WO2012072512A1 - N-cyclobutyl-imidazopyridine or -pyrazolopyridine carboxamides as trpv1 antagonists - Google Patents
N-cyclobutyl-imidazopyridine or -pyrazolopyridine carboxamides as trpv1 antagonists Download PDFInfo
- Publication number
- WO2012072512A1 WO2012072512A1 PCT/EP2011/071035 EP2011071035W WO2012072512A1 WO 2012072512 A1 WO2012072512 A1 WO 2012072512A1 EP 2011071035 W EP2011071035 W EP 2011071035W WO 2012072512 A1 WO2012072512 A1 WO 2012072512A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oxy
- cyclobutyl
- pyridine
- carboxamide
- frans
- Prior art date
Links
- 0 *c1cccnc1* Chemical compound *c1cccnc1* 0.000 description 6
- YGUOUZOUTDREDM-UHFFFAOYSA-N CC(C)(C)OC(NC(C1)CC1Oc(cc1)ccc1Cl)=O Chemical compound CC(C)(C)OC(NC(C1)CC1Oc(cc1)ccc1Cl)=O YGUOUZOUTDREDM-UHFFFAOYSA-N 0.000 description 1
- WVMDSNGINQNHLN-UHFFFAOYSA-N CC(C)(C)OC(NOS(c1c(C)cc(C)cc1C)(=O)=O)=O Chemical compound CC(C)(C)OC(NOS(c1c(C)cc(C)cc1C)(=O)=O)=O WVMDSNGINQNHLN-UHFFFAOYSA-N 0.000 description 1
- YGUOUZOUTDREDM-AULYBMBSSA-N CC(C)(C)OC(N[C@H](C1)C[C@@H]1Oc(cc1)ccc1Cl)=O Chemical compound CC(C)(C)OC(N[C@H](C1)C[C@@H]1Oc(cc1)ccc1Cl)=O YGUOUZOUTDREDM-AULYBMBSSA-N 0.000 description 1
- MOOJLOULTXIPKL-HAQNSBGRSA-N CC(C)(C)OC(N[C@H](C1)C[C@@H]1Oc1c(C)c(F)ccc1)=O Chemical compound CC(C)(C)OC(N[C@H](C1)C[C@@H]1Oc1c(C)c(F)ccc1)=O MOOJLOULTXIPKL-HAQNSBGRSA-N 0.000 description 1
- MLAGMXYZWBNRQY-UHFFFAOYSA-N CC(C)(C)c(c(F)cc(/[O]=C(\C1)/C[C@H]1NC(c1cnc2[n]1cccc2)=O)c1)c1F Chemical compound CC(C)(C)c(c(F)cc(/[O]=C(\C1)/C[C@H]1NC(c1cnc2[n]1cccc2)=O)c1)c1F MLAGMXYZWBNRQY-UHFFFAOYSA-N 0.000 description 1
- DMBJHRKFWWSSOC-UHFFFAOYSA-N CC(C)(C)c(c(F)cc(O)c1)c1F Chemical compound CC(C)(C)c(c(F)cc(O)c1)c1F DMBJHRKFWWSSOC-UHFFFAOYSA-N 0.000 description 1
- LTCFZQNXOGBZRP-JDVJWJCPSA-N CC(C)c(cc1)cc(C(C)C)c1O[C@H](C1)C[C@@H]1C(CC(c1c(cccc2)[n]2nc1)=O)NC Chemical compound CC(C)c(cc1)cc(C(C)C)c1O[C@H](C1)C[C@@H]1C(CC(c1c(cccc2)[n]2nc1)=O)NC LTCFZQNXOGBZRP-JDVJWJCPSA-N 0.000 description 1
- BJHHCGRWVQLRAI-UHFFFAOYSA-N Cc(c(OC(C1)CC1NC(c1cnc2[n]1cccc2)=O)ccc1)c1F Chemical compound Cc(c(OC(C1)CC1NC(c1cnc2[n]1cccc2)=O)ccc1)c1F BJHHCGRWVQLRAI-UHFFFAOYSA-N 0.000 description 1
- CHKQALUEEULCPZ-UHFFFAOYSA-N Cc(cc1C)cc(C)c1S(ON)(=O)=O Chemical compound Cc(cc1C)cc(C)c1S(ON)(=O)=O CHKQALUEEULCPZ-UHFFFAOYSA-N 0.000 description 1
- VFDZZAXVMDKSML-UHFFFAOYSA-N O=C(c1c(cccc2)[n]2nc1)NC(C1)CC1Oc(cccc1)c1OC(F)(F)F Chemical compound O=C(c1c(cccc2)[n]2nc1)NC(C1)CC1Oc(cccc1)c1OC(F)(F)F VFDZZAXVMDKSML-UHFFFAOYSA-N 0.000 description 1
- UQBONSCSDNPNCQ-OTVXOJSOSA-N O=C(c1cnc2[n]1cccc2)N[C@H](C1)C[C@H]1Oc(cc1)ccc1Cl Chemical compound O=C(c1cnc2[n]1cccc2)N[C@H](C1)C[C@H]1Oc(cc1)ccc1Cl UQBONSCSDNPNCQ-OTVXOJSOSA-N 0.000 description 1
- VLPFWXBUHIUDJE-UHFFFAOYSA-N OC1CC(CNC(c2c(cccc3)[n]3nc2)=O)C1 Chemical compound OC1CC(CNC(c2c(cccc3)[n]3nc2)=O)C1 VLPFWXBUHIUDJE-UHFFFAOYSA-N 0.000 description 1
- VPGHGXKEPUYSDU-UHFFFAOYSA-N OCc([n]1nc2)cccc1c2C(NC(C1)CC1Oc(cc1)ccc1Cl)=O Chemical compound OCc([n]1nc2)cccc1c2C(NC(C1)CC1Oc(cc1)ccc1Cl)=O VPGHGXKEPUYSDU-UHFFFAOYSA-N 0.000 description 1
- XJPZMQGRRRBNSY-KOMQPUFPSA-N OCc1c[n]2ncc(C(N[C@H](C3)C[C@@H]3Oc(cc3)ccc3Cl)=O)c2cc1 Chemical compound OCc1c[n]2ncc(C(N[C@H](C3)C[C@@H]3Oc(cc3)ccc3Cl)=O)c2cc1 XJPZMQGRRRBNSY-KOMQPUFPSA-N 0.000 description 1
- YNAYKRRIMZFURA-KOMQPUFPSA-N OCc1ccc[n]2c1ncc2C(N[C@H](C1)C[C@@H]1Oc(cc1)ccc1Cl)=O Chemical compound OCc1ccc[n]2c1ncc2C(N[C@H](C1)C[C@@H]1Oc(cc1)ccc1Cl)=O YNAYKRRIMZFURA-KOMQPUFPSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
Definitions
- the present invention relates to novel compounds, being TRPV1 antagonists having pharmacological activity, to pharmaceutical compositions comprising the compounds and to the use of the compounds in medicine, especially in the treatment of rhinitis or the treatment of asthma.
- vanilloid compounds or vanilloid receptor modulators are capsaicin or trans 8-methyl-N-vanillyl-6-nonenamide which is isolated from the pepper plant, capsazepine (Tetrahedron, 53, 1997, 4791) and olvanil or - N- (4-hydroxy-3-methoxybenzyl)oleamide (J. Med. Chem., 36, 1993, 2595).
- TRPV1 Transient Receptor Potential Vanilloid subfamily member 1
- TRPV1 is a calcium-permeable, ligand gated ion channel which is highly expressed in sensory neurones (Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD and Julius D (1997) Nature 389, 816-824) whose function is modulated by such Vanilloid compounds.
- TRPV1 has been studied and is extensively reviewed by Szallasi and Blumberg (The American Society for Pharmacology and Experimental Therapeutics, 1999, Vol. 51 , No. 2.).
- the channel pore Upon activation, the channel pore opens and allows influx of cations which depolarises the nerve membrane and triggers neuronal axonal firing and/or local release of neurotransmitters such as Substance P and CGRP.
- Activation may be caused by a single trigger, such as pH, but may be caused by integration of different triggers acting in concert on the channel.
- the role of TRPV1 in disease has been studied extensively in pain models where a role in both thermal and post-inflammatory hyperalgesia is well established (Chizh et al, Jara-Oseguera et al, 2008).
- TRPV1 has also been implicated in other diseases where symptoms are potentially driven wholly or in part by neuronal hypersensitivity or hyperactivity, because of its role in sensory signalling in peripheral nerves. Such diseases include asthma, rhinitis, overactive bladder, reflux oesophagitis, irritable bowel syndrome and migraine. TRPV1 has been implicated in inflammatory responses occurring in dry eye syndrome (Pan, Wang, Yang, Zhang & Reinach (2010), TRPV1 Activation is Required for Hypertonicity Stimulated Inflammatory Cytokine Release in Human Corneal Epithelial Cells, Manuscript IOVS, 10-5801).
- TRPV1 is also implicated to play a role in metabolic diseases such as diabetes and obesity (Motter AL & Ahern GP (2008) FEBS Letters 582, 2257-2262; Suri & Szallasi A (2007), The emerging role of TRPV1 in diabetes and obesity, Trends in Pharm Sci; Rasavi et al (2006) Cell 127, 1123-1135.)
- Patent Application Number WO 03/022809 discloses urea derivatives including ⁇ /-(2- Bromophenyl)-/V'-[((f?)-1-(5-trifluoromethyl-2-pyridyl)pyrrolidin-3-yl)]urea and A/-(3-methyl-5- isoquinolinyl))-/V'-[(3f?)-1-(5-trifluoromethyl-2-pyridyl)pyrrolidin-3-yl)]urea or pharmaceutically acceptable salts or solvates thereof and their use in the treatment of diseases associated with the activity of TRPV1.
- Patent Application Number WO 10/026129 discloses A/-(2-Bromophenyl)-/V-[((ft)-1-(5- trifluoromethyl-2-pyridyl) pyrrolidin-3-yl)] urea for use in the treatment of rhinitis.
- Patent Application Number WO 10/026128 discloses A/-(3-methyl-5-isoquinolinyl))-/V-[(3ft)-1-(5- trifluoromethyl-2-pyridyl) pyrrolidin-3-yl)] urea for use in the treatment of rhinitis.
- X 1 represents a hydrogen atom, or a CH 2 OH group
- X 2 represents a hydrogen atom, a fluorine atom, an OCH 3 group or a CH 2 OH group, and at least one of X 1 and X 2 is hydrogen
- Y represents a carbon atom and Z represents a nitrogen atom
- Y represents a nitrogen atom and Z represents a carbon atom
- R 1 represents a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group
- R 2 and R 3 are each independently selected from a hydrogen atom, a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group
- the invention further provides a method for the treatment or prophylaxis of disorders in which antagonism of TRPV1 is beneficial, in a human, which comprises administering a human in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
- the invention also provides a method for the treatment of asthma which comprises administering to a human in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
- the invention provides for the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof in the manufacture of a medicament for the treatment of conditions in which an antagonist of TRPV1 is indicated, particularly rhinitis or asthma.
- the compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof is used in the treatment of non allergic rhinitis.
- a compound of formula (I) may be prepared by methods described herein.
- Y represents a carbon atom and Z represents a nitrogen atom
- the present invention covers compounds of formula (I) as the free base and as salts thereof, for example as a pharmaceutically acceptable salt thereof.
- the invention relates to compounds of formula (I) or a pharmaceutically acceptable salt thereof.
- salts of the compounds of formula (I) are desirably pharmaceutically acceptable.
- suitable pharmaceutically acceptable salts can include acid addition salts.
- a pharmaceutically acceptable salt may be readily prepared by using a desired acid or base as appropriate. The resultant salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent.
- a pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of formula (I) with a suitable inorganic or organic acid (such as hydrobromic, hydrochloric, sulphuric, nitric, phosphoric, succinic, maleic, acetic, propionic, fumaric, citric, tartaric, lactic, benzoic, salicylic, glutamaic, aspartic, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic, or hexanoic acid), optionally in a suitable solvent such as an organic solvent, to give the salt which is usually isolated for example by crystallisation and filtration.
- a suitable inorganic or organic acid such as hydrobromic, hydrochloric, sulphuric, nitric, phosphoric, succinic, maleic, acetic,
- a pharmaceutically acceptable acid addition salt of a compound of formula (I) can comprise or be for example a hydrobromide, hydrochloride, sulphate, nitrate, phosphate, succinate, maleate, acetate, propionate, fumarate, citrate, tartrate, lactate, benzoate, salicylate, glutamate, aspartate, p- toluenesulfonate, benzenesulfonate, methanesulfonate, ethanesulphonate, naphthalenesulfonate (e.g. 2-naphthalenesulfonate) or hexanoate salt.
- Other non-pharmaceutically acceptable salts e.g. formates, oxalates or trifluoroacetates, may be used, for example in the isolation of the compounds of formula (I), and are included within the scope of this invention.
- Separation of isomers may be achieved by conventional techniques known to those skilled in the art, e.g. by fractional crystallisation, chromatography or HPLC.
- X 1 represents a hydrogen atom.
- Y represents C and Z represents N. In another embodiment, Y represents N and Z represents C.
- R 1 represents a 1 , 1-dimethylethyl group and R 2 and R 3 independently represent a fluorine atom or a hydrogen atom.
- R 1 represents a 1 , 1-dimethylethyl group at the para position relative to the ether group
- R 2 represents a fluorine atom at a meta position relative to the ether group
- R 3 represents a fluorine or a hydrogen atom at the other meta position to the ether group.
- the compound of formula (I) is selected from:
- TRPV1 antagonist activity and are believed to be of potential use for the treatment or prophylaxis of certain disorders, or treatment of the pain associated with them, such as: respiratory diseases, asthma, cough, COPD, bronchoconstriction, rhinitis, inflammatory disorders, pain, such as acute pain, chronic pain, neuropathic pain, postoperative pain, postrheumatoid arthritic pain, osteoarthritic pain, back pain, visceral pain, cancer pain, algesia, neuralgia, dental pain, headache, migraine, neuropathies, carpal tunnel syndrome, diabetic neuropathy, HlV-related neuropathy, post-herpetic neuralgia, fibromyalgia, neuritis, sciatica, nerve injury, ischaemia, neurodegeneration, stroke, post stroke pain, multiple sclerosis, oesophagitis, heart burn, Barrett's metaplasia, dysphagia, gastroeosophageal reflux disorder (GER)
- disorders of particular interest are respiratory diseases, asthma, cough, COPD,
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention, for use in the treatment of a condition for which a TRPV1 antagonist is indicated.
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention, for use in the treatment of rhinitis.
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention, for use in the treatment of asthma.
- the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention in the manufacture of a medicament for the treatment of a condition for which a TRPV1 antagonist is indicated.
- the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention in the manufacture of a medicament for the treatment of rhinitis. In a further aspect the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention in the manufacture of a medicament for the treatment of asthma.
- the invention provides a method for the treatment or prophylaxis of disorders in which antagonism of TRPV1 is beneficial in a human comprising administering to the human in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention.
- the invention provides a method for the treatment of rhinitis in a human in need thereof comprising administering to the human a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention.
- the invention provides a method for the treatment of asthma in a human in need thereof comprising administering to the human a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention.
- R 1 , R 2 and R 3 are defined as above for compounds of formula (I).
- This coupling may be conducted, for example, using HATU (0-(7-azabenzotriazol-1-yl)-/V, N, N', ⁇ /'-tetramethyluronium hexafluorophosphate) in the presence of a suitable base such as triethylamine or DIPEA (A/,A/-diisopropylethylamine) in a suitable solvent such as DMF (N,N- dimethylformamide).
- a suitable base such as triethylamine or DIPEA (A/,A/-diisopropylethylamine)
- DMF N,N- dimethylformamide
- the cycloaddition reaction may be performed, for example, by reacting a compound of formula (V) with methyl or ethyl propiolate in the presence of a suitable base such as potassium hydroxide or potassium carbonate in a suitable solvent such as DMF or DMSO. Where the substituent X 1 in compound (V) is not hydrogen the cycloaddition may give a mixture of regioisomeric cycloaddition products which may be separated for example by chromatography.
- Compounds of formula (V) are either commercially available or may be prepared by reaction of the corresponding pyridines (VI) with O-(mesitylsulphonyl) hydroxylamine (VII) in a suitable solvent, for example, DME.
- Compound of formula (VII) may be generated by reaction of 1 , 1- dimethylethyl ⁇ [(2, 4, 6-trimethylphenyl) sulfonyl] oxy ⁇ carbamate (VIII) with TFA.
- P is a suitable protecting group such as a BOC (tert-butyloxycarbonyl) group, followed by removal of the protecting group.
- BOC tert-butyloxycarbonyl
- This ether formation may be conducted for example using Mitsunobu conditions by reacting the phenol (X) with the protected amincyclobutanol (XI) in the presence of diisopropylazodicarboxylate and triphenylphosphine in a suitable solvent such as THF.
- the protecting group is a BOC group this can then be removed under acidic conditions, for example, using hydrochloric acid in dioxane, or TFA in dichloromethane to give compounds of formula (III).
- X 1 , X 2 , Y and Z are as defined above for compounds of formula (I) and Q is, for example, methyl, with a phenol of formula (X)
- a base such as potassium f-butoxide, potassium carbonate or A/,A/-diisopropylethylamine in DMF.
- the reaction is usually conducted at elevated temperatures via conventional heating, e.g. 70°C or via microwave irradiation e.g. 200°C
- Compounds of formula (XII) can be prepared from compounds of formula (XIII):
- reaction can be conducted using methodology described in the literature, for example by reaction of the alcohol (XIII) with a sulfonyl chloride, such as methanesulfonyl chloride, in a suitable solvent such as DMF in the presence of a base such as triethylamine.
- a sulfonyl chloride such as methanesulfonyl chloride
- a suitable solvent such as DMF
- a base such as triethylamine
- This coupling may be conducted, for example, using HATU (0-(7-azabenzotriazol-1-yl)-/V, N, N', ⁇ /'-tetramethyluronium hexafluorophosphate) in the presence of a suitable base such as triethylamine or DIPEA (N, /V-diisopropylethylamine) in a suitable solvent such as DMF.
- HATU ((7-azabenzotriazol-1-yl)-/V, N, N', ⁇ /'-tetramethyluronium hexafluorophosphate
- a suitable base such as triethylamine or DIPEA (N, /V-diisopropylethylamine)
- a suitable solvent such as DMF.
- the coupling may also be conducted using alternative, conventional conditions for amide bond formation known in the art.
- Crystallisation may be performed using conventional solvents such as methanol, ethanol, or butanol, or aqueous mixtures thereof. It will be appreciated that specific reaction times temperatures may typically be determined by reaction-monitoring techniques, for example thin-layer chromatography and LC-MS.
- the absolute stereochemistry of compounds may be determined using conventional methods, such as X-ray crystallography.
- Compounds (III), (XI), (XII), (XIII) and (XIV) can exist as stereoisomers, cis and trans, both isomers are either commercially available or may be derived from compounds that are commercially available and/or may be prepared using methodology described in the literature.
- the synthesis may result in mixtures of cis I trans isomers which can be separated by chromatographic techniques or may give the individual cis or trans isomers. Subsequent reactions as described and known to a person skilled in the art can result in inversion of the stereochemistry.
- Compounds of formula (I) may be in crystalline or non-crystalline form.
- a compound of formula (I) can form pharmaceutically acceptable salts.
- pharmaceutically acceptable salts are those used conventionally in the art and include those described in Berge, J. Pharm. Sci., 1977, 66, 1-19.
- pharmaceutically acceptable acid addition salts include salts with inorganic acids such, for example, as hydrochloric acid, hydrobromic acid, orthophosphoric acid or sulphuric acid, or with organic acids such, for example as methanesulphonic acid, toluenesulphonic acid, acetic acid, propionic acid, lactic acid, citric acid, fumaric acid, malic acid, succinic acid, salicylic acid, maleic acid, glycerophosphoric acid or acetylsalicylic acid.
- the compound of formula (I) is in the form of a free base.
- a compound of formula (I) or a pharmaceutically acceptable salt thereof may be formulated with one or more pharmaceutically acceptable excipients to provide a pharmaceutical composition.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention and one or more pharmaceutically acceptable carriers or excipients.
- compositions for the treatment or prophylaxis of disorders in which antagonism of TRPV1 is beneficial comprising a compound as defined in the first aspect of the present invention or a pharmaceutically acceptable salt or solvate thereof and a pharmaceutically acceptable carrier.
- compositions for the treatment or prophylaxis of rhinitis comprising a compound as defined in the first aspect of the present invention or a pharmaceutically acceptable salt or solvate thereof and a pharmaceutically acceptable carrier.
- compositions for the treatment or prophylaxis of asthma comprising a compound as defined in the first aspect of the present invention or a pharmaceutically acceptable salt or solvate thereof and a pharmaceutically acceptable carrier.
- Pharmaceutical compositions may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual), rectal, inhaled, intranasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal) route.
- Such compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier(s) or excipient(s).
- the invention provides a formulation for intranasal administration comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- a compound of formula (I) or a pharmaceutically acceptable salt thereof would typically be in a particle-size-reduced form, which may be prepared by conventional techniques, for example, microfluidisation, micronisation and milling e.g. wet bead milling.
- the size-reduced (e.g. micronised) compound of formula (I) or a pharmaceutically acceptable salt thereof can be defined by a D 50 value of about 0.1 to 10 microns such as about 0.5 to 10 microns, more particularly about 2 to 4 microns (for example as measured using laser diffraction).
- the proportion of a compound of formula (I) or a pharmaceutically acceptable salt thereof will depend on the precise type of composition to be prepared, but will generally be within the range of from about 0.01 to 20% (w/w), based on the total weight of the composition.
- the proportion used will be within the range of from about 0.05 to 10% (w/w), such as about 0.1 to 5% (w/w).
- the dose of a compound of formula (I) or a pharmaceutically acceptable salt thereof will vary in the usual way with the seriousness of the disease to be treated and other factors such as the weight of the sufferer.
- suitable unit doses may be about between 0.005 and 1 mg for example between 0.005 and 0.5mg per dose.
- Such unit doses may be administered once a day, or more than once a day, for example two or three times a day. Such therapy may extend for a number of weeks or months.
- Combination therapies according to the present invention thus comprise the administration of at least one compound of formula (I) or a pharmaceutically acceptable salt thereof, and the use of at least one other pharmaceutically active agent.
- combination therapies according to the present invention comprise the administration of at least one compound of formula (I) or a pharmaceutically acceptable salt thereof, and at least one other pharmaceutically active agent.
- the compound(s) of the invention and the other pharmaceutically active agent(s) may be administered together in a single pharmaceutical composition or separately and, when administered separately this may occur simultaneously or sequentially in any order.
- the amounts of the compound(s) of the invention and the other pharmaceutically active agent(s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect.
- a combination comprising a compound of the invention and at least one other pharmaceutically active agent.
- composition is an aqueous pharmaceutical composition
- a further active ingredient may be incorporated into the aqueous pharmaceutical composition, particularly one used in the treatment of rhinitis and suitable for intranasal administration such as an antihistamine or a corticosteroid.
- anti-histamines include azelastine, olopatadine, bepotastine or a compound selected from:
- corticosteroids include fluticasone propionate (which is marketed as an intranasal formulation under the trade name Flixonase®), beclomethasone dipropionate (which is marketed as an intranasal formulation under the trade name Beconase®) or fluticasone furoate (which is marketed under the trade name
- the present invention provides for an aqueous
- composition comprising a compound of formula (I). or a pharmaceutically acceptable salt thereof and fluticasone furoate.
- the proportion of the further active ingredient will generally be in the range from about 0.05 to 10% (w/w), such as about 0.1 to 5% (w/w).
- Aqueous pharmaceutical compositions of the invention may include one or more pharmaceutically acceptable excipients selected from the group consisting of suspending agents, thickening agents, preservatives, wetting agents and isotonicity adjusting agents.
- an aqueous pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and a suspending agent.
- an aqueous pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and a preservative.
- compositions of the invention may be protected from microbial or fungal contamination and growth by inclusion of a preservative.
- pharmaceutically acceptable anti-microbial agents or preservatives may include quaternary ammonium compounds (e.g. benzalkonium chloride, benzethonium chloride, cetrimide and
- cetylpyridinium chloride mercurial agents (e.g. phenylmercuric nitrate, phenylmercuric acetate and thimerosal), alcoholic agents (e.g. chlorobutanol, phenylethyl alcohol and benzyl alcohol), antibacterial esters (e.g. esters of para-hydroxybenzoic acid), chelating agents such as disodium ethylenediaminetetraacetate (EDTA) and other anti-microbial agents such as chlorhexidine, chlorocresol, sorbic acid and its salts (such as potassium sorbate) and polymyxin.
- EDTA disodium ethylenediaminetetraacetate
- examples of pharmaceutically acceptable anti-fungal agents or preservatives may include sodium benzoate.
- the preservative if included, may be present in an amount of between about 0.001 and 1 % (w/w), such as about 0.015% (w/w), based on the total weight of the
- an aqueous pharmaceutical composition which is preservative free.
- an aqueous pharmaceutical composition comprising .a compound of formula (I) or a pharmaceutically acceptable salt thereof and a wetting agent.
- Compositions which contain a suspended medicament may include a pharmaceutically acceptable wetting agent which functions to wet the particles of medicament to facilitate dispersion thereof in the aqueous phase of the composition.
- wetting agent include fatty alcohols, esters and ethers, such as polyoxyethylene (20) sorbitan monooleate (Polysorbate 80).
- the wetting agent may be present in an amount of between about 0.001 and 1.0% (w/w), such as between about 0.001 and 0.05% (w/w), for example about 0.025% (w/w), based on the total weight of the composition.
- an aqueous pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and an isotonicity adjusting agent.
- An isotonicity adjusting agent may be included to achieve isotonicity with body fluids e.g. fluids of the nasal cavity, resulting in reduced levels of irritancy.
- body fluids e.g. fluids of the nasal cavity
- isotonicity adjusting agents include sodium chloride, dextrose, xylitol and calcium chloride.
- An isotonicity adjusting agent may be included in an amount of between about 0.1 and 10% (w/w), such as about 5.0% (w/w), based on the total weight of the composition.
- aqueous pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof may be buffered by the addition of suitable buffering agents such as sodium citrate, citric acid, phosphates such as disodium phosphate (for example the dodecahydrate, heptahydrate, dihydrate and anhydrous forms) or sodium phosphate and mixtures thereof.
- suitable buffering agents such as sodium citrate, citric acid, phosphates such as disodium phosphate (for example the dodecahydrate, heptahydrate, dihydrate and anhydrous forms) or sodium phosphate and mixtures thereof.
- compositions of the invention e.g. those suitable for intranasal administration may also optionally contain other excipients, such as antioxidants (for example sodium metabisulphite), taste-masking agents (such as menthol) and sweetening agents (for example dextrose, glycerol, saccharin and/or sorbitol).
- antioxidants for example sodium metabisulphite
- taste-masking agents such as menthol
- sweetening agents for example dextrose, glycerol, saccharin and/or sorbitol
- Aqueous pharmaceutical compositions according to the invention can be prepared using standard procedures that are familiar to the person skilled in the art e.g. by admixture of the various components, suitably at ambient temperature and atmospheric pressure.
- the aqueous pharmaceutical compositions of the invention are suitable for intranasal administration.
- Intranasal compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof may permit the compound(s) to be delivered to all areas of the nasal cavities (the target tissue) and further, may permit the compound(s) to remain in contact with the target tissue for longer periods of time.
- a suitable dosing regimen for intranasal compositions would be for the patient to inhale slowly through the nose subsequent to the nasal cavity being cleared. During inhalation the composition would be administered to one nostril (for example, as a spray or drops) while the other is manually compressed. This procedure would then be repeated for the other nostril.
- compositions of the invention may be provided in a suitable container.
- Aqueous pharmaceutical compositions which are non-pressurized and adapted to be administered topically to the nasal cavity are of particular interest.
- Aqueous compositions may also be administered to the nose by nebulisation.
- a container comprising an aqueous pharmaceutical composition comprising a compound of formula (I) suitable for delivering said composition to the nasal cavities.
- a suitable container being a fluid dispenser e.g. a multi-dose container with a nasal applicator, wherein the dose is capable of being metered by volume.
- Such a fluid dispenser may typically have a dispensing nozzle or dispensing orifice through which a metered dose of the fluid composition is dispensed upon the application of a user- applied force to a pump mechanism of the fluid dispenser.
- Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid composition, the doses being dispensable upon sequential pump actuations.
- the dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid composition into the nasal cavity.
- a fluid dispenser of the aforementioned type is described and illustrated in WO05/044354 the entire content of which is hereby incorporated herein by reference.
- the dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid composition.
- the housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the composition out of a pump stem through a nasal nozzle of the housing.
- the fluid dispenser is of the general type illustrated in Figures 30 - 40 of WO05/044354.
- UV Detection Range 210 to 350nm
- Mass spectrum Recorded on a mass spectrometer using alternative-scan positive and negative mode electrospray ionisation.
- Solvents A: 0.1 % v/v formic acid in water
- UV detection range 210 to 350nm
- Mass spectrum Recorded on a mass spectrometer using alternative-scan positive and negative mode electrospray ionisation Solvents: A: 10mM ammonium bicarbonate in water adjusted to pH10 with ammonia solution
- UV Detection Range 210 to 350nm
- Mass spectrum Recorded on a mass spectrometer using alternative-scan positive and negative mode electrospray ionisation.
- Solvents A: 0.1 % v/v formic acid in water
- Mass spectrum Recorded on a mass spectrometer using alternative-scan positive and negative mode electrospray ionisation.
- Solvents A: 0.1 % v/v TFA in water
- UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
- Method B was conducted on a Sunfire C 18 column (typically 150mm x 30mm i.d. 5 ⁇ packing diameter) at ambient temperature.
- the solvents employed were:
- Method A was conducted on an XBridge Ci 8 column (typically 150mm x 19mm i.d. 5 ⁇ packing diameter) at ambient temperature.
- the solvents employed were:
- A 10 mM aqueous ammonium bicarbonate adjusted to pH 10 with ammonia solution.
- Method C was conducted on a Sunfire Ci 8 column (typically 150mm x 30mm i.d. 5 ⁇ packing diameter) at ambient temperature.
- the solvents employed were:
- Trifluoroacetic acid (20ml_, 260mmol) was added to solid 1 , 1-dimethylethyl ⁇ [(2,4,6- trimethylphenyl)sulfonyl]oxy ⁇ carbamate ⁇ Org. Proc. Res. Dev. 2009, 13(2), 263) (7.851 g, 24.89mmol) and the mixture stirred at room temperature under nitrogen for 2 hours and then poured onto a water/ice mixture (200ml_) and stirred until the ice had melted. The precipitate was collected by filtration under nitrogen and dissolved in anhydrous DCM (25m L) and dried using a hydrophobic frit.
- a solution of potassium hydroxylamine-O-sulfonate (prepared by neutralising hydroxylaminesulfonic acid (3.33g, 0.03mol) in water (16ml) with a solution of potassium hydroxide (1.68g, 0.3mol) in water (8ml) at 0°C) was added dropwise to a solution of 2- pyridinylmethanol (3.27g, 0.3mol) in water (12ml) at 60°C over a period of 10 minutes. The reaction mixture was heated at 70°C for 4 hours then concentrated at 50°C to give a brown solid.
- the material was dissolve din DMF (50ml) and potassium carbonate (4.17g, 0.045mol) and methyl 2-propynoate (3.78g, 0.045mol) were added.
- the reaction mixture was stirred overnight at room temperature then added to water (10ml) and extracted with ethyl acetate (50ml x 3).
- the organic phases were washed with brine (10ml x 3), dried (Na 2 S0 4 ) then concentrated.
- the residue was purified by chromatography on silica eluting with petroleum ether / ethyl acetate (4: 1) to give the title compound as a yellow solid (0.92g).
- Zirconium (IV) chloride 13.44g, 57.69mmol was added to a stirred mixture of 3,5- difluorophenol (15g, 115.38mmol) in dry fe/f-butyl methyl ether (9.82g, 115.38mmol) under argon and the mixture stirred at room temperature overnight. The mixture was then quenched with saturated ammonium chloride and 2M hydrochloric acid and extracted with DCM (250ml_). The organic extract was washed with 2M sodium hydroxide solution and the aqueous layer then acidified with 2M hydrochloric acid and extracted with DCM (2 x 250m L) and the DCM extract dried over sodium sulphate and evaporated. The residue was purified by silica gel chromatography using DCM:hexane 2:98 as eluant to afford the title compound as a white solid (5.6g)
- the reaction was filtered through a pad of Celite and the cake washed with THF (100 mL), The filtrate was concentrated in vacuo and the residue dissolved in dichloromethane (100 mL) and washed with 2M aqueous sodium hydroxide (100 mL). The organic phase was dried using a hydrophobic frit and concentrated in vacuo.
- the sample was purified by chromatography on silica (2 x 100g) using a gradient of 0-50% ethyl acetate- cyclohexane gradient over 40 mins. The appropriate fractions were combined and evaporated in vacuo to give the title compound as white solid (3.17g).
- Example 3 A/-( ra/7s-3- ⁇ r2-Bromo-4-(1 , 1-dimethylethyl)phenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de
- Example 5 /V-( ra/7s-3- ⁇ [4-(1 , 1-Dimethylethyl)-3-fluorophenyl1oxy)cvclobutyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de
- the reaction was transferred into a microwave vial and heated in the microwave at 200°C for 40min.
- the reaction was concentrated in vacuo and the crude product was purified by MDAP
- Example 7 A/-( ra/7s-3- ⁇ r2-Chloro-4-(1 , 1-dimethylethyl)phenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de
- Example 13 /V-( ra/7s-3- ⁇ [3-(1-Methylethyl)phenyl1oxy)cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide
- Example 14 /V-( ra/7s-3- ⁇ r4-(1-Methylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide
- Example 15 A/- ⁇ ra/7s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide
- Example 16 A/- ⁇ ra/7s-3-r(3,4,5-Trifluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide
- Example 17 A/-( ra/7s-3-fr4-(1.1-Dimethylethvn-3.5- difluorophenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-3-carboxamide
- Example 18 /V-( ra/7s-3- ⁇ [4-(1 , 1-Dimethylethyl)-2-fluorophenyl1oxy)cvclobutyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de
- Example 19 A/- ⁇ frans-3-[(4-Chloro-3-fluorophenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide
- Example 22 A/- ⁇ ra/7s-3-r(3-Fluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide
- Potassium carbonate (heaped microspatula approximately 0.028g, 0.2mmol) was and the reaction heated to 80°C for 18 hours. An additional portion of 3-methylphenol was added and the solution heated for a further 18hrs at 80°C. The solution was filtered and DMSO (0.3ml) was added. The solution was purified by MDAP HPLC (Method A) and the appropriate fractions evaporated in vacuo using the Genevac to give the title compound (3.3mg).
- Example 24 A/- ⁇ ra/7s-3-[(2-Propylphenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide
- Example 26 A/- ⁇ ra/7s-3-[(2,3-Dimethylphenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide
- Example 27 A/- ⁇ frans-3-r(2,3-Difluorophenyl)oxylcvclobutyl)pyrazoloH ,5-alpyridine-3- carboxamide
- Example 28 A/- ⁇ ra/7s-3-r(4-Chloro-3,5-difluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine- 3-carboxamide
- Example 29 /V- ⁇ ra/7s-3-[(3-Fluoro-2-methylphenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide
- Example 30 A/- ⁇ ra/7s-3-[(2-Chlorophenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide
- Example 32 A/- ⁇ frans-3-r(2-Chloro-3-fluorophenyl)oxylcvclobutyl)pyrazoloH ,5-alpyridine-3- carboxamide
- Example 33 A/- ⁇ frans-3-r(3,4-Difluorophenyl)oxylcvclobutyl)pyrazoloH ,5-alpyridine-3- carboxamide
- Example 34 A/- ⁇ ra/7s-3-r(3-Fluoro-4-methylphenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide
- Example 35 A/- ⁇ ra/7s-3-r(2-Chloro-3,5-difluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-
- Example 36 A/-( ra/7s-3- ⁇ r4-Methyl-2-(1-methylethyl)phenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de
- Example 37 A/-( ra/7s-3-(r2,4-£)/s(1-Methylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine- 3-carboxamide
- Example 38 /V-[ ra/7s-3-( ⁇ 2-[(Trifluoromethyl)oxylphenyl)oxy)cvclobutyllpyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de
- Example 39 A/-( ra/7s-3- ⁇ r2-(1 ,1-Dimethylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-
- Example 40 A/-(c/s-3- ⁇ r4-(1 ,1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de
- Example 41 Ay- ⁇ ra/7s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)-6-(hvdroxymethyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de
- Example 43 /V-( ra/7s-3- ⁇ [4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl1oxy)cvclobutyl)-7- (methyloxy)pyrazoloH ,5-alpyridine-3-carboxamide
- Example 45 A/-(fra/?s-3-(r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de
- Example 46 /V- ⁇ ra/7s-3-[(3,5-Difluorophenyl)oxy1cvclobutyl)-7-(methyloxy)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de
- Example 47 /V- ⁇ ra/7s-3-[(3,5-Difluorophenyl)oxy1cvclobutyl)-7-(hvdroxymethyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de
- Example 48 A/- ⁇ ra/7s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)imidazori ,2-alpyridine-3- carboxamide
- Example 52 A/- ⁇ frans-3-[(4-chlorophenyl)oxy1cvclobutyl)imidazo[1 ,2-a]pyridine-3-carboxamide
- Example 53 A/- ⁇ ra/7s-3-r(4-Chlorophenyl)oxylcvclobutyl)-7-(hvdroxymethyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and ⁇ frans-3-[(4-chlorophenyl)oxy]cyclobutyl ⁇ amine hydrochloride
- Example 55 A/- ⁇ c/s-3-[(3,5-Difluorophenyl)oxy1cvclobutyl)-7-(hvdroxymethyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de
- Example 60 A/- ⁇ c/s-3-[(4-Chlorophenyl)oxy1cvclobutyl)imidazo[1 ,2-a]pyridine-3-carboxamide
- Example 62 A/- ⁇ c/s-3-[(4-Chlorophenyl)oxy1cvclobutyl)-7-(hvdroxymethyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de
- Example 65 A/-(c/s-3- ⁇ r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)-7-
- Example 66 /V-(c/s-3-(r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de
- Example 68 A/- ⁇ ra/7s-3-r(2,3-Dichlorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide Prepared similarly to Example 49 from pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and trans-3- [(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride.
- Example 69 /V- ⁇ ra/7s-3-[(2,3-Dichlorophenyl)oxy1cvclobutyl)-7-(hvdroxymethyl)pyrazolo[1 ,5- a] pyri d i n e- 3- carboxa m i de
- Example 70 A/- ⁇ c/s-3-[(2,3-Dichlorophenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide
- Example 71 A/- ⁇ c/s-3-[(2,3-Dichlorophenyl)oxy1cvclobutyl)imidazo[1 ,2-alpyridine-3- carboxamide
- Example 72 A/- ⁇ c/s-3-r(2,3-Dichlorophenyl)oxylcvclobutyl)-7-(hvdroxymethyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de
- Example 73 A/- ⁇ ra/7s-3-r(2,3-Dichlorophenyl)oxylcvclobutyl)-8-(hvdroxymethyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and frans-3-[(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride..
- Example 75 A/-( ra/7s-3- ⁇ r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)-8- (hvdroxymethyl)imidazoH ,2-alpyridine-3-carboxamide
- Example 76 A/- ⁇ c/s-3-r(2,3-Dichlorophenyl)oxylcvclobutyl)-8-(hvdroxymethyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and c/ ' s-3-[(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride..
- Example 78 A/- ⁇ ra/7s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)-8-(hvdroxymethyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de
- Example 79 A/- ⁇ ra/7s-3-r(4-Chlorophenyl)oxylcvclobutyl)-8-(hvdroxymethyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de
- Example 8 j A/-( ra/7s-3- ⁇ r4-Fluoro-3-(trifluoromethyl)phenylloxy)cvclobutyl)-8-
- Example 82 /V-( ra/7s-3- ⁇ r4-Fluoro-3-(trifluoromethyl)phenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de
- Example 83 /V-( ra/7s-3- ⁇ [2-Chloro-3-(trifluoromethyl)phenyl1oxy)cvclobutyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de
- Example 84 A/-( ra/7s-3- ⁇ r2-Chloro-3-(trifluoromethyl)phenylloxy)cvclobutyl)-7-
- Example 85 A/-( ra/7s-3- ⁇ r4-Fluoro-3-(trifluoromethyl)phenylloxy)cvclobutyl)-7-
- Example 86 /V-( ra/7s-3- ⁇ [4-(1 , 1-Dimethvlethvl)-3,5-difluorophenvl1oxv)cvclobutyl)-8- fluoroimidazo[1 ,2-a]pyridine-3-carboxamide
- Example 87 A/-(c/s-3- ⁇ r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)-8- fluoroimidazoH ,2-alpyridine-3-carboxamide
- HEK-293 cells stably expressing mitochondrial targeted Aequorin were transfected with TRPV1 receptor bacmam at scale for cryopreservation in 1 ml vial aliquots.
- Cells can be stored at -140°C for up to 18 months. 18-20 hours before assay, cells were rapidly thawed in a water bath at 37°C and transferred to a 50ml Falcon tube. Cells were resuspended in 9mls of M1 'generic' media (DMEM/F12 with 10% dialysed FBS -Invitrogen 041-95750V) for every 1 ml cells and then centrifuged for 5min at l OOOrpm.
- M1 'generic' media DMEM/F12 with 10% dialysed FBS -Invitrogen 041-95750V
- the cell pellet was resuspended in ⁇ 10mls loading buffer (Tyrodes Base Buffer* + 0.1 % Pluronic Acid F68 solution + 0.1 % BSA) and the pH was adjusted to 7.4 for Capsaicin stimulus assay or 6.7 for Acid stimulus assay.
- Cell density was calculated using the Trypan Blue stain method and adjusted to 2.5 x 10e6 cells /ml using loading buffer.
- Coelentrazine (DiscoverX Cat. No 0-0084L - 500uM stock made in 100% ethanol) was added to a final concentration of 5uM and the falcon tube was covered in foil (to protect from light) and placed on a windmill rotator at room temperature for approximately 20 hours.
- Base Buffer Sigma kit T2145 was dissolved in deionised water, 20ml_ HEPES solution (Sigma H0887) and 13.4ml_ of NaHC03 (Sigma S8761) and made up to 1 L.
- Capsaicin stimulus assay cells were diluted to 1.25x10e5 cells/ml using dilution buffer (Tyrodes Base Buffer + 0.1 % Pluronic Acid F68 solution) at pH 7.4
- Example 3 had a plC 50 of 8.0 in the Capsaicin assay
- Example 5 had a plC 50 of 8.0 in the Capsaicin assay
- Example 6 had a plC 50 of 8.2 in the Capsaicin assay
- Example 7 had a plC 50 of 8.1 in the Capsaicin assay
- Example 17 had a pICso Of 8.4 in the Capsaicin assay
- Example 40 had a pICso Of 8.7 in the Capsaicin assay
- Example 42 had a pICso of 8.2 in the Capsaicin assay
- Example 45 had a pICso of 8.2 in the Capsaicin assay
- Example 65 had a pICso of 8.4 in the Capsaicin assay
- Example 66 had a pICso of 8.4 in the Capsaicin assay
- Example 67 had a pICso of 8.7 in the Capsaicin assay
- Example 68 had a pICso of 8.2 in the Capsaicin assay
- Example 69 had a pICso of 8.2 in the Capsaicin assay
- Example 73 had a pICso of 8.8 in the Capsaicin assay
- Example 74 had a pICso of 9.1 in the Capsaicin assay
- Example 75 had a pICso of 8.0 in the Capsaicin assay
- Example 77 had a plC 50 of 8.3 in the Capsaicin assay
- Example 80 had a plC 50 of 8.1 in the Capsaicin assay
- Example 81 had a plC 50 of 8.2 in the Capsaicin assay
- Example 82 had a plC 50 of 8.0 in the Capsaicin assay
- Example 86 had a plC 50 of 8.7 in the Capsaicin assay
- Example 87 had a plC 50 of 8.9 in the Capsaicin assay
- Acid stimulus assay cells were diluted to 2.5x10e5 cells/ml using dilution buffer at pH 6.7. The cells and the compound plates were added to the LumiluxTM reader (Perkin Elmer) with on-board liquid handling.
- AUC data was exported from the reader and data analysis was performed using 4 parameter logistic model, with data normalised to nominal high and low controls within plate
- Each of Examples 1 to 87 had a plC 50 of greater than 6.0 in this acid stimulus assay.
- Example 3 had a plC 50 of 8.1 in the pH assay
- Example 5 had a plC 50 of 8.2 in the pH assay
- Example 6 had a plC 50 of 8.3 in the pH assay
- Example 7 had a plC 50 of 8.0 in the pH assay
- Example 17 had a plC 50 of 8.4 n the pH assay
- Example 40 had a plC 50 of 8.9 n the pH assay
- Example 42 had a plC 50 of 8.3 n the pH assay
- Example 45 had a plC 50 of 8.5 n the pH assay
- Example 65 had a plC 50 of 8.8 n the pH assay
- Example 66 had a plC 50 of 8.8 n the pH assay
- Example 67 had a plC 50 of 8.5 n the pH assay
- Example 68 had a plC 50 of 8.4 n the pH assay
- Example 69 had a pICso of 8.7 in the PH assay
- Example 73 had a pICso of 8.9 in the PH assay
- Example 74 had a pICso of 9.2 in the PH assay
- Example 75 had a pICso of 8.3 in the PH assay
- Example 77 had a pICso of 8.6 in the PH assay
- Example 80 had a pICso of 8.5 in the PH assay
- Example 81 had a pICso of 8.6 in the PH assay
- Example 84 had a pICso of 8.1 in the PH assay
- Example 85 had a pICso of 8.1 in the PH assay
- Example 86 had a pICso of 8.9 in the PH assay
- Example 87 had a pICso of 9.3 in the PH assay.
- aqueous pharmaceutical compositions of the invention may be prepared according to the following general method.
- the isotonicity adjusting agent(s) is charged into a suitable mixing vessel containing purified water and dissolved with stirring.
- Preservative(s) is pre-dissolved in purified water in a separate vessel, optionally with heating, for example to 50 - 60 °C depending on the preservative chosen, to aid dissolution, and then added to the isotonicity adjusting agent(s) with continuous stirring.
- the suspending agent(s) is then charged into the mixing vessel and dispersed throughout the solution.
- the resulting suspending vehicle is allowed to hydrate for an appropriate period of time to ensure cross-linkage and gelation, which may take 60 minutes or longer.
- the wetting agent(s) is mixed with purified water which optionally may be heated, for example to about 50 - 60 °C as appropriate depending on the wetting agent(s) chosen, and stirred to dissolve.
- a slurry of the compound or a pharmaceutically acceptable salt thereof (alone or in combination with a further active ingredient) is then prepared by adding the resultant wetting agent(s) solution to the active compound(s), which may be particle size reduced for example micronised, and mixed prior to homogenising/refining.
- additional preservative(s) if needed, may be diluted with purified water and stirred to mix.
- the slurry of active compound(s) is added to the mixing vessel containing the suspending agent and dispersed with stirring.
- any additional preservative may be added to the bulk suspension and dispersed with continuous stirring.
- the suspension is made to its final mass by adding water and stirred.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Neurology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
A compound of formula (I) wherein X1 represents a hydrogen atom, or a CH2OH group X2 represents a hydrogen atom, a fluorine atom, an OCH3 group or a CH2OH group, and at least one of X1 and X2 is hydrogen Y represents a carbon atom and Z represents a nitrogen atom or Y represents a nitrogen atom and Z represents a carbon atom; R1 represents a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group, and R2 are R3 each independently selected from a hydrogen atom, a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group; or a pharmaceutically acceptable salt or solvate thereof.
Description
TITLE OF THE INVENTION
N-CYCLOBUTYL-IMIDAZOPYRIDINE OR -PYRAZOLOPYRIDINE CARBOXAMIDES AS
TRPV1 ANTAGONISTS
FIELD OF THE INVENTION
The present invention relates to novel compounds, being TRPV1 antagonists having pharmacological activity, to pharmaceutical compositions comprising the compounds and to the use of the compounds in medicine, especially in the treatment of rhinitis or the treatment of asthma.
BACKGROUND OF THE INVENTION
Vanilloids are a class of natural and synthetic compounds that are characterised by the presence of a vanillyl (4-hydroxy 3-methoxybenzyl) group or a functionally equivalent group. A wide variety of Vanilloid compounds of different structures are known in the art, for example those disclosed in European Patent Application Numbers, EP 0 347 000 and EP 0 401 903, UK Patent Application Number GB 2226313 and International Patent Application, Publication Number WO 92/09285. Particularly notable examples of vanilloid compounds or vanilloid receptor modulators are capsaicin or trans 8-methyl-N-vanillyl-6-nonenamide which is isolated from the pepper plant, capsazepine (Tetrahedron, 53, 1997, 4791) and olvanil or - N- (4-hydroxy-3-methoxybenzyl)oleamide (J. Med. Chem., 36, 1993, 2595).
Vanilloid Receptor (VR-1) has now been renamed as TRPV1 (Transient Receptor Potential Vanilloid subfamily member 1). TRPV1 is a calcium-permeable, ligand gated ion channel which is highly expressed in sensory neurones (Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD and Julius D (1997) Nature 389, 816-824) whose function is modulated by such Vanilloid compounds. TRPV1 has been studied and is extensively reviewed by Szallasi and Blumberg (The American Society for Pharmacology and Experimental Therapeutics, 1999, Vol. 51 , No. 2.). TRPV1 plays a key role in peripheral neuronal signalling where it mediates depolarising, excitatory responses to noxious stimuli such as heat, acid and capsaicin, the pungent component in chilli peppers (Szallasi et al, Nature Reviews Drug Discovery, 6, 357-372 (2007). TRPV1 acts as a polymodal receptor, responding in an integrative manner to an extensive array of activators including products of inflammation such as histamine, prostaglandins and bradykinin (which activate indirectly via protein kinase A and protein kinase C) as well as eicosanoid derivatives such as HPETEs, anandamide and environmental irritants. Upon activation, the channel pore opens and allows influx of cations which depolarises the nerve
membrane and triggers neuronal axonal firing and/or local release of neurotransmitters such as Substance P and CGRP. Activation may be caused by a single trigger, such as pH, but may be caused by integration of different triggers acting in concert on the channel. The role of TRPV1 in disease has been studied extensively in pain models where a role in both thermal and post-inflammatory hyperalgesia is well established (Chizh et al, Jara-Oseguera et al, 2008). TRPV1 has also been implicated in other diseases where symptoms are potentially driven wholly or in part by neuronal hypersensitivity or hyperactivity, because of its role in sensory signalling in peripheral nerves. Such diseases include asthma, rhinitis, overactive bladder, reflux oesophagitis, irritable bowel syndrome and migraine. TRPV1 has been implicated in inflammatory responses occurring in dry eye syndrome (Pan, Wang, Yang, Zhang & Reinach (2010), TRPV1 Activation is Required for Hypertonicity Stimulated Inflammatory Cytokine Release in Human Corneal Epithelial Cells, Manuscript IOVS, 10-5801). TRPV1 is also implicated to play a role in metabolic diseases such as diabetes and obesity (Motter AL & Ahern GP (2008) FEBS Letters 582, 2257-2262; Suri & Szallasi A (2007), The emerging role of TRPV1 in diabetes and obesity, Trends in Pharm Sci; Rasavi et al (2006) Cell 127, 1123-1135.)
TRPV1 expression is not limited only to peripheral sensory nerves, but is also expressed in spinal cord and in various regions of the central nervous system. TRPV1 is also found in non- neuronal cells and tissues including various types of epithelial cell and immune cells such as mast cells and dendritic cells (Khairatkar-Joshi N & Szallasi A (2008) Trends in Molecular Medicine.
International Patent Applications, Publication Numbers WO 02/08221 , WO 02/16317, WO 02/16318 and WO 02/16319 each disclose certain TRPV1 antagonists and their use in the treatment of diseases associated with the activity of TRPV1.
Patent Application Number WO 03/022809 discloses urea derivatives including Λ/-(2- Bromophenyl)-/V'-[((f?)-1-(5-trifluoromethyl-2-pyridyl)pyrrolidin-3-yl)]urea and A/-(3-methyl-5- isoquinolinyl))-/V'-[(3f?)-1-(5-trifluoromethyl-2-pyridyl)pyrrolidin-3-yl)]urea or pharmaceutically acceptable salts or solvates thereof and their use in the treatment of diseases associated with the activity of TRPV1.
Patent Application Number WO 10/026129 discloses A/-(2-Bromophenyl)-/V-[((ft)-1-(5- trifluoromethyl-2-pyridyl) pyrrolidin-3-yl)] urea for use in the treatment of rhinitis. Patent
Application Number WO 10/026128 discloses A/-(3-methyl-5-isoquinolinyl))-/V-[(3ft)-1-(5- trifluoromethyl-2-pyridyl) pyrrolidin-3-yl)] urea for use in the treatment of rhinitis.
It is an object of the invention to provide further TRPV1 antagonists.
BRIEF SUMMARY OF THE INVENTION
In a first aspect of the invention there is provided a compound of formula (I)

wherein
X1 represents a hydrogen atom, or a CH2OH group
X2 represents a hydrogen atom, a fluorine atom, an OCH3 group or a CH2OH group, and at least one of X1 and X2 is hydrogen
Y represents a carbon atom and Z represents a nitrogen atom or
Y represents a nitrogen atom and Z represents a carbon atom;
R1 represents a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group, and R2 and R3 are each independently selected from a hydrogen atom, a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group;
or a pharmaceutically acceptable salt or solvate thereof.
Compounds of formula (I) and their pharmaceutically acceptable salts or solvates have TRPV1 antagonist activity and are believed to be of potential use for the treatment or prophylaxis of certain disorders, or treatment of the pain associated with them.
Accordingly, in another aspect, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect for use medicine.
According to another aspect of the invention there is provided a pharmaceutical composition comprising a compound according to the first aspect, or a pharmaceutically acceptable salt or solvate thereof and one or more pharmaceutically acceptable carriers or excipients. The invention also provides a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, for use in the treatment of a condition for which a TRPV1 antagonist is indicated, in particular in the treatment and/or prophylaxis of rhinitis or of asthma.
The invention further provides a method for the treatment or prophylaxis of disorders in which antagonism of TRPV1 is beneficial, in a human, which comprises administering a human in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
In particular, the invention provides a method for the treatment of rhinitis which comprises administering to a human in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
The invention also provides a method for the treatment of asthma which comprises administering to a human in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
The invention provides for the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof in the manufacture of a medicament for the treatment of conditions in which an antagonist of TRPV1 is indicated, particularly rhinitis or asthma.
In another aspect of the invention, there is provided a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof in the manufacture of a medicament for use in the treatment of rhinitis. Where used herein the term rhinitis is to be understood to include both allergic and non allergic rhinitis. Examples of non-allergic rhinitis include vasomotor rhinitis, irritant rhinitis, occupational rhinitis and NARES (non allergic rhinitis with eosinophils).
In one embodiment the compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof is used in the treatment of non allergic rhinitis.
A compound of formula (I) may be prepared by methods described herein.
DETAILED DESCRIPTION OF THE INVENTION In one aspect of the invention rmula (I)

wherein:
X1 represents a hydrogen atom, or a CH2OH group
X2 represents a hydrogen atom, a fluorine atom, an OCH3 group or a CH2OH group, and at least one of X1 and X2 is hydrogen
Y represents a carbon atom and Z represents a nitrogen atom or
Y represents a nitrogen atom and Z represents a carbon atom;
R1 represents a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group, and R2 and R3 are each independently selected from a hydrogen atom, a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group;
or a pharmaceutically acceptable salt or solvate thereof.
It will be appreciated that the present invention covers compounds of formula (I) as the free base and as salts thereof, for example as a pharmaceutically acceptable salt thereof. In one embodiment the invention relates to compounds of formula (I) or a pharmaceutically acceptable salt thereof.
Because of their potential use in medicine, salts of the compounds of formula (I) are desirably pharmaceutically acceptable. Suitable pharmaceutically acceptable salts can include acid addition salts. For a review on suitable salts see Berge et al., J. Pharm. Sci., 66: 1-19, (1977). Typically, a pharmaceutically acceptable salt may be readily prepared by using a desired acid or base as appropriate. The resultant salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent.
A pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of formula (I) with a suitable inorganic or organic acid (such as hydrobromic, hydrochloric, sulphuric, nitric, phosphoric, succinic, maleic, acetic, propionic, fumaric, citric, tartaric, lactic, benzoic, salicylic, glutamaic, aspartic, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic, or hexanoic acid), optionally in a suitable solvent such as an organic solvent, to give the salt which is usually isolated for example by crystallisation and filtration. A pharmaceutically acceptable acid addition salt of a compound of formula (I) can comprise or be for example a hydrobromide, hydrochloride, sulphate, nitrate, phosphate, succinate, maleate, acetate, propionate, fumarate, citrate, tartrate, lactate, benzoate, salicylate, glutamate, aspartate, p- toluenesulfonate, benzenesulfonate, methanesulfonate, ethanesulphonate, naphthalenesulfonate (e.g. 2-naphthalenesulfonate) or hexanoate salt. Other non-pharmaceutically acceptable salts, e.g. formates, oxalates or trifluoroacetates, may be used, for example in the isolation of the compounds of formula (I), and are included within the scope of this invention.
The invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the salts of the compounds of formula (I).
It will be appreciated that many organic compounds can form complexes with solvents in which they are reacted or from which they are precipitated or crystallized. These complexes are known as "solvates". For example, a complex with water is known as a "hydrate". Solvents with high boiling points and/or capable of forming hydrogen bonds such as water, xylene, /V-methyl pyrrolidinone, methanol and ethanol may be used to form solvates. Methods for identification of solvates include, but are not limited to, NMR and microanalysis. Solvates of the compounds of formula (I) are within the scope of the invention. The invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the solvates of the compounds of formula (I).
The compounds of formula (I) may be in crystalline or amorphous form. Furthermore, some of the crystalline forms of the compounds of formula (I) may exist as polymorphs, which are included within the scope of the present invention. Polymorphic forms of compounds of formula (I) may be characterized and differentiated using a number of conventional
analytical techniques, including, but not limited to, X-ray powder diffraction (XRPD) patterns, infrared (IR) spectra, Raman spectra, differential scanning calorimetry (DSC),
thermogravimetric analysis (TGA) and solid state nuclear magnetic resonance (SSNMR). Certain of the compounds described herein can exists as stereoisomers, i.e. cis and trans isomers may be formed. Accordingly, the present invention encompasses all isomers of the compounds of formula (I) whether as individual isomers isolated such as to be substantially free of the other isomer (i.e. pure) or as mixtures. An individual isomer isolated such as to be substantially free of the other isomer (i.e. pure) may be isolated such that less than 10%, particularly less than about 1 %, for example less than about 0.1 % of the other isomer is present.
Separation of isomers may be achieved by conventional techniques known to those skilled in the art, e.g. by fractional crystallisation, chromatography or HPLC.
It will be appreciated from the foregoing that included within the scope of the invention are solvates, isomers and polymorphic forms of the compounds of formula (I) and salts thereof.
In one embodiment, X1 represents a hydrogen atom.
In one embodiment, X2 represents a hydrogen atom or a CH2OH group.
In one embodiment Y represents C and Z represents N. In another embodiment, Y represents N and Z represents C.
In one embodiment, R1 represents a 1 , 1-dimethylethyl group and R2 and R3 independently represent a fluorine atom or a hydrogen atom. In one embodiment, R1 represents a 1 , 1-dimethylethyl group at the para position relative to the ether group, R2 represents a fluorine atom at a meta position relative to the ether group, and R3 represents a fluorine or a hydrogen atom at the other meta position to the ether group.
In one embodiment, R1 represents a 1 , 1-dimethylethyl group at the para position relative to the ether group and both R2 and R3 each represent a fluorine atom at the meta positions relative to the ether group.
In another embodiment R1 represents a methyl group at an ortho position relative to the ether group, R2 represents a fluorine atom at a meta position relative to the ether group and R3 represents a hydrogen atom.
In one embodiment the compound of formula (I) is selected from:
A/-(frans-3-{[4-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(c/s-3-{[4-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide; A/-(frans-3-{[2-Bromo-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-3-fluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-2-methylphenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
A/-(frans-3-{[2-Chloro-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
A/-(frans-3-{[3-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-[frans-3-({3-[(Trifluoromethyl)oxy]phenyl}oxy)cyclobutyl]pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[2-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[3-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-[frans-3-({4-[(Trifluoromethyl)oxy]phenyl}oxy)cyclobutyl]pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[3-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[4-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; A/-{frans-3-[(3,4,5-Trifluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5- a]pyridine-3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-2-fluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(4-Chloro-3-fluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(4-Chlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-(frans-3-{[2-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(3-Fluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-{frans-3-[(3-Methylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-{frans-3-[(2-Propylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-{frans-3-[(2-Fluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-{frans-3-[(2,3-Dimethylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; A/-{frans-3-[(2,3-Difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-{frans-3-[(4-Chloro-3,5-difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(2-Chlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-{frans-3-[(2-Methylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxarriide;
A/-{frans-3-[(2-Chloro-3-fluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(3,4-Difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; A/-{frans-3-[(3-Fluoro-4-methylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(2-Chloro-3,5-difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[4-Methyl-2-(1-methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[2,4- 5/'s(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-[frans-3-({2-[(Trifluoromethyl)oxy]phenyl}oxy)cyclobutyl]pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(frans-3-{[2-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(c/s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
A/-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-6-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-6- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (methyloxy)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2- a]pyridine- 3-carboxamide;
A/-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-7-(methyloxy)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
A/-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide; A/-(c/'s-3-{[2-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide; A/-{c/s-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-{c/s-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; A/-{frans-3-[(4-chlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine- 3-carboxamide;
A/-{frans-3-[(4-Chlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
/\/-{c/'s-3-[(3,5-difluorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine- 3-carboxamide;
A/-{c/'s-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3- carboxamide;
A/-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5- a]pyridine-3-carboxamide;
A/-{c/s-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5- a]pyridine- 3-carboxamide;
A/-{c/'s-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide; /\/-{c/'s-3-[(4-Chlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
A/-{c/s-3-[(4-Chlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-{c/'s-3-[(4-Chlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{c/s-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-6-(hydroxymethyl)pyrazolo[1 ,5- a]pyridine-3-carboxamide;
A/-{frans-3-[(4-Chlorophenyl)oxy]cyclobutyl}-6-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-(c/s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-(c/s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine- 3-carboxamide;
A/-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide; A/-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine- 3-carboxamide; A/-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
A/-{c/'s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxarnide;
A/-{c/'s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine- 3-carboxamide;
A/-{c/'s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
A/-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine- 3-carboxamide;
A/-(c/s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-
(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-
(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
A/-{c/'s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
A/-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2- a]pyridine- 3-carboxamide;
A/-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
A/-{frans-3-[(4-Chlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
A/-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
A/-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
A/-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy
carboxamide;
A/-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridi carboxamide;
A/-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
A/-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a]pyridine-3-carboxamide:
A/-(c/s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de
or a pharmaceutically acceptable salt or solvate thereof. In one embodiment the compound of formula (I) is selected from:
N-(frans-3-{[2-Bromo-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3-fluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-2-methylphenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-6- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridin carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2- a]pyridine-3-carboxamide;
N-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8-(hydroxymethyl)i a]pyridine-3-carboxamide;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8-(hydroxymethyl)im a]pyridine-3-carboxamide;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a]pyridine-3-carboxamide:
N-(c/s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de
or a pharmaceutically acceptable salt or solvate thereof.
In a further aspect, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof according to the first aspect of the invention for use in therapy
Compounds of formula (I) and their pharmaceutically acceptable salts have TRPV1 antagonist activity and are believed to be of potential use for the treatment or prophylaxis of certain disorders, or treatment of the pain associated with them, such as: respiratory diseases, asthma, cough, COPD, bronchoconstriction, rhinitis, inflammatory disorders, pain, such as acute pain, chronic pain, neuropathic pain, postoperative pain, postrheumatoid arthritic pain, osteoarthritic pain, back pain, visceral pain, cancer pain, algesia, neuralgia, dental pain, headache, migraine, neuropathies, carpal tunnel syndrome, diabetic neuropathy, HlV-related neuropathy, post-herpetic neuralgia, fibromyalgia, neuritis, sciatica, nerve injury, ischaemia, neurodegeneration, stroke, post stroke pain, multiple sclerosis, oesophagitis, heart burn, Barrett's metaplasia, dysphagia, gastroeosophageal reflux disorder (GERD),
stomach and duodenal ulcers, functional dyspepsia, irritable bowel syndrome, inflammatory bowel disease, colitis, Crohn's disease, pelvic hypersensitivity, pelvic pain, menstrual pain, renal colic, urinary incontinence, cystitis, burns, itch, psoriasis, pruritis andemesis, ocular disorders, dry eye disease.
Disorders of particular interest are respiratory diseases, asthma, cough, COPD,
bronchoconstriction and inflammatory disorders.
In a further aspect the invention provides a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention, for use in the treatment of a condition for which a TRPV1 antagonist is indicated.
In a further aspect the invention provides a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention, for use in the treatment of rhinitis.
In a further aspect the invention provides a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention, for use in the treatment of asthma.
In a further aspect the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention in the manufacture of a medicament for the treatment of a condition for which a TRPV1 antagonist is indicated.
In a further aspect the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention in the manufacture of a medicament for the treatment of rhinitis. In a further aspect the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention in the manufacture of a medicament for the treatment of asthma.
In a further aspect the invention provides a method for the treatment or prophylaxis of disorders in which antagonism of TRPV1 is beneficial in a human comprising administering to the human in need thereof a therapeutically effective amount of a compound of formula (I)
or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention.
In a further aspect the invention provides a method for the treatment of rhinitis in a human in need thereof comprising administering to the human a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention.
In a further aspect the invention provides a method for the treatment of asthma in a human in need thereof comprising administering to the human a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention.
Also disclosed is a process for the preparation of compounds of formula (I) comprising coupling of a carboxylic acid of formula (II):

Wherein X1 , X2, Y, and Z are defined above for compounds of formula (I), with an amine of formula (III)

This coupling may be conducted, for example, using HATU (0-(7-azabenzotriazol-1-yl)-/V, N, N', Λ/'-tetramethyluronium hexafluorophosphate) in the presence of a suitable base such as triethylamine or DIPEA (A/,A/-diisopropylethylamine) in a suitable solvent such as DMF (N,N-
dimethylformamide). The coupling may also be conducted using alternative, conventional conditions for amide bond formation known in the art.
Compounds of formula (II) are either commercially available or may be derived from compounds that are commercially available and/or may be prepared using methodology described in the literature.
For example compounds of formula (II) may be prepared by hydrolysis of the corresponding esters (IV):

(IV)
Wherein X1 , X2, Y and Z are as defined above for compounds of formula (I) and R is an alkyl group for example methyl or ethyl.
The hydrolysis may be conducted, for example, using aqueous sodium hydroxide in THF.
Compounds of formula (IV) wherein Y is carbon, Z is nitrogen may be prepared by cycloaddition reaction of an N-amino pyridine of formula (V) with an alkyl propiolate.

(V)
The cycloaddition reaction may be performed, for example, by reacting a compound of formula (V) with methyl or ethyl propiolate in the presence of a suitable base such as potassium hydroxide or potassium carbonate in a suitable solvent such as DMF or DMSO. Where the substituent X1 in compound (V) is not hydrogen the cycloaddition may give a mixture of regioisomeric cycloaddition products which may be separated for example by chromatography.
Compounds of formula (V) are either commercially available or may be prepared by reaction of the corresponding pyridines (VI) with O-(mesitylsulphonyl) hydroxylamine (VII) in a suitable solvent, for example, DME. Compound of formula (VII) may be generated by reaction of 1 , 1- dimethylethyl {[(2, 4, 6-trimethylphenyl) sulfonyl] oxy} carbamate (VIII) with TFA.

The particular compound of formula (II) where Y is N, Z is C and X2 is a CH2OH group may be prepared by hydrolysis of the commercially available benzyl ether (IX),

Compounds of formula (III) may be prepared by reaction of phenols of formula (X),

(X) wherein R1 , R2 and R3 are as defined above for compounds of formula (I) with a cyclobutanol derivative of formula (XI),

wherein P is a suitable protecting group such as a BOC (tert-butyloxycarbonyl) group, followed by removal of the protecting group. Examples of other protecting groups that may be employed in the synthetic routes described herein and the means for their removal can be found in T. W. Greene 'Protective Groups in Organic Synthesis', 4th Edition, J. Wiley and Sons, 2006, incorporated herein by reference as it relates to such procedures.
This ether formation may be conducted for example using Mitsunobu conditions by reacting the phenol (X) with the protected amincyclobutanol (XI) in the presence of diisopropylazodicarboxylate and triphenylphosphine in a suitable solvent such as THF. Where the protecting group is a BOC group this can then be removed under acidic conditions, for example, using hydrochloric acid in dioxane, or TFA in dichloromethane to give compounds of formula (III).
Also disclosed is a process for the preparation of compounds of formula (I) comprising of reaction of sulfonates of formula (XII):

(XII)
Wherein X1 , X2, Y and Z are as defined above for compounds of formula (I) and Q is, for example, methyl, with a phenol of formula (X)
The ether formation can be conducted by reacting the alkylsulfonate derivative (XII) (Q = Me) with the phenol (X) in the presence of a base such as potassium f-butoxide, potassium carbonate or A/,A/-diisopropylethylamine in DMF. The reaction is usually conducted at elevated temperatures via conventional heating, e.g. 70°C or via microwave irradiation e.g. 200°C
Compounds of formula (XII) can be prepared from compounds of formula (XIII):

(XIII)
Wherein X1 , X2, Y, and Z are as defined above for compounds of formula (I)
The reaction can be conducted using methodology described in the literature, for example by reaction of the alcohol (XIII) with a sulfonyl chloride, such as methanesulfonyl chloride, in a suitable solvent such as DMF in the presence of a base such as triethylamine. Compounds of formula (XIII) may be prepared by reaction of compounds of formula (II) with compounds of formula (XIV).

(XIV)
This coupling may be conducted, for example, using HATU (0-(7-azabenzotriazol-1-yl)-/V, N, N', Λ/'-tetramethyluronium hexafluorophosphate) in the presence of a suitable base such as triethylamine or DIPEA (N, /V-diisopropylethylamine) in a suitable solvent such as DMF. The coupling may also be conducted using alternative, conventional conditions for amide bond formation known in the art. Compounds of formulae (VI), (VIII), (IX), (X), (XI), (XIV) are either commercially available, for example from Sigma-Aldrich UK, or may be prepared using methodology reported in the literature, or may be prepared by analogy with known procedures, for example those disclosed in standard reference texts of synthetic methodology such as J. March, Advanced Organic Chemistry, 6th Edition (2007), WileyBlackwell, or Comprehensive Organic Synthesis (Trost B.M. and Fleming I., (Eds.), Pergamon Press, 1991), each incorporated herein by reference as it relates to such procedures.
For any of the hereinbefore described reactions or processes, conventional methods of heating and cooling may be employed, for example temperature-regulated oil-baths or
temperature-regulated hot-blocks, and ice/salt baths or dry ice/acetone baths respectively. Conventional methods of isolation, for example extraction from or into aqueous or nonaqueous solvents may be used. Conventional methods of drying organic solvents, solutions, or extracts, such as shaking with magnesium sulphate, or sodium sulphate, or passing through a hydrophobic frit, may be employed. Conventional methods of purification, for example crystallisation and chromatography, for example silica chromatography or reverse- phase chromatography, may be used as required. Crystallisation may be performed using conventional solvents such as methanol, ethanol, or butanol, or aqueous mixtures thereof. It will be appreciated that specific reaction times temperatures may typically be determined by reaction-monitoring techniques, for example thin-layer chromatography and LC-MS.
Where appropriate individual isomeric forms of the compounds of the invention may be prepared as individual isomers using conventional procedures such as the fractional crystallisation of diastereoisomeric derivatives or chiral high performance liquid chromatography (chiral H PLC).
The absolute stereochemistry of compounds may be determined using conventional methods, such as X-ray crystallography. Compounds (III), (XI), (XII), (XIII) and (XIV) can exist as stereoisomers, cis and trans, both isomers are either commercially available or may be derived from compounds that are commercially available and/or may be prepared using methodology described in the literature. The synthesis may result in mixtures of cis I trans isomers which can be separated by chromatographic techniques or may give the individual cis or trans isomers. Subsequent reactions as described and known to a person skilled in the art can result in inversion of the stereochemistry.
Compounds of formula (I) may be in crystalline or non-crystalline form. A compound of formula (I) can form pharmaceutically acceptable salts. Suitable
pharmaceutically acceptable salts are those used conventionally in the art and include those described in Berge, J. Pharm. Sci., 1977, 66, 1-19.
Suitable pharmaceutically acceptable salts include acid addition salts. Suitable
pharmaceutically acceptable acid addition salts include salts with inorganic acids such, for example, as hydrochloric acid, hydrobromic acid, orthophosphoric acid or sulphuric acid, or
with organic acids such, for example as methanesulphonic acid, toluenesulphonic acid, acetic acid, propionic acid, lactic acid, citric acid, fumaric acid, malic acid, succinic acid, salicylic acid, maleic acid, glycerophosphoric acid or acetylsalicylic acid. In one embodiment the compound of formula (I) is in the form of a free base.
It will be appreciated that crystalline forms may be optionally hydrated or solvated. This invention includes within the scope of compounds of formula (I) stoichiometric hydrates as well as compounds of formula (I) containing variable amounts of water. Suitable solvates include pharmaceutically acceptable solvates, such as hydrates. Solvates include
stoichiometric solvates and non-stoichiometric solvates.
For use in this invention a compound of formula (I) or a pharmaceutically acceptable salt thereof may be formulated with one or more pharmaceutically acceptable excipients to provide a pharmaceutical composition.
Thus, in a further aspect, the invention provides a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to the first aspect of the invention and one or more pharmaceutically acceptable carriers or excipients.
In a further aspect there is provided a pharmaceutical composition for the treatment or prophylaxis of disorders in which antagonism of TRPV1 is beneficial comprising a compound as defined in the first aspect of the present invention or a pharmaceutically acceptable salt or solvate thereof and a pharmaceutically acceptable carrier.
In a further aspect there is provided a pharmaceutical composition for the treatment or prophylaxis of rhinitis comprising a compound as defined in the first aspect of the present invention or a pharmaceutically acceptable salt or solvate thereof and a pharmaceutically acceptable carrier.
In a further aspect there is provided a pharmaceutical composition for the treatment or prophylaxis of asthma comprising a compound as defined in the first aspect of the present invention or a pharmaceutically acceptable salt or solvate thereof and a pharmaceutically acceptable carrier.
Pharmaceutical compositions may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual), rectal, inhaled, intranasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal) route. Such compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier(s) or excipient(s).
In one embodiment, the invention provides a formulation for intranasal administration comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof.
In a further aspect of the present invention, there is provided an aqueous pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof, in particular a composition adapted for intranasal administration. The aqueous pharmaceutical composition of the invention may be in the form of an aqueous suspension or an aqueous solution. In one embodiment, the aqueous pharmaceutical composition of the invention is in the form of an aqueous suspension.
The aqueous component is preferably a high grade quality of water, in particular purified water.
For use in this invention a compound of formula (I) or a pharmaceutically acceptable salt thereof would typically be in a particle-size-reduced form, which may be prepared by conventional techniques, for example, microfluidisation, micronisation and milling e.g. wet bead milling. Generally, the size-reduced (e.g. micronised) compound of formula (I) or a pharmaceutically acceptable salt thereof can be defined by a D50 value of about 0.1 to 10 microns such as about 0.5 to 10 microns, more particularly about 2 to 4 microns (for example as measured using laser diffraction). The proportion of a compound of formula (I) or a pharmaceutically acceptable salt thereof will depend on the precise type of composition to be prepared, but will generally be within the range of from about 0.01 to 20% (w/w), based on the total weight of the composition.
Generally, however for most types of preparations the proportion used will be within the range of from about 0.05 to 10% (w/w), such as about 0.1 to 5% (w/w).
The dose of a compound of formula (I) or a pharmaceutically acceptable salt thereof will vary in the usual way with the seriousness of the disease to be treated and other factors such as the weight of the sufferer. As a general guide suitable unit doses may be about between 0.005 and 1 mg for example between 0.005 and 0.5mg per dose. Such unit doses may be administered once a day, or more than once a day, for example two or three times a day. Such therapy may extend for a number of weeks or months.
The compounds of the invention may be employed alone or in combination with other therapeutic agents. Combination therapies according to the present invention thus comprise the administration of at least one compound of formula (I) or a pharmaceutically acceptable salt thereof, and the use of at least one other pharmaceutically active agent. Preferably, combination therapies according to the present invention comprise the administration of at least one compound of formula (I) or a pharmaceutically acceptable salt thereof, and at least one other pharmaceutically active agent. The compound(s) of the invention and the other pharmaceutically active agent(s) may be administered together in a single pharmaceutical composition or separately and, when administered separately this may occur simultaneously or sequentially in any order. The amounts of the compound(s) of the invention and the other pharmaceutically active agent(s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect. Thus in a further aspect, there is provided a combination comprising a compound of the invention and at least one other pharmaceutically active agent.
Where the composition is an aqueous pharmaceutical composition, optionally a further active ingredient may be incorporated into the aqueous pharmaceutical composition, particularly one used in the treatment of rhinitis and suitable for intranasal administration such as an antihistamine or a corticosteroid.
For use in combination, suitable examples of anti-histamines include azelastine, olopatadine, bepotastine or a compound selected from:
A/-[2-((2 )-2-{[4-[(4-chlorophenyl) methyl]-1-oxo-2(1 H)-phthalazinyl] methyl}-1 -pyrrolidinyl) ethyl]-4-(methyloxy) butanamide (as disclosed in patent application WO2008/74803);
4-[(4-chlorophenyl) methyl]-2-({(2f?)-1-[4-(4-{[3-(hexahydro-1 /-/-azepin-1-yl)propyl]oxy} phenyl) butyl]-2-pyrrolidinyl}methyl)-1 (2/-/)-phthalazinone (as disclosed in patent application WO2007/122156); or
A/-(4-{4-[(6-butyl-8-quinolinyl)oxy]-1-piperidinyl}butyl)ethanesulfonamide (as disclosed in patent application PCT/EP2008/060622, published as WO2009/021965).
For use in combination, suitable examples of corticosteroids include fluticasone propionate (which is marketed as an intranasal formulation under the trade name Flixonase®), beclomethasone dipropionate (which is marketed as an intranasal formulation under the trade name Beconase®) or fluticasone furoate (which is marketed under the trade name
Veramyst®). In one embodiment the present invention provides for an aqueous
pharmaceutical composition comprising a compound of formula (I). or a pharmaceutically acceptable salt thereof and fluticasone furoate. When present the proportion of the further active ingredient will generally be in the range from about 0.05 to 10% (w/w), such as about 0.1 to 5% (w/w).
Aqueous pharmaceutical compositions of the invention, such as intranasal compositions, may include one or more pharmaceutically acceptable excipients selected from the group consisting of suspending agents, thickening agents, preservatives, wetting agents and isotonicity adjusting agents.
Accordingly in one embodiment, there is provided an aqueous pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and a suspending agent.
The suspending agent, if included, will typically be present in an amount of between about 0.1 and 5% (w/w), such as between about 1.5% and 2.5% (w/w), based on the total weight of the composition. Examples of suspending agents include Avicel®, carboxymethylcellulose, veegum, tragacanth, bentonite, methylcellulose and polyethylene glycols, e.g. microcrystalline cellulose or carboxy methylcellulose sodium.
In one embodiment, there is provided an aqueous pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and a preservative.
For stability purposes, the compositions of the invention may be protected from microbial or fungal contamination and growth by inclusion of a preservative. Examples of pharmaceutically acceptable anti-microbial agents or preservatives may include quaternary ammonium compounds (e.g. benzalkonium chloride, benzethonium chloride, cetrimide and
cetylpyridinium chloride), mercurial agents (e.g. phenylmercuric nitrate, phenylmercuric acetate and thimerosal), alcoholic agents (e.g. chlorobutanol, phenylethyl alcohol and benzyl
alcohol), antibacterial esters (e.g. esters of para-hydroxybenzoic acid), chelating agents such as disodium ethylenediaminetetraacetate (EDTA) and other anti-microbial agents such as chlorhexidine, chlorocresol, sorbic acid and its salts (such as potassium sorbate) and polymyxin. Examples of pharmaceutically acceptable anti-fungal agents or preservatives may include sodium benzoate. The preservative, if included, may be present in an amount of between about 0.001 and 1 % (w/w), such as about 0.015% (w/w), based on the total weight of the composition.
In another embodiment, there is provided an aqueous pharmaceutical composition which is preservative free.
In one embodiment, there is provided an aqueous pharmaceutical composition comprising .a compound of formula (I) or a pharmaceutically acceptable salt thereof and a wetting agent. Compositions which contain a suspended medicament may include a pharmaceutically acceptable wetting agent which functions to wet the particles of medicament to facilitate dispersion thereof in the aqueous phase of the composition. Typically, the amount of wetting agent used will not cause foaming of the dispersion during mixing. Examples of wetting agents include fatty alcohols, esters and ethers, such as polyoxyethylene (20) sorbitan monooleate (Polysorbate 80). The wetting agent may be present in an amount of between about 0.001 and 1.0% (w/w), such as between about 0.001 and 0.05% (w/w), for example about 0.025% (w/w), based on the total weight of the composition.
In one embodiment, there is provided an aqueous pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and an isotonicity adjusting agent.
An isotonicity adjusting agent may be included to achieve isotonicity with body fluids e.g. fluids of the nasal cavity, resulting in reduced levels of irritancy. Examples of isotonicity adjusting agents include sodium chloride, dextrose, xylitol and calcium chloride. An isotonicity adjusting agent may be included in an amount of between about 0.1 and 10% (w/w), such as about 5.0% (w/w), based on the total weight of the composition.
Further, the aqueous pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof may be buffered by the addition of suitable buffering agents such as sodium citrate, citric acid, phosphates such as disodium phosphate (for
example the dodecahydrate, heptahydrate, dihydrate and anhydrous forms) or sodium phosphate and mixtures thereof.
Compositions of the invention e.g. those suitable for intranasal administration may also optionally contain other excipients, such as antioxidants (for example sodium metabisulphite), taste-masking agents (such as menthol) and sweetening agents (for example dextrose, glycerol, saccharin and/or sorbitol).
In one embodiment there is provided an aqueous pharmaceutical composition which comprises:
(i) an aqueous suspension of a compound of formula (I) or a pharmaceutically acceptable salt thereof;
(ii) one or more suspending agents;
(iii) one or more preservatives;
(iv) one or more wetting agents; and
(v) one or more isotonicity adjusting agents.
Aqueous pharmaceutical compositions according to the invention can be prepared using standard procedures that are familiar to the person skilled in the art e.g. by admixture of the various components, suitably at ambient temperature and atmospheric pressure.
In one embodiment, the aqueous pharmaceutical compositions of the invention are suitable for intranasal administration. Intranasal compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof may permit the compound(s) to be delivered to all areas of the nasal cavities (the target tissue) and further, may permit the compound(s) to remain in contact with the target tissue for longer periods of time. A suitable dosing regimen for intranasal compositions would be for the patient to inhale slowly through the nose subsequent to the nasal cavity being cleared. During inhalation the composition would be administered to one nostril (for example, as a spray or drops) while the other is manually compressed. This procedure would then be repeated for the other nostril. Typically, one or two sprays per nostril would be administered by the above procedure up to two or three times each day, ideally once daily.
The compositions of the invention may be provided in a suitable container. Aqueous pharmaceutical compositions which are non-pressurized and adapted to be administered topically to the nasal cavity are of particular interest. Aqueous compositions may also be administered to the nose by nebulisation.
Accordingly, there is provided a container comprising an aqueous pharmaceutical composition comprising a compound of formula (I) suitable for delivering said composition to the nasal cavities. Typically the composition of the present invention will be packaged into a suitable container being a fluid dispenser e.g. a multi-dose container with a nasal applicator, wherein the dose is capable of being metered by volume.
Such a fluid dispenser may typically have a dispensing nozzle or dispensing orifice through which a metered dose of the fluid composition is dispensed upon the application of a user- applied force to a pump mechanism of the fluid dispenser. Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid composition, the doses being dispensable upon sequential pump actuations. The dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid composition into the nasal cavity. A fluid dispenser of the aforementioned type is described and illustrated in WO05/044354 the entire content of which is hereby incorporated herein by reference. The dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid composition. The housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the composition out of a pump stem through a nasal nozzle of the housing. In one embodiment, the fluid dispenser is of the general type illustrated in Figures 30 - 40 of WO05/044354. Abbreviations
The following list provides definitions of certain abbreviations as used herein. It will be appreciated that the list is not exhaustive, but the meaning of those abbreviations not herein below defined will be readily apparent to those skilled in the art.
DCM Dichloromethane
DMF N, N-Dimethylformamide
DMSO Dimethylsulphoxide
EtOAc Ethyl acetate
Et20 Diethyl ether
HCI Hydrochloric acid
HPLC High performance liquid chromatography
ISCO Companion Automated flash chromatography equipment with
fraction analysis by UV absorption available from
Presearch Limited, Basingstoke, Hants., RG24 8PZ,
UK
MDAP HPLC Reverse phase HPLC on a Ci8 column using a
two-solvent gradient and analysis of the fractions by
electrospray mass spectroscopy.
SPE Solid phase extraction
MeOH Methanol
mins minutes
Stripped Removal of solvent under reduced pressure
TFA Trifluoroacetic acid
iPr iso- Propyl
t-Bu tert-Butyl
Ms Mesyl
Ac Acetyl
n-Bu n- Butyl
Ph Phenyl
Experimental Details
NMR
1 H NMR spectra were recorded in either CDCI3 or DMSO-d6 on either a Bruker DPX 400 or Bruker Avance DRX or Varian Unity 400 spectrometer all working at 400 MHz. The internal standard used was either tetramethylsilane or the residual protonated solvent at 7.25 ppm for CDCI3 or 2.50 ppm for DMSO-d6.
Experimental details of HPLC systems as referred to herein are as follows:
System A
Column: 50mm x 2.1 mm ID, 1.7μΐ ι Acquity UPLC BEH C18
Flow Rate: 1 mL/min.
Temp.: 40°C
UV Detection Range: 210 to 350nm
Mass spectrum: Recorded on a mass spectrometer using alternative-scan positive and negative mode electrospray ionisation.
Solvents: A: 0.1 % v/v formic acid in water
B: 0.1 % v/v formic acid in acetonitrile
Gradient: Time (min.) A% B%
0 97 3
0.1 97 3
1.4 0 100
1.9 0 100
2.0 97 3
System B
Column: 50mm x 4.6mm ID, 3.5μηι XBridge C18 column
Flow Rate: 3mL/min.
Temp: 30°C
UV detection range: 210 to 350nm
Mass spectrum: Recorded on a mass spectrometer using alternative-scan positive and negative mode electrospray ionisation Solvents: A: 10mM ammonium bicarbonate in water adjusted to pH10 with ammonia solution
B: acetonitrile
Gradient: Time (min.') A% B%
0 99 1
0.1 99 1
4.0 3 97
5.0 3 97
System C
Column: 30mm x 4.6mm ID, 3.5μηι Sunfire C18
Flow Rate: 3mL/min.
Temp.: 30°C
UV Detection Range: 210 to 350nm
Mass spectrum: Recorded on a mass spectrometer using alternative-scan positive and negative mode electrospray ionisation.
Solvents: A: 0.1 % v/v formic acid in water
B: 0.1 % v/v formic acid in acetonitrile
Gradient: Time (min.) A% B%
0 97 3
0.1 97 3
4.2 0 100
4.8 0 100
4.9 97 3
5.0 97 3
System D
Column: 50mm x 4.6mm ID, 3.5μηι Sunfire C18
Flow Rate: 2mL/min.
Temp.: 40°C
UV Detection Range: 214 / 254 nm
Mass spectrum: Recorded on a mass spectrometer using alternative-scan positive and negative mode electrospray ionisation.
Solvents: A: 0.1 % v/v TFA in water
B: 0.1 % v/v TFA in acetonitrile
Gradient: Time (min.) A%
0 97 3
0.1 97 3
1.4 0 100
1.9 0 100
2.0 97 3
Mass Directed Autopreparative HPLC (MDAP)
Mass directed autopreparative HPLC was undertaken under the conditions given below. The UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
Method A
Method B was conducted on a Sunfire C18 column (typically 150mm x 30mm i.d. 5μηι packing diameter) at ambient temperature. The solvents employed were:
A = 0.1 % v/v solution of formic acid in water
B = 0.1 % v/v solution of formic acid in acetonitrile.
Method B
Method A was conducted on an XBridge Ci8 column (typically 150mm x 19mm i.d. 5μηι packing diameter) at ambient temperature. The solvents employed were:
A = 10 mM aqueous ammonium bicarbonate adjusted to pH 10 with ammonia solution.
B = acetonitrile.
Method C
Method C was conducted on a Sunfire Ci8 column (typically 150mm x 30mm i.d. 5μηι packing diameter) at ambient temperature. The solvents employed were:
A = 0.1 % v/v solution of trifluoroacetic acid in water
B = 0.1 % v/v solution of trifluoroacetic acid in acetonitrile. The invention is illustrated by reference to, but is in no way limited by, the following Examples.
Intermediate 1 : Ethyl 6-(hvdroxymethyl)pyrazolo[1 ,5-alpyridine-3-carboxylate

Trifluoroacetic acid (20ml_, 260mmol) was added to solid 1 , 1-dimethylethyl {[(2,4,6- trimethylphenyl)sulfonyl]oxy}carbamate {Org. Proc. Res. Dev. 2009, 13(2), 263) (7.851 g, 24.89mmol) and the mixture stirred at room temperature under nitrogen for 2 hours and then poured onto a water/ice mixture (200ml_) and stirred until the ice had melted. The precipitate was collected by filtration under nitrogen and dissolved in anhydrous DCM (25m L) and dried using a hydrophobic frit. To the filtrate was added more DCM (50ml_) and 3-pyridinylmethanol (2.6ml_, 26.8mmol) and the mixture stirred under nitrogen at ambient temperature for 22 hours. The mixture was then diluted with diethyl ether (150ml_) and the solid collected to give 1-amino-3-(hydroxymethyl)pyridinium 2,4,6-trimethylbenzenesulfonate (5.4g) which was used directly. This material was dissolved in DMSO (50ml_) and ethyl 2-propynoate (0.316ml_, 3.12mmol), potassium hydroxide (0.091 g, 1.623mmol) and potassium carbonate (0.259g, 1.873mmol) were then added and the mixture stirred under nitrogen at room temperature for 18 hours. The mixture was then diluted with ethyl acetate (200ml_) and washed with water (3 x 200ml_), 0.1 M hydrochloric acid (200ml_) and brine (200ml_), dried over magnesium sulphate, filtered and the solvent evaporated in vacuo. The residue was dissolved in DCM and purified on a silica cartridge (100g) using a 0-25% ethyl acetate-DCM gradient over 100 min. The appropriate fractions were combined and evaporated in vacuo to give the title compound as a white solid (1 16mg).
LC-MS (System A): tRET = 0.67 min; MH+ 221
The isomeric ethyl 4-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylate (174mg) was also isolated from this reaction.
LC-MS (System A): tRET = 0.71 min; MH+ 221 Intermediate 2: 6-(Hvdroxymethyl)pyrazolo[1 ,5-alpyridine-3-carboxylic acid

Intermediate 3: 7-(Methyloxy)pyrazoloH ,5-alpyridine-3-carboxylic acid

LCMS (System A): rt =0.53 mins, MH+ = 193
Intermediate 4: Methyl 7-(hvdroxymethyl)pyrazolo[1 ,5-alpyridine-3-carboxylate

A solution of potassium hydroxylamine-O-sulfonate (prepared by neutralising hydroxylaminesulfonic acid (3.33g, 0.03mol) in water (16ml) with a solution of potassium hydroxide (1.68g, 0.3mol) in water (8ml) at 0°C) was added dropwise to a solution of 2- pyridinylmethanol (3.27g, 0.3mol) in water (12ml) at 60°C over a period of 10 minutes. The reaction mixture was heated at 70°C for 4 hours then concentrated at 50°C to give a brown solid. The material was dissolve din DMF (50ml) and potassium carbonate (4.17g, 0.045mol)
and methyl 2-propynoate (3.78g, 0.045mol) were added. The reaction mixture was stirred overnight at room temperature then added to water (10ml) and extracted with ethyl acetate (50ml x 3). The organic phases were washed with brine (10ml x 3), dried (Na2S04) then concentrated. The residue was purified by chromatography on silica eluting with petroleum ether / ethyl acetate (4: 1) to give the title compound as a yellow solid (0.92g).
LC-MS (System D) rt = 1.13min, MH+ = 207.
Intermediate 5: 7-(Hvdroxymethyl)pyrazolori ,5-alpyridine-3-carboxylic acid

To a solution of methyl 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylate(5.4g, 26.2mmol) in methanol (150ml) was added aqueous potassium hydroxide (50ml, 10%). The reaction mixture was stirred at 50°C for 14 hours. The reaction mixture was concentrated and acidified to pH 2-3 with aqueous hydrochloric acid (2N). The material was collected and washed with water (20ml) and dried in vacuo to give the title compound as a yellow solid (3.78g).
LC-MS (System A) rt = 0.44min, MH+ = 193 Intermediate 6: 8-(Hvdroxymethyl)imidazo[1 ,2-alpyridine-3-carboxylic acid

To a 500ml round bottomed flask fitted with a stirrer bar was added 8- {[(phenylmethyl)oxy]methyl}imidazo[1 ,2-a]pyridine-3-carboxylic acid (Ellanova) (8.0g, 0.028mol) followed by concentrated hydrochloric acid (180ml). The mixture was heated with a heat gun to dissolve the starting material and then stirred at room temperature for 1 hour. The mixture was evaporated to dryness and the residual HCI azeotroped with toluene. Ether was added to the residue, the solid filtered and washed with additional ether to give the title compound as a cream solid (5.5g).
1 H NMR (400 MHz, DMSO-d6) δ ppm 7.49 - 7.58 (m, 2 H) 7.86 - 7.95 (m, 2 H) 8.71 (s, 2 9.35 (d, J=6.5 Hz, 2 H)
Intermediate 7: c/s-3-Aminocvclobutanol
1 , 1-Dimethylethyl (c/'s-3-hydroxycyclobutyl)carbamate (3g, 16.02 mmol) was stirred in HCI in diethyl ether (45 ml, 2M solution, 90 mmol) over the weekend. TLC (10% MeOH in DCM, visualisation with KMn04 dip), showed no starting material remained. The reaction was concentrated in vacuo (water bath at ambient temperature). The crude material was washed with diethyl ether (40ml_) to afford the title compound.
1 H NMR (400 MHz, Methanol-d4) δ ppm 1.97 - 2.09 (m, 2 H) 2.65 - 2.75 (m, 2 H) 3.25 - 3.37 (m, 2 H) 4.01 - 4.11 (m, 1 H)
Intermediate: 8 /V-(c/s-3-Hvdroxycvclobutyl)pyrazoloH ,5-alpyridine-3-carboxamide

LC-MS (System A): rt = 0.48 mins, MH+ = 232
Intermediate 9: c/s-3-r(Pyrazolori ,5-alpyridin-3-ylcarbonyl)aminolcvclobutyl methanesulfonate

Triethylamine (1 1.77 mL, 84 mmol) and mesyl chloride (3.29 mL, 42.2 mmol) were added to a solution of N-(c/'s-3-hydroxycyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide (4.88g, 21.10 mmol) in Ν,Ν-dimethylformamide (DMF) (40 mL). The mixture was stirred at room temperature for 30min. The reaction was concentrated in vacuo and the residue partitioned between DCM (70mL) and water (70mL). The organic layer was concentrated in vacuo. The residue was purified by column chromatography (20g Si column) eluting with a gradient: 0- 15% MeOH/DCM over 60min. Fractions which contained the product were concentrated in vacuo to give the title compound
LCMS (System A): rt = 0.64 mins, MH+ = 310 lntermediate10: 1 , 1-Dimethylethyl( ra/7s-3-{r4-(trifluoromethyl)phenylloxy)cvclobutyl)carbamate

To a stirred solution of 4-(trifluoromethyl)phenol (216 mg, 1.335 mmol), triphenylphosphine (525 mg, 2.003 mmol) and 1 , 1-dimethylethyl (c/'s-3-hydroxycyclobutyl)carbamate (WO2005/1 16009) (250 mg, 1.335 mmol) in anhydrous tetrahydrofuran (THF) (10 mL) at ambient temperature was added neat diisopropylazodicarboxylate (0.389 mL, 2.003 mmol). The reaction was warmed to 45°C under a nitrogen atmosphere for 30 h. The reaction was concentrated in vacuo. The residue was loaded in dichloromethane and purified on silica cartridge (50g) using a gradient 0-25% ethyl acetate-cyclohexane over 40 mins. The appropriate fractions were combined and evaporated in vacuo to give the title compound as a white solid (362mg).
1 H NMR (400 MHz, CDCI3) δ ppm 1.46 (s, 9 H) 2.34 - 2.50 (m, 2 H) 2.52 - 2.65 (m, 2 H) 4.22 - 4.41 (m, 1 H) 4.68 - 4.91 (m, 2 H) 6.83 (d, J=8.8 Hz, 2 H) 7.53 (d, J=8.8 Hz, 2 H)
Intermediate 11 : frans-3-{r4-(Trifluoromethyl)phenylloxy)cvclobutanamine hydrochloride

1 , 1-Dimethylethyl (trans-3-{[4-(trifluoromethyl)phenyl]oxy}cyclobutyl)carbamate (362 mg, 1.093 mmol) was stirred in 4M HCI in dioxane (2ml_, 8.00 mmol) for 5h. The reaction mixture was concentrated in vacuo to give the title compound (289mg).
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.62 (dd, J=8.4, 3.9 Hz, 2 H) 2.70 - 2.83 (m, 2 H) 3.95 - 4.06 (m, 1 H) 5.03 - 5.12 (m, 1 H) 6.95 (d, J=8.5 Hz, 2 H) 7.55 (d, J=8.8 Hz, 2 H) Intermediate 12: 1 , 1-Dimethylethyl (c/s-3-{[4-(trifluoromethyl)phenyl1oxy)cvclobutyl)carbamate

To a stirred solution of 4-(trifluoromethyl)phenol (519 mg, 3.20 mmol), triphenylphosphine (1261 mg, 4.81 mmol) and 1 , 1-dimethylethyl (frans-3-hydroxycyclobutyl)carbamate (WO2007/62332) (600 mg, 3.20 mmol) in anhydrous tetrahydrofuran (THF) (10 ml_) at ambient temperature was added neat diisopropylazodicarboxylate (0.935 ml_, 4.81 mmol). The reaction was warmed to 45°C under a nitrogen atmosphere overnight. The reaction was concentrated in vacuo, and the residue partitioned between sodium hydroxide (50ml_) and DCM (50ml_). The organic layer was concentrated in vacuo. The residue was loaded in dichloromethane and purified on a silica cartridge (50g) using a gradient of 0-25% ethyl acetate-cyclohexane over 40 mins. The appropriate fractions were combined and evaporated in vacuo to give the title compound as a white solid (732mg).
1 H NMR (400 MHz, DMSO-cfe) δ ppm 1.37 (s, 9 H) 1.92 - 2.05 (m, 2 H) 2.53 (none, 0 H) 2.73 - 2.84 (m, 2 H) 3.65 - 3.77 (m, 1 H) 4.43 - 4.52 (m, 1 H) 7.02 (d, J=8.8 Hz, 2 H) 7.16 - 7.25 (m, 1 H) 7.62 (d, J=8.8 Hz, 2 H)
Intermediate 13: c/s-3-{[4-(Trifluoromethyl)phenyl1oxy)cyclobutanamine hydrochloride

Prepared similarly to intermediate 1 1 from 1 , 1-dimethylethyl (c/'s-3-{[4- (trifluoromethyl)phenyl]oxy}cyclobutyl)carbamate.
1 H NMR (400 MHz, DMSO-cfe) δ ppm 1.37 (s, 9 H) 1.92 - 2.05 (m, 2 H) 2.53 (none, 0 H) 2.73 - 2.84 (m, 2 H) 3.65 - 3.77 (m, 1 H) 4.43 - 4.52 (m, 1 H) 7.02 (d, J=8.8 Hz, 2 H) 7.16 - 7.25 (m, 1 H) 7.62 (d, J=8.8 Hz, 2 H)
Intermediate14: 1 , 1-Dimethylethyl (tra ns-3-{[2-bromo-4-(1 , 1-dimethylethyl)phenvHoxy) cyclobutvDcarbamate

Prepared similarly to intermediate 10 from 2-bromo-4-(1 , 1-dimethylethyl)phenol and 1 , 1- dimethylethyl (cis-3-hydroxycyclobutyl)carbamate.
1 H NMR (400 MHz, CDCI3) δ ppm 1.29 (s, 9 H) 1.46 (s, 9 H) 2.34 - 2.45 (m, 2 H) 2.58 - 2.68 (m, 2 H) 4.27 - 4.38 (m, 1 H) 4.67 - 4.85 (m, 2 H) 6.59 (d, J=8.5 Hz, 1 H) 7.18 - 7.24 (m, 1 H) 7.54 (d, J=2.3 Hz, 1 H) Intermediate 15: ra/7s-3-{r2-Bromo-4-(1 , 1-dimethylethyl)phenylloxy)cyclobutanamine hydrochloride

Intermediate 16: 4-(1 , 1-Dimethylethyl)-3,5-difluorophenol

Zirconium (IV) chloride (13.44g, 57.69mmol) was added to a stirred mixture of 3,5- difluorophenol (15g, 115.38mmol) in dry fe/f-butyl methyl ether (9.82g, 115.38mmol) under argon and the mixture stirred at room temperature overnight. The mixture was then quenched with saturated ammonium chloride and 2M hydrochloric acid and extracted with DCM (250ml_). The organic extract was washed with 2M sodium hydroxide solution and the aqueous layer then acidified with 2M hydrochloric acid and extracted with DCM (2 x 250m L) and the DCM extract dried over sodium sulphate and evaporated. The residue was purified by silica gel chromatography using DCM:hexane 2:98 as eluant to afford the title compound as a white solid (5.6g)
LC-MS (System A): tRET = 1.13 min; MH+ 187
Intermediate 17: 1.1-Dimethylethyl (c/s-3-(r4-(1.1-dimethylethyl)-3.5- difluorophenylloxylcyclobutvDcarbamate

To a stirred solution of 4-(1 , 1-dimethylethyl)-3,5-difluorophenol (4.97 g, 26.7 mmol), polymer bound triphenylphosphine (13.35 g, 40.1 mmol) and 1 , 1-dimethylethyl (trans-3- hydroxycyclobutyl)carbamate (5 g, 26.7 mmol) in anhydrous tetrahydrofuran (THF) (250 ml_) at ambient temperature was added neat diisopropylazodicarboxylate (7.79 ml_, 40.1 mmol). The reaction was warmed to 50°C under a nitrogen atmosphere for 18 hours. The reaction was vacuum filtered through a plug of Celite (10 g) and the filtrate concentrated in vacuo to give 19.8 g of viscous oil. The oil was taken up in ethyl acetate (400 ml_) and washed with 2M aqueous sodium hydroxide (400 ml_). The resultant organic was dried (MgS04), filtered and concentrated in vacuo to give an oil (18.6 g) which solidified on standing. Trituration was
attempted with diethyl ether (ca. 200 mL); the resulting solid was collected by filtration and dried in vacuo (0.27g) and the filtrate was concentrated in vacuo to give an oil (17.8 g). The sample was loaded in dichloromethane and purified by chromatography on silica (2 x 100g cartridges) eluting with a gradient of 0-25% ethyl acetate-cyclohexane over 60 mins. The appropriate fractions were combined and evaporated in vacuo to give the title compound as a white solid (6.90 g)
1 H NMR (400 MHz,CDCI3) δ ppm 1.38 - 1.52 (m, 18 H) 1.94 - 2.04 (m, 2 H) 2.88 - 3.00 (m, 2 H) 3.84 - 3.99 (m, 1 H) 4.29 (s, 1 H) 4.63 - 4.76 (m, 1 H) 6.26 (d, J=12.5 Hz, 2 H) Intermediate 18: c/s-3-{[4-(1 ,1-dimethylethyl)-3,5-difluorophenyl1oxy)cyclobutanamine

To a stirred solution of 1 , 1-dimethylethyl (c/'s-3-{[4-(1 , 1-dimethylethyl)-3,5- difluorophenyl]oxy}cyclobutyl)carbamate (6.85 g, 19.27 mmol) in 1 ,4-dioxane (50 mL) was added a solution of hydrogen chloride in 1 ,4-dioxane (50 ml, 4M, 200 mmol) in one charge. The reaction vessel was sealed and the reaction stirred at ambient temperature for 16 h. The reaction was concentrated to approx. half volume in vacuo and the resultant slurry was diluted with diethyl ether (200 mL) and the mixture stirred rapidly for 10 min. The precipitate was collected by filtration and dried in vacuo to give the title compound as a white solid (3.39 g). The mother liquors were concentrated in vacuo to give a yellow sticky solid. Trituration with diethyl ether (ca 20 mL) gave a white precipitate, the precipitate was collected by filtration and dried in vacuo to give a further batch of the title compound (1.035 g)
1 H NMR (400 MHz, Methanol-d4) δ ppm 1.41 (t, 9 H) 2.19 - 2.30 (m, 2 H) 2.97 (d, J=7.0 Hz, 2 H) 3.55 (s, 1 H) 4.56 (s, 1 H) 6.40 (d, J=12.8 Hz, 2 H)
Intermediate 19: 1 , 1-Dimethylethyl {fra/?s-3-r(3,5-difluorophenyl)oxylcvclobutyl)carbamate

To a stirred solution of 3,5-difluorophenol (1.65 g, 12.68 mmol), triphenylphosphine (4.20 g,
16.02 mmol) and 1 , 1-dimethylethyl (c/'s-3-hydroxycyclobutyl)carbamate (2 g, 10.68 mmol) in anhydrous tetrahydrofuran (THF) (100 mL) at ambient temperature was added neat diisopropylazodicarboxylate (3.12 mL, 16.02 mmol). The reaction was warmed to 50°C under a nitrogen atmosphere for 6 h. The reaction was concentrated in vacuo. The residue was dissolved in dichloromethane (200 mL) and washed with 2M aqueous sodium hydroxide (200 mL), dried using a hydrophobic frit and concentrated in vacuo to give a yellow oil. The sample was loaded in dichloromethane and purified on a silica cartridge (100g) using a gradient of 0-25% ethyl acetate-cyclohexane over 60 mins. The appropriate fractions were combined and evaporated in vacuo to give the title compound as a white solid (1.898g).
1 H NMR (400 MHz, Methanol-d4) δ ppm 1.43 (s, 9 H) 2.34 - 2.50 (m, 4 H) 4.13 - 4.25 (m, 1 H) 4.73 - 4.81 (m, 1 H) 6.36 - 6.51 (m, 3 H)
Intermediate 20: frans-3-[(3,5-Difluorophenyl)oxy1cvclobutanamine hydrochloride

To a stirred solution of 1 , 1-dimethylethyl {trans-3-[(3,5- difluorophenyl)oxy]cyclobutyl}carbamate (1.895 g, 6.33 mmol) in 1 ,4-dioxane (10 mL) was added a solution of hydrochloric acid 4M in 1 ,4-dioxane (20 mL, 80 mmol) in one charge. The reaction vessel was sealed and the reaction stirred at ambient temperature for 16 h. A further aliquot of hydrochloric acid 4M in 1 ,4 dioxane (10 mL, 40 mmol) was added and stirring continued for 6 h. The reaction was diluted with diethyl ether (50 mL). The white solid was collected by filtration and dried in vacuo to give the title compound as a white solid (1.303g). 1 H NMR (400 MHz, Methanol-d4) δ ppm 2.54 - 2.72 (m, 4 H) 3.93 - 4.03 (m, 1 H) 4.90 - 4.98 (m, 1 H) 6.44 (dd, J=9.0, 2.3 Hz, 2 H) 6.50 - 6.58 (m, 1 H)
Intermediate 21j 1.1-Dimethylethyl (frans-3-(r4-(1.1-dimethylethyl)-3.5- difluorophenylloxylcyclobutvDcarbamate

1 H NMR (400 MHz, CDCI3) δ ppm 1.36 - 1.55 (m, 18 H) 2.31 - 2.45 (m, 2 H) 2.49 - 2.62 (m, 2 H) 4.19 - 4.37 (m, 1 H) 4.65 - 4.82 (m, 1 H) 6.18 - 6.28 (m, 2 H) Intermediate 22: ra/7s-3-{[4-(1 , 1-dimethylethyl)-3,5-difluorophenyl1oxy)cyclobutanamine hydrochloride

LC-MS (System A) rt = 0.88min, MH+ = 256
Intermediate 23: 1 , 1-Dimethylethyl (c/s-3-{r2-(1-methylethyl)phenylloxy)cvclobutyl)carbamate

To a stirred solution of 2-(1-methylethyl)phenol (0.072 mL, 0.534 mmol), triphenylphosphine (210 mg, 0.801 mmol) and 1 , 1-dimethylethyl (trans-3-hydroxycyclobutyl)carbamate (100 mg, 0.534 mmol) in anhydrous tetrahydrofuran (THF) (5 mL) at ambient temperature was added neat diisopropylazodicarboxylate (0.156 mL, 0.801 mmol). The reaction was warmed to 45°C
under a nitrogen atmosphere overnight. Additional diisopropylazodicarboxylate (0.156 ml_, 0.801 mmol) and triphenylphosphine (210 mg, 0.801 mmol) were added and the reaction was left to stir at 45 °C for a further 24 hours. The reaction was concentrated in vacuo and the crude product was purified by Mass Directed HPLC using H20 + 10mM Ammonium Bicarbonate adjusted to pH 10 with Ammonia and CH3CN as eluents. Appropriate fractions were combined and evaporated to give the title compound (38mg).
LC-MS (System B) rt = 3.65min, MH+ = 306
Intermediate 24: c/s-3-{r2-(1-Methylethyl)phenylloxy)cvclobutanamine hydrochloride

Prepared similarly to intermediate 1 1 from 1 , 1-dimethylethyl (c/'s-3-{[2-(1- methylethyl)phenyl]oxy}cyclobutyl)carbamate
1 H NMR (400 MHz, Methanol-d4) δ ppm 1.19 (d, 6 H) 2.29 (br. s., 2 H) 2.99 (br. s., 2 H) 3.50 - 3.61 (m, 1 H) 4.52 - 4.61 (m, 1 H) 6.74 (d, J=8.0 Hz, 1 H) 6.86 - 6.93 (m, 1 H) 7.06 - 7.12 (m, 1 H) 7.16 - 7.21 (m, 1 H) Intermediate 25: 1 , 1-Dimethylethyl {c/s-3-r(3-fluoro-2-methylphenyl)oxylcvclobutyl)carbamate

Prepared similarly to intermediate 12 from 1 , 1-dimethylethyl (trans-3- hydroxycyclobutyl)carbamate and 3-fluoro-2-methylphenol
1 H NMR (400 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 1.91 - 2.07 (m, 5 H) 2.71 - 2.83 (m, 2 H) 3.64 - 3.76 (m, 1 H) 4.34 - 4.44 (m, 1 H) 6.61 - 6.67 (m, 1 H) 6.70 - 6.77 (m, 1 H) 7.08 - 7.22 (m, 2 H) Intermediate 26: c/s-3-r(3-Fluoro-2-methylphenyl)oxylcvclobutanamine hydrochloride

Prepared similarly to Intermediate 11 from 1 , 1-dimethylethyl {c/'s-3-[(3-fluoro-2- methylphenyl)oxy]cyclobutyl}carbamate.
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.08 (d, J=1.8 Hz, 3 H) 2.26 - 2.39 (m, 2 H) 2.93 - 3.06 (m, 2 H) 3.52 - 3.65 (m, 1 H) 4.51 - 4.63 (m, 1 H) 6.57 (d, J=8.3 Hz, 1 H) 6.63 (t, J=8.8 Hz, 1 H) 7.08 (d, J=7.0 Hz, 1 H)
Intermediate 27: 1 , 1-Dimethylethyl {frans-3-r(4-chlorophenyl)oxylcvclobutyl)carbamate

Prepared similarly to Intermediate 19 from 1 , 1-dimethylethyl (c/s-3- hydroxycyclobutyl)carbamate and 4-chlorophenol.
1 H NMR (400 MHz, Methanol-d4) δ ppm 1.43 (s, 9 H) 2.33 - 2.47 (m, 4 H) 4.13 - 4.23 (m, 1 H) 4.72 - 4.79 (m, 1 H) 6.73 - 6.79 (m, 2 H) 7.19 - 7.24 (m, 2 H)
Intermediate 28: frans-3-[(4-Chlorophenyl)oxy1cyclobutanamine hydrochloride

Prepared similarly to Intermediate 20 from 1 , 1-dimethylethyl {trans-3-[(4- chlorophenyl)oxy]cyclobutyl}carbamate.
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.53 - 2.70 (m, 4 H) 3.92 - 4.01 (m, 1 H) 4.59 (br. s., 1 H) 6.76 - 6.83 (m, 2 H) 7.22 - 7.29 (m, 2 H)
Intermediate 29: 1 ,1-Dimethylethyl {frans-3-[(3-fluoro-2-methylphenyl)oxy1cvclobutyl) carbamate

Prepared similarly to intermediate 10 from 1 , 1-dimethylethyl (c/s-3- hydroxycyclobutyl)carbamate and 3-fluoro-2-methylphenol.
1 H NMR (400 MHz, Methanol-d4) δ ppm 1.43 (s, 9 H) 2.10 (d, J=1.8 Hz, 3 H) 2.34 - 2.51 (m, 4 H) 4.14 - 4.26 (m, 1 H) 4.49 - 4.65 (m, 1 H) 6.43 - 6.49 (m, 1 H) 6.58 - 6.66 (m, 1 H) 7.01 - 7.1 1 (m, 1 H)
Intermediate 30: frans-3-[(3-Fluoro-2-methylphenyl)oxy1cvclobutanamine hydrochloride

To 1 , 1-dimethylethyl {trans-3-[(3-fluoro-2-methylphenyl)oxy]cyclobutyl}carbamate (331 mg, 1.121 mmol) was added hydrochloric acid 4M in dioxane (5 mL, 20.00 mmol) and the reaction mixture was left to stand at ambient temperature for 66 hours. The reaction mixture was then diluted in 40-60°C Petroleum Ether (500 mL), filtered and dried in vacuo to give the title compound as a white solid (170 mg)
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.1 1 (d, J=2.0 Hz, 3 H) 2.55 - 2.73 (m, 4 H) 3.94 - 4.03 (m, 1 H) 4.53 - 4.63 (m, 1 H) 4.94 - 5.01 (m, 1 H) 6.48 (d, J=8.3 Hz, 1 H) 6.67 (dd, 1 H) 7.06 - 7.14 (m, 1 H)
Intermediate 31 : 1 , 1-Dimethylethyl {c/s-3-[(4-chlorophenyl)oxy1cvclobutyl)carbamate

Prepared similarly to Intermediate 19 from 1 , 1-dimethylethyl (trans-3- hydroxycyclobutyl)carbamate and 4-chlorophenol
1 H NMR (400 MHz, Methanol-d4) d ppm 1.42 (s, 9 H) 1.94 - 2.04 (m, 2 H) 2.80 - 2.90 (m, 2 H) 3.70 - 3.83 (m, 1 H) 4.32 - 4.41 (m, 1 H) 6.77 - 6.83 (m, 2 H) 7.18 - 7.24 (m, 2 H)
Intermediate 32: c/s-3-[(4-Chlorophenyl)oxy1cyclobutanamine hydrochloride

Prepared similarly to Intermediate 20 from 1 , 1-dimethylethyl {c/'s-3-[(4- chlorophenyl)oxy]cyclobutyl}carbamate.
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.20 - 2.30 (m, 2 H) 2.93 - 3.02 (m, 2 H) 3.50 - 3.60 (m, 1 H) 4.52 - 4.60 (m, 1 H) 6.80 - 6.86 (m, 2 H) 7.22 - 7.28 (m, 2 H)
Intermediate 33: 1 , 1-Dimethylethyl {frans-3-[(2,3-dichlorophenyl)oxy1cvclobutyl)carbamate

1 H NMR (400 MHz, DMSO-d6) δ ppm 1.38 (s, 9 H) 2.29 - 2.45 (m, 4 H) 4.06 - 4.17 (m, 1 H) 4.86 - 4.94 (m, 1 H) 6.86 - 6.91 (m, 1 H) 7.17 - 7.22 (m, 1 H) 7.26 - 7.35 (m, 2 H) Intermediate 34: frans-3-r(2,3-dichlorophenyl)oxylcyclobutanamine hydrochloride

Prepared similarly to Intermediate 20 from 1 , 1-dimethylethyl {trans-3-[(2,3- dichlorophenyl)oxy]cyclobutyl}carbamate.
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.63 - 2.70 (m, 4 H) 4.01 (t, J=7.2 Hz, 1 H) 4.99 - 5.06 (m, 1 H) 6.79 (dd, J=8.3, 1.3 Hz, 1 H) 7.12 - 7.16 (m, 1 H) 7.19 - 7.25 (m, 1 H)
Intermediate 35: 1 , 1-Dimethylethyl {c/s-3-r(2,3-dichlorophenyl)oxylcvclobutyl)carbamate

Prepared similarly to Intermediate 19 from 1 , 1-dimethylethyl {trans-3- hydroxycyclobutyl)carbamate and 2,3-dichlorophenol
1 H NMR (400 MHz, Methanol-d4) δ ppm 1.43 (s, 9 H) 2.02 - 2.12 (m, 2 H) 2.85 - 2.94 (m, 2 H) 3.73 - 3.86 (m, 1 H) 4.43 - 4.52 (m, 1 H) 6.87 (dd, J=8.3, 1.3 Hz, 1 H) 7.06 - 7.10 (m, 1 H) 7.15 - 7.21 (m, 1 H)
Intermediate 36: c/s-3-r(2,3-Dichlorophenyl)oxylcyclobutanamine hydrochloride

Prepared similarly to Intermediate 20 from 1 , 1-dimethylethyl {cis- 3- [(2,3- dichlorophenyl)oxy]cyclobutyl}carbamate.
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.28 - 2.38 (m, 2 H) 2.98 - 3.08 (m, 2 H) 3.56 (t, J=8.0 Hz, 1 H) 3.65 (s, 2 H) 4.67 (t, J=7.0 Hz, 1 H) 6.88 (dd, J=8.3, 1.3 Hz, 1 H) 7.12 - 7.16 (m, 1 H) 7.19 - 7.25 (m, 1 H)
Intermediate37: 1 , 1-Dimethylethyl ( ra/7s-3-{[2-chloro-3-(trifluoromethyl)phenyl1oxy)cvclobutyl) carbamate

Prepared similarly to Intermediate 19 from 1 , 1-dimethylethyl (c/s-3- hydroxycyclobutyl)carbamate and 2-chloro-3-(trifluoromethyl)phenol
1 H NMR (400 MHz, Methanol-d4) δ ppm 1.43 (s, 9 H) 2.40 - 2.57 (m, 4 H) 4.19 - 4.29 (m, 1 H) 4.88 - 4.95 (m, 1 H) 7.06 - 7.11 (m, 1 H) 7.29 - 7.42 (m, 2 H)
Intermediate 38: ra/7s-3-{r2-Chloro-3-(trifluoromethyl)phenylloxy)cyclobutanamine
hydrochloride

Prepared similarly to Intermediate 20 from 1 , 1-dimethylethyl (frans-3-{[2-chloro-3- (trifluoromethyl)phenyl]oxy}cyclobutyl)carbamate.
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.62 - 2.77 (m, 4 H) 3.98 - 4.08 (m, 1 H) 5.05 - 5.12 (m, 1 H) 7.12 (d, J=8.0 Hz, 1 H) 7.35 - 7.46 (m, 2 H)
Intermediate 39: 1 , 1-Dimethylethyl ( ra/7s-3-{[4-fluoro-3-(trifluoromethyl)phenyl1oxy)cvclobutyl) carbamate

To a stirred suspension of 4-fluoro-3-(trifluoromethyl)phenol (2.365 g, 13.13 mmol), 1 , 1- dimethylethyl (cis-3-hydroxycyclobutyl)carbamate (2.2 g, 11.75 mmol) and triphenylphosphine polymer bound (5.87 g, 17.62 mmol) in dry tetrahydrofuran (THF) (100 mL) was added neat diisopropyl azodicarboxylate (3.43 mL, 17.62 mmol) in one charge. The reaction was stirred at 50°C for 22 h. The reaction was filtered through a pad of Celite and the cake washed with THF (100 mL), The filtrate was concentrated in vacuo and the residue dissolved in dichloromethane (100 mL) and washed with 2M aqueous sodium hydroxide (100 mL). The organic phase was dried using a hydrophobic frit and concentrated in vacuo. The sample was purified by chromatography on silica (2 x 100g) using a gradient of 0-50% ethyl acetate- cyclohexane gradient over 40 mins. The appropriate fractions were combined and evaporated in vacuo to give the title compound as white solid (3.17g).
1 H NMR (400 MHz, CDCI3) δ ppm 1.46 (s, 9 H) 2.35 - 2.46 (m, 2 H) 2.50 - 2.61 (m, 2 H) 4.24 - 4.37 (m, 1 H) 4.71 - 4.81 (m, 1 H) 6.87 - 6.99 (m, 2 H) 7.06 - 7.14 (m, 1 H)
Intermediate 40: ra/7s-3-{r4-Fluoro-3-(trifluoromethyl)phenylloxy)cyclobutanamine
hydrochloride

Prepared similarly to Intermediate 11 from 1 , 1-dimethylethyl (frans-3-{[4-fluoro-3- (trifluoromethyl)phenyl]oxy}cyclobutyl)carbamate.
1 H NMR (400 MHz, Methanol-d4) δ ppm 2.56 - 2.72 (m, 4 H) 3.94 - 4.04 (m, 1 H) 4.94 - 5.02 (m, 1 H) 7.04 - 7.12 (m, 2 H) 7.22 - 7.30 (m, 1 H)
Example 1 : /V-( ra/7s-3-{[4-(Trifluoromethyl)phenyl1oxy)cvclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide

To a stirred suspension of pyrazolo[1 ,5-a]pyridine-3-carboxylic acid (20 mg, 0.123 mmol) and 0-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluroniumhexafluorophosphate (50 mg, 0.131 mmol) in anhydrous Ν,Ν-dimethylformamide (DMF) (1 mL) was added neat triethylamine (52 μΙ, 0.373 mmol). The reaction was stirred at ambient temperature for 15 min then a solution of trans-3-{[4-(trifluoromethyl)phenyl]oxy}cyclobutanamine hydrochloride (35 mg, 0.131 mmol) in anhydrous Ν,Ν-dimethylformamide (DMF) (1 mL) was added. The reaction was stirred at ambient temperature for a further 5.5 h then the reaction was concentrated in vacuo. The sample was dissolved in 1 : 1 MeOH:DMSO (1 mL) and purified by MDAP HPLC (Method A). The solvent was dried under a stream of nitrogen in the Radleys blowdown apparatus to give the title compound (33 mg)
LCMS (System A) rt = HOmins, MH+ = 376 Example 2: A/-(c/s-3-{r4-(Trifluoromethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

LC-MS (System A) rt = 1.12min, MH+ = 376
Example 3: A/-( ra/7s-3-{r2-Bromo-4-(1 , 1-dimethylethyl)phenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de

To a stirred solution of pyrazolo[1 ,5-a]pyridine-3-carboxylic acid (79 mg, 0.487 mmol) and O- (7-azabenzotriazol-1-yl)-/\/,A/,A/',A/'-tetramethyluroniumhexafluorophosphate (204 mg, 0.536 mmol) in anhydrous A/,A/-dimethylformamide (DMF) (5 mL) was added neat triethylamine (0.204 mL, 1.461 mmol). The reaction was stirred at ambient temperature for 15 mins. A solution of trans-3-{[2-bromo-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutanamine hydrochloride (163 mg, 0.487 mmol) in A/,A/-dimethylformamide (DMF) (2 mL) was added and the reaction stirred at ambient temperature for 3 h. The reaction was concentrated in vacuo and the residue dissolved in ethyl acetate (20 mL) and washed sequentially with water (20 mL), saturated aqueous sodium carbonate (20 mL), saturated aqueous citric acid (20 mL) and brine (20 mL). The resultant organic was dried using a hydrophobic frit and concentrated in vacuo. The sample was loaded in dichloromethane and purified by chromatography on silica (20g) using a gradient 0-100% ethyl acetate-cyclohexane over 40 mins. The appropriate fractions were combined and evaporated in vacuo to give the title compound as a white foam (143 mg).
LC-MS (System A) rt = 1.28min, MH+ = 442 / 444
Example 4: /V-( ra/7s-3-{r4-(1 , 1-Dimethylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

Example 5: /V-( ra/7s-3-{[4-(1 , 1-Dimethylethyl)-3-fluorophenyl1oxy)cvclobutyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

To a solution of 4-(1 ,1-dimethylethyl)-3-fluorophenol (35.2 mg, 0.209 mmol) in N,N- dimethylformamide (DMF) (1 mL) was added potassium carbonate (48.3 mg, 0.349 mmol).
The reaction was left to stir at room temperature for 30min. A solution of c/'s-3-[(pyrazolo[1 ,5- a]pyridin-3-ylcarbonyl)amino]cyclobutyl methanesulfonate (54 mg, 0.175 mmol) in N,N- dimethylformamide (DMF) (1 mL) was added and the mixture was heated at 70°C overnight.
The reaction was transferred into a microwave vial and heated in the microwave at 200°C for 40min. The reaction was concentrated in vacuo and the crude product was purified by MDAP
HPLC (Method A). Fractions which contain product were concentrated in vacuo to give the title compound (23mg).
LC-MS (System A) rt = 1.29min, MH+ = 382 Example 6: /V-( ra/7s-3-{[4-(1 , 1-Dimethylethyl)-2-methylphenyl1oxy)cvclobutyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

A mixture of c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3-ylcarbonyl)amino]cyclobutyl methanesulfonate (30 mg, 0.097 mmol), 4-(1 , 1-dimethylethyl)-2-methylphenol (15.93 mg, 0.097 mmol) and potassium carbonate (26.8 mg, 0.194 mmol) in A/,A/-dimethylformamide (DMF) (1 ml_) was heated in the microwave at 200°C for 40min. The reaction was concentrated in vacuo and the crude product was dissolved in 1 : 1 DMSO/MeOH (0.5ml) and purified by MDAP HPLC (Method A). Fractions which contained product were concentrated in vacuo to give the title compound (2.5mg).
LC-MS (System A) rt = 1.28min, MH+ = 378
Example 7: A/-( ra/7s-3-{r2-Chloro-4-(1 , 1-dimethylethyl)phenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de

A mixture of c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3-ylcarbonyl)amino]cyclobutyl methanesulfonate (73 mg, 0.236 mmol), 2-chloro-4-(1 , 1-dimethylethyl)phenol (43.6 mg, 0.236 mmol) and potassium tert-butoxide (53.0 mg, 0.472 mmol) in Ν, Ν-dimethylformamide (DMF) (3.00 ml_) was heated at 70°C for 3 h. Additional potassium tert-butoxide (53.0 mg, 0.472 mmol) was added and the reaction was stirred at 70 °C over the weekend. The reaction was concentrated in vacuo. The crude product was purified by Mass Directed HPLC (Method B) and fractions which contain product were concentrated to give the title compound.
LC-MS (System B) rt = 3.35min, MH+ = 378
Example 8 /V-( ra/7s-3-{r3-(1 , 1-Dimethylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

LC-MS (System A) rt = 1.19min, MH+ = 364
Example 9: /V-[ ra/7s-3-({3-[(Trifluoromethyl)oxylphenyl)oxy)cvclobutyllpyrazolo[1 ,5-alpyridine- 3-carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 3-[(trifluoromethyl)oxy]phenol
LC-MS (System A) rt = 1.1 1 min, MH+ = 392
Example 10: /V-( ra/7s-3-{[2-(Trifluoromethyl)phenyl1oxy)cvclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2-(trifluoromethyl)phenol
LC-MS (System A) rt = 1.08min, MH+ = 376
Example 11 : A/-( rans-3-{[3-(Trifluoromethyl)phenyl1oxy)cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 3-(trifluoromethyl)phenol
LC-MS (System A) rt = 1.09min, MH+ = 376 Example 12: A/-r ra/7s-3-({4-r(Trifluoromethyl)oxylphenyl)oxy)cvclobutyllpyrazolori ,5- al pyri d i n e- 3- carboxa m i de

LC-MS (System A) rt = 1.11 min, MH+ = 392
Example 13: /V-( ra/7s-3-{[3-(1-Methylethyl)phenyl1oxy)cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 3-(1-methylethyl)phenol
LC-MS (System A) rt = 1.14min, MH+ = 350
Example 14: /V-( ra/7s-3-{r4-(1-Methylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 4-(1-methylethyl)phenol
LC-MS (System A) rt = 1.15min, MH+ = 350
Example 15: A/-{ ra/7s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 3,5-difluorophenol
LC-MS (System A) rt = 1.02min, MH+ = 344
Example 16: A/-{ ra/7s-3-r(3,4,5-Trifluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 3,4,5-trifluorophenol
LC-MS (System A) rt = 1.05min, MH+ = 362
Example 17: A/-( ra/7s-3-fr4-(1.1-Dimethylethvn-3.5- difluorophenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-3-carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 4-(1 , 1-dimethylethyl)-3,5-difluorophenol LC-MS (System A) rt = 1.12min, MH+ = 400
Example 18: /V-( ra/7s-3-{[4-(1 , 1-Dimethylethyl)-2-fluorophenyl1oxy)cvclobutyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 4-(1 , 1-dimethylethyl)-2-fluorophenol LC-MS (System A) rt = 1.22min, MH+ = 382
Example 19: A/-{frans-3-[(4-Chloro-3-fluorophenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 4-chloro-3-fluorophenol
LC-MS (System A) rt = 1.07min, MH+ = 360 Example 20; A/-{ ra/7s-3-r(4-Chlorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 4-chlorophenol
LC-MS (System A) rt = 1.05min, MH+ = 342 / 344
Example 21 : /V-( ra/7s-3-{[2-(1-Methylethyl)phenyl1oxy)cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 8 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2-(1-methylethyl)phenol
LC-MS (System A) rt = 1.18 min, MH+ = 350
Example 22: A/-{ ra/7s-3-r(3-Fluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

An aliquot of c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3-ylcarbonyl)amino]cyclobutyl methanesulfonate in DMF (0.25ml equivalent to O. l mmol, prepared from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate (1.17g, 3.8mmol) was dissolved in DMF (9.5ml)) was added to 3-fluorophenol (O. l mmol) in DMF (0.25ml). Potassium carbonate (heaped microspatula approximately 0.028g, 0.2mmol) was added and the reaction heated to 80°C for 18 hours. The solution was filtered and DMSO (0.3ml) was added. The solution was purified by MDAP HPLC (Method A) and the appropriate fractions evaporated in vacuo using the Genevac to give the title compound (2.9mg)
LC-MS (System A) rt = 0.98 min, MH+ = 326
Example 23; A/-{ ra/7s-3-[(3-Methylphenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

An aliquot of c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3-ylcarbonyl)amino]cyclobutyl methanesulfonate in DMF (0.25ml equivalent to O. l mmol, prepared from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate (1.17g, 3.8mmol) was dissolved in DMF (9.5ml)) was added to 3-methylphenol (O. l mmol) in DMF (0.25ml). Potassium carbonate (heaped microspatula approximately 0.028g, 0.2mmol) was and the reaction heated to 80°C for 18 hours. An additional portion of 3-methylphenol was added and the solution heated for a further 18hrs at 80°C. The solution was filtered and DMSO (0.3ml) was added. The solution was purified by MDAP HPLC (Method A) and the appropriate fractions evaporated in vacuo using the Genevac to give the title compound (3.3mg).
LC-MS (System A) rt = 1.01 min, MH+ = 322
Example 24: A/-{ ra/7s-3-[(2-Propylphenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 23 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2-propylphenol
LC-MS (System A) rt = 1.20 min, MH+ = 350
Example 25; A/-{ ra/7s-3-r(2-Fluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

Prepared similarly to Example 23 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2-fluorophenol
LC-MS (System A) rt = 0.94 min, MH+ = 326
Example 26: A/-{ ra/7s-3-[(2,3-Dimethylphenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 23 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2,3-dimethylphenol
LC-MS (System A) rt = 1.10 min, MH+ = 336
Example 27: A/-{frans-3-r(2,3-Difluorophenyl)oxylcvclobutyl)pyrazoloH ,5-alpyridine-3- carboxamide

Prepared similarly to Example 22 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2,3-difluorophenol
LC-MS (System A) rt = 0.99 min, MH+ = 344
Example 28: A/-{ ra/7s-3-r(4-Chloro-3,5-difluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine- 3-carboxamide

Prepared similarly to Example 22 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 4-chloro-3,5-difluorophenol
LC-MS (System A) rt = 1.09 min, MH+ = 378 / 380
Example 29: /V-{ ra/7s-3-[(3-Fluoro-2-methylphenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 22 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 3-fluoro-2-methylphenol
LC-MS (System A) rt = 1.07 min, MH+ = 340
Example 30: A/-{ ra/7s-3-[(2-Chlorophenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 22 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2-chlorophenol
LC-MS (System A) rt = 1.02 min, MH+ = 342 / 344 Example 31j /V-{ ra/7s-3-[(2-Methylphenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 23 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2-methylphenol
LC-MS (System A) rt = 1.04 min, MH+ = 322
Example 32: A/-{frans-3-r(2-Chloro-3-fluorophenyl)oxylcvclobutyl)pyrazoloH ,5-alpyridine-3- carboxamide

Prepared similarly to Example 22 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2-chloro-3-fluorophenol
LC-MS (System A) rt = 1.05 min, MH+ = 360 / 362
Example 33: A/-{frans-3-r(3,4-Difluorophenyl)oxylcvclobutyl)pyrazoloH ,5-alpyridine-3- carboxamide

Prepared similarly to Example 22 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 3,4-difluorophenol
LC-MS (System A) rt = 1.00 min, MH+ = 344
Example 34: A/-{ ra/7s-3-r(3-Fluoro-4-methylphenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

LC-MS (System A) rt = 1.05 min, MH+ = 340
Example 35: A/-{ ra/7s-3-r(2-Chloro-3,5-difluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-
3-carboxamide

LC-MS (System A) rt = 1.09 min, MH+ = 378 / 380
Example 36: A/-( ra/7s-3-{r4-Methyl-2-(1-methylethyl)phenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 23 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 4-methyl-2-(1-methylethyl)phenol
LC-MS (System A) rt = 1.23 min, MH+ = 364
Example 37: A/-( ra/7s-3-(r2,4-£)/s(1-Methylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine- 3-carboxamide

Prepared similarly to Example 23 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2,4-bis(1-methylethyl)phenol
LC-MS (System A) rt = 1.36 min, MH+ = 392
Example 38: /V-[ ra/7s-3-({2-[(Trifluoromethyl)oxylphenyl)oxy)cvclobutyllpyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 23 from c/'s-3-[(pyrazolo[1 ,5-a]pyridin-3- ylcarbonyl)amino]cyclobutyl methanesulfonate and 2-[(trifluoromethyl)oxy]phenol
LC-MS (System A) rt = 1.09 min, MH+ = 392
Example 39: A/-( ra/7s-3-{r2-(1 ,1-Dimethylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-
3-carboxamide

LC-MS (System A) rt = 1.24 min, MH+ = 364
Example 40: A/-(c/s-3-{r4-(1 ,1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de

To a stirred solution of 4-(1 ,1-dimethylethyl)-3,5-difluorophenol (76 mg, 0.406 mmol), triphenylphosphine (160 mg, 0.610 mmol) and N-(frans-3-hydroxycyclobutyl)pyrazolo[1 ,5- a]pyridine-3-carboxamide (94 mg, 0.406 mmol) in anhydrous tetrahydrofuran (THF) (5 mL) at ambient temperature was added neat diisopropylazodicarboxylate (0.119 mL, 0.610 mmol). The reaction was warmed to 45°C under a nitrogen atmosphere overnight. Additional
triphenylphosphine (160 mg, 0.610 mmol) and diisopropylazodicarboxylate (0.1 19 mL, 0.610 mmol) were added and the reaction was left to stir at 45 °C over the weekend.
The reaction was concentrated in vacuo and the residue partitioned between sodium hydroxide (20mL) and DCM (20mL). The organic layer was concentrated in vacuo and the crude product was purified by MDAP HPLC (Method A) in two batches. Fractions which contained product were concentrated to give the title compound (2mg).
LC-MS (System A) rt = 1.29 min, MH+ = 400
Example 41 : Ay-{ ra/7s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)-6-(hvdroxymethyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de

To a solution of 6-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid (20 mg, 0.104 mmol) in Ν,Ν-Dimethylformamide (DMF) (1 mL) was added triethylamine (0.036 mL, 0.260 mmol) and 0-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (40 mg, 0.105 mmol) and the reaction mixture was left stirring for 5 minutes. trans-3-[(3,5- Difluorophenyl)oxy]cyclobutanamine hydrochloride (24.53 mg, 0.104 mmol) was added and the reaction mixture was left stirring at ambient temperature for 16 hours. The solvent was evaporated and the sample was dissolved in 1 : 1 MeOH:DMSO (1 mL) and purified by MDAP HPLC (Method A). The solvent was dried under a stream of nitrogen in the Radleys blowdown apparatus to give the title compound (1.26mg).
LC-MS (System A) rt = 0.93 min, MH+ = 374 Example 42: Ay-( ra/7S-3-{r4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl1oxy)cvclobutyl)-6- (hvdroxymethyl)pyrazolo[1 ,5-alpyridine-3-carboxamide

Example 43: /V-( ra/7s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl1oxy)cvclobutyl)-7- (methyloxy)pyrazoloH ,5-alpyridine-3-carboxamide

An aliquot of a solution of HATU (0.1 ml, O. l mmol - solution prepared from HATU (1.4 g) in DMF (3.6 ml)) was added to 7-(methyloxy)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid (0.100 mmol). Diisopropylethylamine (50 μΙ, 0.286 mmol) was added and the vial was shaken for 2 mins at room temperature. An aliquot of a solution of frans-3-{[4-(1 , 1-dimethylethyl)-3,5- difluorophenyl]oxy}cyclobutanamine hydrochloride (0.1 ml, O. l mmol - solution prepared by dissolving frans-3-{[4-(1 , 1-dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutanamine hydrochloride (522mg) in DMF (1.8ml)) was added. The vial was capped and shaken to aid dissolution then allowed to stand at room temperature for 18 hours. A mixture of 1 : 1 MeOH:DMSO (0.5 ml) was added and the solution purified by MDAP HPLC (Method A). The solvent was evaporated in vacuo in a plate drier to give the title compound (15.8mg)
LC-MS (System A) rt = 1.28 min, MH+ = 430 Example 44: A/-( ra/7s-3-{r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)-7-
(hvdroxymethyl)pyrazolori ,5-alpyridine-3-carboxamide

Example 45: A/-(fra/?s-3-(r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 43 from imidazo[1 ,2-a]pyridine-3-carboxylic acid and trans-3- {[4-(1 , 1-dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutanamine hydrochloride
LC-MS (System A) rt = 1.18 min, MH+ = 400
Example 46: /V-{ ra/7s-3-[(3,5-Difluorophenyl)oxy1cvclobutyl)-7-(methyloxy)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 43 from 7-(methyloxy)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and {frans-3-[(3,5-difluorophenyl)oxy]cyclobutyl}amine hydrochloride
LC-MS (System A) rt = 1.02 min, MH+ = 374
Example 47: /V-{ ra/7s-3-[(3,5-Difluorophenyl)oxy1cvclobutyl)-7-(hvdroxymethyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and {frans-3-[(3,5-difluorophenyl)oxy]cyclobutyl}amine hydrochloride
LC-MS (System A) rt = 0.98 min, MH+ = 374
Example 48: A/-{ ra/7s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)imidazori ,2-alpyridine-3- carboxamide

LC-MS (System A) rt = 0.88 min, MH+ = 344
Example 49: A/-(c/s-3-{r2-(1-Methylethyl)phenylloxy)cvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

LC-MS (System A) rt = 1.13 min, MH+ = 350
Example 50; A/-{c/s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide

LC-MS (System A) rt = 0.99 min, MH+ = 344. Example 51 : A/-{c/s-3-r(3-Fluoro-2-methylphenyl)oxylcvclobutyl)pyrazoloH ,5-alpyridine-3- carboxamide

Prepared similarly to Example 41 from pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and c/'s-3-[(3- fluoro-2-methylphenyl)oxy]cyclobutanamine hydrochloride
LC-MS (System A) rt = 1.05 min, MH+ = 340
Example 52: A/-{frans-3-[(4-chlorophenyl)oxy1cvclobutyl)imidazo[1 ,2-a]pyridine-3-carboxamide

Prepared similarly to Example 43 from imidazo[1 ,2-a]pyridine-3-carboxylic acid and {trans-3- [(4-chlorophenyl)oxy]cyclobutyl}amine hydrochloride
LC-MS (System A) rt = 0.90 min, MH+ = 342 / 344
Example 53: A/-{ ra/7s-3-r(4-Chlorophenyl)oxylcvclobutyl)-7-(hvdroxymethyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de
 Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and {frans-3-[(4-chlorophenyl)oxy]cyclobutyl}amine hydrochloride
Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and {frans-3-[(4-chlorophenyl)oxy]cyclobutyl}amine hydrochloride
LC-MS (System A) rt = 1.01 min, MH+ = 372 / 374
Example 54: A/-{c/s-3-[(3,5-difluorophenyl)oxy1cvclobutyl)imidazo[1 ,2-alpyridine-3- carboxamide

Prepared similarly to Example 43 from imidazo[1 ,2-a]pyridine-3-carboxylic acid and {trans-3- [(3,5-difluorophenyl)oxy]cyclobutyl}amine hydrochloride
LC-MS (System A) rt = 0.84 min, MH+ = 344
Example 55: A/-{c/s-3-[(3,5-Difluorophenyl)oxy1cvclobutyl)-7-(hvdroxymethyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and {frans-3-[(3,5-difluorophenyl)oxy]cyclobutyl}amine hydrochloride
LC-MS (System A) rt = 0.96 min, MH+ = 374
Example 56: /V-{ ra/7s-3-[(3-Fluoro-2-methylphenyl)oxy1cvclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide

Prepared similarly to Example 43 from imidazo[1 ,2-a]pyridine-3-carboxylic acid and trans-3- [(3-fluoro-2-methylphenyl)oxy]cyclobutanamine hydrochloride
LC-MS (System A) rt = 0.91 min, MH+ = 340 Example 57; A/-{frans-3-[(3-Fluoro-2-methylphenyl)oxy1cvclobutyl)-7-
(hvdroxymethyl)pyrazolo[1 ,5-alpyridine-3-carboxamide

Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and frans-3-[(3-fluoro-2-methylphenyl)oxy]cyclobutanamine hydrochloride.
LC-MS (System A) rt = 1.05 min, MH+ = 370
Example 58: A/-{c/s-3-[(3-Fluoro-2-methylphenyl)oxy1cvclobutyl)-7-
(hvdroxymethyl)pyrazolo[1 ,5-alpyridine-3-carboxamide

Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and {c/'s-3-[(3-fluoro-2-methylphenyl)oxy]cyclobutyl}amine hydrochloride
LC-MS (System A) rt = 1.02 min, MH+ = 370
Example 59: A/-{c/s-3-[(3-Fluoro-2-methylphenyl)oxy1cvclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide

LC-MS (System A) rt = 0.88 min, MH+ = 340
Example 60: A/-{c/s-3-[(4-Chlorophenyl)oxy1cvclobutyl)imidazo[1 ,2-a]pyridine-3-carboxamide

Prepared similarly to Example 43 from imidazo[1 ,2-a]pyridine-3-carboxylic acid and c/'s-3-[(4- chlorophenyl)oxy]cyclobutanamine hydrochloride
LC-MS (System A) rt = 0.88 min, MH+ = 342 / 344
Example 61 : A/-{c/s-3-r(4-Chlorophenyl)oxylcvclobutyl)pyrazoloH ,5-alpyridine-3-carboxamide

Prepared similarly to Example 43 from pyrazolo[1 ,5-a]pyridine-3-carboxylic acid
and c/'s-3-[(4-chlorophenyl)oxy]cyclobutanamine hydrochloride
LC-MS (System A) rt = 1.08 min, MH+ = 342 / 344
Example 62: A/-{c/s-3-[(4-Chlorophenyl)oxy1cvclobutyl)-7-(hvdroxymethyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and c/'s-3-[(4-chlorophenyl)oxy]cyclobutanamine hydrochloride.
LC-MS (System A) rt = 1.02 min, MH+ = 372 / 374.
Example 63: A/-{c/s-3-[(3-Fluoro-2-methylphenyl)oxy1cvclobutyl)-6-

LC-MS (System A) rt = 0.98 min, MH+ = 370. Example 64: A/-{ ra/7s-3-r(4-Chlorophenyl)oxylcvclobutyl)-6-(hvdroxymethyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 43 from 6-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and frans-3-[(4-chlorophenyl)oxy]cyclobutanamine hydrochloride.
LC-MS (System A) rt = 0.97 min, MH+ = 372 / 374.
Example 65; A/-(c/s-3-{r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)-7-
(hvdroxymethyl)pyrazolo[1 ,5-alpyridine-3-carboxamide

Prepared similarly to Example 43 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and c/'s-3-{[4-(1 , 1-dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutanamine hydrochloride. LC-MS (System A) rt = 1.25 min, MH+ = 430.
Example 66: /V-(c/s-3-(r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 43 imidazo[1 ,2-a]pyridine-3-carboxylic acid and cis- 3-{[4-( 1 , 1- dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutanamine hydrochloride.
LC-MS (System A) rt = 1.15 min, MH+ = 400
Example 67: /V-{ ra/7s-3-[(2,3-Dichlorophenyl)oxy1cvclobutyl)imidazo[1 ,2-alpyridine-3- carboxamide

Prepared similarly to Example 43 imidazo[1 ,2-a]pyridine-3-carboxylic acid and trans-3-[(2,3- dichlorophenyl)oxy]cyclobutanamine hydrochloride.
LC-MS (System A) rt = 0.95 min, MH+ = 376 / 378
Example 68: A/-{ ra/7s-3-r(2,3-Dichlorophenyl)oxylcvclobutyl)pyrazolori ,5-alpyridine-3- carboxamide
 Prepared similarly to Example 49 from pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and trans-3- [(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride.
Prepared similarly to Example 49 from pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and trans-3- [(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride.
LC-MS (System A) rt = 1.13 min, MH+ = 376 / 378.
Example 69: /V-{ ra/7s-3-[(2,3-Dichlorophenyl)oxy1cvclobutyl)-7-(hvdroxymethyl)pyrazolo[1 ,5- a] pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 49 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and frans-3-[(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride..
LC-MS (System A) rt = 1.06 min, MH+ = 406 / 408.
Example 70: A/-{c/s-3-[(2,3-Dichlorophenyl)oxy1cvclobutyl)pyrazolo[1 ,5-alpyridine-3- carboxamide

Prepared similarly to Example 49 from pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and c/s-3- [(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride.
LC-MS (System A) rt = 1.06 min, MH+ = 376 / 378.
Example 71 : A/-{c/s-3-[(2,3-Dichlorophenyl)oxy1cvclobutyl)imidazo[1 ,2-alpyridine-3- carboxamide

Prepared similarly to Example 49 from imidazo[1 ,2-a]pyridine-3-carboxylic acid and c/s-3- [(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride.
LC-MS (System A) rt = 0.91 min, MH+ = 376 / 378.
Example 72: A/-{c/s-3-r(2,3-Dichlorophenyl)oxylcvclobutyl)-7-(hvdroxymethyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 49 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and c/'s-3-[(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride..
LC-MS (System A) rt = 1.03 min, MH+ = 406 / 408.
Example 73: A/-{ ra/7s-3-r(2,3-Dichlorophenyl)oxylcvclobutyl)-8-(hvdroxymethyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de
 Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and frans-3-[(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride..
Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and frans-3-[(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride..
LC-MS (System A) rt = 0.92 min, MH+ = 406 / 408. Example 74: A/-(c/s-3-{r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)-8-
(hvdroxymethyl)imidazoH ,2-alpyridine-3-carboxamide

Example 75: A/-( ra/7s-3-{r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)-8- (hvdroxymethyl)imidazoH ,2-alpyridine-3-carboxamide

Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and frans-3-{[4-(1 , 1-dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutanamine hydrochloride..
LC-MS (System A) rt = 1.1 1 min, MH+ = 430.
Example 76: A/-{c/s-3-r(2,3-Dichlorophenyl)oxylcvclobutyl)-8-(hvdroxymethyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de
 Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and c/'s-3-[(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride..
Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and c/'s-3-[(2,3-dichlorophenyl)oxy]cyclobutanamine hydrochloride..
LC-MS (System A) rt = 0.89 min, MH+ = 406 / 408. Example 77: A/-{ ra/7s-3-r(3-Fluoro-2-methylphenyl)oxylcvclobutyl)-8-
(hvdroxymethyl)imidazoH ,2-alpyridine-3-carboxamide

LC-MS (System A) rt = 0.87 min, MH+ = 370.
Example 78: A/-{ ra/7s-3-r(3,5-Difluorophenyl)oxylcvclobutyl)-8-(hvdroxymethyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and frans-3-[(3,5-difluorophenyl)oxy]cyclobutanamine hydrochloride
LC-MS (System A) rt = 0.84 min, MH+ = 374.
Example 79: A/-{ ra/7s-3-r(4-Chlorophenyl)oxylcvclobutyl)-8-(hvdroxymethyl)imidazori ,2- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and frans-3-[(4-chlorophenyl)oxy]cyclobutanamine hydrochloride
LC-MS (System A) rt = 0.86 min, MH+ = 372 / 374.
Example 80: /V-( ra/7s-3-{[2-Chloro-3-(trifluoromethyl)phenyl1oxy)cvclobutyl)-8-
(hvdroxymethyl)imidazo[1 ,2-alpyridine-3-carboxamide

Prepared similarly to Example 49 from 8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxylic acid and frans-3-{[2-chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutanamine hydrochloride LC-MS (System A) rt = 0.98 min, MH+ = 440 / 442.
Example 8 j A/-( ra/7s-3-{r4-Fluoro-3-(trifluoromethyl)phenylloxy)cvclobutyl)-8-
(hvdroxymethyl)imidazori ,2-alpyridine-3-carboxamide

Example 82: /V-( ra/7s-3-{r4-Fluoro-3-(trifluoromethyl)phenylloxy)cvclobutyl)pyrazolori ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 49 from pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and trans-3- {[4-fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutanamine hydrochloride
LC-MS (System B) rt = 2.94 min, MH+ = 394.
Example 83: /V-( ra/7s-3-{[2-Chloro-3-(trifluoromethyl)phenyl1oxy)cvclobutyl)pyrazolo[1 ,5- al pyri d i n e- 3- carboxa m i de

Prepared similarly to Example 49 from pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and trans-3- {[2-chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutanamine hydrochloride
LC-MS (System A) rt = 1.17 min, MH+ = 410 / 413.
Example 84: A/-( ra/7s-3-{r2-Chloro-3-(trifluoromethyl)phenylloxy)cvclobutyl)-7-
(hvdroxymethyl)pyrazolori ,5-alpyridine-3-carboxamide
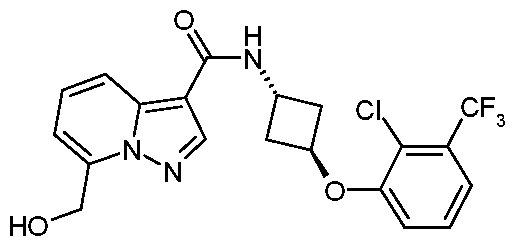
Prepared similarly to Example 49 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and frans-3-{[2-chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutanamine hydrochloride LC-MS (System A) rt = 1.1 1 min, MH+ = 440 / 442.
Example 85: A/-( ra/7s-3-{r4-Fluoro-3-(trifluoromethyl)phenylloxy)cvclobutyl)-7-
(hvdroxymethyl)pyrazoloH ,5-alpyridine-3-carboxamide

Prepared similarly to Example 49 from 7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxylic acid and frans-3-{[4-fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutanamine hydrochloride LC-MS (System A) rt = 1.07 min, MH+ = 424.
Example 86: /V-( ra/7s-3-{[4-(1 , 1-Dimethvlethvl)-3,5-difluorophenvl1oxv)cvclobutyl)-8- fluoroimidazo[1 ,2-a]pyridine-3-carboxamide

A mixture of frans-3-{[4-(1 , 1-dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutanamine hydrochloride (162 mg, 0.555 mmol) and DIPEA (0.194 mL, 1.1 10 mmol) was stirred at room temperature for 5min. 8-Fluoroimidazo[1 ,2-a]pyridine-3-carboxylic acid (100 mg, 0.555 mmol) (Ellanova Laboraties) and HATU (317 mg, 0.833 mmol) were added and the reaction mixture was left to stir at room temperature for 30 min. The reaction was concentrated in vacuo and the residue partitioned between EtOAc and water. The organic layer was dried using a hydrophobic frit and concentrated in vacuo. The residue was purified by MDAP HPLC (Method A) in three batches. The fractions which contained product were evaporated in vacuo to give the title compound as a white solid (85mg).
LC-MS (System A) rt = 1.33 min, MH+ = 418.
Example 87: A/-(c/s-3-{r4-(1 , 1-Dimethylethyl)-3,5-difluorophenylloxy)cvclobutyl)-8- fluoroimidazoH ,2-alpyridine-3-carboxamide

Prepared similarly to Example 86 from 8-fluoroimidazo[1 ,2-a]pyridine-3-carboxylic c/'s-3-{[4-fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutanamine hydrochloride
LC-MS (System A) rt = 1.32 min, MH+ = 418.
Biological Assays
Assays for the functional inhibition of TRPV1 Ion Channel by test compound using Capsaicin or Acid Stimulus challenge.
Compound Preparation
Compounds were dissolved in DMSO to 1 mM. An 11 point 4 fold serial dilution was prepared and 0.5ul dispensed into Greiner black clear bottom 384 well plates.
Preparation of recombinant HEK-293 cells expressing TRPV1 for assay
HEK-293 cells stably expressing mitochondrial targeted Aequorin were transfected with TRPV1 receptor bacmam at scale for cryopreservation in 1 ml vial aliquots. Cells can be stored at -140°C for up to 18 months. 18-20 hours before assay, cells were rapidly thawed in a water bath at 37°C and transferred to a 50ml Falcon tube. Cells were resuspended in 9mls of M1 'generic' media (DMEM/F12 with 10% dialysed FBS -Invitrogen 041-95750V) for every 1 ml cells and then centrifuged for 5min at l OOOrpm. The cell pellet was resuspended in ~10mls loading buffer (Tyrodes Base Buffer* + 0.1 % Pluronic Acid F68 solution + 0.1 % BSA) and the pH was adjusted to 7.4 for Capsaicin stimulus assay or 6.7 for Acid stimulus assay. Cell density was calculated using the Trypan Blue stain method and adjusted to 2.5 x 10e6 cells /ml using loading buffer. Coelentrazine (DiscoverX Cat. No 0-0084L - 500uM stock made in 100% ethanol) was added to a final concentration of 5uM and the falcon tube was covered in foil (to protect from light) and placed on a windmill rotator at room temperature for approximately 20 hours.
*Base Buffer = Sigma kit T2145 was dissolved in deionised water, 20ml_ HEPES solution (Sigma H0887) and 13.4ml_ of NaHC03 (Sigma S8761) and made up to 1 L.
After loading, a cell count was taken and cell density adjusted accordingly depending on assay stimulus.
Capsaicin Assay for TRPV1 receptor Antagonism
For Capsaicin stimulus assay cells were diluted to 1.25x10e5 cells/ml using dilution buffer (Tyrodes Base Buffer + 0.1 % Pluronic Acid F68 solution) at pH 7.4
To the compound plates, the following additions were made:
20ul dilution buffer at pH 7.4 followed by 20ul cells were added to the test compound plate and any agonist activity measured as luminescence AUC counts. The compound/cell mix was incubated for 15 minutes and then challenged with 20ul of a 4xEC50 concentration of Capsaicin (calculated on the day of assay), with concomitant luminescence detection.
AUC data was exported from the reader and data analysis was performed using 4 parameter logistic model, with data normalised to nominal high and low controls within plate. Each of Examples 1 to 87 had a plC50 of greater than 5.8 in this assay.
Each of Examples 1 , 3, 4, 5, 6, 7, 8, 9, 10, 1 1 13, 14, 15, 16, 17, 18, 19, 20, 21 , 24, 26, 28, 29, 32, 35, 36, 37, 38, 39, 40, 42, 43, 44, 45, 47, 48, 52, 56, 57, 58, 65, 66, 67, 68, 69, 71 , 72, 73, 74, 75, 76, 77, 78, 79, 80, 81 , 82, 83, 84, 85, 86, 87 namely
N-(frans-3-{[4-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-(frans-3-{[2-Bromo-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3-fluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-2-methylphenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[3-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-[frans-3-({3-[(Trifluoromethyl)oxy]phenyl}oxy)cyclobutyl]pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[3-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[3-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-{frans-3-[(3,4,5-Trifluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-2-fluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(4-Chloro-3-fluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(4-Chlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[2-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(2-Propylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-{frans-3-[(2,3-Dimethylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(4-Chloro-3,5-difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(2-Chloro-3-fluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(2-Chloro-3,5-difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-Methyl-2-(1-methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2,4-bis(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-[frans-3-({2-[(Trifluoromethyl)oxy]phenyl}oxy)cyclobutyl]pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(fraA7S-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-6-
(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7-
(methyloxy)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(4-chlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5- a]pyridine-3-carboxamide;
N-{c/'s-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-
3-carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{c/s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxarTiide;
N-{c/'s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{c/'s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de ;
N-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(4-Chlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8-(hydroxymethyl)imidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de ;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8-(hydroxymethyl)imidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de ;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-7-(hydroxymethyl)pyrazolo[1 ,5- a]pyridine-3-carboxamide;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-7-(hydroxymethyl)pyraz^ a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a]pyridine-3-carboxamide:
N-(c/s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de
had a plC50 of greater than 6.9 in the Capsaicin assay.
Each of Examples 3, 5, 6, 7, 17, 40, 42, 45, 65, 66, 67, 68, 69, 73, 74, 75, 77, 80, 81 , 82, 86, 87 namely
N-(frans-3-{[2-Bromo-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3-fluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-2-methylphenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-6- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyrid carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de ;
N-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8-(hydroxymethyl)imidazo[ a]pyridine-3-carboxamide;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy
a] py ri d i n e- 3- carboxa m i de ;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 -Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroirnidazo[1 ,2- a]pyridine-3-carboxamide:
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de
had a plC50 of greater than 7.9 in the Capsaicin assay
Example 3 had a plC50 of 8.0 in the Capsaicin assay
Example 5 had a plC50 of 8.0 in the Capsaicin assay
Example 6 had a plC50 of 8.2 in the Capsaicin assay
Example 7 had a plC50 of 8.1 in the Capsaicin assay
Example 17 had a pICso Of 8.4 in the Capsaicin assay
Example 40 had a pICso Of 8.7 in the Capsaicin assay
Example 42 had a pICso of 8.2 in the Capsaicin assay
Example 45 had a pICso of 8.2 in the Capsaicin assay
Example 65 had a pICso of 8.4 in the Capsaicin assay
Example 66 had a pICso of 8.4 in the Capsaicin assay
Example 67 had a pICso of 8.7 in the Capsaicin assay
Example 68 had a pICso of 8.2 in the Capsaicin assay
Example 69 had a pICso of 8.2 in the Capsaicin assay
Example 73 had a pICso of 8.8 in the Capsaicin assay
Example 74 had a pICso of 9.1 in the Capsaicin assay
Example 75 had a pICso of 8.0 in the Capsaicin assay
Example 77 had a plC50 of 8.3 in the Capsaicin assay
Example 80 had a plC50 of 8.1 in the Capsaicin assay
Example 81 had a plC50 of 8.2 in the Capsaicin assay
Example 82 had a plC50 of 8.0 in the Capsaicin assay
Example 86 had a plC50 of 8.7 in the Capsaicin assay
Example 87 had a plC50 of 8.9 in the Capsaicin assay
Acid Stimulus Assay for TRPV1 receptor Antagonism
For Acid stimulus assay, cells were diluted to 2.5x10e5 cells/ml using dilution buffer at pH 6.7. The cells and the compound plates were added to the Lumilux™ reader (Perkin Elmer) with on-board liquid handling.
To the compound plates, the following additions were made:
20ul dilution buffer at pH 6.7 followed by 20ul cells were added to the test compound plate. The compound/cell mix was incubated for 15 minutes and then challenged with 20ul of Acid Stimulus Buffer (NaCI 145mM 8.48g/L, KCL 2.5mM 0.18g/L, CaCI2 2mM 0.294g/L, MgCI2 1 mM 0.203g/L, Glucose 10mM 1.81 g/L, Sucrose 10mM 8.76g/L) + 30ul 1 M HCL for every 10ml Acid Stimulus Buffer (3mM), with concomitant luminescence detection.
AUC data was exported from the reader and data analysis was performed using 4 parameter logistic model, with data normalised to nominal high and low controls within plate
Each of Examples 1 to 87 had a plC50of greater than 6.0 in this acid stimulus assay.
Example 3 had a plC50 of 8.1 in the pH assay
Example 5 had a plC50 of 8.2 in the pH assay
Example 6 had a plC50 of 8.3 in the pH assay
Example 7 had a plC50 of 8.0 in the pH assay
Example 17 had a plC50 of 8.4 n the pH assay
Example 40 had a plC50 of 8.9 n the pH assay
Example 42 had a plC50 of 8.3 n the pH assay
Example 45 had a plC50 of 8.5 n the pH assay
Example 65 had a plC50 of 8.8 n the pH assay
Example 66 had a plC50 of 8.8 n the pH assay
Example 67 had a plC50 of 8.5 n the pH assay
Example 68 had a plC50 of 8.4 n the pH assay
Example 69 had a pICso of 8.7 in the PH assay
Example 73 had a pICso of 8.9 in the PH assay
Example 74 had a pICso of 9.2 in the PH assay
Example 75 had a pICso of 8.3 in the PH assay
Example 77 had a pICso of 8.6 in the PH assay
Example 80 had a pICso of 8.5 in the PH assay
Example 81 had a pICso of 8.6 in the PH assay
Example 84 had a pICso of 8.1 in the PH assay
Example 85 had a pICso of 8.1 in the PH assay
Example 86 had a pICso of 8.9 in the PH assay
Example 87 had a pICso of 9.3 in the PH assay.
Preparation of an aqueous pharmaceutical composition The following illustrates the preparation of the aqueous pharmaceutical compositions and use thereof in accordance with this invention and is to be considered illustrating and not limiting the scope of the disclosure in any way.
The aqueous pharmaceutical compositions of the invention may be prepared according to the following general method.
The isotonicity adjusting agent(s) is charged into a suitable mixing vessel containing purified water and dissolved with stirring. Preservative(s) is pre-dissolved in purified water in a separate vessel, optionally with heating, for example to 50 - 60 °C depending on the preservative chosen, to aid dissolution, and then added to the isotonicity adjusting agent(s) with continuous stirring. The suspending agent(s) is then charged into the mixing vessel and dispersed throughout the solution. The resulting suspending vehicle is allowed to hydrate for an appropriate period of time to ensure cross-linkage and gelation, which may take 60 minutes or longer.
In a separate mixing vessel, the wetting agent(s) is mixed with purified water which optionally may be heated, for example to about 50 - 60 °C as appropriate depending on the wetting agent(s) chosen, and stirred to dissolve. A slurry of the compound or a pharmaceutically acceptable salt thereof (alone or in combination with a further active ingredient) is then prepared by adding the resultant wetting agent(s) solution to the active compound(s), which may be particle size reduced for example micronised, and mixed prior to
homogenising/refining. Additionally, in a separate mixing vessel, additional preservative(s), if needed, may be diluted with purified water and stirred to mix.
Following the dispersion and gelation the slurry of active compound(s) is added to the mixing vessel containing the suspending agent and dispersed with stirring. Following the addition of the slurry of active compound(s), any additional preservative may be added to the bulk suspension and dispersed with continuous stirring. Finally, the suspension is made to its final mass by adding water and stirred.
Claims
1 A compound of formu
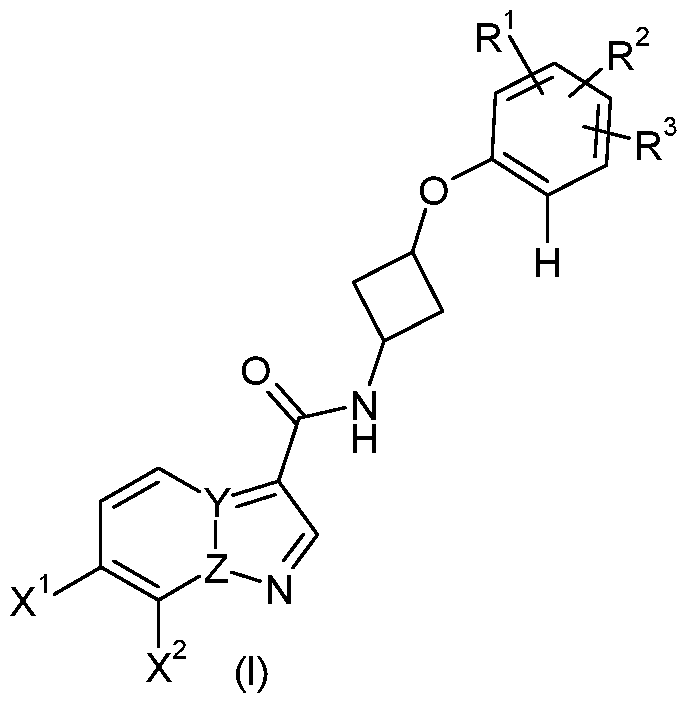
wherein
X1 represents a hydrogen atom, or a CH2OH group
X2 represents a hydrogen atom, a fluorine atom, a OCH3 group or a CH2OH group, and at least one of X1 and X2 is hydrogen
Y represents a carbon atom and Z represents a nitrogen atom or
Y represents a nitrogen atom and Z represents a carbon atom;
R1 represents a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group, and R2 and R3 are each independently selected from a hydrogen atom, a halogen atom, a C1-4 alkyl group, a trifluoromethyl group or a trifluoromethoxy group;
or a pharmaceutically acceptable salt or solvate thereof 2 A compound according to claim 1 or a pharmaceutically acceptable salt or solvate thereof wherein X1 represents a hydrogen atom.
3 A compound according to claim 1 claim 2 or a pharmaceutically acceptable salt or solvate thereof wherein X2 represents a hydrogen atom or a CH2OH group.
4 A compound according to any one of claims 1 to 3 or a pharmaceutically acceptable salt or solvate thereof wherein Y represents C and Z represents N
5 A compound according to any one of claims 1 to 3 or a pharmaceutically acceptable salt or solvate thereof wherein Y represents N and Z represents C
6 A compound according to any one of claims 1 to 5 or a pharmaceutically acceptable salt or solvate thereof wherein R1 represents a 1 , 1-dimethylethyl group and R2 and R3
independently represent a fluorine atom or a hydrogen atom.
A compound according to claim 6 or a pharmaceutically acceptable salt or solvate thereof wherein R1 represents a 1 , 1-dimethylethyl group at the para position relative to the ether group, R2 represents a fluorine atom at a meta position relative to the ether group, and R3 represents a fluorine or a hydrogen atom at the other meta position relative to the ether group.
A compound according to claim 7 or a pharmaceutically acceptable salt or solvate thereof wherein both R2 and R3 each represent a fluorine atom at the meta positions relative to the ether group.
A compound according to any one of claims 1 to 5 or a pharmaceutically acceptable salt or solvate thereof wherein R1 represents a methyl group at an ortho position relative to the ether group, and R2 represents a fluorine atom at a meta position relative to the ether group and R3 represents a hydrogen atom.
A compound according to claim 9 which is
N-(frans-3-{[4-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Bromo-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3-fluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-2-methylphenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-4-(1 , 1-dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[3-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-[frans-3-({3-[(Trifluoromethyl)oxy]phenyl}oxy)cyclobutyl]pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[3-(Trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[3-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-(frans-3-{[4-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-{frans-3-[(3,4,5-Trifluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5- a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-2-fluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(4-Chloro-3-fluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(4-Chlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[2-(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(2-Propylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-{frans-3-[(2,3-Dimethylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(4-Chloro-3,5-difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(2-Chloro-3-fluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(2-Chloro-3,5-difluorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[4-Methyl-2-(1-methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2,4-bis(1-Methylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-[frans-3-({2-[(Trifluoromethyl)oxy]phenyl}oxy)cyclobutyl]pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-(1 , 1-Dimethylethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-6- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (methyloxy)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine- 3-carboxamide;
N-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(4-chlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide; N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5- a] py ri d i n e- 3- carboxa m i de ;
N-{c/'s-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5- a]pyridine-3-carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxamide; N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}pyrazolo[1 ,5-a]pyridine-3-carboxamide; N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine- 3-carboxamide;
N-{c/s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}imidazo[1 ,2-a]pyridine-3-carboxarTiide;
N-{c/'s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-7-(hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-{frans-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-(c/'s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-{c/'s-3-[(2,3-Dichlorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(3-Fluoro-2-methylphenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2- a]pyridine- 3-carboxamide;
N-{frans-3-[(3,5-Difluorophenyl)oxy]cyclobutyl}-8-(hydroxymethyl)imidazo[1 ,2-a]pyridine-3- carboxamide;
N-{frans-3-[(4-Chlorophenyl)oxy]cyclob
carboxamide;
N-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-8- (hydroxymethyl)imidazo[1 ,2-a]pyridine-3-carboxamide;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)pyrazolo[1 ,5-a]pyridine-3- carboxamide;
N-(frans-3-{[2-Chloro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-Fluoro-3-(trifluoromethyl)phenyl]oxy}cyclobutyl)-7- (hydroxymethyl)pyrazolo[1 ,5-a]pyridine-3-carboxamide;
N-(frans-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a]pyridine-3-carboxamide:
N-(c/s-3-{[4-(1 , 1-Dimethylethyl)-3,5-difluorophenyl]oxy}cyclobutyl)-8-fluoroimidazo[1 ,2- a] py ri d i n e- 3- carboxa m i de
or a pharmaceutically acceptable salt or solvate thereof.
11 A pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to any one of claims 1 to 10 and one or more pharmaceutically acceptable carriers or excipients.
12 A compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to any one of claims 1 to 10 for use in therapy.
13 A compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to any one of claims 1 to 10, for use in the treatment of a condition for which a TRPV1 antagonist is indicated.
14 A method for the treatment or prophylaxis of disorders in which antagonism of TRPV1 is beneficial in a human comprising administering to the human in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to any one of claims 1 to 10
15 A pharmaceutical composition for the treatment or prophylaxis of a disorder in which antagonism of TRPV1 is beneficial comprising a compound as defined in any one of claims 1 to 12 or a pharmaceutically acceptable salt or solvate thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US41753110P | 2010-11-29 | 2010-11-29 | |
| US61/417,531 | 2010-11-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012072512A1 true WO2012072512A1 (en) | 2012-06-07 |
Family
ID=45092346
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2011/071035 WO2012072512A1 (en) | 2010-11-29 | 2011-11-25 | N-cyclobutyl-imidazopyridine or -pyrazolopyridine carboxamides as trpv1 antagonists |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2012072512A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015018814A1 (en) * | 2013-08-08 | 2015-02-12 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-a]-pyridine-3-carboxamides and use thereof |
| WO2016087342A1 (en) | 2014-12-02 | 2016-06-09 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-a]pyridines and imidazo[1,2-a]pyrazines and their use |
| WO2016124565A1 (en) | 2015-02-05 | 2016-08-11 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-a]-pyridine-3-carboxamides and use thereof |
Citations (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0347000A2 (en) | 1988-06-17 | 1989-12-20 | The Procter & Gamble Company | The use of vanilloids for the manufacture of a medicament for treating herpes simplex infections |
| GB2226313A (en) | 1988-12-23 | 1990-06-27 | Sandoz Products Ltd | Capsaicin derivatives |
| EP0401903A2 (en) | 1989-06-08 | 1990-12-12 | The Procter & Gamble Company | The use of vanilloids for the treatment of respiratory diseases or disorders |
| WO1992009285A1 (en) | 1990-11-27 | 1992-06-11 | The Procter & Gamble Company | Combinations of vanilloid compounds and phosphonate antiviral compounds for treatment of herpes infections |
| WO2002008221A2 (en) | 2000-07-20 | 2002-01-31 | Neurogen Corporation | Capsaicin receptor ligands |
| WO2002016318A1 (en) | 2000-08-21 | 2002-02-28 | Pacific Corporation | Novel thiourea derivatives and the pharmaceutical compositions containing the same |
| WO2002016317A1 (en) | 2000-08-21 | 2002-02-28 | Pacific Corporation | Novel thiocarbamic acid derivatives and the pharmaceutical compositions containing the same |
| WO2002016319A1 (en) | 2000-08-21 | 2002-02-28 | Pacific Corporation | Novel thiourea compounds and the pharmaceutical compositions containing the same |
| WO2003022809A2 (en) | 2001-09-13 | 2003-03-20 | Smithkline Beecham P.L.C. | Urea-compounds active as vanilloid receptor antagonists for the treatment of pain |
| WO2005044354A1 (en) | 2003-11-03 | 2005-05-19 | Glaxo Group Limited | A fluid dispensing device |
| WO2005105798A1 (en) * | 2004-04-30 | 2005-11-10 | Grünenthal GmbH | Substituted imidazo[1,2-a]pyridine compounds and drugs containing substituted imidazo[1,2-a]pyridine compounds |
| WO2005116009A1 (en) | 2004-05-18 | 2005-12-08 | Schering Corporation | Substituted 2-quinolyl-oxazoles useful as pde4 inhibitors |
| WO2007062332A2 (en) | 2005-11-18 | 2007-05-31 | Glaxo Group Limited | Novel intermediates and their use |
| WO2007122156A1 (en) | 2006-04-20 | 2007-11-01 | Glaxo Group Limited | 2-substituted 4-benzylphthalazinone derivatives as histamine h1 and h3 antagonists |
| WO2008074803A2 (en) | 2006-12-20 | 2008-06-26 | Glaxo Group Limited | 4-benzyl-l ( 2h) -phthalazinones as h1 receptor antagonists |
| WO2008113559A2 (en) | 2007-03-21 | 2008-09-25 | Schwarz Pharma Ag | Indolizines and aza-analog derivatives thereof as cns active compounds |
| WO2009021965A2 (en) | 2007-08-15 | 2009-02-19 | Glaxo Group Limited | Substituted quinoline derivatives as h1 receptor antagonists |
| WO2010011914A1 (en) * | 2008-07-25 | 2010-01-28 | Smithkline Beecham Corporation | Trpv4 antagonists |
| WO2010026129A1 (en) | 2008-09-02 | 2010-03-11 | Glaxo Group Limited | The trpvl antagonist sb-705498 for treating rhinitis |
| WO2010026128A1 (en) | 2008-09-02 | 2010-03-11 | Glaxo Group Limited | N-(3-methyl-5-isoquinolinyl)-n'-((3r)-1-[5-(trifluoromethyl)-2-pyridinyl]-3-pyrrolidinyl) urea for the treatment of rhinitis |
| WO2010070452A1 (en) * | 2008-12-15 | 2010-06-24 | Glenmark Pharmaceuticals, S.A. | Fused imidazole carboxamides as trpv3 modulators |
-
2011
- 2011-11-25 WO PCT/EP2011/071035 patent/WO2012072512A1/en active Application Filing
Patent Citations (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0347000A2 (en) | 1988-06-17 | 1989-12-20 | The Procter & Gamble Company | The use of vanilloids for the manufacture of a medicament for treating herpes simplex infections |
| GB2226313A (en) | 1988-12-23 | 1990-06-27 | Sandoz Products Ltd | Capsaicin derivatives |
| EP0401903A2 (en) | 1989-06-08 | 1990-12-12 | The Procter & Gamble Company | The use of vanilloids for the treatment of respiratory diseases or disorders |
| WO1992009285A1 (en) | 1990-11-27 | 1992-06-11 | The Procter & Gamble Company | Combinations of vanilloid compounds and phosphonate antiviral compounds for treatment of herpes infections |
| WO2002008221A2 (en) | 2000-07-20 | 2002-01-31 | Neurogen Corporation | Capsaicin receptor ligands |
| WO2002016318A1 (en) | 2000-08-21 | 2002-02-28 | Pacific Corporation | Novel thiourea derivatives and the pharmaceutical compositions containing the same |
| WO2002016317A1 (en) | 2000-08-21 | 2002-02-28 | Pacific Corporation | Novel thiocarbamic acid derivatives and the pharmaceutical compositions containing the same |
| WO2002016319A1 (en) | 2000-08-21 | 2002-02-28 | Pacific Corporation | Novel thiourea compounds and the pharmaceutical compositions containing the same |
| WO2003022809A2 (en) | 2001-09-13 | 2003-03-20 | Smithkline Beecham P.L.C. | Urea-compounds active as vanilloid receptor antagonists for the treatment of pain |
| WO2005044354A1 (en) | 2003-11-03 | 2005-05-19 | Glaxo Group Limited | A fluid dispensing device |
| WO2005105798A1 (en) * | 2004-04-30 | 2005-11-10 | Grünenthal GmbH | Substituted imidazo[1,2-a]pyridine compounds and drugs containing substituted imidazo[1,2-a]pyridine compounds |
| WO2005116009A1 (en) | 2004-05-18 | 2005-12-08 | Schering Corporation | Substituted 2-quinolyl-oxazoles useful as pde4 inhibitors |
| WO2007062332A2 (en) | 2005-11-18 | 2007-05-31 | Glaxo Group Limited | Novel intermediates and their use |
| WO2007122156A1 (en) | 2006-04-20 | 2007-11-01 | Glaxo Group Limited | 2-substituted 4-benzylphthalazinone derivatives as histamine h1 and h3 antagonists |
| WO2008074803A2 (en) | 2006-12-20 | 2008-06-26 | Glaxo Group Limited | 4-benzyl-l ( 2h) -phthalazinones as h1 receptor antagonists |
| WO2008113559A2 (en) | 2007-03-21 | 2008-09-25 | Schwarz Pharma Ag | Indolizines and aza-analog derivatives thereof as cns active compounds |
| WO2009021965A2 (en) | 2007-08-15 | 2009-02-19 | Glaxo Group Limited | Substituted quinoline derivatives as h1 receptor antagonists |
| WO2010011914A1 (en) * | 2008-07-25 | 2010-01-28 | Smithkline Beecham Corporation | Trpv4 antagonists |
| WO2010026129A1 (en) | 2008-09-02 | 2010-03-11 | Glaxo Group Limited | The trpvl antagonist sb-705498 for treating rhinitis |
| WO2010026128A1 (en) | 2008-09-02 | 2010-03-11 | Glaxo Group Limited | N-(3-methyl-5-isoquinolinyl)-n'-((3r)-1-[5-(trifluoromethyl)-2-pyridinyl]-3-pyrrolidinyl) urea for the treatment of rhinitis |
| WO2010070452A1 (en) * | 2008-12-15 | 2010-06-24 | Glenmark Pharmaceuticals, S.A. | Fused imidazole carboxamides as trpv3 modulators |
Non-Patent Citations (16)
| Title |
|---|
| BERGE ET AL., J. PHARM. SCI, vol. 66, 1977, pages 1 - 19 |
| BERGE, J. PHARM. SCI., vol. 66, 1977, pages 1 - 19 |
| CATERINA MJ; SCHUMACHER MA; TOMINAGA M; ROSEN TA; LEVINE JD; JULIUS D, NATURE, vol. 389, 1997, pages 816 - 824 |
| J. MARCH: "Advanced Organic Chemistry, 6th Edition", 2007, WILEYBLACKWELL |
| J. MED. CHEM., vol. 36, 1993, pages 2595 |
| KHAIRATKAR-JOSHI N; SZALLASI A, TRENDS IN MOLECULAR MEDICINE, 2008 |
| MOTTER AL; AHERN GP, FEBS LETTERS, vol. 582, 2008, pages 2257 - 2262 |
| ORG. PROC. RES. DEV., vol. 13, no. 2, 2009, pages 263 |
| PAN; WANG; YANG; ZHANG; REINACH: "TRPV1 Activation is Required for Hypertonicity Stimulated Inflammatory Cytokine Release in Human Corneal Epithelial Cells", MANUSCRIPT IOVS, 2010, pages 10 - 5801 |
| RASAVI ET AL., CELL, vol. 127, 2006, pages 1123 - 1135 |
| SURI; SZALLASI A: "The emerging role of TRPV1 in diabetes and obesity", TRENDS IN PHARM SCI, 2007 |
| SZALLASI ET AL., NATURE REVIEWS DRUG DISCOVERY, vol. 6, 2007, pages 357 - 372 |
| SZALLASI; BLUMBERG, THE AMERICAN SOCIETY FOR PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 51, no. 2, 1999 |
| T. W. GREENE: "Protective Groups in Organic Synthesis; 4th Edition,", 2006, J. WILEY AND SONS |
| TETRAHEDRON, vol. 53, 1997, pages 4791 |
| TROST B.M. AND FLEMING: "Comprehensive Organic Synthesis", 1991, PERGAMON PRESS |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015018814A1 (en) * | 2013-08-08 | 2015-02-12 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-a]-pyridine-3-carboxamides and use thereof |
| US9422285B2 (en) | 2013-08-08 | 2016-08-23 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-A]-pyridine-3-carboxamides and use thereof |
| JP2016529249A (en) * | 2013-08-08 | 2016-09-23 | バイエル・ファルマ・アクティエンゲゼルシャフト | Substituted pyrazolo [1,5-a] pyridine-3-carboxamide and uses thereof |
| CN105992765A (en) * | 2013-08-08 | 2016-10-05 | 拜耳制药股份公司 | Substituted pyrazolo[1,5-a]-pyridine-3-carboxamides and use thereof |
| WO2016087342A1 (en) | 2014-12-02 | 2016-06-09 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-a]pyridines and imidazo[1,2-a]pyrazines and their use |
| US10174021B2 (en) | 2014-12-02 | 2019-01-08 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-A]pyridines and their use |
| WO2016124565A1 (en) | 2015-02-05 | 2016-08-11 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-a]-pyridine-3-carboxamides and use thereof |
| US10214526B2 (en) | 2015-02-05 | 2019-02-26 | Bayer Pharma Aktiengesellschaft | Substituted pyrazolo[1,5-a]-pyridine-3-carboxamides and use thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8895544B2 (en) | Indazoles | |
| CA2763960C (en) | Bicyclic and tricyclic compounds as kat ii inhibitors | |
| JP2024045225A (en) | Heterocyclic compound, intermediate, preparation method therefor and application therefor | |
| WO2017066014A1 (en) | Bruton's tyrosine kinase inhibitors | |
| AU2005262330B2 (en) | Piperidine derivatives as NK1 antagonists | |
| AU2014230816A1 (en) | Furopyridines as bromodomain inhibitors | |
| EP3209296B1 (en) | Ethyl n-boc piperidinyl pyrazolo pyridones as janus kinase inhibitors | |
| AU2002230392A1 (en) | AZA-and polyaza-naphthalenyl ketones useful as HIV integrase inhibitors | |
| AU2010207494A1 (en) | New adenosine receptor ligands and uses thereof | |
| MX2014006724A (en) | Kinase inhibitors. | |
| AU2005275213A1 (en) | Hydantoin derivatives for the treatment of inflammatory disorders | |
| JP2012506396A (en) | Pyrimidine- and triazine-sulfonamide derivatives as bradykinin B1 receptor (B1R) inhibitors for the treatment of pain | |
| WO2011008597A1 (en) | Dihydroimidazoisoquinoline derivatives useful as pde10 inhibitors | |
| CA2579059A1 (en) | Chemical compounds | |
| AU2019353144B2 (en) | Compounds and compositions for treating conditions associated with APJ receptor activity | |
| US8754101B2 (en) | N-cyclobutyl-imidazopyridine-methylamine as TRPV1 antagonists | |
| EP2731932B1 (en) | Aminomethyl quinolone compounds | |
| CN112752757B (en) | Tyrosine amide derivatives as RHO-kinase inhibitors | |
| WO2014146249A1 (en) | Geminally substituted cyanoethylpyrazolo pyridones as janus kinase inhibitors | |
| WO2012072512A1 (en) | N-cyclobutyl-imidazopyridine or -pyrazolopyridine carboxamides as trpv1 antagonists | |
| US20070185079A1 (en) | Therapeutic agents I | |
| TW202328102A (en) | Pyridazinyl amino derivatives as alk5 inhibitors | |
| WO2012045729A1 (en) | Imidazo [1, 2 -a] pyridine and pyrazolo [1, 5 -a] pyridine derivatives as trpv1 antagonists | |
| EP1866305A1 (en) | 1,7-naphthyridines as pde4 inhibitors | |
| AU2013280599A1 (en) | Sulfonamide derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11790765 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11790765 Country of ref document: EP Kind code of ref document: A1 |