WO2022222963A1 - 含咪唑稠环类衍生物、其制备方法及其在医药上的应用 - Google Patents
含咪唑稠环类衍生物、其制备方法及其在医药上的应用 Download PDFInfo
- Publication number
- WO2022222963A1 WO2022222963A1 PCT/CN2022/087947 CN2022087947W WO2022222963A1 WO 2022222963 A1 WO2022222963 A1 WO 2022222963A1 CN 2022087947 W CN2022087947 W CN 2022087947W WO 2022222963 A1 WO2022222963 A1 WO 2022222963A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- methyl
- formula
- heteroaryl
- substituted
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 186
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 title claims description 21
- 239000003814 drug Substances 0.000 title claims description 10
- 150000001875 compounds Chemical class 0.000 claims abstract description 212
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 claims abstract description 69
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 54
- 201000010099 disease Diseases 0.000 claims abstract description 36
- 239000000651 prodrug Substances 0.000 claims abstract description 32
- 229940002612 prodrug Drugs 0.000 claims abstract description 32
- 150000003839 salts Chemical class 0.000 claims abstract description 28
- 239000012453 solvate Substances 0.000 claims abstract description 23
- 208000035475 disorder Diseases 0.000 claims abstract description 17
- 108010040716 Neurokinin-3 Receptors Proteins 0.000 claims abstract description 15
- 230000001419 dependent effect Effects 0.000 claims abstract description 13
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 12
- 102000002003 Neurokinin-3 Receptors Human genes 0.000 claims abstract description 11
- 208000008589 Obesity Diseases 0.000 claims abstract description 8
- 208000028017 Psychotic disease Diseases 0.000 claims abstract description 8
- 239000003163 gonadal steroid hormone Substances 0.000 claims abstract description 8
- 235000020824 obesity Nutrition 0.000 claims abstract description 8
- 208000002193 Pain Diseases 0.000 claims abstract description 6
- 230000001850 reproductive effect Effects 0.000 claims abstract description 6
- 208000019901 Anxiety disease Diseases 0.000 claims abstract description 5
- 201000000980 schizophrenia Diseases 0.000 claims abstract description 5
- 208000024827 Alzheimer disease Diseases 0.000 claims abstract description 4
- 208000006096 Attention Deficit Disorder with Hyperactivity Diseases 0.000 claims abstract description 4
- 208000020925 Bipolar disease Diseases 0.000 claims abstract description 4
- 206010010904 Convulsion Diseases 0.000 claims abstract description 4
- 208000018737 Parkinson disease Diseases 0.000 claims abstract description 4
- 206010047700 Vomiting Diseases 0.000 claims abstract description 4
- 208000010877 cognitive disease Diseases 0.000 claims abstract description 4
- 230000036461 convulsion Effects 0.000 claims abstract description 4
- 201000011461 pre-eclampsia Diseases 0.000 claims abstract description 4
- 230000008673 vomiting Effects 0.000 claims abstract description 4
- -1 R 21 Chemical compound 0.000 claims description 373
- 125000000217 alkyl group Chemical group 0.000 claims description 133
- 125000000623 heterocyclic group Chemical group 0.000 claims description 117
- 125000001072 heteroaryl group Chemical group 0.000 claims description 116
- 125000003118 aryl group Chemical group 0.000 claims description 104
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 83
- 229910052736 halogen Inorganic materials 0.000 claims description 74
- 150000002367 halogens Chemical class 0.000 claims description 74
- 229910052757 nitrogen Inorganic materials 0.000 claims description 72
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 71
- 125000001424 substituent group Chemical group 0.000 claims description 71
- 125000003342 alkenyl group Chemical group 0.000 claims description 64
- 125000000304 alkynyl group Chemical group 0.000 claims description 60
- 125000004043 oxo group Chemical group O=* 0.000 claims description 57
- 238000000034 method Methods 0.000 claims description 30
- 125000003545 alkoxy group Chemical group 0.000 claims description 28
- 125000001188 haloalkyl group Chemical group 0.000 claims description 28
- 229910052739 hydrogen Inorganic materials 0.000 claims description 27
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 25
- 125000004432 carbon atom Chemical group C* 0.000 claims description 24
- 229910052805 deuterium Inorganic materials 0.000 claims description 24
- 125000003282 alkyl amino group Chemical group 0.000 claims description 22
- 229910052801 chlorine Inorganic materials 0.000 claims description 21
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 21
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 21
- 229910052794 bromium Inorganic materials 0.000 claims description 20
- 229910052731 fluorine Inorganic materials 0.000 claims description 20
- 229910052740 iodine Inorganic materials 0.000 claims description 19
- 125000002950 monocyclic group Chemical group 0.000 claims description 17
- 229910052717 sulfur Inorganic materials 0.000 claims description 17
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 16
- 230000000155 isotopic effect Effects 0.000 claims description 15
- 239000002207 metabolite Substances 0.000 claims description 15
- 229910052760 oxygen Inorganic materials 0.000 claims description 15
- 125000002619 bicyclic group Chemical group 0.000 claims description 14
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 12
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 10
- 238000006467 substitution reaction Methods 0.000 claims description 10
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 9
- 208000004403 Prostatic Hyperplasia Diseases 0.000 claims description 9
- 125000002947 alkylene group Chemical group 0.000 claims description 9
- 239000003098 androgen Substances 0.000 claims description 9
- 125000004193 piperazinyl group Chemical group 0.000 claims description 9
- 125000003386 piperidinyl group Chemical group 0.000 claims description 9
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 9
- 208000024891 symptom Diseases 0.000 claims description 9
- 125000002757 morpholinyl group Chemical group 0.000 claims description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 8
- 125000001544 thienyl group Chemical group 0.000 claims description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 7
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 claims description 7
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims description 7
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 7
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims description 7
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims description 7
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 claims description 7
- 125000005045 dihydroisoquinolinyl group Chemical group C1(NC=CC2=CC=CC=C12)* 0.000 claims description 7
- 125000002541 furyl group Chemical group 0.000 claims description 7
- 125000002883 imidazolyl group Chemical group 0.000 claims description 7
- 125000001041 indolyl group Chemical group 0.000 claims description 7
- 125000001624 naphthyl group Chemical group 0.000 claims description 7
- 125000002971 oxazolyl group Chemical group 0.000 claims description 7
- 125000006239 protecting group Chemical group 0.000 claims description 7
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 7
- 125000004076 pyridyl group Chemical group 0.000 claims description 7
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 7
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 claims description 7
- 125000000335 thiazolyl group Chemical group 0.000 claims description 7
- 125000004306 triazinyl group Chemical group 0.000 claims description 7
- YNGDWRXWKFWCJY-UHFFFAOYSA-N 1,4-Dihydropyridine Chemical compound C1C=CNC=C1 YNGDWRXWKFWCJY-UHFFFAOYSA-N 0.000 claims description 6
- UUAVYXKRUSVCDJ-UHFFFAOYSA-N 1-cycloheptylazepane Chemical compound C1CCCCCC1N1CCCCCC1 UUAVYXKRUSVCDJ-UHFFFAOYSA-N 0.000 claims description 6
- QUIJMHDIAFOPRK-UHFFFAOYSA-N 1-cyclooctylazocane Chemical compound C1CCCCCCC1N1CCCCCCC1 QUIJMHDIAFOPRK-UHFFFAOYSA-N 0.000 claims description 6
- VSWICNJIUPRZIK-UHFFFAOYSA-N 2-piperideine Chemical compound C1CNC=CC1 VSWICNJIUPRZIK-UHFFFAOYSA-N 0.000 claims description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 6
- 206010028980 Neoplasm Diseases 0.000 claims description 6
- 208000027030 Premenstrual dysphoric disease Diseases 0.000 claims description 6
- 125000003725 azepanyl group Chemical group 0.000 claims description 6
- 125000002618 bicyclic heterocycle group Chemical group 0.000 claims description 6
- 125000005044 dihydroquinolinyl group Chemical group N1(CC=CC2=CC=CC=C12)* 0.000 claims description 6
- 201000010066 hyperandrogenism Diseases 0.000 claims description 6
- 206010020718 hyperplasia Diseases 0.000 claims description 6
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 claims description 6
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 claims description 6
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 claims description 6
- 230000002611 ovarian Effects 0.000 claims description 6
- 125000000160 oxazolidinyl group Chemical group 0.000 claims description 6
- 201000010065 polycystic ovary syndrome Diseases 0.000 claims description 6
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 6
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 6
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 claims description 6
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 claims description 6
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 claims description 6
- 210000003684 theca cell Anatomy 0.000 claims description 6
- 125000001113 thiadiazolyl group Chemical group 0.000 claims description 6
- 125000004305 thiazinyl group Chemical group S1NC(=CC=C1)* 0.000 claims description 6
- 125000001984 thiazolidinyl group Chemical group 0.000 claims description 6
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 5
- 208000033830 Hot Flashes Diseases 0.000 claims description 5
- 206010060800 Hot flush Diseases 0.000 claims description 5
- WCZVZNOTHYJIEI-UHFFFAOYSA-N cinnoline Chemical compound N1=NC=CC2=CC=CC=C21 WCZVZNOTHYJIEI-UHFFFAOYSA-N 0.000 claims description 5
- 125000002632 imidazolidinyl group Chemical group 0.000 claims description 5
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 5
- 230000035900 sweating Effects 0.000 claims description 5
- 125000001698 2H-pyranyl group Chemical group O1C(C=CC=C1)* 0.000 claims description 4
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 4
- 206010027304 Menopausal symptoms Diseases 0.000 claims description 4
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 4
- 125000002393 azetidinyl group Chemical group 0.000 claims description 4
- 208000002173 dizziness Diseases 0.000 claims description 4
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 claims description 4
- 239000005556 hormone Substances 0.000 claims description 4
- 229940088597 hormone Drugs 0.000 claims description 4
- 150000002460 imidazoles Chemical class 0.000 claims description 4
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 claims description 4
- 208000000509 infertility Diseases 0.000 claims description 4
- 231100000535 infertility Toxicity 0.000 claims description 4
- 230000036512 infertility Effects 0.000 claims description 4
- 230000002265 prevention Effects 0.000 claims description 4
- 150000004867 thiadiazoles Chemical class 0.000 claims description 4
- 150000003557 thiazoles Chemical class 0.000 claims description 4
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 3
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 claims description 3
- 208000002874 Acne Vulgaris Diseases 0.000 claims description 3
- 208000005641 Adenomyosis Diseases 0.000 claims description 3
- 201000004384 Alopecia Diseases 0.000 claims description 3
- 201000005670 Anovulation Diseases 0.000 claims description 3
- 206010002659 Anovulatory cycle Diseases 0.000 claims description 3
- 206010006187 Breast cancer Diseases 0.000 claims description 3
- 208000026310 Breast neoplasm Diseases 0.000 claims description 3
- 206010006482 Bronchospasm Diseases 0.000 claims description 3
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 3
- 206010011224 Cough Diseases 0.000 claims description 3
- 206010013908 Dysfunctional uterine bleeding Diseases 0.000 claims description 3
- 208000005171 Dysmenorrhea Diseases 0.000 claims description 3
- 206010013935 Dysmenorrhoea Diseases 0.000 claims description 3
- 208000010254 HAIR-AN syndrome Diseases 0.000 claims description 3
- 206010020112 Hirsutism Diseases 0.000 claims description 3
- 208000008454 Hyperhidrosis Diseases 0.000 claims description 3
- 206010022489 Insulin Resistance Diseases 0.000 claims description 3
- 208000035771 Malignant Sertoli-Leydig cell tumor of the ovary Diseases 0.000 claims description 3
- 206010027514 Metrorrhagia Diseases 0.000 claims description 3
- 206010033128 Ovarian cancer Diseases 0.000 claims description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 3
- 206010033557 Palpitations Diseases 0.000 claims description 3
- 208000002200 Respiratory Hypersensitivity Diseases 0.000 claims description 3
- 208000024313 Testicular Neoplasms Diseases 0.000 claims description 3
- 206010057644 Testis cancer Diseases 0.000 claims description 3
- 206010067269 Uterine fibrosis Diseases 0.000 claims description 3
- 206010046798 Uterine leiomyoma Diseases 0.000 claims description 3
- 206010047486 Virilism Diseases 0.000 claims description 3
- 201000010272 acanthosis nigricans Diseases 0.000 claims description 3
- 206010000496 acne Diseases 0.000 claims description 3
- 208000024447 adrenal gland neoplasm Diseases 0.000 claims description 3
- 230000010085 airway hyperresponsiveness Effects 0.000 claims description 3
- 229910021529 ammonia Inorganic materials 0.000 claims description 3
- 206010068168 androgenetic alopecia Diseases 0.000 claims description 3
- 231100000552 anovulation Toxicity 0.000 claims description 3
- 208000006673 asthma Diseases 0.000 claims description 3
- 230000007885 bronchoconstriction Effects 0.000 claims description 3
- 239000003433 contraceptive agent Substances 0.000 claims description 3
- 230000002254 contraceptive effect Effects 0.000 claims description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 3
- SXZIXHOMFPUIRK-UHFFFAOYSA-N diphenylmethanimine Chemical compound C=1C=CC=CC=1C(=N)C1=CC=CC=C1 SXZIXHOMFPUIRK-UHFFFAOYSA-N 0.000 claims description 3
- 201000009274 endometriosis of uterus Diseases 0.000 claims description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 3
- 230000003325 follicular Effects 0.000 claims description 3
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 3
- 125000005350 hydroxycycloalkyl group Chemical group 0.000 claims description 3
- 125000002636 imidazolinyl group Chemical group 0.000 claims description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- 125000003965 isoxazolidinyl group Chemical group 0.000 claims description 3
- 201000010260 leiomyoma Diseases 0.000 claims description 3
- 208000017717 malignant Sertoli-Leydig cell tumor of ovary Diseases 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 230000035800 maturation Effects 0.000 claims description 3
- 230000001404 mediated effect Effects 0.000 claims description 3
- 208000007106 menorrhagia Diseases 0.000 claims description 3
- 208000010658 metastatic prostate carcinoma Diseases 0.000 claims description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 3
- 150000004866 oxadiazoles Chemical class 0.000 claims description 3
- 150000002916 oxazoles Chemical class 0.000 claims description 3
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- 201000003120 testicular cancer Diseases 0.000 claims description 3
- 150000003852 triazoles Chemical class 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 3
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 3
- 229920002554 vinyl polymer Polymers 0.000 claims description 3
- 208000028698 Cognitive impairment Diseases 0.000 claims description 2
- 206010061218 Inflammation Diseases 0.000 claims description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 2
- 230000004054 inflammatory process Effects 0.000 claims description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 239000011593 sulfur Chemical group 0.000 claims description 2
- 125000000446 sulfanediyl group Chemical group *S* 0.000 claims 35
- SCVJRXQHFJXZFZ-KVQBGUIXSA-N 2-amino-9-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purine-6-thione Chemical compound C1=2NC(N)=NC(=S)C=2N=CN1[C@H]1C[C@H](O)[C@@H](CO)O1 SCVJRXQHFJXZFZ-KVQBGUIXSA-N 0.000 claims 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N benzopyrrole Natural products C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims 2
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 claims 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims 2
- DHHVAGZRUROJKS-UHFFFAOYSA-N phentermine Chemical compound CC(C)(N)CC1=CC=CC=C1 DHHVAGZRUROJKS-UHFFFAOYSA-N 0.000 claims 2
- USPWKWBDZOARPV-UHFFFAOYSA-N pyrazolidine Chemical compound C1CNNC1 USPWKWBDZOARPV-UHFFFAOYSA-N 0.000 claims 2
- 125000002755 pyrazolinyl group Chemical group 0.000 claims 2
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 claims 2
- 208000036864 Attention deficit/hyperactivity disease Diseases 0.000 claims 1
- 201000004458 Myoma Diseases 0.000 claims 1
- 230000005856 abnormality Effects 0.000 claims 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims 1
- 208000015891 sexual disease Diseases 0.000 claims 1
- 208000027866 inflammatory disease Diseases 0.000 abstract description 3
- 239000002464 receptor antagonist Substances 0.000 abstract description 2
- 229940044551 receptor antagonist Drugs 0.000 abstract description 2
- 239000003550 marker Substances 0.000 abstract 1
- 208000020016 psychiatric disease Diseases 0.000 abstract 1
- 208000011580 syndromic disease Diseases 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 340
- 238000006243 chemical reaction Methods 0.000 description 326
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 275
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 270
- 239000000243 solution Substances 0.000 description 251
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 180
- 239000007787 solid Substances 0.000 description 143
- 238000005481 NMR spectroscopy Methods 0.000 description 138
- 235000019439 ethyl acetate Nutrition 0.000 description 114
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 112
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 112
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 91
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 85
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 76
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 72
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 63
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 62
- 239000000706 filtrate Substances 0.000 description 60
- 230000002829 reductive effect Effects 0.000 description 58
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 57
- 239000003208 petroleum Substances 0.000 description 56
- 239000012071 phase Substances 0.000 description 56
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 53
- 238000004128 high performance liquid chromatography Methods 0.000 description 52
- 230000002441 reversible effect Effects 0.000 description 50
- 239000000741 silica gel Substances 0.000 description 50
- 229910002027 silica gel Inorganic materials 0.000 description 50
- 239000002904 solvent Substances 0.000 description 42
- 239000012074 organic phase Substances 0.000 description 38
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 36
- 229910000027 potassium carbonate Inorganic materials 0.000 description 36
- WPJHMVKYTPHBGI-SECBINFHSA-N C[C@H](C(N(CC1)C(C2=NC(C)=NS2)=N2)=C2Br)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C(N(CC1)C(C2=NC(C)=NS2)=N2)=C2Br)N1C(C(C=C1)=CC=C1F)=O WPJHMVKYTPHBGI-SECBINFHSA-N 0.000 description 35
- 235000011181 potassium carbonates Nutrition 0.000 description 35
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 35
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 29
- SRWFWQFRAOFHNK-UHFFFAOYSA-N (4-fluorophenyl)methanone Chemical compound FC1=CC=C([C+]=O)C=C1 SRWFWQFRAOFHNK-UHFFFAOYSA-N 0.000 description 28
- 239000000047 product Substances 0.000 description 28
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 27
- 239000012043 crude product Substances 0.000 description 25
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 23
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 23
- 238000000746 purification Methods 0.000 description 23
- 239000000460 chlorine Substances 0.000 description 22
- 125000004149 thio group Chemical group *S* 0.000 description 22
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 21
- 238000004440 column chromatography Methods 0.000 description 21
- CKJNUZNMWOVDFN-UHFFFAOYSA-N methanone Chemical compound O=[CH-] CKJNUZNMWOVDFN-UHFFFAOYSA-N 0.000 description 21
- 239000000203 mixture Substances 0.000 description 21
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical class [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 19
- 235000002639 sodium chloride Nutrition 0.000 description 19
- 238000003756 stirring Methods 0.000 description 19
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 18
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 17
- 238000002953 preparative HPLC Methods 0.000 description 17
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 16
- OSBZGWVELDSHNH-SECBINFHSA-N C[C@H](C1=C(N)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O OSBZGWVELDSHNH-SECBINFHSA-N 0.000 description 16
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- 238000001914 filtration Methods 0.000 description 15
- 239000011541 reaction mixture Substances 0.000 description 15
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 13
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 13
- 229920006395 saturated elastomer Polymers 0.000 description 13
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 238000000605 extraction Methods 0.000 description 12
- 239000007788 liquid Substances 0.000 description 12
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 12
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 11
- 238000010791 quenching Methods 0.000 description 11
- 238000002390 rotary evaporation Methods 0.000 description 11
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 10
- 229910000024 caesium carbonate Inorganic materials 0.000 description 10
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 10
- 125000001183 hydrocarbyl group Chemical group 0.000 description 10
- 238000000926 separation method Methods 0.000 description 10
- JRHPOFJADXHYBR-HTQZYQBOSA-N (1r,2r)-1-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CN[C@@H]1CCCC[C@H]1NC JRHPOFJADXHYBR-HTQZYQBOSA-N 0.000 description 9
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 9
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 9
- 239000012046 mixed solvent Substances 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 238000010898 silica gel chromatography Methods 0.000 description 9
- 229910000104 sodium hydride Inorganic materials 0.000 description 9
- NGYCJCKQJNBHTQ-UHFFFAOYSA-N CC(C1=CN=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=CN=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O NGYCJCKQJNBHTQ-UHFFFAOYSA-N 0.000 description 8
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- 210000003169 central nervous system Anatomy 0.000 description 8
- 125000005842 heteroatom Chemical group 0.000 description 8
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 8
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 8
- 239000012141 concentrate Substances 0.000 description 7
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 7
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 7
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- GNZMAFXEWPQWNQ-CQSZACIVSA-N C[C@H](C1=C(NC(C2=CC=CC=C2)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(NC(C2=CC=CC=C2)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O GNZMAFXEWPQWNQ-CQSZACIVSA-N 0.000 description 6
- LTJCXXQGLJXOGJ-LLVKDONJSA-N C[C@H](C1=C(NC(C2CC2)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(NC(C2CC2)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O LTJCXXQGLJXOGJ-LLVKDONJSA-N 0.000 description 6
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 6
- 150000001204 N-oxides Chemical class 0.000 description 6
- NHXYSAFTNPANFK-HDMCBQFHSA-N Neurokinin B Chemical compound C([C@@H](C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCSC)NC(=O)[C@@H](N)CC(O)=O)C1=CC=CC=C1 NHXYSAFTNPANFK-HDMCBQFHSA-N 0.000 description 6
- 102000046798 Neurokinin B Human genes 0.000 description 6
- 101800002813 Neurokinin-B Proteins 0.000 description 6
- 102100029409 Neuromedin-K receptor Human genes 0.000 description 6
- 102100024304 Protachykinin-1 Human genes 0.000 description 6
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 6
- 239000012346 acetyl chloride Substances 0.000 description 6
- 150000001721 carbon Chemical group 0.000 description 6
- 238000003818 flash chromatography Methods 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 6
- 238000012544 monitoring process Methods 0.000 description 6
- 230000002093 peripheral effect Effects 0.000 description 6
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 6
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 6
- 239000012312 sodium hydride Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- UTUQJIYKGCKQHD-UHFFFAOYSA-N (3-methylpyrazin-2-yl)methanamine Chemical compound CC1=NC=CN=C1CN UTUQJIYKGCKQHD-UHFFFAOYSA-N 0.000 description 5
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 5
- WUZZVDDVPKHJJM-UHFFFAOYSA-N CC(C1=C(C(O)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(C(O)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O WUZZVDDVPKHJJM-UHFFFAOYSA-N 0.000 description 5
- HFZPXUOGZJLHST-UHFFFAOYSA-N CC(C1=C(N(C)C(C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(N(C)C(C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O HFZPXUOGZJLHST-UHFFFAOYSA-N 0.000 description 5
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 5
- KAXDDEGPGAFAQQ-CYBMUJFWSA-N C[C@H](C1=C(C(C=CN=C2C)=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C(C=CN=C2C)=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O KAXDDEGPGAFAQQ-CYBMUJFWSA-N 0.000 description 5
- CSSOOOTWLLMLLG-CYBMUJFWSA-N C[C@H](C1=C(C2=CC(C)=NC=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=CC(C)=NC=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O CSSOOOTWLLMLLG-CYBMUJFWSA-N 0.000 description 5
- QRGQYYDCDJMIPL-GFCCVEGCSA-N C[C@H](C1=C(N(C)C(CNC)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N(C)C(CNC)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O QRGQYYDCDJMIPL-GFCCVEGCSA-N 0.000 description 5
- DDUZMENMBWPCSI-LLVKDONJSA-N C[C@H](C1=C(N(S(C)(=O)=O)S(C)(=O)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N(S(C)(=O)=O)S(C)(=O)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O DDUZMENMBWPCSI-LLVKDONJSA-N 0.000 description 5
- YDQQVPNYHRMYPP-SNVBAGLBSA-N C[C@H](C1=C(NC(C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(NC(C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O YDQQVPNYHRMYPP-SNVBAGLBSA-N 0.000 description 5
- NGYCJCKQJNBHTQ-SNVBAGLBSA-N C[C@H](C1=CN=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=CN=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O NGYCJCKQJNBHTQ-SNVBAGLBSA-N 0.000 description 5
- RDKDCHBXACLVOU-ZCFIWIBFSA-N C[C@H]1NCCN2C(C3=NC(C)=NS3)=NC=C12 Chemical compound C[C@H]1NCCN2C(C3=NC(C)=NS3)=NC=C12 RDKDCHBXACLVOU-ZCFIWIBFSA-N 0.000 description 5
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 5
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 5
- 239000012230 colorless oil Substances 0.000 description 5
- ZCPNCZPHOXIIGW-UHFFFAOYSA-N methyl imidazo[1,5-a]pyrazine-1-carboxylate Chemical compound COC(=O)C=1N=CN2C1C=NC=C2 ZCPNCZPHOXIIGW-UHFFFAOYSA-N 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 238000003419 tautomerization reaction Methods 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- IYRKPHRAECMQTJ-UHFFFAOYSA-N 7-bromo-4-fluoro-1-benzofuran Chemical compound FC1=CC=C(Br)C2=C1C=CO2 IYRKPHRAECMQTJ-UHFFFAOYSA-N 0.000 description 4
- KLJLOIUBDVYTKR-UHFFFAOYSA-N CC(C1=C(C(C=C2)=CC=C2C#N)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(C(C=C2)=CC=C2C#N)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O KLJLOIUBDVYTKR-UHFFFAOYSA-N 0.000 description 4
- KIVYPRPBARNEMT-UHFFFAOYSA-N CC(C1=C(C2=CC=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(C2=CC=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O KIVYPRPBARNEMT-UHFFFAOYSA-N 0.000 description 4
- IOOBIPWMUMCPRF-UHFFFAOYSA-N CC(C1=C(N2C=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(N2C=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O IOOBIPWMUMCPRF-UHFFFAOYSA-N 0.000 description 4
- KNCUCSRRDKLDPM-CYBMUJFWSA-N C[C@H](C1=C(C(C=C2)=CC=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C(C=C2)=CC=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O KNCUCSRRDKLDPM-CYBMUJFWSA-N 0.000 description 4
- KCYHUNQRFZBVRO-CQSZACIVSA-N C[C@H](C1=C(C(C=C2)=CC=C2OC)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C(C=C2)=CC=C2OC)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O KCYHUNQRFZBVRO-CQSZACIVSA-N 0.000 description 4
- XVCZKNVTTRXCOU-GFCCVEGCSA-N C[C@H](C1=C(C(C=CN=C2OC)=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C(C=CN=C2OC)=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O XVCZKNVTTRXCOU-GFCCVEGCSA-N 0.000 description 4
- KPFCCDGEGMLLBF-CYBMUJFWSA-N C[C@H](C1=C(C2=CC(OC)=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=CC(OC)=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O KPFCCDGEGMLLBF-CYBMUJFWSA-N 0.000 description 4
- FDTFFSBNGHKTDX-CQSZACIVSA-N C[C@H](C1=C(C2=CC=CC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=CC=CC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O FDTFFSBNGHKTDX-CQSZACIVSA-N 0.000 description 4
- YQAICPIURFPKFV-CYBMUJFWSA-N C[C@H](C1=C(C2=NC=CC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=NC=CC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O YQAICPIURFPKFV-CYBMUJFWSA-N 0.000 description 4
- XFBPLJHHRNQZPW-GFCCVEGCSA-N C[C@H](C1=C(N(CC(OC)=C2)C2=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N(CC(OC)=C2)C2=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O XFBPLJHHRNQZPW-GFCCVEGCSA-N 0.000 description 4
- ODLJLHVHHCQNLP-CYBMUJFWSA-N C[C@H](C1=C(N2CCOCC2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N2CCOCC2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O ODLJLHVHHCQNLP-CYBMUJFWSA-N 0.000 description 4
- ZMERBFMWEBBLQT-LLVKDONJSA-N C[C@H](C1=C(NC(C=C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(NC(C=C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O ZMERBFMWEBBLQT-LLVKDONJSA-N 0.000 description 4
- CVLUKQVXWLVSHL-SNVBAGLBSA-N C[C@H](C1=CN=C(C2=NC(C)=NS2)N1CC1)N1C(C(C1=C2C=CO1)=CC=C2F)=O Chemical compound C[C@H](C1=CN=C(C2=NC(C)=NS2)N1CC1)N1C(C(C1=C2C=CO1)=CC=C2F)=O CVLUKQVXWLVSHL-SNVBAGLBSA-N 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- 108010040718 Neurokinin-1 Receptors Proteins 0.000 description 4
- 102000002002 Neurokinin-1 Receptors Human genes 0.000 description 4
- 108010040722 Neurokinin-2 Receptors Proteins 0.000 description 4
- FTWCDIHXLGUHDL-UHFFFAOYSA-N O=C(C(C1=C2C=CO1)=CC=C2F)Cl Chemical compound O=C(C(C1=C2C=CO1)=CC=C2F)Cl FTWCDIHXLGUHDL-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 210000002569 neuron Anatomy 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 4
- XUWHAWMETYGRKB-UHFFFAOYSA-N piperidin-2-one Chemical compound O=C1CCCCN1 XUWHAWMETYGRKB-UHFFFAOYSA-N 0.000 description 4
- 239000011698 potassium fluoride Substances 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- QDZOEBFLNHCSSF-PFFBOGFISA-N (2S)-2-[[(2R)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-1-[(2R)-2-amino-5-carbamimidamidopentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-N-[(2R)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]pentanediamide Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CCCNC(N)=N)C1=CC=CC=C1 QDZOEBFLNHCSSF-PFFBOGFISA-N 0.000 description 3
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 description 3
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 3
- IZNZBAWKQJZTLY-UHFFFAOYSA-N 1-bromo-2-(3,3-diethoxypropoxy)-4-fluorobenzene Chemical compound CCOC(OCC)CCOC1=CC(F)=CC=C1Br IZNZBAWKQJZTLY-UHFFFAOYSA-N 0.000 description 3
- HQYGCYJUSJWSBU-UHFFFAOYSA-N 2-(chloromethyl)-3-methylpyrazine Chemical compound CC1=NC=CN=C1CCl HQYGCYJUSJWSBU-UHFFFAOYSA-N 0.000 description 3
- RQUJTDXBBMPCFO-UHFFFAOYSA-N 3-methyl-N-propylpentan-3-amine Chemical compound C(C)C(C)(NCCC)CC RQUJTDXBBMPCFO-UHFFFAOYSA-N 0.000 description 3
- XDADSGBUZWZQQQ-UHFFFAOYSA-N 4-bromopyrimidine Chemical compound BrC1=CC=NC=N1 XDADSGBUZWZQQQ-UHFFFAOYSA-N 0.000 description 3
- CZKLEJHVLCMVQR-UHFFFAOYSA-N 4-fluorobenzoyl chloride Chemical compound FC1=CC=C(C(Cl)=O)C=C1 CZKLEJHVLCMVQR-UHFFFAOYSA-N 0.000 description 3
- SYEGQVXXGOEAMS-UHFFFAOYSA-N 5-fluorofuran-2-carboxylic acid Chemical compound OC(=O)C1=CC=C(F)O1 SYEGQVXXGOEAMS-UHFFFAOYSA-N 0.000 description 3
- HYOQZTMSZZSNSR-UHFFFAOYSA-N CC(C1=C(C(N2CCOCC2)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(C(N2CCOCC2)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O HYOQZTMSZZSNSR-UHFFFAOYSA-N 0.000 description 3
- CNHLWSQFOYDDMK-UHFFFAOYSA-N CC(C1=C(N=C(C2=CC=CC=C2)C2=CC=CC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(N=C(C2=CC=CC=C2)C2=CC=CC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O CNHLWSQFOYDDMK-UHFFFAOYSA-N 0.000 description 3
- KGXXBRWLXIAPIT-UHFFFAOYSA-N CC(C1=C(NC)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(NC)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O KGXXBRWLXIAPIT-UHFFFAOYSA-N 0.000 description 3
- DJUUYOKWTPHPOX-UHFFFAOYSA-N CC(C1=C(NC2=CC=CN=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(NC2=CC=CN=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O DJUUYOKWTPHPOX-UHFFFAOYSA-N 0.000 description 3
- XRKZYSYCXLJVNZ-LLVKDONJSA-N C[C@H](C1=C(C2=CC(F)=NC(F)=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=CC(F)=NC(F)=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O XRKZYSYCXLJVNZ-LLVKDONJSA-N 0.000 description 3
- AMDUGHOMNJHDFA-GFCCVEGCSA-N C[C@H](C1=C(C2=CC(F)=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=CC(F)=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O AMDUGHOMNJHDFA-GFCCVEGCSA-N 0.000 description 3
- LMFFVDLUOXGBRV-LLVKDONJSA-N C[C@H](C1=C(C2=CC(F)=NC=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=CC(F)=NC=C2F)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O LMFFVDLUOXGBRV-LLVKDONJSA-N 0.000 description 3
- KUOIKOZJFLCYID-CYBMUJFWSA-N C[C@H](C1=C(C2=CC=CN=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=CC=CN=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O KUOIKOZJFLCYID-CYBMUJFWSA-N 0.000 description 3
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- 101000831616 Homo sapiens Protachykinin-1 Proteins 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 239000005909 Kieselgur Substances 0.000 description 3
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 3
- HEAUFJZALFKPBA-YRVBCFNBSA-N Neurokinin A Chemical compound C([C@@H](C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)C(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CC=1NC=NC=1)C(C)O)C1=CC=CC=C1 HEAUFJZALFKPBA-YRVBCFNBSA-N 0.000 description 3
- 101800000399 Neurokinin A Proteins 0.000 description 3
- 102400000097 Neurokinin A Human genes 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 229910002666 PdCl2 Inorganic materials 0.000 description 3
- 101150003085 Pdcl gene Proteins 0.000 description 3
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 3
- 101800003906 Substance P Proteins 0.000 description 3
- 102100037342 Substance-K receptor Human genes 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 101150013502 TACR3 gene Proteins 0.000 description 3
- 102000003141 Tachykinin Human genes 0.000 description 3
- 102000007124 Tachykinin Receptors Human genes 0.000 description 3
- 108010072901 Tachykinin Receptors Proteins 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 125000005605 benzo group Chemical group 0.000 description 3
- 239000012965 benzophenone Substances 0.000 description 3
- CEOXKBUCSHIFFW-UHFFFAOYSA-N benzyl 5-fluorofuran-2-carboxylate Chemical compound O1C(F)=CC=C1C(=O)OCC1=CC=CC=C1 CEOXKBUCSHIFFW-UHFFFAOYSA-N 0.000 description 3
- YZMDGRGKHFIBLH-UHFFFAOYSA-N benzyl 5-nitrofuran-2-carboxylate Chemical compound O1C([N+](=O)[O-])=CC=C1C(=O)OCC1=CC=CC=C1 YZMDGRGKHFIBLH-UHFFFAOYSA-N 0.000 description 3
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 3
- 229910002091 carbon monoxide Inorganic materials 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- JCWIWBWXCVGEAN-UHFFFAOYSA-L cyclopentyl(diphenyl)phosphane;dichloropalladium;iron Chemical compound [Fe].Cl[Pd]Cl.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1 JCWIWBWXCVGEAN-UHFFFAOYSA-L 0.000 description 3
- 239000006274 endogenous ligand Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 3
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 3
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- 239000000543 intermediate Substances 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 125000005647 linker group Chemical group 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- 239000002480 mineral oil Substances 0.000 description 3
- 235000010446 mineral oil Nutrition 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 3
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 3
- 235000011056 potassium acetate Nutrition 0.000 description 3
- 235000003270 potassium fluoride Nutrition 0.000 description 3
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 3
- 125000006413 ring segment Chemical group 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 108060008037 tachykinin Proteins 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 125000006708 (C5-C14) heteroaryl group Chemical group 0.000 description 2
- 238000004293 19F NMR spectroscopy Methods 0.000 description 2
- OXQOBQJCDNLAPO-UHFFFAOYSA-N 2,3-Dimethylpyrazine Chemical compound CC1=NC=CN=C1C OXQOBQJCDNLAPO-UHFFFAOYSA-N 0.000 description 2
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethyl-pyridine Natural products CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- CUYKNJBYIJFRCU-UHFFFAOYSA-N 3-aminopyridine Chemical compound NC1=CC=CN=C1 CUYKNJBYIJFRCU-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 2
- UJIAFLBUSAICKJ-UHFFFAOYSA-N 4-(trifluoromethyl)-1,3-thiazole-2-carboxylic acid Chemical compound OC(=O)C1=NC(C(F)(F)F)=CS1 UJIAFLBUSAICKJ-UHFFFAOYSA-N 0.000 description 2
- GCNTZFIIOFTKIY-UHFFFAOYSA-N 4-hydroxypyridine Chemical compound OC1=CC=NC=C1 GCNTZFIIOFTKIY-UHFFFAOYSA-N 0.000 description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- IPOCOANFUVSCLZ-UHFFFAOYSA-N 6-fluoropyridine-3-carbonyl chloride Chemical compound FC1=CC=C(C(Cl)=O)C=N1 IPOCOANFUVSCLZ-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- WPJHMVKYTPHBGI-UHFFFAOYSA-N CC(C(N(CC1)C(C2=NC(C)=NS2)=N2)=C2Br)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C(N(CC1)C(C2=NC(C)=NS2)=N2)=C2Br)N1C(C(C=C1)=CC=C1F)=O WPJHMVKYTPHBGI-UHFFFAOYSA-N 0.000 description 2
- VJCPQBREPSCXOL-UHFFFAOYSA-N CC(C1=C(C(N(C)C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(C(N(C)C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O VJCPQBREPSCXOL-UHFFFAOYSA-N 0.000 description 2
- IXJLVYVIOHYGHL-UHFFFAOYSA-N CC(C1=CN=C(C2=NC(C)=CS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=CN=C(C2=NC(C)=CS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O IXJLVYVIOHYGHL-UHFFFAOYSA-N 0.000 description 2
- GBHJNPYUJZZMJG-UHFFFAOYSA-N CC1=NSC(C(Cl)=O)=N1 Chemical compound CC1=NSC(C(Cl)=O)=N1 GBHJNPYUJZZMJG-UHFFFAOYSA-N 0.000 description 2
- RDKDCHBXACLVOU-UHFFFAOYSA-N CC1NCCN2C(C3=NC(C)=NS3)=NC=C12 Chemical compound CC1NCCN2C(C3=NC(C)=NS3)=NC=C12 RDKDCHBXACLVOU-UHFFFAOYSA-N 0.000 description 2
- ULKBDKWYVMHGCX-UHFFFAOYSA-N COC(=O)c1ccc(F)c2ccoc12 Chemical compound COC(=O)c1ccc(F)c2ccoc12 ULKBDKWYVMHGCX-UHFFFAOYSA-N 0.000 description 2
- HFZPXUOGZJLHST-LLVKDONJSA-N C[C@H](C1=C(N(C)C(C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N(C)C(C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O HFZPXUOGZJLHST-LLVKDONJSA-N 0.000 description 2
- IIBIHYWSWUCKFU-OAHLLOKOSA-N C[C@H](C1=C(N(C)C(C2=CC=CC=C2)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N(C)C(C2=CC=CC=C2)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O IIBIHYWSWUCKFU-OAHLLOKOSA-N 0.000 description 2
- STRKVFNVYFGVHY-CYBMUJFWSA-N C[C@H](C1=C(N(C)C(CCOC)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N(C)C(CCOC)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O STRKVFNVYFGVHY-CYBMUJFWSA-N 0.000 description 2
- SJJBNKFITJOZME-GFCCVEGCSA-N C[C@H](C1=C(N(C)C(COC)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(N(C)C(COC)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O SJJBNKFITJOZME-GFCCVEGCSA-N 0.000 description 2
- RJMQKWZCSIDKDV-LLVKDONJSA-N C[C@H](C1=C(NC(CNC)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(NC(CNC)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O RJMQKWZCSIDKDV-LLVKDONJSA-N 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- 201000009273 Endometriosis Diseases 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 101001125071 Homo sapiens Neuromedin-K receptor Proteins 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 230000036506 anxiety Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- MNFORVFSTILPAW-UHFFFAOYSA-N azetidin-2-one Chemical compound O=C1CCN1 MNFORVFSTILPAW-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- YVHPHQBRUPLYOS-UHFFFAOYSA-N dichloromethane;methane Chemical compound C.ClCCl YVHPHQBRUPLYOS-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 239000000262 estrogen Substances 0.000 description 2
- 229940011871 estrogen Drugs 0.000 description 2
- PSTFYCCCZHOTHW-UHFFFAOYSA-N ethyl 4-(trifluoromethyl)-1,3-thiazole-2-carboxylate Chemical compound CCOC(=O)C1=NC(C(F)(F)F)=CS1 PSTFYCCCZHOTHW-UHFFFAOYSA-N 0.000 description 2
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 2
- 230000008713 feedback mechanism Effects 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000006317 isomerization reaction Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- 125000004944 pyrazin-3-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 2
- XSCHRSMBECNVNS-UHFFFAOYSA-N quinoxaline Chemical compound N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000007789 sealing Methods 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- XHFLOLLMZOTPSM-UHFFFAOYSA-M sodium;hydrogen carbonate;hydrate Chemical class [OH-].[Na+].OC(O)=O XHFLOLLMZOTPSM-UHFFFAOYSA-M 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- ADNPLDHMAVUMIW-CUZNLEPHSA-N substance P Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CCCN=C(N)N)C1=CC=CC=C1 ADNPLDHMAVUMIW-CUZNLEPHSA-N 0.000 description 2
- 238000000967 suction filtration Methods 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 230000024883 vasodilation Effects 0.000 description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- LJEFUOGGCMPGHN-UHFFFAOYSA-N (2,3-difluoropyridin-4-yl)boronic acid Chemical compound OB(O)C1=CC=NC(F)=C1F LJEFUOGGCMPGHN-UHFFFAOYSA-N 0.000 description 1
- CFUGAJDUGLGADA-UHFFFAOYSA-N (2,5-difluoropyridin-4-yl)boronic acid Chemical compound OB(O)C1=CC(F)=NC=C1F CFUGAJDUGLGADA-UHFFFAOYSA-N 0.000 description 1
- WXGBZJJAGLSBPR-UHFFFAOYSA-N (2-fluoropyridin-4-yl)boronic acid Chemical compound OB(O)C1=CC=NC(F)=C1 WXGBZJJAGLSBPR-UHFFFAOYSA-N 0.000 description 1
- DHQMUJSACXTPEA-UHFFFAOYSA-N (2-methoxypyridin-4-yl)boronic acid Chemical compound COC1=CC(B(O)O)=CC=N1 DHQMUJSACXTPEA-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- JUSWZYFYLXTMLJ-JTQLQIEISA-N (2s)-1-(benzenesulfonyl)pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1S(=O)(=O)C1=CC=CC=C1 JUSWZYFYLXTMLJ-JTQLQIEISA-N 0.000 description 1
- RQYKQWFHJOBBAO-JTQLQIEISA-N (2s)-1-benzoylpyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1C(=O)C1=CC=CC=C1 RQYKQWFHJOBBAO-JTQLQIEISA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- KSERCJHSQPNSJM-UHFFFAOYSA-N (3-methylpyridin-4-yl)boronic acid Chemical compound CC1=CN=CC=C1B(O)O KSERCJHSQPNSJM-UHFFFAOYSA-N 0.000 description 1
- MKPDAJWEBQRQCO-UHFFFAOYSA-N (4-aminophenyl)boronic acid Chemical compound NC1=CC=C(B(O)O)C=C1 MKPDAJWEBQRQCO-UHFFFAOYSA-N 0.000 description 1
- CAYQIZIAYYNFCS-UHFFFAOYSA-N (4-chlorophenyl)boronic acid Chemical compound OB(O)C1=CC=C(Cl)C=C1 CAYQIZIAYYNFCS-UHFFFAOYSA-N 0.000 description 1
- VOAAEKKFGLPLLU-UHFFFAOYSA-N (4-methoxyphenyl)boronic acid Chemical compound COC1=CC=C(B(O)O)C=C1 VOAAEKKFGLPLLU-UHFFFAOYSA-N 0.000 description 1
- 125000006714 (C3-C10) heterocyclyl group Chemical group 0.000 description 1
- 239000001211 (E)-4-phenylbut-3-en-2-one Substances 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- YGTAZGSLCXNBQL-UHFFFAOYSA-N 1,2,4-thiadiazole Chemical compound C=1N=CSN=1 YGTAZGSLCXNBQL-UHFFFAOYSA-N 0.000 description 1
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 1
- KEQGZUUPPQEDPF-UHFFFAOYSA-N 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione Chemical compound CC1(C)N(Cl)C(=O)N(Cl)C1=O KEQGZUUPPQEDPF-UHFFFAOYSA-N 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- 125000006218 1-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 1
- DNCYBUMDUBHIJZ-UHFFFAOYSA-N 1h-pyrimidin-6-one Chemical compound O=C1C=CN=CN1 DNCYBUMDUBHIJZ-UHFFFAOYSA-N 0.000 description 1
- UZYQSNQJLWTICD-UHFFFAOYSA-N 2-(n-benzoylanilino)-2,2-dinitroacetic acid Chemical compound C=1C=CC=CC=1N(C(C(=O)O)([N+]([O-])=O)[N+]([O-])=O)C(=O)C1=CC=CC=C1 UZYQSNQJLWTICD-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- LILXDMFJXYAKMK-UHFFFAOYSA-N 2-bromo-1,1-diethoxyethane Chemical compound CCOC(CBr)OCC LILXDMFJXYAKMK-UHFFFAOYSA-N 0.000 description 1
- HUVAOAVBKOVPBZ-UHFFFAOYSA-N 2-bromo-5-fluorophenol Chemical compound OC1=CC(F)=CC=C1Br HUVAOAVBKOVPBZ-UHFFFAOYSA-N 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 description 1
- JJKWHOSQTYYFAE-UHFFFAOYSA-N 2-methoxyacetyl chloride Chemical compound COCC(Cl)=O JJKWHOSQTYYFAE-UHFFFAOYSA-N 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 1
- BKKXHEBTUYSEHU-UHFFFAOYSA-N 2h-pyrazine-1-carbonitrile Chemical compound N#CN1CC=NC=C1 BKKXHEBTUYSEHU-UHFFFAOYSA-N 0.000 description 1
- LKFYWEGWCBDTTB-UHFFFAOYSA-N 3,4,4a,8a-tetrahydro-2h-isoquinolin-1-one Chemical compound C1=CC=CC2C(=O)NCCC21 LKFYWEGWCBDTTB-UHFFFAOYSA-N 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- ONZQYZKCUHFORE-UHFFFAOYSA-N 3-bromo-1,1,1-trifluoropropan-2-one Chemical compound FC(F)(F)C(=O)CBr ONZQYZKCUHFORE-UHFFFAOYSA-N 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- XXFNWMYNGDPHGX-UHFFFAOYSA-N 3-methyl-1,2,4-thiadiazole-5-carboxylic acid Chemical compound CC1=NSC(C(O)=O)=N1 XXFNWMYNGDPHGX-UHFFFAOYSA-N 0.000 description 1
- ZBOTZOCIHCIMQA-RXMQYKEDSA-N 3-methyl-5-[(8r)-8-methyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazin-3-yl]-1,2,4-thiadiazole Chemical compound N([C@@H]1C)CCN2C1=NN=C2C1=NC(C)=NS1 ZBOTZOCIHCIMQA-RXMQYKEDSA-N 0.000 description 1
- 125000003542 3-methylbutan-2-yl group Chemical group [H]C([H])([H])C([H])(*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- CUQOVOZIDNPNHO-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-(trifluoromethyl)pyridine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=NC(C(F)(F)F)=C1 CUQOVOZIDNPNHO-UHFFFAOYSA-N 0.000 description 1
- HOPDTPGXBZCBNP-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(C#N)C=C1 HOPDTPGXBZCBNP-UHFFFAOYSA-N 0.000 description 1
- MOCRCEBISJMOGG-UHFFFAOYSA-N 4-bromo-3-fluoro-2-methylpyridine Chemical compound BrC1=C(C(=NC=C1)C)F MOCRCEBISJMOGG-UHFFFAOYSA-N 0.000 description 1
- MENOXSCJWFSFHL-UHFFFAOYSA-N 4-bromo-5-fluoro-2-methylpyridine Chemical compound CC1=CC(Br)=C(F)C=N1 MENOXSCJWFSFHL-UHFFFAOYSA-N 0.000 description 1
- LBUNNMJLXWQQBY-UHFFFAOYSA-N 4-fluorophenylboronic acid Chemical compound OB(O)C1=CC=C(F)C=C1 LBUNNMJLXWQQBY-UHFFFAOYSA-N 0.000 description 1
- POILWHVDKZOXJZ-UHFFFAOYSA-N 4-hydroxypent-3-en-2-one Chemical compound CC(O)=CC(C)=O POILWHVDKZOXJZ-UHFFFAOYSA-N 0.000 description 1
- XIDJCOCVZDQNCL-UHFFFAOYSA-N 4-methoxy-1,3-dihydropyrrol-2-one Chemical compound COC1=CNC(=O)C1 XIDJCOCVZDQNCL-UHFFFAOYSA-N 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- SGXFKGABJPKDAK-UHFFFAOYSA-N 4-methyl-1,3-thiazole-2-carbonyl chloride Chemical compound CC1=CSC(C(Cl)=O)=N1 SGXFKGABJPKDAK-UHFFFAOYSA-N 0.000 description 1
- GNGDWDFLILPTKL-UHFFFAOYSA-N 4-methyl-1,3-thiazole-2-carboxylic acid Chemical compound CC1=CSC(C(O)=O)=N1 GNGDWDFLILPTKL-UHFFFAOYSA-N 0.000 description 1
- ODQJQEROUCVZNG-UHFFFAOYSA-N 5-fluoropyridine Chemical compound FC1=C=NC=C[CH]1 ODQJQEROUCVZNG-UHFFFAOYSA-N 0.000 description 1
- IFBZBMDHYAUEJV-UHFFFAOYSA-N 5-fluoropyridine-2-carbonyl chloride Chemical compound FC1=CC=C(C(Cl)=O)N=C1 IFBZBMDHYAUEJV-UHFFFAOYSA-N 0.000 description 1
- JTKFIIQGMVKDNZ-UHFFFAOYSA-N 5-fluoropyridine-2-carboxylic acid Chemical compound OC(=O)C1=CC=C(F)C=N1 JTKFIIQGMVKDNZ-UHFFFAOYSA-N 0.000 description 1
- XQURBTZGKJENJS-UHFFFAOYSA-N 5-fluorothiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=C(F)S1 XQURBTZGKJENJS-UHFFFAOYSA-N 0.000 description 1
- IODMEDPPCXSFLD-UHFFFAOYSA-N 5-nitrofuran-2-carboxylic acid Chemical compound OC(=O)C1=CC=C([N+]([O-])=O)O1 IODMEDPPCXSFLD-UHFFFAOYSA-N 0.000 description 1
- UJDLCTNVHJEBDG-UHFFFAOYSA-N 6-fluoropyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=C(F)N=C1 UJDLCTNVHJEBDG-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- JRLTTZUODKEYDH-UHFFFAOYSA-N 8-methylquinoline Chemical group C1=CN=C2C(C)=CC=CC2=C1 JRLTTZUODKEYDH-UHFFFAOYSA-N 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 1
- SJXSCUKEIASMFO-UHFFFAOYSA-N C(=O)(S(=O)Cl)Cl Chemical compound C(=O)(S(=O)Cl)Cl SJXSCUKEIASMFO-UHFFFAOYSA-N 0.000 description 1
- YXBSOQIDSJOJRD-UHFFFAOYSA-N C(C=C1)=CC=C1P(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1.Br Chemical compound C(C=C1)=CC=C1P(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1.Br YXBSOQIDSJOJRD-UHFFFAOYSA-N 0.000 description 1
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 description 1
- 125000005865 C2-C10alkynyl group Chemical group 0.000 description 1
- 125000005915 C6-C14 aryl group Chemical group 0.000 description 1
- YDQQVPNYHRMYPP-UHFFFAOYSA-N CC(C1=C(NC(C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound CC(C1=C(NC(C)=O)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O YDQQVPNYHRMYPP-UHFFFAOYSA-N 0.000 description 1
- IYQKDYHBTDYFDX-GFCCVEGCSA-N C[C@H](C1=C(C2=CC(C(F)(F)F)=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O Chemical compound C[C@H](C1=C(C2=CC(C(F)(F)F)=NC=C2)N=C(C2=NC(C)=NS2)N1CC1)N1C(C(C=C1)=CC=C1F)=O IYQKDYHBTDYFDX-GFCCVEGCSA-N 0.000 description 1
- 229920002284 Cellulose triacetate Polymers 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 208000020401 Depressive disease Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- 108010065372 Dynorphins Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 102000006771 Gonadotropins Human genes 0.000 description 1
- 108010086677 Gonadotropins Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 206010058359 Hypogonadism Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000013599 Kisspeptins Human genes 0.000 description 1
- 108010012048 Kisspeptins Proteins 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- 102000009493 Neurokinin receptors Human genes 0.000 description 1
- 108050000302 Neurokinin receptors Proteins 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 102100024622 Proenkephalin-B Human genes 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 206010047139 Vasoconstriction Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- NNLVGZFZQQXQNW-ADJNRHBOSA-N [(2r,3r,4s,5r,6s)-4,5-diacetyloxy-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6s)-4,5,6-triacetyloxy-2-(acetyloxymethyl)oxan-3-yl]oxyoxan-2-yl]methyl acetate Chemical compound O([C@@H]1O[C@@H]([C@H]([C@H](OC(C)=O)[C@H]1OC(C)=O)O[C@H]1[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1)OC(C)=O)COC(=O)C)[C@@H]1[C@@H](COC(C)=O)O[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O NNLVGZFZQQXQNW-ADJNRHBOSA-N 0.000 description 1
- RSMCZIMSRVMJEB-UHFFFAOYSA-N [Cl].CCOC(C)=O Chemical compound [Cl].CCOC(C)=O RSMCZIMSRVMJEB-UHFFFAOYSA-N 0.000 description 1
- 206010000059 abdominal discomfort Diseases 0.000 description 1
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 1
- 229960004373 acetylcholine Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- HFBMWMNUJJDEQZ-UHFFFAOYSA-N acryloyl chloride Chemical compound ClC(=O)C=C HFBMWMNUJJDEQZ-UHFFFAOYSA-N 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical group C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 210000004727 amygdala Anatomy 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- METKIMKYRPQLGS-UHFFFAOYSA-N atenolol Chemical compound CC(C)NCC(O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-UHFFFAOYSA-N 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- UENWRTRMUIOCKN-UHFFFAOYSA-N benzyl thiol Chemical compound SCC1=CC=CC=C1 UENWRTRMUIOCKN-UHFFFAOYSA-N 0.000 description 1
- 229930008407 benzylideneacetone Natural products 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical class C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 150000001719 carbohydrate derivatives Chemical class 0.000 description 1
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 239000007806 chemical reaction intermediate Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 150000001804 chlorine Chemical class 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000037020 contractile activity Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- WJTCGQSWYFHTAC-UHFFFAOYSA-N cyclooctane Chemical compound C1CCCCCCC1 WJTCGQSWYFHTAC-UHFFFAOYSA-N 0.000 description 1
- 239000004914 cyclooctane Substances 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 1
- WMSPXQIQBQAWLL-UHFFFAOYSA-N cyclopropanesulfonamide Chemical compound NS(=O)(=O)C1CC1 WMSPXQIQBQAWLL-UHFFFAOYSA-N 0.000 description 1
- 230000006240 deamidation Effects 0.000 description 1
- 125000004855 decalinyl group Chemical group C1(CCCC2CCCCC12)* 0.000 description 1
- 230000015961 delipidation Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000005959 diazepanyl group Chemical group 0.000 description 1
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical compound CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 1
- 125000005046 dihydronaphthyl group Chemical group 0.000 description 1
- 230000010339 dilation Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 1
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 210000005064 dopaminergic neuron Anatomy 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 210000003372 endocrine gland Anatomy 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- RSOJSFYJLQADDM-UHFFFAOYSA-N ethanesulfonamide Chemical compound [CH2]CS(N)(=O)=O RSOJSFYJLQADDM-UHFFFAOYSA-N 0.000 description 1
- FRYHCSODNHYDPU-UHFFFAOYSA-N ethanesulfonyl chloride Chemical compound CCS(Cl)(=O)=O FRYHCSODNHYDPU-UHFFFAOYSA-N 0.000 description 1
- MSMGXWFHBSCQFB-UHFFFAOYSA-N ethyl cyanoformate Chemical compound CCOC(=O)C#N MSMGXWFHBSCQFB-UHFFFAOYSA-N 0.000 description 1
- 230000002964 excitative effect Effects 0.000 description 1
- 210000003499 exocrine gland Anatomy 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000002622 gonadotropin Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005549 heteroarylene group Chemical group 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- VIPHVHVAGBKHGR-UHFFFAOYSA-N hydron;pyridine-2-carbonyl chloride;chloride Chemical compound Cl.ClC(=O)C1=CC=CC=N1 VIPHVHVAGBKHGR-UHFFFAOYSA-N 0.000 description 1
- BNTRVUUJBGBGLZ-UHFFFAOYSA-N hydron;pyridine-4-carbonyl chloride;chloride Chemical compound Cl.ClC(=O)C1=CC=NC=C1 BNTRVUUJBGBGLZ-UHFFFAOYSA-N 0.000 description 1
- 230000001077 hypotensive effect Effects 0.000 description 1
- 230000004185 hypothalamic-pituitary-gonadal axis Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- UTCSSFWDNNEEBH-UHFFFAOYSA-N imidazo[1,2-a]pyridine Chemical compound C1=CC=CC2=NC=CN21 UTCSSFWDNNEEBH-UHFFFAOYSA-N 0.000 description 1
- MLXWMEFKFOSOCP-UHFFFAOYSA-N imidazo[1,5-a]pyridine Chemical compound C1=CC=CC2=C=N[CH]N21 MLXWMEFKFOSOCP-UHFFFAOYSA-N 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- PXZQEOJJUGGUIB-UHFFFAOYSA-N isoindolin-1-one Chemical compound C1=CC=C2C(=O)NCC2=C1 PXZQEOJJUGGUIB-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- KAHDONZOCXSKII-NJVVDGNHSA-N kisspeptin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H]1N(CCC1)C(=O)[C@H]1N(CCC1)C(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)O)C1=CN=CN1 KAHDONZOCXSKII-NJVVDGNHSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940050176 methyl chloride Drugs 0.000 description 1
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 1
- KTMKRRPZPWUYKK-UHFFFAOYSA-N methylboronic acid Chemical compound CB(O)O KTMKRRPZPWUYKK-UHFFFAOYSA-N 0.000 description 1
- UIUXUFNYAYAMOE-UHFFFAOYSA-N methylsilane Chemical compound [SiH3]C UIUXUFNYAYAMOE-UHFFFAOYSA-N 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- VSEAAEQOQBMPQF-UHFFFAOYSA-N morpholin-3-one Chemical compound O=C1COCCN1 VSEAAEQOQBMPQF-UHFFFAOYSA-N 0.000 description 1
- XCVNDBIXFPGMIW-UHFFFAOYSA-N n-ethylpropan-1-amine Chemical compound CCCNCC XCVNDBIXFPGMIW-UHFFFAOYSA-N 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000005305 organ development Effects 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- UCUUFSAXZMGPGH-UHFFFAOYSA-N penta-1,4-dien-3-one Chemical compound C=CC(=O)C=C UCUUFSAXZMGPGH-UHFFFAOYSA-N 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 150000004965 peroxy acids Chemical class 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- UXCDUFKZSUBXGM-UHFFFAOYSA-N phosphoric tribromide Chemical compound BrP(Br)(Br)=O UXCDUFKZSUBXGM-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- XIPFMBOWZXULIA-UHFFFAOYSA-N pivalamide Chemical compound CC(C)(C)C(N)=O XIPFMBOWZXULIA-UHFFFAOYSA-N 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- FYRHIOVKTDQVFC-UHFFFAOYSA-M potassium phthalimide Chemical compound [K+].C1=CC=C2C(=O)[N-]C(=O)C2=C1 FYRHIOVKTDQVFC-UHFFFAOYSA-M 0.000 description 1
- 210000002442 prefrontal cortex Anatomy 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004353 pyrazol-1-yl group Chemical group [H]C1=NN(*)C([H])=C1[H] 0.000 description 1
- 125000004289 pyrazol-3-yl group Chemical group [H]N1N=C(*)C([H])=C1[H] 0.000 description 1
- 125000004497 pyrazol-5-yl group Chemical group N1N=CC=C1* 0.000 description 1
- UMLDUMMLRZFROX-UHFFFAOYSA-N pyridin-2-ylboronic acid Chemical compound OB(O)C1=CC=CC=N1 UMLDUMMLRZFROX-UHFFFAOYSA-N 0.000 description 1
- ABMYEXAYWZJVOV-UHFFFAOYSA-N pyridin-3-ylboronic acid Chemical compound OB(O)C1=CC=CN=C1 ABMYEXAYWZJVOV-UHFFFAOYSA-N 0.000 description 1
- QLULGIRFKAWHOJ-UHFFFAOYSA-N pyridin-4-ylboronic acid Chemical compound OB(O)C1=CC=NC=C1 QLULGIRFKAWHOJ-UHFFFAOYSA-N 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- VVDCRJGWILREQH-UHFFFAOYSA-N tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC(B2OC(C)(C)C(C)(C)O2)=C1 VVDCRJGWILREQH-UHFFFAOYSA-N 0.000 description 1
- IPISOFJLWYBCAV-UHFFFAOYSA-N tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole-1-carboxylate Chemical compound C1=NN(C(=O)OC(C)(C)C)C=C1B1OC(C)(C)C(C)(C)O1 IPISOFJLWYBCAV-UHFFFAOYSA-N 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- 210000001103 thalamus Anatomy 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001331 thermoregulatory effect Effects 0.000 description 1
- 239000003451 thiazide diuretic agent Substances 0.000 description 1
- 125000005556 thienylene group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- ARYHTUPFQTUBBG-UHFFFAOYSA-N thiophen-2-ylboronic acid Chemical compound OB(O)C1=CC=CS1 ARYHTUPFQTUBBG-UHFFFAOYSA-N 0.000 description 1
- BWHOZHOGCMHOBV-BQYQJAHWSA-N trans-benzylideneacetone Chemical compound CC(=O)\C=C\C1=CC=CC=C1 BWHOZHOGCMHOBV-BQYQJAHWSA-N 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- LENLQGBLVGGAMF-UHFFFAOYSA-N tributyl([1,2,4]triazolo[1,5-a]pyridin-6-yl)stannane Chemical compound C1=C([Sn](CCCC)(CCCC)CCCC)C=CC2=NC=NN21 LENLQGBLVGGAMF-UHFFFAOYSA-N 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- 125000005455 trithianyl group Chemical group 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 230000025033 vasoconstriction Effects 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5355—Non-condensed oxazines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- the invention belongs to the field of pharmaceutical synthesis, and in particular relates to imidazole-containing fused ring derivatives represented by general formula (I), a preparation method thereof, and uses of a pharmaceutical composition containing the derivatives as therapeutic agents, and use thereof as selective NK Use of -3 receptor (NK-3R) antagonists for the treatment and/or prevention of a broad range of CNS and peripheral diseases.
- general formula (I) imidazole-containing fused ring derivatives represented by general formula (I)
- a preparation method thereof and uses of a pharmaceutical composition containing the derivatives as therapeutic agents, and use thereof as selective NK Use of -3 receptor (NK-3R) antagonists for the treatment and/or prevention of a broad range of CNS and peripheral diseases.
- NK-3R -3 receptor
- Tachykinin receptors are targets of a family of structurally related peptides collectively named "tachykinins” that include substance P (SP), neurokinin A (NKA), and neurokinin B (NKB). Tachykinins are synthesized in the central nervous system (CNS) and peripheral tissues, where they exert a variety of biological activities. There are currently three known tachykinin receptors, named neurokinin-1 (NK-1) receptor, neurokinin-2 (NK-2) receptor, and neurokinin-3 (NK-3) receptor ) receptors. Tachykinin receptors belong to the rhodopsin-like seven-transmembrane G protein-coupled receptors.
- NK-1 receptors, NK-2 receptors and NK-3 receptors have been identified in different species. NK-1 receptors and NK-2 receptors are expressed in various peripheral tissues and NK-1 receptors are also expressed in the CNS, while NK-3 receptors are mainly expressed in the CNS.
- Neurokinin receptors mediate a variety of biological effects stimulated by tachykinins, including the transmission of excitatory neuronal signals (eg, pain) in the CNS and the periphery, modulation of smooth muscle contractile activity, modulation of immune and inflammatory responses, via dilation peripheral vasculature to induce hypotensive effects and stimulate secretion from endocrine and exocrine glands.
- excitatory neuronal signals eg, pain
- modulation of smooth muscle contractile activity eg, modulation of smooth muscle contractile activity
- modulation of immune and inflammatory responses via dilation peripheral vasculature to induce hypotensive effects and stimulate secretion from endocrine and exocrine glands.
- the NK-3 receptor is encoded by the TACR3 gene and is involved in the regulation of the hypothalamic-pituitary-gonadal axis. Both TACR3 knockout or mutant mice showed abnormal reproductive organ development, low sex hormone levels, and severely reduced reproductive capacity. Carrying a mutation in the TACR3 gene can cause abnormal gonadotropin release in patients, resulting in sexual immaturity and infertility in patients, and a considerable part of familial hypogonadism is caused by mutations in the TACR3 gene.
- Kisspeptin/neurokinin B/dynorphin (KNDy) neurons are involved in the gonadotropin-releasing hormone (GnRH) signaling pathway, and promote the production of estrogen through the GnRH neuron-pituitary-sex organ pathway, and this signaling pathway It is regulated by a negative feedback mechanism to keep the hormone levels in the body within a certain reasonable range.
- KNDy neurons are also related to the thermoregulatory signaling pathway, by releasing NKB ligands, binding to NK-3 receptors on the median preoptic nucleus, regulating body temperature by inhibiting shaking and vasoconstriction, promoting sweating and vasodilation within a certain range.
- NK-3 receptors are expressed in regions including the medial prefrontal cortex, hippocampus, thalamus, and amygdala. Furthermore, NK-3 receptors are expressed on dopaminergic neurons. Activation of NK-3 receptors has been shown to modulate the release of dopamine, acetylcholine, and serotonin, suggesting the therapeutic utility of NK-3 receptor modulators in the treatment of a variety of conditions, including psychotic disorders, anxiety disorders, depression schizophrenia, as well as obesity, pain or inflammation.
- the present invention provides a NK-3R antagonist with a novel structure, and found that it has such a structure
- the compounds have good activity and can effectively treat a series of CNS and peripheral diseases.
- the present invention provides a compound represented by formula (I), its stereoisomers, tautomers, isotopic labels, nitrogen oxides, solvates, polymorphs, and pharmaceutically acceptable salts or prodrugs,
- ring A is a heterocyclic group containing at least two N atoms; n is an integer of 1-5;
- R 1 is selected from H, halogen, -CN, -NO 2 , -OR 11 , -CO-R 12 , -CS-R 12 , -COO-R 13 , -O-CO-R 14 , -NR 15 R 16 , -CONR 17 R 18 , -NR 19 CO-R 20 , -S(O) a -R 21 , -S(O) a NR 22 R 23 , -NR 24 S(O) a -R 25 , -SR 26 , or unsubstituted or optionally substituted by one or more R a of the following groups: alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocyclyl;
- R 2 is selected from halogen, -CN, -NO 2 , -OR 11 , -CO-R 12 , -CS-R 12 , COO-R 13 , -O-CO-R 14 , -NR 15 R 16 , -CONR 17 R 18 , -NR 19 CO-R 20 , -S(O) a -R 21 , -S(O) a NR 22 R 23 , -NR 24 S(O) a -R 25 , -SR 26 , or The following groups, unsubstituted or optionally substituted with one or more R a : alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocyclyl;
- R a is the same or different and independently selected from H, D (deuterium), oxo, thio, halogen, -CN, -NO 2 , -OR 11 , -CO-R 12 , -CS-R 12 , - COO-R 13 , -O-CO-R 14 , -NR 15 R 16 , -CONR 17 R 18 , -NR 19 CO-R 20 , -S(O) a -R 21 , -S(O) a NR 22 R 23 , -NR 24 S(O) a -R 25 , -SR 26 , or the following groups unsubstituted or optionally substituted with one or more R c : alkyl, alkenyl, alkynyl, cycloalkane base, aryl, heteroaryl, heterocyclyl;
- R 3 is the same or different and independently selected from H, oxo, thio, halogen, -CN, -NO 2 , -OR 31 , -CO-R 32 , -COO-R 33 , -OCO-R 34 , -NR 35 R 36 , -CONR 37 R 38 , -NR 39 CO-R 40 , -S(O) a -R 41 , -S(O) a NR 42 R 43 , -NR 44 S(O) a - R 45 , -SR 46 , the following groups unsubstituted or optionally substituted by one or more R d : alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocyclyl;
- R' is selected from H, -L-Ar, -LR 10 ;
- R 10 is alkyl, alkenyl, alkynyl;
- L is absent or selected from -( CH2 ) m -CO-, -(CH2) m -CS-, -( CH2 ) m - SO2- , -( CH2 ) m -SO-, alkylene ; m is 0, 1, 2, 3;
- Ar is aryl, heteroaryl, cycloalkyl, or heterocyclyl, which may optionally be substituted with one or more R4 ; alternatively, any two adjacent or non-adjacent R 4 are linked to form an unsubstituted or optionally substituted cycloalkyl, heterocyclyl, aryl or heteroaryl group with one or more R ;
- R 4 is the same or different and independently selected from H, oxo, thio, halogen, -CN, -NO 2 , -OR 11 , -CO-R 12 , -CS-R 12 , -COO-R 13 , -O-CO-R 14 , -NR 15 R 16 , -CONR 17 R 18 , -NR 19 CO-R 20 , -S(O) a -R 21 , -S(O) a NR 22 R 23 , - NR 24 S(O) a -R 25 , -SR 26 , or the following groups unsubstituted or optionally substituted with one or more R b : alkyl, alkenyl, alkynyl, cycloalkyl, aryl, Heteroaryl, heterocyclyl;
- R 11 , R 12 , R 13 , R 14 , R 15 , R 16 , R 17 , R 18 , R 19 , R 20 , R 21 , R 22 , R 23 , R 24 , R 25 , R 26 are the same or different , independently selected from the group consisting of H, oxo, thio, halogen, -OH, unsubstituted or optionally substituted by one or more R b : alkyl, alkenyl, alkynyl, cycloalkyl, Aryl, heteroaryl, heterocyclic group; R 15 and R 16 , R 17 and R 18 , R 19 and R 20 , R 22 and R 23 may form a ring to form a heterocyclic group or heterocyclic group containing at least one N atom Aryl (such as a heterocyclyl or heteroaryl group containing one N atom and optionally 1-3 N, O, S); the heterocyclyl or heteroaryl group may optionally be replaced by one or
- R b , R c are the same or different and are independently selected from H, D (deuterium), oxo, thio, halogen, -CN, -NO 2 , -OR 31 , -CO-R 32 , -COO-R 33 , -O-CO-R 34 , -NR 35 R 36 , -CONR 37 R 38 , -NR 39 CO-R 40 , -S(O) a -R 41 , -S(O) a NR 42 R 43 , -NR 44 S(O) a -R 45 , -SR 46 , or the following groups unsubstituted or optionally substituted with one or more R d : alkyl, alkenyl, alkynyl, cycloalkyl, aryl base, heteroaryl, heterocyclyl;
- R d are the same or different and independently selected from H, oxo, thio, halogen, -CN, -NO 2 , -OR 31 , -CO-R 32 , -COO-R 33 , -O-CO-R 34 , -NR 35 R 36 , -CONR 37 R 38 , -NR 39 CO-R 40 , -S(O) a -R 41 , -S(O) a NR 42 R 43 , -NR 44 S(O) a -R 45 , -SR 46 , alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocyclyl;
- a is 1 or 2;
- R 31 , R 32 , R 33 , R 34 , R 35 , R 36 , R 37 , R 38 , R 39 , R 40 , R 41 , R 42 , R 43 , R 44 , R 45 , R 46 are the same or different , independently of each other selected from H, halogen, -OH, alkyl, cycloalkyl, aryl, heteroaryl, heterocyclyl.
- the compound represented by the formula (I) is the compound represented by the following formula (I') and formula (I"):
- Rings A, Ar, L, R 10 , R 1 , R 2 , R 3 , n are as defined above.
- L is selected from -( CH2 ) m -CO-, -( CH2 ) m -CS-, -( CH2 ) m - SO2- or C1-4 alkylene base.
- the compound represented by the formula (I) is the compound represented by the following formula (IA) or formula (IB):
- R 1 , R 2 , R 3 , R' are as defined above; p is 1, 2 or 3.
- the compound represented by the formula (I) is a compound represented by the following formula (II) or formula (II'):
- Ar, L, R 10 , R 1 , R 2 , R 3 are as defined above; p is 1, 2 or 3.
- the compound represented by the formula (I) is the compound represented by the following formula (III-1), formula (III-2), formula (III-3), and formula (III-4) :
- R 10 , R 1 , R 2 , R 3 are as defined above; p is 1, 2 or 3.
- said R 2 is a substituted or unsubstituted heteroaryl, such as substituted or unsubstituted thiadiazole, substituted or unsubstituted thiazole, substituted or unsubstituted imidazole, substituted or unsubstituted Unsubstituted triazole, substituted or unsubstituted oxazole, substituted or unsubstituted oxadiazole, specifically, R 2 is alkyl or haloalkyl substituted thiadiazole, alkyl or haloalkyl substituted thiazole.
- the R 2 is selected from the following groups:
- R 5 and R 6 are the same or different, and are independently selected from H, D (deuterium), oxo, thio, halogen, -CN, -NO 2 , -OR 11 , -CO-R 12 , -CS- R 12 , -COO-R 13 , -O-CO-R 14 , -NR 15 R 16 , -CONR 17 R 18 , -NR 19 CO-R 20 , -S(O) a -R 21 , -S( O) a NR 22 R 23 , -NR 24 S(O) a -R 25 , -SR 26 , the following groups, unsubstituted or optionally substituted by one or more R c : alkyl, alkenyl, alkynyl , cycloalkyl, aryl, heteroaryl, heterocyclyl; wherein each substituent is as defined above.
- the R 2 is haloalkyl, eg, trifluoromethyl, hexafluoroethyl, -CH 2 -CF 3 .
- the R 1 is H, D (deuterium), halogen, -CN, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, -cycloalkyl-alkyl , -Cycloalkyl-CO-alkyl, -CO-alkyl, -CO-alkenyl, -CO-alkynyl, -CO-aryl, -CO-heteroaryl, -CO-cycloalkyl, - CO-heterocyclyl, -COO-alkyl, -O-CO-alkyl, -NH 2 , alkylamino, dialkylamino, -CONH 2 , -CONH-alkyl, -CO-N(alkyl ) 2 , -NHCO-alkyl, -NHCOO-alkyl, -N(alkyl)(-CO-alkyl), -S(O)
- the R 1 is -NR 15 R 16 , wherein R 15 and R 16 are the same or different, and are independently selected from H, alkyl, cycloalkyl, hydroxyalkyl, hydroxy Cycloalkyl, -Alkyl-O-Alkyl, -Alkyl-O-Cycloalkyl, -CO-Alkyl, -CO-Cycloalkyl, -CO-Alkenyl, -CO-Alkynyl, -CO -Aryl, -CO-heteroaryl, -CO-heterocyclyl, -CO-alkyl-O-alkyl, -CO-alkyl-NH 2 , -CO-alkyl-NH-alkyl, - CO-alkyl-N(alkyl) 2 , -CO-O-alkyl, -S(O) 2 -alkyl, -S(O) 2 -
- the R 1 is -NR 15 R 16 , wherein R 15 and R 16 are connected to form a heterocyclic group containing at least one N atom, preferably one N atom and optionally further containing 1-3 N, O, S 4-10-membered monocyclic or bicyclic heterocyclic groups; specific examples are 4-membered rings, such as azetidinyl; 5-membered rings, such as pyrrolidinyl, pyrrolidinyl , imidazolidinyl, imidazolidinyl, pyrazolidinyl, pyrazolidinyl, oxazolidinyl, isoxazolidinyl, thiazolidinyl, isothiazolidinyl; or 6-membered ring, such as piperidinyl, tetra Hydropyridine, dihydropyridine, piperazinyl, morpholinyl, oxazinyl, thiazinyl; or 7
- the N-containing heterocycle may be substituted by the following 1-5 or 1-3 substituents: H, oxo, thio, F, Cl, Br, I, -OH, -CN, -NO 2 , alkyl, alkoxy, haloalkyl, haloalkoxy, -NH-CO-alkyl, -CO-NH-alkyl, -CO-N(alkyl) 2 , -CO-alkyl, -O-CO-alkyl, -CO-O-alkyl, -NH 2 , alkylamino, dialkylamino; optionally, adjacent or non-adjacent substituents on the N-containing heterocycle Can form a ring to form a substituted or unsubstituted cycloalkyl, heterocyclyl, aryl or heteroaryl group, exemplary, R 1 is unsubstituted or substituted by R d the following groups: indolinyl, iso Indolinyl,
- the R 1 is an aryl group, such as phenyl, naphthyl, anthracenyl, and the aryl group may be substituted by one or more of the following substituents (eg, 1-5 substituents) Substitution: H, oxo, thio, halogen, -CN, -NO 2 , -OR 31 , -CO-R 32 , -COO-R 33 , -O-CO-R 34 , -NR 35 R 36 , - CONR 37 R 38 , -NR 39 CO-R 40 , -S(O) a -R 41 , -S(O) a NR 42 R 43 , -NR 44 S(O) a -R 45 , -SR 46 , The following groups, either unsubstituted or optionally substituted with one or more Rd : alkyl, alkenyl, alkynyl, cycloalkyl,
- the aryl group may be substituted with 1-5 or 1-3 substituents as follows: H, oxo, thio, F, Cl, Br, I, -OH, -CN, -NO 2 , alkyl, alkoxy, haloalkyl, haloalkoxy, -NH-CO-alkyl, -CO-NH 2 , -CO-NH-alkyl, -CO-N(alkyl) 2 , -CO- Alkyl, -O-CO-alkyl, -CO-O-alkyl, -NH2 , alkylamino, dialkylamino.
- the R 1 is a heteroaryl group, preferably, optionally containing 1-5 4-10-membered monocyclic or bicyclic heteroaryl groups selected from N, O, S; specific , such as pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, triazinyl, quinolinyl, quinazolinyl, cinnoline, pyrrolyl, indolyl, imidazolyl, benzimidazolyl, pyrazole base, benzopyrazolyl, thienyl, benzothienyl, thiazolyl, thiadiazolyl, oxazolyl, benzoxazolyl, furyl, benzofuranyl, etc.
- the heteroaryl group may be Substituted with one or more of the following substituents (eg 1-5 substituents): H, D, oxo, thio, halogen
- the heteroaryl group may be substituted with 1-5 or 1-3 of the following: H, D, oxo, thio, F, Cl, Br, I, -OH, -CN, -NO 2 , alkyl, alkoxy, haloalkyl, haloalkoxy, -NH-CO-alkyl, -CO-NH-alkyl, -CO-alkyl, -O-CO-alkyl, -CO-O -Alkyl, -NH2 , alkylamino, dialkylamino.
- the R 1 is a heterocyclic group, preferably, optionally containing 1-5 4-10-membered monocyclic or bicyclic heterocyclic groups selected from N, O, S; specific , such as morpholinyl, piperidinyl, dihydropyridine, tetrahydropyridine, piperazinyl, dihydropyrrole, pyrrolidinyl, 2H-pyranyl, N heterocyclobutane, N heterocycloheptane, N hetero Cyclooctane, imidazolinyl, indolinyl, isoindolinyl, dihydroquinolinyl, tetrahydroquinolinyl, dihydroisoquinolinyl, tetrahydroisoquinolinyl, oxazolidinyl, oxazinyl, azepanyl, azabicyclooctyl, thiazinyl, thiazolid
- the heterocyclyl group may be substituted with 1-5 or 1-3 substituents as follows: H, D, oxo, thio, F, Cl, Br, I, -OH, -CN, -NO 2 , alkyl, alkoxy, haloalkyl, haloalkoxy, -NH-CO-alkyl, -CO-NH-alkyl, -CO-alkyl, -O-CO-alkyl, -CO -O-Alkyl, -NH2 , alkylamino, dialkylamino.
- Ar is an aryl group, eg, phenyl, naphthyl, anthracenyl, which may be substituted with one or more of the following substituents (eg, 1-5 substituents): H , oxo, thio, halogen, -CN, -NO 2 , -OR 31 , -CO-R 32 , -COO-R 33 , -O-CO-R 34 , -NR 35 R 36 , -CONR 37 R 38 , -NR 39 CO-R 40 , -S(O) a -R 41 , -S(O) a NR 42 R 43 , -NR 44 S(O) a -R 45 , -SR 46 , or unsubstituted or the following groups optionally substituted with one or more Rd : alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, hetero
- the aryl group may be substituted with 1-5 or 1-3 substituents as follows: H, oxo, thio, F, Cl, Br, I, -OH, -CN, -NO 2 , alkyl, alkoxy, haloalkyl, haloalkoxy, -NH-CO-alkyl, -CO-NH-alkyl, -CO-alkyl, -O-CO-alkyl, -CO-O- Alkyl, -NH 2 , alkylamino, dialkylamino.
- Ar is a heteroaryl group, preferably, optionally containing 1-5 4-10-membered monocyclic or bicyclic heteroaryl groups selected from N, O, S; for example, pyridyl, pyrimidine base, pyridazinyl, pyrazinyl, triazinyl, quinolinyl, quinazolinyl, cinnoline, pyrrolyl, indolyl, imidazolyl, benzimidazolyl, pyrazolyl, benzopyrazole base, thienyl, benzothienyl, thiazolyl, thiadiazolyl, oxazolyl, benzoxazolyl, furyl, benzofuranyl, etc.
- the heteroaryl group may be substituted by one or more of the following Substituted by groups (eg 1-5 substituents): H, oxo, thio, halogen, -CN,
- the heteroaryl group may be substituted by 1-5 or 1-3 of the following: H, oxo, thio, F, Cl, Br, I, -OH, -CN, -NO 2 , Alkyl, alkoxy, haloalkyl, haloalkoxy, -NH-CO-alkyl, -CO-NH-alkyl, -CO-alkyl, -O-CO-alkyl, -CO-O-alkane group, -NH 2 , alkylamino, dialkylamino.
- Ar is a cycloalkyl group, preferably a cycloalkyl group containing 3-10 carbon atoms, more preferably a cycloalkyl group containing 3-6 carbon atoms, and most preferably a cyclopropyl group .
- R 3 is the same or different and independently selected from H, oxo, thio, halogen, -CN, -NO 2 , alkyl, alkoxy, haloalkyl, haloalkoxy .
- R 10 is a C1-6 alkyl group, a C2-6 alkenyl group, or a C2-6 alkynyl group, such as methyl, ethyl, n-propyl, isopropyl, n-butyl base, isobutyl, tert-butyl, vinyl, ethynyl.
- the compound represented by the formula (I) is the compound represented by the following formula (IV):
- R 1 , R 2 are as defined above.
- the compound of formula (IV) may be a compound of formula (IV-A), formula (IV-B) as follows:
- the compound represented by the formula (I) is the compound represented by the following formula (V), formula (V'), and formula (V"):
- M is N or CR 6 ;
- R 5 and R 6 are the same or different, and are independently selected from H, halogen, -CN, -NO 2 , -OR 11 , -CO-R 12 , -CS-R 12 , -COO-R 13 , -O-CO -R 14 , -NR 15 R 16 , -CONR 17 R 18 , -NR 19 CO-R 20 , -S(O) a -R 21 , -S(O) a NR 22 R 23 , -NR 24 S( O) a -R 25 , -SR 26 , the following groups, unsubstituted or optionally substituted by one or more R c : alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, hetero cyclyl; wherein each substituent is as defined above;
- Ar, R 1 , R 10 are as defined above.
- the compound of formula (V) can be the compound of formula (V-A) and formula (V-B) as follows:
- the compound of formula (I) may be a compound of formula (VI) as follows:
- R 1 , R 5 are as defined above.
- the compound of formula (VI) can be the following compound of formula (VI-A) and formula (VI-B):
- the compound of formula (I) may be the following specific compound or a pharmaceutically acceptable salt thereof:
- the compound of formula (I) may be the following specific compound:
- the present invention also provides a method for preparing the compound of the above formula (I), comprising: reacting the compound of the formula (X) with the compound of the formula (XI) in the presence of a base (such as triethylamine) to obtain the compound of the formula (I);
- a base such as triethylamine
- R 1 , R 2 , R 3 , R′, ring A, and n are as defined above;
- R 7 is halogen (eg chlorine, bromine), hydroxyl.
- the compound of formula (I-4) can also be prepared by the following method, comprising: combining formula (I-2) with a protecting group of ammonia (such as benzophenone amine), and then remove the protecting group to obtain the compound of formula (I-3); then, the compound of formula (I-3) reacts with R 9 -X to obtain the compound of formula (I-4);
- a protecting group of ammonia such as benzophenone amine
- ring A, R 2 , R 3 , R', n are as defined above, X is halogen, R 9 is the same or different, and is independently selected from H, -OH, -CO-R 20 , -S(O) a -R 25 , the following groups unsubstituted or optionally substituted with one or more R b : alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocyclyl.
- the compound of formula (I-5) can also be prepared by the following method, including: reacting formula (I-2) with R 1a -H to obtain formula (I- 5) Compounds.
- ring A, R 2 , R 3 , R' and n are as defined above, X is halogen, R 1a is -NR 15 R 16 or heteroaryl, R 15 and R 16 form a ring to form a heterocyclic ring containing N or heteroaryl; the heterocyclyl or heteroaryl may be optionally substituted with one or more R b , and adjacent or non-adjacent R b may be linked into a ring to form unsubstituted or be substituted by one or more Cycloalkyl, heterocyclyl, aryl or heteroaryl substituted by R d .
- the compound of formula (I-5) is obtained by the catalytic coupling of the formula (I-2) and R 1a -H through copper or pd.
- the compound of formula (I-6) can also be prepared by the following method, including: reacting formula (I-2) with ) and R 1b -Y to obtain formula ( I-6) Compounds.
- ring A, R 2 , R 3 , R' and n are as defined above, X is halogen, R 1b is cyano, alkyl, aryl, heteroaryl, Y is -B(OH) 2 , -Sn (alkyl) 3 , or Wait;
- the compound of formula (I-2) can be prepared by the following method, comprising: combining the compound of formula (X) with the compound of formula (XI-1) in the presence of a base (eg, triethylamine) The reaction is carried out to obtain the compound of formula (I-1), which is then reacted with a halogenated reagent to obtain the compound of formula (I-2).
- a base eg, triethylamine
- R 7 , R 2 , R 3 , R', ring A, n, and X are as defined above.
- Any group of the reactants or intermediates in the above schemes can be protected with a protecting group if necessary. After the reaction is completed, select a suitable method to remove the protecting group.
- the raw materials of the reactants in the above scheme can be synthesized by the methods reported in the literature, or obtained by purchasing. Starting materials are typically from commercial sources such as Aldrich or can be readily prepared using methods well known to those skilled in the art (obtained via SciFinder, Reaxys online database).
- suitable reaction conditions and raw materials can be selected according to each situation.
- suitable reaction conditions and raw materials can be selected according to each situation.
- only one substituent can be replaced with another substituent according to the present invention, or in the same reaction step, the Other substituents replace multiple substituents.
- the individual compounds are not obtainable via the above-mentioned routes, they can be prepared by derivatizing other compounds of formula (I) or by routine variations of the described synthetic routes.
- the compound represented by the formula (I) can also be obtained by using the compound prepared in the above scheme, using conventional methods in the art, and peripheral modification to obtain other described compounds represented by the formula (I) compound of.
- the present invention also provides an intermediate whose structure is such as the compound of formula (XI),
- R 1 , R 2 , R 3 , ring A, and n are as defined above.
- the compound of formula (XI) is a compound of formula (XII) below,
- R 1 , R 2 , R 3 , and p are as defined above.
- the compound of formula (XI) is a compound of formula (XIII) below,
- R 2 , R 3 , and p are as defined above.
- the compound of formula (XI) is a compound of formula (XIV) below,
- R5 is as defined above.
- the compound of formula (XI) is a compound of formula (XV) below,
- the compound of formula (XI) is a compound of formula (XV-A) or formula (XV-B) as follows,
- the present invention also provides a method for preparing formula (XIII), comprising any of the following methods,
- Method 1 the method comprises the steps:
- R 2 , R 3 and p are as defined above;
- R 8 is a substituted or unsubstituted alkyl group, a substituted or unsubstituted aralkyl group (such as 4-methoxybenzyl, 2,4-dimethoxyl benzyl);
- X is halogen;
- Method 2 the method comprises the steps of: hydrogenating the compound of formula (XVIII) in the presence of a catalyst to obtain the compound of formula (XIII);
- R 2 , R 3 , and p are as defined above.
- the compound of formula (XVIII) can be prepared by the following method, including: reacting the compound of formula (XX) with R 2 COOH to obtain the compound of formula (XIX), and then cyclizing the compound of formula (XIX) to obtain the compound of formula ( XVIII) compounds;
- R 2 , R 3 , and p are as defined above.
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising the compound represented by the above formula (I), its stereoisomers, tautomers, isotopic labels, nitrogen oxides, solvates, polymorphs, One, two or more of pharmaceutically acceptable salts, metabolites, prodrugs;
- the pharmaceutical composition may also optionally comprise at least one pharmaceutically acceptable auxiliary material;
- the pharmaceutical composition may also optionally include at least one additional active ingredient; specifically, the pharmaceutical composition may also include compounds other than those represented by formula (I), their stereoisomers One or more active ingredients other than tautomers, isotopic labels, nitrogen oxides, solvates, polymorphs, pharmaceutically acceptable salts, metabolites, prodrugs;
- the dosage of acceptable salts, metabolites, and prodrugs above can be a therapeutically effective amount
- compositions of the present invention can be formulated into dosage forms suitable for administration by methods known in the art.
- the present invention also provides the compound represented by the above formula (I), its stereoisomers, tautomers, isotopic labels, nitrogen oxides, solvates, polymorphs, pharmaceutically acceptable salts, metabolites One, two or more of the products, prodrugs, or the use of the pharmaceutical composition in the preparation of medicines;
- the drug is an NK-3 receptor antagonist.
- the medicament is used for the prevention and/or treatment of diseases mediated by NK-3 receptors;
- the medicament is useful for the prevention and/or treatment of depression, anxiety, psychosis, schizophrenia, psychotic disorders, bipolar disorder, cognitive disorders, Parkinson's disease, Alzheimer's disease disease, attention deficit hyperactivity disorder (ADHD), pain, convulsions, obesity, inflammatory diseases, vomiting, preeclampsia, airway-related diseases, reproductive disorders, contraceptive and sex hormone-dependent diseases or gynecological diseases-related diseases.
- depression anxiety, psychosis, schizophrenia, psychotic disorders, bipolar disorder, cognitive disorders, Parkinson's disease, Alzheimer's disease disease, attention deficit hyperactivity disorder (ADHD), pain, convulsions, obesity, inflammatory diseases, vomiting, preeclampsia, airway-related diseases, reproductive disorders, contraceptive and sex hormone-dependent diseases or gynecological diseases-related diseases.
- ADHD attention deficit hyperactivity disorder
- the sex hormone-dependent diseases include, but are not limited to, benign prostatic hyperplasia (BPH), benign prostatic hyperplasia, metastatic prostate cancer, testicular cancer, breast cancer, ovarian cancer, androgen-dependent acne, male pattern baldness, endometriosis Puberty disorders, uterine fibrosis, uterine fibroids, hormone-dependent cancers, hyperandrogenemia, hirsutism, virilization, polycystic ovary syndrome (PCOS), premenstrual dysphoric disorder (PMDD), HAIR-AN syndrome symptoms (hyperandrogenemia, insulin resistance, and acanthosis nigricans), ovarian theca cell hyperplasia (HAIR-AN with luteinizing theca cell hyperplasia in the ovarian stroma), other manifestations of high intraovarian androgen concentrations (such as Follicular maturation arrest, atresia, anovulation, dysmenorrhea, dysfunctional uterine bleeding,
- Such airway-related diseases include chronic obstructive pulmonary disease, asthma, airway hyperresponsiveness, bronchoconstriction, and cough.
- the medicament is used for the treatment and/or prevention of diseases related to menopausal syndrome, which includes symptoms such as hot flashes, sweating, palpitations, dizziness, and obesity.
- the present invention also provides a method for treating and/or preventing a disorder or disease mediated by NK3 receptors, the method comprising administering to a subject a therapeutically effective amount of a compound represented by formula (I), a stereoisomer thereof, one, two or more of tautomers, isotopic labels, nitrogen oxides, solvates, polymorphs, pharmaceutically acceptable salts, metabolites, prodrugs;
- a compound represented by formula (I) a stereoisomer thereof, one, two or more of tautomers, isotopic labels, nitrogen oxides, solvates, polymorphs, pharmaceutically acceptable salts, metabolites, prodrugs
- the condition or disease is depression, anxiety, psychosis, schizophrenia, psychotic disorder, bipolar disorder, cognitive impairment, Parkinson's disease, Alzheimer's disease, attention deficit hyperactivity disorder (ADHD), pain, convulsions, obesity, inflammatory disorders, vomiting, preeclampsia, airway-related disorders, reproductive disorders, contraceptive and sex hormone-dependent disorders or disorders related to gynecological disorders.
- ADHD attention deficit hyperactivity disorder
- the sex hormone-dependent diseases include, but are not limited to, benign prostatic hyperplasia (BPH), benign prostatic hyperplasia, metastatic prostate cancer, testicular cancer, breast cancer, ovarian cancer, androgen-dependent acne, male pattern baldness, endometriosis Puberty disorders, uterine fibrosis, uterine fibroids, hormone-dependent cancers, hyperandrogenemia, hirsutism, virilization, polycystic ovary syndrome (PCOS), premenstrual dysphoric disorder (PMDD), HAIR-AN syndrome symptoms (hyperandrogenemia, insulin resistance, and acanthosis nigricans), ovarian theca cell hyperplasia (HAIR-AN with luteinizing theca cell hyperplasia in the ovarian stroma), other manifestations of high intraovarian androgen concentrations (such as Follicular maturation arrest, atresia, anovulation, dysmenorrhea, dysfunctional uterine bleeding,
- Such airway-related diseases include chronic obstructive pulmonary disease, asthma, airway hyperresponsiveness, bronchoconstriction, and cough.
- the disorder or disease is a disease associated with a menopausal syndrome comprising symptoms such as hot flashes, sweating, palpitations, dizziness, and obesity.
- the method comprises administering to a patient in need thereof a therapeutically effective amount of a compound of formula (I), its stereoisomers, tautomers, isotopic labels, nitrogen oxides, solvates, polymorphs , one, two or more of pharmaceutically acceptable salts, metabolites, prodrugs.
- the patient is a warm-blooded animal, more preferably a human.
- halogen refers to fluorine (F), chlorine (Cl), bromine (Br) or iodine (I).
- alkyl Unless otherwise stated, the definitions of terms herein apply equally to groups containing that term, eg the definitions of alkyl also apply to alkoxy, alkylamino, dialkylamino, haloalkyl, haloalkoxy, etc. alkyl.
- substituted means that one or more hydrogen atoms in a given structure have been replaced with a specified substituent.
- the substituents are independent of each other, that is, the one or more substituents may be different from each other or the same of.
- a substituent group may be substituted at various substitutable positions of the substituted group. When more than one position in a given formula can be substituted with one or more substituents selected from a particular group, the substituents can be substituted identically or differently at each position.
- the description method "...independently selected from” used in the present invention should be understood in a broad sense, meaning that the described individuals are independent of each other, and can be are independently selected from the same or different specific groups.
- the description mode "...independently selected from” can either mean that in different groups, the specific options expressed between the same symbols do not affect each other; it can also mean that in the same group, the same symbols The specific options expressed between them do not affect each other.
- C1-6 alkyl specifically refers to the independently disclosed C1 alkyl, C2 alkyl, C3 alkyl, C4 alkyl, C5 alkyl or C6 alkyl .
- linking substituents are described.
- the Markush variables listed for that group should be understood to be the linking group.
- the Markush group definition for that variable recites “alkyl” or “aryl”, it should be understood that the “alkyl” or “aryl” respectively represents the linking An alkylene group or an arylene group.
- alkyl or alkylene refers to a straight or branched chain saturated hydrocarbon group having 1 to 40 carbon atoms.
- C 1-10 alkyl means straight and branched chains having 1, 2, 3, 4, 5 or 6 carbon atoms alkyl.
- the alkyl group may be optionally substituted with one or more substituents described herein.
- the alkyl or alkylene group contains 1-12 carbon atoms; in other embodiments, the alkyl or alkylene group contains 1-6 carbon atoms; in still other embodiments , the alkyl or alkylene group contains 1-4 carbon atoms.
- alkyl groups include, but are not limited to, methyl, ethyl, propyl, butyl, pentyl, hexyl, isopropyl, isobutyl, sec-butyl, tert-butyl, isopentyl, 2-methylbutyl, 1-methylbutyl, 1-ethylpropyl, 1,2-dimethylpropyl, neopentyl, 1,1-dimethylpropyl, 4-methylpentyl , 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 2-ethylbutyl, 1-ethylbutyl, 3,3-dimethylbutyl, 2,2 -Dimethylbutyl, 1,1-dimethylbutyl, 2,3-dimethylbutyl, 1,3-dimethylbutyl or 1,2-dimethylbutyl, etc. or their isomer.
- alkylene group include, but are not limited to, m
- alkenyl refers to a straight or branched chain hydrocarbon group containing 2 to 40 carbon atoms in which there is at least one site of unsaturation, i.e., a carbon-carbon sp double bond, which can be separated from each other or co- yoke. wherein the alkenyl group may be optionally substituted with one or more substituents described herein, including the “cis” and “tans” positions, or the "E” and “Z” positions, "C 2-10 alkenyl” and “C 2-6 alkenyl” are preferred.
- C 2-6 alkenyl is understood to mean preferably a straight or branched monovalent hydrocarbon radical comprising one or more double bonds and having 2, 3, 4, 5 or 6 carbon atoms, in particular 2 or 3 carbon atoms ("C 2-3 alkenyl").
- alkenyl groups include, but are not limited to, vinyl, allyl, (E)-2-methylvinyl, (Z)-2-methylvinyl, (E)-butan-2- Alkenyl, (Z)-but-2-enyl, (E)-but-1-enyl, (Z)-but-1-enyl, pent-4-enyl, (E)-pent-3 -Alkenyl, (Z)-pent-3-enyl, (E)-pent-2-enyl, (Z)-pent-2-enyl, (E)-pent-1-enyl, (Z )-pent-1-enyl, hex-5-enyl, (E)-hex-4-enyl, (Z)-hex-4-enyl, (E)-hex-3-enyl, ( Z)-hex-3-enyl, (E)-hex-2-enyl, (Z)-hex-2-enyl, (Z)-
- alkynyl is understood to mean a straight-chain or branched hydrocarbon group containing one or more triple bonds and having 2 to 40 carbon atoms, preferably "C2 - C10 -alkynyl""C2 - C 6 -alkynyl".
- C 2 -C 6 -alkynyl is to be understood as meaning preferably a straight-chain or branched monovalent hydrocarbon radical comprising one or more triple bonds and having 2, 3, 4, 5 or 6 carbon atoms, in particular is 2 or 3 carbon atoms ("C2-C3-alkynyl").
- Said C2 - C6 -alkynyl is, for example, ethynyl, prop-1-ynyl, prop-2-ynyl, but-1-ynyl, but-2-ynyl, but-3-ynyl, Pent-1-ynyl, pent-2-ynyl, pent-3-ynyl, pent-4-ynyl, hex-1-ynyl, hex-2-ynyl, hex-3-ynyl, hexyl -4-ynyl, hex-5-ynyl, 1-methylprop-2-ynyl, 2-methylbut-3-ynyl, 1-methylbut-3-ynyl, 1-methyl But-2-ynyl, 3-methylbut-1-ynyl, 1-ethylprop-2-ynyl, 3-methylpent-4-ynyl, 2-methylpent-4-ynyl , 1-methylpent-4-ynyl,
- cycloalkyl is understood to mean a saturated or partially unsaturated monocyclic, bicyclic hydrocarbon ring or polycyclic cycloalkane having 3 to 40 carbon atoms, preferably a " C3-10 cycloalkyl".
- C 3-10 cycloalkyl is understood to mean a saturated or partially unsaturated monocyclic, bicyclic hydrocarbon ring or polycyclic cycloalkane having 3, 4, 5, 6, 7, 8, 9 or 10 carbon atom.
- the C 3-10 cycloalkyl group can be a monocyclic hydrocarbon group, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl or cyclodecyl, or a bicyclic Hydrocarbyl such as decalin ring.
- heterocyclyl is understood to mean a saturated or partially unsaturated monocyclic, bicyclic hydrocarbon ring or polycyclic cycloalkane containing 1 to 5 total ring atoms of heteroatoms independently selected from N, O and S.
- heterocyclyl means a saturated or partially unsaturated monocyclic, bicyclic hydrocarbon ring or polycyclic alkane containing 1-5, preferably 1-3, independently selected from N, O and S Heteroatoms such as 1, 2, 3 heteroatoms independently selected from N, O and S.
- the heterocyclyl group can be attached to the remainder of the molecule through any of the carbon atoms or a nitrogen atom, if present.
- the heterocyclic group may include, but is not limited to: 4-membered ring, such as azetidinyl, oxetanyl; 5-membered ring, such as tetrahydrofuranyl, dioxolyl, pyrrole Alkyl, imidazolidinyl, pyrazolidinyl, pyrrolinyl; or 6-membered ring, such as tetrahydropyranyl, piperidinyl, morpholinyl, dithianyl, thiomorpholinyl, tetrahydropyridine group, 2H-pyranyl, piperazinyl, or trithianyl; or a 7-membered ring such as diazepanyl.
- 4-membered ring such as azetidinyl, oxetanyl
- 5-membered ring such as tetrahydrofuranyl, dioxolyl, pyrrole Alkyl, imidazo
- the heterocyclyl group can be benzo-fused.
- the heterocyclyl group may be bicyclic, such as, but not limited to, a 5,5 membered ring, such as a hexahydrocyclopento[c]pyrrole-2(1H)-yl ring, or a 5,6 membered bicyclic ring, such as a hexahydropyrrole
- the [1,2-a]pyrazin-2(1H)-yl ring may be partially unsaturated, i.e.
- it may contain one or more double bonds such as, but not limited to, 2,5-dihydro-1H-pyrrolyl, 4H-[1,3,4]thiadiene oxazinyl, 4,5-dihydrooxazolyl, or 4H-[1,4]thiazinyl, alternatively, it may be benzo-fused, such as, but not limited to, dihydroisoquinolinyl.
- the heterocyclic group is non-aromatic.
- the carbon atom on the 3-20-membered heterocyclic group can be connected with other groups, or it can be a 3-20-membered heterocyclic group Heterocyclic atoms on the ring are attached to other groups.
- the 3-20 membered heterocyclic group is selected from piperazinyl
- the nitrogen atom on the piperazinyl may be attached to other groups.
- the 3-20-membered heterocyclic group is selected from piperidinyl
- the nitrogen atom on the piperidinyl ring and the carbon atom in the para position are connected to other groups.
- aryl is understood to mean an aromatic or partially aromatic monocyclic, bicyclic or tricyclic hydrocarbon ring having 6 to 20 carbon atoms, preferably a " C6-14 aryl".
- C 6-14 aryl is to be understood as preferably denoting a monovalent aromatic or partially aromatic monocyclic, bicyclic or Tricyclic hydrocarbon rings ("C 6-14 aryl"), especially rings with 6 carbon atoms ("C 6 aryl”), such as phenyl; or biphenyl, or those with 9 carbon atoms a ring (“C 9 aryl”) such as indanyl or indenyl, or a ring having 10 carbon atoms (“C 10 aryl”) such as tetrahydronaphthyl, dihydronaphthyl or naphthyl, Either a ring with 13 carbon atoms (“C13aryl”), such as fluorenyl, or a ring with 14 carbon atoms (“C14aryl”)
- heteroaryl is understood to include monocyclic, bicyclic or tricyclic aromatic ring systems, including aromatic or partially aromatic, having 5 to 20 ring atoms and containing 1 to 5 atoms independently selected from the group consisting of Heteroatoms of N, O and S, eg "5-14 membered heteroaryl".
- the term “5-14 membered heteroaryl” is understood to include monovalent monocyclic, bicyclic or tricyclic aromatic ring systems having 5, 6, 7, 8, 9, 10, 11, 12, 13 or 14 ring atoms, especially 5 or 6 or 9 or 10 carbon atoms, and which contain 1 to 5, preferably 1 to 3, heteroatoms each independently selected from N, O and S, and, in addition, in each may be benzo-fused.
- heteroaryl is selected from the group consisting of thienyl, furyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiazolyl oxazolyl, thi-4H-pyrazolyl, etc.
- the 5-20 membered heteroaryl When the 5-20 membered heteroaryl is substituted, it can be mono- or polysubstituted. Moreover, there is no restriction on the substitution site, for example, the hydrogen attached to the carbon atom on the heteroaryl ring may be substituted, or the hydrogen attached to the heteroatom on the heteroaryl ring may be substituted.
- a heterocyclyl, heteroaryl or heteroarylene group includes all possible isomeric forms thereof, such as positional isomers thereof.
- thienyl or thienylene includes thien-2-yl, thien-2-yl, thien-3-yl and thien-3 - base; pyrazol-1-yl, pyrazol-3-yl, pyrazol-4-yl, pyrazol-5-yl.
- alkoxy is to be understood as -O-alkyl, as defined above.
- haloalkyl is understood to mean that the H on the alkyl group, as defined above, is partially or fully substituted by halogen.
- haloalkoxy is to be understood as -O-haloalkyl, as defined above.
- Stereochemical definitions and rules used in the present invention generally follow S.P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; and Eliel, E. and Wilen, S., "Stereo chemistry of Organic Compounds”, John Wiley & Sons, Inc., New York, 1994.
- Stereoisomers refer to compounds that have the same chemical structure, but differ in the arrangement of atoms or groups in space. Stereoisomers include enantiomers, diastereomers, conformational isomers (rotamers), geometric isomers (cis/trans), atropisomers, etc. .
- Enantiomer refers to two nonsuperimposable, but mirror-image isomers of a compound.
- Diastereomer refers to a stereoisomer having two or more centers of chirality and whose molecules are not mirror images of each other. Diastereomers have different physical properties such as melting point, boiling point, spectral properties and reactivity. Diastereomeric mixtures can be separated by high resolution analytical procedures such as electrophoresis and chromatography, eg HPLC.
- any asymmetric atom (eg, carbon, etc.) of the compounds disclosed herein may exist in racemic or enantiomerically enriched forms, such as (R)-, (S)- or (R,S)-configurations exist.
- each asymmetric atom has at least 0% enantiomeric excess in the (R)- or (S)-configuration, at least 60% enantiomeric excess, at least 70% enantiomeric excess, at least 80% enantiomeric excess, at least 90% enantiomeric excess, at least 95% enantiomeric excess, or at least 99% enantiomeric excess.
- the resulting mixtures of any stereoisomers may be separated into pure or substantially pure geometric isomers, enantiomers, diastereomers on the basis of differences in the physicochemical properties of the components, for example, by chromatography and/or fractional crystallization.
- diastereomers are prepared from the mixture by reaction with an optically active resolving agent.
- optically active acids such as the R and S forms of tartaric acid, diacetyltartaric acid, dibenzoyltartaric acid, mandelic acid, malic acid, lactic acid, suitable N-protected amino acids such as N- Benzoylproline or N-benzenesulfonylproline) or various optically active camphorsulfonic acids.
- optically active resolving agents such as dinitrobenzoylphenylglycine, cellulose triacetate or other carbohydrate derivatives or chiral derivatized methacrylate polymers immobilized on silica gel. Chromatographic enantiomeric resolution is advantageously performed.
- Suitable eluents for this purpose are aqueous or alcoholic solvent mixtures, eg hexane/isopropanol/acetonitrile.
- tautomer refers to structural isomers of different energies that are interconvertible through a low energy barrier.
- a chemical equilibrium of tautomers can be achieved if tautomerism is possible (eg, in solution).
- proton tautomers also known as prototro pictautomers
- Valence tautomers include interconversions by recombination of some of the bonding electrons.
- keto-enol tautomerism is the interconversion of pentane-2,4-dione and 4-hydroxypent-3-en-2-one tautomers.
- tautomerism is phenol-ketone tautomerism.
- a specific example of phenol-ketone tautomerism is the interconversion of pyridin-4-ol and pyridin-4(lH)-one tautomers. Unless otherwise indicated, all tautomeric forms of the compounds of the present invention are within the scope of the present invention.
- pharmaceutically acceptable refers to molecular entities and compositions that are physiologically tolerable when administered to humans and generally do not produce allergic or similar inappropriate reactions, such as gastrointestinal upset, dizziness, and the like.
- carrier refers to a diluent, adjuvant, excipient or base with which the compound is administered.
- These pharmaceutical carriers can be sterile liquids such as water and oils, including those of petroleum, animal, vegetable or synthetic origin, such as peanut oil, soybean oil, mineral oil, sesame oil and the like.
- Water and Aqueous Solutions Saline solutions and aqueous dextrose and glycerol solutions are preferred for use as carriers, especially for injectable solutions. Suitable pharmaceutical carriers are described in "Remington's Pharmaceutical Sciences” by E.W. Martin.
- prodrug refers to the conversion of a compound into a compound of formula (I) in vivo. Such conversion is effected by hydrolysis of the prodrug in blood or enzymatic conversion to the parent structure in blood or tissue.
- the prodrug compounds of the present invention can be esters.
- esters that can be used as prodrugs include phenyl esters, aliphatic (C 1-24 ) esters, acyloxymethyl esters, carbonate esters , carbamates and amino acid esters.
- a compound of the present invention contains a hydroxyl group, which can be acylated to give the compound in prodrug form.
- prodrug forms include phosphates, such as these phosphates are phosphorylated by the hydroxyl group on the parent.
- phosphates such as these phosphates are phosphorylated by the hydroxyl group on the parent.
- a complete discussion of prodrugs can be found in the following literature: T. Higuchi and V. Stella, Pro-drugsas Novel Delivery Systems, Vol. 14 of the ACSSymposium Series, Edward BR°Che, ed., Bioreversible Carriersin Drug Design, American Pharmaceutical Ass Ciation and Pergamon Press, 1987, J.
- metabolite refers to a product obtained by metabolism of a specific compound or its salt in vivo. Metabolites of a compound can be identified by techniques well known in the art, and their activity can be characterized using assays as described herein. Such products may be obtained by subjecting the administered compound to oxidation, reduction, hydrolysis, amidation, deamidation, esterification, delipidation, enzymatic cleavage, and the like. Accordingly, the present invention includes metabolites of compounds, including metabolites produced by contacting a compound of the present invention with a mammal for a sufficient period of time.
- Pharmaceutically acceptable salts may be, for example, acid addition salts of sufficiently basic compounds of the present invention having a nitrogen atom in the chain or ring.
- a “solvate” of the present invention refers to an association of one or more solvent molecules with a compound of the present invention.
- the term “hydrate” refers to an association in which the solvent molecule is water.
- N-oxide in the present invention means that when the compound contains several amine functional groups, one or more nitrogen atoms can be oxidized to form an N-oxide.
- N-oxides are N-oxides of tertiary amines or N-oxides containing nitrogen heterocyclic nitrogen atoms.
- the corresponding amines can be treated with oxidizing agents such as hydrogen peroxide or peracids (eg, peroxycarboxylic acids) to form N-oxides (see Advanced Organic Chemistry, Wiley Interscience, 4th edition, Jerry March, pages).
- oxidizing agents such as hydrogen peroxide or peracids (eg, peroxycarboxylic acids) to form N-oxides (see Advanced Organic Chemistry, Wiley Interscience, 4th edition, Jerry March, pages).
- oxidizing agents such as hydrogen peroxide or peracids (eg, peroxycarboxylic acids) to form N-oxides (see Advanced Organic Chemistry, Wiley Interscience, 4th edition, Jerry March
- isotopic labels includes, but is not limited to, isotopes of hydrogen, carbon, nitrogen, oxygen, fluorine, sulfur and chlorine (eg 2H, 3H , 13C , 14C , 15N , 18O , 17O , 18F , 35 S and 36 Cl) labeled compounds of the present invention.
- Isotopically labeled compounds of the present invention are useful in assays for the tissue distribution of the compounds and their prodrugs and metabolites; preferred isotopes for such assays include3H and14C .
- substitution with heavier isotopes such as deuterium (2H or D) can provide increased metabolic stability, which provides therapeutic advantages such as increased in vivo half-life or reduced dosage requirements.
- Isotopically labeled compounds of the present invention can generally be prepared according to the methods described herein by substituting an isotopically labeled reagent for a non-isotopically labeled reagent.
- any disease or disorder in some embodiments refers to ameliorating the disease or disorder (ie, slowing or arresting or alleviating the development of the disease or at least one clinical symptom thereof). In other embodiments, “treating” refers to alleviating or improving at least one physical parameter, including a physical parameter that may not be perceived by a patient. In other embodiments, “treating” refers to modulating a disease or disorder physically (eg, stabilizing an observable symptom) or physiologically (eg, stabilizing a physical parameter), or both. In other embodiments, “treating” refers to preventing or delaying the onset, occurrence or worsening of a disease or disorder.
- an effective amount refers to an amount of a compound of the present invention sufficient to achieve the intended application, including but not limited to disease treatment as defined below.
- a therapeutically effective amount may vary depending on the intended application (in vitro or in vivo), or the subject and disease condition being treated such as the weight and age of the subject, the severity of the disease condition and the mode of administration, etc., which It can be easily determined by one of ordinary skill in the art.
- the specific dose will vary depending on the particular compound chosen, the dosing regimen followed, whether it is administered in combination with other compounds, the timing of administration, the tissue to which it is administered, and the physical delivery system on which it is carried.
- the structures of the compounds of the present invention are determined by nuclear magnetic resonance (NMR) or/and liquid chromatography-mass spectrometry (LC-MS). NMR chemical shifts ([delta]) are given in parts per million (ppm). NMR was measured by Bruker AVANCE-400 nuclear magnetic instrument, and the solvent was deuterated dimethyl sulfoxide (DMSO-d 6 ), deuterated methanol (CD 3 OD) and deuterated chloroform (CDCl 3 ), and the internal standard was four Methylsilane (TMS).
- DMSO-d 6 dimethyl sulfoxide
- CD 3 OD deuterated methanol
- CDCl 3 deuterated chloroform
- TMS Methylsilane
- the thin layer chromatography silica gel plate uses Yantai Huanghai HSGF254 or Qingdao GF254 silica gel plate, the specifications used for TLC are 0.15mm ⁇ 0.20mm, and the specifications used for TLC separation and purification products are 0.4mm ⁇ 0.5mm.
- Column chromatography generally uses Yantai Huanghai silica gel 200-300 mesh silica gel as the carrier.
- the starting materials in the examples of the present invention are known and commercially available, or can be synthesized using or according to methods known in the art.
- 2,3-Dimethylpyrazine 001a (10g, 92.47mmol) was added to carbon tetrachloride (250mL) solvent, followed by N-chlorosuccinimide (14.83g, 110.96mmol), peroxide Benzoyl (224 mg, 9.25 mmol).
- the reaction solution was reacted at 80°C for 16 hours under nitrogen protection. After the completion of the reaction monitored by LCMS, the solvent was spin-dried and water (150 mL) and dichloromethane (3 ⁇ 100 mL) were added to extract and separate the liquid, then washed with saturated sodium chloride solution (50 mL), dried over anhydrous sodium sulfate, and filtered with suction.
- the seventh step preparation of 5-(7-(4-methoxybenzyl)-8-methyl-5,6,7,8-tetrahydroimidazo[1,5-a]pyrazin-3-yl )-3-methyl-1,2,4-thiadiazole
- the SFC split conditions are:
- HPLC 254nm (95.03%), 214nm (95.95%).
- HPLC 254nm (100%), 214nm (100%).
- reaction solution was concentrated under reduced pressure and purified by reverse phase column (55% acetonitrile/water) to obtain 7-(4-fluorobenzoyl)-N,8-dimethyl-3-(3-methyl-1, 2,4-thiadiazol-5-yl)-5,6,7,8-tetrahydroimidazo[1,5-a]pyrazine-1-carboxamide (007) (2.19 mg, white solid), Yield: 10%.
- HPLC 254nm (99.4%), 214nm (97.4%).
- the first step preparation of [1-[(diphenylmethylene)amino]-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5, 6-Dihydroimidazo[1,5-a]pyrazin-7(8H)-yl](4-fluorophenyl)methanone
- HPLC 254nm (99.2%), 214nm (99.6%).
- reaction solution was extracted with dichloromethane (10 mL), washed with saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered with suction, the filtrate was concentrated under reduced pressure, and the obtained residue was purified by reverse phase column (40% acetonitrile/water). to give (1-bromo-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6-dihydroimidazo[1,5-a]pyridine Azin-7(8H)-yl)(4-fluorophenyl)methanone 009 (16 mg, white solid), yield: 26%.
- the first step preparation of N-(7-(4-fluorobenzoyl)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6 ,7,8-Tetrahydroimidazo[1,5-a]pyrazin-1-yl)acetamide
- HPLC 254nm (98.4%), 214nm (98.2%).
- HPLC 254nm (99.6%), 214nm (99.9%).
- HPLC 99.25% (214nm), 99.62% (254nm).
- the first step preparation (R)-(1-(4-aminophenyl)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5, 6-Dihydroimidazo[1,5-a]pyrazin-7(8H)-yl)(4-fluorophenyl)methanone
- HPLC 98.60% (214nm), 98.40% (254nm).
- HPLC 100% (214nm), 99.51% (254nm).
- HPLC 96.85% (214nm), 96.81% (254nm).
- reaction solution was heated to 90°C for 4 hours, then the reaction solution was cooled to room temperature and 30 mL of water was added, then extracted with ethyl acetate (2 ⁇ 40 mL), the organic phases were combined, and anhydrous sodium sulfate was used. Dry and concentrate.
- HPLC 98.45% (214nm), 98.68% (254nm).
- the first step add (4-fluorophenyl)(8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl) to a 10 mL microwave tube equipped with a stirring bar -5,6-Dihydroimidazo[1,5-a]pyrazin-7(8H)-ylmethanone 024a (60 mg, 0.14 mmol), methylboronic acid (25 mg, 0.42 mmol), Pd(dppf)Cl 2 (15 mg, 0.032 mmol) and potassium carbonate (58 mg, 0.42 mmol) followed by the solvent dioxane/water (5:1, 2/0.4 mL).
- the obtained racemic product 024b was prepared by SFC (CO 2 /MeOH(0.2%NH 4 ⁇ OH)) to obtain (R)-(1,8-dimethyl-3-(3-methyl-) 1,2,4-Thiadiazol-5-yl)-5,6-dihydroimidazo[1,5-a]pyrazin-7(8H)-yl)(4-fluorophenyl)methanone 024 (6.63 mg, white solid), yield: 23%.
- HPLC 254nm (96.3%), 214nm (98.6%).
- HPLC 254nm (96.1%), 214nm (95.2%).
- 4-oxopyrimidine 027a (800 mg, 8.33 mmol) was dissolved in 8 mL of acetonitrile, phosphorus oxybromide (1.53 g, 10.00 mmol) was added, the reaction solution was heated to 100 ° C for 4 hours, the reaction solution was cooled to room temperature, and added Quenched with 20 mL of water, extracted with ethyl acetate (2 x 40 mL), the organic phases were sequentially combined, dried over anhydrous sodium sulfate and concentrated. 4-Bromopyrimidine 027b was obtained (380 mg, orange oil), yield: 27%.
- 4-Bromopyrimidine 027b (318 mg, 2.00 mmol) was dissolved in dioxane, followed by adding hexamethyldistin (655 mg, 2.00 mmol) and Pd(PPh 3 ) 4 (116 mg, 0.10 mmol) under nitrogen protection , the reaction solution was heated to 110° C. for 2 hours. After the reaction was completed, the reaction solution was cooled to room temperature, diatomaceous earth was added, and the solvent was removed by rotary evaporation. /petroleum ether) after purification to obtain 4-(trimethyltinyl)pyrimidine 027c (180 mg, colorless oil), yield: 36%.
- the third step preparation of 4-[(4R)-5-[(4-fluorophenyl)carbonyl]-4-methyl-1-(3-methyl-1,2,4-thiadiazole-5- yl)-4H,6H,7H-imidazo[1,5-a]pyrazin-3-yl]pyrimidine
- HPLC 99.34% (214nm), 98.34% (254nm).
- reaction solution was cooled to room temperature and concentrated, and the obtained residue was separated by silica gel column (33% ethyl acetate/petroleum ether), and purified by reverse phase column (54% acetonitrile/water) to obtain (1 -(Benzylthio)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6-dihydroimidazo[1,5-a] Pyrazine-7(8H))-(4-fluorophenyl)methanone 038b (85 mg, pale yellow solid), yield: 58%.
- the third step preparation of 7-(4-fluorobenzoyl)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6,7, 8-Tetrahydroimidazo[1,5-a]pyrazine-1-sulfonamide
- HPLC 99.72% (214nm), 99.18% (254nm).
- the first step preparation of 7-(4-fluorobenzoyl)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6,7, Methyl 8-tetrahydroimidazo[1,5-a]pyrazine-1-carboxylate
- the third step preparation of 7-(4-fluorobenzoyl)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6,7, 8-Tetrahydroimidazo[1,5-a]pyrazine-1-carboxamide
- HPLC 254nm (98.6%), 214nm (98.6%).
- reaction solution was stirred at 100 degrees Celsius for 16 hours under nitrogen protection, and the reaction solution was sequentially extracted with ethyl acetate (20 mL), washed with saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered with suction, and the filtrate was decompressed.
- HPLC 254nm (90.1%), 214nm (95.4%).
- the seventh step preparation (R)-N-(7-(4-fluorobenzofuran-7-carbonyl)-8-methyl-3-(3-methyl-1,2,4-thiadiazole- 5-yl)-5,6,7,8-tetrahydroimidazo[1,5-a]pyrazin-1-yl)acetamide and (R)-N-acetyl-N-(7-(4 -Fluorobenzofuran-7-carbonyl)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6,7,8-tetrahydroimidazole Io[1,5-a]pyrazin-1-yl)acetamide
- reaction solution was stirred at 90° C. for 3 hours under nitrogen protection. After the reaction solution was filtered, the filtrate was concentrated, and the obtained residue was purified by pre-HPLC (acetonitrile/water) to obtain compound (053) (8.5 mg, white solid), yield: 26%.
- 5-Fluoropicolinic acid 055a 80 mg, 0.57 mmol was dissolved in dichloromethane (3 mL), then oxalyl chloride (360 mg, 2.84 mmol) was added dropwise, followed by a drop of N,N-dimethylformamide. The reaction mixture was stirred at room temperature for 1 hour. The reaction solution was concentrated to dryness to obtain 055b, which was directly used in the next reaction.
- the first step preparation of 6-fluoronicotinic acid chloride
- 6-Fluoropyridine-3-carboxylic acid 056a (50 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), then oxalyl chloride (225 mg, 1.75 mmol) was added dropwise to it, followed by a drop of N,N-dimethylmethane formamide. The reaction mixture was stirred at room temperature for 1 hour. The reaction solution was concentrated to dryness to obtain 056b, which was directly used in the next reaction.
- the second step is to prepare (R)-(6-fluoropyridin-3-yl)(8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5, 6-Dihydroimidazo[1,5-a]pyrazin-7(8H)-yl)methanone
- 6-Fluoronicotinic acid chloride (30.5 mg, 0.19 mmol) 056b was dissolved in dichloromethane (2 mL) and added dropwise to (R)-3-methyl-5-(8-methyl-5,6, 7,8-Tetrahydroimidazo[1,5-a]pyrazin-3-yl)-1,2,4-thiadiazole (30 mg, 0.1275 mmol) and triethylamine (32.0 mg, 0.312 mmol) The solution in methyl chloride (2 mL) was stirred at room temperature for 0.5 h.
- reaction solution was quenched with saturated ammonium chloride (2 mL) and saturated sodium bicarbonate (2 mL), extracted with ethyl acetate (2 ⁇ 10 mL), and then the organic layers were combined successively, dried over sodium sulfate, filtered, and the organic phase was concentrated.
- 5-Nitrofuran-2-carboxylic acid 058a (3.00 g, 19.10 mmol) was added to N,N-dimethylformamide (30 mL), followed by benzyl bromide (4.90 g, 28.60 mmol), potassium carbonate (7.92 g, 57.30 mmol). The reaction was carried out at room temperature for 16 hours.
- Benzyl 5-nitrofuran-2-carboxylate 058b (2.50 g, 10.01 mmol) was added to sulfolane (30 mL), followed by potassium fluoride (2.93 g, 50.4 mmol), tetraphenylphosphine bromide (420 mg) , 1.0 mmol), the reaction solution was reacted at 140 °C for 2 hours.
- Benzyl 5-fluorofuran-2-carboxylate 058c (200 mg, 0.91 mmol) was dissolved in methanol (5 mL), and Pd/C (10 mg, 0.091 mmol) was added thereto. The reaction solution was reacted at 25°C for half an hour in a hydrogen atmosphere. After the reaction was monitored by LCMS, the catalyst was filtered off, methanol was removed by rotary evaporation, and the obtained residue was added to acetonitrile and water to lyophilize to obtain 5-fluorofuran-2-carboxylic acid 058d (100 mg, white body), yield: 78%.
- the first step preparation of (R)-N-(7-(4-fluorobenzoyl)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl) -5,6,7,8-Tetrahydroimidazo[1,5-a]pyrazin-1-yl)-N-(methylsulfonyl)methanesulfonamide
- Trimethylacetamide 066a (2.02 g, 20.00 mmol) and chlorocarbonylsulfinyl chloride (2.88 g, 22.00 mmol) were dissolved in dry toluene (15 mL), and the reaction solution was reacted at 120° C. for 3 hours. Cool to room temperature and add saturated sodium bicarbonate solution (50 mL), then extract with ethyl acetate (2 x 50 mL). The organic phases were combined, dried over anhydrous sodium sulfate, and concentrated to obtain a reaction intermediate. This intermediate was dissolved in toluene (15 mL), and ethyl cyanoformate (3.96 g, 40.00 mmol) was added thereto.
- Ethyl 3-tert-butyl-1,2,4-thiadiazole-5-carboxylate 066b (1.24 g, 5.79 mmol) was dissolved in a mixed solvent of tetrahydrofuran (10 mL) and water (2.5 mL), and added to To this was added lithium hydroxide monohydrate (291 mg, 6.94 mmol). The reaction solution was reacted at 25°C for 1 hour. The tetrahydrofuran was removed by rotary evaporation, and 1M hydrochloric acid was added dropwise to the residue to neutralize the excess lithium hydroxide. The mixture was lyophilized to give 3-tert-butyl-1,2,4-thiadiazole-5-carboxylic acid 066c (1.08 g, white solid), yield: 98%.
- the seventh step preparation of 3-tert-butyl-5- ⁇ 4-methyl-4H, 5H, 6H, 7H imidazo[1,5-a]pyrazin-1-yl ⁇ -1,2,4-thione oxadiazole
- the eighth step preparation (3-(3-tert-butyl-1,2,4-thiadiazol-5-yl)-8-methyl-5,6-dihydroimidazo[1,5-a] Pyrazin-7(8H)-yl)(4-fluorophenyl)methanone
- HPLC 98.61% (214nm), 97.08% (254nm).
- HPLC 99.81% (214nm), 100% (254nm).
- the reaction was heated and stirred at 120°C for 16 hours under nitrogen protection. After the reaction was completed, water was added to quench, extracted with ethyl acetate (3 ⁇ 10 mL), the organic phases were combined and washed with saturated brine (20 mL). The organic phases were combined, dried over anhydrous sodium sulfate, and concentrated.
- reaction solution was heated to 110°C for 16 hours. After the reaction was completed, it was quenched by adding water and extracted with ethyl acetate (3 x 10 mL). The organic phases were combined, washed with saturated brine (20 mL), dried over anhydrous sodium sulfate, and concentrated.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Description
















































































































































































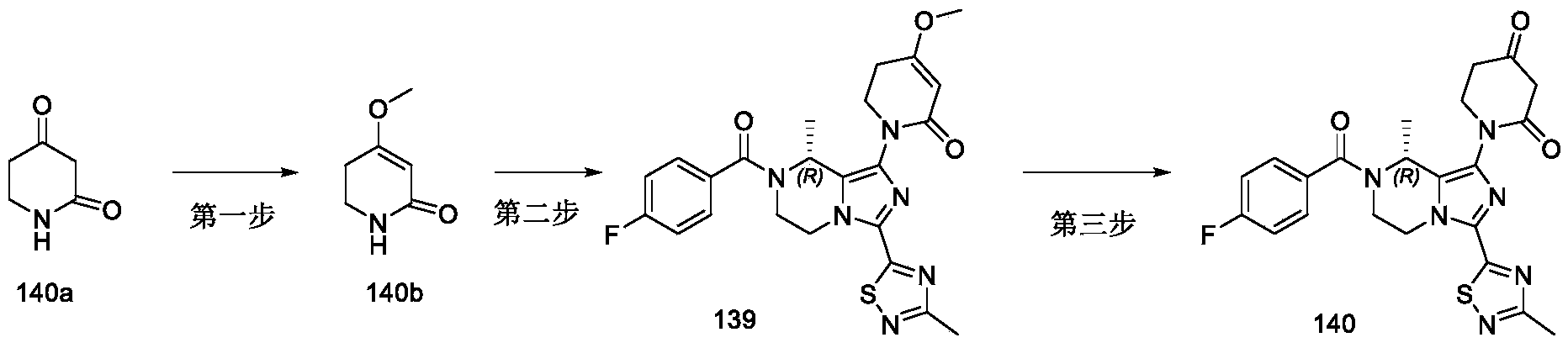































































| 化合物编号 | IC 50(nM) |
| Fezolinetant | 734.3 |
| 005 | 64.63 |
| 010 | 33.42 |
| 026 | 34.96 |
| 027 | 29.69 |
| 028 | 56.35 |
| 033 | 50.58 |
| 034 | 30.44 |
| 040 | 51.71 |
| 043 | 29.65 |
| 044 | 17.01 |
| 046 | 7.02 |
| 053 | 20.15 |
| 060 | 26.94 |
| 063 | 47.63 |
| 064 | 80.11 |
| 068 | 8.36 |
| 069 | 7.43 |
| 072 | 16.82 |
| 074 | 13.64 |
| 077 | 14.54 |
| 080 | 84.47 |
| 084 | 23.70 |
| 085 | 12.80 |
| 089 | 58.63 |
| 096 | 67.93 |
| 100 | 51.87 |
| 101 | 20.24 |
| 103 | 5.70 |
| 104 | 30.98 |
| 107 | 15.39 |
| 114 | 28.72 |
| 115 | 22.73 |
| 116 | 11.92 |
| 117 | 72.52 |
| 118 | 51.68 |
| 120 | 18.81 |
| 123 | 23.01 |
| 128 | 84.62 |
| 129 | 42.50 |
| 132 | 16.11 |
| 133 | 18.60 |
| 169 | 65.69 |
| 184 | 46.5 |
| 186 | 88.05 |
| 197 | 215.7 |
| 204 | 109 |
| 211 | 283.6 |
| 214 | 246.3 |
| 218 | 224.7 |
| 220 | 28.01 |



Claims (16)
- 一种如式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,
 其中,环A为至少含有两个N原子的杂环基;n为1-5的整数;R 1选自H、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R a取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 2选自卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R a取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R a相同或不同,彼此独立地选自H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 3相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-OCO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46,无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基。R’选自H、-L-Ar、-L-R 10;R 10为烷基、烯基、炔基;L不存在或选自-(CH 2) m-CO-、-(CH 2) m-CS-、-(CH 2) m-SO 2-、-(CH 2) m-SO-、亚烷基;m为0,1,2,3;Ar为芳基、杂芳基、环烷基或杂环基,所述芳基、杂芳基、环烷基和杂环基,任选地可以被一个或多个R 4取代;或者,任意两个相邻或不相邻的R 4连接形成未取代或任选被一个或多个R b取代的环烷基、杂环基、芳基或杂芳基;R 4相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 11、R 12、R 13、R 14、R 15、R 16、R 17、R 18、R 19、R 20、R 21、R 22、R 23、R 24、R 25、R 26相同或不同,彼此独立地选自H、氧代、硫代、卤素、-OH、无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 15和R 16、R 17和R 18、R 19和R 20、R 22和R 23可以成环,形成至少含有一个N原子的杂环基或杂芳基;所述杂环基或杂芳基可以任选被一个或多个R b取代,并且相邻或不相邻的R b可以连接成环形成环烷基、杂环基、芳基或杂芳基,所述环烷基、杂环基、芳基或杂芳基可以进一步被一个或多个R d所取代;R b、R c相同或不同,彼此独立地选自H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、 -O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46,或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R d相同或不同,彼此独立的选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;a为1或2;R 31、R 32、R 33、R 34、R 35、R 36、R 37、R 38、R 39、R 40、R 41、R 42、R 43、R 44、R 45、R 46相同或不同,彼此独立地选自H、卤素、-OH、烷基、环烷基、芳基、杂芳基、杂环基;优选地,式(I)中R’为-L-Ar;环A为至少含有两个N原子的杂环基;n为1-5的整数;R 1选自H、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R a取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 2选自卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R a取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R a相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 3相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-OCO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46,无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基。L为-(CH 2) m-CO-、-(CH 2) m-CS-、-(CH 2) m-SO 2-;m为0,1,2,3;Ar为芳基、杂芳基、环烷基或杂环基,所述芳基、杂芳基、环烷基和杂环基,任选地可以被一个或多个R 4取代;或者,任意两个相邻或不相邻的R 4连接形成未取代或任选被一个或多个R b取代的环烷基、杂环基、芳基或杂芳基;R 4相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 11、R 12、R 13、R 14、R 15、R 16、R 17、R 18、R 19、R 20、R 21、R 22、R 23、R 24、R 25、R 26相同或不同,彼此独立地选自H、氧代、硫代、卤素、-OH、无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 15和R 16、R 17和R 18、R 19和R 20、R 22和R 23可以成环,形成至少含有一个N原子的杂环基或杂芳基;所述杂环基或杂芳基可以任选被一个或多个R b取代,并且相邻或不相邻的R b可以连接成环形成环烷基、杂环基、芳基或杂芳基,所述环烷基、杂环基、芳基或杂芳基可以进一步被一个或多个R d所取代;R b、R c相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46,或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R d相同或不同,彼此独立的选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;a为1或2;R 31、R 32、R 33、R 34、R 35、R 36、R 37、R 38、R 39、R 40、R 41、R 42、R 43、R 44、R 45、R 46相同或不同,彼此独立地选自H、卤素、-OH、烷基、环烷基、芳基、杂芳基、杂环基。
其中,环A为至少含有两个N原子的杂环基;n为1-5的整数;R 1选自H、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R a取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 2选自卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R a取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R a相同或不同,彼此独立地选自H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 3相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-OCO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46,无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基。R’选自H、-L-Ar、-L-R 10;R 10为烷基、烯基、炔基;L不存在或选自-(CH 2) m-CO-、-(CH 2) m-CS-、-(CH 2) m-SO 2-、-(CH 2) m-SO-、亚烷基;m为0,1,2,3;Ar为芳基、杂芳基、环烷基或杂环基,所述芳基、杂芳基、环烷基和杂环基,任选地可以被一个或多个R 4取代;或者,任意两个相邻或不相邻的R 4连接形成未取代或任选被一个或多个R b取代的环烷基、杂环基、芳基或杂芳基;R 4相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 11、R 12、R 13、R 14、R 15、R 16、R 17、R 18、R 19、R 20、R 21、R 22、R 23、R 24、R 25、R 26相同或不同,彼此独立地选自H、氧代、硫代、卤素、-OH、无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 15和R 16、R 17和R 18、R 19和R 20、R 22和R 23可以成环,形成至少含有一个N原子的杂环基或杂芳基;所述杂环基或杂芳基可以任选被一个或多个R b取代,并且相邻或不相邻的R b可以连接成环形成环烷基、杂环基、芳基或杂芳基,所述环烷基、杂环基、芳基或杂芳基可以进一步被一个或多个R d所取代;R b、R c相同或不同,彼此独立地选自H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、 -O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46,或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R d相同或不同,彼此独立的选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;a为1或2;R 31、R 32、R 33、R 34、R 35、R 36、R 37、R 38、R 39、R 40、R 41、R 42、R 43、R 44、R 45、R 46相同或不同,彼此独立地选自H、卤素、-OH、烷基、环烷基、芳基、杂芳基、杂环基;优选地,式(I)中R’为-L-Ar;环A为至少含有两个N原子的杂环基;n为1-5的整数;R 1选自H、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R a取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 2选自卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R a取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R a相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 3相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-OCO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46,无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基。L为-(CH 2) m-CO-、-(CH 2) m-CS-、-(CH 2) m-SO 2-;m为0,1,2,3;Ar为芳基、杂芳基、环烷基或杂环基,所述芳基、杂芳基、环烷基和杂环基,任选地可以被一个或多个R 4取代;或者,任意两个相邻或不相邻的R 4连接形成未取代或任选被一个或多个R b取代的环烷基、杂环基、芳基或杂芳基;R 4相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,或者无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 11、R 12、R 13、R 14、R 15、R 16、R 17、R 18、R 19、R 20、R 21、R 22、R 23、R 24、R 25、R 26相同或不同,彼此独立地选自H、氧代、硫代、卤素、-OH、无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R 15和R 16、R 17和R 18、R 19和R 20、R 22和R 23可以成环,形成至少含有一个N原子的杂环基或杂芳基;所述杂环基或杂芳基可以任选被一个或多个R b取代,并且相邻或不相邻的R b可以连接成环形成环烷基、杂环基、芳基或杂芳基,所述环烷基、杂环基、芳基或杂芳基可以进一步被一个或多个R d所取代;R b、R c相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46,或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;R d相同或不同,彼此独立的选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;a为1或2;R 31、R 32、R 33、R 34、R 35、R 36、R 37、R 38、R 39、R 40、R 41、R 42、R 43、R 44、R 45、R 46相同或不同,彼此独立地选自H、卤素、-OH、烷基、环烷基、芳基、杂芳基、杂环基。 - 如权利要求1所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,式(I)所示的化合物为如下式(I’)、式(I”)所示的化合物:
 环A、Ar、L、R 10、R 1、R 2、R 3、n具有权利要求1所述的定义;优选地,L选自-(CH 2) m-CO-、-(CH 2) m-CS-、-(CH 2) m-SO 2-或C 1-4的亚烷基;优选地,式(I)所示的化合物为如下式(IA)或式(IB)所示的化合物:
环A、Ar、L、R 10、R 1、R 2、R 3、n具有权利要求1所述的定义;优选地,L选自-(CH 2) m-CO-、-(CH 2) m-CS-、-(CH 2) m-SO 2-或C 1-4的亚烷基;优选地,式(I)所示的化合物为如下式(IA)或式(IB)所示的化合物: R 1、R 2、R 3、R’如权利要求1所定义;p为1,2,或3;优选地,式(I)所示的化合物为如下式(II)或式(II’)所示的化合物:
R 1、R 2、R 3、R’如权利要求1所定义;p为1,2,或3;优选地,式(I)所示的化合物为如下式(II)或式(II’)所示的化合物: Ar、L、R 10、R 1、R 2、R 3如权利要求1所定义;p为1,2,或3;优选地,式(I)所示的化合物为如下式(III-1)、式(III-2)、式(III-3)、式(III-4)所示的化合物:
Ar、L、R 10、R 1、R 2、R 3如权利要求1所定义;p为1,2,或3;优选地,式(I)所示的化合物为如下式(III-1)、式(III-2)、式(III-3)、式(III-4)所示的化合物: Ar、R 10、R 1、R 2、R 3如权利要求1所定义;p为1,2,或3;优选地,式(I)所示的化合物为如下式(II)所示的化合物:
Ar、R 10、R 1、R 2、R 3如权利要求1所定义;p为1,2,或3;优选地,式(I)所示的化合物为如下式(II)所示的化合物: Ar、L、R 1、R 2、R 3如权利要求1所定义;p为1,2,或3;优选地,式(I)所示的化合物为如下式(III-1)所示的化合物:
Ar、L、R 1、R 2、R 3如权利要求1所定义;p为1,2,或3;优选地,式(I)所示的化合物为如下式(III-1)所示的化合物: Ar、R 1、R 2、R 3如权利要求1所定义;p为1,2,或3。
Ar、R 1、R 2、R 3如权利要求1所定义;p为1,2,或3。 - 如权利要求1或2所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,所述R 2为取代或未取代的杂芳基,例如为取代或未取代的噻二唑、取代或未取代的噻唑、取代或未取代的咪唑、取代或未取代的三唑、取代或未取代的噁唑、取代或未取代的噁二唑;优选的,所述R 2选自如下基团:
 其中,R 5、R 6相同或不同,独立的选自H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;其中,各取代基如上所定义;或者,所述R 2为卤代烷基,例如为三氟甲基、六氟乙基、-CH 2-CF 3;优选地,所述R 2为取代或未取代的杂芳基,例如为取代或未取代的噻二唑、取代或未取代的噻唑、取代或未取代的咪唑、取代或未取代的三唑、取代或未取代的噁唑、取代或未取代的噁二唑;优选的,所述R 2选自如下基团:
其中,R 5、R 6相同或不同,独立的选自H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;其中,各取代基如上所定义;或者,所述R 2为卤代烷基,例如为三氟甲基、六氟乙基、-CH 2-CF 3;优选地,所述R 2为取代或未取代的杂芳基,例如为取代或未取代的噻二唑、取代或未取代的噻唑、取代或未取代的咪唑、取代或未取代的三唑、取代或未取代的噁唑、取代或未取代的噁二唑;优选的,所述R 2选自如下基团: 其中,R 5、R 6相同或不同,独立的选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;其中,各取代基如上所定义;或者,所述R 2为卤代烷基,例如为三氟甲基、六氟乙基、-CH 2-CF 3。
其中,R 5、R 6相同或不同,独立的选自H、氧代、硫代、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-CS-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;其中,各取代基如上所定义;或者,所述R 2为卤代烷基,例如为三氟甲基、六氟乙基、-CH 2-CF 3。 - 如权利要求1-3任一所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,所述R 1为H、D、卤素、-CN、烷基、烯基、炔基、烷氧基、环烷基、-环烷基-烷基、-环烷基-CO-烷基、-CO-烷基、-CO-烯基、-CO-炔基、-CO-芳基、-CO-杂芳基、-CO-环烷基、-CO-杂环基、-COO-烷基、-O-CO-烷基、-NH 2、烷基氨基、二烷基氨基、-CONH 2、-CONH-烷基、-CO-N(烷基) 2、-NHCO-烷基、-NHCOO-烷基、-N(烷基)(-CO-烷基)、-S(O) 2-烷基、-S(O) 2NH 2、-S(O) 2NH-烷基、-S(O) 2N(烷基) 2、-NHS(O) 2-烷基、-SH、-S-烷基,上述烷基、烯基、炔基、环烷基可以被一个或多个卤素、氧代、羟基、烷基、烷氧基取代;或者,所述R 1为-NR 15R 16,其中,R 15、R 16相同或不同,彼此独立的选自H、烷基、环烷基、羟基烷基、羟基环烷基、-烷基-O-烷基、-烷基-O-环烷基、-CO-烷基、-CO-环烷基、-CO-烯基、-CO-炔基、-CO-芳基、-CO-杂芳基、-CO-杂环基、-CO-烷基-O-烷基、-CO-烷基-NH 2、-CO-烷基-NH-烷基、-CO-烷基-N(烷基) 2、-CO-O-烷基、-S(O) 2-烷基、-S(O) 2-环烷基,上述烷基、烯基、炔基、环烷基可以被一个或多个卤素、氧代、羟基取代;或者,所述R 1为-NR 15R 16,其中,R 15、R 16相连形成至少含有一个N原子的杂环基,优选为含有一个N原子且任选再含有1-3个N、O、S的4-10元单环或双环杂环基;具体的,例如为4元环,如氮杂环丁烷基;5元环,如吡咯烷基、吡咯啉基、咪唑烷基、咪唑啉基、吡唑烷基、吡唑啉基、噁唑烷基、异噁唑烷基、噻唑烷基、异噻唑烷基;或6元环,如哌啶基、四氢吡啶、二氢吡啶、哌嗪基、吗啉基、噁嗪烷基、噻嗪烷基;或7元环,如氮杂环庚烷基;或8元环,如氮杂环辛烷基;所述含N的杂环可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述含N的杂环可以被如下1-5个或1-3个取代基所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-N(烷基) 2、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;任选地,所述含N的杂环上相邻或不相邻的取代基可以成环形成取代的或未取代的环烷基、杂环基、芳基或杂芳基,示例性的,R 1为未取代或被R d取代的如下基团:吲哚啉基、异吲哚啉基、二氢喹啉基、四氢喹啉基、二氢异喹啉基、四氢异喹啉基、氮氧杂环辛烷基、氮杂双环己基、氮杂双环庚烷、氮杂双环辛烷、氮氧杂双环己基、氮氧杂双环庚烷、氮氧杂双环辛烷;或者,所述R 1为芳基,例如苯基、萘基、蒽基,所述芳基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述芳基可以被如下1-5个或1-3个取代基所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH 2、-CO-NH-烷基、-CO-N(烷基) 2、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;或者,所述R 1为杂芳基,优选的,任选含有1-5个选自N、O、S的4-10元单环或双环杂芳基;例如吡啶基、嘧啶基、哒嗪基、吡嗪基、三嗪基、喹啉基、喹唑啉基、噌啉基、吡咯基、吲哚基、咪唑基、苯并咪唑基、吡唑基、苯并吡唑基、噻吩基、苯并噻吩基、噻唑基、噻二唑基、噁唑基、苯并噁唑基、呋喃基、苯并呋喃基等,所述杂芳基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述杂芳基可以被如下1-5个或1-3个所取代:H、D、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;或者,所述R 1为杂环基,优选的,任选含有1-5个选自N、O、S的4-10元单环或双环杂环基;例如吗啉基、哌啶基、二氢吡啶、四氢吡啶、哌嗪基、二氢吡咯、吡咯烷基、2H-吡喃基、N杂环丁烷、N杂环庚烷、N杂环辛烷、咪唑啉基、吲哚啉基、异吲哚啉基、二氢喹啉基、四氢喹啉基、二氢异喹啉基、四氢异喹啉基、噁唑烷基、噁嗪烷基、氮氧杂环庚烷基、氮氧杂环辛烷基、噻嗪烷基、噻唑烷基、异噻唑烷基、氮杂双环己基、氮杂双环庚烷基、氮杂双环辛烷基、氮氧杂环己烷基、氮氧杂环庚烷基、氮杂双环辛烷基,所述杂环基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、D、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-OCO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述杂环基可以被如下1-5个或1-3个取代基所取代:H、D、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;优选地,所述R 1为H、卤素、-CN、烷基、烷氧基、-CO-烷基、-CO-烯基、-CO-炔基、-CO-芳基、-CO-杂芳基、-CO-环烷基、-CO-杂环基、-COO-烷基、-O-CO-烷基、-NH 2、烷基氨基、二烷基氨基、-CONH 2、-CONH-烷基、-CO-N(烷基) 2、-NHCO-烷基、-NHCOO-烷基、-N(烷基)CO-烷基、-S(O) 2-烷基、-S(O) 2NH 2、-S(O) 2NH-烷基、-S(O) 2N(烷基) 2、-NHS(O) 2-烷基、-SH、-S-烷基,上述烷基、烯基、炔基、环烷基可以被一个或多个卤素、羟基取代;或者,所述R 1为-NR 15R 16,其中,R 15、R 16相同或不同,彼此独立的选自H、烷基、环烷基、羟基烷基、羟基环烷基、-烷基-O-烷基、-烷基-O-环烷基、-CO-烷基、-CO-环烷基、-CO-烯基、-CO-炔基、-CO-芳基、-CO-杂芳基、-CO-杂环基、-CO-烷基-O-烷基、-CO-烷基-NH 2、-CO-烷基-NH-烷基、-CO-烷基-N(烷基) 2、-CO-O-烷基、-S(O) 2-烷基、-S(O) 2-环烷基,上述烷基、烯基、炔基、环烷基可以被一个或多个卤素、羟基取代;或者,所述R 1为-NR 15R 16,其中,R 15、R 16相连形成至少含有一个N原子的杂环基,优选为含有一个N原子且任选再含有1-3个N、O、S的4-10元单环或双环杂环基;具体的,例如为4元环,如氮杂环 丁烷基;5元环,如吡咯烷基、吡咯啉基、咪唑烷基、咪唑啉基、吡唑烷基、吡唑啉基、噁唑烷基、异噁唑烷基、噻唑烷基、异噻唑烷基;或6元环,如哌啶基、四氢吡啶、二氢吡啶、哌嗪基、吗啉基、噁嗪烷基、噻嗪烷基;或7元环,如氮杂环庚烷基;或8元环,如氮杂环辛烷基;所述含N的杂环可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述含N的杂环可以被如下1-5个或1-3个取代基所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-N(烷基) 2、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;任选地,所述含N的杂环上相邻或不相邻的取代基可以成环形成取代的或未取代的环烷基、杂环基、芳基或杂芳基,示例性的,R 1为未取代或被R d取代的如下基团:吲哚啉基、异吲哚啉基、二氢喹啉基、四氢喹啉基、二氢异喹啉基、四氢异喹啉基、氮氧杂环辛烷基、氮杂双环己基、氮杂双环庚烷、氮杂双环辛烷、氮氧杂双环己基、氮氧杂双环庚烷、氮氧杂双环辛烷;或者,所述R 1为芳基,例如苯基、萘基、蒽基,所述芳基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述芳基可以被如下如下1-5个或1-3个取代基所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH 2、-CO-NH-烷基、-CO-N(烷基) 2、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;或者,所述R 1为杂芳基,优选的,任选含有1-5个选自N、O、S的4-10元单环或双环杂芳基;例如吡啶基、嘧啶基、哒嗪基、吡嗪基、三嗪基、喹啉基、喹唑啉基、噌啉基、吡咯基、吲哚基、咪唑基、苯并咪唑基、吡唑基、苯并吡唑基、噻吩基、苯并噻吩基、噻唑基、噻二唑基、噁唑基、苯并噁唑基、呋喃基、苯并呋喃基等,所述杂芳基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述杂芳基可以被如下1-5个或1-3个所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;或者,所述R 1为杂环基,优选的,任选含有1-5个选自N、O、S的4-10元单环或双环杂环基;例如吗啉基、哌啶基、二氢吡啶、四氢吡啶、哌嗪基、二氢吡咯、吡咯烷基、2H-吡喃基、N杂环丁烷、N杂环庚烷、N杂环辛烷、咪唑啉基、吲哚啉基、异吲哚啉基、二氢喹啉基、四氢喹啉基、二氢异喹啉基、四氢异喹啉基、噁唑烷基、噁嗪烷基、氮氧杂环庚烷基、氮氧杂环辛烷基、噻嗪烷基、噻唑烷基、异噻唑烷基、氮杂双环己基、氮杂双环庚烷基、氮杂双环辛烷基、氮氧杂环己烷基、氮氧杂环庚烷基、氮杂双环辛烷基,所述杂环基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-OCO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述杂环基可以被如下1-5个或1-3个取代基所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基。
- 如权利要求1-4任一所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,Ar为芳基,例如苯基、萘基、蒽基,所述芳基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述芳基可以被如下1-5个或1-3个取代基所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;或者,Ar为杂芳基,例如吡啶基、嘧啶基、哒嗪基、吡嗪基、三嗪基、喹啉基、喹唑啉基、噌啉基、吡咯基、吲哚基、咪唑基、苯并咪唑基、吡唑基、苯并吡唑基、噻吩基、苯并噻吩基、噻唑基、噻二唑基、噁唑基、苯并噁唑基、呋喃基、苯并呋喃基等,所述杂芳基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述杂芳基可以被如下1-5个或1-3个所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;或者,Ar为环烷基,优选为含有3-10个碳原子的环烷基,更优选为含有3-6个碳原子的环烷基,最优选为环丙基;优选地,Ar为芳基,例如苯基、萘基、蒽基,所述芳基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述芳基可以被如下如下1-5个或1-3个取代基所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基;或者,Ar为杂芳基,例如吡啶基、嘧啶基、哒嗪基、吡嗪基、三嗪基、喹啉基、喹唑啉基、噌啉基、吡咯基、吲哚基、咪唑基、苯并咪唑基、吡唑基、苯并吡唑基、噻吩基、苯并噻吩基、噻唑基、噻二唑基、噁唑基、苯并噁唑基、呋喃基、苯并呋喃基等,所述杂芳基可以被如下一个或多个取代基(例如1-5个取代基)所取代:H、氧代、硫代、卤素、-CN、-NO 2、-OR 31、-CO-R 32、-COO-R 33、-O-CO-R 34、-NR 35R 36、-CONR 37R 38、-NR 39CO-R 40、-S(O) a-R 41、-S(O) aNR 42R 43、-NR 44S(O) a-R 45、-S-R 46、或无取代或任选被一个或多个R d取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;示例性的,所述杂芳基可以被如下1-5个或1-3个所取代:H、氧代、硫代、F、Cl、Br、I、-OH、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基、-NH-CO-烷基、-CO-NH-烷基、-CO-烷基、-O-CO-烷基、-CO-O-烷基、-NH 2、烷基氨基、二烷基氨基。
- 如权利要求1-5任一所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,R 3相同或不同,彼此独立地选自H、氧代、硫代、卤素、-CN、-NO 2、烷基、烷氧基、卤代烷基、卤代烷氧基;优选地,R 10为C1-6的烷基、C2-6的烯基或C2-6的炔基,如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、乙烯基、乙炔基。
- 如权利要求1-6任一项所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,式(I)所示的化合物为如下式(IV)所示的化合物:
 优选的,所述式(IV)化合物可以为如下式(IV-A)或式(IV-B)化合物:
优选的,所述式(IV)化合物可以为如下式(IV-A)或式(IV-B)化合物: Ar、R 1、R 2如权利要求1-6任一项所述。
Ar、R 1、R 2如权利要求1-6任一项所述。 - 如权利要求1-7任一项所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,式(I)所示的化合物为如下式(V)、式(V’)、式(V”)所示的化合物:
 优选的,所述式(V)化合物可以为如下式(V-A)或式(V-B)化合物:
优选的,所述式(V)化合物可以为如下式(V-A)或式(V-B)化合物: 其中,M为N或CR 6;R 5、R 6相同或不同,独立的选自H、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;Ar、R 1、R 10、R 11~R 26和a如权利要求1-7任一项所定义;优选地,式(I)所示的化合物为如下式(V)所示的化合物:
其中,M为N或CR 6;R 5、R 6相同或不同,独立的选自H、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;Ar、R 1、R 10、R 11~R 26和a如权利要求1-7任一项所定义;优选地,式(I)所示的化合物为如下式(V)所示的化合物: 优选的,所述式(V)化合物可以为如下式(V-A)或式(V-B)化合物:
优选的,所述式(V)化合物可以为如下式(V-A)或式(V-B)化合物: 其中,M为N或CR 6;R 5、R 6相同或不同,独立的选自H、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;Ar、R 1、R 11~R 26和a如权利要求1-7任一项所定义。
其中,M为N或CR 6;R 5、R 6相同或不同,独立的选自H、卤素、-CN、-NO 2、-OR 11、-CO-R 12、-COO-R 13、-O-CO-R 14、-NR 15R 16、-CONR 17R 18、-NR 19CO-R 20、-S(O) a-R 21、-S(O) aNR 22R 23、-NR 24S(O) a-R 25、-S-R 26,无取代或任选被一个或多个R c取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;Ar、R 1、R 11~R 26和a如权利要求1-7任一项所定义。 - 如权利要求1-8任一项所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,式(I)化合物可以为如下式(VI)化合物:
 优选的,所述式(I)化合物可以为如下式(VI-A)或式(VI-B)化合物:
优选的,所述式(I)化合物可以为如下式(VI-A)或式(VI-B)化合物: 其中,M为N或CR 6;R 1、R 5、R 6如权利要求1-8任一项所述。
其中,M为N或CR 6;R 1、R 5、R 6如权利要求1-8任一项所述。 - 如权利要求1所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐或前药,其中,式(I)所示的化合物为如下具体化合物:















- 一种权利要求1-10任一项所述的化合物的制备方法,包括:将式(X)化合物与式(XI)化合物在碱的存在下进行反应,得到式(I)化合物;
 其中,R 1、R 2、R 3、R’、环A、n如权利要求1-10任一项所定义;R 7为卤素、羟基。
其中,R 1、R 2、R 3、R’、环A、n如权利要求1-10任一项所定义;R 7为卤素、羟基。 - 一种权利要求1-10任一项所述的化合物的制备方法,其中,式(I-4)化合物通过如下方法制备得到,包括:将式(I-2)与带保护基的氨(例如二苯甲酮亚胺)进行反应,再脱去保护基,得到式(I-3)化合物;然后,式(I-3)化合物与R 9-X反应得到式(I-4)化合 物;
 其中,环A、R 2、R 3、R’、n如权利要求1-10任一项所定义,X为卤素;R 9相同或不同,独立的选自H、-OH、-CO-R 20、-S(O) a-R 25、无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;或者式(I-5)化合物来通过如下方法制备得到,包括:将式(I-2)与R 1a-H进行反应,得到式(I-5)化合物,
其中,环A、R 2、R 3、R’、n如权利要求1-10任一项所定义,X为卤素;R 9相同或不同,独立的选自H、-OH、-CO-R 20、-S(O) a-R 25、无取代或任选被一个或多个R b取代的下列基团:烷基、烯基、炔基、环烷基、芳基、杂芳基、杂环基;或者式(I-5)化合物来通过如下方法制备得到,包括:将式(I-2)与R 1a-H进行反应,得到式(I-5)化合物, 其中,环A、R 2、R 3、R’、n如权利要求1-10任一项所定义,X为卤素,R 1a为-NR 15R 16或杂芳基;R 15和R 16成环,形成含有N的杂环基或杂芳基;所述杂环基或杂芳基可以任选被一个或多个R b取代,并且相邻或不相邻的R b可以连接成环形成未取代或被一个或多个R d所取代的环烷基、杂环基、芳基或杂芳基;或者式(I-6)化合物通过如下方法制备得到,包括:将式(I-2)与R 1b-Y进行反应,得到式(I-6)化合物,
其中,环A、R 2、R 3、R’、n如权利要求1-10任一项所定义,X为卤素,R 1a为-NR 15R 16或杂芳基;R 15和R 16成环,形成含有N的杂环基或杂芳基;所述杂环基或杂芳基可以任选被一个或多个R b取代,并且相邻或不相邻的R b可以连接成环形成未取代或被一个或多个R d所取代的环烷基、杂环基、芳基或杂芳基;或者式(I-6)化合物通过如下方法制备得到,包括:将式(I-2)与R 1b-Y进行反应,得到式(I-6)化合物, 其中,环A、R 2、R 3、R’、n如权利要求1-10任一项所定义,X为卤素,R 1b为氰基、烷基、芳基、杂芳基,Y为-B(OH) 2、-Sn(烷基) 3、或等。
其中,环A、R 2、R 3、R’、n如权利要求1-10任一项所定义,X为卤素,R 1b为氰基、烷基、芳基、杂芳基,Y为-B(OH) 2、-Sn(烷基) 3、或等。
- 一种中间体,其结构如式(XI)化合物,
 其中,R 1、R 2、R 3、环A、n如权利要求1-10任一项所定义;优选的,所述式(XI)化合物为如下式(XII)化合物,
其中,R 1、R 2、R 3、环A、n如权利要求1-10任一项所定义;优选的,所述式(XI)化合物为如下式(XII)化合物, 其中,R 1、R 2、R 3、p如权利要求1-10任一项所定义;优选的,所述式(XI)化合物为如下式(XIII)化合物,
其中,R 1、R 2、R 3、p如权利要求1-10任一项所定义;优选的,所述式(XI)化合物为如下式(XIII)化合物, 其中,R 2、R 3、p如权利要求1-10任一项所定义;优选的,所述式(XI)化合物为如下式(XIV)化合物,
其中,R 2、R 3、p如权利要求1-10任一项所定义;优选的,所述式(XI)化合物为如下式(XIV)化合物, 其中,R 5如权利要求1-10任一项所定义;优选的,所述式(XI)化合物为如下式(XV)化合物,
其中,R 5如权利要求1-10任一项所定义;优选的,所述式(XI)化合物为如下式(XV)化合物, 优选的,所述式(XI)化合物为如下式(XV-A)或式(XV-B)化合物,
优选的,所述式(XI)化合物为如下式(XV-A)或式(XV-B)化合物,
- 一种药物组合物,其包含权利要求1-10任一项所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐、代谢产物、前药中的一种、两种或更多种,和至少一种药学上可接受的辅料。
- 权利要求1-10任一项所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐、代谢产物、前药中的一种、两种或更多种,或权利要求14所述的药物组合物在制备药物中的用途,所述药物用于预防和/或治疗由NK-3受体介导的疾病。
- 权利要求1-10任一项所述的式(I)所示的化合物、其立体异构体、互变异构体、同位素标记物、氮氧化物、溶剂化物、多晶型物、药学上可接受的盐、代谢产物、前药中的一种、两种或更多种,或权利要求14所述的药物组合物在制备药物中的用途,所述药物用于预防和/或治疗抑郁症、焦虑症、精神病、精神分裂症、精神病性障碍、双相型障碍、认知障碍、帕金森病、阿尔茨海默氏病、注意力不足多动症(ADHD)、疼痛、惊厥、肥胖、炎性疾病、呕吐、先兆子痫、气道相关疾病、生殖障碍、避孕和性激素依赖性疾病、妇科疾病相关疾病、更年期综合征相关疾病;优选的,所述性激素依赖性疾病包括但不限于良性前列腺增生(BPH)、前列腺增生、转移性前列腺癌、睾丸癌、乳腺癌、卵巢癌、雄激素依赖性痤疮、男性型秃发、子宫内膜异位症、青春期异常、子宫纤维化、子宫纤维瘤、激素依赖性癌症、高雄激素血症、多毛症、男性化、多囊卵巢综合征(PCOS)、经前烦躁症(PMDD)、HAIR-AN综合征(高雄激素血症、胰岛素抵抗以及黑棘皮病)、卵巢卵泡膜细胞增生症(HAIR-AN伴有卵巢间质中黄体化卵泡膜细胞增生)、高卵巢内雄激素浓度的其它表现(例如卵泡成熟停滞、闭锁、无排卵、痛经、功能失调性子宫出血、不孕症)、产生雄激素的肿瘤(男性化卵巢瘤或肾上腺瘤)、月经过多以及子宫腺肌症;优选的,所述气道相关疾病包括慢性阻塞性肺病、哮喘、气道高反应性、支气管收缩以及咳嗽;优选的,所述的更年期综合征包含潮热、出汗、心悸、眩晕及肥胖等症状。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202280028628.5A CN117177974A (zh) | 2021-04-21 | 2022-04-20 | 含咪唑稠环类衍生物、其制备方法及其在医药上的应用 |
| KR1020237032058A KR20230146639A (ko) | 2021-04-21 | 2022-04-20 | 이미다졸 함유 축합고리계 유도체, 이의 제조방법 및 이의 의약적 용도 |
| CA3214215A CA3214215A1 (en) | 2021-04-21 | 2022-04-20 | Imidazole-containing condensed ring derivative, preparation method therefor, and application thereof in medicine |
| JP2023563227A JP2024514007A (ja) | 2021-04-21 | 2022-04-20 | イミダゾール含有縮合環系誘導体、その調製方法及びその医薬上の応用 |
| US18/553,898 US20240228501A1 (en) | 2021-04-21 | 2022-04-20 | Imidazole-containing condensed ring derivative, preparation method therefor, and application thereof in medicine |
| AU2022263070A AU2022263070A1 (en) | 2021-04-21 | 2022-04-20 | Imidazole-containing condensed ring derivative, preparation method therefor, and application thereof in medicine |
| EP22791069.2A EP4293023A1 (en) | 2021-04-21 | 2022-04-20 | Imidazole-containing condensed ring derivative, preparation method therefor, and application thereof in medicine |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110432744.0 | 2021-04-21 | ||
| CN202110432744 | 2021-04-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022222963A1 true WO2022222963A1 (zh) | 2022-10-27 |
Family
ID=83723518
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2022/087947 WO2022222963A1 (zh) | 2021-04-21 | 2022-04-20 | 含咪唑稠环类衍生物、其制备方法及其在医药上的应用 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20240228501A1 (zh) |
| EP (1) | EP4293023A1 (zh) |
| JP (1) | JP2024514007A (zh) |
| KR (1) | KR20230146639A (zh) |
| CN (1) | CN117177974A (zh) |
| AU (1) | AU2022263070A1 (zh) |
| CA (1) | CA3214215A1 (zh) |
| WO (1) | WO2022222963A1 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116332861A (zh) * | 2023-03-21 | 2023-06-27 | 甘肃皓天医药科技有限责任公司 | 一种n-(3-甲基吡嗪-2-)甲基三氟乙酰胺的制备方法 |
| WO2024083150A1 (zh) * | 2022-10-19 | 2024-04-25 | 长春金赛药业有限责任公司 | 一种nk3r拮抗剂的晶型及其制备方法和应用 |
| WO2024188143A1 (zh) * | 2023-03-10 | 2024-09-19 | 山东绿叶制药有限公司 | Nk-3受体拮抗剂化合物、药物组合物、其制备方法和应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101568542A (zh) * | 2006-12-22 | 2009-10-28 | 埃科特莱茵药品有限公司 | 5,6,7,8-四氢-咪唑并[1,5-a]吡嗪衍生物 |
| CN101686989A (zh) * | 2007-06-21 | 2010-03-31 | 卡拉治疗学股份有限公司 | 取代的咪唑并杂环 |
| CN102906093A (zh) * | 2010-04-02 | 2013-01-30 | 欧洲筛选有限公司 | 新型nk-3受体选择性拮抗剂化合物、药物组合物以及在nk-3受体介导的疾病中的使用方法 |
| CN103906750A (zh) * | 2011-10-03 | 2014-07-02 | 欧洲筛选有限公司 | 作为选择性NK-3受体拮抗剂的新型手性N-酰基-5,6,7,(8-取代的)-四氢-[1,2,4]三唑并[4,3-a]吡嗪、药物组合物、用于NK-3受体介导的疾病中的方法及其手性合成 |
| CN107405347A (zh) * | 2015-03-16 | 2017-11-28 | 欧歌达有限公司 | 用于治疗处理或美容处理过量体脂肪的nk‑3受体拮抗剂 |
| CN111278827A (zh) * | 2017-07-24 | 2020-06-12 | 艾科生物科技有限公司 | 突变型异柠檬酸脱氢酶抑制剂、组合物及其方法 |
-
2022
- 2022-04-20 WO PCT/CN2022/087947 patent/WO2022222963A1/zh active Application Filing
- 2022-04-20 US US18/553,898 patent/US20240228501A1/en active Pending
- 2022-04-20 EP EP22791069.2A patent/EP4293023A1/en active Pending
- 2022-04-20 CA CA3214215A patent/CA3214215A1/en active Pending
- 2022-04-20 CN CN202280028628.5A patent/CN117177974A/zh active Pending
- 2022-04-20 KR KR1020237032058A patent/KR20230146639A/ko unknown
- 2022-04-20 AU AU2022263070A patent/AU2022263070A1/en active Pending
- 2022-04-20 JP JP2023563227A patent/JP2024514007A/ja active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101568542A (zh) * | 2006-12-22 | 2009-10-28 | 埃科特莱茵药品有限公司 | 5,6,7,8-四氢-咪唑并[1,5-a]吡嗪衍生物 |
| CN101686989A (zh) * | 2007-06-21 | 2010-03-31 | 卡拉治疗学股份有限公司 | 取代的咪唑并杂环 |
| CN102906093A (zh) * | 2010-04-02 | 2013-01-30 | 欧洲筛选有限公司 | 新型nk-3受体选择性拮抗剂化合物、药物组合物以及在nk-3受体介导的疾病中的使用方法 |
| CN103906750A (zh) * | 2011-10-03 | 2014-07-02 | 欧洲筛选有限公司 | 作为选择性NK-3受体拮抗剂的新型手性N-酰基-5,6,7,(8-取代的)-四氢-[1,2,4]三唑并[4,3-a]吡嗪、药物组合物、用于NK-3受体介导的疾病中的方法及其手性合成 |
| CN107405347A (zh) * | 2015-03-16 | 2017-11-28 | 欧歌达有限公司 | 用于治疗处理或美容处理过量体脂肪的nk‑3受体拮抗剂 |
| CN111278827A (zh) * | 2017-07-24 | 2020-06-12 | 艾科生物科技有限公司 | 突变型异柠檬酸脱氢酶抑制剂、组合物及其方法 |
Non-Patent Citations (10)
| Title |
|---|
| "McGraw-Hill Dictionary of Chemical Terms", 1984, MCGRAW-HILL BOOK COMPANY |
| BI°C HEM, vol. 11, 1972, pages 942 - 944 |
| DATABASE REGISTRY 25 October 2011 (2011-10-25), ANONYMOUS : "-Methanone, (4-fluorophenyl)[3-[2-(4-fluorophenyl)-4-thiazolyl]-5,6- dihydroimidazo[1,5-a]pyrazin-7(8H)-yl]-(CA INDEX NAME)", XP055978842, retrieved from STN Database accession no. 1338374-72-3 * |
| ELIEL, E.WILEN, S.: "Stereo chemistry of Organic Compounds", 1994, JOHN WILEY & SONS, INC. |
| J. RAUTIO ET AL.: "Prodrugs: Designand Clinical Applications", NATURE REVIEW DRUG DISCOVERY, vol. 7, 2008, pages 255 - 270 |
| MICHAEL B. SMITHJERRY MARCH: "March's Advanced Organic Chemistry", 2007, JOHN WILEY & SONS |
| S. J. HECKER ET AL.: "Prodrugs of Phosphatesand Phosphonates", JOURNAL OF MEDICINAL CHEMISTRY, vol. 51, 2008, pages 2328 - 2345 |
| SYN. COMM., vol. 7, 1977, pages 509 - 514 |
| T. HIGUCHIANDV. STELLA: "A.C.S. Symposium Series", vol. 14, 1987, PERGAMON PRESS, article "Pro-drugsas Novel Delivery Systems" |
| THOMAS SORRELL: "Organic Chemistry", 1999, UNIVERSITY SCIENCE BOOKS |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024083150A1 (zh) * | 2022-10-19 | 2024-04-25 | 长春金赛药业有限责任公司 | 一种nk3r拮抗剂的晶型及其制备方法和应用 |
| WO2024188143A1 (zh) * | 2023-03-10 | 2024-09-19 | 山东绿叶制药有限公司 | Nk-3受体拮抗剂化合物、药物组合物、其制备方法和应用 |
| CN116332861A (zh) * | 2023-03-21 | 2023-06-27 | 甘肃皓天医药科技有限责任公司 | 一种n-(3-甲基吡嗪-2-)甲基三氟乙酰胺的制备方法 |
| CN116332861B (zh) * | 2023-03-21 | 2023-10-27 | 甘肃皓天医药科技有限责任公司 | 一种n-(3-甲基吡嗪-2-)甲基三氟乙酰胺的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4293023A1 (en) | 2023-12-20 |
| CA3214215A1 (en) | 2022-10-27 |
| JP2024514007A (ja) | 2024-03-27 |
| CN117177974A (zh) | 2023-12-05 |
| AU2022263070A1 (en) | 2023-10-26 |
| KR20230146639A (ko) | 2023-10-19 |
| US20240228501A1 (en) | 2024-07-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI848954B (zh) | 作為hpk1抑制劑的吡咯並[2,3-b]吡啶或吡咯並[2,3-b]吡嗪及其用途 | |
| CN109923114B (zh) | 作为hpk1调节剂的吡唑并吡啶衍生物和其用于治疗癌症的用途 | |
| CN106170486B (zh) | 新吲唑甲酰胺、其制备方法、包含其的药物制剂及其用于制备药物的用途 | |
| CN109475531B (zh) | Ptpn11的杂环抑制剂 | |
| CN107253963B (zh) | 吡啶酮和氮杂吡啶酮化合物及使用方法 | |
| CN109970743B (zh) | 为jak抑制剂的5-氯-2-二氟甲氧基苯基吡唑并嘧啶化合物 | |
| WO2022222963A1 (zh) | 含咪唑稠环类衍生物、其制备方法及其在医药上的应用 | |
| TWI677499B (zh) | Pde9抑制劑及其用途 | |
| WO2021143680A1 (zh) | 杂芳基类衍生物及其制备方法和用途 | |
| TWI773651B (zh) | 醫藥化合物 | |
| WO2019020070A1 (zh) | 哌嗪并杂芳基类衍生物、其制备方法及其在医药上的应用 | |
| CN104781259A (zh) | 抑制bet蛋白的5-芳基三唑并氮杂* | |
| CN102803265A (zh) | 吡咯并吡嗪激酶抑制剂 | |
| BR112014007652B1 (pt) | N-acil-5,6,7, (8-substituído) - tetraidro-[1,2,4] triazolo [4,3 -a] pirazinas quirais inovadoras como antagonistas de receptor de nk-3 seletivos, composição farmacêutica, métodos para usar em distúrbios mediados por receptor de nk-3 e síntese quiral dos mesmos | |
| CN114341127A (zh) | 作为hpk1抑制剂的氨基吡嗪化合物及其用途 | |
| CN114728962A (zh) | 血浆激肽释放酶抑制剂及其用途 | |
| WO2017101796A1 (zh) | 酞嗪酮衍生物、其制备方法及用途 | |
| WO2020143763A1 (zh) | 卤代烯丙基胺类化合物及其应用 | |
| CN114728975A (zh) | 唑稠合的哒嗪-3(2h)-酮衍生物 | |
| WO2022166642A1 (zh) | 含氮多环稠环类化合物,其药物组合物、制备方法和用途 | |
| CN115260180A (zh) | 含三氮唑稠环类衍生物、药物组合物及其制备方法和应用 | |
| KR20230035036A (ko) | 이중 키나제-브로모도메인 억제제 | |
| WO2019062657A1 (zh) | 氮杂环类衍生物、其制备方法及其医药用途 | |
| JP2016535097A (ja) | 4−(インドール−3−イル)−ピラゾール誘導体、医薬組成物および使用のための方法 | |
| WO2020224656A1 (zh) | 双杂环羰基取代的二氢吡唑类化合物,其制法与医药上的用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22791069 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 22791069 Country of ref document: EP Ref document number: 2022791069 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20237032058 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020237032058 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2022791069 Country of ref document: EP Effective date: 20230913 |
|
| ENP | Entry into the national phase |
Ref document number: 3214215 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18553898 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2022263070 Country of ref document: AU Ref document number: 2022263070 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202317068677 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023563227 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2022263070 Country of ref document: AU Date of ref document: 20220420 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023122985 Country of ref document: RU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |