WO2020202183A1 - The process for the preparation of upadacitinib and its intermediates - Google Patents
The process for the preparation of upadacitinib and its intermediates Download PDFInfo
- Publication number
- WO2020202183A1 WO2020202183A1 PCT/IN2020/050280 IN2020050280W WO2020202183A1 WO 2020202183 A1 WO2020202183 A1 WO 2020202183A1 IN 2020050280 W IN2020050280 W IN 2020050280W WO 2020202183 A1 WO2020202183 A1 WO 2020202183A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- give
- group
- sodium
- Prior art date
Links
- 0 CCC(C*(C1)P)=C1C(*)=O Chemical compound CCC(C*(C1)P)=C1C(*)=O 0.000 description 4
- LKPALAXKZXXURJ-PZORYLMUSA-N CC[C@H](CN(C1)C(OCc2ccccc2)=O)C1C(O)=O Chemical compound CC[C@H](CN(C1)C(OCc2ccccc2)=O)C1C(O)=O LKPALAXKZXXURJ-PZORYLMUSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
Definitions
- the present invention relates to an improved process for the preparation of Upadacitinib and its intermediates.
- Upadacitinib having a chemical name: (3S,4R)-3-ethyl-4-(3H-imidazo[l,2-a]pyrrolo[2,3- e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- 1-carboxamide and structure as below.
- Upadacitinib also known as ABT-494, is a JAK1- selective inhibitor approved for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. It was developed by AbbVie.
- US 8426411 B2 discloses the synthetic preparation of Upadacitinib in a general route as mentioned in Scheme-II.
- W02020043033 discloses the process for the preparation of Upadacitinib intermediates.
- the present invention provides an improved process for preparing Upadacitinib by using novel intermediates.
- the main object of the present invention is to provide novel intermediates and their preparation in the process of the preparation of Upadacitinib.
- the present invention provides an improved process for the preparation of compounds of Formula-D, below, comprising the steps of:
- P is an amine protecting group
- R is H or alkyl
- both R’s can be the same or different
- X is halogen
- the present invention provides a process for the preparation of compounds of Formula-D comprising the steps of:
- P is an amine protecting group
- R is H or alkyl
- X is halogen
- the present invention provides an alternate process for the0 preparation of compounds of Formula-D comprising the steps of: a) reacting a compound of Formula-A with a compound of Formula-0 to give a compound of Formula-I
- the present invention provides a process for the preparation of an acid compound of Formula- A comprising the steps of: a) reacting a compound of Formula-J with ethyl acrylate and converting into an alkaline metal salt of Formula-K;
- P is an amine protecting group
- R is H or alkyl
- both of the R’s can be the same or different
- X is halogen
- the present invention provides an improved process for the preparation of Upadacitinib and its intermediates.
- the present invention provides an improved process for the preparation of Formula-D comprising the steps of:
- P is an amine protecting group
- R is H or alkyl
- both of the R’ s can be the same or different
- X is halogen.
- P is an amine protecting group.
- Each“R” may be, independently, a hydrogen (H) or an alkyl moiety
- “X” is halogen, including F, -Cl, -Br, and -I.
- Amine protecting groups are well known to those skilled in the art. Examples of suitable amine protecting groups, as well as suitable conditions for protecting and deprotecting can be found in prior art, such as J.F.W. McOmie (Ed.), Protective Groups in Organic Chemistry, Plenum Press, London (1973) and Greene's Protective Groups in Organic Synthesis, 5th Edition, Peter G. M. Wuts, John Wiley & Sons, Inc., Hoboken, New Jersey (2014), which are incorporated herein by reference in their entirety.
- suitable protecting groups include, but are not limited to, carbonyls (e.g., benzyloxy carbonyl; methyl carbamate; 9- fluorenylmethyoxycarbonyl (Fmoc); trichloroethoxycarbonyl (Troc); tert-butyloxycarbonyl (BOC); 2-trimethylsilylethyloxycarbonyl (Teoc); allyloxycarbonyl (Alloc); p-methoxybenzyl carbonyl (Moz); and carboxybenzyl (Cbz)); sulfonyls (e.g., p-toluenesufonyl (Ts); trimethylsilylethanesulfoyl (Ses); tert-butylsulfonyl (Bus); 4-methoxyphenylsulfonyl; and 4- nitrobenzenesulfonyl (nosyl)); and trityl
- a compound of Formula- A is reacted with Formula- H (wherein each R can be, independently, H or alkyl) or salts thereof in the presence of a condensing agent and solvent to give a chiral amine of Formula B .
- Suitable condensing agents include, but are not limited to, l-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), Carbonyl diimidazole (CDI) , (l-[Bis(dimethylamino)methylene]-lH- 1,2,3 -triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, Hexafluorophosphate Azabenzotriazole Tetramethyl Uranium (HATU), (Benzotriazol-1- yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), and (benzotriazol-l-yl- oxytripyrrolidinophosphonium hexafluorophosphate) (PyBOP).
- Suitable solvents include, but are not limited to, chloroform, tetrahydrofuran, toluene, 2-Methyl tetrahydrofuran, methyltertbutylether, methylisobutylketone, and acetone.
- the conversion of Formula-A may be carried out by halogenating Formula-A to give an acid halide of Formula-G (wherein X is halogen and P is an amine protecting group) in the presence of a halogenating agent and solvent.
- Suitable halogenating agents include, but are not limited to, thionyl chloride, oxalyl chloride, phosphorousoxy trichloride, N-halo succinamides (such as N-chloro succinimide and N-bromo succinimide), chlorine, and bromine.
- Suitable solvents include, but are not limited to, ethers and hydrocarbons, such as tetrahydrofuran, toluene, 2-methyl tetrahydrofuran, diethyl ether, ethyl methyl ether, 1,4-dioxane, and methyltertbutylether.
- Formula-A is converted into a mixed anhydride of Formula-A by reacting Formula-A with reagents like ethyl chloroformate or pivaloyl chloride to give a mixed anhydride of Formula-A. which is further reacted with Formula-H or salts thereof to give Formula-B by following the processes known in the art.
- the compound of Formula-B is converted to give Formula-C.
- the conversion may include reacting Formula-B with Grignard reagent (CFFMgX, wherein X is halo) in a suitable solvent, such as, but not limited to tetrahydrofuran, 2-Methyl tetrahydrofuran, 1,4-dioxane, or toluene.
- CFFMgX Grignard reagent
- the resulting compound of Formula-C is converted into Formula-D by reacting Formula-C with a halogenating reagent, such as, but not limited to, N-halo succinimides (such as N-chloro succinimide or N-bromo succinimide), chlorine, bromine, or sulfuryl chloride in a suitable solvent.
- a halogenating reagent such as, but not limited to, N-halo succinimides (such as N-chloro succinimide or N-bromo succinimide), chlorine, bromine, or sulfuryl chloride in a suitable solvent.
- suitable solvents include, but are not limited to tetrahydrofuran, toluene, 2-methyl tetrahydrofuran, diethylether, ethyl methyl ether, 1,4-dioxane, and methyltertbutylether.
- the present invention provides a process for the preparation of Formula-D comprising the steps of:
- Formula-E Formula-D wherein P is an amine protecting group; R is H or alkyl; and X is halogen.
- the compound of Formula-A is esterified to give a compound of Formula- E.
- This reaction may be carried out by reacting Formula-A with an alcohol, such as methanol, ethanol, 1 -propanol, isopropanol, butanol, in a solvent, optionally in the presence of a condensing agent.
- Suitable solvents include, but are not limited to tetrahydrofuran, 2-Methyl tetrahydrofuran, 1,4-dioxane, chloroform, dichloromethane, acetonitrile, acetone, dimethyl formamide, dimethyl acetamide, toluene, and methyl tertiary butyl ether.
- Condensing agents include, but are not limited to EDC, CDI, HATU, BOP, and PyBOP.
- the above resulting Formula-E is further converted into Formula-D.
- the conversion may be carried out by reacting Formula-E with 2-haloacetate, including, but not limited to, sodium chloroacetate, sodium bromoacetate, sodium iodoacetate, lithium chloroacetate, lithium bromoacetate, potassium chloroacetate and potassium bromoacetate in the presence of Grignard reagent (RMgX, wherein R is alkyl and X is halogen) and a suitable solvent, such as, but not limited, to tetrahydrofuran, 2-Methyl tetrahydrofuran, methyl tertiary butyl ether, 1,4- dioxane, and toluene.
- 2-haloacetate including, but not limited to, sodium chloroacetate, sodium bromoacetate, sodium iodoacetate,
- One more embodiment of the present invention provides an alternate process for the preparation of Formula-D comprising the steps of: a) reacting a compound of Formula-A with a compound of Formula-0 to give a compound of Formula-I
- P is an amine protecting group
- Ri is O or S
- X is halogen
- the conversion of Formula-A may be carried out by halogenating Formula-A to give an acid halide compound of Formula-G (wherein P is an amine protecting group and X is a halogen) in the presence of a halogenating agent and solvent.
- Suitable halogenating agents include, but are not limited to, thionyl chloride, oxalyl chloride, phosphorous oxy trichloride, N-halo succinimides (such as N-chloro succinimide and N-bromo succinimide), chlorine, and bromine.
- Suitable solvents include, but are not limited to, tetrahydrofuran, toluene, 2-methyl tetrahydrofuran, methyltertbutylether, and methylisobutylketone. The resulting Formula-G is further reacted with Formula-0 to give Formula-I in the presence of a condensing agent and solvent.
- Suitable condensing agents include, but are not limited to EDC, CDI, HATU, BOP, and PyBOP.
- Suitable solvents include, but are not limited to chloroform, tetrahydrofuran, toluene, 2-Methyl tetrahydrofuran, methyltertbutylether, methylisobutylketone, and acetone
- Formula-A is reacted with reagents like ethyl chloroformate or pivaloyl chloride to give a mixed anhydride of Formula-A, which is further reacted with Formula-0 in the presence of a condensing agent and solvent to give Formula-I.
- a condensing agent include, but are not limited to EDC, CDI, HATU, BOP, PyBOP.
- Suitable solvents include, but are not limited to chloroform, tetrahydrofuran, toluene, 2-Methyl tetrahydrofuran, methyltertbutylether, methylisobutylketone, and acetone
- the compound of Formula-I is converted to Formula-C.
- the conversion may include reacting Formula-I with a Grignard reagent, i.e., CH3MgX (wherein X is halogen), in a suitable solvent, such as, but not limited to, tetrahydrofuran, 2-Methyl tetrahydrofuran, 1,4-dioxane, and toluene.
- a Grignard reagent i.e., CH3MgX (wherein X is halogen)
- suitable solvent such as, but not limited to, tetrahydrofuran, 2-Methyl tetrahydrofuran, 1,4-dioxane, and toluene.
- Suitable halogenating agents include, but are not limited to, N-halo succinimides (such as N-chloro succinimide and N-bromo succinimide), chlorine, bromine, and sulfuryl chloride.
- Suitable solvents include, but are not limited to, tetrahydrofuran, toluene, 2-methyl tetrahydrofuran, diethyl ether, ethyl methyl ether, 1,4-dioxane, and methyltertbutylether.
- the present invention provides a process for the preparation of acid compounds of Formula-A comprising the steps of: a) reacting a compound of Formula-J with ethyl acrylate and converting it to an alkaline metal salt of Formula-K;
- Formula-N Formula-A wherein P is an amine protecting group; R is H or alkyl; and X is halogen.
- Formula-J is reacted with ethyl acrylate in the presence of a suitable base and solvent.
- Suitable bases include but are not limited to sodium-t-butoxide, magnesium-t-butoxide, and potassium-t-butoxide.
- Suitable solvents include, but are not limited to, tetrahydrofuran, toluene, methyl tert-butyl ether, methyl isobutyl ketone, acetone, and diethyl ether.
- the above resulting compound is further reacted with an alkaline metal hydroxide in a suitable solvent to yield a compound of Formula-K.
- Suitable alkaline metal hydroxides include, but are not limited to, potassium hydroxide and lithium hydroxide. Lithium hydroxide is preferred. Suitable solvents include, but are not limited to methanol, ethanol, propanol, butanol, and iso propyl alcohol.
- the alkaline metal salts of Formula-K can be selected from K, Li ; preferably Li salt. The obtained Formula-K is reacted with triflic anhydride to give Formula-L as per the processes known in the prior art WO 2017066775.
- Formula-L is then reacted with diethyl methoxy borane in the presence of a base and solvent to give Formula-M.
- bases include, but are not limited to, potassium carbonate, sodium carbonate, sodium bicarbonate, and potassium bicarbonate.
- Suitable solvents include but are not limited to tetrahydrofuran, toluene, methyl tert-butyl ether, methyl isobutyl ketone, acetone, and diethyl ether.
- the conversion of Formula-L to Formula-M may be carried out as per the process known in WO 2017066775 by using diethyl methoxy borane as a reagent.
- the reaction of Formula-L with diethyl methoxy borane may be carried out in the presence of a catalyst, including, but not limited to palladium dichloride diphenyl phosphinoferrocene dichloromethane complex (PdCh(dppf). DCM).
- a catalyst including, but not limited to palladium dichloride diphenyl phosphinoferrocene dichloromethane complex (PdCh(dppf). DCM).
- Formula-M may be further converted into Formula-A as per the process known in WO 2017066775.
- the Formula-M may be subjected to chiral reduction by following the analogous process disclosed in US 8426411 B2 and WO 2017066775.
- Formula-A may be purified by treating with an amine by forming corresponding amine salts by following procedures known in WO 2017066775.
- the resulting Formula-A can be converted into Formula-D as per the present invention or any of the processes known in the art.
- the intermediates of the present invention can be converted into Upadacitinib by processes known in the art, such as US 8426411 and WO2017066775.
- the present invention provides the following novel intermediates designated Formula-B and Formula-I,
- P is an amine protecting group
- R is H or alkyl
- both R’s can be the same or different
- Ri is O or S.
- novel intermediates designated Formula-B and Formula-I can be further converted into Upadacitinib.
- P is benzyloxy carbonyl group
- a suspension of Formula-A (5.0 g, 0.018 moles, 1.0 eq.), N-(3-Dimethyaminopropyl)-N’ -ethyl carbodiimide hydrochloride (EDC.HCl) (5.2 g, 0.027 moles, 1.5 eq.), and Hydroxy benzotriazole (HOBt) (3.65 g, 0.027 moles, 1.5 eq.) in Dichloromethane (50 ml, 10.0 vol.) was cooled to 0 °C, and N-Methylmorpholine (5.46 g, 0.054 moles, 3.0 eq.) was added dropwise.
- EDC.HCl N-(3-Dimethyaminopropyl)-N’ -ethyl carbodiimide hydrochloride
- HOBt Hydroxy benzotriazole
- P is benzyloxy carbonyl group
- X is Br
- a solution of Formula-C (2.0 g, 0.0073 moles, 1.0 eq.) in methanol (20 ml, 10.0 vol.) was acidified with four drops of aqueous Hydrobromic acid and then bromine (2.94 g, 0.018 moles, 2.5 eq.) was added dropwise at room temperature.
- the reaction mass was stirred at room temperature for 4 hours.
- the reaction mixture was quenched with cold water (20 ml).
- the aqueous layer was extracted twice with ethyl acetate (2 X 20 ml) and the organic layer was washed with 2% sodium thiosulphate solution (50 ml) followed by water (20 ml) and concentrated under vacuum to get the crude Formula-D.
- the Formula-D was purified by column chromatography (eluent: 30% ethyl acetate in Hexanes) to achieve the pure Formula-D as a yellow oil.
- R is methyl
- Aqueous citric acid (7.6 g in 23 ml water) was then added to the reaction mixture at below 10° C. Layers were separated and the organic layer was washed with 10% aqueous sodium bicarbonate solution (20 ml) followed by brine solution (20 ml) and concentrated under vacuum to get the crude Formula-D as a pale brown oil. The Formula-D was then purified by column chromatography and the pure chloro intermediate was eluted by 20% Ethyl acetate in hexanes.
- the oil compound was taken in isopropyl ether (1000 ml), a solution of Lithium Hydroxide (86.40 g, 2.11 moles, 1.0 eq.) in methanol (1500 ml) was added at room temperature and stirred for about 15 hours. The resulting slurry was cooled to 0-5°C, stirred for about 1 hour, filtered, and dried to get crude cyclized CBZ compound methanol solvate. The methanol solvate was taken in toluene (6000 ml), stirred at about 50°C for about 4 hours, filtered and dried to get the desired Formula-K as a pale-yellow solid.
- R is ethyl
- reaction mass was quenched with acetic acid (28.3 g, 0.47 moles, 1.0 eq.) and stirred for about 2 hours at the same temperature. Then water (1400 ml) was added, at below 10°C, stirred for about 30 minutes and layers were separated. The organic layer was washed with 10% sodium chloride solution (1400 ml X 3) followed by brine (280 ml). To the organic layer was charged activated carbon (14 g) and stirred at 10-20°C for about 60 minutes. The reaction mixture was filtered and concentrated to give the crude product as an oil.
- acetic acid 28.3 g, 0.47 moles, 1.0 eq.
- P is benzyloxy carbonyl group
- P is benzyloxy carbonyl group
- R is Ethyl
- R is Ethyl
- Formula-L 60.0 g, 0.14 moles, 1.0 eq.
- Dethyl methoxyborane 50% solution in THF, 180 ml, 0.63 moles, 4.5 eq.
- Toluene 1200 ml
- potassium carbonate 58.72 g, 0.42 moles, 3.0 eq.
- Palladium dichloride diphenyl phosphinoferrocene dichloromethane complex (1.8 g, 0.03 w/w) was added, degassed with Argon for additional 30 minutes heated to 95-105°C and stirred for about 3-5 hours. After completion of the reaction by TLC, the reaction mass was cooled to 25-30°C, filtered on Hyflo and concentrated to a total volume of 600 ml. To the above concentrated mass, water (600 ml) was added, stirred for 60 minutes and the layers were separated.
- the organic layer was washed with 0.5 N HC1 (630 ml), water (300 ml X 2) followed by brine (180 ml) and then treated with activated carbon (6 g) and concentrated to get crude Formula-M as oil.
- the Formula-M was treated again with activated carbon (6 g) in Toluene (36 ml) and diluted with heptane (600 ml), stirred at room temperature for about 1 hour, filtered and concentrated to get the pure Formula- M as light brown oil.
- P is benzyloxy carbonyl group Formula-N when
- R is ethyl P is benzyloxy carbonyl group
- P is benzyloxy carbonyl group
- the crude oil was dissolved in methyl tert-Butyl ether (470.0 ml), aqueous sodium hydroxide (20.5 g, 0.51 moles, 1.5 eq.) in 100 ml of water was added and stirred for about 60 minutes and layers were separated.
- the organic layer was extracted with an additional quantity of aqueous sodium hydroxide (6.83 g, 0.17 moles, 0.5 eq.) solution in water (100 ml).
- the aqueous layers were combined, washed with methyl tert-butyl ether (188 ml X 2), cooled to 50 °C and acidified with 5.5 N hydrochloric acid (-200.0 ml) to -2-4 pH.
- the product was then extracted from aqueous layer into methyl tert-butyl ether (376.0 ml X 2) twice.
- the organic layer was washed with water (376.0 ml X 2) and concentrated to get the crude Formula-A.
- the oily crude product of Formula-A (88. Og, 0.31 moles, l.Oeq.) was dissolved in acetonitrile (440.0 ml) and a solution of R-l-( Naphthalen-lyl) ethyl amine (54.7 lg, 0.31 moles, l.Oeq.) in acetonitrile (440.0 ml) was added.
- the reaction mass temperature was raised to 75 0 C and stirred at the same temperature for about one hour.
- the reaction mass was cooled to 0-5° C and allowed for complete precipitation for one hour. After precipitation, the reaction mass was filtered and dried to get naphthyl ethylamine salt of chiral acid.
- the salt was suspended in methyl tert-butyl ether (1070.0 ml), and 0.5 M orthophosphoric acid (535 ml) was added at room temperature and stirred for about 60 minutes. After separating the layers, the organic layer was washed with 0.25 M orthophosphoric acid (535 ml) and brine (357.0 ml) and concentrated to get the Formula-A.
- P is benzyloxy carbonyl group when P is benzyloxy carbonyl group
- a suspension of Formula-A (1.0 g, 0.0036 moles, 1.0 eq.), N-(3-dimethyaminopropyl)-N’- ethyl carbodiimide hydrochloride (EDC.HC1) (1.04 g, 0.0054 moles, 1.5 eq.), and Hydroxy benzotriazole (HOBt) (0.83 g, 0.0054 moles, 1.5 eq.) in dichloromethane (10 ml, 10.0 vol.) was cooled to 0°C and N-methylmorpholine (1.11 g, 0.011 moles, 3.0 eq.) was added dropwise. The reaction mixture was stirred at the same temperature for 30 minutes and further cooled to -10°C.
- EDC.HC1 N-(3-dimethyaminopropyl)-N’- ethyl carbodiimide hydrochloride
- HOBt Hydroxy benzotriazole
- R j is N
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The present invention provides an improved process for the preparation Upadacitinib by using novel intermediates. The present invention also provides processes for the preparation of novel intermediates of Upadacitinib.
Description
THE PROCESS FOR THE PREPARATION OF UPADACITINIB AND ITS
INTERMEDIATES
CROSS-REFERENCE TO RELATED APPLICATIONS:
This application claims the benefit of the filing date of Indian Provisional Application No. 201941012320 filed on March 29, 2019, the contents of which are incorporated herein in their entirety by reference.
FIELD OF INVENTION
The present invention relates to an improved process for the preparation of Upadacitinib and its intermediates.
BACKGROUND ART:
Upadacitinib, having a chemical name: (3S,4R)-3-ethyl-4-(3H-imidazo[l,2-a]pyrrolo[2,3- e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- 1-carboxamide and structure as below.

Upadacitinib
Upadacitinib, also known as ABT-494, is a JAK1- selective inhibitor approved for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. It was developed by AbbVie.
US 8426411 B2 discloses the synthetic preparation of Upadacitinib in a general route as mentioned in Scheme-II.
W02020043033 discloses the process for the preparation of Upadacitinib intermediates.
The present invention provides an improved process for preparing Upadacitinib by using novel intermediates.
OBJECT AND SUMMARY OF THE INVENTION.
The main object of the present invention is to provide novel intermediates and their preparation in the process of the preparation of Upadacitinib.
In one embodiment, the present invention provides an improved process for the preparation of compounds of Formula-D, below, comprising the steps of:
a) converting a compound of Formula-A into a compound of Formula-B ;

Formula-A Formula-B
b) converting the compound of Formula-B into the compound of Formula-C; and

Formula-B Formula-C
c) converting the compound of Formula-C to give a compound of Formula-D

In another embodiment, the present invention provides a process for the preparation of compounds of Formula-D comprising the steps of:
a) esterifying a compound of Formula-A to give a compound of Formula-E;

Formula-A Formula-E
b) converting the compound of Formula-E into a compound of Formula-D; and

cj Formula-E Formula-D
c) isolating Formula-D,
wherein P is an amine protecting group; R is H or alkyl; and X is halogen.
In one more embodiment, the present invention provides an alternate process for the0 preparation of compounds of Formula-D comprising the steps of: a) reacting a compound of Formula-A with a compound of Formula-0 to give a compound of Formula-I

b) converting the compound of Formula-I into a compound of Formula-C; and

5 Formula-I Formula-C
c) halogenating the compound of Formula-C to give a compound of Formula-D.

Formula-C
Formula-D
wherein P is an amine protecting group; Ri is O or S; X is halogen.
In one more embodiment, the present invention provides a process for the preparation of an acid compound of Formula- A comprising the steps of: a) reacting a compound of Formula-J with ethyl acrylate and converting into an alkaline metal salt of Formula-K;

Formula-J Formula-K
b) reacting the compound of Formula-K with triflic anhydride to give a compound of Formula-L;

c) reacting the compound of Formula-L with diethyl methoxy borane to give a compound of Formula-M;

d) hydrolysing the compound of Formula-M to give a compound of Formula-N; and

Formula-M Formula-N
e) reducing the compound of Formula-N to isolate a compound of Formula- A.
 wherein P is an amine protecting group; R is H or alkyl; and X is halogen
In another embodiment, the present invention provides novel intermediates of Formula-B and Formula-I.
wherein P is an amine protecting group; R is H or alkyl; and X is halogen
In another embodiment, the present invention provides novel intermediates of Formula-B and Formula-I.

DETAILED DESCRIPTION OF THE INVENTION
The present invention provides an improved process for the preparation of Upadacitinib and its intermediates.
In one embodiment, the present invention provides an improved process for the preparation of Formula-D comprising the steps of:
a) converting a compound of Formula-A into a compound of Formula-B ;

Formula-A Formula-B
b) converting the compound of Formula-B into a compound of Formula-C; and

Formula-B Formula-C
c) converting the compound of Formula-C to give a compound of Formula D.

Formula-C
Formula-D
wherein P is an amine protecting group; R is H or alkyl; both of the R’ s can be the same or different; and X is halogen.
Within the context of the reactions depicted and disclosed herein“P” is an amine protecting group. Each“R” may be, independently, a hydrogen (H) or an alkyl moiety,“X” is halogen, including F, -Cl, -Br, and -I.
Amine protecting groups (“P”) are well known to those skilled in the art. Examples of suitable amine protecting groups, as well as suitable conditions for protecting and deprotecting can be found in prior art, such as J.F.W. McOmie (Ed.), Protective Groups in Organic Chemistry, Plenum Press, London (1973) and Greene's Protective Groups in Organic Synthesis, 5th Edition, Peter G. M. Wuts, John Wiley & Sons, Inc., Hoboken, New Jersey (2014), which are incorporated herein by reference in their entirety. For example, suitable protecting groups include, but are not limited to, carbonyls (e.g., benzyloxy carbonyl; methyl carbamate; 9- fluorenylmethyoxycarbonyl (Fmoc); trichloroethoxycarbonyl (Troc); tert-butyloxycarbonyl (BOC); 2-trimethylsilylethyloxycarbonyl (Teoc); allyloxycarbonyl (Alloc); p-methoxybenzyl carbonyl (Moz); and carboxybenzyl (Cbz)); sulfonyls (e.g., p-toluenesufonyl (Ts); trimethylsilylethanesulfoyl (Ses); tert-butylsulfonyl (Bus); 4-methoxyphenylsulfonyl; and 4- nitrobenzenesulfonyl (nosyl)); and trityl (trt), benzyl (Bn), 3,4-dimethyoxybenzyl (Dmpm), p- methoxybenzyl (PMB), p-methoxyphenyl (PMP), acetyl (Ac), formyl, trifluoroacetyl (Tfa), benzoyl (Bz), or 2-nitrophenylsulfenyl (Nps) groups. In some embodiments, the protecting group is methyl carbamate.
Within the scope of the above embodiment, a compound of Formula- A is reacted with Formula- H (wherein each R can be, independently, H or alkyl) or salts thereof in the presence of a condensing agent and solvent to give a chiral amine of Formula B .

Formula -H
Suitable condensing agents include, but are not limited to, l-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), Carbonyl diimidazole (CDI) , (l-[Bis(dimethylamino)methylene]-lH- 1,2,3 -triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, Hexafluorophosphate Azabenzotriazole Tetramethyl Uranium (HATU), (Benzotriazol-1- yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), and (benzotriazol-l-yl- oxytripyrrolidinophosphonium hexafluorophosphate) (PyBOP). Suitable solvents include, but
are not limited to, chloroform, tetrahydrofuran, toluene, 2-Methyl tetrahydrofuran, methyltertbutylether, methylisobutylketone, and acetone.
Within the context of the present invention, the conversion of Formula-A may be carried out by halogenating Formula-A to give an acid halide of Formula-G (wherein X is halogen and P is an amine protecting group) in the presence of a halogenating agent and solvent.

Formula-G
Suitable halogenating agents include, but are not limited to, thionyl chloride, oxalyl chloride, phosphorousoxy trichloride, N-halo succinamides (such as N-chloro succinimide and N-bromo succinimide), chlorine, and bromine. Suitable solvents include, but are not limited to, ethers and hydrocarbons, such as tetrahydrofuran, toluene, 2-methyl tetrahydrofuran, diethyl ether, ethyl methyl ether, 1,4-dioxane, and methyltertbutylether. The resulting Formula-G is further reacted with Formula- FI to give Formula-B .
In one embodiment of the present invention, Formula-A is converted into a mixed anhydride of Formula-A by reacting Formula-A with reagents like ethyl chloroformate or pivaloyl chloride to give a mixed anhydride of Formula-A. which is further reacted with Formula-H or salts thereof to give Formula-B by following the processes known in the art.
The compound of Formula-B is converted to give Formula-C. The conversion may include reacting Formula-B with Grignard reagent (CFFMgX, wherein X is halo) in a suitable solvent, such as, but not limited to tetrahydrofuran, 2-Methyl tetrahydrofuran, 1,4-dioxane, or toluene.
The resulting compound of Formula-C is converted into Formula-D by reacting Formula-C with a halogenating reagent, such as, but not limited to, N-halo succinimides (such as N-chloro succinimide or N-bromo succinimide), chlorine, bromine, or sulfuryl chloride in a suitable solvent. Suitable solvents include, but are not limited to tetrahydrofuran, toluene, 2-methyl tetrahydrofuran, diethylether, ethyl methyl ether, 1,4-dioxane, and methyltertbutylether.
In another embodiment, the present invention provides a process for the preparation of Formula-D comprising the steps of:
a) esterifying a compound of Formula-A to give a compound of Formula-E; and

Formula- A Formula-E
b) converting Formula-E into Formula-D

Formula-E Formula-D wherein P is an amine protecting group; R is H or alkyl; and X is halogen.
As depicted above, the compound of Formula-A is esterified to give a compound of Formula- E. This reaction may be carried out by reacting Formula-A with an alcohol, such as methanol, ethanol, 1 -propanol, isopropanol, butanol, in a solvent, optionally in the presence of a condensing agent. Suitable solvents include, but are not limited to tetrahydrofuran, 2-Methyl tetrahydrofuran, 1,4-dioxane, chloroform, dichloromethane, acetonitrile, acetone, dimethyl formamide, dimethyl acetamide, toluene, and methyl tertiary butyl ether. Condensing agents include, but are not limited to EDC, CDI, HATU, BOP, and PyBOP. The above resulting Formula-E is further converted into Formula-D. The conversion may be carried out by reacting Formula-E with 2-haloacetate, including, but not limited to, sodium chloroacetate, sodium bromoacetate, sodium iodoacetate, lithium chloroacetate, lithium bromoacetate, potassium chloroacetate and potassium bromoacetate in the presence of Grignard reagent (RMgX, wherein R is alkyl and X is halogen) and a suitable solvent, such as, but not limited, to tetrahydrofuran, 2-Methyl tetrahydrofuran, methyl tertiary butyl ether, 1,4- dioxane, and toluene.
One more embodiment of the present invention provides an alternate process for the preparation of Formula-D comprising the steps of: a) reacting a compound of Formula-A with a compound of Formula-0 to give a compound of Formula-I

b) converting the compound of Formula-I into a compound of Formula-C; and

Formula-I Formula-C
c) halogenating the compound of Formula-C to give a compound of Formula-D

Formula-C
Formula-D
wherein P is an amine protecting group; Ri is O or S; and X is halogen.
Within the context of the present invention, the conversion of Formula-A may be carried out by halogenating Formula-A to give an acid halide compound of Formula-G (wherein P is an amine protecting group and X is a halogen) in the presence of a halogenating agent and solvent.

Suitable halogenating agents include, but are not limited to, thionyl chloride, oxalyl chloride, phosphorous oxy trichloride, N-halo succinimides (such as N-chloro succinimide and N-bromo succinimide), chlorine, and bromine. Suitable solvents include, but are not limited to, tetrahydrofuran, toluene, 2-methyl tetrahydrofuran, methyltertbutylether, and methylisobutylketone. The resulting Formula-G is further reacted with Formula-0 to give Formula-I in the presence of a condensing agent and solvent.
Suitable condensing agents include, but are not limited to EDC, CDI, HATU, BOP, and PyBOP. Suitable solvents include, but are not limited to chloroform, tetrahydrofuran, toluene, 2-Methyl tetrahydrofuran, methyltertbutylether, methylisobutylketone, and acetone
In another embodiment of the present invention, Formula-A is reacted with reagents like ethyl chloroformate or pivaloyl chloride to give a mixed anhydride of Formula-A, which is further reacted with Formula-0 in the presence of a condensing agent and solvent to give Formula-I. Suitable condensing agents include, but are not limited to EDC, CDI, HATU, BOP, PyBOP. Suitable solvents include, but are not limited to chloroform, tetrahydrofuran, toluene, 2-Methyl tetrahydrofuran, methyltertbutylether, methylisobutylketone, and acetone
The compound of Formula-I is converted to Formula-C. The conversion may include reacting Formula-I with a Grignard reagent, i.e., CH3MgX (wherein X is halogen), in a suitable solvent, such as, but not limited to, tetrahydrofuran, 2-Methyl tetrahydrofuran, 1,4-dioxane, and toluene.
The resulting compound of Formula-C is halogenated with a suitable halogenating agent in a solvent to get Formula-D. Suitable halogenating agents include, but are not limited to, N-halo succinimides (such as N-chloro succinimide and N-bromo succinimide), chlorine, bromine, and sulfuryl chloride. Suitable solvents include, but are not limited to, tetrahydrofuran, toluene, 2-methyl tetrahydrofuran, diethyl ether, ethyl methyl ether, 1,4-dioxane, and methyltertbutylether.
In one more embodiment, the present invention provides a process for the preparation of acid compounds of Formula-A comprising the steps of: a) reacting a compound of Formula-J with ethyl acrylate and converting it to an alkaline metal salt of Formula-K;

Formula-J Formula-K
b) reacting Formula-K with triflic anhydride to give a compound of Formula-L;
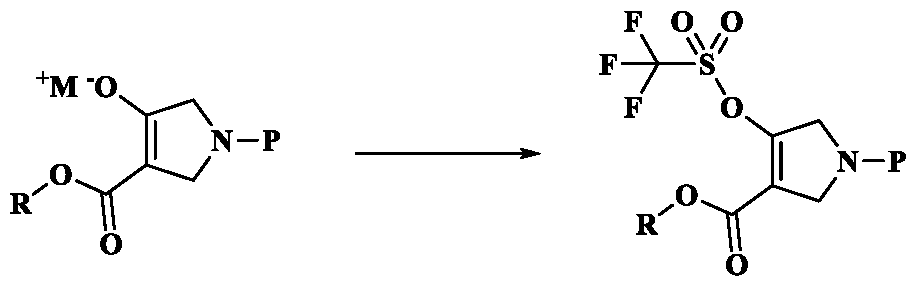
Formula-K Formula-L
c) reacting Formula-L with diethyl methoxy borane to give a compound of Formula-M;

d) hydrolysing Formula-M to give a compound of Formula-N; and

Formula-M Formula-N
e) reducing Formula-N to isolate a compound of Formula-A.

Formula-N Formula-A wherein P is an amine protecting group; R is H or alkyl; and X is halogen.
As per the above embodiment, Formula-J is reacted with ethyl acrylate in the presence of a suitable base and solvent. Suitable bases include but are not limited to sodium-t-butoxide, magnesium-t-butoxide, and potassium-t-butoxide. Suitable solvents include, but are not limited to, tetrahydrofuran, toluene, methyl tert-butyl ether, methyl isobutyl ketone, acetone, and diethyl ether. In this embodiment, the above resulting compound is further reacted with an alkaline metal hydroxide in a suitable solvent to yield a compound of Formula-K. Suitable alkaline metal hydroxides include, but are not limited to, potassium hydroxide and lithium hydroxide. Lithium hydroxide is preferred. Suitable solvents include, but are not limited to methanol, ethanol, propanol, butanol, and iso propyl alcohol. The alkaline metal salts of Formula-K can be selected from K, Li ; preferably Li salt.
The obtained Formula-K is reacted with triflic anhydride to give Formula-L as per the processes known in the prior art WO 2017066775.
Formula-L is then reacted with diethyl methoxy borane in the presence of a base and solvent to give Formula-M. Suitable bases include, but are not limited to, potassium carbonate, sodium carbonate, sodium bicarbonate, and potassium bicarbonate. Suitable solvents include but are not limited to tetrahydrofuran, toluene, methyl tert-butyl ether, methyl isobutyl ketone, acetone, and diethyl ether.
The conversion of Formula-L to Formula-M may be carried out as per the process known in WO 2017066775 by using diethyl methoxy borane as a reagent. The reaction of Formula-L with diethyl methoxy borane may be carried out in the presence of a catalyst, including, but not limited to palladium dichloride diphenyl phosphinoferrocene dichloromethane complex (PdCh(dppf). DCM).
Formula-M may be further converted into Formula-A as per the process known in WO 2017066775. Within the context of the present invention, the Formula-M may be subjected to chiral reduction by following the analogous process disclosed in US 8426411 B2 and WO 2017066775.
According to the present invention, Formula-A may be purified by treating with an amine by forming corresponding amine salts by following procedures known in WO 2017066775. In one embodiment, the resulting Formula-A can be converted into Formula-D as per the present invention or any of the processes known in the art.
In one more embodiment, the intermediates of the present invention can be converted into Upadacitinib by processes known in the art, such as US 8426411 and WO2017066775.
In one more embodiment, the present invention provides the following novel intermediates designated Formula-B and Formula-I,

In one more embodiment, the novel intermediates designated Formula-B and Formula-I can be further converted into Upadacitinib.
The examples mentioned below explain various aspects of the present invention. The examples are given to illustrate the details of the invention and should not be construed to limit the scope of the present invention.
Examples:
Example 1:

Formula-B when both R's are -CH3
Formula-A when P is benzyloxy carbonyl group
P is benzyloxy carbonyl group
A suspension of Formula-A (5.0 g, 0.018 moles, 1.0 eq.), N-(3-Dimethyaminopropyl)-N’ -ethyl carbodiimide hydrochloride (EDC.HCl) (5.2 g, 0.027 moles, 1.5 eq.), and Hydroxy benzotriazole (HOBt) (3.65 g, 0.027 moles, 1.5 eq.) in Dichloromethane (50 ml, 10.0 vol.) was cooled to 0 °C, and N-Methylmorpholine (5.46 g, 0.054 moles, 3.0 eq.) was added dropwise. The reaction mixture was stirred at the same temperature for 30 minutes and further cooled to -10 °C. N, O-Diemthylhydroxylamine hydrochloride (1.76 g, 0.018 moles, 1.0 eq.) was added to the reaction mixture in lots at below 0°C, and stirring was continued for additional 120 minutes. After completion of the reaction, water (50 ml) was added to the reaction mass. The organic and aqueous layers were separated, and the aqueous layer was extracted with additional dichloromethane (25 ml). Organic layers were combined and washed with 10% aqueous sodium bicarbonate solution (25 ml) followed by water (25 ml) and evaporated to get the Formula-B. The crude Formula-B was purified by column chromatography. (Eluent: 30% Ethyl acetate in Hexanes) to get the pure Amide as colorless oil.
Yield: 2.5 g (71%)
Example 2:

Formula-B when both R's are -CH3 Formula-C when P is benzyloxy carbonyl group
P is benzyloxy carbonyl group
To a solution of Formula-B (2.5 g, 0.0078 moles, 1.0 eq.) in tetrahydrofuran (15.0 ml, 6.0 vol.) was added 3M methyl magnesium chloride in THF solution (3.89 ml, 0.012 moles, 1.5 eq.) at 0-5°C. The reaction mass was stirred at the same temperature for about 120 minutes and quenched with 10% aqueous ammonium chloride solution (20 ml). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (25 ml). Both the organic layers were combined, washed with water (25 ml) and concentrated under vacuum to get the crude Formula-C. Formula-C was purified by column chromatography (eluent: 40% ethyl acetate in hexanes) to achieve pure Formula-C as light-yellow oil.
Yield: 1.5 g (70%).
Example 3:

Formula-C when P is benzyloxy carbonyl group Formula-D when P is benzyloxy carbonyl group;
X is Br A solution of Formula-C (2.0 g, 0.0073 moles, 1.0 eq.) in methanol (20 ml, 10.0 vol.) was acidified with four drops of aqueous Hydrobromic acid and then bromine (2.94 g, 0.018 moles, 2.5 eq.) was added dropwise at room temperature. The reaction mass was stirred at room temperature for 4 hours. After completion of the reaction, the reaction mixture was quenched with cold water (20 ml). The aqueous layer was extracted twice with ethyl acetate (2 X 20 ml) and the organic layer was washed with 2% sodium thiosulphate solution (50 ml) followed by water (20 ml) and concentrated under vacuum to get the crude Formula-D. The Formula-D was
purified by column chromatography (eluent: 30% ethyl acetate in Hexanes) to achieve the pure Formula-D as a yellow oil.
Example 4:

Formula-E when P is benzyloxy carbonyl group
Formula-A when P is benzyloxy carbonyl group
R is methyl
To a solution of carbonyl diimidazole (4.39 g, 0.027 moles, 1.5 eq.) in tetrahydrofuran (12.5 ml, 2.5 vol.) was added a solution of Formula-A (5.0 g, 0.018 moles, 1.0 eq.) in tetrahydrofuran (25 ml, 5 vol.) at room temperature. The solution was stirred for one hour at the same temperature and then it was quenched with methanol (50 ml, 10 vol.). The resulting reaction mass was stirred for additional two hours at room temperature and solvents were removed by distillation under vacuum. To the resulting crude was charged water (25 ml, 5 vol.) and isopropyl ether (25 ml, 5 vol.); stirred for about 15 minutes and layers were separated. The organic layer was washed with water (25 ml) and concentrated under vacuum to get Formula- E as an oil.
Yield: 4.5 g (86%) Example 5: t-BuMgCl,

Formula-E when P is benzyloxy carbonyl group Formula-D when P is benzyloxy carbonyl group;
R is methyl X is Clr
To a mixture of Formula-E (4.0 g, 0.014 moles, 1.0 eq.), sodium chloroacetate (4.1 g, 0.035 moles, 2.5 eq.); molecular sieves (4A°) (5.2 g) and triethylamine (2.8 g, 0.028 moles, 2.0 eq.) in tetrahydrofuran (40 ml, 10 vol.) was added tert-butyl magnesium chloride (1 M in THF) (35 ml, 0.035 moles, 2.5 eq.) at -10 to 0° C and the mixture was stirred at the same temperature for about one hour. Aqueous citric acid (7.6 g in 23 ml water) was then added to the reaction
mixture at below 10° C. Layers were separated and the organic layer was washed with 10% aqueous sodium bicarbonate solution (20 ml) followed by brine solution (20 ml) and concentrated under vacuum to get the crude Formula-D as a pale brown oil. The Formula-D was then purified by column chromatography and the pure chloro intermediate was eluted by 20% Ethyl acetate in hexanes.
Example 6:

To a solution of Formula-J (500.0 g, 2.11 moles, 1.0 eq.) and ethyl acrylate (211 g, 2.11 moles, 1.0 eq.) in tetrahydrofuran (5000 ml, 10 vol.) was added Sodium-t- butoxide (202.52 g, 2.11 moles, 1.0 eq.) in lots slowly at 0-5 °C. The reaction mass was stirred for 3 hours at the same temperature and then it was warmed to 25 °C, further maintained for about 15 hours. After completion of the reaction, the solvent was evaporated under vacuum. To the resulting crude was added dichloromethane (3500 ml) followed by Acetic acid (127.7 g, 2.11 moles, 1.0 eq.) at 0-5 °C. The resulting reaction mass was stirred for about 2 hours at the same temperature; water (4500 ml) was added, and stirring continued for additional 45 minutes. The layers were separated, and the organic layer was washed with water (2 X 3500 ml) followed by brine solution (1000 ml). The organic layer was then treated with activated Carbon (PS-133, 25.0 g), filtered and concentrated under vacuum to get crude compound as oil. The oil compound was taken in isopropyl ether (1000 ml), a solution of Lithium Hydroxide (86.40 g, 2.11 moles, 1.0 eq.) in methanol (1500 ml) was added at room temperature and stirred for about 15 hours. The resulting slurry was cooled to 0-5°C, stirred for about 1 hour, filtered, and dried to get crude cyclized CBZ compound methanol solvate. The methanol solvate was taken in toluene (6000 ml), stirred at about 50°C for about 4 hours, filtered and dried to get the desired Formula-K as a pale-yellow solid.
Yield: 320 g
Example 7:

Formula-K Formula-L when when M is Li P is benzyloxy carbonyl group
P is benzyloxy carbonyl R is ethyl
group
R is ethyl
To a suspension of Formula-K (140 g, 0.47 moles, 1.0 eq.) in Dichloromethane (1400 ml, 10 vol) was added triflic anhydride (199.4 g, 0.71 moles, 1.5 eq.) at 0-5°C. The reaction mass was stirred at the same temperature for about 30 minutes. Then, a solution of diisopropylethylamine (78.4 g, 0.61 moles, 1.3 eq.) in dichloromethane (140 ml) was added at 0-5°C and the stirring continued. After completion of the reaction, the reaction mass was quenched with acetic acid (28.3 g, 0.47 moles, 1.0 eq.) and stirred for about 2 hours at the same temperature. Then water (1400 ml) was added, at below 10°C, stirred for about 30 minutes and layers were separated. The organic layer was washed with 10% sodium chloride solution (1400 ml X 3) followed by brine (280 ml). To the organic layer was charged activated carbon (14 g) and stirred at 10-20°C for about 60 minutes. The reaction mixture was filtered and concentrated to give the crude product as an oil. The crude oil was dissolved in a mixture of ethyl acetate (560 ml) and isopropyl ether (560 ml) and washed with 10% aqueous sodium chloride solution (1400 ml X 2) followed by brine (280 ml). The organic layer was then treated with activated carbon (14 g), filtered and concentrated to get pure Formula-L as a brown oil.
Yield: 172 g
Example 8:

Formula-M when
Formula-L when
P is benzyloxy carbonyl group
P is benzyloxy carbonyl group
R is Ethyl
R is Ethyl
To a solution of Formula-L (60.0 g, 0.14 moles, 1.0 eq.) and Dethyl methoxyborane (50% solution in THF, 180 ml, 0.63 moles, 4.5 eq.) in Toluene (1200 ml) was added potassium carbonate (58.72 g, 0.42 moles, 3.0 eq.) at room temperature. The reaction mass was degassed with Argon for about 60 minutes. Palladium dichloride diphenyl phosphinoferrocene dichloromethane complex (PdC12(dppf).DCM) (1.8 g, 0.03 w/w) was added, degassed with Argon for additional 30 minutes heated to 95-105°C and stirred for about 3-5 hours. After completion of the reaction by TLC, the reaction mass was cooled to 25-30°C, filtered on Hyflo and concentrated to a total volume of 600 ml. To the above concentrated mass, water (600 ml) was added, stirred for 60 minutes and the layers were separated. The organic layer was washed with 0.5 N HC1 (630 ml), water (300 ml X 2) followed by brine (180 ml) and then treated with activated carbon (6 g) and concentrated to get crude Formula-M as oil. The Formula-M was treated again with activated carbon (6 g) in Toluene (36 ml) and diluted with heptane (600 ml), stirred at room temperature for about 1 hour, filtered and concentrated to get the pure Formula- M as light brown oil.
Yield: 31.0 g
Example 9:

® Formula-M when
P is benzyloxy carbonyl group Formula-N when
R is ethyl P is benzyloxy carbonyl group
To a solution of Formula-M (71.0 g, 0.23 moles, 1.0 eq.) in tetrahydrofuran (710 ml) was added sodium hydroxide (14.1 g, 0.35 moles, 1.5 eq.) in water (355 ml) at room temperature, and the resulting mixture was warmed to 50 °C and stirred for about 24 hours. After completion of the reaction, THF was evaporated from the reaction mixture under vacuum at below 40°C and co distilled with methyl tertbutyl ether (142 ml). The resulting aqueous layer was washed with methyl tertbutyl ether (142 ml X 2) and cooled to about 10 °C before addition of 2N Hydrochloric acid (264 ml). The reaction mixture was further cooled to 0-5 °C, stirred for about 2 hours and filtered. The wet compound was washed with water and dried to get the Formula- N as solid.
Yield: 49.6 g
Example 10:

Formula-A when P is benzyloxy carbonyl group
Formula-N when
P is benzyloxy carbonyl group
To the suspension of Formula-N (94.0 g,0.34 moles, 1.0 eq.) in methanol (660 ml) was added triethylamine (39.7 g, 0.39 moles, 1.15 eq.) and stirred at room temperature for complete dissolution. This solution was taken in autoclave flask. Ruthenium diacetate (S) Segphos catalyst (0.66 g, 0.007 w/w) was added and stirred under hydrogen pressure (15 Kgs) at 80°C for about 24 hours. After completion of the reaction, the reaction mass was filtered through hyflo bed and concentrated under vacuum to get the crude chiral acid as an oil. The crude oil was dissolved in methyl tert-Butyl ether (470.0 ml), aqueous sodium hydroxide (20.5 g, 0.51 moles, 1.5 eq.) in 100 ml of water was added and stirred for about 60 minutes and layers were separated. The organic layer was extracted with an additional quantity of aqueous sodium hydroxide (6.83 g, 0.17 moles, 0.5 eq.) solution in water (100 ml). The aqueous layers were combined, washed with methyl tert-butyl ether (188 ml X 2), cooled to 50 °C and acidified with 5.5 N hydrochloric acid (-200.0 ml) to -2-4 pH. The product was then extracted from aqueous layer into methyl tert-butyl ether (376.0 ml X 2) twice. The organic layer was washed with water (376.0 ml X 2) and concentrated to get the crude Formula-A.
Yield: 88.0 g.
Example 11:
The oily crude product of Formula-A (88. Og, 0.31 moles, l.Oeq.) was dissolved in acetonitrile (440.0 ml) and a solution of R-l-( Naphthalen-lyl) ethyl amine (54.7 lg, 0.31 moles, l.Oeq.) in acetonitrile (440.0 ml) was added. The reaction mass temperature was raised to 75 0 C and stirred at the same temperature for about one hour. The reaction mass was cooled to 0-5° C and allowed for complete precipitation for one hour. After precipitation, the reaction mass was filtered and dried to get naphthyl ethylamine salt of chiral acid. The salt was suspended in methyl tert-butyl ether (1070.0 ml), and 0.5 M orthophosphoric acid (535 ml) was added at room temperature and stirred for about 60 minutes. After separating the layers, the organic
layer was washed with 0.25 M orthophosphoric acid (535 ml) and brine (357.0 ml) and concentrated to get the Formula-A.
Yield: 70.0 g Example 12:

Formula-A when Formula-O, when Rt is N Formula-I
P is benzyloxy carbonyl group when P is benzyloxy carbonyl group
R! is N
A suspension of Formula-A (1.0 g, 0.0036 moles, 1.0 eq.), N-(3-dimethyaminopropyl)-N’- ethyl carbodiimide hydrochloride (EDC.HC1) (1.04 g, 0.0054 moles, 1.5 eq.), and Hydroxy benzotriazole (HOBt) (0.83 g, 0.0054 moles, 1.5 eq.) in dichloromethane (10 ml, 10.0 vol.) was cooled to 0°C and N-methylmorpholine (1.11 g, 0.011 moles, 3.0 eq.) was added dropwise. The reaction mixture was stirred at the same temperature for 30 minutes and further cooled to -10°C. Formula-0 (0.38 g, 0.0043 moles, 1.2 eq.) was added at below 0°C and stirring was continued for additional 120 minutes. After completion of the reaction, water (50 ml) was added to the reaction mass and layers were separated. An aqueous layer was extracted with additional dichloromethane (25 ml). Both the organic layers were combined, washed with 10% aqueous sodium bicarbonate solution (25 ml) followed by water (25 ml) and evaporated to get the crude Formula-I. The crude product was purified by column chromatography. (Eluent: 30% Ethyl acetate in Hexanes) to get the pure Formula-I as an oil. Yield: 0.80 g (64%).
Example 13:

Formula-I
when P is benzyloxy carbonyl group
Formula-C when
Rj is N
P is benzyloxy carbonyl group
To a solution of Formula-I (0.50 g, 0.0014 moles, 1.0 eq.) in tetrahydrofuran (5.0 ml, 10.0 vol.) was added 3M methyl magnesium chloride in THF solution (0.70 ml, 0.0021 moles, 1.5 eq.) at 0-5°C. The reaction mass was stirred at the same temperature for about 120 minutes and quenched with 10% aqueous ammonium chloride solution (5 ml). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (10 ml). Both the organic layers were combined, washed with water (10 ml) and concentrated under vacuum to get the crude Formula-C. The crude Formula-C was purified by column chromatography (eluent: 40% ethyl acetate in hexanes) to achieve pure Formula-C as light-yellow oil.
Yield: 0.30 g (76%)
Claims
1. A process for the preparation of Formula-D comprising the steps of: a. esterifying a compound of Formula-A to give a compound of Formula-E; and

Formula-A Formula-E
b. converting Formula-E into Formula-D;

Formula-E Formula-D
wherein P is an amine protecting group; R is H or alkyl; and X is halogen.
2. A process for the preparation of a compound of Formula-D comprising reacting a compound of Formula-E with 2-haloacetate in a solvent

1. Formula-E Formula-D
wherein P is an amine protecting group; R is H or alkyl; and X is halogen.
3. The process as claimed in claim 1, wherein the compound of Formula-A is esterified in the presence of a condensing agent.
4. The process as claimed in claim 1, wherein the compound of Formula-A is esterified by treating with an alcohol.
5. The process as claimed in claim 4, wherein the alcohol comprises one or more alcohols selected from the group consisting of methanol, ethanol, 1 -propanol, isopropanol and butanol.
6. The process as claimed in claim 1, wherein the compound of Formula-E is treated with a 2-haloacetate in the presence of a Grignard reagent to give Formula-D.
7. The process as claimed in either claim 6 or claim 2, wherein the 2-haloacetate is selected from the group consisting of sodium chloroacetate, sodium bromoacetate, sodium iodoacetate, lithium chloroacetate, lithium bromoacetate, potassium chloroacetate and potassium bromoacetate.
8. The process as claimed in either claim 1 or claim 2, wherein the compound of formula D is further converted into upadacitinib.
9. A process for the preparation of an acid compound of Formula-A comprising the steps of:
a) reacting a compound of Formula-J with ethyl acrylate and converting the product into an alkaline metal salt of Formula-K

Formula-J Formula-K
b) reacting Formula-K with triflic anhydride to give a compound of Formula-L;

c) reacting Formula-L with diethyl methoxy borane to give a compound of Formula-M;

d) hydrolysing Formula-M to give a compound of Formula-N; and

Formula-M Formula-N
e) reducing Formula-N to isolate Formula-A.

wherein P is an amine protecting group; R is H or alkyl; and X is halogen
10. The process as claimed in claim 9, wherein the Formula-J is reacted with ethyl acrylate in the presence of a base and a solvent.
11. The process as claimed in claim 10, wherein the base is selected from the group consisting of sodium-t-butoxide, magnesium-t-butoxide and potassium-t-butoxide.
12. The process as claimed in claim 10, wherein the solvent is selected from the group consisting of tetrahydrofuran, toluene, methyl tert-butyl ether, methyl isobutyl ketone, acetone and diethyl ether.
13. The process as claimed in claim 9, wherein the alkaline metal salt of Formula-K is a lithium salt or a potassium salt.
14. The process as claimed in claim 13, wherein the metal salt of Formula-K is a lithium salt.
15. The process as claimed in claim 9, wherein the reaction of Formula-L with diethyl methoxy borane is carried out in the presence of a base, a solvent and a catalyst.
16. The process as claimed in claim 15, wherein the base is selected from the group consisting of potassium carbonate, sodium carbonate, sodium bicarbonate and potassium bicarbonate.
17. The process as claimed in claim 15, wherein the solvent is selected from the group consisting of tetrahydrofuran, toluene, methyl tert-butyl ether, methyl isobutyl ketone, acetone and diethyl ether.
18. The process as claimed in claim 15, wherein the catalyst is Palladium dichloride diphenyl phosphinoferrocene dichloromethane complex (PdC12(dppf). DCM).
19. The process as claimed in claim 9, wherein the compound of Formula-A is further converted into upadacitinib.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201941012320 | 2019-03-29 | ||
| IN201941012320 | 2019-03-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020202183A1 true WO2020202183A1 (en) | 2020-10-08 |
Family
ID=70465204
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2020/050280 WO2020202183A1 (en) | 2019-03-29 | 2020-03-25 | The process for the preparation of upadacitinib and its intermediates |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2020202183A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210323971A1 (en) * | 2018-08-31 | 2021-10-21 | Suzhou Pengxu Pharmatech Co., Ltd | Synthesis methods for upadacitinib and intermediate thereof |
| CN115417803A (en) * | 2022-08-30 | 2022-12-02 | 四川同晟生物医药有限公司 | 5363 Synthesis method of intermediate (3R, 4S) -1-benzyloxycarbonyl-4-ethylpyrrolidine-3-carboxylic acid of Wu Pati Ni |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8426411B2 (en) | 2008-06-10 | 2013-04-23 | Abbott Laboratories | Tricyclic compounds |
| WO2017066775A1 (en) | 2015-10-16 | 2017-04-20 | Abbvie Inc. | PROCESSES FOR THE PREPARATION OF (3S,4R)-3-ETHYL-4-(3H-IMIDAZO[1,2-a]PYRROLO[2,3-e]-PYRAZIN-8-YL)-N-(2,2,2-TRIFLUOROETHYL)PYRROLIDINE-1-CARBOXAMIDE AND SOLID STATE FORMS THEREOF |
| WO2018218104A1 (en) * | 2017-05-26 | 2018-11-29 | Imara, Inc. | Methods of making and using pde9 inhibitors |
| WO2020043033A2 (en) | 2018-08-31 | 2020-03-05 | 苏州鹏旭医药科技有限公司 | Synthesis methods for upadacitinib and intermediate thereof |
| CN110872250A (en) * | 2018-08-31 | 2020-03-10 | 苏州鹏旭医药科技有限公司 | Two compounds, preparation methods thereof and application thereof in synthesis of ursitinib |
-
2020
- 2020-03-25 WO PCT/IN2020/050280 patent/WO2020202183A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8426411B2 (en) | 2008-06-10 | 2013-04-23 | Abbott Laboratories | Tricyclic compounds |
| WO2017066775A1 (en) | 2015-10-16 | 2017-04-20 | Abbvie Inc. | PROCESSES FOR THE PREPARATION OF (3S,4R)-3-ETHYL-4-(3H-IMIDAZO[1,2-a]PYRROLO[2,3-e]-PYRAZIN-8-YL)-N-(2,2,2-TRIFLUOROETHYL)PYRROLIDINE-1-CARBOXAMIDE AND SOLID STATE FORMS THEREOF |
| WO2018218104A1 (en) * | 2017-05-26 | 2018-11-29 | Imara, Inc. | Methods of making and using pde9 inhibitors |
| WO2020043033A2 (en) | 2018-08-31 | 2020-03-05 | 苏州鹏旭医药科技有限公司 | Synthesis methods for upadacitinib and intermediate thereof |
| CN110872250A (en) * | 2018-08-31 | 2020-03-10 | 苏州鹏旭医药科技有限公司 | Two compounds, preparation methods thereof and application thereof in synthesis of ursitinib |
Non-Patent Citations (3)
| Title |
|---|
| "Protective Groups in Organic Chemistry", 1973, PLENUM PRESS |
| PETER G. M. WUTS: "Greene's Protective Groups in Organic Synthesis", 2014, JOHN WILEY & SONS, INC. |
| Z. GAJDOSIK: "Upadacitinib tartrate. Tyrosine-protein kinase JAK1 inhibitor, Treatment of autoimmune inflammatory diseases, Treatment of rheumatoid arthritis", DRUGS OF THE FUTURE, vol. 43, no. 10, 1 January 2018 (2018-01-01), ES, pages 731, XP055675942, ISSN: 0377-8282, DOI: 10.1358/dof.2018.043.10.2849626 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210323971A1 (en) * | 2018-08-31 | 2021-10-21 | Suzhou Pengxu Pharmatech Co., Ltd | Synthesis methods for upadacitinib and intermediate thereof |
| US11926633B2 (en) * | 2018-08-31 | 2024-03-12 | Suzhou Pengxu Pharmatech Co., Ltd | Synthesis methods for upadacitinib and intermediate thereof |
| CN115417803A (en) * | 2022-08-30 | 2022-12-02 | 四川同晟生物医药有限公司 | 5363 Synthesis method of intermediate (3R, 4S) -1-benzyloxycarbonyl-4-ethylpyrrolidine-3-carboxylic acid of Wu Pati Ni |
| CN115417803B (en) * | 2022-08-30 | 2023-10-03 | 四川同晟生物医药有限公司 | Synthesis method of Wu Pa tenib intermediate (3R, 4S) -1-benzyloxycarbonyl-4-ethylpyrrolidine-3-carboxylic acid |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8378142B2 (en) | Intermediate compounds and their use in preparation of lacosamide | |
| US9029504B2 (en) | Fluorene compound | |
| WO1999001420A1 (en) | Process for the preparation of 2-aminomalonic acid derivatives and intermediates used in the process | |
| DE69717974T2 (en) | METHOD FOR PRODUCING GANCICLOVIR DERIVATIVES | |
| EP2480524A1 (en) | Processes for the preparation of lacosamide and intermediates thereof | |
| WO2020202183A1 (en) | The process for the preparation of upadacitinib and its intermediates | |
| EP1870411B1 (en) | Process for the preparation and purification of valgancyclovir | |
| KR20120128667A (en) | A process for the preparation of lacosamide | |
| WO2017163257A1 (en) | Process for preparing pure lh-pyrazolo[3,4-d] pyrimidine derivative | |
| JPH07258222A (en) | Substituted n-ethylglycine derivative for producing pna and pna/dna hybrid | |
| US20020120136A1 (en) | Method of preparing cephalosporins using 4-hydroxyphenylglycine derivatives | |
| JP4294121B2 (en) | Process for producing pyridonecarboxylic acid derivatives and intermediates thereof | |
| US10329325B2 (en) | Process for the preparation of (S)-4-methyl-N-((S)-1-(((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxo-pentan-2-yl) amino)-1-oxo-3-phenylpropan-2-yl)-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido) pentanamide | |
| KR100766578B1 (en) | A process for preparing rebamipide | |
| WO2022044037A1 (en) | An improved process for the preparation of upadacitinib intermediate | |
| JP3193597B2 (en) | Method for producing glycine derivative | |
| US20030065207A1 (en) | Processes and compositions for the production of chiral amino-nitriles | |
| JP4181233B2 (en) | Method for producing pyrrolidine-2,4-dione derivative | |
| JP2008184388A (en) | Method for producing tetra-substituted pyrrolidines | |
| KR100450607B1 (en) | Process for preparing Galamin | |
| US20230174569A1 (en) | PROCESS PER PREPARING (3a,5a)-20-OXOPREGNAN-3-YL GLYCYL-L-VALINATE HYDROCHLORIDE | |
| RU2022109586A (en) | CONJUGATE OF 3-HYDROXY-3-METHYLGLUTARYL-COA REDUCTASE INHIBITOR WITH AN ASIALOGLYCOPROTEIN RECEPTOR LIGAND | |
| JPH0118916B2 (en) | ||
| JP2023179354A (en) | Method for producing morpholino nucleic acid using h-phosphonate technique | |
| JP2006022019A (en) | Sphingomyelin analogue and method for producing the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20721822 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20721822 Country of ref document: EP Kind code of ref document: A1 |