WO2012006552A1 - Combinations of kinase inhibitors for the treatment of cancer - Google Patents
Combinations of kinase inhibitors for the treatment of cancer Download PDFInfo
- Publication number
- WO2012006552A1 WO2012006552A1 PCT/US2011/043401 US2011043401W WO2012006552A1 WO 2012006552 A1 WO2012006552 A1 WO 2012006552A1 US 2011043401 W US2011043401 W US 2011043401W WO 2012006552 A1 WO2012006552 A1 WO 2012006552A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nhc
- cancer
- her2
- compound
- carcinoma
- Prior art date
Links
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 116
- 201000011510 cancer Diseases 0.000 title claims abstract description 71
- 238000011282 treatment Methods 0.000 title claims description 33
- 229940043355 kinase inhibitor Drugs 0.000 title description 3
- 239000003757 phosphotransferase inhibitor Substances 0.000 title description 3
- 238000000034 method Methods 0.000 claims abstract description 124
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 claims abstract description 116
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 claims abstract description 116
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 claims abstract description 115
- 150000001875 compounds Chemical class 0.000 claims abstract description 115
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 claims abstract description 113
- 239000003112 inhibitor Substances 0.000 claims abstract description 72
- 210000004027 cell Anatomy 0.000 claims description 170
- 108020004459 Small interfering RNA Proteins 0.000 claims description 132
- 150000007523 nucleic acids Chemical class 0.000 claims description 84
- 102000039446 nucleic acids Human genes 0.000 claims description 77
- 108020004707 nucleic acids Proteins 0.000 claims description 77
- 229940126062 Compound A Drugs 0.000 claims description 64
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 claims description 64
- 125000003729 nucleotide group Chemical group 0.000 claims description 61
- 239000002773 nucleotide Substances 0.000 claims description 58
- 108020004999 messenger RNA Proteins 0.000 claims description 50
- 230000000692 anti-sense effect Effects 0.000 claims description 49
- 239000002136 L01XE07 - Lapatinib Substances 0.000 claims description 28
- 230000000295 complement effect Effects 0.000 claims description 28
- 229960004891 lapatinib Drugs 0.000 claims description 27
- 108091027967 Small hairpin RNA Proteins 0.000 claims description 26
- 230000014509 gene expression Effects 0.000 claims description 26
- 239000013598 vector Substances 0.000 claims description 23
- 206010006187 Breast cancer Diseases 0.000 claims description 20
- 208000009956 adenocarcinoma Diseases 0.000 claims description 20
- 230000000694 effects Effects 0.000 claims description 20
- 239000004055 small Interfering RNA Substances 0.000 claims description 20
- 208000026310 Breast neoplasm Diseases 0.000 claims description 19
- 229960000575 trastuzumab Drugs 0.000 claims description 18
- 206010025323 Lymphomas Diseases 0.000 claims description 14
- 206010039491 Sarcoma Diseases 0.000 claims description 14
- 239000002299 complementary DNA Substances 0.000 claims description 12
- 239000012634 fragment Substances 0.000 claims description 12
- 102000040430 polynucleotide Human genes 0.000 claims description 12
- 108091033319 polynucleotide Proteins 0.000 claims description 12
- 239000002157 polynucleotide Substances 0.000 claims description 12
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 11
- 208000005017 glioblastoma Diseases 0.000 claims description 11
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 11
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 11
- 201000009030 Carcinoma Diseases 0.000 claims description 10
- 206010033128 Ovarian cancer Diseases 0.000 claims description 10
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 10
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 10
- 108091070501 miRNA Proteins 0.000 claims description 9
- 239000002679 microRNA Substances 0.000 claims description 9
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 8
- 206010024612 Lipoma Diseases 0.000 claims description 8
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 8
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 8
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 8
- 201000001441 melanoma Diseases 0.000 claims description 8
- 206010060862 Prostate cancer Diseases 0.000 claims description 7
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 6
- 206010009944 Colon cancer Diseases 0.000 claims description 6
- 206010014733 Endometrial cancer Diseases 0.000 claims description 6
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 6
- 201000008808 Fibrosarcoma Diseases 0.000 claims description 6
- 206010043276 Teratoma Diseases 0.000 claims description 6
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 6
- 210000004369 blood Anatomy 0.000 claims description 6
- 239000008280 blood Substances 0.000 claims description 6
- 201000010881 cervical cancer Diseases 0.000 claims description 6
- 206010016629 fibroma Diseases 0.000 claims description 6
- 201000011066 hemangioma Diseases 0.000 claims description 6
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 6
- 230000001225 therapeutic effect Effects 0.000 claims description 6
- 208000015634 Rectal Neoplasms Diseases 0.000 claims description 5
- 208000029742 colonic neoplasm Diseases 0.000 claims description 5
- 206010017758 gastric cancer Diseases 0.000 claims description 5
- 206010038038 rectal cancer Diseases 0.000 claims description 5
- 201000001275 rectum cancer Diseases 0.000 claims description 5
- 201000003076 Angiosarcoma Diseases 0.000 claims description 4
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 4
- 208000032612 Glial tumor Diseases 0.000 claims description 4
- 206010018338 Glioma Diseases 0.000 claims description 4
- 208000001258 Hemangiosarcoma Diseases 0.000 claims description 4
- 208000018142 Leiomyosarcoma Diseases 0.000 claims description 4
- 208000034578 Multiple myelomas Diseases 0.000 claims description 4
- 206010041067 Small cell lung cancer Diseases 0.000 claims description 4
- 208000033781 Thyroid carcinoma Diseases 0.000 claims description 4
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 4
- 208000008383 Wilms tumor Diseases 0.000 claims description 4
- 208000002458 carcinoid tumor Diseases 0.000 claims description 4
- 208000010749 gastric carcinoma Diseases 0.000 claims description 4
- 210000003734 kidney Anatomy 0.000 claims description 4
- 201000010260 leiomyoma Diseases 0.000 claims description 4
- 206010027191 meningioma Diseases 0.000 claims description 4
- 201000008968 osteosarcoma Diseases 0.000 claims description 4
- 210000001672 ovary Anatomy 0.000 claims description 4
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 4
- 201000000498 stomach carcinoma Diseases 0.000 claims description 4
- 201000002510 thyroid cancer Diseases 0.000 claims description 4
- 208000013077 thyroid gland carcinoma Diseases 0.000 claims description 4
- 206010007279 Carcinoid tumour of the gastrointestinal tract Diseases 0.000 claims description 3
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 claims description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 3
- 230000037396 body weight Effects 0.000 claims description 3
- 208000019065 cervical carcinoma Diseases 0.000 claims description 3
- 230000001684 chronic effect Effects 0.000 claims description 3
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 claims description 3
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 claims description 3
- 210000004185 liver Anatomy 0.000 claims description 3
- 201000005202 lung cancer Diseases 0.000 claims description 3
- 208000020816 lung neoplasm Diseases 0.000 claims description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 3
- 201000002528 pancreatic cancer Diseases 0.000 claims description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 3
- 229960002087 pertuzumab Drugs 0.000 claims description 3
- 201000001514 prostate carcinoma Diseases 0.000 claims description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 2
- 206010001233 Adenoma benign Diseases 0.000 claims description 2
- 206010003571 Astrocytoma Diseases 0.000 claims description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 206010004146 Basal cell carcinoma Diseases 0.000 claims description 2
- 206010073106 Bone giant cell tumour malignant Diseases 0.000 claims description 2
- 208000009458 Carcinoma in Situ Diseases 0.000 claims description 2
- 206010008263 Cervical dysplasia Diseases 0.000 claims description 2
- 201000005262 Chondroma Diseases 0.000 claims description 2
- 208000005243 Chondrosarcoma Diseases 0.000 claims description 2
- 201000009047 Chordoma Diseases 0.000 claims description 2
- 208000006332 Choriocarcinoma Diseases 0.000 claims description 2
- 206010048832 Colon adenoma Diseases 0.000 claims description 2
- 206010010356 Congenital anomaly Diseases 0.000 claims description 2
- 208000007033 Dysgerminoma Diseases 0.000 claims description 2
- 208000000471 Dysplastic Nevus Syndrome Diseases 0.000 claims description 2
- 201000009051 Embryonal Carcinoma Diseases 0.000 claims description 2
- 206010014967 Ependymoma Diseases 0.000 claims description 2
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 2
- 208000007659 Fibroadenoma Diseases 0.000 claims description 2
- 206010053717 Fibrous histiocytoma Diseases 0.000 claims description 2
- 208000000527 Germinoma Diseases 0.000 claims description 2
- 208000007569 Giant Cell Tumors Diseases 0.000 claims description 2
- 201000005409 Gliomatosis cerebri Diseases 0.000 claims description 2
- 206010018404 Glucagonoma Diseases 0.000 claims description 2
- 206010018691 Granuloma Diseases 0.000 claims description 2
- 208000002927 Hamartoma Diseases 0.000 claims description 2
- 206010019629 Hepatic adenoma Diseases 0.000 claims description 2
- 208000017604 Hodgkin disease Diseases 0.000 claims description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 2
- 208000005726 Inflammatory Breast Neoplasms Diseases 0.000 claims description 2
- 206010021980 Inflammatory carcinoma of the breast Diseases 0.000 claims description 2
- 208000005045 Interdigitating dendritic cell sarcoma Diseases 0.000 claims description 2
- 208000037396 Intraductal Noninfiltrating Carcinoma Diseases 0.000 claims description 2
- 206010073094 Intraductal proliferative breast lesion Diseases 0.000 claims description 2
- 208000002260 Keloid Diseases 0.000 claims description 2
- 208000002404 Liver Cell Adenoma Diseases 0.000 claims description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 2
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 claims description 2
- 208000007054 Medullary Carcinoma Diseases 0.000 claims description 2
- 208000000172 Medulloblastoma Diseases 0.000 claims description 2
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 2
- 208000014767 Myeloproliferative disease Diseases 0.000 claims description 2
- 206010029260 Neuroblastoma Diseases 0.000 claims description 2
- 201000004404 Neurofibroma Diseases 0.000 claims description 2
- 201000010133 Oligodendroglioma Diseases 0.000 claims description 2
- 208000000035 Osteochondroma Diseases 0.000 claims description 2
- 208000007641 Pinealoma Diseases 0.000 claims description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 2
- 201000004681 Psoriasis Diseases 0.000 claims description 2
- 201000000582 Retinoblastoma Diseases 0.000 claims description 2
- 208000005678 Rhabdomyoma Diseases 0.000 claims description 2
- 201000010208 Seminoma Diseases 0.000 claims description 2
- 208000000097 Sertoli-Leydig cell tumor Diseases 0.000 claims description 2
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 claims description 2
- 208000009311 VIPoma Diseases 0.000 claims description 2
- 206010048214 Xanthoma Diseases 0.000 claims description 2
- 206010048215 Xanthomatosis Diseases 0.000 claims description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 claims description 2
- 230000001154 acute effect Effects 0.000 claims description 2
- 208000002718 adenomatoid tumor Diseases 0.000 claims description 2
- 210000004100 adrenal gland Anatomy 0.000 claims description 2
- 208000001119 benign fibrous histiocytoma Diseases 0.000 claims description 2
- 210000000988 bone and bone Anatomy 0.000 claims description 2
- 201000009480 botryoid rhabdomyosarcoma Diseases 0.000 claims description 2
- 210000004556 brain Anatomy 0.000 claims description 2
- 210000000481 breast Anatomy 0.000 claims description 2
- 201000003149 breast fibroadenoma Diseases 0.000 claims description 2
- 208000003362 bronchogenic carcinoma Diseases 0.000 claims description 2
- 201000002143 bronchus adenoma Diseases 0.000 claims description 2
- 230000000747 cardiac effect Effects 0.000 claims description 2
- 210000003679 cervix uteri Anatomy 0.000 claims description 2
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 2
- 201000005217 chondroblastoma Diseases 0.000 claims description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 208000009060 clear cell adenocarcinoma Diseases 0.000 claims description 2
- 201000010305 cutaneous fibrous histiocytoma Diseases 0.000 claims description 2
- 208000028715 ductal breast carcinoma in situ Diseases 0.000 claims description 2
- 201000007273 ductal carcinoma in situ Diseases 0.000 claims description 2
- 201000009409 embryonal rhabdomyosarcoma Diseases 0.000 claims description 2
- 201000003914 endometrial carcinoma Diseases 0.000 claims description 2
- 210000003238 esophagus Anatomy 0.000 claims description 2
- 230000002496 gastric effect Effects 0.000 claims description 2
- 208000015419 gastrin-producing neuroendocrine tumor Diseases 0.000 claims description 2
- 201000000052 gastrinoma Diseases 0.000 claims description 2
- 201000003115 germ cell cancer Diseases 0.000 claims description 2
- 230000002489 hematologic effect Effects 0.000 claims description 2
- 208000006359 hepatoblastoma Diseases 0.000 claims description 2
- 201000002735 hepatocellular adenoma Diseases 0.000 claims description 2
- 201000004933 in situ carcinoma Diseases 0.000 claims description 2
- 201000004653 inflammatory breast carcinoma Diseases 0.000 claims description 2
- 206010022498 insulinoma Diseases 0.000 claims description 2
- 210000002570 interstitial cell Anatomy 0.000 claims description 2
- 206010073095 invasive ductal breast carcinoma Diseases 0.000 claims description 2
- 201000010985 invasive ductal carcinoma Diseases 0.000 claims description 2
- 206010073096 invasive lobular breast carcinoma Diseases 0.000 claims description 2
- 210000001117 keloid Anatomy 0.000 claims description 2
- 208000022013 kidney Wilms tumor Diseases 0.000 claims description 2
- 208000032839 leukemia Diseases 0.000 claims description 2
- 206010024627 liposarcoma Diseases 0.000 claims description 2
- 210000004072 lung Anatomy 0.000 claims description 2
- 201000004593 malignant giant cell tumor Diseases 0.000 claims description 2
- 201000000289 malignant teratoma Diseases 0.000 claims description 2
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 claims description 2
- 210000002418 meninge Anatomy 0.000 claims description 2
- 201000010879 mucinous adenocarcinoma Diseases 0.000 claims description 2
- 208000010492 mucinous cystadenocarcinoma Diseases 0.000 claims description 2
- 208000025113 myeloid leukemia Diseases 0.000 claims description 2
- 208000009091 myxoma Diseases 0.000 claims description 2
- 201000008026 nephroblastoma Diseases 0.000 claims description 2
- 210000000653 nervous system Anatomy 0.000 claims description 2
- 208000007538 neurilemmoma Diseases 0.000 claims description 2
- 201000004662 neurofibroma of spinal cord Diseases 0.000 claims description 2
- 208000004649 neutrophil actin dysfunction Diseases 0.000 claims description 2
- 208000003388 osteoid osteoma Diseases 0.000 claims description 2
- 208000008798 osteoma Diseases 0.000 claims description 2
- 210000003101 oviduct Anatomy 0.000 claims description 2
- 210000000496 pancreas Anatomy 0.000 claims description 2
- 208000021255 pancreatic insulinoma Diseases 0.000 claims description 2
- 208000024724 pineal body neoplasm Diseases 0.000 claims description 2
- 201000004123 pineal gland cancer Diseases 0.000 claims description 2
- 210000002307 prostate Anatomy 0.000 claims description 2
- 208000029922 reticulum cell sarcoma Diseases 0.000 claims description 2
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 2
- 206010039667 schwannoma Diseases 0.000 claims description 2
- 208000004548 serous cystadenocarcinoma Diseases 0.000 claims description 2
- 210000003625 skull Anatomy 0.000 claims description 2
- 210000002784 stomach Anatomy 0.000 claims description 2
- 208000001608 teratocarcinoma Diseases 0.000 claims description 2
- 210000001550 testis Anatomy 0.000 claims description 2
- 206010044412 transitional cell carcinoma Diseases 0.000 claims description 2
- 201000007423 tubular adenocarcinoma Diseases 0.000 claims description 2
- 208000022271 tubular adenoma Diseases 0.000 claims description 2
- 210000003708 urethra Anatomy 0.000 claims description 2
- 210000004291 uterus Anatomy 0.000 claims description 2
- 210000001215 vagina Anatomy 0.000 claims description 2
- 208000009540 villous adenoma Diseases 0.000 claims description 2
- 210000003905 vulva Anatomy 0.000 claims description 2
- 206010031149 Osteitis Diseases 0.000 claims 1
- 125000003275 alpha amino acid group Chemical group 0.000 claims 1
- 208000018339 bone inflammation disease Diseases 0.000 claims 1
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 claims 1
- 150000003839 salts Chemical class 0.000 abstract description 27
- 239000012453 solvate Substances 0.000 abstract description 7
- -1 hydroxy, cyano, amino Chemical group 0.000 description 299
- 125000000217 alkyl group Chemical group 0.000 description 166
- 239000001257 hydrogen Substances 0.000 description 164
- 229910052739 hydrogen Inorganic materials 0.000 description 164
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 140
- 125000003342 alkenyl group Chemical group 0.000 description 72
- 125000003545 alkoxy group Chemical group 0.000 description 72
- 108091000080 Phosphotransferase Proteins 0.000 description 60
- 102000020233 phosphotransferase Human genes 0.000 description 60
- 125000005843 halogen group Chemical group 0.000 description 59
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 59
- 125000004005 formimidoyl group Chemical group [H]\N=C(/[H])* 0.000 description 50
- 125000001072 heteroaryl group Chemical group 0.000 description 49
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 47
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 46
- 108090000623 proteins and genes Proteins 0.000 description 44
- 125000003118 aryl group Chemical group 0.000 description 43
- 102000005962 receptors Human genes 0.000 description 43
- 108020003175 receptors Proteins 0.000 description 43
- NCTHHJRIJGFPTG-UHFFFAOYSA-N 6,7-bis(aziridin-1-yl)-4-[7-[[6,7-bis(aziridin-1-yl)-5,8-dioxoquinazolin-4-yl]amino]heptylamino]quinazoline-5,8-dione Chemical compound C1CN1C=1C(=O)C2=C(NCCCCCCCNC=3C=4C(=O)C(N5CC5)=C(N5CC5)C(=O)C=4N=CN=3)N=CN=C2C(=O)C=1N1CC1 NCTHHJRIJGFPTG-UHFFFAOYSA-N 0.000 description 40
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 35
- 125000003282 alkyl amino group Chemical group 0.000 description 31
- 239000002502 liposome Substances 0.000 description 30
- 125000004663 dialkyl amino group Chemical group 0.000 description 29
- 125000004985 dialkyl amino alkyl group Chemical group 0.000 description 28
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 28
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 27
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 26
- 102100036721 Insulin receptor Human genes 0.000 description 25
- 235000018102 proteins Nutrition 0.000 description 25
- 102000004169 proteins and genes Human genes 0.000 description 25
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 24
- 101710196759 Macrophage-stimulating protein receptor Proteins 0.000 description 24
- 102100021435 Macrophage-stimulating protein receptor Human genes 0.000 description 24
- 239000013592 cell lysate Substances 0.000 description 24
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 24
- 102000007665 Extracellular Signal-Regulated MAP Kinases Human genes 0.000 description 23
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 description 23
- 108060003951 Immunoglobulin Proteins 0.000 description 23
- 108010055717 JNK Mitogen-Activated Protein Kinases Proteins 0.000 description 23
- 238000003119 immunoblot Methods 0.000 description 23
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 23
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 23
- 102000001291 MAP Kinase Kinase Kinase Human genes 0.000 description 22
- 108060006687 MAP kinase kinase kinase Proteins 0.000 description 22
- 239000003795 chemical substances by application Substances 0.000 description 22
- 125000004438 haloalkoxy group Chemical group 0.000 description 22
- 125000001188 haloalkyl group Chemical group 0.000 description 22
- 102000018358 immunoglobulin Human genes 0.000 description 22
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 21
- 101100262697 Mus musculus Axl gene Proteins 0.000 description 21
- 125000000753 cycloalkyl group Chemical group 0.000 description 21
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 21
- 125000005885 heterocycloalkylalkyl group Chemical group 0.000 description 20
- 108020004414 DNA Proteins 0.000 description 19
- 102100023600 Fibroblast growth factor receptor 2 Human genes 0.000 description 19
- 101710182389 Fibroblast growth factor receptor 2 Proteins 0.000 description 19
- 108010031794 IGF Type 1 Receptor Proteins 0.000 description 19
- 125000004103 aminoalkyl group Chemical group 0.000 description 19
- 239000000427 antigen Substances 0.000 description 19
- 108091007433 antigens Proteins 0.000 description 19
- 102000036639 antigens Human genes 0.000 description 19
- 201000010099 disease Diseases 0.000 description 19
- 230000005764 inhibitory process Effects 0.000 description 19
- 108090000765 processed proteins & peptides Proteins 0.000 description 19
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- 125000003710 aryl alkyl group Chemical group 0.000 description 18
- 230000027455 binding Effects 0.000 description 18
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 18
- 210000004408 hybridoma Anatomy 0.000 description 17
- 239000007924 injection Substances 0.000 description 17
- 238000002347 injection Methods 0.000 description 17
- 239000000203 mixture Substances 0.000 description 17
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 17
- 102000004196 processed proteins & peptides Human genes 0.000 description 17
- 101000852815 Homo sapiens Insulin receptor Proteins 0.000 description 16
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 16
- 241001465754 Metazoa Species 0.000 description 15
- 108091034117 Oligonucleotide Proteins 0.000 description 15
- 238000004458 analytical method Methods 0.000 description 15
- 230000002401 inhibitory effect Effects 0.000 description 15
- 239000013612 plasmid Substances 0.000 description 15
- 210000001519 tissue Anatomy 0.000 description 15
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 14
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 14
- 239000008194 pharmaceutical composition Substances 0.000 description 14
- 125000003302 alkenyloxy group Chemical group 0.000 description 13
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 13
- 125000002947 alkylene group Chemical group 0.000 description 13
- 229920001223 polyethylene glycol Polymers 0.000 description 13
- 229920001184 polypeptide Polymers 0.000 description 13
- 239000013603 viral vector Substances 0.000 description 13
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 12
- 150000001413 amino acids Chemical group 0.000 description 12
- 125000004093 cyano group Chemical group *C#N 0.000 description 12
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 12
- 238000013518 transcription Methods 0.000 description 12
- 230000035897 transcription Effects 0.000 description 12
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 11
- 239000002202 Polyethylene glycol Substances 0.000 description 11
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical group C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 11
- 239000002609 medium Substances 0.000 description 11
- VMWJCFLUSKZZDX-UHFFFAOYSA-N n,n-dimethylmethanamine Chemical compound [CH2]N(C)C VMWJCFLUSKZZDX-UHFFFAOYSA-N 0.000 description 11
- 125000004043 oxo group Chemical group O=* 0.000 description 11
- 239000000546 pharmaceutical excipient Substances 0.000 description 11
- 239000000126 substance Substances 0.000 description 11
- 238000001890 transfection Methods 0.000 description 11
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 10
- 108091026890 Coding region Proteins 0.000 description 10
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 10
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 10
- 108091007960 PI3Ks Proteins 0.000 description 10
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 10
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 10
- 125000004414 alkyl thio group Chemical group 0.000 description 10
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 10
- 238000003753 real-time PCR Methods 0.000 description 10
- 125000004204 2-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 9
- 101000652725 Drosophila melanogaster Transcription initiation factor TFIID subunit 5 Proteins 0.000 description 9
- 101000752722 Homo sapiens Apoptosis-stimulating of p53 protein 1 Proteins 0.000 description 9
- 101001120056 Homo sapiens Phosphatidylinositol 3-kinase regulatory subunit alpha Proteins 0.000 description 9
- 101001120097 Homo sapiens Phosphatidylinositol 3-kinase regulatory subunit beta Proteins 0.000 description 9
- 101001116549 Homo sapiens Protein CBFA2T2 Proteins 0.000 description 9
- 101001000998 Homo sapiens Protein phosphatase 1 regulatory subunit 12C Proteins 0.000 description 9
- 101000927796 Homo sapiens Rho guanine nucleotide exchange factor 7 Proteins 0.000 description 9
- 108010001127 Insulin Receptor Proteins 0.000 description 9
- 102100026169 Phosphatidylinositol 3-kinase regulatory subunit alpha Human genes 0.000 description 9
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 9
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 9
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 230000009368 gene silencing by RNA Effects 0.000 description 9
- 230000012010 growth Effects 0.000 description 9
- 239000001963 growth medium Substances 0.000 description 9
- 238000002372 labelling Methods 0.000 description 9
- 239000003446 ligand Substances 0.000 description 9
- 238000004519 manufacturing process Methods 0.000 description 9
- 238000003752 polymerase chain reaction Methods 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 150000003254 radicals Chemical class 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- 230000014616 translation Effects 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 229940125497 HER2 kinase inhibitor Drugs 0.000 description 8
- 125000005090 alkenylcarbonyl group Chemical group 0.000 description 8
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 description 8
- 125000002431 aminoalkoxy group Chemical group 0.000 description 8
- 239000003153 chemical reaction reagent Substances 0.000 description 8
- 125000004122 cyclic group Chemical group 0.000 description 8
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 8
- 208000035475 disorder Diseases 0.000 description 8
- 239000003814 drug Substances 0.000 description 8
- 239000012528 membrane Substances 0.000 description 8
- 210000004379 membrane Anatomy 0.000 description 8
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 description 8
- 230000014207 opsonization Effects 0.000 description 8
- 229920000642 polymer Polymers 0.000 description 8
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical group OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 8
- 208000011580 syndromic disease Diseases 0.000 description 8
- 238000013519 translation Methods 0.000 description 8
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 7
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 7
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 238000007792 addition Methods 0.000 description 7
- 125000000304 alkynyl group Chemical group 0.000 description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 7
- 230000010261 cell growth Effects 0.000 description 7
- 125000001309 chloro group Chemical group Cl* 0.000 description 7
- 238000001114 immunoprecipitation Methods 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 238000001802 infusion Methods 0.000 description 7
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 7
- 239000006166 lysate Substances 0.000 description 7
- 201000000050 myeloid neoplasm Diseases 0.000 description 7
- 125000006413 ring segment Chemical group 0.000 description 7
- 210000004881 tumor cell Anatomy 0.000 description 7
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 6
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 description 6
- 125000004207 3-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(OC([H])([H])[H])=C1[H] 0.000 description 6
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 6
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 6
- 102100030322 Ephrin type-A receptor 1 Human genes 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- 108091028043 Nucleic acid sequence Proteins 0.000 description 6
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 6
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000000074 antisense oligonucleotide Substances 0.000 description 6
- 238000012230 antisense oligonucleotides Methods 0.000 description 6
- 238000003491 array Methods 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 6
- 125000000262 haloalkenyl group Chemical group 0.000 description 6
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 6
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 6
- 150000002632 lipids Chemical class 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 6
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 6
- 230000037361 pathway Effects 0.000 description 6
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 6
- 229920001451 polypropylene glycol Polymers 0.000 description 6
- 239000000651 prodrug Substances 0.000 description 6
- 229940002612 prodrug Drugs 0.000 description 6
- 239000000523 sample Substances 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 241000894007 species Species 0.000 description 6
- 238000010561 standard procedure Methods 0.000 description 6
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 6
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 6
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 5
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 5
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 5
- 241000894006 Bacteria Species 0.000 description 5
- 101000938354 Homo sapiens Ephrin type-A receptor 1 Proteins 0.000 description 5
- 239000012828 PI3K inhibitor Substances 0.000 description 5
- 102100038332 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform Human genes 0.000 description 5
- 102100024952 Protein CBFA2T1 Human genes 0.000 description 5
- NFGODEMQGQNUKK-UHFFFAOYSA-M [6-(diethylamino)-9-(2-octadecoxycarbonylphenyl)xanthen-3-ylidene]-diethylazanium;chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCCOC(=O)C1=CC=CC=C1C1=C2C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C21 NFGODEMQGQNUKK-UHFFFAOYSA-M 0.000 description 5
- 235000001014 amino acid Nutrition 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 229960005069 calcium Drugs 0.000 description 5
- 229910052791 calcium Inorganic materials 0.000 description 5
- 239000000969 carrier Substances 0.000 description 5
- 238000004113 cell culture Methods 0.000 description 5
- 238000010367 cloning Methods 0.000 description 5
- 239000003184 complementary RNA Substances 0.000 description 5
- 125000004966 cyanoalkyl group Chemical group 0.000 description 5
- 125000006254 cycloalkyl carbonyl group Chemical group 0.000 description 5
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 5
- 230000001419 dependent effect Effects 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 239000012091 fetal bovine serum Substances 0.000 description 5
- 125000002140 imidazol-4-yl group Chemical group [H]N1C([H])=NC([*])=C1[H] 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- 230000004083 survival effect Effects 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 241000701161 unidentified adenovirus Species 0.000 description 5
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 4
- 239000013607 AAV vector Substances 0.000 description 4
- 239000004215 Carbon black (E152) Substances 0.000 description 4
- 108020004635 Complementary DNA Proteins 0.000 description 4
- 108020004394 Complementary RNA Proteins 0.000 description 4
- 241000701022 Cytomegalovirus Species 0.000 description 4
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical group OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 4
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 4
- 101000605639 Homo sapiens Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform Proteins 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 4
- 150000001204 N-oxides Chemical class 0.000 description 4
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 4
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 229920002873 Polyethylenimine Polymers 0.000 description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 4
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 4
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 4
- 108020005093 RNA Precursors Proteins 0.000 description 4
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 4
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 4
- 239000002671 adjuvant Substances 0.000 description 4
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 description 4
- 230000006907 apoptotic process Effects 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 239000003623 enhancer Substances 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 235000019441 ethanol Nutrition 0.000 description 4
- 238000009396 hybridization Methods 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- 229940072221 immunoglobulins Drugs 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 4
- 210000003292 kidney cell Anatomy 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 231100000682 maximum tolerated dose Toxicity 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- TXXHDPDFNKHHGW-UHFFFAOYSA-N muconic acid Chemical group OC(=O)C=CC=CC(O)=O TXXHDPDFNKHHGW-UHFFFAOYSA-N 0.000 description 4
- 230000035772 mutation Effects 0.000 description 4
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 4
- 230000002018 overexpression Effects 0.000 description 4
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 4
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Chemical group OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 238000002823 phage display Methods 0.000 description 4
- 239000006187 pill Substances 0.000 description 4
- 229920001282 polysaccharide Polymers 0.000 description 4
- 239000005017 polysaccharide Substances 0.000 description 4
- 150000004804 polysaccharides Chemical class 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 238000012827 research and development Methods 0.000 description 4
- 229960004889 salicylic acid Drugs 0.000 description 4
- 238000012163 sequencing technique Methods 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 125000004426 substituted alkynyl group Chemical group 0.000 description 4
- 125000003107 substituted aryl group Chemical group 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 230000003442 weekly effect Effects 0.000 description 4
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 3
- 125000006582 (C5-C6) heterocycloalkyl group Chemical group 0.000 description 3
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 3
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 3
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 3
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 3
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 3
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 3
- 125000004195 4-methylpiperazin-1-yl group Chemical group [H]C([H])([H])N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 108091033380 Coding strand Proteins 0.000 description 3
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 241000588724 Escherichia coli Species 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 3
- 101001010819 Homo sapiens Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 3
- 102100029098 Hypoxanthine-guanine phosphoribosyltransferase Human genes 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 3
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- 101710163270 Nuclease Proteins 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 3
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 3
- 238000002123 RNA extraction Methods 0.000 description 3
- 238000011529 RT qPCR Methods 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 108091081021 Sense strand Proteins 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 150000001408 amides Chemical group 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 3
- 210000003719 b-lymphocyte Anatomy 0.000 description 3
- 125000001246 bromo group Chemical group Br* 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 229960001948 caffeine Drugs 0.000 description 3
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 239000002738 chelating agent Substances 0.000 description 3
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 3
- 229960001231 choline Drugs 0.000 description 3
- 235000015165 citric acid Nutrition 0.000 description 3
- 238000004590 computer program Methods 0.000 description 3
- 239000000470 constituent Substances 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 3
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 238000009510 drug design Methods 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 239000012737 fresh medium Substances 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 102000051957 human ERBB2 Human genes 0.000 description 3
- 125000002349 hydroxyamino group Chemical group [H]ON([H])[*] 0.000 description 3
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 description 3
- 230000001939 inductive effect Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 3
- 125000004498 isoxazol-4-yl group Chemical group O1N=CC(=C1)* 0.000 description 3
- 125000005647 linker group Chemical group 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 230000003278 mimic effect Effects 0.000 description 3
- 210000000865 mononuclear phagocyte system Anatomy 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical group CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 238000010647 peptide synthesis reaction Methods 0.000 description 3
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 3
- 230000026731 phosphorylation Effects 0.000 description 3
- 238000006366 phosphorylation reaction Methods 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 150000003212 purines Chemical class 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 230000009870 specific binding Effects 0.000 description 3
- 210000000952 spleen Anatomy 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 229940032147 starch Drugs 0.000 description 3
- 125000005017 substituted alkenyl group Chemical group 0.000 description 3
- 125000000547 substituted alkyl group Chemical group 0.000 description 3
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 150000008163 sugars Chemical class 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 2
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 2
- 125000006528 (C2-C6) alkyl group Chemical group 0.000 description 2
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 2
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 2
- XXJGBENTLXFVFI-UHFFFAOYSA-N 1-amino-methylene Chemical compound N[CH2] XXJGBENTLXFVFI-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 2
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 2
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 2
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical group C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 2
- XLZYKTYMLBOINK-UHFFFAOYSA-N 3-(4-hydroxybenzoyl)benzoic acid Chemical compound OC(=O)C1=CC=CC(C(=O)C=2C=CC(O)=CC=2)=C1 XLZYKTYMLBOINK-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 2
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 2
- 125000004208 3-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(*)=C1[H] 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- RJWBTWIBUIGANW-UHFFFAOYSA-N 4-chlorobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Cl)C=C1 RJWBTWIBUIGANW-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- 229920000936 Agarose Polymers 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 206010003445 Ascites Diseases 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N Betaine Natural products C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 208000003174 Brain Neoplasms Diseases 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 241000699800 Cricetinae Species 0.000 description 2
- 241000699802 Cricetulus griseus Species 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Chemical group OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 241000255925 Diptera Species 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 102100038132 Endogenous retrovirus group K member 6 Pro protein Human genes 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 description 2
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 241000206672 Gelidium Species 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Chemical group OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 2
- 101000779418 Homo sapiens RAC-alpha serine/threonine-protein kinase Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical group OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 241000713666 Lentivirus Species 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- TXXHDPDFNKHHGW-CCAGOZQPSA-N Muconic acid Chemical group OC(=O)\C=C/C=C\C(O)=O TXXHDPDFNKHHGW-CCAGOZQPSA-N 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-O N,N,N-trimethylglycinium Chemical compound C[N+](C)(C)CC(O)=O KWIUHFFTVRNATP-UHFFFAOYSA-O 0.000 description 2
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 2
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 2
- 229910003827 NRaRb Inorganic materials 0.000 description 2
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- 108091092724 Noncoding DNA Proteins 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- 102000014160 PTEN Phosphohydrolase Human genes 0.000 description 2
- 108010011536 PTEN Phosphohydrolase Proteins 0.000 description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 108010039918 Polylysine Proteins 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 2
- 102100029981 Receptor tyrosine-protein kinase erbB-4 Human genes 0.000 description 2
- 108091028664 Ribonucleotide Proteins 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 241000607720 Serratia Species 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 241000906446 Theraps Species 0.000 description 2
- 101710120037 Toxin CcdB Proteins 0.000 description 2
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 2
- 241000711975 Vesicular stomatitis virus Species 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- HMNZFMSWFCAGGW-XPWSMXQVSA-N [3-[hydroxy(2-hydroxyethoxy)phosphoryl]oxy-2-[(e)-octadec-9-enoyl]oxypropyl] (e)-octadec-9-enoate Chemical compound CCCCCCCC\C=C\CCCCCCCC(=O)OCC(COP(O)(=O)OCCO)OC(=O)CCCCCCC\C=C\CCCCCCCC HMNZFMSWFCAGGW-XPWSMXQVSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 238000001042 affinity chromatography Methods 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 238000003349 alamar blue assay Methods 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 125000005193 alkenylcarbonyloxy group Chemical group 0.000 description 2
- 125000005092 alkenyloxycarbonyl group Chemical group 0.000 description 2
- 125000005083 alkoxyalkoxy group Chemical group 0.000 description 2
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 2
- 125000004949 alkyl amino carbonyl amino group Chemical group 0.000 description 2
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 description 2
- 125000005195 alkyl amino carbonyloxy group Chemical group 0.000 description 2
- 125000004471 alkyl aminosulfonyl group Chemical group 0.000 description 2
- 125000006350 alkyl thio alkyl group Chemical group 0.000 description 2
- RMRFFCXPLWYOOY-UHFFFAOYSA-N allyl radical Chemical compound [CH2]C=C RMRFFCXPLWYOOY-UHFFFAOYSA-N 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000001028 anti-proliverative effect Effects 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 229960003121 arginine Drugs 0.000 description 2
- 125000004566 azetidin-1-yl group Chemical group N1(CCC1)* 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 238000002869 basic local alignment search tool Methods 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 2
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- 229960003237 betaine Drugs 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 235000013985 cinnamic acid Nutrition 0.000 description 2
- 229930016911 cinnamic acid Natural products 0.000 description 2
- 238000011260 co-administration Methods 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 210000004748 cultured cell Anatomy 0.000 description 2
- 238000012258 culturing Methods 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 238000002716 delivery method Methods 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 125000004473 dialkylaminocarbonyl group Chemical group 0.000 description 2
- 125000004472 dialkylaminosulfonyl group Chemical group 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical group CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 229940088679 drug related substance Drugs 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 229940012017 ethylenediamine Drugs 0.000 description 2
- 238000002509 fluorescent in situ hybridization Methods 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 235000011087 fumaric acid Nutrition 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 150000002270 gangliosides Chemical class 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000030279 gene silencing Effects 0.000 description 2
- 239000000174 gluconic acid Chemical group 0.000 description 2
- 235000012208 gluconic acid Nutrition 0.000 description 2
- 229960002442 glucosamine Drugs 0.000 description 2
- 235000001727 glucose Nutrition 0.000 description 2
- 239000004220 glutamic acid Chemical group 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 239000003966 growth inhibitor Substances 0.000 description 2
- 238000003306 harvesting Methods 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 229960002885 histidine Drugs 0.000 description 2
- 229920002674 hyaluronan Polymers 0.000 description 2
- 229960003160 hyaluronic acid Drugs 0.000 description 2
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 229920001477 hydrophilic polymer Polymers 0.000 description 2
- 125000005113 hydroxyalkoxy group Chemical group 0.000 description 2
- 238000012872 hydroxylapatite chromatography Methods 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 125000000336 imidazol-5-yl group Chemical group [H]N1C([H])=NC([H])=C1[*] 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 230000002163 immunogen Effects 0.000 description 2
- 238000003364 immunohistochemistry Methods 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 238000001361 intraarterial administration Methods 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 229960003646 lysine Drugs 0.000 description 2
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 description 2
- 229920001427 mPEG Polymers 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 229960002510 mandelic acid Drugs 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 229910021645 metal ion Inorganic materials 0.000 description 2
- TWXDDNPPQUTEOV-FVGYRXGTSA-N methamphetamine hydrochloride Chemical compound Cl.CN[C@@H](C)CC1=CC=CC=C1 TWXDDNPPQUTEOV-FVGYRXGTSA-N 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 239000003607 modifier Substances 0.000 description 2
- 238000010369 molecular cloning Methods 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 235000015097 nutrients Nutrition 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Chemical group CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 229920001542 oligosaccharide Chemical class 0.000 description 2
- 150000002482 oligosaccharides Chemical class 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 150000008300 phosphoramidites Chemical group 0.000 description 2
- 125000004574 piperidin-2-yl group Chemical group N1C(CCCC1)* 0.000 description 2
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 2
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 2
- 229920000962 poly(amidoamine) Polymers 0.000 description 2
- 229940068917 polyethylene glycols Drugs 0.000 description 2
- 229920000656 polylysine Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 2
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 2
- 229960004919 procaine Drugs 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 229960004063 propylene glycol Drugs 0.000 description 2
- 235000013772 propylene glycol Nutrition 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 125000004940 pyridazin-4-yl group Chemical group N1=NC=C(C=C1)* 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 229940107700 pyruvic acid Drugs 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 238000003127 radioimmunoassay Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 239000002336 ribonucleotide Substances 0.000 description 2
- 125000002652 ribonucleotide group Chemical group 0.000 description 2
- 229910052701 rubidium Inorganic materials 0.000 description 2
- 230000003248 secreting effect Effects 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008117 stearic acid Chemical group 0.000 description 2
- 239000007929 subcutaneous injection Substances 0.000 description 2
- 238000010254 subcutaneous injection Methods 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- 229960004559 theobromine Drugs 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- 238000011830 transgenic mouse model Methods 0.000 description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 229960000281 trometamol Drugs 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 238000009736 wetting Methods 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- ICLYJLBTOGPLMC-KVVVOXFISA-N (z)-octadec-9-enoate;tris(2-hydroxyethyl)azanium Chemical compound OCCN(CCO)CCO.CCCCCCCC\C=C/CCCCCCCC(O)=O ICLYJLBTOGPLMC-KVVVOXFISA-N 0.000 description 1
- XLOVPKCQAPHUKK-UHFFFAOYSA-N 1,2,3,4,4a,5,8,8a-octahydronaphthalene Chemical compound C1C=CCC2CCCCC21 XLOVPKCQAPHUKK-UHFFFAOYSA-N 0.000 description 1
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 1
- HKWJHKSHEWVOSS-OMDJCSNQSA-N 1,2-dihexadecanoyl-sn-glycero-3-phospho-(1D-myo-inositol-3,4-bisphosphate) Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP(O)(=O)O[C@H]1[C@H](O)[C@@H](O)[C@H](OP(O)(O)=O)[C@@H](OP(O)(O)=O)[C@H]1O HKWJHKSHEWVOSS-OMDJCSNQSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- 125000004486 1-methylpiperidin-3-yl group Chemical group CN1CC(CCC1)* 0.000 description 1
- 125000004778 2,2-difluoroethyl group Chemical group [H]C([H])(*)C([H])(F)F 0.000 description 1
- UZURTQHPMXADGG-UHFFFAOYSA-N 2,3,3a,4,7,7a-hexahydro-1h-indene Chemical compound C1C=CCC2CCCC21 UZURTQHPMXADGG-UHFFFAOYSA-N 0.000 description 1
- PHXGAJLBHUUAKB-UHFFFAOYSA-N 2,3,3a,5,6,6a-hexahydrofuro[3,2-b]furan Chemical compound O1CCC2OCCC21 PHXGAJLBHUUAKB-UHFFFAOYSA-N 0.000 description 1
- VQSJAWPFQCXIOB-VODLGYORSA-N 2,3-dihydroxypropyl [(1r,2r,3s,4r,5r,6s)-2,3,6-trihydroxy-4,5-diphosphonooxycyclohexyl] hydrogen phosphate Chemical compound OCC(O)COP(O)(=O)O[C@@H]1[C@H](O)[C@H](O)[C@@H](OP(O)(O)=O)[C@H](OP(O)(O)=O)[C@H]1O VQSJAWPFQCXIOB-VODLGYORSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- 125000004638 2-oxopiperazinyl group Chemical group O=C1N(CCNC1)* 0.000 description 1
- 125000004637 2-oxopiperidinyl group Chemical group O=C1N(CCCC1)* 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 108020005345 3' Untranslated Regions Proteins 0.000 description 1
- SFDGJDBLYNJMFI-UHFFFAOYSA-N 3,1-benzoxazin-4-one Chemical compound C1=CC=C2C(=O)OC=NC2=C1 SFDGJDBLYNJMFI-UHFFFAOYSA-N 0.000 description 1
- 125000004189 3,4-dichlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(Cl)C([H])=C1* 0.000 description 1
- QOXOZONBQWIKDA-UHFFFAOYSA-N 3-hydroxypropyl Chemical group [CH2]CCO QOXOZONBQWIKDA-UHFFFAOYSA-N 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- SXIFAEWFOJETOA-UHFFFAOYSA-N 4-hydroxy-butyl Chemical group [CH2]CCCO SXIFAEWFOJETOA-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- 108020003589 5' Untranslated Regions Proteins 0.000 description 1
- XZIIFPSPUDAGJM-UHFFFAOYSA-N 6-chloro-2-n,2-n-diethylpyrimidine-2,4-diamine Chemical compound CCN(CC)C1=NC(N)=CC(Cl)=N1 XZIIFPSPUDAGJM-UHFFFAOYSA-N 0.000 description 1
- SNZSSCZJMVIOCR-UHFFFAOYSA-N 7-azabicyclo[2.2.1]heptane Chemical compound C1CC2CCC1N2 SNZSSCZJMVIOCR-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 241000256118 Aedes aegypti Species 0.000 description 1
- 241000256173 Aedes albopictus Species 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 101000716807 Arabidopsis thaliana Protein SCO1 homolog 1, mitochondrial Proteins 0.000 description 1
- IYMAXBFPHPZYIK-BQBZGAKWSA-N Arg-Gly-Asp Chemical compound NC(N)=NCCC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(O)=O IYMAXBFPHPZYIK-BQBZGAKWSA-N 0.000 description 1
- 241000228212 Aspergillus Species 0.000 description 1
- 241000351920 Aspergillus nidulans Species 0.000 description 1
- 241000228245 Aspergillus niger Species 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 241001203868 Autographa californica Species 0.000 description 1
- 108091012583 BCL2 Proteins 0.000 description 1
- 241000194108 Bacillus licheniformis Species 0.000 description 1
- 235000014469 Bacillus subtilis Nutrition 0.000 description 1
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 1
- 241000255789 Bombyx mori Species 0.000 description 1
- 241000409811 Bombyx mori nucleopolyhedrovirus Species 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 101100356682 Caenorhabditis elegans rho-1 gene Proteins 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-VANFPWTGSA-N D-mannopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@@H]1O AEMOLEFTQBMNLQ-VANFPWTGSA-N 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 101710177611 DNA polymerase II large subunit Proteins 0.000 description 1
- 101710184669 DNA polymerase II small subunit Proteins 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 1
- 241000255601 Drosophila melanogaster Species 0.000 description 1
- 206010013710 Drug interaction Diseases 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 1
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- 102000001301 EGF receptor Human genes 0.000 description 1
- 108060006698 EGF receptor Proteins 0.000 description 1
- 201000011001 Ebola Hemorrhagic Fever Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000588914 Enterobacter Species 0.000 description 1
- 101710091045 Envelope protein Proteins 0.000 description 1
- 101710116744 Ephrin type-A receptor 1 Proteins 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 241000588698 Erwinia Species 0.000 description 1
- 241000588722 Escherichia Species 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 101150054472 HER2 gene Proteins 0.000 description 1
- 102100021455 Histone deacetylase 3 Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 1
- 101000899282 Homo sapiens Histone deacetylase 3 Proteins 0.000 description 1
- 101001076715 Homo sapiens RNA-binding protein 39 Proteins 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 101710116034 Immunity protein Proteins 0.000 description 1
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 1
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102100034349 Integrase Human genes 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 241000588748 Klebsiella Species 0.000 description 1
- 241000235649 Kluyveromyces Species 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 1
- 239000012098 Lipofectamine RNAiMAX Substances 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 229940124647 MEK inhibitor Drugs 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- YALRCXHVQYBSJC-UHFFFAOYSA-N Mammea A/AB Chemical compound C12=C(O)C(C(=O)C(C)CC)=C(O)C(CC=C(C)C)=C2OC(=O)C=C1C1=CC=CC=C1 YALRCXHVQYBSJC-UHFFFAOYSA-N 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- 102000004232 Mitogen-Activated Protein Kinase Kinases Human genes 0.000 description 1
- 108090000744 Mitogen-Activated Protein Kinase Kinases Proteins 0.000 description 1
- 241000714177 Murine leukemia virus Species 0.000 description 1
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 206010029113 Neovascularisation Diseases 0.000 description 1
- 241000221960 Neurospora Species 0.000 description 1
- 241000221961 Neurospora crassa Species 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- 208000010191 Osteitis Deformans Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000027067 Paget disease of bone Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- 241000228143 Penicillium Species 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 241000577979 Peromyscus spicilegus Species 0.000 description 1
- 101710093328 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform Proteins 0.000 description 1
- 241000235648 Pichia Species 0.000 description 1
- 101150063858 Pik3ca gene Proteins 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108091008611 Protein Kinase B Proteins 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 101710188315 Protein X Proteins 0.000 description 1
- 102000012515 Protein kinase domains Human genes 0.000 description 1
- 108050002122 Protein kinase domains Proteins 0.000 description 1
- 241000588769 Proteus <enterobacteria> Species 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 101710113459 RAC-alpha serine/threonine-protein kinase Proteins 0.000 description 1
- 101150111584 RHOA gene Proteins 0.000 description 1
- 206010037742 Rabies Diseases 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 102100023361 SAP domain-containing ribonucleoprotein Human genes 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 1
- 241000235347 Schizosaccharomyces pombe Species 0.000 description 1
- 241000311088 Schwanniomyces Species 0.000 description 1
- 241001123650 Schwanniomyces occidentalis Species 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 241000607768 Shigella Species 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 241000256251 Spodoptera frugiperda Species 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- 229930182558 Sterol Natural products 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 108091008874 T cell receptors Proteins 0.000 description 1
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 1
- 241000255588 Tephritidae Species 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical group OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 108091036066 Three prime untranslated region Proteins 0.000 description 1
- 108010034949 Thyroglobulin Proteins 0.000 description 1
- 102100033504 Thyroglobulin Human genes 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 241001149964 Tolypocladium Species 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 241000223259 Trichoderma Species 0.000 description 1
- 101710162629 Trypsin inhibitor Proteins 0.000 description 1
- 229940122618 Trypsin inhibitor Drugs 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- 102100037236 Tyrosine-protein kinase receptor UFO Human genes 0.000 description 1
- 108091023045 Untranslated Region Proteins 0.000 description 1
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical group O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 1
- 244000000188 Vaccinium ovalifolium Species 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 241000235013 Yarrowia Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- MMXKIWIWQPKTIK-KPRKPIBOSA-N [(2r,3s,5r)-3-hydroxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl [(2r,3s,5r)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)-2-(phosphonooxymethyl)oxolan-3-yl] hydrogen phosphate Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)O[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(O)=O)[C@@H](O)C1 MMXKIWIWQPKTIK-KPRKPIBOSA-N 0.000 description 1
- DSODRWWHAUGSGD-UHFFFAOYSA-N [5-(carbamimidoylsulfanylmethyl)thiophen-2-yl]methyl carbamimidothioate;dihydrochloride Chemical compound Cl.Cl.NC(=N)SCC1=CC=C(CSC(N)=N)S1 DSODRWWHAUGSGD-UHFFFAOYSA-N 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 230000004931 aggregating effect Effects 0.000 description 1
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229940037003 alum Drugs 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 238000012870 ammonium sulfate precipitation Methods 0.000 description 1
- 238000005571 anion exchange chromatography Methods 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000005875 antibody response Effects 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 108010072041 arginyl-glycyl-aspartic acid Proteins 0.000 description 1
- 238000007080 aromatic substitution reaction Methods 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 108010023337 axl receptor tyrosine kinase Proteins 0.000 description 1
- 125000004567 azetidin-3-yl group Chemical group N1CC(C1)* 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 244000052616 bacterial pathogen Species 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- DZBUGLKDJFMEHC-UHFFFAOYSA-N benzoquinolinylidene Chemical group C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 229960004217 benzyl alcohol Drugs 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 238000002306 biochemical method Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 238000005460 biophysical method Methods 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 208000016738 bone Paget disease Diseases 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010376 calcium ascorbate Nutrition 0.000 description 1
- 239000011692 calcium ascorbate Substances 0.000 description 1
- 229940047036 calcium ascorbate Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 229960002713 calcium chloride Drugs 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 239000004227 calcium gluconate Substances 0.000 description 1
- 235000013927 calcium gluconate Nutrition 0.000 description 1
- 229960004494 calcium gluconate Drugs 0.000 description 1
- MKJXYGKVIBWPFZ-UHFFFAOYSA-L calcium lactate Chemical compound [Ca+2].CC(O)C([O-])=O.CC(O)C([O-])=O MKJXYGKVIBWPFZ-UHFFFAOYSA-L 0.000 description 1
- 239000001527 calcium lactate Substances 0.000 description 1
- 235000011086 calcium lactate Nutrition 0.000 description 1
- 229960002401 calcium lactate Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- BLORRZQTHNGFTI-ZZMNMWMASA-L calcium-L-ascorbate Chemical compound [Ca+2].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] BLORRZQTHNGFTI-ZZMNMWMASA-L 0.000 description 1
- NEEHYRZPVYRGPP-UHFFFAOYSA-L calcium;2,3,4,5,6-pentahydroxyhexanoate Chemical compound [Ca+2].OCC(O)C(O)C(O)C(O)C([O-])=O.OCC(O)C(O)C(O)C(O)C([O-])=O NEEHYRZPVYRGPP-UHFFFAOYSA-L 0.000 description 1
- BHRQIJRLOVHRKH-UHFFFAOYSA-L calcium;2-[bis[2-[bis(carboxylatomethyl)amino]ethyl]amino]acetate;hydron Chemical compound [Ca+2].OC(=O)CN(CC(O)=O)CCN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O BHRQIJRLOVHRKH-UHFFFAOYSA-L 0.000 description 1
- 239000003560 cancer drug Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 150000004653 carbonic acids Chemical class 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 239000000679 carrageenan Substances 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 229940113118 carrageenan Drugs 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 238000005277 cation exchange chromatography Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 108091092328 cellular RNA Proteins 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 230000003034 chemosensitisation Effects 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 238000011098 chromatofocusing Methods 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229960004106 citric acid Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 239000005289 controlled pore glass Substances 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000003235 crystal violet staining Methods 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000003436 cytoskeletal effect Effects 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 125000005507 decahydroisoquinolyl group Chemical group 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000005202 dialkylaminocarbonyloxy group Chemical group 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- 230000002222 downregulating effect Effects 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 108700020302 erbB-2 Genes Proteins 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000012226 gene silencing method Methods 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 229940084910 gliadel Drugs 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 125000004994 halo alkoxy alkyl group Chemical group 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 230000009851 immunogenic response Effects 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 150000002484 inorganic compounds Chemical class 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 108010078480 insect defensin A Proteins 0.000 description 1
- 238000009830 intercalation Methods 0.000 description 1
- 230000002687 intercalation Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 125000004284 isoxazol-3-yl group Chemical group [H]C1=C([H])C(*)=NO1 0.000 description 1
- 125000004499 isoxazol-5-yl group Chemical group O1N=CC=C1* 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- BQINXKOTJQCISL-GRCPKETISA-N keto-neuraminic acid Chemical compound OC(=O)C(=O)C[C@H](O)[C@@H](N)[C@@H](O)[C@H](O)[C@H](O)CO BQINXKOTJQCISL-GRCPKETISA-N 0.000 description 1
- 108010045069 keyhole-limpet hemocyanin Proteins 0.000 description 1
- 229960001320 lapatinib ditosylate Drugs 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000005265 lung cell Anatomy 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 108010082117 matrigel Proteins 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 238000002493 microarray Methods 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- AZBFJBJXUQUQLF-UHFFFAOYSA-N n-(1,5-dimethylpyrrolidin-3-yl)pyrrolidine-1-carboxamide Chemical compound C1N(C)C(C)CC1NC(=O)N1CCCC1 AZBFJBJXUQUQLF-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- CERZMXAJYMMUDR-UHFFFAOYSA-N neuraminic acid Natural products NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO CERZMXAJYMMUDR-UHFFFAOYSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 108091027963 non-coding RNA Proteins 0.000 description 1
- 102000042567 non-coding RNA Human genes 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- OIPZNTLJVJGRCI-UHFFFAOYSA-M octadecanoyloxyaluminum;dihydrate Chemical compound O.O.CCCCCCCCCCCCCCCCCC(=O)O[Al] OIPZNTLJVJGRCI-UHFFFAOYSA-M 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 125000005061 octahydroisoindolyl group Chemical group C1(NCC2CCCCC12)* 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000004287 oxazol-2-yl group Chemical group [H]C1=C([H])N=C(*)O1 0.000 description 1
- 125000003145 oxazol-4-yl group Chemical group O1C=NC(=C1)* 0.000 description 1
- 125000004304 oxazol-5-yl group Chemical group O1C=NC=C1* 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000005968 oxazolinyl group Chemical group 0.000 description 1
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 238000004091 panning Methods 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- LCLHHZYHLXDRQG-ZNKJPWOQSA-N pectic acid Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)O[C@H](C(O)=O)[C@@H]1OC1[C@H](O)[C@@H](O)[C@@H](OC2[C@@H]([C@@H](O)[C@@H](O)[C@H](O2)C(O)=O)O)[C@@H](C(O)=O)O1 LCLHHZYHLXDRQG-ZNKJPWOQSA-N 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NMHMNPHRMNGLLB-UHFFFAOYSA-N phloretic acid Chemical compound OC(=O)CCC1=CC=C(O)C=C1 NMHMNPHRMNGLLB-UHFFFAOYSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 150000008104 phosphatidylethanolamines Chemical group 0.000 description 1
- 150000003910 phosphatidylinositol 3-phosphates Chemical class 0.000 description 1
- 150000003911 phosphatidylinositol 4-phosphates Chemical class 0.000 description 1
- 150000003905 phosphatidylinositols Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- DCWXELXMIBXGTH-QMMMGPOBSA-N phosphonotyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-QMMMGPOBSA-N 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 1
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical compound OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 1
- 125000005545 phthalimidyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 229920000191 poly(N-vinyl pyrrolidone) Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 239000010318 polygalacturonic acid Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003140 primary amides Chemical class 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 238000000734 protein sequencing Methods 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 230000012743 protein tagging Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 239000002213 purine nucleotide Substances 0.000 description 1
- 125000004289 pyrazol-3-yl group Chemical group [H]N1N=C(*)C([H])=C1[H] 0.000 description 1
- 125000004497 pyrazol-5-yl group Chemical group N1N=CC=C1* 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- DRYRBWIFRVMRPV-UHFFFAOYSA-N quinazolin-4-amine Chemical compound C1=CC=C2C(N)=NC=NC2=C1 DRYRBWIFRVMRPV-UHFFFAOYSA-N 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 108091006082 receptor inhibitors Proteins 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000001603 reducing effect Effects 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 230000008521 reorganization Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000002342 ribonucleoside Substances 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 238000013391 scatchard analysis Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 210000000717 sertoli cell Anatomy 0.000 description 1
- 239000012679 serum free medium Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- ILJOYZVVZZFIKA-UHFFFAOYSA-M sodium;1,1-dioxo-1,2-benzothiazol-3-olate;hydrate Chemical compound O.[Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 ILJOYZVVZZFIKA-UHFFFAOYSA-M 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 229940035044 sorbitan monolaurate Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 235000003702 sterols Nutrition 0.000 description 1
- 150000003432 sterols Chemical class 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000002344 surface layer Substances 0.000 description 1
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000004114 suspension culture Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 125000006318 tert-butyl amino group Chemical group [H]N(*)C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000004495 thiazol-4-yl group Chemical group S1C=NC(=C1)* 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000002769 thiazolinyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004001 thioalkyl group Chemical group 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 239000011573 trace mineral Substances 0.000 description 1
- 235000013619 trace mineral Nutrition 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229940117013 triethanolamine oleate Drugs 0.000 description 1
- 229940073585 tromethamine hydrochloride Drugs 0.000 description 1
- 230000010415 tropism Effects 0.000 description 1
- 239000002753 trypsin inhibitor Substances 0.000 description 1
- 230000010304 tumor cell viability Effects 0.000 description 1
- 229940094060 tykerb Drugs 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000028973 vesicle-mediated transport Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/498—Pyrazines or piperazines ortho- and peri-condensed with carbocyclic ring systems, e.g. quinoxaline, phenazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39558—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against tumor tissues, cells, antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/34—Identification of a linear epitope shorter than 20 amino acid residues or of a conformational epitope defined by amino acid residues
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
Definitions
- This invention relates to methods of treating cancer with a quinaxoline PI3K inhibitor in combination with one or more additional kinase inhibitors.
- Phosphatidylinositol 3-kinase is composed of a 1 10 kDa catalytic subunit (encoded by the PIK3CA gene) and an 85 kDa regulatory subunit.
- the catalytic subunit uses ATP to phosphorylate Ptdlns, PtdIns4P, and PtdIns(4,5)P2 to create the second messengers PtdIns3P, PtdIns(3,4)P2, and PtdIns(3,4,5)P3 (PIP3).
- PTEN a tumor suppressor which inhibits cell growth through multiple mechanisms, can dephosphorylate PIP3, the major product of PIK3CA.
- PIP3 in turn, is required for translocation of protein kinase B (AKT1, PKB) to the cell membrane, where it is phosphorylated and activated by upstream kinases.
- AKT1 protein kinase B
- PKB protein kinase B
- PI3Koc has been implicated in the control of cytoskeletal reorganization, apoptosis, vesicular trafficking, proliferation, and differentiation processes.
- Increased copy number and expression of PIK3CA or activating mutations in PIK3CA are associated with a number of malignancies such as ovarian cancer (Campbell et al., Cancer Res 2004, 64, 7678- 7681 ; Levine et al., Clin Cancer Res 2005, 11, 2875-2878; Wang et al., Hum Mutat 2005, 25, 322; Lee et al., Gynecol Oncol 2005, 97, 26-34), cervical cancer, breast cancer (Bachman, et al.
- combining treatments with different mechanisms of action may lead to enhanced anti-tumor activity as compared to single treatments administered alone.
- activation of the PI3K pathway may contribute to the resistance of human tumor cells to certain chemotherapeutic agents, such as microtubule stabilizing agents like taxol (Brognard, J., et. al. Cancer Res 2001, 61, 3986-3997; Clark, A. S., et. al. Mol Cancer Ther 2002, 1, 707-717; Kraus, A. C, et. al. Oncogene 2002, 21, 8683-8695; Krystal, G. W., et. al. Mol Cancer Ther 2002, 1, 913-922; and Yuan, Z. Q., et. al. / Biol Chem 2003, 278, 23432- 23440).
- compositions of the invention are used to treat diseases associated with abnormal and or unregulated cellular activities.
- Disease states which can be treated by the methods and compositions provided herein include cancer.
- the invention is directed to methods of treating these diseases by administering a compound of formula I or 1(a) in combination with one or more treatments.
- One aspect of the invention is directed to a method of treating cancer which method comprises administering to a patient a therapeutically effective amount of a compound of formula I:
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula I and a pharmaceutically acceptable carrier, excipient, or diluent in combination with one or more inhibitors of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2, where the compound of formula I is that wherein:
- R 51 is hydrogen or alkyl
- R 52 is hydrogen or halo
- R 50 , R 53 , and R 54 are independently hydrogen, alkyl, alkenyl, halo, haloalkyl, haloalkenyl, hydroxy, alkoxy, alkenyloxy, haloalkoxy, nitro, amino, alkylamino, dialkylamino, -N(R 55 )C(0)-Ci-C 6 -alkylene-N(R 55a )R 55 , alkylcarbonyl, alkenylcarbonyl, carboxy, alkoxycarbonyl, cyano, alkylthio, -S(0) 2 NR 55 R 55a , or alkylcarbonylamino, and R 55 and R 55b are independently hydrogen, alkyl, or alkenyl, and R a is hydrogen, alkyl, alkenyl, hydroxy, or alkoxy; or R 53 and R 54 together with the carbons to which they are attached form a 5- or 6-membered heteroaryl or 5- or 6-membered heterocyclo
- B is phenyl substituted with R 3a and optionally further substituted with one, two, or three R 3 ; or
- B is heteroaryl optionally substituted with one, two, or three R 3 ;
- R 3a is cyano, hydroxyamino, carboxy, alkoxycarbonyl, alkylamino, dialkylamino,
- alkylcarbonyl haloalkoxy, alkylsulfonyl, aminoalkyloxy, alkylaminoalkyloxy, or dialkylaminoalkyloxy; or
- R 7 is hydrogen, alkyl, or alkenyl
- R 7a and R 7b are independently hydrogen, alkyl, alkenyl, hydroxyalkyl, haloalkyl, alkoxy, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroarylalkyl, aryl, arylalkyl, or arylalkyloxy, and where the aryl, cycloalkyl, heterocycloalkyl and heteroaryl rings in R 7a and R 7b (either alone or as part of arylalkyl, cycloalkylalkyl, heterocycloalkylalkyl, where the aryl, cycloalkyl, heterocycloalkyl and heteroaryl rings in R 7a and R 7b (either alone or as
- R 8 is hydrogen, hydroxy, alkoxy, alkyl, alkenyl, haloalkyl, or haloalkoxy
- R 8a is hydrogen, alkyl, alkenyl, hydroxyalkyl, cyanoalkyl, alkoxyalkyl, alky!thioalkyl, heterocycloalkyl, heterocycloalkylalkyl, cycloalkyl, cycloalkylalkyl, heteroaryl, heteroarylalkyl, aryl, or arylalkyl, and where the aryl, cycloalkyl, heteroaryl, and heterocycloalkyl rings in R 8a (either alone or as part of arylalkyl, cycloalkylalkyl, heterocycloalkylalkyl and heteroarylalkyl) are independently optionally substituted with 1 , 2, or 3 groups independently selected from alkyl, alkenyl, alkoxy,
- R 9 is hydrogen, hydroxy, alkoxy, alkyl, alkenyl, haloalkyl, or haloalkoxy
- R 9a is hydrogen, C2-C6-alkyl, alkenyl, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroarylalkyl, aryl, or arylalkyl, and where the aryl, cycloalkyl, heteroaryl, and heterocycloalkyl rings in R 9a (either alone or as part of arylalkyl, cycloalkylalkyl, heterocycloalkylalkyl, and heteroarylalkyl) are independently optionally substituted with 1, 2, or 3 groups independently selected from alkyl, alkenyl, alkoxy, hydroxy, hydroxyalkyl, hal
- R IOa is hydrogen, hydroxy, alkoxy, alkyl, alkenyl, haloalkyl, aminoalkyi, alkylaminoalkyl, dialkylaminoalkyl, or hydroxyalkyl
- R 10 and R 10 are independently hydrogen, alkyl, alkenyl, haloalkyl, aminoalkyi, alkylaminoalkyl, dialkylaminoalkyl, or hydroxyalkyl;
- R 1 la is hydrogen, alkyl, alkenyl, hydroxy, or alkoxy
- R" and R l lb are independently hydrogen, alkyl, alkenyl, aminoalkyi, alkylaminoalkyl, or dialkylaminoalkyl
- R 12 is heterocycloalkyl optionally substituted with 1, 2, or 3 groups selected from alkyl, oxo, amino, alkylamino, and heterocycloalkylalkyl;
- R 13 is hydrogen, alkyl, or alkenyl, and R 13a is aminoalkyi, alkylaminoalkyl, dialkylaminoalkyl, aryl, or arylalkyi;
- R 14 , R 14a , and R l4b are independently hydrogen, alkyl, or alkenyl;
- heteroaryl optionally substituted with one or two aminoalkyi, alkylaminoalkyl, or dialkylaminoalkyl;
- R 18a is hydrogen, alkyl, alkenyl, or alkoxy
- R 18 and R l8b are independently hydrogen, alkyl, or alkenyl
- R l9a is amino, alkylamino, dialkylamino, or heterocycloalkyl
- R 20 is hydrogen, alkyl, or alkenyl
- R 20a is cycloalkyl or heterocycloalkyl
- R 21a and R 21b are independently hydrogen, alkyl, or alkenyl; q) -N(R 22 )C(0)-C r C 6 -alkylene-N(R 22b )-N(R 22c )(R 22a ), where R", R ⁇ a and R" 2D are independently hydrogen, alkyl, or alkenyi;
- R 24 is hydrogen, alkyl, or alkenyi
- R 24a is alkoxyalkyl or aryl optionally substituted with one or two halo or alkyl
- each of the alkylene in R 3a is independently optionally further substituted with 1 , 2, 3, 4, or 5 groups selected from halo, hydroxy, amino, alkylamino, and dialkylamino
- each R 3 (when R 3 is present) is independently alkyl, alkenyi, alkynyl, halo, hydroxy, oxo, alkoxy, cyano, hydroxyamino, carboxy, alkoxycarbonyl, amino, alkylamino, dialkylamino, alkylcarbonyl, haloalkoxy, alkylsulfonyl, aminoalkyloxy,
- alkylaminoalkyloxy or dialkylaminoalkyloxy
- R 7 is hydrogen, alkyl, or alkenyi
- R 7a and R 7b are independently hydrogen, alkyl, alkenyi, hydroxyalkyl, haloalkyl, alkoxy, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl, cycloalkyl, cycloalkylalkyi, heterocycloalkyi, heterocycloalkylalkyl, heteroaryl, heteroarylalkyl, aryl, arylalkyl, or arylalkyloxy, and where the aryl, cycloalkyl, heterocycloalkyi and heteroaryl rings in R 7a and R 7b (either alone or as part of arylalkyl, cycloalkylalkyi, heterocycloalkylalkyl,
- R 8 is hydrogen, hydroxy, alkoxy, alkyl, alkenyi, haloalkyl, or haloalkoxy
- R 8a is hydrogen, alkyl, alkenyi, hydroxyalkyl, cyanoalkyl, alkoxyalkyl, alkylthioalkyl, heterocycloalkyi, heterocycloalkylalkyl, cycloalkyl, cycloalkylalkyi, heteroaryl, heteroarylalkyl, aryl, or arylalkyl, and where the aryl, cycloalkyl, heteroaryl, and heterocycloalkyi rings in R 8a (either alone or as part of arylalkyl, cycloalkylalkyi, heterocycloalkylalkyl and heteroarylalkyl) are independently optionally substituted with 1 , 2, or 3 groups independently selected from alkyl, alkenyi, alkoxy,
- R 9 is hydrogen, hydroxy, alkoxy, alkyl, alkenyi, haloalkyl, or haloalkoxy
- R 9a is hydrogen, C 2 -C6-alkyl, alkenyi, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyi, heterocycloalkyi, heterocycloalkylalkyl, heteroaryl, heteroarylalkyl, aryl, or arylalkyl, and where the aryl, cycloalkyl, heteroaryl, and heterocycloalkyl rings in R 9a (either alone or as part of arylalkyl, cycloalkylalkyl, heterocycloalkyialkyl and heteroarylalkyl) are independently optionally substituted with 1, 2, or 3 groups independently selected from alkyl, alkenyl, alkoxy, hydroxy, hydroxyalkyl, hal
- R IOa is hydrogen, hydroxy, alkoxy, alkyl, alkenyl, haloalkyl, or hydroxyalkyl
- R 10 and R IOb are independently hydrogen, alkyl, alkenyl, haloalkyl, or hydroxyalkyl
- R 1 la is hydrogen, alkyl, alkenyl, hydroxy, or alkoxy
- R 11 and R l lb are independently hydrogen, alkyl, alkenyl, aminoalkyi
- alkylaminooalkyl or dialkylaminoalkyl
- R 12 is heterocycloalkyl optionally substituted with 1, 2, or 3 groups selected from alkyl, oxo, amino, alkylamino, and heterocycloalkyialkyl;
- R 13 is hydrogen, alkyl, or alkenyl and R 13a is aminoalkyi, alkylaminoalkyl, dialkylaminoalkyl, aryl, or arylalkyl);
- R 14 , R 1 a , and R 1 b are independently hydrogen, alkyl, or alkenyl;
- heteroaryl optionally substituted with one or two aminoalkyi, alkylaminoalkyl, or dialkylaminoalkyl;
- R 18a is hydrogen, alkyl, alkenyl, or alkoxy
- R 18 and R 18b are independently hydrogen, alkyl, or alkenyl
- R 19a is amino, alkylamino, dialkylamino, or heterocycloalkyl
- R 20 is hydrogen, alkyl, or alkenyl
- R 20a is cycloalkyl or heterocycloalkyl; p) -NR 2, S(0) 2 -Ci-C6-alkylene-N(R 2l )R 2la , where R 21 is hydrogen, alkyl, or alkenyl, and
- R 2l and R 21b are independently hydrogen, alkyl, or alkenyl
- R 22 , R 22a and R 22b are independently hydrogen, alkyl, or alkenyl;
- R 23 , R 23a and R 23b are independently hydrogen, alkyl, or alkenyl; or
- R 24 is hydrogen, alkyl, or alkenyl, and R 24a is alkoxyalkyl or aryl optionally substituted with one or two halo or alkyl;
- each of the alkylene in R 3 is independently optionally further substituted with 1 , 2, 3,
- R 50 and R 52 are hydrogen, R 51 is hydrogen or methyl, R 53 is hydrogen or methoxy, and R 54 is hydrogen or methoxy, then B is not 2,3-dihydro-l ,4-benzodioxinyl, thien-2-yl, or thien-2-yl, substituted with one R 3 , where R 3 is halo.
- an inhibitor of HER3, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), or MEKK kinases/kinase receptors can be a functional nucleic acid, while in another it can be an antibody or protein display scaffold.
- an inhibitor of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, or FGFR2 can be a functional nucleic acid, lapatinib, while in another it can be an antibody or protein display scaffold.
- methods for treating a patient with HER2 non- overexpressing cancer, comprising administering to the patient a therapeutically effective amount of compound A and a therapeutically effective amount of MM- 121.
- the cancer comprises a non-HER2 amplified tumor.
- the combination exhibits therapeutic synergy in the treatment of HER2 non-overexpressing cancer.
- the combination effects a Iog
- the cancer comprises a HER2 non-overexpressing cancer.
- the cancer comprises a non-HER2 amplified tumor.
- a therapeutically effective amount of Compound A is coadministered with a therapeutically effective amount of MM- 121.
- the combination of Compound A and MM121 exhibits therapeutic synergy in the treatment of cancer.
- the combination of Compound A and MM 121 exhibits effects a logio cell kill of at least 2.8, at least 2.9 or at least 3.0.
- the cancer is lung cancer.
- Figure 1 A siRNA screen to identify kinases that potentially compensate for PI3K inhibition.
- HCCI 37 breast cancer cells were treated with the indicated amounts of compound A for 6, 24, or 72 hours in medium containing 2.5% FBS. Cell lysates analyzed by immunoblotting with the indicated antibodies.
- Figure IB siRNA screen to identify kinases that potentially compensate for PI3K inhibition.
- HCC1937 cells were seeded in triplicate in a 96-well plate and treated with DMSO or the indicated concentration of compound A for 72 hours. Cell viability was measured by the alamar blue assay.
- Figure 2 Scheme of the RNAi screen.
- the Dharmacon RTF Protein Kinase siRNA library which contains SMARTpool siRNAs targeting 779 kinases, was used.
- HCC1937 cells were reverse-transfected in 96-well plates with 5 pmol siRNA/well. Plates were split 1 :6 and treated with DMSO or 10 ⁇ compound A for 72 hours. Cell viability was measured using the alamar blue assay. The screen has been performed twice.
- FIG. 3A and 3B PI3K inhibition induces phosphorylation of RTKs and intracellular kinases.
- HCC1937 cells were treated with 10 ⁇ compound A in medium containing 2.5% FBS for 0, 8, 24, or 48 hours. Media and drugs were replenished every 24 hours. Lysates were used to probe phospho-RTK arrays (Fig. 3A) or phospho-kinase arrays (Fig. 3B) (R&D systems). Corner spots are positive controls.
- Candidate kinases from the siRNA screen that are also phosphorylated upon treatment with compound A are shown in red.
- FIG. 4A and 4B HCC1 37 cells were treated with 10 ⁇ compound A in medium containing 2.5% FBS for 0, 8, 24, or 48 hours. Media and drugs were replenished every 24 hours. Lysates were used to probe phospho-RTK arrays (A) or phospho-kinase arrays (B) (R&D systems). Corner spots are positive controls. Candidate kinases from the siRNA screen that are also phosphorylated upon treatment with compound A are shown in red.
- FIG. 1 HCC1937 cells were treated with 10 ⁇ compound A in medium containing 2.5% FBS for 0, 2, 8, or 24 hours. RNA was isolated with Trizol and purified using the RNeasy column (Qiagen). Gene expression from three independent experiments was measured using the Gene titan 3' microarray. *, p ⁇ 0.05; **, p ⁇ 0.01 versus 0 hours.
- FIG. 6A HER3 siRNA enhances the anti-proliferative effect of a PI3K inhibitor.
- BT474 Cells were transfected with control or HER3 siRNA oligonucleotides followed by treatment with 6 ⁇ compound A in medium containing 2.5% FBS. Cells were harvested for proliferation assay (A).
- FIG. 6B HER3 siRNA enhances the anti-proliferative efffect of a PI3K inhibitor. Crystal violet staining on day 6 post-transfection.
- FIG. 7 BT-474 cells were treated with 6 uM compound A. Fresh medium and inhibitor were replenished every 24 hours. At the indicated times, cells were harvested in NP- 40 lysis buffer containing protease and phosphatase inhibitors. Lysates were prepared, separated by SDS-PAGE, and subjected to immunoblot analysis with the indicated antibodies.
- FIG. 8 A and 8B MDA-453 (A) and SKBR-3 (B) cells were transfected with 10 nM HER3 or control siRNA duplexes in the presence of lipofectamine RNAiMAX as described in Fig. 7.
- Figure 9A depicts a bar chart representing the growth of SKBR3, BT474, HCC1937, MDAMMB453, HCC1954, UACC893, and SUM190 cells in the presence of compound A.
- Figure 9B depicts photomicrographs of BT474, HCC 1937, MDA453, and HCC1954 cells cultured in Matrigel in the absence and presence of 0-20 ⁇ compound A.
- Figure 10A depicts a bar graph of percent of cell number relative to the initial amount of plated cells as provided in Figure 9A above was calculated for each breast cancer cell line treated with the indicated concentrations of compound A. Numbers below 100% (straight black line) are indicative of apoptosis.
- Figure 10B depicts immunoblots wherein cells were treated with 0-20 uM compound A for 24 hours. Cell lysates were prepared and subjected to immunob!otting with the antibodies indicated to the left of the panels. The upper arrow indicates total PARP and lower arrow indicates cleaved PARP.
- Figure 1 1 depicts immunoblots of various cell lines treated overnight in serum- free medium with 0-20 uM compound A. Cells were harvested and lysates were used for immunobloting analysis with the indicated antibodies.
- Figure 12A depicts a bar chart of timed HER3 mRNA transcription in BT474 cells which were treated with 6 uM compound A for the indicated times prior to RNA isolation and real-time qPCR analysis with HER3-specific primers.
- Figure 12B depicts a bar chart of timed HER3 mRNA transcription in BT474 cells which were treated with 2 uM 5J8, 20 uM LY294002, and 50 nM rapamycin for 10 hours prior to RNA isolation and qPCR analysis with HER3-specific primers.
- Figure 12C depicts a bar chart of HER3 mRNA transcription in MDA453 and SKBR3 cells which were treated with 6 ⁇ compound A over the indicated time course up to 48 hours prior to RNA isolation and qPCR analysis with HER3-specific primers.
- Figure 12D depicts a photomicrrograph of immunoblots of cell lysates from BT474 and MDA453 cells which were treated with DMSO (control), 6 uM compound A, or 2 uM 5J8 for 4 hours.
- Nuclear (Nu) and cytoplasmic (Cy) extracts were isolated and subjected to immunoblotting with FoxOl and Fox03a antibodies. Loading controls for Nu: HDAC3; for Cy: RhoA (BT474) and MEK1/2 (MDA453). Arrow indicates the Fox03a-specific band.
- Figure 12E depicts a bar chart representing BT474, MDA453, and SKBR3 cells which were transfected with either control or FoxOl and Fox03a-specific siRNA duplexes. Two days after transfection cells were treated with compound A for 6 hours prior to harvesting, RNA preparation, and qPCR analysis for HER3.
- Figure 12F depicts a bar chart representing BT474, MDA453 and SKBR3 cells which were transfected with either control or FoxOl and Fox03a-specific siRNA duplexes. Two days after transfection cells were treated with compound A for 6 hours prior to harvesting, RNA preparation, and qPCR analysis for FoxOl and Fox03a.
- Figure 13A depicts a photomicrrograph of immunoblots of cell lysates from BT474 cells which were treated with 6 uM compound A over a time course up to 72 hours and subjected to immunoblotting using the various antibodies listed on the Y-axis, compound A and media were replenished every 24 hours.
- Figure 13B depicts a photomicrrograph of immunoblots of cell lysates from BT474 cells which were treated with 6 ⁇ or 20 uM compound A for 0-48 hours. Cell lysates were subjected to immunobloting with the indicated antibodies.
- Figure 13C depicts a photomicrrograph of immunoblots of cell lysates from BT474 cells which were treated with 0-20 uM compound A for 24 hours. Cells were then lysed, and 0.5 mg of lysate subjected to immunoprecipitation (IP) with a p85 antibody followed by p85 and pTyr.
- IP immunoprecipitation
- Figure 13D depicts a photomicrrograph of immunoblots of cell lysates from BT474 cells which were treated with 0-20 uM compound A for 24 hours. Cells were then lysed, and 0.5 mg of lysate subjected to immunoprecipitation ( ⁇ ) with a p85 antibody followed by p85 and HER3.
- Figure 13E depicts a photomicrrograph of immunoblots of cell lysates from BT474 cells which were transfected with control or HER3-specific siRNA duplexes, followed by treatment with 6 uM compound A for 24 hours.
- Cell lysates were prepared, and 0.5 mg subjected to IP with a p85 antibody. Immune complexes were separated by SDS- PAGE and immunoblotted with p85 and HER3 antibodies.
- Figure 13F depicts a photomicrrograph of immunoblots of cell lysates from BT474 cells which were transfected with control or HER3-specific siRNA duplexes, followed by treatment with 6 ⁇ compound A for 24 hours.
- Cell lysates were prepared, and 0.5 mg subjected to IP with a p85 antibody.
- Immune complexes were separated by SDS- PAGE and immunoblotted with p85 and pHER3 YI 197 antibodies.
- Figure 13G depicts a bar-graph representing BT474 cells which were transfected with HER3-specific siRNA and one day post-transfection treated with DMSO or 2 ⁇ compound A. Growth medium and inhibitor were replenished every 3 days. Cells were harvested for counting on day 6.
- Figure 13H depicts a photomicrograph representing BT474 cells which were transfected with HER3-specific siRNA and one day post-transfection treated with DMSO or 2 ⁇ compound A. Growth medium and inhibitor were replenished every 3 days. Cells were crystal violet stained on day 6 and photographed.
- Figure 14A depicts a bar-graph representing number of PI stained BT474 cells after transfection with HER3 siRNA or control duplexes and then treated with 6 ⁇ compound A for 3 days. At this time, cells were washed, harvested, and prepared for cell cycle analysis.
- Figure 14B depicts a photomicrograph of an immunoblot of cell lysates from BT474 cells which were transfected with control or HER3-specific siRNA duplexes, followed by treatment with 6 uM compound A and then immunoblotted with HER3 and PARP antibodies.
- Figure 15A depicts a photomicrograph of immunoblots of cell lysates from
- MDA453 cells which were transfected with control or HER3-specific siRNA duplexes, followed by treatment with 6 uM compound A for 24 hours. Cell lysates were prepared and subjected to immunoblotting with the indicated antibodies on the Y-axis.
- Figure 15B depicts a bar graph illustrating the growth or decline of MDA453 cells as indicated in Fig. 7A, except cells were allowed to grow while replenishing fresh medium and compound A (2uM) every 3 days and counted on day 10.
- Figure 15C depicts a photomicrograph of immunoblots of cell lysates from
- SKBR3 cells which were transfected with control or HER3-specific siRNA duplexes, followed by treatment with 6 uM compound A for 24 hours.
- Cell lysates were prepared and subjected to immunoblotting with the indicated antibodies on the Y-axis.
- Figure 15D depicts a bar graph illustrating the growth or decline of SKBR3 cells as indicated in Fig. 7C, except cells were allowed to grow while replenishing fresh medium and compound A (2uM) every 3 days and counted on day 10.
- Figure 16A depicts a bar-graph illustrating the cell growth or inhibition of BT474 cells which were treated in the absence or presence of 2 uM compound A alone or in combination with 0.1 uM lapatinib (Lap).
- Figure 16B depicts a bar-graph illustrating the cell growth or inhibition of BT474 cells which were treated in the absence or presence of 2 ⁇ compound A alone or in combination with 10 g/ml trastuzumab (Tras).
- Figure 16C depicts a photomicrograph of immunoblots for biomarkers of apoptosis and G
- Figure 16D depicts a bar-graph indicating real-time qPCR analysis of HER3 mRNA in cells treated with compound A (6 uM), lapatinib (1 ⁇ ), trastuzumab (10 ⁇ ), or the indicated combinations for 10 hours.
- Figure 16E depicts photomicrograph of an immunoblot of cell lysates from BT474 cells which measured the presence of HER3 and phosphorylated HER3 in the cell lysates after treatment with compound A (6 uM), lapatinib (1 uM), trastuzumab (10 ⁇ ), or the indicated combinations over a time course (0-24 hours).
- Figure 17A depicts a bar-graph illustrating the cell growth or inhibition of HER2- dependent MDAMB453 cells treated with 0.1 uM lapatinib, or 2 uM compound A either alone or in the indicated combinations for a total of 6 days.
- Figure 17B depicts a bar-graph illustrating the cell growth or inhibition of HER2- dependent MDAMB453 cells treated with 10 ⁇ g ml trastuzumab, or 2 ⁇ compound A either alone or in the indicated combinations for a total of 6 days.
- Figure 17C depicts a bar-graph illustrating the cell growth or inhibition of HER2- dependent SKBR3 cells treated with 0.1 ⁇ lapatinib, or 2 uM compound A either alone or in the indicated combinations for a total of 6 days.
- Figure 17D depicts a bar-graph illustrating the cell growth or inhibition of HER2- dependent SKBR3 cells treated with 10 ⁇ g/ml trastuzumab, or 2 ⁇ compound A either alone or in the indicated combinations for a total of 6 days.
- Figure 18B depicts photomicrographs of immunohistochemistry sections from formalin-fixed, paraffin-embedded tumor blocks. Xenografts were harvested on day 28, 1 hour after the last dose of lapatinib and/or compound A. Antibodies used were against HER3, pHER3 YI289 , and pAKT 5473 . Photographs were taken at 400x magnification (scale bar: 50 ⁇ ).
- Figure 18C depicts bar graphs representing the Histoscore (H-score) analysis of immunostained sections as provided in Fig. 18B.
- Figure 19A depicts a photomicrograph of an immunoblot representing BT474 cells which were transfected with HER3 siRNA duplexes and treated with compound A for 24 hours.
- Cell lysates were prepared and 0.5 mg was immunoprecipitated with a p85 antibody. Immune complexes were next subjected to immunoblotting with antibodies indicated to the right of the panel. Cell lysates from BT474 cells treated with and without 1 uM lapatinib for 6 hours were used as controls (lanes 1 & 2).
- FIG. 19B depicts a photomicrograph of BT474 cells were treated with 6 uM compound A over a time course up to 24 hours as indicated.
- Cell lysates were prepared, and 200 ⁇ g of total protein were hybridized to arrays containing probes for 42 different receptors of tyrosine kinase (RTKs).
- RTKs tyrosine kinase
- FIG. 19C depicts a photomicrograph of BT474 cells were treated with 6 ⁇ compound A over a time course up to 24 hours as indicated.
- Cell lysates were prepared, and 500 ⁇ g of total protein were hybridized to arrays containing probes for 42 different receptors of tyrosine kinase (RTKs)
- RTKs tyrosine kinase
- Figure 1 D depicts a bar-graph representing the amount of RTK mRNA as identified using real-time qPCR analysis of the indicated RTKs in RNA collected from BT474 cells treated with either DMSO or 10 ⁇ compound A for 6 hours.
- Figure 1 E depicts a bar-graph representing the amount of IGF-IR, InsR, and FGFR2 mRNA as identified using real-time qPCR in RNA extracted from BT474 cells transfected with FoxOl and Fox03a siRNA duplexes and then treated with 10 uM compound A for 6 hours.
- Figure 20A depicts a bar-graph representing IGF1R mRNA levels in BT474, MDA453, and MCF7 cells which were transfected with IGF-IR siRNA or control duplexes. Forty-eight hours later, RNA was isolated and subjected to qPCR analysis for IGF- IRmRNA levels.
- Figure 20B depicts a bar-graph representing InsR mRNA levels in BT474, MDA453, and MCF7 cells which were transfected with InsR siRNA or control duplexes. Forty-eight hours later, RNA was isolated and subjected to qPCR analysis for InsR mRNA levels.
- Figure 20C depicts a bar-graph representing inhibition of BT474 cells which were transfected with either IGF-IR siRNA or InsR siRNA or control duplexes. Forty-eight hours later they were treated with 10 ⁇ compound A for a total of 4 days. At this time, the cells were counted.
- Figure 20D depicts a bar-graph representing inhibition of MDAMB453 cells which were transfected with either IGF-IR siRNA, InsR siRNA, or control duplexes. Forty- eight hours later they were treated with 10 ⁇ compound A for a total of 4 days. At this time, the cells were counted.
- Figure 20E depicts a bar-graph representing inhibition of MCF7 cells which were transfected with either IGF-IR siRNA, InsR siRNA, or control duplexes. Forty-eight hours later they were treated with 10 ⁇ compound A for a total of 4 days. At this time, the cells were counted.
- administering and variants thereof (e.g., “administering” a compound) in reference to a compound of the invention means introducing the compound or a prodrug of the compound into the system of the animal in need of treatment.
- a compound of the invention or prodrug thereof is provided in combination with one or more other active agents (e.g., surgery, radiation, chemotherapy, etc.)
- “administration” and its variants are each understood to include concurrent and sequential introduction of the compound or prodrug thereof and other agents.
- alkenyl or "lower alkenyl” means a straight or branched hydrocarbon radical having from 2 to 6 carbon atoms and at least one double bond. Representative examples include ethenyl, propenyl, l-but-3-enyl, 1 -pent-3-enyl, l-hex-5-enyl, and the like.
- alkenylcarbonyl means a C(0)R group, where R is alkenyl, as defined herein.
- alkenyloxy or “lower alkenyloxy” means an -OR group where R is alkenyl, as defined herein. Representative examples include methoxy, ethoxy, 1 -methoxyprop- l-en-3-yl, propoxy, isopropoxy, cyclopropyloxy, cyclohexyloxy, and the like.
- Alkoxy or “lower alkoxy” means an -OR group where R is alkyl, as defined herein. Representative examples include methoxy, ethoxy, I -methoxyprop-l -en-3-yl, propoxy, isopropoxy, cyclopropyloxy, cyclohexyloxy, and the like.
- Alkoxyalkyl means an alkyl group, as defined herein, substituted with one, two, or three alkoxy groups, as defined herein.
- Akoxycarbonyl means a -C(0)OR group, where R is alkyl, as defined herein.
- Alkoxyycarbonylalkyl means an alkyl group, as defined herein, substituted with one, two, or three alkoxycarbonyl groups, as defined herein.
- Alkyl or "lower alkyl” means a linear or branched hydrocarbon group having one to six carbon atoms. Examples of lower alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, s-butyl, f-butyl, isobutyl, pentyl, hexyl, and the like.
- a "Co” alkyl (as in “Co-Ce-alkyl”) is a covalent bond.
- Ce alkyl refers to, for example, n-hexyl, wo-hexyl, and the like.
- Alkylamino means a -NHR radical, where R is alkyl, as defined herein, or an N-oxide derivative thereof, e.g., methylamino, ethylamino, n-, /. o-propylamino, «-, iso-, tert- butylamino, methylamino-N-oxide, and the like.
- Alkylaminoalkyl means an alkyl group substituted with one or two alkylamino groups, as defined herein.
- Alkylaminoalkyloxy means an -OR group where R is alkylaminoalkyl, as defined herein.
- Alkylcarbonyl means a C(0)R group where R is alkyl, as defined herein.
- Alkylcarbonylamino means a -NRC(0)R' group, where R is hydrogen or alkyl, as defined herein, and R' is alkyl, as defiend herein.
- Alkylene refers to straight or branched divalent hydrocarbon, containing no unsaturation and having from two to eight carbon atoms.
- alkylene include eth- diyl (-CH2CH2-), prop-1,3-diyl (-CH 2 CH 2 CH 2 -), 2,2-dimethylprop-l ,3-diyl
- Alkylsulfonyl means a -S(0) 2 R group, where R is alkyl, as defined herein.
- Alkylthio means a -SR group, where R is alkyl, as defined herein.
- alkylthio include methylthio, ethylthio, and the like.
- Alkylthioalkyl means an alkyl group substituted with one or two alkylthio groups, as defined herein, e.g. 2-(methylthio)-ethyl and 2-(ethylthio)-ethyl.
- Alkynyl or “lower alkynyl” means a straight or branched hydrocarbon radical having from 2 to 6 carbon atoms and at least one triple bond. Representative examples include ethynyl, propynyl, butynyl, pentyn-2-yl, and the like.
- Amino means a -NH 2 group.
- aminoalkyl means an alkyl group substituted with at least one, for example one, two, or three, amino groups.
- aminoalkyloxy means an -OR group, where R is aminoalkyl, as defined herein.
- Antisense or “antisense oligonucleotide” refers to a nucleic acid molecule complementary to a portion of a particular gene transcript that can hybridize to the transcript and block its translation.
- An antisense oligonucleotide may comprise RNA or DNA.
- Aryl means a monovalent six- to fourteen-membered, mono- or bi-carbocyclic ring, wherein the monocyclic ring is aromatic and at least one of the rings in the bicyclic ring is aromatic. Representative examples include phenyl, naphthyl, and indanyl, and the like.
- Arylalkyl means an alkyl group, as defined herein, substituted with one or two aryl groups, as defined herein. Examples include benzyl, phenethyl, phenylvinyl, phenylallyl, and the like.
- Aryloxy means a -OR group, where R is aryl, as defined herein.
- Arylalkyloxy means a -OR group, where R is arylalkyl, as defined herein.
- Arylsulfonyl means a -SO2R group, where R is aryl, as defined herein.
- Carboxyalkyl means an alkyl group, as defined herein, substituted with one, two, or three -C(0)OH groups.
- Carboxy ester means a -C(0)OR group, where R is lower alkyl, lower alkenyl, lower alkynyl, cycloalkyl, aryl, or arylalkyl, each of which is defined herein. Representative examples include methoxycarbonyl, ethoxycarbonyl, benzyloxycarbonyl, and the like.
- Compound A is a compound of formula I and of Table I, having the structure:
- Cyanoalkyl means an alkyl, alkenyl, or alkynyl radical, as defined herein, substituted with at least one, for example one, two, or three, cyano groups.
- Cycloalkyl means a monocyclic or polycyclic hydrocarbon radical having three to thirteen carbon atoms.
- the cycloalkyl can be saturated or partially unsaturated, but cannot contain an aromatic ring. Cycloalkyl includes fused, bridged, and spiro ring systems.
- radicals examples include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- Cycloalkylalkyl means alkyl group substituted with one or two cycloalkyl groups, as defined herein. Representative examples include cyclopropylmethyl,
- Cycloalkylcarbonyl means a -C(0)R group, where R is cycloalkyl, as defined herein.
- Dialkylamino means a -NRR' radical, where R and R' are independently alkyl, as defined herein, or an N-oxide derivative, or a protected derivative thereof, e.g., dimethylamino, diethylamino, N,N-methylpropylamino, N,N-methylethylamino, and the like.
- Dialkylaminoalkyl means an alkyl group substituted with one or dialkylamino groups, as defined herein.
- Dialkylaminoalkyloxy means an -OR group, where R is dialkylaminoalkyl, as defined herein.
- fused ring system and "fused ring” refer to a polycyclic ring system that contains bridged or fused rings; that is, where two rings have more than one shared atom in their ring structures.
- fused-polycyclics and fused ring systems are not necessarily all aromatic ring systems.
- fused-polycyclics share a vicinal set of atoms, for example naphthalene or 1 ,2,3,4-tetrahydro-naphthalene.
- a spiro ring system is not a fused-polycyclic by this definition, but fused polycyclic ring systems of the invention may themselves have spiro rings attached thereto via a single ring atom of the fused-polycyclic.
- two adjacent groups on an aromatic system may be fused together to form a ring structure.
- the fused ring structure may contain heteroatoms and may be optionally substituted with one or more groups. It should additionally be noted that saturated carbons of such fused groups (i.e. saturated ring structures) can contain two substitution groups.
- Haloalkoxy means an -OR' group, where R' is haloalkyl as defined herein, e.g., trifluoromethoxy, 2,2,2-trifluoroethoxy, and the like.
- Haloalkoxyalkyl means an alkyl group, as defined herein, substituted with one, two, or three haloalkoxy, as defined herein.
- Halogen or "halo" means fluoro, chloro, bromo, or iodo.
- Haloalkenyl means an alkenyl group, as defined herein, substituted with one or more halogens, for example one to five halo atoms.
- Haloalkyl means an alkyl group, as defined herein, substituted with one or more halogens, for example one to five halo atoms. Representative examples includes
- Heteroaryl means a monocyclic, fused bicyciic, or fused tricyclic, monovalent radical of 5 to 14 ring atoms containing one or more, for example one, two, three, or four ring heteroatoms independently selected from -0-, -S(0) n - (n is 0, 1, or 2), -N-, -N(R X )-, and the remaining ring atoms being carbon, wherein the ring comprising a monocyclic radical is aromatic, and wherein at least one of the fused rings comprising a bicyciic or tricyclic radical is aromatic.
- R x is hydrogen, alkyl, hydroxy, alkoxy, acyl, or alkylsulfonyl.
- Fused bicyciic radical includes bridged ring systems.
- the valency may be located on any atom of any ring of the heteroaryl group, valency rules permitting. In particular, when the point of valency is located on the nitrogen, R is absent.
- heteroaryl includes, but is not limited to, 1,2,4-triazolyl, 1 ,3,5-triazolyl, phthalimidyl, pyridinyl, pyrrolyl, imidazolyl, thienyl, furanyl, indolyl, 2,3-dihydro-lH-indolyl (including, for example, 2,3-dihydro-lH- indol-2-yl, 2,3-dihydro- li/-indol-5-yl, and the like), isoindolyl, indolinyl, isoindolinyl, benzimidazolyl, benzodioxol-4-yl, benzofuranyl, cinnolinyl, indolizinyl, naphthyridin-3-yl, phthalazin-3-yl, phthalazin-4-yl, pteridinyl, purinyl, quin
- Hetereoarylalkyl means an alkyl group substituted with one or two heteroaryl groups, as defined herein.
- Fused bicyciic radical includes bridged ring systems.
- the valency of the group may be located on any atom of any ring within the radical, valency rules permitting. In particular, when the point of valency is located on a nitrogen atom, R is absent.
- heterocycloalkyl includes, but is not limited to, azetidinyl, pyrrolidinyl, 2-oxopyrrolidinyl, 2,5-dihydro-lH-pyrrolyl, piperidinyl, 4-piperidonyl, morpholinyl, piperaziny!, 2-oxopiperazinyl, tetrahydropyranyl, 2- oxopiperidinyl, thiomorpholinyl, thiamorpholinyl, perhydroazepinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, dihydropyridinyl, tetrahydropyridinyl, oxazolinyl, oxazolidinyl, isoxazolidinyl, thiazolinyl, thiazolidinyl, quinuclidinyl, isothiazolidinyl, octahydro
- Heterocycloalkylalkyl means an alkyl group, as defined herein, substituted with one or two heterocycloalkyl groups, as defined herein.
- Hydroxyalkyl means an alkyl radical, as defined herein, substituted with at least one, for example one, two, or three, hydroxy groups, provided that if two hydroxy groups are present they are not both on the same carbon atom.
- Representative examples include, but are not limited to, hydroxy methyl, 2-hydroxyethyl, 2-hydroxypropyl, 3-hydroxypropyl, l -(hydroxymethyl)-2-methylpropyl, 2-hydroxybutyl, 3-hydroxybutyl, 4-hydroxybutyl,
- Hydroxyamino means a -NH(OH) group.
- nucleic acid molecules and the term “polynucleotides” denote polymeric forms of nucleotides of any length, either ribonucleotides or deoxynucleotides. They include single-, double-, or multi-stranded DNA or RNA, genomic DNA, cDNA, DNA- RNA hybrids, or a polymer comprising purine and pyrimidine bases or other natural, chemically or biochemically modified, non-natural, or derivatized nucleotide bases.
- the backbone of a polynucleotide can comprise sugars and phosphate groups (as may typically be found in RNA or DNA), or modified or substituted sugar or phosphate groups.
- the backbone of the polynucleotide can comprise a polymer of synthetic subunits such as phosphoramidites and thus can be an oligodeoxynucleoside phosphoramidate or a mixed phosphoramidate-phosphodiester oligomer.
- a polynucleotide may comprise modified nucleotides, such as methylated nucleotides and nucleotide analogs, uracyl, other sugars, and linking groups such as fluororibose and thioate, and nucleotide branches.
- a polynucleotide may be further modified, such as by conjugation with a labeling component.
- modifications include caps, substitution of one or more of the naturally occurring nucleotides with an analog, and introduction of means for attaching the polynucleotide to proteins, metal ions, labeling components, other polynucleotides, or a solid support.
- Optionally substituted alkyl means an alkyl radical, as defined herein, optionally substituted with one or more groups, for example one, two, three, four, or five groups, independently selected from alkylcarbonyl, alkenylcarbonyl, cycloalkylcarbonyl, alkylcarbonyloxy, alkenylcarbonyloxy, amino, alkylamino, dialkylamino, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, cyano, cyanoalkylaminocarbonyl, alkoxy, alkenyloxy, hydroxy, hydroxyalkoxy, carboxy, alkylcarbonylamino, alkylcarbonyloxy, alkyl- S(0)o-2-, alkenyl-S(0)o-2-, aminosulfonyl, alkylaminosulfonyl, dialkylaminosulfonyl, a!kylsulfonyl-NR c -
- dialkylaminocarbonylamino, alkoxyalkyloxy, and -C(0)NR a R b (where R a and R b are independently hydrogen, alkyl, optionally substituted alkenyl, optionally substituted alkynyl, hydroxy, alkoxy, alkenyloxy, or cyanoalkyl).
- Optionally substituted alkenyl means an alkenyl radical, as defined herein, optionally substituted with one or more groups, for example one, two, or three groups, independently selected from alkylcarbonyl, alkenylcarbonyl, cycloalkylcarbonyl, alkylcarbonyloxy, alkenylcarbonyloxy, amino, alkylamino, dialkylamino, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, cyano, cyanoalkylaminocarbonyl, alkoxy, alkenyloxy, hydroxy, hydroxyalkoxy, carboxy, alkylcarbonylamino, alkylcarbonyloxy, alkyl-S(0)o-2-, alkenyl-S(0)o-2-, aminosulfonyl, alkylaminosulfonyl, dialkylaminosulfonyl, alkylsulfonyl-NR c - (where R
- alkoxyalkyloxy and -C(0)NR a R b (where R a and R b are independently hydrogen, optionally substituted alkyl, alkenyl, optionally substituted alkynyl, hydroxy, alkoxy, or alkenyloxy).
- Optionally substituted aryl means an aryl group, as defined herein, which is optionally substituted with one, two, three, four, or five groups selected from halo, haloalkyi, haloalkoxy, hydroxy, lower alkyl, lower alkenyl, lower alkynyl, alkoxy, carboxy, carboxy ester, amino, alkylamino, dialkylamino, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted heteroaryl, -C(0)NR'R" (where R' is hydrogen or alkyl, and R" is hydrogen, alkyl, aryl, heteroaryl, or heterocycloalkyl), -NR'C(0)R" (where R' is hydrogen or alkyl and R" is alkyl, aryl, heteroaryl, or heterocycloalkyl), -NR'C(0)R" (where R' is hydrogen or alkyl and R" is alkyl, aryl, hetero
- heterocycloalkyl and -NHS(0)2R' (where R' is alkyl, aryl, or heteroaryl).
- Optionally substituted heteroaryl means a heteroaryl group, as defined herein, optionally substituted with one, two, three, four, or five groups selected from halo, haloalkyi, haloalkoxy, lower alkyl, lower alkenyl, lower alkynyl, alkoxy, hydroxy, oxo (valency rules permitting), carboxy, carboxy ester, amino, alkylamino, dialkylamino, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, heteroaryl, optionally substituted aryl, -C(0)NR'R" (where R' is hydrogen or alkyl and R" is hydrogen, alkyl, aryl, heteroaryl, or heterocycloalkyl), -NR'C(0)R" (where R' is hydrogen or alkyl and R" is alkyl, aryl, heteroaryl, or heterocycloalkyl), and -NHS(0) 2 R' (where R' is alkyl,
- Optionally substituted heterocycloalkyl means a heterocycloalkyl, as defined herein, optionally substituted with one, two, three, four, or five groups selected from halo, haloalkyi, haloalkoxy, hydroxy, oxo, lower alkyl, lower alkenyl, lower alkynyl, alkoxy, optionally substituted cycloalkyl, heterocycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, alkylaminoalkyl, dialkylaminoalkyl, carboxy, carboxy ester,
- -C(0)NR'R (where R' is hydrogen or alkyl, and R" is hydrogen, alkyl, aryl, heteroaryl, or heterocycloalkyl)
- -NR'C(0)R (where R' is hydrogen or alkyl, and R" is alkyl, aryl, heteroaryl, or heterocycloalkyl)
- amino, alkylamino, dialkylamino, and -NHS(0)2R' where R' is alkyl, aryl, or heteroaryl).
- saturated bridged ring system refers to a bicyclic or polycyclic ring system that is not aromatic. Such a system may contain isolated or conjugated unsaturation, but not aromatic or heteroaromatic rings in its core structure (but may have aromatic substitution thereon). For example, hexahydro-furo[3,2-b]furan, 2,3,3a,4,7,7a-hexahydro-1H-indene, 7-aza-bicyclo[2.2.1]heptane, and 1,2,3,4,4a,5,8,8a-octahydro-naphthalene are all included in the class "saturated bridged ring system.”
- siRNA short interfering RNA
- RNAi RNA interference
- Spirocyclyl or "spirocyclic ring” refers to a ring originating from a particular annular carbon of another ring.
- a ring atom of a saturated bridged ring system (rings C and C), but not a bridgehead atom, can be a shared atom between the saturated bridged ring system and a spirocyclyl (ring D) attached thereto.
- a spirocyclyl can be carbocyclic or heteroalicyclic.
- Yield for each of the reactions described herein is expressed as a percentage of the theoretical yield.
- Antibody includes any immunoglobulin molecule that recognizes and specifically binds to a target, such as a protein, polypeptide, peptide, carbohydrate, polynucleotide, lipid, etc., through at least one antigen recognition site within the variable region of the immunoglobulin molecule.
- an antibody can be of any the five major classes of immunoglobulins: IgA, IgD, IgE, IgG, and IgM, or subclasses (isotypes) thereof (e.g.
- IgGl, IgG2, IgG3, IgG4, IgAl and IgA2) based on the identity of their heavy-chain constant domains referred to as alpha, delta, epsilon, gamma, and mu, respectively.
- the different classes of immunoglobulins have different and well known subunit structures and three- dimensional configurations.
- Antibodies can be naked or conjugated to other molecules such as cytotoxics, toxins, radioisotopes, etc.
- Antibody fragment can refer to a portion of an intact antibody.
- Examples of antibody fragments include, but are not limited to, linear antibodies, single-chain antibody molecules, Fc or Fc' peptides, Fab and Fab fragments, and multispecific antibodies formed from antibody fragments.
- Chimeric antibodies refers to antibodies wherein the amino acid sequence of the immunoglobulin molecule is derived from two or more species. Typically, the variable region of both light and heavy chains corresponds to the variable region of antibodies derived from one species of mammals (e.g. mouse, rat, rabbit, etc) with the desired specificity, affinity, and capability while the constant regions are homologous to the sequences in antibodies derived from another (usually human) to avoid eliciting an immune response in that species.
- mammals e.g. mouse, rat, rabbit, etc
- Humanized forms of non-human (e.g., rabbit) antibodies include chimeric antibodies that contain minimal sequence, or no sequence, derived from non-human immunoglobulin.
- humanized antibodies are human immunoglobulins (recipient antibody) in which residues from a hypervariable region of the recipient are replaced by residues from a hypervariable region of a non-human species (donor antibody) such as mouse, rat, rabbit, or nonhuman primate having the desired specificity, affinity, and capacity.
- donor antibody such as mouse, rat, rabbit, or nonhuman primate having the desired specificity, affinity, and capacity.
- humanized antibodies can comprise residues that are not found in the recipient antibody or in the donor antibody. Most often, the humanized antibody can comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a nonhuman immunoglobulin and all or substantially all of the FR residues are those of a human immunoglobulin sequence.
- the humanized antibody can also comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin.
- Hybrid antibodies can include immunoglobulin molecules in which pairs of heavy and light chains from antibodies with different antigenic determinant regions are assembled together so that two different epitopes or two different antigens can be recognized and bound by the resulting tetramer.
- epitopes or "antigenic determinant” are used interchangeably herein and refer to that portion of an antigen capable of being recognized and specifically bound by a particular antibody.
- the antigen is a polypeptide
- epitopes can be formed both from contiguous amino acids and noncontiguous amino acids juxtaposed by tertiary folding of a protein. Epitopes formed from contiguous amino acids are typically retained upon protein denaturing, whereas epitopes formed by tertiary folding are typically lost upon protein denaturing.
- An epitope typically includes at least 3-5, and more usually, at least 5 or 8-10, amino acids in a unique spatial conformation.
- protein display scaffold refers to binding reagents such as affibodies, immunity proteins, defensin A, T-cell receptors and the like, as described in Hosse et al., Protein Sci., 15( 1 ): 14-27 (2006), which is incorporated herein by reference.
- Specifically binds" to or shows “specific binding" towards an epitope means that the antibody reacts or associates more frequently, and/or more rapidly, and/or greater duration, and/or with greater affinity with the epitope than with alternative substances.
- Inhibitors of HER2 also known as NEU, NGL, Human Epidermal growth factor Receptor 2 (or ErbB2), pi 85 and CD340
- HER3 also known as c-ErbB3
- ERBB4 also known as HER4, pl80erbB4
- MSPR Macrophage-stimulating protein receptor
- Axl AXL Receptor Tyrosine Kinase
- MAP3K Mitogen-activated protein kinase
- ERK extracellular signal-regulated kinase
- JNK C-jun N-terminal kinase
- p38MAPK mitogen-activated protein kinase
- MEKK MEK kinase- 1
- Insulin growth factor 1 receptor IGF- IR, or CD221
- EphAl also known as ephrin type-A receptor 1, EPH receptor A 1 , EPH, EPHT1
- Cancer refers to cellular-proliferative disease states, including but not limited to: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hanlartoma, inesothelioma; Breast: ductal carcinoma in situ, infiltrating ductal carcinoma, medullary carcinoma, infiltrating lobular carcinoma, tubular carcinoma, mucinous carcinoma, inflammatory breast cancer: Gastrointestinal: esophagus (squamous cell
- Genitourinary tract kidney (adenocarcinoma, Wilm's tumor [nephroblastoma], lymphoma, leukemia), bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma), prostate (adenocarcinoma, sarcoma), testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma); Liver: hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma, angiosarcoma, hepatocellular adenoma, hemangioma; Bone: osteogenic sarcoma (osteosarcoma), fibrosarcoma, malignant fibrous histiocytom
- glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors), spinal cord neurofibroma, meningioma, glioma, sarcoma);
- Gynecological uterus (endometrial carcinoma), cervix (cervical carcinoma, pre-tumor cervical dysplasia), ovaries (ovarian carcinoma [serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma], granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma), vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma], fallopian tubes (carcinoma); Hematologic: blood (myeloid leukemia [acute and chronic], acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myel
- phosphatases can also play a role in "kinase-dependent diseases or conditions" as cognates of kinases; that is, kinases phosphorylate and phosphatases dephosphorylate, for example lipid substrates. Therefore compounds of the invention, while modulating kinase activity as described herein, may also modulate, either directly or indirectly, phosphatase activity. This additional modulation, if present, may be synergistic (or not) to activity of compounds of the invention toward a related or otherwise interdependent kinase or kinase family.
- the compounds of the invention when used with the combination of other kinase receptor inhibitors, are useful for treating diseases characterized in part by abnormal levels of cell proliferation (i.e., tumor growth), programmed cell death (apoptosis), cell migration and invasion and angiogenesis associated with tumor growth.
- a cancer which "overexpresses" an HER receptor is one which has significantly higher levels of an HER receptor, such as HER2, at the cell surface thereof, compared to a noncancerous cell of the same tissue type.
- Such overexpression may be caused by gene amplification or by increased transcription or translation.
- HER receptor overexpression may be determined in a diagnostic or prognostic assay by evaluating increased levels of the HER protein present on the surface of a cell (e.g. via an immunohistochemistry assay; 1HC). Alternatively, or additionally, one may measure levels of HER-encoding nucleic acid in the cell, e.g.
- FISH fluorescent in situ hybridization
- PCR polymerase chain reaction
- RT-PCR real time quantitative PCR
- HER receptor overexpression by measuring shed antigen (e.g., HER extracellular domain) in a biological fluid such as serum (see, e.g., U.S. Pat. No. 4,933,294, issued Jun. 12, 1990; W091/05264, published Apr. 18, 1991; U.S. Pat. No. 5,401,638, issued Mar. 28, 1995; and Sias et al. J. Immunol. Methods 132: 73-80 (1990)).
- various in vivo assays are available to the skilled practitioner.
- a detectable label e.g. a radioactive isotope
- Overexpression of HER2 at the 3+ level which leads to ligand-independent activation of the tyrosine kinase (Hudziak et al., Proc. Natl. Acad. Sci.
- Non-HER2 amplified tumors consist essentially of cells possessing a normal (i.e. wildtype) copy number of the HER2 gene.
- "Patient,” for the purposes of the present invention includes humans and other animals, particularly mammals, and other organisms. Thus the methods are applicable to both human therapy and veterinary applications. In another embodiment the patient is a mammal, and in another embodiment the patient is human.
- a "pharmaceutically acceptable salt” of a compound means a salt that is pharmaceutically acceptable and that possesses the desired pharmacological activity of the parent compound. It is understood that the pharmaceutically acceptable salts are non-toxic. Additional information on suitable pharmaceutically acceptable salts can be found in Remington's Pharmaceutical Sciences, 17 th ed., Mack Publishing Company, Easton, PA, 1985, or S. M. Berge, et al., "Pharmaceutical Salts," J. Pharm. Sci., 1977;66: 1-19, both of which are incorporated herein by reference.
- Examples of pharmaceutically acceptable acid addition salts include those formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like; as well as organic acids such as acetic acid, trifluoroacetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, 3-(4-hydroxybenzoyl)benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2-ethanedisuIfonic acid,
- 2-naphthalenesulfonic acid 4-toluenesulfonic acid, camphorsulfonic acid, glucoheptonic acid, 4,4'-methylenebis-(3-hydroxy-2-ene- l-carboxylic acid), 3-phenyIpropionic acid, trimethylacetic acid, tertiary butylacetic acid, lauryl sulfuric acid, gluconic acid, glutamic acid, hydroxynaphthoic acid, salicylic acid, stearic acid, muconic acid, p-toluenesulfonic acid, salicylic acid, and the like.
- Examples of a pharmaceutically acceptable base addition salts include those formed when an acidic proton present in the parent compound is replaced by a metal ion, such as sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Preferable salts are the ammonium, potassium, sodium, calcium, and magnesium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include, but are not limited to, salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins. Examples of organic bases include isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine,
- Exemplary organic bases are isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexylamine, choline, and caffeine.
- Prodrug refers to compounds that are transformed (typically rapidly) in vivo to yield the parent compound of the above formulae, for example, by hydrolysis in blood.
- esters of the compounds of this invention include, but are not limited to, alkyl esters (for example, with between about one and about six carbons) the alkyl group is a straight or branched chain. Acceptable esters also include cycloalkyl esters and arylalkyl esters such as, but not limited to benzyl.
- pharmaceutically acceptable amides of the compounds of this invention include, but are not limited to, primary amides, and secondary and tertiary alkyl amides (for example with between about one and about six carbons).
- Amides and esters of the compounds of the present invention may be prepared according to conventional methods. A thorough discussion of prodrugs is provided in T. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," Vol 14 of the A.C.S.
- therapeutic synergy is used when the combination of two products at given doses is more efficacious than the best of the two products alone considering the same doses.
- therapeutic synergy can be evaluated by comparing a combination to the best single agent using estimates obtained from a two-way analysis of variance with repeated measurements (Time factor) on parameter tumor volume.
- the maximum tolerated dose of the combination can be compared with the maximum tolerated dose of each of the isolated constituents in the study under consideration. This effectiveness can be quantified, for example, by the logio cell kill, which is determined according to the following equation:
- T-C represents the delay in growth of the cells, which is the average time, in days, for the tumors of the treated group (T) and the tumors of the control group (C) to have reached a predetermined value (1 g for example), and T d represents the time, in days, necessary for the volume of the tumor to double in the control animals [T.H. Corbett et al., Cancer, 40, 2660.2680 (1977); F.M. Schabel et al., Cancer Drug Development, Part B, Methods in Cancer Research, 17, 3-51, New York, Academic Press Inc. (1979)].
- a product is considered to be active if logio cell kill is greater than or equal to 0.7.
- a product is considered to be very active if logio cell kill is greater than 2.8.
- the combination, used at its own maximum tolerated dose, in which each of the constituents is present at a dose generally less than or equal to its maximum tolerated dose, will show therapeutic synergy when the logio cell kill is greater than the value of the logio cell kill of the best constituent when it is administered alone, and in particular has a superiority of at least one log cell kill.
- “Therapeutically effective amount” is an amount of a compound of formula I and/or an inhibitor of a kinase or receptor, for example, HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2, that when co-administered to a patient, ameliorates a symptom of the disease.
- the amount of a compound of formula I, or inhibitor of a kinase of the invention, which constitutes a "therapeutically effective amount” will vary depending on the compound, inhibitor, the disease state and its severity, the bioavailability characteristics of the compound and/or inhibitor, the age of the patient to be treated, and the like.
- the therapeutically effective amount can be determined routinely by one of ordinary skill in the art having regard to their knowledge and to this disclosure.
- the dosage or dosages comprising the therapeutically effective amounts are not toxic and confer to accepted medical practices commensurate with an appropriate risk/benefit ratio.
- Treating" or "treatment” of a disease, disorder, or syndrome includes: (i) preventing the disease, disorder, or syndrome from occurring in a human, i.e. causing the clinical symptoms of the disease, disorder, or syndrome not to develop in an animal that may be exposed to or predisposed to the disease, disorder, or syndrome but does not yet experience or display symptoms of the disease, disorder, or syndrome; (ii) inhibiting the disease, disorder, or syndrome, i.e., arresting its development; and (iii) relieving the disease, disorder, or syndrome, i.e., causing regression of the disease, disorder, or syndrome.
- Co-administration are meant to include modes of administration of the selected active, therapeutic agents to a single patient and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
- Co-administration can also include delivery of the active ingredients in a "fixed combination,” e.g.
- a compound of formula I and an inhibitor for example, a functional nucleic acid, lapatinib, and/or an antibody against any one of kinases or kinase receptors: HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, 1GF-IR, and FGFR2, are both administered to a patient simultaneously in the form of a single entity or dosage.
- an inhibitor for example, a functional nucleic acid, lapatinib, and/or an antibody against any one of kinases or kinase receptors: HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, 1GF-IR, and FGFR2, are both administered to a patient simultaneously in the form of a single entity or dosage.
- non-fixed combination means that the
- a compound of formula I and an inhibitor (for example, a functional nucleic acid or an antibody) against any one or more of the kinases or receptors: HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2, are both administered to a patient as separate entities either simultaneously, concurrently, or sequentially with no specific time limits, such that the administration provides therapeutically effective levels of the combination of active agents in the body of the patient.
- the embodiment includes both the recited compounds as well as individual isomers and mixtures of isomers.
- the embodiment optionally includes the pharmaceutically acceptable salts, hydrates, and/or solvates of the recited compounds and any individual isomers or mixture of isomers thereof.
- the compound of formula I can, for example, be compound A or be selected from a compound in Table 1.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, where growth and/or survival of tumor cells of the cancer is enhanced, at least in part, by the activity of PI3K; in combination with one or more inhibitors of HER3, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), and MEKK kinases/kinase receptors.
- the inhibitor can be a functional nucleic acid, while in another it is an antibody.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, in combination with one or more inhibitors of HER3, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK) and MEKK kinases/kinase receptors; where the cancer is selected from breast cancer, colon cancer, rectal cancer, endometrial cancer, gastric carcinoma (including gastrointestinal carcinoid tumors and gastrointestinal stromal tumors), glioblastoma, hepatocellular carcinoma, small cell lung cancer, non-small cell lung cancer (NSCLC), melanoma, ovarian cancer, cervical cancer, pancreatic cancer, prostate carcinoma, acute myelogenous leukemia (AML), chronic myelogenous leukemia (CML), non-Hodgkin's lymphoma, and thyroid carcinoma.
- the inhibitor can be a functional nucleic acid
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, in combination with one or more inhibitors of HER3, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), and MEKK kinases/kinase receptors; where the cancer is selected from prostate cancer, NSCLC, ovarian cancer, cervical cancer, breast cancer, colon cancer, rectal cancer, and glioblastoma.
- the inhibitor can be a functional nucleic acid, while in another it is an antibody.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, in combination with one or more inhibitors of HER3, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), and MEKK kinases/kinase receptors; where the cancer is selected from NSCLC, breast cancer, prostate cancer, glioblastoma, and ovarian cancer.
- the inhibitor can be a functional nucleic acid, while in another it is an antibody.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, in combination with a treatment where the treatment includes a therapeutically effective amount of one or more antibodies operable to inhibit the activity and/or expression of HER3 receptor tyrosine kinase.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, where growth and/or survival of tumor cells of the cancer is enhanced, at least in part, by the activity of PI3K; in combination with one or more inhibitors of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2 kinases/kinase receptors.
- the inhibitor can be a functional nucleic acid, or lapatanib, while in another it is an antibody.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER3 inhibitor.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER2 inhibitor.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER3 inhibitor and a HER2 inhibitor.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with lapatinib.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with trastuzumab.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with lapatinib and trastuzumab.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER3 inhibitor and lapatinib.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER3 inhibitor and trastuzumab.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER2 inhibitor and lapatinib.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER2 inhibitor and trastuzumab.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER3 inhibitor, a HER2 inhibitor, and lapatinib.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER3 inhibitor, a HER2 inhibitor, and trastuzumab.
- the method of the invention comprises administering to a patient a therapeutically effective amount of a compound of formula I in combination with a HER3 inhibitor, a HER2 inhibitor, lapatinib, and trastuzumab.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, in combination with one or more inhibitors of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2 kinases/kinase receptors; where the cancer is selected from the group consisting of breast cancer (including HER2+ or HER2 overexpressing breast cancer), colon cancer, rectal cancer, endometrial cancer, gastric carcinoma (including gastrointestinal carcinoid tumors and gastrointestinal stromal tumors), glioblastoma, hepatocellular carcinoma, small cell lung cancer, non-small cell lung cancer (NSCLC), melanoma, ovarian cancer, cervical cancer, pancreatic cancer, prostate carcinoma, acute myelogenous leukemia (AML
- the inhibitor can be a functional nucleic acid, while in another, the inhibitor can be lapatinib. In another embodiment, it is an antibody, while in another, the inhibitor can include a combination of functional nucleic acids, and/or lapatinib, and/or a combination of antibodies.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, in combination with one or more inhibitors of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2 kinases/kinase receptors; where the cancer is selected from the group consisting of prostate cancer, NSCLC, ovarian cancer, cervical cancer, gastric cancer, breast cancer (including HER2+ or HER2 overexpressing breast cancer), colon cancer, rectal cancer, and glioblastoma.
- the inhibitor can be a functional nucleic acid, while in another, the inhibitor can be lapatinib. In another embodiment, it is an antibody, while in another, the inhibitor can include a combination of functional nucleic acids, and/or lapatinib, and/or a combination of antibodies.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, in combination with one or more inhibitors of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2 kinases/kinase receptors; where the cancer is selected from the group consisting of NSCLC, breast cancer, prostate cancer, glioblastoma, and ovarian cancer.
- the inhibitor can be a functional nucleic acid, in another, the inhibitor can be lapatinib, while in another it is an antibody, while in another embodiment, the inhibitor can include a combination of functional nucleic acids, and/or lapatinib and/or a combination of antibodies.
- the invention is directed to a method of treating cancer, comprising administering to a patient a therapeutically effective amount of a compound of formula I, as defined in the Summary, in combination with a treatment that includes a therapeutically effective amount of one or more functional nucleic acids and/or a therapeutically effective amount of one or more antibodies operable to inhibit the activity and/or expression of HER2 receptor tyrosine kinase.
- the compound of formula I is selected from any of the following embodiments, including from the representative compounds in Table 1.
- R 50 is hydrogen, alkyl, alkenyl, halo, haloalkyl, haloalkenyl, hydroxy, alkoxy, alkenyloxy, haloalkoxy, nitro, amino, alkylamino, dialkylamino, -N(R 55 )C(0)-C C 6 -alkylene-N(R 55a )R 55b , alkylcarbonyl, alkenylcarbonyl, carboxy, alkoxycarbonyl, cyano, alkylthio, -S(0) 2 NR 55 R 55a , or alkylcarbonylamino; where R 55 and R 55b are indepedently hydrogen, alkyl, or alkenyl, and R 55a is hydrogen, alkyl, alkenyl, hydroxy, or alkoxy; and all other groups are as defined in the Summary.
- R 50 is hydrogen.
- R 51 is hydrogen or alkyl
- R 52 is hydrogen or halo; and all other groups are as defined in the Summary.
- R 52 is hydrogen or fluoro. More particularly, R 52 is hydrogen.
- R 53 is hydrogen, alkyl, alkenyl, halo, haloalkyl, haloalkenyl, hydroxy, alkoxy, alkenyloxy, haloalkoxy, nitro, amino, alkylamino, dialkylamino, -N(R 55 )C(0)-Ci-C 6 -alkylene-N(R 55a )R 55b , alkylcarbonyl, alkenylcarbonyl, carboxy, alkoxycarbonyl, cyano, alkylthio, -S(0) 2 NR 55 R 55a , or alkylcarbonylamino; where R 55 and R 55b are indepedently hydrogen, alkyl, or alkenyl, and R 55a is hydrogen, alkyl, alkenyl, hydroxy, or alkoxy; and all other groups are as defined in the Summary.
- R 53 is hydrogen, alkoxy, nitro, amino, or -N(R 55 )C(0)-C
- R 53 is hydrogen, methoxy, nitro, amino, or -NHC(0)CH 2 N(CH 3 ) 2 . More particularly, R 53 is hydrogen or methoxy.
- R 54 is hydrogen, alkyl, alkenyl, halo, haloalkyl, haloalkenyl, hydroxy, alkoxy, alkenyloxy, haloalkoxy, nitro, amino, alkylamino, dialkylamino, -N(R 55 )C(0)-C,-C6-alkylene-N(R 55a )R 55b , alkylcarbonyl, alkenylcarbonyl, carboxy, alkoxycarbonyl, cyano, alkylthio, -S(0) 2 NR 55 R 55a , or alkylcarbonylamino; where R 55 and R 55b are indepedently hydrogen, alkyl, or alkenyl, and R 55a is hydrogen, alkyl, alkenyl, hydroxy, or alkoxy; and all other groups are as defined in the Summary.
- R 54 is hydrogen, alkyl, alkoxy, or halo. In yet another embodiment, R 54 is hydrogen, methyl, methoxy, bromo, or chloro. More particularly, R 54 is hydrogen, methoxy, or chloro.
- R 50 , R 52 , and R 53 aTe hydrogen, and R 54 is halo or alkoxy; R 50 , R 52 , and R 54 are hydrogen, and R 53 is alkoxy; or R 50 and R 52 are hydrogen, and R 53 and R 54 together with the carbons to which they are attached form a 6-membered heteroaryl; and all other groups are as defined in the Summary.
- R 50 , R 52 , and R 53 are hydrogen, and R 54 is chloro or methoxy; R 50 , R 52 , and R 54 are hydrogen, and R 53 is methoxy; or R 50 and R 52 are hydrogen, and R 53 and R 54 together with the carbons to which they are attached form pyridinyl.
- R 50 , R 52 , and R 53 are hydrogen, and R 54 is chloro or methoxy; or R 50 , R 52 , and R 54 is hydrogen and R 53 is methoxy. More particularly, R 51 is methyl.
- B is phenyl substituted with R and optionally further substituted with one, two, or three R 3 ; and all other groups are as defined in the Summary. In a further embodiment, B is phenyl substituted with R 3a . More particularly, the compound of formula I is a compound of formula 1(a):
- B is phenyl substituted with R , as depicted in formula 1(a), and is not further substituted with R 3 .
- B is heteroaryl optionally substituted with one, two, or three R 3 .
- B is thien-3-yl, pyridinyl, pyrimidinyl, pyridazinyl, pyrazinyl, oxazolyl, isoxazolyl, pyrrolyl, imidazolyl, pyrazolyl, or thiazolyl, each of which is optionally substituted with one or two R 3 .
- B is thien-3-yl, pyridin-2-yl, pyridin-3-yl, pyridin-4-yl, oxazol-2-yl, oxazol-4-yl, oxazol-5-yl, isoxazol-3-yl, isoxazol-4-yl, isoxazol-5-yl, imidazol-2-yl, pyrrol-2-yl, pyrrol-3-yl, imidazol-4-yl, imidazol- 5-yl, pyrazol-3-yl, pyrazol-4-yl, or pyrazol-5-yl, each of which is optionally substituted with one or two R 3 .
- B is thien-3-yl, pyridin-3-yl, pyridin-4-yl, isoxazol-4-yl, or pyrazol-4-yl, each of which is optionally substituted with one or two R 3 .
- B is pyridin-3-yl, 2-hydroxy-pyridin-5-yl, isoxazol-4-yl, or pyrazol-4-yl, each of which is optionally substituted with one or two R 3 .
- R a is cyano, hydroxyamino, carboxy, alkylsulfonyl, aminoalkyloxy, alkylaminoalkyloxy, dialkylaminoalkyloxy, -N(R 7 )C(0)-Ci-C 6 -alkylene- N(R 7a )(R 7b ), -CiOJNR ⁇ 83 , -NR C(0)R 3 ⁇ 4 , -C(O)N(R 10 )-C,-C 6 -alkylene-N(R 10a )R l0b , -NR 1 1 C(0)NR l la R l lb where R l la , -C(0)R 12 , -NR l3 C(0)OR 13a , -C(0)N(R ,4 )N(R 14a )(R l b ), -S(0) 2 N(R 15 )-Ci-C 6 -alkylene-N(R
- R 3a is:
- NHC(0)CH 2 (2-phenylpyrrolidin-1-yl), -NHC(0)CH 2 (mc ⁇ holin-4-yl), - C(0)NHCH(CH3)CH 2 N(CH 3 )2, -C(0)NHCH 2 CH 2 N(CH3)2, -C(0)NH(pyrrolidin-3-yl), - C(0)NHCH 2 CH2(pyrrolidin-1-yl), -C(0)NHCH 2 CH 2 NH 2 , -C(0)N(CH 3 )CH2CH 2 N(CH3)2, - C(0)NHCH 2 (piperidin-2-yl), -C(0)NH(l-melhylazetidin-3-yl), -C(0)NHCH 2 CH 2 (piperidin- 1 -yl), -C(0)NHCH2CH 2 N(CH 2 CH3) 2 , -C(0)NH( l-methylpiperidin-3-yl),
- R 3a is hydroxyamino, -N(R 7 )C(0)-C
- -NHC(0)CH 2 (azetidin- 1 -yl), -NHC(0)(pyrrolidin-2-yl), -NHC(0)CH(NH 2 )CH 2 OH, -NHC(0)(azetidin-4-yl), -NHC(0)C(CH 3 ) 2 NH(CH 3 ), -NH 2 , -NHC(0)CH 2 NH(CH2CH 2 CH 3 ), -NHC(0)CH 2 CH 2 NH2, -NHOH, or - HC(0)(piperidin-3-yl).
- -C -alkyIene-N(R 7a )(R 7b ); and R 7 is hydrogen or alkyl, and R 7a and R 7b are independently hydrogen, alkyl, aminoalkyl, alkylaminoalkyl, or dialkylaminoalkyl; and all other groups are as defined in the Summary.
- R 3a is -NHC(0)CH 2 NH(CH 3 ), -NHC(0)CH(CH 3 )NH 2 ,
- each R 3 is independently halo, cyano, alkyl, alkenyl, alkoxy, hydroxyamino, carboxy, alkylsulfonyl, aminoalkyloxy, alkylaminoalkyloxy,
- dialkylaminoalkyloxy -N(R 7 )C(0)-C,-C 6 -alkylene-N(R 7a )(R 7b ), -C(0)NR 8 R 8a , -NR 9 C(0)R 3 ⁇ 4 , -C(O)N(R 10 )-C
- each of the alkylene in R 3 is independently optionally further substituted with 1, 2, 3, 4, or 5 groups selected from halo, hydroxy, amino, alkylamino, and dialkylamino; and all other groups are as defined in the Summary.
- each R 3 is independently methyl, bromo, chloro, fluoro, -NHC(0)CH 2 NH(CH 3 ), -NHC(0)CH 2 NH(CH 2 CH 3 ), -NHC(0)CH(CH 3 )NH 2>
- -NHC(0)CH 2 (4-ethylpiperazin-1-yl), -NHC(0)(thien-2-yI), -NHC(0)(3-fluoro-2- methylphenyl), -NHC(0)(2-bromo-thien-3-yl), -NHC(0)(4-fluorophenyl), -NHC(0)CH 2 (3- methylpiperidin-1-yl), -NHC(0)CH(CH 3 ) 2 , -NHC(0)(CH 2 ) 3 CH 3 , - HC(0)CH 2 OCH 2 CH3, -NHC(0)CH 2 NH(2-fluorophenyl), -NHC(0)(3-dimethylaminophenyl), -NHC(0)CH 2 (4- methylpiperidin- 1 -yl), -NHC(0)CH 2 NH(2-n-propy lphenyl), -NHC(0)phenyl,
- NHC(0)CH 2 (2-phenylpyrrolidin- 1 -yl), -NHC(0)CH 2 (morpholin-4-yl),
- each R 3 is independently halo, alkyl, hydroxyamino, -N(R 7 )C(0)-Ci-C 6 -alkylene-N(R 7a )(R 7b ), -C(0)NR 8 R 8a , -NR 9 C(0)R 9a , -C(O)N(R 10 )-C
- R 3 is alky] or -N(R 7 )C(0)-Ci-C 6 -alkylene-N(R 7a )(R 7 ); and R 7 is hydrogen or alkyl, and R 7a and R 7 are independently hydrogen, alkyl, aminoalkyl, alkylaminoalkyl, or dialkylaminoalkyl; and all other groups are as defined in the Summary. More particularly, each R 3 is independently methyl, -NHC(0)CH 2 NH(CH 3 ),
- B is phenyl, R 3 is not present or R 3 is halo, alkyl, or alkoxy;
- R 3a is -C(0)NR 8 R 8a , -NR 9 C(0)R 9a , -N(R 7 )C(0)-Ci-C 6 -alkylene-N(R 7a )(R 7b ), or -C(O)N(R ,0 )-C,-C 6 -alkylene-N(R 10a )R 10b ; where each of the alkylene in R a is independently optionally further substituted with 1 , 2, 3, 4, or 5 groups selected from halo, hydroxy, and amino; and all other groups are as defined in the Summary.
- R 50 , R 52 , and R 53 are hydrogen, and R 54 is halo or alkoxy; R 50 , R 52 , and R 54 are hydrogen, and R 53 is alkoxy; or R 50 and R 52 are hydrogen, and R 53 and R 54 together with the carbons to which they are attached form a 6-membered heteroaryl; and all other groups are as defined in the Summary.
- R 50 , R 52 , and R 53 are hydrogen, and R 54 is halo or alkoxy; or R 50 , R 52 , and R 54 are hydrogen, and R 53 is alkoxy. More particularly, R 51 is methyl.
- R 3 is not present or R 3 is alkyl
- R 3a is -N(R 7 )C(0)-Ci-C 6 -alkylene-N(R 7a )(R 7 ), -C(0)NR 8 R 8a , -NR 9 C(0)R 9a , or -C(O)N(R ,0 )-C l -C 6 -alkylene-N(R 10a )R ,0 ; where each of the alkylene in R 3a is independently optionally further substituted with 1 , 2, 3, 4, or 5 groups selected from halo, hydroxy, and amino; and all other groups are as defined in the Summary.
- R 3 is not present or is methyl. More particularly, R 3 is not present.
- R 7 is hydrogen or alkyl
- R 7a and R 7b are
- dialkylaminoalkyl R 8 is hydrogen or alkyl, and R 8a is heterocycloalkyl or
- R 9 is hydrogen or alkyl
- R 9a is hydrogen, heterocycloalkyl, or heterocycloalkylalkyl
- R 10 , R 10a , and R IOb are independently hydrogen, alkyl, hydroxyalkyl, aminoalkyl, alkylaminoalkyl, or dialkylaminoalkyl.
- R 50 , R 52 , and R 53 are hydrogen, and R 54 is halo or alkoxy; or R 50 , R 52 , and R 54 are hydrogen, and R 53 is alkoxy; or R 50 and R 52 are hydrogen, and R 53 and R 54 together with the carbons to which they are attached form a 6-membered heteroaryl.
- R 50 , R 52 , and R 53 are hydrogen, and R 54 is halo or alkoxy; or R 50 , R 52 , and R 54 are hydrogen, and R 53 is alkoxy. More particularly, R 51 is methyl.
- B is heteroaryl
- one R 3 is halo, alkyl, or alkoxy
- a second R 3 is -QOJNR ⁇ 83 , -NR 9 C(0)R 9a , -N(R 7 )C(0)-C,-C 6 -alkylene-N(R 7a )(R 7b ), or -C(0)N(R 10 )- C
- each of the alkylene in R 3 is independently optionally further substituted with 1, 2, 3, 4, or 5 groups selected from halo, hydroxy, and amino; and all other groups are as defined in the Summary.
- R 7 is hydrogen or alkyl
- R 7a and R 7 are
- dialkylaminoalkyl R 8 is hydrogen or alkyl, and R 8a is heterocycloalkyl or
- R 9 is hydrogen or alkyl
- R 9a is hydrogen, heterocycloalkyl, or heterocycloalkylalkyl
- R 10 , R 10a , and R 10b are independently hydrogen, alkyl, hydroxyalkyl, aminoalkyl, alkylaminoalkyl, or dialkylaminoalkyl.
- B is:
- R 3 is independently halo, alkyl, alkoxy, aminoalkyloxy, alkylaminoalkyloxy, dialkylaminoalkyloxy, alkylamino, dialkylamino, -C(0)NR R ⁇ -NR C(0)R 9a , -N(R 7 )C(0)-C,-C 6 -alkylene-N(R 7a )(R 7b ), or -C(O)N(R 10 )-C,-C 6 -alkylene- N(R IOa )R l0b ; and all other groups are as defined in the Summary.
- R 50 , R 52 , and R 53 are hydrogen, and R 54 is halo or alkoxy; R 50 , R 52 , and R 54 are hydrogen, and R 53 is alkoxy; or R 50 and R 52 are hydrogen, and R 53 and R 54 together with the carbons to which they are attached form a 6-membered heteroaryl; and all other groups are as defined in the Summary.
- R 50 , R 52 , and R 53 are hydrogen, and R 54 is halo or alkoxy; or R 50 , R 52 , and R 54 are hydrogen and R 53 is alkoxy. More particularly, R 51 is methyl.
- R 7 is hydrogen or alkyl
- R 7 and R 7b are independently hydrogen, alkyl, hydroxyalkyl, aminoalkyl, alkylaminoalkyl, or
- dialkylaminoalkyl R 8 is hydrogen or alkyl, and R 8a is heterocycloalkyl or
- R 9 is hydrogen or alkyl
- R 9a is hydrogen, heterocycloalkyl, or heterocycloalkylalkyl
- R 10 , R 10a , and R 10b are independently hydrogen, alkyl, hydroxyalkyl, aminoalkyl, alkylaminoalkyl, or dialkylaminoalkyl
- R 50 is hydrogen
- R 51 is hydrogen or alkyl
- R 52 is hydrogen
- R 53 is hydrogen, alkoxy, nitro, amino, or -N(R 55 )C(0)-C C 6 -alkyIene-N(R 53 ⁇ 4 )R 5b ; and R 54 is hydrogen, alkyl, alkoxy, or halo; or R 53 and R 54 together with the carbons to which they are attached form a 6-membered heteroaryl;
- B is phenyl substituted with R 3a and optionally further substituted with one R 3 ;
- B is heteroaryl optionally substituted with one or two R 3 ;
- R 3a is cyano, hydroxyamino, carboxy, alkylsulfonyl, aminoalkyloxy, alkylaminoalkyloxy, dialkylaminoalkyIoxy, -N(R 7 )C(0)-Ci-C 6 -alkylene-N ⁇ R 7a )(R 7b ), -C(0)NR 8 R 8a , -NR 9 C(0)R 9a , -C(O)N(R l0 )-C,-C 6 -alkylene-N(R 10a )R 10b , - R"C(0)NR l la R l lb where R I la , -C(0)R 12 ; -NR 13 C(0)OR l3a , -C(0)N(R l4 )N(R l4a )(R 14 ), -S(0) 2 N(R 15 )-C
- each R 3 (when R 3 is present) is independently halo, cyano, alkyl, alkenyl, alkoxy,
- R 50 , R 53 , and R 54 are independently hydrogen, alkyl, alkenyl, halo, haloalkyi, haloalkenyl, hydroxy, alkoxy, alkenyloxy, haloalkoxy, nitro, amino, alkylamino, dialkylamino, -N(R 55 )C(0)-C r C6-alkylene-N(R S5a )R 53b , alkylcarbonyl, alkenylcarbonyl, carboxy, alkoxycarbonyl, cyano, alkylthio, -S(0)2NR 55 R 55a , or alkylcarbonylamino and where R 55 and R 55 are indepedently hydrogen, alkyl, or alkenyl and R 55a is hydrogen, alkyl, alkenyl, hydroxy, or alkoxy; or R 53 and R 54 together with the carbons to which they are attached form a 5- or 6-membered heteroaryl
- R 53 and R 54 together with the carbons to which they are attached form a 5- or 6-membered heteroaryl or 5- or 6-membered heterocycloalkyl.
- W 1 , W 2 ,W 3 , and W 4 are -C(H)-;
- R 50 is hydrogen
- R 51 is methyl
- R 52 is hydrogen
- R 53 is hydrogen or alkoxy
- R 54 is hydrogen, alkyl, alkoxy, or halo; or R 53 and R 54 together with the carbons to which they are attached form a 6-membered heteroaryl;
- R 3 is halo or methyl
- R 3a is -N(R 7 )C(0)-C,-C -alkylene-N(R 7a )(R 7b ) where R 7 is hydrogen and R 7a and R 7b are independently hydrogen, alkyl, aminoalkyl, alkylaminoalkyl, or
- R 51 is methyl; and R 50 , R 52 , and R 53 are hydrogen and R 54 is halo or alkoxy or R 50 , R 52 , and R 54 are hydrogen and R 5 is alkoxy; or a single stereoisomer or mixture of stereoisomers thereof and optionally as a pharmaceutically acceptable salt thereof.
- R 3a is
- the compound of Formula 1(a) is:
- the compound of Formula 1(a) is:
- the compounds in Table 1 can be prepared as pharmaceutically acceptable salts, solvates, hydrates, and/or isomers thereof. All such salt, solvate, hydrate, and isomer combinations of the compounds in Table 1 can be used to practice the invention.
- the invention can be practiced with one or two pharmaceutically acceptable salts of a compound of Table 1, wherein the salt(s) are formed with one or two acids independently selected from hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, acetic acid, trifluoroacetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, 3-(4- hydroxybenzoyl)benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2-ethanedisulfonic acid, 2-hydroxyethanesulfonic acid, benzenesulfonic acid,
- the invention can be practiced with one or two pharmaceutically acceptable salts of a compound of Table 1, wherein the salt(s) are formed with one or two bases independently selected from sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum, isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, 2-dimethylaminoethanol, 2-diethylaminoethanol, dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine, purines, piperazine, piperidine, N- ethylpiperidine, tromethamine, and N-methylglucamine.
- Any individual compound (and any optional salt, optional solvate, and optional hydrate thereof) in Table 1 can be used in combination with any of the above embodiment
- the compound of formula I of the present invention can be co-administered with an inhibitor of a kinase/kinase receptor which is upregulated by administration of the formula
- HER2 MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors,
- INSR, IGF-IR, and FGFR2 kinases/kinase receptors can be co-administered with a formula I compound to a patient undergoing treatment for cancer.
- inhibitors of HER2 and/or HER3 can be co-administered with a formula I compound to a patient undergoing treatment for cancer.
- an inhibitor of HER2 and/or HER3 can be co-administered with a formula I compound to a patient undergoing treatment for cancer.
- HER3 can be co-administered with a formula I compound to a patient undergoing treatment for cancer.
- an inhibitor of HER2 and or HER3 can be co-administered with a formula I compound to a patient undergoing treatment for cancer, wherein the cancer is a cancer overexpressing HER2, for example, a HER2 overexpressing breast cancer.
- the HER2 inhibitor is lapatinib (N-[3-chloro-4-[(3- fluorophenyl)methoxy]phenyl]-6- 5-[(2-methylsulfonylethylamino)methyl]-2-furyl] quinazolin-4-amine, for example, in the form of lapatinib ditosylate, commercially available under the tradename TYKERB®.
- Lapatinib has the chemical structure:
- the lapatinib dosage used can be directly determined by a prescribing physician using routine skill in the art.
- the dosage of lapatinib administered in combination with a compound of formula I can range from about 0.1 mg/kg to about 100 mg/kg per day, for example a daily dose ranging from about 100, or 200, or 300, or 500, or 600, or 700, or 800, or 900, or 1 ,000, to about 1 ,500, or about 1 ,600, or about 2,000 mg per day.
- the inhibitor can be a functional nucleic acid.
- the category of “functional nucleic acids” encompasses siRNA molecules, shRNA molecules, miRNA molecules, and antisense nucleic acid molecules.
- siRNA refers to small inhibitory RNA duplexes that induce the RNA interference (RNAi) pathway. These molecules can vary in length (generally 18-30 base pairs) and contain varying degrees of complementarity to their target mRNA in the antisense strand. Some, but not all, siRNA have unpaired overhanging bases on the 5' or 3' end of the sense strand and/or the antisense strand.
- siRNA includes duplexes of two separate strands, as well as single strands that can form hairpin structures comprising a duplex region.
- the functional nucleic acid targets the gene or transcript of a kinase receptor, such as the extracellular domain at which ligand binding occurs.
- the functional nucleic acid targets the intracellular protein kinase domain.
- the functional nucleic acid targets the heterodimer binding site (i.e. the amino-acid sequence that is involved in binding with other EGF receptor family members which form heterodimers with HER2 operable to stabilize ligand binding and enhance kinase- mediated activation of downstream signaling pathways.
- suitable functional nucleic acids are commercially available. See e.g. Qiagen, Valencia, CA.
- functional nucleic acids can be identified and/or synthesized through experimentation or though rational design based on nucleotide sequence information on the target
- kinase/receptor using well-known methods in the Field.
- the sequences can be determined by reference to bioinformatic databases (for example GENBANK, NCBI, PKR, and the like) that disclose the coding regions of genes known to express the particular kinase of interest, (target) i.e. HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), ME K kinases/kinase receptors, INSR, 1GF-IR, and FGFR2.
- bioinformatic databases for example GENBANK, NCBI, PKR, and the like
- bioinformatic databases for example GENBANK, NCBI, PKR, and the like
- kinases and kinase receptor mRNA and amino acid sequences for which inhibition is contemplated herein for example, HER3, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2 can also be identified using the above referenced databases.
- the generation of functional nucleic acids that can hybridize to an mRNA encoding one of several functional HER2 polypeptides is provided in SEQ ID NO: 1.
- SEQ ID NO: 1 is the mRNA transcript variant NM 004448 Version NM 004448.2; GI 54792095.
- the rational design process can involve the use of a computer program to evaluate the criteria for every sequence of 18-30 base pairs or only sequences of a fixed length, e.g., 19 base pairs.
- the computer program is designed such that it provides a report ranking of all of the potential siRNAs 18-30 base pairs, ranked according to which sequences generate the highest value. A higher value refers to a more efficient siRNA for a particular target gene.
- the computer program that may be used may be developed in any computer language that is known to be useful for scoring nucleotide sequences, or it may be developed with the assistance of commercially available product, such as Microsoft's product.net.
- BLAST Basic Local Alignment Search Tool
- the targeted kinase is HER2 and/or HER3, HER2 and/or HER3 mutant, or alternative splice variants thereof, particularly of human HER2 and/or HER3.
- target is used in a variety of different forms throughout this disclosure and is defined by the context in which it is used.
- target mRNA refers to a messenger RNA to which a given siRNA can be directed against.
- target sequence and “target site” refer to a sequence within the mRNA to which the sense strand of an siRNA shows varying degrees of homology and the antisense strand exhibits varying degrees of complementarity.
- siRNA target can refer to the gene, mRNA, or protein against which an siRNA is directed.
- target silencing can refer to the state of a gene, or the corresponding mRNA or protein.
- the siRNA can comprise partially purified RNA, substantially pure RNA, synthetic RNA, or recombinantly produced RNA, as well as altered RNA that differs from naturally-occurring RNA by the addition, deletion, substitution, and/or alteration of one or more nucleotides.
- Such alterations can include addition of non-nucleotide material, such as to the end(s) of the siRNA or to one or more internal nucleotides of the siRNA, including modifications that make the siRNA resistant to nuclease digestion.
- the siRNA can be targeted to any stretch of approximately 18-30 contiguous nucleotides in any of the target mRNA sequences, for example, 18, 19, 20, 21 , 22, 23, 24, 25, 26, 27, 28, 29, or 30 nucleotides in each strand, wherein one of the strands is substantially complementary to, e.g., at least 80% (or more, e.g., 85%, 90%, 95%, or 100%) complementary to, e.g., having for example 3, 2, 1 , or 0 mismatched nucleotide(s), a target mRNA sequence.
- Techniques for selecting target sequences for siRNA are provided, for example, in Tuschl T et al., "The siRNA User Guide," revised Oct.
- the sense strand of the present siRNA comprises a nucleotide sequence identical to any contiguous stretch of about 18 to about 30 nucleotides in the target mRNA.
- one or both strands of the siRNA can also comprise a 3' overhang.
- a "3' overhang” refers to at least one unpaired nucleotide extending from the 3'-end of a duplexed RNA strand.
- the siRNA comprises at least one 3' overhang of from 1 to about 6 nucleotides (which includes ribonucleotides or deoxynucleotides) in length, preferably from 1 to about 5 nucleotides in length, more preferably from 1 to about 4 nucleotides in length, and particularly preferably from about 2 to about 4 nucleotides in length.
- the length of the overhangs can be the same or different for each strand.
- the 3' overhang is present on both strands of the siRNA, and is 2 nucleotides in length.
- each strand of the siRNA can comprise 3' overhangs of dithymidylic acid ("TT") or diuridylic acid ("uu").
- the siRNA can be obtained using a number of techniques known to those of skill in the art.
- the siRNA can be chemically synthesized or recombinantly produced using methods known in the art, such as the Drosophila in vitro system described in U.S. published application 2002/0086356 of Tuschl et al., the entire disclosure of which is herein incorporated by reference.
- the siRNA are chemically synthesized using appropriately protected ribonucleoside phosphoramidites and a conventional DNA/RNA synthesizer.
- the siRNA can be synthesized as two separate, complementary RNA molecules, or as a single RNA molecule with two complementary regions.
- Commercial suppliers of synthetic RNA molecules or synthesis reagents include Proligo (Hamburg, Germany), Dharmacon Research (Lafayette, Colo. USA), Pierce Chemical (part of Perbio Science, Rockford, 111. USA), Glen Research (Sterling, Va. USA), ChemGenes (Ashland, Mass. USA), and Cruachem (Glasgow, UK).
- siRNA can also be expressed from recombinant circular or linear DNA plasmids using any suitable promoter.
- suitable promoters for expressing siRNA from a plasmid include, for example, the U6 or HI RNA pol III promoter sequences and the cytomegalovirus promoter. Selection of other suitable promoters is within the skill in the art.
- the recombinant plasmids of the invention can also comprise inducible or regulatable promoters for expression of the siRNA in a particular tissue or in a particular intracellular environment.
- siRNA expressed from recombinant plasmids can either be isolated from cultured cell expression systems by standard techniques, or can be expressed intracellularly at or near the area of neovascularization in vivo.
- the use of recombinant plasmids to deliver siRNA to cells in vivo is discussed in more detail below.
- siRNA can be expressed from a recombinant plasmid either as two separate, complementary RNA molecules, or as a single RNA molecule with two complementary regions.
- Methods of selecting plasmids suitable for expressing siRNA, methods for inserting nucleic acid sequences for expressing the siRNA into the plasmid, and methods for delivering the recombinant plasmid to the cells of interest are within the skill in the art. See, e.g., Tuschl, T. (2002), Nat. Biotechnol, 20: 446-448; Brumme!kamp T R et al. (2002), Science 296: 550-553; Miyagishi M et al. (2002), Nat.
- That plasmid comprises a sense RNA strand coding sequence in operable connection with a polyT termination sequence under the control of a human U6 RNA promoter and an antisense RNA strand coding sequence in operable connection with a polyT termination sequence under the control of a human U6 RNA promoter.
- the plasmid pAAVsiRNA is ultimately intended for use in producing an recombinant adeno-associated viral vector comprising the same nucleic acid sequences for expressing an siRNA.
- in operable connection with a polyT termination sequence means that the nucleic acid sequences encoding the sense or antisense strands are immediately adjacent to the polyT termination signal in the 5' direction. During transcription of the sense or antisense sequences from the plasmid, the polyT termination signals act to terminate transcription.
- promoter under the control of a promoter means that the nucleic acid sequences encoding the sense or antisense strands are located 3' of the promoter, so that the promoter can initiate transcription of the sense or antisense coding sequences.
- the siRNA can also be expressed from recombinant viral vectors intracellularly at or near the area of the HER2 and/or HER3 gene expressed tumor in vivo.
- the recombinant viral vectors of the invention comprise sequences encoding the siRNA and any suitable promoter for expressing the siRNA sequences. Suitable promoters include, for example, the U6 or HI RNA pol III promoter sequences and the cytomegalovirus promoter. Selection of other suitable promoters is within the skill in the art.
- the recombinant viral vectors of the invention can also comprise inducible or regulatable promoters for expression of the siRNA in a particular tissue or in a particular intracellular environment.
- the siRNA can be expressed from a recombinant viral vector either as two separate, complementary RNA molecules, or as a single RNA molecule with two complementary regions.
- Any viral vector capable of accepting the coding sequences for the siRNA molecule(s) to be expressed can be used, for example vectors derived from adenovirus (AV), adeno-associated virus (AAV), retroviruses (e.g, lentiviruses (LV), Rhabdoviruses, murine leukemia virus), herpes virus, and the like.
- the tropism of the viral vectors can also be modified by pseudotyping the vectors with envelope proteins or other surface antigens from other viruses.
- an AAV vector of the invention can be pseudotyped with surface proteins from vesicular stomatitis virus (VSV), rabies, Ebola, Mokola, and the like.
- Methods for selecting of recombinant viral vectors suitable for use in the invention, methods for inserting nucleic acid sequences for expressing the siRNA into the vector, and methods for delivering the viral vector to the cells of interest are within the skill in the art. See, for example, Dornburg R (1995), Gene Therap. 2: 301 -310; Eglitis M A ( 1988), Biotechniques 6: 608-614; Miller A D ( 1990), Hum Gene Therap. 1 : 5-14; and Anderson W F ( 1998), Nature 392: 25-30, the entire disclosures of which are herein incorporated by reference.
- a commercially available viral delivery system can be used (e.g., vectors for siRNA delivery that are available from Ambion, Austin, Tex.).
- Other methods for delivery are known to those in the art (e.g., siRNA Delivery Centre, sirna.dk/index .htm 1).
- Preferred viral vectors are those derived from AV and AAV.
- the siRNA is expressed as two separate, complementary single- stranded RNA molecules from a recombinant AAV vector comprising, for example, either the U6 or H 1 RNA promoters, or the cytomegalovirus (CMV) promoter.
- CMV cytomegalovirus
- a suitable AV vector for expressing the siRNA, a method for constructing the recombinant AV vector, and a method for delivering the vector into target cells, are described in Xia H et al. (2002), Nat. Biotech. 20: 1006-1010, the entire disclosures of which are herein incorporated by reference.
- Suitable AAV vectors for expressing the siRNA, methods for constructing the recombinant AAV vector, and methods for delivering the vectors into target cells are described in Samulski R et al. ( 1987), J. Virol. 61 : 3096-3101 ; Fisher K J et al. ( 1996), J. Virol., 70: 520-532; Samulski R et al. ( 1989), J. Virol. 63: 3822-3826; U.S. Pat. No.
- siRNA containing a given target sequence can be evaluated using standard techniques for measuring the levels of RNA or protein in cells.
- siRNA can be delivered to cultured cells, and the levels of target mRNA can be measured by Northern blot or dot blotting techniques or by quantitative RT-PCR.
- the levels of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2 kinases/kinase receptor protein in the infected cells can be measured by ELISA or Western blot.
- a suitable cell culture system for measuring the effect of the present siRNA on target mRNA or protein levels is described in the Examples below.
- siRNAs for downregulating the activity and/or expression of HER2 and/or HER3 in vitro and/or in vivo can be synthesized.
- Illustrative siRNA pairs for HER2 inhibition can include the sequence pairs: SEQ ID NO: 3- :CCUGGAACUCACCUACCUGdTdT/ SEQ ID NO: 4- CAGGUAGGUGAGUUCCAGGdTdT; SEQ ID NO: 5- CUACCUUUCUACGGACGUGdTdT/ SEQ ID NO: 6- CACGUCCGUAGAAAGGUAGdTdT; and SEQ ID NO: 7- GAUCCGGAAGUACACGAUGdTdT/ SEQ ID NO: 8-
- CAUCGUGUACUUCCGGAUCdTdT can be chemically synthesized and annealed. These rationally designed siRNAs can be synthesized and commercially available from Dharmacon.
- siRNAs shRNAs, and lentiviral vectors capable of expressing shRNA sequences useful in inhibiting HER2 and/or HER3 expression and/or activity are commercially available under Catalog Nos.: sc- 156048, and sc-29405 from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
- the efficacy of the functional nucleic acids useful herein can be increased when the functional nucleic acids, for example, siRNAs are tumor targeted, delivered systemically, repeatedly, and safely.
- Low transfection efficiency, nuclease degeneration, poor tissue penetration, and non-specific immune degradation can be overcome when the functional nucleic acids are incorporated into protective and functional vehicles, for example, viral vectors, liposomes complexed with polyethyleneimine (PEI), linked with vascular endothelial growth factor (VEGF) receptor-2, and PEI that was PEGylated with an RGD peptide ligand at the distal end, protamine-antibody fusion protein, and tumor- targeting immunol iposome complexes.
- PI polyethyleneimine
- VEGF vascular endothelial growth factor
- the functional nucleic acid is administered to the subject either as naked siRNA, in conjunction with a delivery reagent, or as a recombinant plasmid or viral vector which expresses the siRNA.
- Suitable delivery reagents for administration in conjunction with the present siRNA include the Mirus Transit TKO lipophilic reagent, lipofectin, lipofectamine, cellfectin, or polycations (e.g., polylysine), bacterial minicells, or liposomes.
- a preferred delivery reagent is a liposome.
- Liposomes can aid in the delivery of the siRNA to a particular tissue, such as a tumor tissue, and can also increase the blood half-life of the siRNA.
- Liposomes suitable for use in the invention are formed from standard vesicle-forming lipids, which generally include neutral or negatively charged phospholipids and a sterol, such as cholesterol. The selection of lipids is generally guided by consideration of factors such as the desired liposome size and half-life of the liposomes in the blood stream. A variety of methods are known for preparing liposomes, for example as described in Szoka et al. ( 1980), Ann. Rev. Biophys. Bioeng. 9: 467; and U.S. Pat. Nos. 4,235,871 , 4,501 ,728, 4,837,028, and 5,019,369, the entire disclosures of which are herein incorporated by reference.
- the liposomes encapsulating the present siRNA comprises a ligand molecule that can target the liposome to a particular cell or tissue at or near the site of angiogenesis.
- Ligands which bind to receptors prevalent in HER2 and/or HER3 overexpressing tumors such as monoclonal antibodies that bind to tumor antigens, are preferred.
- the liposomes encapsulating the present siRNA are modified so as to avoid clearance by the mononuclear macrophage and reticuloendothelial systems, for example by having opsonization-inhibition moieties bound to the surface of the structure.
- a liposome of the invention can comprise both opsonization-inhibition moieties and a ligand.
- opsonization-inhibiting moieties for use in preparing the liposomes of the invention are illustratively large hydrophilic polymers that are bound to the liposome membrane.
- an opsonization inhibiting moiety is "bound" to a liposome membrane when it is chemically or physically attached to the membrane, e.g., by the intercalation of a lipid-soluble anchor into the membrane itself, or by binding directly to active groups of membrane lipids.
- opsonization-inhibiting hydrophilic polymers form a protective surface layer which significantly decreases the uptake of the liposomes by the macrophage-monocyte system ("MMS") and reticuloendothelial system ("RES"); e.g., as described in U.S. Pat. No. 4,920,016, the entire disclosure of which is herein incorporated by reference.
- MMS macrophage-monocyte system
- RES reticuloendothelial system
- Liposomes modified with opsonization-inhibition moieties thus remain in the circulation much longer than unmodified liposomes. These liposomes are sometimes called “concealed” liposomes.
- Concealed liposomes are known to accumulate in tissues fed by porous or "leaky” microvasculature.
- liposomes of the invention that are modified with opsonization-inhibition moieties can deliver the present siRNA to tumor cells as exemplified herein.
- Opsonization inhibiting moieties suitable for modifying liposomes are preferably water-soluble polymers with a number-average molecular weight from about 500 to about 40,000 daltons, and more preferably from about 2,000 to about 20,000 daltons.
- Such polymers include polyethylene glycol (PEG) or polypropylene glycol (PPG) derivatives; e.g., methoxy PEG or PPG, and PEG or PPG stearate; synthetic polymers such as polyacrylamide or poly N-vinyl pyrrolidone; linear, branched, or dendrimeric polyamidoamines; polyacrylic acids; polyalcohols, e.g., polyvinylalcohol and polyxylitol to which carboxylic or amino groups are chemically linked, and gangliosides, such as ganglioside GM
- Copolymers of PEG, methoxy PEG, or methoxy PPG, or derivatives thereof, are also suitable.
- the opsonization inhibiting polymer can be a block copolymer of PEG and either a polyamino acid, polysaccharide, polyamidoamine, polyethyleneamine, or polynucleotide.
- the opsonization inhibiting polymers can also be natural polysaccharides containing amino acids or carboxylic acids, e.g., galacturonic acid, glucuronic acid, mannuronic acid, hyaluronic acid, pectic acid, neuraminic acid, alginic acid, carrageenan, aminated polysaccharides or oligosaccharides (linear or branched), or carboxylated polysaccharides or oligosaccharides, e.g., reacted with derivatives of carbonic acids with resultant linking of carboxylic groups.
- the opsonization-inhibiting moiety is a PEG, PPG, or derivatives thereof.
- Liposomes modified with PEG or PEG-derivatives are sometimes called "PEGylated liposomes.”
- the opsonization inhibiting moiety can be bound to the liposome membrane by any one of numerous well-known techniques.
- an N-hydroxysuccinimide ester of PEG can be bound to a phosphatidyl-ethanolamine lipid-soluble anchor, and then bound to a membrane.
- a dextran polymer can be derivatized with a stearylamine lipid- soluble anchor via reductive amination using Na(CN)BH 3 and a solvent mixture such as tetrahydrofuran and water in a 30: 12 ratio at 60°C.
- Illustrative recombinant plasmids which can express siRNA are discussed above. Such recombinant plasmids can also be administered directly or in conjunction with a suitable delivery reagent, including the Mirus Transit LT1 lipophilic reagent, lipofectin, lipofectamine, cellfectin, polycations (e.g., polylysine), bacterial minicells, or liposomes.
- a suitable delivery reagent including the Mirus Transit LT1 lipophilic reagent, lipofectin, lipofectamine, cellfectin, polycations (e.g., polylysine), bacterial minicells, or liposomes.
- Recombinant viral vectors which express siRNA are also discussed above, and methods for delivering such vectors to a solid tumor in a patient are within the skill in the art.
- the delivery method can include sterotactic injection of the vector into the tumor, or the surrounding tissue.
- the siRNA can be administered to the subject by any means suitable for delivering the siRNA to the cells of the tumor tissue at or near the tumor.
- the siRNA can be administered by gene gun, electroporation, stereotactic injection, or by other suitable parenteral or enteral administration routes.
- Suitable enteral administration routes include oral, rectal, transdermal, or intranasal delivery.
- Suitable parenteral administration routes include intravascular administration (e.g. intravenous bolus injection, intravenous infusion, intra-arterial bolus injection, intra-arterial infusion and catheter instillation into the vasculature), peri- and intra-tissue administration (e.g., peri-tumoral and intra-tumoral injection), subcutaneous injection or deposition including subcutaneous infusion (such as by osmotic pumps), direct (e.g., injection) application to the area at or near the site of the tumor, for example by a catheter, syringe, or other placement device (e.g., an implant comprising a porous, non-porous, or gelatinous material), inhalation, or stereotactic injection.
- intravascular administration e.g. intravenous bolus injection, intravenous infusion, intra-arterial bolus injection, intra-arterial infusion and catheter instillation into the vasculature
- peri- and intra-tissue administration e
- injections or infusions of the siRNA are given at or near the site of the HER2 overexpressing tumor. More preferably, the siRNA is administered directly by injection to the tumor or into the vascualture supplying nutrients to the tumor.
- the siRNA can be administered in a single dose or in multiple doses. Where the administration of the siRNA is by infusion, the infusion can be a single sustained dose or can be delivered by multiple infusions. Injection of the agent directly into the tumor tissue is at or near the site of the HER2 overexpressing tumor/cancer, for example, breast cancer. Multiple injections of the agent into the tumor tissue or at or near the site of the tumor are particularly preferred.
- the siRNA can be administered to the subject once, such as by a single injection or deposition at, into, or near the tumor.
- the siRNA can be administered to a subject multiple times daily or weekly.
- the siRNA can be administered to a subject once weekly for a period of from about three to about twenty-eight weeks, more preferably from about seven to about ten weeks.
- the siRNA is injected into or at or near the tumor site (e.g., stereotactic injection) once a week for seven weeks.
- the siRNA is administered intravenously, using a single or multiple daily, or weekly, or monthly doses. It is understood that periodic administrations of the siRNA for an indefinite length of time may be necessary for subjects suffering from a chronic HER2 overexpressing cancer disease, such as HER2 overexpressing breast cancer, that is naive or is refractory or non-responsive to another cancer treatment.
- a dosage regimen comprises multiple administrations
- the effective amount of siRNA administered to the subject can comprise the total amount of siRNA administered over the entire dosage regimen.
- the siRNA are preferably formulated as pharmaceutical compositions prior to administering to a subject, according to techniques known in the art.
- Pharmaceutical compositions of the present invention are characterized as being at least, therapeutically effective, sterile, pyrogen-free, and that are pharmaceutically effective, meaning that there is a reasonable salt solutions, 0.4% saline, 0.3% glycine, hyaluronic acid, and the like.
- compositions can also comprise conventional pharmaceutical excipients and/or additives.
- Suitable pharmaceutical excipients include stabilizers, antioxidants, osmolality adjusting agents, buffers, and pH adjusting agents.
- Suitable additives include physiologically biocompatible buffers (e.g., tromethamine hydrochloride), additions of chelants (such as, for example, DTPA or DTPA-bisamide) or calcium chelate complexes (as for example calcium DTPA, CaNaDTPA-bisamide), and optionally, additions of calcium or sodium salts (for example, calcium chloride, calcium ascorbate, calcium gluconate, or calcium lactate).
- Pharmaceutical compositions of the invention can be packaged for use in liquid form or can be lyophilized.
- solid compositions conventional nontoxic solid carriers can be used; for example, pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharin, talcum, cellulose, glucose, sucrose, magnesium carbonate, and the like.
- a solid pharmaceutical composition for oral administration can comprise any of the carriers and excipients listed above and 10-95%, preferably 25%-75%, of one or more siRNA.
- a pharmaceutical composition for aerosol (inhalational) administration can comprise 0.01-20% by weight, preferably 1 %-10% by weight, of one or more siRNA encapsulated in a liposome as described above, and a propellant.
- a carrier can also be included as desired; e.g., lecithin for intranasal delivery.
- a shRNA nucleic acid molecule refers to an RNA agent having a stem-loop structure, comprising a first and second region of complementary sequence, the degree of complementarity and orientation of the regions being sufficient such that base pairing occurs between the regions, the first and second regions being joined by a loop region, the loop resulting from a lack of base pairing between nucleotides (or nucleotide analogs) within the loop region.
- One portion or segment of a duplex stem of the shRNA structure is anti-sense strand or complementary, e.g., fully complementary, to a section of about 18 to about 40 or more nucleotides of the mRNA of the target gene, for example, HER2 and/or HER3.
- miRNAs are noncoding RNAs of approximately 22 nucleotides which can regulate gene expression at the post transcriptional or translational level during plant and animal development.
- miRNAs are all excised from an approximately 70 nucleotide precursor RNA stem-loop termed pre-miRNA, probably by Dicer, an RNase Ill-type enzyme, or a homolog thereof.
- pre-miRNA Naturally-occurring miRNA precursors
- pre-miRNA have a single strand that forms a duplex stem including two portions that are generally complementary, and a loop, that connects the two portions of the stem.
- the stem includes one or more bulges, e.g., extra nucleotides that create a single nucleotide "loop" in one portion of the stem, and/or one or more unpaired nucleotides that create a gap in the hybridization of the two portions of the stem to each other.
- Short hairpin RNAs, or engineered RNA precursors, of the invention are artificial constructs based on these naturally occurring pre-miRNAs, but which are engineered to deliver desired RNAi agents (e.g., siRNAs of the invention).
- the shRNA molecules of the invention are designed to produce any of the siRNAs described above when processed in a cell e.g., by Dicer present within the cell.
- the requisite elements of a shRNA molecule include a first portion and a second portion, having sufficient complementarity to anneal or hybridize to form a duplex or double-stranded stem portion. The two portions need not be fully or perfectly
- the first and second “stem” portions are connected by a portion having a sequence that has insufficient sequence complementarity to anneal or hybridize to other portions of the shRNA. This latter portion is referred to as a "loop" portion in the shRNA molecule.
- the shRNA molecules are processed to generate siRNAs.
- shRNAs can also include one or more bulges, i.e., extra nucleotides that create a small nucleotide "loop" in a portion of the stem, for example a one-, two-, or three-nucleotide loop.
- the stem portions can be the same length, or one portion can include an overhang of, for example, -5 nucleotides.
- the overhanging nucleotides can include, for example, uracils (Us), e.g., all Us. Such Us are notably encoded by thymidines (Ts) in the shRNA-encoding DNA which signal the termination of transcription.
- Us uracils
- Ts thymidines
- RNA interference RNA interference
- the antisense portion can be on the 5' or 3' end of the stem.
- the stem portions of a shRNA are preferably about 15 to about 50 nucleotides in length.
- the two stem portions are about 18 or 19 to about 25, 30, 35, 37, 38, 39, or 40 or more nucleotides in length.
- the length of the stem portions should be less than about 30 nucleotides to avoid provoking non-specific responses, such as the interferon pathway.
- the stem can be longer than 30 nucleotides.
- a stem portion can include much larger sections complementary to the target mRNA (up to, and including the entire mRNA).
- the two portions of the duplex stem must be sufficiently complementary to hybridize to form the duplex stem.
- the two portions can be, but need not be, fully or perfectly complementary.
- the loop in the shRNAs or engineered RNA precursors may differ from natural pre-miRNA sequences by modifying the loop sequence to increase or decrease the number of paired nucleotides, or replacing all or part of the loop sequence with a tetraloop or other loop sequences.
- the loop portion in the shRNA can be about 2 to about 20 nucleotides in length, i.e., about 2, 3, 4, 5, 6, 7, 8, 9, or more, e.g., 15 or 20, or more nucleotides in length.
- a preferred loop consists of or comprises a "tetraloop" sequences.
- Exemplary tetraloop sequences include, but are not limited to, the sequences GNRA, where N is any nucleotide and R is a purine nucleotide, GGGG, and UUUU.
- shRNAs of the invention include the sequences of a desired siRNA molecule described above.
- the sequence of the antisense portion of a shRNA can be designed essentially as described above or generally by selecting an 18, 19, 20, or 21 nucleotide, or longer, sequence from within the target RNA (e.g., HER2 and/or HER3 mRNA), for example, from a region 100 to 200 or 300 nucleotides upstream or downstream of the start of translation.
- the sequence can be selected from any portion of the target RNA (e.g., mRNA) including the 5' UTR (untranslated region), coding sequence, or 3' UTR, provided said portion is distant from the site of the gain-of- function mutation.
- This sequence can optionally follow immediately after a region of the target gene containing two adjacent AA nucleotides.
- the last two nucleotides of the nucleotide sequence can be selected to be UU.
- This 21 or so nucleotide sequence is used to create one portion of a duplex stem in the shRNA.
- This sequence can replace a stem portion of a wild-type pre-miRNA sequence, e.g., enzymatically, or is included in a complete sequence that is synthesized.
- DNA oligonucleotides that encode the entire stem-loop engineered RNA precursor, or that encode just the portion to be inserted into the duplex stem of the precursor, and using restriction enzymes to build the engineered RNA precursor construct, e.g., from a wild-type pre-miRNA.
- Specific functional nucleic acids e.g. shRNA clones which target mRNA of HER2 and/or HER3, for example, HER2 NM_ 004448.1 (based on Product ID B0017), are also commercially available from GeneCopoeiaTM (GeneCopoeia, Rockville, MD, USA) as Product ID No. HSH004969, which are shRNA expression constructs.
- the hairpin consists of a 7 base loop and 19-29 base stem optimized for the specific gene sequence of target mRNA Accession No. NM_004448.2 (shown as SEQ ID NO: 1 )
- Isolated functional nucleic acid molecules that are antisense to a HER2 and/or HER3 nucleotide sequence are useful for reducing activity or expression of the HER2 and/or HER3 mRNA or polypeptide.
- An "antisense" functional nucleic acid (antisense) is useful for reducing activity or expression of the HER2 and/or HER3 mRNA or polypeptide.
- oligonucleotide can include a nucleotide sequence that is complementary to a "sense" nucleic acid encoding a protein, e.g., complementary to the coding strand of a double- stranded cDNA molecule, or complementary to an mRNA sequence, for example, as provided in SEQ ID NO: 1.
- the antisense nucleic acid can be complementary to an entire HER2 and/or HER3 coding strand, or to only a portion thereof (e.g., coding region of a human HER2 and/or HER3 nucleotide sequence).
- the antisense nucleic acid molecule is antisense to a "noncoding region" of the coding strand of a nucleotide sequence encoding a HER2 and/or HER3 polypeptide (e.g., the 5' or 3' untranslated regions).
- An antisense nucleic acid can be designed such that it is complementary to the entire coding region of HER2 and/or HER3 mRNA, but in general, is an oligonucleotide that is antisense to only a portion of the coding or noncoding region of HER2 and/or HER3 mRNA.
- the antisense oligonucleotide can be complementary to the region surrounding the translation start site of HER2 and/or HER3 mRNA, e.g., between the -10 and +10 regions of the target gene nucleotide sequence of interest.
- An antisense oligonucleotide can be, e.g., about 5-100, or from about 7, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, or 80 or more nucleotides in length.
- an antisense nucleic acid can be constructed using chemical synthesis and enzymatic ligation reactions using procedures known in the art.
- an antisense nucleic acid e.g., an antisense oligonucleotide
- an antisense nucleic acid can be chemically synthesized using naturally occurring nucleotides or variously modified nucleotides designed to increase the biological stability of the molecules or to increase the physical stability of the duplex formed between the antisense and sense nucleic acids, e.g., phosphorothioate derivatives and acridine substituted nucleotides can be used (see, e.g., Protocols for Oligonucleotide Conjugates.
- the antisense nucleic acid also can be produced biologically using an expression vector into which a nucleic acid has been subcloned in an antisense orientation (i.e., RNA transcribed from the inserted nucleic acid will be of an antisense orientation to a target nucleic acid of interest).
- Antisense nucleic acids can also be produced from synthetic methods such as phosphoramidite methods, H-phosphonate methodology, and phosphite trimester methods.
- Antisense nucleic acids can also be produced by PCR methods. Such methods produce cDNA and cRNA sequences complementary to the mRNA.
- antisense molecules can be modified or unmodified RNA, DNA, or mixed polymer oligonucleotides and primarily function by specifically binding to matching sequences resulting in inhibition of peptide synthesis, for example, inhibition in the expression of one or more of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2.
- antisense molecules can be modified or unmodified RNA, DNA, or mixed polymer oligonucleotides and primarily function by specifically binding to matching sequences resulting in inhibition of peptide synthesis, for example, inhibition in the expression of HER2 and or HER3.
- the antisense oligonucleotide binds to target RNA by Watson Crick base-pairing and blocks gene expression by preventing ribosomal translation of the bound sequences either by steric blocking or by activating RNase H enzyme.
- Antisense molecules can also alter protein synthesis by interfering with RNA processing or transport from the nucleus into the cytoplasm.
- An antisense nucleic acid can be an a-anomeric nucleic acid molecule.
- an a- anomeric nucleic acid molecule forms specific double-stranded hybrids with complementary RNA in which, contrary to the usual ⁇ -units, the strands run parallel to each other.
- the antisense nucleic acid molecule can also comprise a 2'-0-methylribonucleotideor a chimeric RNA-DNA analog and can have mixed internucleoside linkages (see, e.g., Protocols for Oligonucleotide Conjugates. Totowa N.J.: Humana Press, 1994).
- methods for treating a cancer comprise administering to a patient a combination of compound 1 and antisense nucleic acids.
- antisense nucleic acid molecules are typically administered to a subject (e.g., by direct injection at a tissue site) or are generated in situ such that they hybridize with or bind to cellular RNA (e.g., mRNA) and/or genomic DNA encoding a HER2 and/or HER3 protein, for example, as provided in SEQ ID NO: 2 or splice variants thereof, to thereby inhibit expression of the protein, e.g., by inhibiting transcription and/or translation.
- antisense nucleic acid molecules can be modified to target selected cells and then administered systemically.
- antisense molecules can be modified such that they specifically bind to receptors or antigens expressed on a selected cell surface, e.g., by linking the antisense nucleic acid molecules to peptides or antibodies that bind to cell surface receptors or antigens present on tumor cells, for example, HER2 over-expressing tumor cells.
- the antisense nucleic acid molecules can also be delivered to tumor cells using the vectors described herein.
- vector constructs in which the antisense nucleic acid molecule is placed under the control of a strong pol II or pol III promoter are also known in the art (e.g., Wacheck et al.
- HER2 and/or HER3 gene expression can be inhibited by targeting nucleotide sequences complementary to the regulatory region of the HER2 and/or HER3 (e.g., the HER2 and/or HER3 promoter and or enhancers) to form triple helical structures that prevent transcription of the HER2 and/or HER3 gene in target cells.
- nucleotide sequences complementary to the regulatory region of the HER2 and/or HER3 e.g., the HER2 and/or HER3 promoter and or enhancers
- Switchback molecules are synthesized in an alternating 5'-3 ⁇ 3 -5' manner, such that they base pair with first one strand of a duplex and then the other, eliminating the necessity for a sizeable stretch of either purines or pyrimidines to be present on one strand of a duplex.
- a HER2 and/or HER3 functional nucleic acid molecule can be modified at the base moiety, sugar moiety or phosphate backbone to improve, e.g., the stability, hybridization, or solubility of the molecule.
- the deoxyribose phosphate backbone of the nucleic acid molecules can be modified to generate peptide nucleic acids (see Hyrup et al. ( 1996) Bioorgan. Med. Chem.4:5-23).
- peptide nucleic acid refers to a nucleic acid mimic, e.g., a DNA mimic, in which the deoxyribose phosphate backbone is replaced by a pseudopeptide backbone and only the four natural nucleobases are retained.
- the neutral backbone of a PNA can allow for specific hybridization to DNA and RNA under conditions of low ionic strength.
- the synthesis of PNA oligomers can be performed using standard solid phase peptide synthesis protocols as described in Hyrup et al. (1996, supra) and Perry-O'Keefe et al. (1996) Proc. Natl. Acad. Sci. U.S.A. 93: 14670-14675).
- PNAs of HER2 and/or HER3 nucleic acid molecules can be used in therapeutic and diagnostic applications.
- PNAs can be used as antisense or antigene agents for sequence-specific modulation of gene expression by, for example, inducing transcription or translation arrest or inhibiting replication.
- PNAs of HER2 and/or HER3 nucleic acid molecules can also be used in the analysis of single base pair mutations in a gene, (e.g., by PNA-directed PCR clamping), as "artificial restriction enzymes" when used in combination with other enzymes, (e.g., SI nucleases (Hyrup ( 1996, supra)), or as probes or primers for DNA sequencing or hybridization (Hyrup et al. (1996, supra); Perry-O'Keefe et al. (1996, supra)).
- the inhibitor can be an antibody or fragments thereof.
- the antibody can inhibit the upregulated kinase activity by, for example, binding to the extracellular domain of the kinase receptor.
- suitable antibodies are known in the field and commercially available.
- antibodies to each of the HER2, HER3, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK) and MEKK kinases can be obtained from R&D Systems (Minneapolis, MN). See, e.g. R&D Systems Catalog: for MSPR (catalog Nos.
- AF691 , FAB691 A, BAF6 1 , FAB691 F, MAB691 , FAB691 P DYC 1947E, DYC 1947- 2, DYC 1947-5, DYC691-2, DYC691-5, AF431, BAF431, 1947-MS-050 and 431 -MS- 100); for Axl (catalog Nos. AF 154 BAF 154, DY 154, MAB 154 and FAB 1541 ); for MAP3k (catalog Nos: 4540-KS-010, MAB4540, MAB6095); for MEKK (catalog No. MAB6095); for HER2 (catalog Nos.
- U3- 1287 (U3 Pharma, Martinsried, Germany), and MM-121 , which is antibody Ab#6 in WO 08/100624, (Merrimack Pharmaceuticals, Cambridge, MA) are additional examples of well-known anti-HER3 antibodies. Further anti-HER3 antibodies are disclosed in WO 97/35885, EP 1414494, WO 08/100624, US 7705130, US2010/0047829, and Chen et al., J. Biol. Chem., 271:7620-7629 ( 1996), all of which are hereby incorporated by reference.
- the HER3 inhibitor is an anti-ErbB3 antibody that binds to ErbB3 with a KD of at least 4 nM as measured using a surface plasmon resonance assay or a cell binding assay.
- such an antibody comprises the following CDRs:
- VH CDR3- SEQ ID NO: 1 1 VH CDR3- SEQ ID NO: 1 1 :
- such an antibody comprises a heavy chain variable region having an amino acid sequence of - SEQ ID NO: 15:
- such an antibody is MM-121 , which comprises a heavy chain with the following amino acid sequence - SEQ ID NO: 17:
- the antibody inhibitor of HER2 can include trastuzumab, commercially available as HERCEPTIN® ((huMAb4D5-8, rhuMAb HER2, U.S. Pat. No. 5,821 ,337) Genentech, Inc., San Francisco, Calif.).
- trastuzumab is administered to the subject in combination with a compound of formula I. The dosage of trastuzumab for such administration can be readily determined by a prescribing physician without undue experimentation.
- the weekly dosage of trastuzumab to be administered with the combination of a compound of formula I can range from about 0.01 mg/kg to about 100 mg/kg, or from about 0.1 mg/kg to about 100 mg/kg, or from about 1 mg/kg to about 100 mg/kg, or from about 0.1 mg/kg to about 50 mg/kg, or from about 0.1 mg/kg to about 10 mg/kg per week, preferably administered intravenously.
- the HER2 antibody inhibitor includes the antibody pertuzumab (disclosed in U.S. Patent No. 7,449,184 and incorporated herein in its entirety) administered in one or more doses, in an amount ranging from about 100 mg per dose to about 1500 mg per dose, administered approximately every week, approximately every 2 weeks, approximately every 3 weeks, or approximately every 4 weeks.
- Anti-HER2 antibodies of murine origin and their humanized and chimeric versions are suitable for use in the methods of the present invention.
- Examples of such HER2 antibodies include, but are not limited to, the 4D5 antibody (described in U.S. Pat. Nos. 5,677,171 and 5,772,997) and the 520C9 antibody and its functional equivalents (W093/21319), designated 452F2, 736G9, 741F8, 758G5, and 761 B IO (described in U.S. Pat. No. 6,054,561 ); all of these patents and patent applications herein incorporated by reference.
- HER2 antibody inhibitors can include one or more antibodies selected from humanized anti-HER2 antibodies, for example, huMAb4D5- l, huMAb4D5-2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7, and 4D5-8 as described in Table 3 of U.S. Pat. No. 5,821 ,337, which is incorporated herein by reference in its entirety, and humanized 2C4 antibodies.
- suitable antibodies can be prepared readily using well-known techniques, as described below.
- Polyclonal antibodies are preferably raised in animals by multiple subcutaneous (sc) or intraperitoneal (ip) injections of the relevant antigen and an adjuvant.
- antigen may be injected directly into the animal's lymph node (see Kilpatrick et al.,
- An improved antibody response may be obtained by conjugating the relevant antigen to a protein that is immunogenic in the species to be immunized, e.g., keyhole limpet hemocyanin, serum albumin, bovine thyroglobulin, or soybean trypsin inhibitor using a bifunctional or derivatizing agent, for example, maleimidobenzoyl sulfosuccinimide ester (conjugation through cysteine residues), N- hydroxysuccinimide (through lysine residues), glutaraldehyde, succinic anhydride, or other agents known in the art.
- a protein that is immunogenic in the species to be immunized e.g., keyhole limpet hemocyanin, serum albumin, bovine thyroglobulin, or soybean trypsin inhibitor
- a bifunctional or derivatizing agent for example, maleimidobenzoyl sulfosuccinimide ester (conjugation through cysteine
- Animals are immunized against the antigen, immunogenic conjugates, or derivatives by combining, e.g., 100 ⁇ g of the protein or conjugate (for mice) with 3 volumes of Freund's complete adjuvant and injecting the solution intradermally at multiple sites.
- the animals are boosted with 1/5 to 1/10 the original amount of peptide or conjugate in Freund's complete adjuvant by subcutaneous injection at multiple sites.
- the animals are bled and the serum is assayed for antibody titer. Animals are boosted until the titer plateaus.
- the animal is boosted with the conjugate of the same antigen, but conjugated through a different cross-linking reagent.
- Conjugates also can be made in recombinant cell culture as protein fusions. Also, aggregating agents such as alum are suitably used to enhance the immune response.
- Monoclonal antibodies can be made using the hybridoma method first described by Kohler et a!., Nature, 256:495 (1975), or by recombinant DNA methods.
- a mouse or other appropriate host animal such as rats, hamster, or macaque monkey, is immunized to elicit lymphocytes that produce or are capable of producing antibodies that will specifically bind to the protein used for immunization.
- lymphocytes may be immunized in vitro.
- Lymphocytes then are fused with myeloma cells using a suitable fusing agent, such as polyethylene glycol, to form a hybridoma cell (Goding, Monoclonal Antibodies: Principles and Practice, pp. 59-103 (Academic Press, 1986)).
- a suitable fusing agent such as polyethylene glycol
- the hybridoma cells thus prepared are seeded and grown in a suitable culture medium that preferably contains one or more substances that inhibit the growth or survival of the unfused, parental myeloma cells.
- the culture medium for the hybridomas typically will include hypoxanthine, aminopterin, and thymidine (HAT medium), which substances prevent the growth of HGPRT-deficient cells.
- HGPRT hypoxanthine guanine phosphoribosyl transferase
- Preferred myeloma cells are those that fuse efficiently, support stable high-level production of antibody by the selected antibody-producing cells, and are sensitive to a medium.
- Human myeloma and mouse-human heteromyeloma cell lines also have been described for the production of human monoclonal antibodies (Kozbor, J. Immunol., 133: 3001 (1984); Brodeur et al., Monoclonal Antibody Production Techniques and Applications, pp. 51 -63 (Marcel Dekker, Inc., New York, 1987)).
- Exemplary murine myeloma lines include those derived from MOP-21 and M. C.-1 1 mouse tumors available from the Salk Institute Cell Distribution Center, San Diego, Calif.
- the binding specificity of monoclonal antibodies produced by hybridoma cells is determined by immunoprecipitation or by an in vitro binding assay, such as radioimmunoassay (RIA) or enzyme-linked immunoabsorbent assay (ELISA).
- RIA radioimmunoassay
- ELISA enzyme-linked immunoabsorbent assay
- the binding affinity of the monoclonal antibody can be determined, for example, by BIAcore or Scatchard analysis (Munson et al., Anal. Biochem., 107:220 (1980)).
- the clones can be subcloned by limiting dilution procedures and grown by standard methods (Goding, Monoclonal Antibodies: Principles and Practice, pp. 59-103 (Academic Press, 1986)). Suitable culture media for this purpose include, for example, D-MEMO or RPMI 1640 medium.
- the hybridoma cells can be grown in vivo as ascites tumors in an animal.
- the monoclonal antibodies secreted by the subclones are suitably separated from the culture medium, ascites fluid, or serum by conventional immunoglobulin purification procedures, such as protein A-Sepharose, hydroxylapatite chromatography, gel electrophoresis, dialysis, or affinity chromatography.
- amino acid sequence of an immunoglobulin of interest can be determined by direct protein sequencing, and suitable encoding nucleotide sequences can be designed according to a universal codon table.
- DNA encoding the monoclonal antibodies can be isolated and sequenced from the hybridoma cells using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the monoclonal antibodies). Sequence determination will generally require isolation of at least a portion of the gene or cDNA of interest. Usually this requires cloning the DNA or mRNA encoding the monoclonal antibodies. Cloning is carried out using standard techniques (see, e.g., Sambrook et al. (1 89) Molecular Cloning: A Laboratory Guide, Vols 1-3, Cold Spring Harbor Press, which is incorporated herein by reference).
- a cDNA library can be constructed by reverse transcription of polyA+ mRNA, preferably membrane-associated mRNA, and the library screened using probes specific for human immunoglobulin polypeptide gene sequences.
- the polymerase chain reaction PCR
- PCR polymerase chain reaction
- the amplified sequences can be cloned readily into any suitable vector, e.g., expression vectors, minigene vectors, or phage display vectors. It will be appreciated that the particular method of cloning used is not critical, so long as it is possible to determine the sequence of some portion of the immunoglobulin polypeptide of interest.
- RNA used for cloning and sequencing is a hybridoma produced by obtaining a B cell from the transgenic mouse and fusing the B cell to an immortal cell.
- An advantage of using hybridomas is that they can be easily screened, and a hybridoma that produces a human monoclonal antibody of interest selected.
- RNA can be isolated from B cells (or whole spleen) of the immunized animal.
- sources other than hybridomas it may be desirable to screen for sequences encoding immunoglobulins or immunoglobulin polypeptides with specific binding characteristics.
- One method for such screening is the use of phage display technology.
- Phage display is described in e.g., Dower et al., WO 91/17271, McCafferty et al., WO 92/01047, and Caton and Koprowski, Proc. Natl. Acad. Sci. USA, 87:6450-6454 (1990), each of which is incorporated herein by reference.
- cDNA from an immunized transgenic mouse e.g., total spleen cDNA
- PCR is used to amplify cDNA sequences that encode a portion of an immunoglobulin polypeptide, e.g., CDR regions, and the amplified sequences are inserted into a phage vector.
- cDNAs encoding peptides of interest are identified by standard techniques such as panning.
- the sequence of the amplified or cloned nucleic acid is then determined.
- sequence encoding an entire variable region of the immunoglobulin polypeptide is determined, however, sometimes only a portion of a variable region need be sequenced, for example, the CDR-encoding portion.
- sequenced portion will be at least 30 bases in length, and more often bases coding for at least about one-third or at least about one-half of the length of the variable region will be sequenced.
- Sequencing can be carried out on clones isolated from a cDNA library or, when PCR is used, after subcloning the amplified sequence or by direct PCR sequencing of the amplified segment. Sequencing is carried out using standard techniques (see, e.g., Sambrook et al. (1989) Molecular Cloning: A Laboratory Guide, Vols 1-3, Cold Spring Harbor Press, and Sanger, F. et al. (1977) Proc. Natl. Acad. Sci. USA 74: 5463-5467, which is incorporated herein by reference).
- the DNA may be operably linked to expression control sequences or placed into expression vectors, which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to direct the synthesis of monoclonal antibodies in the recombinant host cells.
- host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to direct the synthesis of monoclonal antibodies in the recombinant host cells.
- Expression control sequences denote DNA sequences necessary for the expression of an operably linked coding sequence in a particular host organism.
- the control sequences that are suitable for prokaryotes include a promoter, optionally an operator sequence, and a ribosome-binding site.
- Eukaryotic cells are known to utilize promoters, polyadenylation signals, and enhancers.
- Nucleic acid is operably linked when it is placed into a functional relationship with another nucleic acid sequence.
- DNA for a presequence or secretory leader is operably linked to DNA for a polypeptide if it is expressed as a preprotein that participates in the secretion of the polypeptide;
- a promoter or enhancer is operably linked to a coding sequence if it affects the transcription of the sequence; or a ribosome-binding site is operably linked to a coding sequence if it is positioned so as to facilitate translation.
- operably linked means that the DNA sequences being linked are contiguous, and, in the case of a secretory leader, contiguous and in reading phase. However, enhancers do not have to be contiguous. Linking can be accomplished by ligation at convenient restriction sites. If such sites do not exist, synthetic oligonucleotide adaptors or linkers can be used in accordance with conventional practice.
- Cell, cell line, and cell culture are often used interchangeably, and all such designations include progeny.
- Transformants and transformed cells include the primary subject cell and cultures derived therefrom without regard for the number of transfers. It also is understood that all progeny may not be precisely identical in DNA content, due to deliberate or inadvertent mutations. Mutant progeny that have the same function or biological activity as screened for in the originally transformed cell are included.
- Isolated nucleic acids also are provided that encode specific antibodies, optionally operably linked to control sequences recognized by a host cell, vectors, and host cells comprising the nucleic acids, and recombinant techniques for the production of the antibodies, which may comprise culturing the host cell so that the nucleic acid is expressed and, optionally, recovering the antibody from the host cell culture or culture medium.
- Vector components can include one or more of the following: a signal sequence (that, for example, can direct secretion of the antibody), an origin of replication, one or more selective marker genes (that, for example, can confer antibiotic or other drug resistance, complement auxotrophic deficiencies, or supply critical nutrients not available in the media), an enhancer element, a promoter, and a transcription termination sequence, all of which are well known in the art.
- Suitable host cells include prokaryote, yeast, or higher eukaryote cells.
- Suitable prokaryotes include eubacteria, such as Gram-negative or Gram-positive organisms, for example, Enterohacteriaceae such as Escherichia, e.g., E. coli, Enterobacter, Erwinia, Klebsiella, Proteus, Salmonella, e.g., Salmonella typhimurium, Serratia, e.g., Serratia marcescans, and Shigella, as well as Bacilli such as B. subtilis and B. licheniformis, Pseudomonas, and Streptomyces.
- Enterohacteriaceae such as Escherichia, e.g., E. coli, Enterobacter, Erwinia, Klebsiella, Proteus
- Salmonella e.g., Salmonella typhimurium
- Serratia e.g.,
- eukaryotic microbes such as filamentous fungi or yeast are suitable cloning or expression hosts for antibody-encoding vectors.
- Saccharomyces cerevisiae or common baker's yeast, is the most commonly used among lower eukaryotic host microorganisms.
- a number of other genera, species, and strains are commonly available, such as Pichia, e.g. P. pastoris, Schizosaccharomyces pombe; Kluyveromyces, Yarrowia; Candida; Trichoderma reesia; Neurospora crassa;
- Schwanniomyces such as Schwanniomyces occidentalis
- filamentous fungi such as, e.g., Neurospora, Penicillium, Tolypocladium, and Aspergillus hosts such as A. nidulans and A. niger.
- Suitable host cells for the expression of glycosylated antibodies are derived from multicellular organisms.
- examples of invertebrate cells include plant and insect cells.
- baculoviral strains and variants and corresponding permissive insect host cells from hosts such as Spodoptera frugiperda (caterpillar), Aedes aegypti (mosquito), Aedes albopictus (mosquito), Drosophila melanogaster (fruitfly), and Bombyx mori have been identified.
- a variety of viral strains for transfection of such cells are publicly available, e.g., the L-I variant of Autographa californica NPV and the Bm-5 strain of Bombyx mori NPV.
- the host cells can be cultured in a variety of media.
- Commercially available media such as Ham's F10 (Sigma), Minimal Essential Medium ((MEM), (Sigma), RPMI- 1640 (Sigma), and Dulbecco's Modified Eagle's Medium ((DMEM), Sigma), are suitable for culturing the host cells.
- 4,657,866; 4,927,762; 4,560,655; or 5, 122,469; WO90103430; WO 87/00195; or U.S. Pat. Re. No. 30,985 can be used as culture media for the host cells. Any of these media can be supplemented as necessary with hormones and/or other growth factors (such as insulin, transferrin, or epidermal growth factor), salts (such as sodium chloride, calcium, magnesium, and phosphate), buffers (such as HEPES), nucleotides (such as adenosine and thymidine), antibiotics (such as GentamycinTM.
- hormones and/or other growth factors such as insulin, transferrin, or epidermal growth factor
- salts such as sodium chloride, calcium, magnesium, and phosphate
- buffers such as HEPES
- nucleotides such as adenosine and thymidine
- antibiotics such as GentamycinTM.
- the culture conditions such as temperature, pH, and the like, are those previously used with the host cell selected for expression, and will be apparent to the artisan.
- the antibody composition can be purified using, for example, hydroxylapatite chromatography, cation or anion exchange chromatography, or preferably affinity chromatography, using the antigen of interest or protein A or protein G as an affinity ligand.
- Protein A can be used to purify antibodies that are based on human .gamma.1, .gamma.2, or .gamma.4 heavy chains (Lindmark et al., J. Immunol. Meth. 62: 1 -13 (1983)).
- Protein G is recommended for all mouse isotypes and for human .gamma.3 (Guss et al., 20 EMBO J. 5: 15671575 (1986)).
- the matrix to which the affinity ligand is attached is most often agarose, but other matrices are available.
- Mechanically stable matrices such as controlled pore glass or poly(styrenedivinyl)benzene allow for faster flow rates and shorter processing times than can be achieved with agarose.
- the antibody comprises a CH3 domain
- the Bakerbond ABXTM resin J. T. Baker, Phillipsburg, 25 NJ.
- Other techniques for protein purification such as ethanol precipitation, Reverse Phase HPLC,
- Fragments of the anti- HER2 and/or HER3 antibodies are suitable for use in the methods of the invention so long as they retain the desired affinity of the full-length antibody.
- a fragment of an anti- HER2 and/or HER3 antibody will retain the ability to bind to the HER2 and/or HER3 receptor protein.
- Fragments of an antibody comprise a portion of a full- length antibody, generally the antigen binding or variable region thereof. Examples of antibody fragments include, but are not limited to, Fab, Fab'F(ab') 2 , and Fv fragments and single-chain antibody molecules.
- the Fv polypeptide further comprises a polypeptide linker between the V H and V L domains that enables the sFv to form the desired structure for antigen binding.
- Antibodies or antibody fragments can be isolated from antibody phage libraries generated using the techniques described in McCafferty et al. (1990) Nature 348:552-554 (1990). Clackson et al. (1991) Nature 352:624-628 and Marks et al. (1991) J. Mol. Biol. 222:581-597 describe the isolation of murine and human antibodies, respectively, using phage libraries. Subsequent publications describe the production of high affinity (nM range) human antibodies by chain shuffling (Marks et al.
- a humanized antibody has one or more amino acid residues introduced into it from a source that is non-human. These non-human amino acid residues are often referred to as "donor” residues, which are typically taken from a "donor” variable domain. Humanization can be essentially performed following the method of Winter and co-workers (Jones et al. (1986) Nature 321 :522-525; Riechmann et al. (1988) Nature 332:323-327; Verhoeyen et al. (1988) Science 239: 1534-1536), by substituting rodent CDRs or CDR sequences for the corresponding sequences of a human antibody. See, for example, U.S. Pat. Nos.
- humanized antibodies may include antibodies wherein substantially less than an intact human variable domain has been substituted by the corresponding sequence from a non- human species.
- humanized antibodies are typically human antibodies in which some CDR residues and possibly some framework residues are substituted by residues from analogous sites in rodent antibodies. See, for example, U.S. Pat. Nos. 5,225,539; 5,585,089; 5,693,761 ; 5,693,762; 5,859,205.
- anti- HER2 and/or HER3 antibodies suitable for use in the methods of the present invention. See, for example, the methods disclosed in WO 98/52976, which is herein incorporated by reference. Anti- HER2 and/or HER3 antibodies generated using such a method are encompassed by the term "Anti- HER2 and/or HER3 antibody" as used herein.
- any of the previously described anti- HER2 and/or HER3 antibodies may be conjugated prior to use in the methods of the present invention. Such conjugated antibodies are available in the art.
- the anti- HER2 and/or HER3 antibody may be labeled using an indirect labeling or indirect labeling approach.
- indirect labeling or “indirect labeling approach” is intended that a chelating agent is covalently attached to an antibody and at least one radionuclide is inserted into the chelating agent. See, for example, the chelating agents and radionuclides described in Srivagtava and Mease (1991) Nucl. Med. Bio. 18: 589-603, which is herein incorporated by reference.
- the anti- HER2 and/or HER3 antibody may be labeled using "direct labeling” or a "direct labeling approach,” where a radionuclide is covalently attached directly to an antibody (typically via an amino acid residue).
- a radionuclide is covalently attached directly to an antibody (typically via an amino acid residue).
- Preferred radionuclides are provided in Srivagtava and Mease (1991) supra.
- the indirect labeling approach is particularly preferred.
- the invention provides pharmaceutical compositions comprising an inhibitor of PI3K according to the invention and a pharmaceutically acceptable carrier, excipient, or diluent in combination with an antibody, administered in the same or separate vehicles.
- administration may specifically be by the oral route.
- Administration of the compounds of the invention, or their pharmaceutically acceptable salts, in pure form or in an appropriate pharmaceutical composition, can be carried out via any of the accepted modes of administration or agents for serving similar utilities.
- administration can be, for example, orally, nasally, parenterally (intravenous, intramuscular, or subcutaneous), topically, transdermally, intravaginally, intravesically, intracistemally, or rectal!y, in the form of solid, semi-solid, lyophilized powder, or liquid dosage forms, such as for example, tablets, suppositories, pills, soft elastic and hard gelatin capsules, powders, solutions, suspensions, or aerosols, or the like, specifically in unit dosage forms suitable for simple administration of precise dosages.
- the administration may specifically be by placing a gliadel, a dissolvable material that contains the chemotherapy drug (in particular BCNU), directly into brain tumors during an operation.
- compositions will include a compound of formula I or la as the/an active agent and can include a conventional pharmaceutical carrier or excipient and in addition may include other medicinal agents and pharmaceutical agents that are generally administered to a patient being treated for cancer.
- Adjuvants include preserving, wetting, suspending, sweetening, flavoring, perfuming, emulsifying, and dispensing agents. Prevention of the action of microorganisms can be ensured by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for example sugars, sodium chloride, and the like. Prolonged absorption of the injectable pharmaceutical form can be brought about by the use of agents delaying absorption, for example, aluminum monostearate and gelatin.
- a pharmaceutical composition of the invention may also contain minor amounts of auxiliary substances, such as wetting or emulsifying agents, pH buffering agents, antioxidants, and the like, such as citric acid, sorbitan monolaurate, triethanolamine oleate, butylalted hydroxytoluene, etc.
- auxiliary substances such as wetting or emulsifying agents, pH buffering agents, antioxidants, and the like, such as citric acid, sorbitan monolaurate, triethanolamine oleate, butylalted hydroxytoluene, etc.
- the choice of formulation depends on various factors, such as the mode of drug administration (e.g., for oral administration, formulations in the form of tablets, pills, or capsules) and the bioavailability of the drug substance.
- pharmaceutical formulations have been developed especially for drugs that show poor bioavailability based upon the principle that bioavailability can be increased by increasing the surface area, i.e., decreasing particle size.
- U.S. Pat. No. 4,107,288 describes a pharmaceutical formulation having particles in the size range from 10 to 1 ,000 nm, in which the active material is supported on a crosslinked matrix of macromolecules.
- 5, 145,684 describes the production of a pharmaceutical formulation in which the drug substance is pulverized to nanoparticles (average particle size of 400 run) in the presence of a surface modifier and then dispersed in a liquid medium to give a pharmaceutical formulation that exhibits remarkably high bioavailability.
- compositions suitable for parenteral injection may comprise physiologically acceptable sterile aqueous or nonaqueous solutions, dispersions, suspensions, or emulsions, and sterile powders for reconstitution into sterile injectable solutions or dispersions.
- aqueous and nonaqueous carriers, diluents, solvents or vehicles examples include water, ethanol, polyols (propyleneglycol, polyethyleneglycol, glycerol, and the like), suitable mixtures thereof, vegetable oils (such as olive oil), and injectable organic esters (such as ethyl oleate).
- a coating such as lecithin
- surfactants such as surfactants.
- One specific route of administration is oral, using a convenient daily dosage regimen that can be adjusted according to the degree of severity of the disease-state to be treated.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules.
- the active compound is admixed with at least one inert customary excipient (or carrier), such as sodium citrate or dicalcium phosphate, or (a) fillers or extenders, as for example, starches, lactose, sucrose, glucose, mannitol, and silicic acid, (b) binders, as for example, cellulose derivatives, starch, alignates, gelatin, polyvinylpyrrolidone, sucrose, and gum acacia, (c) humectants, as for example, glycerol, (d) disintegrating agents, as for example, agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, croscarmellose sodium, complex silicates, and sodium carbonate, (e) solution retarders, as for example paraffin, (f) absorption accelerators, as for example paraffin, (f) ab
- Solid dosage forms as described above can be prepared with coatings and shells, such as enteric coatings and others well known in the art. They may contain pacifying agents, and can also be of such composition that they release the active compound or compounds in a certain part of the intestinal tract in a delayed manner. Examples of embedded compositions that can be used are polymeric substances and waxes. The active compounds can also be in microencapsulated form, if appropriate, with one or more of the above-mentioned excipients.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs. Such dosage forms are prepared, for example, by dissolving, dispersing, etc., a compound(s) of the invention, or a
- a carrier such as water, saline, aqueous dextrose, glycerol, ethanol, and the like; solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propyleneglycol, 1 ,3-butyleneglycol,
- oils such as cottonseed oil, groundnut oil, corn germ oil, olive oil, castor oil, sesame oil, and the like; glycerol; tetrahydrofurfuryl alcohol;
- polyethyleneglycols polyethyleneglycols; and fatty acid esters of sorbitan; or mixtures of these substances, and the like, to thereby form a solution or suspension.
- suspensions may contain suspending agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar, and tragacanth, or mixtures of these substances, and the like.
- suspending agents as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar, and tragacanth, or mixtures of these substances, and the like.
- compositions for rectal administrations are, for example, suppositories that can be prepared by mixing the compounds of the present invention with for example suitable non- irritating excipients or carriers such as cocoa butter, polyethyleneglycol or a suppository wax, which are solid at ordinary temperatures but liquid at body temperature and therefore melt while in a suitable body cavity and release the active component therein.
- suitable non- irritating excipients or carriers such as cocoa butter, polyethyleneglycol or a suppository wax, which are solid at ordinary temperatures but liquid at body temperature and therefore melt while in a suitable body cavity and release the active component therein.
- Dosage forms for topical administration of a compound of this invention include ointments, powders, sprays, and inhalants.
- the active component is admixed under sterile conditions with a physiologically acceptable carrier and any preservatives, buffers, or propellants as may be required.
- Ophthalmic formulations, eye ointments, powders, and solutions are also contemplated as being within the scope of this invention.
- Compressed gases may be used to disperse a compound of this invention in aerosol form.
- Inert gases suitable for this purpose are nitrogen, carbon dioxide, etc.
- compositions will contain about 1% to about 99% by weight of a compound(s) of the invention, or a pharmaceutically acceptable salt or solvate thereof, and 99% to 1% by weight of a suitable pharmaceutical excipient.
- the composition will be between about 5% and about 75% by weight of a compound(s) of the invention, or a pharmaceutically acceptable salt or solvate thereof, with the rest being suitable pharmaceutical excipients.
- composition to be administered will, in any event, contain a therapeutically effective amount of a compound of the invention, or a pharmaceutically acceptable salt or solvate thereof, for treatment of a disease-state in accordance with the teachings of this invention.
- the compounds of the invention are administered in a therapeutically effective amount which will vary depending upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of the compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular disease-states, and the host undergoing therapy.
- the compounds of the present invention can be administered to a patient at dosage levels in the range of about 0.1 to about 1,000 mg per day. For a normal human adult having a body weight of about 70 kilograms, a dosage in the range of about 0.01 to about 100 mg per kilogram of body weight per day is an example. The specific dosage used, however, can vary.
- the dosage can depend on a number of factors including the requirements of the patient, the severity of the condition being treated, and the pharmacological activity of the compound being used.
- the determination of optimum dosages for a particular patient is well known to one of ordinary skill in the art.
- the inhibitors of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR-2 kinases/kinase receptors can be formulated into solid or liquid forms using pharmaceutically acceptable excipients for oral or parenteral administration, as described above.
- Methods and dosage forms for administering functional nucleic acids are readily known, for example, they may be formulated in sterile aqueous reagents specifically designed for the mode of administering the functional nucleic acid in question.
- the functional nucleic acid may be in the form of a prodrug.
- Oligonucleotides are, by virtue, negatively charged ions. Due to the lipophilic nature of cell membranes the cellular uptake of oligonucleotides are reduced compared to neutral or lipophilic equivalents. This polarity "hindrance" can be avoided by using the pro-drug approach. In this approach the oligonucleotides are prepared in a protected manner so that the oligo is neutral when it is administered. These protecting groups are designed in such a way that so they can be removed, and then the oligo is taken up be the cells. Examples of such protecting groups are S-acetylthioethyl (SATE) or S-pivaloylthioethyl (t-butyl-SATE). These protecting groups are nuclease resistant and are selectively removed intracellulary.
- SATE S-acetylthioethyl
- t-butyl-SATE S-pivaloylthioethyl
- Functional nucleic acids can be linked to ligands/conjugates to facilitate delivery to the intended target site and enhance activity of the functional nucleic acid, i.e. to increase the cellular uptake of functional nucleic acid.
- This conjugation can take place at the terminal positions 5V3'-OH, but the ligands may also take place at the sugars and/or the bases.
- Other examples of conjugates ligands are cholesterol moieties, liposomes, neutral lipids, duplex intercalators, such as acridine, poIy-L-lysine, "end-capping" with one or more nuclease- resistant linkage groups.
- the functional nucleic acids can comprise two or more different functional nucleic acids.
- Pharmaceutically acceptable binding agents and adjuvants may comprise part of the formulated drug.
- Capsules, tablets, pills, etc. may contain, for example, the following compounds: microcrystalline cellulose, gum or gelatin as binders; starch or lactose as excipients; stearates as lubricants; and various sweetening or flavoring agents.
- the dosage unit may contain a liquid carrier, such as fatty oils.
- coatings of sugar or enteric agents may be part of the dosage unit.
- the oligonucleotide formulations may also be emulsions of the active pharmaceutical ingredients and a lipid forming a micellular emulsion.
- An oligonucleotide of the invention may be mixed with any material that do not impair the desired action, or with material that supplement the desired action. These could include other drugs including other nucleotide compounds.
- the formulation may include a sterile diluent, buffers, regulators of tonicity, and antibacterials.
- Functional nucleic acids may be prepared with carriers that protect against degradation or immediate elimination from the body, for example antinuclease activity, including implants or microcapsules with controlled release properties.
- the preferred carriers are physiological saline or phosphate buffered saline.
- a functional nucleic acid is included in a unit formulation, such as in a pharmaceutically acceptable carrier or diluent in an amount sufficient to deliver to a patient a therapeutically effective amount without causing serious side effects in the treated patient.
- the functional nucleic acid co-administered with a compound of formula I of the present invention can be administered in a number of ways depending upon whether local or systemic treatment is desired and upon the area to be treated. Administration may be: (a) oral; (b) pulmonary, e.g., by inhalation or insufflation of powders or aerosols, including by nebulizer; intratracheal, or intranasal; (c) topical including epidermal, transdermal, ophthalmic, and to mucous membranes including vaginal and rectal delivery; (d) parenteral including intravenous, intraarterial, subcutaneous, intraperitoneal, or intramuscular injection or infusion; or (e) intracranial, e.g., intrathecal or intraventricular, administration.
- Administration may be: (a) oral; (b) pulmonary, e.g., by inhalation or insufflation of powders or aerosols, including by nebulizer; intratracheal, or
- the active oligo is administered IV, IP, orally, topically, as a bolus injection, or administered directly in to the target organ.
- the functional nucleic acid is administered via stereaotactic injection directly into the affected tissue or intratumoral injection with or without the aid of scanning devices such as CT scans, PET scanning devices, and MRI scanning devices.
- Compositions and formulations for parenteral, intrathecal, or intratumoral administration may include sterile aqueous solutions which may also contain buffers, diluents, and other suitable additives such as, but not limited to, penetration enhancers, carrier compounds, and other pharmaceutically acceptable carriers or excipients.
- Antibodies of the present invention can be administered to a cancer patient in any standard medically accepted way. Such compositions may be conveniently administered in unit dose and may be prepared in accordance with methods known in the pharmaceutical art. See Remington's Pharmaceutical Sciences, (Mack Publishing Co., Easton Pa., (1980)). By the term “unit dose” is meant a therapeutic composition of the present invention employed in a physically discrete unit suitable as unitary dosages for a primate such as a human, each unit containing a pre-determined quantity of active material calculated to produce the desired therapeutic effect in association with the required diluent or carrier.
- the unit dose will depend on a variety of factors, including the type and severity of the cancer to be treated, capacity of the subject's blood to deliver the antibody, and the ability of the patient to tolerate sufficiently high doses without intolerable side effects. Precise amounts of the antibody to be administered typically will be guided by judgment of the practitioner, typically titrating from a high dose to a tolerable lower dose having comparable tumoricidal activity however, the unit dose will generally depend on the route of administration and be in the range of 10 ng kg body weight to 100 mg/kg body weight per day, more typically in the range of 100 ng kg body weight to about 40 mg/kg body weight per day. Suitable regimens for booster administration are also variable but are typified by an initial administration followed by repeated doses at one or more hour intervals by a subsequent injection or other
- continuous or intermittent intravenous infusions may be made sufficient to maintain concentrations of at least from about 10 nanomolar to 10 micromolar of the antibody in the blood.
- kits for preparing an cancer treatment composition comprising a first container comprising: (a) a therapeutically effective amount of a compound of formula I; (b) a second container comprising a therapeutically effective amount of a solution of antibodies, such as anti- HER2 and/or HER3 antibodies (alternatively in lyophilized form); (c) optionally, one or more medicament delivery devices, for example syringes and the like; (d) optionally, a diluent for resuspending the anti- HER2 and/or HER3 antibody; and (e) a set of instructions for preparing the composition for treatment of cancer in the patient.
- a first container comprising: (a) a therapeutically effective amount of a compound of formula I; (b) a second container comprising a therapeutically effective amount of a solution of antibodies, such as anti- HER2 and/or HER3 antibodies (alternatively in lyophilized form); (c) optionally, one or more medicament delivery devices, for example sy
- Compounds of formula I have been tested using the assay described in Biological Example 1 of WO2007044729 and have been determined to be PI3K inhibitors. As such, compounds of formula I are useful for treating diseases, particularly cancer in which PDKactivity contributes to the pathology and/or symptomatology of the disease.
- cancer in which PI3K activity contributes to its pathology and/or symptomatology include breast cancer, colon cancer, rectal cancer, endometrial cancer, gastric carcinoma, glioblastoma, hepatocellular carcinoma, small cell lung cancer, non-small cell lung cancer, melanoma, ovarian cancer, pancreatic cancer, prostate carcinoma, acute myelogenous leukemia (AML), chronic myelogenous leukemia (CML), and thyroid carcinoma, and the like.
- AML acute myelogenous leukemia
- CML chronic myelogenous leukemia
- methods for identifying a compensatory kinase pathway can be used to design novel cancer therapeutics.
- the present invention provides a method for identifying a compensatory kinase pathway in a cancer cell which attenuates the effect of a PI3 inhibitor.
- the method includes the steps: (a) contacting the cancer cell with a composition comprising a compound of formula I; (b) incubating the mixture of cancer cells in the presence and absence of the composition; (c) measuring the expression of a plurality of kinase enzymes in the presence and absence of the composition after the period of incubation; and (d) determining whether a kinase enzyme within the group of tested kinase enzymes displays an increase in either kinase expression or kinase activity when compared to the kinase enzyme in the cancer cell incubated in the absence of the composition containing the compound of formula I.
- the identified kinase enzyme is an indication of the identified kinase enzyme being involved in a compensatory kinase pathway which attenuates the effect of the PI3K inhibitor.
- Suitable in vitro assays for measuring PI3K activity and the inhibition thereof by compounds are known. Typically, the assay will measure PI3K-induced ATP consumption.
- an in vitro assay for measuring PI3K activity see Biological Examples, Example 1 infra.
- Cellular activity can be determined using assays as described in Biological Examples 2, 3, and 4 of WO2007044729.
- Suitable in vivo models of cancer are known to those of ordinary skill in the art.
- Examples 5-10, of WO2007044729 Examples describing the administration of a compound of formula I in combination with anticancer agents are described in Biological Examples 1 1 -14, of WO2007044729. Following the examples disclosed herein, as well as that disclosed in the art, a person of ordinary skill in the art can determine what combinations of a compound of formula I and anti-cancer agents would be effective for treating cancer.
- the starting materials and reagents used in preparing these compounds are either available from commercial suppliers such as Aldrich Chemical Co. (Milwaukee, Wis.), or Bache (Torrance, Calif.), or are prepared by methods known to those skilled in the art following procedures set forth in references such as Fisher and Fisher's Reagents for Organic Synthesis, Volumes 1-17 (John Wiley and Sons, 1991); Rod's Chemistry of Carbon Compounds, Volumes 1 -5 and Supplemental (Elsevier Science Publishers, 1989); Organic Reactions, Volumes 1 -40 (John Wiley and Sons, 1991), March's Advanced Organic Chemistry, (John Wiley and Sons, 4 l Edition), and Larch's Comprehensive Organic Transformations (VICHY Publishers Inc., 1989).
- the reactions described herein take place at atmospheric pressure and over a temperature range from about -78 °C to about 150°C, in another embodiment from about 0°C to about 125 °C ,and more specifically at about room (or ambient) temperature, e.g., about 20 °C. Unless otherwise stated (as in the case of a hydrogenation), all reactions are performed under an atmosphere of nitrogen.
- Prodrugs can be prepared by techniques known to one skilled in the art. These techniques generally modify appropriate functional groups in a given compound. These modified functional groups regenerate original functional groups by routine manipulation or in vivo. Amides and esters of the compounds of the present invention may be prepared according to conventional methods. A thorough discussion of prodrugs is provided in T. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," Vol 14 of the A.C.S.
- the compounds of the invention may have asymmetric carbon atoms or quaternized nitrogen atoms in their structure.
- Compounds of formula I that may be prepared through the syntheses described herein may exist as single stereoisomers, racemates, and as mixtures of enantiomers and diastereomers.
- the compounds may also exist as geometric isomers. All such single stereoisomers, racemates, and mixtures thereof, and geometric isomers are intended to be within the scope of this invention.
- Some of the compounds of the invention may exist as tautomers.
- the molecule may exist in the enol form; where an amide is present, the molecule may exist as the imidic acid; and where an enamine is present, the molecule may exist as an imine. All such tautomers are within the scope of the invention.
- B can be 2-hydroxy-pyridinyl, also described as its structure:
- Both 2-hydroxy-pyridinyl and the above structure 14 include, and are equivalent to, pyridin- 2 ⁇ ltf)-one and its structure 15:
- the present invention also includes N-oxide derivatives and protected derivatives of compounds of formula I.
- compounds of formula I when compounds of formula I contain an oxidizable nitrogen atom, the nitrogen atom can be converted to an N-oxide by methods well known in the art.
- compounds of formula I When compounds of formula I contain groups such as hydroxy, carboxy, thiol, or any group containing a nitrogen atom(s), these groups can be protected with a suitable "protecting group” or "protective group.”
- a comprehensive list of suitable protective groups can be found in T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, Inc. 1991 , the disclosure of which is incorporated herein by reference in its entirety.
- the protected derivatives of compounds of formula I can be prepared by methods well known in the art.
- stereoisomers from racemic mixtures or non-racemic mixtures of stereoisomers are well known in the art.
- optically active (R)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques.
- Enantiomers may be resolved by methods known to one of ordinary skill in the art, for example by: formation of diastereoisomeric salts or complexes which may be separated, for example, by crystallization; via formation of diastereoisomeric derivatives which may be separated, for example, by crystallization; selective reaction of one enantiomer with an enantiomer-specific reagent, for example enzymatic oxidation or reduction, followed by separation of the modified and unmodified enantiomers; or gas-liquid or liquid
- enantiomer may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts, or solvents, or by converting on enantiomer to the other by asymmetric transformation.
- the major component enantiomer may be further enriched (with concomitant loss in yield) by recrystallization.
- the compounds of the present invention can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like.
- pharmaceutically acceptable solvents such as water, ethanol, and the like.
- the solvated forms are considered equivalent to the unsolvated forms for the purposes of the present invention.
- each LG 1 is a leaving group (in one embodiment halo, in another embodiment chloro), and all other groups are as defined in the Detailed Description.
- an intermediate of formula 3 can be prepared by briefly heating commercially available 2,3-dichloroquinoxaline and an intermediate of formula 2 (which are commercially available or can be prepared by one of ordinary skill in the art), a base, such as K2CO3, in a solvent, such as DMF or DMSO. Upon completion (about 2 hours), the reaction mixture is then poured into water and followed by 2 N HCl. The product is then extracted into a solvent, such as ethyl acetate, and washed with water and brine. The organic layers are combined and dried over a drying agent such as sodium sulfate, filtered, and concentrated under vacuum.
- a base such as K2CO3
- a solvent such as DMF or DMSO
- the intermediate of formula 3 is then treated with an intermediate of formula 4 in a solvent, such as DMF or p-xylene, at reflux temperature. Upon completion of the reaction (about 16 hours or less), the reaction is allowed to cool, extracted into DCM, washed with 2 N HCl and brine, dried over a drying agent such as sodium sulfate or magnesium sulfate, filtered, and concentrated to give a compound of formula I.
- a solvent such as DMF or p-xylene
- quinoxaline derivatives are known to one skilled in the art and include, but are not limited to S. V. Litvinenko, V. I. Savich, D. D. Bobrovnik, Chem. Heterocycl. Compd. (Engl. Transl), 1994, 30, 340 and W. C. Lumma, R. D. Hartman, J. Med. Chem. 1981, 24, 3.
- LG is a leaving group such as chloro. 5 is reacted with NHR a R b or HO-C
- a base such as KHCO3
- a solvent such as DMF.
- the reaction is carried out in the presence of a base such as NaH in a solvent such as DMF.
- alkylene in R 3 and R 3a are independently optionally substituted as described in the Summary can be prepared according to Scheme 4 by reacting with an intermediate of formula 9(a), 9(b), 9(c), 9(d), 9(e), 9(0, or 9(g):
- R 100 in Scheme 4 is -C(0)R 9a , -C(0)NR l la R l lb , -C(0)OR ,3a , -C(0)-C,-C 6 -alkylene- N(R l 8b )C(0)R ll!a , -C(O)-C,-C 6 -alkylene-C(O)R 0il , or -S(0) 2 -C,.C 6 -aIkylene-N(R 2,b )R a .
- the reaction is carried out under standard amide coupling conditions known to one of ordinary skill in the art.
- reaction is carried out in the presence of a coupling agent, such as HATU, a base, such as DIEA, and in a solvent, such as DMF.
- a coupling agent such as HATU
- a base such as DIEA
- a solvent such as DMF.
- the N-protecting group is then removed using procedures known to one of ordinary skill in the art, such as treating with acid where PG is Boc.
- LG is a leaving group such as bromo or chloro. 12 is reacted with NH(R 7b )R 7a
- a base such as DIEA
- a solvent such as ACN
- LG in Scheme 6 is a leaving group, such as chloro.
- the reaction can be carried out by irradiating in a solvent, such as DMA.
- the reaction can be carried out in presence of acetic acid in a solvent, such as DMA and by heating.
- Preparative reverse-phase HPLC was used to isolate the desired product directly from the crude reaction mixture.
- a Waters Fractionlynx preparative reverse-phase HPLC equipped with a Waters SunFire Prep CI 8, OCD 5 ⁇ , 30 X 70 mm column and running a 5-100 % gradient with a binary solvent system of 25 mM ammonium acetate in water/acetonitrile was used to carry out the purification.
- a Waters Fractionlynx preparative reverse-phase HPLC (equipped with a Waters SunFire Prep CI 8, OCD 5 ⁇ , 30 X 70 mm column and running a 5-100 % gradient with a binary solvent system of 25 mM ammonium acetate in water/acetonitrile) was used to carry out the purification.
- a CEM microwave reaction vessel was charged with N-(3-(N-(3- chloroquinoxalin-2-yl)sulfamoyl)phenyI)-2-(dimethylamino)acetamide (30 mg, 0.071 mmol), prepared using standard procedures, the desired aniline (16 mg, 0.14 mmol, 2 eq), and 0.5 mL of dimethylacetamide.
- the vessel was sealed, and the reaction mixture was heated under microwave radiation for 70 minutes at 140 °C in a CEM Discover microwave instrument. The solvent was then removed by rotary-evaporation. Purification of the final product was accomplished by preparatory reverse-phase HPLC with the eluents 25 mM aqueous
- a CEM microwave reaction vessel was charged with N-(3-(N-(3- chloroquinoxalin-2-yl)sulfamoyl)phenyl)-2-(dimethylamino)acetamide (62 mg, 0.147 mmol), prepared using standard procedures, the desired aniline (0.567 mmol, 4 eq), and 1.0 mL of toluene.
- the vessel was sealed, and the reaction mixture was heated under microwave radiation for 60 minutes at 180 °C in a CEM Discover microwave instrument. The solvent was removed on a rotary-evaporator. Purification of the final product was done by preparatory HPLC with N L Ac/ACN as eluent to yield the desired product.
- N-(3-(N-(3-(3,5-dimedioxy-phenylamino)quinoxalin-2-yl)- sulfamoyl)phenyl)azetidine-3-carboxamide (125 mg, 0.23 mmol), prepared using standard procedures, was dissolved into 5 mL DCE in a 10 mL round-bottom flask.
- DEEA 1.17 mmol, 5.0 equiv.
- acid chloride (0.47 mmol, 2.0 equiv.
- the reaction was then stirred at room temperature for 1 hour or until complete as indicated by LCMS.
- the solvent was subsequently removed under reduced pressure on a rotary evaporator.
- Preparative reverse-phase HPLC was used to isolate the desired product.
- a Waters Fractionlynx preparative reverse-phase HPLC (equipped with a Waters SunFire Prep CI 8, OCD 5 ⁇ , 30 X 70 mm column and running a 5-100 % gradient with a binary solvent system of 25 mM ammonium acetate in water/acetonitrile) was used to carry out the purification.
- siRNA screen is employed targeting 779 human kinases in order to determine if downregulation of specific kinases increases the sensitivity of HCCI 937 human breast cancer cells to the PI3K inhibitor compound A, which a compound of formula I and of Table 1.
- Compound A is an ATP-mimetic that inhibits the p 1 10a catalytic subunit of PI3K with an in vitro IC 50 of 39 nM.
- HCCI 937 cells lack PTEN, the negative regulator of the PI3K pathway, and are growth inhibited by compound A with an IC50 of 10 ⁇ .
- IC50 10 ⁇ .
- cells are treated with either DMSO or 10 ⁇ compound A for 72 hours in a medium containing 2.5% fetal bovine serum (Figs. 1 A and 2) and the alamar blue assay is used to measure cell viability Fig. IB.
- gene expression is performed using microarrays, phospho-receptor tyrosine kinase (RTK) arrays, and phospho- intracellular kinase arrays (Figs.
- RNA or lysates from HCCI 937 cells ⁇ compound A in order to determine which kinases and pathways are upregulated upon inhibition of PI3K.
- Compound A treatment results in increased expression and/or phosphorylation of many RTKs and intracellular kinases within 8-48 hours. Eight and 24 hour treatments with compound A suppressed phospho-Akt at both S473 and T308, but at 48 hours, phosphorylation at S473 is partially restored. From these approaches, several kinases and pathways that are upregulated by compound A are identified and that, when
- siRNA downregulated by siRNA, increased sensitivity to compound A.
- RTKs such as HER3, MSPR, and Axl
- members of the MAP kinase signaling pathway such as several MAP3Ks/MEKKs.
- Western blotting confirms that compound A treatment decreases phospho-Akt at S473 and T308 and increases phospho- and total HER3.
- Compound A also increases phosphorylation of the MAP kinases ERK, JNK, and p38 MAPK.
- HCCI 937 cells were treated with 10 ⁇ compound A in medium containing 2.5% FBS for 0, 2, 8, or 24 hours.
- RNA was isolated with Trizol and purified using the RNeasy column (Qiagen). Gene expression from three independent experiments was measured using the Gene titan 3' microarray. *, p ⁇ 0.05; **, p ⁇ 0.01 versus 0 hours, as shown in Fig. 5.
- Combined treatment with compound A and HER3 siRNA inhibits HCCI 937 cell growth to a larger degree than each intervention alone (Fig.
- MST1-R Macrophage stimulating 1 receptor
- the antitumor effect of the PI3K inhibitor compound A is examined in a panel of human breast cancer cells with mutational activation of the PBK/Akt pathway (as a result of PIK3CA mutations, PTEN loss, and/or HER2 gene amplification).
- Site-specific antibodies reveals HER3 phosphorylation at Yl 197 and Y 1289, two of the six p85 binding sites in HER3.
- an anti-HER3 Ab can delay or abrogate feedback upregulation of HER3 in human breast cancer cells treated with a PI3K inhibitor
- human breast cancer cells with or without overexpression of HER2 are treated with compound A ⁇ a saturating concentration of an anti-HER3 Ab, such as MM- 121.
- Experimental endpoints are: (1) association of p85 with HER3, (2) growth in 3D-MatrigeI, (3) colony formation in soft agar, (4) apoptosis assays in both serum-free conditions and in suspension (to induce anoikis), (5) motility in wound closure and transwell assays, (6) PI3K activity in HA pull downs subjected to an in vitro PI3K reaction, and (7) total HER3 and P-HER3, P-Akt, P-Erk, P-S6, P-4EBP-1 by immunoblot.
- Xenografts in athymic mice are established according to known methods. Once tumors reach a volume ⁇ 250 mm3, they are randomized to four treatment arms: (1) control IgGl (30 mg kg x2/week i.p.). (2) compound A ( 100 mg kg/day via orogastric gavage), (3) MM- 121 (30 mg/kg x2/week i.p.), and (4) compound A plus an anti-HER3 Ab, such as MM- 121 (Merrimack Pharmaceuticals, Cambridge, MA, USA). Treatment is delivered for up to five weeks, and tumor growth is monitored thereafter. Study endpoints are assessed after one and five weeks of treatment and following discontinuation of therapy.
- All cell lines were purchased from the American Type Culture Collection. Media and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA, USA). The following growth media were used: HCC1937, HCC1954: RPMI-1640/10% FBS; for BT474: IMEM/10% FBS; SKBR3: McCoy's 5A/15% FBS; UACC893: DMEM/10% FBS; MDA453: DMEM/F12 (l:l)/20% FBS; and SUM 190: DMEM F12 (1 : l)/5% FBS. All cells were grown in a humidified 5% C0 2 incubator at 37°C.
- FBS fetal bovine serum
- lapatinib GW-572016, LC Laboratories
- trastuzumab Genetech, San Francisco, CA, USA
- LY294002 Calbiochem
- the allosteric AKTl/2 inhibitor 5J8 rapamycin
- compound A a compound of formula I (Exelixis, Inc.).
- Cytoplasmic and nuclear extracts were prepared using the Nuclear Extract Kit from Active Motif. Briefly, cells were plated in 100-mm dishes, treated with inhibitors at the indicated concentrations for 3-4 hours, washed with ice-cold PBS/phosphatase inhibitors, lysed in hypotonic buffer and centrifuged for 30 seconds at 14,000 rpm at 4°C to collect the supernatant (cytoplasmic extract). The pellet (nuclear fraction) was resuspended in complete lysis buffer and centrifuged for 10 minutes at 14,000 rpm at 4°C to collect the supernatant (nuclear fraction).
- PI3K phosphatidylinositol-3 kinase
- RTKs ligand-activated receptor tyrosine kinases
- class I A PI3K has been implicated in cancer.
- Class I A PI3K is a heterodimer consisting of a regulatory (p85) and a pi 10 catalytic subunit. PI3 is activated by phosphorylated adaptors or receptors containing YXXM motifs that engage the N-SH2 domain of p85.
- PIP2 phosphatidylinositol-4,5-bisphosphate
- PIP3K phosphorylates PIP2 to produce the second messenger phosphatidylinositol-3,4,5- trisphosphate (PIP3).
- Negative regulation of this pathway is mediated by the PIP3 lipid phosphatase PTEN (phosphatase and tensin homologue at chromosome ten).
- PTEN phosphatase and tensin homologue at chromosome ten.
- a subset of pleckstrin homology (PH) domain-containing proteins, including AKT and PDK1 bind to PIP3 at the plasma membrane.
- AKT phosphorylates a host of cellular proteins, including GSK3a, GSK3P, FoxO
- AKT phosphorylates and inactivates Tuberin, a GTPase-activating protein (GAP) for the Ras homologue Rheb. Inactivation of Tuberin allows GTP bound-Rheb to accumulate and activate the mTOR Raptor (TORC1) complex, which ultimately regulates protein synthesis and cell growth.
- GAP GTPase-activating protein
- P13K/AKT is arguably the most commonly altered signaling pathway in human cancers.
- gain of function mutations in PIK3CA the gene encoding pi 10a, are present in high frequency in multiple human tumors.
- the phosphatase PTEN is a tumor suppressor gene frequently inactivated by mutation, gene deletion, targeting by microRNA, and promoter methylation.
- PI3K is potently activated by oncogenes like mutant Ras and many tyrosine kinases such as Bcr-Abl, HER2 (ErbB2), MET, KIT, etc., which themselves are the targets of mutational activation and/or gene amplification.
- PI3K pathway is also activated by the Aktl or Akt2 gene amplification.
- PI3K pathway antagonists have been developed and are the subject of recent reviews. Some of these compounds are ATP-mimetics that bind competitively and reversibly in the ATP-binding pocket of the kinase domain in pi 10 and are also active against oncogenic mutant forms of this enzyme.
- the present Examples have examined the effect of compound A (from Exelixis, Inc.) against a panel of human breast cancer cell lines harboring a molecular alteration indicative of PI3K dependence, such as HER2 gene amplification, a PIK3CA (pi 10a) activating mutation, and/or loss of PTEN.
- Compound A is an ATP- competitive reversible PI3K inhibitor with an IC 50 against pi 10a of 39 nM which has just completed phase I clinical development.
- HER2+ cells phosphorylation of the upregulated HER3 co-receptor was maintained by the HER2 tyrosine kinase resulting, in turn, in partial recovery of pAKT and, thus, limiting the antitumor action of PI3K inhibitors.
- therapies targeted against HER2/HER3 should be added to PI3K inhibitors in HER2- dependent cells in order to maximally disable PI3K AKT signaling.
- the present Examples have provided insight into the cellular and molecular effects of an inhibitor of phosphatidiIinositol-3 kinase (PI3K), compound A, against human breast cancer cell lines with constitutive PI3K activation.
- PI3K phosphatidiIinositol-3 kinase
- Treatment with compound A resulted in dose-dependent inhibition of cell growth and levels of pAKT and pS6, signal transducers in the PI3K AKT TOR pathway.
- pAKT and pS6, signal transducers in the PI3K AKT TOR pathway In HER2-overexpressing cells, simultaneous with inhibition of PI3K, there was upregulation of expression and phosphorylation of multiple RTKs, including HER3.
- PI3K antagonists will inhibit AKT and relieve suppression of RTK expression and their activity. Relief of this feedback limits the sustained inhibition of the pathway and attenuates the response to these agents.
- PI3K inhibitors should be used in combination with HER2-HER3 antagonists.
- Immunoprecipitation, immunoblotting, and RTK assays were performed according to published methods.
- Primary antibodies included AKT, pAKT S473 , pAKTTM 8 , ERK, pERKTM 04 , pHER2 YI248 , pHER3 Ymv , pHER3 Y1222 , pHER3 Y1289 , S6, P S6 S240/244 , p27, pRb S780 , FoxOI, Fox03a, MEK1/2 (Cell Signaling Technology, Danvers, MA, USA ), p85, 4G 10 pTyr (Millipore, Billerica, MA, USA), HER3, CyclinDl , RhoA, HDAC3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), PARP (BD Transduction Laboratories), ⁇ -actin (Sigma, St.
- siRNA duplexes for mismatch control and human HER3 were described previously (Wang, S.E., et al. (2008) Mol. Cell. Biol. 28:5605-5620, incorporated by reference herein in its entirety); Human FoxOl and Fox03a-specific Silencer ® Select siRNA duplexes were purchased from Ambion (Applied Biosystems, Austin, TX, USA) (FoxOl sense strand, SEQ ID NO: 19: CCAUGGACAACAACAGUAAtt; Fox03a sense strand, SEQ ID NO:20: GCCUUGUCGAAUUCUGUCAtt).
- IGF-IR and InsR siRNA duplexes were obtained from Qiagen (Valencia, CA, USA) (IGF-IR sense strand, SEQ ID NO:21 : GGAGAAUAAUCCAGUCCUAtt; InsR sense strand, SEQ ID NO:22:
- VUMC Institutional Animal Care Committee of Vanderbilt University Medical Center
- Mice were housed in the accredited Animal Care Facility of the VUMC.
- a 17p-estradiol pellet (Innovative Research of America) was injected subcutaneously (s.c.) into each 6- to 7-week-old athymic female mice (Harlan Sprague Dawley, Inc.) the day before tumor cell injection.
- a panel of breast cancer cell lines with dysregulated PI3K activity was treated with increasing doses of compound A under two- and three-dimensional (2D and 3D) growth conditions. All experiments were conducted in 2.5% FBS-containing media, unless otherwise stated. Treatment with compound A inhibited the 2D growth of all cell lines in a dose- dependent manner as shown in Fig. 9A. A similar IC50 of ⁇ 6 uM was observed in cells tested in 3D as shown in Fig. 9B, consistent with the fact that all of the selected cell lines in the present experiments harbor a molecular alteration in the PI3K pathway.
- compound A induced a reduction in cyclin Dl and pRB and an increase in levels of the CDK inhibitor p27 KIP1 but no change in levels of total or cleaved PARP, a biomarker of cell death (see Fig. 10B).
- PI3K pathway inhibitors induced a similar effect: 5J8, an allosteric inhibitor of AKT1/2 and the PI3K inhibitor LY294002 but not the mTOR inhibitor rapamycin also upregulated HER3 mRNA levels, as shown in Fig. 12B.
- the FoxO family of Forkhead transcription factors were examined, as AKT regulates the subcellular localization of these molecules by phosphorylation thereby preventing them from translocating to the nucleus and regulating transcription.
- the FoxO family consists of three members, FoxOl, Fox03a, and Fox04 (also known as FKHR, FKHR-L1 , and AFX, respectively) which bind as monomers through the consensus sequence TTGTTTAC at the promoter of target genes. In absence of AKT activity, FoxO is believed to be predominantly nuclear and therefore can activate transcription. Further, using the PROMO database
- siRNAs reduced 70-80% the levels of both FoxO mRNAs in all three cell lines as shown in Fig. 12F. Finally, simultaneous knockdown of FoxOl and Fox03a abrogated the compound A-induced increase in HER3 mRNA in BT474, SKBR3, and MDA453 cells (see Fig. 12E).
- Example 18
- pHER3 is known to activate the extracellular signal regulated kinase (ER /MAPK) via its interaction through the adaptor protein She, recovery of pERK upon feedback reactivation of HER3 was not detected consistently as shown in Fig. 13 A. Even though recovery of pAKT was less, feedback upregulation of total HER3 and pHER3 was more noticeable with a supra-pharmacological dose of 20 ⁇ , further suggesting inhibition of PI3K causes feedback activation of HER3, illustrated in Fig. 13B.
- ER /MAPK extracellular signal regulated kinase
- HER2 overexpressing cells although not bound by any particular theory, it is believed that the main mechanism of activation of PI3K is the coupling of pHER3 to an SH2 domain in the N-terminus of p85, the regulatory subunit of PI3K.
- the main tyrosine phosphorylated protein precipitated with p85 antibodies is pHER3; this association between HER3 and p85 (PI3K) depends on the catalytic activity of HER2 as it is disrupted by HER2 tyrosine kinase inhibitors (TKI).
- TKI tyrosine kinase inhibitors
- BT474 cells were treated with increasing concentrations of compound A followed by pulldown with p85 antibodies and subsequent pTyr and HER3 immunoblot analyses. Following treatment with compound A, there was a dose-dependent increase of approximately 200-kDa main pTyr band as well as other smaller and less abundant pTyr proteins as shown in Fig. 13C, marked with arrows. Consistent with previous studies, the p85-associated approximately 200-kDa band was also detected with HER3 and pHER3 Y1 197 antibodies as shown in Fig. 13D and I3F.
- the HER2 receptor is the main tyrosine kinase that phosphorylates HER3 which, in turn, directly couples to and activates PI3K. Since compound A does not affect the catalytic activity or autophosphorylation of HER2 (see for example. Fig. 1 1 ), it is logical to speculate that in HER2-overexpressing cells, HER2 remains as the tyrosine kinase maintaining and/or increasing the phosphorylation of HER3 upon inhibition of PI3K.
- Lapatinib alone markedly induced HER3 mRNA this action, as in compound A, is the result of PI3K inhibition followed by derepression of FoxO-mediated transcription.
- the effect of lapatinib was more prominent when used in combination with compound A as shown in Fig. 16D, probably as the result of a more pronounced inhibition of PI3K/AKT compared to either agent alone.
- treatment with the combination of compound A plus lapatinib or compound A plus trastuzumab for 24 h attenuated the recovery of pHER3 compared to cells treated with compound A alone as shown in Fig. 16E, suggesting that inhibition of the HER2 kinase with lapatinib or of ligand-independent HER2/HER3 dimers with trastuzumab limits the activating input of HER2 to HER3.
- Fig. 13C shows an increase in detectable levels of p85-associated pTyr proteins upon compound A-mediated inhibition of PI3K in BT474 cells.
- Results provided herein also demonstrate a prominent approximately 200-kDa pTyr band that coprecipitates with p85 (PI3K) is recognized by HER3 and pHER3 antibodies as shown in Figs. 13D-13F, and is eliminated upon knockdown of HER3 with siRNAs as shown in Figs. 13E and 13F.
- Fig. 13C shows an increase in p85-associated proteins other than HER3.
- results provided in Fig. 19A show that upon knockdown of HER3 with siRNA, there is still an increase in the p85-associated approximately 200-kDa band, leading further to speculation of the presence of other compensatory p85-associated tyrosine kinases and/or adaptors aimed at partially maintaining PI3K active.
- Treatment with compound A resulted in an increase in not only HER3, but also multiple other RTKs including EGFR, ERBB4 HER4, fibroblast growth factor receptor (FGFR)-l , -2, -3, and -4, insulin receptor (InsR), insulin-like growth factor-I receptor (IGF-IR), ephrin receptor A 1 (EphAl), endothelium-specific receptor tyrosine kinase 2 (Tie2), neurotrophic tyrosine kinase receptor type 1 (TrkA), Fms like tyrosine kinase 3 receptor (Flt3), tyrosine-protein kinase Mer (MER) and macrophage- stimulating protein receptor (MSPR), as shown in Figs.
- InsR insulin receptor
- IGF-IR insulin-like growth factor-I receptor
- EphAl ephrin receptor A 1
- Tie2 endothelium-specific receptor tyros
- RTKs migrate at approximately 200 kDa, such as EGFR, ERBB4, and InsR, and may explain the persistent high-MW pTyr band associated with p85 in cells where HER3 has been knocked down as shown in Fig. 19A.
- qPCR was performed and analyzed to determine whether upregulation of these RTKs occurred at the transcriptional level.
- BT474 cells following treatment with compound A there was an increase in ERBB4, IGF-IR, InsR, EphAl, FGFR2, and FGFR3 mRNAs, with IGF-IR and InsR being the most prominent, as shown in Fig. 19D.
- HER2-overexpressing BT474 and MDA453 cells and PI3K-mutant MCF7 cells were transfected with siRNA duplexes against IGF-IR and InsR followed by treatment with compound A. Knockdown of both RTKs was effective in all three cell lines as shown in Figs. 20A and 20B. Depletion of either RTK sensitized all three cells to compound A-mediated growth inhibition as shown in Figs. 20C-20E. Taken together, these data suggest that cancer cells limit the inhibition of PI3K by feedback mechanisms that upregulate multiple RTKs or adaptors capable of engaging p85 and thus activating PI3K. In turn, these molecules partially maintain
- the present invention is not limited to a particular mechanism. Indeed, an understanding of the mechanism is not necessary to practice the present invention.
- Cellular effects of PI3K inhibition with the small molecule compound A in a panel of cancer cell lines suggest PI3K dependence.
- the illustrative experiments provided herein have examined PI3K inhibition and its cellular effects in breast cancer cells with HER2 gene amplification.
- pAKT, pS6 and cell growth inhibition of PI3K with compound A resulted in time-dependent, feedback upregulation of HER3 expression and phosphorylation.
- pHER3 engaged p85, activated PI3K, and induced partial recovery of pAKT and pS6 as shown in Figs.
- AKT has been shown to phosphorylate the FoxO family of transcription factors and thereby prevent their nuclear translocation. Therefore, the initial inhibition of AKT resulted in accumulation of Fox03a and FoxOl proteins in the nucleus as shown in Fig. 12D. Knockdown of both FoxO proteins suppressed the induction of HER3 mRNA upon inhibition of PI3K/AKT with compound A (Fig. 12E).
- HER3 is downregulated by PI3K AKT and are consistent with a recent observation in ovarian cancer cells where the PI3K inhibitor GDC-0941 blocked downregulation of the HER3 mRNA upon treatment with the HER3 activating ligand heregulin.
- HER3 expression and phosphorylation limited the antitumor effect of the PI3K inhibitor. This is supported by the fact that siRNA-mediated knockdown of HER3 sensitized to compound A-induced cell death, as shown in Fig. 14, and enhanced compound A-mediated inhibition of pAKT and pS6, as shown in Figs. 15A and 15C. This result has particular relevance for HER2-overexpressing cells where the kinase-deficient HER3 co-receptor, once dimerized with and activated by the HER2 kinase, is the key mechanism engaging p85 and activating PI3K.
- breast cancer cells with HER2 amplification are particularly sensitive to apoptosis induced by PI3K inhibitors.
- this association of HER2/HER3 dimer with p85 has been found by others to be essential for the viability of HER2-dependent cells and, therefore, sustained inhibition of the output of HER2/HER3 to PI3K is required for the antitumor effect of HER2 inhibitors.
- HER3 phosphorylation was maintained and further increased, remaining associated with p85 in compound A-treated cells as shown in Figs. 13D-13F.
- Addition of the HER2 antibody trastuzumab or the HER2 TKI lapatinib attenuated the rebound of pHER3 upon treatment with compound A as shown in Fig. 16E, even though HER3 transcription was further increased by the combinations of inhibitors compared to each inhibitor alone (see for example, Fig. 16D).
- both combinations of compound A with each HER2 antagonist were more effective than each inhibitor alone at reducing pHER3 and pAKT levels in HER2-overexpressing xenografts and inhibiting their growth as shown in Fig. 18.
- PI3K inhibitors should be used in combination with HER2 antagonists in breast cancers of this subtype.
- the most appropriate anti-cancer agents to combine with PI3K/AKT inhibitors in other PI3K-dependent cancers without HER2 gene amplification will depend on the main compensatory feedback mechanisms that are activated upon inhibition of this pathway.
- the present study was designed to evaluate the efficacy of a combination of compound A and MM-121 in treating non-HER2 amplified tumors.
- the lung cancer cell line A549 was selected for this study.
- A549 cells do not express high HER2 levels, as shown in Bunn et al., Clinical Cancer Research, 7:3239-3250 (2001 ).
- Lung A549 cells were implanted s.c. in female Swiss nude mice using standard techniques.
- the A549 xenografts were treated when the tumors reached a mean volume of 152.9 ⁇ 94.7 mm3, with four combination conditions, employing two doses of MM-121 and compound A, using seven mice per group. Control groups were left untreated.
- Compound A was administered at 100 or 30 mg kg/adm, by PO daily administrations, for 32 consecutive days (from D37 to D68: QlDx32).
- MM-121 was administered at 5 or 30 mg/kg/inj by IP route once every 3 days (D37, D40, D43, D46, D49, D52, D55, D58, D61, D64 and D67; Q3D l 1).
- the four combination conditions were as follows: compound A at 100 mg/kg adm, daily administered with MM-121 at 30 mg/kg once a day every three days; compound A at 100 mg/kg adm, daily administered with MM- 121 at 5 mg/kg once a day every three days; compound A at 30 mg/kg/adm, daily administered with MM-121 at 30 mg kg once a day every three days; and compound A at 30 mg/kg/adm, daily administered with MM- 121 at 5 mg/kg once a day every three days.
- ⁇ /AC (delta T/delta C) x 100.
- a dose is considered therapeutically active when. ⁇ /AC is lower than 40% and very active when ⁇ /AC is lower than 10%. If ⁇ /AC is lower than 0, the dose is considered as highly active, and the percentage of regression is dated.
- Complete regression (CR): Complete regression is achieved when tumor volume 0 mm 3 (CR is considered when tumor volume cannot be recorded).
- TFS Tumor free is defined as the animals with undetectable tumors at the end of the study (>100 days post last treatment).
- mice treated with compound A were treated with 30 and 100 mg kg (-7.3 and -10.3 % at nadir, respectively).
- mice treated with MM- 121 were treated with 5 and 30 mg kg (-3.9 and -6.2 % at nadir, respectively) followed by a complete recovery.
- a dose dependant body weight loss was observed for mice treated with compound A at 30 and 100 mg/kg in combination with M -121 at 5 or 30 mg/kg when compared to mice treated with compound A or MM-121 administered alone at the same respective doses (- 8.4, -1 1.4, -12.4 and -12.9 % at nadir, respectively). No significant difference on body weight loss was observed for compound A at 100 mg/kg in combination with MM- 121 at 5 or 30 mg/kg compared to compound A at the same dose alone. Compound A and MM- 121 combination did not induce added toxicity.
- MM-121 was marginally active at 30 mg/kg with a AT/AC of 31 % and inactive at 5 mg/kg with a ⁇ /AC of 66%.
- Compound A was very active at 30 mg/kg with a AT/AC of 5% (p ⁇ 0.05 vs control) and highly active at 100 mg/kg with a AT/AC of -4% (p ⁇ 0.001 ) and tumor regression of 15%.
- Compound A at 100 mg/kg in combination with MM-121 at 30 mg/kg was highly active with a AT/AC of -20% (p ⁇ 0.001 vs control) and tumor regression of 39%.
- This combination was more active than the best single agent (compound A at 100 mg/kg, p ⁇ 0.01) indicating a therapeutic synergy in these combination conditions.
- the combination of compound A at 100 mg/kg with MM-121 at 5mg/kg was highly active with a AT/AC of - 17% (p ⁇ 0.001 vs control) and tumor regression of 41 %.
- the combination of compound A at 30 mg/kg with MM-121 at 30 mg/kg was highly active with a AT/AC of -7% (p ⁇ 0.001 vs control) and tumor regression of 15%.
- This combination was more active than the best single agent (compound A at 30 mg/kg, p ⁇ 0.01 ) indicating a therapeutic synergy in these combination conditions.
- the combination of compound A at 30 mg/kg with MM- 121 at 5 mg/kg was active with a AT/AC of 16% (p ⁇ 0.01 vs control).
- MM-121 single agent exhibited a marginal activity at 30 mg/kg, and compound A single agent exhibited high activity at 30 and 100 mg/kg.
- a therapeutic synergy was observed with the combination between compound A at 100 mg/kg and MM-121 at 30 mg/kg and with the combination between compound A at 30 mg/kg and MM-121 at 30 mg kg compared to best single agent for both conditions (compound A at 100 mg/kg, p ⁇ 0.01 and compound A at 30mg kg, p ⁇ 0.01 , respectively).
- mice bearing measurable lung A549 tumors implanted SC have been left untreated or treated either by MM 121 as a single agent (at dose of 5 mg/kg or 30 mg/kg, one administration) or by compound A as a single agent (at dose of 30mg/kg or lOOmg/kg, daily administrations) or by a combination of both (at 30 mg/kg for MM 121 and 100 mg/kg for compound A).
- the HER3 expression level in xenografted tumors has been followed for up to 96 hours.
- a method for treating cancer in a patient comprising co-administering a therapeutically effective amount of a compound of formula I and a therapeutically effective amount of a HER2 and/or HER3 inhibitor to said patient.
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula I and a pharmaceutically acceptable carrier, excipient, or diluent in combination with one or more inhibitors of HER3, HER2, MSPR, Axl, MAP3K (ERK, JNK, and p38 MAPK), MEKK kinases/kinase receptors, INSR, IGF-IR, and FGFR2, where the compound of formula I is that wherein:
- R 51 is hydrogen or alkyl
- R 52 is hydrogen or halo
- R 50 , R 53 , and R 54 are independently hydrogen, alkyl, alkenyl, halo, haloalkyl, haloalkenyl, hydroxy, alkoxy, alkenyloxy, haloalkoxy, nitro, amino, alkylamino, dialkylamino, -N(R 55 )C(0)-C r C6-alkylene-N ⁇ R 55a )R 55b , alkylcarbonyl, alkenylcarbonyl, carboxy, alkoxycarbonyl, cyano, alkylthio, -S(0) 2 NR 55 R 55a , or alkylcarbonylamino, and R 55 and R 55b are indepedently hydrogen, alkyl, or alkenyl, and R 55a is hydrogen, alkyl, alkenyl, hydroxy, or alkoxy; or R 53 and R 54 together with the carbons to which they are attached form a 5- or 6-membered heteroaryl or 5- or 6-membere
- B is heteroaryl optionally substituted with one, two, or three R 3 ;
- R 3a is cyano, hydroxyamino, carboxy, alkoxycarbonyl, alkylamino, dialkylamino,
- alkylcarbonyl haloalkoxy, alkylsulfonyl, aminoalkyloxy, alkylaminoalkyloxy, or dialkylaminoalkyloxy; or
- R 7 is hydrogen, alkyl, or alkenyl
- R 7a and R 7b are independently hydrogen, alkyl, alkenyl, hydroxyalkyl, haloalkyl, alkoxy, alkoxyalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroarylalkyl, aryl, arylalkyl, or arylalkyloxy, and where the aryl, cycloalkyl, heterocycloalkyl and heteroaryl rings in R 7a and R 7b (either alone or as part of arylalkyl, cycloalkylalkyl, heterocycloalkylalkyl,
- R 8 is hydrogen, hydroxy, alkoxy, alkyl, alkenyl, haloalkyl, or haloalkoxy
- R 8a is hydrogen, alkyl, alkenyl, hydroxyalkyl, cyanoalkyl, alkoxyalkyl, alkylthioalkyl, heterocycloalkyl, heterocycloalkylalkyl, cycloalkyl, cycloalkylalkyl, heteroaryl, heteroarylalkyl, aryl, or arylalkyl, and where the aryl, cycloalkyl, heteroaryl, and heterocycloalkyl rings in R 8a (either alone or as part of arylalkyl, cycloalkylalkyl, heterocycloalkylalkyl and heteroarylalkyl) are independently optionally substituted with 1, 2, or 3 groups independently selected from alkyl, alkenyl, alkoxy, hal
- R 9 is hydrogen, hydroxy, alkoxy, alkyl, alkenyl, haloalkyl, or haloalkoxy
- R 3 ⁇ 4 is hydrogen, C2-C6-alkyl, alkenyl, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyl, heterocycloalkylalkyl, heteroaryl, heteroarylalkyl, aryl, or arylalkyl, and where the aryl, cycloalkyl, heteroaryl, and heterocycloalkyl rings in R 9a (either alone or as part of arylalkyl, cycloalkylalkyl, heterocycloalkylalkyl, and heteroarylalkyl) are independently optionally substituted with 1, 2, or 3 groups independently selected from alkyl, alkenyl, alkoxy, hydroxy, hydroxyalkyl, hal
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
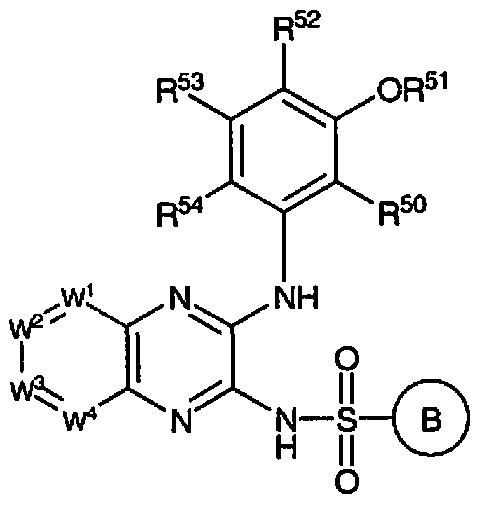













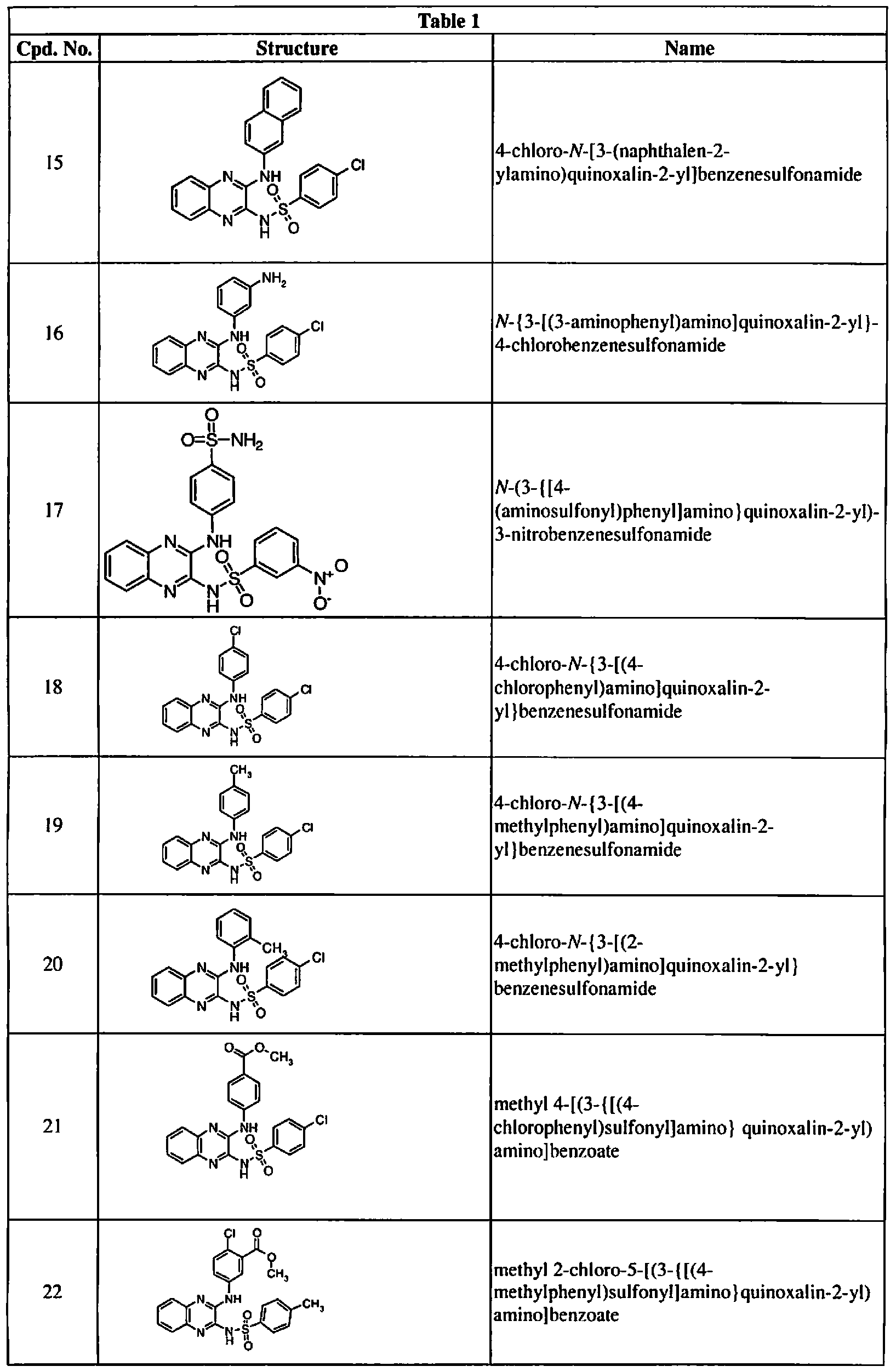












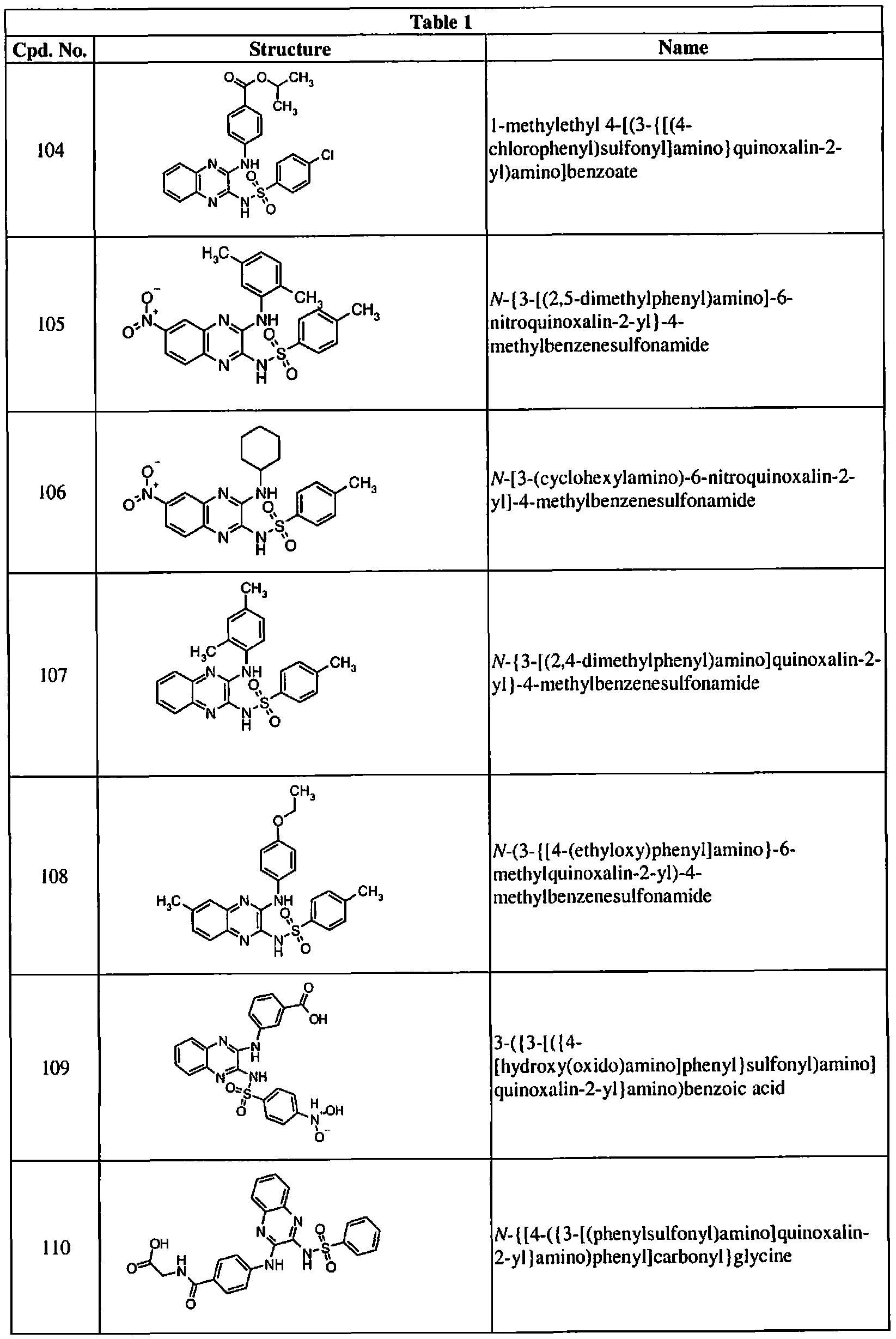
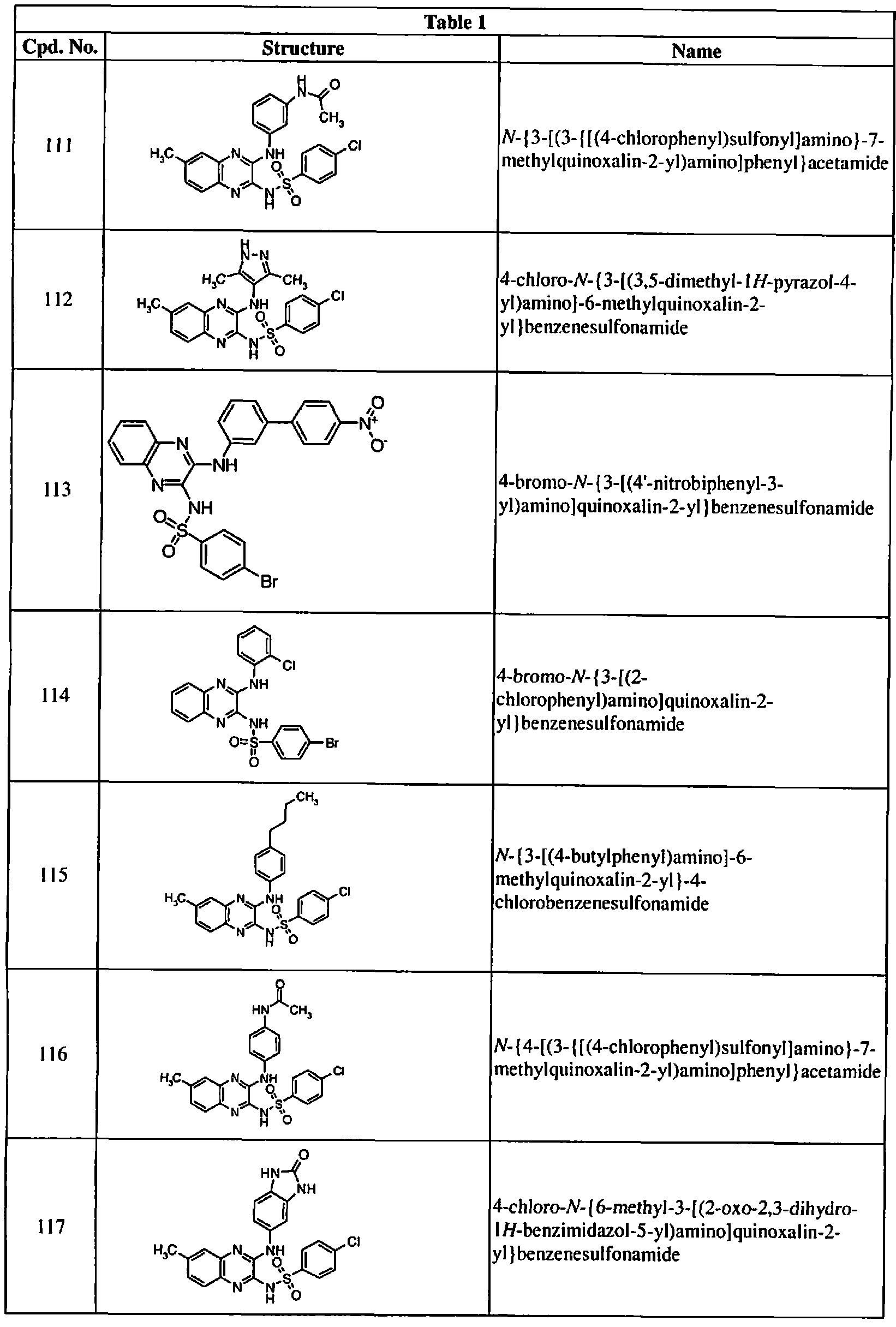



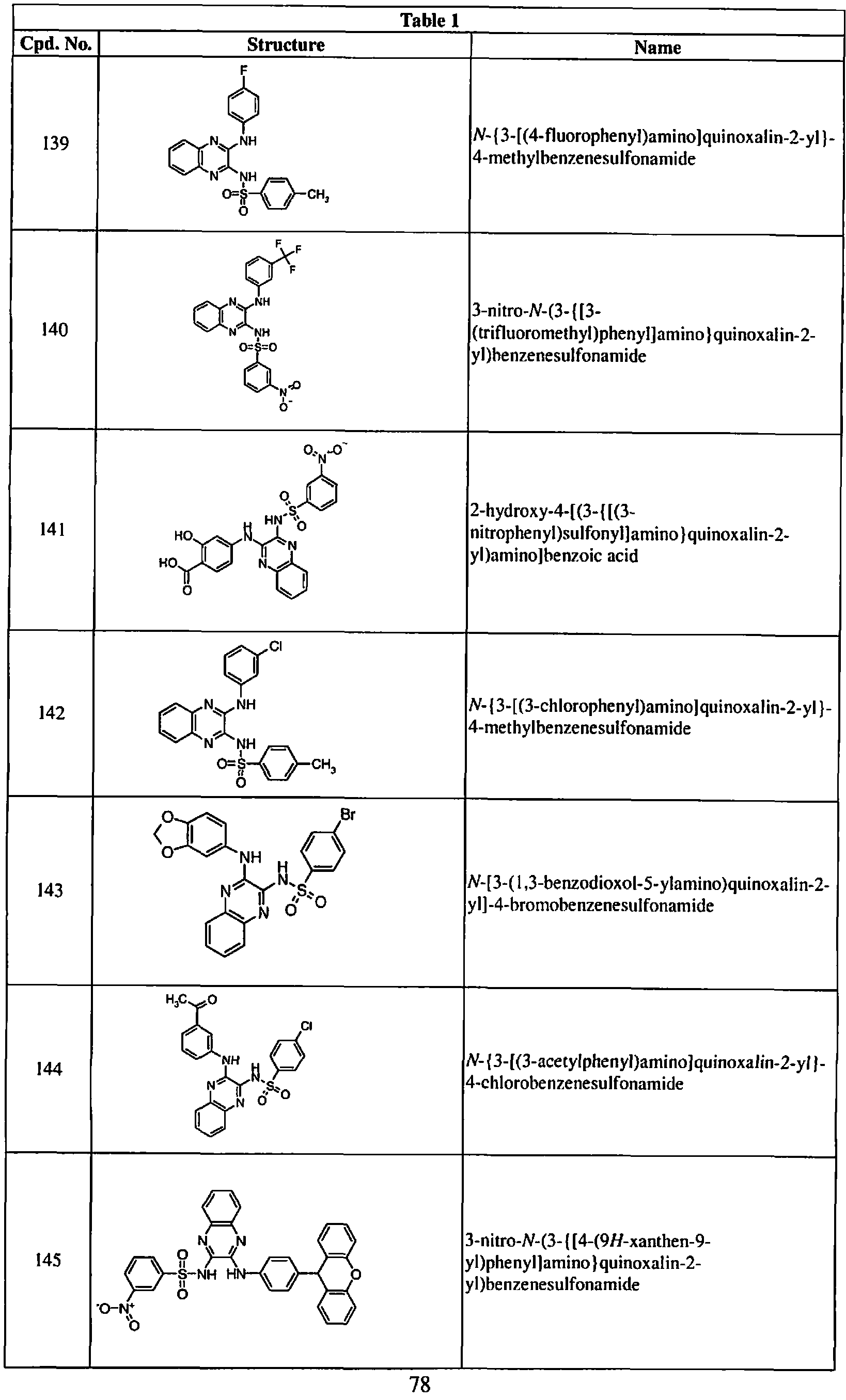







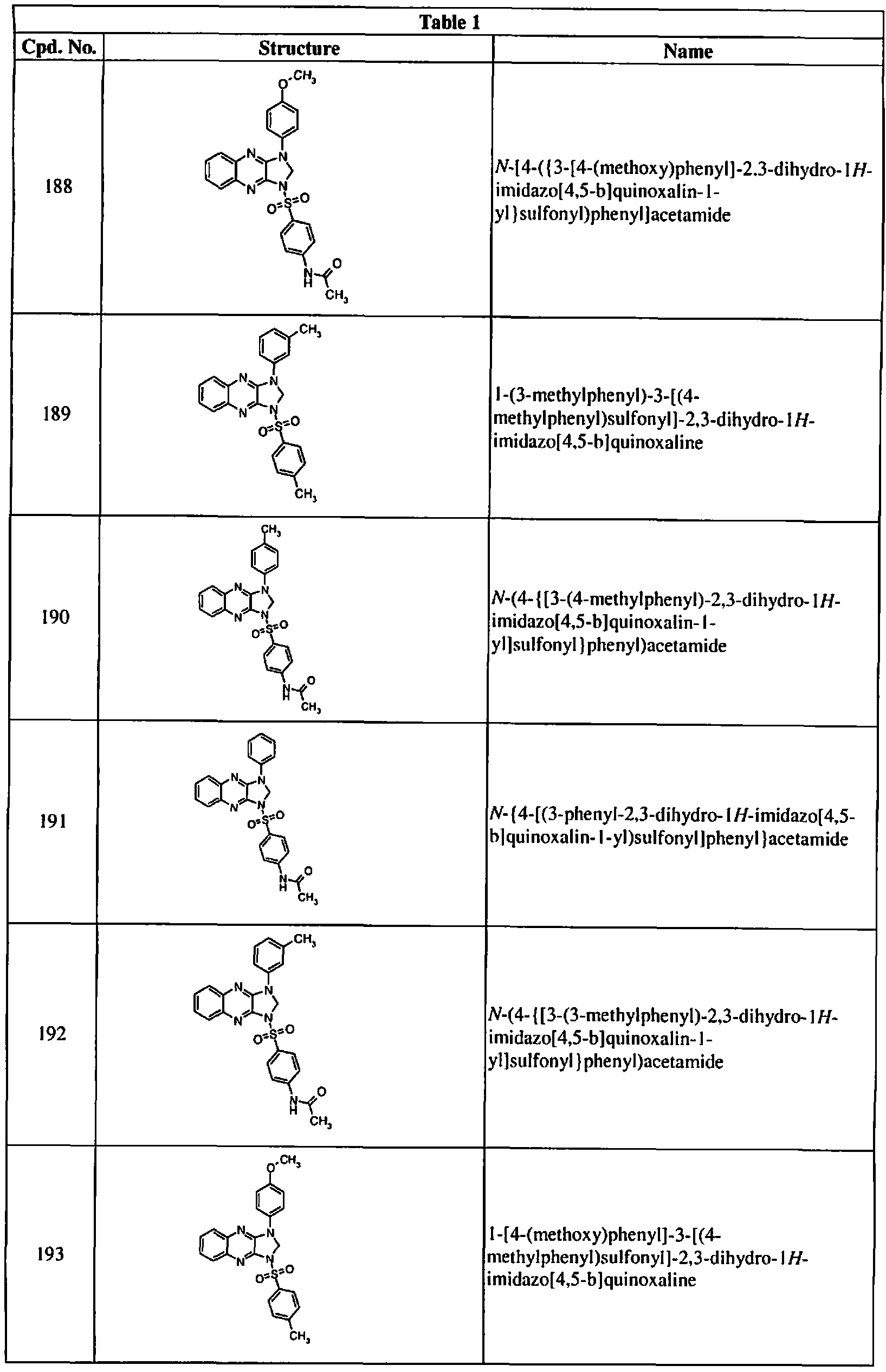





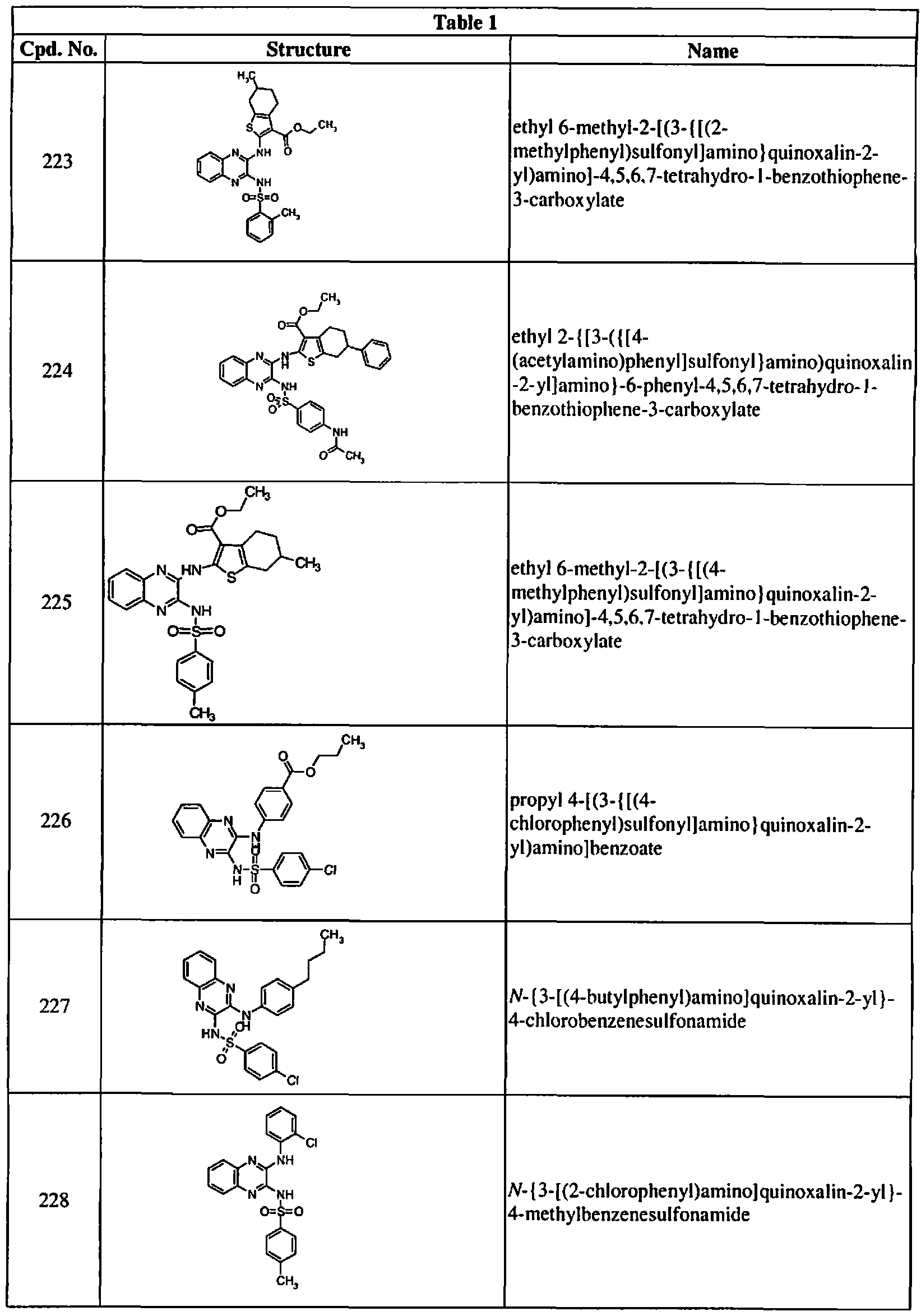











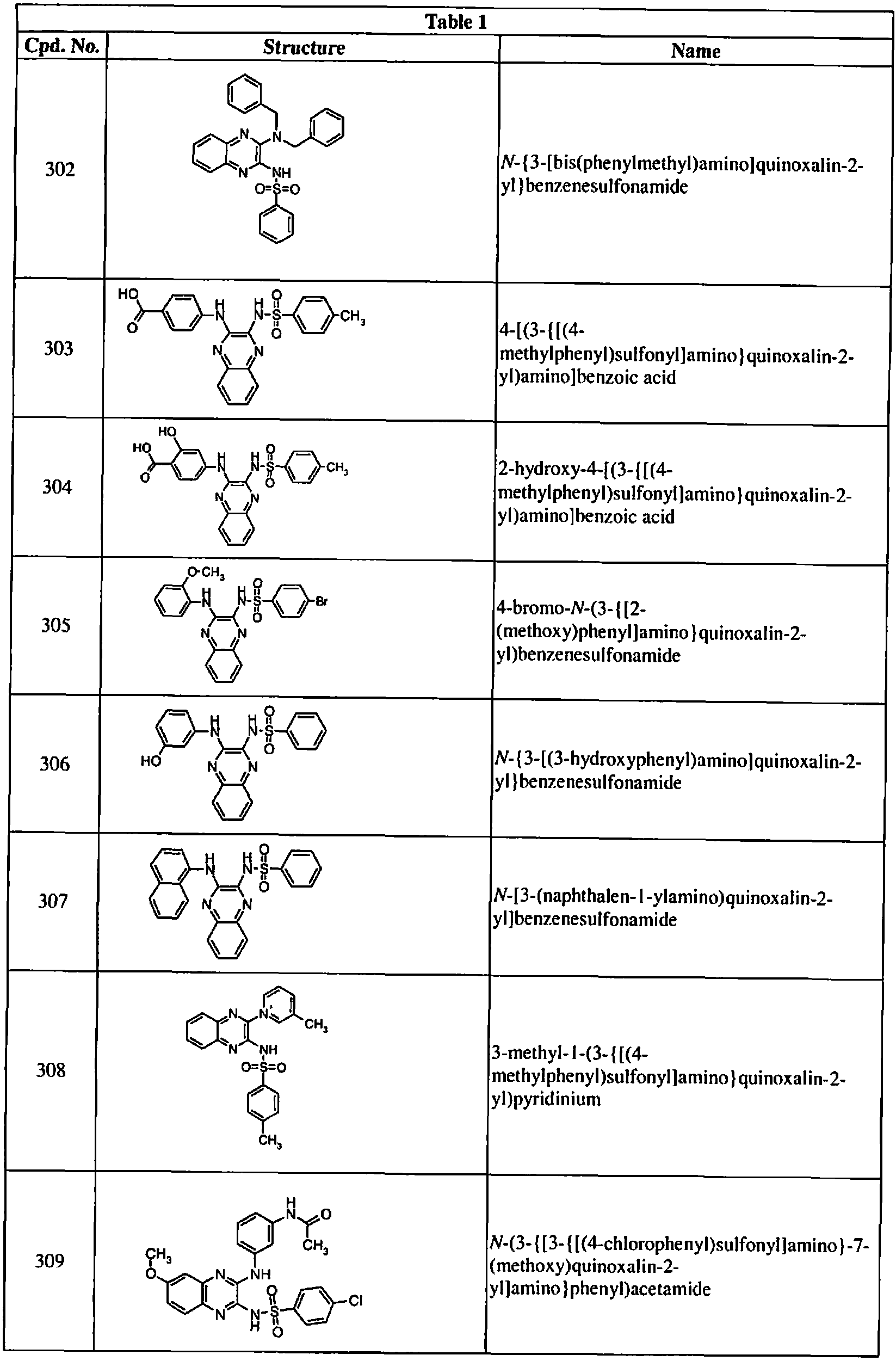


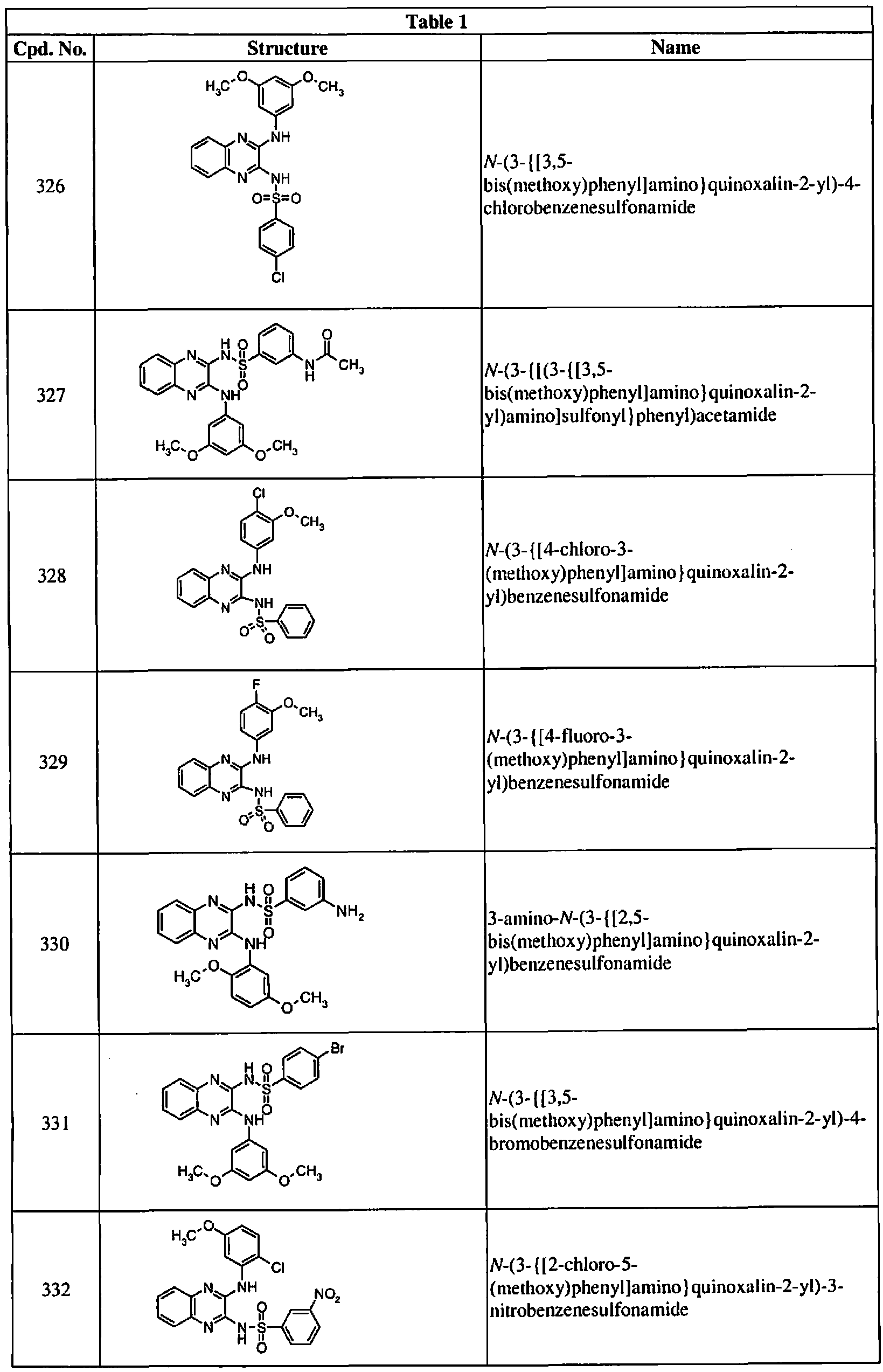












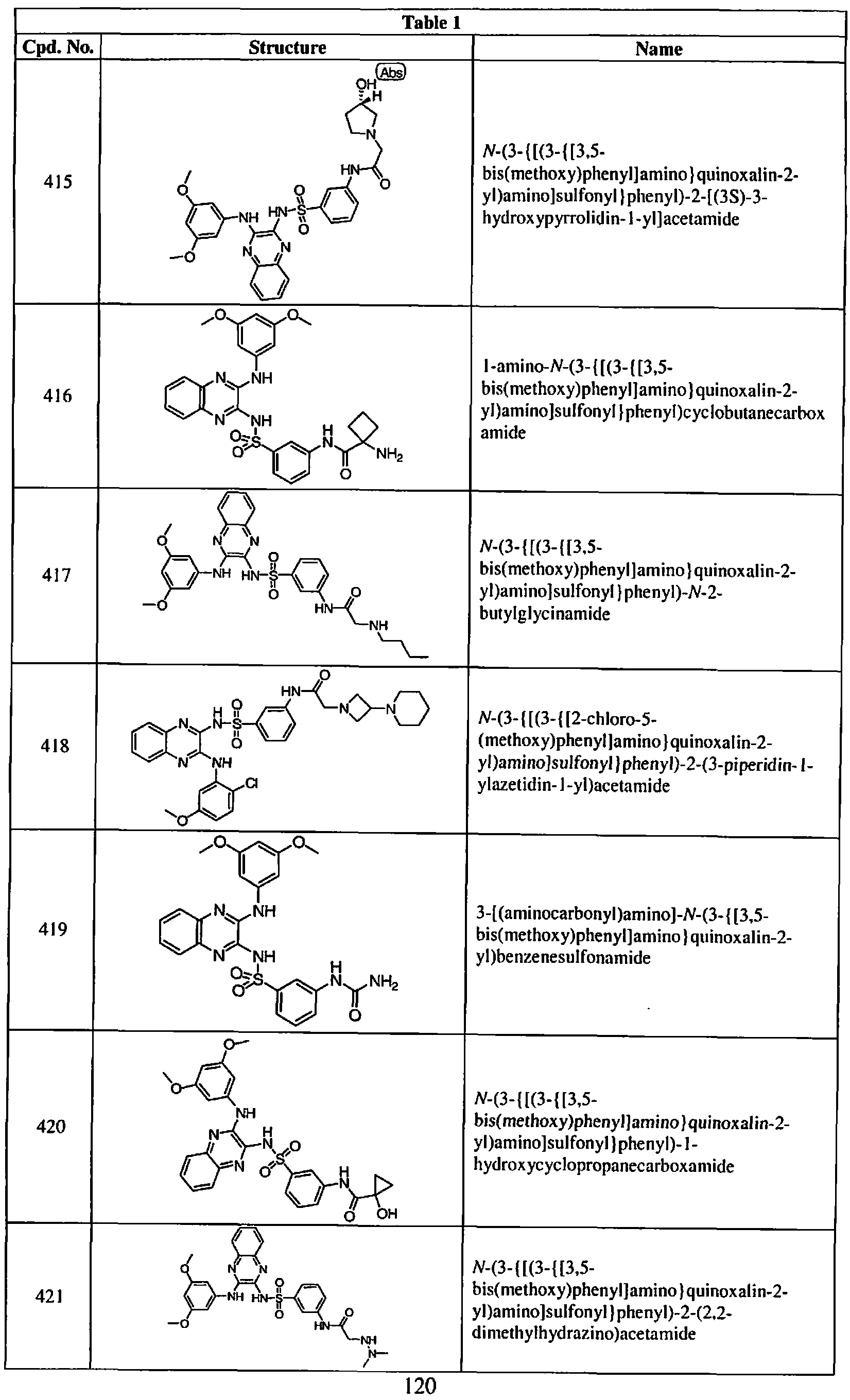


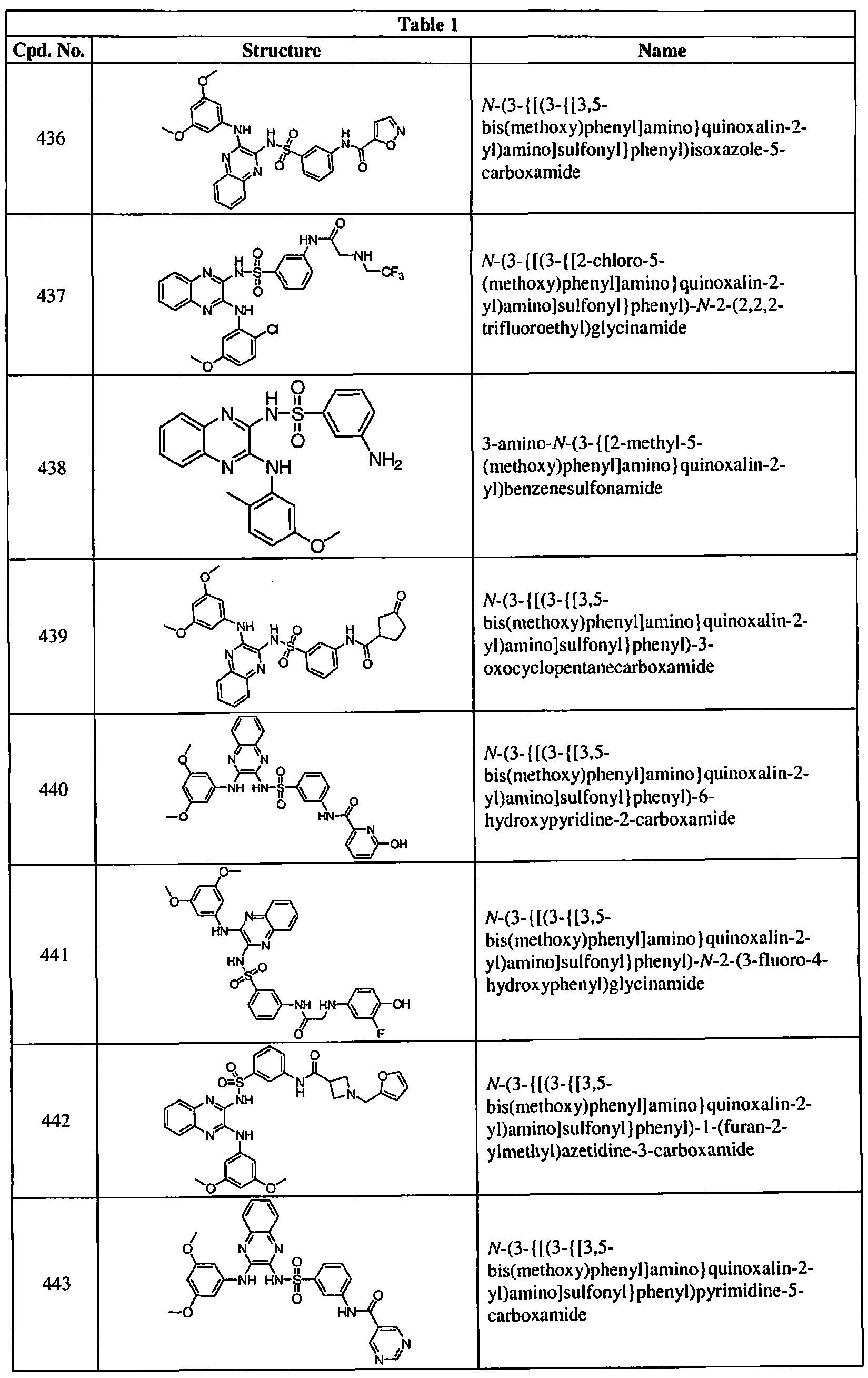




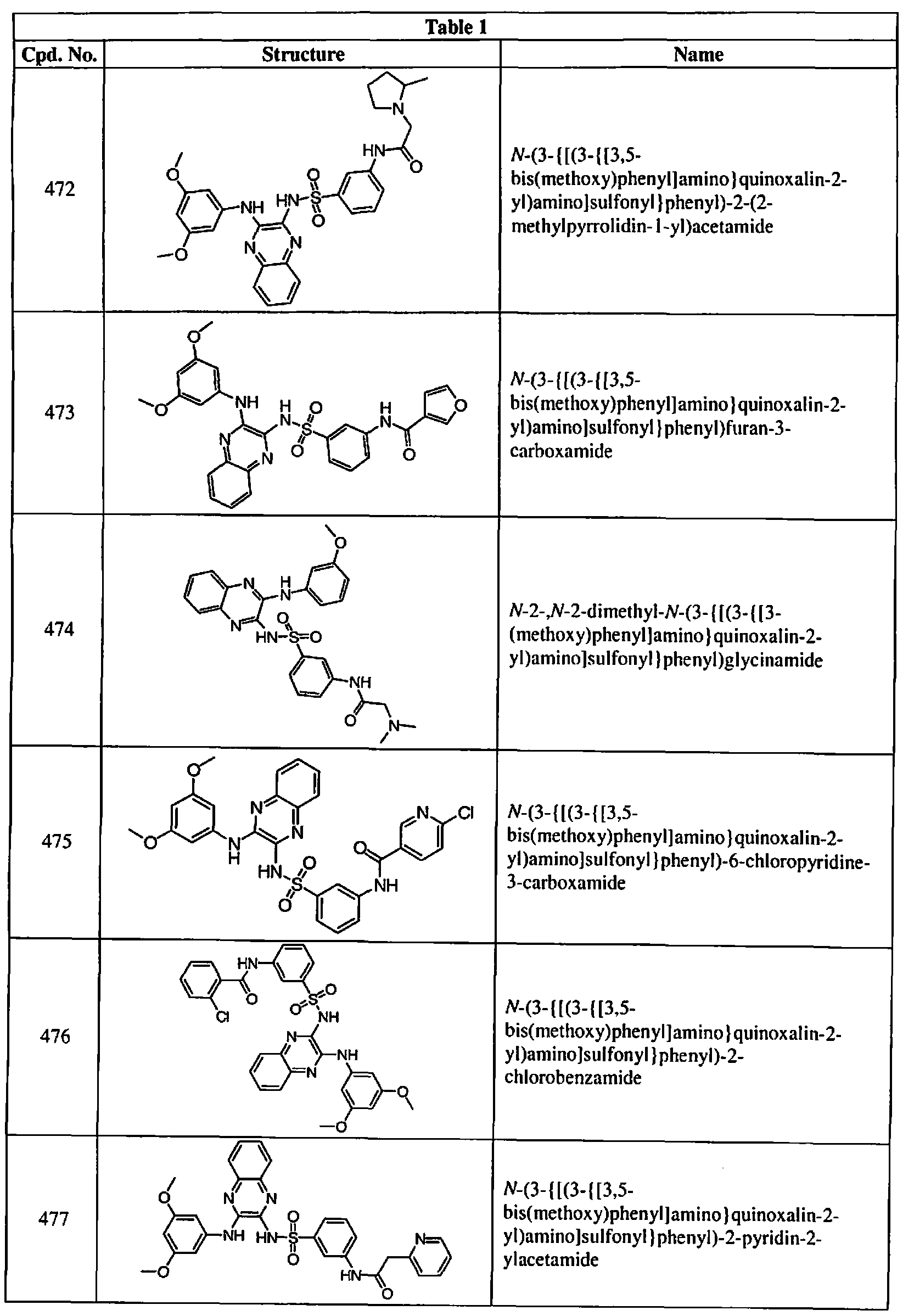















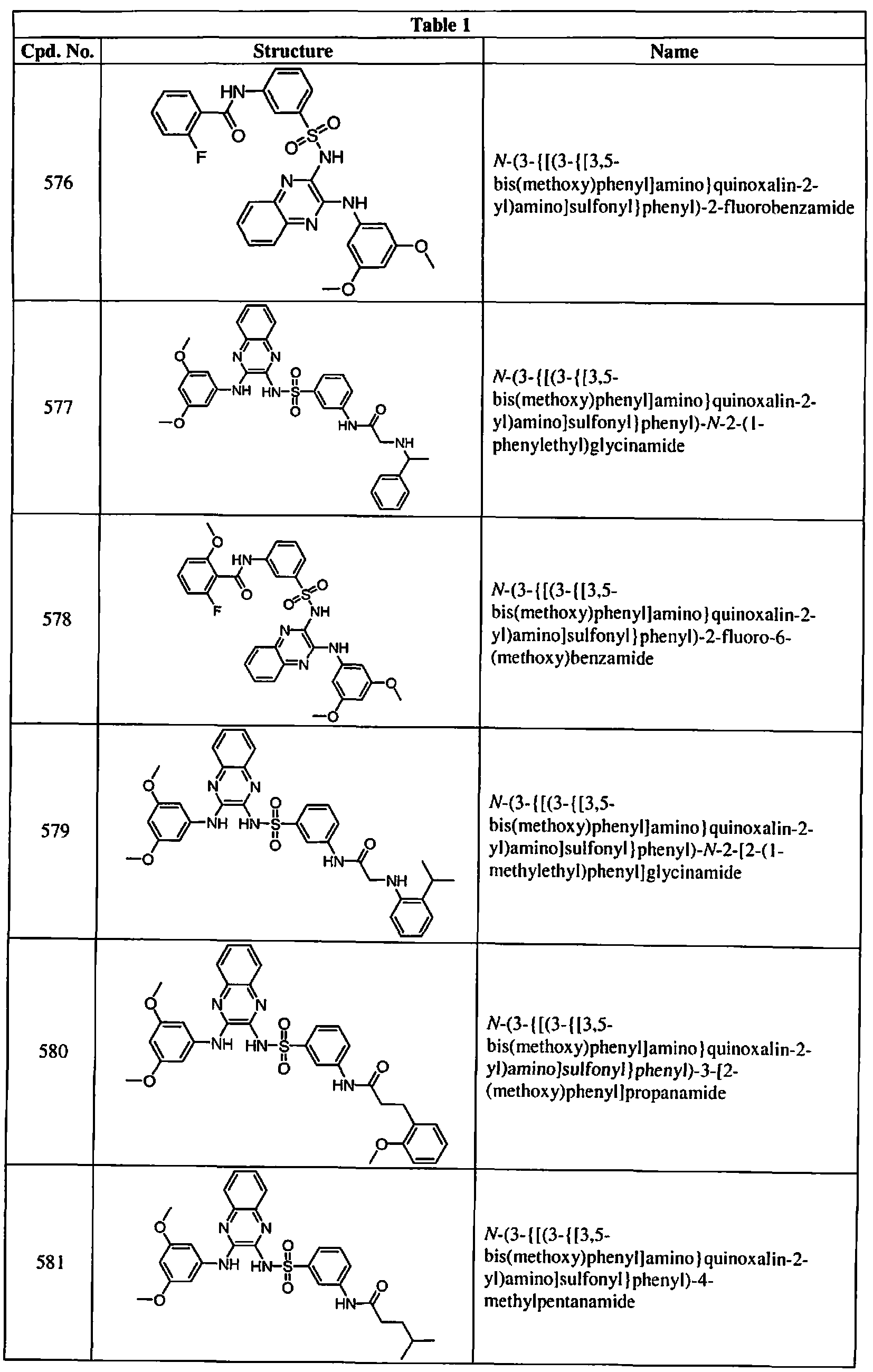
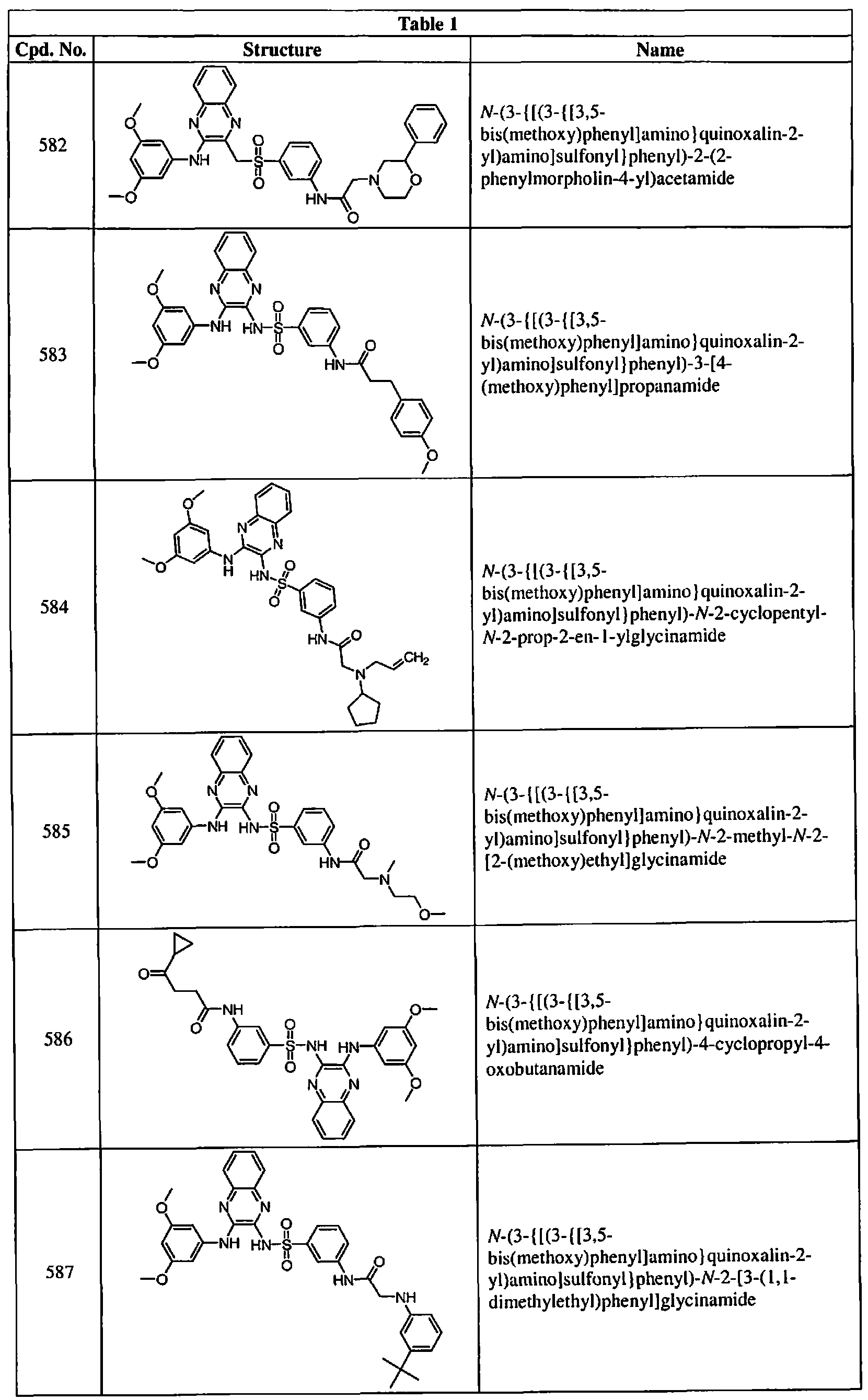





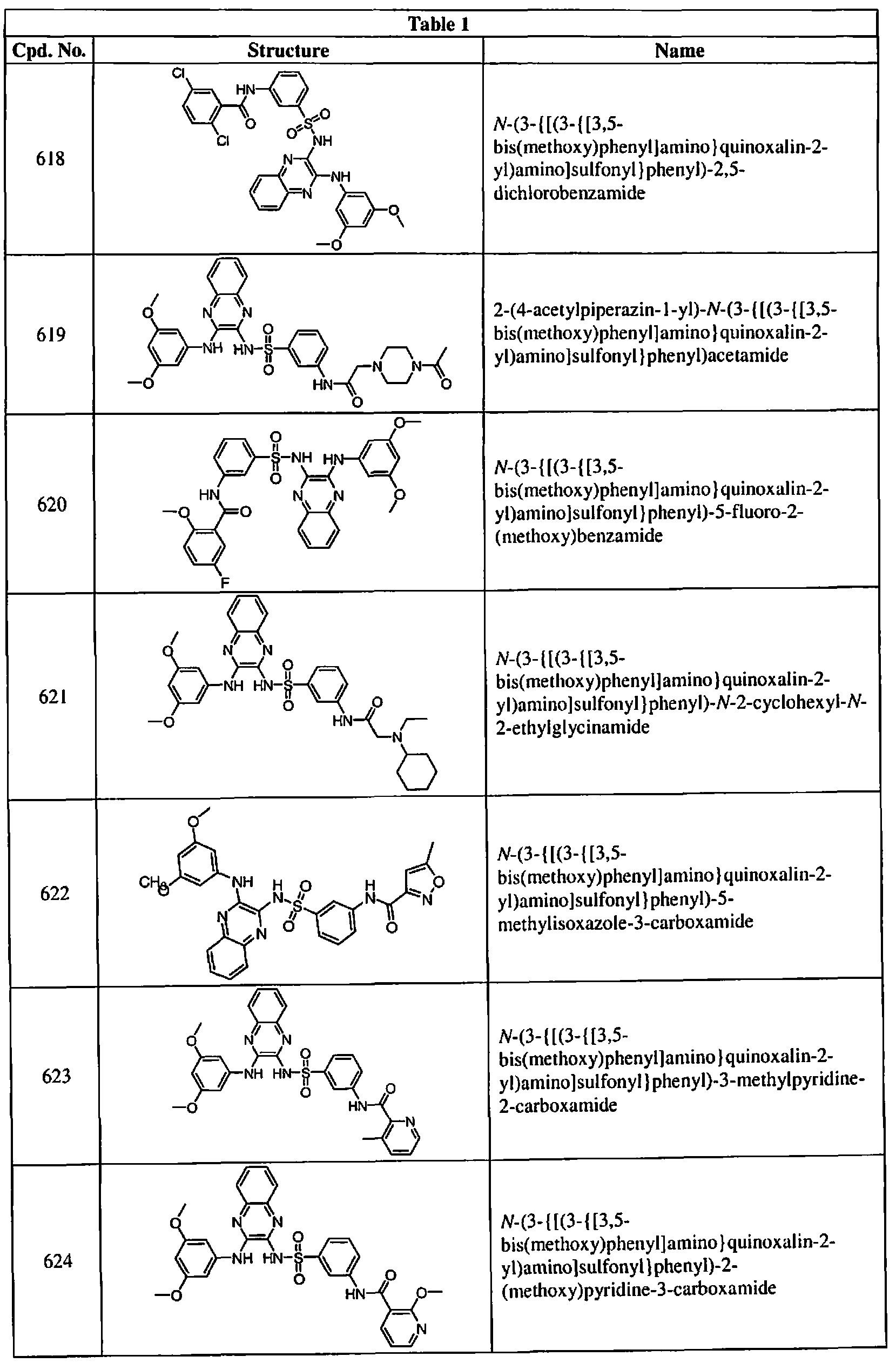












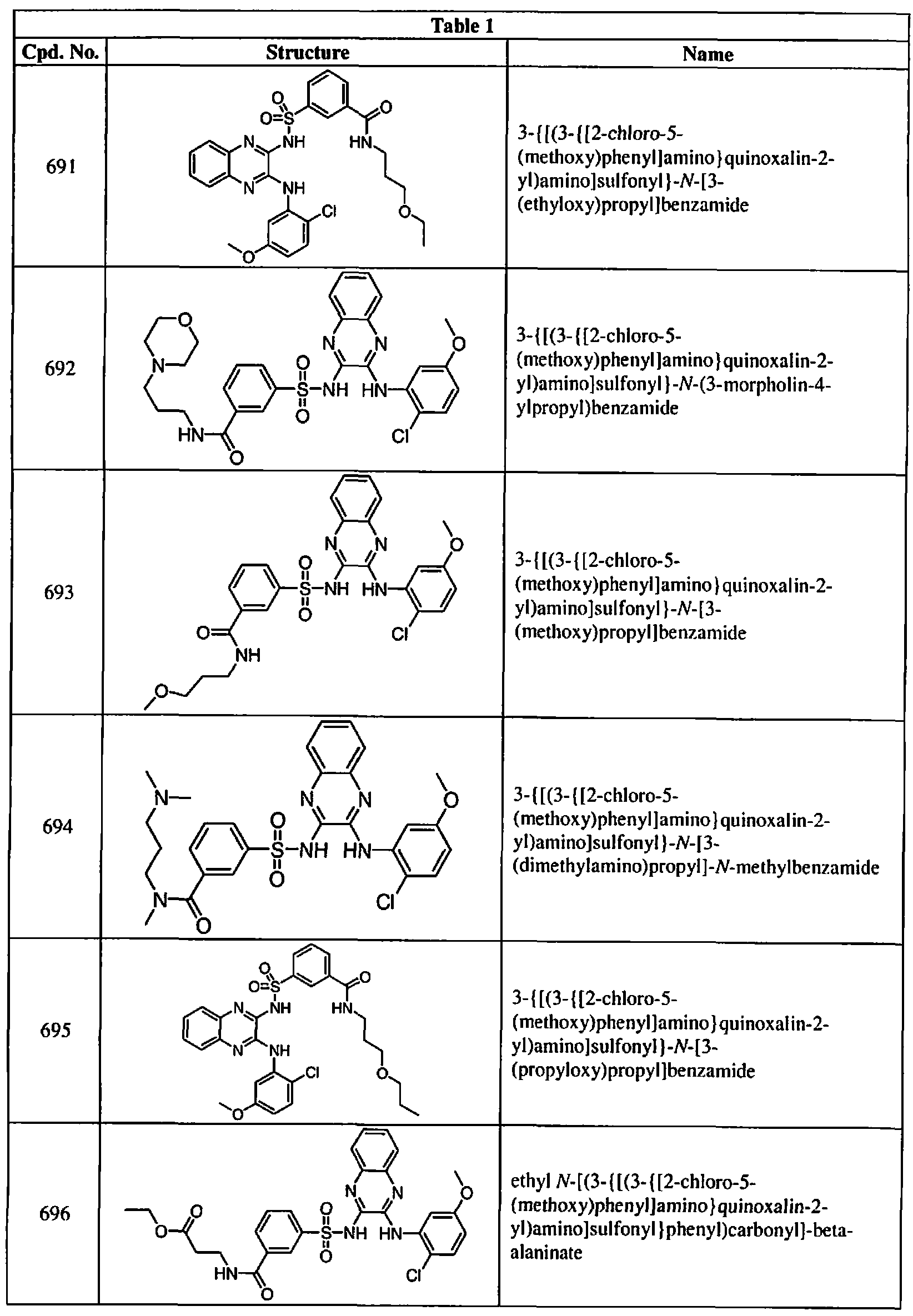












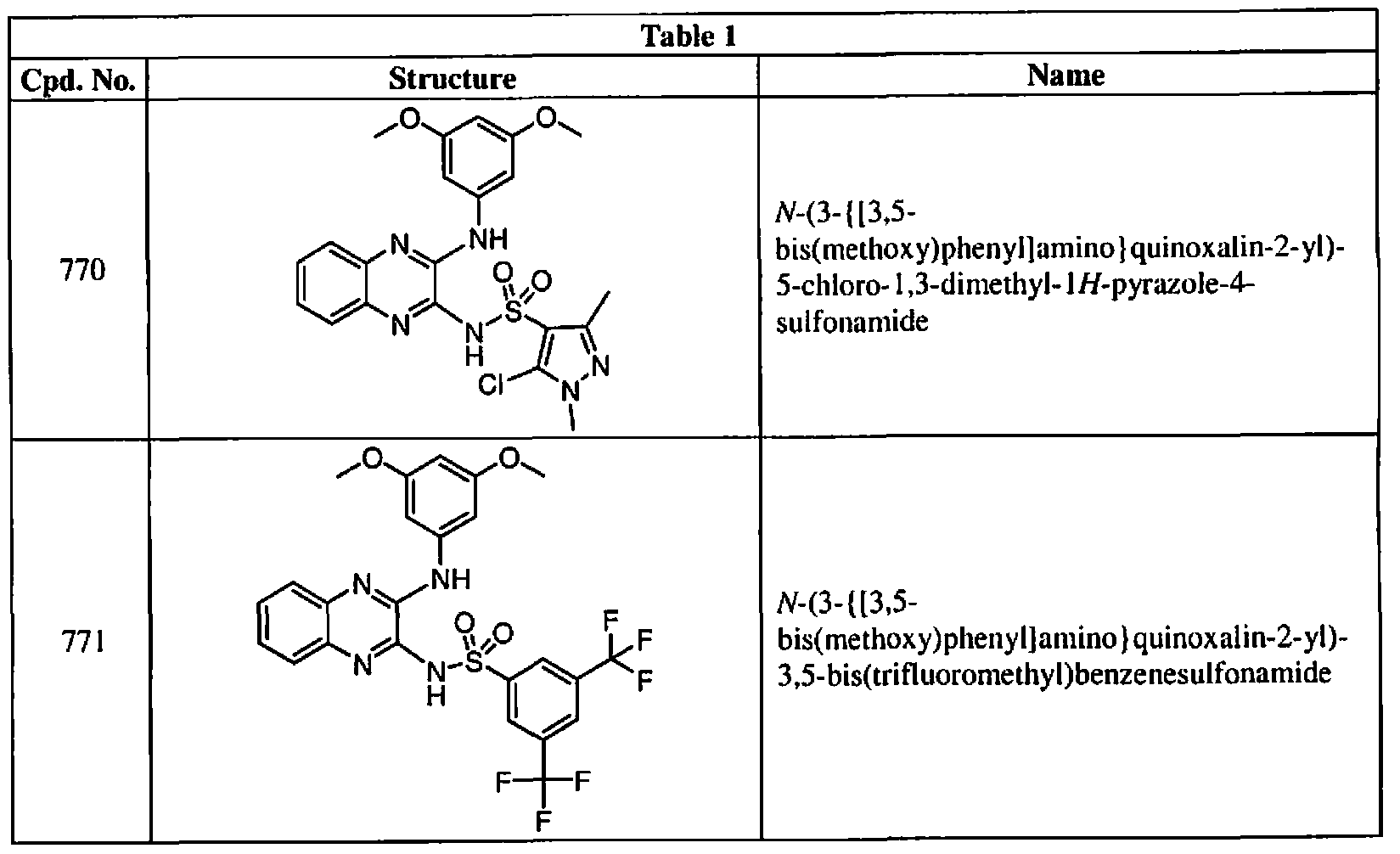


















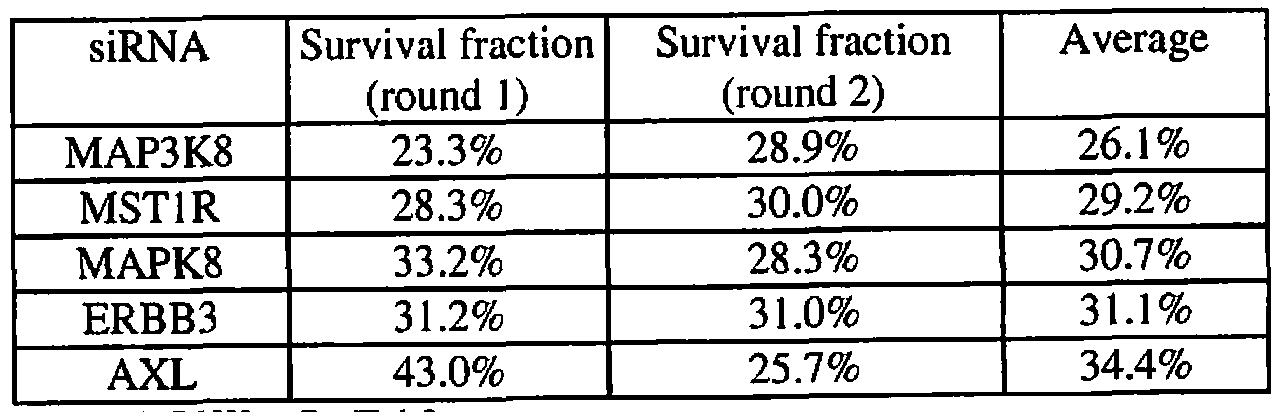


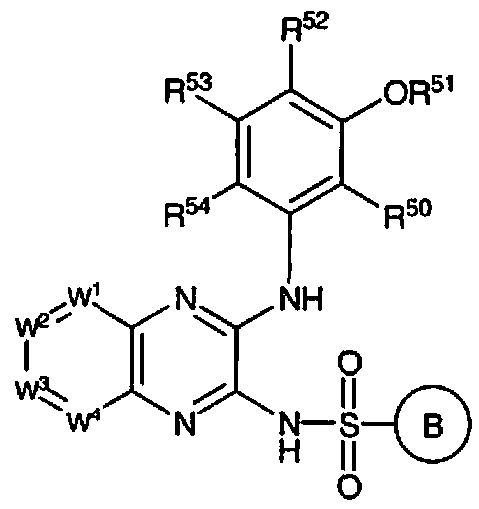





Claims
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR112013000651A BR112013000651A2 (en) | 2010-07-09 | 2011-07-08 | kinase inhibitor combinations for cancer treatment. |
| AU2011274510A AU2011274510A1 (en) | 2010-07-09 | 2011-07-08 | Combinations of kinase inhibitors for the treatment of cancer |
| US13/809,002 US9011863B2 (en) | 2010-07-09 | 2011-07-08 | Combinations of kinase inhibitors for the treatment of cancer |
| CA2803900A CA2803900A1 (en) | 2010-07-09 | 2011-07-08 | Combinations of kinase inhibitors for the treatment of cancer |
| JP2013518865A JP2013539453A (en) | 2010-07-09 | 2011-07-08 | Combination of kinase inhibitors for the treatment of cancer |
| EP11731617.4A EP2590654B1 (en) | 2010-07-09 | 2011-07-08 | Combinations of kinase inhibitors for the treatment of cancer |
| SG2013000088A SG186889A1 (en) | 2010-07-09 | 2011-07-08 | Combinations of kinase inhibitors for the treatment of cancer |
| EA201390085A EA201390085A1 (en) | 2010-07-09 | 2011-07-08 | COMPOSITIONS OF KINAZ INHIBITORS FOR CANCER TREATMENT |
| KR1020137003497A KR20130098301A (en) | 2010-07-09 | 2011-07-08 | Combinations of kinase inhibitors for the treatment of cancer |
| TNP2012000622A TN2012000622A1 (en) | 2010-07-09 | 2012-12-25 | Combinations of kinase inhibitors for the treatment of cancer |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US36307410P | 2010-07-09 | 2010-07-09 | |
| US61/363,074 | 2010-07-09 | ||
| US42192910P | 2010-12-10 | 2010-12-10 | |
| US61/421,929 | 2010-12-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012006552A1 true WO2012006552A1 (en) | 2012-01-12 |
Family
ID=44343802
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/043401 WO2012006552A1 (en) | 2010-07-09 | 2011-07-08 | Combinations of kinase inhibitors for the treatment of cancer |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US9011863B2 (en) |
| EP (1) | EP2590654B1 (en) |
| JP (1) | JP2013539453A (en) |
| KR (1) | KR20130098301A (en) |
| AR (1) | AR084469A1 (en) |
| AU (1) | AU2011274510A1 (en) |
| BR (1) | BR112013000651A2 (en) |
| CA (1) | CA2803900A1 (en) |
| EA (1) | EA201390085A1 (en) |
| SG (1) | SG186889A1 (en) |
| TN (1) | TN2012000622A1 (en) |
| TW (1) | TW201244719A (en) |
| UY (1) | UY33498A (en) |
| WO (1) | WO2012006552A1 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2841102A4 (en) * | 2012-04-23 | 2015-05-06 | Univ Ramot | Methods and compositions for treating cancer |
| WO2015073644A1 (en) | 2013-11-13 | 2015-05-21 | Novartis Ag | Mtor inhibitors for enhancing the immune response |
| WO2015090230A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Human mesothelin chimeric antigen receptors and uses thereof |
| WO2015090229A1 (en) | 2013-12-20 | 2015-06-25 | Novartis Ag | Regulatable chimeric antigen receptor |
| WO2015100459A3 (en) * | 2013-12-27 | 2015-08-20 | Merrimack Pharmaceuticals, Inc. | Biomarker profiles for predicting outcomes of cancer therapy with erbb3 inhibitors and/or chemotherapies |
| WO2015142675A2 (en) | 2014-03-15 | 2015-09-24 | Novartis Ag | Treatment of cancer using chimeric antigen receptor |
| WO2015157252A1 (en) | 2014-04-07 | 2015-10-15 | BROGDON, Jennifer | Treatment of cancer using anti-cd19 chimeric antigen receptor |
| WO2016014553A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Sortase synthesized chimeric antigen receptors |
| WO2016014530A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Combinations of low, immune enhancing. doses of mtor inhibitors and cars |
| WO2016025880A1 (en) | 2014-08-14 | 2016-02-18 | Novartis Ag | Treatment of cancer using gfr alpha-4 chimeric antigen receptor |
| WO2016044605A1 (en) | 2014-09-17 | 2016-03-24 | Beatty, Gregory | Targeting cytotoxic cells with chimeric receptors for adoptive immunotherapy |
| WO2016057705A1 (en) | 2014-10-08 | 2016-04-14 | Novartis Ag | Biomarkers predictive of therapeutic responsiveness to chimeric antigen receptor therapy and uses thereof |
| WO2016164580A1 (en) | 2015-04-07 | 2016-10-13 | Novartis Ag | Combination of chimeric antigen receptor therapy and amino pyrimidine derivatives |
| WO2016168595A1 (en) | 2015-04-17 | 2016-10-20 | Barrett David Maxwell | Methods for improving the efficacy and expansion of chimeric antigen receptor-expressing cells |
| WO2016172583A1 (en) | 2015-04-23 | 2016-10-27 | Novartis Ag | Treatment of cancer using chimeric antigen receptor and protein kinase a blocker |
| WO2018067992A1 (en) | 2016-10-07 | 2018-04-12 | Novartis Ag | Chimeric antigen receptors for the treatment of cancer |
| WO2018201056A1 (en) | 2017-04-28 | 2018-11-01 | Novartis Ag | Cells expressing a bcma-targeting chimeric antigen receptor, and combination therapy with a gamma secretase inhibitor |
| KR20190122764A (en) * | 2017-02-28 | 2019-10-30 | 다이호야쿠힌고교 가부시키가이샤 | Antitumor Effect Enhancers Using Pyrazolo [3,4-d] pyrimidine Compounds |
| WO2019210153A1 (en) | 2018-04-27 | 2019-10-31 | Novartis Ag | Car t cell therapies with enhanced efficacy |
| WO2019213282A1 (en) | 2018-05-01 | 2019-11-07 | Novartis Ag | Biomarkers for evaluating car-t cells to predict clinical outcome |
| CN110958877A (en) * | 2017-07-09 | 2020-04-03 | 江苏英科贝塔医药科技有限公司 | Fluorine-containing compound, use thereof and method for producing same |
| EP3660042A1 (en) | 2014-07-31 | 2020-06-03 | Novartis AG | Subset-optimized chimeric antigen receptor-containing t-cells |
| EP3712171A1 (en) | 2014-08-19 | 2020-09-23 | Novartis AG | Treatment of cancer using a cd123 chimeric antigen receptor |
| EP3722316A1 (en) | 2014-07-21 | 2020-10-14 | Novartis AG | Treatment of cancer using a cd33 chimeric antigen receptor |
| US11365252B2 (en) | 2016-07-20 | 2022-06-21 | University Of Utah Research Foundation | CD229 CAR T cells and methods of use thereof |
| CN115989030A (en) * | 2020-05-29 | 2023-04-18 | 康涅狄格大学 | Inhibitors of porcine reproductive and respiratory syndrome virus |
| RU2799006C2 (en) * | 2017-02-28 | 2023-06-30 | Тайхо Фармасьютикал Ко., Лтд. | ANTITUMOR EFFECT STRENGTHENING AGENT USING PYRAZOLO[3,4-d]PYRIMIDINE COMPOUND |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT2129396E (en) * | 2007-02-16 | 2013-11-18 | Merrimack Pharmaceuticals Inc | Antibodies against erbb3 and uses thereof |
| EP2544680B1 (en) | 2010-03-11 | 2015-01-14 | Merrimack Pharmaceuticals, Inc. | Use of erbb3 inhibitors in the treatment of triple negative breast cancer |
| AR082985A1 (en) | 2010-09-14 | 2013-01-23 | Exelixis Inc | PI3K-d INHIBITORS AND METHODS OF USE AND PREPARATION |
| EP2683741A2 (en) * | 2011-03-11 | 2014-01-15 | Merrimack Pharmaceuticals, Inc. | Use of inhibitors of egfr-family receptors in the treatment of hormone refractory breast cancers |
| MX2014005249A (en) | 2011-11-01 | 2015-03-05 | Exelixis Inc | N- (3- { [ (3- { [2-chloro-5- (methoxy) phenyl] amino} quinoxalin- 2 -yl) amino] sulfonyl} phe nyl) - 2 -methylalaninamide as phosphatidylinositol 3 - kinase inhibitor for the treatment of lymphoproliferative malignancies. |
| CN105985435B (en) * | 2015-01-30 | 2019-10-15 | 嘉和生物药业有限公司 | The mutant antibodies and its encoding gene of full source of people HER2 antibody and application |
| MA45420A (en) * | 2015-04-17 | 2019-05-01 | Merrimack Pharmaceuticals Inc | COMBINED TREATMENTS WITH SERIBANTUMAB |
| US10184006B2 (en) | 2015-06-04 | 2019-01-22 | Merrimack Pharmaceuticals, Inc. | Biomarkers for predicting outcomes of cancer therapy with ErbB3 inhibitors |
| JP2022500396A (en) * | 2018-09-18 | 2022-01-04 | メタクリン,インク. | Farnesoid X receptor agonist for the treatment of disease |
Citations (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US30985A (en) | 1860-12-18 | Thomas l | ||
| US4107288A (en) | 1974-09-18 | 1978-08-15 | Pharmaceutical Society Of Victoria | Injectable compositions, nanoparticles useful therein, and process of manufacturing same |
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| WO1987000195A1 (en) | 1985-06-28 | 1987-01-15 | Celltech Limited | Animal cell culture |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| WO1990003430A1 (en) | 1988-09-23 | 1990-04-05 | Cetus Corporation | Cell culture medium for enhanced cell growth, culture longevity and product expression |
| US4920016A (en) | 1986-12-24 | 1990-04-24 | Linear Technology, Inc. | Liposomes with enhanced circulation time |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| US4933294A (en) | 1984-01-30 | 1990-06-12 | Icrf Patents Limited | Method of detecting truncated epidermal growth factor receptors |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| WO1991005264A1 (en) | 1989-09-29 | 1991-04-18 | Oncogenetics Partners | Detection and quantification of neu related proteins in the biological fluids of humans |
| US5019369A (en) | 1984-10-22 | 1991-05-28 | Vestar, Inc. | Method of targeting tumors in humans |
| WO1991017271A1 (en) | 1990-05-01 | 1991-11-14 | Affymax Technologies N.V. | Recombinant library screening methods |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| US5139941A (en) | 1985-10-31 | 1992-08-18 | University Of Florida Research Foundation, Inc. | AAV transduction vectors |
| US5145684A (en) | 1991-01-25 | 1992-09-08 | Sterling Drug Inc. | Surface modified drug nanoparticles |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| US5252479A (en) | 1991-11-08 | 1993-10-12 | Research Corporation Technologies, Inc. | Safe vector for gene therapy |
| WO1993021319A1 (en) | 1992-04-08 | 1993-10-28 | Cetus Oncology Corporation | HUMANIZED C-erbB-2 SPECIFIC ANTIBODIES |
| US5260203A (en) | 1986-09-02 | 1993-11-09 | Enzon, Inc. | Single polypeptide chain binding molecules |
| WO1993024641A2 (en) | 1992-06-02 | 1993-12-09 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Adeno-associated virus with inverted terminal repeat sequences as promoter |
| WO1994013788A1 (en) | 1992-12-04 | 1994-06-23 | University Of Pittsburgh | Recombinant viral vector system |
| US5401638A (en) | 1986-06-04 | 1995-03-28 | Oncogene Science, Inc. | Detection and quantification of neu related proteins in the biological fluids of humans |
| US5585089A (en) | 1988-12-28 | 1996-12-17 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| WO1997035885A1 (en) | 1996-03-27 | 1997-10-02 | Genentech, Inc. | ErbB3 ANTIBODIES |
| US5677171A (en) | 1988-01-12 | 1997-10-14 | Genentech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| WO1998045479A1 (en) | 1997-04-04 | 1998-10-15 | Albany Medical College | Method for assessing prostate cancer |
| WO1998052976A1 (en) | 1997-05-21 | 1998-11-26 | Biovation Limited | Method for the production of non-immunogenic proteins |
| US5856456A (en) | 1992-11-20 | 1999-01-05 | Enzon, Inc. | Linker for linked fusion polypeptides |
| US5859205A (en) | 1989-12-21 | 1999-01-12 | Celltech Limited | Humanised antibodies |
| US6054561A (en) | 1984-02-08 | 2000-04-25 | Chiron Corporation | Antigen-binding sites of antibody molecules specific for cancer antigens |
| US20020086356A1 (en) | 2000-03-30 | 2002-07-04 | Whitehead Institute For Biomedical Research | RNA sequence-specific mediators of RNA interference |
| EP1414494A1 (en) | 2001-08-09 | 2004-05-06 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Inhibitors of her3 activity |
| WO2007044729A2 (en) | 2005-10-07 | 2007-04-19 | Exelixis, Inc. | N- (3-amino-quinoxalin-2-yl) -sulfonamide derivatives and their use as phosphatidylinositol 3-kinase inhibitors |
| WO2008100624A2 (en) | 2007-02-16 | 2008-08-21 | Merrimack Pharmaceuticals, Inc. | Antibodies against erbb3 and uses thereof |
| WO2008127594A2 (en) | 2007-04-11 | 2008-10-23 | Exelixis, Inc. | Combination therapies comprising quinoxaline inhibitors of pi3k-alpha for use in the treatment of cancer |
| US7449184B2 (en) | 2005-01-21 | 2008-11-11 | Genentech, Inc. | Fixed dosing of HER antibodies |
| US20100047829A1 (en) | 2006-11-28 | 2010-02-25 | U3 Pharma Gmbh | Activated her3 as a marker for predicting therapeutic efficacy |
| US7705130B2 (en) | 2005-12-30 | 2010-04-27 | U3 Pharma Gmbh | Antibodies directed to HER-3 and uses thereof |
-
2011
- 2011-07-08 JP JP2013518865A patent/JP2013539453A/en not_active Ceased
- 2011-07-08 AR ARP110102469A patent/AR084469A1/en unknown
- 2011-07-08 TW TW100124282A patent/TW201244719A/en unknown
- 2011-07-08 CA CA2803900A patent/CA2803900A1/en not_active Abandoned
- 2011-07-08 BR BR112013000651A patent/BR112013000651A2/en not_active Application Discontinuation
- 2011-07-08 SG SG2013000088A patent/SG186889A1/en unknown
- 2011-07-08 EA EA201390085A patent/EA201390085A1/en unknown
- 2011-07-08 US US13/809,002 patent/US9011863B2/en active Active
- 2011-07-08 WO PCT/US2011/043401 patent/WO2012006552A1/en active Application Filing
- 2011-07-08 KR KR1020137003497A patent/KR20130098301A/en not_active Application Discontinuation
- 2011-07-08 AU AU2011274510A patent/AU2011274510A1/en not_active Abandoned
- 2011-07-08 EP EP11731617.4A patent/EP2590654B1/en active Active
- 2011-07-08 UY UY0001033498A patent/UY33498A/en not_active Application Discontinuation
-
2012
- 2012-12-25 TN TNP2012000622A patent/TN2012000622A1/en unknown
Patent Citations (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US30985A (en) | 1860-12-18 | Thomas l | ||
| US4107288A (en) | 1974-09-18 | 1978-08-15 | Pharmaceutical Society Of Victoria | Injectable compositions, nanoparticles useful therein, and process of manufacturing same |
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4933294A (en) | 1984-01-30 | 1990-06-12 | Icrf Patents Limited | Method of detecting truncated epidermal growth factor receptors |
| US6054561A (en) | 1984-02-08 | 2000-04-25 | Chiron Corporation | Antigen-binding sites of antibody molecules specific for cancer antigens |
| US5019369A (en) | 1984-10-22 | 1991-05-28 | Vestar, Inc. | Method of targeting tumors in humans |
| WO1987000195A1 (en) | 1985-06-28 | 1987-01-15 | Celltech Limited | Animal cell culture |
| US5139941A (en) | 1985-10-31 | 1992-08-18 | University Of Florida Research Foundation, Inc. | AAV transduction vectors |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| US5401638A (en) | 1986-06-04 | 1995-03-28 | Oncogene Science, Inc. | Detection and quantification of neu related proteins in the biological fluids of humans |
| US5260203A (en) | 1986-09-02 | 1993-11-09 | Enzon, Inc. | Single polypeptide chain binding molecules |
| US5455030A (en) | 1986-09-02 | 1995-10-03 | Enzon Labs, Inc. | Immunotheraphy using single chain polypeptide binding molecules |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US4920016A (en) | 1986-12-24 | 1990-04-24 | Linear Technology, Inc. | Liposomes with enhanced circulation time |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| US5772997A (en) | 1988-01-12 | 1998-06-30 | Genentech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| US5677171A (en) | 1988-01-12 | 1997-10-14 | Genentech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| WO1990003430A1 (en) | 1988-09-23 | 1990-04-05 | Cetus Corporation | Cell culture medium for enhanced cell growth, culture longevity and product expression |
| US5693761A (en) | 1988-12-28 | 1997-12-02 | Protein Design Labs, Inc. | Polynucleotides encoding improved humanized immunoglobulins |
| US5693762A (en) | 1988-12-28 | 1997-12-02 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5585089A (en) | 1988-12-28 | 1996-12-17 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| WO1991005264A1 (en) | 1989-09-29 | 1991-04-18 | Oncogenetics Partners | Detection and quantification of neu related proteins in the biological fluids of humans |
| US5859205A (en) | 1989-12-21 | 1999-01-12 | Celltech Limited | Humanised antibodies |
| WO1991017271A1 (en) | 1990-05-01 | 1991-11-14 | Affymax Technologies N.V. | Recombinant library screening methods |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| US5145684A (en) | 1991-01-25 | 1992-09-08 | Sterling Drug Inc. | Surface modified drug nanoparticles |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| US5252479A (en) | 1991-11-08 | 1993-10-12 | Research Corporation Technologies, Inc. | Safe vector for gene therapy |
| WO1993021319A1 (en) | 1992-04-08 | 1993-10-28 | Cetus Oncology Corporation | HUMANIZED C-erbB-2 SPECIFIC ANTIBODIES |
| WO1993024641A2 (en) | 1992-06-02 | 1993-12-09 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Adeno-associated virus with inverted terminal repeat sequences as promoter |
| US5856456A (en) | 1992-11-20 | 1999-01-05 | Enzon, Inc. | Linker for linked fusion polypeptides |
| WO1994013788A1 (en) | 1992-12-04 | 1994-06-23 | University Of Pittsburgh | Recombinant viral vector system |
| WO1997035885A1 (en) | 1996-03-27 | 1997-10-02 | Genentech, Inc. | ErbB3 ANTIBODIES |
| WO1998045479A1 (en) | 1997-04-04 | 1998-10-15 | Albany Medical College | Method for assessing prostate cancer |
| WO1998052976A1 (en) | 1997-05-21 | 1998-11-26 | Biovation Limited | Method for the production of non-immunogenic proteins |
| US20020086356A1 (en) | 2000-03-30 | 2002-07-04 | Whitehead Institute For Biomedical Research | RNA sequence-specific mediators of RNA interference |
| EP1414494A1 (en) | 2001-08-09 | 2004-05-06 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Inhibitors of her3 activity |
| US7449184B2 (en) | 2005-01-21 | 2008-11-11 | Genentech, Inc. | Fixed dosing of HER antibodies |
| WO2007044729A2 (en) | 2005-10-07 | 2007-04-19 | Exelixis, Inc. | N- (3-amino-quinoxalin-2-yl) -sulfonamide derivatives and their use as phosphatidylinositol 3-kinase inhibitors |
| US7705130B2 (en) | 2005-12-30 | 2010-04-27 | U3 Pharma Gmbh | Antibodies directed to HER-3 and uses thereof |
| US20100047829A1 (en) | 2006-11-28 | 2010-02-25 | U3 Pharma Gmbh | Activated her3 as a marker for predicting therapeutic efficacy |
| WO2008100624A2 (en) | 2007-02-16 | 2008-08-21 | Merrimack Pharmaceuticals, Inc. | Antibodies against erbb3 and uses thereof |
| WO2008127594A2 (en) | 2007-04-11 | 2008-10-23 | Exelixis, Inc. | Combination therapies comprising quinoxaline inhibitors of pi3k-alpha for use in the treatment of cancer |
Non-Patent Citations (111)
| Title |
|---|
| "Bioreversible Carriers in Drug Design", 1987, AMERICAN PHARMACEUTICAL ASSOCIATION AND PERGAMON PRESS |
| "Organic Reactions", vol. 1-40, 1991, JOHN WILEY AND SONS |
| "Protocols for Oligonucleotide Conjugates", 1994, HUMANA PRESS |
| "Remington's Pharmaceutical Sciences", 1980, MACK PUBLISHING CO. |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING COMPANY |
| "Rod's Chemistry of Carbon Compounds", vol. 1-S, 1989, ELSEVIER SCIENCE PUBLISHERS |
| A CHAKRABARTY ET AL: "H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3", ONCOGENE, vol. 29, no. 37, 28 June 2010 (2010-06-28), pages 5193 - 5203, XP055005313, ISSN: 0950-9232, DOI: 10.1038/onc.2010.257 * |
| ANDERSON W F, NATURE, vol. 392, 1998, pages 25 - 30 |
| BACHMAN ET AL., CANCER BIOL THER, vol. 3, 2004, pages 772 - 775 |
| BARNES ET AL., ANAL. BIOCHEM., vol. 102, 1980, pages 255 |
| BRENNAN ET AL., SCIENCE, vol. 229, 1985, pages 81 |
| BRODEUR ET AL.: "Monoclonal Antibody Production Techniques and Applications", 1987, MARCEL DEKKER, INC., pages: 51 - 63 |
| BROGNARD, J., CANCER RES, vol. 61, 2001, pages 3986 - 3997 |
| BRUMMELKAMP T R ET AL., SCIENCE, vol. 296, 2002, pages 550 - 553 |
| BUNN ET AL., CLINICAL CANCER RESEARCH, vol. 7, 2001, pages 3239 - 3250 |
| BYUN ET AL., INT J CANCER, vol. 104, 2003, pages 318 - 327 |
| CAMPBELL ET AL., CANCER RES, vol. 64, 2004, pages 7678 - 7681 |
| CARTER ET AL., BIO/TECHNOLOGY, vol. 10, 1992, pages 163 - 167 |
| CATON, KOPROWSKI, PROC. NATL. ACAD. SCI. USA, vol. 87, 1990, pages 6450 - 6454 |
| CHEN ET AL., J. BIOL. CHEM., vol. 271, 1996, pages 7620 - 7629 |
| CLACKSON ET AL., NATURE, vol. 352, pages 624 - 628 |
| CLARK, A. S., MOL CANCER THER, vol. 1, 2002, pages 707 - 717 |
| DORNBURG R, GENE THERAP., vol. 2, 1995, pages 301 - 310 |
| EGLITIS M A, BIOTECHNIQUES, vol. 6, 1988, pages 608 - 614 |
| F.M. SCHABEL ET AL.: "Methods in Cancer Research", vol. 17, 1979, ACADEMIC PRESS INC., article "Cancer Drug Development, Part B", pages: 3 - 51 |
| FARRE, D. ET AL., NUCLEIC ACIDS RES., vol. 31, 2003, pages 3651 - 3653 |
| FISHER K J ET AL., J. VIROL., vol. 70, 1996, pages 520 - 532 |
| FISHER, FISHER'S: "Reagents for Organic Synthesis", vol. 1-17, 1991, JOHN WILEY AND SONS |
| GODING: "Monoclonal Antibodies: Principles and Practice", 1986, ACADEMIC PRESS, pages: 59 - 103 |
| GOULDING, H. ET AL., HUM. PATHOL., vol. 26, 1995, pages 291 - 294 |
| GRAHAM ET AL., J. GEN VIROL., vol. 36, 1977, pages 59 |
| GUSS ET AL., EMBO J., vol. 5, 20 December 1985 (1985-12-20), pages 15671575 |
| HAM ET AL., METH. ENZ., vol. 58, 1979, pages 44 |
| HARTMANN ET AL., ACTA NEUROPATHOL (BERL, vol. 109, 2005, pages 639 - 642 |
| HELENE ET AL., ANN. N.Y. ACAD. SCI., vol. 660, 1992, pages 27 - 36 |
| HELENE, ANTICANCER DRUG DES., vol. 6, no. 6, 1991, pages 569 - 84 |
| HICKEY, COTTER, J BIOL CHEM, vol. 281, 2006, pages 2441 - 2450 |
| HOSSE ET AL., PROTEIN SCI., vol. 15, no. 1, 2006, pages 14 - 27 |
| HUDZIAK ET AL., PROC. NATL. ACAD. SCI. USA, vol. 84, 1987, pages 7159 7163 |
| HURST, H.C., BREAST CANCER RES, vol. 3, 2001, pages 395 - 398 |
| HYRUP ET AL., BIOORGAN. MED. CHEM., vol. 4, 1996, pages 5 - 23 |
| JANG JI-YOUNG ET AL: "Degradation of HER2/neu by ANT2 shRNA suppresses migration and invasiveness of breast cancer cells", BMC CANCER, BIOMED CENTRAL, LONDON, GB, vol. 10, no. 1, 23 July 2010 (2010-07-23), pages 391, XP021075213, ISSN: 1471-2407, DOI: 10.1186/1471-2407-10-391 * |
| JONES ET AL., NATURE, vol. 321, 1986, pages 522 - 525 |
| KILPATRICK ET AL., HYBRIDOMA, vol. 16, 1997, pages 381 - 389 |
| KOHLER ET AL., NATURE, vol. 256, 1975, pages 495 |
| KOZBOR, J. IMMUNOL., vol. 133, 1984, pages 3001 |
| KRAUS, A. C., ONCOGENE, vol. 21, 2002, pages 8683 - 8695 |
| KRYSTAL, G. W., MOL CANCER THER, vol. 1, 2002, pages 913 - 922 |
| LARCH'S: "Comprehensive Organic Transformations", 1989, VICHY PUBLISHERS INC. |
| LEE ET AL., GYNECOL ONCOL, vol. 97, 2005, pages 26 - 34 |
| LEE ET AL., ONCOGENE, vol. 24, 2005, pages 1477 - 1480 |
| LEE N S ET AL., NAT. BIOTECHNOL., vol. 20, 2002, pages 500 - 505 |
| LEVINE ET AL., CLIN CANCERRES, vol. 11, 2005, pages 2875 - 2878 |
| LI ET AL., BREAST CANCER RES TREAT, vol. 96, 2006, pages 91 - 95 |
| LINDMARK ET AL., J. IMMUNOL. METH., vol. 62, 1983, pages 1 - 13 |
| MAHER, BIOASSAYS, vol. 14, 1992, pages 807 - 15 |
| MARCH'S: "Advanced Organic Chemistry", JOHN WILEY AND SONS |
| MARKS ET AL., BIO/TECHNOLOGY, vol. 10, 1992, pages 779 - 783 |
| MARKS ET AL., J. MOL. BIOL., vol. 222, 1991, pages 581 - 597 |
| MASSION ET AL., AM J RESPIR CRIT CARE MED, vol. 170, 2004, pages 1088 - 1094 |
| MATHER ET AL., ANNALS N.Y. ACAD. SCI., vol. 383, 1982, pages 44 - 68 |
| MATHER, BIOL. REPROD., vol. 23, 1980, pages 243 - 251 |
| MCCAFFERTY ET AL., NATURE, vol. 348, 1990, pages 552 - 554 |
| MESSEGUER, X. ET AL., BIOINFORMATICS, vol. 18, 2002, pages 333 - 334 |
| MILLER A D, HUM GENE THERAP., vol. 1, 1990, pages 5 - 14 |
| MIYAGISHI M ET AL., NAT. BIOTECHNOL., vol. 20, 2002, pages 497 - 500 |
| MORIMOTO ET AL., JOURNAL OF BIOCHEMICAL AND BIOPHYSICAL METHODS, vol. 24, 1992, pages 107 - 117 |
| MUNSON ET AL., ANAL. BIOCHEM., vol. 107, 1980, pages 220 |
| ODA ET AL., CANCER RES., vol. 65, 2005, pages 10669 - 10673 |
| PADDISON P J ET AL., GENES DEV., vol. 16, 2002, pages 948 - 958 |
| PAUL C P ET AL., NAT. BIOTECHNOL., vol. 20, 2002, pages 505 - 508 |
| PERRY-O'KEEFE ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 93, 1996, pages 14670 - 14675 |
| PLUCKTHUN: "The Pharmacology of Monoclonal Antibodies", vol. 113, 1994, SPRINGER-VERLAG, pages: 269 - 315 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 327 |
| S. H. DANDEGAONKER, C. K. MESTA, J. MED. CHEM., vol. 8, 1965, pages 884 |
| S. M. BERGE ET AL.: "Pharmaceutical Salts", J. PHARM. SCI., vol. 66, 1977, pages 1 - 19, XP002675560, DOI: doi:10.1002/jps.2600660104 |
| S. V. LITVINENKO, V. I. SAVICH, D. D. BOBROVNIK, CHEM. HELEROCYCL. COMPD. (ENGL. TRANSL, vol. 30, 1994, pages 340 |
| SAAL ET AL., CANCER RES, vol. 65, 2005, pages 2554 - 2559 |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Guide", vol. 1-3, 1989, COLD SPRING HARBOR PRESS |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Guide, Vois", 1989, COLD SPRING HARBOR PRESS, pages: 1 - 3 |
| SAMUELS ET AL., SCIENCE, vol. 304, 2004, pages 554 |
| SAMUELS, VELCULESCU, CELL CYCLE, vol. 3, 2004, pages 1221 - 1224 |
| SAMULSKI R ET AL., J. VIROL., vol. 61, 1987, pages 3096 - 3101 |
| SAMULSKI R, J. VIROL., vol. 63, 1989, pages 3822 - 3826 |
| SANGER, F. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 74, 1977, pages 5463 - 5467 |
| SIAS ET AL., J. IMMUNOL. METHODS, vol. 132, 1990, pages 73 - 80 |
| SLAMON ET AL., SCIENCE, vol. 235, 1987, pages 177 182 |
| SLAMON ET AL., SCIENCE, vol. 244, 1989, pages 707 712 |
| SRIVAGTAVA, MEASE, NUCL. MED. BIO., vol. 18, 1991, pages 589 - 603 |
| SUJOBERT ET AL., BLOOD, vol. 106, 1997, pages 1063 - 1066 |
| SZOKA ET AL., ANN. REV. BIOPHYS. BIOENG., vol. 9, 1980, pages 467 |
| T. HIGUCHI, V. STELLA: "Pro-drugs as Novel Delivery Systems", A.C.S. SYMPOSIUM SERIES, vol. 14 |
| T. HIGUCHI, V. STELLA: "Pro-drugs as Novel Delivery Systems", vol. 14, A.C.S. SYMPOSIUM SERIES |
| T.H. CORBETT ET AL., CANCER, vol. 40, 1977 |
| T.W. GREENE: "Protective Groups in Organic Synthesis", 1991, JOHN WILEY & SONS, INC. |
| TANG ET AL., LUNG CANCER, vol. 51, 2006, pages 181 - 191 |
| TUSCHL T ET AL., THE SIRNA USER GUIDE |
| TUSCHL, T., NAT. BIOTECHNOL, vol. 20, 2002, pages 446 - 448 |
| URLAUB ET AL., PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4216 |
| VELHO ET AL., EURJ CANCER, vol. 41, 2005, pages 1649 - 1654 |
| VERHOEYEN ET AL., SCIENCE, vol. 239, 1988, pages 1534 - 1536 |
| W. C. LUMMA, R. D. HARTMAN, J. MED. CHEM., vol. 24, 1981, pages 93 |
| WACHECK ET AL.: "Chemosensitisation of malignant melanoma by BCL2 amisellse therapy", LANCET, vol. 356, 2000, pages 1728 - 1733 |
| WANG ET AL., HUM MUTAT, vol. 25, 2005, pages 322 |
| WANG, S.E. ET AL., MOL. CELL. BIOL., vol. 28, 2008, pages 5605 - 5620 |
| WATERHOUSE ET AL., NUCLEIC. ACIDS RES., vol. 21, 1993, pages 2265 - 2266 |
| WEBB ET AL.: "BCL2 antisense therapy in patients with non-Hodgkin lymphoma", LANCET, vol. 349, 1997, pages 1137 - 1141, XP004267082, DOI: doi:10.1016/S0140-6736(96)11103-X |
| WU ET AL., J CLIN ENDOCRINOL METAB, vol. 90, 2005, pages 4688 - 4693 |
| XIA H ET AL., NAT. BIOTECH., vol. 20, 2002, pages 1006 - 1010 |
| YUAN, Z. Q., J BIOL CHEM, vol. 278, 2003, pages 23432 - 23440 |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2841102A4 (en) * | 2012-04-23 | 2015-05-06 | Univ Ramot | Methods and compositions for treating cancer |
| WO2015073644A1 (en) | 2013-11-13 | 2015-05-21 | Novartis Ag | Mtor inhibitors for enhancing the immune response |
| WO2015090230A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Human mesothelin chimeric antigen receptors and uses thereof |
| EP4026909A1 (en) | 2013-12-19 | 2022-07-13 | Novartis AG | Human mesothelin chimeric antigen receptors and uses thereof |
| WO2015090229A1 (en) | 2013-12-20 | 2015-06-25 | Novartis Ag | Regulatable chimeric antigen receptor |
| EP4420663A2 (en) | 2013-12-20 | 2024-08-28 | Novartis AG | Regulatable chimeric antigen receptor |
| WO2015100459A3 (en) * | 2013-12-27 | 2015-08-20 | Merrimack Pharmaceuticals, Inc. | Biomarker profiles for predicting outcomes of cancer therapy with erbb3 inhibitors and/or chemotherapies |
| WO2015142675A2 (en) | 2014-03-15 | 2015-09-24 | Novartis Ag | Treatment of cancer using chimeric antigen receptor |
| WO2015157252A1 (en) | 2014-04-07 | 2015-10-15 | BROGDON, Jennifer | Treatment of cancer using anti-cd19 chimeric antigen receptor |
| EP3888674A1 (en) | 2014-04-07 | 2021-10-06 | Novartis AG | Treatment of cancer using anti-cd19 chimeric antigen receptor |
| EP4406610A2 (en) | 2014-04-07 | 2024-07-31 | Novartis AG | Treatment of cancer using anti-cd19 chimeric antigen receptor |
| EP3722316A1 (en) | 2014-07-21 | 2020-10-14 | Novartis AG | Treatment of cancer using a cd33 chimeric antigen receptor |
| WO2016014530A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Combinations of low, immune enhancing. doses of mtor inhibitors and cars |
| WO2016014553A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Sortase synthesized chimeric antigen receptors |
| EP4205749A1 (en) | 2014-07-31 | 2023-07-05 | Novartis AG | Subset-optimized chimeric antigen receptor-containing cells |
| EP3660042A1 (en) | 2014-07-31 | 2020-06-03 | Novartis AG | Subset-optimized chimeric antigen receptor-containing t-cells |
| WO2016025880A1 (en) | 2014-08-14 | 2016-02-18 | Novartis Ag | Treatment of cancer using gfr alpha-4 chimeric antigen receptor |
| EP3712171A1 (en) | 2014-08-19 | 2020-09-23 | Novartis AG | Treatment of cancer using a cd123 chimeric antigen receptor |
| WO2016044605A1 (en) | 2014-09-17 | 2016-03-24 | Beatty, Gregory | Targeting cytotoxic cells with chimeric receptors for adoptive immunotherapy |
| EP3967709A1 (en) | 2014-09-17 | 2022-03-16 | Novartis AG | Targeting cytotoxic cells with chimeric receptors for adoptive immunotherapy |
| WO2016057705A1 (en) | 2014-10-08 | 2016-04-14 | Novartis Ag | Biomarkers predictive of therapeutic responsiveness to chimeric antigen receptor therapy and uses thereof |
| WO2016164580A1 (en) | 2015-04-07 | 2016-10-13 | Novartis Ag | Combination of chimeric antigen receptor therapy and amino pyrimidine derivatives |
| WO2016168595A1 (en) | 2015-04-17 | 2016-10-20 | Barrett David Maxwell | Methods for improving the efficacy and expansion of chimeric antigen receptor-expressing cells |
| EP4234685A2 (en) | 2015-04-17 | 2023-08-30 | Novartis AG | Methods for improving the efficacy and expansion of chimeric antigen receptor-expressing cells |
| WO2016172583A1 (en) | 2015-04-23 | 2016-10-27 | Novartis Ag | Treatment of cancer using chimeric antigen receptor and protein kinase a blocker |
| US11365252B2 (en) | 2016-07-20 | 2022-06-21 | University Of Utah Research Foundation | CD229 CAR T cells and methods of use thereof |
| WO2018067992A1 (en) | 2016-10-07 | 2018-04-12 | Novartis Ag | Chimeric antigen receptors for the treatment of cancer |
| EP3590516A4 (en) * | 2017-02-28 | 2020-12-09 | Taiho Pharmaceutical Co., Ltd. | Antitumor-effect enhancer using pyrazolo[3,4-d]pyrimidine compound |
| RU2799006C2 (en) * | 2017-02-28 | 2023-06-30 | Тайхо Фармасьютикал Ко., Лтд. | ANTITUMOR EFFECT STRENGTHENING AGENT USING PYRAZOLO[3,4-d]PYRIMIDINE COMPOUND |
| US11696917B2 (en) | 2017-02-28 | 2023-07-11 | Taiho Pharmaceutical Co., Ltd. | Agent enhancing antitumor effect using pyrazolo[3,4-d]pyrimidine compound |
| KR20190122764A (en) * | 2017-02-28 | 2019-10-30 | 다이호야쿠힌고교 가부시키가이샤 | Antitumor Effect Enhancers Using Pyrazolo [3,4-d] pyrimidine Compounds |
| KR102674711B1 (en) | 2017-02-28 | 2024-06-12 | 다이호야쿠힌고교 가부시키가이샤 | Antitumor effect enhancer using pyrazolo[3,4-d]pyrimidine compound |
| WO2018201056A1 (en) | 2017-04-28 | 2018-11-01 | Novartis Ag | Cells expressing a bcma-targeting chimeric antigen receptor, and combination therapy with a gamma secretase inhibitor |
| CN110958877A (en) * | 2017-07-09 | 2020-04-03 | 江苏英科贝塔医药科技有限公司 | Fluorine-containing compound, use thereof and method for producing same |
| WO2019210153A1 (en) | 2018-04-27 | 2019-10-31 | Novartis Ag | Car t cell therapies with enhanced efficacy |
| WO2019213282A1 (en) | 2018-05-01 | 2019-11-07 | Novartis Ag | Biomarkers for evaluating car-t cells to predict clinical outcome |
| CN115989030A (en) * | 2020-05-29 | 2023-04-18 | 康涅狄格大学 | Inhibitors of porcine reproductive and respiratory syndrome virus |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112013000651A2 (en) | 2016-05-31 |
| US20140271665A1 (en) | 2014-09-18 |
| SG186889A1 (en) | 2013-02-28 |
| KR20130098301A (en) | 2013-09-04 |
| CA2803900A1 (en) | 2012-01-12 |
| EP2590654A1 (en) | 2013-05-15 |
| EA201390085A1 (en) | 2013-06-28 |
| UY33498A (en) | 2013-01-03 |
| TW201244719A (en) | 2012-11-16 |
| AR084469A1 (en) | 2013-05-22 |
| AU2011274510A1 (en) | 2013-01-24 |
| AU2011274510A8 (en) | 2013-04-18 |
| JP2013539453A (en) | 2013-10-24 |
| US9011863B2 (en) | 2015-04-21 |
| EP2590654B1 (en) | 2016-12-21 |
| TN2012000622A1 (en) | 2014-04-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9011863B2 (en) | Combinations of kinase inhibitors for the treatment of cancer | |
| CN106659772B (en) | Compositions and methods for modulating renalase in the treatment of diseases and disorders | |
| KR20180100539A (en) | Therapeutic compositions, methods of use in conjunction and application | |
| CN109196121A (en) | Treatment and diagnostic method for cancer | |
| JP5389675B2 (en) | Monoclonal antibodies for the treatment of cancer | |
| TW200824696A (en) | Combinations and methods of using an immunomodulatory oligodeoxynucleotide | |
| BRPI0918178B1 (en) | MONOCLONAL ANTIBODIES FOR CANCER TREATMENT | |
| US20190008957A1 (en) | Combination therapies including inhibitors of the extracellular domain of e-cadherin | |
| JP2022527825A (en) | Ionic liquids for drug delivery | |
| JP2019502664A (en) | Drugs for the treatment of diseases associated with unwanted cell proliferation | |
| WO2017139468A1 (en) | Combination of ifn-gamma with erbb inhibitor for the treatment of cancers | |
| CN111344013A (en) | Use of NOX inhibitors for the treatment of cancer | |
| Shervington et al. | Glioma: what is the role of c-Myc, hsp90 and telomerase? | |
| US20050222059A1 (en) | Egfrviii specific monoclonal antibody and egfrviii ribozymes and use to detect, treat or prevent egfrviii associated cancer | |
| US20210353639A1 (en) | Mast1 and uses for diagnosing and treating cancer | |
| KR20230156936A (en) | How to Treat Disease Using Gremlin1 Antagonists | |
| JP2016515132A (en) | Combination and use of MEK inhibitor compounds with HER3 / EGFR inhibitor compounds | |
| JP2022548978A (en) | Dosing for Treatment with Drugs Anti-TIGIT and Anti-PD-L1 Antagonist Antibodies | |
| US20160324962A1 (en) | Compositions and methods for treating sarcoma | |
| US20240139198A1 (en) | Method and combination for treating tumors | |
| WO2023235691A1 (en) | Btk reducing molecules for treatment of cancers and immune system disorders | |
| Zavaleta-Monestel et al. | Amivantamab: A Novel Advance in the Treatment of Non-small Cell Lung Cancer | |
| CN110603447A (en) | Compositions and methods for treating cancer with anti-renalase antibodies and anti-PD 1 antibodies | |
| JP2024513246A (en) | Combination treatment with RAF inhibitor and PD-1 axis inhibitor | |
| US20180280423A1 (en) | Compositions and methods for combating drug-resistant cancers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11731617 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2803900 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 223827 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 0173713 Country of ref document: KE Ref document number: 2013518865 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 000032-2013 Country of ref document: PE Ref document number: MX/A/2013/000275 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013000090 Country of ref document: CL Ref document number: 12013500064 Country of ref document: PH |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011731617 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011731617 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2011274510 Country of ref document: AU Date of ref document: 20110708 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13016350 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2013-000052 Country of ref document: CR |
|
| ENP | Entry into the national phase |
Ref document number: 20137003497 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201390085 Country of ref document: EA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013000651 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13809002 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 112013000651 Country of ref document: BR Kind code of ref document: A2 Effective date: 20130109 |