作为RHO激酶抑制剂的异喹啉磺酰衍生物
技术领域
本发明涉及一类作为RHO激酶抑制剂的异喹啉磺酰衍生物及其药物组合物,并涉及其制药用途。具体地,本发明涉及式(I)所示化合物或其药学上可接受的盐。
背景技术
法舒地尔是一种具有广泛药理作用的新型药物,为RHO激酶抑制物,通过增加肌球蛋白轻链磷酸酶的活性扩张血管,降低内皮细胞的张力,改善脑组织微循环,不产生和加重脑的盗血,同时可拮抗炎性因子,保护神经抗凋亡,促进神经再生。本结果表明盐酸法舒地尔对促进神经功能的恢复,减轻临床症状,减少病残率有一定疗效。因此对于在基层由于受经济条件的制约以及对疾病认识程度,超早期溶栓治疗不能实现,但为减少疾病的进一步进展,在治疗时间窗内重建局部血循环显得至关重要,而盐酸法舒地尔具有对缺血性脑血管病的显著神经保护和治疗作用,值得在临床尤其是基层的使用,减少致残率,提高生活质量。
WO2004106325公开了一类化合物,属于法舒地尔的前药,其结构通式如式(B-Ⅰ)所示:
尽管现有技术中存在上述化合物可作为RHO激酶抑制剂,但是它们在活性、溶解性、药代动力学、成药性的等方面有待改进。
发明内容
本发明的目的在于提供式(I)所示化合物或其药学上可接受的盐,
其特征是,
R1、X分别独立地选自H、F、Cl、Br、I、CN、OH、NH2、C1-3烷基、C1-3烷基氧基、C1-3烷基氨基、N,N-二(C1-3烷基)氨基,所述C1-3烷基任选地被R01所取代,R01选自F、Cl、Br、I、OH、NH2,R01的数目为1、2或3;
W、W’分别独立地选自N(Rw1)、C(Rw2)(Rw3)或单键;
L、Z分别独立地选自单键或C(Rz1)(Rz2);
P、P’分别选自(CH2)q1;Q、Q’分别选自(CH2)q2;
q1、q2分别独立地选自0、1、2、3或4;
当q1、q2选自0时,(CH2)q1、(CH2)q2表示单键;
P、W、Q三者中至多两个同时为单键;
P’、W’、Q’三者中至多两个同时为单键;
R3a、R3b、R3、Rw1、Rw2、Rw3、Rz1、Rz2分别独立地选自H、F、Cl、Br、I、CN、OH、NH2、C1-3烷基、C1-3烷基氧基、C1-3烷基氨基、N,N-(二C1-3烷基)氨基,所述C1-3烷基任选地被R01所取代。
本发明的一个方案中,R3a、R3b、R3、Rw1、Rw2、Rw3选自卤代、羟代和/或胺代甲基、乙基或丙基;
R01选自F、Cl、Br、I、OH、NH2,R01的数目为1、2或3。
本发明的一个方案中,所述R1选自H、F、Cl、Br、I、甲基、二氟甲基、三氟甲基、甲氧基;X选自H或OH。
本发明的一个方案中,所述R
2选自
其中A、B中一个为单键,另一个为亚甲基;R4选自H、F、Cl、Br、I、CN、OH、NH
2、C
1-3烷基、C
1-3烷基氧基、C
1-3烷基氨基、N,N二(C
1-3烷基)氨基,所述C
1-3烷基任选地被R
01所取代,R
01选自F、Cl、Br、I、OH、NH
2,R
01的数目为1、2或3;优选地,R4选自NH2、乙氨基;
本发明的一个方案中,上述化合物或其药学上可接受的盐,所述R
2选自
或
其中,D、E中一个为单键,另一个为亚甲基;G选自(CH
2)
g;T选自(CH
2)
t;g、t分别独立地选自0、1、2、3或4;
本发明的一个方案中,g为1、2、3或4,t为0或1;
本发明的一个方案中,R3b选自NH2;
本发明的一个方案中,R2选自
本发明的一个方案中,上述R
2选自
其中,Y选自(CH
2)
y;M选自(CH
2)
m;y选自0、1、2或3;m选自0或1;
本发明的一个方案中,上述R2选自
本发明的一个方案中,上述化合物或其药学上可接受的盐选自:
本发明的另一个目的在于提供一种药物组合物,其含有治疗有效量的上述化合物或其药学上可接受的盐和药学上可接受的载体。
本发明的另一个目的在于提供上述化合物或其药学上可接受的盐或上述药物组合物在制备治疗血管收缩引起的相关各种病症的药物中的应用,其中所述相关的各种病症包括脑栓塞、脑缺血、脑损伤、椎底动脉供血不足、蛛网膜下腔出血所引起的脑血管痉挛、心绞痛、青光眼、高血压、纤维化。
这里所采用的术语“药学上可接受的”,是针对那些化合物、材料、组合物和/或剂型而言,它们在可靠的医学判断的范围之内,适用于与人类和动物的组织接触使用,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。
术语“药学上可接受的盐”是指本发明化合物的盐,由本发明发现的具有特定取代基的化合物与相对无毒的酸或碱制备。当本发明的化合物中含有相对酸性的功能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱与这类化合物的中性形式接触的方式获得碱加成盐。药学上可接受的碱加成盐包括钠、钾、钙、铵、有机氨或镁盐或类似的盐。当本发明的化合物中含有相对碱性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的酸与这类化合物的中性形式接触的方式获得酸加成盐。药学上可接受的酸加成盐的实例包括无机酸盐,所述无机酸包括例如盐酸、氢溴酸、硝酸、碳酸,碳酸氢根,磷酸、磷酸一氢根、磷酸二氢根、硫酸、硫酸氢根、氢碘酸、亚磷酸等;以及有机酸盐,所述有机酸包括如乙酸、丙酸、异丁酸、马来酸、丙二酸、苯甲酸、琥珀酸、辛二酸、反丁烯二酸、乳酸、扁桃酸、邻苯二甲酸、苯磺酸、对甲苯磺酸、柠檬酸、酒石酸和甲磺酸等类似的酸;还包括氨基酸(如精氨酸等)的盐,以及如葡糖醛酸等有机酸的盐(参见Berge et al.,"Pharmaceutical Salts",Journal of Pharmaceutical Science 66:1-19(1977))。本发明的某些特定的化合物含有碱性和酸性的官能团,从而可以被转换成任一碱或酸加成盐。
优选地,以常规方式使盐与碱或酸接触,再分离母体化合物,由此再生化合物的中性形式。化合物的母体形式与其各种盐的形式的不同之处在于某些物理性质,例如在极性溶剂中的溶解度不同。
本文所用的“药学上可接受的盐”属于本发明化合物的衍生物,其中,通过与酸成盐或与碱成盐的方式修饰所述母体化合物。药学上可接受的盐的实例包括但不限于:碱基比如胺的无机酸或有机酸盐、酸根比如羧酸的碱金属或有机盐等等。药学上可接受的盐包括常规的无毒性的盐或母体化合物的季铵盐,例如无毒的无机酸或有机酸所形成的盐。常规的无毒性的盐包括但不限于那些衍生自无机酸和有机酸的盐,所述的无机酸或有机酸选自2-乙酰氧基苯甲酸、2-羟基乙磺酸、乙酸、抗坏血酸、苯磺酸、苯甲酸、碳酸氢根、碳酸、柠檬酸、依地酸、乙烷二磺酸、乙烷磺酸、富马酸、葡庚糖、葡糖酸、谷氨酸、乙醇酸、氢溴酸、盐酸、氢碘酸盐、羟基、羟萘、羟乙磺酸、乳酸、乳糖、十二烷基磺酸、马来酸、苹果酸、扁桃酸、甲烷磺酸、硝酸、草酸、双羟萘酸、泛酸、苯乙酸、磷酸、多聚半乳糖醛、丙酸、水杨酸、硬脂酸、亚乙酸、琥珀酸、氨基磺酸、对氨基苯磺酸、硫酸、单宁、酒石酸和对甲苯磺酸。
本发明的药学上可接受的盐可由含有酸根或碱基的母体化合物通过常规化学方法合成。一般情况下,这样的盐的制备方法是:在水或有机溶剂或两者的混合物中,经由游离酸或碱形式的这些化合物与化学计量的适当的碱或酸反应来制备。一般地,优选醚、乙酸乙酯、乙醇、异丙醇或乙腈等非水介质。
除了盐的形式,本发明所提供的化合物还存在前药形式。本文所描述的化合物的前药容易地在生理条件下发生化学变化从而转化成本发明的化合物。此外,前体药物可以在体内环境中通过化学或生化方法被转换到本发明的化合物。
本发明的某些化合物可以以非溶剂化形式或者溶剂化形式存在,包括水合物形式。一般而言,溶剂化形式与非溶剂化的形式相当,都包含在本发明的范围之内。本发明的某些化合物可以以多晶或无定形形式存在。
本发明的某些化合物可以具有不对称碳原子(光学中心)或双键。外消旋体、非对映异构体、几何异构体和单个的异构体都包括在本发明的范围之内。
本文中消旋体、ambiscalemic and scalemic或者对映体纯的化合物的图示法来自Maehr,J.Chem.Ed.1985,62:114-120。1985年,62:114-120。除非另有说明,用楔形键和虚线键表示一个立体中心的绝对构型。当本文所述化合物含有烯属双键或其它几何不对称中心,除非另有规定,它们包括E、Z几何异构体。同样地,所有的互变异构形式均包括在本发明的范围之内。
本发明的化合物可以存在特定的几何或立体异构体形式。本发明设想所有的这类化合物,包括顺式和反式异构体、(-)-和(+)-对对映体、(R)-和(S)-对映体、非对映异构体、(D)-异构体、(L)-异构体,及其外消旋混合物和其他混合物,例如对映异构体或非对映体富集的混合物,所有这些混合物都属于本发明的范围之内。烷基等取代基中可存在另外的不对称碳原子。所有这些异构体以及它们的混合物,均包括在本发明的范围之内。
可以通过的手性合成或手性试剂或者其他常规技术制备光学活性的(R)-和(S)-异构体以及D和L异构体。如果想得到本发明某化合物的一种对映体,可以通过不对称合成或者具有手性助剂的衍生作用来制备,其中将所得非对映体混合物分离,并且辅助基团裂开以提供纯的所需对映异构体。或者,当分子中含有碱性官能团(如氨基)或酸性官能团(如羧基)时,与适当的光学活性的酸或碱形成非对映异构体的盐,然后通过本领域所公知的分步结晶法或色谱法进行非对映异构体拆分,然后回收得到纯的对映体。此外,对映异构体和非对映异构体的分离通常是通过使用色谱法完成的,所述色谱法采用手性固定相,并任选地与
化学衍生法相结合(例如由胺生成氨基甲酸盐)。
本发明的化合物可以在一个或多个构成该化合物的原子上包含非天然比例的原子同位素。例如,可用放射性同位素标记化合物,比如氚(3H),碘-125(125I)或C-14(14C)。本发明的化合物的所有同位素组成的变换,无论放射性与否,都包括在本发明的范围之内。
术语“药学上可接受的载体”是指能够递送本发明有效量活性物质、不干扰活性物质的生物活性并且对宿主或者患者无毒副作用的任何制剂或载体介质代表性的载体包括水、油、蔬菜和矿物质、膏基、洗剂基质、软膏基质等。这些基质包括悬浮剂、增粘剂、透皮促进剂等。它们的制剂为化妆品领域或局部药物领域的技术人员所周知。关于载体的其他信息,可以参考Remington:The Science and Practice of Pharmacy,21st Ed.,Lippincott,Williams&Wilkins(2005),该文献的内容通过引用的方式并入本文。
术语“赋形剂”通常是指配制有效的药物组合物所需要载体、稀释剂和/或介质。
针对药物或药理学活性剂而言,术语“有效量”或“治疗有效量”是指无毒的但能达到预期效果的药物或药剂的足够用量。对于本发明中的口服剂型,组合物中一种活性物质的“有效量”是指与该组合物中另一种活性物质联用时为了达到预期效果所需要的用量。有效量的确定因人而异,取决于受体的年龄和一般情况,也取决于具体的活性物质,个案中合适的有效量可以由本领域技术人员根据常规试验确定。
术语“活性成分”、“治疗剂”,“活性物质”或“活性剂”是指一种化学实体,它可以有效地治疗目标紊乱、疾病或病症。
术语“被取代的”是指特定原子上的任意一个或多个氢原子被取代基取代,包括重氢和氢的变体,只要特定原子的价态是正常的并且取代后的化合物是稳定的。当取代基为酮基(即=O)时,意味着两个氢原子被取代。酮取代不会发生在芳香基上。术语“任选被取代的”是指可以被取代,也可以不被取代,除非另有规定,取代基的种类和数目在化学上可以实现的基础上可以是任意的。
当任何变量(例如R)在化合物的组成或结构中出现一次以上时,其在每一种情况下的定义都是独立的。因此,例如,如果一个基团被0-2个R所取代,则所述基团可以任选地至多被两个R所取代,并且每种情况下的R都有独立的选项。此外,取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。
当其中一个变量选自单键时,表示其连接的两个基团直接相连,比如A-L-Z中L代表单键时表示该结构实际上是A-Z。
当一个取代基的键可以交叉连接到一个环上的两个原子时,这种取代基可以与这个环上的任意原子相键合。当所列举的取代基中没有指明其通过哪一个原子连接到化学结构通式中包括但未具体提及的化合物时,这种取代基可以通过其任何原子相键合。取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。
烷基和杂烷基原子团(包括通常被称为亚烷基、链烯基、亚杂烷基、杂烯基、炔基、环烷基、杂环烷基、环烯基和杂环烯基的那些基团)的取代基一般被称为“烷基取代基”,它们可以选自但不限于下列基团中的一个或多个:-R’、-OR’、=O、=NR’、=N-OR’、-NR’R”、-SR’、卤素、-SiR’R”R”’、OC(O)R’、-C(O)R’、-CO2R’、-CONR’R”、-OC(O)NR’R”、-NR”C(O)R’、NR’C(O)NR”R”’、-NR”C(O)2R’、-NR””’-C(NR’R”R’”)=NR””、NR””C(NR’R”)=NR’”、-S(O)R’、-S(O)2R’、-S(O)2NR’R”、NR”SO2R’、-CN、
–NO2、-N3、-CH(Ph)2和氟代(C1-C4)烷基,取代基的数目为0~(2m’+1),其中m’是这类原子团中碳原子的总数。R'、R”、R”'、R””和R””’各自独立地优选氢、被取代或未被取代的杂烷基、被取代或未被取代的芳基(例如被1~3个卤素取代芳基)、被取代或未被取代的烷基、烷氧基、硫代烷氧基基团或芳烷基。当本发明的化合物包括一个以上的R基团时,例如,每一个R基团是独立地加以选择的,如同当存在一个以上的R'、R”、R”'、R””和R””’基团时的每个这些基团。当R'和R”附着于同一个氮原子时,它们可与该氮原子结合形成5-,6-或7-元环。例如,-NR'R“意在包括但不仅限于1-吡咯烷基和4-吗啉基。根据上述关于取代基的讨论中,本领域技术人员可以理解,术语“烷基”意在包括碳原子键合于非氢基团所构成的基团,如卤代烷基(例如-CF3、-CH2CF3)和酰基(例如-C(O)CH3、-C(O)CF3、-C(O)CH2OCH3等)。
与烷基原子团所述取代基相似,芳基和杂芳基取代基一般统称为“芳基取代基”,选自例如-R’、-OR’、-NR’R”、-SR’、-卤素,-SiR’R”R”’、OC(O)R’、-C(O)R’、-CO2R’、-CONR’R”、-OC(O)NR’R”、-NR”C(O)R’、NR’C(O)NR”R”’、-NR”C(O)2R’、-NR””’-C(NR’R”R’”)=NR””、NR””C(NR’R”)=NR’”、-S(O)R’、-S(O)2R’、-S(O)2NR’R”、NR”SO2R’、-CN、–NO2、-N3、-CH(Ph)2、氟(C1-C4)烷氧基和氟(C1-C4)烷基等,取代基的数量为0到芳香环上开放化合价的总数之间;其中R’、R”、R”’、R””和R””’独立地优选自氢、被取代或未被取代的烷基、被取代或未被取代的杂烷基、被取代或未被取代的芳基和被取代或未被取代的杂芳基。当本发明的化合物包括一个以上的R基团时,例如,每个R基团是独立地加以选择的,如同当存在一个以上R’、R”、R”’、R””和R””’基团时的每个这些基团。
芳基或杂芳基环的相邻原子上的两个取代基可以任选地被通式为–T-C(O)-(CRR’)q-U-的取代基所取代,其中T和U独立地选自-NR-、-O-、CRR'-或单键,q是0到3的整数。作为替代选择,芳基或杂芳基环的相邻原子上的两个取代基可以任选地被通式为–A(CH2)r B-的取代基所取代,其中A和B独立的选自–CRR’-、-O-、-NR-、-S-、-S(O)-、S(O)2-、-S(O)2NR’-或单键,r是1~4的整数。任选地,由此形成的新环上的一个单键可以替换为双键。作为替代选择,芳基或杂芳基环的相邻原子上的两个取代基可以任选地被通式为–A(CH2)r B-的取代基所取代,其中s和d分别独立的选自0~3的整数,X是–O-、-NR’、-S-、-S(O)-、-S(O)2-或–S(O)2NR’-。取代基R、R’、R”和R”’分别独立地优选自氢和被取代或未被取代的(C1-C6)烷基。
除非另有规定,术语“卤代素”或“卤素”本身或作为另一取代基的一部分表示氟、氯、溴或碘原子。此外,术语“卤代烷基”意在包括单卤代烷基和多卤代烷基。例如,术语“卤代(C1-C4)烷基”意在包括但不仅限于三氟甲基、2,2,2-三氟乙基、4-氯丁基和3-溴丙基等等。
卤代烷基的实例包括但不仅限于:三氟甲基、三氯甲基、五氟乙基,和五氯乙基。“烷氧基”代表通过氧桥连接的具有特定数目碳原子的上述烷基。C1-6烷氧基包括C1、C2、C3、C4、C5和C6的烷氧基。烷氧基的例子包括但不限于:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、正戊氧基和S-戊氧基。“环烷基”包括饱和环基,如环丙基、环丁基或环戊基。3-7环烷基包括C3、C4、C5、C6和C7环烷基。“链烯基”包括直链或支链构型的烃链,其中该链上任何的稳定位点上存在一个或多个碳-碳双键,例如乙烯基和丙烯基。
术语“卤”或“卤素”是指氟、氯、溴和碘。
除非另有规定,术语“杂”表示杂原子或杂原子团(即含有杂原子的原子团),包括碳(C)和氢(H)以
外的原子以及含有这些杂原子的原子团,例如包括氧(O)、氮(N)、硫(S)、硅(Si)、锗(Ge)、铝(Al)、硼(B)、-O-、-S-、=O、=S、-C(=O)O-、-C(=O)-、-C(=S)-、-S(=O)、-S(=O)2-,以及任选被被取代的-C(=O)N(H)-、-N(H)-、-C(=NH)-、-S(=O)2N(H)-或-S(=O)N(H)-。
除非另有规定,“环”表示被取代或未被取代的环烷基、杂环烷基、环烯基、杂环烯基、环炔基、杂环炔基、芳基或杂芳基。所谓的环包括单环、联环、螺环、并环或桥环。环上原子的数目通常被定义为环的元数,例如,“5~7元环”是指环绕排列5~7个原子。除非另有规定,该环任选地包含1~3个杂原子。因此,“5~7元环”包括例如苯基吡啶和哌啶基;另一方面,术语“5~7元杂环烷基环”包括吡啶基和哌啶基,但不包括苯基。术语“环”还包括含有至少一个环的环系,其中的每一个“环”均独立地符合上述定义。
除非另有规定,术语“杂环”或“杂环基”意指稳定的含杂原子或杂原子团的单环、双环或三环,它们可以是饱和的、部分不饱和的或不饱和的(芳族的),它们包含碳原子和1、2、3或4个独立地选自N、O和S的环杂原子,其中上述任意杂环可以稠合到一个苯环上形成双环。氮和硫杂原子可任选被氧化(即NO和S(O)p)。氮原子可以是被取代的或未取代的(即N或NR,其中R是H或本文已经定义过的其他取代基)。该杂环可以附着到任何杂原子或碳原子的侧基上从而形成稳定的结构。如果产生的化合物是稳定的,本文所述的杂环可以发生碳位或氮位上的取代。杂环中的氮原子任选地被季铵化。一个优选方案是,当杂环中S及O原子的总数超过1时,这些杂原子彼此不相邻。另一个优选方案是,杂环中S及O原子的总数不超过1。如本文所用,术语“芳族杂环基团”或“杂芳基”意指稳定的5、6、7元单环或双环或7、8、9或10元双环杂环基的芳香环,它包含碳原子和1、2、3或4个独立地选自N、O和S的环杂原子。氮原子可以是被取代的或未取代的(即N或NR,其中R是H或本文已经定义过的其他取代基)。氮和硫杂原子可任选被氧化(即NO和S(O)p)。值得注意的是,芳香杂环上S和O原子的总数不超过1。桥环也包含在杂环的定义中。当一个或多个原子(即C、O、N或S)连接两个不相邻的碳原子或氮原子时形成桥环。优选的桥环包括但不限于:一个碳原子、两个碳原子、一个氮原子、两个氮原子和一个碳-氮基。值得注意的是,一个桥总是将单环转换成三环。桥环中,环上的取代基也可以出现在桥上。
杂环化合物的实例包括但不限于:吖啶基、吖辛因基、苯并咪唑基、苯并呋喃基、苯并巯基呋喃基、苯并巯基苯基、苯并恶唑基、苯并恶唑啉基、苯并噻唑基、苯并三唑基、苯并四唑基、苯并异恶唑基、苯并异噻唑基、苯并咪唑啉基、咔唑基、4aH-咔唑基、咔啉基、苯并二氢吡喃基、色烯、噌啉基十氢喹啉基、2H,6H-1,5,2-二噻嗪基、二氢呋喃并[2,3-b]四氢呋喃基、呋喃基、呋咱基、咪唑烷基、咪唑啉基、咪唑基、1H-吲唑基、吲哚烯基、二氢吲哚基、中氮茚基、吲哚基、3H-吲哚基、isatino基、异苯并呋喃基、吡喃、异吲哚基、异二氢吲哚基、异吲哚基、吲哚基、异喹啉基、异噻唑基、异恶唑基、亚甲二氧基苯基、吗啉基、萘啶基,八氢异喹啉基、恶二唑基、1,2,3-恶二唑基、1,2,4-恶二唑基、1,2,5-恶二唑基、1,3,4-恶二唑基、恶唑烷基、恶唑基、异恶唑基、羟吲哚基、嘧啶基、菲啶基、菲咯啉基、吩嗪、吩噻嗪、苯并黄嘌呤基、酚恶嗪基、酞嗪基、哌嗪基、哌啶基、哌啶酮基、4-哌啶酮基、胡椒基、蝶啶基、嘌呤基、吡喃基、吡嗪基、吡唑烷基、吡唑啉基、吡唑基、哒嗪基、吡啶并恶唑、吡啶并咪唑、吡啶并噻唑、吡啶基、嘧啶基、吡咯烷基、吡咯啉基、2H-吡咯基、吡咯基、吡唑基、喹唑啉基、喹啉基、4H-喹嗪基、喹喔啉基、奎宁环基、四氢呋喃基、四氢异喹啉基、四氢喹啉基、四唑基,6H-1,2,5-噻二嗪基、1,2,3-噻二唑基、1,2,4-噻二唑基、1,2,5-噻二唑基、1,3,4-噻二唑基、噻蒽基、噻唑基、异噻唑基噻吩基、噻吩基、噻吩并恶唑基、噻
吩并噻唑基、噻吩并咪唑基、噻吩基、三嗪基、1,2,3-三唑基、1,2,4-三唑基、1,2,5-三唑基、1,3,4-三唑基和呫吨基。还包括稠环和螺环化合物。
除非另有规定,术语“烃基”或者其下位概念(比如烷基、烯基、炔基、苯基等等)本身或者作为另一取代基的一部分表示直链的、支链的或环状的烃原子团或其组合,可以是完全饱和的、单元或多元不饱和的,可以是单取代、二取代或多取代的,可以包括二价或多价原子团,具有指定数量的碳原子(如C1-C10表示1至10个碳)。“烃基”包括但不限于脂肪烃基和芳香烃基,所述脂肪烃基包括链状和环状,具体包括但不限于烷基、烯基、炔基,所述芳香烃基包括但不限于6-12元的芳香烃基,例如苯、萘等。在一些实施例中,术语“烷基”表示直链的或支链的原子团或它们的组合,可以是完全饱和的、单元或多元不饱和的,可以包括二价和多价原子团。饱和烃原子团的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、叔丁基、异丁基、仲丁基、异丁基、环己基、(环己基)甲基、环丙基甲基,以及正戊基、正己基、正庚基、正辛基等原子团的同系物或异构体。不饱和烷基具有一个或多个双键或三键,其实例包括但不限于乙烯基、2-丙烯基、丁烯基、巴豆基、2-异戊烯基、2-(丁二烯基)、2,4-戊二烯基、3-(1,4-戊二烯基)、乙炔基、1-和3-丙炔基,3-丁炔基,以及更高级的同系物和异构体。
除非另有规定,术语“杂烃基”或者其下位概念(比如杂烷基、杂烯基、杂炔基、杂芳基等等)本身或者与另一术语联合表示稳定的直链的、支链的或环状的烃原子团或其组合,有一定数目的碳原子和至少一个杂原子组成。在一些实施例中,术语“杂烷基”本身或者与另一术语联合表示稳定的直链的、支链的烃原子团或其组合物,有一定数目的碳原子和至少一个杂原子组成。在一个典型实施例中,杂原子选自B、O、N和S,其中氮和硫原子任选地被氧化,氮杂原子任选地被季铵化。杂原子B、O、N和S可以位于杂烃基的任何内部位置(包括该烃基附着于分子其余部分的位置)。实例包括但不限于-CH2-CH2-O-CH3、-CH2-CH2-NH-CH3、-CH2-CH2-N(CH3)-CH3、-CH2-S-CH2-CH3、-CH2-CH2、-S(O)-CH3、-CH2-CH2-S(O)2-CH3、-CH=CH-O-CH3、-CH2-CH=N-OCH3和–CH=CH-N(CH3)-CH3。至多两个杂原子可以是连续的,例如-CH2-NH-OCH3。
术语“烷氧基”、“烷氨基”和“烷硫基”(或硫代烷氧基)属于惯用表达,是指分别通过一个氧原子、氨基或硫原子连接到分子的其余部分的那些烷基基团。
除非另有规定,术语“环烃基”、“杂环烃基”或者其下位概念(比如芳基、杂芳基、环烷基、杂环烷基、环烯基、杂环烯基、环炔基、杂环炔基等等)本身或与其他术语联合分别表示环化的“烃基”、“杂烃基”。此外,就杂烃基或杂环烃基(比如杂烷基、杂环烷基)而言,杂原子可以占据该杂环附着于分子其余部分的位置。环烷基的实例包括但不限于环戊基、环己基、1-环己烯基、3-环己烯基、环庚基等。杂环基的非限制性实例包括1-(1,2,5,6-四氢吡啶基)、1-哌啶基、2-哌啶基,3-哌啶基、4-吗啉基、3-吗啉基、四氢呋喃-2-基、四氢呋喃吲哚-3-基、四氢噻吩-2-基、四氢噻吩-3-基,1-哌嗪基和2-哌嗪基。
除非另有规定,术语“芳基”表示多不饱和的芳族烃取代基,可以是单取代、二取代或多取代的,它可以是单环或多环(优选1至3个环),它们稠合在一起或共价连接。术语“杂芳基”是指含有一至四个杂原子的芳基(或环)。在一个示范性实例中,杂原子选自B、N、O和S,其中氮和硫原子任选地被氧化,氮原子任选地被季铵化。杂芳基可通过杂原子连接到分子的其余部分。芳基或杂芳基的非限制性实施例包括苯基、1-萘基、2-萘基、4-联苯基、1-吡咯基、2-吡咯基、3-吡咯基、3-吡唑基、2-咪唑基、4-咪唑基、吡嗪
基、2-恶唑基、4-恶唑基、2-苯基-4-恶唑基、5-恶唑基、3-异恶唑基、4-异恶唑基、5-异恶唑基、2-噻唑基、4-噻唑基、5-噻唑基、2-呋喃基、3-呋喃基、2-噻吩基、3-噻吩基、2-吡啶基、3-吡啶基、4-吡啶基、2-嘧啶基、4-嘧啶基、5-苯并噻唑基、嘌呤基、2-苯并咪唑基、5-吲哚基、1-异喹啉基、5-异喹啉基、2-喹喔啉基、5-喹喔啉基、3-喹啉基和6-喹啉基。上述任意一个芳基和杂芳基环系的取代基选自下文所述的可接受的取代基。
为简便起见,芳基在与其他术语联合使用时(例如芳氧基、芳硫基、芳烷基)包括如上定义的芳基和杂芳基环。因此,术语“芳烷基”意在包括芳基附着于烷基的那些原子团(例如苄基、苯乙基、吡啶基甲基等),包括其中碳原子(如亚甲基)已经被例如氧原子代替的那些烷基,例如苯氧基甲基、2-吡啶氧甲基3-(1-萘氧基)丙基等。
术语“离去基团”是指可以被另一种官能团或原子通过取代反应(例如亲和取代反应)所取代的官能团或原子。例如,代表性的离去基团包括三氟甲磺酸酯;氯、溴、碘;磺酸酯基,如甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯、对甲苯磺酸酯等;酰氧基,如乙酰氧基、三氟乙酰氧基等等。
术语“保护基”包括但不限于“氨基保护基”、“羟基保护基”或“巯基保护基”。术语“氨基保护基”是指适合用于阻止氨基氮位上副反应的保护基团。代表性的氨基保护基包括但不限于:甲酰基;酰基,例如链烷酰基(如乙酰基、三氯乙酰基或三氟乙酰基);烷氧基羰基,如叔丁氧基羰基(Boc);芳基甲氧羰基,如苄氧羰基(Cbz)和9-芴甲氧羰基(Fmoc);芳基甲基,如苄基(Bn)、三苯甲基(Tr)、1,1-二-(4'-甲氧基苯基)甲基;甲硅烷基,如三甲基甲硅烷基(TMS)和叔丁基二甲基甲硅烷基(TBS)等等。术语“羟基保护基”是指适合用于阻止羟基副反应的保护基。代表性羟基保护基包括但不限于:烷基,如甲基、乙基和叔丁基;酰基,例如链烷酰基(如乙酰基);芳基甲基,如苄基(Bn),对甲氧基苄基(PMB)、9-芴基甲基(Fm)和二苯基甲基(二苯甲基,DPM);甲硅烷基,如三甲基甲硅烷基(TMS)和叔丁基二甲基甲硅烷基(TBS)等等。
本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。
本发明采用下述缩略词:aq代表水;HATU代表O-7-氮杂苯并三唑-1-基)-N,N,N',N'-四甲基脲六氟磷酸盐;EDC代表N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺盐酸盐;m-CPBA代表3-氯过氧苯甲酸;eq代表当量、等量;CDI代表羰基二咪唑;DCM代表二氯甲烷;PE代表石油醚;DIAD代表偶氮二羧酸二异丙酯;DMF代表N,N-二甲基甲酰胺;DMSO代表二甲亚砜;EtOAc代表乙酸乙酯;EtOH代表乙醇;MeOH代表甲醇;CBz代表苄氧羰基,是一种胺保护基团;BOC代表叔丁基羰基是一种胺保护基团;HOAc代表乙酸;NaCNBH3代表氰基硼氢化钠;r.t.代表室温;O/N代表过夜;THF代表四氢呋喃;Boc2O代表二-叔丁基二碳酸酯;TFA代表三氟乙酸;DIPEA代表二异丙基乙基胺;SOCl2代表氯化亚砜;CS2代表二硫化碳;TsOH代表对甲苯磺酸;NFSI代表N-氟-N-(苯磺酰基)苯磺酰胺;NCS代表1-氯吡咯烷-2,5-二酮;n-Bu4NF代表氟化四丁基铵;iPrOH代表2-丙醇;mp代表熔点。
化合物经手工或者
软件命名,市售化合物采用供应商目录名称。
与现有技术相比,本发明化合物高效、低毒,在活性、半衰期、溶解度和药代动力学等方面均取得了
显著甚至预料不到的进步,更适合于制药。
具体实施方式
下面通过实施例对本发明进行详细描述,但并不意味着对本发明任何不利限制。本文已经详细地描述了本发明,其中也公开了其具体实施例方式,对本领域的技术人员而言,在不脱离本发明精神和范围的情况下针对本发明具体实施方式进行各种变化和改进将是显而易见的。
实施例1
5-((六氢吡咯并[3,2-b]吡咯-1(2H)-基)磺酰基)异喹啉
第一步
将异喹啉1a(47.5mL,405mmol)缓慢加入到22mL浓硫酸中,充分搅拌成小块,然后加入到200mL 20%的发烟硫酸中,室温放置两天。然后倒入700g冰水中并静置过夜,得到的悬浊液过滤,滤饼用水洗涤两遍(100mL x 2),干燥,得到异喹啉-5-磺酸1b(50g,产率:60%),直接用于下一步反应
1H NMR(400MHz,D2O):δ9.66(s,1H),8.94-8.92(m,1H),8.62-8.60(m,2H),8.58-8.56(m,1H),8.50-8.48(m,1H),7.99-7.95(m,1H).
MS-ESI计算值[M+H]+210,实测值210.
第二步
室温下将异喹啉-5-磺酸1b(4.0g,0.019mol)加入到25mL的氯化亚砜中,然后加入0.1mL的N,N-二甲基甲酰胺。反应加热回流两小时后,减压蒸掉多余的氯化亚砜,蒸发残渣用冷的二氯甲烷洗涤(10mL x 2)。干燥得到目标产物异喹啉-5-磺酰氯1c(3.9g,黄色固体,产率:100%)。
MS-ESI计算值[M+H]+227实测值227。
第三步
将异喹啉-5-磺酰氯1c(150mg,0.330mmol)溶解于2mL二氯甲烷中,在氮气保护下0℃先后加入顺式-六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯1d(80mg,0.38mmol,市售商品)和N,N-二异丙基乙基胺(0.25mL,1.14mmol)。所得反应液室在温搅拌16小时直到反应结束。用二氯甲烷(10mL)和水(10mL)稀释,二氯甲烷萃取(10mL x 2),合并有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱(0-100%乙酸乙酯/石油醚)纯化得目标化合物4-(异喹啉-5-基磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯1e(100mg,无色油状物,产率:65%)。
MS-ESI计算值[M+H]+404,实测值404.
第四步
将4-(异喹啉-5-基磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯1e(100mg,0.250mmol)溶解于5mL的乙酸乙酯中,于0℃滴加10mL乙酸乙酯的饱和氯化氢溶液,反应液于室温反应0.5小时。把得到的悬浊液过滤,滤饼用乙酸乙酯洗涤(10mL x2),干燥得目标化合物5-((六氢吡咯并[3,2-b]吡咯-1(2H)-基)磺酰基)异喹啉1(60mg,白色固体,产率:79%)。
1H NMR(400MHz,D2O):δ9.82(s,1H),9.10(d,J=6.8Hz,1H),8.84(d,J=7.6Hz,1H),8.77(d,J=7.6Hz,1H),8.68(d,J=6.8Hz,1H),8.15(t,J=7.6Hz,1H),4.55-4.40(m,1H),4.35-4.25(m,1H),3.62-3.55(m,1H),3.50-3.45(m,1H),3.39-3.30(m,2H),2.35-2.00(m,4H).
MS-ESI计算值[M+H]+304,实测值304.
实施例2
5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)异喹啉
第一步
将1-苄基-1H-吡咯-2,5-二酮2a(74.8g,0.400mol)、2-氯乙胺(58g,0.5mol)和三乙胺(40g,0.40mmol)溶解在400mL的1,4-二氧六环中,加热回流16小时。反应结束后冷却到室温,然后减压浓缩,粗产物用色谱硅胶柱(0-100%乙酸乙酯/石油醚)纯化得到1-苄基-3-((2-氯乙基)氨基)吡咯烷-2,5-二酮2b(102g,产率:96%)。
1H NMR(400MHz,CDCl3):δ7.40-7.25(m,5H),4.66(s,2H),3.82-3.77(m,1H),3.67-3.63(m,2H),3.01-2.94(m,3H),2.55-2.50(m,1H),2.21-2.15(m,1H).
第二步
氮气保护下,0℃将氢化钠(7.74g,358mmol)溶解于700mL N,N-二甲基甲酰胺中,于30℃的条件下加入1-苄基-3-((2-氯乙基)氨基)吡咯烷-2,5-二酮2b(40g,0.14mol)的50mL N,N-二甲基甲酰胺中。反应液室温
搅拌1小时,反应完全后倒入水中(1L),乙酸乙酯萃取(500mL×3),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残余物用色谱硅胶柱纯化(0-100%甲醇/乙酸乙酯)得到5-苄基四氢吡咯并[3,4-b]吡咯-4,6(2H,5H)-二酮2c(20g,无色油状液体,产率:59%)。
1H NMR(400MHz,CDCl3):δ7.37-7.28(m,5H),4.65(s,2H),4.13-4.11(m,1H),3.30-3.27(m,1H),3.07-3.05(m,1H),2.57-2.56(m,1H),2.15-2.05(m,3H).
第三步
在氮气保护下,0℃将四氢铝锂(7.19g,0.18mol)分批加入250mL的四氢呋喃中,0℃下滴加5-苄基四氢吡咯并[3,4-b]吡咯-4,6(2H,5H)-二酮2c(20g,0.086mol)的250mL四氢呋喃溶液。滴加结束后,反应体系逐渐升温至回流3小时。反应体系冷却至0℃,然后依次向反应液里逐滴滴加7.2mL水,7.2mL 15%氢氧化钠,21.6mL水。反应液继续搅拌0.5小时,过滤,浓缩得到5-苄基八氢吡咯并[3,4-b]吡咯2d(16g,无色油状液体,产率:92%)。
MS-ESI计算值[M+H]+203,实测值203.
第四步
将5-苄基八氢吡咯并[3,4-b]吡咯2d(15g,0.074mol)溶于300mL的二氯甲烷中,依次加入N,N-二异丙基乙基胺(19g,148mmol)和二碳酸二叔丁酯(17.8g,0.081mol)。反应于室温条件下搅拌4小时,待反应结束后直接浓缩,残余物用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到5-苄基六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2e(18g,无色油状物,产率:81%)。MS-ESI计算值[M+H]+310,实测值310.
第五步
将5-苄基六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2e(12g,39.7mmol)溶解于1L的四氢呋喃中,氩气保护下加入干氢氧化钯碳(1.8g,10%)。在3MPa氢气氛中和70℃条件下搅拌16小时。反应完全后冷却至室温,反应体系硅藻土过滤除去固体催化剂,滤液浓缩得到顺式-六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2f(8g,淡黄色液体,产率:95%)。
1H NMR(400MHz,CD3OD):δ4.21-4.17(m,1H),3.51-3.36(m,2H),3.05-2.90(m,3H),2.89-2.77(m,2H),2.06-2.02(m,1H),1.77-1.71(m,1H),1.48(s,9H).
第六步
顺式-六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2f(212mg,1.00mmol)和异喹啉-5-磺酰氯1c(250mg,1.10mmol)按照实施例一化合物1e的合成方法得到5-(异喹啉-5-基磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2g,直接用于下一步反应。
第七步
5-(异喹啉-5-基磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2g(50.0mg,0.130mmol)按照实施例一化合物1的合成方法得到5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)异喹啉2(19mg,白色固体,产率:50%)。
1H NMR(400MHz,CD3OD):δ9.39(s,1H),8.68-8.61(m,2H),8.48-8.38(m,2H),7.88-7.84(m,1H),3.83-3.74(m,1H),3.25-3.12(m,4H),2.82-2.77(m,3H),1.91-1.86(m,1H),1.60-1.56(m,1H).
MS-ESI计算值[M+H]+304,实测值304.
实施例3
5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)异喹啉
第一步
顺式-八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯3a(100mg,0.44mmol,市售商品)和异喹啉-5-磺酰氯1c(114mg,0.500mmol)按照实施例一化合物1e的合成方法顺利得到6-(异喹啉-5-基磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯3b,直接用于下一步反应。
第二步
6-(异喹啉-5-基磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯3b(80.0mg,0.190mmol)按照实施例一化合物1的合成方法得到5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)异喹啉3(40mg,白色固体,产率:65%)。
1H NMR(400MHz,CD3OD):δ9.35(s,1H),8.67-8.57(m,2H),8.47(d,J=7.2Hz,1H),8.37(d,J=8.0Hz,1H),7.81(t,J=8.0Hz,1H),3.47-3.38(m,3H),3.34-3.26(m,2H),2.78-2.71(m,1H),2.55-2.46(m,1H),2.17-2.07(m,1H),1.65-1.30(m,4H).
MS-ESI计算值[M+H]+318,实测值318.
实施例4
5-(3,6-二氮二环[3.2.0]庚烷-3-基磺酰基)异喹啉
第一步
3-(异喹啉-5-基磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯4a(66mg,0.33mmol,市售商品)和异喹啉-5-磺酰氯1c(107mg,0.400mmol)按照实施例一化合物1e的合成方法得到3-(异喹啉-5-基磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯4b(110mg,黄色油状液体,产率:85%)。
MS-ESI计算值[M+H]+390,实测值390。
第二步
3-(异喹啉-5-基磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯4b(30mg,0.77mmol)按照实施例一化合物1的合成方法得到5-(3,6-二氮杂双环[3.2.0]庚烷-3-磺酰基)异喹啉4(15mg,黄色固体,产率:67%)。
1H NMR(400MHz,D2O):δ9.74(s,1H),9.17(d,J=7.2Hz,1H),8.78(d,J=7.2Hz,1H),8.7(d,J=8.4Hz,1H),8.6(d,J=8.4Hz,1H),8.09(t,J=7.2Hz,1H),4.15-4.05(m,1H),4.05-3.95(m,1H),3.75-3.69(m,3H),3.29-3.24(m,1H),2.96-2.93(m,1H),2.80-2.78(m,1H)。
MS-ESI计算值[M+H]+290,实测值290。
实施例5
6-(异喹啉-5-基磺酰基)十氢-1,6-二氮杂萘
第一步
将N-叔丁基氧酯-4-哌啶酮5a(10g,0.050mol)溶于250mL甲苯中,加入1-甲基苄胺5b(6.05g,50.0mmol).反应体系使用分水蒸馏头110℃下回流反应3小时,冷却至室温,然后减压浓缩得到4-((1-苯乙基)亚氨基)哌啶-1-羧酸叔丁酯5c(15.1g,黄色油状液体,100%),产物不需纯化直接进行下一步反应。
第二步
在氮气氛围下,-10℃下向二异丙胺(7.4g,0.075mol)的100mL四氢呋喃溶液中缓慢滴加正丁基锂(34.4mL,0.086mol,2M)的正己烷溶液。在此温度下搅拌反应20分钟,冷却至-30℃然后向该反应液中缓慢滴加4-((1-苯乙基)亚氨基)哌啶-1-羧酸叔丁酯5c(15.1g,0.05mol)的100mL四氢呋喃溶液,滴加完毕后在此温度下继
续反应30分钟,然后降温至-65℃,向该反应液中缓慢滴加1-溴-3-氯丙烷的四氢呋喃溶液(9.45g,60.0mmolin 50mL THF)。滴加结束后反应体系逐步升温至室温反应2小时,然后回流反应4小时。反应体系冷却到室温,减压浓缩,加水(100mL),然后用甲基叔丁基醚萃取(200mL×3),合并有机相,用饱和氯化钠水洗涤(100mL),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩得到1-(1-苯乙基)-1,2,3,4,7,8-六氢-1,6-萘啶-6(5H)-羧酸叔丁酯5d(17g,黄色油状液体),产物不经纯化直接进行下一步反应。
MS-ESI计算值[M+H]+343,实测值343。
第三步
将1-(1-苯乙基)-1,2,3,4,7,8-六氢-1,6-萘啶-6(5H)-羧酸叔丁酯5d(7g,0.02mol)溶于600mL四氢呋喃中,加入10%钯碳(700mg,10%),在3MPa氢气压力下于40℃搅拌反应14小时。反应结束后,硅藻土过滤除去钯碳,滤液减压浓缩,粗产品用色谱硅胶柱(0-100%乙酸乙酯/石油醚)纯化得到1-(1-苯乙基)八氢-1,6-萘啶-6(2H)-羧酸叔丁酯5e(2.26g,黄色油状物,产率:33%)。
MS-ESI计算值[M+H]+345,实测值345。
第四步
将1-(1-苯乙基)八氢-1,6-萘啶-6(2H)-羧酸叔丁酯5e(500mg,1.45mmol)溶于5mL乙酸乙酯,加入30mL乙酸乙酯的饱和氯化氢溶液于室温下搅拌1.5小时。待反应结束后减压浓缩得到1-(1-苯乙基)十氢-1,6-二氮杂萘5f(296mg,白色固体,产率:84%)。
MS-ESI计算值[M+H]+245,实测值245。
第五步
将1-(1-苯乙基)十氢-1,6-二氮杂萘5f(296mg,1.21mmol)和异喹啉-5-磺酰氯1c(550mg,2.42mmol)按照实施例1化合物1e的合成方法得到6-(异喹啉-5-磺酰基)-1-(1-苯乙基)十氢-1,6-二氮杂萘5g(96mg,黄色固体,产率:18%)。
MS-ESI计算值[M+H]+436,实测值436。
第六步
将6-(异喹啉-5-基磺酰基)-1-(1-苯乙基)十氢-1,6-二氮杂萘5g(30mg,0.069mmol)溶于1mL三氟乙酸,于100℃微波反应1小时。待反应结束后减压浓缩,用制备高压液相色谱纯化得目标化合物6-(异喹啉-5-基磺酰基)十氢-1,6-二氮杂萘5(7mg,白色固体,产率:31%)。
1H NMR(400MHz,CD3OD):δ9.41(s,1H),8.66-8.62(m,1H),8.59-8.54(m,1H),8.50-8.43(m,2H),7.87(t,J=8.0Hz,1H),4.06-3.50(m,4H),3.14-3.00(m,3H),2.24-2.10(m,1H),1.99-1.63(m,6H)。
MS-ESI计算值[M+H]+332,实测值332。
实施例6
5-((八氢-1H-吡咯并[3,2-b]吡啶-1-基)磺酰基)异喹
第一步
将1H-吡咯并[3,2-b]吡啶6a(2.00g,16.9mmol)溶于30mL二氯甲烷中,加入4-二甲氨基吡啶(2.06g,16.9mmol),三乙胺(2.05g,20.3mmol)和二碳酸二叔丁酯(4.42g,20.3mmol),室温搅拌过夜。待反应结束后将反应液减压浓缩,用色谱硅胶柱以洗脱剂体系(20%乙酸乙酯/石油醚/)纯化得1H-吡咯并[3,2-b]吡啶-1-羧酸叔丁酯6b(3.5g,白色固体,产率:95%)。
MS-ESI计算值[M+H]+219,实测值219。
第二步
将1H-吡咯并[3,2-b]吡啶-1-羧酸叔丁酯6b(1.00g,4.59mmol)溶于30mL乙酸中,加入二氧化铂(104mg,0.460mmol),在4MPa氢气压力下于50℃搅拌反应24小时。反应结束后冷却至室温然后过滤,滤液减压浓缩,得到八氢-1H-吡咯并[3,2-b]吡啶-1-羧酸叔丁酯6c(1.00g,无色油状液体,96%),不需纯化直接进行下一部反应。
MS-ESI计算值[M+H]+227,实测值227。
第三步
氮气保护下将八氢-1H-吡咯并[3,2-b]吡啶-1-羧酸叔丁酯6c(1.00g,4.42mmol)溶于30mL二氯甲烷中,0℃时加入N,N-二异丙基乙胺(2.4mL,13.3mmol)和氯羧酸苄酯(1.13g,6.63mmol),室温搅拌过夜。待反应结束后,减压浓缩,加水(20mL),然后用乙酸乙酯萃取(30mL×3),合并有机相,用饱和氯化钠水溶液(30mL)洗涤,无水硫酸钠干燥,浓缩,粗产物用色谱硅胶柱纯化(10%乙酸乙酯/石油醚)得到六氢-1H-吡咯并[3,2-b]吡啶-1,4(2H)-二羧酸4-苄酯-1-叔丁酯6d(1.35g,黄色油状液体,产率:85%)。
MS-ESI计算值[M+H]+361,实测值361。
第四步
六氢-1H-吡咯并[3,2-b]吡啶-1,4(2H)-二羧酸4-苄酯-1-叔丁酯6d(1.35g,3.75mmol)溶于5mL乙酸乙酯,加入40mL乙酸乙酯的饱和氯化氢溶液于室温下搅拌45分钟。待反应结束后,减压浓缩得到六氢-1H-吡咯
并[3,2-b]吡啶-4(2H)-羧酸苄酯6e(1.0g,白色固体,产率:90%)。
MS-ESI计算值[M+H]+261,实测值261。
第五步
将六氢-1H-吡咯并[3,2-b]吡啶-4(2H)-羧酸苄酯6e(132mg,0.51mmol)和异喹啉-5-磺酰氯1c(141,0.62mmol)按照实施例1化合物1e的合成方法得到1-(异喹啉-5-磺酰基)六氢-1H-吡咯并[3,2-b]吡-4(2H)-羧酸苄酯6f(185mg,黄色油状物,产率:81%)。
MS-ESI计算值[M+H]+452,实测值452。
第六步
将1-(异喹啉-5-磺酰基)六氢-1H-吡咯并[3,2-b]吡-4(2H)-羧酸苄酯6f(150mg,0.332mmol)按照实施例一化合物5的合成方法得到5-((八氢-1H-吡咯并[3,2-b]吡啶-1-基)磺酰基)异喹啉6(64mg,白色固体,产率:61%)。
1H NMR(400MHz,CD3OD):δ9.42(s,1H),8.69-8.64(m,2H),8.50-8.40(m,2H),7.88(t,J=8.0Hz,1H),3.83-3.79(m,1H),3.72-3.54(m,3H),3.16-3.10(m,1H),2.99-2.93(m,1H),2.27-2.23(m,1H),2.10-2.01(m,2H),1.92-1.84(m,1H),1.77-1.69(m,1H),1.64-1.59(m,1H)。
MS-ESI计算值[M+H]+318,实测值318。
实施例7
5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)异喹啉
第一步
顺式-六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯7a(105mg,0.49mmo,市售商品)和异喹啉-5-磺酰氯1c(136mg,0.600mmol)按照实施例1化合物1e的合成方法得到5-(异喹啉-5-基磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯7b,直接用于下一步反应。
第二步
5-(异喹啉-5-基磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯7b(34mg,0.082mmol)按照实施例一化合物1的合成方法得到5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)异喹啉7(15mg,白色固体,产率:60%)。
1H NMR(400MHz,CD3OD):δ9.40(s,1H),8.70-8.62(m,2H),8.48-8.44(m,2H),7.87(t,J=8.0Hz,1H),3.28-3.14(m,4H),3.13-3.01(m,2H),2.83-2.70(m,2H),2.64-2.52(m,2H)。
实施例8
5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)异喹啉
第一步
将3,4-二羧酸吡啶8a(30.0g,180mmol)溶解在250mL醋酸酐中然后加热回流3-4个小时直至反应液变澄清,然后冷却至室温,减压蒸去剩余的醋酸酐得到粗产品3,4-二羧酸酐吡啶8b直接用于下一步反应。
第二步
在0℃下将苄胺(28.9g,270mmol)逐滴滴加到固体3,4-二羧酸酐吡啶8b(第一步粗产物)中,然后自然升至室温并静置1小时。所得粘稠液体4-苄胺基甲酰基烟碱酸8c直接用于下一步反应。
第三步
将上一步粗产品4-苄胺基甲酰基烟碱酸8c小心地溶解于150mL的醋酸酐中,所得溶液加热至110℃反应4小时直到原料消失。冷却至室温后减压蒸去剩下的醋酸酐,将残余物用水(100mL)和乙酸乙酯(100mL)稀释,乙酸乙酯萃取(100mL×2),有机相依次用饱和碳酸氢钠水溶液(100mL)、水(100mL)和饱和食盐水(100mL)洗涤,无水硫酸钠干燥,过滤,浓缩,粗产品硅胶柱分离(30%-50%乙酸乙酯/石油醚)得到白色固体2-苄基-1H-吡咯[3,4-并]吡啶-1,3(2H)-二酮8d(29.5g,3步69%的总产率)。
1H NMR(400MHz,CDCl3):δ9.17(s,1H),9.07(d,J=4Hz,1H),7.76(d,J=4Hz,1H),7.50-7.40(m,2H),7.30-7.36(m,3H),4.88(s,2H)。
第四步
将2-苄基-1H-吡咯[3,4-并]吡啶-1,3(2H)-二酮8d(9.60g,40.3mmol)和湿钯碳(2.0g,20%)加入300mL甲醇中,将此反应液置于3MPa的氢气体系中并加热至60℃反应过夜。当反应结束后将反应液冷却至室温,硅藻土滤除钯碳,所得滤液浓缩得到淡黄色液体2-苄基-1H-吡咯并[3,4-c]六氢吡啶-1,3(2H)-二酮8e直接用于下一步反应。
第五步
在氮气保护和0℃下将氢化锂铝(3.06g,80.6mmol)分批加入到搅拌着的2-苄基-1H-吡咯并[3,4-c]六氢吡啶-1,3(2H)-二酮8e(9.84g,40.3mmol)的120mL四氢呋喃溶液中。加入完毕后将反应液加热回流2小时直到原
料消失,冷却至室温,然后冰水浴冷却至0℃,顺序逐滴滴加水(3mL),NaOH溶液(3mL,15%水溶液),水(9mL)淬灭反应,混合物升温至室温搅拌30min,然后过滤,滤液浓缩得到淡黄绿色2-苄基8氢-1H-吡咯并[3,4-c]吡啶8f直接用于下一步反应。
第六步
在氮气保护和0℃下,将二碳酸二叔丁酯溶液(13.06g,60.45mmol溶于15mL二氯甲烷)逐滴滴加到搅拌着的2-苄基8氢-1H-吡咯并[3,4-c]吡啶8f(8.70g,40.3mmol)和二异丙基乙基胺(11.4mL,80.6mmol)的100mL二氯甲烷溶液中。滴加完毕后移去冰水浴让反应液自然升至室温并持续搅拌2小时直到原料消失。向反应体系中加水(100mL),二氯甲烷萃取(2×50mL),合并有机相然后依次用水(50mL)和饱和食盐水(50mL)洗涤,无水硫酸钠干燥,过滤,浓缩得到粗产品后经硅胶柱分离(2%甲醇/二氯甲烷)得到淡黄色液体2-苄基六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8g。
MS-ESI计算值[M+H]+317,实测值317。
第七步
将2-苄基六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8g(1.5g,4.7mmol)和300mg干氢氧化钯(10%)的120mL的四氢呋喃溶液置于3MPa的氢气氛中并加热到60℃反应24小时。冷却至室温,滤除氢氧化钯,滤液浓缩,经硅胶柱分离(50%-100%甲醇/二氯甲烷)得到六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8h(0.5g,产率:50%)。
MS-ESI计算值[M+H]+227,实测值227。
第八步
六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8h(90mg,0.40mmol)和异喹啉-5-磺酰氯1c(116mg,0.510mmol)按照实施例1化合物1e的合成方法得到2-(异喹啉-5-基磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8i,直接用于下一步。
第九步
2-(异喹啉-5-基磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8i(36mg,0.85mmol)按照实施例1化合物1的合成方法得到5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)异喹啉8(17mg,淡黄色固体,产率:63%)。
1H NMR(400MHz,CD3OD):δ9.42(s,1H),8.69-8.59(m,2H),8.53-8.35(m,3H),7.87(t,J=8.0Hz,1H),3.53-3.46(m,2H),3.44-3.37(m,2H),3.28-3.22(m,1H),3.19-2.95(m,3H),2.62-2.44(m,2H),1.92-1.86(m,1H),1.74-1.63(m,1H).
MS-ESI计算值[M+H]+318,实测值318。
实施例9
第一步
将1H-吡咯并[2,3-c]吡啶9a(1.00g,8.47mmol)溶于20mL二氯甲烷中,0℃时加入2mL三乙胺和二碳酸二叔丁酯溶液(2.00g,9.17mmol,溶于10mL二氯甲烷),反应体系升至室温搅拌16小时。待反应结束后,减压浓缩得到1H-吡咯并[2,3-c]吡啶-1-羧酸叔丁酯9b,直接用于下一步反应。
第二步
将1H-吡咯并[2,3-c]吡啶-1-羧酸叔丁酯9b(1.2g,5.5mmol)溶于15mL醋酸中,加入二氧化铂(0.3g,1.3mmol),所得混合物在4MPa氢气压力下室温搅拌12小时。反应结束后过滤除去二氧化铂,滤液减压浓缩,粗产物由色谱硅胶柱纯化(0-100%甲醇/二氯甲烷)得到八氢-1H-吡咯并[2,3-c]吡啶-1-羧酸叔丁酯9c(600mg,产率:50%)。
MS-ESI计算值[M+H]+227,实测值227。
第三步
将八氢-1H-吡咯并[2,3-c]吡啶-1-羧酸叔丁酯9c(100mg,0.44mmol)和异喹啉-5-磺酰氯1c(120mg,0.51mmol)按照实施例一化合物1e的合成方法得到6-(异喹啉-5-基磺酰基)八氢-1H-吡咯并[2,3-c]吡啶-1-羧酸叔丁酯9d粗品,不需纯化直接用于下一步。
MS-ESI计算值[M+H]+418,实测值418。
第四步
将6-(异喹啉-5-基磺酰基)八氢-1H-吡咯并[2,3-c]吡啶-1-羧酸叔丁酯9d(100mg,0.24mmol)按照实施例1化合物1的合成方法得到5-((六氢-1H-吡咯并[2,3-c]吡啶-6(2H)-基)磺酰基)异喹啉9(60mg,产率:79%)。
1H NMR(400MHz,CD3OD):δ9.63(s,1H),8.90(d,J=6.0Hz,1H),8.69(d,J=6.0Hz,1H),8.65-8.55(m,2H),8.01(t,J=7.8Hz,1H),4.00-3.90(m,1H),3.84-3.76(m,1H),3.70-3.65(m,1H),3.51-3.36(m,2H),3.15-2.98(m,1H),2.65-2.60(m,1H),2.31(brs,1H),2.23-2.05(m,1H),1.96-1.75(m,2H),1.65-1.55(m,1H)。
MS-ESI计算值[M+H]+318,实测值318。
实施例10
5-(异喹啉-5-磺酰基)-5-氮杂螺[2.4]庚-7-胺
第一步
将5-氮杂螺[2.4]庚烷-7-基氨基羧酸叔丁酯10a(50mg,0.25mmol)和异喹啉-5-磺酰氯1c(75mg,0.32mmol)按照实施例1化合物1e的合成方法得到(5-(异喹啉-5-基磺酰基)-5-氮杂螺[2.4]庚烷-7-基)氨基羧酸叔丁酯10b(95mg,黄色油状液体,产率:95%)。
MS-ESI计算值[M+H]+404,实测值404。
第二步
将(5-(异喹啉-5-基磺酰基)-5-氮杂螺[2.4]庚烷-7-基)氨基羧酸叔丁酯10b(30mg,0.074mmol)按照实施例1化合物1的合成方法得到5-(异喹啉-5-磺酰基)-5-氮杂螺[2.4]庚-7-胺10(10mg,黄色固体,产率:44%)。
1H NMR(400MHz,D2O):δ9.40(s,1H),8.60-8.58(m,2H),8.55-8.45(m,2H),7.88(t,J=8.0Hz,1H),3.79-3.69(m,3H),3.43-3.41(m,1H),3.15-3.10(m,1H),0.96-0.89(m,1H),0.85-0.75(m,1H),0.68-0.61(m,1H),0.48-0.42(m,1H)。
MS-ESI计算值[M+H]+304,实测值304。
实施例11
N-乙基-5-(异喹啉-5-磺酰基)-5-氮杂螺[2.4]庚-7-胺
第一步
将(5-(异喹啉-5-磺酰基)-5-氮杂螺[2.4]庚烷-7-基)氨基羧酸叔丁酯10b(65mg,0.16mmol,实施例10)溶于4mL无水N,N-二甲基甲酰胺中,在0℃氮气保护下加入氢化钠(4.6mg,0.19mmol),反应液在0℃搅拌10分钟后加入碘乙烷(30mg,0.19mmol),然后升至室温并搅拌1小时。待反应结束后加入20mL饱和氯化钠水溶液后用乙酸乙酯(30mL×2)萃取,无水硫酸钠干燥,过滤,滤液减压浓缩,粗产物用硅胶柱(0-100%乙酸乙酯/石油醚)纯化得到(5-(异喹啉-5-磺酰基)-5-氮杂螺[2.4]庚烷-7-基)氨基羧酸叔丁酯11a(15mg,黄色油状液体,产率:22%)。
MS-ESI计算值[M+H]+432,实测值432。
第二步
将(5-(异喹啉-5-磺酰基)-5-氮杂螺[2.4]庚烷-7-基)氨基羧酸叔丁酯11a(15mg,0.035mmol)按照实施例1化合物1的合成方法得到N-乙基-5-(异喹啉-5-磺酰基)-5-氮杂螺[2.4]庚-7-胺11(10mg,黄色固体,产率:87%)。
1H NMR(400MHz,D2O):δ9.50(s,1H),8.73(d,J=6.4Hz,1H),8.58-8.48(m,3H),7.92(d,J=8.0Hz,1H),3.85-3.80(m,1H),3.72-3.61(m,2H),3.35-3.30(m,1H),3.10-3.29(m,3H)1.15(t,J=7.2Hz,3H),1.03-0.94(m,1H),0.85-0.75(m,1H),0.62-0.51(m,1H),0.41-0.32(m,1H)。
MS-ESI计算值[M+H]+332,实测值332。
实施例12
7-(异喹啉-5-基磺酰基)八氢吡咯并[3,4-b]吖庚因12;2-(异喹啉-5-基磺酰基)八氢吡咯并[3,4-c]吖庚因12’
第一步
将环己烯酮12a(5.00g,52.1mmol)溶于50mL二氯甲烷中,加入N-甲氧基甲基-N-(三甲基硅烷)苄基胺12b(7.74g,34.7mmol)和0.5mL三氟乙酸,在氮气保护下反应液于室温搅拌12小时。待反应结束后,反应液用饱和碳酸氢钠水溶液(20mL)淬灭,二氯甲烷(20mL×2)萃取,饱和碳酸氢钠水溶液(20mL)洗涤,合并有机相用无水硫酸钠干燥,减压浓缩即为产物2-苄基六氢-1H-异吲哚-4(2H)-酮12c不经纯化直接进行下一步反应。
MS-ESI计算值[M+H]+230,实测值230。
第二步
将2-苄基六氢-1H-异吲哚-4(2H)-酮12c(4.00g,17.5mmol)溶于50mL氯仿中,0℃条件下加入叠氮化钠
(2.28g,35.0mmol)和甲磺酸(1.68g,17.5mmol),在氮气保护下反应液于室温搅拌12小时。待反应结束后,反应体系加水(50mL)稀释,二氯甲烷(30mL×2)萃取,饱和碳酸氢钠水溶液(30mL)洗涤,合并有机相用无水硫酸钠干燥,浓缩蒸干即为产物7-苄基八氢吡咯并[3,4-b]吖庚因-2(1H)-酮12d1和2-苄基八氢吡咯并[3,4-c]吖庚因-4(2H)-酮12d2的混合物,产物不经纯化直接进行下一步反应。
MS-ESI计算值[M+H]+245,实测值245。
第三步
氮气保护下将7-苄基八氢吡咯并[3,4-b]吖庚因-2(1H)-酮12d1和2-苄基八氢吡咯并[3,4-c]吖庚因-4(2H)-酮12d2的混合物(2.80g,11.5mmol)溶于40mL四氢呋喃中,0℃条件下加入四氢锂铝(872mg,22.8mmol),反应体系于60℃条件下搅拌2小时,待反应结束后,0℃条件下加入1mL水,然后1mL 15%的氢氧化钠水溶液和3mL水。室温搅拌30分钟,反应液过滤,滤液用乙酸乙酯(20mL×2)萃取,饱和氯化钠水溶液(30mL)洗涤,合并有机相用无水硫酸钠干燥,浓缩蒸干即为产物7-苄基十氢吡咯并[3,4-b]吖庚因12e1和2-苄基十氢吡咯并[3,4-c]吖庚因12e2的混合物,产物不经纯化直接进行下一步反应。
第四步
将7-苄基十氢吡咯并[3,4-b]吖庚因12e1和2-苄基十氢吡咯并[3,4-c]吖庚因12e2的混合物(2.62g,11.4mmol)溶于40mL四氢呋喃中,室温条件下加入二碳酸二叔丁酯(3.73g,17.1mmol)和三乙胺(1.73g,17.1mmol),氮气保护下室温搅拌2小时。待反应结束后,反应体系加水(50mL)稀释,乙酸乙酯(30mL×2)萃取,饱和碳酸氢钠水溶液(30mL)洗涤,有机相用无水硫酸钠干燥,浓缩蒸干即为产物7-苄基八氢吡咯并[3,4-b]吖庚因-1(2H)-羧酸叔丁酯12f1和2-苄基八氢吡咯并[3,4-c]吖庚因-5(1H)-羧酸叔丁酯12f2的混合物,直接用于下一步反应。
MS-ESI计算值[M+H]+331,实测值331。
第五步
将7-苄基八氢吡咯并[3,4-b]吖庚因-1(2H)-羧酸叔丁酯12f1和2-苄基八氢吡咯并[3,4-c]吖庚因-5(1H)-羧酸叔丁酯12f2(1.90g,5.74mmol)溶于50mL甲醇中,加入100mg湿钯碳,在氢气(1atm)条件下反应液于50℃条件下搅拌2小时。待反应结束后,反应液直接过滤,滤液减压浓缩得八氢吡咯并[3,4-b]吖庚因-1(2H)-羧酸叔丁酯12g1和八氢吡咯并[3,4-c]吖庚因-5(1H)-羧酸叔丁酯12g2的混合物,不经纯化直接进行下一步反应。
MS-ESI计算值[M+H]+241,实测值241。
第六步
八氢吡咯并[3,4-b]吖庚因-1(2H)-羧酸叔丁酯12g1和八氢吡咯并[3,4-c]吖庚因-5(1H)-羧酸叔丁酯12g2的混合物(800mg,3.33mmol)和异喹啉-5-磺酰氯1c(910mg,4.00mmol)按照实施例一化合物1e的合成方法得到7-(异喹啉-5-基磺酰基)八氢吡咯并[3,4-b]吖庚因-1(2H)-羧酸叔丁酯12h1和2-(异喹啉-5-基磺酰基)八氢吡咯并[3,4-c]吖庚因-5(1H)-羧酸叔丁酯12h2的混合物,产物不经纯化直接进行下一步反应。
MS-ESI计算值[M+H]+432,实测值432。
第七步
7-(异喹啉-5-基磺酰基)八氢吡咯并[3,4-b]吖庚因-1(2H)-羧酸叔丁酯12h1和tert-butyl 2-(异喹啉-5-基磺酰基)
八氢吡咯并[3,4-c]吖庚因-5(1H)-羧酸叔丁酯12h2的混合物(1.43g,3.30mmol)按照实施例一化合物1的合成方法得到7-(异喹啉-5-基磺酰基)八氢吡咯并[3,4-b]吖庚因12(284mg,产率:26%)和2-(异喹啉-5-基磺酰基)十氢吡咯并[3,4-c]吖庚因12’(32mg)。
12:1H NMR(400MHz,CDCl3):δ9.28(s,1H),8.62(m,1H),8.52(m,1H),8.33(m,1H),8.17(d,J=8.0Hz,1H),7.66(t,J=8.0Hz,1H),3.79-3.74(m,1H),3.59-3.55(m,1H),3.49-3.44(m,1H),3.10-3.07(m,1H),2.86-2.77(m,3H),2.43-2.36(m,2H),1.70-1.63(m,3H),1.37-1.32(m,3H).
12’:1H NMR(400MHz,CDCl3):δ9.35(s,1H),8.66(m,1H),8.53(m,1H),8.35(m,1H),8.18(d,J=8.0Hz,1H),7.68(t,J=8.0Hz,1H),3.72-3.70(m,1H),3.64-3.62(m,1H),3.07-3.00(m,1H),2.98-2.93(m,3H),2.84(m,1H),1.99(m,1H),1.85-1.83(m,2H),1.70-1.67(m,3H),1.66-1.51(m,1H),1.16-1.14(m,1H).
实施例13
4-(异喹啉-5-基磺酰基)-4-氮杂螺[2.4]庚-7-胺
第一步
氮气保护下将二异丙胺(41.6g,0.41mol)加入到1.5L四氢呋喃中,冷却至-78℃。于氮气保护条件下慢慢的滴加正丁基锂(150mL,0.375mol),滴加完毕继续在此温度下反应一小时,然后滴加丙稀腈(22.78g,0.34mol)。加完丙烯腈后反应体系相同温度下继续搅拌一小时,然后滴加2-溴乙酸乙酯13a(56.8g,0.34mol)。滴加完毕后反应体系在-78℃继续反应一小时至反应完全,然后向反应液中加入饱和氯化铵水溶液(2.0L)淬灭反应,用乙酸乙酯(500mL×3)萃取,合并有机相用水(500mL)和饱和食盐水(500mL)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,残余物用色谱硅胶柱(0-100%乙酸乙酯/石油醚)纯化得到乙基3-氰基戊-4-烯酸甲酯13b(13.5g,无色油状物,产率:34%)。
1H NMR(400MHz,CDCl3):δ5.83-5.71(m,1H),5.51(m,1H),5.34(m,1H),4.20(m,2H),3.79-3.71(m,1H),2.81-2.73(m,1H),2.69-2.61(m,1H),1.28(m,3H).
第二步
将乙基3-氰基戊-4-烯酸甲酯13b(6g,39.2mmol)溶解于1L无水乙醚中,于氮气保护条件下加入钛酸四异丙酯(11.76mL,39.2mmol),然后于室温条件下缓慢滴加乙基溴化镁(21.6mL,64.8mmol,3M四氢呋喃溶液)。室温下反应体系搅拌三小时至原料消失,然后滴加36mL水淬灭反应,得到的悬浊液过滤,滤液减压浓缩,残渣用色谱硅胶柱(0-100%乙酸乙酯/石油醚)纯化得到7-乙烯基-4-氮杂螺[2.4]庚烷-5-酮13c(3.3g,白色固体,产率:63%)。
1H NMR(400MHz,CDCl3):δ6.09(brs,1H),5.68-5.60(m,1H),5.08-5.00(m,2H),3.0-2.96(m,1H),2.70-2.64(m,1H),2.40-2.36(m,1H),0.83-0.75(m,2H),0.65-0.58(m,2H).
MS-ESI计算值[M+H]+138,实测值138。
第三步
将7-乙烯基-4-氮杂螺[2.4]庚烷-5-酮13c(1.2g,8.75mol)溶解于29mL无水乙腈中依次加入4-N,N-二甲基吡啶(110mg,0.875mmol),二碳酸二叔丁酯乙腈溶液(2.86g,13.1mmol,10mL乙腈),室温搅拌5小时。反应结束后将反应液倒入水(25mL)中淬灭,用乙酸乙酯(30mL×3)萃取,合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩,残余物用色谱硅胶柱(0-100%乙酸乙酯/石油醚)纯化得到5-氧代-7-乙烯基-4-氮杂螺[2.4]庚烷-4-羧酸叔丁酯13d(1.2g,棕色固体,产率:55%)。
1H NMR(400MHz,CDCl3):δ5.66-5.57(m,1H),5.13-5.06(m,2H),2.77-2.72(m,2H),2.48-2.43(m,1H),1.62-1.60(m,1H),1.51(s,9H),1.43-1.41(m,1H),0.67-0.65(m,1H),0.54-0.51(m,1H).
MS-ESI计算值[M+H]+238,实测值238。
第四步
将5-氧代-7-乙烯基-4-氮杂螺[2.4]庚烷-4-羧酸叔丁酯13d(3.4g,14.3mmol)溶解于34mL甲醇和51mL水的混合溶液中,于室温条件下加入高碘酸钠(9.2g,43mmol)和四氧化锇(55mg,0.22mmol),室温搅拌4小时。反应完全后,加水(40mL)稀释,乙酸乙酯(40mL×3)萃取,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,残余物用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到7-甲酰基-5-氧代-4-氮杂螺[2.4]庚烷-4-羧酸叔丁酯13e(2.6g,棕色油状物,产率:76%)。
MS-ESI计算值[M+H]+240,实测值240。
第五步
将7-甲酰基-5-氧代-4-氮杂螺[2.4]庚烷-4-羧酸叔丁酯13e(2.60g,10.9mmol)溶解于87mL叔丁醇和87mL四氢呋喃的混合溶液中,于0℃条件下依次加入2-丁烯(22.7mL),亚氯酸钠(979mg,10.9mmol)和两水合磷酸二氢钠(3.39g,21.8mmol)的63mL水溶液。反应液室温搅拌16小时,反应完全,稀盐酸调节酸碱度至pH为4,然后乙酸乙酯(50mL×3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液减压浓缩得到4-(叔丁氧基羰基)-5-氧代-4-氮杂螺[2.4]庚烷-7-羧酸13f(2g,淡黄色固体,产率:75%)。
1H NMR(400MHz,DMSO):δ12.76(brs,1H),2.83-2.72(m,2H),2.56-2.55(m,1H),1.69-1.66(m,1H),1.41(s,9H),1.35-1.33(m,1H),0.79-0.73(m,2H).
MS-ESI计算值[M+H]+256,实测值256。
第六步
将4-(叔丁氧基羰基)-5-氧代-4-氮杂螺[2.4]庚烷-7-羧酸13f(2.31g,9.02mmol)溶解于30mL甲苯中,于0℃下加
入N,N-二异丙基乙基胺(1.51g,11.7mmol)和叠氮磷酸二苯酯(3.23g,11.7mmol)。加热至90℃反应半小时,冷却至室温并加入苄醇(1.07g,9.92mmol)。室温搅拌16小时,反应完全后倒入水中(40mL)并用乙酸乙酯(40mL×3)萃取。有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残渣用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到7-(((苄氧基)羰基)氨基)-5-氧代-4-氮杂螺[2.4]庚烷-4-羧酸叔丁酯13g(1.8g,白色固体,产率:56%)。
MS-ESI计算值[M+H]+361,实测值361。
第七步
氮气保护下将7-(((苄氧基)羰基)氨基)-5-氧代-4-氮杂螺[2.4]庚烷-4-羧酸叔丁酯13g(200mg,0.56mmol)溶解于3mL的四氢呋喃中,于0℃滴加硼烷的二甲硫醚溶液(3.9mL,11.7mmol,3M二甲硫醚溶液),反应液加热到60℃反应一小时。反应完全后,倒入冰水(20mL)中淬灭反应,用乙酸乙酯(20mL×3)萃取,有机相用无水硫酸钠干燥。过滤,滤液减压浓缩,残渣用色谱柱纯化(0-100%乙酸乙酯/石油醚)得到7-(((苄氧基)羰基)氨基)-4-氮杂螺[2.4]庚烷-4-羧酸叔丁酯13h(30mg,无色油状液体,产率:16%)。
MS-ESI计算值[M+H]+347,实测值347。
第八步
将7-(((苄氧基)羰基)氨基)-4-氮杂螺[2.4]庚烷-4-羧酸叔丁酯13h(100mg,0.39mmol)按照实施例五化合物5f的合成方法得到4-氮杂螺[2.4]庚烷-7-基氨基羧酸苄酯13i(101mg,白色固体,产率:100%)。
MS-ESI计算值[M+H]+247,实测值247。
第九步
将4-氮杂螺[2.4]庚烷-7-基氨基羧酸苄酯13i(70mg,0.27mmol)和异喹啉-5-磺酰氯1c(80mg,0.35mmol)按照实施例一化合物1e的合成方法得到4-(异喹啉-5-基磺酰基)-4-氮杂螺[2.4]庚烷-7-基氨基羧酸苄酯13j(80mg,无色油状液体,产率:68%)。
MS-ESI计算值[M+H]+438,实测值438。
第十步
将4-(异喹啉-5-基磺酰基)-4-氮杂螺[2.4]庚烷-7-基氨基羧酸苄酯13j(40mg,0.098mmol)按照实施例五化合物5的合成方法得到4-(异喹啉-5-基磺酰基)-4-氮杂螺[2.4]庚-7-胺13(11mg,黄色固体,产率:50%)。
1H NMR(400MHz,CD3OD):δ9.42(s,1H),8.65(d,J=6.4Hz,1H),8.54(d,J=8.0Hz,1H),8.47(d,J=8.0Hz,1H),8.38(d,J=6.4Hz,1H),7.87(t,J=8.0Hz,1H),4.16-4.12(m,1H),3.67-3.64(m,1H),3.41-3.38(m,1H),2.53-2.51(m,1H),2.09-2.07(m,1H),1.24-1.21(m,1H),1.15-1.13(m,1H),0.80-0.73(m,2H).
MS-ESI计算值[M+H]+304,实测值304。
实施例14
1-(异喹啉-5-磺酰基)十氢吡咯并[3,2-b]吖庚因
第一步
将1,3-环己二酮14a(5g,44.6mmol)溶于10mL四氢呋喃中,在室温下加入乙醇胺(3.27g,53.5mmol)。将所得悬浮液倒入装有分水器和90mL甲苯的圆底烧瓶中,加热回流过夜,反应体系冷却至室温,然后减压浓缩,残余物用色谱硅胶柱(15%乙酸乙酯/石油醚)纯化得到3-(2-羟基-乙基氨基)-环己-2-烯酮14b(4.30g,黄色固体,产率:62%)。
1H NMR(400MHz,DMSO-d6):δ4.80(s,1H),3.47-3.57(m,2H),3.14-3.22(m,2H),3.05-2.95(m,1H),2.27-2.35(m,2H),2.09-2.03(m,1H),1.68-1.82(m,2H).
第二步
将3-(2-羟基-乙基氨基)-环己-2-烯酮14b(3.70g,23.9mmol)溶于120mL N,N二甲基甲酰胺,在氮气保护和室温下加入四三苯基磷钯(552mg,0.48mmol),三甲基溴苯(4.76g,23.9mmol),碳酸钾(6.60g,47.8mmol),所得反应液加热到150℃回流2个小时,冷却至室温后减压浓缩,残余物用乙酸乙酯(100mL)稀释,饱和氯化钠水溶液(100mL)洗涤,无水硫酸钠干燥,过滤,浓缩,残余物用色谱硅胶柱以洗脱剂体系(15%乙酸乙酯/石油醚)纯化得到1,5,6,7-四氢-吲哚-4-酮14c(2.66g,白色固体,产率:82%)。
MS-ESI计算值[M+H]+136,实测值136。
第三步
将1,5,6,7-四氢-吲哚-4-酮14c(4.66g,34.5mmol)溶于120mL乙腈中,在室温下加入N,N-二异丙基乙基胺(9.3mL,51.7mmol),二碳酸二叔丁酯(8.27g,37.9mmol)和N,N-二甲基吡啶(82mg,0.69mmol)。反应液在室温下搅拌过夜。反应完毕后将反应液浓缩,残余物用色谱硅胶柱以洗脱剂体系(10%乙酸乙酯/石油醚)纯化得到4-氧代-4,5,6,7-四氢-吲哚-1-羧酸叔丁酯14d(7g,白色固体,产率:86%)
第四步
将4-氧代-4,5,6,7-四氢-吲哚-1-羧酸叔丁酯14d(500mg,2.13mmol)溶于20mL甲醇中,室温加入催化量冰醋酸和50mg二氧化铂。反应体系抽真空下置换氢气,在1MPa氢气压力和50℃下搅拌3个小时。反应完毕后冷却至室温然后过滤,滤液浓缩,残余物用色谱硅胶柱以洗脱剂(50%乙酸乙酯/石油醚)纯化得到4-羟基八氢-1H-吲哚-1-羧酸叔丁酯14e(400mg,无色油状液体,产率:78%)。
第五步
将4-羟基八氢-1H-吲哚-1-羧酸叔丁酯14e(700mg,2.9mmol)溶于35mL二氯甲烷,在0℃下加入戴斯-马丁氧化剂(2.46g,5.80mmol)。在室温下反应液搅拌30分钟。反应结束后用饱和碳酸氢钠水溶液(50mL)淬灭反应,二氯甲烷(30mL×3)萃取。合并有机相,用饱和氯化钠水溶液(50mL)洗涤,无水硫酸钠干燥,过滤、浓缩,残余物用色谱硅胶柱以洗脱剂(25%乙酸乙酯/石油醚)纯化得到4-氧代八氢-1H-吲哚-1-羧酸叔丁酯14f(470mg,无色油状液体,产率:68%)。
MS-ESI计算值[M+H–C4H8]+184,实测值184。
第六步
将4-氧代八氢-1H-吲哚-1-羧酸叔丁酯14f(470mg,1.97mmol)溶于15mL氯仿,0℃下加入叠氮化钠(250mg,3.85mmol)和甲磺酸(1.51g,15.8mmol),在70℃下反应液搅拌过夜。反应结束后将反应液倒入饱和碳酸氢钠(20mL)水溶液,水相用乙酸乙酯(40mL)洗涤,然后在真空下浓缩得到八氢吡咯并[3,2-b]吖庚因-5(1H)-酮14g不经纯化,直接进行下一步反应。
第七步
将八氢吡咯并[3,2-b]吖庚因-5(1H)-酮14g(303mg,1.97mmol)溶于15mL水中,在0℃下加入氯甲酸苄酯(1.69g,9.85mmol)和碳酸钾(150mg,1.08mmol),然后升至室温并搅拌4个小时。反应结束后反应液用乙酸乙酯(20mL×3)萃取,饱和氯化钠水溶液(20mL)洗涤,用无水硫酸钠干燥,过滤,浓缩,用薄层层析板纯化(1/2乙酸乙酯/石油醚)残余物得到5-氧代八氢吡咯并[3,2-b]吖庚因-1(2H)-羧酸苄酯14h(130mg,无色油状物,产率:23%)。
1H NMR(400MHz,CDCl3):δ7.40-7.30(m,5H),5.80(s,1H),5.20-5.06(m,2H),4.08-4.00(m,1H),3.70-3.55(m,1H),3.47-3.30(m,3H),3.22-3.11(m,1H),2.51-1.37(m,6H)
第八步
将5-氧代八氢吡咯并[3,2-b]吖庚因-1(2H)-羧酸苄酯14h(180mg,0.63mmol)溶于5mL四氢呋喃,在-78℃下缓慢滴加硼烷(0.63mL,1.86mmol,3M二甲硫醚溶液)。滴加完毕后将反应液升温至50℃并搅拌4个小时,直到反应结束。冷却后用3mL甲醇淬灭反应,将所得的混合物浓缩得到八氢吡咯并[3,2-b]吖庚因-1(2H)-羧酸苄酯14i,不经纯化直接进行一下步反应。
第九步
将八氢吡咯并[3,2-b]吖庚因-1(2H)-羧酸苄酯14i(171mg,0.625mmol)溶于3mL二氯甲烷中,在室温下依次加入N,N二异丙基乙胺(161mg,1.25mmol)、4-N,N-二甲基吡啶(8mg,0.025mmol)和二碳酸二叔丁酯(273mg,1.25mmol),所得反应液搅拌过夜。待反应结束后倒入水中淬灭反应,然后用二氯甲烷(10mL×3)萃取,合并有机相饱和氯化钠水溶液(20mL)洗涤,用无水硫酸钠干燥,过滤、浓缩,用薄层层析板纯化(1/1乙酸乙酯/石油醚)残余物得到六氢吡咯并[3,2-b]吖庚因-1,4(2H,5H)-二羧酸1-苄酯4-叔丁基酯14j(180mg,
无色油状液体,产率:77%)。
MS-ESI计算值[M+H–C4H8]+319,实测值319。
第十步
将六氢吡咯并[3,2-b]吖庚因-1,4(2H,5H)-二羧酸1-苄酯4-叔丁基酯14j(180mg,0.48mmol)溶于20mL四氢呋喃中,在室温和氮气保护下,加入18mg干钯碳,室温下反应液在1atm氢气氛中搅拌2个小时。待反应结束后将反应液过滤,滤液浓缩,得到八氢吡咯并[3,2-b]吖庚因-4(2H)-羧酸叔丁酯14k(30mg,无色油状液体,产率:26%)。
第十一步
将八氢吡咯并[3,2-b]吖庚因-4(2H)-羧酸叔丁酯14k(30mg,0.12mmol)异喹啉-5-磺酰氯1c(32mg,0.14mmol)按照实施例一化合物1e的合成方法得到1-(异喹啉-5-基磺酰基)八氢吡咯并[3,2-b]吖庚因-4(2H)-羧酸叔丁酯14l(30mg,无色油状液体,产率:56%)。
MS-ESI计算值[M+H]+432,实测值432。
第十二步
1-(异喹啉-5-基磺酰基)八氢吡咯并[3,2-b]吖庚因-4(2H)-羧酸叔丁酯14l(30mg,0.07mmol)按照实施例一化合物1的合成方法得到1-(异喹啉-5-磺酰基)十氢吡咯并[3,2-b]吖庚因14(15mg,白色固体,产率:65%)。
1H NMR(400MHz,D2O):δ9.81-9.73(m,1H),9.11-9.04(m,1H),8.84-8.76(m,1H),8.74-8.70(m,1H),8.69-8.65(m,1H),8.15-8.07(m,1H),4.05-3.95(m,1H),3.57-3.53(m,1H),3.50-3.40(m,1H),3.45-3.38(m,1H),3.34-3.23(m,1H),3.35-3.25(m,1H),2.95-2.90(m,1H),2.70-2.57(m,1H),2.17-2.07(m,1H),2.03-1.90(m,3H),1.78-1.65(m,2H).
MS-ESI calc'd.[M+H]+332,found 332。
实施例15
5-((六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-基)磺酰基)异喹啉
第一步
将5-氧代八氢环戊烷并[c]吡咯-2(1H)-羧酸叔丁酯15a(2g,8.89mmol)溶于10mL氯仿中,然后向反应液
中依次加入叠氮化钠(1.2g,18.4mmol)和甲基磺酸(8.5g,88.9mmol)。将所得混合物在70℃下搅拌2小时。反应完全后冷却至室温,饱和碳酸氢钠溶液调节至pH=7,然后二氯甲烷(20mL×2)萃取,合并有机相用无水硫酸钠干燥,浓缩,得到黄色油状液体六氢-1H-吡咯并[3,4-c]吡啶-6(2H)-酮15b。该黄色油状物直接用于下一步反应。
第二步
将六氢-1H-吡咯并[3,4-c]吡啶-6(2H)-酮15b(1.22g,8.72mmol)加入到40mL 10%的氢氧化钠水溶液和四氢呋喃的混合溶液中(v/v=1/1),然后加入二碳酸二叔丁酯(3.87g,17.8mmol)。加完后,将混合物在室温搅拌3小时直到反应结束。将反应混合物用乙酸乙酯(30mL×3)萃取,合并有机相用无水硫酸钠干燥,减压浓缩,粗产物用色谱硅胶柱纯化(0-10%甲醇/乙酸乙酯)得到6-氧代六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-羧酸叔丁酯15c(0.9g,黄色油状液体,产率:43%)。
第三步
将6-氧代六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-羧酸叔丁酯15c(600mg,2.5mmol)溶在20mL四氢呋喃溶液中,然后在-10℃加入四氢铝锂(190mg,50mmol)的10mL四氢呋喃溶液,加完后将混合物在室温搅拌3小时。缓慢加入0.19mL水淬灭反应,随后依次滴加0.19mL 15%的氢氧化钠水溶液和0.51mL的水。搅拌30分钟后将混合物过滤,滤液减压浓缩,得到黄色油状液体六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-羧酸叔丁酯15d直接用于下一步反应。
第四步
将六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-羧酸叔丁酯15d(200mg,0.88mmol,上一步粗产物)和异喹啉-5-磺酰氯1c(280mg,1.25mmol)按照实施例一化合物1e的合成方法得到5-(异喹啉-5-基磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-羧酸叔丁酯15e(80mg,无色油状液体,收率:22%)。
MS-ESI计算值[M+H-56]+362,实测值362。
第五步
将5-(异喹啉-5-基磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-羧酸叔丁酯15e(80mg,0.19mmol)按照实施例一化合物1的合成方法得到5-((六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-基)磺酰基)异喹啉15(51mg,黄色油状物,收率:85%)。
1H NMR(400MHz,CD3OD):δ9.39(s,1H),8.62(d,J=8.0Hz,1H),8.55(d,J=8.0Hz,1H),8.52-8.39(m,3H),7.85(t,J=8.0Hz,1H),3.70-3.58(m,2H),3.41-3.34(m,1H),3.21-3.04(m,4H),2.80-2.75(m,1H),2.57-2.48(m,1H),2.42-2.32(m,1H),1.85-1.77(m,1H),1.61-1.51(m,1H).
MS-ESI计算值[M+H]+318,实测值318。
实施例16
4-氯-5-(六氢-吡咯并[3,2-b]吡咯-1-磺酰基)-异喹啉
第一步
在-5℃下将硝酸钾(7.98g,79mmol)的浓硫酸(69.0mL)溶液滴加到4-氯异喹啉16a(10.0g,61mmol)的55mL浓硫酸溶液中。混合物在0℃下搅拌1小时后升至室温搅拌过夜。待反应结束后将反应液缓慢加入到300mL冰水中,用固体碳酸钠调节pH至8,并用乙酸乙酯(200mL×2)萃取,合并有机相,无水硫酸钠干燥,过滤,浓缩滤液,并用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到4-氯-5-硝基异喹啉16b(11.2g,白色固体,产率:88%)。
MS-ESI计算值[M+H]+209,实测值209。
第二步
在0℃下将二氯化锡水合物(13g,57.7mmol)加入到4-氯-5-硝基异喹啉16b(2.00g,9.62mmol)的34mL浓盐酸溶液中,加热到100℃搅拌过夜。冷却至室温然后用固体碳酸氢钠调节反应液的pH至8,用乙酸乙酯(100mL×2)萃取,合并有机相,用无水硫酸钠干燥,过滤,浓缩滤液,并用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到4-氯异喹啉-5-胺16c(1.57g,黄色固体,产率:92%)。
MS-ESI计算值[M+H]+179,实测值179。
第三步
在-5℃下将亚硝酸钠(620mg,8.82mmol)的2mL水溶液加入到4-氯异喹啉-5-胺(1.57g,8.82mmol)的14mL浓盐酸溶液中,所得反应液在-5℃下继续搅拌1小时。然后将该反应液迅速倒入已经加了氯化亚铜水溶液(224mg,2.20mmol,2mL水)的30mL饱和二氧化硫醋酸溶液中,室温搅拌至无气泡生成。反应结束后加冰水(100mL)稀释,用饱和碳酸氢钠水溶液将反应液的pH调至8,然后用二氯甲烷(100mL×2)萃取,合并有机相,用无水硫酸钠干燥,过滤,浓缩滤液,得到4-氯异喹啉-5-磺酰氯16d(1.07g,黄色固体,产率:46%)。
MS-ESI计算值[M+H]+262,实测值262。
第四步
顺式-六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯1d(30mg,0.142mmol)和4-氯异喹啉-5-磺酰氯16d(55.6mg,0.212mmol)按照实施例一化合物1e的合成方法得到4-(4-氯异喹啉-5-磺酰基)-六氢-吡咯并[3,2-b]吡咯-1-羧酸叔丁酯16e(30mg,黄色油状液体,产率:48%)。
MS-ESI计算值[M+H]+438,实测值438。
第五步
将4-(4-氯异喹啉-5-磺酰基)-六氢-吡咯并[3,2-b]吡咯-1-羧酸叔丁酯16e(30mg,0.092mmol)按照实施例一化合物1的合成方法得到4-氯-5-(六氢-吡咯并[3,2-b]吡咯-1-磺酰基)-异喹啉16(20mg,白色固体,产率:96%)。
1H NMR(400MHz,D2O):δ9.21(s,1H),8.63(s,1H),8.40-8.30(m,2H),7.80(t,J=8.0Hz,1H),4.77(s,1H),4.53-4.50(m,1H),3.71-3.66(m,1H),3.35-3.31(m,2H),3.47-3.44(m,1H),2.54-2.44(m,1H),2.28-2.23(m,1H),2.16-2.11(m,2H).
MS-ESI计算值[M+H]+338,实测值338。
实施例17
4-氯-5-(六氢-吡咯并[3,4-b]吡咯-5-磺酰基)-异喹啉
第一步
将六氢-吡咯并[3,4-b]吡咯-1-羧酸叔丁基酯2f(30mg,0.142mmol,实施例2)和4-氯异喹啉-5-磺酰氯16d(56mg,0.212mmol)按照实施例一化合物1e的合成方法得到5-(4-氯异喹啉-5-磺酰基)-六氢-吡咯并[3,4-b]吡咯-1-羧酸叔丁基酯17e(40mg,黄色油状液体,产率:65%)。
MS-ESI计算值[M+H]+438,实测值438。
第二步
5-(4-氯异喹啉-5-磺酰基)-六氢-吡咯并[3,4-b]吡咯-1-羧酸叔丁基酯17e(45mg,0.092mmol)按照实施例一化合物1的合成方法得到4-氯-5-(六氢-吡咯并[3,4-b]吡咯-5-磺酰基)-异喹啉17(30mg,白色固体,产率:96%)。
1H NMR(400MHz,D2O):δ9.25(s,1H),8.64(s,1H),8.45-8.35(m,2H),7.81(t,J=8.0Hz,1H),4.43-4.39(m,1H),3.80-3.75(m,2H),3.65-3.60(m,1H),3.47-3.44(m,1H),3.37-3.23(m,3H),2.27-2.17(m,1H),1.96-1.89(m,1H).
MS-ESI计算值[M+H]+338,实测值338。
实施例18
4-氯-5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)异喹啉
第一步
将八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯3a(30mg,0.13mmol,实施例3)和4-氯异喹啉-5-磺酰氯16d按照化合物1e的合成方法6-(4-氯异喹啉-5-磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯18a(56mg,黄色油状物,产率:93%)。
MS-ESI计算值[M+H]+452,实测值452。
第二步
6-(4-氯异喹啉-5-磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯18a(15mg,0.035mmol)按照实施例一化合物1的合成方法得到4-氯-5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)异喹啉18(10mg,黄色固体,产率:87%)。
1H NMR(400MHz,D2O):δ9.25(s,1H),8.63-8.61(s,1H),8.39-8.36(m,1H),8.26-8.24(m,1H),7.80(t,J=8.0Hz,1H),3.98-3.97(m,1H),3.85-3.76(m,2H),3.51(t,J=10.0Hz,2H),3.33-3.31(m,1H),3.04-2.99(m,2H),1.87-1.74(m,4H).
MS-ESI计算值[M+H]+352,实测值352。
实施例19
5-(3,6-二氮杂双环[3.2.0]庚烷-3-基磺酰基)-4-氯异喹啉
第一步
3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯4a(30mg,0.15mmol)和4-氯异喹啉-5-磺酰氯16d(44mg,0.17mmol)按照实施例一化合物1e的合成方法得到3-((4-氯异喹啉-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯19a(25mg,黄色油状液体,产率:39%)。
MS-ESI计算值[M+Na]418,实测值418。
第二步
3-((4-氯异喹啉-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯19a(25mg,0.059mmol)按照实施例一化合物1的合成方法得到5-(3,6-二氮杂双环[3.2.0]庚烷-3-基磺酰基)-4-氯异喹啉19(8mg,白色固体,产率:38%)。
1H NMR(400MHz,D2O):δ9.27(s,1H),8.70(brs,1H),8.49-8.43(m,2H),7.85(t,J=8.0Hz,1H),5.06-5.01(m,1H),4.26-4.21(m,1H),4.10-4.07(m,1H),3.83-3.91(m,2H),3.69-3.64(m,1H),3.61-3.54(m,1H),3.52-3.48(m,1H).
MS-ESI计算值[M+H]+318,实测值318。
实施例20
6-((4-氯异喹啉-5-基)磺酰基)十氢-1,6-二氮杂萘
第一步
将1-(1-苯乙基)-1,2,3,4,7,8-六氢-1,6-二氮杂萘-6(5H)-羧酸叔丁酯5e(15g,0.044mol)溶于600mL四氢呋喃中,加入1.5g氢氧化钯(20%炭负载),在3MPa氢气氛中于80℃搅拌反应16小时。冷却至室温过滤,减压浓缩滤液,用色谱硅胶柱纯化(50%甲醇/乙酸乙酯)得到八氢-1,6-二氮杂萘-6(2H)-羧酸叔丁酯20a(320mg,黄色油状液体,产率:3%)。
MS-ESI计算值[M+H]+241,实测值241
第二步
将八氢-1,6-二氮杂萘-6(2H)-羧酸叔丁酯20a(320mg,1.33mmol)溶于5mL二氯甲烷中,0℃时加入N,N-二异丙基乙基胺(516mg,3.99mmol)和氯羧酸苄酯(340mg,2.0mmol),室温搅拌1小时。反应结束后,减压浓缩然后加水(20mL)稀释,用乙酸乙酯(30mL×2)萃取,合并有机相,用饱和氯化钠水溶液(50mL)洗涤,无水硫酸钠干燥,浓缩,用制备硅胶板(3/1石油醚/乙酸乙酯)纯化得到六氢-1,6-二氮杂萘-1,6(2H,7H)-1-苄基6-叔丁基二甲酯20b(275mg,黄色油状液体,产率:86%)。
MS-ESI计算值[M+H]+375,实测值375。
第三步
将六氢-1,6-二氮杂萘-1,6(2H,7H)-1-苄基6-叔丁基二甲酯20b(275mg,0.73mmol)按照实施例一化合物1的合成方法得到八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯20c(200mg,白色固体,产率:96%)。
MS-ESI计算值[M+H]+275,实测值275。
第四步
将八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯20c(40mg,0.15mmol)和4-氯异喹啉-5-磺酰氯16d(42mg,0.16mmol)按照实施例一化合物1e的合成方法得到6-((4-氯异喹啉-5-基)磺酰基)八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯20d(20mg,黄色油状液体,产率:27%)。
MS-ESI计算值[M+H]+501,实测值501。
第五步
将6-((4-氯异喹啉-5-基)磺酰基)八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯20d(20mg,0.04mmol)按照实施例五化合物5的合成方法得到6-((4-氯异喹啉-5-基)磺酰基)十氢-1,6-二氮杂萘20(10mg,白色固体,产率:68%)。
1H NMR(400MHz,CD3OD):δ9.30(s,1H),8.72(s,1H),8.49-8.34(m,2H),7.90-7.85(m,1H),4.04-3.68(m,3H),3.54-3.40(m,1H),3.27-3.03(m,3H),2.34-2.18(m,1H),2.13-1.88(m,3H),1.87-1.60(m,3H)。
MS-ESI计算值[M+H]+366,实测值366。
实施例21
4-氯-5-(八氢-吡咯并[3,2-b]吡啶-1-磺酰基)-异喹啉
第一步
八氢-吡咯并[3,2-b]吡啶-4-羧酸苄酯6e(30mg,0.12mmol,实施例六)和4-氯异喹啉-5-磺酰氯16d(45mg,0.17mmol)按照实施例一化合物1e的合成方法得到1-(4-氯异喹啉-5-磺酰基)-八氢吡咯并[3,2-b]吡啶-4-羧酸苄酯21a(30mg,黄色油状液体,产率:36%)。
MS-ESI计算值[M+H]+486,实测值486。
第二步
将1-(4-氯异喹啉-5-磺酰基)-八氢吡咯并[3,2-b]吡啶-4-羧酸苄酯21a(30mg,0.062mmol)按照实施例五化合物5的合成方法得到4-氯-5-(八氢-吡咯并[3,2-b]吡啶-1-磺酰基)-异喹啉21(15mg,白色固体,产率:71%)。
1H NMR(400MHz,D2O):δ9.33(s,1H),8.78(s,1H),8.55-8.50(m,2H),7.91(t,J=8.0Hz,1H),4.64(s,1H),4.18-4.13(m,1H),4.03-3.98(m,1H),3.80-3.75(m,2H),3.17-3.04(m,1H),2.41-2.36(m,2H),1.97-1.80(m,3H),1.58-1.55(m,1H).
MS-ESI计算值[M+H]+352,实测值352。
实施例22
4-氯-5-(六氢吡咯并[3,4-c]吡咯-2(1H)-磺酰基)异喹啉
第一步
顺式-六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯7a(30mg,0.14mmol)和4-氯异喹啉-5-磺酰氯16d(131mg,0.58mmol)按照实施例一化合物1e的合成方法得到5-(4-氯异喹啉-5-磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯22a(50mg,黄色油状液体,产率:81%)。
MS-ESI计算值[M+H]+438,实测值438。
第二步
将5-(4-氯异喹啉-5-磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯22a(50mg,0.11mmol)按照实施例一化合物1的合成方法得到4-氯-5-(六氢吡咯并[3,4-c]吡咯-2(1H)-磺酰基)异喹啉22(35mg,黄色固体,产率:91%)。
1H NMR(400MHz,D2O):δ9.23-9.21(m,1H),8.64(s,1H),8.78(d,J=8.0Hz,1H),8.43-8.38(m,2H),3.61-3.54(m,4H),3.45-3.40(m,2H),3.21-3.19(m,2H),3.05-3.00(m,2H)。
MS-ESI计算值[M+H]+338,实测值338。
实施例23
4-氯-5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)异喹啉
第一步
将六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8h(35mg,0.16mmol)和4-氯异喹啉-5-磺酰氯16d(45mg,0.17mmol)按照化合物1e的合成方法得到2-((4-氯异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯23a(15mg,黄色油状液体,产率:21%)。
MS-ESI计算值[M+Na]474,实测值474。
第二步
2-((4-氯异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯23a(15mg,0.033mmol)按照化合物1的合成方法得到4-氯-5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)异喹啉23(10mg,白色固体,产率:86%)。
1H NMR(400MHz,D2O):δ9.28(s,1H),8.70-8.68(m,1H),8.45-8.41(m,2H),7.85(t,J=8.0Hz,1H),3.70-3.60(m,2H),3.52-3.47(m,2H),3.41-3.36(m,1H),3.25-3.14(m,3H),2.89-2.75(m,2H),2.08-2.02(m,1H),1.90-1.82(m,1H)。
MS-ESI计算值[M+H]+352,实测值352。
实施例24
4-氟-5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)异喹啉
在-65℃条件下将4-溴异喹啉24a(20g,96.6mmol)的144mL四氢呋喃溶液滴加到正丁基锂(133mL,332.5mmol,2.5M四氢呋喃溶液)的760mL四氢呋喃溶液中,滴加完毕后-65℃继续反应30分钟。然后-65℃在1小时内向反应液中加入N-氟二苯磺酰胺(66.68g,211.7mmol)的216mL四氢呋喃溶液,滴加完毕后继续搅拌1小时,然后缓慢升至室温。待反应结束后,将饱和氯化铵水溶液(300mL)缓慢加入到反应液中,并用乙酸乙酯(300mL×3)萃取,有机相用饱和氯化钠水溶液(300mL)洗涤,干燥,浓缩蒸干,并用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到4-氟异喹啉24b(8.5g,红色油状,产率:60%)。
第二步
4-氟异喹啉24b(8.5g,57.8mmol)参照实施例16化合物4-氯-5-硝基异喹啉16b合成方法得到4-氟-5-硝基异喹啉24c(7.2g,黄色油状液体,产率:64.8%)。
第三步
4-氟-5-硝基异喹啉24c(7.2g,37.5mmol)参照实施例16化合物4-氯异喹啉-5-胺16c合成方法得到4-氟异喹啉-5-胺24d(5.5g,黄色固体,产率:90%)。
第四步
4-氟异喹啉-5-胺24d(5.5g,33.95mmol)按照实施例16化合物4-氯异喹啉-5-磺酰氯16d的合成方法得到4-氟异喹啉-5-磺酰氯24e(4.5g,白色固体,产率:54%)。
1H NMR(400MHz,CDCl3):δ9.32-9.20(m,1H),8.79-8.67(m,2H),8.48-8.37(m,1H),7.85(t,J=8.0Hz,1H).
第五步
六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯1d(30mg,0.14mmol和4-氟异喹啉-5-磺酰氯24e(35mg,0.14mmol)按照实施例一化合物4-(异喹啉-5-基磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯1e的合成方法得到4-((4-氟异喹啉-5-基)磺酰基)六氢吡咯并[3,2-b吡咯-1(2H)-羧酸叔丁酯24f(56mg,黄色油状液体,产率:94%)。
MS-ESI计算值[M+Na]444,实测值444。
第六步
4-((4-氟异喹啉-5-基)磺酰基)六氢吡咯并[3,2-b吡咯-1(2H)-羧酸叔丁酯24f(56mg,0.13mmol按照实施例一的合成方法得到4-氟-5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)异喹啉24(20mg,白色固体,产率:42%)。
1H NMR(400MHz,D2O):δ9.24(s,1H),8.56-8.46(m,3H),7.88(t,J=8.0Hz,1H),4.47(m,1H),3.68(m,1H),3.53-3.43(m,1H),3.37-3.24(m,3H),2.45-2.31(m,1H),2.23-2.08(m,3H).
MS-ESI计算值[M+H]+322,实测值322。
实施例25
4-氟-5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)异喹啉
第一步
顺式-六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2f(55mg,0.24mmol,实施例二)4-氟异喹啉-5-磺酰氯24e(50mg,0.2mmol,实施例24)按照实施例一的合成方法得到5-((4-氟异喹啉-5-基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯25a(55mg,黄色固体,收率:58%)。
MS-ESI计算值[M+H-56]+366,实测值366。
第二步
5-((4-氟异喹啉-5-基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯25a(50mg,0.085mmol)按照实施例一的合成方法得到4-氟-5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)异喹啉25(30mg,白色固体,78%)。
1H NMR(400MHz,D2O):δ9.26(s,1H),8.60-8.49(m,3H),7.89(t,J=7.2Hz,1H),4.41(t,J=6.4Hz,1H),36.76-3.25(m,,7H),2.26-2.20(m,1H),1.92-1.87(m,1H).
MS-ESI计算值[M+H]+322,实测值322。
实施例26
4-氟-5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)异喹啉
第一步
4-氟-异喹啉-5-磺酰氯24e(45mg,0.2mmol,实施例24)和顺式-八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯3a(50mg,0.22mmol,实施例三)按照实施例一的合成方法得到6-((4-氟异喹啉-5-基)磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯26a(50mg,无色油状液体,产率:52%)。
第二步
6-((4-氟异喹啉-5-基)磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯26a(50mg,0.11mmol)按照实施例一的合成方法得到4-氟-5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)异喹啉26(20mg,白色粉末固体,产率:42%)。
1H NMR(400MHz,CD3OD):δ9.46(s,1H),8.75-8.67(m,1H),8.65-8.56(m,2H),8.05-7.96(m,1H),4.02-3.96(m,1H),3.95-3.88(m,1H),3.79-3.70(m,2H),3.55-3.50(m,1H),3.36-3.31(m,2H),3.12-3.03(m,1H),2.94-2.84(m,1H),1.97-1.88(m,1H),1.86-1.76(m,2H).
实施例27
5-(3,6-二氮杂双环[3.2.0]庚烷-3-基磺酰基)-4-氟异喹啉
第一步
3-(异喹啉-5-基磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯4a(30mg,0.15mmol)和4-氟-异喹啉-5-磺酰氯24e(41mg,0.17mmol,实施例24)按照实施例一的合成方法得到3-((4-氟异喹啉-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯27a(52mg,黄色油状液体,产率:84%)。
MS-ESI计算值[M+Na]+430,实测值430。
第二步
3-((4-氟异喹啉-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯27a(52mg,0.13mmol)按照实施例一
的合成方法得到5-(3,6-二氮杂双环[3.2.0]庚烷-3-基磺酰基)-4-氟异喹啉27(20mg,白色固体,产率:44%)。
1H NMR(400MHz,D2O):δ9.42(s,1H),8.71(d,J=8.0Hz,1H),8.66(d,J=4.0Hz,1H),8.59(d,J=8.0Hz,1H),7.99(t,J=8.0Hz,1H),4.99-4.95(m,1H),4.22-4.17(m,1H),4.00(d,J=12.4Hz,1H),3.85(d,J=11.2Hz,1H),3.78-3.74(m,1H),3.55-3.48(m,2H),3.38-3.34(m,1H).
MS-ESI计算值[M+H]+308,实测值308。
实施例28
4-氟-5-((八氢-1H-吡咯并[3,2-c]吡啶-1-基)磺酰基)异喹啉
第一步
六氢-1H-吡咯并[3,2-b]吡啶-4(2H)-羧酸苄酯6e(50mg,0.17mmol,实施例六)和4-氟-异喹啉-5-磺酰氯24e(50mg,0.2mmol,实施例24)按照实施例一的合成方法得到1-((4-氟异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,2-c]吡啶-5(6H)-羧酸苄酯28a(72mg,黄色油状液体,产率:91%)。
MS-ESI计算值[M+Na]+492,实测值492。
第二步
1-((4-氟异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,2-c]吡啶-5(6H)-羧酸苄酯28a(72mg,0.15mmol)按照实施例五化合物6-(异喹啉-5-基磺酰基)十氢-1,6-二氮杂萘5的合成方法得到4-氟-5-((八氢-1H-吡咯并[3,2-c]吡啶-1-基)磺酰基)异喹啉28(25mg,白色固体,产率:49%)。
1H NMR(400MHz,CD3OD):δ9.28(s,1H),8.64(d,J=8.0Hz,1H),8.60(d,J=4.0Hz,1H),8.51(d,J=8.0Hz,1H),7.91(t,J=8.0Hz,1H),4.16-4.11(m,1H),3.89-3.83(m,1H),3.81-3.66(m,2H),3.14-3.06(m,1H),3.04-2.93(m,1H),2.34-2.20(m,2H),2.04-1.98(m,1H),1.85-1.82(m,1H),1.55-1.50(m,2H)。
MS-ESI计算值[M+H]+336,实测值336。
实施例29
4-氟-5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)异喹啉
第一步
顺式-六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯7a(30mg,0.14mmol,实施例七)和4-氟-异喹啉-5-磺酰氯24e(35mg,0.14mmol)按照实施例一的合成方法得到5-((4-氟异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯29a(55mg,黄色油状液体,产率:92%)。
MS-ESI计算值[M+Na]+444,实测值444。
第二步
5-((4-氟异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯29a(55mg,0.13mmol)按照实施例一的合成方法得到4-氟-5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)异喹啉29(26mg,白色固体,产率:55%)。
1H NMR(400MHz,D2O):δ9.26(brs,1H),8.58-8.53(m,2H),8.49(d,J=8.0Hz,1H),7.89(t,J=8.0Hz,1H),3.61-3.56(m,2H),3.54-3.47(m,2H),3.45-3.42(m,2H),3.19-3.14(m,2H),2.98-2.91(m,2H).
MS-ESI计算值[M+H]+322,实测值322。
实施例30
4-氟-5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)异喹啉
第一步
六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8h(30mg,0.13mmol,实施例八)和4-氟-异喹啉-5-磺酰氯24e(33mg,0.13mmol,实施例24)按照实施例1的合成方法得到2-((4-氟异喹啉-5-基)磺酰基)六氢-1H-吡
咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯30a(17mg,黄色油状液体,产率:29%)。
MS-ESI计算值[M+H]+436,实测值436。
第二步
2-((4-氟异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯30a(17mg,0.039mmol)按照实施例1的合成方法得到4-氟-5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)异喹啉30(6mg,白色固体,产率:46%)。
1H NMR(400MHz,D2O):δ9.33(s,1H),8.59(d,J=5.6Hz,1H),8.53-8.49(m,2H),7.92(t,J=8.0Hz,1H),3.67-3.60(m,2H),3.50-3.40(m,2H),3.35-3.30(m,1H),3.20–3.08(m,3H),2.81-2.70(m,2H),2.04-1.95(m,1H),1.83-1.74(m,1H).
MS-ESI计算值[M+H]+336,实测值336。
实施例31
4-(二氟甲基)-5-((六氢吡咯并[3,2-b]吡咯-1(2H)-基)磺酰基)异喹啉
第一步
将4-溴异喹啉24a(2.0g,9.6mmol)溶于重蒸的30mL四氢呋喃中,并用干冰丙酮浴降温至-65℃,在氮气保护下缓慢滴加正丁基锂(4.0mL,10mmol,2.5M四氢呋喃溶液),滴加完毕后,-65℃搅拌约0.5小时.然后在-65℃逐滴加入N,N-二甲基甲酰胺(730mg,10mmol),反应继续在此温度下搅拌1小时。待反应结束后加入氯化铵的饱和水溶液(100mL)淬灭反应,用乙酸乙酯(50mL×3)萃取,合并有机层用饱和氯化钠水溶液(100mL)洗涤,无水硫酸钠干燥,减压浓缩,粗产物通过色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到异喹啉-4-甲醛31a(550mg,黄色固体,产率:36%)。
1H NMR(400MHz,CDCl3):δ10.41(s,1H),9.45(s,1H),9.22(d,J=8.4Hz,1H),8.96(s,1H),8.10(d,J=8.4Hz,1H),7.96-7.92(m,1H),7.76(t,J=8.4Hz,1H).
MS-ESI计算值[M+H3O]+176,实测值176。
第二步
将异喹啉-4-甲醛31a(100mg,0.63mmol)溶在干燥的5mL二氯甲烷中,氮气保护下于0℃逐滴加入二乙胺基三氟化硫(1.1g,6.3mmol),滴加完毕后,升至室温继续反应5小时至原料消失。将反应混合物缓慢滴加到30mL冰水中,并慢慢加入饱和碳酸氢钠水溶液,直到混合液的pH为8-9,然后用二氯甲烷(20mL×3)萃取,合并有机相,无水硫酸钠干燥,过滤,减压浓缩,粗产物通过色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚),得到4-二氟甲基异喹啉31b(80mg,黄色油状液体,产率:70%)。
1H NMR(400MHz,CDCl3):δ9.36(s,1H),8.66(s,1H),8.21(d,J=8.4Hz,1H),8.07(d,J=8.4Hz,1H),7.86-7.73(m,1H),7.75-7.65(m,1H),7.07(t,J=54.4Hz,1H).
MS-ESI计算值[M+H]+180,实测值180。
第三步
4-二氟甲基异喹啉31b(1.3g,7.2mmol)参照实施例16的方法得到4-(二氟甲基)-5-硝基异喹啉31c(1.16g,黄色固体,产率:71%)。
1H NMR(400MHz,CD3OD):δ9.60(s,1H),8.94(s,1H),8.60(d,J=8.0Hz,1H),8.45(d,J=8.0Hz,1H),7.94(t,J=8.0Hz,1H),7.24(t,J=56.0Hz,1H).
MS-ESI计算值[M+H]+225,实测值225。
第四步
将4-(二氟甲基)-5-硝基异喹啉31c(100mg,0.45mmol)溶于30mL无水乙醇中,在氮气保护下加入10%湿钯碳30mg和85%的水合肼(22mg,0.45mmol)将混合物在90℃下回流1小时,然后将反应液冷却至室温并过滤。将滤液浓缩,粗产物通过色谱硅胶柱纯化(30%乙酸乙酯/石油醚)得到4-(二氟甲基)异喹啉-5-胺31d(62mg,黄色固体,产率:78.5%)。
1H NMR(400MHz,CDCl3):δ9.25(s,1H),8.70(s,1H),7.78-7.64(m,1H),7.54-7.49(m,2H),7.15-7.10(m,1H).MS-ESI计算值[M+H]+195,实测值195。
第五步
4-(二氟甲基)异喹啉-5-胺31d(230mg,1.18mmol)按照实施例16化合物4-氯异喹啉-5-磺酰氯16d的合成方法得到4-(二氟甲基)异喹啉-5-磺酰氯31e(130mg,黄色固体,收率:39%)。
MS-ESI计算值[M+H]+278,实测值278。
第六步
顺式-六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯1d(37mg,0.24mmol,实施例1)和4-(二氟甲基)异喹啉-5-磺酰氯31e(68mg,0.24mmol)按照实施例1的合成方法得到4-((4-(二氟甲基)异喹啉-5-基)磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯31f(50mg,黄色固体,收率:46%)。
MS-ESI计算值[M+H]+454,实测值454。
第七步
4-((4-(二氟甲基)异喹啉-5-基)磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯31f(50mg,0.11mmol)按照实施例1的合成方法得到4-(二氟甲基)-5-((六氢吡咯并[3,2-b]吡咯-1(2H)-基)磺酰基)异喹啉31(25mg,白色固体,53%)。
1H NMR(400MHz,D2O):δ9.69(s,1H),9.06(s,1H),8.63-8.60(m,2H),8.21-7.94(m,2H),4.66-4.48(m,2H),3.64-3.61(m,1H),3.54-3.49(m,1H),3.32-3.28(m,2H),2.44-2.39(m,1H),2.24-2.22(m,1H),2.07-2.05(m,2H).
MS-ESI计算值[M+H]+354,实测值354。
实施例32
4-(二氟甲基)-5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)异喹啉
第一步
顺式-六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2f(37mg,0.24mmol,实施例2)和4-(二氟甲基)异喹啉-5-磺酰氯31e(68mg,0.24mmol,实施例31)按照实施例1的合成方法得到5-((4-(二氟甲基)异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-1(2H)-羧酸叔丁酯32a(45mg,黄色固体,产率:41%)。
MS-ESI计算值[M+H]+454,实测值454。
第二步
5-((4-(二氟甲基)异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-1(2H)-羧酸叔丁酯32a(45mg,0.1mmol)按照实施例1的合成方法得到4-(二氟甲基)-5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)异喹啉32(25mg,白色固体,产率:60%)。
1H NMR(400MHz,D2O):δ9.63(s,1H),9.04(s,1H),8.71-8.56(m,2H),8.18-7.92(m,2H),4.41-4.38(m,1H),3.69-3.67(m,2H),3.53-3.50(m,1H),3.36-3.21(m,4H),2.24-2.18(m,1H),1.90-1.76(m,1H).
MS-ESI计算值[M+H]+354,实测值354。
实施例33
4-(二氟甲基)-5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)异喹啉
第一步
顺式-八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯3a(30mg,0.18mmol,实施例3)和4-(二氟甲基)异喹啉-5-磺酰氯31e(50mg,0.18mmol,实施例31)按照实施例1的合成方法得到6-((4-(二氟甲基)异喹啉-5-基)磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯33a(40mg,黄色固体,收率:47%)。
MS-ESI计算值[M+H]+468,实测值468。
第二步
6-((4-(二氟甲基)异喹啉-5-基)磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯33a(40mg,0.085mmol)按照实施例1的合成方法得到4-(二氟甲基)-5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)异喹啉33(10mg,白色固体,27%)。
1H NMR(400MHz,D2O):δ9.64-9.59(m,1H),9.03(s,1H),8.57-8.47(m,2H),8.21-7.92(m,2H),3.97(s,1H),3.79-3.74(m,2H),3.64-3.61(m,1H),3.46(t,J=10.2Hz,1H),3.32-3.28(m,1H),3.00-2.92(m,2H),1.84-1.72(m,4H).
MS-ESI计算值[M+H]+368,实测值368。
实施例34
5-(3,6-二氮杂双环[3.2.0]庚烷-3-基磺酰基)-4-(二氟甲基)异喹啉
第一步
3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯4a(30mg,0.18mmol,实施例4)和4-(二氟甲基)异喹啉-5-磺酰氯31e(50mg,0.18mmol,实施例31)按照实施例1的合成方法得到3-((4-(二氟甲基)异喹啉-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯34a(40mg,黄色固体,收率:50%)。
MS-ESI计算值[M+H]+440,实测值440。
第二步
3-((4-(二氟甲基)异喹啉-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯34a(40mg,0.085mmol)按照实施例1的合成方法得到
5-(3,6-二氮杂双环[3.2.0]庚烷-3-基磺酰基)-4-(二氟甲基)异喹啉34(10mg,白色固体,26%)。
1H NMR(400MHz,D2O):δ9.60(s,1H),9.07(s,1H),8.65-8.60(m,2H),8.20(t,J=53.2Hz,1H),7.94(t,J=8.0Hz,1H),5.00-4.95(m,1H),4.20-4.15(m,1H),4.00-3.95(m,1H),3.95-3.70(m,2H),3.54-3.50(m,2H),3.35-3.31(m,1H).
MS-ESI计算值[M+H]+340,实测值340。
实施例35
4-(二氟甲基)-5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)异喹啉
第一步
顺式-六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯7a(37mg,0.24mmol,实施例7)和4-(二氟甲基)异喹啉-5-磺酰氯31e(68mg,0.24mmol,实施例31)按照实施例1的合成方法得到5-((4-(二氟甲基)异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯35a(52mg,黄色固体,收率:48%)。
MS-ESI计算值[M+H]+454,实测值454。
第二步
5-((4-(二氟甲基)异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯35a(52mg,0.11mmol)按照实施例1的合成方法得到4-(二氟甲基)-5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)异喹啉35(25mg,白色固体,51%)。
1H NMR(400MHz,D2O):δ9.59(s,1H),9.05(s,1H),8.66-8.54(m,2H),8.24-7.91(m,2H),4.56-4.46(m,4H),3.35-3.31(m,2H),3.20-3.15(m,2H),3.00-2.90(m,2H).
MS-ESI计算值[M+H]+354,实测值354。
实施例36
第一步
六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8h(56mg,0.25mmol,实施例8)和4-(二氟甲基)异喹啉-5-磺酰氯31e(75mg,0.27mmol,实施例31)按照实施例1的合成方法得到2-((4-(二氟甲基)异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯36a(45mg,浅黄色油状液体,产率:45%)。
MS-ESI计算值[M+Na]+490,实测值490。
第二步
2-((4-(二氟甲基)异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯36a(45mg,0.096mmol)按照实施例1的合成方法得到4-(二氟甲基)-5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)异喹啉36(30mg,白色固体,产率:86%)。
1H NMR(400MHz,CD3OD):δ9.78(s,1H),9.18(s,1H),8.76-8.73(m,1H),8.71-8.66(m,1H),8.39(t,J=54.0Hz,1H),8.08(t,J=8.0Hz,1H),3.66-3.59(m,2H),3.55-3.48(m,2H),3.43-3.36(m,1H),3.27-3.23(m,1H),3.24-3.14(m,2H),2.88-2.70(m,2H),2.13-2.02(m,1H),1.97-1.85(m,1H).
MS-ESI计算值[M+H]+368,实测值368。
实施例37
5-((六氢吡咯并[3,2-b]吡咯-1(2H)-基)磺酰基)-4-甲基异喹啉
第一步
氮气保护下向4-溴异喹啉24a(15.0g,72.5mmol),甲基硼酸(8.8g,146mmol),K3PO4(62.0g,292mmol)的
350mL甲苯溶液中加入三(二亚苄基丙酮)二钯(6.6g,7.2mmol)和二环己基(2',6'-二甲氧基-[1,1'-联苯]-2-基)膦(5.9g,14.3mmol),所得混合物加热回流20小时。待反应结束后将反应液倒入水中(300mL),用乙酸乙酯(100mL×2)萃取,何并有机相用无水硫酸钠干燥,浓缩,用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到4-甲基异喹啉37a(10.7g,黄色油状液体,产率:94%)。
第二步
4-甲基异喹啉37a(10.0g,69.9mmol)按照实施例16的合成方法得到得到4-甲基-5-硝基异喹啉37b(10g,黄色油状液体,产率:76%)。
第三步
4-甲基-5-硝基异喹啉37b(9.00g,47.8mmol)按照实施例16的合成方法得到4-甲基异喹啉-5-胺37c(6.0g,黄色固体,产率:71%)。
第四步
4-甲基异喹啉-5-胺37c(500mg,3.16mmol)按照实施例16的合成方法得到4-甲基异喹啉-5-磺酰氯37d(300mg,绿色固体,产率:40%)。
1H NMR(400MHz,DMSO-d6):δ9.78(brs.,1H),8.89-8.90(m,1H),8.55-8.50(m,2H),8.01-7.88(m,1H),3.34(s,3H).
第五步
顺式-六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯1d(33mg,0.14mmol)和4-甲基异喹啉-5-磺酰氯37d(68mg,0.28mmol)按照实施例1的合成方法得到4-(4-甲基异喹啉-5-磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯37e(15mg,黄色油状液体,产率:25%)。
MS-ESI计算值[M+H]+418,实测值418。
第六步
4-(4-甲基异喹啉-5-磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯37e(15mg,0.036mmol)按照实施例1的合成方法得到5-((六氢吡咯并[3,2-b]吡咯-1(2H)-基)磺酰基)-4-甲基异喹啉37(10mg,黄色固体,产率:67%)。
1H NMR(400MHz,D2O):δ9.58(s,1H),8.64-8.62(m,1H),8.51-8.50(m,2H),8.02(t,J=8.0,1H),4.85-4.81(m,2H),4.62-4.59(m,1H),3.79-3.73(m,1H),3.66-3.60(m,1H),3.48-3.40(m,2H),3.06(s,3H),2.64-2.54(m,1H),2.34-2.25(m,2H).
MS-ESI计算值[M+H]+318,实测值318。
实施例38
5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)-4-甲基异喹啉
第一步
顺式-六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2f(50mg,0.24mmol,实施例2)和4-甲基异喹啉-5-磺酰氯37d(85mg,0.35mmol,实施例37)按照实施例1的合成方法得到5-((4-甲基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯38a(40mg,无色油状液体,产率:40%)。
第二步
5-((4-甲基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯38a(40mg,0.095mmol)按照实施例1的合成方法得到5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)-4-甲基异喹啉38(28mg,白色固体,产率:75%)。
1H NMR(400MHz,D2O):δ9.52(s,1H).8.66-8.42(m,3H),7.96(t,J=7.8Hz,1H),4.46(brs.,1H),3.94-3.80(m,2H),3.75-3.70(m,1H),3.55-3.50(m,1H),3.45-3.26(m,3H),3.01(brs,3H),2.35-2.22(m,1H),2.00-1.95(m,1H).
实施例39
5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)-4-甲基异喹啉-1-醇
在0℃条件下向5-((4-甲基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯38a(100mg,0.24mmol)的5mL二氯甲烷溶液中加入间氯过氧苯羧酸(83mg,0.48mmol),室温搅拌2小时直至反应结束。直接蒸干溶剂后用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到5-((1-(叔丁氧基羰基)六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)-4-甲基异喹啉-2-氧化物39a(75mg,黄色油状液体,产率:73%)。
第二步
将5-((1-(叔丁氧基羰基)六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)-4-甲基异喹啉-2-氧化物39a(75mg,0.17mmol)溶于1mL乙酸酐中在100℃下搅拌2小时直到反应结束。反应液减压浓缩除去溶剂后加入6mL四氢呋喃,2mL水和碳酸钠(37mg,0.35mmol)并室温搅拌30分钟。然后二氯甲烷(20mL×3)萃取,有机相
用无水硫酸钠干燥,浓缩,用色谱硅胶柱纯化(0-100%乙酸乙酯/石油醚)得到5-((1-羟基-4-甲基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯39b(35mg,黄色油状液体,产率:47%)。
第三步
5-((1-羟基-4-甲基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯39b(35mg,0.081mmol)按照实施例1的合成方法得到5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)-4-甲基异喹啉-1-醇39(13mg,白色固体,产率:48%)。
1H NMR(400MHz,D2O):δ8.57(d,J=7.2Hz,1H),8.14(d,J=7.2Hz,1H),7.62-7.57(m,1H),7.19(brs,1H),3.76(brs,2H),3.44(brs,2H),3.40-3.30(m,3H),2.52(s,3H),2.27(brs,1H),1.96(brs,2H).
实施例40
5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)-4-甲基异喹啉
第一步
顺式-八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯3a(50mg,0.22mmol,实施例3)和4-甲基异喹啉-5-磺酰氯37d(106mg,0.44mmol,实施例37)按照实施例1的合成方法得到6-(4-甲基异喹啉-5-磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1–羧酸叔丁酯40a(30mg,黄色油状液体,产率:31%)。
MS-ESI计算值[M+H]+432,实测值432。
第二步
6-(4-甲基异喹啉-5-磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1–羧酸叔丁酯40a(30mg,0.074mmol)按照实施例1的合成方法得到5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)-4-甲基异喹啉40(20mg,白色固体,产率:87%)。
1H NMR(400MHz,CDCl3):δ9.55(s,1H),8.62(d,J=8.0Hz,1H),8.52-8.44(m,2H),8.00(t,J=8.0Hz,1H),4.06(s,1H),3.97-3.91(m,1H),3.89-3.84(m,1H),3.80-3.75(m,1H),3.65-3.60(m,1H),3.40-3.35(m,1H),3.10(s,1H),3.07(s,3H),2.04-1.73(m,5H).
MS-ESI计算值[M+H]+332,实测值332。
实施例41
第一步
3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯4a(30mg,0.15mmol,实施例4)和4-甲基异喹啉-5-磺酰氯37d(73mg,0.31mmol,实施例37)按照实施例1的合成方法得到3-(4-甲基异喹啉-5-磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯41a(45mg,黄色油状液体,产率:74%)。
MS-ESI计算值[M+H]+404,实测值404。
第二步
3-(4-甲基异喹啉-5-磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯41a(45mg,0.11mmol)按照实施例1的合成方法得到5-(3,6-二氮杂双环[3.2.0]庚烷-3-磺酰基)-4-甲基异喹啉41(25mg,黄色固体,产率:74%)。
1H NMR(400MHz,D2O):δ9.49(s,1H),8.57-8.55(m,1H),8.47-8.46(m,2H),7.94(t,J=8.0Hz,1H),4.24-4.16(m,3H),3.90(d,J=8.0Hz,1H),3.82-3.78(m,1H),3.73-3.68(m,1H),3.53(d,J=8.0Hz,2H),3.06(s,3H)。
MS-ESI计算值[M+H]+304,实测值304。
实施例42
4-甲基-5-((八氢-1H-吡咯并[3,2-b]吡啶-1-基)磺酰基)异喹啉
第一步
六氢-1H-吡咯并[3,2-b]吡啶-4(2H)-羧酸苄酯6e(30mg,0.12mmol,实施例6)和4-甲基异喹啉-5-磺酰氯37d
(139mg,0.58mmol,实施例37)按照实施例1的合成方法得到1-(4-甲基异喹啉-5-磺酰基)六氢-1H-吡咯并[3,2-b]吡啶-4(2H)羧酸苄酯42a(30mg,黄色液体,产率:56%)。
MS-ESI计算值[M+H]+466,实测值466。
第二步
1-(4-甲基异喹啉-5-磺酰基)六氢-1H-吡咯并[3,2-b]吡啶-4(2H)羧酸苄酯42a(30mg,0.065mmol)按照实施例5化合物6-(异喹啉-5-基磺酰基)十氢-1,6-二氮杂萘5的合成方法得到4-甲基-5-((八氢-1H-吡咯并[3,2-b]吡啶-1-基)磺酰基)异喹啉42(5mg,黄色固体,产率:23%)。
1H NMR(400MHz,D2O):δ8.98(s,1H),8.30(s,1H),8.24-8.21(m,2H),7.64(t,J=8.0,1H),4.16-4.11(m,1H),4.06-4.03(m,1H),3.70-3.65(m,2H),3.15-3.03(m,2H),2.78(s,3H),2.40-2.34(m,2H),1.80-1.72(m,3H),1.52-1.50(m,1H)。
MS-ESI计算值[M+H]+332,实测值332。
实施例43
5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)-4-甲基异喹啉
第一步
顺式-六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯7a(30mg,0.14mmol,实施例7)和4-甲基异喹啉-5-磺酰氯37d(68mg,0.28mmol,实施例37)按照实施例1的合成方法得到5-((4-甲基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯43a(28mg,黄色油状液体,产率:48%)。
MS-ESI计算值[M+H]+418,实测值418。
第二步
5-((4-甲基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯43a(28mg,0.067mmol)按照实施例1的合成方法得到5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)-4-甲基异喹啉43(8mg,白色固体,产率:34%)。
1H NMR(400MHz,D2O):δ8.99(brs,1H),8.39-8.28(m,2H),8.21(m,1H),7.64(t,J=8.0Hz,1H),3.68-3.59(m,4H),3.49-3.47(m,2H),3.30-3.23(m,2H),3.12-3.04(m,2H),2.84-2.76(brs,3H).
MS-ESI计算值[M+H]+318,实测值318。
实施例44
5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)-4-甲基异喹啉
第一步
六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8h(30mg,0.13mmol,实施例8)和4-甲基异喹啉-5-磺酰氯37d(48mg,0.2mmol,实施例37)按照实施例1的合成方法得到2-(4-甲基异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯44a(40mg,无色油状液体,产率:70%)。
第二步
2-(4-甲基异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯44a(40mg,0.47mmol)按照实施例1的合成方法得到5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)-4-甲基异喹啉44(15mg,白色固体,产率:40%)。
1H NMR(400MHz,D2O):δ9.50(s,1H),8.58(d,J=8.0Hz,1H),8.52-8.44(m,2H),7.96(t,J=8.0Hz,1H)3.70-3.65(m,2H),3.57-3.48(m,2H),3.40-3.35(m,1H),3.26-3.10(m,3H),3.03(s,3H),2.89-2.73(m,2H),2.08-1.97(m,1H),1.89-1.77(m,1H).
实施例45
6-((4-甲基异喹啉-5-基)磺酰基)十氢-1,6-二氮杂萘
第一步
八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯20c(30mg,0.11mmol,实施例20)4-甲基异喹啉-5-磺酰氯37d(40mg,0.16mmol,实施例37)按照实施例1的合成方法得到6-((4-甲基异喹啉-5-基)磺酰基)八氢-1,6-二氮杂萘-1(2H)-羧酸叔丁酯45a(40mg,无色油状液体,产率:76%)。
第二步
6-((4-甲基异喹啉-5-基)磺酰基)八氢-1,6-二氮杂萘-1(2H)-羧酸叔丁酯45a(40mg,0.084mmol)按照实施例5化合物6-(异喹啉-5-基磺酰基)十氢-1,6-二氮杂萘5的合成方法得到得到6-((4-甲基异喹啉-5-基)磺酰基)十氢-1,6-二氮杂萘45(12mg,白色固体,产率:41%)。
1H NMR(400MHz,D2O):δ9.03(brs,1H),8.36-8.30(m,2H),8.21(brs,1H),7.65(brs,1H),3.98-3.74(m,2H),3.60-3.55(m,1H),3.45-3.40(m,1H),3.34-2.88(m,4H),2.80(brs,3H),2.35-2.02(m,2H),1.96-1.70(m,4H),1.59(brs,1H).
实施例46
5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)-4-(三氟甲基)异喹啉
第一步
将4-溴异喹啉24a(10g,48.3mmol)按照实施例24化合物4-氟-5-硝基异喹啉24c的合成方法得到4-溴-5-硝基异喹啉46a(8.5g,黄色固体,产率85%),产物不经纯化直接进行下一步反应。
第二步
将4-溴-5-硝基异喹啉46a(1.00g,3.95mmol)溶于40mL1-甲基吡咯烷酮,氮气保护下依次加入氟化钾(459mg,7.90mmol)、三氟甲基三甲基硅烷(2.80g,19.7mmol)和碘化亚铜(1.13g,5.93mmol),所得混合物100℃搅拌8小时,反应结束后冷却至室温,加入300mL乙酸乙酯稀释,过滤除去固体杂质。滤液依次用水(20mL)、饱和氯化钠水溶液(20mL)洗涤,用无水硫酸钠干燥,过滤,滤液减压浓缩,用色谱硅胶柱纯化(30%乙酸乙酯/石油醚)得5-硝基-4-三氟甲基异喹啉46b(500mg,黄色固体,产率:52%)。
1H NMR(400MHz,DMSO-d6):δ9.82(s,1H),9.20(s,1H),8.71(d,J=7.2Hz,1H),8.57(d,J=7.2Hz,1H),
8.05-8.01(m,1H).
MS-ESI计算值[M+H]+243,实测值243。
第三步
5-硝基-4-三氟甲基异喹啉46b(1.00g,4.15mmol)按照实施例31化合物4-(二氟甲基)异喹啉-5-胺31d的合成方法得4-三氟甲基异喹啉-5-胺46c(523mg,黄色固体,产率:59%)。
1H NMR(400MHz,DMSO):δ9.41(s,1H),8.76(s,1H),7.61-7.55(m,2H),7.10-7.05(m,1H),5.45(s,2H).
MS-ESI计算值[M+H]+213,实测值213。
第四步
4-三氟甲基异喹啉-5-胺46c(300mg,1.41mmol)按照实施例16化合物4-氯异喹啉-5-磺酰氯16d的合成方法得到4-三氟甲基喹啉-5-磺酰氯46d(74mg,浅黄色固体,产率:18%)。
1H NMR(400MHz,DMSO-d6):δ9.53(s,1H),8.89(s,1H),8.60(d,J=8.0Hz,1H),8.27(d,J=8.0Hz,1H),7.81-7.77(m,1H).
第五步
4-三氟甲基异喹啉-5-磺酰氯46d(45mg,0.15mmol)和顺式-六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2f(32mg,0.15mmol,实施例2)按照实施例1的合成方法得到5-((4-(三氟甲基)异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯46e(30mg,黄色固体,产率:42%)。
MS-ESI计算值[M+H]+472,实测值472。
第六步
5-((4-(三氟甲基)异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯46e(30mg,0.063mmol)按照实施例1的合成方法得到5-((六氢吡咯并[3,4-b]吡咯-5(1H)-基)磺酰基)-4-(三氟甲基)异喹啉46(14mg,白色固体,产率:60%)。
1H NMR(400MHz,CD3OD):δ9.62(s,1H),9.04(s,1H),8.58-8.55(m,2H),8.0-7.98(m,1H),4.35-4.30(m,1H),3.75-3.70(m,1H),3.46-3.35(m,3H),3.27-3.12(m,3H),2.28-2.23(m,1H),1.99-1.93(m,1H).
MS-ESI计算值[M+H]+372,实测值372。
实施例47
5-((六氢吡咯并[3,2-b]吡咯-1(2H)-基)磺酰基)-4-甲氧基异喹啉
第一步
4-溴-5-硝基异喹啉46a(3.00g,11.8mmol,实施例46)按照实施例16化合物4-氯异喹啉-5-胺16c的合成方法得到4-溴异喹啉-5-胺47a(1.9g,浅黄色固体,产率:73%)。
1H NMR(400MHz,DMSO):δ9.03(s,1H),8.42(s,1H),7.46-7.42(m,1H),7.36(d,J=7.2Hz,1H),7.06(d,J=7.2Hz,1H),6.17(s,2H).
第二步
将4-溴异喹啉-5-胺47a(1.90g,8.52mmol)按照实施例16化合物4-氯异喹啉-5-磺酰氯16d的合成方法得到4-溴异喹啉-5-磺酰氯47b(500mg,浅黄色固体,产率:20%)。
1H NMR(400MHz,DMSO):δ9.57(s,1H),8.91(s,1H),8.40(d,J=7.2Hz,1H),8.39(d,J=7.2Hz,1H),7.86-7.82(m,1H).
第三步
4-溴异喹啉-5-磺酰氯47b(57mg,0.19mmol)和顺式-六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯1d(40mg,0.19mmol,实施例1)按照实施例1的合成方法得到4-((4-溴异喹啉-5-基)磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸酸叔丁酯47c(62mg,黄色固体,产率:68%)。
1H NMR(400MHz,CDCl3):δ9.23(s,1H),9.02(s,1H),8.37(d,J=7.2Hz,1H),8.22(d,J=7.2Hz,1H),7.73-7.69(m,1H),4.67-4.47(m,2H),3.81-3.63(m,2H),3.50-3.37(m,3H),2.39-2.21(m,2H),2.05-1.93(m,1H),1.45(s,9H).
MS-ESI计算值[M+H]+482,实测值482。
第四步
4-((4-溴异喹啉-5-基)磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸酸叔丁酯47c(62mg,0.13mmol)溶于1mL无水甲醇中,氮气保护下依次加入吡啶(0.2mg,0.003mmol),甲醇钠(300mg,1.28mmol)和CuI(12mg,0.064mmol)。所得混合物在100℃搅拌3小时,冷却至室温,加入30mL乙酸乙酯稀释,过滤除去固体杂质,滤液依次用饱和氯化铵水溶液(20mL)、水(20mL)、饱和氯化钠水溶液(20mL)洗涤,用无水硫酸钠干燥,过滤,滤液减压浓缩,用薄层色谱硅胶板(100%乙酸乙酯)纯化得4-((4-甲氧基异喹啉-5-基)磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯47d(33mg,白色固体,产率:60%)。
MS-ESI计算值[M+H]+434,实测值434。
第五步
4-((4-甲氧基异喹啉-5-基)磺酰基)六氢吡咯并[3,2-b]吡咯-1(2H)-羧酸叔丁酯47d(33mg,0.063mmol)按照实施例1的合成方法得到5-((六氢吡咯并[3,2-b]吡咯-1(2H)-基)磺酰基)-4-甲氧基异喹啉47(26mg,白色固体,产率:83%)。
1H NMR(400MHz,D2O):δ9.31(s,1H),8.61(d,J=7.6Hz,1H),8.50(d,J=7.6Hz,1H),8.34(s,1H),8.05-8.01(m,1H),4.81-4.75(m,1H),4.58-4.57(m,1H),4.16(s,3H),3.80-3.70(m,1H),3.67-3.64(m,1H),3.41-3.38(m,2H),2.53-2.50(m,1H),2.35-2.34(m,1H),2.25-2.21(m,2H).
MS-ESI计算值[M+H]+334,实测值334。
实施例48
5-((六氢吡咯并[3,2-c]吡咯-5(1H)-基)磺酰基)-4-甲氧基异喹啉
第一步
顺式-六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯2f(76mg,0.36mmol,实施例2)和4-溴异喹啉-5-磺酰氯47b(94mg,0.30mmol,实施例47)按照实施例1的合成方法得到5-((4-溴异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯48a(120mg,白色固体,产率81%)
第二步
5-((4-溴异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯48a(120mg,0.250mmol)按照实施例47的合成方法得到5-((4-甲氧基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯48b(86mg,白色固体,收率:80%)。
MS-ESI计算值[M+H]+434,实测值434。
第三步
5-((4-甲氧基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-b]吡咯-1(2H)-羧酸叔丁酯48b(86mg,0.2mmol)按照实施例1的合成方法得到5-((六氢吡咯并[3,2-c]吡咯-5(1H)-基)磺酰基)-4-甲氧基异喹啉48(25mg,白色固体,收率:38%)。
1H NMR(400MHz,D2O):δ9.28(s,1H),8.56-8.53(m,2H),8.28(s,1H),8.00-7.96(m,1H),4.39-4.36(m,1H),4.10(s,3H),3.84-3.81(m,1H),3.75-3.74(m,1H),3.62-3.60(m,1H),3.55-3.54(m,1H),3.31-3.27(m,3H),2.28-2.22(m,1H),1.95-1.92(m,1H).
MS-ESI计算值[M+H]+334,实测值334。
实施例49
5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)-4-甲氧基异喹啉
第一步
4-溴异喹啉-5-磺酰氯47b(54mg,0.18mmol,实施例47)和顺式-八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯3a(40mg,0.18mmol,)按照实施例1的合成方法得到6-((4-溴异喹啉-5-基)磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯49a(80mg,白色固体,收率:91%)。
1H NMR(400MHz,CDCl3):δ9.21(s,1H),8.99(s,1H),8.34-8.32(m,1H),8.20-8.18(m,1H),7.72-7.68(m,1H),4.01-3.98(m,1H),3.56-3.51(m,2H),3.48-3.41(m,2H),2.80-2.75(m,1H),2.28-2.24(m,1H),1.83-1.77(m,2H),1.60-1.57(m,3H),1.43(s,9H).
MS-ESI计算值[M+H]+496,实测值496。
第二步
6-((4-溴异喹啉-5-基)磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯49a(80mg,0.16mmol)按照实施例47的合成方法得到6-((4-甲氧基异喹啉-5-基)磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯49b(46mg,白色固体,收率:64%)。
MS-ESI计算值[M+H]+448,实测值448。
第三步
6-((4-甲氧基异-5-基)磺酰基)八氢-1H-吡咯并[3,4-b]吡啶-1-羧酸叔丁酯49b(46mg,0.1mmol)按照实施例1的合成方法得到5-((六氢-1H-吡咯并[3,4-b]吡啶-6(2H)-基)磺酰基)-4-甲氧基异喹啉49(20mg,白色固体,收率:56%)。
1H NMR:(400MHz,D2O):δ9.21(s,1H),8.51-8.50(m,1H),8.36-8.34(m,1H),8.29(s,1H),7.95-7.91(m,1H),4.08(s,3H),3.96-3.94(m,1H),3.83-3.79(m,2H),3.67-3.65(m,1H),3.54-3.49(m,1H),3.31-3.28(m,1H),3.01-2.93(m,2H),1.90-1.65(m,4H).
MS-ESI计算值[M+H]+348,实测值348。
实施例50
5-(3,6-二氮杂双环[3.2.0]庚烷-3-基磺酰基)-4-甲氧基异喹啉
第一步
4-溴异喹啉-5-磺酰氯47b(57mg,0.19mmol,实施例47)和3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯4a(40mg,0.12mmol,实施例4)按照实施例1的合成方法得到3-((4-溴异喹啉-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯50a(82mg,黄色固体,产率:87%)。
1H NMR(400MHz,CDCl3):δ9.22(s,1H),9.02(s,1H),8.69(d,J=7.6Hz,1H),8.23(d,J=7.6Hz,1H),7.73-7.69(m,1H),4.78-4.76(m,1H),4.10-3.92(m,2H),3.75-3.56(m,2H),3.44-3.12(m,3H),1.39(s,9H).
MS-ESI计算值[M+H]+468,实测值468。
第二步
3-((4-溴异喹啉-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯50a(82mg,0.18mmol)按照实施例47的合成方法得到3-((4-甲氧基异唑-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯50b(30mg,白色固体,产率42%)。
MS-ESI计算值[M+H]+420,实测值420。
第三步
3-((4-甲氧基异唑-5-基)磺酰基)-3,6-二氮杂双环[3.2.0]庚烷-6-羧酸叔丁酯50b(30mg,0.071mmol)按照实施例1的合成方法得到5-(3,6-二氮杂双环[3.2.0]庚烷-3-基磺酰基)-4-甲氧基异喹啉50(20mg,白色固体,产率86%)。
1H NMR(400MHz,D2O):δ9.32(s,1H),8.62-8.57(m,2H),8.33(s,1H),8.03-7.99(m,1H),5.01(t,J=6.0Hz,1H),4.28-4.23(m,1H),4.17(s,3H),4.11(d,J=13.6Hz,1H),3.96(d,J=13.6Hz,1H),3.84-3.79(m,1H),3.62-3.52(m,2H),3.46-3.42(m,1H).
MS-ESI计算值[M+H]+320,实测值320。
实施例51
4-甲氧基-5-((八氢-1H-吡咯并[3,2-b]吡啶-1-基)磺酰基)异喹啉
第一步
4-溴异喹啉-5-磺酰氯47b(59mg,0.19mol,实施例47)和六氢-1H-吡咯并[3,2-b]吡啶-4(2H)-羧酸苄酯6e(50mg,19mmol,实施例6)按照实施例1的合成方法得到1-((4-溴异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,2-b]吡啶-4(2H)-羧酸叔丁酯51a(90mg,白色固体,收率:88%)。
1H NMR(400MHz,CDCl3):δ9.21(s,1H),8.99(s,1H),8.20-8.18(m,2H),7.77-7.70(m,1H),7.38(brs,5H),5.18(s,2H),4.07-4.05(m,1H),3.89-3.86(m,1H),3.68-3.63(m,3H),3.45-3.43(m,1H),2.87-2.84(m,1H),2.24-2.16(m,3H),1.69-1.65(m,1H),1.40-1.34(m,1H).
MS-ESI计算值[M+H]+530,实测值530。
第二步
1-((4-溴异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,2-b]吡啶-4(2H)-羧酸叔丁酯51a(90mg,0.17mmol)按照实施例47的合成方法得到1-((4-甲氧基异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,2-b]吡啶-4(2H)-羧酸甲酯51b(30mg,白色固体,收率:44%)。
MS-ESI计算值[M+H]+406,实测值406。
第三步
向1-((4-甲氧基异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,2-b]吡啶-4(2H)-羧酸甲酯51b(30mg,0.074mmol)和甲醇钠(40mg,0.74mmol)的2mL甲醇溶液中加入0.1m L的25%NaOH水溶液。将混合物在氮气氛围下微波加热至90℃2小时。待反应结束后冷却至室温,将混合物用乙酸乙酯(50mL)稀释,并用饱和氯化钠水溶液(50mL)洗涤,有机相用无水硫酸钠干燥,减压浓缩,残余物用制备薄层色谱纯化(50%甲醇/乙酸乙酯)得到化合物4-甲氧基-5-((八氢-1H-吡咯并[3,2-b]吡啶-1-基)磺酰基)异喹啉51(20mg,白色固体,收率:78%)。
1H NMR(400MHz,CD3OD):δ9.01(s,1H),8.51-8.49(m,1H),8.40-8.38(m,1H),8.36(s,1H),7.86-7.82(m,1H),4.15(s,3H),4.14-4.12(m,1H),3.93-3.92(m,1H),3.80-3.76(m,2H),3.13-3.11(m,1H),3.02-3.00(m,1H),2.34-2.28(m,2H),1.99-1.89(m,2H),1.65-1.54(m,2H).
MS-ESI计算值[M+H]+348,实测值348。
实施例52
5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)-4-甲氧基异喹啉
第一步
4-溴异喹啉-5-磺酰氯47b(58mg,0.19mmol,实施例47)和顺式-六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯7a(40mg,0.19mmol,实施例7)按照实施例1的合成方法得到5-((4-溴异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯52a(80mg,白色固体,收率:88%)。
1H NMR(400MHz,CDCl3):δ9.21(s,1H),9.00(s,1H),8.43-8.41(m,1H),8.22-8.20(m,1H),7.73-7.69(m,1H),3.76-3.72(m,2H),3.38-3.34(m,4H),3.08-3.03(m,4H),1.61(s,9H).
MS-ESI计算值[M+H]+482,实测值482。
第二步
5-((4-溴异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯52a(80mg,0.17mmol)按照实施例47的合成方法得到5-((4-甲氧基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯52b(45mg,白色固体,收率:63%)。
MS-ESI计算值[M+H]+434,实测值434。
第三步
5-((4-甲氧基异喹啉-5-基)磺酰基)六氢吡咯并[3,4-c]吡咯-2(1H)-羧酸叔丁酯52b(45mg,0.1mmol)按照实施例1的合成方法得到5-((六氢吡咯并[3,4-c]吡咯-2(1H)-基)磺酰基)-4-甲氧基异喹啉52(20mg,白色固体,收率:58%)。
1H NMR(400MHz,D2O):δ9.22(s,1H),8.53-8.49(m,2H),8.28(s,1H),7.97-7.93(m,1H),4.10(s,3H),3.64-3.60(m,4H),3.56-3.53(m,2H),3.24-3.22(m,2H),3.08-3.05(m,2H).
MS-ESI计算值[M+H]+334,实测值334。
实施例53
5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)-4-甲氧基异喹啉
第一步
4-溴异喹啉-5-磺酰氯47b(54mg,0.17mmol,实施例47)和六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯8h(40mg,0.17mmol,实施例8)按照实施例1的合成方法得到2-((4-溴异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯53a(40mg,黄色固体,产率:46%)。
MS-ESI计算值[M+H]+496,实测值496。
第二步
2-((4-溴异喹啉-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯53a(40mg,0.081mmol)按照实施例47的合成方法得到2-((4-甲氧基异唑-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯53b(20mg,白色固体,产率:55%)。
MS-ESI计算值[M+H]+448,实测值448。
第三步
2-((4-甲氧基异唑-5-基)磺酰基)六氢-1H-吡咯并[3,4-c]吡啶-5(6H)-羧酸叔丁酯53b(20mg,0.044mmol)按照实施例1的合成方法得到5-((六氢-1H-吡咯并[3,4-c]吡啶-2(3H)-基)磺酰基)-4-甲氧基异喹啉53(14mg,白色固体,产率:77%)。
1H NMR(400MHz,D2O):δ9.26(s,1H),8.57(d,J=7.2Hz,1H),8.46(d,J=7.2Hz,1H),8.33(s,1H),8.01-7.97(m,1H),4.15(s,3H),3.72-3.66(m,2H),3.59-3.53(m,2H),3.42-3.37(m,1H),3.25-3.14(m,3H),2.87-2.77(m,2H),2.10-2.02(m,1H),1.88-1.82(m,1H).
MS-ESI计算值[M+H]+348,实测值348。
实施例54
6-((4-甲氧基异喹啉-5-基)磺酰基)十氢-1,6-二氮杂萘55
第一步
4-溴异喹啉-5-磺酰氯47b(49mg,0.16mmol,实施例47)和八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯20c(50mg,0.16mmol,实施例20)按照实施例1的合成方法得到6-((4-溴异喹啉-5-基)磺酰基)八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯54a(46mg,黄色固体,产率:53%)。
1H NMR(400MHz,CDCl3):δ9.22(s,1H),9.01(s,1H),8.34-8.12(m,2H),7.72-7.68(m,1H),7.37-7.32(m,5H),5.15(s,2H),4.12-3.84(m,2H),3.71-3.49(m,2H),3.26-2.67(m,2H),2.05-1.67(m,4H),1.54-1.45(m,4H).
MS-ESI计算值[M+H]+544,实测值544。
第二步
6-((4-溴异喹啉-5-基)磺酰基)八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯54a(46mg,0.084mmol)按照实施例47的合成方法得到6-((4-甲氧基异喹啉-5-基)磺酰基)八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯55b(20mg,白色固体,产率:47%)。
MS-ESI计算值[M+H]+496,实测值496。
第三步
6-((4-甲氧基异喹啉-5-基)磺酰基)八氢-1,6-二氮杂萘-1(2H)-羧酸苄酯55b(20mg,0.04mmol)按照实施例5化合物6-(异喹啉-5-基磺酰基)十氢-1,6-二氮杂萘5的合成方法得到6-((4-甲氧基异喹啉-5-基)磺酰基)十氢-1,6-二氮杂萘55(3mg,白色固体,产率:21%)。
1H NMR(400MHz,CD3OD):δ8.97(s,1H),8.39-8.33(m,2H),8.31(s,1H),7.80-7.76(m,1H),4.11(s,3H),3.95-3.56(m,2H),3.41-3.35(m,1H),3.20-3.03(m,2H),2.83-2.49(m,2H),1.98-1.84(m,1H),1.80-1.48(m,6H).
MS-ESI计算值[M+H]+362,实测值362。
实施例55
3-(异喹啉-5-基磺酰基)-3-氮杂二环[3.2.0]庚烷-1-胺
第一步
氮气保护下,-78℃向5,6-二氢-2H-吡喃-2-酮55a(300mg,3.06mmol)和N-苄基-1-甲氧基-N-((三甲基硅基)甲基)甲胺55b(1.09g,4.59mmol)的15mL二氯甲烷溶液中滴加入三氟乙酸(520mg,4.59mmol)的0.6mL二氯甲烷溶液。将所得混合物升温至25℃搅拌4小时,然后用50mL二氯甲烷稀释,并用饱和碳酸钾水溶液(50mL x 2)和饱和氯化钠水溶液(50mL x 1)洗涤,有机相用无水硫酸钠干燥,过滤,减压浓缩。残余物用制备薄层层析板纯化(50%乙酸乙酯/石油醚)得到2-苄基六氢吡喃并[4,3-c]吡咯-4(6H)-酮55c(533
mg,无色油状液体,收率:75%)。
1H NMR(400MHz,CDCl3):δ7.26-7.18(m,5H),4.34-4.33(m,2H),4.18-4.15(m,1H),3.57-3.46(m,2H),2.89-2.85(m,2H),2.77-2.73(m,1H),2.71-2.61(m,1H),2.23-2.19(m,1H),1.94-1.93(m,1H),1.60-1.59(m,1H).
MS-ESI计算值[M+H]+232,实测值232。
第二步
0℃下向2-苄基六氢吡喃并[4,3-c]吡咯-4(6H)-酮55c(9.00g,38.9mmol)的100mL乙醇溶液中通入新鲜制备的溴化氢,并在此温度下反应3小时,然后在25℃下搅拌24小时。待反应结束后将反应液浓缩,粗产物在乙醇中重结晶,得到1-基-4-(2-溴乙基)吡咯烷-3-羧酸乙酯55d(9.0g,白色固体,产率:85%)。
1H NMR(400MHz,CDCl3):δ7.69-7.67(m,2H),7.42-7.41(m,3H),4.41-4.37(m,2H),4.26-4.19(m,2H),3.93-3.88(m,1H),3.72-3.62(m,1H),3.42-3.39(m,2H),3.33-3.29(m,2H),3.05-2.95(m,1H),2.82-2.79(m,1H),1.92-1.87(m,2H),1.33-1.27(m,3H).
MS-ESI计算值[M+H]+340,实测值340。
第三步
氮气保护下1-苄基-4-(2-溴乙基)吡咯烷-3-羧酸乙酯55d(1.00g,2.94mmol)溶于100mL无水四氢呋喃中,于-78℃下滴加六甲基二硅基氨基锂(17.6mL,17.6mmol,1.0M的四氢呋喃溶液)。然后将反应液升温至20℃并反应18小时。待反应结束后将反应液用乙酸乙酯(100mL)稀释,依次用饱和氯化铵水溶液(100mL x3)、饱和氯化钠水溶液(100mL x 1)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残余物用制备薄层层析板纯化(50%乙酸乙酯/石油醚)得到3-苄基-3-氮杂双环[3.2.0]庚烷-1-羧酸乙酯55e(130mg,白色固体,产率:17%)。
1H NMR(400MHz,CDCl3):δ7.41-7.39(m,2H),7.34-7.31(m,2H),7.26-7.23(m,1H),4.17-4.12(m,2H),3.71-3.67(m,2H),2.96-2.94(m,2H),2.85-2.82(m,1H),2.45-2.26(m,3H),2.14-2.09(m,2H),1.77-1.76(m,1H),1.27-1.24(m,3H).
MS-ESI计算值[M+H]+260,实测值260。
第四步
将3-苄基-3-氮杂双环[3.2.0]庚烷-1-羧酸乙酯55e(130mg,0.5mmol)和一水合氢氧化锂(63mg,1.5mmol)溶于7mL四氢呋喃/乙醇/水(v/v/v=4/2/1)的混合溶剂中并加热至40℃搅拌6小时。待反应结束后将反应液冷却到0℃,用6N盐酸将反应液pH调至2,加入30mL饱和氯化钠水溶液,乙酸乙酯和四氢呋喃混合溶剂(v/v=4/1,100mL×3)萃取,有机相依次用水(100mL×2)、饱和氯化钠水溶液洗涤(100mL x 2),用无水硫酸钠干燥,过滤,滤液减压浓缩,得粗品3-苄基-3-氮杂双环[3.2.0]庚烷-1-羧酸55f(30mg,白色固体,产率:95%)。产物不经纯化直接进行下一步反应。
1H NMR(400MHz,DMSO-d6):δ11.01(brs,1H),7.62-7.47(m,5H),4.45(s,2H),2.44-2.42(m,7H),2.19-1.92(m,2H).
MS-ESI计算值[M+H]+232,实测值232。
第五步
将3-苄基-3-氮杂双环[3.2.0]庚烷-1-羧酸55f(50mg,0.22mmol),N,N-二异丙基乙胺(56mg,0.43mmol)和
叠氮磷酸二苯酯(65mg,0.24mmol)溶于2mL叔丁醇中,所得反应液在40℃搅拌2小时,然后升至80℃搅拌12小时。待反应结束后将反应液冷却至室温,用50mL饱和氯化钠水溶液稀释,乙酸乙酯萃取(100mLx 3),有机相依次用水(100mL)和饱和氯化钠水溶液(100mL)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得到3-苄基-3-氮杂双环[3.2.0]庚烷-1-基氨基羧酸叔丁酯55g(20mg,白色固体,产率:31%)。产物不经纯化直接进行下一步反应。
MS-ESI计算值[M+H]+303,实测值303。
第六步
将3-苄基-3-氮杂双环[3.2.0]庚烷-1-基氨基羧酸叔丁酯55g(20mg,0.066mmol)溶于5mL四氢呋喃中,在氮气保护下加入Pd(OH)2/C(5mg,20%wt)。反应液在氢气氛(40Psi)下30℃搅拌48小时直至反应结束。待反应液冷却至室温后,过滤,滤液减压浓缩得到3-氮杂双环[3.2.0]庚烷-1-基氨基羧酸叔丁酯55h(10mg,白色固体,产率:71%)。产物不经纯化直接进行下一步反应。
MS-ESI计算值[M+H]+213,实测值213。
第七步
将3-氮杂双环[3.2.0]庚烷-1-基氨基羧酸叔丁酯55h(20mg,0.094mmol)和异喹啉-5-磺酰氯1c(22mg,0.094mmol)按照实施例1的合成方法得到3-(异喹啉-5-基磺酰基)-3-氮杂二环[3.2.0]庚烷-1-基氨基甲酸55i(15mg,白色固体,产率:40%)。产物不经纯化直接进行下一步反应。
MS-ESI计算值[M+H]+404,实测值404。
第八步
3-(异喹啉-5-基磺酰基)-3-氮杂二环[3.2.0]庚烷-1-基氨基甲酸55i(30mg,0.074mmol)溶于3mL无水二氯甲烷中,0℃下滴加三氟乙酸(0.5mL溶于1mL二氯甲烷的溶液),氮气保护下搅拌3小时直到原料消失。将反应液用二氯甲烷(50mL)稀释,依次用饱和碳酸氢钠水溶液(50mL x 3)、饱和氯化钠水溶液(50mL)洗涤,用无水硫酸钠干燥,过滤,滤液减压浓缩用制备HPLC纯化得产物3-(异喹啉-5-基磺酰基)-3-氮杂二环[3.2.0]庚烷-1-胺55(3mg,白色固体,产率:13%)。1H NMR(400MHz,CD3OD):δ9.49(s,1H),8.76-8.74(m,1H),8.70-8.68(m,1H),8.56-8.53(m,2H),7.96-7.92(m,1H),3.84-3.82(m,1H),3.56-3.54(m,1H),3.15-3.11(m,1H),2.99-2.96(m,1H),2.31-2.21(m,3H),1.65-1.63(m,1H),1.39-1.38(m,1H).
MS-ESI计算值[M+H]+304,实测值304。
实施例56
3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-胺
第一步
将3-氮杂双环[3.1.0]己烷-1-基氨基羧酸叔丁酯56a(50mg,0.25mmol)和异喹啉-5-磺酰氯1c(75mg,0.25mmol,实施例1)按照实施例1的合成方法得到(3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-基)氨基羧酸叔丁酯56b(80mg,黄色油状液体,产率:82%)。
MS-ESI计算值[M+H]+390,实测值390。
第二步
(3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-基)氨基羧酸叔丁酯56b(30mg,0.077mmol)按照实施例1的合成方法得到3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-胺56(7mg,黄色固体,产率:29%)。
1H NMR(400MHz,D2O):δ9.70(s,1H),8.90-8.85(m,1H),8.75-8.70(m,1H),8.65-8.60(m,1H),8.60-8.55(m,1H),8.05-8.00(m,1H),3.82-3.80(m,1H),3.53-3.47(m,1H),3.45-3.36(m,2H),1.95-1.87(m,1H),1.15-1.13(m,1H),0.83-0.77(m,1H).
MS-ESI计算值[M+H]+290,实测值290。
实施例57
N-乙基-3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-胺
第一步
(3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-基)氨基羧酸叔丁酯56b(50mg,0.13mmol)按照实施例11的合成方法得到乙基(3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-基)氨基甲酸叔丁酯57a(33mg,黄色油状液物,产率:62%)。
MS-ESI计算值[M+H]+418,实测值418。
第二步
乙基(3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-基)氨基甲酸叔丁酯57a(33mg,0.079mmol)按照实施例1的合成方法得到N-乙基-3-(异喹啉-5-磺酰基)-3-氮杂二环[3.1.0]己烷-1-胺57(10mg,黄色固体,产率:
40%)。
1H NMR(400MHz,D2O):δ9.59(s,1H),8.71(d,J=6.8Hz,1H),8.63-8.54(m,3H),7.96(d,J=8.0Hz,1H),3.88(d,J=9.2Hz,1H),3.54-3.41(m,3H),3.09-2.99(m,2H),2.05-2.00(m,1H),1.25(t,J=8.4Hz,1H),1.14(t,J=7.2Hz,3H),0.90-0.85(m,1H)。
MS-ESI计算值[M+H]+318,实测值318。
实验例1:体外评价ROCK蛋白激酶抑制活性
实验目的:检测化合物的ROCK蛋白激酶抑制IC50值。
实验材料:
测定緩沖溶液:20mM Hepes(pH 7.5),10mM MgCl2,1mM EGTA,0.02%Brij35,0.02mg/ml BSA,0.1mMNa3VO4,2mM DTT,1%DMSO
实验操作:
将新鲜制备的缓冲溶液中加入ROCK蛋白激酶底物Long S6Kinase substrate peptide,浓度20μM.然后加入1nM ROCK蛋白激酶,均匀搅拌.使用Echo550加入含有待测化合物的系列DMSO稀释液(始于10μM,按3倍系列稀释).室温下预温育20分钟,加入33P-ATP(放射强度10μCi/μL)引发反应,室温反应两小时。然后使用P81离子交换纸(Whatman#3698-915)过滤,用0.75%磷酸洗涤。使用Filter-Binding方法检测放射强度。
化合物的蛋白激酶抑制活性表达为相对空白底物(单纯DMSO)残存的蛋白激酶活性。利用Prism软件包(GraphPad Software,San Diego California,USA)计算IC50值和曲线。
实验结果:
表1蛋白激酶抑制活性测试结果
| 供试品(各实施例所制得的化合物) |
蛋白激酶抑制活性 |
| 实施例1 |
++ |
| 实施例2 |
++ |
| 实施例3 |
++ |
| 实施例4 |
++ |
| 实施例5 |
++ |
| 实施例6 |
++ |
| 实施例7 |
++ |
| 实施例8 |
++ |
| 实施例9 |
+ |
| 实施例10 |
-- |
| 实施例11 |
-- |
| 实施例12/12’ |
++/++ |
| 实施例13 |
+++ |
| 实施例14 |
++ |
| 实施例15 |
++ |
| 实施例16 |
++ |
| 实施例17 |
++ |
| 实施例18 |
++ |
| 实施例19 |
+++ |
| 实施例20 |
++ |
| 实施例21 |
++ |
| 实施例22 |
++ |
| 实施例23 |
++ |
| 实施例24 |
+ |
| 实施例25 |
++ |
| 实施例26 |
++ |
| 实施例27 |
++ |
| 实施例28 |
++ |
| 实施例29 |
++ |
| 实施例30 |
++ |
| 实施例31 |
-- |
| 实施例32 |
-- |
| 实施例33 |
-- |
| 实施例34 |
-- |
| 实施例35 |
-- |
| 实施例36 |
-- |
| 实施例37 |
++ |
| 实施例38 |
+++ |
| 实施例39 |
++ |
| 实施例40 |
+++ |
| 实施例41 |
+++ |
| 实施例42 |
+++ |
| 实施例43 |
+++ |
| 实施例44 |
+++ |
| 实施例45 |
+ |
| 实施例46 |
-- |
| 实施例47 |
+ |
| 实施例48 |
+ |
| 实施例49 |
+ |
| 实施例50 |
+ |
| 实施例51 |
+ |
| 实施例52 |
+ |
| 实施例53 |
+ |
| 实施例54 |
+ |
| 实施例55 |
+ |
| 实施例56 |
++ |
| 实施例57 |
+ |
注:1μM≤+<5μM;0.1μM≤++<1μM;+++<0.1μM;--N/A
结论:本发明化合物具有显著甚至意料不到的蛋白酶抑制活性。











































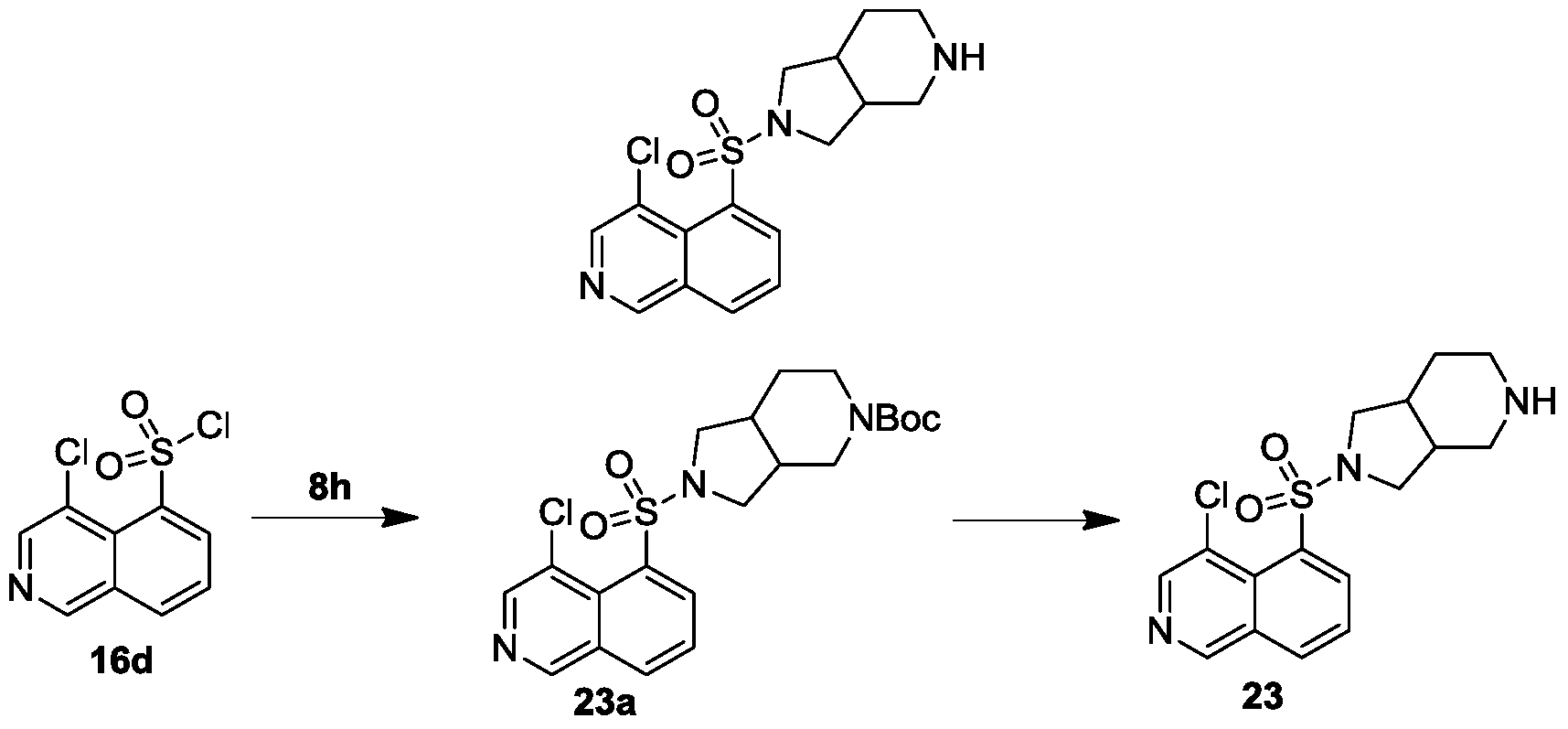










































 其特征是,R1、X分别独立地选自H、F、Cl、Br、I、CN、OH、NH2、C1-3烷基、C1-3烷基氧基、C1-3烷基氨基、N,N-二(C1-3烷基)氨基,所述C1-3烷基任选地被R01所取代;R2选自
其特征是,R1、X分别独立地选自H、F、Cl、Br、I、CN、OH、NH2、C1-3烷基、C1-3烷基氧基、C1-3烷基氨基、N,N-二(C1-3烷基)氨基,所述C1-3烷基任选地被R01所取代;R2选自 W、W’分别独立地选自N(Rw1)、C(Rw2)(Rw3)或单键;L、Z分别独立地选自单键或C(Rz1)(Rz2);P、P’分别选自(CH2)q1;Q、Q’分别选自(CH2)q2;q1、q2分别独立地选自0、1、2、3或4;R3a、R3b、R3、Rw1、Rw2、Rw3、Rz1、Rz2分别独立地选自H、F、Cl、Br、I、CN、OH、NH2、C1-3烷基、C1-3烷基氧基、C1-3烷基氨基、N,N-二(C1-3烷基)氨基,所述C1-3烷基任选地被R01所取代;优选地,R3a、R3b、R3、Rw1、Rw2、Rw3选自卤代、羟代和/或胺代甲基、乙基或丙基;R01选自F、Cl、Br、I、OH、NH2,R01的数目为1、2或3。
W、W’分别独立地选自N(Rw1)、C(Rw2)(Rw3)或单键;L、Z分别独立地选自单键或C(Rz1)(Rz2);P、P’分别选自(CH2)q1;Q、Q’分别选自(CH2)q2;q1、q2分别独立地选自0、1、2、3或4;R3a、R3b、R3、Rw1、Rw2、Rw3、Rz1、Rz2分别独立地选自H、F、Cl、Br、I、CN、OH、NH2、C1-3烷基、C1-3烷基氧基、C1-3烷基氨基、N,N-二(C1-3烷基)氨基,所述C1-3烷基任选地被R01所取代;优选地,R3a、R3b、R3、Rw1、Rw2、Rw3选自卤代、羟代和/或胺代甲基、乙基或丙基;R01选自F、Cl、Br、I、OH、NH2,R01的数目为1、2或3。


 优选地,R4选自NH2、乙氨基;进一步优选地,R2选自
优选地,R4选自NH2、乙氨基;进一步优选地,R2选自
 其中,D、E中一个为单键,另一个为亚甲基;G选自(CH2)g;T选自(CH2)t;g、t分别独立地选自0、1、2、3或4;R3b如权利要求1所定义;
其中,D、E中一个为单键,另一个为亚甲基;G选自(CH2)g;T选自(CH2)t;g、t分别独立地选自0、1、2、3或4;R3b如权利要求1所定义; 优选地,g为1、2、3或4,t为0或1;优选地,R3b选自NH2;进一步优选地,R2选自
优选地,g为1、2、3或4,t为0或1;优选地,R3b选自NH2;进一步优选地,R2选自
 优选地,R2选自
优选地,R2选自


