Nos. 63/278,754, filed November 12, 2021, 63/325,988 filed March 31, 2022, and 63/379,562 filed October 14, 2022, the entire contents of each of which are hereby incorporated by reference in their entireties. TECHNICAL FIELD [002] The present disclosure is directed to modulators of proprotein convertase subtilisin/ kexin type 9 (PCSK9) useful in the treatment of diseases or disorders. Specifically, the disclosure is concerned with compounds, compositions, and methods of treating diseases or disorders associated with PCSK9. BACKGROUND [003] Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a member of the secretory subtilase, subtilisin serine protease family, and is expressed in many tissues and cell types. The PCSK9 protein contains a signal sequence, a prodomain, a catalytic domain containing a conserved triad of residues (D186, H226 and S386), and a C-terminal domain and is synthesized as a soluble 74-kDa precursor that undergoes autocatalytic cleavage in the endoplasmic reticulum. The autocatalytic activity has been shown to be required for secretion. [004] PCSK9 has pronounced effects on plasma low density lipoprotein cholesterol (LDL-C) levels via its modulation of hepatic low density lipoprotein receptors (LDLR), the main route by which cholesterol is removed from the circulation. PCSK9 binds the LDLR and directs it to lysosomal degradation, thereby increasing plasma LDL-C levels and, in turn, coronary heart disease (CHD) risk. (Maxwell K. N., Proc. Natl. Acad. Sci., 101, 2004, 7100-7105; Park, S. W., J. Biol. Chem.279, 2004, 50630-50638; Lagace T.A., et. al. J. Clin. Invest.2006, 116(11):2995-3005). Overexpression of mouse or human PCSK9 in mice has been shown to elevate total and LDL-C levels and dramatically reduce hepatic LDLR protein, without an observed effect on the levels of mRNA, SREBP, or SREBP protein nuclear to cytoplasmic ratio. (Maxwell K. N., Proc. Natl. Acad. Sci.101, 2004, 7100-7105). Moreover, mutations in PCSK9 that cause loss of PCSK9 function in mouse models have also been shown to lower total and LDL-C levels. (Cohen, J. C., et al., N. Engl. J. Med., 354, 2006, 1264-1272). Thus, the results indicate that modulation of PCSK9 results in a reduction of LDLR protein levels. [005] Gene deletion of PCSK9 has also been conducted in mice. PCSK9 knockout mice show an approximate 50% reduction in plasma cholesterol levels and enhanced sensitivity to statins in reducing
plasma cholesterol (Rashid, S., et al., Proc. Natl. Acad. Sci., 2005, 102:5374-5379). Human genetic data strongly support the role of PCSK9 in LDL homeostasis. The link between PCSK9 and plasma LDL-C levels was first established by the discovery of PCSK9 missense mutations in patients with an autosomal dominant form of familial hypercholesterolemia (Abifadel, M., et al., Nature, 2003, 34:154-6). Patients carrying PCSK9 gain-of-function alleles have increased plasma LDL-C levels and premature CHD, whereas those with PCSK9 loss-of-function alleles have markedly reduced plasma LDL-C and are protected from CHD.
[006] PCSK9 also plays a role in Lipoprotein (a) (Lp(a)) metabolism. Lp(a) is a proatherogenic lipoprotein comprised of an LDL particle covalently linked to apoLp(a). Human genetic studies indicate that Lp(a) is causally associated with CHD risk. PCSK9 therapeutic antibodies have been shown to significantly reduce Lp(a) levels in patients with hypercholesterolemia. (Desai, N.R., et. al., Circulation. 2013, 128(9):962-969; Lambert, G., et. al., Clin. Sci., 2017, 131, 261-268). Patients receiving statin therapy treated with a monoclonal antibody against PCSK9 have shown up to 32% reduction in Lp(a) levels compared to placebo. (Desai N.R., et. al., Circulation. 2013, 128(9):962-969).
[007] In addition to having cardiovascular effects, PCSK9 plays an important role in sepsis, a lifethreatening condition caused by a body’s response to infection. Overexpression of PSCK9 in septic mice has been shown to aggravate sepsis by increasing inflammation, while inhibition of PCSK9 has been shown to reduce mortality. (Dwivedi, D. J., et al., Shock, 2016, 46(6), 672-680). Moreover, flow cytometry studies in human HepG2 cells have shown that PCSK9 negatively regulates gram-negative lipopolysaccharide (LPS) uptake by hepatocytes through the regulation of the LDLR-mediated bacterial lipid uptake of lipoteichoic acid (LTA) and LPS through an LDL-dependent mechanism. (Grin, P.M., et al., Nature, 2018, 8(1): 10496) Thus, inhibition of PCSK9 has the potential to treat sepsis by reducing the body’s immune response to an infection.
[008] Inhibition of PCSK9 with a small molecule inhibitor has the potential to be a treatment for a range of diseases. For these reasons, there remains a need for small molecule inhibitors of PCSK9.
SUMMARY
[009] In a first aspect, the disclosure relates to a compound of Formula (I):

2
or a pharmaceutically acceptable salt thereof wherein:
A is a 5 or 6 membered heterocycle or heteroaryl containing at least one N and at least one oxo at a ring carbon and is optionally substituted with (C1-C6)alkyl;

X is N, 0, or S;
Y is CR1 or N;
Z is CR2, NR3, 0, or S;
R1 and R2 are each, independently selected from H, halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6- C10)aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from 0 and N, wherein the (C1- C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1- C6)haloalkyl, (C1-C6)alkoxy, (C3-C6)cycloalkyl, -SO2(C1-C6)alkyl, -COOH, and -COO(C1-C6)alkyl;
R3 is absent, H or (C1-C6)alkyl; and
R4 is H, (C1-C6)alkyl, (C1-C6)haloalkyl, (C1-C6)alkoxy or (C3-C6)cycloalkyl, wherein the (C1-C6)alkoxy is optionally substituted with halogen.
[010] In another aspect, the present disclosure relates to a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable carriers.
[Oil] In another aspect, the present disclosure relates to a combination comprising a compound of Formula I or a pharmaceutically acceptable salt thereof and one or more pharmaceutical agents.
[012] In another aspect, the present disclosure relates to a method for treating a disease or disorder comprising administering to a patient in need thereof a therapeutically effective amount of a compound of Formula I or a pharmaceutically acceptable salt thereof.
[013] In some embodiments, the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides. In some embodiments, the disease or disorder is selected from sepsis, psoriasis and cancer.
[014] In yet another aspect, the present disclosure relates to a method of modulating PCSK9 comprising administering to a patient in need thereof a compound of Formula I or a pharmaceutically acceptable salt thereof. In still another aspect, the present disclosure relates to a method of inhibiting PCSK9 comprising administering to a patient in need thereof a compound of Formula I or a pharmaceutically acceptable salt thereof.
[015] In another aspect, the present disclosure relates to a compound of Formula I or a pharmaceutically acceptable salt thereof for use as a medicament.
[016] In another aspect, the present disclosure relates to a compound of Formula I or a pharmaceutically acceptable salt thereof for use in the treatment of a disease or disorder.
[017] In yet another aspect, the present disclosure relates to a compound of Formula I for use in the manufacture of a medicament for treating a disease or disorder.
[018] In still another aspect, the present disclosure relates to use of a compound of Formula I or a pharmaceutically acceptable salt thereof in the treatment of a disease or disorder.
[019] Other features and advantages of the disclosure will be apparent from the following detailed description and claims.
DETAILED DESCRIPTION
[020] In certain aspects, the disclosure provides substituted diaminocyclopentylpyridine compounds, and pharmaceutical compositions thereof. In particular, such substituted compounds are useful as PCSK9 inhibitors, and thus can be used to treat or prevent a disease or condition.
[021] The details of the disclosure are set forth in the accompanying description below. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of the present disclosure, illustrative methods and materials are now described. Other features, objects, and advantages of the disclosure will be apparent from the description and from the claims. In the specification and the appended claims, the singular forms also include the plural unless the context clearly dictates otherwise. Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure belongs. All patents and publications cited in this specification are incorporated herein by reference in their entireties.
Compounds
[022] In one aspect, the disclosure therefore provides a compound of formula (I)

A is a 5 or 6 membered heterocycle or heteroaryl containing at least one N and at least one oxo at a ring carbon and is optionally substituted with (C1-C6)alkyl;

X is N, 0, or S;
Y is CR1 or N;
Z is CR2, NR3, 0, or S;
R1 and R2 are each, independently selected from H, halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl, (C5- C10)aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from 0 and N, wherein the (C1- C6)alkyl, (C3-C6)cycloalkyl, ( C6- C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1- C6)haloalkyl, (C1-C6)alkoxy, (C3-C6)cycloalkyl, -SO2(C1-C6)alkyl, -COOH, and -COO(C1-C6)alkyl;
R3 is absent, H or (C1-C6)alkyl; and
R4 is H, (C1-C6)alkyl, (C1-C6)haloalkyl, (C1-C6)alkoxy or (C3-C6)cycloalkyl, wherein the (C1-C6)alkoxy is optionally substituted with halogen.
[023] Unless specified otherwise, the term “compounds of the present disclosure” or “compound of the present disclosure” refers to compounds of formula (I) thereof, and exemplified compounds, and salts thereof, as well as all stereoisomers (including diastereoisomers and enantiomers), rotamers, tautomers and isotopically labeled compounds (including deuterium substitutions), as well as inherently formed moieties.
[024] Various embodiments of the disclosure are described herein. It will be recognized that features specified in each embodiment may be combined with other specified features of other embodiments to provide further embodiments.
[025] In some embodiments, the compound is a compound of formula II
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
[026] In some embodiments, the compound is a compound of formula III
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
[027] In some embodiments, at least one of X, Y and Z is N. In some embodiments, when X is O or S, Z is CR2 or N. In some embodiments, when Z is O or S, X is N.
[028] In some embodiments, X is N. In some embodiments, X is N, Y is CR1, and Z is O or S
[029] In some embodiments, B is
 In some embodiments, B is
In some embodiments, B is

 , particularly B is or , more
, particularly B is or , more

 particularly B is
particularly B is

[030] In some embodiments, B is
 . In some embodiments, B is
. In some embodiments, B is

, or
 [031] In some embodiments, A is
[031] In some embodiments, A is

 , or
, or
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is

 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is

 , particularly A is
, particularly A is

[032] In some embodiments, R1 is H, (C1-C6)alkyl, (C3-C6)cycloalkyl, ( C6- C10)aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from 0 and N, wherein the (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1-C6)haloalkyl, (C1-C6)alkoxy, (C3- C6)cycloalkyl, -SO2(C1-C6)alkyl, -COOH, and -COO(C1-C6)alkyl. In some embodiments, R1 is H, (C1- C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, a 4 to 6 membered heterocyclyl comprising 1 heteroatom selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1 or 3 heteroatoms selected from N, wherein the (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1 C6)alkyl, (C1-C6)haloalkyl, (C1-C6)alkoxy, (C3-C6)cycloalkyl, -SO2(C1-C6)alkyl, -COOH, and -COO(C1- C6)alkyl.
[033] In some embodiments, R1 is H, (C1-C6)alkyl, (C3-C6)cycloalkyl, or (C6-C10)aryl, wherein the (C1- C6)alkyl, (C3-C6)cycloalkyl, or (C6-C10)aryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1-C6)haloalkyl, and (C1-C6)alkoxy. In some embodiments, R1 is H, (C1-C6)alkyl optionally substituted with one or more halogen, -CN, (C3- C6)cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C1- C6)alkyl and (C1-C6)haloalkyl, or (C6-C10)aryl optionally substituted one or more with halogen or CN. In some embodiments, R1 is (C1-C6)alkyl optionally substituted with one or more halogen or -CN, (C3-
C6)cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C1- C6)alkyl and (C1-C6)haloalkyl, or (C6-C10)aryl optionally substituted with one or more halogen or -CN. In some embodiments, R1 is H, (C1-C6)alkyl optionally substituted with one or more halogen, (C3- C6)cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C1- C6)alkyl and (C1-C6)haloalkyl, or (C6-C10)aryl optionally substituted one or more with halogen. In some embodiments, R1 is (C1-C6)alkyl optionally substituted with one or more halogen, (C3-C6)cyclo alkyl optionally substituted with one or more substituents selected from halogen, -OH, (C1-C6)alkyl and (C1- C6)haloalkyl, or (C6-C10)aryl optionally substituted with one or more halogen. In some embodiments, R1 is H, (C1-C6)alkyl optionally substituted with one or more fluoro, (C3-C6)cyclo alkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C1-C6)alkyl and (C1-C6)fluoroalkyl, or ( C6- C10)aryl optionally substituted one or more with fluorine. In some embodiments, R1 is (C1-C6)alkyl optionally substituted with fluoro, (C3-C6)cycloalkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C1-C6)alkyl and (C1-C6)haloalkyl, or (C6-C10)aryl optionally substituted with one or more fluoro. In some embodiments, R1 is (C1-C6)alkyl optionally substituted with one or more fluoro or -CN. In some embodiments, R1 is (C1-C6)alkyl optionally substituted with one or more fluoro. In some embodiments, R1 is unsubstituted (C1-C6)alkyl. In some embodiments, R1 is (C3-C6)cycloalkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C1-C6)alkyl and (C1- C6)fluoroalkyl. In certain embodiments, R1 is (C3-C6)cycloalkyl.
[034] In some embodiments, R1 is phenyl optionally substituted with one or more fluoro.
[035] In some embodiments, R1 is -CH3, -CH2CH3, -CHF2, -CF3, -CF2CH3, -CH2CF3, -CH2CN,
 or In certain embodiments, R1 is -CH3, -CH2CH3, -CHF2, -CF3, -CF2CH3, or
or In certain embodiments, R1 is -CH3, -CH2CH3, -CHF2, -CF3, -CF2CH3, or
 . In
. In

 certain embodiments, R1 is -CH3, -CH2CH3, -CHF2, -CF3, or -CF2CH3. In certain preferred embodiments,
certain embodiments, R1 is -CH3, -CH2CH3, -CHF2, -CF3, or -CF2CH3. In certain preferred embodiments,
R1 is -CH3, -CHF2, or -CF3.
[036] , In some embodimen .ts, R 1. is


 In Some embodiments, R1 is
In Some embodiments, R1 is


[037] In some embodiments, R1 is
 In some embodiments, R is
In some embodiments, R is

.
In some embodiments, R1 is
 . In some embodiments, R1 is
. In some embodiments, R1 is

[038] In some embodiments, R2 is H, halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl, or (C6-C10)aryl. In some embodiments, R2 is H, fluoro, chloro, (C1-C6)alkyl, (C3-C6)cycloalkyl, or (C6-C10)aryl. In some embodiments, R2is halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl, or phenyl. In some embodiments, R2 is fluoro, chloro, (C1-C6)alkyl, (C3-C6lcycloalkyl, or phenyl. In some embodiments, R2 is -Cl, -CH?.
 or
or
 [039] In some preferred embodiments, R3 is H.
[039] In some preferred embodiments, R3 is H.
In some embodiments, R4 is (C1-C6)alkyl or (C3-C6) cycloalkyl. In some embodiments, R4 is (C1-C6)alkyl.
In some embodiments, R4 is methyl, ethyl, or cyclopropyl. In some embodiments, R4 is methyl or ethyl. In some embodiments, R4 is (C1-C6)alkoxy optionally substituted with halogen. In some embodiments, R4 is -OCF3.
[040] In some embodiments, B is



. In some embodiments,

B is . In some
 embodiments, B is
embodiments, B is
 , or
, or
1-
. In certain embodiments, B is

[041] Embodiment 1. A compound of formula (I), or a pharmaceutically acceptable salt thereof, as described above.
[042] Embodiment 2. A compound according to embodiment 1 or a pharmaceutically acceptable salt thereof, wherein at least one of X, Y and Z is N.
[043] Embodiment 3. A compound according to embodiment 1 or embodiment 2 or a pharmaceutically acceptable salt thereof, wherein when X is 0 or S, Z is CR2 or N.
[044] Embodiment 4. A compound according to any one of embodiments 1 to 3 or a pharmaceutically acceptable salt thereof, wherein when Z is 0 or S, X is N.
[045] Embodiment 5. A compound according to embodiment 1 or a pharmaceutically acceptable salt thereof, wherein R4 is methyl, ethyl, or cyclopropyl.
[046] Embodiment 6. A compound according to any one of embodiments 1 to 5 or a pharmaceutically acceptable salt thereof, wherein A is


[047] Embodiment 7. A compound according to any one of embodiments 1 to 6 or a pharmaceutically acceptable salt thereof, wherein A is

[048] Embodiment 8. A compound according to any one of embodiments 1 to 7 or a pharmaceutically acceptable salt thereof, wherein X is N.
[049] Embodiment 9. A compound according to any one of embodiments 1 to 8 or a pharmaceutically acceptable salt thereof, wherein X is N, Y is CR1, and Z is 0 or S.
[050] Embodiment 10. A compound according to any one of embodiments 1 to 9 or a pharmaceutically acceptable salt thereof, wherein X is N, Y is CR1, and Z is O.
[051] Embodiment 11. A compound according to any one of embodiments 1 to 10 or a pharmaceutically acceptable salt thereof, wherein R1 is H, (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from O and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from O and N, wherein the (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1-C6)haloalkyl, (C1- C6)alkoxy, (C3-C6)cycloalkyl, -SO2(C1-C6)alkyl, -COOH, and -COO(C1-C6)alkyl.
[052] Embodiment 12. A compound according to any one of embodiments 1 to 11 or a pharmaceutically acceptable salt thereof, wherein R1 is H, (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, a 4 to 6 membered heterocyclyl comprising 1 heteroatom selected from O and N, or a 5 or 6 membered heteroaryl comprising 1 or 3 heteroatoms selected from N, wherein the (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1-C6)haloalkyl, (C1-C6)alkoxy, (C3- C6)cycloalkyl, -SO2(C1-C6)alkyl, -COOH, and -COO(C1-C6)alkyl.
[053] Embodiment 13. A compound according to any one of embodiments 1 to 12 or a pharmaceutically acceptable salt thereof, wherein R1 is H, (C1-C6)alkyl optionally substituted with one or more halogen, -CN, (C3-C6)cycloalkyl optionally substituted with one or more substituents selected from
halogen, -OH, (C1-C6)alkyl and (C1-C6)haloalkyl, or (C6-C10)aryl optionally substituted one or more with halogen or -CN.
[054] Embodiment 14. A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R1 is (C1-C6)alkyl optionally substituted with one or more halogen or -CN, (C3-C6)cyclo alkyl optionally substituted with one or more substituents selected from halogen, -OH, (C1-C6)alkyl and (C1-C6)haloalkyl, or (C6-C10)aryl optionally substituted with one or more halogen or -CN.
[055] Embodiment 15. A compound according to embodiment 13 or a pharmaceutically acceptable salt thereof, wherein each halogen is fluoro.
[056] Embodiment 16. A compound according to any one of embodiments 1 to 15 or a pharmaceutically acceptable salt thereof, wherein R1 is (C1-C6)alkyl optionally substituted with one or more fluoro or -CN.
[057] Embodiment 17. A compound according to any one of embodiments 1 to 16, wherein R1 is -CH3,

[058] Embodiment 18. A compound according to any one of embodiments 1 to 17, wherein R1 is -CH3, -CHF2, or -CF3.
[059] Embodiment 19. A compound according to any one of embodiments 1 to 14 or a pharmaceutically acceptable salt thereof, wherein R1 is (C3-C6)cycloalkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C1-C6)alkyl and (C1-C6)fluoroalkyl.
[060] Embodiment 20. A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R1 is


[061] Embodiment 21. A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R1 is phenyl optionally substituted with one or more fluoro.
[062] Embodiment 22. A compound according to embodiments 21 or a pharmaceutically acceptable salt thereof, wherein R1 is

[063] Embodiment 23. A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R1 is

Embodiment 24. A compound according to any one of embodiments 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R1 is ,

[064] Embodiment 25. A compound according to any one of embodiments 1 to 3, 5 to 8, and 11 to 24 or a pharmaceutically acceptable salt thereof, wherein R2 is H, halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl, or (C6-C10)aryl.
[065] Embodiment 26. A compound according to any one of embodiments Ito 3, 5 to 8, and 11 to 22 or a pharmaceutically acceptable salt thereof, wherein R2 is halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl. or phenyl
[066] Embodiment 27. A compound according to any one of embodiments 1 to 3, 5 to 8, and 11 to 26 or a pharmaceutically acceptable salt thereof, wherein R2 is -Cl, -CH3.

[067] Embodiment 28. A compound according to any one of embodiments 1 to 3, 5 to 8, and 11 to 27 or a pharmaceutically acceptable salt thereof, wherein R3 is H.
[068] Embodiment 29. A compound according to any one of embodiments 1 to 4 or a pharmaceutically acceptable salt thereof, wherein B is



[069] Embodiment 30. A compound according to any one of embodiments 1 to 4 or a pharmaceutically acceptable salt thereof,, wherein B is or


[070] Embodiment 31. A compound according to any one of embodiments 1 to 4 or a pharmaceutically acceptable salt thereof, wherein B is

[071] Embodiments 32 and 33. A compound according to embodiment 1 or a pharmaceutically acceptable salt thereof selected from:









 [072] Additionally, Applicants have surprisingly and unexpectedly found that compounds of Formula (I) exhibit low or no inhibition of the human Ether-a-go-go-Related Gene (hERG). As discussed herein, some aspects of cardiovascular toxicity of a compound can be measured using a hERG assay. The hERG gene encodes the inward rectifying voltage gated potassium channel in the heart known as KV11.1, which is involved in cardiac repolarization. Inhibition of the hERG current causes QT interval prolongation resulting in potentially fatal ventricular tachyarrhythmia. hERG inhibition an important antitarget that must be avoided. In some embodiments, compounds of Formula I exhibited no measurable hERG inhibition. This lack of cardiovascular toxicity by the compounds of Formula I is surprising and unexpected, especially considering the inhibition of hERG by 2-(6-(((lS,3S)-3-((5- cyclopropylpyrimidin-2-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one and 3-(6- ((( IS, 3 S)-3 -((5 -(difluoromethoxy )pyrimidin-2-yl)am ino)cyclopentyl)am ino)pyridin-3-y 1)-1- methylimidazolidine-2, 4-dione.
[072] Additionally, Applicants have surprisingly and unexpectedly found that compounds of Formula (I) exhibit low or no inhibition of the human Ether-a-go-go-Related Gene (hERG). As discussed herein, some aspects of cardiovascular toxicity of a compound can be measured using a hERG assay. The hERG gene encodes the inward rectifying voltage gated potassium channel in the heart known as KV11.1, which is involved in cardiac repolarization. Inhibition of the hERG current causes QT interval prolongation resulting in potentially fatal ventricular tachyarrhythmia. hERG inhibition an important antitarget that must be avoided. In some embodiments, compounds of Formula I exhibited no measurable hERG inhibition. This lack of cardiovascular toxicity by the compounds of Formula I is surprising and unexpected, especially considering the inhibition of hERG by 2-(6-(((lS,3S)-3-((5- cyclopropylpyrimidin-2-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one and 3-(6- ((( IS, 3 S)-3 -((5 -(difluoromethoxy )pyrimidin-2-yl)am ino)cyclopentyl)am ino)pyridin-3-y 1)-1- methylimidazolidine-2, 4-dione.
[073] As used herein, the terms “salt” or “salts” refers to an acid addition or base addition salt of a compound of the present disclosure. “Salts” include in particular “pharmaceutical acceptable salts”. The term “pharmaceutically acceptable salts” refers to salts that retain the biological effectiveness and properties of the compounds of this disclosure and, which typically are not biologically or otherwise undesirable. In many cases, the compounds of the present disclosure are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto. When both a basic group and an acid group are present in the same molecule, the compounds of the present disclosure may also form internal salts, e.g., zwitterionic molecules.
[074] Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids.
[075] Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
[076] Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like.
[077] Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
[078] Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table. In certain embodiments, the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
[079] Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like. C6rtain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
[080] In another aspect, the present disclosure provides compounds of the present disclosure in acetate, ascorbate, adipate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfonate, caprate, chloride/hydrochloride, chlortheophyllonate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, glutamate, glutarate, glycolate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, mucate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, polygalacturonate, propionate, sebacate, stearate, succinate, sulfosalicylate, sulfate, tartrate, tosylate trifenatate, trifluoroacetate or xinafoate salt form.
[081] Any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms of the compounds. Isotopically labeled compounds have structures depicted by the formulae given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number. Isotopes that can be incorporated into compounds of the disclosure include, for example, isotopes of hydrogen.
[082] In another aspect, the disclosure provides a compound of formula (la)
 or a pharmaceutically acceptable salt thereof, wherein each R10a, R10b, R10c, Rlla, Rllb, R12a, R12b, R12c, R12d, R12e and R12 is independently selected from H or deuterium; and A and B are as defined herein.
or a pharmaceutically acceptable salt thereof, wherein each R10a, R10b, R10c, Rlla, Rllb, R12a, R12b, R12c, R12d, R12e and R12 is independently selected from H or deuterium; and A and B are as defined herein.
[083] Further, incorporation of certain isotopes, particularly deuterium (i.e., 2H or D) may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements or an improvement in therapeutic index or tolerability. It is understood that deuterium in this context is regarded as a substituent of a compound of the present disclosure. The
concentration of deuterium, may be defined by the isotopic enrichment factor. The term "isotopic enrichment factor" as used herein means the ratio between the isotopic abundance and the natural abundance of a specified isotope. If a substituent in a compound of this disclosure is denoted as being deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation). It should be understood that the term “isotopic enrichment factor” can be applied to any isotope in the same manner as described for deuterium.
[084] Other examples of isotopes that can be incorporated into compounds of the disclosure include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 3H, nC, 13C, 14C, 15N, 18F 31P, 32P, 35S, 36C1, 123I, 1241, 125I respectively. Accordingly it should be understood that the disclosure includes compounds that incorporate one or more of any of the aforementioned isotopes, including for example, radioactive isotopes, such as 3H and 14C, or those into which non-radio active isotopes, such as 2H and 13C are present. Such isotopically labelled compounds are useful in metabolic studies (with 14C), reaction kinetic studies (with, for example 2H or 3H), detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in radioactive treatment of patients. In particular, an 18F or labeled compound may be particularly desirable for PET or SPECT studies. Isotopically-labeled compounds of the present disclosure can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
[085] Any asymmetric atom (e.g., carbon or the like) of the compound(s) of the present disclosure can be present in racemic or enantiomerically enriched, for example the (R~)-, (5)- or (R, 5)- configuration. In certain embodiments, each asymmetric atom has at least 50 % enantiomeric excess, at least 60 % enantiomeric excess, at least 70 % enantiomeric excess, at least 80 % enantiomeric excess, at least 90 % enantiomeric excess, at least 95 % enantiomeric excess, or at least 99 % enantiomeric excess in the (R~)- or (5)- configuration. Substituents at atoms with unsaturated double bonds may, if possible, be present in cis- (Z)- or trans- (E)- form.
[086] Accordingly, as used herein a compound of the present disclosure can be in the form of one of the possible stereoisomers, rotamers, atropisomers, tautomers or mixtures thereof, for example, as
substantially pure geometric (cis or trans) stereoisomers, diastereomers, optical isomers (antipodes), racemates or mixtures thereof.
[087] Any resulting mixtures of stereoisomers can be separated on the basis of the physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization.
[088] Any resulting racemates of compounds of the present disclosure or of intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the diastereomeric salts thereof, obtained with an optically active acid or base, and liberating the optically active acidic or basic compound. In particular, a basic moiety may thus be employed to resolve the compounds of the present disclosure into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl tartaric acid, diacetyl tartaric acid, di-G.G'-/)-toluoyl tartaric acid, mandelic acid, malic acid or camphor- 10-sulfonic acid. Racemic compounds of the present disclosure or racemic intermediates can also be resolved by chiral chromatography, e.g., high pressure liquid chromatography (HPLC) using a chiral adsorbent.
Pharmaceutical Compositions
[089] In another aspect, the present disclosure provides a pharmaceutical composition comprising a compound of the present disclosure, or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable carriers. In a further embodiment, the composition comprises at least two pharmaceutically acceptable carriers, such as those described herein.
[090] In some embodiments, pharmaceutical composition further comprises at least one additional pharmaceutically active agent. In some embodiments, the additional pharmaceutically active agent is selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) 0-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA: diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl- like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9, an inhibitory nucleic acid targeting Lp(a), an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, anti-obesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9, an immune checkpoint inhibitor and combinations thereof. In certain preferred embodiments, the additional pharmaceutically active agent is selected from bempedoic acid, statins, ezetimibe, inclisiran, pelacarsen, evolocumab, PD-1, PD-L1, PD-L2 and combinations thereof
[091] The pharmaceutical composition can be formulated for particular routes of administration such as oral administration, parenteral administration (e.g. by injection, infusion, transdermal or topical administration), and rectal administration. Topical administration may also pertain to inhalation or intranasal application. The pharmaceutical compositions of the present disclosure can be made up in a solid form (including, without limitation, capsules, tablets, pills, granules, powders or suppositories), or in a liquid form (including, without limitation, solutions, suspensions or emulsions). Tablets may be either film coated or enteric coated according to methods known in the art. Typically, the pharmaceutical compositions are tablets or gelatin capsules comprising the active ingredient together with one or more of: a) diluents, e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine; b) lubricants, e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or polyethyleneglycol; for tablets also c) binders, e.g., magnesium aluminum silicate, starch paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; if desired d) disintegrants, e.g., starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and e) absorbents, colorants, flavors and sweeteners.
[092] Liquid, particularly injectable, compositions can, for example, be prepared by dissolution, dispersion, etc. For example, the disclosed compound is dissolved in or mixed with a pharmaceutically acceptable solvent such as, for example, water, saline, aqueous dextrose, glycerol, ethanol, and the like, to thereby form an injectable isotonic solution or suspension. Proteins such as albumin, chylomicron particles, or serum proteins can be used to solubilize the disclosed compounds.
[093] The disclosed compounds can be also formulated as a suppository that can be prepared from fatty emulsions or suspensions; using polyalkylene glycols such as propylene glycol, as the carrier.
[094] Parental injectable administration is generally used for subcutaneous, intramuscular or intravenous injections and infusions. Injectables can be prepared in conventional forms, either as liquid solutions or suspensions or solid forms suitable for dissolving in liquid prior to injection.
[095] Compositions can be prepared according to conventional mixing, granulating or coating methods, respectively, and the present pharmaceutical compositions can contain from about 0.1% to about 99%, from about 5% to about 90%, or from about 1% to about 20% of the disclosed compound by weight or volume.
[096] The dosage regimen utilizing the disclosed compound is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; the renal or hepatic function of the patient; and the particular disclosed compound employed. A physician or veterinarian of ordinary skill in the art can
readily determine and prescribe the effective amount of the drug required to prevent, counter or arrest the progress of the condition.
[097] The pharmaceutical composition or combination of the present disclosure may, for example, be in unit dosage of about 1-1000 mg of active ingredient(s) for a subject of about 50-70 kg. In one embodiment, the compositions are in the form of a tablet that can be scored. The therapeutically effective dosage of a compound, the pharmaceutical composition, or the combinations thereof, is dependent on the species of the subject, the body weight, age and individual condition, the disorder or disease or the severity thereof being treated.
[098] Embodiment 34. A pharmaceutical composition comprising a compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable carriers.
[099] Embodiment 35. The pharmaceutical composition of embodiment 34, further comprising at least one additional pharmaceutically active agent.
[100] Embodiment 36. The pharmaceutical composition of embodiment 35, wherein the additional pharmaceutically active agent is selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression, an inhibitory nucleic acid targeting Lp(a) protein expression, an inhibitory nucleic acid targeting apoB 100, apoA-I up- regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, anti-obesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9, an immune checkpoint inhibitor and combinations thereof.
Activity of the Compounds
[101] The activity of compounds according to the present disclosure as PCSK9 inhibitors can be assessed using a time resolved fluorescence resonance energy transfer (TR-FRET) assay. This TR-FRET assay measures the ability of a compounds of the present disclosure to compete for binding with Alexa Fluor 647 labeled probe in a known region of human PCSK9. The assay provides measures of both potency (IC50) and efficacy (Amax).
[102] Solutions of varying concentrations are prepared by diluting a compound of the disclosure in dimethylsulfoxide (DMSO) and the resulting solutions are pipetted into a plate. DMSO is used as a no displacement inactive control. An intermediate plate is prepared in by transferring a known amount of
each compound solution and of the control from the compound plate into a corresponding well containing assay buffer and mixing thoroughly. A third plate is then prepared to be used for the assay by adding Terbium labeled human PCSK9, followed by a known amount of each solution from the intermediate plate. Unlabeled human PCSK9 in assay buffer containing DMSO is used as a control for the assay. Following incubation, Alexa Fluor 647 labeled probe is added to each well of the assay plate and the resulting mixture is incubated for an additional period of time. The TR-FRET signal is measured and the FRET ratio (FRET/Terbium) is used to calculate the IC50 and Amax of the compounds.
Method of Synthesizing the Compounds
[103] The compounds of the present disclosure may be made by a variety of methods, including standard chemistry. Suitable synthetic routes are depicted in the Schemes given below.
[104] The compounds of Formula (I) may be prepared by methods known in the art of organic synthesis as set forth in part by the following synthetic schemes. In the schemes described below, it is well understood that protecting groups for sensitive or reactive groups are employed where necessary in accordance with general principles or chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection processes, as well as the reaction conditions and order of their execution, shall be consistent with the preparation of compounds of Formula (I).
[105] Those skilled in the art will recognize if a stereocenter exists in the compounds of Formula (I). Accordingly, the present disclosure includes both possible stereoisomers (unless specified in the synthesis) and includes not only racemic compounds but the individual enantiomers and/or diastereomers as well. When a compound is desired as a single enantiomer or diastereomer, it may be obtained by stereospecific synthesis or by resolution of the final product or any convenient intermediate. Resolution of the final product, an intermediate, or a starting material may be affected by any suitable method known in the art. See, for example, "Stereochemistry of Organic Compounds" by E. L. Eliel, S. H. Wilen, and L. N. Mander (Wiley-lnterscience, 1994).
[106] The compounds described herein may be made from commercially available starting materials or synthesized using known organic, inorganic, and/or enzymatic processes.
Methods of Use
[107] In yet another aspect, the present disclosure is directed to a method of treating or preventing a disease or disorder comprising administering to a patient in need thereof an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof or a pharmaceutical composition
comprising a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, and a pharmaceutically acceptable carrier.
[108] In another aspect, the disclosure is directed to a method of modulating PCSK9 comprising administering to a patient in need thereof a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
[109] In another aspect, the disclosure is directed to a method of inhibiting PCSK9. The method involves administering to a patient in need thereof an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
[110] Another aspect of the disclosure relates to a method of treating, preventing, inhibiting, or eliminating a disease or disorder in which PCSK9 plays a role. The method comprises administering to a patient in need of a treatment for diseases or disorders in which PCSK9 plays a role an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
[111] Another aspect of the present disclosure relates to a method of treating, preventing, inhibiting, or eliminating a disease or disorder in a patient associated with the inhibition of PCSK9, the method comprising administering to a patient in need thereof an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
[112] In another aspect, the present disclosure relates to a method of treating, preventing, inhibiting, or eliminating a PCSK9-mediated disease or disorder. The method comprises administering to a patient in need of a treatment for a PCSK9-mediated disease or disorder an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, and a pharmaceutically acceptable carrier.
[113] In another aspect, the present disclosure relates to a method of reducing Lp(a), reducing Lp(a) plasma levels, reducing Lp(a) serum levels, reducing serum TRL or LDL levels, reducing serum triglyceride levels, reducing LDL-C, reducing total plasma apoB concentrations, reducing LDL apoB, reducing TRL apoB, or reducing non HDL-C. The method comprises administering to a patient in need thereof an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
[114] In another aspect, the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof for use a medicament.
[115] Another aspect of the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of a PCSK9-mediated disease or disorder.
[116] In another aspect, the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of a disease or disorder in which PCSK9 plays a role.
[117] In another aspect, the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of a disease or disorder , wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, , elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides. In another aspect, the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of a disease or disorder , the disease or disorder is selected from sepsis, psoriasis, psoriasis and cancer.
[118] Another aspect of the present disclosure relates to the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier in the manufacture of a medicament for treating of a PCSK9-mediated disease or disorder.
[119] Another aspect of the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
Another aspect of the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable thereof, and a pharmaceutically acceptable carrier for use in the treatment, prevention, inhibition, or elimination of sepsis, psoriasis, and cancer.
[120] Another aspect of the present disclosure relates to a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, and a pharmaceutically acceptable carrier for use in the manufacture of a medicament for treating a disease in which PCSK9 plays a role.
[121] In another aspect, the present disclosure relates to the use of a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, and a pharmaceutically acceptable carrier in the manufacture of a medicament treating a PCSK9-mediated disease or disorder.
[122] In another aspect, the present disclosure relates to the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier in the manufacture of a medicament for treating, preventing, inhibiting, or eliminating hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides. In another aspect, the present disclosure relates to the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier in the manufacture of a medicament for treating, preventing, inhibiting, or eliminating sepsis, psoriasis, and cancer.
[123] In certain embodiments, the disease or disorder is a PCSK9-mediated disease or disorder. In some embodiments, the PCSK9-mediated disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides. In some embodiments, the PCSK9-mediated disease or disorder is selected from sepsis, psoriasis, and cancer.
[124] In some embodiments, the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart
disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides. In some embodiments, the disease or disorder is selected from sepsis, psoriasis, and cancer.
[125] The disclosed compounds of the disclosure can be administered in effective amounts to treat or prevent a disorder and/or prevent the development thereof in subjects.
[126] Embodiment 37. The pharmaceutical composition of any one of embodiments 34 to 36 for use in the treatment of a PCSK9-mediated disease or disorder.
[127] Embodiment 38. The pharmaceutical composition of any one of embodiments 34 to 36 for use in the treatment of a disease or disorder, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
[128] Embodiment 41. A method for treating or preventing a disease or disorder comprising administering to a patient in need thereof a therapeutically effective amount of a compound according to any one of the embodiments 1 to 33, or a pharmaceutically acceptable salt thereof.
[129] Embodiment 42. The method of embodiment 41, wherein the disease or disorder is a PCSK9- mediated disease or disorder.
[130] Embodiment 43. The method of embodiments 42, wherein the PCSK9-mediated disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
[131] Embodiment 44. A method for treating a disease or disorder comprising administering to a patient in need thereof a therapeutically effective amount of a compound according to any one of the embodiments 1-33, or a pharmaceutically acceptable salt thereof, wherein the disease or disorder is selected from selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
[132] Embodiment 45. A method of modulating PCSK9 comprising administering to a patient in need thereof a compound of any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof.
[133] Embodiment 46. A method of inhibiting PCSK9 comprising administering to a patient in need thereof a compound of any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof.
[134] Embodiment 47. The method of any one of embodiments 41 to 46, wherein administering the compound is oral, parental, subcutaneous, by injection, or by infusion.
[135] Embodiment 48. A compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof, for use as a medicament.
[136] Embodiment 49. A compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof, for use in the treatment of a PCSK9-mediated disease or disorder.
[137] Embodiment 50. The compound of embodiment 49, wherein the PCSK9-mediated disease or disorder selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
[138] Embodiment 51. A compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof, for use in the treatment of a disease or disorder, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
[139] Embodiment 52. A compound according to any one of the embodiments 1 to 33, or a pharmaceutically acceptable salt thereof, for use in the manufacture of a medicament for treating of a PCSK9-mediated disease or disorder.
[140] Embodiment 53. The compound for use in the manufacture of a medicament of embodiment 52, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
[141] Embodiment 54. Use of a compound according to any one of embodiments 1 to 33, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating a PCSK9- mediated disease or disorder.
[142] Embodiment 55. The use of embodiment 54, wherein said PCSK9-mediated disease or disorder selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
[143] Embodiment 56. Use of a compound according to any one of embodiments 1 to 33, or a pharmaceutically acceptable salt thereof, in the treatment of a disease or disorder, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
Combination Therapy
[144] The compounds of the disclosure can be administered in therapeutically effective amounts in a combinational therapy with one or more therapeutic agents (pharmaceutical combinations) or modalities, e.g., non-drug therapies. For example, synergistic effects can occur with other cardiovascular agents, antihypertensive agents, coronary vasodilators, and diuretic substances. Where the compounds of the application are administered in conjunction with other therapies, dosages of the co-administered compounds will of course vary depending on the type of co-drug employed, on the specific drug employed, on the condition being treated and so forth.
[145] The compounds of the present disclosure may be administered either simultaneously with, or before or after, one or more other therapeutic agent. The compound of the present disclosure may be administered separately, by the same or different route of administration, or together in the same pharmaceutical composition as the other agents. A therapeutic agent is, for example, a chemical compound, peptide, antibody, antibody fragment or nucleic acid, which is therapeutically active or enhances the therapeutic activity when administered to a patient in combination with a compound of the present disclosure.
[146] In one embodiment, the disclosure provides a product comprising a compound of the present disclosure and at least one other therapeutic agent as a combined preparation for simultaneous, separate or sequential use in therapy. In one embodiment, the therapy is the treatment of a disease or condition mediated by PCSK9. Products provided as a combined preparation include a composition comprising the compound of the present disclosure and the other therapeutic agent(s) together in the same pharmaceutical composition, or the compound of the present disclosure and the other therapeutic agent(s) in separate form, e.g. in the form of a kit.
[147] In another aspect, the disclosure includes a compound of Formula (I) or a pharmaceutically acceptable salt thereof, for use in a combination therapy. A compound, composition, medicament and compounds for use of Formula according to any one of embodiments 1 to 33 or embodiments 41 to 44, or any embodiment of Formula (I) or a pharmaceutically acceptable salt thereof, may also be used to advantage in combination with one or more other therapeutic agents.
[148] Another aspect of the disclosure is directed to pharmaceutical compositions comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, a pharmaceutically acceptable carrier, and one or more therapeutic agents. The pharmaceutical acceptable carrier may further include an excipient, diluent, or surfactant.
[149] Combination therapy includes the administration of the subject compounds in further combination with other biologically active ingredients (such as, but not limited to, a second agent such as, but not limited to, a cardiovascular agent, an adrenergic blocker, an antihypertensive agent, an angiotensin system inhibitor, an angiotensin-converting enzyme (ACE) inhibitor, a coronary vasodilator, a diuretic, or an adrenergic stimulant or a second agent that targets PCSK9) and non-drug therapies (such as, but not limited to, surgery or radiation treatment). For instance, the compounds of the application can be used in combination with other pharmaceutically active compounds, preferably compounds that are able to enhance the effect of the compounds of the application. The compounds of the application can be administered simultaneously (as a single preparation or separate preparation) or sequentially to the other drug therapy or treatment modality. In general, a combination therapy envisions administration of two or more drugs during a single cycle or course of therapy.
[150] In some embodiments, compounds of the application can be used in combination with agents known to be beneficial for reducing cholesterol, including LDL-C, non-HDL-C, triglyceride-lowering agents, and total cholesterol and/or raising HDL-C.
[151] Exemplary therapeutic agents that may be used in combination with the compounds of the disclosure, include, but are not limited to, hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression ( e.g., inclisiran), an inhibitory nucleic acid targeting Lp(a) protein expression (e.g., pelacarsen, an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, anti-obesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9 (e.g., evolocumab), an immune checkpoint inhibitor (e.g., PD-1, PD-L1, PD- L2) and combinations thereof.
[152] Examples of hypolipidemic agents that may be used in combination with the compounds of the disclosure include, but are not limited to, an HMG-CoA reductase inhibitor, squalene synthase inhibitors, LXR agonist, FXR agonist, fibrates, cholesterol absorption inhibitors, nicotinic acid bile acid binding resins, bempedoic acid, nicotinic acid and other GPR109 agonists, and aspirin.
[153] HMG-CoA reductase inhibitors (i.e., statins) are a class of drugs used to lower cholesterol levels by inhibiting the enzyme HMG-CoA reductase, which plays a central role in the production of cholesterol in the liver. Increased cholesterol levels have been associated with cardiovascular diseases and statins are therefore used in the prevention of these diseases. Exemplary statins include, but are not limited to, atorvastatin, cerivastatin, compactin, dalvastatin, dihydrocompactin, fluindostatin, fluvastatin, lovastatin, pitavastatin, mevastatin, pravastatin, rivastatin, simvastatin, and velostatin, or pharmaceutically acceptable salts thereof.
[154] Fibrates or fibric acid derivatives lower triglycerides and raise HDL cholesterol. They may have little effect on LDL cholesterol. For example, Gemfibrozil or fenofibrate is prescribed for people who have very high triglycerides or who have low HDL and high triglycerides. Gemfibrozil may be used to reduce the risk of heart attack in people with coronary artery disease (CAD) who have low HDL and high triglycerides. Examples of fibrates include, but are not limited to, clofibrate, gemfibrozil, fenofibrate, ciprofibrate, and bezafibrate.
[155] Cholesterol absorption inhibitors are a class of compounds that prevents the uptake of cholesterol from the small intestine into the circulatory system, and, in turn, reduce plasma LDL-C concentrations. Increased cholesterol levels are associated with increased CVD risk; thus, cholesterol absorption inhibitors are used with the goal of reducing CVD risk. A non-limiting example of a cholesterol absorption inhibitor is Ezetimibe.
[156] Examples of bile acid sequestrants that may be used in combination with the compounds of the disclosure include, but are not limited to, cholestyramine, colestipol, and colesvelam.
[157] A non-limiting example of a thyroid hormone mimetic that may be used in combination with the compounds of the disclosure is compound KB2115.
[158] A non-limiting example of a thyroid hormone receptor (THR) P-selective agonist that may be used in combination with the compounds of the disclosure is MGL-3196.
[159] DGAT is an enzyme that catalyzes the last step in triacylglycerol biosynthesis. DGAT catalyzes the coupling of a 1,2-diacylglycerol with a fatty acyl-CoA resulting in Coenzyme A and triacylglycerol. Two enzymes that display DGAT activity have been identified: DGAT1 (acyl coA-diacylglycerol acyl transferase 1, see Cases et al, Proc. Natl. Acad. Sci. 95: 13018-13023, 1998) and DGAT2 (acyl coA- diacylglycerol acyl transferase 2, see Cases et al, J. Biol. Chem. 276:38870-38876, 2001). DGAT1 and DGAT2 do not share significant protein sequence homology. Importantly, DGAT1 knockout mice are protected from high fat diet-induced weight gain and insulin resistance (Smith et al, Nature Genetics 25:87-90, 2000). The phenotype of the DGAT1 knockout mice suggests that a DGAT1 inhibitor has utility for the treatment of obesity and obesity-associated complications. DGAT1 inhibitors useful in said combination are compounds and analogs generically and specifically disclosed e.g. in WO2007/126957
and W02009/040410, in particular in the compound claims and the final products of the working examples, the subject-matter of the final products, the pharmaceutical preparations and the claims.
[160] Examples of DGAT1 inhibitors suitable for use in combination with compounds of the present disclosure, include but are not limited to, {4-[4-(3-Methoxy-5-phenylamino-pyridin-2-yl)-phenyl]- cyclohexyl} -acetic acid, (4-{4-[5-(l-Methyl-lH-pyrazol-3-ylamino)-pyridin-2-yl]-phenyl}-cyclohexyl)- acetic acid, (4-{4-[5-(5-Fluoro-6-methoxy-pyridin-3-ylamino)-pyridin-2-yl]-phenyl}-cyclohexyl)-acetic acid, (4-{5-[5-(6-Trifluoromethyl-pyridin-3-ylamino)-pyridin-2-yl]-spirocyclohexylidenyl-l,l ’-indanyl}- acetic acid, (4-{4-[5-(Benzooxazol-2-ylamino)-pyridin-2-yl]-phenyl}-cyclohexyl)-acetic acid, 4-(4-{4-[2- (3 -Chlorophenylamino)-oxazol-5 -yl] -phenyl} -cyclohexylj-butyric acid, (4- { 4- [5 -(6-Trifluoromethyl- pyridin-3-ylamino)-pyridin-2-yl]-phenyl}-cyclohexyl)-acetic acid, (6-{4-[4-(2H-Tetrazol-5-ylmethyl)- cyclohexyl]-phenyl}-pyridazin-3-yl)-(6-trifluoromethyl-pyridin-3-yl)-amine, 3-(4-{4-[6-(6- Trifluoromethyl-pyridin-3 -ylamino)-pyridazin-3 -yl] -phenyl } -cyclohexylmethyl)-4H- [ 1 ,2,4]oxadiazol-5 - one, (l-{4-[6-(3-Trifluoromethyl-phenylamino)-pyridazin-3-yl]-phenyl}-piperidin-4-yl)-acetic acid, (4- {4-[4-Methyl-6-(6-trifluoromethyl-pyridin-3-ylamino)-pyridazin-3-yl]-phenyl}-cyclohexyl)-acetic acid, (4-{4-[5-(6-Trifluoromethyl-pyridin-3-ylamino)-pyrazin-2-yl]-phenyl}-cyclohexyl)-acetic acid, 6-[5-(4- Chloro-phenyl)-[l,3,4]oxadiazol-2-yl]-2-(2,6-dichloro-phenyl)-lH-benzoimidazole, 6-(5-Cyclohexyl-
[ 1 ,3 ,4]oxadiazol-2-yl)-2-(2,6-dichloro-phenyl)- 1 H-benzoimidazole, 6-(5 -Butyl- [ 1 ,3,4]oxadiazol-2-yl)-2- (2,6-dichloro-phenyl)-l H-benzoimidazole, 2-(2,6-Dichloro-phenyl)-6-[5-(5-methyl-pyridin-3-yl)- [1, 3 ,4]oxadiazol-2-yl]-l H-benzoimidazole, 6-[5-(4-Chloro-phenyl)-[l,3,4]oxadiazol-2-yl]-2-(2,6- dimethyl-4-morpholin-4-yl-phenyl)-l H-benzoimidazole, 6-[5-(4-Chloro-phenyl)-[l,3,4]oxadiazol-2-yl]- 2-(3 ,5 -dichloro-pyridin-4-yl)- 1 H-benzoimidazole, 3 -(4- { 5 - [5 -(4-Methoxy-phenyl)-[ 1 ,3 ,4]oxadiazol-2-yl] - lH-benzoimidazol-2-yl} -3, 5-dimethyl-phenyl)-2,2-dimethyl -propionic acid, 3-(4-{6-[5-(4-Methoxy- phenyl)- [1 ,3 ,4]oxadiazol-2-yl] - 1 H-benzoimidazol-2-yl} -3 ,5 -dimethyl-phenyl)-prop ionic acid, 3 -(4- { 6- [5 - (4-methoxyphenylamino)- [ 1 ,3 ,4]oxadiazol-2-yl] - 1 H-benzimidazol-2-yl} -3 ,5 -dimethylphenylj-propionic acid, [3 -(4- { 6- [5 -(4-Chloro-phenyl)- [ 1 ,3 ,4]oxadiazol-2-yl] - 1 H-benzoimidazol-2-yl } -3 ,5 -dim ethyl - phenyl)-propyl]-phosphonic acid, 2-(2,6-Dichloro-phenyl)-6-(4,5-diphenyl-oxazol-2-yl)-lH- benzoimidazole, (4- { 6- [5 -(4-Chloro-phenyl)- [ 1 ,3 ,4]oxadiazol-2-yl] - 1 H-benzoimidazol-2-yl } -3 ,5 - dimethyl-phenoxyj-acetic acid, 2-(2,6-Dichloro-phenyl)-6-(5-pyrrolidin-l-yl-[l,3,4]oxadiazol-2-yl)-lH- benzoimidazole, and 3 ,5 -Dimethyl-4-{ 6- [5 -(4-trifluoromethyl-phenylamino)-[ 1 ,3 ,4]oxadiazol-2-yl] - 1 H- benzoimidazol-2-yl } -phenol.
[161] A non-limiting example of a Niemann Pick Cl-like 1 (NPC1-L1) inhibitor that may be used in combination with the compounds of the disclosure is ezetimibe.
[162] Apolipoprotein A-I is a protein that in humans is encoded by the APOA1 gene. It has a specific role in lipid metabolism. Apolipoprotein A-I is the major protein component of high density lipoprotein
(HDL) in plasma. Chylomicrons secreted from enterocytes also contain ApoA-I but it is quickly transferred to HDL in the bloodstream. The protein promotes cholesterol efflux from tissues to the liver for excretion. It is a cofactor for lecithin cholesterolacyltransferase (LCAT) which is responsible for the formation of most plasma cholesteryl esters. Infusion of a variant of apoA-I in humans has been shown to regress atherosclerotic plaque, as assessed by intravascular ultrasound; thus, apoA-I reduces CVD risk and has the ability to both slow progression and induce regression of atherosclerosis. A non-limiting example of an apoA-I up-regulator/inducer is RVX208.
[163] ATP-binding cassette transporter, ABCA1 (member 1 of human transporter sub-family ABCA), also known as the cholesterol efflux regulatory protein (CHRP) is a protein which in humans is encoded by the ABCA1 gene. This transporter is a major regulator of cellular cholesterol and phospholipid homeostasis. A non-limiting example of an ABCA1 regulator is Probucol. Probucol lowers the level of cholesterol in the bloodstream by increasing the rate of LDL catabolism. Additionally, probucol may inhibit cholesterol synthesis and delay cholesterol absorption. Probucol is a powerful antioxidant, which inhibits the oxidation of cholesterol in LDLs; this slows the formation of foam cells, which contribute to atherosclerotic plaques.
[164] The liver X receptor (LXR) is a member of the nuclear receptor family of transcription factors and is closely related to nuclear receptors such as PPAR, FXR and RXR. Liver X receptors (LXRs) are important regulators of cholesterol, fatty acids and glucose homeostasis. LXR agonists are effective for treatment of murine models of atherosclerosis, diabetes, anti-inflammation and Alzheimer’s disease. Treatment with LXR agonists (including but not limited to, hypocholamide, T0901317, GW3965, or N,N- dimethyl-3-beta-hydroxy-cholenamide (DMHCA)) lowers the cholesterol level in serum and liver and inhibits the development of atherosclerosis in murine disease models. Examples of LXR agonists include, but are not limited to, GW3965 (a synthetic nonsteroidal liver X receptor (LXR) agonist/activator) and T0901317 (a dual LXR, FXR agonist).
[165] The famesoid X receptor (FXR), also known as NR1H4 (nuclear receptor subfamily 1, group H, member 4) is a nuclear hormone receptor with activity similar to that seen in other steroid receptors such as estrogen or progesterone but more similar in form to PPAR, LXR and RXR. Activation of the nuclear receptor FXR is known to improve hyperglycemia and hyperlipidemia. A non-limiting example of a FXR agonist is GW4064 (3-(2,6-Dichlorophenyl)-4-(3'-carboxy-2-chlorostilben-4-yl)oxymethyl-5- isopropylisoxazole).
[166] Phospholipid transfer protein (PLTP) is a protein that in humans is encoded by the PLTP gene. The protein encoded by this gene is one of at least two lipid transfer proteins found in human plasma, with CETP being the other. The encoded protein transfers phospholipids from triglyceride-rich lipoproteins to HDL. In addition to regulating the size of HDL particles, this protein may be involved in
cholesterol metabolism. At least two transcript variants encoding different isoforms have been found for this gene. Because PLTP influences the metabolism of both triglyceride-rich lipoproteins and HDL, modulation of this transfer protein has the potential to alter cardiovascular disease risk.
[167] Fish oil is derived from the tissues of oily fish. Fish oils contain the omega-3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), precursors of eicosanoids that are known to have many health benefits. Fish oil and other omega-3 sources are most highly recommended for the following conditions: hypertriglyceridemia, secondary cardiovascular disease and prevention of high blood pressure. For example, Lovaza ® is used along with a low-fat and low-cholesterol diet to lower very high triglycerides (fats) in your blood. Examples of omega-3 fatty acids that may be used in combination with the compounds of the disclosure include, but are not limited to Lovaza ® and Vascepa ® (icosapent ethyl).
[168] Examples of anti-diabetic agents that may be used in combination with the compounds of the disclosure include, but are not limited to, insulin, insulin derivatives and mimetics; insulin secretagogues such as the sulfonylureas; insulinotropic sulfonylurea receptor ligands such as meglitinides, e.g., nateglinide and repaglinide; protein tyrosine phosphatase- IB (PTP-1B) inhibitors including, but not limited to, PTP-112; GSK3 (glycogen synthase kinase-3) inhibitors including, but not limited to, SB- 517955, SB-4195052, SB-216763, NN-57-05441 and NN-57-05445; RXR ligands including, but not limited to, GW-0791 and AGN- 194204; sodium -dependent glucose cotransporter inhibitors including, but not limited to, T-1095; glycogen phosphorylase A inhibitors including, but not limited to, BAY R3401; biguanides including, but not limited to, metformin; alpha-glucosidase inhibitors including, but not limited to, acarbose; GLP-1 (glucagon like peptide-1), GLP-1 analogs including, but not limited to, Exendin-4 and GLP-1 mimetics; and DPPIV (dipeptidyl peptidase IV) inhibitors including, but not limited to, vildagliptin; GLP-1 receptor agonists inhibitors including, but not limited to semaglutide.
[169] Examples of sulfonylureas include, but are not limited to, tolbutamide, chlorpropamide, tolazamide, acetohexamide, 4-chloro-V-[(l-pyrolidinylamino)carbonyl]-benzenesulfonamide (glycopyramide), glibenclamide (glyburide), gliclazide, l-butyl-3-metanilylurea, carbutamide, glibonuride, glipizide, gliquidone, glisoxepid, glybuthiazole, glibuzole, glyhexamide, glymidine, glypinamide, phenbutamide, amaryl, and tolylcyclamide, or pharmaceutically acceptable salts thereof.
[170] DPP-IV (dipeptidyl peptidase IV) is responsible for inactivating GLP-1. More particularly, DPPIV generates a GLP-1 receptor antagonist and thereby shortens the physiological response to GLP-1. GLP-1 is a major stimulator of pancreatic insulin secretion and has direct beneficial effects on glucose disposal.
[171] The DPP-IV inhibitor can be peptidic or, preferably, non-peptidic. Examples of DPP-IV inhibitors also include, but are not limited to, generically and specifically DPP-IV inhibitors disclosed in
WO 98/19998, DE 196 16 486 Al, WO 00/34241 and WO 95/15309, in each case in particular in the compound claims and the final products of the working examples, the subject-matter of the final products, the pharmaceutical preparations and the claims are hereby incorporated into the present application by reference to these publications.
[172] GLP-1 (glucagon like peptide-1) is an insulinotropic protein which is described, e.g., by W.E. Schmidt et al. in Diabetologia, 28, 1985, 704-707 and in US 5,705,483. The term “GLP-1 agonists” includes variants and analogs of GLP-1 (7-36)NHz which are disclosed in particular in U.S. 5,120,712, U.S. 5,118666, U.S. 5,512,549, WO 91/11457 and by C. Orskov, et al, in J. Biol. Chem., 264 (1989) 12826. Further examples include GLP-l(7-37), in which compound the carboxy-terminal amide functionality of Arg36 is displaced with Gly at the 37th position of the GLP-1 (7-36)NHz molecule and variants and analogs thereof including GLN9-GLP-l(7-37), D-GLN9-GLP-l(7-37), acetyl LYS9-GLP-1(7- 37), LYS18-GLP-l(7-37) and, in particular, GLP-1 (7-37)OH, VAL8-GLP-l(7-37), GLY8-GLP-l(7-37), THR8-GLP-l(7-37), MET8-GLP-l(7-37) and 4-imidazopropionyl-GLP-l. Special preference is also given to the GLP agonist analog exendin-4, described by Greig, et al., in Diabetologia, 1999, 42, 45-50.
[173] Also included in the definition “anti-diabetic agent” are insulin sensitivity enhancers which restore impaired insulin receptor function to reduce insulin resistance and consequently enhance the insulin sensitivity. Examples include hypoglycemic thiazolidinedione derivatives (e.g., glitazone, (5)- ((3 ,4-dihydro-2-(phenyl-methyl)-2W- 1 -benzopyran-6-yl)methyl-thiazolidine-2, 4-dione (englitazone), 5- {[4-(3-(5-methyl-2-phenyl-4-oxazolyl)-l-oxopropyl)-phenyl]-methyl}-thiazolidine-2, 4-dione (darglitazone), 5-{ [4-(l -methyl-cyclohexyl)methoxy)-phenyl]methyl}-thiazolidine-2, 4-dione (ciglitazone), 5 - { [4 - (2 - ( 1 -indolyl)ethoxy)phenyl]methyl } -thiazolidine-2, 4-dione (DRF2189), 5-{4-[2-(5- methyl-2-phenyl-4-oxazolyl)-ethoxy)]benzyl } -thiazolidine-2, 4-dione (BM- 13.1246), 5 -(2- naphthylsulfonyl)-thiazolidine-2, 4-dione (AY -31637), bis{4-[(2,4-dioxo-5- thiazolidinyl)methyl]phenyl}methane (YM268), 5-{4-[2-(5-methyl-2-phenyl-4-oxazolyl)-2- hydroxyethoxy]benzyl}-thiazolidine-2, 4-dione (AD-5075), 5 -[4-(l -phenyl- 1- cyclopropanecarbonylamino)-benzyl]-thiazolidine-2, 4-dione (DN-108) 5-{[4-(2-(2,3-dihydroindol-l- yl)ethoxy)phenyl]methyl}-thiazolidine-2, 4-dione, 5-[3-(4-chloro-phenyl])-2-propynyl]-5- phenylsulfonyl)thiazolidine-2, 4-dione, 5-[3-(4-chlorophenyl])-2-propynyl]-5-(4-fluorophenyl- sulfonyl)thiazolidine-2, 4-dione, 5-{[4-(2-(methyl-2-pyridinyl-amino)-ethoxy)phenyl]methyl}- thiazolidine-2, 4-dione (rosiglitazone), 5-{[4-(2-(5-ethyl-2-pyridyl)ethoxy)phenyl]-methyl}thiazolidine- 2, 4-dione (pioglitazone), 5-{[4-((3,4-dihydro-6-hydroxy-2,5,7,8-tetramethyl-2W-l-benzopyran-2- yl)methoxy)-phenyl]-methyl}-thiazolidine-2, 4-dione (troglitazone), 5-[6-(2-fluoro-benzyloxy)naphthalen- 2 -ylmethyl] -thiazolidine-2, 4-dione (MCC555), 5-{[2-(2-naphthyl)-benzoxazol-5-yl]-methyl}thiazolidine-
2,4-dione (T-174) and 5-(2,4-dioxothiazolidin-5-ylmethyl)-2-methoxy-N-(4-trifluoromethyl- benzyl)benzamide (KRP297)).
[174] Examples of anti-obesity agents that may be used in combination with the compounds of the disclosure include, but are not limited to, semaglutide, GDF15, orlistat, sibutramine, phentermine and Cannabinoid Receptor 1 (CB1) antagonists e.g., rimonabant.
[175] Examples of agonists of peroxisome proliferator-activator receptors that may be used in combination with the compounds of the disclosure include, but are not limited to, fenofibrate, pioglitazone, rosiglitazone, tesaglitazar, BMS-298585, L-796449, the compounds specifically described in the patent application WO 2004/103995 i.e. compounds of examples 1 to 35 or compounds specifically listed in claim 21, or the compounds specifically described in the patent application WO 03/043985 i.e. compounds of examples 1 to 7 or compounds specifically listed in claim 19 and especially (R)-l-{4-[5- methyl-2-(4-trifluorom ethyl -phenyl)-oxazol-4-ylm ethoxy] -benzenesulfonyl} -2, 3 -dihydro- lH-indole-2- carboxylic or a salt thereof.
[176] Examples of anti-hypertensive agents that may be used in combination with the compounds of the disclosure include, but are not limited to, loop diuretics; angiotensin converting enzyme (ACE); inhibitors of the Na-K-ATPase membrane pump; neutralendopeptidase (NEP) inhibitors; ACE/NEP inhibitors; angiotensin II antagonists; renin inhibitors; 0-adrenergic receptor blockers; inotropic agents; calcium channel; aldosterone receptor antagonists; and aldosterone synthase inhibitors.
[177] Examples of loop diuretics that may be used in combination with the compounds of the disclosure include, but are not limited to, ethacrynic acid, furosemide and torsemide.
[178] The term “ACE-inhibitor” (also called angiotensin converting enzyme inhibitors) includes molecules that interrupt the enzymatic degradation of angiotensin I to angiotensin II. Such compounds may be used for the regulation of blood pressure and for the treatment of congestive heart failure. Examples include, but are not limited to, alacepril, benazepril, benazeprilat, captopril, ceronapril, cilazapril, delapril, enalapril, enaprilat, fosinopril, imidapril, lisinopril, moexipril, moveltopril, perindopril, quinapril, ramipril, spirapril, temocapril, and trandolapril, or a pharmaceutically acceptable salt thereof.
[179] A non-limiting example of an inhibitor of the Na-K-ATPase membrane pump is digoxin.
[180] The term “NEP inhibitor” refers to a compound that inhibits neutral endopeptidase (NEP). Examples include, but are not limited to, Candoxatril, Candoxatrilat, Dexecadotril, Ecadotril, Racecadotril, Sampatrilat, Fasidotril, Omapatrilat, Gemopatrilat, Daglutril, SCH-42495, SCH-32615, UK- 447841, AVE-0848, PL-37, and (2R,4s)-5-Biphenyl-4-yl-4-(3-carboxy-propionylamino)-2-methyl- pentanoic acid ethyl ester, or a pharmaceutically acceptable salt thereof. NEP inhibitors also include Phosphono/biaryl substituted dipeptide derivatives, as disclosed in U.S. Patent 5,155,100. NEP inhibitors
also include N-mercaptoacyl phenylalanine derivative as disclosed in PCT application WO 2003/104200. NEP inhibitors also include dual-acting antihypertensive agents as disclosed in PCT applications WO 2008/133896, WO 2009/035543, or WO 2009/134741. Other examples include compounds disclosed in U.S. applicationsl2/788,794; 12/788,766, and 12/947,029. NEP inhibitors also include compounds disclosed in WO 2010/136474, WO 2010/136493, WO 2011/061271, WO 2012/065953, WO 2012/065956, WO 2014/126979, and WO 2014/015965. Other examples of NEP inhibitors are compounds disclosed in WO2015116786, W02015116760, WO2014138053, WO2014025891, WO2013184934, WO2013067163, WO2012166389, WO2012166387, WO2012112742, and WO2012082853.
[181] The term “ACE/NEP inhibitors” refers to a compound that inhibits both angiotensin converting enzyme(ACE) and neutral endopeptidase (NEP). Examples of ACE/NEP inhibitors that may be used in combination with the compounds of the disclosure include, but are not limited to, omapatrilat, sampatrilat, and fasidotril.
[182] The class of angiotensin II antagonists or ATi receptor antagonists comprises compounds having differing structural features, essentially preferred are the non-peptidic ones. Examples of angiotensin II antagonists that may be used in combination with the compounds of the disclosure include, but are not limited to, valsartan, losartan, candesartan, eprosartan, irbesartan, saprisartan, tasosartan, telmisartan, the compounds with the designation E-1477 and ZD-8731 of the following formulae

SC-52458 ZD-8731 or, in each case, a pharmaceutically acceptable salt thereof.
[183] The term “renin inhibitor” includes ditekiren (chemical name: [1S-[1R,2R,4R(1R,2R)]]- l-[(l,l-dimethylethoxy)carbonyl]-L-proly l-L-phenylalanyl-N-[2-hydroxy-5-methyl-l-(2-methylpropyl)- 4-[[[2-methyl-l-[[(2 pyridinylmrthyl)amino]carbonyl]butyl]amino]carbonyl]hexyl]-N-alfa-methyl-L- histidinamide); terlakiren (chemical name: [R-(R,S)]-N-(4-morpholinylcarbonyl)-L-phenylalanyl-N-[l- (cyclohexylmethyl)-2-hydroxy-3-(l-methylethoxy)-3-oxopropyl]-S-methyl-L-cysteineamide); Aliskiren (chemical name: (2S,4s,5S,7S)-5-amino-N-(2-carbamoyl-2,2-dimethylethyl)-4-hydroxy-7-{[4-methoxy-
3-(3-methoxypropoxy)phenyl]methyl}-8-methyl-2-(propan-2-yl)nonanamide) and zankiren (chemical name: [lS-[lR[R(R)],2S,3r]]-N-[l-(cyclohexylmethyl)-2,3-dihydroxy-5-m ethylhexyl]-alfa-[[2-[[(4- methyl-l-piperazinyl)sulfonyl]methyl]-l-oxo-3-phenylpropyl]-amino]-4-thiazolepropanamide), or, hydrochloride salts thereof, or, SPP630, SPP635 and SPP800 as developed by Speedel, or RO 66-1132 and RO 66-1168 of Formula (A) and (B):
 and
and
 or pharmaceutically acceptable salts thereof. The term “aliskiren”, if not defined specifically, is to be understood both as the free base and as a salt thereof, especially a pharmaceutically acceptable salt thereof, most preferably a hemi-fumarate salt thereof.
or pharmaceutically acceptable salts thereof. The term “aliskiren”, if not defined specifically, is to be understood both as the free base and as a salt thereof, especially a pharmaceutically acceptable salt thereof, most preferably a hemi-fumarate salt thereof.
[184] Examples of (3-adrenergic receptor blockers that may be used in combination with the compounds of the disclosure include, but are not limited to, acebutolol, atenolol, betaxolol, bisoprolol, metoprolol, nadolol, propranolol, sotalol, and timolol.
[185] Examples of inotropic agents that may be used in combination with the compounds of the disclosure include, but are not limited to, digoxin, dobutamine, and milrinone; Inotropes as used herein include, for example, dobutamine, isoproterenol, milrinone, amirinone, levosimendan, epinephrine, norepinephrine, isoproterenol, and digoxin.
[186] Examples of calcium channel blockers that may be used in combination with the compounds of the disclosure include, but are not limited to, amlodipine, bepridil, diltiazem, felodipine, nicardipine, nimodipine, nifedipine, nisoldipine and verapamil.
[187] The class of aldosterone synthase inhibitors comprises both steroidal and non-steroidal aldosterone synthase inhibitors, the latter being most preferred. The class of aldosterone synthase inhibitors comprises compounds having differing structural features. Examples of aldosterone synthase inhibitor that can be used in combination with the compounds of the present disclosure include, but are not limited to, the (+)-enantiomer of the hydrochloride of fadrozole (U.S. patents 4,617,307 and 4,889,861) of formula
 or, if appropriable, a pharmaceutically acceptable salt thereof; and compounds and analogs generically and specifically disclosed e.g. in US2007/0049616, in particular in the compound claims and the final products of the working examples, the subject-matter of the final products, the pharmaceutical preparations and the claims are hereby incorporated into the present application by reference to this publication. Examples of aldosterone synthase inhibitors that can be used in combination with the compounds of the present disclosure include, but are not limited to, without limitation 4-(6,7-dihydro-5H- pyrrolo[l,2-c]imidazol-5-yl)-3-m ethylbenzonitrile; 5-(2-chloro-4-cyanophenyl)-6,7-dihydro-5H- pyrrolo[l,2-c]imidazole-5-carboxylic acid (4-methoxybenzyl)methylamide; 4 ’-fluoro -6 -(6, 7,8,9- tetrahydro-5H-imidazo[l,5-a]azepin-5-yl)biphenyl-3-carbonitrile; 5-(4-Cyano-2-methoxyphenyl)-6,7- dihydro-5H-pyrrolo[l,2-c]imidazole-5-carboxylic acid butyl ester; 4-(6,7-Dihydro-5H-pyrrolo[l,2- c]imidazol-5-yl)-2-m ethoxybenzonitrile; 5-(2-Chloro-4-cyanophenyl)-6,7-dihydro-5H-pyrrolo[l,2- c]imidazole-5-carboxylic acid 4-fluorobenzyl ester; 5-(4-Cyano-2-trifluoromethoxyphenyl)-6,7-dihydro- 5H-pyrrolo[l,2-c]imidazole-5-carboxylic acid methyl ester; 5-(4-Cyano-2-methoxyphenyl)-6,7-dihydro- 5H-pyrrolo[l,2-c]imidazole-5-carboxylic acid 2 -isopropoxyethyl ester; 4-(6,7-Dihydro-5H-pyrrolo[l,2- c]imidazol-5-yl)-2-methylbenzonitrile; 4-(6,7-dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl)-3- fluorobenzonitrile; 4-(6,7-Dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl)-2-methoxybenzonitrile; 3-Fluoro-4- (7-methylene-6,7-dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl)benzonitrile; cis-3-Fluoro-4-[7-(4-fluoro- benzyl)-5,6,7,8-tetrahydro-imidazo[l,5-a]pyridin-5-yl]benzonitrile; 4’-Fluoro-6-(9-methyl-6,7,8,9- tetrahydro-5H-imidazo[l,5-a]azepin-5-yl)biphenyl-3-carbonitrile; 4’-Fluoro-6-(9-methyl-6,7,8,9- tetrahydro-5H-imidazo[l,5-a]azepin-5-yl)biphenyl-3-carbonitrile or in each case, the (R) or (S) enantiomer thereof; or if appropriable, a pharmaceutically acceptable salt thereof.
or, if appropriable, a pharmaceutically acceptable salt thereof; and compounds and analogs generically and specifically disclosed e.g. in US2007/0049616, in particular in the compound claims and the final products of the working examples, the subject-matter of the final products, the pharmaceutical preparations and the claims are hereby incorporated into the present application by reference to this publication. Examples of aldosterone synthase inhibitors that can be used in combination with the compounds of the present disclosure include, but are not limited to, without limitation 4-(6,7-dihydro-5H- pyrrolo[l,2-c]imidazol-5-yl)-3-m ethylbenzonitrile; 5-(2-chloro-4-cyanophenyl)-6,7-dihydro-5H- pyrrolo[l,2-c]imidazole-5-carboxylic acid (4-methoxybenzyl)methylamide; 4 ’-fluoro -6 -(6, 7,8,9- tetrahydro-5H-imidazo[l,5-a]azepin-5-yl)biphenyl-3-carbonitrile; 5-(4-Cyano-2-methoxyphenyl)-6,7- dihydro-5H-pyrrolo[l,2-c]imidazole-5-carboxylic acid butyl ester; 4-(6,7-Dihydro-5H-pyrrolo[l,2- c]imidazol-5-yl)-2-m ethoxybenzonitrile; 5-(2-Chloro-4-cyanophenyl)-6,7-dihydro-5H-pyrrolo[l,2- c]imidazole-5-carboxylic acid 4-fluorobenzyl ester; 5-(4-Cyano-2-trifluoromethoxyphenyl)-6,7-dihydro- 5H-pyrrolo[l,2-c]imidazole-5-carboxylic acid methyl ester; 5-(4-Cyano-2-methoxyphenyl)-6,7-dihydro- 5H-pyrrolo[l,2-c]imidazole-5-carboxylic acid 2 -isopropoxyethyl ester; 4-(6,7-Dihydro-5H-pyrrolo[l,2- c]imidazol-5-yl)-2-methylbenzonitrile; 4-(6,7-dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl)-3- fluorobenzonitrile; 4-(6,7-Dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl)-2-methoxybenzonitrile; 3-Fluoro-4- (7-methylene-6,7-dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl)benzonitrile; cis-3-Fluoro-4-[7-(4-fluoro- benzyl)-5,6,7,8-tetrahydro-imidazo[l,5-a]pyridin-5-yl]benzonitrile; 4’-Fluoro-6-(9-methyl-6,7,8,9- tetrahydro-5H-imidazo[l,5-a]azepin-5-yl)biphenyl-3-carbonitrile; 4’-Fluoro-6-(9-methyl-6,7,8,9- tetrahydro-5H-imidazo[l,5-a]azepin-5-yl)biphenyl-3-carbonitrile or in each case, the (R) or (S) enantiomer thereof; or if appropriable, a pharmaceutically acceptable salt thereof.
[188] The term aldosterone synthase inhibitors also include, but are not limited to, compounds and analogs disclosed in W02008/076860, W02008/076336, W02008/076862, W02008/027284, W02004/046145, W02004/014914, and WO2001/076574.
[189] Furthermore, Aldosterone synthase inhibitors also include, but are not limited to, compounds and analogs disclosed in U.S. patent applications US2007/0225232, US2007/0208035, US2008/0318978, US2008/0076794, US2009/0012068, US20090048241 and in PCT applications W02006/005726, WO2006/128853, WO2006128851, WO2006/128852, W02007065942, W02007/116099, W02007/116908, W02008/119744 and in European patent application EP 1886695.
Preferred aldosterone synthase inhibitors suitable for use in the present disclosure include, without limitation 8-(4-Fluorophenyl)-5,6-dihydro-8H-imidazo[5,l-cl[l ,41oxazine; 4-(5,6-Dihydro-8H- imidazo[5,l-c][l ,4]oxazin-8-yl)-2-fluorobenzonitrile; 4-(5,6-Dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8- yl)-2,6-difluorobenzonitrile; 4-(5,6-Dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8-yl)-2-methoxybenzonitrile; 3-(5,6-Dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8-yl)benzonitrile; 4-(5,6-Dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8-yl)phthalonitrile; 4-(8-(4-Cyanophenyl)-5,6-dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8- yljbenzonitrile; 4-(5,6-Dihydro-8H-imidazo[5,l-c][l ,4]oxazin-8-yl)benzonitrile; 4-(5,6-Dihydro-8H- imidazo [5 , 1 -c] [ 1 ,4]oxazin-8 -yljnaphthalene- 1 -carbonitrile; 8 - [4-( 1 H-T etrazol-5 -yljphenyl 1-5 ,6-dihvdro- 8H-imidazo[5,l-c][l ,4]oxazine as developed by Speedel or in each case, the (R) or (S) enantiomer thereof; or if appropriable, a pharmaceutically acceptable salt thereof.
[190] Aldosterone synthase inhibitors useful in said combination include, but are not limited to, compounds and analogs generically and specifically disclosed e.g. in WO 2009/156462 and WO 2010/130796, in particular in the compound claims and the final products of the working examples, the subject-matter of the final products, the pharmaceutical preparations and the claims. Preferred Aldosterone Synthase inhibitors suitable for combination in the present disclosure include, 3-(6-Fluoro-3- methyl-2-pyridin-3-yl-lH-indol-l-ylmethyl)-benzonitrile hydrochloride, l-(4-Methanesulfonyl-benzyl)-3- methyl -2 -pyridin-3 -yl- 1 H-indole, 2-(5 -Benzyloxy -pyridin-3 -yl)-6-chloro- 1 -methyl- 1 H-indole, 5-(3- Cyano-1 -methyl- lH-indol-2-yl)-nicotinic acid ethyl ester, N-[5-(6-chloro-3-cyano-l-methyl-lH-indol-2- yl)-pyridin-3-y Im ethyl] -ethanesulfonamide, Pyrrolidine- 1 -sulfonic acid 5-(6-chloro-3-cyano-l-methyl- lH-indol-2-yl)-pyridin-3-yl ester, N -Methyl-N-[5-(l -methyl- lH-indol-2-yl)-pyridin-3-ylm ethyl]- methanesulfonamide, 6-Chloro-l-methyl-2-{5-[(2-pyrrolidin-l-yl-ethylamino)-methyl]-pyridin-3-yl}-lH- indole-3 -carbonitrile, 6-Chloro-2-[5 -(4-m ethanesulfonyl -piperazin- 1 -ylmethyl)-pyridin-3-yl]- 1 -methyl- lH-indole-3 -carbonitrile, 6-Chloro- l-methyl-2-{5-[(l -methyl -p iperidin-4-ylamino)-m ethyl] -pyridin-3 - yl} - 1 H-indole-3 -carbonitrile, Morpholine-4-carboxylic acid [5 -(6-chloro-3 -cyano- 1 -methyl- 1 H-indol-2- yl)-pyridin-3-y Im ethyl] -amide, N-[5-(6-Chloro- 1 -methyl- lH-indol-2-yl)-pyridin-3-ylmethyl]- ethanesulfonamide, C,C,C-Trifhioro-N-[5-(l-methyl-lH-indol-2-yl)-pyridin-3-yhnethyl]- methanesulfonamide, N-[5-(3-Chloro-4-cyano-phenyl)-pyridin-3-yl]-4-trifluoromethyl- benzenesulfonamide, N-[5 -(3 -Chloro-4-cyano-phenyl)-pyridin-3 -yl] - 1 -phenyl-methanesulfonamide, N- (5 -(3 -chloro -4 -cyanophenyl)pyridin-3 -yljbutane- 1 -sulfonamide, N-( 1 -(5 -(4-cyano-3 - methoxyphenyl)pyridin-3-yl)ethyl)ethanesulfonamide, N-((5-(3-chloro-4-cyanophenyl)pyridin-3- yl)(cyclopropyl)methyl)ethanesulfonamide, N -(cyclopropyl(5 -( 1 H-indol-5 -yl)pyridin-3 - yl)methyl)ethanesulfonamide, N-(cyclopropyl(5 -naphtalen- 1 -yl-pyridin-3 -yl)methyl)ethanesulfonamide, Ethanesulfonic acid [5-(6-chloro-l-methyl-lH-pyrrolo[2,3-b]pyridin-2-yl)-pyridin-3-ylmethyl]-amide and Ethanesulfonic acid {[5 -(3 -chloro-4-cyano-phenyl)-pyridin-3-yl] -cyclopropyl -methyl }-ethyl-amide.
[191] Lipid-lowering agents are known in the art, and described, e.g., in Goodman and Gilman ’s The Pharmacological Basis of Therapeutics, 11th Ed., Brunton, Lazo and Parker, Eds., McGraw-Hill (2006); 2009 Physicians ’ Desk Reference (PDR), for example, in the 63rd (2008) Eds., Thomson PDR.
[192] “Combination therapy” is intended to embrace administration of these therapeutic agents in a sequential manner, wherein each therapeutic agent is administered at a different time and in any order, or in alternation and in any order, as well as administration of these therapeutic agents, or at least two of the therapeutic agents, in a substantially simultaneous manner. Substantially simultaneous administration can be accomplished, for example, by administering to the subject a single capsule having a fixed ratio of each therapeutic agent or in multiple, single capsules for each of the therapeutic agents. Sequential or substantially simultaneous administration of each therapeutic agent can be effected by any appropriate route including, but not limited to, oral routes, intravenous routes, intramuscular routes, and direct absorption through mucous membrane tissues. The therapeutic agents can be administered by the same route or by different routes. For example, a first therapeutic agent of the combination selected may be administered by intravenous injection while the other therapeutic agents of the combination may be administered orally. Alternatively, for example, all therapeutic agents may be administered orally or all therapeutic agents may be administered by intravenous injection. The sequence in which the therapeutic agents are administered is not narrowly critical.
[193] Embodiment 39. A combination comprising of a compound according to any one of embodiments 1 to 33 or a pharmaceutically acceptable salt thereof and one or more additional pharmaceutical agents.
[194] Embodiment 40. The combination of embodiment 39, where the one or more agents additional pharmaceutically active agent is selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA: diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression, an inhibitory nucleic acid targeting Lp(a) protein expression, an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, anti-obesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9, an immune checkpoint inhibitor and combinations thereof
[195] In accordance with the foregoing, the present disclosure also provides a therapeutic combination, e.g., a kit, kit of parts, e.g., for use in any method as defined herein, comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, to be used concomitantly or in sequence with at least
one pharmaceutical composition comprising at least another therapeutic agent, selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression ( e.g., inclisiran), an inhibitory nucleic acid targeting Lp(a) protein expression (e.g., pelacarsen, an inhibitory nucleic acid targeting apoB 100, apoA-I up- regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, anti-obesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9 (e.g., evolocumab), an immune checkpoint inhibitor (e.g., PD-1, PD-L1, PD-L2) and combinations thereof. The kit may comprise instructions for its administration. The combination can be a fixed combination (e.g. in the same pharmaceutical composition) or a free combination (e.g. in separate pharmaceutical compositions).
[196] Similarly, the present disclosure provides a kit of parts comprising: (i) a pharmaceutical composition of the disclosure; and (ii) a pharmaceutical composition comprising a compound selected from a hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA: diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl- like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression ( e.g., inclisiran), an inhibitory nucleic acid targeting Lp(a) protein expression (e.g., pelacarsen, an inhibitory nucleic acid targeting apoB 100, apoA-I up- regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, anti-obesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9 (e.g., evolocumab), an immune checkpoint inhibitor (e.g., PD-1, PD-L1, PD-L2) and combinations thereof, in the form of two separate units of the components (i) to (ii).
[197] Likewise, the present disclosure provides a method as defined above comprising coadministration, e.g., concomitantly or in sequence, of a therapeutically effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a second drug substance, said second drug substance being hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression ( e.g., inclisiran), an inhibitory nucleic acid
targeting Lp(a) protein expression (e.g., pelacarsen, an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, anti-obesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9 (e.g., evolocumab), an immune checkpoint inhibitor (e.g., PD-1, PD-L1, PD-L2) and combinations thereof, e.g., as indicated above.
Definitions
[198] Terms not specifically defined herein should be given the meanings that would be given to them by one of skill in the art in light of the disclosure and the context. For purposes of interpreting this specification, the following definitions will apply unless specified otherwise and whenever appropriate, terms used in the singular will also include the plural and vice versa.
[199] It must be noted that as used herein and in the appended claims, the singular forms “a”, “an” and
“the” include plural referents unless the context clearly dictates otherwise. Thus, for example, reference to “the pharmaceutical formulation” includes reference to one or more pharmaceutical formulations; and so forth.
[200] The term “acyl”, as used herein, refers to a group represented by the general formula hydrocarbylC(O) — , preferably alkylC(O) — .
[201] The term “acyloxy”, as used herein, refers to a group represented by the general formula hydrocarbylC(O)O — , preferably alkylC(O)O — .
[202] The term “alkenyl”, as used herein, refers to an aliphatic group containing at least one double bond and is intended to include both “unsubstituted alkenyls” and “substituted alkenyls”, the latter of which refers to alkenyl moieties having substituents replacing a hydrogen on one or more carbons of the alkenyl group. Such substituents may occur on one or more carbons that are included or not included in one or more double bonds. Moreover, such substituents include all those contemplated for alkyl groups, as discussed below, except where stability is prohibitive. For example, substitution of alkenyl groups by one or more alkyl, carbocyclyl, aryl, heterocyclyl, or heteroaryl groups is contemplated. Examples of alkenyl groups include ethenyl, propenyl, n-butenyl, iso-butenyl, pentenyl, or hexenyl.
[203] The term “alkoxy”, as used herein, refers to an alkyl group, preferably a lower alkyl group, having an oxygen attached thereto, e.g., -O(alkyl). Representative alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, tert-butoxy and the like. Representative substituted alkoxy groups include, but are not limited to, — OCF3 and the like.
[204] An “alkyl” group or “alkane” is a straight chained or branched non-aromatic hydrocarbon which is completely saturated. Typically, a straight chained or branched, alkyl group has from 1 to about 20
carbon atoms, preferably from 1 to about 10 unless otherwise defined. Examples of straight chained and branched alkyl groups include methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, tert-butyl, pentyl, hexyl, pentyl and octyl. A C1-C6 straight chained or branched alkyl group is also referred to as a 'lower alkyl’’ group. Moreover, the term '“alkyl” (or ‘‘lower alkyl”) as used throughout the specification, examples, and claims is intended to include both “unsubstituted alkyls’’ and “substituted alkyls”, the latter of which refers to alkyl moselles having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone. Such substituents, if not otherwise specified, can include, for example, a halogen., a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or .heteroaromatic moiety. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate. For instance, the substituents of a substituted alkyl may include substituted and unsubstituted forms of ammo, azido, imino, amido, phosphoryl (including phosphonate and. phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), CF,, CN and the like. Exemplary substituted alkyls are described below. Cycloalkyls can be further substituted with alkyls, alkenyls, alkoxys. alkylthios, aminoalkyls, carbonyl-substituted alkyls, CF3, CN, and the like.
[205] The term “alkynyl”, as used herein, refers to an aliphatic group containing at least one triple bond and is intended to include both, “unsubstituted alkynyls” and “substituted alkynyls”, the latter of which refers to alkynyl moieties having substituents replacing a hy drogen on one or more carbons of the alkynyl group. Such substituents may occur on one or more carbons that are included or not included in one or more triple bonds. Moreover, such substituents include all those contemplated for alkyl groups, as discussed above, except where stability is prohibitive. For example, substitution of alkynyl groups by one or more alkyl, carbocycly 1, aryl, heterocyclyl, or heteroaryl groups is contemplated. Examples of alkenyl groups include ethynyl, propargyl, n-butynyl, iso-butynyl, pentynyl, or hexynyl.
[206] The term “aryl”, as used herein, include substituted or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon. Preferably the ring is a 5- to 7-membered ring, more preferably a 6-membered ring. The term “aryl” also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cyclo alkynyls, aryls, heteroaryls, and/or heterocyclyls. Aryl groups include, but are not limited to, phenyl, biphenyl, naphthyl, anthracenyl,
phenalenyl, phenanthrenyl, indanyl, indenyl, tetrahydronaphthalenyl, tetrahydrobenzoannulenyl, and the like.
[207] The term “Cx.y” when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups that contain from x to y carbons in the chain. For example, the term “Cx.yalkyl” refers to substituted or unsubstituted saturated hydrocarbon groups, including straight-chain alkyl and branched-chain alkyl groups that contain from x to y carbons in the chain, including haloalkyl groups such as trifluoromethyl and 2,2,2-trifluoroethyl, etc. Co alkyl indicates a hydrogen where the group is in a terminal position, a bond if internal. The terms “C2-yalkenyl” and “C2- yalkynyl” refer to substituted or unsubstituted unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
[208] The terms “carbocycle”, and “carbocyclic”, as used herein, refers to a saturated or unsaturated ring in which each atom of the ring is carbon. The term carbocycle includes both aromatic carbocycles and non-aromatic carbocycles. Non-aromatic carbocycles include both cycloalkane rings, in which all carbon atoms are saturated, and cycloalkene rings, which contain at least one double bond. “Carbocycle” includes 5-7 membered monocyclic and 8-12 membered bicyclic rings. Each ring of a bicyclic carbocycle may be selected from saturated, unsaturated and aromatic rings. Carbocycle includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings. The term “fused carbocycle” refers to a bicyclic carbocycle in which each of the rings shares two adjacent atoms with the other ring. Each ring of a fused carbocycle may be selected from saturated, unsaturated and aromatic rings. In an exemplary embodiment, an aromatic ring, e.g., phenyl, may be fused to a saturated or unsaturated ring, e.g., cyclohexane, cyclopentane, or cyclohexene. Any combination of saturated, unsaturated and aromatic bicyclic rings, as valence permits, is included in the definition of carbocyclic. Exemplary “carbocycles” include cyclopentane, cyclohexane, bicyclo[2.2.1]heptane, 1,5 -cyclooctadiene, 1, 2,3,4- tetrahydronaphthalene, bicyclo[4.2.0]oct-3-ene, naphthalene and adamantane. Exemplary fused carbocycles include decalin, naphthalene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]octane, 4, 5,6,7- tetrahydro-lH-indene and bicyclo[4.1.0]hept-3-ene. “Carbocycles” may be susbstituted at any one or more positions capable of bearing a hydrogen atom.
[209] A “cycloalkyl” group is a cyclic hydrocarbon which is completely saturated. “Cycloalkyl” includes monocyclic and bicyclic rings. Typically, a monocyclic cycloalkyl group has from 3 to about 10 carbon atoms, more typically 3 to 8 carbon atoms unless otherwise defined. The second ring of a bicyclic cycloalkyl may be selected from saturated, unsaturated and aromatic rings. Cycloalkyl includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings. The term “fused cycloalkyl” refers to a bicyclic cycloalkyl in which each of the rings shares two adjacent atoms with the
other ring. The second ring of a fused bicyclic cycloalkyl may be selected from saturated, unsaturated and aromatic rings.
[210] The terms “halo” and “halogen”, as used herein, means halogen and includes chloro, fluoro, bromo, and iodo.
[211] The term "haloalkyl", as used herein, refers to an alkyl group substituted by one or more halo. Examples of (C1-6)haloalkyl include, but are not limited to, trifluoromethyl, difluoromethyl, fluoromethyl, trichloromethyl, 2,2,2-trifluoroethyl, l,3-dibromopropan-2-yl, 3-bromo-2-fluoropropyl and 1,4,4- trifluorobutan-2-yl.
[212] The terms “hetaralkyl” and “hetero aralkyl”, as used herein, refers to an alkyl group substituted with a hetaryl group.
[213] The term “hetero alkyl”, as used herein, refers to a saturated or unsaturated chain of carbon atoms and at least one heteroatom, wherein no two heteroatoms are adjacent.
[214] The terms “heteroaryl” and “hetaryl” include substituted or unsubstituted aromatic single ring structures, preferably 5- to 7 -membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The terms “heteroaryl” and “hetaryl” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heteroaryl groups include, but are not limited to, furyl, thienyl, pyrrolyl, pyridyl, pyridyl N-oxide, pyrazolyl, pyrimidinyl, imidazolyl, isoxazolyl, oxazolyl, oxadiazolyl, pyrazinyl, indolyl, thiophen-2-yl, quinolyl, benzopyranyl, isothiazolyl, thiazolyl, thiadiazole, indazole, benzimidazolyl, thieno[3,2-b]thiophene, triazolyl, triazinyl, imidazo[l,2-b]pyrazolyl, furo[2,3-c]pyridinyl, imidazo[l,2-a]pyridinyl, indazolyl, pyrrolo[2,3-c]pyridinyl, pyrrolo[3,2-c]pyridinyl, pyrazolo[3,4- c]pyridinyl, thieno[3,2-c]pyridinyl, thieno[2,3-c]pyridinyl, thieno[2,3-b]pyridinyl, benzothiazolyl, indolyl, indolinyl, indolinonyl, dihydrobenzothiophenyl, dihydrobenzofuranyl, benzofuran, chromanyl, thiochromanyl, tetrahydroquinolinyl, dihydrobenzothiazine, dihydrobenzoxanyl, quinolinyl, isoquinolinyl, 1,6-naphthyridinyl, benzo[de]isoquinolinyl, pyrido[4,3-b][l,6]naphthyridinyl, thieno[2,3- b]pyrazinyl, quinazolinyl, tetrazo lo[l, 5 -a]pyridinyl, [l,2,4]triazolo[4,3-a]pyridinyl, isoindolyl, pyrrolo[2,3-b]pyridinyl, pyrrolo[3,4-b]pyridinyl, pyrrolo[3,2-b]pyridinyl, imidazo[5,4-b]pyridinyl, pyrrolo[l,2-a]pyrimidinyl, tetrahydropyrrolo[l,2-a]pyrimidinyl, 3,4-dihydro-2H-lA2-pyrrolo[2,l- b]pyrimidine, dibenzo [b,d]thiophene, pyridin-2-one, furo[3,2-c]pyridinyl, furo[2,3-c]pyridinyl, 1H- pyrido[3,4-b][l,4]thiazinyl, benzooxazolyl, benzoisoxazolyl, furo[2,3-b]pyridinyl, benzothiophenyl, 1,5- naphthyridinyl, furo[3,2-b]pyridine, [l,2,4]triazolo[l,5-a]pyridinyl, benzo[l,2,3]triazolyl, imidazo[l,2- a]pyrimidinyl, [l,2,4]triazolo[4,3-b]pyridazinyl, benzo[c][l,2,5]thiadiazolyl, benzo[c][l,2,5]oxadiazole,
1 ,3 -dihydro-2H-benzo [d]imidazol-2-one, 3 ,4-dihydro-2H-pyrazolo [ 1 ,5 -b] [ 1 ,2]oxazinyl, 4,5,6,?- tetrahydropyrazolo[l,5-a]pyridinyl, thiazolo[5,4 d]thiazolyl, imidazo[2,l-b][l,3,4]thiadiazolyl, thieno[2,3-b]pyrrolyl, 3H-indolyl, indolinyl, indolinonyl, dihydrobenzothiophenyl, dihydrobenzofuran, chromanyl, thiochromanyl, tetrahydroquinolinyl, dihydrobenzothiazine, 3 ,4-dihydro-lH-isoquinolinyl, 2,3- dihydrobenzofuran, indolinyl, indolyl, and dihydrobenzoxanyl.
[215] The term “heteroatom” as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur.
[216] The terms “heterocyclyl”, “heterocycle”, and “heterocyclic” refer to substituted or unsubstituted non-aromatic ring structures, preferably 3- to 10-membered rings, more preferably 3- to 7-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The terms “heterocyclyl” and “heterocyclic” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heterocyclyl groups include, for example, piperidine, piperazine, pyrrolidine, morpholine, lactones, lactams, and the like. Heterocyclyl groups can also be substituted by oxo groups. For example, “heterocyclyl” encompasses both pyrrolidine and pyrrolidinone.
[21?] The term “hydroxyalkyl”, as used herein, refers to an alkyl group substituted with a hydroxy group.
[218] “Haloalkyl”, as used herein, refers to an alkyl group substituted with one or more halogens.
Examples of haloalkyl groups include, but are not limited to, trifluoromethyl, difluoromethyl, pentafluoroethyl, trichloromethyl, etc.
[219] As used herein, the term " ox_ o"XxOo” refers to a carbonyl group. When an oxo substituent occurs on an otherwise saturated group, such as with an oxo-substituted cycloalkyl group (e.g., 3-oxo-cyclobutyl), the substituted group is still intended to be a saturated group. When a group is referred to as being substituted by an “oxo” group, this can mean that a carbonyl moiety (i.e., — C(=0) — ) replaces a methylene unit (i.e., — CH2— ).
[220] The term “optionally substituted” means that a given chemical moiety (e.g., an alky 1 group) can (but is not required to) be bonded other substituents (e.g., heteroatoms). For instance, an alkyl group that is optionally substituted can be a fully saturated alkyl chain (e.g., a pure hydrocarbon).
Alternatively, the same optionally substituted alkyl group can have substituents different from hydrogen. For instance, it can, at any point along the chain be bounded to a halogen atom, a hydroxyl group, or any other substituent described herein. Thus, the term “optionally substituted” means that a given chemical moiety has the potential to contain other functional groups, but does not necessarily have any further
functional groups. Suitable substituents used in the optional substitution of the described groups include, without limitation, halogen, oxo, -OH, -CN, -COOH, -CH2CN, -O-(C1-C6)alkyl, (C1-C6)alkyl, (C1- C6)alkoxy, (C1-C6)haloalkyl, (C1-C6)haloalkoxy, -O-(C2-C6)alkenyl, -O-(C2-C6)alkynyl, (C2-C6)alkenyl, (C2-C6)alkynyl, -OH, -OP(O)(OH)2, -OC(O)(C1-C6)alkyl, -C(O)(C1-C6)alkyl, -OC(O)O(C1-C6)alkyl, - NH2, -NH((C1-C6)alkyl), -N((C1-C6)alkyl)2, -NHC(O)(C1-C6)alkyl, -C(O)NH(C1-C6)alkyl, -S(O)2(C1- C6)alkyl, -S(O)NH(C1-C6)alkyl, and S(O)N((C1-C6)alkyl)2. The substituents can themselves be optionally substituted. “Optionally substituted” as used herein also refers to substituted or unsubstituted whose meaning is described below.
[221] The term “substituted” means that the specified group or moiety bears one or more suitable substituents wherein the substituents may connect to the specified group or moiety at one or more positions. For example, an aryl substituted with a cycloalkyl may indicate that the cycloalkyl connects to one atom of the aryl with a bond or by fusing with the aryl and sharing two or more common atoms.
[222] The term “unsubstituted” means that the specified group bears no substituents.
[223] “Pharmaceutically-acceptable acid addition salt” means those salts which retain the biological effectiveness and properties of the free bases and which are not biologically or otherwise undesirable, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, sulfamic acid, nitric acid, phosphoric acid, and the like, and organic acids such as acetic acid, trichloroacetic acid, trifluoroacetic acid, adipic acid, alginic acid, ascorbic acid, aspartic acid, benzenesulfonic acid, benzoic acid, 2-acetoxybenzoic acid, butyric acid, camphoric acid, camphorsulfonic acid, cinnamic acid, citric acid, digluconic acid, ethanesulfonic acid, glutamic acid, glycolic acid, glycerophosphoric acid, hemisulfic acid, heptanoic acid, hexanoic acid, formic acid, fumaric acid, 2- hydroxyethanesulfonic acid (isethionic acid), lactic acid, maleic acid, hydroxymaleic acid, malic acid, malonic acid, mandelic acid, mesitylenesulfonic acid, methanesulfonic acid, naphthalenesulfonic acid, nicotinic acid, 2 -naphthalenesulfonic acid, oxalic acid, pamoic acid, pectinic acid, phenylacetic acid, 3- phenylpropionic acid, picric acid, pivalic acid, propionic acid, pyruvic acid, pyruvic acid, salicylic acid, stearic acid, succinic acid, sulfanilic acid, tartaric acid, p-toluenesulfonic acid, undecanoic acid, and the like.
[224] “Pharmaceutically-acceptable base addition salt” means those salts which retain the biological effectiveness and properties of the free acids and which are not biologically or otherwise undesirable, formed with inorganic bases such as ammonia or hydroxide, carbonate, or bicarbonate of ammonium or a metal cation such as sodium, potassium, lithium, calcium, magnesium, iron, zinc, copper, manganese, aluminum, and the like. Particularly preferred are the ammonium, potassium, sodium, calcium, and magnesium salts. Salts derived from pharmaceutically-acceptable organic nontoxic bases include salts of primary, secondary, and tertiary amines, quaternary amine compounds, substituted amines including
naturally occurring substituted amines, cyclic amines and basic ion-exchange resins, such as methylamine, dimethylamine, trimethylamine, ethylamine, diethylamine, triethylamine, isopropylamine, tripropylamine, tributylamine, ethanolamine, diethanolamine, 2-dimethylaminoethanol, 2- diethylaminoethanol, dicyclohexylamine, lysine, arginine, histidine, caffeine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine, purines, piperazine, piperidine, A'-cthylpipcridinc. tetramethylammonium compounds, tetraethylammonium compounds, pyridine, N,N- dimethylaniline, A'-mcthylpipcridinc. A'-mcthylmorpholinc. dicyclohexylamine, dibenzylamine, N,N- dibenzylphenethylamine, 1 -ephenamine, N,N ’-dibenzylethylenediamine, polyamine resins, and the like. Particularly preferred organic nontoxic bases are isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexylamine, choline, and caffeine.
[225] A “patient” or “subject” is a mammal, e.g., a human, mouse, rat, guinea pig, dog, cat, horse, cow, pig, or nonhuman primate, such as a monkey, chimpanzee, baboon or, rhesus. In certain embodiments, the subject is a primate. In yet other embodiments, the subject is a human.
[226] The terms “pharmaceutically effective amount” or “therapeutically effective amount” or “effective amount” means an amount of a compound according to the disclosure which, when administered to a patient in need thereof, is sufficient to effect treatment for disease-states, conditions, or disorders for which the compounds have utility. Such an amount would be sufficient to elicit the biological or medical response of a tissue, system, or patient that is sought by a researcher or clinician. The amount of a compound according to the disclosure which constitutes a therapeutically effective amount will vary depending on such factors as the compound and its biological activity, the composition used for administration, the time of administration, the route of administration, the rate of excretion of the compound, the duration of treatment, the type of disease-state or disorder being treated and its severity, drugs used in combination with or coincidentally with the compounds of the disclosure, and the age, body weight, general health, sex, and diet of the patient. Such a therapeutically effective amount can be determined routinely by one of ordinary skill in the art having regard to their own knowledge, the prior art, and this disclosure.
[227] As used herein, the term “pharmaceutical composition” refers to a compound of the disclosure, or a pharmaceutically acceptable salt, hydrate, solvate, stereoisomer, or tautomer thereof, together with at least one pharmaceutically acceptable carrier, in a form suitable for oral or parenteral administration.
[228] “Carrier” encompasses carriers, excipients, and diluents and means a material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting a pharmaceutical agent from one organ, or portion of the body, to another organ, or portion of the body of a subject.
[229] “Combination” refers to either a fixed combination in one dosage unit form, or a combined administration where a compound of the present disclosure and at least one combination partner (e.g. another drug as explained below, also referred to as “therapeutic agent” or “co-agent”) may be administered independently at the same time or separately within time intervals, especially where these time intervals allow that the combination partners show a beneficial effect from the co-action of these therapeutic agents. The beneficial effect of the combination includes, but is not limited to, a cooperative, e.g., synergistic, effect and/or a pharmacokinetic or pharmacodynamic co-action, or any combination thereof, resulting from the combination of therapeutic agents. In one embodiment, administration of these therapeutic agents in combination is carried out over a defined time period (e.g., minutes, hours, days or weeks depending upon the combination selected). “
[230] The single components may be packaged in a kit or separately. One or both of the components (e.g., powders or liquids) may be reconstituted or diluted to a desired dose prior to administration. The terms “co -administration” or “combined administration” or the like as utilized herein are meant to encompass administration of the selected combination partner to a single subject in need thereof (e.g. a patient), and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
[231] The term “pharmaceutical combination” as used herein means a product that results from the mixing or combining of more than one therapeutic agent and includes both fixed and non-fixed combinations of the therapeutic agents. The term “fixed combination” means that the therapeutic agents, e.g., a compound of the present disclosure and a combination partner, are both administered to a patient simultaneously in the form of a single entity or dosage. The term “non-fixed combination” means that the therapeutic agents, e.g., a compound of the present disclosure and a combination partner, are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the two compounds in the body of the patient. The latter also applies to cocktail therapy, e.g. the administration of three or more therapeutic agents.
[232] A subject is “in need of’ a treatment if such subject would benefit biologically, medically, or in quality of life from such treatment (preferably, a human).
[233] The term “PCSK9” or “proprotein convertase subtilisin/kexin type 9” interchangeably refer to a naturally-occurring human proprotein convertase belonging to the proteinase K subfamily of the secretory subtilase family. PCSK9 is synthesized as a soluble zymogen that undergoes autocatalytic intramolecular processing in the endoplasmic reticulum, and is thought to function as a proprotein convertase. PCSK9 plays a role in cholesterol homeostasis and may have a role in the differentiation of cortical neurons.
Mutations in the PCSK9 gene are a cause of autosomal dominant familial hypercholesterolemia. (Burnett and Hooper, Clin. Biochem. Rev. (2008) 29(1): 11 -26)
[234] As used herein, the term “inhibit”, “inhibition”, or “inhibiting” refers to the reduction or suppression of a given condition, symptom, or disorder, or disease, or a significant decrease in the baseline activity of a biological activity or process.
[235] As used herein, the term “treat”, “treating", or "treatment" of any disease or disorder refers to alleviating or ameliorating the disease or disorder (i.e., slowing or arresting the development of the disease or at least one of the clinical symptoms thereof); or alleviating or ameliorating at least one physical parameter or biomarker associated with the disease or disorder, including those which may not be discernible to the patient.
[236] As used herein, the term “prevent”, “preventing", or “prevention” of any disease or disorder refers to the prophylactic treatment of the disease or disorder; or delaying the onset or progression of the disease or disorder.
[237] “Pharmaceutically acceptable” means that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
[238] “Disorder” means, and is used interchangeably with, the terms disease, condition, or illness, unless otherwise indicated.
[239] ‘ ‘Administer”, “administering”, or “administration” means to either directly administering a disclosed compound or pharmaceutically acceptable salt of the disclosed compound or a composition to a subject, or administering a prodrug derivative or analog of the compound or pharmaceutically acceptable salt of the compound or composition to the subject, which can form an equivalent amount of active compound within the subject’s body.
[240] “Compounds of the present disclosure”, “Compounds of Formula (I)”, “compounds of the disclosure”, and equivalent expressions (unless specifically identified otherwise) refer to compounds of Formulae (I) and (la) as herein described including the salts particularly the pharmaceutically acceptable salts thereof, where the context so permits thereof, as well as all stereoisomers (including diastereoisomers and enantiomers), rotamers, tautomers, and isotopically labelled compounds (including deuterium (“D”) substitutions).
[241] In a specific embodiment, the term “about” or “approximately” means within 20%, preferably within 10%, and more preferably within 5% of a given value or range.
[242] As used herein, a "modulator of PCSK9" refers to a compound or molecule that is able to modulate PCSK9 biological activity or function, and/or downstream pathway(s) mediated by PCSK9 activity.
[243] As used herein, an "inhibitor of PCSK9" refers to a compound or molecule that is able to inhibit PCSK9 biological activity or function, and/or downstream pathway(s) mediated by PCSK9 signaling. An inhibitor of PCSK9 activity encompasses compounds that block, antagonize, suppress or reduce (to any degree including significantly) PCSK9 biological activity, including downstream pathways mediated by PCSK9 activity.
[244] As used herein, “disorders or diseases responsive to the inhibition of PCSK9,” “disorders and conditions responsive to the inhibition of PCSK9,” “disorders and conditions responsive to the inhibition of PCSK9 activity,” “disorders responsive to the inhibition of PCSK9,” “disorders responsive to the inhibition of PCSK9 activity,” “disorders in which PCSK9 plays a role,” “PCSK9-mediated disease or disorder” and like terms include, but are not limited to, hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, sepsis, cancer, psoriasis, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
[245] As used herein, “Inhibition of PCSK9 activity,” or “inhibition of PCSK9,” refers to a decrease in the PCSK9 activity, e.g., by administration of the compound of the disclosure.
[246] The term “hypercholesterolemia” or “dyslipidemia” includes, e.g., familial and non-familial hypercholesterolemia. Familial hypercholesterolemia (FH) is an autosomal dominant disorder characterized by elevation of serum cholesterol bound to low density lipoprotein (LDL). Familial hypercholesterolemia includes both heterozygous FH and homozygous FH. Hypercholesterolemia (or dyslipidemia) is the presence of high levels of cholesterol in the blood. It is a form of hyperlipidemia (elevated levels of lipids in the blood) and hyperlipoproteinemia (elevated levels of lipoproteins in the blood).
[247] Hyperlipidemia is an elevation of lipids in the bloodstream. These lipids include cholesterol, cholesterol esters, phospholipids and triglycerides. Hyperlipidemia includes for example, type I, Ila, lib, III, IV and V.
[248] Hypertriglyceridemia denotes high blood levels of triglycerides. Elevated levels of triglycerides are associated with atherosclerosis, even in the absence of hypercholesterolemia, and predispose to cardiovascular disease.
[249] “Sitosterolemia” or “phytosterolemia” is a rare autosomal recessively inherited lipid metabolic disorder characterized by hyperab sorption of sitosterol from the gastrointestinal tract and decreased biliary excretion of dietary sterols (i.e., leading to hypercholesterolemia, tendon and tuberous xanthomas, premature development of atherosclerosis) and altered cholesterol synthesis.
[250] “Atherosclerosis” includes hardening of arteries associated with deposition of fatty substances, cholesterol, cellular waste products, calcium and fibrin in the inner lining of an artery. The buildup that results is called plaque.
[251] ‘ ‘Atherosclerosis” or “arteriosclerotic vascular disease (ASVD)” is a specific form of arteriosclerosis involving thickening, hardening and loss of elasticity of the walls of arteries as a result of invasion and accumulation of white blood cells, containing both living, active white blood cells (producing inflammation) and remnants of dead cells, including cholesterol and triglycerides. Atherosclerosis is therefore a syndrome affecting arterial blood vessels due to a chronic inflammatory response of white blood cells in the walls of arteries.
[252] “Coronary heart disease,” also known as atherosclerotic artery disease, atherosclerotic cardiovascular disease, coronary heart disease or ischemic heart disease is the most common type of heart disease and cause of heart attacks. The disease is caused by plaque building up along the inner walls of the arteries of the heart, which narrows the lumen of arteries and reduces blood flow to the heart.
[253] ‘ ‘Xanthoma” is a cutaneous manifestation of lipidosis in which lipids accumulate in large foam cells within the skin. Xanthomas are associated with hyperlipidemias.
[254] The term "elevated Lp(a) concentration", as used herein, refers to a serum Lp(a) concentration above 30 mg/dl (75 nmol/L). "Elevated serum Lp(a)" means a serum Lp(a) level greater than about 14 mg/dL. In certain embodiments, a patient is considered to exhibit elevated serum Lp(a) if the level of serum Lp{a) measured in the patient is greater than about 15 mg/dL, about 20 mg/dL, about 25 mg/dL, about 30 mg/dL, about 35 mg/dL, about 40 mg/dL, about 45 mg/dL, about 50 mg/dL, about 60 mg/dL, about 70 mg/dL, about 80 mg/dL, about 90 mg/dL, about 100 mg/dL, about 20 mg/dL, about 140 mg/dL, about 150 mg dL, about 180 mg/dL, or about 200 mg/dL The serum Lp(a) level can be measured in a patient post-prandial. In some embodiments, the Lp(a) level is measured after a period of time of fasting (e.g., after fasting for 8 hrs, 8 hrs, 10 hrs, 12 hrs or more). Exemplary methods for measuring serum Lp(a) in a patient include, but are not limited to, rate immunonephelometry, ELISA, nephelometry, immunoturbidimetry, and dissociation-enhanced lanthanide fluorescent immunoassay, although any clinically acceptable diagnostic method can be used in the context of the present disclosure.
[255] By “elevated triglyceride levels” or “ETL” is meant any degree of triglyceride levels that is determined to be undesirable or is targeted for modulation.
[256] “Sepsis” is a systemic reaction characterized by arterial hypotension, metabolic acidosis, decreased systemic vascular resistance, tachypnea, and organ dysfunction. Sepsis can result from septicemia (i.e., organisms, their metabolic end-products or toxins in the blood stream), including bacteremia (i.e., bacteria in the blood), as well as toxemia (i.e., toxins in the blood), including endotoxemia (i.e., endotoxin in the blood). The term "sepsis" also encompasses systemic reactions
resulting from fungemia (i.e., fungi in the blood), viremia (i.e., viruses or virus particles in the blood), and parasitemia (i.e., helminthic or protozoan parasites in the blood). Thus, septicemia and septic shock (acute circulatory failure resulting from septicemia often associated with multiple organ failure and a high mortality rate) may be caused by a number of organisms.
Examples
[257] The disclosure is further illustrated by the following examples and synthesis schemes, which are not to be construed as limiting this disclosure in scope or spirit to the specific procedures herein described. It is to be understood that the examples are provided to illustrate certain embodiments and that no limitation to the scope of the disclosure is intended thereby. It is to be further understood that resort may be had to various other embodiments, modifications, and equivalents thereof which may suggest themselves to those skilled in the art without departing from the spirit of the present disclosure and/or scope of the appended claims.
[258] Compounds of the present disclosure may be prepared by methods known in the art of organic synthesis. In all of the methods it is understood that protecting groups for sensitive or reactive groups may be employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Green and P. G. M. Wuts (1999) Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art.
[259] Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton nuclear magnetic resonance (NMR) spectra were obtained on either Bruker Avance spectrometer or Varian Oxford 400 MHz spectrometer unless otherwise noted. NMR spectra are given in ppm (5) and coupling constants, J, are reported in Hertz. Tetramethylsilane (TMS) was used as an internal standard. Chemical shifts are reported in ppm relative to dimethyl sulfoxide (5 2.50), methanol (5 3.31), chloroform (5 7.26) or other solvent as indicated in NMR spectral data. A small amount of dry sample (2-5 mg) is dissolved in an appropriate deuterated solvent (1 mL). Mass spectra (ESI-MS) were collected using a Waters System (Acquity UPLC and a Micromass ZQ mass spectrometer) or Agilent-1260 Infinity (6120 Quadrupole); all masses reported are the m/z of the protonated parent ions unless recorded otherwise. The chemical names were generated using ChemBioDraw Ultra vl4 from CambridgeSoft.
[260] Temperatures are given in degrees C6lsius. As used herein, unless specified otherwise, the term "room temperature" or "ambient temperature" means a temperature of from 15 degrees centigrade to 30 degrees centigrade, such as of from 20 degrees centigrade to 30 degrees centigrade, such as of from 20 degrees centigrade to 25 degrees centigrade. If not mentioned otherwise, all evaporations are performed under reduced pressure, typically between about 15 mm Hg and 100 mm Hg (= 20-133 mbar). The
structure of final products, intermediates and starting materials is confirmed by standard analytical methods, e.g., microanalysis and spectroscopic characteristics, e.g., MS, IR, NMR. Abbreviations used are those conventional in the art.
[261] All starting materials, building blocks, reagents, acids, bases, dehydrating agents, solvents, and catalysts utilized to synthesis the compounds of the present disclosure are either commercially available or can be produced by organic synthesis methods known to one of ordinary skill in the art.
Table 1. Abbreviations used in the following examples and elsewhere herein are:


General synthetic routes
[262] Typically, the compounds of formula (I) can be prepared according to the Schemes provided infra.
[263] Scheme 1 represents the general synthesis of a compound of formula (I).
 wherein R1, A are as defined in herein.
wherein R1, A are as defined in herein.
Step A: tert-butyl (3-aminocyclopentyl)carbamate is reacted with la in the presence of a suitable base such as diisopropylethylamine at a suitable temperature such as 120 °C to form lb.
Step B: Deprotection of the protecting group to form 1c in the presence of a strong acid such as hydrochloric acid or trifluoroacetic acid.
Step C: Cyanamide Id can be formed through a nucleophilic substitution reaction of 1c with cyanogen bromide and a suitable base such as sodium acetate.
Step D: Cyanamide Id is reacted with hydroxyamine to form A'-hydroxyguanidinc le.
Step E: A'-hydroxyguanidinc le can be converted to the target compound 1,2,4-oxadiazole 1g by the following two conditions.
Condition E-l: Acylation with various acid If using coupling reagents such as HATU in presence of a suitable base such as A'. A'-diisopropylcthylaminc.
Condition E-2: Cyclization at a suitable temperature such as 100 °C to form 1g.
[264] Scheme 2 represents the alternative synthesis of a compound of formula (I) in Scheme 1.
 wherein R1, A are as defined in herein.
wherein R1, A are as defined in herein.
Step A: A'-hydroxyguanidinc le can be converted to the target compound 1,2,4-oxadiazole 1g by reacting with an orthoester 2a.
[265] Scheme 3 represents the alternative synthesis of a compound of formula (I) in Scheme 1.
 wherein R1, A are as defined in herein.
wherein R1, A are as defined in herein.
Step A: Amine 1c is reacted with 3-chloro-l,2,4-thiadiazole 3a to form the target compound 1g in the presence of a suitable base such as A'. A'-diisopropylcthylaminc at a suitable temperature such as 100 °C.
[266] Scheme 4 represents the general synthesis of a compound of formula (I).
 wherein R2, A are as defined in herein.
wherein R2, A are as defined in herein.
Step A: Amine 1c is reacted with 5-chloro-l,2,4-thiadiazole 4a to form the target compound 4b in the presence of a suitable base such as A'. A'-diisopropylcthylaminc at a suitable temperature such as 110 °C.
[267] Scheme 5 represents the general synthesis of a compound of Formula (I).
 wherein R1, A are as defined in herein.
wherein R1, A are as defined in herein.
Step A: Amine 1c is reacted with l,3,4-oxadiazol-2)(3H )-one 5a to form the target compound 5b with a coupling reagent such as PyBroP in the presence of a suitable base such as A'. A'-diisopropylcthylaminc.
[268] Scheme 6 represents the general synthesis of a compound of Formula (I).
 wherein R4, A are as defined in herein.
wherein R4, A are as defined in herein.
Step A: Amine 1c is reacted with 3 -(methylsulfinyl)- 1,2, 4-triazine 6a or 3 -(methylsulfonyl)- 1, 2, 4-triazine 6a’ to form the target compound 6b in the presence of a suitable base such as sodium carbonate.
[269] Scheme 7 represents the general synthesis of a compound of formula (I).
 wherein A is as defined herein.
wherein A is as defined herein.
Step A: Cyanamide Id reacted with sodium azide to form the target compound 7a.
[270] Scheme 8 represents the general synthesis of a compound of formula (I).
 wherein X, Y, Z, and A are as defined in herein.
wherein X, Y, Z, and A are as defined in herein.
Step A: Intermediate 8a can be formed by either of the following conditions
Scheme 1 Step C, D, and E
Scheme 2 Step A
Scheme 3 Step A
Scheme 4 Step A
Scheme 5 Step A
Scheme 6 Step A
Step B: Deprotection of the protecting group to form amine 8b in the presence of a strong acid such as hydrochloric acid or trifluoro acetic acid.
Step C: Amine 8b is reacted with la to form the target compound 8c following Scheme 1 Step A.
[271] Scheme 9 represents the general synthesis of a compound of formula (I).
 wherein R4, and A are as defined in in herein.
wherein R4, and A are as defined in in herein.
Step A: Intermediate 9a is formed following Scheme 6 Step A.
Step B: Deprotection of the protecting group to form 9 b in the presence of a strong acid such as hydrochloric acid or trifluoroacetic acid.
Step C: Amine 9b is reacted with la to form the target compound 9c following Scheme 1 Step A.
Example 1: Synthesis of 2-(6-(((1S,3S) -3-((5-methvl-l,2,4-oxadiazol-3-vl) amino)cyclopentyl)amino) pyridin-3-vl)pvridazin-3(2H)-one

Step-1; Synthesis of 2-(6-fluoropyridin-3-yl)pyridazin-3(2H)-one (Int-1)
[272] To a solution of 2-fluoro-5-iodopyridine (10 g, 44.84 mmol) in DMSO (150 mL), potassium carbonate (12.4 g, 89.69 mmol), pyridazin-3(2H )-one (4.52 g, 47.09 mmol) were added and the reaction mixture was purged with argon for 15 min at room temperature, then Cui (0.85 g, 4.48 mmol) and N,N- dim ethylcyclohexane- 1,2-diamine (0.95 g, 6.72 mmol) were added and purged with argon for another 10 min. After stirring at 130 °C for 16 h, the reaction was quenched with water and the product was extracted with EtOAc. The organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-70% EtOAc in hexane) to afford the title compound as a yellow solid (5.5 g, 64%). ESI-MS m/z: 191.7 [M+H]+. *H NMR (400 MHz, CD3OD) 5 8.50 - 8.49 (m,lH), 8.24 - 8.19 (m,lH), 8.05 (dd, J = 4.0, 1.6 Hz, 1H), 7.48 (dd, J = 9.6, 3.6 Hz, 1H), 7.21 - 7.18 (m, 1H), 7.09 (dd, J= 9.6, 1.6 Hz, 1H).
Step-2; Synthesis of tert-butyl ((LS',3.S')-3-((5-(6-oxopyridazin-l(6H )-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-2)
[273] To a solution of tert-butyl ((1S,35)-3-aminocyclopentyl)carbamate (2.5 g, 12.48 mmol) in DMSO
(30 mL), DIPEA (6.5 mL, 37.44 mmol) and Int-1 (2.86 g, 14.97 mmol) were added. After stirring at 120
°C for 16 h, the reaction mixture was quenched with water and the product was extracted with EtOAc.
The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-70% EtOAc in hexane) to afford the title compound as an off-white solid (2.5 g, 54%), ESI-MS m/z; 372.05 [M+H]+.
Step-3; Synthesis of 2-(6-(((1S,3S)-3-aminocyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one hydrochloride (Int-3)
[274] A mixture of Int-2 (1.2 g, 3.23 mmol) in 4 M HC1 in 1,4-dioxane (15 mL) was stirred for 2 h at room temperature. The mixture was concentrated under reduced pressure. The residue was triturated three times with n-pentane (10 mL) to afford the title compound as a brown solid (1.2 g), which was used in the next step without further purification. ESI-MS m/z: 271.8 [M+H]+.
Step-4; Synthesis of /V-((LS',3.S')-3-((5-(6-oxopyridazin-l(6H )-yl)pyridin-2- yl)amino)cyclopentyl)cyanamide (Int-4)
[275] To a solution of Int-3 (1.5 g, 4.87 mmol) in THE (50 mL) were added NaOAc (1.2 g, 14.62 mmol) and cyanogen bromide (1.55 g, 14.62 mmol) at 0 °C. The reaction mixture was allowed to warm to room temperature and was stirred for 16 h, then filtered through a C6lite® pad, the filtrate was concentrated under reduced pressure to afford the crude title compound as a yellow sticky solid (1.8 g), which was used in the next step without further purification. ESI-MS m/z: 296.85 [M+H]+.
Step-5; Synthesis of 2-hydroxy-l-((1S,3S)-3-((5-(6-oxopyridazin-l(6H )-yl)pyridin-2-yl)amino) cyclopentyl)guanidine (Int-5)
[276] A mixture of Int-4 (1.11 g, 3 mmol), hydroxylamine hydrochloride (1.67 g, 24 mmol) and TEA (3.35 mL, 24 mmol) in EtOH (20 mL) was stirred at 50 °C overnight. The reaction mixture was concentrated under reduced pressure, and the residue was purified by reverse phase flash column chromatography on C18 (eluent: 10-80% 0.1% NH4OH in ACN/water) to afford the title compound as a yellow solid (680 mg, 69%), ESI-MS m/z: 330.1 [M+H]+.
Step-6; Synthesis of 2-(6-(((1S,3S')-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino) pyridin-3-yl)pyridazin-3(2H)-one (Example 1)
[277] To a mixture of Int-5 (150 mg, 90 wt%, 0.41 mmol) in 1,1,1 -trim ethoxyethane (12 mL, 94 mmol) was added acetic acid (24 μM, 0.41 mmol). After stirring at 60 °C for 3 h, the reaction mixture was concentrated under reduced pressure. The residue was purified by reverse phase flash column chromatography on Cl 8 (eluent: 10-80% ACN/water with 0.1% NH4OH) to provide the title compound as a yellow solid (120 mg, 81%). ESI-MS m/z: 354.1 [M+H]+. *H NMR (400 MHz, CD3OD) 5 8.15 (d, J = 2.4 Hz, 1H), 8.02 (dd, J= 3.6, 1.6 Hz, 1H), 7.61 (dd, J = 9.3, 2.8 Hz, 1H), 7.46 (dd, J = 9.3, 3.6 Hz,
1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.59 (d, J= 9.6 Hz, 1H), 4.37 - 4.30 (m, 1H), 4.01 - 3.95 (m, 1H), 2.41 (s, 3H), 2.29 - 2.19 (m, 2H), 2.10 - 2.02 (m, 1H), 1.99 - 1.90 (m, 1H), 1.66 - 1.55 (m, 2H).
Example 2: Synthesis of 2-(6-(((1S,3S)-3-((5-cvclopropvl-l,2,4-oxadiazol-3-vl)amino)cvclopentvl) amino)pyridin-3-vl) pyridazin-3(2H)-one

[278] To a solution of cyclopropanecarboxylic acid (30 mg, 0.34 mmol) in DCM (4 mL), was added HATU (0.132 g, 0.34 mmol) followed by DIPEA (70 pL, 0.41 mmol). After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (0.138 g, 0.41 mmol) in DCM (6 mL) and NMP (0.2 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, the residue was dissolved in DCE (5 mL). The mixture was heated under stirring at 100 °C for 2 h, then concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (20-40% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 20 mm), 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (35 mg, 27%), ESI-MS m/z: 380.15 [M+H]+. 1H NMR (400 MHz, CD3OD) 5 8.15 - 8.14 (m, 1H), 8.02 (dd, J= 3.6, 1.6 Hz, 1H), 7.60 (dd, J= 8.8, 2.4 Hz, 1H), 7.45 (dd, J= 9.2, 3.6 Hz, 1H), 7.06 (dd, J= 9.2, 1.6 Hz, 1H), 6.58 (d, J= 8.8 Hz, 1H), 4.28 - 4.19 (m, 1H), 3.90 - 3.82 (m, 1H), 2.10 - 2.05 (m, 2H), 2.01 - 1.91 (m, 2H), 1.78 - 1.70 (m, 1H), 1.58 - 1.43 (m, 2H), 1.18 - 1.14 (m, 2H), 1.11 - 1.08 (m, 2H).
Example 3: Synthesis of 2-(6-(((1S,3S)-3-((5-(l,l-difluoroethvl)-l,2,4-oxadiazol-3-vl) amino)cvclopentvl)amino)pyridin-3-vl)pyridazin-3(2H) -one

[279] To a solution of 1 -fluorocyclopropane- 1 -carboxylic acid (40 mg, 0.36 mmol) in DCM (4 mL), was added HATU (0.138 g, 0.36 mmol) followed by DIPEA (70 pL, 0.43 mmol) at room temperature. After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (0.144 g, 0.43 mmol) in DCM (6 mL) and NMP (0.2 mL). After stirring at room temperature for 1
h, the mixture was concentrated under reduced pressure, the residue was dissolved in DCE (5 mL) and heated at 100 °C for 2 h. The mixture was concentrated under reduced pressure and purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The obtained crude product was purified by preparative HPLC (20-40% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 20 mm), 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (40 mg, 27%), ESI-MS m/z: 404.05 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.14 (dd, J= 2.8, 0.8 Hz, 1H), 8.02 (dd, J= 4.0, 2.0 Hz, 1H), 7.59 (dd, J= 9.2, 2.8 Hz, 1H), 7.45 (dd, J= 9.2, 4.0 Hz, 1H), 7.05 (dd, J = 9.2, 1.6 Hz, 1H), 6.59 (d, J = 8.8 Hz, 1H), 4.36 - 4.32 (m, 1H), 4.04 - 4.00 (m, 1H), 2.27 - 2.22 (m, 2H), 2.11 - 2.07 (m, 1H), 2.04 (t, J= 18.8 Hz, 3H), 1.98 - 1.94 (m, 1H), 1.67 - 1.58 (m, 2H).
Example 4: Synthesis of 2-(6-(( -3-((5-(difluoromethvl)-l,2,4-oxadiazol-3-
 vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H) -one
vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H) -one

!NT-5 Example 4
[280] To a solution of 2,2-difluoroacetic acid (25 mg, 0.26 mmol) in DCM (4 mL) was added HATH (100 mg, 0.26 mmol) followed by DIPEA (50 pL, 0.31 mmol). After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (103 mg, 0.31 mmol) in DCM (6 mL) and NMP (0.2 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, the residue was dissolved in DCE (5 mL). After stirring at 100 °C for 2 h, the mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (20-60% acetonitrile/0.02% NH4OH in water; Gemini NX, 250 mm x 20 mm, 5.0 μm column, flow rate 20 mL/min) to afford the title compound as a yellow oil (27 mg, 33%), ESI-MS m/z: 390.15 [M+H]+. ' H NMR (400 MHz, CD3OD) 5 8.15 (dd, J= 2.4, 0.4 Hz, 1H), 8.02 (dd, J= 4.0, 1.6 Hz, 1H), 7.60 (dd, J = 8.8, 2.8 Hz, 1H), 7.45 (dd, J = 9.2, 4.0 Hz, 1H), 7.05 (dd, J= 9.2, 1.6 Hz, 1H), 6.91 (t, J = 52.0 Hz, 1H), 6.59 (d, J = 8.8 Hz, 1H), 4.37 - 4.32 (m, 1H), 4.04 - 4.00 (m, 1H), 2.29 - 2.22 (m, 2H), 2.11 - 2.06 (m, 1H), 1.99 - 1.93 (m, 1H), 1.69 - 1.57 (m, 2H).
Example 5: Synthesis of 2-(6-(((1S,3S)-3-((5-cvclobutvl-l,2,4-oxadiazol-3-yl)amino) cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one

[281] To a solution of cyclobutanoic acid (20 mg, 0.19 mmol) in DCM (4 mL), was added HATU (75 mg, 0.19 mmol) followed by DIPEA (0.1 mL, 0.23 mmol). After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (78 mg, 0.19 mmol) in DCM (6 mL) and NMP (0.2 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, and the residue was dissolved in DCE (5 mL). After stirring at 100 °C for 2 h, the mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The obtained crude product was purified by preparative HPLC (20- 50% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 20 mm), 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (20 mg, 21%), ESLMS m/z: 394.2 [M+H]+. ‘H NMR (400 MHz, CD3OD) 5 8.15 (d, J= 2.8 Hz, 1H), 8.03 (dd, J= 4.0, 1.6 Hz, 1H), 7.60 (dd, J= 9.2, 2.8 Hz, 1H), 7.46 (dd, J= 9.2, 3.6 Hz, 1H), 7.06 (dd, J= 9.2, 1.6 Hz, 1H), 6.59 (d, J= 9.2 Hz, 1H), 4.37 - 4.32 (m, 1H), 4.02 - 3.97 (m, 1H), 3.67 - 3.60 (m, 1H), 2.43 - 2.37 (m, 4H), 2.27 - 2.21 (m, 2H), 2.14 - 1.94 (m, 4H), 1.69 - 1.56 (m, 2H).
Example 6: Synthesis of 2-(6-(((1S,3S)-3-((5-(l-methvlcvclopropvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pvridazin-3(2H) -one

[282] To a solution 1-methylcyclopropane-l-carboxylic acid (36 mg, 0.35 mmol) in DCM (2 mL), was added HATU (0.137 g, 0.35 mmol) followed by DIPEA (0.1 mL, 0.43 mmol). After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (0.15 g, 0.43 mmol) in DCM (6 mL) and NMP (0.2 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, and the residue was dissolved in DCE (5 mL). After stirring at 100 °C for 2 h, the mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The obtained crude product was purified by preparative HPLC (20-50% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 20 mm), 5.0 μm column, flow rate 15 mL/min) to provide the title compound as a yellow solid (50
mg, 28%). ESI-MS m/z: 394.15 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.14 (dd, J= 2.4, 0.4 Hz, 1H), 8.02 (dd, .7= 4.0, 1.6 Hz, 1H), 7.59 (dd, J = 9.2, 2.8 Hz, 1H), 7.45 (dd, J = 9.6, 4.0 Hz, 1H), 7.05 (dd, J = 9.6, 1.6 Hz, 1H), 6.58 (d, J= 8.8 Hz, 1H), 4.35 - 4.28 (m, 1H), 3.98 - 3.92 (m, 1H), 2.26 - 2.21 (m, 2H), 2.05 - 2.01 (m, 1H), 1.96 - 1,91 (m, 1H), 1.64 - 1.54 (m, 2H), 1.46 (s, 3H), 1.29 (dd, J = 7.2, 3.6 Hz, 2H), 0.97 (dd, J= 6.8, 4.0 Hz, 2H).
Example 7: Synthesis of 2-(6-(((1S,3S)-3-((5-isopropvl-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pvridin-3-vl)pvridazin-3(2H) -one

[283] To a solution of isobutyric acid (40 mg, 0.45 mmol) in DCM (5 mL), were added HATH (0.172 g, 0.45 mmol) followed by DIPEA (0.1 mL, 0.54 mmol). After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (0.179 g, 0.45 mmol) in DCM (6 mL) and NMP (0.2 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, and the residue was dissolved in DCE (5 mL). The mixture was heated under stirring at 100 °C for 2 h, then concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The obtained crude product was purified by preparative HPLC (20-60% acetonitrile/0.02% NH4OH in water; Gemini NX, 250 mm x 20 mm, 5.0 μm column, flow rate 20 mL/min) to afford the title compound as a yellow solid (59 mg, 28%), ESI-MS m/z: 381.95 [M+H]+. XH NMR (400 MHz, CD3OD) 5 8.15 (dd, J= 2.8, 0.8 Hz, 1H), 8.02 (dd, .7= 4.0, 1.6 Hz, 1H), 7.60 (dd, J= 9.2, 2.8 Hz, 1H), 7.45 (dd, J= 9.2, 3.6 Hz, 1H), 7.06 (dd, J= 5.6, 1.6 Hz, 1H), 6.59 (d, J= 8.8 Hz, 1H), 4.37 - 4.31 (m, 1H), 4.01 - 3.95 (m, 1H), 3.11 - 3.01 (m, 1H), 2.28 - 2.26 (m, 2H), 2.08 - 2.03 (m, 1H), 1.98 - 1.92 (m, 1H), 1.67 - 1.57 (m, 2H), 1.32 (d, J= 7.2 Hz, 6H).
Example 8: Synthesis of 2-(6-(((1S,3S)-3-((5-(l-fluorocvclopropvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-yl)pvridazin-3(H) -one

[284] To a solution of 1 -fluorocyclopropane- 1 -carboxylic acid (29 mg, 0.27 mmol) in DCM (4 mL), was added HATH (106 mg, 0.27 mmol) followed by DIPEA (60 pL, 0.33 mmol) at room temperature.
After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (0.11 g, 0.33 mmol) in DCM (6 mL) and NMP (0.2 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, and the residue was dissolved in DCE (5 mL). After stirring at 100 °C for 2 h, the mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (20-70% acetonitrile/0.05% NH4OH in water; WATERS XB RIDGE (150 mm x 20 mm), 5.0 μm column, flow rate 15 mL/min) to provide the title compound as a yellow solid (30 mg, 27%). ESI-MS m/z: 398.10 [M+H]+. ' H NMR (400 MHz, CD3OD) 5 8.15 (dd, J = 2.4, 0.4 Hz, 1H), 8.03 (dd, J= 4.0, 1.6 Hz, 1H), 7.60 (dd, J= 5.2, 2.8 Hz, 1H), 7.46 (dd, J = 9.2, 3.6 Hz, 1H), 7.06 (dd, J= 9.6, 2.0 Hz, 1H), 6.59 (d, J= 8.8 Hz, 1H), 4.36 - 4.31 (m, 1H), 4.02 - 3.96 (m, 1H), 2.28 - 2.21 (m, 2H), 2.08 - 2.02 (m, 1H), 1.98 - 1.93 (m, 1H), 1.69 - 1.59 (m, 4H), 1.47 - 1.42 (m, 2H).
Example 9: Synthesis of 2-(6-(((1S,3S)-3-((3-phenvl-l,2,4-thiadiazol-5-vl)amino) cvclonentvl)amino)pvridin-3-vl)pyridazin-3(2H))-one

[285] To a solution of Int-3 (60 mg, 0.19 mmol) in DMA (1 mL), DIPEA (0.1 mL, 0.58 mmol) was added followed by 5-chloro-3-phenyl-l,2,4-thiadiazole (57 mg, 0.29 mmol). The resulting reaction mixture was stirred at 80 °C for 16 h. After quenching with water, the product was extracted with EtOAc. The combined organic phases were washed with byine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM) to provide the title compound as a yellow solid (30 mg, 36%). ESI-MS m/z: 432.25 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.18 - 8.17 (m, 1H), 8.14 - 8.10 (m, 2H), 8.03 (dd, .7= 4.0, 1.6 Hz, 1H), 7.61 (dd, J = 8.8, 2.4 Hz, 1H), 7.48 - 7.40 (m, 4H), 7.06 (dd, J= 9.6, 2.0 Hz, 1H), 6.61 (d, J= 8.8 Hz, 1H), 4.43 - 4.38 (m, 1H), 4.30 - 4.23 (m, 1H), 2.38 - 2.28 (m, 2H), 2.21 - 2.05 (m, 2H), 1.79 - 1.61 (m, 2H).
Example 10: Synthesis of l-methyl-3-(6-((((1S,3S-)3-((5-methvl-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pyridin-3-vl)imidazolidine-2, 4-dione

Step-1; Synthesis of 4-nitrophenyl(6-fluoropyridin-3-yl)carbamate (Int-6)
[286] To a solution of 6-fluoropyridin-3 -amine (5 g, 44.64 mmol) in acetonitrile (100 mL), was added 4-nitrophenyl chloroformate (9.89 g, 49.1 mmol) in acetonitrile (10 mL) dropwise. After stirring at room temperature for 30 min, the precipitation was collected by filtration and washed with acetonitrile to afford the title compound as a brown solid (7 g, 56%), ESI-MS m/z: 278.1 [M+H]+.
Step-2; Synthesis of 3-(6-fhioropyridin-3-yl)-l-methylimidazolidine-2, 4-dione (Int-7)
[287] To a mixture of ethyl methylglycinate hydrochloride (2.95 g, 25.27 mmol) in acetonitrile (120 mL) was added DIPEA (13.2 mL, 75.81 mmol). After stirring for 15 min, the mixture was added Int-6 (7 g, 25.27 mmol) portion wise. After stirring at room temperature for 20 min, the reaction mixture was concentrated under reduced pressure, the residue was purified by flash column chromatography on silica gel (eluent: 0-60% EtOAc in hexane) to afford the title compound as a brown oil (4 g, 75%). ESI-MS m/z: 210.05 [M+H]+. xH NMR (400 MHz, CD3OD) 5 8.31-8.30 (m, 1H), 8.05-8.00 (m, 1H), 7.21-7.18 (m, 1H), 4.13 (s, 3H), 3.04 (s, 3H).
Step-3; Synthesis of tert-butyl ((1S,3S)-3-((5-(3-methyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-8)
[288] To a solution of tert-butyl ((lS,35)-3-aminocyclopentyl)carbamate (0.8 g, 3.99 mmol) in DMSO (10 mL), were added DIPEA (2.1 mL, 11.98 mmol) and Int-7 (1 g, 4.79 mmol) at room temperature. After stirring at 120 °C for 16 h, the mixture was quenched with water and extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-60%
EtOAc in hexane) to afford the title compound as an off white solid (0.2 g, 13%). ESI-MS m/z: 390.2 [M+H]+.
Step-4; Synthesis of 3-(6-(((1S,3S)-3-aminocyclopentyl)amino)pyridin-3-yl)-l-methylimidazolidine- 2, 4-dione hydrochloride (Int-9)
[289] A mixture of Int-8 (0.2 g, 0.51 mmol) in 4M HC1 in 1,4-dioxane (2 mL) was stirred for 2 h at room temperature. The reaction mixture was concentrated under reduced pressure. The residue was triturated three times with n-pentane to provide the crude title compound as a brown solid (0.2 g), which was used in next step without purification. ESI-MS m/z: 290.15 [M+H]+.
Step-5; Synthesis of N-((1S,3S)-3-((5-(3-methyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)cyanamide (Int-10)
[290] To a solution of Int-9 (200 mg, 0.65 mmol) in THE (15 mL), were added NaOAc (160 mg, 1.95 mmol) and cyanogen bromide (205 mg, 1.95 mmol) at 0 °C. After stirring at room temperature for 16h, the reaction mixture was filtered through a C6lite® pad and the filtrate was concentrated under reduced pressure to afford the crude title compound as a yellow sticky solid (0.24 g, 80%), which was used in next step without purification. ESI-MS m/z: 315.2 [M+H]+.
Step-6; Synthesis of 2-hydroxy-l-((1S,3S)-3-((5-(3-methyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)guanidine (Int-11)
[291] To a solution of Int-10 (0.2 g, 0.64 mmol) in EtOH (10 mL), was added TEA (0.18 mL, 1.27 mmol) followed by hydroxylamine hydrochloride (49 mg, 0.7 mmol). The mixture was heated at 50 °C for 1 h, then concentrated under reduced pressure to afford the crude title compound as a yellow gum. (0.3 g), which was used in the next step without further purification. ESI-MS m/z: 348.25 [M+H]+.
Step-7: Synthesis of l-methyl-3-(6-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)imidazolidine-2, 4-dione (Example 10)
[292] A mixture of Int-11 (0.3 g, 0.86 mmol) in trimethyl orthoacetate (3 mL), DCE (3 mL) and NMP (0.2 mL), was added acetic acid (20 pL). The resulting mixture was stirred at 80 °C for 2 h. After concentration under reduced pressure, the residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (10-50% acetonitrile/0.02% NH4OH in water; Gemini NX, 250 mm x 20 mm, 5.0 μm column, flow rate 20 mL/min) to afford the title compound as a colorless solid (10 mg, 3%) ESI-MS m/z: 371.95 [M+H]+. JH NMR (400 MHz, CD3OD) 5 7.81 (d, J= 2.0 Hz, 1H), 7.27 (dd, J= 9.2, 2.8 Hz, 1H), 6.47 (dd, J= 9.2, 0.4 Hz, 1H), 4.23 - 4.18 (m, 1H), 3.98 (s, 2H), 3.91 - 3.83 (m, 1H), 2.96 (s, 3H), 2.31 (s, 3H), 2.19 - 2.07 (m, 2H), 2.00 - 1.91 (m, 1H), 1.85 - 1.78 (m, 1H), 1.59 - 1.41 (m, 2H).
Example 11: Synthesis of 2-(6-(((1S,3S)-3-((5-cvclonentvl-l,2,4-oxadiazol-3-yl)amino) cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one

[293] To a solution of cyclopentanecarboxylic acid (60 mg, 0.53 mmol) in DCM (3 mL), was added HATU (0.2 g, 0.53 mmol) followed by DIPEA (0.11 mL, 0.63 mmol) at room temperature. After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (0.21 g, 0.631 mmol) in DCM (7 mL) and NMP (0.5 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, the residue was dissolved in DCE (5 mL). After stirring at 100 °C for 2 h, the mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (20-60% acetonitrile/0.02% NH4OH in water; Gemini NX, 250 mm x 20 mm, 5.0 μm column, flow rate 20 mL/min) to afford the title compound as a yellow solid (65 mg, 25%). ESI-MS m/z: 408.15 [M+H]+. 1H NMR (400 MHz, CD3OD) 5 8.15 (dd, J= 2.8, 0.4 Hz, 1H), 8.02 (dd, J= 4.0, 1.6 Hz, 1H), 7.60 (dd, J= 8.8, 2.4 Hz, 1H), 7.45 (dd, J= 9.2, 3.6 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.59 (d, J = 9.2 Hz, 1H), 4.38 - 4.30 (m, 1H), 4.02 - 3.95 (m, 1H), 3.18 - 3.17 (m, 1H), 2.30 - 2.19 (m, 2H), 2.12 - 2.03 (m, 3H), 1.98 - 1.75 (m, 5H), 1.73 - 1.54 (m, 4H).
Example 12: Synthesis of 2-(6-(((1S,3S)-3-((5-(tert-butvl)-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one

[294| To a solution of pivalic acid (50 mg, 0.49 mmol) in DCM (3 mL), was added HATU (186 mg, 0.49 mmol) followed by DIPEA (0.1 mL, 0.59 mmol). After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (192 mg, 0.587 mmol) in DCM (5 mL) and NMP (0.5 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, the residue was dissolved in DCE (5 mL). After stirring at 100°C for 2 h, the mixture was concentrated under reduced pressure, was purified by flash column chromatography on silica gel (eluent:
0-10% MeOH in DCM). The crude product was purified by preparative HPLC (20-60% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 20 mm), 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (70 mg, 30%) ESI-MS m/z: 396.2 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.15 (dd, J= 2.8, 0.8 Hz, 1H), 8.02 (dd, J= 3.6, 1.6 Hz, 1H), 7.60 (dd, J = 9.2, 2.8 Hz, 1H), 7.46 (dd, J = 9.2, 4.0 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.60 - 6.58 (m, 1H), 4.38 - 4.29 (m, 1H), 4.01 - 3.94 (m, 1H), 2.30 - 2.18 (m, 2H), 2.11 - 2.03 (m, 1H), 1.98 - 1.89 (m, 1H), 1.70 - 1.52 (m, 2H), 1.37 (s, 9H).
Example 13: Synthesis of 2-(6-(((1S,3S) -3-((5-(3-hvdroxvcvclobutvl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-vl)pyridazin-3(2H)-one

[295] To a solution of 3 -hydroxycyclobutane- 1 -carboxylic acid (50 mg, 0.43 mmol) in DCM (4 mL), was added HATH (0.133 g, 0.43 mmol) followed by DIPEA (90 pL, 0.52 mmol). After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (0.17 g, 0.52 mmol) in DCM (6 mL) and NMP (0.5 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, the residue was dissolved in DCE (5 mL). After stirring at 100 °C for 2 h, the mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM) The crude product was purified by preparative HPLC (10-50% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 20 mm), 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (60 mg, 28%), ESI-MS m/z: 410.05 [M+H]+. 1H NMR (400 MHz, CD3OD) 5 8.05 (dd, .7= 2.8, 0.8 Hz, 1H), 7.92 (dd, J = 3.6, 1.6 Hz, 1H), 7.50 (dd, J= 9.2, 2.8 Hz, 1H), 7.35 (dd, J= 9.6, 4.0 Hz, 1H), 6.96 (dd, J= 9.6, 1.6 Hz, 1H), 6.48 (d, J= 9.2 Hz, 1H), 4.28 - 4.20 (m, 1H), 4.17 - 4.08 (m, 1H), 3.92 - 3.85 (m, 1H), 3.02 - 2.92 (m, 1H), 2.63 - 2.55 (m, 2H), 2.20 - 2.09 (m, 4H), 2.01 - 1.92 (m, 1H), 1.78 - 1.70 (m, 1H), 1.60 - 1.42 (m, 2H).
Example 14: Synthesis of 2-(6-(((1S,3S) -3-((5-ethvl-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pyridin-3-vl)pyridazin-3(2H)-one

[296] To a solution of propionic acid (35 mg, 0.47 mmol) in DCM (2 mL), was added HATU (180 mg, 0.47 mmol) followed by DIPEA (90 pL, 0.56 mmol). After stirring at room temperature for 30 min, the reaction mixture was added dropwise to a solution of Int-5 (154 mg, 0.467 mmol) in DCM (5 mL) and NMP (0.5 mL). After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure, the residue was dissolved in DCE (5 mL). The mixture was heated at 100 °C for 2 h, then concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (10-75% acetonitrile/0.05% NH4OH in water; Gemini NX, 250 mm x 20 mm, 5.0 μm column, flow rate 18 mL/min) to afford the title compound as a yellow solid (45 mg, 22%), ESI-MS m/z: 368.15 [M+H]+. JH NMR (400 MHz, CD3OD) 5 8.15 (dd, J= 2.8, 0.8 Hz, 1H), 8.02 (dd, J= 4.0, 1.6 Hz, 1H), 7.60 (dd, J = 9.2, 2.8 Hz, 1H), 7.46 (dd, J= 9.6, 4.0 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.58 (d, .7= 9.2 Hz, 1H), 4.38 - 4.29 (m, 1H), 4.02 - 3.95 (m, 1H), 2.77 (q, J= 7.6 Hz, 2H), 2.28 - 2.18 (m, 2H), 2. 11 - 2.03 (m, 1H), 1.98 - 1.90 (m, 1H), 1.60 - 1.52 (m, 2H), 1.30 (t, J= 7.6 Hz, 3H).
Example 15: Synthesis of 2-(6-(((1S,3S)-3-((5-(l-(trifluoromethvl)cvclopropvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pyridin-3-yl)pyridazin-3(2H)-one

Step-1; Synthesis of 2,5-dioxopyrrolidin-l-yl l-(trifluoromethyl)cyclopropane-l-carboxylate (Int- 12)
[297] To a solution of 1 -hydroxypyrrolidine-2,5 -dione (1 g, 8.69 mmol) in DCM (10 mL), were added l-(trifluoromethyl)cyclopropane-l-carboxylic acid (1.3 g, 8.69 mmol), DMAP (106 mg, 0.87 mmol) and DIC (1.3 mL, 8.69 mmol) dropwise at room temperature. After stirring for 12 h, the reaction mixture was filtered through a C6lite® pad, washed with DCM. The filtrate was concentrated under reduced pressure.
The residue was purified by flash column chromatography on silica gel (eluent: 0-60% EtOAc in hexane) to provide the title compound as an off-white solid (1.5 g, 66%). 'H NMR (300 MHz, CDC13) 5 2.84 (s, 4H), 1.79-1.72 (m, 2H), 1.64-1.58 (m, 2H).
Step-2; Synthesis of 2-(6-(((15,3S')-3-((5-(l-(trifhioromethyl)cyclopropyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 15)
[298] To a solution of 2,5-dioxopyrrolidin-l-yl l-(trifluoromethyl)cyclopropane-l -carboxylate (114 mg, 0.45 mmol) in NMP (3 mL), was added Int-5 (150 mg, 0.453 mmol). After stirring at room temperature for 4 h, then the mixture was heated to 70 °C and stirred for 16 h. The reaction mixture was quenched with water and extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by preparative HPLC (10-75% acetonitrile/0.05% NH4OH in water; Gemini NX, 250 mm x 20 mm, 5.0 μm column, flow rate 18 mL/min) to provide the title compound as a yellow solid (50 mg, 25%). ESI-MS m/z: 448.15 [M+H]+. ‘H NMR (400 MHz, CD3OD) 5 8.14 (dd, J= 2.8, 0.8 Hz, 1H), 8.01 (dd, J= 4.0, 1.6 Hz, 1H), 7.59 (dd, J = 9.2, 2.8 Hz, 1H), 7.45 (dd, J = 9.6, 4.0 Hz, 1H), 7.05 (dd, J= 9.2, 1.6 Hz, 1H), 6.57 (d, J= 9.2 Hz, 1H), 4.35 - 4.28 (m, 1H), 4.01 - 3.92 (m, 1H), 2.30 - 2.18 (m, 2H), 2.11 - 2.03 (m, 1H), 2.97 - 2.87 (m, 1H), 1.60 - 1.52 (m, 6H).
Example 16: Synthesis of 2-(6-(((1S,3S)-3-((5-chloro-l,2,4-thiadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one

[299] To a solution 2-(6-(((lS,35)-3-aminocyclopentyl)amino)pyridin-3-yl)pyridazin-3(2Z7)-one hydrochloride (60 mg, 0.19 mmol) in DMA (2 mL), DIPEA (0.1 mL, 0.58 mmol) and 3,5-dichloro-l,2,4- thiadiazole (60 mg, 0.38 mmol) were added at room temperature. After stirring at 80 °C for 16 h, the reaction mixture was quenched with water and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM) to provide the title compound as a pale yellow solid (30 mg, 39%), ESI-MS m/z: 390.10 [M+H]+. ‘H NMR (400 MHz, CD3OD) 5 8.14 (d, J= 2.4 Hz, 1H), 8.01 (dd, J= 4.0, 1.6 Hz, 1H), 7.58 (dd, J= 8.8, 2.4 Hz, 1H), 7.44 (dd, J= 9.2, 4.0 Hz, 1H), 7.04 (dd, J= 9.2, 1.6 Hz, 1H), 6.57 (d, J= 8.8 Hz, 1H), 4.37 - 4.31 (m, 1H), 4.16 - 4.12 (m, 1H), 2.30 - 2.22 (m, 2H), 2.10 - 1.98 (m, 2H), 1.68 - 1.59 (m, 2H).
Example 17: Synthesis of 2-(6-(((1S,3S)-3-((3-cvclopropyl-l,2,4-thiadiazol-5- vl)amino)cvclopentvl)amino)pvridin-3-yl)pvridazin-3(2H)-one

[300] To a solution of Int-5 (0.1 g, 0.32 mmol) in DMSO (5 mL), was added DIPEA (0.17 mL, 0.97 mmol) followed by 5-chloro-3-cyclopropyl-l,2,4-thiadiazole (78 mg, 0.48 mmol). After stirring at 110 °C for 16 h, the reaction mixture was quenched with water and extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (20-40% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 19 mm), 5.0 μm column, flow rate 15 mL/min) to provide the title compound as a yellow solid (40 mg, 31%), ESI-MS m/z: 396.25 [M+H]+. 1H NMR (400 MHz, CD3OD) 5 8.16 (dd, J= 2.8, 0.8 Hz, 1H), 8.02 (dd, J= 4.0, 2.0 Hz, 1H), 7.60 (dd, J= 8.8, 2.4 Hz, 1H), 7.45 (dd, J = 8.8, 4.0 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.60 - 6.58 (m, 1H), 4.38 - 4.31 (m, 1H), 4.15 - 4.08 (m, 1H), 2.31 - 2.21 (m, 2H), 2.12 - 2.04 (m, 1H), 2.04 - 1.95 (m, 2H), 1.71 - 1.58 (m, 2H), 1.00 - 0.96 (m, 2H), 0.94 - 0.90 (m, 2H).
Example 18: Synthesis of 2-(6-(((1S,3S) 3-((2H)-tetrazol-5-yl)amino) cyclopentyl) amino)pyridin-3- vl)pyridazin-3(2H)-one

[301] To a solution of Int-4 (0.2 g, 0.67 mmol) in DMF (5 mL), were added sodium azide (0.44 g, 6.74 mmol) and ammonium chloride (0.36 g, 6.74 mmol). After stirring at 90 °C for 16 h, the reaction mixture was quenched with water and extracted with DCM. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (10-40% acetonitrile/0.05% NH4OH in water; YMC (150 mm x 21.2 mm), 5.0 μm column, flow rate 18 mL/min) to afford the title compound as a yellow solid (40 mg, 17%). ESI-MS m/z: 340.15 [M+H]+. XH NMR (400 MHz, CD3OD) 5 8.17 (dd, J= 2.8, 0.8 Hz, 1H), 8.02 (dd, J= 4.0, 1.6 Hz, 1H), 7.62 (dd, J = 9.2, 2.8 Hz, 1H), 7.46 (dd, J = 9.6, 4.0 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.61 (d, J= 9.2 Hz, 1H), 4.88 - 4.80 (m, 1H), 4.59 - 4.50 (m, 1H), 2.58 - 2.50 (m, 1H), 2.48 - 2.39 (m, 2H), 2.22 - 2.08 (m, 2H), 1.82 - 1.71 (m, 1H).
Example 19: Synthesis of 2-(6-(((1S,3S) -3-((5-methvl-l,3,4-oxadiazol-2-vl)amino) cyclopentyl) amino)pvridin-3-vl)pyridazin-3(2H)-one

[302] To a solution of Int-3 (80 mg, 0.25 mmol) in DMF (1 mb), DIPEA (80 pL, 0.518 mmol), 5- methyl-l,3,4-oxadiazol-2(3H))-one (13 mg, 0.12 mmol) and PyBrop (13 mg, 0.31 mmol) were added sequentially. After stirring at room temperature for 16 h, the reaction mixture was quenched with water and extracted with DCM. The combined organic phases were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (10-60% acetonitrile/0.02% NH4OH in water; WATERS XBRIDGE (150 mm x 19 mm), 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (4 mg, 4%). ESI-MS m/z: 353.90 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.15 (dd, J= 2.4, 0.4 Hz, 1H), 8.02 (dd, J= 4.0, 1.6 Hz, 1H), 7.59 (dd, J= 8.8, 2.8 Hz, 1H), 7.45 (dd, J= 9.6, 4.0 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.58 (d, J= 9.2 Hz, 1H), 4.39 - 4.32 (m, 1H), 4.12 - 4.06 (m, 1H), 2.36 (s, 3H), 2.29 - 2.21 (m, 2H), 2.12 - 1.93 (m, 2H), 1.69 - 1.57 (m, 2H).
Example 20: Synthesis of l-(6-(((1S,3S) -3-((5-methvl-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pyridin-3-vl)pyrrolidin-2-one

Step-1-. Synthesis of tert-butyl ((1S,3S)-3-((5-iodopyridm-2-yl)ammo)cyclopentyl)carbamate (Int-13)
[303] To a solution of tert-butyl ((lS,35)-3-aminocyclopentyl)carbamate (1.5 g, 7.48 mmol) in DMSO (20 mL), was added DIPEA (3.9 mL, 22.46 mmol) and 2-fluoro-5-iodopyridine (2 g, 8.98 mmol). After stirring at 120 °C for 16 h, the reaction mixture was quenched with water and extracted with EtOAc. The combined organic phases were washed with water dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-25% EtOAc in hexane) to afford the title compound as a colorless solid (2 g, 66%). ESI-MS m/z: 403.95 [M+H]+.
Step-2-. Synthesis of tert-butyl ((1S,3S)-3-((5-(2-oxopyrrolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-14)
[304] To a solution of Int-13 (0.5 g, 1.23 mmol) in IPA (5 mL), K3PO4 (0.79 g, 3.71 mmol) and pyrrolidin-2-one (0.21 g, 0.47 mmol) were added. The reaction mixture was purged with argon for 15 min at room temperature. Then Cui (0.12 g, 0.61 mmol) and N,Ndim ethylcyclohexane- 1,2-diamine (88 mg, 0.61 mmol) were added and purged with argon for another 10 min. After stirring at 110 °C for 16 h, the mixture was quenched with water and extracted with EtOAc. The combined organic phases were washed with water, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 20-70% EtOAc in hexane) to afford the title compound as an off white solid (0.4 g, 89%). ESI-MS m/z: 361.15 [M+H]+.
Step-3; Synthesis of l-(6-(((lS,3S)-3-aminocyclopentyl) amino) pyridin-3-yl) pyrrolidine-2-one hydrochloride (Int-15)
[305] A mixture of Int-14 (0.4 g, 1.1 mmol) in 4 M HC1 in 1,4-dioxane (5 mL) was stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure. The residue was triturated three times with n-pentane to provide the crude title compound as a brown solid (0.4 g), which was carried to the next step without further purification. ESI-MS m/z: 260.8 [M+H]+.
Step-4; Synthesis of V-((l.S’.3.S’)-3-((5-(2-o\opyrroli(lin-l-yl)pyriilin-2- yl)amino)cyclopentyl)cyanamide (Int-16)
[306] To a solution of Int-15 (0.4 g, 1.34 mmol) in THF (20 mL), was added NaOAc (0.33 g, 4.04 mmol) and cyanogen bromide (0.43 g, 4.04 mmol) at 0 °C. After stirring at RT for 16 h, the reaction mixture was filtered through a C6lite® pad, washed with EtOAc. The filtrate was concentrated under reduced pressure to compound as a yellow gum(0.5 g), which was used in the next step without further purification. ESI-MS m/z: 286.15 [M+H]+.
Step-5; Synthesis of 2-hydroxy-l-((15,35)-3-((5-(2-oxopyrrolidin-l-yl)pyridin-2- yl)amino)cyclopentyl) guanidine (Int-17)
[307] To a solution of Int-16 (0.5 g, 1.75 mmol) in EtOH (10 mL), was added hydroxylamine hydrochloride (0.13 g, 1.92 mmol) and TEA (0.5 mL, 3.5 mmol). After stirring at 50 °C for 1 h, the reaction mixture was concentrated under reduced pressure. The residue was purified with reverse phase flash column chromatography on C18 (eluent: 2-30% MeCN in water) to afford the title compound as a colorless solid (0.18 g, 33%). ESI-MS m/z: 318.9 [M+H]+.
Step-6; Synthesis of l-(6-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino) cyclopentyl)amino)pyridin-3-yl)pyrrolidin-2-one (Example 20)
[308] To a solution of Int-17 (90 mg, 0.28 mmol) in trimethyl orthoacetate (10 mL), was added acetic acid (50 pL). After stirring at 60 °C for 30 min. the reaction mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (10-40% acetonitrile/0.05% NELOH in water; WATERS XSelect (250 mm x 19 mm), 5.0 μm column, flow rate 18 mL/min) to afford the title compound as a colorless solid (2 mg, 2%). ESI-MS m/z: 343.3 [M+H]+. 1 H NMR (400 MHz, CD3OD) 5 8.07 (dd, J = 2.8, 0.8 Hz, 1H), 7.62 (dd, J = 9.2, 2.8 Hz, 1H), 6.54 (d, J= 9.2 Hz, 1H), 4.30 - 4.21 (m, 1H), 3.99 - 3.92 (m, 1H), 3.83 (t, J= 7.0 Hz, 2H), 2.55 (t, J= 8.0 Hz, 2H), 2.41 (s, 3H), 2.27 - 2.12 (m, 4H), 2.08 - 1.99 (m, 1H), 1.93 - 1.86 (m, 1H), 1.68 - 1.50 (m, 2H).
Example 21: Synthesis of 3-methyl-l-(6-(((1S,3S)-3-((5-methvl-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)imidazolidine-2,4-dione

Step-1; Synthesis of tert-butyl ((1S,3S)-3-((5-(3-methyl-2,4-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-18)
[309] To a solution of Int-13 (500 mg, 1.24 mmol) in IP A (6 mL), K3PO4 (790 mg, 3.72 mmol) and 3- methylimidazolidine-2, 4-dione (283 mg, 2.48 mmol) were added. The resulting reaction mixture was purged with argon for 15 min. Cui (118 mg, 0.62 mmol) and N ,N -dimethylcyclohexane-l,2-diamine (88 mg, 0.62 mmol) were added and purged with argon for another 10 min. After stirring at 110 °C for 16 h, the reaction mixture was quenched with water and extracted with EtOAc. The combined organic phases were washed with water, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 20-70% EtOAc in hexane) to afford the title compound as an off white solid (0.4 g, 83%). ESI-MS m/z: 390.2 [M+H]+. Step-2; Synthesis of l-(6-(((1S,3S)-3-aminocyclopentyl)amino)pyridin-3-yl)-3-methylimidazolidine- 2, 4-dione hydrochloride (Int-19)
[310] A mixture of Int-18 (400 mg, 1.03 mmol) in 4M HC1 in 1,4-dioxane (5 mL) was stirred for 2 h at room temperature. The reaction mixture was concentrated under reduced pressure. The residue was triturated three times with n-pentane to provide the crude title compound as a brown solid (0.43 g), which was used in the next step without further purification. ESI-MS m/z: 289.85 [M+H]+.
Step-3; Synthesis of A-((1S,3S)-3-((5-(3-methyl-2,4-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)cyan amide
[311] To a solution of Int-19 (430 mg, 1.32 mmol) in THE (15 mL), was added NaOAc (325 mg, 3.96 mmol) and cyanogen bromide (420 mg, 3.96 mmol). After stirring at room temperature for 16 h, the reaction mixture was filtered through a C6lite® pad, washed with EtOAc. The filtrate was concentrated under reduced pressure to afford the crude title compound as a brown sticky solid (0.395 g), which was used in the next step without further purification. ESI-MS m/z: 314.85 [M+H]+.
Step-4; Synthesis of 2-hydroxy-l-((15,35)-3-((5-(3-methyl-2,4-dioxoimidazolidine-l-yl)pyridin-2- yl)amino)cyclopentyl)guanidine (Int-21)
[312] To a solution of Int-20 (395 mg, 1.26 mmol) in EtOH (10 mL), was added hydroxylamine hydrochloride (96 mg, 1.38 mmol) and TEA (0.35 mL, 2.51 mmol). The mixture was heated under stirring at 50 °C for 1 h, then concentrated under reduced pressure. The residue was purified by reverse phase flash column chromatography on Cl 8 (eluent: 2-30% MeCN in water) to afford the title compound as an off-white solid (76 mg, 17%). ESLMS m/z: 348.15 [M+H]+.
Step-51. Synthesis of 3-methyl-l-(6-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)imidazolidine-2, 4-dione (Example 21)
[313] To a solution of Int-21 (70 mg, 0.2 mmol) in NMP (1 mL), was added 2,5-dioxopyrrolidin-l-yl acetate (24.4 mg, 0.18 mmol). After stirring at room temperature for 3 h, the reaction mixture was heated at 70 °C for 16 h. The reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were washed with water, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by preparative HPLC (10-40% acetonitrile/0.05% NH4OH in water; WATERS XSelect (250 mm x 19 mm), 5.0 μm column, flow rate 18 mL/min) to afford the title compound as a colorless solid (7 mg, 9%). ESI-MS m/z: 372.15 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.10 (dd, J= 2.8, 0.4 Hz, 1H), 7.64 (dd, J= 9.2, 2.8 Hz, 1H), 6.54 (d, J= 9.2 Hz, 1H), 4.33 (s, 2H), 4.30 - 4.21 (m, 1H), 3.99 - 3.91 (m, 1H), 3.03 (s, 3H), 2.40 (s, 3H), 2.28 - 2.15 (m, 2H), 2.07 - 1.99 (m, 1H), 1.92 - 1.83 (m, 1H), 1.68 - 1.49 (m, 2H).
Example 22: Synthesis of 2-(6-(((1S,3S)-3-((5-(l-hvdroxvcvclopentvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pvridazin-3(2H)-one

Step-11. Synthesis of 2,5-dioxopyrrolidin-l-yl 1-hydroxycyclopentane-l-carboxylate
[314] To a solution of 1 -hydroxycyclopentane- 1 -carboxylic acid (100 mg, 0.768 mmol) and 1- hydroxypyrrolidine-2,5-dione (124 mg, 1.08 mmol) in THF (5 mL) was added a solution of EDC hydrochloride (177 mg, 0.92 mmol) in DCM (2 mL) dropwise at 0 °C. The mixture was allowed to warm to room temperature and was stirred for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between water and EtOAc. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure to provide crude title compound as off-white solid (0.2 g), which was used in the next step without further purification. XH NMR (300 MHz, CDCh) 5 2.82 (s, 4H), 2.41-2.31 (m, 2H), 2.05-1.82 (m, 6H).
Step-21. Synthesis of 2-(6-(((1S,3S)-3-((5-(l-hydroxycyclopentyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 22)
[315] To a solution of Int-22 (124 mg, 0.55 mmol) in NMP (3 mL) was added Int-5 (180 mg, 0.55 mmol). After stirring at room temperature for 4 h, the reaction mixture was heated under stirring to 70 °C for 16 h. After quenching with water, the product was extracted with EtOAc. The combined organic phases were washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by preparative HPLC (10-40% acetonitrile/0.05% NEUOH in water; WATERS XSelect (250 mm x 19 mm), 5.0 μm column, flow rate 18 mL/min) to afford the title compound as yellow solid (9 mg, 4%). ESI-MS m/z: 424.1 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.14 (dd, J= 2.8, 0.4 Hz,lH), 8.02 (dd, J= 4.0, 1.6 Hz, 1H), 7.59 (dd, J= 9.2, 2.8 Hz, 1H), 7.45 (dd, J= 9.6, 4.0 Hz, 1H), 7.05 (dd, J= 9.6, 2.0 Hz, 1H), 6.58 (d, J = 9.2 Hz, 1H), 4.38 - 4.30 (m, 1H), 4.03 - 3.96 (m, 1H), 2.30 - 2.19 (m, 2H), 2.18 - 1.88 (m, 8H), 1.86 - 1.78 (m, 2H), 1.70 - 1.52 (m, 2H).
Example 23: Synthesis of 2-(6-(((1S,3S)-3-((5-(l-hvdroxvcvcloDentvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pvridazin-3(2H)-one

Step-11. Synthesis of 6'-fhioro-27/-[l,3'-bipyridin]-2-one (Int-23)
[316] To a solution of pyridin-2(177)-one (5 g, 24.4 mmol) in DCM (150 mL), was added pyridine (2.65 g, 33.6 mmol), (6-fluoropyridin-3-yl)boronic acid (3.79 g, 26.9 mmol). After stirring at room temperature for 10 min, Cu(OAc)z (4.068 g, 22.4 mmol) was added to the mixture. The resulting mixture was stirred at room temperature with oxygen balloon for 16 h. After quenching with water, the product was extracted with DCM. The combined organic phases were dried over sodium sulfate, filtered, and concentrated
under reduced pressure. The residue was purified with flash column chromatography on silica gel (eluent: 0-70% EtOAc in hexane) to afford the title compound as a yellow solid (0.68 g, 16%). ESI-MS m/z: 190.5 [M+H]+.
Step-2; Synthesis of tert-butyl ((l1$',31S')-3-((2-oxo-27/-[l,3,-bipyridin]-6,-yl)amino) cyclopentyl)carbamate
[317] To a solution of tert-butyl ((lS,35)-3-aminocyclopentyl)carbamate (500 mg, 2.5 mmol) in NMP (20 mL) was added DIPEA (1.3 mL, 7.5 mmol) and Int-23 (573 mg, 3 mmol). The mixture was heating under stirring in a microwave reactor at 140 °C for 3 h, then quenched with water and extracted with EtOAc. The combined organic phases were washed with brine, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified with flash column chromatography on silica gel (eluent: 0-70% EtOAc in hexane) to afford the title compound as an off white solid (0.28 g, 30%). ESI-MS m/z: 371.2 [M+H]+.
Step-3; Synthesis of 6'-(((1S,3S)-3-aminocyclopentyl)amino)-27/-[l,3'-bipyridin]-2-one hydrochloride (Int-25)
[318] To a mixture of Int-24 (0.28 g, 0.756 mmol) in DCM (10 mL) was added 4M HC1 in 1,4-dioxane (2.3 mL). After stirring for 2 h at room temperature, the reaction mixture was concentrated under reduced pressure. The residue was triturated three times with n-pentane to provide crude title compound as a brown solid (0.3 g), which was used in the next step without further purification. ESI-MS m/z: 271.05 [M+H]+.
Step-4: Synthesis of A-((1S,3S)-3-((2-oxo-277-[l,3'-bipyridin]-6'-yl)amino)cyclopentyl)cyanamide (Int-26)
[319] To a solution of Int-25 (0.3 g, 0.98 mmol) in THE (10 mL) was added NaOAc (241 mg, 2.94 mmol) and cyanogen bromide (308 mg, 2.94 mmol) at 0 °C. After stirring at room temperature for 16 h, the reaction mixture was filtered through a C6lite® pad, and the reside was washed with EtOAc. The filtrate was wash fractions were combined and concentrated under reduced pressure to afford the crude title compound as a brown gum(0.4 g), which was used in next step without further purification. ESI-MS m/z: 296.1 [M+H]+.
Step-5; Synthesis of 2-hydroxy-l-((1S,3S)-3-((2-oxo-27/-[l,3'-bipyridin]-6'- yl)amino)cyclopentyl)guanidine (Int-27)
[320] To a solution of Int-26 (400 mg, 1.36 mmol) in EtOH (10 mL), were added hydroxylamine hydrochloride (102 mg, 1.49 mmol) and TEA (0.3 mL, 2.71 mmol). The mixture was heated under stirring at 50 °C for 1 h, then concentrated under reduced pressure. The residue was purified by reverse
phase flash column chromatography on Cl 8 (eluent: 2-30% acetonitrile in water) to afford the title compound as a yellow gum (0.18 g, 41%). ESI-MS m/z: 329.1 [M+H]+.
Step-6; Synthesis of bMflLS'vLS'l-d-flS-methyl-LZH-oxadiazol-d-yllamino) cyclo pentyl )am in o)-2//- [l,3'-bipyridin]-2-one (Example 23)
[321] To a solution of acetic acid (8 mg, 0.25 mmol) in DCM (4 mL), was added HATU (95 mg, 0.25 mmol) followed by DIPEA (39 mg, 0.3 mmol). After stirring for 30 min, the reaction mixture was added dropwise to a solution of Int-27 (99 mg, 0.3 mmol) in DCM (5 mL) and NMP (0.5 mL). After stirring at room temperature for 1 h, the reaction mixture was concentrated under reduced pressure and the residue was dissolved in DCE (5 mL). The mixture was heated under stirring at 100 °C for 2 h, then concentrated under reduced pressure. The residue was purified with flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (15-55% acetonitrile/0.05% NH4OH in water; Kinetex® EVO (250 mm x 19 mm), 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a colorless solid (7 mg, 7%). ESI-MS m/z: 353.05 [M+H]+. XH NMR (400 MHz, CD3OD) 5 7.93 (dd, J= 2.8, 0.8 Hz 1H), 7.62 - 7.56 (m, 2H), 7.42 (dd, J= 9.2, 2.8 Hz, 1H), 6.62 - 6.58 (m, 2H), 6.47 - 6.43 (m, 1H), 4.37 - 4.30 (m, 1H), 4.01 - 3.93 (m, 1H), 2.40 (s, 3H), 2.38 - 2.16 (m, 2H), 2.10 - 2.01 (m, 1H), 1.96 - 1.88 (m 1H), 1.68 - 1.51 (m, 2H).
Example 24: Synthesis of 2-(6-(((1S,3S)-3-((5-cvclopropvl-l,2,4-thiadiazol-3- vl)amino)cvclonentvl)amino)pvridin-3-vl)pyridazin-3(27D-one

[322] To a solution of Int-3 (10 mg, 0.029 mmol) and 3-chloro-5-cyclopropyl-l,2,4-thiadiazole (14 mg, 0.087 mmol) in NMP (0.5 mL) was added DIPEA (0.5 mL, 2.96 mmol). The mixture was heated under stirring at 160 °C for 6 h, then purified by reverse phase flash column chromatography on Cl 8 (eluent: 10-80% ACN/water with 0.1% NH4OH) to provide the title compound as a yellow solid (4.4 mg, 28%). ESI-MS m/z: 396.2 [M+H]+. XH NMR (400 MHz, CD3OD) 5 8.27 (d, 1H, J = 2.4 Hz), 8.11 (dd, 1H, J = 2.4, 9.8 Hz), 7.98 (dd, 1H, J= 1.5, 3.9 Hz), 7.39 (dd, 1H, J= 3.9, 9.3 Hz), 7.15-7.10 (m, 1H), 6.61 (d, J = 9.2 Hz, 1H) 4.28-4.19 (m, 2H), 2.42-2.19 (m, 3H), 2.15-2.08 (m, 2H), 1.75-1.69 (m, 2H), 1.27-1.16 (m, 2H), 1.07-0.92 (m, 2H).
Example 25: Synthesis of 2-(6-(((l1S,,31S)-3-((5-(4-fluorophenvl)-l,2,4-thiadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one

[323] To a solution of Int-3 (22.94 mg, 0.075 mmol) and 3-chloro-5-(4-fluorophenyl)-l,2,4-thiadiazole (8 mg, 0.037 mmol) in NMP (0.5 mL) was added DIPEA (0.5 mL, 2.96 mmol). The mixture was heated under stirring at 180 °C for 1.5 h, then purified by reverse phase flash column chromatography on Cl 8 (eluent: 10-80% ACN/water with 0.1% NH4OH) to provide the title compound as a yellow solid (1.2 mg, 7%). ESI-MS m/z: 450.7 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.25 (d, 1H, J= 2.4 Hz), 8.04 (dd, 1H, J= 2.4, 9.8 Hz), 7.97 (dd, 1H, J= 1.5, 3.9 Hz), 7.92-7.88 (m, 2H), 7.39 (dd, 1H, J= 3.9, 9.8 Hz), 7.19- 7.13 (m, 2H), 7.05-6.98 (m, 2H), 4.31-4.29 (m, 1H), 4.21-4.16 (m, 1H), 2.32-2.22 (m, 2H), 2.11-2.08 (m, 2H) 1.71-1.64 (m, 2H)
Example 26: Synthesis of 2-(6-(((1S,3S) -3-((5-(l-hvdroxvcvclonropvl)-l,2,4-oxadiazol-3- vl)amino)cvclonentvl)amino)pvridin-3-vl)pyridazin-3(277)-one

[324] A mixture of 1-hydroxycyclopropane-l -carboxylic acid (2.48 mg, 0.024 mmol), HATU (9.24 mg, 0.024 mmol) and DIPEA (12.73 pL, 0.073 mmol) in DMF (0.5 mL) was stirred at room temperature for 20 min, then Int-5 (10 mg, 0.024 mmol) was added to the mixture. After stirring at room temperature for another 30 min., the reaction mixture was heated under stirring at 100 °C for 1 h. After cooling to room temperature, the mixture was purified by reverse phase flash column chromatography on Cl 8 (eluent: 10- 80% ACN/water with 0. 1% NH4OH) to provide the title compound as a yellow solid (0.3 mg, 3%). ESIMS m/z: 396.2 [M+H]+. *H NMR (400 MHz, CD3OD) 5 8.70-8.69 (m, 1H), 8.09 (d, 1H, J= 9.0 Hz), 7.92 (d, 1H, J= 4.3 Hz), 7.47 (d, 1H, J = 9.6 Hz), 7.09 (dd, 1H, J = 4.3, 9.6 Hz), 6.86 (d, 1H, J = 9.0 Hz), 4.14-4.08 (m, 2H), 2.29-2.25 (m, 1H,), 2.17-2.10 (m, 3H), 1.65-1.53 (m, 2H), 1.17-1.12 (m, 2H), 0.96- 0.90 (m, 2H).
Example 27: Synthesis of 2-(6-(((1S,3ty)-3-((5-(trifluoromethvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one

[325] To a mixture of Int-5 (11 mg, 0.033 mmol) in pyridine (0.5 mL) was added TFAA (7.08 pL, 0.05 mmol) at 0 °C. After stirring at room temperature for 1 h, the mixture was heated at 60 °C for 2 h. After cooling to rt, the mixture was purified by reverse phase flash column chromatography on Cl 8 (eluent: 10- 80% ACN/water with 0. 1% NFUOH) to provide the title compound as a yellow solid (0.4 mg, 3%). ESIMS m/z: 408.2 [M+H]+?H NMR (400 MHz, CD3OD) 5 8.69-8.67 (m, 1H), 8.09 (d, 1H, J= 9.0 Hz), 7.92 (d, 1H, J= 4.3 Hz), 7.47 (d, 1H, J = 9.6 Hz), 7.09 (dd, 1H, J = 4.3, 9.6 Hz), 6.86 (d, 1H, J = 9.0 Hz), 4.31-4.10 (m, 2H), 2.27-2.05 (m, 4H), 1.65-1.53 (m, 2H).
Example 28: Synthesis of 3-(6-((2-(6'-(((1S,3S)-3-((5-cvcloDroDvlDvrimidin-2- vl)amino)cvcloDentvl)amino)-2-oxo-27/-[l,3'-biDvridine]-5-carboxamido)ethvl)amino)-6-oxohexvl)-
2-((l£',3£')-5-((£')-3,3-dimethvl-5-sulfo-l-(3-sulfoDroDvl)mdolin-2-vlidene)Denta-l,3-dien-l-vl)-3- methvl-5-sulfo-l-(4-sulfobutyl)-37/-indol-l-ium

Step-1 : Synthesis of 2-chloro-5-cyclopropylpyrimidine (Int-28)
[326] To a solution of 5-bromo-2-chloropyrimidine (3 g, 15.5 mmol) in 1,4-dioxane (50 mL) was added cyclopropyl boronic acid (1.6 g, 18.61 mmol) followed by CS2CO3 (7.6 g, 23.26 mmol) at room temperature. After degassing with argon for 15 min, Pd(dppf)Ch.DCM (0.38 g, 0.46 mmol) was added to the mixture and the mixture was further purged with argon for 10 min at room temperature. The mixture was heated under stirring at 90 °C for 16 h, then filtered through a C6lite® pad and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica
gel (eluent: 0-20% EtOAc in hexane) to afford the title compound as a yellow solid (1.8 g, 75%). ESI-MS m/z: 155.10 [M+H]+.
Step-2; Synthesis of tert-butyl ((15,35)-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)carbamate (Int-29)
[327] To a solution of Int-28 (0.8 g, 5.17 mmol) in DMSO (20 mL), were added DIPEA (2.7 mL, 15.52 mmol) and teri-butyl ((lS,35)-3aminocyclopentyl) carbamate (1.09 g, 5.43 mmol) at room temperature. The mixture was heated under stirring at 110 °C for 16 h, then cooled to rt and quenched with water. The product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-40% EtOAc in hexane) to afford the title compound as a colorless solid (0.8 g, 49%). ESI-MS m/z: 318.85 [M+H]+.
Step-3; Synthesis of (15,3S')-/V1-(5-cyclopropylpyrimidin-2-yl)cyclopentane-l,3-diamine hydrochloride (Int-30)
[328] A mixture of Int-29 (0.8 g, 2.51 mmol) in 4 M HC1 in 1,4-dioxane (8 mL) was stirred for 2 h. The mixture was concentrated under reduced pressure. The residue was triturated three times with n-pentane (10 mL) to afford the title compound as a colorless solid (0.8 g), which was used in the next step without further purification. ESI-MS m/z: 218.75 [M+H]+.
Step-4; Synthesis of tert-butyl (2-(6-oxo-l,6-dihydropyridine-3-carboxamido)ethyl)carbamate (Int- 31)
[329] To a solution of 6-oxo- l,6-dihydropyridine-3 -carboxy lie acid (1 g, 7.18 mmol) in DMF (10 mL) were added HATU (3.5 g, 9.35 mmol) and DIPEA (4.9 mL, 28.77 mmol) followed by teri-butyl (2- aminoethyl) carbamate (1.3 g, 7.9 mmol) at 0 °C. The mixture was allowed to warm to room temperature and was stirred for 16 h. The mixture was quenched with water. The product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-20% EtOAc in hexane) to afford the title compound as a yellow solid (1.4 g, 69%). ESI-MS m/z: 281.75 [M+H]+.
Step-5; Synthesis ooff tert-butyl (2-(6'-fluoro-2-o\o-2//-| 1.3'-bipyridine|-5- carboxamido)ethyl)carbamate (Int-32)
[330] To a stirred solution of Int-31 (1 g, 3.55 mmol) in DCM (20 mL) was added (6-fluoropyridin-3- yl)boronic acid (601 mg, 4.26 mmol) at room temperature. The mixture was purged with oxygen for 10 min., then TEA (1.5 mL, 10.66 mmol) and copper acetate (1.9 g, 10.66 mmol) were added at room temperature. After stirring under O2 balloon for 48 h at room temperature, the reaction mixture was
diluted with cold water. The product was extracted with DCM. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-20% EtOAc in hexane) to afford the title compound as yellow sticky solid (0.4 g, 30%). ESI-MS m/z: 376.85 [M+H]+.
Step-6; Synthesis of tert-butyl (2-(6'-(((1S,3S)-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)amino)-2-oxo-27/-[l,3'-bipyridine]-5-carboxamido)ethyl)carbamate (Int-33)
[331] To a stirred solution of Int-32 (0.23 g, 0.61 mmol) in DMSO (5 mL), was added DIPEA (0.31 mL, 1.83 mmol), followed by Int-30 (0.156 g, 0.61 mmol). The reaction mixture was heated under stirring at 120 °C for 16 h, then cooled to rt. The mixture was quenched with water and extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM) to afford the title compound as a brown solid (80 mg, 23%). ESI-MS m/z: 575.35 [M+H]+.
Step-7; Synthesis of 2V-(2-aminoethyl)-6'-(((lS',3S')-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)amino)-2-oxo-27/-[l,3'-bipyridine]-5-carboxamide (Int-34)
[332] 4 M HC1 in 1,4-dioxane (2 mL) was added to a mixture of Int-33 (0.8 g, 2.51 mmol) in MeOH (2 mL) at room temperature. After stirring at room temperature for 2 h, the mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (10-50% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (19 mm xl50 mm), 5.0 μm column, flow rate 20 mL/min) to afford the title compound as an off-white solid (20 mg, 30%). ESI-MS m/z: 475.1 [M+H]+. ' H NMR (400 MHz, CD3OD) 5 8.24 (d, J= 2.4 Hz, 1H), 8.07 (s, 2H), 8.01 - 7.97 (m, 2H), 7.47 (dd, J= 8.8, 2.4 Hz, 1H), 6.62 (dd, J= 10.0, 2.4 Hz, 2H), 4.41 - 4.31 (m, 2H), 3.47 (t, J= 6.0 Hz, 2H), 2.91 (t, J= 6.4 Hz, 2H), 2.32 - 2.19 (m, 2H), 1.99 (t, J= 6.8 Hz, 2H), 1.79 - 1.72 (m, 1H), 1.65 - 1.53 (m, 2H), 0.94 - 0.87 (m, 2H), 0.63 - 0.58 (m, 2H).
Step-8; Synthesis of 3-(6-((2-(6'-(((1S,3S)-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)amino)-2-oxo-27/-[l,3'-bipyridine]-5-carboxamido)ethyl)amino)-6-oxohexyl)- 2-((12j,32j)-5-((2j)-3,3-dimethyl-5-sulfo-l-(3-sulfopropyl)indolin-2-ylidene)penta-l,3-dien-l-yl)-3- methyl-5-sulfo-l-(4-sulfobutyl)-37/-indol-l-ium
[333] A solution of Alexa Fluor® 647 NHS ester (21 mg, 0.016 mmol) in acetonitrile (1 mL) was added to Int-33 (7.82 mg, 0.016 mmol), followed by DIPEA (57.6 pL, 0.33 mmol) at room temperature. The reaction vial was wrapped in aluminum foil. After stirring at room temperature for 4 h, the mixture was purified by reverse phase flash column chromatography on Cl 8 (eluent: 10-80% ACN/water with 0.1%
NH4OH) to provide the ammonium salt of the title compound as a blue solid (15.6 mg, 67%). ESI-MS m/z: 665.22 [M+2H]2+.
Example 29: Synthesis of 2-(6-(((1S,3S)-3-((6-methvl-l,2,4-triazin-3-vl)amino) cvclopentvl)amino)pvridin-3-vl)pvridazin-3(277)-one

Step-1; Synthesis of 6-methyl-3-(methylthio)-l,2,4-triazine (Int-35)
[334] To a stirred solution of methyl hydrazinecarbimidothioate hydrogen iodide (5 g, 21.45 mmol) in EtOH (100 mL), was added l,l-dimethoxypropan-2-one (2.5 g, 21.45 mmol) at room temperature. After stirring at 70 °C for 4 h, the reaction mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-20% EtOAc in hexane) to provide the title compound as a brown oil (2.3 g, 76%). ESI-MS m!z; 141.95 [M+H]+.
Step-2; Synthesis of 6-methyl-3-(methylsulfonyl)-l,2,4-triazine (Int-36)
[335] To a solution of Int-35 (2.3 g, 16.29 mmol) in DCM (50 mL), was added mCPBA (8.4 g, 48.87 mmol) portion wise at 0 °C. The reaction mixture was allowed to warm to room temperature and was stirred for 2 h. The reaction mixture was concentrated under reduced pressure, and then the residue was purified by flash column chromatography on silica gel (eluent: 20-80% EtOAc in hexane) to provide the title compound as an off white solid (1.3 g, 61%). ESI-MS m!z; 173.95 [M+H]+.
Step-3; Synthesis of 2-(6-(((1S,3S)-3-((6-methyl-l,2,4-triazin-3-yl)amino) cyclopentyl)amino)pyridin- 3-yl)pyridazin-3(2H)-one (Example 29)
[336] To a solution of Int-3 (1 g, 3.69 mmol) in NMP (10 mL), was added NazCCL (0.78 g, 7.37 mmol) followed by Int-36 (0.96 g, 5.53 mmol). After stirring at room temperature for 16 h, the reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% DCM in MeOH). The obtained crude product was purified by preparative HPLC (10-45% acetonitrile/0.02% NELOH in water; WATERS XSELECT (250 mm x 20 mm), 5.0 μm column, flow rate 18 mL/min) to afford the title compound as a yellow solid (60 mg, 4.4%). ESI-MS m!z; 364.9 [M+H]+. *H NMR (400 MHz, CD3OD) 5 8.18 (s, 1H), 8.14 (d, J = 2.8 Hz, 1H), 8.01 (dd, J= 4.0, 1.6 Hz, 1H), 7.59 (dd, J= 9.2, 2.8 Hz, 1H), 7.45 (dd, J= 9.6,
4.0 Hz, 1H), 7.05 (dd, J= 9.6, 1.6 Hz, 1H), 6.59 (d, J= 9.2 Hz, 1H), 4.49 - 4.32 (m, 2H), 2.43 (s, 3H),
2.32 - 2.22 (m, 2H), 2.10 - 1.99 (m, 2H), 1.71 - 1.55 (m, 2H).
Example 30: Synthesis of 2-(6-(((1S,3S)-3-aminocvcloDentvl) amino)Dvridin-3-yl)-4- methvlpvridazin-3(27O-one

Step-1; Synthesis of 2-(6-fluoropyridin-3-yl)-4-methylpyridazin-3(2H)-one (Int-37)
[337] To a solution of 2 -fluoro-5 -iodopyridine (5 g, 22.42 mmol) in DMSO (100 mL), was added potassium carbonate (6.1 g, 44.84 mmol) followed by 4-methylpyridazin-3(2Z/)-one (2.5 g, 23.54 mmol). The reaction mixture was purged with argon for 15 min at room temperature, then Cui (0.42 g, 2.24 mmol) and trans-N, A-dimethylcyclohexane-l,2-diamine (0.31 g, 2.24 mmol) were added and purged with argon for another 10 min. After stirring at 130 °C for 16 h, the reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-60% EtOAc in hexane) to afford the title compound as an off white solid (1.5 g, 32%). ESI-MS m!z; 205.9 [M+H]+.
Step-2; Synthesis of tert-butyl ((ES',3,S')-3-((5-(5-methyl-6-oxopyridazin- l(6//)-yl) pyridine-2-yl) amino) cyclopentyl) carbamate (lnt-38)
[338] To a solution of tert-butyl ((lS,35)-3-aminocyclopentyl)carbamate (1 g, 4.99 mmol) in DMSO (20 mL), were added DIPEA (2.56 mL, 14.98 mmol) and Int-37(1.1 g, 5.49 mmol). After stirring at 120 °C for 16 h, the reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-70% EtOAc in hexane) to afford the title compound as an off-white solid (1.5 g, 78%), ESI-MS m/z; 386.0 [M+H]+.
StepS: Synthesis of 2-(6-(((15',35')-3-aminocyclopentyl)amino)pyridin-3-yl)-4-methylpyridazin- 3(2//)-one (Int-39)
[339] To a mixture of Int-38 (1.5 g, 3.89 mmol) in MeOH (10 ml) was added 4 M HC1 in 1,4-dioxane (6 mL). After stirring for 2 h at room temperature, the mixture was concentrated under reduced pressure. The crude product was treated with Amberlyst® A21 ion exchange resin (3 g) in MeOH (20 mL) and stirred for 15 min. Then the mixture was filtered to remove the resin and the resin was washed with methanol. The filtrate was concentrated under reduced pressure to afford the title compound as a brown solid (1.3 g, crude), which was used in the next step without further purification. ESI-MS m/z: 285.95 [M+H]+.
Step-4: Synthesis of 4-methyl-2-(6-(((1S,3S)-3-((6-methyl-l,2,4-triazin-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 30)
[340] To a solution of Int-39 (0.3 g, 1.05 mmol) in NMP (5 mL), was added NazCOs (0.22 g, 2.1 mmol) followed by Int-36 (0. 18 g, 1.05 mmol). After stirring at rt for 16 h, the reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (20-40% acetonitrile/0.02% NH4OH in water; WATERS XSELECT (250 mm x 20 mm), 5.0 μm column, flow rate 18 mL/min) to provide the title compound as a yellow solid (23 mg, 5.7%), ESI-MS m/z: 379.05 [M+H] +. JH NMR (400 MHz, CD3OD) 5 8.19 (s, 1H), 8.13 (dd, J= 2.8, 0.4 Hz, 1H), 7.89 (d, .7= 4.0, Hz, 1H), 7.58 (dd, J= 9.2, 2.8 Hz, 1H), 7.33 - 7.32 (m, 1H), 6.60 (dd, J= 8.8, 0.4 Hz, 1H), 4.49 - 4.33 (m, 2H), 2.44 (s, 3H), 2.32 - 2.24 (m, 2H), 2.22 (s, 3H), 2.12 - 1.98 (m, 2H), 1.69 - 1.60 (m, 2H).
Example 31: Synthesis of 2-(6-(((1S,3S)-3-((6-cvclopropvl-l,2,4-triazin-3- vl)amino)cvdopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one

Step-1; Synthesis of 3-(methylsulfinyl)-l,2,4-triazine (Int-40)
[341] To a solution of 3-(methylthio)-l,2,4-triazine (3 g, 23.59 mmol) in DCM (300 mL) was added mCPBA (12.21 g, 70.77 mmol) portion wise at -50°C. After stirring at -50°C for 4 h, the reaction mixture was filtered through a C6lite® pad, the filtrate was concentrated under reduced pressure. The residue was
purified by flash column chromatography on silica gel (eluent: 20-80% EtOAc in Hexane) to provide title compound as a yellow sticky solid (2.5 g, 74%). ESI-MS m/z: 143.9 [M+H] +.
Step-2; Synthesis of tert-butyl ((l1S',31S')-3-((l,2,4-triazin-3-yl)amino)cyclopentyl) carbamate (Int-41)
[342] To a solution of tert-butyl ((lS,35)-3-aminocyclopentyl)carbamate (2 g, 9.98 mmol) in NMP (20 mL) were added NazCOs (3.17 g, 29.96 mmol) and Int-40 (2.14 g, 14.98 mmol). After stirring for 16 h, the reaction mixture was quenched with cold water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM) to afford the title compound as an off-white solid (2.3 g, 82%), ESI-MS m!z; 280.0 [M+H]+.
Step-3; Synthesis of tert-butyl ((f1S',31S')-3-((6-bromo-l,2,4-triazin-3-yl)amino)cyclopentyl)carbamate (Int-42)
[343] To a stirred solution of Int-41 (3.5 g, 12.52 mmol) in acetonitrile (80 mL) and water (120 mL) was added NBS (2.3 g, 13.14 mmol) portion wise. After stirring for 8 h, the reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM) to afford the title compound as a yellow solid (2.9 g, 65%), ESI-MS m!z; 359.75 [M+H]+.
Step-4; Synthesis of tert-butyl ((LS',3.S')-3-((6-cyclopropyl-l,2,4-triazin-3-yl)amino) cyclopentyl)carbamate (Int-43)
[344] To a solution of Int-42 (1 g, 2.79 mmol) in a mixture of 1,4-dioxane and water (30 mL, 3: 1), were added cyclopropyl boronic acid (1.2 g, 13.95 mmol) and CS2CO3 (2.72 g, 8.37 mmol) and the reaction mixture was purged with argon for 15 min, then PdlOAcf (63 mg, 0.27 mmol) and tricyclohexyl phosphine (0.16 g 0.55 mmol) were added and purged with argon for another 10 min. After stirring at 100 °C for 16 h, the reaction mixture was filtered through a C6lite® pad, the filtrate was quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM) to afford the title compound as a brown solid (0.65 g, 72.8%). ESI-MS m!z; 319.95 [M+H]+.
Step-5; Synthesis of (15',35')-A7-(6-cyclopropyl-l,2,4-triazin-3-yl)cyclopentane-l,3-diamine (Int-44)
[345] A mixture of Int-43 (0.65 g, 2.03 mmol) in 4 M HC1 in 1,4-dioxane (7 mL) was stirred for 2 h at room temperature. The mixture was concentrated under reduced pressure. The crude was treated with Amberlyst® A21 ion exchange resin (1.3 g) in MeOH (20 mL) and stirred for 15 min. The mixture was
filtered to remove the resin and the resin was washed with methanol. The filtrate was concentrated under reduced pressure to afford the title compound as a brown solid (0.6 g, crude), which was used in the next step without further purification. ESI-MS m/z: 220.0 [M+H]+.
Step-6: Synthesis of 2-(6-(((1S,3S)-3-((6-cyclopropyl-l,2,4-triazin-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 31)
[346] To a solution of Int-44 (450 mg, 2.05 mmol) in NMP (3 mL), were added DIPEA (0.58 mL, 6.15 mmol) and Int-1 (470 mg, 2.46 mmol). After stirring at 140 °C for 3 h under microwave irradiation, the reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM). The crude product was purified by preparative HPLC (20-60% acetonitrile/0.02% NH4OH in water; YMC (150 mm x 21.2 mm), 5.0 μm column, flow rate 15 mL/min) to provide the title compound as a yellow solid (35 mg, 4.3%), ESI-MS m/z: 391.2 [M+H] +. 'H NMR (400 MHz, CD3OD) 5 8.16 (s, 1H), 8.15 (dd, J= 2.8, 0.8 Hz, 1H), 8.03 (dd, J= 3.6, 1.6 Hz, 1H), 7.60 (dd, J= 9.2, 2.8 Hz, 1H), 7.46 (dd, J= 9.2, 3.6 Hz, 1H), 7.06 (dd, J= 9.2, 1.6 Hz, 1H), 6.60 (dd, J= 8.8, 0.4 Hz, 1H), 4.45 - 4.32 (m, 2H), 2.31 - 2.25 (m, 2H), 2.09 - 2.00 (m, 3H), 1.68 - 1.62 (m, 2H), 1.05 - 0.95 (m, 4H).
Example 32: Synthesis of 2-(6-(((1S,3S)-3-((6-cvclopropvl-l,2,4-triazin-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)-4-methvlpyridazin-3(2ZD-one

[347] To a solution of Int-44 (200 mg, 0.91 mmol) in NMP (2 mL), DIPEA (0.4 mL, 2.73 mmol) was added followed by Int-37 (220 mg, 1.09 mmol). After stirring at 140 °C for 3 h under microwave irradiation, the reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% MeOH in DCM) to provide the title compound as a brown solid (30 mg, 8.1%), ESI-MS m/z: 405.05 [M+H] +. ‘H NMR (400 MHz, CD3OD) 5 8.16 (s, 1H), 8.13 (d, J= 2.4 Hz, 1H), 7.89 (d, J= 4.0 Hz, 1H), 7.58 (dd, J= 9.2, 2.8 Hz, 1H), 7.33 - 7.32 (m, 1H), 6.59 (d, J= 9.2 Hz, 1H), 4.47 - 4.32 (m, 2H), 2.32 - 2.26 (m, 2H), 2.22 (s, 3H), 2.10 - 2.01 (m, 3H), 1.68 - 1.59 (m, 2H), 1.06 - 0.95 (m, 4H).
Example 33: Synthesis of 2-(6-(((l1S,,31S)-3-((6-ethvl-l,2,4-triazin-3-yl)amino)cvclopentvl) amino)pvridin-3-vl)-4-methvlpyridazin-3(277)-one

Step-1; Synthesis of methyl 2,2-dimethoxyacetate (lnt-45)
[348] To a solution of 2-oxoacetic acid (20 g, 270.1 mmol) in trimethoxy methane (150 mL) was added cone. H2SO4 (2 mL). After stirring at 100 °C for 4 h, the reaction mixture was quenched with cold water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide the crude title compound as a pale yellow liquid (15 g, 41%). 'H NMR (300 MHz, CDCI3) 5 4.82 (s, 1H), 3.80 (s, 3H), 3.42 (s, 6H).
Step-2; Synthesis of /V,2,2-trimethoxy-/V-methylacetamide (Int-46)
[349] To a solution of N, O-dimethyl hydroxylamine hydrochloride (15.2 g, 156.7 mmol) in THF (200 mL) was added 0.5 M isopropyl magnesium chloride in THF (156 mL, 313.4 mmol) at -78 °C. After stirring at -78 °C for 30 min, lnt-45 (15 g, 111.9 mmol) was added dropwise at -78 °C to the reaction mixture. After stirring at -78 °C for 1.5 h, the reaction mixture was quenched with saturated ammonium chloride solution and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-25% EtOAc in hexane) to provide the title compound as a pale yellow liquid (6 g, 33%). 'H NMR (400 MHz, DMSO-cfc) 5 5.07 (s, 1H), 3.66 (s, 3 H), 3.32 (s, 6H), 3.11 (s, 3H).
Step-3; Synthesis of l,l-dimethoxybutan-2-one (Int-47)
[350] To a solution of Int-46 (6 g, 3.68 mmol) in THF (250 mL) was added 3 M ethyl magnesium bromide in ether (2.4 mL, 7.36 mmol) at -78 °C. After stirring at -78 °C for 1 h, the reaction mixture was quenched with saturated ammonium chloride solution and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-30% EtOAc in hexane) to provide the title compound as a pale yellow liquid (2.6 g, 53%). 1 H NMR (400 MHz, CDCh) 5 4.48 (s, 1H), 3.40 (s, 6H), 2.57 (q, J= 7.2 Hz, 2H), 1.05 (t, J= 7.2 Hz, 3H).
Step-4; Synthesis of 6-ethyl-3-(methylthio)-l,2,4-triazine (lnt-48)
[351] A mixture of methyl hydrazinecarbimidothioate hydroiodide (5 g, 21.4 mmol) and Int-47 (2.5 g, 21.4 mmol) in EtOH (100 mL) was heated at 70 °C for 4 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-25% EtOAc in hexane) to provide the title compound as a pale brown solid (1 g, 30%), ESI-MS m/z: 155.95 [M+H] +.
Step-5; Synthesis of 6-ethyl-3-(methylsulfonyl)-l, 2, 4-triazine and 6-ethyl-3-(methylsulfinyl)-l,2,4- triazine (Int-49 / Int-49a)
[352] To a solution of Int-48 (1 g, 7.09 mmol) in DCM (15 mL) was added mCPBA (6 g, 35.4 mmol) portion wise at 0°C. After stirring at rt for 2 h, the reaction mixture was filtered through a C6lite® pad, the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-20% EtOAc in hexane and 0-5% MeOH in DCM) to provide both 6-ethyl-3 -(methylsulfonyl)- 1,2, 4-triazine as pale-brown solid (0.48 g, 40%), ESI-MS m/z: 187.85 [M+H], and 6-ethyl -3 -(methylsulfinyl)- 1,2, 4-triazine as pale-brown solid (0.2 g, 18%), ESI-MS m/z: 171.90 [M+H] +.
Step-6; Synthesis of 2-(6-(((lS,3S)-3-((6-ethyl-l,2,4-triazin-3-yl)amino)cyclopentyl) amino)pyridin-3- yl)-4-methylpyridazin-3(2H)-one (Example 33)
[353] Batch-1: To a solution of Int-39 (330 mg, 1.16 mmol) in NMP (5 mL), was added NazCOs (243 mg, 2.32 mmol) followed by Int-49a (200 mg, 1.16 mmol). The reaction mixture was stirred at rt for 16 h. Batch-2; To the stirred solution of Int-39 (732 mg, 2.56 mmol) in NMP (5 mL), was added NazCOs (537 mg, 5.12 mmol) followed by Int-49 (480 mg, 2.56 mmol). The reaction mixture was stirred at rt for 16 h. The combined reaction mixtures were quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% DCM in MeOH). The obtained crude product was purified by preparative HPLC (10- 50% acetonitrile/0.02% NH4OH in water; WATERS XB RIDGE (150 mm x 19 mm), 5.0 μm column, flow rate 10 mL/min) to afford the title compound as a yellow solid (90 mg, 6.5%). ESI-MS m!z; 393.5 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.22 (s, 1H), 8.13 (d, J= 2.0 Hz, 1H), 7.88 (d, J= 4.4 Hz, 1H), 7.59 (dd, J= 9.2, 2.8 Hz, 1H), 7.33 - 7.32 (m, 1H), 6.60 (d, J= 8.8 Hz, 1H), 4.49 - 4.34 (m, 2H), 2.77 (q, J= 7.6 Hz, 2H), 2.33 - 2.28 (m, 2H), 2.22 (s, 3H), 2.10 - 2.01 (m, 2H), 1.69 - 1.60 (m, 2H), 1.28 (t, J = 7.6 Hz, 3H).
Example 34: Synthesis of 2-(6-(((lS,3S)-3-((6-ethyl-l,2,4-triazin-3-vl)amino)cvclopentvl) amino)pvridin-3-vl)pyridazin-3(277)-one

[354] To a solution of Int-3 (0.4 g, 1.474 mmol) in NMP (5 mb), was added Na3CO3 (0.31 g, 2.95 mmol) followed by Int-49 (0.27 g, 1.47 mmol). After stirring at rt for 16 h, the reaction mixture was quenched with water and the product was extracted with EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% DCM in MeOH). The obtained crude product was purified by preparative HPLC (50% EtOH: MeOH (1:1) / hexane, Chiralpak IH (250 mm x 21.2 mm), 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a brown solid (6 mg, 1.0%). ESI-MS m/z: 378.9 [M+H]+. XH NMR (400 MHz, CD3OD) 5 8.21 (s, IH), 8.15 (dd, J= 2.8, 0.4 Hz, IH), 8.03 (dd, J= 4.0, 2.0 Hz, IH), 7.59 (dd, J= 8.8, 2.8 Hz, IH), 7.46 (dd, J= 9.2, 3.6 Hz, IH), 7.06 (dd, .7= 9.6, 1.6 Hz, IH), 6.60 (dd, J= 9.2, 0.8 Hz, IH), 4.49 - 4.43 (m,lH), 4.39 - 4.35 (m, IH), 2.77 (q, J= 7.6 Hz, 2H), 2.32 - 2.25 (m, 2H), 2.09 - 2.01 (m, 2H), 1.68 - 1.61 (m, 2H), 1.28 (t, J= 7.6 Hz, 3H).
Example 35: Synthesis Of 2-(6-(((15',35')-3-((5-(l-hvdroxvcvcloDentvl)-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-yl)Dvridazin-3(2H)-one

Step-1; Synthesis of 2,5-dioxopyrrolidin-l-yl 1-hydroxycyclopentane-l-carboxylate (Int-35) lnt-60 was synthesized following the Int-12 starting from commercially available 1 -hydroxycyclopentane- 1 -carboxylic acid. 'H NMR (300 MHz, CDC13) 5 2.86 (s, 4H), 2.61 (s, IH), 2.38 - 2.33 (m, 2H), 2.04 - 1.84 (m, 6H).
Step-2; Synthesis of 2-(6-(((1S,3S)-3-((5-(l-hydroxycyclopentyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 35)
Example 35 was synthesized following the Example 15 starting from Int-5. ESI-MS m/z: 424.1 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.14 (d, J= 2.8 Hz, IH), 8.02 (dd, J= 4.0, 1.6 Hz, IH), 7.59 (dd, J = 9.2, 2.8 Hz, IH), 7.45 (dd, J= 9.6, 4.0 Hz, IH), 7.05 (dd, J= 9.6, 2.0 Hz, IH), 6.58 (d, J= 9.2 Hz, IH), 4.38 - 4.30 (m, IH), 4.01 - 3.93 (m, IH), 2.30 - 2.21 (m, 2H), 2.16 - 1.86 (m, 8H), 1.83 - 1.74 (m, 2H), 1.66 - 1.52 (m, 2H).
Example 36: Synthesis of l,5,5-trimethyl-3-(6-(((l1S',31S)-3-((5-methyl-l,2,4-oxadiazol-3- vl)amino)cvdopentvl)amino)pyridin-3-vl)imidazolidine-2, 4-dione

Step-1; Synthesis of tert-butyl ((15',35')-3-((5-(3,4,4-trimethyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-61)
Int-61 was synthesized following the Int-14 starting from commercially available 1,5,5- trimethylimidazolidine-2, 4-dione, ESI-MS m/z: 418.25 [M+H]+.
Step-2; Synthesis ooff 3-(6-(((LS',3.S')-3-aminocyclopentyl) amino) pyridin-3-yl)-l ,5,5- trimethylimidazolidine-2, 4-dione hydrochloride (Int-62)
Int-62 was synthesized following the Int-3 starting from Int-61, ESI-MS m/z: 318.15 [M+H]+.
Step-3; Synthesis ooff N-((1S,3S)-3-((5-(3,4,4-trimethyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)cyanamide (Int-63)
Int-63 was synthesized following the Int-4 starting from Int-62, ESI-MS m/z: 343.15 [M+H]+.
Step-4; Synthesis of 2-hydroxy-l-((15,3^)-3-((5-(3,4,4-trimethyl-2,5-dioxoimidazolidin-l-yl)pyridin- 2-yl)amino)cyclopentyl)guanidine (Int-64)
Int-64 was synthesized following the Int-5 starting from Int-63, ESI-MS m/z: 375.95 [M+H]+.
Step-5; Synthesis of l,5,5-trimethyl-3-(6-(((15,35)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)imidazolidine-2, 4-dione (Example 36)
Example 36 was synthesized following the Example 1 starting from Int-64. ESI-MS m/z: 400.1 [M+H]+. ‘HNMR (400 MHz, CD3OD) 5 7.10 (d, J= 2.0 Hz, 1H), 6.56 (dd, J= 9.2, 2.8 Hz, 1H), 5.76 (d, J= 9.2 Hz,
1H), 3.51 - 3.48 (m, 1H), 3.17 - 3.14 (m, 1H), 2.15 (s, 3H), 1.60 (s, 3H), 1.44 - 1.38 (m, 2H), 1.26 - 1.21 (m, 1H), 1.14 - 1.02 (m, 1H), 0.85 - 0.73 (m, 2H), 0.67 (s, 6H).
Example 37: Synthesis of 4-methyl-2-(6-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-vl)Dvridazin-3(27Q-one

Step-1; Synthesis of 2-(6-fluoropyridin-3-yl)-4-methylpyridazin-3(2H)-one (Int-65)
Int-65 was synthesized following the Int-1 starting from commercially available 4-methylpyridazin- 3(2H)-one, ESI-MS m/z: 205.70 [M+H]+.
Step-2; Synthesis ooff tert-butyl ((1S,3S)-3-((5-(5-methyl-6-oxopyridazin-l(67/)-yl)pyridin-2- yl)amino)cyclopentyl)carbamate (Int-66)
Int-66 was synthesized following the Int-2 starting from Int-65, ESI-MS m/z: 385.75 [M+H]+.
Step-3; Synthesis of 2-(6-(((1S,3S)-3-aminocyclopentyl) amino) pyridin-3-yl)-4-methylpyridazin- 3(2H)-one hydrochloride (lnt-67) lnt-67 was synthesized following the Int-3 starting from Int-66, ESI-MS m/z: 286.10 [M+H]+.
Step-4; Synthesis ooff AL((1S,3S)-3-((5-(5-methyl-6-oxopyridazin-l(67/)-yl) pyridin-2- yl)amino)cyclopentyl)cyanamide (Int-68)
Int-68 was synthesized following the Int-4 starting from lnt-67, ESI-MS m/z: 310.95 [M+H]+.
Step-5; Synthesis of 2-hydroxy-l-((1S,3S)-3-((5-(5-methyl-6-oxopyridazin-l(67/)-yl)pyridin-2- yl)amino)cyclopentyl)guanidine (Int-69)
Int-69 was synthesized following the Int-5 starting from Int-68, ESI-MS m/z: 344.20 [M+H]+.
Step-6; Synthesis ooff 4-methyl-2-(6-(((15,35)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 37)
Example 37 was synthesized following the Example 1 starting from Int-69. ESI-MS m/z: 368.15 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.13 (d, J = 2.4 Hz, 1H), 7.89 (d, J = 4.0 Hz, 1H), 7.59 (dd, J = 9.2, 2.8 Hz, 1H), 7.33 - 7.32 (m, 1H), 6.59 (d, J= 9.2 Hz, 1H), 4.35 - 4.32 (m, 1H), 3.99 - 3.96 (m, 1H), 2.41 (s, 3H), 2.27 - 2.19 (m, 2H), 2.21 (s, 3H), 2.10 - 2.03 (m, 1H), 1.97 - 1.90 (m, 1H), 1.67 - 1.56 (m, 2H).
Example 38: Synthesis of 2-(6-(((15',35')-3-((5-(3-methvloxetan-3-vl)-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-yl)Dvridazin-3(27D-one

Example 38 was synthesized following the Example 2 starting from Int-5 with commercially available 3- methyloxetane-3 -carboxylic acid. ESI-MS m/z: 410.10 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.15 (d, J = 2.4 Hz, 1H), 8.03 (dd, J = 3.6, 1.6 Hz, 1H), 7.60 (dd, J = 9.2, 2.8 Hz, 1H), 7.46 (dd, J = 9.6, 4.0 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.59 (d, J= 9.2 Hz, 1H), 5.00 (d, J= 8.0 Hz, 2H), 4.57 (d, J = 6.0 Hz, 2H), 4.37 - 4.33 (m, 1H), 4.03 - 3.99 (m, 1H), 2.28 - 2.22 (m, 2H), 2.12 - 2.03 (m, 1H), 1.99 - 1.93 (m, 1H), 1.69 (s, 3H), 1.67 - 1.58 (m, 2H).
Example 39: Synthesis Of 2-(6-(((15',35')-3-((5-(cvcloDroDvlmethvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(27D-one

Example 39 was synthesized following the Example 2 starting from Int-5 with commercially available 2- cyclopropylacetic acid. ESI-MS m/z: 394.15 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.15 (d, J= 2.4 Hz, 1H), 8.03 (dd, J= 4.0, 1.6 Hz, 1H), 7.60 (dd, J= 9.2, 2.8 Hz, 1H), 7.46 (dd, J= 9.2, 4.0 Hz, 1H), 7.06 (dd,
J= 9.6, 1.6 Hz, 1H), 6.59 (d, J= 9.2 Hz, 1H), 4.36 - 4.33 (m, 1H), 4.01 - 3.96 (m, 1H), 5 2.66 (d, J= 7.2 Hz, 2H), 2.28 - 2.22 (m, 2H), 2.12 - 2.03 (m, 1H), 1.98 - 1.95 (m, 1H), 1.67 - 1.52 (m, 2H), 1.17 - 1.08 (m, 1H), 0.61 - 0.57 (m, 2H), 0.30 - 0.26 (m, 2H).
Example 40: Synthesis of 2-(6-(((1S,3S)-3-((5-(2-hydroxypropan-2-yl)-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-vl)Dvridazin-3(2H)-one

Step-1; Synthesis of Synthesis of 2-acetoxy-2-methylpropanoic acid (Int-70)
The mixture of 2 -hydroxy-2 -methylpropanoic acid (1.0 g, 9.60 mmol) in acetyl chloride (1.5 g, 19.21 mmol) was stirred at rt for 16 h. The reaction mixture was concentrated under reduced pressure to dryness. The residue was purified by flash column chromatography on silica gel (eluent: 0-10% EtOAc in hexane) to afford the title compound as a colorless liquid (1.3 g, 93%). 'H NMR: (DMSO- de, 400 MHz): 5 12.54 (br s, 1H), 1.99 (s, 3H), 1.45 (s, 6H).
Step-2; Synthesis of 2,5-dioxopyrrolidin-l-yl 2-acetoxy-2-methylpropanoate (Int-71)
Int-71 was synthesized following the Int-12 starting from Int-71. ' H NMR: (CD3OD, 400 MHz): 5 2.82 (s, 4H), 2.08 (s, 3H), 1.69 (s, 6H).
Step-3; Synthesis of 2-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(67/)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)propan-2-yl acetate (Int-72)
Int-72 was synthesized following the Example 15 starting from Int-5 and Int-71. ESI-MS m/z: 439.95 [M+H]+.
Step-4; Synthesis ooff 2-(6-(((15,35)-3-((5-(2-hydroxypropan-2-yl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 40)
To a stirred solution of Int-72 (180 mg, 0.40 mmol) in MeOH (3.0 mL) was added potassium carbonate (113 mg, 0.81 mmol) at 0°C. After stirring at rt for 3 h, the reaction was quenched with water and the product was extracted with EtOAc. The organic phases were combined, dried over anhydrous sodium
sulfate, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (10-40% acetonitrile/0.02% NH4OH in water; WATER X BRIDGE, 150 mm x 20 mm, 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (30 mg, 18%), ESI-MS m/z: 398.10 [M+H]+. ‘H NMR (400 MHz, CD3OD) 5 8.15 (d, J= 2.4 Hz, 1H), 8.03 (dd, J= 3.6, 1.6 Hz, 1H), 7.60 (dd, .7= 8.8, 2.4 Hz, 1H), 7.46 (dd, J= 9.2, 3.6 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.59 (d, J = 9.2 Hz, 1H), 4.36 - 4.33 (m, 1H), 4.00 - 3.98 (m, 1H), 2.29 - 2.19 (m, 2H), 2.12 - 2.03 (m, 1H), 1.98 - 1.95 (m, 1H), 1.68 - 1.52 (m, 2H), 1.56 (s, 6H).
Example 41: Synthesis of 2-(6-(((15',35')-3-((5-(l-hvdroxvcvclobutvl)-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-vl)Dvridazin-3(2Zfi-one

Step-1; Synthesis of 2-acetoxy-2-methylpropanoic acid (Int-73)
Int-73 was synthesized following the Int-70 starting from commercially available 1-hydroxycyclobutane- 1 -carboxylic acid. 'H NMR: (CDC13, 300MHz): 5 2.82 - 2.68 (m, 2H), 2.44 - 2.30 (m, 2H), 2.11 (s, 3H), 2.08 - 1.89 (m, 2H).
Step-2; Synthesis of 2,5-dioxopyrrolidin-l-yl 1-acetoxycyclobutane-l-carboxylate (Int-74)
Int-74 was synthesized following the Int-12 starting from Int-73. 1 H NMR: (CDCh, 300MHz): 5 2.95 - 2.92 (m, 2H), 2.82 (s, 4H), 2.53 - 2.42 (m, 2H), 2.14 (s, 3H), 2.12 - 1.92 (m, 2H).
Step-3; Synthesis of l-(3-(((l1S',31S')-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)cyclobutyl acetate (Int-75)
Int-75 was synthesized following the Example 15 starting from Int-5 and Int-74. ESI-MS m/z: 452.10 [M+H]+.
Step-41. Synthesis oOff 2-(6-(((1S,3S)-3-((5-(l-hydroxycyclobutyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 41)
Example 41 was synthesized following the Example 41 starting from Int-75. ESI-MS m/z: 410.15 [M+H]+. 'H NMR (400 MHz, DMSO-d6) 5 7.34 (d, J= 2.4 Hz, 1H), 7.22 (dd, J= 3.6, 1.2 Hz, 1H), 6.79
(dd, J = 9.2, 2.8 Hz, 1H), 6.65 (dd, J = 9.6, 4.0 Hz, 1H), 6.25 (dd, J= 9.6, 1.6 Hz, 1H), 5.78 (d, J= 8.8 Hz, 1H), 3.55 - 3.52 (m, 1H), 3.22 - 3.18 (m, 1H), 1.84 - 1.78 (m, 2H), 1.59 - 1.41 (m, 4H), 1.30 - 1.21 (m, 1H), 1.20 - 1.06 (m, 3H), 0.86 - 0.77 (m, 2H).
Example 42: Synthesis of 2-(6-(((1S,3S)-3-((5-cyclohexyl-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(277)-one
 p
p
Example 42 was synthesized following the Example 2 starting from Int-5 with commercially available cyclohexanecarboxylic acid. ESI-MS m/z: 422.40 [M+H]+. 1H NMR (400 MHz, CD3OD) 5 8.15 (d, J = 2.4 Hz, 1H), 8.03 (dd, .7= 4.0, 1.6 Hz, 1H), 7.60 (dd, J= 9.2, 2.8 Hz, 1H), 7.46 (dd, J = 9.2, 3.6 Hz, 1H), 7.06 (dd, J = 9.6, 1.6 Hz, 1H), 6.59 (d, J = 8.8 Hz, 1H), 4.39 - 4.31 (m, 1H), 4.01 - 3.97 (m, 1H), 2.86 - 2.69 (m, 1H), 2.30 - 2.19 (m, 2H), 2.11 - 1.90 (m, 4H), 1.85 - 1.78 (m, 2H), 1.73 - 1.52 (m, 5H), 1.49 - 1.28 (m, 3H),
Example 43: 2-(6-(((1S,3S)-3-((5-phenvl-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pyridin-3- vl)pyridazin-3(277)-one

Example 43 was synthesized following the Example 2 starting from Int-5 with commercially available benzoic acid. ESI-MS m/z: 416.0 [M+H]+. xH NMR (400 MHz, CD3OD) 5 8.15 (d, J= 2.4 Hz, 1H), 8.08 - 8.02 (m, 3H), 7.65 - 7.59 (m, 2H), 7.57 - 7.53 (m, 2H), 7.46 (dd, J = 9.6, 4.0 Hz, 1H), 7.06 (dd, J = 9.6, 1.6 Hz, 1H), 6.61 (d, J = 9.2 Hz, 1H), 4.41 - 4.37 (m, 1H), 4.11 - 4.07 (m, 1H), 2.32 - 2.25 (m, 2H), 2.15 - 2.10 (m, 1H), 2.03 - 1.98 (m, 1H), 1.72 - 1.61 (m, 2H).
Example 44: 2-(6-(((1S,3S)-3-((5-(pvridin-3-vl)-l,2,4-oxadiazol-3- vl)amino)cvclonentvl)amino)pvridin-3-vl)pyridazin-3(277)-one

Example 44 was synthesized following the Example 2 starting from Int-5 with commercially available nicotinic acid. ESI-MS m/z: 416.95 [M+H]+. 1H NMR (400 MHz, CD3OD) 5 9.19 (d, J= 1.6 Hz, 1H), 8.77 - 8.75 (m, 1H), 8.46 - 8.43 (m, 1H), 5 8.20 (d, J= 2.0 Hz, 1H), 5 8.03 (dd, J= 4.0, 1.6 Hz, 1H), 7.74 - 7.72 (m, 1H), 7.64 - 7.61 (m, 1H), 7.46 (dd, J= 9.2, 3.6 Hz, 1H), 7.O6 (dd, J= 9.6, 1.6 Hz, 1H), 6.71 (d, J= 8.8 Hz, 1H), 4.41 - 4.33 (m, 1H), 4.14 - 4.08 (m, 1H), 2.35 - 2.26 (m, 2H), 2.18 - 2.11 (m, 1H), 2.06 - 2.01 (m, 1H), 1.75 - 1.62 (m, 2H).
Example 45: Synthesis of 2-(6-(((15',35')-3-((5-(Dvridin-2-vl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(277)-one

Example 45 was synthesized following the Example 2 starting from Int-5 with commercially available picolinic acid. ESI-MS m/z: 417.10 [M+H]+. *H NMR (400 MHz, CD3OD) 5 8.73 - 8.72 (m, 1H), 8.21 - 8.16 (m, 2H), 8.07 - 8.02 (m, 2H), 7.66 - 7.60 (m, 2H), 7.45 (dd, J = 9.6, 4.0 Hz, 1H), 7.05 (dd, J = 9.6, 4.0 Hz, 1H), 6.61 (d, J = 9.2 Hz, 1H), 4.41 - 4.35 (m, 1H), 4.15 - 4.08 (m, 1H), 2.32 - 2.25 (m, 2H), 2.18 - 2.11 (m, 1H), 2.05 - 1.98 (m, 1H), 1.75 - 1.60 (m, 2H).
Example 46: Synthesis of 2-(6-(((1S,3S)-3-((5-((methvlsulfonvl)methvl)-l,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-vl)Dvridazin-3(2H)-one

Example 46 was synthesized following the Example 2 starting from Int-5 with commercially available 2- (methylsulfonyl)acetic acid. ESI-MS m/z: 432.05 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.15 (d, J = 2.8 Hz, 1H), 8.03 (dd, J = 4.0, 1.6 Hz, 1H), 5 7.60 (dd, J= 9.2, 2.8 Hz, 1H), 7.46 (dd, J = 9.2, 3.6 Hz, 1H), 7.06
(dd, J= 9.2, 1.6 Hz, 1H), 6.59 (d, J= 8.8 Hz, 1H), 4.89 (s, 2 H), 4.37 - 4.32 (m, 1H), 4.04 - 4.00 (m, 1H), 3.19 (s, 3 H), 2.28 - 2.17 (m, 2H), 2.11 - 2.02 (m, 1H), 1.99 - 1.94 (m, 1H), 1.70 - 1.53 (m, 2H).
Example 47: 2-(6-(((1S,3S)-3-((5-(l-hvdroxv-2-methvlpropan-2-vl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(27O-one

Step-1 : Synthesis of 3-acetoxy-2,2-dimethylpropanoic acid (Int-76)
Int-76 was synthesized following the Int-70 starting from commercially available 3-hydroxy-2,2- dimethylpropanoic acid. 'H NMR (300 MHz, CDC13) 5 4.18 (s, 2H), 2.08 (s, 3 H), 1.35 (s, 6 H).
Step-2; Synthesis of 2,5-dioxopyrrolidin-l-yl 3-acetoxy-2,2-dimethylpropanoate (Int-77)
Int-77 was synthesized following the Int-12 starting from Int-76. ’H NMR: (400MHz, DMSO-de) 5 4.15 (s, 2H), 3.32 (s, 4 H), 2.03 (s, 3 H), 1.33 (s, 6H)
Step-3; Synthesis of 2-methyl-2-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)propyl acetate (Int-78)
Int-78 was synthesized following the Example 15 starting from Int-5 and Int-77. ESI-MS m/z: 454.10 [M+H]+.
Step-4; Synthesis of Synthesis of 2-(6-(((15,35)-3-((5-(l-hydroxy-2-methylpropan-2-yl)-l,2,4- oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one (Example 47)
Example 47 was synthesized following the Example 40 starting from Int-78. ESI-MS m/z: 412.15 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.15 (d, J = 2.8 Hz, 1H), 8.03 (dd, J = 3.6, 1.6 Hz, 1H), 5 7.60 (dd, J= 8.8, 2.4 Hz, 1H), 7.46 (dd, J= 9.6, 4.0 Hz, 1H), 7.06 (dd, J= 9.2, 1.6 Hz, 1H), 6.59 (d, J= 9.2 Hz, 1H), 4.38 - 4.31 (m, 1H), 4.02 - 3.97 (m, 1H), 3.66 (s, 2 H), 2.30 - 2.19 (m, 2H), 2.11 - 2.04 (m, 1H), 1.98 - 1.92 (m, 1H), 1.68 - 1.53 (m, 2H), 1.33 (s, 6 H).
Example 48: Synthesis ooff 2-(6-(((1S,3S)-3-((5-(2,2,2-trifluoroethvl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(27O-one

Example 48 was synthesized following the Example 2 starting from Int-5 with commercially available 3,3,3-trifluoropropanoic acid. ESI-MS m/z: 422.15 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.18 (d, J = 2.8 Hz, 1H), 8.03 (dd, J= 4.0, 1.6 Hz, 1H), 5 7.68 (dd, J= 9.2, 2.4 Hz, 1H), 7.47 (dd, J= 9.6, 4.0 Hz, 1H), 7.07 (dd, J = 9.2, 1.6 Hz, 1H), 6.65 (d, J= 9.2 Hz, 1H), 4.38 - 4.31 (m, 1H), 4.03 - 3.98 (m, 1H), 3.92-3.86 (m, 2 H), 2.34 - 2.21 (m, 2H), 2.18 - 2.07 (m, 1H), 2.01 - 1.97 (m, 1H), 1.70 - 1.58 (m, 2H).
Example _ 49: _ methyl _ l-(3-(((l1$',31S')-3-((5-(6-oxopvridazin-l(6H)-vl)pvridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)cvclopropane-l-carboxylate

Example 49 was synthesized following the Example 2 starting from Int-5 with commercially available 1- (methoxycarbonyl)cyclopropane-l -carboxylic acid. ESI-MS m/z: 438.15 [M+H]+. XH NMR (400 MHz, CD3OD) 5 8.15 (d, .7 = 2.8 Hz, 1H), 8.03 (dd, J = 4.0, 1.6 Hz, 1H), 5 7.60 (dd, J = 8.8, 2.4 Hz, 1H), 7.46 (dd, J= 9.2, 3.6 Hz, 1H), 7.06 (dd, J= 9.2, 1.6 Hz, 1H), 6.59 (d, J= 9.2 Hz, 1H), 4.39 - 4.31 (m, 1H), 4.03 - 3.98 (m, 1H), 3.74 (s, 3 H), 2.29 - 2.20 (m, 2H), 2.11 - 2.03 (m, 1H), 1.98 - 1.89 (m, 1H), 1.72 - 1.55 (m, 6H).
Example 50: Synthesis of l-methyl-6'-(((15,35)-3-((5-methyl-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)-[3,3'-bipyridin]-2(lZ7)-one
 Step-1; Synthesis of Synthesis of tert-butyl ((15,35)-3-((5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl) pyridin-2-yl) amino) cyclopentyl) carbamate (Int-79)
Step-1; Synthesis of Synthesis of tert-butyl ((15,35)-3-((5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl) pyridin-2-yl) amino) cyclopentyl) carbamate (Int-79)
To a solution of Int-13 (0.4 g, 0.992 mmol) in DMSO (10 mL) were added bispinacolatodiborane (0.5 g, 1.984 mmol), KOAc (0.49 g, 4.959 mmol) and Pd(dppf)C12.DCM (81 mg, 0.099 mmol). After stirred at 80°C for 16 h, the reaction mixture was quenched with water and extracted with EtOAc. The combined organic phases were washed with water dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide crude product (0.7 g, crude) as a brown liquid. ESI-MS m/z: 322.15 [M+H]+, boronic acid mass.
Step-2-. Synthesis of tert-butyl ((lS,35)-3-((r-methyl-2'-oxo-r,2'-dihydro-[3,3'-bipyridin]-6-yl) amino) cyclopentyl) carbamate (Int-80)
To a solution of 3-bromo-l-methylpyridin-2(lH)-one (0.1 g, 0.532 mmol) in dioxane (4 mL) and water (1 ml) were added Int-79 (0.32 g, 0.798 mmol), NazCOs (0. 11 g, 1.064 mmol) and Pd(dppf)Ch.DCM (43 mg, 0.053 mmol). After stirred at 100°C for 16 h, the reaction mixture was quenched with water and extracted with EtOAc. The combined organic phases were washed with water dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: 0-3% MeOH in DCM) to afford the title compound as a brown sticky solid (0.1 g, 49%). ESI-MS m/z: 385.15 [M+H]+.
Step-3; Synthesis of 6'-(((LS',3>S')-3-aminocyclopcntyl) amino)-l-methyl-[3,3'-bipyridin]-2(17/)-one hydrochloride (Int-81)
Int-81 was synthesized following the Int-3 starting from Int-80, ESI-MS m/z: 285.10 [M+H]+.
Step-4-. Synthesis of N-((15,35)-3-((l'-methyl-2'-oxo-l',2'-dihydro-[3,3'-bipyridin]-6-yl) amino) cyclopentyl) cyanamide (Int-82)
Int-82 was synthesized following the Int-4 starting from Int-81, ESI-MS m/z: 310.05 [M+H]+.
Step-5; Synthesis of 2-hydroxy-l-((15,35)-3-((l'-methyl-2'-oxo-l',2'-dihydro-[3,3'-bipyridin]-6- yl)amino)cyclopentyl)guanidine (Int-83)
Int-83 was synthesized following the Int-5 starting from Int-82, ESI-MS m/z: 343.15 [M+H]+.
Step-6; Synthesis of l-methyl-6'-(((1S,3S)-3-((5-methyl-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)-[3,3'-bipyridin]-2(17/)-one (Example 50)
Example 50 was synthesized following the Example 1 starting from Int-83. ESI-MS m/z: 367.15 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.27 (d, J= 2.4 Hz, 1H), 7.78 (dd, J= 9.2, 2.4 Hz, 1H), 5 7.59 (d, J= 6.8
Hz, 2H), 6.57 (d, J = 9.2 Hz, 1H), 6.43 (t, .7= 6.8 Hz, 1H), 4.35 - 4.27 (m, 1H), 4.03 - 3.96 (m, 1H), 3.61 (s, 3H), 2.41 (s, 3H), 2.29 - 2.18 (m, 2H), 2.11 - 2.03 (m, 1H), 1.98 - 1.89 (m, 1H), 1.69 - 1.52 (m, 2H).
Example 51: Synthesis of 2-methyl-2-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(67f)-yl) pyridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-yl)propanenitrile

Example 51 was synthesized following the Example 2 starting from Int-5 with commercially available 2- cyano-2-methylpropanoic acid. ESI-MS m/z: 407.0 [M+H]+. *H NMR (400 MHz, CD3OD) 5 8.15 (d, J = 2.4 Hz, 1H), 8.03 (dd, J= 4.0, 1.6 Hz, 1H), 5 7.60 (dd, J= 9.2, 2.8, Hz, 1H), 7.46 (dd, J= 9.6, 4.0 1H), 7.06 (dd, .7= 9.2, 1.6 Hz, 1H), 5 6.59 (d, J= 9.2 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.03 - 3.96 (m, 1H), 2.30 - 2.20 (m, 2H), 2.12 - 2.06 (m, 1H), 2.0 - 1.91 (m, 1H), 1.80 (s, 6H), 1.70 - 1.57 (m, 2H).
Example 52: Synthesis of 2-(6-(((15',35')-3-((5-cvclopropvl-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)-4-methvlpyridazin-3(27D-one

Example 52 was synthesized following the Example 2 starting from Int-69 with commercially available cyclopropane carboxylic acid. ESI-MS m/z: 394.15 [M+H]+. ' H NMR (400 MHz, CD3OD) 5 8.13 (d, J = 2.4 Hz, 1H), 7.89 (d, J= 4.0 Hz, 1H), 5 7.58 (dd, J = 9.2, 2.8, Hz, 1H), 7.33 - 7.32 (m, 1H), 6.58 (d, J= 9.2 Hz, 1H), 4.38 - 4.30 (m, 1H), 4.01 - 3.94 (m, 1H), 2.30 - 2.19 (m, 5H), 2.11 - 2.03 (m, 2H), 1.98 - 1.90 (m, 1H), 1.67 - 1.54 (m, 2H), 1.20 - 1.15 (m, 2H), 1.12 - 1.05 (m, 2H).
Example 53: Synthesis of l-(3-(((15',35')-3-((5-(6-oxopyridazin-l(6Zf)-yl)pyridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)cvclopropane-l-carboxylic acid

To a solution of Example 49 (200 mg, 0.45 mmol) in MeOH (3.0 mL) and THF(7.0 mL) was added LiOH.H2O (56 mg, 1.37 mmol) in H2O (1.0 mL). After stirred for 2 h, the reaction was quenched with water and the product was extracted with EtOAc. The organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. . The residue was purified by preparative HPLC (20-75% acetonitrile/0.1% formic acid in water; Gemini NX, 250 mm x 20 mm, 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (30 mg, 16%), ESLMS m/z: 424.10 [M+H]+. XH NMR (400 MHz, CD3OD) 5 8.15 (d, J= 2.8 Hz, 1H), 8.03 (dd, J= 4.0, 1.6 Hz, 1H), 7.61 (dd, J= 9.2, 2.8 Hz, 1H), 7.46 (dd, J = 9.6, 4.0 Hz, 1H), 7.06 (dd, J= 9.2, 1.2 Hz, 1H), 6.60 (d, J= 9.2 Hz, 1H), 4.39 - 4.31 (m, 1H), 4.03 - 3.98 (m, 1H), 2.29 - 2.20 (m, 2H), 2.12 - 2.03 (m, 1H), 1.99 - 1.89 (m, 1H), 1.71 - 1.55 (m, 4H), 1.53 - 1.49 (m, 2H).
Example 54: Synthesis of 2-(6-(((1S,3S)-3-((5-(l-methvl-lH-l,2,4-triazol-3-vl)-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)pyridazin-3(2H)-one

Step-1; Synthesis of l-methyl-lH-l,2,4-triazole-3-carboxylic acid (Int-84)
To a solution of methyl l-methyl-lH-l,2,4-triazole-3-carboxylate (0.3 g, 2.126 mmol) in MeOH (1 mL) and water (1 mL) was added KOH (0. 13 g, 2.338 mmol). ). After stirred for 2 h, the reaction was quenched with IN HC1 to pH=3-4 and concentrated under reduced pressure. . The residue was purified by preparative HPLC (0-15% acetonitrile/0.1% formic acid in water, Kinetex EVO, 250 mm x 21.2 mm, 5.0 μm column, flow rate 18 mL/min) to afford the title compound as an off-white solid (150 mg, 55%), ’H NMR (300 MHz, DMSO-d6) 5 8.55 (s, 1H), 3.91 (s, 3H).
Step-2; Synthesis ooff 2-(6-(((1S,3S)-3-((5-(l-methyl-lH-l,2,4-triazol-3-yl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2Z/)-one (Example 54)
Example 54 was synthesized following the Example 2 starting from Int-5 and Int-84. ESI-MS m/z: 421.10 [M+H]+. ‘H NMR (400 MHz, CD3OD) 5 8.59 (s, 1H), 8.16 (d, J = 2.4 Hz, 1H), 8.03 (dd, J = 3.6, 1.6 Hz, 1H), 5 7.61 (dd, J= 9.2, 2.8 Hz, 1H), 7.46 (dd, J= 9.2, 3.6 Hz, 1H), 7.06 (dd, J = 9.6, 1.6 Hz, 1H), 6.60 (d, J= 8.8 Hz, 1H), 4.41 - 4.33 (m, 1H), 4.13 - 4.07 (m, 1H), 4.05 (s, 3H), 2.34 - 2.23 (m, 2H), 2.20 - 2.10 (m, 1H), 2.08 - 1.96 (m, 1H), 1.72 - 1.59 (m, 2H).
Example 55: Synthesis of 2-(3-(((LS',3.S')-3-((5-(6-oxopvridazin-l(6//)-vl)i)vridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)acetonitrile

Step-1; Synthesis of 2,5-dioxopyrrolidin-l-yl 2-cyanoacetate (Int-85)
Int-85 was synthesized following the Int-12 starting from commercially available 2-cyanoacetic acid. ’H NMR (300 MHz, CD3OD) 5 3.68 (s, 2H), 2.67 (s, 4H).
Step-2; Synthesis of 2-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(67/)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l ,2,4-oxadiazol-5-yl)acetonitrile (Example 55)
Example 55 was synthesized following the Example 15 starting from Int-5 and Int-85. ESI-MS m/z: 378.90 [M+H]+. ‘H NMR (400 MHz, CDC13) 5 8.33 (d, J = 2.4 Hz, 1H), 8.18 (dd, J= 3.6, 1.6 Hz, 1H), 5 7.70 (dd, J= 8.8, 2.8 Hz, 1H), 7.24 (dd, J= 9.6, 4.0 Hz, 1H), 7.04 (dd, J= 9.6, 1.6 Hz, 1H), 6.43 (d, J= 8.8 Hz, 1H), 4.89 - 4.80 (m, 1H), 4.64 - 4.60 ( m, 1H), 4.33 - 4.26 (m, 1H), 4.13 - 4.05 (m, 1H), 3.91 (s, 2H), 2.35 - 2.28 (m, 2H), 2.15 - 2.08 (m, 1H), 2.04 - 1.98 (m, 1H), 1.68 - 1.52 (m, 2H).
Example 56: Synthesis of 2 - (6 -((( l1S',31S')-3-((5-eth yl-1 ,2,4-oxadiazol-3- vl)amino)cvcloDentvl)amino)Dvridin-3-vl)-4-methvlpvridazin-3(2ZD-one

Example 56 was synthesized following the Example 2 starting from Int-69 with commercially available propionic acid. ESI-MS m/z: 382.20 [M+H]+. *H NMR (400 MHz, CD3OD) 5 8.12 (d, J= 2.4 Hz, 1H), 7.88 (d, J = 4.0 Hz, 1H), 7.57 (dd, J = 8.8, 2.4 Hz, 1H), 7.33 - 7.31 (m, 1H), 6.58 (d, J = 9.2 Hz, 1H), 4.37 -
4.31 (m, 1H), 4.02 - 3.96 (m, 1H), 2.75 (q, J= 7.6 Hz, 2H), 2.29 - 2.21 (m, 2H), 2.20 (s, 3H), 2.09 - 2.03 (m, 1H), 1.99 - 1.90 (m, 1H), 1.66 - 1.56 (m, 2H), 1.30 (t, J = 7.6 Hz, 3H).
Example 57: Synthesis of l-(3-(((1S,3S)-3-((5-(6-oxoDvridazin-l(6Zn-vl)Dvridin-2- vl)amino)cvcloDentvl)amino)-l,2,4-oxadiazol-5-vl)cvcloDropane-l-carbonitrile

Int-5 Example 57
Example 57 was synthesized following the Example 2 starting from Int-5 with commercially available 1- cy anocyclopropane- 1 -carboxylic acid. ESI-MS m/z: 405.02 [M+H]+. 'HNMR (400 MHz, CD3OD) 5 8.14 (d, J = 2.8 Hz, 1H), 8.02 (dd, J = 4.0, 1.6 Hz, 1H), 7.59 (dd, J = 9.2, 2.8 Hz, 1H), 7.45 (dd, J = 9.6, 4.0 Hz, 1H), 7.05 (dd, J = 9.6, 1.6 Hz, 1H), 6.58 (d, .7= 9.2 Hz, 1H), 4.37 - 4.31 (m, 1H), 3.98 - 3.95 (m, 1H), 2.29 - 2.19 (m, 2H), 2.09 - 2.03 (m, 1H), 1.96 - 1.89 (m, 3H), 1.85 - 1.82 (m, 2H), 1.66 - 1.52 (m, 2H).
Example 58: Synthesis of ethyl l-(3-(((f1S',31S')-3-((5-(6-oxopvridazin-l(677)-vl)pvridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)cvclobutane-l-carboxylate

Example 58 was synthesized following the Example 2 starting from Int-5 with commercially available 1- (ethoxycarbonyl)cyclobutane-l -carboxylic acid. ESI-MS m/z: 466.0 [M+H]+. ’ H NMR (400 MHz, CDsOD) 5 8.15 (dd, .7= 2.4, 0.4 Hz, 1H), 8.03 (dd, J= 4.0, 1.6 Hz, 1H), 7.60 (dd, J = 9.2, 2.8 Hz, 1H), 7.47 (dd, J= 9.2, 4.0 Hz, 1H), 7.06 (dd, .7= 9.2, 1.6 Hz, 1H), 6.59 (dd, J = 8.8, 0.4 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.21 (q, J = 7.2 Hz, 2H), 4.04 - 3.98 (m, 1H), 2.82 - 2.72 (m, 2H), 2.70 - 2.62 (m, 2H), 2.31 - 2.20 (m, 2H), 2.12 - 2.04 (m, 3H), 1.99 - 1.91 (m, 1H), 1.69 - 1.55 (m, 2H), 1.24 (t, J = 7.2 Hz, 3H).
Example 59: Synthesis of l-(3-(((1S,3S)-3-((5-(6-oxopvridazin-l(6Z7)-vl)pvridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-vl)cvclobutane-l-carboxylic acid

Example 59 was synthesized following the Example 53 starting from Example 58. ESI-MS m/z: 437.95 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.16 (d, J= 2.4 Hz, 1H), 8.03 (dd, J= 3.6, 1.6 Hz, 1H), 7.62 (dd, .7= 9.2, 2.8 Hz, 1H), 7.46 (dd, J = 9.6, 4.0 Hz, 1H), 7.06 (dd, J= 9.6, 1.6 Hz, 1H), 6.61 (d, J = 9.2 Hz, 1H), 4.38 - 4.31 (m, 1H), 4.05 - 3.98 (m, 1H), 2.81 - 2.71 (m, 2H), 2.69 - 2,61 (m, 2H), 2.30 - 2.19 (m, 2H), 2.14 - 2.02 (m, 3H), 2.01 - 1.93 (m, 1H), 1.69 - 1.55 (m, 2H).
Example 60: Synthesis of l-methyl-3-(6-(((l1$',31S')-3-((5-(l-methvlcvclonropvl)-l,2,4-oxadiazol-3- vl)amino)cvclonentvl)amino)pyridin-3-vl)imidazolidine-2,4-dione

Example 60 was synthesized following the Example 2 starting from Int-11 with commercially available 1 -methylcyclopropane- 1 -carboxylic acid. ESI-MS m/z: 412.25 [M+H]+. ’ H NMR (400 MHz, CD3OD) 5 7.90 (dd, J= 2.4, 0.4 Hz, 1H), 7.35 (dd, J= 9.2, 2.8 Hz, 1H), 6.56 (dd, J= 9.2, 0.8 Hz, 1H), 4.31 - 4.25 (m, 1H), 4.07 (s, 2H), 3.98 - 3.91 (m, 1H), 3.01 (s, 3H), 2.28 - 2.13 (m, 2H), 2.08 - 2.00 (m, 1H), 1.93 - 1.86 (m, 1H), 1.65 - 1.51 (m, 2H), 1.45 (s, 3H), 1.32 - 1.27 (m, 2H), 0.98 - 0.92 (m, 2H).
Example 61: Synthesis of 3-(6-(((l1$',31S')-3-((5-cvclobutvl-l,2,4-oxadiazol-3-vl)amino) cvclopentvl)amino)pyridin-3-vl)-l-methvlimidazolidine-2, 4-dione

Example 61 was synthesized following the Example 2 starting from Int-11 with commercially available cyclobutanecarboxylic acid. ESI-MS m/z: 412.25 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 7.90 (d, J= 2.8 Hz, 1H), 7.35 (dd, J = 8.8, 2.4 Hz, 1H), 6.56 (dd, J = 9.2, 0.8 Hz, 1H), 4.32 - 4.27 (m, 1H), 4.07 (s, 2H), 4.01 - 3.93 (m, 1H), 3.65 - 3.59 (m, 1H), 3.01 (s, 3H), 2.42 - 2.34 (m, 4H), 2.25 - 2.18 (m, 2H), 2.15 - 2.08 (m, 1H), 2.07 - 1.98 (m, 2H), 1.95 - 1.89 (m, 1H), 1.65 - 1.52 (m, 2H).
Example 62: Synthesis of l,5,5-trimethyl-3-(6-(((1S,3S)-3-((5-(l-methvlcvcloDroDvl)-l,2,4-oxadiazol-
3-vl)amino)cvcloDentvl)amino)Dvridin-3-yl)imidazolidine-2,4-dione

Example 62 was synthesized following the Example 2 starting from Int-64 with commercially available 1 -methylcyclopropane- 1 -carboxylic acid. ESI-MS m/z: 440.0 [M+H]+. JH NMR (400 MHz, CD3OD) 57.91 - 7.90 (m, 1H), 7.37 (dd, J = 9.2, 2.8 Hz, 1H), 6.57 (dd, J = 9.2, 0.8 Hz, 1H), 4.32 - 4.26 (m, 1H), 3.99 - 3.92 (m, 1H), 2.95 (s, 3H), 2.28 - 2.17 (m, 2H), 2.08 - 2.01 (m, 1H), 1.93 - 1.88 (m, 1H), 1.65 - 1.52 (m, 2H), 1.48 (s, 6H), 1.47 (s, 3H), 1.31 1.28 (m, 2H), 0.98 - 0.96 (m, 2H). Example 63: Synthesis of l-(3-(((15,,35,)-3-((5-(6-oxopvridazin-l(6H)-vl)pyridin-2- vl)amino)cvclopentvl)amino)-l,2,4-oxadiazol-5-yl)cvclobutane-l-carbonitrile

Step-1: Synthesis ooff Synthesis ooff l-(3-(((LS',3.S')-3-((5-(6-oxopyridazin-l(6//)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)cyclobutane-l-carboxamide (Int-86)
[355] To a solution of Example 59 (35 mg, 0.08 mmol) in DMF (1.0 mL) were added ammonium chloride (21 mg, 0.40 mmol), HATH (45 mg, 0.12 mmol) and DIPEA ( 0.06 mL, 0.40 mmol). After stirring for 48 h, the reaction was quenched with water and the product was extracted with EtOAc. The organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide crude product as a pale brown sticky solid (50 mg, Crude) . ESI-MS m/z: 436.95 [M+H]+.
Step-2: Synthesis oOff l-(3-(((1S,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)cyclobutane-l-carbonitrile (Example 63)
To a solution of Int-86 (40 mg, 0.09 mmol) in DCM (1.0 mL) was added triethylamine (0.05 mL,0.36 mmol) and trifluoracetic anhydride (0.01 mL,0.11 mmol) at 0°C. The mixture was allowed to warm up to rt, and stirred for 1 h. After methanol (0.5 mL) and K2CO3 (86 mg, 0.63 mmol) were added, the reaction mixture was stirred for 48 h. After quenched with water, the product was extracted with EtOAc. The organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated under
reduced pressure. The residue was purified by preparative HPLC (30-75% acetonitrile/0.02% NH40H in water; WATERS X BRIDGE, 250 mm x 20 mm, 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (7 mg, 18%), ESI-MS m/z: 419.05 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 8.15 (d, J= 2.8 Hz, 1H), 8.03 (dd, J= 4.0, 2.0 Hz, 1H), 7.60 (dd, J= 9.2, 2.8 Hz, 1H), 7.46 (dd, J= 9.6, 4.0 Hz, 1H), 7.06 (dd, .7= 9.6, 1.6 Hz, 1H), 6.59 (d, J = 9.2 Hz, 1H), 4.39 - 4.31 (m, 1H), 4.05 - 3.97 (m, 1H), 2.88 - 2.81 (m, 4H), 2.40 - 2.20 (m, 4H), 2.16 - 2.06 (m, 1H), 1.99 - 1.90 (m, 1H), 1.68 - 1.58 (m, 2H).
Example 64: Synthesis of 3-(6-(((lS'.3Sr)-3-((5-cvclopropvl-1.2.4-oxadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)-l,5,5-trimethylimidazolidine-2,4-dione

Example 64 was synthesized following the Example 2 starting from Int-64 with commercially available cyclopropanecarboxylic acid. ESI-MS m/z: 426.2 [M+H]+. 'H NMR (400 MHz, CD3OD) 5 7.91 (d, J= 2.8 Hz, 1H), 7.37 (dd, J= 9.2, 2.8 Hz, 1H), 6.57 (dd, .7= 8.8, 0.4 Hz, 1H), 4.32 - 4.25 (m, 1H), 3.99 - 3.91 (m, 1H), 2.95 (s, 3H), 2.28 - 2.18 (m, 2H), 2.10 - 2.00 (m, 2H), 1.95 - 1.87 (m, 1H), 1.65 - 1.52 (m, 2H), 1.48 (s, 6H), 1.19 - 1.12 (m, 2H), 1.11 - 1.08 (m, 2H).
Example 65: Synthesis of 3-(6-(((lS,,3S)-3-((5-cyclopropyl-l,2,4-oxadiazol-3- vl)amino)cvclopentvl)amino)pyridin-3-vl)-l-methvlimidazolidine-2,4-dione

Example 65 was synthesized following the Example 2 starting from Int-11 with commercially available 1 -methylcyclopropane- 1 -carboxylic acid. ESI-MS m/z: 398.25 [M+H]+. ’ H NMR (400 MHz, CD3OD) 5 7.91 (d, .7= 2.8 Hz, 1H), 7.36 (dd, J= 8.8, 2.4 Hz, 1H), 6.56 (dd, J= 9.2, 0.8 Hz, 1H), 4.31 - 4.26 (m, 1H), 4.08 (s, 2H), 3.99 - 3.92 (m, 1H), 3.02 (s, 3H), 2.27 - 2.18 (m, 2H), 2.10 - 2.01 (m, 2H), 1.97 - 1.88 (m, 1H), 1.65 - 1.52 (m, 2H), 1.19 - 1.12 (m, 2H), 1.11 - 1.07 (m, 2H).
Example 66: Synthesis of 2-(6-(((1S,3S)-3-((5-cvclopropvl-l,2,4-thiadiazol-3- vl)amino)cvclopentvl)amino)pvridin-3-vl)-4-methvlpyridazin-3(2ZD-one

Example 66 was synthesized following the Example 24 starting from Int-67. ESI-MS m/z: 410. 1 [M+H]+. ‘HNMR (400 MHz, CD3OD) 5 8.12 (d, J= 2.4 Hz, 1H), 7.87 (d, J= 4.0 Hz, 1H), 7.57 (dd, J= 9.2, 2.8 Hz, 1H), 7.32 - 7.31 (m, 1H), 6.58 (d, J= 8.8 Hz, 1H), 4.35 - 4.29 (m, 1H), 4.28 - 4.21 (m, 1H), 2.37 - 2.30 (m, 1H), 2.29 - 2.21 (m, 5H), 2.05 - 1.91 (m, 2H), 1.63 - 1.52 (m, 2H), 1.22 - 1.18 (m, 2H), 1.10 - 1.07 (m, 2H).
Example 67: Synthesis Of 3-(6-(((ES'.3.S')-3-((5-(l-hvdroxvcvcloi)roi)vl)-1.2.4-oxadiazol-3- vl)amino)cvdoDentvl)amino)Dvridin-3-vl)-l-methylimidazolidine-2,4-dione

Step-1; Synthesis of 1-acetoxycyclopropane-l-carboxylic acid (Int-76)
Int-76 was synthesized following the Int-70 starting from commercially available 1 -hydroxy cyclopropane- 1 -carboxylic acid. *H NMR (400 MHz, DMSO-ds) 5 12.86 (s, 1H), 2.01 (s, 3H), 1.36 - 1.33 (m, 2H), 1.17
- 1.13 (m, 2H).
Step-2; Synthesis ooff l-(3-(((1S,3S)-3-((5-(3-methyl-2,5-dioxoimidazolidin-l-yl)pyridin-2- yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5-yl)cyclopropyl acetate (Int-77)
Int-77 was synthesized following the Example 2 starting from Int-76 and Int-11. ESI-MS m/z: 456.10 [M+H]+.
Step-3; Synthesis ooff 3-(6-(((LS',3.S')-3-((5-(l-hydroxycyclopropyl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)-l-methylimidazolidine-2, 4-dione (Example 67)
To a solution of Int-77 (60 mg, 0.13 mmol) in THE (L0 mL) was added 6N HCI (1.0 mL) at 0°C. The mixture was allowed to warm up to rt and stirred for 48 h. After quenched with saturated sodium
bicarbonate solution until pH=7-8, the product was extracted with EtOAc. The organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by preparative HPLC (10-40% acetonitrile/0.1% formic acid in water; ZODIAC, Cl 8, 250 mm x 19 mm, 5.0 μm column, flow rate 15 mL/min) to afford the title compound as a yellow solid (3 mg, 6%), ESI-MS m/z: 413.95 [M+H]+. *HNMR (400 MHz, CD3OD) 5 7.91 (d, J= 2.8 Hz, 1H), 7.43 (dd, J= 8.8, 2.4 Hz, 1H), 6.62 (d, J= 8.8 Hz, 1H), 4.31 - 4.25 (m, 1H), 4.09 (s, 2H), 4.01 - 3.94 (m, 1H), 3.02 (s, 3H), 2.28 - 2.18 (m, 2H), 2.09 - 2.02 (m, 1H), 1.98 - 1.91 (m, 1H), 1.65 - 1.56 (m, 2H), 1.34 - 1.25 (m, 4H).
Example 68: PCSK9 Ligand Binding Assay
[356] The PCSK9 binding of the compounds of the disclosure was measured using a time resolved fluorescence resonance energy transfer (TR-FRET) assay. This TR-FRET assay measures the ability of a compounds of the present disclosure to compete for binding with Alexa Fluor 647 labeled probe in a known region of human PCSK9. The assay provides measures of both potency (IC50) and efficacy (Amax).
[357] Materials
Human PCSK9
Human PCSK9 Terbium
• Alexa Fluor 647 labeled probe, prepared as described in Example 28.
• Proxi plate-low volume assay plate (PerkinElmer #6008280)
• Greiner V-bottom (Greiner BioOne #781280)
• Assay Buffer o 20 mM HEPES, pH 7.5 o 150 mM NaCl o 1 mM CaCh o 0.01% v/v Tween20 o 0.01% w/v BSA
[358] A master compound plate was prepared in a Greiner V bottom plate by diluting compounds of the disclosure in dimethylsulfoxide to the correct concentration for the desired top concentration based on the desired final concentration: for a 30 pM final concentration the master plate concentration is 1.5 mM (68 pL DMSO + 12 pL 10 mM of a compound of the disclosure), for a 10 pM final concentration the master plate concentration is 0.5 mM (76 pL DMSO + 4 pL 10 mM of a compound of the disclosure), for a 3 pM final concentration the master plate concentration is 150 pM (69 pL DMSO + 1 pL 10 mM of a compound of the disclosure). These solutions were pipetted into columns 1 and 11 of the compound plate.
Threefold serial dilutions were generated in columns 2-10 and 12-20 of the compound plate by transferring 10 pL into 20 pL of DMSO. Columns 21 and 22 of the compound plate were negative controls containing DMSO alone.
[359] An intermediate plate was generated in a Greiner V bottom plate by transferring 8 pL from each well of the master plate into a corresponding well containing 92 pL of assay buffer and mixing thoroughly.
[360] A Proxi plate-low volume assay plate was used for the assay. To all wells of the plate was added 10 pL of 7 nM Human PCSK9 Terbium, followed by 5 pL from the intermediate plate. For the competition displacement control wells in columns 23 and 24 of the plate, 5 pL of unlabeled human PCSK9 was added at 4 pM in assay buffer containing 8% DMSO. Following a 30 minute incubation, 5 pL of 120 pM Alexa Fluor 647 labeled probe was added and the mixture was incubated for an additional 2 hours.
[361] The TR-FRET signal was measured on an EnVision instrument with a 60 ms delay, 330 nm excitation and 665 nm emission (FRET), and 330 nM excitation and 615 nm (Terbium). The FRET ratio (FRET/ Terbium) was used for calculations.
DATA ANALYSIS
[362] No inhibition (0%) was observed from the wells containing DMSO (Control) in columns 21 and 22 of the compound plate. Full inhibition (100%) was observed from the wells containing 1 pM human PCSK9 (Control) in columns 23 and 24 of the plate. Data is expressed as percent inhibition: (value - 0%)/(100% - 0%) - Table 2.
Table 2: PCSK9 Ligand Binding Assay



Example 69: hERG Qpatch Assay
[363] The cardiovascular toxicity of certain compounds was measured using a hERG Qpatch assay. Briefly, hERG expressing cell lines were produced using CH0-K1 T-Rex inducible plasmid system (Invitrogen) as described previously (Cao et al, Assay Drug Dev. Technol. 2010, 8, 766-780). C6ll lines were maintained in Ham’s F12 nutrient mixture containing 10% FBS, blasticidin (10 mg/mL; InvivoGen), hygromycin B (200 mg/mL;InvivoGen), Zeocin (200 mg/mL, Invitrogen), and neomycin (200 mg/mL, Invitrogen) using SelecT automated cell culture system (TAP Biosystems, Cambridge, U.K.). hERG and hCavl.2 channels expression was induced with tetracycline (0.25-1 μg/mL, Invitrogen) at least 24 h prior to the experiment. hERG currents were recorded using the Qpatch automated patch clamp systems (Sophion Bioscience Inc., North Brunswick, NJ) in the whole (single) cell configuration. hERG expressing CHO-K1 cells were harvested with Detachin (Genlantis) and stored in the modified serum-free SFM-2 media (Life Technologies) at room temperature. The extracellular solution contained (in mM) NaCl (145), KC1 (4), MgC12 (1), CaC12 (2), and HEPES (10), pH 7.4, with NaOH. The intracellular solution contained KC1 (135), MgC12 (1.75), CaC12 (5.4), EGTA (10), K2-ATP (4), and HEPES (10), pH 7.2, with KOH.
[364] After whole cell configuration was achieved, the cell was held at -90 mV, and a 0.1 s pulse to -50 mV was delivered to measure the leaking current, which was subtracted from the tail current online. Then the cell was depolarized to +20 mV for 4 s (prepulse), followed by a 4 s test pulse to -50 mV to reveal the hERG tail current. To monitor changes in the current amplitude, this voltage protocol was repeatedly applied every 20 s. Test compounds were first diluted in DMSO for six dose-response experiments and then dissolved in the extracellular solution using Freedom EVO liquid handling robotic system (Tecan, Mannedorf, Switzerland). The final DMSO concentration in samples was 0.3% v/v. Amitriptyline (Sigma) was tested as a positive control. Data were analyzed using a MatLab-based program and using Sophion QPatch Assay software. The data, shown in Table 3, represent the results of a single experiment.
Table 3: hERG Qpatch Assay

Reference Compound 1 is 2-(6-(((lS,3S)-3-((5-cyclopropylpyrimidin-2- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one. The compound was prepared following the procedure reported in W02020150473. Reference Compound 2 is 3-(6-(((lS,3S)-3-((5- (difluoromethoxy)pyrimidin-2-yl)amino)cyclopentyl)amino)pyridin-3-yl)-l-methylimidazolidine-2,4- dione The compound was prepared following the procedure reported in W02020150473.
[365] As show in Table 3, Examples 1, 2, 5, 6, 10, 24, 29 and 31 showed unexpectedly reduced inhibition of the hERG channel, compared to the reference compounds (Reference compounds 1 and 2). Reduced inhibition of the hERG channel may translate into an improved safety profile for the compounds as compared to reference compounds.
Equivalents
[366] Those skilled in the art will recognize, or be able to ascertain, using no more than routine experimentation, numerous equivalents to the specific embodiments described specifically herein. Such equivalents are intended to be encompassed in the scope of the following claims.
Claims
1. A compound of formula (I):
 or a pharmaceutically acceptable salt thereof wherein:
or a pharmaceutically acceptable salt thereof wherein:
A is a 5 or 6 membered heterocycle or heteroaryl containing at least one N and at least one oxo at a ring carbon and is optionally substituted with (C1-C6)alkyl;

X is N, 0, or S;
V is CR1 or N;
Z is CR2, NR3, 0, or S;
R1 and R2 are each, independently selected from H, halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6- C10)aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from 0 and N, wherein the (C1- C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1- C6)haloalkyl, (C1-C6)alkoxy, (C3-C6)cycloalkyl, -SO2(C1-C6)alkyl, -COOH, and -COO(C1-C6)alkyl;
R3 is absent, H or (C1-C6)alkyl; and
R4 is H, (C1-C6)alkyl, (C1-C6)haloalkyl, (C1-C6)alkoxy or (C3-C6)cycloalkyl, wherein the (C1-C6)alkoxy is optionally substituted with halogen.
2. A compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein at least one of X, Y and Z is N.
3. A compound according to claim 1 or claim 2 or a pharmaceutically acceptable salt thereof, wherein when X is 0 or S, Z is CR2 or N.
4. A compound according to any one of claims 1 to 3 or a pharmaceutically acceptable salt thereof, wherein when Z is 0 or S, X is N.
5. A compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein R4 is methyl, ethyl, or cyclopropyl.
6. A compound according to any one of claims 1 to 5 or a pharmaceutically acceptable salt thereof, wherein

7. A compound according to any one of claims 1 to 6 or a pharmaceutically acceptable salt thereof, wherein

8. A compound according to any one of claims 1 to 7 or a pharmaceutically acceptable salt thereof, wherein X is N.
9. A compound according to any one of claims 1 to 8 or a pharmaceutically acceptable salt thereof, wherein X is N, Y is CR1, and Z is 0 or S.
10. A compound according to any one of claims 1 to 9 or a pharmaceutically acceptable salt thereof, wherein X is N, Y is CR1, and Z is 0.
11. A compound according to any one of claims 1 to 10 or a pharmaceutically acceptable salt thereof, wherein R1 is H, (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, a 4 to 6 membered heterocyclyl comprising 1, 2 or 3 heteroatoms selected from O and N, or a 5 or 6 membered heteroaryl comprising 1, 2 or 3 heteroatoms selected from O and N, wherein the (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1-C6)haloalkyl, (C1-C6)alkoxy, (C3-C6)cycloalkyl, - SO2(C1-C6)alkyl, -COOH, and -COO(C1-C6)alkyl.
12. A compound according to any one of claims 1 to 11 or a pharmaceutically acceptable salt thereof, wherein R1 is H, (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, a 4 to 6 membered heterocyclyl
comprising 1 heteroatom selected from 0 and N, or a 5 or 6 membered heteroaryl comprising 1 or 3 heteroatoms selected from N, wherein the (C1-C6)alkyl, (C3-C6)cycloalkyl, (C6-C10)aryl, heterocyclyl, or heteroaryl are each independently optionally substituted with one or more substituents selected from halogen, -OH, -CN, (C1-C6)alkyl, (C1-C6)haloalkyl, (C1-C6)alkoxy, (C3-C6)cycloalkyl, -SO2(C1-C6)alkyl, -COOH, and -COO(C1-C6)alkyl.
13. A compound according to any one of claims 1 to 12 or a pharmaceutically acceptable salt thereof, wherein R1 is H, (C1-C6)alkyl optionally substituted with one or more halogen, -CN, (C3-C6)cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C1-C6)alkyl and (C1- C6)haloalkyl, or (C6-C10)aryl optionally substituted one or more with halogen.
14. A compound according to any one of claims 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R1 is (C1-C6)alkyl optionally substituted with one or more halogen or -CN, (C3-C6)cycloalkyl optionally substituted with one or more substituents selected from halogen, -OH, (C1-C6)alkyl and (C1- C6)haloalkyl, or (C6-C10)aryl optionally substituted with one or more halogen or -CN.
15. A compound according to claim 13 or a pharmaceutically acceptable salt thereof, wherein each halogen is fluoro.
16. A compound according to any one of claims 1 to 15 or a pharmaceutically acceptable salt thereof, wherein R1 is (C1-C6)alkyl optionally substituted with one or more fluoro or -CN.
17. A compound according to any one of claims 1 to 16, wherein R1 is -CH3, -CH2CH3, -CHF2, -CF3,

18. A compound according to any one of claims 1 to 17, wherein R1 is -CH3, -CHF2, or -CF3.
19. A compound according to any one of claims 1 to 14 or a pharmaceutically acceptable salt thereof, wherein R1 is (C3-C6)cycloalkyl optionally substituted with one or more substituents selected from fluoro, -OH, (C1-C6)alkyl and (C1-C6)fluoroalkyl.
20. A compound according to any one of claims 1 to 14 or a pharmaceutically acceptable salt thereof, wherein R1 is


21. A compound according to any one of claims 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R1 is phenyl optionally substituted with one or more fluoro.
22. A compound according to claim 21 or a pharmaceutically acceptable salt thereof, wherein R1 is or
23. A compound according to any one of claims 1 to 13 or a pharmaceutically acceptable salt thereof, wherein

24. A compound according to any one of claims 1 to 13 or a pharmaceutically acceptable salt thereof, wherein R1 is ,

25. A compound according to any one of claims 1 to 3, 5 to 8, and 11 to 24 or a pharmaceutically acceptable salt thereof, wherein R2 is H, halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl, or (C6-C10)aryl.
26. A compound according to any one of claims Ito 3, 5 to 8, and 11 to 25 or a pharmaceutically acceptable salt thereof, wherein R2 is halogen, (C1-C6)alkyl, (C3-C6)cycloalkyl, or phenyl
27. A compound according to any one of claims 1 to 3, 5 to 8, and 11 to 26 or a pharmaceutically acceptable salt thereof, wherein R2 is -Cl, -CH3,

28. A compound according to any one of claims 1 to 3, 5 to 7, and 11 to 27 or a pharmaceutically acceptable salt thereof, wherein R3 is H.
29. A compound according to any one of claims 1 to 4 or a pharmaceutically acceptable salt thereof,
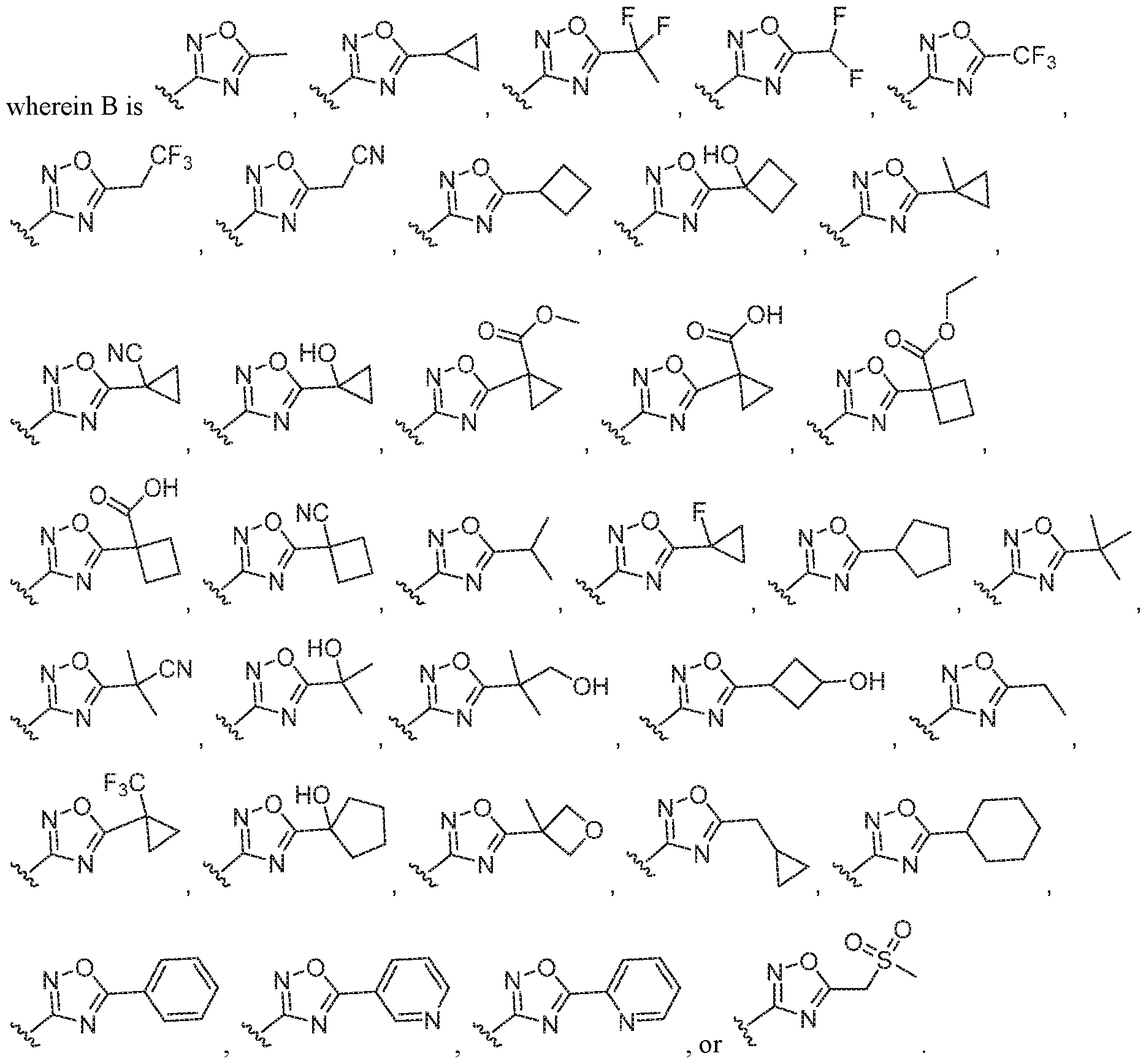
30. A compound according to any one of claims 1 to 4 or a pharmaceutically acceptable salt thereof,, wherein

31. A compound according to any one of claims 1 to 4 or a pharmaceutically acceptable salt thereof, wherein

32. A compound according to claim 1 or a pharmaceutically acceptable salt thereof selected from: 2-(6-(((lS,3S)-3-((5-cyclobutyl-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin- 3(2H)-one;
2-(6-(((lS,3S)-3-((5-cyclopropyl-l,2,4-thiadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin- 3(2H)-one;
2-(6-(((lS,3S)-3-((5-(3-hydroxycyclobutyl)-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)pyridazin-3 (2H)-one;
2-(6-(((lS,3S)-3-((3-chloro-l,2,4-thiadiazol-5-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)- one;
2-(6-((( 1 S,3 S)-3 -((5 -( 1 -methylcyclopropyl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
3-methyl-l-(6-(((lS,3S)-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)imidazolidine-2, 4-dione;
2 - (6 - ((( 1 S , 3 S ) -3 - ((5 -( 1 , 1 -difluoroethyl)- 1 ,2,4-oxadiazol-3 -yl) amino) cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
2-(6-((( IS, 3 S)-3 -((5 -(1 -fluorocyclopropyl)-!, 2, 4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)pyridazin-3 (2H)-one;
2-(6-(((lS,3S)-3-((5-ethyl-l,2,4-oxadiazol-3-yl)amino) cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)- one;
2-(6-(((lS,3S)-3-((3-cyclopropyl-l,2,4-thiadiazol-5-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-
3(2H)-one;
2-(6-(((lS,3S)-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino) cyclopentyl) amino) pyridin-3-yl) pyridazin-
3(2H)-one;
2-(6-(((lS,3S)-3-((5-(trifluoromethyl)-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)pyridazin-3 (2H)-one;
2-(6-(((l S,3 S)-3 -((5 -( 1 -hydroxycyclopropyl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
2-(6-(((lS,3S)-3-((5-(difluoromethyl)-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)pyridazin-3 (2H)-one;
2-(6-((( 1 S,3 S)-3 -((5 -cyclopentyl- 1 ,2,4-oxadiazol-3 -yl)amino) cyclopentyl)amino)pyridin-3 -yl)pyridazin- 3(2H)-one;
2-(6-(((l S,3S)-3-((5-cyclopropyl-l,2,4-oxadiazol-3-yl)amino) cyclopentyl) amino)pyridin-3-yl) pyridazin-3 (2H)-one;
2-(6-(((l S,3 S)-3 -((3 -phenyl- 1 ,2,4-thiadiazol-5 -yl)amino)cyclopentyl)amino)pyridin-3 -yl)pyridazin-
3(2H)-one;
2-(6-(((lS,3S)-3-((5-isopropyl-l,2,4-oxadiazol-3-yl)amino) cyclopentyl)amino)pyridin-3-yl)pyridazin-
3(2H)-one;
2-(6-(((lS,3S)-3-((5-(tert-butyl)-l,2,4-oxadiazol-3-yl)amino) cyclopentyl)amino)pyridin-3-yl)pyridazin-
3(2H)-one;
2-(6-(((lS,3S)-3-((5-(4-fluorophenyl)-l,2,4-thiadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)pyridazin-3 (2H)-one;
2-(6-(((lS,3S)-3-((2H-tetrazol-5-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-;
2-(6-((( 1 S,3 S)-3 -((5 -methyl- 1 ,3 ,4-oxadiazol-2-yl)amino)cyclopentyl)amino)pyridin-3 -yl)pyridazin-
3(2H)-one;
1-methyl-3-(6-(((lS,3S)-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)imidazolidine-2, 4-dione;
2-(6-(((l S,3 S)-3 -((5 -( 1 -(trifluoromethyl)cyclopropyl)- 1 ,2,4-oxadiazol-3 - yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one; l-(6-(((lS,3S)-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino) cyclopentyl)amino)pyridin-3-yl)pyrrolidin-2- one;
2-(6-((( 1 S,3 S)-3 -((5 -( 1 -hydroxycyclopentyl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
6 ' - ((( 1 S,3 S)-3 -((5 -methyl- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)-2H- [ 1 ,3 '-bipyridin] -2-one;
2-(6-(((lS,3S)-3-((6-methyl-l,2,4-triazin-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)- one;
4-methyl-2-(6-(((lS,3S)-3-((6-methyl-l,2,4-triazin-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-
3(2H)-one;
2-(6-(((lS,3S)-3-((6-cyclopropyl-l,2,4-triazin-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-
3(2H)-one;
2-(6-(((lS,3S)-3-((6-cyclopropyl-l,2,4-triazin-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)-4- methylpyridazin-3(2H)-one;
2-(6-(((lS,3S)-3-((6-ethyl-l,2,4-triazin-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)-4-methylpyridazin-
3(2H)-one;
2-(6-((( 1 S,3 S)-3 -((6-ethyl- 1 , 2, 4 -tri azin-3 -yl)amino)cyclopentyl)amino)pyridin-3 -yl)pyridazin-3 (2H)-one :
2-(6-((( 1 S,3 S)-3 -((5 -( 1 -hydroxycyclopentyl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
1 ,5 ,5 -trimethyl-3 -(6-(((l S,3 S)-3 -((5 -methyl- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)imidazolidine-2, 4-dione;
4-methyl-2-(6-(((lS,3S)-3-((5-methyl-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)pyridazin-3 (2H)-one;
2-(6-(((l S,3 S)-3 -((5 -(3 -methyloxetan-3 -yl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
2-(6-(((lS,3S)-3-((5-(cyclopropylmethyl)-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)pyridazin-3 (2H)-one;
2-(6-((( 1 S,3 S)-3 -((5 -(2 -hydroxypropan -2 -yl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
2-(6-(((l S,3 S)-3 -((5 -( 1 -hydroxycyclobutyl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
2-(6-(((lS,3S)-3-((5-cyclohexyl-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-
3(2H)-one;
2-(6-(((l S,3 S)-3 -((5 -phenyl- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 -yl)pyridazin-
3(2H)-one;
2-(6-((( 1 S,3 S)-3 -((5 -(pyridin-3 -yl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
2-(6-((( 1 S,3 S)-3 -((5 -(pyridin-2-yl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one;
2-(6-(((lS,3S)-3-((5-((methylsulfonyl)methyl)-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3- yl)pyridazin-3 (2H)-one;
2-(6-(((lS,3S)-3-((5-(l-hydroxy-2-methylpropan-2-yl)-l,2,4-oxadiazol-3- yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one;
2-(6-(((l S,3 S)-3 -((5 -(2,2,2-trifluoroethyl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)pyridazin-3 (2H)-one; methyl 1 -(3 -((( 1 S,3 S)-3 -((5 -(6-oxopyridazin- 1 (6H)-yl)pyridin-2-yl)amino)cyclopentyl)amino)- 1,2,4- oxadiazol-5-yl)cyclopropane-l-carboxylate;
1 -methyl-6'-((( 1 S,3 S)-3 -((5 -methyl- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)-[3 ,3 '-bipyridin] - 2(lH)-one;
2-methyl-2-(3-(((lS,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2-yl)amino)cyclopentyl)amino)-l,2,4- oxadiazol-5-yl)propanenitrile;
2-(6-(((l S,3 S)-3 -((5 -cyclopropyl- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 -yl)-4- methylpyridazin-3(2H)-one;
1-(3-(((lS,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2-yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5- yl)cyclopropane- 1 -carboxylic acid;
2-(6-((( 1 S,3 S)-3 -((5 -( 1 -methyl- 1 H- 1 ,2,4-triazol-3 -yl)- 1 ,2,4-oxadiazol-3 - yl)amino)cyclopentyl)amino)pyridin-3-yl)pyridazin-3(2H)-one;
2-(3-(((lS,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2-yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5- yl)acetonitrile;
2-(6-(((lS,3S)-3-((5-ethyl-l,2,4-oxadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)-4- methylpyridazin-3(2H)-one; l-(3-(((lS,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2-yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5- yl)cyclopropane- 1 -carbonitrile; ethyl 1 -(3 -((( 1 S,3 S)-3 -((5 -(6-oxopyridazin- 1 (6H)-yl)pyridin-2-yl)amino)cyclopentyl)amino)- 1,2,4- oxadiazol-5-yl)cyclobutane-l -carboxylate; l-(3-(((lS,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2-yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5- yl)cyclobutane-l -carboxylic acid;
1 -methyl-3 -(6-((( 1 S,3 S)-3 -((5 -( 1 -methylcyclopropyl)- 1 ,2,4-oxadiazol-3 - yl)amino)cyclopentyl)amino)pyridin-3-yl)imidazolidine-2, 4-dione;
3 -(6-(((l S,3 S)-3 -((5 -cyclobutyl- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 -yl)- 1 - methylimidazolidine-2, 4-dione;
1 ,5 ,5 -trimethyl-3 -(6-((( 1 S,3 S)-3 -((5 -(1 -methylcyclopropyl)- 1 ,2,4-oxadiazol-3 - yl)amino)cyclopentyl)amino)pyridin-3-yl)imidazolidine-2, 4-dione;
1-(3-(((lS,3S)-3-((5-(6-oxopyridazin-l(6H)-yl)pyridin-2-yl)amino)cyclopentyl)amino)-l,2,4-oxadiazol-5- yl)cyclobutane- 1 -carbonitrile;
3 -(6-(((l S,3 S)-3 -((5 -cyclopropyl- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 -yl)- 1,5,5- trimethylimidazolidine-2, 4-dione;
3 -(6-(((l S,3 S)-3 -((5 -cyclopropyl- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 -yl)- 1 - methylimidazolidine-2, 4-dione;
2-(6-(((lS,3S)-3-((5-cyclopropyl-l,2,4-thiadiazol-3-yl)amino)cyclopentyl)amino)pyridin-3-yl)-4- methylpyridazin-3(2H)-one; and
3 -(6-(((l S,3 S)-3 -((5 -( 1 -hydroxycyclopropyl)- 1 ,2,4-oxadiazol-3 -yl)amino)cyclopentyl)amino)pyridin-3 - yl)- 1 -methylimidazolidine-2, 4-dione.
33. A compound according to claim 1 or a pharmaceutically acceptable salt thereof selected from:





34. A pharmaceutical composition comprising a compound according to any one of claims 1 to 33 or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable carriers.
35. The pharmaceutical composition of claim 34, further comprising at least one additional pharmaceutically active agent.
36. The pharmaceutical composition of claim 35, wherein the additional pharmaceutically active agent is selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression, an inhibitory nucleic acid targeting Lp(a) protein expression, an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1
stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, antiobesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9, an immune checkpoint inhibitor and combinations thereof.
37. The pharmaceutical composition of any one of claims 34 to 36 for use in the treatment of a PCSK9-mediated disease or disorder.
38. The pharmaceutical composition of any one of claims 34 to 36 for use in the treatment of a disease or disorder, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
39. A combination comprising of a compound according to any one of claims 1 to 33 or a pharmaceutically acceptable salt thereof and one or more additional pharmaceutical agents.
40. The combination of claim 39, where the one or more agents additional pharmaceutically active agent is selected from hypolipidemic agents, niacin and analogs thereof, bile acid sequestrants, a thyroid hormone mimetic, thyroid hormone receptor (THR) P-selective agonist, a microsomal triglyceride transfer protein (MTP) inhibitor, an acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) inhibitor, a Niemann Pick Cl-like 1 (NPC1-L 1) inhibitor, an agonist of ATP Binding Cassette (ABC) proteins G5 or G8, an inhibitory nucleic acid targeting PCSK9 protein expression, an inhibitory nucleic acid targeting Lp(a) protein expression, an inhibitory nucleic acid targeting apoB 100, apoA-I up-regulator/inducer, ABCA 1 stabilizer or inducer, phospholipid transfer protein (PL TP) inhibitor, fish oil, anti-diabetic agent, antiobesity agent, agonists of peroxisome proliferator-activator receptors, ATP citrate lyase (ACL) inhibitor, and anti-hypertensive agents, an antibody targeting PCSK9, an immune checkpoint inhibitor and combinations thereof
41. A method for treating or preventing a disease or disorder comprising administering to a patient in need thereof a therapeutically effective amount of a compound according to any one of the claims 1 to 33, or a pharmaceutically acceptable salt thereof.
42. The method of claim 41, wherein the disease or disorder is a PCSK9-mediated disease or disorder.
43. The method of claim 42, wherein the PCSK9-mediated disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
44. A method for treating a disease or disorder comprising administering to a patient in need thereof a therapeutically effective amount of a compound according to any one of the claims 1 to 33, or a pharmaceutically acceptable salt thereof, wherein the disease or disorder is selected from selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
45. A method of modulating PCSK9 comprising administering to a patient in need thereof a compound of any one of claims 1 to 33 or a pharmaceutically acceptable salt thereof.
46. A method of inhibiting PCSK9 comprising administering to a patient in need thereof a compound of any one of claims 1 to 33 or a pharmaceutically acceptable salt thereof.
47. The method of any one of claims 41 to 46, wherein administering the compound is oral, parental, subcutaneous, by injection, or by infusion.
48. A compound according to any one of claims 1 to 33 or a pharmaceutically acceptable salt thereof, for use as a medicament.
49. A compound according to any one of claims 1 to 33 or a pharmaceutically acceptable salt thereof, for use in the treatment of a PCSK9-mediated disease or disorder.
50. The compound of claim 48, wherein the PCSK9-mediated disease or disorder selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
51. A compound according to any one of claims 1 to 33 or a pharmaceutically acceptable salt thereof, for use in the treatment of a disease or disorder, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
52. A compound according to any one of the claims 1 to 33, or a pharmaceutically acceptable salt thereof, for use in the manufacture of a medicament for treating of a PCSK9-mediated disease or disorder.
53. The compound for use in the manufacture of a medicament of claim 52, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
54. Use of a compound according to any one of claims 1 to 33, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating a PCSK9-mediated disease or disorder.
55. The use of claim 54, wherein said PCSK9-mediated disease or disorder selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
56. Use of a compound according to any one of claims 1 to 33, or a pharmaceutically acceptable salt thereof, in the treatment of a disease or disorder, wherein the disease or disorder is selected from hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, sitosterolemia, atherosclerosis, arteriosclerosis, coronary heart disease, peripheral vascular disease, vascular inflammation, xanthoma, peripheral arterial disease, elevated Lp(a), elevated LDL, elevated TRL, and elevated triglycerides.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163278754P | 2021-11-12 | 2021-11-12 | |
| US63/278,754 | 2021-11-12 | ||
| US202263325988P | 2022-03-31 | 2022-03-31 | |
| US63/325,988 | 2022-03-31 | ||
| US202263379562P | 2022-10-14 | 2022-10-14 | |
| US63/379,562 | 2022-10-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023084449A1 true WO2023084449A1 (en) | 2023-05-19 |
Family
ID=84362640
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2022/060845 WO2023084449A1 (en) | 2021-11-12 | 2022-11-10 | Diaminocyclopentylpyridine derivatives for the treatment of a disease or disorder |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202333563A (en) |
| WO (1) | WO2023084449A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024062090A1 (en) * | 2022-09-23 | 2024-03-28 | Astrazeneca Ab | Pcsk9 inhibitors and methods of use thereof |
| WO2024078620A1 (en) * | 2022-10-14 | 2024-04-18 | 上海翰森生物医药科技有限公司 | Nitrogen-containing heterocyclic derivative inhibitor, preparation method therefor and use thereof |
Citations (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4617307A (en) | 1984-06-20 | 1986-10-14 | Ciba-Geigy Corporation | Substituted imidazo[1,5-A]pyridine derivatives as aromatase inhibitors |
| US4889861A (en) | 1982-12-21 | 1989-12-26 | Ciba-Geigy Corp. | Substituted imidazo[1,5-a]pyridine derivatives and other substituted bicyclic derivatives and their use as aromatase inhibitors |
| WO1991011457A1 (en) | 1990-01-24 | 1991-08-08 | Buckley Douglas I | Glp-1 analogs useful for diabetes treatment |
| US5118666A (en) | 1986-05-05 | 1992-06-02 | The General Hospital Corporation | Insulinotropic hormone |
| US5120712A (en) | 1986-05-05 | 1992-06-09 | The General Hospital Corporation | Insulinotropic hormone |
| US5155100A (en) | 1991-05-01 | 1992-10-13 | Ciba-Geigy Corporation | Phosphono/biaryl substituted dipeptide derivatives |
| WO1995015309A1 (en) | 1993-12-03 | 1995-06-08 | Ferring B.V. | Dp-iv-serine protease inhibitors |
| US5512549A (en) | 1994-10-18 | 1996-04-30 | Eli Lilly And Company | Glucagon-like insulinotropic peptide analogs, compositions, and methods of use |
| DE19616486A1 (en) | 1996-04-25 | 1997-10-30 | Knoell Hans Forschung Ev | Method of lowering blood glucose levels in mammals |
| US5705483A (en) | 1993-12-09 | 1998-01-06 | Eli Lilly And Company | Glucagon-like insulinotropic peptides, compositions and methods |
| WO1998019998A2 (en) | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2000034241A1 (en) | 1998-12-10 | 2000-06-15 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2001076574A2 (en) | 2000-04-12 | 2001-10-18 | Novartis Ag | Novel medical use of aldosterone synthase inhibitors alone or in combination with at1-receptor antagonists |
| WO2003043985A1 (en) | 2001-11-21 | 2003-05-30 | Novartis Ag | Heterocyclic compounds and methods of use |
| WO2003104200A1 (en) | 2002-06-07 | 2003-12-18 | Glaxo Group Limited | N-mercaptoacyl phenyalanine derivatives, process for their preparation, and pharmaceutical compositions containing them |
| WO2004014914A1 (en) | 2002-08-07 | 2004-02-19 | Novartis Ag | Organic compounds as agents for the treatment of aldosterone mediated conditions |
| WO2004046145A1 (en) | 2002-11-18 | 2004-06-03 | Novartis Ag | Imidazo[1, 5a]pyridine derivatives and methods for treating aldosterone mediated diseases |
| WO2004103995A1 (en) | 2003-05-20 | 2004-12-02 | Novartis Ag | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2006005726A2 (en) | 2004-07-09 | 2006-01-19 | Speedel Experimenta Ag | Heterocyclic compounds |
| WO2006128851A1 (en) | 2005-05-31 | 2006-12-07 | Speedel Experimenta Ag | Fused imidazole derivatives and use thereof as aldosterone synthase inhibitors |
| WO2006128852A2 (en) | 2005-05-31 | 2006-12-07 | Speedel Experimenta Ag | Heterocyclic spiro-compounds as aldosterone synthase inhibitors |
| WO2006128853A1 (en) | 2005-05-31 | 2006-12-07 | Speedel Experimenta Ag | Heterocyclic spiro-compounds as aldosterone synthase inhibitors |
| US20070049616A1 (en) | 2005-08-25 | 2007-03-01 | Ksander Gary M | Organic compounds |
| WO2007065942A2 (en) | 2005-12-09 | 2007-06-14 | Speedel Experimenta Ag | Bis-heterocyclic imidazolyl compounds |
| US20070208035A1 (en) | 2004-05-28 | 2007-09-06 | Honda Motor Co., Ltd. | Heterocyclic compounds and their use as aldosterone synthase inhibitors |
| US20070225232A1 (en) | 2004-05-28 | 2007-09-27 | Peter Herold | Tetrahydro-Imidazo [1,5-A] Pyridyin Derivatives as Aldosterone Synthase Inhibitors |
| WO2007116099A1 (en) | 2006-04-12 | 2007-10-18 | Speedel Experimenta Ag | Imidazo compounds |
| WO2007116908A1 (en) | 2006-04-04 | 2007-10-18 | Taiyo Nippon Sanso Corporation | Method for separation of methane, methane separator, and methane utilization system |
| WO2007126957A2 (en) | 2006-03-31 | 2007-11-08 | Novartis Ag | New compounds |
| EP1886695A1 (en) | 2006-06-27 | 2008-02-13 | Speedel Experimenta AG | Pharmaceutical combination of an aldosterone synthase inhibitor and a glucocorticoid receptor antagonist or a cortisol synthesis inhibitor or a corticotropin releasing factor antagonist |
| WO2008027284A1 (en) | 2006-08-25 | 2008-03-06 | Novartis Ag | Fused imidazole derivatives for the treatment of disorders mediated by aldosterone synthase and/or 11-beta-hydroxylase and/or aromatase |
| US20080076794A1 (en) | 2004-05-28 | 2008-03-27 | Peter Herold | Heterocyclic Compounds And Their Use As Aldosterone Synthase Inhibitors |
| WO2008076336A2 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | Imidazoles as aldosterone synthase inhibitors |
| WO2008076860A1 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | 4-imidazolyl-1,2,3,4-tetrahydroquinoline derivatives and their use as aldosterone/11-beta-hydroxylase inhibitors |
| WO2008076862A2 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | 1-substituted imidazole derivatives and their use as aldosterone synthase inhibitors |
| WO2008119744A1 (en) | 2007-03-29 | 2008-10-09 | Novartis Ag | Heterocyclic spiro-compounds |
| WO2008133896A2 (en) | 2007-04-24 | 2008-11-06 | Theravance, Inc. | Dual-acting antihypertensive agents |
| US20090012068A1 (en) | 2006-04-12 | 2009-01-08 | Peter Herold | Condensed Imidazole Derivatives as Aldosterone Synthase Inhibitors |
| WO2009035543A1 (en) | 2007-09-07 | 2009-03-19 | Theravance, Inc. | Dual-acting antihypertensive agents |
| WO2009040410A1 (en) | 2007-09-28 | 2009-04-02 | Novartis Ag | Oxadiazole- and oxazole-substituted benzimidazole- and indole-derivatives as dgat1 inhibitors |
| WO2009134741A1 (en) | 2008-04-29 | 2009-11-05 | Theravance, Inc. | Dual-acting antihypertensive agents |
| WO2009156462A2 (en) | 2008-06-27 | 2009-12-30 | Novartis Ag | Organic compounds |
| WO2010130796A1 (en) | 2009-05-15 | 2010-11-18 | Novartis Ag | Aryl pyridine as aldosterone synthase inhibitors |
| WO2010136493A1 (en) | 2009-05-28 | 2010-12-02 | Novartis Ag | Substituted aminopropionic derivatives as neprilysin inhibitors |
| WO2010136474A2 (en) | 2009-05-28 | 2010-12-02 | Novartis Ag | Substituted aminobutyric derivatives as neprilysin inhibitors |
| WO2011061271A1 (en) | 2009-11-20 | 2011-05-26 | Novartis Ag | Substituted carbamoylmethylamino acetic acid derivatives as novel nep inhibitors |
| WO2012065953A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Substituted carbamoylcycloalkyl acetic acid derivatives as nep inhibitors |
| WO2012065956A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Substituted amino bisphenyl pentanoic acid derivatives as nep inhibitors |
| WO2012082853A1 (en) | 2010-12-15 | 2012-06-21 | Theravance, Inc. | Neprilysin inhibitors |
| WO2012112742A1 (en) | 2011-02-17 | 2012-08-23 | Theravance, Inc. | Substituted aminobutyric derivatives as neprilysin inhibitors |
| WO2012166387A1 (en) | 2011-05-31 | 2012-12-06 | Theravance, Inc. | Neprilysin inhibitors |
| WO2012166389A1 (en) | 2011-05-31 | 2012-12-06 | Theravance, Inc. | Neprilysin inhibitors |
| WO2013067163A1 (en) | 2011-11-02 | 2013-05-10 | Theravance, Inc. | Neprilysin inhibitors |
| WO2013184934A1 (en) | 2012-06-08 | 2013-12-12 | Theravance, Inc. | Neprilysin inhibitors |
| WO2014015965A1 (en) | 2012-07-21 | 2014-01-30 | Daimler Ag | Structure of an interior decor element |
| WO2014025891A1 (en) | 2012-08-08 | 2014-02-13 | Theravance, Inc. | Neprilysin inhibitors |
| WO2014126979A1 (en) | 2013-02-14 | 2014-08-21 | Novartis Ag | Substituted bisphenyl butanoic phosphonic acid derivatives as nep (neutral endopeptidase) inhibitors |
| WO2014138053A1 (en) | 2013-03-05 | 2014-09-12 | Theravance Biopharma R&D Ip, Llc | Neprilysin inhibitors |
| WO2015116786A1 (en) | 2014-01-30 | 2015-08-06 | Theravance Biopharma R&D Ip, Llc | Neprilysin inhibitors |
| WO2015116760A1 (en) | 2014-01-30 | 2015-08-06 | Theravance Biopharma R&D Ip, Llc | 5-biphenyl-4-heteroarylcarbonylamino-pentanoic acid derivatives as neprilysin inhibitors |
| WO2020150474A1 (en) * | 2019-01-18 | 2020-07-23 | Dogma Therapeutics, Inc. | Pcsk9 inhibitors and methods of use thereof |
| WO2020150473A2 (en) | 2019-01-18 | 2020-07-23 | Dogma Therapeutics, Inc. | Pcsk9 inhibitors and methods of use thereof |
-
2022
- 2022-11-10 WO PCT/IB2022/060845 patent/WO2023084449A1/en unknown
- 2022-11-10 TW TW111142990A patent/TW202333563A/en unknown
Patent Citations (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4889861A (en) | 1982-12-21 | 1989-12-26 | Ciba-Geigy Corp. | Substituted imidazo[1,5-a]pyridine derivatives and other substituted bicyclic derivatives and their use as aromatase inhibitors |
| US4617307A (en) | 1984-06-20 | 1986-10-14 | Ciba-Geigy Corporation | Substituted imidazo[1,5-A]pyridine derivatives as aromatase inhibitors |
| US5118666A (en) | 1986-05-05 | 1992-06-02 | The General Hospital Corporation | Insulinotropic hormone |
| US5120712A (en) | 1986-05-05 | 1992-06-09 | The General Hospital Corporation | Insulinotropic hormone |
| WO1991011457A1 (en) | 1990-01-24 | 1991-08-08 | Buckley Douglas I | Glp-1 analogs useful for diabetes treatment |
| US5155100A (en) | 1991-05-01 | 1992-10-13 | Ciba-Geigy Corporation | Phosphono/biaryl substituted dipeptide derivatives |
| WO1995015309A1 (en) | 1993-12-03 | 1995-06-08 | Ferring B.V. | Dp-iv-serine protease inhibitors |
| US5705483A (en) | 1993-12-09 | 1998-01-06 | Eli Lilly And Company | Glucagon-like insulinotropic peptides, compositions and methods |
| US5512549A (en) | 1994-10-18 | 1996-04-30 | Eli Lilly And Company | Glucagon-like insulinotropic peptide analogs, compositions, and methods of use |
| DE19616486A1 (en) | 1996-04-25 | 1997-10-30 | Knoell Hans Forschung Ev | Method of lowering blood glucose levels in mammals |
| WO1998019998A2 (en) | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2000034241A1 (en) | 1998-12-10 | 2000-06-15 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2001076574A2 (en) | 2000-04-12 | 2001-10-18 | Novartis Ag | Novel medical use of aldosterone synthase inhibitors alone or in combination with at1-receptor antagonists |
| WO2003043985A1 (en) | 2001-11-21 | 2003-05-30 | Novartis Ag | Heterocyclic compounds and methods of use |
| WO2003104200A1 (en) | 2002-06-07 | 2003-12-18 | Glaxo Group Limited | N-mercaptoacyl phenyalanine derivatives, process for their preparation, and pharmaceutical compositions containing them |
| WO2004014914A1 (en) | 2002-08-07 | 2004-02-19 | Novartis Ag | Organic compounds as agents for the treatment of aldosterone mediated conditions |
| WO2004046145A1 (en) | 2002-11-18 | 2004-06-03 | Novartis Ag | Imidazo[1, 5a]pyridine derivatives and methods for treating aldosterone mediated diseases |
| WO2004103995A1 (en) | 2003-05-20 | 2004-12-02 | Novartis Ag | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| US20080076794A1 (en) | 2004-05-28 | 2008-03-27 | Peter Herold | Heterocyclic Compounds And Their Use As Aldosterone Synthase Inhibitors |
| US20070208035A1 (en) | 2004-05-28 | 2007-09-06 | Honda Motor Co., Ltd. | Heterocyclic compounds and their use as aldosterone synthase inhibitors |
| US20070225232A1 (en) | 2004-05-28 | 2007-09-27 | Peter Herold | Tetrahydro-Imidazo [1,5-A] Pyridyin Derivatives as Aldosterone Synthase Inhibitors |
| US20080318978A2 (en) | 2004-05-28 | 2008-12-25 | Speedel Experimenta Ag | Heterocyclic compounds and their use as aldosterone synthase inhibitors |
| WO2006005726A2 (en) | 2004-07-09 | 2006-01-19 | Speedel Experimenta Ag | Heterocyclic compounds |
| WO2006128851A1 (en) | 2005-05-31 | 2006-12-07 | Speedel Experimenta Ag | Fused imidazole derivatives and use thereof as aldosterone synthase inhibitors |
| WO2006128852A2 (en) | 2005-05-31 | 2006-12-07 | Speedel Experimenta Ag | Heterocyclic spiro-compounds as aldosterone synthase inhibitors |
| WO2006128853A1 (en) | 2005-05-31 | 2006-12-07 | Speedel Experimenta Ag | Heterocyclic spiro-compounds as aldosterone synthase inhibitors |
| US20070049616A1 (en) | 2005-08-25 | 2007-03-01 | Ksander Gary M | Organic compounds |
| WO2007065942A2 (en) | 2005-12-09 | 2007-06-14 | Speedel Experimenta Ag | Bis-heterocyclic imidazolyl compounds |
| WO2007126957A2 (en) | 2006-03-31 | 2007-11-08 | Novartis Ag | New compounds |
| WO2007116908A1 (en) | 2006-04-04 | 2007-10-18 | Taiyo Nippon Sanso Corporation | Method for separation of methane, methane separator, and methane utilization system |
| WO2007116099A1 (en) | 2006-04-12 | 2007-10-18 | Speedel Experimenta Ag | Imidazo compounds |
| US20090048241A1 (en) | 2006-04-12 | 2009-02-19 | Speedel Experimenta Ag | Condensed Imidazole Derivatives as Aromatase Inhibitors |
| US20090012068A1 (en) | 2006-04-12 | 2009-01-08 | Peter Herold | Condensed Imidazole Derivatives as Aldosterone Synthase Inhibitors |
| EP1886695A1 (en) | 2006-06-27 | 2008-02-13 | Speedel Experimenta AG | Pharmaceutical combination of an aldosterone synthase inhibitor and a glucocorticoid receptor antagonist or a cortisol synthesis inhibitor or a corticotropin releasing factor antagonist |
| WO2008027284A1 (en) | 2006-08-25 | 2008-03-06 | Novartis Ag | Fused imidazole derivatives for the treatment of disorders mediated by aldosterone synthase and/or 11-beta-hydroxylase and/or aromatase |
| WO2008076336A2 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | Imidazoles as aldosterone synthase inhibitors |
| WO2008076862A2 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | 1-substituted imidazole derivatives and their use as aldosterone synthase inhibitors |
| WO2008076860A1 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | 4-imidazolyl-1,2,3,4-tetrahydroquinoline derivatives and their use as aldosterone/11-beta-hydroxylase inhibitors |
| WO2008119744A1 (en) | 2007-03-29 | 2008-10-09 | Novartis Ag | Heterocyclic spiro-compounds |
| WO2008133896A2 (en) | 2007-04-24 | 2008-11-06 | Theravance, Inc. | Dual-acting antihypertensive agents |
| WO2009035543A1 (en) | 2007-09-07 | 2009-03-19 | Theravance, Inc. | Dual-acting antihypertensive agents |
| WO2009040410A1 (en) | 2007-09-28 | 2009-04-02 | Novartis Ag | Oxadiazole- and oxazole-substituted benzimidazole- and indole-derivatives as dgat1 inhibitors |
| WO2009134741A1 (en) | 2008-04-29 | 2009-11-05 | Theravance, Inc. | Dual-acting antihypertensive agents |
| WO2009156462A2 (en) | 2008-06-27 | 2009-12-30 | Novartis Ag | Organic compounds |
| WO2010130796A1 (en) | 2009-05-15 | 2010-11-18 | Novartis Ag | Aryl pyridine as aldosterone synthase inhibitors |
| WO2010136493A1 (en) | 2009-05-28 | 2010-12-02 | Novartis Ag | Substituted aminopropionic derivatives as neprilysin inhibitors |
| WO2010136474A2 (en) | 2009-05-28 | 2010-12-02 | Novartis Ag | Substituted aminobutyric derivatives as neprilysin inhibitors |
| WO2011061271A1 (en) | 2009-11-20 | 2011-05-26 | Novartis Ag | Substituted carbamoylmethylamino acetic acid derivatives as novel nep inhibitors |
| WO2012065953A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Substituted carbamoylcycloalkyl acetic acid derivatives as nep inhibitors |
| WO2012065956A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Substituted amino bisphenyl pentanoic acid derivatives as nep inhibitors |
| WO2012082853A1 (en) | 2010-12-15 | 2012-06-21 | Theravance, Inc. | Neprilysin inhibitors |
| WO2012112742A1 (en) | 2011-02-17 | 2012-08-23 | Theravance, Inc. | Substituted aminobutyric derivatives as neprilysin inhibitors |
| WO2012166387A1 (en) | 2011-05-31 | 2012-12-06 | Theravance, Inc. | Neprilysin inhibitors |
| WO2012166389A1 (en) | 2011-05-31 | 2012-12-06 | Theravance, Inc. | Neprilysin inhibitors |
| WO2013067163A1 (en) | 2011-11-02 | 2013-05-10 | Theravance, Inc. | Neprilysin inhibitors |
| WO2013184934A1 (en) | 2012-06-08 | 2013-12-12 | Theravance, Inc. | Neprilysin inhibitors |
| WO2014015965A1 (en) | 2012-07-21 | 2014-01-30 | Daimler Ag | Structure of an interior decor element |
| WO2014025891A1 (en) | 2012-08-08 | 2014-02-13 | Theravance, Inc. | Neprilysin inhibitors |
| WO2014126979A1 (en) | 2013-02-14 | 2014-08-21 | Novartis Ag | Substituted bisphenyl butanoic phosphonic acid derivatives as nep (neutral endopeptidase) inhibitors |
| WO2014138053A1 (en) | 2013-03-05 | 2014-09-12 | Theravance Biopharma R&D Ip, Llc | Neprilysin inhibitors |
| WO2015116786A1 (en) | 2014-01-30 | 2015-08-06 | Theravance Biopharma R&D Ip, Llc | Neprilysin inhibitors |
| WO2015116760A1 (en) | 2014-01-30 | 2015-08-06 | Theravance Biopharma R&D Ip, Llc | 5-biphenyl-4-heteroarylcarbonylamino-pentanoic acid derivatives as neprilysin inhibitors |
| WO2020150474A1 (en) * | 2019-01-18 | 2020-07-23 | Dogma Therapeutics, Inc. | Pcsk9 inhibitors and methods of use thereof |
| WO2020150473A2 (en) | 2019-01-18 | 2020-07-23 | Dogma Therapeutics, Inc. | Pcsk9 inhibitors and methods of use thereof |
Non-Patent Citations (22)
| Title |
|---|
| "Goodman and Gilman's The Pharmacological Basis of Therapeutics", 2006, MCGRAW-HILL |
| "Physicians' Desk Reference (PDR)", 2009 |
| ABIFADEL, M. ET AL., NATURE, vol. 34, 2003, pages 154 - 6 |
| BURNETTHOOPER, CLIN. BIOCHEM. REV, vol. 29, no. 1, 2008, pages 11 - 26 |
| C. ORSKOV ET AL., J. BIOL. CHEM., vol. 264, 1989, pages 12826 |
| CAO ET AL., ASSAY DRUG DEV. TECHNOL., vol. 8, 2010, pages 766 - 780 |
| CASES ET AL., J. BIOL. CHEM., vol. 276, 2001, pages 38870 - 38876 |
| CASES ET AL., PROC. NATL. ACAD. SCI., vol. 95, 1998, pages 13018 - 13023 |
| COHEN, J. C. ET AL., N. ENGL. J. MED., vol. 354, 2006, pages 1264 - 1272 |
| DESAI, N.R., CIRCULATION, vol. 128, no. 9, 2013, pages 962 - 969 |
| DWIVEDI, D. J. ET AL., SHOCK, vol. 46, no. 6, 2016, pages 672 - 680 |
| E. L. ELIELS. H. WILENL. N. MANDER: "Stereochemistry of Organic Compounds", 1994, WILEY-LNTERSCIENCE |
| GREIG ET AL., DIABETOLOGIA, vol. 42, 1999, pages 45 - 50 |
| GRIN, P.M. ET AL., NATURE, vol. 8, no. 1, 2018, pages 10496 |
| LAGACE T.A., J. CLIN. INVEST., vol. 116, no. 11, 2006, pages 2995 - 3005 |
| LAMBERT, G., CLIN. SCI., vol. 131, 2017, pages 261 - 268 |
| MAXWELL K. N., PROC. NATL. ACAD. SCI., vol. 101, 2004, pages 7100 - 7105 |
| PARK, S. W., J. BIOL. CHEM., vol. 279, 2004, pages 50630 - 50638 |
| RASHID, S. ET AL., PROC. NATL. ACAD. SCI., vol. 102, 2005, pages 5374 - 5379 |
| SMITH ET AL., NATURE GENETICS, vol. 25, 2000, pages 87 - 90 |
| T. W. GREENP. G. M. WUTS: "Protective Groups in Organic Synthesis", 1999, JOHN WILEY & SONS |
| W.E. SCHMIDT ET AL., DIABETOLOGIA, vol. 28, 1985, pages 704 - 707 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024062090A1 (en) * | 2022-09-23 | 2024-03-28 | Astrazeneca Ab | Pcsk9 inhibitors and methods of use thereof |
| WO2024078620A1 (en) * | 2022-10-14 | 2024-04-18 | 上海翰森生物医药科技有限公司 | Nitrogen-containing heterocyclic derivative inhibitor, preparation method therefor and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202333563A (en) | 2023-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2023084449A1 (en) | Diaminocyclopentylpyridine derivatives for the treatment of a disease or disorder | |
| CN102666493B (en) | As the carbamo, lmethyl aminobenzoic acid derivative of the replacement of nep inhibitor | |
| JP6295277B2 (en) | Substituted bisphenylbutanoic acid phosphonic acid derivatives as NEP (neutral endopeptidase) inhibitors | |
| AU2009279874B2 (en) | Oxazole derivatives useful as inhibitors of FAAH | |
| EP2852590B1 (en) | Cyclic bridgehead ether dgat1 inhibitors | |
| JP6310581B2 (en) | Methods for treating atherosclerosis in subjects with high triglycerides | |
| JP2014532066A (en) | Oxazine derivatives and their use in the treatment of neurological disorders | |
| JP2010520199A (en) | Piperidine derivatives and methods of use thereof | |
| EP3887363A1 (en) | Cyclic pentamer compounds as proprotein convertase subtilisin/kexin type 9 (pcsk9) inhibitors for the treatment of metabolic disorder | |
| JP2020502058A (en) | Indazole derivatives useful as inhibitors of diacylglyceride O-acyltransferase 2 | |
| US11813306B2 (en) | Cyclic tetramer compounds as proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors for the treatment of metabolic disorders | |
| EP3887388A1 (en) | Cyclic peptides as proprotein convertase subtilisin/kexin type 9 (pcsk9) inhibitors for the treatment of metabolic disorders | |
| JP6301371B2 (en) | Substituted bisphenylbutanoic acid derivatives as NEP inhibitors with improved in vivo efficacy | |
| JP7577656B2 (en) | Cyclic tetramer compounds as proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors for the treatment of metabolic disorders | |
| JP2018505885A (en) | Substituted pyrazolo [1,5-a] -pyridine-3-carboxamide and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22812779 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |