WO2011062503A1 - Parenteral formulations of gemcitabine derivatives - Google Patents
Parenteral formulations of gemcitabine derivatives Download PDFInfo
- Publication number
- WO2011062503A1 WO2011062503A1 PCT/NO2010/000417 NO2010000417W WO2011062503A1 WO 2011062503 A1 WO2011062503 A1 WO 2011062503A1 NO 2010000417 W NO2010000417 W NO 2010000417W WO 2011062503 A1 WO2011062503 A1 WO 2011062503A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutical composition
- gemcitabine
- active ingredient
- cancer
- solubilizer
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/24—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing atoms other than carbon, hydrogen, oxygen, halogen, nitrogen or sulfur, e.g. cyclomethicone or phospholipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
- A61K9/1075—Microemulsions or submicron emulsions; Preconcentrates or solids thereof; Micelles, e.g. made of phospholipids or block copolymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/09—Pyrimidine radicals with arabinosyl as the saccharide radical
Definitions
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising certain long chain saturated and monounsaturated fatty acid derivatives of 2',2'-difluorodeoxy- cytidine (gemcitabine) as the active ingredient.
- the present invention relates to a pharmaceutical composition and the method of preparation thereof, suitable for parenteral administration of therapeutically effective doses of the said derivatives in order to ameliorate compliance in treatment of cancer.
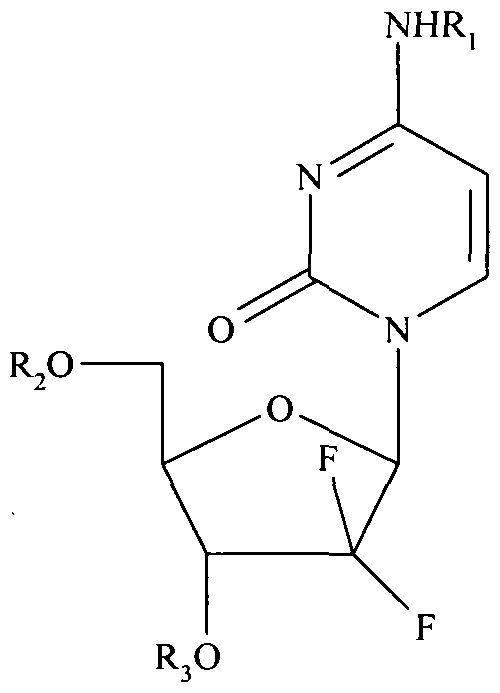
- Gemcitabine which is a well known cytostatic compound, marketed under the trade name Gemzar by Eli Lilly & Co., has the formula:
- the active ingredients of the pharmaceutical composition of the present invention comprise gemcitabine derivatives of the formula I:
- Ri, R 2 and R 3 are independently selected from hydrogen and Ci 8 - and C 20 - saturated and monounsaturated acyl groups, with the proviso that R l 5 R 2 and R 3 cannot all be hydrogen.
- nucleosides and nucleoside analogues such as gemcitabine occurs mainly via the selective Nucleoside Transport (NT) receptor. Modulation/inhibition of this receptor may be seen as resistance to the drug in a clinical situation. This phenomenon can be observed in-vitro through addition of NT inhibitors.
- NT inhibitors We have previously reported that our derivatives are not influenced by the presence of NT inhibitors, since the cytostatic activity of the preferred derivatives is conserved in the presence of such inhibitors (WO 98/32762).
- Multi drug resistance is one of the principal reasons for failure of otherwise effective drugs. We have found that the derivatives of this invention seem not to be substrates for the MDR-pump, and hence circumvent this problem.
- the derivatives of this invention are poor substrates for the deaminating enzyme. Consequently, the derivatives of this invention are more suited than gemcitabine itself for systemic or local treatment of malignant tumours.
- formulation of a therapeutically effective amount of the poorly soluble derivatives of formula (I) into a pharmaceutical composition suitable for parenteral administration represents a problem.
- the composition of the excipients should be selected so that the said derivatives were solubilised or formed nanosized particles.
- the gemcitabine derivatives of formula (I) are amphiphilic and have poor solubility both in water and in oils, which limits the choice of potential excipients that can solubilise them.
- the formulation is a particulate system, there are certain requirements for the size of the particles in the formulations for intravenous administration.
- parenteral products must be sterile and often sterile filtration is the only viable method for pharmaceutical particulate systems. This means that the particle size of these formulations must be smaller than 220 nm (0.22 ⁇ ), which is the pore size of the sterile filters. In practice and for an industrial scale process, the particles should be much smaller to avoid filter clogging.
- gemzar is administered intravenously (i.v.) at a dose of 1000 mg/m 2 (3.3 mmol/m 2 of active), and the recommended dose for intravenous gemcitabine-5'- elaidic acid ester is 1250 mg/m 2 (2.4 mmol/m of active). This means that for an average patient with a surface area of 1.8 m 2 , the total dose of gemcitabine-5'- elaidic acid ester will be 2250 mg as a monotherapy.
- the derivatives of formula (I) are prone to hydrolytic degradation in physiological pH, the rate of which depends on the type of the derivative and the buffer.
- gemcitabine-5'- elaidic acid ester has a half life of approximately 30 minutes in pH 7.4. This represents further challenges both to the formulation and to the manufacturing process parameters. It is normally preferred that a pharmaceutical product be ready-to-use, but it is also possible to freeze dry the formulation in order to avoid degradation during the products shelf-life period. If ready-to-use, then the said derivatives should be protected from hydrolytic degradation in the aqueous environment of the parenteral formulation during its entire shelf-life period.
- the present invention presents a solution to all the above problems.
- a pharmaceutical composition suitable for parenteral administration and a method of preparation for gemcitabine derivatives of formula (I) that results in ready-to-use high drug load aqueous nanoparticulate formulation based on phospholipids, with a drug to lipid molar ratio as high as 1 :2, even more preferably as high as 1 : 1., where the said lipid nanoparticles protect the said derivative from hydrolytic degradation to gemcitabine for at least 38 months when stored at 2-8°C under nitrogen blanket.
- the method uses natural phospholipids derived from egg yolk, does not incorporate any surfactant and results in micelle-like nanoparticles with volume based D( VO i,o.99) diameter of 5-20 nm, or intensity based Z-average based diameter of 30-50 nm as determined using instrumentation designed for small particle size analysis such as the Malvern Zetasizer Nano. Particles of this size can be easily sterile-filtered. Additionally, the method of preparation is an industrially scalable one, suitable for manufacture of aqueous sterile products.
- a pharmaceutical composition comprising a gemcitabine derivative of formula (I):
- R l s R 2 and R 3 are independently selected from hydrogen and Ci 8 - and C 20 - saturated and monounsaturated acyl groups, with the proviso that R ⁇ , R 2 and R 3 cannot all be hydrogen, or a pharmaceutically acceptable salt thereof as the active ingredient; wherein the active ingredient is dissolved or dispersed in phospholipids, is provided.
- the active ingredient is prepared into a formulation comprising:
- a solubilizer phospholipid selected from the group consisting of
- phosphatidylcholine phosphatidylglycerol, phosphatidylethanolamine
- phosphatidylinositol phosphatidylserine, phosphatidic acid, lysophospholipids, sphingomyelin and cardiolipin in any form, including salted or desalted, hydrogenated or partially hydrogenated, natural, semisynthetic or synthetic;
- a co-solubilizer selected from the group consisting of charged phospholipids; c) an isotonicity agent and;
- the active ingredient to phospholipid molar ratio is between 1 :5 to 1 :1 and the formulation has an average D (VO i ) particle size ranging between 2.5 - 30 nm.
- the gemcitabine derivative of formula (I) has R] and R 3 as hydrogen and R 2 is a C 18 - or C 20 - saturated or monounsaturated acyl group.
- Gemcitabine has three derivatisable functions, namely the 5'- and 3'-hydroxyl groups and the N4- amino group. Each group can selectively be transformed into an ester or amide derivative, but di-adducts (di-esters or ester-amides) and tri-adducts may be formed as well. In the case of the di- and tri-adducts the acyl substituent groups need not necessarily be the same.
- the mono-acyl derivatives i.e. with two of Ri, R 2 and R 3 being hydrogen, are preferred for use as the active ingredient of the present pharmaceutical composition. It is especially preferred that the monosubstitution with the acyl group should be in the 3'-0 and 5'-0 positions of the sugar moiety, with 5'-0 substitution being most preferred.
- the double bond of the mono-unsaturated acyl groups may be in either the cis or the trans configuration, although the therapeutic effect may differ depending on which configuration is used.
- esters, ester-amides and amides derived from oleic acid CI 8: 1 ⁇ -9, cis
- elaidic acid CI 8: 1 ⁇ -9, trans
- eicosenoic acid(s) C20: l co-9, cis
- C20: l co-9, trans eicosenoic acid(s)
- C20: l co-9, trans eicosenoic acid(s)
- amides and 5 '-esters are currently the most preferred derivatives.
- Esters, ester-amides and amides of gemcitabine derived from stearic acid (CI 8:0) and eicosanoic acid (C20:0) are advantageously used in some cases.
- Gemcitabine (N4)- elaidic acid amide, gemcitabine-5'- elaidic acid ester and gemcitabine-3'- elaidic acid ester are among the most preferred derivatives and according to a preferred embodiment of the invention gemcitabine-5'- elaidic acid ester is the active ingredient of the pharmaceutical composition.
- the pharmaceutical composition of the present invention is described herein as an aqueous formulation containing the active pharmaceutical ingredient (as described above), a solubilizer, a co-solubilizer and an isotonicity agent.
- the pharmaceutical composition comprises gemcitabine-5'- elaidic acid ester, phosphatidylcholine, phosphatidylglycerol, glycerol and water.
- the phospholipids of the said pharmaceutical composition comprise a neutrally charged phospholipid alone, or in combination with other phospholipids where at least one is a negatively charged phospholipid.
- Phospholipids are natural components of cell membranes and are highly biocompatible. Phospholipids are amphiphilic molecules that spontaneously form bilayers in contact with water and upon further dilution turn into micro- and nanosized particles called liposomes. Lipophilic and amphiphilic molecules can be solubilised in the bilayers of phospholipids to a certain molar ratio without compromising the structure of the liposomes.
- the maximum drug concentration in such a formulation is dependent on the type and concentration of the phospholipids and the physicochemical characteristics of the active substance.
- the frequently used molar ratio of the drug to phospholipids in such formulations is in the range of 1 :20.
- Liposomes are prepared from natural or synthetic phospholipids, mainly phosphatidylcholine.
- a smaller amount of a negatively charged phospholipid such as phosphatidylglycerol may also be incorporated.
- the electrostatic repulsion due to the negative charge of the particles provides an effective barrier to aggregation and formation of larger paticles.
- Phospholipids can also form (mixed) micelles when combined with a surfactant such as bile salts, glycocholic acid, taurocholic acid, poloxamer, polysorbates, cremophore, sorbitan monolaurate, etc.
- a surfactant such as bile salts, glycocholic acid, taurocholic acid, poloxamer, polysorbates, cremophore, sorbitan monolaurate, etc.
- the amphiphilic or lipophilic drug may be solubilised in the micelles in higher molar ratios than expected from liposomes.
- micelles permit a greater concentration of phospholipids, and hence the amphiphilic/lipophilic drug, per unit volume of their nanoparticle than liposomes do.
- surfactants are generally considered to exhibit concentration-dependent toxic adverse effects, the addition of these excipients to pharmaceutical formulations should be limited to a minimum.
- the lipid nanostructures of the formulation may comprise, but are not restricted to, the following phospholipids, which function as solubilizers, bilayer-forming or micelle- forming excipients: phosphatidylcholine, phosphatidylglycerol, phosphatidylethanolamine, phosphatidylinositol, phosphatidylserine, phosphatidic acid, lysophospholipids, sphingomyelin, cardiolipin.
- the phospholipids may be in any form, including salted or desalted, hydrogenated or partially hydrogenated, natural, semisynthetic or synthetic. Also, attachment of hydrophilic polymers such as polyethyleneglycol (PEG) to the phospholipids in order to avoid rapid clearance by the reticuloendothelial system (RES) is possible.
- PEG polyethyleneglycol
- natural unsaturated phospholipids derived from hen egg are used alone or in combination as the solubilizer.
- the natural egg phospholipids comprise a zwitterionic phospholipid which is neutral in the pH range of 6-8 such as egg phosphatidylcholine.
- the solubilizer is purified hen egg phosphatidylcholine which is more than 96.0% phosphatidylcholine and not more than 1.0% lysophosphtidylcholine, not more than 1.0% sphingomyelin, and not more than 0.1% phosphatidylethanolamine. .
- the formulations of the present invention contemplate use of a co-solubilizer.
- the co-solubilizer may be any suitable charged phospolipid.
- the phospholipid is negatively charged.
- the co-solubilizer phospholipid is negatively charged in the pH range of 6-8, such as hen egg phosphatidylglycerol which is more than 98.0% phosphatidylglycerol sodium salt, not more than 1.5% phosphatidic acid, not more than 0.5% lysophosphatidylglycerol, and not more than 0.5% phosphatidylcholine
- an isotonic agent is included in the pharmaceutical composition.
- An isotonic agent includes, but is not limited to, glycerol, propyleneglycol, polyethyleneglycol, poloxamers, polyols, carbohydrates, sugars, dextrans, aminoacids or proteins, organic or inorganic salts, and a mixture thereof.
- the isotonicity agent is glycerol.
- a sterol is added.
- this sterol is cholesterol.
- antioxidants more preferably a- tocopherol, or fatty acids are added.
- a cryoprotectant is added in the pharmaceutical formulation to facilitate freeze drying.
- a cryoprotectant includes, but is not limited to, maltose, cellobiose, lactose, xylobiose, sucrose, trehalose, mannitol or dextran.
- the isotonic agent is also a cryoprotectant and is added in the pharmaceutical formulation both for isotonicity and to facilitate freeze drying.
- this isotonic and cryoprotectant agent is a disaccharide such as lactose, trehalose, sucrose, mannitol and the like.
- the composition of the excipients, the drug to lipid ratio and the method for manufacture is selected to favour a liposomal structure.
- the said parameters are selected to favour micellar nanoparticles, or a combination of micelles and liposomes.
- excipients of the pharmaceutical composition are selected to solubilise or to increase the solubility of the compounds of formula (I).
- the said excipient might be removed from the final product.
- this excipient is ethanol and is largely removed from the final product.
- the pharmaceutical composition according to the invention is solid, semi-solid or liquid, preferably in liquid form, and may be presented in discrete units such as vials, infusion bags or the like.
- the pharmaceutical form of the final composition is a nanosized suspension or dispersion, either liposomes or micelle-like nanoparticles, or a combination of both.
- HDLCs high drug:lipid complexes
- final pharmaceutical composition refers to the prepared pharmaceutical composition that can be directly administered to the patient. This means that if the pharmaceutical composition is a freeze dried solid, the final pharmaceutical composition would refer to the reconstituted solution of the said formulation according to presettled instructions.
- therapeutically effective amount refers to from about 0.001 to 10 grams per day of a gemcitabine derivative of formula (I) or a pharmaceutically acceptable salt thereof, more preferred from about 10 mg to 6 grams per day of a gemcitabine derivative of formula (I) or a pharmaceutically acceptable salt thereof, in a formulation containing 0.001 - 80% of the said derivative or salt thereof formulated for parenteral administration.
- the amount of the phospholipid phase in the final pharmaceutical composition may vary from about 0.1% to 50%, preferably 1 - 15%, and more preferably 5 - 12%. In the most preferred but not limiting embodiment, the amount of the phospholipid phase is 9.5-10%) of the final pharmaceutical composition. All subranges from 0.1% to 50% are included as part of the invention.
- the molar ratio of the gemcitabine derivative of formula (I) to the total amount of the phospholipids in the final pharmaceutical composition may vary from 1 : 130 to 1 : 1, preferably 1 :70 to 1 :2.
- Another preferred range includes 1 :6.6 (corresponding to 10 mg/ml gemcitabine derivative to lipid in final formulation) to 1 :1.9 (corresponding to 35 mg/ml gemcitabine derivative to lipid in final formulation).
- Another preferred range includes 1 :5.3 (corresponding to 12.5 mg/ml gemcitabine derivative to lipid in final formulation) to 1 :2.2 (corresponding to 30 mg/ml gemcitabine derivative to lipid in final formulation).
- Another preferred range is 1 :5 to 1 :2.
- the most preferable range is 1 :5 to 1 :1.
- Another most preferred molar ratio of the gemcitabine derivative of formula (I) to the total amount of the phospholipids is 1 :4.4 (corresponding to 15 mg/ml gemcitabine derivative to lipid in final formulation). All subranges between 1 :130 and 1 : 1 are included as part of the invention.
- the molar ratio of egg phosphatidylcholine to egg phosphatidylglycerol in the composition may vary from 1 : 1 to 99:1, preferably 2: 1 to 80: 1, with the most preferable ratio being 25: 1.
- Another preferred range includes 15:1 to 40: 1 , more preferably 20: 1 to 30:1, even more preferably 23:1 to 27:1. All subranges between 1 : 1 and 99: 1 are included as a part of the invention.
- Cholesterol may be added to the phospholipids in a molar ratio of 0.05: 1 to 1 : 1, more preferably 0.2: 1 to 0.5: 1.
- the amount of glycerol as the isotonic agent is adjusted to provide isoosmolar condition in the final pharmaceutical composition, and may or may not be added. If added, the amount may vary between approximately 50mM to 350mM depending on the other constituents of the formulation. In a preferred embodiment, the amount of glycerol is 260-300 mM, especially preferred amount is 285mM. All subranges between 50 and 350 mM are included as a part of the invention.
- the amount of the disaccharide used as isotonic/cryoprotective agent may vary between 1 to 50% of the final pharmaceutical composition, more preferably 5 tol5% and most preferably 7-10%. All subranges between 1 and 50% are included as part of the invention.
- the molar ratio of the isotonic/cryoprotective agent to total phospholipids is between 10:1 and 1 :5, more preferably 5:1 to 1 : 1. All subranges between 10: 1 and 1 :5 are included as part of invention.
- the present invention also provides a process for the preparation of a pharmaceutical composition as mentioned above.
- the said process comprises the steps of dissolving the phospholipids and the gemcitabine derivative of formula (I) in a suitable water-miscible organic solvent.
- water-miscible solvents are acetone, acetonitrile, dimethylformamide, ethylene glycol, glycerol, methanol, 1-propanol, 2-propanol, ethanol and DMSO.
- Phospholipids and the gemcitabine derivative of formula (I) may be dissolved in the same or in different water-miscible organic solvents as long as both organic solutions can be mixed together.
- the said organic solution is then injected into an aqueous solution whereupon the lipid nanoparticles are formed.
- the size and structure of the nanoparticles in the said "intermediate bulk solution" are determined by the formulation and the injection parameters.
- One important parameter is the type and concentration of the organic solvent in the intermediate bulk solution.
- the organic solvent is ethanol used in an amount of 5 to 40%, more preferably 10 to 30% of the intermediate bulk solution. All intermediate values between 5 and 40% is covered by the present invention.
- the intermediate bulk solution is subjected to homogenization and removal of the organic solvent.
- the active pharmaceutical ingredient, solubilizer (preferably purified phosphatidylcholine) and co-solubilizer (preferably phosphatidylglycerol) are dissolved in ethanol and then injected into an aqueous solution containing water and the isotonicity agent (preferably glycerol) to form an intermediate bulk solution.
- solubilizer preferably purified phosphatidylcholine
- co-solubilizer preferably phosphatidylglycerol
- the intermediate bulk solution is homogenised using conventional equipment until the aimed particle size is achieved, and then the bulk solution is concentrated to the final volume by tangential flow filtration and the organic solvent is removed by further diafiltration.
- the said bulk solution is first concentrated to the final or an intermediate volume, thereafter homogenised using conventional techniques and equipment until the desired particle size is achieved. The bulk solution is thereafter, if necessary, concentrated further and the organic solution is finally removed by diafiltration. It is also possible to combine the final concentration and diafiltration in one step.
- the intermediate bulk solution is subjected to high pressure homogenization (typically between 625 - 1000 bar) for several passes until no further change is particle size is observed is subsequent passes.
- the particle size may be conveniently measured by laser light scattering to produce a volume-based diameter measeurement - D( VO i).
- the particles formed according to the manufacturing process above have a D( vo i) (measured as the percentage of particles that have a size smaller than the indicated range) as follows:
- the particle size as measured by the laser light scattering technique may also be expressed as an intensity based mean hydrodynamic volume (Z-average).
- Z-average mean hydrodynamic volume
- FIG. 1 provides sample transmission electron micrographs (TEMs) of drug product prepared as described in Example 1.
- Figure 3 shows an enlargement of the image from Figure 2.
- a pharmaceutical wetting agent may initially be added to the active substance before mixing with the lipid excipients.
- the wetting agents are polymers, surfactants, carbohydrates, polysaccharides, mineral solids, oils, alcohols or acids, organic or inorganic.
- the nanoparticles of the final pharmaceutical composition are either liposome-like, meaning vesicles surrounded by phospholipid bilayer, or micelle-like, or a combination of both.
- the particle size distribution may be monomodal, bimodal or even multimodal, provided this does not impact the sterile filterability of the bulk solution.
- the particle size of the final pharmaceutical composition as determined by laser light scattering and expressed in volume based diameter units may be in a range of 2.5 nm to 220 nm. In one embodiment, the particle size distribution is monomodal and the average size ranges between 2.5 nm to 30nm, or between 30nm and 220nm. In another embodiment, the particle size distribution is bimodal and has 2 distributions, one 2.5nm-30nm and the second 30-220nm.
- the particle size distribution is multimodal and the distributions have averages between 2.5 nm and 220 nm.
- the particle size distribution of the final pharmaceutical composition is bimodal, where the main particle fraction has an average size of 2.5-25nm, and the second minor particle fraction has an average particle size of 40-120nm. All intermediate particle size values between 2.5 and 220nm are covered by this invention. Alternative measurement techniques or expression units may result in small changes in the absolute ranges described above, however the measured particle sizes are consistently less than 200 nm in diameter.
- compositions of this invention are useful in treating a wide variety of cancers and specifically including metastatic pancreatic cancer, non-metastatic pancreatic cancer, metastatic breast cancer, non-metastatic breast cancer, non-small cell lung cancer, uterine cancer, ovarian cancer, cervical cancer, prostate cancer, biliary tract cancer, head and neck cancer, lymphomas, myelomas, and soft tissue sarcomas.
- the pharmaceutical composition may also be used as monotherapy or in combination with other approved or experimental cancer therapies.
- the present invention provides a pharmaceutical composition defined as aforesaid for use in treatment of cancer, and in particular treatment of the cancers indicated above.
- the preferred dosing schedule for intravenous administration is 1250 mg/m 2 once weekly for daily for 3 weeks out of 4 weeks.
- Alternative dosing schedules may be appropriate for specific cancer types, or when gemcitabine-5'-elaidate is used in conjuction with other therapeutic agents.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- the ethanol solution was thereafter injected into the glycerol/water (2.6% w/w) solution at 250 ml/min under stirring.
- the weight ratio of ethanol solution to the glycerol solution was 1 :7.6.
- the bulk solution was concentrated by tangential flow filtration, and the concentrated bulk was processed 6 times through a homogeniser at 15-20°C.
- the resulting product was further concentrated by tangential flow filtration to the final batch volume of 20L and final gemcitabine-5 '-elaidate concentration of 15 mg/niL.
- the residual ethanol was then removed through a washing step by diafiltration, the final product was sterile filtered and aseptically filled in sterile vials, purged with nitrogen and sealed.
- the vials were stored at 2-8°C protected from light, and the stability of the batch was monitored up to 38 months. During the course of this stability study, no changes in the content of gemcitabine-5'- elaidic acid ester was observed. The amount of the main degradation product, gemcitabine, was 0.03% after 38 months. The batch showed a bimodal particle size distribution; after 38 months the main fraction (99.7%) had a mean size of 4.3 nm, and the other fraction (0.3%) a mean size of 69 nm. At the time of manufacturing the D (VO i,o.99) for this product at was measured to be 11 nm, and the Z-average was measured to be 47 nm.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- the ethanol solution was thereafter injected into the glycerol/water (2.6% w/w) solution under stirring.
- the weight ratio of ethanol solution to the glycerol solution was 1 :8.7.
- the bulk solution was homogenized 2 times and then concentrated by tangential flow filtration.
- the concentrated bulk was then processed 4 times through a homogenizer.
- the resulting product was further concentrated by tangential flow filtration to the final batch volume and gemcitabine-5 '-elaidate concentration of 35 mg/mL.
- the residual ethanol was then removed through a washing step by diafiltration, the final product was sterile filtered and filled in vials, purged with nitrogen and sealed.
- the measured D( VO i,o.99) for the batch was 7.2 nm.
- the measured Z- average intensity based particle size was 46 nm.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- the ethanol solution was thereafter injected into the glycerol/water (2.6% w/w) solution immediately upstream of a homogenizer.
- the weight ratio of ethanol solution to the glycerol solution was 1 :5.3.
- the homogenizer was utilized to both mix and reduce particle size under these operational conditions.
- the bulk solution was concentrated by tangential flow filtration, and the concentrated bulk was processed 12 times through a homogeniser.
- the resulting product was further concentrated by tangential flow filtration to the final batch volume and gemcitabine-5 '-elaidate concentration of 35 mg/mL.
- the residual ethanol was then removed through a washing step by diafiltration and the final product was sterile filtered.
- the batch showed a bimodal volume particle size distribution; the main fraction (99.9%) had a size of 3.9 ran, and the other fraction (0.1%) a mean size of 79 ran.
- the measured Z-average intensity based particle size was 61 ran.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- the ethanol solution was thereafter injected into the glycerol/water (2.6% w/w) solution immediately upstream of a homogenizer.
- the weight ratio of ethanol solution to the glycerol solution was 1 :2.2.
- the homogenizer was utilized to both mix and reduce particle size under these operational conditions.
- the bulk solution was processed 3 times through a homogeniser.
- the resulting product was concentrated by tangential flow filtration to the final batch volume and gemcitabine-5 '-elaidate concentration of 35 mg/mL.
- the bulk solution was then processed an additional 6 times through a homogenizer.
- the residual ethanol was then removed through a washing step by diafiltration and the final product was sterile filtered.
- the batch showed a bimodal volume particle size distribution; the main fraction (99.9%) had a size of 2.9 ran, and the other fraction (0.1%) a mean size of 42 ran.
- the measured Z-average intensity based particle size was 15 ran.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- the ethanol solution was thereafter injected into the glycerol/water (2.6% w/w) solution immediately upstream of a homogenizer.
- the weight ratio of ethanol solution to the glycerol solution was 1 :3.
- the homogenizer was utilized to both mix and reduce particle size under these operational conditions.
- the bulk solution was processed 3 times through a homogeniser.
- the resulting product was concentrated by tangential flow filtration to the final batch volume and gemcitabine-5 '-elaidate concentration of 35 mg/mL.
- the bulk solution was then processed an additional 6 times through a homogenizer.
- the residual ethanol was then removed through a washing step by diafiltration and the final product was sterile filtered.
- the batch showed a bimodal volume particle size distribution; the main fraction (99.9%) had a size of 3.9 nm, and the other fraction (0.1%) a mean size of 44 nm.
- the measured Z-average intensity based particle size was 14 nm.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- the ethanol solution was thereafter injected into the glycerol/water (2.6% w/w) solution immediately upstream of a homogenizer.
- the weight ratio of ethanol solution to the glycerol solution was 1 :8.5.
- the homogenizer was utilized to both mix and reduce particle size under these operational conditions.
- the bulk solution was passed 2 times through a homogenizer and then concentrated by tangential flow filtration to a gemcitabine-5 '-elai date concentration of 40 mg/mL and residual ethanol removed via diafiltration.
- the bulk solution was then processed an additional 4 times through a homogenizer and sterile filtered and filled in vials, purged with nitrogen and sealed
- the measured D( VO i,o.99) for the batch was 9.1 nm.
- the measured Z-average intensity based particle size was 33 nm.
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- the ethanol solution was thereafter injected into the glycerol/water (2.6% w/w) solution immediately upstream of a homogenizer.
- the weight ratio of ethanol solution to the glycerol solution was 1 :8.5.
- the homogenizer was utilized to both mix and reduce particle size under these operational conditions.
- the bulk solution was passed 6 times through a homogenizer and then concentrated by tangential flow filtration to a gemcitabine-5 '-elaidate concentration of 60 mg/mL and residual ethanol removed via diafiltration.
- the bulk solution was then sterile filtered and filled in vials, purged with nitrogen and sealed.
- the measured Z-average intensity based particle size was 44 nm.
- EXAMPLE 8 Thermal analysis by Differential Scanning Calorimetry (DSC) of the formulation described in Example 1 was performed to confirm the storage and shipment temperature of the product. It was shown that the feezing point was low at -22°C, probably due to supercooling of water. The melting point was at approximately -3°C. This suggested that a storage and shipment temperature of 2-8°C would not cause melting or freezing of the phospholipids and hence would not pose any negative impact on the structure of the particles.
- DSC Differential Scanning Calorimetry
- EPC egg phosphatidylcholine
- EPG egg phosphatidylglycerol
- Another batch was manufactured by reducing the starting concentrations of the lipids and the drug by 20%, while keeping the rest of the parameters constant.
- the particle size of this batch was 53 nm (Z-average) and the polydispersity index 0.27.
- the ethanol solution was thereafter injected into a water for injection solution with vigorous mixing.
- the weight ratio of ethanol solution to the glycerol solution was 1 :12.
- the mixture appeared to contain conglomerates as determined by visual inspection. Attempts at homogenization were unsuccessful indicating that gemcitabine-5 '-elaidate was not capable of forming stable particles in the absence of phospholipids.
- Liposomes were prepared by means of dissolution of gemcitabine-5 '-elaidate, egg lecithin, and oleic acid in a molar ratio of 1 : 14.4:2 in ethanol. The solvent was evaporated and the residual solids dispersed into 2.6% glycerol in water using an Ultraturrax followed by high pressure homogenization. Stable formulations could be achieved up to 3 mg/mL gemcitabine-5 '-elaidate. Formulations with higher concentrations of gemcitabine-5 '-elaidate were found to contain undissolved solids. EXAMPLE 13
- Formula II compound provides unexpectedly unique attributes of high loading and stability to the described lipid based formulations.
- EXAMPLE 1 The intravenous formulation of gemcitabine-5'- elaidic acid ester as described in Example 1 was used in a phase I, first in human clinical study. The aims of this study were to determine the safety, toxicity, MTD (Maximum Tolerated Dose) and the RD (Recommended Dose) of gemcitabine-5'- elaidic acid ester, to describe its pharmacokinetic (PK) characteristics, and to assess its preliminary antitumor activity.
- MTD Maximum Tolerated Dose
- RD Recommended Dose
- Gemcitabine-5'- elaidic acid ester was administered on days (d) 1, 8 and 15 every 4 week by a 30 min IV infusion.
- the dose range was from 30 to 1600 mg/m2/d.
- 43 patients were enrolled and the RD was established at 1250 mg/m 2 /d.
- the drug was well tolerated and the most frequent toxicities included nausea, fatigue, vomiting and anorexia, mostly of mild intensity.
- Stabilisation of disease > 3 months
- 7 patients pancreas, colon and ovarian cancer
- One patient with ovarian cancer had a 28.3% reduction in tumor mass.
- Gemcitabine-5'- elaidic acid ester was detected in plasma up to 24 hrs post-dosing.
- AUC for gemcitabine (dFdC) exposure was significantly higher than reported when gemcitabine was administered intravenousely at comparable dose levels.
- Urinary excretion of the main metabolite, dFdU, during the first 24 hrs was 48-71% of the dose.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Dispersion Chemistry (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Neurology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Physical Education & Sports Medicine (AREA)
- Dermatology (AREA)
- Pulmonology (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Gastroenterology & Hepatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Reproductive Health (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description






Claims


Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2010800522764A CN102740833A (en) | 2009-11-20 | 2010-11-15 | Parenteral formulations of gemcitabine derivatives |
| MX2012005677A MX2012005677A (en) | 2009-11-20 | 2010-11-15 | Parenteral formulations of gemcitabine derivatives. |
| JP2012539841A JP2013511516A (en) | 2009-11-20 | 2010-11-15 | Parenteral preparations of gemcitabine derivatives |
| RU2012125350/04A RU2012125350A (en) | 2009-11-20 | 2010-11-15 | Parenteral preparations of gemcitabine derivatives |
| KR1020127015947A KR20120086729A (en) | 2009-11-20 | 2010-11-15 | Parenteral formulations of gemcitabine derivatives |
| AU2010322516A AU2010322516A1 (en) | 2009-11-20 | 2010-11-15 | Parenteral formulations of gemcitabine derivatives |
| US13/121,660 US20110281815A1 (en) | 2009-11-20 | 2010-11-15 | Parental formulations of gemcitabine derivatives |
| EP10831847A EP2501364A4 (en) | 2009-11-20 | 2010-11-15 | Parenteral formulations of gemcitabine derivatives |
| BR112012011784A BR112012011784A2 (en) | 2009-11-20 | 2010-11-15 | "parenteral formulations of gemcitabine derivatives". |
| CA2778432A CA2778432A1 (en) | 2009-11-20 | 2010-11-15 | Parenteral formulations of gemcitabine derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US26299909P | 2009-11-20 | 2009-11-20 | |
| US61/262,999 | 2009-11-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011062503A1 true WO2011062503A1 (en) | 2011-05-26 |
Family
ID=43467050
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/NO2010/000417 WO2011062503A1 (en) | 2009-11-20 | 2010-11-15 | Parenteral formulations of gemcitabine derivatives |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20110281815A1 (en) |
| EP (1) | EP2501364A4 (en) |
| JP (1) | JP2013511516A (en) |
| KR (1) | KR20120086729A (en) |
| CN (1) | CN102740833A (en) |
| AU (1) | AU2010322516A1 (en) |
| BR (1) | BR112012011784A2 (en) |
| CA (1) | CA2778432A1 (en) |
| GB (1) | GB201019703D0 (en) |
| MX (1) | MX2012005677A (en) |
| RU (1) | RU2012125350A (en) |
| TW (1) | TW201124425A (en) |
| WO (1) | WO2011062503A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102600077A (en) * | 2012-03-29 | 2012-07-25 | 江苏豪森药业股份有限公司 | Gemcitabine or gemcitabine salt nano-emulsion injecta and preparation method thereof |
| EP2591770A2 (en) | 2011-11-14 | 2013-05-15 | Silenseed Ltd | Compositions for siRNA delivery and methods of manufacturing and using same |
| US8889642B2 (en) | 2012-04-19 | 2014-11-18 | Silenseed Ltd. | Methods and compositions for RNAi-based cancer treatment |
| US8956613B2 (en) | 2012-11-13 | 2015-02-17 | BoYen Therapeutics, Inc. | Gemcitabine prodrugs and uses thereof |
| US9006199B2 (en) | 2011-11-14 | 2015-04-14 | Silenseed Ltd. | Methods and compositions for treating prostate cancer |
| CN106470672A (en) * | 2014-06-25 | 2017-03-01 | 努卡那生物医药有限责任公司 | Preparation including gemcitabine prodrug |
| WO2018216014A1 (en) | 2017-05-24 | 2018-11-29 | Silenseed Ltd. | Compositions and methods for cancer immunotherapy |
| US10463684B2 (en) | 2014-01-29 | 2019-11-05 | Board Of Regents, The Uneversety Of Texas System | Nucleobase analogue derivatives and their applications |
| US10662213B2 (en) | 2014-06-25 | 2020-05-26 | NuCana plc | Gemcitabine prodrugs |
| CN111249252A (en) * | 2020-03-08 | 2020-06-09 | 中国医学科学院医药生物技术研究所 | Albumin nanoparticle composition and preparation method thereof |
| EP4248949A1 (en) | 2022-03-21 | 2023-09-27 | R.G.C.C. Holdings AG | Liposomal compositions |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013009701A2 (en) | 2011-07-08 | 2013-01-17 | The University Of North Carolina At Chapel Hill | Metal bisphosphonate nanoparticles for anti-cancer therapy and imaging and for treating bone disorders |
| US10517822B2 (en) | 2013-11-06 | 2019-12-31 | The University Of Chicago | Nanoscale carriers for the delivery or co-delivery of chemotherapeutics, nucleic acids and photosensitizers |
| EP3138555B1 (en) | 2014-04-30 | 2020-10-28 | FUJIFILM Corporation | Liposome composition and production method therefor |
| JP7090034B2 (en) | 2016-05-20 | 2022-06-23 | ザ ユニバーシティ オブ シカゴ | Nanoparticles for chemotherapy, targeted therapy, photodynamic therapy, immunotherapy and any combination thereof |
| WO2019028250A1 (en) | 2017-08-02 | 2019-02-07 | The University Of Chicago | Nanoscale metal-organic layers and metal-organic nanoplates for x-ray induced photodynamic therapy, radiotherapy, rodiodynamic therapy, chemotherapy, immunotherapy, and any combination thereof |
| CN111107842B (en) * | 2017-09-22 | 2021-10-08 | 杭州景杰生物科技股份有限公司 | Capecitabine polymer-lipid hybrid nanoparticles utilizing micro-mixing and capecitabine amphiphilic properties |
| JP7364561B2 (en) * | 2018-06-20 | 2023-10-18 | 富士フイルム株式会社 | Combination drug containing a liposome composition containing gemcitabine and an immune checkpoint inhibitor |
| CN109998996B (en) * | 2019-05-05 | 2021-02-26 | 中国医学科学院医药生物技术研究所 | Lipid composition and method for improving antitumor activity of drug |
| CN112898277B (en) * | 2019-11-19 | 2022-04-22 | 扬子江药业集团有限公司 | Preparation method of afatinib intermediate |
| CN113307824B (en) * | 2021-04-26 | 2022-05-27 | 浙江大学 | Amphiphilic material and application thereof in preparation of liposome |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997005154A1 (en) * | 1995-07-25 | 1997-02-13 | Norsk Hydro Asa | Improved therapeutic agents |
| WO1998032762A1 (en) * | 1997-01-24 | 1998-07-30 | Norsk Hydro Asa | Gemcitabine derivatives |
| US20020156062A1 (en) * | 2001-04-11 | 2002-10-24 | Boch Ronald Erwin | Drug delivery system for hydrophobic drugs |
| WO2004017944A1 (en) * | 2002-08-23 | 2004-03-04 | Neopharm, Inc. | Liposomal gemcitabine compositions for better drug delivery |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5785976A (en) * | 1993-03-05 | 1998-07-28 | Pharmacia & Upjohn Ab | Solid lipid particles, particles of bioactive agents and methods for the manufacture and use thereof |
| WO2010039039A1 (en) * | 2008-10-03 | 2010-04-08 | Clavis Pharma Asa | Oral formulations of gemcitabine derivatives |
-
2010
- 2010-11-15 JP JP2012539841A patent/JP2013511516A/en active Pending
- 2010-11-15 US US13/121,660 patent/US20110281815A1/en not_active Abandoned
- 2010-11-15 CA CA2778432A patent/CA2778432A1/en not_active Abandoned
- 2010-11-15 WO PCT/NO2010/000417 patent/WO2011062503A1/en active Application Filing
- 2010-11-15 BR BR112012011784A patent/BR112012011784A2/en not_active IP Right Cessation
- 2010-11-15 AU AU2010322516A patent/AU2010322516A1/en not_active Abandoned
- 2010-11-15 KR KR1020127015947A patent/KR20120086729A/en not_active Application Discontinuation
- 2010-11-15 CN CN2010800522764A patent/CN102740833A/en active Pending
- 2010-11-15 EP EP10831847A patent/EP2501364A4/en not_active Withdrawn
- 2010-11-15 RU RU2012125350/04A patent/RU2012125350A/en not_active Application Discontinuation
- 2010-11-15 MX MX2012005677A patent/MX2012005677A/en not_active Application Discontinuation
- 2010-11-16 TW TW099139312A patent/TW201124425A/en unknown
- 2010-11-19 GB GBGB1019703.6A patent/GB201019703D0/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997005154A1 (en) * | 1995-07-25 | 1997-02-13 | Norsk Hydro Asa | Improved therapeutic agents |
| WO1998032762A1 (en) * | 1997-01-24 | 1998-07-30 | Norsk Hydro Asa | Gemcitabine derivatives |
| US20020156062A1 (en) * | 2001-04-11 | 2002-10-24 | Boch Ronald Erwin | Drug delivery system for hydrophobic drugs |
| WO2004017944A1 (en) * | 2002-08-23 | 2004-03-04 | Neopharm, Inc. | Liposomal gemcitabine compositions for better drug delivery |
Non-Patent Citations (2)
| Title |
|---|
| CANCER CHEMOTHER. PHARMACOL., vol. 61, 2008, pages 395 - 405, XP008155689 * |
| See also references of EP2501364A4 * |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2591770A2 (en) | 2011-11-14 | 2013-05-15 | Silenseed Ltd | Compositions for siRNA delivery and methods of manufacturing and using same |
| US9006199B2 (en) | 2011-11-14 | 2015-04-14 | Silenseed Ltd. | Methods and compositions for treating prostate cancer |
| US9764035B2 (en) | 2011-11-14 | 2017-09-19 | Silenseed Ltd. | Methods and compositions for treating prostate cancer |
| CN102600077B (en) * | 2012-03-29 | 2013-06-05 | 江苏豪森药业股份有限公司 | Gemcitabine or gemcitabine salt nano-emulsion injecta and preparation method thereof |
| CN102600077A (en) * | 2012-03-29 | 2012-07-25 | 江苏豪森药业股份有限公司 | Gemcitabine or gemcitabine salt nano-emulsion injecta and preparation method thereof |
| US8889642B2 (en) | 2012-04-19 | 2014-11-18 | Silenseed Ltd. | Methods and compositions for RNAi-based cancer treatment |
| US9080173B2 (en) | 2012-04-19 | 2015-07-14 | Silenseed Ltd. | Methods and compositions for RNAi-based cancer treatment |
| US9890189B2 (en) | 2012-11-13 | 2018-02-13 | BoYen Therapeutics, Inc. | Gemcitabine prodrugs and uses thereof |
| US8956613B2 (en) | 2012-11-13 | 2015-02-17 | BoYen Therapeutics, Inc. | Gemcitabine prodrugs and uses thereof |
| US9540410B2 (en) | 2012-11-13 | 2017-01-10 | BoYen Therapeutics, Inc. | Gemcitabine prodrugs and uses thereof |
| US10463684B2 (en) | 2014-01-29 | 2019-11-05 | Board Of Regents, The Uneversety Of Texas System | Nucleobase analogue derivatives and their applications |
| US11883423B2 (en) | 2014-01-29 | 2024-01-30 | Board Of Regents, The University Of Texas System | Nucleobase analogue derivatives and their applications |
| US11219633B2 (en) | 2014-01-29 | 2022-01-11 | Board Of Regents, The University Of Texas System | Nucleobase analogue derivatives and their applications |
| US11707477B2 (en) | 2014-06-25 | 2023-07-25 | NuCana plc | Formulation comprising a gemcitabine-prodrug |
| CN106470672A (en) * | 2014-06-25 | 2017-03-01 | 努卡那生物医药有限责任公司 | Preparation including gemcitabine prodrug |
| US10662213B2 (en) | 2014-06-25 | 2020-05-26 | NuCana plc | Gemcitabine prodrugs |
| US10786523B2 (en) | 2014-06-25 | 2020-09-29 | NuCana plc | Formulation comprising a gemcitabine-prodrug |
| US11040051B2 (en) | 2014-06-25 | 2021-06-22 | NuCana plc | Formulation comprising a gemcitabine-prodrug |
| US11629164B2 (en) | 2014-06-25 | 2023-04-18 | NuCana plc | Gemcitabine prodrugs |
| WO2018216014A1 (en) | 2017-05-24 | 2018-11-29 | Silenseed Ltd. | Compositions and methods for cancer immunotherapy |
| CN111249252A (en) * | 2020-03-08 | 2020-06-09 | 中国医学科学院医药生物技术研究所 | Albumin nanoparticle composition and preparation method thereof |
| CN111249252B (en) * | 2020-03-08 | 2021-09-14 | 中国医学科学院医药生物技术研究所 | Albumin nanoparticle composition and preparation method thereof |
| EP4248949A1 (en) | 2022-03-21 | 2023-09-27 | R.G.C.C. Holdings AG | Liposomal compositions |
| WO2023180183A1 (en) | 2022-03-21 | 2023-09-28 | R.G.C.C. Holdings AG | Liposomal compositions |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201124425A (en) | 2011-07-16 |
| AU2010322516A1 (en) | 2012-05-17 |
| CA2778432A1 (en) | 2011-05-26 |
| US20110281815A1 (en) | 2011-11-17 |
| EP2501364A1 (en) | 2012-09-26 |
| CN102740833A (en) | 2012-10-17 |
| JP2013511516A (en) | 2013-04-04 |
| RU2012125350A (en) | 2013-12-27 |
| KR20120086729A (en) | 2012-08-03 |
| BR112012011784A2 (en) | 2019-09-24 |
| EP2501364A4 (en) | 2012-10-24 |
| GB201019703D0 (en) | 2011-01-05 |
| MX2012005677A (en) | 2012-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20110281815A1 (en) | Parental formulations of gemcitabine derivatives | |
| ES2199338T3 (en) | PHARMACEUTICAL COMPOSITIONS IN EMULSION, CONTAINING (3'-DESOXI-3'-OXO-MEBMT) 1- (VAL) 2-CYCLOSPORIN. | |
| EP2086513B1 (en) | Submicron nanoparticle of poorly water soluble camptothecin derivatives and process for preparation thereof | |
| KR101890503B1 (en) | Membrane-adherent self-assembled systems for treatment of ocular disorders | |
| US9700866B2 (en) | Surfactant systems for delivery of organic compounds | |
| US20110275705A1 (en) | Stable injectable oil-in-water docetaxel nanoemulsion | |
| WO2012028101A1 (en) | Liquid compositions of insoluble drugs and preparation methods thereof | |
| ES2316489T3 (en) | PHARMACEUTICAL COMPOSITIONS OF FENRETINIDA THAT HAVE INCREASED BIODISPONIBILITY AND METHODS OF THE SAME USE. | |
| JPWO2006098241A1 (en) | Pharmaceutical composition containing poorly water-soluble drug | |
| US8912162B2 (en) | Parenteral formulations of elacytarabine derivatives | |
| KR20110056042A (en) | Nano particles for tumor-targeting and processes for the preparation thereof | |
| EP3138557A1 (en) | Liposome composition and method for producing same | |
| US20050049209A1 (en) | Pharmaceutical compositions for delivering macrolides | |
| US8481589B2 (en) | Taxoid-based compositions | |
| WO2019186444A1 (en) | Oral liquid formulations of abiraterone | |
| EP4417204A1 (en) | Composition containing antitumor drug, and preparation method therefor and use thereof | |
| WO2017097197A1 (en) | Moexitecan pharmaceutical composition | |
| US20240366521A1 (en) | Nanoparticles and methods of use | |
| WO2024155728A1 (en) | Nanoparticles and methods of use | |
| CN111249252A (en) | Albumin nanoparticle composition and preparation method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080052276.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10831847 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012539841 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010322516 Country of ref document: AU Ref document number: 2778432 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010831847 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2012/005677 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2010322516 Country of ref document: AU Date of ref document: 20101115 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20127015947 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012125350 Country of ref document: RU |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012011784 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012011784 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120517 |