WO2013107820A1 - Novel fap inhibitors - Google Patents
Novel fap inhibitors Download PDFInfo
- Publication number
- WO2013107820A1 WO2013107820A1 PCT/EP2013/050845 EP2013050845W WO2013107820A1 WO 2013107820 A1 WO2013107820 A1 WO 2013107820A1 EP 2013050845 W EP2013050845 W EP 2013050845W WO 2013107820 A1 WO2013107820 A1 WO 2013107820A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- halo
- fap
- independently selected
- Prior art date
Links
- 0 CCNC(C*)C1(C)C(C)(**)C(C)(C)CCCCCC1C Chemical compound CCNC(C*)C1(C)C(C)(**)C(C)(C)CCCCCC1C 0.000 description 2
- ASBHQXYJHGISBG-XMBNJWBXSA-N C/C=C(\C(\C)=C\C=C)/C(NCC1(CC1)N(CCC1)[C@@H]1C#N)=O Chemical compound C/C=C(\C(\C)=C\C=C)/C(NCC1(CC1)N(CCC1)[C@@H]1C#N)=O ASBHQXYJHGISBG-XMBNJWBXSA-N 0.000 description 1
- VBFCMPQWRIJLLB-INIZCTEOSA-N CN[C@H](CCC1)N1C(CNC(c1c(cc(cc2)OC)c2ncc1)=O)=O Chemical compound CN[C@H](CCC1)N1C(CNC(c1c(cc(cc2)OC)c2ncc1)=O)=O VBFCMPQWRIJLLB-INIZCTEOSA-N 0.000 description 1
- ALYYADQHQGUFHD-NSHDSACASA-N N#C[C@H](CCC1)N1C(CNC(c1ccnc2cccc(Cl)c12)=O)=O Chemical compound N#C[C@H](CCC1)N1C(CNC(c1ccnc2cccc(Cl)c12)=O)=O ALYYADQHQGUFHD-NSHDSACASA-N 0.000 description 1
- UOYPBFHJMPKZTB-LBPRGKRZSA-N N[C@H](CC(C1)(F)F)N1C(CNC(c1ccnc2c1c(Br)ccc2)=O)=O Chemical compound N[C@H](CC(C1)(F)F)N1C(CNC(c1ccnc2c1c(Br)ccc2)=O)=O UOYPBFHJMPKZTB-LBPRGKRZSA-N 0.000 description 1
- QXCOHSRHFCHCHN-UHFFFAOYSA-N OC(c1cc(Cl)ncc1)=O Chemical compound OC(c1cc(Cl)ncc1)=O QXCOHSRHFCHCHN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present invention relates to novel inhibitors having high selectivity and specificity for FAP (fibroblast activation protein). Said inhibitors are useful as a human and/or veterinary medicine, in particular for the treatment and/or prevention of FAP-related disorders such as but not limited to proliferative disorders.
- FAP fibroblast activation protein
- Fibroblast activation protein (FAP, FAP-alpha, seprase, alpha2 antiplasmin converting enzyme) is a Clan SC protease of the prolyl oligopeptidase subfamily S9b, occurring as a cell surface homodimer. FAP has been demonstrated to possess both dipeptidyl peptidase and endopeptidase activity, catalyzed by the same active center. Its expression is associated with activated stromal fibroblasts and pericytes of over 90% of human epithelial tumors examined and with normal or excessive wound healing, e.g. in tissue remodeling sites or during chronic inflammation. The enzyme is generally not expressed in normal adult tissues and in nonmalignant tumors.
- inhibitors belonging to the scaffold type described here have remarkable stability both in aqueous solution and in human plasma and retain activity and selectivity for FAP within the latter media.
- WO2007085895, WO2007005991 , WO2010083570, WO2006125227 and WO0238590 all disclose FAP inhibitors having a general structure closely relating to the compounds of the present invention. However, none of them actually discloses - -
- N-containing aromatic or non-aromatic mono- or bicyclic heterocycle wherein there are exactly 2 ring atoms between the N atom and X.
- said feature is relevant for providing the compounds of the present invention with the FAP activity and selectivity as defined herein.
- FAP FAP's status
- PT-100 dipeptide derived boronic acid talabostat
- Val-boroPro close analogues
- talabostat has been evaluated as a drug in various clinical trials up to phase I I, for the treatment of, i.a. metastatic kidney cancer, chronic lymphocytary leukemia, pancreatic adenocarcinoma and non-small cell lung cancer. While talabostat in several of these trials was able to induce clinical response, questions were raised with regards to the safety profile of the compound, potentially related to its well-known lack of selectivity with respect to other Subfamily S9B proteases. 7 - -
- FAP and diseases involving tissue remodeling and/or chronic inflammation including but not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation, atherosclerotic disease and Chron's disease
- FAP expression was found to be significantly increased on keloid fibroblasts compared to normal skin fibroblasts and inhibition of FAP activity with the albeit unselective (with respect to phylogenetically related dipeptidyl peptidases) irreversible inhibitor Gly-Pro (P) (OPh) 2 was found to lead to a decrease in invasiveness. 10
- FAP expression and activity was also shown to be associated with rheumatoid arthritis and osteoarthritis: FAP-activity on the surface of chondrocytes and elevated expression and activity in cartilage affected by osteoarthritis were demonstrated. FAP was also found to be present in synovial tissue of affected joints, and elevated expression is detected in the murine collagen induced arthritis model. An additional pathway by which FAP could be operating in the pathogenesis and progression of arthritis, has been proposed to imply proteolytic cleavage of alpha2-antiplasmin, ultimately leading to fibrin deposition in the joint.
- FAP was found to be overexpressed in enteric strictures of patients with Chron ' s disease (CD) and the protein was observed to be upregulated on strictured CD myofibroblasts by profibrogenic stimuli, leading the authors of this study to propose FAP as a potential target for the treatment of fibrostenosing CD. 12
- FAP and diseases involving endocrinological disorder including but not limited to disorders of glucose metabolism
- diseases involving blood clotting disorders including but not limited to disorders of glucose metabolism
- the prime aim underlying our effort to establish detailed SAR data for /V-acylated aminoacyl pyrrolidine inhibitors of FAP was to identify compounds with significantly improved (a) chemical stability and (b) selectivity characteristics when compared to known FAP inhibitors, while retaining high affinity for the target enzyme.
- SAR Structure-Activity Relationship
- FAP-inhibitors of formula I exhibit good chemical stability and high selectivity for FAP, rendering them very suitable for the preparation of a medicine for the treatment of various FAP-related disorders.
- our invention has the potential to deliver compounds with high solubility and low LogD-values, a feature that is far from evident for dipeptide-derived compounds lacking a basic amino terminus and that is accounted for by the presence of heteroatoms introduced at specific positions of the P3 substituent.
- the present invention provides a compound of Formula I or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof, - -
- Ri and R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- Ci -6 alkyl, S-Ci -6 alkyl;
- -C C-S(0) 2 aryl, -C0 2 H, -S0 3 H, -S0 2 NH 2 ,-P0 3 H 2 , and 5-tetrazolyl;
- R 4 is selected from the group comprising -H, -C 1 -6 alkyl, -0-C 1 -6 alkyl, -S-C 1-6 alkyl, - ⁇ , and -C ⁇ 6aralkyl; each of said -C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - Ci -6 alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl, -NR 8 R 9 , -OR 12 -Het 2 and -Ar 2 ; each of said Ci -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 8 , R 9 and R 12 are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -0-d_ 6 alkyl, -S-Ci -6 alkyl, and -Ar 3
- Rio, Rii , Ri3 and R 14 are each independently selected from the group comprising -H, -OH, -halo, -
- ⁇ , Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said ⁇ , Ar 2 and Ar 3 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , - d-ealkyl, -0-Ci -6 alkyl, -S-Ci -6 alkyl;
- Het 2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said Het 2 being optionally substituted with from 1 to 3 substituents selected from -NR 13 R 14 , -C 1 -6 alkyl, -0-C 1-6 alkyl, -S-C 1 -6 alkyl;
- bicyclic heterocycle wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom - -
- the present invention provides a compound according to form ula I, wherein
- F and R 2 are each independently selected from the group comprising -H , and -halo;
- Fl 3 is -CN , or -B(OH) 2
- R 4 is selected from the group comprising -H or -C ⁇ alkyl ; each of said -C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , ;
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH , -oxo, -halo, -
- R 8 and R 9 are each independently selected from the group comprising -H and -Ar 3
- Ar 2 and Ar 3 are each independently -phenyl optionally substituted with from 1 to 3 -O-C ⁇ alkyl ;
- N represents a 5 to 1 0- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
- the present invention provides a compound according to formula I, wherein
- Ri and R 2 are each independently selected from the group comprising -H , and -F;
- R 3 is -CN ,and -B(OH) 2
- R 4 is -H ;
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -oxo, -halo, -Ci_ 6alkyl, and -0-CF 3 ;
- N represents a 5 to 1 0- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
- the current invention further provides a compound of Form ula I I or a stereoisomer, tautomer, racem ic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof, - -
- R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- C ⁇ alkyl, S-C ⁇ alkyl;
- R 4 is selected from the group comprising -H, -C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl, - ⁇ , and -C ⁇ 6aralkyl; each of said -C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - Gi_ 6 alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 8 and R 9 , R 10 and R are each independently selected from the group comprising -H, -OH, -halo, -C ⁇ alkyl, -O-C ⁇ alkyl, and -S-C ⁇ alkyl;
- R 10 and Rn are each independently selected from the group comprising -H, -OH, -halo, -C ⁇ alkyl, - O-d-ealkyl, -S-C ⁇ alkyl, and -Ar 3;
- ⁇ , Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said An , and Ar 2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , - C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl;
- n 0, 1 , 2, or 3
- N and S represents a 5 to 6- membered N-containing aromatic or non-aromatic monocyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S.
- the current invention provides a compound according to formula II, wherein
- RT and R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- d-ealkyl, S-C ⁇ alkyl;
- R 3 is selected from the group comprising -H, -CN, and -B(OH) 2
- R 4 is -H ; - -
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C 1-6 alkyl, -0-C 1 -6 alkyl, -S-C 1 -6 alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 8 , R 9 , R 10 and R are each independently selected from the group comprising -H, -OH, -halo, -C ⁇
- Ar 2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar 2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , -C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl;
- n 0, 1 , 2, or 3
- R 5 and R 6 are each -H ;
- R 7 is selected from the group comprising - H, -OH, -oxo, -halo, -C 1 -6 alkyl, -0-C 1-6 alkyl, -S-C 1-6 alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo; and
- R 7 is attached to position 2 or 3, in particular position 2, as represented in
- the present invention provides a compound of formula I lia, ll lb or ll lc or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof, - -
- Ri and R 2 are each independently selected from the group comprising -H, OH, -halo, C 1-6 alkyl, -0- C 1-6 alkyl, S-C 1-6 alkyl;
- R 4 is selected from the group comprising -H, -d- 6 alkyl, -O-d-ealkyl, -S-d-ealkyl, -An, and -d- 6aralkyl; each of said -Ci -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H, -OH, -oxo, -halo, - C 1-6 alkyl, -0-C 1-6 alkyl, -S-C 1-6 alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C 1-6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 8 and R 9 are each independently selected from the group comprising -H, -OH, -halo, -d-ealkyl, -
- Rio and Rn are each independently selected from the group comprising -H, -OH, -halo, -d-ealkyl, - O-d-ealkyl, and -S-Ci -6 alkyl;
- Ar 1t Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said ⁇ , and Ar 2 being - -
- n 0, 1 , 2, or 3
- N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from O, N and S.
- the present invention provides a compound according to formula I II, wherein
- R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- d-ealkyl, S-C ⁇ alkyl;
- R 3 is selected from the group comprising -H, -CN, and -B(OH) 2 ;
- R 4 is -H ;
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 8 , R 9 , Rio and R are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -O-C ⁇ alkyl, and -S-C ⁇ alkyl;
- Ar 2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar 2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , -C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl;
- n 0, 1 , 2, or 3
- R 5 is preferably attached to position 2 or 3, in particular position 3, as represented in
- the current invention further provides a compound of formula I, II, Il ia, ll lb or l llc as defined above, as well as pharmaceutical compositions comprising said compounds, for use as a human or veterinary medicine.
- the present invention provides the use of a compound as defined above, as well as pharmaceutical compositions comprising said compounds, in the manufacture of a medicament for the prevention and/or treatment of a FAP-related disorder.
- FAP-related disorders can include proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal - -
- FAP-related disorders that are envisaged here, includes diseases characterised by tissue remodeling and/or chronic inflammation. These include but are not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation, atherosclerotic disease and Chron's disease. Furthermore, FAP related disorders involving endocrinological dysfunction (including but not limited to disorders of glucose metabolism) and diseases involving blood clotting disorders are part of this list.
- the present invention provides the use of a compound as defined above, as well as pharmaceutical compositions comprising said compounds, for the prevention and/or treatment of a FAP-related disorder.
- FAP-related disorders can include proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma.
- the list of FAP-related disorders that are envisaged here includes diseases characterised by tissue remodeling and/or chronic inflammation.
- FAP related disorders involving endocrinological dysfunction including but not limited to disorders of glucose metabolism
- diseases involving blood clotting disorders are part of this list.
- the present invention provides a method for the prevention and/or treatment of a FAP-related disorder.
- FAP-related disorders can include proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma.
- proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma.
- the list of FAP-related disorders that are envisaged here includes diseases characterised by tissue remodeling and/or chronic inflammation.
- fibrotic disease include but are not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation, atherosclerotic disease and Chron's disease.
- FAP related disorders involving endocrinological dysfunction including but not limited to disorders of glucose metabolism
- diseases involving blood clotting disorders are part of this list.
- Inhibitors 3,4 and 5 shared substantial FAP affinity (IC 50 ⁇ 5 ⁇ ), decoupled from PREP binding potential.
- the common structural feature that can be held accountable for this profile, is an (azaheterocyclyl)ac(et-)yl group as the scaffold substituent.
- PREP's S1 pocket seems even more limited than for FAP: only in the case of the fluorinated compounds, introduction of a 4-substituent does not completely delete enzyme affinity. Taking into account its positive effect on FAP-inhibitory activity, (di-)fluorination of the 4-position of the pyrrolidine ring could be regarded upon as a viable strategy to improve FAP-selectivity of promising inhibitors.
- Table 2 Affinity/selectivity data for selected A-acyl-glycyl-(2-cyano)pyrrolidines.
- Essential to both affinity and selectivity for FAP in this scaffold type is the presence of at least one nitrogen hetero-atom that is part of a cyclic system in P3 of these compounds. Equally essential for both parameters is the relative 1 ,4-positioning of (1 ) the N-atom and (2) the fragment that links the P3 cyclic framework to the P2 amino function. All of these aspects of the invention will be demonstrated using the enzymatic evaluation results both of compounds that correspond and of - -
- the present invention provides compounds of Formula I, including a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof
- Ri and R 2 are each independently selected from the group comprising -H , OH , -halo, C ⁇ alkyl, -0- C ⁇ alkyl, S-C ⁇ alkyl ;
- R 4 is selected from the group comprising -H , -C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl , -An , and -Ci_ 6aralkyl ; each of said -C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , -halo
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH , -oxo, -halo, - C 1-6 alkyl, -0-C 1 -6 alkyl, -S-C 1 -6 alkyl, -N R 8 R 9 , -OR 12 -Het 2 and -Ar 2 ; each of said C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , -halo
- R 8 , R 9 and R 12 are each independently selected from the group comprising -H , -OH , -halo, -Ci_ 6alkyl, -0-Ci -6 alkyl, -S-C ⁇ alkyl , and -Ar 3
- R 10 , Ri i , Ri3 and R 14 are each independently selected from the group comprising -H , -OH , -halo, - d_ 6 alkyl, -0-Ci -6 alkyl, and -S-Ci -6 alkyl ;
- ⁇ , Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said ⁇ , Ar 2 and Ar 3 being optionally and independently substituted with from 1 to 3 substituents selected from -N R 10 Rn , - C 1-6 alkyl, -0-C 1-6 alkyl, -S-C 1 -6 alkyl ;
- Het 2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S ; said Het 2 being optionally substituted with from 1 to 3 substituents selected from -N R 13 R 14 , -C ⁇ alkyl, -0-Ci -6 alkyl, -S-Ci -6 alkyl ;
- n 0, 1 , 2, or 3 represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
- said embodiment discloses a compound of Formula X, including a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof
- Ri and R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- d_ 6 alkyl, S-C ⁇ alkyl;
- -C C-S(0) 2 aryl, -C0 2 H, -S0 3 H, -S0 2 NH 2 ,-P0 3 H 2 , and 5-tetrazolyl;
- R 4 is selected from the group comprising -H, -C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl, - ⁇ , and -C ⁇
- each of said -C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, -
- R 8 , R 9 and R 12 are each independently selected from the group comprising -H, -OH, -halo, -Ci_
- Rio, Ri i , Ri3 and R 14 are each independently selected from the group comprising -H, -OH, -halo, -
- ⁇ , Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said ⁇ , Ar 2 and Ar 3 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , - d-ealkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl; - -
- Het 2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said Het 2 being optionally substituted with from 1 to 3 substituents selected from -NR 13 R 14 , -C 1 -6 alkyl, -0-C 1-6 alkyl, -S-C 1 -6 alkyl;
- N-containing aromatic or non-aromatic mono- or bicyclic heterocycle wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S;
- the present invention also provides a compound of formula (I), a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof
- Ri and R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- d_ 6 alkyl, S-Ci -6 alkyl;
- R 4 is selected from the group comprising -H, -C 1 -6 alkyl, -0-C 1 -6 alkyl, -S-C 1-6 alkyl, - ⁇ , and -C ⁇ 6aralkyl; each of said -C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - Ci -6 alkyl, -O-C ⁇ alkyl, -S-Ci -6 alkyl, -NR 8 R 9 , -OR 12 -Het 2 and -Ar 2 ; each of said C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 8 , R 9 and R 12 are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -0-C 1 -6 alkyl, -S-C 1 -6 alkyl, and -Ar 3
- R 10 , Rii , Ri3 and R 14 are each independently selected from the group comprising -H, -OH, -halo, - C 1-6 alkyl, -0-C 1-6 alkyl, and -S-C 1-6 alkyl;
- ⁇ , Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said ⁇ , Ar 2 and Ar 3 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , - Ci -6 alkyl, -0-Ci -6 alkyl, -S-Ci -6 alkyl; - -
- Het 2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said Het 2 being optionally substituted with from 1 to 3 substituents selected from -NR 13 R 14 , -C 1 -6 alkyl, -0-C 1-6 alkyl, -S-C 1 -6 alkyl;
- N-containing aromatic or non-aromatic mono- or bicyclic heterocycle wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
- the present invention provides a compound of formula (I), a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof
- Ri and R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- Ci -6 alkyl, S-d_ 6 alkyl;
- -C C-S(0) 2 aryl, -C0 2 H, -S0 3 H, -S0 2 NH 2 ,-P0 3 H 2 , and 5-tetrazolyl;
- R 4 is selected from the group comprising -H, -C 1 -6 alkyl, -0-C 1 -6 alkyl, -S-C 1-6 alkyl, - ⁇ , and -C ⁇
- each of said -C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, -
- R 8 , R 9 and R 12 are each independently selected from the group comprising -H, -OH, -halo, -C ⁇ 6alkyl, -0-C 1 -6 alkyl, -S-C 1 -6 alkyl, and -Ar 3
- Rio. Rii , Ri3 and R 14 are each independently selected from the group comprising -H, -OH, -halo, -
- Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said ⁇ , Ar 2 and Ar 3 being - -
- Het 2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said Het 2 being optionally substituted with from 1 to 3 substituents selected from -NR 13 R 14 , -C 1 -6 alkyl, -0-C 1-6 alkyl, -S-C 1 -6 alkyl;
- n 3 represents a 6 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
- alkyl by itself or as part of another substituent refers to a fully saturated hydrocarbon of Formula C x H 2x+1 wherein x is a number greater than or equal to 1 .
- alkyl groups of this invention comprise from 1 to 6 carbon atoms.
- Alkyl groups may be linear or branched and may be substituted as indicated herein.
- a subscript is used herein following a carbon atom , the subscript refers to the number of carbon atoms that the named group may contain.
- C 1-4 alkyl means an alkyl of one to four carbon atoms.
- alkyl groups are methyl, ethyl, n-propyl, i-propyl, butyl, and its isomers (e.g. n-butyl, i-butyl and t-butyl) ; pentyl and its isomers, hexyl and its isomers, heptyl and its isomers, octyl and its isomers, nonyl and its isomers; decyl and its isomers.
- Ci -6 alkyl includes all linear, branched, or cyclic alkyl groups with between 1 and 6 carbon atoms, and thus includes methyl, ethyl, n-propyl, i-propyl, butyl and its isomers (e.g. n-butyl, i-butyl and t-butyl) ; pentyl and its isomers, hexyl and its isomers, cyclopentyl, 2-, 3-, or 4-methylcyclopentyl, cyclopentylmethylene, and cyclohexyl.
- aralkyl as a group of part of a group refers to an alkyl moiety, as detailed above, wherein at least one -H atom is replaced by an aryl moeity.
- aryl as a group of part of a group is generic for a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said aryl further being optionally substituted with from 1 to 3 substituents as defined herein.
- halo or halogen as a group or part of a group is generic for fluoro, chloro, bromo or iodo. Unless a context dictates otherwise, asterisks are used herein to indicate the point at which a mono- or bivalent radical depicted is connected to the structure to which it relates and of which the - -
- radical forms part.
- the aforementioned graphical representation has no bearing as to the actual orientation of said groups in the remainder of the molecule.
- the term 'compounds of the invention' or a similar term is meant to include the compounds of general Formula I or any subgroup thereof. This term also refers to a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof.
- a compound means one compound or more than one compound.
- the present invention provides a compound of formula I, or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt hydrate or solvate thereof, wherein
- F and R 2 are each independently selected from the group comprising -H, and -halo;
- Fl 3 is -CN, or -B(OH) 2
- R 4 is selected from the group comprising -H or -C ⁇ alkyl ; each of said -C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH,;
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C ⁇ alkyl, -O-C ⁇ alkyl, Ar 2 and -NR 8 R 9 ; each of said C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
- R 8 and R 9 are each independently selected from the group comprising -H and -Ar 3
- Ar 2 and Ar 3 are each independently -phenyl optionally substituted with from 1 to 3 -O-C ⁇ alkyl;
- N represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from O, N and S; and X represents a C atom
- the present invention provides a compound of formula I, or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt hydrate or solvate thereof, wherein
- RT and R 2 are each independently selected from the group comprising -H, and -F;
- R 3 is -CN,and -B(OH) 2
- R 4 is -H ; - -
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H, -oxo, -halo, -Ci_
- N represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
- a 10-membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S;
- R 5 , R 6 and R 7 is selected from the group comprising -H, -OH, -oxo, -halo, -C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl, -NR 8 R 9 , -OR 12 -Het 2 and -Ar 2 ; each of said C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
- R 5 , R 6 or R 7 is preferably attached at position 3, 6, 7 or 8 according to the following formula:
- I a particular embodiment represents a 9 to 10-membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S; such as for example:
- N-containing aromatic or non-aromatic monocyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S selected from the list comprising:
- the current invention provides a compound according to formula II, or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,
- R and R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- C ⁇ alkyl, S-C ⁇ alkyl;
- -C C-S(0) 2 aryl, -C0 2 H, -S0 3 H, -S0 2 NH 2 ,-P0 3 H 2 , and 5-tetrazolyl
- R 4 is selected from the group comprising -H, -C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl, -An , and -Ci_
- each of said -Ci -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, -
- R 8 and R 9 , R 10 and R are each independently selected from the group comprising -H, -OH, -halo,
- -d_ 6 alkyl -0-Ci -6 alkyl, and -S-Ci -6 alkyl
- Rio and Rn are each independently selected from the group comprising -H, -OH, -halo, -C ⁇ alkyl, -
- ⁇ , Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said ⁇ , and Ar 2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , - d_ 6 alkyl, -0-Ci -6 alkyl, -S-Ci -6 alkyl;
- n 0, 1 , 2, or 3
- n 1 or 2
- ⁇ 3 ⁇ 4 m ⁇ * represents a 5 to 6- membered N-containing aromatic or non-aromatic monocyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S.
- the present invention provides a compound according to formula I I, wherein
- Ri and R 2 are each independently selected from the group comprising -H, OH, -halo, C ⁇ alkyl, -0- C 1-6 alkyl, S-C 1-6 alkyl;
- R 3 is selected from the group comprising -H, -CN, and -B(OH) 2
- R 4 is -H
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H, -OH, -oxo, -halo, - Ci -6 alkyl, -O-C ⁇ alkyl, -S-Ci -6 alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C ⁇ alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 8 , R 9 , Rio and R are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -0-C 1 -6 alkyl, and -S-C 1-6 alkyl;
- Ar 2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar 2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , -C 1-6 alkyl, -0-C 1-6 alkyl, -S-C 1 -6 alkyl;
- n 0, 1 , 2, or 3
- n 1 or 2
- HN 3 ⁇ 4 m ⁇ * is a 5- or 6-membered aromatic or non-aromatic monocylic heterocycle optionally nd S; selected from the list comprising
- R 5 and R 6 are each -H ;
- R 7 is selected from the group comprising -H, -OH, -oxo, -halo, -C 1-6 alkyl, -0-C 1 -6 alkyl, -S-C 1 -6 alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo; and R 7 is attached to position 2 or 3, in particular position 2, as represented in
- the present invention provides a compound of formula I lia, l llb or ll lc or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,
- R-i and R 2 are each independently selected from the group comprising -H, OH , -halo, C 1 -6 alkyl, -0- C 1-6 alkyl, S-C 1-6 alkyl;
- R 4 is selected from the group comprising -H, -d- 6 alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl, -An , and -Ci_ 6aralkyl; each of said -Ci -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C 1-6 alkyl, -0-C 1 -6 alkyl, -S-C 1 -6 alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- R 8 and R 9 are each independently selected from the group comprising -H, -OH, -halo, -C 1 -6 alkyl, -
- Rio and Rn are each independently selected from the group comprising -H, -OH, -halo, -C ⁇ alkyl, - 0-Ci -6 alkyl, and -S-Ci -6 alkyl ;
- Ar 1 t Ar 2 and Ar 3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said An , and Ar 2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , - C 1-6 alkyl, -0-C 1-6 alkyl, -S-C 1 -6 alkyl;
- n O, 1 , 2, or 3
- N and S represent a 9 to 10- membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S.
- the current invention provides a compound according to formula II I, wherein
- Ri and R 2 are each independently selected from the group comprising -H, OH , -halo, C ⁇ alkyl, -0- Ci -6 alkyl, S-Ci -6 alkyl;
- R 3 is selected from the group comprising -H, -CN, and -B(OH) 2 ;
- R 4 is -H ;
- R 5 , R 6 and R 7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C 1-6 alkyl, -0-C 1 -6 alkyl, -S-C 1 -6 alkyl, -NR 8 R 9 , and -Ar 2 ; each of said C 1 -6 alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo - -
- R 8 , R 9 , Rio and R are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -O-C ⁇ alkyl, and -S-C ⁇ alkyl;
- Ar 2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar 2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR 10 Rn , -C ⁇ alkyl, -O-C ⁇ alkyl, -S-C ⁇ alkyl;
- n 0, 1 , 2, or 3
- R 5 is attached to position 2 or 3, in particular position 3, as represented in:
- R 6 is -H and R 7 is attached to position 7 as represented in: - -
- R 6 and R 7 may be present at any suitable position, i.e. position 6, 7, 8 or 9 as represented in:
- this invention provides a compound according to this invention for use as a human or veterinary medicine.
- This invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound according to this invention; said composition being suitable for use as a human or veterinary medicine.
- the compounds of the invention may be used as a free acid or base, and/or in the form of a pharmaceutically acceptable acid-addition and/or base-addition salt (e.g. obtained with non-toxic organic or inorganic acid or base), in the form of a hydrate, solvate and/or complex, and/or in the form or a pro-drug or pre-drug, such as an ester.
- a pharmaceutically acceptable acid-addition and/or base-addition salt e.g. obtained with non-toxic organic or inorganic acid or base
- solvate includes any combination which may be formed by a compound of this invention with a suitable inorganic solvent (e.g. hydrates) or organic solvent, such as but not limited to alcohols, ketones, esters and the like.
- suitable inorganic solvent e.g. hydrates
- organic solvent such as but not limited to alcohols, ketones, esters and the like.
- the pharmaceutically acceptable salts of the compounds according to the invention include the conventional non-toxic salts or the quaternary ammonium salts which are formed, e.g., from inorganic or organic acids or bases.
- acid addition salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2-naphthalene-sulfonate, - -
- Base salts include ammonium salts, alkali metal salts such as sodium and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases such as dicyclohexylamine salts, N-methyl-D-glucamine, and salts with amino acids such as arginine, lysine, and so forth.
- the basic nitrogen-containing groups may be quaternized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl ; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl-bromides and others.
- Other pharmaceutically acceptable salts include the sulfate salt ethanolate and sulfate salts.
- the compounds of this invention may be formulated as a pharmaceutical preparation or pharmaceutical composition comprising at least one compound of the invention and at least one pharmaceutically acceptable carrier, diluent or excipient and/or adjuvant.
- such a formulation may be in a form suitable for oral administration, parenteral administration (such as by intravenous, intramuscular or subcutaneous injection or intravenous infusion), for topical administration (including ocular), for administration by inhalation, by a skin patch, by an implant, by a suppository, etc..
- parenteral administration such as by intravenous, intramuscular or subcutaneous injection or intravenous infusion
- topical administration including ocular
- inhalation by a skin patch, by an implant, by a suppository, etc.
- Such suitable administration forms which may be solid, semi-solid or liquid, depending on the manner of administration - as well as methods and carriers, diluents and excipients for use in the preparation thereof, will be clear to the skilled person; reference is again made to for instance US-A-6,372,778, US-A-6,369,086, US-A- 6,369,087 and US-A-6,372,733, as well as to the standard handbooks, such as the latest edition of Remington's Pharmaceutical Sciences.
- Such preparations include tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosols, ointments, creams, lotions, soft and hard gelatin capsules, suppositories, eye drops, sterile injectable solutions and sterile packaged powders (which are usually reconstituted prior to use) for administration as a bolus and/or for continuous administration, which may be formulated with carriers, excipients, and diluents that are suitable per se for such formulations, such as lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, polyethylene glycol, cellulose, (sterile) water, methylcellulose, methyl- and propy
- the formulations can optionally contain other pharmaceutically active substances (which may or may not lead to a synergistic effect with the compounds of the invention) and other substances that are commonly used in pharmaceutical formulations, such as lubricating agents, wetting agents, emulsifying and suspending agents, dispersing agents, desintegrants, bulking agents, fillers, - -
- compositions may also be formulated so as to provide rapid, sustained or delayed release of the active compound(s) contained therein, for example using liposomes or hydrophilic polymeric matrices based on natural gels or synthetic polymers.
- liposomes or hydrophilic polymeric matrices based on natural gels or synthetic polymers.
- co-solvents such as alcohols may improve the solubility and/or the stability of the compounds.
- addition of salts of the compounds of the invention can be more suitable due to their increased water solubility.
- the preparations may be prepared in a manner known per se, which usually involves mixing at least one compound according to the invention with the one or more pharmaceutically acceptable carriers, and, if desired, in combination with other pharmaceutical active compounds, when necessary under aseptic conditions.
- the pharmaceutical preparations of the invention are preferably in a unit dosage form, and may be suitably packaged, for example in a box, blister, vial, bottle, sachet, ampoule or in any other suitable single-dose or multi-dose holder or container (which may be properly labeled) ; optionally with one or more leaflets containing product information and/or instructions for use.
- unit dosages will contain between 1 and 1000 mg, and usually between 5 and 500 mg, of the at least one compound of the invention, e.g. about 10, 25, 50, 100, 200, 300 or 400 mg per unit dosage.
- the compounds can be administered by a variety of routes including the oral, rectal, ocular, transdermal, subcutaneous, intravenous, intramuscular or intranasal routes, depending mainly on the specific preparation used and the condition to be treated or prevented, and with oral and intravenous administration usually being preferred.
- the at least one compound of the invention will generally be administered in an "effective amount", by which is meant any amount of a compound of the Formula I or I I, upon suitable administration, is sufficient to achieve the desired therapeutic or prophylactic effect in the individual to which it is administered.
- such an effective amount will usually be between 0.01 to 1000 mg per kilogram body weight day of the patient per day, more often between 0.1 and 500 mg, such as between 1 and 250 mg, for example about 5, 10, 20, 50, 100, 150, 200 or 250 mg, per kilogram body weight day of the patient per day, which may be administered as a single daily dose, divided over one or more daily doses, or essentially continuously, e.g. using a drip infusion.
- the amount(s) to be administered, the route of administration and the further treatment regimen may be determined by the treating clinician, - -
- said pharmaceutical composition can be administered separately at different times during the course of therapy or concurrently in divided or single combination forms.
- the present invention is therefore to be understood as embracing all such regimes of simultaneous or alternating treatment and the term "administering" is to be interpreted accordingly.
- compositions of the present invention can be mixed with suitable additives, such as excipients, stabilizers, or inert diluents, and brought by means of the customary methods into the suitable administration forms, such as tablets, coated tablets, hard capsules, aqueous, alcoholic, or oily solutions.
- suitable inert carriers are gum arabic, magnesia, magnesium carbonate, potassium phosphate, lactose, glucose, or starch, in particular, corn starch.
- the preparation can be carried out both as dry and as moist granules.
- Suitable oily excipients or solvents are vegetable or animal oils, such as sunflower oil or cod liver oil.
- Suitable solvents for aqueous or alcoholic solutions are water, ethanol, sugar solutions, or mixtures thereof.
- Polyethylene glycols and polypropylene glycols are also useful as further auxiliaries for other administration forms.
- these compositions may contain microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants known in the art.
- compositions When administered by nasal aerosol or inhalation, these compositions may be prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
- Suitable pharmaceutical formulations for administration in the form of aerosols or sprays are, for example, solutions, suspensions or emulsions of the compounds of the invention or their physiologically tolerable salts in a pharmaceutically acceptable solvent, such as ethanol or water, or a mixture of such solvents.
- the formulation can also additionally contain other pharmaceutical auxiliaries such as surfactants, emulsifiers and stabilizers as well as a propellant.
- auxiliaries such as surfactants, emulsifiers and stabilizers as well as a propellant.
- the compound according to the invention if desired with the substances customary therefore such as solubilizers, emulsifiers or further auxiliaries are brought into solution, suspension, or emulsion.
- the compounds of the invention can also be lyophilized and the lyophilizates obtained used, for example, for the production of injection or infusion preparations.
- Suitable solvents are, for example, water, physiological saline solution or alcohols, e.g. ethanol, propanol, glycerol, in addition also sugar solutions such as glucose or mannitol solutions, or alternatively mixtures of the various solvents mentioned.
- Suitable non-toxic, parenterally-acceptable diluents or solvents such as mannitol, 1 ,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- these formulations When rectally administered in the form of suppositories, these formulations may be prepared by mixing the compounds according to the invention with a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquefy and/or dissolve in the rectal cavity to release the drug.
- a suitable non-irritating excipient such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquefy and/or dissolve in the rectal cavity to release the drug.
- the compounds of the present invention are useful in human or veterinary medicine, in particular for use as FAP (fibroblast activation protein) inhibitors.
- FAP inhibition as used in the context of this invention is to include inhibition of endopeptidase activity and/or exopeptidase activity of FAP.
- the present invention further provides the use of a compound as defined hereinbefore or the use of a composition comprising said compound, as a human or veterinary medicine, in particular for the prevention and/or treatment of FAP-related disorders.
- Said inhibition may be effected in vitro and/or in vivo, and when effected in vivo, is preferably effected in a selective manner, as defined herein.
- Particular reference is given to compounds of Formula I or any subgroup thereof which are at least 100x more selective for FAP compared to DPPIV, DPP9 and DPP2; in particular at least 1000x more selective for FAP compared to DPPIV, DPP9 and DPP2 .
- compounds of Formula I or any subgroup thereof which are at least 1 0x more selective for FAP compared to PREP, in particular at least 20 to 50x more selective for FAP compared to PREP.
- FAP-related disorder means any disease or other deleterious condition in which FAP is known to play a role.
- FAP-related disorder also means those diseases - -
- FAP-related disorders can include proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma.
- proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma.
- the list of FAP-related disorders that are envisaged here includes diseases characterised by tissue remodeling and/or chronic inflammation.
- the invention also provides methods for the prevention and/or treatment of a FAP-related disorder; said method comprising administering to a subject in need thereof a compound according to this invention, or a composition comprising said compound.
- the compounds of the present invention can be prepared according to the reaction schemes provided in the examples hereinafter, but those skilled in the art will appreciate that these are only illustrative for the invention and that the compounds of this invention can be prepared by any of several standard synthetic processes commonly used by those skilled in the art of organic chemistry.
- the invention will now be illustrated by means of the following synthetic and biological examples, which do not limit the scope of the invention in any way.
- Boc-am inocarboxylate (A) with compound (B) can be done using standard techniques for peptide coupling, known to a person skilled in the art, to afford compound (C) .
- This is either pyrrolidine or a pyrrolidine derivative.
- an additional dehydration step e.g . using trifluoroacetic anhydride and pyridine, vide infra
- the chosen protecting group of (C), including the Boc- and Z-group can be deprotected using a suitable acid or other deprotecting agent, known to a person skilled in the art, to generate compound (D) .
- reaction of compound (D) with compound (E) can be done using standard peptide coupling procedures, or using a corresponding acyl halide or active ester of (E) which is made in situ or in a separate reaction using procedures known to a person skilled in the art.
- compounds of form ula (I) can also be prepared by converting one or more groups F3 ⁇ 4, R3, R 4 , R5, Re or R 7 of another compound of formula (I) obtained as mentioned above, into a desired substituent.
- the selected conversion method will depend on the kind of substituents desired. For example, it can be carried out by the following methodologies (Method a to Method e)..
- a compound of type (I) in which R 5 , R 6 or R 7 in formula (I) is a hydrogen atom can be prepared by elim inating a protective group from a corresponding Compound (I) in which R 5 , R 6 or R 7 is a protective group. Removal of the protective group can be carried out by conventional methodology, known to a person skilled in the art (e.g. acid treatment, base treatment, catalytic reduction, etc.) . - -
- a compound of type (I) in which R 3 is a boronic acid function can be prepared by removing a protective group from a corresponding compound of formula (I) in which R 3 is a protected boronate (e.g. a dialkyl ester, a pinanediol diester) 15
- Method c A compound of type (I) in which R 3 is a carbonitrile function, can be prepared by dehydration of a corresponding compound of formula (I) in which R 3 is a carboxamide function.
- a compound of type (I) in which R 4 is a proteinogenic or non-proteinogenic amino acid side chain can be obtained by eliminating a protective group from a corresponding Compound (I) in which R 4 is a protected proteinogenic or non-proteinogenic amino acid side chain. Removal of the protective group can be carried out by conventional methodology, known to a person skilled in the art (e.g. acid treatment, base treatment, catalytic reduction, etc.).
- a compound of type (I) can be made by reacting compound (II) with a suitable protected compound (V), using standard peptide coupling procedures, or using a corresponding acyl halide or active ester of (I I) which is made in situ or in a separate reaction using procedures known to a person skilled in the art.
- reaction with (IV) using standard peptide coupling procedures, or using a corresponding acyl halide or active ester of (I I) which is made in situ or in a separate reaction using procedures known to a person skilled in the art.
- (S)-4,4-difluoropyrrolidine-2-carboxamide hydrochloride can be made as described in general procedure A. This is starting from (S)-1 -iert-butyl 2-methyl 4-oxopyrrolidine-1 ,2- dicarboxylate which is fluorinated with diethylaminosulfur trifluoride (DAST) and subsequently hydrolyzed with potassium hydroxide. The amide is made from this acid, reacting it with dicyclohexylcarbodiimide and /V-hydroxysuccinimide, subsequent removal of the Boc group yields the hydrochloride salt (B).
- DAST diethylaminosulfur trifluoride
- prolinamide was bought from commercial suppliers (Fluorochem).
- pinanediol ester of pyrrolidineboronic acid can be made as described in reference
- (2S,4S)-4-fluoropyrrolidine-2-carboxamide can be prepared as described analogously to (S)-4,4- difluoropyrrolidine-2-carboxamide, starting from 1 -ferf-butyl 4-hydroxypyrrolidine-1 ,2 dicarboxylate. - -
- the quinoline-4-carboxylic acid [E] can be prepared using the classical Sandmeyer isatin synthesis followed by the Pfitzinger reacton.
- the aniline was reacted with chloral hydrate and hydroxylamine hydrochloride to afford the substituted isonitrosoacetanilide which was cyclized using a suitable acid catalyst to afford the isatin as described in.
- the isatin can be bought from commercial sources.
- the isatin can then be converted into the corresponding substituted quinoline- 4-carboxylic acid using the Pfitzinger reaction as in reference 17.
- the Pfitzinger reaction yielded a quinoline-2,4-dicarboxylic acid which was decarboxylated to the quinoline-4- carboxylic acid by reacting it in water for 2h using a pressured tube at 200 °C. - -
- the 2-aminopyridine-4-carboxylic acids can be prepared by nucleophilic aromatic substitution of 2- chloropyridine-4-carboxylic acid with the corresponding primary or secondary amines. rboxylate derivatives.
- the 2-(hetero)aryl-4-carboxylate derivatives can be prepared by the palladium catalyzed coupling of 2-bromopyridine-4-carboxylic acid with the corresponding boronic acid.
- Method I In 1 .75 min from 95% A, 5%B to 95%B, 5% A , then 0.25min 95% B, 5% A. The wavelength for UV detection was 254nm.
- Method I I In 4.75 min from 95% A, 5%B to 95%B, 5% A, then 0.25min 95% B, 5% A. The wavelength for UV detection was 214nm.
- HRMS The dry samples were dissolved in 1 ml methanol and diluted 1 /100 in CH 3 CN/H 2 0 0.1 %formic acid. 10 ⁇ of each sample was injected using the CapLC system (Waters, Manchester, U K) and electrosprayed through the Nanomate (Advion, Ithaca, NY) nanoelectrospray source.
- the Nanomate was operated in positive ion mode at an electrospray potential of 1 .5 kV. Samples were injected with an interval of 3 minutes Positive ion mode accurate mass spectra were acquired using a Q-TOF II instrument (Waters, Manchester, UK). The MS was calibrated prior to use with a 0.2% H3PO4 solution. The spectra were lock mass corrected using the know mass of the nearest H3PO4 cluster or the phthalate background ions. The Waters acquity U PLC system coupled to a waters TQD ESI mass spectrometer was also used for LC/MS/MS measurements.
- Step 1 2-(fert-Butoxycarbonylamino)acetic acid.
- Step2 2,5-Dioxopyrrolidine-1-yl 2-(fert-butoxycarbonylamino)acetate.
- Step 3 (S)-fert-Butyl 2-(2-carbamoylpyrrolidine-1-yl)-2-oxoethylcarbamate.
- Step 4 (S)-fert-butyl 2-(2-cyanopyrrolidine-1-yl)-2-oxoethylcarbamate.
- the amide obtained from step 3 (1 .77 g, 6.52 mmol) was dissolved in 80 mL of dichloromethane. Pyridine (5.27 m L, 65.2 mmol) was added to the cooled solution (-15°C), followed by the dropwise addition of trifluoroacetic anhydride (1 .012 m L, 7.1 8 mmol) solution in 15m L of dichloromethane. The resulting transparent yellowish solution was stirred for 90 min. The mixture was washed with 1 M HCI (3 x 30 m L), saturated sodium bicarbonate (1 x 40 mL) and brine. The organic layer was dried over Na 2 S0 4 , and concentrated to afford the crude product which was purified using column chromatography (hexane/ethyl acetate gradient) to afford the product as a yellowish oil.
- Step 5 (S)-1-(2-aminoacetyl)pyrrolidine-2-carbonitrile 2,2,2-trifluoroacetate.
- nitrile obtained from step 4 (1 .21 g, 4.78 mmol) was dissolved in acetonitrile (9.95 mL, 191 .3 mmol) and cooled to 0°C. Trifluoroacetic acid (7.1 mL, 96.63 mmol) was added dropwise. The solution was stirred overnight, concentrated and washed with ether (2 x 15 m L) to yield the crude product as an orange oily substance Yield: 2.00 g, 99%. - -
- Step 1 (S)-1-fert-butyl 2-methyl 4-oxopyrrolidine-1 ,2-dicarboxylate 1 ,3,5-Trichloro-1 ,3,5-triazinane-2,4,6-trione (3.78 g, 1 6.27 mmol) was added to a cooled (0 ⁇ ) solution of (S)-1 -ieri-butyl 2-methyl 4-oxopyrrolidine-1 ,2-dicarboxylate (3.8 g, 15.49 mmol) in DCM (25 mL), followed by the addition of catalytic TEMPO (0.024 g, 0.155 mmol). After 5 min the mixture was allowed to reach room temperature, stirred for another 30 minutes and filtrated over Celite. The organic layer was washed with 20m L saturated potassium carbonate solution, washed with brine, dried over anhydrous sodium sulfate, filtrated and evaporated. The crude compound 1 was used without further purification.
- Step 2 (S)-1-fert-butyl 2-methyl 4,4-difluoropyrrolidine-1 ,2-dicarboxylate
- Step 4 (S)-ferf-butyl 2-carbamoyl-4,4-difluoropyrrolidine-1-carboxylate
- Step 6 (S)-fert-butyl 2-(2-carbamoyl-4,4-difluoropyrrolidin-1-yl)-2-oxoethylcarbamate
- HATU (12.47 g, 32.8 mmol) was dissolved in 20 mL DMF and added to a solution of 2- ⁇ tert- butoxycarbonylamino)acetic acid (5.75 g, 32.8 mmol), as obtained in Scheme 4 from step 1 , and DIPEA (5.43 ml, 32.65 mmol) in 30 m L DCM .
- DIPEA 5.43 ml, 32.65 mmol
- a solution of (S)-4,4-difluoropyrrolidine- 2-carboxamide hydrochloride 5.1 g, 27.3 mmol
- DIPEA 9.06 ml, 54.38 mmol
- Step 7 (S)-fert-butyl 2-(2-cyano-4,4-difluoropyrrolidin-1-yl)-2-oxoethylcarbamate
- Step 8 (S)-1-(2-aminoacetyl)-4,4-difluoropyrrolidine-2-carbonitrile hydrochloride.
- the title compound was prepared in a manner similar to that described in Example 3, using commercially available 2-methylquinoline-4-carboxylic acid.
- the title compound was prepared in a manner similar to that described in Example 3 using pyridine-4-carboxylic acid.
- the title compound was prepared in a manner similar to that described in Example 3 using quinoline-8-carboxylic acid.
- the title compound was prepared in a manner similar to that described in Example 3 using 3- hydroxychinaldin-4-carboxylic acid.
- This compound was prepared relying on the general approach, consisting of coupling an intermediate of formula (D) and a non-commercially available intermediate of formula (E).
- the latter was obtained from a Pfitzinger-type reaction on a commercially available isatin ('stepl '), followed by decarboxylation of the quinoline dicarboxylate product of the Pfitzinger-type reaction.
- This approach is summarized in Scheme 5.
- Step 1 6-Fluoroquinoline-2,4-dicarboxylic acid.
- Step 2 6-Fluoroquinoline-4- carboxylic acid.
- 6-Fluoroquinoline-2,4- dicarboxylic acid (0.103 g, 0.437 mmol) was transferred in a pressure tube, 6 mL water was added. The closed tube was heated to 200 ⁇ for 4h. After slow cooling of the tube, the resulting precipitate was filtered and washed with water to yield white crystals
- Step 3 (3 ⁇ 4-A ⁇ (2-(2-Cyanopyrrolidin-1-yl)-2-oxoethyl)-6-fluoroquinoline-4-carboxamide
- 6-Fluoroquinoline-4- carboxylic acid (17) (0.054 g, 0.282 mmol) was dissolved in a 1 :1 mixture of dry DCM and THF (5 m L). 1 -chloro-/V,/V,2-trimethylprop-1 -en-1 -amine (0.052 mL, 0.395 mmol) was added to this solution, and the mixture was stirred for 30minut.es at rt.
- Step 1 6-Chloroquinoline-2,4-dicarboxylic acid.
- Step 2 6-Chloroquinoline-4-carboxylic acid.
- Step 3 (S)-6-Chloro-AA(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide.
- Step 1 6-(Trifluoromethoxy)quinoline-2,4- dicarboxylic acid.
- Step 3 (3 ⁇ 4-AA(2-(2-Cyanopyrrolidin-1-yl)-2-oxoethyl)-6-(trifluoromethoxy)quinoline-4- carboxamide.
- Step 1 8-Chloroquinoline-2,4-dicarboxylic acid.
- Step 2 8-Chloroquinoline-4-carboxylic acid.
- Step 1 6-methoxyquinoline-2,4-dicarboxylic acid
- the starting material was reacted in a pressured tube for 1.5 hour at 200°C with stirring. And worked up as described in example 9.
- Step 3 (S)-7-bromo-N-(2-(2-cyanopyrrolidin-1 -yl)-2-oxoethyl)quinoline-4-carboxamide
- Step 1 7-chloroquinoline-2,4-dicarboxylic acid
- the starting material was reacted in a pressured tube for 1.5 hour at 200 ⁇ with stirring. And worked up as described in example 9.
- 2-bromoisonicotinic acid (0.210 g, 1 .040 mmol) was dissolved in degassed DME (Volume: 8 ml) under nitrogen. Tetrakis(triphenylphosphine)palladium(0) (0.060 g, 0.052 mmol) was added, the resulting reaction mixture was stirred for 15min.Then aqueous potassium carbonate (4.16 ml, 8.32 mmol) and phenylboronic acid (0.171 g, 1 .403 mmol) were added subsequently. The resulting RM was refluxed at 95 °C for 18h and then cooled to rt. After filtration over celite the reaction mixture was acidified to pH 3-4 and the white precipitate was filtered off and washed with water.
- Step 1 2-(3,4-dimethoxyphenyl)isonicotinic acid
- Step 2 (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-(3,4-dimethoxyphenyl)isonicotinamide
- Step 2 (S)-2-(4-cyanophenyl)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)isonicotinamide
- the title compound was prepared in a manner similar to that described in Example 3 using 1- methyl-1 H-imidazole-5-carboxylic acid.
- the title compound was prepared in a manner similar to that described in Example 3 using 4- methylthiazole-5-carboxylic acid.
- the title compound was prepared in a manner similar to that described in Example 3 using 4- methyloxazole-5-carboxylic acid.
- Step 1 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid
- Step 2 (S)-tert-butyl 4-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethylcarbamoyl)piperidine-1- carboxylate
- MeOD MeOD: ⁇ 178.10, 169.80, 119.55, 47.9, 46.90, 46.11, 43.66, 42.70, 42.48, 30.97, 29.85, 26.13, 23.76.
- Step 1 5-chloroquinoline-2,4-dicarboxylic acid
- Step 1 5-bromoquinoline-2,4-dicarboxylic acid - -
- the starting material was reacted in a pressured tube for 50 minutes at 205 °C with stirring.
- Example 27 (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-5-methylquinoline-4-carboxamide
- the title compound was prepared in a manner similar to that described in Example 3.
- the title compound was prepared in a manner similar to that described in Example 1 .
- reaction mixture was diluted with CH2CI2 and washed with saturated aqueous NaHC03 solution (10 mL), 0.2N aqueous citric acid solution (10 mL) and brine (10 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The mixture was purified using column chromatography with dichloromethane-methanol as an eluent.
- Step 1 tert-butyl (R)-3-(tert-butyldimethylsilyloxy)-1-((S)-2-cyanopyrrolidin-1-yl)-1- oxopropan-2-ylcarbamate
- Tosic Acid (0,174 g, 0,91 6 mmol) was added to a cold (0 S C) solution of tert-butyl (R)-3-(tert- butyldimethylsilyloxy)-1 -((S)-2-cyanopyrrolidin-1 -yl)-1 -oxopropan-2-ylcarbamate (0,260 g, 0,654 mmol) in acetonitrile(about 0.4M) at 0 S C. after 30min the mixture was allowed to warm till room temperature and stirred for 24h.
- Step 3 N-((R)-1-((S)-2-cyanopyrrolidin-1-yl)-3-hydroxy-1-oxopropan-2-yl)quinoline-4- carboxamide
- Example 37 N-((R)-1-((S)-2-cyanopyrrolidin-1-yl)-1-oxopropan-2-yl)quinoline-4-carboxamide The title compound was prepared in a manner similar to that described in Example 36.
- Step 1 tert-butyl (R)-1-((S)-2-cyanopyrrolidin-1-yl)-1-oxopropan-2-ylcarbamate
- Step 3 N-((R)-1-((S)-2-cyanopyrrolidin-1-yl)-1-oxopropan-2-yl)quinoline-4-carboxamide
- Step 1 N-(2-oxo-2-((2R)-2-((3aS,7aR)-3a,5,5-trimethylhexahydro-4,6-methanobenzo [d][1 ,3,2]dioxaborol-2-yl)pyrrolidin-1-yl)ethyl)quinoline-4-carboxamide
- DPP IV, DPP I I, DPP8 and DPP9 were obtained as described in reference 18.
- Recombinant murine FAP was purified from the culture supernatant of HEK293 human embryonic kidney cellline as described in reference 19.
- Recombinant human PREP was expressed in and purified from E coli as described before in reference 20.
- Enzyme activities were determined kinetically in a final volume of 200 ⁇ for 10 minutes at 37°C by measuring the initial velocities of pNA release (405 nm) from the substrate using a Spectramax plus microtiterplate reader (Molecular devices).
- One unit of enzyme activity was defined as the amount of enzyme that catalyzes the release of 1 ⁇ pNA from the substrate per minute under assay conditions.
- IC 50 value was defined as the inhibitor concentration, which caused a 50% decrease of the activity under assay conditions.
- the chromogenic substrate Gly-Pro-p-nitroanilide (100 ⁇ / ⁇ ) was used at pH 8.3 for DPP IV, Lys- Ala-p-nitroanilide (1 mmol/l) at pH 5.5 for DPP II, Ala-Pro-p-nitroanilide (300 ⁇ / ⁇ ) at pH 7.4 for DPP9 and Ala-Pro-p-nitroanilide (2 mmol/l) at pH 7.4 for FAP activity measurement.
- the endopeptidase activity of FAP and the influence of inhibitors thereon, Z-Gly-Pro-AMC and Z- Gly-Pro-p-nitroanilide were used at a final concentration of 300 and 100 ⁇ / ⁇ , respectively.
- the substrate concentrations were chosen around the Km value obtained under the assay conditions used.
- Buffer compositions for the DPP assays were reported before in the purification articles - vide supra.
- the FAP assay buffer consisted of 50 mM Tris pH7.4 containing 1 00 mmol/l NaCI and 0.1 mg/ml bovine serum albumin.
- the PREP activity was measured as described by Brandt et al.
- Test compounds were dissolved and diluted in DMSO (final concentration DMSO during assay 5% v/v) except for FAP where dilution of the inhibitor was done in water.
- Inhibitors are pre-incubated with the enzyme for 1 5 min at 37 °C before starting the assay by the addition of substrate. The concentration of enzyme and of inhibitor during the preincubation is double of the final concentration during activity measurement.
- Z-Gly-Pro-AMC was used as a substrate at a concentration of 260 ⁇ / ⁇ in phosphate buffer pH 7.5 containing 1 mmol/l NaN 3 , 1 mmol/l EDTA with or without 10 mmol/l dithiothreitol (DTT). Final dilution of the plasma in the assay is 20 times.
- the 'total' activity (FAP AN D PREP) is measured when DTT is present, while in the absence of DTT, only FAP activity can be measured.
- the endogenous PREP activity is calculated as the difference between the 'total' activity and the FAP activity.
- DPPIV dipeptidyl peptidase-4
- DPP9 dipeptidyl peptidase-2
- DPP9 dipeptidyl peptidase-2
- PREP prolyl endopeptidase
- Table 4 represents evaluation data for compounds of the general formula that structurally accord to t
- FAP displays both endo-and exopeptidase activity, mediated by the same active center.
- the following data provide experimental evidence that the inhibitors that structurally accord to this invention, inhibit both activity types of FAP to the same extent (exemplified using Cmpd ⁇ from table 4).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to novel inhibitors having high selectivity and specificity for FAP (fibroblast activation protein). Said inhibitors are useful as a human and/or veterinary medicine, in particular for the treatment and/or prevention of FAP-related disorders such as but not limited to proliferative disorders.
Description
NOVEL FAP INHIBITORS
FIELD OF THE INVENTION
The present invention relates to novel inhibitors having high selectivity and specificity for FAP (fibroblast activation protein). Said inhibitors are useful as a human and/or veterinary medicine, in particular for the treatment and/or prevention of FAP-related disorders such as but not limited to proliferative disorders.
BACKGROUND TO THE INVENTION
1. Introduction
Fibroblast activation protein (FAP, FAP-alpha, seprase, alpha2 antiplasmin converting enzyme) is a Clan SC protease of the prolyl oligopeptidase subfamily S9b, occurring as a cell surface homodimer. FAP has been demonstrated to possess both dipeptidyl peptidase and endopeptidase activity, catalyzed by the same active center. Its expression is associated with activated stromal fibroblasts and pericytes of over 90% of human epithelial tumors examined and with normal or excessive wound healing, e.g. in tissue remodeling sites or during chronic inflammation. The enzyme is generally not expressed in normal adult tissues and in nonmalignant tumors.1 Several studies have tried to map the physiological substrate spectrum of FAP, including very recent reports that identify i.a. a/p/?a2-antiplasmin, type I collagen and gelatin as in vitro substrates of the endopeptidase activity of FAP.2 Analogously, Neuropeptide Y, B-type natriuretic peptide, substance P and peptide YY have been identified as in vitro substrates of the dipeptidyl peptidase activity of FAP.3 Nonetheless, the relevance of these findings under in vivo conditions remains debatable and the unambiguous definition of FAP's physiological substrate spectrum remains untouched matter so far.
Through structure-based design studies combined with extensive synthetic and biochemical effort, we were able to establish a Structure-Activity Relationship (SAR) of /V-acylated aminoacyl pyrrolidine inhibitors of fibroblast activation protein. This has led to the discovery of a novel scaffold type that has the potential to deliver inhibitors of FAP that combine low nanomolar activity with unprecedented selectivity toward related Clan SC proteases (dipeptidyl peptidases IV, I I, 8/9 and the endopeptidase prolyl oligopeptidase (PREP, PO). When compared to most other classes of reported inhibitors of FAP, inhibitors belonging to the scaffold type described here have remarkable stability both in aqueous solution and in human plasma and retain activity and selectivity for FAP within the latter media. For example, WO2007085895, WO2007005991 , WO2010083570, WO2006125227 and WO0238590 all disclose FAP inhibitors having a general structure closely relating to the compounds of the present invention. However, none of them actually discloses
- -
compounds wherein
 as defined in the present invention, is a 5 to 10- membered
as defined in the present invention, is a 5 to 10- membered
N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X. As further detailed herein below, in particular said feature is relevant for providing the compounds of the present invention with the FAP activity and selectivity as defined herein.
Based on FAP's role in (patho-)fysiology, documented extensively in literature, we reasonably foresee potential applications of our inhibitors in disease domains characterised by: (a) proliferation (including but not limited to cancer) (b) tissue remodelling and/or chronic inflammation (including but not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation) and (c) endocrinological disorders (including but not limited to disorders of glucose metabolism). The relationship of FAP with said pathological processes is described in more detail hereafter. (a) FAP and proliferative diseases (including but not limited to cancer).
During the last decade, numerous reports have been published that claim an important role for FAP in tumor growth and proliferation. The exact mechanism by which FAP takes part in these processes is unknown, but direct modulation of tumor growth, angiogenesis or disease progression by proteolytic processing of growth factors, cytokines, collagenase activity regulating proteins and even collagen derived proteins, is currently the subject of intensive research.
While awaiting the detailed functional characterization of the enzyme in these processes, several groups currently focus on FAP's status as a potential cancer biomarker which presence or activity in tumors could also be used for site-directed delivery of oncology drugs.4 Equally important, FAP or its activity are being targeted by several groups as a direct way to reduce tumor growth and proliferation by means of immunotherapeutic and small molecule inhibitor approaches.5 For the latter, a number of in vivo proof-of-concept studies are present. These all involve the dipeptide derived boronic acid talabostat (PT-100, Val-boroPro) or close analogues, and report significant activity on tumor stromagenesis and growth.6 In addition, talabostat has been evaluated as a drug in various clinical trials up to phase I I, for the treatment of, i.a. metastatic kidney cancer, chronic lymphocytary leukemia, pancreatic adenocarcinoma and non-small cell lung cancer. While talabostat in several of these trials was able to induce clinical response, questions were raised with regards to the safety profile of the compound, potentially related to its well-known lack of selectivity with respect to other Subfamily S9B proteases.7
- -
(b) FAP and diseases involving tissue remodeling and/or chronic inflammation (including but not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation, atherosclerotic disease and Chron's disease)
Multiple reports on occurrence of significantly increased FAP expression and/or activity both in physiological processes and in several clearly distinct disease domains, indicate that the enzyme might play an important role during events characterized by tissue remodeling and/or inflammation. Although the exact mechanism by which FAP is alleged to do so has to date not been clarified, the most straightforward hypothesis involves the enzyme's capability of processing collagenase activity regulating proteins and even collagen derived proteins, thereby altering the composition and structure of the extracellular matrix (ECM) of tissues. This effect could be supplemented by influences on the proteolytic processing of peptide growth factors and cytokines. Similar arguments are summoned to describe the of FAP's role in proliferative disease (vide supra).
Significant FAP expression has been confirmed for reactive fibroblasts in granulation tissue of healing wounds, on stellate cells at the tissue remodeling interface in hepatic cirrhosis, and in lung tissue in idiopathic pulmonary fibrosis.8 For hepatic cirrhosis (the pathological state characterized by fibrosis in which FAP's involvement has been best characterized) elevated expression of FAP was observed regardless of the etiology of the disease (viral hepatitis-induced, alcohol-induced, biliary cirrhosis). This given might suggest broad applicability of FAP-targeted therapy, e.g. using small molecule inhibitors, in disease area's involving fibrotic liver degeneration.9
FAP expression was found to be significantly increased on keloid fibroblasts compared to normal skin fibroblasts and inhibition of FAP activity with the albeit unselective (with respect to phylogenetically related dipeptidyl peptidases) irreversible inhibitor Gly-Pro(P)(OPh)2 was found to lead to a decrease in invasiveness.10
FAP expression and activity was also shown to be associated with rheumatoid arthritis and osteoarthritis: FAP-activity on the surface of chondrocytes and elevated expression and activity in cartilage affected by osteoarthritis were demonstrated. FAP was also found to be present in synovial tissue of affected joints, and elevated expression is detected in the murine collagen induced arthritis model. An additional pathway by which FAP could be operating in the pathogenesis and progression of arthritis, has been proposed to imply proteolytic cleavage of alpha2-antiplasmin, ultimately leading to fibrin deposition in the joint. Notably, in a Phase 1 clinical dosing study with a humanized anti-FAP antibody (sibrotuzumab) for advanced and metastatic cancer, the antibody in three patients not only localized to tumors, but also to the knees and shoulders. This observation has been connected to early-stage arthritis, offering initial support for the in vivo validation of FAP as a target for arthritis and related diseases.1 1
Recently, significantly increased expression of FAP was reported for human Type IV-Type V aortic atheromata, compared to type I I I atheromata and healthy aortae. Additionally, thin-cap human
- -
coronary atheromata were found to contain more FAP than thick-cap lesions. The enzyme's occurrence was found to be concentrated on smooth muscle and endothelial cells, and it could not be detected on macrophages. Nonetheless, macrophage burden did correlate with total FAP expression in the plaques. Furthermore, in vitro zymography revealed that FAP-mediated collagenase activity was neutralized by an antibody directed to the enzyme's catalytic domain both in human atherosclerotic smooth muscle cells and in fibrous caps of atherosclerotic plaques.2b
In a very recent publication, FAP was found to be overexpressed in enteric strictures of patients with Chron's disease (CD) and the protein was observed to be upregulated on strictured CD myofibroblasts by profibrogenic stimuli, leading the authors of this study to propose FAP as a potential target for the treatment of fibrostenosing CD.12
In general, no in vivo or clinical results (apart from the mentioned) have so far been disclosed dealing with the application of FAP-targeting small molecules or immunotherapeutic strategies in disease domains mentioned under this part. Nonetheless, mounting in vitro evidence from literature can certainly be considered compelling to initiate such investigations.
(c) FAP and diseases involving endocrinological disorder (including but not limited to disorders of glucose metabolism) and diseases involving blood clotting disorders.
A recent patent application by Gorrell et al. claims the utility of FAP inhibitors in the prevention and treatment of metabolic abnormalities characterized by abnormal glucose metabolism, including diabetes mellitus and new onset diabetes. This claim is however not otherwise documented in the literature. 13
Finally, blocking the activity of the soluble form of FAP (alpha2-antiplasmin cleaving enzyme, APCE) occurring in plasma, using small molecule inhibitors was found to cause enhanced fibrinolysis and to lead to a decrease of plasminogen activator induced clot lysis time. This observation led the authors to state that APCE-inhibition might constitute a novel approach in thrombolytic therapy without significant risk of bleeding.14
2. Inhibitor design
The prime aim underlying our effort to establish detailed SAR data for /V-acylated aminoacyl pyrrolidine inhibitors of FAP, was to identify compounds with significantly improved (a) chemical stability and (b) selectivity characteristics when compared to known FAP inhibitors, while retaining high affinity for the target enzyme.
(a) Limited chemical stability due to intramolecular cyclisation is a well known problem of several currently available highly potent dipeptide derived boronic acids (e.g. Val-boroPro). This property, caused by the combined presence of a nucleophilic amino terminus and an electrophilic boronic
- -
acid, puts constraints e.g. on the applicability of this compound and its analogues at physiological pH both in vitro and in vivo.15
(b) Selectivity with respect to related S9b proteases (DPP IV, DPP8/9, DPP I I, PREP) is a potential point of concern for all FAP inhibitors. Due to the high degree of phylogenetic relationship between the S9b proteases, pharmacophores of their inhibitors generally display substantial overlap. This problem is well documented for a number of described FAP inhibitors, including the well known ValboroPro. Noteworthy however, for most reported FAP inhibitors incomplete and in some cases even no selectivity data have been reported, rendering existing knowledge as a starting point for selective FAP inhibitor discovery inadequate. Nonetheless, taking into account the importance of inhibitor selectivity in the framework of potential compound toxicity and off-target effects, we deemed the preparation of selective compounds an important goal of our endeavours.1
With the number of reported FAP-inhibitors being small and most of them belonging to the class of boronic acids, we initially decided to focus on compounds that contain a carbonitrile warhead in place of the boronic acid, but conserve an overall dipeptide derived architecture. The latter is a hallmark of most chemotypes of published Subfamily S9B inhibitors. The carbonitrile function itself is also a popular affinity-enhancing moiety in reported series of inhibitors of DPP IV, DPP8, DPP9 and PREP. Compared to other warheads that are used in serine protease inhibitor design (e.g. - B(OH)2, -CHO, chloromethylketones, ketoamides, ...) the relatively mildly electrophilic carbonitrile could account for making the inhibitor more selective in vivo, a hypothesis that has been raised in literature earlier.1 In addition, the projected structures' overall architecture does in principle not impose fundamental limitations with respect to in vivo use, as e.g. illustrated by the EMA-approved vildagliptin and the FDA approved saxagliptin, both inhibitors of DPP IV. Three other publications are known to us that also contain carbonitrile-based inhibitors of FAP, all of them including incomplete FAP affinity and selectivity data or, in one case, even no affinity at all.16
Using the boundary assumptions described above, we decided to start an in depth investigation of the Structure-Activity Relationship (SAR) of A/-acylaminoacyl(2-cyanopyrrolidines) as inhibitors of FAP and their selectivity toward dipeptidyl peptidases and PREP. Three main structural fragments within this structure were marked for investigation and elaboration of the SAR:

Val-boroPro Generic structure of
talabostat /V-acyl-glycyl(2-cyano)pyrrolidines
- -
(a) the P3 moiety: By attaching this moiety (via an acyl linkage) to the aminoacyl(2- cyanopyrrolidine) backbone of the inhibitor, we wanted to make the P2 residue non-basic and non- nucleophilic, thus increasing the likeliness of inhibitor selectivity and higher stability with respect to the S9b dipeptidyl peptidases. Some literature evidence existed for peptide derived boronic acid inhibitors that this approach might be viable, although no systematic studies in this direction have been carried out. In addition, a substantial number of these literature FAP inhibitors have been reported with only limited or even without selectivity data for the related dipeptidyl peptidases. Additionally, while one might anticipate affinity toward dipeptidyl peptidases to be smaller, blocking the amino terminus does substantially increase the risk of selectivity problems with respect to the endopeptidase PREP. Again, very limited literature information was present dealing with FAP to PREP selectivity of inhibitors with an acylated P2 amine function.1
(b) the P2 moiety: while several acylated qlvcyl(2-borono)pvrrolidines have been reported in literature, almost no data exist on the influence of other amino acid residues at the P2 position in acylated compounds. At the outset of our activities, substrate kinetics studies nonetheless indicated a rather strict preference of FAP for a P2-glycine residue in substrates containing an acylated P2 amino function. This given is in sharp contrast with a series of dipeptide-derived substrates and/or inhibitors (e.g. ValboroPro) with a free amino terminus, where the number of tolerated P2 residues is known to be much larger.
(c) The P1 moiety: We decided to investigate the influence on activity and selectivity of substituting the pyrrolidine ring in compounds with structure 1. To this end, we selected a number of different functional groups with different bulk size and electronic effects.
In addition, we expected the obtained SAR-information poised to be applicable to analogous inhibitor types containing specific other warhead types or even no warhead, a hypothesis that we later on showed to be correct.
We have now surprisingly found that FAP-inhibitors of formula I exhibit good chemical stability and high selectivity for FAP, rendering them very suitable for the preparation of a medicine for the treatment of various FAP-related disorders. In addition, our invention has the potential to deliver compounds with high solubility and low LogD-values, a feature that is far from evident for dipeptide-derived compounds lacking a basic amino terminus and that is accounted for by the presence of heteroatoms introduced at specific positions of the P3 substituent.
SUMMARY OF THE INVENTION
In a first aspect, the present invention provides a compound of Formula I or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,
- -

Wherein
Ri and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- Ci-6alkyl, S-Ci-6alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl,
-C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl;
R4 is selected from the group comprising -H, -C1 -6alkyl, -0-C1 -6alkyl, -S-C1-6alkyl, -Αη , and -C^ 6aralkyl; each of said -C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - Ci-6alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, -OR12 -Het2 and -Ar2; each of said Ci-6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9 and R12 are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -0-d_6alkyl, -S-Ci-6alkyl, and -Ar3
Rio, Rii , Ri3 and R14 are each independently selected from the group comprising -H, -OH, -halo, -
C1-6alkyl, -0-C1-6alkyl, and -S-C1-6alkyl;
Αη , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , Ar2 and Ar3 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , - d-ealkyl, -0-Ci-6alkyl, -S-Ci-6alkyl;
Het2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said Het2 being optionally substituted with from 1 to 3 substituents selected from -NR13R14, -C1 -6alkyl, -0-C1-6alkyl, -S-C1 -6alkyl;
n

bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
- -
In a preferred embodiment, the present invention provides a compound according to form ula I, wherein
F and R2 are each independently selected from the group comprising -H , and -halo;
Fl3 is -CN , or -B(OH)2
R4 is selected from the group comprising -H or -C^alkyl ; each of said -C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , ;
R5, R6 and R7 are each independently selected from the group comprising -H , -OH , -oxo, -halo, -
Ci_6alkyl, -O-C^alkyl, Ar2 and -N R8R9; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , -halo;
R8 and R9 are each independently selected from the group comprising -H and -Ar3
Ar2 and Ar3 are each independently -phenyl optionally substituted with from 1 to 3 -O-C^alkyl ;

In yet another preferred embodiment, the present invention provides a compound according to formula I, wherein
Ri and R2 are each independently selected from the group comprising -H , and -F;
R3 is -CN ,and -B(OH)2
R4 is -H ;
R5, R6 and R7 are each independently selected from the group comprising -H , -oxo, -halo, -Ci_ 6alkyl, and -0-CF3;

The current invention further provides a compound of Form ula I I or a stereoisomer, tautomer, racem ic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,
- -

wherein
and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- C^alkyl, S-C^alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl, -C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl
R4 is selected from the group comprising -H, -C^alkyl, -O-C^alkyl, -S-C^alkyl, -Αη , and -C^ 6aralkyl; each of said -C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - Gi_6alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8 and R9, R10 and R are each independently selected from the group comprising -H, -OH, -halo, -C^alkyl, -O-C^alkyl, and -S-C^alkyl;
R10 and Rn are each independently selected from the group comprising -H, -OH, -halo, -C^alkyl, - O-d-ealkyl, -S-C^alkyl, and -Ar3;
Αη , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said An , and Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , - C^alkyl, -O-C^alkyl, -S-C^alkyl;
n is 0, 1 , 2, or 3

In a preferred embodiment, the current invention provides a compound according to formula II, wherein
RT and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- d-ealkyl, S-C^alkyl;
R3 is selected from the group comprising -H, -CN, and -B(OH)2
R4 is -H ;
- -
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C1-6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, -NR8R9, and -Ar2; each of said C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9, R10 and R are each independently selected from the group comprising -H, -OH, -halo, -C^
6alkyl, -0-C1 -6alkyl, and -S-C1-6alkyl;
Ar2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -C^alkyl, -O-C^alkyl, -S-C^alkyl;
n is 0, 1 , 2, or 3


In said embodiment, preferably, R5 and R6 are each -H ; R7 is selected from the group comprising - H, -OH, -oxo, -halo, -C1 -6alkyl, -0-C1-6alkyl, -S-C1-6alkyl, -NR8R9, and -Ar2; each of said C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo; and
R7 is attached to position 2 or 3, in particular position 2, as represented in

In yet a further embodiment, the present invention provides a compound of formula I lia, ll lb or ll lc or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,
- -

Illc
wherein
Ri and R2 are each independently selected from the group comprising -H, OH, -halo, C1-6alkyl, -0- C1-6alkyl, S-C1-6alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2„ -C(0)alkyl, -C(0)aryl-, -C=C- C(0)aryl, -C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl
R4 is selected from the group comprising -H, -d-6alkyl, -O-d-ealkyl, -S-d-ealkyl, -An, and -d- 6aralkyl; each of said -Ci-6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
R5, R6 and R7 are each independently selected from the group comprising -H, -OH, -oxo, -halo, - C1-6alkyl, -0-C1-6alkyl, -S-C1-6alkyl, -NR8R9, and -Ar2; each of said C1-6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8 and R9, are each independently selected from the group comprising -H, -OH, -halo, -d-ealkyl, -
O-d-ealkyl, -S-Ci-6alkyl, and -Ar3
Rio and Rn are each independently selected from the group comprising -H, -OH, -halo, -d-ealkyl, - O-d-ealkyl, and -S-Ci-6alkyl;
Ar1t Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη, and Ar2 being
- -
optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -
C^alkyl, -O-C^alkyl, -S-C^alkyl;
n is 0, 1 , 2, or 3

represent a 9 to 10- membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from O, N and S.
In a preferred embodiment, the present invention provides a compound according to formula I II, wherein
and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- d-ealkyl, S-C^alkyl;
R3 is selected from the group comprising -H, -CN, and -B(OH)2;
R4 is -H ;
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C^alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9, Rio and R are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -O-C^alkyl, and -S-C^alkyl;
Ar2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -C^alkyl, -O-C^alkyl, -S-C^alkyl;
n is 0, 1 , 2, or 3


In said embodiment, R5 is preferably attached to position 2 or 3, in particular position 3, as represented in

The current invention further provides a compound of formula I, II, Il ia, ll lb or l llc as defined above, as well as pharmaceutical compositions comprising said compounds, for use as a human or veterinary medicine.
In a further aspect, the present invention provides the use of a compound as defined above, as well as pharmaceutical compositions comprising said compounds, in the manufacture of a medicament for the prevention and/or treatment of a FAP-related disorder. A non-limiting list of examples of FAP-related disorders can include proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal
- -
cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma. In addition, the list of FAP-related disorders that are envisaged here, includes diseases characterised by tissue remodeling and/or chronic inflammation. These include but are not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation, atherosclerotic disease and Chron's disease. Furthermore, FAP related disorders involving endocrinological dysfunction (including but not limited to disorders of glucose metabolism) and diseases involving blood clotting disorders are part of this list.
It further provides the use of a compound as defined above, as well as pharmaceutical compositions comprising said compounds for inhibiting the activity of FAP.
In a further aspect, the present invention provides the use of a compound as defined above, as well as pharmaceutical compositions comprising said compounds, for the prevention and/or treatment of a FAP-related disorder. A non-limiting list of examples of FAP-related disorders can include proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma. In addition, the list of FAP-related disorders that are envisaged here, includes diseases characterised by tissue remodeling and/or chronic inflammation. These include but are not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation, atherosclerotic disease and Chron's disease. Furthermore, FAP related disorders involving endocrinological dysfunction (including but not limited to disorders of glucose metabolism) and diseases involving blood clotting disorders are part of this list.
In a final aspect, the present invention provides a method for the prevention and/or treatment of a FAP-related disorder. A non-limiting list of examples of FAP-related disorders can include proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma. In addition, the list of FAP-related disorders that are envisaged here, includes diseases characterised by tissue remodeling and/or chronic inflammation. These include but are not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation, atherosclerotic disease and Chron's disease. Furthermore, FAP related disorders involving endocrinological dysfunction (including but not limited to disorders of glucose metabolism) and diseases involving blood clotting disorders are part of this list.
- -
DETAILED DESCRIPTION OF THE INVENTION
The present invention will now be further described. In the following passages, different aspects of the invention are defined in more detail. Each aspect so defined may be combined with any other aspect or aspects unless clearly indicated to the contrary. In particular, any feature indicated as being preferred or advantageous may be combined with any other feature or features indicated as being preferred or advantageous.
Results of our SAR investigations that were directly instructive for the invention will be summarized here along with the corresponding modification types mentioned in the former part, that were subject of this SAR study.
(a) The P3 moiety:
An initial amount of SAR data for the P3 position was obtained by preparing and evaluating a series of compounds with a common Gly-(2-cyano)pyrrolidine backbone, carrying different P3 acyl- or sulfonamide type substituents at the free amino terminus. From this first series of compounds, the following inhibitors deserve special mentioning (Table 1 ) :
Inhibitors 3,4 and 5 shared substantial FAP affinity (IC50 < 5 μΜ), decoupled from PREP binding potential. The common structural feature that can be held accountable for this profile, is an (azaheterocyclyl)ac(et-)yl group as the scaffold substituent. Evidently, both the scope of this assumption and the possibility to improve FAP affinity were the subject of further investigation (vide infra).
Compound 6, containing a 1 -naphthoyl substituent, was found to have equally good FAP inhibitiory activity compared to compounds 3, 4 and 5, however it was far less selective towards FAP, compared to the other inhibitors of the first series. Our selection of the 1 -naphthoyl residue was based on a patent by Bachovchin et al., in which the activity of A/-(1 -naphthoyl)-substituted Gly- boroPro was claimed to possess superior FAP-affinity relative to the /V-benzoyl substituted congener, an observation we also found to hold for the corresponding nitriles. In addition, compound 6 was also reported in a recent publication by Tsai et al., with comparable FAP potency, but not including PREP assay data.14
Table 1 : Affinity/selectivity data for selected W-acyl-glycyl-(2-cyano)pyrrolidines.i

aDPP9 potencies reported can reasonably be expected to be indicative for inhibitor affinities toward the highly homologous D PP8 These findings were used as the starting point of extended research, aiming at the identification of optimised P3 residues with the potential to yield FAP inhibitors with maximal activity and selectivity. This was supported by a modelling study in which potential interactions of the 1 -naphthyl and aza- heteroaryl residues with the active sites of FAP, PREP and the S9b dipeptidyl peptidases were investigated together with the potential effect of modification of the aryl rings (e.g. substitution, introduction of hetero-atoms) . Combining these modelling data with our experimental findings for i.a. compounds 3-6, led to the proposal of a general P3 moiety structure that has the combined potential to deliver highly active and selective inhibitors of FAP. b) The P1 moiety:
A set of sterically and electronically diverse substituent types was chemically introduced at the pyrrolidine ring. Summarizing, available space in the part of FAP's active center accommodating the P1 pyrrolidine ring turned out to be very lim ited. Inhibitors 8-10, having a 4-fluoro- or 4,4- difluorosubstituent, were the only compounds found to outperform FAP-potency of their non- substitued analogues. (Table 2) No significant difference could be observed between the mono- and difluorinated compounds. With regards to the FAP/PREP selectivity issue, available space in
- -
PREP's S1 pocket seems even more limited than for FAP: only in the case of the fluorinated compounds, introduction of a 4-substituent does not completely delete enzyme affinity. Taking into account its positive effect on FAP-inhibitory activity, (di-)fluorination of the 4-position of the pyrrolidine ring could be regarded upon as a viable strategy to improve FAP-selectivity of promising inhibitors.
Table 2: Affinity/selectivity data for selected A-acyl-glycyl-(2-cyano)pyrrolidines.

After thorough examination and evaluation of both the experimental and modeling data we generated, we were able to inventively design a novel scaffold type according to formula I that has the potential to deliver stable FAP inhibitors that possess low nanomolar affinities for the target enzyme and that have very high selectivity indices with respect to the dipeptidyl peptidases and PREP. In addition, we were able to experimentally confirm these compounds' excellent potential with regards to FAP affinity, selectivity and stability, in a biologically relevant matrix (plasma).
Essential to both affinity and selectivity for FAP in this scaffold type is the presence of at least one nitrogen hetero-atom that is part of a cyclic system in P3 of these compounds. Equally essential for both parameters is the relative 1 ,4-positioning of (1 ) the N-atom and (2) the fragment that links the P3 cyclic framework to the P2 amino function. All of these aspects of the invention will be demonstrated using the enzymatic evaluation results both of compounds that correspond and of
- -
compounds that do not correspond to the proposed scaffold type. To the best of our knowledge this finding is unprecedented in literature.
Therefore, in a first aspect the present invention provides compounds of Formula I, including a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof

Wherein
Ri and R2 are each independently selected from the group comprising -H , OH , -halo, C^alkyl, -0- C^alkyl, S-C^alkyl ;
R3 is selected from the group comprising -H , -CN , -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl, -C=C-S(0)2aryl, -C02H , -S03H , -S02N H2,-P03H2, and 5-tetrazolyl ;
R4 is selected from the group comprising -H , -C^alkyl, -O-C^alkyl, -S-C^alkyl , -An , and -Ci_ 6aralkyl ; each of said -C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , -halo
R5, R6 and R7 are each independently selected from the group comprising -H , -OH , -oxo, -halo, - C1-6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, -N R8R9, -OR12 -Het2 and -Ar2; each of said C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , -halo
R8, R9 and R12 are each independently selected from the group comprising -H , -OH , -halo, -Ci_ 6alkyl, -0-Ci-6alkyl, -S-C^alkyl , and -Ar3
R10, Ri i , Ri3 and R14 are each independently selected from the group comprising -H , -OH , -halo, - d_6alkyl, -0-Ci-6alkyl, and -S-Ci-6alkyl ;
Αη , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , Ar2 and Ar3 being optionally and independently substituted with from 1 to 3 substituents selected from -N R10Rn , - C1-6alkyl, -0-C1-6alkyl, -S-C1 -6alkyl ;
Het2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S ; said Het2 being optionally substituted with from 1 to 3 substituents selected from -N R13R14, -C^alkyl, -0-Ci-6alkyl, -S-Ci-6alkyl ;
n is 0, 1 , 2, or 3
 represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
In an alternative representation, said embodiment discloses a compound of Formula X, including a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof

Ri and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- d_6alkyl, S-C^alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl,
-C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl;
R4 is selected from the group comprising -H, -C^alkyl, -O-C^alkyl, -S-C^alkyl, -Αη , and -C^
6aralkyl; each of said -C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, -
Ci_6alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, -OR12 -Het2 and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9 and R12 are each independently selected from the group comprising -H, -OH, -halo, -Ci_
6alkyl, -O-C^alkyl, -S-C^alkyl, and -Ar3
Rio, Ri i , Ri3 and R14 are each independently selected from the group comprising -H, -OH, -halo, -
C^alkyl, -O-C^alkyl, and -S-C^alkyl;
Αη , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , Ar2 and Ar3 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , - d-ealkyl, -O-C^alkyl, -S-C^alkyl;
- -
Het2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said Het2 being optionally substituted with from 1 to 3 substituents selected from -NR13R14, -C1 -6alkyl, -0-C1-6alkyl, -S-C1 -6alkyl;
3
 represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S;
represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S;
The present invention also provides a compound of formula (I), a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof

Wherein one or more of the following restrictions apply:
Ri and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- d_6alkyl, S-Ci-6alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl, -C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl;
R4 is selected from the group comprising -H, -C1 -6alkyl, -0-C1 -6alkyl, -S-C1-6alkyl, -Αη , and -C^ 6aralkyl; each of said -C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - Ci-6alkyl, -O-C^alkyl, -S-Ci-6alkyl, -NR8R9, -OR12 -Het2 and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9 and R12 are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, and -Ar3
R10, Rii , Ri3 and R14 are each independently selected from the group comprising -H, -OH, -halo, - C1-6alkyl, -0-C1-6alkyl, and -S-C1-6alkyl;
Αη , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , Ar2 and Ar3 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , - Ci-6alkyl, -0-Ci-6alkyl, -S-Ci-6alkyl;
- -
Het2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said Het2 being optionally substituted with from 1 to 3 substituents selected from -NR13R14, -C1 -6alkyl, -0-C1-6alkyl, -S-C1 -6alkyl;
3
 represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
represents a 5 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
In a particular embodiment, the present invention provides a compound of formula (I), a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof

Wherein one or more of the following restrictions apply:
Ri and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- Ci-6alkyl, S-d_6alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl,
-C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl;
R4 is selected from the group comprising -H, -C1 -6alkyl, -0-C1 -6alkyl, -S-C1-6alkyl, -Αη , and -C^
6aralkyl; each of said -C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, -
Ci-6alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, -0R12 -Het2 and -Ar2; each of said Ci-6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9 and R12 are each independently selected from the group comprising -H, -OH, -halo, -C^ 6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, and -Ar3
Rio. Rii , Ri3 and R14 are each independently selected from the group comprising -H, -OH, -halo, -
C1-6alkyl, -0-C1-6alkyl, and -S-C1-6alkyl;
An , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , Ar2 and Ar3 being
- -
optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -
C^alkyl, -O-C^alkyl, -S-C^alkyl;
Het2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said Het2 being optionally substituted with from 1 to 3 substituents selected from -NR13R14, -C1 -6alkyl, -0-C1-6alkyl, -S-C1 -6alkyl;
n 3
 represents a 6 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
represents a 6 to 10- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
When describing the compounds of the invention, the terms used are to be construed in accordance with the following definitions, unless a context dictates otherwise: The term "alkyl" by itself or as part of another substituent refers to a fully saturated hydrocarbon of Formula CxH2x+1 wherein x is a number greater than or equal to 1 . Generally, alkyl groups of this invention comprise from 1 to 6 carbon atoms. Alkyl groups may be linear or branched and may be substituted as indicated herein. When a subscript is used herein following a carbon atom , the subscript refers to the number of carbon atoms that the named group may contain. Thus, for example, C1-4alkyl means an alkyl of one to four carbon atoms. Examples of alkyl groups are methyl, ethyl, n-propyl, i-propyl, butyl, and its isomers (e.g. n-butyl, i-butyl and t-butyl) ; pentyl and its isomers, hexyl and its isomers, heptyl and its isomers, octyl and its isomers, nonyl and its isomers; decyl and its isomers. Ci-6 alkyl includes all linear, branched, or cyclic alkyl groups with between 1 and 6 carbon atoms, and thus includes methyl, ethyl, n-propyl, i-propyl, butyl and its isomers (e.g. n-butyl, i-butyl and t-butyl) ; pentyl and its isomers, hexyl and its isomers, cyclopentyl, 2-, 3-, or 4-methylcyclopentyl, cyclopentylmethylene, and cyclohexyl.
The term "aralkyl" as a group of part of a group refers to an alkyl moiety, as detailed above, wherein at least one -H atom is replaced by an aryl moeity.
The term "aryl" as a group of part of a group is generic for a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; said aryl further being optionally substituted with from 1 to 3 substituents as defined herein.
The term "halo" or "halogen" as a group or part of a group is generic for fluoro, chloro, bromo or iodo. Unless a context dictates otherwise, asterisks are used herein to indicate the point at which a mono- or bivalent radical depicted is connected to the structure to which it relates and of which the
- -
radical forms part. The aforementioned graphical representation has no bearing as to the actual orientation of said groups in the remainder of the molecule.
Whenever used in the present invention, the term 'compounds of the invention' or a similar term is meant to include the compounds of general Formula I or any subgroup thereof. This term also refers to a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof.
As used in the specification and the appended claims, the singular forms "a", "an" and "the" include plural referents unless the context clearly dictates otherwise. By way of example, "a compound" means one compound or more than one compound.
In a preferred embodiment, the present invention provides a compound of formula I, or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt hydrate or solvate thereof, wherein
F and R2 are each independently selected from the group comprising -H, and -halo;
Fl3 is -CN, or -B(OH)2
R4 is selected from the group comprising -H or -C^alkyl ; each of said -C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH,;
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C^alkyl, -O-C^alkyl, Ar2 and -NR8R9; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
R8 and R9 are each independently selected from the group comprising -H and -Ar3
Ar2 and Ar3 are each independently -phenyl optionally substituted with from 1 to 3 -O-C^alkyl;

In yet another preferred embodiment, the present invention provides a compound of formula I, or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt hydrate or solvate thereof, wherein
RT and R2 are each independently selected from the group comprising -H, and -F;
R3 is -CN,and -B(OH)2
R4 is -H ;
- -
R5, R6 and R7 are each independently selected from the group comprising -H, -oxo, -halo, -Ci_
6alkyl, and -0-CF3;

In a particular embodiment
 represents a 10-membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S; wherein
represents a 10-membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S; wherein
at least one of R5, R6 and R7 is selected from the group comprising -H, -OH, -oxo, -halo, -C^alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, -OR12 -Het2 and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
and wherein said R5, R6 or R7 is preferably attached at position 3, 6, 7 or 8 according to the following formula:
I
a particular embodiment represents a 9 to 10-membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S; such as for example:





As a further object, the current invention provides a compound according to formula II, or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,

wherein
R and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- C^alkyl, S-C^alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl,
-C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl
R4 is selected from the group comprising -H, -C^alkyl, -O-C^alkyl, -S-C^alkyl, -An , and -Ci_
6aralkyl; each of said -Ci-6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, -
C1-6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, -NR8R9, and -Ar2; each of said C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8 and R9, R10 and R are each independently selected from the group comprising -H, -OH, -halo,
-d_6alkyl, -0-Ci-6alkyl, and -S-Ci-6alkyl;
Rio and Rn are each independently selected from the group comprising -H, -OH, -halo, -C^alkyl, -
0-C1 -6alkyl, -S-C1 -6alkyl, and -Ar3;
Αη , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , and Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , - d_6alkyl, -0-Ci-6alkyl, -S-Ci-6alkyl;
n is 0, 1 , 2, or 3
m is 1 or 2
ηνΓ¾ m~* represents a 5 to 6- membered N-containing aromatic or non-aromatic monocyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S.
In a preferred embodiment, the present invention provides a compound according to formula I I, wherein
Ri and R2 are each independently selected from the group comprising -H, OH, -halo, C^alkyl, -0- C1-6alkyl, S-C1-6alkyl;
R3 is selected from the group comprising -H, -CN, and -B(OH)2
R4 is -H;
R5, R6 and R7 are each independently selected from the group comprising -H, -OH, -oxo, -halo, - Ci-6alkyl, -O-C^alkyl, -S-Ci-6alkyl, -NR8R9, and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9, Rio and R are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -0-C1 -6alkyl, and -S-C1-6alkyl;
Ar2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -C1-6alkyl, -0-C1-6alkyl, -S-C1 -6alkyl;
n is 0, 1 , 2, or 3
m is 1 or 2
HN¾ m~* is a 5- or 6-membered aromatic or non-aromatic monocylic heterocycle optionally nd S; selected from the list comprising


In said particular embodiment, preferably, R5 and R6 are each -H ; R7 is selected from the group comprising -H, -OH, -oxo, -halo, -C1-6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, -NR8R9, and -Ar2; each of said C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo; and R7 is attached to position 2 or 3, in particular position 2, as represented in

In yet another aspect, the present invention provides a compound of formula I lia, l llb or ll lc or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,

l llc
- -
wherein
R-i and R2 are each independently selected from the group comprising -H, OH , -halo, C1 -6alkyl, -0- C1-6alkyl, S-C1-6alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2„ -C(0)alkyl, -C(0)aryl-, -C=C- C(0)aryl, -C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl
R4 is selected from the group comprising -H, -d-6alkyl, -O-C^alkyl, -S-C^alkyl, -An , and -Ci_ 6aralkyl; each of said -Ci-6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C1-6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, -NR8R9, and -Ar2; each of said C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8 and R9, are each independently selected from the group comprising -H, -OH, -halo, -C1 -6alkyl, -
0-C1 -6alkyl, -S-C1 -6alkyl, and -Ar3
Rio and Rn are each independently selected from the group comprising -H, -OH, -halo, -C^alkyl, - 0-Ci-6alkyl, and -S-Ci-6alkyl ;
Ar1 t Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said An , and Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , - C1-6alkyl, -0-C1-6alkyl, -S-C1 -6alkyl;
n is O, 1 , 2, or 3

represent a 9 to 10- membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S.
In a particular embodiment, the current invention provides a compound according to formula II I, wherein
Ri and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- Ci-6alkyl, S-Ci-6alkyl;
R3 is selected from the group comprising -H, -CN, and -B(OH)2;
R4 is -H ;
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C1-6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, -NR8R9, and -Ar2; each of said C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
- -
R8, R9, Rio and R are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -O-C^alkyl, and -S-C^alkyl;
Ar2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -C^alkyl, -O-C^alkyl, -S-C^alkyl;
n is 0, 1 , 2, or 3

In said particular embodiment, preferably, R5 is attached to position 2 or 3, in particular position 3, as represented in:
Furthermore, for 9-membered aromatic or non-aromatic bicylic heterocycles, preferably R6 is -H and R7 is attached to position 7 as represented in:
- -
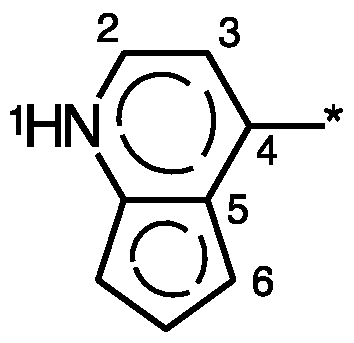
For 10-membered aromatic or non-aromatic bicylic heterocycles, R6 and R7 may be present at any suitable position, i.e. position 6, 7, 8 or 9 as represented in:

As a further object, this invention provides a compound according to this invention for use as a human or veterinary medicine.
This invention also provides a pharmaceutical composition comprising a compound according to this invention; said composition being suitable for use as a human or veterinary medicine.
For example, the compounds of the invention may be used as a free acid or base, and/or in the form of a pharmaceutically acceptable acid-addition and/or base-addition salt (e.g. obtained with non-toxic organic or inorganic acid or base), in the form of a hydrate, solvate and/or complex, and/or in the form or a pro-drug or pre-drug, such as an ester. As used herein and unless otherwise stated, the term "solvate" includes any combination which may be formed by a compound of this invention with a suitable inorganic solvent (e.g. hydrates) or organic solvent, such as but not limited to alcohols, ketones, esters and the like. Such salts, hydrates, solvates, etc. and the preparation thereof will be clear to the skilled person; reference is for instance made to the salts, hydrates, solvates, etc. described in US-A-6,372,778, US-A-6,369,086, US-A-6,369,087 and US-A-6,372,733.
The pharmaceutically acceptable salts of the compounds according to the invention, i.e. in the form of water-, oil-soluble, or dispersible products, include the conventional non-toxic salts or the quaternary ammonium salts which are formed, e.g., from inorganic or organic acids or bases. Examples of such acid addition salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2-naphthalene-sulfonate,
- -
nicotinate, oxalate, palmoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, tosylate, and undecanoate. Base salts include ammonium salts, alkali metal salts such as sodium and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases such as dicyclohexylamine salts, N-methyl-D-glucamine, and salts with amino acids such as arginine, lysine, and so forth. In addition, the basic nitrogen-containing groups may be quaternized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl ; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl-bromides and others. Other pharmaceutically acceptable salts include the sulfate salt ethanolate and sulfate salts.
Generally, the compounds of this invention may be formulated as a pharmaceutical preparation or pharmaceutical composition comprising at least one compound of the invention and at least one pharmaceutically acceptable carrier, diluent or excipient and/or adjuvant.
By means of non-limiting examples, such a formulation may be in a form suitable for oral administration, parenteral administration (such as by intravenous, intramuscular or subcutaneous injection or intravenous infusion), for topical administration (including ocular), for administration by inhalation, by a skin patch, by an implant, by a suppository, etc.. Such suitable administration forms - which may be solid, semi-solid or liquid, depending on the manner of administration - as well as methods and carriers, diluents and excipients for use in the preparation thereof, will be clear to the skilled person; reference is again made to for instance US-A-6,372,778, US-A-6,369,086, US-A- 6,369,087 and US-A-6,372,733, as well as to the standard handbooks, such as the latest edition of Remington's Pharmaceutical Sciences.
Some preferred, but non-limiting examples of such preparations include tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosols, ointments, creams, lotions, soft and hard gelatin capsules, suppositories, eye drops, sterile injectable solutions and sterile packaged powders (which are usually reconstituted prior to use) for administration as a bolus and/or for continuous administration, which may be formulated with carriers, excipients, and diluents that are suitable per se for such formulations, such as lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, polyethylene glycol, cellulose, (sterile) water, methylcellulose, methyl- and propylhydroxybenzoates, talc, magnesium stearate, edible oils, vegetable oils and mineral oils or suitable mixtures thereof. The formulations can optionally contain other pharmaceutically active substances (which may or may not lead to a synergistic effect with the compounds of the invention) and other substances that are commonly used in pharmaceutical formulations, such as lubricating agents, wetting agents, emulsifying and suspending agents, dispersing agents, desintegrants, bulking agents, fillers,
- -
preserving agents, sweetening agents, flavoring agents, flow regulators, release agents, etc.. The compositions may also be formulated so as to provide rapid, sustained or delayed release of the active compound(s) contained therein, for example using liposomes or hydrophilic polymeric matrices based on natural gels or synthetic polymers. In order to enhance the solubility and/or the stability of the compounds of a pharmaceutical composition according to the invention, it can be advantageous to employ α-, β- or γ-cyclodextrins or their derivatives.
In addition, co-solvents such as alcohols may improve the solubility and/or the stability of the compounds. In the preparation of aqueous compositions, addition of salts of the compounds of the invention can be more suitable due to their increased water solubility.
The preparations may be prepared in a manner known per se, which usually involves mixing at least one compound according to the invention with the one or more pharmaceutically acceptable carriers, and, if desired, in combination with other pharmaceutical active compounds, when necessary under aseptic conditions. Reference is again made to US-A-6, 372,778, US-A-6,369,086, US-A-6,369,087 and US-A-6,372,733 and the further prior art mentioned above, as well as to the standard handbooks, such as the latest edition of Remington's Pharmaceutical Sciences.
The pharmaceutical preparations of the invention are preferably in a unit dosage form, and may be suitably packaged, for example in a box, blister, vial, bottle, sachet, ampoule or in any other suitable single-dose or multi-dose holder or container (which may be properly labeled) ; optionally with one or more leaflets containing product information and/or instructions for use. Generally, such unit dosages will contain between 1 and 1000 mg, and usually between 5 and 500 mg, of the at least one compound of the invention, e.g. about 10, 25, 50, 100, 200, 300 or 400 mg per unit dosage.
The compounds can be administered by a variety of routes including the oral, rectal, ocular, transdermal, subcutaneous, intravenous, intramuscular or intranasal routes, depending mainly on the specific preparation used and the condition to be treated or prevented, and with oral and intravenous administration usually being preferred. The at least one compound of the invention will generally be administered in an "effective amount", by which is meant any amount of a compound of the Formula I or I I, upon suitable administration, is sufficient to achieve the desired therapeutic or prophylactic effect in the individual to which it is administered. Usually, depending on the condition to be prevented or treated and the route of administration, such an effective amount will usually be between 0.01 to 1000 mg per kilogram body weight day of the patient per day, more often between 0.1 and 500 mg, such as between 1 and 250 mg, for example about 5, 10, 20, 50, 100, 150, 200 or 250 mg, per kilogram body weight day of the patient per day, which may be administered as a single daily dose, divided over one or more daily doses, or essentially continuously, e.g. using a drip infusion. The amount(s) to be administered, the route of administration and the further treatment regimen may be determined by the treating clinician,
- -
depending on factors such as the age, gender and general condition of the patient and the nature and severity of the disease/symptoms to be treated. Reference is again made to US-A- 6,372,778, US-A-6,369, 086, US-A-6,369,087 and US-A-6,372,733 and the further prior art mentioned above, as well as to the standard handbooks, such as the latest edition of Remington's Pharmaceutical Sciences.
In accordance with the method of the present invention, said pharmaceutical composition can be administered separately at different times during the course of therapy or concurrently in divided or single combination forms. The present invention is therefore to be understood as embracing all such regimes of simultaneous or alternating treatment and the term "administering" is to be interpreted accordingly.
For an oral administration form , the compositions of the present invention can be mixed with suitable additives, such as excipients, stabilizers, or inert diluents, and brought by means of the customary methods into the suitable administration forms, such as tablets, coated tablets, hard capsules, aqueous, alcoholic, or oily solutions. Examples of suitable inert carriers are gum arabic, magnesia, magnesium carbonate, potassium phosphate, lactose, glucose, or starch, in particular, corn starch. In this case, the preparation can be carried out both as dry and as moist granules. Suitable oily excipients or solvents are vegetable or animal oils, such as sunflower oil or cod liver oil. Suitable solvents for aqueous or alcoholic solutions are water, ethanol, sugar solutions, or mixtures thereof. Polyethylene glycols and polypropylene glycols are also useful as further auxiliaries for other administration forms. As immediate release tablets, these compositions may contain microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants known in the art. When administered by nasal aerosol or inhalation, these compositions may be prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art. Suitable pharmaceutical formulations for administration in the form of aerosols or sprays are, for example, solutions, suspensions or emulsions of the compounds of the invention or their physiologically tolerable salts in a pharmaceutically acceptable solvent, such as ethanol or water, or a mixture of such solvents. If required, the formulation can also additionally contain other pharmaceutical auxiliaries such as surfactants, emulsifiers and stabilizers as well as a propellant. For subcutaneous administration, the compound according to the invention, if desired with the substances customary therefore such as solubilizers, emulsifiers or further auxiliaries are brought into solution, suspension, or emulsion. The compounds of the invention can also be lyophilized and the lyophilizates obtained used, for example, for the production of injection or infusion preparations. Suitable solvents are, for example, water, physiological saline solution or alcohols, e.g. ethanol, propanol, glycerol, in addition also sugar solutions such as glucose or mannitol solutions, or alternatively mixtures of the various solvents mentioned. The injectable solutions or suspensions
- -
may be formulated according to known art, using suitable non-toxic, parenterally-acceptable diluents or solvents, such as mannitol, 1 ,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
When rectally administered in the form of suppositories, these formulations may be prepared by mixing the compounds according to the invention with a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquefy and/or dissolve in the rectal cavity to release the drug. In a preferred embodiment, the compounds of the present invention are useful in human or veterinary medicine, in particular for use as FAP (fibroblast activation protein) inhibitors.
It is generally known that FAP exhibits both endo-and exopeptidase activity, mediated by the same active center. As further detailed in the examples that follow hereinafter, the newly developed inhibitors are capable of inhibiting both said endo-and exopeptidase activity. Therefore, FAP inhibition as used in the context of this invention is to include inhibition of endopeptidase activity and/or exopeptidase activity of FAP.
The present invention further provides the use of a compound as defined hereinbefore or the use of a composition comprising said compound, as a human or veterinary medicine, in particular for the prevention and/or treatment of FAP-related disorders.
In the invention, particular preference is given to compounds of Formula I or any subgroup thereof which inhibit FAP activity with an IC50 value of less than 10 μΜ, preferably less than 1 μΜ, most preferably less than 0.1 μΜ.
Said inhibition may be effected in vitro and/or in vivo, and when effected in vivo, is preferably effected in a selective manner, as defined herein. Particular reference is given to compounds of Formula I or any subgroup thereof which are at least 100x more selective for FAP compared to DPPIV, DPP9 and DPP2; in particular at least 1000x more selective for FAP compared to DPPIV, DPP9 and DPP2 .
Particular reference is also given to compounds of Formula I or any subgroup thereof which are at least 1 0x more selective for FAP compared to PREP, in particular at least 20 to 50x more selective for FAP compared to PREP.
The term "FAP-related disorder" as used herein, means any disease or other deleterious condition in which FAP is known to play a role. The term " FAP-related disorder" also means those diseases
- -
or conditions that are alleviated by treatment with a FAP inhibitor. Accordingly, another embodiment of the present invention relates to treating or lessening the severity of one or more diseases in which FAP is known to play a role. A non-limiting list of examples of FAP-related disorders can include proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma. In addition, the list of FAP-related disorders that are envisaged here, includes diseases characterised by tissue remodeling and/or chronic inflammation. These include but are not limited to fibrotic disease, wound healing, keloid formation, osteoarthritis, rheumatoid arthritis and related disorders involving cartilage degradation, atherosclerotic disease and Chron's disease. Furthermore, FAP related disorders involving endocrinological dysfunction (including but not limited to disorders of glucose metabolism) and diseases involving blood clotting disorders are part of this list. The invention also provides methods for the prevention and/or treatment of a FAP-related disorder; said method comprising administering to a subject in need thereof a compound according to this invention, or a composition comprising said compound.
The compounds of the present invention can be prepared according to the reaction schemes provided in the examples hereinafter, but those skilled in the art will appreciate that these are only illustrative for the invention and that the compounds of this invention can be prepared by any of several standard synthetic processes commonly used by those skilled in the art of organic chemistry. The invention will now be illustrated by means of the following synthetic and biological examples, which do not limit the scope of the invention in any way.
- -
EXAMPLES
Compounds of formula (I) can be prepared as indicated in general Scheme 1 . The variables are as defined above for formula I Scheme 1. General scheme for the synthesis of compounds defined by formula I

This can be carried out as mentioned below,
The reaction of Boc-am inocarboxylate (A) with compound (B) can be done using standard techniques for peptide coupling, known to a person skilled in the art, to afford compound (C) . This is either pyrrolidine or a pyrrolidine derivative. In cases where the pyrrolidine is a pyrrolidinecarboxam ide an additional dehydration step (e.g . using trifluoroacetic anhydride and pyridine, vide infra) has to follow the coupling step. The chosen protecting group of (C), including the Boc- and Z-group can be deprotected using a suitable acid or other deprotecting agent, known to a person skilled in the art, to generate compound (D) .
The reaction of compound (D) with compound (E) can be done using standard peptide coupling procedures, or using a corresponding acyl halide or active ester of (E) which is made in situ or in a separate reaction using procedures known to a person skilled in the art.
Alternatively, compounds of form ula (I) can also be prepared by converting one or more groups F¾, R3, R4, R5, Re or R7 of another compound of formula (I) obtained as mentioned above, into a desired substituent. The selected conversion method will depend on the kind of substituents desired. For example, it can be carried out by the following methodologies (Method a to Method e)..
(Method a) : A compound of type (I) in which R5, R6 or R7 in formula (I) is a hydrogen atom can be prepared by elim inating a protective group from a corresponding Compound (I) in which R5, R6 or R7 is a protective group. Removal of the protective group can be carried out by conventional methodology, known to a person skilled in the art (e.g. acid treatment, base treatment, catalytic reduction, etc.) .
- -
(Method b) : A compound of type (I) in which R3 is a boronic acid function, can be prepared by removing a protective group from a corresponding compound of formula (I) in which R3 is a protected boronate (e.g. a dialkyl ester, a pinanediol diester)15
(Method c) : A compound of type (I) in which R3 is a carbonitrile function, can be prepared by dehydration of a corresponding compound of formula (I) in which R3 is a carboxamide function.
(Method d) : A compound of type (I) in which R4 is a proteinogenic or non-proteinogenic amino acid side chain, can be obtained by eliminating a protective group from a corresponding Compound (I) in which R4 is a protected proteinogenic or non-proteinogenic amino acid side chain. Removal of the protective group can be carried out by conventional methodology, known to a person skilled in the art (e.g. acid treatment, base treatment, catalytic reduction, etc.).
(Method e) A compound of type (I) can be made by reacting compound (II) with a suitable protected compound (V), using standard peptide coupling procedures, or using a corresponding acyl halide or active ester of (I I) which is made in situ or in a separate reaction using procedures known to a person skilled in the art. Followed by reaction with (IV) using standard peptide coupling procedures, or using a corresponding acyl halide or active ester of (I I) which is made in situ or in a separate reaction using procedures known to a person skilled in the art.
Compounds of formula (B) are commercially available or can be prepared according to methods known to one skilled in the art or by methods disclosed herein.
For example, (S)-4,4-difluoropyrrolidine-2-carboxamide hydrochloride can be made as described in general procedure A. This is starting from (S)-1 -iert-butyl 2-methyl 4-oxopyrrolidine-1 ,2- dicarboxylate which is fluorinated with diethylaminosulfur trifluoride (DAST) and subsequently hydrolyzed with potassium hydroxide. The amide is made from this acid, reacting it with dicyclohexylcarbodiimide and /V-hydroxysuccinimide, subsequent removal of the Boc group yields the hydrochloride salt (B).
For example, prolinamide was bought from commercial suppliers (Fluorochem).
For example, the pinanediol ester of pyrrolidineboronic acid can be made as described in reference
12.
(2S,4S)-4-fluoropyrrolidine-2-carboxamide can be prepared as described analogously to (S)-4,4- difluoropyrrolidine-2-carboxamide, starting from 1 -ferf-butyl 4-hydroxypyrrolidine-1 ,2 dicarboxylate.
- -
Compounds of formula (E) of the present invention, can for example be obtained directly from commercial sources or be prepared from commercially available compounds as indicated in Scheme 2a for substituted quinoline-4-carboxylates or in Scheme 2b for the preparation of 2- aminopyridine-4-carboxylate derivatives and Scheme 2c for 2-(hetero)aryl-4-carboxylate derivatives.
Non-limiting list of commercially available compounds for preparing a compound of formula E:


Scheme 2a. Synthesis of compounds of Type II with a substituted quinoline-4-carboxylate basic structure.

The quinoline-4-carboxylic acid [E] can be prepared using the classical Sandmeyer isatin synthesis followed by the Pfitzinger reacton. The aniline was reacted with chloral hydrate and hydroxylamine hydrochloride to afford the substituted isonitrosoacetanilide which was cyclized using a suitable acid catalyst to afford the isatin as described in. Alternatively the isatin can be bought from commercial sources. The isatin can then be converted into the corresponding substituted quinoline- 4-carboxylic acid using the Pfitzinger reaction as in reference 17. In some instances the Pfitzinger reaction yielded a quinoline-2,4-dicarboxylic acid which was decarboxylated to the quinoline-4- carboxylic acid by reacting it in water for 2h using a pressured tube at 200 °C.
- -
Scheme 2b: Synthesis of 2-aminopyridine-4-carboxylate derivatives.

The 2-aminopyridine-4-carboxylic acids can be prepared by nucleophilic aromatic substitution of 2- chloropyridine-4-carboxylic acid with the corresponding primary or secondary amines. rboxylate derivatives.

The 2-(hetero)aryl-4-carboxylate derivatives can be prepared by the palladium catalyzed coupling of 2-bromopyridine-4-carboxylic acid with the corresponding boronic acid.
EXPERIMENTAL SECTION The experimental section is divided into three parts:
1 . Synthetic procedures
2. In vitro and in vivo assay protocols
3. Biochemical evaluation results
1 Synthetic procedures
Reagents were obtained from Sigma-Aldrich, Acros organics or Fluorochem and were used without further purification, unless otherwise mentioned. Characterization of all compounds was done with 1 H NMR and mass spectrometry. 1 H NMR spectra were recorded on a 400 MHz Bruker Avance II I nanobay spectrometer with ultrashield. In some NMR spectra, only the most important rotamer shown. Chemical shifts are in ppm and coupling constants are in Hz. ES mass spectra were obtained from an Esquire 3000plus iontrap mass spectrometer from Bruker Daltonics. Purity was verified using two diverse HPLC systems using, respectively, a mass and UV-detector. Water (A) and CH3CN (B) were used as eluents. LC-MS spectra were recorded on an Agilent 1 100 Series HPLC system using a Alltech Prevail C18 column (2.1 χ 50 mm , 3 μητι) coupled with an Esquire 3000plus as MS detector and a 'method A' 5-100% B, 20 min gradient was used with a
- -
flow rate from 0.2 m L/min. Formic acid 0.1 % was added to solvents A and B. When necessary, the products were purified with flash chromatography on a Biotage ® ISOLERA One flash system equipped with a internal variable dual-wavelength diode array detector(200-400nm). For normal phase purifications SNAP cartridges (10-340g; flow rate 10mL/min.-100ml_/min.) were used, reversed phase purifications were done making use of KP-C18 containing cartridges. Dry sample loading was done by self packing samplet ® cartridges using silica or Celite 545 respectively for normal -and reversed phase purifications. Gradients used varied by purification. However typical gradients used for normal phase were 30min. gradient of 0-50% EtOAc in hexane to 100 hexane or 0-5% MeOH in DCM to 20%MeOH in DCM and for reversed phase a gradient of 10%ACN in water to 50% ACN in water.. Waters acquity UPLC system coupled to a waters TQD ESI mass spectrometer and waters TUV detector. A waters acquity UPLC BEH C18 1 .7μιη 2.1 x 50 mm column was used. Solvent A: water with 0.1 % formic acid, Solvent B: acetonitrile with 0.1 % formic acid. Method I : In 1 .75 min from 95% A, 5%B to 95%B, 5% A , then 0.25min 95% B, 5% A. The wavelength for UV detection was 254nm. Method I I: In 4.75 min from 95% A, 5%B to 95%B, 5% A, then 0.25min 95% B, 5% A. The wavelength for UV detection was 214nm. HRMS: The dry samples were dissolved in 1 ml methanol and diluted 1 /100 in CH3CN/H20 0.1 %formic acid. 10μΙ of each sample was injected using the CapLC system (Waters, Manchester, U K) and electrosprayed through the Nanomate (Advion, Ithaca, NY) nanoelectrospray source. The Nanomate was operated in positive ion mode at an electrospray potential of 1 .5 kV. Samples were injected with an interval of 3 minutes Positive ion mode accurate mass spectra were acquired using a Q-TOF II instrument (Waters, Manchester, UK). The MS was calibrated prior to use with a 0.2% H3PO4 solution. The spectra were lock mass corrected using the know mass of the nearest H3PO4 cluster or the phthalate background ions. The Waters acquity U PLC system coupled to a waters TQD ESI mass spectrometer was also used for LC/MS/MS measurements.
This section will be further divided into two parts:
-1 .1 : Synthesis of intermediates of formula (D), via intermediates of formula (A),(B) and (C), as defined in Scheme 1 .
-1 .2: Synthesis of target products and intermediates of formula (I), via intermediates (D) and (E), as defined in Scheme 1 .
- -
1.1 : Synthesis of Intermediates of Formula (D), , via intermediates of formula (A),(B) and (C)
1 .1 .1 Synthesis of (S)-1 -(2-Aminoacetyl)pyrrolidine-2-carbonitrile 2,2,2-trifluoroacetate.
The synthetic pathway toward this compound is depicted in Scheme 3.
Sche yl(2-cyanopyrrolidine) intermediate of formula (D)


Step 1 : 2-(fert-Butoxycarbonylamino)acetic acid.
Glycine (3 g, 40.0 mmol) was dissolved in a dioxane-water (1 :1 ) (1 00 m L) mixture. Triethylamine (5.54 m L, 40.0 mmol) and di-ferf-butyl dicarbonate (8.72 g, 40.0 mmol) were added and the mixture was stirred for 2h at room temperature. After evaporation of the volatiles, redissolving in water, the solution was washed with diethylether (25 m L). The pH was set to 2 with 2N HCI. The mixture was extracted with ethyl acetate (2 x 5mL), dried over sodium sulfate and concentrated in vacuo, yielding a white crystalline product. Yield: 6.9g, 99%
MS (ESI) m/z 198.1 [M + Na]+.
Step2: 2,5-Dioxopyrrolidine-1-yl 2-(fert-butoxycarbonylamino)acetate.
/V-Boc-glycine (7.31 g, 41 .7 mmol) was dissolved in 100 mL of DCM and to the cooled (15°C) solution /V-hydroxysuccinimide (5.28 g, 45.9 mmol) was added. Λ/,Λ/'-dicyclohexylcarbodiimide (9.47 g, 45.9 mmol) was added to the formed suspension under vigorous stirring. After a few seconds, a cloudy white suspension formed, the mixture was allowed to reach room temperature and stirred for 1 h. It was subsequently filtrated over celite, washed with 50m L saturated sodium
- -
bicarbonate, dried over sodium sulfate and concentrated in vacuo to yield a crystalline powder. Yield: 7.02 g, 61 .8%
1 H NMR (400 MHz, DMSO -cfe) : δ 1 .38 (s, 9H), 2.80 (s, 4H), 4.07 (d, J = 6 Hz, 2H), 7.43 (br s, 1 H). MS (ESI) m/z 567.2 [2M + Na]+.
Step 3: (S)-fert-Butyl 2-(2-carbamoylpyrrolidine-1-yl)-2-oxoethylcarbamate.
A solution of the N-hydroxysuccinimidate obtained from step 2 (8.22g, 46.9mmol) in dichloromethane (100 mL) was cooled in a cooling bath to 15°C. After 15 min, a solution of (S)- pyrrolidine-2-carboxamide (5.1 g, 44.7 mmol, bought from commercial supplier Fluorochem) and 2.1 eq of Hunig's base in 50 m L of dichloromethane were added. After 3h, the resulting solution was washed with 1 N hydrochloric acid, with saturated sodium bicarbonate, brine, dried over sodium sulfate, filtrated and evaporated to dryness. The crude mixture was purified using flash chromatography with 5% methanol in DCM.
Yield: 8.61 g, 71 %
1 H NMR (400 MHz, DMSO -cfe): δ 1 .38 (s, 9H), 1 .72-2.22 (m , 4H), 3.38-3.45 (m,1 H), 3.49-3.55 (m,1 H), 3,74 (d, J = 5.7 Hz, 2H), 4.18 (dd, J =8.8, 2.8 Hz, 1 H), 5.43 (br s, 1 H), 5.54 (br_s, 1 H) , 6.83 (br s, 1 H)
MS (ESI) m/z 294.2 [M + Na]+.
Step 4: (S)-fert-butyl 2-(2-cyanopyrrolidine-1-yl)-2-oxoethylcarbamate.
The amide obtained from step 3 (1 .77 g, 6.52 mmol) was dissolved in 80 mL of dichloromethane. Pyridine (5.27 m L, 65.2 mmol) was added to the cooled solution (-15°C), followed by the dropwise addition of trifluoroacetic anhydride (1 .012 m L, 7.1 8 mmol) solution in 15m L of dichloromethane. The resulting transparent yellowish solution was stirred for 90 min. The mixture was washed with 1 M HCI (3 x 30 m L), saturated sodium bicarbonate (1 x 40 mL) and brine. The organic layer was dried over Na2S04, and concentrated to afford the crude product which was purified using column chromatography (hexane/ethyl acetate gradient) to afford the product as a yellowish oil.
Yield: (1 .21 g, 73%).
1 H NMR (400 MHz, DMSO -cfe): δ 1 .37 (s, 9H), 1 .73-1 .89 (m , 3H), 1 .94-2.00 (m, 1 H), 3.34-3.43 (m , 1 H), 3.49-3.53 (m, 1 H), 3.73 (d, J = 5.6 Hz, 2H), 4.23 (dd, J = 36, 7.6 Hz, 1 H), 6.68 (br s, 1 H). MS (ESI) m/z 276.1 [M + Na]+.
Step 5: (S)-1-(2-aminoacetyl)pyrrolidine-2-carbonitrile 2,2,2-trifluoroacetate.
The nitrile obtained from step 4 (1 .21 g, 4.78 mmol) was dissolved in acetonitrile (9.95 mL, 191 .3 mmol) and cooled to 0°C. Trifluoroacetic acid (7.1 mL, 96.63 mmol) was added dropwise. The solution was stirred overnight, concentrated and washed with ether (2 x 15 m L) to yield the crude product as an orange oily substance Yield: 2.00 g, 99%.
- -
1 H NMR (400 MHz, DMSO-d6): δ 1 .92-2.09 (m, 2H), 2.10-2.21 (m , 2H), 3.39-3.47 (m , 1 H), 3.57- 3.64 (m , 1 H), 3.86 (q, J = 18.2 Hz, 2H), 4.83 (dd, J = 7.0, 4.34 Hz, 1 H), 8.2 (br s, 3H)
MS (ESI) m/z 154.2 [M+H]+ 1 .1 .2 Synthesis of (S)-1 -(2-aminoacetyl)-4,4-difluoropyrrolidine-2-carbonitrile 2,2,2-trifluoroacetate starting from (2S,4f?)-1 -ferf-butyl 2-methyl 4-hvdroxypyrrolidine-l ,2-dicarboxylate.
The synthetic pathway toward this compound is depicted in Scheme 4. formula (D)

Step 1 : (S)-1-fert-butyl 2-methyl 4-oxopyrrolidine-1 ,2-dicarboxylate 1 ,3,5-Trichloro-1 ,3,5-triazinane-2,4,6-trione (3.78 g, 1 6.27 mmol) was added to a cooled (0Ό) solution of (S)-1 -ieri-butyl 2-methyl 4-oxopyrrolidine-1 ,2-dicarboxylate (3.8 g, 15.49 mmol) in DCM (25 mL), followed by the addition of catalytic TEMPO (0.024 g, 0.155 mmol). After 5 min the mixture was allowed to reach room temperature, stirred for another 30 minutes and filtrated over Celite. The organic layer was washed with 20m L saturated potassium carbonate solution, washed with brine, dried over anhydrous sodium sulfate, filtrated and evaporated. The crude compound 1 was used without further purification.
Yield: 2.77g,74%.
1 H NMR (400 MHz, CDCI3) : δ 1 .46 (s, 9H), 2.57 (dd, J = 18.8, 2.4, 1 H), 2.90 (s, 1 H), 3.75 (s, 3H) , 3.88 (br s, 2H), 4.77 (dd, J = 36.8, 8 Hz, 1 H).
MS (ESI) m/z 376.2 [M + MeOH + H]+.
- -
Step 2: (S)-1-fert-butyl 2-methyl 4,4-difluoropyrrolidine-1 ,2-dicarboxylate
A solution of the (S)-1 -ieri-butyl 2-methyl 4-oxopyrrolidine-1 ,2-dicarboxylate (0.23 g, 0.946 mmol), obtained from step 1 , in CH2CI2 (3.0 m l_), in a 25-mL flask equipped with a N2 inlet tube and stirring bar, was treated with a solution of diethylaminosulfur trifluoride (0.197 ml, 1 .607 mmol) in CH2CI2 (2.0 m l_) at room temperature. Ethanol (0.01 1 ml, 0.1 89 mmol) was added (for in situ generation of catalytic quantities of HF) and the mixture was stirred for 18h at room temperature. The solution was poured into saturated sodium bicarbonate and after C02 evolution ceased it was extracted into CH2CI2 (3 x 15 ml_), dried (Na2S04), filtered, and evaporated in vacuo. Chromatography on silica gel in DCM afforded a yellowish oil.
Yield: 0.150g, 61 %.
1 H NMR (400 MHz, CDCI3): δ 1 .44 (br s, 9H), 2.45 (dq, J= 13.6, 5.2 Hz, 1 H), 2.61 -2.81 (m, 1 H), 3.75 (s, 3H), 3.60-3.90 (m , 2H), 4.45-4.55 (m, 1 H).
MS (ESI) m/z 266.1 [M + H]+
Step 3: (S)-1-(iert-butoxycarbonyl)-4,4-difluoropyrrolidine-2-carboxylic acid
(S)-1 -ieri-butyl 2-methyl 4,4-difluoropyrrolidine-1 ,2-dicarboxylate (1 .51 g, 5.69 mmol), obtained from step 2, was dissolved in 6 m l_ of 1 M potassium hydroxide solution. The solution was stirred overnight. The mixture was washed with ether, acidified, extracted with ethyl acetate, washed with brine, dried over sodium sulfate, filtered and evaporated to yield slightly brownish crystals. The crude mixture was used without further purification. Yield: 1 ,23g, 86%
1 H NMR (400 MHz, DMSO-c¾) : δ 1 .38 (s, 9H), 2.32 - 2,48 (m , 1 H), 2.78-2.98 (m, 1 H), 3.65 - 3.77 (m, 2H), 4.31 -4.37 (m , 1 H), 13.0 (br s, 1 H).
MS (ESI) m/z 250.8 [M-H]~
LC-MS(I) Rt 1 .51 min, m/z 252.5 [M+H]+ (90%).
Step 4: (S)-ferf-butyl 2-carbamoyl-4,4-difluoropyrrolidine-1-carboxylate
In a 50 m L round-bottomed flask (S)-1 -(ieri-butoxycarbonyl)-4,4-difluoropyrrolidine-2-carboxylic acid (1 .6 g, 6.37 mmol), obtained from step 4, was dissolved in 10 mL of dichloromethane at 15°C. Then 1 -hydroxypyrrolidine-2,5-dione (0.806 g, 7.01 mmol) was added. To the formed suspension Λ/,/V-dicyclohexylcarbodiimide (1 .445 g, 7.01 mmol) was added at vigorous stirring. In a few seconds a cloudy white suspension forms. The mixture was allowed to reach RT and stirred for 30 min, followed by the addition of 7N ammonia in methanol (2.002 ml, 14.01 mmol) and stirring for another 20 min. Before evaporation of volatile components 1 spoon of Celite was added to the flask. Cold ethyl acetate was added to the residue and filtered over Celite. The filtrate was washed with saturated sodium bicarbonate. The formed slightly yellowish crystals were used without further purification.
Yield: 1 .15g, 72%
- -
1 H NMR (400 MHz, CDCI3) : δ 1 .45 (s, 9H), 2.77 - 2.44 (m , 1 H), 2.82 - 3.02 (m , 1 H), 3.56 - 3.78 (m, 1 H), 3.80 - 4.00 (m, 1 H), 4.52 (br_s, 1 H), 5.54 (br s, 1 H), 6.77 (br_s, 1 H).
LC-MS(I) Rt 1 .34 min, m/z 251 .5 [M+H]+ (89%).
MS (ESI) m/z 251 .2 [M + H]+
Step 5: (S)-4,4-difluoropyrrolidine-2-carboxamide trifluoroacetate
9.54 mL of trifluoroacetic acid was added to a solution of (S)-iert-butyl 2-carbamoyl-4,4- difluoropyrrolidine-1 -carboxylate (1 ,25 g, 5 mmol) in 10 mL of dichloromethane. The solution was stirred for 1 hour before evaporation. The residue was washed with ether to yield white crystals. Yield: 0.84g, 90%
1 H NMR (400 MHz, DMSO-cfe) : δ 2.42 - 2.61 (m, 1 H), 2.86 - 3.00 (m , 1 H), 3.71 (dd, J = 19.54, 7.48 Hz, 2H), 4.46 (t, J = 8.59 Hz, 1 H), 7.82 (s, 1 H), 8.07 (s, 1 H), 10.01 (br_s, 2H).
MS (ESI) m/z 155.2 [M + H]+
Step 6: (S)-fert-butyl 2-(2-carbamoyl-4,4-difluoropyrrolidin-1-yl)-2-oxoethylcarbamate
HATU (12.47 g, 32.8 mmol) was dissolved in 20 mL DMF and added to a solution of 2-{tert- butoxycarbonylamino)acetic acid (5.75 g, 32.8 mmol), as obtained in Scheme 4 from step 1 , and DIPEA (5.43 ml, 32.65 mmol) in 30 m L DCM . After 15min a solution of (S)-4,4-difluoropyrrolidine- 2-carboxamide hydrochloride (5.1 g, 27.3 mmol) and DIPEA (9.06 ml, 54.38 mmol) in 40 m L DCM was added. After 3h the cloudy mixture was filtered of. The filtrate was cooled and filtrated again. The combined residues were washed with DCM and water.
Yield: 8.39g, 77% 1 H NMR (400 MHz, DMSO-cfe) δ 1 .39 (s, 9H), 2.30 - 2.43 (m , 1 H), 2.65 - 2.78 (m , 1 H), 3.71 (dd, J = 17.2, 5.32 Hz, 1 H), 3.81 (dd, J = 1 7.2, 5.68 Hz, 1 H), 3.96 (dd, J = 24.58, 1 1 .16 Hz, 1 H), 4.09 (dd, J = 25.40, 12.76 Hz, 1 H), 4.46 (dd, J = 9.72, 4.13 Hz, 1 H), 6.89 (br tr J = 5.6 Hz), 7.13 - 7.73 (m, 2H).
LC-MS(I) Rt 1 .28 min, m/z 308.5 [M+H]+ (94%).
MS (ESI) m/z 308.1 [M + H]+
Step 7: (S)-fert-butyl 2-(2-cyano-4,4-difluoropyrrolidin-1-yl)-2-oxoethylcarbamate
In a 50 m L round-bottomed flask (S)-iert-butyl 2-(2-carbamoyl-4,4-difluoropyrrolidin-1 -yl)-2- oxoethylcarbamate (41) (0.720 g, 2.343 mmol) was dissolved in dry THF at -15 °C. Then pyridine was added, followed by the dropwise addition of the solution of 2,2,2-trifluoroacetic anhydride (0.094 ml, 0.664 mmol) in 5 m L of DCM after the complete addition, the mixture was allowed to reach RT. The reaction mixture was stirred for 90 minutes. The reaction mixture was washed with 1 M solution of aqueous solution of hydrochloric acid. Then the organic layer was washed three times with saturated sodium bicarbonate, brine, dried over sodium sulfate and evaporated. The
- -
crude mixture was purified using column chromatography (hexane-ethyl acetate 2-3) yielding a yellowish oil Yield: 0.678g, 70%
1 H NMR (400 MHz, CDCI3) δ 1 .45 (s, 9H), 2.70 - 2.83 (m, 2H), 3.80 - 4.04 (m, 4H), 4.94 - 5.00 (m , 1 H), 5.29 ( br s, 1 H).
LC-MS(I) Rt 1 .52 min, m/z 290.6 [M+H]+ (94%).
MS (ESI) m/z 290.1 [M + H]+
Step 8: (S)-1-(2-aminoacetyl)-4,4-difluoropyrrolidine-2-carbonitrile hydrochloride.
(S)-1 -(2-aminoacetyl)-4,4-difluoropyrrolidine-2-carbonitrile trifluoroacetate
To a cooled (0°C) solution of (S)-iert-butyl 2-(2-cyano-4,4-difluoropyrrolidin-1 -yl)-2- oxoethylcarbamate (7.3 g, 25.2 mmol), obtained from step 7, in 1 50 ml_ of ACN was added trifluoroacetic acid (38.9 m l_, 0.505 mol) dropwise. The mixture was stirred overnight and the volatiles were evaporated. The residue was washed with ether.
(S)-1 -(2-aminoacetyl)-4,4-difluoropyrrolidine-2-carbonitrile hydrochloride
The residue was stirred for 30min at 0Ό in diethylether and dry HCI was bubbled in the reaction flask. A white solid precipitated out. The solid was collected via vacuum filtration and rinsed with cold ether and dried under vacuum to yield a whitish powder.
1 H NMR (400MHz, D20) : (5/1 mixture of trans/cis amide rotamers) 6 2.84-3.05 (m, 2H), 4.03 - 4.24 (m, 2H), 4.01 (s, 2H), 5.17 (dd, J = 8.4, 4.1 Hz, 0.8H), 5.37 (d, J = 8.4 Hz, 0.2H)
Yield: 70%
MS (ESI) m/z: 190.2 [M+H]+
2.2 Synthesis of final products of formula (I), via intermediates (D) and (E), as defined in Scheme 1. -(2-(2-cyano-4,4-difluoropyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide

Commercially available quinoline-4-carboxylic acid (0.065 g, 0.377 mmol) was dissolved in 7 ml of DMF, A/-ethyl-/V-isopropylpropan-2-amine (0.210 ml, 1 .168 mmol), HOBT (0.058 g, 0.377 mmol) and TBTU (0.121 g, 0.377mmol) were added. After 15 min a solution of (S)-1 -(2-aminoacetyl)-4,4- difluoropyrrolidine-2-carbonitrile trifluoroacetate (0.085g, 0.377mmol) (prepared as described under A.2 of the experimental part) in DMF was added. The mixture was stirred overnight at room
- -
temperature. The volatiles were evaporated, the residue was dissolved in ethyl acetate and washed with 1 N citric acid, saturated sodium bicarbonate and brine. The solution was dried over sodium sulfate, filtrated and evaporated. It was purified using column chromatography (1-4 hexane- ethyl acetate). Yield: 64mg, 45%
1H NMR (400 MHz, CDCI3): (8.5/1.5 mixture of trans/cis amide rotamers) δ 2.72 - 2.83 (m, 2H), 3.91 -4.07 (m, 2H), 4.21 (dd, J= 17.4, 4.2 Hz, 0.85H), 4.33 (dd, J= 17.4, 4.3 Hz, 0.15H), 4.39 (dd, J= 17.4, 5.6 Hz, 0.85H), 4.70 (dd, J= 17.4, 5.7 Hz, 0.15H), 4.92-4.99 (m, 0.85H), 5.15 (d, J = 9 Hz, 0.15H), 7.30 (s, 1 H), 7.49 (dd, J= 10.11 , 4.30 Hz, 1 H), 7.60 (dd, J = 11.22, 4.11 Hz, 1 H), 7.74 (t, J= 7.69 Hz, 1H), 8.12 (d, J= 8.42 Hz, 1H), 8.23 (t, J= 9.99 Hz, 1H), 8.96 - 8.86 (m, 1H).
MS (ESI) m/z 345.0 [M+1]+
LC-MS (l-B) Rt10.8 min, m/z 345.0 [M+H]+ (98%).
Example 2: (S)-A-(2-(2-cyano-4,4-difluoropyrrolidin-1-yl)-2-oxoethyl)-1-naphthamide
Reference compound N°1

The title compound was prepared in a manner similar to that described in Example 1.
Yield: 43 mg,36%
1H NMR (400 MHz, CDCI3): δ 2.61-2.73 (m,2H), 3.83-4.07 (m, 3H), 4.34 (dd, J= 17.6, 6.0 Hz, 1H), 4.85-4.95 (m, 1H), 7.10 (t, J= 5.6 Hz, 1H), 7.40 (dd, J=8.4 Hz, J'= 7.2 Hz, 1H), 7.50 -7.57(m, 2H), 7.65 (dd, J= 7.0 Hz, J = 10 Hz, 1H), 7.86 (dd, J= 7.2, 2.16 Hz, 1H), 7.90 (d, J= 8.2 Hz, 1H), 8.32 (dd, J = 7.52 Hz, J = 1.92 Hz, 1H)
LC-MS (l-B) Rt15.9 min, m/z 344.1 [M+H]+ (97%) -A-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide

Commercially available quinoline-4-carboxylic acid (0.078g, 0.449 mmol) was dissolved in 7 ml of DMF, A/-ethyl-/V-isopropylpropan-2-amine (0.242ml, 1.347 mmol), HOBT (0.069 g, 0.449 mmol) and TBTU (0.144g, 0.449mmol) were added. After 15 min a solution of (S)-1-(2- aminoacetyl)pyrrolidine-2-carbonitrile 2,2,2-trifluoroacetate (0.120 g, 0.449 mmol) (prepared as described under A.1 of the experimental part) in DMF was added. The mixture was stirred overnight at room temperature. The volatiles were evaporated, the residue was dissolved in ethyl
- -
acetate and extracted with 1 N citric acid, saturated sodium bicarbonate and brine. The solution was dried over sodium sulfate, filtrated and evaporated. It was purified using column chromatography (1 -5 hexane-ethyl acetate).
Yield: 66 mg, 38%
1 H NMR (400 MHz, CDCI3) : (8.5/1 .5 mixture of trans/cis amide rotamers) δ 2.12 - 2.38 (m, 4H), 3.50 - 3.58 (m, 1 H), 3.68-3.74 (m , 1 H), 4.27 (dd, J= 1 7.6, 4.1 Hz, 0.85H), 4.33 (dd, J= 17.3, 4.3 Hz, 0.15H), 4.43 (dd, J= 18.0, 5.0 Hz, 0.85H), 4.60 (dd, J= 17.3, 5.4 Hz, 0.15H), 4.75 - 4.78 (m, 1 H), 7.14 (br s, 1 H), 7.49 (d, J = 6 Hz, 1 H), 7.62 (d tr, J = 7.7 Hz, J= 1 .3 Hz, 1 H), 7.77 (d tr, J = 7.6, 1 .4 Hz, 1 H), 8.15 (d, J= 8.49 Hz, 1 H), 8.28 (dd, J = 8.07, 0.79 Hz, 1 H) , 8.95 (d, J = 4.3 Hz, 1 H) MS (ESI) m/z 331 .1 [M+Na]+
LC-MS (l-B) Rt 9.7 min, m/z 309.0 [M+H]+ (97%). -A-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-methylquinoline-4-carboxamide

The title compound was prepared in a manner similar to that described in Example 3, using commercially available 2-methylquinoline-4-carboxylic acid.
Yield: 58mg, 37% 1 H NMR (400 MHz, CDCI3) : (8.5/1 .5 mixture of trans/cis amide rotamers) δ 2.23 - 2.41 (m, 4H), 2.78 (s, 3H), 3.53 - 3.58 (m, 1 H), 3.68 - 3.74 (m , 1 H), 4.26 (dd, J= 18.0, 4.1 Hz, 0.85H), 4.33 (dd, J= 18.0, 4.3 Hz, 0.15H), 4.42 (dd, J= 1 8.0, 5.1 Hz, 0.85H), 4.60 (dd, J= 18.0, 5.4 Hz, 0.15H), 4.76 (d, J = 9 Hz, 0.15H), 4.78 - 4.83 (m , 0.85H), 7.02 (br s, 1 H), 7.43 (s, 1 H), 7.55 (tr, J = 7.7 Hz, 1 H), 7.74 (d tr, J = 7.6, 1 .4 Hz, 1 H), 8.06 (br d, J= 8.0 Hz, 1 H), 8.28 (br d, J = 8.3 Hz, 1 H) , 8.95 (d, J = 4.3 Hz, 1 H)
MS (ESI) m/z 323.1 [M+H]+
LC-MS (l-B) Rt 8.5 min, m/z 323.1 [M+H]+ (95%).
Example 5: (S)-A-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-3-carboxamide
Reference compound N ° 3

The title compound was prepared in a manner similar to that described in Example 3, using commercially available quinoline-3-carboxylic acid.
Yield: 47m g, 36%
1H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) 52.18 - 2.39 (m, 4H), 3.50- 3.59 (m, 1H), 3.70 - 3.78 (m, 1H), 4.18 (dd, J= 17.7, 3.6 Hz, 0.9H), 4.26 (dd, J= 17.7, 3.6 Hz, 0.1 H), 4.35 (dd, J= 18.0, 4.3 Hz, 0.1 H), 4.56 (dd, J= 17.7, 6.0 Hz, 0.9H), 4.77 - 4.82 (m, 1H), 7.56 (d tr, J = 7.5, 1.1 Hz, 1H), 7.73 - 7.79 (m, 2H), 7.96 (br_s, 1H),8.05 (br_d, J= 8.0 Hz, 1H),8.53 (d, J =2.0 Hz, 1H) , 9.23 (d, J=2.0 Hz, 1H)
MS (ESI) m/z 331.1 [M+Na]+
LC-MS (l-B) Rt 11.2 min, m/z 309.1 [M+H]+ (98%). -(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)isonicotinamide

The title compound was prepared in a manner similar to that described in Example 3 using pyridine-4-carboxylic acid.
Yield: 41mg,34%
1H NMR (400 MHz, CDCI3): 52.13 - 2.36 (m, 4H), 3.45 - 3.54 (m, 1 H), 3.66 - 3.74 (m, 1 H), 4.05 - 4.12 (m, 1H), 4.46 - 4.53 (m, 1H), 4.72 - 4.76 (m, 1H), 7.61 (d, J= 6 Hz, 2H), 8.63 (d, J= 6 Hz, 2H) MS (ESI) m/z 259.1 [M+H]+
LC-MS (l-B) R,3.7 min, m/z 259.1 [M+H]+ (96%)
Example 7: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-8-carboxamide
Reference compound N°6
The title compound was prepared in a manner similar to that described in Example 3 using quinoline-8-carboxylic acid.
, 48%

1H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) 52.08-2.41 (m, 4H), 3.66 - 3.55 (m, 1H), 3.71 -3.79 (m, 1H), 4.36 (dd, J = 17.8, 4.2 Hz, 1H), 4.50 - 4.62 (dd, J = 17.8, 5.1 Hz, 0.9H), 4.72 (dd, J = 18.0, 4.2 Hz, 0.1 H), 4.86 (br_d, J = 6.2 Hz, 1 H), 5.03 (d, J = 7 Hz, 0.1 H), 7.51 (dd, J = 8.3, 4.3 Hz, 1 H), 7.68 (t, J = 8 Hz 1 H), 7.99 (dd, J = 8.1 , 1.5 Hz, 1 H), 8.28 (dd, J = 8.3, 1.8 Hz, 1 H), 8.84 (dd, J = 7.4, 1.6 Hz, 1 H), 9.06 (dd, J = 4.3, 1.8 Hz, 1 H), 11.97 - 12.07 (br_s, 1 H). UPLC I (ESI) Rt 1.31 min, m/z 309.5 [M+H]+ (96%).
- -
Example 8: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-3-hydroxy-2-methylquinoline-4- carboxamide hydrochloride
The title compound was prepared in a manner similar to that described in Example 3 using 3- hydroxychinaldin-4-carboxylic acid.
29%

1 H NMR (400 MHz, MeOD) : δ 2.19-2.39 (m, 4H), 2.95 (s, 3H), 3.64-3.68 (m, 1 H), 3.79-3.88 (m , 1 H), 4.44 (d, J = 5.8 Hz, 2H), 4.86 - 4.90 (m , 1 H), 7.84 - 7.98 (m , 2H), 8.1 0 (d, J = 7.9 Hz, 1 H), 8.30 (d, J = 7.5 Hz, 1 H).
UPLC I (ESI) Rt 1 .1 1 min, m/z 339.6 [M+H]+ (95%). -(2-(2-Cyanopyrrolidin-1-yl)-2-oxoethyl)-6-fluoroquinoline-4-carboxamide.

This compound was prepared relying on the general approach, consisting of coupling an intermediate of formula (D) and a non-commercially available intermediate of formula (E). The latter was obtained from a Pfitzinger-type reaction on a commercially available isatin ('stepl '), followed by decarboxylation of the quinoline dicarboxylate product of the Pfitzinger-type reaction. This approach is summarized in Scheme 5.
Scheme 5. Synthesis of compounds of formula (I) involving a Pfitzinger-type condensation/decarboxylation.

Step 1 : 6-Fluoroquinoline-2,4-dicarboxylic acid.
To 5-fluoroindoline-2,3-dione (0.454 g, 2.75 mmol) was a 3M NaOH (5.46 m L, 16.52 mmol) solution added. The reaction mixture was heated until refluxing, and 2-oxopropanoate (0.364 g, 3.30 mmol) was added. After refluxing at 1 10°C for 4h, the mixture was cooled to room temperature. The pH was adjusted to 3-4 with 1 M HCI, and the precipitate was filtered and washed with water to yield whitish crystals
Yield: 0.453g, 70%
1 H NMR (400 MHz, DMSO-c¾): 6 7.89 (ddd, J = 9.27, 8.15, 2.92 Hz, 1 H), 8.35 (dd, J = 9.33, 5.81 Hz, 1 H), 8.57 (s, 1 H), 8.60 (dd, J = 1 1 .05, 2.86 Hz, 1 H).
MS (ESI) m/z 236.1 [M+H]+.
Step 2: 6-Fluoroquinoline-4- carboxylic acid.
6-Fluoroquinoline-2,4- dicarboxylic acid (0.103 g, 0.437 mmol) was transferred in a pressure tube, 6 mL water was added. The closed tube was heated to 200Ό for 4h. After slow cooling of the tube, the resulting precipitate was filtered and washed with water to yield white crystals
Yield: 0.076 g, 90%
1 H NMR (400 MHz, DMSO-d6) : δ 7.79 (ddd, J = 9.24, 8.20, 2.94 Hz, 1 H), 8.03 (d, J = 4.31 Hz, 1 H), 8.21 (dd, J = 5.86 Hz, J' = 9.27 Hz, 1 H), 8.52 (dd, J = 1 1 .19, 2.90 Hz, 1 H), 9.05 (d, J = 4.38 Hz, 1 H).
MS (ESI) m/z 192.1 [M+H]+, 189.9 [M-H]\
Step 3: (¾-A^(2-(2-Cyanopyrrolidin-1-yl)-2-oxoethyl)-6-fluoroquinoline-4-carboxamide
6-Fluoroquinoline-4- carboxylic acid (17) (0.054 g, 0.282 mmol) was dissolved in a 1 :1 mixture of dry DCM and THF (5 m L). 1 -chloro-/V,/V,2-trimethylprop-1 -en-1 -amine (0.052 mL, 0.395 mmol) was added to this solution, and the mixture was stirred for 30minut.es at rt. Then, a solution of (S)-1 -(2- aminoacetyl)pyrrolidine-2-carbonitril 2,2,2-trifluoroacetate (0.075 g, 0.282 mmol) prepared as described in general procedure B and triethylamine (80 iL, 0.571 mmol) in 3 m L dry THF was added, and the mixture was stirred for 2h. After evaporation of volatiles, the residue was dissolved in DCM (15 m L), washed with saturated sodium bicarbonate and brine. The organic layer was dried over sodium sulfate, filtrated and purified using flash chromatography (95-5 ethyl acetate - methanol) to yield white crystals
Yield: 0.019g, 20%
1 H NMR (400 MHz, CDCI3) (9/1 mixture of trans/cis amide rotamers) 62.21 - 2.41 (m, 4H), 3.57 (m, 1 H), 3.73 (m , 1 H), 4.27 (dd, J = 17.81 , 3.90 Hz, 1 H), 4.42 (dd, J = 17.86, 4.81 Hz, 1 H), 4.73 (d, J = 8.8 Hz, 0.1 H), 4.81 (m , 0.9H), 7.06 (br_s, 1 H), 7.51 - 7.56 (m , 1 H), 7.56 - 7.59 (m , 1 H), 8.01 (dd, J = 10.00, 2.81 Hz, 1 H), 8.17 (dd, J = 9.25, 5.45 Hz, 1 H), 8.95 (d, J = 4.36 Hz, 1 H).
UPLC I (ESI) Rt 1 .29 min, m/z 325.3 [M-H]+ (100%).
- -
MS (ESI) m/z 327.2 [M+H]+. -6-Chloro-AA(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide

The title compound was prepared in a manner similar to that described in Example 9
Step 1 : 6-Chloroquinoline-2,4-dicarboxylic acid.
1H NMR (400 MHz, DMSO-d6): δ 7.82 (dd, J = 9.0, 2.4 Hz, 1 H), 8.20 (d, J = 9.0 Hz, 1 H), 8.37 (s, 1H), 9.01 (d, J = 2.3 Hz, 1H).
MS (ESI) m/z 252.6 [M+H]+.
Step 2: 6-Chloroquinoline-4-carboxylic acid.
1H NMR (400 MHz, DMSO-c¾): δ 7.57 (d, J = 4.32 Hz, 1 H), 7.66 (dd, J = 8.96, 2.53 Hz, 1 H), 7.95 (d, J= 8.99 Hz, 1H), 8.80 (d, J= 4.33 Hz, 1H), 8.96 (d, J= 2.47 Hz, 1H).
MS (ESI) m/z 205.8 [M-H]+.
Step 3: (S)-6-Chloro-AA(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide.
Yield: 0.011 g, 10%. 1H NMR (400 MHz, CDCI3): 62.24-2.44 (m, 4H), 3.59 (m, 1H), 3.75 (m, 1H), 4.31 (dd, J= 17.95, 3.96 Hz, 1 H), 4.45 (dd, J = 17.89, 4.76 Hz, 1 H), 4.83 (m, 1 H), 7.07 (br_s, 1 H), 7.59 (d, J = 4.32 Hz, 1H), 7.74 (dd, J= 9.00, 2.33 Hz, 1H), 8.12 (d, J= 8.98 Hz, 1H), 8.36 (d, J= 2.23 Hz, 1H), 9.00 (d, J = 4.34 Hz, 1H).
UPLC I (ESI) Rt 1.41 min, m/z 343.5 [M+H]+ (95%).
MS (ESI) m/z 343.5 [M+H]+.
Example 11 : (S)-AA(2-(2-Cyanopyrrolidin-1-yl)-2-oxoethyl)-6-(trifluoromethoxy)quinoline-4-

The title compound was prepared in a manner similar to that described in Example 9
Step 1 : 6-(Trifluoromethoxy)quinoline-2,4- dicarboxylic acid.
Yield: 0.190g, 73%
1 H NMR (400 MHz, DMSO-d6): δ 7.95 (d, J = 9.38 Hz, 1 H), 8.41 (d, J = 9.23 Hz, 1 H), 8.58 (s, 1 H), 8.96 (s, 1 H).
MS (ESI) m/z 323.9 [M+Na]+ Step 2: 6-(Trifluoromethoxy)quinoline-4- carboxylic acid.
Yield: 0.132g, 86%
1 H NMR (400 MHz, DMSO-c¾) : δ 7.64 (d, J = 5.3 Hz, 1 H), 8.01 (dd, J = 2.80 Hz, J' = 9.0 Hz, 1 H), 8.27 (d, J = 8.8 Hz 1 H), 8.74 (d, J = 4.5 Hz 1 H), 9.03 (d, J = 2.8 Hz, 1 H).
MS (ESI) m/z 258.0 [M+H]+, 255.7 [M-H]+.
Step 3: (¾-AA(2-(2-Cyanopyrrolidin-1-yl)-2-oxoethyl)-6-(trifluoromethoxy)quinoline-4- carboxamide.
Yield: 0.015 g, 1 5%
1 H NMR (400 MHz, CDCI3) : (9/1 mixture of trans/cis amide rotamers) δ 2.20 - 2.42 (m, 4H), 3.57 (m, 1 H), 3.7 (m, 1 H), 4.30 (dd, J = 17.9, 3.9 Hz, 1 H), 4.43 (dd, J = 17.9, 4.8 Hz, 1 H), 4.74 (d, J = 8.8 Hz, 0.1 H), 4.81 (m, 0.9H), 7.20 (br_s, 1 H), 7.70 - 7.74 (m, 1 H), 7.75 (d, J = 4.6 Hz, 1 H), 8.28 (s, 1 H), 8.34 (d, J = 9.3 Hz, 1 H), 9.1 1 , (d, J = 4.6 Hz, 1 H). UPLC I (ESI) R, 1 .57 min, m/z 393.5 [M+H]+ (100%).
MS (ESI) m/z 393.5 [M+H]+.
Example 12: (S)-8-Chloro-A-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide

The title compound was prepared in a manner similar to that described in Example 9
Step 1 : 8-Chloroquinoline-2,4-dicarboxylic acid.
Yield: 0.1 88g, 68% 1 H NMR (400 MHz, DMSO-d6) : δ 7.70 (dd, J = 8.4, 8.04 Hz, 1 H), 8.1 1 (dd, J = 8.04, 1 .8 Hz, 1 H), 8.70 (ddd, J = 8.04, 2.63, 1 .8 Hz, 1 H), 8.74 (d, J = 2.63 Hz, 1 H).
MS (ESI) m/z 273.9 [M+Na]+.
Step 2: 8-Chloroquinoline-4-carboxylic acid.
Yield: 0.081 g, 54%
- -
1H NMR (400 MHz, DMSO-c¾): δ 7.71 (dd, J= 8.58, 7.57 Hz, 1H), 8.03 (d, J = 4.34 Hz, 1H), 8.06 (dd, J= 7.53, 1.21 Hz, 1H), 8.67 (dd, J= 8.62, 1.22 Hz, 1H), 9.16 (d, J = 4.34 Hz, 1H).
MS (ESI) m/z 208.5 [M+H]+. Step 3: (¾-8-Chloro-A^(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide.
Yield: 0.019 g, 20%
1H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) 62.32 (m, 4H), 3.56 (m, 1H), 3.73 (m, 1H), 4.29 (dd, J= 17.9, 3.9 Hz, 1H), 4.41 (dd, J= 17.81, 4.83 Hz, 1H), 4.69 (d, J= 8.8 Hz, 0.1 H), 4.81 (m, 0.9H), 7.02 (br s, 1 H), 7.54- 7.58 (m, 1 H), 7.62 (d, J = 4.3 Hz, 1 H), 7.92 (dd, J = 7.5, 1.2 Hz, 1H), 8.24 (dd, J= 8.5, 1.2 Hz, 1H), 9.12 (d, J=4.30 Hz, 1H). UPLC I (ESI) R,1.36 min, m/z 343.8 [M+H]+ (96%).
MS (ESI) m/z 343.8 [M+H]+.
Example 13: (S)-8-Bromo-A-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide

The title compound was prepared in a manner similar to that described in Example 9 Step 1
Yield: 0.350g, 85%
1H NMR (400 MHz, DMSO-d6): δ 7.70 - 7.74 (m, 1 H), 8.29 - 8.31 (m, 1 H), 8.50 (s, 1 H), 8.79 - 8.81 (dd, J= 8.6, 1.2 Hz, 1 H),14.02 (s, 2H)
MS (ESI) m/z 297.3 [M+H]+
Step 2
Yield: 0.21 Og, 90%
1H NMR (400 MHz, CDCI3): 58.04 (s, 1H), 8.15 (d, J= 7.52 Hz, 1H), 8.70 (s, 1H), 8.87 (d, J= 8.61 Hz, 1 H), 9.20 (d,J= 4.39 Hz, 1H)
MS (ESI) m/z 253.4 [M+H]+
(S)-8-Bromo-A-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide.
Yield: 0.011 g, 12%.
- -
1 H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) 6 2.16 - 2.45 (m, 4H), 3.59 (m, 1 H), 3.75 (m , 1 H), 4.31 (dd, J = 17.85, 4.01 Hz, 1 H), 4.44 (dd, J = 17.86, 4.80 Hz, 1 H), 4.72 (d, J = 7.1 Hz, 0.1 H), 4.83 (d, J = 5.15 Hz, 0.9H), 7.04 (br s, 1 H), 7.52 (dd, J = 13.81 , 6.28 Hz , 1 H) , 7.63 (d, J = 4.30 Hz, 1 H), 8.16 (dd, J = 7.49, 1 .21 Hz, 1 H), 8.31 (dd, J = 8.47, 1 .20 Hz, 1 H), 9.14 (d, J = 4.29 Hz, 1 H).
UPLC I (ESI) Rt 1 .39 min, m/z 386.2 [M+H]+ (100%).
MS (ESI) m/z 388.1 [M+H]+.
Example 14: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-6-methoxyquinoline-4-

The title compound was prepared in a manner similar to that described in Example 9
Step 1 : 6-methoxyquinoline-2,4-dicarboxylic acid
1 H NMR (400 MHz, DMSO-d6): δ 3.95 (s, 3H), 7.57 - 7.63 (dd, J = 9.3, 2.8 Hz, 1 H), 8.13 - 8.1 9 (d, J = 9.2 Hz, 1 H), 8.24 - 8.29 (d, J = 2.8 Hz, 1 H), 8.47 - 8.54 (s, 1 H)
UPLC I (ESI) Rt 1 .01 min, m/z 248.5 [M+H]+ (99%).
Step 2: 6-methoxyquinoline-4-carboxylic acid
The starting material was reacted in a pressured tube for 1.5 hour at 200°C with stirring. And worked up as described in example 9.
1 H NMR (400 MHz, DMSO-d6): δ 3.85 - 3.96 (s, 3H), 7.45 - 7.55 (dd, J = 9.0, 3.0 Hz, 1 H), 7.88 - 7.99 (d, J = 4.5 Hz, 1 H), 7.99 - 8.08 (d, J = 9.2 Hz, 1 H), 8.13 - 8.23 (d, J = 3.0 Hz, 1 H),8.83 - 8.92 (d, J = 4.5 Hz, 1 H).
UPLC I (ESI) R, 0.44 min, m/z 204.5 [M+H]+ (99%).
Step 3: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-6-methoxyquinoline-4-carboxamide
Yield: 32mg, 34%
1 H NMR (400 MHz, CDCI3) δ 2.22 - 2.42 (m, 4H), 3.51 - 3.60 (m, 1 H), 3.70 - 3.78 (m, 1 H), 3.93 - 3.99 (s, 3H), 4.28 - 4.36 (dd, J = 17.8, 4.2 Hz, 1 H), 4.36 - 4.44 (dd, J = 17.9, 4.7 Hz, 1 H), 4.77 - 4.82 (m , 1 H), 7.01 - 7.07 (s, 1 H), 7.39 - 7.44 (dd, J = 9.2, 2.8 Hz, 1 H), 7.49 - 7.53 (d, J = 4.4 Hz, 1 H), 7.66 - 7.70 (d, J = 2.8 Hz, 1 H), 8.01 - 8.07 (d, J = 9.2 Hz, 1 H), 8.79 - 8.85 (d, J = 4.4 Hz, 1 H) . UPLC I (ESI) Rt 1 .18 min, m/z 339.7 [M+H]+ (95%).
- -
Example 15: (S)-7-bromo-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide

The title compound was prepared in a manner similar to that described in Example 9 Step 1 : 7-bromoquinoline-2,4-dicarboxylic acid
1 H NMR (400 MHz, DMSO-cfe): δ 7.90 - 8.04 (dd, J = 9.1 , 2.1 Hz, 1 H), 8.40 - 8.53 (m , 2H), 8.74 - 8.79 (d, J = 9.2 Hz, 1 H)
UPLC I (ESI) Rt 1 .27 min, m/z 296.8, 298.8 [M+H]+ (99%). Step 2: 7-bromo-4-quinoline-carboxylic acid
The starting material was reacted in a pressured tube for 50 min at 200 °C with stirring. And worked up as described in example 9
1 H NMR (400 MHz, DMSO-d6) : δ 7.54 - 7.60 (d, J = 4.4 Hz, 1 H), 7.69 - 7.75 (dd, J = 8.4, 7.5 Hz, 1 H), 7.98 - 8.05 (dd, J = 7.6, 1 .2 Hz, 1 H), 8.07 - 8.16 (dd, J = 8.4, 1 .2 Hz, 1 H), 8.91 - 9.01 (d, J = 4.3 Hz, 1 H).
UPLC I (ESI) Rt 1 .19 min, m/z 252.4, 254.4 [M+H]+ (99%).
Step 3: (S)-7-bromo-N-(2-(2-cyanopyrrolidin-1 -yl)-2-oxoethyl)quinoline-4-carboxamide
Yield: 33mg, 54%
1 H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) 52.1 1 - 2.40 (m, 4H), 3.48 - 3.58 (m , 1 H), 3.65 - 3.74 (ddd, J = 9.8, 7.4, 3.0 Hz, 1 H), 4.20 - 4.29 (dd, J = 17.8, 4.0 Hz, 1 H), 4.28 - 4.34 (dd, J= 17.8 Hz, J = 4.1 Hz, 0.1 H), 4.32 - 4.42 (dd, J = 17.8, 4.8 Hz, 1 H), 4.52-4.58 (dd, J= 17.5 Hz, J = 5.3 Hz, 0.1 H), 4.70 (d, J = 8.0 Hz, 0.1 H), 4.74 - 4.80 (m , 0.9H), 7.01 - 7.07 (m, 1 H), 7.50 -7.55 (d, J = 4.4 Hz, 1 H), 7.65 -7.71 (dd, J = 9.0, 2.0 Hz, 1 H), 8.15 - 8.22 (d, J = 9.0 Hz, 1 H), 8.29 - 8.34 (d, J = 2.0 Hz, 1 H), 8.93 - 8.97 - (d, J = 4.3 Hz, 1 H).
13C NMR (101 MHz, CDCI3) δ 166.84, 166.74, 1 50.88, 149.41 , 140.77, 132.24, 131 .39, 126.77, 124.46, 123.16, 1 19.05, 1 17.71 , 46.75, 45.63, 42.50, 29.95, 25.09.
UPLC I (ESI) Rt 1 .44 min, m/z 386.4, 389.4 [M+H]+ (99%).
- -
Example 16: (S)-7-chloro-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide

The title compound was prepared in a manner similar to that described in Example 9
Step 1 : 7-chloroquinoline-2,4-dicarboxylic acid
1 H NMR (400 MHz, DMSO-cfe) : δ 7.85 - 7.92 (dd, J = 9.1 , 2.3 Hz, 1 H), 8.28 -8.33 (d, J = 2.3 Hz,
1 H), 8.45 -8.53 (s, 1 H), 8.82 - 8.89 (d, J = 9.2 Hz, 1 H).
UPLC I (ESI) R, 1 .24 min, m/z 252.3 [M+H]+ (96%).
Step 2: 7-chloroquinoline-4-carboxylic acid
The starting material was reacted in a pressured tube for 1.5 hour at 200 Ό with stirring. And worked up as described in example 9.
1 H NMR (400 MHz, DMSO-cfe) : δ 7.74 - 7.83 (dd, J = 9.2, 2.3 Hz, 1 H), 7.93 - 8.02 (d, J = 4.4 Hz, 1 H), 8.15 - 8.24 (d, J = 2.3 Hz, 1 H), 8.71 - 8.83 (d, J = 9.1 Hz, 1 H), 9.06 - 9.15 (d, J = 4.4 Hz, 1 H), 13.34 - 14.27 (br s, 1 H).
UPLC I (ESI) Rt 1 .15 min, m/z 208.4 [M+H]+ (98%).
Step 3: (S)-7-chloro-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide
Yield: 43mg, 62%
1 H NMR (400 MHz, DMSO-d6): (9/1 mixture of trans/cis amide rotamers) δ 1 .92 - 2.34 (m, 4H), 3.50 - 3.59 (td, J = 9.0, 6.8 Hz, 1 H), 3.70 - 3.78 (ddd, J = 9.4, 7.6, 3.7 Hz, 1 H), 4.14 - 4.22 (dd, J = 17.0, 5.7 Hz, 1 H), 4.22 - 4.30 (dd, J = 17.0, 6.2 Hz, 0.9H), 4.36 - 4.42 (dd, J = 16.8, 5.8 Hz, 0.1 H), 4.83 - 4.88 (dd, J = 7.3, 3.6 Hz, 0.9H), 5.28 - 5.30 (dd, J = 7.0, 2.4 Hz, 0.1 H), 7.59 - 7.65 (d, J = 4.3 Hz, 1 H), 7.72 - 7.79 (dd, J = 9.0, 2.3 Hz, 1 H), 8.13 - 8.21 (d, J = 2.2 Hz, 1 H), 8.38 - 8.45 (d, J = 8.9 Hz, 1 H), 9.02 - 9.07 (d, J = 4.3 Hz, 1 H), 9.1 1 - 9.17 (t, J = 5.9 Hz, 1 H).
13C NMR (101 MHz, CDCI3) δ 166.89, 166.76, 1 50.94, 149.21 , 140.69, 136.16, 128.88 (2carbon atoms), 126.78, 122.88, 1 18.92, 1 1 7.72, 46.75, 45.63, 42.50, 29.95, 25.09.
UPLC I (ESI) Rt 1 .40 min, m/z 343.6 [M+H]+ (99%).
- -
Example 17: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-5-carboxamide
Reference compound N° 7

The title compound was prepared in a manner similar to that described in Example 3 starting from the commercially available quinoline-5-carboxylic acid
Yield: 24mg, 40%
1 H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) δ 2.12 - 2.43 (m , 4H), 3.52 - 3.60 (td, J = 8.5, 6.1 Hz, 1 H), 3.68 - 3.77 (ddd, J = 9.6, 7.1 , 2.8 Hz, 1 H), 4.21 - 4.31 (dd, J = 17.8, 4.0 Hz, 1 H), 4.37 - 4.46 (dd, J = 1 7.8, 4.9 Hz, 0.9H), 4.56 - 4.63 (dd, J = 17.2, 5.6 Hz, 0.1 H), 4.76 - 4.83 (m , 1 H), 6.98 - 7.01 (s, OH), 7.01 - 7.10 (s, 1 H), 7.45 - 7.50 (dd, J = 8.6, 4.1 Hz, 1 H), 7.67 - 7.74 (m , 1 H), 7.78 - 7.84 (dd, J = 7.2, 1 .4 Hz, 1 H), 8.18 - 8.26 (m , 1 H), 8.76 - 8.85 (dt, J = 8.5, 1 .3 Hz, 1 H), 8.93 - 9.00 (dd, J = 4.1 , 1 .7 Hz, 1 H).
UPLC I (ESI) Rt 0.94min, m/z 309.6 [M+H]+ (98%).
Example 18: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-phenylisonicotinamide
The title compound is prepared as described in scheme 2b
Stepl : 2-phenylisonicotinic acid
2-bromoisonicotinic acid (0.210 g, 1 .040 mmol) was dissolved in degassed DME (Volume: 8 ml) under nitrogen. Tetrakis(triphenylphosphine)palladium(0) (0.060 g, 0.052 mmol) was added, the resulting reaction mixture was stirred for 15min.Then aqueous potassium carbonate (4.16 ml, 8.32 mmol) and phenylboronic acid (0.171 g, 1 .403 mmol) were added subsequently. The resulting RM was refluxed at 95 °C for 18h and then cooled to rt. After filtration over celite the reaction mixture was acidified to pH 3-4 and the white precipitate was filtered off and washed with water.
This resulted in a white powder after recrystallization from 2-methoxyethanol.
Yield: 0.1 06 g, 57%.
1 H NMR (400 MHz, DMSO-c¾): δ 7.44 - 7.60 (m, 3H), 7.71 - 7.86 (dd, J = 4.9, 1 .5 Hz, 1 H), 8.05 - 8.19 (m, 2H), 8.23 - 8.35 (t, J = 1 .2 Hz, 1 H), 8.79 - 8.93 (dd, J = 5.1 , 0.8 Hz, 1 H), 13.56 - 13.97 (s, 1 H).
UPLC I (ESI) Rt 1 .37 min, m/z 200.5 [M+H]+ (92%).
- -
Step 2: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-phenylisonicotinamide
 1H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) δ 2.15 - 2.43 (m, 4H), 3.49 - 3.58 (td, J = 8.6, 7.6, 5.9 Hz, 1H), 3.67 - 3.75 (ddd, J = 14.9, 8.4, 4.0 Hz, 1H), 4.17 - 4.26 (dd, J = 18.0, 3.7 Hz, 1 H), 4.31 - 4.40 (dd, J = 18.0, 4.7 Hz, 0.9H), 4.46 - 4.53 (dd, J = 17.6, 5.2 Hz, 0.1 H), 4.71 - 4.75 (d, J = 8.3 Hz, 0.1 H), 4.77 - 4.83 (dd, J = 8.0, 2.4 Hz, 0.9H), 7.36 - 7.41 (s, 1 H), 7.42 - 7.53 (m, 3H), 7.56 - 7.60 (dd, J = 5.0, 1.6 Hz, 1H), 8.02 - 8.07 (m, 2H), 8.10 - 8.13 (dd, J = 1.6, 0.9 Hz, 1 H), 8.79 - 8.83 (dd, J = 5.0, 0.9 Hz, 1 H)
1H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) δ 2.15 - 2.43 (m, 4H), 3.49 - 3.58 (td, J = 8.6, 7.6, 5.9 Hz, 1H), 3.67 - 3.75 (ddd, J = 14.9, 8.4, 4.0 Hz, 1H), 4.17 - 4.26 (dd, J = 18.0, 3.7 Hz, 1 H), 4.31 - 4.40 (dd, J = 18.0, 4.7 Hz, 0.9H), 4.46 - 4.53 (dd, J = 17.6, 5.2 Hz, 0.1 H), 4.71 - 4.75 (d, J = 8.3 Hz, 0.1 H), 4.77 - 4.83 (dd, J = 8.0, 2.4 Hz, 0.9H), 7.36 - 7.41 (s, 1 H), 7.42 - 7.53 (m, 3H), 7.56 - 7.60 (dd, J = 5.0, 1.6 Hz, 1H), 8.02 - 8.07 (m, 2H), 8.10 - 8.13 (dd, J = 1.6, 0.9 Hz, 1 H), 8.79 - 8.83 (dd, J = 5.0, 0.9 Hz, 1 H)
UPLC I (ESI) Rt 1.45 min, m/z 335.6 [M+H]+ (98%).
Example 19: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-(3,4- dimethoxyphenyl)isonicotinamide
Yield: 0.130 g, 49%.
The title compound was prepared in a manner similar to that described in Example 14.
Step 1 : 2-(3,4-dimethoxyphenyl)isonicotinic acid
1H NMR (400 MHz, DMSO-d6): δ 3.78 - 3.84 (s, 3H), 3.85 - 3.89 (s, 3H), 7.01 - 7.14 (d, J = 8.4 Hz, 1 H), 7.61 - 7.78 (m, 3H), 8.20 - 8.33 (t, J = 1.2 Hz, 1 H), 8.72 - 8.87 (dd, J = 5.0, 0.9 Hz, 1 H), 13.53 - 13.88 (s, 1H).
UPLC I (ESI) Rt 1.21 min, m/z 260.5 [M+H]+ (93%).
Step 2: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-(3,4-dimethoxyphenyl)isonicotinamide

1H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) 52.16-2.42 (m, 4H), 3.50 - 3.60 (m, 1H), 3.68-3.78 (m, 1H), 3.92-3.97 (s, 3H), 3.99-4.03 (s, 3H), 4.21 (dd, J= 18.0, 3.7 Hz, 1H), 4.36 (dd, J= 18.0, 4.7 Hz, 0.9H), 4.46 - 4.52 (dd, J= 17.3, 5.1 Hz, 0.1 H), 4.72-4.74 (d, J = 8.1 Hz, 0.1 H), 4.76-4.83 (dd, J = 8.1, 2.8 Hz, 0.9H), 6.97 (d, J= 8.4 Hz, 1H), 7.33-7.40 (t, J = 3.9 Hz, 1H), 7.48-7.52 (dd, J= 5.1, 1.6 Hz, 1H), 7.57-7.61 (dd, J= 8.4, 2.1 Hz, 1H), 7.66-7.73 (d, J= 2.1 Hz, 1 H), 8.00 - 8.11 (s, 1 H), 8.69 - 8.82 (dd, J= 5.0, 0.9 Hz, 1 H).
UPLC I (ESI) Rt1.34 min, m/z 395.5 [M+H]+ (100%).
Yield: 0.034 g, 42%.
- -
Example 20: (S)-2-(4-cyanophenyl)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)isonicotinamide
The title compound was prepared in a manner similar to that described in Example 14.
Step 1 : 2-(4-cyanophenyl)isonicotinic acid
Yield: 0.154 g, 58%.
1H NMR (400 MHz, DMSO-c¾): δ 7.82 - 7.91 (dd, J = 4.8, 1.1 Hz, 1H), 7.95 - 8.06 (m, 2H), 8.30 - 8.39 (m, 2H), 8.39 - 8.45 (t, J = 1.1 Hz, 1H), 8.87 - 8.94 (d, J = 4.8 Hz, 1H), 13,83 - 14.15 (br_s, 1H).
UPLC I (ESI) R, 1.44 min, m/z 225.4 [M+H]+ (96%).
Step 2: (S)-2-(4-cyanophenyl)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)isonicotinamide
Yield: 0.045 g, 44%.

1H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) δ 2.18 - 2.42 (m, 4H),3.51 - 3.58 (m, 1H), 3.70 - 3.81 (m, 1H), 4.13 - 4.23 (dd, J = 17.8, 3.5 Hz, 0.9H), 4.28 - 4.34 (dd, J = 17.4, 3.6 Hz, 0.1 H), 4.43-4.53 (dd, J= 17.8, 5.7 Hz, 1H), 4.71 -4.73 (dd, J = 9.6, 1.9 Hz, 0.1 H), 4.74-4.84 (m, 0.9H), 7.61 (dd, J= 5.0, 1.5 Hz, 1H), 7.72-7.81 (m, 3H), 8.05-8.16 (m, 3H), 8.76 (d, J= 5.0 Hz, 1H).
UPLC I (ESI) Rt1.44 min, m/z 360.6 [M+H]+ (100%).
Example 21 : (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-1-methyl-1H-imidazole-5- carboxamide
The title compound was prepared in a manner similar to that described in Example 3 using 1- methyl-1 H-imidazole-5-carboxylic acid.
Yield: 46mg, 59%

1H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) δ 2.12 - 2.40 (m, 4H)), 3.44 - 3.54 (td, J = 9.0, 8.4, 6.3 Hz, 1 H), 3.64 - 3.71 (ddd, J = 9.4, 7.0, 3.0 Hz, 1 H), 3.84 - 3.97 (s, 3H), 4.05 - 4.13 (dd, J = 17.7, 3.9 Hz, 0.9H), 4.15 - 4.23 (m, 0.1 H), 4.24 - 4.33 (dd, J = 17.7, 5.2 Hz,
- -
0.9H), 4.34 - 4.43 (dd, J = 17.3, 5.2 Hz, 0.1 H), 4.68 - 4.72 (dd, J = 7.8, 1 .9 Hz, 0.1 H), 4.75 - 4.81 (m, 0.9H), 7.02 - 7.08 (d, J = 5.6 Hz, 1 H), 7.44 - 7.51 (s, 1 H), 7.51 - 7.57 (s, 1 H).
UPLC I (ESI) Rt 0.29 min, m/z 262.6 [M+H]+ (98%) Example 22: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-4-methylthiazole-5-carboxamide
The title compound was prepared in a manner similar to that described in Example 3 using 4- methylthiazole-5-carboxylic acid.
Yield: 57mg, 48%

1 H NMR (400 MHz, CDCI3): (9/1 mixture of trans/cis amide rotamers) δ 2.14 - 2.38 (m, 4H), 2.73 - 2.76 (s, 3H), 3.44 - 3.54 (m, 1 H), 3.64 - 3.71 (ddd, J = 9.3, 7.2, 3.3 Hz, 1 H), 4.05 - 4.16 (dd, J = 18.0, 3.4 Hz, 0.9H), 4.18 - 4.26 (dd, J = 1 7.4, 3.5 Hz, 0.1 H), 4.27 - 4.38 (dd, J = 17.8, 5.1 Hz, 0.9H), 4.40 - 4.47 (dd, J = 17.4, 5.1 Hz, 0.1 H), 4.71 - 4.74 (d, J = 7.7 Hz, 0.1 H), 4.74 - 4.79 (d, J = 7.7 Hz, 0.9H), 7.04 - 7.13 (d, J = 7.3 Hz, 1 H), 8.67 - 8.71 (s, 1 H).
UPLC I (ESI) Rt 1 .08 min, m/z 279.6 [M+H]+ (100%)
Example 23: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-4-methyloxazole-5-carboxamide
The title compound was prepared in a manner similar to that described in Example 3 using 4- methyloxazole-5-carboxylic acid.
Yield: 34mg, 36%

1 H NMR (400 MHz, CDCI3) : (9/1 mixture of trans/cis amide rotamers) δ 2.18 - 2.40 (m, 4H, 2.45 - 2.58 (s, 3H), 3.43 - 3.56 (m, 1 H), 3.64 - 3.72 (ddd, J = 9.5, 7.1 , 2.8 Hz, 1 H), 4.09 - 4.16 (dd, J = 17.9, 4.0 Hz, 0.9H), 4.17 - 4.22 (m , 0.1 H), 4.22 - 4.31 (dd, J = 17.9, 4.9 Hz, 0.9H), 4.39 - 4.46 (dd, J = 17.3, 5.6 Hz, 0.1 H), 4.74 - 4.83 (d, 8.0 Hz, 0.1 H), 4.76 - 4.83 (dd, J = 8.0, 2.6 Hz, 0.9H), 7.12 - 7.19 (s, 1 H), 7.75 - 7.83 (s, 1 H).
UPLC I (ESI) R, 1 .04 min, m/z 263.4 [M+H]+ (96%)
- -
(S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)piperidine-4-carboxamide

Step 1 : 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid
The piperidine-4-carboxylic acid (1 g, 7.74 mmol) was added to a stirred solution of guanidine hydrochloride (0.1 1 1 g, 1 .161 mmol)and di-tert-butyl dicarbonate (4.22 g, 19.36 mmol) in EtOH (7 m l_), at 35-40 °C. The mixture was stirred overnight. Then the Ethanol was evaporated and the residue was dissolved in CH2CI2 (or EtOAc) and filtered to separate the catalyst, the filtrate was evaporated and washed with hexane to yield a white powder.
1 H NMR (400 MHz, MeOD) : δ 1 .38 - 1 .52 (s, 9H), 1 .47 - 1 .61 (m , 2H), 1 .83 - 1 .94 (dq, J = 13.9, 3.6 Hz, 2H), 2.43 - 2.55 (tt, J = 1 1 .0, 4.0 Hz, 1 H), 2.83 - 2.96 (br s, 2H), 3.94 - 4.02 (td, J = 4.0, 1 .3 Hz, 2H).
MS (ESI) m/z 230.4 [M+H]+
Step 2: (S)-tert-butyl 4-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethylcarbamoyl)piperidine-1- carboxylate
The title compound was prepared in a manner similar to that described in Example 3 using 1 -(tert- butoxycarbonyl)piperidine-4-carboxylic acid
UPLC I (ESI) Rt 1 .47 min, m/z 365.6 [M+H]+ (90%)
Step 3: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)piperidine-4-carboxamide
To a solution of (S)-tert-butyl 4-(2-(2-cyanopyrrolidin-1 -yl)-2-oxoethylcarbamoyl)piperidine-1 - carboxylate (0.100 g, 0,274 mmol) in 2ml Acetonitrile was 4-methylbenzenesulfonic acid hydrate (0,073 g, 0,384 mmol) added and the mixture was stirred for 24h. After evaporation of the volatiles, the sample was purified by preparative HPLC to yield an reddish oil.
Yield: 20mg, 35%
1 H NMR (400 MHz, MeOD) : (9/1 mixture of trans/cis amide rotamers) δ 1 .58 - 1 .76 (m , 2H), 1 .77 - 1 .89 (m , 2H), 2.1 1 - 2.22 (m, 2H), 2.19 - 2.31 (m, 2H), 2.39 - 2.54 (tt, J = 1 1 .6, 3.8 Hz, 1 H), 2.58 - 2.73 (td, J = 12.5, 2.9 Hz, 2H), 3.06 - 3.12 (t, J = 3.3 Hz, 1 H), 3.09 - 3.15 (t, J = 3.4 Hz, 1 H), 3.48 - 3.61 (dt, J = 9.5, 7.5 Hz, 1 H), 3.63 - 3.77 (m, 1 H), 3.92 - 4.04 (d, J = 17.1 Hz, 1 H), 3.99 - 4.12 (d, J = 17.1 Hz, 1 H), 4.72 - 4.80 (dd, J = 6.1 , 4.6 Hz, 0.1 H) , 5.03 - 5.07 (m , 0.1 H).13C NMR (101 MHz,
- -
MeOD): δ 178.10, 169.80, 119.55, 47.9, 46.90, 46.11, 43.66, 42.70, 42.48, 30.97, 29.85, 26.13, 23.76.
UPLC I (ESI) Rt0.26 min, m/z 265.5 [M+H]+ (97%) Example 25: (S)-5-chloro-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide
The title compound was prepared in a manner similar to that described in Example 9

Step 1 : 5-chloroquinoline-2,4-dicarboxylic acid
1H NMR (400 MHz, DMSO-c¾) δ 8.22 - 8.16 (dd, J= 7.3, 2.4 Hz, 1H), 7.98 - 7.90 (s, 1H), 7.88 - 7.81 (m, 2H);
UPLC I (ESI) Rt0.60 min, m/z 252.4 [M+H]+ (95%)
Step 2: 5-chloroquinoline-4-carboxylic acid
1H NMR (400 MHz, DMSO-d6) δ 8.83 - 8.76 (d, J = 4.3 Hz, 1 H), 7.98 - 7.92 (dd, J = 7.4, 2.4 Hz, 1H), 7.70 - 7.63 (m, 2H), 7.25 - 7.19 (d, J = 4.4 Hz, 1H); UPLC I (ESI) Rt 1.13 min, m/z 208.5 [M+H]+ (98%)
(S)-5-chloro-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide
Yield: 58mg, 42%
1H NMR (400MHz, CDCI3) δ 8.97 (d, J = 4.27 Hz, 1H), 8.12 (t, J = 4.86 Hz, 1H), 7.68 (dd, J = 4.89, 0.86 Hz, 2H), 7.47 (d, J = 4.27 Hz, 1 H), 6.89 (s, 1 H), 4.79 (d, J = 6.74 Hz, 1 H), 4.32 (br d, J = 20.2H), 3.77 - 3.67 (m, 1 H), 3.60 - 3.51 (m, 1 H), 2.41 - 2.20 (m, 4H);
UPLC I (ESI) Rt1.26 min, m/z 343.6 [M+H]+ (96%); LC-MS (l-B) Rt 11.6 min, m/z 343.1 [M+H]+ (96%).
Example 26: (S)-5-bromo-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide
The title compound was prepared in a manner similar to that described in Example 9

Step 1 : 5-bromoquinoline-2,4-dicarboxylic acid
- -
1H NMR (400 MHz, DMSO-c¾) 58.27 (dd, J= 8.5, 1.1 Hz, 1H), 8.16 (dd, J= 7.6, 1.1 Hz, 1H), 8.07 (s, 1H), 7.83 (dd, J= 8.4, 7.7 Hz, 1H); UPLC I (ESI) Rt1.02 min, m/z 296.5, 298.5 [M+H]+ (95%) Step 2: 5-bromoquinoline-4-carboxylic acid
The starting material was reacted in a pressured tube for 50 minutes at 205 °C with stirring.
1H NMR (400 MHz, DMSO-d6) δ 9.01 - 8.91 (d, J = 4.3 Hz, 1H), 8.16 - 8.07 (dd, J= 8.4, 1.2 Hz, 1 H), 8.05 - 7.98 (dd, J = 7.6, 1.2 Hz, 1 H), 7.75 - 7.69 (dd, J = 8.4, 7.5 Hz, 1 H), 7.60 - 7.54 (d, J = 4.4 Hz, 1 H); UPLC I (ESI) Rt 1.36 min, m/z 252.4,254.4 [M+H]+ (98%).
Step 3: (S)-5-bromo-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)quinoline-4-carboxamide
Yield: 39mg, 42%
1H NMR (400MHz, CDCI3) δ 8.95 (d, J=4.24 Hz, 1H), 8.16 (dd, J= 8.51, 1.25 Hz, 1H), 7.90 (dd, J = 7.51, 1.33 Hz, 1H), 7.60 (dd, J= 8.47, 7.51 Hz, 1H), 7.49 (d, J= 4.25 Hz, 1H), 6.97 (s, 1H), 4.74- 4.81 (m, 1H), 4.34 (s, 2H), 3.72 (ddd, J= 11.35, 6.78, 2.48 Hz, 1H), 3.59 - 3.50 (m, 1H), 2.43 - 2.13 (m, 4H);
UPLC I (ESI) Rt 1.29 min, m/z 387.5, 389.5 [M+H]+ (96%); LC-MS (l-B) Rt 11.9 min, m/z 387.1 ,388.9 [M+H]+ (96%); HRMS calcd for: C17H16N402Br [M + H]+, 387.0457; found, 387.0448.
Example 27: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-5-methylquinoline-4-carboxamide The title compound was prepared in a manner similar to that described in Example 3.
Yield: 72mg, 47%
1H NMR (400 MHz, CDCI3) δ 8.93 - 8.84 (m, 1 H), 8.04 - 7.96 (m, 1 H), 7.62 (dd, J = 7.06, 8.49 Hz, 1 H), 7.38 (dt, J = 1.20, 7.04 Hz, 1 H), 7.35 (d, J = 4.24 Hz, 1 H), 7.06 (t, J = 4.50 Hz, 1 H), 4.75 (dd, J = 1.99, 7.79 Hz, 0.1 H), 4.72 - 4.67 (m, 0.9H), 4.56 (dd, J = 5.53, 17.38 Hz, 0.1 H), 4.37 (dd, J = 4.95, 17.82 Hz, 0.9H), 4.32 (dd, J = 5.53, 17.38 Hz, 0.1 H) 4.23 (dd, J = 3.95, 17.81 Hz, 0.9H), 3.70 (ddd, J = 2.77, 7.09, 11.25 Hz, 1H), 3.52 (dt, J = 7.63, 10.69 Hz, 1H), 2.65 (s, 3H), 2.42 - 2.09 (m, 4H).
13C NMR (101 MHz, CDCI3) δ 170.38, 166.83, 149.68, 149.13, 141.90, 134.49, 130.19, 129.73, 128.76, 123.43, 119.96, 117.89,46.78,45.74,42.61,30.05,25.19,21.31.
UPLC I (ESI) Rt 1.07 min, m/z 323.5 [M+H]+ (99%); LC-MS (l-B) Rt10.0 min, m/z 323.1 [M+H]+ (98%)
- -
Example 28: (S)-N-(2-(2-cyano-4,4-difluoropyrrolidin-1-yl)-2-oxoethyl)-6-methoxyquinoline-4- carboxamide
The title compound was prepared in a manner similar to that described in Example 1 .
Yield: 61 mg, 33%

1 H NMR (400 MHz, CDCI3) (9/1 mixture of trans/cis amide rotamers) δ 8.63 (d, J = 4.43 Hz, 1 H), 7.90 (d, J = 9.19 Hz, 1 H), 7.54 (d, J = 2.75 Hz, 1 H), 7.47 - 7.40 (br s, 1 H), 7.35 (d, J = 4.40 Hz, 1 H), 7.31 (dd, J = 2.78, 9.24 Hz, 1 H), 5.1 9 - 5.12 (m, 0.1 H), 4.91 (dd, J = 4.58, 8.37 Hz, 0.9H), 4.52 (d, J = 5.77 Hz, 0.1 H), 4.30 (dd, J = 5.71 , 17.35 Hz, 0.9H), 4.12 (dd, J = 4.73, 17.33 Hz, 1 H), 4.08 - 3.86 (m , 2H), 3.86 (s, 3H), 2.83 - 2.65 (m , 4H).
13C NMR (101 MHz, CDCI3) δ 1 68.18, 167.71 , 158.77, 147.05, 144.85, 139.04, 131 .13, 127.88, 125.54, 123.14, 1 19.15, 1 16.40, 102.85, 55.83,52.05 (t, J = 32.25 Hz), 44.41 , 42.20, 37.26 (t, J = 25.29 Hz).
UPLC I (ESI) R, 1 .28 min, m/z 375.6 [M+H]+ (99%) ; LC-MS (l-B) (ESI) R, 1 1 .6 min, m/z 374.9 [M+H]+ (98%).
Example 29: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-5-phenylquinoline-4-carboxamide

To a solution containing this acid 5-phenylquinoline-4-carboxylic acid (0,04 g, 0,160 mmol) and HOBT( Hydroxybenzotriazoie ) (0,027 g, 0,177 mmol) in 1 ,4-dioxane (5 m L) was added a solution of EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide HCI) (0,034 g, 0,1 77 mmol) in CH2CI2 (5 mL). The mixture was stirred for 10 min at room temperature. To the resulting solution was added the appropriate amine (S)-1 -(2-aminoacetyl)pyrrolidine-2-carbonitrile 4-methylbenzenesulfonate (0,063 g, 0,193 mmol) and D IPEA (0,059 ml, 0,337 mmol) in CH2CI2 (4 mL), with stirring.
After 2 h, the reaction mixture was diluted with CH2CI2 and washed with saturated aqueous NaHC03 solution (10 mL), 0.2N aqueous citric acid solution (10 mL) and brine (10 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The mixture was purified using column chromatography with dichloromethane-methanol as an eluent.
Yield: 52mg, 84%
- -
9/1 mixture of trans/cis amide rotamers, major rotamer: 1H NMR (400 MHz, CDCI3) δ 8.98 (d, J = 4.21 Hz, 1H), 8.20 (dd, J = 1.30, 8.46 Hz, 1H), 7.80 (dd, J = 7.14, 8.47 Hz, 1H), 7.52 (dd, J = 1.35, 7.16 Hz, 1H), 7.49 - 7.46 (m, 1H), 7.43 (s, 1H), 7.33 (dd, J = 8.72, 11.73 Hz, 2H), 7.05 (s, 1H), 6.49 - 6.43 (m, 2H), 4.77 - 4.71 (m, 1H), 4.43 (dd, J = 4.80, 17.69 Hz, 1H), 4.29 (dd, J = 4.01, 17.76 Hz, 1H), 3.44 (ddd, J = 3.19, 7.76, 10.27 Hz, 1H), 3.32 - 3.10 (m, 1H), 2.44 -2.10 (m, 4H).
minor rotamer: 1H NMR (400 MHz, CDCI3) δ 8.95 (d, J = 4.26 Hz, 1H), 8.16 (dd, J = 1.24, 8.46 Hz, 1 H), 7.60 (dd, J = 7.55, 8.47 Hz, 1 H), 7.55 (d, J = 4.33 Hz, 1 H), 7.49 (d, J = 4.27 Hz, 1 H), 7.43 (s, 1 H), 7.33 (dd, J = 8.72, 11.73 Hz, 2H), 6.94 (s, 1 H), 6.49 - 6.43 (m, 2H), 4.79 (d, J = 5.81 Hz, 1 H), 4.43 (dd, J = 4.80, 17.69 Hz, 1H), 4.29 (dd, J = 4.01, 17.76 Hz, 1H), 3.76 - 3.66 (m, 1H), 3.62 - 3.50 (m, 1H), 2.44 -2.10 (m, 4H).
13C NMR (101 MHz, CDCI3) δ 167.91, 166.54, 149.70, 141.36, 139.43, 133.70, 131.05, 129.81, 129.32, 128.11, 127.02, 122.45, 121.11, 118.86, 117.70, 100.00, 46.55, 45.37, 42.20, 29.98, 24.97.
LC-MS(I-B) (ESI) Rt 13.3 min, m/z 385.1 [M+H]+ (96%).
Example 30: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-7-(phenylamino)quinoline-4- carboxamide
The title compound was prepared in a manner similar to that described in Example 29.
Yield: 78mg, 73%

1H NMR (400 MHz, CDCI3) δ 8.67 (d, J = 4.43 Hz, 1H), 8.07 (d, J = 9.10 Hz, 1H), 7.57 (d, J = 2.39 Hz, 1H), 7.42 (t, J = 4.78 Hz, 1H), 7.33 - 7.27 (m, 2H), 7.20 (ddd, J = 1.60, 2.58, 8.15 Hz, 4H), 7.02 (tt, J = 1.21, 7.35 Hz, 1H), 6.53 (s, 1H), 4.75 (dd, J = 1.94, 7.81 Hz, 0.1 H), 4.72 - 4.66 (m, 0.9H), 4.50 (dd, J = 5.62, 17.21 Hz, 0.1 H), 4.33 (dd, J = 5.36, 17.59 Hz, 0.9H), 4.24 (dd, J = 3.96, 17.21 Hz, 0.1 H), 4.13 (dd, J = 4.18, 17.59 Hz, 0.9H), 3.62 (ddd, J = 3.23, 7.04, 9.02 Hz, 1H), 3.44 (td, J = 4.49, 8.52, 9.05 Hz, 1 H), 2.32 - 2.08 (m, 4H).
13C NMR (101 MHz, CDCI3) δ 167.88, 167.29, 150.41, 150.23, 145.40, 141.28, 140.57, 129.56, 126.41, 122.81, 120.71, 119.91, 119.07, 118.12, 115.97, 110.03, 46.77, 45.72, 42.42, 29.93, 25.16.
LC-MS(I-B) (ESI) R,11.8 min, m/z 400.0 [M+H]+ (98%).
- -
Example 31 : (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-7-phenylquinoline-4-carboxamide
The title compound was prepared in a manner similar to that described in Example 29.
Yield: 86mg, 74%

1 H NMR (400 MHz, CDCI3) (9/1 mixture of trans/cis amide rotamers) δ 8.96 (d, J = 4.35 Hz, 1 H), 8.38 - 8.33 (m, 2H),7.89 (dd, J = 2.01 , 8.66 Hz, 1 H), 7.79 - 7.71 (m , 2H), 7.55 - 7.46 (m , 3H), 7.47 - 7.37 (m, 1 H), 7.22 (t, J = 4.58 Hz, 1 H), 7.12 (t, J = 4.28 Hz, 0.1 H), 4.77 (dd, J = 2.41 , 7.67 Hz, 1 H), 4.60 (dd, J = 5.54, 17.33 Hz, 0.1 H), 4.43 (dd, J = 5.07, 17.75 Hz, 0.9H), 4.34 (dd, J = 3.51 , 17.28 Hz, 0.1 H), 4.26 (dd, J = 4.02, 1 7.77 Hz, 0.9H), 3.72 (ddd, J = 2.95, 7.09, 9.42 Hz, 1 H), 3.59 - 3.50 (m, 1 H), 2.51 - 2.12 (m, 4H).
13C NMR (101 MHz, CDCI3) δ 167.35, 166.97, 150.32, 149.12, 142.68, 140.57, 139.78, 129.07, 128.16, 127.50, 127.47, 127.39, 125.73, 123.49, 1 18.69, 1 17.83, 46.72, 45.65, 42.48, 29.94, 25.10.
LC-MS (I-B) (ESI) R, 14.6 min, m/z 385.1 [M+H]+ (95%).
Example 32: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-7-methylquinoline-4-carboxamide
The title compound was prepared in a manner similar to that described in Example 3.
Yield: 55mg, 56%

1 H NMR (400 MHz, CDCI3) δ 8.89 (d, J = 4.36 Hz, 1 H), 8.17 (d, J = 8.64 Hz, 1 H), 7.92 - 7.88 (m , 1 H), 7.47 - 7.41 (m, 2H), 7.16 (t, J = 4.64 Hz, 1 H)„ 4.79 - 4.73 (m , 1 H), 4.58 (dd, J = 5.36, 17.37 Hz, 0.1 H), 4.40 (dd, J = 5.05, 17.76 Hz, 0.9H), 4.31 (dd, J = 3.70, 17.23 Hz, 0.1 H), 4.24 (dd, J = 3.98, 17.77 Hz, 0.9H), 3.70 (ddd, J = 2.85, 6.84, 10.97 Hz, 1 H), 3.58 - 3.49 (m , 1 H), 2.57 (s, 3H), 2.39 - 2.17 (m, 4H).
13C NMR (101 MHz, CDCI3) δ 167.66, 167.14, 149.91 , 149.1 0, 140.66, 140.60, 130.21 , 128.98, 124.96, 122.52, 1 18.18, 1 17.96, 46.81 , 45.75, 42.56, 30.03, 25.20, 21 .93.
UPLC I (ESI) Rt 1 .14 min, m/z 323.5 [M+H]+ (98%) ; LC-MS (l-B) Rt 10.1 min, m/z 323.0 [M+H]+ (97%)
- -
Example 33: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-(4-methoxyphenyl)-3H- imidazo[4,5-b]pyridine-7-carboxamide
The title compound was prepared in a manner similar to that described in Example 3.
Yield: 67mg, 75%

1 H NMR (400 MHz, DMSO) (9/1 mixture of trans/cis amide rotamers) δ 10.15 (s, 1 H), 8.44 (d, J = 5.02 Hz, 1 H), 8.37 (d, J = 8.74 Hz, 2H), 7.72 (d, J = 4.99 Hz, 1 H), 7.17 (d, J = 8.78 Hz, 2H), 5.32 - 5.26 (m, 0.1 H), 4.89 (dd, J = 4.13, 6.70 Hz, 0.9H), 4.58 (dd, J = 4.45, 17.77 Hz, 0.1 H), 4.45 (dd, J = 4.92, 17.95 Hz, 0.9H), 4.36 (dd, J = 4.78, 17.81 Hz, 1 H), 3.88 (s, 3H), 3.74 (ddd, J = 3.71 , 7.64, 1 1 .08 Hz, 1 H), 3.55 (td, J = 6.92, 9.1 1 Hz, 1 H), 2.23 - 1 .96 (m, 4H).
13C NMR (101 MHz, DMSO) δ 170.68, 166.32, 162.77, 161 .25, 153.30, 149.94, 142.98, 132.23, 128.45, 120.49, 1 18.63, 1 15.75, 1 14.05, 54.98, 45.82, 44.80, 41 .88, 29.03, 24.26.
UPLC I (ESI) Rt 1 .50 min, m/z 405.6 [M+H]+ (96%) ; LC-MS (l-B) (ESI) Rt 14.6 min, m/z 405.0 [M+H]+ (97%).
Example 34: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-1 H-pyrrolo[2,3-b]pyridine-4- carboxamide
The title compound was prepared in a manner similar to that described in Example 3.
Yield: 50mg, 35%

1 H NMR (400 MHz, DMSO) (9/1 mixture of trans/cis amide rotamers) δ 1 1 .88 (s, 1 H), 8.68 (t, J = 5.69 Hz, 1 H), 8.33 (d, J = 4.91 Hz, 1 H), 7.61 (dd, J = 2.51 , 3.44 Hz, 1 H), 7.42 (d, J = 4.92 Hz, 1 H), 6.86 (dd, J = 1 .86, 3.43 Hz, 1 H), 5.29 (d, J = 7.22 Hz, 0.1 H), 4.80 (dd, J = 3.62, 7.39 Hz, 0.9H), 4.38 (dd, J = 5.71 , 16.63 Hz, 0.1 H), 4.22 (dd, J = 6.09, 16.95 Hz, 0.9H), 4.14 (dd, J = 5.47, 16.93 Hz, 1 H), 3.73 (ddd, J = 3.72, 7.57, 9.60 Hz, 1 H), 3.54 (td, J = 6.79, 8.95 Hz, 1 H), 2.34 - 2.01 (m, 4H).
UPLC I (ESI) Rt 1 .08 min, m/z 298.6 [M+H]+ (96%) ; LC-MS (l-B) (ESI) Rt 9.2 min, m/z 298.0 [M+H]+ (97%).
- -
Example 35: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-[1 ,2,4]triazolo[1 ,5-a]pyrimidine-7- carboxamide
The title compound was prepared in a manner similar to that described in Example 3.
Yield: 54mg, 57%

1 H NMR (400 MHz, DMSO) δ 9.59 (d, J = 6.96 Hz, 1 H), 9.28 (s, 1 H), 8.86 (s, 1 H), 7.87 (d, J = 6.95 Hz, 1 H), 4.79 (dd, J = 3.72, 7.35 Hz, 1 H), 4.18 (d, J = 5.1 1 Hz, 2H), 3.76 - 3.67 (m , 1 H), 3.52 (q, J = 8.33 Hz, 1 H), 2.36 - 1 .97 (m, 4H).
UPLC I (ESI) R, 1 .02 min, m/z 300.6 [M+H]+ (96%) ; LC-MS (l-B) (ESI) R, 9.0 min, m/z 300.0 [M+H]+ (97%).
Example 36: N-((R)-1-((S)-2-cyanopyrrolidin-1-yl)-3-hydroxy-1-oxopropan-2-yl)quinoline-4-

Step 1 : tert-butyl (R)-3-(tert-butyldimethylsilyloxy)-1-((S)-2-cyanopyrrolidin-1-yl)-1- oxopropan-2-ylcarbamate
A mixture of DIPEA (0,542 ml, 3,1 1 mmol) and (S)-pyrrolidine-2-carbonitrile 4- methylbenzenesulfonate (0,269 g, 1 ,002 mmol) in DCM was added to a mixture of (R)-2-(tert- butoxycarbonylamino)-3-(tert-butyldimethylsilyloxy)propanoic acid (0,320 g, 1 ,002 mmol) and HATU (0,381 g, 1 ,002 mmol) in DMF. After 3h the mixture was washed with 1 N citric acid and saturated sodium bicarbonate and brine. The organic layer was dried sodium sulfate, filtered and purified by column chromatography using hexane-ethylacetate as eluents.
1 H NMR (400 MHz, CDCI3) δ 5.31 - 5.22 (d, J = 8.4 Hz, 1 H), 4.72 - 4.66 (m , 1 H), 4.66 - 4.56 (td, J = 8.9, 5.3 Hz, 1 H), 3.86 - 3.70 (m , 3H), 3.69 - 3.61 (t, J = 9.1 Hz, 1 H), 2.36 - 2.07 (m , 4H), 1 .50 - 1 .37 (s, 9H), 0.88 - 0.78 (s, 9H), 0.03 - 0.02 (s, 3H), 0.015 - 0.00 (s, 3H)
UPLC I (ESI) Rt 2.25 min, m/z 398.67 [M+H]+ (96%).
- -
Step 2: (S)-1-((R)-2-amino-3-hydroxypropanoyl)pyrrolidine-2-carbonitrile 4- methylbenzenesulfonate
Tosic Acid (0,174 g, 0,91 6 mmol) was added to a cold (0SC) solution of tert-butyl (R)-3-(tert- butyldimethylsilyloxy)-1 -((S)-2-cyanopyrrolidin-1 -yl)-1 -oxopropan-2-ylcarbamate (0,260 g, 0,654 mmol) in acetonitrile(about 0.4M) at 0SC. after 30min the mixture was allowed to warm till room temperature and stirred for 24h. The volatiles were evaporated and the mixture was washed with cold ethyl acetate to remove N-tert-butyl acetamide, the white precipitate (S)-1 -((R)-2-amino-3- hydroxypropanoyl)pyrrolidine-2-carbonitrile 4-methylbenzenesulfonate (0.220, 0.619mmol, 95%yield)
1 H NMR (400 MHz, D20) δ 7.73 - 7.66 (m , 2H), 7.40 - 7.35 (m, 2H), 4.84 - 4.81 (m , 1 H), 4.43 (dd, J= 5.5, 4.3 Hz, 1 H), 4.02 - 3.91 (m , 1 H), 3.99 (dd, J = 12.6, 4.3 Hz, 1 H), 3.94 (dd, J = 12.6, 5.6 Hz, 1 H), 3.62 (dt, J = 9.8, 7.9 Hz, 1 H), 2.41 (s, 3H), 2.39 - 2.16 (m, 4H).
UPLC I (ESI) Rt 0.20 min, m/z 184.4 [M+H]+ (94%).
Step 3: N-((R)-1-((S)-2-cyanopyrrolidin-1-yl)-3-hydroxy-1-oxopropan-2-yl)quinoline-4- carboxamide
Quinoline-4-carbonyl chloride hydrochloride (0,071 g, 0,31 0 mmol) was added to a solution of the amine tosyl salt (S)-1 -((R)-2-amino-3-hydroxypropanoyl)pyrrolidine-2-carbonitrile 4- methylbenzenesulfonate (0,1 10 g, 0,310 mmol) and DIPEA (0,168 ml, 0,959 mmol) in DCM and stirred until completion as followed with LC-MS (usually 2 to 3hours). On completion of the reaction, it was diluted with DCM-isopropanol (4-1 ), washed with saturated aqueous NaHC03 solution (1 0 mL), 0.1 N aqueous citric acid solution (1 0 mL), and brine (10 m L). The organic layer was dried over Na2S04, filtered, and concentrated and purified with flash chromatography using an ethylacetate - methanol gradient.
Yield: 42mg, 40%
1 H NMR (400 MHz, CDCI3) (10/3 mixture of trans/cis amide rotamers) δ 8.91 (d, J = 4.35 Hz, 0.3H), 8.89 (d, J = 4.33 Hz, 1 H), 8.19 (dd, J = 1 .29, 8.69 Hz, 1 H), 8.19 - 8.16 (m, 0.3H), 8.13 (d, J = 8.75 Hz, 0.3H), 8.1 1 (dd, J = 1 .09, 8.75 Hz, 1 H), 7.77-7.74 (m , 0.3H), 7.73 (ddd, J = 1 .39, 6.87, 8.40 Hz, 1 H), 7.63 - 7.59 (d, J = 1 .35 Hz, 0.3H), 7.58 (ddd, J = 1 .30, 6.82, 8.36 Hz, 1 H), 7.51 (d, J = 7.87 Hz, 1 H), 7.46 (d, J = 4.31 Hz, 1 .3H), 7.43 (d, J = 7.26 Hz, 0.3H), 5.62 (dd, J = 2.08, 7.73 Hz, 0.3H), 5.1 1 (dt, J = 4.58, 7.87 Hz, 1 H), 4.94 (dt, J = 3.82, 7.23 Hz, 0.3H), 4.60 - 4.55 (m, 1 H), 4.15 (dd, J = 3.43, 1 1 .74 Hz, 0.3H), 4.02 (dd, J = 4.18, 1 1 .36 Hz, 1 ,3H), 3.97 - 3.89 (m , 2.3H), 3.73 - 3.62 (m , 2H), 3.59 - 3.50 (m, 0.6H), 2.46 - 2.10 (m, 5.2H).
major conformer: 13C NMR (101 MHz, CDCI3) δ 169.42, 167.55, 149.86, 148.70, 140.63, 130.31 , 129.98, 128.14, 125.17, 124.37, 1 1 9.00, 1 17.90, 77.36, 63.36, 53.19, 47.01 , 30.08, 25.24.
minor conformer: 13C NMR (101 MHz, CDCI3) δ 170.61 , 168.09, 149.86, 148.76, 140.21 , 130.34, 130.10, 128.18, 125.02, 124.37, 1 1 9.07, 1 17.90, 77.36, 62.35, 52.1 1 , 46.65, 32.30, 23.27.
- -
UPLC I (ESI) Rt 1 .10 min, m/z 339.6 [M+H]+ (95%) ; LC-MS (l-B) (ESI) Rt 10.5 min, m/z 339.0 [M+H]+ (95%).
Example 37: N-((R)-1-((S)-2-cyanopyrrolidin-1-yl)-1-oxopropan-2-yl)quinoline-4-carboxamide The title compound was prepared in a manner similar to that described in Example 36.

Step 1 : tert-butyl (R)-1-((S)-2-cyanopyrrolidin-1-yl)-1-oxopropan-2-ylcarbamate
A mixture of DIPEA (1 ,145 ml, 6,55 mmol) and (S)-pyrrolidine-2-carbonitrile 4- methylbenzenesulfonate (0,567 g, 2,1 14 mmol) in DCM was added to a mixture of (R)-2-(tert- butoxycarbonylamino)propanoic acid (0,4 g, 2,1 14 mmol)and HATU (0,804 g, 2,1 14 mmol)in DMF. After 3h the mixture was washed with 1 N citric acid and saturated sodium bicarbonate and brine. The organic layer was dried sodium sulfate, filtered and purified by column chromatography with a gradient DCM to DCM-MeOH (95-5). (0.47g, 83%)
1 H NMR (400 MHz, CDCI3) δ 5.26 (d, J = 7.9 Hz, 1 H) , 4.78 (dd, J = 6.8, 3.7 Hz, 1 H), 4.47 - 4.37 (m, 1 H), 3.77 - 3.58 (m, 2H), 2.33 - 2.12 (m, 4H), 1 .42 (s, J = 6.3 Hz, 9H), 1 .35 (d, J = 6.9 Hz, 3H). UPLC I (ESI) Rt 1 .44 min, m/z 268.5 [M+H]+ (96%). Step 2: (S)-1-((R)-2-aminopropanoyl)pyrrolidine-2-carbonitrile 4-methylbenzenesulfonate
1 H NMR (400 MHz, D20) δ 7.73 - 7.65 (d, J = 8.0 Hz, 3H), 7.40 - 7.34 (d, J = 7.9 Hz, 3H), 4.79-4- 75 (m , 1 H), 4.39 - 4.29 (q, J = 7.1 Hz, 1 H), 3.83 - 3.73 (dt, J = 10.6, 5.7 Hz, 1 H), 3.60 - 3.49 (q, J = 8.8 Hz, 1 H), 2.45 - 2.36 (s, 3H), 2.37 - 2.12 (m , 4H), 1 .54 - 1 .44 (dd, J = 7.1 Hz, 3H).
UPLC I (ESI) Rt 0.25 min, m/z 168.4 [M+H]+ (95%).
Step 3: N-((R)-1-((S)-2-cyanopyrrolidin-1-yl)-1-oxopropan-2-yl)quinoline-4-carboxamide
Quinoline-4-carbonyl chloride hydrochloride (0,067 g, 0,295 mmol) was added to a mixture of DIPEA (0,160 ml, 0,913 mmol) and (S)-1 -((R)-2-aminopropanoyl)pyrrolidine-2-carbonitrile 4- methylbenzenesulfonate (0,100 g, 0,295 mmol) in DCM. After 3h the mixture was washed with 1 N citric acid and saturated sodium bicarbonate and brine. The organic layer was dried sodium sulfate, filtered and purified by column chromatography using a mixture of ethylacetate and methanol as eluents.
Yield: 48mg, 50%
- -
1H NMR (400 MHz, CDCI3) (10/3 mixture of trans/cis amide rotamers) δ 8.94 (d, J = 4.31 Hz, 1H), 8.24 (dd, J = 1.16, 8.99 Hz, 1H), 8.16 - 8.11 (m, 1H), 7.76 (ddd, J = 1.41, 6.93, 8.38 Hz, 1H), 7.62 (ddd, J = 1.36, 6.84, 8.32 Hz, 1 H), 7.49 (d, J = 4.29 Hz, 1 H), 7.19 (d, J = 7.55 Hz, 1 H), 5.04 (qd, J = 2.10, 6.79 Hz, 1H), 4.68 (dd, J = 2.00, 8.01 Hz, 1H), 4.00 - 3.91 (m, 1H), 3.59 - 3.51 (m, 1H), 2.49 - 2.10 (m, 4H), 1.52 (d, J = 6.83 Hz, 3H).
13C NMR (101 MHz, CDCI3) δ 171.10, 166.71, 149.92, 148.79, 141.13, 130.17, 130.02, 127.96, 125.29, 124.50, 118.86, 117.97, 47.67, 47.06, 46.59, 30.15, 25.23, 18.31.
minor conformer: 1H NMR (400 MHz, CDCI3) δ 8.93 (d, J = 4.30 Hz, 1H), 8.20 (dd, J = 1.15, 8.41 Hz, 1H), 8.16 - 8.11 (m, 1H), 7.76 (ddd, J = 1.41 , 6.93, 8.38 Hz, 1H), 7.62 (ddd, J = 1.36, 6.84, 8.32 Hz, 1 H), 7.46 (d, J = 4.29 Hz, 1 H), 6.93 (d, J = 7.24 Hz, 1 H), 5.38 (dd, J = 2.16, 7.68 Hz, 1 H), 5.01 - 4.95 (m, 1H), 4.00 - 3.91 (m, 1H), 3.68 - 3.61 (m, 1H), 2.49 - 2.10 (m, 4H), 1.62 (d, J = 6.87 Hz, 3H).
13C NMR (101 MHz, CDCI3) δ 171.07, 166.63, 149.88, 148.76, 141.09, 130.23, 130.07, 128.03, 125.12, 124.43, 118.91, 117.97, 47.56, 47.06, 46.59, 29.82, 23.23, 17.93.
UPLC I (ESI) Rt 1.18 min, m/z 323.6 [M+H]+ (96%); LC-MS (l-B) (ESI) Rt 10.2 min, m/z 323.0 [M+H]+ (97%).
Example 38: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-1,7-naphthyridine-4-carboxamide
The title compound was prepared in a manner similar to that described in Example 29.
Yield: 58mg, 57%

Mixtrure of cis/trans amide conformers (1.5/8.5) Major rotamer: 1H NMR (400 MHz, CDCI3) δ 9.44 (d, J = 1.02 Hz, 1H), 8.97 (d, J = 4.29 Hz, 1H), 8.59 (d, J = 5.90 Hz, 1H), 8.12 (dd, J = 0.98, 5.86 Hz, 1H), 7.72 (d, J = 4.34 Hz, 1H), 7.64 - 7.57 (m, 1H), 4.76 - 4.69 (m, 1H), 4.41 (dd, J = 5.58, 17.60 Hz, 1H), 4.19 (dd, J = 4.00, 17.59 Hz, 1H), 3.69 (ddd, J = 3.37, 6.58, 11.84 Hz, 1H), 3.58 - 3.46 (m, 1H), 2.39 - 2.07 (m, 4H). Minor rotamer: 1H NMR (400 MHz, CDCI3) δ 9.48 (d, J = 0.94 Hz, 1H), 9.02 (d, J = 4.29 Hz, 1H), 8.61 (d, J = 4.26 Hz, 1H), 8.10 (d, 5.86 Hz, 1H), 7.70 (d, J = 4.12 Hz, 1H), 7.44 (t, J = 4.78 Hz, 1H), 4.79 (dd, J = 2.04, 7.77 Hz, 1H), 4.54 (dd, J = 5.44, 17.23 Hz, 1H), 4.29 (dd, J = 3.98, 17.27 Hz, 1H), 3.69 (ddd, J = 3.37, 6.58, 11.84 Hz, 1H), 3.58 - 3.46 (m, 1H), 2.39 -2.07 (m, 4H).
13C NMR (101 MHz, CDCI3) δ 167.24, 166.00, 154.45, 151.46, 144.79, 143.45, 139.40, 127.89, 122.74, 118.00, 117.76, 46.82, 45.79, 42.45, 29.91, 25.16.
UPLC I (ESI) Rt1.03 min, m/z 310.7 [M+H]+ (96%); LC-MS (l-B) Rt 9.0 min, m/z 309.9 [M+H]+ (96%)
- -
Example 39: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-(pyridin-4-yl)acetamide
The title compound was prepared in a manner similar to that described in Example 3.
Yield: 40 mg, 29%

1 H NMR (400 MHz, MeOD) δ 8.49 - 8.44 (d, J = 4.5, J' = 1 .6 Hz, 2H), 7.46 - 7.41 (d, J = 4.5, J' = 1 .6 Hz, 2H), 5.05 -5.02 (m, 0.14H), 4.79 - 4.74 (t, J = 5,4 Hz, 0.86H), 4.26 - 4.1 6 (m , 0.2H), 4.12 - 4.00 (m , 1 .8H), 3.73 - 3.66 (m , 3H), 3.57 - 3.49 (dt, J = 9.6, 7.6 Hz, 1 H), 2.28 - 2.12 (m , 4H).
13C NMR (101 MHz, MeOD) δ 172.5, 169.6, 150.1 , 147.3, 126..4, 1 1 9.5, 48..0, -46.9, 42.9, 42.5, 31 .0, 26.1 .
UPLC I (ESI) Rt 0.28 min, m/z 273.6 [M+H]+ (97%) ; LC-MS (l-B) R, 1 .5 min, m/z 273.0 [M+H]+ (97%)
Example 40: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-2-(1 H-imidazol-4-yl)acetamide
The title compound was prepared in a manner similar to that described in Example 3.
Yield: 72mg, 66%

1 H NMR (400 MHz, MeOD) δ 7.68 - 7.63 (s, 1 H), 7.05 - 6.99 (s, 1 H), 4.78 - 4.73 (t, J = 5.3 Hz, 1 H), 4.10 - 3.98 (m , 2H), 3.71 - 3.64 (ddd, J = 9.7, 6.4, 4.7 Hz, 1 H), 3.63 - 3.58 (s, 2H), 3.56 - 3.47 (dt, J = 9.5, 7.5 Hz, 1 H), 2.36 - 2.06 (m, 4H).
UPLC I (ESI) Rt 0.29 min, m/z 262.6 [M+H]+ (99%) ; LC-MS (l-B) Rt 1 .2 min, m/z 262.0 [M+H]+ (96%) -cyanopyrrolidin-1 -yl)-2-oxoethyl)-1 H-1 ,2,3-triazole-5-carboxamide

1 H-1 ,2,3-triazole-4-carboxylic acid (0,092 g, 0,814 mmol) was dispersed in dry dioxane (3m L) in a round bottom flask with nitrogen. To this 1 -chloro-N,N,2-trimethylprop-1 -en-1 -amine (0,151 ml, 1 ,139 mmol) was added and the reaction was stirred for 30 minutes at room temperature. The starting material dissolves over time, Then (S)-1 -(2-aminoacetyl)pyrrolidine-2-carbonitrile hydrochloride (0,247 g, 1 ,302 mmol) with N-ethyl-N-isopropylpropan-2-amine (0,307 ml, 1 ,709 mmol) was added and the mixture was stirred for 2h, evaporated to dryness and redissolved in ethyl acetate. Followed by washing with 0.1 N citric acid and saturated sodium bicarbonate and
- -
brine. After drying over sodium sulfate, filtration and evaporation, the product was purified using column chromatography.
Yield: 34mg, 40%
1 H NMR (400 MHz, DMSO) (9/1 mixture of trans/cis amide rotamers) δ 15.53 (s, 1 H), 8.58 - 8.44 (s, 1 H), 8.39 (s, 1 H), 5.28 - 5.22 (m, 0.1 H), 4.77 (dd, J = 3.76, 7.32 Hz, 1 H), 4.30 (dd, J = 5.61 , 16.77 Hz, 0.2H), 4.10 (d, J = 5.76 Hz, 2H), 3.68 (ddd, J = 4.04, 7.68, 9.39 Hz, 1 H), 3.50 (td, J = 6.84, 9.09 Hz, 1 H), 2.31 - 1 .88 (m, 4H).
UPLC I (ESI) Rt 0.72 min, m/z 249.6 [M+H]+ (96%) ; LC-MS (l-B) Rt 4.0 min, m/z 249.0 [M+H]+ (96%)
Example 42: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-1 H-1 ,2,4-triazole-3-carboxamide
The title compound was prepared in a manner similar to that described in Example 3.
Yield: 53mg, 40%

1 H NMR (400 MHz, D20) (9/1 mixture of trans/cis amide rotamers) δ 8.56 (s, 1 H), 5.12 (dd, J = 2.05, 7.76 Hz, 0.1 H), 4.85 - 4.81 (m, 0.9H), 4.43 (d, J = 2.63 Hz, 0.2H), 4.32 (s, 2H), 3.79 (ddd, J = 4.53, 7.02, 9.82 Hz, 1 H), 3.63 (dt, J = 7.65, 9.63 Hz, 1 H), 2.48 - 2.1 0 (m , 4H).
UPLC I (ESI) Rt 0.77 min, m/z 271 .5 [M+Na]+ (97%) ; LC-MS (l-B) Rt 2.8 min, m/z 249.0 [M+H]+ (97%)
Example 43: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-3-hydroxyquinoline-4-carboxamide

To a 50m L round bottom flask, was added 3-hydroxyquinoline-4-carboxylic acid (0,1 g, 0,529 mmol), acetonitrile (Volume: 10 ml) and N-ethyl-N-isopropylpropan-2-amine (0,157 g, 1 ,216 mmol) at room temperature with stirring until a solution was observed. Then, di(1 H-imidazol-1 - yl)methanone (0,094 g, 0,581 mmol) was added in one portion and the mixture was held for 4h. Then the amine(S)-1 -(2-aminoacetyl)pyrrolidine-2-carbonitrile 2,2,2-trifluoroacetate (0,198 g, 0,740 mmol) was added in one portion. The mixture was heated to 75 °C, held at 75Ό for 5 h, cooled to room temperature, and stirred at room temperature overnight. The volatiles were evaporated and the residue was redissolved in EtOAc, washed with 0.3N citric acid, saturated sodium bicarbonate,
- -
brine, dried over sodium sulfate, filtered and evaporated. The crude product was purified using column chromatography with ethyl acetate-methanol (95-5) mixture to yield the product (0.076g, 44%) 1 H NMR (400 MHz, CDCI3) : (8.5/1 .5 mixture of trans/cis amide rotamers) δ 8.77 (d, J = 3.51 Hz, 1 H), 8.24 - 8.1 7 (m, 1 H), 8.10 (dd, J = 1 .49, 7.97 Hz, 1 H), 7.64 (ddd, J = 8.47, 7.06,1 .58 Hz, 1 H), 7.59 (ddd, J = 8.28, 6.92, 1 .48Hz, 1 H), 7.41 (s, 1 H), 4.82 (d, J = 7.01 Hz, 1 H) , 4.77 - 4.71 (m, 0.2H), 4.47 (dd, J = 17.1 , 4.8 Hz, 0.15H), 4.47 (dd, J = 17.76, 4.8 Hz, 0.85H), 4.42 - 4.36 (m, 0.15H), 4.28 (dd, J = 17.84, 3.76 Hz, 0.85H), 3.78 - 3.69 (m , 1 H), 3.57 (m , 1 H), 2.43 - 2.20 (m , 4H) ; UPLC I (ESI) R, 1 .19 min, m/z 325.6 [M+H]+ (98%)
Example 44 (S)-5-bromo-N-(2-(2-cyano-4,4-difluoropyrrolidin-1-yl)-2-oxoethyl)quinoline-4- carboxamide
The title compound was prepared in a manner similar to that described in Example 1 and Example 26.
Yield: 37mg, 30%

1 H NMR (400 MHz, CDCI3) (9/1 mixture of trans/cis amide rotamers) δ 9.10 (dd, J = 4.46, 6.43 Hz, 0.9H), 8.88 (s, 0.1 H), 8.41 (d, J = 8.76 Hz, 1 H), 8.05 (d, J = 8.73 Hz, 1 H), 7.96 (d, J = 8.55 Hz, 1 H), 7.84 (dd, J = 1 .06, 7.58 Hz, 1 H), 7.55 (dd, J = 7.47, 8.49 Hz, 1 H), 5.34 (d, J = 8.83 Hz, 0.1 H), 5.03 (dd, J = 5.05, 8.22 Hz, 0.9H), 4.64 (dd, J = 6.13, 17.08 Hz, 0.1 H), 4.56 (dd, J = 6.74, 17.53 Hz, 0.9H), 4.30 (dd, J = 4.51 , 17.10 Hz, 0.1 H), 4.1 7 (dd, J = 4.17, 17.48 Hz, 0.9H), 4.14 - 4.05 (m , 1 H), 4.04 - 3.95 (m, 1 H), 2.84 - 2.73 (m , 2H). UPLC I (ESI) Rt 1 .83 min, m/z 423.5, 425.3 [M+H]+ (96%) ; LC-MS (l-B) Rt 15.8 min, m/z 265.9, 267.9 (95%)
Example 45: (S)-N-(2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)-5-methoxyquinoline-4- carboxamide
The title compound was prepared in a manner similar to that described in Example 29.
Yield: 22m g, 44%


Step 1 : N-(2-oxo-2-((2R)-2-((3aS,7aR)-3a,5,5-trimethylhexahydro-4,6-methanobenzo [d][1 ,3,2]dioxaborol-2-yl)pyrrolidin-1-yl)ethyl)quinoline-4-carboxamide
(2R)-2-((3aSJaR)-3a,5,5-trimethylhexahydro-4,6-methanobenzo[d][1 ,3,2]dioxaborol-2- yl)pyrrolidine hydrochloride (0,190 g, 0,665 mmol) was added to a mixture of 2-(quinoline-4- carboxamido)acetic acid (0,153 g, 0,665 mmol), HATU (0,253 g, 0,665 mmol) and DIPEA (0,360 ml, 2,062 mmol) in dichloromethane (5m L). The mixture was stirred for 3h and washed with 0.5N citric acid, saturated sodium bicarbonate and brine, followed by filtration and evaporation. Purification was done using column chromatography DCM-MeOH 0-6% MeOH .
1 H NMR (400 MHz, CDCI3) δ 8.97 (d, J = 4.36 Hz, 1 H), 8.30 (ddd, J = 0.72, 1 .48, 8.50 Hz, 1 H), 8.17 (dd, J = 1 .25, 8.28 Hz, 1 H), 7.77 (ddd, J = 1 .44, 6.92, 8.47 Hz, 1 H), 7.62 (ddd, J = 1 .29, 6.94, 8.30 Hz, 1 H), 7.53 (d, J = 4.34 Hz, 1 H), 7.19 (t, J = 4.10 Hz, 1 H), 4.32 - 4.27 (m, 3H), 3.61 - 3.39 (m, 2H), 3.21 (dd, J = 6.93, 10.00 Hz, 1 H), 2.50 - 2.25(m, 1 H), 2.25 - 1 .95 (m , 5H), 1 .94 - 1 .78 (m, 3H), 1 .41 (s, 3H), 1 .28 - 1 .24 (m, 4H), 0.82 (s, 3H).
13C NMR (101 MHz, CDCI3) δ 167.01 , 165.44, 149.56, 130.42, 129.57, 128.03, 125.63, 124.73, 1 19.02, 86.32, 78.05, 77.36, 53.57, 51 .35, 45.86, 42.35, 39.61 , 38.75, 38.35, 35.62, 28.59, 27.53, 27.34, 27.20, 26.41 , 24.15.
UPLC I (ESI) Rt 1 .82 min, m/z 462.7 [M+H]+ (91 %) ;
Step 2: (R)-1-(2-(quinoline-4-carboxamido)acetyl)pyrrolidin-2-ylboronic acid
To a stirred solution of the N-(2-oxo-2-((2R)-2-((3aS,7aR)-3a,5,5-trimethylhexahydro-4,6- methanobenzo[d][1 ,3,2]dioxaborol-2-yl)pyrrolidin-1 -yl)ethyl)quinoline-4-carboxamide (0,27 g, 0,585 mmol) in water (8 mL) at pH=3 (adjusting as necessary with 2N aq HCI) was added phenylboric acid (0,143 g, 1 ,17 mmol) and methyl-tert-butylether (7 mL). The mixture was stirred for 2 days
- -
(pH was 4) water and organic layer were seperated, the water layer was adjusted to pH 3, and the water layer was extrarcted with a mixture 4-1 of DCM- isopropanol. The DCM layer was dried over sodium sulfate, filtrated evaporated and further purified using column chormatography (DCM- MeOH 0-10% MeOH) to yield a clear oil of 0.1 19g (79%) (R)-1 -(2-(quinoline-4- carboxamido)acetyl)pyrrolidin-2-ylboronic acid.
1 H NMR (400 MHz, D20) δ 8.92 - 8.84 (m , 1 H), 8.15 (ddd, J = 0.64, 1 .43, 8.58 Hz, 1 H), 8.09 - 8.00 (m, 1 H), 7.84 (ddd, J = 1 .41 , 6.92, 8.47 Hz, 1 H), 7.70 (ddd, J = 1 .18, 6.94, 8.29 Hz, 1 H), 7.64 (d, J = 4.46 Hz, 1 H), 4.32 - 4.27 (m , 2H), 3.67 (ddd, J = 3.33, 8.37, 10.14 Hz, 1 H), 3.55 (ddd, J = 6.54, 8.44, 10.90 Hz, 1 H), 3.12 (dd, J = 7.01 , 10.34 Hz, 1 H) , 2.13 (tdd, J = 3.42, 7.02, 9.03 Hz, 2H), 2.08 - 1 .87 (m, 1 H), 1 .82 - 1 .68 (m , 1 H).
13C NMR (1 01 MHz, D20) δ 170.63, 1 68.1 1 , 150.41 , 147.54, 142.15, 131 .50, 128.92, 128.64, 125.66, 124.61 , 1 19.99, 48.94(13C-B splitting), 47.22, 42.32, 27.48, 27.43.
UPLC I (ESI) Rt 1 .01 min, m/z 328.6 [M+H]+ (99%) ; LC-MS (l-B) Rt 9.3 min, m/z 328.0 [M+H]+ (98%)
2 In vitro and in vivo assay protocols. 2.1 Enzymatic assays.
2.1 .1 Enzymes
DPP IV, DPP I I, DPP8 and DPP9 were obtained as described in reference 18. Recombinant murine FAP was purified from the culture supernatant of HEK293 human embryonic kidney cellline as described in reference 19. Recombinant human PREP was expressed in and purified from E coli as described before in reference 20.
2.1 .2 IC^n measurements
Enzyme activities were determined kinetically in a final volume of 200 μΙ for 10 minutes at 37°C by measuring the initial velocities of pNA release (405 nm) from the substrate using a Spectramax plus microtiterplate reader (Molecular devices). One unit of enzyme activity was defined as the amount of enzyme that catalyzes the release of 1 μιηοΙ pNA from the substrate per minute under assay conditions.
All measurements were carried out in duplicate. The IC50 value was defined as the inhibitor concentration, which caused a 50% decrease of the activity under assay conditions.
IC50, purified enzymes
The chromogenic substrate Gly-Pro-p-nitroanilide (100 μιηοΙ/Ι) was used at pH 8.3 for DPP IV, Lys- Ala-p-nitroanilide (1 mmol/l) at pH 5.5 for DPP II, Ala-Pro-p-nitroanilide (300 μιτιοΙ/Ι) at pH 7.4 for DPP9 and Ala-Pro-p-nitroanilide (2 mmol/l) at pH 7.4 for FAP activity measurement. To evaluate
- -
the endopeptidase activity of FAP and the influence of inhibitors thereon, Z-Gly-Pro-AMC and Z- Gly-Pro-p-nitroanilide were used at a final concentration of 300 and 100 μιηοΙ/Ι, respectively. The substrate concentrations were chosen around the Km value obtained under the assay conditions used. Buffer compositions for the DPP assays were reported before in the purification articles - vide supra. The FAP assay buffer consisted of 50 mM Tris pH7.4 containing 1 00 mmol/l NaCI and 0.1 mg/ml bovine serum albumin. The PREP activity was measured as described by Brandt et al. using the chromogenic substrate Z-Gly-Pro-p-nitroanilide (0.25 mmol/l) at pH 7.5 in the presence of 10 mmol/l DTT.18Test compounds were dissolved and diluted in DMSO (final concentration DMSO during assay 5% v/v) except for FAP where dilution of the inhibitor was done in water. Inhibitors are pre-incubated with the enzyme for 1 5 min at 37 °C before starting the assay by the addition of substrate. The concentration of enzyme and of inhibitor during the preincubation is double of the final concentration during activity measurement.
IC50, plasma
For the measurements of endogenous FAP AND PREP activity in plasma, Z-Gly-Pro-AMC was used as a substrate at a concentration of 260 μιηοΙ/Ι in phosphate buffer pH 7.5 containing 1 mmol/l NaN3, 1 mmol/l EDTA with or without 10 mmol/l dithiothreitol (DTT). Final dilution of the plasma in the assay is 20 times. The 'total' activity (FAP AN D PREP) is measured when DTT is present, while in the absence of DTT, only FAP activity can be measured. The endogenous PREP activity is calculated as the difference between the 'total' activity and the FAP activity.
3 Biochemical Evaluation results
3.1 Inhibtory potency/selectivity of compounds
A set of reference compounds was prepared and evaluated that structurally are not of Formula (I), but nonetheless are close analogues of the compounds that correspond to Formula (I). These reference compounds serve as a control for the validity of our novel SAR data that form the basis of the present invention. Results, given as IC50-values, are summarised in Table 3.
Relevance of the N position
With FAP-affinities spanning almost three orders of magnitude, evaluation results of the compounds as depicted in table 3, nonetheless reveal a pivotal importance of the nitrogen's position. Of all the positional isomers synthesized, the 4-quinolinoyl ring clearly displays the best results and takes in a singular position within this series. The 4-isoquinolinoyl and 8-quinolinoyl derivatives are characterized by very low FAP-affinity.
All compounds were used in inhibition assays for FAP (DPPIV (dipeptidyl peptidase-4), DPP9, DPP2 and PREP (prolyl endopeptidase). DPP9 potencies reported can reasonably be expected to be indicative for inhibitor affinities toward the highly homologous DPP8.
- -
Table 3: Reference compounds


Table 4 represents evaluation data for compounds of the general formula that structurally accord to t

Table 4: Compounds according to this invention





Example 38
- -

As evident from Table 4, all compounds according to this invention have an IC50 for FAP which is less than 10 μΜ. Furthermore, most of the compounds according to this invention have an IC50 for FAP which is less than 1 μΜ, whereas most of the used reference compounds have an IC50 which is at least 10-fold higher (see table 2). In addition, apart from compound 8 and reference compound 5, most of the exemplified compounds have an IC50 value for DPPIV, DPP9 and DPP2 which is more than 100 μΜ, indicating that all of said compounds have a higher selectivity for FAP than for DPPIV, DPP8, DPP9 and DPP2. Furthermore, in comparison to the used reference compounds, most compounds according to this invention have an at least 50x higher selectivity for FAP than for PREP.
3.2 Evaluation of the inhibitory potency toward the endo-and exopeptidase activity of FAP
As explained in the 'background to the invention', FAP displays both endo-and exopeptidase activity, mediated by the same active center. The following data provide experimental evidence that the inhibitors that structurally accord to this invention, inhibit both activity types of FAP to the same extent (exemplified using Cmpd Ν from table 4).
- -
Inhibition of endopeptidase activity
Cmpd N °1 (table 4) : IC50 value using Z-Gly-Pro-AMC as a substrate 3.2 nmol/l
IC50 value using Z-Gly-Pro-p-NA as a substrate 2.3 nmol/l Compared to IC50 value using Ala-Pro-p-NA 3.2 nmol/l
Conclusion : In this in vitro setting, Cmpd Ν inhibits endo- end exopeptidase activity to the same extent.
3.3 Functional stability in plasma
As explained hereinbefore, several known FAP inhibitors, including the clinically relevant Val- boroPro (talabostat), suffer from poor stability both in aqueous solution and in plasma. The 'functional' stability of compound Cmpd Ν was determined in rat as well as in human plasma and serves as an example of the excellent potential of the compounds according to the present invention with regards to aqueous/plasma stability. Conclusion : Preincubation of Cmpd N °1 in rat or human plasma for over 12 h at 37 °C did not result in a shift of IC50 values. Final concentrations of 2.5 and 5 nmol/l all resulted in inhibition of 50 % or more in an inhibition assay as described above. The compound is very stable in plasma. (Or in case metabolites are formed, they are equally potent)
3.4 Inhibition of plasma FAP activity.
The potential of compounds according to the present invention to inhibit FAP activity in biologically relevant matrices is exemplified using IC50-values for Cmpd Ν and Cmpd N °2 in plasma. These are similar to the ones observed in the assays with purified enzyme (Table 4).
The IC50 values for inhibition of endogenous FAP activity in plasma :
Cmpd N °2 : IC50 value using Z-Gly-Pro-AMC as a substrate : 22.6 nmol/l
Cmpd N °1 : IC50 value using Z-Gly-Pro-AMC as a substrate :. 4.4 nmol/l
Conclusion :
Very limited plasma shifts were observed for Cmpd N and Cmpd N °2. 3.5 Selectivity of Cmpd N °1 in plasma
The potential of compounds according to the present invention to selectively inhibit FAP activity in biologically relevant matrices is exemplified using IC50-values for Cmpd Ν and Cmpd N °2 in plasma. These are similar to the ones observed in the assays with purified enzyme (Table 4).
Conclusion
- -
In the presence of 100 nmol/l Cmpd Ν , plasma FAP activity is inhibited > 90 %, while PREP activity remains > 95% of the control without Cmpd N °1 .
3.6 Preclinical AD ME of selected compounds
Selected compounds were tested on LOG D, pH 7.4, kinetic solubility, plasma stability in mouse and human, as well as metabolic stability in mouse and stability in plasma (Table 6). The results of all tests until now are satisfactory. Table 6. Preclinical ADME of selected compounds
Compound N ° LOG D Kinetic Plasma Plasma Metabolic Stability solubility stability stability stability in PBS
(mouse) (human) (mouse) buffer
90% after
N ° 1 1 >200 μΜ >24h >24h
24h
N ° 2 0.51 >200 μΜ >24h
Ν 4 0.8 >6h
70% after
N °22 0.7 >6h >6h
24h
- -
REFERENCES
1) Wolf, B.B.;Quan, C; Tran, T.; Wiesman, C; Sutherlin, C. Mini-Rev. Med; Chem.2008, 8, 719-727
2) Lee, K.N.; Jackson, K.W.; Christiaensen, V.J.; Lee, J.S.; Chun, J.G.; McKee, P.A. Blood
2006, 107, 1397-1404. (b) Brokopp, C.E.; Schoenauer, R.; Richards, P.; Bauer, S.; Lohmann, C; Emmert, M.Y.; Weber, B.; Winnik, S.; Aikawa, E.; Graves, K.; Genoni, M.; Vogt, P.; Luscher, T.F.; Renner, C; Hoerstrup, S.P.; Matter, CM. Eur. Heart J. 2011, published online doi 10.1093/eurheartj/ehq519 (c) Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. J. Biol. Chem.1999, 274, 36505-36512. (d) Huang, C.H.; Suen, C.S.; Lin, C.T.; Chien, C.H.; Lee, H.Y.; Chung, K.M.; Tsai, T.Y.; Jiaang, W.T.; Hwang, M.J.; Chen, X. J. Biochem. 2011, published online doi:10.1093/jb/mvr017
3) Keene, F.M.; Nadvi, N.A.; Yao, T.W.; Gorrell, M.D. FEBS journal 2011, 278, 8, 1316-1332.
4) Lo, P.C.; Chen, J.; Stefflova, K.; Warren, M.S.; Navab, R.; Bandarchi, B.; Mullins, S.; Tsao, M.; Cheng, J.D.; Zheng, G.; J. Med. Chem.2009, 52, 358-368.
5) (b) Loeffler, M.; KrQger, J.A.; Niethammer, A.G.; Reisfeld, R.A. J. Clin. Invest.2006, 166, 1955-1962. (b) Hofheinz, R.D.; Al-Batran, S.E.; Hartmann, F.; Hartung, D.; Jager, G.; Renner, C; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, A.; Stehle, G. Onkologie 2003, 26, 44-48.
6) (a) Cheng, J.D.; Valianou, M.; Canutescu, A.A.; Jaffe, E.K.; Lee, H.O.; Wang, H.; Lai, J.H.;
Bachovchin, W.W.; Weiner, L.M. Mol. Cancer Ther. 2005, 4, 351-360. (b) Cheng, J.D.; Dunbrack, R.L., Jr.; Valianou, M.; Rogatko, A.; Alpaugh, R.K.; Weiner, L.M. Cancer Res.2002, 62, 4767-4772. (c) Santos, A.M.; Jung, J.; Aziz, N.; Kissil, J.L. Pure, E.; J. Clin. Invest.2009, 109, 3613-3625.
7) Cunningham, C; Pavlick, A.C.; Khan, K.D.; Frenette, G.. O'Day, S.; Stephenson, J.;
Gonzalez, R.; Yang, Z.; Vrhovac, V.; Uprichard, M.J. J.Clin. Oncol.2006, 24, 462s
8) Acharya, P.S.; Zukas, A.; Chandan, V.; Katzenstein, A.L.; Pure, E. Hum. Pathol.2006, 37, 3, 352-360
9) Levy, M.T.; McCaughan, G.W.; Abbott, C.A. Park, J.E.; Cunningham, A.M.; Muller, E.;
Rettig, W.J.; Gorrell, M.D. Hepatology 1999, 291768-1778
10) Dienus, K.; Bayat, A.; Gilmore, F.; Seifert, O. Arch. Dermatol. Res.2010, 302, 710-725.
11) Milner, J.; Patel, A.; Rowan, A.D. Arthritis & Rheumatism 2008, 58, 3644-3656.
12) Rovedatti, L.; Sabatino, A.F.; Knowles, C.H.; Sengupta, N.; Bioancheri, P.; Corazza, G.R.;
MacDonald, T.T. Inflamm. Bowel Dis.2011, 17, 5, 1251-1253.
- - ) Gorrell, M.; Song, S.; Xin, W. WO 2010/083570A1
) Lee, K.N.; Jackson, K.W.; Christiansen, V.J.; Dolence, E.K.; McKee, P.A. J.Thromb. Haemostas.2011, 9, 987-996.
) Coutts, S.J.; Kelly, T.A.; Snow, R.J.; Kennedy, C.A.; Boston, R.W.; Adams, J.; Krolikowski, D.A.; Freeman, D.M.; Campbell, S.J.; Ksiazek, J.F.; Bachovchin, W.W. J. Med. Chem. 1996, 39, 2087-2094
) (a) Bachovchin, W.W.; Lai, H.S. U.S. Patent 2007, PCT/US2006/026258, (b) Evans, D.M.; Horton, J.; Trim, J.E. WO 2007085895 A220070802 (c) Tsai, T.Y; Yeh, T.K.; Chen, X.; Hsu, T.; Jao, Y.C.; Huang, C.H.; Song, J.S.; Huang, Y.C.; Chien, C.H.; Chiu, Y.H.; Yen, S.C.; Tang, H.K.; Chao, Y.S.; Jiaang, W.K. J. Med. Chem.2010, 53, 6573-6583.
) Kaila, N.; Janz, K.; De Bernardo, S.; Bedard, P.W.; Camphausen, R.T.; Tarn, S.; Tsao, D.H.H.; Keith, J.C.; Nicherson-Nutter, C.C.; Shilling, A.; Young-Sciame, R.; Wang, Q. J. Med. Chem.2007, 50, 21-39.
) Van Goethem, S.; Matheeussen V.; Joossens, J.; Lambeir, A.M.; Chen, X.; De Meester, I.; Haemers, A.; Augustyns, K.; Van der Veken, P.; J. Med. Chem. 2011, doi 10.1021 /jm200383j
) Cheng J.; Dunbrack R.L.; Valianou M.; Rogatko, A.; Alpaugh, R.K.; Weiner, L.M. Cancer Research 2002; 62: 4767-4772
) Brandt I, Gerard M, Sergeant K, Devreese B, Baekelandt V, Augustyns K, Scharpe S, Engelborghs Y, Lambeir AM. Peptides 2008, 29, 1472-1478
Claims
1 . A compound of Form ula I or a stereoisomer, tautomer, racem ic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,

Wherein
Ri and R2 are each independently selected from the group comprising -H , OH , -halo, C^alkyl, -0- d-ealkyl, S-C^alkyl ;
R3 is selected from the group comprising -H , -CN , -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl,
-C=C-S(0)2aryl, -C02H , -S03H , -S02N H2,-P03H2, and 5-tetrazolyl ;
R4 is selected from the group comprising -H , -C^alkyl, -O-C^alkyl, -S-C^alkyl , -Αη , and -C^
6aralkyl ; each of said -C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , -halo
R5, R6 and R7 are each independently selected from the group comprising -H , -OH , -oxo, -halo, -
Ci_6alkyl, -O-C^alkyl, -S-C^alkyl, -N R8R9, -OR12 -Het2 and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH , -halo
R8, R9 and R12 are each independently selected from the group comprising -H , -OH , -halo, -Ci_
6alkyl, -O-C^alkyl, -S-C^alkyl , and -Ar3
Rio, Ri i , Ri3 and R14 are each independently selected from the group comprising -H , -OH , -halo, -
C^alkyl, -O-C^alkyl, and -S-C^alkyl ;
Αη , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , Ar2 and Ar3 being optionally and independently substituted with from 1 to 3 substituents selected from -N R10Rn , - d-ealkyl, -O-C^alkyl, -S-C^alkyl ;
Het2 is a 5- or 6-membered non-aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S ; said Het2 being optionally substituted with from 1 to 3 substituents selected from -N R13R14, -C1 -6alkyl, -O-C^alkyl, -S-C^alkyl ;
n 3  represents a 5 to 1 0- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
represents a 5 to 1 0- membered N-containing aromatic or non-aromatic mono- or bicyclic heterocycle, wherein there are exactly 2 ring atoms between the N atom and X; said heterocycle optionally further comprising 1 , 2 or 3 heteroatoms selected from 0, N and S; and X represents a C atom
2. A compound according to claim 1 , wherein
R-i and R2 are each independently selected from the group comprising -H, and -halo;
R3 is -CN, or -B(OH)2
R4 is selected from the group comprising -H or -d-6alkyl ; each of said -C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH,;
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, -
Ci_6alkyl, -O-C^alkyl, Ar2 and -NR8R9; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
R8 and R9 are each independently selected from the group comprising -H and -Ar3
Ar2 and Ar3 are each independently -phenyl optionally substituted with from 1 to 3 -O-C^alkyl;

3. A compound according to claim 1 , wherein
R-i and R2 are each independently selected from the group comprising -H, and -F;
R3 is -CN,and -B(OH)2
R4 is -H ;
R5, R6 and R7 are each independently selected from the group comprising -H, -oxo, -halo, -Ci_
6alkyl, and -0-CF3;

4. A compound of Formula I I or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,

wherein
F and R2 are each independently selected from the group comprising -H, OH, -halo, C1 -6alkyl, -0- C1-6alkyl, S-C1-6alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2, -C(0)alkyl, -C(0)aryl-, -C=C-C(0)aryl,
-C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl
R4 is selected from the group comprising -H, -d-6alkyl, -O-C^alkyl, -S-Ci-6alkyl, -An , and -Ci_
6aralkyl; each of said -Ci-6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R5, R6 and R7 are each independently selected from the group comprising -H, -OH, -oxo, -halo, -
C1-6alkyl, -0-C1 -6alkyl, -S-C1 -6alkyl, -NR8R9, and -Ar2; each of said C1 -6alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8 and R9, R10 and R are each independently selected from the group comprising -H, -OH, -halo,
-Ci-6alkyl, -0-Ci-6alkyl, and -S-Ci-6alkyl;
Rio and Rn are each independently selected from the group comprising -H, -OH, -halo, -C^alkyl, -
O-d-ealkyl, -S-Ci-6alkyl, and -Ar3;
Αη , Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , and Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -
C1-6alkyl, -0-C1-6alkyl, -S-C1 -6alkyl;
n is 0, 1 , 2, or 3

5. A compound according to claim 4, wherein
Ri and R2 are each independently selected from the group comprising -H, OH, -halo, C^alkyl, -0-
Ci-6alkyl, S-Ci-6alkyl;
R3 is selected from the group comprising -H, -CN, and -B(OH)2 R4 is -H ;
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - C^alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9, R10 and R are each independently selected from the group comprising -H, -OH, -halo, -C^ 6alkyl, -O-C^alkyl, and -S-C^alkyl;
Ar2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -C^alkyl, -O-C^alkyl, -S-C^alkyl;
n is 0, 1 , 2, or 3


6. A compound according to anyone of claims 4 or 5, wherein
R5 and R6 are each -H ;
R7 is selected from the group comprising -H, -OH, -oxo, -halo, -C^alkyl, -O-C^alkyl, -S-C^alkyl, - NR8R9, and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo; and
R7 is attached to position 2 or 3, in particular position 2, as represented in
3 
7. A compound of Formula Il ia, ll lb or l llc or a stereoisomer, tautomer, racemic, metabolite, pro- or predrug, salt, hydrate, or solvate thereof,

l llc
wherein
Ri and R2 are each independently selected from the group comprising -H, OH, -halo, C^alkyl, -0- d_6alkyl, S-C^alkyl;
R3 is selected from the group comprising -H, -CN, -B(OH)2„ -C(0)alkyl, -C(0)aryl-, -C=C-
C(0)aryl, -C=C-S(0)2aryl, -C02H, -S03H, -S02NH2,-P03H2, and 5-tetrazolyl
R4 is selected from the group comprising -H, -C^alkyl, -O-C^alkyl, -S-C^alkyl, -Αη , and -C^
6aralkyl; each of said -C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo;
R5, R6 and R7 are each independently selected from the group comprising -H, -OH, -oxo, -halo, -
Ci_6alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8 and R9, are each independently selected from the group comprising -H, -OH, -halo, -C^alkyl, -
O-d-ealkyl, -S-C^alkyl, and -Ar3
R10 and Rn are each independently selected from the group comprising -H, -OH, -halo, -C^alkyl, -
O-C^alkyl, and -S-C^alkyl ; Ar1 t Ar2 and Ar3 are each independently a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; each of said Αη , and Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , - C^alkyl, -O-C^alkyl, -S-C^alkyl;
n is 0, 1 , 2, or 3

represent a 9 to 10- membered N-containing aromatic or non-aromatic bicyclic heterocycle optionally further comprising 1 or 2 heteroatoms selected from 0, N and S.
8. A compound according to claim 7, wherein
and R2 are each independently selected from the group comprising -H, OH , -halo, C^alkyl, -0- d-ealkyl, S-C^alkyl;
R3 is selected from the group comprising -H, -CN, and -B(OH)2;
R4 is -H ;
R5, R6 and R7 are each independently selected from the group comprising -H , -OH, -oxo, -halo, - Ci_6alkyl, -O-C^alkyl, -S-C^alkyl, -NR8R9, and -Ar2; each of said C^alkyl being optionally substituted with from 1 to 3 substituents selected from -OH, -halo
R8, R9, Rio and R are each independently selected from the group comprising -H, -OH, -halo, -Ci_ 6alkyl, -O-C^alkyl, and -S-C^alkyl;
Ar2 is a 5- or 6-membered aromatic monocycle optionally comprising 1 or 2 heteroatoms selected from 0, N and S; Ar2 being optionally and independently substituted with from 1 to 3 substituents selected from -NR10Rn , -C^alkyl, -O-C^alkyl, -S-C^alkyl;
n is 0, 1 , 2, or 3


9. A compound according to anyone of claims 7 or 8, wherein R5 is attached to position 2 or 3, in particular position 3, as represented in

10. A compound as defined in any one of claims 1 to 9, for use as a human or veterinary medicine.
1 1 . Use of a compound as defined in any one of claims 1 to 9 in the manufacture of a medicament for the prevention and/or treatment of a FAP-related disorder.
12. A pharmaceutical composition comprising a compound as defined in any one of claims 1 to 9, suitable for use as a human or veterinary medicine.
13. Use of a compound as defined in any one of claims 1 to 9, or a composition as defined in claim 12, suitable for inhibiting the activity of FAP (Fibroblast Activation Protein).
14. Use of a compound as defined in any one of claims 1 to 9, or a composition as defined in claim 12, for the prevention and/or treatment of a FAP-related disorder.
15. A method for the prevention and/or treatment of a FAP-related disorder; said method comprising administering to a subject in need thereof a compound according to any one of claims 1 to 9 or a composition as defined in claim 12.
16. The use according to anyone of claims 1 1 or 14, or the method according to claim 15, wherein the FAP-related disorder is selected from the list comprising proliferative diseases selected from the group comprising breast cancer, colorectal cancer, ovarian cancer, prostate cancer, pancreatic cancer, kidney cancer, lung cancer, melanoma, fibrosarcoma, bone and connective tissue sarcomas, renal cell carcinoma, giant cell carcinoma, squamous cell carcinoma, and adenocarcinoma; diseases characterised by tissue remodeling and/or chronic inflammation such as fibrotic diseases, wound healing disorders, keloid formation disorders, osteoarthritis, rheumatoid arthritis, cartilage degradation disorders, atherosclerotic disease and Chron's disease; disorders involving endocrinological dysfunction, such as disorders of glucose metabolism ; and blood clotting disorders.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13701410.6A EP2804859B1 (en) | 2012-01-17 | 2013-01-17 | Novel fap inhibitors |
| US14/372,798 US9346814B2 (en) | 2012-01-17 | 2013-01-17 | FAP inhibitors |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1200705.0A GB201200705D0 (en) | 2012-01-17 | 2012-01-17 | Novel fap inhibitors |
| GB1200705.0 | 2012-01-17 | ||
| GB1220458.2 | 2012-11-14 | ||
| GBGB1220458.2A GB201220458D0 (en) | 2012-11-14 | 2012-11-14 | Novel FAP inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013107820A1 true WO2013107820A1 (en) | 2013-07-25 |
Family
ID=47605504
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2013/050845 WO2013107820A1 (en) | 2012-01-17 | 2013-01-17 | Novel fap inhibitors |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US9346814B2 (en) |
| EP (1) | EP2804859B1 (en) |
| WO (1) | WO2013107820A1 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103467374A (en) * | 2013-08-30 | 2013-12-25 | 江苏弘和药物研发有限公司 | Synthesis method of 8-bromine-4-carboxyl quinoline |
| WO2018129497A1 (en) | 2017-01-09 | 2018-07-12 | Bioxcel Therapeutics, Inc. | Predictive and diagnostic methods for prostate cancer |
| WO2019083990A2 (en) | 2017-10-23 | 2019-05-02 | The Johns Hopkins University | IMAGING AND RADIOTHERAPEUTICS AGENTS TARGETING FIBROBLAST-ACTIVATION PROTEIN-α (FAP-α) |
| WO2019118932A1 (en) | 2017-12-15 | 2019-06-20 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| WO2019154886A1 (en) * | 2018-02-06 | 2019-08-15 | Universität Heidelberg | Fap inhibitor |
| WO2019154859A1 (en) * | 2018-02-06 | 2019-08-15 | Universität Heidelberg | Fap inhibitor |
| WO2020132661A2 (en) | 2018-12-21 | 2020-06-25 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| EP3763726A1 (en) | 2019-07-08 | 2021-01-13 | 3B Pharmaceuticals GmbH | Compounds comprising a fibroblast activation protein ligand and use thereof |
| WO2021005131A1 (en) | 2019-07-08 | 2021-01-14 | 3B Pharmaceuticals Gmbh | Compounds comprising a fibroblast activation protein ligand and use thereof |
| WO2021005125A1 (en) | 2019-07-08 | 2021-01-14 | 3B Pharmaceuticals Gmbh | Compounds comprising a fibroblast activation protein ligand and use thereof |
| WO2021160825A1 (en) | 2020-02-12 | 2021-08-19 | Philochem Ag | Fibroblast activation protein ligands for targeted delivery applications |
| WO2021197519A1 (en) * | 2020-03-30 | 2021-10-07 | Ustav Organicke Chemie A Biochemie Av Cr, V. V. I. | Compounds for inhibition of fibroblast activation protein |
| US20210316018A1 (en) * | 2016-12-14 | 2021-10-14 | Purdue Research Foundation | Fibroblast activation protein (fap)-targeted imaging and therapy |
| WO2021224438A1 (en) | 2020-05-07 | 2021-11-11 | Institut Curie | Antxr1 as a biomarker of immunosuppressive fibroblast populations and its use for predicting response to immunotherapy |
| JP2022516555A (en) * | 2019-01-04 | 2022-02-28 | プラクシス バイオテック エルエルシー | Inhibitor of fibroblast-activated protein |
| WO2022148843A1 (en) | 2021-01-07 | 2022-07-14 | 3B Pharmaceuticals Gmbh | Compounds comprising a fibroblast activation protein ligand and use thereof |
| WO2022148851A1 (en) | 2021-01-07 | 2022-07-14 | 3B Pharmaceuticals Gmbh | Compounds comprising a fibroblast activation protein ligand and use thereof |
| CN114796533A (en) * | 2018-10-24 | 2022-07-29 | Scv-特种化学品销售有限公司 | Mark precursors with squaric acid linkage |
| EP4043452A1 (en) | 2021-02-12 | 2022-08-17 | Philochem AG | Bivalent fibroblast activation protein ligands for targeted delivery applications |
| WO2022171811A1 (en) | 2021-02-12 | 2022-08-18 | Philochem Ag | Bivalent fibroblast activation protein ligands for targeted delivery applications |
| US11426472B2 (en) | 2018-10-17 | 2022-08-30 | Purdue Research Foundation | Fibroblast activation protein (FAP) targeted imaging and therapy in fibrosis |
| WO2023057457A1 (en) | 2021-10-04 | 2023-04-13 | Philochem Ag | Radiolabelled fibroblast activation protein ligands |
| RU2797409C2 (en) * | 2018-02-06 | 2023-06-05 | Университэт Хайдельберг | Fap inhibitor |
| WO2023144379A1 (en) | 2022-01-30 | 2023-08-03 | Philochem Ag | High-affinity ligands of fibroblast activation protein for targeted delivery applications |
| WO2023233023A1 (en) | 2022-06-03 | 2023-12-07 | Universiteit Antwerpen | Inhibitors of fibroblast activation protein |
| US11858924B2 (en) | 2020-12-17 | 2024-01-02 | Astrazeneca | N-(2-(4-cyanothiazolidin-3-yl)-2-oxoethyl)-quinoline-4-carboxamides |
| WO2024052333A1 (en) | 2022-09-06 | 2024-03-14 | Philochem Ag | Multivalent fibroblast activation protein ligands for targeted delivery applications |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102274077B1 (en) * | 2019-03-29 | 2021-07-09 | 보령제약 주식회사 | Pyrrolidine derivatives as inhibitor of fibroblast activation protein (fap) and pharmaceutical composition including the same |
| CN114380795A (en) * | 2020-10-22 | 2022-04-22 | 四川大学华西医院 | Deuterated FAP inhibitor and application thereof |
| CN117255685A (en) * | 2021-04-02 | 2023-12-19 | 约翰霍普金斯大学 | Heterodivalent and homodivalent agents targeting fibroblast activation protein alpha and/or prostate specific membrane antigen |
| CN113527266A (en) * | 2021-06-23 | 2021-10-22 | 上海健康医学院 | FAP-targeted hydrogen peroxide-responsive prodrug and preparation method and application thereof |
| TW202317541A (en) | 2021-10-28 | 2023-05-01 | 行政院原子能委員會核能研究所 | Compound or its salt thereof targeting fibroblast activation protein, its preparation methods and its uses thereof |
| CN115304582B (en) * | 2022-07-20 | 2023-05-12 | 北京法伯新天医药科技有限公司 | FAP-alpha specific tumor diagnostic imaging agent |
| CN115368342B (en) * | 2022-08-24 | 2024-01-23 | 西南医科大学附属医院 | Fibroblast active protein inhibitor, radionuclide marker, preparation method and application thereof |
| WO2024183618A1 (en) * | 2023-03-03 | 2024-09-12 | 晶核生物医药科技(南京)有限公司 | Nitrogen-containing heterocyclic compound, preparation method therefor, and use thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995032948A1 (en) * | 1994-05-27 | 1995-12-07 | Smithkline Beecham Farmaceutici S.P.A. | Quinoline derivatives as tachykinin nk3 receptor antagonists |
| WO2007085895A2 (en) * | 2005-09-02 | 2007-08-02 | Ferring B.V. | Fap inhibitors |
| US20110230462A1 (en) * | 2010-03-22 | 2011-09-22 | Robert Than Hendricks | Pyrrolopyrazine Kinase Inhibitors |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8754107B2 (en) * | 2006-11-17 | 2014-06-17 | Abbvie Inc. | Aminopyrrolidines as chemokine receptor antagonists |
| US20120053222A1 (en) | 2009-01-23 | 2012-03-01 | Mark Gorrell | Novel Metabolic Disease Therapy |
-
2013
- 2013-01-17 WO PCT/EP2013/050845 patent/WO2013107820A1/en active Application Filing
- 2013-01-17 EP EP13701410.6A patent/EP2804859B1/en active Active
- 2013-01-17 US US14/372,798 patent/US9346814B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995032948A1 (en) * | 1994-05-27 | 1995-12-07 | Smithkline Beecham Farmaceutici S.P.A. | Quinoline derivatives as tachykinin nk3 receptor antagonists |
| WO2007085895A2 (en) * | 2005-09-02 | 2007-08-02 | Ferring B.V. | Fap inhibitors |
| US20110230462A1 (en) * | 2010-03-22 | 2011-09-22 | Robert Than Hendricks | Pyrrolopyrazine Kinase Inhibitors |
Non-Patent Citations (4)
| Title |
|---|
| DATABASE REGISTRY [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 1 February 2000 (2000-02-01), XP002695552, Database accession no. 1097629-86-1 * |
| NINOSLAV OPAC IC ET AL: "-Amino Acid Derivatives of Hydroxyurea and Hydantoins: Synthesis, X-ray Crystal Structure Study, and Cytostatic and Antiviral Activity Evaluations", JOURNAL OF MEDICINAL CHEMISTRY, vol. 48, no. 2, 1 January 2005 (2005-01-01), pages 475 - 482, XP055053198, ISSN: 0022-2623, DOI: 10.1021/jm040869i * |
| SUZUKI M ET AL: "Synthesis and Central Nervous System Actions of Thyrotropin-Releasing Hormone Analogues Containing a Dihydroorotic Acid Moiety", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 33, no. 8, 1 January 1990 (1990-01-01), pages 2130 - 2137, XP002186256, ISSN: 0022-2623, DOI: 10.1021/JM00170A014 * |
| THEODORE J. NITZ ET AL: "An excursion into the synthesis of potential angiotensin converting enzyme inhibitors", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 47, no. 21, 1 October 1982 (1982-10-01), pages 4029 - 4032, XP055053192, ISSN: 0022-3263, DOI: 10.1021/jo00142a005 * |
Cited By (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103467374A (en) * | 2013-08-30 | 2013-12-25 | 江苏弘和药物研发有限公司 | Synthesis method of 8-bromine-4-carboxyl quinoline |
| US11872291B2 (en) | 2016-12-14 | 2024-01-16 | Purdue Research Foundation | Fibroblast activation protein (FAP)-targeted imaging and therapy |
| US20210316018A1 (en) * | 2016-12-14 | 2021-10-14 | Purdue Research Foundation | Fibroblast activation protein (fap)-targeted imaging and therapy |
| WO2018129497A1 (en) | 2017-01-09 | 2018-07-12 | Bioxcel Therapeutics, Inc. | Predictive and diagnostic methods for prostate cancer |
| US11938201B2 (en) | 2017-10-23 | 2024-03-26 | The Johns Hopkins University | Imaging and radiotherapeutics agents targeting fibroblast-activation protein-alpha (FAP-alpha) |
| EP3700580A4 (en) * | 2017-10-23 | 2021-06-23 | The Johns Hopkins University | Imaging and radiotherapeutics agents targeting fibroblast-activation protein- (fap- ) |
| WO2019083990A2 (en) | 2017-10-23 | 2019-05-02 | The Johns Hopkins University | IMAGING AND RADIOTHERAPEUTICS AGENTS TARGETING FIBROBLAST-ACTIVATION PROTEIN-α (FAP-α) |
| US12115233B2 (en) | 2017-10-23 | 2024-10-15 | The Johns Hopkins University | Imaging and radiotherapeutics agents targeting fibroblast-activation protein-α (FAP-α) |
| IL275333B2 (en) * | 2017-12-15 | 2023-09-01 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| AU2018386298B2 (en) * | 2017-12-15 | 2023-09-07 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| IL275333B1 (en) * | 2017-12-15 | 2023-05-01 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| EP3723753A4 (en) * | 2017-12-15 | 2022-01-26 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| US11780821B2 (en) | 2017-12-15 | 2023-10-10 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| WO2019118932A1 (en) | 2017-12-15 | 2019-06-20 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| CN111699181A (en) * | 2018-02-06 | 2020-09-22 | 海德堡大学 | FAP inhibitors |
| WO2019154886A1 (en) * | 2018-02-06 | 2019-08-15 | Universität Heidelberg | Fap inhibitor |
| AU2019219057B2 (en) * | 2018-02-06 | 2022-11-24 | Universität Heidelberg | FAP inhibitor |
| RU2797409C2 (en) * | 2018-02-06 | 2023-06-05 | Университэт Хайдельберг | Fap inhibitor |
| JP2021512949A (en) * | 2018-02-06 | 2021-05-20 | ウニベルジテート ハイデルベルク | FAP inhibitor |
| WO2019154859A1 (en) * | 2018-02-06 | 2019-08-15 | Universität Heidelberg | Fap inhibitor |
| US20230112012A1 (en) * | 2018-02-06 | 2023-04-13 | Universität Heidelberg | Fap inhibitor |
| IL276594B2 (en) * | 2018-02-06 | 2023-11-01 | Univ Heidelberg | Fap inhibitor |
| IL276594B1 (en) * | 2018-02-06 | 2023-07-01 | Univ Heidelberg | Fap inhibitor |
| US11426472B2 (en) | 2018-10-17 | 2022-08-30 | Purdue Research Foundation | Fibroblast activation protein (FAP) targeted imaging and therapy in fibrosis |
| US11890357B2 (en) | 2018-10-17 | 2024-02-06 | Purdue Research Foundation | Fibroblast activation protein (FAP) targeted imaging and therapy in fibrosis |
| US11833229B2 (en) | 2018-10-24 | 2023-12-05 | Scv Spezial-Chemikalien-Vertriebs Gmbh | Labeling precursors with squaric acid coupling |
| CN114796533A (en) * | 2018-10-24 | 2022-07-29 | Scv-特种化学品销售有限公司 | Mark precursors with squaric acid linkage |
| CN114796533B (en) * | 2018-10-24 | 2024-01-26 | Scv-特种化学品销售有限公司 | Marking precursors with squaric acid linkages |
| EP4082581A1 (en) * | 2018-10-24 | 2022-11-02 | SCV - SpezialChemikalienVertrieb GmbH | Marking precursor with squaric acid coupling |
| JP2022514352A (en) * | 2018-12-21 | 2022-02-10 | プラクシス バイオテック エルエルシー | Inhibitor of fibroblast-activated protein |
| CN113811529A (en) * | 2018-12-21 | 2021-12-17 | 普拉西斯生物技术有限责任公司 | Fibroblast activation protein inhibitor |
| WO2020132661A2 (en) | 2018-12-21 | 2020-06-25 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| EP3898654A4 (en) * | 2018-12-21 | 2022-10-26 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| US11504364B2 (en) | 2018-12-21 | 2022-11-22 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| EP3906024A4 (en) * | 2019-01-04 | 2022-10-26 | Praxis Biotech LLC | Inhibitors of fibroblast activation protein |
| CN114126597A (en) * | 2019-01-04 | 2022-03-01 | 普拉西斯生物技术有限责任公司 | Fibroblast activation protein inhibitor |
| JP2022516555A (en) * | 2019-01-04 | 2022-02-28 | プラクシス バイオテック エルエルシー | Inhibitor of fibroblast-activated protein |
| RU2802426C2 (en) * | 2019-01-04 | 2023-08-28 | ПРАКСИС БАЙОТЕК ЭлЭлСи | Fibroblast activation protein inhibitors |
| WO2021005125A1 (en) | 2019-07-08 | 2021-01-14 | 3B Pharmaceuticals Gmbh | Compounds comprising a fibroblast activation protein ligand and use thereof |
| WO2021005131A1 (en) | 2019-07-08 | 2021-01-14 | 3B Pharmaceuticals Gmbh | Compounds comprising a fibroblast activation protein ligand and use thereof |
| EP3763726A1 (en) | 2019-07-08 | 2021-01-13 | 3B Pharmaceuticals GmbH | Compounds comprising a fibroblast activation protein ligand and use thereof |
| WO2021160825A1 (en) | 2020-02-12 | 2021-08-19 | Philochem Ag | Fibroblast activation protein ligands for targeted delivery applications |
| EP4019507A1 (en) | 2020-02-12 | 2022-06-29 | Philochem AG | Fibroblast activation protein ligands for targeted delivery applications |
| JP2023519752A (en) * | 2020-03-30 | 2023-05-12 | ウスタフ オルガニッケ ヘミエ アー ビオヘミエ アカデミエ ヴェド ツェーエル,ヴェー.ヴェー.イー | Compounds for inhibiting fibroblast activation protein |
| WO2021197519A1 (en) * | 2020-03-30 | 2021-10-07 | Ustav Organicke Chemie A Biochemie Av Cr, V. V. I. | Compounds for inhibition of fibroblast activation protein |
| AU2021248368B2 (en) * | 2020-03-30 | 2023-04-06 | Ustav Organicke Chemie A Biochemie Av Cr, V. V. I. | Compounds for inhibition of fibroblast activation protein |
| JP7368637B2 (en) | 2020-03-30 | 2023-10-24 | ウスタフ オルガニッケ ヘミエ アー ビオヘミエ アカデミエ ヴェド ツェーエル,ヴェー.ヴェー.イー | Compounds for inhibiting fibroblast activation proteins |
| WO2021224438A1 (en) | 2020-05-07 | 2021-11-11 | Institut Curie | Antxr1 as a biomarker of immunosuppressive fibroblast populations and its use for predicting response to immunotherapy |
| US11858924B2 (en) | 2020-12-17 | 2024-01-02 | Astrazeneca | N-(2-(4-cyanothiazolidin-3-yl)-2-oxoethyl)-quinoline-4-carboxamides |
| EP4050018A1 (en) | 2021-01-07 | 2022-08-31 | 3B Pharmaceuticals GmbH | Compounds comprising a fibroblast activation protein ligand and use thereof |
| WO2022148851A1 (en) | 2021-01-07 | 2022-07-14 | 3B Pharmaceuticals Gmbh | Compounds comprising a fibroblast activation protein ligand and use thereof |
| WO2022148843A1 (en) | 2021-01-07 | 2022-07-14 | 3B Pharmaceuticals Gmbh | Compounds comprising a fibroblast activation protein ligand and use thereof |
| EP4043452A1 (en) | 2021-02-12 | 2022-08-17 | Philochem AG | Bivalent fibroblast activation protein ligands for targeted delivery applications |
| WO2022171811A1 (en) | 2021-02-12 | 2022-08-18 | Philochem Ag | Bivalent fibroblast activation protein ligands for targeted delivery applications |
| WO2023057457A1 (en) | 2021-10-04 | 2023-04-13 | Philochem Ag | Radiolabelled fibroblast activation protein ligands |
| WO2023144379A1 (en) | 2022-01-30 | 2023-08-03 | Philochem Ag | High-affinity ligands of fibroblast activation protein for targeted delivery applications |
| WO2023233023A1 (en) | 2022-06-03 | 2023-12-07 | Universiteit Antwerpen | Inhibitors of fibroblast activation protein |
| WO2024052333A1 (en) | 2022-09-06 | 2024-03-14 | Philochem Ag | Multivalent fibroblast activation protein ligands for targeted delivery applications |
Also Published As
| Publication number | Publication date |
|---|---|
| US20140357650A1 (en) | 2014-12-04 |
| EP2804859B1 (en) | 2019-06-12 |
| EP2804859A1 (en) | 2014-11-26 |
| US9346814B2 (en) | 2016-05-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2804859B1 (en) | Novel fap inhibitors | |
| KR102642203B1 (en) | Multicyclic compounds and methods for targeted cleavage of rapidly progressive fibrosarcoma polypeptides | |
| KR102090780B1 (en) | Substituted pyrrolidines as factor xia inhibitors for the treatment thromboembolic diseases | |
| CN112262139B (en) | Aminopyrrolotriazines as kinase inhibitors | |
| JP5847284B2 (en) | Hepatitis C virus inhibitor | |
| TWI475019B (en) | Hepatitis c virus inhibitors | |
| EP4132655A1 (en) | Compounds and methods for targeted degradation of kras | |
| JP2023530267A (en) | Antiviral compounds for treating coronavirus, picornavirus and norovirus infections | |
| JP5612661B2 (en) | Hepatitis C virus inhibitor | |
| KR20200035435A (en) | Compounds and methods for targeted decomposition of androgen receptors | |
| CA3189027A1 (en) | Antiviral heteroaryl ketone derivatives | |
| KR20200020978A (en) | Compounds and methods for the targeted degradation of androgen receptor | |
| KR20110124774A (en) | Hepatitis c virus inhibitors | |
| KR20100124290A (en) | Hepatitis c virus inhibitors | |
| KR20140045903A (en) | Hepatitis c virus inhibitors | |
| JP6337750B2 (en) | Compound | |
| KR20220050942A (en) | enzyme inhibitor | |
| KR20160064100A (en) | Substituted phenylalanine derivatives | |
| CN115484948A (en) | Compound and use thereof | |
| WO2023232776A1 (en) | Haloindole macrocyclic compounds for the treatment of cancer | |
| WO2017006295A1 (en) | Hydroxy formamide derivatives and their use | |
| EP4058448A1 (en) | Substituted hydantoinamides as adamts7 antagonists | |
| KR20210075120A (en) | Inhibition of fatty acid synthase (FASN) | |
| WO2024199266A1 (en) | Compounds for degradation and inhibition of kras (g12d) protein | |
| OA20440A (en) | Nitrile-containing antiviral compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13701410 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013701410 Country of ref document: EP Ref document number: 14372798 Country of ref document: US |